
IL−6レセプター・IL−6直結融合蛋白質
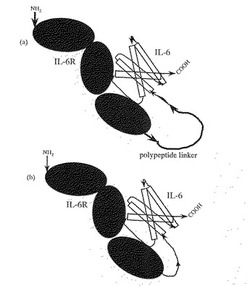
【課題】IL−6RとIL−6がリンカーを介することなく直接に結合しているIL−6R・IL−6融合蛋白質等を提供することを目的とするものである。
【解決手段】IL−6レセプターを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質
【解決手段】IL−6レセプターを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、リンカー配列を介さずに直結されたインターロイキン−6レセプター(以下IL−6Rと略す)・インターロイキン−6(以下IL−6と略す)融合蛋白質、該融合蛋白質をコードする遺伝子、該遺伝子を含有する発現ベクターで形質転換された宿主、該宿主を培養する方法、該宿主の培養物から該融合蛋白質を精製する方法、該融合蛋白質を含むことを特徴とする新規な造血幹細胞のex vivo増幅剤、及び該融合蛋白質を主成分として含む新規な血小板増多剤に関するものである。
【背景技術】
【0002】
IL−6タイプのサイトカインに属するIL−6、IL−11、Ciliary neurotropic factor、Leukemia inhibitory factor、oncostatin−M、Cardiotropin−1は、いずれも、少なくとも1つはシグナル伝達蛋白質gp130を含むレセプター複合体を介してシグナルを伝えることが知られている。IL−6を例にとると、IL−6はIL−6Rに結合し、その結果生じたIL−6・IL−6R複合体がgp130に結合する。
【0003】
造血系はIL−6が重要な役割を示す生体防御系のひとつである。造血系の細胞におけるIL−6Rの発現様式はヒトとマウスでは異なる。メチルセルロース培地上で顆粒球・マクロファージコロニー、赤芽球コロニー、巨核球コロニー、及び混合コロニーを形成することができるヒトの未分化な造血前駆細胞は、十分な数のIL−6Rを発現していない(Tajimaら、J.Exp.Med.184、1996年参照)。したがって、ヒトの未分化な造血前駆細胞はIL−6にはほとんど反応しないが、IL−6・IL−6R複合体には強く反応する。一方、マウスの未分化な造血前駆細胞は十分な数のIL−6Rを発現している。さらに、中畑と本発明者らはヒトの巨核球の前駆細胞は十分な数のIL−6Rを発現していなことを発見した(平成9年特許願第325847号)。これらの知見は、IL−6をマウスに投与すると血小板数の有意な増多が観察されるが、IL−6をヒトに投与すると効果が限定されることと合致する。
【0004】
IL−6と可溶性IL−6Rの結合定数は5x10−9Mであることが報告されている(Yasukawaら、J.Biochem.108巻、673頁、1990年参照)。これは200ng/ml(1x10−8M)のIL−6(分子量2万)と500ng/ml(1x10−8M)の可溶性IL−6R(分子量5万)を混合すると、半数の分子が単体で存在することを意味する。実際、IL−6Rを発現していない細胞に十分に作用させるには1000ng/ml以上の可溶性IL−6Rを必要とする。
【0005】
遺伝子工学を用いると、天然ではそれぞれ別の蛋白質として存在する2種類の蛋白質を1本のポリペプチド鎖から成る融合蛋白質として発現させることができる。互いに結合する性質をもつ2種類の蛋白質を融合蛋白質として発現させた場合、該2種類の蛋白質が融合された状態でそれぞれ本来の構造(生理活性を発現しうる構造)をとり得れば両者の結合は強固となり、解離は融合されていない場合に比較して起こりにくくなることが考えられる。
【0006】
融合蛋白質において2種類の蛋白質が本来の構造をとるためには、2種類の蛋白質間に立体障害が起こらないことが必要である。更には、融合させた本来の構造を有する2種類の蛋白質が互いに結合できる程度の自由度を有していることも必要である。このため従来は、主にグリシン残基やセリン残基のような自由度の高いアミノ酸残基を5〜20個つないでリンカーとし、リンカーを介して蛋白質を融合するのは普通である。このような、融合される蛋白質には含まれていない、本来無関係のリンカーを介して間接的に融合することで、2種類の蛋白質間に立体障害を生じさせず、かつ、互いに結合可能となる程度の自由度を与えることができる。
【0007】
例えば、互いに結合する性質をもつ抗体のH鎖のV領域とL鎖のV領域を融合蛋白質として発現させる場合は、GGGGSGGGGSGGGGS(Gはグリシン残基、Sはセリン残基)がリンカー配列として報告されている(Houstonら、Proc.Natl.Acad.Sci.USA、85、5879頁、1988年参照)。
【0008】
互いに結合する性質をもつIL−6RとIL−6についても、最近になってリンカーを介して両者を結合した融合蛋白質(図1(a)はイラスト図)が報告されている。一つはIL−6Rの323番目のアラニン残基のC末端側に配列番号60で示される、RGGGGSGGGGSVEというIL−6及びIL−6Rには含まれていない、本来無関係なリンカーを結合し、更にそのC末端にIL−6を結合した融合蛋白質である(Fisherら、Nature Biotech、15、142頁、1997年参照)。2番目は、IL−6Rの356番目のバリン残基のC末端にEFM(Eはグルタミン酸残基、Fはフェニルアラニン残基、Mはメチオニン残基)というリンカーを結合し、さらにそのC末端側にIL−6が結合した融合蛋白質である(Chebathら、Eur.CytokineNetw.、8、359頁、1997年参照)。また本出願人らも、IL−6Rの344番目のロイシン残基のC末端にSSELV(Lはロイシン残基、Vはバリン酸残基)というリンカーを結合し、更にそのC末端側にIL−6が結合した融合蛋白質を発明している(平成10年特許願第2921号)。
【0009】
ところで、一般的に、異種蛋白質を遺伝子組換えで作製する場合、宿主が天然に発現するプロテアーゼの作用により発現された異種蛋白質が切断されることが指摘されている。従って、IL−6R・IL−6融合蛋白質においても、宿主中で発現された後に宿主のプロテアーゼによる切断作用を受ける可能性がある。特にピキア・パストリス種の酵母は多種のプロテアーゼを発現することが知られているが、前述した報告ではこの点について述べられていない。仮に宿主が発現するプロテアーゼの切断作用によってIL−6R・IL−6融合蛋白質が切断されているのであれば、かかる切断作用に抵抗性のIL−6R・IL−6融合蛋白質を提供することにより、従来に比較してより多量のIL−6R・IL−6融合蛋白質をピキア・パストリス種の酵母や他の宿主の培養液から取得することが可能となる。
【発明の概要】
【発明が解決しようとする課題】
【0010】
融合蛋白質は2種類の蛋白質の結合状態を強固に維持することが可能と考えられるため、IL−6RとIL−6のように、これら2種類の蛋白質が結合した状態でIL−6のシグナル伝達系を形成する蛋白質を医薬品としてヒト等の体内に投与する場合に特に有効と考えられる。
【0011】
融合蛋白質を体内に持続的に投与する医薬品として開発する場合、リンカーは結合される蛋白質には含まれていない、これら蛋白質とは無関係の配列であり、しかも独自の立体構造をとるために上記免疫反応が惹起される可能性が高いことが課題となる。従って、一般的にリンカーはできるだけ短い配列であることが好ましく、更には全くリンカーが存在しないことが最も好ましい。
【0012】
しかし、前述したように融合蛋白質には、2種類の蛋白質間に立体障害等が起こらないこと、及び、融合された本来の構造を有する2種類の蛋白質が互いに結合できる程度の自由度を有していることが必要である。例えばリンカーを使用せずにIL−6RとIL−6を直接融合させる場合、両者の立体障害を起こさず、かつ両者が互いに結合できる程度の自由度を有し得るようにするためには、融合させる蛋白質の順序、N末端側蛋白質のどのアミノ酸残基にどのC末端側アミノ酸残基を結合するか等、決定しなければならない事項が数多くある。
【0013】
IL−6RとIL−6の融合蛋白質については、前記した3種類のリンカーを用いた例しか報告されておらず、リンカーを介して両者が結合したものしか知られていない。図1(a)はリンカーを介して両者が結合した融合蛋白質のイラスト図である。一方、リンカーを介することなく両者が結合されたものについては報告されていない。
【0014】
そこで本発明は、図1(b)に示したような、IL−6RとIL−6がリンカーを介することなく直接に結合しているIL−6R・IL−6融合蛋白質等を提供することを目的とするものである。また本発明は、宿主、特にピキア・パストリス種の酵母が発現するプロテアーゼの切断作用に対して抵抗性を有するIL−6R・IL−6融合蛋白質、及び該融合蛋白質をコードする遺伝子を提供することをも目的とする。
【課題を解決するための手段】
【0015】
上記目的を達成するため、本発明者らはIL−6R・IL−6融合蛋白質について鋭意検討した結果、リンカーを介することなくN末端側にIL−6Rが、C末端側にIL−6が配置された融合蛋白質を完成するに至った。即ち本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ残基の一つが直接に結合していることを特徴とするIL−6R・IL−6融合蛋白質である。また本発明者らは、IL−6のN末端37番目のリジン残基のC末端側がプロテアーゼの切断作用を受けることを見いだし、一次構造中のプロテアーゼ切断部位に変異が挿入され、プロテアーゼに対する抵抗性を有することを特徴とするプロテアーゼ抵抗性のIL−6R・IL−6融合蛋白質、中でも、IL−6RのC末端側に、N末端28番目のアラニン残基から37番目のリジン残基までの10アミノ酸残基が欠失したIL−6が結合した、IL−6R・IL−6融合蛋白質と、これをコードする遺伝子を完成するに至った。
【0016】
また本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子である。
【0017】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母である。
【0018】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス種の酵母を培養し、当該培養物からIL−6R・IL−6融合蛋白質を分泌型蛋白質として採取することを特徴とするIL−6R・IL−6融合蛋白質の製造法である。
【0019】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス種の酵母をメタノールを含まず天然物由来の炭素源を含む培地で培養し、途中でメタノールを添加して培養することを特徴とするIL−6R・IL−6融合蛋白質の製造法である。
【0020】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質を含有する溶液を、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲルろ過クロマトグラフィーの3種類のクロマトグラフィーにかけ、IL−6R・IL−6融合蛋白質を採取することを特徴とするIL−6R・IL−6融合蛋白質の製造法である。
【0021】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質を含むことを特徴とする造血幹細胞のex vivo増幅剤である。
【0022】
そして本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質を主成分として含む血小板増多剤である。以下本発明を詳細に説明する。
【0023】
本発明の融合蛋白質は、そのN末端側にIL−6Rが位置し、C末端側にIL−6が位置し、両蛋白質の間がリンカーを介することなく直接結合されていることを特徴とするものである。前記したように、プロテアーゼに対する抵抗性付与のみを目的とする場合は、両蛋白質の間はポリペプチドリンカーを介して結合しても良いが、投与の際の抗原性低減等をも考慮するのであれば、ポリペプチドリンカーを介することなく直接結合されていることが好ましい。リンカーを用いる場合には、例えば公知のリンカー配列(Fisherら、Nature Biotech、15、142頁、1997年;Chebathら、Eur.Cytokine Netw.、8、359頁、1997年)を使用したり、IL−6Rの344番目のロイシン残基のC末端にセリン残基−セリン残基−グルタミン酸残基−ロイシン残基−バリン酸残基というリンカーを結合し、更にそのC末端側にIL−6を結合させることが例示できる。本発明のIL−6R・IL−6融合蛋白質を構成するIL−6Rは、全長468アミノ酸残基で構成される膜蛋白質で、シグナル領域、細胞外領域、膜貫通領域及び細胞内領域から成る(Yamasakiら、Science、241、825頁、1988年参照)。ヒトIL−6Rの場合、シグナル領域はN末端1番目のメチオニン残基付近から19番目のアラニン残基付近まで、細胞外領域は20番目のロイシン残基付近から358番目のアスパラギン酸残基付近まで、膜貫通領域は359番目のセリン残基付近から386番目のロイシン残基付近まで、細胞内領域はおよそ387番目のアルギニン残基付近から468番目のアルギニン残基付近までと考えられている。細胞外領域はイムノグロブリン様領域とサイトカインレセプター領域に分けられ、イムノグロブリン様領域は20番目のロイシン残基付近から111番目のアスパラギン酸残基付近まで、サイトカインレセプター領域は112番目のバリン残基付近から323番目のアラニン残基付近までと考えられている。
【0024】
IL−6Rにおいて、IL−6との結合に必須なのはサイトカインレセプター領域であり、イムノグロブリン様領域は不要であることが知られている。なおサイトカインレセプター領域は、7つのβシートから構成されるバレル(樽)様の構造体が短い2個つながった構造体である(Yawataら、EMBO J.、12、1705頁、1993年参照)。
【0025】
本発明では、全長のIL−6Rはもちろんのこと、その細胞外領域全体又はサイトカインレセプター領域のいずれか、即ち部分的IL−6Rを用いることもできる。IL−6と結合してそのシグナル伝達系を構成するのはサイトカインレセプター領域であり、細胞外領域は該領域を含む領域だからである。
【0026】
本発明者らの知見によれば、具体的にはIL−6RのN末端として、N末端20番目のロイシン残基、N末端112番目のバリン残基、及びN末端116番目のグルタミン酸残基が例示できる。
【0027】
また本発明者らの知見によれば、具体的にはIL−6RのC末端として、N末端323番目のアラニン残基から361番目のセリン残基までの39個のアミノ酸残基のうちのいずれか一つ、特に好ましくは323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、及び343番目のイソロイシン残基の6個のアミノ酸残基のいずれか一つを用いることが例示できる。この部分的IL−6RのC末端側に、IL−6のN末端とを結合させるのである。なおIL−6RのN末端は、融合蛋白質のシグナル伝達における作用効果を勘案して適宜削除することができる。
【0028】
IL−6レセプター・IL−6融合蛋白質を構成するIL−6は、4つのαヘリックスから構成される全長212アミノ酸残基の分泌型蛋白質で(Hiranoら、Nature,324,731巻、1986年参照)。IL−6が活性を示すためには、これら4つのαヘリックス全てが必要であることが知られている。従って、本発明で使用するIL−6としては、4つのαヘリックスすべてを有するものであれば、特に制限はない。即ち、全長のIL−6はもちろんのこと、例えばN末端やC末端の一部アミノ酸残基が削除された部分的IL−6であってもよい。より具体的には、分泌型IL−6の構造として知られているN末端28番目のアラニン残基又は29番目のプロリン残基から212番目のメチオニン残基までのIL−6を例示することができる。またその他にも、IL−6の発現例(例えばYasukawaら、Biotech.Lett.、12,419頁、1990年)やIL−6R・IL−6融合蛋白質の発現例(Fisherら、Nature Biotech.、15,142頁、1997年)を参照にして、部分的IL−6としていかなる配列のものを使用するか決定することができる。
【0029】
一次構造中のプロテアーゼ切断部位に変異が挿入され、プロテアーゼに対する抵抗性が付与された本発明の融合蛋白質では、プロテアーゼの切断部位が種類毎に異なるため、その一次構造において、どの部分に変異を導入する、即ちどの部分を欠失させ、どの部分に本来存在しないアミノ酸残基を付加し、或いはどの部分のアミノ酸残基を置換してプロテアーゼによる切断作用を受け難くするか、を適宜決定する。置換による変異の導入では、置換により蛋白質分子が本来の立体構造をとらないようにすることにより、プロテアーゼの切断作用を受けない、或いは受け難くする。この場合、目的とするアミノ酸残基を分子サイズの小さいアミノ酸残基であるグリシンやセリンに置換することが好ましい置換として例示できる。欠失による変異の導入では、目的とするアミノ酸残基を削除することでプロテアーゼが切断作用部位を認識できないようにしたり、蛋白質分子が本来の立体構造をとらないようにすることにより、プロテアーゼの切断作用を受けない、或いは受け難いようにする。
【0030】
変異の導入は、遺伝子工学的にIL−6R・IL−6融合蛋白質をコードする遺伝子に対して行う等すれば良い。また前記アミノ酸残基の欠失、アミノ酸残基の挿入、アミノ酸残基の置換のいずれを行っても良いが、操作が比較的容易であり、しかも本来存在しないアミノ酸残基の置換による抗原性の変化等の影響が小さいと考えられることから、プロテアーゼの切断部位近傍の連続する1以上のアミノ酸残基を削除して欠失させることが特に好ましい。
【0031】
より具体的な選択の基準は、本願発明のIL−6R・IL−6融合蛋白質を遺伝子工学的に製造する際に使用する宿主細胞が分泌するプロテアーゼに対する抵抗性を付与することである。即ち、後の実施例に示したように、遺伝子工学的に製造しようとするIL−6R・IL−6融合蛋白質を選定した宿主細胞で発現させ、通常の液体クロマトグラフィーを用いる方法等で部分的に精製する過程でプロテアーゼによる分解産物と思われるピークを採取したり、通常のSDS−PAGEを用いる方法でプロテアーゼによる分解産物と思われるバンドを採取し、N末端のアミノ酸配列を解析することにより、前記選定した宿主細胞が分泌するプロテアーゼによりIL−6R・IL−6融合蛋白質のいずれの部分が切断作用を受けているかを知ることができる。
【0032】
以上のようにしてIL−6R・IL−6融合蛋白質の一次構造におけるプロテアーゼ切断部位を検出したならば、当該切断部位付近のアミノ酸残基を欠失等してプロテアーゼの切断作用を受けない、或いは受け難くすることにより、プロテアーゼに対する抵抗性を有するIL−6R・IL−6融合蛋白質を得ることが可能になる。
前記以外に例えば、医薬品等のようにプロテアーゼ存在下でIL−6R・IL−6融合蛋白質の生物活性を長時間持続させる目的でも、本願発明のプロテアーゼ抵抗性IL−6R・IL−6融合蛋白質は有効である。このようなIL−6R・IL−6融合蛋白質を製造するためには、IL−6R・IL−6融合蛋白質が生物活性を発揮すべき環境に共存するプロテアーゼを取得し、該プロテアーゼにより変異導入前のIL−6R・IL−6融合蛋白質においてどの部位が切断作用を受けるかを検出し、当該部分に前記のように変異させれば良い。
【0033】
前記のようにして変異を導入したIL−6R・IL−6融合蛋白質については、変異導入後、生物活性を維持していることを確認することが好ましい。この目的のためには、変異を導入したIL−6R・IL−6融合蛋白質を調製し、その生物活性を、例えば後の実施例に示したようなBAF130細胞株を用いる確認に供することが例示できる。この確認により変異を導入したIL−6R・IL−6融合蛋白質に生物活性が認められなかった場合には、別の変異を導入して同様の確認を行う操作を繰り返し行えば良い。
【0034】
引き続き、変異を導入したIL−6R・IL−6融合蛋白質については、プロテアーゼ抵抗性であることを確認することが好ましい。この目的のためには、変異を導入したIL−6R・IL−6融合蛋白質を一定時間プロテアーゼと共存させたものをSDS−PAGEとウエスタンブロッティングを組み合わせた解析に供する等すれば良い。この結果プロテアーゼに対する抵抗性が認められなかった場合には、別の変異を導入して同様の確認を行う操作を繰り返し行えば良い。なお、宿主細胞が分泌するプロテアーゼに対する抵抗性を付与したIL−6R・IL−6融合蛋白質においては、該宿主細胞を用いてこれを調製した後、細胞培養液について前述のような確認操作を行えば良い。
