
LIFおよびBMPによる腫瘍性幹細胞の潜在的腫瘍形成能力の抑制
本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を、治療または予防するための方法および組成物、例えば腫瘍を治療する方法を含む。該方法は、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬を含有する医薬組成物の使用を含む。本開示はまた、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤を含み、また、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤を同定する方法を含む。本開示はさらに、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬を含有する医薬組成物を含む。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療する医薬の製造におけるBMP調製物および方法に関する。
【背景技術】
【0002】
発明の背景
本明細書におけるいかなる先行技術の引用も、その先行技術があらゆる国に共通の一般知識の一部を成すと認識するものでも何らかの形で示唆するものでもなく、またそのような認識や示唆とみなされるべきものでもない。本明細書に引用された参照文献はすべて、引用によってその全体が本願に明確に組み込まれる。
【0003】
神経幹細胞
従来、幹細胞は、分化細胞が最も失われやすく、かつ頻繁に置き換わる必要のある組織、たとえば皮膚(非特許文献1)、腸管上皮(非特許文献2)および血液(非特許文献3)にのみあると考えられていた。確かに、成体幹細胞の最もよく知られている例は造血幹細胞(HSC)であり、造血幹細胞は骨髄に見出され、動物の一生を通じてすべての種類の血液細胞の生成に根本的に関与している(非特許文献3〜5)。成人の中枢神経系(CNS)では大量のニューロンが死ぬことはなく、かつ再生能はないと考えられたので、神経幹細胞は存在しそうもなく、かつ不必要に思われた。しかしながら、1992年に2つの独立したグループが、新しいニューロンを生じさせる能力を持った成体哺乳類CNS内の前駆細胞の存在を実証することに成功した(非特許文献6および7)。
【0004】
新しいニューロンの供給源は、成体哺乳類CNSの脳室の神経軸全体の内側を覆っている幹細胞であると同定された(非特許文献6)。他の組織で見つかった幹細胞のように、CNSの幹細胞(あるいは神経幹細胞(NSC))は、増殖、大規模な自己複製、多数の子孫細胞の生成、多系列分化能というインビトロにおける決定的な幹細胞の特性(非特許文献8および非特許文献2)、ならびに損傷後の組織再生というインビボにおける特性を示すことが明らかになった。
【0005】
幹細胞の役割のうちの1つは、分裂することと、多くの未分化細胞を増殖および生成する能力を備えた、より分化決定の進んだ前駆細胞を供給することである。究極的には、分化した子孫細胞を生じるのは前記のより分化決定の進んだ前駆細胞種の子孫細胞である。したがって幹細胞は、動物の寿命全体を通じて分裂する能力を備え、従って広範囲に及ぶ増殖能を備えた分化決定されていない細胞の、比較的活動性の低い貯蔵所と見なすことができる。一方、前駆細胞はより分化決定が進んでおり、より頻繁に分裂するが、時間とともに増殖能は制限される。発生中および成体のいずれにおいても、幹細胞および前駆細胞の増殖は細胞発生の基礎となる。
【0006】
特定の形態学的特徴、分子的特徴、あるいは抗原性の特徴を欠いているため、幹細胞は機能上の基準に基づいて同定される。従って、幹細胞の制御についてインビトロで検討するためには、幹細胞の分裂を引き起こす組織培養法を開発しなければならない。しかしながらそのような方法はほとんどなく、神経系ではニューロスフェア法(Neurosphere Assay)(NA)(非特許文献6)と呼ばれる培養法が、NSCをインビトロで同定し、増殖させ、かつ計数するために一般に使用される。簡潔に述べると、NAは、胚から成人までのCNS組織を顕微解剖した後に、細胞間の接着を破壊し、単個細胞の浮遊液を生成させることを含む。細胞を、組織培養器において規定の無血清培地中で、少なくとも1つの増殖誘導性の成長因子(すなわち上皮成長因子[EGF]、塩基性繊維芽
細胞成長因子[bFGF]など)の存在下で(典型的には低密度で)播種する。この条件下では、2〜5日以内に多能性NSCは分裂し、ニューロスフェアと呼ばれるクローン由来の未分化細胞集合体を生じる。増殖を誘導する因子が持続的に存在する状態では、ニューロスフェア中の細胞は分裂を続け、その結果ニューロスフェアを構成する細胞の数が増大し、従ってニューロスフェアが大きくなる。ニューロスフェアを回収し、破壊して単個細胞浮遊液とし、その細胞を再度培養液中に播種して新しいニューロスフェアを生成することができる。このようにしてNSCを継代すると、成育可能なCNS前駆細胞が算術的に増加する。NA法により規定の条件でNSCを単離および増殖させることが可能となり、この推定上の幹細胞の挙動を様々な実験条件下で研究することが可能である。NAは哺乳類NSCを単離する標準的な方法となっており、神経系における幹細胞の細胞分子生物学を理解するために用いられる多くの分析法の中核を成している。
【0007】
腫瘍の増大および増殖に寄与する幹細胞の特徴を備えた細胞の小集団から腫瘍が発生するという概念は、がん生物学の分野において目新しいことではない。この概念は1970年代の初めに提案され、存在頻度の低い腫瘍起源細胞が正常な造血幹細胞(HSC)に似ていることが実証された急性骨髄性白血病(AML)に関する研究で、実験的に確認された。これらの研究は、白血病幹細胞がHSCの直接の子孫であるか、あるいはHSCの特徴を獲得したより分化の進んだ細胞の生産物であることを示唆した。血液系以外の幹細胞の発見から、固形組織のがんにも幹細胞様の細胞が含まれている可能性が高まった。固形腫瘍中の腫瘍の起源となる幹細胞様の細胞の存在および単離は、ヒト乳がん組織において最初に実証され、その手法はCNSの腫瘍にも適用されている。
【0008】
最近、いくつかのグループから、ヒト神経膠腫組織由来の細胞が培養中にニューロスフェア状の細胞を生成する能力について報告され、CNS腫瘍内のNSCの存在が示唆されている。興味深いことに、蛍光活性化細胞選別法(FACS)による「副集団(side−population)」細胞の単離に基づいて、確立されたC6神経膠腫細胞株に、生体内での悪性度を保持するニューロスフェア形成性の細胞の小集団が含まれていることが実証された。ガルリおよび共同研究者(非特許文献9)は、胚CNSおよび成体CNS由来のNSCとほぼ同一の機能特性を示す、ヒト多形膠芽腫(GBM)由来の腫瘍神経幹細胞(tNSC)の単離、増殖および累代移植について報告した。これらのGBM由来tNSCは、インビトロで重要な神経幹細胞の特徴を示す、プロミニン陽性の前駆細胞であり、安定した方法で増殖可能であり、かつ、累代移植−培養サイクル全体を通じて元の腫瘍誘発性を再現する。同時にこれらの研究は、CNSの腫瘍が腫瘍の発生および悪性度に関与する可能性のある幹細胞集団を含んでいる、という仮説を強く支持するものである。tNSCは、CD133のtNSC上での発現を利用したFACSを用いて、他のGBM細胞から選別することができる(非特許文献10)。
【0009】
GBMは、平均生存時間9〜12か月の、最も一般的な成人の悪性脳腫瘍である。大多数の患者は診断から2年までに死亡する。本質的に治癒することはなく、管理療法は一般に手術、放射線療法および化学療法の組合せに基づくものである。生存率は、遺伝子療法、抗血管形成療法、免疫療法および低分子伝達阻害剤のような新しい治療法の活発な探究が促進されてきた30年以上の間、ほとんど変わらなかった。
【0010】
LIF
白血病抑制因子(LIF)は、その誘導産物が多数の組織、恐らくはすべての組織に生じる可能性がある、多機能性の糖タンパク質サイトカインである。LIFはコリン作動性分化因子(CDF)と呼ばれることもある。LIFは、低親和性のLIF特異的レセプタ(LIFR)と、インターロイキン6、オンコスタチンM、カルジオトロフィン−1および毛様体神経栄養因子のレセプタとしても使用されるgpl30レセプタ鎖とで構成されたヘテロダイマーの細胞膜レセプタに結合することにより、反応性細胞に対して作用する
。LIFは、胚盤胞移植、ならびに海馬および嗅覚レセプタのニューロンの正常な発生に不可欠である。LIFは、胚性幹細胞の分化全能性を保持させるという重要な能力から、実験生物学において広範囲に使用されている。LIFは、血小板形成の刺激剤としての作用、いくつかの造血細胞の増殖、骨形成、脂肪細胞の脂質輸送、副腎皮質刺激ホルモン生産、ニューロンの生存および形成、筋衛星細胞の増殖、ならびに肝細胞による急性期産生などの多くの作用を有している(非特許文献11)。
【0011】
BMP
骨形成タンパク質(BMP)は、TGFhスーパーファミリーのメンバーである(非特許文献12)。20を超えるメンバーが知られており、その配列ホモロジーに従ってサブグループに分けることができる(非特許文献12および13)。BMPは胚発生の間に重大な役割を果たす。例えば、BMPは原腸形成、神経新生、アポトーシスおよび造血に影響を及ぼす(総説として非特許文献14を参照のこと)。BMPレセプタは以降BMPRと称する。ヒト由来のBMPRにはBMPR1a、BMPR1bおよびBMPR2が含まれる。
【0012】
本開示のとおり、LIFおよびBMPが、前駆細胞および幹細胞の生存、自己複製、増殖および/または分化を制御し、特にがん組織中の増殖細胞の数を減少させうることがここで解明された。
【非特許文献1】ヒュールスケン(Huelsken)ら、Cell 第105巻、p.533−45、2001年
【非特許文献2】ポッテン(Potten)ら、Development 第110巻、p.1001−20、1990年
【非特許文献3】モリソン(Morrison)ら、Annu Rev Cell Dev Biol 第11巻、p.35−71、1995年
【非特許文献4】ワイスマン(Weissman)、Cell 第100巻、p.157−68、2000年
【非特許文献5】ワイスマン(Weissman)、Science 第287巻、p.1442−6、2000年
【非特許文献6】レノルズ(Reynolds)およびワイス(Weiss)、Science 第255巻、p.1707−10、1992年
【非特許文献7】リチャーズ(Richards)ら、Proc Natl Acad Sci USA 第89巻、p.8591−5、1992年
【非特許文献8】ホール(Hall)ら、Development 第106巻、p.619−33、1989年
【非特許文献9】ガルリ(Galli)ら、Cancer Research 第64巻、p.7011−7021、2004年
【非特許文献10】シン(Singh)ら、Nature 第432巻、p.396−401、2004年
【非特許文献11】総説としてメタカルフ(Metacalf)、Stem Cells 第21巻、p.5−14、2003年
【非特許文献12】フッドレス(Hoodless)ら、Cell 第85巻、p.489−500、1996年
【非特許文献13】ウォズニー(Wozney)ら、J Cell Sci、追補13、p.149−156、1990年
【非特許文献14】ノーエ(Nohe)ら、Cellular Signalling 第16巻、p.291−299、2004年(発明の開示) 1つの態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を、治療または予防するための方法を提供する。該方法は、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に、治療上有効な量の白血病抑制因子(LIF)調製物および少なくとも1つの骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を投与することを含む。
【0013】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、治療上有効な量の白血病抑制因子(LIF)調製物および骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を前記腫瘍に投与することを含む方法を提供する。ヒト患者にBMP−4調製物を投与することにより、脳腫瘍(例えば多形膠芽腫)などのヒト患者の腫瘍の成長を低減する方法が含まれる。
【0014】
さらなる態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、該腫瘍をLIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0015】
別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞中で、LIFレセプタ(LIFR)を介したシグナル伝達またはBMPレセプタ(BMPR)を介したシグナル伝達をそれぞれ増大させることができる、LIF調製物およびBMP調製物を提供する。
【0016】
別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFレセプタ(LIFR)を介したシグナル伝達を増大させることができる、本明細書では以降「LIFRシグナル伝達活性化剤」および「LIFレセプタシグナル伝達活性化剤」と称する作用薬を提供する。
【0017】
別の態様では、本開示は、腫瘍におけるLIFRまたはBMPRを介したシグナル伝達を増大させることにより、前記腫瘍の成長を低減する方法を提供する。LIFRを介したシグナル伝達は、例えば、LIF調製物およびLIFRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能であり、BMPRを介したシグナル伝達は、例えば、BMP調製物およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能である。
【0018】
別の態様では、本開示は、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤を同定するための方法を提供する。
別の態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を、治療または予防するための方法を提供する。該方法は、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に、治療上有効な量の、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を投与することを含む。
【0019】
別の態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、該腫瘍を、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0020】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、前記腫瘍に治療上有効な量のLIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を投与することを含む方法を提供する。
【0021】
別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬と接触させることを含む方法を提供する。
【0022】
別の態様では、本開示は、連続的に継代された神経幹細胞中の神経幹細胞の存在頻度および神経前駆細胞の存在頻度を低減する方法であって、神経幹細胞をLIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0023】
別の態様では、本開示は、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬を含有する医薬組成物を提供する。
【0024】
さらなる態様では、腫瘍の治療のための医薬の製造におけるBMPまたはLIF調製物の使用方法、たとえば脳腫瘍(例えば多形膠芽腫)の治療的処置および予防的処置のうち少なくともいずれか一方のための医薬の製造におけるBMP−4調製物の使用方法が開示される。
【0025】
さらなる態様では、本開示は、腫瘍性幹細胞を含む腫瘍を治療するための方法であって、腫瘍性幹細胞を、腫瘍性幹細胞の分化を誘導する作用薬と接触させることを含む方法を提供する。適切な分化作用薬には、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤が含まれる。例えば、多形膠芽腫は、開示の方法に従って、腫瘍中の腫瘍性神経幹細胞(または、腫瘍の減量手術後の切除腔内残留物)を、腫瘍性神経幹細胞の分化を誘導するのに十分な量のBMP−4調製物と接触させることにより、治療可能である。
【発明を実施するための最良の形態】
【0026】
本開示について詳細に説明する前に、当然のことであるが、別途定めのないかぎり本開示内容は、変更可能なものである処方成分、製造法、投与計画などについて特定のものに限定されない。同様に当然のことであるが、本明細書中で使用される用語は、単に特別な実施形態を説明するためのものであり、限定を意図するものではない。
【0027】
本明細書中で使用されるように、単数形の名詞は、文脈上そうでないことが明らかでないかぎり、複数形も含んでいることに留意すべきである。したがって、例えば、「作用薬」という場合には、単一の作用薬だけでなく2以上の作用薬を含んでおり;「幹細胞」という場合には、単一の幹細胞だけでなく2以上の幹細胞を含んでいる;などである。
【0028】
本明細書中で使用されるように、「治療上有効な量」とは、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を、治療または管理するのに十分な治療薬の量、および好ましくは、原発がん組織、局所がん組織または転移がん組織を破壊、改変、制御、または除去するのに十分な量を指す。治療上有効な量は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の発症を、遅延させるかまたは最小限にする、例えば、がんの蔓延または腫瘍の成長を遅延させるかまたは最小限にするのに十分な治療薬の量を指す場合もある。また治療上有効な量は、腫瘍またはがんの治療または管理において治療上の利点を提供する治療薬の量を指す場合もある。さらに、本開示の治療薬に関しての治療上有効な量とは、細胞過剰増殖性の疾病またはがんの治療または管理において治療上の利点を提供する、単独での、または他の療法との併用における治療薬の量を意味する。該用語は、治療法全体を改善する量、有害反応を低減もしくは回避する量、または別の治療薬の治療効果もしくは別の治療薬との相乗作用を増強する量を包含しうる。
【0029】
用語「作用薬」、「化合物」、「活性作用薬」、「活性化合物」、「治療薬」、「薬理学的に活性な作用薬」、「医薬」、「活性体」および「薬物」は、所望の薬理学的かつ/または生理的な効果を引き起こす物質を指すために本明細書において互換的に使用される
。該用語は、本明細書に具体的に記載された活性作用薬の、医薬として許容可能かつ薬理学的に活性な有効成分、例えば限定するものではないが塩、エステル、アミド、プロドラッグ、活性代謝物、アナログおよびその他同種のものも包含する。用語「作用薬」、「化合物」、「活性作用薬」、「薬理学的に活性な作用薬」、「医薬」、「活性体」および「薬物」が用いられる場合、当然ながら同用語は、活性作用薬それ自身だけでなく、医薬として許容可能で、薬理学的に活性な塩、エステル、アミド、プロドラッグ、代謝産物、アナログなどを包含する。本開示の作用薬は、ペプチド、ポリペプチド、およびタンパク質のような任意のタンパク質性分子であってもよいし、核酸分子のような非タンパク質分子であってもよく、天然または合成的に誘導された有機および無機の低分子ないし高分子であってよい。該作用薬は一般には血液脳関門を横断することが可能であり、あるいはCNSへの直接投与に好適な場合もある。
【0030】
本明細書において「治療」という場合、存在している疾病または症状の重症度の低減を意味する場合がある。用語「治療」は、疾病または症状の発症を防止する「予防的処置」を包含するとも解釈される。用語「治療」は、対象が完全に回復するまで治療されることを必ずしも示唆するものではない。同様に、「予防的処置」は、対象が最終的に疾病または症状を発症しないということを必ずしも意味するものではない。
【0031】
本明細書において使用される場合、「幹細胞」は、(a)増殖が可能、(b)長期間にわたる自己複製が可能で、(c)多くの子孫を生成することができ、かつ(d)その幹細胞が得られた組織のすべての種類の細胞を生じさせる能力を有する、未分化細胞を指す。
【0032】
本明細書において使用されるように、「腫瘍性幹細胞」は腫瘍から得られた幹細胞である。腫瘍性幹細胞は、(a)増殖が可能、(b)長期間にわたる自己複製が可能で、(c)多くの子孫を生成することができ、かつ(d)その腫瘍性幹細胞が得られた腫瘍のすべての種類の細胞を生じさせる能力を有する。本明細書において「tNSC」とも呼ばれる「腫瘍性神経幹細胞」は、CNSの腫瘍から得られた腫瘍性幹細胞を指す。
【0033】
本明細書において使用される場合、「前駆細胞」は、(a)増殖が可能で、(b)限定的な自己複製能力を有し、(c)限られた数の子孫を生成することができ、かつ(d)少なくとも1種類の子孫を生じさせる能力を有する、未分化細胞を指す。
【0034】
本明細書において使用されるように、「腫瘍前駆細胞」は腫瘍から得られた前駆細胞である。腫瘍前駆細胞は、(a)増殖が可能で、(b)限定的な自己複製能力を有し、(c)限られた数の子孫を生成することができ、かつ(d)その腫瘍前駆細胞が得られた腫瘍に見出される少なくとも1種類の細胞を生じさせる能力を有する。
【0035】
LIF調製物およびBMP調製物
1つの態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の治療または予防のための方法を提供する。該方法は、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に、治療上有効な量の白血病抑制因子(LIF)調製物および少なくとも1つの骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を投与することを含む。
【0036】
好ましくは、過度の増殖を特徴とする障害は良性腫瘍または悪性腫瘍(がん)である。例えば、腫瘍は、脳腫瘍、例えば限定するものではないが聴神経腫、腺腫、星状細胞腫、若年性毛様細胞性星状細胞腫、脳幹部神経膠腫、脊索腫、脈絡叢、頭蓋咽頭腫、脳室上衣腫、神経節膠腫、神経節膠腫神経細胞、多形膠芽腫(GBM)、神経膠腫、リンパ腫、髄芽細胞腫、髄膜腫、乏突起膠腫、視神経膠腫、下垂体部腫瘍、松果体部腫瘍、または松果体芽腫であってよい。好ましい実施形態では、脳腫瘍はGBMである。
【0037】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、前記腫瘍に、治療上有効な量の白血病抑制因子(LIF)調製物および少なくとも1つの骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を投与することを含む方法を提供する。いくつかの実施形態では、治療上有効な量のBMP−4調製物が、GBMの成長を低減するためにヒト患者のGBMに投与される。
【0038】
さらなる態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、該腫瘍を、LIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。理論または仮説によって限定されるものではないが、腫瘍へと投与された時、LIF調製物およびBMP調製物はLIFRまたはBMPRを介したシグナル伝達の増大をもたらし、その結果次の腫瘍性幹細胞または腫瘍前駆細胞の特性、例えば限定するものではないが細胞の生存、自己複製、対称分裂、増殖、および/または分化についての特性のうちのいずれか1つ以上を調節するものと考えられる。理論または仮説によって限定されるものではないが、特に、LIFRまたはBMPRを介したシグナル伝達が増大する結果、幹細胞および前駆細胞の増殖特性が低下し、特に増殖している幹細胞または前駆細胞による対称分裂の可能性が低下することによって該細胞の数が低下すると考えられる。従って、別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0039】
別の態様では、本開示は、腫瘍におけるLIFRまたはBMPRを介したシグナル伝達を増大させることにより、前記腫瘍の成長を低減する方法を提供する。LIFRを介したシグナル伝達は、例えば、LIF調製物およびLIFRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能であり(後述)、BMPRを介したシグナル伝達は、例えば、BMP調製物およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能である(後述)。
【0040】
従来の治療法を使用して腫瘍形成性の細胞を根絶することを目指した現在の治療は、細胞周期が急速に回転している細胞の除去を意図したものである。例えば、従来の化学療法薬は、分裂している細胞に対して最も有効性が高い。tNSCは、対応するその非形質転換体と同じように細胞周期の回転が低頻度であるため、治療の毒性作用を免れ、治療後に腫瘍の増大を容易に再開しうる。成体幹細胞が本質的に寿命が長いこと、また本来薬剤抵抗性遺伝子および抗アポトーシス遺伝子を発現する能力を有することは、該細胞に対応する悪性の細胞においても見出される場合があり、腫瘍性幹細胞の根絶を目指した有効な治療戦略を開発する際の困難の度を増している。本明細書に開示された方法および組成物は、tNSC細胞を標的とすることにより上記の困難を克服する。理論または仮説によって制限を受けるものではないが、本明細書に開示の方法および組成物は、tNSCに対する分化促進効果を有し(実施例に示すように、神経の分化マーカー、特に星状膠細胞の抗原のアップレギュレーションによって証明された)、したがって細胞生存率への影響やアポトーシス誘発を伴わずに幹細胞群を永続的に縮小させると考えられる。その結果(以下の実施例に示すように)、本開示の組成物(特にGBM治療用のBMP−4組成物)への一時的な曝露でも、tNSCの腫瘍形成能が不可逆的に抑制される。
【0041】
腫瘍細胞を死滅させようとするのではなく分化を誘導することは、がん治療への全く新しいアプローチである。したがって、別の態様では、本開示は、腫瘍性幹細胞を含む腫瘍を治療する方法であって、腫瘍性幹細胞を、腫瘍性幹細胞の分化を誘導する作用薬(BMP調製物など)と接触させることを含む方法を提供する。1つのそのような実施形態では、多形膠芽腫のような脳腫瘍中の腫瘍性神経幹細胞を、該細胞の分化を誘導するためにB
MP−4と接触させる。
【0042】
用語「LIF調製物」および「BMP調製物」には、LIFポリペプチドまたはBMPポリペプチドであって、天然に、好ましくはヒトにおいて産生された、任意の翻訳後修飾を有するものまたは翻訳後修飾されていないものが含まれる。この用語は、ヒトLIFの場合、GenBank受入番号NM_002309のmRNAによってコードされるポリペプチドを含んでいる。ヒトBMPの場合、この用語は、ヒト遺伝子BMP1、BMP2、BMP3、BMP4、BMP5、BMP6、BMP7、BMP8A、BMP8B、GDF10(BMP−3b)、GDF11(BMP11)、GDF2(BMP9)、BMP10、BMP15によってコードされるBMPポリペプチド、およびGenBank受入番号NM_001719(BMP7);NM_001201(BMP3);NM_001200(BMP2);NM_005448(BMP15);NM_001720(BMP8B);NM_014482(BMP10);NM_006132(BMP1−4);NM_006131(BMP1−5);NM_006130(BMP1−6);NM_006129(BMP1−3):NM_006128(BMP1−2):NM_001718(BMP6);NM_001199(BMP1−1);NM_130851(BMP4−3);NM_130850(BMP4−2);NM_001202(BMP4−1);NM_181809(BMP8A);NM_021073(BMP5)のmRNAによってコードされるポリペプチドを含んでいる。BMP−4ポリペプチドはBMP−2Bと呼ばれる場合もあることに留意されたい。
【0043】
本開示の方法および組成物において使用される好ましいBMPには、BMP−2、BMP−4、BMP−5、BMP−6、BMP−7およびBMP−8bが挙げられる。特に、BMP−4への曝露は、ヒトGBMから単離された細胞の成熟を強化するとともに全体的な生存度およびアポトーシスには影響しないことが以下の実施例において示されている。このことから、ニューロンマーカーおよび神経膠マーカーのアップレギュレーションが引き起こされ、増殖能力の顕著な低下ももたらされる。BMP−4への曝露は、一時的であっても、GBM培養物中のGBM tNSC集団(CD133+のGBM細胞)を大幅に縮小し、GBM細胞のコロニー形成の指標を大幅に低減し、GBM tNSCの増大の速度論を劇的に低減することが以下の実施例において示されている。これらの効果は不可逆的であり、ヒトGBM細胞のインビボにおける腫瘍発生能力を消滅させる。
【0044】
本明細書で使用される用語「LIF調製物」または「BMP調製物」はまた、LIFもしくはBMPのポリペプチドもしくは糖ポリペプチドのフラグメントであって、本開示の分析法および治療方法において過度の細胞増殖を減弱する能力を少なくとも部分的に保持している、例えば、本開示の分析法または治療法において完全長LIFまたは完全長BMPの活性の1〜100%を保持しているものも含む。そのようなフラグメントは、本開示の分析法または治療方法において完全長LIFまたは完全長BMPよりも高い活性を有していてもよい。そのようなフラグメントは、完全長タンパク質と比較して、アミノ末端またはカルボキシ末端、あるいは両末端から残基が欠失したひと続きのものであってもよい。フラグメントは、構造ドメインまたは機能ドメインを特徴とするもの、例えばαヘリックス領域およびαヘリックス形成領域、βシート領域およびβシート形成領域、ターン領域またはターン形成領域、コイル領域およびコイル形成領域、親水性領域、疎水性領域、両親媒性α領域、両親媒性β領域、フレキシブル領域、表面を形成する領域、および基質結合領域を含むフラグメントであってもよい。フラグメントは、ペプチド合成技術によって生産されたものでも、あるいは完全長のLIFまたはBMPポリペプチドの切断によって生じたものでもよい。フラグメントは、そのN末端、C末端、あるいはN末端およびC末端の両方が、他のポリペプチド配列に連結されることにより、融合タンパク質を形成する場合もある。
【0045】
本明細書において使用される用語「LIF調製物」または「BMP調製物」はまた、LIFもしくはBMPポリペプチド、または上述のようなそのフラグメントのアミノ酸配列と部分的に相同なアミノ酸配列を有し、かつ本開示の分析法および治療方法において過度の細胞増殖を減弱する能力を少なくとも部分的に保持しているポリペプチドまたは糖ポリペプチドも含む。相同体は、LIFもしくはBMPまたはそのフラグメントと50%、70%、80%、80.6%、83%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99.1%、99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%または99.9%同一であってよい。
【0046】
用語「LIF調製物」または「BMP調製物」はまた、LIFまたはBMPの完全長ポリペプチドのバリアント、およびLIPまたはBMPのフラグメントのバリアントも含む。そのようなバリアントは、本開示の分析法および治療方法において過度の細胞増殖を減弱する能力を少なくとも部分的に保持している。バリアントは、当技術分野で既知の一般的な法則に従って活性に対する影響がほとんどないように選択された欠失、挿入、逆位、反復、および置換を含んでいてもよい。例えば、表現型の上では表に出ないアミノ酸置換を行う方法に関する手引きはボウイ(Bowie)ら、Science 第247巻、p.1306−1310、1990年に提供されており、同文献はその全体が参照によって本願に組み込まれる。例えば、バリアントは部位特異的突然変異法またはアラニン走査突然変異法(分子中のすべての残基に単一アラニン変異を導入)(カニングハム(Cunningham)およびウェルズ(Wells)、Science 第244巻、p.1081−1085、1989年)によって得ることができる。バリアントはまた、例えば、そのタンパク質配列またはペプチド配列中に1以上の非ペプチド結合を(ペプチド結合の代わりとして)含むアミノ酸置換を有していてもよい。バリアントはさらに、天然に存在するLアミノ酸以外のアミノ酸残基、例えばDアミノ酸または非天然もしくは合成アミノ酸、例えばBアミノ酸もしくはyアミノ酸、を含む置換を有していてもよい。バリアントはさらに、ポリペプチドに立体構造上の制約を課する架橋基を含んでいてもよい。バリアントはさらに、グリコシル化、アセチル化、リン酸化およびその他同種のものを含んでいてもよい。バリアントはまた、(i)1以上の非保存アミノ酸残基による置換を含んでいてもよく、該置換アミノ酸残基は遺伝子コードによってコードされたアミノ酸残基であってもそうでなくてもよく、あるいは(ii)置換基を有する1以上のアミノ酸残基による置換を含んでいてもよく、あるいは(iii)成熟ポリペプチドが別の化合物と、例えばLIFまたはBMP調製物の安定性および/または溶解度を増大させる化合物(例えばポリエチレングリコール)、あるいはLIFまたはBMP調製物が特定の種類の細胞(腫瘍性神経幹細胞など)を標的とするための化合物、あるいはLIFまたはBMP調製物が血液脳関門(BBB)および/または血液腫瘍関門(BTB;blood−tumor barrier)を横断可能とするための化合物、と融合していてもよく、あるいは(iv)該ポリペプチドと、付加アミノ酸もしくは付加ペプチドもしくは付加ポリペプチドと融合していてもよく、あるいは(v)細胞毒性を有する作用薬、例えば毒素または放射活性化合物と融合していてもよく、あるいは(vi)画像化を目的として使用されるマーカー、例えば放射標識)と融合していてもよい。
【0047】
本開示のLIF調製物およびBMP調製物は、任意の適切な方法で、例えば天然に存在するポリペプチドの単離により、組換え技術により、ポリペプチド合成技術により、あるいはこれらの方法の組合せにより、調製することができる。そのようなポリペプチドを調製する方法は、当技術分野ではよく了解されている。LIF調製物またはBMP調製物は、融合タンパク質のようなより大きなタンパク質の形態であってもよい。分泌配列またはリーダー配列、プロ配列、ポリヒスチジン残基のような精製を支援する配列、あるいは組換え体生産中の安定性のための付加配列など、付加アミノ酸配列を備えていると有利な場合が多い。
【0048】
本開示のLIF調製物およびBMP調製物は、単離された形態で提供されることが好ましく、十分に精製されることが好ましい。組換え法によって生成されたLIFまたはBMP調製物は、本明細書に記載された技法または当技術分野で周知の他の方法、たとえばスミス(Smith)およびジョンソン(Johnson)、Gene 第67巻、p.31−40、1988年に記載されているワンステップ法を用いて、十分に精製することが可能である。本開示のLIFまたはBMPの調製物はまた、天然、合成または組換えの供給源から、当技術分野で周知のプロトコールを用いて、例えば完全長のLIFまたはBMPに対して作製された本開示の抗体を使用して精製することもできる。
【0049】
本開示のいくつかの実施形態では、内在する腫瘍性幹細胞および腫瘍前駆細胞がインビボで制御されるように、LIF調製物およびBMP調製物のうち少なくともいずれか一方が対象に直接投与される場合がある。例えば、BMP−4調製物はヒト患者体内のGBMのような脳腫瘍に投与される場合がある。本開示の別の実施形態では、腫瘍性幹細胞および腫瘍前駆細胞に、本開示の作用薬をインビトロで接触させる場合がある。例えば、腫瘍性幹細胞と腫瘍前駆細胞とを含む単離された腫瘍に、本開示の作用薬をインビトロで接触させる場合がある。
【0050】
LIF調製物およびBMP調製物のうち少なくともいずれか一方を、LIF調製物およびBMP調製物のうち少なくともいずれか一方を含む医薬組成物とともに、対象に、例えば対象者の腫瘍に投与する方法については、「投与および医薬組成物」と題した項で後述する。
【0051】
LIFレセプタシグナル伝達活性化剤およびBMPレセプタシグナル伝達活性化剤
別の態様の実施形態では、本開示は、本明細書中では以降「LIFRシグナル伝達活性化剤」および「LIFレセプタシグナル伝達活性化剤」とも称する、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFレセプタ(LIFR)を介したシグナル伝達を増大させることのできる作用薬を提供する。本開示はまた、そのようなLIFRシグナル伝達活性化剤を同定するための方法、およびそのようなLIFRシグナル伝達活性化剤を含有する医薬組成物を提供する。本開示のLIFRシグナル伝達活性化剤は、LIFRを直接活性化することにより幹細胞または前駆細胞におけるLIFRを介したシグナル伝達を増大させてもよいし(例えばアゴニスト)、あるいは間接的に、例えば腫瘍性幹細胞または腫瘍前駆細胞中で第2の分子もしくは化合物の発現または活性を増大させて(例えば、LIF自体の発現を増大させて、あるいはJAKまたはSTATのようなLIFRを介したシグナル伝達の下流成分の活性または発現を増加させて)ひいては腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRを介したシグナル伝達を増大させてもよい。
【0052】
追加の態様では、本開示は、本明細書中では以降「BMPRシグナル伝達活性化剤」および「BMPレセプタシグナル伝達活性化剤」とも称する、腫瘍性幹細胞または腫瘍前駆細胞におけるBMPレセプタ(BMPR)を介したシグナル伝達を増大させることのできる作用薬を提供する。本開示はまた、そのようなBMPRシグナル伝達活性化剤を同定するための方法、およびそのようなBMPRシグナル伝達活性化剤を含有する医薬組成物を提供する。本開示のBMPRシグナル伝達活性化剤は、BMPRを直接活性化することにより腫瘍性幹細胞または腫瘍前駆細胞におけるBMPRを介したシグナル伝達を増大させてもよいし(例えばアゴニスト)、あるいは間接的に、例えば腫瘍性幹細胞または腫瘍前駆細胞中で第2の分子もしくは化合物の発現または活性を増大させて(例えば、BMP自体の発現を増大させて、あるいはBMPRを介したシグナル伝達の下流成分の活性または発現を増加させて)ひいては腫瘍性幹細胞または腫瘍前駆細胞におけるBMPRを介したシグナル伝達を増大させてもよい。
【0053】
本明細書中で「LIFR」という場合、LIFRのホモログ、パラログ、オーソログ、誘導体、フラグメントおよび機能的等価物など、LIFRのすべての形態を指す。本明細書中で「BMPR」という場合、BMPRのホモログ、パラログ、オーソログ、誘導体、フラグメントおよび機能的等価物など、BMPRのすべての形態を指す。
【0054】
本開示の文脈においては、LIFRまたはBMPRを介したシグナル伝達の増大とは、LIFRまたはBMPRを介したシグナル伝達の正常なレベルの1〜約1000%の増大を意味する。別例として、LIFRまたはBMPRのシグナル伝達活性化剤は、LIFRまたはBMPRを介したシグナル伝達のレベルを、シグナル伝達が正常よりも低い場合に回復させることができる。
【0055】
好ましくは、LIFRまたはBMPRを介したシグナル伝達の増大により、腫瘍性幹細胞および腫瘍前駆細胞の特性のうちいずれか1つ以上、例えば限定されるものではないが生存、自己複製、増殖、対称分裂および分化のうち少なくともいずれかの特性が調節される。最も好ましくは、LIFRまたはBMPRを介したシグナル伝達の増大は、腫瘍性幹細胞および腫瘍前駆細胞の分裂特性を変化させ、特に対称分裂の可能性の低減あるいは増殖している腫瘍性幹細胞および腫瘍前駆細胞によって示される細胞周期回転数の低減により、腫瘍性幹細胞および腫瘍前駆細胞の数の低下をもたらす。
【0056】
本開示のLIFRおよびBMPRシグナル伝達活性化剤は、ペプチド、ポリペプチドおよびタンパク質のような任意のタンパク質性分子であってもよいし、非タンパク質性の分子であってもよい。LIFRおよびBMPRシグナル伝達活性化剤の単離のための方法は本明細書において提供される。
【0057】
本開示に関して、ミメティックはLIFRおよびBMPRシグナル伝達活性化剤の特に有用な一群である。該用語は、ミメティックが模倣する例えばLIFなどの分子に対してある種の化学的類似性を有する物質であって、例えばLIFRのような標的とLIFとの相互作用をアゴナイズする(模倣する)物質を指すように意図されている。ペプチドミメティックはミメティックの1種であり、タンパク質二次構造の構成要素を模倣するペプチド含有分子であってもよい(ジョンソン(Johnson)ら、「Peptide Turn Mimetics in Biotechnology and Pharmacy」、ペズート(Pezzuto)ら編、チャップマン・アンド・ホール(Chapman and Hall)、ニューヨーク、1993年)。ペプチドミメティックの使用の背景にある論理的根拠は、タンパク質のペプチド・バックボーンの存在が主として、分子の相互作用、たとえば抗体と抗原との相互作用、酵素と基質との相互作用または足場タンパク質の相互作用を促進するようにアミノ酸側鎖を正しい向きにするためにあるということである。したがってペプチドミメティックは、天然の分子に似た分子相互作用が可能なように設計される。
【0058】
薬学的に活性な化合物を目指したミメティックの設計は、「リード」化合物に基づいた薬剤開発への既知のアプローチである。上記アプローチは、活性化合物の合成が困難であるか費用のかかる場合、あるいは特定の投与方法に適さない場合(例えば、ペプチドは消化管中のプロテアーゼによって急速に分解される傾向があるので経口組成物には不適当な活性作用薬である)、望ましいかもしれない。ミメティックの設計、合成および試験は、一般に、標的の特性に対して多数の分子をランダムにスクリーニングするのを回避するために使用される。BMP−4のミメティック、例えばペプチドミメティックなどについて、本明細書中で具体的に検討する。
【0059】
合理的薬物設計の目標は、対象とする生物学的に活性なポリペプチドまたは該ポリペプチドが相互作用する小分子の構造アナログを、例えばより高活性または安定な形態のポリ
ペプチドである薬物、あるいは例えばインビボでポリペプチドの機能を増強または妨害する薬物を作るために、作製することである(例えばホジスン(Hodgson)、Bio/Technology 第9巻、p.19−21、1991年を参照のこと)。1つの手法では、X線結晶解析、コンピュータモデリング、または最も典型的には手法の組み合わせによって、対象とするタンパク質の三次元構造を最初に決定する。ポリペプチドの構造に関する有用な情報は、相同タンパク質の構造に基づいたモデリングにより得られることもある。合理的薬物設計の一例は、HIVプロテアーゼインヒビターの開発である(エリクソン(Erickson)ら、Science 第249巻、p.527−533、1990年)。
