
LTP阻害の抑制方法
長期増強(LTP)のアミロイド媒介性阻害を抑制するための方法および組成物が提供される。
【発明の詳細な説明】
【技術分野】
【0001】
アミロイド形成タンパク質は複数の疾患状態の病理学に関与している。アミロイド形成タンパク質の異常沈着から生じる疾患は、限定されるものでないがアルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、プリオンにより全部若しくは部分的に引き起こされる疾患(クロイツフェルト・ヤコブ病、スクレイピーおよびウシ海綿状脳症のような)、ならびに遺伝性アミロイドーシスおよび全身性アミロイドーシス双方を包含するアミロイドーシスを挙げることができる。
【背景技術】
【0002】
アルツハイマー病(AD)は、米国単独で400万人を苦しめている老人性認知症をもたらす進行性神経変性疾患である(全般として、非特許文献1;特許文献1;非特許文献2;非特許文献3;非特許文献4を参照されたい)。大まかに言って、該疾患は2種類、すなわち高齢(65歳超)で発生する晩発性;および老年期の十分に前すなわち35と60歳の間で発症する早発性に分類される。双方の型の疾患で病状は同一であるが、しかしより若年齢で開始する症例で異常がより重篤かつ広まる傾向がある。該疾患は、脳中の少なくとも2型の病変、すなわち老人斑および神経原線維変化を特徴とする。神経原線維変化は、対で相互の周囲で捻られた2本のフィラメントよりなる微小管結合τタンパク質の細胞内沈着物である。老人斑は直径150ミクロンまでの混乱した神経線維網の領域であり(脳組織の切片の顕微鏡分析により見える)、そして中央に細胞外アミロイド沈着物を有する。こうした斑の主成分はAβペプチドである(非特許文献5を参照されたい)。斑中で見出される付加的なタンパク質は、非特許文献6により記述されるところのラミニン、アポE、アセチルコリンエステラーゼ、および非特許文献7により記述されるところのヘパリン硫酸プロテオグリカンを包含する。Aβペプチドは、アミロイド前駆体タンパク質(APP)と命名される前駆体タンパク質の39〜43アミノ酸の内部フラグメントである。APPタンパク質内の数種の突然変異がアルツハイマー病の存在と相互に関連付けられている(非特許文献8(バリン717からイソロイシン);非特許文献9(バリン717からグリシン);非特許文献10(バリン717からフェニルアラニン);非特許文献11(リシン595メチオニン596をアスパラギン595ロイシン596に変える二重突然変異))。こうした突然変異は、AβへのAPPの増大された若しくは変えられたプロセシング、とりわけ、増大された量の長い形態のAβ(すなわちA1−42およびA1−43)へのAPPのプロセシングによりアルツハイマー病を引き起こすと考えられている。プレセニリン遺伝子PS1およびPS2のような他の遺伝子中の突然変異は、APPのプロセシングに間接的に影響して増大された量の長い形態のAβを生成すると考えられている(非特許文献12)。これらの観察結果は、Aβおよびとりわけその長い形態がアルツハイマー病における原因要素であることを示す(非特許文献13)。
【0003】
研究者は、シナプス不全がADの発症の基礎にあると仮定している。シナプス減少がADの早期事象でありかつ認知障害と構造的相関関係にあるためである。研究者は、ADにおける臨床的認知症の潜行性発症に先立つ軽度認知障害が、大スケールの神経変性に先立つシナプス不全から生じるとさらに仮定している。長期増強(LTP)は、学習および記憶の細胞モデルであると広く仮定されているシナプスの可塑性の一形態である。
【0004】
LTPはシナプス伝達の効率の持続的な使用依存性の増大である。大部分の研究でLTPは高頻度シナプス刺激(HFS)の送達により実験的に誘導する。しかしながら他の馴化プロトコルが存在し、それらのいくつかは性質が薬理学的でありかつシナプス刺激を伴わない。さらに、複数の形態のLTPが同定されている。げっ歯類脳薄片での研究は、とりわけ海馬のCA3−CA1シナプスでLTPの多くの局面を解明した。LTPの4つの
特徴は、共同性、連合性、持続性および入力特異性である。
【0005】
シナプス効率の増大を開始する早期事象を構成するLTP誘導の過程は、LTPのその後の持続的発現と機構的に異なる。LTP誘導の間、領域CA3から領域CA1に突出する線維へのHFSの送達がシナプス中にグルタミン酸を放出し、そしてシナプス後ニューロンを脱分極する。高頻度の刺激(例えば100Hz)により、連続的興奮性シナプス後電位(EPSP)により誘発される脱分極が重なり、そして一連のHFSの間の累積的脱分極が実質的になり得る。樹状突起に後方伝播するシナプス後活動電位の発生が付加的な脱分極に寄与する。グルタミン酸放出および脱分極は原因として関係するとは言え、実験的に、それらを分離しかつLTPの誘導が双方の事象を必要とする(「共同性」と命名されている関係)ことを示すことが可能である。従って、シナプス後膜が馴化の間に直接過分極される場合、HFSはLTPを誘導することに失敗する。逆に、電流注入でシナプス後膜を直接脱分極することは、低頻度シナプス刺激でさえLTPを誘導することを可能にする。
【0006】
第21染色体の余分の1コピーを有するダウン症候群のほとんど全部の個体は、彼らが彼らの40歳代まで生き延びる場合に、アルツハイマー病で見られるものに類似の神経病理学的変化を示す。これは、第21染色体上のAPP遺伝子によりコードされるβアミロイドタンパク質の過剰産生に帰されている。
【0007】
数種のタンパク質がAβとの可能な相互作用について検討されている。これらは、高度な糖化最終生成物、RAGE(非特許文献14を参照されたい)、スカベンジャー受容体(非特許文献15;および非特許文献16)、小胞体関連アミロイドβ結合タンパク質(ERAB)(非特許文献17)、α4若しくはα7ニコチン性アセチルコリン受容体(非特許文献18および非特許文献19)、ならびに低親和性p75 NGF受容体(非特許文献20を参照されたい)を包含する。加えて、Aβは、非特許文献21および非特許文献22により、プレート上に被覆される場合にβ1インテグリンサブユニット依存性の様式で細胞の接着を媒介することが報告されている。
【0008】
Aβと相互作用しうる多様な機能の多様な分子の数を考慮すると、Aβが神経変性を媒介しうる機序は不明なままである。該過程で役割を有しうる他の細胞タンパク質の存在および性質もまた不明である。
【0009】
島アミロイドは、世紀の始まり以来II型糖尿病における病理学的実体として認識されている。それは、その独特の成分として、インスリンと共分泌される島β細胞ペプチド、島アミロイドポリペプチド(IAPP)すなわちアミリンを有する。この独特の成分に加え、島アミロイドは、アポリポタンパク質E、およびヘパリン硫酸プロテオグリカン、パールカンのような他のタンパク質を含有し、それらは典型的に他の形態の全身性および局在性アミロイドで観察される。島アミロイドは、II型糖尿病を伴う個体の大多数において病理学的検査で観察されるが、しかし糖代謝の混乱を伴わないヒトで稀に観察される。げっ歯類からのIAPPと対照的にヒトIAPPはin vitroでアミロイド原線維を形成することが示されている。全ヒト被験体がアミロイド形成性の形態のIAPPを産生かつ分泌し、それでもなお全部が島アミロイドを発生するわけではないため、いくつかの他の因子が島アミロイド形成に関与していることがありそうである。1仮説は、IAPPの産生、プロセシングおよび/若しくは分泌の変化をもたらすβ細胞機能の変化が、ヒト糖尿病における島アミロイド原線維の初期形成に関与することである。アミロイド原線維のこの形成がその後IAPP含有原線維の進行的蓄積を可能にする。β細胞塊のアミロイドによる最終的な置換が高血糖症の発症に寄与している。
【0010】
アミロイド形成の開始をもたらすβ細胞の変化を生じることに関与しうる1因子は、食
物脂肪の増大された消費である。食物脂肪は島β細胞ペプチドの産生、プロセシングおよび分泌を変えることが知られており、また、ヒトIAPPを発現するトランスジェニックマウスでの研究はこの機序の作動を裏付ける。これおよび他のモデルを使用するさらなる検討が、島アミロイド形成に関与する機序への洞察を提供しそしてアミロイド原線維形成を阻害若しくは復帰する治療薬の開発を可能にするはずであり、その目標はII型糖尿病でβ細胞の機能を保存しかつグルコース制御を改善することである。非特許文献23。
【0011】
伝染性海綿状脳症すなわちプリオン病は、後天的、遺伝性若しくは特発性(「散発性」)疾患として症状発現し得る伝染性の迅速に進行する必ず致死的な神経変性疾患の一群を構成する。それらは、ヒトにおけるクロイツフェルト・ヤコブ病ならびに動物におけるスクレイピーおよびウシ海綿状脳症(BSE)を包含し、そして、実験的若しくは偶発的伝播後に数十年まで持続しうる長い潜伏期を特徴とする。プリオン病の古典的病理学的特徴は海綿状変化、神経膠症およびニューロン減少を包含する。ウイルスによる引き起こされる感染性疾患で典型的に見られるものと対照的に、プリオン病は有意の炎症応答を欠く(非特許文献24、非特許文献25)。
【0012】
プリオン病は伝染性病原体「プリオン」の独特の特性によりかなりの科学的注目を集めている(非特許文献26)。該感染性病原体は非常に小さく、そして核酸を破壊しかつ慣習的ウイルスを不活性化する処置に対し極めて抵抗性である(非特許文献26)が、しかしタンパク質を変性させる処置に感受性である。該感染性病原体を精製する試みは、プリオンタンパク質(PrP)と命名された、これまで未知のタンパク質について高度に濃縮された画分を生じた(非特許文献27:非特許文献28:非特許文献29)。病原体特異的核酸はこれらの調製物中で見出されておらず(非特許文献30;非特許文献31);むしろ、プリオンタンパク質は宿主ゲノム中でコードされる(非特許文献29;非特許文献32;非特許文献33)。冒された個体の脳中には、PrPScと命名されたプリオンタンパク質の特異的な疾患関連アイソフォームの特徴的な蓄積が見出される(図1)。PrPScは、コンホメーション変化を伴う明確に定義されていない翻訳後過程によりプリオンタンパク質の正常な細胞アイソフォーム(PrPc)から派生する(非特許文献25)。PrPcおよびPrPScは同一アミノ酸配列を有する(非特許文献34)が、しかしながらそれらはコンホメーションが異なる。PrPScは、βシート構造のその高含量(非特許文献35)、大型凝集物を形成するその傾向、およびプロテイナーゼKでの消化に対するその部分的抵抗性によりPrPcと識別し得る。
【0013】
遺伝性アミロイドーシスは、細胞外マトリックス中の不溶性タンパク質原線維の沈着物を特徴とする常染色体優性の遺伝性疾患の臨床上および遺伝的に不均一な一群を含んでなる。これらの疾患は、硝子体混濁および腎不全によりときに付随される、多発神経炎、手根幹症候群、自律神経障害、心筋症の症状および胃腸の特徴を典型的に提示する。他の表現型は、ニューロパシー、胃潰瘍、脳神経障害および格子状角膜ジストロフィーを特徴とする。稀に軟髄膜若しくは脳の構造もまた臨床像に関与している。発症時年齢は17歳くらい早くかつ78歳くらい遅い。アミロイド原線維の基礎構成要素は、遺伝子で決定されるコンホメーション変化によりアミロイド形成性となる生理学的タンパク質である。以前はプレアルブミンと命名されていた変異トランスサイレチン(TTR)が遺伝性アミロイドーシスの最も頻繁な攻撃者(offender)である。同所性肝移植(OLT)は、アミロイド形成タンパク質の主産生部位を除去することにより、そうでなければ一般的に致死性である該疾患の進行を停止する。OLTの適応症およびその成功は、手術時点での心血管系および自律神経障害の進行度、齢、併存症ならびに突然変異の型に依存する。TTRの天然の四量体コンホメーションを安定化しかつ原線維形成を阻害する薬物を用いる代替処置モダリティーが現在集中的に研究されている。
【0014】
全身性アミロイドーシスは、実質器官;血管;皮下、粘膜下および腱周囲の脂肪;心;
眼;ならびに髄膜の原線維タンパク質凝集の細胞外沈着物を特徴とする。加えて、神経幹、神経叢、ならびに知覚および自律神経節を包含する末梢神経系のいずれの部分も関与しうる。抹消神経では、アミロイド沈着物は、通常は斑状および限局性の分布で神経上膜、神経周膜若しくは神経内膜に存在する。慣習的染色を用いる光学顕微鏡検査で、アミロイド沈着物は均一なエオシン好性の外見を有する。コンゴーレッド染色で、それらは偏光下に特徴的な黄緑色の複屈折を示す。
【0015】
多様なタンパク質がアミロイド形成の原因であり;事実、合計18種のアミロイド形成タンパク質がヒトアミロイドーシスで同定されている。非遺伝性の全身性アミロイドーシスは、免疫グロブリンL鎖(AL型、形質細胞悪液質における)、血清アミロイドAのフラグメント、急性期タンパク質(AA型、慢性炎症性疾患における)、トランスサイレチン(TTR;老人性全身性アミロイドーシスにおける)およびβ2−ミクログロブリン(尿毒症および透析を伴う患者における)により引き起こされ得る。遺伝性アミロイドーシスは、TTR、およびはるかにより稀にはアポリポタンパク質A1、リゾチーム、フィブリノーゲン、ゲルゾリン、アミロイドβおよびシスタチンCを包含する生理学的タンパク質の遺伝子バリアントによる。以前はプレアルブミンと呼ばれたTTRは、血清チロキシンおよび網膜結合タンパク質の輸送に関与する正常の四量体血清タンパク質である。それは第18染色体上の単一遺伝子によりコードされ、それの51個の異なる部位に存在する70以上の常染色体優性遺伝性点突然変異が記述されている。これらのなかで、位置30のメチオニン(Met30)によるバリンの置換はこれまで最も頻繁でありかつ地理的に最も広く散在している。
【0016】
パーキンソン病は、振戦、筋固縮、ならびに平衡および協調障害により特徴付けられる進行性神経障害である。化学物質ドーパミンを産生する脳細胞の破壊がこれらの症状の基礎にある。これらの病的な細胞はレビー小体と呼ばれるタンパク質沈着物によってもまた特徴付けられる。細胞がなぜ死ぬか、若しくはレビー小体がそれらを死滅させるのを助けるかどうかは誰も知らない。パーキンおよびα−シヌクレインと呼ばれる2種のタンパク質の遺伝子中の突然変異が、稀な形態の遺伝性パーキンソン病を分離するために結び付けられている。しかし、パーキンおよびα−シヌクレインの双方が、全パーキンソン病患者の脳中に蓄積するレビー小体中で見出される。
【0017】
最近の知見は、パーキンが、α−シヌクレインおよびシンフィリンを包含する脳中のレビー小体と関連するタンパク質の調節において重要な役割を演じていることを示唆している。正常には、パーキンは破壊のため他のタンパク質を「標識する」のにユビキチンと呼ばれるなお別のタンパク質を使用する。しかし、これらのタンパク質のあいだの関係で何かがうまくいかない場合、これはパーキンソン病で見られる細胞死の基礎作りをし得る。パーキンおよびα−シヌクレインの双方が、レビー小体関連タンパク質のユビキチン化を伴う共通の発病機序でシンフィリン−1と結合される。非特許文献36。従って、パーキンとのその相互作用を考えれば、α−シヌクレインでの問題は、遺伝性および一般的形態の双方のパーキンソン病の中心にありうる。非特許文献36。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】Hardyら、第WO 92/13069号明細書
【非特許文献】
【0019】
【非特許文献1】Sloe、TINS、16:403−409(1993)
【非特許文献2】Sloe、J.Neuropathol.Exp.Neurol.、53:438−447(1994)
【非特許文献3】Duffら、Nature、373:476−477(1995)
【非特許文献4】Gamesら、Nature、373:523(1995)
【非特許文献5】Forsyth Phys.Ther.、78:1325−1331(1998)
【非特許文献6】Murtomakiら、J.Neurosci.Res.、32:261−273(1992)
【非特許文献7】Yanら、Biochim.Biophys.Acta、1502:145−57(2000)
【非特許文献8】Goateら、Nature、349:704−06(1991)
【非特許文献9】Harlinら、Nature、353:844−46(1991)
【非特許文献10】Murrellら、Science、254:97−99(1991)
【非特許文献11】Mullanら、Nature Genet.、1:345−47(1992)
【非特許文献12】Hardy、TINS、20:154(1997)
【非特許文献13】Velez−Pardoら、Gen.Pharm.、31(5):675−81(1998)
【非特許文献14】Yanら、Nature、382:685−91(1996)
【非特許文献15】Khouryら、Nature、382:716−719(1996)
【非特許文献16】Paresceら、Neuron 17:553−65(1996)
【非特許文献17】Yanら、Nature、389:689−695(1997)
【非特許文献18】Wangら、J.Neurochem.、75:1155−1161(2000)
【非特許文献19】Wangら、J.Biol.Chem.、275:5626−5632(2000)
【非特許文献20】Yaarら、J.Clin.Invest.、100:2333−2340(1997)
【非特許文献21】Ghisoら、Biochem.J.、288:1053−59(1992)
【非特許文献22】Matterら、J.Cell Bio.、141:1019−1030(1998)
【非特許文献23】Diabetes、48:241−253(1999)
【非特許文献24】Prusiner、Arch.Neurol.、50:1129−1153(1953)
【非特許文献25】Prusiner、Proc.Natl.Acad.Sci.U.S.A.、95:13363−13383(1998)
【非特許文献26】Prusiner、Science、216:136−144(1982)
【非特許文献27】Boltonら、Science、218:1309−1311(1982)
【非特許文献28】Prusinerら、Cell、38:127−134(1983)
【非特許文献29】Oeschら、Cell、40:735−746(1985)
【非特許文献30】Kellingsら、J.Gen.Virol.、73:1025−1029(1992)
【非特許文献31】Riesnerら、Dev.Biol.Stand.、80:173−181(1993)
【非特許文献32】Chesebroら、Nature、315:331−333(1985)
【非特許文献33】Baslerら、Cell、46:417−428(1986)
【非特許文献34】Stahlら、Biochemistry、32:1991−2002(1993)
【非特許文献35】Panら、Proc.Natl.Acad.Sci.U.S.A.、90:10962−10966(1993)
【非特許文献36】Dawsonら、Nature Medicine、7:1144−1150(2001)
【発明の概要】
【0020】
[発明の要約]
1種若しくはそれ以上の剤が長期増強(LTP)のアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する該1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、LTPのアミロイド媒介性阻害の抑制方法が提供される。該方法の一態様において、インテグリンサブユニットαvに結合する最低2剤の有効投薬量が投与される。該方法の一態様において、該剤は、Aβ産生の阻害剤、Aβ沈着の阻害剤、Aβ消失のメディエーター、アミロイド斑消失のメディエーター、Aβ神経毒性の阻害剤、Aβ凝集の阻害剤およびAβ解離のメディエーターよりなる群から選ばれる第二の剤とともに投与される。一態様において、Aβ産生の阻害剤はγ分泌酵素阻害剤である。一態様において、Aβ産生の阻害剤はβ分泌酵素阻害剤である。該方法の一態様において、該剤はAβに対する抗体とともに投与される。該方法の一態様において、該剤はRGD(Arg−Gly−Asp)モチーフを含んでなるペプチドである。該方法の一態様において、該剤はαvβ1インテグリンのリガンドである。該方法の一態様において、該剤はフィブロネクチン若しくはスーパーフィブロネクチンである。該方法の一態様において、該剤はαvインテグリンサブユニット発現細胞のビトロネクチン若しくはフィブロネクチンへの接着を阻害する。該方法の一態様において、該剤はαvインテグリンサブユニット発現細胞のオステオポンチンへの接着を阻害する。該方法の一態様において、該剤はモノクローナル若しくはポリクローナル抗体である。該方法の一態様において、該剤は、18C7、20A9および17E6から選択される抗体と同一のエピトープを認識する抗体である。一態様において、該抗体はヒト化抗体、キメラ抗体およびナノボディ(nanobody)から選択される。該方法の一態様において、該剤は18C7、20A9および17E6から選択される抗体である。該方法の一態様において、該剤は、インテグリンサブユニットαvへの結合について18C7、20A9および17E6から選ばれる抗体と競合する。
【0021】
該方法のさらなる一態様において、該剤は、それらの立体異性体、若しくはそれらの立体異性体の混合物、またはそれらの製薬学的に許容できる塩の形態を包含する、式IaおよびIb
【0022】
【化1】

【0023】
の化合物から選択される化合物であり、ここで
X1およびX3は窒素若しくは炭素から独立に選択され;
R1は
【0024】
【化2】
【0025】
から選択され;
ここで、上の複素環は、NH2、ハロゲン、NO2、CN、CF3、C1−C4アルコキシ、C1−C6アルキルおよびC3−C7シクロアルキルよりなる群から選択される0〜2置換基で場合によっては置換されており;
Uは−(CH2)n−、−(CH2)tQ(CH2)m−および−C(=O)(CH2)
n−1−から選択され、ここでメチレン基の1個は場合によってはR7で置換されており;
Qは、1,2−フェニレン、1,3−フェニレン、2,3−ピリジニレン、3,4−ピリジニレンおよび2,4−ピリジニレンから選択され;
R6はH、C1−C4アルキルおよびベンジルから選択され;
R7はC1−C6アルキル、C3−C7シクロアルキル、C4−C11シクロアルキルアルキル、アリール、アリール(C1−C6アルキル)、ヘテロアリールおよびヘテロアリール(C1−C6アルキル)から選択され;
R10は、H、ハロゲン、CO2R17、CONR17R20、0〜1個のR15若しくは0〜1個のR21で置換されているC1−C6アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR15若しくは0〜1個のR21で置換されているC3−C7シクロアルキル、0〜1個のR15若しくは0〜1個のR21で置換されているC4−C11シクロアルキルアルキル、および0〜1個のR15若しくは0〜2個のR11若しくは0〜1個のR21で置換されているアリール(C1−C6アルキル)−から選択され;
R11は、H、ハロゲン、CF3、CN、NO2、ヒドロキシ、NR2R3、0〜1個のR21で置換されているC1−C4アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR21で置換されているアリール、0〜1個のR21で置換されているアリール(C1−C6アルキル)−、0〜1個のR21で置換されている(C1−C4アルコキシ)カルボニル、0〜1個のR21で置換されている(C1−C4アルキル)カルボニル、0〜1個のR21で置換されているC1−C4アルキルスルホニル、および0〜1個のR21で置換されているC1−C4アルキルアミノスルホニルから選択され;
Wは−C(=O)−N(R13)−であり;
Xは−CH(R14)−CH(R15)−であり;
R13はHおよびCH3から選択され;
R14は、H、C1−C10アルキル、アリールおよびヘテロアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ハロ、シアノ、アミノ、CF3およびNO2から選択される0〜3置換基で場合によっては置換されており;
R15はHおよびR16から選択され;
Yは−COR19であり;
R16は
−NH(R20)−C(=O)−R17、
−N(R20)−C(=O)−R17、
−N(R20)−C(=O)−NH−R17、
―N(R20)SO2−R17、および
−N(R20)SO2−N(R20)R17
から選択され、
R17は、C1−C10アルキル、C3−C11シクロアルキル、アリール(C1−C6アルキル)−、(C1−C6アルキル)アリール、ヘテロアリール(C1−C6アルキル)−、(C1−C6アルキル)ヘテロアリール、ビアリール(C1−C6アルキル)−、ヘテロアリール若しくはアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ヘテロアリール、ハロ、シアノ、アミノ、CF3およびNO2よりなる群から選択される0〜3置換基で場合によっては置換されており;
R19は−O−(CH2)kN+(R22)(R23)(R24)Z−であり;
Z−は、ハロゲン化イオン、重硫酸イオン、硫酸イオン、リン酸水素イオン、リン酸イオン、トルエンスルホン酸イオン、メタンスルホン酸イオン、エタンスルホン酸イオン、酢酸イオン、トリフルオロ酢酸イオン、クエン酸イオン、シュウ酸イオン、コハク酸イオン
およびマロン酸イオンから選択される製薬学的に許容できる陰イオンであり;
R22、R23およびR24はH、C1−C4アルキルおよびC4−C11シクロアルキルアルキルから独立に選択されるか;
あるいは、R22およびR23は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する5〜7員の複素環系を形成し得、ならびにR24は上のとおり定義されるか、または、R22、R23およびR24は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する複素二環系を形成し得;
R20はHおよびCH3から選択され;
R21はCOOHおよびNR62から選択され;
kは2であり;
mは0および1から選択され;
nは1〜4であり;ならびに
tは0および1から選択される。
【0026】
該方法の一態様において、該剤は式II:
【0027】
【化3】
【0028】
の化合物であり、
ここでR19は−H、−CH3および−CH2CH2N+(CH3)3から選ばれる。一態様において、R19は−Hである。別の態様において、R19は−CH3である。別の態様において、R19は−CH2CH2N+(CH3)3である。
【0029】
該方法の別の態様において、該剤はディスインテグリンである。該方法の別の態様において、該剤はエキスタチンである。該方法の別の態様において、該剤はヒト抗体である。該方法の別の態様において、該剤はヒト化抗体である。該方法の別の態様において、該剤はキメラ抗体である。該方法の別の態様において、該剤はナノボディである。該方法の別の態様において、該剤は抗体フラグメントである。該方法の別の態様において、該剤は、抗体の1個若しくはそれ以上のH鎖、L鎖、F(ab)、F(ab)2、F(ab)c若しくはF(v)、またはそれらのいずれかの組合せを含んでなる。該方法の別の態様において、該剤は抗体でありかつ該抗体のアイソタイプはIgG1若しくはIgG4である。該方法の別の態様において、該剤は抗体でありかつ該抗体のアイソタイプはIgG2若しくはIgG3である。該方法の別の態様において、該剤は1抗体鎖である。該方法の別の態様において、該剤は抗体でありかつ該抗体は2対のLおよびH鎖を含んでなる。
【0030】
該方法の別の態様において、該剤は患者に投与される。一態様において、該剤は抗体でありかつ該抗体の投薬量は患者体重1kgあたり約0.01から約10mgまでの範囲に
わたる。一態様において、該剤は製薬学的組成物として担体とともに投与される。一態様において、該剤は、腹腔内、経口、鼻内、皮下、くも膜下腔内、筋肉内、局所若しくは静脈内で投与される。一態様において、患者はアミロイド形成疾患に罹っている。一態様において、該疾患は、アルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、アミロイドーシス、ダウン症候群、およびプリオン感染により全部若しくは部分的に引き起こされる疾患よりなる群から選ばれる。
【0031】
該方法の別の態様において、該剤をコードする核酸が投与される。該方法の別の態様において、該剤は、アンチセンスRNA分子、アンチセンスDNA分子、リボザイム、RNAiおよびジンクフィンガータンパク質よりなる群から選ばれる。別の態様において、該方法はアミロイド沈着物の形成を阻害することをさらに含んでなる。別の態様において、該方法はアミロイド毒性を阻害することをさらに含んでなる。該方法の別の態様において、該剤はLTPの維持期を阻害しない。該方法の別の態様において、該剤は培養中の薄片調製物でのLTPのアミロイド媒介性阻害を抑制する。該方法の別の態様において、該剤は可溶性AβによるLTPの阻害を抑制する。
【0032】
ある剤をインテグリンサブユニットαv結合剤と同定すること;および該同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することを決定することを含んでなる、LTPのアミロイド媒介性阻害を抑制する剤の同定方法もまた提供される。該方法の一態様において、剤を同定する段階は、直接結合アッセイ、競合結合アッセイおよび細胞接着アッセイの1種若しくはそれ以上を含んでなり;また、該同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することを決定する段階は、第一の神経回路に高頻度刺激を導入することおよびLTPの誘導を測定すること、Aβの存在下の第二の神経回路に高頻度刺激を導入することおよびLTP誘導の阻害を測定すること、ならびにAβおよび該剤の存在下の第三の神経回路に高頻度刺激を導入することおよびLTP誘導の阻害の抑制を測定することを含んでなる。
【0033】
該方法により同定されるLTPのアミロイド媒介性阻害を抑制する剤もまた提供される。一態様において、該剤は抗体である。
【0034】
該剤および製薬学的に許容できる担体を含んでなる組成物がさらに提供される。
【0035】
該方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法がさらに提供される。
【0036】
該方法により同定される、LTPのアミロイド媒介性阻害を抑制する剤もまた提供される。一態様において、該剤は抗体である。
【0037】
該剤および製薬学的に許容できる担体を含んでなる組成物もまた提供される。
【0038】
該方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法もまた提供される。
【0039】
αv拮抗剤若しくはαv媒介性細胞接着の阻害剤を長期増強(LTP)のアミロイド媒介性阻害を抑制するのに有効な量で投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置方法もまた提供される。一態様において、アミロイド形成疾患はアルツハイマー病である。別の態様において、アミロイド形成疾患は軽度認知障害である。
【0040】
1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下で
インテグリンサブユニットαvに結合する1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置若しくは予防方法もまた提供される。一態様において、アミロイド形成疾患はアルツハイマー病若しくは軽度認知障害である。一態様において、アミロイド形成疾患はパーキンソン病若しくはびまん性レビー小体病である。
【発明を実施するための形態】
【0041】
[発明の詳細な記述]
定義
本発明の治療薬は、典型的には望ましくない汚染物質から実質的に精製されている。これは、ある剤が典型的には最低約50w/w(重量)%純度であり、ならびに妨害するタンパク質および汚染物質を実質的に含まないことを意味している。ときに、該剤は最低約60%、70%、80%、90%若しくは95w/w%純度である。慣習的タンパク質精製技術を使用して、最低99w/w%の均質なペプチドもまた得ることができる。
【0042】
2実体間の特異的結合は最低106、107、108、109若しくは1010M−1の親和性を意味している。一態様において、親和性は約108M−1以上である。
【0043】
「抗体」若しくは「免疫グロブリン」という用語は、無傷の抗体およびそれらの結合フラグメントを包含するのに使用する。典型的に、フラグメントは、抗原フラグメントへの特異的結合について、それらが由来した無傷の抗体と競合し、個別のH鎖、L鎖Fab、Fab’、F(ab’)2、F(ab)cおよびFvを包含する。フラグメントは組換えDNA技術、または無傷の免疫グロブリンの酵素的若しくは化学的分離により製造しうる。「抗体」という用語は、他のタンパク質に化学的に複合されるか若しくは他のタンパク質との融合タンパク質として発現される1種若しくはそれ以上の免疫グロブリン鎖もまた包含する。「抗体」という用語は二特異性抗体もまた包含する。二特異性若しくは二機能性抗体は、2種の異なるH/L鎖対および2種の異なる結合部位を有する人工的ハイブリッド抗体である。二特異性抗体は、ハイブリドーマの融合若しくはFab’フラグメントの連結を包含する多様な方法により製造し得る(例えばSongsivilaiとLachmann、Clin.Exp.Immunol.、79:315−321(1990);Kostelnyら、J.Immunol.、148:1547−53(1992)を参照されたい)。
【0044】
APP695、APP751およびAPP770は、ヒトAPP遺伝子によりコードされるそれぞれ695、751および770アミノ酸残基長のポリペプチドを指す。Kangら、Nature、325:733−36(1987);Ponteら、Nature、331:525−27(1988);およびKitaguchiら、Nature、331:530−32(1988)を参照されたい。ヒトアミロイド前駆体タンパク質(APP)内のアミノ酸は、APP770アイソフォームの配列に従って番号を割り当てられている。Aβ39、Aβ40、Aβ41、Aβ42およびAβ43のような用語は、アミノ酸残基1−39、1−40、1−41、1−42および1−43を含有するAβペプチドを指す。Aβ42は配列(配列番号1):
H2N−Asp−Ala−Glu−Phe−Arg−His−Asp−Ser−Gly−Tyr−Glu−Val−His−His−Gln−Lys−Leu−Val−Phe−Phe−Ala−Glu−Asp−Val−Gly−Ser−Asn−Lys−Gly−Ala−Ile−Ile−Gly−Leu−Met−Val−Gly−Gly−Val−Val−Ile−Ala−OHを有する。Aβ41(配列番号3)、Aβ40(配列番号4)およびAβ39(配列番号5)は、C末端からのそれぞれAla、Ala−IleおよびAla−Ile−Valの脱落によりAβ42(配列番号1)と異なる。Aβ43(配列番号2)はC末端のトレオニン残基の存在によりAβ42(配列番号1)と異なる。
【0045】
「アミリン」は、当該技術分野で公知のタンパク質、またはそのペプチド若しくはポリペプチド若しくはフラグメント、あるいは該タンパク質、ペプチド若しくはポリペプチドの前駆体タンパク質若しくはポリマーを指す。該用語は島アミロイドポリペプチドを包含する。アミリンの記述は、当該技術分野でCooperら、Proc.Natl.Acad.Sci.U.S.A.、85:7763(1988)およびLeightonら、Nature、335:632(1988)のような場所で見出しうる。
【0046】
「アミロイドペプチド若しくはタンパク質」という用語は、アミリンおよびAβを包含するアミロイド様沈着物を形成するペプチドおよびタンパク質のファミリーを指す。
【0047】
「アミロイド若しくはアミロイド様沈着物」という句は、アミロイド原線維、ならびに、老人性アミロイドーシス(例えばAβ)、プリオン関連脳症(例えばPrP)および糖尿病患者の腎若しくは膵(例えばアミリン)での沈着物などのような、アミロイド若しくはアミロイド様であると当該技術分野で認識されている構造が原線維若しくは非原線維の他のアミロイド若しくはアミロイド様沈着物を包含する。慣習的染色を用いる光学顕微鏡検査でこうした沈着物は均一なエオシン好性の外見を有する。コンゴーレッド染色でそれらは偏光下で特徴的な黄緑色の複屈折を示す。該用語は、アミロイド沈着物と異なりコンゴーレッドで染色しないプレアミロイド沈着物もまた包含する。
【0048】
「アミロイド形成疾患」という用語は、タンパク質若しくはペプチドの不要な沈着を特徴とする疾患を包含することを意図している。該用語は、Kahnら、Diabetes、48:241−253(1999);およびJohnsonら、Laboratory
Investigation、66(5):522−535(1992)により記述されるところのII型糖尿病(例えばアミリン)、アルツハイマー病(例えばAβ)、多発性骨髄腫および関節リウマチのような、アミロイドペプチドの不要な沈着を特徴とする疾患をとりわけ包含する。該用語は、トランスサイレチン(TTR)沈着により媒介されるものを包含する、Hundら、Neurology、56:431−435(2001)により記述されるところのパーキンソン病または遺伝性若しくは全身性アミロイドーシスのようなアミロイド形成タンパク質の不要な沈着を特徴とする疾患もまたとりわけ包含する。該用語はびまん性レビー小体病もまた包含する。さらに、該用語は、クロイツフェルト・ヤコブ病のようなプリオンへの感染により全部若しくは部分的に引き起こされる疾患を包含する。こうしたプリオン媒介性疾患は、Gieseら、Curr.Topics Microbiology and Immunology、253:203−217(2001)により記述されるところのプリオンタンパク質の蓄積を特徴とする。約言すれば、該用語は、周囲の細胞の健康および満足のいく状態に悪影響を及ぼす不要なタンパク質若しくはペプチド沈着物により病状が媒介される全疾患を包含することを意味している。
【0049】
「抗原」は抗体が特異的に結合する実体である。
【0050】
「エピトープ」若しくは「抗原決定基」という用語は、Bおよび/若しくはT細胞が応答する抗原上の1部位を指す。B細胞エピトープは、連続するアミノ酸、若しくはタンパク質の三次フォールディングにより並置される連続しないアミノ酸の双方から形成され得る。連続するアミノ酸から形成されるエピトープは変性溶媒への曝露に際して典型的に保持される一方、三次フォールディングにより形成されるエピトープは変性溶媒での処理に際して典型的に喪失される。エピトープは、典型的に、独特の空間的コンホメーション中に最低3、およびより通常は最低5若しくは8〜10アミノ酸を包含する。エピトープの空間的コンホメーションの決定方法は、例えばx線結晶学および二次元核磁気共鳴を包含する。例えば、Methods in Molecular Biology、Vol.