【0035】
本発明のIL−6R・IL−6融合蛋白質は、これをコードする遺伝子を用いて遺伝子組換え操作を行うことにより容易に作製することができる。IL−6R又はIL−6をコードする遺伝子は既に単離されており、その塩基配列もよく知られている。従って、本発明の融合蛋白質を作製する際には、融合蛋白質を構成するIL−6RとIL−6のアミノ酸配列から必要な遺伝子配列を調製し、これを制限酵素を用いて結合させておけば良い。またここで、天然の遺伝子配列を用いる以外にコドンの縮合を勘案し、任意のコドンを同一のアミノ酸残基をコードするが塩基配列の異なるものに置換するなどしても良い。遺伝子組換えにより宿主に蛋白質を発現させる場合、特定のコドンを使うと発現率や翻訳率が向上することがあるからである。
【0036】
本発明の融合蛋白質を遺伝子組換えで作製する場合に使用する宿主に特別の制限はなく、従来の報告を参考にしつつ、通常の遺伝子組換え操作で使用されている大腸菌やCHO細胞等に代表される動物細胞を使用することができる(Yasukawaら、J.Biotech.、108,673頁、1990年参照)。中でも、本実施例に示したピキア・パストリス種の酵母(Pichia pastoris)は、メタノールを唯一の炭素源として生育できる酵母で、CHO細胞等のような動物細胞と比較して安価に培養できることから特に好ましい宿主として例示できる。
【0037】
IL−6R・IL−6融合蛋白質を遺伝子工学的に製造する際に好適な宿主細胞であるピキア・パストリス種の酵母(Pichia pastoris)が分泌するプロテアーゼに抵抗性であるIL−6R・IL−6融合蛋白質として、IL−6Rと少なくともN末端28番目のアラニン残基からN末端37番目のリジン残基までが欠失したIL−6との融合蛋白質を例示することができる。プロテアーゼ抵抗性を有するIL−6R・IL−6融合蛋白質をコードする遺伝子を調製し、これを組み込んで発現ベクターを調製し、宿主細胞を形質転換すれば、形質転換宿主細胞を培養することにより本願発明のプロテアーゼ抵抗性を有するIL−6R・IL−6融合蛋白質を大量に製造することができる。前述のように宿主細胞が分泌するプロテアーゼに対する抵抗性を付与したIL−6R・IL−6融合蛋白質では、宿主細胞で発現された後に該プロテアーゼにより切断されない、或いは切断され難いため、抵抗性を有していないIL−6R・IL−6融合蛋白質に比較して、より大量の蛋白質を得ることが可能となる。
【0038】
前述した、本発明の融合蛋白質を遺伝子組換えで作製するための遺伝子は、宿主に導入する(形質転換する)際には、発現ベクターの中に組み込んで使用する。発現ベクターは当該遺伝子の他に、発現制御遺伝子や形質転換された宿主の選択のための指標となる遺伝子等を組み込むが、かかる遺伝子は使用する宿主との関係で適宜選択して使用すれば良い。例えばピキア・パストリス種の酵母を宿主として使用するのであれば、その染色体DNA中にIL−6R・IL−6融合蛋白質をコードする遺伝子を導入するためのアルコールオキシダーゼ遺伝子の上流配列と下流配列、選択の指標となるヒスチジン合成遺伝子そして発現制御のためのアルコールオキシダーゼ遺伝子のプロモーター配列等が、大腸菌を宿主として使用するのであれば選択の指標となるアンピシリン耐性遺伝子や発現制御のためのLacプロモーター/オペレーター配列等が例示できる。なお、市販の発現ベクター(例えばpPIC9、ピキア・パストリス種の酵母用の発現ベクター、インビトロジェン社製)に本発明の遺伝子を導入して使用することもできる。
【0039】
本発明において、好ましくピキア・パストリス種の酵母をIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターで形質転換して融合蛋白質を作製する場合は、発現ベクター中に、融合蛋白質のシグナルペプチドとしてIL−6R本来のシグナルペプチドやα因子のシグナルぺプチドを組み込むことが好ましく、特に高発現を実現できることからα因子のシグナルぺプチドを用いることが好ましい。
【0040】
前述の形質転換された宿主を適当な条件下で培養し、発現ベクター中の発現制御遺伝子との関係で必要に応じて融合蛋白質の発現を誘導すれば融合蛋白質を作製することができる。本発明において、好ましくピキア・パストリス種の酵母を宿主とする場合は、ジャーファーメンターを用いる方法が例示できる。より具体的には、100mLの培地が仕込まれた500mL容量の振とうフラスコに、あらかじめ作製しておいたIL−6R・IL−6融合蛋白質を発現するピキア・パストリス種の酵母の20%グリセロール凍結菌株を接種し、28〜30℃で20時間振とう培養する。次に6〜9Lの培地が仕込まれた16L容量のジャーファーメンターに上記培養液100mLを接種し、28〜30℃にて通気撹拌培養を開始する。培養中の酵母の状態をモニタリングするために、OD600、pH、溶存酸素濃度、撹拌速度、温度をモニタリングしてかつ制御することが好ましい。培地は天然物由来の炭素源を含むものであれば特に種別は問わないが、具体的組成は実施例を参考にすればよい。また、該培養を実施するためのジャーファーメンターとしては市販の装置を使用することができる。培養開始後、培養液中のグリセロールが枯渇した段階でメタノールを添加すればよい。グリセロールの枯渇は溶存酸素をモニタリングすることにより知ることができる。メタノールの添加は枯渇後5時間以内に行うことが望ましい。メタノールの添加量は多すぎると酵母に毒性を示すが、少なすぎるとアルコールオキシダーゼ遺伝子のプロモーター配列が十分に機能しないことを勘案すると、0.5%以上5%以下(質量/容量)が好ましい。
【0041】
培養により作製された融合蛋白質は、適当な方法で培地等から取得することが可能である。宿主として大腸菌を用いた場合は、発現された融合蛋白質は大腸菌内に不溶性塊として蓄積されることから、菌体を破砕後、適当な条件下でリフォールディングや精製操作を行えばよい。好ましくピキア・パストリス種の酵母を宿主として用いた場合には、その培養上清から融合蛋白質を精製して取得することが可能である。精製原料は融合蛋白質を含む溶液であれば特に制限はなく、例えば融合蛋白質を含むピキア・パストリス種の酵母の培養液を例示することができる。該溶液はそのまま用いてもよく、またはそれらを緩衝液あるいは純水により希釈したり、限外ろ過膜あるいは硫酸アンモニウム等により濃縮した後に用いてもよい。精製操作としては、液体クロマトグラフィー操作を例示することができる。好ましくはイオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲルろ過クロマトグラフィーの3種類のクロマトグラフィーの組み合わせを例示することができる。
【0042】
ピキア・パストリス種の酵母の培養上清は容量が大きいため、最初にイオン交換クロマトグラフィーにかけることが好ましい。イオン交換クロマトグラフィーは陽イオン交換クロマトグラフィーと陰イオン交換クロマトグラフィーに分かれるが、除蛋白質効率を考慮するして適宜選択することができる。前者の例としては陽イオン交換基としてSPを、後者の例としては陰イオン交換基としてDEAEを例示することができる。ここで流速を毎分100ml以上に上げられることと、1μm以下のフィルターを通していないサンプルを添加することができることを勘案すると、後の実施例に示すように、陽イオンクロマトグラフィーとしては吸着流動床Streamline SP C−50カラム(アマシャムファルマシア社製)を好ましく例示することができる。上記方法により得た融合蛋白質を含む画分は、次に疎水性クロマトグラフィーにかけ、さらに融合蛋白質の純度がより高い画分を得ることができる。この画分は、後の実施例に示すように、例えば陽イオン交換クロマトグラフィーで濃縮することにより、ゲルろ過クロマトグラフィーを効率よく行うことができる。
【0043】
本発明者らは、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質の幹細胞増幅効果について鋭意検討した結果、後の実施例に示すように、該融合蛋白質と幹細胞因子(SCF)存在下でCD34陽性細胞をメチルセルロースプレート上で培養すると、従来報告されているIL−6、IL−6R、及びSCF存在下で培養したときよりも、著しくコロニー形成能が高くなることを発見した。本発明はこれらの新たに見出されたIL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質が有する造血幹細胞のex vivo増幅に基づきなされたものであり、該融合蛋白質を主成分として含む造血幹細胞のex vivo増幅剤又は造血幹細胞のex vivo増幅法である。
【0044】
本発明の造血幹細胞のex vivo増幅剤は、本発明の融合蛋白質単独でも若干の効果を示すので実用価値はあるが、SCF、FLK2リガンド等のチロシンキナーゼを刺激するサイトカイン類のうちいずれかひとつを、特に好ましくはSCFを添加すると効果は顕著なものとなる。更にインターロイキン−3(IL−3)や血小板増殖因子(TPO)等を加えてもよい。造血幹細胞は臍帯血、末梢血、又は骨髄からCD34選択により、造血幹細胞を含む画分として得ることができる。本発明の融合蛋白質は他のサイトカインを入れた個々の容器を含むキットとして提供することもできるし、または単一の容器の中の混合物として提供することもできる。造血幹細胞のex vivo増幅は、例えばプラスチックバッグのような容器の中で、造血幹細胞を含む画分を、本発明の融合蛋白質と例えばSCFを添加した無血清培地で37℃で1〜3週間培養すればよい。増幅された造血幹細胞を患者に戻す場合は、現行の末梢血幹細胞移植と同じ方法を採用すればよい。
【0045】
本発明者らは、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質の血小板増多効果について鋭意検討した結果、後の実施例に示すように、該融合蛋白質をマウスに投与すると血小板が有意に増多することと、該融合蛋白質を予め制癌剤を投与したマウスに投与すると血小板回復が有意に早くなることを発見した。本発明はこれらの新たに見出されたIL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質が有する血小板の増多あるいは回復に基づきなされたものであり、該融合蛋白質を主成分として含む血小板増多剤又は血小板増多方法である。本発明の血小板増多剤は、好ましくは非経口投与により、例えば静脈内投与、筋肉内投与、経皮投与等により投与することが好ましい。投与量は、血小板の不足症状を示す疾患の種類、患者の状態等により適宜選択されるが、一般に1〜500μg/kg/日の範囲であり、血小板の増多の様子により継続的に投与すれば良い。本発明の血小板増多剤は、常用の賦形剤、例えば生理食塩水、ブドウ糖液、マンニトール、メチルセルロース、ゼラチン、ヒト血清アルブミン等の賦活剤と混合して製剤化することができる。また本発明の血小板増多剤は凍結乾燥品とすることも可能であり、凍結乾燥した場合には使用直前に生理食塩水、ブドウ糖液、リンゲル液等の等張液により再溶解すれば良い。
【発明の効果】
【0046】
本発明で提供されるリンカー配列をもたないIL−6R・IL−6融合蛋白質は、従来報告されているリンカー配列を有するIL−6R・IL−6融合蛋白質に比べ、高い薬効と抗原性の著しい低下が期待される。このことは本融合蛋白質は造血領域における新規治療薬として大きな意義をもち、特に造血幹細胞のexvivo増幅剤や血小板増多剤としての開発が期待される。
【0047】
宿主細胞が分泌するプロテーゼに対する抵抗性を付与したものでは、遺伝子工学的に宿主細胞を形質転換して製造する工程で、該プロテアーゼによる切断を受ない等の理由により、従来のものに比較してより大量に製造することが可能となる。また前記プロテアーゼが共存した状態で精製工程に供しても切断されない等のために最終的には収量を増加することができ、しかもプロテアーゼを操作に先立って失活させるといった操作を省略することが可能であるため、精製操作を簡便化できるという効果もある。更には最終精製物中にプロテアーゼによって切断された分子が混じることがないため、より均一な精製品とすることも可能となる。
【0048】
上記効果に加えて、生物活性を発揮させようとする環境に共存が予想されるプロテアーゼに対する抵抗性を有するものでは、生物活性を持続して発揮するという効果を予想することができる。
【図面の簡単な説明】
【0049】
【図1】図1は、(a)リンカー配列を介してIL−6RとIL−6が融合したIL−6R・IL−6融合蛋白質と(b)リンカーを介さずにIL−6RとIL−6が直接融合した本発明のIL−6R・IL−6融合蛋白質のイラストを示す図である。
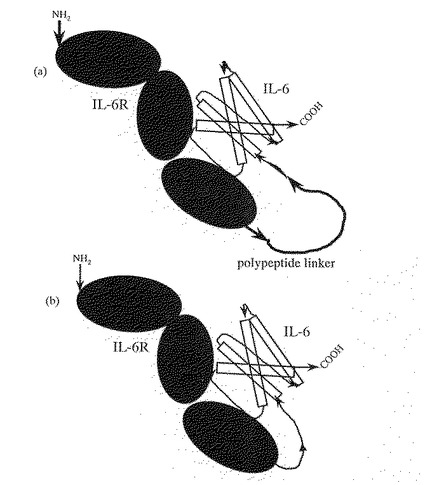
【図2】図2は、実施例1で作製したプラスミドpBS6R6Sの構造を示す図である。ampはアンピシリン耐性遺伝子を、oriは転写開始部位を、IL−6はIL−6遺伝子を、IL−6RはIL−6R遺伝子をそれぞれ示す。
【図3】図3は、図2に示したプラスミドpBS6R6Sに、実施例2で示す方法でオリゴヌクレオチド2種をアニールさせたものを挿入する手順を示す図である。
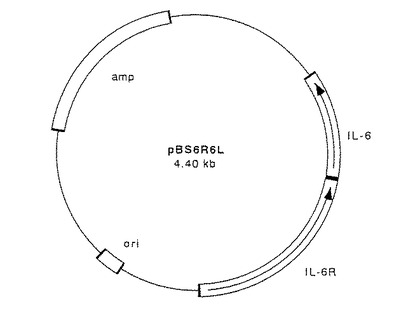
【図4】図4は、実施例1で作製したプラスミドpBS6R6Lの構造を示す図である。ampはアンピシリン耐性遺伝子を、oriは転写開始部位を、IL−6はIL−6遺伝子を、IL−6RはIL−6R遺伝子をそれぞれ示す。
【図5】図5は、図4に示したプラスミドpBS6R6Lに、実施例2で示す方法でオリゴヌクレオチド2種をアニールさせたものを挿入する手順を示す図である。
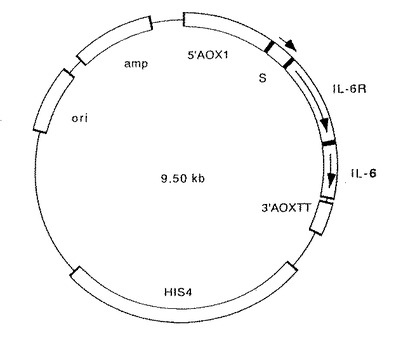
【図6】図6は、実施例2で作製した誘導体333AΔAの発現プラスミドの構造を示す図である。図中、ampはアンピシリン耐性遺伝子を、oriは転写開始部位を、HIS4はヒスチジン合成遺伝子を、3’AOXTTはターミネーターを、IL−6はIL−6遺伝子を、IL−6RはIL−6レセプター遺伝子を、Sはシグナル配列をコードする遺伝子を、5’AOX1はプロモーター配列を含むアルコールオキシダーゼ遺伝子の上流領域をそれぞれ示す。
【図7】図7は、各融合蛋白質の誘導体ごとに、(A)IL−6R領域のC末端のアミノ酸残基の種類と番号、(B)リンカーのアミノ酸配列(「−」はリンカーがないことを示す。)、(C)IL−6領域のN末端のアミノ酸残基の種類と番号、(D)実施例4で得られた216種類の培養上清それぞれに対し、実施例5に示す方法で得られた生物活性値平均値をそれぞれ示した図である。
【図8】図8は、実施例6に示す方法で培養を行ったときの経時変化を示す図である。図中、MeOH conc.はメタノール濃度(単位は%(質量/容量))を、EIAはサンドイッチイムノアッセーを、Bio Assayは生物活性をそれぞれ示す。
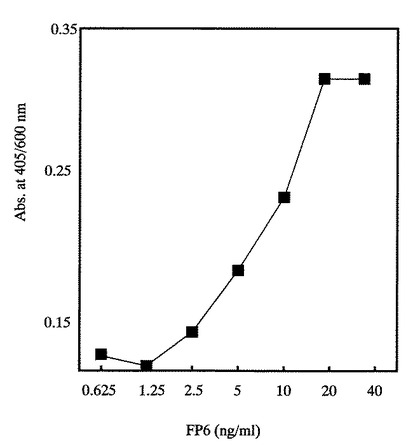
【図9】図9は、実施例8に示す方法でFP6の生物活性を求めた図である。
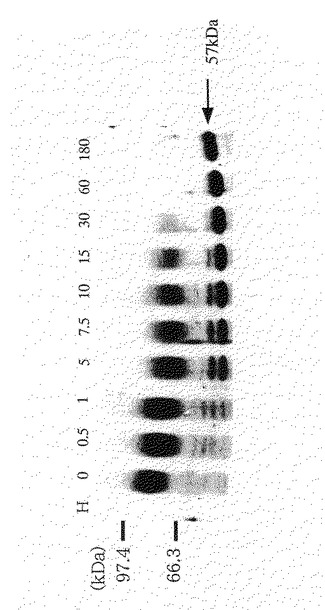
【図10】図10は、実施例10に示す方法でエンドグリコシダーゼHで0〜180分処理したFP6をSDSポリアクリルアミドゲル電気泳動に供したときの結果を示す図である。図中、Hと示したレーンはエンドグリコシダーゼHのみを、0から180の数字を付したレーンはそれぞれの数字の時間(分)だけエンドグリコシダーゼH処理されたFP6を示す。図中の97.4と66.3の数値を付したバーは、分子量マーカー蛋白質(分子量が97.4kDaと66.3kDa)がそれぞれ検出された位置である。
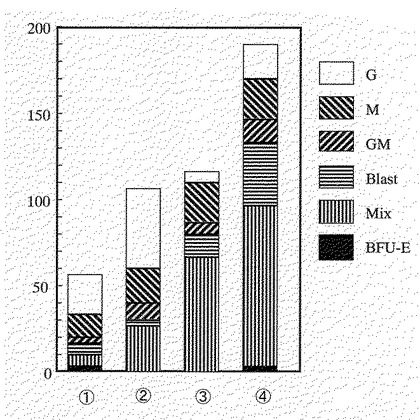
【図11】図11は、実施例11に示す方法で、1 IL−6(100ng/ml)とIL−6R(100ng/ml)の組み合わせ、2 IL−6(100ng/ml)とIL−6R(200ng/ml)の組み合わせ、3 FP6(300ng/ml)、4 FP6(600ng/ml)の500個のCD34陽性細胞に対するコロニー形成能を求めた図である。図中、Gは顆粒球コロニーを、Mはマクロファージコロニーを、GMは顆粒球・マクロファージの混合コロニーを、Blastは芽球コロニーを、Mixは顆粒球・マクロファージ・赤芽球の混合コロニーを、BFU−Eは赤芽球コロニーをそれぞれ示す。
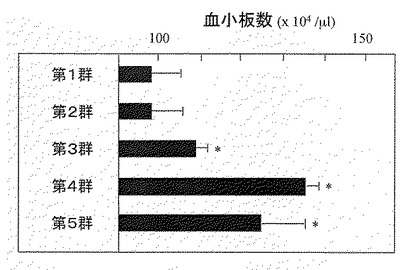
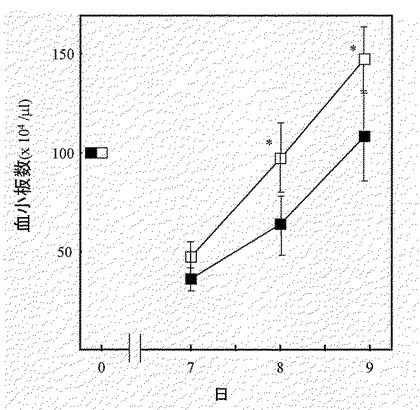
【図12】図12は、実施例12に示す方法でC57BL6マウスに対し、FP6を投与しなかったとき(第1群)と投与したとき(第2−5群)の血小板数の平均値を示す図である。図中、星印は第1群のマウスの血小板数との有意差(P<0.05)を示す。
【図13】図13は、実施例13に示す方法で予め5FUを投与したC57BL6マウスに対し、FP6を投与しなかったとき(第1群、黒塗りの四角)と投与したとき(第2群、白い四角)の7〜9日目の血小板数の平均値を示す図である。図中、星印は同日の第1群のマウスの血小板数との有意差(P<0.05)を示す。
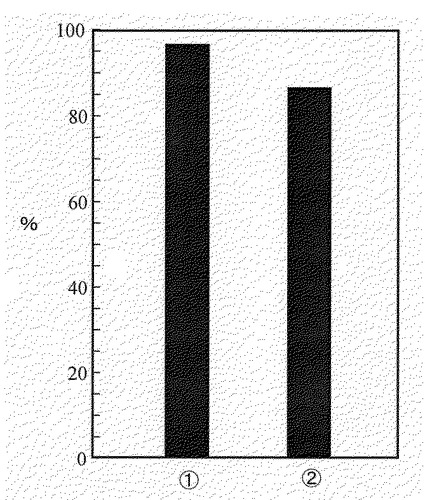
【図14】図14は、実施例14に示す方法で得られた(1)融合蛋白質112VAAを発現するmuts菌9株と(2)融合蛋白質116EAAを発現するmuts菌11に対し、 実施例5に示す方法で得られた生物活性値平均値を、333AΔAを発現するmuts菌11株のうち最も発現量の高かった株であるIII−108株に対する相対値(%)として、それぞれ示した図である。
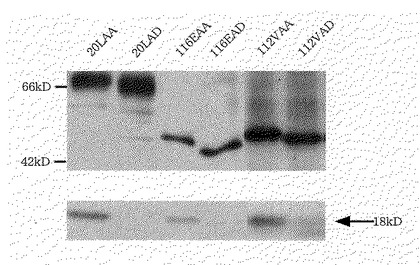
【図15】図15は、(a)20LAA、(b)20LAD、(c)116EAA、(d)116EAD、(e)112VAA、(f)112VADを含む培養上清を実施例8に示す方法でウエスタンブロットしたときの結果を示す図である。
【発明を実施するための形態】
【0050】
以下に、発明を更に詳細に説明するために実施例を示すが、本発明はこれら実施例に限定されるものではない。
【0051】
実施例1 中間体プラスミドの作製各種リンカーをコードするオリゴヌクレオチドを挿入することにより、各種リンカーを介して融合されたIL−6R・IL−6融合蛋白質をコードする遺伝子(cDNA)を作製可能とすべく、中間体プラスミドpBS6R6SとpBS6R6Lを以下の方法で作製した。
【0052】
まず、クローニングベクターであるpBluescript II KS(−)(東洋紡(株)製)を制限酵素KpnIで切断し、クレノウフラグメントで処理した後、ライゲーション反応を行ってKpnIサイトが消失したプラスミドpBSを作製した。
次に、プライマーp6RAB20L(配列番号45)とプライマーp6RF320S(配列番号46)を用いてIL−6R遺伝子(cDNA)を増幅し、XhoIで切断した。これを、予めXhoIとEcoRVで切断したpBSに挿入することにより、プラスミドpBS6Rを得た。
【0053】
次に、プライマーpIL6B2(配列番号47)とプライマーpIL6F(配列番号48)により、IL−6遺伝子(cDNA)を増幅し、Bgl IIとNotIで切断した。これを、予めBgl IIとNotIで切断したプラスミドpBS6Rに挿入することにより、pBS6R6Sを得た。得られたpBS6R6Sの構造を図2に示す。
次に、オリゴマー320S333Ab(配列番号49)と320S333Af(配列番号50)を通常の方法でアニールさせた。これを、予めBgl IIとKpn Iで切断したプラスミドpBS6R6Sに挿入することにより、pBS6R6Lを得た。得られたpBS6R6Lの構造を図4に示す。
【0054】
実施例2 発現プラスミドの作製予めBgl IIとKpnIで切断したpBS6R6Sに、下記のようにそれぞれ2種類のオリゴヌクレオチドをアニールしたアニール配列1〜5をそれぞれ挿入し、IL−6R・IL−6融合蛋白質をコードする遺伝子をインサートにもつプラスミド5種類を作製した。
【0055】