【0060】
本開示のLIFRおよびBMPRシグナル伝達活性化剤がタンパク質性であれ非タンパク質性であれ、該活性化体が幹細胞または前駆細胞中でLIFRもしくはBMPRと相互作用し、かつ/またはLIFRもしくはBMPRを介したシグナル伝達を(直接的または間接的に)増大させる能力は、当業者に周知のいくつかのスクリーニング法によって評価することができる。該方法は、天然物ライブラリ、化学合成物ライブラリ、ならびにコンビナトリアルライブラリ、ファージディスプレイライブラリおよびインビトロトランスレーション系ライブラリのスクリーニングを含みうる。
【0061】
LIFRおよびBMPRに対して作製された抗体は、LIFおよびBMPの活性構造をそれぞれ模倣するアゴニストとして特に有用であり得る。適切な抗体には、ポリクローナル抗体、モノクローナル抗体、一価の抗体、二重特異性抗体、ヘテロ結合体抗体、多重特異性抗体、ヒト抗体、ヒト化またはキメラ抗体、一本鎖抗体、Fabフラグメント、F(ab’)フラグメント、Fab発現ライブラリによって生成されたフラグメント、抗イディオタイプ(抗Id)抗体、および上記のうちいずれかのエピトープ結合フラグメントがある。本明細書で用いられる用語「抗体」は、免疫グロブリン分子および免疫グロブリン分子の免疫学的に活性な部分、すなわち免疫学的に特異的に抗原に結合する抗原結合部位を含んでいる分子を指す。免疫グロブリン分子は、免疫グロブリン分子の任意の種類(例えばIgG、IgE、IgM、IgD、IgAおよびIgY)、クラス(例えばIgG1、IgG2、IgG3、IgG4、IgA1およびIgA2)またはサブクラスのものであってよい。さらに、用語「抗体」(Ab)あるいは「モノクローナル抗体」(Mab)は、完全な分子だけでなくタンパク質に特異的に結合することができる抗体フラグメント(例えば、FabおよびF(ab’)2フラグメント)をも含むことを意味する。FabおよびF(ab’)2フラグメントは、完全な抗体のFcフラグメントを欠き、完全な抗体に比べて動物または植物の循環系からより急速に消滅し、完全な抗体よりも組織への非特異的な結合が小さい場合がある(ウォール(Wahl)ら、J.Nucl.Med.第24巻、p.316−325、1983年)。抗体アゴニストを作製する方法は、例えば国際公開公報第96/40281号パンフレット;米国特許第5,811,097号;デング(Deng)ら、Blood 第92巻(6)、p.1981−1988(1998);チェン(Chen)ら、Cancer Res.第58巻(16)、p.3668−3678(1998);ハロップ(Harrop)ら、J.Immunol.第161巻(4)、p.1786−1794(1998);チュー(Zhu)ら、Cancer Res.第58巻(15)、p.3209−3214(1998);ユーン(Yoon)ら、J.Immunol.第160巻(7)、p.3170−3179(1998);プラト(Prat)ら、J.Cell.Sci.第111巻(Pt2)、p.237−247(1998);ピタール(Pitard)ら、J.Immunol.Methods 第205巻(2)、p.177−190(1997);リアウタール(Liautard)ら、Cytokine 第9巻(4)、p.233−241(1997);カールソン(Carlson)ら、J.Biol.Chem.第272巻(17)、p.11295−11301(1997);タリーマン(Taryman)ら、Neuron 第14巻(4)、p.755−762(1995);ミュラー(Muller)ら、Structu
re 第6巻(9)、p.1153−1167(1998);バルツネク(Bartunek)ら、Cytokine 第8巻(1)、p.14−20(1996);ハーロー(Harlow)ら、「Antibodies: A Laboratory Manual」(コールド・スプリング・ハーバー研究所出版社(第2版)、1988);ハマーリング(Hammerling)ら、「Monoclonal Antibodies and T−CeIl Hybridomas」中のp.563−681(エルゼビア(Elsevier)、ニューヨーク、1981)(これらはすべて参照によって全体が本願に組み込まれる)。
【0062】
核酸リガンド(「アプタマー」としても知られている)も、LIFおよびBMPの活性構造をそれぞれ模倣するアゴニストとして特に有用な場合がある。例えば、アプタマーはSELEX(指数関数的な富化によるリガンドの系統的進化(Systematic Evolution of Ligands by Exponential Enrichment))法(ツエルク(Tuerk)およびゴールド(Gold)、Science 第249巻、p.505−510、1990年、この文献は参照によって全体が本願に組み込まれる)を使用して選択することができる。SELEX法では、大きな核酸分子ライブラリ(例えば1015個の異なる分子)を作製し、かつ/または標的分子(この場合はBMPR、LIFRまたはその一部分)でスクリーニングする。標的分子を、ヌクレオチド配列のライブラリとともに一定期間インキュベートする。その後、混合物中の非結合分子から標的アプタマー分子を物理的に単離するためにいくつかの方法を使用することが可能であり、非結合分子は廃棄することができる。その後、標的分子への親和性が最も高いアプタマーを精製して標的分子から分離し、標的分子に結合することができるアプタマーが十分に富化された新たなライブラリを作製するために、酵素を用いて増幅させることができる。その後、この富化されたライブラリを用いて、選択、分離および増幅の新しいサイクルを開始することができる。この選択、分離および増幅のプロセスを5〜15サイクル行った後、ライブラリは、標的分子に強固に結合する少数のアプタマーに縮小される。その後、混合物中の個々の分子を単離し、該分子のヌクレオチド配列を決定し、該分子の結合親和性および結合特異性を測定して比較することができる。その後、単離されたアプタマーをさらに洗練して、標的への結合および/またはアプタマー構造に寄与しないあらゆるヌクレオチドを除去する(つまり、アプタマーを結合コアドメインまで切り詰める)ことができる。アプタマー技術の総説としては、例えばジャヤセナ(Jayasena)、Clin.Chem.第45巻、p.1628−1650、1999年を参照のこと。同文献の教示内容は全て参照によって本願に組込まれる。
【0063】
本質的にいかなる化学化合物もLIFRまたはBMPRシグナル伝達活性化剤の候補として使用することができる。ハイスループットスクリーニング法は、そのような候補活性化体の検出用として特に想定される。そのようなハイスループットスクリーニング法は、典型的には多数の潜在的な治療用化合物(例えばリガンドまたはモジュレータ化合物)を含んでいるコンビナトリアル化学ライブラリまたはペプチドライブラリを用意することを必要とする。その後、そのようなコンビナトリアル化学ライブラリまたはリガンドライブラリを1以上の分析法でスクリーニングして、所望の特徴的活性を示すライブラリ構成メンバー(例えば特定の化学種またはサブクラス)を同定する。このようにして同定された化合物は、従来のリード化合物として役立てることもできるし、それ自体を治療薬候補または実際の治療薬として使用することもできる。
【0064】
コンビナトリアル化学ライブラリは、多くの化学ビルディングブロック(すなわちアミノ酸のような試薬)を組み合わせることにより、化学合成あるいは生物学的合成のいずれかによって生成された種々の化学化合物の集まりである。一例として、直鎖コンビナトリアルライブラリ、例えばポリペプチドまたはペプチドのライブラリは、化合物の所定の長さ(すなわちポリペプチド化合物またはペプチド化合物中のアミノ酸の数)について可能
なあらゆる方法で1セットの化学ビルディングブロックを組み合わせることにより形成される。化学ビルディングブロックをそのように組み合わせて混合することによって、何百万もの化学化合物を合成することができる。
【0065】
コンビナトリアル化学ライブラリの作製およびスクリーニングは関連技術分野の技術者にはよく知られている。コンビナトリアルライブラリには、限定するものではないがペプチドライブラリが挙げられる(例えば米国特許第5,010,175号;フルカ(Furka)、Int.J.Pept.Prot.Res.、第37巻、p.487−493、1991年;ならびにホートン(Houghton)ら、Nature 第354巻、p.84−88、1991年)。多様な化学ライブラリを作製するためのその他の化学手法も使用することができる。多様な化学ライブラリの化学成分の非限定的な例としては、ペプチド(国際公開公報第91/019735号パンフレット)、コード化されたペプチド(国際公開公報第93/20242号パンフレット)、ランダムなバイオオリゴマー(国際公開公報第92/00091号パンフレット)、ベンゾジアゼピン(米国特許第5,288,514号)、ヒダントイン、ベンゾジアゼピンおよびジペプチドのようなダイバーソマー(ホッブズ(Hobbs)ら、1993年、Proc.Natl.Acad.Sci. USA 第90巻、p.6909−6913)、ビニル性ポリペプチド(ハギハラ(Hagihara)ら、1992年、J.Amer.Chem.Soc.、第114巻、p.6568)、グルコース骨格を有する非ペプチド性のペプチドミメティック(ヒルシュマン(Hirschmann)ら、1992年、J.Amer.Chem.Soc.、第114巻、p.9217−9218)、小化合物のアナログ有機合成ライブラリ(analogous organic synthesis of small compound libraries)(チェン(Chen)ら、1994年、J.Amer.Chem.Soc、第116巻、p.2661)、オリゴカルバメート(チョ(Cho)ら、1993年、Science 第261巻、p.1303)かつ/またはペプチジルホスホネート(キャンベル(Campbell)ら、1994年、J.Org.Chem
第59巻、p.658)、核酸ライブラリ(いずれも上述のオースベル(Ausubel)、バーガー(Berger)およびサムブルック(Sambrook)の文献を参照のこと)、ペプチド核酸ライブラリ(米国特許第5,539,083号)、抗体ライブラリ(例えばヴォーン(Vaughn)ら、1996年、Nature Biotechnology 第14巻(3)、p.309−314)およびPCT/US96/10287)、炭水化物ライブラリ(例えばリアン(Liang)ら、1996年、Science、第274巻、p.1520−1522および米国特許第5,593,853号)、有機小分子ライブラリ(例えばベンゾジアゼピン、Baum C&EN、1993年1月18日、p.33および米国特許第5,288,514号;イソプレノイド、米国特許第5,569,588号;チアゾリジノンおよびメタチアザノン、米国特許第5,549,974号);ピロリジン、米国特許第5,525,735号および同第5,519,134号;モルホリノ化合物、米国特許第5,506,337号など)が挙げられる。
【0066】
コンビナトリアルライブラリ作製用のデバイスは市販されている(例えば、米国ケンタッキー州ルイビル所在のアドバンスドケムテック(Advanced Chem Tech)の357MPS、390MPS;米国マサチューセッツ州ウォーバーン所在のレイニン(Rainin)のSymphony(TM);米国カリフォルニア州フォスターシティー所在のアプライド・バイオシステムズ(Applied Biosystems)の433A;米国マサチューセッツ州ベッドフォード所在のミリポア(Millipore)の9050Plus)。さらに、多くのコンビナトリアルライブラリが市販されている(例えば、米国ニュージャージー州プリンストン所在のコムジェネックス(ComGenex);ロシア国モスクワ所在のアシネックス(Asinex);米国ミズーリ州セントルイス所在のトリポス社(Tripos,Inc.;ロシア国モスクワ所在のケムスター株式会社(ChemStar,Ltd.);米国ペンシルバニア州エクストン所在の3D
ファーマシューティカルズ(3D Pharmaceuticals);米国メリーランド州コロンビア所在のマーテック・バイオサイエンシズ(Martek Biosciences)など)。
【0067】
LIFRおよびBMPRシグナル伝達活性化剤の候補は、結合分析を使用して、LIFRまたはBMPRに対する、またはLIFRもしくはBMPRシグナル伝達経路の下流成分に対する結合能力について最初にスクリーニングし、結合する活性化体候補を次いで機能分析でスクリーニングすることができる。適切な結合分析には、蛍光を用いるサーマルシフトアッセイ(米国ペンシルバニア州エクストン所在の3Dファーマシューティカルズ社(3−Dimensional Pharmaceuticals, Inc.)、3DP)が挙げられる(パントリアノ(Pantoliano)らの米国特許第6,020,141号および6,036,920号に記載;さらにJ.ジマーマン(J.Zimmerman)、2000年、Gen.Eng.News 第20巻(8)も参照のこと)。
【0068】
LIFRおよびBMPRシグナル伝達活性化剤の候補を機能に関してスクリーニングする方法の一例は、下記の工程:
(i) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のサンプルを単離する工程;
(ii) 前記腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを適切な容器に入れる工程;
(iii) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを、特定の期間、特定の条件の下で候補作用薬に曝露する工程;ならびに
(iv) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方への形態学的、生理学的、および遺伝学的変化をスクリーニングする工程
を含む。
【0069】
形態学的、生理学的、および遺伝学的変化には、生存、自己複製、増殖および分化のうち少なくともいずれかの状態に関するスクリーニングが含まれる。使用することができる分析法の一例は、参照により全体が本願に組込まれる米国特許出願第2005/0112546号に記載の神経コロニー形成細胞法(Neural Colony Forming Cell Assay)(NCFCA)である。NCFCAは、いずれも増殖力を有し浮遊培養法(ニューロスフェア分析法)で球体を形成するかまたはNCFCAにおいてコロニーを形成することができる幹細胞と前駆細胞とを、区別することができる。簡潔に述べると、腫瘍から得られた初代細胞または培養細胞を、マイトジェンであるFGF2およびEGFを含んだ無血清3Dコラーゲンマトリックスに播種する。この培養状態下では、増殖力を有する幹細胞および前駆細胞のみが分裂して輪郭の明確なコロニーを形成し、このコロニーの大きさを1〜4週後に計測することができる。コロニーの大きさの違いは元の細胞の増殖力と正に相関し、幹細胞および前駆細胞の存在頻度の計測値を提供する。この条件下では、直径2mmを越えるコロニーだけが幹細胞に由来するものであり、直径2mm未満のものは前駆細胞に由来するものである。幹細胞および前駆細胞の、意味のある、かつ正確な計測値から、これら2種類の細胞の存在頻度を変化させる遺伝的・後成的な要素をスクリーニングすることが可能になる。
【0070】
レセプタLIFRおよびBMPRのスクリーニングに使用可能な、生存、自己複製、増殖および分化のうち少なくともいずれかの分析法の別例は、以下のように実施される。最初に、参照により本願に組込まれるグリッティ(Gritti)ら、J.Neurosci.(1996)第16巻(3)、p.109−1100)に記載のように、分散させた多形膠芽腫腫瘍由来の細胞を、マイトジェンであるFGF2(繊維芽細胞成長因子2)およびEGF(上皮成長因子)を含んだ無血清培地中に播種する。この培養系は、初代腫瘍培養物から分化中の細胞/分化細胞を選択除去し、指数関数的に増殖拡大して初代ニュー
ロスフェアを形成できる腫瘍性幹細胞だけを残す。この初代ニューロスフェアを個々に分散させ、マイクロタイタープレート中EGFおよびFGF2の存在下、クローン化しうる密度で無血清培地中に再度播種することができる。LIFRおよびBMPRシグナル伝達活性化剤の候補をマイクロタイタープレートの各ウェルに加え、該プレートを、未処置の細胞が十分に増殖可能な期間インキュベートする。インキュベーションを終えたらニューロスフェアを再び個々に分散させ、上記プロセスを、LIFRおよびBMPRシグナル伝達活性化剤候補の存在下でさらに所定の継代数だけ繰り返すことができる。所定の継代数を終えたら、顕微鏡を使用してニューロスフェアの存在についてマイクロタイタープレートのウェルを検査し、ニューロスフェアの数および大きさを測定し、幹細胞および前駆細胞に対するLIFRまたはBMPRシグナル伝達活性化剤候補の作用の尺度を得る。各継代の終了時の幹細胞および前駆細胞の数を決定するために実施例1の数学的アルゴリズムを使用することができる。連続的に継代された未処置の細胞との比較により、LIFRおよびBMPRシグナル伝達活性化剤候補(例えば幹細胞および前駆細胞の増殖特性を減弱化する作用薬)の同定が可能となる。その後、LIFRおよびBMPRシグナル伝達活性化剤候補を、分化した細胞または分化中の細胞で分析し、その候補作用薬の作用が全般的な細胞毒性ではなく腫瘍性幹細胞に特異的かどうかを判断することができる。
【0071】
LIFRおよびBMPRのシグナル伝達活性化剤としては、RNA干渉(RNAi)分子、リボザイム、あるいはアンチセンスオリゴヌクレオチドも含まれうる。そのような分子は、LIFRおよびBMPRシグナル伝達のインヒビターの発現を低減し、したがってLIFRおよびBMPRシグナル伝達を活性化する作用を有しうる。
【0072】
本開示のLIFRおよびBMPRシグナル伝達活性化剤は、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRまたはBMPRを介したシグナル伝達を増大させるのに役立つ。従って、本開示は、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRまたはBMPRを介したシグナル伝達を増大させる方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤と、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRまたはBMPRを介したシグナル伝達を増大させるのに十分な時間および条件の下で接触させることを含む方法を提供する。LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤は、本明細書に開示されたようなLIF調製物およびBMP調製物のうち少なくともいずれか一方と組み合わせて使用されてもよい。
【0073】
本開示はまた、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾患または障害を治療または予防するための方法を提供する。該方法は、治療上有効な量の、LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤を、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に投与することを含む。好ましくは、過度な細胞増殖を特徴とする障害は脳障害であり、より好ましくは脳腫瘍、例えば限定するものではないが聴神経腫、腺腫、星状細胞腫、若年性毛様細胞性星状細胞腫、脳幹部神経膠腫、脊索腫、脈絡叢、頭蓋咽頭腫、脳室上衣腫、神経節膠腫、神経節膠腫神経細胞、多形膠芽腫(GBM)、神経膠腫、リンパ腫、髄芽細胞腫、髄膜腫、乏突起膠腫、視神経膠腫、下垂体部腫瘍、松果体部腫瘍、あるいは松果体芽腫である。LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤は、本明細書に開示されたようなLIF調製物およびBMP調製物のうち少なくともいずれか一方と組み合わせて(同時にまたは時を異にして)投与することも可能である。
【0074】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、前記腫瘍に、治療上有効な量のLIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を投与することを含む方法、を提供する。
【0075】
さらなる態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍を、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方と接触させることを含む方法を提供する。理論または仮説によって限定されるものではないが、腫瘍に投与される場合、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤は、LIFまたはBMPを介したシグナル伝達の増大をもたらし、その結果として腫瘍性幹細胞または腫瘍前駆細胞の次の特性、例えば限定するものではないが細胞の生存、自己複製、対称分裂、増殖または分化のうち少なくともいずれかの特性の1つ以上を調節すると考えられる。特に、LIFRまたはBMPRを介したシグナル伝達の増大が幹細胞および前駆細胞の増殖特性を低下させ、特に増殖中の幹細胞または前駆細胞が示す対称分裂の可能性を低下させることによって該細胞の数が低減すると考えられる。従って、別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、BMPRシグナル伝達活性化剤およびLIFRシグナル伝達活性化剤のうち少なくともいずれか一方と接触させることを含む方法、を提供する。
【0076】
本開示のいくつかの実施形態では、内在する腫瘍性幹細胞および腫瘍前駆細胞がインビボで制御されるように、LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤が対象に直接投与されてもよい。本開示の代替実施形態では、腫瘍性幹細胞および腫瘍前駆細胞を、本開示の作用薬とインビトロで接触させる場合もある。
【0077】
腫瘍などの対象に、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を含む医薬組成物とともに投与する方法については、「投与および医薬組成物」と題した項で後述する。
【0078】
投与および医薬組成物
上述のように、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を治療または予防するために、治療上有効な量のLIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかを、とりわけ、対象または組織に投与することができる。例えば、一実施形態では、治療上有効な量のBMP−4調製物またはBMP−4ミメティックを、GBMに罹患しているヒト患者に投与する。上記の腫瘍およびがんに加えて、本明細書に開示された方法によって治療または予防され得るその他のがんには、癌腫、リンパ腫、芽細胞腫、肉腫および白血病が挙げられるがこれらに限定はされない。そのようながんのより具体的な例としては、扁平上皮がん、肺がん(小細胞肺がん、非小細胞肺がん、肺の腺癌および肺の扁平上皮癌を含む)、腹膜のがん、肝細胞がん、消化器がんもしくは胃がん(胃腸のがんを含む)、膵臓がん、子宮頸がん、卵巣がん、肝臓がん、膀胱がん、ヘパトーマ、乳がん、結腸がん、結腸直腸がん、子宮内膜もしくは子宮の癌腫、唾液腺癌腫、腎臓がん、肝臓がん、前立腺がん、陰門がん、黒色腫、甲状腺がん、肝癌および様々なタイプの頭頸部がん、ならびにB細胞リンパ腫(例えば、低悪性度/濾胞性の非ホジキンリンパ腫(NHL);小リンパ球性(SL)NHL;中悪性度/濾胞性のNHL;中悪性度の播種性NHL;高悪性度の免疫芽細胞性NHL;高悪性度のリンパ芽球性NHL;高悪性度のバーキット型(small non−cleaved cell)NHL;巨大腫瘤病変NHL;マントル細胞リンパ腫;AIDS関連リンパ腫;およびヴァルデンストレームマクログロブリン血症);慢性リンパ球性白血病(CLL);急性リンパ芽球性白血病(ALL);ヘアリー・セル白血病;慢性骨髄芽球白血病;ならびに、移植後リンパ増殖性疾患(PTLD)がある。
【0079】
一実施形態では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤および
BMPRシグナル伝達活性化剤のうち少なくともいずれかを、医薬組成物の形態で対象に投与する。従って、別の態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の治療または予防に有用な医薬組成物を提供する。本開示の医薬組成物は、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬を含有する。例えば、一実施形態では、治療上有効な量のBMP−4調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−2調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−5調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−6調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−7調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−8b調製物を含む医薬組成物がGBMの治療に提供される。
【0080】
別の態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の治療または予防のための医薬の製造における、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤、およびBMPRシグナル伝達活性化剤のうち少なくともいずれかの使用方法を開示する。例えば、多形膠芽腫の治療のための医薬の製造における、BMP−4調製物またはBMP−4ミメティックの使用について、具体的に検討する。
【0081】
本開示の医薬組成物は単一の作用薬を含んでいてもよいし、あるいは複数の前述の作用薬の任意の組み合わせ(例えばLIFおよびBMP−4の組み合わせ)を含んでいてもよい。さらに、医薬組成物は、特定の種類の2以上の作用薬、例えば2つの異なるLIF調製物、または2つの異なるBMPRシグナル伝達活性化剤(例えばBMP−2およびBMP−4)を含んでいてもよい。
【0082】
医薬組成物は少なくとも1つの医薬として許容可能な担体を含むことが好ましい。そのような医薬組成物では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかが「活性化合物」を形成する。本明細書で使用されるように、「医薬として許容可能な担体」という言葉は、薬務行政に適合した溶媒、分散媒、コーティング剤、抗菌物質および抗真菌物質、等張剤および吸収遅延剤などを含んでいる。追加の活性化合物を組成物に組み入れることもできる。医薬組成物はその意図した投与経路に適合するように製剤化される。投与経路の例として、非経口、例えば、静脈内、皮内、皮下、経口(例えば吸入)、経皮(局所)、経粘膜、および直腸内投与が挙げられる。非経口、皮内、または皮下への適用に使用される溶液または懸濁物は、下記成分、すなわち、注射用蒸留水、生理食塩水、固定油、ポリエチレングリコール、グリセリン、プロピレングリコールまたはその他の合成溶剤のような無菌の希釈剤;ベンジルアルコールまたはメチルパラベンのような抗菌物質;アスコルビン酸または亜硫酸水素ナトリウムのような酸化防止剤;エチレンジアミン四酢酸のようなキレート剤;酢酸塩、クエン酸塩またはリン酸塩のようなバッファ、ならびに塩化ナトリウムまたはデキストロースのような張度調整のための作用薬;を含むことができる。pHは、塩酸または水酸化ナトリウムのような酸または塩基で調整することができる。非経口の調製物は、ガラス製またはプラスチック製のアンプル、ディスポーザブルシリンジまたは多人数用バイアル中に入れることができる。
【0083】
過度な細胞増殖または誤制御された細胞増殖が生じていると思われる組織または細胞を、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかによりインビトロで治療する実施形態において、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝
達活性化剤のうち少なくともいずれかは、医薬組成物の形態であってもよいし、そうでなくてもよいことに注意されたい。
【0084】
本明細書において使用されるように対象とは、ヒトおよびヒト以外の霊長類(例えばゴリラ、マカク、マーモセット)、家畜動物(例えばヒツジ、畜牛、ウマ、ロバ、ブタ)、コンパニオン・アニマル(例えばイヌ、ネコ)、実験動物(例えばマウス、ウサギ、ラット、モルモット、ハムスター)、捕獲された野生動物(例えばキツネ、シカ)、および本開示の作用薬から利益を享受しうるその他の任意の生物を指す。ここで述べる作用薬から利益を享受し得る動物の種類に制限はない。本開示の中で最も好ましい対象はヒトである。ヒトかヒト以外の生物かどうかにかかわらず、対象を、患者、個体、動物、宿主またはレシピエントと称する場合がある。
【0085】
注射可能な使用に適した医薬組成物には、無菌の水溶液(水に可溶な場合)または分散液、あるいは注射可能な無菌の溶液または分散液を即席に調製するための無菌の散剤が挙げられる。静脈内投与については、適切な担体として生理食塩水、静菌水、Cremophor EL(TM)(米国ニュージャージー州パーシッパニー所在のバスフ(BASF))またはリン酸緩衝生理食塩水(PBS)が挙げられる。いずれの場合にも、組成物は無菌でなければならず、簡単にシリンジ操作ができる程度の流動性を有するべきである。組成物は製造および貯蔵の条件下で安定しているべきであり、細菌および真菌のような微生物の混入に対抗した状態に維持されなければならない。担体は、例えば、水、エタノール、多価アルコール(例えばグリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)およびこれらの適切な混合物を含んでいる溶媒または分散媒でありうる。適切な流動性は、例えば、レシチンのようなコーティング剤の使用により、分散液の場合は要求される粒度を維持することにより、また界面活性剤の使用により、維持することができる。微生物の活動の防止は、様々な抗菌性物質および抗真菌物質(例えばパラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなど)によって達成することができる。多くの場合、組成物中に等張剤、例えば糖、多価アルコール例えばマンニトール、ソルビトール、塩化ナトリウムを含むことが望ましいだろう。注射可能な組成物の吸収の延長は、組成物中に吸収を遅延させる作用薬(例えばモノステアリン酸アルミニウムおよびゼラチン)を含めることにより成される場合がある。
【0086】
無菌注射剤溶液は、必要に応じて上に列挙された成分を1つまたは組み合わせて含有する適切な溶媒中に、必要な量の活性化合物を組み入れた後、ろ過滅菌することにより調製することができる。一般に、分散液は、基本の分散媒と、上に列挙された成分からの必要なその他の成分とを含んでいる無菌のビヒクルに、活性化合物を組み入れることにより調製される。無菌注射剤溶液を調製するための無菌の散剤の場合、好ましい調製法は、あらかじめろ過滅菌された、有効成分と任意の所望の付加成分との溶液から、有効成分および所望の付加成分の粉末を生じる、真空乾燥および凍結乾燥である。
【0087】
経口組成物は一般に不活性の希釈剤または食用に適した担体を含んでいる。経口による治療投与の目的では、活性化合物を賦形剤と合わせて、錠剤、トローチ剤またはカプセル剤(例えばゼラチン・カプセル剤)の形態で使用することができる。経口組成物は、口内洗浄液として使用するための流動性の担体を用いて調製することもできる。薬学的に適した結着剤、または補助材のうち少なくともいずれか一方を、組成物の一部として含めることができる。錠剤、丸剤、カプセル剤、トローチ剤などは、次の成分、すなわち微結晶性セルロース、トラガカントゴムまたはゼラチンのような結合剤;デンプンまたはラクトースのような賦形剤、アルギン酸、Primogel(登録商標)またはトウモロコシデンプンのような崩壊剤;ステアリン酸マグネシウムまたはSterotes(登録商標)のような潤滑剤;コロイド状二酸化ケイ素のような流動促進剤;スクロースまたはサッカリンのような甘味料;あるいはペパーミント、サリチル酸メチルまたはオレンジ香味料のよ
うな香料、あるいは同様の性質の化合物のうちの任意のものを含むことができる。
【0088】
吸入による投与については、化合物は、適切な噴射剤(例えば二酸化炭素のようなガス)の入った加圧容器またはディスペンサ、あるいは噴霧器から、エアゾルスプレーの形態で送達される。
【0089】
経粘膜的手段または経皮的手段によって全身投与することも可能である。経粘膜投与または経皮投与の場合、浸透すべき障壁に適した浸透剤を製剤中に使用する。そのような浸透剤は当技術分野で一般に知られており、例えば、経粘膜投与については界面活性剤、胆汁酸塩およびフシジン酸誘導体が挙げられる。経粘膜投与は、鼻内噴霧剤または坐剤の使用により遂行することができる。経皮投与については、活性化合物を、当技術分野で一般に知られているような軟膏剤、サルベ、ゲル剤あるいはクリーム剤に製剤化する。化合物を、直腸送達のために、坐剤(例えばココアバターおよび他のグリセリドのような従来の坐剤基剤を用いる)または停留浣腸剤の形態に調製することもできる。
【0090】
一実施形態では、活性化合物は、植込剤およびマイクロカプセル化送達システムなどの制御放出製剤のように、化合物が身体から急速に消失するのを防ぐ担体を用いて調製される。エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステルおよびポリ乳酸のような生分解性で生体適合性のポリマーを使用することができる。そのような製剤を調製する方法は当業者には明白であろう。材料も、アルザコーポレイション(Alza Corporation)およびノバ・ファーマシューティカルズ・インコーポレイテッド(Nova Pharmaceuticals, Inc.)から商業的に入手することができる。リポソーム懸濁液(細胞特異抗原に対するモノクローナル抗体を用いて標的を感染細胞に設定したリポソームなど)も、医薬として許容可能な担体として使用することができる。これらは、例えば米国特許第4,522,811号に述べられているように、当業者に周知の方法によって調製することができる。
【0091】
投与し易く、かつ投与量を均一にするために経口または非経口の組成物を投与量単位の形態に製剤化すると有利である。本明細書で使用される場合、投与量単位の形態とは、治療すべき対象についての単一量として適した物理的に個別の単位を指し;各々の単位は、所望の治療効果を生じるために計算された所定量の活性化合物を、必要な製薬担体とともに含んでいる。
【0092】
そのような化合物の毒性および治療上の有効性は、例えばLD50(集団の50%が死亡する用量)およびED50(集団の50%に治療上有効な用量)を決定するための、細胞培養または実験動物における標準的な薬学的手法により決定することができる。毒性作用および治療効果の間の用量比が治療係数であり、治療係数はLD50/ED50比として表すことができる。高い治療係数を示す化合物が好ましい。毒性副作用を示す化合物を使用することは可能であるが、非感染細胞が損傷する可能性を最小限にすることによって副作用を低減するために、そのような化合物の標的を罹患組織の部位に設定する送達システムを設計するように配慮すべきである。
【0093】
細胞培養分析および動物実験から得られたデータを、ヒトで用いる用量範囲を策定するのに使用することができる。そのような化合物の用量は、毒性をほとんどまたは全く伴わないED50を含めた血中濃度範囲にあることが好ましい。用量は、この範囲内で、使用される剤形および利用される投与経路に依存して変わりうる。本開示の方法において使用される任意の化合物について、治療上有効な用量は、最初に細胞培養分析から推測することができる。用量は、細胞培養で決定されるようなIC50(すなわち症状の最大限の抑制を2分の1達成する試験化合物の濃度)を含む血漿中濃度範囲を達成するように動物モデルで策定することができる。そのような情報は、ヒトでの有用な用量をより正確に決定
するために使用することができる。血漿中濃度は、例えば高速液体クロマトグラフィによって測定することができる。
【0094】
本明細書において定義される場合、治療上有効な量のタンパク質またはポリペプチド(すなわち有効な用量)は、体重1kgあたり約0.001〜30mg、好ましくは体重1kgあたり約0.01〜25mg、より好ましくは体重1kgあたり約0.1〜20mg、より一層好ましくは体重1kgあたり約1〜10mg、2〜9mg、3〜8mg、4〜7mg、または5〜6mgの範囲にありうる。タンパク質またはポリペプチドを、1週間に1回として、約1〜10週間、好ましくは約2〜8週間、より好ましくは約3〜7週間、より一層好ましくは約4、5または6週間投与することができる。当業者には当然のことであるが、一定の要因、例えば限定するものではないが疾病または障害の重症度、以前の治療、対象の全体的な健康状態および/または対象の年齢、ならびにその他の疾病の存在などが、対象を有効に治療するのに必要な用量および時期に影響しうる。さらに、治療上有効な量のタンパク質、ポリペプチドまたは抗体を用いた対象の治療は、単回治療を含むものでもよいが、好ましくは一連の複数の治療を含むこともできる。
【0095】
本開示の実施形態において、脳の増殖性障害、例えばGBMが治療される場合、血液脳関門(BBB)および血液腫瘍関門(BTB)のうち少なくともいずれか一方を乗り越えるために、医薬組成物および投与方法のうち少なくともいずれか一方を調整することが望ましい。BBBを越えて医薬組成物を送達する方法は、当技術分野で周知である。例えば、参照により全体が本願に組込まれる、ミスラ(Misra)ら、2003年、J Pharm Pharm Sci 第6巻、p.252−273を参照のこと。適切な方法としては、限定するものではないが、脳内移植、脳室内(ICV)注入、またはCED(convection enhanced diffusion)法のような経頭蓋的脳内薬物送達法が挙げられる。脳内移植は、例えば、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかを含浸させた、ポリマー製ビーズ(ヘパリンアクリルビーズなど)またはポリマー製ウエハ(ポリフェプロサン(polifeprosan)20など)を使用して実行可能である。例えば、脳内移植は、BMP−4を装荷したヘパリンアクリルビーズを腫瘍内または切除腔内に定位的に注入することにより実行可能である。
【0096】
本開示の医薬組成物、例えばBMP−2、BMP−4、BMP−5、BMP−6、BMP−7またはBMP−8b調製物(またはこれらの組み合わせ)を含有する医薬組成物を、上記の方法を使用して未切除の腫瘍に投与してもよいし、あるいは、医薬組成物を腫瘍切除後に切除腔に投与してもよい。いくつかの実施形態では、医薬組成物を最初に腫瘍内に投与し、次に、腫瘍切除後に術後投与する。
【0097】
いくつかの実施形態では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかが、血液脳関門(BBB)レセプタ仲介輸送(RMT)システムの構成分子上の外表面エピトープと結合することができる分子と会合している。この方法では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかは、内在性のRMTシステムを使用してBBBを越えて輸送されうる。例えば、LIFまたはBMPの調製物をトランスフェリンレセプタ(TfR)に対するモノクローナル抗体(OX26など)と結合させて、該結合体の膜透過輸送を可能にする。例えば、参照により全体が本願に組み込まれるパルドリッジ(Pardridge)、Neurorx 第2巻(1)、p.3−14(2005)を参照されたい。例えばOX26との結合によってナノ粒子がBBBを超えるようにすることも可能であり、そのようなナノ粒子を、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかと結合させてもよい。例えば、参照により全体が本願に組
み込まれるオリヴィエ(Olivier)ら、Pharm Res.(2002)第19巻(8)、p.1137−43を参照されたい。さらに、例えばOX26に結合させたリポソームを用いて、カプセル封入されたLIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかを、BBBを越えて送達させることもできる。参照により全体が本願に組み込まれるヒュイラー(Huwyler)ら、Proc Natl Acad Sci USA.(1996)第93巻(24)、p.14164−9を参照されたい。さらに、BBBおよびBTBを破壊する作用薬および処置法を用いて、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤またはBMPRシグナル伝達活性化剤のうち少なくともいずれかを、BBBまたはBTBに通過させることもできる。例えば、血管作用薬ブラジキニンの頸動脈内注入は、脳腫瘍毛細血管の透過性を選択的に増大させることができる。参照により全体が本願に組み込まれるマツカド(Matsukado)ら、Brain Res.(1998)第792巻(1)、p.10−5を参照されたい。
【0098】
一実施形態では、LIFおよびBMP調製物、またはポリペプチド性およびペプチド性のLIFRもしくはBMPRシグナル伝達活性化剤、または、LIFRもしくはBMPシグナル伝達活性化剤であるリボザイム、RNAi分子およびアンチセンス分子、をコードする核酸分子をベクターに挿入し、遺伝子療法ベクターとして使用する。遺伝子療法ベクターは、例えば静脈注射、局所投与(米国特許第5,328,470号を参照)、または定位注入(例えば、いずれも参照により全体が本願に組み込まれるチェン(Chen)ら(1994)、Proc.Natl.Acad.Sci. USA 第91巻、p.3054−3057;ボーグ(Voges)ら(2003)、Ann.Neurol.第54巻、p.479−487を参照)によって対象に送達することができる。遺伝子療法ベクターの医薬品製剤は、許容可能な希釈剤中またはカプセル材料(ベクターを含むリポソームなど)中に入った遺伝子療法ベクターを含むものでもよいし、あるいは遺伝子送達手段が埋め込まれた遅延放出マトリックスからなるものでもよい。別例として、完全な遺伝子送達ベクターが組換え細胞からそのまま生産される、例えばレトロウイルス・ベクターの場合、医薬品製剤は、遺伝子送達システムを生産する1以上の細胞を含むことができる。一実施形態では、LIFおよびBMP調製物をコードする、またはポリペプチド性およびペプチド性のLIFRもしくはBMPRシグナル伝達活性化剤をコードする核酸分子は、参照により全体が本願に組み込まれるコンシグリオ(Consiglio)ら、Proc
