
Mnk−1タンパク質およびMnk−2タンパク質の結晶学的構造
【課題】結晶ヒトMnk−2タンパク質、結晶ヒトMnk−1、およびそれらの調製物の製造方法を提供する。
【解決手段】スペースグループP43212と、a=93.5Å、b=93.5Åおよびc=175.2Åであるユニットセル寸法を有する、結晶ヒトMnk−1キナーゼ。前記Mnk−1キナーゼが、大腸菌中の融合タンパク質として発現され、結晶が拡散蒸着によって成長される、前記結晶ヒトMnk−1キナーゼ調製物の製造方法。
【解決手段】スペースグループP43212と、a=93.5Å、b=93.5Åおよびc=175.2Åであるユニットセル寸法を有する、結晶ヒトMnk−1キナーゼ。前記Mnk−1キナーゼが、大腸菌中の融合タンパク質として発現され、結晶が拡散蒸着によって成長される、前記結晶ヒトMnk−1キナーゼ調製物の製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、結晶Mnk−1タンパク質ならびにMnk−2タンパク質に関し、より詳細には、Mnk−1およびMnk−2キナーゼ領域の結晶構造に関する。
【背景技術】
【0002】
ヒトにおいては、500を超えるキナーゼが知られており、それらはヌクレオチドからタンパク質基質までのリン酸基の伝達に介在する。タンパク質キナーゼによる基質識別、制御ならびに触媒作用の詳細な理解は、高度に多様な生物学的経路の完全なイメージを描くための基本であり、それらの多くは広範囲に及ぶ疾病に直接関連している。cAMP依存性キナーゼの結晶構造は、タンパク質キナーゼの分子構造の第1の高解像度画像を提供した(Knighton等、Sicence 235(5018)(1991)407−414)。
【0003】
他のヒトのタンパク質キナーゼの結晶構造により、貴重な触媒および調節メカニズムへの識見が提供され、特定の阻害因子の設計が助成される。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Knighton等、Sicence 235(5018)(1991)407−414
【発明の概要】
【課題を解決するための手段】
【0005】
したがって、本発明の主題は、結晶ヒトMnk−2タンパク質と結晶ヒトMnk−1、およびそれらの調製方法と用途に関するものである。
【0006】
第1の実施態様において、本発明は、ヒトのセリン−トレオニンキナーゼマイトジェン活性化キナーゼ(MAP)相互作用性キナーゼ−2に関するものであり、これはMnk−2タンパク質とも称される。ヒトにおいて4つのMnkタンパク質が発見されており、すなわち、2つのアイソフォームMnk−1とMnk−2であり、後者は2つのスプライスバリアントMnk−2aとMnk−2bとして存在する。スプライスバリアントMnk−1bもまた開示されている。Mnk−2aとMnk−2bのキナーゼのドメINは同一である。Mnkタンパク質がMAPキナーゼファミリーの1つによって活性化できることが示されている。具体的には、ストレス誘発p38タンパク質と、マイトジェン活性化Erk1/2タンパク質が、この機能を果たすことができる。Mnk−1とMnk−2は、同様の経路を経て活性化され、同様の基質特異性を呈する。キナーゼドメIN内におけるそれらのアミノ酸配列は、ほぼ同じであり、かつ、以下に記載のアミノ酸と同一である。そのため、Mnkキナーゼは、これら2つのMAPキナーゼ経路の集合点を構成し得る。
【0007】
Mnkタンパク質は、タンパク質キナーゼのMAPキナーゼ活性化タンパク質キナーゼ(MAPKAPK)ファミリーのサブファミリーであり、これらタンパク質キナーゼは、Ca/カドミウム調節キナーゼ(CAMK)グループに属する。
【0008】
Mnkは、3つのMAPKカスケードのうちの2つ、すなわち、成長因子誘導Ras細胞外信号調節タンパク質キナーゼ(ERK)1/2およびストレス誘発p38経路(福永等のEmbo J.(16)(1997)1921−1933;Embo J.(16)(1997)1909−1910)によるリン酸化によって活性化される。2つのほ乳類MnkアイソフォームであるMnk−1とMnk−2は、真核細胞開始因子4E(elF4E)をin vitroならびにin vivoでリン酸化する(シェパー(Scheper)等のEur J.Biochem.(269)(2001)5350−5359、上田等のMol.Cell Biol.(24)(2004)6539−6549、ヴァスキエヴィッツ(Waskiewicz)等のMol. Cell Biol.(19)(1999)1871−1880)。elF4Eは、翻訳開始複合体の必須成分であり、真核細胞のメッセンジャーRNAのCAP構造を結合する(マルコトリジアーノ(Marcotrigiano)等のCell(89)(1997)951−961)。Mnk介在性elF4Eリン酸化により、例えばRFLAT−1やウィルス性転写の特定のmRNAの翻訳が刺激されることは明らかである(ニコルシェバ(Nikolcheva)等のClin.Invest.(110)(2002)119−126、ウォルシュ(Walsh)等のGenet.Dev.18(2004)660−672)。さらに、Mnk1は、hnRNPA1による腫瘍壊死因子アルファ(TNF−α)の翻訳を減じるため、炎症性疾患において役割を果たす(バクセード(Buxade)等のImmunity 23,177−189)。脂質代謝、炎症ならびにウィルス性翻訳におけるMnkの改善により、製薬的介入のためのターゲットとしてそれらは定義される。
【0009】
Mnkは、3つのMAPKカスケードのうちの2つ、すなわち、成長因子誘導Ras細胞外信号調節タンパク質キナーゼ(ERK)1/2およびストレス誘発p38経路(福永等のEmbo J.(16)(1997)1921−1933;Embo J.(16)(1997)1909−1910)によるリン酸化によって活性化される。2つのほ乳類MnkアイソフォームであるMnk−1とMnk−2は、真核細胞開始因子4E(elF4E)をin vitroならびにin vivoでリン酸化する(シェパー(Scheper)等のEur J.Biochem.(269)(2001)5350−5359、上田等のMol.Cell Biol.(24)(2004)6539−6549、ヴァスキエヴィッツ(Waskiewicz)等のMol. Cell Biol.(19)(1999)1871−1880)。elF4Eは、翻訳開始複合体の必須成分であり、真核細胞のメッセンジャーRNAのCAP構造を結合する(マルコトリジアーノ(Marcotrigiano)等のCell(89)(1997)951−961)。Mnk介在性elF4Eリン酸化により、例えばRFLAT−1やウィルス性転写の特定のmRNAの翻訳が刺激されることは明らかである(ニコルシェバ(Nikolcheva)等のClin.Invest.(110)(2002)119−126、ウォルシュ(Walsh)等のGenet.Dev.18(2004)660−672)。さらに、Mnk1は、hnRNPA1による腫瘍壊死因子アルファ(TNF−α)の翻訳を減じるため、炎症性疾患において役割を果たす(バクセード(Buxade)等のImmunity 23,177−189)。脂質代謝、炎症ならびにウィルス性翻訳におけるMnkの改善により、製薬的介入のためのターゲットとしてそれらは定義される。
【0010】
CAMLグループのその他の部分と一列に並ぶ配列は、Mnkタンパク質の無比のいくつかの特徴を明らかにした。構造的および機能的見地におけるこの観察の結果を明らかにするため、Mnk−2に対する結晶学的な研究が行われた。本発明により、Mnk−2のキナーゼドメINの2.1Å結晶構造が得られた。その結果、Mnk−2のApo酵素が、活性ループとP+1ループのC末端を含む、ハンクス(Hanks)のスキームのサブドメINXIIIに対応するセグメントの例外的な開放高次構造を示すことが示されている(ハンクス(Hanks)等のMethods Enzymol.200(1991)38−62)。P+1ループは、基質結合に対して重要であることが知られている。
【0011】
DFDとしてMnkタンパク質中で保存される、マグネシウム結合DFGモチーフの同等物は、ATP結合ポケット中に突出し、ヌクレオチド結合を妨害する。したがって、活性化ループの開始時に保存されたDF(G/D)は、ATP結合を阻害する立体配座を受け入れる(DF(G/D)OUT立体配座と称す)。これはヌクレオチド結合を調節する阻害メカニズムが、ATP結合の割り接ぎが、非リン酸化apo酵素中で可能であるCAMKグループの既知の構造の他のキナーゼ(DF(G/D)IN立体配座)とは対照的であることを明らかにしている。これは、Ser/Thrキナーゼアポ酵素中におけるDF(G/D)OUT立体配置の第1の観察である。
【0012】
さらに、Cループ中の亜鉛配位モチーフ、タンパク質キナーゼにおいては開示しなかったが、が発見された。Mnk−2キナーゼドメINは、Mnkタンパク質中の長さと配列中に保存されるが他のキナーゼでは欠けている、Cループ中に15の残基の挿入を包含している。この挿入中の4つの保存されたシステINは、ここで開示するMnk−2構造によって明らかにされるように、亜鉛イオン結合部位として機能する。
【0013】
したがって本発明のMnk−2構造は、キナーゼアーキテクチャと調節の新規な態様を明らかにするものであり、これは論理的な阻害設計に用いることができる。
【0014】
特に好ましくは、本発明は、結晶ヒトMnk2aまたはMnk−2bタンパク質に関するものである。Mnk−2aは、真核細胞開始因子4e(elF4E)のリン酸化による翻訳機構をターゲットとする、ヒトタンパク質キナーゼである。
【0015】
トランスリン酸化反応中に包含されることが知られている残基は、CAMKキナーゼグループ内で保存される(テイラー(Taylor)等の、Structure 2(5)(1994)345−355、ハンクス(Hanks)等の、Science 241(4861)(1988)42−52)。これらの残基は、
(A)Lys113と、
(B)推定受容体ベースAsp205を含有する触媒ループ(残基205−210)と、
(C)γリン酸の活性に必要なマグネシウムイオンを配位する、DF(G/D)モチーフの第1のAsp226と、
である。
【0016】
しかしながら、Mnkタンパク質を他のタンパク質キナーゼから区別するいくつかの特徴がある。すなわち、活性ループのDFGモチーフN末端中に保存されたグリシンは、全てのMnkタンパク質中のアスパルテートで置換され、その結果、DFDモチーフ中に存在する(DF(G/D)と称される)。この単一のアミノ酸置換は、その他のCAMKグループ中には見られない。さらには、Mnkタンパク質は、全て長さが保存される3つの異なる箇所アミノ酸挿入部を含む。10個のアミノ酸の第1の挿入部(l1)は、DFDモチーフに続く活性化セグメントのN末端に位置する。第2の挿入部(l2)は、へリックスFの上流にあり、おおよそ5つのアミノ酸を含む。挿入部3(l3)は、15のアミノ酸のストレッチであり、Mnkのサブファミリー内に高度に保存されたパターンを呈し、CローブのGとHのへリックスを接続するループのN末端に位置する。4つのシステINのクラスタは、全てのMnkにおいては不変のl3内に存在する。
【0017】
一実施態様においては、本発明による、結晶ヒトMnk−2タンパク質、特に結晶Mnk−2aタンパク質は、完全なタンパク質である。他の実施態様では、これもまた好ましく、フルレングスタンパク質ではなく一部欠失形、特に、少なくともアミノ酸残基72−385を含む一部欠失形であり、キナーゼドメIN(KD)を含む。数字は、エントリーAAG26337(Mnk−2b)とAAG26336(Mnk2a)を示す。特に好ましくは、20Åより良好な、より好ましくは10Åより良好な、さらに好ましくは3Å未満より良好な解像度を有するX線構造解析が可能な結晶が関連する。
【0018】
本発明による結晶調合物は、スペースグループP3221と、a=104.5ű3Å、b=104.5ű3Åおよびc=72.35ű3Åであるユニットセル寸法を有することが好ましい。本発明によれば、2Åに対して回折可能な結晶が調製され、それによって、その構造は、分子置換によって溶解されて0.21(Rfree=0.25)のR因子に改良することができた。特に好ましいのは、本発明により不活性形態のヒトMnk−2タンパク質の結晶が検討されることである。
【0019】
さらに好ましいのは、非リン酸化Apo配座のMnk−2結晶ドメINが検討されることである。
【0020】
本発明によりわかるように、活性化セグメントとそのC末端のへリックスαF(αF:残基270−290)までの延長部は、異常なオープン構造にある(Mnk−2アミノ酸残基のナンバリングは、Entrez Entry AAG26336命名法に対応する)。この領域はサブドメINXIII分類に該当する。活性化セグメントは、活性キナーゼのリン酸化ターゲットである残基を担持し、19−32残基とは独立した、2つの保存モチーフであるDF(G/D)とAPEの間に配置される領域として定義されている。
【0021】
従来開示のキナーゼ構造との際だって対照的に、ヒトMnk−2タンパク質のサブドメINXIIIは、キナーゼコアから突き出ている。サブドメINXIIIは、リン酸化部位Thr249とAPEモチーフとの間に位置するP+1ループを含む。P+1ループは、触媒作用のためにペプチド基質の位置を決める。
【0022】
サブドメINXIIIの突出部は、基質認識、基質位置決め、および活性化体機構に影響を与えるMnkタンパク質中の位相転位に向いている。
【0023】
さらには、ATP加水分解に関連する残基とリン酸塩転移とは、タンパク質キナーゼ中で十分に不変である。触媒活性に関連する領域は、Lys113、Glu129、Asp205、およびAsn210を含む。本発明によって得られる構造データから確認できるように、結晶ヒトMnk−2タンパク質構造は、ATPまたは関連する化合物の影響を受けない。したがって、本発明による結晶は、不活性形態のヒトMnk−2タンパク質の結晶体にあって好ましい。本発明のMakキナーゼ中のDF(G/D)OUT立体配座と一致するDF(G/D)OUT立体配座p38が、一定の化学物質によって誘導されることが示されている(パーゲリス(Pargellis)等、nature structrual biology,vol.9,no.4(2002)268−272)。DF(G/D)OUT配座により、ATP結合クレフトをターゲットとする化合物に加え、ジアリール尿素阻害剤などの代替物質クラスの使用を包含する広範な薬理学的用途を伴う新規なアロステリック結合部位が提供される。さらには、DF(G/D)OUT配座の安定化により、酵素が阻害される。
【0024】
したがって、本明細書で提供されるデータにより、Mnk−2aのDFDモチーフが、基質のリン酸化が要求される際に、得られるATP結合、すなわちATP結合と不適合である配座を呈することができることが示される。すなわち、それは、非リン酸化Mnk−2が、ATPと結合できないか、あるいは、ATP結合を可能にするためにDFDモチーフ中での配座変化をまず生じさせなければならないことから生じる。Mnk−2の決定された配座は、該タンパク質(DFGモチーフに代わるDFDモチーフ)の特定の配列に起因して、その他全てのキナーゼとは異なる。この情報により、Mnk−2の阻害物質ならびに異性体、および非生産的DFD配座を認識して安定化するその他のタンパク質キナーゼを特定することができる。この上方により、Mnk−2の阻害因子ならびにアイソフォームおよび非生産性DFD立体配座を認識して安定化する、他のタンパク質キナーゼを特定することができる。また、これはMnk−2、Mnk−2キナーゼドメINそれぞれに特異な阻害因子を提供することができるが、他のキナーゼは認識しない。これは、DFGモチーフを呈するその他のキナーゼが異なる配列を有するために可能である。
【0025】
したがって、とりわけ、ATP結合ポケット(ここでは、DFDOUTポケットとも称する)ならびに他のポケット(ここでは、DFDINポケットとも称する)は、本発明による構造データによって決定することができる。活性形態において、ATPポケットは、ATPの結合部位を提供する。該ポケットは、特に、アミノ酸残記Glu129とAsp205、ならびにアミノ酸残記Lys113とAsn210によって定義される。認識され得る第2のポケットは、本発明により特に関心の対象となるものである。該第2のポケット、すなわちDFDINポケットは、DFDモチーフのPheが活性構造中で位置する部位である。不活性形態において、ATPポケットは、少なくとも部分的にDFDモチーフ、特にDFDモチーフのPheによって占められている。この不活性形態は、DFDINポケットを占有すること、特に、活性化セグメントまたはその他の分子、特に、阻害因子として機能する小さい分子でDFDINポケットを占有することによってロックすることができる。DFDINポエットを占有することにより、ATPが、この形態においては少なくとも部分的にDFDモチーフによって占有されているATPポケットにアクセスできないため、キナーゼ活性の阻害因子が作用する。DFDINポケットは、特に、アミノ酸残記Lue133、His203、Ile142、およびIle224によって定義される。該DFDINポケットをブロッキングすることによって、不活性構造がロックされる。したがって、本発明の主題は、前記ポケットを占有することができることにより、Mnkの選択的阻害因子の役割を果たす分子を提供することである。すなわち、そのようなDFDINポケット内へ結合することができる阻害因子が、本発明のその他の主題を提供する。Mnkにおいて、活性セグメント、特に活性化セグメントの挿入部12、より詳細にはアミノ酸残基Phe265は、DFDINポケットをブロックするため、適当な阻害因子は、例えば、小さい活性化セグメントの少なくとも部分的な配列を有するペプチドである。活性化セグメントは、アミノ酸Asp226〜Cyc275からなり、特に、アミノ酸263〜267まで延在する挿入部12を含む。したがって、Mnkの適当ペプチド阻害因子は、活性セグメントの配列または少なくとも4つ、特に少なくとも5つ、好ましくは少なくとも6つ、より好ましくは8つのアミノ酸を有するその連続的なフラグメントを有するペプチドである。そのような阻害因子の例とは、(258)APEVVEAFSEEA(269)または(260)EVVEAFS(266)である。
【0026】
本発明によって提供される、アロステリックな結合部位に対抗する阻害因子を提供する可能性により、さらには、顕著に改善された選択性を有する阻害因子が得られる。キナーゼのATP結合部位に対抗する標準的なキナーゼ阻害因子は、キナーゼの高い相互ホモロジーに起因した大きなクロス反応可能性を有する。したがって、ATP結合部位に対抗する阻害因子は、通常、わずかな選択性しか有さないため、強力に阻害して選択的阻害因子の発達を制限する。しかしながら、本発明によれば、Mnkのアロステリック結合部位における選択的な阻害因子の結合を提供することが可能となる。
【0027】
本発明により用いることが可能な阻害因子の1つは、BIRB796(パーゲリス(Pargellis)等の、nature structural biology,vol.9,no.4(2002),268−272)である。その他の阻害因子は、ジアリール尿素ベースの阻害因子(1−(5−tert−ブチル−2−メチル−2H−ピラゾール−3−イル)−3−(4−クロロフェニル)尿素)である。
【0028】
さらに、Mnkタンパク質は、Mnkタンパク質をその他のCAMKグループのキナーゼから区別する4つのシステINの不変クラスタを含むαFとαGとの間に挿入部を有する。分子のフレキシブルループ中のこれら4つのシステINクラスタは、亜鉛結合部位を形成する。そのため、この挿入部は亜鉛のフINガー状構造、タンパク質キナーゼの特異的なフINガープリントをマークする。さらに、4つの保存されたグリシンは、この挿入部中に存在しており(Gly297、Gly300、Gly304およびGly308)、これらは、このヘアピン状モジュールに折りたたむのに必要な領域にねじれフレキシビリティを与える。亜鉛フINガーモジュールは、変化を有する核酸またはタンパク質結合モジュールであることが知られている(クリシュナ(Krishna)等の、Nucleic Acids Res. 31(2)(2003)532−550)。このドメINは、その他のタンパク質、特に、基質や調整器のアダプタモジュールである。本発明による結晶ヒトMnk−2キナーゼはまた、変異体、好ましくは、天然Mnk−2キナーゼの少なくとも1つのアミノ酸、特に、少なくとも2つのアミノ酸が、別のアミノ酸で置き換えられたタンパク質を含んでなる。そのような変異体の結晶は、特に、機構学的研究ならびに結合ポケットの研究および配位子、基質あるいは阻害因子との相互作用を研究するのに遊離に用いることができる。この結果、適切に、結合能に対する相互作用または影響が推定あるいは予測される位置にある個々のアミノ酸を選択的に変化させる。この目的のため、例えば、20までの、好ましくは10までの、より好ましくは5までの、さらにこのましくは最大1つの突然変異を有する結晶ヒトMnk−2キナーゼは好ましいものとなり得る。結晶ヒトMnk−2キナーゼ変異D228Gが特に好ましい。さらに好ましい実施態様において、配位子、基質および/または阻害因子、特に、阻害因子スタウロスポリンとの組み合わせた結晶ヒトMnk−2キナーゼ変異D228Gが考慮される。
【0029】
好ましい変異体は、位置Asp226,Phe227またはAsp228で交換させたアミノ酸を有する。
【0030】
本発明は、さらに、表1に示す構造的配位の全てのまたは選択された部分で決定される三次元構造を有する結晶ヒトMnk−2タンパク質に関する。この表1に示す配位は、ここで説明する例のように得られたものである。
【0031】
一実施態様において、本発明は、同属のタンパク質キナーゼ阻害因子スタウロスポリンと共結晶されたヒトMnk−2−D228G変異体の結晶構造をさらに提供する。この構造において、DFG/DIN配座中へのDFGモチーフフリップにより、ATP結合ポケット内におけるその同属の結合部位においてスタウロスポリンを結合させることができる。この配座は表3に示す。阻害因子のいずれも有さない、特に、阻害因子スタウロスポリンを有さない、ヒトMnk−2キナーゼD228G変異体の結晶構造がさらに提供される。その配剤は表1aに示す。
【0032】
本発明による結晶ヒトMnk−2タンパク質の特徴は、例えば、
i.細胞、例えば大腸菌中におけるヒトMnk−2タンパク質の発現;
ii.Mnk−2タンパク質祖調製物を回収するための細胞を溶解すること;
iii.Mnk−2タンパク質祖調製物を、例えば、親和性タグクロマトグラフィーによって精製すること;および
iv.精製されたヒトMnk−2タンパク質を、例えば、拡散蒸着によって結晶化すること、
