
L−スレオ−3−ヒドロキシアスパラギン酸の製造方法及び触媒組成物の組合せ
【課題】L−スレオ−3−ヒドロキシアスパラギン酸を効率的に製造する方法を開発すること。
【解決手段】本発明は、(1)L−アスパラギンをL−アスパラギン水酸化酵素に接触させてL−ヒドロキシアスパラギンを生成するステップと、(2)前記L−ヒドロキシアスパラギンをL−アスパラギナーゼに接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するステップとを含む、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法を提供する。本発明は、L−アスパラギン水酸化酵素を含む第1の触媒組成物と、L−アスパラギナーゼを含む第2の触媒組成物とを含む触媒組成物の組合せであって、L−アスパラギンを第1の触媒組成物に接触させて生成されるL−ヒドロキシアスパラギンを、第2の触媒組成物に接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するための触媒組成物の組合せを提供する。
【解決手段】本発明は、(1)L−アスパラギンをL−アスパラギン水酸化酵素に接触させてL−ヒドロキシアスパラギンを生成するステップと、(2)前記L−ヒドロキシアスパラギンをL−アスパラギナーゼに接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するステップとを含む、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法を提供する。本発明は、L−アスパラギン水酸化酵素を含む第1の触媒組成物と、L−アスパラギナーゼを含む第2の触媒組成物とを含む触媒組成物の組合せであって、L−アスパラギンを第1の触媒組成物に接触させて生成されるL−ヒドロキシアスパラギンを、第2の触媒組成物に接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するための触媒組成物の組合せを提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法と、触媒組成物の組合せとに関し、具体的には、L−アスパラギン水酸化酵素とL−アスパラギナーゼとの2段階反応を利用する、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法と、触媒組成物の組合せとに関する。
【背景技術】
【0002】
ヒドロキシアスパラギン酸は、その立体異性体がシス体又はトランス体(幾何異性体)か、アスパラギン酸がD体又はL体(光学異性体)かによって4種類の立体異性体が存在する。ヒドロキシアスパラギン酸のうち、L−スレオ−3−ヒドロキシアスパラギン酸(以下、「L−THA」という。)は、抗生物質、皮膚外用剤、グルタミン酸トランスポーターのブロッカー、機能性ゲル、樹脂などの原料として有用な物質である。
【0003】
L−THAの製造方法としては、マレイン酸又はフマル酸か、これらの塩かと、過酸化水素とをエポキシ化触媒の存在下で反応させてエポキシコハク酸を得るステップと、前記エポキシコハク酸にアンモニアを作用させることによりスレオ−3−ヒドロキシアスパラギン酸を得るステップとを含む方法が開示されている(特許文献1)。しかし、前記方法ではL−体及びD−体が等しく生成されるため、L−THAを単離するために複雑な精製処理が必要となる。糸状菌によってL−THAを発酵生産する方法も開示されているが、培養液から微量のL−THAを精製するには多段階のカラムクロマトグラフィ処理が必要となる(特許文献2及び3)。
【0004】
近年、アスパラギン水酸化酵素が遺伝子工学的に改変されて、アスパラギン酸水酸化活性が付与された改変酵素が報告された(特許文献4及び非特許文献1)。前記酵素を用いることによって、1段階反応でL−アスパラギン酸からL−THAを位置及び立体選択的に合成可能である。しかし前記改変酵素は、安定性及び活性などに問題がある。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平11−222471号公報
【特許文献2】特開平7−170992号公報
【特許文献3】特開平7−107990号公報
【特許文献4】国際公開第WO 2008/125080号公報
【非特許文献】
【0006】
【非特許文献1】Strieker、M.ら、Chembiochem.、9:374−6(2008).
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、産業上有用なL−スレオ−3−ヒドロキシアスパラギン酸を効率的に製造する方法を開発する必要がある。
【課題を解決するための手段】
【0008】
本発明はL−スレオ−3−ヒドロキシアスパラギン酸の製造方法を提供する。前記製造方法は、(1)L−アスパラギンをL−アスパラギン水酸化酵素に接触させてL−ヒドロキシアスパラギンを生成するステップと、(2)前記L−ヒドロキシアスパラギンをL−アスパラギナーゼに接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するステップとを含む。
【0009】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質の場合がある。
【0010】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0011】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0012】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、同一の細胞の表面又は内部に存在する場合がある。
【0013】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素は、(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0014】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギナーゼは、(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0015】
本発明は、L−アスパラギン水酸化酵素を含む第1の触媒組成物と、L−アスパラギナーゼを含む第2の触媒組成物とを含む触媒組成物の組合せであって、L−アスパラギンを第1の触媒組成物に接触させて生成されるL−ヒドロキシアスパラギンを、第2の触媒組成物に接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するための触媒組成物の組合せを提供する。
【0016】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質の場合がある。
【0017】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0018】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0019】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞の表面又は内部に存在する場合がある。
【0020】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素は、(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0021】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギナーゼは、(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0022】
本明細書では、ヒドロキシアスパラギン(酸)の立体異性体は、D体又はL体(光学異性体)かで区別され、L−スレオ−3−ヒドロキシアスパラギン(酸)、D−スレオ−3−ヒドロキシアスパラギン(酸)、L−エリスロ−3−ヒドロキシアスパラギン(酸)及びD−エリスロ−3−ヒドロキシアスパラギン(酸)と表される。本発明の方法で製造されるL−スレオ−3−ヒドロキシアスパラギン酸は、(2S,3S)−3−ヒドロキシアスパラギン酸とも表され、以下では「L−THA」という。本発明のヒドロキシアスパラギン(酸)の立体異性体は、以下の実施例2で説明されるHPLC法の他、X線結晶構造解析を含むが、これに限定されない、当業者に知られたいずれかの方法によって分離分析が可能である。
【0023】
本発明の製造方法と、触媒組成物の組合せとは、L−アスパラギンを反応基質としてL−スレオ−3−ヒドロキシアスパラギンを反応産物とする第1の化学反応と、L−スレオ−3−ヒドロキシアスパラギンを反応基質としてL−スレオ−3−ヒドロキシアスパラギン酸(L−THA)を反応産物とする第2の化学反応とを共役させることを特徴とする。したがって、本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、それぞれ、L−アスパラギン及びL−スレオ−3−ヒドロキシアスパラギンを反応基質とし、L−THAを反応産物とする化学反応を触媒することを条件として、いかなる生物種に由来するものであっても、いかなるアミノ酸配列を有するものであってもかまわない。前記L−アスパラギン水酸化酵素は、(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。前記L−アスパラギナーゼは、(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0024】
本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、そのアミノ酸配列をエンコードするヌクレオチド配列からなるDNAを、無生物発現系か、宿主生物及び発現ベクターを使用する発現系かで発現させることにより産生される。前記宿主生物は、大腸菌、枯草菌等のような原核生物と、酵母、菌類、植物、動物等のような真核生物とを含む。本発明の宿主生物及び発現ベクターを使用する発現系は、細胞や組織のような生物の一部か、生物の個体全体かの場合がある。本発明のL−アスパラギン水酸化酵素又はL−アスパラギナーゼは、それぞれ、L−アスパラギン水酸化酵素又はL−アスパラギナーゼの活性を有することを条件として、無生物発現系又は宿主生物及び発現ベクターを使用する発現系の他の成分が混在する状態で本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法に使用される場合がある。本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼを前記宿主生物及び発現ベクターを使用する発現系で発現させる場合には、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼを発現する宿主生物、例えば本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼの遺伝子を発現するベクターを含む形質転換体が生きた状態で本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造に用いられる場合がある。このとき、本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造は、菌体反応系や発酵法によって実行される場合がある。あるいは、前記酵素タンパク質は、精製された状態で本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法に使用されてもよい。
【0025】
本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、形質転換体で産生された後、単離精製された状態で、本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法に使用される場合がある。あるいは、本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、該酵素を発現する細胞すなわち形質転換体の、表面又は内部に存在する場合がある。前記細胞の表面及び内部とは、細菌又は植物の細胞壁、細胞の外膜及び内膜、ペリプラズムと、細胞膜の内側とを含む。遠心分離、膜濾過その他の方法で物理的に細胞を分離するときに、イオン強度や界面活性剤の有無等の条件を適切に設定すると、ある酵素が細胞と挙動をともにすることができる場合に、該酵素は前記細胞の表面又は内部に存在するといえる。
【0026】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法と、触媒組成物の組合せとにおいては、L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち少なくともL−アスパラギン水酸化酵素が前記細胞の表面又は内部に存在することが好ましい。L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が前記細胞の表面又は内部に存在する場合がある。この場合には、L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞に共発現することがあり、あるいは、L−アスパラギン水酸化酵素とL−アスパラギナーゼとが別々の細胞で発現していることがある。L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞に共発現するとき、L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が強制発現させられる場合があり、あるいは、いずれか一方のみが強制発現させられ、他方は前記細胞自体がもともと保持する遺伝子が発現する場合がある。L−アスパラギン水酸化酵素及びL−アスパラギナーゼの発現が強制されるとき、形質転換体が増殖する間はこれらの酵素遺伝子の発現は抑制されていて、形質転換体が十分に増殖してから、人工的に遺伝子発現が誘導されることが好ましい。かかる遺伝子発現の誘導及び抑制は、大腸菌ラクトースオペロンの遺伝子発現制御システムとイソプロピルチオ−β−D−チオガラクトピラノシドとを含むが、これらに限定されない、当業者に周知ないずれかの遺伝子発現制御システム及び誘導物質を用いて実現される場合がある。