66、Glenn E.Morris編(1996)中Epitope Mapping
Protocolsを参照されたい。同一の若しくは重なるエピトープを認識する抗体は、標的抗原への別の抗体の結合を阻止する1抗体の能力を示す単純なイムノアッセイで同定し得る。
【0051】
「裸のポリヌクレオチド」若しくは「裸のDNA」という用語は、コロイド状物質例えばタンパク質と複合体形成されていないポリヌクレオチドを指す。裸のポリヌクレオチドはときにプラスミドベクターにクローン化される。
【0052】
「患者」という用語は予防的若しくは治療的処置のいずれかを受領するヒトおよび他の哺乳動物被験体を包含する。
【0053】
「予防する」、「予防すること」および「予防」という用語は、疾患若しくは状態の最低1症状(例えば長期増強の抑制および/若しくは神経変性)を究極的に明示しうるがしかし未だそうしていない個体への予防的(prophylactic)若しくは予防的(preventative)な治療の投与を指す。こうした個体は、該疾患のその後の発生と相関することが既知である危険因子に基づき同定しうる。あるいは、予防治療は予防対策として危険因子の事前の同定を伴わず投与しうる。疾患若しくは状態の最低1症状の発生を遅らせることもまた予防(prevention)若しくは予防(prophylaxis)とみなしうる。
【0054】
本明細書で使用されるところの「処置する」、「処置すること」若しくは「処置」という用語は、疾患若しくは状態の最低1症状(例えば長期増強の抑制および/若しくは神経変性)を既に明示している個体への治療の投与を指す。
【0055】
剤および第二の剤の「共投与」は、in vitro系、若しくは患者でのようなin
vivoで時間が重なる剤および第二の剤の治療濃度を達成するためのいかなる投薬計画による投与も包含する。従って、例えば、共投与は、剤および第二の剤双方を含んでなる製剤の投与、ならびに一方が該剤を含んでなりかつ別のものが第二の剤を含んでなる別個の製剤の投与を包含する。別個の製剤を投与する場合、投与は同時であっても連続してもよい。連続する場合、該剤は次々に投与しうるか、あるいは、該剤の投与と第二の剤の投与の間の時間は、例えば1時間まで、2時間まで、4時間まで、6時間まで、12時間まで、または1日若しくは数日まででありうる。
【0056】
神経細胞は、in vivoで起こるAPPのAβへの天然のプロセシングの結果として、若しくは、in vitroアッセイでのAβの調製物と神経細胞の接触の結果として、Aβペプチドに曝露し得る。Aβペプチドへの曝露は薬物への曝露前、後若しくはそれと同時に発生し得る。
【0057】
Aβペプチドのアミロイド沈着物は、図1Aに示されるようなin vitro若しくはin vivoで皮質細胞上および周囲に生じる原線維をおそらく包含するAβペプチドの凝集物を指す。
【0058】
文脈から別の方法で明らかでない限り、フィブロネクチンへの言及はスーパーフィブロネクチンを包含する。
【0059】
抗体間の競合は、試験中の免疫グロブリンが共通抗原への参照抗体の特異的結合を阻害するアッセイにより測定する。多数の型の競合結合アッセイ、例えば固相直接若しくは間接ラジオイムノアッセイ(RIA)、固相直接若しくは間接エンザイムイムノアッセイ(EIA)、サンドイッチ競合アッセイ(Stahliら、Methods in Enz
ymology、9:242−53(1983)を参照されたい);固相直接ビオチン−アビジンEIA(Kirklandら、J.Immunol.、137:3614−19(1986)を参照されたい);固相直接標識アッセイ、固相直接標識サンドイッチアッセイ(HarlowとLane、”Antibodies,A Laboratory Manual”、Cold Spring Harbor Press(1988)を参照されたい);I−125標識を使用する固相直接標識RIA(Morelら、Molec.Immunol.、25(1):7−15(1988)を参照されたい);固相直接ビオチン−アビジンEIA(Cheungら、Virology、176:546−52(1990));および直接標識RIA(Moldenhauerら、Scand.J.Immunol.、32:77−82(1990))が既知である。典型的に、こうしたアッセイは、未標識の試験免疫グロブリン若しくは標識参照免疫グロブリンいずれかを持つ固体表面若しくは細胞に結合した精製抗原の使用を必要とする。競合阻害は、試験免疫グロブリンの存在下で固体表面若しくは細胞に結合される標識の量を測定することにより測定する。通常、試験免疫グロブリンは過剰に存在する。競合アッセイにより同定される抗体(競合抗体)は、参照抗体と同一エピトープに結合する抗体、および参照抗体により結合されるエピトープの十分に近位の隣接するエピトープに結合して立体障害を起こす抗体を包含する。通常、競合抗体が過剰に存在する場合、それは参照抗体の共通抗原への特異的結合を最低50若しくは75%阻害することができる。
【0060】
1種若しくはそれ以上の列挙される要素を「含んでなる」組成物若しくは方法は、具体的に列挙されない他の要素を包含しうる。例えば、抗体を含んでなる組成物は、単独若しくは他の成分と組合せの該抗体を含有しうる。
【0061】
I.方法
本発明は、アミリンおよびAβペプチドのようなアミロイドタンパク質の細胞外網目構造の形成の阻害若しくは予防方法、こうしたタンパク質の毒性効果の媒介方法、ならびに該方法での使用のための剤を提供する。該方法は、アルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、全身性および遺伝性アミロイドーシス、ならびにプリオン感染により全部若しくは部分的に引き起こされる疾患を処置若しくは予防するのに使用し得る。これらの方法での使用に有効な剤は、β1、α2、α6若しくはαvのようなインテグリンサブユニットに結合する抗体および他の剤を包含する。これらのサブユニットはヘテロ二量体受容体として会合してインテグリン、例えばα2β1、α6β1およびαvβ1を形成する。上の剤は、インテグリンとAβペプチドの間の相互作用を阻害するために個別で若しくは組合せで使用し得る。αvβ1およびα2β1双方のインテグリンとAβの間の相互作用を阻害する剤(1種若しくは複数)の使用が好ましい。フィブロネクチン(インテグリンαvβ1のリガンド)もまた、上の方法でラミニン(αvβ1のリガンド)に対する抗体がし得るように剤として使用し得る。本発明は、部分的に、α2、αv、α6およびβ1インテグリンサブユニットに対する抗体がアミリンおよびAβペプチドのようなアミロイドタンパク質の細胞外網目構造の形成を阻害するという観察結果を前提とする。それにより、こうした抗体はアミロイドタンパク質の毒性を阻害する。αvβ1リガンド、フィブロネクチンもまた網目構造形成を阻害する。α2β1リガンド、ラミニンは網目構造形成を阻害しないが、しかしラミニンに対する抗体は網目構造形成および毒性を阻害する。
【0062】
本発明は、部分的に、αvインテグリンサブユニットに対する選択的抗体がin vitroおよびin vivo双方でAβによるLTPの阻害を抑制するという観察結果を前提とする。αv含有インテグリンの小分子非ペプチド拮抗剤、ならびにインテグリンの2種の他の拮抗的リガンド、スーパーフィブロネクチンおよびディスインテグリン、エキスタチンもまたLTPのAβ阻害を抑制する。従って、αvインテグリンサブユニットに結合する剤はLTPのAβ阻害を抑制し、ならびに、アミリンおよびAβペプチドのよう
なアミロイドタンパク質の細胞外網目構造の形成を阻害若しくは予防する。
【0063】
従って、本発明は、長期増強(LTP)のアミロイド媒介性阻害を抑制するのに有効な量のαv拮抗剤若しくはαv媒介性細胞接着の阻害剤を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法、およびAβ沈着を特徴とするアミロイド形成疾患の処置若しくは予防方法、ならびに、Aβの沈着を特徴とするアミロイド形成疾患の処置方法もまた提供する。該方法は、限定されるものでないがアルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、全身性および遺伝性アミロイドーシスを挙げることができる疾患若しくは状態、ならびにプリオン感染により全部若しくは部分的に引き起こされる疾患を処置若しくは予防するのに有用である。これらの方法での使用に有効な剤は、限定されるものでないが、抗体、および例えばインテグリンサブユニットαvに結合するSM256のような他の剤を挙げることができる。上の剤は、インテグリンとAβペプチドの間の相互作用を阻害するのに個別で若しくは組合せで使用し得る。
【0064】
II.インテグリン
インテグリンは、細胞外マトリックスおよび他細胞の双方への細胞の接着を制御する、細胞表面接着ヘテロ二量体膜貫通受容体のスーパーファミリーである。接着は成長、移動および分化のための固定およびシグナルを提供する。インテグリンは、約15種の既知のα鎖の1種の約8種の既知のβ鎖の1種との会合により形成される。赤血球を除く全部のヒト細胞が1種若しくはそれ以上のインテグリンを発現する。
【0065】
インテグリンサブユニットα2、αv、α6およびβ1は全部公知である。例示的ヒト配列はそれぞれGenBank受託番号AF062039、M14648、X59512およびX07979から取出し可能である。別の方法で示されない限り、α2、αv、α6、β1への言及は、これらの例示的配列、それらのアレルバリアントおよび他の種からの同族バリアントを包含する。天然の配列のリガンドへの特異的結合について天然の配列と競合するのに十分な天然の配列に対する配列同一性を有するこれらの配列の誘導されるバリアントもまた、いくつかの方法で使用し得る。αv、およびβサブユニットβ1、β3、β5、β6若しくはβ8の1種を含有するインテグリンはRGDモチーフを持つリガンドを認識するが、しかし結合特異性はどのβサブユニットが存在するかに依存して変動する。αvβ1は、ビトロネクチン(GenBank受託番号X03168)、フィブロネクチン(GenBank受託番号M26179)およびオステオポンチン(GenBank受託番号J04765)を認識することが既知である。フィブロネクチンは、結合組織中、細胞表面上、ならびに血漿および他の体液中で見出される大型多ドメイン糖タンパク質である。フィブロネクチンは、細胞骨格および細胞外マトリックスの成分、血液凝固応答、線維素溶解の急性期および補体系に関与する循環成分を包含する多様な巨大分子、ならびに線維芽細胞、ニューロン、食細胞および細菌を包含する多様な細胞上の細胞表面受容体と作用する。
【0066】
α2およびβ1サブユニットを含有するインテグリンは、VLA−2(最晩期抗原(very late activation antigen)2)、GPIa−IIa(血小板上糖タンパク質Ia−IIa)およびECMRII(細胞外マトリックス受容体II)として知られる。α2β1インテグリンはコラーゲンIないしVI、ラミニンおよびおそらくフィブロネクチンを結合する。該受容体は、BおよびTリンパ球、血小板、線維芽細胞、内皮細胞ならびに黒色腫細胞上で発現され、そしてコラーゲンおよびラミニンをリガンドとして特異的に認識する。ラミニンは共通の構造的構成をもつ大型多ドメインタンパク質である。ラミニン分子は、コイルドコイルドメインにより一緒に結合されたα、βおよびγ鎖サブユニットを有する。最低5種のα鎖、2種のβ鎖および3種のγ鎖が既知であり、そしてこれらの鎖の異なる組合せを有する最低12種のラミニンが報告されて
いる(第WO 00/66730号明細書)。ラミニンはアルツハイマー病において斑を包含する細胞外マトリックス中で見出される(Murtomakiら、J.Neuro.Res.、32:261−73(1992);Bronfinanら、Int.J.Exp.Clin.Invest.、5:16−23(1997);およびCastilloら、J.Neuro.Res.、62:451−62(2000))。コラーゲンは哺乳動物で最も豊富なタンパク質であり、そして皮膚、骨、腱、軟骨および歯の主要線維成分である。23種以上の既知のコラーゲン遺伝子が存在する(Adamsら、Am.J.Respir.Cell.Molec.Biol.、1:161−168(1989))。
【0067】
α6/β1インテグリンは、血小板、リンパ球、単球、胸腺細胞および内皮細胞上で発現され、それら上でそれはin vivoでラミニン−1、ラミニン−2およびラミニン−4のラミニン受容体として機能する。それはまたラミニン−5の受容体でもあるが、しかしin vivoででない。ラミニン−1について、結合部位はこの細胞外マトリックス分子のE8ドメイン中に突き止められている。この受容体は最晩期抗原6(VLA−6)および糖タンパク質Ic−IIa(血小板上GPIc−IIa)としてもまた知られている。
【0068】
インテグリンは、セレクチンおよび免疫グロブリン(Ig)スーパーファミリーメンバーもまた包含する接着タンパク質として知られるタンパク質の大きな一分類の一例である(Springer、Nature、346:425(1990);Osborn、Cell、62:3(1990);Hynes、Cell、69:11(1992)(全部の目的上そっくりそのまま引用することにより組み込まれる)を参照されたい)。抗体、および接着タンパク質若しくはそれらのリガンドに結合しかつ/または該2者間の相互作用を阻止する他の剤を、下述されるスクリーニング方法でAβ沈着物の蓄積の予防若しくは阻害における活性についてスクリーニングし得る。下述される方法によるスクリーニングに適する他のセレクチンおよびそれらのリガンドの例は、インテグリンα2β5、αvβ5、α6β5、α2β6、αvβ6およびα6β6を包含する。コラーゲンに結合するα2β1を除く他のリガンドもまたスクリーニングしうる。
【0069】
III.剤
本発明の治療薬は、α2、αv、α6およびβ1インテグリンサブユニットに特異的に結合する抗体を包含する。結合は、場合によっては固相に固定された単離されたインテグリンサブユニット若しくはそれらのフラグメント、または細胞の表面上で発現されたインテグリンサブユニットのいずれを用いても評価し得る。しばしば、結合はヘテロ二量体インテグリンを発現する細胞を使用して分析される。例えば、ある剤が唯一のインテグリンとしてα2β1を発現する細胞に結合する場合には、該剤がα2若しくはβ1またはいずれのサブユニット単独にも結合することなくα2β1に結合すると結論付け得る。これらの可能性は、異なるヘテロ二量体インテグリンを持つ細胞への同一の剤の結合を試験することにより識別し得る。例えば、同一の剤が、存在する唯一のインテグリンとしてαvβ1を持つ細胞に特異的に結合する場合には、該剤がβ1サブユニットに結合していることがありそうである。インテグリンおよびインテグリンサブユニットに対する多様な抗体が商業的に入手可能であり、それらのいくつかを実施例に記述する。
【0070】
モノクローナル若しくはポリクローナル抗体を本発明の方法で使用し得る。好ましい抗体は、それらの天然のリガンドの1種若しくはそれ以上とのこれらのインテグリンサブユニットの相互作用を阻止する。すなわち、αvβ1に対する阻止抗体は、フィブロネクチン、オステオポンチンおよび/若しくはビトロネクチンとのこのインテグリンの相互作用を阻止する。例えば、第WO 99/37683号明細書により記述される14D9.F8抗体はαvのフィブロネクチンへの結合を阻止する。α2β1に対する阻止抗体はこのインテグリンのコラーゲン若しくはラミニンとの相互作用を阻止する。抗体若しくは他の
剤の阻止する能力は、抗体(若しくは他の剤)の存在若しくは非存在下で、リガンドで被覆したプレートへの接着についてインテグリンを発現する細胞を試験する単純なアッセイにより認識し得る。プレートへの細胞結合の量の最低約30%、40%、50%、60%、70%、80%、90%若しくは100%の減少は、該抗体がインテグリンに関してモル濃度過剰で存在する場合に阻止抗体(若しくは他の剤)を同定する。インテグリンサブユニットの他の組合せに対する剤の阻止能力のさらなる分析は、ヘテロ二量体インテグリンのどのサブユニットが阻止されているかを特定し得る。抗体若しくは他の剤の結合特異性もまた、インテグリンサブユニット若しくはそれを持つ細胞への結合について所望のエピトープ特異性を有することが既知の参照抗体と試験抗体が競合する競合アッセイにより決定し得る。試験および参照抗体が競合する場合には、それらは、一方の抗体の結合が他方の結合を妨害する十分に近位の同一のエピトープ(1個若しくは複数)に結合する。いくつかの態様において、トランスフェクトした細胞は単一の型のインテグリンを発現する。
【0071】
本発明での使用のための数種の抗体はただ1つの型のインテグリンサブユニットに結合する。数種の抗体は2種若しくはそれ以上のインテグリンサブユニットに特異的に結合する。数種の抗体は、インテグリンのサブユニットがヘテロ二量体インテグリンとして会合している場合にのみ結合する。例えば、数種の抗体はα2若しくはβ1いずれか単独に結合することなくα2β1に結合する。数種の抗体はαv若しくはβ1いずれか単独に結合することなくαvβ1に結合する。数種の抗体はαvインテグリンサブユニットに結合する。数種の抗体は、遊離の形態で、および該サブユニットがヘテロ二量体インテグリンの一成分である場合の双方でサブユニットに結合する。上の抗体の同一の結合特異性を有するペプチドおよび小分子もまた使用し得る。
【0072】
本発明での使用のための他の治療薬は、フィブリノーゲン、オステオポンチン、ビトロネクチン、それらのフラグメント、およびαvβ1への結合についてフィブリノーゲン若しくはビトロネクチンと競合するRGDペプチドモチーフを含有する他の天然の若しくは合成ペプチドを包含する。αvβ1への結合についてフィブリノーゲン、ビトロネクチン若しくはオステオポンチンと競合する小分子模倣物もまた使用し得る。他の治療薬は、ラミニンに対する抗体、ならびに同一の結合特異性をもつペプチドおよび小分子を包含する。
【0073】
候補治療薬は以下のスクリーニングの1種若しくはそれ以上を実施することにより評価し得る。典型的には、インテグリンサブユニットα2、αv、α6若しくはβ1、および/またはヘテロ二量体インテグリンα2β1、αvβ1 α6β1、あるいはラミニンへの特異的結合について剤を最初に評価する。適する剤は、典型的に最低107、108、109若しくは1010M−1の特異的親和性で結合する。
【0074】
その後、候補は場合によっては特定のエピトープ特異性について評価する。これは、参照剤を用いる競合アッセイ、上述されたところの機能的プレートブロッキングアッセイ、または、抗原の一連の欠失変異体を結合するその能力についウエスタンブロッティング若しくはELISAにより抗体若しくは他の剤を評価するエピトープマッピング実験により決定し得る。抗体若しくは他の剤への特異的結合を示すための最小フラグメントが該抗体若しくは他の剤のエピトープを規定する。あるいは、若しくは加えて、候補剤をアミロイドペプチドの細胞外網目構造の形成を阻害する能力について評価する。適する剤は、典型的に、剤の存在下でアミリン若しくはAβのようなアミロイドペプチドでの処理から生じる毒性を対照に関して最低約10%、20%、30%、40%、50%、60%、70%、80%、90%若しくは100%またはそれ以上低下させる。
【0075】
候補化合物は、アミロイド形成疾患になりやすいトランスジェニック動物で予防的およ
び治療的有効性についてもまた試験し得る。こうした動物は、例えば、Gamesら、上記により記述されるAPPの717突然変異を持つマウス、ならびにMcConlogueら、米国特許第US 5,612,486号明細書;Hsiaoら、Science、274:99(1996);Sturchler−Plerratら、Proc.Natl.Acad.Sci.U.S.A.、94:13287−92(1997);およびBorcheltら、Neuron、19:939−45(1997)により記述されるようなAPPの670/671スウェーデン型突然変異を持つマウスを包含する。トランスジェニックマウスで活性を示す剤をその後ヒト臨床試験で評価し得る。アルツハイマー病患者でヒト臨床試験を実施するための例示的形式が第WO 98/24678号明細書(引用することにより本明細書に組み込まれる)に記述されている。
【0076】
長期増強(LTP)のアミロイド媒介性阻害の抑制方法での使用のための候補化合物の場合、該化合物をLTPのin vitroおよび/若しくはin vivoモデルで試験しうる。例えば、候補化合物をin vitroモデルで最初に試験することができ、そしてその後、該化合物がそのモデルで長期増強(LTP)のアミロイド媒介性阻害を抑制する場合は、in vivoモデルでその後試験しうる。
【0077】
A.抗体
1.免疫グロブリンの一般的特徴
基本的抗体構造ユニットはサブユニットの四量体を含んでなることが知られている。各四量体はポリペプチド鎖の2個の同一の対から構成され、各対は1本の「L」(約25kDa)および1本の「H」鎖(約50〜70kDa)を有する。各鎖のアミノ末端部分は主に抗原認識を司る約100ないし110若しくはそれ以上のアミノ酸の可変領域を包含する。各鎖のカルボキシ末端部分は主にエフェクター機能を司る定常領域を規定する。
【0078】
L鎖はκ若しくはλいずれかに分類される。H鎖はγ、μ、α、δ若しくはεに分類され、そしてそれぞれIgG、IgM、IgA、IgDおよびIgEのような抗体のアイソタイプを規定する。LおよびH鎖内で、可変および定常領域は約12若しくはそれ以上のアミノ酸の「J」領域により結合され、H鎖は約10より多いアミノ酸の「D」領域もまた包含する。(全般として、Fundamental Immunology(Paul,W.編、第2版、Raven Press、ニューヨーク、1989)、第7章(全部の目的上そっくりそのまま引用することにより組み込まれる)を参照されたい)。
【0079】
各L/H鎖対の可変領域は抗体の結合部位を形成する。従って無傷の抗体は2個の結合部位を有する。二機能性若しくは二特異性抗体でを除き該2結合部位は同一である。該鎖は全部、相補性決定領域若しくはCDRともまた呼ばれる3個の超可変領域により結合される比較的保存された枠組み領域(FR)の同一の一般的構造を表す。各対の2本の鎖からのCDRは枠組み領域により整列されて、特異的エピトープへの結合を可能にする。N末端からC末端へ、LおよびH鎖双方はドメインFR1、CDR1、FR2、CDR2、FR3、CDR3およびFR4を含んでなる。各ドメインへのアミノ酸の割り当ては、Kabat、Seqeunces of Proteins of Immunological Interest(国立保健研究所、メリーランド州ベセスダ、1987および1991)、若しくはChothiaとLesk、J.Mol.Biol.、196:901−17(1987);Chothiaら、Nature、342:878−83(1989)の定義に従う。
【0080】
2.ヒト以外の抗体の製造
ヒト以外のモノクローナル抗体(例えばマウス、モルモット、霊長類、ウサギ若しくはラット)の製造は、例えば、インテグリン、そのサブユニット若しくはそれらのフラグメント、またはインテグリン若しくはそのサブユニットを持つ細胞で動物を免疫することに
より達成し得る。ラミニンに対する抗体を生成するための免疫原としてラミニンもまた使用し得る。HarlowとLane、Antibodies,A Laboratory
Manual(Cold Spring Harbor Press、ニューヨーク、1988(全部の目的上引用することにより本明細書に組み込まれる))を参照されたい。こうした免疫原は、天然の供給源から、ペプチド合成により、若しくは組換え発現により得ることができる。場合によっては、免疫原は下述されるところの担体タンパク質と融合若しくは別の方法で複合体形成して投与し得る。場合によっては、免疫原はアジュバントとともに投与し得る。いくつかの型のアジュバントを下述されるとおり使用し得る。フロイントの完全アジュバント、次いで不完全アジュバントが実験動物の免疫化に好ましい。ウサギ、ヤギ、ヒツジ若しくはモルモットを、ポリクローナル抗体を作成するのに典型的に使用する。マウスはモノクローナル抗体を作成するのに典型的に使用する。抗体を、意図しているインテグリン若しくはそのサブユニット、またはラミニンのような他の抗原への特異的結合についてスクリーニングする。抗体は上述されたところのそのリガンドへのインテグリンの結合を阻止する能力についてもまたスクリーニングし得る。上述された他のスクリーニング手順もまた実施し得る。
【0081】
3.キメラおよびヒト化抗体
キメラおよびヒト化抗体は、キメラ若しくはヒト化抗体の構築のための出発原料を提供するマウス若しくは他のヒト以外の抗体と同一の若しくは類似の結合特異性および親和性を有しうる。数種のキメラ若しくはヒト化抗体はマウスのものの2倍、5倍若しくは10倍の係数内の親和性を有する。キメラ抗体は、そのLおよびH鎖遺伝子が、異なる種に属する免疫グロブリン遺伝子セグメントから典型的には遺伝子工学により構築された抗体である。例えば、マウスモノクローナル抗体からの遺伝子の可変(V)セグメントをIgG1、IgG2、IgG3若しくはIgG4のようなヒト定常(C)セグメントに結合しうる。典型的なキメラ抗体は、従って、マウス抗体からのVすなわち抗原結合ドメインおよびヒト抗体からのCすなわちエフェクタードメインよりなるハイブリッドタンパク質である。
【0082】
ヒト化抗体は、実質的にヒト抗体(アクセプター抗体と命名される)からの可変領域枠組み残基、および実質的にマウス抗体のようなヒト以外の抗体(ドナー免疫グロブリンと称される)からの相補性決定領域を有する。Queenら、Proc.Natl.Acad.Sci.U.S.A.、86:10029−33(1989)および第WO 90/07861号、米国特許第US 5,693,762号、同第US 5,693,761号、同第US 5,585,089号、同第US 5,530,101号、およびWinter、米国特許第US 5,225,539号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)を参照されたい。定常領域(存在する場合)もまた実質的に若しくは完全にヒト免疫グロブリンからである。ヒト可変ドメインは通常、CDRが由来したマウス可変領域ドメインとの高程度の配列同一性をその枠組み配列が表すヒト抗体から選ばれる。HおよびL鎖可変領域枠組み残基は同一若しくは異なるヒト抗体配列に由来し得る。ヒト抗体配列は天然に存在するヒト抗体の配列であり得るか、若しくは数種のヒト抗体のコンセンサス配列であり得る。Carterら、第WO 92/22653号明細書を参照されたい。ヒト可変領域枠組み残基からのあるアミノ酸を、CDRコンホメーションおよび/若しくは抗原への結合に対するそれらの可能な影響に基づき置換のため選択する。こうした可能な影響の検討は、モデル化、特定の場所のアミノ酸の特徴の検査、または特定のアミノ酸の置換若しくは突然変異誘発の影響の経験的観察による。
【0083】
例えば、あるアミノ酸がマウス可変領域枠組み残基と選択したヒト可変領域枠組み残基の間で異なる場合、該ヒト枠組みアミノ酸は通常、該アミノ酸が:
(1)抗原を直接非共有結合する、
(2)CDR領域に隣接する、
(3)CDR領域と別の方法で相互作用する(例えばCDR領域の約6Å以内にある)、若しくは
(4)VL−VH界面に参画する
ことが合理的に期待される場合は、マウス抗体からの同等の枠組みアミノ酸により置換すべきである。
【0084】
置換の他の候補は、その位置でヒト免疫グロブリンにとって異常であるアクセプターヒト枠組みアミノ酸である。これらのアミノ酸を、ドナー抗体の同等の位置若しくはより典型的なヒト免疫グロブリンの同等の位置からのアミノ酸で置換し得る。ヒト化免疫グロブリンの可変領域枠組みは通常、ヒト可変領域枠組み配列若しくはこうした配列のコンセンサスに対し最低85%の配列同一性を示す。
【0085】
4.ヒト抗体
上のインテグリン若しくはラミニンに対するヒト抗体は下述される多様な技術により提供される。数種のヒト抗体は、競合結合実験により、若しくは、そうでなければ、実施例に記述されるマウスモノクローナル抗体の1種のような特定の1マウス抗体と同一のエピトープ特異性を有するように選択する。ヒト抗体は、免疫原としてインテグリン若しくはラミニンの1フラグメントのみを使用することにより、および/またはインテグリンの欠失変異体の集合物に対し抗体をスクリーニングすることにより、特定の1エピトープ特異性についてもまたスクリーニングし得る。
【0086】
a.トリオーマの方法論
基本的アプローチ、および本アプローチでの使用のための例示的1細胞融合パートナーSPAZ−4が、Oestbergら、Hybridoma、2:361−67(1983);Oestberg、米国特許第4,634,664号明細書;およびEnglemanら、米国特許第4,634,666号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)により記述されている。この方法により得られる抗体産生細胞株は、それらが3種の細胞(2種のヒトおよび1種のマウス)を祖先とするためトリオーマと呼ばれる。最初に、マウス骨髄腫株をヒトBリンパ球と融合して、Oestberg、上記により記述されるSPAZ−4細胞株のような抗体を産生しない異種ハイブリッド細胞を得る。該異種細胞をその後、免疫ヒトBリンパ球と融合して抗体産生トリオーマ細胞株を得る。トリオーマはヒト細胞から作成される通常のハイブリドーマより安定に抗体を産生することが見出されている。
【0087】
免疫Bリンパ球はヒトドナーの血液、脾、リンパ節若しくは骨髄から得る。特定の1抗原若しくはエピトープに対する抗体が望ましい場合は、免疫化にその抗原若しくはその1エピトープを使用することが好ましい。免疫化はin vivo若しくはin vitroのいずれでもあり得る。in vivo免疫化のため、B細胞を、典型的に、Aβ、そのフラグメント、Aβ若しくはフラグメントを含有するより大きいペプチド、またはAに対する抗体に対する抗イディオタイプ抗体で免疫したヒトから単離する。いくつかの方法において、B細胞は抗体療法を最終的に投与されるはずである同一患者から単離する。in vitro免疫化のためには、Bリンパ球は、典型的に、10%ヒト血漿を補充したRPMI−1640(Engleman、上記を参照されたい)のような培地中で7〜14日間抗原に曝露する。
【0088】
免疫Bリンパ球を公知の方法によりSPAZ−4のような異種ハイブリッド細胞に融合する。例えば、細胞をMW1000〜4000の40〜50%ポリエチレングリコールで約37℃で約5〜10分間処理する。細胞を融合混合物から分離し、そして所望のハイブリッドについて選択的な培地(例えばヒポキサンチン+アメトプテリン+チミジン(HA
T培地)若しくはアメトプテリン+ヒポキサンチン(AH培地)を含有する)中で増殖させる。必要とされる結合特異性を有する抗体を分泌するクローンは、Aβ若しくはそのフラグメントに結合する能力についてトリオーマ培地をアッセイすることにより同定する。所望の特異性を有するヒト抗体を産生するトリオーマを限界希釈技術によりサブクローニングし、そして培地中in vitroで増殖させる。得られるトリオーマ細胞株をその後Aβ若しくはそのフラグメントを結合する能力について試験する。
【0089】
トリオーマは遺伝子的に安定であるとは言え、それらは非常に高レベルで抗体を産生しない。発現レベルは、抗体遺伝子をトリオーマから1種若しくはそれ以上の発現ベクターにクローン化すること、および該ベクターを標準的哺乳動物、細菌若しくは酵母細胞株に形質転換することにより増大させ得る。
【0090】
b.トランスジェニックのヒト以外の哺乳動物
インテグリン若しくはラミニンに対するヒト抗体は、ヒト免疫グロブリン遺伝子座の最低1セグメントをコードする導入遺伝子を有するヒト以外のトランスジェニック哺乳動物からもまた製造し得る。通常、こうしたトランスジェニック哺乳動物の内因性免疫グロブリン遺伝子座は機能的に不活性化されている。好ましくは、ヒト免疫グロブリン遺伝子座のセグメントは、HおよびL鎖コンポーネントの再配列されない配列を包含する。内因性免疫グロブリン遺伝子の不活性化および外因性免疫グロブリン遺伝子の導入の双方を、標的を定めた相同的組換え、若しくは酵母人工染色体(YAC)の導入により達成し得る。この方法から生じるトランスジェニック哺乳動物は、免疫グロブリンコンポーネントの配列を機能的に再配列すること、および内因性免疫グロブリン遺伝子を発現することなくヒト免疫グロブリン遺伝子によりコードされる多様なアイソタイプの抗体のレパートリーを発現することが可能である。これらの特性を有する哺乳動物の製造および特性は、例えばLonbergら、第W093/12227号(1993);第US 5,877,397号、同第US 5,874,299号、同第US 5,814,318号、同第US5,789,650号、同第US 5,770,429号、同第US 5,661,016号、同第US 5,633,425号、同第US 5,625,126号、同第US 5,569,825号、同第US 5,545,806号明細書、Nature 148、1547−53(1994)、Fishwildら、Nature Biotechnology、14、845−51(1996)、Kucherlapati、第WO 91/10741号明細書(1991)(それらのそれぞれは全部の目的上そっくりそのまま引用することにより組み込まれる)により詳細に記述されている。トランスジェニックマウスはとりわけ適する。抗インテグリン若しくは抗ラミニン抗体は、Lonberg若しくはKucherlapati、上記により記述されたようなトランスジェニックのヒト以外の哺乳動物をインテグリンまたはそのサブユニット若しくはフラグメントで免疫することにより得られる。モノクローナル抗体は、例えば、慣習的なKohler−Milsteinの技術を使用してこうした哺乳動物からのB細胞を適する骨髄腫細胞株に融合することにより製造する。ヒトポリクローナル抗体は、免疫原性剤で免疫したヒトからの血清の形態でもまた提供し得る。場合によっては、こうしたポリクローナル抗体は、インテグリン若しくはラミニンをアフィニティー試薬として使用するアフィニティー精製により濃縮し得る。
【0091】
c.ファージディスプレイ法
ヒト抗インテグリン若しくは抗ラミニン抗体を得るためのさらなる1アプローチは、Huseら、Science、246:1275−81(1989)により概説される一般的プロトコルに従ってヒトB細胞からのDNAライブラリーをスクリーニングすることである。トリオーマの方法論について記述されたとおり、こうしたB細胞は、インテグリン、サブユニット若しくはそれらのフラグメント、またはラミニンおよびそのフラグメントで免疫したヒトから得ることができる。場合によっては、こうしたB細胞は抗体処置を最
終的に受領するはずである患者から得る。目的の抗原若しくはそのフラグメントに結合する抗体を選択する。こうした抗体(若しくは結合フラグメント)をコードする配列をその後クローン化しかつ増幅する。Huseにより記述されるプロトコルはファージディスプレイ技術と組合せでより効率的にされる。例えば、Dowerら、第WO 91/17271号、およびMcCaffertyら、同第WO 92/01047号、米国特許第US 5,877,218号、同第US 5,871,907号、同第US 5,858,657号、同第US 5,837,242号、同第US 5,733,743号、同第US 5,565,332号、同第US 5,969,108号、同第US 6,172,197号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)を参照されたい。抗体、若しくは特定の1リガンドに結合する他のタンパク質の付加的な選択および標識方法は、第US 5,994,519号および同第US 6,180,336号明細書により記述されている。
【0092】
ファージディスプレイ法において、メンバーがそれらの外表面に多様な抗体を表示するファージのライブラリーを製造する。抗体は通常Fv若しくはFabフラグメントとして表示される。所望の特異性をもつ抗体を表示するファージを、インテグリン、サブユニット若しくはそれらのフラグメントに対するアフィニティー濃縮により選択する。
【0093】
ファージディスプレイ法の1変形において、選択したマウス抗体の結合特異性を有するヒト抗体を製造し得る。Winter、第WO 92/20791号明細書を参照されたい。この方法では、選択したマウス抗体のH若しくはL鎖可変領域のいずれかを出発原料として使用する。例えばL鎖可変領域を出発原料として選択する場合、メンバーが同一L鎖の可変領域(すなわちマウス出発原料)および異なるH鎖可変領域を表示するファージライブラリーが構築される。H鎖可変領域は再配列されたヒトH鎖可変領域のライブラリーから得る。Aβに対する強い特異的結合(例えば最低約108若しくは最低約109M−1)を示すファージを選択する。このファージからのヒトH鎖可変領域がその後、さらなるファージライブラリーを構築するための出発原料としてはたらく。このライブラリー中で、各ファージは同一のH鎖可変領域(すなわち最初のディスプレイライブラリーから同定される領域)および異なるL鎖可変領域を表示する。L鎖可変領域は、再配列されたヒト可変L鎖領域のライブラリーから得られる。再度、所望のインテグリンに対する強い特異的結合を示すファージを選択する。これらのファージは完全にヒトの抗インテグリン抗体の可変領域を表示する。これらの抗体は通常、マウス出発原料と同一若しくは類似のエピトープ特異性を有する。
【0094】
5.定常領域の選択
キメラ、ヒト化若しくはヒト抗体のHおよびL鎖可変領域をヒト定常領域の少なくとも一部分に連結し得る。定常領域の選択は、部分的に、抗体依存性補体および/若しくは細胞媒介性毒性が望ましいかどうかに依存する。例えば、アイソタイプIgG1およびIgG3は補体活性を有し、また、アイソタイプIgG2およびIgG4は有しない。アイソタイプの選択はまた抗体の脳への通過に影響を及ぼし得る。L鎖定常領域はλ若しくはκであり得る。抗体は、2本のLおよび2本のH鎖を含有する四量体として、個別のH鎖、L鎖として、Fab、Fab’、F(ab’)2およびFv、若しくはHおよびL鎖可変ドメインがスペーサーにより連結されている一本鎖抗体として、発現され得る。
【0095】
6.組換え抗体の発現
キメラ、ヒト化およびヒト抗体は組換え発現により典型的に製造される。組換えポリヌクレオチド構築物は典型的に、天然に関連した若しくは異種のプロモーター領域を包含する抗体鎖のコーディング配列に作動可能に連結された発現制御配列を包含する。好ましくは、発現制御配列は、真核生物宿主細胞を形質転換若しくはトランスフェクトすることが可能なベクター中の真核生物プロモーター系である。ベクターを適切な宿主に一旦組み込
めば、該ヌクレオチド配列の高レベル発現ならびに交差反応抗体の収集および精製に適する条件下で該宿主を維持する。
【0096】
これらの発現ベクターは、典型的に、エピソームとして、若しくは宿主染色体DNAの一体部分としてのいずれかで宿主生物体中で複製する。一般に、発現ベクターは、所望のDNA配列で形質転換された細胞の検出を可能にするための選択マーカー、例えばアンピシリン耐性若しくはハイグロマイシン耐性を含有する。
【0097】
大腸菌(Escherichia coli)は本発明のDNA配列をクローン化するのにとりわけ有用な1種の原核生物宿主である。酵母のような微生物もまた発現に有用である。サッカロミセス属(Saccharomyces)は好ましい酵母宿主であり、適するベクターは発現制御配列、複製起点、終止配列などを所望のとおり有する。典型的なプロモーターは、3−ホスホグリセリン酸キナーゼプロモーター、および他の解糖酵素からのプロモーターを包含する。誘導可能な酵母プロモーターは、とりわけ、アルコール脱水素酵素、イソチトクロームC、ならびに麦芽糖およびガラクトース利用を司る酵素からのプロモーターを包含する。
【0098】
哺乳動物細胞は、免疫グロブリン若しくはそれらのフラグメントをコードするヌクレオチドセグメントを発現するのに好ましい宿主である。Winnacker、From Genes to Clones、(VCH Publishers、ニューヨーク、1987)を参照されたい。無傷の異種タンパク質を分泌することが可能な多数の適する宿主細胞株が当該技術分野で開発されており、そしてCHO細胞株、多様なCOS細胞株、HeLa細胞、L細胞および骨髄腫細胞株を包含する。好ましくは細胞はヒト以外である。これらの細胞の発現ベクターは、複製起点、プロモーター、エンハンサー(Queenら、Immunol.Rev.、89:49−68(1986))のような発現制御配列、ならびにリボソーム結合部位、RNAスプライス部位、ポリアデニル化部位のような必要なプロセシング情報部位、および転写終止配列を包含し得る。好ましい発現制御配列は、内因性遺伝子、サイトメガロウイルス、SV40、アデノウイルス、ウシパピローマウイルスなど由来のプロモーターである。Coら、J.Immunol.、148:1149−54(1992)を参照されたい。
【0099】
あるいは、抗体コーディング配列は、トランスジェニック動物のゲノムへの導入および該トランスジェニック動物の乳中でのその後の発現のため導入遺伝子に組み込み得る(例えば、米国特許第US 5,741,957号、同第US 5,304,489号、同第US 5,849,992号明細書を参照されたい)。適する導入遺伝子は、カゼイン若しくはβラクトグロブリンのような乳腺特異的遺伝子からのプロモーターおよびエンハンサーと作動可能な連結にあるLおよび/若しくはH鎖のコーディング配列を包含する。
【0100】
目的のDNAセグメントを含有するベクターを、細胞宿主の型に依存して公知の方法により宿主細胞に移入し得る。例えば、塩化カルシウムトランスフェクションは原核生物細胞に一般に利用される一方、リン酸カルシウム処理、電気穿孔法、リポフェクション、遺伝子銃若しくはウイルスに基づくトランスフェクションを他の細胞宿主に使用し得る。哺乳動物細胞を形質転換するのに使用される他の方法は、ポリブレン、プロトプラスト融合、リポソームおよび微小注入法の使用を包含する。トランスジェニック動物の製造のため、導入遺伝子を受精卵母細胞に微小注入し得るか若しくは胚性幹細胞のゲノム中に組み込み得、そしてこうした細胞の核を除核卵母細胞に移入し得る。
【0101】
一旦発現されれば、抗体は、HPLC精製、カラムクロマトグラフィー、ゲル電気泳動などを包含する当該技術分野で既知の標準的手順に従って精製し得る(全般として、Scopes、Protein Purification(Springer−Verla
g、ニューヨーク、1982)を参照されたい)。
【0102】
7.ナノボディ
ナノボディは単一可変ドメイン(VHH)および2個の定常ドメイン(CH2およびCH3)を含有するH鎖抗体である。クローン化かつ単離されたVHHドメインは、元のH鎖抗体の完全な抗原結合能力を持つ安定なポリペプチドである。
【0103】
8.他の抗体
抗体は、米国特許出願公開第20040038304号、同第20070020685号、同第20060257396号、同第20060160184号、同第20060134098号、同第20050255552号、同第20050008625号、同第20040132066号、同第20040038317号、同第20030198971号、および同第20030157579号明細書に記述されるもののような方法によってもまた同定かつ/若しくは製造しうる。
【0104】
B.他の剤
剤は天然に存在する若しくは合成の分子であり得る。スクリーニングされるべき剤は、例えば海洋微生物、藻類、植物および真菌のような天然の供給源からもまた得ることができる。例えば、米国特許第US 6,096,707号明細書は、クサリヘビ、ジャララカ(Bothrops jararaca)からのメタロプロテイナーゼ、ジャララギン由来のペプチドを提供する。これらのペプチドはアミノ酸モチーフArg−Lys−Lys(RKK)を含有し、そしてヒトα2β1インテグリンのコラーゲンとの相互作用を低下させる。あるいは、スクリーニングされるべき剤は、ペプチド若しくは小分子を包含する剤のコンビナトリアルライブラリーから、または産業で、例えば化学、製薬、環境、農業、海洋、化粧品、薬物およびバイオテクノロジー産業により合成される化合物の既存のレパートリーからであり得る。剤は、例えば、医薬品、治療薬、環境、農業、若しくは工業的剤、汚染物質、薬用化粧品、薬物、有機化合物、脂質、グルココルチコイド、抗生物質、ペプチド、タンパク質、糖、炭水化物およびキメラ分子を包含し得る。
【0105】
