
LXR−アルファ過剰発現に起因する疾病の予防用及び治療用の1,2−ジチオールチオン誘導体を含有する薬学組成物
1,2−ジチオールチオン誘導体を含有する薬学組成物であって、肝Xレセプター(LXR α)又はステロール調節エレメント結合タンパク質(SREBP−1)の過剰活性に起因する疾病の予防及び治療に有用な薬学組成物を提供する。具体的には、前記薬学組成物は、4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン、3−メチル−1,2−ジチオール−3−チオン又は5−(6−メトキシピラジニル)−4−メチル−1,2−ジチオール−3−チオンなどの1,2−ジチオールチオン誘導体を含有する。前記薬学組成物は、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症、脂肪肝、高トリグリセリド血症、高レニン血症の予防及び治療に有効である。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、肝臓Xレセプターα(LXR α)の発現又は活性の抑制、及び、ステロール調節エレメント結合タンパク質−1(SREBP−1:sterol response element binding protein)の発現又は活性を抑制する1,2−ジチオールチオン(1,2−dithiolthione)誘導体に関する。本発明はまた、1,2−ジチオールチオン誘導体を含む薬学組成物に関する。該薬学組成物は、LXR α又はSREBP−1の過剰発現に起因する疾病の予防又は治療に有効である。このような疾病としては、例えば、脂肪肝、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症(aldosteronism)、副腎白質ジストロフィー(adrenoleukodystrophy)、糸球体硬化症、蛋白尿症、腎症(nephropathy)を含む。
【0002】
本発明は、教育科学技術部優秀研究センター(ERC)事業の一環として行われた研究から導き出されたものである[課題固有番号R11−2007−107−01001−0、課題名:代謝及び炎症疾患新薬開発研究センター]。
【背景技術】
【0003】
肝臓Xレセプター(LXR)、ペルオキシソーム増殖因子活性化レセプター(PPAR:peroxisome proliferator-activated receptor)、及びファルネソイドXレセプター(FXR:farnesoid X receptor)は、第2型受容体スーパーファミリー(typeII receptor superfamily)に属する核ホルモン受容体(nuclear hormone receptor)である。これらの受容体は、レチノイドXレセプター(RXR:retinoid X receptor)とヘテロダイマーを形成して、DNAに結合する。リガンドが結合しない場合には、前記ヘテロダイマーがDNAに結合し、コリプレッサー蛋白質と複合体を形成し、一方、リガンドが結合する場合には、構造的変化が生じ、コリプレッサー蛋白質が分離し、補助活性蛋白質(coactivator protein)が結合し、標的遺伝子の転写が促進される(非特許文献1)。核ホルモン受容体のうち、LXRは、コレステロール代謝及び恒常性と関連がある遺伝子の転写調節に重要な役割を果たしている。このような遺伝子の例として、アポリポプロテインE(apoE)、ABCA1、ABCG1、ABCG5、ABCG8、コレステロール7α−ヒドロキシラーゼ(cholesterol 7α-hydroxylase)及びスカベンジャー受容体classB タイプI(scavenger receptor classB typeI)が含まれる(非特許文献2)。またLXRは、SREBP−1c遺伝子に直接に作用し、脂質代謝を調節する(非特許文献3)。
【0004】
LXRには、LXR αとLXR βの2つの異性体が存在する。LXR αは、主に肝臓に存在し、LXR βは、ほとんどの臓器に存在する。LXR αは、天然リガンドであるオキシステロール類、高濃度のグルコース及び人工リガンドであるT0901017、GW3965によって活性化され、脂質生成に関連する遺伝子の発現、及びコレステロール恒常性を調節する。肝臓で脂質が合成されるとき、LXR αは、脂質センサとして作用し、脂質合成遺伝子の発現を調節する重要な転写因子であるSREBP−1cの発現及び活性を著しく増加させることにより、肝組織内の脂肪酸合成を促進し、血中トリグリセリドの量を増加させる。
【0005】
LXR αが誘発する、非アルコール性脂肪肝症の発症には、2つの異なる経路があり、それらはSREBP−1c依存的経路と、SREBP−1c非依存的経路である。SREBP−1c依存的脂肪肝は、LXR α媒介性SREBP−1cの転写活性化を介して、脂質生成遺伝子の発現が上向き調節されて現れる。SREBP−1c非依存的脂肪肝は、LXR αの活性化が、遊離脂肪酸のキャリアであるCD36タンパク質の発現を増加させることによって、より多くの脂肪酸が肝臓へ移動するために現れる。このように、LXR α活性化は、非アルコール性脂肪肝症の発症を促進する。しかしながら、今のところLXR αの活性を調節することによって、脂肪肝を抑制する薬物は知られていない。
【0006】
SREBPは、ステロールによって調節される遺伝子の転写調節部位であるステロール応答エレメント(SRE:sterol response element)に結合する蛋白質であり、SREBP蛋白質は、三種のイソ型(isoform)で存在し、それらは、SREBP−1a、SREBP−1c及びSREBP−2である。SREBP−1a及びSREBP−1cは、同一遺伝子から転写され、SREBP−2は、これとは異なる遺伝子から発現される。SREBP−1cは、脂肪酸合成に関連した遺伝子の転写を調節する転写因子であり、SREBP−2は、コレステロール合成に関連した遺伝子の転写を調節する転写因子である。SREBPは、静止状態では、小胞体膜蛋白質として存在し、サイズは、125キロダルトン(kDa)である。SREBPは、ステロール枯渇などの刺激によって活性化される前には、不活性化形態で膜に結合している。SREBPは、活性化されるときにゴルジ体に移動し、切断され、65キロダルトン・サイズの活性型蛋白質になる。SREBPは、活性化されるとき、核内に移動して標的遺伝子のSREに結合し、脂質合成遺伝子の発現を増加させる。SREBP−1cの標的遺伝子としては、脂肪酸合成を促進する酵素である脂肪酸合成酵素(FAS)、アセチルCoAカルボキシラーゼ(ACC)、ステアロイルCoA不飽和酵素(SCD)などがある。血中から肝臓に流入する遊離脂肪酸と、肝臓で直接生成する脂肪酸との量が、超低密度脂タンパク(VLDL)形態で排出されたり、又はベータ酸化される脂肪酸の量より多い場合、肝臓での脂質代謝均衡が崩れて脂肪肝症が発症する。そのため、肝臓での脂肪合成を促進する蛋白質であるFAS、ACC、SCDを誘導し、及び調節するSREBP−1cは、アルコール性又は非アルコール性の脂肪肝症(Liver steatosis)の重要要因である(非特許文献4及び非特許文献5)。脂肪肝は、脂肪が肝臓の全重量の5%以上を超える病的状態を意味するが、脂肪肝症を含む肝疾患は、40〜50代の成人の死亡原因のうち、癌の次に最もよくある原因である。先進国では、人口のうち約30%は、すでに脂肪肝症状を示しており、それらのうち20%は、肝線維化を経て肝硬変症に進む。このような肝硬変患者の50%は、肝硬変と診断された後、10年以内に肝疾患で死亡する。非アルコール性脂肪肝は、現在、西欧型高脂肪食摂取の増加と運動不足とによって発病率が上昇している。現在のところ、食習慣などの生活習慣を改善することが唯一の治療方法である。
【0007】
現在、脂肪肝を薬物学的に治療するのに有用な薬剤は、ほとんどない状態であるので、運動と食餌療法とが勧められているに過ぎないが、実際に、かような方法による脂肪肝の治療効率は非常に低く、有効な治療剤開発への要求が切実である状況である。薬物補助療法として、ベタイン、グルクロネート、メチオニン、コリン、脂肪親和性(lipotrophic)製剤が補助的に利用されもするが、それらの医学的又は薬学的効果が完全に証明されているわけではない。従って、副作用がなく、脂肪肝を効果的に治療する薬剤の開発が切望されている。
【0008】
一方、SREBP−1及びSREBP−2は、老人では、腎臓での発現が増加し、増加したSREBP−1及びSREBP−2によって、腎臓での脂質合成、及びトリグリセリドとコレステロールとの蓄積が増加するが、これは、糸球体硬化症、タンパク尿症及び腎症の原因となりえる(非特許文献6)。
【0009】
LXR αは、腎臓でのレニンの分泌において、重要な役割を担うと報告されている。LXR αとLXR βは、いずれもレニンを生成する傍糸球体細胞に豊富に発現される。Morelloらによれば、LXR αのアゴニストであるT0901017及びGW3965が、腎臓でのレニンのmRNAの発現を増加させ、血中レニン活性を増加させる(非特許文献7)。血中に過度のレニンが存在すると、高レニン血症を招き、これによって、高血圧及びアルドステロン症が現れる。
【0010】
LXRはまた、in vivoで超長鎖脂肪酸(VLCFA)が分解されずに脳に入って行き、神経細胞を破壊する稀な疾患である副腎白質ジストロフィー(ALD)と関連したABCD2遺伝子の発現を調節するので、LXRの阻害剤は、ALDの治療に有用であると報告されている(非特許文献8)。
【0011】
硫黄含有化合物であるジチオールチオンは、アブラナ科(Brassicaceae)植物に存在することが発見され、それらのいくつかの置換体は、肝臓の保護効果があるものもあると知られている。1,2−ジチオールチオンの代表的な化合物は、オルチプラズ(Oltipraz)(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン)であり、1980年代初めに、住血吸虫(schistosomiasis)治療剤として使われた薬剤であって、癌の化学的予防薬、及び肝硬変治療薬として研究されている。オルチプラズは、in vivo組織の、細胞内チオール含有量を増加させ、グルタチオン(GSH)プールの維持に関連した酵素の発現を誘発するほか、求電子性物質の解毒に関与する酵素の発現をも誘導する。オルチプラズによって、その活性が上昇する酵素としては、NAD(P)H:キノン還元酵素、ミクロソームエポキシドヒドロラーゼ、グルタチオン S−トランスフェラーゼ(GST)及びUDP−GTなどを挙げることができ、特に、GSTは、四塩化炭素やアセトアミノフェンなどの毒性物質による肝毒性を防御する酵素である。
【0012】
本発明の発明者らは、オルチプラズが、TGF βの発現を抑制することを発見し、肝線維化及び肝硬化の予防及び治療のための製薬組成物に関する特許を獲得した(特許文献1)。本発明の発明者らはまた、オルチプラズが、C/EBP β−LIPの発現を増加させ、またC/EBP α遺伝子及びPPAR γ遺伝子の発現を抑制することを発見し、肥満の予防又は治療用薬物に関する特許を獲得した(特許文献2)。また本発明の発明者らは、オルチプラズを始めとする1,2−ジチオールチオン化合物が、RSK1(p90 リボソームS6キナーゼ1)のキナーゼ活性を増進させることを発見し、糖尿病及びその合併症の予防及び治療のための、1,2−ジチオールチオン含有薬剤に関する特許も獲得している(特許文献3)。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】韓国特許第10−0404303号公報
【特許文献2】韓国特許第10−0576157号公報
【特許文献3】韓国特許第10−0590818号公報
【非特許文献】
【0014】
【非特許文献1】Hermanson et al., Trends Endocrinol. Metab., 2002, 13: 55-60
【非特許文献2】Schwartz et al., Biochem. Biophys. Res. Commun., 2000, 274: 794-802
【非特許文献3】Yoshikawa et al., Mol. Cell. Biol., 2001, 21: 2991-3000
【非特許文献4】Kohijima et al., Int.J.Mol.Med.,2008,21(4): 507-511
【非特許文献5】Donohue, World J.Gastroenterol.2007,13(37): 4974-4978
【非特許文献6】Jiang et al.,Kidney Int.,2005,68(6): 2608-2620
【非特許文献7】Morello et al.,J.Clin.Invest.,2005,115: 1913-1922
【非特許文献8】Weinhofer et al.,J.Biol.Chem.,2005,280: 41243-41251
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明では、LXR α又はSREBP−1の阻害剤を提供する。本発明ではさらに、LXR α又はSREBP−1の過剰発現に起因する疾病の予防及び治療のための薬学組成物を提供する。