【0027】
本明細書においてヌクレオチド配列の相同性は、本発明のヌクレオチド配列と、比較対象のヌクレオチド配列との間でヌクレオチド配列が一致する部分が最も多くなるように整列させて、ヌクレオチド配列が一致する部分のヌクレオチドの数を本発明のヌクレオチド配列のヌクレオチドの総数で割った商の百分率で表される。同様に、本明細書においてアミノ酸配列の相同性は、本発明のアミノ酸配列と、比較対象のアミノ酸配列との間で配列が一致するアミノ酸残基の数が最も多くなるように整列させて、配列が一致するアミノ酸残基の数の合計を本発明のアミノ酸配列のアミノ酸残基の総数で割った商の百分率で表される。本発明のヌクレオチド配列及びアミノ酸配列の相同性は、当業者に周知の配列整列プログラムCLUSTALWを使用することにより算出することができる。
【0028】
本明細書において「ストリンジェントな条件」とは、Sambrook、J.及びRussell、D.W.、Molecular Cloning A Laboratory Manual 3rd Edition,Cold Spring Harbor Laboratory Press(2001)に説明されるサザンブロット法で以下の実験条件で行うことを指す。比較対象のヌクレオチド配列からなるポリヌクレオチドをアガロース電気泳動によりバンドを形成させた上で毛管現象又は電気泳動によりニトロセルロースフィルターその他の固相に不動化する。6× SSC及び0.2% SDSからなる溶液で前洗浄する。本発明のヌクレオチド配列からなるポリヌクレオチドを放射性同位元素その他の標識物質で標識したプローブと前記固相に不動化された比較対象のポリヌクレオチドとの間のハイブリダイゼーション反応を6× SSC及び0.2% SDSからなる溶液中で65°C、終夜行う。その後前記固相を1× SSC及び0.1% SDSからなる溶液中で65°C、各30分ずつ2回洗浄し、0.2× SSC及び0.1% SDSからなる溶液中で65°C、各30分ずつ2回洗浄する。最後に前記固相に残存するプローブの量を前記標識物質の定量により決定する。本明細書において「ストリンジェントな条件」でハイブリダイゼーションをするとは、比較対象のヌクレオチド配列からなるポリヌクレオチドを不動化した固相に残存するプローブの量が、本発明のヌクレオチド配列からなるポリヌクレオチドを不動化した陽性対照実験の固相に残存するプローブの量の少なくとも25%、好ましくは少なくとも50%、より好ましくは少なくとも75%以上であることを指す。
【0029】
本発明の融合タンパク質は、特異的結合タグペプチドが本発明のアスパラギン水酸化酵素及び/又はL−アスパラギナーゼのアミノ末端又はカルボキシル末端に連結したものである。
【0030】
本発明の特異的結合タグペプチドは、本発明のL−アスパラギン水酸化酵素及び/又はL−アスパラギナーゼを調製する際に、発現したタンパク質の検出、分離又は精製をより容易に行うことを可能にするために、他のタンパク質、多糖類、糖脂質、核酸及びこれらの誘導体、樹脂等と特異的に結合するポリペプチドである。特異的結合タグと結合するリガンドは、水溶液中に溶解した遊離状態の場合も固体支持体に不動化される場合もある。そこで、本発明の融合タンパク質は固体支持体に不動化されたリガンドに特異的に結合するため、発現系の他の成分を洗浄除去することができる。その後、遊離状態のリガンドを添加したり、pH、イオン強度その他の条件を変えることにより、固体支持体から前記融合タンパク質を分離して回収することができる。本発明の特異的結合タグは、Hisタグ、mycタグ、HAタグ、インテインタグ、MBP、GSTその他これらに類するポリペプチドが含まれるがこれらに限定されない。本発明の特異的結合タグは、融合タンパク質がL−アスパラギン水酸化酵素及び/又はL−アスパラギナーゼ活性を保持することを条件として、いかなるアミノ酸配列を有してもかまわない。
【0031】
本発明のL−アスパラギン水酸化酵素及び/又はL−アスパラギナーゼの活性は、本発明の酵素と、反応基質であるL−アスパラギン及び/又はL−ヒドロキシアスパラギンと、2−オキソグルタル酸と、2価鉄イオンと、L−アスコルビン酸とを含む反応液中で前記タンパク質をL−アスパラギン又はL−ヒドロキシアスパラギンに対して作用させることにより生成される反応産物である、L−スレオ−3−ヒドロキシアスパラギン、L−スレオ−3−ヒドロキシアスパラギン酸等を定量することにより評価される場合がある。
【0032】
L−アスパラギン及びL−アスパラギン酸と、L−ヒドロキシアスパラギン及び/又はL−ヒドロキシアスパラギン酸の立体異性体とを含むが、これらに限定されない化合物は、LC/MS等のような当業者に周知の分析機器の使用により分離分析される場合がある。これらの化合物の定量分析の感度を増強するために、発色団、蛍光色素その他の増感剤との誘導体化を行った後に、前記分析機器に適用される場合がある。
【0033】
本発明の製造方法のステップ(1)において、L−アスパラギンは、適当な組成の反応混合液中でL−アスパラギン水酸化酵素に接触させられる。前記反応混合液は、前記L−アスパラギン水酸化酵素の活性を阻害しないことを条件として、いかなる組成であってもかまわないが、pH6.0ないしpH8.0、好ましくは、pH6.5ないしpH7.5の範囲に調整された、リン酸緩衝液、Hepes−NaOH緩衝液を含むが、これらに限定されない緩衝液と、適当なイオン強度の塩とを含む場合がある。また、本発明のL−アスパラギン水酸化酵素は2−オキソグルタル酸依存型酵素であるから、2−オキソグルタル酸及び2価鉄イオンが必要な場合がある。また、抗酸化剤としてアスコルビン酸等が添加される場合がある。これらの化合物の反応混合液中の濃度と、反応温度と、反応時間とは、当業者が適宜設定することができる。
【0034】
本発明の製造方法のステップ(2)において、前記ステップ(1)で生成されたL−ヒドロキシアスパラギンは、適当な組成の反応混合液中でL−アスパラギナーゼと接触させられる。前記反応混合液は、前記L−アスパラギナーゼの活性を阻害しないことを条件として、いかなる組成であってもかまわないが、pH6.0ないしpH8.0、好ましくは、pH6.5ないしpH7.5の範囲に調整された、リン酸緩衝液、Hepes−NaOH緩衝液を含むが、これらに限定されない緩衝液と、適当なイオン強度の塩とを含む場合がある。これらの化合物の反応混合液中の濃度と、反応温度と、反応時間とは、当業者が適宜設定することができる。
【0035】
本明細書において「発現ベクター」とは、所望の機能を有するタンパク質を宿主生物において発現させるために使用される、該所望の機能を有するタンパク質をエンコードするポリヌクレオチドが組み込まれたベクターである。
【0036】
本明細書において「ベクター」とは、所望の機能を有するタンパク質をエンコードするポリヌクレオチドを組み込み宿主生物へ導入することにより、該所望の機能を有するタンパク質を該宿主生物において複製及び発現させるために用いられる遺伝因子であり、プラスミド、ウイルス、ファージ、コスミド等を含むがこれらに限定されない。好ましくは前記ベクターはプラスミドの場合がある。さらに好ましくは前記ベクターは、pET−21a(+)プラスミドの場合がある。
【0037】
本発明の発現ベクターは、制限酵素、DNA連結酵素等を使用する当業者に周知の遺伝子工学手法を用いて本発明のタンパク質をエンコードするポリヌクレオチドといずれかのベクターとを連結することにより作製される場合がある。
【0038】
本明細書において「形質転換体」とは、本発明のタンパク質をエンコードするポリヌクレオチドが組み込まれた発現ベクターが導入され、所望の機能を有するタンパク質に関連する所望の形質を表すことができるようになった生物である。
【0039】
本明細書において「宿主生物」とは、形質転換体の作製において、所望の機能を有するタンパク質をエンコードするポリヌクレオチドが組み込まれた組換えベクターが導入される生物である。前記宿主生物は、大腸菌、枯草菌等のような原核生物と、酵母、菌類、植物、動物等のような真核生物とを含む。前記宿主生物は大腸菌の場合がある。
【0040】
本発明の形質転換体は、本発明の発現ベクターをいずれかの適切な宿主生物に導入することにより作製される。発現ベクターの導入は、電気刺激で細胞膜に空隙を作るエレクトロポレーション法、カルシウムイオン処理と併せて行うヒートショック法等を含む当業者に周知のさまざまな手法により実施される場合がある。
【図面の簡単な説明】
【0041】
【図1】ヒドロキシアスパラギン酸の光学異性体を単独で前記HPLC法で分析したクロマトグラムを重ね合わせた波形図。
【図2】AsnO(黒丸)と、AsnO−D241N(白丸)とについて、各反応温度での酵素活性をプロットしたグラフ。
【図3】AsnO(黒丸)と、AsnO−D241N(白丸)とについて、各温度で30分間温度処理を施された酵素の残存活性をプロットしたグラフ。
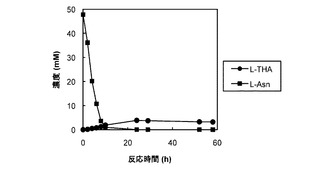
【図4】AsnO−D241Nのタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【図5】L−アスパラギン水酸化酵素及びL−アスラパギナーゼの2段階反応の反応式。
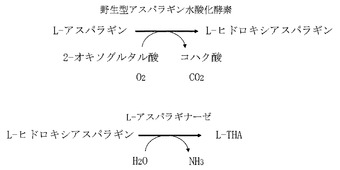
【図6A】第1のインキュベーション前に回収された反応混合液のHPLC溶出パターンのクロマトグラム。
【図6B】第1のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラム。
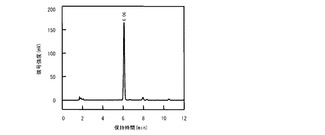
【図6C】第2のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラム。
【図7】AsnOのタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【図8】CAB92259のタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【図9】AsnOのタンパク質を発現する大腸菌の菌体が反応混合液に懸濁された場合の反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【発明を実施するための形態】
【0042】
以下に説明する本発明の実施例は例示のみを目的とし、本発明の技術的範囲を限定するものではない。本発明の技術的範囲は特許請求の範囲の記載によってのみ限定される。本発明の趣旨を逸脱しないことを条件として、本発明の変更、例えば、本発明の構成要件の追加、削除及び置換を行うことができる。本明細書において言及される全ての文献はその全体が引用により本明細書に取り込まれる。
【実施例1】
【0043】
本発明に用いられる酵素遺伝子のクローニング及び発現
放線菌ストレプトマイセス・セリカラー(Streptomyces coelicolor)A3(2)株は独立行政法人製品評価技術基盤機構のNBRC(NITE Biological Resource Center)から購入され、ISP No.2培地(イーストエクストラクト 4g/L、モルトエクストラクト 10g/L、グルコース 4g/L)を用いて、28°C、200rpmで2日間培養され、菌体が遠心により回収された。
【0044】
既知アスパラギン水酸化酵素と、本発明の新規アスパラギン水酸化酵素とをエンコードする遺伝子(以下、それぞれ「AsnO」及び「CAB92259」という。)は、前記菌体の染色体DNAを鋳型として、GC-RICH PCR システム(ロッシュ)により増幅された。AsnO及びCAB92259のヌクレオチド配列は、それぞれ、配列番号1及び2に列挙される。AsnO増幅用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号6及び7に列挙され、CAB92259増幅用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号8及び9に列挙される。反応は、始めに95°C、3分間、その後、95°C、10秒間、50°C、15秒間及び72°C、1分間を30回繰り返し、最後に72°C、7分間という条件で行われた。
【0045】
前記PCR反応の増幅産物は、順方向プライマー中のNdeI部位と、逆方向プライマー中のXhoI部位とで切断され、同じ酵素で二重消化されたpET−21a(+)ベクター(Novagen)に連結された。こうして、AsnO及びCAB92259がエンコードするタンパク質のカルボキシル末端にヒスチジンタグペプチドが連結した融合タンパク質を大腸菌で発現するプラスミドコンストラクトpETAsnO及びpECAB92259が作成された。
【0046】