Natl Acad Sci USA、2004年10月12日、第101巻(41)、p.14835−14840に述べられているように、レンチウイルスベクターによって哺乳類のがん性神経幹細胞に導入される。
【0099】
いくつかの実施形態では、本開示による単一の活性化合物、例えば単一のLIF調製物または単一のBMP調製物が投与される。他の実施形態では、複数の活性化合物、例えば2つの異なるLIF調製物;またはLIF調製物とBMP調製物;またはBMPRシグナル伝達活性化剤と2つの異なるBMP調製物、が同時投与される。さらに、本開示の活性化合物は、化学療法剤、放射線増感剤、放射線治療薬などのような他の薬剤と同時投与されることもある。本明細書で「同時投与」という場合、同一の製剤として、または2つの異なる製剤として、同一経路または異なる経路で同時に投与すること、あるいは同一経路または異なる経路で連続的に投与することを意味する。本明細書で「連続的に」投与するという場合、2種類の作用薬および/または医薬組成物の投与の間の時差が、数秒、数分、数時間または数日であることを意味する。作用薬および/または医薬組成物の同時投与は任意の順序で行われうる。
【0100】
本明細書に開示された治療方法および医薬組成物は、他の治療、たとえば化学療法(カルマスティン、シスプラチン、パクリタクセル、テモゾロミド、PCV(プロカルバジン、ロムスチンおよびビンクリスチン)およびIL13−PE38QQRなど)、放射線治
療薬(放射標識抗体または放射標識核酸リガンドなど)を用いた治療、放射線療法、および手術(腫瘍の切除/減量手術など)などと併用されてもよい。例えば、BMP−4調製物を、GBM治療用の腫瘍切除手術および放射線治療後の化学療法剤とともに同時投与してもよい。これらの付加的な治療は、本明細書に開示された方法による治療の前、治療中、または治療後のうちいずれにおいても使用できる。
【0101】
実施例
本開示について、以下の非限定的な実施例によりさらに説明する。実施例19−23については、列挙された結果を得るために使用されたプロトコールが実施例24−29に提示されていることに注意されたい。
【0102】
実施例1:ヒト胎児神経幹細胞の連続継代および数学的モデル化に基づく幹細胞と前駆細胞の存在頻度の分析
連続的に継代されたニューロスフェア培養物中に存在する神経幹細胞の数および神経前駆細胞の数を計算するためにアルゴリズムを導き出した。このアルゴリズムにために、すべての細胞を以下の3つのカテゴリーに分類する。
【0103】
1.幹細胞は、(a)増殖が可能、(b)長期間にわたる自己複製が可能、(c)多くの子孫を生じることが可能で、かつ(d)その幹細胞が得られた組織のすべての種類の細胞を生じる能力を有する、未分化細胞として定義する。
【0104】
2.前駆細胞は、(a)増殖が可能で、(b)限定的な自己複製能力を有し、(c)限られた数の子孫を生じることが可能で、かつ(d)少なくとも1種類の子孫を生じる能力を有する、未分化細胞として定義する。
【0105】
3.分化細胞は、増殖能力が限定されているかまたは増殖能力がなく、細胞系統特異的マーカーおよび成熟した機能特性のいずれをも発現している細胞として定義する。
用語「幹細胞」およびNSCは本明細書中で互換的に使用されることがある。同様に、用語「前駆細胞」およびNPCも本明細書中で互換的に使用されることがある。このアルゴリズムについて説明する際に、以下のいくつかの仮定が為される。
【0106】
1.ニューロスフェアは、幹細胞、前駆細胞および分化細胞で構成される。
2.すべてのニューロスフェアは単一の幹細胞または単一の前駆細胞のいずれかから成長する。
【0107】
3.幹細胞の寿命は無限であり、前駆細胞の寿命は有限である。有限の寿命を継代数lと定義する。
4.幹細胞は常にニューロスフェアを形成可能であり、前駆細胞はその寿命の終わりに達していなければニューロスフェアを形成する。
【0108】
5.各ニューロスフェアは、合計c個の細胞を有し、これらの細胞は2つの可能な組成のうちの1つである。可能な組成とは、
a.単一の幹細胞に由来する各ニューロスフェアにおいて、組成は幹細胞s個、前駆細胞p個、残りは分化細胞であり;
b.単一の前駆細胞に由来する各ニューロスフェアにおいて、組成は前駆細胞p個と残りの分化細胞である。
【0109】
継代数nにおける細胞の総数Tnを表すアルゴリズムを導き出した。実験の初めに、幹細胞由来のニューロスフェアを個々に分散させる、すなわち、幹細胞がs個、前駆細胞がp個、および分化細胞がc−s−p個存在する。この幹細胞および前駆細胞を増殖させて
、合計s+p個のニューロスフェアになるように成長させ、分化細胞は死滅する。その結果、最初の継代の時点T1での総細胞数は、ニューロスフェアの総数と各ニューロスフェア中の細胞数との積:
【0110】
【数1】
【0111】
で与えられる。
上記方程式の第2の等式は興味深い。というのも、scは幹細胞由来のニューロスフェア中の総細胞数を表し、pcは前駆細胞由来のニューロスフェア中の総細胞数を表しているからである。
【0112】
前駆細胞が2世代生きると仮定し、ここでこれらのニューロスフェアを個々に分散させる。最初の継代では、幹細胞由来のs個のニューロスフェアが存在し、その各々がs個の幹細胞、p個の前駆細胞およびc−s−p個の分化細胞を含むはずである。同様に、最初の継代では、前駆細胞由来のp個のニューロスフェアが存在し、今度はその各々がp個の前駆細胞およびc−p個の分化細胞を含む。ここで、新たに個々に分散させたこれらの細胞は、死滅する分化細胞以外はその細胞独自のニューロスフェアへと成長することになる。その結果、2番目の継代の時点T2での総細胞数は、
【0113】
【数2】
【0114】
【数3】
【0115】
で与えられる。
等式の右辺の第1のカラムは、幹細胞由来のニューロスフェアを表す項を含んでいる。第2のカラムは、前駆細胞由来のニューロスフェアを表している。
【0116】
T2の方程式中の第2の等式は、総細胞数を展開式で表すものである。s2c項は、幹
細胞由来のニューロスフェア中の、次世代で幹細胞由来のニューロスフェアになる細胞の総数を示す。第2項spcは、幹細胞由来のニューロスフェア中の、次世代で前駆細胞由来のニューロスフェアになる細胞の総数を示す。最後の項p2cは、その寿命に依存して
前駆細胞由来のニューロスフェアになるかまたは死滅する、前駆細胞由来のニューロスフェアを表す。
【0117】
ここでこれらの細胞を継代して第3の世代を生じさせる。この世代の総細胞数は、前駆細胞の寿命に左右される。寿命がわずか2世代であれば、前駆細胞由来の前駆細胞は新しいニューロスフェアを生成せずにここで死滅することになる。この仮定の下では、3番目の継代の時点T3における総細胞数は、
【0118】
【数4】
【0119】
【数5】
【0120】
【数6】
【0121】
で与えられる。
同様に、4番目の継代の時点における総細胞数を決定することができる。これらの方程式を書き直すと、
【0122】
【数7】
【0123】
【数8】
【0124】
【数9】
【0125】
【数10】
【0126】
となる。
したがって、導き出すと、n番目の継代における総細胞数は、
【0127】
【数11】
【0128】
で与えられることになる。
この方程式は前駆細胞の寿命が2世代であるという仮定に基づいていることを繰り返しておかなければならない。
【0129】
前駆細胞の寿命が2世代より長い場合、3番目の継代の時点T3における総細胞数は、
【0130】
【数12】
【0131】
【数13】
【0132】
【数14】
【0133】
で与えられる。
前駆細胞の寿命が3世代である場合、第4世代までの総細胞数は、
【0134】
【数15】
【0135】
【数16】
【0136】
【数17】
【0137】
【数18】
【0138】
で与えられる。
したがって、この特定の仮定を用いて導き出すと、n番目の継代における総細胞数は、
【0139】
【数19】
【0140】
で与えられる。
一般に、同様の論拠によって、前駆細胞の寿命がl世代である場合のn番目の継代の時点における総細胞数は、
【0141】
【数20】
【0142】
【数21】
【0143】
で与えられる。
上記方程式中の総和については単純化がよく知られている。
【0144】
【数22】
【0145】
従ってTnを表す式は、
【0146】
【数23】
【0147】
【数24】
【0148】
に単純化される。
所与の種類の細胞について、p、s、c、およびlは一定であるため、角括弧内の項を定数に置き換えて、
【0149】
【数25】
【0150】
および
【0151】
【数26】
【0152】
とすることができる。
この方程式は、式(25)の対数をとることにより一次式で表して
【0153】
【数27】
【0154】
とすることができる。
方程式(27)を検討すると、この直線の傾きはlog sで、y切片はlog Bである。実験上の観点から見ると、このことは、総細胞数の対数を継代数に対してプロットすると、このプロットの傾きを調べることによりニューロスフェア中の幹細胞の数を計算することができ、y切片を調べることにより前駆細胞の数を計算することができる、ということを意味する。このプロットを構築する場合、継代数が前駆細胞の寿命より大きいときのデータポイントのみを含めるように注意しなければならない。前駆細胞の寿命は、l≧nの場合に所与の継代とその前の継代とにおける総細胞数の比率は一定((Tn+1/
Tn)=s)であるが、l<nの場合はこの比率は一定ではないことに注意して計算される。従ってl=n+1であり、ここでnは上記の比率がsと等しくならない最大の整数である。この手法はノイズの多いデータを処理するのに適している場合がある。
【0155】
注意すべき重要なことは、最初のl−1継代まで、すなわち(1,logT1),・・・,(l−1,logTl−1)は、直線にのらないことである。このことは方程式(23)および(24)を調べればわかる。
【0156】
方程式(27)から、ニューロスフェア中の前駆細胞の数pは、直線の傾きには影響しないことがわかる。傾きが変わる場合、これは幹細胞の数が変わることを意味するはずである。傾きが0である場合、つまり、直線が水平の場合、ニューロスフェア中には幹細胞が1つだけ存在し、従って、各継代における細胞の総数は増大しない。
【0157】
ニューロスフェアの成長条件が変わる場合、例えば成長因子がEGFのみからEGF+FGFのみに変わる場合、直線の傾きは依然としてニューロスフェア中の幹細胞の数によってのみ決定される。この主張の証拠は、本項の残りの部分で述べる。
【0158】
r番目の継代後に条件が変わり、生じる幹細胞の数がqに、前駆細胞の数がwに、前駆細胞の寿命がm世代に変わると仮定する(便宜上、m≦lとするがm>lの場合も同じである)。r+l回の継代後の総細胞数は、
【0159】
【数28】
【0160】
で与えられる。
r+m−1回の継代後には、総数は
【0161】
【数29】
【0162】
であり、r+m回の継代後には、総数は
【0163】
【数30】
【0164】
である。
導き出すと、r+N回の継代後の総細胞数は、
【0165】
【数31】
【0166】
【数32】
【0167】
となり、ここで
【0168】
【数33】
【0169】
である。
線形化すると、
【0170】
【数34】
【0171】
である。
したがって、上述のように、条件の変更後この直線の傾きは幹細胞の数によってのみ影響されることが分かる。
【0172】
上記のアルゴリズムに基づいて、幹細胞および前駆細胞の存在頻度を以下の工程、すなわち
1. ニューロスフェア分析法において細胞を連続的に継代し、各継代で生成された細胞の総数を、その前の継代の総細胞数と、現在の継代で生成された細胞における増加倍数とを乗じることに基づいてプロットする工程;
2. 各継代で生成された細胞の総数の対数をとることにより、対数増殖カーブを線形化する工程;
3. 最も良くフィットする直線を計算し、次いで直線式(y=mx+b)を用いて直線の傾きとy切片を計算し、次には上記の式を使用して幹細胞の数と前駆細胞の数とを明らかにする工程
から計算することができる。
【0173】
実施例2:ヒト胎児神経幹細胞の連続継代および数学的モデル化に基づく幹細胞と前駆細胞の存在頻度の分析
実施例1の数理モデルを、連続的に継代したヒト胎児神経幹細胞に適用した。ヒト胎児神経幹細胞を、参照により全体が本願に組み込まれるベスコビ(Vescovi)ら、1999年、Exp.Neurol.第156巻、p.71−83に記載されているような従来の技術(すなわち、細胞を、その前の継代由来の細胞の一部を用いて、かつマイトジ
ェンであるEGFおよびFGF2を補足した無血清培地を用いて、各継代につきクローン化しうる密度で播種する)を使用して、連続的に継代した。各継代の終了時に、細胞を個々に分散させて単個細胞浮遊液とし、この単個細胞浮遊液の細胞を計数し、トリパンブルー排除法によって生細胞および死細胞の数を計数することにより、総細胞数を決定した。各継代終了時の理論上の総細胞数(継代の終了時に計数された総細胞数と、計数されたその細胞を生成させるために播種された継代直前の細胞の一部との関数である)を継代数に対してプロットして(図1A)成長曲線を得た。各継代の終了時の総細胞数の対数も継代数に対してプロットした(図1B)。線形の対数プロットについて最もフィットする傾向線を作成し(図1B)、直線式(y=mx+b)を用いて傾きとy切片とを決定した。その後、幹細胞および前駆細胞の存在頻度を、実施例1の方程式(26)および(27)によって計算した(n=1、l=n+1=2、c=1000とした;これらの値は以降全ての実施例でも使用)。幹細胞の存在頻度は細胞集団全体の0.24%であると算出され、前駆細胞の存在頻度は細胞集団全体の1.15%であると算出された。
【0174】
実施例3:神経コロニー形成細胞法におけるヒト胎児神経幹細胞の幹細胞および前駆細胞の存在頻度の分析
神経コロニー形成細胞法(NCFCA)は、神経コロニーの大きさの分析に基づいて幹細胞および前駆細胞の存在頻度を決定するために使用することができる。NCFCAは、参照によってその全体が本願に組み込まれる2005年5月26日に公開された米国特許出願番号第2005/0112546号に述べられている。簡潔に述べると、NCFCAは、軟寒天培地、好ましくはコラーゲンベースまたはメチルセルロースベース(IMDM、DMEM/F12、マッコイ(McKoy’s)、イスコフ(Iscoves))の軟寒天培地中に神経細胞を浮遊させることにより行なわれる。軟寒天培地は、神経細胞を培養するために使用されるのと同じ適切な培地(例えばNeurocult(TM)[ステムセルテクノロジーズ社(StemCell Technologies, Inc.]無血清培地でサイトカインを含まずNeurocult(TM)増殖サプリメントとEGFを含むもの)にコラーゲンまたはメチルセルロースを加えて成るものでよい。培地は血清を含まないことが好ましい。軟寒天培地中の細胞は、データの統計学的解析に十分な数のコロニーを生じる濃度(例えば35mmの培養ディッシュ当たりの細胞数が1,000〜25,000個、好ましくは2,500〜7,500個)に播種する。形成されるコロニーは、単一の細胞すなわち神経幹細胞または前駆細胞いずれかから生じる。このコロニーを、コロニー間の大きさおよび差異が識別可能になるまで(例えば約10〜30日)培養し、次にコロニーを計数し、かつスコアリング用ディッシュのグリッドを使用してコロニーの大きさを推定する。単一の神経幹細胞から生成されたコロニーは時間とともに大きく成長し続けるが、神経前駆細胞から生成されたコロニーは成長力に限界があり、従って時間とともに大きく成長し続けることはない。コロニーの大きさから、増殖力の高いNSC(HPP−NSC)、増殖力の低いNSC(LPP−NSC)、および神経前駆細胞が区別される。したがって、生成されたコロニーの大きさは、そのコロニーが神経幹細胞から生成したか神経前駆細胞から生成したか、さらにはそのNSCの増殖力が高いか低いかの指標となりうる。特に、(ディッシュ上の他のコロニーに比べて)より大きなコロニーは、増殖力の高い神経幹細胞を示し、中型のコロニーは増殖力の低い神経前駆細胞を示し、より小さなコロニーは神経前駆細胞を示す。「より大きなコロニー」または「より小さなコロニー」の実際の直径は、コロニーを培養する時間の長さなど多くの要因によって変化する。例えば、細胞2,500個/ディッシュで14〜28日間培養した後、コロニーを直径に基づいて4つのカテゴリー、すなわち(1)>2.0mm、(2)1−2mm、(3)0.5−1mmおよび(4)<0.5mmのうちの1つに分類した。したがって、コロニーを少なくとも14日間培養すると仮定すると、直径が2.0mmを超えると神経幹細胞から生成されたコロニーを示す。NCFCAにおいて細胞の種類は、その細胞種が形成する形態に基づいて区別することもできる。波状のコロニーは、神経幹細胞によって生成され、周囲が滑らかなコロニーは神経前駆細胞によって生成される。NCFCAを行
なうためのキットはステムセルテクノロジーズ社から市販で入手可能である。
【0175】
ヒト胎児神経幹細胞に関する幹細胞および前駆細胞の存在頻度を、NCFCAにおいて細胞をプレート培養することにより計算した。継代数12の胎児神経幹細胞の子孫を個々に分散させて単個細胞浮遊液とし、細胞密度を2000個/mlとして20ng/mlのEGFおよび10ng/mlのbFGFとともにプレート培養した。細胞を軟寒天培地において3週間培養した後、コロニーの存在頻度および大きさを計算した。プレート培養した全細胞の2%未満が幹細胞の特性を示すコロニーを形成した。したがって、実施例2における数学的分析法からの結果はNCFCAの結果と一致している。
【0176】
実施例4:白血病抑制因子(LIF)は、連続的に継代されたヒト胎児神経幹細胞における幹細胞および前駆細胞の存在頻度を低下させる
連続的に継代されたヒト胎児神経幹細胞を、実施例2のような通常の増殖条件(マイトジェンFGF2およびEGFを含む)で、20ng/mlのヒトLIFを添加したものと添加していないものについて培養した。いずれの群についても成長曲線を作成し(図2A)、各継代の終了時に生成された細胞の理論上の総数の対数をとることにより均等目盛に変換し(実施例2を参照)、最も良くフィットする傾向線に基づいて傾きおよびy切片を決定した(図2B)。実施例1の方法による幹細胞および前駆細胞の存在頻度の分析から、LIF存在下では幹細胞の存在頻度が48%、前駆細胞の存在頻度が18%低下することが明らかとなった(図2C;y軸は対照の値に対する割合(%)を表す)。従って、LIFは連続的に継代されたヒト神経幹細胞中の神経幹細胞および神経前駆細胞の存在頻度を低下させるために使用することができる。
【0177】
実施例5:骨形成タンパク質2(BMP−2)は、連続的に継代されたヒト胎児神経幹細胞における幹細胞および前駆細胞の存在頻度を低下させる
連続的に継代されたヒト胎児神経幹細胞を、実施例2のような通常の増殖条件(マイトジェンFGF2およびEGFを含む)で、20ng/mlのヒトBMP−2タンパク質を添加したものと添加していないものについて培養した。いずれの群についても成長曲線を作成し、各継代の終了時に生成された細胞の理論上の総数の対数をとることにより均等目盛に変換し、最も良くフィットする傾向線に基づいて傾きおよびy切片を決定した。実施例1の方法による幹細胞および前駆細胞の存在頻度を分析すると、BMP−2が存在するといずれの種類の細胞においても低下することが明らかとなった。従って、BMPは連続的に継代されたヒト神経幹細胞中の神経幹細胞の存在頻度および神経前駆細胞の存在頻度を低下させるために使用することができる。
【0178】
実施例6:連続的に継代された多形膠芽腫(GBM)由来の細胞は幹細胞の重要な特徴を示す
ヒトGBMには、神経幹細胞が生体条件外(ex vivo)で増殖できるように調整された条件下で増殖する、腫瘍性神経幹細胞(tNSC)が含まれている。参照によって全体が本願に組み込まれるガルリ(Galli)ら、2004年、Cancer Research 第64巻、p.7011−7021を参照されたい。tNSCは、上述のガルリ(Galli)らの文献(2004年)および参照によって全体が本願に組み込まれるベスコビ(Vescovi)ら、Exp.Neurol.第156巻、p.71−83、1999年の方法に従って腫瘍サンプルから単離することができる。簡潔に述べると、tNSCを単離するには、最初に、腫瘍サンプルを、07.mg/mLのオボムコイドを含んでいるダルベッコ改変イーグル培地(DMEM)/F12の中で粉砕して個々に分離する。細胞を遠心分離によって回収し、成長因子を含まず化学的組成の明らかなNS−A培地(カナダ国ブリティッシュコロンビア州バンクーバー所在のステムセルテクノロジーズ(Stem Cell Technologies))に2mMグルタミン、0.6%グルコース、9.6gm/mLプトレッシン、6.3ng/mLプロゲステロン、5.2
ng/mL亜セレン酸ナトリウム、0.025mg/mLインシュリン、0.1mg/mLトランスフェリンを含めたものに再浮遊させる。その後、生細胞を、化学的組成の明らかな同じNS−A培地に20ng/mLのEGFおよび20ng/mLのFGF2を加えた中に、細胞密度を2,500−5,000個/cm2として25cm2組織培養フラスコ中に播種する。必要ならば培地を24−48時間ごとに新鮮な培地に交換する。播種の5〜20日後に、参照によって全体が本願に組み込まれるレノルズ(Reynolds)およびワイス(Weiss)、1992年、Science 第255巻、p.1707−1710によって記述されるように、正常な神経幹細胞によってインビトロで形成される古典的なニューロスフェアに似た腫瘍細胞のニューロスフェアを観察することができる(図3Aを参照:図3Aは20倍の位相差観察像である)。腫瘍細胞のニューロスフェアを個々に分散させて同じ条件下で連続的に再継代すると、tNSCは新しい腫瘍細胞ニューロスフェアを構築する。tNSCは、培養下で安定的に自己複製して増殖する(2つの異なるGBM細胞株について各分裂における理論上の総細胞数を示す図3Bを参照されたい)。tNSCの子孫は分化条件下でニューロン、乏突起膠細胞および星状細胞を生じるので、tNSCはインビトロにおいて多能性である。例えば、抗β−チューブリン抗体、抗グリア線維性酸性タンパク質(GFAP)抗体(GFAPは星状膠細胞への分化のマーカーである)、および抗ガラクトセレブロシド(GalC)抗体による、分化した子孫の染色が観察された(GalCは乏突起膠細胞のマーカーである)。
【0179】
免疫不全マウスの脳へ移植すると、tNSCは、ちょうど典型的なヒトGBMと同じように、星状膠細胞特異的マーカーとの免疫反応性を備えたGBMを生じる。これらの腫瘍そのものを取り出してtNSCを再培養すると、次世代のtNSCは免疫不全マウスの脳内に移植されるとGBMを再生成する。したがって、ヒトのtNSCはインビボでは単能性で、インビトロでは多能性であり、クローンのレベルに至るまで腫瘍の基となる細胞として働く可能性があり、連続的な再移植の後でさえヒトの疾病の重要な特徴に非常によく似た腫瘍をマウスにおいて創り出すことができる。したがって、ヒトのtNSCは、ヒトGBMの増殖および再発に関係するように思われる。
【0180】
実施例7:腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を計算するための連続的に継代されたGBM腫瘍細胞の数学的モデル化の使用
幹細胞および前駆細胞の存在頻度を、実施例1の分析法を使用して3つのGBM細胞株について測定した。これらの細胞株は、3人の異なる患者の腫瘍(C.ベスタ神経学研究所(Neurological Institute C. Besta)から入手し、世界保健機構(WHO)のガイドラインによって分類した腫瘍)に由来するものである。腫瘍細胞を実施例6の方法を使用して連続的に継代し、生成された細胞の理論上の総数を、各細胞株(627、913、1022)について継代ごとにプロットした(図4Aを参照)。各継代の終了時に生成された細胞の理論上の総数の対数を計算し、最も良くフィットする傾向線に基づいて傾きおよびy切片を計算した(図4Bを参照)。幹細胞および前駆細胞の数についての度数は実施例1の方法を使用して計算した。腫瘍性幹細胞の存在頻度は細胞全体の0.44%であり、腫瘍前駆細胞の存在頻度は細胞全体の1.56%であった(図4Cに3つのGBM細胞株各々について示した)。
【0181】
実施例8:神経コロニー形成細胞法(NCFCA)における、GBM腫瘍性幹細胞由来の子孫細胞からの腫瘍性幹細胞および腫瘍前駆細胞の存在頻度の算出
継代数15のGBM細胞を、実施例3に記載のように神経コロニー形成細胞法(NCFCA)でプレート培養した。培養物に毎週新鮮な培地を供給し、3週後にコロニーの直径を測定した。この分析法では、最も大きなコロニーだけが、軟寒天培地からの抽出およびサブクローニングの後で幹細胞の特性を示す。播種した細胞全体の0.25%〜0.65%が大きなコロニーを形成した。図5の主グラフを参照すると、播種した細胞全体に対する、直径が1,500−2,000μm;1,000−1,500μm;500−1,0
00μm;および<500μmのコロニーを形成するものの割合(%)(y軸)を示している。図5の主グラフに挿入されたグラフは、播種した細胞全体に対する、直径が1,500−2,000μmおよび1,000−1,500μmの区分のコロニーを形成するものの割合(%)(y軸)を、主グラフとは異なるy軸目盛りで示している。したがって、NCFCAおよび実施例7の数学的分析から、GBM腫瘍細胞株中の腫瘍性幹細胞について同様の存在頻度が得られる。
【0182】
実施例9:初代腫瘍サンプルおよびtNSCにおけるすべての種類のBMPレセプタの存在
3種のBMPレセプタ(BMPR1a、BMPR1bおよびBMPR2)の発現を、以下のものについてリアルタイムPCRで測定した。
【0183】
1. 初代ヒト脳腫瘍標本:未分化星細胞腫(AA031217);膠芽腫(GBM050203、GBM040114、GBM040202、GBM050207、GBM050208);および胚芽異形成性神経上皮新生物(NND040115);
2. 正常なヒト胎児神経幹細胞(fNSC);および
3. 実施例6の方法によって調製されたヒト膠芽腫の腫瘍性神経幹細胞(tNSC)の細胞株(GBM010627、GBM020913、GBM021022)。
【0184】
以下の方法を用いた。最初に、TRIzol試薬(米国メリーランド州ロックヴィル所在のライフ・テクノロジーズ(Life Technologies))を使用してfNSC、tNSCs、および初代腫瘍サンプルから全RNAを単離し、次に、SuperScript(登録商標)RnaseH逆転写酵素(米国メリーランド州ロックヴィル所在のライフ・テクノロジーズ)を使用して逆転写した。鋳型として使用したcDNAはすべて、ハウスキーピング遺伝子としてグリセルアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)および陽性対照としてMCF−7細胞株を使用して、あらかじめ標準化されたものとした。その後、各BMPレセプタ特異的プライマーを使用し、Brilliant(登録商標)SYBR(登録商標)グリーンQPCRコア試薬キット(米国カリフォルニア州ラ・ホーヤ所在のストラタジーン(STRATAGENE))を使用して、定量的リアルタイムPCR反応を3連で実施した。SYBRグリーン色素はあらゆるPCR生成物に結合するため、配列特異的プローブを使用する必要がない。Brilliant SYBRグリーンのマスターミックスは、UNGによるコンタミネーション除去プロトコールに使用するdUTPを含んでいる。使用したプライマー配列は以下のとおり、すなわち
プライマー配列×逆転写(RT)PCR
hBMPR−1A Fw:5’−AATGGAGTAACCTTAGCACCAGAG−3’
hBMPR−1A Rw:5’−AGCTGAGTCCAGGAACCTGTAC−3’
hBMPR−1B Fw:5’−GGTTGCCTGTGGTCACTTCTGG−3’
hBMPR−1B Rw:5’−TAGTCTGTGATTAGGTACAACTGG−3’
hBMPR−2 Fw:5’−TCAGATATATGGCACCAGAAGTG−3’
hBMPR−2 Rw:5’−GTGGAGAGGCTGGTGACACTTG−3’
プライマー配列×リアルタイムPCR
hBMPR1a Fw:5’−caggttcctggactcagctc−3’
hBMPR1a Rw:5’−ctttccttgggtgccataaa−3’
hBMPR1b Fw:5’−aaaggtcgctatggggaagt−3’
hBMPR1b Rw:5’−gcagcaatgaaacccaaaat−3’
hBMPR2 Fw:5’−gctaaaatttggcagcaagc−3’
hBMPR2 Rw:5’−cttgggccctatgtgtcact−3’
である。
【0185】
蛍光性の発光をリアルタイムで記録した(Chromo4(TM)4色リアルタイムPCR検出器、MJリサーチ(MJ Research)、バイオラッド(BIO−RAD)、米国)。遺伝子発現の解析は、相対的な定量法の比較Ct法(ヒグチ(Higuchi)ら)を使用して完了した。各遺伝子について、相対的なRNA量を2つの内在性の対照すなわちGAPDHおよび18s rRNAに対して標準化した。3連実験のそれぞれを標準化し、平均の相対量(RQ)を各遺伝子について報告する。平均で何倍変化したかを、3連実験の標準偏差および95%信頼区間と共に計算した。
【0186】
図6に結果を示す。この結果は、悪性度の異なる様々な腫瘍が特有のBMPR発現プロファイルを有することを示している。したがって腫瘍のBMPR発現プロファイルは、腫瘍の特徴を解析し、腫瘍の悪性度を予測するための診断法において使用することができる。
【0187】
実施例10:白血病抑制因子(LIF)およびBMPは、連続的に継代されたヒトGBM細胞株における幹細胞の存在頻度を低減する
3人の異なる患者から得られたGBM腫瘍細胞を、LIF(20ng/ml)またはBMP−4(20ng/ml)のいずれかを含むかまたは含まないコントロール培地(マイトジェンEGF+FGF2を含む;実施例6を参照のこと)の中で連続的に継代した。実施例1の方法を使用した幹細胞および前駆細胞の存在頻度の分析から、LIFまたはBMP−4の添加により腫瘍性幹細胞の数が著しく減少し(統計的有意性 p<0.01)、BMP−4の添加により腫瘍前駆細胞の数が著しく減少する(統計的有意性 p<0.05)が、LIFを添加しても腫瘍前駆細胞集団の存在頻度は有意には変化しないことが明らかとなった。これらの結果は、BMP分子が腫瘍性幹細胞および腫瘍前駆細胞の増殖を低減し、LIFは腫瘍性幹細胞の数を選択的に低減し腫瘍前駆細胞群に対しては影響しないことを示している。図7は、結果をグラフで(未処置対照の値に対する割合(%)として)表している。これらの結果は、LIFおよびBMPがそれぞれ、マイトジェンEGFおよびFGF2の存在下であってもtNSCの増殖を低減するのに有効であることを示している。これは、LIFおよびBMPのうち少なくともいずれか一方による処置がヒトの脳腫瘍の治療に有効となることを示している。
【0188】
実施例11:LIFおよびBMP−2を連続的に継代されたGBM由来の細胞に適用すると、幹細胞および前駆細胞の存在頻度が低下する
コントロール培地(マイトジェンEGF+FGF2を含む;実施例6を参照のこと)中で連続的に継代したGBM細胞(実施例6を参照)を、LIF、BMP−2またはBMP−2+LIFの組み合わせで処理した。連続的に継代した対照のGBM細胞は、どちらのタンパク質による処理も行わなかった。幹細胞および前駆細胞の存在頻度を計算するために実施例1の方法を使用して成長曲線を比較した。データから、LIFおよびBMP−2を一緒に使用すると、単独で使用した時よりも腫瘍性幹細胞および腫瘍前駆細胞の存在頻度が大きく低下することが明らかである。図8は、結果をグラフで示すものである(y軸は、未処理対照の値に対する割合(%)を表す)。これらの結果は、LIFおよびBMP−2の同時投与が、マイトジェンEGFおよびFGF2の存在下であってもtNSCの増殖を低減するのに有効であること、またLIFおよびBMP−2の同時投与はヒトの脳腫瘍を治療するのに有効であろうことを示している。
【0189】
実施例12:培養したGBM細胞をBMP−2で一時的に処理すると幹細胞および前駆細胞の存在頻度が永続的に低下する
3人の異なる患者から得られたGBM腫瘍細胞を、コントロール培地(EGF+FGF2を含む;実施例6を参照のこと)中、BMP−2(20ng/ml)を添加するか(図9に「BMP」と示す)または1回の継代時にBMP−2に一時的に曝露して(図9に「BMP後」と示す)、連続的に継代した。実施例1の方法を使用した腫瘍性幹細胞および腫瘍前駆細胞の存在頻度の分析から、いずれのBMP処理群においても腫瘍性幹細胞および腫瘍前駆細胞の数が著しく減少することが明らかになった。これらの結果は、BMP−2に一時的に曝露すると、マイトジェンEGFおよびFGF2の存在下であっても腫瘍性幹細胞および腫瘍前駆細胞の存在頻度が永続的に低下することを示している。これらの結果は、BMP−2治療がヒトの脳腫瘍の増殖に永続的効果を有すると思われることを示している。
【0190】
実施例13:LIFおよびBMPを用いた事前処理後の培養GBM細胞における腫瘍形成性の低下
免疫不全マウスの脳(腹側外側の線条体)に、ヒトGBM由来の腫瘍性神経幹細胞100,000個を定位注入により移植した。腫瘍性神経幹細胞はマウスにおいてGBMを発症させた。これらのGBM病変は、正常組織とは明白に区別される、核が過剰に存在する広範な領域として明らかになった(図10の上側パネルを参照)。同じ腫瘍性神経幹細胞を移植に先立って100ng/mLのBMP−4または100ng/mLのLIFで48時間前処理すると、腫瘍の形成は著しく(ほぼ80%)低減され、核が過剰に存在する領域は検出困難なものが多い(図10の下側パネルを参照)。これらの結果は、BMPおよびLIFのうち少なくともいずれか一方による治療がヒト脳腫瘍の治療に有効であることをさらに示すものである。補足例については、実施例22も参照されたい。
【0191】
実施例14:LIFおよびBMP−2処理後の乳癌組織における腫瘍性幹細胞の存在頻度の分析
腫瘍切除のために生検を受ける患者から同意のもとに腫瘍標本を入手し、1:1コラゲナーゼ/ヒアルロニダーゼ溶液中37℃にて1時間酵素消化してから40μmフィルタで濾過し、細胞を、マイトジェンEGFおよびFGF2(20ng/ml)を含んだ無血清DMEM/F12ホルモン混合物(NeuroCult(登録商標)、ステムセルテクノロジーズ(Stem Cell Technologies))中にクローン化しうる密度で播種した。3−5日後、クローンから生じた細胞塊が懸濁浮遊しているのが観察される。これらの細胞を回収し、酵素的に個々に分散させ、新鮮な増殖培地に再度播種する。このようにして4−7日ごとに継代した細胞は、生成される総細胞数が幾何学的に増加する。
【0192】
連続的に継代した乳房腫瘍由来の幹細胞を、LIFおよびBMP−2のうち少なくともいずれか一方に曝露し、対照の培養物と比較する。神経コロニー形成細胞法において乳房性球状細胞塊(mammary sphere)を分散させた細胞を播種し、実施例1の数学的モデルを用いて一連の成長曲線を分析することにより、処理群および対照群の腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を分析する。
【0193】
実施例15:LIFおよびBMP−2処理後の前立腺癌組織における腫瘍性幹細胞の存在頻度の分析
リンパ節転移から前立腺がん細胞を入手し、5%の血清と幹細胞マイトジェンEGFおよびFGF2とを添加したDMEM/F12ホルモン混合物(ステムセルテクノロジーズ)で培養する。クローンから生じた球状塊をトリプシン処理してから連続的に継代し、継代した細胞(未処理のもの、またはLIFおよびBMP−2のうち少なくともいずれか一方で処理したもの)由来の増殖データを用いて、実施例1の数学的モデルを用いて腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を計算する。
【0194】
実施例16;LIFおよびBMP−2処理後の黒色腫組織における腫瘍性幹細胞の存在頻度の分析
切除された腫瘍から黒色腫細胞を得て、哺乳類の皮膚から多能性幹細胞を培養するために使用された方法(参照により全体が本願に組み込まれるトマ(Toma)ら、Nat Cell Biol.2001年9月、第3巻(9)、p.778−84を参照のこと)に従って幹細胞を単離する。簡潔に述べると、組織を小片(<2mm)に切断し、HEMで洗浄し、0.1トリプシンで37℃にて30分間消化する。サンプルをPBSですすぎ、機械的に分散させて単個細胞浮遊液とし、40um細胞ストレーナでろ過し、ヌンク(Nunc)のT−80組織培養フラスコ中で1ml当たり細胞50,000個の密度で播種する。増殖培地は、幹細胞マイトジェンEGFおよびFGF2を添加したホルモン混合物含有無血清DMEM/F12(ステムセルテクノロジーズ)である。7−10回の分裂の後、クローンから生じた細胞塊が培養物中に確認される。これらのメラノスフェアを回収し、分散させて単個細胞浮遊液とし、幹細胞マイトジェンを含む新鮮な培地を使用して、再度播種する。このように継代した培養物をLIFまたはBMP−2で処理し、経時的に生成された細胞の総数を対数目盛でプロットする。治療群および対照群の腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を、実施例1の数学的モデルを使用して分析する。
【0195】
実施例17:腫瘍切除後のCED法によるLIF投与を用いた再発性多形膠芽腫の治療
再発性多形膠芽腫の患者を選出する。腫瘍切除に続いて、切除腔を囲んでいる脳実質に、2〜3個のカテーテルを、脳溝または脳室内に入らないように画像誘導を使用して配置する。該カテーテルを通して、72mLのヒトLIF(1μg/mL)を、シリンジ・ポンプを使用して96時間かけて注入する。
【0196】
実施例18:ポリマービーズを用いたBMP−4投与による再発性多形膠芽腫の治療
再発性多形膠芽腫の患者を選出する。腫瘍切除に続いて、BMP−4で飽和させたポリアクリル酸ビーズ(1週間以上BMP−4を放出する)を切除部位に埋め込む。
【0197】
実施例19:GBM標本由来の細胞におけるBMPRの発現
GBM組織由来の細胞におけるBMPおよびそのレセプタ(BMPR)の転写物およびタンパク質の発現を、GBM組織由来のCD133+tNSC集団について評価した(シン(Singh)、S.K.ら、Nature 第432巻、p.396−401、2004年)。下記のインビトロのデータは、1つの代表的なGBM標本由来の細胞から得たデータを例証するものであるが、同等の結果は別の4つのサンプルでも得られている。該サンプルは選別されたCD133+細胞または非選別細胞を含み、該細胞はGBMから急性単離したものであるかまたはマイトジェンとともに短時間培養した後(培養した細胞)であるかのいずれかである(ガルリ(Galli)ら、Cancer Res 第64巻、p.7011−21、2004年;シン(Singh)、S.K.ら、Nature 第432巻、p.396−401、2004年)。BMPRの3つのサブタイプ(BMPR1A、−1B、−2)およびBMPのすべての転写物が、急性単離したGBM細胞および培養したGBM細胞のいずれにおいても見出された。図11Aを参照すると、同図においてレーン1は陰性対照、レーン2は急性単離したGBMのCD133+細胞、レーン3は培養したGBMのCD133+細胞、レーン4は陽性対照のMCF7細胞である。さらに、同系統のレセプタおよびBMP−4タンパク質がいずれの集団においても、CD133+画分およびCD133−画分のいずれにおいても見出された。図11B−Gを参照すると、単離直後(図11B−D)および培養した(図11E−G)CD133+GBM細胞における記載のタンパク質の免疫蛍光が示されている。
【0198】
最も有効なリガンドのうちの1つであるBMP−4を飽和濃度(100ng/ml)で添加した後にSmad1−5−8のリン酸化が観察されたので、BMPRは機能性を有しており、一方p38MAPK経路の活性化は検出されなかった。図11H−Jを参照する
と、GBM細胞における、記載の時間(分)での、レセプタに活性化されたSmadタンパク質のリン酸化および核移行を示す(抗リン酸化Smad1,5,8)。
【0199】
図11K−Pは、記載のタンパク質のウエスタンブロット解析を示す。図11Kは、急性単離した(左側のレーン)、および培養した(右側のレーン)CD133+GBM細胞におけるBMP−4タンパク質を示す。図11Lは、BMP−4処理した培養細胞でSmad1のレベルは変化しなかったことを示している(左側のレーンが対照、右側のレーンがBMP−4処理)。図11M−Nは、単離直後の細胞(図11M)および培養した細胞(図11N)におけるBMP−4存在下でのSmad1,5,8のリン酸化の増大を示している(左側のレーンが対照、右側のレーンがBMP−4処理)。図11O−Pは、培養したGBM細胞(図11O)および急性単離したGBM細胞(図11P)におけるBMP−4処理後のSmad4発現の増大を示している(左側のレーンが対照;右側のレーンがBMP−4処理)。
【0200】
この結果は、Smad1,5,8複合体もまたGBMの治療に有用な治療標的であり得ることを示すものでもある。例えば、該複合体のリン酸化を増大させる治療薬はtNSCに対してBMP−4と同じ効果を有するであろう。本開示の他の箇所に開示したスクリーニング法は、そのような治療薬を得るために使用されてもよい。
【0201】
実施例20:GBMのtNSCに対するBMP−4曝露の作用に関する検討
GBMから単離された細胞に対するBMP−4の作用の性質について検討した。他の細胞系(ハラハン(Hallahan)ら、Nat Med 第9巻、p.1033−8、2003年;ズザルテ−ルイ(Zuzarte−Luis)およびヒューレ(Hurle)、Semin Cell Dev Biol 第16巻、p.261−9、2005年;フルスカ(Hruska)ら、Circ Res 第97巻、p.105−14、2005年)、例えば髄芽細胞腫(グラハム(Graham)ら、Mol Cell Neurosci 第8巻、p.76−83、1996年)などとは異なり、BMP−4は細胞死(ヨウ化プロピジウム排除法により測定)またはアポトーシス(TUNEL法により測定)を生じなかったが、マイトジェンに応答するGBM細胞の増殖を著しく低減した(Ki67免疫蛍光法により測定)。図12Aは、GBM培養物におけるBMP−4の作用をグラフで示している(白抜きのカラムは対照の培養物、暗色のカラムはBMP−4処理物;平均+SE、n=3;*p、0.005;PI=ヨウ化プロピジウム)。
【0202】
増殖に対する他のBMPの作用も分析した。図15を参照すると、GBM細胞の増殖に対する他のBMP(100ng/ml)の作用が示されている。BMP−2、−4、−5、−6、−7、−8bは細胞増殖を抑制するが、BMP1、−3および−3bは、TGFβ1、2およびTGFβ1,2(TGFβアゴニストのキメラポリペプチド)と同様に効果がないようであった(すべて100ng/ml)。*BMPと対照、p<0.005、平均±SE、n=3、両側スチューデントt検定;**BMP4と対照、p<0.001、平均±SE、n=3、両側スチューデントt検定。したがって、BMP−2、−4、−5、−6、−7、−8bも、特にGBMの治療のための本開示の方法および組成物に有用であろう。
【0203】
