
LEWISy抗原及びLEWISb抗原に対する抗癌抗体
本発明は、LewisyとLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、かつ、前記抗体又は結合断片が自然にはマルチマーを形成しない、結合複合体を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、癌の治療に有用な抗体又は抗原結合断片を含む結合複合体に関する。特に、本発明は、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にある、結合複合体を提供する。前記結合複合体は、Lewisy及びLewisbを過剰発現している腫瘍細胞において細胞死を誘導する。
【背景技術】
【0002】
Lewisy及びLewisbは、乳癌、肺癌、結腸癌、及び卵巣癌によって過剰発現されている複合糖質である。したがって、それらは、モノクローナル抗体(mAb)治療の良好な標的である。Lewisyハプテンは、二型血液型オリゴ糖で発見されたジフコシル化四糖類(Fucα1−2Galβ1−4(Fucα1−3)GlcNAc)である。この抗原は、Lewisbハプテン(Fucα1−2Galβ1−3(Fucα1−4)GlNAc))の位置異性体及びLewisxハプテンのフコシル化誘導体である(Abe et al.(1986)Cancer Reseach 46:2639;Kim et al.(1986)Cancer Reseach 46:5985)。Lewisyは胸部、気管支、脾臓、秘尿生殖器、及び胃腸管の深部にある腺内において発現する。対照的に、Lewisbは、表面上皮によって発現する(Sakamoto et al.(1989)Cancer Research 49:745−52;Kitamura et al.(1994)Proc.Natl.Acad.Sci.91:12957−61)。
【0003】
IgMマウスmAbであるC14は、標準的な融合方法を用いて原発結腸直腸腫瘍細胞に対して作製し、LewisyとLewisbとの双方の(伸長又は非伸長(extended or non−extended))抗原に結合する(Brown et al.(1983)Biosci.Rep.3:163;Brown et al.(1984)Int.J.Cancer 33:727;Durrant et al.(1993)Hybridoma 12:647−60)。C14抗体は、78%の結腸直腸癌に結合したが(Durrant et al.(1989)J.Natl.Cancer Inst.81:688−95)、マウスIgMなので、in vivo試験には適さなかった。その抗体のIgGバリアントを生産するために、C14mAbでラットを免疫して、生産されたラットの抗C14を精製した。この抗血清及びC14gp200抗原でマウスに免疫した後に、それらの脾細胞をマウスのミエローマと融合させて、5種のIgG mAb、すなわち、2種のIgG3(SC101/23、SC101/29 mAb)及び3種のIgG1(SC101/33、SC101/42、及びSC101/43)(これらの5種の抗体は既に「692」抗体として公開している)を生産させた。全てのIgGバリアントは、Lewisy及びLewisb抗原を認識し、C14と同一の特異性を示した。さらに、これらの抗体は、薄層クロマトグラフィー及びELISAによって、伸長及び非伸長Lewisy及びLewisbハプテンに結合するが、Lewisx又はH血液型ハプテンには結合しないことが示された(Brown et al.(1983)Biosci.Rep.3:163)。
【0004】
LewisyとLewisbとの双方に結合する抗体は既知であるが、SC101 mAbは、Lewisy決定基とLewisb決定基との双方を認識するという能力において特異なものである。他のmAbはこれらの2つの分子の同様の面を認識せず、また、1つの珍しいレクチンのみがこれらの2つの分子の同様の面を認識する。Lewis y/bは、糖脂質としてセラミド骨格上で大部分が発現する。最近の結晶学的研究によって、Lewisyに特異的な抗体は、ハプテンのN−アセチルグルコサミン又はフコースのいずれかに適応する(Ramsland et al.(2004)J.Mol.Biol.340:809−18)非常に異なる結合部位を有することが示された。SC101は、LewisyとLewisbとが互いに立体異性体であるため非常に珍しいことに、LewisyとLewisbとの双方の面に結合部位が適応するため異なる。更に興味深いことに、これらの抗体は、おそらくセラミド又は他の密接に関連する分子からの立体障害のため、胃腸管を含む大半の通常の組織における糖脂質を認識することができない。このことにより、SC101抗体は特有の組織分布を有するが、広範な上皮腫瘍に非常に強力に結合する(Brown et al.(1983)Biosci.Rep.3:163)。
【0005】
脱凝集された原発結腸直腸腫瘍に対する抗体結合の最初の特性決定の間に、それらが細胞死を誘導することが認められた。細胞死は、アポトーシス及びオンコーシスを含む多数の機構によって生じ得る。アポトーシスはカスパーゼ依存性であり、最終的には細胞のアポトーシス小体への分割を生じさせる細胞収縮、クロマチンの凝縮及び辺縁趨向、並びに細胞膜のラッフリングを特徴とする。オンコーシスは、早期の細胞壊死であり、膜透過性の増大期の結果としての細胞膨張を特徴とする。壊死は、細胞死後に現れ、アポトーシス又はオンコーシスの後に生じる形態変化である。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Abe et al.(1986)Cancer Reseach 46:2639
【非特許文献2】Kim et al.(1986)Cancer Reseach 46:5985
【非特許文献3】Sakamoto et al.(1989)Cancer Research 49:745−52
【非特許文献4】Kitamura et al.(1994)Proc.Natl.Acad.Sci.91:12957−61
【非特許文献5】Brown et al.(1983)Biosci.Rep.3:163
【非特許文献6】Brown et al.(1984)Int.J.Cancer 33:727
【非特許文献7】Durrant et al.(1993)Hybridoma 12:647−60
【非特許文献8】Durrant et al.(1989)J.Natl.Cancer Inst.81:688−95
【非特許文献9】Brown et al.(1983)Biosci.Rep.3:163
【非特許文献10】Ramsland et al.(2004)J.Mol.Biol.340:809−18
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明者は、SC101/29 mAbによる腫瘍細胞の殺滅を実証し、当該抗体が、古典的なオンコーシスと多数の類似点を有する特有の機構によって、in vitroとin vivoの双方においてLewisy及びLewisbを過剰発現する腫瘍細胞を直接殺滅することを実証した。
【課題を解決するための手段】
【0008】
本発明の第一の態様では、LewisyとLewisbとの双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にある、結合複合体を提供する。
【0009】
本発明の第二の態様では、本発明の第一の態様に係る結合複合体の有効量を対象に投与する工程を含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞における細胞死の誘導を提供する。
【0010】
本発明の第三の態様では、本発明の第一の態様に係る結合複合体の治療上有効量を対象に投与する工程を含む、治療の必要がある対象の癌を治療するための方法を提供する。
【0011】
本発明の第四の態様では、本発明の第一の態様に係る結合組成物を製薬学的に許容される担体又は希釈剤と共に含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞における細胞死を誘導するための医薬組成物を提供する。
【図面の簡単な説明】
【0012】
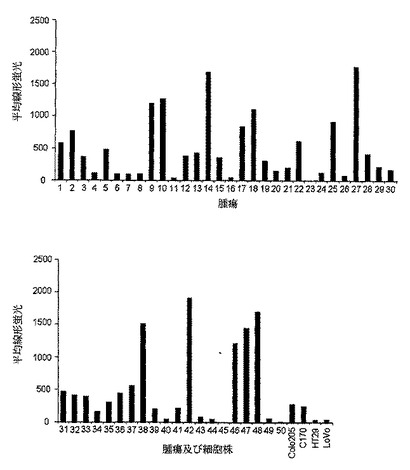
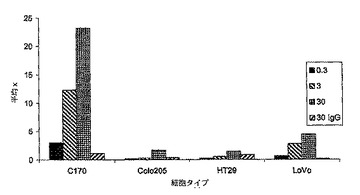
【図1a】間接免疫蛍光によってアッセイし、フローサイトメトリーで分析した、新しく脱凝集させた結腸直腸腫瘍細胞に対するSC101の結合を示す。各々のバーは、個々の腫瘍の平均蛍光を示す。結腸直腸腫瘍細胞株であるColo205、C170、HT29、及びLoVoに対するSC101の結合を比較のために示している。
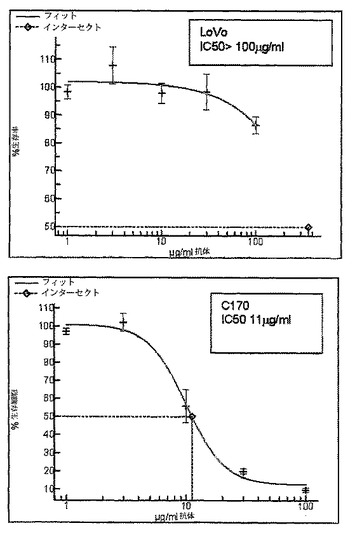
【図1b】SC101/29によるLoVo及びC170細胞のIC50増殖阻害曲線を示す。増殖阻害の%を、SC101/29mAbに曝露した細胞数/対照mAbに曝露した細胞数として表示した。増殖阻害曲線からの生細胞数を、490nmの吸光度の読み取るMTSによって決定した。
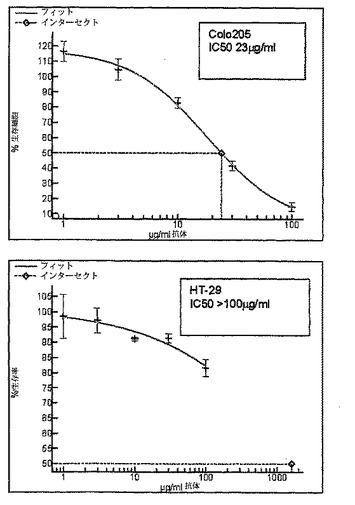
【図1c】SC101/29によるColo205及びHT−29のIC50増殖阻害曲線を示す。増殖の阻害%は、SC101/29 mAbに曝露された細胞数/対照mAbに曝露された細胞数として表示される。増殖阻害曲線からの生細胞数は、490nmにおける吸光度を読取るMTSによって決定した。
【図2a】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のColo205細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
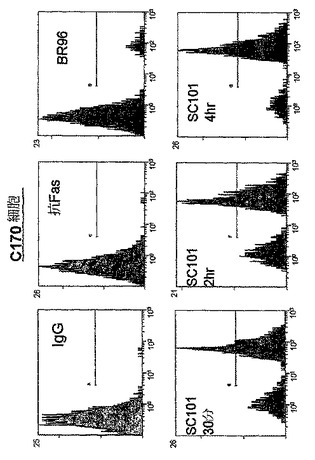
【図2b】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のC170細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
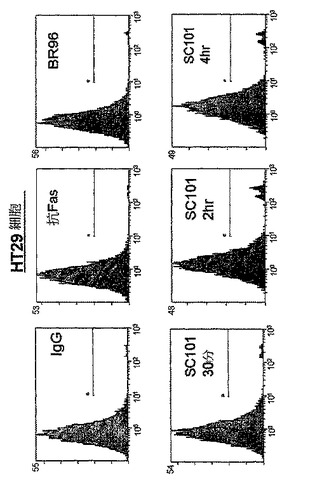
【図2c】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のHT−29細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
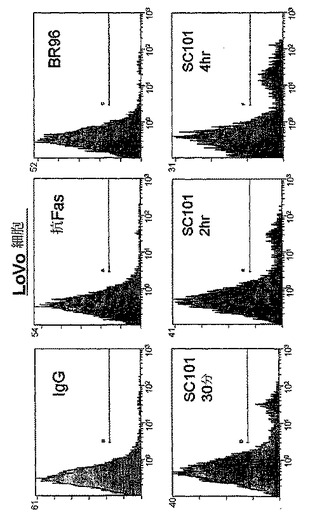
【図2d】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のLoVo細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
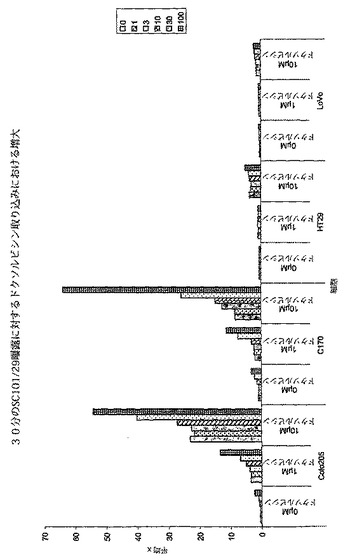
【図2e】100μg/mlのSC101/29 mAbを用いて30分に亘って0から10μMドクソルビシンの存在下において処理した2×105のColo205、C170、HT−29、又はLoVo細胞を示す。細胞は、488nmでFC500フローサイトメーター(発光 575nm FL2)を用いて分析した。
【図2f】細胞をp−糖タンパク質(0.3から30μg/ml)を認識するmAbを用いて染色し、488nmでFC500フローサイトメーター(発光 525 FL1)を用いて分析した。
【図3a】SC101/29、SC101/43、又はそれらの双方のmAbのいずれかを用いて1時間に亘って処理した2×105のC170細胞を示す。細胞は、PIを用いて15分に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620 FL3)を用いて分析した。
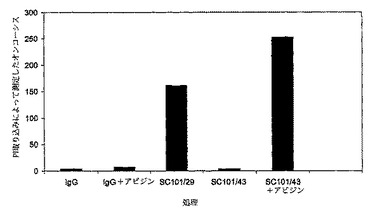
【図3b】100μg/mlのIgG、アビジンと混合した100μg/mlのIgG、100μg/mlのSC101/29、100μg/mlのSC101/43−ビオチン、又はアビジンと架橋した100μg/mlのビオチン化SC101/43のいずれかを用いて1時間に亘って処理した2×105のC170細胞を示す。細胞は、PIを用いて15分に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620 FL3)を用いて分析した。
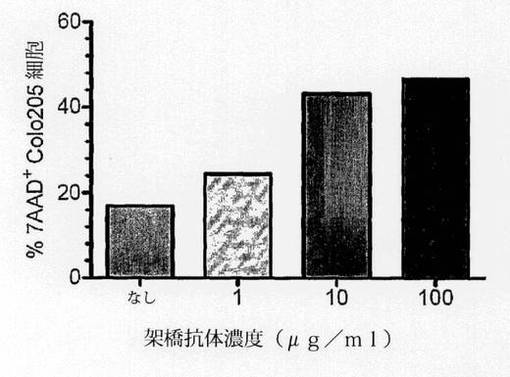
【図3c】100μg/mlのマウスSC101/29ヒトIgG2キメラ抗体で30分間に亘って処理し、続いて架橋抗ヒトIgG抗体(0−100μg/ml)を用いて処理した1×105のColo205細胞を示す。3時間室温でインキュベートした後に、細胞を7AADを用いて20分間に亘って暗所で染色し、488nmでCell Quanta SC MPLを用いて分析した。2つのサンプルにおける死(7ADD+)細胞の平均割合を示す。
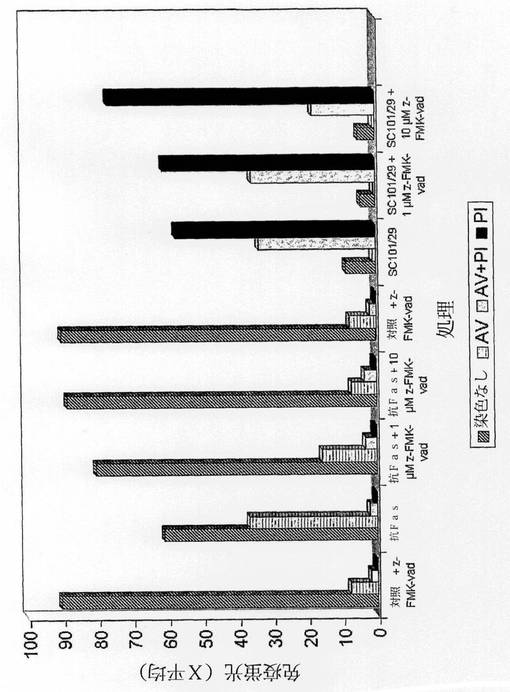
【図3d】100μg/mlのアイソタイプネガティブmAb、100ng/mlの抗Fas、又は100μg/mlのSC101/29 mAbを用いて、0、1、又は10μMのいずれかのz−FMK−vad pan−カスパーゼインヒビターの存在下において、約12時間に亘って処理した5×105のC170細胞を示す。細胞は、アネキシンV及びPIを用いて15分に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 525nm FL1、620nm FL3)を用いて分析した。
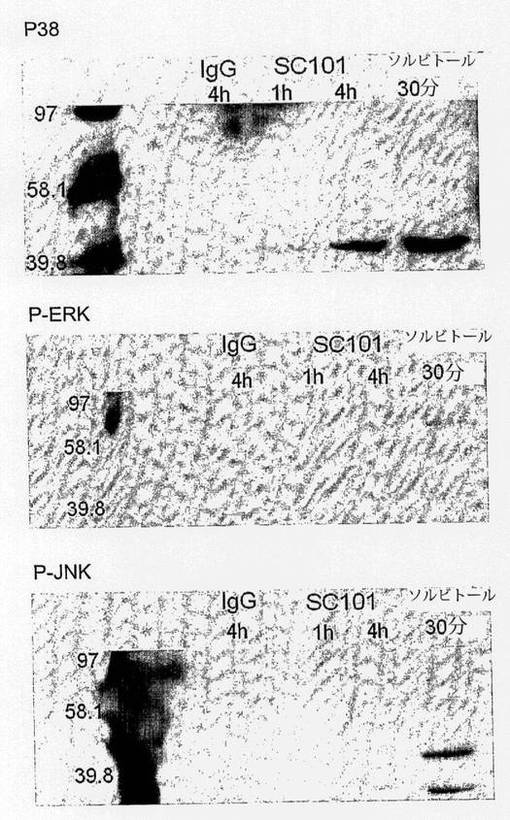
【図3e】IgG(30μg/ml)ネガティブコントロールに4時間、SC101に1から4時間、又は0.5Mのソルビトールに30分間(ポジティブコントロール)曝露した2×106のC170細胞を示す。細胞は、氷冷Tris抽出干渉液+0.5mM NaVO4中で抽出し、75μgの総タンパク質をSDS−PAGEで1レーン毎に載せた。10%ゲルを130Vで90分間に亘って分離させ、BioRad semi−dry blotter(12V 定電圧)で70分間に亘ってPVDF膜に転写させた。膜をブロッキングして、2mlあたり1μlのP−p38、P−JNK、又はP−ERK(1% BSA TBS−T、ECL Plus+を用いて洗浄及び現像した)で探索した。
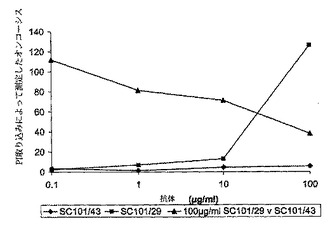
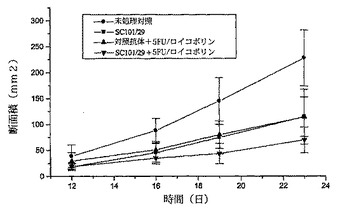
【図4a】ヌードマウスにおけるC170異種移植片の増殖に対するSC101/29 mAb、5−FU/ロイコボリン、又はSC101/29mAbと5−FU/ロイコボリンとの組み合わせの効果を表すグラフを示す。C170異種移植片の増殖は、動物をSC191/29 mAb ip(0.2mg)、対照抗体 ip(0.2mg)、及び5−FU/ロイコボリン(12.5mg/kg;iv)を用いて処理した際に、断面積(mm2)の測定によって、12、16、19、及び23日目に測定した。23日目に得られた結果の分散分析は、SC101/29 mAbを未処理の対照群と比較した際に、p<0.004の有意な値を示し、SC101/29 mAB+5−FU/ロイコボリンに対する対照群+5−FU/ロイコボリンについてはp<0.020の有意な値を示した。