多様な方法がペプチドライブラリーを製造するのに利用可能である(例えば、Lamら、Nature、354:92、1991および第WO 92/00091号明細書;Geysenら、J.Immunol.Meth.、102:259(1987);Houghtenら、Nature、354:84(1991);第WO 92/09300号明細書;ならびにLeblら、Int.J.Pept.Prot.Res.、41:201(1993)を参照されたい)。ペプチドライブラリーはファージディスプレイ法によってもまた生成し得る。例えばDevlin、第W0 91/18980号明細書を参照されたい。
【0106】
段階的様式で合成し得る多くの型の化合物についてコンビナトリアルライブラリーを製造し得る(例えば、EllmanとBunin、J.Amer.Chem.Soc.、114:10997、1992(ベンゾジアゼピン鋳型)、第WO 95/32184号明細書(オキサゾロンおよびアミニジン鋳型)、第WO 95/30642号明細書(ジヒドロベンゾピラン鋳型)ならびに第WO 95/35278号明細書(ピロリジン鋳型)を参照されたい)。化合物のライブラリーは通常固相化学により合成する。しかしながら溶液相ライブラリー合成もまた有用である。コンビナトリアル合成の戦略はDolleとNelson、J.Combinatorial Chemistry、1:235−282(1999)(全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)により記述されている。合成は、典型的には、異なる単量体若しくは他の成分を合成の各回に添加する周期的様式で実施する。いくつかの方法は初期プールを連続的に分画することにより実施する。例えば、第1回の合成は全部の支持体上で実施する。支持体
をその後2プールに分割し、そして別個の合成反応を各プールで実施する。該2プールをその後それぞれさらなる2プールにさらに分割し、などである。他の方法は分割および再プールの双方を使用する。例えば、初回の合成後に、化合物のプールを第2回の別個の合成のため2個に分割する。その後、別個のプールからのアリコートを第3回の合成のため再組合せする。分割およびプール法は混合化合物のプールをもたらす。これらの方法は下により詳細に記述されるとおり標識にとりわけ従いやすい。こうした方法により生成されるライブラリーの大きさは、2種の異なる化合物から106若しくは1010またはその間のいずれかの範囲まで変動し得る。
【0107】
コードされるライブラリーの製造法は、Needelsら、Proc.Natl.Acad.Sci.U.S.A.、90:10700(1993);Niら、J.Med.Chem.、39:1601(1996)、第WO 95/12608号、第WO 93/06121号、第WO 94/08051号、第WO 95/35505号、および第WO 95/30642号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)を包含する多様な刊行物に記述されている。コードされるライブラリーの合成方法は、典型的に、ランダムコンビナトリアルアプローチならびに単量体単位の化学的および/若しくは酵素的集成を必要とする。例えば、該方法は、典型的に、(a)複数の反応容器のあいだで複数の固体支持体を割り当てる段階;(b)各異なる反応容器中の異なる第一の単量体および標識の組合せを使用して第一の単量体および第一の標識を各反応容器中の支持体に結合する段階;(c)支持体をプールする段階;(d)複数の反応容器のあいだで該支持体を割り当てる段階;(e)各異なる反応容器中で、異なる第二の単量体および第二の標識の組合せを使用して、第一の単量体に第二の単量体を結合する段階、および固体支持体若しくは第一の標識のいずれかに第二の標識を結合する段階;ならびに、場合によっては、異なる標識および異なる単量体を用いて結合および割り当て段階を1ないし20回若しくはそれ以上反復する段階を包含する。単量体の組は段階ごとに拡張若しくは縮小し得るか;または、単量体の組は次の段階について完全に変更し得る(例えば、一段階でアミノ酸、別の段階でヌクレオシド、別の段階で炭水化物)。例えばペプチド合成のための単量体単位は単一アミノ酸若しくはより大きいペプチド単位または双方を包含し得る。
【0108】
こうした方法により合成可能な化合物は、ポリペプチド、βターン模倣物、多糖、リン脂質、ホルモン、プロスタグランジン、ステロイド、芳香族化合物、複素環化合物、ベンゾジアゼピン、オリゴマーN−置換グリシンおよびオリゴカルバメートを包含する。製造されるコンビナトリアルライブラリーは商業的供給源(例えばChemRx、カリフォルニア州サウスサンフランシスコ)からもまた入手可能である。
【0109】
コンビナトリアルライブラリーおよび他の化合物は、α2β1、α6β1若しくはαvβ1インテグリンまたはラミニンに結合するそれらの能力を測定することにより適合性について最初にスクリーニングする。上述された付加的なスクリーニング手順もまた使用し得る。
【0110】
それらの立体異性体、若しくはそれらの立体異性体の混合物、またはそれらの製薬学的に許容できる塩の形態を包含する、式IaおよびIbの化合物は、LTPのAβ誘導性阻害の抑制方法で有用であり
【0111】
【化4】
【0112】
ここで
X1およびX3は窒素若しくは炭素から独立に選択され;
R1は
【0113】
【化5】
【0114】
から選択され;
ここで、上の複素環は、NH2、ハロゲン、NO2、CN、CF3、C1−C4アルコキシ、C1−C6アルキルおよびC3−C7シクロアルキルよりなる群から選択される0〜2置換基で場合によっては置換されており;
Uは−(CH2)n−、−(CH2)tQ(CH2)m−および−C(=O)(CH2)n−1−から選択され、ここでメチレン基の1個は場合によってはR7で置換されており;
Qは、1,2−フェニレン、1,3−フェニレン、2,3−ピリジニレン、3,4−ピリジニレンおよび2,4−ピリジニレンから選択され;
R6はH、C1−C4アルキルおよびベンジルから選択され;
R7はC1−C6アルキル、C3−C7シクロアルキル、C4−C11シクロアルキルアルキル、アリール、アリール(C1−C6アルキル)、ヘテロアリールおよびヘテロアリール(C1−C6アルキル)から選択され;
R10は、H、ハロゲン、CO2R17、CONR17R20、0〜1個のR15若しくは0〜1個のR21で置換されているC1−C6アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR15若しくは0〜1個のR21で置換されているC3−C7シクロアルキル、0〜1個のR15若しくは0〜1個のR21で置換されているC4−C11シクロアルキルアルキル、および0〜1個のR15若しくは0〜2個のR11若しくは0〜1個のR21で置換されているアリール(C1−C6アルキル)−から選択され;
R11は、H、ハロゲン、CF3、CN、NO2、ヒドロキシ、NR2R3、0〜1個のR21で置換されているC1−C4アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR21で置換されているアリール、0〜1個のR21で置換されているアリール(C1−C6アルキル)−、0〜1個のR21で置換されている(C1−C4アルコキシ)カルボニル、0〜1個のR21で置換されている(C1−C4アルキル)カルボニル、0〜1個のR21で置換されているC1−C4アルキルスルホニル、および0〜1個のR21で置換されているC1−C4アルキルアミノスルホニルから選択され;
Wは−C(=O)−N(R13)−であり;
Xは−CH(R14)−CH(R15)−であり;
R13はHおよびCH3から選択され;
R14は、H、C1−C10アルキル、アリールおよびヘテロアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ハロ、シアノ、アミノ、CF3およびNO2から選択される0〜3置換基で場合によっては置換されており;
R15はHおよびR16から選択され;
Yは−COR19であり;
R16は
−NH(R20)−C(=O)−R17、
−N(R20)−C(=O)−R17、
−N(R20)−C(=O)−NH−R17、
―N(R20)SO2−R17、および
−N(R20)SO2−N(R20)R17
から選択され、
R17は、C1−C10アルキル、C3−C11シクロアルキル、アリール(C1−C6アルキル)−、(C1−C6アルキル)アリール、ヘテロアリール(C1−C6アルキル)−、(C1−C6アルキル)ヘテロアリール、ビアリール(C1−C6アルキル)−、ヘテロアリール若しくはアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ヘテロアリール、ハロ、シアノ、アミノ、CF3およびNO2よりなる群から選択される0〜3置換基で場合によっては置換されており;
R19は−O−(CH2)kN+(R22)(R23)(R24)Z−であり;
Z−は、ハロゲン化イオン、重硫酸イオン、硫酸イオン、リン酸水素イオン、リン酸イオン、トルエンスルホン酸イオン、メタンスルホン酸イオン、エタンスルホン酸イオン、酢
酸イオン、トリフルオロ酢酸イオン、クエン酸イオン、シュウ酸イオン、コハク酸イオンおよびマロン酸イオンから選択される製薬学的に許容できる陰イオンであり;
R22、R23およびR24はH、C1−C4アルキルおよびC4−C11シクロアルキルアルキルから独立に選択されるか;
あるいは、R22およびR23は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する5〜7員の複素環系を形成し得、ならびにR24は上のとおり定義されるか、または、R22、R23およびR24は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する複素二環系を形成し得;
R20はHおよびCH3から選択され;
R21はCOOHおよびNR62から選択され;
kは2であり;
mは0および1から選択され;
nは1〜4であり;ならびに
tは0および1から選択される。
【0115】
式Iaの化合物の例は、限定されるものでないが式II:
【0116】
【化6】
【0117】
の化合物を挙げることができ;ここでR19は−H、−CH3および−CH2CH2N+(CH3)3から選ばれる。一態様において、H19は−Hである(式IIの化合物はSM256である)。
【0118】
LTPのAβ誘導性阻害の抑制方法で有用な付加的な化合物は、米国特許第6,214,834号明細書に開示される化合物を包含する。その特許は、αvβ3に拮抗するためのSM256(式II)および他の化合物の使用を開示する。従って、1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する化合物を同定するためにαvβ3拮抗剤をスクリーニングしうる。そのようにスクリーニングされうる例示的αvβ3拮抗剤は、限定されるものでないが米国特許第6,214,834号明細書に開示されるαvβ3拮抗剤を挙げることができる。
【0119】
C.遺伝子抑制剤
遺伝子発現を抑制する剤を使用して、インテグリンサブユニットβ1、α2、α6若しくはαvをコードする遺伝子の発現を抑制し得る。アンチセンス剤もまた、ラミニンのようなそれに対するある種のリガンドの発現を抑制するのに使用し得る。ラミニン発現の抑制は、ラミニンに対する抗体での処置に類似の効果を達成し得る。標的細胞若しくは患者への本発明のアンチセンス試薬の投与は、上のインテグリン遺伝子若しくはそのリガンド
の1種の低下された活性をもたらす。アンチセンスポリヌクレオチドに関する一般的方法については、例えばAntisense RNA and DNA、(1988)、D.A.Melton編、Cold Spring Harbor Laboratory、ニューヨーク州コールドスプリングハーバー);Dagleら、Nucleic Acids Research、19:1805(1991);Uhlmannら、Chem.Reviews、90:543−584(1990)を参照されたい。リボザイムは遺伝子発現を抑制し得る別のアンチセンス剤である。
【0120】
アンチセンスオリゴヌクレオチドは、センスmRNAに結合しかつその翻訳を妨害すること、mRNAをヌクレアーゼ消化に対し感受性にすること、転写を妨害すること、RNA前駆体のプロセシング若しくは局在化を妨害すること、mRNAの転写を抑制すること、若しくはいくつかの他の機序により作用することにより抑制を引き起こし得る。アンチセンス分子が発現を低下させる特定の機序は重要でない。
【0121】
典型的に、アンチセンスポリヌクレオチドは、遺伝子のmRNAからの配列に特異的にハイブリダイズする最低7ないし10ないし典型的に20若しくはそれ以上のヌクレオチドのアンチセンス配列を含んでなる。数種のアンチセンスポリヌクレオチドは、長さが約10から約50ヌクレオチドまで、若しくは長さが約14から約35ヌクレオチドまでである。数種のアンチセンスポリヌクレオチドは約100ヌクレオチド未満若しくは約200ヌクレオチド未満のポリヌクレオチドである。一般に、アンチセンスポリヌクレオチドは安定な二重鎖を形成するのに十分長くあるべきであるが、しかし、所望の場合はin vivoで投与するために送達様式に依存して十分に短くあるべきである。標的配列への特異的ハイブリダイゼーションに必要とされるポリヌクレオチドの最小長さは、とりわけ、G/C含量、不適正塩基(あれば)の配置、標的ポリヌクレオチドの集団に関しての該配列の全体的差違、およびポリヌクレオチドの化学的性質(例えばメチルホスホネートバックボーン、ペプチド核酸、ホスホロチオエート)のようないくつかの因子に依存する。
【0122】
相補核酸分子をハイブリダイズするのに適する条件は当業者に公知である。例えば、典型的な高緊縮性条件下のハイブリダイゼーションは、5×SSPE、5×Denhart溶液、0.5%SDS(w/v)および100μg/mlサケ精子DNAを含有する混合物中で実施しうる。DNAは、約68℃で指定される時間ハイブリダイズさせる。典型的には膜若しくはフィルターに結合されているハイブリダイズされたDNAをその後、2×SSPE、0.1%SDS(w/v)中室温で10分間2回洗浄する。膜(若しくはフィルター)をその後、1×SSPE、0.1%SDS(w/v)の溶液に68℃で15分間、そして最後に1×SSPE、0.1%SDS(w/v)の溶液に68℃で15分間浸漬する。
【0123】
特異的ハイブリダイゼーションを確実にするため、アンチセンス配列は標的mRNA若しくはそれをコードする遺伝子に少なくとも実質的に相補的である。数種のアンチセンス配列はそれらの意図している標的配列に正確に相補的である。アンチセンスポリヌクレオチドは、しかしながら、RNA若しくはその遺伝子に対応する適切な標的配列への特異的結合が該ポリヌクレオチドの機能的特性として保持される限りは、ヌクレオチドの置換、付加、欠失、トランジション、転位若しくは修飾、または他の核酸配列若しくは核酸以外の部分もまた包含し得る。
【0124】
数種のアンチセンス配列はmRNAの相対的に接近可能な配列に相補的である(例えば二次構造を相対的に欠く)。これは、予測されるRNA二次構造を例えばMFOLDプログラム(Genetics Computer Group、ウィスコンシン州マディソン)を使用して解析すること、および当該技術分野で既知であるとおりin vitro若しくはin vivoで試験することにより決定し得る。効果的なアンチセンス組成物
の別の有用な同定方法はオリゴヌクレオチドのコンビナトリアルアレイを使用する(例えば、Milnerら、Nature Biotechnology、15:537(1997)を参照されたい)。
【0125】
遺伝子発現を阻害するための一技術は、阻害的RNA(RNAi)ともまた称される二本鎖RNAの細胞中への導入を必要とする。RNAiは、二本鎖RNA分子を形成するよう相互にアニーリングされたRNAの2本の相補鎖(センス鎖およびアンチセンス鎖)を含んでなる。RNAiは、典型的には阻害の標的とされている遺伝子のエキソンすなわちコーディング配列に由来する。RNAiは該RNAi分子の配列に相補的なmRNAの破壊をもたらす。RNAiおよび生存生物体でのそれらの使用の例は、例えばFireら、Nature、391:806−811(1998);Nykaenenら、Cell、107:309−321(2001);および第WO 01/29058号、第WO 01/75164号および第WO 99/32619号明細書により記述される。いくつかの方法において、RNAiは約100bpと1000bpの間、例えば約100、200、300、400、500、600、700、800、900、1000若しくはそれ以上の塩基対である。いくつかの方法においてRNAiはエキソン由来である。他の方法において、RNAiはイントロン若しくはシグナル伝達配列由来である。
【0126】
いくつかの方法において、アンチセンスポリヌクレオチドは、アンチセンス配列に加え、アンチセンス配列の発現をもたらすプロモーターおよび他の制御配列を包含する配列を有する。プロモーターならびに好ましくは終止およびポリアデニル化シグナルが適正に配置されていることを条件に、非コーディング鎖に対応する挿入された配列の鎖が転写され、そしてアンチセンスオリゴヌクレオチドとして作用する。いくつかの方法において、該ポリヌクレオチドはアンチセンス配列より本質的になるか若しくはアンチセンス配列である。アンチセンス核酸(DNA、RNA、修飾、アナログなど)は、本明細書に開示される化学合成および組換え法のような核酸のいずれかの適する製造方法を使用して作成し得る。例えば、アンチセンスRNA分子はデノボ化学合成若しくはクローニングにより製造し得る。
【0127】
β1、α2、α6若しくはαvインテグリンサブユニットをコードする遺伝子の発現を抑制するため、ジンクフィンガータンパク質を、アンチセンスポリヌクレオチドの代替として若しくはそれに加えて使用し得る。ジンクフィンガータンパク質は、ラミニンのようなこれらのインテグリンサブユニットのある種のリガンドの発現を抑制するのにもまた使用し得る。ジンクフィンガータンパク質は、本方法で剤としてそれら自身を使用し得るフィブロネクチンのような他のリガンドの発現を活性化する若しくは高めるのにもまた使用し得る。ジンクフィンガータンパク質は標的遺伝子内のいずれかの所望の標的部位に結合するよう工作若しくは選択し得る。いくつかの方法において標的部位はプロモーター若しくはエンハンサー内にある。他の方法において標的部位は構造遺伝子内にある。いくつかの方法において、ジンクフィンガータンパク質はヒトKOX−1タンパク質からのKRAB抑制領域のような転写リプレッサーに連結される(Thiesenら、New Biologist、2、363−374(1990);Margolinら、Proc.Natl.Acad.Sci.U.S.A.、91、4509−4513(1994));Pengueら、Nucl.Acids Res.、22:2908−2914(1994);Witzgallら、Proc.Natl.Acad.Sci.U.S.A.、91、4514−4518(1994)。活性化を達成するために好ましいドメインは、HSV VP16活性化ドメイン(例えばHagmannら、J.Virol.、71:5952−5962(1997)を参照されたい)核ホルモン受容体(例えばTorchiaら、Curr.Opin.Cell.Biol.、10:373−383(1998)を参照されたい);核因子κBのp65サブユニット(BitkoとBarik、J.Virol.、72:5610−5618(1998)およびDoyleとHunt、Neuroreport、8:2937−2942(1997));Liuら、Cancer Gene Ther.、5:3−28(1998))、若しくはVP64のような人工的キメラ機能ドメイン(Seifpalら、EMBO J.、11:4961−4968(1992))を包含する。ジンクフィンガータンパク質により標的を定めるのに適する標的部位の選択方法、および選択された標的部位に結合するためのジンクフィンガータンパク質の設計方法は、第WO 00/00388号明細書に記述されている。標的に結合するジンクフィンガータンパク質のファージディスプレイを使用する選択方法は、第EP 95908614A号明細書により記述されている。内因性遺伝子を調節するためのジンクフィンガータンパク質の使用方法は第WO 00/00409号明細書に記述されている。ジンクフィンガータンパク質は、タンパク質として、若しくはジンクフィンガーをコードしかつ適切な制御配列を有する核酸の形態でのいずれでも投与し得る。
【0128】
D.治療薬をコードする核酸
抗体若しくは他のペプチド試薬は、抗体鎖若しくはペプチドをコードする核酸の形態で投与し得る。こうした核酸は、典型的には、患者の意図している標的細胞中での該DNAセグメントの発現を可能にするプロモーターおよびエンハンサーのような調節エレメントに連結されている。L若しくはH鎖免疫グロブリン遺伝子からのプロモーターおよびエンハンサーエレメント、またはサイトメガロウイルス(CMV)主要中初期プロモーター(major intermediate early promoter)およびエンハンサーが直接発現に適する。いくつかの方法において、脳中で発現を引き起こすプロモーターを使用する。血小板由来増殖因子(PDGF)、プリオン若しくは神経エノラーゼのプロモーターのようなプロモーターが適する。
【0129】
連結された調節エレメントおよびコーディング配列はしばしば1ベクターにクローン化される。二本鎖抗体の投与のため、該2鎖を同一若しくは別個のベクターにクローン化し得る。
【0130】
レトロウイルス系(例えばLawrieとTumin、Curr.Opin.Genet.Develop.、2:102−109(1993)を参照されたい);アデノウイルスベクター(例えばBettら、J.Virol.、67:5911(1993)を参照されたい);アデノ随伴ウイルスベクター(例えばZhouら、J.Exp.Med.、179:1867−75(1994)を参照されたい)、ワクシニアウイルスおよびトリポックスウイルスを包含するポックス科からのウイルスベクター、シンドビスおよびセムリキ森林ウイルス(例えばDubenskyら、J.Virol.、70:508−19(1996)を参照されたい)、ベネズエラウマ脳炎ウイルス(米国特許第US 5,643,576号明細書を参照されたい)由来のもののようなアルファウイルス属、水疱性口内炎ウイルス(第WO 96/34625号明細書を参照されたい)のようなラブドウイルス、ならびにパピローマウイルス(Oheら、Human Gene Therapy、6:325−33(1995);Wooら、第WO 94/12629号明細書;およびXiaoとBrandsma、Nucleic Acids.Res.、24:2630−22(1996))からのウイルスベクターを包含する多数のウイルスベクター系が利用可能である。
【0131】
DNAはリポソーム中にパッケージングし得る。適する脂質および関連アナログは、米国特許第US 5,208,036号、同第5,264,618号、同第5,279,833号および同第5,283,185号明細書により記述されている。免疫原をコードするベクターおよびDNAはまた微粒子担体に吸着若しくはそれらと会合もし得、それらの例はポリメチルメタクリレートポリマー、ポリラクチドおよびポリ(ラクチドコグリコリド)を包含する。
【0132】
遺伝子治療ベクター若しくは裸のDNAは、典型的に全身投与(例えば静脈内、腹腔内、鼻、胃、皮内、筋肉内、くも膜下腔内、皮下若しくは頭蓋内注入)または局所適用(例えば米国特許第US 5,399,346号明細書を参照されたい)による個々の患者への投与によりin vivoで送達し得る。こうしたベクターはブピバカインのような促進剤をさらに包含し得る(米国特許第US 5,593,970号明細書)。DNAは遺伝子銃を使用してもまた投与し得る。XiaoとBrandsma、上記を参照されたい。DNAを微細な金属ビーズの表面上に沈殿させる。微小発射体をショック波若しくは膨張するヘリウムガスで加速し、そして数細胞層の深さまで組織を貫通させる。例えば、Agacetus,Inc.、ウィスコンシン州ミドルトンにより製造されるAccelTM遺伝子送達装置が適する。あるいは、裸のDNAは、単純に化学若しくは機械的刺激で皮膚上にDNAをスポットすることにより、皮膚を通り血流に移行し得る(第WO 95/05853号明細書を参照されたい)。
【0133】
さらなる一変形において、核酸を、個々の患者から外植した細胞(例えばリンパ球、骨髄吸引物、組織生検)若しくは普遍的ドナーの造血幹細胞のような細胞にex vivoで送達し得、次いで、通常はベクターを組み込んだ細胞についての選択後に細胞を患者に再移植し得る。
【0134】
E.LTPのAβ媒介性阻害を抑制する剤の同定
インテグリンサブユニットαvに結合する小分子剤は、1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下で投与しうる。適する小分子剤は、例えば、インテグリンサブユニットαv結合アッセイおよびLTPのアミロイド媒介性阻害抑制アッセイを含んでなる方法、若しくはLTPのアミロイド媒介性阻害抑制アッセイを含んでなるがしかしインテグリンサブユニットαv結合アッセイを含まない方法により同定する。
【0135】
インテグリンサブユニットαv結合アッセイは、例えば、単量体若しくは二量体いずれかとしてのインテグリンサブユニットαvへの剤の特異的結合を同定するいずれかのアッセイである。例示的アッセイは、支持体への直接若しくは間接結合によるように固定されうるインテグリンサブユニットαvに結合されている標識した剤の使用によるように結合を直接検出する。さらなる例示的アッセイは結合を間接的に検出する。一態様において競合結合アッセイを使用する。競合結合は、例えば、本明細書に開示されるか若しくは当該技術分野で既知のもののような別のインテグリンサブユニットαv結合剤、または本明細書に開示されるアッセイにより事前に同定された結合剤を使用して実施しうる。インテグリンサブユニットαv結合アッセイでの使用に適する結合剤は、限定されるものでないが、フィブロネクチン若しくはスーパーフィブロネクチン、モノクローナル若しくはポリクローナル抗体、18C7、20A9および17E6から選ばれる抗体と同一のエピトープを認識する抗体、ならびに18C7、20A9および17E6から選ばれる抗体、インテグリンサブユニットαvへの結合について18C7、20A9および17E6から選ばれる抗体と競合する剤、ならびにSM256のような式1の化合物を挙げることができる。
【0136】
LTPのアミロイド媒介性阻害抑制アッセイは、限定されるものでないがLTPを実験的に誘導する系を使用するアッセイを挙げることができる。適する系は、in vivo、または海馬領域若しくは海馬の小領域を含んでなる脳薄片のようなin vitroでありうる。例示的アッセイを実施例10〜13に記述する。
【0137】
それらの実施例で、LTPをHFS若しくは薬理学的刺激により誘導する。LTPは、HFS若しくは薬理学的刺激の投与の前、それらと同時に若しくは後に該系に添加されるAβペプチドにより阻害される。候補剤によるLTPのアミロイド媒介性阻害の抑制をその後、該候補剤の存在および非存在下のAβペプチドによるLTPの阻害を比較すること
によりアッセイする。抑制の程度は、対照系(Aβペプチドの非存在下のLTPの誘導後の系および/若しくはAβペプチドによるLTPの阻害後の系のいずれでもあり得る)のHFS若しくは薬理学的刺激後に観察される興奮性シナプス後場電位(EPSP)の振幅に関して測定する。
【0138】
IV.第二の剤
本発明は1種若しくはそれ以上の剤および1種若しくはそれ以上の第二の剤の共投与にさらに向けられる。例えば、適する第二の剤は、限定されるものでないが、Aβ産生の阻害剤、Aβ沈着の阻害剤、Aβ消失のメディエーター、アミロイド斑消失のメディエーター、Aβ神経毒性の阻害剤、Aβ凝集の阻害剤、Aβ解離のメディエーター、およびAβに対する抗体、ならびにそれらのいずれかの組合せから選択される第二の剤を挙げることができる。Aβ産生の適する阻害剤は、限定されるものでないがγ分泌酵素阻害剤およびβ分泌酵素阻害剤を挙げることができる。
【0139】
例示的γ分泌酵素阻害剤は、限定されるものでないが、米国特許第6,992,081号、同第6,982,264号、同第6,962,934号および同第6,610,493号明細書、ならびに米国特許出願公開第20060287306号、同第20060154926号、同第20050159460号、同第20020016320号および同第20030148392号明細書に開示されるもののようなγ分泌酵素阻害剤を挙げることができる。
【0140】
例示的β分泌酵素阻害剤は、限定されるものでないが、米国特許第7,115,410号、同第7,109,017号、同第7,067,271号、同第6,864,240号、同第6,852,482号、同第6,627,739号、同第6,321,163号、同第6,221,645号、同第5,942,400号および同第5,744,346号明細書、ならびに米国特許出願公開第20050196839号、同第20050182138号、同第20050177888号、同第20050170489号、同第20050164327号、同第20050164294号、および同第200402655965号明細書に開示されるもののようなβ分泌酵素阻害剤を挙げることができる。
【0141】
Aβに対する例示的抗体は、限定されるものでないが、米国特許第7,014,855号、同第6,982,084号、同第6,972,127号、同第6,962,707号、同第6,946,135号、同第6,913,745号、同第6,905,686号、同第6,890,535号、同第6,875,434号、同第6,866,850号、同第6,866,849号、同第6,818,218号、同第6,808,712号、同第6,787,637号、同第6,787,523号、同第6,787,144号、同第6,787,143号、同第6,787,140号、同第6,787,139号、同第6,787,138号、同第6,761,888号、同第6,750,324号、同第6,743,427号および同第6,710,226号明細書に開示されるもののようなAβに対する抗体を挙げることができる。
【0142】
V.長期増強
長期増強はin vivo若しくはin vitroで自然に発生し、およびin vitro若しくはin vivoモデル系で実験的に誘導しうる。本明細書に開示されるある種の方法は、該実験系のAβペプチドへの投与若しくは曝露により阻害し得るLTPの実験的誘導を企図している。それらの方法では剤を使用してAβペプチドによるLTPのその阻害を実験的に抑制する。天然に存在するLTPは多様なアミロイド形成疾患状態の発症の経過でAβペプチドにより阻害される。本明細書に開示される剤は、例えば、AβペプチドによるLTPの阻害をin vivoで処置かつ/若しくは阻害するため、ならびに/または該疾患状態を処置かつ/若しくは予防するための製薬学的製剤として患者
に投与しうる。
【0143】
LTPはHFSの送達若しくは薬理学的方法により実験的に誘導しうる。LTP阻害の実験モデルに、実験操作に応答してLTPを表す神経回路が提供される。それはマウス若しくはラットのようなげっ歯類若しくは霊長類のような哺乳動物のような動物から選ばれた生物体で行い得る。in vitroで培養した霊長類若しくはげっ歯類脳薄片のような哺乳動物脳薄片、またはin vivoで哺乳動物の脳中でLTPを実験的に誘導するための技術が当該技術分野で既知である。
【0144】
LTPの強さは、誘導後の指定される時点で基礎(誘導の非存在下)に関して観察されるEPSP上昇の大きさにより定量化し得る。その大きさは150%のような基礎に関するパーセンテージとして表しうる。Aβペプチドの投与後に観察されるLTP阻害は、基礎に関するEPSP上昇の大きさの低下をもたらす。
【0145】
例えば、Aβペプチドは、HFS後に観察されるEPSPが基礎と統計学的に異ならない点までLTPを完全に阻害する。その阻害が抑制される場合、LTPはAβの存在下であってさえ観察し得る。その抑制は、Aβの非存在下でLTPの誘導後に観察されるEPSPの大きさに関して定量化し得る。例えば、抑制は完全であることができ、その結果、Aβおよびαv結合剤の存在下でLTPの誘導後のデータ組全体で観察されるEPSPの大きさは、Aβの非存在下でLTPの誘導後に観察されるEPSPの大きさと統計学的に識別可能である。他の態様において、Aβおよびαv結合剤の存在下でLTPの誘導後のデータ組全体で観察されるEPSPの大きさは、例えば、Aβの非存在下でLTPの誘導後に観察されるEPSPの大きさの25%以上、50%以上、75%以上、85%以上、90%以上若しくは95%以上である。
【0146】
VI.処置可能な患者
本方法は、進行性記憶障害を特徴とする疾患若しくは障害を包含する記憶障害若しくは記憶障害の可能性を特徴とする疾患若しくは障害の予防的若しくは治療的処置に有用である。例示的疾患若しくは障害は、限定されるものでないが、アミリン若しくはAβペプチドのようなアミロイドタンパク質の沈着物の存在を特徴とするアミロイド形成疾患および状態を挙げることができる。こうした疾患は、アルツハイマー病、ダウン症候群および認知障害、II型糖尿病、パーキンソン病、びまん性レビー小体病、遺伝性若しくは全身性アミロイドーシスのようなアミロイドーシス、ならびにプリオン感染により全部若しくは部分的に引き起こされる疾患を包含する。
【0147】
処置可能な患者は、疾患の危険にさらされているがしかし症状を示さない個体、ならびに現在症状を示している患者を包含する。アルツハイマー病の場合、事実上誰でも彼若しくは彼女が十分に長く生きる場合にアルツハイマー病に罹る危険にさらされている。本方法は、アルツハイマー病の既知の遺伝的危険性を有する個体にとりわけ有用である。こうした個体は、この疾患を経験した親族を有する者、および遺伝子若しくは生化学的マーカーの分析によりその危険性が決定される者を包含する。アルツハイマー病に対する危険性の遺伝子マーカーは、APP遺伝子中の突然変異、例えばそれぞれハーディ型およびスウェーデン型突然変異と称される位置717ならびに位置670および671の突然変異を包含する(Hardy、TINS、上記を参照されたい)。危険性の他のマーカーは、プレセニリン遺伝子PS1およびPS2ならびにApoE4中の突然変異、ADの家族歴、高コレステロール血症若しくは動脈硬化症である。現在アルツハイマー病に罹っている個体は、特徴的な認知症ならびに上述された危険因子の存在から認識し得る。加えて、ADを有する個体を同定するための多数の診断試験が利用可能である。これらは脳脊髄液(CSF)のτおよびAβ42濃度の測定を包含する。上昇されたτおよび低下されたAβ42濃度はADの存在を示す。アルツハイマー病に罹っている個体はADRDA基準によっ
てもまた診断し得る。無症候性患者で処置はいかなる齢(例えば約10、約20、約30歳)でも開始し得る。通常、しかしながら患者が約40、約50、約60、約70、約80若しくは約90歳に達するまで処置を開始することは必要でない。処置は、典型的にはある期間にわたる複数投薬量を必要とする。ダウン症候群患者の場合には、母親に治療薬を投与することにより出産前に処置を開始し得るか、若しくは処置を出生直後に開始しうる。
【0148】
VII.処置計画
予防的応用において、製薬学的組成物若しくは医薬品は、疾患、その合併症および該疾患の発症の間に見つかる中間的病理学的表現型の生化学的、組織学的および/若しくは行動的症状を包含する該疾患の危険性を排除若しくは低下、その重症度を低下、またはその発症を遅延させるのに十分な量で、アミロイド形成疾患に感受性の若しくはそうでなければそれを発症する危険にさらされている患者に投与する。
【0149】
治療的応用において、組成物若しくは医薬品は、その合併症および中間的病理学的表現型を包含する該疾患の症状(生化学的、組織学的および/若しくは行動的)を治癒若しくは少なくとも部分的に停止するのに十分な量で、こうした疾患が疑われる若しくは既に罹っている患者に投与する。治療的若しくは予防的処置を達成するのに十分な量を治療的若しくは予防的有効用量と定義する。治療計画において、該剤は通常、該疾患の症状が消失若しくは有意に低下するまで間隔を置いて投与する。場合によっては、再発を予防するために投与を継続し得る。予防計画において、剤は通常間隔を置いて、いくつかの例では患者の生涯の残りの間もまた投与する。処置は、投与した剤の濃度をアッセイすること若しくは患者の応答をモニターすることによりモニターし得る。応答は、ADRDA基準、および患者の脳中の斑の画像化によりモニターし得る(第WO 00/14810号明細書を参照されたい)。
【0150】
上述された状態の処置のための本発明の組成物の有効用量は、投与手段、標的部位、患者の生理学的状態、患者がヒトであるか動物であるか、投与されている他の医薬品、および処置が予防的であるか治療的であるかを包含する多くの異なる因子に依存して変動する。通常、患者はヒトであり;トランスジェニック哺乳動物を包含するヒト以外の哺乳動物もまた処置し得る。処置投薬量は、典型的には安全性および有効性を最適化するよう用量設定する。
【0151】
抗体、ペプチドおよび小分子の投薬量は、約0.0001から約100mg/kgまで、およびより通常は約0.01ないし約20mg/kg宿主体重の範囲にわたる。例えば、投薬量は約1mg/kg体重若しくは約20mg/kg体重または約1ないし約10mg/kgの範囲内にあり得る。例示的処置レジメンは、2週ごとあたり1回若しくは1か月に1回または3ないし6か月ごとに1回の投与を必要とする。いくつかの方法において、異なる結合特異性をもつ2、3、4種若しくはそれ以上のモノクローナル抗体を同時に投与し、この場合投与される各抗体の投薬量は示される範囲内にある。例えば、いくつかの方法において、β1インテグリン、α2インテグリンおよびαvインテグリンサブユニットの2種若しくは全3種に対する抗体を同時に投与する。いくつかの方法において、α6インテグリンサブユニットに対する抗体もまた投与する。抗体は通常複数の機会に投与する。単一投薬量の間の間隔は週、月若しくは年であり得る。間隔は、患者中のインテグリンに対する抗体の血中濃度を測定することにより示されるように不規則でもまたあり得る。いくつかの方法において、抗体の投薬量は、約1ないし約1000μg/ml、およびいくつかの方法においては約25ないし約300μg/mlの血漿抗体濃度を達成するように調節する。あるいは、抗体は徐放製剤として投与し得、この場合はより少なく頻繁な投与を必要とする。投薬量および頻度は患者中の抗体の半減期に依存して変動する。一般に、ヒト抗体は最長半減期を示し、次いでヒト化抗体、キメラ抗体およびヒト以外の抗
体である。投薬量および投与頻度は、処置が予防的であるか治療的であるかに依存して変動し得る。予防的応用において、比較的低用量を長期間にわたり比較的頻繁でない間隔で投与する。若干の患者は彼らの生涯の残りの間処置を受領することを継続する。治療的応用においては、疾患の進行が低下若しくは終了するまで、および好ましくは患者が該疾患の症状の部分的若しくは完全な軽減を示すまで、比較的短い間隔での比較的高投薬量をときに必要とする。その後、該患者に予防的レジメンを投与し得る。
【0152】
剤をコードする核酸の用量は、患者あたり約10ngから1gまで、約100ngないし約100mg、約1μgないし約10mg、若しくは約30ないし約300μgのDNAの範囲にわたる。感染性ウイルスベクターの用量は、用量あたり約10から約100、若しくは約103、約104、約105、約106、約107、約108、約109、約1010若しくはそれ以上のビリオンまで変動しうる。
【0153】
本発明の剤は、予防的および/若しくは治療的処置のため非経口、局所、静脈内、経口、皮下、くも膜下腔内、動脈内、頭蓋内、腹腔内、鼻内若しくは筋肉内手段により投与し得る。いくつかの方法において、剤は沈着物が蓄積した特定の組織に直接注入する(例えば頭蓋内注入)。いくつかの方法において、筋肉内注入若しくは静脈内注入を抗体の投与に使用する。いくつかの方法において、特定の治療抗体を頭蓋中に直接注入する。いくつかの方法において、抗体を徐放組成物、若しくはMedipadTM装置のような装置として投与する。
【0154】
本発明の剤は、場合によっては、アミロイド形成疾患の処置で少なくとも部分的に有効である他の剤とともに投与し得る。アミロイド沈着物が脳中に存在するアルツハイマー病およびダウン症候群の症例においては、本発明の剤は、血液脳関門を横断する本発明の剤の通過を増大させる他の剤とともにもまた投与し得る。
【0155】
本発明の剤は、しばしば、有効治療薬および多様な他の製薬学的に許容できる成分を含んでなる組成物として投与する。Remington’s Pharmaceutical Science(第15版、Mack Publishing Company、ペンシルバニア州イーストン、1980)を参照されたい。使用される特定の製剤は意図している投与様式および治療的応用に依存する。該組成物は、所望の製剤に依存して、動物若しくはヒト投与のための製薬学的組成物を処方するのに通例使用されるベヒクルと定義される、製薬学的に許容できる非毒性の担体若しくは希釈剤もまた包含し得る。希釈剤は該組合せの生物学的活性に悪影響を及ぼさないように選択する。こうした希釈剤の例は、限定されるものでないが、蒸留水、生理学的リン酸緩衝生理的食塩水、リンゲル液、ブドウ糖溶液およびハンクス液を挙げることができる。加えて、製薬学的組成物若しくは製剤は、他の担体、補助物質、若しくは非毒性の非治療的非免疫原性安定剤などもまた包含しうる。
【0156】
製薬学的組成物は、タンパク質、キトサンのような多糖、ポリ乳酸、ポリグリコール酸、コポリマー(ラテックス官能性化SepharoseTMビーズ、アガロース、セルロースなどのような)、ポリマーアミノ酸、アミノ酸コポリマー、および脂質凝集物(油滴若しくはリポソームのような)のような、大型のゆっくりと代謝される巨大分子もまた包含し得る。
【0157】
非経口投与のため、本発明の剤は、水、油、生理的食塩水、グリセロール若しくはエタノールのような無菌の液体であり得る製薬学的担体を含む生理学的に許容できる希釈剤中の物質の溶液若しくは懸濁液の注入可能な投薬量として投与し得る。ヒト投与のための非経口組成物は無菌、実質的に等張であり、そしてGMP条件下で作成される。加えて、湿潤若しくは乳化剤、界面活性剤、pH緩衝物質などのような補助物質が組成物中に存在し
得る。製薬学的組成物の他成分は、石油、動物、植物若しくは合成起源のもの、例えばラッカセイ油、ダイズ油および鉱物油である。一般に、プロピレングリコール若しくはポリエチレングリコールのようなグリコールは、とりわけ注入可能な溶液に好ましい液体担体である。抗体は、有効成分の徐放を可能にするような様式で処方し得るデポー注射剤若しくは植込み製剤の形態で投与し得る。例示的一組成物は、HClで適するpHに調節された、50mM L−ヒスチジン(任意)、150mM NaClを含有する水性緩衝液中で処方された5mg/mLのモノクローナル抗体を含んでなる。
【0158】
典型的に、組成物は液体の溶液若しくは懸濁液いずれかのような注射液として製造し;注入前の液体ベヒクル中の溶液若しくは懸濁液に適する固体の形態もまた製造し得る。該製剤は、上で論考されたとおり、高められたアジュバント効果のため、乳化、あるいはリポソーム、またはポリラクチド、ポリグリコリド若しくはコポリマーのような微小粒子中に被包化もまたし得る(Langer、Science、249:1527−33(1990)およびHanesら、Advanced Drug Delivery Reviews、28:97−119(1997)を参照されたい)。本発明の剤は、有効成分の徐放若しくはパルス型放出を可能にするような様式で処方し得るデポー注射剤若しくは植込み製剤の形態で投与し得る。
【0159】
他の投与様式に適する付加的な製剤は、経口、鼻内および肺製剤、坐剤ならびに経皮適用を包含する。坐剤について、結合剤および担体は例えばポリアルキレングリコール若しくはトリグリセリドを包含し;こうした坐剤は、約0.5%ないし約10%、若しくは約1%ないし約2%の範囲の有効成分を含有する混合物から形成し得る。経口製剤は、製薬学的等級のマンニトール、乳糖、デンプン、ステアリン酸マグネシウム、サッカリンナトリウム、セルロースおよび炭酸マグネシウムのような賦形剤を包含するがしかしこれらに限定されない。これらの組成物は、典型的に、溶液、懸濁剤、錠剤、丸剤、カプセル剤、徐放製剤若しくは散剤の形態をとり、そして約10%ないし約95%、若しくは約25%ないし約70%の有効成分を含有する。
【0160】