大腸菌アスパラギナーゼをエンコードする遺伝子(ansA)は、大腸菌K−12株の染色体DNAを鋳型として、KOD−Plus−(東洋紡)により増幅された。ansAのヌクレオチド配列は配列番号5に列挙される。アスパラギナーゼ遺伝子増幅用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号14及び15に列挙される。反応は、始めに95°C、3分間、その後、95°C、10秒間、50°C、15秒間及び72°C、1分間を30回繰り返し、最後に72°C、7分間という条件で行われた。
【0047】
前記PCR反応の増幅産物は、順方向プライマー中のNdeI部位と、逆方向プライマー中のXhoI部位とで切断され、同じ酵素で二重消化されたpET−21a(+)ベクター(Novagen)に連結された。こうして、大腸菌アスパラギナーゼ遺伝子のカルボキシル末端にヒスチジンタグペプチドが連結した融合タンパク質を大腸菌で発現するプラスミドコンストラクトpEEcAnsAが作成された。
【0048】
アスパラギン水酸化酵素遺伝子の改変は、発現ベクターpEAsnO及びpECAB92259を鋳型とするインバースPCR法により部位特異的な突然変異導入によって行われた。耐熱性DNAポリメラーゼは、KOD−Plus−(東洋紡)が用いられた。AsnO改変用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号10及び11に列挙され、CAB92259改変用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号12及び13に列挙される。PCR反応の温度条件は、始めに94°C、2分間、その後、98°C、10秒間及び68°C、7分間を10回繰り返すという条件で行われた。AsnO及びCAB92259遺伝子と、pET−21a(+)ベクターとには、制限酵素DpnIの切断部位がそれぞれ1カ所ずつ存在するが、突然変異導入によりAsnO及びCAB92259遺伝子内のDpnIの切断部位はなくなる。そこで前記PCR反応産物は制限酵素DpnIで消化された後、DNAリガーゼ処理され、突然変異体DNAのみが自己環化された。前記DNAリガーゼ処理の反応産物は大腸菌JM109株に形質転換され、アンピシリン選択によりプラスミドが単離された。前記プラスミドの塩基配列決定により、AsnO遺伝子の241番目のアスパラギン酸残基がアスパラギン残基に置換した変異体と、CAB92259遺伝子の246番目のスパラギン酸残基がアスパラギン残基に置換した変異体とが確認された(以下、それぞれ、「AsnO−D241N」及び「CAB92259−D246N」という。AsnO−D241N及びCAB92259−D246Nのヌクレオチド配列は、それぞれ、配列番号3及び4に列挙される。また、変異体遺伝子AsnO−D241N及びCAB92259−D246Nがエンコードするタンパク質のカルボキシル末端にヒスチジンタグペプチドが連結した融合タンパク質を大腸菌で発現するプラスミドコンストラクトは、それぞれ、pETAsnO−D241N及びpECAB92259−D246Nという。
【0049】
発現コンストラクトpEAsnO、pECAB92259、pEAsnO−D241N及びpECAB92259−D246Nは、大腸菌Rosetta2(DE3)に形質転換された。形質転換体の単離コロニーは、50μg/mL アンピシリン及び34μg/mL クロラムフェニコールが添加されたLB液体培地(以下、「LB−AC液体培地」という。)5mLに接種され、37°C、150rpmで16時間振盪培養された。その後、前記液体培地1mLは新鮮なLB−AC液体培地100mLに接種され、37°C、120rpmでO.D.660が0.5に達するまで振盪培養された。前記培地に終濃度0.1mMのイソプロピル−β−d−チオガラクトピラノシド(IPTG)が添加され遺伝子発現が誘導され、25°C、120rpmで6時間振盪培養された。その後、遠心分離(4°C、5000×g、10分間)によって回収された遺伝子発現誘導菌体は、5mLの酵素反応液に懸濁して菌体反応系として利用されるか、発現タンパク質の精製に供された。
【0050】
AsnO、CAB92259、AsnO−D241N及びCAB92259−D246Nの遺伝子産物のカルボキシル末端にHisタグが連結された融合タンパク質(以下、「タンパク質」という。)の精製は常法に従って行われた。すなわち、前記遺伝子発現誘導菌体は、20mM リン酸カリウム緩衝液(pH6.5)5mLに懸濁され、超音波破砕装置(トミー、UD−200、出力:200W、発振周波数:20kHz)で3分間超音波破砕された。遠心分離(4°C、20000×g、30分間)によって残渣が除去された後の上清が無細胞抽出液として回収された。前記無細胞抽出液はNi2+アフィニティーカラム(His−trap、GEヘルスケア)に適用された。その後、溶出バッファー(20mM リン酸カリウム緩衝液(pH6.5)及び500mM イミダゾール)を用いて溶出された。溶出された分画は、脱塩カラムPD−10(GE Healthcare)を用いて脱塩処理が施された後、分注された(以下、「精製標品」という。)。
【実施例2】
【0051】
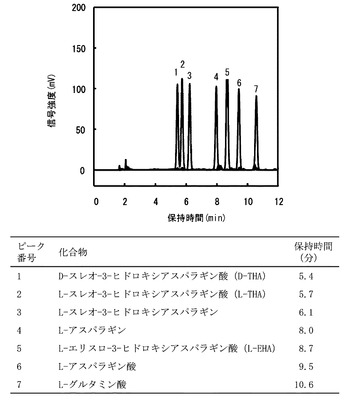
ヒドロキシアスパラギン酸の光学異性体の同定
方法
L−アスパラギン酸、L−アスパラギン、L−スレオ−3−ヒドロキシアスパラギン酸(L−THA)、L−エリスロ−3−ヒドロキシアスパラギン酸(L−EHA)、D−スレオ−3−ヒドロキシアスパラギン酸(D−THA)、L−スレオ−3−ヒドロキシアスパラギン及びL−グルタミン酸の0.5mM溶液100μL、0.5M ホウ酸−NaOH緩衝液(pH9.0)50μL、10mM Nα−(5−フルオロ−2,4−ジニトロフェニル)−L−アラニンアミド100μLとを混合させ、40°C、1時間反応させた後、1N HClを50μL添加して反応を停止させて、それぞれのアミノ酸の誘導体が得られた。前記アミノ酸誘導体のそれぞれは、COSMOSIL 5C18−AR−II Packed Column (内径:4.6mm、長さ:150mm)(ナカライテスク)が装着されたHPLC装置(L−7000系、日立)により分析された。カラムオーブンは40°Cに設定され、340nmにおける吸光度によって検出された。溶離液A(45mM リン酸緩衝液(pH2.7)、5% メタノール及び5% アセトニトリル)と、溶離液B(30mM リン酸緩衝液(pH2.7)、5% メタノール及び35% アセトニトリル)の直線的グラジェントにより化合物が分離された。グラジェントプログラムはA/B:70/30(0分)からA/B:20/80(12分)に設定された。
【0052】
結果
図1は、ヒドロキシアスパラギン酸の光学異性体を単独で前記HPLC法で分析したクロマトグラムを重ね合わせた波形図である。図1の波形図のピーク1はD−THA(保持時間5.4分)、ピーク2はL−THA(保持時間5.7分)、ピーク3はL−スレオ−3−ヒドロキシアスパラギン(保持時間6.1分)、ピーク4はL−アスパラギン(保持時間8.0分)、ピーク5はL−EHA(保持時間8.7分)、ピーク6はL−アスパラギン酸(保持時間9.5分)、ピーク7はL−グルタミン酸(保持時間10.6分)であった。以上の結果から、前記HPLC法によってヒドロキシアスパラギン酸の光学異性体を同定することが可能となった。また、前記ヒドロキシアスパラギン酸の光学異性体の濃度と、HPLCのクロマトグラムの信号強度との較正曲線を作成することにより、前記ヒドロキシアスパラギン酸の光学異性体を定量測定することも可能となった(データは示されない)。
【実施例3】
【0053】
アスパラギン水酸化活性の評価
方法
アスパラギン水酸化反応混合液として、50mM リン酸カリウム緩衝液(pH6.5)中に、5mM L−アスパラギンと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.05mg/mLのAsnO又はCAB92259タンパク質精製標品とを含む反応混合液が調製された。対照実験では、前記アスパラギン水酸化反応混合液から、L−アスパラギンか、L−アスコルビン酸か、FeSO4か、前記タンパク質精製標品かのいずれかが欠けた反応混合液が用いられた。前記アスパラギン水酸化反応混合液又は対照実験の反応混合液は25°Cで30分間インキュベーションされた。その後、生成されたヒドロキシアスパラギンの量が実施例2で説明されたHPLC法によって分析された。
【0054】
結果
前記HPLC法による分析の結果、CAB92259タンパク質精製標品及びAsnOタンパク質精製標品の生成するヒドロキシアスパラギンは同一の保持時間であった。そこで、CAB92259タンパク質も、AsnOタンパク質と同じく、L−アスパラギンを基質としてL−スレオ−3−ヒドロキシアスパラギンを生成する水酸化反応を触媒する活性を有することが明らかになった。表1は、前記アスパラギン水酸化反応混合液又は対照実験の反応混合液が、実施例2のHPLC法によって分析された結果をまとめた表である。
【0055】
【表1】

【0056】
表1から明らかなとおり、CAB92259タンパク質精製標品は、25°Cで30分間の反応で、5mMのL−アスパラギンから4.3mMのL−スレオ−3−ヒドロキシアスパラギンを生成することができた。また、CAB92259タンパク質によるL−アスパラギンの水酸化反応は、2−オキソグルタル酸、L−アスコルビン酸及び2価鉄イオンに依存するので、CAB92259遺伝子産物は2−オキソグルタル酸依存型ジオキシゲナーゼ活性を有することが示された。
【実施例4】
【0057】
アスパラギン水酸化酵素の改変
方法
Strieker、M.ら(ChemBioChem、9:374−376(2008))は、アスパラギン水酸化活性を有するAsnO遺伝子の241番目のアスパラギン酸残基がアスパラギン残基に置換されるように部位特異的突然変異を導入することによって、アスパラギン酸を水酸化できるように改変した。そこで、CAB92259遺伝子の246番目のスパラギン酸残基がアスパラギン残基に置換されるように部位特異的突然変異が導入された。
【0058】
アスパラギン酸水酸化反応反応混合液として、50mM リン酸カリウム緩衝液(pH6.5)中に、5mM L−アスパラギン酸と、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.05mg/mLのAsnO−D241N又はCAB92259−D246Nタンパク質精製標品とを含む反応混合液が調製された。対照実験では、前記アスパラギン酸水酸化反応混合液から、L−アスパラギン酸か、L−アスコルビン酸か、FeSO4か、前記タンパク質精製標品かのいずれかが欠けた反応混合液が用いられた。前記アスパラギン酸水酸化反応混合液又は対照実験の反応混合液は25°Cで30分間インキュベーションされた。その後、生成されたヒドロキシアスパラギン酸の量が実施例2で説明されたHPLC法によって分析された。
【0059】
結果
前記HPLC法による分析の結果、CAB92259−D246タンパク質精製標品の生成するヒドロキシアスパラギン酸はL−THAと同一の保持時間であり、他の異性体は含まれなかった。そこで、CAB92259−D246N遺伝子産物も、AsnO−D241遺伝子産物と同じく、L−アスパラギン酸を基質としてL−スレオ−3−ヒドロキシアスパラギン酸を生成する水酸化反応を触媒する活性を有することが明らかになった。また、対照実験の結果、CAB92259−D246Nタンパク質によるL−アスパラギン酸の水酸化反応は、2−オキソグルタル酸、L−アスコルビン酸及び2価鉄イオンに依存するので、CAB92259−D246N遺伝子産物も2−オキソグルタル酸依存型ジオキシゲナーゼ活性を有することが示された。しかし、CAB92259−D246Nタンパク質精製標品は、25°Cで30分間の反応で、5mMのL−アスパラギン酸から0.07mMのL−スレオ−3−ヒドロキシアスパラギンしか生成することができなかった。これは、AsnO−D241Nタンパク質と比較して低い活性であった。
【0060】
ここで、AsnO及びCAB92259のアスパラギン水酸化酵素と、AsnO−D241N及びCAB92259−D246Nのアスパラギン酸水酸化酵素とについて、水酸化反応による基質の転換率が比較された。水酸化反応混合液として、50mM リン酸カリウム緩衝液(pH6.5)中に、5mM L−アスパラギン又はL−アスパラギン酸と、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.05mg/mLのAsnO、AsnO−D241N、CAB92259、又はCAB92259−D246Nタンパク質とを含む反応混合液が調製された。前記水酸化反応混合液は25°Cで30分間インキュベーションされた。その後、生成されたヒドロキシアスパラギン又はヒドロキシアスパラギン酸の量が実施例2で説明されたHPLC法によって分析された(表2)。
【0061】
【表2】
【0062】
表2は、それぞれの酵素について、基質の反応開始時のモル濃度に対する、前記水酸化反応混合液中で25°C、30分間のインキュベーションで生成した反応産物のモル濃度の百分率(転換率)を比較した表である。表から明らかなとおり、AsnO−D241N及びCAB92259−D246Nのアスパラギン酸水酸化酵素は、改変のもととなったAsnO及びCAB92259のアスパラギン水酸化酵素と比べて非常に転換率が低かった。
【0063】
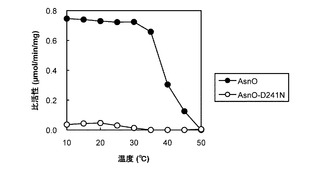
野生型酵素と改変酵素との最適反応温度の比較