【課題を解決するための手段】
【0016】
本発明者らは、前記課題を解決するために、さまざまな薬物の効果をスクリーニングした。その結果、本発明者らは、オルチプラズを始めとする1,2−ジチオールチオン誘導体を含有する薬物投与によって、LXR α及びLXR α依存性SREBP−1の発現及び活性化が抑制されることを発見した。また1,2−ジチオールチオン誘導体によって、SREBP−1が抑制されると、標的遺伝子である脂質生成遺伝子の発現が顕著に抑制され、さらに、高脂質食餌によって誘発される肝組織での中性脂質蓄積が抑制されることを発見した。これらの発見に基づいて、本発明では、LXR α又はSREBP−1の過剰発現に起因する疾病の予防及び治療のための、1,2−ジチオールチオン誘導体を含有する、薬学組成物を提供する。
【発明の効果】
【0017】
本発明による薬学組成物は、有効成分として1,2−ジチオールチオン誘導体を含有する。前記薬学組成物は、LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病の予防及び治療に効果的である。1,2−ジチオールチオン誘導体の例としては、オルチプラズ(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン 4−methyl−5−(2−pyrazinyl) −1,2−dithiol−3−thione)、3−メチル−1,2−ジチオール−3−チオン(3−methyl−1,2−dithiol−3−thion)、5−(6−メトキシピラジニル)−4−メチル−1,2−ジチオール−3−チオン(5−(6−methoxypyrazinyl) −4−methyl−1,2−dithiol−3−thione)が挙げられる。前記薬学組成物を投与すると、SREBP−1の発現及び活性が抑制される。前記SREBP−1は、LXR αの活性化を調節して、脂質生成酵素遺伝子の発現を調節する重要な転写因子である。さらに脂質生成遺伝子の発現が抑制されることにより、代謝異常による脂肪肝症による、肝組織内のトリグリセリドの蓄積が抑制される。したがって、1,2−ジチオールチオン誘導体を有効成分として含有する本発明の薬学組成物は、脂肪肝症を予防及び治療に有効である。また本発明による薬学組成物は、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症の予防及び治療に有効である。
【0018】
さらに、本発明は、本発明で提供する化合物又は組成物を使用して、LXR α又はSREBP−1の発現又は活性を阻害する方法を提供する。前記阻害は、LXR α又はSREBP−1の過剰発現又は過剰活性と関連した疾病又は状態の抑制又は治療となりえる。
【0019】
また、本発明は、LXR α又はSREBP−1の過剰発現又は過剰活性と関連した疾病又は状態、例えば、脂肪肝症、高中性脂肪血症、高レニン血症、レニンに起因した高血圧、アンドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎障害を予防又は治療する方法を提供する。
【図面の簡単な説明】
【0020】
【図1】オルチプラズ投与スケジュールを示す。
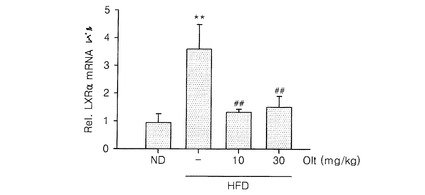
【図2】高脂質食餌で発現が増加したLXR αに対するオルチプラズ治療の結果である。結果は、正常食餌群であるLXR α mRNAに対する値で示した(ND:正常食餌、HFD:高脂質食餌,Olt:オルチプラズ,**:ND群と比較してp<0.01,##:HFD治療群のみと比較してp<0.01)。
【図3】LXR α過剰発現によって上昇したLXR αの活性に対するオルチプラズの治療効果を示す(S.S:スーパーシフト)。
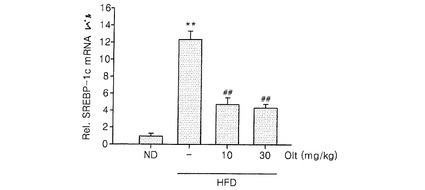
【図4】高脂質食餌で発現が増加したSREBP−1に対するオルチプラズの治療結果を示す(ND:正常食餌、HFD:高脂質食餌,Olt:オルチプラズ、**:ND群と比較してp<0.01,##:HFD治療群のみと比較してp<0.01)。
【図5】LXR α活性化剤であるT0901317で、肝細胞株であるH4IIE、HepG2及びラットの初代培養した肝細胞を治療した後での、SREBP−1タンパク質の発現に対するオルチプラズの治療結果を示す。SREBP−1タンパク質はウェスタンブロット法によって確認した(S.E:短時間フィルム露光、L.E:長時間フィルム露光、A:全細胞抽出溶解液、B:HepG2 cellの核分画。
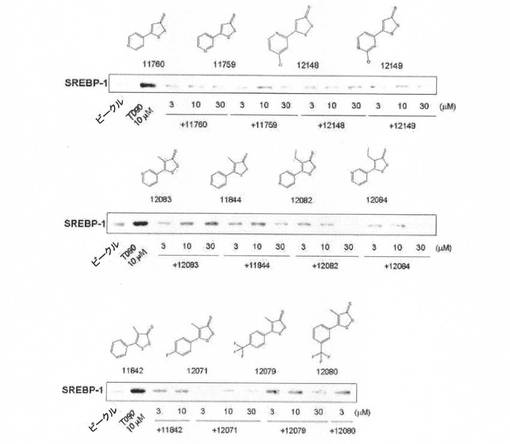
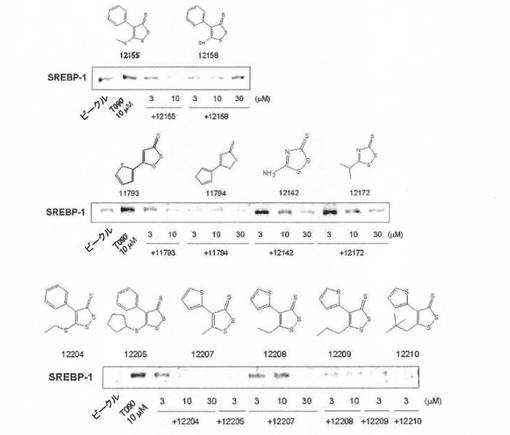
【図6】LXR α活性化剤処理で発現が増加したSREBP−1に対する1,2−ジチオールチオン誘導体のウェスタンブロットの結果である。
【図7】LXR α活性化剤処理で発現が増加したSREBP−1に対する1,2−ジチオールチオン誘導体のウェスタンブロットの結果である。
【図8】LXR α活性化剤処理で発現が増加したSREBP−1に対する1,2−ジチオールチオン誘導体のウェスタンブロットの結果である。
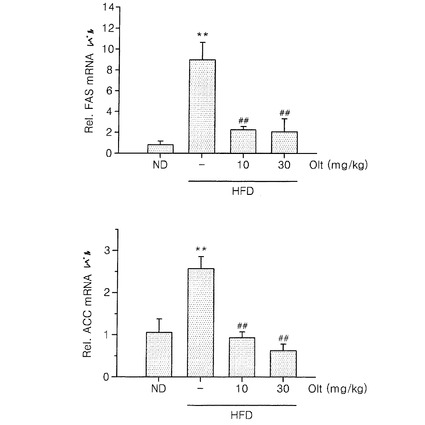
【図9】高脂質食餌によって誘導された脂肪肝動物モデルにオルチプラズを投与した場合の、高脂質食餌によって誘導された脂質生成遺伝子(FAS、ACC)の肝組織内発現量を比較した結果である。
【図10】高脂質食餌によって誘導された脂肪肝動物モデルにオルチプラズを投与したときの、前記高脂質食餌によって誘導された脂肪肝動物モデルの肝組織におけるトリグリセリド量に対するオルチプラズの治療結果を示す。
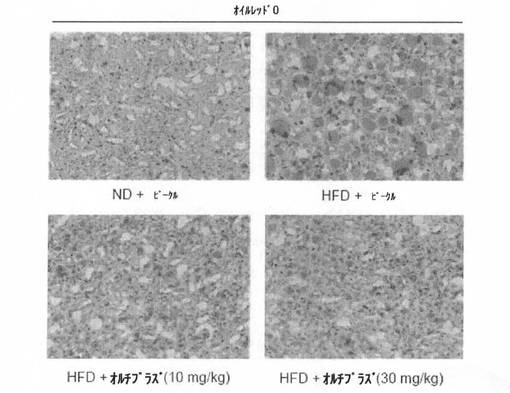
【図11】高脂質食餌によって誘導された脂肪肝動物モデルにオルチプラズを投与したときの、オイルレッドOで染色した肝組織の画像である。
【発明を実施するための形態】
【0021】
本発明は、オルチプラズなどの1,2−ジチオールチオン誘導体がLXR αの活性を抑制し、脂質生成遺伝子発現を調節する細胞タンパク質である、ステロール調節エレメント結合タンパク質−1(SREBP−1)の発現及び活性を抑制するということを発見したことに基づいたものである。本発明の発明者らは、高脂質食餌で発現が増加したマウスのLXR αの発現が、オルチプラズによって抑制され(図2)、LXR結合DNAエレメントに対するLXR αの結合能も、オルチプラズによって弱まるということを発見した(図3)。また本発明の発明者らは、LXR αの活性化剤として知られているT0901317を肝細胞株に付与したときに増加するSREBP−1の発現が、オルチプラズ又は他の1,2−ジチオールチオン誘導体によって抑制されることを発見した(図4〜図8)。
【0022】
オルチプラズなどの1,2−ジチオールチオン誘導体の投与は、LXR αの活性化を調節して、脂質生成酵素の遺伝子発現を調節する重要な転写因子であるSREBP−1の発現及び活性を抑制し、さらに脂質生成遺伝子の発現を抑制することにより、代謝異常による脂肪肝症による肝組織内のトリグリセリドの蓄積を抑制する。したがって、1,2−ジチオールチオン誘導体を有効成分として含有する本発明の薬学組成物は、脂肪肝症の予防及び治療に有効である。本発明の発明者らは、高脂質食餌の投与によって、肝組織内にトリグリセリドの含有量が増加したマウスに、オルチプラズを投与した場合、前記トリグリセリドの含有量が有意に減少し(図10及び図11)、また、前記高脂質食餌投与マウスで増加した脂肪酸合成酵素である脂肪酸合成酵素(FAS)及びアセチルCoAカルボキシラーゼ(ACC)の発現がオルチプラズの投与によって有意に抑制されることを発見した(図9)。
【0023】
本発明の発明者らによる発見に基づき、本発明は、LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病の予防用及び治療用の薬学組成物を提供する。前記薬学組成物は、下記式1で表される1,2−ジチオールチオン誘導体、その薬学的に許容される塩、それらの溶媒物又は水和物、又はそれらのプロドラッグを有効成分として含有し、及び薬学的に許容される担体を含む薬学組成物である。
【0024】
【化1】
【0025】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2はそれぞれ独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル、ヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され、前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択された少なくとも1つのヘテロ原子を含有する五員環又は六員環の化合物であり、前記フェニル及びヘテロアリールは、置換されていても非置換であってもよく、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、前記フェニル及びヘテロアリールが置換される場合には、一置換又は多置換であってよい。前記フェニル及びヘテロアリールはそれぞれ独立に、少なくとも1つのベンゼン又は前述のヘテロアリールと縮合してもよく、前記縮合したフェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。
【0026】
前記LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病としては、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症、脂肪肝、高トリグリセリド血症、高レニン血症などを挙げることができるが、これに限定されるものではない。よって、本発明は、レニンに起因した高血圧、アンドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症又は腎症、脂肪肝、高トリグリセリド血症又は高レニン血症の予防用及び治療用の薬学組成物であって、式1で表される1,2−ジチオールチオン誘導体を有効成分として含有し、薬剤学的に許容される担体を含む薬学組成物を提供する。
【0027】
本発明はまた、下記式1で表される1,2−ジチオールチオン誘導体、その薬学的に許容可能な塩、それらのプロドラッグ、それらの溶媒和物又は水和物を有効成分として含有し、及び薬剤学的に許容される担体を含む薬学組成物を投与することを含む、試験管内(in vitro)又は生体内(in vivo)で、LXR α又はSREBP−1の発現又は活性を阻害する方法を提供する。
【0028】
【化2】
【0029】
ここで、Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2はそれぞれ独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル、ヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択される。前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含有する五員環又は六員環化合物を意味する。前記フェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルから構成された群(基)から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。前記フェニル及びヘテロアリールはそれぞれ独立に、少なくとも1つのベンゼン又は前述のヘテロアリールと縮合してもよく、前記縮合したフェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。
【0030】
本発明はまた、下記式1で表される1,2−ジチオールチオン誘導体、その薬学的に許容可能な塩、それらのプロドラッグ、それらの溶媒和物又は水和物を有効成分として含有し、また、薬剤学的に許容される担体を含む薬学組成物を投与することを含む、LXR α又はSREBP−1の過剰発現又は過剰活性と関連した疾病又は状態を予防又は治療する方法を提供する。
【0031】
【化3】
【0032】
ここで、Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2はそれぞれ独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル、ヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択される。前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含有する五員環又は六員環化合物を意味する。前記フェニル及びヘテロアリールは、置換されていても、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。前記フェニル及びヘテロアリールはそれぞれ独立に、少なくとも1つのベンゼン又は前述のヘテロアリールと縮合していてもよい。前記縮合されたフェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。