アニール配列1(323AG4SA);センス側は配列番号1のオリゴヌクレオチド、アンチセンス側は配列番号2のオリゴヌクレオチド。
アニール配列2(323AG4S2A);センス側は配列番号3のオリゴヌクレオチド、アンチセンス側は配列番号4のオリゴヌクレオチド。
アニール配列3(334LPA);センス側は配列番号5のオリゴヌクレオチド、アンチセンス側;配列番号6のオリゴヌクレオチド。
アニール配列4(333AΔA);センス側は配列番号7のオリゴヌクレオチド、アンチセンス側;配列番号8のオリゴヌクレオチド。
アニール配列5(323AΔA);センス側は配列番号9のオリゴヌクレオチド、アンチセンス側;配列番号10のオリゴヌクレオチド。
【0056】
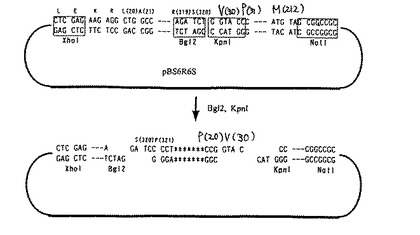
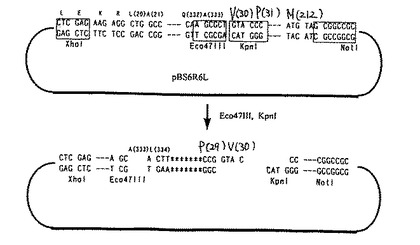
なお、アニール配列1(323AG4SA)は、IL−6RのN末端323番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列2(323AG4S2A)は、IL−6RのN末端323番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列3はIL−6RのN末端334番目のロイシンとIL−6のN末端28番目のアラニンをPというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列4(333AΔA)は、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、そしてアニール配列5(323AΔA)は、IL−6RのN末端323番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものである。
【0057】
次に、予めEco47 IIIとKpnIで切断したpBS6R6Lに、下記のようにそれぞれ2種類のオリゴヌクレオチドをアニールしたアニール配列6〜22をそれぞれ挿入し、IL−6R・IL−6融合蛋白質をコードする遺伝子をインサートにもつプラスミド17を作製した。
【0058】
アニール配列6(333AG4SA);センス側は配列番号11のオリゴヌクレオチド、アンチセンス側;配列番号12のオリゴヌクレオチド。
アニール配列7(333AG4S2A);センス側は配列番号13のオリゴヌクレオチド、アンチセンス側は配列番号14のオリゴヌクレオチド。
アニール配列8(343IG4S2A);センス側は配列番号15のオリゴヌクレオチド、アンチセンス側は配列番号16のオリゴヌクレオチド。
アニール配列9(333AG4S3A);センス側は配列番号17のオリゴヌクレオチド、アンチセンス側は配列番号18のオリゴヌクレオチド。
アニール配列10(333AG2A);センス側は配列番号19のオリゴヌクレオチド、アンチセンス側は配列番号20のオリゴヌクレオチド。
アニール配列11(333AG4A);センス側は配列番号21のオリゴヌクレオチド、アンチセンス側は配列番号22のオリゴヌクレオチド。
アニール配列12(343IG2A);センス側は配列番号23のオリゴヌクレオチド、アンチセンス側は配列番号24のオリゴヌクレオチド。
アニール配列13(343IG4A);1センス側は配列番号25のオリゴヌクレオチド、アンチセンス側は配列番号26のオリゴヌクレオチド。
アニール配列14(361SΔA);センス側は配列番号27のオリゴヌクレオチド、アンチセンス側は配列番号28のオリゴヌクレオチド。
アニール配列15(358DΔA);センス側は配列番号29のオリゴヌクレオチド、アンチセンス側は配列番号30のオリゴヌクレオチド。
アニール配列16(352TΔA);センス側は配列番号31のオリゴヌクレオチド、アンチセンス側は配列番号32のオリゴヌクレオチド。
アニール配列17(346RΔA);センス側は配列番号33のオリゴヌクレオチド、アンチセンス側は配列番号34のオリゴヌクレオチド。
アニール配列18(343IΔA);センス側は配列番号35のオリゴヌクレオチド、アンチセンス側は配列番号36のオリゴヌクレオチド。
アニール配列19(338KΔA);センス側は配列番号37のオリゴヌクレオチド、アンチセンス側は配列番号38のオリゴヌクレオチド。
アニール配列20(335TΔA);センス側は配列番号39のオリゴヌクレオチド、アンチセンス側は配列番号40のオリゴヌクレオチド。
アニール配列21(343IΔP);センス側は配列番号41のオリゴヌクレオチド、アンチセンス側は配列番号42のオリゴヌクレオチド。
アニール配列22(338KΔP);センス側は配列番号43のオリゴヌクレオチド、 アンチセンス側は配列番号44のオリゴヌクレオチド。
【0059】
なお、アニール配列6(333AG4SA)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列7(333AG4S2A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列8(343IG4S2A)はIL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列9(333AG4S3A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列10(333AG2A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列11(333AG4A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列12(343IG2A)はIL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列13(343IG4A)はIL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをGGGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列14(361SΔA)は、IL−6RのN末端361番目のセリンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列15(358DΔA)は、IL−6RのN末端358番目のアスパラギン酸とIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列16(352TΔA)は、IL−6RのN末端352番目のトレオニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列17(346RΔA)は、IL−6RのN末端346番目のアルギニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列18(343IΔA)は、IL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列19(338KΔA)は、IL−6RのN末端338番目のリジンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列20(335TΔA)は、IL−6RのN末端335番目のトレオニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列21(343IΔP)は、IL−6RのN末端343番目のイソロイシンとIL−6のN末端29番目のプロリンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、そしてアニール配列22(338KΔP)は、IL−6RのN末端338番目のリジンとIL−6のN末端29番目のプロリンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものである。
【0060】
pBS6R6L自体がIL−6R・IL−6融合蛋白質をコードする遺伝子をインサートにもつが、該遺伝子により発現する融合蛋白質(334LΔV)は、IL−6RのN末端334番目のロイシンとIL−6のN末端30番目のバリンをリンカーを介さずに直接融合した本発明の融合蛋白質である。
【0061】
最後に、上記方法で作製したプラスミド23種及びpBS6R6LをそれぞれXhoIとNotIで切断してIL−6R・IL−6融合蛋白質をコードする遺伝子を取得し、予めXhoIとNotIで切断したpPIC9に挿入して24種類の発現プラスミドを作製した。これら発現プラスミドの一例として、333AΔAの発現プラスミドの構造を図6に示す。
【0062】
実施例3 形質転換体の作製Pichia pastoris GS115株(インビトロジェン社)から、EasyComp Transformation Kit(インビトロジェン社)を用いてコンピテントセルを調製し、Bgl IIによって線状化した各発現プラスミドを導入した。形質転換菌は最小栄養培地で培養し、ヒスチジン要求性を失った形質転換菌を選別した。
【0063】
次に、得られた形質転換体をMDプレート(1.34%(W/V)YNB wo AA(Yeast Nitrogen Base Without Amino Acid)、0.00004%(W/V)ビオチン、2%(W/V)グルコース)とMMプレート(1.34%(W/V)YNB wo AA、0.00004%(W/V)ビオチン、0.5%(V/V)メタノール)にそれぞれ接種し、各形質転換体に対し、mut+であるかmutsであるかを調べた。
各誘導体ごとの、得られた形質転換体の数とmuts菌株の数を表1に示す。各発現プラスミドで形質転換された菌株ごとに、平均9.4個のmuts菌株(最低4種、最高17種、合計216種)を取得した。
【0064】
【表1】

【0065】
実施例4 形質転換体の培養実施例3で取得されたmuts菌(合計216種)とIL−6RのN末端344番目のロイシンとIL−6のN末端28番目のアラニンをSSELVというリンカーを介して融合した融合蛋白質を発現するmuts菌(平成10年特許願第2921号に記載)をそれぞれ試験管を用いて5%(V/V)のメタノールを含むBMGY培地(1%(W/V)酵母エキス、2%(W/V)ペプトン、1.34%(W/V)YNB wo AA(Yeast Nitrogen BaseWithout Amino Acid)、0.4mg/lビオチン、100mMリン酸カリウム(pH6.0)、1%(W/V)グリセロール3mlで、30℃で120時間培養した。各培養液から120時間目の培養液をサンプリングし、遠心により上清を取得した。
【0066】
実施例5 生物活性の測定実施例4により得られた培養上清中のIL−6R・IL−6融合蛋白質の生物活性を、BAF130細胞を用いた生物活性評価法により測定した。BAF130は、本来ヒトgp130蛋白質を発現していないマウス細胞BAF(Hatakeyamaら、Cell、63、154頁、1989年参照)にヒトgp130蛋白質をコードする遺伝子を導入して形質転換させ、当該蛋白質を発現させた細胞である。このため、生理活性を有するIL−6R及びIL−6共存下で増殖活性を示す。
【0067】
BAF130の懸濁液を、96穴プレートに2x104細胞/穴となるように添加し、216種類の培養上清をそれぞれ、1%、0.25%、0.061%、0.015%に希釈して添加した。2日後、Cell Counting Kit(和光純薬工業(株)製)を用いて、参照波長を600nmとしたときの405nmの吸光度を得た。培養上清の代わりに、1μg/mlのIL−6を各種濃度に希釈して、それぞれ100ng/mlの可溶性IL−6Rとともに添加したときの吸光度と同じ濃度依存性を示す培養上清の活性を1unit/mlと定義した。図7には、各融合蛋白質ごとに、同一の融合蛋白質発現ベクターで形質転換されたmuts菌4〜17種類の培養上清の生物活性の平均値を示した。
【0068】
図7から、リンカー配列をもたない融合蛋白質12種類の全てに関し、同一の融合蛋白質発現ベクターで形質転換された、少なくとも一つ以上のmuts菌の培養上清中に活性が認められたことが分かる。これは、IL−6RのN末端323番目のアラニン残基から361番目のセリン残基までの39個のアミノ酸残基のいずれか一つのアミノ酸残基のC末端にIL−6のN末端のアミノ酸残基が結合している本発明のIL−6R・IL−6融合蛋白質がIL−6のシグナル伝達活性を有することを示す。
【0069】
更に図7から、323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、又は343番目のイソロイシン残基のいずれかのC末端にIL−6のN末端のアミノ酸残基が結合したIL−6R・IL−6融合蛋白質である323AΔA、333AΔA、334LΔV、335TΔA 、338KΔP、338KΔA、343IΔP又は343IΔAは、先に報告されているIL−6RのN末端344番目のロイシンとIL−6のN末端28番目のアラニンをSSELVというリンカーを介して融合した融合蛋白質である334LSSELVA(平成10年特許願第2921号に記載)よりも強い活性をもつことが示された。
更に図7から、323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、又は343番目のイソロイシン残基のいずれかのC末端にIL−6のN末端のアミノ酸残基が結合したIL−6R・IL−6融合蛋白質である323AΔA、333AΔA、334LΔV、335TΔA 、338KΔP、338KΔA、343IΔP又は343IΔAは、グリシン残基やセリン残基のような自由度の高いアミノ酸残基が5から15個つながったペプチドから成るリンカーを介して融合されたIL−6R・IL−6融合蛋白質である323AG4SA、333AG4SA、323AG4S2A、333AG4S2A、343IG4S2A、333AG4S3Aや、アミノ酸残基が1から4個つながったペプチドから成るリンカーを介して融合されたIL−6R・IL−6融合蛋白質である334LPA、333AG2A、333AG4A、343IG2A、343IG4Aと比較して、ほぼ同等かやや低い程度の活性を有することが分かる。これにより、323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、343番目のイソロイシン残基の6個のアミノ酸残基のいずれか一つのアミノ酸残基のC末端にIL−6のN末端のアミノ酸残基が結合した本発明のIL−6R・IL−6融合蛋白質が、中でも特に好ましいことが分かる。
【0070】
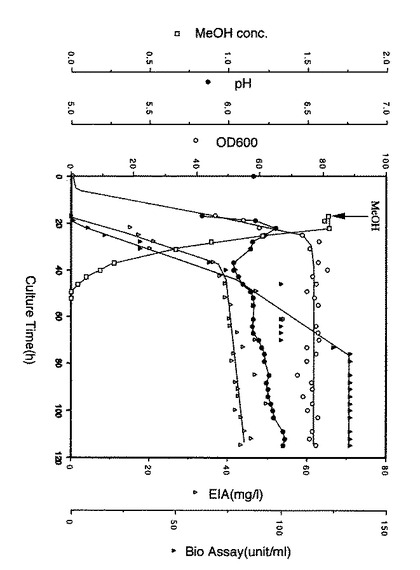
実施例6 融合蛋白質を発現するmuts菌の大量培養実施例3に記載の333AΔAを発現するmuts菌11株のうち、実施例4と5に記載の方法で最も発現量の高かった株であるIII−108株を用いて、16リットルジャーを用いた培養を行った。III−108株のグリセロールストックを100mlのBMGY(Bacto Yeast Extract 10g/l,Bacto Peptone 20g/l,Yeast Nitrogen Base without Amino Acid 1.34g/l,100mM リン酸カリウム緩衝液pH6.0,グリセロール 10g/l,ビオチン 0.4mg/l)培地に接種し、G−20振とう培養器(NBS社製)にて30℃、200rpmの条件で24時間前培養を行った。この培養液全量を8LのBMGY(Bacto Yeast Extract 10g/l,Bacto Peptone 20g/l,Yeast Nitrogen Base without Amino Acid 1.34g/l,100mM リン酸カリウム緩衝液 pH6.0,グリセロール 10g/l,ビオチン 0.4mg/l)培地に接種し、16リットルジャーSF−116(NBS社製)を用いて温度28℃、攪拌速度350rpm、通気量1vvmの条件で培養を行った。培養開始から16時間後、メタノール240ml、Bacto YeastExtract 50g/l、及びBacto Peptone 100g/lの混合液1.6リットルを添加し、融合蛋白質の発現誘導を開始した。図に本培養の培養経過を示す。
【0071】
培養液中のメタノール濃度はガスクロマトグラフィーで測定した。培養液中のpHはpHメーターで測定した。培養液中のOD600(菌体密度の指標)は、培養液を生理食塩水で100倍希釈し、分光光度計で測定した。培養液中の融合蛋白質の濃度は、抗ヒトIL−6Rモノクローナル抗体MT−18(Hirataら、J.Immunol.、143巻、2900、1989年参照)を固相抗体に、抗ヒトIL−6ポリクローナル抗体(ジェンザイム社製)を検出用抗体に、実施例7に示す方法で取得した融合蛋白質精製品を標準物質にそれぞれ用いたサンドイッチエンザイムイムノアッセーで測定した。また、融合蛋白質の生物活性は実施例5に示すBAF130細胞を用いた方法で測定した。
【0072】
実施例7 融合蛋白質の精製実施例6で選ばれた培養液を遠心分離操作により菌体と上清に分離した。得られた上清(11.6L)を蒸留水で、導電率がNaCl濃度換算で約50mMになるまで希釈した(66.05L)。次に酢酸を用いてpHを4.5に調製した。
【0073】
20mM酢酸緩衝液(pH4.5)で平衡化した吸着流動床Streamline SP C−50カラム(50mmID×100cm、ゲル量300ml)(アマシャムファルマシア社製)に、上記希釈調製液をカラム下部から上方送液することにより添加した(線速毎時300cm)。添加終了後、平衡化緩衝液をさらに上方送液することで洗浄した。次に送液方向を変え、カラム上部から溶出緩衝液(500mM NaCl、5%グリセロール、20mMリン酸緩衝液(pH6.5))を送液して(線速毎時150cm)、溶出画分(Streamline溶出フラクション、300mL)を得た。なお、溶出画分の検出は280nmによる吸光度を測定することにより行った。
【0074】
Streamline溶出フラクションに−20℃に冷却した硫酸アンモニウムを2Mになるように溶解させ、2M硫酸アンモニウム、20mMリン酸緩衝液(pH6.5)で平衡化したTSKgel Phenyl−5PWカラム(21.5mmID×15cm)(東ソー製)に添加した(流速毎分5ml)。添加終了後、平衡化緩衝液をさらに送液し、吸着基と疎水性相互作用の弱い蛋白質を溶出させた。次いで、緩衝液中の硫酸アンモニウム濃度を徐々に下げ、0.4Mの硫酸アンモニウム濃度にて溶出される画分を融合蛋白質の溶出画分として集めた(Phenyl−5PW溶出フラクション、67ml)。融合蛋白質の検出は実施例5に示すBAF130細胞の増殖活性を指標とした。
【0075】
Phenyl−5PW溶出フラクションを、5%グリセロールを含む20mM酢酸緩衝液(pH4.5)で透析し脱塩した。脱塩した溶液を同緩衝液で平衡化したTSKgel SP−5PWカラム(7.5mmID×7.5cm)(東ソー製)に添加した(流速毎分2ml)。添加終了後、平衡化緩衝液をさらに送液し、吸着基と相互作用の弱い蛋白質を溶出させた。次いで、500mMNaClを含む20mMリン酸緩衝液(pH6.5)を送液し(毎分1ml)、融合蛋白質を高濃度に含む画分を得た(SP−5PW溶出フラクション、10ml)。融合蛋白質の検出は280nmにおける吸光度を測定することによって行った。
【0076】
SP−5PW溶出フラクションを、100mM NaClを含む20mMリン酸緩衝液(pH6.5)で平衡化したTSKgel G3000SWカラム(21.5mmID×30cm)(東ソー製)に数回に分け添加し(毎分5ml)、280nmにおける吸光度を測定し融合蛋白質を分取した。
【0077】
実施例8 融合蛋白質の活性測定実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0078】
各種濃度のFP6(0.625〜40ng/ml)の生物活性を、実施例5に示すBAF130細胞を用いた生物活性評価法により測定した。図9から明らかなように、FP6は濃度依存性を示した。
【0079】
また図には示さないが、1μg/mlのFP6を各種濃度(1〜1024倍)に希釈して得た濃度依存曲線と5μg/mlのIL−6を各種濃度(1〜1024倍)に希釈して得た濃度依存曲線がほぼ一致した。一方、実施例5に記載しているように、1μg/mlのIL−6を各種濃度に希釈して、それぞれ100ng/mlの可溶性IL−6Rとともに添加したときに得られる濃度依存曲線と同じ濃度依存曲線を示す培養上清の活性を1unit/mlと定義した。従って、1μgのFP6は5unitに相当することが示された。