表2の結果から、比較的転換率が高かったAsnO(以下、「野生型酵素」という。)と、AsnO−D241N(以下、「改変酵素」という。)との最適反応温度が比較検討された。酵素反応混合液として、50mM HEPES−NaOH緩衝液(pH7.5)中に、前記タンパク質精製標品0.1mg/mLと、5mM L−アスパラギン(AsnO又はCAB92259の場合)か、L−アスパラギン酸(AsnO−D241N又はCAB92259−D246Nの場合)かと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とを含む反応混合液が調製された。反応は、10°Cないし50°Cの温度で1時間行われた。前記反応混合液中に生成された水酸化アミノ酸が実施例2で説明されたHPLC法によって分析され、野生型酵素とその改変酵素とで比較された。
【0064】
図2は、AsnO(黒丸(●))と、AsnO−D241N(白丸(○))とについて、各反応温度での酵素活性をプロットしたグラフである。図2から明らかなとおり、野生型酵素は10°Cから45°Cまでの温度範囲ではほぼ同じ活性を示したが、改変酵素は35°C以上では活性がなくなった。また、野生型酵素に比べて改変酵素は酵素活性が著しく低く、改変酵素で活性が高い10°Cないし30°Cでの酵素活性でも、野生型酵素で活性が急激に低下する50°Cでの酵素活性と同程度であった。
【0065】
改変酵素の温度安定性
野生型酵素と、改変酵素とについて酵素の温度安定性が比較検討された。AsnO、AsnO−D241N、CAB92259又はCAB92259−D246Nのタンパク質精製標品は、10°Cないし50°Cの温度で30分間インキュベーションされる温度処理の後、30°C1時間の酵素反応に供された。酵素反応混合液として、50mM HEPES−NaOH緩衝液(pH7.5)中に、前記温度処理が施されたタンパク質精製標品0.1mg/mLと、5mM L−アスパラギン(AsnO又はCAB92259の場合)か、L−アスパラギン酸(AsnO−D241N又はCAB92259−D246Nの場合)かと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とを含む反応混合液が調製された。前記反応混合液中に生成された水酸化アミノ酸が実施例2で説明されたHPLC法によって分析され、それぞれの野生型酵素とその改変酵素とで比較された。
【0066】
図3は、AsnO(黒丸(●))と、AsnO−D241N(白丸(○))とについて、各温度で30分間温度処理を施された酵素の残存活性をプロットしたグラフである。図3から明らかなとおり、野生型酵素は10°Cから35°Cまでの温度範囲ではほぼ安定であったが、改変酵素は10°Cでも活性がほとんどなかった。
【0067】
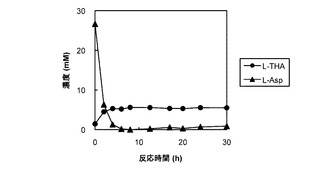
菌体反応系を用いる1段階反応の検討
発現コンストラクトpEAsnO−D241Nが形質転換された大腸菌Rosetta2(DE3)株が、実施例1で説明される手順で振盪培養された。すなわち、終夜培養1mLが100mLの新鮮なLB液体培地に接種され、37°C、120rpmでO.D.660が0.5に達するまで振盪培養された。前記培地に終濃度0.1mMのイソプロピル−β−d−チオガラクトピラノシド(IPTG)が添加され遺伝子発現が誘導され、25°C、120rpmで6時間振盪培養された。その後、遠心分離(4°C、5000×g、10分間)によって回収された遺伝子発現誘導菌体は、5mLの酵素反応液に懸濁された。前記反応混合液は、50mM リン酸緩衝液(pH6.5)中に、5mM L−アスパラギンと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とが含まれた。前記反応混合液は25°C、280rpmで振盪しながらインキュベーションされた。前記反応混合液の一部が経時的にサンプリングされ、実施例2に説明されるHPLC法で分析された。
【0068】
図4は、AsnO−D241Nのタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフである。図4では、反応基質のL−アスパラギン酸の濃度の経時変化が黒い三角(▲)のグラフで表され、反応産物のL−THAの濃度の経時変化が黒い丸(●)のグラフで表された。反応基質のL−アスパラギン酸の濃度は反応開始から8時間後にはほぼ0となり、反応産物のL−THAの濃度は反応開始から6時間後にはほぼプラトーに達し、反応開始から30時間後でも5.5mMであった。なお、反応の副産物として、L−グルタミン酸が反応開始から30時間後に7.2mMも蓄積していた(データ示さず)。したがって、改変酵素による1段階反応は、菌体反応系では副産物の生成のため、効率に限界があった。
【実施例5】
【0069】
L−アスパラギン水酸化酵素及びL−アスラパギナーゼの2段階反応
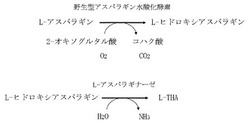
実施例4に詳しく説明されたとおり、L−アスパラギン水酸化酵素の改変により1段階でL−アスパラギン酸からL−THAを生成させるアプローチでは、酵素の活性自体が非常に低く、室温でも酵素の安定性が低く、菌体反応系では反応副産物が大量に生成するため、工業的なL−THAの生産には問題が多い。そこで、L−アスパラギン水酸化酵素をL−アスラパギナーゼと共役させる2段階反応を検討した。図5は、L−アスパラギン水酸化酵素及びL−アスラパギナーゼの2段階反応の反応式である。第1段階の反応は、AsnO及びCAB92259のようなL−アスパラギン水酸化酵素によって触媒される。第2段階の反応は、AnsAのようなL−アスパラギナーゼによって触媒される。L−アスパラギナーゼが、L−アスパラギンだけでなく、L−ヒドロキシアスパラギンも基質として加水分解反応を触媒することができることは本願で明らかにされる新知見である。
【0070】
実施例1で説明されたL−アスパラギン水酸化酵素(AsnO及びCAB92250)と、L−アスパラギナーゼ(AnsA)とのタンパク質精製標品を用いて、L−アスパラギンからL−THAを生成させた。まず、アスパラギン水酸化反応反応混合液として、20mM リン酸緩衝液(pH6.5)と、5mM L−アスパラギンと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.1mg/mLのAsnOタンパク質精製標品とを含む反応混合液が調製された。前記反応混合液が、25°C、16時間の第1のインキュベーションに供された。その後、前記反応混合液にL−アスパラギナーゼ(AnsA)のタンパク質精製標品0.005mg/mLが添加され、37°C、1時間の第2のインキュベーションに供された。第1のインキュベーションの前と、第1のインキュベーションの後と、第2のインキュベーションの後とに、反応混合液の一部が回収され、実施例2で説明されたHPLC法により、反応基質及び反応産物が分析された。
【0071】
図6Aは第1のインキュベーション前に回収された反応混合液のHPLC溶出パターンのクロマトグラムであり、図6Bは第1のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラムであり、図6Cは第2のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラムである。図1と対比すると明かなとおり、第1のインキュベーションの前ではL−アスパラギンが反応混合液に存在していたことが図6Aからわかる。第1のインキュベーションの後ではL−アスパラギンは完全に消失して、かわりにL−スレオ−3−ヒドロキシアスパラギンが生成したことが図6Bからわかる。さらに第2のインキュベーションの後ではL−スレオ−3−ヒドロキシアスパラギンが完全に消失して、かわりにL−THAが生成したことが図6Cからわかる。これらの結果から、精製酵素を触媒とする場合には、L−アスパラギン水酸化酵素をL−アスラパギナーゼと共役させる2段階反応系によって、効率的にL−アスパラギンからL−THAを生成できることが示された。
【0072】
発現コンストラクトpEAsnO又はpECAB92259が形質転換された大腸菌Rosetta2(DE3)株が実施例1で説明される手順で振盪培養された。すなわち、終夜培養1mLが100mLの新鮮なLB液体培地に接種され、37°C、120rpmでO.D.660が0.5に達するまで振盪培養された。前記培地に終濃度0.1mMのイソプロピル−β−d−チオガラクトピラノシド(IPTG)が添加され遺伝子発現が誘導され、25°C、120rpmで6時間振盪培養された。その後、遠心分離(4°C、5000×g、10分間)によって回収された遺伝子発現誘導菌体は、5mLの酵素反応液に懸濁された。前記反応混合液は、50mM リン酸緩衝液(pH6.5)中に、45mM L−アスパラギンと、90mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とが含まれた。前記反応混合液は25°C、280rpmで振盪しながらインキュベーションされた。前記反応混合液の一部が経時的にサンプリングされ、実施例2に説明されるHPLC法で分析された。
【0073】
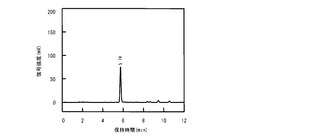
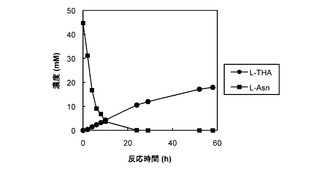
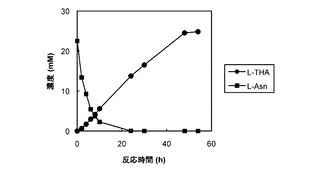
図7及び8は、それぞれ、AsnO及びCAB92259のタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフである。図7及び8では、反応基質のL−アスパラギンの濃度の経時変化が黒い四角(■)のグラフで表され、反応産物のL−THAの濃度の経時変化が黒い丸(●)のグラフで表された。反応基質のL−アスパラギンの濃度は反応開始から24時間後にはほぼ0となったが、反応産物のL−THAの濃度は反応開始から58時間後でもまだ少しずつ上昇中であった。なお、反応の副産物の蓄積はほとんどみられなかった(データ示さず)。AsnOのタンパク質を発現する大腸菌では、反応開始から58時間後の反応産物L−THAの濃度は18mM、CAB92259のタンパク質を発現する大腸菌では、反応開始から58時間後の反応産物L−THAの濃度は3.2mMであった。
【0074】
図9は、AsnOのタンパク質を発現する大腸菌の菌体が反応混合液(L−アスパラギン濃度を25mM、2−オキソグルタル酸濃度を50mMとした他は、図7及び8の実験と同じ組成)に懸濁された場合の反応基質及び反応産物の濃度の経時変化をプロットしたグラフである。図9では、反応基質のL−アスパラギンの濃度の経時変化が黒い四角(■)のグラフで表され、反応産物のL−THAの濃度の経時変化が黒い丸(●)のグラフで表された。反応基質のL−アスパラギンの濃度は反応開始から24時間後にはほぼ0となったが、反応産物のL−THAの濃度は反応開始から54時間後で25mMに達した。なお、この実験では、反応混合液に添加された反応基質のL−アスパラギンの濃度は25mMであったので、反応産物のモル収率はほぼ100%であった。また、反応の副産物の蓄積はほとんどみられなかった(データ示さず)。
【0075】
糸状菌によるL−THAの発酵生産では、ジャーファーメンターによって18Lの培養液を48時間培養して、純度82%の粉末が17.4gしか得られなかった。改変酵素による1段階反応を菌体反応系で行う場合では、50mMのL−アスパラギン酸から5.5mMのL−THAが得られたが、副生物として7.2mMものL−グルタミン酸が蓄積した。これに対し、本発明の2段階反応では、最適化を行うと、100%のモル収率を達成することができた。したがって、本発明のL−THAの製造方法は産業上の利用性が非常に高い。
【技術分野】
【0001】
本発明は、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法と、触媒組成物の組合せとに関し、具体的には、L−アスパラギン水酸化酵素とL−アスパラギナーゼとの2段階反応を利用する、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法と、触媒組成物の組合せとに関する。
【背景技術】
【0002】
ヒドロキシアスパラギン酸は、その立体異性体がシス体又はトランス体(幾何異性体)か、アスパラギン酸がD体又はL体(光学異性体)かによって4種類の立体異性体が存在する。ヒドロキシアスパラギン酸のうち、L−スレオ−3−ヒドロキシアスパラギン酸(以下、「L−THA」という。)は、抗生物質、皮膚外用剤、グルタミン酸トランスポーターのブロッカー、機能性ゲル、樹脂などの原料として有用な物質である。
【0003】
L−THAの製造方法としては、マレイン酸又はフマル酸か、これらの塩かと、過酸化水素とをエポキシ化触媒の存在下で反応させてエポキシコハク酸を得るステップと、前記エポキシコハク酸にアンモニアを作用させることによりスレオ−3−ヒドロキシアスパラギン酸を得るステップとを含む方法が開示されている(特許文献1)。しかし、前記方法ではL−体及びD−体が等しく生成されるため、L−THAを単離するために複雑な精製処理が必要となる。糸状菌によってL−THAを発酵生産する方法も開示されているが、培養液から微量のL−THAを精製するには多段階のカラムクロマトグラフィ処理が必要となる(特許文献2及び3)。
【0004】
近年、アスパラギン水酸化酵素が遺伝子工学的に改変されて、アスパラギン酸水酸化活性が付与された改変酵素が報告された(特許文献4及び非特許文献1)。前記酵素を用いることによって、1段階反応でL−アスパラギン酸からL−THAを位置及び立体選択的に合成可能である。しかし前記改変酵素は、安定性及び活性などに問題がある。
【先行技術文献】
【特許文献】
【0005】
【特許文献1】特開平11−222471号公報
【特許文献2】特開平7−170992号公報
【特許文献3】特開平7−107990号公報
【特許文献4】国際公開第WO 2008/125080号公報
【非特許文献】
【0006】
【非特許文献1】Strieker、M.ら、Chembiochem.、9:374−6(2008).