BMP−4の抗増殖作用は、BMP−4に応答してG0/G1期のGBM細胞の数が著しく増加し、S期の細胞の割合(%)が減少することを示した細胞周期分析によって確証された。GBM細胞に見出されるTGFβ(ケルマン(Kjellman)ら、Int J Cancer 第89巻、p.251−8、2000年)は、BMPのシグナル伝達経路とオーバーラップするシグナル伝達経路を用いて(カナリ(Canalis)ら、Endocr Rev 第2巻、p.218−35、2003年;ゴレステーン(Golestaneh)ら、Oncogene 第24巻、p.5722−30、2005年)、
促進性の作用または反促進性の作用を誘発する(ジェニングス(Jennings)ら、J Neurooncol 第36巻、p.123−40(1998))が、BMP4とは異なり、本明細書に記載のBMP4作用の特異性の基礎をなすGBM細胞の増殖への影響は示さなかった。
【0204】
古典的な2つの分析法、すなわち1つはクローン形成の指標を測定し、従ってクローンを形成するニューロン前駆体の割合(%)を計測する分析法(グリッティ(Gritti)ら、J Neurosci 第16巻、p.1091−100、1996年;レノルズ(Reynolds)ら、Dev Biol 第175巻、p.1−13、1996年)ならびに第2には増殖速度論データに基づいてNSC集団の大きさの増大を測定する分析法(ガルリ(Galli)ら、Development 第129巻、p.1633−44、2002年;レノルズ(Reynolds)ら、Nat Methods 第2巻、p.333−6、2005年)により、BMP−4の細胞増殖抑制作用がGBM細胞中のtNSC集団に影響を与えたことが確認された。BMP−4に48時間曝露すると、GBM細胞におけるクローン形成の指標(播種した細胞の合計に対する形成クローンの割合(%))はインビトロで70%を超えて低減された(図12Bを参照すると、急性単離したGBM細胞および培養したGBM細胞の両方についての、対照群およびBMP−4処理群に関するクローン形成の指標がグラフで示されている;平均±SE、n=3;*はp<0.005)。さらに、初代腫瘍標本からの単離直後にGBM細胞をBMP−4に曝露すると、同細胞が培養時に増殖する能力が消失し、一方、既にインビトロで増殖中のGBM細胞にBMP4を添加すると同細胞の成長速度が大きく低下した。ニューロスフェア分析法を使用してインビトロでのGBM細胞の増殖をグラフに示す図12Cを参照すると、急性単離したGBM組織からの細胞はBMP−4の存在下では連続的に二次培養できなかったことが明らかである(黒い点線に黒塗り三角形)。図12Cはまた、同じ条件下で短時間増殖させた後(菱形)、急性単離した同じ組織に由来する細胞をBMP−4で処理しても、成長曲線の傾きが著しく小さくなった(正方形)ことも示している。同様の結果は、BMP−4で処理したヒト胎児NSCについても見られ(図12C参照、黒い星印(対照)と黒丸(BMP−4))、ヒト神経膠腫細胞株U87(BMPRを持たない)は影響を受けなかった(図12Cを参照、白抜き三角形(対照)とバツ印(BMP−4))。
【0205】
細胞蛍光測光法から、BMP4処理の結果、急性単離したGBM細胞および培養したGBM細胞のいずれにおいても、CD133+集団の大きさがほとんど半分になること(図12Aを参照)が示された。これらのデータはともに、BMP−4がGBM細胞のtNSC集団を標的とすることができることを示している。
【0206】
GBM細胞をBMP−4に曝露すると、インビトロで明らかな形態学的変化が生じた。マイトジェン単独で増殖させた細胞に比べ、さらにBMP4を与えられた細胞はより分化の進んだ(扁平、濃密な様相、複雑な突起を有する)形態を呈した。対照の細胞を示す図13A、およびBMP−4に48時間曝露した後の細胞を示す図13Bを参照されたい。これに応じて、星状膠細胞のマーカーの発現(GFAP免疫反応性[IR])のかなりの増加と一緒に、それほど大きくはないがニューロンのマーカー(βIIIチューブリンおよびMAP5)または乏突起膠細胞のマーカー(GalC)に関する標識の増大が観察された。図13C−Hを参照されたい。具体的には、図13C(対照)および図13D(BMP−4)はGFAP−IRを示し;図13E(対照)および図13F(BMP−4)は、βIIIチューブリン−IRを示し;図13G(対照)および図13H(BMP−4)はGalC−IRを示す。
【0207】
特定の分化マーカーを発現している細胞の数を数えることによるBMP4の作用の定量化は、増殖している未分化のGBM培養物中および同細胞内でニューロンの抗原および神経膠の抗原が異常に発現しているという、正常な神経幹細胞では観察されない2つの現象
のため、困難であった。したがって、細胞蛍光測光法を用いて蛍光信号強度を計測し、同方法により、対照に比べてBMP−4で処理した培養物ではGFAP−IRが2倍以上増大すること(図13Iを参照)、そして同様に、ただし顕著ではないがβIIIチューブリン(図13J)およびGalC(図13K)の免疫反応性が増大することが見出された(MEFL=フルオレセイン1分子相当の蛍光量;MEFE=フィコエリスリン1分子相当の蛍光量)。
【0208】
理論または仮説によって制限されるものではないが、ここで述べるBMP−4の作用は、それ自体tNSCである2つの同一の娘細胞を生じる対称分裂の回数の減少によるものであるか、あるいはtNSC群の一部が分化してもはや幹細胞の特性を保持することができないことによるものと考えられる。要するにこのことは、BMP−4が細胞分裂促進性の刺激を無効にして、tNSCに、より成熟した腫瘍形成性の低い表現型を獲得させることができることを示唆している。BMP−4はヒトES細胞に対し、幹細胞集団および増殖速度を増大させる抗分化作用を有するので、上記の結果は予想外である。
【0209】
実施例21:BMP−4はGBSCの腫瘍形成能を抑制し、インビボに送達可能である
急性単離したCD133+GBM細胞および培養されたCD133+GBM細胞の両方を、培養下で48時間BMP−4に曝露した後、免疫不全のscid/bgマウスの片側の線条体へ注入した(3×105個の生細胞)。腫瘍の形成および増大を、同一の条件下だがBMP−4を伴わずに維持されたGBM細胞を与えた対照動物のものと比較した。
【0210】
未処理のGBM細胞を与えた動物はすべて、注入された側に確立した腫瘤を生じた(図14Aを参照)。この腫瘤は、膠芽腫の特徴、たとえば顕著な核異型性、異常な神経膠成分の発現、広範囲な新血管新生および高い有系分裂活動(ガルリ(Galli)ら、Cancer Res 第64巻、p.7011−21、2004年)を示し、側脳室、第三脳室および第四脳室に侵入した。これに対し、BMP4で処理した細胞は侵襲性の腫瘍を形成せず、小さくて範囲の定まった病変を形成し、該病変は注入部位に制限され、分裂指数が低く、脳室への侵入を示さなかった(図14Bを参照)。注入後3〜4か月の間に対照の動物はすべて死亡したが、予めBMP−4処理した細胞を与えたほぼすべてのマウスは生き残った(図14Jを参照。同図は前処理(左側パネル)、同時処理(中心パネル)、および後処理(右側パネル)した例における生存をグラフで示している(ログランク検定、それぞれp<0.001、p<0.001、およびp<0.005)。同じ結果は、BMP−4処理したGBM培養物からの残りのCD133+画分をFACSで精製し、その腫瘍形成性を、対照のGBM細胞から精製した同数のCD133+細胞(動物1匹あたり1.5×105個のCD133+細胞)の腫瘍形成性と比較した場合にも観察された。さらに、従来示されるように(ガルリ(Galli)ら、Cancer Res 第64巻、p.7011−21、2004年;シン(Singh)ら、Nature 第432巻、p.396−401、2004年)、急性単離した対照GBM細胞を与えたマウスの脳からCD133+tNSCを再培養することは常に可能であった(平均のクローン形成頻度:9.0±1.3%[n=2、移植後90日])。これらは、scid/bgマウスの脳に再び脳内移植されると大きな二次腫瘍を生じさせた。反対に、上記のことは、最初の移植においてBMP−4で前処理したGBM細胞を与えた動物では不可能であり、また同じマウスの一次腫瘍から急性単離した細胞3×105個を直接注入しても二次腫瘍を確立することは不可能であった。
【0211】
総合すると、これらの所見は、BMP−4への一時的な曝露であっても、GBMのtNSC集団を激減させ、GBM細胞のインビボにおける腫瘍発生能力を顕著に低下させることを実証するものである。さらに実施例13を参照されたい。
【0212】
実施例22:BMP−4のインビボ送達により脳内腫瘍の確立および増大が防止される
次に、脳内腫瘍の確立および増大を防止する治療法としての、BMP−4のインビボ送達について評価した。GBMを移植する際、細胞と同時(同時処理例)または10日後(後処理例)に、ビヒクル(対照)またはBMP−4で飽和させたポリアクリル酸ビーズ(1週間以上BMP−4を放出)を、細胞移植部位に注入した。双方の処理群における組織学的な一連の再構成から、BMP−4処理した動物では対照に対して腫瘤の最大増量が著しく減少することが明らかになった。具体的には、すべての実験設定において、対照のビーズを与えた動物は大きな悪性腫瘍を生じ(図14C(培養したGBM細胞、注入後4週間、同時処理例);図14E(単離直後のGBM細胞、細胞注入後30日、後処理例))かつすぐに死亡した(図14J)が、BMP4放出ビーズを注入したマウスでは小さく制限された病変が見られ(図14D(培養したGBM細胞、注入後4週間、同時処理例);図14F(単離直後のGBM細胞、細胞注入後30日、後処理例);そして有意に長く生存した(図14J)。対照の腫瘍は、多形態性で、新生物形成性の高い要素を、反応性の染色質および悪性の高い浸潤性の細胞と共に含んでいた(図14G)。反対に、BMP−4処理した動物は、新生物形成性の低い細胞、多数の高度に分化した要素および多数のマクロファージを含む病変を示した(図14H)。分裂指数は、BMP−4処理した動物に対して対照で有意に高かった(同時処理:対照の3.8±0.2に対しBMP−4では0.20±0.1;p<0.01。後処理:対照の4.3±0.3に対しBMP−4では0.7±0.3;p<0.05。平均±SE、n=4、両側スチューデントt検定)。インビボにおける免疫蛍光法から、対照の腫瘍およびBMP4処理した腫瘍のいずれにおいても、星状細胞およびネスチン陽性細胞は存在するが、乏突起膠細胞やニューロン細胞は存在しないことが明らかとなった。
【0213】
これらの結果は、BMP−4を放出するか、そうでなければBMP−4を送達するデバイスの脳内移植が、ヒトにおけるGBMの治療に有効と思われることを示唆している。
実施例23:BMP−4抗体中和実験
実施例19は、内在性のBMP4および他のBMPがGBM細胞中に見出されることを示している。先の実施例で示された所見と一致して、抗BMP4中和抗体はtNSCの増殖を増大させる。これは、内在性のBMPはインビボで生理的にGBM細胞に作用しうるが、その影響は腫瘍の成長を停止させるのには不十分であることを示唆している。この現象の背景にあるメカニズム、例えば腫瘍中に内在性BMPアンタゴニストが存在する可能性について検討することは(カナリ(Canalis)ら、Endocr Rev 第2巻、p.218−35、2003年)、GBM治療のための別の戦略を指し示し得る。さらに、BMP、BMPの同系統レセプタおよびBMPに関連する細胞内の伝達メカニズム、特にSmad経路は、GBMの確立および増殖の主因である細胞に対してより特異的に照準を定める治療法の有望な標的として浮上する。
【0214】
実施例24:初代培養、培養増殖、クローニングおよび細胞株の確立
GBM細胞は、ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021に記述されるように腫瘍サンプルを処理して得た。急性単離した細胞を、CD133への免疫反応性について選別し(以下を参照)、NeuroCult(登録商標)NS−A無血清培地(ステムセルテクノロジーズ)中で最終密度を1cm2あたり細胞2500個として25cm2組織培養フラスコに播種した。培養増殖、クローン形成の分析および集団の解析は、ガルリ(Galli)ら、Development 第129巻、p.1633−44(2002)にすでに記載されているのと同一の条件を使用して実施した。急性単離したCD133+GBM細胞のBMP4による前処理については、これらの細胞を、同じ培地でマイトジェンを欠くもの(対照)または100ng/mlのBMP4を含むものの中で、移植に先立って48時間プレート培養した。
【0215】
実施例25:免疫細胞化学
2.5×104個/cm2のGBM細胞を、FGF2/EGFの存在下でMatrig
el(TM)コーティングされたガラス製カバーガラス上に播種し、BMP4(100ng/ml)で48時間処理した。その後、細胞を洗浄し、4%のパラホルムアルデヒド中で固定した(10分)。ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021に記述されるようにして、神経抗原(GFAP、ダコ・コーポレーション(Dako Corporation);Tuj1、バブコ(Babco);Galc、ケミコン(Chemicon))について多重免疫蛍光法を実施した。Ki67染色(1:1000、英国ニューカースル所在のノボカストラ(NovoCastra))により、増殖している細胞を検出した。
【0216】
4%パラホルムアルデヒド中で固定した後、BMPR−1A、−1Bおよび−2について、製造業者の指示書に従って免疫染色した(1:50 R&Dシステムズ)。リン酸化Smad1について染色する場合(1:100 米国マサチューセッツ州ベヴァリー所在のセルシグナリング(Cell Signaling))、細胞をBMP−4(100ng/ml)で様々な時間(5分〜2時間)処理した。適切なアイソタイプ対照または陰性対照を、これらの手順全体にわたって常に含めた。オリジナルのTUNEL法に、ガブリエリ(Gavrieli)ら、J Cell Biol 第119巻、p.493−501(1992)によるジゴキシゲニンに基づいた変更を加えて、フルオレセイン−dUTP TUNEL法(In Situ細胞死検出キット、フルオレセイン、ロッシュ・アプライドサイエンス(Roche Applied Science))を使用してアポトーシスの細胞を検出した。簡潔に述べると、12mmのカバーガラス上で増殖させた細胞を、4%パラホルムアルデヒド中で室温にて10分間固定し、次にPBSですすいだ。その後、細胞を氷中で2分間透過処理してからTUNEL反応液50ulで標識し、パラフィルムカバーガラス下の湿潤チャンバ内で37℃にて1時間インキュベートした。PBSで洗浄した後、スライドを、DAPIを含んだVectashield(TM)にマウントし、蛍光顕微鏡法で調べた。ヨウ化プロピジウム(PI)染色については、細胞を4%パラホルムアルデヒド中で室温にて10分固定し、PBSですすぎ、PI(1ug/ml)とともに室温で5分間インキュベートした。洗浄後、カバーガラスをDAPI含有Vectashield(TM)にマウントし、蛍光顕微鏡法で調べた。PI排除法により生細胞を示した。実施例19の分析法については、サンプルを各試験条件につき6連で実施した。
【0217】
実施例26:従来型PCRおよびリアルタイムPCR
全RNAを、TRIzol試薬(米国メリーランド州ロックヴィル所在のライフ・テクノロジーズ)を使用して培養GBM細胞および急性単離したGBM細胞から単離し、Superscript(登録商標)RNAseH逆転写酵素(ライフ・テクノロジーズ)を使用して逆転写した。従来型のPCR反応で鋳型として使用したcDNAの量は、グリセリンアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)に関して標準化した。MCF−7細胞株をBMPRに関する陽性対照として使用した。PCR生成物は、臭化エチジウムで染色したアガロース(1%)ゲル中での電気泳動によって視覚化した。
【0218】
定量的RT−PCR反応は、Brilliant(登録商標)SYBR(登録商標)グリーンQPCRコア試薬キット(米国カリフォルニア州ラ・ホーヤ所在のストラタジーン(Stratagene))を用いて3連で実施した。SYBRグリーン色素はあらゆるPCR生成物に結合し、したがって配列特異的プローブを使用する必要がない。蛍光性の発光をリアルタイムで記録した(Chromo4(TM)4色リアルタイムPCR検出器、MJリサーチ(MJ Research)、バイオラッド(BIO−RAD))。遺伝子発現の解析は、相対的な定量法の比較Ct法を使用して完了した。相対的なRNA量を2つの内在性の対照すなわちGAPDHおよび18SリボソームRNA(18S rRNA)に対して標準化した。
【0219】
従来型PCRについては、次のプライマー、すなわち、BMP4、順方向:5’−cttcagtctggggaggag−3’、逆方向:5’−gatgaggtgcccaggcac−3’;BMPR1A、順方向:5’−aatggagtaaccttagcaccagag−3’、逆方向:5’−agctgagtccaggaacctgtac−3’;BMPR1B、順方向:5’−ggttgcctgtggtcacttctgg−3’、逆方向:5’−tagtctgtgattaggtacaactgg−3’;BMPR2、順方向:5’−tcagatatatggcaccagaagtg−3’、逆方向:5’−gtggagaggctggtgacacttg−3’;GAPDH、順方向:5’−cggagtcaacggatttggtcgtat−3’、逆方向:5’−agccttctccatggtggtgaagac−3’を使用した。PCR増幅条件は、56℃でプライマーをアニーリングする35サイクルで構成されるものとした。
【0220】
RT−PCRについては、次のプライマー、すなわち、BMPR1A、順方向:5’−caggttcctggactcagctc−3’、逆方向:5’−ctttccttgggtgccataaa−3’;BMPR1B、順方向:5’−aaaggtcgctatggggaagt−3’、逆方向:5’−gcagcaatgaaacccaaaat−3’;BMPR2、順方向:5’−gctaaaatttggcagcaagc−3’、逆方向:5’−cttgggccctatgtgtcact−3’;GAPDH:従来型PCRについて記載したのと同じプライマーを使用;18S rRNA、順方向:5’−agtccctgccctttgtacaca−3’、逆方向:5’−gatccgagggcctcactaaac−3’を使用した。プライマーの特異性は、解離曲線分析(Opticon(登録商標)2およびChromo4(TM)リアルタイム・システム・ソフトウェア、MJリサーチ(MJ Research))により、PCRを実施するごとに確認した。RT−PCR増幅条件は、56℃でプライマーをアニーリングする40サイクルで構成されるものとした。
【0221】
実施例27:ウェスタンブロッティング
培養GBM細胞および急性単離されたGBM細胞を冷PBSで洗浄し、500μlの1×サンプル・バッファー(62.5mMトリスHCl、25℃でpH6.8;2%(w/v)SDS;10%グリセロール;50mM DTT;0.01%(w/v)ブロモフェノールブルー)で溶解することにより、タンパク質を採取した。サンプルを氷中でインキュベートし、−20℃で保存した。アリコートを5分間沸騰させ、氷中でインキュベートし、SDS−PAGEゲル(10cm×10cm)に20μl/レーンとして載せた。その後、タンパク質をニトロセルロース・メンブレンに転写した。メンブレンを、TBS中5%粉乳/0.1%トゥイーンで室温にて1時間ブロッキング処理し、15mlのTBS/0.1%トゥイーンで3回洗浄した。その後、ブロットを、TBS中5%粉乳/0.1%トゥイーンの中で、抗Smad1−5−8抗体(1:1000;セルシグナリング(Cell Signaling))、抗リン酸化Smad1−5−8抗体(1:1000;セルシグナリング)、抗Smad4抗体(1:200;サンタクルーズ(Santa Cruz))、抗BMP4抗体(1:400、ケミコン(Chemicon))とともにインキュベートした。すべてのブロットについて、メンブレンを15mlのTBS/0.1%トゥイーンで3回洗浄し、次いでホースラディッシュ結合型の適切な二次抗体(1:1000、アマシャム(Amersham))とともに、TBS中5%粉乳/0.1%トゥイーンの中で室温にて1時間インキュベートした。バンドを化学発光(ECL;アマシャム)によって視覚化した。
【0222】
実施例28:フローサイトメトリー
p38およびSmad1−5−8のリン酸化状態を決定するために、細胞調製物を遠心分離し、0.5mlのPBSおよび0.5mlの4%パラホルムアルデヒド中に37℃で
10分間再浮遊させた。その後、あらかじめ冷却したGBM細胞を優しく攪拌しながら同細胞に氷冷100%メタノールをゆっくり添加して、終濃度90%のメタノールとすることにより、細胞を透過処理した。4℃で30分間のインキュベーションおよび遠心分離に続いて、細胞をPBS中0.5%のウシ血清アルブミン(BSA、シグマ)3mlで2回洗浄し、150ulのPBSに再浮遊させ、室温で10分間インキュベートした。インキュベーション後、1:50希釈の抗リン酸化Smad1−5−8ウサギポリクローナル抗体(セルシグナリング)または1:50希釈のリン酸化p38 MAPキナーゼ・ウサギポリクローナル抗体(セルシグナリング)に対し、暗所にて室温で1時間曝露した。十分に洗浄した後、1:800希釈のヤギ抗ウサギIg FITC標識抗体(BD、ファーミンゲン(BD,Pharmingen))を添加し、各チューブを暗所にて室温で30分間インキュベートした。PBS中0.5%BSA(シグマ)3mlで2回洗浄した後、細胞を0.5mlのPBSに再浮遊させ、フローサイトメトリーによって分析した。自己蛍光およびアイソタイプの対照について、上記の分析法すべてについて常に実施した。
【0223】
細胞周期分析については、1サンプルあたり100万個の培養GBM細胞および急性単離したGBM細胞を、BMP4(100ng/ml)で指定の時間処理した。その後、GBM細胞を、氷冷PBSおよび100%エタノールの等容量混合物に再浮遊させ、氷中で30分間インキュベートした。遠心分離した後、細胞ペレットをPBSで3回洗浄し、5分間遠心分離した。その後、GBM細胞を、RNAse(12.5ug/ml;シグマ)およびヨウ化プロピジウム(3ug/ml;シグマ)を含んだ1mlのPBS中で暗所にて一晩インキュベートし、フローサイトメトリーによって分析した。
【0224】
CD133発現の定量化については、生細胞を区別するために7−アミノアクチノマイシンD(7AAD)を使用して二重染色フローサイトメトリーを実施した。PBSで洗浄した後、GBM細胞を7AAD標識バッファ(0.15M NaCl、5mM EDTA、0.5%BSAおよび0.004%サポニンを含む0.1Mリン酸−クエン酸バッファ、pH6.0)中に再浮遊させた後、トバ(Toba)ら、J Immunol Methods 第182巻、p.193−207(1995)に記述されるように7AADを終濃度20uMとなるように添加した。5〜7分の7AADインキュベーションに続いて、GBM細胞をCD133/1(CD133)−PE結合型モノクローナル抗体(1:40、ミルテニーバイオテク(Miltenyi Biotec))とともに4℃で30分間インキュベートし、1mlの増殖培地で洗浄した。その後、細胞を500×gで5分間遠心分離し、0.5mlの増殖培地中に再浮遊させ、フローサイトメトリーで分析した。同じ分析を、さらにCD133/2(293C3)−PE結合型モノクローナル抗体を用いて実施し、同一の結果を得た。
【0225】
GBM細胞の選別(MoFlo(TM)高速セルソーター、ダコサイトメーション(DakoCytomation))は、同じ希釈率のCD133/1(CD133)−PE結合型抗体およびCD133/2(293C3)−PE結合型抗体を使用して実施した。シン(Singh)らの文献(Nature(2004)第432巻、p.396−401)に記述されているように、選別した細胞を、同じ抗体を使用してFACSCalibur(TM)装置(BDバイオサイエンシズ(BD Biosciences))を用いたフローサイトメトリーによって純度分析した。純度はCD133陽性および陰性のフラクションのいずれについても少なくとも87%であった。
【0226】
グリア線維性酸性タンパク質(GFAP)、βIIIチューブリンおよびガラクトセレブロシドC(GalC、「GC」と称されることもある)の定量化については、3.0−3.4umのレインボーキャリブレーション粒子混合物(8ピーク)(BDバイオサイエンシズ)をキャリブレーションに使用し、細胞の標識強度を、フィコエリスリン1分子相当の蛍光量(MEPE)またはフルオレセイン1分子相当の蛍光量(MEFL)として表
した。簡潔に述べると、細胞内染色については、細胞を、0.5mlのCytofix/Cytoperm(TM)溶液(BDバイオサイエンシズ)中で室温にて20分間透過処理した。細胞を2mlのBD Perm/Wash(TM)1×(BDバイオサイエンシズ)で洗浄し、室温で10分間インキュベートした。遠心分離した後、該細胞を、適切な一次抗体混合物を含んだ0.2mlのBD Perm/Wash(TM)1×(BDバイオサイエンシズ)中に再浮遊させた。膜抗原については、細胞を0.2mlの増殖培地中に再浮遊させ、次いで以下の一次抗体、すなわち1:400の抗GFAPポリクローナル抗体(ダコ・コーポレーション)、抗βIIIチューブリンモノクローナル抗体(バブコ)、および抗galCモノクローナル抗体(ケミコン)、とともに4℃で30分間インキュベートした。その後、細胞を洗浄し、4℃で30分間二次抗体に曝露した。細胞内の抗原の場合には、二次抗体は1:800のヤギ抗ウサギIg FITC標識抗体またはヤギ抗マウスIgG R−PE標識抗体(BDファーミンゲン)であり、膜抗原については、1:1000のFITC結合型F(ab’)2ヤギ抗マウスIgMまたはFITC結合型ヤギ抗マウスIgM(ジャクソン・イムノリサーチ(Jackson ImmunoResearch))を使用した。十分に洗浄した後、細胞を再浮遊させてフローサイトメトリーによって分析した。
【0227】
上記のすべての分析法について、分析は、CellQuest(TM)ソフトウェア(BDバイオサイエンシズ)を使用してフローサイトメトリー(FACSCalibur、BDバイオサイエンシズ)によって実施した。バックグラウンドの蛍光は、特異的一次抗体を特異的アイソタイプ対照で代用することにより概算した。自己蛍光の計測も、各試験条件について常に実施した。
【0228】
実施例29:同所注入および免疫組織化学による腫瘍形成性の評価
腫瘍形成性は、コントロール条件の下で、または100ng/mlのBMP−4をさらに追加した条件で48時間培養したGBM細胞の同所移植によって測定した。移植に先立って、細胞を洗浄しPBS中に再浮遊させた(108個/ml)。3マイクロリットルを、ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021にすでに記載されているようにして免疫不全マウスの右側線条体に定位的に注入した。ポリマーを用いたBMP−4の送達は、BMP−4を装荷したヘパリン・アクリル樹脂ビーズ(動物1匹あたり100個のビーズ;シグマ・アルドリッチ(Sigma−Aldrich))を使用して実施した。移植に先立って、PBS単独または0.65μg/μlのBMP4を含むPBS中でビーズを37℃にて1時間インキュベートし、そしてPBSで2×3回徹底的にすすいでから移植した。ヘマトキシリン−エオジン染色および免疫組織化学分析を、ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021にすでに記載されているようにして処理した、パラフィン包埋した4um厚のミクロトーム切片について実施した。
【0229】
期間中(X軸)の生存者の累積的な見込み(Y軸)の下方傾斜プロットは、ソフトウェアMedCalc(ベルギー国所在のマリアケルク(Mariakerke))を使用して実施した。生存の有意差はログランク検定によって測定した。
【図面の簡単な説明】
【0230】
【図1A】ヒト神経幹細胞について、各継代の終了時における理論上の総細胞数を継代数に対してプロットした図。
【図1B】各継代の終了時における総細胞数の対数を継代数に対してプロットし、その線形対数プロットに最も良くフィットする傾向線を示す図。
【図2A】ヒト神経幹細胞にLIFを添加したものとしていないものについて、各継代の終了時における理論上の総細胞数を継代数に対してプロットした図。
【図2B】各継代の終了時における総細胞数の対数を継代数に対してプロットし、その線形対数プロットに最も良くフィットする傾向線を示す図。
【図2C】LIFの存在下における幹細胞および前駆細胞の存在頻度の低下を示すグラフ。
【図3A】腫瘍細胞ニューロスフェアの20×での位相差観察像を示す図。
【図3B】2つの異なるGBM細胞株について各分裂時の理論上の総細胞数を示す図。
【図4A】連続的に継代したGBM腫瘍細胞について、各継代の終了時における理論上の総細胞数を継代数に対してプロットした図。
【図4B】各継代の終了時における総細胞数の対数を継代数に対してプロットし、その線形対数プロットに最も良くフィットする傾向線を示す図。
【図4C】腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を示すグラフ。
【図5】連続的に継代したGBM腫瘍細胞について、神経コロニー形成細胞法(NCFCA)で直径が次の範囲の大きさ:1,500−2,000μm;1,000−1,500μm;500−1,000μm;および<500μmのコロニーを形成する細胞の、播種した細胞全体に対する割合(%)(y軸)を示す図(主グラフ)。主グラフに挿入されたグラフは、直径が1,500−2,000μmおよび1,000−1,500μmの区分にあるコロニーを形成する細胞の、播種した細胞全体に対する割合(%)(y軸)を、主グラフとは異なるy軸目盛りで示している。
【図6】ヒト初代腫瘍標本およびヒト腫瘍神経幹細胞株におけるBMPR1a、BMPR1bおよびBMPR2についてのリアルタイムPCRの結果を示す図。
【図7】LIFまたはBMP−4で処理したGBM細胞株中の幹細胞および前駆細胞の割合(%)を示す図。
【図8】LIF、BMP−4、またはLIF+BMP−4で処理したGBM細胞株中の幹細胞および前駆細胞の割合(%)を示す図。
【図9】連続継代中持続的にBMP−2で処理したGBM細胞株、または1回の継代について一時的にBMP−2で処理したGBM細胞株(「BMP後」)における幹細胞および前駆細胞の割合(%)を示す図。
【図10】ヒトGBM由来の腫瘍性神経幹細胞の移植によって免疫不全マウスに引き起こされたGBMを示す図(上側パネル)。ならびにヒト腫瘍性神経幹細胞をBMP−4またはLIFで処理してから移植した場合はGBM形成が低減されることを示す図(下側パネル)。
【図11】11A:急性単離したGBM細胞および培養したGBM細胞からの細胞について、BMPR1A、BMPR1B、BMPR2およびBMP−4の転写物レベルを示す図。11B−D:単離直後のGBM細胞におけるBMPR1A、BMPR1BおよびBMPR2の免疫反応性を示す図。11E−G:培養したGBM細胞におけるBMPR1A、BMPR1BおよびBMPR2の免疫反応性を示す図。11H−I:GBM細胞におけるリン酸化Smad1,5,8の免疫反応性を示す図。11K−P:GBM細胞におけるBMP−4、Smad1、リン酸化Smad1,5,8およびSmad4のウエスタンブロット解析を示す図。
【図12A】BMP−4の存在下および非存在下におけるGBM培養物中の細胞死、アポトーシス、Ki67免疫反応性およびCD133免疫反応性の測定結果を示す図。
【図12B】BMP−4の存在下および非存在下におけるGBM細胞のクローン形成の指標を示す図。
【図12C】ニューロスフェア分析法におけるBMP−4の存在下および非存在下でのGBM細胞の増殖を示す図。
【図13A】BMP−4非存在下におけるGBM細胞を示す図。
【図13B】BMP−4とともに培養したGBM細胞を示す図。
【図13C】GFAPの免疫反応性(IR)を示す図(対照GBM細胞)。
【図13D】GFAPの免疫反応性(IR)を示す図(BMP−4処理したGBM細胞)。
【図13E】βIIIチューブリンのIRを示す図(対照GBM細胞)。
【図13F】βIIIチューブリンのIRを示す図(BMP−4処理したGBM細胞)。
【図13G】GalCのIRを示す図(対照GBM細胞)。
【図13H】GalCのIRを示す図(BMP−4処理したGBM細胞)。
【図13I】対照GBM細胞およびBMP−4処理したGBM細胞の、細胞蛍光測光法によるGFAPのIRについての解析を示す図。
【図13J】対照GBM細胞およびBMP−4処理したGBM細胞の、βIIIチューブリンのIRについての解析を示す図。
【図13K】対照GBM細胞およびBMP−4処理したGBM細胞の、GalCのIRについての解析を示す図。
【図14A】未処理のGBM細胞を注入したマウスにおける腫瘤を示す図。
【図14B】BMP−4処理したGBM細胞を注入したマウスには比較できるほどの腫瘤が存在しないことを示す図。
【図14C】BMP−4を欠いた対照ビーズで同時処理したマウスにおける腫瘍を示す図。
【図14D】BMP−4ビーズで同時処理したマウスには比較できるほどの腫瘍が存在しないことを示す図。
【図14E】対照ビーズで後処理したマウスにおける腫瘍を示す図。
【図14F】BMP−4ビーズで後処理したマウスには比較できるほどの腫瘍が存在しないことを示す図。
【図14G】マウスにおける未処理のGBM腫瘍の細胞形態を示す図。
【図14H】マウスにおけるBMP−4処理したGBM腫瘍の細胞形態を示す図。
【図14J】対照ビーズまたはBMP−4ビーズのいずれかを用いて、GBM注入の前(左側パネル)、同時(中央パネル)および後(右側パネル)に処理したGBM注入マウスについて生存グラフを示す図。
【図15】GBM細胞の増殖に対する様々なBMPの影響を示す図。
【技術分野】
【0001】
本発明は過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療する医薬の製造におけるBMP調製物および方法に関する。
【背景技術】
【0002】
発明の背景
本明細書におけるいかなる先行技術の引用も、その先行技術があらゆる国に共通の一般知識の一部を成すと認識するものでも何らかの形で示唆するものでもなく、またそのような認識や示唆とみなされるべきものでもない。本明細書に引用された参照文献はすべて、引用によってその全体が本願に明確に組み込まれる。
【0003】
神経幹細胞
従来、幹細胞は、分化細胞が最も失われやすく、かつ頻繁に置き換わる必要のある組織、たとえば皮膚(非特許文献1)、腸管上皮(非特許文献2)および血液(非特許文献3)にのみあると考えられていた。確かに、成体幹細胞の最もよく知られている例は造血幹細胞(HSC)であり、造血幹細胞は骨髄に見出され、動物の一生を通じてすべての種類の血液細胞の生成に根本的に関与している(非特許文献3〜5)。成人の中枢神経系(CNS)では大量のニューロンが死ぬことはなく、かつ再生能はないと考えられたので、神経幹細胞は存在しそうもなく、かつ不必要に思われた。しかしながら、1992年に2つの独立したグループが、新しいニューロンを生じさせる能力を持った成体哺乳類CNS内の前駆細胞の存在を実証することに成功した(非特許文献6および7)。
【0004】
新しいニューロンの供給源は、成体哺乳類CNSの脳室の神経軸全体の内側を覆っている幹細胞であると同定された(非特許文献6)。他の組織で見つかった幹細胞のように、CNSの幹細胞(あるいは神経幹細胞(NSC))は、増殖、大規模な自己複製、多数の子孫細胞の生成、多系列分化能というインビトロにおける決定的な幹細胞の特性(非特許文献8および非特許文献2)、ならびに損傷後の組織再生というインビボにおける特性を示すことが明らかになった。
【0005】
幹細胞の役割のうちの1つは、分裂することと、多くの未分化細胞を増殖および生成する能力を備えた、より分化決定の進んだ前駆細胞を供給することである。究極的には、分化した子孫細胞を生じるのは前記のより分化決定の進んだ前駆細胞種の子孫細胞である。したがって幹細胞は、動物の寿命全体を通じて分裂する能力を備え、従って広範囲に及ぶ増殖能を備えた分化決定されていない細胞の、比較的活動性の低い貯蔵所と見なすことができる。一方、前駆細胞はより分化決定が進んでおり、より頻繁に分裂するが、時間とともに増殖能は制限される。発生中および成体のいずれにおいても、幹細胞および前駆細胞の増殖は細胞発生の基礎となる。
【0006】
特定の形態学的特徴、分子的特徴、あるいは抗原性の特徴を欠いているため、幹細胞は機能上の基準に基づいて同定される。従って、幹細胞の制御についてインビトロで検討するためには、幹細胞の分裂を引き起こす組織培養法を開発しなければならない。しかしながらそのような方法はほとんどなく、神経系ではニューロスフェア法(Neurosphere Assay)(NA)(非特許文献6)と呼ばれる培養法が、NSCをインビトロで同定し、増殖させ、かつ計数するために一般に使用される。簡潔に述べると、NAは、胚から成人までのCNS組織を顕微解剖した後に、細胞間の接着を破壊し、単個細胞の浮遊液を生成させることを含む。細胞を、組織培養器において規定の無血清培地中で、少なくとも1つの増殖誘導性の成長因子(すなわち上皮成長因子[EGF]、塩基性繊維芽
細胞成長因子[bFGF]など)の存在下で(典型的には低密度で)播種する。この条件下では、2〜5日以内に多能性NSCは分裂し、ニューロスフェアと呼ばれるクローン由来の未分化細胞集合体を生じる。増殖を誘導する因子が持続的に存在する状態では、ニューロスフェア中の細胞は分裂を続け、その結果ニューロスフェアを構成する細胞の数が増大し、従ってニューロスフェアが大きくなる。ニューロスフェアを回収し、破壊して単個細胞浮遊液とし、その細胞を再度培養液中に播種して新しいニューロスフェアを生成することができる。このようにしてNSCを継代すると、成育可能なCNS前駆細胞が算術的に増加する。NA法により規定の条件でNSCを単離および増殖させることが可能となり、この推定上の幹細胞の挙動を様々な実験条件下で研究することが可能である。NAは哺乳類NSCを単離する標準的な方法となっており、神経系における幹細胞の細胞分子生物学を理解するために用いられる多くの分析法の中核を成している。
【0007】
腫瘍の増大および増殖に寄与する幹細胞の特徴を備えた細胞の小集団から腫瘍が発生するという概念は、がん生物学の分野において目新しいことではない。この概念は1970年代の初めに提案され、存在頻度の低い腫瘍起源細胞が正常な造血幹細胞(HSC)に似ていることが実証された急性骨髄性白血病(AML)に関する研究で、実験的に確認された。これらの研究は、白血病幹細胞がHSCの直接の子孫であるか、あるいはHSCの特徴を獲得したより分化の進んだ細胞の生産物であることを示唆した。血液系以外の幹細胞の発見から、固形組織のがんにも幹細胞様の細胞が含まれている可能性が高まった。固形腫瘍中の腫瘍の起源となる幹細胞様の細胞の存在および単離は、ヒト乳がん組織において最初に実証され、その手法はCNSの腫瘍にも適用されている。
【0008】
最近、いくつかのグループから、ヒト神経膠腫組織由来の細胞が培養中にニューロスフェア状の細胞を生成する能力について報告され、CNS腫瘍内のNSCの存在が示唆されている。興味深いことに、蛍光活性化細胞選別法(FACS)による「副集団(side−population)」細胞の単離に基づいて、確立されたC6神経膠腫細胞株に、生体内での悪性度を保持するニューロスフェア形成性の細胞の小集団が含まれていることが実証された。ガルリおよび共同研究者(非特許文献9)は、胚CNSおよび成体CNS由来のNSCとほぼ同一の機能特性を示す、ヒト多形膠芽腫(GBM)由来の腫瘍神経幹細胞(tNSC)の単離、増殖および累代移植について報告した。これらのGBM由来tNSCは、インビトロで重要な神経幹細胞の特徴を示す、プロミニン陽性の前駆細胞であり、安定した方法で増殖可能であり、かつ、累代移植−培養サイクル全体を通じて元の腫瘍誘発性を再現する。同時にこれらの研究は、CNSの腫瘍が腫瘍の発生および悪性度に関与する可能性のある幹細胞集団を含んでいる、という仮説を強く支持するものである。tNSCは、CD133のtNSC上での発現を利用したFACSを用いて、他のGBM細胞から選別することができる(非特許文献10)。
【0009】
GBMは、平均生存時間9〜12か月の、最も一般的な成人の悪性脳腫瘍である。大多数の患者は診断から2年までに死亡する。本質的に治癒することはなく、管理療法は一般に手術、放射線療法および化学療法の組合せに基づくものである。生存率は、遺伝子療法、抗血管形成療法、免疫療法および低分子伝達阻害剤のような新しい治療法の活発な探究が促進されてきた30年以上の間、ほとんど変わらなかった。
【0010】
LIF
白血病抑制因子(LIF)は、その誘導産物が多数の組織、恐らくはすべての組織に生じる可能性がある、多機能性の糖タンパク質サイトカインである。LIFはコリン作動性分化因子(CDF)と呼ばれることもある。LIFは、低親和性のLIF特異的レセプタ(LIFR)と、インターロイキン6、オンコスタチンM、カルジオトロフィン−1および毛様体神経栄養因子のレセプタとしても使用されるgpl30レセプタ鎖とで構成されたヘテロダイマーの細胞膜レセプタに結合することにより、反応性細胞に対して作用する
。LIFは、胚盤胞移植、ならびに海馬および嗅覚レセプタのニューロンの正常な発生に不可欠である。LIFは、胚性幹細胞の分化全能性を保持させるという重要な能力から、実験生物学において広範囲に使用されている。LIFは、血小板形成の刺激剤としての作用、いくつかの造血細胞の増殖、骨形成、脂肪細胞の脂質輸送、副腎皮質刺激ホルモン生産、ニューロンの生存および形成、筋衛星細胞の増殖、ならびに肝細胞による急性期産生などの多くの作用を有している(非特許文献11)。
【0011】
BMP
骨形成タンパク質(BMP)は、TGFhスーパーファミリーのメンバーである(非特許文献12)。20を超えるメンバーが知られており、その配列ホモロジーに従ってサブグループに分けることができる(非特許文献12および13)。BMPは胚発生の間に重大な役割を果たす。例えば、BMPは原腸形成、神経新生、アポトーシスおよび造血に影響を及ぼす(総説として非特許文献14を参照のこと)。BMPレセプタは以降BMPRと称する。ヒト由来のBMPRにはBMPR1a、BMPR1bおよびBMPR2が含まれる。
【0012】
本開示のとおり、LIFおよびBMPが、前駆細胞および幹細胞の生存、自己複製、増殖および/または分化を制御し、特にがん組織中の増殖細胞の数を減少させうることがここで解明された。
【非特許文献1】ヒュールスケン(Huelsken)ら、Cell 第105巻、p.533−45、2001年
【非特許文献2】ポッテン(Potten)ら、Development 第110巻、p.1001−20、1990年
【非特許文献3】モリソン(Morrison)ら、Annu Rev Cell Dev Biol 第11巻、p.35−71、1995年
【非特許文献4】ワイスマン(Weissman)、Cell 第100巻、p.157−68、2000年
【非特許文献5】ワイスマン(Weissman)、Science 第287巻、p.1442−6、2000年
【非特許文献6】レノルズ(Reynolds)およびワイス(Weiss)、Science 第255巻、p.1707−10、1992年
【非特許文献7】リチャーズ(Richards)ら、Proc Natl Acad Sci USA 第89巻、p.8591−5、1992年
【非特許文献8】ホール(Hall)ら、Development 第106巻、p.619−33、1989年
