
Kv1.3カリウムチャネルの選択的阻害におけるShKトキシンの類似体ならびにその使用方法
【課題】リンパ球のKv1.3チャネルを選択的に阻害しながら、Kv1.1チャネルまたは他のカリウムチャネルを阻害する作用を最小限に抑えた新規のShK類似体を提供する。
【解決手段】アニオン電荷を有する有機または無機化学物質を付加したShKを含んでなる組成物が提供される。
【解決手段】アニオン電荷を有する有機または無機化学物質を付加したShKを含んでなる組成物が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、a)新規な組成物、b)Tリンパ球およびBリンパ球ならびに他のタイプの細胞におけるKv1.3チャネルのin vivoおよび/またはin vitro阻害のための方法およびキット、ならびに、c)ヒトまたは動物において自己免疫疾患またはその他の障害を治療するための方法を提供する。
【背景技術】
【0002】
(関連出願)
本出願は、2004年10月7日出願の米国仮特許出願第60/617,395号の優先権を主張するものであり、この仮出願の内容全体を参照により本明細書に明確に組み込む。
【0003】
(発明の背景)
細胞膜は真核細胞の外側表面を形成する。細胞膜を通る受動拡散により、種々のイオン(たとえば、ナトリウム、カリウム、カルシウムなど)が細胞に出入りする。細胞に出入りするこのようなイオンの拡散は、細胞膜内に存在する「イオンチャネル」によって促進される。イオンチャネルは、細胞膜内に埋め込まれたタンパク質であり、細胞膜を横切るイオンの選択的な流れを制御しており、これによって細胞内部の内容物と細胞周囲の細胞外液との間に濃度勾配を形成させる。イオン濃度は、興奮性細胞(たとえば神経細胞)の電気的活動に直接関与するため、イオンチャネルの機能(または機能不全)は当該細胞の電気的特性および挙動を実質的に制御することができる。実際に、幅広く「チャネル病」と呼ばれる種々の疾患は、イオンチャネルの機能不全または機能異常に関連していると考えられている。
【0004】
イオンチャネルは、開閉可能な場合は「ゲート型」と呼ばれる。基本タイプのゲート型イオンチャネルには、a)リガンドゲート型チャネル、b)機械ゲート型チャネルおよびc)電位差ゲート型チャネルがある。特に、電位差ゲート型チャネルは神経細胞、筋細胞、およびリンパ球などの非興奮性細胞にみられる。これらのチャネルは、細胞膜を通る電荷の変化に反応して開閉する。
【0005】
Kv1.3チャネルと自己免疫疾患
多発性硬化症(MS)、1型糖尿病(T1DM)、関節リウマチ(RA)および乾癬などの自己免疫疾患は、世界で数億人が罹患している。これらの障害では、特異的自己反応性T細胞−たとえば、MS患者ではミエリン特異的T細胞−が疾患の経過中に自己抗原刺激を繰り返し受け、慢性的に活性化された記憶細胞に分化し、これが炎症性組織に遊走してサイトカインを分泌することにより病因に寄与すると考えられている(ヴィグリエッタ(Viglietta)ら、2002年;ヴィサース(Vissers)ら、2002年;ウルフ(Wulff)ら、2003年b)。慢性的に活性化された記憶T細胞を優先的に標的とする治療法があれば、自己免疫疾患に対し重要な価値を有すると考えられる。
【0006】
記憶T細胞は、ケモカイン受容体CCR7およびホスファターゼCD45RAの発現に基づき、セントラル記憶(TCM)およびエフェクター記憶(TEM)の2つのサブセットに分けられる(ゲギナト(Geginat)ら、2001年;サルスト(Sallusto)ら、1999年)。ナイーブおよびTCM細胞は、炎症部位に遊走する前はリンパ節に存在し、一方、TEM細胞は炎症部位に直接存在し、その場で大量のIFN−βおよびTNF−αを分泌し、即時型エフェクター機能を示す。最近になって、MS患者ではミエリン特異的自己反応性T細胞は大部分が、活性化したTEM細胞であることが示され(Wulffら、2003年b)、ミエリン特異的活性化ラットTEM細胞をナイーブなレシピエントに養子移入すると重度のEAEを誘発することが示された(ビートン(Beeton)ら、2001年a;ビートン(Beeton)ら、2001年b)。電位差ゲート型Kv1.3K+チャネルは、TEM細胞の免疫調節に対する非常に刺激的な新しい治療標的である。TEM細胞は活性化されるとKv1.3チャネルをアップレギュレートし、その抗原誘発性増殖はKv1.3ブロッカーにきわめて感受性が高い(ウルフ(Wulff)ら、2003年b)。対照的にナイーブおよびTCM細胞は、そもそもKv1.3ブロッカーへの感受性がかなり弱く、カルシウム活性化K+チャネルIKCa1をアップレギュレートすることによりKv1.3遮断に対し急速に抵抗性となる(ガンシャニ(Ghanshani)ら、2000年;ウルフ(Wulff)ら、2003年b)。
【0007】
TEM細胞においてKv1.3が優性であることは、特異的Kv1.3阻害薬により該サブセットの活性を操作する強力な方法が提供されることになる。該チャネルの組織分布が機能的に制限されていることと、in vivoでのKv1.3遮断が、動物モデルにおいて、TEM媒介性EAE、歯周病の骨吸収および遅延型過敏性反応を明白な副作用を引き起こさずに改善するという事実が、治療標的としてのKv1.3の魅力をいっそう高めてきた(ビートン(Beeton)ら、2001年b;クー(Koo)ら、1997年;ヴァルヴェルデ(Valverde)ら、2004年)。Kv1.3ブロッカーは活性化されたTEM細胞(たとえば、ワクチン抗原に特異的なTEM細胞)をすべて抑制すると考えられるが、Kv1.3に基づいた治療法は、免疫系全体を幅広く無差別に調節する現行の治療法よりもかなり改善された治療法になると考えられる。Kv1.3ブロッカーのこのほかの利点は、可逆性であることである。このため、症状が治まるまで数カ月かかる化学療法薬とは異なり、必要時にKv1.3ブロッカーの治療効果を適正に増減し、感染に直面したら治療を中止することもできる。
【0008】
Kv1.3チャネルと肥満
Kv1.3チャネルはエネルギーホメオスタシスとエネルギーバランスに関与していることが見出された(非特許文献1)。Kv1.3チャネルを遺伝学的にノックアウトしたマウスは、脂肪食を摂取しても体重は増加しなかったが、同一の食餌を与えた対照マウスは過体重となった。Kv1.3チャネルの薬理学的遮断は、Kv1.3チャネルの遺伝子ノックアウトの上記効果を再現した。したがって、Kv1.3ブロッカーは、肥満の管理に使用できる可能性が高い。
【0009】
Kv1.3チャネルと2型糖尿病
Kv1.3チャネルは、肝臓および筋肉などの末梢標的器官におけるインスリン感受性の調節に関与している(非特許文献2)。マウスでは、Kv1.3チャネルの遺伝子ノックアウトが肝臓および筋肉のインスリンに対する感受性を増強した。したがって、Kv1.3ブロッカーは、インスリンの末梢作用を増強し、これによって血糖値を低下させることにより、2型糖尿病の治療に使用できる可能性がある。
【0010】
Kv1.3チャネルを阻害することが知られている天然ポリペプチド
最も強力なKv1.3阻害薬はカリブイソギンチャクStichodactyla helianthus由来のペプチドShKである。ShKは、3つのジスルフィド架橋により連結された35残基のポリペプチドである。ShKは、pMレベルの濃度でKv1.3を遮断して(Kd=11pM)TEM細胞の増殖を抑制し、さらに、ミエリン特異的TEM細胞の養子移入により誘発したラットの実験的自己免疫性脳脊髄炎(EAE)を改善する。考えられるShKの欠点は、神経細胞のKv1.1チャネルに対し低pM濃度の親和性がある点である(Kd=28pM)。EAE試験ではShKに副作用はまったく観察されなかったが、MSにおいて血液脳関門に障害があると起こる可能性があるように、高濃度のShKが脳内に進入すれば好ましくない神経毒性を招くと考えられる。したがって、特異性の高いKv1.3阻害薬の開発が必要である。製薬産業および学術機関の広範囲に及ぶ努力により、中位のnM範囲でKv1.3を阻害する小分子がいくつか産出されたが、これらの物質は実行可能な薬剤候補となるような選択性および力価を有していない。
【0011】
これまでにShKの断片化ペプチド類似体がいくつか報告されている。これらのShK類似体の1つでは、天然の配列が断片化され、続いて追加の共有結合(1つの非天然ジスルフィドおよび2つのラクタムブリッジ)を導入することにより安定化された。このほかの類似体では、ジスルフィドおよび/またはラクタムブリッジにより安定化された非天然骨格構造が、天然トキシン由来の重要なアミノ酸残基を含むように修飾された。これらのShK類似体は、程度がさまざまなKv1.3阻害活性および特異性を示した(非特許文献3)。
【先行技術文献】
【非特許文献】
【0012】
【非特許文献1】Hum Mol Genet.2003年、第12巻、p.551−9
【非特許文献2】Proc Natl Acad USA.2004年、第102巻、p.3112−7
【非特許文献3】ラニガン、エム.ディー.(Lanigan,M.D.)ら「カリウムチャネルブロッカーShKトキシンの設計したペプチド類似体(Designed Pepetide Analogues of the Potassium Channel Blocker ShK Toxin)」、Biochemistry,25;40(51):15528−37(2001年12月)
【発明の概要】
【発明が解決しようとする課題】
【0013】
当技術分野には、リンパ球のKv1.3チャネルを選択的に阻害しながら、Kv1.1チャネルまたは他のカリウムチャネルを阻害する作用を最小限に抑えた、もしくはその作用がまったくない新規のShK類似体を開発する必要が依然として存在する。
【課題を解決するための手段】
【0014】
本発明は、アニオン電荷を有する有機または無機化学物質(たとえば、原子、分子、化学基、残基、化合物、分子部分など)を付加した(たとえば、結合した、リンカーにより連結した、または別の方法で結びつけた)ShKトキシンを含んでなる新規組成物(本明細書で「ShK類似体」と呼ぶ)を提供する。
【0015】
さらに本発明に基づき、カリウムチャネルを阻害する方法、および/または本発明のShK類似体の有効量をヒトもしくは動物の対象者に投与することにより該対象者において疾患または障害を治療する方法を提供する。一部の実施形態では、ShKトキシンに付加する化学物質は、他のカリウムチャネル(たとえばKv1.1チャネル)よりも特定のカリウムチャネル(たとえばKv1.3チャネル)に対し選択的阻害を提供するように選ぶことができる。
【0016】
またさらに本発明によれば、前記の特徴を有するShK類似体は蛍光団の標識を有することができ、当該蛍光団で標識した本発明のShK類似体は、単独で、もしくは自己反応性細胞を検出できるクラスII四量体と併用して、フローサイトメトリーに使用することができる。
【0017】
以下に述べる詳細な説明および実施例を読むことにより、当業者には本発明のさらなる態様、要素および詳細が明らかになるはずである。
【図面の簡単な説明】
【0018】
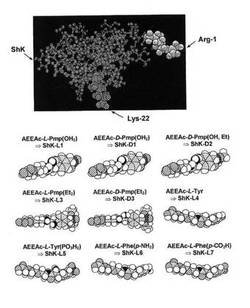
【図1】本発明のいくつかのShK類似体の化学構造を示す図。
【図2A】発表されているNMR構造に基づくShKの分子モデルを示す図。図中、チャネルの遮断に重要なLys22は灰色の影をつけて強調されている。Aeeaリンカー(右)を介してShKのArg1のαアミノ基(灰色の第2の影をつけて強調した)にL−pTyrを接続した。該リンカーおよびL−pTyrの構造をHyperchem(登録商標)のAM1でモデル化した。
【図2B】安定にトランスフェクトした細胞におけるKv1.3およびKv1.1電流に対するShK(上段)およびShK(L5)(下段)の作用を示す図。
【図2C】ShK(暗)およびShK(L5)(明)によるKv1.3(白記号)およびKv1.1(黒記号)の用量依存的阻害を示す図。Kv1.3に対するKdは10±1pM(ShK)および69±5pM(ShK(L5))、Kv1.1に対するKdは28±6pM(ShK)および7.4±0.8nM(ShK(L5))である。
【図2D】Kv1.3に対するShK(L5)のウォッシュインおよびウォッシュアウトの時間経過を示す図。細胞を保持電位80mVで保持し、30秒ごとに200ミリ秒にわたって40mVまで脱分極した。
【図2E】ShK類似体によるKv1.3およびKv1.1の阻害についてKd値を示す図。ShK−F6CAおよびShK−Dap22のKd値は発表文献に基づくものである。
【図3A】CD3、CD45RAおよびCCR7に対する抗体で染色したヒトPBMCのCD3+ゲート型母集団におけるフローサイトメトリーで測定したCD45RAおよびCCR7の染色強度を示すグラフ。
【図3B】CD3、CD45RAおよびCCR7に対する抗体で染色したヒトTEM細胞株の細胞のCD3+ゲート型母集団におけるフローサイトメトリーで測定したCD45RAおよびCCR7の染色強度を示すグラフ。
【図3C】抗CD3抗体で48時間刺激したPBMC(白記号、ナイーブ/TCM細胞の混合物)およびTEM細胞(黒記号)による[3H]チミジン取込みの、ShK(暗灰色)およびShK(L5)(明灰色)による阻害作用を示すグラフ。
【図3D】KCa3.1の発現をアップレギュレートする、事前に活性化したヒトPBMC(ナイーブ/TCM細胞)は、抗CD3抗体で再活性化するとShK(L5)阻害に対して抵抗性となることを示すグラフ。当該細胞は、KCa3.1に特異的な阻害薬であるTRAM−34に感受性となることがこれまでに報告されている。
【図4A】フローサイトメトリーにより検出したラット脾臓T細胞(左)およびPAS T細胞(右)のCD45RC染色を示すグラフ。
【図4B】静止期の(上段)およびミエリン抗原で活性化した(下段)PAS T細胞により示されるKv1.3電流を示すグラフ。
【図4C】静止期の(上段)およびミエリン抗原で活性化した(下段)PAS T細胞におけるShK‐F6CA染色のフローサイトメトリープロフィールを示すグラフ。ShK‐F6CAで染色されなかった細胞(黒線)および染色された細胞(明灰色に塗りつぶした領域)。未標識のShK(L5)によるShK‐F6CA染色の競合は、暗灰色に塗りつぶした領域で示されている。
【図4D】静止期の(上段)およびミエリン抗原で活性化した(下段)PAS T細胞におけるKv1.3免疫染色の共焦点画像を示す図。マンホットニー(Mann−Whitney)のU検定を用いて統計解析を行なった。
【図4E】ConA(1μg/ml)で活性化したラット(左)ナイーブ/TCM細胞(白記号)およびTEM(黒記号)による[3H]チミジン取込みのShK(暗線)およびShK(L5)(明線)による用量依存的阻害を示す図。
【図4F】MBPによる刺激から7時間後のPAS T細胞によるIL2分泌のShK(暗線)およびShK(L5)(明線)による用量依存的阻害を示す図。
【図4G】PAS T細胞によるミエリン抗原誘発性[3H]チミジン取込みのShK(L5)による阻害(白記号)は、20μg/mlのIL2の添加により逆転する(黒記号)ことを示す図。
【図5A】L929細胞で安定に発現されるKv1.3チャネルについて測定したShK(L5)のKv1.3遮断活性を示すグラフ。
【図5B】ラット4匹に200mg/kgのShK(L5)を単回皮下注射した後の種々の時点での血中ShK(L5)濃度を示すグラフ。表記の時間に血液を採取し、パッチクランプ法により血清を試験してShK(L5)の量を測定した。
【図5C】図5Bのデータを単一指数関数減衰にフィッティングしたグラフ。半減期は約50分であることが示されている。
【図5D】10μg/kg/日のShK(L5)の単回皮下注射を5日間受けたLewisラット5匹における血中ShK(L5)濃度を示すグラフ。血液を毎朝(前回の注射から24時間後に)採取し、パッチクランプ法によりKv1.3チャネルに対する遮断活性について試験した。
【図5E】皮下投与(白棒;n=4)または静脈内投与(黒棒;n=4)のいずれかで10mg/kgのShK(L5)を単回投与した後の種々の時点におけるラットの血清中ShK(L5)濃度を示すグラフ。表記の時間に血液を採取し、パッチクランプ法により血清を試験して血中のShK(L5)量を測定した。単回皮下注射からほぼ24時間後には、ShK(L5)は300pMの定常状態濃度を維持した。該濃度は、TEM細胞の機能を選択的に阻害するのに十分な濃度である。
【図5F】ShK(L5)の半遮断投与量をラット血漿または2%ラット血漿を含有するPBSに添加し、種々の時間にわたって37℃でインキュベートした後のShK(L5)の回収率(%)を示すグラフ。表記の時間に等分量を採取し、Kv1.3チャネルに対する遮断活性を測定した。ShK(L5)は血漿中できわめて安定である。
【図6A】スコア化したEAEの予防効果を示すグラフ。PAS T細胞をin vitroで活性化し、洗浄し、第0日に腹腔内に注射した。EAEの臨床スコア:0=臨床徴候なし、0.5=尾遠位部のひきずり、1=尾のひきずり、2=軽度の両側不全麻痺または運動失調、3=中等度の両側不全麻痺、4=完全な後脚麻痺、5=4+失禁、6=死亡。ラットを1群あたりn=6とし、第0日から第5日まで、賦形剤単独(n=6)またはShK(L5)(n=6;10mg/kg/日)を皮下注射した。
【図6B】スコア化したEAEの治療効果を示すグラフ。PAS T細胞をin vitroで活性化し、洗浄し、第0日に腹腔内に注射した。ラットがEAEの臨床徴候を発現したとき、10mg/kg/日でShK(L5)による治療を開始し、3日間継続した。
【図6C】卵アルブミンに対してラットに現れるDTH反応の指標としての耳の厚さを示すグラフ。ShK(L5)10mg/kg/日で2日間にわたって動物(1群あたりn=6)を治療し、その後、耳の腫脹を測定した。マンホットニーのU検定を用いて統計解析を行なった。
【図7A】ShK(L5)の構造を示す図、ならびにTEM細胞におけるKv1.3チャネルの阻害をShK(L5)濃度の関数として示したグラフ。各データ点は3つの測定値の平均を示す。
【図7B】Kv1.3を含むシグナル伝達複合体の概略図。
【図7C】ISにおけるCD4、Kv1.3、Kvβ2、SAP97、ZIPおよびp56lckの共局在性を示す図。
【図7D】明らかなTEM−APC接触がない状態におけるCD4およびKv1.3の染色を示す図。
【図7E】MBPを取り付けたAPCに曝されたGAD65特異的TEM細胞におけるCD4およびKv1.3の染色を示す図。
【図7F】ShK(L5)100nMはIS形成を妨げないことを示す図。
【図7G】ShK(L5)100nMはISを崩壊させないことを示す図。
【図8A】ShK(L5)の不在下(黒)またはShK(L5)0.1nM(暗灰色)、1nM(中灰色)もしくは100nM(明灰色)の存在下で抗CD3抗体+架橋結合二次抗体(矢印)により誘発された、T1DM患者3例からのGAD特異的TEM細胞におけるカルシウムシグナル伝達を示すグラフ。
【図8B】T1DMおよびRAを有する患者からのナイーブ/TCM細胞およびTEM細胞(左)ならびにナイーブ/TCMエフェクターおよびTEMエフェクター(右)による[3H]チミジン取込みを示すグラフ。TEM細胞:T1DM患者3例からのGAD65で活性化したTEMクローンおよびRA患者3例からの抗CD3抗体で活性化したSF−TEM細胞。ナイーブ/TCM細胞:同一のRA患者3例からの抗CD3抗体で活性化したPB−ナイーブ/TCM細胞。
【図8C】図8Bで用いたTEM細胞およびナイーブ/TCM細胞によるサイトカイン産生を示す一連の棒グラフ。
【図8D】MS、T1DMおよびRAにおける疾患関連性および疾患無関連性の自己反応性T細胞の表現型を示す図。
【図8E】ShK(L5)がカルシウムシグナル伝達、リンパ球増殖およびサイトカイン産生を阻害し、IS形成を阻害しない様式を示す概略図。
【図9】エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルを表す概略図。
【図10】エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルにおけるShK(L5)の治療プロトコルを示す概略図。
【図11】ナイーブおよびセントラル記憶T細胞またはB細胞の機能を損なわずに、ラットにおいてin vivoでShK(L5)によりエフェクター記憶反応が特異的に抑制されることを表す概略図。
【図12A】新規発症1型糖尿病(T1DM)患者、健康な対照者および多発性硬化症患者からのGAD65特異的、インスリン特異的およびミエリン特異的T細胞におけるKv1.3電流(上段)およびチャネル数/細胞(下段)を示す図。
【図12B】上記患者からの個々のT細胞のKv1.3染色強度(上段)および蛍光強度(下段)を示す図。
【図12C】相対的細胞数とCCR7染色強度を対比させたグラフ。高濃度のKv1.3を発現している細胞は、CCR7陰性、すなわちTEMエフェクターである。低濃度のKv1.3を発現している細胞は、CCR7陽性、すなわちナイーブ細胞もしくはTCM細胞のいずれかである。
【図12D】T1DMおよびMSを有する患者(左)、罹患期間が5年を超えるT1DM患者(中)および非自己免疫性の2型DM患者からの自己反応性T細胞における1細胞あたりのKv1.3の数を示す図。
【図12E】新規発症T1DMの患者からのCD4+GAD65四量体+T細胞におけるKv1.3の数を示す図。
【図13A】RA患者の末梢血T細胞および滑液T細胞ならびにOA患者の滑液T細胞における細胞1個あたりのKv1.3チャネルの数を示す図。
【図13B】図13Aに示した細胞におけるKv1.3染色(明灰色)およびKvβ2染色(暗灰色)の共焦点画像を示す図。
【図13C】相対的細胞数とCCR7染色強度を対比させたグラフ。
【図13D】抗CD3抗体または抗Kv1.3抗体で染色し、ヘマトキリン/エオシンで対比染色した、RAおよびOA患者からの滑膜の顕微鏡写真(上)(40倍)ならびに炎症指数を示す棒グラフ(下)。
【発明を実施するための形態】
【0019】
以下に記載する本発明の詳細な説明および添付図面は、本発明の一部の実施例または実施形態だけを説明することを意図しており、必ずしも全部を説明することを意図したものではない。本詳細な説明および添付図面は、決して本発明の範囲を限定するものではない。
【0020】
本発明は、ShKの新規類似体と、当該組成物を作製する方法のほか、ヒトまたは動物の細胞におけるKv1.3チャネル(または他のイオンチャネル)を阻害するための、さらにはT細胞媒介性自己免疫疾患などの疾患および障害を治療または予防するための当該組成物の使用方法を提供する。本発明の組成物は、アニオン電荷を有する有機または無機化学物質(たとえば、原子、分子、化学基、残基、化合物、分子部分など)を付加した(たとえば、結合した、リンカーにより連結した、または別の方法で結びつけた)ShKトキシンよりなる。本発明の少なくとも一部の実施形態では、Kv1.1チャネルよりもKv1.3チャネルを阻害するために該組成物の親和性を増大または最適化できるように、アニオン電荷を有する有機または無機化学物質を選択することができる。本発明による、ShKと連結または結合しうる、アニオン電荷を有する有機または無機の分子もしくは化学基の例として、アミノ酸、ポリペプチド、アミノ酸残基、非天然アミノ酸残基、スレオニン、スレオニン誘導体、ホスホスレオニン、セリン、セリン誘導体、ホスホセリン、グルタミン酸、グルタミン酸誘導体、γカルボキシグルタミン酸、アスパラギン酸、アスパラギン酸誘導体、無機化合物または無機化学基、有機化合物または有機化学基、無水コハク酸、および無水フタル酸が挙げられるが、必ずしもこれらに限定されない。