【発明の開示】
【発明が解決しようとする課題】
【0007】
そこで、産業上有用なL−スレオ−3−ヒドロキシアスパラギン酸を効率的に製造する方法を開発する必要がある。
【課題を解決するための手段】
【0008】
本発明はL−スレオ−3−ヒドロキシアスパラギン酸の製造方法を提供する。前記製造方法は、(1)L−アスパラギンをL−アスパラギン水酸化酵素に接触させてL−ヒドロキシアスパラギンを生成するステップと、(2)前記L−ヒドロキシアスパラギンをL−アスパラギナーゼに接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するステップとを含む。
【0009】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質の場合がある。
【0010】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0011】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0012】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、同一の細胞の表面又は内部に存在する場合がある。
【0013】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギン水酸化酵素は、(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0014】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法において、前記L−アスパラギナーゼは、(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0015】
本発明は、L−アスパラギン水酸化酵素を含む第1の触媒組成物と、L−アスパラギナーゼを含む第2の触媒組成物とを含む触媒組成物の組合せであって、L−アスパラギンを第1の触媒組成物に接触させて生成されるL−ヒドロキシアスパラギンを、第2の触媒組成物に接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するための触媒組成物の組合せを提供する。
【0016】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質の場合がある。
【0017】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0018】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在する場合がある。
【0019】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞の表面又は内部に存在する場合がある。
【0020】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギン水酸化酵素は、(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0021】
本発明の触媒組成物の組合せにおいて、前記L−アスパラギナーゼは、(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上、85%以上、90%以上又は95%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0022】
本明細書では、ヒドロキシアスパラギン(酸)の立体異性体は、D体又はL体(光学異性体)かで区別され、L−スレオ−3−ヒドロキシアスパラギン(酸)、D−スレオ−3−ヒドロキシアスパラギン(酸)、L−エリスロ−3−ヒドロキシアスパラギン(酸)及びD−エリスロ−3−ヒドロキシアスパラギン(酸)と表される。本発明の方法で製造されるL−スレオ−3−ヒドロキシアスパラギン酸は、(2S,3S)−3−ヒドロキシアスパラギン酸とも表され、以下では「L−THA」という。本発明のヒドロキシアスパラギン(酸)の立体異性体は、以下の実施例2で説明されるHPLC法の他、X線結晶構造解析を含むが、これに限定されない、当業者に知られたいずれかの方法によって分離分析が可能である。
【0023】
本発明の製造方法と、触媒組成物の組合せとは、L−アスパラギンを反応基質としてL−スレオ−3−ヒドロキシアスパラギンを反応産物とする第1の化学反応と、L−スレオ−3−ヒドロキシアスパラギンを反応基質としてL−スレオ−3−ヒドロキシアスパラギン酸(L−THA)を反応産物とする第2の化学反応とを共役させることを特徴とする。したがって、本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、それぞれ、L−アスパラギン及びL−スレオ−3−ヒドロキシアスパラギンを反応基質とし、L−THAを反応産物とする化学反応を触媒することを条件として、いかなる生物種に由来するものであっても、いかなるアミノ酸配列を有するものであってもかまわない。前記L−アスパラギン水酸化酵素は、(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。前記L−アスパラギナーゼは、(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含む場合がある。
【0024】
本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、そのアミノ酸配列をエンコードするヌクレオチド配列からなるDNAを、無生物発現系か、宿主生物及び発現ベクターを使用する発現系かで発現させることにより産生される。前記宿主生物は、大腸菌、枯草菌等のような原核生物と、酵母、菌類、植物、動物等のような真核生物とを含む。本発明の宿主生物及び発現ベクターを使用する発現系は、細胞や組織のような生物の一部か、生物の個体全体かの場合がある。本発明のL−アスパラギン水酸化酵素又はL−アスパラギナーゼは、それぞれ、L−アスパラギン水酸化酵素又はL−アスパラギナーゼの活性を有することを条件として、無生物発現系又は宿主生物及び発現ベクターを使用する発現系の他の成分が混在する状態で本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法に使用される場合がある。本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼを前記宿主生物及び発現ベクターを使用する発現系で発現させる場合には、前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼを発現する宿主生物、例えば本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼの遺伝子を発現するベクターを含む形質転換体が生きた状態で本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造に用いられる場合がある。このとき、本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造は、菌体反応系や発酵法によって実行される場合がある。あるいは、前記酵素タンパク質は、精製された状態で本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法に使用されてもよい。
【0025】
本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、形質転換体で産生された後、単離精製された状態で、本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法に使用される場合がある。あるいは、本発明のL−アスパラギン水酸化酵素及びL−アスパラギナーゼは、該酵素を発現する細胞すなわち形質転換体の、表面又は内部に存在する場合がある。前記細胞の表面及び内部とは、細菌又は植物の細胞壁、細胞の外膜及び内膜、ペリプラズムと、細胞膜の内側とを含む。遠心分離、膜濾過その他の方法で物理的に細胞を分離するときに、イオン強度や界面活性剤の有無等の条件を適切に設定すると、ある酵素が細胞と挙動をともにすることができる場合に、該酵素は前記細胞の表面又は内部に存在するといえる。
【0026】
本発明のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法と、触媒組成物の組合せとにおいては、L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち少なくともL−アスパラギン水酸化酵素が前記細胞の表面又は内部に存在することが好ましい。L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が前記細胞の表面又は内部に存在する場合がある。この場合には、L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞に共発現することがあり、あるいは、L−アスパラギン水酸化酵素とL−アスパラギナーゼとが別々の細胞で発現していることがある。L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞に共発現するとき、L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が強制発現させられる場合があり、あるいは、いずれか一方のみが強制発現させられ、他方は前記細胞自体がもともと保持する遺伝子が発現する場合がある。L−アスパラギン水酸化酵素及びL−アスパラギナーゼの発現が強制されるとき、形質転換体が増殖する間はこれらの酵素遺伝子の発現は抑制されていて、形質転換体が十分に増殖してから、人工的に遺伝子発現が誘導されることが好ましい。かかる遺伝子発現の誘導及び抑制は、大腸菌ラクトースオペロンの遺伝子発現制御システムとイソプロピルチオ−β−D−チオガラクトピラノシドとを含むが、これらに限定されない、当業者に周知ないずれかの遺伝子発現制御システム及び誘導物質を用いて実現される場合がある。
【0027】
本明細書においてヌクレオチド配列の相同性は、本発明のヌクレオチド配列と、比較対象のヌクレオチド配列との間でヌクレオチド配列が一致する部分が最も多くなるように整列させて、ヌクレオチド配列が一致する部分のヌクレオチドの数を本発明のヌクレオチド配列のヌクレオチドの総数で割った商の百分率で表される。同様に、本明細書においてアミノ酸配列の相同性は、本発明のアミノ酸配列と、比較対象のアミノ酸配列との間で配列が一致するアミノ酸残基の数が最も多くなるように整列させて、配列が一致するアミノ酸残基の数の合計を本発明のアミノ酸配列のアミノ酸残基の総数で割った商の百分率で表される。本発明のヌクレオチド配列及びアミノ酸配列の相同性は、当業者に周知の配列整列プログラムCLUSTALWを使用することにより算出することができる。
【0028】
本明細書において「ストリンジェントな条件」とは、Sambrook、J.及びRussell、D.W.、Molecular Cloning A Laboratory Manual 3rd Edition,Cold Spring Harbor Laboratory Press(2001)に説明されるサザンブロット法で以下の実験条件で行うことを指す。比較対象のヌクレオチド配列からなるポリヌクレオチドをアガロース電気泳動によりバンドを形成させた上で毛管現象又は電気泳動によりニトロセルロースフィルターその他の固相に不動化する。6× SSC及び0.2% SDSからなる溶液で前洗浄する。本発明のヌクレオチド配列からなるポリヌクレオチドを放射性同位元素その他の標識物質で標識したプローブと前記固相に不動化された比較対象のポリヌクレオチドとの間のハイブリダイゼーション反応を6× SSC及び0.2% SDSからなる溶液中で65°C、終夜行う。その後前記固相を1× SSC及び0.1% SDSからなる溶液中で65°C、各30分ずつ2回洗浄し、0.2× SSC及び0.1% SDSからなる溶液中で65°C、各30分ずつ2回洗浄する。最後に前記固相に残存するプローブの量を前記標識物質の定量により決定する。本明細書において「ストリンジェントな条件」でハイブリダイゼーションをするとは、比較対象のヌクレオチド配列からなるポリヌクレオチドを不動化した固相に残存するプローブの量が、本発明のヌクレオチド配列からなるポリヌクレオチドを不動化した陽性対照実験の固相に残存するプローブの量の少なくとも25%、好ましくは少なくとも50%、より好ましくは少なくとも75%以上であることを指す。
【0029】
本発明の融合タンパク質は、特異的結合タグペプチドが本発明のアスパラギン水酸化酵素及び/又はL−アスパラギナーゼのアミノ末端又はカルボキシル末端に連結したものである。
【0030】
本発明の特異的結合タグペプチドは、本発明のL−アスパラギン水酸化酵素及び/又はL−アスパラギナーゼを調製する際に、発現したタンパク質の検出、分離又は精製をより容易に行うことを可能にするために、他のタンパク質、多糖類、糖脂質、核酸及びこれらの誘導体、樹脂等と特異的に結合するポリペプチドである。特異的結合タグと結合するリガンドは、水溶液中に溶解した遊離状態の場合も固体支持体に不動化される場合もある。そこで、本発明の融合タンパク質は固体支持体に不動化されたリガンドに特異的に結合するため、発現系の他の成分を洗浄除去することができる。その後、遊離状態のリガンドを添加したり、pH、イオン強度その他の条件を変えることにより、固体支持体から前記融合タンパク質を分離して回収することができる。本発明の特異的結合タグは、Hisタグ、mycタグ、HAタグ、インテインタグ、MBP、GSTその他これらに類するポリペプチドが含まれるがこれらに限定されない。本発明の特異的結合タグは、融合タンパク質がL−アスパラギン水酸化酵素及び/又はL−アスパラギナーゼ活性を保持することを条件として、いかなるアミノ酸配列を有してもかまわない。
【0031】
本発明のL−アスパラギン水酸化酵素及び/又はL−アスパラギナーゼの活性は、本発明の酵素と、反応基質であるL−アスパラギン及び/又はL−ヒドロキシアスパラギンと、2−オキソグルタル酸と、2価鉄イオンと、L−アスコルビン酸とを含む反応液中で前記タンパク質をL−アスパラギン又はL−ヒドロキシアスパラギンに対して作用させることにより生成される反応産物である、L−スレオ−3−ヒドロキシアスパラギン、L−スレオ−3−ヒドロキシアスパラギン酸等を定量することにより評価される場合がある。