【0033】
LXR α又はSREBP−1の過発現又は過活性と関連した疾病又は状態を予防又は治療する方法の一例として、前記疾病又は状態は、例えば、脂肪肝、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症である。実施形態では、本明細書に開示される化合物は、これらの疾病又は状態の1つ又はそれ以上を改善し、及び/又は発現を遅らせるために使用される。
本発明の薬学組成物に含まれる1,2−ジチオールチオン誘導体には、1,2−ジチオール−3−チオン及び、二環式分子を含む有機化合物と、それらの誘導体有機化合物とが含まれる。二環式分子は、ピラジン、ピリダジン、ピリミジン、チアゾール又はチオフェンであってよい(表1)。
【0034】
【表1】
【0035】
本発明は、前記化合物が形成されうる薬学的に許容可能な塩、又はそれらの溶媒和物、水和物又はプロドラッグを含んでよい。薬剤学的に許容される付加塩は、薬剤学的に許容される酸付加塩、及び薬剤学的に許容される塩基付加塩を含む。本明細書に記載の「薬剤学的に許容される酸付加塩」には、式1で表される化合物を用いて形成されうる非毒性酸付加塩が含まれ、それは治療的に活性である。式1の化合物は元来、塩基性特性を有しており、適当な酸で処理することにより、薬剤学的に許容される酸付加塩に転換されうる。適当な酸は、無機酸又は有機酸でありうる。無機酸としては、例えば、塩酸又はブロム酸などのハロゲン化水素酸、硫酸、硝酸、リン酸などを挙げることができる。有機酸としては、例えば、酢酸、トリフルオロ酢酸、プロパン酸、ヒドロキシ酢酸、乳酸、ピルビン酸、シュウ酸、マロン酸、コハク酸(すなわち、ブタン二酸)、マレイン酸、フマル酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、シクラミン酸、サリチル酸、p−アミノ−サリチル酸、パモ酸などを挙げることができる。酸性特性を有する前記転換された化合物は、適当な有機又は無機の塩基で処理することによって、その薬剤学的に許容される塩基付加塩に転換されうる。適当な塩基付加塩としては、例えば、アンモニウム塩;アルカリ及びアルカリ土類金属塩、例えば、リチウム塩、ナトリウム塩、カリウム塩、マグネシウム塩、カルシウム塩;ベンザチン塩、N−メチル−D−グルカミン塩、ヒドラバミン塩(hydrabamie salt)などの有機塩基との塩;アルギニン、リシンなどのアミノ酸との塩を含む。
【0036】
本発明の組成物は、薬剤学的分野で一般的な方法によって、経口投与に適した単位投与型の製剤及び注射剤に剤形化することができ、これを投与できる。単位投与型の製剤には、硬質及び軟質のカプセル剤、錠剤、散剤、懸濁剤、シロップ剤が含まれる。経口投与に適した単位投与型の製剤には、 少なくとも1つの薬物学的活性成分を含み、またそれ以外に、少なくとも1つの薬剤学的に不活性である一般的な担体が含まれていてよい。少なくとも1つの薬剤学的に不活性である一般的な担体としては、例えば、賦形剤、結合剤、崩壊剤、潤滑剤が含まれる。賦形剤としては、例えば、粉末、ラクトース、カルボキシメチルセルロース、カオリンが挙げられる。結合剤としては、例えば、水、ゼラチン、アルコール、グルコース、アラビアゴム(Arabia rubber)、トラガカントゴムが挙げられる。崩壊剤としては、例えば、粉末、デキストリン、アルギン酸ナトリウムが挙げられる。潤滑剤としては、例えば、タルク、ステアリン酸(staric acid)、ステアリン酸マグネシウム、流動パラフィンが挙げられる。本発明ではまた、溶解のための溶解補助剤をさらに含んでもよい。
【0037】
本発明の組成物の1日投与容量は、投与しようとする対象の疾病の進行程度、発病時期、年齢、健康状態、合併症などによって異なる。例えば、成人に対して、1日あたり、本発明の組成物を1〜500mg、特に、30〜200mg投与できる。投与は1日1回又は数回分割して投与できる。
【0038】
以下、本発明について、実験例によって詳細に説明する。下記実施例は、本発明について具体的に例示するだけであり、本発明の内容が、下記実施例に限定されるものではない。
【0039】
(参考例1:実験動物及び食餌)
実験動物として使われた雄C57BL/6マウス(平均体重25〜30g)は、Charles River Orient(韓国・ソウル)から購入した。実験に使用する前、少なくとも1週間、55±5%の湿度、22±2℃の温度及び換気が調節されたソウル大学校薬科大学動物実験研究棟で動物を適応させた。午前7時と午後7時を基準として、12時間周期で明暗を変えた。実験期間中、マウスが摂取した食餌量及び飲料水量には、有意的な変化が観察されなかった。動物の重さと状態とを毎週1回検査した。2つのグループのマウスは、高脂質食餌(Dyets Inc.,Bethlehem)又は正常食餌でそれぞれ10週間飼育した。それぞれのグループについて、最後の4週間の間、オルチプラズ(10又は30mg/kg、3回/週)をマウスに投与した(図1)。各グループは、全部で10匹のマウスから構成された。
【0040】
(参考例2:試料準備)
オルチプラズ及び1,2−ジチオールチオン誘導体は、株式会社CJから提供を受けた。本発明に使われた1,2−ジチオールチオン誘導体は、韓国特許第10−0604261号公報に記載された方法によって製造されうる。脂肪肝誘発のための高脂質食餌は、米国・ダイエット社(Dyet Co.)から購入した。オルチプラズを40%PEG200で希釈して、所望の濃度に調製した。
【0041】
(参考例3:リアルタイム−RT PCR)
マウスの肝臓から抽出した全RNA(2μg)、d(T)16プライマー、及びAMV逆転写酵素を使用し、cDNAを得た。遺伝子の相対的な量は、サイバーグリーン染料(CyBr green dye)を使用したリアルタイムRT−PCR法によって定量した。リアルタイム RT−PCRは、ロッシュ社(マンハイム、ドイツ)のLight-cycler2.0を利用した。メーカーの方法によって、PCRを行い、Light-cycler software4.0プログラムを使用し、各遺伝子の相対的な量を分析した。
【0042】
(参考例4:ウェスタンブロット)
Laemmli UK法(1970)によって、Mighty Small II SE250装置を使用し、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS−PAGE)を行った。肝試料の溶解分画を、サンプル希釈緩衝液[63mM Tris(pH.6.8)、10%グリセロール、2%SDS、0.0013%ブロモフェノールブルー、5%β−メルカプトエタノール]に希釈し、7.5%及び9%のゲルを使用し、電極緩衝液(1L溶液中にTris 15g、グリシン72g、SDS 5g含む)内で電気泳動した。電気泳動が終わったゲルは、電気泳動装置を利用し、緩衝液[25mM Tris、192mMグリシン、20%v/v メタノール(pH.8.3)]内で、190mAmpsで1時間、ニトロセルロース紙にタンパク質を転移させた。Anti−SREBP−1を一次抗体として反応させた後、二次抗体として西洋わさびペルオキシダーゼ−ヤギ抗ウサギIgG(horseradish peroxidase-conjugated goat anti-rabbit IgG)を1時間反応させた。その後、ECL 化学発光分析システム(Amersham、Gaithesberg、MA)を使用して免疫反応性蛋白質を可視化させた。各試料中の蛋白質量の同質性は、anti−β−actin抗体(Sigma、セントルイス、MO)を使用して確認した。
【0043】
(参考例5:分析方法)
下記実施例で提示した資料は、薬物学的計算プログラムを利用して分析したものであり、多様な実験群間の有意性を一方向平方偏差分析法(Fisher,R.A.,Statistical Methods for Research Workers,Edinburgh: Oliver & Boyd,1925)で検定した後、ニューマン・クールズ検査(Norman GR et al.,Biostatistics: The Bare Essentials,2000)で判定した(*p<0.05、**p<0.01)。
【0044】
(実験例1:発現が増加したLXR αに対するオルチプラズ処置の効果)
マウスを、高脂質食餌及び正常食餌で10週間飼育した。高脂質食餌で飼育したマウス群に、最後の4週間の間、オルチプラズ(10−30mg/kg、3回/週)、すなわち、1,2−ジチオールチオン化合物、を投与したマウスの肝組織のLXR αの発現を測定した。肝組織からmRNAを単離し、RT−PCRを介してcDNAを合成した後、特定プライマー(mouse LXR、5’−TGCCATCAGCATCTTCTCTG−3’(sense)及び5’−GGCTCACCAGCTTCATTAGC−3’(antisense))を使用し、リアルタイムPCRを行った。正常食餌群(ND)のLXR α mRNAの発現レベルを1としたときの、高脂質食餌群、又は、高脂質食餌かつオルチプラズ投与群の相対的な発現レベルを測定し、その結果を図2に示した。これにより、細胞内脂質センサであるLXR αの発現が、高脂質食餌によって有意に増加し(p<0.01)、LXR α発現の増加は、オルチプラズ投与によって抑制される(p<0.01)ということが明らかになった。
【0045】
(実験例2:オルチプラズ処置によるLXR αの活性抑制効果)
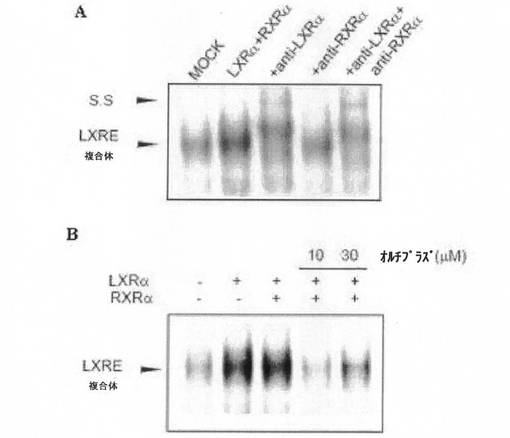
LXR αは、RXRαとダイマーを形成し、標的遺伝子プロモーターに存在する特定領域(LXRE)に結合することにより、遺伝子の発現を調節する。オルチプラズ処置によって、LXRE結合能が変化するか否かを、ゲルシフトアッセイで確認した。SREBP−1c遺伝子のLXRE二本鎖オリゴヌクレオチドを、[γ−32P]ATPとT4 ポリヌクレオチドキナーゼとを使用し、5’−末端を放射性同位元素で標識した。その後、標識したプローブ(1ml、>106cpm)と核分画タンパク質とを結合緩衝液中で反応させた。反応液を4%ポリアクリルアミド・ゲルで電気泳動した後、オートラジオグラフィーで分析した。実験に使われたLXREオリゴヌクレオチド配列は、5’−CAGTGACCGCCAGTAACCCCAGC−3’である。DNA結合特異性は、コールドプローブ滴定(cold probe titration)及びスーパーシフトアッセイにより確認した。コールドプローブ滴定のために、20倍多い(モル基準)非標識オリゴヌクレオチドをあらかじめ反応させた。スーパーシフトのために、LXR α抗体又はRXRα抗体(2μg)を反応混合物と、室温で約30分間反応させ、そこに、放射線同位元素で標識されたプローブを加え、さらに30分間反応させた後、電気泳動した。
【0046】
LXR α及びRXRαが過剰発現した場合、対照群(mock)に比べて移動の遅い泳動バンドの密度が高まった(図3Aの最初のバンド及び2番目のバンドを参照のこと)。LXR α及びRXRαの抗体を使用し、スーパーシフトを行ったとき、LXR α抗体及びRXRα抗体によって、DNAタンパク結合が減少し、スーパーシフトされたバンドが観察された(図3A、3番目のバンド〜5番目のバンド)。これは、LXR α/RXRα複合体の、DNA結合特異性を支持する結果である。
【0047】
オルチプラズが投与されると、移動が遅延するバンド(図3の最初のバンド〜3番目のバンド)の密度が低くなった(図3Bの4番目のバンド及び5番目のバンド)。これは、LXR αの、DNA結合能がオルチプラズ処置によって実質的に弱まるということを示している。
【0048】
(実験例3:発現が増加したSREBP−1に対するオルチプラズ処置の効果)
実験例1で使用したマウスの肝組織におけるSREBP−1の発現レベルを測定した。肝組織からmRNAを単離し、RT−PCRによってcDNAを合成した後、これに特定プライマー(mouse SREBP−1、5’−AACGTCACTTCCAGCTAGAC−3’(sense)及び5’−CCACTAAGGTGCCTACAGAGC−3’(antisense))を用いて、リアルタイムPCRを行った。正常食餌群(ND)のSREBP−1mRNAの発現レベルを1としたとき、高脂質食餌群、又は、高脂質食餌かつオルチプラズ投与群の相対的な発現レベルを測定した。その結果を図4に示した。SREBP−1の発現が高脂質食餌によって有意に増加し(p<0.01)、SREBP−1発現の増加は、オルチプラズ投与によって抑制される(p<0.01)ということが明らかになった。
【0049】
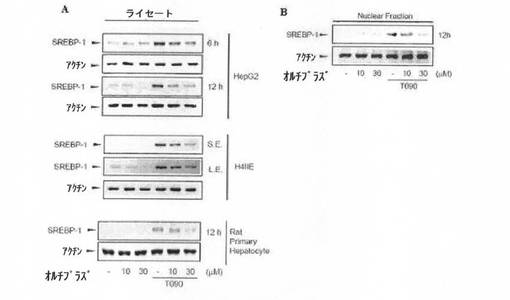
(実験例4:オルチプラズ処置によるSREBP−1の発現及び活性の抑制効果)
肝細胞株であるH4IIE、HepG2、及びラット初代培養肝細胞を、LXR α活性化剤であるT0901317で処置し、ウェスタンブロット法によって、SREBP−1タンパク質を確認した。SREBP−1の発現は、T0901317で処置した後、12時間以内に顕著に増加した(図5Aの各ゲル写真の4番目のカラム)。SREBP−1タンパク質の発現の増加は、オルチプラズ処置を行うと、濃度依存的に減少した(図5Aの各ゲル写真の5番目及び6番目のカラム)。これは、SREBP−1の発現がオルチプラズによって抑制されることを示す。