局所適用は経皮若しくは皮内送達をもたらし得る。局所投与は、コレラ毒素若しくは解毒誘導体またはそれらのサブユニット、あるいは他の類似の細菌毒素との剤の共投与により助長し得る(Glennら、Nature、391:851(1988)を参照されたい)。共投与は、成分を混合物として、または化学的架橋若しくは発現により融合タンパク質として得られた結合分子として使用することにより達成し得る。あるいは、経皮送達は皮膚貼付剤を使用して若しくはトランスフェロソーム(transferosome)を使用して達成し得る(Paulら、Eur.J.Immunol.、25:3521−24(1995);Cevcら、Biochem.Biophys.Acta、1368:201−15(1998))。
【0161】
本明細書で引用される全部の文書はそっくりそのまま引用することによりここに本明細書に組み込まれる。
【図面の簡単な説明】
【0162】
【図1】ヒト皮質培養物(HCC)(上)若しくはポリエチレンイミン(PEI)(下)被覆プレートでのAβ網目構造を具体的に説明する。
【図2】HCCでのAβ網目構造形成および神経毒性双方に対するβ1インテグリンサブユニットの効果を具体的に説明する。図2AはHCCでのβ1インテグリンサブユニット発現を具体的に説明する。図2Bは抗β1抗体1965の非存在(上)若しくは存在(下)下のHCCでの72時間Aβ網目構造形成を具体的に説明する。図2Cは、β1インテグリンサブユニット阻止抗体とプレインキュベートしたHCCでの神経毒性の阻害を具体的に説明する。エラーバーは(n=3)ウェルからの標準偏差を表す。
【図3A】HCCでのAβ網目構造形成および神経毒性に対するα2およびαvインテグリンサブユニットの効果を具体的に説明する。
【図3B】抗α2(中)若しくは抗αv(下)抗体の非存在(上)若しくは存在下でプレインキュベートしたHCCでの72時間Aβ網目構造形成を具体的に説明する。
【図3C】β1インテグリンサブユニット阻止抗体とプレインキュベートしたHCCでの神経毒性を具体的に説明する。
【図4】HCCでのα2およびαv発現を具体的に説明する。
【図5】Aβ網目構造形成および神経毒性の阻害における抗ラミニン抗体の効果を具体的に説明する。図5Aは抗ラミニン抗体の非存在(上)若しくは存在(下)下でプレインキュベートしたHCCでの72時間Aβ網目構造形成を具体的に説明する。図5Bは抗ラミニン抗体とプレインキュベートしたHCCでの神経毒性を具体的に説明する。エラーバーは(n=3)ウェルからの標準偏差を表す。図5Cは抗コラーゲン抗体とプレインキュベートしたHCCでの神経毒性を具体的に説明する。エラーバーは(n=3)ウェルからの標準偏差を表す。
【図6】HCCでのAβシグナル伝達経路の活性化を具体的に説明する。図6Aは、Aβ処理したHCCでの接着斑キナーゼ(Fak)と会合したパキシリンのチロシンリン酸化を具体的に説明する。図6Bは、Aβ処理したHCCでのプロリンリッチチロシンキナーゼ(Pyk2)と会合したパキシリンのチロシンリン酸化を具体的に説明する。
【図7】ヒト皮質ニューロンを1時間播種し、次いで吸引かつ可溶性アミリンで処理する場合の1日後の毒性を具体的に説明する。インテグリン若しくはインテグリンサブユニット抗体を種(seed)および可溶性アミリンの存在下で細胞に添加して毒性を阻害し得る。種および可溶性アミリン単独は毒性でない。しかしながら、細胞を1時間播種し次いで吸引かつ可溶性アミリンで処理する場合、該アミリンは毒性である。
【図8】インテグリンおよびインテグリンサブユニット抗体、とりわけ抗ラミニン、抗β1、抗αvおよび抗α2抗体がアミリン毒性に対し保護することを具体的に説明する。
【図9A−9B】細胞を種アミリンに1時間曝露した後のアミリン2成分毒性の阻害パーセントにより示されるところの、アミリン毒性に対する保護におけるインテグリン、若しくは抗αvβ3抗αvを包含するインテグリンサブユニット抗体;およびサイトカラシンDの効果を具体的に説明する。
【図10A】抗αvインテグリン抗体18C7がin vitroで歯状回中の合成AβによるLTPの阻害を抑制することを具体的に説明する。
【図10B】抗αvインテグリン抗体20A9がin vitroで歯状回中の合成AβによるLTPの阻害を抑制することを具体的に説明する。
【図10C】抗αvインテグリン抗体17E6がin vitroで歯状回中の合成AβによるLTPの阻害を抑制することを具体的に説明する。
【図11A】対照抗αv抗体27/1がAβによるLTPの抑制を予防しなかったことを具体的に説明する。
【図11B】対照抗αv抗体7H10がAβによるLTPの抑制を予防しなかったことを具体的に説明する。
【図12A】可溶性Aβがin vivoでCA1領域でLTPを阻害することを具体的に説明する。
【図12B】抗αvインテグリン抗体17E6がin vivoでCA1領域での可溶性AβによるLTPの阻害を抑制することを具体的に説明する。
【図12C】SM256がin vivoでCA1領域での可溶性AβによるLTPの阻害を抑制することを具体的に説明する。
【図13A】αv含有インテグリンリガンドSM256がAβによるLTPの阻害を抑制することを具体的に説明する。
【図13B】αv含有インテグリンリガンド、スーパーフィブロネクチンがAβによるLTPの阻害を抑制することを具体的に説明する。
【図13C】αv含有インテグリンリガンド、エキスタチンがAβによるLTPの阻害を抑制することを具体的に説明する。
【実施例】
【0163】
実施例1〜5の材料および方法
抗体の供給源
【0164】
【表1】
【0165】
組織培養
組織培養プレートを150mMホウ酸ナトリウム、pH8.5中ポリエチレンイミン(PEI)で被覆し、そして室温で一夜インキュベートした。細胞を添加する前にウェルをPBSで洗浄し、そして細胞がプレーティングの準備ができるまで最小必須培地(10%FBSを含むMEM)を添加した。ヒト胎児大脳皮質(E13−E16)をハンクス平衡塩溶液(HBSS)ですすいだ。組織をHBSS中1mgのDNアーゼ中で摩砕した。この懸濁液を100ミクロンのナイロン細胞濾過器で濾過し、そして250×gで5分間遠心した。細胞をトリプシンに再懸濁しかつ37℃で20分間インキュベートした。改変最小必須培地(10%FBSおよび1mgのDNアーゼを含むMMEM)を添加しそして細胞を再懸濁し;その後遠心分離により再度収集した。B27を含有するMMEMに細胞を再懸濁し、そして、洗浄したPEI被覆プレート中、96ウェルプレートに125,000細胞/ウェルで、若しくは6ウェルプレートに250万細胞/ウェルで蒔いた。ヒト皮質培養物(HCC)は処理前に2週ごとの培地交換を伴い3週間インキュベートした。
【0166】
Aβ生成
Aβは、二重蒸留水(ddH2O)をAβに添加して1mMストックを構成することにより生成した。これを37℃で3日間エイジングし、アリコートに分割しそして−20℃で凍結保存した。可溶性Aβは、DMSOをAβに添加して7.5mMストックを作成すること、30分間超音波処理すること、アリコート分割することおよび−20℃で凍結す
ることにより作成した。神経毒性Aβは、ddH2OをAβに添加すること、アリコート分割することおよび−20℃で凍結することにより生成した。
【0167】
HCCライセートからのインテグリン免疫沈降
6ウェルプレート中のHCCを、メチオニンを含まない培地中100μCi/mlの35S−メチオニンで一夜標識した。細胞を洗浄し、25mM Hepes、pH7.5、1%Triton X−100、0.1%SDS、150mM NaCl、0.5mM EDTA、0.5mM EGTAで溶解し、そして26ゲージ針を3回通過させた。不溶性物質を15,000rpmで4℃で15分間の遠心分離により除去した。ライセートを、プロテインAビーズに結合したウサギ抗マウス(RAM)抗体で前浄化し、そしてインテグリンサブユニット特異的抗体(β1についてLia1/2、α1についてTS2/7、α2についてGi9、α3についてP1B5、α4についてAN100226m、α5についてAb0771、α6についてGoH3、α9についてY9A2およびαvについてVNR147)で免疫沈降した。免疫沈降物を1mlの25mM Hepes、pH7.5、1%Triton X−100 150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで3回洗浄した。免疫沈降したサンプルを8%トリス−グリシンゲル(Novex)で分離しかつ固定し;ゲルを乾燥し、そしてゲル中の35S標識タンパク質をオートラジオグラフィーにより可視化した。
【0168】
Aβ免疫蛍光
Aβで72時間処理したHCCを4%パラホルムアルデヒドで固定し、5μg/mlの抗Aβ−3D6−ビオチンで染色し、そして10μg/mlのストレプトアビジン−FITC(Jackson)で可視化した。
【0169】
ヒト皮質ニューロン中のAβ神経毒性
HCCを、グルタミンおよびペニシリン/ストレプトマイシンを補充した神経培地(MEM)(基礎培地)中で抗体若しくはリガンドで30分間処理した。基礎培地中1μモル濃度のAβを1時間添加した。培地を除去し、そしてHCCを抗体若しくはリガンドおよび基礎培地中20μM可溶性Aβで3日間処理した。3日目に、基礎培地中10%アラマーブルー中で2時間インキュベートすることにより毒性を測定した。蛍光レベルを三重で対照およびAβ処理したウェルに関して測定した。
【0170】
インテグリンヘテロ二量体会合
N−2(BottensteinのN−2処方、例えばカタログ番号17502、Invitrogen Corp.、カリフォルニア州カールズバッド)を補充したMMEM培地中の6ウェルプレート中のHCCを湿潤氷上に置き、PBSで洗浄し、25mM Hepes、pH7.5、1%Triton X−100、0.1%SDS、150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで溶解し、そして26ゲージ針を3回通過させた。不溶性物質を15,000rpmで4℃で15分間の遠心分離により除去した。ライセートをプロテインAビーズで前浄化し、そして抗β1インテグリンLia1/2(Immunotech)およびRAM/プロテインGビーズ(Pharmacia)を使用してβ1インテグリンを免疫沈降した。免疫沈降物を1mlの25mM
Hepes、pH7.5、1%Triton X−100 150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで3回洗浄した。免疫沈降したサンプルを4〜12%トリス−グリシンゲル(Novex)で分離し、そして抗α2インテグリン(ChemiconからのAB 1936)若しくは抗αvインテグリンMAB 1960(Chemicon)でウエスタンブロットした。
【0171】
パキシリンリン酸化のAβ誘導
神経毒性Aβを、N−2を補充した基礎培地中の6ウェルプレート中HCCに0分ない
し24時間添加した。HCCを湿潤氷上に置き、PBSで洗浄し、25mM Hepes、pH7.5、1%Triton X−100、0.1%SDS、150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで溶解し、そして26ゲージ針を3回通過させた。不溶性物質を15,000rpmで4℃で15分間の遠心分離により除去した。ライセートをプロテインAビーズで前浄化し、そして抗Fak(UBI)若しくは抗Pyk2抗体(UBI)およびプロテインAビーズを使用してそれぞれFak若しくはPykを免疫沈降した。免疫沈降物を1mlの25mM Hepes、pH7.5、1%Triton X−100、150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで3回洗浄した。免疫沈降したサンプルを8%トリス−グリシンゲル(Novex)で分離し、そして抗ホスホチロシン(Transduction labsからのRC20)および抗パキシリン(Transduction labs)でウエスタンブロットした。
【実施例1】
【0172】
ヒト皮質ニューロンでの免疫蛍光パターン
本実施例は、アルツハイマー病(AD)の海馬および皮質ニューロンで生じかつ関連した神経毒性を表すAβ斑のin vitro組織培養物モデルを生じる。該モデルは、ADで遂げられるニューロンを可能な限り緊密に表す初代ヒト皮質ニューロン培養物を使用する。これらの培養物へのAβの添加は、ニューロンで1〜3日にわたり生じかつその後ニューロンで毒性を引き起こす、再現可能なAβ網目構造をもたらす。HCCを伴わないプレート上でインキュベートしたAβもまた網目構造として染色したが、しかしHCCで見られるものより短く薄くかつより直線状であった伸長を伴うより均一なパターンを一貫して示した。図1Aおよび1BはHCCの存在および非存在下の網目構造を比較する。
【実施例2】
【0173】
β1インテグリンはAβ網目構造および神経毒性を媒介する
網目構造がインテグリンにより形成されるもののような細胞外マトリックスに似ていたため、インテグリンがHCC中に存在するかどうか;およびその場合、インテグリンがHCCでのAβ網目構造形成を助長したかどうかを検討した。ゲル電気泳動はβ1インテグリンがHCCで発現されていることを示した。MAB1965を包含するβ1インテグリン阻止抗体が、Aβ網目構造パターンがHCCで形成されることを阻止し得たこともまた見出された(図2A(抗体なし)を図2B(抗体を伴う)と比較されたい)。これらの培養物でAβにより生成される毒性に該網目構造パターンが必要であったかどうかもまた検討した。これを行うため、Aβ網目構造を阻止することが示されたβ1インテグリン阻止抗体(AIIB2およびMAB1965)とHCCをインキュベートした。これらの抗体はAβ誘発性の毒性もまた用量依存性の様式で阻害した(図2C)。抗体AIIB2はAβ毒性の非常に強力な阻害剤であり、170ng/mlすなわち1nMのIC50を表した。対照的に、対照抗体は毒性に対する影響を有しなかった。
【実施例3】
【0174】
α2およびαvインテグリンに結合する剤は網目構造およびAB媒介性神経毒性を阻害する
β1インテグリンは数種のαサブユニットと対形成して多様なヘテロ二量体を形成し得る。従って、どのαインテグリンサブユニットがHCC中に存在したかを試験した。α2、α3、α4、α5、α6およびαvインテグリンがHCC中で発現された。α1およびα9インテグリンはHCC中で発現されなかった。全部のこれらのαインテグリンサブユニットに対する阻害抗体を、Aβ網目構造形成を阻害しかつその神経毒性を阻害するそれらの能力について試験した。α2およびαvに対する阻害抗体はAβ網目構造形成を阻害した(図3A)。これらの抗体はHCCでのAβの神経毒性効果も阻害した(図3B)。これらの特定のインテグリンに対する特異性を示すため、2〜3種の阻害抗体をこれらの
インテグリンのそれぞれおよび同様に他のインテグリンサブユニットに対し試験した。網目構造形成および神経毒性の双方を媒介するα1、α2およびαvインテグリンに対する非常に明瞭な特異性が見出された(表1)。
【0175】
【表2】
【0176】
毒性に対する抗α6抗体の弱いがしかし再現可能な効果が観察された。最後に、これらの影響がインテグリンに向けられかつAβ重合を非特異的に妨害しなかったことを確認するため、Aβ毒性をヒトおよびマウス皮質培養物で並べて分析した。これらのアッセイで使用した抗体はマウスインテグリンと交差反応しない。抗ヒトインテグリン抗体はヒト培養物でAβ毒性を阻害し得たがしかしマウス培養物ではし得ず、該抗体が毒性を阻害するためにAβと非特異的に相互作用しなかったことを示唆した。HCCライセートをβ1抗体で免疫沈降すること、ならびにその後沈降した物質をα2およびαvに対する抗体でブロッティングすることにより、α2およびαvがHCC細胞中でβ1インテグリンと会合したことが確認された。これらの結果は、α2β1およびαvβ1のヘテロ二量体が、A
β網目構造形成および神経毒性の機能的メディエーターであることを示す。
【実施例4】
【0177】
フィブロネクチンおよび抗ラミニン抗体はAB網目構造形成および神経毒性を阻害する
インテグリン/細胞外網目構造の他成分をAβ網目構造形成および神経毒性の媒介への関与について検討した。これらの他成分は、αvβ1インテグリンリガンド、フィブロネクチン、およびスーパーフィブロネクチン(網目構造を形成するフィブロネクチンドメインの多量体)、ならびにα2β1リガンド、コラーゲンおよびラミニンを包含した。ラミニンは2鎖β1およびγ1を有し、それらの双方はアルツハイマー病斑中で上昇している(Murtomakiら、J.Neur.Res.、32:261−73(1992))。フィブロネクチンおよびスーパーフィブロネクチンはAβ網目構造形成および神経毒性を阻害することが可能であった(表1)。この結果はαvβ1機能に対する影響についてAβと競合するフィブロネクチンにより説明し得る。対照的に、αvβ1リガンド、ラミニンは、Aβ網目構造若しくは神経毒性を阻害することが可能でなかった(表1)。あるαvβ1リガンドがそうでなかった場合にあるαvβ1リガンドがなぜ保護的であったかを確定するため、抗ラミニン抗体を網目構造形成および神経毒性アッセイで試験した。2種のラミニン抗体はAβ網目構造形成(図5A)およびAβ媒介性神経毒性(図5B)双方で高度に保護的であった。抗ラミニン抗体番号AB19012は1nM未満のIC50を示した。対照的に、抗コラーゲン抗体はAβ網目構造形成および神経毒性に対する影響を有しなかった(図5C)。
【実施例5】
【0178】
Aβはパキシリンのチロシンリン酸化(インテグリンシグナル伝達経路の早期事象)を活性化する
細胞外マトリックスリガンドによるインテグリン活性化はFakのような接着斑キナーゼの活性化およびその基質パキシリンのチロシンリン酸化に至る。Aβがインテグリンシグナル伝達経路を同様に刺激していたかどうかを確定するため、HCCへのAβ添加に際してのFak会合パキシリンのチロシンリン酸化パターン(図6A)を分析した。Fak会合パキシリンのチロシンリン酸化の一貫した活性化は見出されなかった。しかしながら、Aβ刺激後のPyk2会合パキシリンのチロシンリン酸化の一貫した増大が観察された。Pyk2もまた接着斑キナーゼであり、そして神経毒性につながる異常なAβ/インテグリンシグナル伝達経路を媒介しているかもしれない(図6B)。FakよりむしろPyk2会合パキシリンのチロシンリン酸化の活性化は、これらの状態で毒性応答を引き起こすものでありうる。いずれの場合も、Aβはインテグリンシグナル伝達経路の早期事象であるパキシリンのチロシンリン酸化を活性化し、Aβ神経毒性シグナルがα2β1およびαvβ1インテグリンシグナル伝達経路の直接の関与により媒介されうることを示す。
【実施例6】
【0179】
アミリン2成分毒性
CPR,Inc.からの種(seed)若しくは凝集アミリン(1mM)(641−80、ロットNG−0213)を、200μl水/mg粉末を添加することおよびその後該溶液を37℃で3日間エイジングすることにより作成した。可溶性アミリン(5mM)は、40μl DMSO/mg粉末を添加すること、および該混合物を水浴中で30分間超音波処理することにより調製した。双方のストック溶液をアリコート分割しかつ使用の準備ができるまで凍結した。可溶性アミリンストックは、使用直前に培地で20μMに希釈し、そして水で前洗浄したAmicon 30フィルターで濾過した。濾過した物質をその後その適切な濃度に希釈した。
【0180】
ヒト皮質ニューロン(125,000細胞/96ウェル)を5μMの種アミリン(100μl/ウェル)で1時間処理した。細胞を吸引し、そして可溶性アミリンを100μl
/ウェルあたり5μMで戻した。化合物研究のため、可溶性アミリンを添加する前に50μlの2×化合物を添加した。
【0181】
図6はヒト皮質ニューロンを1時間播種し次いで吸引しかつ可溶性アミリンで処理した1日後の毒性を示す。インテグリン抗体を種および可溶性アミリンの存在下で細胞に添加して毒性を阻害した。図7は、数種のインテグリン抗体すなわち2034抗ラミニン、1965抗β1インテグリン、1958抗αv(VNR)および1950抗α2が、アミリン2成分の毒性に対し細胞を保護したことを示す。図8は、アミリン2成分の毒性が、抗αvβ3および抗αvを包含する付加的なインテグリン抗体;ならびにサイトカラシンDによりさらに阻害されることを示す。
【0182】
実施例7〜11の材料および方法
薄片の調製
全実験はラット海馬(雄性、3〜4週齢、重量40〜80g)若しくはマウス(雄性、3〜4月齢)の横断薄片で実施した。断頭後迅速に脳を取り出しかつ冷酸素添加(95%O2/5%CO2)培地に入れた。Intracell Plus 1000を使用して350μmの厚さで薄片を切断し、そして室温(20〜22℃)の酸素添加培地を含有する貯蔵容器に1時間入れた。薄片をその後浸積薄片のための記録チャンバーに移し、そして30〜32℃で5〜6ml/分の速度で連続灌流した。対照培地は(mMで)NaCl、120;KCl 2.5、NaH2PO4、1.25;NaHCO3 26;MgSO4、2.0;CaCl2、2.0;D−グルコース 10を含有した。全溶液はGABAA媒介性活性を阻害するため100μMピクロトキシン(Sigma)を含有した。
【0183】
in vitro電気生理学的技術
標準的電気生理学的技術を使用して電場電位を記録した。双極絶縁タングステンワイヤ電極を使用して歯状回の内側貫通路にシナプス前刺激を適用し、また、ガラス電極を用い、歯状回の分子層の中1/3から0.033Hzの対照試験周波数で興奮性シナプス後場電位(EPSP)を記録した。歯状回の内刃(inner blade)を全研究で使用した。各実験で、入力−出力曲線(求心性刺激強度対EPSP振幅)を試験周波数でプロットした。全実験について、試験EPSPの振幅を最大の1/3(約1.2mV)に調節した。LTPを8連の高頻度刺激(HFS)により惹起し(8刺激のそれぞれは200Hz、連続間間隔2s)、正常の試験EPSP振幅の2倍の連続の初期EPSPを惹起するようにHFSの間に刺激電圧を増大した。LTPの対照(ベヒクル単独)および実験レベルを同一海馬から調製した薄片で測定した。p−CLAMP(Axon Instruments、米国カリフォルニア州)を使用して記録を解析した。本明細書に報告する値はn薄片の平均±S.E.M.であった。Studentの両側t検定を統計学的比較に使用した。
【0184】
in vivo電気生理学
実験はウレタン(エチルカルバメート、1.5gm/kg i.p.)麻酔した雄性Wistarラット(250〜300g)で実施した。体温は37〜37.3℃に維持した。動物の世話および実験プロトコルはアイルランド共和国保健省(Department
of Health、Republic of Ireland)により承認された。
【0185】
電極は以前に記述された(Klyubinら、2004、2005)とおり作成しかつ植込んだ。簡潔には、捻りワイヤ双極電極をTeflon被覆タングステンワイヤ(62.5μm内核径、75μm外径)から構築した。興奮性シナプス後場電位(EPSP)の単一経路記録を、同側のSchaffer側枝−交連経路の刺激に応答する右海馬半球のCA1領域中の放線状層から行った。電極植込み部位は十字縫合に関する定位座標を使用して同定し、記録部位は十字縫合の後方約3.4mmかつ正中線の右約2.5mmに位置
し、また、刺激電極は十字縫合の後方約4.2mmかつ正中線の右約3.8に位置した。背側海馬のCA1領域の放線状層中のワイヤ電極の最適深度は電気生理学的基準を使用して決定し、かつ、死後確認した。試験EPSPは、0.033Hzの周波数および最大の50%のEPSP振幅を与えるように調節した刺激強度で惹起した。LTPを誘導するためのHFSプロトコルは、10連続の20刺激、刺激間間隔5ms(200Hz)、連続間間隔2秒よりなった。強度は最大振幅の約75%のEPSPを与えるようにHFSの間に増大させた。
【0186】
サンプルを注入するため、ステンレス鋼製ガイドカニューレ(22ゲージ、0.7mm外径、13mm長さ)を、電極植込み直前に右側脳室の上(正中線の側方約1mmおよび硬膜の表面の下約4mm)に植込んだ。内部カニューレ(28ゲージ、0.36mm外径)を介して脳室内(i.c.v.)注入を行った。カニューレの配置の確認はi.c.v.注入後のインク色素の広がりを確認することにより死後に実施した。本明細書に報告する値はn薄片の平均±S.E.M.であった。Studentの両側t検定を統計学的比較に使用した。
【0187】
剤
合成Aβ(1−42)をBachemから得た。in vitro実験のため、合成Aβ1−42を水酸化アンモニウム(0.1%)中50μMのストック溶液として調製し、−20℃で保存し、そしてその後各実験の直前に生理学的媒体に添加した。in vivo実験のため、合成Aβ(1−42)(Bachem)を氷冷milliQ水若しくはTeplowに再懸濁した。アリコートを取り出し、そして100,000gで3時間(原線維およびプロトフィブリル(protofibril)をペレットにすることが既知の条件)(Klyubinら、2004)遠心分離した。遠心分離後、アミノ酸分析により測定されるところの30若しくは64μMの可溶性Aβの最終濃度を有した上清を、小アリコートで−80℃で保存した。
【0188】
以下のインテグリンαv抗体(全部IgG1アイソタイプ)すなわち18C7、17E6および20A9(全部Calbiochemから)を研究で使用した。使用した対照抗体は、ヒトICAM−1に対するIgG2aマウス抗体7H10およびIgG1アイソタイプ27/1であった。使用した他の成分は、エキスタチン(Source)、Van Maesら、1994の方法に従ってElan Pharmaceuticalsにより製造されたSM256(3−[1−[3−(N−イミダゾル−2−イルアミノ)プロピル]インダゾル−5−イルカルボニルアミノ]−2(S)−(2,4,6−トリメチルベンゼンスルホニルアミノ)プロピオン酸トリフルオロ酢酸塩)(ELN 151993として記録される)、スーパーフィブロネクチン(Sigma)およびファロイジン(Calbiochem)であった。in vivo実験において、SM256は生理的食塩水中Tween 80(Sigma)の懸濁液中でi.p投与のため調製した(15v/v%)。
【実施例7】
【0189】
抗インテグリンαv抗体はin vitroで歯状回でのLTPのAβ媒介性阻害を予防する
in vitroの歯状回での短いHFSによるLTPの誘導は、HFS前30分間のAβの前灌流により予防され、以前の研究(Wangら、2004a、b、2005)を確認した。従って、Aβ(500nM、LTPの最大阻害を引き起こすことが以前に見出された濃度(Wangら、2004a))の存在下でのLTPは、HFSの1時間後に基礎の105±3%であり(図10A)(n=5、P<0.001)、152±5%であった(図10A)対照LTPと比較して有意に低下された。
【0190】
αvインテグリンサブユニットに対する選択的抗体を灌流することの効果をLTPのAβ媒介性阻害に対し検討した。18C7、20A9および17E6と命名される、αV含有インテグリンに対する3種の異なる抗体を検討した。該抗体のいずれも基礎のEPSP若しくはLTP誘導に対するいかなる影響も有しなかった。しかしながら、LTPのAβ媒介性阻害はαv抗体のそれぞれの灌流により予防された。図10A〜10Cは、全3種のαv抗体が、介在対照実験と比較してLTPに対するAβの阻害効果の予防において有効であったことを示す。従って、LTPは、Aβならびに抗インテグリン抗体18C7、20A9および17E6の存在下で144±6%、137±7%および153±11%であった。該値は対照LTPと比較して有意に異ならなかった(各実験についてn=5、P>0.01)。各抗体実験と平行して実施した介在薄片において、LTPはAβ単独の存在下で105±3%、105±6%および103±3%であり、値はLTPに対するAβ単独のものと有意に異ならなかった(各実験についてn=5、P>0.01)。
【0191】
対照的に、2種の対照抗体はLTPのAβ媒介性阻害を有意に予防しなかった。LTPは、Aβおよび対照抗体27/1の存在下で105±6%(図11A)、ならびに、Aβおよび対照抗体7H10(アイソタイプ対照)の存在下で107±8%であった(図11B)(n=5、P>0.05)。
【実施例8】
【0192】
抗αvインテグリン抗体はin vivoでCA1でのLTPのAβ媒介性阻害を予防する
ウレタン麻酔したラットのCA1でのLTPの誘導は、HFS10分前の可溶性の原線維を含まないAβのi.c.v.注入により予防され、以前の研究(Klyubinら、2004)を確認した。対照ベヒクル注入動物で、LTPはHFSの3時間後に基礎の140±5%であった(n=6)。可溶性の原線維を含まないAβ(5μl中50pmol、n=5)の存在下でLTPは強く阻害され、105±5%であり(図12A)、対照LTPと比較して有意に低下された(P<0.001)。αvインテグリンサブユニットに対する選択的抗体17E6(10μl中27.9μg)はLTPのAβ媒介性阻害を予防し、135±8%であった(n=5、ベヒクルおよびAβに比較してP<0.01;ベヒクルおよびベヒクルに比較してP>0.05)。アイソタイプ(IgG1マウス)対照抗体27/1(10μl中27.9μg)はLTPのAβ媒介性阻害に影響を及ぼすことに失敗した(103±6%、n=6、ベヒクルおよびAβに比較してP>0.05;ベヒクルおよびベヒクルに比較してP<0.01)(図12B)。
【実施例9】
【0193】
αv含有インテグリンの小分子非ペプチド拮抗剤はin vitroで歯状回およびin
vivoでCA1でのLTPのAβ媒介性阻害を予防する
SM256は強力なαv拮抗剤でありかつαv媒介性の細胞接着を阻害する非ペプチド剤である(Van Waesら、2000)。SM256(10μM)単独はLTPを阻害せず、148±7%であった。しかしながら、SM256はLTPのAβ媒介性阻害を予防し、129±5%であった(図13A)(n=5、P<0.01)。
【0194】
同様に、SM256の全身前投与は、可溶性の原線維を含まないAβのi.c.v.注入により引き起こされるin vivoのCA1でのLTPの阻害を無効にした。選ばれたSM256の用量(1.2mlベヒクル中40mg、i.p.)は基礎のシナプス伝達若しくは対照LTPに対する認識可能な影響を有しなかった(図12Cおよびデータは示されない)。SM256の40分後かつHFSの10分前に注入した可溶性Aβ(50pmol、i.c.v.)はLTPを阻害することに失敗し、133±5%であり(図12C、n=4;HFS前基礎に比較してP<0.01;末梢ベヒクルおよび中枢ベヒクル(144±9%、n=3)に比較してP>0.05)、また、ベヒクルの末梢注入次いでA
βの中枢注入を与えた動物のLTPの大きさと有意に異ならなかった(103±10%、n=4、P<0.05)。
【実施例10】
【0195】
スーパーフィブロネクチンはin vitroで歯状回でのLTPのAβ媒介性阻害を予防する
スーパーフィブロネクチンはαvβ1のリガンドである(Morlaら、1994)。スーパーフィブロネクチン(1μM)単独はLTPを阻害せず、155±2%であった。しかしながら、スーパーフィブロネクチンはLTPのAβ媒介性阻止を予防し、147±6%であった(図13B)(n=5、P<0.01)。
【実施例11】
【0196】
ディスインテグリンはin vitroで歯状回でのLTPのAβ媒介性阻害を予防する
ディスインテグリン、エキスタチンの効果もまたLTPのAβ惹起性の阻害に対して検討した。ディスインテグリンは、非常に高い親和性でかつRGDペプチドより強くインテグリンに結合する、インテグリンのアンタゴニストである蛇毒から単離された小型の4〜10kDaのRGDを含有するシステイン豊富なペプチドである(Ganら、1988)。エキスタチンはαv/β3およびα5/β1を包含するRGD依存性インテグリンを阻害することが示された(Kumarら、1997)1種のこうしたディスインテグリンである。エキスタチン(50nM)単独はLTPを阻害せず、158±5%であった。しかしながら、エキスタチンはLTPのAβ媒介性阻止を予防し、143±6%、n=5、P<0.01であった(図13C)。
【0197】
上で引用された全部の刊行物、特許および特許出願は、各個々の刊行物若しくは特許出願が引用することによりそのように組み込まれることをとりわけかつ個々に示された場合と同一の程度まで全部の目的上そっくりそのまま引用することにより組み込まれる。本発明は明瞭さおよび理解の目的上具体的説明および実施例として若干詳細に記述されたとは言え、付随する請求の範囲の範囲内である種の変更および改変を実施しうることが明らかであろう。
【技術分野】
【0001】
アミロイド形成タンパク質は複数の疾患状態の病理学に関与している。アミロイド形成タンパク質の異常沈着から生じる疾患は、限定されるものでないがアルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、プリオンにより全部若しくは部分的に引き起こされる疾患(クロイツフェルト・ヤコブ病、スクレイピーおよびウシ海綿状脳症のような)、ならびに遺伝性アミロイドーシスおよび全身性アミロイドーシス双方を包含するアミロイドーシスを挙げることができる。
【背景技術】
【0002】
アルツハイマー病(AD)は、米国単独で400万人を苦しめている老人性認知症をもたらす進行性神経変性疾患である(全般として、非特許文献1;特許文献1;非特許文献2;非特許文献3;非特許文献4を参照されたい)。大まかに言って、該疾患は2種類、すなわち高齢(65歳超)で発生する晩発性;および老年期の十分に前すなわち35と60歳の間で発症する早発性に分類される。双方の型の疾患で病状は同一であるが、しかしより若年齢で開始する症例で異常がより重篤かつ広まる傾向がある。該疾患は、脳中の少なくとも2型の病変、すなわち老人斑および神経原線維変化を特徴とする。神経原線維変化は、対で相互の周囲で捻られた2本のフィラメントよりなる微小管結合τタンパク質の細胞内沈着物である。老人斑は直径150ミクロンまでの混乱した神経線維網の領域であり(脳組織の切片の顕微鏡分析により見える)、そして中央に細胞外アミロイド沈着物を有する。こうした斑の主成分はAβペプチドである(非特許文献5を参照されたい)。斑中で見出される付加的なタンパク質は、非特許文献6により記述されるところのラミニン、アポE、アセチルコリンエステラーゼ、および非特許文献7により記述されるところのヘパリン硫酸プロテオグリカンを包含する。Aβペプチドは、アミロイド前駆体タンパク質(APP)と命名される前駆体タンパク質の39〜43アミノ酸の内部フラグメントである。APPタンパク質内の数種の突然変異がアルツハイマー病の存在と相互に関連付けられている(非特許文献8(バリン717からイソロイシン);非特許文献9(バリン717からグリシン);非特許文献10(バリン717からフェニルアラニン);非特許文献11(リシン595メチオニン596をアスパラギン595ロイシン596に変える二重突然変異))。こうした突然変異は、AβへのAPPの増大された若しくは変えられたプロセシング、とりわけ、増大された量の長い形態のAβ(すなわちA1−42およびA1−43)へのAPPのプロセシングによりアルツハイマー病を引き起こすと考えられている。プレセニリン遺伝子PS1およびPS2のような他の遺伝子中の突然変異は、APPのプロセシングに間接的に影響して増大された量の長い形態のAβを生成すると考えられている(非特許文献12)。これらの観察結果は、Aβおよびとりわけその長い形態がアルツハイマー病における原因要素であることを示す(非特許文献13)。
【0003】
研究者は、シナプス不全がADの発症の基礎にあると仮定している。シナプス減少がADの早期事象でありかつ認知障害と構造的相関関係にあるためである。研究者は、ADにおける臨床的認知症の潜行性発症に先立つ軽度認知障害が、大スケールの神経変性に先立つシナプス不全から生じるとさらに仮定している。長期増強(LTP)は、学習および記憶の細胞モデルであると広く仮定されているシナプスの可塑性の一形態である。
【0004】
LTPはシナプス伝達の効率の持続的な使用依存性の増大である。大部分の研究でLTPは高頻度シナプス刺激(HFS)の送達により実験的に誘導する。しかしながら他の馴化プロトコルが存在し、それらのいくつかは性質が薬理学的でありかつシナプス刺激を伴わない。さらに、複数の形態のLTPが同定されている。げっ歯類脳薄片での研究は、とりわけ海馬のCA3−CA1シナプスでLTPの多くの局面を解明した。LTPの4つの
特徴は、共同性、連合性、持続性および入力特異性である。
【0005】
シナプス効率の増大を開始する早期事象を構成するLTP誘導の過程は、LTPのその後の持続的発現と機構的に異なる。LTP誘導の間、領域CA3から領域CA1に突出する線維へのHFSの送達がシナプス中にグルタミン酸を放出し、そしてシナプス後ニューロンを脱分極する。高頻度の刺激(例えば100Hz)により、連続的興奮性シナプス後電位(EPSP)により誘発される脱分極が重なり、そして一連のHFSの間の累積的脱分極が実質的になり得る。樹状突起に後方伝播するシナプス後活動電位の発生が付加的な脱分極に寄与する。グルタミン酸放出および脱分極は原因として関係するとは言え、実験的に、それらを分離しかつLTPの誘導が双方の事象を必要とする(「共同性」と命名されている関係)ことを示すことが可能である。従って、シナプス後膜が馴化の間に直接過分極される場合、HFSはLTPを誘導することに失敗する。逆に、電流注入でシナプス後膜を直接脱分極することは、低頻度シナプス刺激でさえLTPを誘導することを可能にする。
【0006】
第21染色体の余分の1コピーを有するダウン症候群のほとんど全部の個体は、彼らが彼らの40歳代まで生き延びる場合に、アルツハイマー病で見られるものに類似の神経病理学的変化を示す。これは、第21染色体上のAPP遺伝子によりコードされるβアミロイドタンパク質の過剰産生に帰されている。
【0007】
数種のタンパク質がAβとの可能な相互作用について検討されている。これらは、高度な糖化最終生成物、RAGE(非特許文献14を参照されたい)、スカベンジャー受容体(非特許文献15;および非特許文献16)、小胞体関連アミロイドβ結合タンパク質(ERAB)(非特許文献17)、α4若しくはα7ニコチン性アセチルコリン受容体(非特許文献18および非特許文献19)、ならびに低親和性p75 NGF受容体(非特許文献20を参照されたい)を包含する。加えて、Aβは、非特許文献21および非特許文献22により、プレート上に被覆される場合にβ1インテグリンサブユニット依存性の様式で細胞の接着を媒介することが報告されている。
【0008】
Aβと相互作用しうる多様な機能の多様な分子の数を考慮すると、Aβが神経変性を媒介しうる機序は不明なままである。該過程で役割を有しうる他の細胞タンパク質の存在および性質もまた不明である。
【0009】
島アミロイドは、世紀の始まり以来II型糖尿病における病理学的実体として認識されている。それは、その独特の成分として、インスリンと共分泌される島β細胞ペプチド、島アミロイドポリペプチド(IAPP)すなわちアミリンを有する。この独特の成分に加え、島アミロイドは、アポリポタンパク質E、およびヘパリン硫酸プロテオグリカン、パールカンのような他のタンパク質を含有し、それらは典型的に他の形態の全身性および局在性アミロイドで観察される。島アミロイドは、II型糖尿病を伴う個体の大多数において病理学的検査で観察されるが、しかし糖代謝の混乱を伴わないヒトで稀に観察される。げっ歯類からのIAPPと対照的にヒトIAPPはin vitroでアミロイド原線維を形成することが示されている。全ヒト被験体がアミロイド形成性の形態のIAPPを産生かつ分泌し、それでもなお全部が島アミロイドを発生するわけではないため、いくつかの他の因子が島アミロイド形成に関与していることがありそうである。1仮説は、IAPPの産生、プロセシングおよび/若しくは分泌の変化をもたらすβ細胞機能の変化が、ヒト糖尿病における島アミロイド原線維の初期形成に関与することである。アミロイド原線維のこの形成がその後IAPP含有原線維の進行的蓄積を可能にする。β細胞塊のアミロイドによる最終的な置換が高血糖症の発症に寄与している。
【0010】
アミロイド形成の開始をもたらすβ細胞の変化を生じることに関与しうる1因子は、食
物脂肪の増大された消費である。食物脂肪は島β細胞ペプチドの産生、プロセシングおよび分泌を変えることが知られており、また、ヒトIAPPを発現するトランスジェニックマウスでの研究はこの機序の作動を裏付ける。これおよび他のモデルを使用するさらなる検討が、島アミロイド形成に関与する機序への洞察を提供しそしてアミロイド原線維形成を阻害若しくは復帰する治療薬の開発を可能にするはずであり、その目標はII型糖尿病でβ細胞の機能を保存しかつグルコース制御を改善することである。非特許文献23。
【0011】
伝染性海綿状脳症すなわちプリオン病は、後天的、遺伝性若しくは特発性(「散発性」)疾患として症状発現し得る伝染性の迅速に進行する必ず致死的な神経変性疾患の一群を構成する。それらは、ヒトにおけるクロイツフェルト・ヤコブ病ならびに動物におけるスクレイピーおよびウシ海綿状脳症(BSE)を包含し、そして、実験的若しくは偶発的伝播後に数十年まで持続しうる長い潜伏期を特徴とする。プリオン病の古典的病理学的特徴は海綿状変化、神経膠症およびニューロン減少を包含する。ウイルスによる引き起こされる感染性疾患で典型的に見られるものと対照的に、プリオン病は有意の炎症応答を欠く(非特許文献24、非特許文献25)。
【0012】
プリオン病は伝染性病原体「プリオン」の独特の特性によりかなりの科学的注目を集めている(非特許文献26)。該感染性病原体は非常に小さく、そして核酸を破壊しかつ慣習的ウイルスを不活性化する処置に対し極めて抵抗性である(非特許文献26)が、しかしタンパク質を変性させる処置に感受性である。該感染性病原体を精製する試みは、プリオンタンパク質(PrP)と命名された、これまで未知のタンパク質について高度に濃縮された画分を生じた(非特許文献27:非特許文献28:非特許文献29)。病原体特異的核酸はこれらの調製物中で見出されておらず(非特許文献30;非特許文献31);むしろ、プリオンタンパク質は宿主ゲノム中でコードされる(非特許文献29;非特許文献32;非特許文献33)。冒された個体の脳中には、PrPScと命名されたプリオンタンパク質の特異的な疾患関連アイソフォームの特徴的な蓄積が見出される(図1)。PrPScは、コンホメーション変化を伴う明確に定義されていない翻訳後過程によりプリオンタンパク質の正常な細胞アイソフォーム(PrPc)から派生する(非特許文献25)。PrPcおよびPrPScは同一アミノ酸配列を有する(非特許文献34)が、しかしながらそれらはコンホメーションが異なる。PrPScは、βシート構造のその高含量(非特許文献35)、大型凝集物を形成するその傾向、およびプロテイナーゼKでの消化に対するその部分的抵抗性によりPrPcと識別し得る。
【0013】
遺伝性アミロイドーシスは、細胞外マトリックス中の不溶性タンパク質原線維の沈着物を特徴とする常染色体優性の遺伝性疾患の臨床上および遺伝的に不均一な一群を含んでなる。これらの疾患は、硝子体混濁および腎不全によりときに付随される、多発神経炎、手根幹症候群、自律神経障害、心筋症の症状および胃腸の特徴を典型的に提示する。他の表現型は、ニューロパシー、胃潰瘍、脳神経障害および格子状角膜ジストロフィーを特徴とする。稀に軟髄膜若しくは脳の構造もまた臨床像に関与している。発症時年齢は17歳くらい早くかつ78歳くらい遅い。アミロイド原線維の基礎構成要素は、遺伝子で決定されるコンホメーション変化によりアミロイド形成性となる生理学的タンパク質である。以前はプレアルブミンと命名されていた変異トランスサイレチン(TTR)が遺伝性アミロイドーシスの最も頻繁な攻撃者(offender)である。同所性肝移植(OLT)は、アミロイド形成タンパク質の主産生部位を除去することにより、そうでなければ一般的に致死性である該疾患の進行を停止する。OLTの適応症およびその成功は、手術時点での心血管系および自律神経障害の進行度、齢、併存症ならびに突然変異の型に依存する。TTRの天然の四量体コンホメーションを安定化しかつ原線維形成を阻害する薬物を用いる代替処置モダリティーが現在集中的に研究されている。
【0014】
全身性アミロイドーシスは、実質器官;血管;皮下、粘膜下および腱周囲の脂肪;心;
眼;ならびに髄膜の原線維タンパク質凝集の細胞外沈着物を特徴とする。加えて、神経幹、神経叢、ならびに知覚および自律神経節を包含する末梢神経系のいずれの部分も関与しうる。抹消神経では、アミロイド沈着物は、通常は斑状および限局性の分布で神経上膜、神経周膜若しくは神経内膜に存在する。慣習的染色を用いる光学顕微鏡検査で、アミロイド沈着物は均一なエオシン好性の外見を有する。コンゴーレッド染色で、それらは偏光下に特徴的な黄緑色の複屈折を示す。
【0015】