【0021】
本発明によれば、本発明の組成物の非限定的な一部の実施例であって、アニオン電荷を有する化学物質がアミノ酸残基からなる実施例は、図1および2Cに示されており、以下の表1に示すように、本明細書では英数字による記号で記載されている。
【0022】
【表1】

特に図1を参照すると、チロシンもしくはフェニルアラニン、または荷電したこれらの非天然誘導体を、ShKのN末端(影付き灰色で示した残基Arg1)に付加したリンカーを介してShK(左上)に結合させた。チャネルの遮断に必要なLys22は暗灰色の影で示している。ShKの分子モデルは発表されているNMR構造に基づいており、上記リンカーおよび新規残基の構造をモデル化した。本発明の組成物の上記実施形態は全般に、アニオン電荷を有する少なくとも1つのアミノ酸残基と結合された(たとえば、化学的に結合された、連結された、または別の方法で結び付けられた)ポリペプチドShKトキシンを構成する。アミノ酸残基がキラル中心を有する実施形態では、当該アミノ酸残基のDおよび/またはLエナンチオマーを使用することができる。アニオン電荷を有するアミノ酸残基は、非天然残基であってもよく、ShKポリペプチドのN末端に付加または連結させることができる。一部の実施形態では、アニオン電荷を有するアミノ酸残基は、アミノエチルオキシエチルオキシアセチル・リンカーなどのリンカーを介してShKのN末端に連結させてもよい。これらのShK類似体は、他のカリウムチャネル(たとえばKv1.1)に対する親和性が低下しているため、ShKよりもKv1.3チャネルをより特異的に阻害する。ShKは、当技術分野で周知のように天然源から単離してもよいし、合成してもよい。
【0023】
ShKトキシンの合成
ShKトキシンは任意の適切な方法で合成することができる。そのような方法の1つでは、Arg(Pmc)、Asp(OtBu)、Cys(Trt)、Gln(Trt)、His(Trt)、Lys(Boc)、Ser(tBu)およびThr(tBu)を含む市販のFmoc−アミノ酸(バッケム社(Bachem Feinchemikalien))を入手し、組み立ててShKトキシンを形成する。アプライドバイオシステムズ(Applied Biosystems)431Aペプチド合成装置を用い、0.25mmolスケールでFmoc−Cys(Trt)−Rから開始してアミノ酸の段階的組立てを行ってもよい。残基34〜22を単結合させる。その後、樹脂の等分量(たとえば半量)を分取してさらに適切に混合する。続いて、残った等分量の樹脂に残りのペプチド配列を二重結合させる。カップリングはすべて、2当量の1−ヒドロキシベンゾトリアゾールの存在下でジシクロヘキシルカルボジイミドを介して行なう。最後の2残基もHBTU/DIEA法で結合させる。当該2残基はAeea(Fmoc−アミノエチルオキシエチルオキシ酢酸)と、N末端残基としてのFmoc−Tyr(PO4)モノベンジルエステルである。最終的にFmoc基を除去した後、ペプチド樹脂(2.42g)を樹脂から切断し、同時に試薬Kを用いて室温で2時間にわたって脱保護する。当技術分野では試薬Kは周知であり、文献に記載されている。キング、ディー.エス.(King,D.S.)、フィールズ、シー.ジー.(Fields,C.G.)およびフィールズ、ジー.ビー.(Fields,G.B.)(1990年)Int.J.Peptide Protein Res.第36巻、p.255−266を参照されたい。切断後、ペプチドを濾過して消耗した樹脂ビーズを取り除き、氷冷ジエチルエーテルで沈殿させる。続いて微細なフィルタ漏斗で該ペプチドを収集し、氷冷エーテルで洗浄し、最後に20%AcOH水溶液で抽出する。次に該ペプチド抽出物をH2Oで希釈して2リットルとし、NH4OHでpHを8.0に調整し、室温で36時間にわたって空気酸化させる。ジスルフィド結合を酸化して還元型グルタチオンと酸化型グルタチオンの比が2:1になった後、該ペプチド溶液をpH2.5に酸性化し、レイニン社(Rainin)のDynamax(商標)C18カラム(5.0×30cm)にポンプで送り込む。0.1%TFAを含有するH2Oに混和したアセトニトリル5〜30%を用い、直線濃度勾配で上記試料を溶出する。2つのRP−HPLC分析装置TFAおよびTEAPを用いて、得られた画分を分析する。純粋画分を併せ、凍結乾燥する(ペニントン、エム.ダブリュ.(Pennington,M.W.)、バーンズ、エム.イー.(Byrnes,M.E.)、ザイデンバーグ、アイ.(Zaydenberg,I.)、カイティン、アイ.(Khaytin,I.)、デカストナイ、ジェイ.(de Chastonay,J.)、クラフテ、ディー.(Krafte,D.)、ヒル、アール.(Hill,R.)、マーニル、ブイ.(Mahnir,V.)、ボルバーグ、ダブリュ.エイ.(Volberg,W.A.)、ゴルチカ、ダブリュ.(Gorczyca,W.)およびケム、ダブリュ.アール.(Kem,W.R.)(1995年)Int.J.Peptide Protein Res.第46巻、p.354−358を参照)。
【0024】
別の方法としてBoc−Bzl保護基戦略を用いる固相ペプチド合成を使用して、該ペプチドの一次構造ならびに類似体を組み立ててもよい。次に該ペプチドを無水HFで固相から切断すると、上記のFmocで合成したペプチドと同じく、折り畳み(フォールディング)の準備ができている直線ペプチドが生成される(スチュアート、ジェイ.エム.(Stewart,J.M.)およびヤング、ジェイ.ディー.(Young J.D.)(1984年)「固相ペプチド合成(Solid Phase Peptide Synthesis)」、第2版、ロックフォード(Rockford)所在のピアスケミカルカンパニー(Pierce Chemical Company)、IIを参照)。
【0025】
あるいは、ShKまたは類似体の一次構造を組み立てる別の合成方法として化学的ライゲーション技術が挙げられるが、この方法では、ペプチドは、C末端チオエステルペプチドを有するように設計された一連の断片として作製される。該チオエステルペプチドは、N末端にCys残基を含む別のペプチドと反応して天然ペプチド結合を備えたペプチドを形成することができる。この技術を使用することにより、ShKの一次構造を効果的に組み立てることができると考えられる((4)ウィルケン、ジェイ.(Wilken,J.)およびケント、エス.ビー.エイチ.(Kent S.B.H.)(1998年)「化学的タンパク質合成(Chemical protein synthesis)」、Current Opin.Biotech.第9巻、p.412−426を参照)。
【0026】
また、ShKの一次構造を組み立てるために使用しうる別の合成方法は、アルベリキオ、エフ.(Albericio,F.)、ロイド‐ウィリアムス、ピー.(Lloyd−Williams,P.)およびギラルト、イー.(Giralt,E)(1997年)「コンバージェントペプチド合成(Convergent peptide synthesis)」、ジー.フィールズ(G.Fields)編「Methods in Enzymol.」、ニューヨーク州ニューヨーク所在のアカデミックプレス(Academic Press)に収録(pp313−335)に記載されているように、保護ペプチド断片のコンバージェント法を利用することであると考えられる。当該方法では、直線状の保護断片を完全な側鎖保護断片として組み立てる。続いて、該断片をコンバージェント法で互いに結合させ、ShKまたはその1つの類似体の一次配列を組み立てることができる。該断片の組立ては、固相樹脂を用いてカップリング工程および洗浄工程を容易にすることもできると考えられる。
【0027】
また別の方法として遺伝子組換え法を使用することも可能であり、この方法では、原核生物または真核生物の発現系のいずれかで発現できるように、ShKをコードするcDNA配列を作製することができると考えられる。非標準コドンを利用する装荷済み(プレロード)tRNA分子を活用することにより、非天然アミノ酸を含有する組換えShK類似体も可能である。上記の未使用コドンの1つを使用してホスホチロシン残基のほかAeea残基を付加するように、cDNA構築物を設計することができる。次に、組換え生産したShK類似体の折り畳みは、合成ペプチドに使用する方法と類似した方法で完成させることができると考えられる(ペニントン、エム.ダブリュ.(Pennington,M.W.)、バーンズ、エム.イー.(Byrnes,M.E.)、ザイデンバーグ、アイ.(Zaydenberg,I.)、カイティン、アイ.(Khaytin,I.)、デカストナイ、ジェイ.(de Chastonay,J.)、クラフテ、ディー.(Krafte,D.)、ヒル、アール.(Hill,R.)、マーニル、ブイ.(Mahnir,V.)、ボルバーグ、ダブリュ.エイ.(Volberg,W.A.)、ゴルチカ、ダブリュ.(Gorczyca,W.)およびケム、ダブリュ.アール.(Kem,W.R.)(1995年)Int.J.Peptide Protein Res.第46巻、p.354−358を参照)。
【0028】
アニオン性アミノ酸残基のShKへの付加およびShKの任意の修飾
アニオン性アミノ酸残基は、アミノエチルオキシエチルオキシアセチル・リンカーなどのリンカーにより、もしくは他の何らかの適切な手段により、天然または合成ShKトキシンのN末端に付加してもよい。当該実施例では、図1に示した9つのShK類似体が作製される。まず、Fmoc−Aeea−OHを、上述のように組み立てた合成ShKトキシンのN末端に結合させる。続いて樹脂を9つに等分する。次に、DICおよびHOBTを用いて、Fmoc−Tyr(PO4Bzl)−OH、Fmoc−d−Tyr(PO4Bzl)−OH、Fmoc−Tyr(PO4Me2)−OH、Fmoc−Pmp−OH、Fmoc−d−Pmp−OH、Fmoc−Pmp(Et)−OH、Fmoc−Pmp(Et)2−OH、Fmoc−Tyr(tBu)−OH、またはFmoc−Amp(Boc)−OHのいずれかを等分した樹脂の1つに結合させる。続いて非ブロック化したペプチド樹脂を切断し、5%トリイソプロピルシランを含有する試薬K(キング(King)ら、1990年)を用いて室温で2時間にわたって脱保護する。t−15分時点で切断混合物に固体のNH4Iを加えてMet(O)を還元する(ニコラス(Nicolas)ら、1995年)。Tyr(PO4Me2)−OHを含有するペプチドについては、スカベンジャーとしてチオアニソールを含有する1MのTMSBrのTFA溶液を含有する切断混合物を4℃で18時間にわたって使用した(ティアン(Tian)ら、1993年)。この方法を用いると、メチル保護基の不完全な除去がよくみられ、2つの種(Tyr(PO4)およびTyr(PO4Me))はRP−HPLCで容易に精製される。Tyr(PO4Me2)を含む類似体は標準の試薬Kによる切断を介して切断され、両Me基はそのまま維持される。いずれの場合も、切断混合物を濾過し、粗ペプチドを氷冷ジエチルエーテル中で沈殿させる。沈殿を収集し、樹脂200mgから約75mgのペプチドが産出される。この粗生成物を50%AcOH水溶液20mlに溶解し、0.75LのH2Oで希釈する。NH4OHで該溶液のpHを8.2に調整し、グルタチオン(2mM:1mM)(還元型:酸化型)を加えて一晩放置し、折り畳ませた。類似体はすべて、以前に記載されているようにRP−HPLCを用いて精製する(ペニントン(Pennington)ら、1995年;ペニントン(Pennington)ら、1996年a;ペニントン(Pennington)ら、1996年b)。純粋な画分を併せ、凍結乾燥する。各試料をRP−HPLC、AAAおよびMALDI−TOF MSで確認し、バイオアッセイの前に調整してペプチド含量を明らかにする。
【0029】
本発明の一部の実施形態では、ShK構造のPK/PD特性を改善するため、分解特性に敏感な残基を交換または置換してもよい。このため、21位のMet残基の置換を行なって酸化に対する安定効果を与えてもよい。さらに、C末端の酸官能基をアミドで置換すれば、C末端カルボキシペプチダーゼ酵素に対する安定性を与えることになる。N末端でアニオン性分子と結合させたShKの一次構造に上記の2つの置換を行なった物質はすでに合成され、最も安定で選択性の高いKv1.3ブロッカーを生み出している。非加水分解性リン酸塩置換も、ホスファターゼ酵素に対する安定性だけでなく、リン酸塩の酸性および塩基性加水分解に対する安定効果を与えるはずである。置換について以下に要約する。使用した略語の定義は次のとおり、すなわち:Pmp=p−ホスホノメチル−フェニルアラニン;Ppa=p−ホスファチジル−フェニルアラニンおよびNle=ノルロイシンである。
【0030】
置換:
p−ホスホ−Tyr−Aeea−ShK−Nle21−Cys35−アミド
p−ホスホノ−メチル−フェニルアラニン−Aeea−ShK−Nle21−Cys35−アミド(Pmp)
p−ホスファチジル−Phe−Aeea−ShK−Nle21−Cys35−アミド(Ppa)
p−ホスホ−Tyr−Aeea−ShK−Nle21−Cys35−アシド
p−ホスホノ−メチル−フェニルアラニン−Aeea−ShK−Nle21−Cys35−アシド(Pmp)
p−ホスファチジル−Phe−Aeea−ShK−Nle21−Cys35−アシド(Ppa)
非加水分解性PmpおよびPpaのほかに、p−ホスホノ(ジフルオロ−メチル)−フェニルアラニン(Pfp)およびp−ホスホノ−メチルケト−フェニルアラニン(Pkp)による置換もアニオン性置換であり、以下:
Pfp−Aeea−ShK−Nle21−Cys35アミド
Pkp−Aeea−ShK−Nle21−Cys35アミド
Pfp−Aeea−ShK−Nle21−Cys35アシド
Pkp−Aeea−ShK−Nle21−Cys35アシド
が得られる。
【0031】
N末端置換体の構造は付録Bに示している。本発明の範囲内にある他の構造は、ビートン、シー.(Beeton,C.)らの「自己免疫疾患の治療に向けKv1.3チャネルの選択的ペプチド阻害薬でエフェクター記憶T細胞を標的にする(Targeting Effector Memory T Cells with a Selective Peptide Inhibitor of Kv1.3 Channels for Therapy of Autoimmune Diseases)」、Molecular Pharmacology、第67巻、第4号、p.1369−(2005年)に発表されている。同文献の内容全体は参照により特別に本明細書に組み込まれ、その完全な複写は付録Cとして本明細書に添付される。
【0032】
本発明のShK類似体の治療用途
本発明は、本発明のShK類似体(上記表1に列記した類似体を含むがこれらに限定されない)のみで構成される、または当該類似体を含んでなる薬剤学的に許容可能な製剤の治療有効量(たとえば、症状または疾患進行を軽減または消失させる予防的もしくは効果的な量)を投与することにより、ヒトまたは動物対象者におけるT細胞媒介性障害(たとえば自己免疫疾患、移植片対宿主病、臓器移植の拒絶反応の予防など)、他の炎症性障害、肥満および2型糖尿病などの特定の障害または疾患を治療もしくは予防するための方法を提供する。任意の適切な投与経路(たとえば、経口、経直腸、静脈内、筋肉内、皮下、皮内、鼻腔内、局所、経粘膜、経皮、薬剤供給インプラントによるなど)を使用できる。T細胞媒介性障害の予防または治療に使用する場合は、T細胞膜上のKv1.3チャネルを阻害するのに十分な投与量を使用する。これに関して本発明のShK類似体は、さまざまな種類のT細胞媒介性自己免疫疾患の予防または治療に使用できる可能性を有する。以下に、本発明の方法により予防または治療しうる一部のT細胞媒介性自己免疫疾患の一部の例を、各疾患により主に影響を受ける標的器官に着目して分類して示す。
【0033】
神経系:
多発性硬化症
重症筋無力症
ギラン・バレーなどの自己免疫性末梢神経障害
自己免疫性ブドウ膜炎
胃腸管:
クローン病
潰瘍性大腸炎
原発性胆汁性肝硬変
自己免疫性肝炎
歯周病に随伴する骨吸収
血液:
自己免疫性溶血性貧血
悪性貧血
自己免疫性血小板減少症
内分泌:
1型糖尿病
アジソン病
グレーブス病
橋本甲状腺炎
自己免疫性卵巣炎および精巣炎
血管:
側頭動脈炎
抗リン脂質抗体症候群
ウェゲナー肉芽腫症およびベーチェット病などの血管炎
多臓器および/または筋骨格系:
関節リウマチ(RA)
変形性関節症(OA)
全身性紅斑性狼瘡
強皮症
多発筋炎、皮膚筋炎
強直性脊椎炎などの脊椎関節症
シェーグレン症候群
皮膚:
乾癬
疱疹状皮膚炎
尋常性天疱瘡
白斑
影響を受ける特定の器官とは関係なく、Tリンパ球は自己免疫疾患の発現に寄与していると考えられている。これらの疾患に現在利用可能な治療法は大いに不満足なものであり、グルココルチコイド(メチルプレドニゾロン、プレドニゾンなど)、非ステロイド性抗炎症薬、金塩、メトトレキサート、抗マラリア薬のほか、シクロスポリンおよびFK−506などの他の免疫抑制薬を使用するのが通例である。さらに、本発明の方法により予防または治療しうる別のT細胞媒介性障害は、移植片対宿主病および/または移植臓器の拒絶反応である。実際に、組織型決定、外科技術、および有効な免疫抑制治療の改良技術の開発に伴い、臓器移植の転帰は徐々に改善されてきている。しかし、移植臓器の拒絶反応は未だに重大な問題である。Tリンパ球は免疫応答に中心的な役割を演じており、多くの移植臓器の拒絶反応に大きく関与している。Tリンパ球はいわゆる移植片対宿主病の原因でもあり、該疾患は、移植した骨髄細胞がMHC不適合の宿主組織を認識して破壊する。このため、シクロスポリンおよびFK506などのT細胞性免疫を抑制する薬剤を使用して、移植拒絶反応および移植片対宿主病が予防されている。しかし残念ながら、こうしたT細胞阻害薬は毒性を有し、肝毒性および腎毒性のため、その使用が制限される。このため本発明の方法は、移植片対宿主病もしくは移植拒絶反応の治療または予防に対し、毒性の低い代替選択肢を提供しうる。また、電位差ゲート型Kv1.3カリウムチャネルの阻害薬はエフェクター記憶T細胞の抑制に特に有効であることが示されており、このため本発明の方法は、骨吸収および歯周病、乾癬、関節リウマチ、糖尿病および多発性硬化症など、エフェクター記憶T細胞に関連する疾患の予防または治療に特に有効である可能性がある。T細胞媒介性疾患のほか、Kv1.3チャネルは、エネルギーホメオスタシス、体重および末梢インスリン感受性を調節することが解明されている。このため本発明の方法は、細胞膜上のKv1.3チャネルを阻害することにより、異常なホメオスタシス、体重および末梢インスリン感受性が関わる他の疾患および障害を治療するために使用できる。このような他の疾患および障害には、歯周病における骨吸収、2型糖尿病、メタボリック症候群および肥満が含まれるが、これらに限定されない。
【0034】
フローサイトメトリーにおける本発明のShK類似体の使用
さらに本発明によれば、蛍光団で標識したShK(L5)を単独で、もしくは自己反応性細胞を検出できるクラスII四量体と併用してフローサイトメトリーに使用することにより、in vitroでT細胞媒介性障害を診断するための方法、あるいは、種々の細胞型の選別または識別の方法を提供する。フローサイトメトリーは懸濁液中の細胞を特徴付けるための柔軟な方法であり、蛍光励起細胞分取法を用いて、フローサイトメトリーで測定した特徴に基づき生細胞を選別する。フローサイトメトリーで検出しうる細胞の特徴および機能の種類には、細胞内外のタンパク質の発現、DNAの含量と生存能力とアポトーシスのタイプ、多剤耐性ポンプ活性、酵素活性、T細胞活性化、T細胞受容体特異性、サイトカイン発現、食作用ならびに酸化バースト活性が含まれる。このため、本発明の当該方法では、ShKに付加されるアミノ酸残基には、単独で、もしくは自己反応性細胞を検出できる特異的な自己抗原を取り付けたクラスII四量体と併用してフローサイトメトリーに使用するために、蛍光団の標識が組み込まれてもよい。当該フローサイトメトリーを行ないうる方法の具体的な説明は、ビートン、シー.(Beeton,C)らの「慢性的に活性化されたTリンパ球におけるKv1.3チャネルのアップレギュレーションを検出および研究するための新規蛍光性トキシン(A Novel Fluorescent Toxin to Detect and Investigate Kv1.3 Channel Up−Regulation in Chronically Activated T Lymhocytes)」J.Biol.Chem.、第278巻、第11号、p.9928−9937(2003年3月)に記載されている。一般にフローサイトメーターは、集束レーザー光を使用して、細胞が流体の流れのなかでレーザー光線を通過するときに細胞を照射する。細胞から散乱した光と、目的の細胞に取り付けた蛍光色素から放たれた光をいくつかの検出器で分析し、コンピュータで処理する。細胞は、大きさおよび形のほか、細胞内および細胞表面上のさまざまな分子の存在によっても識別および選別しうる。
【0035】
本発明のShK類似体のカリウムチャネル阻害作用および治療的有用性の例
ShKは神経細胞のKv1.1チャネルとKv1.3チャネルをほぼ同等の力価で遮断する。このため、血液脳関門に障害があり、Kv1.1チャネルを遮断するのに十分な量のShKが進入できる状況下では、神経毒性が懸念される。Kv1.3に特異的な阻害薬を設計するための我々の戦略は、フルオレセイン−6−カルボキシレート(F6CA)が20Å長のAeeaリンカーを介してShKのN末端に付加されているShK−F6CAが、Kv1.1に比べKv1.3に対し80倍の選択性を示したという我々の所見によって導かれた(ビートン(Beeton)ら、2003年)。F6CAは制限されたカルボキシレートとしても、環化したラクトンとしても存在できるため、ShK−F6CAのKv1.3特異性がF6CAの陰電荷によるものか、フルオレセインの大きく嵩高い原子核により作り出される疎水性によるものか、可能性のある平面性p電子スタッキングによるものなのか、もしくは、これらの可能性のある寄与因子がすべて組み合わされたことによるものなのか、はっきりしなかった。これらの可能性を区別するために、かつ非蛍光性のKv1.3選択的阻害薬を開発する意図をもって、N末端を置換した一連の12個の新規ShK類似体を作製し、上記相互作用の一部について探査した。Aeeaリンカーを介してチロシン、フェニルアラニンまたはこれらの誘導体(電荷、大きさおよび疎水性は多岐にわたる)をShKのN末端に付加することにより、電荷および疎水性の影響を探り、F6CA置換でみられた選択性の増強について洞察を得ることができた。
【0036】
Kv1.1阻害に比べ選択的なKv1.3阻害:
図2A〜2Dに示した実施例では、陰電荷(実効電荷2)を有する翻訳後修飾された芳香族アミノ酸であるL−ホスホチロシン(L−pTyr)を、AEEAリンカーを介してShK−Arg1に付加し、新規類似体ShK(L5)を作製した。L929細胞に安定に発現しているKv1.3 およびKv1.1チャネルで、ShKトキシンとShK(L5)を試験した。図2Bは、保持電位80mV〜40mVの200msにわたる脱分極パルスにより誘発したKv1.3およびKv1.1電流に対するShKおよびShK(L5)の作用を示している。いずれのペプチドも、投与量依存的にKv1.3およびKv1.1を可逆性に遮断し、ヒル係数は1であった。Microcal Origin(登録商標)ソフトウェアを用いて示した用量反応曲線から、Kdを判定した。ShKはKv1.3(Kd=10±1pM)およびKv1.1(Kd=28±6pM)を遮断し、予測どおり力価はほぼ同等であった(図1C)。対照的にShK(L5)は、Kv1.1(Kd=7.4±0.8nM)に比べKv1.3(Kd=69±5pM)に対し100倍選択的であった(図1B、1C)。ShK(L5)およびそのウォッシュアウトによるKv1.3電流遮断の時間経過を図1Dに示す。ShK(L5)ウォッシュインの時定数(TON)は131±21秒(n=7)であり、ペプチドウォッシュアウトの時定数(TOFF)は150±28秒(n=4)であった。KON(15×106±0.5×106M−1秒−1)およびKOFF(0.0059±0.0013秒−1)値から算出したKd(57±7pM)は、Microcal Originソフトウェアで判定したKd(69±5pM)と一致している。
【0037】
他のShK類似体もKv1.3およびKv1.1チャネルで試験した。D−ホスホチロシン(D−pTyr)を含むShK(D5)は、Kv1.1に比べKv1.3に対して35倍選択的であったが、ShK(L5)よりは一桁低い力価であった。L−pTyr−モノメチルを含むShK(L6)は、軽度(11倍)のKv1.3特異性を示し、L−pTyr−ジメチルまたはL−Tyrを含むShK類似体はKv1.1と比べてもKv1.3に対し選択的ではなかった。フェニルアラニンまたはその誘導体を含んだ類似体(かさ、p電子密度および電荷が多岐にわたる)は、K1.1と比べた場合のKv1.3に対する特異性が軽度か、または特異性がなかった。K1.1と比べた場合のKv1.3に対するShK(L5)の100倍の特異性は、ShK−F6CA(80倍)、ShK(D5)(35倍)、ShK−Dap22(33倍)または試験した他のいずれのShK類似体よりも大きい。
【0038】
本出願人は、20種類の一群のイオンチャネルについてもShK(L5)の特異性を評価し、そのデータを下記の表2に要約する。
【0039】
【表2】
上記表2のデータから理解されうるように、ShK(L5)はT細胞のKv1.3チャネルを遮断し、クローン化Kv1.3チャネル(69pM)に対するKdと同等のKd(76pM)を示した。ShK(L5)は、Kv1.3に対する選択性がKv1.1に比べ100倍、Kv1.6に比べ260倍、Kv3.2に比べ280倍、Kv1.2に比べ680倍であり、試験した他のすべてのチャネルに比べ1000倍を超えていた。重要なことにShK(L5)は、ヒトナイーブおよびTCM細胞の活性化を調節するカルシウム活性化K+チャネルであるKCa3.1に比べ、Kv1.3選択性が1600倍であった(ウルフ(Wulff)ら、2003年)。天然ShKはShK(L5)より選択性が低かった。ShKは、Kv1.3(Kd=10±1pM)に対する選択性がKv1.1(Kd=28±6pM)に比べ2.8倍、Kv1.6(200±20pM)に比べ20倍、Kv3.2(Kd=5,000±1,000pM)に比べ500倍であり、Kv1.2(10±1nM)およびKCa3.1(Kd=28±3nM)に比べ1000倍を超えていた。特異的Kv1.3阻害薬として売り込まれてきたサソリ毒由来のペプチドであるマーガトキシン(クー(Koo)ら、1997年;リン(Lin)ら、1993年;ミドルトン(Middleton)ら、2003年)も特異的ではなかった。マーガトキシンは、Kv1.3(Kd=110±12pM)に対する選択性がKv1.2(Kd=520±1pM)に比べ5倍、Kv1.1(10±1nM)に比べ9倍であり、Kv1.6およびKv3.2(Kd>100nM)に比べ1000倍を超えていた。Kv1.3阻害薬であるという根拠(レイヒ(Lahey)およびラジャダクーシャ(Rajadhyaksha)、2004年)から自己免疫疾患用に売られている栄養補助食品ルテオリン(www.lutimax.com )は、Kv1.3を弱く遮断し(Kd=65±5mM)、Kv1.1(Kd=77±5mM)、Kv1.2(Kd=63±4mM)およびKv1.5(Kd=41±3mM)に比べて選択性をまったく示さなかった。ShK(L5)のKv1.3に対する優れた選択性と該チャネルに対するpMレベルの親和性が相まって、ShK(L5)の魅力的な免疫抑制薬としての可能性を高めている。
【0040】
ヒトTEM細胞増殖の優先的抑制
図3A〜3Dを参照すると、ShK(L5)のin vitroでの免疫抑制活性を評価するために、本出願人は、ヒトTEM細胞株と、ナイーブおよびTCM細胞の混合物を含むヒトPBMCとの間で、抗CD3抗体で刺激した増殖を抑制するShK(L5)の能力を比較した。フローサイトメトリーにより、検討した2つの母集団の細胞表面の表現型を確認した。図3Aにみられるように、TEM細胞株はCCR7−CD45RA−が90%を超え、一方、図3Bに示すように、PBMCは65%のCCR7+CD45RA+(ナイーブ)および18%のCCR7+CD45RA−(TCM)細胞を含有していた。図3Cは、ShK(L5)およびShKがPMBCの増殖抑制(IC50=5nM、p<0.05)に比べTEM細胞の増殖抑制(IC50=約80pM)において60倍効果が高かったことを示している。PBMCの低い感受性は、これまでに報告されているように(ガンシャニ(Ghanshani)ら、2000年;ウルフ(Wulff)ら、2003年)、ナイーブおよびTCM細胞における、刺激によるKCa3.1チャネルの迅速なアップレギュレーションにより説明されると考えられる。この解釈を踏まえ、図3Dの上の列に示すように、PBMCを48時間活性化してKCa3.1発現をアップレギュレートし、続いて12時間休ませ、さらに抗CD3抗体で再活性化したところ、PBMCはShK(L5)遮断に対して完全に抵抗性となった。第1ラウンドの刺激でShK(L5)により抑制されたPBMCは、洗浄して休ませ、抗CD3抗体で再度誘発すると、同じようにShK(L5)に抵抗性を示した。上記の結果は、ナイーブおよびTCM細胞がKCa3.1チャネルをアップレギュレートすることによりKv1.3阻害薬を免れることを示した早期の研究を確証するものである。以上のように、ShK(L5)は優先的かつ持続的にTEM細胞の増殖を抑制する。
【0041】
ラットTEM細胞増殖の優先的抑制
ShK(L5)の治療的有効性を評価することを前提として、ラットにMS様疾患を引き起こす記憶T細胞株PASの増殖を抑制するShK(L5)の能力について検討した。対照として、本出願人はラット脾臓T細胞を使用した。2つの細胞母集団の分化状態を確認するため、ナイーブT細胞のマーカーであるCD45RCの発現を評価した(バンス(Bunce)およびベル(Bell)、1997年)。図4Aに示すように、ラット脾臓T細胞はCD45RC+が76%であったが(すなわち主にナイーブ細胞)、PAS細胞はCD45RC−であったことから記憶細胞であることが示唆される。PAS細胞がTEM状態にあるか、もしくはTCM状態にあるか判断するために、活性化の前および48時間後にKv1.3発現を調査した。TEM細胞は刺激によりKv1.3レベルを著明にアップレギュレートするが、TCM細胞ではアップレギュレートしないと予測される。図4Bを参照すると、パッチクランプ法実験から、PAS細胞のMBP刺激後にKv1.3電流の振幅が著しく増大することが明らかにされ、TEM細胞であることと一致している。PAS細胞上のKv1.3チャネル数を測定する独自の測定法として、Kv1.3と特異的に結合することが報告されている蛍光標識したShK類似体であるShK−F6CAを用いた。フローサイトメトリーで測定したShK−F6CAの染色強度は、細胞表面に発現したKv1.3四量体の数を反映している。図4Cにみられるように、ShK−F6CA(10nM)の染色強度はPAS細胞のMBP活性化とともに増大し、過剰な非標識ShK(L5)(100nM)がShK−F6CA染色を競合的に阻害した。最終試験として本出願人は、固定してKv1.3特異抗体で染色した静止期PAS細胞およびMBPで刺激したPAS細胞について共焦点顕微鏡検査を行なった。図4Bおよび4Cのデータと一致して、休止期PAS T細胞はKv1.3染色強度が4.4±0.6であり、この値は抗原による活性化後に10.6±2.3(p<0.005)に増大したことから(図4Dを参照)、活性化後にKv1.3タンパク質発現が増加したことを示している。このため、MBPで活性化したPAS細胞は、CD45RC−Kv1.3highTEM細胞であり、一方、実験に用いたラット脾臓T細胞は大部分がナイーブ状態である。
【0042】
ShK(L5)およびShKによる、PAS細胞のMBP誘発増殖の抑制(IC50=約80pM)は、ラット脾臓T細胞のマイトジェン誘発増殖の抑制(IC50=約100nM)よりも約1000倍も効果的であった(図4E参照、p<0.05)。以上の結果は、上に述べたヒトT細胞による所見を確証するものである。図4Gにみられるように、ShK(L5)はPAS細胞によるMBP誘発IL2産生を阻害し(図4F)、外因性IL2はPAS細胞増殖のShK(L5)抑制を部分的に無効にした(図4G)。早期の研究では、ヒト、ラットおよびミニブタのT細胞について特異性の低いKv1.3阻害薬に関する類似した所見が報告された。以上を要約すると、ShK(L5)はヒトおよびラットのTEM細胞の強力かつ選択的な阻害薬であり、したがって自己免疫疾患の病因に寄与するTEM細胞を優先的に標的にすることにより、当該疾患において治療用途を有する可能性がある。
【0043】
血中半減期および安定性
パッチクランプ法によるバイオアッセイを用いて、皮下注射後のShK(L5)の血中濃度がTEM細胞の阻害に十分であるかどうか確認した。これらの実験の結果を図5A〜5Fに示す。
【0044】
Kv1.3チャネルに対する遮断活性について、ShK(L5)投与ラットおよび対照ラットからの血清試料を試験した。対照血清は検出可能な遮断活性を示さなかったことから、内因性チャネルブロッカーは存在しないことが示された。該アッセイを標準化するため、既知量のShK(L5)をラット血清に添加し、当該試料をKv1.3チャネルについて試験した。添加した該血清試料は用量依存的にKv1.3電流を遮断し(Kd=77±9pM)、血清がない場合のShK(L5)の作用と区別できなかった(図4A)。標準曲線と比較することにより、投与した動物のShK(L5)濃度を測定した。ShK(L5)は、200mg/kgの単回皮下注射から5分後に血清中に検出可能であった。30分で最高濃度(12nM)に達し、その後420分かけて約300pMのベースライン値まで低下した。血中からのShK(L5)の消失は、単一指数でフィッティングさせることができた。血中半減期は約50分であると推定された。
【0045】
200mg/kg投与後の最高血清中濃度(12nM)は、Kv1.3チャネルおよびTEM細胞機能を選択的に遮断する必要量を超えていることから、これより少ない投与量を試験した。10mg/kgの単回注射後、30分以内にShK(L5)の最高血清中濃度約500pMに達し(データ省略)、この濃度は90%を超えるKv1.3を遮断するのに十分であり、Kv1.1には影響を及ぼさない濃度である。上記投与量(10mg/kg/日)を1日1回反復投与した結果、約300pMの定常状態濃度に達し(注射後24時間時点で測定、図5D)、これはTEM細胞を60〜70%抑制するのに十分な濃度であり、ナイーブ/TCM細胞にはほとんど影響を及ぼさない。この「定常状態」濃度は、血中半減期が約50分と推定されたことを考えると予期しない結果であり、ShK(L5)は反復投与により「蓄積される」ことを示している。その「貯蔵所」が皮膚なのか、または身体の他の部位なのか判断するために、10mg/kgのShK(L5)を単回静脈内注射または皮下注射したラットの10時間後のShK(L5)の血中濃度を測定した。当該ペプチドは、いずれの投与経路でも同一の経時変化で消失したことから(図5E)、10mg/kg/日の単回注射後に到達したShK(L5)の定常状態濃度300pMには皮膚が関与しておらず(図5D)、貯蔵所はほかにあることが示された。
【0046】
10mg/kg/日の単回注射後にShK(L5)の定常状態濃度300pMにうまく到達したことは、該ペプチドがin vivoで安定であることを示唆している。その安定性を直接検討するために、ラット血漿中またはラット血漿を2%含有するPBS中で、インキュベーション時間をさまざまに変えて37℃でShK(L5)をインキュベートし、Kv1.3の遮断活性を測定した。いずれの添加試料のセット(血漿およびPBS)においても、約5時間でKv1.3遮断活性が50%低下することを観察したが、これはおそらくペプチドの試験管プラスチック表面との結合によるものと思われ、濃度は次の2日間も定常状態を維持していた(図5F)。追加の安定性試験として、ShK(L5)投与ラットからの血清を用いてKv1.3遮断活性とKv1.1遮断活性を比較した。pTyrの脱リン酸化またはAeea−pTyr側鎖の切断のいずれかにより、ShK(L5)がin vivoで修飾されるとすれば、それぞれShK(L4)およびShKが生成されることになり、これらはいずれもKv1.1と比較してKv1.3への選択性がない。ShK(L5)投与動物からの血清試料は、ShK(L5)と同じように、Kv1.1に比べてKv1.3に対する選択性を示したことから、該ペプチドは上記の修飾を受けないことが示された。以上を統合すれば、これらの結果は、ShK(L5)は血漿中できわめて安定であり、毎日の10mg/kgの単回皮下注射後に薬理学的に適切な血清中濃度に達することを示している。
【0047】
無毒性
本出願人は、in vitroおよびin vivo試験をいくつか実施してShK(L5)が毒性を示すかどうか判定した。当該試験の結果を付録Aに要約する。ヒトおよびラットのリンパ系細胞を、Kv1.3半遮断量またはTEM抑制のIC50(70〜80pM)の1200倍を超える濃度のShK(L5)(100nM)とともに48時間インキュベートしたところ、軽微な細胞毒性を示した。同じ高濃度のShK(L5)は、試験株TA97Aを用いたエイムス試験で陰性であったことから、変異原物質ではないことが示唆された。いずれのin vitro試験においても特筆すべき毒性は検出できなかった。
【0048】
薬剤誘発性のKv11.1(HERG)チャネルの遮断は、重大な心毒性に寄与しており、いくつかの薬剤が市場から回収されている。ShK(L5)は、100nM(Kv1.3に対するKdの1430倍を超える)ではKv11.1チャネルに何の影響も及ぼさず、したがって、本出願人が選択した治療処方(10mg/kg/日、定常状態血中濃度300pM)は心毒性を引き起こさないはずである。さらなる試験として本出願人は、第1日に賦形剤(PBS+2%ラット血清)を投与し、第2日にShK(L5)10mg/kg/日を投与した覚醒ラットを用いて、心拍変動解析を行なった。ShK(L5)は、時間および頻度のいずれの領域においても心拍および標準HRV(心拍変動)パラメータ(欧州循環器病協会および北米心拍調節・電気生理学協会の特別委員会、1996年)に何の影響も及ぼさなかった。
【0049】
上記急性毒性実験の余勢を駆って本出願人は、10mg/kgのShK(L5)または賦形剤を2週間にわたって1日1回皮下注射したラットを用いて亜慢性試験を行なった(各群n=6)。ShK(L5)投与動物は、賦形剤投与動物と同じ程度に体重が増加した(付録A)。血液学的および血液生化学的解析により、ShK(L5)投与ラットと賦形剤投与ラットには差がないことが示され、フローサイトメトリー解析により、胸腺細胞およびリンパ球サブセットの割合にも差がないことが示された(付録A)。これらの試験を集合すると、ShK(L5)は安全であることが示唆される。
【0050】
治療的安全指数を判定するため、健康なラットに60倍高い投与量(600mg/kg/日)のShK(L5)を5日間投与したところ、毒性の臨床徴候は観察されず、健康なラットに1000mg/kgのShK(L5)を単回注射したときも毒性はみられなかった。EAEおよびMSで起こるように、血液脳関門に障害がある場合は、この状況は楽天的ではない。ShK(L5)10mg/kg/日を10日間投与されたEAEラットは、毒性の徴候をまったく示さなかった。対照的に、600mg/kg/日を5日間投与したラットは、その40%(5/12)が第5日にEAEの臨床徴候を発現して死亡した(推定LD50=750mg/kg/日)。200mg/kg単回注射後の血清中のShK(L5)最高濃度(12nM)は、50%を超えるKv1.1チャネルを遮断するのに十分な濃度であることから、600mg/kg/日のShK(L5)を投与したEAEラットに観察された毒性は、Kv1.1を遮断するのに十分な量のShK(L5)が脳内に進入したことによるものである可能性が高い。したがって、血液脳関門に障害がない状況(中枢神経系(CNS)に影響を及ぼさない自己免疫疾患にみられる状況)では、ShK(L5)の有効な治療的安全指数は100を十分に超えるが、血液脳関門が破られると治療的安全指数は75である。
【0051】
DTHおよび養子移入性急性EAEの予防
図6A〜6Cを参照すると、2つの動物モデルにおいてin vivoでの免疫抑制活性についてShK(L5)を評価した。本出願人は、MBPで活性化したPAS TEM細胞をLewisラットに移入して誘発した急性EAEを予防および治療する能力のほか、TEM細胞で媒介されるDTH反応を抑制する能力も試験した。PAS細胞をin vitroで48時間にわたってMBPで活性化し、続いてLewisラットに養子移入した(生細胞数:6〜8×106)。次に予防試験では、ラットに生理食塩水(対照)またはShK(L5)(10μg/kg/日)を5日間にわたって皮下注射した。第1の予防試験では、対照ラットは軽度EAEを発現し(平均最大臨床スコア2.0±1.2)、平均発症日は5.6±0.6日であった(データ省略)。ShK(L5)は疾患の重症度を軽減した(平均最大臨床スコア0.7±0.6、p<0.05)。第2の予防試験では、対照ラットはより重度のEAEを発現し(平均最大臨床スコア3.2±0.4)、平均発症日は4.8±0.4日であった(図6A)。ShK(L5)は疾患の重症度を有意に軽減した(平均最大臨床スコア0.6±0.4、p<0.007)が、疾患発症日は有意には遅延させなかった(5.5±0.7日;p=0.07)。これらの試験では毒性の徴候がまったくみられなかった。
【0052】
治療試験(図6B)では、ラットにMBP活性化PAS細胞を注射し、最初にEAEの徴候(尾のひきずり、猫背および24時間で6%以上の体重減少)を発現したときに生理食塩水または10μg/kg/日のShK(L5)を投与し、3日間にわたって治療を続けた。EAEの臨床徴候は、対照群では第6日に最大に達し(スコア=3.9±0.7)、治療群では第7日に最大に達した(スコア=1.9±0.9;p<0.05)。
【0053】
in vivoにおけるShK(L5)の免疫抑制活性の独自の評価法として本出願人は、主に皮膚ホーミング性TEM細胞により媒介されるDTH反応を阻害する上でのShK(L5)の有効性を検討した。卵アルブミンおよびアジュバントで免疫したLewisラットに、7日後に片耳に卵アルブミンを接種し、他方の耳に生理食塩水を接種した。続いてラットに生理食塩水(対照)またはShK(L5)(10μg/kg/日)を注射し、DTHの指標として耳の厚さを測定した。全ての対照ラットが卵アルブミン接種後24時間および48時間の時点で耳の腫脹を発現したが、ShK(L5)投与動物ではDTH反応がかなり軽度であった(図6C)。したがってShK(L5)は、TEM媒介性DTH反応を抑制し、ミエリン活性化TEM細胞により誘発される重度の養子移入性EAEを予防および改善する。
【0054】
Kv1.3は抗原提示によりISに集積するが、Kv1.3を通るK+の流出はISの形成および安定性に必要ない
図7A〜7Gを参照すると、高選択性Kv1.3阻害薬(21)であるShK(L5)は、GAD65特異的TEM細胞においてKv1.3電流を遮断し、Kdは72±3pMであった。ShK(L5)を薬理学的プローブとして使用して、Kv1.3機能を必要とするTEM細胞活性化のステップを明らかにした。生化学的研究から、Kv1.3およびKvb2は、SAP97(シナプス関連タンパク質97)、ZIP(PKCゼータ結合タンパク質、p56lck関連p62タンパク質、A170)、p56lckおよびCD4を含むシグナル伝達複合体に属することが示されている(図7B)。ヒトTEM細胞における当該複合体の存在は、Kv1.3、Kvb2、SAP97、ZIPおよびp56lckとCD4との共キャップ形成を示す本出願人の結果により裏付けられている。さらに、FRET(蛍光エネルギー変換)研究から、Kv1.3をトランスフェクトしたヒトT細胞ではKv1.3がCD3に非常に接近しており、さらに該チャネルは、Kv1.3をトランスフェクトしたヒト細胞傷害性T細胞とその標的との接点に優先的に局在していることが示されている。CD4は、T細胞と抗原提示細胞(APC)が接する領域であるISに移動するため、シグナル伝達複合体のKv1.3および他のタンパク質も抗原提示によりISに局在する可能性がある。この考えを検討するため、T1DM患者からのGAD65特異的Kv1.3highTEMクローンを、GAD65 557Iペプチドを取り付けてDAPIで染色して視覚化を助けたHLA適合APCとともにインキュベートした。20分後、シグナル伝達複合体中のタンパク質を検討するため、APC−TEM結合体を免疫染色した。ISには、Kv1.3、Kvb2、SAD97、ZIPおよびp56lckとともにCD4が局在していた。APCとTEMとの接触のない場合では、CD4およびKv1.3は細胞全体に分布していた。さらに、GAD65特異的TEM細胞がMBP(無関連抗原)を取り付けたAPCに曝された場合は、CD4およびKv1.3は接点に局在できなかったことから、ISの集積は抗原特異的であることが立証された。以上のように、GAD65特異的TEM細胞では、抗原提示により、Kv1.3含有シグナル伝達複合体がCD4とともにISに移動することから、Kv1.3はTEM細胞におけるシグナル変換機構に不可欠な構成要素であることが示唆される。以上の試験に基づけば、Kv1.3チャネルの約99%を遮断する濃度(100nM)のShK(L5)は、ISの集積を妨げず、ひとたび形成されたISを崩壊させなかったことから、Kv1.3チャネルを通るK+の流出はISの形成または安定性に不必要であることが示された。
【0055】
ヒトTEM細胞の抑制