【0050】
また、T0901317を投与したHepG2細胞株におけるSREBP−1の核内移動の増加が、オルチプラズ処置によって濃度依存的に減少した(図5B)。これは、SREBP−1の活性が、オルチプラズによって抑制されることを示す。
【0051】
細胞分画は以下の方法で単離した。低浸透圧緩衝溶液[10mM HEPES(pH7.9)、10mM KCl、0.1mM EDTA、0.5%ノニデットP−40、1mM DTT及び0.5mMフッ化フェニルメチルスルホニル(PMSF)]を肝細胞株に加え、氷中に10分放置した。その後、溶液を7,200gで5分間遠心分離し、上澄み液を細胞質分画として使用した。別途、高浸透圧緩衝溶液[20mM HEPES(pH7.9)、400mM NaCl、1mM EDTA、10mM DTT及び1mM PMSF]を肝細胞株に加え、氷中に1時間放置した。その後、得られた溶液を15,000gで10分間遠心分離して上澄み液を取り、核分画として使用した。別途、溶液緩衝液[10mM HEPES(pH7.9)、100mM NaCl、1mM EDTA、10%グリセロール、0.5%Triton X−100、0.5%ノニデット P−40、1mM DTT及び0.5mM PMSF]をPBSで洗浄した細胞に加え、氷中で1時間溶解させた。その後、得られた溶液を10,000gで10分間遠心分離した後、上澄み液を全細胞抽出液として使用した。これらの細胞分画は、使用時まで−70゜Cに保管した。タンパク質濃度は、ブラッドフォードアッセイ(Bradford assay;Bio−Rad protein assay kit、Hercules、CA、USA)で定量した。
【0052】
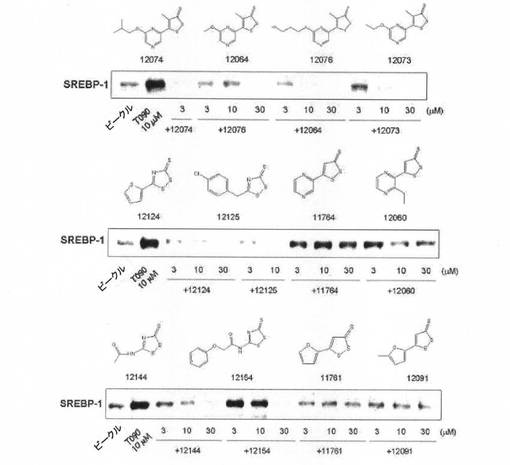
(実験例5:1,2−ジチオールチオン誘導体ら処置によるSREBP−1の発現抑制効果)
LXR α活性化剤(T0901317)によって増加したSREBP−1の発現に対する1,2−ジチオールチオン誘導体の効果を、H4IIE細胞株を利用して評価した。H4IIE細胞株をT0901317で処置することで増加したSREBP−1の発現が、1,2−ジチオールチオン誘導体処置によって抑制された(図6〜図8)。
【0053】
(実験例6:脂肪酸合成酵素FAS及びACCの発現に対する、オルチプラズ処置による抑制効果)
実験例1で使用したマウスの肝組織におけるSREBP−1の標的遺伝子であるFAS及びACCの発現レベルを測定した。肝組織からmRNAを単離し、RT−PCRによりcDNAを合成した後、これを特定プライマー(mouse ACC1、5’−GTCAGCGGATGGGCGGAATG−3’(sense)及び5’−CGCCGGATGCCATGCTCAAC−3’(antisense);mouse FAS、5’−AGCGGCCATTTCCATTGCCC−3’(sense)及び5’−CCATGCCCAGAGGGTGGTTG−3’(antisense))を使用して、リアルタイムPCRを行った。正常食餌群(ND)のFAS及びACCのmRNAの発現レベルを1としたとき、高脂質食餌群、又は、高脂質食餌かつオルチプラズ投与群の相対的なFASmRNA又はACCmRNAの発現レベルを測定した。結果を図9に示した。FAS及びACCの発現が、高脂質食餌によって有意に増加し(p<0.01)、FAS及びACC発現の増加は、オルチプラズ投与によって抑制される(p<0.01)ということが明らかになった。
【0054】
(実験例7:オルチプラズ投与による高脂質食餌に蓄積になった肝組織内のトリグリセリドの含有量抑制効果)
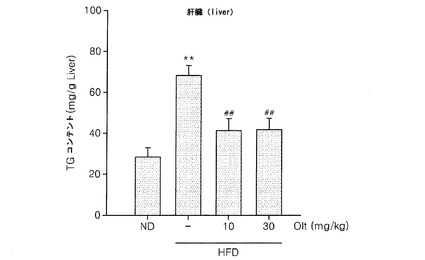
実験例1で使用したマウスの肝組織内の実験例1におけるトリグリセリド含有量に対するオルチプラズの効果を評価した。肝組織内のトリグリセリド含有量は、脂肪肝を示す指標である。オルチプラズを投与した後(Bae et al.,Hepatology,2007,46: 730−739)、肝組織内のトリグリセリド含有量を測定した。高脂質食餌を10週実施したマウスでは、肝組織内のトリグリセリド含有量が、正常食餌群に比べて顕著に増加した(p<0.01)。一方、オルチプラズが投与された場合には、組織内トリグリセリドの含有量が有意に減少した(p<0.01)(図10)。
【0055】
(実験例8:高脂質食餌によって誘導された脂肪肝動物モデルの肝組織におけるオルチプラズ治療効果)
実験例7で使用した、高脂質食餌によって誘導された脂肪肝に対するオルチプラズの治療効果を、脂肪特異染色剤であるオイルレッドO染色法を利用して確認した。脂肪肝の肝組織を、10%中性ホルマリン溶液で固定し、一般的な固定工程及び脱水工程を行った後、工程後の肝組織をパラフィンに包埋した。包埋した組織に対して、4μm厚に組織切片を行い、オイルレッドOで染色した後、光学顕微鏡で観察した。高脂質食餌群では、オイルレッドOで赤く染色された部分が顕著に現れ(HFD+ビークル(vehicle))、オルチプラズが投与された群(HFD+オルチプラズ)では、赤く染色された部分が顕著に減った(図11)。これにより、オルチプラズ治療効果が高いことがわかる。
【0056】
1,2−ジチオールチオン誘導体を有効成分として含有する多様な形態の製剤を調製した。
【0057】
(製造例1)
オルチプラズ25mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0058】
前記の成分を混合し、一般的な錠剤の製造方法によって打錠し、錠剤を製造した。
【0059】
(製造例2)
3−メチル−1,2−ジチオール−3−チオン50mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0060】
前記の成分を混合し、一般的な錠剤の製造方法によって打錠し、錠剤を製造した。
【0061】
(製造例3)
5−(6−メトキシピラジニル)−4−メチル−1,2−ジチオール−3−チオン 100mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0062】
前記の成分を混合し、一般的な散剤の製造方法によって、散剤を製造した。
【0063】
(製造例4)
オルチプラズ250mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0064】
前記の成分を混合し、一般的な散剤の製造方法によって、散剤を製造した。
【0065】
(製造例5)
オルチプラズ25mg
乳糖30mg
澱粉28mg
タルク2mg
ステアリン酸マグネシウム適量
【0066】
前記の成分を混合し、一般的な方法によるカプセル剤の製造方法によってゼラチン軟カプセルに充填し、カプセル剤を製造した。
【0067】
(製造例6)
5−(6−メトキシピラジニル)−4−メチル−1,2−ジチオール−3−チオン50mg
乳糖30mg
澱粉28mg
タルク2mg
ステアリン酸マグネシウム適量
【0068】
前記の成分を混合し、一般的なカプセル剤の製造方法によってゼラチン軟カプセルに充填し、カプセル剤を製造した。
【0069】
(製造例7)
オルチプラズ100mg
異性化糖10g
砂糖30mg
ナトリウムCMC 100mg
レモン香料 適量
精製水 バランス
(精製水適量を加えて全体100mlとする)
【0070】
前記の成分を通常の懸濁剤の製造方法によって懸濁剤を製造し、100ml容量の褐色瓶に充填して滅菌し、懸濁剤を製造した。
【0071】
(製造例8)
3−メチル−1,2−ジチオール−3−チオン250mg
乳糖30mg
澱粉20mg
ステアリン酸マグネシウム適量
【0072】
前記の成分を均一に混合し、ポリエチレンがコーティングされた袋に充填して密閉し、散剤を製造した。
【0073】
(製造例9)
軟質カプセル剤1錠中に以下を含む:
オルチプラズ100mg
ポリエチレングリコール400 400mg
濃グリセリン55mg
精製水35mg
【0074】
ポリエチレングリコールと濃グリセリンとを混合し、ここに精製水を加え、この混合物を約60℃で維持した状態で、1,2−ジチオールチオン誘導体を加えた。約1,500rpmで撹拌して、均一に混合した。その後ゆっくりと撹拌しつつ温度を室温に冷却し、真空ポンプを使用して気泡を除去し、軟質カプセルの内容物とした。軟質カプセルの被膜は、一般的に公知のゼラチン、可塑剤でソフト処方とした。1カプセル当たりゼラチン132mg、濃グリセリン52mg、70%ジソルビトール液6mg、着香剤としてエチルバニリン適量及びコーティング剤としてカルナウバワックスを使用し、通常の調剤法で製造した。
【産業上の利用可能性】
【0075】
本発明による薬学組成物は、LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病の予防又は治療に有効である。前記薬学組成物を投与すると、SREBP−1の発現及び活性が阻害される。SREBP−1は、LXR α活性化調節を介して、脂質生成酵素遺伝子の発現を調節する重要な転写因子であり、さらに脂質生成遺伝子の発現を抑制することにより、代謝障害による脂肪肝症による肝組織内のトリグリセリドの蓄積を抑制する。従って、1,2−ジチオールチオン誘導体を有効成分として含有する本発明の薬学組成物は、脂肪肝症の予防及び治療に有効である。また、本発明による薬学組成物は、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症の予防及び治療に有効である。
【技術分野】
【0001】
本発明は、肝臓Xレセプターα(LXR α)の発現又は活性の抑制、及び、ステロール調節エレメント結合タンパク質−1(SREBP−1:sterol response element binding protein)の発現又は活性を抑制する1,2−ジチオールチオン(1,2−dithiolthione)誘導体に関する。本発明はまた、1,2−ジチオールチオン誘導体を含む薬学組成物に関する。該薬学組成物は、LXR α又はSREBP−1の過剰発現に起因する疾病の予防又は治療に有効である。このような疾病としては、例えば、脂肪肝、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症(aldosteronism)、副腎白質ジストロフィー(adrenoleukodystrophy)、糸球体硬化症、蛋白尿症、腎症(nephropathy)を含む。
【0002】
本発明は、教育科学技術部優秀研究センター(ERC)事業の一環として行われた研究から導き出されたものである[課題固有番号R11−2007−107−01001−0、課題名:代謝及び炎症疾患新薬開発研究センター]。
【背景技術】
【0003】
肝臓Xレセプター(LXR)、ペルオキシソーム増殖因子活性化レセプター(PPAR:peroxisome proliferator-activated receptor)、及びファルネソイドXレセプター(FXR:farnesoid X receptor)は、第2型受容体スーパーファミリー(typeII receptor superfamily)に属する核ホルモン受容体(nuclear hormone receptor)である。これらの受容体は、レチノイドXレセプター(RXR:retinoid X receptor)とヘテロダイマーを形成して、DNAに結合する。リガンドが結合しない場合には、前記ヘテロダイマーがDNAに結合し、コリプレッサー蛋白質と複合体を形成し、一方、リガンドが結合する場合には、構造的変化が生じ、コリプレッサー蛋白質が分離し、補助活性蛋白質(coactivator protein)が結合し、標的遺伝子の転写が促進される(非特許文献1)。核ホルモン受容体のうち、LXRは、コレステロール代謝及び恒常性と関連がある遺伝子の転写調節に重要な役割を果たしている。このような遺伝子の例として、アポリポプロテインE(apoE)、ABCA1、ABCG1、ABCG5、ABCG8、コレステロール7α−ヒドロキシラーゼ(cholesterol 7α-hydroxylase)及びスカベンジャー受容体classB タイプI(scavenger receptor classB typeI)が含まれる(非特許文献2)。またLXRは、SREBP−1c遺伝子に直接に作用し、脂質代謝を調節する(非特許文献3)。
【0004】
LXRには、LXR αとLXR βの2つの異性体が存在する。LXR αは、主に肝臓に存在し、LXR βは、ほとんどの臓器に存在する。LXR αは、天然リガンドであるオキシステロール類、高濃度のグルコース及び人工リガンドであるT0901017、GW3965によって活性化され、脂質生成に関連する遺伝子の発現、及びコレステロール恒常性を調節する。肝臓で脂質が合成されるとき、LXR αは、脂質センサとして作用し、脂質合成遺伝子の発現を調節する重要な転写因子であるSREBP−1cの発現及び活性を著しく増加させることにより、肝組織内の脂肪酸合成を促進し、血中トリグリセリドの量を増加させる。