多様なタンパク質がアミロイド形成の原因であり;事実、合計18種のアミロイド形成タンパク質がヒトアミロイドーシスで同定されている。非遺伝性の全身性アミロイドーシスは、免疫グロブリンL鎖(AL型、形質細胞悪液質における)、血清アミロイドAのフラグメント、急性期タンパク質(AA型、慢性炎症性疾患における)、トランスサイレチン(TTR;老人性全身性アミロイドーシスにおける)およびβ2−ミクログロブリン(尿毒症および透析を伴う患者における)により引き起こされ得る。遺伝性アミロイドーシスは、TTR、およびはるかにより稀にはアポリポタンパク質A1、リゾチーム、フィブリノーゲン、ゲルゾリン、アミロイドβおよびシスタチンCを包含する生理学的タンパク質の遺伝子バリアントによる。以前はプレアルブミンと呼ばれたTTRは、血清チロキシンおよび網膜結合タンパク質の輸送に関与する正常の四量体血清タンパク質である。それは第18染色体上の単一遺伝子によりコードされ、それの51個の異なる部位に存在する70以上の常染色体優性遺伝性点突然変異が記述されている。これらのなかで、位置30のメチオニン(Met30)によるバリンの置換はこれまで最も頻繁でありかつ地理的に最も広く散在している。
【0016】
パーキンソン病は、振戦、筋固縮、ならびに平衡および協調障害により特徴付けられる進行性神経障害である。化学物質ドーパミンを産生する脳細胞の破壊がこれらの症状の基礎にある。これらの病的な細胞はレビー小体と呼ばれるタンパク質沈着物によってもまた特徴付けられる。細胞がなぜ死ぬか、若しくはレビー小体がそれらを死滅させるのを助けるかどうかは誰も知らない。パーキンおよびα−シヌクレインと呼ばれる2種のタンパク質の遺伝子中の突然変異が、稀な形態の遺伝性パーキンソン病を分離するために結び付けられている。しかし、パーキンおよびα−シヌクレインの双方が、全パーキンソン病患者の脳中に蓄積するレビー小体中で見出される。
【0017】
最近の知見は、パーキンが、α−シヌクレインおよびシンフィリンを包含する脳中のレビー小体と関連するタンパク質の調節において重要な役割を演じていることを示唆している。正常には、パーキンは破壊のため他のタンパク質を「標識する」のにユビキチンと呼ばれるなお別のタンパク質を使用する。しかし、これらのタンパク質のあいだの関係で何かがうまくいかない場合、これはパーキンソン病で見られる細胞死の基礎作りをし得る。パーキンおよびα−シヌクレインの双方が、レビー小体関連タンパク質のユビキチン化を伴う共通の発病機序でシンフィリン−1と結合される。非特許文献36。従って、パーキンとのその相互作用を考えれば、α−シヌクレインでの問題は、遺伝性および一般的形態の双方のパーキンソン病の中心にありうる。非特許文献36。
【先行技術文献】
【特許文献】
【0018】
【特許文献1】Hardyら、第WO 92/13069号明細書
【非特許文献】
【0019】
【非特許文献1】Sloe、TINS、16:403−409(1993)
【非特許文献2】Sloe、J.Neuropathol.Exp.Neurol.、53:438−447(1994)
【非特許文献3】Duffら、Nature、373:476−477(1995)
【非特許文献4】Gamesら、Nature、373:523(1995)
【非特許文献5】Forsyth Phys.Ther.、78:1325−1331(1998)
【非特許文献6】Murtomakiら、J.Neurosci.Res.、32:261−273(1992)
【非特許文献7】Yanら、Biochim.Biophys.Acta、1502:145−57(2000)
【非特許文献8】Goateら、Nature、349:704−06(1991)
【非特許文献9】Harlinら、Nature、353:844−46(1991)
【非特許文献10】Murrellら、Science、254:97−99(1991)
【非特許文献11】Mullanら、Nature Genet.、1:345−47(1992)
【非特許文献12】Hardy、TINS、20:154(1997)
【非特許文献13】Velez−Pardoら、Gen.Pharm.、31(5):675−81(1998)
【非特許文献14】Yanら、Nature、382:685−91(1996)
【非特許文献15】Khouryら、Nature、382:716−719(1996)
【非特許文献16】Paresceら、Neuron 17:553−65(1996)
【非特許文献17】Yanら、Nature、389:689−695(1997)
【非特許文献18】Wangら、J.Neurochem.、75:1155−1161(2000)
【非特許文献19】Wangら、J.Biol.Chem.、275:5626−5632(2000)
【非特許文献20】Yaarら、J.Clin.Invest.、100:2333−2340(1997)
【非特許文献21】Ghisoら、Biochem.J.、288:1053−59(1992)
【非特許文献22】Matterら、J.Cell Bio.、141:1019−1030(1998)
【非特許文献23】Diabetes、48:241−253(1999)
【非特許文献24】Prusiner、Arch.Neurol.、50:1129−1153(1953)
【非特許文献25】Prusiner、Proc.Natl.Acad.Sci.U.S.A.、95:13363−13383(1998)
【非特許文献26】Prusiner、Science、216:136−144(1982)
【非特許文献27】Boltonら、Science、218:1309−1311(1982)
【非特許文献28】Prusinerら、Cell、38:127−134(1983)
【非特許文献29】Oeschら、Cell、40:735−746(1985)
【非特許文献30】Kellingsら、J.Gen.Virol.、73:1025−1029(1992)
【非特許文献31】Riesnerら、Dev.Biol.Stand.、80:173−181(1993)
【非特許文献32】Chesebroら、Nature、315:331−333(1985)
【非特許文献33】Baslerら、Cell、46:417−428(1986)
【非特許文献34】Stahlら、Biochemistry、32:1991−2002(1993)
【非特許文献35】Panら、Proc.Natl.Acad.Sci.U.S.A.、90:10962−10966(1993)
【非特許文献36】Dawsonら、Nature Medicine、7:1144−1150(2001)
【発明の概要】
【0020】
[発明の要約]
1種若しくはそれ以上の剤が長期増強(LTP)のアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する該1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、LTPのアミロイド媒介性阻害の抑制方法が提供される。該方法の一態様において、インテグリンサブユニットαvに結合する最低2剤の有効投薬量が投与される。該方法の一態様において、該剤は、Aβ産生の阻害剤、Aβ沈着の阻害剤、Aβ消失のメディエーター、アミロイド斑消失のメディエーター、Aβ神経毒性の阻害剤、Aβ凝集の阻害剤およびAβ解離のメディエーターよりなる群から選ばれる第二の剤とともに投与される。一態様において、Aβ産生の阻害剤はγ分泌酵素阻害剤である。一態様において、Aβ産生の阻害剤はβ分泌酵素阻害剤である。該方法の一態様において、該剤はAβに対する抗体とともに投与される。該方法の一態様において、該剤はRGD(Arg−Gly−Asp)モチーフを含んでなるペプチドである。該方法の一態様において、該剤はαvβ1インテグリンのリガンドである。該方法の一態様において、該剤はフィブロネクチン若しくはスーパーフィブロネクチンである。該方法の一態様において、該剤はαvインテグリンサブユニット発現細胞のビトロネクチン若しくはフィブロネクチンへの接着を阻害する。該方法の一態様において、該剤はαvインテグリンサブユニット発現細胞のオステオポンチンへの接着を阻害する。該方法の一態様において、該剤はモノクローナル若しくはポリクローナル抗体である。該方法の一態様において、該剤は、18C7、20A9および17E6から選択される抗体と同一のエピトープを認識する抗体である。一態様において、該抗体はヒト化抗体、キメラ抗体およびナノボディ(nanobody)から選択される。該方法の一態様において、該剤は18C7、20A9および17E6から選択される抗体である。該方法の一態様において、該剤は、インテグリンサブユニットαvへの結合について18C7、20A9および17E6から選ばれる抗体と競合する。
【0021】
該方法のさらなる一態様において、該剤は、それらの立体異性体、若しくはそれらの立体異性体の混合物、またはそれらの製薬学的に許容できる塩の形態を包含する、式IaおよびIb
【0022】
【化1】
【0023】
の化合物から選択される化合物であり、ここで
X1およびX3は窒素若しくは炭素から独立に選択され;
R1は
【0024】
【化2】
【0025】
から選択され;
ここで、上の複素環は、NH2、ハロゲン、NO2、CN、CF3、C1−C4アルコキシ、C1−C6アルキルおよびC3−C7シクロアルキルよりなる群から選択される0〜2置換基で場合によっては置換されており;
Uは−(CH2)n−、−(CH2)tQ(CH2)m−および−C(=O)(CH2)
n−1−から選択され、ここでメチレン基の1個は場合によってはR7で置換されており;
Qは、1,2−フェニレン、1,3−フェニレン、2,3−ピリジニレン、3,4−ピリジニレンおよび2,4−ピリジニレンから選択され;
R6はH、C1−C4アルキルおよびベンジルから選択され;
R7はC1−C6アルキル、C3−C7シクロアルキル、C4−C11シクロアルキルアルキル、アリール、アリール(C1−C6アルキル)、ヘテロアリールおよびヘテロアリール(C1−C6アルキル)から選択され;
R10は、H、ハロゲン、CO2R17、CONR17R20、0〜1個のR15若しくは0〜1個のR21で置換されているC1−C6アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR15若しくは0〜1個のR21で置換されているC3−C7シクロアルキル、0〜1個のR15若しくは0〜1個のR21で置換されているC4−C11シクロアルキルアルキル、および0〜1個のR15若しくは0〜2個のR11若しくは0〜1個のR21で置換されているアリール(C1−C6アルキル)−から選択され;
R11は、H、ハロゲン、CF3、CN、NO2、ヒドロキシ、NR2R3、0〜1個のR21で置換されているC1−C4アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR21で置換されているアリール、0〜1個のR21で置換されているアリール(C1−C6アルキル)−、0〜1個のR21で置換されている(C1−C4アルコキシ)カルボニル、0〜1個のR21で置換されている(C1−C4アルキル)カルボニル、0〜1個のR21で置換されているC1−C4アルキルスルホニル、および0〜1個のR21で置換されているC1−C4アルキルアミノスルホニルから選択され;
Wは−C(=O)−N(R13)−であり;
Xは−CH(R14)−CH(R15)−であり;
R13はHおよびCH3から選択され;
R14は、H、C1−C10アルキル、アリールおよびヘテロアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ハロ、シアノ、アミノ、CF3およびNO2から選択される0〜3置換基で場合によっては置換されており;
R15はHおよびR16から選択され;
Yは−COR19であり;
R16は
−NH(R20)−C(=O)−R17、
−N(R20)−C(=O)−R17、
−N(R20)−C(=O)−NH−R17、
―N(R20)SO2−R17、および
−N(R20)SO2−N(R20)R17
から選択され、
R17は、C1−C10アルキル、C3−C11シクロアルキル、アリール(C1−C6アルキル)−、(C1−C6アルキル)アリール、ヘテロアリール(C1−C6アルキル)−、(C1−C6アルキル)ヘテロアリール、ビアリール(C1−C6アルキル)−、ヘテロアリール若しくはアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ヘテロアリール、ハロ、シアノ、アミノ、CF3およびNO2よりなる群から選択される0〜3置換基で場合によっては置換されており;
R19は−O−(CH2)kN+(R22)(R23)(R24)Z−であり;
Z−は、ハロゲン化イオン、重硫酸イオン、硫酸イオン、リン酸水素イオン、リン酸イオン、トルエンスルホン酸イオン、メタンスルホン酸イオン、エタンスルホン酸イオン、酢酸イオン、トリフルオロ酢酸イオン、クエン酸イオン、シュウ酸イオン、コハク酸イオン
およびマロン酸イオンから選択される製薬学的に許容できる陰イオンであり;
R22、R23およびR24はH、C1−C4アルキルおよびC4−C11シクロアルキルアルキルから独立に選択されるか;
あるいは、R22およびR23は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する5〜7員の複素環系を形成し得、ならびにR24は上のとおり定義されるか、または、R22、R23およびR24は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する複素二環系を形成し得;
R20はHおよびCH3から選択され;
R21はCOOHおよびNR62から選択され;
kは2であり;
mは0および1から選択され;
nは1〜4であり;ならびに
tは0および1から選択される。
【0026】
該方法の一態様において、該剤は式II:
【0027】
【化3】
【0028】
の化合物であり、
ここでR19は−H、−CH3および−CH2CH2N+(CH3)3から選ばれる。一態様において、R19は−Hである。別の態様において、R19は−CH3である。別の態様において、R19は−CH2CH2N+(CH3)3である。
【0029】
該方法の別の態様において、該剤はディスインテグリンである。該方法の別の態様において、該剤はエキスタチンである。該方法の別の態様において、該剤はヒト抗体である。該方法の別の態様において、該剤はヒト化抗体である。該方法の別の態様において、該剤はキメラ抗体である。該方法の別の態様において、該剤はナノボディである。該方法の別の態様において、該剤は抗体フラグメントである。該方法の別の態様において、該剤は、抗体の1個若しくはそれ以上のH鎖、L鎖、F(ab)、F(ab)2、F(ab)c若しくはF(v)、またはそれらのいずれかの組合せを含んでなる。該方法の別の態様において、該剤は抗体でありかつ該抗体のアイソタイプはIgG1若しくはIgG4である。該方法の別の態様において、該剤は抗体でありかつ該抗体のアイソタイプはIgG2若しくはIgG3である。該方法の別の態様において、該剤は1抗体鎖である。該方法の別の態様において、該剤は抗体でありかつ該抗体は2対のLおよびH鎖を含んでなる。
【0030】
該方法の別の態様において、該剤は患者に投与される。一態様において、該剤は抗体でありかつ該抗体の投薬量は患者体重1kgあたり約0.01から約10mgまでの範囲に
わたる。一態様において、該剤は製薬学的組成物として担体とともに投与される。一態様において、該剤は、腹腔内、経口、鼻内、皮下、くも膜下腔内、筋肉内、局所若しくは静脈内で投与される。一態様において、患者はアミロイド形成疾患に罹っている。一態様において、該疾患は、アルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、アミロイドーシス、ダウン症候群、およびプリオン感染により全部若しくは部分的に引き起こされる疾患よりなる群から選ばれる。
【0031】
該方法の別の態様において、該剤をコードする核酸が投与される。該方法の別の態様において、該剤は、アンチセンスRNA分子、アンチセンスDNA分子、リボザイム、RNAiおよびジンクフィンガータンパク質よりなる群から選ばれる。別の態様において、該方法はアミロイド沈着物の形成を阻害することをさらに含んでなる。別の態様において、該方法はアミロイド毒性を阻害することをさらに含んでなる。該方法の別の態様において、該剤はLTPの維持期を阻害しない。該方法の別の態様において、該剤は培養中の薄片調製物でのLTPのアミロイド媒介性阻害を抑制する。該方法の別の態様において、該剤は可溶性AβによるLTPの阻害を抑制する。
【0032】
ある剤をインテグリンサブユニットαv結合剤と同定すること;および該同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することを決定することを含んでなる、LTPのアミロイド媒介性阻害を抑制する剤の同定方法もまた提供される。該方法の一態様において、剤を同定する段階は、直接結合アッセイ、競合結合アッセイおよび細胞接着アッセイの1種若しくはそれ以上を含んでなり;また、該同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することを決定する段階は、第一の神経回路に高頻度刺激を導入することおよびLTPの誘導を測定すること、Aβの存在下の第二の神経回路に高頻度刺激を導入することおよびLTP誘導の阻害を測定すること、ならびにAβおよび該剤の存在下の第三の神経回路に高頻度刺激を導入することおよびLTP誘導の阻害の抑制を測定することを含んでなる。
【0033】
該方法により同定されるLTPのアミロイド媒介性阻害を抑制する剤もまた提供される。一態様において、該剤は抗体である。
【0034】
該剤および製薬学的に許容できる担体を含んでなる組成物がさらに提供される。
【0035】
該方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法がさらに提供される。
【0036】
該方法により同定される、LTPのアミロイド媒介性阻害を抑制する剤もまた提供される。一態様において、該剤は抗体である。
【0037】
該剤および製薬学的に許容できる担体を含んでなる組成物もまた提供される。
【0038】
該方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法もまた提供される。
【0039】
αv拮抗剤若しくはαv媒介性細胞接着の阻害剤を長期増強(LTP)のアミロイド媒介性阻害を抑制するのに有効な量で投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置方法もまた提供される。一態様において、アミロイド形成疾患はアルツハイマー病である。別の態様において、アミロイド形成疾患は軽度認知障害である。
【0040】
1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下で
インテグリンサブユニットαvに結合する1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置若しくは予防方法もまた提供される。一態様において、アミロイド形成疾患はアルツハイマー病若しくは軽度認知障害である。一態様において、アミロイド形成疾患はパーキンソン病若しくはびまん性レビー小体病である。
【発明を実施するための形態】
【0041】
[発明の詳細な記述]
定義
本発明の治療薬は、典型的には望ましくない汚染物質から実質的に精製されている。これは、ある剤が典型的には最低約50w/w(重量)%純度であり、ならびに妨害するタンパク質および汚染物質を実質的に含まないことを意味している。ときに、該剤は最低約60%、70%、80%、90%若しくは95w/w%純度である。慣習的タンパク質精製技術を使用して、最低99w/w%の均質なペプチドもまた得ることができる。
【0042】
2実体間の特異的結合は最低106、107、108、109若しくは1010M−1の親和性を意味している。一態様において、親和性は約108M−1以上である。
【0043】
「抗体」若しくは「免疫グロブリン」という用語は、無傷の抗体およびそれらの結合フラグメントを包含するのに使用する。典型的に、フラグメントは、抗原フラグメントへの特異的結合について、それらが由来した無傷の抗体と競合し、個別のH鎖、L鎖Fab、Fab’、F(ab’)2、F(ab)cおよびFvを包含する。フラグメントは組換えDNA技術、または無傷の免疫グロブリンの酵素的若しくは化学的分離により製造しうる。「抗体」という用語は、他のタンパク質に化学的に複合されるか若しくは他のタンパク質との融合タンパク質として発現される1種若しくはそれ以上の免疫グロブリン鎖もまた包含する。「抗体」という用語は二特異性抗体もまた包含する。二特異性若しくは二機能性抗体は、2種の異なるH/L鎖対および2種の異なる結合部位を有する人工的ハイブリッド抗体である。二特異性抗体は、ハイブリドーマの融合若しくはFab’フラグメントの連結を包含する多様な方法により製造し得る(例えばSongsivilaiとLachmann、Clin.Exp.Immunol.、79:315−321(1990);Kostelnyら、J.Immunol.、148:1547−53(1992)を参照されたい)。
【0044】
APP695、APP751およびAPP770は、ヒトAPP遺伝子によりコードされるそれぞれ695、751および770アミノ酸残基長のポリペプチドを指す。Kangら、Nature、325:733−36(1987);Ponteら、Nature、331:525−27(1988);およびKitaguchiら、Nature、331:530−32(1988)を参照されたい。ヒトアミロイド前駆体タンパク質(APP)内のアミノ酸は、APP770アイソフォームの配列に従って番号を割り当てられている。Aβ39、Aβ40、Aβ41、Aβ42およびAβ43のような用語は、アミノ酸残基1−39、1−40、1−41、1−42および1−43を含有するAβペプチドを指す。Aβ42は配列(配列番号1):
H2N−Asp−Ala−Glu−Phe−Arg−His−Asp−Ser−Gly−Tyr−Glu−Val−His−His−Gln−Lys−Leu−Val−Phe−Phe−Ala−Glu−Asp−Val−Gly−Ser−Asn−Lys−Gly−Ala−Ile−Ile−Gly−Leu−Met−Val−Gly−Gly−Val−Val−Ile−Ala−OHを有する。Aβ41(配列番号3)、Aβ40(配列番号4)およびAβ39(配列番号5)は、C末端からのそれぞれAla、Ala−IleおよびAla−Ile−Valの脱落によりAβ42(配列番号1)と異なる。Aβ43(配列番号2)はC末端のトレオニン残基の存在によりAβ42(配列番号1)と異なる。
【0045】
「アミリン」は、当該技術分野で公知のタンパク質、またはそのペプチド若しくはポリペプチド若しくはフラグメント、あるいは該タンパク質、ペプチド若しくはポリペプチドの前駆体タンパク質若しくはポリマーを指す。該用語は島アミロイドポリペプチドを包含する。アミリンの記述は、当該技術分野でCooperら、Proc.Natl.Acad.Sci.U.S.A.、85:7763(1988)およびLeightonら、Nature、335:632(1988)のような場所で見出しうる。
【0046】
「アミロイドペプチド若しくはタンパク質」という用語は、アミリンおよびAβを包含するアミロイド様沈着物を形成するペプチドおよびタンパク質のファミリーを指す。
【0047】
「アミロイド若しくはアミロイド様沈着物」という句は、アミロイド原線維、ならびに、老人性アミロイドーシス(例えばAβ)、プリオン関連脳症(例えばPrP)および糖尿病患者の腎若しくは膵(例えばアミリン)での沈着物などのような、アミロイド若しくはアミロイド様であると当該技術分野で認識されている構造が原線維若しくは非原線維の他のアミロイド若しくはアミロイド様沈着物を包含する。慣習的染色を用いる光学顕微鏡検査でこうした沈着物は均一なエオシン好性の外見を有する。コンゴーレッド染色でそれらは偏光下で特徴的な黄緑色の複屈折を示す。該用語は、アミロイド沈着物と異なりコンゴーレッドで染色しないプレアミロイド沈着物もまた包含する。
【0048】
「アミロイド形成疾患」という用語は、タンパク質若しくはペプチドの不要な沈着を特徴とする疾患を包含することを意図している。該用語は、Kahnら、Diabetes、48:241−253(1999);およびJohnsonら、Laboratory
Investigation、66(5):522−535(1992)により記述されるところのII型糖尿病(例えばアミリン)、アルツハイマー病(例えばAβ)、多発性骨髄腫および関節リウマチのような、アミロイドペプチドの不要な沈着を特徴とする疾患をとりわけ包含する。該用語は、トランスサイレチン(TTR)沈着により媒介されるものを包含する、Hundら、Neurology、56:431−435(2001)により記述されるところのパーキンソン病または遺伝性若しくは全身性アミロイドーシスのようなアミロイド形成タンパク質の不要な沈着を特徴とする疾患もまたとりわけ包含する。該用語はびまん性レビー小体病もまた包含する。さらに、該用語は、クロイツフェルト・ヤコブ病のようなプリオンへの感染により全部若しくは部分的に引き起こされる疾患を包含する。こうしたプリオン媒介性疾患は、Gieseら、Curr.Topics Microbiology and Immunology、253:203−217(2001)により記述されるところのプリオンタンパク質の蓄積を特徴とする。約言すれば、該用語は、周囲の細胞の健康および満足のいく状態に悪影響を及ぼす不要なタンパク質若しくはペプチド沈着物により病状が媒介される全疾患を包含することを意味している。
【0049】
「抗原」は抗体が特異的に結合する実体である。
【0050】
「エピトープ」若しくは「抗原決定基」という用語は、Bおよび/若しくはT細胞が応答する抗原上の1部位を指す。B細胞エピトープは、連続するアミノ酸、若しくはタンパク質の三次フォールディングにより並置される連続しないアミノ酸の双方から形成され得る。連続するアミノ酸から形成されるエピトープは変性溶媒への曝露に際して典型的に保持される一方、三次フォールディングにより形成されるエピトープは変性溶媒での処理に際して典型的に喪失される。エピトープは、典型的に、独特の空間的コンホメーション中に最低3、およびより通常は最低5若しくは8〜10アミノ酸を包含する。エピトープの空間的コンホメーションの決定方法は、例えばx線結晶学および二次元核磁気共鳴を包含する。例えば、Methods in Molecular Biology、Vol.
66、Glenn E.Morris編(1996)中Epitope Mapping
Protocolsを参照されたい。同一の若しくは重なるエピトープを認識する抗体は、標的抗原への別の抗体の結合を阻止する1抗体の能力を示す単純なイムノアッセイで同定し得る。
【0051】
「裸のポリヌクレオチド」若しくは「裸のDNA」という用語は、コロイド状物質例えばタンパク質と複合体形成されていないポリヌクレオチドを指す。裸のポリヌクレオチドはときにプラスミドベクターにクローン化される。
【0052】
「患者」という用語は予防的若しくは治療的処置のいずれかを受領するヒトおよび他の哺乳動物被験体を包含する。
【0053】
「予防する」、「予防すること」および「予防」という用語は、疾患若しくは状態の最低1症状(例えば長期増強の抑制および/若しくは神経変性)を究極的に明示しうるがしかし未だそうしていない個体への予防的(prophylactic)若しくは予防的(preventative)な治療の投与を指す。こうした個体は、該疾患のその後の発生と相関することが既知である危険因子に基づき同定しうる。あるいは、予防治療は予防対策として危険因子の事前の同定を伴わず投与しうる。疾患若しくは状態の最低1症状の発生を遅らせることもまた予防(prevention)若しくは予防(prophylaxis)とみなしうる。
【0054】
本明細書で使用されるところの「処置する」、「処置すること」若しくは「処置」という用語は、疾患若しくは状態の最低1症状(例えば長期増強の抑制および/若しくは神経変性)を既に明示している個体への治療の投与を指す。
【0055】
剤および第二の剤の「共投与」は、in vitro系、若しくは患者でのようなin
vivoで時間が重なる剤および第二の剤の治療濃度を達成するためのいかなる投薬計画による投与も包含する。従って、例えば、共投与は、剤および第二の剤双方を含んでなる製剤の投与、ならびに一方が該剤を含んでなりかつ別のものが第二の剤を含んでなる別個の製剤の投与を包含する。別個の製剤を投与する場合、投与は同時であっても連続してもよい。連続する場合、該剤は次々に投与しうるか、あるいは、該剤の投与と第二の剤の投与の間の時間は、例えば1時間まで、2時間まで、4時間まで、6時間まで、12時間まで、または1日若しくは数日まででありうる。
【0056】
神経細胞は、in vivoで起こるAPPのAβへの天然のプロセシングの結果として、若しくは、in vitroアッセイでのAβの調製物と神経細胞の接触の結果として、Aβペプチドに曝露し得る。Aβペプチドへの曝露は薬物への曝露前、後若しくはそれと同時に発生し得る。
【0057】
Aβペプチドのアミロイド沈着物は、図1Aに示されるようなin vitro若しくはin vivoで皮質細胞上および周囲に生じる原線維をおそらく包含するAβペプチドの凝集物を指す。
【0058】
文脈から別の方法で明らかでない限り、フィブロネクチンへの言及はスーパーフィブロネクチンを包含する。
【0059】
抗体間の競合は、試験中の免疫グロブリンが共通抗原への参照抗体の特異的結合を阻害するアッセイにより測定する。多数の型の競合結合アッセイ、例えば固相直接若しくは間接ラジオイムノアッセイ(RIA)、固相直接若しくは間接エンザイムイムノアッセイ(EIA)、サンドイッチ競合アッセイ(Stahliら、Methods in Enz
ymology、9:242−53(1983)を参照されたい);固相直接ビオチン−アビジンEIA(Kirklandら、J.Immunol.、137:3614−19(1986)を参照されたい);固相直接標識アッセイ、固相直接標識サンドイッチアッセイ(HarlowとLane、”Antibodies,A Laboratory Manual”、Cold Spring Harbor Press(1988)を参照されたい);I−125標識を使用する固相直接標識RIA(Morelら、Molec.Immunol.、25(1):7−15(1988)を参照されたい);固相直接ビオチン−アビジンEIA(Cheungら、Virology、176:546−52(1990));および直接標識RIA(Moldenhauerら、Scand.J.Immunol.、32:77−82(1990))が既知である。典型的に、こうしたアッセイは、未標識の試験免疫グロブリン若しくは標識参照免疫グロブリンいずれかを持つ固体表面若しくは細胞に結合した精製抗原の使用を必要とする。競合阻害は、試験免疫グロブリンの存在下で固体表面若しくは細胞に結合される標識の量を測定することにより測定する。通常、試験免疫グロブリンは過剰に存在する。競合アッセイにより同定される抗体(競合抗体)は、参照抗体と同一エピトープに結合する抗体、および参照抗体により結合されるエピトープの十分に近位の隣接するエピトープに結合して立体障害を起こす抗体を包含する。通常、競合抗体が過剰に存在する場合、それは参照抗体の共通抗原への特異的結合を最低50若しくは75%阻害することができる。
【0060】
1種若しくはそれ以上の列挙される要素を「含んでなる」組成物若しくは方法は、具体的に列挙されない他の要素を包含しうる。例えば、抗体を含んでなる組成物は、単独若しくは他の成分と組合せの該抗体を含有しうる。
【0061】
I.方法
本発明は、アミリンおよびAβペプチドのようなアミロイドタンパク質の細胞外網目構造の形成の阻害若しくは予防方法、こうしたタンパク質の毒性効果の媒介方法、ならびに該方法での使用のための剤を提供する。該方法は、アルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、全身性および遺伝性アミロイドーシス、ならびにプリオン感染により全部若しくは部分的に引き起こされる疾患を処置若しくは予防するのに使用し得る。これらの方法での使用に有効な剤は、β1、α2、α6若しくはαvのようなインテグリンサブユニットに結合する抗体および他の剤を包含する。これらのサブユニットはヘテロ二量体受容体として会合してインテグリン、例えばα2β1、α6β1およびαvβ1を形成する。上の剤は、インテグリンとAβペプチドの間の相互作用を阻害するために個別で若しくは組合せで使用し得る。αvβ1およびα2β1双方のインテグリンとAβの間の相互作用を阻害する剤(1種若しくは複数)の使用が好ましい。フィブロネクチン(インテグリンαvβ1のリガンド)もまた、上の方法でラミニン(αvβ1のリガンド)に対する抗体がし得るように剤として使用し得る。本発明は、部分的に、α2、αv、α6およびβ1インテグリンサブユニットに対する抗体がアミリンおよびAβペプチドのようなアミロイドタンパク質の細胞外網目構造の形成を阻害するという観察結果を前提とする。それにより、こうした抗体はアミロイドタンパク質の毒性を阻害する。αvβ1リガンド、フィブロネクチンもまた網目構造形成を阻害する。α2β1リガンド、ラミニンは網目構造形成を阻害しないが、しかしラミニンに対する抗体は網目構造形成および毒性を阻害する。
【0062】
本発明は、部分的に、αvインテグリンサブユニットに対する選択的抗体がin vitroおよびin vivo双方でAβによるLTPの阻害を抑制するという観察結果を前提とする。αv含有インテグリンの小分子非ペプチド拮抗剤、ならびにインテグリンの2種の他の拮抗的リガンド、スーパーフィブロネクチンおよびディスインテグリン、エキスタチンもまたLTPのAβ阻害を抑制する。従って、αvインテグリンサブユニットに結合する剤はLTPのAβ阻害を抑制し、ならびに、アミリンおよびAβペプチドのよう
なアミロイドタンパク質の細胞外網目構造の形成を阻害若しくは予防する。
【0063】
従って、本発明は、長期増強(LTP)のアミロイド媒介性阻害を抑制するのに有効な量のαv拮抗剤若しくはαv媒介性細胞接着の阻害剤を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法、およびAβ沈着を特徴とするアミロイド形成疾患の処置若しくは予防方法、ならびに、Aβの沈着を特徴とするアミロイド形成疾患の処置方法もまた提供する。該方法は、限定されるものでないがアルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、全身性および遺伝性アミロイドーシスを挙げることができる疾患若しくは状態、ならびにプリオン感染により全部若しくは部分的に引き起こされる疾患を処置若しくは予防するのに有用である。これらの方法での使用に有効な剤は、限定されるものでないが、抗体、および例えばインテグリンサブユニットαvに結合するSM256のような他の剤を挙げることができる。上の剤は、インテグリンとAβペプチドの間の相互作用を阻害するのに個別で若しくは組合せで使用し得る。
【0064】
II.インテグリン
インテグリンは、細胞外マトリックスおよび他細胞の双方への細胞の接着を制御する、細胞表面接着ヘテロ二量体膜貫通受容体のスーパーファミリーである。接着は成長、移動および分化のための固定およびシグナルを提供する。インテグリンは、約15種の既知のα鎖の1種の約8種の既知のβ鎖の1種との会合により形成される。赤血球を除く全部のヒト細胞が1種若しくはそれ以上のインテグリンを発現する。
【0065】
インテグリンサブユニットα2、αv、α6およびβ1は全部公知である。例示的ヒト配列はそれぞれGenBank受託番号AF062039、M14648、X59512およびX07979から取出し可能である。別の方法で示されない限り、α2、αv、α6、β1への言及は、これらの例示的配列、それらのアレルバリアントおよび他の種からの同族バリアントを包含する。天然の配列のリガンドへの特異的結合について天然の配列と競合するのに十分な天然の配列に対する配列同一性を有するこれらの配列の誘導されるバリアントもまた、いくつかの方法で使用し得る。αv、およびβサブユニットβ1、β3、β5、β6若しくはβ8の1種を含有するインテグリンはRGDモチーフを持つリガンドを認識するが、しかし結合特異性はどのβサブユニットが存在するかに依存して変動する。αvβ1は、ビトロネクチン(GenBank受託番号X03168)、フィブロネクチン(GenBank受託番号M26179)およびオステオポンチン(GenBank受託番号J04765)を認識することが既知である。フィブロネクチンは、結合組織中、細胞表面上、ならびに血漿および他の体液中で見出される大型多ドメイン糖タンパク質である。フィブロネクチンは、細胞骨格および細胞外マトリックスの成分、血液凝固応答、線維素溶解の急性期および補体系に関与する循環成分を包含する多様な巨大分子、ならびに線維芽細胞、ニューロン、食細胞および細菌を包含する多様な細胞上の細胞表面受容体と作用する。
【0066】
α2およびβ1サブユニットを含有するインテグリンは、VLA−2(最晩期抗原(very late activation antigen)2)、GPIa−IIa(血小板上糖タンパク質Ia−IIa)およびECMRII(細胞外マトリックス受容体II)として知られる。α2β1インテグリンはコラーゲンIないしVI、ラミニンおよびおそらくフィブロネクチンを結合する。該受容体は、BおよびTリンパ球、血小板、線維芽細胞、内皮細胞ならびに黒色腫細胞上で発現され、そしてコラーゲンおよびラミニンをリガンドとして特異的に認識する。ラミニンは共通の構造的構成をもつ大型多ドメインタンパク質である。ラミニン分子は、コイルドコイルドメインにより一緒に結合されたα、βおよびγ鎖サブユニットを有する。最低5種のα鎖、2種のβ鎖および3種のγ鎖が既知であり、そしてこれらの鎖の異なる組合せを有する最低12種のラミニンが報告されて
いる(第WO 00/66730号明細書)。ラミニンはアルツハイマー病において斑を包含する細胞外マトリックス中で見出される(Murtomakiら、J.Neuro.Res.、32:261−73(1992);Bronfinanら、Int.J.Exp.Clin.Invest.、5:16−23(1997);およびCastilloら、J.Neuro.Res.、62:451−62(2000))。コラーゲンは哺乳動物で最も豊富なタンパク質であり、そして皮膚、骨、腱、軟骨および歯の主要線維成分である。23種以上の既知のコラーゲン遺伝子が存在する(Adamsら、Am.J.Respir.Cell.Molec.Biol.、1:161−168(1989))。
【0067】
α6/β1インテグリンは、血小板、リンパ球、単球、胸腺細胞および内皮細胞上で発現され、それら上でそれはin vivoでラミニン−1、ラミニン−2およびラミニン−4のラミニン受容体として機能する。それはまたラミニン−5の受容体でもあるが、しかしin vivoででない。ラミニン−1について、結合部位はこの細胞外マトリックス分子のE8ドメイン中に突き止められている。この受容体は最晩期抗原6(VLA−6)および糖タンパク質Ic−IIa(血小板上GPIc−IIa)としてもまた知られている。
【0068】
インテグリンは、セレクチンおよび免疫グロブリン(Ig)スーパーファミリーメンバーもまた包含する接着タンパク質として知られるタンパク質の大きな一分類の一例である(Springer、Nature、346:425(1990);Osborn、Cell、62:3(1990);Hynes、Cell、69:11(1992)(全部の目的上そっくりそのまま引用することにより組み込まれる)を参照されたい)。抗体、および接着タンパク質若しくはそれらのリガンドに結合しかつ/または該2者間の相互作用を阻止する他の剤を、下述されるスクリーニング方法でAβ沈着物の蓄積の予防若しくは阻害における活性についてスクリーニングし得る。下述される方法によるスクリーニングに適する他のセレクチンおよびそれらのリガンドの例は、インテグリンα2β5、αvβ5、α6β5、α2β6、αvβ6およびα6β6を包含する。コラーゲンに結合するα2β1を除く他のリガンドもまたスクリーニングしうる。
【0069】
III.剤
本発明の治療薬は、α2、αv、α6およびβ1インテグリンサブユニットに特異的に結合する抗体を包含する。結合は、場合によっては固相に固定された単離されたインテグリンサブユニット若しくはそれらのフラグメント、または細胞の表面上で発現されたインテグリンサブユニットのいずれを用いても評価し得る。しばしば、結合はヘテロ二量体インテグリンを発現する細胞を使用して分析される。例えば、ある剤が唯一のインテグリンとしてα2β1を発現する細胞に結合する場合には、該剤がα2若しくはβ1またはいずれのサブユニット単独にも結合することなくα2β1に結合すると結論付け得る。これらの可能性は、異なるヘテロ二量体インテグリンを持つ細胞への同一の剤の結合を試験することにより識別し得る。例えば、同一の剤が、存在する唯一のインテグリンとしてαvβ1を持つ細胞に特異的に結合する場合には、該剤がβ1サブユニットに結合していることがありそうである。インテグリンおよびインテグリンサブユニットに対する多様な抗体が商業的に入手可能であり、それらのいくつかを実施例に記述する。
【0070】
モノクローナル若しくはポリクローナル抗体を本発明の方法で使用し得る。好ましい抗体は、それらの天然のリガンドの1種若しくはそれ以上とのこれらのインテグリンサブユニットの相互作用を阻止する。すなわち、αvβ1に対する阻止抗体は、フィブロネクチン、オステオポンチンおよび/若しくはビトロネクチンとのこのインテグリンの相互作用を阻止する。例えば、第WO 99/37683号明細書により記述される14D9.F8抗体はαvのフィブロネクチンへの結合を阻止する。α2β1に対する阻止抗体はこのインテグリンのコラーゲン若しくはラミニンとの相互作用を阻止する。抗体若しくは他の
剤の阻止する能力は、抗体(若しくは他の剤)の存在若しくは非存在下で、リガンドで被覆したプレートへの接着についてインテグリンを発現する細胞を試験する単純なアッセイにより認識し得る。プレートへの細胞結合の量の最低約30%、40%、50%、60%、70%、80%、90%若しくは100%の減少は、該抗体がインテグリンに関してモル濃度過剰で存在する場合に阻止抗体(若しくは他の剤)を同定する。インテグリンサブユニットの他の組合せに対する剤の阻止能力のさらなる分析は、ヘテロ二量体インテグリンのどのサブユニットが阻止されているかを特定し得る。抗体若しくは他の剤の結合特異性もまた、インテグリンサブユニット若しくはそれを持つ細胞への結合について所望のエピトープ特異性を有することが既知の参照抗体と試験抗体が競合する競合アッセイにより決定し得る。試験および参照抗体が競合する場合には、それらは、一方の抗体の結合が他方の結合を妨害する十分に近位の同一のエピトープ(1個若しくは複数)に結合する。いくつかの態様において、トランスフェクトした細胞は単一の型のインテグリンを発現する。
【0071】
本発明での使用のための数種の抗体はただ1つの型のインテグリンサブユニットに結合する。数種の抗体は2種若しくはそれ以上のインテグリンサブユニットに特異的に結合する。数種の抗体は、インテグリンのサブユニットがヘテロ二量体インテグリンとして会合している場合にのみ結合する。例えば、数種の抗体はα2若しくはβ1いずれか単独に結合することなくα2β1に結合する。数種の抗体はαv若しくはβ1いずれか単独に結合することなくαvβ1に結合する。数種の抗体はαvインテグリンサブユニットに結合する。数種の抗体は、遊離の形態で、および該サブユニットがヘテロ二量体インテグリンの一成分である場合の双方でサブユニットに結合する。上の抗体の同一の結合特異性を有するペプチドおよび小分子もまた使用し得る。
【0072】
本発明での使用のための他の治療薬は、フィブリノーゲン、オステオポンチン、ビトロネクチン、それらのフラグメント、およびαvβ1への結合についてフィブリノーゲン若しくはビトロネクチンと競合するRGDペプチドモチーフを含有する他の天然の若しくは合成ペプチドを包含する。αvβ1への結合についてフィブリノーゲン、ビトロネクチン若しくはオステオポンチンと競合する小分子模倣物もまた使用し得る。他の治療薬は、ラミニンに対する抗体、ならびに同一の結合特異性をもつペプチドおよび小分子を包含する。
【0073】