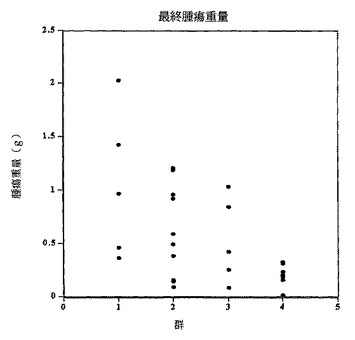
【図4b】SC101/29 mAbのみ(第二群)、5−FU/ロイコボリンのみ(第三群)、若しくはSC101/29 mAbと5−FU/ロイコボリンとの組み合わせ(第四群)を用いて免疫されているか又は未処理(第一群)のマウスの最終腫瘍重量を示す。
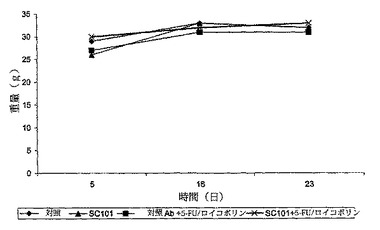
【図4c】SC101/29 mAbのみ、5−FU/ロイコボリンのみ、若しくはSC101/29 mAbと5−FU/ロイコボリンとの組み合わせを用いて免疫されているか又は未処理のマウスの重量を示す。
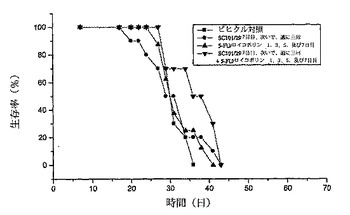
【図4d】SC101/29 mAb ip(0.2mg)、5−FU/ロイコボリン(12.5mg/kg iv)、又はSC101/29 mAb(0.2mg)と5−FU/ロイコボリン(12.5mg/kg iv)との組み合わせを用いて処理したC170異種移植片を有する動物の生存データを示す。SC101/29 mAbを7日目に与え、次いで1週間に3回与えた。5−FU/ロイコボリンは、1、3、5、及び7日目に与えた。SC101/29 mAbは、5−FU/ロイコボリンと併用する際には、ビヒクル対照マウスを超えて生存率を有意に改善した(p=0.0163 Log Rank)。
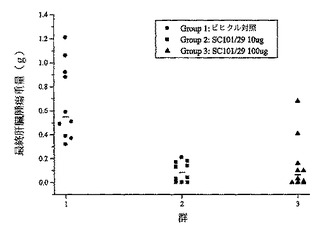
【図5】C170HM2肝臓転移ヌードマウスモデルにおける最終肝臓腫瘍重量(g)に対する10μg若しくは100μgのSC101/29 mAb又はビヒクル対照の効果を表すグラフを示す。
【発明を実施するための形態】
【0013】
本発明者は、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体が、Lewisy及びLewisbを過剰発現している腫瘍細胞における細胞死を誘導することを実証した。
【0014】
本発明の第一の態様では、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にある、結合複合体を提供する。
【0015】
本明細書において使用する用語「抗体」は、4つのポリペプチド鎖、すなわちジスルフィド結合で相互に連結した2つの重(H)鎖及び2つの軽(L)鎖を含む。各重鎖は、重鎖可変領域(HCVR又はVH)及び重鎖定常領域を含む。重鎖定常領域は、3つのドメインとしてCH1、CH2、及びCH3を含む。各軽鎖は、軽鎖可変領域(LCVR又はVL)及び軽鎖定常領域を含む。軽鎖定常領域は、1つのドメインとしてCLを含む。VH及びVL領域は、フレームワーク領域(FR)と称されるより保存された領域が散りばめられた、相補性決定領域(CDR)と称される超可変領域に細分することが可能である。各VH及びVLは、FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4の順番でアミノ末端からカルボキシ末端へと配置された3つのCDR及び4つのFRからなる。
【0016】
本明細書で使用する、抗体の「抗原結合断片」という用語は、抗原に結合する能力を示す免疫グロブリンの1つ又は複数の成分又は誘導体を示す。抗体の抗原結合機能は、全長の抗体の断片によって奏されるものであることが示されている。抗体の「抗原結合断片」という用語の範囲に含まれる結合断片の例は、(i)Fab断片、VL、VH、CL、及びCH1ドメインからなる一価断片;(ii)F(ab’)2断片、ヒンジ領域においてジスルフィド架橋によって結合した2つのFab断片を含む二価断片;(iii)VH及びCH1ドメインからなるFd断片;(iv)抗体の1つの腕のVL及びVHドメインからなるFv断片;(v)1つのVHドメインからなるdAb断片(Ward et al(1989)Nature 341:544−546)又はVLドメイン(van den Beuken et al(2001)J.Mol.Biol,310,591);(vi)単離された相補性決定領域(CDR);並びに(vii)同種のフレームワーク領域によって融合した複数の相補性決定領域、例えばQiu et al.(2007)Nature Biotechnology25(8):921−929に開示されているものを含む。さらに、Fv断片の2つのドメインであるVL及びVHは別々の遺伝子にコードされているが、それらは、VL領域とVH領域が対になって一価分子(単鎖Fv(scFV)としても知られている)を形成する単鎖タンパク質とすることを可能にする合成リンカーによって、組換え法を用いて、連結させてよい(Bird et al.(1988) Science 242:423−426;Huston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883)。その様な単鎖Fvも、抗体の「抗原結合断片」という用語の範囲内含まれることが意図される。単鎖FV及び関連する分子、例えば、二重特異性抗体又は三重特異性抗体も含まれる。二重特異性抗体は、短いために同一の鎖上において2つのドメインの間で対を形成することを可能にするリンカーを使用するがVHドメインとVLドメインとが単独のポリペプチド鎖に発現され、そのドメインを他の鎖の相補的なドメインと対を形成させ、2つの抗原結合部位を生じさせている二価の抗体である(例えば、Holliger et al.(1993)Proc.Natl.Acad.Sci.USA 90:6444−6448;Poljak et al.(1994)Structure 2:1121−1123)。
【0017】
抗体又は抗原結合断片がマルチマーの状態にあることは、本発明に必須の特徴である。抗体又は抗原結合断片は、Lewisy抗原とLewisb抗原との双方に結合して、抗原の架橋を生じさせる。Lewisy抗原とLewisb抗原との架橋は、Lewisy及びLewisbを過剰発現している細胞についての腫瘍細胞死の仲介において重要であり得る。
【0018】
本明細書で使用する用語「マルチマー」は、互いに結合を形成している2つ又はそれ以上の抗体又は抗原結合断片を意味する。さらに、マルチマーは、ホモ又はヘテロマルチマーであってよい。例えば、マルチマーは、ホモ若しくはヘテロダイマー、ホモ若しくはヘテロトリマー、又はそれより高度のマルチマーであってよい。ホモマルチマーは、同一の抗体又は抗原結合断片を含むマルチマーであり、ヘテロマルチマーは、少なくとも2つの異種抗体又は抗原結合断片を含むマルチマーである。しかしながら、マルチマーの組成にかかわらず、各抗体又は抗原結合断片は、Lewisy抗原とLewisb抗原との双方に結合する必要がある。
【0019】
本発明の好ましい実施態様では、マルチマーは、ホモ又はヘテロダイマーである。
【0020】
本発明の更に好ましい実施態様では、抗体が自然にはダイマー又はマルチマーを形成しない。
【0021】
本明細書で使用する用語「Lewisy抗原とLewisb抗原との双方に結合する」は、Lewisy抗原とLewisb抗原とが架橋を形成するように、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を意味する。
【0022】
本明細書で使用する用語「結合する」は、例えば、BIAcore(商標)表面プラズモン共鳴システム及びBIAcore(商標)動力学的評価ソフト(例えば、version 2.1)を使用して表面プラズモン共鳴分析によって測定すると、1μM以下の解離定数(Kd)を有する、抗体の免疫グロブリン可変領域による抗原への結合を意味する。特異的な結合相互作用についての親和性又は解離定数(Kd)は、約500nMから約50pM、より好ましくは約500nM以下、より好ましくは約300nM以下、好ましくは少なくとも約300nMから50pM、約200nMから約50pM、より好ましくは約100nMから約50pM、約75nMから約50pM、約10nMから約50pMである。
【0023】
本発明のある実施態様では、抗体又は抗原結合断片が、二重特異性抗体又は多重特異性抗体である。本発明の他の実施態様では、二重特異性抗体又は多重特異性抗体が修飾されたFcドメインを含む。
【0024】
本発明の他の実施態様では、抗体又は抗原結合断片を含むマルチマーがIgG抗体である。本発明の更なる実施態様では、マルチマーがダイマーのIgG1抗体である。
【0025】
幾つかの抗体(例えば、IgG3)はin vitro及びin vivoでダイマーを形成し、IgG1抗体はモノマー形態で存在する。モノマーの抗体又はその抗原結合断片が、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において腫瘍細胞死を誘導し得るために、Lewisy抗原及びLewisb抗原は、モノマーの抗体又は抗原結合断片のアビジン/ビオチンによる架橋によって架橋される。代替的には、モノマーの抗体又は抗原結合断片は、組換え工学、例えば、モノマーのカルボキシ末端における鎖間ジスルフィド結合の形成を可能にする、CH3領域の遺伝子におけるセリンからシステインへの変異によって、架橋されてよい(Caron et al.(1992)J.Exp.Med.1191−1195)。これが、次いで、Lewisy抗原とLewisb抗原との架橋を容易にする。
【0026】
本発明の第二の態様では、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の有効量を対象に投与する工程を含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための方法であって、前記抗体又は抗原結合断片がマルチマーの形態にある、方法を提供する。
【0027】
本発明の第三の実施態様では、Lewisy抗原とLewisb抗原との双方に結合する本発明の第一の態様に係る結合複合体の治療上有効量を、対象に投与する工程を含む、治療の必要がある対象の癌を治療するための方法を提供する。
【0028】
用語「治療上有効量」は、特定の疾患若しくは障害(本発明の場合には癌)又はその症状の1つ若しくは複数を治療又は緩和し、及び/又は疾患又は障害の発生を予防又は低減させるために十分な、抗体又はその抗原結合断片(抗体又は抗原結合断片を含む医薬組成物を含む)の量を意味する。
【0029】
抗体又はその抗原結合断片(抗体又はその抗原結合断片を含む医薬組成物を含む)の使用及び治療方法に関して使用する際に、「治療の必要がある」個体は、癌であると診断されているか又は過去に治療されている個体であってよい。
【0030】
本発明の好ましい実施態様では、癌は、乳癌、肺癌、結腸癌、及び卵巣癌からなる群から選択されてよい。
【0031】
本発明の第四の態様では、本発明の第一の態様に係る結合複合体を製薬学的に許容される担体又は希釈剤と共に含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための医薬組成物を提供する。
【0032】
「製薬学的に許容される担体又は希釈剤」は、任意の全ての溶媒、分散媒体、コーティング、抗菌剤若しくは抗真菌剤、等張剤、及び吸収遅延剤などの生理的に適合するものを含む。製薬学的に許容される担体の例は、水、生理食塩水、リン酸緩衝生理食塩水、デキストロース、グリセロール、エタノールなど、及びそれらの組み合わせの1つ又は複数を含む。多くの場合において、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール、ソルビトール、又は塩化ナトリウムを組成物に含むことが好ましいであろう。
【0033】
組成物は、液体、半固体、又は固体の剤形、例えば、溶液(例えば、注射可能な溶液及び輸液可能な溶液)、分散物又は懸濁物、錠剤、丸剤、粉末剤、リポソーム、又は座剤を含む各種の形態にあってよい。好ましくは、組成物は、免疫のための注射可能な溶液の形態にある。投与は、静脈内、皮下、腹腔内、筋肉内、経皮、鞘内、及び動脈内であってよい。好ましくは、前記剤形は、約0.001mgから約10mg/kg体重の範囲内で、毎日、毎週、二週若しくは三週に一度、又は毎月投与され、好ましくは約0.05から約5mg/kg体重で毎週投与される。
【0034】
前記組成物は、滅菌した注射可能な溶液の調製のための滅菌粉末として製剤化されてもよい。
【0035】
ある実施態様では、結合複合体が、急速な放出に対して化合物を保護する担体と共に調製されてよく、例えば、インプラント、経皮パッチ、及びマイクロカプセル化送達系を含む制御放出製剤であってよい。適合性のあるポリマーが使用されてよく、例えば、エチレンビニルアセテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、又はポリ乳酸が使用されてよい。
【0036】
前記組成物は、経口投与のために製剤化されてもよい。この実施態様では、抗体は、硬質ゼラチンカプセル又は軟質ゼラチンカプセルに覆われてよく、錠剤に押し固められてよく、又は対象の食料に直接含ませてもよい。
【0037】
前記組成物は、直腸投与のために製剤化されてもよい。
【0038】
本発明の結合複合体は、in vivo及びin vitroにおいて特定の細胞に結合及び同定するか、in vivoにおいて特定の細胞に結合及び破壊するか、又はin vivoにおいて特定の細胞に入り込み、破壊するために投与されてよい。
【0039】
好ましい実施態様では、前記組成物はヒトに投与される。
【0040】
本発明の本質がより明確に理解され得るように、その好ましい態様を、以下の非限定的な実施例を参照して説明する。
【実施例】
【0041】
(実施例1)
材料及び方法
細胞株
C170は、原発腫瘍由来の結腸直腸細胞である(Durrant et al.(1986)Br.J.Cancer 53:37−45)。Colo205、HT−29、及びLoVoは、ATCCから得られた結腸直腸細胞である。全ての細胞は、10%ウシ胎仔血清(FCS F6178;Sigma,Poole,UK)RPMI1640(BE12−702F;Cambrex Bio Science,Berkshire,UK)において培養した。
【0042】
モノクローナル抗体
LewisyハプテンとLewisbハプテンとの双方を認識するSC101 mAbは、過去に開示されている(Brown et al.(1983)Biosci.Rep.3:163;Druurat et al.(1993)Hybridoma 12:647−60)ように精製した。抗Fas(ヒト活性化)クローンCh11(05−201)抗体は、Upstateから得られ、Br96およびIgGネガティブコントロールはSigma(I5381)から得られた。
【0043】
結腸直腸細胞に結合する抗体