【0005】
LXR αが誘発する、非アルコール性脂肪肝症の発症には、2つの異なる経路があり、それらはSREBP−1c依存的経路と、SREBP−1c非依存的経路である。SREBP−1c依存的脂肪肝は、LXR α媒介性SREBP−1cの転写活性化を介して、脂質生成遺伝子の発現が上向き調節されて現れる。SREBP−1c非依存的脂肪肝は、LXR αの活性化が、遊離脂肪酸のキャリアであるCD36タンパク質の発現を増加させることによって、より多くの脂肪酸が肝臓へ移動するために現れる。このように、LXR α活性化は、非アルコール性脂肪肝症の発症を促進する。しかしながら、今のところLXR αの活性を調節することによって、脂肪肝を抑制する薬物は知られていない。
【0006】
SREBPは、ステロールによって調節される遺伝子の転写調節部位であるステロール応答エレメント(SRE:sterol response element)に結合する蛋白質であり、SREBP蛋白質は、三種のイソ型(isoform)で存在し、それらは、SREBP−1a、SREBP−1c及びSREBP−2である。SREBP−1a及びSREBP−1cは、同一遺伝子から転写され、SREBP−2は、これとは異なる遺伝子から発現される。SREBP−1cは、脂肪酸合成に関連した遺伝子の転写を調節する転写因子であり、SREBP−2は、コレステロール合成に関連した遺伝子の転写を調節する転写因子である。SREBPは、静止状態では、小胞体膜蛋白質として存在し、サイズは、125キロダルトン(kDa)である。SREBPは、ステロール枯渇などの刺激によって活性化される前には、不活性化形態で膜に結合している。SREBPは、活性化されるときにゴルジ体に移動し、切断され、65キロダルトン・サイズの活性型蛋白質になる。SREBPは、活性化されるとき、核内に移動して標的遺伝子のSREに結合し、脂質合成遺伝子の発現を増加させる。SREBP−1cの標的遺伝子としては、脂肪酸合成を促進する酵素である脂肪酸合成酵素(FAS)、アセチルCoAカルボキシラーゼ(ACC)、ステアロイルCoA不飽和酵素(SCD)などがある。血中から肝臓に流入する遊離脂肪酸と、肝臓で直接生成する脂肪酸との量が、超低密度脂タンパク(VLDL)形態で排出されたり、又はベータ酸化される脂肪酸の量より多い場合、肝臓での脂質代謝均衡が崩れて脂肪肝症が発症する。そのため、肝臓での脂肪合成を促進する蛋白質であるFAS、ACC、SCDを誘導し、及び調節するSREBP−1cは、アルコール性又は非アルコール性の脂肪肝症(Liver steatosis)の重要要因である(非特許文献4及び非特許文献5)。脂肪肝は、脂肪が肝臓の全重量の5%以上を超える病的状態を意味するが、脂肪肝症を含む肝疾患は、40〜50代の成人の死亡原因のうち、癌の次に最もよくある原因である。先進国では、人口のうち約30%は、すでに脂肪肝症状を示しており、それらのうち20%は、肝線維化を経て肝硬変症に進む。このような肝硬変患者の50%は、肝硬変と診断された後、10年以内に肝疾患で死亡する。非アルコール性脂肪肝は、現在、西欧型高脂肪食摂取の増加と運動不足とによって発病率が上昇している。現在のところ、食習慣などの生活習慣を改善することが唯一の治療方法である。
【0007】
現在、脂肪肝を薬物学的に治療するのに有用な薬剤は、ほとんどない状態であるので、運動と食餌療法とが勧められているに過ぎないが、実際に、かような方法による脂肪肝の治療効率は非常に低く、有効な治療剤開発への要求が切実である状況である。薬物補助療法として、ベタイン、グルクロネート、メチオニン、コリン、脂肪親和性(lipotrophic)製剤が補助的に利用されもするが、それらの医学的又は薬学的効果が完全に証明されているわけではない。従って、副作用がなく、脂肪肝を効果的に治療する薬剤の開発が切望されている。
【0008】
一方、SREBP−1及びSREBP−2は、老人では、腎臓での発現が増加し、増加したSREBP−1及びSREBP−2によって、腎臓での脂質合成、及びトリグリセリドとコレステロールとの蓄積が増加するが、これは、糸球体硬化症、タンパク尿症及び腎症の原因となりえる(非特許文献6)。
【0009】
LXR αは、腎臓でのレニンの分泌において、重要な役割を担うと報告されている。LXR αとLXR βは、いずれもレニンを生成する傍糸球体細胞に豊富に発現される。Morelloらによれば、LXR αのアゴニストであるT0901017及びGW3965が、腎臓でのレニンのmRNAの発現を増加させ、血中レニン活性を増加させる(非特許文献7)。血中に過度のレニンが存在すると、高レニン血症を招き、これによって、高血圧及びアルドステロン症が現れる。
【0010】
LXRはまた、in vivoで超長鎖脂肪酸(VLCFA)が分解されずに脳に入って行き、神経細胞を破壊する稀な疾患である副腎白質ジストロフィー(ALD)と関連したABCD2遺伝子の発現を調節するので、LXRの阻害剤は、ALDの治療に有用であると報告されている(非特許文献8)。
【0011】
硫黄含有化合物であるジチオールチオンは、アブラナ科(Brassicaceae)植物に存在することが発見され、それらのいくつかの置換体は、肝臓の保護効果があるものもあると知られている。1,2−ジチオールチオンの代表的な化合物は、オルチプラズ(Oltipraz)(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン)であり、1980年代初めに、住血吸虫(schistosomiasis)治療剤として使われた薬剤であって、癌の化学的予防薬、及び肝硬変治療薬として研究されている。オルチプラズは、in vivo組織の、細胞内チオール含有量を増加させ、グルタチオン(GSH)プールの維持に関連した酵素の発現を誘発するほか、求電子性物質の解毒に関与する酵素の発現をも誘導する。オルチプラズによって、その活性が上昇する酵素としては、NAD(P)H:キノン還元酵素、ミクロソームエポキシドヒドロラーゼ、グルタチオン S−トランスフェラーゼ(GST)及びUDP−GTなどを挙げることができ、特に、GSTは、四塩化炭素やアセトアミノフェンなどの毒性物質による肝毒性を防御する酵素である。
【0012】
本発明の発明者らは、オルチプラズが、TGF βの発現を抑制することを発見し、肝線維化及び肝硬化の予防及び治療のための製薬組成物に関する特許を獲得した(特許文献1)。本発明の発明者らはまた、オルチプラズが、C/EBP β−LIPの発現を増加させ、またC/EBP α遺伝子及びPPAR γ遺伝子の発現を抑制することを発見し、肥満の予防又は治療用薬物に関する特許を獲得した(特許文献2)。また本発明の発明者らは、オルチプラズを始めとする1,2−ジチオールチオン化合物が、RSK1(p90 リボソームS6キナーゼ1)のキナーゼ活性を増進させることを発見し、糖尿病及びその合併症の予防及び治療のための、1,2−ジチオールチオン含有薬剤に関する特許も獲得している(特許文献3)。
【先行技術文献】
【特許文献】
【0013】
【特許文献1】韓国特許第10−0404303号公報
【特許文献2】韓国特許第10−0576157号公報
【特許文献3】韓国特許第10−0590818号公報
【非特許文献】
【0014】
【非特許文献1】Hermanson et al., Trends Endocrinol. Metab., 2002, 13: 55-60
【非特許文献2】Schwartz et al., Biochem. Biophys. Res. Commun., 2000, 274: 794-802
【非特許文献3】Yoshikawa et al., Mol. Cell. Biol., 2001, 21: 2991-3000
【非特許文献4】Kohijima et al., Int.J.Mol.Med.,2008,21(4): 507-511
【非特許文献5】Donohue, World J.Gastroenterol.2007,13(37): 4974-4978
【非特許文献6】Jiang et al.,Kidney Int.,2005,68(6): 2608-2620
【非特許文献7】Morello et al.,J.Clin.Invest.,2005,115: 1913-1922
【非特許文献8】Weinhofer et al.,J.Biol.Chem.,2005,280: 41243-41251
【発明の概要】
【発明が解決しようとする課題】
【0015】
本発明では、LXR α又はSREBP−1の阻害剤を提供する。本発明ではさらに、LXR α又はSREBP−1の過剰発現に起因する疾病の予防及び治療のための薬学組成物を提供する。
【課題を解決するための手段】
【0016】
本発明者らは、前記課題を解決するために、さまざまな薬物の効果をスクリーニングした。その結果、本発明者らは、オルチプラズを始めとする1,2−ジチオールチオン誘導体を含有する薬物投与によって、LXR α及びLXR α依存性SREBP−1の発現及び活性化が抑制されることを発見した。また1,2−ジチオールチオン誘導体によって、SREBP−1が抑制されると、標的遺伝子である脂質生成遺伝子の発現が顕著に抑制され、さらに、高脂質食餌によって誘発される肝組織での中性脂質蓄積が抑制されることを発見した。これらの発見に基づいて、本発明では、LXR α又はSREBP−1の過剰発現に起因する疾病の予防及び治療のための、1,2−ジチオールチオン誘導体を含有する、薬学組成物を提供する。
【発明の効果】
【0017】
本発明による薬学組成物は、有効成分として1,2−ジチオールチオン誘導体を含有する。前記薬学組成物は、LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病の予防及び治療に効果的である。1,2−ジチオールチオン誘導体の例としては、オルチプラズ(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン 4−methyl−5−(2−pyrazinyl) −1,2−dithiol−3−thione)、3−メチル−1,2−ジチオール−3−チオン(3−methyl−1,2−dithiol−3−thion)、5−(6−メトキシピラジニル)−4−メチル−1,2−ジチオール−3−チオン(5−(6−methoxypyrazinyl) −4−methyl−1,2−dithiol−3−thione)が挙げられる。前記薬学組成物を投与すると、SREBP−1の発現及び活性が抑制される。前記SREBP−1は、LXR αの活性化を調節して、脂質生成酵素遺伝子の発現を調節する重要な転写因子である。さらに脂質生成遺伝子の発現が抑制されることにより、代謝異常による脂肪肝症による、肝組織内のトリグリセリドの蓄積が抑制される。したがって、1,2−ジチオールチオン誘導体を有効成分として含有する本発明の薬学組成物は、脂肪肝症を予防及び治療に有効である。また本発明による薬学組成物は、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症の予防及び治療に有効である。
【0018】
さらに、本発明は、本発明で提供する化合物又は組成物を使用して、LXR α又はSREBP−1の発現又は活性を阻害する方法を提供する。前記阻害は、LXR α又はSREBP−1の過剰発現又は過剰活性と関連した疾病又は状態の抑制又は治療となりえる。
【0019】
また、本発明は、LXR α又はSREBP−1の過剰発現又は過剰活性と関連した疾病又は状態、例えば、脂肪肝症、高中性脂肪血症、高レニン血症、レニンに起因した高血圧、アンドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎障害を予防又は治療する方法を提供する。
【図面の簡単な説明】
【0020】
【図1】オルチプラズ投与スケジュールを示す。
【図2】高脂質食餌で発現が増加したLXR αに対するオルチプラズ治療の結果である。結果は、正常食餌群であるLXR α mRNAに対する値で示した(ND:正常食餌、HFD:高脂質食餌,Olt:オルチプラズ,**:ND群と比較してp<0.01,##:HFD治療群のみと比較してp<0.01)。
【図3】LXR α過剰発現によって上昇したLXR αの活性に対するオルチプラズの治療効果を示す(S.S:スーパーシフト)。
【図4】高脂質食餌で発現が増加したSREBP−1に対するオルチプラズの治療結果を示す(ND:正常食餌、HFD:高脂質食餌,Olt:オルチプラズ、**:ND群と比較してp<0.01,##:HFD治療群のみと比較してp<0.01)。
【図5】LXR α活性化剤であるT0901317で、肝細胞株であるH4IIE、HepG2及びラットの初代培養した肝細胞を治療した後での、SREBP−1タンパク質の発現に対するオルチプラズの治療結果を示す。SREBP−1タンパク質はウェスタンブロット法によって確認した(S.E:短時間フィルム露光、L.E:長時間フィルム露光、A:全細胞抽出溶解液、B:HepG2 cellの核分画。
【図6】LXR α活性化剤処理で発現が増加したSREBP−1に対する1,2−ジチオールチオン誘導体のウェスタンブロットの結果である。