【非特許文献9】ガルリ(Galli)ら、Cancer Research 第64巻、p.7011−7021、2004年
【非特許文献10】シン(Singh)ら、Nature 第432巻、p.396−401、2004年
【非特許文献11】総説としてメタカルフ(Metacalf)、Stem Cells 第21巻、p.5−14、2003年
【非特許文献12】フッドレス(Hoodless)ら、Cell 第85巻、p.489−500、1996年
【非特許文献13】ウォズニー(Wozney)ら、J Cell Sci、追補13、p.149−156、1990年
【非特許文献14】ノーエ(Nohe)ら、Cellular Signalling 第16巻、p.291−299、2004年(発明の開示) 1つの態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を、治療または予防するための方法を提供する。該方法は、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に、治療上有効な量の白血病抑制因子(LIF)調製物および少なくとも1つの骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を投与することを含む。
【0013】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、治療上有効な量の白血病抑制因子(LIF)調製物および骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を前記腫瘍に投与することを含む方法を提供する。ヒト患者にBMP−4調製物を投与することにより、脳腫瘍(例えば多形膠芽腫)などのヒト患者の腫瘍の成長を低減する方法が含まれる。
【0014】
さらなる態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、該腫瘍をLIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0015】
別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞中で、LIFレセプタ(LIFR)を介したシグナル伝達またはBMPレセプタ(BMPR)を介したシグナル伝達をそれぞれ増大させることができる、LIF調製物およびBMP調製物を提供する。
【0016】
別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFレセプタ(LIFR)を介したシグナル伝達を増大させることができる、本明細書では以降「LIFRシグナル伝達活性化剤」および「LIFレセプタシグナル伝達活性化剤」と称する作用薬を提供する。
【0017】
別の態様では、本開示は、腫瘍におけるLIFRまたはBMPRを介したシグナル伝達を増大させることにより、前記腫瘍の成長を低減する方法を提供する。LIFRを介したシグナル伝達は、例えば、LIF調製物およびLIFRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能であり、BMPRを介したシグナル伝達は、例えば、BMP調製物およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能である。
【0018】
別の態様では、本開示は、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤を同定するための方法を提供する。
別の態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を、治療または予防するための方法を提供する。該方法は、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に、治療上有効な量の、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を投与することを含む。
【0019】
別の態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、該腫瘍を、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0020】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、前記腫瘍に治療上有効な量のLIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を投与することを含む方法を提供する。
【0021】
別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬と接触させることを含む方法を提供する。
【0022】
別の態様では、本開示は、連続的に継代された神経幹細胞中の神経幹細胞の存在頻度および神経前駆細胞の存在頻度を低減する方法であって、神経幹細胞をLIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0023】
別の態様では、本開示は、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬を含有する医薬組成物を提供する。
【0024】
さらなる態様では、腫瘍の治療のための医薬の製造におけるBMPまたはLIF調製物の使用方法、たとえば脳腫瘍(例えば多形膠芽腫)の治療的処置および予防的処置のうち少なくともいずれか一方のための医薬の製造におけるBMP−4調製物の使用方法が開示される。
【0025】
さらなる態様では、本開示は、腫瘍性幹細胞を含む腫瘍を治療するための方法であって、腫瘍性幹細胞を、腫瘍性幹細胞の分化を誘導する作用薬と接触させることを含む方法を提供する。適切な分化作用薬には、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤が含まれる。例えば、多形膠芽腫は、開示の方法に従って、腫瘍中の腫瘍性神経幹細胞(または、腫瘍の減量手術後の切除腔内残留物)を、腫瘍性神経幹細胞の分化を誘導するのに十分な量のBMP−4調製物と接触させることにより、治療可能である。
【発明を実施するための最良の形態】
【0026】
本開示について詳細に説明する前に、当然のことであるが、別途定めのないかぎり本開示内容は、変更可能なものである処方成分、製造法、投与計画などについて特定のものに限定されない。同様に当然のことであるが、本明細書中で使用される用語は、単に特別な実施形態を説明するためのものであり、限定を意図するものではない。
【0027】
本明細書中で使用されるように、単数形の名詞は、文脈上そうでないことが明らかでないかぎり、複数形も含んでいることに留意すべきである。したがって、例えば、「作用薬」という場合には、単一の作用薬だけでなく2以上の作用薬を含んでおり;「幹細胞」という場合には、単一の幹細胞だけでなく2以上の幹細胞を含んでいる;などである。
【0028】
本明細書中で使用されるように、「治療上有効な量」とは、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を、治療または管理するのに十分な治療薬の量、および好ましくは、原発がん組織、局所がん組織または転移がん組織を破壊、改変、制御、または除去するのに十分な量を指す。治療上有効な量は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の発症を、遅延させるかまたは最小限にする、例えば、がんの蔓延または腫瘍の成長を遅延させるかまたは最小限にするのに十分な治療薬の量を指す場合もある。また治療上有効な量は、腫瘍またはがんの治療または管理において治療上の利点を提供する治療薬の量を指す場合もある。さらに、本開示の治療薬に関しての治療上有効な量とは、細胞過剰増殖性の疾病またはがんの治療または管理において治療上の利点を提供する、単独での、または他の療法との併用における治療薬の量を意味する。該用語は、治療法全体を改善する量、有害反応を低減もしくは回避する量、または別の治療薬の治療効果もしくは別の治療薬との相乗作用を増強する量を包含しうる。
【0029】
用語「作用薬」、「化合物」、「活性作用薬」、「活性化合物」、「治療薬」、「薬理学的に活性な作用薬」、「医薬」、「活性体」および「薬物」は、所望の薬理学的かつ/または生理的な効果を引き起こす物質を指すために本明細書において互換的に使用される
。該用語は、本明細書に具体的に記載された活性作用薬の、医薬として許容可能かつ薬理学的に活性な有効成分、例えば限定するものではないが塩、エステル、アミド、プロドラッグ、活性代謝物、アナログおよびその他同種のものも包含する。用語「作用薬」、「化合物」、「活性作用薬」、「薬理学的に活性な作用薬」、「医薬」、「活性体」および「薬物」が用いられる場合、当然ながら同用語は、活性作用薬それ自身だけでなく、医薬として許容可能で、薬理学的に活性な塩、エステル、アミド、プロドラッグ、代謝産物、アナログなどを包含する。本開示の作用薬は、ペプチド、ポリペプチド、およびタンパク質のような任意のタンパク質性分子であってもよいし、核酸分子のような非タンパク質分子であってもよく、天然または合成的に誘導された有機および無機の低分子ないし高分子であってよい。該作用薬は一般には血液脳関門を横断することが可能であり、あるいはCNSへの直接投与に好適な場合もある。
【0030】
本明細書において「治療」という場合、存在している疾病または症状の重症度の低減を意味する場合がある。用語「治療」は、疾病または症状の発症を防止する「予防的処置」を包含するとも解釈される。用語「治療」は、対象が完全に回復するまで治療されることを必ずしも示唆するものではない。同様に、「予防的処置」は、対象が最終的に疾病または症状を発症しないということを必ずしも意味するものではない。
【0031】
本明細書において使用される場合、「幹細胞」は、(a)増殖が可能、(b)長期間にわたる自己複製が可能で、(c)多くの子孫を生成することができ、かつ(d)その幹細胞が得られた組織のすべての種類の細胞を生じさせる能力を有する、未分化細胞を指す。
【0032】
本明細書において使用されるように、「腫瘍性幹細胞」は腫瘍から得られた幹細胞である。腫瘍性幹細胞は、(a)増殖が可能、(b)長期間にわたる自己複製が可能で、(c)多くの子孫を生成することができ、かつ(d)その腫瘍性幹細胞が得られた腫瘍のすべての種類の細胞を生じさせる能力を有する。本明細書において「tNSC」とも呼ばれる「腫瘍性神経幹細胞」は、CNSの腫瘍から得られた腫瘍性幹細胞を指す。
【0033】
本明細書において使用される場合、「前駆細胞」は、(a)増殖が可能で、(b)限定的な自己複製能力を有し、(c)限られた数の子孫を生成することができ、かつ(d)少なくとも1種類の子孫を生じさせる能力を有する、未分化細胞を指す。
【0034】
本明細書において使用されるように、「腫瘍前駆細胞」は腫瘍から得られた前駆細胞である。腫瘍前駆細胞は、(a)増殖が可能で、(b)限定的な自己複製能力を有し、(c)限られた数の子孫を生成することができ、かつ(d)その腫瘍前駆細胞が得られた腫瘍に見出される少なくとも1種類の細胞を生じさせる能力を有する。
【0035】
LIF調製物およびBMP調製物
1つの態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の治療または予防のための方法を提供する。該方法は、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に、治療上有効な量の白血病抑制因子(LIF)調製物および少なくとも1つの骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を投与することを含む。
【0036】
好ましくは、過度の増殖を特徴とする障害は良性腫瘍または悪性腫瘍(がん)である。例えば、腫瘍は、脳腫瘍、例えば限定するものではないが聴神経腫、腺腫、星状細胞腫、若年性毛様細胞性星状細胞腫、脳幹部神経膠腫、脊索腫、脈絡叢、頭蓋咽頭腫、脳室上衣腫、神経節膠腫、神経節膠腫神経細胞、多形膠芽腫(GBM)、神経膠腫、リンパ腫、髄芽細胞腫、髄膜腫、乏突起膠腫、視神経膠腫、下垂体部腫瘍、松果体部腫瘍、または松果体芽腫であってよい。好ましい実施形態では、脳腫瘍はGBMである。
【0037】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、前記腫瘍に、治療上有効な量の白血病抑制因子(LIF)調製物および少なくとも1つの骨形成タンパク質(BMP)調製物のうち少なくともいずれか一方を投与することを含む方法を提供する。いくつかの実施形態では、治療上有効な量のBMP−4調製物が、GBMの成長を低減するためにヒト患者のGBMに投与される。
【0038】
さらなる態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、該腫瘍を、LIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。理論または仮説によって限定されるものではないが、腫瘍へと投与された時、LIF調製物およびBMP調製物はLIFRまたはBMPRを介したシグナル伝達の増大をもたらし、その結果次の腫瘍性幹細胞または腫瘍前駆細胞の特性、例えば限定するものではないが細胞の生存、自己複製、対称分裂、増殖、および/または分化についての特性のうちのいずれか1つ以上を調節するものと考えられる。理論または仮説によって限定されるものではないが、特に、LIFRまたはBMPRを介したシグナル伝達が増大する結果、幹細胞および前駆細胞の増殖特性が低下し、特に増殖している幹細胞または前駆細胞による対称分裂の可能性が低下することによって該細胞の数が低下すると考えられる。従って、別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIF調製物およびBMP調製物のうち少なくともいずれか一方と接触させることを含む方法を提供する。
【0039】
別の態様では、本開示は、腫瘍におけるLIFRまたはBMPRを介したシグナル伝達を増大させることにより、前記腫瘍の成長を低減する方法を提供する。LIFRを介したシグナル伝達は、例えば、LIF調製物およびLIFRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能であり(後述)、BMPRを介したシグナル伝達は、例えば、BMP調製物およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を使用して活性化可能である(後述)。
【0040】
従来の治療法を使用して腫瘍形成性の細胞を根絶することを目指した現在の治療は、細胞周期が急速に回転している細胞の除去を意図したものである。例えば、従来の化学療法薬は、分裂している細胞に対して最も有効性が高い。tNSCは、対応するその非形質転換体と同じように細胞周期の回転が低頻度であるため、治療の毒性作用を免れ、治療後に腫瘍の増大を容易に再開しうる。成体幹細胞が本質的に寿命が長いこと、また本来薬剤抵抗性遺伝子および抗アポトーシス遺伝子を発現する能力を有することは、該細胞に対応する悪性の細胞においても見出される場合があり、腫瘍性幹細胞の根絶を目指した有効な治療戦略を開発する際の困難の度を増している。本明細書に開示された方法および組成物は、tNSC細胞を標的とすることにより上記の困難を克服する。理論または仮説によって制限を受けるものではないが、本明細書に開示の方法および組成物は、tNSCに対する分化促進効果を有し(実施例に示すように、神経の分化マーカー、特に星状膠細胞の抗原のアップレギュレーションによって証明された)、したがって細胞生存率への影響やアポトーシス誘発を伴わずに幹細胞群を永続的に縮小させると考えられる。その結果(以下の実施例に示すように)、本開示の組成物(特にGBM治療用のBMP−4組成物)への一時的な曝露でも、tNSCの腫瘍形成能が不可逆的に抑制される。
【0041】
腫瘍細胞を死滅させようとするのではなく分化を誘導することは、がん治療への全く新しいアプローチである。したがって、別の態様では、本開示は、腫瘍性幹細胞を含む腫瘍を治療する方法であって、腫瘍性幹細胞を、腫瘍性幹細胞の分化を誘導する作用薬(BMP調製物など)と接触させることを含む方法を提供する。1つのそのような実施形態では、多形膠芽腫のような脳腫瘍中の腫瘍性神経幹細胞を、該細胞の分化を誘導するためにB
MP−4と接触させる。
【0042】
用語「LIF調製物」および「BMP調製物」には、LIFポリペプチドまたはBMPポリペプチドであって、天然に、好ましくはヒトにおいて産生された、任意の翻訳後修飾を有するものまたは翻訳後修飾されていないものが含まれる。この用語は、ヒトLIFの場合、GenBank受入番号NM_002309のmRNAによってコードされるポリペプチドを含んでいる。ヒトBMPの場合、この用語は、ヒト遺伝子BMP1、BMP2、BMP3、BMP4、BMP5、BMP6、BMP7、BMP8A、BMP8B、GDF10(BMP−3b)、GDF11(BMP11)、GDF2(BMP9)、BMP10、BMP15によってコードされるBMPポリペプチド、およびGenBank受入番号NM_001719(BMP7);NM_001201(BMP3);NM_001200(BMP2);NM_005448(BMP15);NM_001720(BMP8B);NM_014482(BMP10);NM_006132(BMP1−4);NM_006131(BMP1−5);NM_006130(BMP1−6);NM_006129(BMP1−3):NM_006128(BMP1−2):NM_001718(BMP6);NM_001199(BMP1−1);NM_130851(BMP4−3);NM_130850(BMP4−2);NM_001202(BMP4−1);NM_181809(BMP8A);NM_021073(BMP5)のmRNAによってコードされるポリペプチドを含んでいる。BMP−4ポリペプチドはBMP−2Bと呼ばれる場合もあることに留意されたい。
【0043】
本開示の方法および組成物において使用される好ましいBMPには、BMP−2、BMP−4、BMP−5、BMP−6、BMP−7およびBMP−8bが挙げられる。特に、BMP−4への曝露は、ヒトGBMから単離された細胞の成熟を強化するとともに全体的な生存度およびアポトーシスには影響しないことが以下の実施例において示されている。このことから、ニューロンマーカーおよび神経膠マーカーのアップレギュレーションが引き起こされ、増殖能力の顕著な低下ももたらされる。BMP−4への曝露は、一時的であっても、GBM培養物中のGBM tNSC集団(CD133+のGBM細胞)を大幅に縮小し、GBM細胞のコロニー形成の指標を大幅に低減し、GBM tNSCの増大の速度論を劇的に低減することが以下の実施例において示されている。これらの効果は不可逆的であり、ヒトGBM細胞のインビボにおける腫瘍発生能力を消滅させる。
【0044】
本明細書で使用される用語「LIF調製物」または「BMP調製物」はまた、LIFもしくはBMPのポリペプチドもしくは糖ポリペプチドのフラグメントであって、本開示の分析法および治療方法において過度の細胞増殖を減弱する能力を少なくとも部分的に保持している、例えば、本開示の分析法または治療法において完全長LIFまたは完全長BMPの活性の1〜100%を保持しているものも含む。そのようなフラグメントは、本開示の分析法または治療方法において完全長LIFまたは完全長BMPよりも高い活性を有していてもよい。そのようなフラグメントは、完全長タンパク質と比較して、アミノ末端またはカルボキシ末端、あるいは両末端から残基が欠失したひと続きのものであってもよい。フラグメントは、構造ドメインまたは機能ドメインを特徴とするもの、例えばαヘリックス領域およびαヘリックス形成領域、βシート領域およびβシート形成領域、ターン領域またはターン形成領域、コイル領域およびコイル形成領域、親水性領域、疎水性領域、両親媒性α領域、両親媒性β領域、フレキシブル領域、表面を形成する領域、および基質結合領域を含むフラグメントであってもよい。フラグメントは、ペプチド合成技術によって生産されたものでも、あるいは完全長のLIFまたはBMPポリペプチドの切断によって生じたものでもよい。フラグメントは、そのN末端、C末端、あるいはN末端およびC末端の両方が、他のポリペプチド配列に連結されることにより、融合タンパク質を形成する場合もある。
【0045】
本明細書において使用される用語「LIF調製物」または「BMP調製物」はまた、LIFもしくはBMPポリペプチド、または上述のようなそのフラグメントのアミノ酸配列と部分的に相同なアミノ酸配列を有し、かつ本開示の分析法および治療方法において過度の細胞増殖を減弱する能力を少なくとも部分的に保持しているポリペプチドまたは糖ポリペプチドも含む。相同体は、LIFもしくはBMPまたはそのフラグメントと50%、70%、80%、80.6%、83%、85%、90%、91%、92%、93%、94%、95%、96%、97%、98%、99%、99.1%、99.2%、99.3%、99.4%、99.5%、99.6%、99.7%、99.8%または99.9%同一であってよい。
【0046】
用語「LIF調製物」または「BMP調製物」はまた、LIFまたはBMPの完全長ポリペプチドのバリアント、およびLIPまたはBMPのフラグメントのバリアントも含む。そのようなバリアントは、本開示の分析法および治療方法において過度の細胞増殖を減弱する能力を少なくとも部分的に保持している。バリアントは、当技術分野で既知の一般的な法則に従って活性に対する影響がほとんどないように選択された欠失、挿入、逆位、反復、および置換を含んでいてもよい。例えば、表現型の上では表に出ないアミノ酸置換を行う方法に関する手引きはボウイ(Bowie)ら、Science 第247巻、p.1306−1310、1990年に提供されており、同文献はその全体が参照によって本願に組み込まれる。例えば、バリアントは部位特異的突然変異法またはアラニン走査突然変異法(分子中のすべての残基に単一アラニン変異を導入)(カニングハム(Cunningham)およびウェルズ(Wells)、Science 第244巻、p.1081−1085、1989年)によって得ることができる。バリアントはまた、例えば、そのタンパク質配列またはペプチド配列中に1以上の非ペプチド結合を(ペプチド結合の代わりとして)含むアミノ酸置換を有していてもよい。バリアントはさらに、天然に存在するLアミノ酸以外のアミノ酸残基、例えばDアミノ酸または非天然もしくは合成アミノ酸、例えばBアミノ酸もしくはyアミノ酸、を含む置換を有していてもよい。バリアントはさらに、ポリペプチドに立体構造上の制約を課する架橋基を含んでいてもよい。バリアントはさらに、グリコシル化、アセチル化、リン酸化およびその他同種のものを含んでいてもよい。バリアントはまた、(i)1以上の非保存アミノ酸残基による置換を含んでいてもよく、該置換アミノ酸残基は遺伝子コードによってコードされたアミノ酸残基であってもそうでなくてもよく、あるいは(ii)置換基を有する1以上のアミノ酸残基による置換を含んでいてもよく、あるいは(iii)成熟ポリペプチドが別の化合物と、例えばLIFまたはBMP調製物の安定性および/または溶解度を増大させる化合物(例えばポリエチレングリコール)、あるいはLIFまたはBMP調製物が特定の種類の細胞(腫瘍性神経幹細胞など)を標的とするための化合物、あるいはLIFまたはBMP調製物が血液脳関門(BBB)および/または血液腫瘍関門(BTB;blood−tumor barrier)を横断可能とするための化合物、と融合していてもよく、あるいは(iv)該ポリペプチドと、付加アミノ酸もしくは付加ペプチドもしくは付加ポリペプチドと融合していてもよく、あるいは(v)細胞毒性を有する作用薬、例えば毒素または放射活性化合物と融合していてもよく、あるいは(vi)画像化を目的として使用されるマーカー、例えば放射標識)と融合していてもよい。
【0047】
本開示のLIF調製物およびBMP調製物は、任意の適切な方法で、例えば天然に存在するポリペプチドの単離により、組換え技術により、ポリペプチド合成技術により、あるいはこれらの方法の組合せにより、調製することができる。そのようなポリペプチドを調製する方法は、当技術分野ではよく了解されている。LIF調製物またはBMP調製物は、融合タンパク質のようなより大きなタンパク質の形態であってもよい。分泌配列またはリーダー配列、プロ配列、ポリヒスチジン残基のような精製を支援する配列、あるいは組換え体生産中の安定性のための付加配列など、付加アミノ酸配列を備えていると有利な場合が多い。
【0048】
本開示のLIF調製物およびBMP調製物は、単離された形態で提供されることが好ましく、十分に精製されることが好ましい。組換え法によって生成されたLIFまたはBMP調製物は、本明細書に記載された技法または当技術分野で周知の他の方法、たとえばスミス(Smith)およびジョンソン(Johnson)、Gene 第67巻、p.31−40、1988年に記載されているワンステップ法を用いて、十分に精製することが可能である。本開示のLIFまたはBMPの調製物はまた、天然、合成または組換えの供給源から、当技術分野で周知のプロトコールを用いて、例えば完全長のLIFまたはBMPに対して作製された本開示の抗体を使用して精製することもできる。
【0049】
本開示のいくつかの実施形態では、内在する腫瘍性幹細胞および腫瘍前駆細胞がインビボで制御されるように、LIF調製物およびBMP調製物のうち少なくともいずれか一方が対象に直接投与される場合がある。例えば、BMP−4調製物はヒト患者体内のGBMのような脳腫瘍に投与される場合がある。本開示の別の実施形態では、腫瘍性幹細胞および腫瘍前駆細胞に、本開示の作用薬をインビトロで接触させる場合がある。例えば、腫瘍性幹細胞と腫瘍前駆細胞とを含む単離された腫瘍に、本開示の作用薬をインビトロで接触させる場合がある。
【0050】
LIF調製物およびBMP調製物のうち少なくともいずれか一方を、LIF調製物およびBMP調製物のうち少なくともいずれか一方を含む医薬組成物とともに、対象に、例えば対象者の腫瘍に投与する方法については、「投与および医薬組成物」と題した項で後述する。
【0051】
LIFレセプタシグナル伝達活性化剤およびBMPレセプタシグナル伝達活性化剤
別の態様の実施形態では、本開示は、本明細書中では以降「LIFRシグナル伝達活性化剤」および「LIFレセプタシグナル伝達活性化剤」とも称する、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFレセプタ(LIFR)を介したシグナル伝達を増大させることのできる作用薬を提供する。本開示はまた、そのようなLIFRシグナル伝達活性化剤を同定するための方法、およびそのようなLIFRシグナル伝達活性化剤を含有する医薬組成物を提供する。本開示のLIFRシグナル伝達活性化剤は、LIFRを直接活性化することにより幹細胞または前駆細胞におけるLIFRを介したシグナル伝達を増大させてもよいし(例えばアゴニスト)、あるいは間接的に、例えば腫瘍性幹細胞または腫瘍前駆細胞中で第2の分子もしくは化合物の発現または活性を増大させて(例えば、LIF自体の発現を増大させて、あるいはJAKまたはSTATのようなLIFRを介したシグナル伝達の下流成分の活性または発現を増加させて)ひいては腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRを介したシグナル伝達を増大させてもよい。
【0052】
追加の態様では、本開示は、本明細書中では以降「BMPRシグナル伝達活性化剤」および「BMPレセプタシグナル伝達活性化剤」とも称する、腫瘍性幹細胞または腫瘍前駆細胞におけるBMPレセプタ(BMPR)を介したシグナル伝達を増大させることのできる作用薬を提供する。本開示はまた、そのようなBMPRシグナル伝達活性化剤を同定するための方法、およびそのようなBMPRシグナル伝達活性化剤を含有する医薬組成物を提供する。本開示のBMPRシグナル伝達活性化剤は、BMPRを直接活性化することにより腫瘍性幹細胞または腫瘍前駆細胞におけるBMPRを介したシグナル伝達を増大させてもよいし(例えばアゴニスト)、あるいは間接的に、例えば腫瘍性幹細胞または腫瘍前駆細胞中で第2の分子もしくは化合物の発現または活性を増大させて(例えば、BMP自体の発現を増大させて、あるいはBMPRを介したシグナル伝達の下流成分の活性または発現を増加させて)ひいては腫瘍性幹細胞または腫瘍前駆細胞におけるBMPRを介したシグナル伝達を増大させてもよい。
【0053】
本明細書中で「LIFR」という場合、LIFRのホモログ、パラログ、オーソログ、誘導体、フラグメントおよび機能的等価物など、LIFRのすべての形態を指す。本明細書中で「BMPR」という場合、BMPRのホモログ、パラログ、オーソログ、誘導体、フラグメントおよび機能的等価物など、BMPRのすべての形態を指す。
【0054】
本開示の文脈においては、LIFRまたはBMPRを介したシグナル伝達の増大とは、LIFRまたはBMPRを介したシグナル伝達の正常なレベルの1〜約1000%の増大を意味する。別例として、LIFRまたはBMPRのシグナル伝達活性化剤は、LIFRまたはBMPRを介したシグナル伝達のレベルを、シグナル伝達が正常よりも低い場合に回復させることができる。
【0055】
好ましくは、LIFRまたはBMPRを介したシグナル伝達の増大により、腫瘍性幹細胞および腫瘍前駆細胞の特性のうちいずれか1つ以上、例えば限定されるものではないが生存、自己複製、増殖、対称分裂および分化のうち少なくともいずれかの特性が調節される。最も好ましくは、LIFRまたはBMPRを介したシグナル伝達の増大は、腫瘍性幹細胞および腫瘍前駆細胞の分裂特性を変化させ、特に対称分裂の可能性の低減あるいは増殖している腫瘍性幹細胞および腫瘍前駆細胞によって示される細胞周期回転数の低減により、腫瘍性幹細胞および腫瘍前駆細胞の数の低下をもたらす。
【0056】
本開示のLIFRおよびBMPRシグナル伝達活性化剤は、ペプチド、ポリペプチドおよびタンパク質のような任意のタンパク質性分子であってもよいし、非タンパク質性の分子であってもよい。LIFRおよびBMPRシグナル伝達活性化剤の単離のための方法は本明細書において提供される。
【0057】
本開示に関して、ミメティックはLIFRおよびBMPRシグナル伝達活性化剤の特に有用な一群である。該用語は、ミメティックが模倣する例えばLIFなどの分子に対してある種の化学的類似性を有する物質であって、例えばLIFRのような標的とLIFとの相互作用をアゴナイズする(模倣する)物質を指すように意図されている。ペプチドミメティックはミメティックの1種であり、タンパク質二次構造の構成要素を模倣するペプチド含有分子であってもよい(ジョンソン(Johnson)ら、「Peptide Turn Mimetics in Biotechnology and Pharmacy」、ペズート(Pezzuto)ら編、チャップマン・アンド・ホール(Chapman and Hall)、ニューヨーク、1993年)。ペプチドミメティックの使用の背景にある論理的根拠は、タンパク質のペプチド・バックボーンの存在が主として、分子の相互作用、たとえば抗体と抗原との相互作用、酵素と基質との相互作用または足場タンパク質の相互作用を促進するようにアミノ酸側鎖を正しい向きにするためにあるということである。したがってペプチドミメティックは、天然の分子に似た分子相互作用が可能なように設計される。
【0058】
薬学的に活性な化合物を目指したミメティックの設計は、「リード」化合物に基づいた薬剤開発への既知のアプローチである。上記アプローチは、活性化合物の合成が困難であるか費用のかかる場合、あるいは特定の投与方法に適さない場合(例えば、ペプチドは消化管中のプロテアーゼによって急速に分解される傾向があるので経口組成物には不適当な活性作用薬である)、望ましいかもしれない。ミメティックの設計、合成および試験は、一般に、標的の特性に対して多数の分子をランダムにスクリーニングするのを回避するために使用される。BMP−4のミメティック、例えばペプチドミメティックなどについて、本明細書中で具体的に検討する。
【0059】
合理的薬物設計の目標は、対象とする生物学的に活性なポリペプチドまたは該ポリペプチドが相互作用する小分子の構造アナログを、例えばより高活性または安定な形態のポリ
ペプチドである薬物、あるいは例えばインビボでポリペプチドの機能を増強または妨害する薬物を作るために、作製することである(例えばホジスン(Hodgson)、Bio/Technology 第9巻、p.19−21、1991年を参照のこと)。1つの手法では、X線結晶解析、コンピュータモデリング、または最も典型的には手法の組み合わせによって、対象とするタンパク質の三次元構造を最初に決定する。ポリペプチドの構造に関する有用な情報は、相同タンパク質の構造に基づいたモデリングにより得られることもある。合理的薬物設計の一例は、HIVプロテアーゼインヒビターの開発である(エリクソン(Erickson)ら、Science 第249巻、p.527−533、1990年)。
【0060】
本開示のLIFRおよびBMPRシグナル伝達活性化剤がタンパク質性であれ非タンパク質性であれ、該活性化体が幹細胞または前駆細胞中でLIFRもしくはBMPRと相互作用し、かつ/またはLIFRもしくはBMPRを介したシグナル伝達を(直接的または間接的に)増大させる能力は、当業者に周知のいくつかのスクリーニング法によって評価することができる。該方法は、天然物ライブラリ、化学合成物ライブラリ、ならびにコンビナトリアルライブラリ、ファージディスプレイライブラリおよびインビトロトランスレーション系ライブラリのスクリーニングを含みうる。
【0061】
LIFRおよびBMPRに対して作製された抗体は、LIFおよびBMPの活性構造をそれぞれ模倣するアゴニストとして特に有用であり得る。適切な抗体には、ポリクローナル抗体、モノクローナル抗体、一価の抗体、二重特異性抗体、ヘテロ結合体抗体、多重特異性抗体、ヒト抗体、ヒト化またはキメラ抗体、一本鎖抗体、Fabフラグメント、F(ab’)フラグメント、Fab発現ライブラリによって生成されたフラグメント、抗イディオタイプ(抗Id)抗体、および上記のうちいずれかのエピトープ結合フラグメントがある。本明細書で用いられる用語「抗体」は、免疫グロブリン分子および免疫グロブリン分子の免疫学的に活性な部分、すなわち免疫学的に特異的に抗原に結合する抗原結合部位を含んでいる分子を指す。免疫グロブリン分子は、免疫グロブリン分子の任意の種類(例えばIgG、IgE、IgM、IgD、IgAおよびIgY)、クラス(例えばIgG1、IgG2、IgG3、IgG4、IgA1およびIgA2)またはサブクラスのものであってよい。さらに、用語「抗体」(Ab)あるいは「モノクローナル抗体」(Mab)は、完全な分子だけでなくタンパク質に特異的に結合することができる抗体フラグメント(例えば、FabおよびF(ab’)2フラグメント)をも含むことを意味する。FabおよびF(ab’)2フラグメントは、完全な抗体のFcフラグメントを欠き、完全な抗体に比べて動物または植物の循環系からより急速に消滅し、完全な抗体よりも組織への非特異的な結合が小さい場合がある(ウォール(Wahl)ら、J.Nucl.Med.第24巻、p.316−325、1983年)。抗体アゴニストを作製する方法は、例えば国際公開公報第96/40281号パンフレット;米国特許第5,811,097号;デング(Deng)ら、Blood 第92巻(6)、p.1981−1988(1998);チェン(Chen)ら、Cancer Res.第58巻(16)、p.3668−3678(1998);ハロップ(Harrop)ら、J.Immunol.第161巻(4)、p.1786−1794(1998);チュー(Zhu)ら、Cancer Res.第58巻(15)、p.3209−3214(1998);ユーン(Yoon)ら、J.Immunol.第160巻(7)、p.3170−3179(1998);プラト(Prat)ら、J.Cell.Sci.第111巻(Pt2)、p.237−247(1998);ピタール(Pitard)ら、J.Immunol.Methods 第205巻(2)、p.177−190(1997);リアウタール(Liautard)ら、Cytokine 第9巻(4)、p.233−241(1997);カールソン(Carlson)ら、J.Biol.Chem.第272巻(17)、p.11295−11301(1997);タリーマン(Taryman)ら、Neuron 第14巻(4)、p.755−762(1995);ミュラー(Muller)ら、Structu
re 第6巻(9)、p.1153−1167(1998);バルツネク(Bartunek)ら、Cytokine 第8巻(1)、p.14−20(1996);ハーロー(Harlow)ら、「Antibodies: A Laboratory Manual」(コールド・スプリング・ハーバー研究所出版社(第2版)、1988);ハマーリング(Hammerling)ら、「Monoclonal Antibodies and T−CeIl Hybridomas」中のp.563−681(エルゼビア(Elsevier)、ニューヨーク、1981)(これらはすべて参照によって全体が本願に組み込まれる)。
【0062】
核酸リガンド(「アプタマー」としても知られている)も、LIFおよびBMPの活性構造をそれぞれ模倣するアゴニストとして特に有用な場合がある。例えば、アプタマーはSELEX(指数関数的な富化によるリガンドの系統的進化(Systematic Evolution of Ligands by Exponential Enrichment))法(ツエルク(Tuerk)およびゴールド(Gold)、Science 第249巻、p.505−510、1990年、この文献は参照によって全体が本願に組み込まれる)を使用して選択することができる。SELEX法では、大きな核酸分子ライブラリ(例えば1015個の異なる分子)を作製し、かつ/または標的分子(この場合はBMPR、LIFRまたはその一部分)でスクリーニングする。標的分子を、ヌクレオチド配列のライブラリとともに一定期間インキュベートする。その後、混合物中の非結合分子から標的アプタマー分子を物理的に単離するためにいくつかの方法を使用することが可能であり、非結合分子は廃棄することができる。その後、標的分子への親和性が最も高いアプタマーを精製して標的分子から分離し、標的分子に結合することができるアプタマーが十分に富化された新たなライブラリを作製するために、酵素を用いて増幅させることができる。その後、この富化されたライブラリを用いて、選択、分離および増幅の新しいサイクルを開始することができる。この選択、分離および増幅のプロセスを5〜15サイクル行った後、ライブラリは、標的分子に強固に結合する少数のアプタマーに縮小される。その後、混合物中の個々の分子を単離し、該分子のヌクレオチド配列を決定し、該分子の結合親和性および結合特異性を測定して比較することができる。その後、単離されたアプタマーをさらに洗練して、標的への結合および/またはアプタマー構造に寄与しないあらゆるヌクレオチドを除去する(つまり、アプタマーを結合コアドメインまで切り詰める)ことができる。アプタマー技術の総説としては、例えばジャヤセナ(Jayasena)、Clin.Chem.第45巻、p.1628−1650、1999年を参照のこと。同文献の教示内容は全て参照によって本願に組込まれる。
【0063】
本質的にいかなる化学化合物もLIFRまたはBMPRシグナル伝達活性化剤の候補として使用することができる。ハイスループットスクリーニング法は、そのような候補活性化体の検出用として特に想定される。そのようなハイスループットスクリーニング法は、典型的には多数の潜在的な治療用化合物(例えばリガンドまたはモジュレータ化合物)を含んでいるコンビナトリアル化学ライブラリまたはペプチドライブラリを用意することを必要とする。その後、そのようなコンビナトリアル化学ライブラリまたはリガンドライブラリを1以上の分析法でスクリーニングして、所望の特徴的活性を示すライブラリ構成メンバー(例えば特定の化学種またはサブクラス)を同定する。このようにして同定された化合物は、従来のリード化合物として役立てることもできるし、それ自体を治療薬候補または実際の治療薬として使用することもできる。
【0064】
コンビナトリアル化学ライブラリは、多くの化学ビルディングブロック(すなわちアミノ酸のような試薬)を組み合わせることにより、化学合成あるいは生物学的合成のいずれかによって生成された種々の化学化合物の集まりである。一例として、直鎖コンビナトリアルライブラリ、例えばポリペプチドまたはペプチドのライブラリは、化合物の所定の長さ(すなわちポリペプチド化合物またはペプチド化合物中のアミノ酸の数)について可能
なあらゆる方法で1セットの化学ビルディングブロックを組み合わせることにより形成される。化学ビルディングブロックをそのように組み合わせて混合することによって、何百万もの化学化合物を合成することができる。
【0065】
コンビナトリアル化学ライブラリの作製およびスクリーニングは関連技術分野の技術者にはよく知られている。コンビナトリアルライブラリには、限定するものではないがペプチドライブラリが挙げられる(例えば米国特許第5,010,175号;フルカ(Furka)、Int.J.Pept.Prot.Res.、第37巻、p.487−493、1991年;ならびにホートン(Houghton)ら、Nature 第354巻、p.84−88、1991年)。多様な化学ライブラリを作製するためのその他の化学手法も使用することができる。多様な化学ライブラリの化学成分の非限定的な例としては、ペプチド(国際公開公報第91/019735号パンフレット)、コード化されたペプチド(国際公開公報第93/20242号パンフレット)、ランダムなバイオオリゴマー(国際公開公報第92/00091号パンフレット)、ベンゾジアゼピン(米国特許第5,288,514号)、ヒダントイン、ベンゾジアゼピンおよびジペプチドのようなダイバーソマー(ホッブズ(Hobbs)ら、1993年、Proc.Natl.Acad.Sci. USA 第90巻、p.6909−6913)、ビニル性ポリペプチド(ハギハラ(Hagihara)ら、1992年、J.Amer.Chem.Soc.、第114巻、p.6568)、グルコース骨格を有する非ペプチド性のペプチドミメティック(ヒルシュマン(Hirschmann)ら、1992年、J.Amer.Chem.Soc.、第114巻、p.9217−9218)、小化合物のアナログ有機合成ライブラリ(analogous organic synthesis of small compound libraries)(チェン(Chen)ら、1994年、J.Amer.Chem.Soc、第116巻、p.2661)、オリゴカルバメート(チョ(Cho)ら、1993年、Science 第261巻、p.1303)かつ/またはペプチジルホスホネート(キャンベル(Campbell)ら、1994年、J.Org.Chem
第59巻、p.658)、核酸ライブラリ(いずれも上述のオースベル(Ausubel)、バーガー(Berger)およびサムブルック(Sambrook)の文献を参照のこと)、ペプチド核酸ライブラリ(米国特許第5,539,083号)、抗体ライブラリ(例えばヴォーン(Vaughn)ら、1996年、Nature Biotechnology 第14巻(3)、p.309−314)およびPCT/US96/10287)、炭水化物ライブラリ(例えばリアン(Liang)ら、1996年、Science、第274巻、p.1520−1522および米国特許第5,593,853号)、有機小分子ライブラリ(例えばベンゾジアゼピン、Baum C&EN、1993年1月18日、p.33および米国特許第5,288,514号;イソプレノイド、米国特許第5,569,588号;チアゾリジノンおよびメタチアザノン、米国特許第5,549,974号);ピロリジン、米国特許第5,525,735号および同第5,519,134号;モルホリノ化合物、米国特許第5,506,337号など)が挙げられる。