腫瘍検体は、結腸直腸癌の切除時に得た。検体を非常に細かく切って、0.05%のコラゲナーゼ(タイプIV、Boehringer Mannheim,Lewes,UK)を用いて20分間に亘って37℃で脱凝集させて、SC101/29及びヤギ抗マウスFITC(Dako Ltd,Bucks UK;1:100希釈)を用いて間接免疫蛍光によって染色した。腫瘍細胞株は一定分量(105)に分けて、SC101/29 mAb又は抗P−糖タンパク質(多剤耐性を示す、BD Pharmingen,San Diego,California USA)を各種の濃度で用いて、4℃において30分間に亘って染色した。培地(RPMI/10%FCS)で二回洗浄した後に、細胞を二次抗体(ヤギ抗マウスFITC)を用いて4℃で30分間に亘ってインキュベートし、その後に、最終的な洗浄を行い、488nmでFC500フローサイトメーター(発光525nm FL1)において分析した。
【0044】
結腸直腸腫瘍細胞のin vitroにおける殺滅
1×103の結腸直腸C170、Colo205、HT−29、及びLoVo細胞を、平底96ウェルプレートの個々のウェルに一定分量に分けて、一晩37℃で静置して接着させた。翌日、100、30、10、3、1、又は0μg/mlのSC101/29 mAb(100μl/ウェル)を用いて細胞を処理した。アイソタイプに適合したポジティブコントロール又はネガティブコントロールを比較のために使用した。ウェルを3つ重複させて使用した。細胞を抗体の存在下で37℃に5日間に亘って静置した後に、Cell Titer96 Aqueous One Solution(G3580;Promega,Southampton,UK)を各ウェルに添加して、490nmにおける吸光度を測定した。
【0045】
PI取り込み
2×105のColo205、C170、HT−29、又はLoVo細胞を、30μg/mlのIgG、300ng/mlの抗Fas、30μg/mlのBR96、又はSC101/29 mAbのいずれかを30分、2時間、又は4時間に亘って処理した。その後に、PI(630110;BD Biosciences,Oxford,UK)を用いて15分間に亘って暗所で室温において細胞を染色し、488nmでFC500 flow cytometerを用いて分析した(発光620nm FL3)。
【0046】
7AAD取り込み
1×105のColo205を、100μg/mlのマウスSC101/29−ヒトIgG2キメラ抗体とともに室温においてリン酸干渉生理食塩水(PBS)中でインキュベートした。30分後に、各種の濃度の架橋抗ヒトIgG抗体(0−100μg/ml;Sigma)を添加して、細胞を3時間に亘って室温でインキュベートし、次いで、7AAD(Bechman Coulter)を用いて20分間に亘って暗所で細胞を染色し、488nmでQuanta SC MPL(Beckman Coulter;発光フィルター670 LP FL3)を用いて分析した。
【0047】
MDR細胞株における薬剤取り込み
2×105のColo205、C170、HT−29、又はLoVo細胞を、0、1、3、10、30、又は100μg/mlのSC101/29 mAbを用いて30分間に亘って37℃で処理した。次いで、0、1、又は10μMの終濃度でドクソルビシンを前記細胞に添加し、37℃で1時間に亘ってインキュベートした。細胞を氷冷リン酸干渉生理食塩水(PBS)で五回洗浄し、488nmでFC500フローサイトメーター(発光575nm FL2)を用いて蛍光分析した。
【0048】
カスパーゼ依存性細胞死の阻害アッセイ
5×105のC170細胞を、平底滅菌24ウェルプレートの個々のウェルに一定分量に分けて、6時間に亘って37℃で接着させた。前記細胞を、100μg/mlのアイソタイプに適合したネガティブコントロールmAb、100ng/mlの抗Fas、又は100μg/mlのSC101/29 mAb、及び0、1、又は10μMのz−FMK−vad(G7232;Perbio Science,UK)を用いて処理した。細胞を37℃で一晩静置して、その後に、アネキシンV及びPIを用いて15分に亘って染色し、488nmでFC500フローサイトメーター(発光525nm FL1、620nm FL3)を用いて分析した。
【0049】
ストレス関連キナーゼを介するシグナル伝達
2×106のC170細胞を、IgG(30μg/ml)ネガティブコントロールに4時間に亘って、SC101(30μg/ml)に1〜4時間に亘って、又は0.5Mソルビトール(ポジティブコントロール)に30分間に亘って曝露した。細胞は、氷冷抽出干渉液(50mM Tris pH 7.4、2mM EDTA、2mM Na4P2O7、2mM ベンズアミジン、1mM PMSF、0.5mM NaVO4、0.5mM DTT 0.1% Triton−X−100)中で抽出して、標準的なBioRadタンパク質アッセイを用いてタンパク質濃度を測定し、75μgの総タンパク質をSDS−PAGEのレーン毎に載せた。10%ゲルを130Vで90分に亘って分離させて、BioRad semi−dry blotter(12V定電圧)で70分間に亘ってPVDFに転写した。膜を1時間に亘って1%のウシ血清アルブミン(BSA)Tris干渉生理食塩水0.05%Tween−20(TBS−T)中で1時間に亘ってブロッキングし、1% BSA TBS−T中で2mlあたり1μlのP−p38、P−JNK、又はP−ERK(Promega)で探索した。その後に、膜をTBS−T中で三回洗浄して、ヤギ抗ウサギHRP(GαR HRP;Dako,P0448)で探索し、TBS−Tで三回洗浄して、ECL Plus+で現像した。
【0050】
in vivo試験
予防モデル
結腸直腸腫瘍細胞株であるC170を、ヌードマウスにおいて連続継代に維持した。治療のために、マウスを屠殺して、腫瘍を摘出した。前記腫瘍を非常に細かく切って、3mm2片を麻酔(Hypnorm、Roche/Hypnovel、Jannsen)条件下で、無作為に4つの実験群に割り当てた40匹のオスのマウスの皮下に移植した。1つの群のマウスは、5−FU/ロイコボリン(12.5mg/kg)を用いて静脈内(iv)注射で1、3、5、7、21、及び22日目に処理した。一週間に三回、マウスに、0.2mgのSC101/29 mAbを用いる腹腔内(ip)注射も実施した。対照マウスは、SC101/29 mAb、5−FU/ロイコボリン、又はビヒクルのみのいずれかを受けた。腫瘍の大きさはキャリパーによって測定し、腫瘍断面積を12、16、19、及び23日目に計算した。実験の終わりに、腫瘍を秤量して、抗腫瘍効果を評価した。動物を秤量して、治療の毒性を評価した。
【0051】
治療モデル
結腸直腸腫瘍細胞株であるC170を、ヌードマウスにおいて連続継代に維持した。治療のために、マウスを屠殺して、腫瘍を摘出した。前記腫瘍を非常に細かく切って、3mm3片を麻酔条件下で、無作為に4つの実験群に割り当てた40匹のオスのマウスの皮下に移植した。マウスは、C170異種移植片の3mm3片を外植した。前記群のマウスを、5−FU/ロイコボリン(25mg/kg)を用いてiv注射で1日目、3日目、5日目、7日目、適用可能な場合には28日目から繰り返して治療した。一週間に三回、マウスに、0.2mgのSC101/29 mAbを用いてiv注射も実施し、対照マウスは、SC101/29 mAbのみ又は対照マウスIgG抗体を5−FU/ロイコボリンとともに受けた。
【0052】
転移モデル
C170HM2細胞を、37℃、5% CO2で加湿条件下において10%熱不活化FCSを含有するRPMI 1640培養培地中でin vitroに維持した。ほぼコンフルエントな単層から細胞を、0.025% EDTAを用いて回収し、上記の培養培地で二回洗浄し、in vivo投与のために滅菌PBS(pH 7.4)に再懸濁した。1mlの容量における1.5×106の細胞を、30匹のオスのヌードマウスの腹腔に注射した。動物をそれらの治療群に割り当てて、治療を1日目に開始して、試験に間に亘って継続した。前記群のマウスを、10μg若しくは100μgのSC101/29 mAb又はビヒクル対照を用いてiv注射によって、一日目及び次いで一週間に三回、治療した。マウスは40日目に屠殺して、体重及び腫瘍の重量を評価した。
【0053】
統計
統計分析を、PC用のMinitabプログラムのAnalysis of Variance及びLog Rankを用いて実施した。
【0054】
(実施例2)
結果
結腸直腸腫瘍細胞による抗体結合
SC101 mAbは、LewisyハプテンとLewisbハプテンとの双方に結合することが過去に示されている。新しく脱凝集させた結腸直腸腫瘍の強力な染色を、図1aに示している。mAbは、脱凝集した卵巣及び胃の腫瘍にも結合する(Brown et al.(1983)Biosci.Rep.3:163)。さらに、これらの抗体は、原発腫瘍細胞の細胞死の促進を誘導した。SC101/29抗体及びSC101/33抗体を用いた免疫組織化学及びフローサイトメトリー実験は、以下のタイプ:卵巣、胸部、肺、前立腺、及び膵臓の腫瘍細胞株及び腫瘍異種移植片に顕著に結合した(データ示さず)。
【0055】
結腸直腸腫瘍細胞のin vitroにおける殺滅
当該殺滅を調べるために、さらに広範な細胞株を、SC101/29 mAbによる結合(図1a)及び細胞増殖の阻害(図1b及び1c)についてスクリーニングした。C170及びColo205細胞株の双方が、SC101/29 mAbに結合したが、大半の原発性腫瘍が、より強力な結合を示した。対照的に、SC101/29 mAbは、HT−29及びLoVoには原発腫瘍の20%に相当するレベルで弱く結合した。特に興味深いことは、SC101/29が、C170及びColo205細胞の増殖を阻害するが、Lewisy抗原及びLewisb抗原を低レベルで発現するHT−29及びLoVo細胞の増殖を阻害し得ないことである。
【0056】
生存率試験
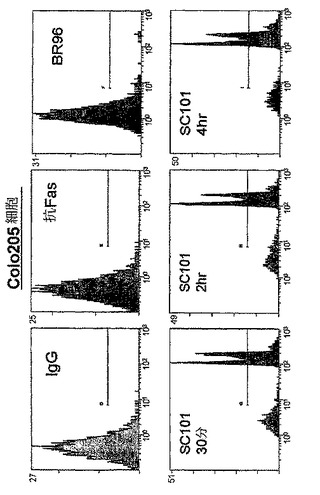
SC101/29 mAbによって仲介される細胞死の機構を評価するために、ヨウ化プロピジウム(PI)という、等張条件下において非生細胞にのみ侵入し得る蛍光化合物をプローブとして使用して、細胞膜の完全性を失った細胞を同定した。図2a及び2bに示すように、SC101/29 mAbは、Colo205及びC170細胞の浸透性を急速に増大させ、75%超の細胞が、早くも抗体処理の30分後にPIで強力に染色された。同一の条件下において、アポトーシス性細胞死を誘導することが既知の抗CD95抗体又は対照マウスIgG抗体を用いて4時間に亘ってインキュベートした際には、10%未満の細胞がPIを取り込んだ。同様に、Lewisbには結合せずLewisyに結合するのみのmAbであるBR96は、4時間に亘って、PIの顕著な取り込みを示し得なかった。細胞増殖の阻害と一致して、SC101/29 mAbは、低い抗原の発現を有するHT−29及びLoVo細胞におけるPI取り込みを誘導し得なかった。
【0057】
SC101/43も、膜透過性を変化する能力について分析したが、SC101/29とは反対に、PI取り込みを誘導し得なかった(図3a)。しかしながら、それは、SC101/29によって誘導されるPI取り込みの阻害において非常に効果的であり、同一又は密接に関連するエピトープに結合することを示している。SC101/43がアビジン/ビオチンを使用して架橋されている場合は、効果的に細胞死を誘導することも可能である。同様に、キメラSC101/29 IgG2抗体の架橋は、結腸腫瘍細胞の直接的な殺滅を誘導する(図3c)。SC101/29は天然にダイマーとして存在するマウスIgG3であり、これらの結果は、Lewisy及びLewisbの架橋がオンコーシスには重要であり、SC101がそのダイマー構造により架橋するが、IgG1であるSC101/43は人工的な架橋が必要であることを示す。
【0058】
MDR細胞株における薬剤取り込み
膜の完全性を失うことによって、低分子が細胞に接近することを可能にする。これは、蛍光薬剤であるドクソルビシンを用いて示されており、SC101/29 mAbに対する広範な細胞の曝露の後に、抗原を高レベルで発現し、且つ、膜の完全性の損失している細胞株においてデュロキセチンの取り込みが向上したが(Colo205及びC170)、低い抗原密度を有する細胞では向上しなかった(HT−29及びLoVo、図2c)。その効果は、p−糖タンパク質を発現する細胞(C170)で更に強力に現れ(図2f)、このことは、SC101/29によって生じた膜の混乱が当該ポンプによる薬剤の排出を阻害したことを示す。
【0059】
カスパーゼ依存性細胞死アッセイの阻害
膜の完全性の急速な損失は、膜に形成される細孔が、大きな細胞質タンパク質、例えば、LDHの放出を可能にするまで、そのサイズを大きくし続けるオンコーシスを生じさせる。PIは完全性の損失を示すが、同時に起こるC170細胞からのLDHの放出は認められなかった。LDH放出が存在しないことによってC170のオンコーシスの発現に疑問を抱いたため、同定したPI染色が、古典的なアポトーシスのアーチファクトによるものではないことを確認するために、全てのカスパーゼのインヒビターであるz−FMK−vadを使用した。要するに、抗Fasに対する曝露は、C170細胞におけるアポトーシスを十分には誘導しないことが過去に認められていたが、前記インヒビターは、前記抗体の効果を完全に逆転させ得た(図3d)。z−FMK−vadの存在下におけるSC101/29 mAbに対する腫瘍細胞の曝露は、認識可能な効果を有さず、このことは、細胞死の様式が古典的なアポトーシス経路からは独立していることを示す。
【0060】
ストレス関連キナーゼを解するシグナル伝達
膜の透過性は、HSP27をリン酸化してアクチン重合を生じさせるp38の活性化と強く関連している。弱いp38活性化が、1時間後にC170細胞において認められ、4時間後により強力な活性化が認められた(図3e)。対照的に、JNK又はERKの活性化は存在しなかった。ストレスによって活性化されるタンパク質キナーゼであるp38は、5−FUを含む広範な化学療法剤によって誘導される。したがって、SC101/29がin vivoで細胞死を誘導するかどうか及びこれが5−FUと相乗効果を奏するかどうかを決定するために、ヒト異種移植片を移植したマウスにそれを投与した。
【0061】
in vivo試験
前記抗体を、C170腫瘍の3mm2抽出物を移植したマウス又は抗体の投与前に5日間に亘って増殖させたC170腫瘍に一週間に三回投与した。前記動物は、SC101/29 mAbのみ、5−FU/ロイコボリン、又は双方の組み合わせのいずれかを用いて3週間に亘って処理した。図4aは、SC101/29と5−FU/ロイコボリンとの双方のみが、新しく外植した腫瘍の腫瘍増殖阻害を生じさせた(p<0.004 ANOVA)。対照的に、双方の組み合わせは更なる増殖阻害を示し、2/10のマウスのみが移植した腫瘍の0.3gという重量を超える任意の増殖を示した(図4b p<0.02 ANOVA)。このSC101/29 mAbの用量は全てのマウスに良好に許容され、体重減少又は任意の他のひどい症状を示さなかった(図4c)SC101/29 mAbが、C170腫瘍を発現するマウスに、25mg/kgの5−FU/ロイコボリンと組み合わせて治療目的で投与される際は、腫瘍増殖を顕著に阻害し、且つ、生存を向上させた(図4d:p<0.0163 Log Rank)。
【0062】