【図7】LXR α活性化剤処理で発現が増加したSREBP−1に対する1,2−ジチオールチオン誘導体のウェスタンブロットの結果である。
【図8】LXR α活性化剤処理で発現が増加したSREBP−1に対する1,2−ジチオールチオン誘導体のウェスタンブロットの結果である。
【図9】高脂質食餌によって誘導された脂肪肝動物モデルにオルチプラズを投与した場合の、高脂質食餌によって誘導された脂質生成遺伝子(FAS、ACC)の肝組織内発現量を比較した結果である。
【図10】高脂質食餌によって誘導された脂肪肝動物モデルにオルチプラズを投与したときの、前記高脂質食餌によって誘導された脂肪肝動物モデルの肝組織におけるトリグリセリド量に対するオルチプラズの治療結果を示す。
【図11】高脂質食餌によって誘導された脂肪肝動物モデルにオルチプラズを投与したときの、オイルレッドOで染色した肝組織の画像である。
【発明を実施するための形態】
【0021】
本発明は、オルチプラズなどの1,2−ジチオールチオン誘導体がLXR αの活性を抑制し、脂質生成遺伝子発現を調節する細胞タンパク質である、ステロール調節エレメント結合タンパク質−1(SREBP−1)の発現及び活性を抑制するということを発見したことに基づいたものである。本発明の発明者らは、高脂質食餌で発現が増加したマウスのLXR αの発現が、オルチプラズによって抑制され(図2)、LXR結合DNAエレメントに対するLXR αの結合能も、オルチプラズによって弱まるということを発見した(図3)。また本発明の発明者らは、LXR αの活性化剤として知られているT0901317を肝細胞株に付与したときに増加するSREBP−1の発現が、オルチプラズ又は他の1,2−ジチオールチオン誘導体によって抑制されることを発見した(図4〜図8)。
【0022】
オルチプラズなどの1,2−ジチオールチオン誘導体の投与は、LXR αの活性化を調節して、脂質生成酵素の遺伝子発現を調節する重要な転写因子であるSREBP−1の発現及び活性を抑制し、さらに脂質生成遺伝子の発現を抑制することにより、代謝異常による脂肪肝症による肝組織内のトリグリセリドの蓄積を抑制する。したがって、1,2−ジチオールチオン誘導体を有効成分として含有する本発明の薬学組成物は、脂肪肝症の予防及び治療に有効である。本発明の発明者らは、高脂質食餌の投与によって、肝組織内にトリグリセリドの含有量が増加したマウスに、オルチプラズを投与した場合、前記トリグリセリドの含有量が有意に減少し(図10及び図11)、また、前記高脂質食餌投与マウスで増加した脂肪酸合成酵素である脂肪酸合成酵素(FAS)及びアセチルCoAカルボキシラーゼ(ACC)の発現がオルチプラズの投与によって有意に抑制されることを発見した(図9)。
【0023】
本発明の発明者らによる発見に基づき、本発明は、LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病の予防用及び治療用の薬学組成物を提供する。前記薬学組成物は、下記式1で表される1,2−ジチオールチオン誘導体、その薬学的に許容される塩、それらの溶媒物又は水和物、又はそれらのプロドラッグを有効成分として含有し、及び薬学的に許容される担体を含む薬学組成物である。
【0024】
【化1】
【0025】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2はそれぞれ独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル、ヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され、前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択された少なくとも1つのヘテロ原子を含有する五員環又は六員環の化合物であり、前記フェニル及びヘテロアリールは、置換されていても非置換であってもよく、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、前記フェニル及びヘテロアリールが置換される場合には、一置換又は多置換であってよい。前記フェニル及びヘテロアリールはそれぞれ独立に、少なくとも1つのベンゼン又は前述のヘテロアリールと縮合してもよく、前記縮合したフェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。
【0026】
前記LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病としては、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症、脂肪肝、高トリグリセリド血症、高レニン血症などを挙げることができるが、これに限定されるものではない。よって、本発明は、レニンに起因した高血圧、アンドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症又は腎症、脂肪肝、高トリグリセリド血症又は高レニン血症の予防用及び治療用の薬学組成物であって、式1で表される1,2−ジチオールチオン誘導体を有効成分として含有し、薬剤学的に許容される担体を含む薬学組成物を提供する。
【0027】
本発明はまた、下記式1で表される1,2−ジチオールチオン誘導体、その薬学的に許容可能な塩、それらのプロドラッグ、それらの溶媒和物又は水和物を有効成分として含有し、及び薬剤学的に許容される担体を含む薬学組成物を投与することを含む、試験管内(in vitro)又は生体内(in vivo)で、LXR α又はSREBP−1の発現又は活性を阻害する方法を提供する。
【0028】
【化2】
【0029】
ここで、Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2はそれぞれ独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル、ヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択される。前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含有する五員環又は六員環化合物を意味する。前記フェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルから構成された群(基)から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。前記フェニル及びヘテロアリールはそれぞれ独立に、少なくとも1つのベンゼン又は前述のヘテロアリールと縮合してもよく、前記縮合したフェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。
【0030】
本発明はまた、下記式1で表される1,2−ジチオールチオン誘導体、その薬学的に許容可能な塩、それらのプロドラッグ、それらの溶媒和物又は水和物を有効成分として含有し、また、薬剤学的に許容される担体を含む薬学組成物を投与することを含む、LXR α又はSREBP−1の過剰発現又は過剰活性と関連した疾病又は状態を予防又は治療する方法を提供する。
【0031】
【化3】
【0032】
ここで、Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2はそれぞれ独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル、ヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択される。前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含有する五員環又は六員環化合物を意味する。前記フェニル及びヘテロアリールは、置換されていても、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。前記フェニル及びヘテロアリールはそれぞれ独立に、少なくとも1つのベンゼン又は前述のヘテロアリールと縮合していてもよい。前記縮合されたフェニル及びヘテロアリールは、置換されていてもよく、又は非置換であってもよく、可能な置換基としては、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ、カルボキシルからなる群から選択され、置換される場合には、前記フェニル及びヘテロアリールは一置換又は多置換であってよい。
【0033】
LXR α又はSREBP−1の過発現又は過活性と関連した疾病又は状態を予防又は治療する方法の一例として、前記疾病又は状態は、例えば、脂肪肝、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症である。実施形態では、本明細書に開示される化合物は、これらの疾病又は状態の1つ又はそれ以上を改善し、及び/又は発現を遅らせるために使用される。
本発明の薬学組成物に含まれる1,2−ジチオールチオン誘導体には、1,2−ジチオール−3−チオン及び、二環式分子を含む有機化合物と、それらの誘導体有機化合物とが含まれる。二環式分子は、ピラジン、ピリダジン、ピリミジン、チアゾール又はチオフェンであってよい(表1)。
【0034】
【表1】
【0035】
本発明は、前記化合物が形成されうる薬学的に許容可能な塩、又はそれらの溶媒和物、水和物又はプロドラッグを含んでよい。薬剤学的に許容される付加塩は、薬剤学的に許容される酸付加塩、及び薬剤学的に許容される塩基付加塩を含む。本明細書に記載の「薬剤学的に許容される酸付加塩」には、式1で表される化合物を用いて形成されうる非毒性酸付加塩が含まれ、それは治療的に活性である。式1の化合物は元来、塩基性特性を有しており、適当な酸で処理することにより、薬剤学的に許容される酸付加塩に転換されうる。適当な酸は、無機酸又は有機酸でありうる。無機酸としては、例えば、塩酸又はブロム酸などのハロゲン化水素酸、硫酸、硝酸、リン酸などを挙げることができる。有機酸としては、例えば、酢酸、トリフルオロ酢酸、プロパン酸、ヒドロキシ酢酸、乳酸、ピルビン酸、シュウ酸、マロン酸、コハク酸(すなわち、ブタン二酸)、マレイン酸、フマル酸、リンゴ酸、酒石酸、クエン酸、メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸、p−トルエンスルホン酸、シクラミン酸、サリチル酸、p−アミノ−サリチル酸、パモ酸などを挙げることができる。酸性特性を有する前記転換された化合物は、適当な有機又は無機の塩基で処理することによって、その薬剤学的に許容される塩基付加塩に転換されうる。適当な塩基付加塩としては、例えば、アンモニウム塩;アルカリ及びアルカリ土類金属塩、例えば、リチウム塩、ナトリウム塩、カリウム塩、マグネシウム塩、カルシウム塩;ベンザチン塩、N−メチル−D−グルカミン塩、ヒドラバミン塩(hydrabamie salt)などの有機塩基との塩;アルギニン、リシンなどのアミノ酸との塩を含む。
【0036】
本発明の組成物は、薬剤学的分野で一般的な方法によって、経口投与に適した単位投与型の製剤及び注射剤に剤形化することができ、これを投与できる。単位投与型の製剤には、硬質及び軟質のカプセル剤、錠剤、散剤、懸濁剤、シロップ剤が含まれる。経口投与に適した単位投与型の製剤には、 少なくとも1つの薬物学的活性成分を含み、またそれ以外に、少なくとも1つの薬剤学的に不活性である一般的な担体が含まれていてよい。少なくとも1つの薬剤学的に不活性である一般的な担体としては、例えば、賦形剤、結合剤、崩壊剤、潤滑剤が含まれる。賦形剤としては、例えば、粉末、ラクトース、カルボキシメチルセルロース、カオリンが挙げられる。結合剤としては、例えば、水、ゼラチン、アルコール、グルコース、アラビアゴム(Arabia rubber)、トラガカントゴムが挙げられる。崩壊剤としては、例えば、粉末、デキストリン、アルギン酸ナトリウムが挙げられる。潤滑剤としては、例えば、タルク、ステアリン酸(staric acid)、ステアリン酸マグネシウム、流動パラフィンが挙げられる。本発明ではまた、溶解のための溶解補助剤をさらに含んでもよい。
【0037】
本発明の組成物の1日投与容量は、投与しようとする対象の疾病の進行程度、発病時期、年齢、健康状態、合併症などによって異なる。例えば、成人に対して、1日あたり、本発明の組成物を1〜500mg、特に、30〜200mg投与できる。投与は1日1回又は数回分割して投与できる。
【0038】
以下、本発明について、実験例によって詳細に説明する。下記実施例は、本発明について具体的に例示するだけであり、本発明の内容が、下記実施例に限定されるものではない。
【0039】
(参考例1:実験動物及び食餌)
実験動物として使われた雄C57BL/6マウス(平均体重25〜30g)は、Charles River Orient(韓国・ソウル)から購入した。実験に使用する前、少なくとも1週間、55±5%の湿度、22±2℃の温度及び換気が調節されたソウル大学校薬科大学動物実験研究棟で動物を適応させた。午前7時と午後7時を基準として、12時間周期で明暗を変えた。実験期間中、マウスが摂取した食餌量及び飲料水量には、有意的な変化が観察されなかった。動物の重さと状態とを毎週1回検査した。2つのグループのマウスは、高脂質食餌(Dyets Inc.,Bethlehem)又は正常食餌でそれぞれ10週間飼育した。それぞれのグループについて、最後の4週間の間、オルチプラズ(10又は30mg/kg、3回/週)をマウスに投与した(図1)。各グループは、全部で10匹のマウスから構成された。