によって調製することができる。ヒトMnk−2タンパク質、特に、本発明による、ヒトMnk−2aタンパク質またはMnk−2bタンパク質、より好ましくは、ヒトMnk−2aタンパク質のキナーゼドメINの結晶化方法は、ヒトMnkタンパク質の結晶構造データの生成に特に用いることができる。特に、配位子、特に阻害因子や基質との結合部位または相互作用部位がそれによって得られる。さらには、タンパク質を活性または不活性形態に維持するために結合部位を特定することができる。さらに、ここで得られるMnk−2タンパク質の結果により、Mnk−1などのMnk−2のアイソフォームの配位子、特に阻害因子や基質もまた特定することができる。本発明による結晶化方法は、好ましくは単結晶であり、より好ましくは少なくとも1μm、さらに好ましくは少なくとも10μm、最も好ましくは少なくとも50μmの端長を有する結晶である。結晶は、X線構造解析を実施可能な方法で配置することが好ましい。したがって、本発明のその他の主題は、ヒトMnk−2タンパク質、特に、表1、表1aまたは表3に示す構造配座の全てまたは選択されたタンパク質によって決定されるヒトMnk−2aタンパク質の結晶構造である。好ましくは、不活性ヒトMnk−2aタンパク質の結晶構造が考慮される。結晶構造は、50Å以上の分解能、より好ましくは10Å以上、さらに好ましくは3Å以上の分解能を有することが望ましい。
【0033】
結晶ヒトMnk−2タンパク質と結晶構造のそれぞれを用いることにより、Mnk−2タンパク質配位子を設計、同定または調製することができる。さらには、タンパク質キナーゼ、ならびに上述したようにMnk−2のアイソフォームのタンパク質キナーゼの調節機構を同定することができる。配位子または調節機構を同定するために、特にコンピュータ支援モデル化プログラムが用いられる。
【0034】
適した配位子は、例えば、ヒトMnk−2タンパク質の相互作用部位と相補的な三次元構造を有する分子を作製することによって同定することができる。特に好ましい配位子は少なくともアミノ酸Asp226,Phe227,Asp228の少なくとも1つと相互作用する。さらに好ましい配位子は、少なくとも1つの原子が、DFDモチーフの原子のいずれかまでの所定の距離内、好ましくは7Åの距離内、さらに好ましくは6Å、特に5Åの距離内にある少なくとも1つのアミノ酸と相互作用する。
【0035】
配位子を同定するためのコンピュータ支援スクリーニングによれば、WO03/037362号公報に開示の方法が、実際に配位子を同定して照合するのに好ましく適用される。
【0036】
表1、表1aまたは表3で与えられるヒトMnk−2タンパク質の結晶構造の構造的配位はまた、ヒトMnk−2タンパク質の結晶構造の三次元表現を作成するのに用いることができる。そして、そのような三次元構造に形成された相互作用ポケットは、それらの三次元構造により、対応する配位子を同定するのに用いることができる。
【0037】
表1、表1aまたは表3に示される、本発明によって提供される構造的配位は、その他のタンパク質の結晶構造を決定するのにも用いることができ、それにより、構造的配位は分子置換に用いられる。
【0038】
ここで提供するデータは、コンピューターで読み取り可能な保存媒体に保存し、適当に提供することが好ましい。
【0039】
本発明は、さらに、Mnk−2タンパク質のアイソフォーム、ならびに結晶化法または結晶構造を用いて得たその他のタンパク質キナーゼの配位子、特に基質または阻害因子に関する。そのような配位子は、好ましくは、薬剤組成物中における活性剤である。そのような薬剤組成物は、処置、特にMnk−2タンパク質の阻害が必要な、例えば、肥満、糖尿病およびメタボリック症候群ならびに癌などの代謝障害などの病気を治療するのに用いることができる。さらに実施態様において、本発明は、結晶ヒトMnk−1タンパク質に関する。
【0040】
Mnk−1キナーゼ領域(Mnk−1−KR)の結晶構造は、Mnk−2−KR異なる立体配座を取るが、触媒ドメINのアミノ酸配列は78%一致する。Mnk−1とMnk−2の構造データの組み合わせにより、Mnkサブファミリーメンバーの活性化に不随する機械的事象のダイナミックな像を描くことができるようになる。
【0041】
また、この実施態様においては、ヒトMnk−1タンパク質の突然変異体は、特に、交換させた少なくとも1つの、特に少なくとも2つのアミノ酸を有する突然変異体に包含される。上記で説明したように、そのような突然変異体は、機構学研究に特に用いることができる。好ましくは、突然変異体は≦20、より好ましくは≦10、さらに好ましくは≦5、最も好ましくは最大1の交換アミノ酸を有する。Mnk−1の場合における交換アミノ酸の好ましい部位は、位置Arg90またはArg93、およびArg191,Phe192またはArg193である。
【0042】
本発明は、N末端ローブ、マグネシウム結合ループ、および活性化セグメントが、徹底的な構造転位を受け、完全活性状態まで自動阻害されることによって得られるMnk活性化モデルにも関する。本発明のさらなる態様は、活性化セグメント介在の官能要素の位置移動によって自動阻害を得るためにMnkを用いることである。
【0043】
その他のタンパク質キナーゼの多くに見られるそのカノニカルな立体配座において、活性化セグメントのC末端部分は折り返され、ショートへリックスα−EFと基質結合P+1ループは、へリックスαF,αGおよび触媒ループによって与えられる環境中でキナーゼコア内に埋没する(ナイトン(Knighton)等の、Science(253)(1991)414−420;ノレン(Nolen)の、Mol. Cell(15)(2004)661−675)。
【0044】
しかしながら、Mnk−1−KRにおいては、α−EFは解かれてペプチド結合溝内へ崩壊するため、N末端ローブ配座と活性部位残基が変化する(図9)。特に、αCへリックス(Arg90:Glu225;Arg93:Glu228)との相互作用が引き上げ力を与えて、αCとNローブの残部を置き換え、それによりローブは終了する(図9A)。相互作用残基Arg90とArg93に対応する残基は、活性状態のタンパク質キナーゼ中のリン酸部分に結合することが知られている(クリュパ(Krupa)等の、Mol. Biol.(339)(2004)1025−1139)。そのため、再構築された活性化セグメントは、活性部位の配座を変更する分子スイッチとして機能するよう予定されている。ナンバリングは、オログレン・エー(O‘Loghlen, A.)、ゴンザレス・ヴィー・エム(Gonzlez,V.M.)、ピネイロ・ディー(Pineiro,D.)、プレッツモルガード・エム・アイ(Prez−Morgado,M.I.)およびマーテIN・エム・イー(Martin,M.E.)(2004)、Mnk1bの同定および分子特徴付け、ヒトMAPキナーゼ相互作用キナーゼMnk1のスプライス異体、ExpCellRes299,343−355に相当する。
【0045】
この活性化セグメントの「ウェッジ」形態のさらなる効果は、リン酸部位の到達可能性を促進し得る、活性化ループをさらすことである。ローブの終了は、Mnk−2構造によって示すように、活性化セグメントと調節Cへリックスとの間の相互作用が途絶える場合に逆戻りさせることができる(図9B)。Mnk−2において、分子本体から突出しているため、活性化セグメントは全く異なる形態を取る。その結果、活性化セグメント:Cへリックス相互作用は緩和されてNローブは突然回復する(図9B)。
【0046】
Mnk−1と比較した場合、Mnk−2のNローブは、おおよそ10°傾き、キナーゼのATP結合口を開く(図9C)。
【0047】
Mnk−1の延長、再構築された活性化セグメントは、その他のCAMKグループメンバーのほとんどには存在しない2つのアミノ酸挿入部を有する。
【0048】
内部ローブ溝における活性化セグメントの新規な配置の結果、Phe230は、Leu98とThr97出発配座αC,Cループの上流のHis168,Ile107,Ile189およびLeu161によって与えられる構造的に保存されたポケット内に存在することになる(Mnk−1アミノ酸残基のナンバリングは、EntrezEntryCAl14764に対応する)。
【0049】
後者のポケットが、活性状態のキナーゼにおいてDGG/Dモチーフのフェニルアラニンの結合部位として機能することが発見された(図10)。この活性DFG/Dモチーフ配座は、DFG/DIN配座と見なされ、その対応する結合部位は、今後、DFG/DINポケットと称する。Mnk−1においては、しかしながら、Phe230のDFG/DINポケットにおける存在は、DFG/Dモチーフのアクセスを制限し、ATP結合部位を立体的にブロックする阻害DFG/DOUTを誘導する(次の段参照)。そのため、再構築された活性化セグメント、特にPhe230は、Mnk固有の調節機構の重要な役割を果たす自動阻害要素を構成する。
【0050】
これまで、自動阻害戦略という意味でのDFG/DINポケットの障害は、c−KITおよびFlt3に見られ、これら2つはタイプIIIレセプターチロシンキナーゼと非常に関連性が高い。c−KITおよびFlt3は、DFG/DOUTの誘導により「in trans」で自動阻害するキナーゼのN末端に位置する傍膜(juxtamembrane;JM)ドメINを含む(グリフィス(Griffith)等の、Mol.Cell(13)(2004)169−178;モル(Mol)等の、J. Biol. Chme. (279)(2004a)31655−31663)。いずれのケースにおいても、JMドメINから出発する残基(Flt3中のLeu576およびc−KIT中のTrp557)は、DFG/DINポケット内へプランジされ、これはDFG/Dモチーフを強制的に阻害DFG/DOUT配座にする(図10A、図10B)。その結果、Mnk−1は、c−KITとFlt3と類似するが、異なる構造要素を利用する自動阻害機構を達成する(図10C)。Mnk−1は、JMドメINの代わりに、その活性が静まるようその再構築された活性化セグメントとPhe230とを採用し、これは、c−KITとFlt3内で、DFG/DINポケットの関与が維持されるよう生じる(図10C)。
【0051】
また、Mnk−1の場合、本発明によって提供されるデータにより、DFDINポケットの定量が可能となる。このポケットは、上述したように、特に、Leu98とThr97、His168、Ile107、Ile189とLeu161によって決定される。上記DFDINポケットを用いることにより、DFDモチーフは、少なくとも部分的にATPポケット内に位置されるため、MnkによりATP結合を阻害することができる。そのため、DFDinポケットをブロックすることにより、キナーゼ活性が阻害される結果となる。したがって、本発明のさらなる態様とは、DFDinポケットに結合し、それによりMnkを阻害する分子を提供することである。活性化セグメントによるMnk−1の自動阻害が、特に、活性化セグメントのDFDINポケット内へのl2挿入部のPhe230の位置によって生じるため、適切な阻害因子は、アミノ酸191〜240からなるMnk−1の活性化セグメントの全配列または部分的配列からなり、特に、アミノ酸228〜232からなる挿入部l2の配列からなる。適切なペプチドは、例えば、(223)APEVVEVFTDQA(234)または(225)EVVEVFT(231)である。
【0052】
タンパク質キナーゼの広大な大部分は、インターローバルクレフトにおいてタンパク質キナーゼのATP結合「口」の「唇」を形成する、活性化セグメント(サブドメINVII)の開始時にAsp−Phe−Gly(DFG)モチーフを担持する(ハンクス(Hanks)の、Genome Biol.(4)(2003)111;ハンクスの、Science(241)(1988)42−52);テイラー(Taylor)の、Structure(2)(1994)345−355)。このモチーフの第1のアスパラートは、触媒活性タンパク質キナーゼ間で不変であり、リン酸転位に必須のマグネシウムイオンと配位することが知られている(図11A)(アダムス(Adams)の、Chme. Rev.(101)(2001)227102290)。そのため、DFGモチーフはマグネシウム結合ループと称される。
【0053】
しかしながら、Mnkは、配位位置でAsp−Phe−Asp(DFD)モチーフを担持する。DFG/DINポケットがPhe230でブロックされた結果、Mnk−1のDFG/Dモチーフは阻害DFG/DOUT配座に適合する(図11B)。DFDモチーフは、活性状態のタンパク質キナーゼのDFG/DIN配座に対して、Asp191のΦ角度(ΦAsp191=−120)について〜180°だけ回転される(例えば、DAPK1ΦAsp161=55°、図11A)。その結果、Phe120は、Val63、Leu108、Phe124(ゲートキーパー残基)および、Lue177で与えられる、通常、ATPのアデノシル部分を受容する疎水性ポケットを占める。DFG/DOUT配座は、Mnk−2に関する説明において上述した。
【0054】
すなわち、Mnk−1とMnk−2の両方がこの特徴を示すため、DFG/DOUTの適合は、不活性Mnkキナーゼの初期(default)状態であり、活性DFG/DIN配座を呈するその他のSer/Thrキナーゼの多くと、それらの縛られない配座において区別される。
【0055】
Mnk−1において、DFDモチーフは、DFG/DOUT配座に対する選択性を説明するイオンネットワークに関与する。不変Asp191とMnk固有Asp193の両方は、活性部位残基との緊密な酸−酸側鎖相互作用に関与する(図11B)。
(i)Asp191はGlu94に結合し;
(ii)Asp193はAsp170に結合する。
【0056】
Glu94とAsp170は、触媒活性タンパク質キナーゼ間で不変の残基に対応する(ハンクスの、Science(241)1988)42−52。Glu−94は、調節へリックスαCから出発し、ATP結合の生成に必要なLys89−Glu94とイオン対を形成することが知られている(アダムスの、Chem. Rev.(101)(2001)2271−2220)。この対形成は、DFG/DモチーフのAsp191がGlu94と(OD−Asp191:OE−Glu94)さらには、Lys78(O−Asp191:Nz−Lys78)と相互作用するため、Mnk−1中で妨害される。Asp193と相互作用するAsp170は、Cループの触媒アスパラートに相当する。
【0057】
酸−酸側鎖相互作用は一見して異常に見えるが、酸側鎖間の相互作用は、タンパク質構造中で観察される場合が多々あり、酵素の触媒中心内で特に多い(フロッコ(Flocco)の、J. Mol. Biol.(254)(1995)96−105)。その結晶化条件のpH(pH5.6)は、これら相互作用の安定化に都合良いが、これらは塩基性環境においても観察され、局所的なpKaの強い変動を示唆する(同じくフロッコ等)。2つのカルボン酸基間のO−O距離は、おおよそ2.6Åと2.5Åにあり、これは、非酸性水素供与/アクセプター対間のO−O距離よりも著しく短い。後者の観察は、陽子共有結合モードによるものである(同じくフロッコ等)。その他のそのような酸−酸相互作用を説明すると、Asp191:Glu94ならびにAsp193:Asp170が、アミン(Lys78)またはアミド(Asn175)それぞれによって安定化される(比較として、例えば、(ワーテン(Werten)の、J. Biol. Chem.(277)(2002)45502−45509)参照)。
【0058】
活性化セグメントはタンパク質キナーゼドメINの構造要素を具体化し、強い立体配座的可塑性を示し、上流部調節によって構造的に修飾される場合が多々ある(ヒューズ(Huse)の、Cell(109)(2002)275−282)。タンパク質キナーゼの多くにおいて、活性化セグメントのフレキシブル部分は、活性化ループと呼ばれるストレッチに制限されており、DFG/Dモチーフ間に位置するため、P+1ループと呼ばれる(ノレン(Nolen)の、Mol. Cell(15)(2004)661−675)。P+1ループは、基質ペプチドのリン酸化部位に隣接する残基と相互作用することが知られているため、基質ペプチド配置において重要な役割を果たす(ナイトン(Knighton)の、Science(253)(1991)414−420)。
【0059】
Mnkサブファミリー内では、しかしながら、活性化セグメントは、2つのアミノ酸挿入部によってその他のCaMKグループキナーゼに対して延長され(図8)、立体配座的可塑性にさらされるストレッチは強く拡張される。フレキシブル部分は、活性化ループだけでなくP+1ループも含むため、該領域はショートへリックスα−EFとα−EF/αFループに対応する。活性化セグメント、P+1ループとα−EFの両方は、報告されたタンパク質キナーゼ構造の非常に大部分の保存された部位を占める。Mnkにおいては、しかしながら、この領域は折りたたまれておらず、Mnk−1とMnk−2との間で異なる、延長された形態に適合する。そのため、Mnkサブファミリー内の活性化セグメントは拡張され、マグネシウム結合DFDモチーフ(他のキナーゼではDFG)からへリックスαFまでの領域に広がるフレキシブル45アミノ酸ストレッチを包含する(図8B)。
【0060】
自動阻害は、タンパク質キナーゼ制御の卓越した戦略であり、個々のケースにおいて別個に導入され、分子の様々な機能部位に影響を与え得る。タンパク質キナーゼのコアの外側の領域に位置する調節ドメINは、例えば、CaMKI(ゴールドバーグ(Goldberg)の、Cell(84)(1996)875−887)、Twitchin(Kobe,Embo J.(15)(1996)6810−6821)、およびc−KIT(Mol,J.Biol.Chem.(279)(2004a)31635−31663)において採用される。
【0061】
c−KITとFlt−3のケースの場合、2つのタイプIIIレセプターチロシンキナーゼ、DFG/DOUT配座の誘導によりN末端JMドメINは自動阻害するため、ATP結合をブロックすることができる。Mnk−1は、同様に、DFG/DOUT配座の誘導により自動阻害される。しかしながら、c−KITとFlt3とは対照的に、JMドメINが自動阻害を「in trans」で緩和する場合、Mnk−1は、再構築された活性化セグメントを介してDFG/DOUTを誘導し、Phe230をDFG/DINポケット内へ挿入し、これは通常DFG/D−Pheを適応する。そのため、Mnk−1の活性化セグメントは、c−KitとFlt−3のJMドメINのアナロジーにおいて内部自動阻害ドメINとして機能する。Mnk−1とMnk−2の構造は、タンパク質キナーゼ調節のためのDFG/Dモチーフの重要性を強調する。今日では、DFG/DOUT配座は、〜50タンパク質キナーゼのフラクション中でのみ観察されており、その構造的データは入手可能である。
【0062】
留意すべきは、タンパク質キナーゼ阻害因子の開発のため、DFG/DOUT配座は重要である。ある小さい分子阻害因子キナーゼ錯体構造において、DFG/DOUT配座は安定化されおよび/またはBirb796:p38(パルゲリス(Pargellis)の、Nat.Struct.Biol.(9)(2002)(2002)269−272)、Cleevec:c−Abl(ナガール(Nagar)の、Mol.Cell(15)2004(661−675)、およびAAL−993:VEGFR−2(マンレイ(Manley)の、Biochem.Biophys.Acta(1679)(2004)17−27)などに誘導され、それにより、酵素が活性化される。Mnk−1とMnk−2の構造は、DFG/DOUTの適合性が、キナーゼ調節の共通する戦略であるということを証明するものであり、一定の系統グループに制限されるものではない。
【0063】
本発明はさらに、4つの状態を含むMnk活性化のモデルに関連する。
(I)阻害された状態;
(II)中間状態;
(III)プライム化状態;および
(IV)活性状態(図12)。
【0064】
理論で縛られるのを望むことなく、状態IとIIは、Mnk−1とMnk−2の構造にそれぞれ示されており、状態IIIは、Mnk−2の突然変異構造に基づいてモデル化することができ、その他の活性状態のキナーゼと状態IVは仮説である(図12)。配列的な相互転換には、活性化セグメント、NローブおよびATP結合部位に影響を与える明白な位相的転位が要求される。状態Iの特徴は、DFG/DOUT配座の誘導であり、さらに再構築された活性化セグメントの新規な配置によって誘導されるローブクロージャとαC置換である。状態IIは、活性化セグメントの隆起によって可能となり、主にへリックスαCの再配置によってローブ内クレフトを開放し、その結果、必須Lys−Gluイオン対を形成する。しかしながら、状態IIは、不活性状態のキナーゼ、例えば、DFG/DOUT配座のいくつかの特徴を示すため、さらなる構造的再配置を必要とする。
【0065】
活性化セグメントの内部スイッチは、例えば、触媒ベースアスパルテート(Asp−170)の、P+1ループ(Ser218)からのSer/Thr残基との相互作用など、保存された分子内接触の形成を可能にするのに必要である。さらに、マグネシウム結合ループは、DFG/DINポケット内へスイッチし、Mnk−2突然変異構造に見られるようにATPポケット阻害を除外しなければならない。Mnk−2の構造は、さらに、Mnkが、同属のα−EFへリックスとP+1ループ、Mnk−1中で完全に解かれる領域を折りたたむ可能性の損失がないことを証明している。上流キナーゼ(同じくワスキーウィッツ(Waskiewicz))によってターゲットとされる2つのリン酸化部位を担持するMnkの活性化ループは、多くのキナーゼで見られるようにリン酸化によってほぼ安定化される(ジョンソン(Johnson)の、Cell(85)(1996)149−158;ノレン(Nolen)の、Mol.Cell(15)(2004)661−675)。
【0066】
その他の例と同様の、主要なリン酸化は、塩基性RDポケットと相互作用させることによって活性化ループ形態を安定化させることができ、それにより、中性化され、RD−ArgとAsp−238(Mnk−2においてはAsp−273)の相互作用を中断し、これは、活性化セグメントのより先端部分の開放形態を不安定化させる。続いて、二次的リン酸化により、活性化ループ形態がさらに変化されて、状態Iに類似しているが、阻害DFG/DOUT配座のない、ATP結合クレフトに近いGlu−Lysイオン対の誘導によってローブクロージャが誘導される。その結果、新たに誘導された二次P部位の陰電荷は、ウェッジ状態のGlu225とGlu228のために、Arg90およびArg93などの塩基性残基と置換基との相互作用による引き上げ力を提供し、その場でαCへリックスを保持した。要するに、状態II/IIIおよび/またはIII/IVとの間における転換にはリン酸化が必要である。第一リン酸塩は、例えば、RD−Argとの相互作用によって処理された状態IIIを安定化し、第二リン酸化により、活性化セグメント形態を受容する基質をさらに安定化し、αCへリックスとの相互作用によってローブの再封鎖を促進する。
【0067】
へリックスα−EFとP+1ループは、Mnk−1内で解かれて活性化セグメントの完全な再構築物となる。Mnk−1は、いくつかのレベルに対して自動阻害される。活性化セグメントは、2つの構造的変化のクロストーキングシリーズによるこの不活性を必要とする。第一に、これは、DFG/DOUT配座の誘導によるATPポケット遮断を誘導するため、間接的にLys−Glu対とN−ローブとを伝達させる。第二に、これは、へリックスαCとの相互作用によりN−ローブの偽活性閉鎖形態を誘導する。