候補治療薬は以下のスクリーニングの1種若しくはそれ以上を実施することにより評価し得る。典型的には、インテグリンサブユニットα2、αv、α6若しくはβ1、および/またはヘテロ二量体インテグリンα2β1、αvβ1 α6β1、あるいはラミニンへの特異的結合について剤を最初に評価する。適する剤は、典型的に最低107、108、109若しくは1010M−1の特異的親和性で結合する。
【0074】
その後、候補は場合によっては特定のエピトープ特異性について評価する。これは、参照剤を用いる競合アッセイ、上述されたところの機能的プレートブロッキングアッセイ、または、抗原の一連の欠失変異体を結合するその能力についウエスタンブロッティング若しくはELISAにより抗体若しくは他の剤を評価するエピトープマッピング実験により決定し得る。抗体若しくは他の剤への特異的結合を示すための最小フラグメントが該抗体若しくは他の剤のエピトープを規定する。あるいは、若しくは加えて、候補剤をアミロイドペプチドの細胞外網目構造の形成を阻害する能力について評価する。適する剤は、典型的に、剤の存在下でアミリン若しくはAβのようなアミロイドペプチドでの処理から生じる毒性を対照に関して最低約10%、20%、30%、40%、50%、60%、70%、80%、90%若しくは100%またはそれ以上低下させる。
【0075】
候補化合物は、アミロイド形成疾患になりやすいトランスジェニック動物で予防的およ
び治療的有効性についてもまた試験し得る。こうした動物は、例えば、Gamesら、上記により記述されるAPPの717突然変異を持つマウス、ならびにMcConlogueら、米国特許第US 5,612,486号明細書;Hsiaoら、Science、274:99(1996);Sturchler−Plerratら、Proc.Natl.Acad.Sci.U.S.A.、94:13287−92(1997);およびBorcheltら、Neuron、19:939−45(1997)により記述されるようなAPPの670/671スウェーデン型突然変異を持つマウスを包含する。トランスジェニックマウスで活性を示す剤をその後ヒト臨床試験で評価し得る。アルツハイマー病患者でヒト臨床試験を実施するための例示的形式が第WO 98/24678号明細書(引用することにより本明細書に組み込まれる)に記述されている。
【0076】
長期増強(LTP)のアミロイド媒介性阻害の抑制方法での使用のための候補化合物の場合、該化合物をLTPのin vitroおよび/若しくはin vivoモデルで試験しうる。例えば、候補化合物をin vitroモデルで最初に試験することができ、そしてその後、該化合物がそのモデルで長期増強(LTP)のアミロイド媒介性阻害を抑制する場合は、in vivoモデルでその後試験しうる。
【0077】
A.抗体
1.免疫グロブリンの一般的特徴
基本的抗体構造ユニットはサブユニットの四量体を含んでなることが知られている。各四量体はポリペプチド鎖の2個の同一の対から構成され、各対は1本の「L」(約25kDa)および1本の「H」鎖(約50〜70kDa)を有する。各鎖のアミノ末端部分は主に抗原認識を司る約100ないし110若しくはそれ以上のアミノ酸の可変領域を包含する。各鎖のカルボキシ末端部分は主にエフェクター機能を司る定常領域を規定する。
【0078】
L鎖はκ若しくはλいずれかに分類される。H鎖はγ、μ、α、δ若しくはεに分類され、そしてそれぞれIgG、IgM、IgA、IgDおよびIgEのような抗体のアイソタイプを規定する。LおよびH鎖内で、可変および定常領域は約12若しくはそれ以上のアミノ酸の「J」領域により結合され、H鎖は約10より多いアミノ酸の「D」領域もまた包含する。(全般として、Fundamental Immunology(Paul,W.編、第2版、Raven Press、ニューヨーク、1989)、第7章(全部の目的上そっくりそのまま引用することにより組み込まれる)を参照されたい)。
【0079】
各L/H鎖対の可変領域は抗体の結合部位を形成する。従って無傷の抗体は2個の結合部位を有する。二機能性若しくは二特異性抗体でを除き該2結合部位は同一である。該鎖は全部、相補性決定領域若しくはCDRともまた呼ばれる3個の超可変領域により結合される比較的保存された枠組み領域(FR)の同一の一般的構造を表す。各対の2本の鎖からのCDRは枠組み領域により整列されて、特異的エピトープへの結合を可能にする。N末端からC末端へ、LおよびH鎖双方はドメインFR1、CDR1、FR2、CDR2、FR3、CDR3およびFR4を含んでなる。各ドメインへのアミノ酸の割り当ては、Kabat、Seqeunces of Proteins of Immunological Interest(国立保健研究所、メリーランド州ベセスダ、1987および1991)、若しくはChothiaとLesk、J.Mol.Biol.、196:901−17(1987);Chothiaら、Nature、342:878−83(1989)の定義に従う。
【0080】
2.ヒト以外の抗体の製造
ヒト以外のモノクローナル抗体(例えばマウス、モルモット、霊長類、ウサギ若しくはラット)の製造は、例えば、インテグリン、そのサブユニット若しくはそれらのフラグメント、またはインテグリン若しくはそのサブユニットを持つ細胞で動物を免疫することに
より達成し得る。ラミニンに対する抗体を生成するための免疫原としてラミニンもまた使用し得る。HarlowとLane、Antibodies,A Laboratory
Manual(Cold Spring Harbor Press、ニューヨーク、1988(全部の目的上引用することにより本明細書に組み込まれる))を参照されたい。こうした免疫原は、天然の供給源から、ペプチド合成により、若しくは組換え発現により得ることができる。場合によっては、免疫原は下述されるところの担体タンパク質と融合若しくは別の方法で複合体形成して投与し得る。場合によっては、免疫原はアジュバントとともに投与し得る。いくつかの型のアジュバントを下述されるとおり使用し得る。フロイントの完全アジュバント、次いで不完全アジュバントが実験動物の免疫化に好ましい。ウサギ、ヤギ、ヒツジ若しくはモルモットを、ポリクローナル抗体を作成するのに典型的に使用する。マウスはモノクローナル抗体を作成するのに典型的に使用する。抗体を、意図しているインテグリン若しくはそのサブユニット、またはラミニンのような他の抗原への特異的結合についてスクリーニングする。抗体は上述されたところのそのリガンドへのインテグリンの結合を阻止する能力についてもまたスクリーニングし得る。上述された他のスクリーニング手順もまた実施し得る。
【0081】
3.キメラおよびヒト化抗体
キメラおよびヒト化抗体は、キメラ若しくはヒト化抗体の構築のための出発原料を提供するマウス若しくは他のヒト以外の抗体と同一の若しくは類似の結合特異性および親和性を有しうる。数種のキメラ若しくはヒト化抗体はマウスのものの2倍、5倍若しくは10倍の係数内の親和性を有する。キメラ抗体は、そのLおよびH鎖遺伝子が、異なる種に属する免疫グロブリン遺伝子セグメントから典型的には遺伝子工学により構築された抗体である。例えば、マウスモノクローナル抗体からの遺伝子の可変(V)セグメントをIgG1、IgG2、IgG3若しくはIgG4のようなヒト定常(C)セグメントに結合しうる。典型的なキメラ抗体は、従って、マウス抗体からのVすなわち抗原結合ドメインおよびヒト抗体からのCすなわちエフェクタードメインよりなるハイブリッドタンパク質である。
【0082】
ヒト化抗体は、実質的にヒト抗体(アクセプター抗体と命名される)からの可変領域枠組み残基、および実質的にマウス抗体のようなヒト以外の抗体(ドナー免疫グロブリンと称される)からの相補性決定領域を有する。Queenら、Proc.Natl.Acad.Sci.U.S.A.、86:10029−33(1989)および第WO 90/07861号、米国特許第US 5,693,762号、同第US 5,693,761号、同第US 5,585,089号、同第US 5,530,101号、およびWinter、米国特許第US 5,225,539号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)を参照されたい。定常領域(存在する場合)もまた実質的に若しくは完全にヒト免疫グロブリンからである。ヒト可変ドメインは通常、CDRが由来したマウス可変領域ドメインとの高程度の配列同一性をその枠組み配列が表すヒト抗体から選ばれる。HおよびL鎖可変領域枠組み残基は同一若しくは異なるヒト抗体配列に由来し得る。ヒト抗体配列は天然に存在するヒト抗体の配列であり得るか、若しくは数種のヒト抗体のコンセンサス配列であり得る。Carterら、第WO 92/22653号明細書を参照されたい。ヒト可変領域枠組み残基からのあるアミノ酸を、CDRコンホメーションおよび/若しくは抗原への結合に対するそれらの可能な影響に基づき置換のため選択する。こうした可能な影響の検討は、モデル化、特定の場所のアミノ酸の特徴の検査、または特定のアミノ酸の置換若しくは突然変異誘発の影響の経験的観察による。
【0083】
例えば、あるアミノ酸がマウス可変領域枠組み残基と選択したヒト可変領域枠組み残基の間で異なる場合、該ヒト枠組みアミノ酸は通常、該アミノ酸が:
(1)抗原を直接非共有結合する、
(2)CDR領域に隣接する、
(3)CDR領域と別の方法で相互作用する(例えばCDR領域の約6Å以内にある)、若しくは
(4)VL−VH界面に参画する
ことが合理的に期待される場合は、マウス抗体からの同等の枠組みアミノ酸により置換すべきである。
【0084】
置換の他の候補は、その位置でヒト免疫グロブリンにとって異常であるアクセプターヒト枠組みアミノ酸である。これらのアミノ酸を、ドナー抗体の同等の位置若しくはより典型的なヒト免疫グロブリンの同等の位置からのアミノ酸で置換し得る。ヒト化免疫グロブリンの可変領域枠組みは通常、ヒト可変領域枠組み配列若しくはこうした配列のコンセンサスに対し最低85%の配列同一性を示す。
【0085】
4.ヒト抗体
上のインテグリン若しくはラミニンに対するヒト抗体は下述される多様な技術により提供される。数種のヒト抗体は、競合結合実験により、若しくは、そうでなければ、実施例に記述されるマウスモノクローナル抗体の1種のような特定の1マウス抗体と同一のエピトープ特異性を有するように選択する。ヒト抗体は、免疫原としてインテグリン若しくはラミニンの1フラグメントのみを使用することにより、および/またはインテグリンの欠失変異体の集合物に対し抗体をスクリーニングすることにより、特定の1エピトープ特異性についてもまたスクリーニングし得る。
【0086】
a.トリオーマの方法論
基本的アプローチ、および本アプローチでの使用のための例示的1細胞融合パートナーSPAZ−4が、Oestbergら、Hybridoma、2:361−67(1983);Oestberg、米国特許第4,634,664号明細書;およびEnglemanら、米国特許第4,634,666号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)により記述されている。この方法により得られる抗体産生細胞株は、それらが3種の細胞(2種のヒトおよび1種のマウス)を祖先とするためトリオーマと呼ばれる。最初に、マウス骨髄腫株をヒトBリンパ球と融合して、Oestberg、上記により記述されるSPAZ−4細胞株のような抗体を産生しない異種ハイブリッド細胞を得る。該異種細胞をその後、免疫ヒトBリンパ球と融合して抗体産生トリオーマ細胞株を得る。トリオーマはヒト細胞から作成される通常のハイブリドーマより安定に抗体を産生することが見出されている。
【0087】
免疫Bリンパ球はヒトドナーの血液、脾、リンパ節若しくは骨髄から得る。特定の1抗原若しくはエピトープに対する抗体が望ましい場合は、免疫化にその抗原若しくはその1エピトープを使用することが好ましい。免疫化はin vivo若しくはin vitroのいずれでもあり得る。in vivo免疫化のため、B細胞を、典型的に、Aβ、そのフラグメント、Aβ若しくはフラグメントを含有するより大きいペプチド、またはAに対する抗体に対する抗イディオタイプ抗体で免疫したヒトから単離する。いくつかの方法において、B細胞は抗体療法を最終的に投与されるはずである同一患者から単離する。in vitro免疫化のためには、Bリンパ球は、典型的に、10%ヒト血漿を補充したRPMI−1640(Engleman、上記を参照されたい)のような培地中で7〜14日間抗原に曝露する。
【0088】
免疫Bリンパ球を公知の方法によりSPAZ−4のような異種ハイブリッド細胞に融合する。例えば、細胞をMW1000〜4000の40〜50%ポリエチレングリコールで約37℃で約5〜10分間処理する。細胞を融合混合物から分離し、そして所望のハイブリッドについて選択的な培地(例えばヒポキサンチン+アメトプテリン+チミジン(HA
T培地)若しくはアメトプテリン+ヒポキサンチン(AH培地)を含有する)中で増殖させる。必要とされる結合特異性を有する抗体を分泌するクローンは、Aβ若しくはそのフラグメントに結合する能力についてトリオーマ培地をアッセイすることにより同定する。所望の特異性を有するヒト抗体を産生するトリオーマを限界希釈技術によりサブクローニングし、そして培地中in vitroで増殖させる。得られるトリオーマ細胞株をその後Aβ若しくはそのフラグメントを結合する能力について試験する。
【0089】
トリオーマは遺伝子的に安定であるとは言え、それらは非常に高レベルで抗体を産生しない。発現レベルは、抗体遺伝子をトリオーマから1種若しくはそれ以上の発現ベクターにクローン化すること、および該ベクターを標準的哺乳動物、細菌若しくは酵母細胞株に形質転換することにより増大させ得る。
【0090】
b.トランスジェニックのヒト以外の哺乳動物
インテグリン若しくはラミニンに対するヒト抗体は、ヒト免疫グロブリン遺伝子座の最低1セグメントをコードする導入遺伝子を有するヒト以外のトランスジェニック哺乳動物からもまた製造し得る。通常、こうしたトランスジェニック哺乳動物の内因性免疫グロブリン遺伝子座は機能的に不活性化されている。好ましくは、ヒト免疫グロブリン遺伝子座のセグメントは、HおよびL鎖コンポーネントの再配列されない配列を包含する。内因性免疫グロブリン遺伝子の不活性化および外因性免疫グロブリン遺伝子の導入の双方を、標的を定めた相同的組換え、若しくは酵母人工染色体(YAC)の導入により達成し得る。この方法から生じるトランスジェニック哺乳動物は、免疫グロブリンコンポーネントの配列を機能的に再配列すること、および内因性免疫グロブリン遺伝子を発現することなくヒト免疫グロブリン遺伝子によりコードされる多様なアイソタイプの抗体のレパートリーを発現することが可能である。これらの特性を有する哺乳動物の製造および特性は、例えばLonbergら、第W093/12227号(1993);第US 5,877,397号、同第US 5,874,299号、同第US 5,814,318号、同第US5,789,650号、同第US 5,770,429号、同第US 5,661,016号、同第US 5,633,425号、同第US 5,625,126号、同第US 5,569,825号、同第US 5,545,806号明細書、Nature 148、1547−53(1994)、Fishwildら、Nature Biotechnology、14、845−51(1996)、Kucherlapati、第WO 91/10741号明細書(1991)(それらのそれぞれは全部の目的上そっくりそのまま引用することにより組み込まれる)により詳細に記述されている。トランスジェニックマウスはとりわけ適する。抗インテグリン若しくは抗ラミニン抗体は、Lonberg若しくはKucherlapati、上記により記述されたようなトランスジェニックのヒト以外の哺乳動物をインテグリンまたはそのサブユニット若しくはフラグメントで免疫することにより得られる。モノクローナル抗体は、例えば、慣習的なKohler−Milsteinの技術を使用してこうした哺乳動物からのB細胞を適する骨髄腫細胞株に融合することにより製造する。ヒトポリクローナル抗体は、免疫原性剤で免疫したヒトからの血清の形態でもまた提供し得る。場合によっては、こうしたポリクローナル抗体は、インテグリン若しくはラミニンをアフィニティー試薬として使用するアフィニティー精製により濃縮し得る。
【0091】
c.ファージディスプレイ法
ヒト抗インテグリン若しくは抗ラミニン抗体を得るためのさらなる1アプローチは、Huseら、Science、246:1275−81(1989)により概説される一般的プロトコルに従ってヒトB細胞からのDNAライブラリーをスクリーニングすることである。トリオーマの方法論について記述されたとおり、こうしたB細胞は、インテグリン、サブユニット若しくはそれらのフラグメント、またはラミニンおよびそのフラグメントで免疫したヒトから得ることができる。場合によっては、こうしたB細胞は抗体処置を最
終的に受領するはずである患者から得る。目的の抗原若しくはそのフラグメントに結合する抗体を選択する。こうした抗体(若しくは結合フラグメント)をコードする配列をその後クローン化しかつ増幅する。Huseにより記述されるプロトコルはファージディスプレイ技術と組合せでより効率的にされる。例えば、Dowerら、第WO 91/17271号、およびMcCaffertyら、同第WO 92/01047号、米国特許第US 5,877,218号、同第US 5,871,907号、同第US 5,858,657号、同第US 5,837,242号、同第US 5,733,743号、同第US 5,565,332号、同第US 5,969,108号、同第US 6,172,197号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)を参照されたい。抗体、若しくは特定の1リガンドに結合する他のタンパク質の付加的な選択および標識方法は、第US 5,994,519号および同第US 6,180,336号明細書により記述されている。
【0092】
ファージディスプレイ法において、メンバーがそれらの外表面に多様な抗体を表示するファージのライブラリーを製造する。抗体は通常Fv若しくはFabフラグメントとして表示される。所望の特異性をもつ抗体を表示するファージを、インテグリン、サブユニット若しくはそれらのフラグメントに対するアフィニティー濃縮により選択する。
【0093】
ファージディスプレイ法の1変形において、選択したマウス抗体の結合特異性を有するヒト抗体を製造し得る。Winter、第WO 92/20791号明細書を参照されたい。この方法では、選択したマウス抗体のH若しくはL鎖可変領域のいずれかを出発原料として使用する。例えばL鎖可変領域を出発原料として選択する場合、メンバーが同一L鎖の可変領域(すなわちマウス出発原料)および異なるH鎖可変領域を表示するファージライブラリーが構築される。H鎖可変領域は再配列されたヒトH鎖可変領域のライブラリーから得る。Aβに対する強い特異的結合(例えば最低約108若しくは最低約109M−1)を示すファージを選択する。このファージからのヒトH鎖可変領域がその後、さらなるファージライブラリーを構築するための出発原料としてはたらく。このライブラリー中で、各ファージは同一のH鎖可変領域(すなわち最初のディスプレイライブラリーから同定される領域)および異なるL鎖可変領域を表示する。L鎖可変領域は、再配列されたヒト可変L鎖領域のライブラリーから得られる。再度、所望のインテグリンに対する強い特異的結合を示すファージを選択する。これらのファージは完全にヒトの抗インテグリン抗体の可変領域を表示する。これらの抗体は通常、マウス出発原料と同一若しくは類似のエピトープ特異性を有する。
【0094】
5.定常領域の選択
キメラ、ヒト化若しくはヒト抗体のHおよびL鎖可変領域をヒト定常領域の少なくとも一部分に連結し得る。定常領域の選択は、部分的に、抗体依存性補体および/若しくは細胞媒介性毒性が望ましいかどうかに依存する。例えば、アイソタイプIgG1およびIgG3は補体活性を有し、また、アイソタイプIgG2およびIgG4は有しない。アイソタイプの選択はまた抗体の脳への通過に影響を及ぼし得る。L鎖定常領域はλ若しくはκであり得る。抗体は、2本のLおよび2本のH鎖を含有する四量体として、個別のH鎖、L鎖として、Fab、Fab’、F(ab’)2およびFv、若しくはHおよびL鎖可変ドメインがスペーサーにより連結されている一本鎖抗体として、発現され得る。
【0095】
6.組換え抗体の発現
キメラ、ヒト化およびヒト抗体は組換え発現により典型的に製造される。組換えポリヌクレオチド構築物は典型的に、天然に関連した若しくは異種のプロモーター領域を包含する抗体鎖のコーディング配列に作動可能に連結された発現制御配列を包含する。好ましくは、発現制御配列は、真核生物宿主細胞を形質転換若しくはトランスフェクトすることが可能なベクター中の真核生物プロモーター系である。ベクターを適切な宿主に一旦組み込
めば、該ヌクレオチド配列の高レベル発現ならびに交差反応抗体の収集および精製に適する条件下で該宿主を維持する。
【0096】
これらの発現ベクターは、典型的に、エピソームとして、若しくは宿主染色体DNAの一体部分としてのいずれかで宿主生物体中で複製する。一般に、発現ベクターは、所望のDNA配列で形質転換された細胞の検出を可能にするための選択マーカー、例えばアンピシリン耐性若しくはハイグロマイシン耐性を含有する。
【0097】
大腸菌(Escherichia coli)は本発明のDNA配列をクローン化するのにとりわけ有用な1種の原核生物宿主である。酵母のような微生物もまた発現に有用である。サッカロミセス属(Saccharomyces)は好ましい酵母宿主であり、適するベクターは発現制御配列、複製起点、終止配列などを所望のとおり有する。典型的なプロモーターは、3−ホスホグリセリン酸キナーゼプロモーター、および他の解糖酵素からのプロモーターを包含する。誘導可能な酵母プロモーターは、とりわけ、アルコール脱水素酵素、イソチトクロームC、ならびに麦芽糖およびガラクトース利用を司る酵素からのプロモーターを包含する。
【0098】
哺乳動物細胞は、免疫グロブリン若しくはそれらのフラグメントをコードするヌクレオチドセグメントを発現するのに好ましい宿主である。Winnacker、From Genes to Clones、(VCH Publishers、ニューヨーク、1987)を参照されたい。無傷の異種タンパク質を分泌することが可能な多数の適する宿主細胞株が当該技術分野で開発されており、そしてCHO細胞株、多様なCOS細胞株、HeLa細胞、L細胞および骨髄腫細胞株を包含する。好ましくは細胞はヒト以外である。これらの細胞の発現ベクターは、複製起点、プロモーター、エンハンサー(Queenら、Immunol.Rev.、89:49−68(1986))のような発現制御配列、ならびにリボソーム結合部位、RNAスプライス部位、ポリアデニル化部位のような必要なプロセシング情報部位、および転写終止配列を包含し得る。好ましい発現制御配列は、内因性遺伝子、サイトメガロウイルス、SV40、アデノウイルス、ウシパピローマウイルスなど由来のプロモーターである。Coら、J.Immunol.、148:1149−54(1992)を参照されたい。
【0099】
あるいは、抗体コーディング配列は、トランスジェニック動物のゲノムへの導入および該トランスジェニック動物の乳中でのその後の発現のため導入遺伝子に組み込み得る(例えば、米国特許第US 5,741,957号、同第US 5,304,489号、同第US 5,849,992号明細書を参照されたい)。適する導入遺伝子は、カゼイン若しくはβラクトグロブリンのような乳腺特異的遺伝子からのプロモーターおよびエンハンサーと作動可能な連結にあるLおよび/若しくはH鎖のコーディング配列を包含する。
【0100】
目的のDNAセグメントを含有するベクターを、細胞宿主の型に依存して公知の方法により宿主細胞に移入し得る。例えば、塩化カルシウムトランスフェクションは原核生物細胞に一般に利用される一方、リン酸カルシウム処理、電気穿孔法、リポフェクション、遺伝子銃若しくはウイルスに基づくトランスフェクションを他の細胞宿主に使用し得る。哺乳動物細胞を形質転換するのに使用される他の方法は、ポリブレン、プロトプラスト融合、リポソームおよび微小注入法の使用を包含する。トランスジェニック動物の製造のため、導入遺伝子を受精卵母細胞に微小注入し得るか若しくは胚性幹細胞のゲノム中に組み込み得、そしてこうした細胞の核を除核卵母細胞に移入し得る。
【0101】
一旦発現されれば、抗体は、HPLC精製、カラムクロマトグラフィー、ゲル電気泳動などを包含する当該技術分野で既知の標準的手順に従って精製し得る(全般として、Scopes、Protein Purification(Springer−Verla
g、ニューヨーク、1982)を参照されたい)。
【0102】
7.ナノボディ
ナノボディは単一可変ドメイン(VHH)および2個の定常ドメイン(CH2およびCH3)を含有するH鎖抗体である。クローン化かつ単離されたVHHドメインは、元のH鎖抗体の完全な抗原結合能力を持つ安定なポリペプチドである。
【0103】
8.他の抗体
抗体は、米国特許出願公開第20040038304号、同第20070020685号、同第20060257396号、同第20060160184号、同第20060134098号、同第20050255552号、同第20050008625号、同第20040132066号、同第20040038317号、同第20030198971号、および同第20030157579号明細書に記述されるもののような方法によってもまた同定かつ/若しくは製造しうる。
【0104】
B.他の剤
剤は天然に存在する若しくは合成の分子であり得る。スクリーニングされるべき剤は、例えば海洋微生物、藻類、植物および真菌のような天然の供給源からもまた得ることができる。例えば、米国特許第US 6,096,707号明細書は、クサリヘビ、ジャララカ(Bothrops jararaca)からのメタロプロテイナーゼ、ジャララギン由来のペプチドを提供する。これらのペプチドはアミノ酸モチーフArg−Lys−Lys(RKK)を含有し、そしてヒトα2β1インテグリンのコラーゲンとの相互作用を低下させる。あるいは、スクリーニングされるべき剤は、ペプチド若しくは小分子を包含する剤のコンビナトリアルライブラリーから、または産業で、例えば化学、製薬、環境、農業、海洋、化粧品、薬物およびバイオテクノロジー産業により合成される化合物の既存のレパートリーからであり得る。剤は、例えば、医薬品、治療薬、環境、農業、若しくは工業的剤、汚染物質、薬用化粧品、薬物、有機化合物、脂質、グルココルチコイド、抗生物質、ペプチド、タンパク質、糖、炭水化物およびキメラ分子を包含し得る。
【0105】
多様な方法がペプチドライブラリーを製造するのに利用可能である(例えば、Lamら、Nature、354:92、1991および第WO 92/00091号明細書;Geysenら、J.Immunol.Meth.、102:259(1987);Houghtenら、Nature、354:84(1991);第WO 92/09300号明細書;ならびにLeblら、Int.J.Pept.Prot.Res.、41:201(1993)を参照されたい)。ペプチドライブラリーはファージディスプレイ法によってもまた生成し得る。例えばDevlin、第W0 91/18980号明細書を参照されたい。
【0106】
段階的様式で合成し得る多くの型の化合物についてコンビナトリアルライブラリーを製造し得る(例えば、EllmanとBunin、J.Amer.Chem.Soc.、114:10997、1992(ベンゾジアゼピン鋳型)、第WO 95/32184号明細書(オキサゾロンおよびアミニジン鋳型)、第WO 95/30642号明細書(ジヒドロベンゾピラン鋳型)ならびに第WO 95/35278号明細書(ピロリジン鋳型)を参照されたい)。化合物のライブラリーは通常固相化学により合成する。しかしながら溶液相ライブラリー合成もまた有用である。コンビナトリアル合成の戦略はDolleとNelson、J.Combinatorial Chemistry、1:235−282(1999)(全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)により記述されている。合成は、典型的には、異なる単量体若しくは他の成分を合成の各回に添加する周期的様式で実施する。いくつかの方法は初期プールを連続的に分画することにより実施する。例えば、第1回の合成は全部の支持体上で実施する。支持体
をその後2プールに分割し、そして別個の合成反応を各プールで実施する。該2プールをその後それぞれさらなる2プールにさらに分割し、などである。他の方法は分割および再プールの双方を使用する。例えば、初回の合成後に、化合物のプールを第2回の別個の合成のため2個に分割する。その後、別個のプールからのアリコートを第3回の合成のため再組合せする。分割およびプール法は混合化合物のプールをもたらす。これらの方法は下により詳細に記述されるとおり標識にとりわけ従いやすい。こうした方法により生成されるライブラリーの大きさは、2種の異なる化合物から106若しくは1010またはその間のいずれかの範囲まで変動し得る。
【0107】
コードされるライブラリーの製造法は、Needelsら、Proc.Natl.Acad.Sci.U.S.A.、90:10700(1993);Niら、J.Med.Chem.、39:1601(1996)、第WO 95/12608号、第WO 93/06121号、第WO 94/08051号、第WO 95/35505号、および第WO 95/30642号明細書(それらのそれぞれは全部の目的上そっくりそのまま引用することにより本明細書に組み込まれる)を包含する多様な刊行物に記述されている。コードされるライブラリーの合成方法は、典型的に、ランダムコンビナトリアルアプローチならびに単量体単位の化学的および/若しくは酵素的集成を必要とする。例えば、該方法は、典型的に、(a)複数の反応容器のあいだで複数の固体支持体を割り当てる段階;(b)各異なる反応容器中の異なる第一の単量体および標識の組合せを使用して第一の単量体および第一の標識を各反応容器中の支持体に結合する段階;(c)支持体をプールする段階;(d)複数の反応容器のあいだで該支持体を割り当てる段階;(e)各異なる反応容器中で、異なる第二の単量体および第二の標識の組合せを使用して、第一の単量体に第二の単量体を結合する段階、および固体支持体若しくは第一の標識のいずれかに第二の標識を結合する段階;ならびに、場合によっては、異なる標識および異なる単量体を用いて結合および割り当て段階を1ないし20回若しくはそれ以上反復する段階を包含する。単量体の組は段階ごとに拡張若しくは縮小し得るか;または、単量体の組は次の段階について完全に変更し得る(例えば、一段階でアミノ酸、別の段階でヌクレオシド、別の段階で炭水化物)。例えばペプチド合成のための単量体単位は単一アミノ酸若しくはより大きいペプチド単位または双方を包含し得る。
【0108】
こうした方法により合成可能な化合物は、ポリペプチド、βターン模倣物、多糖、リン脂質、ホルモン、プロスタグランジン、ステロイド、芳香族化合物、複素環化合物、ベンゾジアゼピン、オリゴマーN−置換グリシンおよびオリゴカルバメートを包含する。製造されるコンビナトリアルライブラリーは商業的供給源(例えばChemRx、カリフォルニア州サウスサンフランシスコ)からもまた入手可能である。
【0109】
コンビナトリアルライブラリーおよび他の化合物は、α2β1、α6β1若しくはαvβ1インテグリンまたはラミニンに結合するそれらの能力を測定することにより適合性について最初にスクリーニングする。上述された付加的なスクリーニング手順もまた使用し得る。
【0110】
それらの立体異性体、若しくはそれらの立体異性体の混合物、またはそれらの製薬学的に許容できる塩の形態を包含する、式IaおよびIbの化合物は、LTPのAβ誘導性阻害の抑制方法で有用であり
【0111】
【化4】
【0112】
ここで
X1およびX3は窒素若しくは炭素から独立に選択され;
R1は
【0113】
【化5】
【0114】
から選択され;
ここで、上の複素環は、NH2、ハロゲン、NO2、CN、CF3、C1−C4アルコキシ、C1−C6アルキルおよびC3−C7シクロアルキルよりなる群から選択される0〜2置換基で場合によっては置換されており;
Uは−(CH2)n−、−(CH2)tQ(CH2)m−および−C(=O)(CH2)n−1−から選択され、ここでメチレン基の1個は場合によってはR7で置換されており;
Qは、1,2−フェニレン、1,3−フェニレン、2,3−ピリジニレン、3,4−ピリジニレンおよび2,4−ピリジニレンから選択され;
R6はH、C1−C4アルキルおよびベンジルから選択され;
R7はC1−C6アルキル、C3−C7シクロアルキル、C4−C11シクロアルキルアルキル、アリール、アリール(C1−C6アルキル)、ヘテロアリールおよびヘテロアリール(C1−C6アルキル)から選択され;
R10は、H、ハロゲン、CO2R17、CONR17R20、0〜1個のR15若しくは0〜1個のR21で置換されているC1−C6アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR15若しくは0〜1個のR21で置換されているC3−C7シクロアルキル、0〜1個のR15若しくは0〜1個のR21で置換されているC4−C11シクロアルキルアルキル、および0〜1個のR15若しくは0〜2個のR11若しくは0〜1個のR21で置換されているアリール(C1−C6アルキル)−から選択され;
R11は、H、ハロゲン、CF3、CN、NO2、ヒドロキシ、NR2R3、0〜1個のR21で置換されているC1−C4アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR21で置換されているアリール、0〜1個のR21で置換されているアリール(C1−C6アルキル)−、0〜1個のR21で置換されている(C1−C4アルコキシ)カルボニル、0〜1個のR21で置換されている(C1−C4アルキル)カルボニル、0〜1個のR21で置換されているC1−C4アルキルスルホニル、および0〜1個のR21で置換されているC1−C4アルキルアミノスルホニルから選択され;
Wは−C(=O)−N(R13)−であり;
Xは−CH(R14)−CH(R15)−であり;
R13はHおよびCH3から選択され;
R14は、H、C1−C10アルキル、アリールおよびヘテロアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ハロ、シアノ、アミノ、CF3およびNO2から選択される0〜3置換基で場合によっては置換されており;
R15はHおよびR16から選択され;
Yは−COR19であり;
R16は
−NH(R20)−C(=O)−R17、
−N(R20)−C(=O)−R17、
−N(R20)−C(=O)−NH−R17、
―N(R20)SO2−R17、および
−N(R20)SO2−N(R20)R17
から選択され、
R17は、C1−C10アルキル、C3−C11シクロアルキル、アリール(C1−C6アルキル)−、(C1−C6アルキル)アリール、ヘテロアリール(C1−C6アルキル)−、(C1−C6アルキル)ヘテロアリール、ビアリール(C1−C6アルキル)−、ヘテロアリール若しくはアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ヘテロアリール、ハロ、シアノ、アミノ、CF3およびNO2よりなる群から選択される0〜3置換基で場合によっては置換されており;
R19は−O−(CH2)kN+(R22)(R23)(R24)Z−であり;
Z−は、ハロゲン化イオン、重硫酸イオン、硫酸イオン、リン酸水素イオン、リン酸イオン、トルエンスルホン酸イオン、メタンスルホン酸イオン、エタンスルホン酸イオン、酢
酸イオン、トリフルオロ酢酸イオン、クエン酸イオン、シュウ酸イオン、コハク酸イオンおよびマロン酸イオンから選択される製薬学的に許容できる陰イオンであり;
R22、R23およびR24はH、C1−C4アルキルおよびC4−C11シクロアルキルアルキルから独立に選択されるか;
あるいは、R22およびR23は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する5〜7員の複素環系を形成し得、ならびにR24は上のとおり定義されるか、または、R22、R23およびR24は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する複素二環系を形成し得;
R20はHおよびCH3から選択され;
R21はCOOHおよびNR62から選択され;
kは2であり;
mは0および1から選択され;
nは1〜4であり;ならびに
tは0および1から選択される。
【0115】
式Iaの化合物の例は、限定されるものでないが式II:
【0116】
【化6】
【0117】
の化合物を挙げることができ;ここでR19は−H、−CH3および−CH2CH2N+(CH3)3から選ばれる。一態様において、H19は−Hである(式IIの化合物はSM256である)。
【0118】
LTPのAβ誘導性阻害の抑制方法で有用な付加的な化合物は、米国特許第6,214,834号明細書に開示される化合物を包含する。その特許は、αvβ3に拮抗するためのSM256(式II)および他の化合物の使用を開示する。従って、1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する化合物を同定するためにαvβ3拮抗剤をスクリーニングしうる。そのようにスクリーニングされうる例示的αvβ3拮抗剤は、限定されるものでないが米国特許第6,214,834号明細書に開示されるαvβ3拮抗剤を挙げることができる。
【0119】
C.遺伝子抑制剤
遺伝子発現を抑制する剤を使用して、インテグリンサブユニットβ1、α2、α6若しくはαvをコードする遺伝子の発現を抑制し得る。アンチセンス剤もまた、ラミニンのようなそれに対するある種のリガンドの発現を抑制するのに使用し得る。ラミニン発現の抑制は、ラミニンに対する抗体での処置に類似の効果を達成し得る。標的細胞若しくは患者への本発明のアンチセンス試薬の投与は、上のインテグリン遺伝子若しくはそのリガンド
の1種の低下された活性をもたらす。アンチセンスポリヌクレオチドに関する一般的方法については、例えばAntisense RNA and DNA、(1988)、D.A.Melton編、Cold Spring Harbor Laboratory、ニューヨーク州コールドスプリングハーバー);Dagleら、Nucleic Acids Research、19:1805(1991);Uhlmannら、Chem.Reviews、90:543−584(1990)を参照されたい。リボザイムは遺伝子発現を抑制し得る別のアンチセンス剤である。
【0120】
アンチセンスオリゴヌクレオチドは、センスmRNAに結合しかつその翻訳を妨害すること、mRNAをヌクレアーゼ消化に対し感受性にすること、転写を妨害すること、RNA前駆体のプロセシング若しくは局在化を妨害すること、mRNAの転写を抑制すること、若しくはいくつかの他の機序により作用することにより抑制を引き起こし得る。アンチセンス分子が発現を低下させる特定の機序は重要でない。
【0121】
典型的に、アンチセンスポリヌクレオチドは、遺伝子のmRNAからの配列に特異的にハイブリダイズする最低7ないし10ないし典型的に20若しくはそれ以上のヌクレオチドのアンチセンス配列を含んでなる。数種のアンチセンスポリヌクレオチドは、長さが約10から約50ヌクレオチドまで、若しくは長さが約14から約35ヌクレオチドまでである。数種のアンチセンスポリヌクレオチドは約100ヌクレオチド未満若しくは約200ヌクレオチド未満のポリヌクレオチドである。一般に、アンチセンスポリヌクレオチドは安定な二重鎖を形成するのに十分長くあるべきであるが、しかし、所望の場合はin vivoで投与するために送達様式に依存して十分に短くあるべきである。標的配列への特異的ハイブリダイゼーションに必要とされるポリヌクレオチドの最小長さは、とりわけ、G/C含量、不適正塩基(あれば)の配置、標的ポリヌクレオチドの集団に関しての該配列の全体的差違、およびポリヌクレオチドの化学的性質(例えばメチルホスホネートバックボーン、ペプチド核酸、ホスホロチオエート)のようないくつかの因子に依存する。
【0122】
相補核酸分子をハイブリダイズするのに適する条件は当業者に公知である。例えば、典型的な高緊縮性条件下のハイブリダイゼーションは、5×SSPE、5×Denhart溶液、0.5%SDS(w/v)および100μg/mlサケ精子DNAを含有する混合物中で実施しうる。DNAは、約68℃で指定される時間ハイブリダイズさせる。典型的には膜若しくはフィルターに結合されているハイブリダイズされたDNAをその後、2×SSPE、0.1%SDS(w/v)中室温で10分間2回洗浄する。膜(若しくはフィルター)をその後、1×SSPE、0.1%SDS(w/v)の溶液に68℃で15分間、そして最後に1×SSPE、0.1%SDS(w/v)の溶液に68℃で15分間浸漬する。
【0123】
特異的ハイブリダイゼーションを確実にするため、アンチセンス配列は標的mRNA若しくはそれをコードする遺伝子に少なくとも実質的に相補的である。数種のアンチセンス配列はそれらの意図している標的配列に正確に相補的である。アンチセンスポリヌクレオチドは、しかしながら、RNA若しくはその遺伝子に対応する適切な標的配列への特異的結合が該ポリヌクレオチドの機能的特性として保持される限りは、ヌクレオチドの置換、付加、欠失、トランジション、転位若しくは修飾、または他の核酸配列若しくは核酸以外の部分もまた包含し得る。
【0124】
数種のアンチセンス配列はmRNAの相対的に接近可能な配列に相補的である(例えば二次構造を相対的に欠く)。これは、予測されるRNA二次構造を例えばMFOLDプログラム(Genetics Computer Group、ウィスコンシン州マディソン)を使用して解析すること、および当該技術分野で既知であるとおりin vitro若しくはin vivoで試験することにより決定し得る。効果的なアンチセンス組成物
の別の有用な同定方法はオリゴヌクレオチドのコンビナトリアルアレイを使用する(例えば、Milnerら、Nature Biotechnology、15:537(1997)を参照されたい)。
【0125】
遺伝子発現を阻害するための一技術は、阻害的RNA(RNAi)ともまた称される二本鎖RNAの細胞中への導入を必要とする。RNAiは、二本鎖RNA分子を形成するよう相互にアニーリングされたRNAの2本の相補鎖(センス鎖およびアンチセンス鎖)を含んでなる。RNAiは、典型的には阻害の標的とされている遺伝子のエキソンすなわちコーディング配列に由来する。RNAiは該RNAi分子の配列に相補的なmRNAの破壊をもたらす。RNAiおよび生存生物体でのそれらの使用の例は、例えばFireら、Nature、391:806−811(1998);Nykaenenら、Cell、107:309−321(2001);および第WO 01/29058号、第WO 01/75164号および第WO 99/32619号明細書により記述される。いくつかの方法において、RNAiは約100bpと1000bpの間、例えば約100、200、300、400、500、600、700、800、900、1000若しくはそれ以上の塩基対である。いくつかの方法においてRNAiはエキソン由来である。他の方法において、RNAiはイントロン若しくはシグナル伝達配列由来である。
【0126】
いくつかの方法において、アンチセンスポリヌクレオチドは、アンチセンス配列に加え、アンチセンス配列の発現をもたらすプロモーターおよび他の制御配列を包含する配列を有する。プロモーターならびに好ましくは終止およびポリアデニル化シグナルが適正に配置されていることを条件に、非コーディング鎖に対応する挿入された配列の鎖が転写され、そしてアンチセンスオリゴヌクレオチドとして作用する。いくつかの方法において、該ポリヌクレオチドはアンチセンス配列より本質的になるか若しくはアンチセンス配列である。アンチセンス核酸(DNA、RNA、修飾、アナログなど)は、本明細書に開示される化学合成および組換え法のような核酸のいずれかの適する製造方法を使用して作成し得る。例えば、アンチセンスRNA分子はデノボ化学合成若しくはクローニングにより製造し得る。
【0127】