【0040】
(参考例2:試料準備)
オルチプラズ及び1,2−ジチオールチオン誘導体は、株式会社CJから提供を受けた。本発明に使われた1,2−ジチオールチオン誘導体は、韓国特許第10−0604261号公報に記載された方法によって製造されうる。脂肪肝誘発のための高脂質食餌は、米国・ダイエット社(Dyet Co.)から購入した。オルチプラズを40%PEG200で希釈して、所望の濃度に調製した。
【0041】
(参考例3:リアルタイム−RT PCR)
マウスの肝臓から抽出した全RNA(2μg)、d(T)16プライマー、及びAMV逆転写酵素を使用し、cDNAを得た。遺伝子の相対的な量は、サイバーグリーン染料(CyBr green dye)を使用したリアルタイムRT−PCR法によって定量した。リアルタイム RT−PCRは、ロッシュ社(マンハイム、ドイツ)のLight-cycler2.0を利用した。メーカーの方法によって、PCRを行い、Light-cycler software4.0プログラムを使用し、各遺伝子の相対的な量を分析した。
【0042】
(参考例4:ウェスタンブロット)
Laemmli UK法(1970)によって、Mighty Small II SE250装置を使用し、ドデシル硫酸ナトリウム−ポリアクリルアミドゲル電気泳動(SDS−PAGE)を行った。肝試料の溶解分画を、サンプル希釈緩衝液[63mM Tris(pH.6.8)、10%グリセロール、2%SDS、0.0013%ブロモフェノールブルー、5%β−メルカプトエタノール]に希釈し、7.5%及び9%のゲルを使用し、電極緩衝液(1L溶液中にTris 15g、グリシン72g、SDS 5g含む)内で電気泳動した。電気泳動が終わったゲルは、電気泳動装置を利用し、緩衝液[25mM Tris、192mMグリシン、20%v/v メタノール(pH.8.3)]内で、190mAmpsで1時間、ニトロセルロース紙にタンパク質を転移させた。Anti−SREBP−1を一次抗体として反応させた後、二次抗体として西洋わさびペルオキシダーゼ−ヤギ抗ウサギIgG(horseradish peroxidase-conjugated goat anti-rabbit IgG)を1時間反応させた。その後、ECL 化学発光分析システム(Amersham、Gaithesberg、MA)を使用して免疫反応性蛋白質を可視化させた。各試料中の蛋白質量の同質性は、anti−β−actin抗体(Sigma、セントルイス、MO)を使用して確認した。
【0043】
(参考例5:分析方法)
下記実施例で提示した資料は、薬物学的計算プログラムを利用して分析したものであり、多様な実験群間の有意性を一方向平方偏差分析法(Fisher,R.A.,Statistical Methods for Research Workers,Edinburgh: Oliver & Boyd,1925)で検定した後、ニューマン・クールズ検査(Norman GR et al.,Biostatistics: The Bare Essentials,2000)で判定した(*p<0.05、**p<0.01)。
【0044】
(実験例1:発現が増加したLXR αに対するオルチプラズ処置の効果)
マウスを、高脂質食餌及び正常食餌で10週間飼育した。高脂質食餌で飼育したマウス群に、最後の4週間の間、オルチプラズ(10−30mg/kg、3回/週)、すなわち、1,2−ジチオールチオン化合物、を投与したマウスの肝組織のLXR αの発現を測定した。肝組織からmRNAを単離し、RT−PCRを介してcDNAを合成した後、特定プライマー(mouse LXR、5’−TGCCATCAGCATCTTCTCTG−3’(sense)及び5’−GGCTCACCAGCTTCATTAGC−3’(antisense))を使用し、リアルタイムPCRを行った。正常食餌群(ND)のLXR α mRNAの発現レベルを1としたときの、高脂質食餌群、又は、高脂質食餌かつオルチプラズ投与群の相対的な発現レベルを測定し、その結果を図2に示した。これにより、細胞内脂質センサであるLXR αの発現が、高脂質食餌によって有意に増加し(p<0.01)、LXR α発現の増加は、オルチプラズ投与によって抑制される(p<0.01)ということが明らかになった。
【0045】
(実験例2:オルチプラズ処置によるLXR αの活性抑制効果)
LXR αは、RXRαとダイマーを形成し、標的遺伝子プロモーターに存在する特定領域(LXRE)に結合することにより、遺伝子の発現を調節する。オルチプラズ処置によって、LXRE結合能が変化するか否かを、ゲルシフトアッセイで確認した。SREBP−1c遺伝子のLXRE二本鎖オリゴヌクレオチドを、[γ−32P]ATPとT4 ポリヌクレオチドキナーゼとを使用し、5’−末端を放射性同位元素で標識した。その後、標識したプローブ(1ml、>106cpm)と核分画タンパク質とを結合緩衝液中で反応させた。反応液を4%ポリアクリルアミド・ゲルで電気泳動した後、オートラジオグラフィーで分析した。実験に使われたLXREオリゴヌクレオチド配列は、5’−CAGTGACCGCCAGTAACCCCAGC−3’である。DNA結合特異性は、コールドプローブ滴定(cold probe titration)及びスーパーシフトアッセイにより確認した。コールドプローブ滴定のために、20倍多い(モル基準)非標識オリゴヌクレオチドをあらかじめ反応させた。スーパーシフトのために、LXR α抗体又はRXRα抗体(2μg)を反応混合物と、室温で約30分間反応させ、そこに、放射線同位元素で標識されたプローブを加え、さらに30分間反応させた後、電気泳動した。
【0046】
LXR α及びRXRαが過剰発現した場合、対照群(mock)に比べて移動の遅い泳動バンドの密度が高まった(図3Aの最初のバンド及び2番目のバンドを参照のこと)。LXR α及びRXRαの抗体を使用し、スーパーシフトを行ったとき、LXR α抗体及びRXRα抗体によって、DNAタンパク結合が減少し、スーパーシフトされたバンドが観察された(図3A、3番目のバンド〜5番目のバンド)。これは、LXR α/RXRα複合体の、DNA結合特異性を支持する結果である。
【0047】
オルチプラズが投与されると、移動が遅延するバンド(図3の最初のバンド〜3番目のバンド)の密度が低くなった(図3Bの4番目のバンド及び5番目のバンド)。これは、LXR αの、DNA結合能がオルチプラズ処置によって実質的に弱まるということを示している。
【0048】
(実験例3:発現が増加したSREBP−1に対するオルチプラズ処置の効果)
実験例1で使用したマウスの肝組織におけるSREBP−1の発現レベルを測定した。肝組織からmRNAを単離し、RT−PCRによってcDNAを合成した後、これに特定プライマー(mouse SREBP−1、5’−AACGTCACTTCCAGCTAGAC−3’(sense)及び5’−CCACTAAGGTGCCTACAGAGC−3’(antisense))を用いて、リアルタイムPCRを行った。正常食餌群(ND)のSREBP−1mRNAの発現レベルを1としたとき、高脂質食餌群、又は、高脂質食餌かつオルチプラズ投与群の相対的な発現レベルを測定した。その結果を図4に示した。SREBP−1の発現が高脂質食餌によって有意に増加し(p<0.01)、SREBP−1発現の増加は、オルチプラズ投与によって抑制される(p<0.01)ということが明らかになった。
【0049】
(実験例4:オルチプラズ処置によるSREBP−1の発現及び活性の抑制効果)
肝細胞株であるH4IIE、HepG2、及びラット初代培養肝細胞を、LXR α活性化剤であるT0901317で処置し、ウェスタンブロット法によって、SREBP−1タンパク質を確認した。SREBP−1の発現は、T0901317で処置した後、12時間以内に顕著に増加した(図5Aの各ゲル写真の4番目のカラム)。SREBP−1タンパク質の発現の増加は、オルチプラズ処置を行うと、濃度依存的に減少した(図5Aの各ゲル写真の5番目及び6番目のカラム)。これは、SREBP−1の発現がオルチプラズによって抑制されることを示す。
【0050】
また、T0901317を投与したHepG2細胞株におけるSREBP−1の核内移動の増加が、オルチプラズ処置によって濃度依存的に減少した(図5B)。これは、SREBP−1の活性が、オルチプラズによって抑制されることを示す。
【0051】
細胞分画は以下の方法で単離した。低浸透圧緩衝溶液[10mM HEPES(pH7.9)、10mM KCl、0.1mM EDTA、0.5%ノニデットP−40、1mM DTT及び0.5mMフッ化フェニルメチルスルホニル(PMSF)]を肝細胞株に加え、氷中に10分放置した。その後、溶液を7,200gで5分間遠心分離し、上澄み液を細胞質分画として使用した。別途、高浸透圧緩衝溶液[20mM HEPES(pH7.9)、400mM NaCl、1mM EDTA、10mM DTT及び1mM PMSF]を肝細胞株に加え、氷中に1時間放置した。その後、得られた溶液を15,000gで10分間遠心分離して上澄み液を取り、核分画として使用した。別途、溶液緩衝液[10mM HEPES(pH7.9)、100mM NaCl、1mM EDTA、10%グリセロール、0.5%Triton X−100、0.5%ノニデット P−40、1mM DTT及び0.5mM PMSF]をPBSで洗浄した細胞に加え、氷中で1時間溶解させた。その後、得られた溶液を10,000gで10分間遠心分離した後、上澄み液を全細胞抽出液として使用した。これらの細胞分画は、使用時まで−70゜Cに保管した。タンパク質濃度は、ブラッドフォードアッセイ(Bradford assay;Bio−Rad protein assay kit、Hercules、CA、USA)で定量した。
【0052】
(実験例5:1,2−ジチオールチオン誘導体ら処置によるSREBP−1の発現抑制効果)
LXR α活性化剤(T0901317)によって増加したSREBP−1の発現に対する1,2−ジチオールチオン誘導体の効果を、H4IIE細胞株を利用して評価した。H4IIE細胞株をT0901317で処置することで増加したSREBP−1の発現が、1,2−ジチオールチオン誘導体処置によって抑制された(図6〜図8)。
【0053】
(実験例6:脂肪酸合成酵素FAS及びACCの発現に対する、オルチプラズ処置による抑制効果)
実験例1で使用したマウスの肝組織におけるSREBP−1の標的遺伝子であるFAS及びACCの発現レベルを測定した。肝組織からmRNAを単離し、RT−PCRによりcDNAを合成した後、これを特定プライマー(mouse ACC1、5’−GTCAGCGGATGGGCGGAATG−3’(sense)及び5’−CGCCGGATGCCATGCTCAAC−3’(antisense);mouse FAS、5’−AGCGGCCATTTCCATTGCCC−3’(sense)及び5’−CCATGCCCAGAGGGTGGTTG−3’(antisense))を使用して、リアルタイムPCRを行った。正常食餌群(ND)のFAS及びACCのmRNAの発現レベルを1としたとき、高脂質食餌群、又は、高脂質食餌かつオルチプラズ投与群の相対的なFASmRNA又はACCmRNAの発現レベルを測定した。結果を図9に示した。FAS及びACCの発現が、高脂質食餌によって有意に増加し(p<0.01)、FAS及びACC発現の増加は、オルチプラズ投与によって抑制される(p<0.01)ということが明らかになった。
【0054】
(実験例7:オルチプラズ投与による高脂質食餌に蓄積になった肝組織内のトリグリセリドの含有量抑制効果)
実験例1で使用したマウスの肝組織内の実験例1におけるトリグリセリド含有量に対するオルチプラズの効果を評価した。肝組織内のトリグリセリド含有量は、脂肪肝を示す指標である。オルチプラズを投与した後(Bae et al.,Hepatology,2007,46: 730−739)、肝組織内のトリグリセリド含有量を測定した。高脂質食餌を10週実施したマウスでは、肝組織内のトリグリセリド含有量が、正常食餌群に比べて顕著に増加した(p<0.01)。一方、オルチプラズが投与された場合には、組織内トリグリセリドの含有量が有意に減少した(p<0.01)(図10)。
【0055】
(実験例8:高脂質食餌によって誘導された脂肪肝動物モデルの肝組織におけるオルチプラズ治療効果)
実験例7で使用した、高脂質食餌によって誘導された脂肪肝に対するオルチプラズの治療効果を、脂肪特異染色剤であるオイルレッドO染色法を利用して確認した。脂肪肝の肝組織を、10%中性ホルマリン溶液で固定し、一般的な固定工程及び脱水工程を行った後、工程後の肝組織をパラフィンに包埋した。包埋した組織に対して、4μm厚に組織切片を行い、オイルレッドOで染色した後、光学顕微鏡で観察した。高脂質食餌群では、オイルレッドOで赤く染色された部分が顕著に現れ(HFD+ビークル(vehicle))、オルチプラズが投与された群(HFD+オルチプラズ)では、赤く染色された部分が顕著に減った(図11)。これにより、オルチプラズ治療効果が高いことがわかる。