【0066】
コンビナトリアルライブラリ作製用のデバイスは市販されている(例えば、米国ケンタッキー州ルイビル所在のアドバンスドケムテック(Advanced Chem Tech)の357MPS、390MPS;米国マサチューセッツ州ウォーバーン所在のレイニン(Rainin)のSymphony(TM);米国カリフォルニア州フォスターシティー所在のアプライド・バイオシステムズ(Applied Biosystems)の433A;米国マサチューセッツ州ベッドフォード所在のミリポア(Millipore)の9050Plus)。さらに、多くのコンビナトリアルライブラリが市販されている(例えば、米国ニュージャージー州プリンストン所在のコムジェネックス(ComGenex);ロシア国モスクワ所在のアシネックス(Asinex);米国ミズーリ州セントルイス所在のトリポス社(Tripos,Inc.;ロシア国モスクワ所在のケムスター株式会社(ChemStar,Ltd.);米国ペンシルバニア州エクストン所在の3D
ファーマシューティカルズ(3D Pharmaceuticals);米国メリーランド州コロンビア所在のマーテック・バイオサイエンシズ(Martek Biosciences)など)。
【0067】
LIFRおよびBMPRシグナル伝達活性化剤の候補は、結合分析を使用して、LIFRまたはBMPRに対する、またはLIFRもしくはBMPRシグナル伝達経路の下流成分に対する結合能力について最初にスクリーニングし、結合する活性化体候補を次いで機能分析でスクリーニングすることができる。適切な結合分析には、蛍光を用いるサーマルシフトアッセイ(米国ペンシルバニア州エクストン所在の3Dファーマシューティカルズ社(3−Dimensional Pharmaceuticals, Inc.)、3DP)が挙げられる(パントリアノ(Pantoliano)らの米国特許第6,020,141号および6,036,920号に記載;さらにJ.ジマーマン(J.Zimmerman)、2000年、Gen.Eng.News 第20巻(8)も参照のこと)。
【0068】
LIFRおよびBMPRシグナル伝達活性化剤の候補を機能に関してスクリーニングする方法の一例は、下記の工程:
(i) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のサンプルを単離する工程;
(ii) 前記腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを適切な容器に入れる工程;
(iii) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを、特定の期間、特定の条件の下で候補作用薬に曝露する工程;ならびに
(iv) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方への形態学的、生理学的、および遺伝学的変化をスクリーニングする工程
を含む。
【0069】
形態学的、生理学的、および遺伝学的変化には、生存、自己複製、増殖および分化のうち少なくともいずれかの状態に関するスクリーニングが含まれる。使用することができる分析法の一例は、参照により全体が本願に組込まれる米国特許出願第2005/0112546号に記載の神経コロニー形成細胞法(Neural Colony Forming Cell Assay)(NCFCA)である。NCFCAは、いずれも増殖力を有し浮遊培養法(ニューロスフェア分析法)で球体を形成するかまたはNCFCAにおいてコロニーを形成することができる幹細胞と前駆細胞とを、区別することができる。簡潔に述べると、腫瘍から得られた初代細胞または培養細胞を、マイトジェンであるFGF2およびEGFを含んだ無血清3Dコラーゲンマトリックスに播種する。この培養状態下では、増殖力を有する幹細胞および前駆細胞のみが分裂して輪郭の明確なコロニーを形成し、このコロニーの大きさを1〜4週後に計測することができる。コロニーの大きさの違いは元の細胞の増殖力と正に相関し、幹細胞および前駆細胞の存在頻度の計測値を提供する。この条件下では、直径2mmを越えるコロニーだけが幹細胞に由来するものであり、直径2mm未満のものは前駆細胞に由来するものである。幹細胞および前駆細胞の、意味のある、かつ正確な計測値から、これら2種類の細胞の存在頻度を変化させる遺伝的・後成的な要素をスクリーニングすることが可能になる。
【0070】
レセプタLIFRおよびBMPRのスクリーニングに使用可能な、生存、自己複製、増殖および分化のうち少なくともいずれかの分析法の別例は、以下のように実施される。最初に、参照により本願に組込まれるグリッティ(Gritti)ら、J.Neurosci.(1996)第16巻(3)、p.109−1100)に記載のように、分散させた多形膠芽腫腫瘍由来の細胞を、マイトジェンであるFGF2(繊維芽細胞成長因子2)およびEGF(上皮成長因子)を含んだ無血清培地中に播種する。この培養系は、初代腫瘍培養物から分化中の細胞/分化細胞を選択除去し、指数関数的に増殖拡大して初代ニュー
ロスフェアを形成できる腫瘍性幹細胞だけを残す。この初代ニューロスフェアを個々に分散させ、マイクロタイタープレート中EGFおよびFGF2の存在下、クローン化しうる密度で無血清培地中に再度播種することができる。LIFRおよびBMPRシグナル伝達活性化剤の候補をマイクロタイタープレートの各ウェルに加え、該プレートを、未処置の細胞が十分に増殖可能な期間インキュベートする。インキュベーションを終えたらニューロスフェアを再び個々に分散させ、上記プロセスを、LIFRおよびBMPRシグナル伝達活性化剤候補の存在下でさらに所定の継代数だけ繰り返すことができる。所定の継代数を終えたら、顕微鏡を使用してニューロスフェアの存在についてマイクロタイタープレートのウェルを検査し、ニューロスフェアの数および大きさを測定し、幹細胞および前駆細胞に対するLIFRまたはBMPRシグナル伝達活性化剤候補の作用の尺度を得る。各継代の終了時の幹細胞および前駆細胞の数を決定するために実施例1の数学的アルゴリズムを使用することができる。連続的に継代された未処置の細胞との比較により、LIFRおよびBMPRシグナル伝達活性化剤候補(例えば幹細胞および前駆細胞の増殖特性を減弱化する作用薬)の同定が可能となる。その後、LIFRおよびBMPRシグナル伝達活性化剤候補を、分化した細胞または分化中の細胞で分析し、その候補作用薬の作用が全般的な細胞毒性ではなく腫瘍性幹細胞に特異的かどうかを判断することができる。
【0071】
LIFRおよびBMPRのシグナル伝達活性化剤としては、RNA干渉(RNAi)分子、リボザイム、あるいはアンチセンスオリゴヌクレオチドも含まれうる。そのような分子は、LIFRおよびBMPRシグナル伝達のインヒビターの発現を低減し、したがってLIFRおよびBMPRシグナル伝達を活性化する作用を有しうる。
【0072】
本開示のLIFRおよびBMPRシグナル伝達活性化剤は、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRまたはBMPRを介したシグナル伝達を増大させるのに役立つ。従って、本開示は、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRまたはBMPRを介したシグナル伝達を増大させる方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤と、腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRまたはBMPRを介したシグナル伝達を増大させるのに十分な時間および条件の下で接触させることを含む方法を提供する。LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤は、本明細書に開示されたようなLIF調製物およびBMP調製物のうち少なくともいずれか一方と組み合わせて使用されてもよい。
【0073】
本開示はまた、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾患または障害を治療または予防するための方法を提供する。該方法は、治療上有効な量の、LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤を、そのような過度な細胞増殖または誤制御された細胞増殖が生じていると思われる対象または組織に投与することを含む。好ましくは、過度な細胞増殖を特徴とする障害は脳障害であり、より好ましくは脳腫瘍、例えば限定するものではないが聴神経腫、腺腫、星状細胞腫、若年性毛様細胞性星状細胞腫、脳幹部神経膠腫、脊索腫、脈絡叢、頭蓋咽頭腫、脳室上衣腫、神経節膠腫、神経節膠腫神経細胞、多形膠芽腫(GBM)、神経膠腫、リンパ腫、髄芽細胞腫、髄膜腫、乏突起膠腫、視神経膠腫、下垂体部腫瘍、松果体部腫瘍、あるいは松果体芽腫である。LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤は、本明細書に開示されたようなLIF調製物およびBMP調製物のうち少なくともいずれか一方と組み合わせて(同時にまたは時を異にして)投与することも可能である。
【0074】
別の態様では、本開示は、腫瘍の成長を低減する方法であって、前記腫瘍に、治療上有効な量のLIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を投与することを含む方法、を提供する。
【0075】
さらなる態様では、本開示は、腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍を、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方と接触させることを含む方法を提供する。理論または仮説によって限定されるものではないが、腫瘍に投与される場合、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤は、LIFまたはBMPを介したシグナル伝達の増大をもたらし、その結果として腫瘍性幹細胞または腫瘍前駆細胞の次の特性、例えば限定するものではないが細胞の生存、自己複製、対称分裂、増殖または分化のうち少なくともいずれかの特性の1つ以上を調節すると考えられる。特に、LIFRまたはBMPRを介したシグナル伝達の増大が幹細胞および前駆細胞の増殖特性を低下させ、特に増殖中の幹細胞または前駆細胞が示す対称分裂の可能性を低下させることによって該細胞の数が低減すると考えられる。従って、別の態様では、本開示は、腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、BMPRシグナル伝達活性化剤およびLIFRシグナル伝達活性化剤のうち少なくともいずれか一方と接触させることを含む方法、を提供する。
【0076】
本開示のいくつかの実施形態では、内在する腫瘍性幹細胞および腫瘍前駆細胞がインビボで制御されるように、LIFRおよびBMPRのうち少なくともいずれか一方のシグナル伝達活性化剤が対象に直接投与されてもよい。本開示の代替実施形態では、腫瘍性幹細胞および腫瘍前駆細胞を、本開示の作用薬とインビトロで接触させる場合もある。
【0077】
腫瘍などの対象に、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれか一方を含む医薬組成物とともに投与する方法については、「投与および医薬組成物」と題した項で後述する。
【0078】
投与および医薬組成物
上述のように、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害を治療または予防するために、治療上有効な量のLIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかを、とりわけ、対象または組織に投与することができる。例えば、一実施形態では、治療上有効な量のBMP−4調製物またはBMP−4ミメティックを、GBMに罹患しているヒト患者に投与する。上記の腫瘍およびがんに加えて、本明細書に開示された方法によって治療または予防され得るその他のがんには、癌腫、リンパ腫、芽細胞腫、肉腫および白血病が挙げられるがこれらに限定はされない。そのようながんのより具体的な例としては、扁平上皮がん、肺がん(小細胞肺がん、非小細胞肺がん、肺の腺癌および肺の扁平上皮癌を含む)、腹膜のがん、肝細胞がん、消化器がんもしくは胃がん(胃腸のがんを含む)、膵臓がん、子宮頸がん、卵巣がん、肝臓がん、膀胱がん、ヘパトーマ、乳がん、結腸がん、結腸直腸がん、子宮内膜もしくは子宮の癌腫、唾液腺癌腫、腎臓がん、肝臓がん、前立腺がん、陰門がん、黒色腫、甲状腺がん、肝癌および様々なタイプの頭頸部がん、ならびにB細胞リンパ腫(例えば、低悪性度/濾胞性の非ホジキンリンパ腫(NHL);小リンパ球性(SL)NHL;中悪性度/濾胞性のNHL;中悪性度の播種性NHL;高悪性度の免疫芽細胞性NHL;高悪性度のリンパ芽球性NHL;高悪性度のバーキット型(small non−cleaved cell)NHL;巨大腫瘤病変NHL;マントル細胞リンパ腫;AIDS関連リンパ腫;およびヴァルデンストレームマクログロブリン血症);慢性リンパ球性白血病(CLL);急性リンパ芽球性白血病(ALL);ヘアリー・セル白血病;慢性骨髄芽球白血病;ならびに、移植後リンパ増殖性疾患(PTLD)がある。
【0079】
一実施形態では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤および
BMPRシグナル伝達活性化剤のうち少なくともいずれかを、医薬組成物の形態で対象に投与する。従って、別の態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の治療または予防に有用な医薬組成物を提供する。本開示の医薬組成物は、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤から成る群から選択された少なくとも1つの作用薬を含有する。例えば、一実施形態では、治療上有効な量のBMP−4調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−2調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−5調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−6調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−7調製物を含む医薬組成物がGBMの治療に提供される。別の実施形態では、治療上有効な量のBMP−8b調製物を含む医薬組成物がGBMの治療に提供される。
【0080】
別の態様では、本開示は、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病または障害の治療または予防のための医薬の製造における、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤、およびBMPRシグナル伝達活性化剤のうち少なくともいずれかの使用方法を開示する。例えば、多形膠芽腫の治療のための医薬の製造における、BMP−4調製物またはBMP−4ミメティックの使用について、具体的に検討する。
【0081】
本開示の医薬組成物は単一の作用薬を含んでいてもよいし、あるいは複数の前述の作用薬の任意の組み合わせ(例えばLIFおよびBMP−4の組み合わせ)を含んでいてもよい。さらに、医薬組成物は、特定の種類の2以上の作用薬、例えば2つの異なるLIF調製物、または2つの異なるBMPRシグナル伝達活性化剤(例えばBMP−2およびBMP−4)を含んでいてもよい。
【0082】
医薬組成物は少なくとも1つの医薬として許容可能な担体を含むことが好ましい。そのような医薬組成物では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかが「活性化合物」を形成する。本明細書で使用されるように、「医薬として許容可能な担体」という言葉は、薬務行政に適合した溶媒、分散媒、コーティング剤、抗菌物質および抗真菌物質、等張剤および吸収遅延剤などを含んでいる。追加の活性化合物を組成物に組み入れることもできる。医薬組成物はその意図した投与経路に適合するように製剤化される。投与経路の例として、非経口、例えば、静脈内、皮内、皮下、経口(例えば吸入)、経皮(局所)、経粘膜、および直腸内投与が挙げられる。非経口、皮内、または皮下への適用に使用される溶液または懸濁物は、下記成分、すなわち、注射用蒸留水、生理食塩水、固定油、ポリエチレングリコール、グリセリン、プロピレングリコールまたはその他の合成溶剤のような無菌の希釈剤;ベンジルアルコールまたはメチルパラベンのような抗菌物質;アスコルビン酸または亜硫酸水素ナトリウムのような酸化防止剤;エチレンジアミン四酢酸のようなキレート剤;酢酸塩、クエン酸塩またはリン酸塩のようなバッファ、ならびに塩化ナトリウムまたはデキストロースのような張度調整のための作用薬;を含むことができる。pHは、塩酸または水酸化ナトリウムのような酸または塩基で調整することができる。非経口の調製物は、ガラス製またはプラスチック製のアンプル、ディスポーザブルシリンジまたは多人数用バイアル中に入れることができる。
【0083】
過度な細胞増殖または誤制御された細胞増殖が生じていると思われる組織または細胞を、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかによりインビトロで治療する実施形態において、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝
達活性化剤のうち少なくともいずれかは、医薬組成物の形態であってもよいし、そうでなくてもよいことに注意されたい。
【0084】
本明細書において使用されるように対象とは、ヒトおよびヒト以外の霊長類(例えばゴリラ、マカク、マーモセット)、家畜動物(例えばヒツジ、畜牛、ウマ、ロバ、ブタ)、コンパニオン・アニマル(例えばイヌ、ネコ)、実験動物(例えばマウス、ウサギ、ラット、モルモット、ハムスター)、捕獲された野生動物(例えばキツネ、シカ)、および本開示の作用薬から利益を享受しうるその他の任意の生物を指す。ここで述べる作用薬から利益を享受し得る動物の種類に制限はない。本開示の中で最も好ましい対象はヒトである。ヒトかヒト以外の生物かどうかにかかわらず、対象を、患者、個体、動物、宿主またはレシピエントと称する場合がある。
【0085】
注射可能な使用に適した医薬組成物には、無菌の水溶液(水に可溶な場合)または分散液、あるいは注射可能な無菌の溶液または分散液を即席に調製するための無菌の散剤が挙げられる。静脈内投与については、適切な担体として生理食塩水、静菌水、Cremophor EL(TM)(米国ニュージャージー州パーシッパニー所在のバスフ(BASF))またはリン酸緩衝生理食塩水(PBS)が挙げられる。いずれの場合にも、組成物は無菌でなければならず、簡単にシリンジ操作ができる程度の流動性を有するべきである。組成物は製造および貯蔵の条件下で安定しているべきであり、細菌および真菌のような微生物の混入に対抗した状態に維持されなければならない。担体は、例えば、水、エタノール、多価アルコール(例えばグリセロール、プロピレングリコール、および液体ポリエチレングリコールなど)およびこれらの適切な混合物を含んでいる溶媒または分散媒でありうる。適切な流動性は、例えば、レシチンのようなコーティング剤の使用により、分散液の場合は要求される粒度を維持することにより、また界面活性剤の使用により、維持することができる。微生物の活動の防止は、様々な抗菌性物質および抗真菌物質(例えばパラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサールなど)によって達成することができる。多くの場合、組成物中に等張剤、例えば糖、多価アルコール例えばマンニトール、ソルビトール、塩化ナトリウムを含むことが望ましいだろう。注射可能な組成物の吸収の延長は、組成物中に吸収を遅延させる作用薬(例えばモノステアリン酸アルミニウムおよびゼラチン)を含めることにより成される場合がある。
【0086】
無菌注射剤溶液は、必要に応じて上に列挙された成分を1つまたは組み合わせて含有する適切な溶媒中に、必要な量の活性化合物を組み入れた後、ろ過滅菌することにより調製することができる。一般に、分散液は、基本の分散媒と、上に列挙された成分からの必要なその他の成分とを含んでいる無菌のビヒクルに、活性化合物を組み入れることにより調製される。無菌注射剤溶液を調製するための無菌の散剤の場合、好ましい調製法は、あらかじめろ過滅菌された、有効成分と任意の所望の付加成分との溶液から、有効成分および所望の付加成分の粉末を生じる、真空乾燥および凍結乾燥である。
【0087】
経口組成物は一般に不活性の希釈剤または食用に適した担体を含んでいる。経口による治療投与の目的では、活性化合物を賦形剤と合わせて、錠剤、トローチ剤またはカプセル剤(例えばゼラチン・カプセル剤)の形態で使用することができる。経口組成物は、口内洗浄液として使用するための流動性の担体を用いて調製することもできる。薬学的に適した結着剤、または補助材のうち少なくともいずれか一方を、組成物の一部として含めることができる。錠剤、丸剤、カプセル剤、トローチ剤などは、次の成分、すなわち微結晶性セルロース、トラガカントゴムまたはゼラチンのような結合剤;デンプンまたはラクトースのような賦形剤、アルギン酸、Primogel(登録商標)またはトウモロコシデンプンのような崩壊剤;ステアリン酸マグネシウムまたはSterotes(登録商標)のような潤滑剤;コロイド状二酸化ケイ素のような流動促進剤;スクロースまたはサッカリンのような甘味料;あるいはペパーミント、サリチル酸メチルまたはオレンジ香味料のよ
うな香料、あるいは同様の性質の化合物のうちの任意のものを含むことができる。
【0088】
吸入による投与については、化合物は、適切な噴射剤(例えば二酸化炭素のようなガス)の入った加圧容器またはディスペンサ、あるいは噴霧器から、エアゾルスプレーの形態で送達される。
【0089】
経粘膜的手段または経皮的手段によって全身投与することも可能である。経粘膜投与または経皮投与の場合、浸透すべき障壁に適した浸透剤を製剤中に使用する。そのような浸透剤は当技術分野で一般に知られており、例えば、経粘膜投与については界面活性剤、胆汁酸塩およびフシジン酸誘導体が挙げられる。経粘膜投与は、鼻内噴霧剤または坐剤の使用により遂行することができる。経皮投与については、活性化合物を、当技術分野で一般に知られているような軟膏剤、サルベ、ゲル剤あるいはクリーム剤に製剤化する。化合物を、直腸送達のために、坐剤(例えばココアバターおよび他のグリセリドのような従来の坐剤基剤を用いる)または停留浣腸剤の形態に調製することもできる。
【0090】
一実施形態では、活性化合物は、植込剤およびマイクロカプセル化送達システムなどの制御放出製剤のように、化合物が身体から急速に消失するのを防ぐ担体を用いて調製される。エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステルおよびポリ乳酸のような生分解性で生体適合性のポリマーを使用することができる。そのような製剤を調製する方法は当業者には明白であろう。材料も、アルザコーポレイション(Alza Corporation)およびノバ・ファーマシューティカルズ・インコーポレイテッド(Nova Pharmaceuticals, Inc.)から商業的に入手することができる。リポソーム懸濁液(細胞特異抗原に対するモノクローナル抗体を用いて標的を感染細胞に設定したリポソームなど)も、医薬として許容可能な担体として使用することができる。これらは、例えば米国特許第4,522,811号に述べられているように、当業者に周知の方法によって調製することができる。
【0091】
投与し易く、かつ投与量を均一にするために経口または非経口の組成物を投与量単位の形態に製剤化すると有利である。本明細書で使用される場合、投与量単位の形態とは、治療すべき対象についての単一量として適した物理的に個別の単位を指し;各々の単位は、所望の治療効果を生じるために計算された所定量の活性化合物を、必要な製薬担体とともに含んでいる。
【0092】
そのような化合物の毒性および治療上の有効性は、例えばLD50(集団の50%が死亡する用量)およびED50(集団の50%に治療上有効な用量)を決定するための、細胞培養または実験動物における標準的な薬学的手法により決定することができる。毒性作用および治療効果の間の用量比が治療係数であり、治療係数はLD50/ED50比として表すことができる。高い治療係数を示す化合物が好ましい。毒性副作用を示す化合物を使用することは可能であるが、非感染細胞が損傷する可能性を最小限にすることによって副作用を低減するために、そのような化合物の標的を罹患組織の部位に設定する送達システムを設計するように配慮すべきである。
【0093】
細胞培養分析および動物実験から得られたデータを、ヒトで用いる用量範囲を策定するのに使用することができる。そのような化合物の用量は、毒性をほとんどまたは全く伴わないED50を含めた血中濃度範囲にあることが好ましい。用量は、この範囲内で、使用される剤形および利用される投与経路に依存して変わりうる。本開示の方法において使用される任意の化合物について、治療上有効な用量は、最初に細胞培養分析から推測することができる。用量は、細胞培養で決定されるようなIC50(すなわち症状の最大限の抑制を2分の1達成する試験化合物の濃度)を含む血漿中濃度範囲を達成するように動物モデルで策定することができる。そのような情報は、ヒトでの有用な用量をより正確に決定
するために使用することができる。血漿中濃度は、例えば高速液体クロマトグラフィによって測定することができる。
【0094】
本明細書において定義される場合、治療上有効な量のタンパク質またはポリペプチド(すなわち有効な用量)は、体重1kgあたり約0.001〜30mg、好ましくは体重1kgあたり約0.01〜25mg、より好ましくは体重1kgあたり約0.1〜20mg、より一層好ましくは体重1kgあたり約1〜10mg、2〜9mg、3〜8mg、4〜7mg、または5〜6mgの範囲にありうる。タンパク質またはポリペプチドを、1週間に1回として、約1〜10週間、好ましくは約2〜8週間、より好ましくは約3〜7週間、より一層好ましくは約4、5または6週間投与することができる。当業者には当然のことであるが、一定の要因、例えば限定するものではないが疾病または障害の重症度、以前の治療、対象の全体的な健康状態および/または対象の年齢、ならびにその他の疾病の存在などが、対象を有効に治療するのに必要な用量および時期に影響しうる。さらに、治療上有効な量のタンパク質、ポリペプチドまたは抗体を用いた対象の治療は、単回治療を含むものでもよいが、好ましくは一連の複数の治療を含むこともできる。
【0095】
本開示の実施形態において、脳の増殖性障害、例えばGBMが治療される場合、血液脳関門(BBB)および血液腫瘍関門(BTB)のうち少なくともいずれか一方を乗り越えるために、医薬組成物および投与方法のうち少なくともいずれか一方を調整することが望ましい。BBBを越えて医薬組成物を送達する方法は、当技術分野で周知である。例えば、参照により全体が本願に組込まれる、ミスラ(Misra)ら、2003年、J Pharm Pharm Sci 第6巻、p.252−273を参照のこと。適切な方法としては、限定するものではないが、脳内移植、脳室内(ICV)注入、またはCED(convection enhanced diffusion)法のような経頭蓋的脳内薬物送達法が挙げられる。脳内移植は、例えば、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかを含浸させた、ポリマー製ビーズ(ヘパリンアクリルビーズなど)またはポリマー製ウエハ(ポリフェプロサン(polifeprosan)20など)を使用して実行可能である。例えば、脳内移植は、BMP−4を装荷したヘパリンアクリルビーズを腫瘍内または切除腔内に定位的に注入することにより実行可能である。
【0096】
本開示の医薬組成物、例えばBMP−2、BMP−4、BMP−5、BMP−6、BMP−7またはBMP−8b調製物(またはこれらの組み合わせ)を含有する医薬組成物を、上記の方法を使用して未切除の腫瘍に投与してもよいし、あるいは、医薬組成物を腫瘍切除後に切除腔に投与してもよい。いくつかの実施形態では、医薬組成物を最初に腫瘍内に投与し、次に、腫瘍切除後に術後投与する。
【0097】
いくつかの実施形態では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかが、血液脳関門(BBB)レセプタ仲介輸送(RMT)システムの構成分子上の外表面エピトープと結合することができる分子と会合している。この方法では、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかは、内在性のRMTシステムを使用してBBBを越えて輸送されうる。例えば、LIFまたはBMPの調製物をトランスフェリンレセプタ(TfR)に対するモノクローナル抗体(OX26など)と結合させて、該結合体の膜透過輸送を可能にする。例えば、参照により全体が本願に組み込まれるパルドリッジ(Pardridge)、Neurorx 第2巻(1)、p.3−14(2005)を参照されたい。例えばOX26との結合によってナノ粒子がBBBを超えるようにすることも可能であり、そのようなナノ粒子を、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかと結合させてもよい。例えば、参照により全体が本願に組
み込まれるオリヴィエ(Olivier)ら、Pharm Res.(2002)第19巻(8)、p.1137−43を参照されたい。さらに、例えばOX26に結合させたリポソームを用いて、カプセル封入されたLIF調製物、BMP調製物、LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤のうち少なくともいずれかを、BBBを越えて送達させることもできる。参照により全体が本願に組み込まれるヒュイラー(Huwyler)ら、Proc Natl Acad Sci USA.(1996)第93巻(24)、p.14164−9を参照されたい。さらに、BBBおよびBTBを破壊する作用薬および処置法を用いて、LIF調製物、BMP調製物、LIFRシグナル伝達活性化剤またはBMPRシグナル伝達活性化剤のうち少なくともいずれかを、BBBまたはBTBに通過させることもできる。例えば、血管作用薬ブラジキニンの頸動脈内注入は、脳腫瘍毛細血管の透過性を選択的に増大させることができる。参照により全体が本願に組み込まれるマツカド(Matsukado)ら、Brain Res.(1998)第792巻(1)、p.10−5を参照されたい。
【0098】
一実施形態では、LIFおよびBMP調製物、またはポリペプチド性およびペプチド性のLIFRもしくはBMPRシグナル伝達活性化剤、または、LIFRもしくはBMPシグナル伝達活性化剤であるリボザイム、RNAi分子およびアンチセンス分子、をコードする核酸分子をベクターに挿入し、遺伝子療法ベクターとして使用する。遺伝子療法ベクターは、例えば静脈注射、局所投与(米国特許第5,328,470号を参照)、または定位注入(例えば、いずれも参照により全体が本願に組み込まれるチェン(Chen)ら(1994)、Proc.Natl.Acad.Sci. USA 第91巻、p.3054−3057;ボーグ(Voges)ら(2003)、Ann.Neurol.第54巻、p.479−487を参照)によって対象に送達することができる。遺伝子療法ベクターの医薬品製剤は、許容可能な希釈剤中またはカプセル材料(ベクターを含むリポソームなど)中に入った遺伝子療法ベクターを含むものでもよいし、あるいは遺伝子送達手段が埋め込まれた遅延放出マトリックスからなるものでもよい。別例として、完全な遺伝子送達ベクターが組換え細胞からそのまま生産される、例えばレトロウイルス・ベクターの場合、医薬品製剤は、遺伝子送達システムを生産する1以上の細胞を含むことができる。一実施形態では、LIFおよびBMP調製物をコードする、またはポリペプチド性およびペプチド性のLIFRもしくはBMPRシグナル伝達活性化剤をコードする核酸分子は、参照により全体が本願に組み込まれるコンシグリオ(Consiglio)ら、Proc
Natl Acad Sci USA、2004年10月12日、第101巻(41)、p.14835−14840に述べられているように、レンチウイルスベクターによって哺乳類のがん性神経幹細胞に導入される。
【0099】
いくつかの実施形態では、本開示による単一の活性化合物、例えば単一のLIF調製物または単一のBMP調製物が投与される。他の実施形態では、複数の活性化合物、例えば2つの異なるLIF調製物;またはLIF調製物とBMP調製物;またはBMPRシグナル伝達活性化剤と2つの異なるBMP調製物、が同時投与される。さらに、本開示の活性化合物は、化学療法剤、放射線増感剤、放射線治療薬などのような他の薬剤と同時投与されることもある。本明細書で「同時投与」という場合、同一の製剤として、または2つの異なる製剤として、同一経路または異なる経路で同時に投与すること、あるいは同一経路または異なる経路で連続的に投与することを意味する。本明細書で「連続的に」投与するという場合、2種類の作用薬および/または医薬組成物の投与の間の時差が、数秒、数分、数時間または数日であることを意味する。作用薬および/または医薬組成物の同時投与は任意の順序で行われうる。
【0100】
本明細書に開示された治療方法および医薬組成物は、他の治療、たとえば化学療法(カルマスティン、シスプラチン、パクリタクセル、テモゾロミド、PCV(プロカルバジン、ロムスチンおよびビンクリスチン)およびIL13−PE38QQRなど)、放射線治
療薬(放射標識抗体または放射標識核酸リガンドなど)を用いた治療、放射線療法、および手術(腫瘍の切除/減量手術など)などと併用されてもよい。例えば、BMP−4調製物を、GBM治療用の腫瘍切除手術および放射線治療後の化学療法剤とともに同時投与してもよい。これらの付加的な治療は、本明細書に開示された方法による治療の前、治療中、または治療後のうちいずれにおいても使用できる。
【0101】
実施例
本開示について、以下の非限定的な実施例によりさらに説明する。実施例19−23については、列挙された結果を得るために使用されたプロトコールが実施例24−29に提示されていることに注意されたい。
【0102】
実施例1:ヒト胎児神経幹細胞の連続継代および数学的モデル化に基づく幹細胞と前駆細胞の存在頻度の分析
連続的に継代されたニューロスフェア培養物中に存在する神経幹細胞の数および神経前駆細胞の数を計算するためにアルゴリズムを導き出した。このアルゴリズムにために、すべての細胞を以下の3つのカテゴリーに分類する。
【0103】
1.幹細胞は、(a)増殖が可能、(b)長期間にわたる自己複製が可能、(c)多くの子孫を生じることが可能で、かつ(d)その幹細胞が得られた組織のすべての種類の細胞を生じる能力を有する、未分化細胞として定義する。
【0104】
2.前駆細胞は、(a)増殖が可能で、(b)限定的な自己複製能力を有し、(c)限られた数の子孫を生じることが可能で、かつ(d)少なくとも1種類の子孫を生じる能力を有する、未分化細胞として定義する。
【0105】
3.分化細胞は、増殖能力が限定されているかまたは増殖能力がなく、細胞系統特異的マーカーおよび成熟した機能特性のいずれをも発現している細胞として定義する。
用語「幹細胞」およびNSCは本明細書中で互換的に使用されることがある。同様に、用語「前駆細胞」およびNPCも本明細書中で互換的に使用されることがある。このアルゴリズムについて説明する際に、以下のいくつかの仮定が為される。
【0106】
1.ニューロスフェアは、幹細胞、前駆細胞および分化細胞で構成される。
2.すべてのニューロスフェアは単一の幹細胞または単一の前駆細胞のいずれかから成長する。
【0107】
3.幹細胞の寿命は無限であり、前駆細胞の寿命は有限である。有限の寿命を継代数lと定義する。
4.幹細胞は常にニューロスフェアを形成可能であり、前駆細胞はその寿命の終わりに達していなければニューロスフェアを形成する。
【0108】
5.各ニューロスフェアは、合計c個の細胞を有し、これらの細胞は2つの可能な組成のうちの1つである。可能な組成とは、
a.単一の幹細胞に由来する各ニューロスフェアにおいて、組成は幹細胞s個、前駆細胞p個、残りは分化細胞であり;
b.単一の前駆細胞に由来する各ニューロスフェアにおいて、組成は前駆細胞p個と残りの分化細胞である。
【0109】
継代数nにおける細胞の総数Tnを表すアルゴリズムを導き出した。実験の初めに、幹細胞由来のニューロスフェアを個々に分散させる、すなわち、幹細胞がs個、前駆細胞がp個、および分化細胞がc−s−p個存在する。この幹細胞および前駆細胞を増殖させて
、合計s+p個のニューロスフェアになるように成長させ、分化細胞は死滅する。その結果、最初の継代の時点T1での総細胞数は、ニューロスフェアの総数と各ニューロスフェア中の細胞数との積:
【0110】
【数1】
【0111】
で与えられる。
上記方程式の第2の等式は興味深い。というのも、scは幹細胞由来のニューロスフェア中の総細胞数を表し、pcは前駆細胞由来のニューロスフェア中の総細胞数を表しているからである。
【0112】
前駆細胞が2世代生きると仮定し、ここでこれらのニューロスフェアを個々に分散させる。最初の継代では、幹細胞由来のs個のニューロスフェアが存在し、その各々がs個の幹細胞、p個の前駆細胞およびc−s−p個の分化細胞を含むはずである。同様に、最初の継代では、前駆細胞由来のp個のニューロスフェアが存在し、今度はその各々がp個の前駆細胞およびc−p個の分化細胞を含む。ここで、新たに個々に分散させたこれらの細胞は、死滅する分化細胞以外はその細胞独自のニューロスフェアへと成長することになる。その結果、2番目の継代の時点T2での総細胞数は、
【0113】
【数2】
【0114】
【数3】
【0115】
で与えられる。
等式の右辺の第1のカラムは、幹細胞由来のニューロスフェアを表す項を含んでいる。第2のカラムは、前駆細胞由来のニューロスフェアを表している。
【0116】
T2の方程式中の第2の等式は、総細胞数を展開式で表すものである。s2c項は、幹
細胞由来のニューロスフェア中の、次世代で幹細胞由来のニューロスフェアになる細胞の総数を示す。第2項spcは、幹細胞由来のニューロスフェア中の、次世代で前駆細胞由来のニューロスフェアになる細胞の総数を示す。最後の項p2cは、その寿命に依存して
前駆細胞由来のニューロスフェアになるかまたは死滅する、前駆細胞由来のニューロスフェアを表す。
【0117】
ここでこれらの細胞を継代して第3の世代を生じさせる。この世代の総細胞数は、前駆細胞の寿命に左右される。寿命がわずか2世代であれば、前駆細胞由来の前駆細胞は新しいニューロスフェアを生成せずにここで死滅することになる。この仮定の下では、3番目の継代の時点T3における総細胞数は、
【0118】
【数4】
【0119】
【数5】
【0120】
【数6】
【0121】
で与えられる。
同様に、4番目の継代の時点における総細胞数を決定することができる。これらの方程式を書き直すと、
【0122】
【数7】
【0123】
【数8】
【0124】
【数9】
【0125】
【数10】
【0126】
となる。
したがって、導き出すと、n番目の継代における総細胞数は、
【0127】
【数11】
【0128】
で与えられることになる。
この方程式は前駆細胞の寿命が2世代であるという仮定に基づいていることを繰り返しておかなければならない。
【0129】
前駆細胞の寿命が2世代より長い場合、3番目の継代の時点T3における総細胞数は、
【0130】
【数12】
【0131】
【数13】
【0132】
【数14】
【0133】
で与えられる。
前駆細胞の寿命が3世代である場合、第4世代までの総細胞数は、
【0134】
【数15】
【0135】
【数16】
【0136】
【数17】
【0137】
【数18】
【0138】
で与えられる。
したがって、この特定の仮定を用いて導き出すと、n番目の継代における総細胞数は、
【0139】
【数19】
【0140】
で与えられる。
一般に、同様の論拠によって、前駆細胞の寿命がl世代である場合のn番目の継代の時点における総細胞数は、
【0141】
【数20】
【0142】
【数21】
【0143】
で与えられる。
上記方程式中の総和については単純化がよく知られている。
【0144】
【数22】
【0145】
従ってTnを表す式は、
【0146】
【数23】
【0147】
【数24】
【0148】
に単純化される。
所与の種類の細胞について、p、s、c、およびlは一定であるため、角括弧内の項を定数に置き換えて、
【0149】
【数25】
【0150】
および
【0151】
【数26】
【0152】
とすることができる。
この方程式は、式(25)の対数をとることにより一次式で表して
【0153】
【数27】
【0154】
とすることができる。
方程式(27)を検討すると、この直線の傾きはlog sで、y切片はlog Bである。実験上の観点から見ると、このことは、総細胞数の対数を継代数に対してプロットすると、このプロットの傾きを調べることによりニューロスフェア中の幹細胞の数を計算することができ、y切片を調べることにより前駆細胞の数を計算することができる、ということを意味する。このプロットを構築する場合、継代数が前駆細胞の寿命より大きいときのデータポイントのみを含めるように注意しなければならない。前駆細胞の寿命は、l≧nの場合に所与の継代とその前の継代とにおける総細胞数の比率は一定((Tn+1/
Tn)=s)であるが、l<nの場合はこの比率は一定ではないことに注意して計算される。従ってl=n+1であり、ここでnは上記の比率がsと等しくならない最大の整数である。この手法はノイズの多いデータを処理するのに適している場合がある。
【0155】
注意すべき重要なことは、最初のl−1継代まで、すなわち(1,logT1),・・・,(l−1,logTl−1)は、直線にのらないことである。このことは方程式(23)および(24)を調べればわかる。
【0156】
方程式(27)から、ニューロスフェア中の前駆細胞の数pは、直線の傾きには影響しないことがわかる。傾きが変わる場合、これは幹細胞の数が変わることを意味するはずである。傾きが0である場合、つまり、直線が水平の場合、ニューロスフェア中には幹細胞が1つだけ存在し、従って、各継代における細胞の総数は増大しない。
【0157】
ニューロスフェアの成長条件が変わる場合、例えば成長因子がEGFのみからEGF+FGFのみに変わる場合、直線の傾きは依然としてニューロスフェア中の幹細胞の数によってのみ決定される。この主張の証拠は、本項の残りの部分で述べる。
【0158】
r番目の継代後に条件が変わり、生じる幹細胞の数がqに、前駆細胞の数がwに、前駆細胞の寿命がm世代に変わると仮定する(便宜上、m≦lとするがm>lの場合も同じである)。r+l回の継代後の総細胞数は、
【0159】