β1、α2、α6若しくはαvインテグリンサブユニットをコードする遺伝子の発現を抑制するため、ジンクフィンガータンパク質を、アンチセンスポリヌクレオチドの代替として若しくはそれに加えて使用し得る。ジンクフィンガータンパク質は、ラミニンのようなこれらのインテグリンサブユニットのある種のリガンドの発現を抑制するのにもまた使用し得る。ジンクフィンガータンパク質は、本方法で剤としてそれら自身を使用し得るフィブロネクチンのような他のリガンドの発現を活性化する若しくは高めるのにもまた使用し得る。ジンクフィンガータンパク質は標的遺伝子内のいずれかの所望の標的部位に結合するよう工作若しくは選択し得る。いくつかの方法において標的部位はプロモーター若しくはエンハンサー内にある。他の方法において標的部位は構造遺伝子内にある。いくつかの方法において、ジンクフィンガータンパク質はヒトKOX−1タンパク質からのKRAB抑制領域のような転写リプレッサーに連結される(Thiesenら、New Biologist、2、363−374(1990);Margolinら、Proc.Natl.Acad.Sci.U.S.A.、91、4509−4513(1994));Pengueら、Nucl.Acids Res.、22:2908−2914(1994);Witzgallら、Proc.Natl.Acad.Sci.U.S.A.、91、4514−4518(1994)。活性化を達成するために好ましいドメインは、HSV VP16活性化ドメイン(例えばHagmannら、J.Virol.、71:5952−5962(1997)を参照されたい)核ホルモン受容体(例えばTorchiaら、Curr.Opin.Cell.Biol.、10:373−383(1998)を参照されたい);核因子κBのp65サブユニット(BitkoとBarik、J.Virol.、72:5610−5618(1998)およびDoyleとHunt、Neuroreport、8:2937−2942(1997));Liuら、Cancer Gene Ther.、5:3−28(1998))、若しくはVP64のような人工的キメラ機能ドメイン(Seifpalら、EMBO J.、11:4961−4968(1992))を包含する。ジンクフィンガータンパク質により標的を定めるのに適する標的部位の選択方法、および選択された標的部位に結合するためのジンクフィンガータンパク質の設計方法は、第WO 00/00388号明細書に記述されている。標的に結合するジンクフィンガータンパク質のファージディスプレイを使用する選択方法は、第EP 95908614A号明細書により記述されている。内因性遺伝子を調節するためのジンクフィンガータンパク質の使用方法は第WO 00/00409号明細書に記述されている。ジンクフィンガータンパク質は、タンパク質として、若しくはジンクフィンガーをコードしかつ適切な制御配列を有する核酸の形態でのいずれでも投与し得る。
【0128】
D.治療薬をコードする核酸
抗体若しくは他のペプチド試薬は、抗体鎖若しくはペプチドをコードする核酸の形態で投与し得る。こうした核酸は、典型的には、患者の意図している標的細胞中での該DNAセグメントの発現を可能にするプロモーターおよびエンハンサーのような調節エレメントに連結されている。L若しくはH鎖免疫グロブリン遺伝子からのプロモーターおよびエンハンサーエレメント、またはサイトメガロウイルス(CMV)主要中初期プロモーター(major intermediate early promoter)およびエンハンサーが直接発現に適する。いくつかの方法において、脳中で発現を引き起こすプロモーターを使用する。血小板由来増殖因子(PDGF)、プリオン若しくは神経エノラーゼのプロモーターのようなプロモーターが適する。
【0129】
連結された調節エレメントおよびコーディング配列はしばしば1ベクターにクローン化される。二本鎖抗体の投与のため、該2鎖を同一若しくは別個のベクターにクローン化し得る。
【0130】
レトロウイルス系(例えばLawrieとTumin、Curr.Opin.Genet.Develop.、2:102−109(1993)を参照されたい);アデノウイルスベクター(例えばBettら、J.Virol.、67:5911(1993)を参照されたい);アデノ随伴ウイルスベクター(例えばZhouら、J.Exp.Med.、179:1867−75(1994)を参照されたい)、ワクシニアウイルスおよびトリポックスウイルスを包含するポックス科からのウイルスベクター、シンドビスおよびセムリキ森林ウイルス(例えばDubenskyら、J.Virol.、70:508−19(1996)を参照されたい)、ベネズエラウマ脳炎ウイルス(米国特許第US 5,643,576号明細書を参照されたい)由来のもののようなアルファウイルス属、水疱性口内炎ウイルス(第WO 96/34625号明細書を参照されたい)のようなラブドウイルス、ならびにパピローマウイルス(Oheら、Human Gene Therapy、6:325−33(1995);Wooら、第WO 94/12629号明細書;およびXiaoとBrandsma、Nucleic Acids.Res.、24:2630−22(1996))からのウイルスベクターを包含する多数のウイルスベクター系が利用可能である。
【0131】
DNAはリポソーム中にパッケージングし得る。適する脂質および関連アナログは、米国特許第US 5,208,036号、同第5,264,618号、同第5,279,833号および同第5,283,185号明細書により記述されている。免疫原をコードするベクターおよびDNAはまた微粒子担体に吸着若しくはそれらと会合もし得、それらの例はポリメチルメタクリレートポリマー、ポリラクチドおよびポリ(ラクチドコグリコリド)を包含する。
【0132】
遺伝子治療ベクター若しくは裸のDNAは、典型的に全身投与(例えば静脈内、腹腔内、鼻、胃、皮内、筋肉内、くも膜下腔内、皮下若しくは頭蓋内注入)または局所適用(例えば米国特許第US 5,399,346号明細書を参照されたい)による個々の患者への投与によりin vivoで送達し得る。こうしたベクターはブピバカインのような促進剤をさらに包含し得る(米国特許第US 5,593,970号明細書)。DNAは遺伝子銃を使用してもまた投与し得る。XiaoとBrandsma、上記を参照されたい。DNAを微細な金属ビーズの表面上に沈殿させる。微小発射体をショック波若しくは膨張するヘリウムガスで加速し、そして数細胞層の深さまで組織を貫通させる。例えば、Agacetus,Inc.、ウィスコンシン州ミドルトンにより製造されるAccelTM遺伝子送達装置が適する。あるいは、裸のDNAは、単純に化学若しくは機械的刺激で皮膚上にDNAをスポットすることにより、皮膚を通り血流に移行し得る(第WO 95/05853号明細書を参照されたい)。
【0133】
さらなる一変形において、核酸を、個々の患者から外植した細胞(例えばリンパ球、骨髄吸引物、組織生検)若しくは普遍的ドナーの造血幹細胞のような細胞にex vivoで送達し得、次いで、通常はベクターを組み込んだ細胞についての選択後に細胞を患者に再移植し得る。
【0134】
E.LTPのAβ媒介性阻害を抑制する剤の同定
インテグリンサブユニットαvに結合する小分子剤は、1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下で投与しうる。適する小分子剤は、例えば、インテグリンサブユニットαv結合アッセイおよびLTPのアミロイド媒介性阻害抑制アッセイを含んでなる方法、若しくはLTPのアミロイド媒介性阻害抑制アッセイを含んでなるがしかしインテグリンサブユニットαv結合アッセイを含まない方法により同定する。
【0135】
インテグリンサブユニットαv結合アッセイは、例えば、単量体若しくは二量体いずれかとしてのインテグリンサブユニットαvへの剤の特異的結合を同定するいずれかのアッセイである。例示的アッセイは、支持体への直接若しくは間接結合によるように固定されうるインテグリンサブユニットαvに結合されている標識した剤の使用によるように結合を直接検出する。さらなる例示的アッセイは結合を間接的に検出する。一態様において競合結合アッセイを使用する。競合結合は、例えば、本明細書に開示されるか若しくは当該技術分野で既知のもののような別のインテグリンサブユニットαv結合剤、または本明細書に開示されるアッセイにより事前に同定された結合剤を使用して実施しうる。インテグリンサブユニットαv結合アッセイでの使用に適する結合剤は、限定されるものでないが、フィブロネクチン若しくはスーパーフィブロネクチン、モノクローナル若しくはポリクローナル抗体、18C7、20A9および17E6から選ばれる抗体と同一のエピトープを認識する抗体、ならびに18C7、20A9および17E6から選ばれる抗体、インテグリンサブユニットαvへの結合について18C7、20A9および17E6から選ばれる抗体と競合する剤、ならびにSM256のような式1の化合物を挙げることができる。
【0136】
LTPのアミロイド媒介性阻害抑制アッセイは、限定されるものでないがLTPを実験的に誘導する系を使用するアッセイを挙げることができる。適する系は、in vivo、または海馬領域若しくは海馬の小領域を含んでなる脳薄片のようなin vitroでありうる。例示的アッセイを実施例10〜13に記述する。
【0137】
それらの実施例で、LTPをHFS若しくは薬理学的刺激により誘導する。LTPは、HFS若しくは薬理学的刺激の投与の前、それらと同時に若しくは後に該系に添加されるAβペプチドにより阻害される。候補剤によるLTPのアミロイド媒介性阻害の抑制をその後、該候補剤の存在および非存在下のAβペプチドによるLTPの阻害を比較すること
によりアッセイする。抑制の程度は、対照系(Aβペプチドの非存在下のLTPの誘導後の系および/若しくはAβペプチドによるLTPの阻害後の系のいずれでもあり得る)のHFS若しくは薬理学的刺激後に観察される興奮性シナプス後場電位(EPSP)の振幅に関して測定する。
【0138】
IV.第二の剤
本発明は1種若しくはそれ以上の剤および1種若しくはそれ以上の第二の剤の共投与にさらに向けられる。例えば、適する第二の剤は、限定されるものでないが、Aβ産生の阻害剤、Aβ沈着の阻害剤、Aβ消失のメディエーター、アミロイド斑消失のメディエーター、Aβ神経毒性の阻害剤、Aβ凝集の阻害剤、Aβ解離のメディエーター、およびAβに対する抗体、ならびにそれらのいずれかの組合せから選択される第二の剤を挙げることができる。Aβ産生の適する阻害剤は、限定されるものでないがγ分泌酵素阻害剤およびβ分泌酵素阻害剤を挙げることができる。
【0139】
例示的γ分泌酵素阻害剤は、限定されるものでないが、米国特許第6,992,081号、同第6,982,264号、同第6,962,934号および同第6,610,493号明細書、ならびに米国特許出願公開第20060287306号、同第20060154926号、同第20050159460号、同第20020016320号および同第20030148392号明細書に開示されるもののようなγ分泌酵素阻害剤を挙げることができる。
【0140】
例示的β分泌酵素阻害剤は、限定されるものでないが、米国特許第7,115,410号、同第7,109,017号、同第7,067,271号、同第6,864,240号、同第6,852,482号、同第6,627,739号、同第6,321,163号、同第6,221,645号、同第5,942,400号および同第5,744,346号明細書、ならびに米国特許出願公開第20050196839号、同第20050182138号、同第20050177888号、同第20050170489号、同第20050164327号、同第20050164294号、および同第200402655965号明細書に開示されるもののようなβ分泌酵素阻害剤を挙げることができる。
【0141】
Aβに対する例示的抗体は、限定されるものでないが、米国特許第7,014,855号、同第6,982,084号、同第6,972,127号、同第6,962,707号、同第6,946,135号、同第6,913,745号、同第6,905,686号、同第6,890,535号、同第6,875,434号、同第6,866,850号、同第6,866,849号、同第6,818,218号、同第6,808,712号、同第6,787,637号、同第6,787,523号、同第6,787,144号、同第6,787,143号、同第6,787,140号、同第6,787,139号、同第6,787,138号、同第6,761,888号、同第6,750,324号、同第6,743,427号および同第6,710,226号明細書に開示されるもののようなAβに対する抗体を挙げることができる。
【0142】
V.長期増強
長期増強はin vivo若しくはin vitroで自然に発生し、およびin vitro若しくはin vivoモデル系で実験的に誘導しうる。本明細書に開示されるある種の方法は、該実験系のAβペプチドへの投与若しくは曝露により阻害し得るLTPの実験的誘導を企図している。それらの方法では剤を使用してAβペプチドによるLTPのその阻害を実験的に抑制する。天然に存在するLTPは多様なアミロイド形成疾患状態の発症の経過でAβペプチドにより阻害される。本明細書に開示される剤は、例えば、AβペプチドによるLTPの阻害をin vivoで処置かつ/若しくは阻害するため、ならびに/または該疾患状態を処置かつ/若しくは予防するための製薬学的製剤として患者
に投与しうる。
【0143】
LTPはHFSの送達若しくは薬理学的方法により実験的に誘導しうる。LTP阻害の実験モデルに、実験操作に応答してLTPを表す神経回路が提供される。それはマウス若しくはラットのようなげっ歯類若しくは霊長類のような哺乳動物のような動物から選ばれた生物体で行い得る。in vitroで培養した霊長類若しくはげっ歯類脳薄片のような哺乳動物脳薄片、またはin vivoで哺乳動物の脳中でLTPを実験的に誘導するための技術が当該技術分野で既知である。
【0144】
LTPの強さは、誘導後の指定される時点で基礎(誘導の非存在下)に関して観察されるEPSP上昇の大きさにより定量化し得る。その大きさは150%のような基礎に関するパーセンテージとして表しうる。Aβペプチドの投与後に観察されるLTP阻害は、基礎に関するEPSP上昇の大きさの低下をもたらす。
【0145】
例えば、Aβペプチドは、HFS後に観察されるEPSPが基礎と統計学的に異ならない点までLTPを完全に阻害する。その阻害が抑制される場合、LTPはAβの存在下であってさえ観察し得る。その抑制は、Aβの非存在下でLTPの誘導後に観察されるEPSPの大きさに関して定量化し得る。例えば、抑制は完全であることができ、その結果、Aβおよびαv結合剤の存在下でLTPの誘導後のデータ組全体で観察されるEPSPの大きさは、Aβの非存在下でLTPの誘導後に観察されるEPSPの大きさと統計学的に識別可能である。他の態様において、Aβおよびαv結合剤の存在下でLTPの誘導後のデータ組全体で観察されるEPSPの大きさは、例えば、Aβの非存在下でLTPの誘導後に観察されるEPSPの大きさの25%以上、50%以上、75%以上、85%以上、90%以上若しくは95%以上である。
【0146】
VI.処置可能な患者
本方法は、進行性記憶障害を特徴とする疾患若しくは障害を包含する記憶障害若しくは記憶障害の可能性を特徴とする疾患若しくは障害の予防的若しくは治療的処置に有用である。例示的疾患若しくは障害は、限定されるものでないが、アミリン若しくはAβペプチドのようなアミロイドタンパク質の沈着物の存在を特徴とするアミロイド形成疾患および状態を挙げることができる。こうした疾患は、アルツハイマー病、ダウン症候群および認知障害、II型糖尿病、パーキンソン病、びまん性レビー小体病、遺伝性若しくは全身性アミロイドーシスのようなアミロイドーシス、ならびにプリオン感染により全部若しくは部分的に引き起こされる疾患を包含する。
【0147】
処置可能な患者は、疾患の危険にさらされているがしかし症状を示さない個体、ならびに現在症状を示している患者を包含する。アルツハイマー病の場合、事実上誰でも彼若しくは彼女が十分に長く生きる場合にアルツハイマー病に罹る危険にさらされている。本方法は、アルツハイマー病の既知の遺伝的危険性を有する個体にとりわけ有用である。こうした個体は、この疾患を経験した親族を有する者、および遺伝子若しくは生化学的マーカーの分析によりその危険性が決定される者を包含する。アルツハイマー病に対する危険性の遺伝子マーカーは、APP遺伝子中の突然変異、例えばそれぞれハーディ型およびスウェーデン型突然変異と称される位置717ならびに位置670および671の突然変異を包含する(Hardy、TINS、上記を参照されたい)。危険性の他のマーカーは、プレセニリン遺伝子PS1およびPS2ならびにApoE4中の突然変異、ADの家族歴、高コレステロール血症若しくは動脈硬化症である。現在アルツハイマー病に罹っている個体は、特徴的な認知症ならびに上述された危険因子の存在から認識し得る。加えて、ADを有する個体を同定するための多数の診断試験が利用可能である。これらは脳脊髄液(CSF)のτおよびAβ42濃度の測定を包含する。上昇されたτおよび低下されたAβ42濃度はADの存在を示す。アルツハイマー病に罹っている個体はADRDA基準によっ
てもまた診断し得る。無症候性患者で処置はいかなる齢(例えば約10、約20、約30歳)でも開始し得る。通常、しかしながら患者が約40、約50、約60、約70、約80若しくは約90歳に達するまで処置を開始することは必要でない。処置は、典型的にはある期間にわたる複数投薬量を必要とする。ダウン症候群患者の場合には、母親に治療薬を投与することにより出産前に処置を開始し得るか、若しくは処置を出生直後に開始しうる。
【0148】
VII.処置計画
予防的応用において、製薬学的組成物若しくは医薬品は、疾患、その合併症および該疾患の発症の間に見つかる中間的病理学的表現型の生化学的、組織学的および/若しくは行動的症状を包含する該疾患の危険性を排除若しくは低下、その重症度を低下、またはその発症を遅延させるのに十分な量で、アミロイド形成疾患に感受性の若しくはそうでなければそれを発症する危険にさらされている患者に投与する。
【0149】
治療的応用において、組成物若しくは医薬品は、その合併症および中間的病理学的表現型を包含する該疾患の症状(生化学的、組織学的および/若しくは行動的)を治癒若しくは少なくとも部分的に停止するのに十分な量で、こうした疾患が疑われる若しくは既に罹っている患者に投与する。治療的若しくは予防的処置を達成するのに十分な量を治療的若しくは予防的有効用量と定義する。治療計画において、該剤は通常、該疾患の症状が消失若しくは有意に低下するまで間隔を置いて投与する。場合によっては、再発を予防するために投与を継続し得る。予防計画において、剤は通常間隔を置いて、いくつかの例では患者の生涯の残りの間もまた投与する。処置は、投与した剤の濃度をアッセイすること若しくは患者の応答をモニターすることによりモニターし得る。応答は、ADRDA基準、および患者の脳中の斑の画像化によりモニターし得る(第WO 00/14810号明細書を参照されたい)。
【0150】
上述された状態の処置のための本発明の組成物の有効用量は、投与手段、標的部位、患者の生理学的状態、患者がヒトであるか動物であるか、投与されている他の医薬品、および処置が予防的であるか治療的であるかを包含する多くの異なる因子に依存して変動する。通常、患者はヒトであり;トランスジェニック哺乳動物を包含するヒト以外の哺乳動物もまた処置し得る。処置投薬量は、典型的には安全性および有効性を最適化するよう用量設定する。
【0151】
抗体、ペプチドおよび小分子の投薬量は、約0.0001から約100mg/kgまで、およびより通常は約0.01ないし約20mg/kg宿主体重の範囲にわたる。例えば、投薬量は約1mg/kg体重若しくは約20mg/kg体重または約1ないし約10mg/kgの範囲内にあり得る。例示的処置レジメンは、2週ごとあたり1回若しくは1か月に1回または3ないし6か月ごとに1回の投与を必要とする。いくつかの方法において、異なる結合特異性をもつ2、3、4種若しくはそれ以上のモノクローナル抗体を同時に投与し、この場合投与される各抗体の投薬量は示される範囲内にある。例えば、いくつかの方法において、β1インテグリン、α2インテグリンおよびαvインテグリンサブユニットの2種若しくは全3種に対する抗体を同時に投与する。いくつかの方法において、α6インテグリンサブユニットに対する抗体もまた投与する。抗体は通常複数の機会に投与する。単一投薬量の間の間隔は週、月若しくは年であり得る。間隔は、患者中のインテグリンに対する抗体の血中濃度を測定することにより示されるように不規則でもまたあり得る。いくつかの方法において、抗体の投薬量は、約1ないし約1000μg/ml、およびいくつかの方法においては約25ないし約300μg/mlの血漿抗体濃度を達成するように調節する。あるいは、抗体は徐放製剤として投与し得、この場合はより少なく頻繁な投与を必要とする。投薬量および頻度は患者中の抗体の半減期に依存して変動する。一般に、ヒト抗体は最長半減期を示し、次いでヒト化抗体、キメラ抗体およびヒト以外の抗
体である。投薬量および投与頻度は、処置が予防的であるか治療的であるかに依存して変動し得る。予防的応用において、比較的低用量を長期間にわたり比較的頻繁でない間隔で投与する。若干の患者は彼らの生涯の残りの間処置を受領することを継続する。治療的応用においては、疾患の進行が低下若しくは終了するまで、および好ましくは患者が該疾患の症状の部分的若しくは完全な軽減を示すまで、比較的短い間隔での比較的高投薬量をときに必要とする。その後、該患者に予防的レジメンを投与し得る。
【0152】
剤をコードする核酸の用量は、患者あたり約10ngから1gまで、約100ngないし約100mg、約1μgないし約10mg、若しくは約30ないし約300μgのDNAの範囲にわたる。感染性ウイルスベクターの用量は、用量あたり約10から約100、若しくは約103、約104、約105、約106、約107、約108、約109、約1010若しくはそれ以上のビリオンまで変動しうる。
【0153】
本発明の剤は、予防的および/若しくは治療的処置のため非経口、局所、静脈内、経口、皮下、くも膜下腔内、動脈内、頭蓋内、腹腔内、鼻内若しくは筋肉内手段により投与し得る。いくつかの方法において、剤は沈着物が蓄積した特定の組織に直接注入する(例えば頭蓋内注入)。いくつかの方法において、筋肉内注入若しくは静脈内注入を抗体の投与に使用する。いくつかの方法において、特定の治療抗体を頭蓋中に直接注入する。いくつかの方法において、抗体を徐放組成物、若しくはMedipadTM装置のような装置として投与する。
【0154】
本発明の剤は、場合によっては、アミロイド形成疾患の処置で少なくとも部分的に有効である他の剤とともに投与し得る。アミロイド沈着物が脳中に存在するアルツハイマー病およびダウン症候群の症例においては、本発明の剤は、血液脳関門を横断する本発明の剤の通過を増大させる他の剤とともにもまた投与し得る。
【0155】
本発明の剤は、しばしば、有効治療薬および多様な他の製薬学的に許容できる成分を含んでなる組成物として投与する。Remington’s Pharmaceutical Science(第15版、Mack Publishing Company、ペンシルバニア州イーストン、1980)を参照されたい。使用される特定の製剤は意図している投与様式および治療的応用に依存する。該組成物は、所望の製剤に依存して、動物若しくはヒト投与のための製薬学的組成物を処方するのに通例使用されるベヒクルと定義される、製薬学的に許容できる非毒性の担体若しくは希釈剤もまた包含し得る。希釈剤は該組合せの生物学的活性に悪影響を及ぼさないように選択する。こうした希釈剤の例は、限定されるものでないが、蒸留水、生理学的リン酸緩衝生理的食塩水、リンゲル液、ブドウ糖溶液およびハンクス液を挙げることができる。加えて、製薬学的組成物若しくは製剤は、他の担体、補助物質、若しくは非毒性の非治療的非免疫原性安定剤などもまた包含しうる。
【0156】
製薬学的組成物は、タンパク質、キトサンのような多糖、ポリ乳酸、ポリグリコール酸、コポリマー(ラテックス官能性化SepharoseTMビーズ、アガロース、セルロースなどのような)、ポリマーアミノ酸、アミノ酸コポリマー、および脂質凝集物(油滴若しくはリポソームのような)のような、大型のゆっくりと代謝される巨大分子もまた包含し得る。
【0157】
非経口投与のため、本発明の剤は、水、油、生理的食塩水、グリセロール若しくはエタノールのような無菌の液体であり得る製薬学的担体を含む生理学的に許容できる希釈剤中の物質の溶液若しくは懸濁液の注入可能な投薬量として投与し得る。ヒト投与のための非経口組成物は無菌、実質的に等張であり、そしてGMP条件下で作成される。加えて、湿潤若しくは乳化剤、界面活性剤、pH緩衝物質などのような補助物質が組成物中に存在し
得る。製薬学的組成物の他成分は、石油、動物、植物若しくは合成起源のもの、例えばラッカセイ油、ダイズ油および鉱物油である。一般に、プロピレングリコール若しくはポリエチレングリコールのようなグリコールは、とりわけ注入可能な溶液に好ましい液体担体である。抗体は、有効成分の徐放を可能にするような様式で処方し得るデポー注射剤若しくは植込み製剤の形態で投与し得る。例示的一組成物は、HClで適するpHに調節された、50mM L−ヒスチジン(任意)、150mM NaClを含有する水性緩衝液中で処方された5mg/mLのモノクローナル抗体を含んでなる。
【0158】
典型的に、組成物は液体の溶液若しくは懸濁液いずれかのような注射液として製造し;注入前の液体ベヒクル中の溶液若しくは懸濁液に適する固体の形態もまた製造し得る。該製剤は、上で論考されたとおり、高められたアジュバント効果のため、乳化、あるいはリポソーム、またはポリラクチド、ポリグリコリド若しくはコポリマーのような微小粒子中に被包化もまたし得る(Langer、Science、249:1527−33(1990)およびHanesら、Advanced Drug Delivery Reviews、28:97−119(1997)を参照されたい)。本発明の剤は、有効成分の徐放若しくはパルス型放出を可能にするような様式で処方し得るデポー注射剤若しくは植込み製剤の形態で投与し得る。
【0159】
他の投与様式に適する付加的な製剤は、経口、鼻内および肺製剤、坐剤ならびに経皮適用を包含する。坐剤について、結合剤および担体は例えばポリアルキレングリコール若しくはトリグリセリドを包含し;こうした坐剤は、約0.5%ないし約10%、若しくは約1%ないし約2%の範囲の有効成分を含有する混合物から形成し得る。経口製剤は、製薬学的等級のマンニトール、乳糖、デンプン、ステアリン酸マグネシウム、サッカリンナトリウム、セルロースおよび炭酸マグネシウムのような賦形剤を包含するがしかしこれらに限定されない。これらの組成物は、典型的に、溶液、懸濁剤、錠剤、丸剤、カプセル剤、徐放製剤若しくは散剤の形態をとり、そして約10%ないし約95%、若しくは約25%ないし約70%の有効成分を含有する。
【0160】
局所適用は経皮若しくは皮内送達をもたらし得る。局所投与は、コレラ毒素若しくは解毒誘導体またはそれらのサブユニット、あるいは他の類似の細菌毒素との剤の共投与により助長し得る(Glennら、Nature、391:851(1988)を参照されたい)。共投与は、成分を混合物として、または化学的架橋若しくは発現により融合タンパク質として得られた結合分子として使用することにより達成し得る。あるいは、経皮送達は皮膚貼付剤を使用して若しくはトランスフェロソーム(transferosome)を使用して達成し得る(Paulら、Eur.J.Immunol.、25:3521−24(1995);Cevcら、Biochem.Biophys.Acta、1368:201−15(1998))。
【0161】
本明細書で引用される全部の文書はそっくりそのまま引用することによりここに本明細書に組み込まれる。
【図面の簡単な説明】
【0162】
【図1】ヒト皮質培養物(HCC)(上)若しくはポリエチレンイミン(PEI)(下)被覆プレートでのAβ網目構造を具体的に説明する。
【図2】HCCでのAβ網目構造形成および神経毒性双方に対するβ1インテグリンサブユニットの効果を具体的に説明する。図2AはHCCでのβ1インテグリンサブユニット発現を具体的に説明する。図2Bは抗β1抗体1965の非存在(上)若しくは存在(下)下のHCCでの72時間Aβ網目構造形成を具体的に説明する。図2Cは、β1インテグリンサブユニット阻止抗体とプレインキュベートしたHCCでの神経毒性の阻害を具体的に説明する。エラーバーは(n=3)ウェルからの標準偏差を表す。
【図3A】HCCでのAβ網目構造形成および神経毒性に対するα2およびαvインテグリンサブユニットの効果を具体的に説明する。
【図3B】抗α2(中)若しくは抗αv(下)抗体の非存在(上)若しくは存在下でプレインキュベートしたHCCでの72時間Aβ網目構造形成を具体的に説明する。
【図3C】β1インテグリンサブユニット阻止抗体とプレインキュベートしたHCCでの神経毒性を具体的に説明する。
【図4】HCCでのα2およびαv発現を具体的に説明する。
【図5】Aβ網目構造形成および神経毒性の阻害における抗ラミニン抗体の効果を具体的に説明する。図5Aは抗ラミニン抗体の非存在(上)若しくは存在(下)下でプレインキュベートしたHCCでの72時間Aβ網目構造形成を具体的に説明する。図5Bは抗ラミニン抗体とプレインキュベートしたHCCでの神経毒性を具体的に説明する。エラーバーは(n=3)ウェルからの標準偏差を表す。図5Cは抗コラーゲン抗体とプレインキュベートしたHCCでの神経毒性を具体的に説明する。エラーバーは(n=3)ウェルからの標準偏差を表す。
【図6】HCCでのAβシグナル伝達経路の活性化を具体的に説明する。図6Aは、Aβ処理したHCCでの接着斑キナーゼ(Fak)と会合したパキシリンのチロシンリン酸化を具体的に説明する。図6Bは、Aβ処理したHCCでのプロリンリッチチロシンキナーゼ(Pyk2)と会合したパキシリンのチロシンリン酸化を具体的に説明する。
【図7】ヒト皮質ニューロンを1時間播種し、次いで吸引かつ可溶性アミリンで処理する場合の1日後の毒性を具体的に説明する。インテグリン若しくはインテグリンサブユニット抗体を種(seed)および可溶性アミリンの存在下で細胞に添加して毒性を阻害し得る。種および可溶性アミリン単独は毒性でない。しかしながら、細胞を1時間播種し次いで吸引かつ可溶性アミリンで処理する場合、該アミリンは毒性である。
【図8】インテグリンおよびインテグリンサブユニット抗体、とりわけ抗ラミニン、抗β1、抗αvおよび抗α2抗体がアミリン毒性に対し保護することを具体的に説明する。
【図9A−9B】細胞を種アミリンに1時間曝露した後のアミリン2成分毒性の阻害パーセントにより示されるところの、アミリン毒性に対する保護におけるインテグリン、若しくは抗αvβ3抗αvを包含するインテグリンサブユニット抗体;およびサイトカラシンDの効果を具体的に説明する。
【図10A】抗αvインテグリン抗体18C7がin vitroで歯状回中の合成AβによるLTPの阻害を抑制することを具体的に説明する。
【図10B】抗αvインテグリン抗体20A9がin vitroで歯状回中の合成AβによるLTPの阻害を抑制することを具体的に説明する。
【図10C】抗αvインテグリン抗体17E6がin vitroで歯状回中の合成AβによるLTPの阻害を抑制することを具体的に説明する。
【図11A】対照抗αv抗体27/1がAβによるLTPの抑制を予防しなかったことを具体的に説明する。
【図11B】対照抗αv抗体7H10がAβによるLTPの抑制を予防しなかったことを具体的に説明する。
【図12A】可溶性Aβがin vivoでCA1領域でLTPを阻害することを具体的に説明する。
【図12B】抗αvインテグリン抗体17E6がin vivoでCA1領域での可溶性AβによるLTPの阻害を抑制することを具体的に説明する。
【図12C】SM256がin vivoでCA1領域での可溶性AβによるLTPの阻害を抑制することを具体的に説明する。
【図13A】αv含有インテグリンリガンドSM256がAβによるLTPの阻害を抑制することを具体的に説明する。
【図13B】αv含有インテグリンリガンド、スーパーフィブロネクチンがAβによるLTPの阻害を抑制することを具体的に説明する。
【図13C】αv含有インテグリンリガンド、エキスタチンがAβによるLTPの阻害を抑制することを具体的に説明する。
【実施例】
【0163】
実施例1〜5の材料および方法
抗体の供給源
【0164】
【表1】
【0165】
組織培養
組織培養プレートを150mMホウ酸ナトリウム、pH8.5中ポリエチレンイミン(PEI)で被覆し、そして室温で一夜インキュベートした。細胞を添加する前にウェルをPBSで洗浄し、そして細胞がプレーティングの準備ができるまで最小必須培地(10%FBSを含むMEM)を添加した。ヒト胎児大脳皮質(E13−E16)をハンクス平衡塩溶液(HBSS)ですすいだ。組織をHBSS中1mgのDNアーゼ中で摩砕した。この懸濁液を100ミクロンのナイロン細胞濾過器で濾過し、そして250×gで5分間遠心した。細胞をトリプシンに再懸濁しかつ37℃で20分間インキュベートした。改変最小必須培地(10%FBSおよび1mgのDNアーゼを含むMMEM)を添加しそして細胞を再懸濁し;その後遠心分離により再度収集した。B27を含有するMMEMに細胞を再懸濁し、そして、洗浄したPEI被覆プレート中、96ウェルプレートに125,000細胞/ウェルで、若しくは6ウェルプレートに250万細胞/ウェルで蒔いた。ヒト皮質培養物(HCC)は処理前に2週ごとの培地交換を伴い3週間インキュベートした。
【0166】
Aβ生成
Aβは、二重蒸留水(ddH2O)をAβに添加して1mMストックを構成することにより生成した。これを37℃で3日間エイジングし、アリコートに分割しそして−20℃で凍結保存した。可溶性Aβは、DMSOをAβに添加して7.5mMストックを作成すること、30分間超音波処理すること、アリコート分割することおよび−20℃で凍結す
ることにより作成した。神経毒性Aβは、ddH2OをAβに添加すること、アリコート分割することおよび−20℃で凍結することにより生成した。
【0167】
HCCライセートからのインテグリン免疫沈降
6ウェルプレート中のHCCを、メチオニンを含まない培地中100μCi/mlの35S−メチオニンで一夜標識した。細胞を洗浄し、25mM Hepes、pH7.5、1%Triton X−100、0.1%SDS、150mM NaCl、0.5mM EDTA、0.5mM EGTAで溶解し、そして26ゲージ針を3回通過させた。不溶性物質を15,000rpmで4℃で15分間の遠心分離により除去した。ライセートを、プロテインAビーズに結合したウサギ抗マウス(RAM)抗体で前浄化し、そしてインテグリンサブユニット特異的抗体(β1についてLia1/2、α1についてTS2/7、α2についてGi9、α3についてP1B5、α4についてAN100226m、α5についてAb0771、α6についてGoH3、α9についてY9A2およびαvについてVNR147)で免疫沈降した。免疫沈降物を1mlの25mM Hepes、pH7.5、1%Triton X−100 150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで3回洗浄した。免疫沈降したサンプルを8%トリス−グリシンゲル(Novex)で分離しかつ固定し;ゲルを乾燥し、そしてゲル中の35S標識タンパク質をオートラジオグラフィーにより可視化した。
【0168】
Aβ免疫蛍光
Aβで72時間処理したHCCを4%パラホルムアルデヒドで固定し、5μg/mlの抗Aβ−3D6−ビオチンで染色し、そして10μg/mlのストレプトアビジン−FITC(Jackson)で可視化した。
【0169】
ヒト皮質ニューロン中のAβ神経毒性
HCCを、グルタミンおよびペニシリン/ストレプトマイシンを補充した神経培地(MEM)(基礎培地)中で抗体若しくはリガンドで30分間処理した。基礎培地中1μモル濃度のAβを1時間添加した。培地を除去し、そしてHCCを抗体若しくはリガンドおよび基礎培地中20μM可溶性Aβで3日間処理した。3日目に、基礎培地中10%アラマーブルー中で2時間インキュベートすることにより毒性を測定した。蛍光レベルを三重で対照およびAβ処理したウェルに関して測定した。
【0170】
インテグリンヘテロ二量体会合
N−2(BottensteinのN−2処方、例えばカタログ番号17502、Invitrogen Corp.、カリフォルニア州カールズバッド)を補充したMMEM培地中の6ウェルプレート中のHCCを湿潤氷上に置き、PBSで洗浄し、25mM Hepes、pH7.5、1%Triton X−100、0.1%SDS、150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで溶解し、そして26ゲージ針を3回通過させた。不溶性物質を15,000rpmで4℃で15分間の遠心分離により除去した。ライセートをプロテインAビーズで前浄化し、そして抗β1インテグリンLia1/2(Immunotech)およびRAM/プロテインGビーズ(Pharmacia)を使用してβ1インテグリンを免疫沈降した。免疫沈降物を1mlの25mM
Hepes、pH7.5、1%Triton X−100 150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで3回洗浄した。免疫沈降したサンプルを4〜12%トリス−グリシンゲル(Novex)で分離し、そして抗α2インテグリン(ChemiconからのAB 1936)若しくは抗αvインテグリンMAB 1960(Chemicon)でウエスタンブロットした。
【0171】
パキシリンリン酸化のAβ誘導
神経毒性Aβを、N−2を補充した基礎培地中の6ウェルプレート中HCCに0分ない
し24時間添加した。HCCを湿潤氷上に置き、PBSで洗浄し、25mM Hepes、pH7.5、1%Triton X−100、0.1%SDS、150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで溶解し、そして26ゲージ針を3回通過させた。不溶性物質を15,000rpmで4℃で15分間の遠心分離により除去した。ライセートをプロテインAビーズで前浄化し、そして抗Fak(UBI)若しくは抗Pyk2抗体(UBI)およびプロテインAビーズを使用してそれぞれFak若しくはPykを免疫沈降した。免疫沈降物を1mlの25mM Hepes、pH7.5、1%Triton X−100、150mM NaCl、0.5mM EDTAおよび0.5mM EGTAで3回洗浄した。免疫沈降したサンプルを8%トリス−グリシンゲル(Novex)で分離し、そして抗ホスホチロシン(Transduction labsからのRC20)および抗パキシリン(Transduction labs)でウエスタンブロットした。
【実施例1】
【0172】
ヒト皮質ニューロンでの免疫蛍光パターン
本実施例は、アルツハイマー病(AD)の海馬および皮質ニューロンで生じかつ関連した神経毒性を表すAβ斑のin vitro組織培養物モデルを生じる。該モデルは、ADで遂げられるニューロンを可能な限り緊密に表す初代ヒト皮質ニューロン培養物を使用する。これらの培養物へのAβの添加は、ニューロンで1〜3日にわたり生じかつその後ニューロンで毒性を引き起こす、再現可能なAβ網目構造をもたらす。HCCを伴わないプレート上でインキュベートしたAβもまた網目構造として染色したが、しかしHCCで見られるものより短く薄くかつより直線状であった伸長を伴うより均一なパターンを一貫して示した。図1Aおよび1BはHCCの存在および非存在下の網目構造を比較する。
【実施例2】
【0173】
β1インテグリンはAβ網目構造および神経毒性を媒介する
網目構造がインテグリンにより形成されるもののような細胞外マトリックスに似ていたため、インテグリンがHCC中に存在するかどうか;およびその場合、インテグリンがHCCでのAβ網目構造形成を助長したかどうかを検討した。ゲル電気泳動はβ1インテグリンがHCCで発現されていることを示した。MAB1965を包含するβ1インテグリン阻止抗体が、Aβ網目構造パターンがHCCで形成されることを阻止し得たこともまた見出された(図2A(抗体なし)を図2B(抗体を伴う)と比較されたい)。これらの培養物でAβにより生成される毒性に該網目構造パターンが必要であったかどうかもまた検討した。これを行うため、Aβ網目構造を阻止することが示されたβ1インテグリン阻止抗体(AIIB2およびMAB1965)とHCCをインキュベートした。これらの抗体はAβ誘発性の毒性もまた用量依存性の様式で阻害した(図2C)。抗体AIIB2はAβ毒性の非常に強力な阻害剤であり、170ng/mlすなわち1nMのIC50を表した。対照的に、対照抗体は毒性に対する影響を有しなかった。
【実施例3】
【0174】
α2およびαvインテグリンに結合する剤は網目構造およびAB媒介性神経毒性を阻害する
β1インテグリンは数種のαサブユニットと対形成して多様なヘテロ二量体を形成し得る。従って、どのαインテグリンサブユニットがHCC中に存在したかを試験した。α2、α3、α4、α5、α6およびαvインテグリンがHCC中で発現された。α1およびα9インテグリンはHCC中で発現されなかった。全部のこれらのαインテグリンサブユニットに対する阻害抗体を、Aβ網目構造形成を阻害しかつその神経毒性を阻害するそれらの能力について試験した。α2およびαvに対する阻害抗体はAβ網目構造形成を阻害した(図3A)。これらの抗体はHCCでのAβの神経毒性効果も阻害した(図3B)。これらの特定のインテグリンに対する特異性を示すため、2〜3種の阻害抗体をこれらの
インテグリンのそれぞれおよび同様に他のインテグリンサブユニットに対し試験した。網目構造形成および神経毒性の双方を媒介するα1、α2およびαvインテグリンに対する非常に明瞭な特異性が見出された(表1)。
【0175】
【表2】
【0176】
毒性に対する抗α6抗体の弱いがしかし再現可能な効果が観察された。最後に、これらの影響がインテグリンに向けられかつAβ重合を非特異的に妨害しなかったことを確認するため、Aβ毒性をヒトおよびマウス皮質培養物で並べて分析した。これらのアッセイで使用した抗体はマウスインテグリンと交差反応しない。抗ヒトインテグリン抗体はヒト培養物でAβ毒性を阻害し得たがしかしマウス培養物ではし得ず、該抗体が毒性を阻害するためにAβと非特異的に相互作用しなかったことを示唆した。HCCライセートをβ1抗体で免疫沈降すること、ならびにその後沈降した物質をα2およびαvに対する抗体でブロッティングすることにより、α2およびαvがHCC細胞中でβ1インテグリンと会合したことが確認された。これらの結果は、α2β1およびαvβ1のヘテロ二量体が、A
β網目構造形成および神経毒性の機能的メディエーターであることを示す。
【実施例4】
【0177】
フィブロネクチンおよび抗ラミニン抗体はAB網目構造形成および神経毒性を阻害する
インテグリン/細胞外網目構造の他成分をAβ網目構造形成および神経毒性の媒介への関与について検討した。これらの他成分は、αvβ1インテグリンリガンド、フィブロネクチン、およびスーパーフィブロネクチン(網目構造を形成するフィブロネクチンドメインの多量体)、ならびにα2β1リガンド、コラーゲンおよびラミニンを包含した。ラミニンは2鎖β1およびγ1を有し、それらの双方はアルツハイマー病斑中で上昇している(Murtomakiら、J.Neur.Res.、32:261−73(1992))。フィブロネクチンおよびスーパーフィブロネクチンはAβ網目構造形成および神経毒性を阻害することが可能であった(表1)。この結果はαvβ1機能に対する影響についてAβと競合するフィブロネクチンにより説明し得る。対照的に、αvβ1リガンド、ラミニンは、Aβ網目構造若しくは神経毒性を阻害することが可能でなかった(表1)。あるαvβ1リガンドがそうでなかった場合にあるαvβ1リガンドがなぜ保護的であったかを確定するため、抗ラミニン抗体を網目構造形成および神経毒性アッセイで試験した。2種のラミニン抗体はAβ網目構造形成(図5A)およびAβ媒介性神経毒性(図5B)双方で高度に保護的であった。抗ラミニン抗体番号AB19012は1nM未満のIC50を示した。対照的に、抗コラーゲン抗体はAβ網目構造形成および神経毒性に対する影響を有しなかった(図5C)。
【実施例5】
【0178】
Aβはパキシリンのチロシンリン酸化(インテグリンシグナル伝達経路の早期事象)を活性化する
細胞外マトリックスリガンドによるインテグリン活性化はFakのような接着斑キナーゼの活性化およびその基質パキシリンのチロシンリン酸化に至る。Aβがインテグリンシグナル伝達経路を同様に刺激していたかどうかを確定するため、HCCへのAβ添加に際してのFak会合パキシリンのチロシンリン酸化パターン(図6A)を分析した。Fak会合パキシリンのチロシンリン酸化の一貫した活性化は見出されなかった。しかしながら、Aβ刺激後のPyk2会合パキシリンのチロシンリン酸化の一貫した増大が観察された。Pyk2もまた接着斑キナーゼであり、そして神経毒性につながる異常なAβ/インテグリンシグナル伝達経路を媒介しているかもしれない(図6B)。FakよりむしろPyk2会合パキシリンのチロシンリン酸化の活性化は、これらの状態で毒性応答を引き起こすものでありうる。いずれの場合も、Aβはインテグリンシグナル伝達経路の早期事象であるパキシリンのチロシンリン酸化を活性化し、Aβ神経毒性シグナルがα2β1およびαvβ1インテグリンシグナル伝達経路の直接の関与により媒介されうることを示す。