【0032】
L−アスパラギン及びL−アスパラギン酸と、L−ヒドロキシアスパラギン及び/又はL−ヒドロキシアスパラギン酸の立体異性体とを含むが、これらに限定されない化合物は、LC/MS等のような当業者に周知の分析機器の使用により分離分析される場合がある。これらの化合物の定量分析の感度を増強するために、発色団、蛍光色素その他の増感剤との誘導体化を行った後に、前記分析機器に適用される場合がある。
【0033】
本発明の製造方法のステップ(1)において、L−アスパラギンは、適当な組成の反応混合液中でL−アスパラギン水酸化酵素に接触させられる。前記反応混合液は、前記L−アスパラギン水酸化酵素の活性を阻害しないことを条件として、いかなる組成であってもかまわないが、pH6.0ないしpH8.0、好ましくは、pH6.5ないしpH7.5の範囲に調整された、リン酸緩衝液、Hepes−NaOH緩衝液を含むが、これらに限定されない緩衝液と、適当なイオン強度の塩とを含む場合がある。また、本発明のL−アスパラギン水酸化酵素は2−オキソグルタル酸依存型酵素であるから、2−オキソグルタル酸及び2価鉄イオンが必要な場合がある。また、抗酸化剤としてアスコルビン酸等が添加される場合がある。これらの化合物の反応混合液中の濃度と、反応温度と、反応時間とは、当業者が適宜設定することができる。
【0034】
本発明の製造方法のステップ(2)において、前記ステップ(1)で生成されたL−ヒドロキシアスパラギンは、適当な組成の反応混合液中でL−アスパラギナーゼと接触させられる。前記反応混合液は、前記L−アスパラギナーゼの活性を阻害しないことを条件として、いかなる組成であってもかまわないが、pH6.0ないしpH8.0、好ましくは、pH6.5ないしpH7.5の範囲に調整された、リン酸緩衝液、Hepes−NaOH緩衝液を含むが、これらに限定されない緩衝液と、適当なイオン強度の塩とを含む場合がある。これらの化合物の反応混合液中の濃度と、反応温度と、反応時間とは、当業者が適宜設定することができる。
【0035】
本明細書において「発現ベクター」とは、所望の機能を有するタンパク質を宿主生物において発現させるために使用される、該所望の機能を有するタンパク質をエンコードするポリヌクレオチドが組み込まれたベクターである。
【0036】
本明細書において「ベクター」とは、所望の機能を有するタンパク質をエンコードするポリヌクレオチドを組み込み宿主生物へ導入することにより、該所望の機能を有するタンパク質を該宿主生物において複製及び発現させるために用いられる遺伝因子であり、プラスミド、ウイルス、ファージ、コスミド等を含むがこれらに限定されない。好ましくは前記ベクターはプラスミドの場合がある。さらに好ましくは前記ベクターは、pET−21a(+)プラスミドの場合がある。
【0037】
本発明の発現ベクターは、制限酵素、DNA連結酵素等を使用する当業者に周知の遺伝子工学手法を用いて本発明のタンパク質をエンコードするポリヌクレオチドといずれかのベクターとを連結することにより作製される場合がある。
【0038】
本明細書において「形質転換体」とは、本発明のタンパク質をエンコードするポリヌクレオチドが組み込まれた発現ベクターが導入され、所望の機能を有するタンパク質に関連する所望の形質を表すことができるようになった生物である。
【0039】
本明細書において「宿主生物」とは、形質転換体の作製において、所望の機能を有するタンパク質をエンコードするポリヌクレオチドが組み込まれた組換えベクターが導入される生物である。前記宿主生物は、大腸菌、枯草菌等のような原核生物と、酵母、菌類、植物、動物等のような真核生物とを含む。前記宿主生物は大腸菌の場合がある。
【0040】
本発明の形質転換体は、本発明の発現ベクターをいずれかの適切な宿主生物に導入することにより作製される。発現ベクターの導入は、電気刺激で細胞膜に空隙を作るエレクトロポレーション法、カルシウムイオン処理と併せて行うヒートショック法等を含む当業者に周知のさまざまな手法により実施される場合がある。
【図面の簡単な説明】
【0041】
【図1】ヒドロキシアスパラギン酸の光学異性体を単独で前記HPLC法で分析したクロマトグラムを重ね合わせた波形図。
【図2】AsnO(黒丸)と、AsnO−D241N(白丸)とについて、各反応温度での酵素活性をプロットしたグラフ。
【図3】AsnO(黒丸)と、AsnO−D241N(白丸)とについて、各温度で30分間温度処理を施された酵素の残存活性をプロットしたグラフ。
【図4】AsnO−D241Nのタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【図5】L−アスパラギン水酸化酵素及びL−アスラパギナーゼの2段階反応の反応式。
【図6A】第1のインキュベーション前に回収された反応混合液のHPLC溶出パターンのクロマトグラム。
【図6B】第1のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラム。
【図6C】第2のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラム。
【図7】AsnOのタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【図8】CAB92259のタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【図9】AsnOのタンパク質を発現する大腸菌の菌体が反応混合液に懸濁された場合の反応基質及び反応産物の濃度の経時変化をプロットしたグラフ。
【発明を実施するための形態】
【0042】
以下に説明する本発明の実施例は例示のみを目的とし、本発明の技術的範囲を限定するものではない。本発明の技術的範囲は特許請求の範囲の記載によってのみ限定される。本発明の趣旨を逸脱しないことを条件として、本発明の変更、例えば、本発明の構成要件の追加、削除及び置換を行うことができる。本明細書において言及される全ての文献はその全体が引用により本明細書に取り込まれる。
【実施例1】
【0043】
本発明に用いられる酵素遺伝子のクローニング及び発現
放線菌ストレプトマイセス・セリカラー(Streptomyces coelicolor)A3(2)株は独立行政法人製品評価技術基盤機構のNBRC(NITE Biological Resource Center)から購入され、ISP No.2培地(イーストエクストラクト 4g/L、モルトエクストラクト 10g/L、グルコース 4g/L)を用いて、28°C、200rpmで2日間培養され、菌体が遠心により回収された。
【0044】
既知アスパラギン水酸化酵素と、本発明の新規アスパラギン水酸化酵素とをエンコードする遺伝子(以下、それぞれ「AsnO」及び「CAB92259」という。)は、前記菌体の染色体DNAを鋳型として、GC-RICH PCR システム(ロッシュ)により増幅された。AsnO及びCAB92259のヌクレオチド配列は、それぞれ、配列番号1及び2に列挙される。AsnO増幅用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号6及び7に列挙され、CAB92259増幅用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号8及び9に列挙される。反応は、始めに95°C、3分間、その後、95°C、10秒間、50°C、15秒間及び72°C、1分間を30回繰り返し、最後に72°C、7分間という条件で行われた。
【0045】
前記PCR反応の増幅産物は、順方向プライマー中のNdeI部位と、逆方向プライマー中のXhoI部位とで切断され、同じ酵素で二重消化されたpET−21a(+)ベクター(Novagen)に連結された。こうして、AsnO及びCAB92259がエンコードするタンパク質のカルボキシル末端にヒスチジンタグペプチドが連結した融合タンパク質を大腸菌で発現するプラスミドコンストラクトpETAsnO及びpECAB92259が作成された。
【0046】
大腸菌アスパラギナーゼをエンコードする遺伝子(ansA)は、大腸菌K−12株の染色体DNAを鋳型として、KOD−Plus−(東洋紡)により増幅された。ansAのヌクレオチド配列は配列番号5に列挙される。アスパラギナーゼ遺伝子増幅用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号14及び15に列挙される。反応は、始めに95°C、3分間、その後、95°C、10秒間、50°C、15秒間及び72°C、1分間を30回繰り返し、最後に72°C、7分間という条件で行われた。
【0047】
前記PCR反応の増幅産物は、順方向プライマー中のNdeI部位と、逆方向プライマー中のXhoI部位とで切断され、同じ酵素で二重消化されたpET−21a(+)ベクター(Novagen)に連結された。こうして、大腸菌アスパラギナーゼ遺伝子のカルボキシル末端にヒスチジンタグペプチドが連結した融合タンパク質を大腸菌で発現するプラスミドコンストラクトpEEcAnsAが作成された。
【0048】
アスパラギン水酸化酵素遺伝子の改変は、発現ベクターpEAsnO及びpECAB92259を鋳型とするインバースPCR法により部位特異的な突然変異導入によって行われた。耐熱性DNAポリメラーゼは、KOD−Plus−(東洋紡)が用いられた。AsnO改変用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号10及び11に列挙され、CAB92259改変用の順方向及び逆方向プライマーの配列は、それぞれ、配列番号12及び13に列挙される。PCR反応の温度条件は、始めに94°C、2分間、その後、98°C、10秒間及び68°C、7分間を10回繰り返すという条件で行われた。AsnO及びCAB92259遺伝子と、pET−21a(+)ベクターとには、制限酵素DpnIの切断部位がそれぞれ1カ所ずつ存在するが、突然変異導入によりAsnO及びCAB92259遺伝子内のDpnIの切断部位はなくなる。そこで前記PCR反応産物は制限酵素DpnIで消化された後、DNAリガーゼ処理され、突然変異体DNAのみが自己環化された。前記DNAリガーゼ処理の反応産物は大腸菌JM109株に形質転換され、アンピシリン選択によりプラスミドが単離された。前記プラスミドの塩基配列決定により、AsnO遺伝子の241番目のアスパラギン酸残基がアスパラギン残基に置換した変異体と、CAB92259遺伝子の246番目のスパラギン酸残基がアスパラギン残基に置換した変異体とが確認された(以下、それぞれ、「AsnO−D241N」及び「CAB92259−D246N」という。AsnO−D241N及びCAB92259−D246Nのヌクレオチド配列は、それぞれ、配列番号3及び4に列挙される。また、変異体遺伝子AsnO−D241N及びCAB92259−D246Nがエンコードするタンパク質のカルボキシル末端にヒスチジンタグペプチドが連結した融合タンパク質を大腸菌で発現するプラスミドコンストラクトは、それぞれ、pETAsnO−D241N及びpECAB92259−D246Nという。
【0049】
発現コンストラクトpEAsnO、pECAB92259、pEAsnO−D241N及びpECAB92259−D246Nは、大腸菌Rosetta2(DE3)に形質転換された。形質転換体の単離コロニーは、50μg/mL アンピシリン及び34μg/mL クロラムフェニコールが添加されたLB液体培地(以下、「LB−AC液体培地」という。)5mLに接種され、37°C、150rpmで16時間振盪培養された。その後、前記液体培地1mLは新鮮なLB−AC液体培地100mLに接種され、37°C、120rpmでO.D.660が0.5に達するまで振盪培養された。前記培地に終濃度0.1mMのイソプロピル−β−d−チオガラクトピラノシド(IPTG)が添加され遺伝子発現が誘導され、25°C、120rpmで6時間振盪培養された。その後、遠心分離(4°C、5000×g、10分間)によって回収された遺伝子発現誘導菌体は、5mLの酵素反応液に懸濁して菌体反応系として利用されるか、発現タンパク質の精製に供された。
【0050】
AsnO、CAB92259、AsnO−D241N及びCAB92259−D246Nの遺伝子産物のカルボキシル末端にHisタグが連結された融合タンパク質(以下、「タンパク質」という。)の精製は常法に従って行われた。すなわち、前記遺伝子発現誘導菌体は、20mM リン酸カリウム緩衝液(pH6.5)5mLに懸濁され、超音波破砕装置(トミー、UD−200、出力:200W、発振周波数:20kHz)で3分間超音波破砕された。遠心分離(4°C、20000×g、30分間)によって残渣が除去された後の上清が無細胞抽出液として回収された。前記無細胞抽出液はNi2+アフィニティーカラム(His−trap、GEヘルスケア)に適用された。その後、溶出バッファー(20mM リン酸カリウム緩衝液(pH6.5)及び500mM イミダゾール)を用いて溶出された。溶出された分画は、脱塩カラムPD−10(GE Healthcare)を用いて脱塩処理が施された後、分注された(以下、「精製標品」という。)。
【実施例2】
【0051】
ヒドロキシアスパラギン酸の光学異性体の同定
方法
L−アスパラギン酸、L−アスパラギン、L−スレオ−3−ヒドロキシアスパラギン酸(L−THA)、L−エリスロ−3−ヒドロキシアスパラギン酸(L−EHA)、D−スレオ−3−ヒドロキシアスパラギン酸(D−THA)、L−スレオ−3−ヒドロキシアスパラギン及びL−グルタミン酸の0.5mM溶液100μL、0.5M ホウ酸−NaOH緩衝液(pH9.0)50μL、10mM Nα−(5−フルオロ−2,4−ジニトロフェニル)−L−アラニンアミド100μLとを混合させ、40°C、1時間反応させた後、1N HClを50μL添加して反応を停止させて、それぞれのアミノ酸の誘導体が得られた。前記アミノ酸誘導体のそれぞれは、COSMOSIL 5C18−AR−II Packed Column (内径:4.6mm、長さ:150mm)(ナカライテスク)が装着されたHPLC装置(L−7000系、日立)により分析された。カラムオーブンは40°Cに設定され、340nmにおける吸光度によって検出された。溶離液A(45mM リン酸緩衝液(pH2.7)、5% メタノール及び5% アセトニトリル)と、溶離液B(30mM リン酸緩衝液(pH2.7)、5% メタノール及び35% アセトニトリル)の直線的グラジェントにより化合物が分離された。グラジェントプログラムはA/B:70/30(0分)からA/B:20/80(12分)に設定された。