細胞殺滅の機構はSC101/29の微小転移細胞に対する結合が非常に効果的であるはずであるため、肝臓転移の予防におけるSC101/29 mAbの治療効果を、C170HM2結腸直腸モデルを使用して評価した。マウスは、低用量である100μg及び10μgのSC101/29 mAbを用いて一週間に三回、六週間に亘って治療した。試験の終わりに、摘出したC170HM2肝臓腫瘍の重量を測定した。各群について中央値を計算して、図5にデータをグラフ化した。各群についての中央値及び平均値は、Mann−Whitney試験によって統計学的有意性について分析した。10μg及び100μg SC101/29 mAb(第二群及び第三群)についての中央値は、ビヒクル対照(第一群)から有意(p<0.001)であることが認められた。SC101/29は当該モデルにおいて低用量であっても非常に効果的であったため、5−FUは必要でなかった。
【0063】
(実施例3)
考察
広範なLewisy抗体が同定されているが、それらは一貫して、Lewisx及びH型2構造と交差反応する。このことが、正常な組織との望ましくない交差反応を生じさせ、臨床試験において後に毒性を誘導し得る。例えば、H抗原と交差反応するマウスmAb BR55−2のフェーズI試験において、6/12の患者において血尿症が存在し、2/9の患者においてdiahoreaが存在し、3人の患者において一過性の皮膚病変の減少が存在していた(Tolcher et al.(1999)J.Clin.Oncol.17:478−84)。Lewisyのみに対するLMB−1イムノトキシン(シュードモナス属のエンドトキシンに結合したB3抗体)は、5/38の患者において反応を生じた(Pai et al.(1996)Nat.Med.2:350−3)。BR96 mAbは、B血液型と交差反応するが、Lewisbとは交差反応しない。胃腸に対する結合は用量制限的であり、抗体結合と関連することが示された(Saleh et al.(2000)J.Clin.Oncol.18:2282−92)。最後に、Lewisy特異的なヒト化抗体である3S193は、結合試験において優れた選択性を示し(Scott et al.(2000)Cancer Research 60:3254−61)、臨床試験に入っている。
【0064】
SC101抗体は、LewisyとLewisbとの双方の特有の面を認識する。SC101抗体は、Lewisy又はLewisbのいずれかを発現する広範な正常組織と強力な交差反応性を示すことを予測し得るが、他のLewis抗体とは対照的に、胃腸管内のムチンの弱い染色のみが認められた(Brown et al.(1983)Biosci.Rep.3:163)。このことは、BR96 mAbを用いて認められた胃腸管の毒性はSC101抗体を用いて回避し得ることを示す。
【0065】
SC101抗体は、免疫エフェクター細胞なしでin vitroで腫瘍細胞死を誘導し、おそらく、IgG3抗体はCDC及びADCCといった媒介因子が非常に乏しいようなin vivoでも誘導する。非常に興味深いことは、腫瘍細胞が、殺滅されるためにLewisy及びLewisbを過剰発現していることを必要としたことである。この発現レベルにおいて、80%の胃腸管及び30%の卵巣/胸部の腫瘍が、細胞の殺滅を受けやすいが、正常な組織は除外され、良好な治療方法を提供する。SC101/29 mAbが膜の完全性の急速な損失を生じさせるように、細胞の殺滅の機構は非常に興味深い。これは、浸透圧刺激及び酸素過剰細胞において認められるオンコーシスによる細胞死と類似する(Garmyn et al.(2001)J.Invest.Dermatol.117:1290−5;Moriguchi et al.(1996)J.Biol.Chem.271:26981−8;Romanshko et al.(2003)Free Radic.Biol.Med.35:978−93;Shen et al.(2002)J.Biol.Chem.277:45776−84;Tilley et al.(1996)FEBS Letters 395:133−6)。しかしながら、伝統的なオンコーシスの進行は、次いで、LDHなどの細胞質の内容物の放出を生じさせる(Chen et al.(2001)Toxicol.Appl.Pharmacol.171:1−11)。よく特徴付けられた抗Lewisy抗体であるBR96が細胞膜に対する作用を示さないので、細胞死の機構はLewisy/Lewisb構造と関連しているようである。Lewisy及びLewisbが、天然のマウスIgG3又はIgG1バリアントのアビジン/ビオチン架橋によって架橋されて、膜の混乱を生じさせることも必要とした。腫瘍細胞死などの腫脹を誘導し得る幾つかの他の抗体が開示されている。これらは、広範に発現する糖タンパク質を認識する抗ポリミン(Porimin)抗体(Ma et al.(2001)Proc.Natl.Acad.Sci.98:9778−83)、腎臓関連糖タンパク質を認識するRE2(Matsuoka(1995)J.Exp.Med.181:2007−2015)、及び新規の糖タンパク質を認識するRAV12を含む。しかしながら、これらの抗体は、特にMDR細胞株において化学療法剤の取り込みを増大させることを示さないため、SC101は独特なものである。
【0066】
細胞におけるPI取り込みは、p38活性化を伴う。p38活性化はマイクロフィラメントダイナミクスを変化させるHSP27のリン酸化と関連しているため(Deschesnes(2001)Mol.Biol.Cell 12:1569−82;Huot et al.(1998)J.Cell Biology 143:1361−73)、これによって膜の透過性が説明される。膜の完全性の損失は急速に起こるものであり、細胞死の上流にある。p38の活性化は、カスパーゼ依存性又は非依存性細胞死を誘導し得る(Deschesnes(2001)Mol.Biol.Cell 12:1569−82)。SC101/29 mAbは、DNA断片化を誘導し得ず(データは示さず)、全てのカスパーゼのインヒビターであるz−FMK−vadを用いるカスパーゼの阻害は細胞死を妨げないようであり、このことは、抗体がカスパーゼ非依存性の細胞死を誘導することを示す。結腸直腸腫瘍は、アポトーシス反応に関与する遺伝子における>70%の変異を示し、当該反応を避ける任意の治療が非常に治療的価値がある(Vogelstein(1988)N.Engl.J.Med.319:525−32;Fulda et al.(2004)Curr.Cancer Drug Targets 4:569−76)。1つのカスパーゼ非依存性の機構は、ミトコンドリアからのアポトーシス誘導因子(AIF)の放出及びその核移行、そこで未知の様式において核濃縮に寄与することを伴う(Susin et al.(2000)J.Exp.Med.192:571−80)。
【0067】
ストレス関連キナーゼであるp38は、広範な化学療法剤によっても誘導され得るものであり、薬剤合成において極めて重要な役割を担っていると提案されている(Olson & Hallahan(2004)Trend Mol Med 10:125−9)。Lewisy及びLewisbは、腫瘍糖タンパク質及び糖脂質上で発現し、これらは、細胞ストレスに反応してアップレギュレートされる。実際、化学療法ストレス(Flieger et al.(2001)Clin.Exp.Immunol.123:9−14)、特に5−FUに対するLewisyのアップレギュレートが過去に報告されている。したがって、5−FU/ロイコボリンは結腸直腸腫瘍に標準的な化学療法であるため、Lewisyをアップレギュレートし、且つ、p38を活性化し得る(Feng et al.(2002) Cancer Research 62:1920−6)。SC101/29及び5−FU/ロイコボリンの組み合わせを、ヌードマウスにおける結腸異種移植変の増殖疎外能力についてin vivoでスクリーニングした。SC101/29 mAbのみ及び5−FU/ロイコボリンとの併用において、小さい腫瘍及び樹立腫瘍の双方の増殖を顕著に阻害した。キメラバージョンのSC101/29 mAbを用いたフェーズI試験が安全であることを示したら、その後に、手術後の結腸直腸癌患者において、薬剤のみとの比較における5−FU/ロイコボリンとSC101/29 mAbとの併用の試験が開始されるであろう。当該併用は、パノレックス/5−FUは直接殺滅する活性を有さず、且つ、CDC及びADCCに依存しており、補体調節分子の過剰発現による固形癌への限定的な成功を有するため、パノレックス/5−FUよりもさらに効果的であり得る(Li et al.(2001)Br.J.Cancer 84:80−6)。
【0068】
本発明者は、SC101/29が、低分子量(<800D)の薬剤を取り込ませるが、LDHなどの大きな細胞内成分を放出させないように膜の透過性を誘導することを示した。SC101/29は、ストレス関連キナーゼであるp38のリン酸化を誘導するが、JNKのリン酸化は誘導せず、最終的にカスパーゼ非依存性細胞死を誘導する。セラミドがp38の活性化及びAIFを含む多数のミトコンドリアタンパク質の放出によって神経細胞死を誘導することが示されているため、このことは特に興味深い(Ghatan et al.(2000)J.Cell.Biol.150:335−47;Stoica et al.(2005)Mol.Cell.Neurosci.29:335−71)。Lewisy/Lewisbは糖脂質上で主に発現するため、その分解は、セラミドの増加及びその後のp38活性化を生じさせ得る。SC101/29は古典的な経路によって細胞死を誘導しないため、古典的なアポトーシス又はオンコーシスに対する耐性機構を発達させている腫瘍細胞において特に効果的であり得る。しかしながら、その経路は、より注意深く解明し、本発明者による殺滅アッセイの更なる検証によって、潜在的な耐性機構の評価及び最適な腫瘍標的の同定をすることが必要である。
【0069】
結論として、SC101/29は、Lewisy及びLewisbを認識し、膜の完全性の損失を誘導することによって細胞死を直接誘導する新規mAbである。本願は、オンコーシスの様式において、腫瘍細胞を選択的に殺滅し得る血液型抗原を認識する抗体を初めて開示するものである。治療における相乗的な反応におけるp38の役割は、次第に明らかになっている。SC101/29は、5−FU/ロイコボリンとの組み合わせにおいて殺滅の促進を示し、これらの薬剤と組み合わせて使用して、毒性を低減し、且つ、癌治療の効果を増大させる。
【0070】
本明細書を通じて、「含む」なる用語は、記載した要素、数字、若しくは工程、又は要素、数字、若しくは工程の群の包含を示すものであり、任意の他の要素、数字、若しくは工程、又は要素、数字、若しくは工程の群の排除を意味するものではないと解されるであろう。
【0071】
本明細書で挙げた全ての文献は、参照によって本明細書に取り込む。本明細書に含まれる文献、行為、材料、装置、又は論文などの任意の議論は、本明細書の内容を提供するためだけのものである。これらが従来技術の基礎の一部を形成し、又は本願の各請求項の優先日前に豪州又は他の地域に存在した本発明に関連する分野の共通する一般的知識であったと認めるものとして解釈されるべきではない。
【0072】
広範に記載したように本発明の精神又は範囲を逸脱すること無く、特定の実施態様に示したように、本発明の多数の変形例及び/又は修飾例を作製し得ることが当業者によって理解されるであろう。本実施態様は、したがって、全ての態様において説明するものであり、限定するものではないと解されるべきである。
【技術分野】
【0001】
本発明は、癌の治療に有用な抗体又は抗原結合断片を含む結合複合体に関する。特に、本発明は、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にある、結合複合体を提供する。前記結合複合体は、Lewisy及びLewisbを過剰発現している腫瘍細胞において細胞死を誘導する。
【背景技術】
【0002】
Lewisy及びLewisbは、乳癌、肺癌、結腸癌、及び卵巣癌によって過剰発現されている複合糖質である。したがって、それらは、モノクローナル抗体(mAb)治療の良好な標的である。Lewisyハプテンは、二型血液型オリゴ糖で発見されたジフコシル化四糖類(Fucα1−2Galβ1−4(Fucα1−3)GlcNAc)である。この抗原は、Lewisbハプテン(Fucα1−2Galβ1−3(Fucα1−4)GlNAc))の位置異性体及びLewisxハプテンのフコシル化誘導体である(Abe et al.(1986)Cancer Reseach 46:2639;Kim et al.(1986)Cancer Reseach 46:5985)。Lewisyは胸部、気管支、脾臓、秘尿生殖器、及び胃腸管の深部にある腺内において発現する。対照的に、Lewisbは、表面上皮によって発現する(Sakamoto et al.(1989)Cancer Research 49:745−52;Kitamura et al.(1994)Proc.Natl.Acad.Sci.91:12957−61)。
【0003】
IgMマウスmAbであるC14は、標準的な融合方法を用いて原発結腸直腸腫瘍細胞に対して作製し、LewisyとLewisbとの双方の(伸長又は非伸長(extended or non−extended))抗原に結合する(Brown et al.(1983)Biosci.Rep.3:163;Brown et al.(1984)Int.J.Cancer 33:727;Durrant et al.(1993)Hybridoma 12:647−60)。C14抗体は、78%の結腸直腸癌に結合したが(Durrant et al.(1989)J.Natl.Cancer Inst.81:688−95)、マウスIgMなので、in vivo試験には適さなかった。その抗体のIgGバリアントを生産するために、C14mAbでラットを免疫して、生産されたラットの抗C14を精製した。この抗血清及びC14gp200抗原でマウスに免疫した後に、それらの脾細胞をマウスのミエローマと融合させて、5種のIgG mAb、すなわち、2種のIgG3(SC101/23、SC101/29 mAb)及び3種のIgG1(SC101/33、SC101/42、及びSC101/43)(これらの5種の抗体は既に「692」抗体として公開している)を生産させた。全てのIgGバリアントは、Lewisy及びLewisb抗原を認識し、C14と同一の特異性を示した。さらに、これらの抗体は、薄層クロマトグラフィー及びELISAによって、伸長及び非伸長Lewisy及びLewisbハプテンに結合するが、Lewisx又はH血液型ハプテンには結合しないことが示された(Brown et al.(1983)Biosci.Rep.3:163)。
【0004】
LewisyとLewisbとの双方に結合する抗体は既知であるが、SC101 mAbは、Lewisy決定基とLewisb決定基との双方を認識するという能力において特異なものである。他のmAbはこれらの2つの分子の同様の面を認識せず、また、1つの珍しいレクチンのみがこれらの2つの分子の同様の面を認識する。Lewis y/bは、糖脂質としてセラミド骨格上で大部分が発現する。最近の結晶学的研究によって、Lewisyに特異的な抗体は、ハプテンのN−アセチルグルコサミン又はフコースのいずれかに適応する(Ramsland et al.(2004)J.Mol.Biol.340:809−18)非常に異なる結合部位を有することが示された。SC101は、LewisyとLewisbとが互いに立体異性体であるため非常に珍しいことに、LewisyとLewisbとの双方の面に結合部位が適応するため異なる。更に興味深いことに、これらの抗体は、おそらくセラミド又は他の密接に関連する分子からの立体障害のため、胃腸管を含む大半の通常の組織における糖脂質を認識することができない。このことにより、SC101抗体は特有の組織分布を有するが、広範な上皮腫瘍に非常に強力に結合する(Brown et al.(1983)Biosci.Rep.3:163)。