【0068】
本発明のMnk−1構造は、したがって、キナーゼアーキテクチャの新規な態様と、合理的な阻害設計に使用可能な制御を明らかにする。
【0069】
特に好ましくは、本発明は、結晶ヒトMnk−1タンパク質キナーゼに関する。Mnk−1は、真核細胞阻害因子4E(elF4E)のリン酸化による翻訳機構をターゲットとするヒトタンパク質キナーゼである。
【0070】
本発明はまた、表2に示す構造配位の全てまたは選択された部分によって定義される三次元構造を有する結晶ヒトMnk−1タンパク質に関する。表2に示す配位は、ここにおける例で説明するように得られたものである。
【0071】
【表1】

【0072】
本発明による結晶ヒトMnk−1タンパク質調製物は、例えば、以下によって調製できる。
i.例えば大腸菌などの細胞中でヒトMnk−1タンパク質の発現;
ii.粗Mnk−1タンパク質調製物を回収するために細胞を溶解する;
iii.粗Mnk−1タンパク質調製物を、例えば、タグクロマトグラフィなどにより精製する;および
iv.例えば核酸蒸着などにより、精製したヒトMnk−1タンパク質を結晶化する。
【0073】
ヒトMnk−1タンパク質の結晶調製物、特に、本発明によるヒトMnk−1タンパク質のキナーゼ領域は、ヒトMnkタンパク質の結晶構造データの生成に用いることができる。特に、阻害因子や基質などの配位子との結合部位または相互作用部位、がそれによって得られる。さらには、タンパク質を活性形態または不活性形態に維持するための結合部位を特定することが可能である。特に、ここでMnk−1に対して与えられる結果により、配位子の同定が可能となる。
【0074】
本発明による結晶調製物は、単結晶であることが好ましく、より好ましくは、少なくとも1μm、さらに好ましくは少なくとも10μm、最も好ましくは50μmのエッジ長さを有する結晶であることが好ましい。結晶は、X線構造解析を行えるようなに配置されていることが好ましい。したがって、本発明のさらなる主題は、表2に示す構造配位の全てまたは選択された部分で定義されるヒトMnk−1タンパク質の結晶構造である。
【0075】
結晶ヒトMnk−1タンパク質と、結晶構造とをそれぞれ用いることにより、Mnk−1タンパク質配位子を設計、同定または調製することができる。また、上述のようにタンパク質キナーゼの制御機構を特定することもできる。配位子または制御機構を特定するために、特に、コンピュータ支援モデル化プログラムを用いる。
【0076】
配位子を同定するコンピューター支援のスクリーニングに加えて、WO03/037362号公報に開示される方法を実際の配位子同定および確認に適用することが好ましい。
【0077】
本発明はさらに、特に、Mnk−1タンパク質のアイソマー、ならびに結晶調製物や結晶構造を用いて得られたその他タンパク質キナーゼの基質や阻害因子などの配位子に関する。そのような配位子は、薬剤組成物中で活性剤であることが好ましい。そのような薬剤組成物は、Mnk−1の操作または特に阻害が必要な、例えば、肥満、糖尿病およびメタボリック症候群ならびに癌などの代謝障害の場合などの病気の治療に用いることができる。
【0078】
ここで提供する結果およびデータは、DFG/DINポケット(Mnk−1中のPhe230を含む)が、汎用的な阻害因子結合部位として機能可能であることを示している。阻害因子は、Mnkに制限されない。したがって、阻害因子はまた、DFG/Dinポケットからなる阻害因子結合部位に関する。
【0079】
本発明はさらに、添付の図面ならびに以下に与えられる例によって例示される。
図1:Mnk2の組織と配列アライメント
(A)(ラベルを付したように)機能ドメINの配置を示す、ヒトMnk2の2つのスプライス変異体の概略的な比較図。ここで調査する領域(Mnk2キナーゼ領域、Mnk2−KR)は四角で囲まれている。代替的なスプライスは、N末端にもキナーゼドメIN、NLS核局在信号、elF4G真核細胞阻害因子4G、翻訳阻害複合体の足場タンパク質に影響を与えず、Mnk1とMnk2に結合する(1999年のパイロネット(Pyronnet)等、2001年のシェーパー(Scheper)等)
(B)ヒトMnk1とMnk2のキナーゼドメINの配列アライメント、DrosophilaおよびC.elegansMnk遺伝子座(Lk6とR166.5のそれぞれ)および既知構造の3つのヒトCaMK群キナーゼ(MAPKAP−MAPキナーゼ活性化タンパク質キナーゼ)Mnk2のナンバリングは、最近報告された配列に基づく(2000年のスレンツ−ケスラー(Slenz−Kesler)等)。Mnk2−KR中に見いだされた二次構造要素は、以下の配列に示される。星印は、リン酸化部位を示している(2001年のシェーパー(Scheper)等)。触媒ループ(i)DFDモチーフ(その他のキナーゼ中のDFG);と(ii)P+1ループとは、(iii)着色されたバーでマークされている。Mnkの挿入部の特徴は四角でかんでいる(l1−l3)。白抜きの円は、グリシンリッチループのGly91とGly93、ATP結合に重要であることが知られているLys113とGlu129(1994年のテイラーとラッジオ−アンゼルム(Taylor and Radzio−Andzelm))、黒塗りの円は、N末端ローブとC末端ローブを分離するヒンジ領域のGly163とGly165を示す。
図2: Mnk−2キナーゼの全位相。キナーゼドメINのコアの外側の構造的部分は削除された。CAMK1(a,1a06.pdb)、DAPK1(b,1jks.pdb)およびMAKPKAPK2(c;1kwp.pdb)のアポ酵素の構造は、Mnk−2(b)上に重ねられ、同様の配向上に示されている。電子密度でトレースできない部分は点線で示されている。
図3:活性化セグメントの開放形態。赤と青で着色された2つの対称性等価Mnk−2分子は、(a)に示される。同分子は、90°だけ回転された、上から示されている。(c)は、1σで着色された2Fo−Fc電子密度とDAPK1の同じ領域(黒)を示している。
図4:ATP結合ポケットの形態。Mnk−2(青)、MAPKAP2(赤)、CaMk1(緑)およびDAPK1(黒)からの触媒作用の重要性を有する領域が示されている。(a)は、Ly113とGlu129(Mnk−2ナンバリング)を示す。(b)において、Cループのバックボーンと、Asp205とAsn210の側鎖とは、MAPKAP2/ADP共構造(1ny3.pdb)由来のADP(黄)と一緒に示されている。(c)は、DFG(DFD)モチーフ周囲のバックボーンを示しており、(d)はこの領域と(b)由来のADPの側鎖を包含している。
図5:亜鉛結合部位。(a)Mnk−2中の推定状の亜鉛部位の領域が、1σ(青)で描かれた2Fo−Fcマップと、5σで描かれたDANOマップとともにバックボーンプロットとして示されている。領域は、我々の結晶中で高度にフレキシブルであり、Trp305からGlu309までの領域は鮮明なバックボーン密度を欠いている。(b)I=ZnKα線、II=ZnKベータ、IIIコンプトン散乱、IV弾性散乱に対応するピークを有する、天然Mnk−2結晶のX線放射スペクトル。
図6:Mnk−2キナーゼドメINとp38との比較
A.同じ配向のMnk−2キナーゼドメIN(左)とp38(右;PDB ID1KV1)のリボンプロット。分子はN末端(青)からC末端(赤)まで七色に着色されており、その他のタンパク質キナーゼでも観察されるように、全て同じ構造的構成を示している。
B.2つのタンパク質の最適グローバルアライメント後のDFD/DFG領域の空間プロット。Mnk−2−石灰;p38−コムギ。DFD/DFGモチーフは、スティック形状で示されており、原子型でカラーコードされている(炭素(Mnk−2)−石灰;炭素(p38)−コムギ;酸素−赤;窒素−青)。Mnk−2のアスパルテート226と228は、ポリペプチド鎖の方向を示すようラベルを付されている。周囲を取り囲む構造要素はリボンで示されている。p38の非定型型DFG形態は、尿素ジアリール型の阻害因子の結合によって誘導される(図示せず;PDB ID1KV1と1KV2)。Mnk−2は、本発明の結晶中で自然に同じ形態に適合する。阻害因子の尿素ジアリールクラスはp38のDFGモチーフとへリックスとの間で結合し、へリックスはバックグラウンド中に表れた。Mnk−2のDFDモチーフは、阻害因子結合ポケットに向かってさらに置き換えられ、阻害因子によって本発明の形態中に同様に捉えることができることを示唆している。
図7:Mnk−2キナーゼドメINに結合する阻害因子のモデル
A.ジアリール尿素ベースの阻害因子(1−(5−tブチル−2−メチル−2H−ピラゾール−3−イル)−3−(4−クロロフェニル)尿素;BMU;PDB ID 1KV1)との錯体であるMnk−2キナーゼドメINの外観。Mnk−2は、その二次構造要素(へリックス−赤、ストランド−青、ループ−グレイ)に従って示される。阻害因子1(炭素−オレンジ)、DFDモチーフ(炭素−ピンク)とその他の薬(炭素−シアン)に接触するMnk−2残基は、スティック形状で示される。モデルは、p38−BMU複合体(PDB ID1KV1)のCα原子位置と、Mnk−2キナーゼドメINのCα配位との最適なスーパーポジションによって作製される。MNU配置は、続いて、手動により示されたMnk−2キナーゼドメINの結合ポケットに適合させた。いくつかのMnk−2残基の側鎖形態も同様に不良接触を排除するよう適合させた。
B.Mnk−2キナーゼドメINのヌクレオチド結合ポケット中に配置されたDAPK1(PDB ID 1lG1)の共晶構造由来の、ATPアナログ(AMPPNP)の詳細な空間図。モデルは、Aで説明したような2つのタンパク質分子の最適なスーパーポジションによって作製された。標準結合モデル中のAMPPNP分子が、本発明の形態のMnk−2のDFDモチーフと立体的に干渉するように見える。この発見は、生産的なMnk−2へのATP結合がDFDモチーフ内での再配置を必要とすることを示唆する。その結果、本発明の形態において、Mnk−2は、ATP結合内において不活性であることが導き出される。別の分子とモチーフは、原子型ごとにカラーコードされている。炭素(AMPPNP)−オレンジ;炭素(DFD)−ピンク;窒素−青;酸素−赤;亜リン酸−石灰。A中に二次構造要素。
C.Mnk−2−BMU複合体モデルの詳細な空間図。BMUは、疎水性ポケットでt−ブチル基と結合し、Phe227(DFDモチーフ由来)とPhe159の芳香環の間のpクロロフェニル環をスライドさせる場合がある。別の分子とモチーフは、原子型ごとにカラーコードされている:炭素(BMU)−オレンジ;炭素(DFD)−ピンク;炭素(疎水性ポケット)−シアン;窒素−青;酸素−赤;塩素−緑。A中のような二次構造要素。
図8:立体的表現(a)と主要配列(b)におけるMnk−1の全構造。他に示さない限り、着色法は以下のスタイルに維持される。Nローブ:グレイ;Cローブ:黒;Cループ:黄;DFG/Dモチーフ:オレンジ;αCへリックスとLys−Glu対:シアン;活性化セグメント:緑。(b)ATPと相互作用すると知られている残基をカラーコードでマークし、DFG/Ginポケットからなる残基:緑の白抜きの円;DFG/DOUTポケットからなる残基:赤の白抜きの円。Mnk固有のアミノ酸挿入部は、四角で囲まれ、官能性の適合性の高いMnk固有残基は、赤い矢印でハイライトしている。リン酸化部位は星印で示している。
図9:活性化セグメントにより誘導されたNローブ移動。Mnk−1(a)とMnk−2(b)の全構造。残基を含むMnk−1(a)は、NローブαC相互作用、Phe239とDFDモチーフに包含された。スティックで表されたPhe。Arg90とArg93は、亜リン酸アミノ酸と相互作用することが知られている残基に対応した(クリュッパ(Krupa)等の、J. Mol. Biol.(339)(2004)1025−1039)。Mnk−2(b)中で対応する残基は、Phe265、Arg123とArg125である。
図10:c−KIT(a,b)とMnk−1(C)における自動阻害c−KITの自動阻害JMドメINは赤で着色されている。
図11:(a)DAPL1(1ig1;(テレスコ(Tereshko)等の、Nat.Struct.Biol.(8)(2001)899−907));(b)Mnk−1;Mnk−2のATP結合ポケット。分子は図8のように同じ配向にあり、ATP結合領域が拡大されている。(a)は活性状態のCaMK群のタンパク質キナーゼを例示し、非クリーブ化ATPアナログANP−PNPと、Mg2+の代わりに官能部位においてMn2+を含む。マグネシウム結合DFGモチーフの許容可能なDFG/Dinポケット形態に留意。Mnk−1(b)とMnk−2のATP部位遮断は、阻害DFG/DOUT配座によって得られる。Mnk−1(b)は、酸−酸側鎖の相互作用がMnk−2中に存在しないことを示している。
図12:Mnk活性化カスケードモデル。
図13:DFDモチーフの近傍
(A)DFD領域とATP結合クレフトの拡大空間図。野生型Mnk2−KRのDFG/DOUT配座は、左上にAsp226、Phe227とAsp228に関して、Phe227とAsp228とがATP結合クレフト内に突き出した状態でスティックにより示されている(炭素、シアン)。DFG/DIN配座(右下;炭素、緑)は、Mnk2−KRのAsp228Gly突然変異体について観察されるように、その他のキナーゼに見られるDFG/DIN配座に従ってモデル化された。Mnk2−KRのバックボーントレースは、半透明なグレイのチューブで示される。DFG/DINまたはOUT配座いずれかにおけるDFDモチーフ周囲の径4Å内の残基は、スティックで示されている(炭素、グレイ)。DFG/DOUT配座を安定化する、タンパク質マトリックスとの直接的な相互作用は、点線で示されている。Phe227は、2つの異なる疎水性ポケット中に2つの異なる配座で存在するようになる。DFG/DIN配座の適合に対する障害は見えない。
(B)((A)におけるのと同様にカラーコードされている)スティックで示されたDFDモチーフの2つの配座をともなう、静電ポテンシャルごとにカラーコードされたMnk2−KRの分子構造の空間図(青、正電荷;赤、負電荷)。ATP結合クレフトが指摘されている。いずれかの配座にあるAsp228は、水溶性溶媒を良好に利用可能である。DFG/DOUT配座は、ATP結合クレフト中にPhe228とAsp228を位置するだけでなく、このクレフトの前からのアクセスを遮断する。分子は、DFDポケット中への遮断されていない図を得るために、水平軸(N末端ローブから後ろ)について、(A)に対して30°回転されている。
(C)DAPK1との共晶構造(PDB ID 1IG1)に見られるようにスーパーINポーズされた、非加水分解性のATPアナログ(アデノシン5’−[β,γ−イミド]三リン酸[AMPPNP]);炭素、ベージュ、亜リン酸、紫)による(A)におけるのと同じ図。DFG/DOUT配座において、アデノシン塩基はPhe227の側鎖とクラッシュし、リン酸基はAsp228の側鎖とクラッシュする。
(D)DFG/DOUT配座のみが示された、(A)と(C)におけるのと同じ図。p38−BMU阻害複合体(PDB ID 1KV1)のDFG領域は、タンパク質構造のグローバルスーパーポジション後に見られるように、比較目的で示されている(マジェンタ色のチューブ;スティックで示されたDFG;炭素、マジェンタ色)。BMU阻害因子(炭素、ベージュ;塩素、緑)は、DFG/DIN結合ポケットの部分をふさぎ、p38中にDFG/DOUT配座を誘導する。
表1:ヒトMnk−2の原子配位を示す。
表1a:ヒトMnk−2突然変異D228Gの原子配位を示す。
表2:ヒトMnk−1の原子配位を示す。
表3:ヒトMnk−2キナーゼ突然変異D228Gと一般的なタンパク質キナーゼ阻害因子スタウロスポリンとの共結晶の原子配位を示す。
【図面の簡単な説明】
【0080】
【図1】図1は、Mnk2の組織と配列アライメントを示している。
【図2】図2は、Mnk−2キナーゼの全位相を示している。
【図3】図3は、活性化セグメントの開放形態を示している。
【図4】図4は、ATP結合ポケットの形態を示している。
【図5】図5は、亜鉛結合部位を示している。
【図6】図6は、Mnk−2キナーゼドメINとp38との比較を示している。
【図7】図7は、Mnk−2キナーゼドメINに結合する阻害因子のモデルを示している。
【図8】図8は、立体的表現(a)と主要配列(b)におけるMnk−1の全構造を示している。
【図9】図9は、活性化セグメントにより誘導されたNローブ移動を示している。
【図10】図10は、c−KIT(a,b)とMnk−1(C)における自動阻害を示している。
【図11】図11は、(a)DAPL1(1ig1;(テレスコ(Tereshko)等の、Nat.Struct.Biol.(8)(2001)899−907));(b)Mnk−1;Mnk−2のATP結合ポケットを示している。
【図12】図12は、Mnk活性化カスケードモデルを示している。
【図13】図13は、DFDモチーフの近傍を示している。
【図14】図14は、原子配置を示す。
【実施例】
【0081】
実施例1
Mnk−2とMnk−1キナーゼ領域のクローニングと精製
従来既知の技術を用いて、アミノ酸残基72〜385に対応し、キナーゼドメIN(KD)を包含するヒトMnk−2のcDNAフラグメントを、F(forward)/R(reverse)プライマー対5’CGGGATCCACCGACAGCTTCTCGGGCAGG / 5’ACGCGTCGACCTACCTCTGCAGGACCATGGGAG (利用した限定部位は下線部)を用いて増幅し、ベクターpGEX−4T1のBamHiとSall部位中にクローン化した(アマーシャム(Amersham)の、スウェーデン、cat.no.27−4580−01)。この構築により、N末端を有する融合タンパク質、トロンビンクリーブ可能なグルタチオンSトランスフェラーゼ(GST)タグとしてのMnk−2キナーゼ領域(KR)の原核細胞発現が可能となる。
【0082】
アミノ酸置換D228Gは、製造元の指示にしたがう、Stratagene Quik Change Site Directed Mutagenesisキットを採用して、GST−Mnk−2KR構造中に導入された。突然変異誘発オリトヌクレオチドは、5’GAAGATCTGT GACTTCGGC CTGGGCAGCG GCATCAAACT Cおよび5’GAGTTTGATG CCGCTGCCCA GGCCGAAGTC ACAGATCTTCであった。Mnk−2KRD228Gの精製は、Mnk−2KRに関して説明したように実施した。
【0083】
アミノ酸残基37〜341に対応し、キナーゼドメIN(KD)を包含するヒトMnk−1のcDNAフラグメントは、F/Rプライマー対5’CGGGATCCACTGACTCCTTGCCAGGAAAG/ 5’ACGCGTCGACCTATCCCTTTTCTGGAGCTTGCCを用いて(利用した限定部位は下線部)を用いて増幅し、ベクターpGEX−4T1のBamHiとSall部位中にクローン化した(アマーシャム(Amersham)の、スウェーデン、cat.no.27−4580−01)。この構築により、N末端を有する融合タンパク質、トロンビンクリーブ可能なグルタチオンSトランスフェラーゼ(GST)タグとしてのMnk−2キナーゼ領域(KR)の原核細胞発現が可能となる。
【0084】
GST−Mnk−2KRまたはGST−Mnk−1KRの発現は、大腸菌BL21(Merck Biosciences, Germany, cat. no. 69449)中である。細胞は、バッフル付の5リットルフラスコ内で、100μg/mlのアンピシリン(シグマ(Sigma)の、ドイツ、cat. no. A-9518)で補ったLBブイヨン中(メルク(Merck)のドイツ、cat. no. 1.10285)で、37℃、毎分130回転(rpm)で成長させた。培地が、0.8のA600に相当する密度に達したとき、同容量の氷温のLB/アンピシリンを加え、培地を25℃にして、1mMのイソプロピルチオガラクトースで誘導した(ロス(Roth)の、ドイツ、cat. no. 2316.4)。細胞を遠心分離で採取した。細胞ペレットは、その湿潤重量1gあたり10mlのリーシスバッファ(50mMトリス/HCl(シグマ(Sigma)の、ドイツ、cat. no. T-5941)、pH7.5、200mMNaCl(シグマ(Sigma)の、ドイツ、cat. no. S-7653)、5mMDTT(ロス(Roth)の、ドイツ、 cat. no. 6908.2))で再希釈した。MS72プローブ付のBadelin sonoplus sonifierによる細胞の崩壊(バデリン(Badelin)の、ドイツ、cat. no. HD207)と、後続のSorvallSS34ロータ(ソルボール(Sorvall)、ドイツの、cat. no. 28020)中における18000rpm/45分/4℃での除去によりライセートを調製した。ライセートは、直列に接続した、リーシススバッファで平衡した2つのGSTPrepFF16/10カラム(アマーシャム(Amersham)の、スウェーデン、cat. no. 17-5234-01)に適用した。洗浄液は、3カラム容量(CV)ウォッシュバッファ(50mMトリス/HCl、pH7.5、100mMNaCl、1mMDTT)、2CVのATPバッファ(50mMトリス/HCl、pH7.5、100mMKCl(ロス(Roth)の、ドイツ、 6781.1))、20mMMgCl2(シグマ(Sigma)の、ドイツ、cat. no. M-2670)、5mMATP(シグマ(Sigma)の、ドイツ、cat. no. A-7699))、およびさらに3CVウォッシュバッファ)であった。Mnk−2KDは、続いて、GSTタグ由来のオンカラムのトロンビン開裂によって溶出した。簡単にいうと、1000ユニットのトロンビン(アマーシャム(Amersham)のスウェーデン、cat. no. 27-0846-01)を、60mlウォッシュバッファに溶解して、2つのカラムにわたって8℃で一晩循環させた。溶出物を、ウォッシュバッファをカラムに適用しながらループを開くことによって回収した。
【0085】
トロンビン溶出物は、50mMトリス/HCl、pH8.0中に1:5に希釈し、直列に接続した5mlのQセファロースHPカラム(アマーシャム(Amersham)の、スウェーデン、cat. no. 17-1154-01)に適用した。溶出物は、塩化ナトリウムの直線上の勾配を有するものであった(50mMトリス/HCl、pH8.0、0−1MNaCl)。フラクションは、10.000ダルトン分子量カットオフ(MWCO)VivaSpin濃縮器中で(ビバサイエンス(VivaScience)、ドイツ、cat. no. VS0403)、おおよそ16mg/mlまで精製かつ濃縮してプールした。濃縮物は、PD10カラム(アマーシャム(Amersham)、cat. no. 17-0851-01)へのゲルろ過により、10mMトリス/HCl、pH7.5、50mMNaCl、1mMDTT中に移した。典型的な最終タンパク質濃度は、おおよそ12mg/mlであった。アリコートは、液体窒素内でショック凍結させ、−80℃で保存した。タンパク質生成量は、細胞ペレット湿潤重量1g当たりのおおよそ2mgのMnk−2キナーゼドメINであった。ERK2によって活性化した後、対応するMnkキナーゼ領域と完全長のMnkタンパク質とは、elF4e(Ser209)リン酸化に基づくキナーゼアッセイにおいて同一の活性を示す。