図8A〜8Eを参照すると、ShK(L5)は、T細胞活性化に不可欠な初期段階であるTEM細胞のカルシウムシグナル伝達を阻害した。T1DM患者からのGAD65特異的TEMクローンに、カルシウム指示薬である色素Fluo3を取り付け、培養液のみで、または濃度を増大させたShK(L5)とともにプレインキュベートし、活性化用抗CD3抗体および架橋用第2抗体の添加前後にフローサイトメトリーにより画像化した。刺激後242±35秒で最高カルシウム上昇が起こり、これがShK(L5)で遮断され、IC50は約200pMであった(図8A)。ShK(L5)は、T1DM患者およびRA患者からのナイーブ/TCM細胞に比べ、同患者からの自己反応性TEM細胞による[3H」チミジン取込みを10倍効果的に抑制した(図8B、左)。第2セットの実験(図8B、右)では、RA−SFおよびRA−PBT細胞を抗CD3抗体で48時間活性化し、それぞれ「TEMエフェクター」および「ナイーブ/TCMエフェクター」を作製した。培地中で細胞を一晩休ませ、ShK(L5)の存在下または非存在下で抗CD3抗体でさらに48時間再刺激し、[3H」チミジン取込みを測定した。RA−SF−TEMエフェクターはShK(L5)阻害に対する感受性を保持していたが、RA−PB−ナイーブ/TCMエフェクターはKv1.3遮断に抵抗性であり(図8B,右)、これはおそらく、該エフェクターがカルシウム活性化KCa3.1/IKCa1チャネルをアップレギュレートしたことによるものである可能性が高く、このチャネルはカルシウム流入の際にKv1.3の代わりになる。ShK(L5)は、T1DMおよびRA患者からのTEM細胞によるインターロイキン2(IL2)およびインターフェロン−g(IFN−g)の産生を大いに抑制したが、これらの患者からのナイーブ/TCM細胞によるIL2およびIFN−gの産生はそれほど影響を受けなかった(図8C)。TEM細胞およびナイーブ/TCM細胞による腫瘍壊死因子−αおよびインターロイキン4の産生は、いずれもShK(L5)に感受性が低かった(図8C)。
【0056】
エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルの検証
図9に示すように、アジュバント中の卵アルブミン(OVA)でラットを免疫した。7日後に片耳にOVAを接種し、他方の耳に生理食塩水を接種した。遅延型過敏症(DTH)の徴候として、24時間後に耳の腫脹を測定した。図9に示したFACSヒストグラムは、OVAを接種した耳のT細胞はCD45RC陰性記憶細胞であり、一方、同一ラットの血液および脾臓のT細胞は大部分がナイーブT細胞であることを示している。
【0057】
エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルにおけるShK(L5)の治療プロトコル
図10に示すように、ナイーブ細胞のエフェクター記憶TEM細胞への分化を妨げるため第0日から第7日まで(初回刺激段階)、もしくはTEM細胞の機能を妨げるため耳に卵アルブミンを接種した後のエフェクター段階で、皮下注射として10μg/kg/日のShK(L5)をラットに投与した。
【0058】
ShK(L5)はナイーブおよびセントラル記憶T細胞またはB細胞の機能を損なわずにラットin vivoにおいてエフェクター記憶反応を特異的に抑制する
図11に示すように、対照ラットは耳の腫脹、すなわち陽性DTH反応を発現した。ShK(L5)は、初回刺激段階で投与するとDTHの抑制に効果がなかったことから、ナイーブおよびセントラル記憶T細胞のエフェクター記憶細胞への分化を抑制しなかったことを示している。ShK(L5)は、エフェクター段階で投与するとDTHを抑制したことから、エフェクター記憶T細胞が耳に達する能力を妨げたか、またはエフェクター記憶T細胞の活性化を抑制したかのうち少なくともいずれかであることを示している。ShK(L5)投与ラットの耳のT細胞数は、賦形剤を投与したラットの耳と同じであったため、第一の可能性は除外された。エフェクター記憶T細胞はKv1.3陰性であったが、賦形剤を投与した動物の耳の記憶T細胞はKv1.3陽性であったため、ShK(L5)は耳におけるエフェクター記憶T細胞の活性化を抑制したのである。上記動物ではIgMおよびIgG B細胞の反応も影響を受けなかった。
【0059】
T1DMまたはMSの患者および健康な対照者からの、GAD65/555−567、インスリン/9−23およびミエリン抗原に特異的なT細胞におけるKv1.3発現
図12Aは、新規発症1型糖尿病患者、健康な対照者および多発性硬化症患者に由来する抗原特異的T細胞からのKv1.3電流(上)およびチャネル数/細胞(下)を示している。各データ点は、3回目の抗原刺激から48時間後に測定した、単一ドナーからの2〜4のT細胞株に由来する細胞20〜50個についての平均値±SEMを示している。T1DM患者および対照者の血中にはインスリンおよびGAD65に特異的なT細胞の頻度が低いため、スプリットウェル法を用いて短期自己抗原特異的CD4+T細胞株を作製することにより、上記母集団を増幅した。対照として、MSには関係しているがT1DMには関係のない無関連自己抗原であるミエリン塩基性タンパク質(MBP)に特異的なT細胞株を作製した。3回目の抗原刺激後に、4pFを超える膜電気容量を有する活性化細胞を用い、ホールセルパッチクランプ法によりKv1.3電流を測定した(細胞直径11μm以上)。代表的なKv1.3電流およびKv1.3チャネル数/T細胞を図12Aに示す。電流は、Kv1.3に特徴的な生物物理学的および薬理学的特性を示した。新規発症T1DM患者由来のインスリン(9〜23)またはGAD65(555〜567)特異的T細胞は、Kv1.3電流が大きく、Kv1.3チャネル数が多かったが、当該患者由来の疾患とは無関連なMBP特異的T細胞はKv1.3lowであった(p=0.001)。比較のため、反対のパターンが観察されたMS患者について本出願人が発表したKv1.3データをプロットした。MS患者では、MBPまたはミエリン乏突起神経膠細胞の糖タンパク質(ペプチド35〜55)もしくはプロテオリピドタンパク質(ペプチド139〜151)に特異的なT細胞はKv1.3highであったが、インスリンおよびGAD65に特異的なT細胞はKv1.3lowであった(p=0.0001)。健康対照者から単離した自己反応性T細胞は、特異性に関係なくKv1.3lowであった。MSおよびT1DMの両方を有する患者1例では、3つの自己抗原に特異的なT細胞がKv1.3highであった。長期にわたるT1DMを有する患者からのGAD65特異的およびインスリン特異的T細胞がKv1.3highであったことは、自己反応性TEM細胞の持続性を反映しているが、非自己免疫性2型DM患者からのGAD65およびインスリン特異的T細胞にはKv1.3lowパターンがみられた。図12Bに、個々の細胞のKv1.3の染色強度(上)および蛍光強度(下)を示す。本出願人は、Kv1.3の免疫染色によりパッチクランプ法のデータを確認した。T1DM患者からのインスリン特異的およびGAD65特異的T細胞、ならびにMS患者からのMBP特異的T細胞は明るく染色されたが、無関連自己抗原に特異的な細胞の染色はかすかであった。図12CはCCR7発現を示している。フローサイトメトリーにより、Kv1.3highT細胞はCCR7−TEM細胞であるが、Kv1.3low細胞はCCR7+ナイーブまたはTCM細胞であることが示された。図12Dは、T1DMおよびMSの両方を有する患者からと、それぞれ5年以上および2年以上のT1DMまたは2型DMを有する患者からの自己反応性T細胞における1細胞あたりのKv1.3の数を示している。図12Eは、新規発症T1DM患者からのCD4+GAD65四量体+T細胞におけるKv1.3数を示している。さらなる対照として、GAD65 557Iペプチドを含有する蛍光性MHCクラスII四量体を使用して、新規発症T1DMを有するDR−0401陽性患者からGAD65特異的CD4+T細胞を単離した。四量体で選別したGAD65活性化T細胞は、T1DM患者からのGAD65特異的T細胞株に観察されたパターンと同じKv1.3highパターンを示した。以上を要約すると、T1DMおよびMSのいずれにおいても、疾患関連性の自己抗原活性化T細胞はKv1.3highCCR7−TEMエフェクターであり、一方、当該患者における疾患無関連性の自己反応性細胞はKv1.3lowCCR7+ナイーブ/TCM細胞である。
【0060】
関節リウマチおよび変形性関節症におけるKv1.3発現
RAでは、罹患関節からの疾患関連T細胞の単離が可能である。本出願人は、抗CD3抗体で刺激後48時間の時点でRA患者7例の滑液(SF)から採取したT細胞についてパッチクランプ法を行なった。図13Aにみられるように、対照として本出願人は、非自己免疫性の退行性変形性関節症(OA)患者7例からのSF−T細胞(同一プロトコルで活性化した)を分析した。RA−SFのT細胞はKv1.3highであり、一方、OA−SFのT細胞はKv1.3lowであった(p<0.0001)。本出願人は、RA患者の末梢血(PB)からの抗CD3活性化T細胞にKv1.3lowパターンを見出した(p<0.0001)が、これは血液中には自己反応性Kv1.3highTEM細胞がまれであるためである。Kv1.3およびその付属のKvβ2サブユニットの免疫染色により、パッチクランプ法のデータの確証を得た。図13Bは、 Kv1.3(図では明灰色)および Kvβ2(図では暗灰色)染色の共焦点画像を示している。RA−SFのT細胞はKv1.3およびKvβ2の両方が明るく染色されたが、OA−SFおよびRA−PBのT細胞は弱い染色を示した。図13CはCCR7発現を示している。フローサイトメトリーにより、Kv1.3highRA−SF T細胞はCCR7−TEM細胞であるが、Kv1.3lowOA−SFおよびRA−PB T細胞はCCR7+ナイーブ/TCM細胞であることが検証された。図13D(上)は、抗CD3抗体または抗Kv1.3抗体で染色し、ヘマトキリン/エオシンで対比染色したRAおよびOA患者からの滑膜の顕微鏡写真を示している(40倍)。さらなる試験として、RA患者5例およびOA患者5例からのパラフィン包埋滑膜組織(ST)をCD3、Kv1.3およびCCR7について免疫染色した。以前に示したとおり、本出願人の染色法は、ナイーブ/TCM細胞ではKv1.3チャネル数が少ないためにKv1.3を検出しない。RA−STではCD3+Kv1.3+CCR7−TEM細胞の優勢がみられたが、OA滑膜ではCD3+細胞がまばらであり、これらの細胞は主にKv1.3−CCR7+ナイーブ/TCM細胞であった。分類法により評価したCD3+、Kv1.3+およびCCR7+細胞による浸潤度を図S2Aに示す。CD3+炎症指数:RA=3.2±0.1;OA=1.1±0.2(p<0.01);Kv1.3+炎症指数:RA=2.8±0.3;OA=0.6±0.3(p<0.01)。以上のように、異なる3つの自己免疫疾患において、我々の結果は、疾患関連性自己反応性T細胞がKv1.3highCCR7−TEMエフェクターであることで一致している。
【0061】
本発明の一部の実施例および実施形態を参照しながら本明細書において本発明を説明してきたが、上記実施例および実施形態には、本発明の趣旨および範囲から逸脱することなく種々の追加、削除、変更および修正を加えうることを理解されたい。たとえば、1つの実施形態または実施例の任意の要素もしくは属性は、実施形態または実施例がその意図した用途に対し不適切なものとならない限り、別の実施形態または実施例に組み込んでもよいし、別の実施形態または実施例とともに使用してもよい。また、方法または手順の工程が特定の順番で列記もしくは記述されている場合、当該工程の順番は、特に定めのない限り、または当該工程の順番の変更が本発明を特許性のないもの、もしくはその意図した用途に対し不適切なものとすることがない限り、変更してもよい。妥当な追加、削除、修正および変更はすべて、説明した実施例および実施形態と同等であるとみなすものとし、特許請求の範囲内に含まれるものとする。
【0062】
【表3】
【0063】
【化1】
【技術分野】
【0001】
本発明は、a)新規な組成物、b)Tリンパ球およびBリンパ球ならびに他のタイプの細胞におけるKv1.3チャネルのin vivoおよび/またはin vitro阻害のための方法およびキット、ならびに、c)ヒトまたは動物において自己免疫疾患またはその他の障害を治療するための方法を提供する。
【背景技術】
【0002】
(関連出願)
本出願は、2004年10月7日出願の米国仮特許出願第60/617,395号の優先権を主張するものであり、この仮出願の内容全体を参照により本明細書に明確に組み込む。
【0003】
(発明の背景)
細胞膜は真核細胞の外側表面を形成する。細胞膜を通る受動拡散により、種々のイオン(たとえば、ナトリウム、カリウム、カルシウムなど)が細胞に出入りする。細胞に出入りするこのようなイオンの拡散は、細胞膜内に存在する「イオンチャネル」によって促進される。イオンチャネルは、細胞膜内に埋め込まれたタンパク質であり、細胞膜を横切るイオンの選択的な流れを制御しており、これによって細胞内部の内容物と細胞周囲の細胞外液との間に濃度勾配を形成させる。イオン濃度は、興奮性細胞(たとえば神経細胞)の電気的活動に直接関与するため、イオンチャネルの機能(または機能不全)は当該細胞の電気的特性および挙動を実質的に制御することができる。実際に、幅広く「チャネル病」と呼ばれる種々の疾患は、イオンチャネルの機能不全または機能異常に関連していると考えられている。
【0004】
イオンチャネルは、開閉可能な場合は「ゲート型」と呼ばれる。基本タイプのゲート型イオンチャネルには、a)リガンドゲート型チャネル、b)機械ゲート型チャネルおよびc)電位差ゲート型チャネルがある。特に、電位差ゲート型チャネルは神経細胞、筋細胞、およびリンパ球などの非興奮性細胞にみられる。これらのチャネルは、細胞膜を通る電荷の変化に反応して開閉する。
【0005】
Kv1.3チャネルと自己免疫疾患
多発性硬化症(MS)、1型糖尿病(T1DM)、関節リウマチ(RA)および乾癬などの自己免疫疾患は、世界で数億人が罹患している。これらの障害では、特異的自己反応性T細胞−たとえば、MS患者ではミエリン特異的T細胞−が疾患の経過中に自己抗原刺激を繰り返し受け、慢性的に活性化された記憶細胞に分化し、これが炎症性組織に遊走してサイトカインを分泌することにより病因に寄与すると考えられている(ヴィグリエッタ(Viglietta)ら、2002年;ヴィサース(Vissers)ら、2002年;ウルフ(Wulff)ら、2003年b)。慢性的に活性化された記憶T細胞を優先的に標的とする治療法があれば、自己免疫疾患に対し重要な価値を有すると考えられる。
【0006】
記憶T細胞は、ケモカイン受容体CCR7およびホスファターゼCD45RAの発現に基づき、セントラル記憶(TCM)およびエフェクター記憶(TEM)の2つのサブセットに分けられる(ゲギナト(Geginat)ら、2001年;サルスト(Sallusto)ら、1999年)。ナイーブおよびTCM細胞は、炎症部位に遊走する前はリンパ節に存在し、一方、TEM細胞は炎症部位に直接存在し、その場で大量のIFN−βおよびTNF−αを分泌し、即時型エフェクター機能を示す。最近になって、MS患者ではミエリン特異的自己反応性T細胞は大部分が、活性化したTEM細胞であることが示され(Wulffら、2003年b)、ミエリン特異的活性化ラットTEM細胞をナイーブなレシピエントに養子移入すると重度のEAEを誘発することが示された(ビートン(Beeton)ら、2001年a;ビートン(Beeton)ら、2001年b)。電位差ゲート型Kv1.3K+チャネルは、TEM細胞の免疫調節に対する非常に刺激的な新しい治療標的である。TEM細胞は活性化されるとKv1.3チャネルをアップレギュレートし、その抗原誘発性増殖はKv1.3ブロッカーにきわめて感受性が高い(ウルフ(Wulff)ら、2003年b)。対照的にナイーブおよびTCM細胞は、そもそもKv1.3ブロッカーへの感受性がかなり弱く、カルシウム活性化K+チャネルIKCa1をアップレギュレートすることによりKv1.3遮断に対し急速に抵抗性となる(ガンシャニ(Ghanshani)ら、2000年;ウルフ(Wulff)ら、2003年b)。
【0007】
TEM細胞においてKv1.3が優性であることは、特異的Kv1.3阻害薬により該サブセットの活性を操作する強力な方法が提供されることになる。該チャネルの組織分布が機能的に制限されていることと、in vivoでのKv1.3遮断が、動物モデルにおいて、TEM媒介性EAE、歯周病の骨吸収および遅延型過敏性反応を明白な副作用を引き起こさずに改善するという事実が、治療標的としてのKv1.3の魅力をいっそう高めてきた(ビートン(Beeton)ら、2001年b;クー(Koo)ら、1997年;ヴァルヴェルデ(Valverde)ら、2004年)。Kv1.3ブロッカーは活性化されたTEM細胞(たとえば、ワクチン抗原に特異的なTEM細胞)をすべて抑制すると考えられるが、Kv1.3に基づいた治療法は、免疫系全体を幅広く無差別に調節する現行の治療法よりもかなり改善された治療法になると考えられる。Kv1.3ブロッカーのこのほかの利点は、可逆性であることである。このため、症状が治まるまで数カ月かかる化学療法薬とは異なり、必要時にKv1.3ブロッカーの治療効果を適正に増減し、感染に直面したら治療を中止することもできる。
【0008】
Kv1.3チャネルと肥満
Kv1.3チャネルはエネルギーホメオスタシスとエネルギーバランスに関与していることが見出された(非特許文献1)。Kv1.3チャネルを遺伝学的にノックアウトしたマウスは、脂肪食を摂取しても体重は増加しなかったが、同一の食餌を与えた対照マウスは過体重となった。Kv1.3チャネルの薬理学的遮断は、Kv1.3チャネルの遺伝子ノックアウトの上記効果を再現した。したがって、Kv1.3ブロッカーは、肥満の管理に使用できる可能性が高い。
【0009】
Kv1.3チャネルと2型糖尿病
Kv1.3チャネルは、肝臓および筋肉などの末梢標的器官におけるインスリン感受性の調節に関与している(非特許文献2)。マウスでは、Kv1.3チャネルの遺伝子ノックアウトが肝臓および筋肉のインスリンに対する感受性を増強した。したがって、Kv1.3ブロッカーは、インスリンの末梢作用を増強し、これによって血糖値を低下させることにより、2型糖尿病の治療に使用できる可能性がある。
【0010】
Kv1.3チャネルを阻害することが知られている天然ポリペプチド
最も強力なKv1.3阻害薬はカリブイソギンチャクStichodactyla helianthus由来のペプチドShKである。ShKは、3つのジスルフィド架橋により連結された35残基のポリペプチドである。ShKは、pMレベルの濃度でKv1.3を遮断して(Kd=11pM)TEM細胞の増殖を抑制し、さらに、ミエリン特異的TEM細胞の養子移入により誘発したラットの実験的自己免疫性脳脊髄炎(EAE)を改善する。考えられるShKの欠点は、神経細胞のKv1.1チャネルに対し低pM濃度の親和性がある点である(Kd=28pM)。EAE試験ではShKに副作用はまったく観察されなかったが、MSにおいて血液脳関門に障害があると起こる可能性があるように、高濃度のShKが脳内に進入すれば好ましくない神経毒性を招くと考えられる。したがって、特異性の高いKv1.3阻害薬の開発が必要である。製薬産業および学術機関の広範囲に及ぶ努力により、中位のnM範囲でKv1.3を阻害する小分子がいくつか産出されたが、これらの物質は実行可能な薬剤候補となるような選択性および力価を有していない。
【0011】
これまでにShKの断片化ペプチド類似体がいくつか報告されている。これらのShK類似体の1つでは、天然の配列が断片化され、続いて追加の共有結合(1つの非天然ジスルフィドおよび2つのラクタムブリッジ)を導入することにより安定化された。このほかの類似体では、ジスルフィドおよび/またはラクタムブリッジにより安定化された非天然骨格構造が、天然トキシン由来の重要なアミノ酸残基を含むように修飾された。これらのShK類似体は、程度がさまざまなKv1.3阻害活性および特異性を示した(非特許文献3)。
【先行技術文献】
【非特許文献】
【0012】
【非特許文献1】Hum Mol Genet.2003年、第12巻、p.551−9
【非特許文献2】Proc Natl Acad USA.2004年、第102巻、p.3112−7
【非特許文献3】ラニガン、エム.ディー.(Lanigan,M.D.)ら「カリウムチャネルブロッカーShKトキシンの設計したペプチド類似体(Designed Pepetide Analogues of the Potassium Channel Blocker ShK Toxin)」、Biochemistry,25;40(51):15528−37(2001年12月)
【発明の概要】
【発明が解決しようとする課題】
【0013】
当技術分野には、リンパ球のKv1.3チャネルを選択的に阻害しながら、Kv1.1チャネルまたは他のカリウムチャネルを阻害する作用を最小限に抑えた、もしくはその作用がまったくない新規のShK類似体を開発する必要が依然として存在する。
【課題を解決するための手段】
【0014】
本発明は、アニオン電荷を有する有機または無機化学物質(たとえば、原子、分子、化学基、残基、化合物、分子部分など)を付加した(たとえば、結合した、リンカーにより連結した、または別の方法で結びつけた)ShKトキシンを含んでなる新規組成物(本明細書で「ShK類似体」と呼ぶ)を提供する。
【0015】
さらに本発明に基づき、カリウムチャネルを阻害する方法、および/または本発明のShK類似体の有効量をヒトもしくは動物の対象者に投与することにより該対象者において疾患または障害を治療する方法を提供する。一部の実施形態では、ShKトキシンに付加する化学物質は、他のカリウムチャネル(たとえばKv1.1チャネル)よりも特定のカリウムチャネル(たとえばKv1.3チャネル)に対し選択的阻害を提供するように選ぶことができる。
【0016】
またさらに本発明によれば、前記の特徴を有するShK類似体は蛍光団の標識を有することができ、当該蛍光団で標識した本発明のShK類似体は、単独で、もしくは自己反応性細胞を検出できるクラスII四量体と併用して、フローサイトメトリーに使用することができる。
【0017】
以下に述べる詳細な説明および実施例を読むことにより、当業者には本発明のさらなる態様、要素および詳細が明らかになるはずである。
【図面の簡単な説明】
【0018】
【図1】本発明のいくつかのShK類似体の化学構造を示す図。
【図2A】発表されているNMR構造に基づくShKの分子モデルを示す図。図中、チャネルの遮断に重要なLys22は灰色の影をつけて強調されている。Aeeaリンカー(右)を介してShKのArg1のαアミノ基(灰色の第2の影をつけて強調した)にL−pTyrを接続した。該リンカーおよびL−pTyrの構造をHyperchem(登録商標)のAM1でモデル化した。
【図2B】安定にトランスフェクトした細胞におけるKv1.3およびKv1.1電流に対するShK(上段)およびShK(L5)(下段)の作用を示す図。