【0005】
脱凝集された原発結腸直腸腫瘍に対する抗体結合の最初の特性決定の間に、それらが細胞死を誘導することが認められた。細胞死は、アポトーシス及びオンコーシスを含む多数の機構によって生じ得る。アポトーシスはカスパーゼ依存性であり、最終的には細胞のアポトーシス小体への分割を生じさせる細胞収縮、クロマチンの凝縮及び辺縁趨向、並びに細胞膜のラッフリングを特徴とする。オンコーシスは、早期の細胞壊死であり、膜透過性の増大期の結果としての細胞膨張を特徴とする。壊死は、細胞死後に現れ、アポトーシス又はオンコーシスの後に生じる形態変化である。
【先行技術文献】
【非特許文献】
【0006】
【非特許文献1】Abe et al.(1986)Cancer Reseach 46:2639
【非特許文献2】Kim et al.(1986)Cancer Reseach 46:5985
【非特許文献3】Sakamoto et al.(1989)Cancer Research 49:745−52
【非特許文献4】Kitamura et al.(1994)Proc.Natl.Acad.Sci.91:12957−61
【非特許文献5】Brown et al.(1983)Biosci.Rep.3:163
【非特許文献6】Brown et al.(1984)Int.J.Cancer 33:727
【非特許文献7】Durrant et al.(1993)Hybridoma 12:647−60
【非特許文献8】Durrant et al.(1989)J.Natl.Cancer Inst.81:688−95
【非特許文献9】Brown et al.(1983)Biosci.Rep.3:163
【非特許文献10】Ramsland et al.(2004)J.Mol.Biol.340:809−18
【発明の概要】
【発明が解決しようとする課題】
【0007】
本発明者は、SC101/29 mAbによる腫瘍細胞の殺滅を実証し、当該抗体が、古典的なオンコーシスと多数の類似点を有する特有の機構によって、in vitroとin vivoの双方においてLewisy及びLewisbを過剰発現する腫瘍細胞を直接殺滅することを実証した。
【課題を解決するための手段】
【0008】
本発明の第一の態様では、LewisyとLewisbとの双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にある、結合複合体を提供する。
【0009】
本発明の第二の態様では、本発明の第一の態様に係る結合複合体の有効量を対象に投与する工程を含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞における細胞死の誘導を提供する。
【0010】
本発明の第三の態様では、本発明の第一の態様に係る結合複合体の治療上有効量を対象に投与する工程を含む、治療の必要がある対象の癌を治療するための方法を提供する。
【0011】
本発明の第四の態様では、本発明の第一の態様に係る結合組成物を製薬学的に許容される担体又は希釈剤と共に含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞における細胞死を誘導するための医薬組成物を提供する。
【図面の簡単な説明】
【0012】
【図1a】間接免疫蛍光によってアッセイし、フローサイトメトリーで分析した、新しく脱凝集させた結腸直腸腫瘍細胞に対するSC101の結合を示す。各々のバーは、個々の腫瘍の平均蛍光を示す。結腸直腸腫瘍細胞株であるColo205、C170、HT29、及びLoVoに対するSC101の結合を比較のために示している。
【図1b】SC101/29によるLoVo及びC170細胞のIC50増殖阻害曲線を示す。増殖阻害の%を、SC101/29mAbに曝露した細胞数/対照mAbに曝露した細胞数として表示した。増殖阻害曲線からの生細胞数を、490nmの吸光度の読み取るMTSによって決定した。
【図1c】SC101/29によるColo205及びHT−29のIC50増殖阻害曲線を示す。増殖の阻害%は、SC101/29 mAbに曝露された細胞数/対照mAbに曝露された細胞数として表示される。増殖阻害曲線からの生細胞数は、490nmにおける吸光度を読取るMTSによって決定した。
【図2a】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のColo205細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
【図2b】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のC170細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
【図2c】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のHT−29細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
【図2d】30μg/mlのIgG、300ng/mlの抗FasクローンCh11、30μg/mlのBR96、又は30μg/mlのSC101/29 mAbのいずれかを用いて30分、2時間、又は4時間処理した2×105のLoVo細胞を示す。細胞は、PIを用いて15分間に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620nm FL3)を用いて分析した。
【図2e】100μg/mlのSC101/29 mAbを用いて30分に亘って0から10μMドクソルビシンの存在下において処理した2×105のColo205、C170、HT−29、又はLoVo細胞を示す。細胞は、488nmでFC500フローサイトメーター(発光 575nm FL2)を用いて分析した。
【図2f】細胞をp−糖タンパク質(0.3から30μg/ml)を認識するmAbを用いて染色し、488nmでFC500フローサイトメーター(発光 525 FL1)を用いて分析した。
【図3a】SC101/29、SC101/43、又はそれらの双方のmAbのいずれかを用いて1時間に亘って処理した2×105のC170細胞を示す。細胞は、PIを用いて15分に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620 FL3)を用いて分析した。
【図3b】100μg/mlのIgG、アビジンと混合した100μg/mlのIgG、100μg/mlのSC101/29、100μg/mlのSC101/43−ビオチン、又はアビジンと架橋した100μg/mlのビオチン化SC101/43のいずれかを用いて1時間に亘って処理した2×105のC170細胞を示す。細胞は、PIを用いて15分に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 620 FL3)を用いて分析した。
【図3c】100μg/mlのマウスSC101/29ヒトIgG2キメラ抗体で30分間に亘って処理し、続いて架橋抗ヒトIgG抗体(0−100μg/ml)を用いて処理した1×105のColo205細胞を示す。3時間室温でインキュベートした後に、細胞を7AADを用いて20分間に亘って暗所で染色し、488nmでCell Quanta SC MPLを用いて分析した。2つのサンプルにおける死(7ADD+)細胞の平均割合を示す。
【図3d】100μg/mlのアイソタイプネガティブmAb、100ng/mlの抗Fas、又は100μg/mlのSC101/29 mAbを用いて、0、1、又は10μMのいずれかのz−FMK−vad pan−カスパーゼインヒビターの存在下において、約12時間に亘って処理した5×105のC170細胞を示す。細胞は、アネキシンV及びPIを用いて15分に亘って室温で暗所において染色し、488nmでFC500フローサイトメーター(発光 525nm FL1、620nm FL3)を用いて分析した。
【図3e】IgG(30μg/ml)ネガティブコントロールに4時間、SC101に1から4時間、又は0.5Mのソルビトールに30分間(ポジティブコントロール)曝露した2×106のC170細胞を示す。細胞は、氷冷Tris抽出干渉液+0.5mM NaVO4中で抽出し、75μgの総タンパク質をSDS−PAGEで1レーン毎に載せた。10%ゲルを130Vで90分間に亘って分離させ、BioRad semi−dry blotter(12V 定電圧)で70分間に亘ってPVDF膜に転写させた。膜をブロッキングして、2mlあたり1μlのP−p38、P−JNK、又はP−ERK(1% BSA TBS−T、ECL Plus+を用いて洗浄及び現像した)で探索した。
【図4a】ヌードマウスにおけるC170異種移植片の増殖に対するSC101/29 mAb、5−FU/ロイコボリン、又はSC101/29mAbと5−FU/ロイコボリンとの組み合わせの効果を表すグラフを示す。C170異種移植片の増殖は、動物をSC191/29 mAb ip(0.2mg)、対照抗体 ip(0.2mg)、及び5−FU/ロイコボリン(12.5mg/kg;iv)を用いて処理した際に、断面積(mm2)の測定によって、12、16、19、及び23日目に測定した。23日目に得られた結果の分散分析は、SC101/29 mAbを未処理の対照群と比較した際に、p<0.004の有意な値を示し、SC101/29 mAB+5−FU/ロイコボリンに対する対照群+5−FU/ロイコボリンについてはp<0.020の有意な値を示した。
【図4b】SC101/29 mAbのみ(第二群)、5−FU/ロイコボリンのみ(第三群)、若しくはSC101/29 mAbと5−FU/ロイコボリンとの組み合わせ(第四群)を用いて免疫されているか又は未処理(第一群)のマウスの最終腫瘍重量を示す。
【図4c】SC101/29 mAbのみ、5−FU/ロイコボリンのみ、若しくはSC101/29 mAbと5−FU/ロイコボリンとの組み合わせを用いて免疫されているか又は未処理のマウスの重量を示す。
【図4d】SC101/29 mAb ip(0.2mg)、5−FU/ロイコボリン(12.5mg/kg iv)、又はSC101/29 mAb(0.2mg)と5−FU/ロイコボリン(12.5mg/kg iv)との組み合わせを用いて処理したC170異種移植片を有する動物の生存データを示す。SC101/29 mAbを7日目に与え、次いで1週間に3回与えた。5−FU/ロイコボリンは、1、3、5、及び7日目に与えた。SC101/29 mAbは、5−FU/ロイコボリンと併用する際には、ビヒクル対照マウスを超えて生存率を有意に改善した(p=0.0163 Log Rank)。
【図5】C170HM2肝臓転移ヌードマウスモデルにおける最終肝臓腫瘍重量(g)に対する10μg若しくは100μgのSC101/29 mAb又はビヒクル対照の効果を表すグラフを示す。
【発明を実施するための形態】
【0013】
本発明者は、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体が、Lewisy及びLewisbを過剰発現している腫瘍細胞における細胞死を誘導することを実証した。
【0014】
本発明の第一の態様では、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にある、結合複合体を提供する。
【0015】
本明細書において使用する用語「抗体」は、4つのポリペプチド鎖、すなわちジスルフィド結合で相互に連結した2つの重(H)鎖及び2つの軽(L)鎖を含む。各重鎖は、重鎖可変領域(HCVR又はVH)及び重鎖定常領域を含む。重鎖定常領域は、3つのドメインとしてCH1、CH2、及びCH3を含む。各軽鎖は、軽鎖可変領域(LCVR又はVL)及び軽鎖定常領域を含む。軽鎖定常領域は、1つのドメインとしてCLを含む。VH及びVL領域は、フレームワーク領域(FR)と称されるより保存された領域が散りばめられた、相補性決定領域(CDR)と称される超可変領域に細分することが可能である。各VH及びVLは、FR1、CDR1、FR2、CDR2、FR3、CDR3、FR4の順番でアミノ末端からカルボキシ末端へと配置された3つのCDR及び4つのFRからなる。
【0016】
本明細書で使用する、抗体の「抗原結合断片」という用語は、抗原に結合する能力を示す免疫グロブリンの1つ又は複数の成分又は誘導体を示す。抗体の抗原結合機能は、全長の抗体の断片によって奏されるものであることが示されている。抗体の「抗原結合断片」という用語の範囲に含まれる結合断片の例は、(i)Fab断片、VL、VH、CL、及びCH1ドメインからなる一価断片;(ii)F(ab’)2断片、ヒンジ領域においてジスルフィド架橋によって結合した2つのFab断片を含む二価断片;(iii)VH及びCH1ドメインからなるFd断片;(iv)抗体の1つの腕のVL及びVHドメインからなるFv断片;(v)1つのVHドメインからなるdAb断片(Ward et al(1989)Nature 341:544−546)又はVLドメイン(van den Beuken et al(2001)J.Mol.Biol,310,591);(vi)単離された相補性決定領域(CDR);並びに(vii)同種のフレームワーク領域によって融合した複数の相補性決定領域、例えばQiu et al.(2007)Nature Biotechnology25(8):921−929に開示されているものを含む。さらに、Fv断片の2つのドメインであるVL及びVHは別々の遺伝子にコードされているが、それらは、VL領域とVH領域が対になって一価分子(単鎖Fv(scFV)としても知られている)を形成する単鎖タンパク質とすることを可能にする合成リンカーによって、組換え法を用いて、連結させてよい(Bird et al.(1988) Science 242:423−426;Huston et al.(1988)Proc.Natl.Acad.Sci.USA 85:5879−5883)。その様な単鎖Fvも、抗体の「抗原結合断片」という用語の範囲内含まれることが意図される。単鎖FV及び関連する分子、例えば、二重特異性抗体又は三重特異性抗体も含まれる。二重特異性抗体は、短いために同一の鎖上において2つのドメインの間で対を形成することを可能にするリンカーを使用するがVHドメインとVLドメインとが単独のポリペプチド鎖に発現され、そのドメインを他の鎖の相補的なドメインと対を形成させ、2つの抗原結合部位を生じさせている二価の抗体である(例えば、Holliger et al.(1993)Proc.Natl.Acad.Sci.USA 90:6444−6448;Poljak et al.(1994)Structure 2:1121−1123)。
【0017】