2.結晶化とデータ収集
初期結晶スクリーニングは、100μl貯留溶液と、200nl〜1μlの範囲の液滴寸法とを用いる96ウェルフォーマット中でMicroSys SQシリーズ4000/4100(デカルト分配システム(Cartesian Dispensing Systems))により実施した。回折研究に用いた結晶は、20℃で懸滴または座滴のいずれかを用いて拡散蒸着によって成長させた。タンパク質溶液は、タンパク質溶液の10倍を超える量までの貯留バッファ(100mMNa−Hepes、pH7.8、22%ポリアクリル酸5100と2%2,3−メタンペンタジオール(MPD))と混合した。結晶は、液体窒素中で凍結させた。回折データは、Mar−Research(ノルデステット(Norderstedt)、ドイツ)CCD検出器の100Kおよびλ=1.05で、HASYLABビームラINBW6(DESY、ドイツハンブルグ)上に回収し、HKLパッケージ(オトウィノスキ・ゼット(Otwinowski,Z)とマイナー・ダブリュー(Minor,W)の、オシレーションモードにおけるX線開設データの処理、Methods Enzymol. 167, 307-326, 1997年9月)で処理した。
3.構造決定と精製
初期相は、研究モデル(PDB ID:1lG1)として死亡関連タンパク質キナーゼ(DAPK)を用いたCCP4パッケージ(Collaborative Computational Project, The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. D 50, 760-763, Dec 1994年)からのMolRep自動化分子置換ルーチンを用いて得た。相情報を有するmtz形式のファイルを、REFMAC(Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S.とDodson, E. J.のFFTを用いた効率的な分子構造の異方性精製、Acta Crystallogr. D Biol. Crystallogr. 55 (Pt 1), 247-255,1999年1月)中で剛性体精製を用いて作製し、arp/warpによる自動化モデルの構築(Morris, R. J., Perrakis, A.とLamzin, V. S. ARP/wARPとタンパク質電子密度マップの自動翻訳;Methods Enzymol. 374, 229-244 (2003年))に用いた。得られたモデルは、Xフィット(McRee, D. E.の、原子座と電子密度を操作するためのXtalView/Xfit-Aバーサタイルプログラム;J. Struct. Biol. 125(2-3), 156-165, 1999年4月)を用いて手動でさらに変更した。精製は、CNS(Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T.およびWarren, G. L.の、結晶学とNMRシステム:マクロ分子構造決定に適した新しいソフトウェア、Acta Crystallogr. D Biol. Crystallogr. 54 (Pt 5), 905-921, 1998年9月)とREFMAC(Murshudov, G. N.等, 1999年、上記参照)を用いて実施した。
4.ゲルろ過と光散乱
Superdex75PC3.2/30カラム(ファーマシア(Pharmacia)製)を用いてSMARTシステムによりゲルろ過クロマトグラフィを実施した。実験は、バッファA(20mMトリスHCl、pH7.5、100mMNaCl、1mMDTT)中で0.04ml/分の流速において室温で行った。Mnk−2KDの分子量は、基準タンパク質(Bio−Rad)を用いて推定した。マルチアングルレーザ光散乱を、UV分光計と、ドーンとオプチラブ(Dawn and Optilab)計器XY(ワイアットテクノロジー社(Wyatt Technology Corp.))に接続したHR−10/30Superdex−200サイズの排除カラム(アマーシャム)で行った。30μmのMnk−2a溶液を、バッファA中で、632.8nm、90度におけるUV吸収、光散乱で、クロマトグラフィによって分離し、溶出プロファイルの屈折差はASTRAソフトウェアパッケージ(Wyatt, P.のマクロ分子の光散乱と絶対化学分析:Anal. Cim. Acta 272, 1-40 (1993年))で監視し、分析した。
【0086】
実施例2
Mnk−2特異阻害因子設計のために誘導されたp38−ジアリール尿素阻害因子共晶構造
タンパク質キナーゼp38の構造は、全体的に、Mnk−2キナーゼドメINの構造に非常に類似している(図1A)。p38は、ATP結合ポケット中の典型的なDFG配列モチーフを特徴付ける。ジアリール尿素足場に基づくp38特定の阻害因子を設計し、p38のこれら2つの阻害因子(BMUとBIRB796、(Pargellis et al. (2002年), Nat. Struct. Biol. 9, 268-272))との共晶構造を解明した(PDB Ids 1KV1と1KV2のそれぞれ)。これらの阻害因子は、p38の非カノニカル形DFG配座(DFGOUTと称す)を誘導し、この配座においては、フェニルアラニンは疎水性ポケットにおけるその標準位置から置き換えられており(DFGINと称す)、アポ酵素とその他タンパク質キナーゼ構造中に存在する(図1B)。DFGモチーフのDFGOUT配座は、立体障害により生産的ATP結合と干渉する。
【0087】
Mnk−2キナーゼ領域は、DFGモチーフに代わりDFDを呈する(残基226−228、図1参照)。非活性化アポ酵素の構造において、このDFDモチーフは、p38の非カノニカル形DFGOUT配座に類似する配座に適合する(図1B)。Mnk−2のPhe227は、クレフト中へ突出し、この場合、p38内はジアリール型阻害因子で占有することができる(図2A)。置換は、p38−阻害因子複合体よりも厳密であるが、Mnk−2キナーゼ領域の結晶化には阻害因子は用いられない(図1B)。Mnk−2キナーゼ領域結晶構造中に見られるDFD配座はまた、立体障害のためにカノニカル形ATP結合とは共存しない(図2B)。観察により、本発明の結晶構造に観察されるDFG/DOUT配座中のDFDモチーフの捕捉が、Mnk−2を不活性にし、リン酸化状態を無視させることが示唆される。
【0088】
ジアリール型阻害因子BMUがMnk−2キナーゼ領域を結合可能であるかどうかを調査した。p38−BMU複合体(PDB ID 1KV1)のMnk−2キナーゼ領域上への全体的なスーパーポジション後、BMU位置におけるわずかな手動調整と、いくつかのMnk−2側鎖配座におけるわずかな再調整とを行い、Mnk−2−BMU複合体モデルを得た(図2Aと図C)。阻害因子は、Phe227とPhe159の環の間に挟まれたそのpクロロフェニル環と結合することが見られた(図2Aと図C)。そのtブチル部分は、Mnk−2の疎水性ポケットに収容可能であった(図2Aと図C)。Mnk−2の異常なAsp228は、このモデル中のBMU分子からかけ離れているが、p38中のその他の領域(p38の、1KV2構造のBIRB796の延長された足場と1KV1構造のBMUとの比較)で示したように、さらなる改質された阻害因子上の化学基によってターゲットとすることができた。この方法において、DFDモチーフへの特異性(Mnk−2のフINガープリント;その他のキナーゼのDFGに代わり)が達成され得る。Mnk−2に対する特異で強力な結合は、Mnk−2の特異的な結合に対する新規な阻害因子に適合させるためのBMUのpクロロフェニルとtブチル基のさらなる改質によって支持することができた。
【0089】
実施例3
Mnk−1−KRの構造決定および全構造
野生型Mnk1−KRの針状結晶を、タンパク質溶液を等容量の20%(w/v)PEG3350と0.2M硫酸アンモニウムと0.1Mクエン酸ナトリウムを含有するpH5.4の貯留溶液とを混合後の拡散蒸着により、20℃で成長させた。結晶は、20%のグリコールで補った貯留溶液中で凍結させた(液体窒素)。回折データを、ビームラINPXII(SLS、ビリンゲン(Villingen)、スイス)に対して、MarResearch(ノルダーシュテット(Norderstedt)、ドイツ)CCD検出器における100Kで収集し、HKLパッケージ(オトウィノスキ・ゼット(Otwinowski,Z)とマイナー・ダブリュー(Minor,W)、1997年)で処理した(表2参照)。
【0090】
Mnk−2−KRの密度変更後の切頭型モデルを用いた分子置換後、翻訳可能な電子密度を得て、モデルを23.5/28.0%のR/Rfree因子に精製することができた(表2)。非対称ユニットは、非クリスタルグラフィックの2倍軸で関連づけられる2つのMnk−1−KR分子を含む。分子Aは、いくつかの領域においてより低温の因子とクリアラ電子密度を呈する。機能的に重要な領域は、しかしながら、分子Aと分子Bとの間で実際上同一である。最終モデルは、Mnk−1のキナーゼドメINの範囲にわたり、残基39−335を含む。Mnk−1−KRは、バイロバル(bilobal)構築を含むキナーゼアーキテクチャのいくつかの広域な特徴を保存する。N末端ローブは、グリシンリッチループやLys−Gluイオン対などのATP結合に必要な重要な要素を担持し、5本撚りのねじれたβシートと調製へリックスαCの形態にされる(図8)。大型で優勢なαらせんC末端ローブは、触媒ループ(Cループ)、マグネシウム結合ループ(DFDモチーフ)、および活性化セグメントなどの、基質結合および亜リン酸転移に必要な要素を含む(図8)。Mnk−1−KR内のセグメントは、強い、配座的なフレキシビリティを呈し、そのため、電子密度では痕跡をたどることはできず、活性化セグメントのコアはP+1ループ(残基197−222)と、へリックスαG(残基261−290)を包含するMnk特異システINクラスタとを含む。
【技術分野】
【0001】
本発明は、結晶Mnk−1タンパク質ならびにMnk−2タンパク質に関し、より詳細には、Mnk−1およびMnk−2キナーゼ領域の結晶構造に関する。
【背景技術】
【0002】
ヒトにおいては、500を超えるキナーゼが知られており、それらはヌクレオチドからタンパク質基質までのリン酸基の伝達に介在する。タンパク質キナーゼによる基質識別、制御ならびに触媒作用の詳細な理解は、高度に多様な生物学的経路の完全なイメージを描くための基本であり、それらの多くは広範囲に及ぶ疾病に直接関連している。cAMP依存性キナーゼの結晶構造は、タンパク質キナーゼの分子構造の第1の高解像度画像を提供した(Knighton等、Sicence 235(5018)(1991)407−414)。
【0003】
他のヒトのタンパク質キナーゼの結晶構造により、貴重な触媒および調節メカニズムへの識見が提供され、特定の阻害因子の設計が助成される。
【先行技術文献】
【非特許文献】
【0004】
【非特許文献1】Knighton等、Sicence 235(5018)(1991)407−414
【発明の概要】
【課題を解決するための手段】
【0005】
したがって、本発明の主題は、結晶ヒトMnk−2タンパク質と結晶ヒトMnk−1、およびそれらの調製方法と用途に関するものである。
【0006】
第1の実施態様において、本発明は、ヒトのセリン−トレオニンキナーゼマイトジェン活性化キナーゼ(MAP)相互作用性キナーゼ−2に関するものであり、これはMnk−2タンパク質とも称される。ヒトにおいて4つのMnkタンパク質が発見されており、すなわち、2つのアイソフォームMnk−1とMnk−2であり、後者は2つのスプライスバリアントMnk−2aとMnk−2bとして存在する。スプライスバリアントMnk−1bもまた開示されている。Mnk−2aとMnk−2bのキナーゼのドメINは同一である。Mnkタンパク質がMAPキナーゼファミリーの1つによって活性化できることが示されている。具体的には、ストレス誘発p38タンパク質と、マイトジェン活性化Erk1/2タンパク質が、この機能を果たすことができる。Mnk−1とMnk−2は、同様の経路を経て活性化され、同様の基質特異性を呈する。キナーゼドメIN内におけるそれらのアミノ酸配列は、ほぼ同じであり、かつ、以下に記載のアミノ酸と同一である。そのため、Mnkキナーゼは、これら2つのMAPキナーゼ経路の集合点を構成し得る。
【0007】
Mnkタンパク質は、タンパク質キナーゼのMAPキナーゼ活性化タンパク質キナーゼ(MAPKAPK)ファミリーのサブファミリーであり、これらタンパク質キナーゼは、Ca/カドミウム調節キナーゼ(CAMK)グループに属する。
【0008】
Mnkは、3つのMAPKカスケードのうちの2つ、すなわち、成長因子誘導Ras細胞外信号調節タンパク質キナーゼ(ERK)1/2およびストレス誘発p38経路(福永等のEmbo J.(16)(1997)1921−1933;Embo J.(16)(1997)1909−1910)によるリン酸化によって活性化される。2つのほ乳類MnkアイソフォームであるMnk−1とMnk−2は、真核細胞開始因子4E(elF4E)をin vitroならびにin vivoでリン酸化する(シェパー(Scheper)等のEur J.Biochem.(269)(2001)5350−5359、上田等のMol.Cell Biol.(24)(2004)6539−6549、ヴァスキエヴィッツ(Waskiewicz)等のMol. Cell Biol.(19)(1999)1871−1880)。elF4Eは、翻訳開始複合体の必須成分であり、真核細胞のメッセンジャーRNAのCAP構造を結合する(マルコトリジアーノ(Marcotrigiano)等のCell(89)(1997)951−961)。Mnk介在性elF4Eリン酸化により、例えばRFLAT−1やウィルス性転写の特定のmRNAの翻訳が刺激されることは明らかである(ニコルシェバ(Nikolcheva)等のClin.Invest.(110)(2002)119−126、ウォルシュ(Walsh)等のGenet.Dev.18(2004)660−672)。さらに、Mnk1は、hnRNPA1による腫瘍壊死因子アルファ(TNF−α)の翻訳を減じるため、炎症性疾患において役割を果たす(バクセード(Buxade)等のImmunity 23,177−189)。脂質代謝、炎症ならびにウィルス性翻訳におけるMnkの改善により、製薬的介入のためのターゲットとしてそれらは定義される。
【0009】
Mnkは、3つのMAPKカスケードのうちの2つ、すなわち、成長因子誘導Ras細胞外信号調節タンパク質キナーゼ(ERK)1/2およびストレス誘発p38経路(福永等のEmbo J.(16)(1997)1921−1933;Embo J.(16)(1997)1909−1910)によるリン酸化によって活性化される。2つのほ乳類MnkアイソフォームであるMnk−1とMnk−2は、真核細胞開始因子4E(elF4E)をin vitroならびにin vivoでリン酸化する(シェパー(Scheper)等のEur J.Biochem.(269)(2001)5350−5359、上田等のMol.Cell Biol.(24)(2004)6539−6549、ヴァスキエヴィッツ(Waskiewicz)等のMol. Cell Biol.(19)(1999)1871−1880)。elF4Eは、翻訳開始複合体の必須成分であり、真核細胞のメッセンジャーRNAのCAP構造を結合する(マルコトリジアーノ(Marcotrigiano)等のCell(89)(1997)951−961)。Mnk介在性elF4Eリン酸化により、例えばRFLAT−1やウィルス性転写の特定のmRNAの翻訳が刺激されることは明らかである(ニコルシェバ(Nikolcheva)等のClin.Invest.(110)(2002)119−126、ウォルシュ(Walsh)等のGenet.Dev.18(2004)660−672)。さらに、Mnk1は、hnRNPA1による腫瘍壊死因子アルファ(TNF−α)の翻訳を減じるため、炎症性疾患において役割を果たす(バクセード(Buxade)等のImmunity 23,177−189)。脂質代謝、炎症ならびにウィルス性翻訳におけるMnkの改善により、製薬的介入のためのターゲットとしてそれらは定義される。
【0010】
CAMLグループのその他の部分と一列に並ぶ配列は、Mnkタンパク質の無比のいくつかの特徴を明らかにした。構造的および機能的見地におけるこの観察の結果を明らかにするため、Mnk−2に対する結晶学的な研究が行われた。本発明により、Mnk−2のキナーゼドメINの2.1Å結晶構造が得られた。その結果、Mnk−2のApo酵素が、活性ループとP+1ループのC末端を含む、ハンクス(Hanks)のスキームのサブドメINXIIIに対応するセグメントの例外的な開放高次構造を示すことが示されている(ハンクス(Hanks)等のMethods Enzymol.200(1991)38−62)。P+1ループは、基質結合に対して重要であることが知られている。
【0011】
DFDとしてMnkタンパク質中で保存される、マグネシウム結合DFGモチーフの同等物は、ATP結合ポケット中に突出し、ヌクレオチド結合を妨害する。したがって、活性化ループの開始時に保存されたDF(G/D)は、ATP結合を阻害する立体配座を受け入れる(DF(G/D)OUT立体配座と称す)。これはヌクレオチド結合を調節する阻害メカニズムが、ATP結合の割り接ぎが、非リン酸化apo酵素中で可能であるCAMKグループの既知の構造の他のキナーゼ(DF(G/D)IN立体配座)とは対照的であることを明らかにしている。これは、Ser/Thrキナーゼアポ酵素中におけるDF(G/D)OUT立体配置の第1の観察である。
【0012】
さらに、Cループ中の亜鉛配位モチーフ、タンパク質キナーゼにおいては開示しなかったが、が発見された。Mnk−2キナーゼドメINは、Mnkタンパク質中の長さと配列中に保存されるが他のキナーゼでは欠けている、Cループ中に15の残基の挿入を包含している。この挿入中の4つの保存されたシステINは、ここで開示するMnk−2構造によって明らかにされるように、亜鉛イオン結合部位として機能する。
【0013】
したがって本発明のMnk−2構造は、キナーゼアーキテクチャと調節の新規な態様を明らかにするものであり、これは論理的な阻害設計に用いることができる。
【0014】
特に好ましくは、本発明は、結晶ヒトMnk2aまたはMnk−2bタンパク質に関するものである。Mnk−2aは、真核細胞開始因子4e(elF4E)のリン酸化による翻訳機構をターゲットとする、ヒトタンパク質キナーゼである。
【0015】
トランスリン酸化反応中に包含されることが知られている残基は、CAMKキナーゼグループ内で保存される(テイラー(Taylor)等の、Structure 2(5)(1994)345−355、ハンクス(Hanks)等の、Science 241(4861)(1988)42−52)。これらの残基は、
(A)Lys113と、
(B)推定受容体ベースAsp205を含有する触媒ループ(残基205−210)と、
(C)γリン酸の活性に必要なマグネシウムイオンを配位する、DF(G/D)モチーフの第1のAsp226と、
である。
【0016】
しかしながら、Mnkタンパク質を他のタンパク質キナーゼから区別するいくつかの特徴がある。すなわち、活性ループのDFGモチーフN末端中に保存されたグリシンは、全てのMnkタンパク質中のアスパルテートで置換され、その結果、DFDモチーフ中に存在する(DF(G/D)と称される)。この単一のアミノ酸置換は、その他のCAMKグループ中には見られない。さらには、Mnkタンパク質は、全て長さが保存される3つの異なる箇所アミノ酸挿入部を含む。10個のアミノ酸の第1の挿入部(l1)は、DFDモチーフに続く活性化セグメントのN末端に位置する。第2の挿入部(l2)は、へリックスFの上流にあり、おおよそ5つのアミノ酸を含む。挿入部3(l3)は、15のアミノ酸のストレッチであり、Mnkのサブファミリー内に高度に保存されたパターンを呈し、CローブのGとHのへリックスを接続するループのN末端に位置する。4つのシステINのクラスタは、全てのMnkにおいては不変のl3内に存在する。
【0017】
一実施態様においては、本発明による、結晶ヒトMnk−2タンパク質、特に結晶Mnk−2aタンパク質は、完全なタンパク質である。他の実施態様では、これもまた好ましく、フルレングスタンパク質ではなく一部欠失形、特に、少なくともアミノ酸残基72−385を含む一部欠失形であり、キナーゼドメIN(KD)を含む。数字は、エントリーAAG26337(Mnk−2b)とAAG26336(Mnk2a)を示す。特に好ましくは、20Åより良好な、より好ましくは10Åより良好な、さらに好ましくは3Å未満より良好な解像度を有するX線構造解析が可能な結晶が関連する。
【0018】
本発明による結晶調合物は、スペースグループP3221と、a=104.5ű3Å、b=104.5ű3Åおよびc=72.35ű3Åであるユニットセル寸法を有することが好ましい。本発明によれば、2Åに対して回折可能な結晶が調製され、それによって、その構造は、分子置換によって溶解されて0.21(Rfree=0.25)のR因子に改良することができた。特に好ましいのは、本発明により不活性形態のヒトMnk−2タンパク質の結晶が検討されることである。
【0019】
さらに好ましいのは、非リン酸化Apo配座のMnk−2結晶ドメINが検討されることである。
【0020】
本発明によりわかるように、活性化セグメントとそのC末端のへリックスαF(αF:残基270−290)までの延長部は、異常なオープン構造にある(Mnk−2アミノ酸残基のナンバリングは、Entrez Entry AAG26336命名法に対応する)。この領域はサブドメINXIII分類に該当する。活性化セグメントは、活性キナーゼのリン酸化ターゲットである残基を担持し、19−32残基とは独立した、2つの保存モチーフであるDF(G/D)とAPEの間に配置される領域として定義されている。
【0021】
従来開示のキナーゼ構造との際だって対照的に、ヒトMnk−2タンパク質のサブドメINXIIIは、キナーゼコアから突き出ている。サブドメINXIIIは、リン酸化部位Thr249とAPEモチーフとの間に位置するP+1ループを含む。P+1ループは、触媒作用のためにペプチド基質の位置を決める。
【0022】
サブドメINXIIIの突出部は、基質認識、基質位置決め、および活性化体機構に影響を与えるMnkタンパク質中の位相転位に向いている。
【0023】
さらには、ATP加水分解に関連する残基とリン酸塩転移とは、タンパク質キナーゼ中で十分に不変である。触媒活性に関連する領域は、Lys113、Glu129、Asp205、およびAsn210を含む。本発明によって得られる構造データから確認できるように、結晶ヒトMnk−2タンパク質構造は、ATPまたは関連する化合物の影響を受けない。したがって、本発明による結晶は、不活性形態のヒトMnk−2タンパク質の結晶体にあって好ましい。本発明のMakキナーゼ中のDF(G/D)OUT立体配座と一致するDF(G/D)OUT立体配座p38が、一定の化学物質によって誘導されることが示されている(パーゲリス(Pargellis)等、nature structrual biology,vol.9,no.4(2002)268−272)。DF(G/D)OUT配座により、ATP結合クレフトをターゲットとする化合物に加え、ジアリール尿素阻害剤などの代替物質クラスの使用を包含する広範な薬理学的用途を伴う新規なアロステリック結合部位が提供される。さらには、DF(G/D)OUT配座の安定化により、酵素が阻害される。