【数28】
【0160】
で与えられる。
r+m−1回の継代後には、総数は
【0161】
【数29】
【0162】
であり、r+m回の継代後には、総数は
【0163】
【数30】
【0164】
である。
導き出すと、r+N回の継代後の総細胞数は、
【0165】
【数31】
【0166】
【数32】
【0167】
となり、ここで
【0168】
【数33】
【0169】
である。
線形化すると、
【0170】
【数34】
【0171】
である。
したがって、上述のように、条件の変更後この直線の傾きは幹細胞の数によってのみ影響されることが分かる。
【0172】
上記のアルゴリズムに基づいて、幹細胞および前駆細胞の存在頻度を以下の工程、すなわち
1. ニューロスフェア分析法において細胞を連続的に継代し、各継代で生成された細胞の総数を、その前の継代の総細胞数と、現在の継代で生成された細胞における増加倍数とを乗じることに基づいてプロットする工程;
2. 各継代で生成された細胞の総数の対数をとることにより、対数増殖カーブを線形化する工程;
3. 最も良くフィットする直線を計算し、次いで直線式(y=mx+b)を用いて直線の傾きとy切片を計算し、次には上記の式を使用して幹細胞の数と前駆細胞の数とを明らかにする工程
から計算することができる。
【0173】
実施例2:ヒト胎児神経幹細胞の連続継代および数学的モデル化に基づく幹細胞と前駆細胞の存在頻度の分析
実施例1の数理モデルを、連続的に継代したヒト胎児神経幹細胞に適用した。ヒト胎児神経幹細胞を、参照により全体が本願に組み込まれるベスコビ(Vescovi)ら、1999年、Exp.Neurol.第156巻、p.71−83に記載されているような従来の技術(すなわち、細胞を、その前の継代由来の細胞の一部を用いて、かつマイトジ
ェンであるEGFおよびFGF2を補足した無血清培地を用いて、各継代につきクローン化しうる密度で播種する)を使用して、連続的に継代した。各継代の終了時に、細胞を個々に分散させて単個細胞浮遊液とし、この単個細胞浮遊液の細胞を計数し、トリパンブルー排除法によって生細胞および死細胞の数を計数することにより、総細胞数を決定した。各継代終了時の理論上の総細胞数(継代の終了時に計数された総細胞数と、計数されたその細胞を生成させるために播種された継代直前の細胞の一部との関数である)を継代数に対してプロットして(図1A)成長曲線を得た。各継代の終了時の総細胞数の対数も継代数に対してプロットした(図1B)。線形の対数プロットについて最もフィットする傾向線を作成し(図1B)、直線式(y=mx+b)を用いて傾きとy切片とを決定した。その後、幹細胞および前駆細胞の存在頻度を、実施例1の方程式(26)および(27)によって計算した(n=1、l=n+1=2、c=1000とした;これらの値は以降全ての実施例でも使用)。幹細胞の存在頻度は細胞集団全体の0.24%であると算出され、前駆細胞の存在頻度は細胞集団全体の1.15%であると算出された。
【0174】
実施例3:神経コロニー形成細胞法におけるヒト胎児神経幹細胞の幹細胞および前駆細胞の存在頻度の分析
神経コロニー形成細胞法(NCFCA)は、神経コロニーの大きさの分析に基づいて幹細胞および前駆細胞の存在頻度を決定するために使用することができる。NCFCAは、参照によってその全体が本願に組み込まれる2005年5月26日に公開された米国特許出願番号第2005/0112546号に述べられている。簡潔に述べると、NCFCAは、軟寒天培地、好ましくはコラーゲンベースまたはメチルセルロースベース(IMDM、DMEM/F12、マッコイ(McKoy’s)、イスコフ(Iscoves))の軟寒天培地中に神経細胞を浮遊させることにより行なわれる。軟寒天培地は、神経細胞を培養するために使用されるのと同じ適切な培地(例えばNeurocult(TM)[ステムセルテクノロジーズ社(StemCell Technologies, Inc.]無血清培地でサイトカインを含まずNeurocult(TM)増殖サプリメントとEGFを含むもの)にコラーゲンまたはメチルセルロースを加えて成るものでよい。培地は血清を含まないことが好ましい。軟寒天培地中の細胞は、データの統計学的解析に十分な数のコロニーを生じる濃度(例えば35mmの培養ディッシュ当たりの細胞数が1,000〜25,000個、好ましくは2,500〜7,500個)に播種する。形成されるコロニーは、単一の細胞すなわち神経幹細胞または前駆細胞いずれかから生じる。このコロニーを、コロニー間の大きさおよび差異が識別可能になるまで(例えば約10〜30日)培養し、次にコロニーを計数し、かつスコアリング用ディッシュのグリッドを使用してコロニーの大きさを推定する。単一の神経幹細胞から生成されたコロニーは時間とともに大きく成長し続けるが、神経前駆細胞から生成されたコロニーは成長力に限界があり、従って時間とともに大きく成長し続けることはない。コロニーの大きさから、増殖力の高いNSC(HPP−NSC)、増殖力の低いNSC(LPP−NSC)、および神経前駆細胞が区別される。したがって、生成されたコロニーの大きさは、そのコロニーが神経幹細胞から生成したか神経前駆細胞から生成したか、さらにはそのNSCの増殖力が高いか低いかの指標となりうる。特に、(ディッシュ上の他のコロニーに比べて)より大きなコロニーは、増殖力の高い神経幹細胞を示し、中型のコロニーは増殖力の低い神経前駆細胞を示し、より小さなコロニーは神経前駆細胞を示す。「より大きなコロニー」または「より小さなコロニー」の実際の直径は、コロニーを培養する時間の長さなど多くの要因によって変化する。例えば、細胞2,500個/ディッシュで14〜28日間培養した後、コロニーを直径に基づいて4つのカテゴリー、すなわち(1)>2.0mm、(2)1−2mm、(3)0.5−1mmおよび(4)<0.5mmのうちの1つに分類した。したがって、コロニーを少なくとも14日間培養すると仮定すると、直径が2.0mmを超えると神経幹細胞から生成されたコロニーを示す。NCFCAにおいて細胞の種類は、その細胞種が形成する形態に基づいて区別することもできる。波状のコロニーは、神経幹細胞によって生成され、周囲が滑らかなコロニーは神経前駆細胞によって生成される。NCFCAを行
なうためのキットはステムセルテクノロジーズ社から市販で入手可能である。
【0175】
ヒト胎児神経幹細胞に関する幹細胞および前駆細胞の存在頻度を、NCFCAにおいて細胞をプレート培養することにより計算した。継代数12の胎児神経幹細胞の子孫を個々に分散させて単個細胞浮遊液とし、細胞密度を2000個/mlとして20ng/mlのEGFおよび10ng/mlのbFGFとともにプレート培養した。細胞を軟寒天培地において3週間培養した後、コロニーの存在頻度および大きさを計算した。プレート培養した全細胞の2%未満が幹細胞の特性を示すコロニーを形成した。したがって、実施例2における数学的分析法からの結果はNCFCAの結果と一致している。
【0176】
実施例4:白血病抑制因子(LIF)は、連続的に継代されたヒト胎児神経幹細胞における幹細胞および前駆細胞の存在頻度を低下させる
連続的に継代されたヒト胎児神経幹細胞を、実施例2のような通常の増殖条件(マイトジェンFGF2およびEGFを含む)で、20ng/mlのヒトLIFを添加したものと添加していないものについて培養した。いずれの群についても成長曲線を作成し(図2A)、各継代の終了時に生成された細胞の理論上の総数の対数をとることにより均等目盛に変換し(実施例2を参照)、最も良くフィットする傾向線に基づいて傾きおよびy切片を決定した(図2B)。実施例1の方法による幹細胞および前駆細胞の存在頻度の分析から、LIF存在下では幹細胞の存在頻度が48%、前駆細胞の存在頻度が18%低下することが明らかとなった(図2C;y軸は対照の値に対する割合(%)を表す)。従って、LIFは連続的に継代されたヒト神経幹細胞中の神経幹細胞および神経前駆細胞の存在頻度を低下させるために使用することができる。
【0177】
実施例5:骨形成タンパク質2(BMP−2)は、連続的に継代されたヒト胎児神経幹細胞における幹細胞および前駆細胞の存在頻度を低下させる
連続的に継代されたヒト胎児神経幹細胞を、実施例2のような通常の増殖条件(マイトジェンFGF2およびEGFを含む)で、20ng/mlのヒトBMP−2タンパク質を添加したものと添加していないものについて培養した。いずれの群についても成長曲線を作成し、各継代の終了時に生成された細胞の理論上の総数の対数をとることにより均等目盛に変換し、最も良くフィットする傾向線に基づいて傾きおよびy切片を決定した。実施例1の方法による幹細胞および前駆細胞の存在頻度を分析すると、BMP−2が存在するといずれの種類の細胞においても低下することが明らかとなった。従って、BMPは連続的に継代されたヒト神経幹細胞中の神経幹細胞の存在頻度および神経前駆細胞の存在頻度を低下させるために使用することができる。
【0178】
実施例6:連続的に継代された多形膠芽腫(GBM)由来の細胞は幹細胞の重要な特徴を示す
ヒトGBMには、神経幹細胞が生体条件外(ex vivo)で増殖できるように調整された条件下で増殖する、腫瘍性神経幹細胞(tNSC)が含まれている。参照によって全体が本願に組み込まれるガルリ(Galli)ら、2004年、Cancer Research 第64巻、p.7011−7021を参照されたい。tNSCは、上述のガルリ(Galli)らの文献(2004年)および参照によって全体が本願に組み込まれるベスコビ(Vescovi)ら、Exp.Neurol.第156巻、p.71−83、1999年の方法に従って腫瘍サンプルから単離することができる。簡潔に述べると、tNSCを単離するには、最初に、腫瘍サンプルを、07.mg/mLのオボムコイドを含んでいるダルベッコ改変イーグル培地(DMEM)/F12の中で粉砕して個々に分離する。細胞を遠心分離によって回収し、成長因子を含まず化学的組成の明らかなNS−A培地(カナダ国ブリティッシュコロンビア州バンクーバー所在のステムセルテクノロジーズ(Stem Cell Technologies))に2mMグルタミン、0.6%グルコース、9.6gm/mLプトレッシン、6.3ng/mLプロゲステロン、5.2
ng/mL亜セレン酸ナトリウム、0.025mg/mLインシュリン、0.1mg/mLトランスフェリンを含めたものに再浮遊させる。その後、生細胞を、化学的組成の明らかな同じNS−A培地に20ng/mLのEGFおよび20ng/mLのFGF2を加えた中に、細胞密度を2,500−5,000個/cm2として25cm2組織培養フラスコ中に播種する。必要ならば培地を24−48時間ごとに新鮮な培地に交換する。播種の5〜20日後に、参照によって全体が本願に組み込まれるレノルズ(Reynolds)およびワイス(Weiss)、1992年、Science 第255巻、p.1707−1710によって記述されるように、正常な神経幹細胞によってインビトロで形成される古典的なニューロスフェアに似た腫瘍細胞のニューロスフェアを観察することができる(図3Aを参照:図3Aは20倍の位相差観察像である)。腫瘍細胞のニューロスフェアを個々に分散させて同じ条件下で連続的に再継代すると、tNSCは新しい腫瘍細胞ニューロスフェアを構築する。tNSCは、培養下で安定的に自己複製して増殖する(2つの異なるGBM細胞株について各分裂における理論上の総細胞数を示す図3Bを参照されたい)。tNSCの子孫は分化条件下でニューロン、乏突起膠細胞および星状細胞を生じるので、tNSCはインビトロにおいて多能性である。例えば、抗β−チューブリン抗体、抗グリア線維性酸性タンパク質(GFAP)抗体(GFAPは星状膠細胞への分化のマーカーである)、および抗ガラクトセレブロシド(GalC)抗体による、分化した子孫の染色が観察された(GalCは乏突起膠細胞のマーカーである)。
【0179】
免疫不全マウスの脳へ移植すると、tNSCは、ちょうど典型的なヒトGBMと同じように、星状膠細胞特異的マーカーとの免疫反応性を備えたGBMを生じる。これらの腫瘍そのものを取り出してtNSCを再培養すると、次世代のtNSCは免疫不全マウスの脳内に移植されるとGBMを再生成する。したがって、ヒトのtNSCはインビボでは単能性で、インビトロでは多能性であり、クローンのレベルに至るまで腫瘍の基となる細胞として働く可能性があり、連続的な再移植の後でさえヒトの疾病の重要な特徴に非常によく似た腫瘍をマウスにおいて創り出すことができる。したがって、ヒトのtNSCは、ヒトGBMの増殖および再発に関係するように思われる。
【0180】
実施例7:腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を計算するための連続的に継代されたGBM腫瘍細胞の数学的モデル化の使用
幹細胞および前駆細胞の存在頻度を、実施例1の分析法を使用して3つのGBM細胞株について測定した。これらの細胞株は、3人の異なる患者の腫瘍(C.ベスタ神経学研究所(Neurological Institute C. Besta)から入手し、世界保健機構(WHO)のガイドラインによって分類した腫瘍)に由来するものである。腫瘍細胞を実施例6の方法を使用して連続的に継代し、生成された細胞の理論上の総数を、各細胞株(627、913、1022)について継代ごとにプロットした(図4Aを参照)。各継代の終了時に生成された細胞の理論上の総数の対数を計算し、最も良くフィットする傾向線に基づいて傾きおよびy切片を計算した(図4Bを参照)。幹細胞および前駆細胞の数についての度数は実施例1の方法を使用して計算した。腫瘍性幹細胞の存在頻度は細胞全体の0.44%であり、腫瘍前駆細胞の存在頻度は細胞全体の1.56%であった(図4Cに3つのGBM細胞株各々について示した)。
【0181】
実施例8:神経コロニー形成細胞法(NCFCA)における、GBM腫瘍性幹細胞由来の子孫細胞からの腫瘍性幹細胞および腫瘍前駆細胞の存在頻度の算出
継代数15のGBM細胞を、実施例3に記載のように神経コロニー形成細胞法(NCFCA)でプレート培養した。培養物に毎週新鮮な培地を供給し、3週後にコロニーの直径を測定した。この分析法では、最も大きなコロニーだけが、軟寒天培地からの抽出およびサブクローニングの後で幹細胞の特性を示す。播種した細胞全体の0.25%〜0.65%が大きなコロニーを形成した。図5の主グラフを参照すると、播種した細胞全体に対する、直径が1,500−2,000μm;1,000−1,500μm;500−1,0
00μm;および<500μmのコロニーを形成するものの割合(%)(y軸)を示している。図5の主グラフに挿入されたグラフは、播種した細胞全体に対する、直径が1,500−2,000μmおよび1,000−1,500μmの区分のコロニーを形成するものの割合(%)(y軸)を、主グラフとは異なるy軸目盛りで示している。したがって、NCFCAおよび実施例7の数学的分析から、GBM腫瘍細胞株中の腫瘍性幹細胞について同様の存在頻度が得られる。
【0182】
実施例9:初代腫瘍サンプルおよびtNSCにおけるすべての種類のBMPレセプタの存在
3種のBMPレセプタ(BMPR1a、BMPR1bおよびBMPR2)の発現を、以下のものについてリアルタイムPCRで測定した。
【0183】
1. 初代ヒト脳腫瘍標本:未分化星細胞腫(AA031217);膠芽腫(GBM050203、GBM040114、GBM040202、GBM050207、GBM050208);および胚芽異形成性神経上皮新生物(NND040115);
2. 正常なヒト胎児神経幹細胞(fNSC);および
3. 実施例6の方法によって調製されたヒト膠芽腫の腫瘍性神経幹細胞(tNSC)の細胞株(GBM010627、GBM020913、GBM021022)。
【0184】
以下の方法を用いた。最初に、TRIzol試薬(米国メリーランド州ロックヴィル所在のライフ・テクノロジーズ(Life Technologies))を使用してfNSC、tNSCs、および初代腫瘍サンプルから全RNAを単離し、次に、SuperScript(登録商標)RnaseH逆転写酵素(米国メリーランド州ロックヴィル所在のライフ・テクノロジーズ)を使用して逆転写した。鋳型として使用したcDNAはすべて、ハウスキーピング遺伝子としてグリセルアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)および陽性対照としてMCF−7細胞株を使用して、あらかじめ標準化されたものとした。その後、各BMPレセプタ特異的プライマーを使用し、Brilliant(登録商標)SYBR(登録商標)グリーンQPCRコア試薬キット(米国カリフォルニア州ラ・ホーヤ所在のストラタジーン(STRATAGENE))を使用して、定量的リアルタイムPCR反応を3連で実施した。SYBRグリーン色素はあらゆるPCR生成物に結合するため、配列特異的プローブを使用する必要がない。Brilliant SYBRグリーンのマスターミックスは、UNGによるコンタミネーション除去プロトコールに使用するdUTPを含んでいる。使用したプライマー配列は以下のとおり、すなわち
プライマー配列×逆転写(RT)PCR
hBMPR−1A Fw:5’−AATGGAGTAACCTTAGCACCAGAG−3’
hBMPR−1A Rw:5’−AGCTGAGTCCAGGAACCTGTAC−3’
hBMPR−1B Fw:5’−GGTTGCCTGTGGTCACTTCTGG−3’
hBMPR−1B Rw:5’−TAGTCTGTGATTAGGTACAACTGG−3’
hBMPR−2 Fw:5’−TCAGATATATGGCACCAGAAGTG−3’
hBMPR−2 Rw:5’−GTGGAGAGGCTGGTGACACTTG−3’
プライマー配列×リアルタイムPCR
hBMPR1a Fw:5’−caggttcctggactcagctc−3’
hBMPR1a Rw:5’−ctttccttgggtgccataaa−3’
hBMPR1b Fw:5’−aaaggtcgctatggggaagt−3’
hBMPR1b Rw:5’−gcagcaatgaaacccaaaat−3’
hBMPR2 Fw:5’−gctaaaatttggcagcaagc−3’
hBMPR2 Rw:5’−cttgggccctatgtgtcact−3’
である。
【0185】
蛍光性の発光をリアルタイムで記録した(Chromo4(TM)4色リアルタイムPCR検出器、MJリサーチ(MJ Research)、バイオラッド(BIO−RAD)、米国)。遺伝子発現の解析は、相対的な定量法の比較Ct法(ヒグチ(Higuchi)ら)を使用して完了した。各遺伝子について、相対的なRNA量を2つの内在性の対照すなわちGAPDHおよび18s rRNAに対して標準化した。3連実験のそれぞれを標準化し、平均の相対量(RQ)を各遺伝子について報告する。平均で何倍変化したかを、3連実験の標準偏差および95%信頼区間と共に計算した。
【0186】
図6に結果を示す。この結果は、悪性度の異なる様々な腫瘍が特有のBMPR発現プロファイルを有することを示している。したがって腫瘍のBMPR発現プロファイルは、腫瘍の特徴を解析し、腫瘍の悪性度を予測するための診断法において使用することができる。
【0187】
実施例10:白血病抑制因子(LIF)およびBMPは、連続的に継代されたヒトGBM細胞株における幹細胞の存在頻度を低減する
3人の異なる患者から得られたGBM腫瘍細胞を、LIF(20ng/ml)またはBMP−4(20ng/ml)のいずれかを含むかまたは含まないコントロール培地(マイトジェンEGF+FGF2を含む;実施例6を参照のこと)の中で連続的に継代した。実施例1の方法を使用した幹細胞および前駆細胞の存在頻度の分析から、LIFまたはBMP−4の添加により腫瘍性幹細胞の数が著しく減少し(統計的有意性 p<0.01)、BMP−4の添加により腫瘍前駆細胞の数が著しく減少する(統計的有意性 p<0.05)が、LIFを添加しても腫瘍前駆細胞集団の存在頻度は有意には変化しないことが明らかとなった。これらの結果は、BMP分子が腫瘍性幹細胞および腫瘍前駆細胞の増殖を低減し、LIFは腫瘍性幹細胞の数を選択的に低減し腫瘍前駆細胞群に対しては影響しないことを示している。図7は、結果をグラフで(未処置対照の値に対する割合(%)として)表している。これらの結果は、LIFおよびBMPがそれぞれ、マイトジェンEGFおよびFGF2の存在下であってもtNSCの増殖を低減するのに有効であることを示している。これは、LIFおよびBMPのうち少なくともいずれか一方による処置がヒトの脳腫瘍の治療に有効となることを示している。
【0188】
実施例11:LIFおよびBMP−2を連続的に継代されたGBM由来の細胞に適用すると、幹細胞および前駆細胞の存在頻度が低下する
コントロール培地(マイトジェンEGF+FGF2を含む;実施例6を参照のこと)中で連続的に継代したGBM細胞(実施例6を参照)を、LIF、BMP−2またはBMP−2+LIFの組み合わせで処理した。連続的に継代した対照のGBM細胞は、どちらのタンパク質による処理も行わなかった。幹細胞および前駆細胞の存在頻度を計算するために実施例1の方法を使用して成長曲線を比較した。データから、LIFおよびBMP−2を一緒に使用すると、単独で使用した時よりも腫瘍性幹細胞および腫瘍前駆細胞の存在頻度が大きく低下することが明らかである。図8は、結果をグラフで示すものである(y軸は、未処理対照の値に対する割合(%)を表す)。これらの結果は、LIFおよびBMP−2の同時投与が、マイトジェンEGFおよびFGF2の存在下であってもtNSCの増殖を低減するのに有効であること、またLIFおよびBMP−2の同時投与はヒトの脳腫瘍を治療するのに有効であろうことを示している。
【0189】
実施例12:培養したGBM細胞をBMP−2で一時的に処理すると幹細胞および前駆細胞の存在頻度が永続的に低下する
3人の異なる患者から得られたGBM腫瘍細胞を、コントロール培地(EGF+FGF2を含む;実施例6を参照のこと)中、BMP−2(20ng/ml)を添加するか(図9に「BMP」と示す)または1回の継代時にBMP−2に一時的に曝露して(図9に「BMP後」と示す)、連続的に継代した。実施例1の方法を使用した腫瘍性幹細胞および腫瘍前駆細胞の存在頻度の分析から、いずれのBMP処理群においても腫瘍性幹細胞および腫瘍前駆細胞の数が著しく減少することが明らかになった。これらの結果は、BMP−2に一時的に曝露すると、マイトジェンEGFおよびFGF2の存在下であっても腫瘍性幹細胞および腫瘍前駆細胞の存在頻度が永続的に低下することを示している。これらの結果は、BMP−2治療がヒトの脳腫瘍の増殖に永続的効果を有すると思われることを示している。
【0190】
実施例13:LIFおよびBMPを用いた事前処理後の培養GBM細胞における腫瘍形成性の低下
免疫不全マウスの脳(腹側外側の線条体)に、ヒトGBM由来の腫瘍性神経幹細胞100,000個を定位注入により移植した。腫瘍性神経幹細胞はマウスにおいてGBMを発症させた。これらのGBM病変は、正常組織とは明白に区別される、核が過剰に存在する広範な領域として明らかになった(図10の上側パネルを参照)。同じ腫瘍性神経幹細胞を移植に先立って100ng/mLのBMP−4または100ng/mLのLIFで48時間前処理すると、腫瘍の形成は著しく(ほぼ80%)低減され、核が過剰に存在する領域は検出困難なものが多い(図10の下側パネルを参照)。これらの結果は、BMPおよびLIFのうち少なくともいずれか一方による治療がヒト脳腫瘍の治療に有効であることをさらに示すものである。補足例については、実施例22も参照されたい。
【0191】
実施例14:LIFおよびBMP−2処理後の乳癌組織における腫瘍性幹細胞の存在頻度の分析
腫瘍切除のために生検を受ける患者から同意のもとに腫瘍標本を入手し、1:1コラゲナーゼ/ヒアルロニダーゼ溶液中37℃にて1時間酵素消化してから40μmフィルタで濾過し、細胞を、マイトジェンEGFおよびFGF2(20ng/ml)を含んだ無血清DMEM/F12ホルモン混合物(NeuroCult(登録商標)、ステムセルテクノロジーズ(Stem Cell Technologies))中にクローン化しうる密度で播種した。3−5日後、クローンから生じた細胞塊が懸濁浮遊しているのが観察される。これらの細胞を回収し、酵素的に個々に分散させ、新鮮な増殖培地に再度播種する。このようにして4−7日ごとに継代した細胞は、生成される総細胞数が幾何学的に増加する。
【0192】
連続的に継代した乳房腫瘍由来の幹細胞を、LIFおよびBMP−2のうち少なくともいずれか一方に曝露し、対照の培養物と比較する。神経コロニー形成細胞法において乳房性球状細胞塊(mammary sphere)を分散させた細胞を播種し、実施例1の数学的モデルを用いて一連の成長曲線を分析することにより、処理群および対照群の腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を分析する。
【0193】
実施例15:LIFおよびBMP−2処理後の前立腺癌組織における腫瘍性幹細胞の存在頻度の分析
リンパ節転移から前立腺がん細胞を入手し、5%の血清と幹細胞マイトジェンEGFおよびFGF2とを添加したDMEM/F12ホルモン混合物(ステムセルテクノロジーズ)で培養する。クローンから生じた球状塊をトリプシン処理してから連続的に継代し、継代した細胞(未処理のもの、またはLIFおよびBMP−2のうち少なくともいずれか一方で処理したもの)由来の増殖データを用いて、実施例1の数学的モデルを用いて腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を計算する。
【0194】
実施例16;LIFおよびBMP−2処理後の黒色腫組織における腫瘍性幹細胞の存在頻度の分析
切除された腫瘍から黒色腫細胞を得て、哺乳類の皮膚から多能性幹細胞を培養するために使用された方法(参照により全体が本願に組み込まれるトマ(Toma)ら、Nat Cell Biol.2001年9月、第3巻(9)、p.778−84を参照のこと)に従って幹細胞を単離する。簡潔に述べると、組織を小片(<2mm)に切断し、HEMで洗浄し、0.1トリプシンで37℃にて30分間消化する。サンプルをPBSですすぎ、機械的に分散させて単個細胞浮遊液とし、40um細胞ストレーナでろ過し、ヌンク(Nunc)のT−80組織培養フラスコ中で1ml当たり細胞50,000個の密度で播種する。増殖培地は、幹細胞マイトジェンEGFおよびFGF2を添加したホルモン混合物含有無血清DMEM/F12(ステムセルテクノロジーズ)である。7−10回の分裂の後、クローンから生じた細胞塊が培養物中に確認される。これらのメラノスフェアを回収し、分散させて単個細胞浮遊液とし、幹細胞マイトジェンを含む新鮮な培地を使用して、再度播種する。このように継代した培養物をLIFまたはBMP−2で処理し、経時的に生成された細胞の総数を対数目盛でプロットする。治療群および対照群の腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を、実施例1の数学的モデルを使用して分析する。
【0195】
実施例17:腫瘍切除後のCED法によるLIF投与を用いた再発性多形膠芽腫の治療
再発性多形膠芽腫の患者を選出する。腫瘍切除に続いて、切除腔を囲んでいる脳実質に、2〜3個のカテーテルを、脳溝または脳室内に入らないように画像誘導を使用して配置する。該カテーテルを通して、72mLのヒトLIF(1μg/mL)を、シリンジ・ポンプを使用して96時間かけて注入する。
【0196】
実施例18:ポリマービーズを用いたBMP−4投与による再発性多形膠芽腫の治療
再発性多形膠芽腫の患者を選出する。腫瘍切除に続いて、BMP−4で飽和させたポリアクリル酸ビーズ(1週間以上BMP−4を放出する)を切除部位に埋め込む。
【0197】
実施例19:GBM標本由来の細胞におけるBMPRの発現
GBM組織由来の細胞におけるBMPおよびそのレセプタ(BMPR)の転写物およびタンパク質の発現を、GBM組織由来のCD133+tNSC集団について評価した(シン(Singh)、S.K.ら、Nature 第432巻、p.396−401、2004年)。下記のインビトロのデータは、1つの代表的なGBM標本由来の細胞から得たデータを例証するものであるが、同等の結果は別の4つのサンプルでも得られている。該サンプルは選別されたCD133+細胞または非選別細胞を含み、該細胞はGBMから急性単離したものであるかまたはマイトジェンとともに短時間培養した後(培養した細胞)であるかのいずれかである(ガルリ(Galli)ら、Cancer Res 第64巻、p.7011−21、2004年;シン(Singh)、S.K.ら、Nature 第432巻、p.396−401、2004年)。BMPRの3つのサブタイプ(BMPR1A、−1B、−2)およびBMPのすべての転写物が、急性単離したGBM細胞および培養したGBM細胞のいずれにおいても見出された。図11Aを参照すると、同図においてレーン1は陰性対照、レーン2は急性単離したGBMのCD133+細胞、レーン3は培養したGBMのCD133+細胞、レーン4は陽性対照のMCF7細胞である。さらに、同系統のレセプタおよびBMP−4タンパク質がいずれの集団においても、CD133+画分およびCD133−画分のいずれにおいても見出された。図11B−Gを参照すると、単離直後(図11B−D)および培養した(図11E−G)CD133+GBM細胞における記載のタンパク質の免疫蛍光が示されている。
【0198】
最も有効なリガンドのうちの1つであるBMP−4を飽和濃度(100ng/ml)で添加した後にSmad1−5−8のリン酸化が観察されたので、BMPRは機能性を有しており、一方p38MAPK経路の活性化は検出されなかった。図11H−Jを参照する
と、GBM細胞における、記載の時間(分)での、レセプタに活性化されたSmadタンパク質のリン酸化および核移行を示す(抗リン酸化Smad1,5,8)。
【0199】
図11K−Pは、記載のタンパク質のウエスタンブロット解析を示す。図11Kは、急性単離した(左側のレーン)、および培養した(右側のレーン)CD133+GBM細胞におけるBMP−4タンパク質を示す。図11Lは、BMP−4処理した培養細胞でSmad1のレベルは変化しなかったことを示している(左側のレーンが対照、右側のレーンがBMP−4処理)。図11M−Nは、単離直後の細胞(図11M)および培養した細胞(図11N)におけるBMP−4存在下でのSmad1,5,8のリン酸化の増大を示している(左側のレーンが対照、右側のレーンがBMP−4処理)。図11O−Pは、培養したGBM細胞(図11O)および急性単離したGBM細胞(図11P)におけるBMP−4処理後のSmad4発現の増大を示している(左側のレーンが対照;右側のレーンがBMP−4処理)。
【0200】
この結果は、Smad1,5,8複合体もまたGBMの治療に有用な治療標的であり得ることを示すものでもある。例えば、該複合体のリン酸化を増大させる治療薬はtNSCに対してBMP−4と同じ効果を有するであろう。本開示の他の箇所に開示したスクリーニング法は、そのような治療薬を得るために使用されてもよい。
【0201】
実施例20:GBMのtNSCに対するBMP−4曝露の作用に関する検討
GBMから単離された細胞に対するBMP−4の作用の性質について検討した。他の細胞系(ハラハン(Hallahan)ら、Nat Med 第9巻、p.1033−8、2003年;ズザルテ−ルイ(Zuzarte−Luis)およびヒューレ(Hurle)、Semin Cell Dev Biol 第16巻、p.261−9、2005年;フルスカ(Hruska)ら、Circ Res 第97巻、p.105−14、2005年)、例えば髄芽細胞腫(グラハム(Graham)ら、Mol Cell Neurosci 第8巻、p.76−83、1996年)などとは異なり、BMP−4は細胞死(ヨウ化プロピジウム排除法により測定)またはアポトーシス(TUNEL法により測定)を生じなかったが、マイトジェンに応答するGBM細胞の増殖を著しく低減した(Ki67免疫蛍光法により測定)。図12Aは、GBM培養物におけるBMP−4の作用をグラフで示している(白抜きのカラムは対照の培養物、暗色のカラムはBMP−4処理物;平均+SE、n=3;*p、0.005;PI=ヨウ化プロピジウム)。
【0202】
増殖に対する他のBMPの作用も分析した。図15を参照すると、GBM細胞の増殖に対する他のBMP(100ng/ml)の作用が示されている。BMP−2、−4、−5、−6、−7、−8bは細胞増殖を抑制するが、BMP1、−3および−3bは、TGFβ1、2およびTGFβ1,2(TGFβアゴニストのキメラポリペプチド)と同様に効果がないようであった(すべて100ng/ml)。*BMPと対照、p<0.005、平均±SE、n=3、両側スチューデントt検定;**BMP4と対照、p<0.001、平均±SE、n=3、両側スチューデントt検定。したがって、BMP−2、−4、−5、−6、−7、−8bも、特にGBMの治療のための本開示の方法および組成物に有用であろう。
【0203】
BMP−4の抗増殖作用は、BMP−4に応答してG0/G1期のGBM細胞の数が著しく増加し、S期の細胞の割合(%)が減少することを示した細胞周期分析によって確証された。GBM細胞に見出されるTGFβ(ケルマン(Kjellman)ら、Int J Cancer 第89巻、p.251−8、2000年)は、BMPのシグナル伝達経路とオーバーラップするシグナル伝達経路を用いて(カナリ(Canalis)ら、Endocr Rev 第2巻、p.218−35、2003年;ゴレステーン(Golestaneh)ら、Oncogene 第24巻、p.5722−30、2005年)、
促進性の作用または反促進性の作用を誘発する(ジェニングス(Jennings)ら、J Neurooncol 第36巻、p.123−40(1998))が、BMP4とは異なり、本明細書に記載のBMP4作用の特異性の基礎をなすGBM細胞の増殖への影響は示さなかった。
【0204】
古典的な2つの分析法、すなわち1つはクローン形成の指標を測定し、従ってクローンを形成するニューロン前駆体の割合(%)を計測する分析法(グリッティ(Gritti)ら、J Neurosci 第16巻、p.1091−100、1996年;レノルズ(Reynolds)ら、Dev Biol 第175巻、p.1−13、1996年)ならびに第2には増殖速度論データに基づいてNSC集団の大きさの増大を測定する分析法(ガルリ(Galli)ら、Development 第129巻、p.1633−44、2002年;レノルズ(Reynolds)ら、Nat Methods 第2巻、p.333−6、2005年)により、BMP−4の細胞増殖抑制作用がGBM細胞中のtNSC集団に影響を与えたことが確認された。BMP−4に48時間曝露すると、GBM細胞におけるクローン形成の指標(播種した細胞の合計に対する形成クローンの割合(%))はインビトロで70%を超えて低減された(図12Bを参照すると、急性単離したGBM細胞および培養したGBM細胞の両方についての、対照群およびBMP−4処理群に関するクローン形成の指標がグラフで示されている;平均±SE、n=3;*はp<0.005)。さらに、初代腫瘍標本からの単離直後にGBM細胞をBMP−4に曝露すると、同細胞が培養時に増殖する能力が消失し、一方、既にインビトロで増殖中のGBM細胞にBMP4を添加すると同細胞の成長速度が大きく低下した。ニューロスフェア分析法を使用してインビトロでのGBM細胞の増殖をグラフに示す図12Cを参照すると、急性単離したGBM組織からの細胞はBMP−4の存在下では連続的に二次培養できなかったことが明らかである(黒い点線に黒塗り三角形)。図12Cはまた、同じ条件下で短時間増殖させた後(菱形)、急性単離した同じ組織に由来する細胞をBMP−4で処理しても、成長曲線の傾きが著しく小さくなった(正方形)ことも示している。同様の結果は、BMP−4で処理したヒト胎児NSCについても見られ(図12C参照、黒い星印(対照)と黒丸(BMP−4))、ヒト神経膠腫細胞株U87(BMPRを持たない)は影響を受けなかった(図12Cを参照、白抜き三角形(対照)とバツ印(BMP−4))。
【0205】
細胞蛍光測光法から、BMP4処理の結果、急性単離したGBM細胞および培養したGBM細胞のいずれにおいても、CD133+集団の大きさがほとんど半分になること(図12Aを参照)が示された。これらのデータはともに、BMP−4がGBM細胞のtNSC集団を標的とすることができることを示している。
【0206】
GBM細胞をBMP−4に曝露すると、インビトロで明らかな形態学的変化が生じた。マイトジェン単独で増殖させた細胞に比べ、さらにBMP4を与えられた細胞はより分化の進んだ(扁平、濃密な様相、複雑な突起を有する)形態を呈した。対照の細胞を示す図13A、およびBMP−4に48時間曝露した後の細胞を示す図13Bを参照されたい。これに応じて、星状膠細胞のマーカーの発現(GFAP免疫反応性[IR])のかなりの増加と一緒に、それほど大きくはないがニューロンのマーカー(βIIIチューブリンおよびMAP5)または乏突起膠細胞のマーカー(GalC)に関する標識の増大が観察された。図13C−Hを参照されたい。具体的には、図13C(対照)および図13D(BMP−4)はGFAP−IRを示し;図13E(対照)および図13F(BMP−4)は、βIIIチューブリン−IRを示し;図13G(対照)および図13H(BMP−4)はGalC−IRを示す。
【0207】
特定の分化マーカーを発現している細胞の数を数えることによるBMP4の作用の定量化は、増殖している未分化のGBM培養物中および同細胞内でニューロンの抗原および神経膠の抗原が異常に発現しているという、正常な神経幹細胞では観察されない2つの現象
のため、困難であった。したがって、細胞蛍光測光法を用いて蛍光信号強度を計測し、同方法により、対照に比べてBMP−4で処理した培養物ではGFAP−IRが2倍以上増大すること(図13Iを参照)、そして同様に、ただし顕著ではないがβIIIチューブリン(図13J)およびGalC(図13K)の免疫反応性が増大することが見出された(MEFL=フルオレセイン1分子相当の蛍光量;MEFE=フィコエリスリン1分子相当の蛍光量)。
【0208】
理論または仮説によって制限されるものではないが、ここで述べるBMP−4の作用は、それ自体tNSCである2つの同一の娘細胞を生じる対称分裂の回数の減少によるものであるか、あるいはtNSC群の一部が分化してもはや幹細胞の特性を保持することができないことによるものと考えられる。要するにこのことは、BMP−4が細胞分裂促進性の刺激を無効にして、tNSCに、より成熟した腫瘍形成性の低い表現型を獲得させることができることを示唆している。BMP−4はヒトES細胞に対し、幹細胞集団および増殖速度を増大させる抗分化作用を有するので、上記の結果は予想外である。
【0209】
実施例21:BMP−4はGBSCの腫瘍形成能を抑制し、インビボに送達可能である
急性単離したCD133+GBM細胞および培養されたCD133+GBM細胞の両方を、培養下で48時間BMP−4に曝露した後、免疫不全のscid/bgマウスの片側の線条体へ注入した(3×105個の生細胞)。腫瘍の形成および増大を、同一の条件下だがBMP−4を伴わずに維持されたGBM細胞を与えた対照動物のものと比較した。
【0210】
未処理のGBM細胞を与えた動物はすべて、注入された側に確立した腫瘤を生じた(図14Aを参照)。この腫瘤は、膠芽腫の特徴、たとえば顕著な核異型性、異常な神経膠成分の発現、広範囲な新血管新生および高い有系分裂活動(ガルリ(Galli)ら、Cancer Res 第64巻、p.7011−21、2004年)を示し、側脳室、第三脳室および第四脳室に侵入した。これに対し、BMP4で処理した細胞は侵襲性の腫瘍を形成せず、小さくて範囲の定まった病変を形成し、該病変は注入部位に制限され、分裂指数が低く、脳室への侵入を示さなかった(図14Bを参照)。注入後3〜4か月の間に対照の動物はすべて死亡したが、予めBMP−4処理した細胞を与えたほぼすべてのマウスは生き残った(図14Jを参照。同図は前処理(左側パネル)、同時処理(中心パネル)、および後処理(右側パネル)した例における生存をグラフで示している(ログランク検定、それぞれp<0.001、p<0.001、およびp<0.005)。同じ結果は、BMP−4処理したGBM培養物からの残りのCD133+画分をFACSで精製し、その腫瘍形成性を、対照のGBM細胞から精製した同数のCD133+細胞(動物1匹あたり1.5×105個のCD133+細胞)の腫瘍形成性と比較した場合にも観察された。さらに、従来示されるように(ガルリ(Galli)ら、Cancer Res 第64巻、p.7011−21、2004年;シン(Singh)ら、Nature 第432巻、p.396−401、2004年)、急性単離した対照GBM細胞を与えたマウスの脳からCD133+tNSCを再培養することは常に可能であった(平均のクローン形成頻度:9.0±1.3%[n=2、移植後90日])。これらは、scid/bgマウスの脳に再び脳内移植されると大きな二次腫瘍を生じさせた。反対に、上記のことは、最初の移植においてBMP−4で前処理したGBM細胞を与えた動物では不可能であり、また同じマウスの一次腫瘍から急性単離した細胞3×105個を直接注入しても二次腫瘍を確立することは不可能であった。
【0211】
総合すると、これらの所見は、BMP−4への一時的な曝露であっても、GBMのtNSC集団を激減させ、GBM細胞のインビボにおける腫瘍発生能力を顕著に低下させることを実証するものである。さらに実施例13を参照されたい。
【0212】
実施例22:BMP−4のインビボ送達により脳内腫瘍の確立および増大が防止される
次に、脳内腫瘍の確立および増大を防止する治療法としての、BMP−4のインビボ送達について評価した。GBMを移植する際、細胞と同時(同時処理例)または10日後(後処理例)に、ビヒクル(対照)またはBMP−4で飽和させたポリアクリル酸ビーズ(1週間以上BMP−4を放出)を、細胞移植部位に注入した。双方の処理群における組織学的な一連の再構成から、BMP−4処理した動物では対照に対して腫瘤の最大増量が著しく減少することが明らかになった。具体的には、すべての実験設定において、対照のビーズを与えた動物は大きな悪性腫瘍を生じ(図14C(培養したGBM細胞、注入後4週間、同時処理例);図14E(単離直後のGBM細胞、細胞注入後30日、後処理例))かつすぐに死亡した(図14J)が、BMP4放出ビーズを注入したマウスでは小さく制限された病変が見られ(図14D(培養したGBM細胞、注入後4週間、同時処理例);図14F(単離直後のGBM細胞、細胞注入後30日、後処理例);そして有意に長く生存した(図14J)。対照の腫瘍は、多形態性で、新生物形成性の高い要素を、反応性の染色質および悪性の高い浸潤性の細胞と共に含んでいた(図14G)。反対に、BMP−4処理した動物は、新生物形成性の低い細胞、多数の高度に分化した要素および多数のマクロファージを含む病変を示した(図14H)。分裂指数は、BMP−4処理した動物に対して対照で有意に高かった(同時処理:対照の3.8±0.2に対しBMP−4では0.20±0.1;p<0.01。後処理:対照の4.3±0.3に対しBMP−4では0.7±0.3;p<0.05。平均±SE、n=4、両側スチューデントt検定)。インビボにおける免疫蛍光法から、対照の腫瘍およびBMP4処理した腫瘍のいずれにおいても、星状細胞およびネスチン陽性細胞は存在するが、乏突起膠細胞やニューロン細胞は存在しないことが明らかとなった。
【0213】
これらの結果は、BMP−4を放出するか、そうでなければBMP−4を送達するデバイスの脳内移植が、ヒトにおけるGBMの治療に有効と思われることを示唆している。
実施例23:BMP−4抗体中和実験