【0080】
実施例9 融合蛋白質のアミノ酸配列の分析実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0081】
FP6をPhenyl−5PW RPカラムで逆相クロマトグラフィーを行い、さらに精製した。溶出は20%アセトニトリル、0.1%トリフルオロ酢酸水溶液から60%アセトニトリル、0.1%トリフルオロ酢酸水溶液へのグラジエントでおこなった。得られた画分を減圧乾固したのち、20%アセトニトリル、0.1%トリフルオロ酢酸水溶液で再溶解して、プロテインシーケンサー477A(アプライドバイオシステムズ社製)に供して分析した。
【0082】
分析の結果、N末端からロイシン、アラニン、プロリン、アルギニンの順に検出され、遺伝子によりコードされるN末端の配列と矛盾していないことを確認した。
実施例10 融合蛋白質の糖鎖部分の分子量の分析実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0083】
0.1mg/mlのFP6を含む150μlの0.1%SDS、100mM塩化ナトリウム、20mMリン酸緩衝液(pH6.0)を5分間煮沸し、水冷した後に、10μlを1unit/mlのエンドグリコシダーゼH(シグマ社製)1μlと混合し、25℃で30秒から3時間反応させた。反応は500mMグリシン塩酸緩衝液(pH2.5)を10μlを加えて停止させた。なお、500mMグリシン塩酸緩衝液(pH2.5)10μl、予め冷却したFP6溶液10μl、及びエンドグリコシダーゼH1μlを順次混合したものを、エンドグリコシダーゼH反応時間0分間の試料とした。
反応が停止した上記各溶液にブロモフェニルブルーで着色した72%グリセロール、10%2−メルカプトエタノールを3μl加えた。これをSDSポリアクリルアミドゲル電気泳動に供した。電気泳動後のゲル中の蛋白質はコマジーブリリアントブルーR250で染色して検出した。
【0084】
図10から明らかなように、エンドグリコシダーゼH未反応のFP6は76〜93kDaの位置にひとつのバンドとして検出された。このことは、FP6の糖鎖部位の分子量は、17kDaの不均一性を有することを示す。また、エンドグリコシダーゼHで完全消化されたFP6は57kDaの位置に検出された。更に、図には示さないが、FP6をSDSで変性後、10倍量のTritonX−100を加え、N−グリコシルダーゼFを加えて18時間反応させた場合にも、FP6は57kDaの位置に検出された。このことは、FP6のNグリコシド結合で結合した糖鎖の分子量は19〜36kDaであることを示す。
【0085】
実施例11 融合蛋白質の幹細胞増幅効果実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0086】
ヒト臍帯血20mlより特願平9−325847号に示す方法に従ってCD34陽性細胞を精製分離した。得られた細胞500個、1.2%メチルセルロース(信越化学)、30%ウシ胎児血清アルブミン(Hyclone Laboratories Inc.)、1%ウシ血清アルブミン(以下BSAと略する)(Sigma社)、0.05mM 2−メルカプトエタノール(Sigma社)、SCF(幹細胞因子)(100ng/ml)、及び以下の4条件のいずれか(1 IL−6(100ng/ml)とIL−6R(100ng/ml)の組み合わせ、2 IL−6(100ng/ml)とIL−6R(200ng/ml)の組み合わせ、3 FP6(300ng/ml)、4 FP6(600ng/ml))をα−MEM(Flow社)1mlを35mm浮遊培養用プラスチック培養皿(Nunc社)に分注し、37℃、5%CO2,湿度100%の条件で培養した。
2週間後、倒立顕微鏡下の観察でコロニーの同定を行った。結果を図11に示す。コロニーは顆粒球コロニー(G)、マクロファージコロニー(M)、顆粒球・マクロファージの混合コロニー(GM)、芽球コロニー(Blast)、顆粒球・マクロファージ・赤芽球の混合コロニー(Mix)、赤芽球コロニー(BFU−E)の6種類に分類した。
図11から明らかなように、既に報告されている条件である1と2では、既存の報告(Suiら、Proc.Natl.Acad.Sci.USA、92、2589頁、1995年参照)とほぼ同等のコロニー形成能が観察された。一方、本発明の条件である3(FP6(300ng/ml))では、同濃度のIL−6とIL−6Rの組み合わせである2(IL−6(100ng/ml)とIL−6R(200ng/ml)を大きく上回るコロニー形成能が観察された。さらに、図には表されていないが、本発明の条件である3あるいは4により形成された混合コロニーのサイズは、既に報告されている条件である1と2のそれよりも、著しく大きいものであった。これらのことは、本発明の融合蛋白質は、既に報告されているIL−6とIL−6Rの組み合わせよりも優れた造血幹細胞のex vivo増幅効果を有することを示す。
【0087】
実施例12 融合蛋白質のマウスに対する血小板増多効果実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0088】
C57BL6マウス(雄性、8週齢)を1群5匹、全5群を用い、第1群をFP6未投与群(対照群)として300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第2群を0.5μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第3群を1μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第4群を2μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第5群を5μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群とし、それぞれ12時間置きに1日2回、5日間連続腹腔内に投与した。6日目に下行静脈より全採血し、血小板数を血球測定器CC−180A(東亜医用電子株式会社製)で計数した。
図12から明らかなように、1μg以上のFP6を投与した群(第3−5群)ではFP6未投与群(第1群)に比べ、有意な血小板増多が観察された。一方、FP6(分子量約84kDa)1μgの2倍の分子数であるIL−6(分子量約21kDa)0.5μgを同条件で投与した場合は有意な血小板増多が観察されなかったことが報告されている(Ishibashiら、Blood、74、1241頁、1989年参照)。このことは、本発明の融合蛋白質は、IL−6よりも優れたin vivoでの血小板増多効果を有することを示す。
【0089】
実施例13 融合蛋白質の5FU投与マウスに対する血小板回復効果実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
C57BL6マウス(雄性、8週齢)を1群15匹、全2群を用い、第1群をFP6未投与群(対照群)として300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第2群を5μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群とした。全群とも150mg/kgの5FUを尾静脈から投与し、上記薬剤を12時間置きに1日2回、2日目から6日目まで5日間連続腹腔内に投与した。7日目にから9日目までの3日間連続で、各群5匹ずつのマウスの下行動脈より全採血し、血小板数を血球測定器CC−180A(東亜医用電子株式会社製)で計数した。
図13から明らかなように、FP6を投与した群(第2群)ではFP6未投与群(第1群)に比べ、8日目と9日目に有意な血小板回復が観察された。このことは、本発明の融合蛋白質は5FU投与による血小板産生機能の低下に対し、in vivoでの回復効果を有することを示す。
【0090】
実施例14 イムノグロブリン様領域欠失型融合蛋白質の発現IL−6R誘導体112VL(IL−6RのN末端112番目のバリンからN末端344番目のロイシンまでの233アミノ酸)をコードする発現プラスミドpPIC9−112VL、及びIL−6R誘導体116EL(IL−6RのN末端116番目のグルタミン酸からN末端344番目のロイシンまでの229アミノ酸)をコードする発現プラスミドpPIC9−116ELを以下の方法で構築した。
【0091】
プライマーp6RAB112V(配列番号51)とプライマーp344F(配列番号52)を用いてIL−6R遺伝子(cDNA)を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvr IIで切断したpPIC9に挿入することにより、pPIC9−112VLを得た。
【0092】
次に、プライマーp6RAB116E(配列番号53)とプライマーp344F(配列番号54)を用いてIL−6R遺伝子(cDNA)を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvr IIで切断したpPIC9に挿入することにより、pPIC9−116ELを得た。
【0093】
次に、イムノグロブリン様領域欠失型融合蛋白質112VAA(N末端がIL−6RのN末端112番目のバリンで、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質)をコードする発現プラスミドpPIC9−112VAA、及び該融合蛋白質116EAA(N末端がIL−6RのN末端116番目のグルタミン酸で、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質)をコードする発現プラスミドpPIC9−116EAAを以下の方法で構築した。
【0094】
pPIC9−112VLをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断した333AΔAの発現プラスミドpPIC9−333AΔAに挿入し、pPIC9−112VAAを作製した。
【0095】
次に、pPIC9−116ELをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断した333AΔAの発現プラスミドpPIC9−333AΔAに挿入し、pPIC9−116EAAを作製した。
【0096】
pPIC9−112VAA、pPIC9−116EAAを用いて実施例3に示す方法で形質転換体を作製した結果、112VAAを発現するmuts菌9株と116EAAを発現するmuts菌11株を樹立した。更に、これを実施例4に示す方法で培養し、実施例5に示す方法で上清中の生物活性を測定した。
図14から明らかなように、イムノグロブリン様領域欠失型融合蛋白質112VAA及び116EAAは、イムノグロブリン様領域を有する融合蛋白質333AΔAとほぼ同等の活性を有した。
【0097】
実施例15 プロテアーゼ切断部位の決定実施例7のようにして得られたStreamline溶出フラクション1.9mlに2%SDS、64%グリセロール、0.25%ブロモフェノールブルーを0.1ml加えたのち煮沸した。これをSDSポリアクリルアミドゲル電気泳動にかけたのち、10%メタノール、10mM CAPS緩衝液(pH11)中でポリビニリデンジフルオリド(PVDF)膜にエレクトロブロッティングした。これをコマジ−ブリリアントブルー G−250で染色し検出した後、検出されたいくつかの蛋白質部分をそれぞれ裁断して回収し、プロテインシーケンサー477A(アプライドバイオシステムズ社製)にかけた。その結果、分子量18kDの蛋白質は、アスパラギン酸残基−バリン残基−アラニン残基−アラニン残基−プロリン残基−ヒスチジン残基という、融合蛋白質の一部であり、IL−6部分のN末端37番目リジン残基と38番目アスパラギン酸残基の間のペプチド結合がピキア酵母由来のプロテアーゼで切断されて生じたものであった。
【0098】
実施例16 プロテアーゼ抵抗性融合蛋白質の発現(1)
実施例1のようにして得られたpBS6R6LからプライマーpKN6B38D(配列番号55)とプライマーpIL6F2(配列番号56)を用いてIL−6遺伝子(cDNA)を増幅し、NruIとNotIで切断した。これを、予めEco47IIIとNotIで切断したpBS6R6Lに挿入することにより、プラスミドpBS6R6L−38Dを得た。
【0099】
pBS6R6L−38DをXhoIとNotIで切断してIL−6R・IL−6融合蛋白質をコードする遺伝子を取得し、予めXhoIとNotIで切断したpPIC9に挿入して、本願発明のプロテアーゼ抵抗性融合蛋白質20LADをコードする発現プラスミドpPIC9−20LADを作製した。
プライマーp6RAB112V(配列番号57)とプライマーp344F(配列番号58)を用いてIL−6R遺伝子を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvrIIで切断したpPIC9に挿入することにより、pPIC9−112VLを得た。pPIC9−112VLをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断したpPIC9−20LAAに挿入し、イムノグロブリン様領域欠失型融合蛋白質112VAA(N末端がIL−6RのN末端112番目のバリンで、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した融合蛋白質)をコードする発現プラスミドpPIC9−112VAAを作製した。
【0100】
次に、プライマーp6RAB116E(配列番号59)とプライマーp344F(配列番号58)を用いてIL−6R遺伝子を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvrIIで切断したpPIC9に挿入することにより、 pPIC9−116ELを得た。pPIC9−116ELをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断したpPIC9−20LAAに挿入し、イムノグロブリン様領域欠失型融合蛋白質116EAA(N末端がIL−6RのN末端116番目のグルタミン酸で、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した融合蛋白質)発現プラスミドpPIC9−116EAAを作製した。
更に、pPIC9−20LADをXhoIとPmaCIで切断してIL−6Rをコードする遺伝子を取得し、予めXhoIとPmaCIで切断したpPIC9−112VAAに挿入することにより、本願発明のプロテアーゼ抵抗性融合蛋白質112VAD(N末端がIL−6RのN末端112番目のバリンで、IL−6RのN末端333番目のアラニンとIL−6のN末端38番目のアスパラギン酸をリンカーを介さずに直接融合した融合蛋白質)をコードする発現プラスミドpPIC9−112VADを、予めXhoIとPmaCIで切断したpPIC9−116EAAに挿入することにより、本願発明のプロテアーゼ抵抗性融合蛋白質116EAD(N末端がIL−6RのN末端116番目のグルタミン酸で、IL−6RのN末端333番目のアラニンとIL−6のN末端38番目のアスパラギン酸をリンカーを介さずに直接融合した融合蛋白質)をコードする発現プラスミドpPIC9−116EADをそれぞれ作製した。
【0101】
実施例17 プロテアーゼ抵抗性融合蛋白質の発現(2)
実施例16に記載の5種類の発現プラスミド(pPIC9−20LAD、pPIC9−112VAA、pPIC9−116EAA、pPIC9−112VAD、pPIC9−116EAD)を実施例3に記載の方法でそれぞれピキア酵母に導入した。その結果、pPIC9−20LADにより形質転換されたmuts株を10株、pPIC9−112VAAにより形質転換されたmuts株を7株、pPIC9−116EAAにより形質転換されたmuts株を10株、pPIC9−112VADにより形質転換されたmuts株を1株、pPIC9−116EADにより形質転換されたmuts株を6株を取得した。これを実施例4に記載の方法で培養して上清を取得し、実施例5に示す方法で生物活性を測定した結果、pPIC9−20LADにより形質転換されたmuts株では10株中10株、pPIC9−112VAAにより形質転換されたmuts株では7株中7株、pPIC9−116EAAにより形質転換されたmuts株では10株中7株、pPIC9−112VADにより形質転換されたmuts株では1株中1株、pPIC9−116EADにより形質転換されたmuts株では6株中6株が生物活性を有していた。この結果は、IL−6のN末端28番目のアラニンから37番目リジンまでを欠失させても生物活性は影響を受けないことを示すものである。
【0102】
実施例18 プロテアーゼ抵抗性の評価実施例3に記載の333AΔA(以下、20LAAと記載する)、実施例17に記載の本願発明のプロテアーゼ抵抗性融合蛋白質20LAD、融合蛋白質112VAA、本願発明のプロテアーゼ抵抗性融合蛋白質112VAD、融合蛋白質116EAA、本願発明のプロテアーゼ抵抗性融合蛋白質116EADをそれぞれ発現する形質転換株を下記の方法により培養した。試験管を用いてBMGY培地(1%(W/V)酵母エキス、2%(W/V)ペプトン、1.34%(W/V)YNB wo AA、0.00004%(W/V)ビオチン、100mMリン酸カリウム(pH6.0)、1%(W/V)グリセロール)3mlで、30℃で48時間培養した後、遠心して集菌し、菌体ペレットをBMMY培地(1%(W/V)酵母エキス、2%(W/V)ペプトン、1.34%(W/V)YNB wo AA、0.00004%(W/V)ビオチン、100mMリン酸カリウム(pH6.0)、0.5%(V/V)メタノール)2mlに懸濁し、30℃で24時間培養した。各培養液から24時間目の培養液をサンプリングして遠心し、培養上清を回収してSDSポリアクリルアミドゲル電気泳動に供した。電気泳動後、ゲル中の蛋白質をPVDF膜に転写し、ウサギ抗IL−6ポリクローナル抗体(ジェンザイム社製)を用いてウエスタンブロッティングを行った。結果を図15に示す。
図15から明らかなように、20LAA、116EAA、112VAAでは、それぞれのIL−6領域のN末端37番目のリジンのC末端で切断されて生じたと思われる分子量18kDのバンドが鮮明に検出されたが、本願発明のプロテアーゼ抵抗性融合蛋白質20LAD、116EAD、112VADでは検出されないか、或いはほとんど検出されなかった。この結果から、IL−6のN末端28番目から37番目までの10アミノ酸を失欠させる変異を導入したことにより、プロテアーゼ抵抗性を付与できたことが分かる。
【技術分野】
【0001】
本発明は、リンカー配列を介さずに直結されたインターロイキン−6レセプター(以下IL−6Rと略す)・インターロイキン−6(以下IL−6と略す)融合蛋白質、該融合蛋白質をコードする遺伝子、該遺伝子を含有する発現ベクターで形質転換された宿主、該宿主を培養する方法、該宿主の培養物から該融合蛋白質を精製する方法、該融合蛋白質を含むことを特徴とする新規な造血幹細胞のex vivo増幅剤、及び該融合蛋白質を主成分として含む新規な血小板増多剤に関するものである。
【背景技術】
【0002】
IL−6タイプのサイトカインに属するIL−6、IL−11、Ciliary neurotropic factor、Leukemia inhibitory factor、oncostatin−M、Cardiotropin−1は、いずれも、少なくとも1つはシグナル伝達蛋白質gp130を含むレセプター複合体を介してシグナルを伝えることが知られている。IL−6を例にとると、IL−6はIL−6Rに結合し、その結果生じたIL−6・IL−6R複合体がgp130に結合する。
【0003】
造血系はIL−6が重要な役割を示す生体防御系のひとつである。造血系の細胞におけるIL−6Rの発現様式はヒトとマウスでは異なる。メチルセルロース培地上で顆粒球・マクロファージコロニー、赤芽球コロニー、巨核球コロニー、及び混合コロニーを形成することができるヒトの未分化な造血前駆細胞は、十分な数のIL−6Rを発現していない(Tajimaら、J.Exp.Med.184、1996年参照)。したがって、ヒトの未分化な造血前駆細胞はIL−6にはほとんど反応しないが、IL−6・IL−6R複合体には強く反応する。一方、マウスの未分化な造血前駆細胞は十分な数のIL−6Rを発現している。さらに、中畑と本発明者らはヒトの巨核球の前駆細胞は十分な数のIL−6Rを発現していなことを発見した(平成9年特許願第325847号)。これらの知見は、IL−6をマウスに投与すると血小板数の有意な増多が観察されるが、IL−6をヒトに投与すると効果が限定されることと合致する。