【0052】
結果
図1は、ヒドロキシアスパラギン酸の光学異性体を単独で前記HPLC法で分析したクロマトグラムを重ね合わせた波形図である。図1の波形図のピーク1はD−THA(保持時間5.4分)、ピーク2はL−THA(保持時間5.7分)、ピーク3はL−スレオ−3−ヒドロキシアスパラギン(保持時間6.1分)、ピーク4はL−アスパラギン(保持時間8.0分)、ピーク5はL−EHA(保持時間8.7分)、ピーク6はL−アスパラギン酸(保持時間9.5分)、ピーク7はL−グルタミン酸(保持時間10.6分)であった。以上の結果から、前記HPLC法によってヒドロキシアスパラギン酸の光学異性体を同定することが可能となった。また、前記ヒドロキシアスパラギン酸の光学異性体の濃度と、HPLCのクロマトグラムの信号強度との較正曲線を作成することにより、前記ヒドロキシアスパラギン酸の光学異性体を定量測定することも可能となった(データは示されない)。
【実施例3】
【0053】
アスパラギン水酸化活性の評価
方法
アスパラギン水酸化反応混合液として、50mM リン酸カリウム緩衝液(pH6.5)中に、5mM L−アスパラギンと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.05mg/mLのAsnO又はCAB92259タンパク質精製標品とを含む反応混合液が調製された。対照実験では、前記アスパラギン水酸化反応混合液から、L−アスパラギンか、L−アスコルビン酸か、FeSO4か、前記タンパク質精製標品かのいずれかが欠けた反応混合液が用いられた。前記アスパラギン水酸化反応混合液又は対照実験の反応混合液は25°Cで30分間インキュベーションされた。その後、生成されたヒドロキシアスパラギンの量が実施例2で説明されたHPLC法によって分析された。
【0054】
結果
前記HPLC法による分析の結果、CAB92259タンパク質精製標品及びAsnOタンパク質精製標品の生成するヒドロキシアスパラギンは同一の保持時間であった。そこで、CAB92259タンパク質も、AsnOタンパク質と同じく、L−アスパラギンを基質としてL−スレオ−3−ヒドロキシアスパラギンを生成する水酸化反応を触媒する活性を有することが明らかになった。表1は、前記アスパラギン水酸化反応混合液又は対照実験の反応混合液が、実施例2のHPLC法によって分析された結果をまとめた表である。
【0055】
【表1】
【0056】
表1から明らかなとおり、CAB92259タンパク質精製標品は、25°Cで30分間の反応で、5mMのL−アスパラギンから4.3mMのL−スレオ−3−ヒドロキシアスパラギンを生成することができた。また、CAB92259タンパク質によるL−アスパラギンの水酸化反応は、2−オキソグルタル酸、L−アスコルビン酸及び2価鉄イオンに依存するので、CAB92259遺伝子産物は2−オキソグルタル酸依存型ジオキシゲナーゼ活性を有することが示された。
【実施例4】
【0057】
アスパラギン水酸化酵素の改変
方法
Strieker、M.ら(ChemBioChem、9:374−376(2008))は、アスパラギン水酸化活性を有するAsnO遺伝子の241番目のアスパラギン酸残基がアスパラギン残基に置換されるように部位特異的突然変異を導入することによって、アスパラギン酸を水酸化できるように改変した。そこで、CAB92259遺伝子の246番目のスパラギン酸残基がアスパラギン残基に置換されるように部位特異的突然変異が導入された。
【0058】
アスパラギン酸水酸化反応反応混合液として、50mM リン酸カリウム緩衝液(pH6.5)中に、5mM L−アスパラギン酸と、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.05mg/mLのAsnO−D241N又はCAB92259−D246Nタンパク質精製標品とを含む反応混合液が調製された。対照実験では、前記アスパラギン酸水酸化反応混合液から、L−アスパラギン酸か、L−アスコルビン酸か、FeSO4か、前記タンパク質精製標品かのいずれかが欠けた反応混合液が用いられた。前記アスパラギン酸水酸化反応混合液又は対照実験の反応混合液は25°Cで30分間インキュベーションされた。その後、生成されたヒドロキシアスパラギン酸の量が実施例2で説明されたHPLC法によって分析された。
【0059】
結果
前記HPLC法による分析の結果、CAB92259−D246タンパク質精製標品の生成するヒドロキシアスパラギン酸はL−THAと同一の保持時間であり、他の異性体は含まれなかった。そこで、CAB92259−D246N遺伝子産物も、AsnO−D241遺伝子産物と同じく、L−アスパラギン酸を基質としてL−スレオ−3−ヒドロキシアスパラギン酸を生成する水酸化反応を触媒する活性を有することが明らかになった。また、対照実験の結果、CAB92259−D246Nタンパク質によるL−アスパラギン酸の水酸化反応は、2−オキソグルタル酸、L−アスコルビン酸及び2価鉄イオンに依存するので、CAB92259−D246N遺伝子産物も2−オキソグルタル酸依存型ジオキシゲナーゼ活性を有することが示された。しかし、CAB92259−D246Nタンパク質精製標品は、25°Cで30分間の反応で、5mMのL−アスパラギン酸から0.07mMのL−スレオ−3−ヒドロキシアスパラギンしか生成することができなかった。これは、AsnO−D241Nタンパク質と比較して低い活性であった。
【0060】
ここで、AsnO及びCAB92259のアスパラギン水酸化酵素と、AsnO−D241N及びCAB92259−D246Nのアスパラギン酸水酸化酵素とについて、水酸化反応による基質の転換率が比較された。水酸化反応混合液として、50mM リン酸カリウム緩衝液(pH6.5)中に、5mM L−アスパラギン又はL−アスパラギン酸と、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.05mg/mLのAsnO、AsnO−D241N、CAB92259、又はCAB92259−D246Nタンパク質とを含む反応混合液が調製された。前記水酸化反応混合液は25°Cで30分間インキュベーションされた。その後、生成されたヒドロキシアスパラギン又はヒドロキシアスパラギン酸の量が実施例2で説明されたHPLC法によって分析された(表2)。
【0061】
【表2】
【0062】
表2は、それぞれの酵素について、基質の反応開始時のモル濃度に対する、前記水酸化反応混合液中で25°C、30分間のインキュベーションで生成した反応産物のモル濃度の百分率(転換率)を比較した表である。表から明らかなとおり、AsnO−D241N及びCAB92259−D246Nのアスパラギン酸水酸化酵素は、改変のもととなったAsnO及びCAB92259のアスパラギン水酸化酵素と比べて非常に転換率が低かった。
【0063】
野生型酵素と改変酵素との最適反応温度の比較
表2の結果から、比較的転換率が高かったAsnO(以下、「野生型酵素」という。)と、AsnO−D241N(以下、「改変酵素」という。)との最適反応温度が比較検討された。酵素反応混合液として、50mM HEPES−NaOH緩衝液(pH7.5)中に、前記タンパク質精製標品0.1mg/mLと、5mM L−アスパラギン(AsnO又はCAB92259の場合)か、L−アスパラギン酸(AsnO−D241N又はCAB92259−D246Nの場合)かと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とを含む反応混合液が調製された。反応は、10°Cないし50°Cの温度で1時間行われた。前記反応混合液中に生成された水酸化アミノ酸が実施例2で説明されたHPLC法によって分析され、野生型酵素とその改変酵素とで比較された。
【0064】
図2は、AsnO(黒丸(●))と、AsnO−D241N(白丸(○))とについて、各反応温度での酵素活性をプロットしたグラフである。図2から明らかなとおり、野生型酵素は10°Cから45°Cまでの温度範囲ではほぼ同じ活性を示したが、改変酵素は35°C以上では活性がなくなった。また、野生型酵素に比べて改変酵素は酵素活性が著しく低く、改変酵素で活性が高い10°Cないし30°Cでの酵素活性でも、野生型酵素で活性が急激に低下する50°Cでの酵素活性と同程度であった。
【0065】
改変酵素の温度安定性
野生型酵素と、改変酵素とについて酵素の温度安定性が比較検討された。AsnO、AsnO−D241N、CAB92259又はCAB92259−D246Nのタンパク質精製標品は、10°Cないし50°Cの温度で30分間インキュベーションされる温度処理の後、30°C1時間の酵素反応に供された。酵素反応混合液として、50mM HEPES−NaOH緩衝液(pH7.5)中に、前記温度処理が施されたタンパク質精製標品0.1mg/mLと、5mM L−アスパラギン(AsnO又はCAB92259の場合)か、L−アスパラギン酸(AsnO−D241N又はCAB92259−D246Nの場合)かと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とを含む反応混合液が調製された。前記反応混合液中に生成された水酸化アミノ酸が実施例2で説明されたHPLC法によって分析され、それぞれの野生型酵素とその改変酵素とで比較された。
【0066】
図3は、AsnO(黒丸(●))と、AsnO−D241N(白丸(○))とについて、各温度で30分間温度処理を施された酵素の残存活性をプロットしたグラフである。図3から明らかなとおり、野生型酵素は10°Cから35°Cまでの温度範囲ではほぼ安定であったが、改変酵素は10°Cでも活性がほとんどなかった。
【0067】
菌体反応系を用いる1段階反応の検討
発現コンストラクトpEAsnO−D241Nが形質転換された大腸菌Rosetta2(DE3)株が、実施例1で説明される手順で振盪培養された。すなわち、終夜培養1mLが100mLの新鮮なLB液体培地に接種され、37°C、120rpmでO.D.660が0.5に達するまで振盪培養された。前記培地に終濃度0.1mMのイソプロピル−β−d−チオガラクトピラノシド(IPTG)が添加され遺伝子発現が誘導され、25°C、120rpmで6時間振盪培養された。その後、遠心分離(4°C、5000×g、10分間)によって回収された遺伝子発現誘導菌体は、5mLの酵素反応液に懸濁された。前記反応混合液は、50mM リン酸緩衝液(pH6.5)中に、5mM L−アスパラギンと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とが含まれた。前記反応混合液は25°C、280rpmで振盪しながらインキュベーションされた。前記反応混合液の一部が経時的にサンプリングされ、実施例2に説明されるHPLC法で分析された。
【0068】
図4は、AsnO−D241Nのタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフである。図4では、反応基質のL−アスパラギン酸の濃度の経時変化が黒い三角(▲)のグラフで表され、反応産物のL−THAの濃度の経時変化が黒い丸(●)のグラフで表された。反応基質のL−アスパラギン酸の濃度は反応開始から8時間後にはほぼ0となり、反応産物のL−THAの濃度は反応開始から6時間後にはほぼプラトーに達し、反応開始から30時間後でも5.5mMであった。なお、反応の副産物として、L−グルタミン酸が反応開始から30時間後に7.2mMも蓄積していた(データ示さず)。したがって、改変酵素による1段階反応は、菌体反応系では副産物の生成のため、効率に限界があった。
【実施例5】
【0069】
L−アスパラギン水酸化酵素及びL−アスラパギナーゼの2段階反応
実施例4に詳しく説明されたとおり、L−アスパラギン水酸化酵素の改変により1段階でL−アスパラギン酸からL−THAを生成させるアプローチでは、酵素の活性自体が非常に低く、室温でも酵素の安定性が低く、菌体反応系では反応副産物が大量に生成するため、工業的なL−THAの生産には問題が多い。そこで、L−アスパラギン水酸化酵素をL−アスラパギナーゼと共役させる2段階反応を検討した。図5は、L−アスパラギン水酸化酵素及びL−アスラパギナーゼの2段階反応の反応式である。第1段階の反応は、AsnO及びCAB92259のようなL−アスパラギン水酸化酵素によって触媒される。第2段階の反応は、AnsAのようなL−アスパラギナーゼによって触媒される。L−アスパラギナーゼが、L−アスパラギンだけでなく、L−ヒドロキシアスパラギンも基質として加水分解反応を触媒することができることは本願で明らかにされる新知見である。
【0070】
実施例1で説明されたL−アスパラギン水酸化酵素(AsnO及びCAB92250)と、L−アスパラギナーゼ(AnsA)とのタンパク質精製標品を用いて、L−アスパラギンからL−THAを生成させた。まず、アスパラギン水酸化反応反応混合液として、20mM リン酸緩衝液(pH6.5)と、5mM L−アスパラギンと、10mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4と、0.1mg/mLのAsnOタンパク質精製標品とを含む反応混合液が調製された。前記反応混合液が、25°C、16時間の第1のインキュベーションに供された。その後、前記反応混合液にL−アスパラギナーゼ(AnsA)のタンパク質精製標品0.005mg/mLが添加され、37°C、1時間の第2のインキュベーションに供された。第1のインキュベーションの前と、第1のインキュベーションの後と、第2のインキュベーションの後とに、反応混合液の一部が回収され、実施例2で説明されたHPLC法により、反応基質及び反応産物が分析された。
【0071】
図6Aは第1のインキュベーション前に回収された反応混合液のHPLC溶出パターンのクロマトグラムであり、図6Bは第1のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラムであり、図6Cは第2のインキュベーション後に回収された反応混合液のHPLC溶出パターンのクロマトグラムである。図1と対比すると明かなとおり、第1のインキュベーションの前ではL−アスパラギンが反応混合液に存在していたことが図6Aからわかる。第1のインキュベーションの後ではL−アスパラギンは完全に消失して、かわりにL−スレオ−3−ヒドロキシアスパラギンが生成したことが図6Bからわかる。さらに第2のインキュベーションの後ではL−スレオ−3−ヒドロキシアスパラギンが完全に消失して、かわりにL−THAが生成したことが図6Cからわかる。これらの結果から、精製酵素を触媒とする場合には、L−アスパラギン水酸化酵素をL−アスラパギナーゼと共役させる2段階反応系によって、効率的にL−アスパラギンからL−THAを生成できることが示された。