【0024】
したがって、本明細書で提供されるデータにより、Mnk−2aのDFDモチーフが、基質のリン酸化が要求される際に、得られるATP結合、すなわちATP結合と不適合である配座を呈することができることが示される。すなわち、それは、非リン酸化Mnk−2が、ATPと結合できないか、あるいは、ATP結合を可能にするためにDFDモチーフ中での配座変化をまず生じさせなければならないことから生じる。Mnk−2の決定された配座は、該タンパク質(DFGモチーフに代わるDFDモチーフ)の特定の配列に起因して、その他全てのキナーゼとは異なる。この情報により、Mnk−2の阻害物質ならびに異性体、および非生産的DFD配座を認識して安定化するその他のタンパク質キナーゼを特定することができる。この上方により、Mnk−2の阻害因子ならびにアイソフォームおよび非生産性DFD立体配座を認識して安定化する、他のタンパク質キナーゼを特定することができる。また、これはMnk−2、Mnk−2キナーゼドメINそれぞれに特異な阻害因子を提供することができるが、他のキナーゼは認識しない。これは、DFGモチーフを呈するその他のキナーゼが異なる配列を有するために可能である。
【0025】
したがって、とりわけ、ATP結合ポケット(ここでは、DFDOUTポケットとも称する)ならびに他のポケット(ここでは、DFDINポケットとも称する)は、本発明による構造データによって決定することができる。活性形態において、ATPポケットは、ATPの結合部位を提供する。該ポケットは、特に、アミノ酸残記Glu129とAsp205、ならびにアミノ酸残記Lys113とAsn210によって定義される。認識され得る第2のポケットは、本発明により特に関心の対象となるものである。該第2のポケット、すなわちDFDINポケットは、DFDモチーフのPheが活性構造中で位置する部位である。不活性形態において、ATPポケットは、少なくとも部分的にDFDモチーフ、特にDFDモチーフのPheによって占められている。この不活性形態は、DFDINポケットを占有すること、特に、活性化セグメントまたはその他の分子、特に、阻害因子として機能する小さい分子でDFDINポケットを占有することによってロックすることができる。DFDINポエットを占有することにより、ATPが、この形態においては少なくとも部分的にDFDモチーフによって占有されているATPポケットにアクセスできないため、キナーゼ活性の阻害因子が作用する。DFDINポケットは、特に、アミノ酸残記Lue133、His203、Ile142、およびIle224によって定義される。該DFDINポケットをブロッキングすることによって、不活性構造がロックされる。したがって、本発明の主題は、前記ポケットを占有することができることにより、Mnkの選択的阻害因子の役割を果たす分子を提供することである。すなわち、そのようなDFDINポケット内へ結合することができる阻害因子が、本発明のその他の主題を提供する。Mnkにおいて、活性セグメント、特に活性化セグメントの挿入部12、より詳細にはアミノ酸残基Phe265は、DFDINポケットをブロックするため、適当な阻害因子は、例えば、小さい活性化セグメントの少なくとも部分的な配列を有するペプチドである。活性化セグメントは、アミノ酸Asp226〜Cyc275からなり、特に、アミノ酸263〜267まで延在する挿入部12を含む。したがって、Mnkの適当ペプチド阻害因子は、活性セグメントの配列または少なくとも4つ、特に少なくとも5つ、好ましくは少なくとも6つ、より好ましくは8つのアミノ酸を有するその連続的なフラグメントを有するペプチドである。そのような阻害因子の例とは、(258)APEVVEAFSEEA(269)または(260)EVVEAFS(266)である。
【0026】
本発明によって提供される、アロステリックな結合部位に対抗する阻害因子を提供する可能性により、さらには、顕著に改善された選択性を有する阻害因子が得られる。キナーゼのATP結合部位に対抗する標準的なキナーゼ阻害因子は、キナーゼの高い相互ホモロジーに起因した大きなクロス反応可能性を有する。したがって、ATP結合部位に対抗する阻害因子は、通常、わずかな選択性しか有さないため、強力に阻害して選択的阻害因子の発達を制限する。しかしながら、本発明によれば、Mnkのアロステリック結合部位における選択的な阻害因子の結合を提供することが可能となる。
【0027】
本発明により用いることが可能な阻害因子の1つは、BIRB796(パーゲリス(Pargellis)等の、nature structural biology,vol.9,no.4(2002),268−272)である。その他の阻害因子は、ジアリール尿素ベースの阻害因子(1−(5−tert−ブチル−2−メチル−2H−ピラゾール−3−イル)−3−(4−クロロフェニル)尿素)である。
【0028】
さらに、Mnkタンパク質は、Mnkタンパク質をその他のCAMKグループのキナーゼから区別する4つのシステINの不変クラスタを含むαFとαGとの間に挿入部を有する。分子のフレキシブルループ中のこれら4つのシステINクラスタは、亜鉛結合部位を形成する。そのため、この挿入部は亜鉛のフINガー状構造、タンパク質キナーゼの特異的なフINガープリントをマークする。さらに、4つの保存されたグリシンは、この挿入部中に存在しており(Gly297、Gly300、Gly304およびGly308)、これらは、このヘアピン状モジュールに折りたたむのに必要な領域にねじれフレキシビリティを与える。亜鉛フINガーモジュールは、変化を有する核酸またはタンパク質結合モジュールであることが知られている(クリシュナ(Krishna)等の、Nucleic Acids Res. 31(2)(2003)532−550)。このドメINは、その他のタンパク質、特に、基質や調整器のアダプタモジュールである。本発明による結晶ヒトMnk−2キナーゼはまた、変異体、好ましくは、天然Mnk−2キナーゼの少なくとも1つのアミノ酸、特に、少なくとも2つのアミノ酸が、別のアミノ酸で置き換えられたタンパク質を含んでなる。そのような変異体の結晶は、特に、機構学的研究ならびに結合ポケットの研究および配位子、基質あるいは阻害因子との相互作用を研究するのに遊離に用いることができる。この結果、適切に、結合能に対する相互作用または影響が推定あるいは予測される位置にある個々のアミノ酸を選択的に変化させる。この目的のため、例えば、20までの、好ましくは10までの、より好ましくは5までの、さらにこのましくは最大1つの突然変異を有する結晶ヒトMnk−2キナーゼは好ましいものとなり得る。結晶ヒトMnk−2キナーゼ変異D228Gが特に好ましい。さらに好ましい実施態様において、配位子、基質および/または阻害因子、特に、阻害因子スタウロスポリンとの組み合わせた結晶ヒトMnk−2キナーゼ変異D228Gが考慮される。
【0029】
好ましい変異体は、位置Asp226,Phe227またはAsp228で交換させたアミノ酸を有する。
【0030】
本発明は、さらに、表1に示す構造的配位の全てのまたは選択された部分で決定される三次元構造を有する結晶ヒトMnk−2タンパク質に関する。この表1に示す配位は、ここで説明する例のように得られたものである。
【0031】
一実施態様において、本発明は、同属のタンパク質キナーゼ阻害因子スタウロスポリンと共結晶されたヒトMnk−2−D228G変異体の結晶構造をさらに提供する。この構造において、DFG/DIN配座中へのDFGモチーフフリップにより、ATP結合ポケット内におけるその同属の結合部位においてスタウロスポリンを結合させることができる。この配座は表3に示す。阻害因子のいずれも有さない、特に、阻害因子スタウロスポリンを有さない、ヒトMnk−2キナーゼD228G変異体の結晶構造がさらに提供される。その配剤は表1aに示す。
【0032】
本発明による結晶ヒトMnk−2タンパク質の特徴は、例えば、
i.細胞、例えば大腸菌中におけるヒトMnk−2タンパク質の発現;
ii.Mnk−2タンパク質祖調製物を回収するための細胞を溶解すること;
iii.Mnk−2タンパク質祖調製物を、例えば、親和性タグクロマトグラフィーによって精製すること;および
iv.精製されたヒトMnk−2タンパク質を、例えば、拡散蒸着によって結晶化すること、
によって調製することができる。ヒトMnk−2タンパク質、特に、本発明による、ヒトMnk−2aタンパク質またはMnk−2bタンパク質、より好ましくは、ヒトMnk−2aタンパク質のキナーゼドメINの結晶化方法は、ヒトMnkタンパク質の結晶構造データの生成に特に用いることができる。特に、配位子、特に阻害因子や基質との結合部位または相互作用部位がそれによって得られる。さらには、タンパク質を活性または不活性形態に維持するために結合部位を特定することができる。さらに、ここで得られるMnk−2タンパク質の結果により、Mnk−1などのMnk−2のアイソフォームの配位子、特に阻害因子や基質もまた特定することができる。本発明による結晶化方法は、好ましくは単結晶であり、より好ましくは少なくとも1μm、さらに好ましくは少なくとも10μm、最も好ましくは少なくとも50μmの端長を有する結晶である。結晶は、X線構造解析を実施可能な方法で配置することが好ましい。したがって、本発明のその他の主題は、ヒトMnk−2タンパク質、特に、表1、表1aまたは表3に示す構造配座の全てまたは選択されたタンパク質によって決定されるヒトMnk−2aタンパク質の結晶構造である。好ましくは、不活性ヒトMnk−2aタンパク質の結晶構造が考慮される。結晶構造は、50Å以上の分解能、より好ましくは10Å以上、さらに好ましくは3Å以上の分解能を有することが望ましい。
【0033】
結晶ヒトMnk−2タンパク質と結晶構造のそれぞれを用いることにより、Mnk−2タンパク質配位子を設計、同定または調製することができる。さらには、タンパク質キナーゼ、ならびに上述したようにMnk−2のアイソフォームのタンパク質キナーゼの調節機構を同定することができる。配位子または調節機構を同定するために、特にコンピュータ支援モデル化プログラムが用いられる。
【0034】
適した配位子は、例えば、ヒトMnk−2タンパク質の相互作用部位と相補的な三次元構造を有する分子を作製することによって同定することができる。特に好ましい配位子は少なくともアミノ酸Asp226,Phe227,Asp228の少なくとも1つと相互作用する。さらに好ましい配位子は、少なくとも1つの原子が、DFDモチーフの原子のいずれかまでの所定の距離内、好ましくは7Åの距離内、さらに好ましくは6Å、特に5Åの距離内にある少なくとも1つのアミノ酸と相互作用する。
【0035】
配位子を同定するためのコンピュータ支援スクリーニングによれば、WO03/037362号公報に開示の方法が、実際に配位子を同定して照合するのに好ましく適用される。
【0036】
表1、表1aまたは表3で与えられるヒトMnk−2タンパク質の結晶構造の構造的配位はまた、ヒトMnk−2タンパク質の結晶構造の三次元表現を作成するのに用いることができる。そして、そのような三次元構造に形成された相互作用ポケットは、それらの三次元構造により、対応する配位子を同定するのに用いることができる。
【0037】
表1、表1aまたは表3に示される、本発明によって提供される構造的配位は、その他のタンパク質の結晶構造を決定するのにも用いることができ、それにより、構造的配位は分子置換に用いられる。
【0038】
ここで提供するデータは、コンピューターで読み取り可能な保存媒体に保存し、適当に提供することが好ましい。
【0039】
本発明は、さらに、Mnk−2タンパク質のアイソフォーム、ならびに結晶化法または結晶構造を用いて得たその他のタンパク質キナーゼの配位子、特に基質または阻害因子に関する。そのような配位子は、好ましくは、薬剤組成物中における活性剤である。そのような薬剤組成物は、処置、特にMnk−2タンパク質の阻害が必要な、例えば、肥満、糖尿病およびメタボリック症候群ならびに癌などの代謝障害などの病気を治療するのに用いることができる。さらに実施態様において、本発明は、結晶ヒトMnk−1タンパク質に関する。
【0040】
Mnk−1キナーゼ領域(Mnk−1−KR)の結晶構造は、Mnk−2−KR異なる立体配座を取るが、触媒ドメINのアミノ酸配列は78%一致する。Mnk−1とMnk−2の構造データの組み合わせにより、Mnkサブファミリーメンバーの活性化に不随する機械的事象のダイナミックな像を描くことができるようになる。
【0041】
また、この実施態様においては、ヒトMnk−1タンパク質の突然変異体は、特に、交換させた少なくとも1つの、特に少なくとも2つのアミノ酸を有する突然変異体に包含される。上記で説明したように、そのような突然変異体は、機構学研究に特に用いることができる。好ましくは、突然変異体は≦20、より好ましくは≦10、さらに好ましくは≦5、最も好ましくは最大1の交換アミノ酸を有する。Mnk−1の場合における交換アミノ酸の好ましい部位は、位置Arg90またはArg93、およびArg191,Phe192またはArg193である。
【0042】
本発明は、N末端ローブ、マグネシウム結合ループ、および活性化セグメントが、徹底的な構造転位を受け、完全活性状態まで自動阻害されることによって得られるMnk活性化モデルにも関する。本発明のさらなる態様は、活性化セグメント介在の官能要素の位置移動によって自動阻害を得るためにMnkを用いることである。
【0043】
その他のタンパク質キナーゼの多くに見られるそのカノニカルな立体配座において、活性化セグメントのC末端部分は折り返され、ショートへリックスα−EFと基質結合P+1ループは、へリックスαF,αGおよび触媒ループによって与えられる環境中でキナーゼコア内に埋没する(ナイトン(Knighton)等の、Science(253)(1991)414−420;ノレン(Nolen)の、Mol. Cell(15)(2004)661−675)。
【0044】
しかしながら、Mnk−1−KRにおいては、α−EFは解かれてペプチド結合溝内へ崩壊するため、N末端ローブ配座と活性部位残基が変化する(図9)。特に、αCへリックス(Arg90:Glu225;Arg93:Glu228)との相互作用が引き上げ力を与えて、αCとNローブの残部を置き換え、それによりローブは終了する(図9A)。相互作用残基Arg90とArg93に対応する残基は、活性状態のタンパク質キナーゼ中のリン酸部分に結合することが知られている(クリュパ(Krupa)等の、Mol. Biol.(339)(2004)1025−1139)。そのため、再構築された活性化セグメントは、活性部位の配座を変更する分子スイッチとして機能するよう予定されている。ナンバリングは、オログレン・エー(O‘Loghlen, A.)、ゴンザレス・ヴィー・エム(Gonzlez,V.M.)、ピネイロ・ディー(Pineiro,D.)、プレッツモルガード・エム・アイ(Prez−Morgado,M.I.)およびマーテIN・エム・イー(Martin,M.E.)(2004)、Mnk1bの同定および分子特徴付け、ヒトMAPキナーゼ相互作用キナーゼMnk1のスプライス異体、ExpCellRes299,343−355に相当する。
【0045】
この活性化セグメントの「ウェッジ」形態のさらなる効果は、リン酸部位の到達可能性を促進し得る、活性化ループをさらすことである。ローブの終了は、Mnk−2構造によって示すように、活性化セグメントと調節Cへリックスとの間の相互作用が途絶える場合に逆戻りさせることができる(図9B)。Mnk−2において、分子本体から突出しているため、活性化セグメントは全く異なる形態を取る。その結果、活性化セグメント:Cへリックス相互作用は緩和されてNローブは突然回復する(図9B)。
【0046】
Mnk−1と比較した場合、Mnk−2のNローブは、おおよそ10°傾き、キナーゼのATP結合口を開く(図9C)。
【0047】
Mnk−1の延長、再構築された活性化セグメントは、その他のCAMKグループメンバーのほとんどには存在しない2つのアミノ酸挿入部を有する。
【0048】
内部ローブ溝における活性化セグメントの新規な配置の結果、Phe230は、Leu98とThr97出発配座αC,Cループの上流のHis168,Ile107,Ile189およびLeu161によって与えられる構造的に保存されたポケット内に存在することになる(Mnk−1アミノ酸残基のナンバリングは、EntrezEntryCAl14764に対応する)。
【0049】
後者のポケットが、活性状態のキナーゼにおいてDGG/Dモチーフのフェニルアラニンの結合部位として機能することが発見された(図10)。この活性DFG/Dモチーフ配座は、DFG/DIN配座と見なされ、その対応する結合部位は、今後、DFG/DINポケットと称する。Mnk−1においては、しかしながら、Phe230のDFG/DINポケットにおける存在は、DFG/Dモチーフのアクセスを制限し、ATP結合部位を立体的にブロックする阻害DFG/DOUTを誘導する(次の段参照)。そのため、再構築された活性化セグメント、特にPhe230は、Mnk固有の調節機構の重要な役割を果たす自動阻害要素を構成する。
【0050】
これまで、自動阻害戦略という意味でのDFG/DINポケットの障害は、c−KITおよびFlt3に見られ、これら2つはタイプIIIレセプターチロシンキナーゼと非常に関連性が高い。c−KITおよびFlt3は、DFG/DOUTの誘導により「in trans」で自動阻害するキナーゼのN末端に位置する傍膜(juxtamembrane;JM)ドメINを含む(グリフィス(Griffith)等の、Mol.Cell(13)(2004)169−178;モル(Mol)等の、J. Biol. Chme. (279)(2004a)31655−31663)。いずれのケースにおいても、JMドメINから出発する残基(Flt3中のLeu576およびc−KIT中のTrp557)は、DFG/DINポケット内へプランジされ、これはDFG/Dモチーフを強制的に阻害DFG/DOUT配座にする(図10A、図10B)。その結果、Mnk−1は、c−KITとFlt3と類似するが、異なる構造要素を利用する自動阻害機構を達成する(図10C)。Mnk−1は、JMドメINの代わりに、その活性が静まるようその再構築された活性化セグメントとPhe230とを採用し、これは、c−KITとFlt3内で、DFG/DINポケットの関与が維持されるよう生じる(図10C)。
【0051】
また、Mnk−1の場合、本発明によって提供されるデータにより、DFDINポケットの定量が可能となる。このポケットは、上述したように、特に、Leu98とThr97、His168、Ile107、Ile189とLeu161によって決定される。上記DFDINポケットを用いることにより、DFDモチーフは、少なくとも部分的にATPポケット内に位置されるため、MnkによりATP結合を阻害することができる。そのため、DFDinポケットをブロックすることにより、キナーゼ活性が阻害される結果となる。したがって、本発明のさらなる態様とは、DFDinポケットに結合し、それによりMnkを阻害する分子を提供することである。活性化セグメントによるMnk−1の自動阻害が、特に、活性化セグメントのDFDINポケット内へのl2挿入部のPhe230の位置によって生じるため、適切な阻害因子は、アミノ酸191〜240からなるMnk−1の活性化セグメントの全配列または部分的配列からなり、特に、アミノ酸228〜232からなる挿入部l2の配列からなる。適切なペプチドは、例えば、(223)APEVVEVFTDQA(234)または(225)EVVEVFT(231)である。
【0052】
タンパク質キナーゼの広大な大部分は、インターローバルクレフトにおいてタンパク質キナーゼのATP結合「口」の「唇」を形成する、活性化セグメント(サブドメINVII)の開始時にAsp−Phe−Gly(DFG)モチーフを担持する(ハンクス(Hanks)の、Genome Biol.(4)(2003)111;ハンクスの、Science(241)(1988)42−52);テイラー(Taylor)の、Structure(2)(1994)345−355)。このモチーフの第1のアスパラートは、触媒活性タンパク質キナーゼ間で不変であり、リン酸転位に必須のマグネシウムイオンと配位することが知られている(図11A)(アダムス(Adams)の、Chme. Rev.(101)(2001)227102290)。そのため、DFGモチーフはマグネシウム結合ループと称される。
【0053】
しかしながら、Mnkは、配位位置でAsp−Phe−Asp(DFD)モチーフを担持する。DFG/DINポケットがPhe230でブロックされた結果、Mnk−1のDFG/Dモチーフは阻害DFG/DOUT配座に適合する(図11B)。DFDモチーフは、活性状態のタンパク質キナーゼのDFG/DIN配座に対して、Asp191のΦ角度(ΦAsp191=−120)について〜180°だけ回転される(例えば、DAPK1ΦAsp161=55°、図11A)。その結果、Phe120は、Val63、Leu108、Phe124(ゲートキーパー残基)および、Lue177で与えられる、通常、ATPのアデノシル部分を受容する疎水性ポケットを占める。DFG/DOUT配座は、Mnk−2に関する説明において上述した。
【0054】
すなわち、Mnk−1とMnk−2の両方がこの特徴を示すため、DFG/DOUTの適合は、不活性Mnkキナーゼの初期(default)状態であり、活性DFG/DIN配座を呈するその他のSer/Thrキナーゼの多くと、それらの縛られない配座において区別される。
【0055】
Mnk−1において、DFDモチーフは、DFG/DOUT配座に対する選択性を説明するイオンネットワークに関与する。不変Asp191とMnk固有Asp193の両方は、活性部位残基との緊密な酸−酸側鎖相互作用に関与する(図11B)。
(i)Asp191はGlu94に結合し;
(ii)Asp193はAsp170に結合する。
【0056】
Glu94とAsp170は、触媒活性タンパク質キナーゼ間で不変の残基に対応する(ハンクスの、Science(241)1988)42−52。Glu−94は、調節へリックスαCから出発し、ATP結合の生成に必要なLys89−Glu94とイオン対を形成することが知られている(アダムスの、Chem. Rev.(101)(2001)2271−2220)。この対形成は、DFG/DモチーフのAsp191がGlu94と(OD−Asp191:OE−Glu94)さらには、Lys78(O−Asp191:Nz−Lys78)と相互作用するため、Mnk−1中で妨害される。Asp193と相互作用するAsp170は、Cループの触媒アスパラートに相当する。
【0057】