【図2C】ShK(暗)およびShK(L5)(明)によるKv1.3(白記号)およびKv1.1(黒記号)の用量依存的阻害を示す図。Kv1.3に対するKdは10±1pM(ShK)および69±5pM(ShK(L5))、Kv1.1に対するKdは28±6pM(ShK)および7.4±0.8nM(ShK(L5))である。
【図2D】Kv1.3に対するShK(L5)のウォッシュインおよびウォッシュアウトの時間経過を示す図。細胞を保持電位80mVで保持し、30秒ごとに200ミリ秒にわたって40mVまで脱分極した。
【図2E】ShK類似体によるKv1.3およびKv1.1の阻害についてKd値を示す図。ShK−F6CAおよびShK−Dap22のKd値は発表文献に基づくものである。
【図3A】CD3、CD45RAおよびCCR7に対する抗体で染色したヒトPBMCのCD3+ゲート型母集団におけるフローサイトメトリーで測定したCD45RAおよびCCR7の染色強度を示すグラフ。
【図3B】CD3、CD45RAおよびCCR7に対する抗体で染色したヒトTEM細胞株の細胞のCD3+ゲート型母集団におけるフローサイトメトリーで測定したCD45RAおよびCCR7の染色強度を示すグラフ。
【図3C】抗CD3抗体で48時間刺激したPBMC(白記号、ナイーブ/TCM細胞の混合物)およびTEM細胞(黒記号)による[3H]チミジン取込みの、ShK(暗灰色)およびShK(L5)(明灰色)による阻害作用を示すグラフ。
【図3D】KCa3.1の発現をアップレギュレートする、事前に活性化したヒトPBMC(ナイーブ/TCM細胞)は、抗CD3抗体で再活性化するとShK(L5)阻害に対して抵抗性となることを示すグラフ。当該細胞は、KCa3.1に特異的な阻害薬であるTRAM−34に感受性となることがこれまでに報告されている。
【図4A】フローサイトメトリーにより検出したラット脾臓T細胞(左)およびPAS T細胞(右)のCD45RC染色を示すグラフ。
【図4B】静止期の(上段)およびミエリン抗原で活性化した(下段)PAS T細胞により示されるKv1.3電流を示すグラフ。
【図4C】静止期の(上段)およびミエリン抗原で活性化した(下段)PAS T細胞におけるShK‐F6CA染色のフローサイトメトリープロフィールを示すグラフ。ShK‐F6CAで染色されなかった細胞(黒線)および染色された細胞(明灰色に塗りつぶした領域)。未標識のShK(L5)によるShK‐F6CA染色の競合は、暗灰色に塗りつぶした領域で示されている。
【図4D】静止期の(上段)およびミエリン抗原で活性化した(下段)PAS T細胞におけるKv1.3免疫染色の共焦点画像を示す図。マンホットニー(Mann−Whitney)のU検定を用いて統計解析を行なった。
【図4E】ConA(1μg/ml)で活性化したラット(左)ナイーブ/TCM細胞(白記号)およびTEM(黒記号)による[3H]チミジン取込みのShK(暗線)およびShK(L5)(明線)による用量依存的阻害を示す図。
【図4F】MBPによる刺激から7時間後のPAS T細胞によるIL2分泌のShK(暗線)およびShK(L5)(明線)による用量依存的阻害を示す図。
【図4G】PAS T細胞によるミエリン抗原誘発性[3H]チミジン取込みのShK(L5)による阻害(白記号)は、20μg/mlのIL2の添加により逆転する(黒記号)ことを示す図。
【図5A】L929細胞で安定に発現されるKv1.3チャネルについて測定したShK(L5)のKv1.3遮断活性を示すグラフ。
【図5B】ラット4匹に200mg/kgのShK(L5)を単回皮下注射した後の種々の時点での血中ShK(L5)濃度を示すグラフ。表記の時間に血液を採取し、パッチクランプ法により血清を試験してShK(L5)の量を測定した。
【図5C】図5Bのデータを単一指数関数減衰にフィッティングしたグラフ。半減期は約50分であることが示されている。
【図5D】10μg/kg/日のShK(L5)の単回皮下注射を5日間受けたLewisラット5匹における血中ShK(L5)濃度を示すグラフ。血液を毎朝(前回の注射から24時間後に)採取し、パッチクランプ法によりKv1.3チャネルに対する遮断活性について試験した。
【図5E】皮下投与(白棒;n=4)または静脈内投与(黒棒;n=4)のいずれかで10mg/kgのShK(L5)を単回投与した後の種々の時点におけるラットの血清中ShK(L5)濃度を示すグラフ。表記の時間に血液を採取し、パッチクランプ法により血清を試験して血中のShK(L5)量を測定した。単回皮下注射からほぼ24時間後には、ShK(L5)は300pMの定常状態濃度を維持した。該濃度は、TEM細胞の機能を選択的に阻害するのに十分な濃度である。
【図5F】ShK(L5)の半遮断投与量をラット血漿または2%ラット血漿を含有するPBSに添加し、種々の時間にわたって37℃でインキュベートした後のShK(L5)の回収率(%)を示すグラフ。表記の時間に等分量を採取し、Kv1.3チャネルに対する遮断活性を測定した。ShK(L5)は血漿中できわめて安定である。
【図6A】スコア化したEAEの予防効果を示すグラフ。PAS T細胞をin vitroで活性化し、洗浄し、第0日に腹腔内に注射した。EAEの臨床スコア:0=臨床徴候なし、0.5=尾遠位部のひきずり、1=尾のひきずり、2=軽度の両側不全麻痺または運動失調、3=中等度の両側不全麻痺、4=完全な後脚麻痺、5=4+失禁、6=死亡。ラットを1群あたりn=6とし、第0日から第5日まで、賦形剤単独(n=6)またはShK(L5)(n=6;10mg/kg/日)を皮下注射した。
【図6B】スコア化したEAEの治療効果を示すグラフ。PAS T細胞をin vitroで活性化し、洗浄し、第0日に腹腔内に注射した。ラットがEAEの臨床徴候を発現したとき、10mg/kg/日でShK(L5)による治療を開始し、3日間継続した。
【図6C】卵アルブミンに対してラットに現れるDTH反応の指標としての耳の厚さを示すグラフ。ShK(L5)10mg/kg/日で2日間にわたって動物(1群あたりn=6)を治療し、その後、耳の腫脹を測定した。マンホットニーのU検定を用いて統計解析を行なった。
【図7A】ShK(L5)の構造を示す図、ならびにTEM細胞におけるKv1.3チャネルの阻害をShK(L5)濃度の関数として示したグラフ。各データ点は3つの測定値の平均を示す。
【図7B】Kv1.3を含むシグナル伝達複合体の概略図。
【図7C】ISにおけるCD4、Kv1.3、Kvβ2、SAP97、ZIPおよびp56lckの共局在性を示す図。
【図7D】明らかなTEM−APC接触がない状態におけるCD4およびKv1.3の染色を示す図。
【図7E】MBPを取り付けたAPCに曝されたGAD65特異的TEM細胞におけるCD4およびKv1.3の染色を示す図。
【図7F】ShK(L5)100nMはIS形成を妨げないことを示す図。
【図7G】ShK(L5)100nMはISを崩壊させないことを示す図。
【図8A】ShK(L5)の不在下(黒)またはShK(L5)0.1nM(暗灰色)、1nM(中灰色)もしくは100nM(明灰色)の存在下で抗CD3抗体+架橋結合二次抗体(矢印)により誘発された、T1DM患者3例からのGAD特異的TEM細胞におけるカルシウムシグナル伝達を示すグラフ。
【図8B】T1DMおよびRAを有する患者からのナイーブ/TCM細胞およびTEM細胞(左)ならびにナイーブ/TCMエフェクターおよびTEMエフェクター(右)による[3H]チミジン取込みを示すグラフ。TEM細胞:T1DM患者3例からのGAD65で活性化したTEMクローンおよびRA患者3例からの抗CD3抗体で活性化したSF−TEM細胞。ナイーブ/TCM細胞:同一のRA患者3例からの抗CD3抗体で活性化したPB−ナイーブ/TCM細胞。
【図8C】図8Bで用いたTEM細胞およびナイーブ/TCM細胞によるサイトカイン産生を示す一連の棒グラフ。
【図8D】MS、T1DMおよびRAにおける疾患関連性および疾患無関連性の自己反応性T細胞の表現型を示す図。
【図8E】ShK(L5)がカルシウムシグナル伝達、リンパ球増殖およびサイトカイン産生を阻害し、IS形成を阻害しない様式を示す概略図。
【図9】エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルを表す概略図。
【図10】エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルにおけるShK(L5)の治療プロトコルを示す概略図。
【図11】ナイーブおよびセントラル記憶T細胞またはB細胞の機能を損なわずに、ラットにおいてin vivoでShK(L5)によりエフェクター記憶反応が特異的に抑制されることを表す概略図。
【図12A】新規発症1型糖尿病(T1DM)患者、健康な対照者および多発性硬化症患者からのGAD65特異的、インスリン特異的およびミエリン特異的T細胞におけるKv1.3電流(上段)およびチャネル数/細胞(下段)を示す図。
【図12B】上記患者からの個々のT細胞のKv1.3染色強度(上段)および蛍光強度(下段)を示す図。
【図12C】相対的細胞数とCCR7染色強度を対比させたグラフ。高濃度のKv1.3を発現している細胞は、CCR7陰性、すなわちTEMエフェクターである。低濃度のKv1.3を発現している細胞は、CCR7陽性、すなわちナイーブ細胞もしくはTCM細胞のいずれかである。
【図12D】T1DMおよびMSを有する患者(左)、罹患期間が5年を超えるT1DM患者(中)および非自己免疫性の2型DM患者からの自己反応性T細胞における1細胞あたりのKv1.3の数を示す図。
【図12E】新規発症T1DMの患者からのCD4+GAD65四量体+T細胞におけるKv1.3の数を示す図。
【図13A】RA患者の末梢血T細胞および滑液T細胞ならびにOA患者の滑液T細胞における細胞1個あたりのKv1.3チャネルの数を示す図。
【図13B】図13Aに示した細胞におけるKv1.3染色(明灰色)およびKvβ2染色(暗灰色)の共焦点画像を示す図。
【図13C】相対的細胞数とCCR7染色強度を対比させたグラフ。
【図13D】抗CD3抗体または抗Kv1.3抗体で染色し、ヘマトキリン/エオシンで対比染色した、RAおよびOA患者からの滑膜の顕微鏡写真(上)(40倍)ならびに炎症指数を示す棒グラフ(下)。
【発明を実施するための形態】
【0019】
以下に記載する本発明の詳細な説明および添付図面は、本発明の一部の実施例または実施形態だけを説明することを意図しており、必ずしも全部を説明することを意図したものではない。本詳細な説明および添付図面は、決して本発明の範囲を限定するものではない。
【0020】
本発明は、ShKの新規類似体と、当該組成物を作製する方法のほか、ヒトまたは動物の細胞におけるKv1.3チャネル(または他のイオンチャネル)を阻害するための、さらにはT細胞媒介性自己免疫疾患などの疾患および障害を治療または予防するための当該組成物の使用方法を提供する。本発明の組成物は、アニオン電荷を有する有機または無機化学物質(たとえば、原子、分子、化学基、残基、化合物、分子部分など)を付加した(たとえば、結合した、リンカーにより連結した、または別の方法で結びつけた)ShKトキシンよりなる。本発明の少なくとも一部の実施形態では、Kv1.1チャネルよりもKv1.3チャネルを阻害するために該組成物の親和性を増大または最適化できるように、アニオン電荷を有する有機または無機化学物質を選択することができる。本発明による、ShKと連結または結合しうる、アニオン電荷を有する有機または無機の分子もしくは化学基の例として、アミノ酸、ポリペプチド、アミノ酸残基、非天然アミノ酸残基、スレオニン、スレオニン誘導体、ホスホスレオニン、セリン、セリン誘導体、ホスホセリン、グルタミン酸、グルタミン酸誘導体、γカルボキシグルタミン酸、アスパラギン酸、アスパラギン酸誘導体、無機化合物または無機化学基、有機化合物または有機化学基、無水コハク酸、および無水フタル酸が挙げられるが、必ずしもこれらに限定されない。
【0021】
本発明によれば、本発明の組成物の非限定的な一部の実施例であって、アニオン電荷を有する化学物質がアミノ酸残基からなる実施例は、図1および2Cに示されており、以下の表1に示すように、本明細書では英数字による記号で記載されている。
【0022】
【表1】
特に図1を参照すると、チロシンもしくはフェニルアラニン、または荷電したこれらの非天然誘導体を、ShKのN末端(影付き灰色で示した残基Arg1)に付加したリンカーを介してShK(左上)に結合させた。チャネルの遮断に必要なLys22は暗灰色の影で示している。ShKの分子モデルは発表されているNMR構造に基づいており、上記リンカーおよび新規残基の構造をモデル化した。本発明の組成物の上記実施形態は全般に、アニオン電荷を有する少なくとも1つのアミノ酸残基と結合された(たとえば、化学的に結合された、連結された、または別の方法で結び付けられた)ポリペプチドShKトキシンを構成する。アミノ酸残基がキラル中心を有する実施形態では、当該アミノ酸残基のDおよび/またはLエナンチオマーを使用することができる。アニオン電荷を有するアミノ酸残基は、非天然残基であってもよく、ShKポリペプチドのN末端に付加または連結させることができる。一部の実施形態では、アニオン電荷を有するアミノ酸残基は、アミノエチルオキシエチルオキシアセチル・リンカーなどのリンカーを介してShKのN末端に連結させてもよい。これらのShK類似体は、他のカリウムチャネル(たとえばKv1.1)に対する親和性が低下しているため、ShKよりもKv1.3チャネルをより特異的に阻害する。ShKは、当技術分野で周知のように天然源から単離してもよいし、合成してもよい。
【0023】
ShKトキシンの合成
ShKトキシンは任意の適切な方法で合成することができる。そのような方法の1つでは、Arg(Pmc)、Asp(OtBu)、Cys(Trt)、Gln(Trt)、His(Trt)、Lys(Boc)、Ser(tBu)およびThr(tBu)を含む市販のFmoc−アミノ酸(バッケム社(Bachem Feinchemikalien))を入手し、組み立ててShKトキシンを形成する。アプライドバイオシステムズ(Applied Biosystems)431Aペプチド合成装置を用い、0.25mmolスケールでFmoc−Cys(Trt)−Rから開始してアミノ酸の段階的組立てを行ってもよい。残基34〜22を単結合させる。その後、樹脂の等分量(たとえば半量)を分取してさらに適切に混合する。続いて、残った等分量の樹脂に残りのペプチド配列を二重結合させる。カップリングはすべて、2当量の1−ヒドロキシベンゾトリアゾールの存在下でジシクロヘキシルカルボジイミドを介して行なう。最後の2残基もHBTU/DIEA法で結合させる。当該2残基はAeea(Fmoc−アミノエチルオキシエチルオキシ酢酸)と、N末端残基としてのFmoc−Tyr(PO4)モノベンジルエステルである。最終的にFmoc基を除去した後、ペプチド樹脂(2.42g)を樹脂から切断し、同時に試薬Kを用いて室温で2時間にわたって脱保護する。当技術分野では試薬Kは周知であり、文献に記載されている。キング、ディー.エス.(King,D.S.)、フィールズ、シー.ジー.(Fields,C.G.)およびフィールズ、ジー.ビー.(Fields,G.B.)(1990年)Int.J.Peptide Protein Res.第36巻、p.255−266を参照されたい。切断後、ペプチドを濾過して消耗した樹脂ビーズを取り除き、氷冷ジエチルエーテルで沈殿させる。続いて微細なフィルタ漏斗で該ペプチドを収集し、氷冷エーテルで洗浄し、最後に20%AcOH水溶液で抽出する。次に該ペプチド抽出物をH2Oで希釈して2リットルとし、NH4OHでpHを8.0に調整し、室温で36時間にわたって空気酸化させる。ジスルフィド結合を酸化して還元型グルタチオンと酸化型グルタチオンの比が2:1になった後、該ペプチド溶液をpH2.5に酸性化し、レイニン社(Rainin)のDynamax(商標)C18カラム(5.0×30cm)にポンプで送り込む。0.1%TFAを含有するH2Oに混和したアセトニトリル5〜30%を用い、直線濃度勾配で上記試料を溶出する。2つのRP−HPLC分析装置TFAおよびTEAPを用いて、得られた画分を分析する。純粋画分を併せ、凍結乾燥する(ペニントン、エム.ダブリュ.(Pennington,M.W.)、バーンズ、エム.イー.(Byrnes,M.E.)、ザイデンバーグ、アイ.(Zaydenberg,I.)、カイティン、アイ.(Khaytin,I.)、デカストナイ、ジェイ.(de Chastonay,J.)、クラフテ、ディー.(Krafte,D.)、ヒル、アール.(Hill,R.)、マーニル、ブイ.(Mahnir,V.)、ボルバーグ、ダブリュ.エイ.(Volberg,W.A.)、ゴルチカ、ダブリュ.(Gorczyca,W.)およびケム、ダブリュ.アール.(Kem,W.R.)(1995年)Int.J.Peptide Protein Res.第46巻、p.354−358を参照)。
【0024】
別の方法としてBoc−Bzl保護基戦略を用いる固相ペプチド合成を使用して、該ペプチドの一次構造ならびに類似体を組み立ててもよい。次に該ペプチドを無水HFで固相から切断すると、上記のFmocで合成したペプチドと同じく、折り畳み(フォールディング)の準備ができている直線ペプチドが生成される(スチュアート、ジェイ.エム.(Stewart,J.M.)およびヤング、ジェイ.ディー.(Young J.D.)(1984年)「固相ペプチド合成(Solid Phase Peptide Synthesis)」、第2版、ロックフォード(Rockford)所在のピアスケミカルカンパニー(Pierce Chemical Company)、IIを参照)。
【0025】
あるいは、ShKまたは類似体の一次構造を組み立てる別の合成方法として化学的ライゲーション技術が挙げられるが、この方法では、ペプチドは、C末端チオエステルペプチドを有するように設計された一連の断片として作製される。該チオエステルペプチドは、N末端にCys残基を含む別のペプチドと反応して天然ペプチド結合を備えたペプチドを形成することができる。この技術を使用することにより、ShKの一次構造を効果的に組み立てることができると考えられる((4)ウィルケン、ジェイ.(Wilken,J.)およびケント、エス.ビー.エイチ.(Kent S.B.H.)(1998年)「化学的タンパク質合成(Chemical protein synthesis)」、Current Opin.Biotech.第9巻、p.412−426を参照)。
【0026】
また、ShKの一次構造を組み立てるために使用しうる別の合成方法は、アルベリキオ、エフ.(Albericio,F.)、ロイド‐ウィリアムス、ピー.(Lloyd−Williams,P.)およびギラルト、イー.(Giralt,E)(1997年)「コンバージェントペプチド合成(Convergent peptide synthesis)」、ジー.フィールズ(G.Fields)編「Methods in Enzymol.」、ニューヨーク州ニューヨーク所在のアカデミックプレス(Academic Press)に収録(pp313−335)に記載されているように、保護ペプチド断片のコンバージェント法を利用することであると考えられる。当該方法では、直線状の保護断片を完全な側鎖保護断片として組み立てる。続いて、該断片をコンバージェント法で互いに結合させ、ShKまたはその1つの類似体の一次配列を組み立てることができる。該断片の組立ては、固相樹脂を用いてカップリング工程および洗浄工程を容易にすることもできると考えられる。
【0027】
また別の方法として遺伝子組換え法を使用することも可能であり、この方法では、原核生物または真核生物の発現系のいずれかで発現できるように、ShKをコードするcDNA配列を作製することができると考えられる。非標準コドンを利用する装荷済み(プレロード)tRNA分子を活用することにより、非天然アミノ酸を含有する組換えShK類似体も可能である。上記の未使用コドンの1つを使用してホスホチロシン残基のほかAeea残基を付加するように、cDNA構築物を設計することができる。次に、組換え生産したShK類似体の折り畳みは、合成ペプチドに使用する方法と類似した方法で完成させることができると考えられる(ペニントン、エム.ダブリュ.(Pennington,M.W.)、バーンズ、エム.イー.(Byrnes,M.E.)、ザイデンバーグ、アイ.(Zaydenberg,I.)、カイティン、アイ.(Khaytin,I.)、デカストナイ、ジェイ.(de Chastonay,J.)、クラフテ、ディー.(Krafte,D.)、ヒル、アール.(Hill,R.)、マーニル、ブイ.(Mahnir,V.)、ボルバーグ、ダブリュ.エイ.(Volberg,W.A.)、ゴルチカ、ダブリュ.(Gorczyca,W.)およびケム、ダブリュ.アール.(Kem,W.R.)(1995年)Int.J.Peptide Protein Res.第46巻、p.354−358を参照)。
【0028】
アニオン性アミノ酸残基のShKへの付加およびShKの任意の修飾
アニオン性アミノ酸残基は、アミノエチルオキシエチルオキシアセチル・リンカーなどのリンカーにより、もしくは他の何らかの適切な手段により、天然または合成ShKトキシンのN末端に付加してもよい。当該実施例では、図1に示した9つのShK類似体が作製される。まず、Fmoc−Aeea−OHを、上述のように組み立てた合成ShKトキシンのN末端に結合させる。続いて樹脂を9つに等分する。次に、DICおよびHOBTを用いて、Fmoc−Tyr(PO4Bzl)−OH、Fmoc−d−Tyr(PO4Bzl)−OH、Fmoc−Tyr(PO4Me2)−OH、Fmoc−Pmp−OH、Fmoc−d−Pmp−OH、Fmoc−Pmp(Et)−OH、Fmoc−Pmp(Et)2−OH、Fmoc−Tyr(tBu)−OH、またはFmoc−Amp(Boc)−OHのいずれかを等分した樹脂の1つに結合させる。続いて非ブロック化したペプチド樹脂を切断し、5%トリイソプロピルシランを含有する試薬K(キング(King)ら、1990年)を用いて室温で2時間にわたって脱保護する。t−15分時点で切断混合物に固体のNH4Iを加えてMet(O)を還元する(ニコラス(Nicolas)ら、1995年)。Tyr(PO4Me2)−OHを含有するペプチドについては、スカベンジャーとしてチオアニソールを含有する1MのTMSBrのTFA溶液を含有する切断混合物を4℃で18時間にわたって使用した(ティアン(Tian)ら、1993年)。この方法を用いると、メチル保護基の不完全な除去がよくみられ、2つの種(Tyr(PO4)およびTyr(PO4Me))はRP−HPLCで容易に精製される。Tyr(PO4Me2)を含む類似体は標準の試薬Kによる切断を介して切断され、両Me基はそのまま維持される。いずれの場合も、切断混合物を濾過し、粗ペプチドを氷冷ジエチルエーテル中で沈殿させる。沈殿を収集し、樹脂200mgから約75mgのペプチドが産出される。この粗生成物を50%AcOH水溶液20mlに溶解し、0.75LのH2Oで希釈する。NH4OHで該溶液のpHを8.2に調整し、グルタチオン(2mM:1mM)(還元型:酸化型)を加えて一晩放置し、折り畳ませた。類似体はすべて、以前に記載されているようにRP−HPLCを用いて精製する(ペニントン(Pennington)ら、1995年;ペニントン(Pennington)ら、1996年a;ペニントン(Pennington)ら、1996年b)。純粋な画分を併せ、凍結乾燥する。各試料をRP−HPLC、AAAおよびMALDI−TOF MSで確認し、バイオアッセイの前に調整してペプチド含量を明らかにする。