【0072】
発現コンストラクトpEAsnO又はpECAB92259が形質転換された大腸菌Rosetta2(DE3)株が実施例1で説明される手順で振盪培養された。すなわち、終夜培養1mLが100mLの新鮮なLB液体培地に接種され、37°C、120rpmでO.D.660が0.5に達するまで振盪培養された。前記培地に終濃度0.1mMのイソプロピル−β−d−チオガラクトピラノシド(IPTG)が添加され遺伝子発現が誘導され、25°C、120rpmで6時間振盪培養された。その後、遠心分離(4°C、5000×g、10分間)によって回収された遺伝子発現誘導菌体は、5mLの酵素反応液に懸濁された。前記反応混合液は、50mM リン酸緩衝液(pH6.5)中に、45mM L−アスパラギンと、90mM 2−オキソグルタル酸と、1mM L−アスコルビン酸と、0.5mM FeSO4とが含まれた。前記反応混合液は25°C、280rpmで振盪しながらインキュベーションされた。前記反応混合液の一部が経時的にサンプリングされ、実施例2に説明されるHPLC法で分析された。
【0073】
図7及び8は、それぞれ、AsnO及びCAB92259のタンパク質を発現する大腸菌の菌体が懸濁された反応混合液での反応基質及び反応産物の濃度の経時変化をプロットしたグラフである。図7及び8では、反応基質のL−アスパラギンの濃度の経時変化が黒い四角(■)のグラフで表され、反応産物のL−THAの濃度の経時変化が黒い丸(●)のグラフで表された。反応基質のL−アスパラギンの濃度は反応開始から24時間後にはほぼ0となったが、反応産物のL−THAの濃度は反応開始から58時間後でもまだ少しずつ上昇中であった。なお、反応の副産物の蓄積はほとんどみられなかった(データ示さず)。AsnOのタンパク質を発現する大腸菌では、反応開始から58時間後の反応産物L−THAの濃度は18mM、CAB92259のタンパク質を発現する大腸菌では、反応開始から58時間後の反応産物L−THAの濃度は3.2mMであった。
【0074】
図9は、AsnOのタンパク質を発現する大腸菌の菌体が反応混合液(L−アスパラギン濃度を25mM、2−オキソグルタル酸濃度を50mMとした他は、図7及び8の実験と同じ組成)に懸濁された場合の反応基質及び反応産物の濃度の経時変化をプロットしたグラフである。図9では、反応基質のL−アスパラギンの濃度の経時変化が黒い四角(■)のグラフで表され、反応産物のL−THAの濃度の経時変化が黒い丸(●)のグラフで表された。反応基質のL−アスパラギンの濃度は反応開始から24時間後にはほぼ0となったが、反応産物のL−THAの濃度は反応開始から54時間後で25mMに達した。なお、この実験では、反応混合液に添加された反応基質のL−アスパラギンの濃度は25mMであったので、反応産物のモル収率はほぼ100%であった。また、反応の副産物の蓄積はほとんどみられなかった(データ示さず)。
【0075】
糸状菌によるL−THAの発酵生産では、ジャーファーメンターによって18Lの培養液を48時間培養して、純度82%の粉末が17.4gしか得られなかった。改変酵素による1段階反応を菌体反応系で行う場合では、50mMのL−アスパラギン酸から5.5mMのL−THAが得られたが、副生物として7.2mMものL−グルタミン酸が蓄積した。これに対し、本発明の2段階反応では、最適化を行うと、100%のモル収率を達成することができた。したがって、本発明のL−THAの製造方法は産業上の利用性が非常に高い。
【特許請求の範囲】
【請求項1】
(1)L−アスパラギンをL−アスパラギン水酸化酵素に接触させてL−ヒドロキシアスパラギンを生成するステップと、
(2)前記L−ヒドロキシアスパラギンをL−アスパラギナーゼに接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するステップとを含むことを特徴とする、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項2】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質であることを特徴とする、請求項1に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項3】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項1に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項4】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項3に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項5】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞の表面又は内部に存在することを特徴とする、請求項4に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項6】
前記L−アスパラギン水酸化酵素は、
(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、
(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項1ないし5のいずれか1つに記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項7】
前記L−アスパラギナーゼは、
(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項1ないし6のいずれか1つに記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項8】
L−アスパラギン水酸化酵素を含む第1の触媒組成物と、L−アスパラギナーゼを含む第2の触媒組成物とを含む触媒組成物の組合せであって、
L−アスパラギンを第1の触媒組成物に接触させて生成されるL−ヒドロキシアスパラギンを、第2の触媒組成物に接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するための触媒組成物の組合せ。
【請求項9】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質であることを特徴とする、請求項8に記載の触媒組成物の組合せ。
【請求項10】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項8に記載の触媒組成物の組合せ。
【請求項11】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項10に記載の触媒組成物の組合せ。
【請求項12】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞の表面又は内部に存在することを特徴とする、請求項11に記載の触媒組成物の組合せ。
【請求項13】
前記L−アスパラギン水酸化酵素は、
(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、
(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項8ないし12のいずれか1つに記載の触媒組成物の組合せ。
【請求項14】
前記L−アスパラギナーゼは、
(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項8ないし13のいずれか1つに記載の触媒組成物の組合せ。
【請求項1】
(1)L−アスパラギンをL−アスパラギン水酸化酵素に接触させてL−ヒドロキシアスパラギンを生成するステップと、
(2)前記L−ヒドロキシアスパラギンをL−アスパラギナーゼに接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するステップとを含むことを特徴とする、L−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項2】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質であることを特徴とする、請求項1に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項3】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項1に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項4】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項3に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項5】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞の表面又は内部に存在することを特徴とする、請求項4に記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項6】
前記L−アスパラギン水酸化酵素は、
(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、
(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項1ないし5のいずれか1つに記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項7】
前記L−アスパラギナーゼは、
(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項1ないし6のいずれか1つに記載のL−スレオ−3−ヒドロキシアスパラギン酸の製造方法。
【請求項8】
L−アスパラギン水酸化酵素を含む第1の触媒組成物と、L−アスパラギナーゼを含む第2の触媒組成物とを含む触媒組成物の組合せであって、
L−アスパラギンを第1の触媒組成物に接触させて生成されるL−ヒドロキシアスパラギンを、第2の触媒組成物に接触させてL−スレオ−3−ヒドロキシアスパラギン酸を生成するための触媒組成物の組合せ。
【請求項9】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼは精製タンパク質であることを特徴とする、請求項8に記載の触媒組成物の組合せ。
【請求項10】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼのうち、少なくともL−アスパラギン水酸化酵素は該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項8に記載の触媒組成物の組合せ。
【請求項11】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が、該酵素を発現する細胞の表面又は内部に存在することを特徴とする、請求項10に記載の触媒組成物の組合せ。
【請求項12】
前記L−アスパラギン水酸化酵素及びL−アスパラギナーゼの両方が同一の細胞の表面又は内部に存在することを特徴とする、請求項11に記載の触媒組成物の組合せ。
【請求項13】
前記L−アスパラギン水酸化酵素は、
(a)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(b)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギン水酸化酵素活性を有するタンパク質と、
(c)配列番号1又は2のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつL−アスパラギン水酸化酵素活性を有するタンパク質と、
(d)特異的結合タグペプチドが前記(a)ないし(c)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項8ないし12のいずれか1つに記載の触媒組成物の組合せ。
【請求項14】
前記L−アスパラギナーゼは、
(e)配列番号5のヌクレオチド配列からなるポリヌクレオチドにエンコードされるタンパク質と、
(f)配列番号5のヌクレオチド配列からなるポリヌクレオチドに1個若しくは数個のヌクレオチドが欠失、置換又は付加されたヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(g)配列番号5のヌクレオチド配列からなるポリヌクレオチドと80%以上の相同性を示すヌクレオチド配列にエンコードされるアミノ酸配列からなり、かつ、L−アスパラギナーゼ活性を有するタンパク質と、
(h)特異的結合タグペプチドが前記(e)ないし(g)のいずれかのタンパク質に連結した融合タンパク質とからなるグループから選択される少なくとも1種類のタンパク質を含むことを特徴とする、請求項8ないし13のいずれか1つに記載の触媒組成物の組合せ。
【図1】

【図2】

【図3】
【図4】
【図5】
【図6A】

【図6B】
【図6C】
【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図7】
【図8】
【図9】
【公開番号】特開2012−45(P2012−45A)
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願番号】特願2010−137138(P2010−137138)
【出願日】平成22年6月16日(2010.6.16)
【出願人】(899000068)学校法人早稲田大学 (602)
【Fターム(参考)】
【公開日】平成24年1月5日(2012.1.5)
【国際特許分類】
【出願日】平成22年6月16日(2010.6.16)
【出願人】(899000068)学校法人早稲田大学 (602)
【Fターム(参考)】
[ Back to top ]