抗体又は抗原結合断片がマルチマーの状態にあることは、本発明に必須の特徴である。抗体又は抗原結合断片は、Lewisy抗原とLewisb抗原との双方に結合して、抗原の架橋を生じさせる。Lewisy抗原とLewisb抗原との架橋は、Lewisy及びLewisbを過剰発現している細胞についての腫瘍細胞死の仲介において重要であり得る。
【0018】
本明細書で使用する用語「マルチマー」は、互いに結合を形成している2つ又はそれ以上の抗体又は抗原結合断片を意味する。さらに、マルチマーは、ホモ又はヘテロマルチマーであってよい。例えば、マルチマーは、ホモ若しくはヘテロダイマー、ホモ若しくはヘテロトリマー、又はそれより高度のマルチマーであってよい。ホモマルチマーは、同一の抗体又は抗原結合断片を含むマルチマーであり、ヘテロマルチマーは、少なくとも2つの異種抗体又は抗原結合断片を含むマルチマーである。しかしながら、マルチマーの組成にかかわらず、各抗体又は抗原結合断片は、Lewisy抗原とLewisb抗原との双方に結合する必要がある。
【0019】
本発明の好ましい実施態様では、マルチマーは、ホモ又はヘテロダイマーである。
【0020】
本発明の更に好ましい実施態様では、抗体が自然にはダイマー又はマルチマーを形成しない。
【0021】
本明細書で使用する用語「Lewisy抗原とLewisb抗原との双方に結合する」は、Lewisy抗原とLewisb抗原とが架橋を形成するように、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を意味する。
【0022】
本明細書で使用する用語「結合する」は、例えば、BIAcore(商標)表面プラズモン共鳴システム及びBIAcore(商標)動力学的評価ソフト(例えば、version 2.1)を使用して表面プラズモン共鳴分析によって測定すると、1μM以下の解離定数(Kd)を有する、抗体の免疫グロブリン可変領域による抗原への結合を意味する。特異的な結合相互作用についての親和性又は解離定数(Kd)は、約500nMから約50pM、より好ましくは約500nM以下、より好ましくは約300nM以下、好ましくは少なくとも約300nMから50pM、約200nMから約50pM、より好ましくは約100nMから約50pM、約75nMから約50pM、約10nMから約50pMである。
【0023】
本発明のある実施態様では、抗体又は抗原結合断片が、二重特異性抗体又は多重特異性抗体である。本発明の他の実施態様では、二重特異性抗体又は多重特異性抗体が修飾されたFcドメインを含む。
【0024】
本発明の他の実施態様では、抗体又は抗原結合断片を含むマルチマーがIgG抗体である。本発明の更なる実施態様では、マルチマーがダイマーのIgG1抗体である。
【0025】
幾つかの抗体(例えば、IgG3)はin vitro及びin vivoでダイマーを形成し、IgG1抗体はモノマー形態で存在する。モノマーの抗体又はその抗原結合断片が、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において腫瘍細胞死を誘導し得るために、Lewisy抗原及びLewisb抗原は、モノマーの抗体又は抗原結合断片のアビジン/ビオチンによる架橋によって架橋される。代替的には、モノマーの抗体又は抗原結合断片は、組換え工学、例えば、モノマーのカルボキシ末端における鎖間ジスルフィド結合の形成を可能にする、CH3領域の遺伝子におけるセリンからシステインへの変異によって、架橋されてよい(Caron et al.(1992)J.Exp.Med.1191−1195)。これが、次いで、Lewisy抗原とLewisb抗原との架橋を容易にする。
【0026】
本発明の第二の態様では、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の有効量を対象に投与する工程を含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための方法であって、前記抗体又は抗原結合断片がマルチマーの形態にある、方法を提供する。
【0027】
本発明の第三の実施態様では、Lewisy抗原とLewisb抗原との双方に結合する本発明の第一の態様に係る結合複合体の治療上有効量を、対象に投与する工程を含む、治療の必要がある対象の癌を治療するための方法を提供する。
【0028】
用語「治療上有効量」は、特定の疾患若しくは障害(本発明の場合には癌)又はその症状の1つ若しくは複数を治療又は緩和し、及び/又は疾患又は障害の発生を予防又は低減させるために十分な、抗体又はその抗原結合断片(抗体又は抗原結合断片を含む医薬組成物を含む)の量を意味する。
【0029】
抗体又はその抗原結合断片(抗体又はその抗原結合断片を含む医薬組成物を含む)の使用及び治療方法に関して使用する際に、「治療の必要がある」個体は、癌であると診断されているか又は過去に治療されている個体であってよい。
【0030】
本発明の好ましい実施態様では、癌は、乳癌、肺癌、結腸癌、及び卵巣癌からなる群から選択されてよい。
【0031】
本発明の第四の態様では、本発明の第一の態様に係る結合複合体を製薬学的に許容される担体又は希釈剤と共に含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための医薬組成物を提供する。
【0032】
「製薬学的に許容される担体又は希釈剤」は、任意の全ての溶媒、分散媒体、コーティング、抗菌剤若しくは抗真菌剤、等張剤、及び吸収遅延剤などの生理的に適合するものを含む。製薬学的に許容される担体の例は、水、生理食塩水、リン酸緩衝生理食塩水、デキストロース、グリセロール、エタノールなど、及びそれらの組み合わせの1つ又は複数を含む。多くの場合において、等張剤、例えば、糖、ポリアルコール、例えば、マンニトール、ソルビトール、又は塩化ナトリウムを組成物に含むことが好ましいであろう。
【0033】
組成物は、液体、半固体、又は固体の剤形、例えば、溶液(例えば、注射可能な溶液及び輸液可能な溶液)、分散物又は懸濁物、錠剤、丸剤、粉末剤、リポソーム、又は座剤を含む各種の形態にあってよい。好ましくは、組成物は、免疫のための注射可能な溶液の形態にある。投与は、静脈内、皮下、腹腔内、筋肉内、経皮、鞘内、及び動脈内であってよい。好ましくは、前記剤形は、約0.001mgから約10mg/kg体重の範囲内で、毎日、毎週、二週若しくは三週に一度、又は毎月投与され、好ましくは約0.05から約5mg/kg体重で毎週投与される。
【0034】
前記組成物は、滅菌した注射可能な溶液の調製のための滅菌粉末として製剤化されてもよい。
【0035】
ある実施態様では、結合複合体が、急速な放出に対して化合物を保護する担体と共に調製されてよく、例えば、インプラント、経皮パッチ、及びマイクロカプセル化送達系を含む制御放出製剤であってよい。適合性のあるポリマーが使用されてよく、例えば、エチレンビニルアセテート、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、又はポリ乳酸が使用されてよい。
【0036】
前記組成物は、経口投与のために製剤化されてもよい。この実施態様では、抗体は、硬質ゼラチンカプセル又は軟質ゼラチンカプセルに覆われてよく、錠剤に押し固められてよく、又は対象の食料に直接含ませてもよい。
【0037】
前記組成物は、直腸投与のために製剤化されてもよい。
【0038】
本発明の結合複合体は、in vivo及びin vitroにおいて特定の細胞に結合及び同定するか、in vivoにおいて特定の細胞に結合及び破壊するか、又はin vivoにおいて特定の細胞に入り込み、破壊するために投与されてよい。
【0039】
好ましい実施態様では、前記組成物はヒトに投与される。
【0040】
本発明の本質がより明確に理解され得るように、その好ましい態様を、以下の非限定的な実施例を参照して説明する。
【実施例】
【0041】
(実施例1)
材料及び方法
細胞株
C170は、原発腫瘍由来の結腸直腸細胞である(Durrant et al.(1986)Br.J.Cancer 53:37−45)。Colo205、HT−29、及びLoVoは、ATCCから得られた結腸直腸細胞である。全ての細胞は、10%ウシ胎仔血清(FCS F6178;Sigma,Poole,UK)RPMI1640(BE12−702F;Cambrex Bio Science,Berkshire,UK)において培養した。
【0042】
モノクローナル抗体
LewisyハプテンとLewisbハプテンとの双方を認識するSC101 mAbは、過去に開示されている(Brown et al.(1983)Biosci.Rep.3:163;Druurat et al.(1993)Hybridoma 12:647−60)ように精製した。抗Fas(ヒト活性化)クローンCh11(05−201)抗体は、Upstateから得られ、Br96およびIgGネガティブコントロールはSigma(I5381)から得られた。
【0043】
結腸直腸細胞に結合する抗体
腫瘍検体は、結腸直腸癌の切除時に得た。検体を非常に細かく切って、0.05%のコラゲナーゼ(タイプIV、Boehringer Mannheim,Lewes,UK)を用いて20分間に亘って37℃で脱凝集させて、SC101/29及びヤギ抗マウスFITC(Dako Ltd,Bucks UK;1:100希釈)を用いて間接免疫蛍光によって染色した。腫瘍細胞株は一定分量(105)に分けて、SC101/29 mAb又は抗P−糖タンパク質(多剤耐性を示す、BD Pharmingen,San Diego,California USA)を各種の濃度で用いて、4℃において30分間に亘って染色した。培地(RPMI/10%FCS)で二回洗浄した後に、細胞を二次抗体(ヤギ抗マウスFITC)を用いて4℃で30分間に亘ってインキュベートし、その後に、最終的な洗浄を行い、488nmでFC500フローサイトメーター(発光525nm FL1)において分析した。
【0044】
結腸直腸腫瘍細胞のin vitroにおける殺滅
1×103の結腸直腸C170、Colo205、HT−29、及びLoVo細胞を、平底96ウェルプレートの個々のウェルに一定分量に分けて、一晩37℃で静置して接着させた。翌日、100、30、10、3、1、又は0μg/mlのSC101/29 mAb(100μl/ウェル)を用いて細胞を処理した。アイソタイプに適合したポジティブコントロール又はネガティブコントロールを比較のために使用した。ウェルを3つ重複させて使用した。細胞を抗体の存在下で37℃に5日間に亘って静置した後に、Cell Titer96 Aqueous One Solution(G3580;Promega,Southampton,UK)を各ウェルに添加して、490nmにおける吸光度を測定した。
【0045】
PI取り込み
2×105のColo205、C170、HT−29、又はLoVo細胞を、30μg/mlのIgG、300ng/mlの抗Fas、30μg/mlのBR96、又はSC101/29 mAbのいずれかを30分、2時間、又は4時間に亘って処理した。その後に、PI(630110;BD Biosciences,Oxford,UK)を用いて15分間に亘って暗所で室温において細胞を染色し、488nmでFC500 flow cytometerを用いて分析した(発光620nm FL3)。
【0046】
7AAD取り込み
1×105のColo205を、100μg/mlのマウスSC101/29−ヒトIgG2キメラ抗体とともに室温においてリン酸干渉生理食塩水(PBS)中でインキュベートした。30分後に、各種の濃度の架橋抗ヒトIgG抗体(0−100μg/ml;Sigma)を添加して、細胞を3時間に亘って室温でインキュベートし、次いで、7AAD(Bechman Coulter)を用いて20分間に亘って暗所で細胞を染色し、488nmでQuanta SC MPL(Beckman Coulter;発光フィルター670 LP FL3)を用いて分析した。
【0047】
MDR細胞株における薬剤取り込み
2×105のColo205、C170、HT−29、又はLoVo細胞を、0、1、3、10、30、又は100μg/mlのSC101/29 mAbを用いて30分間に亘って37℃で処理した。次いで、0、1、又は10μMの終濃度でドクソルビシンを前記細胞に添加し、37℃で1時間に亘ってインキュベートした。細胞を氷冷リン酸干渉生理食塩水(PBS)で五回洗浄し、488nmでFC500フローサイトメーター(発光575nm FL2)を用いて蛍光分析した。
【0048】
カスパーゼ依存性細胞死の阻害アッセイ
5×105のC170細胞を、平底滅菌24ウェルプレートの個々のウェルに一定分量に分けて、6時間に亘って37℃で接着させた。前記細胞を、100μg/mlのアイソタイプに適合したネガティブコントロールmAb、100ng/mlの抗Fas、又は100μg/mlのSC101/29 mAb、及び0、1、又は10μMのz−FMK−vad(G7232;Perbio Science,UK)を用いて処理した。細胞を37℃で一晩静置して、その後に、アネキシンV及びPIを用いて15分に亘って染色し、488nmでFC500フローサイトメーター(発光525nm FL1、620nm FL3)を用いて分析した。
【0049】
ストレス関連キナーゼを介するシグナル伝達
2×106のC170細胞を、IgG(30μg/ml)ネガティブコントロールに4時間に亘って、SC101(30μg/ml)に1〜4時間に亘って、又は0.5Mソルビトール(ポジティブコントロール)に30分間に亘って曝露した。細胞は、氷冷抽出干渉液(50mM Tris pH 7.4、2mM EDTA、2mM Na4P2O7、2mM ベンズアミジン、1mM PMSF、0.5mM NaVO4、0.5mM DTT 0.1% Triton−X−100)中で抽出して、標準的なBioRadタンパク質アッセイを用いてタンパク質濃度を測定し、75μgの総タンパク質をSDS−PAGEのレーン毎に載せた。10%ゲルを130Vで90分に亘って分離させて、BioRad semi−dry blotter(12V定電圧)で70分間に亘ってPVDFに転写した。膜を1時間に亘って1%のウシ血清アルブミン(BSA)Tris干渉生理食塩水0.05%Tween−20(TBS−T)中で1時間に亘ってブロッキングし、1% BSA TBS−T中で2mlあたり1μlのP−p38、P−JNK、又はP−ERK(Promega)で探索した。その後に、膜をTBS−T中で三回洗浄して、ヤギ抗ウサギHRP(GαR HRP;Dako,P0448)で探索し、TBS−Tで三回洗浄して、ECL Plus+で現像した。
【0050】
in vivo試験
予防モデル
結腸直腸腫瘍細胞株であるC170を、ヌードマウスにおいて連続継代に維持した。治療のために、マウスを屠殺して、腫瘍を摘出した。前記腫瘍を非常に細かく切って、3mm2片を麻酔(Hypnorm、Roche/Hypnovel、Jannsen)条件下で、無作為に4つの実験群に割り当てた40匹のオスのマウスの皮下に移植した。1つの群のマウスは、5−FU/ロイコボリン(12.5mg/kg)を用いて静脈内(iv)注射で1、3、5、7、21、及び22日目に処理した。一週間に三回、マウスに、0.2mgのSC101/29 mAbを用いる腹腔内(ip)注射も実施した。対照マウスは、SC101/29 mAb、5−FU/ロイコボリン、又はビヒクルのみのいずれかを受けた。腫瘍の大きさはキャリパーによって測定し、腫瘍断面積を12、16、19、及び23日目に計算した。実験の終わりに、腫瘍を秤量して、抗腫瘍効果を評価した。動物を秤量して、治療の毒性を評価した。