【実施例6】
【0179】
アミリン2成分毒性
CPR,Inc.からの種(seed)若しくは凝集アミリン(1mM)(641−80、ロットNG−0213)を、200μl水/mg粉末を添加することおよびその後該溶液を37℃で3日間エイジングすることにより作成した。可溶性アミリン(5mM)は、40μl DMSO/mg粉末を添加すること、および該混合物を水浴中で30分間超音波処理することにより調製した。双方のストック溶液をアリコート分割しかつ使用の準備ができるまで凍結した。可溶性アミリンストックは、使用直前に培地で20μMに希釈し、そして水で前洗浄したAmicon 30フィルターで濾過した。濾過した物質をその後その適切な濃度に希釈した。
【0180】
ヒト皮質ニューロン(125,000細胞/96ウェル)を5μMの種アミリン(100μl/ウェル)で1時間処理した。細胞を吸引し、そして可溶性アミリンを100μl
/ウェルあたり5μMで戻した。化合物研究のため、可溶性アミリンを添加する前に50μlの2×化合物を添加した。
【0181】
図6はヒト皮質ニューロンを1時間播種し次いで吸引しかつ可溶性アミリンで処理した1日後の毒性を示す。インテグリン抗体を種および可溶性アミリンの存在下で細胞に添加して毒性を阻害した。図7は、数種のインテグリン抗体すなわち2034抗ラミニン、1965抗β1インテグリン、1958抗αv(VNR)および1950抗α2が、アミリン2成分の毒性に対し細胞を保護したことを示す。図8は、アミリン2成分の毒性が、抗αvβ3および抗αvを包含する付加的なインテグリン抗体;ならびにサイトカラシンDによりさらに阻害されることを示す。
【0182】
実施例7〜11の材料および方法
薄片の調製
全実験はラット海馬(雄性、3〜4週齢、重量40〜80g)若しくはマウス(雄性、3〜4月齢)の横断薄片で実施した。断頭後迅速に脳を取り出しかつ冷酸素添加(95%O2/5%CO2)培地に入れた。Intracell Plus 1000を使用して350μmの厚さで薄片を切断し、そして室温(20〜22℃)の酸素添加培地を含有する貯蔵容器に1時間入れた。薄片をその後浸積薄片のための記録チャンバーに移し、そして30〜32℃で5〜6ml/分の速度で連続灌流した。対照培地は(mMで)NaCl、120;KCl 2.5、NaH2PO4、1.25;NaHCO3 26;MgSO4、2.0;CaCl2、2.0;D−グルコース 10を含有した。全溶液はGABAA媒介性活性を阻害するため100μMピクロトキシン(Sigma)を含有した。
【0183】
in vitro電気生理学的技術
標準的電気生理学的技術を使用して電場電位を記録した。双極絶縁タングステンワイヤ電極を使用して歯状回の内側貫通路にシナプス前刺激を適用し、また、ガラス電極を用い、歯状回の分子層の中1/3から0.033Hzの対照試験周波数で興奮性シナプス後場電位(EPSP)を記録した。歯状回の内刃(inner blade)を全研究で使用した。各実験で、入力−出力曲線(求心性刺激強度対EPSP振幅)を試験周波数でプロットした。全実験について、試験EPSPの振幅を最大の1/3(約1.2mV)に調節した。LTPを8連の高頻度刺激(HFS)により惹起し(8刺激のそれぞれは200Hz、連続間間隔2s)、正常の試験EPSP振幅の2倍の連続の初期EPSPを惹起するようにHFSの間に刺激電圧を増大した。LTPの対照(ベヒクル単独)および実験レベルを同一海馬から調製した薄片で測定した。p−CLAMP(Axon Instruments、米国カリフォルニア州)を使用して記録を解析した。本明細書に報告する値はn薄片の平均±S.E.M.であった。Studentの両側t検定を統計学的比較に使用した。
【0184】
in vivo電気生理学
実験はウレタン(エチルカルバメート、1.5gm/kg i.p.)麻酔した雄性Wistarラット(250〜300g)で実施した。体温は37〜37.3℃に維持した。動物の世話および実験プロトコルはアイルランド共和国保健省(Department
of Health、Republic of Ireland)により承認された。
【0185】
電極は以前に記述された(Klyubinら、2004、2005)とおり作成しかつ植込んだ。簡潔には、捻りワイヤ双極電極をTeflon被覆タングステンワイヤ(62.5μm内核径、75μm外径)から構築した。興奮性シナプス後場電位(EPSP)の単一経路記録を、同側のSchaffer側枝−交連経路の刺激に応答する右海馬半球のCA1領域中の放線状層から行った。電極植込み部位は十字縫合に関する定位座標を使用して同定し、記録部位は十字縫合の後方約3.4mmかつ正中線の右約2.5mmに位置
し、また、刺激電極は十字縫合の後方約4.2mmかつ正中線の右約3.8に位置した。背側海馬のCA1領域の放線状層中のワイヤ電極の最適深度は電気生理学的基準を使用して決定し、かつ、死後確認した。試験EPSPは、0.033Hzの周波数および最大の50%のEPSP振幅を与えるように調節した刺激強度で惹起した。LTPを誘導するためのHFSプロトコルは、10連続の20刺激、刺激間間隔5ms(200Hz)、連続間間隔2秒よりなった。強度は最大振幅の約75%のEPSPを与えるようにHFSの間に増大させた。
【0186】
サンプルを注入するため、ステンレス鋼製ガイドカニューレ(22ゲージ、0.7mm外径、13mm長さ)を、電極植込み直前に右側脳室の上(正中線の側方約1mmおよび硬膜の表面の下約4mm)に植込んだ。内部カニューレ(28ゲージ、0.36mm外径)を介して脳室内(i.c.v.)注入を行った。カニューレの配置の確認はi.c.v.注入後のインク色素の広がりを確認することにより死後に実施した。本明細書に報告する値はn薄片の平均±S.E.M.であった。Studentの両側t検定を統計学的比較に使用した。
【0187】
剤
合成Aβ(1−42)をBachemから得た。in vitro実験のため、合成Aβ1−42を水酸化アンモニウム(0.1%)中50μMのストック溶液として調製し、−20℃で保存し、そしてその後各実験の直前に生理学的媒体に添加した。in vivo実験のため、合成Aβ(1−42)(Bachem)を氷冷milliQ水若しくはTeplowに再懸濁した。アリコートを取り出し、そして100,000gで3時間(原線維およびプロトフィブリル(protofibril)をペレットにすることが既知の条件)(Klyubinら、2004)遠心分離した。遠心分離後、アミノ酸分析により測定されるところの30若しくは64μMの可溶性Aβの最終濃度を有した上清を、小アリコートで−80℃で保存した。
【0188】
以下のインテグリンαv抗体(全部IgG1アイソタイプ)すなわち18C7、17E6および20A9(全部Calbiochemから)を研究で使用した。使用した対照抗体は、ヒトICAM−1に対するIgG2aマウス抗体7H10およびIgG1アイソタイプ27/1であった。使用した他の成分は、エキスタチン(Source)、Van Maesら、1994の方法に従ってElan Pharmaceuticalsにより製造されたSM256(3−[1−[3−(N−イミダゾル−2−イルアミノ)プロピル]インダゾル−5−イルカルボニルアミノ]−2(S)−(2,4,6−トリメチルベンゼンスルホニルアミノ)プロピオン酸トリフルオロ酢酸塩)(ELN 151993として記録される)、スーパーフィブロネクチン(Sigma)およびファロイジン(Calbiochem)であった。in vivo実験において、SM256は生理的食塩水中Tween 80(Sigma)の懸濁液中でi.p投与のため調製した(15v/v%)。
【実施例7】
【0189】
抗インテグリンαv抗体はin vitroで歯状回でのLTPのAβ媒介性阻害を予防する
in vitroの歯状回での短いHFSによるLTPの誘導は、HFS前30分間のAβの前灌流により予防され、以前の研究(Wangら、2004a、b、2005)を確認した。従って、Aβ(500nM、LTPの最大阻害を引き起こすことが以前に見出された濃度(Wangら、2004a))の存在下でのLTPは、HFSの1時間後に基礎の105±3%であり(図10A)(n=5、P<0.001)、152±5%であった(図10A)対照LTPと比較して有意に低下された。
【0190】
αvインテグリンサブユニットに対する選択的抗体を灌流することの効果をLTPのAβ媒介性阻害に対し検討した。18C7、20A9および17E6と命名される、αV含有インテグリンに対する3種の異なる抗体を検討した。該抗体のいずれも基礎のEPSP若しくはLTP誘導に対するいかなる影響も有しなかった。しかしながら、LTPのAβ媒介性阻害はαv抗体のそれぞれの灌流により予防された。図10A〜10Cは、全3種のαv抗体が、介在対照実験と比較してLTPに対するAβの阻害効果の予防において有効であったことを示す。従って、LTPは、Aβならびに抗インテグリン抗体18C7、20A9および17E6の存在下で144±6%、137±7%および153±11%であった。該値は対照LTPと比較して有意に異ならなかった(各実験についてn=5、P>0.01)。各抗体実験と平行して実施した介在薄片において、LTPはAβ単独の存在下で105±3%、105±6%および103±3%であり、値はLTPに対するAβ単独のものと有意に異ならなかった(各実験についてn=5、P>0.01)。
【0191】
対照的に、2種の対照抗体はLTPのAβ媒介性阻害を有意に予防しなかった。LTPは、Aβおよび対照抗体27/1の存在下で105±6%(図11A)、ならびに、Aβおよび対照抗体7H10(アイソタイプ対照)の存在下で107±8%であった(図11B)(n=5、P>0.05)。
【実施例8】
【0192】
抗αvインテグリン抗体はin vivoでCA1でのLTPのAβ媒介性阻害を予防する
ウレタン麻酔したラットのCA1でのLTPの誘導は、HFS10分前の可溶性の原線維を含まないAβのi.c.v.注入により予防され、以前の研究(Klyubinら、2004)を確認した。対照ベヒクル注入動物で、LTPはHFSの3時間後に基礎の140±5%であった(n=6)。可溶性の原線維を含まないAβ(5μl中50pmol、n=5)の存在下でLTPは強く阻害され、105±5%であり(図12A)、対照LTPと比較して有意に低下された(P<0.001)。αvインテグリンサブユニットに対する選択的抗体17E6(10μl中27.9μg)はLTPのAβ媒介性阻害を予防し、135±8%であった(n=5、ベヒクルおよびAβに比較してP<0.01;ベヒクルおよびベヒクルに比較してP>0.05)。アイソタイプ(IgG1マウス)対照抗体27/1(10μl中27.9μg)はLTPのAβ媒介性阻害に影響を及ぼすことに失敗した(103±6%、n=6、ベヒクルおよびAβに比較してP>0.05;ベヒクルおよびベヒクルに比較してP<0.01)(図12B)。
【実施例9】
【0193】
αv含有インテグリンの小分子非ペプチド拮抗剤はin vitroで歯状回およびin
vivoでCA1でのLTPのAβ媒介性阻害を予防する
SM256は強力なαv拮抗剤でありかつαv媒介性の細胞接着を阻害する非ペプチド剤である(Van Waesら、2000)。SM256(10μM)単独はLTPを阻害せず、148±7%であった。しかしながら、SM256はLTPのAβ媒介性阻害を予防し、129±5%であった(図13A)(n=5、P<0.01)。
【0194】
同様に、SM256の全身前投与は、可溶性の原線維を含まないAβのi.c.v.注入により引き起こされるin vivoのCA1でのLTPの阻害を無効にした。選ばれたSM256の用量(1.2mlベヒクル中40mg、i.p.)は基礎のシナプス伝達若しくは対照LTPに対する認識可能な影響を有しなかった(図12Cおよびデータは示されない)。SM256の40分後かつHFSの10分前に注入した可溶性Aβ(50pmol、i.c.v.)はLTPを阻害することに失敗し、133±5%であり(図12C、n=4;HFS前基礎に比較してP<0.01;末梢ベヒクルおよび中枢ベヒクル(144±9%、n=3)に比較してP>0.05)、また、ベヒクルの末梢注入次いでA
βの中枢注入を与えた動物のLTPの大きさと有意に異ならなかった(103±10%、n=4、P<0.05)。
【実施例10】
【0195】
スーパーフィブロネクチンはin vitroで歯状回でのLTPのAβ媒介性阻害を予防する
スーパーフィブロネクチンはαvβ1のリガンドである(Morlaら、1994)。スーパーフィブロネクチン(1μM)単独はLTPを阻害せず、155±2%であった。しかしながら、スーパーフィブロネクチンはLTPのAβ媒介性阻止を予防し、147±6%であった(図13B)(n=5、P<0.01)。
【実施例11】
【0196】
ディスインテグリンはin vitroで歯状回でのLTPのAβ媒介性阻害を予防する
ディスインテグリン、エキスタチンの効果もまたLTPのAβ惹起性の阻害に対して検討した。ディスインテグリンは、非常に高い親和性でかつRGDペプチドより強くインテグリンに結合する、インテグリンのアンタゴニストである蛇毒から単離された小型の4〜10kDaのRGDを含有するシステイン豊富なペプチドである(Ganら、1988)。エキスタチンはαv/β3およびα5/β1を包含するRGD依存性インテグリンを阻害することが示された(Kumarら、1997)1種のこうしたディスインテグリンである。エキスタチン(50nM)単独はLTPを阻害せず、158±5%であった。しかしながら、エキスタチンはLTPのAβ媒介性阻止を予防し、143±6%、n=5、P<0.01であった(図13C)。
【0197】
上で引用された全部の刊行物、特許および特許出願は、各個々の刊行物若しくは特許出願が引用することによりそのように組み込まれることをとりわけかつ個々に示された場合と同一の程度まで全部の目的上そっくりそのまま引用することにより組み込まれる。本発明は明瞭さおよび理解の目的上具体的説明および実施例として若干詳細に記述されたとは言え、付随する請求の範囲の範囲内である種の変更および改変を実施しうることが明らかであろう。
【特許請求の範囲】
【請求項1】
1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法。
【請求項2】
1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置若しくは予防方法。
【請求項3】
アミロイド形成疾患がアルツハイマー病若しくは軽度認知障害である、請求項2に記載の方法。
【請求項4】
アミロイド形成疾患がびまん性レビー小体病若しくはパーキンソン病である、請求項2に記載の方法。
【請求項5】
インテグリンサブユニットαvに結合する最低2剤の有効投薬量が投与される、請求項1に記載の方法。
【請求項6】
剤が、Aβ産生の阻害剤、Aβ沈着の阻害剤、Aβ消失のメディエーター、アミロイド斑消失のメディエーター、Aβ神経毒性の阻害剤、Aβ凝集の阻害剤およびAβ解離のメディエーターよりなる群から選ばれる第二の剤とともに投与される、請求項1に記載の方法。
【請求項7】
Aβ産生の阻害剤がγ分泌酵素阻害剤である、請求項6に記載の方法。
【請求項8】
Aβ産生の阻害剤がβ分泌酵素阻害剤である、請求項6に記載の方法。
【請求項9】
剤がAβに対する抗体とともに投与される、請求項1に記載の方法。
【請求項10】
剤がRGD(Arg−Gly−Asp)モチーフを含んでなるペプチドである、請求項1に記載の方法。
【請求項11】
剤がαvβ1インテグリンのリガンドである、請求項1に記載の方法。
【請求項12】
剤がフィブロネクチン若しくはスーパーフィブロネクチンである、請求項1に記載の方法。
【請求項13】
剤が、αvインテグリンサブユニット発現細胞のビトロネクチン若しくはフィブロネクチンへの接着を阻害する、請求項1に記載の方法。
【請求項14】
剤が、αvインテグリンサブユニット発現細胞のオステオポンチンへの接着を阻害する、請求項1に記載の方法。
【請求項15】
剤がモノクローナル若しくはポリクローナル抗体である、請求項1に記載の方法。
【請求項16】
剤が18C7、20A9および17E6から選択される抗体と同一のエピトープを認識する抗体である、請求項1に記載の方法。
【請求項17】
抗体がヒト化抗体、キメラ抗体およびナノボディから選択される、請求項16に記載の方法。
【請求項18】
剤が18C7、20A9および17E6から選択される抗体である、請求項1に記載の方法。
【請求項19】
剤がインテグリンサブユニットαvへの結合について18C7、20A9および17E6から選ばれる抗体と競合する、請求項1に記載の方法。
【請求項20】
剤が、それらの立体異性体、若しくはそれらの立体異性体の混合物、またはそれらの製薬学的に許容できる塩の形態を包含する、式IaおよびIbの化合物
【化1】
ここで
X1およびX3は窒素若しくは炭素から独立に選択され;
R1は
【化2】
から選択され;
ここで、上の複素環は、NH2、ハロゲン、NO2、CN、CF3、C1−C4アルコキシ、C1−C6アルキルおよびC3−C7シクロアルキルよりなる群から選択される0〜2置換基で場合によっては置換されており;
Uは−(CH2)n−、−(CH2)tQ(CH2)m−および−C(=O)(CH2)n−1−から選択され、ここでメチレン基の1個は場合によってはR7で置換されており
;
Qは、1,2−フェニレン、1,3−フェニレン、2,3−ピリジニレン、3,4−ピリジニレンおよび2,4−ピリジニレンから選択され;
R6はH、C1−C4アルキルおよびベンジルから選択され;
R7はC1−C6アルキル、C3−C7シクロアルキル、C4−C11シクロアルキルアルキル、アリール、アリール(C1−C6アルキル)、ヘテロアリールおよびヘテロアリール(C1−C6アルキル)から選択され;
R10は、H、ハロゲン、CO2R17、CONR17R20、0〜1個のR15若しくは0〜1個のR21で置換されているC1−C6アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR15若しくは0〜1個のR21で置換されているC3−C7シクロアルキル、0〜1個のR15若しくは0〜1個のR21で置換されているC4−C11シクロアルキルアルキル、および0〜1個のR15若しくは0〜2個のR11若しくは0〜1個のR21で置換されているアリール(C1−C6アルキル)−から選択され;
R11は、H、ハロゲン、CF3、CN、NO2、ヒドロキシ、NR2R3、0〜1個のR21で置換されているC1−C4アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR21で置換されているアリール、0〜1個のR21で置換されているアリール(C1−C6アルキル)−、0〜1個のR21で置換されている(C1−C4アルコキシ)カルボニル、0〜1個のR21で置換されている(C1−C4アルキル)カルボニル、0〜1個のR21で置換されているC1−C4アルキルスルホニル、および0〜1個のR21で置換されているC1−C4アルキルアミノスルホニルから選択され;
Wは−C(=O)−N(R13)−であり;
Xは−CH(R14)−CH(R15)−であり;
R13はHおよびCH3から選択され;
R14は、H、C1−C10アルキル、アリールおよびヘテロアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ハロ、シアノ、アミノ、CF3およびNO2から選択される0〜3個の置換基で場合によっては置換されており;
R15はHおよびR16から選択され;
Yは−COR19であり;
R16は
−NH(R20)−C(=O)−R17、
−N(R20)−C(=O)−R17、
−N(R20)−C(=O)−NH−R17、
―N(R20)SO2−R17、および
−N(R20)SO2−N(R20)R17
から選択され、
R17は、C1−C10アルキル、C3−C11シクロアルキル、アリール(C1−C6アルキル)−、(C1−C6アルキル)アリール、ヘテロアリール(C1−C6アルキル)−、(C1−C6アルキル)ヘテロアリール、ビアリール(C1−C6アルキル)−、ヘテロアリール若しくはアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ヘテロアリール、ハロ、シアノ、アミノ、CF3およびNO2よりなる群から選択される0〜3置換基で場合によっては置換されており;
R19は−O−(CH2)kN+(R22)(R23)(R24)Z−であり;
Z−は、ハロゲン化イオン、重硫酸イオン、硫酸イオン、リン酸水素イオン、リン酸イオン、トルエンスルホン酸イオン、メタンスルホン酸イオン、エタンスルホン酸イオン、酢酸イオン、トリフルオロ酢酸イオン、クエン酸イオン、シュウ酸イオン、コハク酸イオンおよびマロン酸イオンから選択される製薬学的に許容できる陰イオンであり;
R22、R23およびR24はH、C1−C4アルキルおよびC4−C11シクロアルキルアルキルから独立に選択されるか;
あるいは、R22およびR23は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する5〜7員の複素環系を形成し得、ならびにR24は上のとおり定義されるか、または、R22、R23およびR24は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する複素二環系を形成し得;
R20はHおよびCH3から選択され;
R21はCOOHおよびNR62から選択され;
kは2であり;
mは0および1から選択され;
nは1〜4であり;ならびに
tは0および1から選択される、
から選択される化合物である、請求項1に記載の方法。
【請求項21】
剤が、式II:
【化3】
ここでR19は−H、−CH3および−CH2CH2N+(CH3)3から選ばれる
の化合物である、請求項1に記載の方法。
【請求項22】
H19が−Hである、請求項21に記載の方法。
【請求項23】
H19が−CH3である、請求項21に記載の方法。
【請求項24】
R19が−CH2CH2N+(CH3)3である、請求項21に記載の方法。
【請求項25】
剤がディスインテグリンである、請求項1に記載の方法。
【請求項26】
剤がエキスタチンである、請求項1に記載の方法。
【請求項27】
剤がヒト抗体である、請求項1に記載の方法。
【請求項28】
剤がヒト化抗体である、請求項1に記載の方法。
【請求項29】
剤がキメラ抗体である、請求項1に記載の方法。
【請求項30】
剤がナノボディである、請求項1に記載の方法。
【請求項31】
剤が抗体フラグメントである、請求項1に記載の方法。
【請求項32】
剤が、抗体の1種若しくはそれ以上のH鎖、L鎖、F(ab)、F(ab)2、F(ab)c若しくはF(v)、またはそれらのいずれかの組合せを含んでなる、請求項1に記載の方法。
【請求項33】
剤が抗体でありかつ該抗体のアイソタイプがIgG1若しくはIgG4である、請求項1に記載の方法。
【請求項34】
剤が抗体でありかつ該抗体のアイソタイプがIgG2若しくはIgG3である、請求項1に記載の方法。
【請求項35】
剤が抗体鎖である、請求項1に記載の方法。
【請求項36】
剤が抗体でありかつ該抗体が2対のLおよびH鎖を含んでなる、請求項1に記載の方法。
【請求項37】
剤が患者に投与される、請求項1に記載の方法。
【請求項38】
剤が抗体でありかつ該抗体の投薬量が患者体重1kgあたり約0.01から約10mgまでの範囲にわたる、請求項37に記載の方法。
【請求項39】
剤が製薬学的組成物として担体とともに投与される、請求項37に記載の方法。
【請求項40】
剤が、腹腔内、経口、鼻内、皮下、くも膜下腔内、筋肉内、局所若しくは静脈内に投与される、請求項37に記載の方法。
【請求項41】
患者がアミロイド形成疾患に罹っている、請求項37に記載の方法。
【請求項42】
疾患が、アルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、アミロイドーシス、ダウン症候群、およびプリオン感染により全部若しくは部分的に引き起こされる疾患よりなる群から選ばれる、請求項41に記載の方法。
【請求項43】
剤をコードする核酸が投与される、請求項1に記載の方法。
【請求項44】
剤が、アンチセンスRNA分子、アンチセンスDNA分子、リボザイム、RNAiおよびジンクフィンガータンパク質よりなる群から選ばれる、請求項1に記載の方法。
【請求項45】
アミロイド沈着物の形成を阻害することをさらに含んでなる、請求項1に記載の方法。
【請求項46】
アミロイド毒性を阻害することをさらに含んでなる、請求項1に記載の方法。
【請求項47】
剤がLTPの維持期を阻止しない、請求項1に記載の方法。
【請求項48】
剤が培養中の薄片調製物中のLTPのアミロイド媒介性阻害を抑制する、請求項1に記載の方法。
【請求項49】
剤が可溶性AβによるLTPの阻害を抑制する、請求項1に記載の方法。
【請求項50】
a)剤をインテグリンサブユニットαv結合剤と同定すること;および
b)該同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することを確定すること
を含んでなる、LTPのアミロイド媒介性阻害を抑制する剤の同定方法。
【請求項51】
剤を同定する段階が、直接結合アッセイ、競合結合アッセイおよび細胞接着アッセイの1種若しくはそれ以上を含んでなり;ならびに
同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することの決定段階が、高頻度刺激を第一の神経回路に導入することおよびLTPの誘導を測定すること、高頻度刺激をAβの存在下の第二の神経回路に導入することおよびLTP誘導の阻害を測定すること、ならびにAβおよび剤の存在下の第三の神経回路に高頻度刺激を導入することならびにLTP誘導の阻害の抑制を測定することを含んでなる、
請求項50に記載の方法。
【請求項52】
請求項50に記載の方法により同定される、LTPのアミロイド媒介性阻害を抑制する剤。
【請求項53】
剤が抗体である、請求項52に記載の剤。
【請求項54】
請求項52に記載の剤および製薬学的に許容できる担体を含んでなる組成物。
【請求項55】
請求項50に記載の方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法。
【請求項56】
請求項51に記載の方法により同定される、LTPのアミロイド媒介性阻害を抑制する剤。
【請求項57】
剤が抗体である、請求項56に記載の剤。
【請求項58】
請求項56に記載の剤および製薬学的に許容できる担体を含んでなる組成物。
【請求項59】
請求項51に記載の方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法。
【請求項60】
長期増強(LTP)のアミロイド媒介性阻害を抑制するのに有効な量のαv拮抗剤若しくはαv媒介性の細胞接着の阻害剤を投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置方法。
【請求項61】
アミロイド形成疾患がアルツハイマー病である、請求項60に記載の方法。
【請求項62】
アミロイド形成疾患が軽度認知障害である、請求項60に記載の方法。
【請求項1】
1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法。
【請求項2】
1種若しくはそれ以上の剤がLTPのアミロイド媒介性阻害を抑制するような条件下でインテグリンサブユニットαvに結合する1種若しくはそれ以上の剤の有効投薬量を投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置若しくは予防方法。
【請求項3】
アミロイド形成疾患がアルツハイマー病若しくは軽度認知障害である、請求項2に記載の方法。
【請求項4】
アミロイド形成疾患がびまん性レビー小体病若しくはパーキンソン病である、請求項2に記載の方法。
【請求項5】
インテグリンサブユニットαvに結合する最低2剤の有効投薬量が投与される、請求項1に記載の方法。
【請求項6】
剤が、Aβ産生の阻害剤、Aβ沈着の阻害剤、Aβ消失のメディエーター、アミロイド斑消失のメディエーター、Aβ神経毒性の阻害剤、Aβ凝集の阻害剤およびAβ解離のメディエーターよりなる群から選ばれる第二の剤とともに投与される、請求項1に記載の方法。
【請求項7】
Aβ産生の阻害剤がγ分泌酵素阻害剤である、請求項6に記載の方法。
【請求項8】
Aβ産生の阻害剤がβ分泌酵素阻害剤である、請求項6に記載の方法。
【請求項9】
剤がAβに対する抗体とともに投与される、請求項1に記載の方法。
【請求項10】
剤がRGD(Arg−Gly−Asp)モチーフを含んでなるペプチドである、請求項1に記載の方法。
【請求項11】
剤がαvβ1インテグリンのリガンドである、請求項1に記載の方法。
【請求項12】
剤がフィブロネクチン若しくはスーパーフィブロネクチンである、請求項1に記載の方法。
【請求項13】
剤が、αvインテグリンサブユニット発現細胞のビトロネクチン若しくはフィブロネクチンへの接着を阻害する、請求項1に記載の方法。
【請求項14】
剤が、αvインテグリンサブユニット発現細胞のオステオポンチンへの接着を阻害する、請求項1に記載の方法。
【請求項15】
剤がモノクローナル若しくはポリクローナル抗体である、請求項1に記載の方法。
【請求項16】
剤が18C7、20A9および17E6から選択される抗体と同一のエピトープを認識する抗体である、請求項1に記載の方法。
【請求項17】
抗体がヒト化抗体、キメラ抗体およびナノボディから選択される、請求項16に記載の方法。
【請求項18】
剤が18C7、20A9および17E6から選択される抗体である、請求項1に記載の方法。
【請求項19】
剤がインテグリンサブユニットαvへの結合について18C7、20A9および17E6から選ばれる抗体と競合する、請求項1に記載の方法。
【請求項20】
剤が、それらの立体異性体、若しくはそれらの立体異性体の混合物、またはそれらの製薬学的に許容できる塩の形態を包含する、式IaおよびIbの化合物
【化1】
ここで
X1およびX3は窒素若しくは炭素から独立に選択され;
R1は
【化2】
から選択され;
ここで、上の複素環は、NH2、ハロゲン、NO2、CN、CF3、C1−C4アルコキシ、C1−C6アルキルおよびC3−C7シクロアルキルよりなる群から選択される0〜2置換基で場合によっては置換されており;
Uは−(CH2)n−、−(CH2)tQ(CH2)m−および−C(=O)(CH2)n−1−から選択され、ここでメチレン基の1個は場合によってはR7で置換されており
;
Qは、1,2−フェニレン、1,3−フェニレン、2,3−ピリジニレン、3,4−ピリジニレンおよび2,4−ピリジニレンから選択され;
R6はH、C1−C4アルキルおよびベンジルから選択され;
R7はC1−C6アルキル、C3−C7シクロアルキル、C4−C11シクロアルキルアルキル、アリール、アリール(C1−C6アルキル)、ヘテロアリールおよびヘテロアリール(C1−C6アルキル)から選択され;
R10は、H、ハロゲン、CO2R17、CONR17R20、0〜1個のR15若しくは0〜1個のR21で置換されているC1−C6アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR15若しくは0〜1個のR21で置換されているC3−C7シクロアルキル、0〜1個のR15若しくは0〜1個のR21で置換されているC4−C11シクロアルキルアルキル、および0〜1個のR15若しくは0〜2個のR11若しくは0〜1個のR21で置換されているアリール(C1−C6アルキル)−から選択され;
R11は、H、ハロゲン、CF3、CN、NO2、ヒドロキシ、NR2R3、0〜1個のR21で置換されているC1−C4アルキル、0〜1個のR21で置換されているC1−C4アルコキシ、0〜1個のR21で置換されているアリール、0〜1個のR21で置換されているアリール(C1−C6アルキル)−、0〜1個のR21で置換されている(C1−C4アルコキシ)カルボニル、0〜1個のR21で置換されている(C1−C4アルキル)カルボニル、0〜1個のR21で置換されているC1−C4アルキルスルホニル、および0〜1個のR21で置換されているC1−C4アルキルアミノスルホニルから選択され;
Wは−C(=O)−N(R13)−であり;
Xは−CH(R14)−CH(R15)−であり;
R13はHおよびCH3から選択され;
R14は、H、C1−C10アルキル、アリールおよびヘテロアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ハロ、シアノ、アミノ、CF3およびNO2から選択される0〜3個の置換基で場合によっては置換されており;
R15はHおよびR16から選択され;
Yは−COR19であり;
R16は
−NH(R20)−C(=O)−R17、
−N(R20)−C(=O)−R17、
−N(R20)−C(=O)−NH−R17、
―N(R20)SO2−R17、および
−N(R20)SO2−N(R20)R17
から選択され、
R17は、C1−C10アルキル、C3−C11シクロアルキル、アリール(C1−C6アルキル)−、(C1−C6アルキル)アリール、ヘテロアリール(C1−C6アルキル)−、(C1−C6アルキル)ヘテロアリール、ビアリール(C1−C6アルキル)−、ヘテロアリール若しくはアリールから選択され、前記アリール若しくはヘテロアリール基は、C1−C4アルキル、C1−C4アルコキシ、アリール、ヘテロアリール、ハロ、シアノ、アミノ、CF3およびNO2よりなる群から選択される0〜3置換基で場合によっては置換されており;
R19は−O−(CH2)kN+(R22)(R23)(R24)Z−であり;
Z−は、ハロゲン化イオン、重硫酸イオン、硫酸イオン、リン酸水素イオン、リン酸イオン、トルエンスルホン酸イオン、メタンスルホン酸イオン、エタンスルホン酸イオン、酢酸イオン、トリフルオロ酢酸イオン、クエン酸イオン、シュウ酸イオン、コハク酸イオンおよびマロン酸イオンから選択される製薬学的に許容できる陰イオンであり;
R22、R23およびR24はH、C1−C4アルキルおよびC4−C11シクロアルキルアルキルから独立に選択されるか;
あるいは、R22およびR23は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する5〜7員の複素環系を形成し得、ならびにR24は上のとおり定義されるか、または、R22、R23およびR24は、一緒になって、N、OおよびSから選択される1〜2個のヘテロ原子を含有する複素二環系を形成し得;
R20はHおよびCH3から選択され;
R21はCOOHおよびNR62から選択され;
kは2であり;
mは0および1から選択され;
nは1〜4であり;ならびに
tは0および1から選択される、
から選択される化合物である、請求項1に記載の方法。
【請求項21】
剤が、式II:
【化3】
ここでR19は−H、−CH3および−CH2CH2N+(CH3)3から選ばれる
の化合物である、請求項1に記載の方法。
【請求項22】
H19が−Hである、請求項21に記載の方法。
【請求項23】
H19が−CH3である、請求項21に記載の方法。
【請求項24】
R19が−CH2CH2N+(CH3)3である、請求項21に記載の方法。
【請求項25】
剤がディスインテグリンである、請求項1に記載の方法。
【請求項26】
剤がエキスタチンである、請求項1に記載の方法。
【請求項27】
剤がヒト抗体である、請求項1に記載の方法。
【請求項28】
剤がヒト化抗体である、請求項1に記載の方法。
【請求項29】
剤がキメラ抗体である、請求項1に記載の方法。
【請求項30】
剤がナノボディである、請求項1に記載の方法。
【請求項31】
剤が抗体フラグメントである、請求項1に記載の方法。
【請求項32】
剤が、抗体の1種若しくはそれ以上のH鎖、L鎖、F(ab)、F(ab)2、F(ab)c若しくはF(v)、またはそれらのいずれかの組合せを含んでなる、請求項1に記載の方法。
【請求項33】
剤が抗体でありかつ該抗体のアイソタイプがIgG1若しくはIgG4である、請求項1に記載の方法。
【請求項34】
剤が抗体でありかつ該抗体のアイソタイプがIgG2若しくはIgG3である、請求項1に記載の方法。
【請求項35】
剤が抗体鎖である、請求項1に記載の方法。
【請求項36】
剤が抗体でありかつ該抗体が2対のLおよびH鎖を含んでなる、請求項1に記載の方法。
【請求項37】
剤が患者に投与される、請求項1に記載の方法。
【請求項38】
剤が抗体でありかつ該抗体の投薬量が患者体重1kgあたり約0.01から約10mgまでの範囲にわたる、請求項37に記載の方法。
【請求項39】
剤が製薬学的組成物として担体とともに投与される、請求項37に記載の方法。
【請求項40】
剤が、腹腔内、経口、鼻内、皮下、くも膜下腔内、筋肉内、局所若しくは静脈内に投与される、請求項37に記載の方法。
【請求項41】
患者がアミロイド形成疾患に罹っている、請求項37に記載の方法。
【請求項42】
疾患が、アルツハイマー病、II型糖尿病、パーキンソン病、びまん性レビー小体病、アミロイドーシス、ダウン症候群、およびプリオン感染により全部若しくは部分的に引き起こされる疾患よりなる群から選ばれる、請求項41に記載の方法。
【請求項43】
剤をコードする核酸が投与される、請求項1に記載の方法。
【請求項44】
剤が、アンチセンスRNA分子、アンチセンスDNA分子、リボザイム、RNAiおよびジンクフィンガータンパク質よりなる群から選ばれる、請求項1に記載の方法。
【請求項45】
アミロイド沈着物の形成を阻害することをさらに含んでなる、請求項1に記載の方法。
【請求項46】
アミロイド毒性を阻害することをさらに含んでなる、請求項1に記載の方法。
【請求項47】
剤がLTPの維持期を阻止しない、請求項1に記載の方法。
【請求項48】
剤が培養中の薄片調製物中のLTPのアミロイド媒介性阻害を抑制する、請求項1に記載の方法。
【請求項49】
剤が可溶性AβによるLTPの阻害を抑制する、請求項1に記載の方法。
【請求項50】
a)剤をインテグリンサブユニットαv結合剤と同定すること;および
b)該同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することを確定すること
を含んでなる、LTPのアミロイド媒介性阻害を抑制する剤の同定方法。
【請求項51】
剤を同定する段階が、直接結合アッセイ、競合結合アッセイおよび細胞接着アッセイの1種若しくはそれ以上を含んでなり;ならびに
同定されたαv結合剤がLTPのアミロイド媒介性阻害を抑制することの決定段階が、高頻度刺激を第一の神経回路に導入することおよびLTPの誘導を測定すること、高頻度刺激をAβの存在下の第二の神経回路に導入することおよびLTP誘導の阻害を測定すること、ならびにAβおよび剤の存在下の第三の神経回路に高頻度刺激を導入することならびにLTP誘導の阻害の抑制を測定することを含んでなる、
請求項50に記載の方法。
【請求項52】
請求項50に記載の方法により同定される、LTPのアミロイド媒介性阻害を抑制する剤。
【請求項53】
剤が抗体である、請求項52に記載の剤。
【請求項54】
請求項52に記載の剤および製薬学的に許容できる担体を含んでなる組成物。
【請求項55】
請求項50に記載の方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法。
【請求項56】
請求項51に記載の方法により同定される、LTPのアミロイド媒介性阻害を抑制する剤。
【請求項57】
剤が抗体である、請求項56に記載の剤。
【請求項58】
請求項56に記載の剤および製薬学的に許容できる担体を含んでなる組成物。
【請求項59】
請求項51に記載の方法により同定される剤の有効投薬量を投与することを含んでなる、長期増強(LTP)のアミロイド媒介性阻害の抑制方法。
【請求項60】
長期増強(LTP)のアミロイド媒介性阻害を抑制するのに有効な量のαv拮抗剤若しくはαv媒介性の細胞接着の阻害剤を投与することを含んでなる、Aβ沈着を特徴とするアミロイド形成疾患の処置方法。
【請求項61】
アミロイド形成疾患がアルツハイマー病である、請求項60に記載の方法。
【請求項62】
アミロイド形成疾患が軽度認知障害である、請求項60に記載の方法。
【図1】


【図2A】
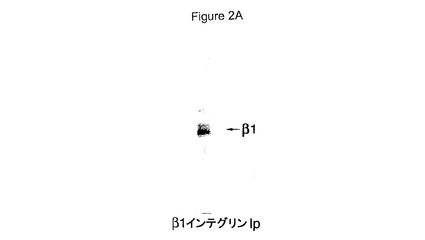
【図2B】

【図2C】
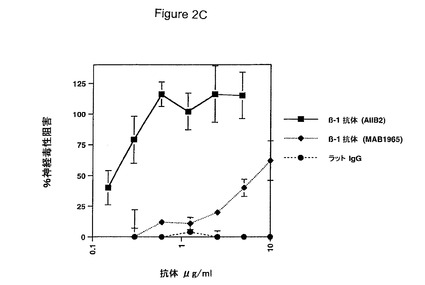
【図3A】
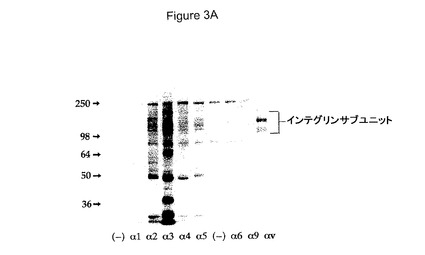
【図3B】
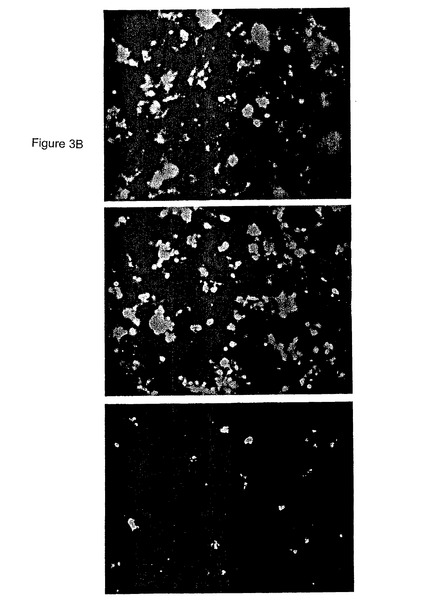
【図3C】
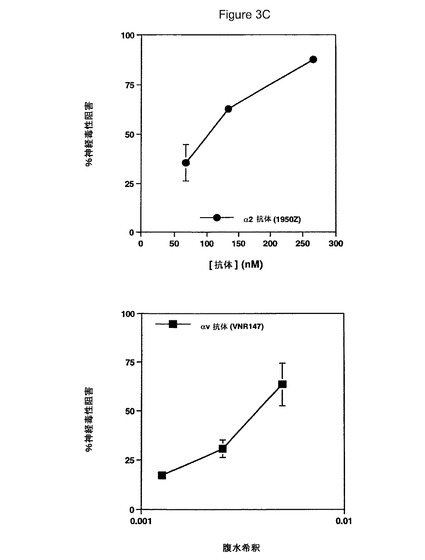
【図4】
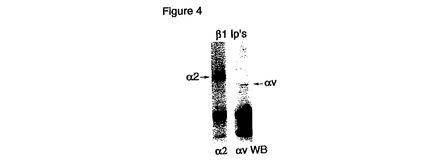
【図5A】
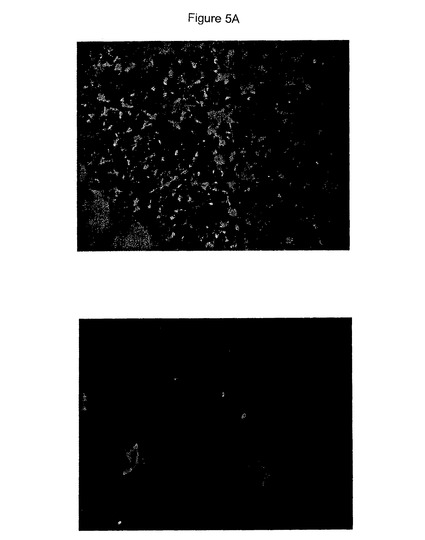
【図5B】
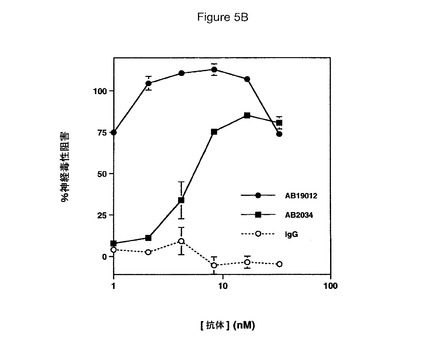
【図5C】
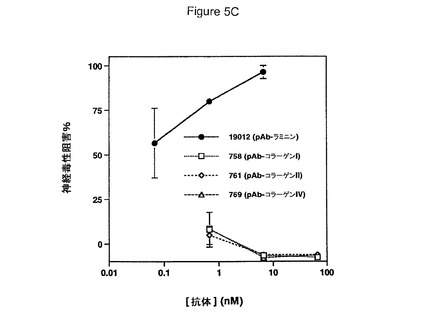
【図6A】
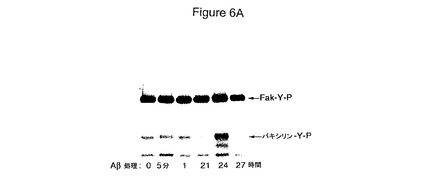
【図6B】
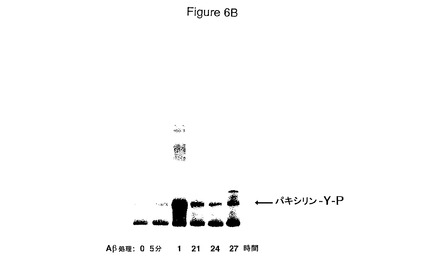
【図7】
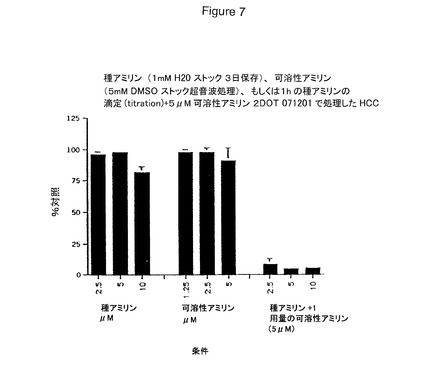
【図8】
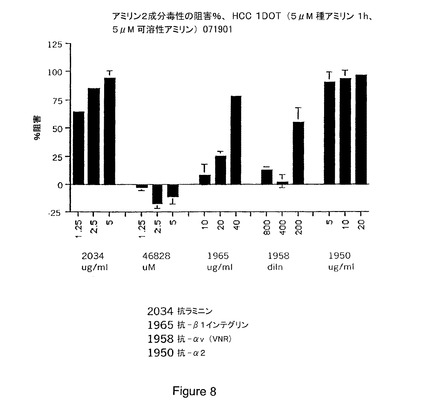
【図9A】
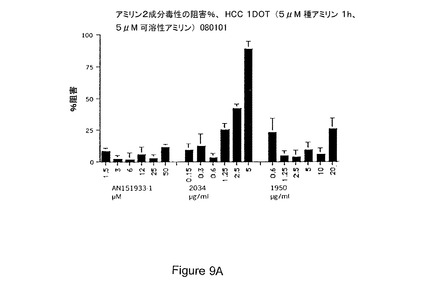
【図9B】
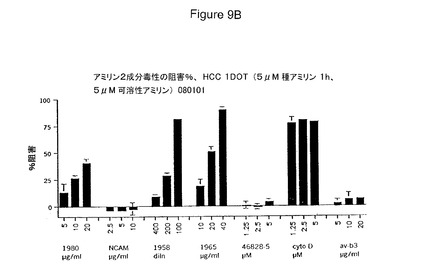
【図10A】
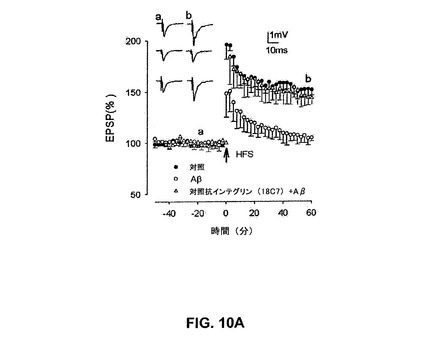
【図10B】
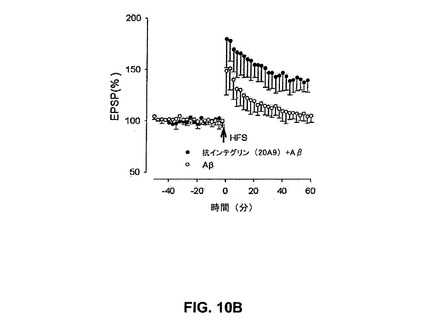
【図10C】
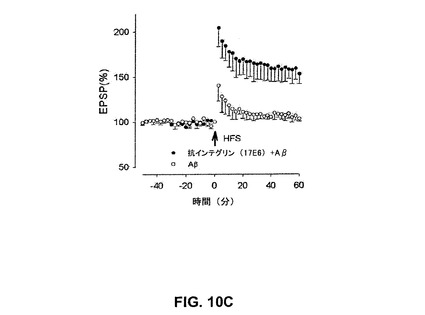
【図11A】
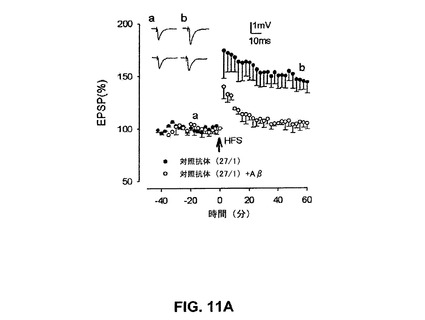
【図11B】
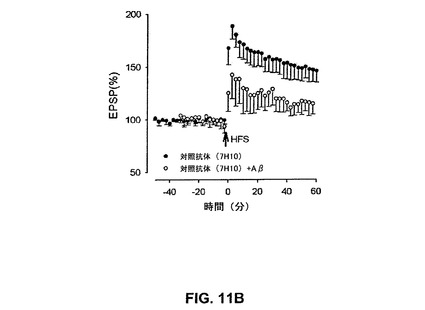
【図12A】
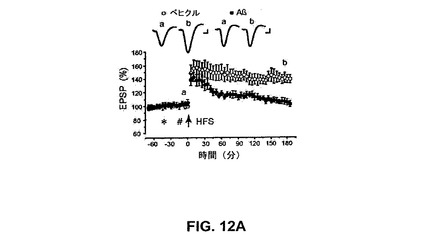
【図12B】
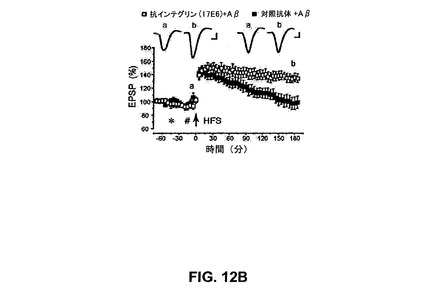
【図12C】
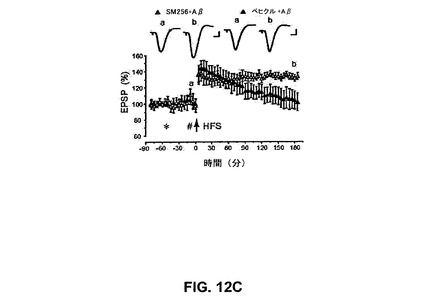
【図13A】
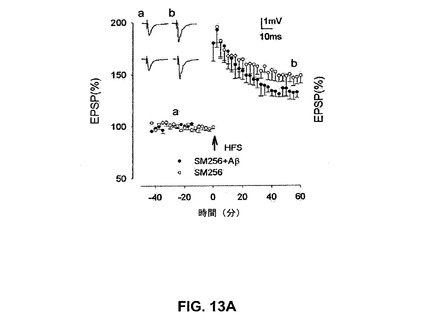
【図13B】
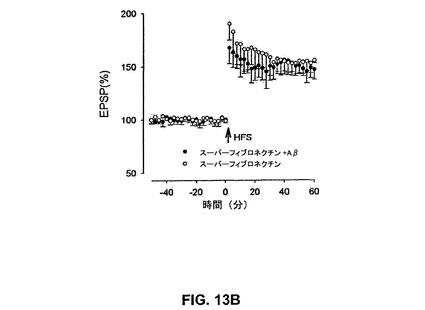
【図13C】
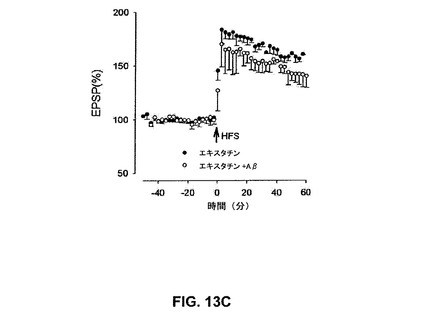
【図2A】
【図2B】
【図2C】
【図3A】
【図3B】
【図3C】
【図4】
【図5A】
【図5B】
【図5C】
【図6A】
【図6B】
【図7】
【図8】
【図9A】
【図9B】
【図10A】
【図10B】
【図10C】
【図11A】
【図11B】
【図12A】
【図12B】
【図12C】
【図13A】
【図13B】
【図13C】
【公表番号】特表2010−524847(P2010−524847A)
【公表日】平成22年7月22日(2010.7.22)
【国際特許分類】
【出願番号】特願2009−554752(P2009−554752)
【出願日】平成20年3月20日(2008.3.20)
【国際出願番号】PCT/US2008/057713
【国際公開番号】WO2008/116100
【国際公開日】平成20年9月25日(2008.9.25)
【出願人】(506209846)エラン・フアーマシユーチカルズ・インコーポレーテツド (4)
【出願人】(509262677)
【Fターム(参考)】
【公表日】平成22年7月22日(2010.7.22)
【国際特許分類】
【出願日】平成20年3月20日(2008.3.20)
【国際出願番号】PCT/US2008/057713
【国際公開番号】WO2008/116100
【国際公開日】平成20年9月25日(2008.9.25)
【出願人】(506209846)エラン・フアーマシユーチカルズ・インコーポレーテツド (4)
【出願人】(509262677)
【Fターム(参考)】
[ Back to top ]