酸−酸側鎖相互作用は一見して異常に見えるが、酸側鎖間の相互作用は、タンパク質構造中で観察される場合が多々あり、酵素の触媒中心内で特に多い(フロッコ(Flocco)の、J. Mol. Biol.(254)(1995)96−105)。その結晶化条件のpH(pH5.6)は、これら相互作用の安定化に都合良いが、これらは塩基性環境においても観察され、局所的なpKaの強い変動を示唆する(同じくフロッコ等)。2つのカルボン酸基間のO−O距離は、おおよそ2.6Åと2.5Åにあり、これは、非酸性水素供与/アクセプター対間のO−O距離よりも著しく短い。後者の観察は、陽子共有結合モードによるものである(同じくフロッコ等)。その他のそのような酸−酸相互作用を説明すると、Asp191:Glu94ならびにAsp193:Asp170が、アミン(Lys78)またはアミド(Asn175)それぞれによって安定化される(比較として、例えば、(ワーテン(Werten)の、J. Biol. Chem.(277)(2002)45502−45509)参照)。
【0058】
活性化セグメントはタンパク質キナーゼドメINの構造要素を具体化し、強い立体配座的可塑性を示し、上流部調節によって構造的に修飾される場合が多々ある(ヒューズ(Huse)の、Cell(109)(2002)275−282)。タンパク質キナーゼの多くにおいて、活性化セグメントのフレキシブル部分は、活性化ループと呼ばれるストレッチに制限されており、DFG/Dモチーフ間に位置するため、P+1ループと呼ばれる(ノレン(Nolen)の、Mol. Cell(15)(2004)661−675)。P+1ループは、基質ペプチドのリン酸化部位に隣接する残基と相互作用することが知られているため、基質ペプチド配置において重要な役割を果たす(ナイトン(Knighton)の、Science(253)(1991)414−420)。
【0059】
Mnkサブファミリー内では、しかしながら、活性化セグメントは、2つのアミノ酸挿入部によってその他のCaMKグループキナーゼに対して延長され(図8)、立体配座的可塑性にさらされるストレッチは強く拡張される。フレキシブル部分は、活性化ループだけでなくP+1ループも含むため、該領域はショートへリックスα−EFとα−EF/αFループに対応する。活性化セグメント、P+1ループとα−EFの両方は、報告されたタンパク質キナーゼ構造の非常に大部分の保存された部位を占める。Mnkにおいては、しかしながら、この領域は折りたたまれておらず、Mnk−1とMnk−2との間で異なる、延長された形態に適合する。そのため、Mnkサブファミリー内の活性化セグメントは拡張され、マグネシウム結合DFDモチーフ(他のキナーゼではDFG)からへリックスαFまでの領域に広がるフレキシブル45アミノ酸ストレッチを包含する(図8B)。
【0060】
自動阻害は、タンパク質キナーゼ制御の卓越した戦略であり、個々のケースにおいて別個に導入され、分子の様々な機能部位に影響を与え得る。タンパク質キナーゼのコアの外側の領域に位置する調節ドメINは、例えば、CaMKI(ゴールドバーグ(Goldberg)の、Cell(84)(1996)875−887)、Twitchin(Kobe,Embo J.(15)(1996)6810−6821)、およびc−KIT(Mol,J.Biol.Chem.(279)(2004a)31635−31663)において採用される。
【0061】
c−KITとFlt−3のケースの場合、2つのタイプIIIレセプターチロシンキナーゼ、DFG/DOUT配座の誘導によりN末端JMドメINは自動阻害するため、ATP結合をブロックすることができる。Mnk−1は、同様に、DFG/DOUT配座の誘導により自動阻害される。しかしながら、c−KITとFlt3とは対照的に、JMドメINが自動阻害を「in trans」で緩和する場合、Mnk−1は、再構築された活性化セグメントを介してDFG/DOUTを誘導し、Phe230をDFG/DINポケット内へ挿入し、これは通常DFG/D−Pheを適応する。そのため、Mnk−1の活性化セグメントは、c−KitとFlt−3のJMドメINのアナロジーにおいて内部自動阻害ドメINとして機能する。Mnk−1とMnk−2の構造は、タンパク質キナーゼ調節のためのDFG/Dモチーフの重要性を強調する。今日では、DFG/DOUT配座は、〜50タンパク質キナーゼのフラクション中でのみ観察されており、その構造的データは入手可能である。
【0062】
留意すべきは、タンパク質キナーゼ阻害因子の開発のため、DFG/DOUT配座は重要である。ある小さい分子阻害因子キナーゼ錯体構造において、DFG/DOUT配座は安定化されおよび/またはBirb796:p38(パルゲリス(Pargellis)の、Nat.Struct.Biol.(9)(2002)(2002)269−272)、Cleevec:c−Abl(ナガール(Nagar)の、Mol.Cell(15)2004(661−675)、およびAAL−993:VEGFR−2(マンレイ(Manley)の、Biochem.Biophys.Acta(1679)(2004)17−27)などに誘導され、それにより、酵素が活性化される。Mnk−1とMnk−2の構造は、DFG/DOUTの適合性が、キナーゼ調節の共通する戦略であるということを証明するものであり、一定の系統グループに制限されるものではない。
【0063】
本発明はさらに、4つの状態を含むMnk活性化のモデルに関連する。
(I)阻害された状態;
(II)中間状態;
(III)プライム化状態;および
(IV)活性状態(図12)。
【0064】
理論で縛られるのを望むことなく、状態IとIIは、Mnk−1とMnk−2の構造にそれぞれ示されており、状態IIIは、Mnk−2の突然変異構造に基づいてモデル化することができ、その他の活性状態のキナーゼと状態IVは仮説である(図12)。配列的な相互転換には、活性化セグメント、NローブおよびATP結合部位に影響を与える明白な位相的転位が要求される。状態Iの特徴は、DFG/DOUT配座の誘導であり、さらに再構築された活性化セグメントの新規な配置によって誘導されるローブクロージャとαC置換である。状態IIは、活性化セグメントの隆起によって可能となり、主にへリックスαCの再配置によってローブ内クレフトを開放し、その結果、必須Lys−Gluイオン対を形成する。しかしながら、状態IIは、不活性状態のキナーゼ、例えば、DFG/DOUT配座のいくつかの特徴を示すため、さらなる構造的再配置を必要とする。
【0065】
活性化セグメントの内部スイッチは、例えば、触媒ベースアスパルテート(Asp−170)の、P+1ループ(Ser218)からのSer/Thr残基との相互作用など、保存された分子内接触の形成を可能にするのに必要である。さらに、マグネシウム結合ループは、DFG/DINポケット内へスイッチし、Mnk−2突然変異構造に見られるようにATPポケット阻害を除外しなければならない。Mnk−2の構造は、さらに、Mnkが、同属のα−EFへリックスとP+1ループ、Mnk−1中で完全に解かれる領域を折りたたむ可能性の損失がないことを証明している。上流キナーゼ(同じくワスキーウィッツ(Waskiewicz))によってターゲットとされる2つのリン酸化部位を担持するMnkの活性化ループは、多くのキナーゼで見られるようにリン酸化によってほぼ安定化される(ジョンソン(Johnson)の、Cell(85)(1996)149−158;ノレン(Nolen)の、Mol.Cell(15)(2004)661−675)。
【0066】
その他の例と同様の、主要なリン酸化は、塩基性RDポケットと相互作用させることによって活性化ループ形態を安定化させることができ、それにより、中性化され、RD−ArgとAsp−238(Mnk−2においてはAsp−273)の相互作用を中断し、これは、活性化セグメントのより先端部分の開放形態を不安定化させる。続いて、二次的リン酸化により、活性化ループ形態がさらに変化されて、状態Iに類似しているが、阻害DFG/DOUT配座のない、ATP結合クレフトに近いGlu−Lysイオン対の誘導によってローブクロージャが誘導される。その結果、新たに誘導された二次P部位の陰電荷は、ウェッジ状態のGlu225とGlu228のために、Arg90およびArg93などの塩基性残基と置換基との相互作用による引き上げ力を提供し、その場でαCへリックスを保持した。要するに、状態II/IIIおよび/またはIII/IVとの間における転換にはリン酸化が必要である。第一リン酸塩は、例えば、RD−Argとの相互作用によって処理された状態IIIを安定化し、第二リン酸化により、活性化セグメント形態を受容する基質をさらに安定化し、αCへリックスとの相互作用によってローブの再封鎖を促進する。
【0067】
へリックスα−EFとP+1ループは、Mnk−1内で解かれて活性化セグメントの完全な再構築物となる。Mnk−1は、いくつかのレベルに対して自動阻害される。活性化セグメントは、2つの構造的変化のクロストーキングシリーズによるこの不活性を必要とする。第一に、これは、DFG/DOUT配座の誘導によるATPポケット遮断を誘導するため、間接的にLys−Glu対とN−ローブとを伝達させる。第二に、これは、へリックスαCとの相互作用によりN−ローブの偽活性閉鎖形態を誘導する。
【0068】
本発明のMnk−1構造は、したがって、キナーゼアーキテクチャの新規な態様と、合理的な阻害設計に使用可能な制御を明らかにする。
【0069】
特に好ましくは、本発明は、結晶ヒトMnk−1タンパク質キナーゼに関する。Mnk−1は、真核細胞阻害因子4E(elF4E)のリン酸化による翻訳機構をターゲットとするヒトタンパク質キナーゼである。
【0070】
本発明はまた、表2に示す構造配位の全てまたは選択された部分によって定義される三次元構造を有する結晶ヒトMnk−1タンパク質に関する。表2に示す配位は、ここにおける例で説明するように得られたものである。
【0071】
【表1】
【0072】
本発明による結晶ヒトMnk−1タンパク質調製物は、例えば、以下によって調製できる。
i.例えば大腸菌などの細胞中でヒトMnk−1タンパク質の発現;
ii.粗Mnk−1タンパク質調製物を回収するために細胞を溶解する;
iii.粗Mnk−1タンパク質調製物を、例えば、タグクロマトグラフィなどにより精製する;および
iv.例えば核酸蒸着などにより、精製したヒトMnk−1タンパク質を結晶化する。
【0073】
ヒトMnk−1タンパク質の結晶調製物、特に、本発明によるヒトMnk−1タンパク質のキナーゼ領域は、ヒトMnkタンパク質の結晶構造データの生成に用いることができる。特に、阻害因子や基質などの配位子との結合部位または相互作用部位、がそれによって得られる。さらには、タンパク質を活性形態または不活性形態に維持するための結合部位を特定することが可能である。特に、ここでMnk−1に対して与えられる結果により、配位子の同定が可能となる。
【0074】
本発明による結晶調製物は、単結晶であることが好ましく、より好ましくは、少なくとも1μm、さらに好ましくは少なくとも10μm、最も好ましくは50μmのエッジ長さを有する結晶であることが好ましい。結晶は、X線構造解析を行えるようなに配置されていることが好ましい。したがって、本発明のさらなる主題は、表2に示す構造配位の全てまたは選択された部分で定義されるヒトMnk−1タンパク質の結晶構造である。
【0075】
結晶ヒトMnk−1タンパク質と、結晶構造とをそれぞれ用いることにより、Mnk−1タンパク質配位子を設計、同定または調製することができる。また、上述のようにタンパク質キナーゼの制御機構を特定することもできる。配位子または制御機構を特定するために、特に、コンピュータ支援モデル化プログラムを用いる。
【0076】
配位子を同定するコンピューター支援のスクリーニングに加えて、WO03/037362号公報に開示される方法を実際の配位子同定および確認に適用することが好ましい。
【0077】
本発明はさらに、特に、Mnk−1タンパク質のアイソマー、ならびに結晶調製物や結晶構造を用いて得られたその他タンパク質キナーゼの基質や阻害因子などの配位子に関する。そのような配位子は、薬剤組成物中で活性剤であることが好ましい。そのような薬剤組成物は、Mnk−1の操作または特に阻害が必要な、例えば、肥満、糖尿病およびメタボリック症候群ならびに癌などの代謝障害の場合などの病気の治療に用いることができる。
【0078】
ここで提供する結果およびデータは、DFG/DINポケット(Mnk−1中のPhe230を含む)が、汎用的な阻害因子結合部位として機能可能であることを示している。阻害因子は、Mnkに制限されない。したがって、阻害因子はまた、DFG/Dinポケットからなる阻害因子結合部位に関する。
【0079】
本発明はさらに、添付の図面ならびに以下に与えられる例によって例示される。
図1:Mnk2の組織と配列アライメント
(A)(ラベルを付したように)機能ドメINの配置を示す、ヒトMnk2の2つのスプライス変異体の概略的な比較図。ここで調査する領域(Mnk2キナーゼ領域、Mnk2−KR)は四角で囲まれている。代替的なスプライスは、N末端にもキナーゼドメIN、NLS核局在信号、elF4G真核細胞阻害因子4G、翻訳阻害複合体の足場タンパク質に影響を与えず、Mnk1とMnk2に結合する(1999年のパイロネット(Pyronnet)等、2001年のシェーパー(Scheper)等)
(B)ヒトMnk1とMnk2のキナーゼドメINの配列アライメント、DrosophilaおよびC.elegansMnk遺伝子座(Lk6とR166.5のそれぞれ)および既知構造の3つのヒトCaMK群キナーゼ(MAPKAP−MAPキナーゼ活性化タンパク質キナーゼ)Mnk2のナンバリングは、最近報告された配列に基づく(2000年のスレンツ−ケスラー(Slenz−Kesler)等)。Mnk2−KR中に見いだされた二次構造要素は、以下の配列に示される。星印は、リン酸化部位を示している(2001年のシェーパー(Scheper)等)。触媒ループ(i)DFDモチーフ(その他のキナーゼ中のDFG);と(ii)P+1ループとは、(iii)着色されたバーでマークされている。Mnkの挿入部の特徴は四角でかんでいる(l1−l3)。白抜きの円は、グリシンリッチループのGly91とGly93、ATP結合に重要であることが知られているLys113とGlu129(1994年のテイラーとラッジオ−アンゼルム(Taylor and Radzio−Andzelm))、黒塗りの円は、N末端ローブとC末端ローブを分離するヒンジ領域のGly163とGly165を示す。
図2: Mnk−2キナーゼの全位相。キナーゼドメINのコアの外側の構造的部分は削除された。CAMK1(a,1a06.pdb)、DAPK1(b,1jks.pdb)およびMAKPKAPK2(c;1kwp.pdb)のアポ酵素の構造は、Mnk−2(b)上に重ねられ、同様の配向上に示されている。電子密度でトレースできない部分は点線で示されている。
図3:活性化セグメントの開放形態。赤と青で着色された2つの対称性等価Mnk−2分子は、(a)に示される。同分子は、90°だけ回転された、上から示されている。(c)は、1σで着色された2Fo−Fc電子密度とDAPK1の同じ領域(黒)を示している。
図4:ATP結合ポケットの形態。Mnk−2(青)、MAPKAP2(赤)、CaMk1(緑)およびDAPK1(黒)からの触媒作用の重要性を有する領域が示されている。(a)は、Ly113とGlu129(Mnk−2ナンバリング)を示す。(b)において、Cループのバックボーンと、Asp205とAsn210の側鎖とは、MAPKAP2/ADP共構造(1ny3.pdb)由来のADP(黄)と一緒に示されている。(c)は、DFG(DFD)モチーフ周囲のバックボーンを示しており、(d)はこの領域と(b)由来のADPの側鎖を包含している。
図5:亜鉛結合部位。(a)Mnk−2中の推定状の亜鉛部位の領域が、1σ(青)で描かれた2Fo−Fcマップと、5σで描かれたDANOマップとともにバックボーンプロットとして示されている。領域は、我々の結晶中で高度にフレキシブルであり、Trp305からGlu309までの領域は鮮明なバックボーン密度を欠いている。(b)I=ZnKα線、II=ZnKベータ、IIIコンプトン散乱、IV弾性散乱に対応するピークを有する、天然Mnk−2結晶のX線放射スペクトル。
図6:Mnk−2キナーゼドメINとp38との比較
A.同じ配向のMnk−2キナーゼドメIN(左)とp38(右;PDB ID1KV1)のリボンプロット。分子はN末端(青)からC末端(赤)まで七色に着色されており、その他のタンパク質キナーゼでも観察されるように、全て同じ構造的構成を示している。
B.2つのタンパク質の最適グローバルアライメント後のDFD/DFG領域の空間プロット。Mnk−2−石灰;p38−コムギ。DFD/DFGモチーフは、スティック形状で示されており、原子型でカラーコードされている(炭素(Mnk−2)−石灰;炭素(p38)−コムギ;酸素−赤;窒素−青)。Mnk−2のアスパルテート226と228は、ポリペプチド鎖の方向を示すようラベルを付されている。周囲を取り囲む構造要素はリボンで示されている。p38の非定型型DFG形態は、尿素ジアリール型の阻害因子の結合によって誘導される(図示せず;PDB ID1KV1と1KV2)。Mnk−2は、本発明の結晶中で自然に同じ形態に適合する。阻害因子の尿素ジアリールクラスはp38のDFGモチーフとへリックスとの間で結合し、へリックスはバックグラウンド中に表れた。Mnk−2のDFDモチーフは、阻害因子結合ポケットに向かってさらに置き換えられ、阻害因子によって本発明の形態中に同様に捉えることができることを示唆している。
図7:Mnk−2キナーゼドメINに結合する阻害因子のモデル
A.ジアリール尿素ベースの阻害因子(1−(5−tブチル−2−メチル−2H−ピラゾール−3−イル)−3−(4−クロロフェニル)尿素;BMU;PDB ID 1KV1)との錯体であるMnk−2キナーゼドメINの外観。Mnk−2は、その二次構造要素(へリックス−赤、ストランド−青、ループ−グレイ)に従って示される。阻害因子1(炭素−オレンジ)、DFDモチーフ(炭素−ピンク)とその他の薬(炭素−シアン)に接触するMnk−2残基は、スティック形状で示される。モデルは、p38−BMU複合体(PDB ID1KV1)のCα原子位置と、Mnk−2キナーゼドメINのCα配位との最適なスーパーポジションによって作製される。MNU配置は、続いて、手動により示されたMnk−2キナーゼドメINの結合ポケットに適合させた。いくつかのMnk−2残基の側鎖形態も同様に不良接触を排除するよう適合させた。
B.Mnk−2キナーゼドメINのヌクレオチド結合ポケット中に配置されたDAPK1(PDB ID 1lG1)の共晶構造由来の、ATPアナログ(AMPPNP)の詳細な空間図。モデルは、Aで説明したような2つのタンパク質分子の最適なスーパーポジションによって作製された。標準結合モデル中のAMPPNP分子が、本発明の形態のMnk−2のDFDモチーフと立体的に干渉するように見える。この発見は、生産的なMnk−2へのATP結合がDFDモチーフ内での再配置を必要とすることを示唆する。その結果、本発明の形態において、Mnk−2は、ATP結合内において不活性であることが導き出される。別の分子とモチーフは、原子型ごとにカラーコードされている。炭素(AMPPNP)−オレンジ;炭素(DFD)−ピンク;窒素−青;酸素−赤;亜リン酸−石灰。A中に二次構造要素。
C.Mnk−2−BMU複合体モデルの詳細な空間図。BMUは、疎水性ポケットでt−ブチル基と結合し、Phe227(DFDモチーフ由来)とPhe159の芳香環の間のpクロロフェニル環をスライドさせる場合がある。別の分子とモチーフは、原子型ごとにカラーコードされている:炭素(BMU)−オレンジ;炭素(DFD)−ピンク;炭素(疎水性ポケット)−シアン;窒素−青;酸素−赤;塩素−緑。A中のような二次構造要素。
図8:立体的表現(a)と主要配列(b)におけるMnk−1の全構造。他に示さない限り、着色法は以下のスタイルに維持される。Nローブ:グレイ;Cローブ:黒;Cループ:黄;DFG/Dモチーフ:オレンジ;αCへリックスとLys−Glu対:シアン;活性化セグメント:緑。(b)ATPと相互作用すると知られている残基をカラーコードでマークし、DFG/Ginポケットからなる残基:緑の白抜きの円;DFG/DOUTポケットからなる残基:赤の白抜きの円。Mnk固有のアミノ酸挿入部は、四角で囲まれ、官能性の適合性の高いMnk固有残基は、赤い矢印でハイライトしている。リン酸化部位は星印で示している。
図9:活性化セグメントにより誘導されたNローブ移動。Mnk−1(a)とMnk−2(b)の全構造。残基を含むMnk−1(a)は、NローブαC相互作用、Phe239とDFDモチーフに包含された。スティックで表されたPhe。Arg90とArg93は、亜リン酸アミノ酸と相互作用することが知られている残基に対応した(クリュッパ(Krupa)等の、J. Mol. Biol.(339)(2004)1025−1039)。Mnk−2(b)中で対応する残基は、Phe265、Arg123とArg125である。
図10:c−KIT(a,b)とMnk−1(C)における自動阻害c−KITの自動阻害JMドメINは赤で着色されている。
図11:(a)DAPL1(1ig1;(テレスコ(Tereshko)等の、Nat.Struct.Biol.(8)(2001)899−907));(b)Mnk−1;Mnk−2のATP結合ポケット。分子は図8のように同じ配向にあり、ATP結合領域が拡大されている。(a)は活性状態のCaMK群のタンパク質キナーゼを例示し、非クリーブ化ATPアナログANP−PNPと、Mg2+の代わりに官能部位においてMn2+を含む。マグネシウム結合DFGモチーフの許容可能なDFG/Dinポケット形態に留意。Mnk−1(b)とMnk−2のATP部位遮断は、阻害DFG/DOUT配座によって得られる。Mnk−1(b)は、酸−酸側鎖の相互作用がMnk−2中に存在しないことを示している。
図12:Mnk活性化カスケードモデル。
図13:DFDモチーフの近傍
(A)DFD領域とATP結合クレフトの拡大空間図。野生型Mnk2−KRのDFG/DOUT配座は、左上にAsp226、Phe227とAsp228に関して、Phe227とAsp228とがATP結合クレフト内に突き出した状態でスティックにより示されている(炭素、シアン)。DFG/DIN配座(右下;炭素、緑)は、Mnk2−KRのAsp228Gly突然変異体について観察されるように、その他のキナーゼに見られるDFG/DIN配座に従ってモデル化された。Mnk2−KRのバックボーントレースは、半透明なグレイのチューブで示される。DFG/DINまたはOUT配座いずれかにおけるDFDモチーフ周囲の径4Å内の残基は、スティックで示されている(炭素、グレイ)。DFG/DOUT配座を安定化する、タンパク質マトリックスとの直接的な相互作用は、点線で示されている。Phe227は、2つの異なる疎水性ポケット中に2つの異なる配座で存在するようになる。DFG/DIN配座の適合に対する障害は見えない。
(B)((A)におけるのと同様にカラーコードされている)スティックで示されたDFDモチーフの2つの配座をともなう、静電ポテンシャルごとにカラーコードされたMnk2−KRの分子構造の空間図(青、正電荷;赤、負電荷)。ATP結合クレフトが指摘されている。いずれかの配座にあるAsp228は、水溶性溶媒を良好に利用可能である。DFG/DOUT配座は、ATP結合クレフト中にPhe228とAsp228を位置するだけでなく、このクレフトの前からのアクセスを遮断する。分子は、DFDポケット中への遮断されていない図を得るために、水平軸(N末端ローブから後ろ)について、(A)に対して30°回転されている。