【0004】
IL−6と可溶性IL−6Rの結合定数は5x10−9Mであることが報告されている(Yasukawaら、J.Biochem.108巻、673頁、1990年参照)。これは200ng/ml(1x10−8M)のIL−6(分子量2万)と500ng/ml(1x10−8M)の可溶性IL−6R(分子量5万)を混合すると、半数の分子が単体で存在することを意味する。実際、IL−6Rを発現していない細胞に十分に作用させるには1000ng/ml以上の可溶性IL−6Rを必要とする。
【0005】
遺伝子工学を用いると、天然ではそれぞれ別の蛋白質として存在する2種類の蛋白質を1本のポリペプチド鎖から成る融合蛋白質として発現させることができる。互いに結合する性質をもつ2種類の蛋白質を融合蛋白質として発現させた場合、該2種類の蛋白質が融合された状態でそれぞれ本来の構造(生理活性を発現しうる構造)をとり得れば両者の結合は強固となり、解離は融合されていない場合に比較して起こりにくくなることが考えられる。
【0006】
融合蛋白質において2種類の蛋白質が本来の構造をとるためには、2種類の蛋白質間に立体障害が起こらないことが必要である。更には、融合させた本来の構造を有する2種類の蛋白質が互いに結合できる程度の自由度を有していることも必要である。このため従来は、主にグリシン残基やセリン残基のような自由度の高いアミノ酸残基を5〜20個つないでリンカーとし、リンカーを介して蛋白質を融合するのは普通である。このような、融合される蛋白質には含まれていない、本来無関係のリンカーを介して間接的に融合することで、2種類の蛋白質間に立体障害を生じさせず、かつ、互いに結合可能となる程度の自由度を与えることができる。
【0007】
例えば、互いに結合する性質をもつ抗体のH鎖のV領域とL鎖のV領域を融合蛋白質として発現させる場合は、GGGGSGGGGSGGGGS(Gはグリシン残基、Sはセリン残基)がリンカー配列として報告されている(Houstonら、Proc.Natl.Acad.Sci.USA、85、5879頁、1988年参照)。
【0008】
互いに結合する性質をもつIL−6RとIL−6についても、最近になってリンカーを介して両者を結合した融合蛋白質(図1(a)はイラスト図)が報告されている。一つはIL−6Rの323番目のアラニン残基のC末端側に配列番号60で示される、RGGGGSGGGGSVEというIL−6及びIL−6Rには含まれていない、本来無関係なリンカーを結合し、更にそのC末端にIL−6を結合した融合蛋白質である(Fisherら、Nature Biotech、15、142頁、1997年参照)。2番目は、IL−6Rの356番目のバリン残基のC末端にEFM(Eはグルタミン酸残基、Fはフェニルアラニン残基、Mはメチオニン残基)というリンカーを結合し、さらにそのC末端側にIL−6が結合した融合蛋白質である(Chebathら、Eur.CytokineNetw.、8、359頁、1997年参照)。また本出願人らも、IL−6Rの344番目のロイシン残基のC末端にSSELV(Lはロイシン残基、Vはバリン酸残基)というリンカーを結合し、更にそのC末端側にIL−6が結合した融合蛋白質を発明している(平成10年特許願第2921号)。
【0009】
ところで、一般的に、異種蛋白質を遺伝子組換えで作製する場合、宿主が天然に発現するプロテアーゼの作用により発現された異種蛋白質が切断されることが指摘されている。従って、IL−6R・IL−6融合蛋白質においても、宿主中で発現された後に宿主のプロテアーゼによる切断作用を受ける可能性がある。特にピキア・パストリス種の酵母は多種のプロテアーゼを発現することが知られているが、前述した報告ではこの点について述べられていない。仮に宿主が発現するプロテアーゼの切断作用によってIL−6R・IL−6融合蛋白質が切断されているのであれば、かかる切断作用に抵抗性のIL−6R・IL−6融合蛋白質を提供することにより、従来に比較してより多量のIL−6R・IL−6融合蛋白質をピキア・パストリス種の酵母や他の宿主の培養液から取得することが可能となる。
【発明の概要】
【発明が解決しようとする課題】
【0010】
融合蛋白質は2種類の蛋白質の結合状態を強固に維持することが可能と考えられるため、IL−6RとIL−6のように、これら2種類の蛋白質が結合した状態でIL−6のシグナル伝達系を形成する蛋白質を医薬品としてヒト等の体内に投与する場合に特に有効と考えられる。
【0011】
融合蛋白質を体内に持続的に投与する医薬品として開発する場合、リンカーは結合される蛋白質には含まれていない、これら蛋白質とは無関係の配列であり、しかも独自の立体構造をとるために上記免疫反応が惹起される可能性が高いことが課題となる。従って、一般的にリンカーはできるだけ短い配列であることが好ましく、更には全くリンカーが存在しないことが最も好ましい。
【0012】
しかし、前述したように融合蛋白質には、2種類の蛋白質間に立体障害等が起こらないこと、及び、融合された本来の構造を有する2種類の蛋白質が互いに結合できる程度の自由度を有していることが必要である。例えばリンカーを使用せずにIL−6RとIL−6を直接融合させる場合、両者の立体障害を起こさず、かつ両者が互いに結合できる程度の自由度を有し得るようにするためには、融合させる蛋白質の順序、N末端側蛋白質のどのアミノ酸残基にどのC末端側アミノ酸残基を結合するか等、決定しなければならない事項が数多くある。
【0013】
IL−6RとIL−6の融合蛋白質については、前記した3種類のリンカーを用いた例しか報告されておらず、リンカーを介して両者が結合したものしか知られていない。図1(a)はリンカーを介して両者が結合した融合蛋白質のイラスト図である。一方、リンカーを介することなく両者が結合されたものについては報告されていない。
【0014】
そこで本発明は、図1(b)に示したような、IL−6RとIL−6がリンカーを介することなく直接に結合しているIL−6R・IL−6融合蛋白質等を提供することを目的とするものである。また本発明は、宿主、特にピキア・パストリス種の酵母が発現するプロテアーゼの切断作用に対して抵抗性を有するIL−6R・IL−6融合蛋白質、及び該融合蛋白質をコードする遺伝子を提供することをも目的とする。
【課題を解決するための手段】
【0015】
上記目的を達成するため、本発明者らはIL−6R・IL−6融合蛋白質について鋭意検討した結果、リンカーを介することなくN末端側にIL−6Rが、C末端側にIL−6が配置された融合蛋白質を完成するに至った。即ち本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ残基の一つが直接に結合していることを特徴とするIL−6R・IL−6融合蛋白質である。また本発明者らは、IL−6のN末端37番目のリジン残基のC末端側がプロテアーゼの切断作用を受けることを見いだし、一次構造中のプロテアーゼ切断部位に変異が挿入され、プロテアーゼに対する抵抗性を有することを特徴とするプロテアーゼ抵抗性のIL−6R・IL−6融合蛋白質、中でも、IL−6RのC末端側に、N末端28番目のアラニン残基から37番目のリジン残基までの10アミノ酸残基が欠失したIL−6が結合した、IL−6R・IL−6融合蛋白質と、これをコードする遺伝子を完成するに至った。
【0016】
また本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子である。
【0017】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母である。
【0018】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス種の酵母を培養し、当該培養物からIL−6R・IL−6融合蛋白質を分泌型蛋白質として採取することを特徴とするIL−6R・IL−6融合蛋白質の製造法である。
【0019】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス種の酵母をメタノールを含まず天然物由来の炭素源を含む培地で培養し、途中でメタノールを添加して培養することを特徴とするIL−6R・IL−6融合蛋白質の製造法である。
【0020】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質を含有する溶液を、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲルろ過クロマトグラフィーの3種類のクロマトグラフィーにかけ、IL−6R・IL−6融合蛋白質を採取することを特徴とするIL−6R・IL−6融合蛋白質の製造法である。
【0021】
更に本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質を含むことを特徴とする造血幹細胞のex vivo増幅剤である。
【0022】
そして本発明は、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質を主成分として含む血小板増多剤である。以下本発明を詳細に説明する。
【0023】
本発明の融合蛋白質は、そのN末端側にIL−6Rが位置し、C末端側にIL−6が位置し、両蛋白質の間がリンカーを介することなく直接結合されていることを特徴とするものである。前記したように、プロテアーゼに対する抵抗性付与のみを目的とする場合は、両蛋白質の間はポリペプチドリンカーを介して結合しても良いが、投与の際の抗原性低減等をも考慮するのであれば、ポリペプチドリンカーを介することなく直接結合されていることが好ましい。リンカーを用いる場合には、例えば公知のリンカー配列(Fisherら、Nature Biotech、15、142頁、1997年;Chebathら、Eur.Cytokine Netw.、8、359頁、1997年)を使用したり、IL−6Rの344番目のロイシン残基のC末端にセリン残基−セリン残基−グルタミン酸残基−ロイシン残基−バリン酸残基というリンカーを結合し、更にそのC末端側にIL−6を結合させることが例示できる。本発明のIL−6R・IL−6融合蛋白質を構成するIL−6Rは、全長468アミノ酸残基で構成される膜蛋白質で、シグナル領域、細胞外領域、膜貫通領域及び細胞内領域から成る(Yamasakiら、Science、241、825頁、1988年参照)。ヒトIL−6Rの場合、シグナル領域はN末端1番目のメチオニン残基付近から19番目のアラニン残基付近まで、細胞外領域は20番目のロイシン残基付近から358番目のアスパラギン酸残基付近まで、膜貫通領域は359番目のセリン残基付近から386番目のロイシン残基付近まで、細胞内領域はおよそ387番目のアルギニン残基付近から468番目のアルギニン残基付近までと考えられている。細胞外領域はイムノグロブリン様領域とサイトカインレセプター領域に分けられ、イムノグロブリン様領域は20番目のロイシン残基付近から111番目のアスパラギン酸残基付近まで、サイトカインレセプター領域は112番目のバリン残基付近から323番目のアラニン残基付近までと考えられている。
【0024】
IL−6Rにおいて、IL−6との結合に必須なのはサイトカインレセプター領域であり、イムノグロブリン様領域は不要であることが知られている。なおサイトカインレセプター領域は、7つのβシートから構成されるバレル(樽)様の構造体が短い2個つながった構造体である(Yawataら、EMBO J.、12、1705頁、1993年参照)。
【0025】
本発明では、全長のIL−6Rはもちろんのこと、その細胞外領域全体又はサイトカインレセプター領域のいずれか、即ち部分的IL−6Rを用いることもできる。IL−6と結合してそのシグナル伝達系を構成するのはサイトカインレセプター領域であり、細胞外領域は該領域を含む領域だからである。
【0026】
本発明者らの知見によれば、具体的にはIL−6RのN末端として、N末端20番目のロイシン残基、N末端112番目のバリン残基、及びN末端116番目のグルタミン酸残基が例示できる。
【0027】
また本発明者らの知見によれば、具体的にはIL−6RのC末端として、N末端323番目のアラニン残基から361番目のセリン残基までの39個のアミノ酸残基のうちのいずれか一つ、特に好ましくは323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、及び343番目のイソロイシン残基の6個のアミノ酸残基のいずれか一つを用いることが例示できる。この部分的IL−6RのC末端側に、IL−6のN末端とを結合させるのである。なおIL−6RのN末端は、融合蛋白質のシグナル伝達における作用効果を勘案して適宜削除することができる。
【0028】
IL−6レセプター・IL−6融合蛋白質を構成するIL−6は、4つのαヘリックスから構成される全長212アミノ酸残基の分泌型蛋白質で(Hiranoら、Nature,324,731巻、1986年参照)。IL−6が活性を示すためには、これら4つのαヘリックス全てが必要であることが知られている。従って、本発明で使用するIL−6としては、4つのαヘリックスすべてを有するものであれば、特に制限はない。即ち、全長のIL−6はもちろんのこと、例えばN末端やC末端の一部アミノ酸残基が削除された部分的IL−6であってもよい。より具体的には、分泌型IL−6の構造として知られているN末端28番目のアラニン残基又は29番目のプロリン残基から212番目のメチオニン残基までのIL−6を例示することができる。またその他にも、IL−6の発現例(例えばYasukawaら、Biotech.Lett.、12,419頁、1990年)やIL−6R・IL−6融合蛋白質の発現例(Fisherら、Nature Biotech.、15,142頁、1997年)を参照にして、部分的IL−6としていかなる配列のものを使用するか決定することができる。
【0029】
一次構造中のプロテアーゼ切断部位に変異が挿入され、プロテアーゼに対する抵抗性が付与された本発明の融合蛋白質では、プロテアーゼの切断部位が種類毎に異なるため、その一次構造において、どの部分に変異を導入する、即ちどの部分を欠失させ、どの部分に本来存在しないアミノ酸残基を付加し、或いはどの部分のアミノ酸残基を置換してプロテアーゼによる切断作用を受け難くするか、を適宜決定する。置換による変異の導入では、置換により蛋白質分子が本来の立体構造をとらないようにすることにより、プロテアーゼの切断作用を受けない、或いは受け難くする。この場合、目的とするアミノ酸残基を分子サイズの小さいアミノ酸残基であるグリシンやセリンに置換することが好ましい置換として例示できる。欠失による変異の導入では、目的とするアミノ酸残基を削除することでプロテアーゼが切断作用部位を認識できないようにしたり、蛋白質分子が本来の立体構造をとらないようにすることにより、プロテアーゼの切断作用を受けない、或いは受け難いようにする。
【0030】
変異の導入は、遺伝子工学的にIL−6R・IL−6融合蛋白質をコードする遺伝子に対して行う等すれば良い。また前記アミノ酸残基の欠失、アミノ酸残基の挿入、アミノ酸残基の置換のいずれを行っても良いが、操作が比較的容易であり、しかも本来存在しないアミノ酸残基の置換による抗原性の変化等の影響が小さいと考えられることから、プロテアーゼの切断部位近傍の連続する1以上のアミノ酸残基を削除して欠失させることが特に好ましい。
【0031】
より具体的な選択の基準は、本願発明のIL−6R・IL−6融合蛋白質を遺伝子工学的に製造する際に使用する宿主細胞が分泌するプロテアーゼに対する抵抗性を付与することである。即ち、後の実施例に示したように、遺伝子工学的に製造しようとするIL−6R・IL−6融合蛋白質を選定した宿主細胞で発現させ、通常の液体クロマトグラフィーを用いる方法等で部分的に精製する過程でプロテアーゼによる分解産物と思われるピークを採取したり、通常のSDS−PAGEを用いる方法でプロテアーゼによる分解産物と思われるバンドを採取し、N末端のアミノ酸配列を解析することにより、前記選定した宿主細胞が分泌するプロテアーゼによりIL−6R・IL−6融合蛋白質のいずれの部分が切断作用を受けているかを知ることができる。
【0032】
以上のようにしてIL−6R・IL−6融合蛋白質の一次構造におけるプロテアーゼ切断部位を検出したならば、当該切断部位付近のアミノ酸残基を欠失等してプロテアーゼの切断作用を受けない、或いは受け難くすることにより、プロテアーゼに対する抵抗性を有するIL−6R・IL−6融合蛋白質を得ることが可能になる。
前記以外に例えば、医薬品等のようにプロテアーゼ存在下でIL−6R・IL−6融合蛋白質の生物活性を長時間持続させる目的でも、本願発明のプロテアーゼ抵抗性IL−6R・IL−6融合蛋白質は有効である。このようなIL−6R・IL−6融合蛋白質を製造するためには、IL−6R・IL−6融合蛋白質が生物活性を発揮すべき環境に共存するプロテアーゼを取得し、該プロテアーゼにより変異導入前のIL−6R・IL−6融合蛋白質においてどの部位が切断作用を受けるかを検出し、当該部分に前記のように変異させれば良い。
【0033】
前記のようにして変異を導入したIL−6R・IL−6融合蛋白質については、変異導入後、生物活性を維持していることを確認することが好ましい。この目的のためには、変異を導入したIL−6R・IL−6融合蛋白質を調製し、その生物活性を、例えば後の実施例に示したようなBAF130細胞株を用いる確認に供することが例示できる。この確認により変異を導入したIL−6R・IL−6融合蛋白質に生物活性が認められなかった場合には、別の変異を導入して同様の確認を行う操作を繰り返し行えば良い。
【0034】
引き続き、変異を導入したIL−6R・IL−6融合蛋白質については、プロテアーゼ抵抗性であることを確認することが好ましい。この目的のためには、変異を導入したIL−6R・IL−6融合蛋白質を一定時間プロテアーゼと共存させたものをSDS−PAGEとウエスタンブロッティングを組み合わせた解析に供する等すれば良い。この結果プロテアーゼに対する抵抗性が認められなかった場合には、別の変異を導入して同様の確認を行う操作を繰り返し行えば良い。なお、宿主細胞が分泌するプロテアーゼに対する抵抗性を付与したIL−6R・IL−6融合蛋白質においては、該宿主細胞を用いてこれを調製した後、細胞培養液について前述のような確認操作を行えば良い。
【0035】
本発明のIL−6R・IL−6融合蛋白質は、これをコードする遺伝子を用いて遺伝子組換え操作を行うことにより容易に作製することができる。IL−6R又はIL−6をコードする遺伝子は既に単離されており、その塩基配列もよく知られている。従って、本発明の融合蛋白質を作製する際には、融合蛋白質を構成するIL−6RとIL−6のアミノ酸配列から必要な遺伝子配列を調製し、これを制限酵素を用いて結合させておけば良い。またここで、天然の遺伝子配列を用いる以外にコドンの縮合を勘案し、任意のコドンを同一のアミノ酸残基をコードするが塩基配列の異なるものに置換するなどしても良い。遺伝子組換えにより宿主に蛋白質を発現させる場合、特定のコドンを使うと発現率や翻訳率が向上することがあるからである。
【0036】
本発明の融合蛋白質を遺伝子組換えで作製する場合に使用する宿主に特別の制限はなく、従来の報告を参考にしつつ、通常の遺伝子組換え操作で使用されている大腸菌やCHO細胞等に代表される動物細胞を使用することができる(Yasukawaら、J.Biotech.、108,673頁、1990年参照)。中でも、本実施例に示したピキア・パストリス種の酵母(Pichia pastoris)は、メタノールを唯一の炭素源として生育できる酵母で、CHO細胞等のような動物細胞と比較して安価に培養できることから特に好ましい宿主として例示できる。
【0037】
IL−6R・IL−6融合蛋白質を遺伝子工学的に製造する際に好適な宿主細胞であるピキア・パストリス種の酵母(Pichia pastoris)が分泌するプロテアーゼに抵抗性であるIL−6R・IL−6融合蛋白質として、IL−6Rと少なくともN末端28番目のアラニン残基からN末端37番目のリジン残基までが欠失したIL−6との融合蛋白質を例示することができる。プロテアーゼ抵抗性を有するIL−6R・IL−6融合蛋白質をコードする遺伝子を調製し、これを組み込んで発現ベクターを調製し、宿主細胞を形質転換すれば、形質転換宿主細胞を培養することにより本願発明のプロテアーゼ抵抗性を有するIL−6R・IL−6融合蛋白質を大量に製造することができる。前述のように宿主細胞が分泌するプロテアーゼに対する抵抗性を付与したIL−6R・IL−6融合蛋白質では、宿主細胞で発現された後に該プロテアーゼにより切断されない、或いは切断され難いため、抵抗性を有していないIL−6R・IL−6融合蛋白質に比較して、より大量の蛋白質を得ることが可能となる。