実施例19は、内在性のBMP4および他のBMPがGBM細胞中に見出されることを示している。先の実施例で示された所見と一致して、抗BMP4中和抗体はtNSCの増殖を増大させる。これは、内在性のBMPはインビボで生理的にGBM細胞に作用しうるが、その影響は腫瘍の成長を停止させるのには不十分であることを示唆している。この現象の背景にあるメカニズム、例えば腫瘍中に内在性BMPアンタゴニストが存在する可能性について検討することは(カナリ(Canalis)ら、Endocr Rev 第2巻、p.218−35、2003年)、GBM治療のための別の戦略を指し示し得る。さらに、BMP、BMPの同系統レセプタおよびBMPに関連する細胞内の伝達メカニズム、特にSmad経路は、GBMの確立および増殖の主因である細胞に対してより特異的に照準を定める治療法の有望な標的として浮上する。
【0214】
実施例24:初代培養、培養増殖、クローニングおよび細胞株の確立
GBM細胞は、ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021に記述されるように腫瘍サンプルを処理して得た。急性単離した細胞を、CD133への免疫反応性について選別し(以下を参照)、NeuroCult(登録商標)NS−A無血清培地(ステムセルテクノロジーズ)中で最終密度を1cm2あたり細胞2500個として25cm2組織培養フラスコに播種した。培養増殖、クローン形成の分析および集団の解析は、ガルリ(Galli)ら、Development 第129巻、p.1633−44(2002)にすでに記載されているのと同一の条件を使用して実施した。急性単離したCD133+GBM細胞のBMP4による前処理については、これらの細胞を、同じ培地でマイトジェンを欠くもの(対照)または100ng/mlのBMP4を含むものの中で、移植に先立って48時間プレート培養した。
【0215】
実施例25:免疫細胞化学
2.5×104個/cm2のGBM細胞を、FGF2/EGFの存在下でMatrig
el(TM)コーティングされたガラス製カバーガラス上に播種し、BMP4(100ng/ml)で48時間処理した。その後、細胞を洗浄し、4%のパラホルムアルデヒド中で固定した(10分)。ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021に記述されるようにして、神経抗原(GFAP、ダコ・コーポレーション(Dako Corporation);Tuj1、バブコ(Babco);Galc、ケミコン(Chemicon))について多重免疫蛍光法を実施した。Ki67染色(1:1000、英国ニューカースル所在のノボカストラ(NovoCastra))により、増殖している細胞を検出した。
【0216】
4%パラホルムアルデヒド中で固定した後、BMPR−1A、−1Bおよび−2について、製造業者の指示書に従って免疫染色した(1:50 R&Dシステムズ)。リン酸化Smad1について染色する場合(1:100 米国マサチューセッツ州ベヴァリー所在のセルシグナリング(Cell Signaling))、細胞をBMP−4(100ng/ml)で様々な時間(5分〜2時間)処理した。適切なアイソタイプ対照または陰性対照を、これらの手順全体にわたって常に含めた。オリジナルのTUNEL法に、ガブリエリ(Gavrieli)ら、J Cell Biol 第119巻、p.493−501(1992)によるジゴキシゲニンに基づいた変更を加えて、フルオレセイン−dUTP TUNEL法(In Situ細胞死検出キット、フルオレセイン、ロッシュ・アプライドサイエンス(Roche Applied Science))を使用してアポトーシスの細胞を検出した。簡潔に述べると、12mmのカバーガラス上で増殖させた細胞を、4%パラホルムアルデヒド中で室温にて10分間固定し、次にPBSですすいだ。その後、細胞を氷中で2分間透過処理してからTUNEL反応液50ulで標識し、パラフィルムカバーガラス下の湿潤チャンバ内で37℃にて1時間インキュベートした。PBSで洗浄した後、スライドを、DAPIを含んだVectashield(TM)にマウントし、蛍光顕微鏡法で調べた。ヨウ化プロピジウム(PI)染色については、細胞を4%パラホルムアルデヒド中で室温にて10分固定し、PBSですすぎ、PI(1ug/ml)とともに室温で5分間インキュベートした。洗浄後、カバーガラスをDAPI含有Vectashield(TM)にマウントし、蛍光顕微鏡法で調べた。PI排除法により生細胞を示した。実施例19の分析法については、サンプルを各試験条件につき6連で実施した。
【0217】
実施例26:従来型PCRおよびリアルタイムPCR
全RNAを、TRIzol試薬(米国メリーランド州ロックヴィル所在のライフ・テクノロジーズ)を使用して培養GBM細胞および急性単離したGBM細胞から単離し、Superscript(登録商標)RNAseH逆転写酵素(ライフ・テクノロジーズ)を使用して逆転写した。従来型のPCR反応で鋳型として使用したcDNAの量は、グリセリンアルデヒド−3−リン酸デヒドロゲナーゼ(GAPDH)に関して標準化した。MCF−7細胞株をBMPRに関する陽性対照として使用した。PCR生成物は、臭化エチジウムで染色したアガロース(1%)ゲル中での電気泳動によって視覚化した。
【0218】
定量的RT−PCR反応は、Brilliant(登録商標)SYBR(登録商標)グリーンQPCRコア試薬キット(米国カリフォルニア州ラ・ホーヤ所在のストラタジーン(Stratagene))を用いて3連で実施した。SYBRグリーン色素はあらゆるPCR生成物に結合し、したがって配列特異的プローブを使用する必要がない。蛍光性の発光をリアルタイムで記録した(Chromo4(TM)4色リアルタイムPCR検出器、MJリサーチ(MJ Research)、バイオラッド(BIO−RAD))。遺伝子発現の解析は、相対的な定量法の比較Ct法を使用して完了した。相対的なRNA量を2つの内在性の対照すなわちGAPDHおよび18SリボソームRNA(18S rRNA)に対して標準化した。
【0219】
従来型PCRについては、次のプライマー、すなわち、BMP4、順方向:5’−cttcagtctggggaggag−3’、逆方向:5’−gatgaggtgcccaggcac−3’;BMPR1A、順方向:5’−aatggagtaaccttagcaccagag−3’、逆方向:5’−agctgagtccaggaacctgtac−3’;BMPR1B、順方向:5’−ggttgcctgtggtcacttctgg−3’、逆方向:5’−tagtctgtgattaggtacaactgg−3’;BMPR2、順方向:5’−tcagatatatggcaccagaagtg−3’、逆方向:5’−gtggagaggctggtgacacttg−3’;GAPDH、順方向:5’−cggagtcaacggatttggtcgtat−3’、逆方向:5’−agccttctccatggtggtgaagac−3’を使用した。PCR増幅条件は、56℃でプライマーをアニーリングする35サイクルで構成されるものとした。
【0220】
RT−PCRについては、次のプライマー、すなわち、BMPR1A、順方向:5’−caggttcctggactcagctc−3’、逆方向:5’−ctttccttgggtgccataaa−3’;BMPR1B、順方向:5’−aaaggtcgctatggggaagt−3’、逆方向:5’−gcagcaatgaaacccaaaat−3’;BMPR2、順方向:5’−gctaaaatttggcagcaagc−3’、逆方向:5’−cttgggccctatgtgtcact−3’;GAPDH:従来型PCRについて記載したのと同じプライマーを使用;18S rRNA、順方向:5’−agtccctgccctttgtacaca−3’、逆方向:5’−gatccgagggcctcactaaac−3’を使用した。プライマーの特異性は、解離曲線分析(Opticon(登録商標)2およびChromo4(TM)リアルタイム・システム・ソフトウェア、MJリサーチ(MJ Research))により、PCRを実施するごとに確認した。RT−PCR増幅条件は、56℃でプライマーをアニーリングする40サイクルで構成されるものとした。
【0221】
実施例27:ウェスタンブロッティング
培養GBM細胞および急性単離されたGBM細胞を冷PBSで洗浄し、500μlの1×サンプル・バッファー(62.5mMトリスHCl、25℃でpH6.8;2%(w/v)SDS;10%グリセロール;50mM DTT;0.01%(w/v)ブロモフェノールブルー)で溶解することにより、タンパク質を採取した。サンプルを氷中でインキュベートし、−20℃で保存した。アリコートを5分間沸騰させ、氷中でインキュベートし、SDS−PAGEゲル(10cm×10cm)に20μl/レーンとして載せた。その後、タンパク質をニトロセルロース・メンブレンに転写した。メンブレンを、TBS中5%粉乳/0.1%トゥイーンで室温にて1時間ブロッキング処理し、15mlのTBS/0.1%トゥイーンで3回洗浄した。その後、ブロットを、TBS中5%粉乳/0.1%トゥイーンの中で、抗Smad1−5−8抗体(1:1000;セルシグナリング(Cell Signaling))、抗リン酸化Smad1−5−8抗体(1:1000;セルシグナリング)、抗Smad4抗体(1:200;サンタクルーズ(Santa Cruz))、抗BMP4抗体(1:400、ケミコン(Chemicon))とともにインキュベートした。すべてのブロットについて、メンブレンを15mlのTBS/0.1%トゥイーンで3回洗浄し、次いでホースラディッシュ結合型の適切な二次抗体(1:1000、アマシャム(Amersham))とともに、TBS中5%粉乳/0.1%トゥイーンの中で室温にて1時間インキュベートした。バンドを化学発光(ECL;アマシャム)によって視覚化した。
【0222】
実施例28:フローサイトメトリー
p38およびSmad1−5−8のリン酸化状態を決定するために、細胞調製物を遠心分離し、0.5mlのPBSおよび0.5mlの4%パラホルムアルデヒド中に37℃で
10分間再浮遊させた。その後、あらかじめ冷却したGBM細胞を優しく攪拌しながら同細胞に氷冷100%メタノールをゆっくり添加して、終濃度90%のメタノールとすることにより、細胞を透過処理した。4℃で30分間のインキュベーションおよび遠心分離に続いて、細胞をPBS中0.5%のウシ血清アルブミン(BSA、シグマ)3mlで2回洗浄し、150ulのPBSに再浮遊させ、室温で10分間インキュベートした。インキュベーション後、1:50希釈の抗リン酸化Smad1−5−8ウサギポリクローナル抗体(セルシグナリング)または1:50希釈のリン酸化p38 MAPキナーゼ・ウサギポリクローナル抗体(セルシグナリング)に対し、暗所にて室温で1時間曝露した。十分に洗浄した後、1:800希釈のヤギ抗ウサギIg FITC標識抗体(BD、ファーミンゲン(BD,Pharmingen))を添加し、各チューブを暗所にて室温で30分間インキュベートした。PBS中0.5%BSA(シグマ)3mlで2回洗浄した後、細胞を0.5mlのPBSに再浮遊させ、フローサイトメトリーによって分析した。自己蛍光およびアイソタイプの対照について、上記の分析法すべてについて常に実施した。
【0223】
細胞周期分析については、1サンプルあたり100万個の培養GBM細胞および急性単離したGBM細胞を、BMP4(100ng/ml)で指定の時間処理した。その後、GBM細胞を、氷冷PBSおよび100%エタノールの等容量混合物に再浮遊させ、氷中で30分間インキュベートした。遠心分離した後、細胞ペレットをPBSで3回洗浄し、5分間遠心分離した。その後、GBM細胞を、RNAse(12.5ug/ml;シグマ)およびヨウ化プロピジウム(3ug/ml;シグマ)を含んだ1mlのPBS中で暗所にて一晩インキュベートし、フローサイトメトリーによって分析した。
【0224】
CD133発現の定量化については、生細胞を区別するために7−アミノアクチノマイシンD(7AAD)を使用して二重染色フローサイトメトリーを実施した。PBSで洗浄した後、GBM細胞を7AAD標識バッファ(0.15M NaCl、5mM EDTA、0.5%BSAおよび0.004%サポニンを含む0.1Mリン酸−クエン酸バッファ、pH6.0)中に再浮遊させた後、トバ(Toba)ら、J Immunol Methods 第182巻、p.193−207(1995)に記述されるように7AADを終濃度20uMとなるように添加した。5〜7分の7AADインキュベーションに続いて、GBM細胞をCD133/1(CD133)−PE結合型モノクローナル抗体(1:40、ミルテニーバイオテク(Miltenyi Biotec))とともに4℃で30分間インキュベートし、1mlの増殖培地で洗浄した。その後、細胞を500×gで5分間遠心分離し、0.5mlの増殖培地中に再浮遊させ、フローサイトメトリーで分析した。同じ分析を、さらにCD133/2(293C3)−PE結合型モノクローナル抗体を用いて実施し、同一の結果を得た。
【0225】
GBM細胞の選別(MoFlo(TM)高速セルソーター、ダコサイトメーション(DakoCytomation))は、同じ希釈率のCD133/1(CD133)−PE結合型抗体およびCD133/2(293C3)−PE結合型抗体を使用して実施した。シン(Singh)らの文献(Nature(2004)第432巻、p.396−401)に記述されているように、選別した細胞を、同じ抗体を使用してFACSCalibur(TM)装置(BDバイオサイエンシズ(BD Biosciences))を用いたフローサイトメトリーによって純度分析した。純度はCD133陽性および陰性のフラクションのいずれについても少なくとも87%であった。
【0226】
グリア線維性酸性タンパク質(GFAP)、βIIIチューブリンおよびガラクトセレブロシドC(GalC、「GC」と称されることもある)の定量化については、3.0−3.4umのレインボーキャリブレーション粒子混合物(8ピーク)(BDバイオサイエンシズ)をキャリブレーションに使用し、細胞の標識強度を、フィコエリスリン1分子相当の蛍光量(MEPE)またはフルオレセイン1分子相当の蛍光量(MEFL)として表
した。簡潔に述べると、細胞内染色については、細胞を、0.5mlのCytofix/Cytoperm(TM)溶液(BDバイオサイエンシズ)中で室温にて20分間透過処理した。細胞を2mlのBD Perm/Wash(TM)1×(BDバイオサイエンシズ)で洗浄し、室温で10分間インキュベートした。遠心分離した後、該細胞を、適切な一次抗体混合物を含んだ0.2mlのBD Perm/Wash(TM)1×(BDバイオサイエンシズ)中に再浮遊させた。膜抗原については、細胞を0.2mlの増殖培地中に再浮遊させ、次いで以下の一次抗体、すなわち1:400の抗GFAPポリクローナル抗体(ダコ・コーポレーション)、抗βIIIチューブリンモノクローナル抗体(バブコ)、および抗galCモノクローナル抗体(ケミコン)、とともに4℃で30分間インキュベートした。その後、細胞を洗浄し、4℃で30分間二次抗体に曝露した。細胞内の抗原の場合には、二次抗体は1:800のヤギ抗ウサギIg FITC標識抗体またはヤギ抗マウスIgG R−PE標識抗体(BDファーミンゲン)であり、膜抗原については、1:1000のFITC結合型F(ab’)2ヤギ抗マウスIgMまたはFITC結合型ヤギ抗マウスIgM(ジャクソン・イムノリサーチ(Jackson ImmunoResearch))を使用した。十分に洗浄した後、細胞を再浮遊させてフローサイトメトリーによって分析した。
【0227】
上記のすべての分析法について、分析は、CellQuest(TM)ソフトウェア(BDバイオサイエンシズ)を使用してフローサイトメトリー(FACSCalibur、BDバイオサイエンシズ)によって実施した。バックグラウンドの蛍光は、特異的一次抗体を特異的アイソタイプ対照で代用することにより概算した。自己蛍光の計測も、各試験条件について常に実施した。
【0228】
実施例29:同所注入および免疫組織化学による腫瘍形成性の評価
腫瘍形成性は、コントロール条件の下で、または100ng/mlのBMP−4をさらに追加した条件で48時間培養したGBM細胞の同所移植によって測定した。移植に先立って、細胞を洗浄しPBS中に再浮遊させた(108個/ml)。3マイクロリットルを、ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021にすでに記載されているようにして免疫不全マウスの右側線条体に定位的に注入した。ポリマーを用いたBMP−4の送達は、BMP−4を装荷したヘパリン・アクリル樹脂ビーズ(動物1匹あたり100個のビーズ;シグマ・アルドリッチ(Sigma−Aldrich))を使用して実施した。移植に先立って、PBS単独または0.65μg/μlのBMP4を含むPBS中でビーズを37℃にて1時間インキュベートし、そしてPBSで2×3回徹底的にすすいでから移植した。ヘマトキシリン−エオジン染色および免疫組織化学分析を、ガルリ(Galli)ら、Cancer Research(2004)第64巻、p.7011−7021にすでに記載されているようにして処理した、パラフィン包埋した4um厚のミクロトーム切片について実施した。
【0229】
期間中(X軸)の生存者の累積的な見込み(Y軸)の下方傾斜プロットは、ソフトウェアMedCalc(ベルギー国所在のマリアケルク(Mariakerke))を使用して実施した。生存の有意差はログランク検定によって測定した。
【図面の簡単な説明】
【0230】
【図1A】ヒト神経幹細胞について、各継代の終了時における理論上の総細胞数を継代数に対してプロットした図。
【図1B】各継代の終了時における総細胞数の対数を継代数に対してプロットし、その線形対数プロットに最も良くフィットする傾向線を示す図。
【図2A】ヒト神経幹細胞にLIFを添加したものとしていないものについて、各継代の終了時における理論上の総細胞数を継代数に対してプロットした図。
【図2B】各継代の終了時における総細胞数の対数を継代数に対してプロットし、その線形対数プロットに最も良くフィットする傾向線を示す図。
【図2C】LIFの存在下における幹細胞および前駆細胞の存在頻度の低下を示すグラフ。
【図3A】腫瘍細胞ニューロスフェアの20×での位相差観察像を示す図。
【図3B】2つの異なるGBM細胞株について各分裂時の理論上の総細胞数を示す図。
【図4A】連続的に継代したGBM腫瘍細胞について、各継代の終了時における理論上の総細胞数を継代数に対してプロットした図。
【図4B】各継代の終了時における総細胞数の対数を継代数に対してプロットし、その線形対数プロットに最も良くフィットする傾向線を示す図。
【図4C】腫瘍性幹細胞および腫瘍前駆細胞の存在頻度を示すグラフ。
【図5】連続的に継代したGBM腫瘍細胞について、神経コロニー形成細胞法(NCFCA)で直径が次の範囲の大きさ:1,500−2,000μm;1,000−1,500μm;500−1,000μm;および<500μmのコロニーを形成する細胞の、播種した細胞全体に対する割合(%)(y軸)を示す図(主グラフ)。主グラフに挿入されたグラフは、直径が1,500−2,000μmおよび1,000−1,500μmの区分にあるコロニーを形成する細胞の、播種した細胞全体に対する割合(%)(y軸)を、主グラフとは異なるy軸目盛りで示している。
【図6】ヒト初代腫瘍標本およびヒト腫瘍神経幹細胞株におけるBMPR1a、BMPR1bおよびBMPR2についてのリアルタイムPCRの結果を示す図。
【図7】LIFまたはBMP−4で処理したGBM細胞株中の幹細胞および前駆細胞の割合(%)を示す図。
【図8】LIF、BMP−4、またはLIF+BMP−4で処理したGBM細胞株中の幹細胞および前駆細胞の割合(%)を示す図。
【図9】連続継代中持続的にBMP−2で処理したGBM細胞株、または1回の継代について一時的にBMP−2で処理したGBM細胞株(「BMP後」)における幹細胞および前駆細胞の割合(%)を示す図。
【図10】ヒトGBM由来の腫瘍性神経幹細胞の移植によって免疫不全マウスに引き起こされたGBMを示す図(上側パネル)。ならびにヒト腫瘍性神経幹細胞をBMP−4またはLIFで処理してから移植した場合はGBM形成が低減されることを示す図(下側パネル)。
【図11】11A:急性単離したGBM細胞および培養したGBM細胞からの細胞について、BMPR1A、BMPR1B、BMPR2およびBMP−4の転写物レベルを示す図。11B−D:単離直後のGBM細胞におけるBMPR1A、BMPR1BおよびBMPR2の免疫反応性を示す図。11E−G:培養したGBM細胞におけるBMPR1A、BMPR1BおよびBMPR2の免疫反応性を示す図。11H−I:GBM細胞におけるリン酸化Smad1,5,8の免疫反応性を示す図。11K−P:GBM細胞におけるBMP−4、Smad1、リン酸化Smad1,5,8およびSmad4のウエスタンブロット解析を示す図。
【図12A】BMP−4の存在下および非存在下におけるGBM培養物中の細胞死、アポトーシス、Ki67免疫反応性およびCD133免疫反応性の測定結果を示す図。
【図12B】BMP−4の存在下および非存在下におけるGBM細胞のクローン形成の指標を示す図。
【図12C】ニューロスフェア分析法におけるBMP−4の存在下および非存在下でのGBM細胞の増殖を示す図。
【図13A】BMP−4非存在下におけるGBM細胞を示す図。
【図13B】BMP−4とともに培養したGBM細胞を示す図。
【図13C】GFAPの免疫反応性(IR)を示す図(対照GBM細胞)。
【図13D】GFAPの免疫反応性(IR)を示す図(BMP−4処理したGBM細胞)。
【図13E】βIIIチューブリンのIRを示す図(対照GBM細胞)。
【図13F】βIIIチューブリンのIRを示す図(BMP−4処理したGBM細胞)。
【図13G】GalCのIRを示す図(対照GBM細胞)。
【図13H】GalCのIRを示す図(BMP−4処理したGBM細胞)。
【図13I】対照GBM細胞およびBMP−4処理したGBM細胞の、細胞蛍光測光法によるGFAPのIRについての解析を示す図。
【図13J】対照GBM細胞およびBMP−4処理したGBM細胞の、βIIIチューブリンのIRについての解析を示す図。
【図13K】対照GBM細胞およびBMP−4処理したGBM細胞の、GalCのIRについての解析を示す図。
【図14A】未処理のGBM細胞を注入したマウスにおける腫瘤を示す図。
【図14B】BMP−4処理したGBM細胞を注入したマウスには比較できるほどの腫瘤が存在しないことを示す図。
【図14C】BMP−4を欠いた対照ビーズで同時処理したマウスにおける腫瘍を示す図。
【図14D】BMP−4ビーズで同時処理したマウスには比較できるほどの腫瘍が存在しないことを示す図。
【図14E】対照ビーズで後処理したマウスにおける腫瘍を示す図。
【図14F】BMP−4ビーズで後処理したマウスには比較できるほどの腫瘍が存在しないことを示す図。
【図14G】マウスにおける未処理のGBM腫瘍の細胞形態を示す図。
【図14H】マウスにおけるBMP−4処理したGBM腫瘍の細胞形態を示す図。
【図14J】対照ビーズまたはBMP−4ビーズのいずれかを用いて、GBM注入の前(左側パネル)、同時(中央パネル)および後(右側パネル)に処理したGBM注入マウスについて生存グラフを示す図。
【図15】GBM細胞の増殖に対する様々なBMPの影響を示す図。
【特許請求の範囲】
【請求項1】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療するための医薬の製造におけるBMP調製物の使用方法。
【請求項2】
前記BMP調製物は、BMP−2調製物、BMP−4調製物、BMP−5調製物、BMP−6調製物、BMP−7調製物およびBMP−8b調製物からなる群から選択される、請求項1に記載の使用方法。
【請求項3】
前記BMP調製物はBMP−4調製物である、請求項1に記載の使用方法。
【請求項4】
前記BMP調製物は、完全長のヒトBMP−4または完全長ヒトBMP−4のフラグメントである、請求項1に記載の使用方法。
【請求項5】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療するための医薬の製造におけるLIF調製物の使用方法。
【請求項6】
前記LIF調製物は、完全長のヒトLIFまたは完全長ヒトLIFのフラグメントである、請求項5に記載の使用方法。
【請求項7】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病は腫瘍である、請求項1〜6のいずれか1項に記載の使用方法。
【請求項8】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病は脳腫瘍である、請求項1〜6のいずれか1項に記載の使用方法。
【請求項9】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病は多形膠芽腫である、請求項1〜6のいずれか1項に記載の使用方法。
【請求項10】
対象における、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療する方法であって、前記対象に、骨形成タンパク質(BMP)調製物、白血病抑制因子(LIF)調製物、白血病抑制因子レセプタ(LIFR)シグナル伝達活性化剤、および骨形成タンパク質レセプタ(BMPR)シグナル伝達活性化剤からなる群から選択される少なくとも1つの作用薬を含有する医薬組成物を投与することを含む方法。
【請求項11】
過度な細胞増殖を特徴とする疾病は腫瘍である、請求項10に記載の方法。
【請求項12】
過度な細胞増殖を特徴とする疾病は脳腫瘍である、請求項10に記載の方法。
【請求項13】
過度な細胞増殖を特徴とする疾病は多形膠芽腫である、請求項10に記載の方法。
【請求項14】
少なくとも1つの作用薬はBMP調製物である、請求項10に記載の方法。
【請求項15】
前記BMP調製物は、BMP−2調製物、BMP−4調製物、BMP−5調製物、BMP−6調製物、BMP−7調製物およびBMP−8b調製物からなる群から選択される、請求項14に記載の方法。
【請求項16】
前記BMP調製物はBMP−2調製物を含む、請求項15に記載の方法。
【請求項17】
前記BMP−2調製物は、完全長のヒトBMP−2または完全長ヒトBMP−2のフラ
グメントを含む、請求項16に記載の方法。
【請求項18】
前記BMP調製物はBMP−4調製物を含む、請求項14に記載の方法。
【請求項19】
前記BMP−4調製物は、完全長のヒトBMP−4または完全長ヒトBMP−4のフラグメントを含む、請求項18に記載の方法。
【請求項20】
腫瘍性幹細胞または腫瘍前駆細胞におけるBMPレセプタ(BMPR)を介したシグナル伝達を増大させる方法であって、腫瘍性幹細胞または腫瘍前駆細胞にBMP調製物を投与することを含む方法。
【請求項21】
腫瘍性幹細胞または腫瘍前駆細胞におけるBMPRを介したシグナル伝達の増大は、細胞の生存、自己複製、対称分裂、増殖、および分化についての特性からなる群から選択される腫瘍性幹細胞または腫瘍前駆細胞の特徴の少なくとも1つを調節する、請求項20に記載の方法。
【請求項22】
腫瘍性幹細胞または腫瘍前駆細胞におけるLIFレセプタ(LIFR)を介したシグナル伝達を増大させる方法であって、腫瘍性幹細胞または腫瘍前駆細胞にLIF調製物を投与することを含む方法。
【請求項23】
腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRを介したシグナル伝達の増大は、細胞の生存、自己複製、対称分裂、増殖、および分化についての特性からなる群から選択される腫瘍性幹細胞または腫瘍前駆細胞の特徴の少なくとも1つを調節する、請求項22に記載の方法。
【請求項24】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をBMP調製物と接触させることを含む方法。
【請求項25】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をBMPRシグナル伝達活性化剤と接触させることを含む方法。
【請求項26】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をLIF調製物と接触させることを含む方法。
【請求項27】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をLIFRシグナル伝達活性化剤と接触させることを含む方法。
【請求項28】
腫瘍の成長を低減する方法であって、前記腫瘍においてBMPRを介したシグナル伝達を活性化することを含む方法。
【請求項29】
腫瘍の成長を低減する方法であって、前記腫瘍においてLIFRを介したシグナル伝達を活性化することを含む方法。
【請求項30】
BMPレセプタ(BMPR)シグナル伝達活性化剤を含有する医薬組成物。
【請求項31】
LIFレセプタ(LIFR)シグナル伝達活性化剤を含有する医薬組成物。
【請求項32】
BMPレセプタ(BMPR)シグナル伝達活性化剤。
【請求項33】
LIFレセプタ(LIFR)シグナル伝達活性化剤。
【請求項34】
少なくとも1つのポリマー製ウエハまたは少なくとも1つのポリマー製ビーズと結合したBMP調製物を含有する医薬組成物。
【請求項35】
前記BMP調製物はBMP−4調製物である、請求項34に記載の医薬組成物。
【請求項36】
LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤を含有する医薬組成物。
【請求項37】
腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤からなる群から選択される少なくとも1つの作用薬と接触させることを含む方法。
【請求項38】
BMPRシグナル伝達活性化剤を候補分子混合物から同定する方法であって、
(i) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のサンプルを単離する工程;
(ii) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを適切な容器に入れる工程;
(iii) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを、特定の期間、特定の条件の下で前記候補分子に曝露する工程;ならびに
(iv) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方の変化をスクリーニングする工程であって、前記変化は形態学的変化、生理学的変化、および遺伝学的変化からなる群から選択されることを特徴とする工程
からなる方法。
【請求項39】
前記変化は生理学的変化である、請求項38に記載の方法。
【請求項40】
生理学的変化は、腫瘍性幹細胞または腫瘍前駆細胞による対称分裂の頻度の低下である、請求項39に記載の方法。
【請求項41】
ヒト患者における多形膠芽腫を治療する方法であって、前記多形膠芽腫を少なくとも部分的に切除して切除腔を形成することと、該切除腔内にBMP−4調製物を放出することとを含む方法。
【請求項42】
前記BMP−4調製物は、少なくとも1つのポリマー製ウエハまたは少なくとも1つのポリマー製ビーズにより放出される、請求項41に記載の方法。
【請求項43】
腫瘍性幹細胞を含む腫瘍を治療する方法であって、腫瘍性幹細胞を、該腫瘍性幹細胞の分化を誘導する作用薬と接触させることを含む方法。
【請求項44】
前記腫瘍は多形膠芽腫であり、前記作用薬はBMP−4調製物である、請求項43に記載の方法。
【請求項1】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療するための医薬の製造におけるBMP調製物の使用方法。
【請求項2】
前記BMP調製物は、BMP−2調製物、BMP−4調製物、BMP−5調製物、BMP−6調製物、BMP−7調製物およびBMP−8b調製物からなる群から選択される、請求項1に記載の使用方法。
【請求項3】
前記BMP調製物はBMP−4調製物である、請求項1に記載の使用方法。
【請求項4】
前記BMP調製物は、完全長のヒトBMP−4または完全長ヒトBMP−4のフラグメントである、請求項1に記載の使用方法。
【請求項5】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療するための医薬の製造におけるLIF調製物の使用方法。
【請求項6】
前記LIF調製物は、完全長のヒトLIFまたは完全長ヒトLIFのフラグメントである、請求項5に記載の使用方法。
【請求項7】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病は腫瘍である、請求項1〜6のいずれか1項に記載の使用方法。
【請求項8】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病は脳腫瘍である、請求項1〜6のいずれか1項に記載の使用方法。
【請求項9】
過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病は多形膠芽腫である、請求項1〜6のいずれか1項に記載の使用方法。
【請求項10】
対象における、過度な細胞増殖または誤制御された細胞増殖を特徴とする疾病を治療する方法であって、前記対象に、骨形成タンパク質(BMP)調製物、白血病抑制因子(LIF)調製物、白血病抑制因子レセプタ(LIFR)シグナル伝達活性化剤、および骨形成タンパク質レセプタ(BMPR)シグナル伝達活性化剤からなる群から選択される少なくとも1つの作用薬を含有する医薬組成物を投与することを含む方法。
【請求項11】
過度な細胞増殖を特徴とする疾病は腫瘍である、請求項10に記載の方法。
【請求項12】
過度な細胞増殖を特徴とする疾病は脳腫瘍である、請求項10に記載の方法。
【請求項13】
過度な細胞増殖を特徴とする疾病は多形膠芽腫である、請求項10に記載の方法。
【請求項14】
少なくとも1つの作用薬はBMP調製物である、請求項10に記載の方法。
【請求項15】
前記BMP調製物は、BMP−2調製物、BMP−4調製物、BMP−5調製物、BMP−6調製物、BMP−7調製物およびBMP−8b調製物からなる群から選択される、請求項14に記載の方法。
【請求項16】
前記BMP調製物はBMP−2調製物を含む、請求項15に記載の方法。
【請求項17】
前記BMP−2調製物は、完全長のヒトBMP−2または完全長ヒトBMP−2のフラ
グメントを含む、請求項16に記載の方法。
【請求項18】
前記BMP調製物はBMP−4調製物を含む、請求項14に記載の方法。
【請求項19】
前記BMP−4調製物は、完全長のヒトBMP−4または完全長ヒトBMP−4のフラグメントを含む、請求項18に記載の方法。
【請求項20】
腫瘍性幹細胞または腫瘍前駆細胞におけるBMPレセプタ(BMPR)を介したシグナル伝達を増大させる方法であって、腫瘍性幹細胞または腫瘍前駆細胞にBMP調製物を投与することを含む方法。
【請求項21】
腫瘍性幹細胞または腫瘍前駆細胞におけるBMPRを介したシグナル伝達の増大は、細胞の生存、自己複製、対称分裂、増殖、および分化についての特性からなる群から選択される腫瘍性幹細胞または腫瘍前駆細胞の特徴の少なくとも1つを調節する、請求項20に記載の方法。
【請求項22】
腫瘍性幹細胞または腫瘍前駆細胞におけるLIFレセプタ(LIFR)を介したシグナル伝達を増大させる方法であって、腫瘍性幹細胞または腫瘍前駆細胞にLIF調製物を投与することを含む方法。
【請求項23】
腫瘍性幹細胞または腫瘍前駆細胞におけるLIFRを介したシグナル伝達の増大は、細胞の生存、自己複製、対称分裂、増殖、および分化についての特性からなる群から選択される腫瘍性幹細胞または腫瘍前駆細胞の特徴の少なくとも1つを調節する、請求項22に記載の方法。
【請求項24】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をBMP調製物と接触させることを含む方法。
【請求項25】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をBMPRシグナル伝達活性化剤と接触させることを含む方法。
【請求項26】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をLIF調製物と接触させることを含む方法。
【請求項27】
腫瘍中の腫瘍性幹細胞および腫瘍前駆細胞のうち少なくともいずれか一方の数を減少させる方法であって、腫瘍をLIFRシグナル伝達活性化剤と接触させることを含む方法。
【請求項28】
腫瘍の成長を低減する方法であって、前記腫瘍においてBMPRを介したシグナル伝達を活性化することを含む方法。
【請求項29】
腫瘍の成長を低減する方法であって、前記腫瘍においてLIFRを介したシグナル伝達を活性化することを含む方法。
【請求項30】
BMPレセプタ(BMPR)シグナル伝達活性化剤を含有する医薬組成物。
【請求項31】
LIFレセプタ(LIFR)シグナル伝達活性化剤を含有する医薬組成物。
【請求項32】
BMPレセプタ(BMPR)シグナル伝達活性化剤。
【請求項33】
LIFレセプタ(LIFR)シグナル伝達活性化剤。
【請求項34】
少なくとも1つのポリマー製ウエハまたは少なくとも1つのポリマー製ビーズと結合したBMP調製物を含有する医薬組成物。
【請求項35】
前記BMP調製物はBMP−4調製物である、請求項34に記載の医薬組成物。
【請求項36】
LIFRシグナル伝達活性化剤およびBMPRシグナル伝達活性化剤を含有する医薬組成物。
【請求項37】
腫瘍性幹細胞または腫瘍前駆細胞が対称分裂を行う可能性を低減する方法であって、腫瘍性幹細胞または腫瘍前駆細胞を、LIF調製物、BMP調製物、BMPRシグナル伝達活性化剤、およびLIFRシグナル伝達活性化剤からなる群から選択される少なくとも1つの作用薬と接触させることを含む方法。
【請求項38】
BMPRシグナル伝達活性化剤を候補分子混合物から同定する方法であって、
(i) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のサンプルを単離する工程;
(ii) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを適切な容器に入れる工程;
(iii) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方のアリコートを、特定の期間、特定の条件の下で前記候補分子に曝露する工程;ならびに
(iv) 腫瘍性幹細胞または腫瘍前駆細胞のうち少なくともいずれか一方の変化をスクリーニングする工程であって、前記変化は形態学的変化、生理学的変化、および遺伝学的変化からなる群から選択されることを特徴とする工程
からなる方法。
【請求項39】
前記変化は生理学的変化である、請求項38に記載の方法。
【請求項40】
生理学的変化は、腫瘍性幹細胞または腫瘍前駆細胞による対称分裂の頻度の低下である、請求項39に記載の方法。
【請求項41】
ヒト患者における多形膠芽腫を治療する方法であって、前記多形膠芽腫を少なくとも部分的に切除して切除腔を形成することと、該切除腔内にBMP−4調製物を放出することとを含む方法。
【請求項42】
前記BMP−4調製物は、少なくとも1つのポリマー製ウエハまたは少なくとも1つのポリマー製ビーズにより放出される、請求項41に記載の方法。
【請求項43】
腫瘍性幹細胞を含む腫瘍を治療する方法であって、腫瘍性幹細胞を、該腫瘍性幹細胞の分化を誘導する作用薬と接触させることを含む方法。
【請求項44】
前記腫瘍は多形膠芽腫であり、前記作用薬はBMP−4調製物である、請求項43に記載の方法。
【図1A】

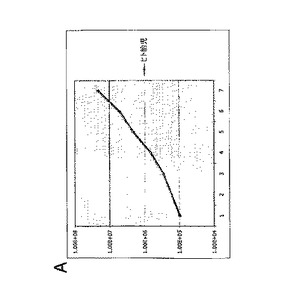
【図1B】
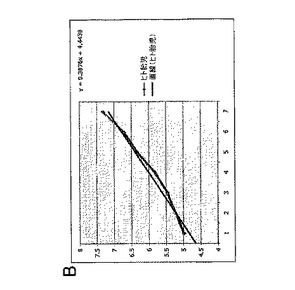
【図2A】
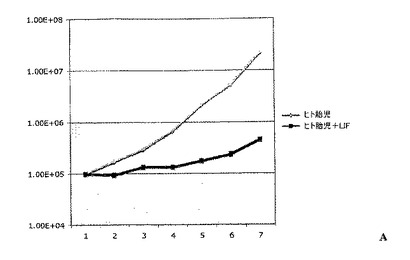
【図2B】
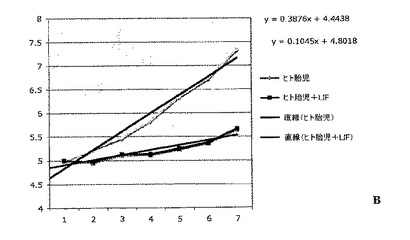
【図2C】
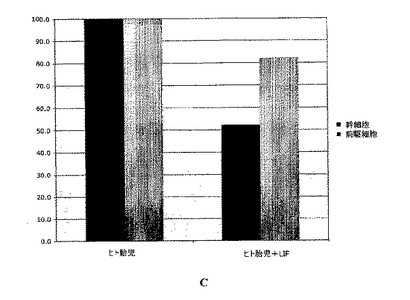

【図3】
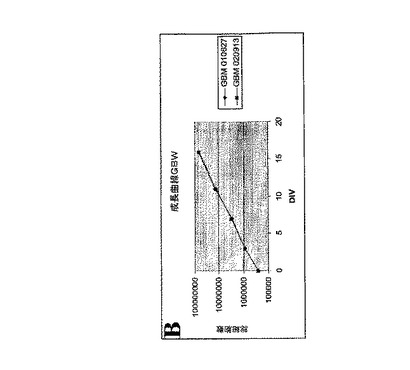
【図4A】
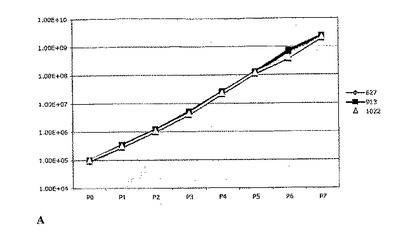
【図4B】
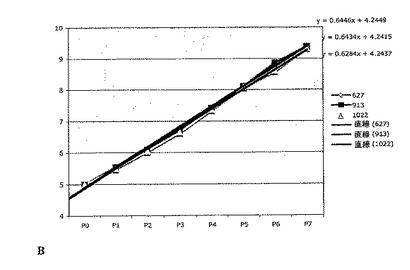
【図4C】
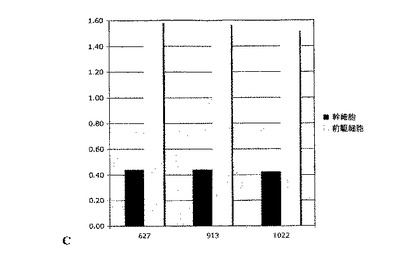
【図5】
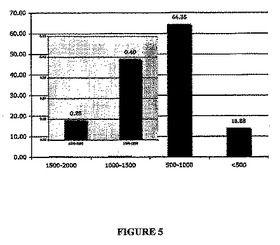
【図6】
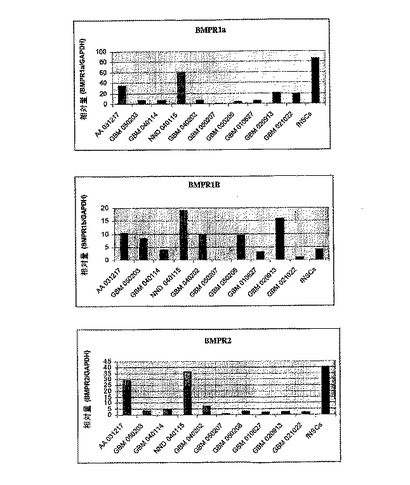
【図7】
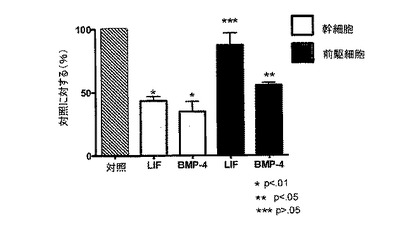
【図8】
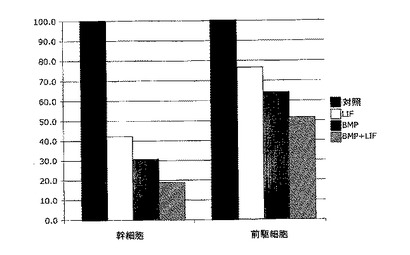
【図9】
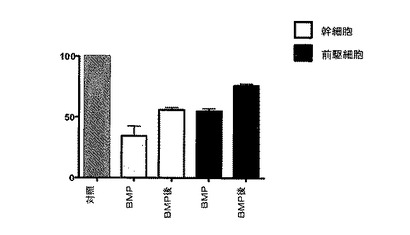
【図10】

【図11】
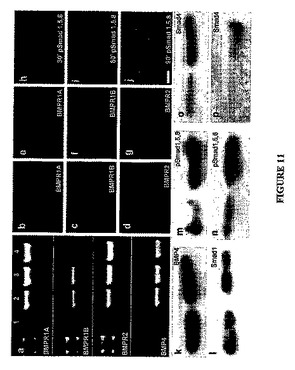
【図12A】
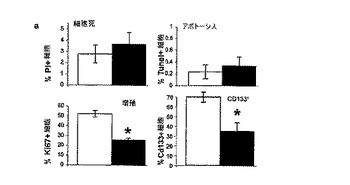
【図12B】
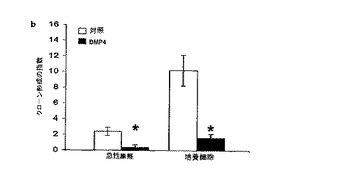
【図12C】
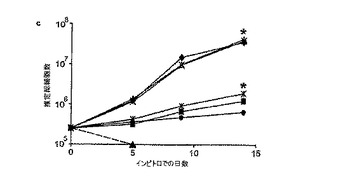
【図13】
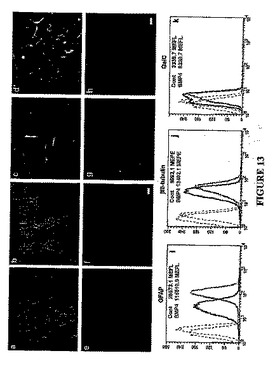
【図13I】
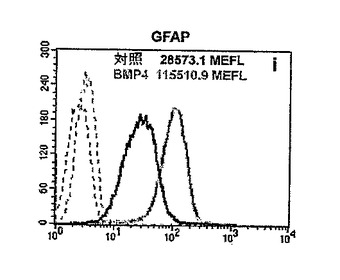
【図13J】
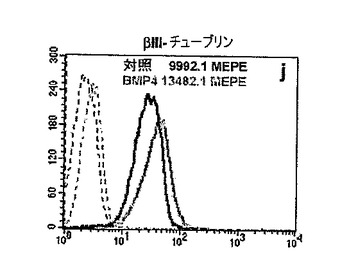
【図13K】
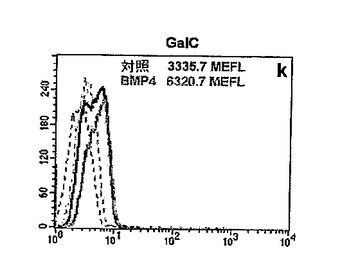
【図14】
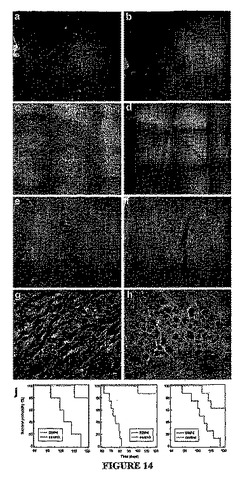
【図14J】

【図15】
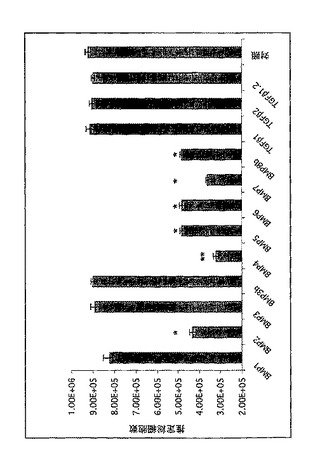
【図1B】
【図2A】
【図2B】
【図2C】
【図3】
【図4A】
【図4B】
【図4C】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12A】
【図12B】
【図12C】
【図13】
【図13I】
【図13J】
【図13K】
【図14】
【図14J】
【図15】
【公表番号】特表2009−502771(P2009−502771A)
【公表日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願番号】特願2008−522091(P2008−522091)
【出願日】平成18年7月19日(2006.7.19)
【国際出願番号】PCT/IB2006/002296
【国際公開番号】WO2007/010394
【国際公開日】平成19年1月25日(2007.1.25)
【出願人】(508018691)ステムジェン ソシエタ ペル アチオニ (2)
【氏名又は名称原語表記】STEMGEN S.P.A.
【Fターム(参考)】
【公表日】平成21年1月29日(2009.1.29)
【国際特許分類】
【出願日】平成18年7月19日(2006.7.19)
【国際出願番号】PCT/IB2006/002296
【国際公開番号】WO2007/010394
【国際公開日】平成19年1月25日(2007.1.25)
【出願人】(508018691)ステムジェン ソシエタ ペル アチオニ (2)
【氏名又は名称原語表記】STEMGEN S.P.A.
【Fターム(参考)】
[ Back to top ]