(C)DAPK1との共晶構造(PDB ID 1IG1)に見られるようにスーパーINポーズされた、非加水分解性のATPアナログ(アデノシン5’−[β,γ−イミド]三リン酸[AMPPNP]);炭素、ベージュ、亜リン酸、紫)による(A)におけるのと同じ図。DFG/DOUT配座において、アデノシン塩基はPhe227の側鎖とクラッシュし、リン酸基はAsp228の側鎖とクラッシュする。
(D)DFG/DOUT配座のみが示された、(A)と(C)におけるのと同じ図。p38−BMU阻害複合体(PDB ID 1KV1)のDFG領域は、タンパク質構造のグローバルスーパーポジション後に見られるように、比較目的で示されている(マジェンタ色のチューブ;スティックで示されたDFG;炭素、マジェンタ色)。BMU阻害因子(炭素、ベージュ;塩素、緑)は、DFG/DIN結合ポケットの部分をふさぎ、p38中にDFG/DOUT配座を誘導する。
表1:ヒトMnk−2の原子配位を示す。
表1a:ヒトMnk−2突然変異D228Gの原子配位を示す。
表2:ヒトMnk−1の原子配位を示す。
表3:ヒトMnk−2キナーゼ突然変異D228Gと一般的なタンパク質キナーゼ阻害因子スタウロスポリンとの共結晶の原子配位を示す。
【図面の簡単な説明】
【0080】
【図1】図1は、Mnk2の組織と配列アライメントを示している。
【図2】図2は、Mnk−2キナーゼの全位相を示している。
【図3】図3は、活性化セグメントの開放形態を示している。
【図4】図4は、ATP結合ポケットの形態を示している。
【図5】図5は、亜鉛結合部位を示している。
【図6】図6は、Mnk−2キナーゼドメINとp38との比較を示している。
【図7】図7は、Mnk−2キナーゼドメINに結合する阻害因子のモデルを示している。
【図8】図8は、立体的表現(a)と主要配列(b)におけるMnk−1の全構造を示している。
【図9】図9は、活性化セグメントにより誘導されたNローブ移動を示している。
【図10】図10は、c−KIT(a,b)とMnk−1(C)における自動阻害を示している。
【図11】図11は、(a)DAPL1(1ig1;(テレスコ(Tereshko)等の、Nat.Struct.Biol.(8)(2001)899−907));(b)Mnk−1;Mnk−2のATP結合ポケットを示している。
【図12】図12は、Mnk活性化カスケードモデルを示している。
【図13】図13は、DFDモチーフの近傍を示している。
【図14】図14は、原子配置を示す。
【実施例】
【0081】
実施例1
Mnk−2とMnk−1キナーゼ領域のクローニングと精製
従来既知の技術を用いて、アミノ酸残基72〜385に対応し、キナーゼドメIN(KD)を包含するヒトMnk−2のcDNAフラグメントを、F(forward)/R(reverse)プライマー対5’CGGGATCCACCGACAGCTTCTCGGGCAGG / 5’ACGCGTCGACCTACCTCTGCAGGACCATGGGAG (利用した限定部位は下線部)を用いて増幅し、ベクターpGEX−4T1のBamHiとSall部位中にクローン化した(アマーシャム(Amersham)の、スウェーデン、cat.no.27−4580−01)。この構築により、N末端を有する融合タンパク質、トロンビンクリーブ可能なグルタチオンSトランスフェラーゼ(GST)タグとしてのMnk−2キナーゼ領域(KR)の原核細胞発現が可能となる。
【0082】
アミノ酸置換D228Gは、製造元の指示にしたがう、Stratagene Quik Change Site Directed Mutagenesisキットを採用して、GST−Mnk−2KR構造中に導入された。突然変異誘発オリトヌクレオチドは、5’GAAGATCTGT GACTTCGGC CTGGGCAGCG GCATCAAACT Cおよび5’GAGTTTGATG CCGCTGCCCA GGCCGAAGTC ACAGATCTTCであった。Mnk−2KRD228Gの精製は、Mnk−2KRに関して説明したように実施した。
【0083】
アミノ酸残基37〜341に対応し、キナーゼドメIN(KD)を包含するヒトMnk−1のcDNAフラグメントは、F/Rプライマー対5’CGGGATCCACTGACTCCTTGCCAGGAAAG/ 5’ACGCGTCGACCTATCCCTTTTCTGGAGCTTGCCを用いて(利用した限定部位は下線部)を用いて増幅し、ベクターpGEX−4T1のBamHiとSall部位中にクローン化した(アマーシャム(Amersham)の、スウェーデン、cat.no.27−4580−01)。この構築により、N末端を有する融合タンパク質、トロンビンクリーブ可能なグルタチオンSトランスフェラーゼ(GST)タグとしてのMnk−2キナーゼ領域(KR)の原核細胞発現が可能となる。
【0084】
GST−Mnk−2KRまたはGST−Mnk−1KRの発現は、大腸菌BL21(Merck Biosciences, Germany, cat. no. 69449)中である。細胞は、バッフル付の5リットルフラスコ内で、100μg/mlのアンピシリン(シグマ(Sigma)の、ドイツ、cat. no. A-9518)で補ったLBブイヨン中(メルク(Merck)のドイツ、cat. no. 1.10285)で、37℃、毎分130回転(rpm)で成長させた。培地が、0.8のA600に相当する密度に達したとき、同容量の氷温のLB/アンピシリンを加え、培地を25℃にして、1mMのイソプロピルチオガラクトースで誘導した(ロス(Roth)の、ドイツ、cat. no. 2316.4)。細胞を遠心分離で採取した。細胞ペレットは、その湿潤重量1gあたり10mlのリーシスバッファ(50mMトリス/HCl(シグマ(Sigma)の、ドイツ、cat. no. T-5941)、pH7.5、200mMNaCl(シグマ(Sigma)の、ドイツ、cat. no. S-7653)、5mMDTT(ロス(Roth)の、ドイツ、 cat. no. 6908.2))で再希釈した。MS72プローブ付のBadelin sonoplus sonifierによる細胞の崩壊(バデリン(Badelin)の、ドイツ、cat. no. HD207)と、後続のSorvallSS34ロータ(ソルボール(Sorvall)、ドイツの、cat. no. 28020)中における18000rpm/45分/4℃での除去によりライセートを調製した。ライセートは、直列に接続した、リーシススバッファで平衡した2つのGSTPrepFF16/10カラム(アマーシャム(Amersham)の、スウェーデン、cat. no. 17-5234-01)に適用した。洗浄液は、3カラム容量(CV)ウォッシュバッファ(50mMトリス/HCl、pH7.5、100mMNaCl、1mMDTT)、2CVのATPバッファ(50mMトリス/HCl、pH7.5、100mMKCl(ロス(Roth)の、ドイツ、 6781.1))、20mMMgCl2(シグマ(Sigma)の、ドイツ、cat. no. M-2670)、5mMATP(シグマ(Sigma)の、ドイツ、cat. no. A-7699))、およびさらに3CVウォッシュバッファ)であった。Mnk−2KDは、続いて、GSTタグ由来のオンカラムのトロンビン開裂によって溶出した。簡単にいうと、1000ユニットのトロンビン(アマーシャム(Amersham)のスウェーデン、cat. no. 27-0846-01)を、60mlウォッシュバッファに溶解して、2つのカラムにわたって8℃で一晩循環させた。溶出物を、ウォッシュバッファをカラムに適用しながらループを開くことによって回収した。
【0085】
トロンビン溶出物は、50mMトリス/HCl、pH8.0中に1:5に希釈し、直列に接続した5mlのQセファロースHPカラム(アマーシャム(Amersham)の、スウェーデン、cat. no. 17-1154-01)に適用した。溶出物は、塩化ナトリウムの直線上の勾配を有するものであった(50mMトリス/HCl、pH8.0、0−1MNaCl)。フラクションは、10.000ダルトン分子量カットオフ(MWCO)VivaSpin濃縮器中で(ビバサイエンス(VivaScience)、ドイツ、cat. no. VS0403)、おおよそ16mg/mlまで精製かつ濃縮してプールした。濃縮物は、PD10カラム(アマーシャム(Amersham)、cat. no. 17-0851-01)へのゲルろ過により、10mMトリス/HCl、pH7.5、50mMNaCl、1mMDTT中に移した。典型的な最終タンパク質濃度は、おおよそ12mg/mlであった。アリコートは、液体窒素内でショック凍結させ、−80℃で保存した。タンパク質生成量は、細胞ペレット湿潤重量1g当たりのおおよそ2mgのMnk−2キナーゼドメINであった。ERK2によって活性化した後、対応するMnkキナーゼ領域と完全長のMnkタンパク質とは、elF4e(Ser209)リン酸化に基づくキナーゼアッセイにおいて同一の活性を示す。
2.結晶化とデータ収集
初期結晶スクリーニングは、100μl貯留溶液と、200nl〜1μlの範囲の液滴寸法とを用いる96ウェルフォーマット中でMicroSys SQシリーズ4000/4100(デカルト分配システム(Cartesian Dispensing Systems))により実施した。回折研究に用いた結晶は、20℃で懸滴または座滴のいずれかを用いて拡散蒸着によって成長させた。タンパク質溶液は、タンパク質溶液の10倍を超える量までの貯留バッファ(100mMNa−Hepes、pH7.8、22%ポリアクリル酸5100と2%2,3−メタンペンタジオール(MPD))と混合した。結晶は、液体窒素中で凍結させた。回折データは、Mar−Research(ノルデステット(Norderstedt)、ドイツ)CCD検出器の100Kおよびλ=1.05で、HASYLABビームラINBW6(DESY、ドイツハンブルグ)上に回収し、HKLパッケージ(オトウィノスキ・ゼット(Otwinowski,Z)とマイナー・ダブリュー(Minor,W)の、オシレーションモードにおけるX線開設データの処理、Methods Enzymol. 167, 307-326, 1997年9月)で処理した。
3.構造決定と精製
初期相は、研究モデル(PDB ID:1lG1)として死亡関連タンパク質キナーゼ(DAPK)を用いたCCP4パッケージ(Collaborative Computational Project, The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. D 50, 760-763, Dec 1994年)からのMolRep自動化分子置換ルーチンを用いて得た。相情報を有するmtz形式のファイルを、REFMAC(Murshudov, G. N., Vagin, A. A., Lebedev, A., Wilson, K. S.とDodson, E. J.のFFTを用いた効率的な分子構造の異方性精製、Acta Crystallogr. D Biol. Crystallogr. 55 (Pt 1), 247-255,1999年1月)中で剛性体精製を用いて作製し、arp/warpによる自動化モデルの構築(Morris, R. J., Perrakis, A.とLamzin, V. S. ARP/wARPとタンパク質電子密度マップの自動翻訳;Methods Enzymol. 374, 229-244 (2003年))に用いた。得られたモデルは、Xフィット(McRee, D. E.の、原子座と電子密度を操作するためのXtalView/Xfit-Aバーサタイルプログラム;J. Struct. Biol. 125(2-3), 156-165, 1999年4月)を用いて手動でさらに変更した。精製は、CNS(Brunger, A. T., Adams, P. D., Clore, G. M., DeLano, W. L., Gros, P., Grosse-Kunstleve, R. W., Jiang, J. S., Kuszewski, J., Nilges, M., Pannu, N. S., Read, R. J., Rice, L. M., Simonson, T.およびWarren, G. L.の、結晶学とNMRシステム:マクロ分子構造決定に適した新しいソフトウェア、Acta Crystallogr. D Biol. Crystallogr. 54 (Pt 5), 905-921, 1998年9月)とREFMAC(Murshudov, G. N.等, 1999年、上記参照)を用いて実施した。
4.ゲルろ過と光散乱
Superdex75PC3.2/30カラム(ファーマシア(Pharmacia)製)を用いてSMARTシステムによりゲルろ過クロマトグラフィを実施した。実験は、バッファA(20mMトリスHCl、pH7.5、100mMNaCl、1mMDTT)中で0.04ml/分の流速において室温で行った。Mnk−2KDの分子量は、基準タンパク質(Bio−Rad)を用いて推定した。マルチアングルレーザ光散乱を、UV分光計と、ドーンとオプチラブ(Dawn and Optilab)計器XY(ワイアットテクノロジー社(Wyatt Technology Corp.))に接続したHR−10/30Superdex−200サイズの排除カラム(アマーシャム)で行った。30μmのMnk−2a溶液を、バッファA中で、632.8nm、90度におけるUV吸収、光散乱で、クロマトグラフィによって分離し、溶出プロファイルの屈折差はASTRAソフトウェアパッケージ(Wyatt, P.のマクロ分子の光散乱と絶対化学分析:Anal. Cim. Acta 272, 1-40 (1993年))で監視し、分析した。
【0086】
実施例2
Mnk−2特異阻害因子設計のために誘導されたp38−ジアリール尿素阻害因子共晶構造
タンパク質キナーゼp38の構造は、全体的に、Mnk−2キナーゼドメINの構造に非常に類似している(図1A)。p38は、ATP結合ポケット中の典型的なDFG配列モチーフを特徴付ける。ジアリール尿素足場に基づくp38特定の阻害因子を設計し、p38のこれら2つの阻害因子(BMUとBIRB796、(Pargellis et al. (2002年), Nat. Struct. Biol. 9, 268-272))との共晶構造を解明した(PDB Ids 1KV1と1KV2のそれぞれ)。これらの阻害因子は、p38の非カノニカル形DFG配座(DFGOUTと称す)を誘導し、この配座においては、フェニルアラニンは疎水性ポケットにおけるその標準位置から置き換えられており(DFGINと称す)、アポ酵素とその他タンパク質キナーゼ構造中に存在する(図1B)。DFGモチーフのDFGOUT配座は、立体障害により生産的ATP結合と干渉する。
【0087】
Mnk−2キナーゼ領域は、DFGモチーフに代わりDFDを呈する(残基226−228、図1参照)。非活性化アポ酵素の構造において、このDFDモチーフは、p38の非カノニカル形DFGOUT配座に類似する配座に適合する(図1B)。Mnk−2のPhe227は、クレフト中へ突出し、この場合、p38内はジアリール型阻害因子で占有することができる(図2A)。置換は、p38−阻害因子複合体よりも厳密であるが、Mnk−2キナーゼ領域の結晶化には阻害因子は用いられない(図1B)。Mnk−2キナーゼ領域結晶構造中に見られるDFD配座はまた、立体障害のためにカノニカル形ATP結合とは共存しない(図2B)。観察により、本発明の結晶構造に観察されるDFG/DOUT配座中のDFDモチーフの捕捉が、Mnk−2を不活性にし、リン酸化状態を無視させることが示唆される。
【0088】
ジアリール型阻害因子BMUがMnk−2キナーゼ領域を結合可能であるかどうかを調査した。p38−BMU複合体(PDB ID 1KV1)のMnk−2キナーゼ領域上への全体的なスーパーポジション後、BMU位置におけるわずかな手動調整と、いくつかのMnk−2側鎖配座におけるわずかな再調整とを行い、Mnk−2−BMU複合体モデルを得た(図2Aと図C)。阻害因子は、Phe227とPhe159の環の間に挟まれたそのpクロロフェニル環と結合することが見られた(図2Aと図C)。そのtブチル部分は、Mnk−2の疎水性ポケットに収容可能であった(図2Aと図C)。Mnk−2の異常なAsp228は、このモデル中のBMU分子からかけ離れているが、p38中のその他の領域(p38の、1KV2構造のBIRB796の延長された足場と1KV1構造のBMUとの比較)で示したように、さらなる改質された阻害因子上の化学基によってターゲットとすることができた。この方法において、DFDモチーフへの特異性(Mnk−2のフINガープリント;その他のキナーゼのDFGに代わり)が達成され得る。Mnk−2に対する特異で強力な結合は、Mnk−2の特異的な結合に対する新規な阻害因子に適合させるためのBMUのpクロロフェニルとtブチル基のさらなる改質によって支持することができた。
【0089】
実施例3
Mnk−1−KRの構造決定および全構造
野生型Mnk1−KRの針状結晶を、タンパク質溶液を等容量の20%(w/v)PEG3350と0.2M硫酸アンモニウムと0.1Mクエン酸ナトリウムを含有するpH5.4の貯留溶液とを混合後の拡散蒸着により、20℃で成長させた。結晶は、20%のグリコールで補った貯留溶液中で凍結させた(液体窒素)。回折データを、ビームラINPXII(SLS、ビリンゲン(Villingen)、スイス)に対して、MarResearch(ノルダーシュテット(Norderstedt)、ドイツ)CCD検出器における100Kで収集し、HKLパッケージ(オトウィノスキ・ゼット(Otwinowski,Z)とマイナー・ダブリュー(Minor,W)、1997年)で処理した(表2参照)。
【0090】
Mnk−2−KRの密度変更後の切頭型モデルを用いた分子置換後、翻訳可能な電子密度を得て、モデルを23.5/28.0%のR/Rfree因子に精製することができた(表2)。非対称ユニットは、非クリスタルグラフィックの2倍軸で関連づけられる2つのMnk−1−KR分子を含む。分子Aは、いくつかの領域においてより低温の因子とクリアラ電子密度を呈する。機能的に重要な領域は、しかしながら、分子Aと分子Bとの間で実際上同一である。最終モデルは、Mnk−1のキナーゼドメINの範囲にわたり、残基39−335を含む。Mnk−1−KRは、バイロバル(bilobal)構築を含むキナーゼアーキテクチャのいくつかの広域な特徴を保存する。N末端ローブは、グリシンリッチループやLys−Gluイオン対などのATP結合に必要な重要な要素を担持し、5本撚りのねじれたβシートと調製へリックスαCの形態にされる(図8)。大型で優勢なαらせんC末端ローブは、触媒ループ(Cループ)、マグネシウム結合ループ(DFDモチーフ)、および活性化セグメントなどの、基質結合および亜リン酸転移に必要な要素を含む(図8)。Mnk−1−KR内のセグメントは、強い、配座的なフレキシビリティを呈し、そのため、電子密度では痕跡をたどることはできず、活性化セグメントのコアはP+1ループ(残基197−222)と、へリックスαG(残基261−290)を包含するMnk特異システINクラスタとを含む。
【特許請求の範囲】
【請求項1】
結晶ヒトMnk−2キナーゼ。
【請求項2】
結晶ヒトMnk−2aキナーゼであることを特徴とする、請求項1記載の結晶ヒトMnk−2キナーゼ。
【請求項3】
前記ヒトMnk−2キナーゼが残基72−385を含むことを特徴とする、請求項1または2のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項4】
前記ヒトMnk−2キナーゼがキナーゼドメINを含むことを特徴とする、請求項1から3のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項5】
ヒトMnk−2キナーゼ突然変異体であることを特徴とする、請求項1から4のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項6】
ヒトMnk−2キナーゼ突然変異D228Gであることを特徴とする、請求項5に記載の結晶ヒトMnk−2キナーゼ。
【請求項7】
結晶ヒトMnk−2キナーゼの配位子、基質および/または阻害因子との複合体であることを特徴とする、請求項1から4のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項8】
前記阻害因子がスタウロスポリンであることを特徴とする、請求項7記載の結晶ヒトMnk−2キナーゼ。
【請求項9】
前記ヒトMnk−2キナーゼが不活性形態であることを特徴とする、請求項1から8のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項10】
単結晶であることを特徴とする、請求項1から9のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項11】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分によって決定される三次元構造を有することを特徴とする、請求項1から10のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項12】
(i)細胞中のヒトMnk−2キナーゼを発現すること、
(ii)粗Mnk−2キナーゼ調製物を回収するために細胞を溶解すること、
(iii)粗Mnk−2キナーゼ調製物を精製すること、
(iV)精製したヒトMnk−2キナーゼを結晶化すること、
を含んでなることを特徴とする、結晶ヒトMnk−2キナーゼ調製物の製造方法。
【請求項13】
前記Mnk−2キナーゼが、大腸菌中の溶融タンパク質として発現されることを特徴とする、請求項12記載の方法。
【請求項14】
前記Mnk−2キナーゼが、融合タグに結合されたカラムを用いて精製されることを特徴とする、請求項12または13のいずれかに記載の方法。
【請求項15】
前記結晶が拡散蒸着によって成長されることを特徴とする、請求項12から14のいずれかに記載の方法。
【請求項16】
ヒトMnkキナーゼの結晶構造データを生成するための、請求項1から11に記載のヒトMnk−2キナーゼまたは請求項12から15のいずれかに記載の方法によって得られたヒトMnk−2キナーゼの結晶調製物の使用。
【請求項17】
配位子、基質および/または阻害因子の結合部位を決定するための、請求項16記載の使用。
【請求項18】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分によって決定されるヒトMnk−2キナーゼの結晶構造。
【請求項19】
不活性ヒトMnk−2キナーゼの結晶構造。
【請求項20】
Mnk−2キナーゼ配位子の設計、同定および/または調製のため、あるいは、タンパク質キナーゼの制御機構の特定のための、請求項1から11のいずれかに記載か、請求項12から15のいずれかによって得られる結晶ヒトMnk−2キナーゼか、および/または請求項18または19のいずれかに記載の結晶構造の使用。