【0038】
前述した、本発明の融合蛋白質を遺伝子組換えで作製するための遺伝子は、宿主に導入する(形質転換する)際には、発現ベクターの中に組み込んで使用する。発現ベクターは当該遺伝子の他に、発現制御遺伝子や形質転換された宿主の選択のための指標となる遺伝子等を組み込むが、かかる遺伝子は使用する宿主との関係で適宜選択して使用すれば良い。例えばピキア・パストリス種の酵母を宿主として使用するのであれば、その染色体DNA中にIL−6R・IL−6融合蛋白質をコードする遺伝子を導入するためのアルコールオキシダーゼ遺伝子の上流配列と下流配列、選択の指標となるヒスチジン合成遺伝子そして発現制御のためのアルコールオキシダーゼ遺伝子のプロモーター配列等が、大腸菌を宿主として使用するのであれば選択の指標となるアンピシリン耐性遺伝子や発現制御のためのLacプロモーター/オペレーター配列等が例示できる。なお、市販の発現ベクター(例えばpPIC9、ピキア・パストリス種の酵母用の発現ベクター、インビトロジェン社製)に本発明の遺伝子を導入して使用することもできる。
【0039】
本発明において、好ましくピキア・パストリス種の酵母をIL−6R・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターで形質転換して融合蛋白質を作製する場合は、発現ベクター中に、融合蛋白質のシグナルペプチドとしてIL−6R本来のシグナルペプチドやα因子のシグナルぺプチドを組み込むことが好ましく、特に高発現を実現できることからα因子のシグナルぺプチドを用いることが好ましい。
【0040】
前述の形質転換された宿主を適当な条件下で培養し、発現ベクター中の発現制御遺伝子との関係で必要に応じて融合蛋白質の発現を誘導すれば融合蛋白質を作製することができる。本発明において、好ましくピキア・パストリス種の酵母を宿主とする場合は、ジャーファーメンターを用いる方法が例示できる。より具体的には、100mLの培地が仕込まれた500mL容量の振とうフラスコに、あらかじめ作製しておいたIL−6R・IL−6融合蛋白質を発現するピキア・パストリス種の酵母の20%グリセロール凍結菌株を接種し、28〜30℃で20時間振とう培養する。次に6〜9Lの培地が仕込まれた16L容量のジャーファーメンターに上記培養液100mLを接種し、28〜30℃にて通気撹拌培養を開始する。培養中の酵母の状態をモニタリングするために、OD600、pH、溶存酸素濃度、撹拌速度、温度をモニタリングしてかつ制御することが好ましい。培地は天然物由来の炭素源を含むものであれば特に種別は問わないが、具体的組成は実施例を参考にすればよい。また、該培養を実施するためのジャーファーメンターとしては市販の装置を使用することができる。培養開始後、培養液中のグリセロールが枯渇した段階でメタノールを添加すればよい。グリセロールの枯渇は溶存酸素をモニタリングすることにより知ることができる。メタノールの添加は枯渇後5時間以内に行うことが望ましい。メタノールの添加量は多すぎると酵母に毒性を示すが、少なすぎるとアルコールオキシダーゼ遺伝子のプロモーター配列が十分に機能しないことを勘案すると、0.5%以上5%以下(質量/容量)が好ましい。
【0041】
培養により作製された融合蛋白質は、適当な方法で培地等から取得することが可能である。宿主として大腸菌を用いた場合は、発現された融合蛋白質は大腸菌内に不溶性塊として蓄積されることから、菌体を破砕後、適当な条件下でリフォールディングや精製操作を行えばよい。好ましくピキア・パストリス種の酵母を宿主として用いた場合には、その培養上清から融合蛋白質を精製して取得することが可能である。精製原料は融合蛋白質を含む溶液であれば特に制限はなく、例えば融合蛋白質を含むピキア・パストリス種の酵母の培養液を例示することができる。該溶液はそのまま用いてもよく、またはそれらを緩衝液あるいは純水により希釈したり、限外ろ過膜あるいは硫酸アンモニウム等により濃縮した後に用いてもよい。精製操作としては、液体クロマトグラフィー操作を例示することができる。好ましくはイオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲルろ過クロマトグラフィーの3種類のクロマトグラフィーの組み合わせを例示することができる。
【0042】
ピキア・パストリス種の酵母の培養上清は容量が大きいため、最初にイオン交換クロマトグラフィーにかけることが好ましい。イオン交換クロマトグラフィーは陽イオン交換クロマトグラフィーと陰イオン交換クロマトグラフィーに分かれるが、除蛋白質効率を考慮するして適宜選択することができる。前者の例としては陽イオン交換基としてSPを、後者の例としては陰イオン交換基としてDEAEを例示することができる。ここで流速を毎分100ml以上に上げられることと、1μm以下のフィルターを通していないサンプルを添加することができることを勘案すると、後の実施例に示すように、陽イオンクロマトグラフィーとしては吸着流動床Streamline SP C−50カラム(アマシャムファルマシア社製)を好ましく例示することができる。上記方法により得た融合蛋白質を含む画分は、次に疎水性クロマトグラフィーにかけ、さらに融合蛋白質の純度がより高い画分を得ることができる。この画分は、後の実施例に示すように、例えば陽イオン交換クロマトグラフィーで濃縮することにより、ゲルろ過クロマトグラフィーを効率よく行うことができる。
【0043】
本発明者らは、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質の幹細胞増幅効果について鋭意検討した結果、後の実施例に示すように、該融合蛋白質と幹細胞因子(SCF)存在下でCD34陽性細胞をメチルセルロースプレート上で培養すると、従来報告されているIL−6、IL−6R、及びSCF存在下で培養したときよりも、著しくコロニー形成能が高くなることを発見した。本発明はこれらの新たに見出されたIL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質が有する造血幹細胞のex vivo増幅に基づきなされたものであり、該融合蛋白質を主成分として含む造血幹細胞のex vivo増幅剤又は造血幹細胞のex vivo増幅法である。
【0044】
本発明の造血幹細胞のex vivo増幅剤は、本発明の融合蛋白質単独でも若干の効果を示すので実用価値はあるが、SCF、FLK2リガンド等のチロシンキナーゼを刺激するサイトカイン類のうちいずれかひとつを、特に好ましくはSCFを添加すると効果は顕著なものとなる。更にインターロイキン−3(IL−3)や血小板増殖因子(TPO)等を加えてもよい。造血幹細胞は臍帯血、末梢血、又は骨髄からCD34選択により、造血幹細胞を含む画分として得ることができる。本発明の融合蛋白質は他のサイトカインを入れた個々の容器を含むキットとして提供することもできるし、または単一の容器の中の混合物として提供することもできる。造血幹細胞のex vivo増幅は、例えばプラスチックバッグのような容器の中で、造血幹細胞を含む画分を、本発明の融合蛋白質と例えばSCFを添加した無血清培地で37℃で1〜3週間培養すればよい。増幅された造血幹細胞を患者に戻す場合は、現行の末梢血幹細胞移植と同じ方法を採用すればよい。
【0045】
本発明者らは、IL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質の血小板増多効果について鋭意検討した結果、後の実施例に示すように、該融合蛋白質をマウスに投与すると血小板が有意に増多することと、該融合蛋白質を予め制癌剤を投与したマウスに投与すると血小板回復が有意に早くなることを発見した。本発明はこれらの新たに見出されたIL−6Rを構成するアミノ酸残基の一つとIL−6を構成するアミノ酸残基の一つが直接に結合しているIL−6R・IL−6融合蛋白質が有する血小板の増多あるいは回復に基づきなされたものであり、該融合蛋白質を主成分として含む血小板増多剤又は血小板増多方法である。本発明の血小板増多剤は、好ましくは非経口投与により、例えば静脈内投与、筋肉内投与、経皮投与等により投与することが好ましい。投与量は、血小板の不足症状を示す疾患の種類、患者の状態等により適宜選択されるが、一般に1〜500μg/kg/日の範囲であり、血小板の増多の様子により継続的に投与すれば良い。本発明の血小板増多剤は、常用の賦形剤、例えば生理食塩水、ブドウ糖液、マンニトール、メチルセルロース、ゼラチン、ヒト血清アルブミン等の賦活剤と混合して製剤化することができる。また本発明の血小板増多剤は凍結乾燥品とすることも可能であり、凍結乾燥した場合には使用直前に生理食塩水、ブドウ糖液、リンゲル液等の等張液により再溶解すれば良い。
【発明の効果】
【0046】
本発明で提供されるリンカー配列をもたないIL−6R・IL−6融合蛋白質は、従来報告されているリンカー配列を有するIL−6R・IL−6融合蛋白質に比べ、高い薬効と抗原性の著しい低下が期待される。このことは本融合蛋白質は造血領域における新規治療薬として大きな意義をもち、特に造血幹細胞のexvivo増幅剤や血小板増多剤としての開発が期待される。
【0047】
宿主細胞が分泌するプロテーゼに対する抵抗性を付与したものでは、遺伝子工学的に宿主細胞を形質転換して製造する工程で、該プロテアーゼによる切断を受ない等の理由により、従来のものに比較してより大量に製造することが可能となる。また前記プロテアーゼが共存した状態で精製工程に供しても切断されない等のために最終的には収量を増加することができ、しかもプロテアーゼを操作に先立って失活させるといった操作を省略することが可能であるため、精製操作を簡便化できるという効果もある。更には最終精製物中にプロテアーゼによって切断された分子が混じることがないため、より均一な精製品とすることも可能となる。
【0048】
上記効果に加えて、生物活性を発揮させようとする環境に共存が予想されるプロテアーゼに対する抵抗性を有するものでは、生物活性を持続して発揮するという効果を予想することができる。
【図面の簡単な説明】
【0049】
【図1】図1は、(a)リンカー配列を介してIL−6RとIL−6が融合したIL−6R・IL−6融合蛋白質と(b)リンカーを介さずにIL−6RとIL−6が直接融合した本発明のIL−6R・IL−6融合蛋白質のイラストを示す図である。
【図2】図2は、実施例1で作製したプラスミドpBS6R6Sの構造を示す図である。ampはアンピシリン耐性遺伝子を、oriは転写開始部位を、IL−6はIL−6遺伝子を、IL−6RはIL−6R遺伝子をそれぞれ示す。
【図3】図3は、図2に示したプラスミドpBS6R6Sに、実施例2で示す方法でオリゴヌクレオチド2種をアニールさせたものを挿入する手順を示す図である。
【図4】図4は、実施例1で作製したプラスミドpBS6R6Lの構造を示す図である。ampはアンピシリン耐性遺伝子を、oriは転写開始部位を、IL−6はIL−6遺伝子を、IL−6RはIL−6R遺伝子をそれぞれ示す。
【図5】図5は、図4に示したプラスミドpBS6R6Lに、実施例2で示す方法でオリゴヌクレオチド2種をアニールさせたものを挿入する手順を示す図である。
【図6】図6は、実施例2で作製した誘導体333AΔAの発現プラスミドの構造を示す図である。図中、ampはアンピシリン耐性遺伝子を、oriは転写開始部位を、HIS4はヒスチジン合成遺伝子を、3’AOXTTはターミネーターを、IL−6はIL−6遺伝子を、IL−6RはIL−6レセプター遺伝子を、Sはシグナル配列をコードする遺伝子を、5’AOX1はプロモーター配列を含むアルコールオキシダーゼ遺伝子の上流領域をそれぞれ示す。
【図7】図7は、各融合蛋白質の誘導体ごとに、(A)IL−6R領域のC末端のアミノ酸残基の種類と番号、(B)リンカーのアミノ酸配列(「−」はリンカーがないことを示す。)、(C)IL−6領域のN末端のアミノ酸残基の種類と番号、(D)実施例4で得られた216種類の培養上清それぞれに対し、実施例5に示す方法で得られた生物活性値平均値をそれぞれ示した図である。
【図8】図8は、実施例6に示す方法で培養を行ったときの経時変化を示す図である。図中、MeOH conc.はメタノール濃度(単位は%(質量/容量))を、EIAはサンドイッチイムノアッセーを、Bio Assayは生物活性をそれぞれ示す。
【図9】図9は、実施例8に示す方法でFP6の生物活性を求めた図である。
【図10】図10は、実施例10に示す方法でエンドグリコシダーゼHで0〜180分処理したFP6をSDSポリアクリルアミドゲル電気泳動に供したときの結果を示す図である。図中、Hと示したレーンはエンドグリコシダーゼHのみを、0から180の数字を付したレーンはそれぞれの数字の時間(分)だけエンドグリコシダーゼH処理されたFP6を示す。図中の97.4と66.3の数値を付したバーは、分子量マーカー蛋白質(分子量が97.4kDaと66.3kDa)がそれぞれ検出された位置である。
【図11】図11は、実施例11に示す方法で、1 IL−6(100ng/ml)とIL−6R(100ng/ml)の組み合わせ、2 IL−6(100ng/ml)とIL−6R(200ng/ml)の組み合わせ、3 FP6(300ng/ml)、4 FP6(600ng/ml)の500個のCD34陽性細胞に対するコロニー形成能を求めた図である。図中、Gは顆粒球コロニーを、Mはマクロファージコロニーを、GMは顆粒球・マクロファージの混合コロニーを、Blastは芽球コロニーを、Mixは顆粒球・マクロファージ・赤芽球の混合コロニーを、BFU−Eは赤芽球コロニーをそれぞれ示す。
【図12】図12は、実施例12に示す方法でC57BL6マウスに対し、FP6を投与しなかったとき(第1群)と投与したとき(第2−5群)の血小板数の平均値を示す図である。図中、星印は第1群のマウスの血小板数との有意差(P<0.05)を示す。
【図13】図13は、実施例13に示す方法で予め5FUを投与したC57BL6マウスに対し、FP6を投与しなかったとき(第1群、黒塗りの四角)と投与したとき(第2群、白い四角)の7〜9日目の血小板数の平均値を示す図である。図中、星印は同日の第1群のマウスの血小板数との有意差(P<0.05)を示す。
【図14】図14は、実施例14に示す方法で得られた(1)融合蛋白質112VAAを発現するmuts菌9株と(2)融合蛋白質116EAAを発現するmuts菌11に対し、 実施例5に示す方法で得られた生物活性値平均値を、333AΔAを発現するmuts菌11株のうち最も発現量の高かった株であるIII−108株に対する相対値(%)として、それぞれ示した図である。
【図15】図15は、(a)20LAA、(b)20LAD、(c)116EAA、(d)116EAD、(e)112VAA、(f)112VADを含む培養上清を実施例8に示す方法でウエスタンブロットしたときの結果を示す図である。
【発明を実施するための形態】
【0050】
以下に、発明を更に詳細に説明するために実施例を示すが、本発明はこれら実施例に限定されるものではない。
【0051】
実施例1 中間体プラスミドの作製各種リンカーをコードするオリゴヌクレオチドを挿入することにより、各種リンカーを介して融合されたIL−6R・IL−6融合蛋白質をコードする遺伝子(cDNA)を作製可能とすべく、中間体プラスミドpBS6R6SとpBS6R6Lを以下の方法で作製した。
【0052】
まず、クローニングベクターであるpBluescript II KS(−)(東洋紡(株)製)を制限酵素KpnIで切断し、クレノウフラグメントで処理した後、ライゲーション反応を行ってKpnIサイトが消失したプラスミドpBSを作製した。
次に、プライマーp6RAB20L(配列番号45)とプライマーp6RF320S(配列番号46)を用いてIL−6R遺伝子(cDNA)を増幅し、XhoIで切断した。これを、予めXhoIとEcoRVで切断したpBSに挿入することにより、プラスミドpBS6Rを得た。
【0053】
次に、プライマーpIL6B2(配列番号47)とプライマーpIL6F(配列番号48)により、IL−6遺伝子(cDNA)を増幅し、Bgl IIとNotIで切断した。これを、予めBgl IIとNotIで切断したプラスミドpBS6Rに挿入することにより、pBS6R6Sを得た。得られたpBS6R6Sの構造を図2に示す。
次に、オリゴマー320S333Ab(配列番号49)と320S333Af(配列番号50)を通常の方法でアニールさせた。これを、予めBgl IIとKpn Iで切断したプラスミドpBS6R6Sに挿入することにより、pBS6R6Lを得た。得られたpBS6R6Lの構造を図4に示す。
【0054】
実施例2 発現プラスミドの作製予めBgl IIとKpnIで切断したpBS6R6Sに、下記のようにそれぞれ2種類のオリゴヌクレオチドをアニールしたアニール配列1〜5をそれぞれ挿入し、IL−6R・IL−6融合蛋白質をコードする遺伝子をインサートにもつプラスミド5種類を作製した。
【0055】
アニール配列1(323AG4SA);センス側は配列番号1のオリゴヌクレオチド、アンチセンス側は配列番号2のオリゴヌクレオチド。
アニール配列2(323AG4S2A);センス側は配列番号3のオリゴヌクレオチド、アンチセンス側は配列番号4のオリゴヌクレオチド。
アニール配列3(334LPA);センス側は配列番号5のオリゴヌクレオチド、アンチセンス側;配列番号6のオリゴヌクレオチド。
アニール配列4(333AΔA);センス側は配列番号7のオリゴヌクレオチド、アンチセンス側;配列番号8のオリゴヌクレオチド。
アニール配列5(323AΔA);センス側は配列番号9のオリゴヌクレオチド、アンチセンス側;配列番号10のオリゴヌクレオチド。
【0056】
なお、アニール配列1(323AG4SA)は、IL−6RのN末端323番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列2(323AG4S2A)は、IL−6RのN末端323番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列3はIL−6RのN末端334番目のロイシンとIL−6のN末端28番目のアラニンをPというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列4(333AΔA)は、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、そしてアニール配列5(323AΔA)は、IL−6RのN末端323番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものである。
【0057】
次に、予めEco47 IIIとKpnIで切断したpBS6R6Lに、下記のようにそれぞれ2種類のオリゴヌクレオチドをアニールしたアニール配列6〜22をそれぞれ挿入し、IL−6R・IL−6融合蛋白質をコードする遺伝子をインサートにもつプラスミド17を作製した。
【0058】
アニール配列6(333AG4SA);センス側は配列番号11のオリゴヌクレオチド、アンチセンス側;配列番号12のオリゴヌクレオチド。
アニール配列7(333AG4S2A);センス側は配列番号13のオリゴヌクレオチド、アンチセンス側は配列番号14のオリゴヌクレオチド。
アニール配列8(343IG4S2A);センス側は配列番号15のオリゴヌクレオチド、アンチセンス側は配列番号16のオリゴヌクレオチド。
アニール配列9(333AG4S3A);センス側は配列番号17のオリゴヌクレオチド、アンチセンス側は配列番号18のオリゴヌクレオチド。
アニール配列10(333AG2A);センス側は配列番号19のオリゴヌクレオチド、アンチセンス側は配列番号20のオリゴヌクレオチド。
アニール配列11(333AG4A);センス側は配列番号21のオリゴヌクレオチド、アンチセンス側は配列番号22のオリゴヌクレオチド。
アニール配列12(343IG2A);センス側は配列番号23のオリゴヌクレオチド、アンチセンス側は配列番号24のオリゴヌクレオチド。
アニール配列13(343IG4A);1センス側は配列番号25のオリゴヌクレオチド、アンチセンス側は配列番号26のオリゴヌクレオチド。
アニール配列14(361SΔA);センス側は配列番号27のオリゴヌクレオチド、アンチセンス側は配列番号28のオリゴヌクレオチド。
アニール配列15(358DΔA);センス側は配列番号29のオリゴヌクレオチド、アンチセンス側は配列番号30のオリゴヌクレオチド。