【0029】
本発明の一部の実施形態では、ShK構造のPK/PD特性を改善するため、分解特性に敏感な残基を交換または置換してもよい。このため、21位のMet残基の置換を行なって酸化に対する安定効果を与えてもよい。さらに、C末端の酸官能基をアミドで置換すれば、C末端カルボキシペプチダーゼ酵素に対する安定性を与えることになる。N末端でアニオン性分子と結合させたShKの一次構造に上記の2つの置換を行なった物質はすでに合成され、最も安定で選択性の高いKv1.3ブロッカーを生み出している。非加水分解性リン酸塩置換も、ホスファターゼ酵素に対する安定性だけでなく、リン酸塩の酸性および塩基性加水分解に対する安定効果を与えるはずである。置換について以下に要約する。使用した略語の定義は次のとおり、すなわち:Pmp=p−ホスホノメチル−フェニルアラニン;Ppa=p−ホスファチジル−フェニルアラニンおよびNle=ノルロイシンである。
【0030】
置換:
p−ホスホ−Tyr−Aeea−ShK−Nle21−Cys35−アミド
p−ホスホノ−メチル−フェニルアラニン−Aeea−ShK−Nle21−Cys35−アミド(Pmp)
p−ホスファチジル−Phe−Aeea−ShK−Nle21−Cys35−アミド(Ppa)
p−ホスホ−Tyr−Aeea−ShK−Nle21−Cys35−アシド
p−ホスホノ−メチル−フェニルアラニン−Aeea−ShK−Nle21−Cys35−アシド(Pmp)
p−ホスファチジル−Phe−Aeea−ShK−Nle21−Cys35−アシド(Ppa)
非加水分解性PmpおよびPpaのほかに、p−ホスホノ(ジフルオロ−メチル)−フェニルアラニン(Pfp)およびp−ホスホノ−メチルケト−フェニルアラニン(Pkp)による置換もアニオン性置換であり、以下:
Pfp−Aeea−ShK−Nle21−Cys35アミド
Pkp−Aeea−ShK−Nle21−Cys35アミド
Pfp−Aeea−ShK−Nle21−Cys35アシド
Pkp−Aeea−ShK−Nle21−Cys35アシド
が得られる。
【0031】
N末端置換体の構造は付録Bに示している。本発明の範囲内にある他の構造は、ビートン、シー.(Beeton,C.)らの「自己免疫疾患の治療に向けKv1.3チャネルの選択的ペプチド阻害薬でエフェクター記憶T細胞を標的にする(Targeting Effector Memory T Cells with a Selective Peptide Inhibitor of Kv1.3 Channels for Therapy of Autoimmune Diseases)」、Molecular Pharmacology、第67巻、第4号、p.1369−(2005年)に発表されている。同文献の内容全体は参照により特別に本明細書に組み込まれ、その完全な複写は付録Cとして本明細書に添付される。
【0032】
本発明のShK類似体の治療用途
本発明は、本発明のShK類似体(上記表1に列記した類似体を含むがこれらに限定されない)のみで構成される、または当該類似体を含んでなる薬剤学的に許容可能な製剤の治療有効量(たとえば、症状または疾患進行を軽減または消失させる予防的もしくは効果的な量)を投与することにより、ヒトまたは動物対象者におけるT細胞媒介性障害(たとえば自己免疫疾患、移植片対宿主病、臓器移植の拒絶反応の予防など)、他の炎症性障害、肥満および2型糖尿病などの特定の障害または疾患を治療もしくは予防するための方法を提供する。任意の適切な投与経路(たとえば、経口、経直腸、静脈内、筋肉内、皮下、皮内、鼻腔内、局所、経粘膜、経皮、薬剤供給インプラントによるなど)を使用できる。T細胞媒介性障害の予防または治療に使用する場合は、T細胞膜上のKv1.3チャネルを阻害するのに十分な投与量を使用する。これに関して本発明のShK類似体は、さまざまな種類のT細胞媒介性自己免疫疾患の予防または治療に使用できる可能性を有する。以下に、本発明の方法により予防または治療しうる一部のT細胞媒介性自己免疫疾患の一部の例を、各疾患により主に影響を受ける標的器官に着目して分類して示す。
【0033】
神経系:
多発性硬化症
重症筋無力症
ギラン・バレーなどの自己免疫性末梢神経障害
自己免疫性ブドウ膜炎
胃腸管:
クローン病
潰瘍性大腸炎
原発性胆汁性肝硬変
自己免疫性肝炎
歯周病に随伴する骨吸収
血液:
自己免疫性溶血性貧血
悪性貧血
自己免疫性血小板減少症
内分泌:
1型糖尿病
アジソン病
グレーブス病
橋本甲状腺炎
自己免疫性卵巣炎および精巣炎
血管:
側頭動脈炎
抗リン脂質抗体症候群
ウェゲナー肉芽腫症およびベーチェット病などの血管炎
多臓器および/または筋骨格系:
関節リウマチ(RA)
変形性関節症(OA)
全身性紅斑性狼瘡
強皮症
多発筋炎、皮膚筋炎
強直性脊椎炎などの脊椎関節症
シェーグレン症候群
皮膚:
乾癬
疱疹状皮膚炎
尋常性天疱瘡
白斑
影響を受ける特定の器官とは関係なく、Tリンパ球は自己免疫疾患の発現に寄与していると考えられている。これらの疾患に現在利用可能な治療法は大いに不満足なものであり、グルココルチコイド(メチルプレドニゾロン、プレドニゾンなど)、非ステロイド性抗炎症薬、金塩、メトトレキサート、抗マラリア薬のほか、シクロスポリンおよびFK−506などの他の免疫抑制薬を使用するのが通例である。さらに、本発明の方法により予防または治療しうる別のT細胞媒介性障害は、移植片対宿主病および/または移植臓器の拒絶反応である。実際に、組織型決定、外科技術、および有効な免疫抑制治療の改良技術の開発に伴い、臓器移植の転帰は徐々に改善されてきている。しかし、移植臓器の拒絶反応は未だに重大な問題である。Tリンパ球は免疫応答に中心的な役割を演じており、多くの移植臓器の拒絶反応に大きく関与している。Tリンパ球はいわゆる移植片対宿主病の原因でもあり、該疾患は、移植した骨髄細胞がMHC不適合の宿主組織を認識して破壊する。このため、シクロスポリンおよびFK506などのT細胞性免疫を抑制する薬剤を使用して、移植拒絶反応および移植片対宿主病が予防されている。しかし残念ながら、こうしたT細胞阻害薬は毒性を有し、肝毒性および腎毒性のため、その使用が制限される。このため本発明の方法は、移植片対宿主病もしくは移植拒絶反応の治療または予防に対し、毒性の低い代替選択肢を提供しうる。また、電位差ゲート型Kv1.3カリウムチャネルの阻害薬はエフェクター記憶T細胞の抑制に特に有効であることが示されており、このため本発明の方法は、骨吸収および歯周病、乾癬、関節リウマチ、糖尿病および多発性硬化症など、エフェクター記憶T細胞に関連する疾患の予防または治療に特に有効である可能性がある。T細胞媒介性疾患のほか、Kv1.3チャネルは、エネルギーホメオスタシス、体重および末梢インスリン感受性を調節することが解明されている。このため本発明の方法は、細胞膜上のKv1.3チャネルを阻害することにより、異常なホメオスタシス、体重および末梢インスリン感受性が関わる他の疾患および障害を治療するために使用できる。このような他の疾患および障害には、歯周病における骨吸収、2型糖尿病、メタボリック症候群および肥満が含まれるが、これらに限定されない。
【0034】
フローサイトメトリーにおける本発明のShK類似体の使用
さらに本発明によれば、蛍光団で標識したShK(L5)を単独で、もしくは自己反応性細胞を検出できるクラスII四量体と併用してフローサイトメトリーに使用することにより、in vitroでT細胞媒介性障害を診断するための方法、あるいは、種々の細胞型の選別または識別の方法を提供する。フローサイトメトリーは懸濁液中の細胞を特徴付けるための柔軟な方法であり、蛍光励起細胞分取法を用いて、フローサイトメトリーで測定した特徴に基づき生細胞を選別する。フローサイトメトリーで検出しうる細胞の特徴および機能の種類には、細胞内外のタンパク質の発現、DNAの含量と生存能力とアポトーシスのタイプ、多剤耐性ポンプ活性、酵素活性、T細胞活性化、T細胞受容体特異性、サイトカイン発現、食作用ならびに酸化バースト活性が含まれる。このため、本発明の当該方法では、ShKに付加されるアミノ酸残基には、単独で、もしくは自己反応性細胞を検出できる特異的な自己抗原を取り付けたクラスII四量体と併用してフローサイトメトリーに使用するために、蛍光団の標識が組み込まれてもよい。当該フローサイトメトリーを行ないうる方法の具体的な説明は、ビートン、シー.(Beeton,C)らの「慢性的に活性化されたTリンパ球におけるKv1.3チャネルのアップレギュレーションを検出および研究するための新規蛍光性トキシン(A Novel Fluorescent Toxin to Detect and Investigate Kv1.3 Channel Up−Regulation in Chronically Activated T Lymhocytes)」J.Biol.Chem.、第278巻、第11号、p.9928−9937(2003年3月)に記載されている。一般にフローサイトメーターは、集束レーザー光を使用して、細胞が流体の流れのなかでレーザー光線を通過するときに細胞を照射する。細胞から散乱した光と、目的の細胞に取り付けた蛍光色素から放たれた光をいくつかの検出器で分析し、コンピュータで処理する。細胞は、大きさおよび形のほか、細胞内および細胞表面上のさまざまな分子の存在によっても識別および選別しうる。
【0035】
本発明のShK類似体のカリウムチャネル阻害作用および治療的有用性の例
ShKは神経細胞のKv1.1チャネルとKv1.3チャネルをほぼ同等の力価で遮断する。このため、血液脳関門に障害があり、Kv1.1チャネルを遮断するのに十分な量のShKが進入できる状況下では、神経毒性が懸念される。Kv1.3に特異的な阻害薬を設計するための我々の戦略は、フルオレセイン−6−カルボキシレート(F6CA)が20Å長のAeeaリンカーを介してShKのN末端に付加されているShK−F6CAが、Kv1.1に比べKv1.3に対し80倍の選択性を示したという我々の所見によって導かれた(ビートン(Beeton)ら、2003年)。F6CAは制限されたカルボキシレートとしても、環化したラクトンとしても存在できるため、ShK−F6CAのKv1.3特異性がF6CAの陰電荷によるものか、フルオレセインの大きく嵩高い原子核により作り出される疎水性によるものか、可能性のある平面性p電子スタッキングによるものなのか、もしくは、これらの可能性のある寄与因子がすべて組み合わされたことによるものなのか、はっきりしなかった。これらの可能性を区別するために、かつ非蛍光性のKv1.3選択的阻害薬を開発する意図をもって、N末端を置換した一連の12個の新規ShK類似体を作製し、上記相互作用の一部について探査した。Aeeaリンカーを介してチロシン、フェニルアラニンまたはこれらの誘導体(電荷、大きさおよび疎水性は多岐にわたる)をShKのN末端に付加することにより、電荷および疎水性の影響を探り、F6CA置換でみられた選択性の増強について洞察を得ることができた。
【0036】
Kv1.1阻害に比べ選択的なKv1.3阻害:
図2A〜2Dに示した実施例では、陰電荷(実効電荷2)を有する翻訳後修飾された芳香族アミノ酸であるL−ホスホチロシン(L−pTyr)を、AEEAリンカーを介してShK−Arg1に付加し、新規類似体ShK(L5)を作製した。L929細胞に安定に発現しているKv1.3 およびKv1.1チャネルで、ShKトキシンとShK(L5)を試験した。図2Bは、保持電位80mV〜40mVの200msにわたる脱分極パルスにより誘発したKv1.3およびKv1.1電流に対するShKおよびShK(L5)の作用を示している。いずれのペプチドも、投与量依存的にKv1.3およびKv1.1を可逆性に遮断し、ヒル係数は1であった。Microcal Origin(登録商標)ソフトウェアを用いて示した用量反応曲線から、Kdを判定した。ShKはKv1.3(Kd=10±1pM)およびKv1.1(Kd=28±6pM)を遮断し、予測どおり力価はほぼ同等であった(図1C)。対照的にShK(L5)は、Kv1.1(Kd=7.4±0.8nM)に比べKv1.3(Kd=69±5pM)に対し100倍選択的であった(図1B、1C)。ShK(L5)およびそのウォッシュアウトによるKv1.3電流遮断の時間経過を図1Dに示す。ShK(L5)ウォッシュインの時定数(TON)は131±21秒(n=7)であり、ペプチドウォッシュアウトの時定数(TOFF)は150±28秒(n=4)であった。KON(15×106±0.5×106M−1秒−1)およびKOFF(0.0059±0.0013秒−1)値から算出したKd(57±7pM)は、Microcal Originソフトウェアで判定したKd(69±5pM)と一致している。
【0037】
他のShK類似体もKv1.3およびKv1.1チャネルで試験した。D−ホスホチロシン(D−pTyr)を含むShK(D5)は、Kv1.1に比べKv1.3に対して35倍選択的であったが、ShK(L5)よりは一桁低い力価であった。L−pTyr−モノメチルを含むShK(L6)は、軽度(11倍)のKv1.3特異性を示し、L−pTyr−ジメチルまたはL−Tyrを含むShK類似体はKv1.1と比べてもKv1.3に対し選択的ではなかった。フェニルアラニンまたはその誘導体を含んだ類似体(かさ、p電子密度および電荷が多岐にわたる)は、K1.1と比べた場合のKv1.3に対する特異性が軽度か、または特異性がなかった。K1.1と比べた場合のKv1.3に対するShK(L5)の100倍の特異性は、ShK−F6CA(80倍)、ShK(D5)(35倍)、ShK−Dap22(33倍)または試験した他のいずれのShK類似体よりも大きい。
【0038】
本出願人は、20種類の一群のイオンチャネルについてもShK(L5)の特異性を評価し、そのデータを下記の表2に要約する。
【0039】
【表2】
上記表2のデータから理解されうるように、ShK(L5)はT細胞のKv1.3チャネルを遮断し、クローン化Kv1.3チャネル(69pM)に対するKdと同等のKd(76pM)を示した。ShK(L5)は、Kv1.3に対する選択性がKv1.1に比べ100倍、Kv1.6に比べ260倍、Kv3.2に比べ280倍、Kv1.2に比べ680倍であり、試験した他のすべてのチャネルに比べ1000倍を超えていた。重要なことにShK(L5)は、ヒトナイーブおよびTCM細胞の活性化を調節するカルシウム活性化K+チャネルであるKCa3.1に比べ、Kv1.3選択性が1600倍であった(ウルフ(Wulff)ら、2003年)。天然ShKはShK(L5)より選択性が低かった。ShKは、Kv1.3(Kd=10±1pM)に対する選択性がKv1.1(Kd=28±6pM)に比べ2.8倍、Kv1.6(200±20pM)に比べ20倍、Kv3.2(Kd=5,000±1,000pM)に比べ500倍であり、Kv1.2(10±1nM)およびKCa3.1(Kd=28±3nM)に比べ1000倍を超えていた。特異的Kv1.3阻害薬として売り込まれてきたサソリ毒由来のペプチドであるマーガトキシン(クー(Koo)ら、1997年;リン(Lin)ら、1993年;ミドルトン(Middleton)ら、2003年)も特異的ではなかった。マーガトキシンは、Kv1.3(Kd=110±12pM)に対する選択性がKv1.2(Kd=520±1pM)に比べ5倍、Kv1.1(10±1nM)に比べ9倍であり、Kv1.6およびKv3.2(Kd>100nM)に比べ1000倍を超えていた。Kv1.3阻害薬であるという根拠(レイヒ(Lahey)およびラジャダクーシャ(Rajadhyaksha)、2004年)から自己免疫疾患用に売られている栄養補助食品ルテオリン(www.lutimax.com )は、Kv1.3を弱く遮断し(Kd=65±5mM)、Kv1.1(Kd=77±5mM)、Kv1.2(Kd=63±4mM)およびKv1.5(Kd=41±3mM)に比べて選択性をまったく示さなかった。ShK(L5)のKv1.3に対する優れた選択性と該チャネルに対するpMレベルの親和性が相まって、ShK(L5)の魅力的な免疫抑制薬としての可能性を高めている。
【0040】
ヒトTEM細胞増殖の優先的抑制
図3A〜3Dを参照すると、ShK(L5)のin vitroでの免疫抑制活性を評価するために、本出願人は、ヒトTEM細胞株と、ナイーブおよびTCM細胞の混合物を含むヒトPBMCとの間で、抗CD3抗体で刺激した増殖を抑制するShK(L5)の能力を比較した。フローサイトメトリーにより、検討した2つの母集団の細胞表面の表現型を確認した。図3Aにみられるように、TEM細胞株はCCR7−CD45RA−が90%を超え、一方、図3Bに示すように、PBMCは65%のCCR7+CD45RA+(ナイーブ)および18%のCCR7+CD45RA−(TCM)細胞を含有していた。図3Cは、ShK(L5)およびShKがPMBCの増殖抑制(IC50=5nM、p<0.05)に比べTEM細胞の増殖抑制(IC50=約80pM)において60倍効果が高かったことを示している。PBMCの低い感受性は、これまでに報告されているように(ガンシャニ(Ghanshani)ら、2000年;ウルフ(Wulff)ら、2003年)、ナイーブおよびTCM細胞における、刺激によるKCa3.1チャネルの迅速なアップレギュレーションにより説明されると考えられる。この解釈を踏まえ、図3Dの上の列に示すように、PBMCを48時間活性化してKCa3.1発現をアップレギュレートし、続いて12時間休ませ、さらに抗CD3抗体で再活性化したところ、PBMCはShK(L5)遮断に対して完全に抵抗性となった。第1ラウンドの刺激でShK(L5)により抑制されたPBMCは、洗浄して休ませ、抗CD3抗体で再度誘発すると、同じようにShK(L5)に抵抗性を示した。上記の結果は、ナイーブおよびTCM細胞がKCa3.1チャネルをアップレギュレートすることによりKv1.3阻害薬を免れることを示した早期の研究を確証するものである。以上のように、ShK(L5)は優先的かつ持続的にTEM細胞の増殖を抑制する。
【0041】
ラットTEM細胞増殖の優先的抑制
ShK(L5)の治療的有効性を評価することを前提として、ラットにMS様疾患を引き起こす記憶T細胞株PASの増殖を抑制するShK(L5)の能力について検討した。対照として、本出願人はラット脾臓T細胞を使用した。2つの細胞母集団の分化状態を確認するため、ナイーブT細胞のマーカーであるCD45RCの発現を評価した(バンス(Bunce)およびベル(Bell)、1997年)。図4Aに示すように、ラット脾臓T細胞はCD45RC+が76%であったが(すなわち主にナイーブ細胞)、PAS細胞はCD45RC−であったことから記憶細胞であることが示唆される。PAS細胞がTEM状態にあるか、もしくはTCM状態にあるか判断するために、活性化の前および48時間後にKv1.3発現を調査した。TEM細胞は刺激によりKv1.3レベルを著明にアップレギュレートするが、TCM細胞ではアップレギュレートしないと予測される。図4Bを参照すると、パッチクランプ法実験から、PAS細胞のMBP刺激後にKv1.3電流の振幅が著しく増大することが明らかにされ、TEM細胞であることと一致している。PAS細胞上のKv1.3チャネル数を測定する独自の測定法として、Kv1.3と特異的に結合することが報告されている蛍光標識したShK類似体であるShK−F6CAを用いた。フローサイトメトリーで測定したShK−F6CAの染色強度は、細胞表面に発現したKv1.3四量体の数を反映している。図4Cにみられるように、ShK−F6CA(10nM)の染色強度はPAS細胞のMBP活性化とともに増大し、過剰な非標識ShK(L5)(100nM)がShK−F6CA染色を競合的に阻害した。最終試験として本出願人は、固定してKv1.3特異抗体で染色した静止期PAS細胞およびMBPで刺激したPAS細胞について共焦点顕微鏡検査を行なった。図4Bおよび4Cのデータと一致して、休止期PAS T細胞はKv1.3染色強度が4.4±0.6であり、この値は抗原による活性化後に10.6±2.3(p<0.005)に増大したことから(図4Dを参照)、活性化後にKv1.3タンパク質発現が増加したことを示している。このため、MBPで活性化したPAS細胞は、CD45RC−Kv1.3highTEM細胞であり、一方、実験に用いたラット脾臓T細胞は大部分がナイーブ状態である。
【0042】
ShK(L5)およびShKによる、PAS細胞のMBP誘発増殖の抑制(IC50=約80pM)は、ラット脾臓T細胞のマイトジェン誘発増殖の抑制(IC50=約100nM)よりも約1000倍も効果的であった(図4E参照、p<0.05)。以上の結果は、上に述べたヒトT細胞による所見を確証するものである。図4Gにみられるように、ShK(L5)はPAS細胞によるMBP誘発IL2産生を阻害し(図4F)、外因性IL2はPAS細胞増殖のShK(L5)抑制を部分的に無効にした(図4G)。早期の研究では、ヒト、ラットおよびミニブタのT細胞について特異性の低いKv1.3阻害薬に関する類似した所見が報告された。以上を要約すると、ShK(L5)はヒトおよびラットのTEM細胞の強力かつ選択的な阻害薬であり、したがって自己免疫疾患の病因に寄与するTEM細胞を優先的に標的にすることにより、当該疾患において治療用途を有する可能性がある。
【0043】
血中半減期および安定性
パッチクランプ法によるバイオアッセイを用いて、皮下注射後のShK(L5)の血中濃度がTEM細胞の阻害に十分であるかどうか確認した。これらの実験の結果を図5A〜5Fに示す。
【0044】
Kv1.3チャネルに対する遮断活性について、ShK(L5)投与ラットおよび対照ラットからの血清試料を試験した。対照血清は検出可能な遮断活性を示さなかったことから、内因性チャネルブロッカーは存在しないことが示された。該アッセイを標準化するため、既知量のShK(L5)をラット血清に添加し、当該試料をKv1.3チャネルについて試験した。添加した該血清試料は用量依存的にKv1.3電流を遮断し(Kd=77±9pM)、血清がない場合のShK(L5)の作用と区別できなかった(図4A)。標準曲線と比較することにより、投与した動物のShK(L5)濃度を測定した。ShK(L5)は、200mg/kgの単回皮下注射から5分後に血清中に検出可能であった。30分で最高濃度(12nM)に達し、その後420分かけて約300pMのベースライン値まで低下した。血中からのShK(L5)の消失は、単一指数でフィッティングさせることができた。血中半減期は約50分であると推定された。
【0045】