【0051】
治療モデル
結腸直腸腫瘍細胞株であるC170を、ヌードマウスにおいて連続継代に維持した。治療のために、マウスを屠殺して、腫瘍を摘出した。前記腫瘍を非常に細かく切って、3mm3片を麻酔条件下で、無作為に4つの実験群に割り当てた40匹のオスのマウスの皮下に移植した。マウスは、C170異種移植片の3mm3片を外植した。前記群のマウスを、5−FU/ロイコボリン(25mg/kg)を用いてiv注射で1日目、3日目、5日目、7日目、適用可能な場合には28日目から繰り返して治療した。一週間に三回、マウスに、0.2mgのSC101/29 mAbを用いてiv注射も実施し、対照マウスは、SC101/29 mAbのみ又は対照マウスIgG抗体を5−FU/ロイコボリンとともに受けた。
【0052】
転移モデル
C170HM2細胞を、37℃、5% CO2で加湿条件下において10%熱不活化FCSを含有するRPMI 1640培養培地中でin vitroに維持した。ほぼコンフルエントな単層から細胞を、0.025% EDTAを用いて回収し、上記の培養培地で二回洗浄し、in vivo投与のために滅菌PBS(pH 7.4)に再懸濁した。1mlの容量における1.5×106の細胞を、30匹のオスのヌードマウスの腹腔に注射した。動物をそれらの治療群に割り当てて、治療を1日目に開始して、試験に間に亘って継続した。前記群のマウスを、10μg若しくは100μgのSC101/29 mAb又はビヒクル対照を用いてiv注射によって、一日目及び次いで一週間に三回、治療した。マウスは40日目に屠殺して、体重及び腫瘍の重量を評価した。
【0053】
統計
統計分析を、PC用のMinitabプログラムのAnalysis of Variance及びLog Rankを用いて実施した。
【0054】
(実施例2)
結果
結腸直腸腫瘍細胞による抗体結合
SC101 mAbは、LewisyハプテンとLewisbハプテンとの双方に結合することが過去に示されている。新しく脱凝集させた結腸直腸腫瘍の強力な染色を、図1aに示している。mAbは、脱凝集した卵巣及び胃の腫瘍にも結合する(Brown et al.(1983)Biosci.Rep.3:163)。さらに、これらの抗体は、原発腫瘍細胞の細胞死の促進を誘導した。SC101/29抗体及びSC101/33抗体を用いた免疫組織化学及びフローサイトメトリー実験は、以下のタイプ:卵巣、胸部、肺、前立腺、及び膵臓の腫瘍細胞株及び腫瘍異種移植片に顕著に結合した(データ示さず)。
【0055】
結腸直腸腫瘍細胞のin vitroにおける殺滅
当該殺滅を調べるために、さらに広範な細胞株を、SC101/29 mAbによる結合(図1a)及び細胞増殖の阻害(図1b及び1c)についてスクリーニングした。C170及びColo205細胞株の双方が、SC101/29 mAbに結合したが、大半の原発性腫瘍が、より強力な結合を示した。対照的に、SC101/29 mAbは、HT−29及びLoVoには原発腫瘍の20%に相当するレベルで弱く結合した。特に興味深いことは、SC101/29が、C170及びColo205細胞の増殖を阻害するが、Lewisy抗原及びLewisb抗原を低レベルで発現するHT−29及びLoVo細胞の増殖を阻害し得ないことである。
【0056】
生存率試験
SC101/29 mAbによって仲介される細胞死の機構を評価するために、ヨウ化プロピジウム(PI)という、等張条件下において非生細胞にのみ侵入し得る蛍光化合物をプローブとして使用して、細胞膜の完全性を失った細胞を同定した。図2a及び2bに示すように、SC101/29 mAbは、Colo205及びC170細胞の浸透性を急速に増大させ、75%超の細胞が、早くも抗体処理の30分後にPIで強力に染色された。同一の条件下において、アポトーシス性細胞死を誘導することが既知の抗CD95抗体又は対照マウスIgG抗体を用いて4時間に亘ってインキュベートした際には、10%未満の細胞がPIを取り込んだ。同様に、Lewisbには結合せずLewisyに結合するのみのmAbであるBR96は、4時間に亘って、PIの顕著な取り込みを示し得なかった。細胞増殖の阻害と一致して、SC101/29 mAbは、低い抗原の発現を有するHT−29及びLoVo細胞におけるPI取り込みを誘導し得なかった。
【0057】
SC101/43も、膜透過性を変化する能力について分析したが、SC101/29とは反対に、PI取り込みを誘導し得なかった(図3a)。しかしながら、それは、SC101/29によって誘導されるPI取り込みの阻害において非常に効果的であり、同一又は密接に関連するエピトープに結合することを示している。SC101/43がアビジン/ビオチンを使用して架橋されている場合は、効果的に細胞死を誘導することも可能である。同様に、キメラSC101/29 IgG2抗体の架橋は、結腸腫瘍細胞の直接的な殺滅を誘導する(図3c)。SC101/29は天然にダイマーとして存在するマウスIgG3であり、これらの結果は、Lewisy及びLewisbの架橋がオンコーシスには重要であり、SC101がそのダイマー構造により架橋するが、IgG1であるSC101/43は人工的な架橋が必要であることを示す。
【0058】
MDR細胞株における薬剤取り込み
膜の完全性を失うことによって、低分子が細胞に接近することを可能にする。これは、蛍光薬剤であるドクソルビシンを用いて示されており、SC101/29 mAbに対する広範な細胞の曝露の後に、抗原を高レベルで発現し、且つ、膜の完全性の損失している細胞株においてデュロキセチンの取り込みが向上したが(Colo205及びC170)、低い抗原密度を有する細胞では向上しなかった(HT−29及びLoVo、図2c)。その効果は、p−糖タンパク質を発現する細胞(C170)で更に強力に現れ(図2f)、このことは、SC101/29によって生じた膜の混乱が当該ポンプによる薬剤の排出を阻害したことを示す。
【0059】
カスパーゼ依存性細胞死アッセイの阻害
膜の完全性の急速な損失は、膜に形成される細孔が、大きな細胞質タンパク質、例えば、LDHの放出を可能にするまで、そのサイズを大きくし続けるオンコーシスを生じさせる。PIは完全性の損失を示すが、同時に起こるC170細胞からのLDHの放出は認められなかった。LDH放出が存在しないことによってC170のオンコーシスの発現に疑問を抱いたため、同定したPI染色が、古典的なアポトーシスのアーチファクトによるものではないことを確認するために、全てのカスパーゼのインヒビターであるz−FMK−vadを使用した。要するに、抗Fasに対する曝露は、C170細胞におけるアポトーシスを十分には誘導しないことが過去に認められていたが、前記インヒビターは、前記抗体の効果を完全に逆転させ得た(図3d)。z−FMK−vadの存在下におけるSC101/29 mAbに対する腫瘍細胞の曝露は、認識可能な効果を有さず、このことは、細胞死の様式が古典的なアポトーシス経路からは独立していることを示す。
【0060】
ストレス関連キナーゼを解するシグナル伝達
膜の透過性は、HSP27をリン酸化してアクチン重合を生じさせるp38の活性化と強く関連している。弱いp38活性化が、1時間後にC170細胞において認められ、4時間後により強力な活性化が認められた(図3e)。対照的に、JNK又はERKの活性化は存在しなかった。ストレスによって活性化されるタンパク質キナーゼであるp38は、5−FUを含む広範な化学療法剤によって誘導される。したがって、SC101/29がin vivoで細胞死を誘導するかどうか及びこれが5−FUと相乗効果を奏するかどうかを決定するために、ヒト異種移植片を移植したマウスにそれを投与した。
【0061】
in vivo試験
前記抗体を、C170腫瘍の3mm2抽出物を移植したマウス又は抗体の投与前に5日間に亘って増殖させたC170腫瘍に一週間に三回投与した。前記動物は、SC101/29 mAbのみ、5−FU/ロイコボリン、又は双方の組み合わせのいずれかを用いて3週間に亘って処理した。図4aは、SC101/29と5−FU/ロイコボリンとの双方のみが、新しく外植した腫瘍の腫瘍増殖阻害を生じさせた(p<0.004 ANOVA)。対照的に、双方の組み合わせは更なる増殖阻害を示し、2/10のマウスのみが移植した腫瘍の0.3gという重量を超える任意の増殖を示した(図4b p<0.02 ANOVA)。このSC101/29 mAbの用量は全てのマウスに良好に許容され、体重減少又は任意の他のひどい症状を示さなかった(図4c)SC101/29 mAbが、C170腫瘍を発現するマウスに、25mg/kgの5−FU/ロイコボリンと組み合わせて治療目的で投与される際は、腫瘍増殖を顕著に阻害し、且つ、生存を向上させた(図4d:p<0.0163 Log Rank)。
【0062】
細胞殺滅の機構はSC101/29の微小転移細胞に対する結合が非常に効果的であるはずであるため、肝臓転移の予防におけるSC101/29 mAbの治療効果を、C170HM2結腸直腸モデルを使用して評価した。マウスは、低用量である100μg及び10μgのSC101/29 mAbを用いて一週間に三回、六週間に亘って治療した。試験の終わりに、摘出したC170HM2肝臓腫瘍の重量を測定した。各群について中央値を計算して、図5にデータをグラフ化した。各群についての中央値及び平均値は、Mann−Whitney試験によって統計学的有意性について分析した。10μg及び100μg SC101/29 mAb(第二群及び第三群)についての中央値は、ビヒクル対照(第一群)から有意(p<0.001)であることが認められた。SC101/29は当該モデルにおいて低用量であっても非常に効果的であったため、5−FUは必要でなかった。
【0063】
(実施例3)
考察
広範なLewisy抗体が同定されているが、それらは一貫して、Lewisx及びH型2構造と交差反応する。このことが、正常な組織との望ましくない交差反応を生じさせ、臨床試験において後に毒性を誘導し得る。例えば、H抗原と交差反応するマウスmAb BR55−2のフェーズI試験において、6/12の患者において血尿症が存在し、2/9の患者においてdiahoreaが存在し、3人の患者において一過性の皮膚病変の減少が存在していた(Tolcher et al.(1999)J.Clin.Oncol.17:478−84)。Lewisyのみに対するLMB−1イムノトキシン(シュードモナス属のエンドトキシンに結合したB3抗体)は、5/38の患者において反応を生じた(Pai et al.(1996)Nat.Med.2:350−3)。BR96 mAbは、B血液型と交差反応するが、Lewisbとは交差反応しない。胃腸に対する結合は用量制限的であり、抗体結合と関連することが示された(Saleh et al.(2000)J.Clin.Oncol.18:2282−92)。最後に、Lewisy特異的なヒト化抗体である3S193は、結合試験において優れた選択性を示し(Scott et al.(2000)Cancer Research 60:3254−61)、臨床試験に入っている。
【0064】
SC101抗体は、LewisyとLewisbとの双方の特有の面を認識する。SC101抗体は、Lewisy又はLewisbのいずれかを発現する広範な正常組織と強力な交差反応性を示すことを予測し得るが、他のLewis抗体とは対照的に、胃腸管内のムチンの弱い染色のみが認められた(Brown et al.(1983)Biosci.Rep.3:163)。このことは、BR96 mAbを用いて認められた胃腸管の毒性はSC101抗体を用いて回避し得ることを示す。
【0065】
SC101抗体は、免疫エフェクター細胞なしでin vitroで腫瘍細胞死を誘導し、おそらく、IgG3抗体はCDC及びADCCといった媒介因子が非常に乏しいようなin vivoでも誘導する。非常に興味深いことは、腫瘍細胞が、殺滅されるためにLewisy及びLewisbを過剰発現していることを必要としたことである。この発現レベルにおいて、80%の胃腸管及び30%の卵巣/胸部の腫瘍が、細胞の殺滅を受けやすいが、正常な組織は除外され、良好な治療方法を提供する。SC101/29 mAbが膜の完全性の急速な損失を生じさせるように、細胞の殺滅の機構は非常に興味深い。これは、浸透圧刺激及び酸素過剰細胞において認められるオンコーシスによる細胞死と類似する(Garmyn et al.(2001)J.Invest.Dermatol.117:1290−5;Moriguchi et al.(1996)J.Biol.Chem.271:26981−8;Romanshko et al.(2003)Free Radic.Biol.Med.35:978−93;Shen et al.(2002)J.Biol.Chem.277:45776−84;Tilley et al.(1996)FEBS Letters 395:133−6)。しかしながら、伝統的なオンコーシスの進行は、次いで、LDHなどの細胞質の内容物の放出を生じさせる(Chen et al.(2001)Toxicol.Appl.Pharmacol.171:1−11)。よく特徴付けられた抗Lewisy抗体であるBR96が細胞膜に対する作用を示さないので、細胞死の機構はLewisy/Lewisb構造と関連しているようである。Lewisy及びLewisbが、天然のマウスIgG3又はIgG1バリアントのアビジン/ビオチン架橋によって架橋されて、膜の混乱を生じさせることも必要とした。腫瘍細胞死などの腫脹を誘導し得る幾つかの他の抗体が開示されている。これらは、広範に発現する糖タンパク質を認識する抗ポリミン(Porimin)抗体(Ma et al.(2001)Proc.Natl.Acad.Sci.98:9778−83)、腎臓関連糖タンパク質を認識するRE2(Matsuoka(1995)J.Exp.Med.181:2007−2015)、及び新規の糖タンパク質を認識するRAV12を含む。しかしながら、これらの抗体は、特にMDR細胞株において化学療法剤の取り込みを増大させることを示さないため、SC101は独特なものである。
【0066】
細胞におけるPI取り込みは、p38活性化を伴う。p38活性化はマイクロフィラメントダイナミクスを変化させるHSP27のリン酸化と関連しているため(Deschesnes(2001)Mol.Biol.Cell 12:1569−82;Huot et al.(1998)J.Cell Biology 143:1361−73)、これによって膜の透過性が説明される。膜の完全性の損失は急速に起こるものであり、細胞死の上流にある。p38の活性化は、カスパーゼ依存性又は非依存性細胞死を誘導し得る(Deschesnes(2001)Mol.Biol.Cell 12:1569−82)。SC101/29 mAbは、DNA断片化を誘導し得ず(データは示さず)、全てのカスパーゼのインヒビターであるz−FMK−vadを用いるカスパーゼの阻害は細胞死を妨げないようであり、このことは、抗体がカスパーゼ非依存性の細胞死を誘導することを示す。結腸直腸腫瘍は、アポトーシス反応に関与する遺伝子における>70%の変異を示し、当該反応を避ける任意の治療が非常に治療的価値がある(Vogelstein(1988)N.Engl.J.Med.319:525−32;Fulda et al.(2004)Curr.Cancer Drug Targets 4:569−76)。1つのカスパーゼ非依存性の機構は、ミトコンドリアからのアポトーシス誘導因子(AIF)の放出及びその核移行、そこで未知の様式において核濃縮に寄与することを伴う(Susin et al.(2000)J.Exp.Med.192:571−80)。