アニール配列16(352TΔA);センス側は配列番号31のオリゴヌクレオチド、アンチセンス側は配列番号32のオリゴヌクレオチド。
アニール配列17(346RΔA);センス側は配列番号33のオリゴヌクレオチド、アンチセンス側は配列番号34のオリゴヌクレオチド。
アニール配列18(343IΔA);センス側は配列番号35のオリゴヌクレオチド、アンチセンス側は配列番号36のオリゴヌクレオチド。
アニール配列19(338KΔA);センス側は配列番号37のオリゴヌクレオチド、アンチセンス側は配列番号38のオリゴヌクレオチド。
アニール配列20(335TΔA);センス側は配列番号39のオリゴヌクレオチド、アンチセンス側は配列番号40のオリゴヌクレオチド。
アニール配列21(343IΔP);センス側は配列番号41のオリゴヌクレオチド、アンチセンス側は配列番号42のオリゴヌクレオチド。
アニール配列22(338KΔP);センス側は配列番号43のオリゴヌクレオチド、 アンチセンス側は配列番号44のオリゴヌクレオチド。
【0059】
なお、アニール配列6(333AG4SA)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列7(333AG4S2A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列8(343IG4S2A)はIL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列9(333AG4S3A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGSGGGGSGGGGSというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列10(333AG2A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列11(333AG4A)はIL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをGGGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列12(343IG2A)はIL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列13(343IG4A)はIL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをGGGGというリンカーを介して融合した融合蛋白質を発現するためのものであり、アニール配列14(361SΔA)は、IL−6RのN末端361番目のセリンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列15(358DΔA)は、IL−6RのN末端358番目のアスパラギン酸とIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列16(352TΔA)は、IL−6RのN末端352番目のトレオニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列17(346RΔA)は、IL−6RのN末端346番目のアルギニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列18(343IΔA)は、IL−6RのN末端343番目のイソロイシンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列19(338KΔA)は、IL−6RのN末端338番目のリジンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列20(335TΔA)は、IL−6RのN末端335番目のトレオニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、アニール配列21(343IΔP)は、IL−6RのN末端343番目のイソロイシンとIL−6のN末端29番目のプロリンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものであり、そしてアニール配列22(338KΔP)は、IL−6RのN末端338番目のリジンとIL−6のN末端29番目のプロリンをリンカーを介さずに直接融合した本発明の融合蛋白質を発現するためのものである。
【0060】
pBS6R6L自体がIL−6R・IL−6融合蛋白質をコードする遺伝子をインサートにもつが、該遺伝子により発現する融合蛋白質(334LΔV)は、IL−6RのN末端334番目のロイシンとIL−6のN末端30番目のバリンをリンカーを介さずに直接融合した本発明の融合蛋白質である。
【0061】
最後に、上記方法で作製したプラスミド23種及びpBS6R6LをそれぞれXhoIとNotIで切断してIL−6R・IL−6融合蛋白質をコードする遺伝子を取得し、予めXhoIとNotIで切断したpPIC9に挿入して24種類の発現プラスミドを作製した。これら発現プラスミドの一例として、333AΔAの発現プラスミドの構造を図6に示す。
【0062】
実施例3 形質転換体の作製Pichia pastoris GS115株(インビトロジェン社)から、EasyComp Transformation Kit(インビトロジェン社)を用いてコンピテントセルを調製し、Bgl IIによって線状化した各発現プラスミドを導入した。形質転換菌は最小栄養培地で培養し、ヒスチジン要求性を失った形質転換菌を選別した。
【0063】
次に、得られた形質転換体をMDプレート(1.34%(W/V)YNB wo AA(Yeast Nitrogen Base Without Amino Acid)、0.00004%(W/V)ビオチン、2%(W/V)グルコース)とMMプレート(1.34%(W/V)YNB wo AA、0.00004%(W/V)ビオチン、0.5%(V/V)メタノール)にそれぞれ接種し、各形質転換体に対し、mut+であるかmutsであるかを調べた。
各誘導体ごとの、得られた形質転換体の数とmuts菌株の数を表1に示す。各発現プラスミドで形質転換された菌株ごとに、平均9.4個のmuts菌株(最低4種、最高17種、合計216種)を取得した。
【0064】
【表1】
【0065】
実施例4 形質転換体の培養実施例3で取得されたmuts菌(合計216種)とIL−6RのN末端344番目のロイシンとIL−6のN末端28番目のアラニンをSSELVというリンカーを介して融合した融合蛋白質を発現するmuts菌(平成10年特許願第2921号に記載)をそれぞれ試験管を用いて5%(V/V)のメタノールを含むBMGY培地(1%(W/V)酵母エキス、2%(W/V)ペプトン、1.34%(W/V)YNB wo AA(Yeast Nitrogen BaseWithout Amino Acid)、0.4mg/lビオチン、100mMリン酸カリウム(pH6.0)、1%(W/V)グリセロール3mlで、30℃で120時間培養した。各培養液から120時間目の培養液をサンプリングし、遠心により上清を取得した。
【0066】
実施例5 生物活性の測定実施例4により得られた培養上清中のIL−6R・IL−6融合蛋白質の生物活性を、BAF130細胞を用いた生物活性評価法により測定した。BAF130は、本来ヒトgp130蛋白質を発現していないマウス細胞BAF(Hatakeyamaら、Cell、63、154頁、1989年参照)にヒトgp130蛋白質をコードする遺伝子を導入して形質転換させ、当該蛋白質を発現させた細胞である。このため、生理活性を有するIL−6R及びIL−6共存下で増殖活性を示す。
【0067】
BAF130の懸濁液を、96穴プレートに2x104細胞/穴となるように添加し、216種類の培養上清をそれぞれ、1%、0.25%、0.061%、0.015%に希釈して添加した。2日後、Cell Counting Kit(和光純薬工業(株)製)を用いて、参照波長を600nmとしたときの405nmの吸光度を得た。培養上清の代わりに、1μg/mlのIL−6を各種濃度に希釈して、それぞれ100ng/mlの可溶性IL−6Rとともに添加したときの吸光度と同じ濃度依存性を示す培養上清の活性を1unit/mlと定義した。図7には、各融合蛋白質ごとに、同一の融合蛋白質発現ベクターで形質転換されたmuts菌4〜17種類の培養上清の生物活性の平均値を示した。
【0068】
図7から、リンカー配列をもたない融合蛋白質12種類の全てに関し、同一の融合蛋白質発現ベクターで形質転換された、少なくとも一つ以上のmuts菌の培養上清中に活性が認められたことが分かる。これは、IL−6RのN末端323番目のアラニン残基から361番目のセリン残基までの39個のアミノ酸残基のいずれか一つのアミノ酸残基のC末端にIL−6のN末端のアミノ酸残基が結合している本発明のIL−6R・IL−6融合蛋白質がIL−6のシグナル伝達活性を有することを示す。
【0069】
更に図7から、323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、又は343番目のイソロイシン残基のいずれかのC末端にIL−6のN末端のアミノ酸残基が結合したIL−6R・IL−6融合蛋白質である323AΔA、333AΔA、334LΔV、335TΔA 、338KΔP、338KΔA、343IΔP又は343IΔAは、先に報告されているIL−6RのN末端344番目のロイシンとIL−6のN末端28番目のアラニンをSSELVというリンカーを介して融合した融合蛋白質である334LSSELVA(平成10年特許願第2921号に記載)よりも強い活性をもつことが示された。
更に図7から、323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、又は343番目のイソロイシン残基のいずれかのC末端にIL−6のN末端のアミノ酸残基が結合したIL−6R・IL−6融合蛋白質である323AΔA、333AΔA、334LΔV、335TΔA 、338KΔP、338KΔA、343IΔP又は343IΔAは、グリシン残基やセリン残基のような自由度の高いアミノ酸残基が5から15個つながったペプチドから成るリンカーを介して融合されたIL−6R・IL−6融合蛋白質である323AG4SA、333AG4SA、323AG4S2A、333AG4S2A、343IG4S2A、333AG4S3Aや、アミノ酸残基が1から4個つながったペプチドから成るリンカーを介して融合されたIL−6R・IL−6融合蛋白質である334LPA、333AG2A、333AG4A、343IG2A、343IG4Aと比較して、ほぼ同等かやや低い程度の活性を有することが分かる。これにより、323番目のアラニン残基、333番目のアラニン残基、334番目のロイシン残基、335番目のトレオニン残基、338番目のリジン残基、343番目のイソロイシン残基の6個のアミノ酸残基のいずれか一つのアミノ酸残基のC末端にIL−6のN末端のアミノ酸残基が結合した本発明のIL−6R・IL−6融合蛋白質が、中でも特に好ましいことが分かる。
【0070】
実施例6 融合蛋白質を発現するmuts菌の大量培養実施例3に記載の333AΔAを発現するmuts菌11株のうち、実施例4と5に記載の方法で最も発現量の高かった株であるIII−108株を用いて、16リットルジャーを用いた培養を行った。III−108株のグリセロールストックを100mlのBMGY(Bacto Yeast Extract 10g/l,Bacto Peptone 20g/l,Yeast Nitrogen Base without Amino Acid 1.34g/l,100mM リン酸カリウム緩衝液pH6.0,グリセロール 10g/l,ビオチン 0.4mg/l)培地に接種し、G−20振とう培養器(NBS社製)にて30℃、200rpmの条件で24時間前培養を行った。この培養液全量を8LのBMGY(Bacto Yeast Extract 10g/l,Bacto Peptone 20g/l,Yeast Nitrogen Base without Amino Acid 1.34g/l,100mM リン酸カリウム緩衝液 pH6.0,グリセロール 10g/l,ビオチン 0.4mg/l)培地に接種し、16リットルジャーSF−116(NBS社製)を用いて温度28℃、攪拌速度350rpm、通気量1vvmの条件で培養を行った。培養開始から16時間後、メタノール240ml、Bacto YeastExtract 50g/l、及びBacto Peptone 100g/lの混合液1.6リットルを添加し、融合蛋白質の発現誘導を開始した。図に本培養の培養経過を示す。
【0071】
培養液中のメタノール濃度はガスクロマトグラフィーで測定した。培養液中のpHはpHメーターで測定した。培養液中のOD600(菌体密度の指標)は、培養液を生理食塩水で100倍希釈し、分光光度計で測定した。培養液中の融合蛋白質の濃度は、抗ヒトIL−6Rモノクローナル抗体MT−18(Hirataら、J.Immunol.、143巻、2900、1989年参照)を固相抗体に、抗ヒトIL−6ポリクローナル抗体(ジェンザイム社製)を検出用抗体に、実施例7に示す方法で取得した融合蛋白質精製品を標準物質にそれぞれ用いたサンドイッチエンザイムイムノアッセーで測定した。また、融合蛋白質の生物活性は実施例5に示すBAF130細胞を用いた方法で測定した。
【0072】
実施例7 融合蛋白質の精製実施例6で選ばれた培養液を遠心分離操作により菌体と上清に分離した。得られた上清(11.6L)を蒸留水で、導電率がNaCl濃度換算で約50mMになるまで希釈した(66.05L)。次に酢酸を用いてpHを4.5に調製した。
【0073】
20mM酢酸緩衝液(pH4.5)で平衡化した吸着流動床Streamline SP C−50カラム(50mmID×100cm、ゲル量300ml)(アマシャムファルマシア社製)に、上記希釈調製液をカラム下部から上方送液することにより添加した(線速毎時300cm)。添加終了後、平衡化緩衝液をさらに上方送液することで洗浄した。次に送液方向を変え、カラム上部から溶出緩衝液(500mM NaCl、5%グリセロール、20mMリン酸緩衝液(pH6.5))を送液して(線速毎時150cm)、溶出画分(Streamline溶出フラクション、300mL)を得た。なお、溶出画分の検出は280nmによる吸光度を測定することにより行った。
【0074】
Streamline溶出フラクションに−20℃に冷却した硫酸アンモニウムを2Mになるように溶解させ、2M硫酸アンモニウム、20mMリン酸緩衝液(pH6.5)で平衡化したTSKgel Phenyl−5PWカラム(21.5mmID×15cm)(東ソー製)に添加した(流速毎分5ml)。添加終了後、平衡化緩衝液をさらに送液し、吸着基と疎水性相互作用の弱い蛋白質を溶出させた。次いで、緩衝液中の硫酸アンモニウム濃度を徐々に下げ、0.4Mの硫酸アンモニウム濃度にて溶出される画分を融合蛋白質の溶出画分として集めた(Phenyl−5PW溶出フラクション、67ml)。融合蛋白質の検出は実施例5に示すBAF130細胞の増殖活性を指標とした。
【0075】
Phenyl−5PW溶出フラクションを、5%グリセロールを含む20mM酢酸緩衝液(pH4.5)で透析し脱塩した。脱塩した溶液を同緩衝液で平衡化したTSKgel SP−5PWカラム(7.5mmID×7.5cm)(東ソー製)に添加した(流速毎分2ml)。添加終了後、平衡化緩衝液をさらに送液し、吸着基と相互作用の弱い蛋白質を溶出させた。次いで、500mMNaClを含む20mMリン酸緩衝液(pH6.5)を送液し(毎分1ml)、融合蛋白質を高濃度に含む画分を得た(SP−5PW溶出フラクション、10ml)。融合蛋白質の検出は280nmにおける吸光度を測定することによって行った。
【0076】
SP−5PW溶出フラクションを、100mM NaClを含む20mMリン酸緩衝液(pH6.5)で平衡化したTSKgel G3000SWカラム(21.5mmID×30cm)(東ソー製)に数回に分け添加し(毎分5ml)、280nmにおける吸光度を測定し融合蛋白質を分取した。
【0077】
実施例8 融合蛋白質の活性測定実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0078】
各種濃度のFP6(0.625〜40ng/ml)の生物活性を、実施例5に示すBAF130細胞を用いた生物活性評価法により測定した。図9から明らかなように、FP6は濃度依存性を示した。
【0079】
また図には示さないが、1μg/mlのFP6を各種濃度(1〜1024倍)に希釈して得た濃度依存曲線と5μg/mlのIL−6を各種濃度(1〜1024倍)に希釈して得た濃度依存曲線がほぼ一致した。一方、実施例5に記載しているように、1μg/mlのIL−6を各種濃度に希釈して、それぞれ100ng/mlの可溶性IL−6Rとともに添加したときに得られる濃度依存曲線と同じ濃度依存曲線を示す培養上清の活性を1unit/mlと定義した。従って、1μgのFP6は5unitに相当することが示された。
【0080】
実施例9 融合蛋白質のアミノ酸配列の分析実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0081】
FP6をPhenyl−5PW RPカラムで逆相クロマトグラフィーを行い、さらに精製した。溶出は20%アセトニトリル、0.1%トリフルオロ酢酸水溶液から60%アセトニトリル、0.1%トリフルオロ酢酸水溶液へのグラジエントでおこなった。得られた画分を減圧乾固したのち、20%アセトニトリル、0.1%トリフルオロ酢酸水溶液で再溶解して、プロテインシーケンサー477A(アプライドバイオシステムズ社製)に供して分析した。
【0082】
分析の結果、N末端からロイシン、アラニン、プロリン、アルギニンの順に検出され、遺伝子によりコードされるN末端の配列と矛盾していないことを確認した。
実施例10 融合蛋白質の糖鎖部分の分子量の分析実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0083】
0.1mg/mlのFP6を含む150μlの0.1%SDS、100mM塩化ナトリウム、20mMリン酸緩衝液(pH6.0)を5分間煮沸し、水冷した後に、10μlを1unit/mlのエンドグリコシダーゼH(シグマ社製)1μlと混合し、25℃で30秒から3時間反応させた。反応は500mMグリシン塩酸緩衝液(pH2.5)を10μlを加えて停止させた。なお、500mMグリシン塩酸緩衝液(pH2.5)10μl、予め冷却したFP6溶液10μl、及びエンドグリコシダーゼH1μlを順次混合したものを、エンドグリコシダーゼH反応時間0分間の試料とした。
反応が停止した上記各溶液にブロモフェニルブルーで着色した72%グリセロール、10%2−メルカプトエタノールを3μl加えた。これをSDSポリアクリルアミドゲル電気泳動に供した。電気泳動後のゲル中の蛋白質はコマジーブリリアントブルーR250で染色して検出した。
【0084】
図10から明らかなように、エンドグリコシダーゼH未反応のFP6は76〜93kDaの位置にひとつのバンドとして検出された。このことは、FP6の糖鎖部位の分子量は、17kDaの不均一性を有することを示す。また、エンドグリコシダーゼHで完全消化されたFP6は57kDaの位置に検出された。更に、図には示さないが、FP6をSDSで変性後、10倍量のTritonX−100を加え、N−グリコシルダーゼFを加えて18時間反応させた場合にも、FP6は57kDaの位置に検出された。このことは、FP6のNグリコシド結合で結合した糖鎖の分子量は19〜36kDaであることを示す。
【0085】
実施例11 融合蛋白質の幹細胞増幅効果実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0086】
ヒト臍帯血20mlより特願平9−325847号に示す方法に従ってCD34陽性細胞を精製分離した。得られた細胞500個、1.2%メチルセルロース(信越化学)、30%ウシ胎児血清アルブミン(Hyclone Laboratories Inc.)、1%ウシ血清アルブミン(以下BSAと略する)(Sigma社)、0.05mM 2−メルカプトエタノール(Sigma社)、SCF(幹細胞因子)(100ng/ml)、及び以下の4条件のいずれか(1 IL−6(100ng/ml)とIL−6R(100ng/ml)の組み合わせ、2 IL−6(100ng/ml)とIL−6R(200ng/ml)の組み合わせ、3 FP6(300ng/ml)、4 FP6(600ng/ml))をα−MEM(Flow社)1mlを35mm浮遊培養用プラスチック培養皿(Nunc社)に分注し、37℃、5%CO2,湿度100%の条件で培養した。