200mg/kg投与後の最高血清中濃度(12nM)は、Kv1.3チャネルおよびTEM細胞機能を選択的に遮断する必要量を超えていることから、これより少ない投与量を試験した。10mg/kgの単回注射後、30分以内にShK(L5)の最高血清中濃度約500pMに達し(データ省略)、この濃度は90%を超えるKv1.3を遮断するのに十分であり、Kv1.1には影響を及ぼさない濃度である。上記投与量(10mg/kg/日)を1日1回反復投与した結果、約300pMの定常状態濃度に達し(注射後24時間時点で測定、図5D)、これはTEM細胞を60〜70%抑制するのに十分な濃度であり、ナイーブ/TCM細胞にはほとんど影響を及ぼさない。この「定常状態」濃度は、血中半減期が約50分と推定されたことを考えると予期しない結果であり、ShK(L5)は反復投与により「蓄積される」ことを示している。その「貯蔵所」が皮膚なのか、または身体の他の部位なのか判断するために、10mg/kgのShK(L5)を単回静脈内注射または皮下注射したラットの10時間後のShK(L5)の血中濃度を測定した。当該ペプチドは、いずれの投与経路でも同一の経時変化で消失したことから(図5E)、10mg/kg/日の単回注射後に到達したShK(L5)の定常状態濃度300pMには皮膚が関与しておらず(図5D)、貯蔵所はほかにあることが示された。
【0046】
10mg/kg/日の単回注射後にShK(L5)の定常状態濃度300pMにうまく到達したことは、該ペプチドがin vivoで安定であることを示唆している。その安定性を直接検討するために、ラット血漿中またはラット血漿を2%含有するPBS中で、インキュベーション時間をさまざまに変えて37℃でShK(L5)をインキュベートし、Kv1.3の遮断活性を測定した。いずれの添加試料のセット(血漿およびPBS)においても、約5時間でKv1.3遮断活性が50%低下することを観察したが、これはおそらくペプチドの試験管プラスチック表面との結合によるものと思われ、濃度は次の2日間も定常状態を維持していた(図5F)。追加の安定性試験として、ShK(L5)投与ラットからの血清を用いてKv1.3遮断活性とKv1.1遮断活性を比較した。pTyrの脱リン酸化またはAeea−pTyr側鎖の切断のいずれかにより、ShK(L5)がin vivoで修飾されるとすれば、それぞれShK(L4)およびShKが生成されることになり、これらはいずれもKv1.1と比較してKv1.3への選択性がない。ShK(L5)投与動物からの血清試料は、ShK(L5)と同じように、Kv1.1に比べてKv1.3に対する選択性を示したことから、該ペプチドは上記の修飾を受けないことが示された。以上を統合すれば、これらの結果は、ShK(L5)は血漿中できわめて安定であり、毎日の10mg/kgの単回皮下注射後に薬理学的に適切な血清中濃度に達することを示している。
【0047】
無毒性
本出願人は、in vitroおよびin vivo試験をいくつか実施してShK(L5)が毒性を示すかどうか判定した。当該試験の結果を付録Aに要約する。ヒトおよびラットのリンパ系細胞を、Kv1.3半遮断量またはTEM抑制のIC50(70〜80pM)の1200倍を超える濃度のShK(L5)(100nM)とともに48時間インキュベートしたところ、軽微な細胞毒性を示した。同じ高濃度のShK(L5)は、試験株TA97Aを用いたエイムス試験で陰性であったことから、変異原物質ではないことが示唆された。いずれのin vitro試験においても特筆すべき毒性は検出できなかった。
【0048】
薬剤誘発性のKv11.1(HERG)チャネルの遮断は、重大な心毒性に寄与しており、いくつかの薬剤が市場から回収されている。ShK(L5)は、100nM(Kv1.3に対するKdの1430倍を超える)ではKv11.1チャネルに何の影響も及ぼさず、したがって、本出願人が選択した治療処方(10mg/kg/日、定常状態血中濃度300pM)は心毒性を引き起こさないはずである。さらなる試験として本出願人は、第1日に賦形剤(PBS+2%ラット血清)を投与し、第2日にShK(L5)10mg/kg/日を投与した覚醒ラットを用いて、心拍変動解析を行なった。ShK(L5)は、時間および頻度のいずれの領域においても心拍および標準HRV(心拍変動)パラメータ(欧州循環器病協会および北米心拍調節・電気生理学協会の特別委員会、1996年)に何の影響も及ぼさなかった。
【0049】
上記急性毒性実験の余勢を駆って本出願人は、10mg/kgのShK(L5)または賦形剤を2週間にわたって1日1回皮下注射したラットを用いて亜慢性試験を行なった(各群n=6)。ShK(L5)投与動物は、賦形剤投与動物と同じ程度に体重が増加した(付録A)。血液学的および血液生化学的解析により、ShK(L5)投与ラットと賦形剤投与ラットには差がないことが示され、フローサイトメトリー解析により、胸腺細胞およびリンパ球サブセットの割合にも差がないことが示された(付録A)。これらの試験を集合すると、ShK(L5)は安全であることが示唆される。
【0050】
治療的安全指数を判定するため、健康なラットに60倍高い投与量(600mg/kg/日)のShK(L5)を5日間投与したところ、毒性の臨床徴候は観察されず、健康なラットに1000mg/kgのShK(L5)を単回注射したときも毒性はみられなかった。EAEおよびMSで起こるように、血液脳関門に障害がある場合は、この状況は楽天的ではない。ShK(L5)10mg/kg/日を10日間投与されたEAEラットは、毒性の徴候をまったく示さなかった。対照的に、600mg/kg/日を5日間投与したラットは、その40%(5/12)が第5日にEAEの臨床徴候を発現して死亡した(推定LD50=750mg/kg/日)。200mg/kg単回注射後の血清中のShK(L5)最高濃度(12nM)は、50%を超えるKv1.1チャネルを遮断するのに十分な濃度であることから、600mg/kg/日のShK(L5)を投与したEAEラットに観察された毒性は、Kv1.1を遮断するのに十分な量のShK(L5)が脳内に進入したことによるものである可能性が高い。したがって、血液脳関門に障害がない状況(中枢神経系(CNS)に影響を及ぼさない自己免疫疾患にみられる状況)では、ShK(L5)の有効な治療的安全指数は100を十分に超えるが、血液脳関門が破られると治療的安全指数は75である。
【0051】
DTHおよび養子移入性急性EAEの予防
図6A〜6Cを参照すると、2つの動物モデルにおいてin vivoでの免疫抑制活性についてShK(L5)を評価した。本出願人は、MBPで活性化したPAS TEM細胞をLewisラットに移入して誘発した急性EAEを予防および治療する能力のほか、TEM細胞で媒介されるDTH反応を抑制する能力も試験した。PAS細胞をin vitroで48時間にわたってMBPで活性化し、続いてLewisラットに養子移入した(生細胞数:6〜8×106)。次に予防試験では、ラットに生理食塩水(対照)またはShK(L5)(10μg/kg/日)を5日間にわたって皮下注射した。第1の予防試験では、対照ラットは軽度EAEを発現し(平均最大臨床スコア2.0±1.2)、平均発症日は5.6±0.6日であった(データ省略)。ShK(L5)は疾患の重症度を軽減した(平均最大臨床スコア0.7±0.6、p<0.05)。第2の予防試験では、対照ラットはより重度のEAEを発現し(平均最大臨床スコア3.2±0.4)、平均発症日は4.8±0.4日であった(図6A)。ShK(L5)は疾患の重症度を有意に軽減した(平均最大臨床スコア0.6±0.4、p<0.007)が、疾患発症日は有意には遅延させなかった(5.5±0.7日;p=0.07)。これらの試験では毒性の徴候がまったくみられなかった。
【0052】
治療試験(図6B)では、ラットにMBP活性化PAS細胞を注射し、最初にEAEの徴候(尾のひきずり、猫背および24時間で6%以上の体重減少)を発現したときに生理食塩水または10μg/kg/日のShK(L5)を投与し、3日間にわたって治療を続けた。EAEの臨床徴候は、対照群では第6日に最大に達し(スコア=3.9±0.7)、治療群では第7日に最大に達した(スコア=1.9±0.9;p<0.05)。
【0053】
in vivoにおけるShK(L5)の免疫抑制活性の独自の評価法として本出願人は、主に皮膚ホーミング性TEM細胞により媒介されるDTH反応を阻害する上でのShK(L5)の有効性を検討した。卵アルブミンおよびアジュバントで免疫したLewisラットに、7日後に片耳に卵アルブミンを接種し、他方の耳に生理食塩水を接種した。続いてラットに生理食塩水(対照)またはShK(L5)(10μg/kg/日)を注射し、DTHの指標として耳の厚さを測定した。全ての対照ラットが卵アルブミン接種後24時間および48時間の時点で耳の腫脹を発現したが、ShK(L5)投与動物ではDTH反応がかなり軽度であった(図6C)。したがってShK(L5)は、TEM媒介性DTH反応を抑制し、ミエリン活性化TEM細胞により誘発される重度の養子移入性EAEを予防および改善する。
【0054】
Kv1.3は抗原提示によりISに集積するが、Kv1.3を通るK+の流出はISの形成および安定性に必要ない
図7A〜7Gを参照すると、高選択性Kv1.3阻害薬(21)であるShK(L5)は、GAD65特異的TEM細胞においてKv1.3電流を遮断し、Kdは72±3pMであった。ShK(L5)を薬理学的プローブとして使用して、Kv1.3機能を必要とするTEM細胞活性化のステップを明らかにした。生化学的研究から、Kv1.3およびKvb2は、SAP97(シナプス関連タンパク質97)、ZIP(PKCゼータ結合タンパク質、p56lck関連p62タンパク質、A170)、p56lckおよびCD4を含むシグナル伝達複合体に属することが示されている(図7B)。ヒトTEM細胞における当該複合体の存在は、Kv1.3、Kvb2、SAP97、ZIPおよびp56lckとCD4との共キャップ形成を示す本出願人の結果により裏付けられている。さらに、FRET(蛍光エネルギー変換)研究から、Kv1.3をトランスフェクトしたヒトT細胞ではKv1.3がCD3に非常に接近しており、さらに該チャネルは、Kv1.3をトランスフェクトしたヒト細胞傷害性T細胞とその標的との接点に優先的に局在していることが示されている。CD4は、T細胞と抗原提示細胞(APC)が接する領域であるISに移動するため、シグナル伝達複合体のKv1.3および他のタンパク質も抗原提示によりISに局在する可能性がある。この考えを検討するため、T1DM患者からのGAD65特異的Kv1.3highTEMクローンを、GAD65 557Iペプチドを取り付けてDAPIで染色して視覚化を助けたHLA適合APCとともにインキュベートした。20分後、シグナル伝達複合体中のタンパク質を検討するため、APC−TEM結合体を免疫染色した。ISには、Kv1.3、Kvb2、SAD97、ZIPおよびp56lckとともにCD4が局在していた。APCとTEMとの接触のない場合では、CD4およびKv1.3は細胞全体に分布していた。さらに、GAD65特異的TEM細胞がMBP(無関連抗原)を取り付けたAPCに曝された場合は、CD4およびKv1.3は接点に局在できなかったことから、ISの集積は抗原特異的であることが立証された。以上のように、GAD65特異的TEM細胞では、抗原提示により、Kv1.3含有シグナル伝達複合体がCD4とともにISに移動することから、Kv1.3はTEM細胞におけるシグナル変換機構に不可欠な構成要素であることが示唆される。以上の試験に基づけば、Kv1.3チャネルの約99%を遮断する濃度(100nM)のShK(L5)は、ISの集積を妨げず、ひとたび形成されたISを崩壊させなかったことから、Kv1.3チャネルを通るK+の流出はISの形成または安定性に不必要であることが示された。
【0055】
ヒトTEM細胞の抑制
図8A〜8Eを参照すると、ShK(L5)は、T細胞活性化に不可欠な初期段階であるTEM細胞のカルシウムシグナル伝達を阻害した。T1DM患者からのGAD65特異的TEMクローンに、カルシウム指示薬である色素Fluo3を取り付け、培養液のみで、または濃度を増大させたShK(L5)とともにプレインキュベートし、活性化用抗CD3抗体および架橋用第2抗体の添加前後にフローサイトメトリーにより画像化した。刺激後242±35秒で最高カルシウム上昇が起こり、これがShK(L5)で遮断され、IC50は約200pMであった(図8A)。ShK(L5)は、T1DM患者およびRA患者からのナイーブ/TCM細胞に比べ、同患者からの自己反応性TEM細胞による[3H」チミジン取込みを10倍効果的に抑制した(図8B、左)。第2セットの実験(図8B、右)では、RA−SFおよびRA−PBT細胞を抗CD3抗体で48時間活性化し、それぞれ「TEMエフェクター」および「ナイーブ/TCMエフェクター」を作製した。培地中で細胞を一晩休ませ、ShK(L5)の存在下または非存在下で抗CD3抗体でさらに48時間再刺激し、[3H」チミジン取込みを測定した。RA−SF−TEMエフェクターはShK(L5)阻害に対する感受性を保持していたが、RA−PB−ナイーブ/TCMエフェクターはKv1.3遮断に抵抗性であり(図8B,右)、これはおそらく、該エフェクターがカルシウム活性化KCa3.1/IKCa1チャネルをアップレギュレートしたことによるものである可能性が高く、このチャネルはカルシウム流入の際にKv1.3の代わりになる。ShK(L5)は、T1DMおよびRA患者からのTEM細胞によるインターロイキン2(IL2)およびインターフェロン−g(IFN−g)の産生を大いに抑制したが、これらの患者からのナイーブ/TCM細胞によるIL2およびIFN−gの産生はそれほど影響を受けなかった(図8C)。TEM細胞およびナイーブ/TCM細胞による腫瘍壊死因子−αおよびインターロイキン4の産生は、いずれもShK(L5)に感受性が低かった(図8C)。
【0056】
エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルの検証
図9に示すように、アジュバント中の卵アルブミン(OVA)でラットを免疫した。7日後に片耳にOVAを接種し、他方の耳に生理食塩水を接種した。遅延型過敏症(DTH)の徴候として、24時間後に耳の腫脹を測定した。図9に示したFACSヒストグラムは、OVAを接種した耳のT細胞はCD45RC陰性記憶細胞であり、一方、同一ラットの血液および脾臓のT細胞は大部分がナイーブT細胞であることを示している。
【0057】
エフェクター記憶T細胞により引き起こされる遅延型過敏症(DTH)のラットモデルにおけるShK(L5)の治療プロトコル
図10に示すように、ナイーブ細胞のエフェクター記憶TEM細胞への分化を妨げるため第0日から第7日まで(初回刺激段階)、もしくはTEM細胞の機能を妨げるため耳に卵アルブミンを接種した後のエフェクター段階で、皮下注射として10μg/kg/日のShK(L5)をラットに投与した。
【0058】
ShK(L5)はナイーブおよびセントラル記憶T細胞またはB細胞の機能を損なわずにラットin vivoにおいてエフェクター記憶反応を特異的に抑制する
図11に示すように、対照ラットは耳の腫脹、すなわち陽性DTH反応を発現した。ShK(L5)は、初回刺激段階で投与するとDTHの抑制に効果がなかったことから、ナイーブおよびセントラル記憶T細胞のエフェクター記憶細胞への分化を抑制しなかったことを示している。ShK(L5)は、エフェクター段階で投与するとDTHを抑制したことから、エフェクター記憶T細胞が耳に達する能力を妨げたか、またはエフェクター記憶T細胞の活性化を抑制したかのうち少なくともいずれかであることを示している。ShK(L5)投与ラットの耳のT細胞数は、賦形剤を投与したラットの耳と同じであったため、第一の可能性は除外された。エフェクター記憶T細胞はKv1.3陰性であったが、賦形剤を投与した動物の耳の記憶T細胞はKv1.3陽性であったため、ShK(L5)は耳におけるエフェクター記憶T細胞の活性化を抑制したのである。上記動物ではIgMおよびIgG B細胞の反応も影響を受けなかった。
【0059】
T1DMまたはMSの患者および健康な対照者からの、GAD65/555−567、インスリン/9−23およびミエリン抗原に特異的なT細胞におけるKv1.3発現
図12Aは、新規発症1型糖尿病患者、健康な対照者および多発性硬化症患者に由来する抗原特異的T細胞からのKv1.3電流(上)およびチャネル数/細胞(下)を示している。各データ点は、3回目の抗原刺激から48時間後に測定した、単一ドナーからの2〜4のT細胞株に由来する細胞20〜50個についての平均値±SEMを示している。T1DM患者および対照者の血中にはインスリンおよびGAD65に特異的なT細胞の頻度が低いため、スプリットウェル法を用いて短期自己抗原特異的CD4+T細胞株を作製することにより、上記母集団を増幅した。対照として、MSには関係しているがT1DMには関係のない無関連自己抗原であるミエリン塩基性タンパク質(MBP)に特異的なT細胞株を作製した。3回目の抗原刺激後に、4pFを超える膜電気容量を有する活性化細胞を用い、ホールセルパッチクランプ法によりKv1.3電流を測定した(細胞直径11μm以上)。代表的なKv1.3電流およびKv1.3チャネル数/T細胞を図12Aに示す。電流は、Kv1.3に特徴的な生物物理学的および薬理学的特性を示した。新規発症T1DM患者由来のインスリン(9〜23)またはGAD65(555〜567)特異的T細胞は、Kv1.3電流が大きく、Kv1.3チャネル数が多かったが、当該患者由来の疾患とは無関連なMBP特異的T細胞はKv1.3lowであった(p=0.001)。比較のため、反対のパターンが観察されたMS患者について本出願人が発表したKv1.3データをプロットした。MS患者では、MBPまたはミエリン乏突起神経膠細胞の糖タンパク質(ペプチド35〜55)もしくはプロテオリピドタンパク質(ペプチド139〜151)に特異的なT細胞はKv1.3highであったが、インスリンおよびGAD65に特異的なT細胞はKv1.3lowであった(p=0.0001)。健康対照者から単離した自己反応性T細胞は、特異性に関係なくKv1.3lowであった。MSおよびT1DMの両方を有する患者1例では、3つの自己抗原に特異的なT細胞がKv1.3highであった。長期にわたるT1DMを有する患者からのGAD65特異的およびインスリン特異的T細胞がKv1.3highであったことは、自己反応性TEM細胞の持続性を反映しているが、非自己免疫性2型DM患者からのGAD65およびインスリン特異的T細胞にはKv1.3lowパターンがみられた。図12Bに、個々の細胞のKv1.3の染色強度(上)および蛍光強度(下)を示す。本出願人は、Kv1.3の免疫染色によりパッチクランプ法のデータを確認した。T1DM患者からのインスリン特異的およびGAD65特異的T細胞、ならびにMS患者からのMBP特異的T細胞は明るく染色されたが、無関連自己抗原に特異的な細胞の染色はかすかであった。図12CはCCR7発現を示している。フローサイトメトリーにより、Kv1.3highT細胞はCCR7−TEM細胞であるが、Kv1.3low細胞はCCR7+ナイーブまたはTCM細胞であることが示された。図12Dは、T1DMおよびMSの両方を有する患者からと、それぞれ5年以上および2年以上のT1DMまたは2型DMを有する患者からの自己反応性T細胞における1細胞あたりのKv1.3の数を示している。図12Eは、新規発症T1DM患者からのCD4+GAD65四量体+T細胞におけるKv1.3数を示している。さらなる対照として、GAD65 557Iペプチドを含有する蛍光性MHCクラスII四量体を使用して、新規発症T1DMを有するDR−0401陽性患者からGAD65特異的CD4+T細胞を単離した。四量体で選別したGAD65活性化T細胞は、T1DM患者からのGAD65特異的T細胞株に観察されたパターンと同じKv1.3highパターンを示した。以上を要約すると、T1DMおよびMSのいずれにおいても、疾患関連性の自己抗原活性化T細胞はKv1.3highCCR7−TEMエフェクターであり、一方、当該患者における疾患無関連性の自己反応性細胞はKv1.3lowCCR7+ナイーブ/TCM細胞である。
【0060】
関節リウマチおよび変形性関節症におけるKv1.3発現
RAでは、罹患関節からの疾患関連T細胞の単離が可能である。本出願人は、抗CD3抗体で刺激後48時間の時点でRA患者7例の滑液(SF)から採取したT細胞についてパッチクランプ法を行なった。図13Aにみられるように、対照として本出願人は、非自己免疫性の退行性変形性関節症(OA)患者7例からのSF−T細胞(同一プロトコルで活性化した)を分析した。RA−SFのT細胞はKv1.3highであり、一方、OA−SFのT細胞はKv1.3lowであった(p<0.0001)。本出願人は、RA患者の末梢血(PB)からの抗CD3活性化T細胞にKv1.3lowパターンを見出した(p<0.0001)が、これは血液中には自己反応性Kv1.3highTEM細胞がまれであるためである。Kv1.3およびその付属のKvβ2サブユニットの免疫染色により、パッチクランプ法のデータの確証を得た。図13Bは、 Kv1.3(図では明灰色)および Kvβ2(図では暗灰色)染色の共焦点画像を示している。RA−SFのT細胞はKv1.3およびKvβ2の両方が明るく染色されたが、OA−SFおよびRA−PBのT細胞は弱い染色を示した。図13CはCCR7発現を示している。フローサイトメトリーにより、Kv1.3highRA−SF T細胞はCCR7−TEM細胞であるが、Kv1.3lowOA−SFおよびRA−PB T細胞はCCR7+ナイーブ/TCM細胞であることが検証された。図13D(上)は、抗CD3抗体または抗Kv1.3抗体で染色し、ヘマトキリン/エオシンで対比染色したRAおよびOA患者からの滑膜の顕微鏡写真を示している(40倍)。さらなる試験として、RA患者5例およびOA患者5例からのパラフィン包埋滑膜組織(ST)をCD3、Kv1.3およびCCR7について免疫染色した。以前に示したとおり、本出願人の染色法は、ナイーブ/TCM細胞ではKv1.3チャネル数が少ないためにKv1.3を検出しない。RA−STではCD3+Kv1.3+CCR7−TEM細胞の優勢がみられたが、OA滑膜ではCD3+細胞がまばらであり、これらの細胞は主にKv1.3−CCR7+ナイーブ/TCM細胞であった。分類法により評価したCD3+、Kv1.3+およびCCR7+細胞による浸潤度を図S2Aに示す。CD3+炎症指数:RA=3.2±0.1;OA=1.1±0.2(p<0.01);Kv1.3+炎症指数:RA=2.8±0.3;OA=0.6±0.3(p<0.01)。以上のように、異なる3つの自己免疫疾患において、我々の結果は、疾患関連性自己反応性T細胞がKv1.3highCCR7−TEMエフェクターであることで一致している。
【0061】
本発明の一部の実施例および実施形態を参照しながら本明細書において本発明を説明してきたが、上記実施例および実施形態には、本発明の趣旨および範囲から逸脱することなく種々の追加、削除、変更および修正を加えうることを理解されたい。たとえば、1つの実施形態または実施例の任意の要素もしくは属性は、実施形態または実施例がその意図した用途に対し不適切なものとならない限り、別の実施形態または実施例に組み込んでもよいし、別の実施形態または実施例とともに使用してもよい。また、方法または手順の工程が特定の順番で列記もしくは記述されている場合、当該工程の順番は、特に定めのない限り、または当該工程の順番の変更が本発明を特許性のないもの、もしくはその意図した用途に対し不適切なものとすることがない限り、変更してもよい。妥当な追加、削除、修正および変更はすべて、説明した実施例および実施形態と同等であるとみなすものとし、特許請求の範囲内に含まれるものとする。