【0067】
ストレス関連キナーゼであるp38は、広範な化学療法剤によっても誘導され得るものであり、薬剤合成において極めて重要な役割を担っていると提案されている(Olson & Hallahan(2004)Trend Mol Med 10:125−9)。Lewisy及びLewisbは、腫瘍糖タンパク質及び糖脂質上で発現し、これらは、細胞ストレスに反応してアップレギュレートされる。実際、化学療法ストレス(Flieger et al.(2001)Clin.Exp.Immunol.123:9−14)、特に5−FUに対するLewisyのアップレギュレートが過去に報告されている。したがって、5−FU/ロイコボリンは結腸直腸腫瘍に標準的な化学療法であるため、Lewisyをアップレギュレートし、且つ、p38を活性化し得る(Feng et al.(2002) Cancer Research 62:1920−6)。SC101/29及び5−FU/ロイコボリンの組み合わせを、ヌードマウスにおける結腸異種移植変の増殖疎外能力についてin vivoでスクリーニングした。SC101/29 mAbのみ及び5−FU/ロイコボリンとの併用において、小さい腫瘍及び樹立腫瘍の双方の増殖を顕著に阻害した。キメラバージョンのSC101/29 mAbを用いたフェーズI試験が安全であることを示したら、その後に、手術後の結腸直腸癌患者において、薬剤のみとの比較における5−FU/ロイコボリンとSC101/29 mAbとの併用の試験が開始されるであろう。当該併用は、パノレックス/5−FUは直接殺滅する活性を有さず、且つ、CDC及びADCCに依存しており、補体調節分子の過剰発現による固形癌への限定的な成功を有するため、パノレックス/5−FUよりもさらに効果的であり得る(Li et al.(2001)Br.J.Cancer 84:80−6)。
【0068】
本発明者は、SC101/29が、低分子量(<800D)の薬剤を取り込ませるが、LDHなどの大きな細胞内成分を放出させないように膜の透過性を誘導することを示した。SC101/29は、ストレス関連キナーゼであるp38のリン酸化を誘導するが、JNKのリン酸化は誘導せず、最終的にカスパーゼ非依存性細胞死を誘導する。セラミドがp38の活性化及びAIFを含む多数のミトコンドリアタンパク質の放出によって神経細胞死を誘導することが示されているため、このことは特に興味深い(Ghatan et al.(2000)J.Cell.Biol.150:335−47;Stoica et al.(2005)Mol.Cell.Neurosci.29:335−71)。Lewisy/Lewisbは糖脂質上で主に発現するため、その分解は、セラミドの増加及びその後のp38活性化を生じさせ得る。SC101/29は古典的な経路によって細胞死を誘導しないため、古典的なアポトーシス又はオンコーシスに対する耐性機構を発達させている腫瘍細胞において特に効果的であり得る。しかしながら、その経路は、より注意深く解明し、本発明者による殺滅アッセイの更なる検証によって、潜在的な耐性機構の評価及び最適な腫瘍標的の同定をすることが必要である。
【0069】
結論として、SC101/29は、Lewisy及びLewisbを認識し、膜の完全性の損失を誘導することによって細胞死を直接誘導する新規mAbである。本願は、オンコーシスの様式において、腫瘍細胞を選択的に殺滅し得る血液型抗原を認識する抗体を初めて開示するものである。治療における相乗的な反応におけるp38の役割は、次第に明らかになっている。SC101/29は、5−FU/ロイコボリンとの組み合わせにおいて殺滅の促進を示し、これらの薬剤と組み合わせて使用して、毒性を低減し、且つ、癌治療の効果を増大させる。
【0070】
本明細書を通じて、「含む」なる用語は、記載した要素、数字、若しくは工程、又は要素、数字、若しくは工程の群の包含を示すものであり、任意の他の要素、数字、若しくは工程、又は要素、数字、若しくは工程の群の排除を意味するものではないと解されるであろう。
【0071】
本明細書で挙げた全ての文献は、参照によって本明細書に取り込む。本明細書に含まれる文献、行為、材料、装置、又は論文などの任意の議論は、本明細書の内容を提供するためだけのものである。これらが従来技術の基礎の一部を形成し、又は本願の各請求項の優先日前に豪州又は他の地域に存在した本発明に関連する分野の共通する一般的知識であったと認めるものとして解釈されるべきではない。
【0072】
広範に記載したように本発明の精神又は範囲を逸脱すること無く、特定の実施態様に示したように、本発明の多数の変形例及び/又は修飾例を作製し得ることが当業者によって理解されるであろう。本実施態様は、したがって、全ての態様において説明するものであり、限定するものではないと解されるべきである。
【特許請求の範囲】
【請求項1】
LewisyとLewisbとの双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、結合複合体。
【請求項2】
前記マルチマーがホモ又はヘテロマルチマーである、請求項1に記載の結合複合体。
【請求項3】
前記マルチマーがホモ又はヘテロダイマーである、請求項1又は2に記載の結合複合体。
【請求項4】
前記マルチマーがIgG抗体を含む、請求項1から3のいずれか一項に記載の結合複合体。
【請求項5】
前記マルチマーがヘテロダイマーのIgG2/IgG3抗体である、請求項1から4のいずれか一項に記載の結合複合体。
【請求項6】
前記マルチマーが、同一の種に由来しない抗体又は抗原結合断片を含む、請求項1から5のいずれか一項に記載の結合複合体。
【請求項7】
前記マルチマーがダイマーのIgG抗体である、請求項1から4のいずれか一項に記載の結合複合体。
【請求項8】
前記抗原結合断片が、多重特異性抗体、ドメイン抗体、Fv断片、Fd断片、Fab断片、F(ab’)2断片、相補性決定領域(CDR)、及び同種のフレームワーク領域によって融合した複数のCDRからなる群から選択される、請求項1から7のいずれか一項に記載の結合複合体。
【請求項9】
前記多重特異性抗体が二重特異性抗体又は三重特異性抗体である、請求項8に記載の結合複合体。
【請求項10】
前記多重特異性抗体が二重特異性抗体である、請求項9に記載の結合複合体。
【請求項11】
前記多重特異性抗体が修飾Fcドメインをさらに含む、請求項8から10のいずれか一項に記載の結合複合体。
【請求項12】
Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための方法であって、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の有効量を対象に投与する工程を含み、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、方法。
【請求項13】
Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量を対象に投与する工程を含む、対象における癌を治療するための方法であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、方法。
【請求項14】
前記癌が、乳癌、肺癌、結腸癌、卵巣癌、前立腺癌、及び膵臓癌からなる群から選択される、請求項13に記載の方法。
【請求項15】
前記結合複合体が、対象に投与するための医薬組成物として製剤化されている、請求項12から14のいずれか一項に記載の方法。
【請求項16】
前記結合複合体が注射可能な溶液として投与される、請求項12から15のいずれか一項に記載の方法。
【請求項17】
前記結合複合体が静脈内に投与される、請求項12から16のいずれか一項に記載の方法。
【請求項18】
前記結合複合体がヒトに投与される、請求項12から17のいずれか一項に記載の方法。
【請求項19】
Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量を製薬学的に許容される担体と共に含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための医薬組成物であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、医薬組成物。
【請求項20】
Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための医薬の製造における、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量の使用であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、使用。
【請求項21】
対象において癌を治療するための医薬の製造における、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量の使用であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、使用。
【請求項1】
LewisyとLewisbとの双方に結合する抗体又は抗原結合断片を含む結合複合体であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、結合複合体。
【請求項2】
前記マルチマーがホモ又はヘテロマルチマーである、請求項1に記載の結合複合体。
【請求項3】
前記マルチマーがホモ又はヘテロダイマーである、請求項1又は2に記載の結合複合体。
【請求項4】
前記マルチマーがIgG抗体を含む、請求項1から3のいずれか一項に記載の結合複合体。
【請求項5】
前記マルチマーがヘテロダイマーのIgG2/IgG3抗体である、請求項1から4のいずれか一項に記載の結合複合体。
【請求項6】
前記マルチマーが、同一の種に由来しない抗体又は抗原結合断片を含む、請求項1から5のいずれか一項に記載の結合複合体。
【請求項7】
前記マルチマーがダイマーのIgG抗体である、請求項1から4のいずれか一項に記載の結合複合体。
【請求項8】
前記抗原結合断片が、多重特異性抗体、ドメイン抗体、Fv断片、Fd断片、Fab断片、F(ab’)2断片、相補性決定領域(CDR)、及び同種のフレームワーク領域によって融合した複数のCDRからなる群から選択される、請求項1から7のいずれか一項に記載の結合複合体。
【請求項9】
前記多重特異性抗体が二重特異性抗体又は三重特異性抗体である、請求項8に記載の結合複合体。
【請求項10】
前記多重特異性抗体が二重特異性抗体である、請求項9に記載の結合複合体。
【請求項11】
前記多重特異性抗体が修飾Fcドメインをさらに含む、請求項8から10のいずれか一項に記載の結合複合体。
【請求項12】
Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための方法であって、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の有効量を対象に投与する工程を含み、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、方法。
【請求項13】
Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量を対象に投与する工程を含む、対象における癌を治療するための方法であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、方法。
【請求項14】
前記癌が、乳癌、肺癌、結腸癌、卵巣癌、前立腺癌、及び膵臓癌からなる群から選択される、請求項13に記載の方法。
【請求項15】
前記結合複合体が、対象に投与するための医薬組成物として製剤化されている、請求項12から14のいずれか一項に記載の方法。
【請求項16】
前記結合複合体が注射可能な溶液として投与される、請求項12から15のいずれか一項に記載の方法。
【請求項17】
前記結合複合体が静脈内に投与される、請求項12から16のいずれか一項に記載の方法。
【請求項18】
前記結合複合体がヒトに投与される、請求項12から17のいずれか一項に記載の方法。
【請求項19】
Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量を製薬学的に許容される担体と共に含む、Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための医薬組成物であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、医薬組成物。
【請求項20】
Lewisy抗原及びLewisb抗原を過剰発現している腫瘍細胞において細胞死を誘導するための医薬の製造における、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量の使用であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、使用。
【請求項21】
対象において癌を治療するための医薬の製造における、Lewisy抗原とLewisb抗原との双方に結合する抗体又は抗原結合断片を含む結合複合体の治療上有効量の使用であって、前記抗体又は抗原結合断片がマルチマーの形態にあり、且つ、前記抗体又は抗原結合断片が自然にはマルチマーを形成しない、使用。
【図1a】


【図1b】
【図1c】
【図2a】
【図2b】
【図2c】
【図2d】
【図2e】
【図2f】
【図3a】
【図3b】
【図4a】
【図4b】
【図4c】
【図4d】
【図5】
【図3c】
【図3d】
【図3e】
【図1b】
【図1c】
【図2a】
【図2b】
【図2c】
【図2d】
【図2e】
【図2f】
【図3a】
【図3b】
【図4a】
【図4b】
【図4c】
【図4d】
【図5】
【図3c】
【図3d】
【図3e】
【公表番号】特表2010−504289(P2010−504289A)
【公表日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願番号】特願2009−528552(P2009−528552)
【出願日】平成19年9月20日(2007.9.20)
【国際出願番号】PCT/AU2007/001386
【国際公開番号】WO2008/034181
【国際公開日】平成20年3月27日(2008.3.27)
【出願人】(500468537)アラーナ・テラピューティクス・リミテッド (16)
【Fターム(参考)】
【公表日】平成22年2月12日(2010.2.12)
【国際特許分類】
【出願日】平成19年9月20日(2007.9.20)
【国際出願番号】PCT/AU2007/001386
【国際公開番号】WO2008/034181
【国際公開日】平成20年3月27日(2008.3.27)
【出願人】(500468537)アラーナ・テラピューティクス・リミテッド (16)
【Fターム(参考)】
[ Back to top ]