2週間後、倒立顕微鏡下の観察でコロニーの同定を行った。結果を図11に示す。コロニーは顆粒球コロニー(G)、マクロファージコロニー(M)、顆粒球・マクロファージの混合コロニー(GM)、芽球コロニー(Blast)、顆粒球・マクロファージ・赤芽球の混合コロニー(Mix)、赤芽球コロニー(BFU−E)の6種類に分類した。
図11から明らかなように、既に報告されている条件である1と2では、既存の報告(Suiら、Proc.Natl.Acad.Sci.USA、92、2589頁、1995年参照)とほぼ同等のコロニー形成能が観察された。一方、本発明の条件である3(FP6(300ng/ml))では、同濃度のIL−6とIL−6Rの組み合わせである2(IL−6(100ng/ml)とIL−6R(200ng/ml)を大きく上回るコロニー形成能が観察された。さらに、図には表されていないが、本発明の条件である3あるいは4により形成された混合コロニーのサイズは、既に報告されている条件である1と2のそれよりも、著しく大きいものであった。これらのことは、本発明の融合蛋白質は、既に報告されているIL−6とIL−6Rの組み合わせよりも優れた造血幹細胞のex vivo増幅効果を有することを示す。
【0087】
実施例12 融合蛋白質のマウスに対する血小板増多効果実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
【0088】
C57BL6マウス(雄性、8週齢)を1群5匹、全5群を用い、第1群をFP6未投与群(対照群)として300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第2群を0.5μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第3群を1μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第4群を2μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第5群を5μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群とし、それぞれ12時間置きに1日2回、5日間連続腹腔内に投与した。6日目に下行静脈より全採血し、血小板数を血球測定器CC−180A(東亜医用電子株式会社製)で計数した。
図12から明らかなように、1μg以上のFP6を投与した群(第3−5群)ではFP6未投与群(第1群)に比べ、有意な血小板増多が観察された。一方、FP6(分子量約84kDa)1μgの2倍の分子数であるIL−6(分子量約21kDa)0.5μgを同条件で投与した場合は有意な血小板増多が観察されなかったことが報告されている(Ishibashiら、Blood、74、1241頁、1989年参照)。このことは、本発明の融合蛋白質は、IL−6よりも優れたin vivoでの血小板増多効果を有することを示す。
【0089】
実施例13 融合蛋白質の5FU投与マウスに対する血小板回復効果実施例5に記載の本発明の融合蛋白質333AΔAを実施例7に記載した方法で精製したもの(以下FP6と略記)を用いて以下の実験を行った。
C57BL6マウス(雄性、8週齢)を1群15匹、全2群を用い、第1群をFP6未投与群(対照群)として300μlのPBS、1%牛血清アルブミン(BSA)を投与する群、第2群を5μgのFP6を含む300μlのPBS、1%牛血清アルブミン(BSA)を投与する群とした。全群とも150mg/kgの5FUを尾静脈から投与し、上記薬剤を12時間置きに1日2回、2日目から6日目まで5日間連続腹腔内に投与した。7日目にから9日目までの3日間連続で、各群5匹ずつのマウスの下行動脈より全採血し、血小板数を血球測定器CC−180A(東亜医用電子株式会社製)で計数した。
図13から明らかなように、FP6を投与した群(第2群)ではFP6未投与群(第1群)に比べ、8日目と9日目に有意な血小板回復が観察された。このことは、本発明の融合蛋白質は5FU投与による血小板産生機能の低下に対し、in vivoでの回復効果を有することを示す。
【0090】
実施例14 イムノグロブリン様領域欠失型融合蛋白質の発現IL−6R誘導体112VL(IL−6RのN末端112番目のバリンからN末端344番目のロイシンまでの233アミノ酸)をコードする発現プラスミドpPIC9−112VL、及びIL−6R誘導体116EL(IL−6RのN末端116番目のグルタミン酸からN末端344番目のロイシンまでの229アミノ酸)をコードする発現プラスミドpPIC9−116ELを以下の方法で構築した。
【0091】
プライマーp6RAB112V(配列番号51)とプライマーp344F(配列番号52)を用いてIL−6R遺伝子(cDNA)を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvr IIで切断したpPIC9に挿入することにより、pPIC9−112VLを得た。
【0092】
次に、プライマーp6RAB116E(配列番号53)とプライマーp344F(配列番号54)を用いてIL−6R遺伝子(cDNA)を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvr IIで切断したpPIC9に挿入することにより、pPIC9−116ELを得た。
【0093】
次に、イムノグロブリン様領域欠失型融合蛋白質112VAA(N末端がIL−6RのN末端112番目のバリンで、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質)をコードする発現プラスミドpPIC9−112VAA、及び該融合蛋白質116EAA(N末端がIL−6RのN末端116番目のグルタミン酸で、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した本発明の融合蛋白質)をコードする発現プラスミドpPIC9−116EAAを以下の方法で構築した。
【0094】
pPIC9−112VLをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断した333AΔAの発現プラスミドpPIC9−333AΔAに挿入し、pPIC9−112VAAを作製した。
【0095】
次に、pPIC9−116ELをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断した333AΔAの発現プラスミドpPIC9−333AΔAに挿入し、pPIC9−116EAAを作製した。
【0096】
pPIC9−112VAA、pPIC9−116EAAを用いて実施例3に示す方法で形質転換体を作製した結果、112VAAを発現するmuts菌9株と116EAAを発現するmuts菌11株を樹立した。更に、これを実施例4に示す方法で培養し、実施例5に示す方法で上清中の生物活性を測定した。
図14から明らかなように、イムノグロブリン様領域欠失型融合蛋白質112VAA及び116EAAは、イムノグロブリン様領域を有する融合蛋白質333AΔAとほぼ同等の活性を有した。
【0097】
実施例15 プロテアーゼ切断部位の決定実施例7のようにして得られたStreamline溶出フラクション1.9mlに2%SDS、64%グリセロール、0.25%ブロモフェノールブルーを0.1ml加えたのち煮沸した。これをSDSポリアクリルアミドゲル電気泳動にかけたのち、10%メタノール、10mM CAPS緩衝液(pH11)中でポリビニリデンジフルオリド(PVDF)膜にエレクトロブロッティングした。これをコマジ−ブリリアントブルー G−250で染色し検出した後、検出されたいくつかの蛋白質部分をそれぞれ裁断して回収し、プロテインシーケンサー477A(アプライドバイオシステムズ社製)にかけた。その結果、分子量18kDの蛋白質は、アスパラギン酸残基−バリン残基−アラニン残基−アラニン残基−プロリン残基−ヒスチジン残基という、融合蛋白質の一部であり、IL−6部分のN末端37番目リジン残基と38番目アスパラギン酸残基の間のペプチド結合がピキア酵母由来のプロテアーゼで切断されて生じたものであった。
【0098】
実施例16 プロテアーゼ抵抗性融合蛋白質の発現(1)
実施例1のようにして得られたpBS6R6LからプライマーpKN6B38D(配列番号55)とプライマーpIL6F2(配列番号56)を用いてIL−6遺伝子(cDNA)を増幅し、NruIとNotIで切断した。これを、予めEco47IIIとNotIで切断したpBS6R6Lに挿入することにより、プラスミドpBS6R6L−38Dを得た。
【0099】
pBS6R6L−38DをXhoIとNotIで切断してIL−6R・IL−6融合蛋白質をコードする遺伝子を取得し、予めXhoIとNotIで切断したpPIC9に挿入して、本願発明のプロテアーゼ抵抗性融合蛋白質20LADをコードする発現プラスミドpPIC9−20LADを作製した。
プライマーp6RAB112V(配列番号57)とプライマーp344F(配列番号58)を用いてIL−6R遺伝子を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvrIIで切断したpPIC9に挿入することにより、pPIC9−112VLを得た。pPIC9−112VLをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断したpPIC9−20LAAに挿入し、イムノグロブリン様領域欠失型融合蛋白質112VAA(N末端がIL−6RのN末端112番目のバリンで、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した融合蛋白質)をコードする発現プラスミドpPIC9−112VAAを作製した。
【0100】
次に、プライマーp6RAB116E(配列番号59)とプライマーp344F(配列番号58)を用いてIL−6R遺伝子を増幅し、XhoIとXbaIで切断した。これを予めXhoIとAvrIIで切断したpPIC9に挿入することにより、 pPIC9−116ELを得た。pPIC9−116ELをXhoIとPmaCIで切断して得られた断片を予めXhoIとPmaCIで切断したpPIC9−20LAAに挿入し、イムノグロブリン様領域欠失型融合蛋白質116EAA(N末端がIL−6RのN末端116番目のグルタミン酸で、IL−6RのN末端333番目のアラニンとIL−6のN末端28番目のアラニンをリンカーを介さずに直接融合した融合蛋白質)発現プラスミドpPIC9−116EAAを作製した。
更に、pPIC9−20LADをXhoIとPmaCIで切断してIL−6Rをコードする遺伝子を取得し、予めXhoIとPmaCIで切断したpPIC9−112VAAに挿入することにより、本願発明のプロテアーゼ抵抗性融合蛋白質112VAD(N末端がIL−6RのN末端112番目のバリンで、IL−6RのN末端333番目のアラニンとIL−6のN末端38番目のアスパラギン酸をリンカーを介さずに直接融合した融合蛋白質)をコードする発現プラスミドpPIC9−112VADを、予めXhoIとPmaCIで切断したpPIC9−116EAAに挿入することにより、本願発明のプロテアーゼ抵抗性融合蛋白質116EAD(N末端がIL−6RのN末端116番目のグルタミン酸で、IL−6RのN末端333番目のアラニンとIL−6のN末端38番目のアスパラギン酸をリンカーを介さずに直接融合した融合蛋白質)をコードする発現プラスミドpPIC9−116EADをそれぞれ作製した。
【0101】
実施例17 プロテアーゼ抵抗性融合蛋白質の発現(2)
実施例16に記載の5種類の発現プラスミド(pPIC9−20LAD、pPIC9−112VAA、pPIC9−116EAA、pPIC9−112VAD、pPIC9−116EAD)を実施例3に記載の方法でそれぞれピキア酵母に導入した。その結果、pPIC9−20LADにより形質転換されたmuts株を10株、pPIC9−112VAAにより形質転換されたmuts株を7株、pPIC9−116EAAにより形質転換されたmuts株を10株、pPIC9−112VADにより形質転換されたmuts株を1株、pPIC9−116EADにより形質転換されたmuts株を6株を取得した。これを実施例4に記載の方法で培養して上清を取得し、実施例5に示す方法で生物活性を測定した結果、pPIC9−20LADにより形質転換されたmuts株では10株中10株、pPIC9−112VAAにより形質転換されたmuts株では7株中7株、pPIC9−116EAAにより形質転換されたmuts株では10株中7株、pPIC9−112VADにより形質転換されたmuts株では1株中1株、pPIC9−116EADにより形質転換されたmuts株では6株中6株が生物活性を有していた。この結果は、IL−6のN末端28番目のアラニンから37番目リジンまでを欠失させても生物活性は影響を受けないことを示すものである。
【0102】
実施例18 プロテアーゼ抵抗性の評価実施例3に記載の333AΔA(以下、20LAAと記載する)、実施例17に記載の本願発明のプロテアーゼ抵抗性融合蛋白質20LAD、融合蛋白質112VAA、本願発明のプロテアーゼ抵抗性融合蛋白質112VAD、融合蛋白質116EAA、本願発明のプロテアーゼ抵抗性融合蛋白質116EADをそれぞれ発現する形質転換株を下記の方法により培養した。試験管を用いてBMGY培地(1%(W/V)酵母エキス、2%(W/V)ペプトン、1.34%(W/V)YNB wo AA、0.00004%(W/V)ビオチン、100mMリン酸カリウム(pH6.0)、1%(W/V)グリセロール)3mlで、30℃で48時間培養した後、遠心して集菌し、菌体ペレットをBMMY培地(1%(W/V)酵母エキス、2%(W/V)ペプトン、1.34%(W/V)YNB wo AA、0.00004%(W/V)ビオチン、100mMリン酸カリウム(pH6.0)、0.5%(V/V)メタノール)2mlに懸濁し、30℃で24時間培養した。各培養液から24時間目の培養液をサンプリングして遠心し、培養上清を回収してSDSポリアクリルアミドゲル電気泳動に供した。電気泳動後、ゲル中の蛋白質をPVDF膜に転写し、ウサギ抗IL−6ポリクローナル抗体(ジェンザイム社製)を用いてウエスタンブロッティングを行った。結果を図15に示す。
図15から明らかなように、20LAA、116EAA、112VAAでは、それぞれのIL−6領域のN末端37番目のリジンのC末端で切断されて生じたと思われる分子量18kDのバンドが鮮明に検出されたが、本願発明のプロテアーゼ抵抗性融合蛋白質20LAD、116EAD、112VADでは検出されないか、或いはほとんど検出されなかった。この結果から、IL−6のN末端28番目から37番目までの10アミノ酸を失欠させる変異を導入したことにより、プロテアーゼ抵抗性を付与できたことが分かる。
【特許請求の範囲】
【請求項1】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質。
【請求項2】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質をコードする遺伝子。
【請求項3】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母。
【請求項4】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするL−6レセプター・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母を培養し、当該培養物からIL−6レセプター・IL−6融合蛋白質を分泌型蛋白質として採取することを特徴とするIL−6レセプター・IL−6融合蛋白質の製造法。
【請求項5】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母をメタノールを含まず天然物由来の炭素源を含む培地で培養し、途中でメタノールを添加して培養することを特徴とするIL−6レセプター・IL−6融合蛋白質の製造法。
【請求項6】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質を含有する溶液を、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲルろ過クロマトグラフィーの3種類のクロマトグラフィーにかけ、IL−6レセプター・IL−6融合蛋白質を採取することを特徴とするIL−6レセプター・IL−6融合蛋白質の製造法。
【請求項7】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質を含むことを特徴とする造血幹細胞のex vivo増幅剤。
【請求項8】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質を主成分として含む血小板増多剤。
【請求項1】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質。
【請求項2】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質をコードする遺伝子。
【請求項3】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母。
【請求項4】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするL−6レセプター・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母を培養し、当該培養物からIL−6レセプター・IL−6融合蛋白質を分泌型蛋白質として採取することを特徴とするIL−6レセプター・IL−6融合蛋白質の製造法。
【請求項5】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質をコードする遺伝子を含有する発現ベクターにより形質転換されたピキア・パストリス(Pichia pastoris)種の酵母をメタノールを含まず天然物由来の炭素源を含む培地で培養し、途中でメタノールを添加して培養することを特徴とするIL−6レセプター・IL−6融合蛋白質の製造法。
【請求項6】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質を含有する溶液を、イオン交換クロマトグラフィー、疎水性クロマトグラフィー、ゲルろ過クロマトグラフィーの3種類のクロマトグラフィーにかけ、IL−6レセプター・IL−6融合蛋白質を採取することを特徴とするIL−6レセプター・IL−6融合蛋白質の製造法。
【請求項7】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質を含むことを特徴とする造血幹細胞のex vivo増幅剤。
【請求項8】
IL−6レセプターのN末端112番目のバリンから333番目のアラニン残基にIL−6のN末端から38番目のアスパラギン残基が直接に結合していることを特徴とするIL−6レセプター・IL−6融合蛋白質を主成分として含む血小板増多剤。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】

【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公開番号】特開2010−142247(P2010−142247A)
【公開日】平成22年7月1日(2010.7.1)
【国際特許分類】
【出願番号】特願2010−28220(P2010−28220)
【出願日】平成22年2月10日(2010.2.10)
【分割の表示】特願平11−188650の分割
【原出願日】平成11年7月2日(1999.7.2)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
【公開日】平成22年7月1日(2010.7.1)
【国際特許分類】
【出願日】平成22年2月10日(2010.2.10)
【分割の表示】特願平11−188650の分割
【原出願日】平成11年7月2日(1999.7.2)
【出願人】(000003300)東ソー株式会社 (1,901)
【Fターム(参考)】
[ Back to top ]