【0056】
1,2−ジチオールチオン誘導体を有効成分として含有する多様な形態の製剤を調製した。
【0057】
(製造例1)
オルチプラズ25mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0058】
前記の成分を混合し、一般的な錠剤の製造方法によって打錠し、錠剤を製造した。
【0059】
(製造例2)
3−メチル−1,2−ジチオール−3−チオン50mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0060】
前記の成分を混合し、一般的な錠剤の製造方法によって打錠し、錠剤を製造した。
【0061】
(製造例3)
5−(6−メトキシピラジニル)−4−メチル−1,2−ジチオール−3−チオン 100mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0062】
前記の成分を混合し、一般的な散剤の製造方法によって、散剤を製造した。
【0063】
(製造例4)
オルチプラズ250mg
乳糖50mg
澱粉10mg
ステアリン酸マグネシウム適量
【0064】
前記の成分を混合し、一般的な散剤の製造方法によって、散剤を製造した。
【0065】
(製造例5)
オルチプラズ25mg
乳糖30mg
澱粉28mg
タルク2mg
ステアリン酸マグネシウム適量
【0066】
前記の成分を混合し、一般的な方法によるカプセル剤の製造方法によってゼラチン軟カプセルに充填し、カプセル剤を製造した。
【0067】
(製造例6)
5−(6−メトキシピラジニル)−4−メチル−1,2−ジチオール−3−チオン50mg
乳糖30mg
澱粉28mg
タルク2mg
ステアリン酸マグネシウム適量
【0068】
前記の成分を混合し、一般的なカプセル剤の製造方法によってゼラチン軟カプセルに充填し、カプセル剤を製造した。
【0069】
(製造例7)
オルチプラズ100mg
異性化糖10g
砂糖30mg
ナトリウムCMC 100mg
レモン香料 適量
精製水 バランス
(精製水適量を加えて全体100mlとする)
【0070】
前記の成分を通常の懸濁剤の製造方法によって懸濁剤を製造し、100ml容量の褐色瓶に充填して滅菌し、懸濁剤を製造した。
【0071】
(製造例8)
3−メチル−1,2−ジチオール−3−チオン250mg
乳糖30mg
澱粉20mg
ステアリン酸マグネシウム適量
【0072】
前記の成分を均一に混合し、ポリエチレンがコーティングされた袋に充填して密閉し、散剤を製造した。
【0073】
(製造例9)
軟質カプセル剤1錠中に以下を含む:
オルチプラズ100mg
ポリエチレングリコール400 400mg
濃グリセリン55mg
精製水35mg
【0074】
ポリエチレングリコールと濃グリセリンとを混合し、ここに精製水を加え、この混合物を約60℃で維持した状態で、1,2−ジチオールチオン誘導体を加えた。約1,500rpmで撹拌して、均一に混合した。その後ゆっくりと撹拌しつつ温度を室温に冷却し、真空ポンプを使用して気泡を除去し、軟質カプセルの内容物とした。軟質カプセルの被膜は、一般的に公知のゼラチン、可塑剤でソフト処方とした。1カプセル当たりゼラチン132mg、濃グリセリン52mg、70%ジソルビトール液6mg、着香剤としてエチルバニリン適量及びコーティング剤としてカルナウバワックスを使用し、通常の調剤法で製造した。
【産業上の利用可能性】
【0075】
本発明による薬学組成物は、LXR αの過剰発現又は過剰活性、又はSREBP−1の過剰発現又は過剰活性に起因する疾病の予防又は治療に有効である。前記薬学組成物を投与すると、SREBP−1の発現及び活性が阻害される。SREBP−1は、LXR α活性化調節を介して、脂質生成酵素遺伝子の発現を調節する重要な転写因子であり、さらに脂質生成遺伝子の発現を抑制することにより、代謝障害による脂肪肝症による肝組織内のトリグリセリドの蓄積を抑制する。従って、1,2−ジチオールチオン誘導体を有効成分として含有する本発明の薬学組成物は、脂肪肝症の予防及び治療に有効である。また、本発明による薬学組成物は、高トリグリセリド血症、高レニン血症、レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症の予防及び治療に有効である。
【特許請求の範囲】
【請求項1】
肝Xレセプター(LXR α)の過剰発現又は過剰活性に起因する疾病の予防又は治療のための薬学組成物であって、
前記薬学組成物は、有効成分として下記式1で表される化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物, そのプロドラッグを含む薬学組成物。
【化1】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項2】
ステロール調節エレメント結合タンパク質−1c(SREBP−1c)の過剰発現又は過剰活性に起因する疾病の予防又は治療のための薬学組成物であって、
前記薬学組成物は、下記式1の化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【化2】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項3】
レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症、脂肪肝、高トリグリセリド血症又は高レニン血症の予防又は治療のための薬学組成物であって、
前記薬学組成物は、下記式1の化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【化3】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項4】
脂肪肝の予防又は治療のための薬学組成物であって、
前記薬学組成物は、下記式1の化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【化4】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項5】
式1で表される前記化合物が、オルチプラズ(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン)である、請求項1から請求項4のいずれか1項に記載の薬学組成物。
【請求項6】
脂肪肝の予防又は治療のための薬学組成物であって、オルチプラズ(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン)、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【請求項1】
肝Xレセプター(LXR α)の過剰発現又は過剰活性に起因する疾病の予防又は治療のための薬学組成物であって、
前記薬学組成物は、有効成分として下記式1で表される化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物, そのプロドラッグを含む薬学組成物。
【化1】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項2】
ステロール調節エレメント結合タンパク質−1c(SREBP−1c)の過剰発現又は過剰活性に起因する疾病の予防又は治療のための薬学組成物であって、
前記薬学組成物は、下記式1の化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【化2】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項3】
レニンに起因した高血圧、アルドステロン症、副腎白質ジストロフィー、糸球体硬化症、タンパク尿症、腎症、脂肪肝、高トリグリセリド血症又は高レニン血症の予防又は治療のための薬学組成物であって、
前記薬学組成物は、下記式1の化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【化3】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項4】
脂肪肝の予防又は治療のための薬学組成物であって、
前記薬学組成物は、下記式1の化合物、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【化4】
Xは、炭素又は窒素であり、Qは、硫黄、酸素又は−S=Oであり、R1及びR2は各々独立に、水素原子、C1−7アルキル、C3−7シクロアルキル、C1−7ハロアルキル、C1−7アルコキシ、C3−7シクロアルコキシ、C1−7アルキルチオ、C3−7シクロアルキルチオ、C1−7アルケニル、C1−7アルキニル、C1−7アルキルスルホニル、C1−7アルキルアミノカルボニル、HO−C1−7アルキル、HS−C1−7アルキル、ヒドロキシ、チオール、ハロゲン、カルボキシル、ニトロ、シアノ、C1−7アルキルカルボニル、C1−7アルコキシカルボニル、C1−7アルキルカルボニルオキシ、C1−4アルキルカルボニル−C1−4アルキル、C1−4アルコキシ−C1−4アルキル、C1−4アルキルチオ−C1−4アルキル、アミノ、C1−7アルキルアミノ、C1−7アルキルカルボニルアミノ、C1−4アルコキシ−C1−4アルキルアミノ、C1−4アルキルチオ−C1−4アルキルアミノ、C1−4アルキルスルホンアミノ、フェニル、ヘテロアリール、フェニル−C1−4アルキル、ヘテロアリール−C1−4アルキル、フェニル−C1−4アルコキシ−C1−4アルキル、フェニル−C1−4アルキルチオ−C1−4アルキル、フェノキシ−C1−4アルキル、フェニルチオ−C1−4アルキル、フェニルカルボニルアミノ、フェノキシ−C1−4アルキルカルボニルアミノ、フェニル−C1−4アルコキシ−C1−4アルキルカルボニルアミノ、ヘテロアリールオキシ−C1−4アルキル、ヘテロアリールチオ−C1−4アルキル及びヘテロアリール−C1−4アルキルチオ−C1−4アルキルからなる群から選択され;前記ヘテロアリールは、窒素、硫黄及び酸素からなる群から選択される少なくとも1つのヘテロ原子を含む五員環又は六員環化合物であり、;前記フェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは一置換又は多置換であり;前記フェニル及びヘテロアリールは各々独立に、少なくとも1つのベンゼン又は前記ヘテロアリールと縮合され;また、前記縮合されたフェニル及びヘテロアリールは、置換又は非置換であり、可能な置換基は、ハロゲン、C1−7アルキル、C1−7アルコキシ、C1−7ハロアルキル、C1−7アルキルチオ、C1−7アルケニルオキシ、C1−4アルキルカルボニル、C1−4アルキルアミノ、ニトロ、アミノ、シアノ、HO−C1−4アルキル、HS−C1−4アルキル、HO−C1−7アルコキシ、HO−C1−7アルキルチオ、HS−C1−7アルキルチオ、HS−C1−7アルコキシ、チオール、ヒドロキシ及びカルボキシルからなる群から選択され、置換される場合、前記フェニル及びヘテロアリールは、一置換又は多置換である。
【請求項5】
式1で表される前記化合物が、オルチプラズ(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン)である、請求項1から請求項4のいずれか1項に記載の薬学組成物。
【請求項6】
脂肪肝の予防又は治療のための薬学組成物であって、オルチプラズ(4−メチル−5−(2−ピラジニル)−1,2−ジチオール−3−チオン)、その薬学的に許容可能な塩、その溶媒和物、その水和物又はそのプロドラッグを有効成分として含む薬学組成物。
【図1】

【図2】
【図4】
【図9】
【図10】
【図3】
【図5】
【図6】
【図7】
【図8】
【図11】
【図2】
【図4】
【図9】
【図10】
【図3】
【図5】
【図6】
【図7】
【図8】
【図11】
【公表番号】特表2011−529047(P2011−529047A)
【公表日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願番号】特願2011−520000(P2011−520000)
【出願日】平成21年7月30日(2009.7.30)
【国際出願番号】PCT/KR2009/004242
【国際公開番号】WO2010/016681
【国際公開日】平成22年2月11日(2010.2.11)
【出願人】(508369906)エスエヌユー アール アンド ディービー ファウンデーション (11)
【Fターム(参考)】
【公表日】平成23年12月1日(2011.12.1)
【国際特許分類】
【出願日】平成21年7月30日(2009.7.30)
【国際出願番号】PCT/KR2009/004242
【国際公開番号】WO2010/016681
【国際公開日】平成22年2月11日(2010.2.11)
【出願人】(508369906)エスエヌユー アール アンド ディービー ファウンデーション (11)
【Fターム(参考)】
[ Back to top ]