【0062】
【表3】
【0063】
【化1】
【特許請求の範囲】
【請求項1】
アニオン電荷を有する有機または無機化学物質を付加したShKを含んでなる組成物。
【請求項2】
前記化学物質が、アミノ酸、ポリペプチド、アミノ酸残基、非天然アミノ酸残基、スレオニン、スレオニン誘導体、ホスホスレオニン、セリン、セリン誘導体、ホスホセリン、グルタミン酸、グルタミン酸誘導体、γカルボキシグルタミン酸、アスパラギン酸、アスパラギン酸誘導体、無機化合物または無機化学基、有機化合物または有機化学基、無水コハク酸、および無水フタル酸からなる群から選択される請求項1に記載の組成物。
【請求項3】
前記ShKトキシンが天然源から入手される請求項1に記載の組成物。
【請求項4】
前記ShKトキシンが合成物である請求項1に記載の組成物。
【請求項5】
前記化学物質がShKポリペプチドのN末端に付加されている請求項1に記載の組成物。
【請求項6】
前記化学物質が結合分子または結合基を介してShKのN末端に付加されている請求項5に記載の組成物。
【請求項7】
前記化学物質がアミノエチルオキシエチルオキシアセチル・リンカーによりShKのN末端に付加されている請求項5に記載の組成物。
【請求項8】
前記化学物質が蛍光団の標識を含んでいる請求項1に記載の組成物。
【請求項9】
前記化学物質がAEEAc−L−Pmp(OH2)である請求項1に記載の組成物。
【請求項10】
前記化学物質がAEEAc−D−Pmp(OH2)である請求項1に記載の組成物。
【請求項11】
前記化学物質がAEEAc−D−Pmp(OH,Et)である請求項1に記載の組成物。
【請求項12】
前記化学物質がAEEAc−L−Pmp(Et2)である請求項1に記載の組成物。
【請求項13】
前記化学物質がAEEAc−D−Pmp(Et2)である請求項1に記載の組成物。
【請求項14】
前記化学物質がAEEAc−L−Tyrである請求項1に記載の組成物。
【請求項15】
前記化学物質がAEEAc−L−Tyr(PO3H2)である請求項1に記載の組成物。
【請求項16】
前記化学物質がAEEAc−L−Phe(p−NH2)である請求項1に記載の組成物。
【請求項17】
前記化学物質がAEEAc−L−Phe(p−CO2H)である請求項1に記載の組成物。
【請求項18】
前記化学物質がAEEAc−L−アスパラギン酸塩である請求項1に記載の組成物。
【請求項19】
前記化学物質がAEEAc−D−アスパラギン酸塩である請求項1に記載の組成物。
【請求項20】
前記化学物質がAEEAc−L−グルタミン酸塩である請求項1に記載の組成物。
【請求項21】
前記化学物質がAEEAc−D−グルタミン酸塩である請求項1に記載の組成物。
【請求項22】
ヒトまたは動物対象者におけるKv1.3カリウムチャネルの阻害を引き起こす方法であって、前記方法は、
(A)アニオン電荷を有する有機または無機化学物質を付加したShKトキシンを含んでなる組成物を、Kv1.3カリウムチャネルの阻害に有効な様式および量で前記対象者に投与する工程を含むことを特徴とする方法。
【請求項23】
自己免疫疾患を予防または治療するために実施する請求項22に記載の方法。
【請求項24】
前記自己免疫疾患が、多発性硬化症、重症筋無力症、ギラン・バレーなどの自己免疫性末梢神経障害、自己免疫性ブドウ膜炎、クローン病、潰瘍性大腸炎、原発性胆汁性肝硬変、自己免疫性肝炎、自己免疫性血小板減少症、1型糖尿病、アジソン病、グレーブス病、橋本甲状腺炎、自己免疫性卵巣炎および精巣炎、ベーチェット病、関節リウマチ、歯周病に随伴する骨吸収、全身性紅斑性狼瘡、強皮症、多発筋炎、皮膚筋炎、尋常性天疱瘡、強直性脊椎炎などの脊椎関節症、およびシェーグレン症候群からなる群から選択される請求項23に記載の方法。
【請求項25】
移植片対宿主病を予防または治療するために実施する請求項22に記載の方法。
【請求項26】
移植した組織または臓器の拒絶反応を予防または治療するために実施する請求項22に記載の方法。
【請求項27】
メタボリック症候群を予防または治療するために実施する請求項22に記載の方法。
【請求項28】
2型糖尿病を治療または予防するために実施する請求項22に記載の方法。
【請求項29】
肥満を治療または予防するために実施する請求項22に記載の方法。
【請求項30】
歯周病に随伴する骨吸収を治療または予防するために実施する請求項22に記載の方法。
【請求項31】
前記組成物が、アミノ酸、ポリペプチド、アミノ酸残基、非天然アミノ酸残基、スレオニン、スレオニン誘導体、ホスホスレオニン、セリン、セリン誘導体、ホスホセリン、グルタミン酸、グルタミン酸誘導体、γカルボキシグルタミン酸、アスパラギン酸、アスパラギン酸誘導体、無機化合物または無機化学基、有機化合物または有機化学基、無水コハク酸、および無水フタル酸からなる群から選択される化学物質を付加したShKトキシンを含んでなる請求項22に記載の方法。
【請求項32】
前記化学物質がAEEAc−L−Pmp(OH2)である請求項22に記載の方法。
【請求項33】
前記化学物質がAEEAc−D−Pmp(OH2)である請求項22に記載の方法。
【請求項34】
前記化学物質がAEEAc−D−Pmp(OH,Et)である請求項22に記載の方法。
【請求項35】
前記化学物質がAEEAc−L−Pmp(Et2)である請求項22に記載の方法。
【請求項36】
前記化学物質がAEEAc−D−Pmp(Et2)である請求項22に記載の方法。
【請求項37】
前記化学物質がAEEAc−L−Tyrである請求項22に記載の方法。
【請求項38】
前記化学物質がAEEAc−L−Tyr(PO3H2)である請求項22に記載の方法。
【請求項39】
前記化学物質がAEEAc−L−Phe(p−NH2)である請求項22に記載の方法。
【請求項40】
前記化学物質がAEEAc−L−Phe(p−CO2H)である請求項22に記載の方法。
【請求項41】
前記化学物質がAEEAc−L−アスパラギン酸塩である請求項22に記載の方法。
【請求項42】
前記化学物質がAEEAc−D−アスパラギン酸塩である請求項22に記載の方法。
【請求項43】
前記化学物質がAEEAc−L−グルタミン酸塩である請求項22に記載の方法。
【請求項44】
前記化学物質がAEEAc−D−グルタミン酸塩である請求項22に記載の方法。
【請求項45】
フローサイトメトリーを実施するための方法であって、
(A)アニオン電荷および蛍光団標識を有する有機または無機化学物質を付加したShKを含んでなる組成物を提供する工程と、
(B)工程Aで提供された組成物を細胞と混合する工程と、
(C)工程Aで提供された組成物に対し親和性を有する細胞を、フローサイトメーターを用いて、計数、単離、または識別する工程と
を含むことを特徴とする方法。
【請求項46】
前記工程Cがフローサイトメーターを用いてTリンパ球を計数、単離または識別することからなる請求項45に記載の方法。
【請求項47】
前記ShKが21位におけるMet残基の置換により修飾されている請求項1に記載の組成物。
【請求項48】
前記21位Met残基の置換が酸化を阻止するようになされた請求項47に記載の組成物。
【請求項49】
前記ShKがC末端酸官能基をアミドで置換することにより修飾されている請求項1に記載の組成物。
【請求項50】
前記C末端酸官能基のアミドへの置換が、C末端カルボキシペプチダーゼ酵素に対する安定性を与えるようになされた請求項49に記載の組成物。
【請求項51】
前記ShKが21位におけるMet残基の置換により修飾されている請求項22または45に記載の方法。
【請求項52】
前記21位Met残基の置換が酸化を阻止するようになされた請求項51に記載の方法。
【請求項53】
前記ShKがC末端酸官能基をアミドで置換することにより修飾されている請求項22または45に記載の方法。
【請求項54】
前記C末端酸官能基のアミドへの置換が、C末端カルボキシペプチダーゼ酵素に対する安定性を与えるようになされた請求項53に記載の方法。
【請求項1】
アニオン電荷を有する有機または無機化学物質を付加したShKを含んでなる組成物。
【請求項2】
前記化学物質が、アミノ酸、ポリペプチド、アミノ酸残基、非天然アミノ酸残基、スレオニン、スレオニン誘導体、ホスホスレオニン、セリン、セリン誘導体、ホスホセリン、グルタミン酸、グルタミン酸誘導体、γカルボキシグルタミン酸、アスパラギン酸、アスパラギン酸誘導体、無機化合物または無機化学基、有機化合物または有機化学基、無水コハク酸、および無水フタル酸からなる群から選択される請求項1に記載の組成物。
【請求項3】
前記ShKトキシンが天然源から入手される請求項1に記載の組成物。
【請求項4】
前記ShKトキシンが合成物である請求項1に記載の組成物。
【請求項5】
前記化学物質がShKポリペプチドのN末端に付加されている請求項1に記載の組成物。
【請求項6】
前記化学物質が結合分子または結合基を介してShKのN末端に付加されている請求項5に記載の組成物。
【請求項7】
前記化学物質がアミノエチルオキシエチルオキシアセチル・リンカーによりShKのN末端に付加されている請求項5に記載の組成物。
【請求項8】
前記化学物質が蛍光団の標識を含んでいる請求項1に記載の組成物。
【請求項9】
前記化学物質がAEEAc−L−Pmp(OH2)である請求項1に記載の組成物。
【請求項10】
前記化学物質がAEEAc−D−Pmp(OH2)である請求項1に記載の組成物。
【請求項11】
前記化学物質がAEEAc−D−Pmp(OH,Et)である請求項1に記載の組成物。
【請求項12】
前記化学物質がAEEAc−L−Pmp(Et2)である請求項1に記載の組成物。
【請求項13】
前記化学物質がAEEAc−D−Pmp(Et2)である請求項1に記載の組成物。
【請求項14】
前記化学物質がAEEAc−L−Tyrである請求項1に記載の組成物。
【請求項15】
前記化学物質がAEEAc−L−Tyr(PO3H2)である請求項1に記載の組成物。
【請求項16】
前記化学物質がAEEAc−L−Phe(p−NH2)である請求項1に記載の組成物。
【請求項17】
前記化学物質がAEEAc−L−Phe(p−CO2H)である請求項1に記載の組成物。
【請求項18】
前記化学物質がAEEAc−L−アスパラギン酸塩である請求項1に記載の組成物。
【請求項19】
前記化学物質がAEEAc−D−アスパラギン酸塩である請求項1に記載の組成物。
【請求項20】
前記化学物質がAEEAc−L−グルタミン酸塩である請求項1に記載の組成物。
【請求項21】
前記化学物質がAEEAc−D−グルタミン酸塩である請求項1に記載の組成物。
【請求項22】
ヒトまたは動物対象者におけるKv1.3カリウムチャネルの阻害を引き起こす方法であって、前記方法は、
(A)アニオン電荷を有する有機または無機化学物質を付加したShKトキシンを含んでなる組成物を、Kv1.3カリウムチャネルの阻害に有効な様式および量で前記対象者に投与する工程を含むことを特徴とする方法。
【請求項23】
自己免疫疾患を予防または治療するために実施する請求項22に記載の方法。
【請求項24】
前記自己免疫疾患が、多発性硬化症、重症筋無力症、ギラン・バレーなどの自己免疫性末梢神経障害、自己免疫性ブドウ膜炎、クローン病、潰瘍性大腸炎、原発性胆汁性肝硬変、自己免疫性肝炎、自己免疫性血小板減少症、1型糖尿病、アジソン病、グレーブス病、橋本甲状腺炎、自己免疫性卵巣炎および精巣炎、ベーチェット病、関節リウマチ、歯周病に随伴する骨吸収、全身性紅斑性狼瘡、強皮症、多発筋炎、皮膚筋炎、尋常性天疱瘡、強直性脊椎炎などの脊椎関節症、およびシェーグレン症候群からなる群から選択される請求項23に記載の方法。
【請求項25】
移植片対宿主病を予防または治療するために実施する請求項22に記載の方法。
【請求項26】
移植した組織または臓器の拒絶反応を予防または治療するために実施する請求項22に記載の方法。
【請求項27】
メタボリック症候群を予防または治療するために実施する請求項22に記載の方法。
【請求項28】
2型糖尿病を治療または予防するために実施する請求項22に記載の方法。
【請求項29】
肥満を治療または予防するために実施する請求項22に記載の方法。
【請求項30】
歯周病に随伴する骨吸収を治療または予防するために実施する請求項22に記載の方法。
【請求項31】
前記組成物が、アミノ酸、ポリペプチド、アミノ酸残基、非天然アミノ酸残基、スレオニン、スレオニン誘導体、ホスホスレオニン、セリン、セリン誘導体、ホスホセリン、グルタミン酸、グルタミン酸誘導体、γカルボキシグルタミン酸、アスパラギン酸、アスパラギン酸誘導体、無機化合物または無機化学基、有機化合物または有機化学基、無水コハク酸、および無水フタル酸からなる群から選択される化学物質を付加したShKトキシンを含んでなる請求項22に記載の方法。
【請求項32】
前記化学物質がAEEAc−L−Pmp(OH2)である請求項22に記載の方法。
【請求項33】
前記化学物質がAEEAc−D−Pmp(OH2)である請求項22に記載の方法。
【請求項34】
前記化学物質がAEEAc−D−Pmp(OH,Et)である請求項22に記載の方法。
【請求項35】
前記化学物質がAEEAc−L−Pmp(Et2)である請求項22に記載の方法。
【請求項36】
前記化学物質がAEEAc−D−Pmp(Et2)である請求項22に記載の方法。
【請求項37】
前記化学物質がAEEAc−L−Tyrである請求項22に記載の方法。
【請求項38】
前記化学物質がAEEAc−L−Tyr(PO3H2)である請求項22に記載の方法。
【請求項39】
前記化学物質がAEEAc−L−Phe(p−NH2)である請求項22に記載の方法。
【請求項40】
前記化学物質がAEEAc−L−Phe(p−CO2H)である請求項22に記載の方法。
【請求項41】
前記化学物質がAEEAc−L−アスパラギン酸塩である請求項22に記載の方法。
【請求項42】
前記化学物質がAEEAc−D−アスパラギン酸塩である請求項22に記載の方法。
【請求項43】
前記化学物質がAEEAc−L−グルタミン酸塩である請求項22に記載の方法。
【請求項44】
前記化学物質がAEEAc−D−グルタミン酸塩である請求項22に記載の方法。
【請求項45】
フローサイトメトリーを実施するための方法であって、
(A)アニオン電荷および蛍光団標識を有する有機または無機化学物質を付加したShKを含んでなる組成物を提供する工程と、
(B)工程Aで提供された組成物を細胞と混合する工程と、
(C)工程Aで提供された組成物に対し親和性を有する細胞を、フローサイトメーターを用いて、計数、単離、または識別する工程と
を含むことを特徴とする方法。
【請求項46】
前記工程Cがフローサイトメーターを用いてTリンパ球を計数、単離または識別することからなる請求項45に記載の方法。
【請求項47】
前記ShKが21位におけるMet残基の置換により修飾されている請求項1に記載の組成物。
【請求項48】
前記21位Met残基の置換が酸化を阻止するようになされた請求項47に記載の組成物。
【請求項49】
前記ShKがC末端酸官能基をアミドで置換することにより修飾されている請求項1に記載の組成物。
【請求項50】
前記C末端酸官能基のアミドへの置換が、C末端カルボキシペプチダーゼ酵素に対する安定性を与えるようになされた請求項49に記載の組成物。
【請求項51】
前記ShKが21位におけるMet残基の置換により修飾されている請求項22または45に記載の方法。
【請求項52】
前記21位Met残基の置換が酸化を阻止するようになされた請求項51に記載の方法。
【請求項53】
前記ShKがC末端酸官能基をアミドで置換することにより修飾されている請求項22または45に記載の方法。
【請求項54】
前記C末端酸官能基のアミドへの置換が、C末端カルボキシペプチダーゼ酵素に対する安定性を与えるようになされた請求項53に記載の方法。
【図1】

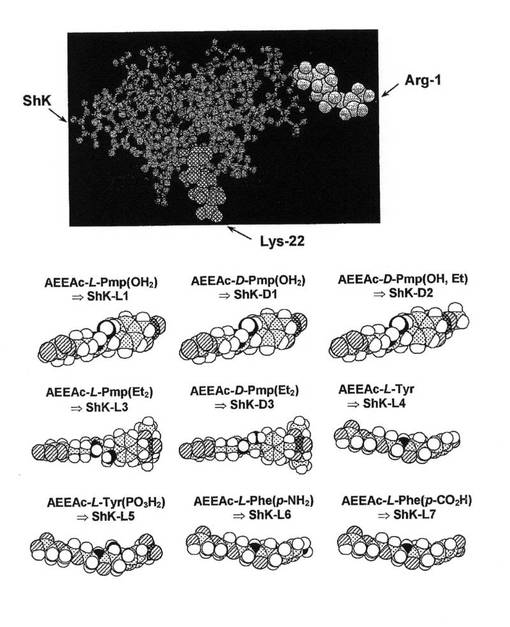
【図2A】

【図2B】
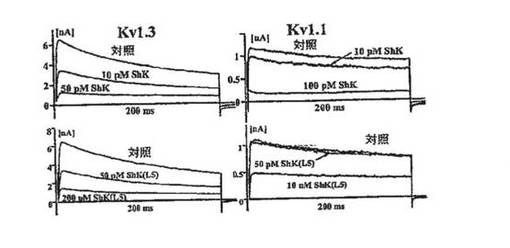
【図2C】
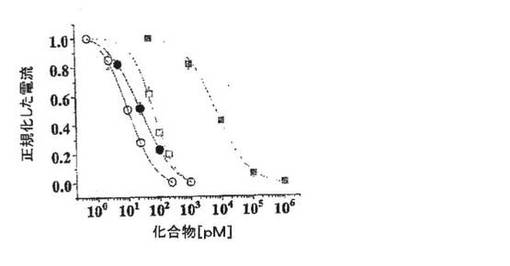
【図2D】
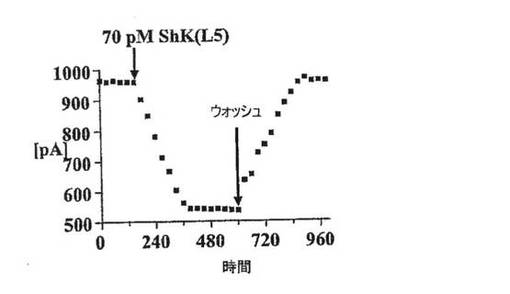
【図2E】
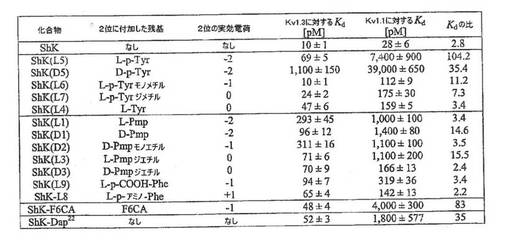
【図3A】

【図3B】

【図3C】
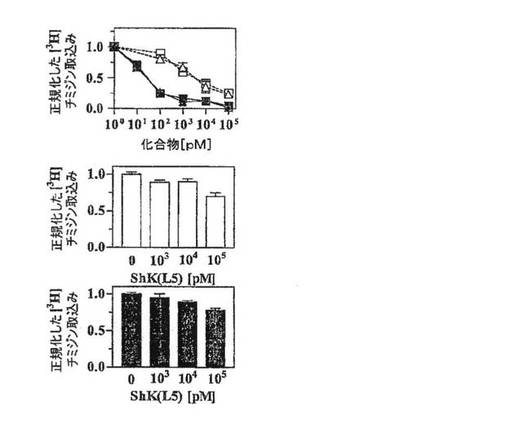
【図3D】
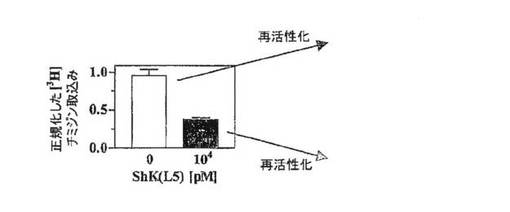
【図4A】
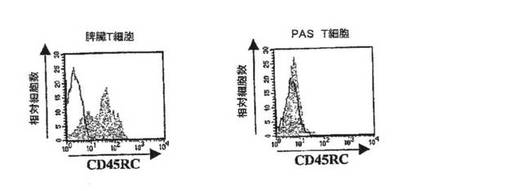
【図4B】
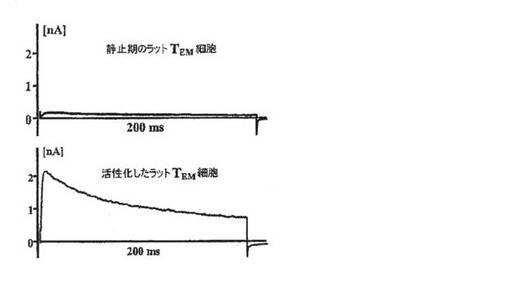
【図4C】
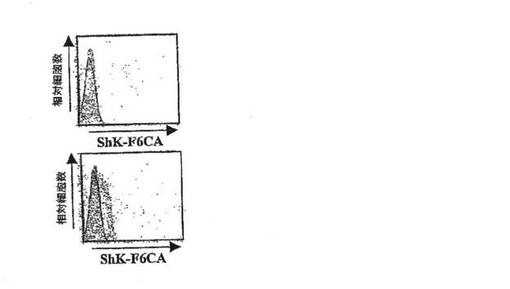
【図4D】

【図4E】
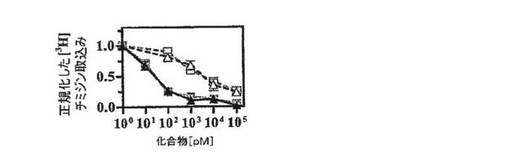
【図4F】
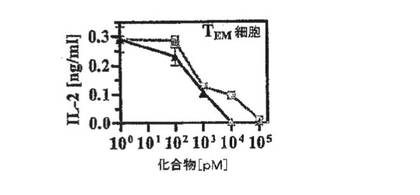
【図4G】
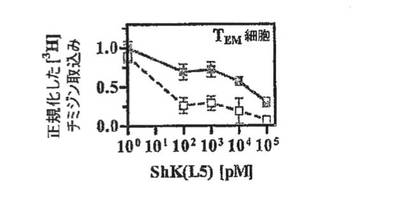
【図5A】
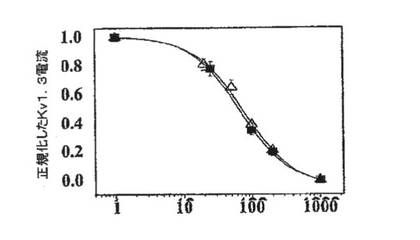
【図5B】
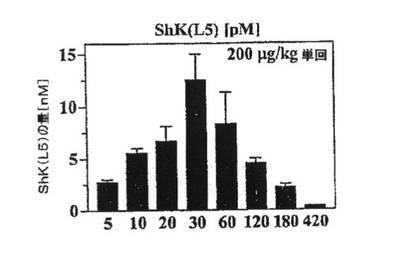
【図5C】
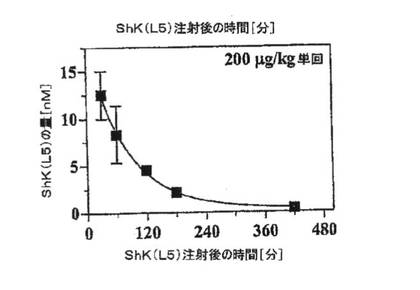
【図5D】
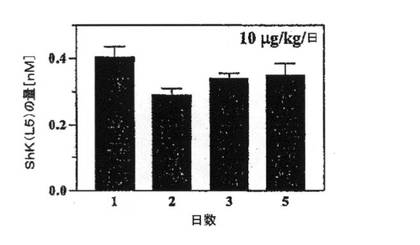
【図5E】
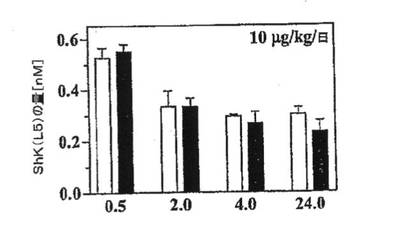
【図5F】
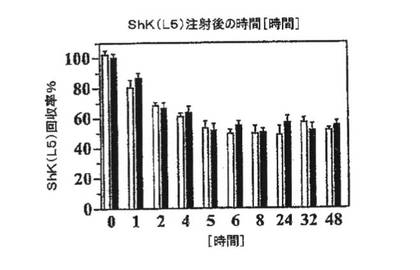
【図6A】
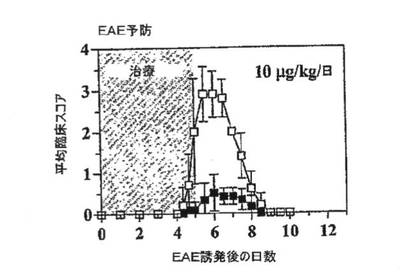
【図6B】
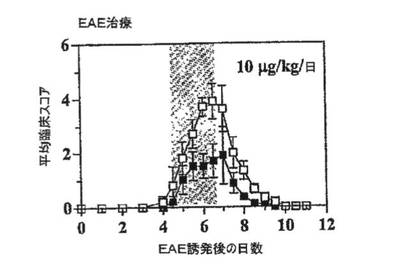
【図6C】
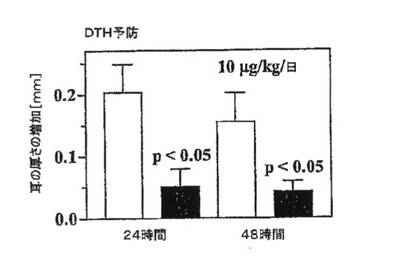
【図7A】
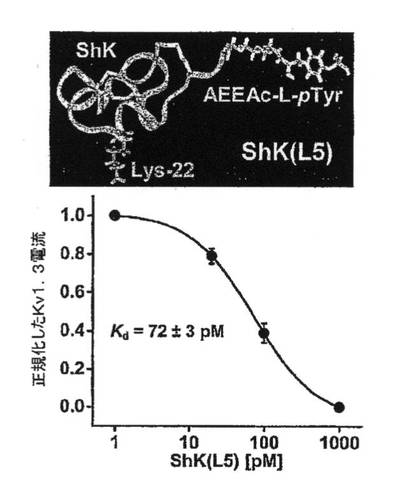
【図7B】
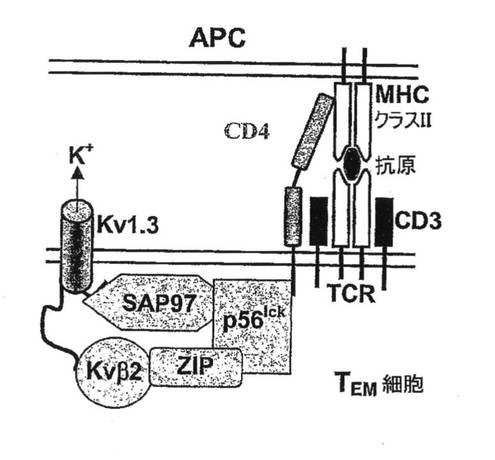
【図7C】
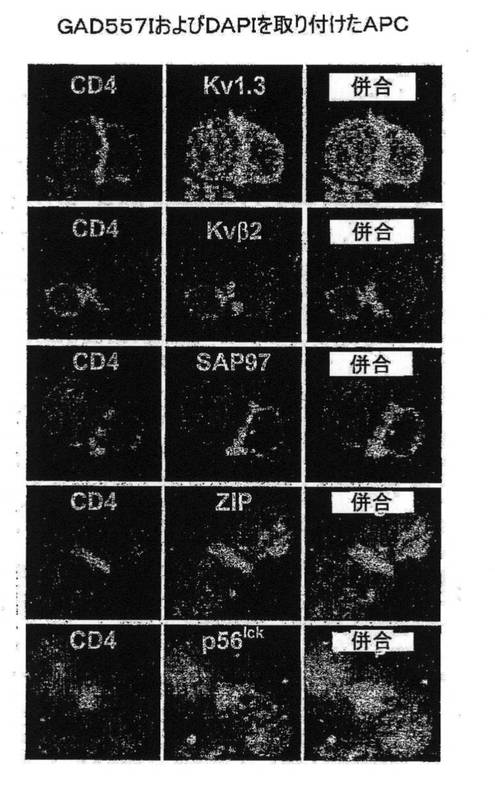
【図7D】
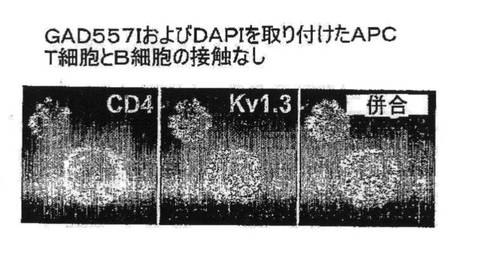
【図7E】
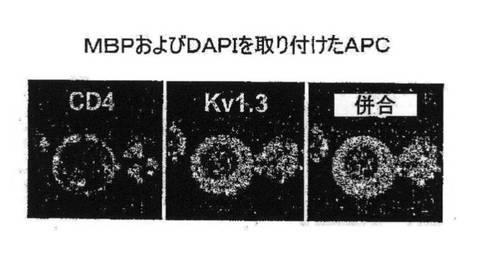
【図7F】

【図7G】
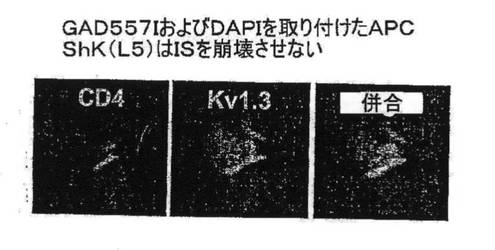
【図8A】
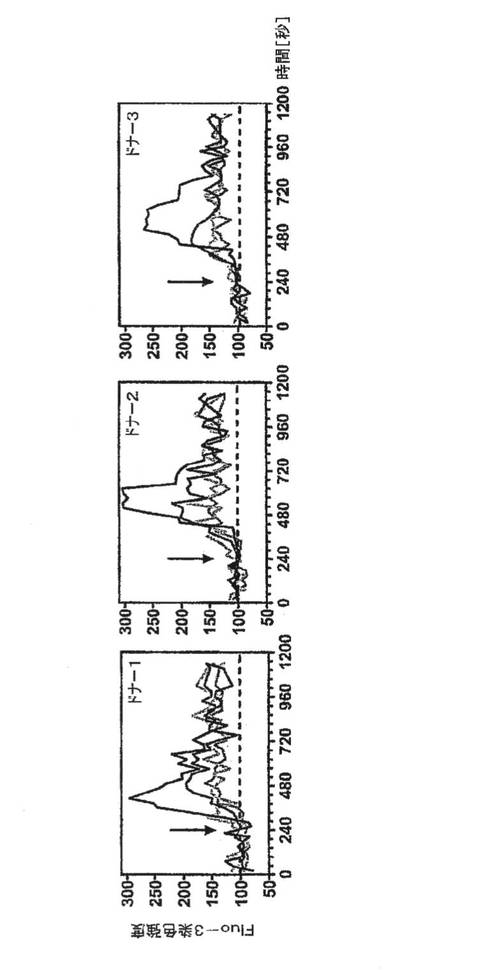
【図8B】
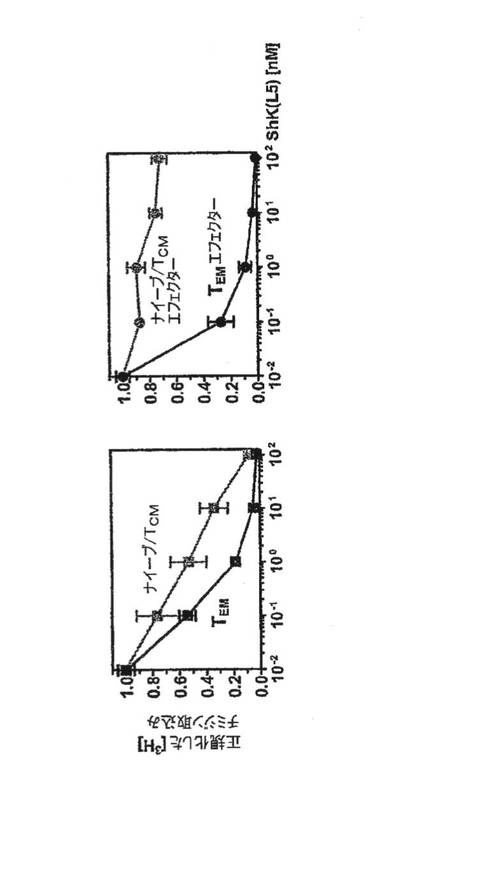
【図8C】
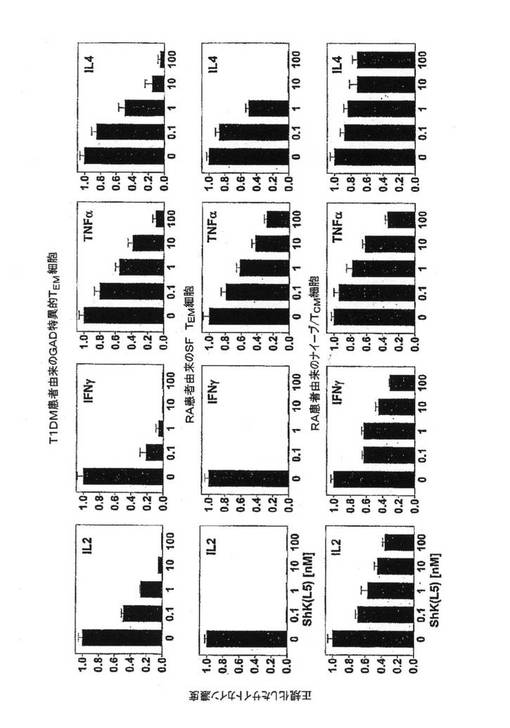
【図8D】
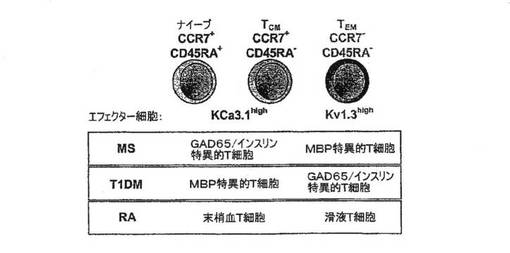
【図8E】
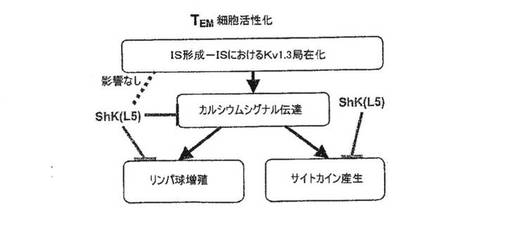
【図9】
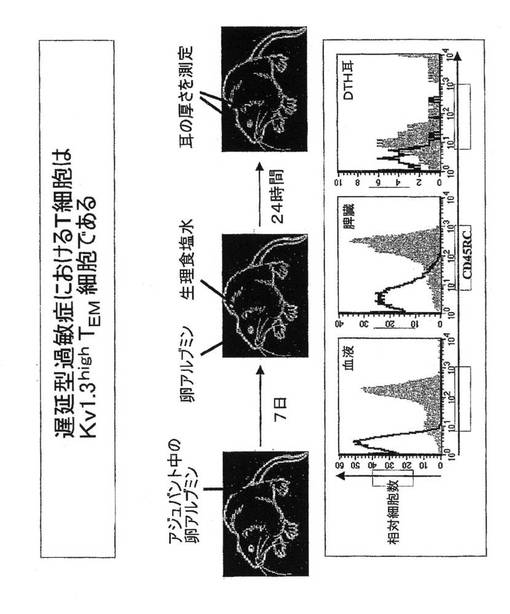
【図10】
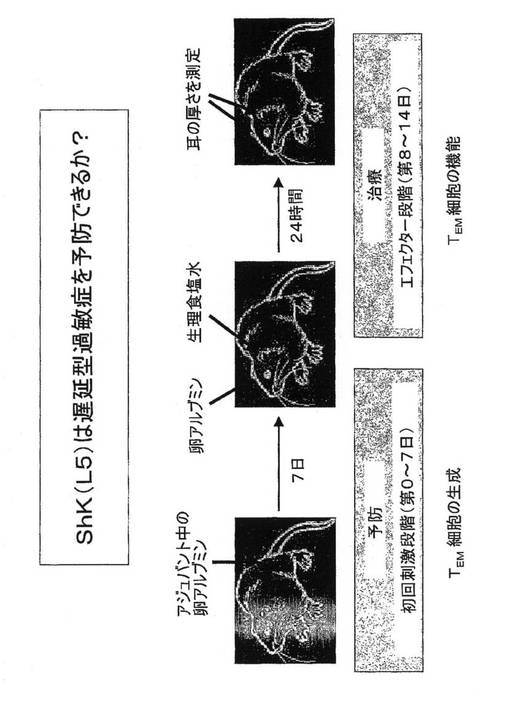
【図11】

【図12A】
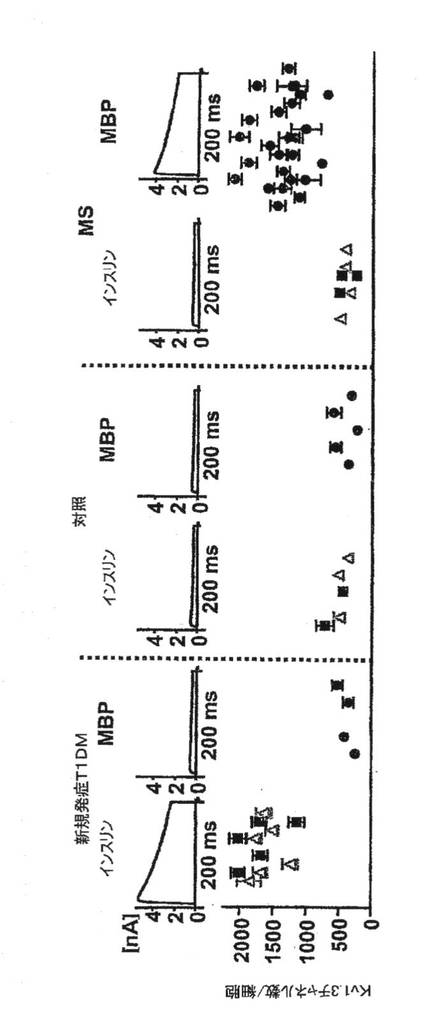
【図12B】
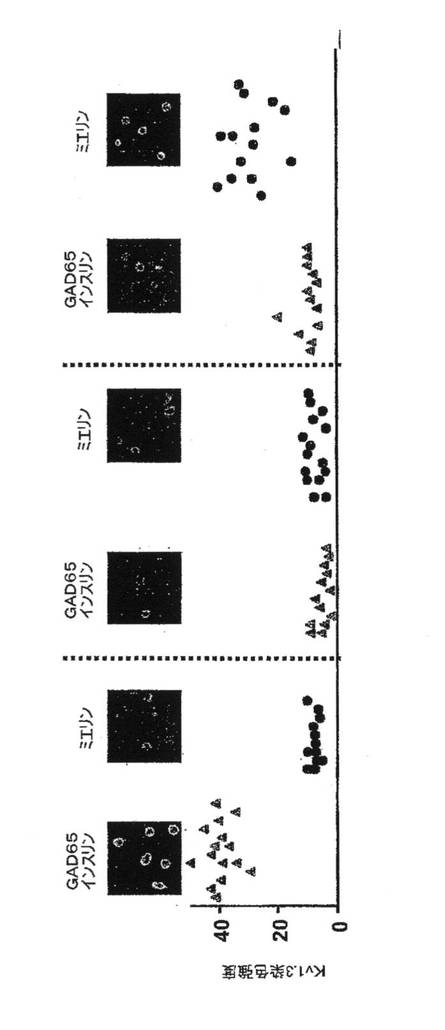
【図12C】
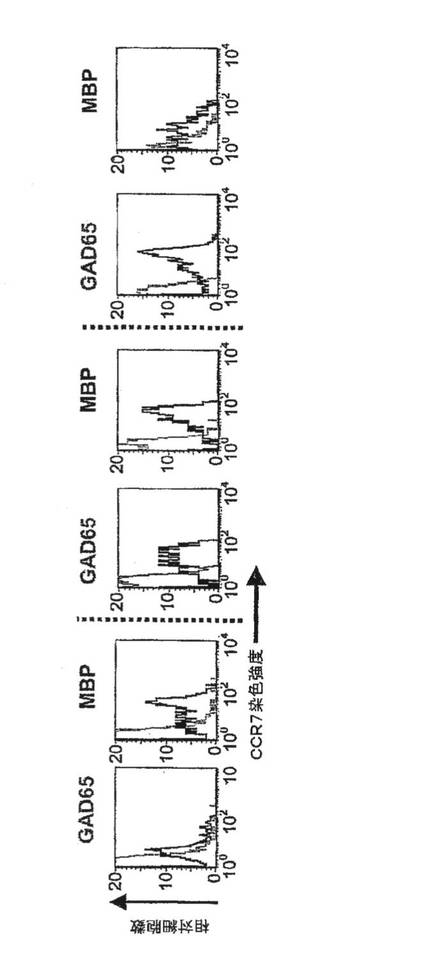
【図12D】
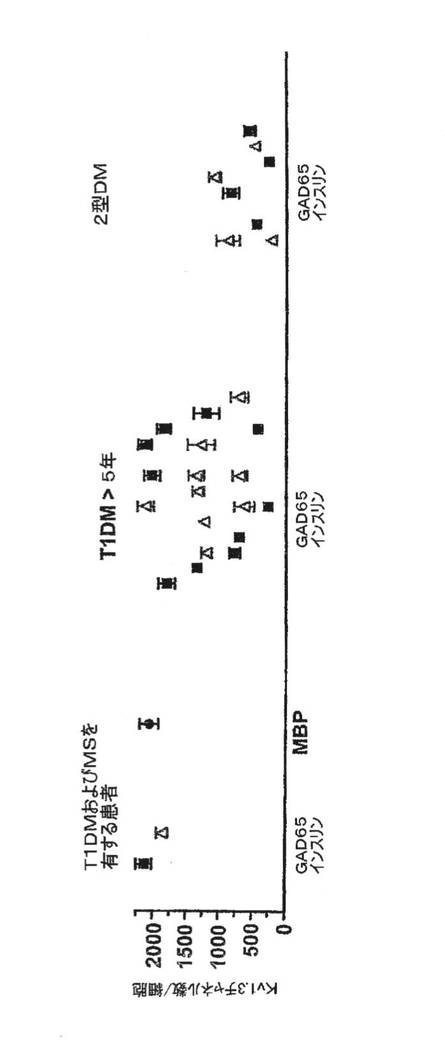
【図12E】
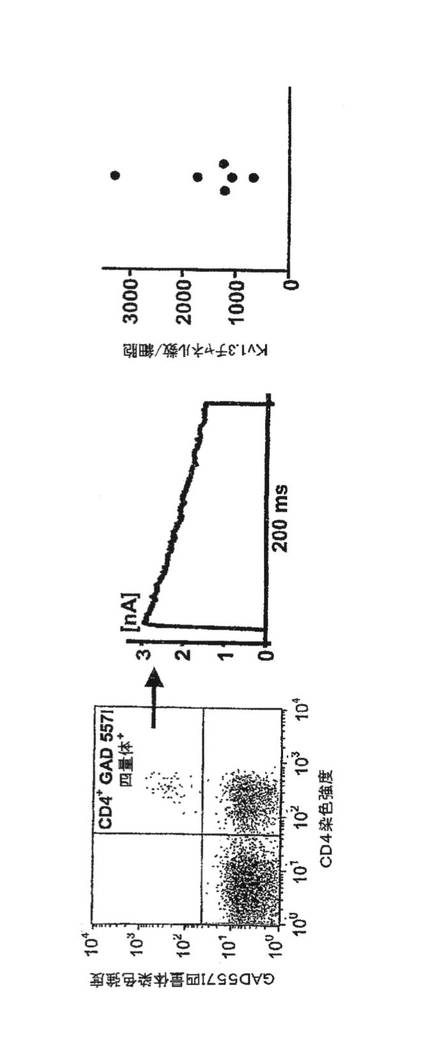
【図13A】
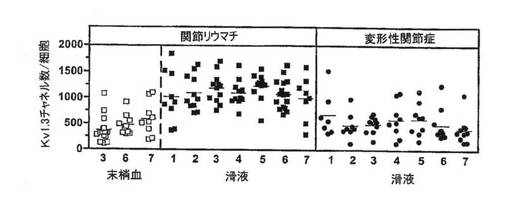
【図13B】
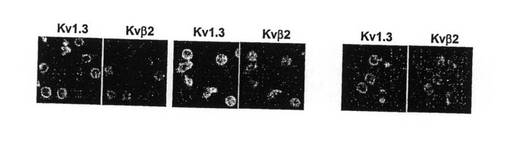
【図13C】
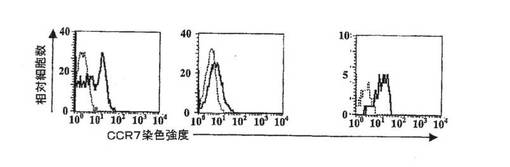
【図13D】
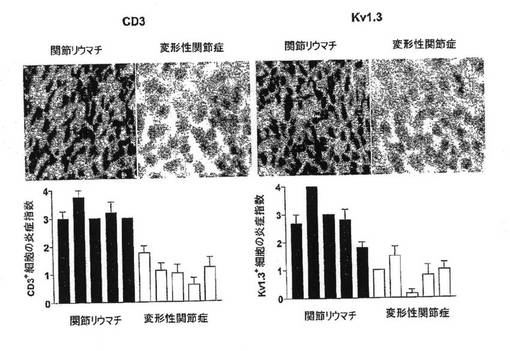
【図2A】
【図2B】
【図2C】
【図2D】
【図2E】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【図4C】
【図4D】
【図4E】
【図4F】
【図4G】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図6A】
【図6B】
【図6C】
【図7A】
【図7B】
【図7C】
【図7D】
【図7E】
【図7F】
【図7G】
【図8A】
【図8B】
【図8C】
【図8D】
【図8E】
【図9】
【図10】
【図11】
【図12A】
【図12B】
【図12C】
【図12D】
【図12E】
【図13A】
【図13B】
【図13C】
【図13D】
【公開番号】特開2012−180374(P2012−180374A)
【公開日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願番号】特願2012−134524(P2012−134524)
【出願日】平成24年6月14日(2012.6.14)
【分割の表示】特願2007−535855(P2007−535855)の分割
【原出願日】平成17年10月7日(2005.10.7)
【出願人】(500210903)ザ、リージェンツ、オブ、ザ、ユニバーシティ、オブ、カリフォルニア (31)
【氏名又は名称原語表記】THE REGENTS OF THE UNIVERSITY OF CALIFORNIA
【出願人】(507114727)バッケム バイオサイエンシズ インコーポレイテッド (2)
【氏名又は名称原語表記】BACHEM BIOSCIENCES,INC.
【Fターム(参考)】
【公開日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願日】平成24年6月14日(2012.6.14)
【分割の表示】特願2007−535855(P2007−535855)の分割
【原出願日】平成17年10月7日(2005.10.7)
【出願人】(500210903)ザ、リージェンツ、オブ、ザ、ユニバーシティ、オブ、カリフォルニア (31)
【氏名又は名称原語表記】THE REGENTS OF THE UNIVERSITY OF CALIFORNIA
【出願人】(507114727)バッケム バイオサイエンシズ インコーポレイテッド (2)
【氏名又は名称原語表記】BACHEM BIOSCIENCES,INC.
【Fターム(参考)】
[ Back to top ]