【請求項21】
配位子分子の設計にコンピュータ支援モデル化プログラムを用いることを特徴とする、請求項20記載の使用。
【請求項22】
前記配位子が、ヒトMnk−2キナーゼの相互作用部位と相補的な三次元構造を有することを特徴とする、請求項20または21のいずれかに記載の使用。
【請求項23】
前記配位子が、アミノ酸Asp226、Phe227およびAsp228のうちの少なくともいずれか1つと相互作用することを特徴とする、請求項22記載の使用。
【請求項24】
前記配位子が、亜鉛結合部位のシステIN残基の少なくとも1つと相互作用することを特徴とする、請求項22記載の使用。
【請求項25】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分を提供することと、
該結晶構造の三次元表現を構築するために該構造配座を使用することと、
を含んでなることを特徴とする、ヒトMnk−2キナーゼの結晶構造の三次元表現を得る方法。
【請求項26】
請求項25に記載のヒトMnk−2キナーゼの三次元表現を提供することと、
前記化合物の三次元表現を提供することと、
前記化合物の三次元表現と、ヒトMnk−2キナーゼの該三次元表現と、を適合させることと、
を含んでなることを特徴とする、ヒトMnk−2キナーゼとの化合物の相互作用の分析のためのコンピュータベースの方法。
【請求項27】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分を提供することと、
前記タンパク質の結晶構造を得る分子置換のために、該構造配座を使用することと、
を含んでなることを特徴とする、タンパク質、特に、キナーゼの結晶構造を決定する方法。
【請求項28】
コンピュータ読み取り可能データを有するデータ保存媒体からなる該データを含んでなり、該データは表1、表1aまたは表3に示す構造配座の全てまたは選択された部分を含んでなることを特徴とする、コンピュータ読み取り可能な保存媒体。
【請求項29】
請求項1から11のいずれかに記載の結晶ヒトMnk−2キナーゼ、請求項18または19のいずれかに記載のヒトMnk−2キナーゼの結晶構造データ、および/または請求項20から22のいずれかに記載の方法の使用を用いて得られたヒトMnk−2キナーゼのための配位子。
【請求項30】
Mnk−2キナーゼの基質または阻害因子であることを特徴とする、請求項29記載の配位子。
【請求項31】
Mnk−2キナーゼのアロステリック阻害因子であることを特徴とする、請求項29または30のいずれかに記載の配位子。
【請求項32】
請求項29から31のいずれかに記載の配位子を含む薬剤組成物。
【請求項33】
体重制限または発生熱に関連する病気または障害の治療、緩和および/または防止のための、請求項32記載の薬剤組成物。
【請求項34】
メタボリック症候群、摂食障害、カヘキシー、糖尿病、高血圧、冠状脈心臓疾患、高コレステロール血症、異常脂質血症、変性関節疾患、胆石、睡眠中無呼吸症、神経変性疾患障害または癌の治療、緩和および/または防止のための請求項32または33のいずれかに記載の薬剤組成物。
【請求項35】
炎症または炎症徴候の治療、緩和および/または防止のための請求項32記載の薬剤組成物。
【請求項36】
アロステリックタンパク質キナーゼ阻害因子の同定を可能にする構造モチーフの同定のための方法。
【請求項37】
前記キナーゼがp38、Mnk−1またはMnk−2から選択されることを特徴とする、請求項33記載の方法。
【請求項38】
結晶ヒトMnk−1キナーゼ。
【請求項39】
前記比とMnk−1キナーゼが、残基37から341を含むことを特徴とする、結晶ヒトMnk−1キナーゼ。
【請求項40】
前記ヒトMnk−1キナーゼが、前記キナーゼドメINを含むことを特徴とする、請求項38または39のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項41】
ヒトMnk−1キナーゼ突然変異体であることを特徴とする、請求項38から40のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項42】
結晶ヒトMnk−1キナーゼの配位子、基質および/または阻害因子との複合体であることを特徴とする、請求項38から41のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項43】
前記ヒトMnk−1キナーゼが不活性形態であることを特徴とする、請求項38から42のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項44】
単結晶であることを特徴とする、請求項38から43のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項45】
表2に示す構造配座の全てまたは選択された部分によって決定された三次元表現を有することを特徴とする、請求項38から44のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項46】
(i)細胞中のヒトMnk−1キナーゼを発現すること、
(ii)粗Mnk−1キナーゼ調製物を回収するために細胞を溶解すること、
(iii)粗Mnk−1キナーゼ調製物を精製すること、
(iV)精製したヒトMnk−1キナーゼを結晶化すること、
を含んでなることを特徴とする、結晶ヒトMnk−1キナーゼ調製物の製造方法。
【請求項47】
前記Mnk−1キナーゼを大腸菌中の融解タンパク質として発現させることを特徴とする、請求項46記載の方法。
【請求項48】
前記Mnk−1キナーゼを、融合タグに結合したカラムを用いて精製することを特徴とする、請求項46または47のいずれかに記載の方法。
【請求項49】
前記結晶が拡散蒸着によって成長させることを特徴とする、請求項46から48のいずれかに記載の方法。
【請求項50】
ヒトMnkキナーゼの結晶構造データを生成するための、請求項38から45に記載のヒトMnk−1キナーゼまたは請求項46から49のいずれかに記載の方法によって得られたヒトMnk−1キナーゼの結晶調製物の使用。
【請求項51】
配位子、基質および/または阻害因子の結合部位を決定するための、請求項50記載の使用。
【請求項52】
表2に示す構造配座の全てまたは選択された部分によって決定されるヒトMnk−21キナーゼの結晶構造。
【請求項53】
不活性ヒトMnk−1キナーゼの結晶構造。
【請求項54】
Mnk−1キナーゼ配位子の設計、同定および/または調製のため、あるいは、タンパク質キナーゼの制御機構の特定のための、請求項38から45のいずれかに記載か、請求項46から49のいずれかによって得られる結晶ヒトMnk−1キナーゼか、および/または請求項52または53のいずれかに記載の結晶構造の使用。
【請求項55】
配位子分子の設計にコンピュータ支援モデル化プログラムを用いることを特徴とする、請求項54記載の使用。
【請求項56】
前記配位子が、ヒトMnk−1キナーゼの相互作用部位と相補的な三次元構造を有することを特徴とする、請求項54または55のいずれかに記載の使用。
【請求項57】
前記配位子が、アミノ酸Arg90またはARg93の少なくともいずれか一方と相互反応することを特徴とする、請求項56記載の使用。
【請求項58】
表2に示す構造配座の全てまたは選択された部分を提供することと、
該結晶構造の三次元表現を構築するために該構造配座を使用することと、
を含んでなることを特徴とする、ヒトMnk−1キナーゼの結晶構造の三次元表現を得る方法。
【請求項59】
請求項58に記載のヒトMnk−1キナーゼの三次元表現を提供することと、
前記化合物の三次元表現を提供することと、
前記化合物の三次元表現と、ヒトMnk−1キナーゼの該三次元表現と、を適合させることと、
を含んでなることを特徴とする、ヒトMnk−1キナーゼとの化合物の相互作用の分析のためのコンピュータベースの方法。
【請求項60】
表2に示す構造配座の全てまたは選択された部分を提供することと、
前記タンパク質の結晶構造を得る分子置換のために、該構造配座を使用することと、
を含んでなることを特徴とする、タンパク質、特に、キナーゼの結晶構造を決定する方法。
【請求項61】
コンピュータ読み取り可能データを有するデータ保存媒体からなる該データを含んでなり、該データは表2に示す構造配座の全てまたは選択された部分を含んでなることを特徴とする、コンピュータ読み取り可能な保存媒体。
【請求項62】
請求項38から45のいずれかに記載の結晶ヒトMnk−1キナーゼ、請求項52または53のいずれかに記載のヒトMnk−1キナーゼの結晶構造データ、および/または請求項54から56のいずれかに記載の方法の使用を用いて得られたヒトMnk−1キナーゼのための配位子。
【請求項63】
Mnk−1キナーゼの基質または阻害因子であることを特徴とする、請求項62記載の配位子。
【請求項64】
Mnk−1キナーゼのアロステリック阻害因子であることを特徴とする、請求項62または63のいずれかに記載の配位子。
【請求項65】
請求項62から64のいずれかに記載の配位子を含む薬剤組成物。
【請求項66】
体重制限または発生熱に関連する病気または障害の治療、緩和および/または防止のための、請求項65記載の薬剤組成物。
【請求項67】
メタボリック症候群、摂食障害、カヘキシー、糖尿病、高血圧、冠状脈心臓疾患、高コレステロール血症、異常脂質血症、変性関節疾患、胆石、睡眠中無呼吸症、神経変性疾患障害または癌の治療、緩和および/または防止のための請求項65または66のいずれかに記載の薬剤組成物。
【請求項68】
炎症または炎症徴候の治療、緩和および/または防止のための請求項64記載の薬剤組成物。
【請求項1】
結晶ヒトMnk−2キナーゼ。
【請求項2】
結晶ヒトMnk−2aキナーゼであることを特徴とする、請求項1記載の結晶ヒトMnk−2キナーゼ。
【請求項3】
前記ヒトMnk−2キナーゼが残基72−385を含むことを特徴とする、請求項1または2のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項4】
前記ヒトMnk−2キナーゼがキナーゼドメINを含むことを特徴とする、請求項1から3のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項5】
ヒトMnk−2キナーゼ突然変異体であることを特徴とする、請求項1から4のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項6】
ヒトMnk−2キナーゼ突然変異D228Gであることを特徴とする、請求項5に記載の結晶ヒトMnk−2キナーゼ。
【請求項7】
結晶ヒトMnk−2キナーゼの配位子、基質および/または阻害因子との複合体であることを特徴とする、請求項1から4のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項8】
前記阻害因子がスタウロスポリンであることを特徴とする、請求項7記載の結晶ヒトMnk−2キナーゼ。
【請求項9】
前記ヒトMnk−2キナーゼが不活性形態であることを特徴とする、請求項1から8のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項10】
単結晶であることを特徴とする、請求項1から9のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項11】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分によって決定される三次元構造を有することを特徴とする、請求項1から10のいずれかに記載の結晶ヒトMnk−2キナーゼ。
【請求項12】
(i)細胞中のヒトMnk−2キナーゼを発現すること、
(ii)粗Mnk−2キナーゼ調製物を回収するために細胞を溶解すること、
(iii)粗Mnk−2キナーゼ調製物を精製すること、
(iV)精製したヒトMnk−2キナーゼを結晶化すること、
を含んでなることを特徴とする、結晶ヒトMnk−2キナーゼ調製物の製造方法。
【請求項13】
前記Mnk−2キナーゼが、大腸菌中の溶融タンパク質として発現されることを特徴とする、請求項12記載の方法。
【請求項14】
前記Mnk−2キナーゼが、融合タグに結合されたカラムを用いて精製されることを特徴とする、請求項12または13のいずれかに記載の方法。
【請求項15】
前記結晶が拡散蒸着によって成長されることを特徴とする、請求項12から14のいずれかに記載の方法。
【請求項16】
ヒトMnkキナーゼの結晶構造データを生成するための、請求項1から11に記載のヒトMnk−2キナーゼまたは請求項12から15のいずれかに記載の方法によって得られたヒトMnk−2キナーゼの結晶調製物の使用。
【請求項17】
配位子、基質および/または阻害因子の結合部位を決定するための、請求項16記載の使用。
【請求項18】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分によって決定されるヒトMnk−2キナーゼの結晶構造。
【請求項19】
不活性ヒトMnk−2キナーゼの結晶構造。
【請求項20】
Mnk−2キナーゼ配位子の設計、同定および/または調製のため、あるいは、タンパク質キナーゼの制御機構の特定のための、請求項1から11のいずれかに記載か、請求項12から15のいずれかによって得られる結晶ヒトMnk−2キナーゼか、および/または請求項18または19のいずれかに記載の結晶構造の使用。
【請求項21】
配位子分子の設計にコンピュータ支援モデル化プログラムを用いることを特徴とする、請求項20記載の使用。
【請求項22】
前記配位子が、ヒトMnk−2キナーゼの相互作用部位と相補的な三次元構造を有することを特徴とする、請求項20または21のいずれかに記載の使用。
【請求項23】
前記配位子が、アミノ酸Asp226、Phe227およびAsp228のうちの少なくともいずれか1つと相互作用することを特徴とする、請求項22記載の使用。
【請求項24】
前記配位子が、亜鉛結合部位のシステIN残基の少なくとも1つと相互作用することを特徴とする、請求項22記載の使用。
【請求項25】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分を提供することと、
該結晶構造の三次元表現を構築するために該構造配座を使用することと、
を含んでなることを特徴とする、ヒトMnk−2キナーゼの結晶構造の三次元表現を得る方法。
【請求項26】
請求項25に記載のヒトMnk−2キナーゼの三次元表現を提供することと、
前記化合物の三次元表現を提供することと、
前記化合物の三次元表現と、ヒトMnk−2キナーゼの該三次元表現と、を適合させることと、
を含んでなることを特徴とする、ヒトMnk−2キナーゼとの化合物の相互作用の分析のためのコンピュータベースの方法。
【請求項27】
表1、表1aまたは表3に示す構造配座の全てまたは選択された部分を提供することと、
前記タンパク質の結晶構造を得る分子置換のために、該構造配座を使用することと、
を含んでなることを特徴とする、タンパク質、特に、キナーゼの結晶構造を決定する方法。
【請求項28】
コンピュータ読み取り可能データを有するデータ保存媒体からなる該データを含んでなり、該データは表1、表1aまたは表3に示す構造配座の全てまたは選択された部分を含んでなることを特徴とする、コンピュータ読み取り可能な保存媒体。
【請求項29】
請求項1から11のいずれかに記載の結晶ヒトMnk−2キナーゼ、請求項18または19のいずれかに記載のヒトMnk−2キナーゼの結晶構造データ、および/または請求項20から22のいずれかに記載の方法の使用を用いて得られたヒトMnk−2キナーゼのための配位子。
【請求項30】
Mnk−2キナーゼの基質または阻害因子であることを特徴とする、請求項29記載の配位子。
【請求項31】
Mnk−2キナーゼのアロステリック阻害因子であることを特徴とする、請求項29または30のいずれかに記載の配位子。
【請求項32】
請求項29から31のいずれかに記載の配位子を含む薬剤組成物。
【請求項33】
体重制限または発生熱に関連する病気または障害の治療、緩和および/または防止のための、請求項32記載の薬剤組成物。
【請求項34】
メタボリック症候群、摂食障害、カヘキシー、糖尿病、高血圧、冠状脈心臓疾患、高コレステロール血症、異常脂質血症、変性関節疾患、胆石、睡眠中無呼吸症、神経変性疾患障害または癌の治療、緩和および/または防止のための請求項32または33のいずれかに記載の薬剤組成物。
【請求項35】
炎症または炎症徴候の治療、緩和および/または防止のための請求項32記載の薬剤組成物。
【請求項36】
アロステリックタンパク質キナーゼ阻害因子の同定を可能にする構造モチーフの同定のための方法。
【請求項37】
前記キナーゼがp38、Mnk−1またはMnk−2から選択されることを特徴とする、請求項33記載の方法。
【請求項38】
結晶ヒトMnk−1キナーゼ。
【請求項39】
前記比とMnk−1キナーゼが、残基37から341を含むことを特徴とする、結晶ヒトMnk−1キナーゼ。
【請求項40】
前記ヒトMnk−1キナーゼが、前記キナーゼドメINを含むことを特徴とする、請求項38または39のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項41】
ヒトMnk−1キナーゼ突然変異体であることを特徴とする、請求項38から40のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項42】
結晶ヒトMnk−1キナーゼの配位子、基質および/または阻害因子との複合体であることを特徴とする、請求項38から41のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項43】
前記ヒトMnk−1キナーゼが不活性形態であることを特徴とする、請求項38から42のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項44】
単結晶であることを特徴とする、請求項38から43のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項45】
表2に示す構造配座の全てまたは選択された部分によって決定された三次元表現を有することを特徴とする、請求項38から44のいずれかに記載の結晶ヒトMnk−1キナーゼ。
【請求項46】
(i)細胞中のヒトMnk−1キナーゼを発現すること、
(ii)粗Mnk−1キナーゼ調製物を回収するために細胞を溶解すること、
(iii)粗Mnk−1キナーゼ調製物を精製すること、
(iV)精製したヒトMnk−1キナーゼを結晶化すること、
を含んでなることを特徴とする、結晶ヒトMnk−1キナーゼ調製物の製造方法。
【請求項47】
前記Mnk−1キナーゼを大腸菌中の融解タンパク質として発現させることを特徴とする、請求項46記載の方法。
【請求項48】
前記Mnk−1キナーゼを、融合タグに結合したカラムを用いて精製することを特徴とする、請求項46または47のいずれかに記載の方法。
【請求項49】
前記結晶が拡散蒸着によって成長させることを特徴とする、請求項46から48のいずれかに記載の方法。
【請求項50】
ヒトMnkキナーゼの結晶構造データを生成するための、請求項38から45に記載のヒトMnk−1キナーゼまたは請求項46から49のいずれかに記載の方法によって得られたヒトMnk−1キナーゼの結晶調製物の使用。
【請求項51】
配位子、基質および/または阻害因子の結合部位を決定するための、請求項50記載の使用。
【請求項52】
表2に示す構造配座の全てまたは選択された部分によって決定されるヒトMnk−21キナーゼの結晶構造。
【請求項53】
不活性ヒトMnk−1キナーゼの結晶構造。
【請求項54】
Mnk−1キナーゼ配位子の設計、同定および/または調製のため、あるいは、タンパク質キナーゼの制御機構の特定のための、請求項38から45のいずれかに記載か、請求項46から49のいずれかによって得られる結晶ヒトMnk−1キナーゼか、および/または請求項52または53のいずれかに記載の結晶構造の使用。
【請求項55】
配位子分子の設計にコンピュータ支援モデル化プログラムを用いることを特徴とする、請求項54記載の使用。
【請求項56】
前記配位子が、ヒトMnk−1キナーゼの相互作用部位と相補的な三次元構造を有することを特徴とする、請求項54または55のいずれかに記載の使用。
【請求項57】
前記配位子が、アミノ酸Arg90またはARg93の少なくともいずれか一方と相互反応することを特徴とする、請求項56記載の使用。
【請求項58】
表2に示す構造配座の全てまたは選択された部分を提供することと、
該結晶構造の三次元表現を構築するために該構造配座を使用することと、
を含んでなることを特徴とする、ヒトMnk−1キナーゼの結晶構造の三次元表現を得る方法。
【請求項59】
請求項58に記載のヒトMnk−1キナーゼの三次元表現を提供することと、
前記化合物の三次元表現を提供することと、
前記化合物の三次元表現と、ヒトMnk−1キナーゼの該三次元表現と、を適合させることと、
を含んでなることを特徴とする、ヒトMnk−1キナーゼとの化合物の相互作用の分析のためのコンピュータベースの方法。
【請求項60】
表2に示す構造配座の全てまたは選択された部分を提供することと、
前記タンパク質の結晶構造を得る分子置換のために、該構造配座を使用することと、
を含んでなることを特徴とする、タンパク質、特に、キナーゼの結晶構造を決定する方法。
【請求項61】
コンピュータ読み取り可能データを有するデータ保存媒体からなる該データを含んでなり、該データは表2に示す構造配座の全てまたは選択された部分を含んでなることを特徴とする、コンピュータ読み取り可能な保存媒体。
【請求項62】
請求項38から45のいずれかに記載の結晶ヒトMnk−1キナーゼ、請求項52または53のいずれかに記載のヒトMnk−1キナーゼの結晶構造データ、および/または請求項54から56のいずれかに記載の方法の使用を用いて得られたヒトMnk−1キナーゼのための配位子。
【請求項63】
Mnk−1キナーゼの基質または阻害因子であることを特徴とする、請求項62記載の配位子。
【請求項64】
Mnk−1キナーゼのアロステリック阻害因子であることを特徴とする、請求項62または63のいずれかに記載の配位子。
【請求項65】
請求項62から64のいずれかに記載の配位子を含む薬剤組成物。
【請求項66】
体重制限または発生熱に関連する病気または障害の治療、緩和および/または防止のための、請求項65記載の薬剤組成物。
【請求項67】
メタボリック症候群、摂食障害、カヘキシー、糖尿病、高血圧、冠状脈心臓疾患、高コレステロール血症、異常脂質血症、変性関節疾患、胆石、睡眠中無呼吸症、神経変性疾患障害または癌の治療、緩和および/または防止のための請求項65または66のいずれかに記載の薬剤組成物。
【請求項68】
炎症または炎症徴候の治療、緩和および/または防止のための請求項64記載の薬剤組成物。
【図1】

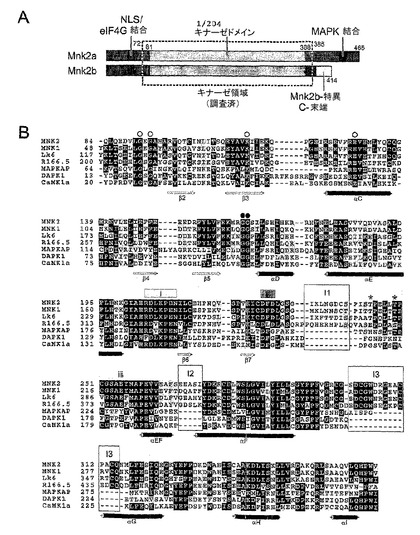
【図2】
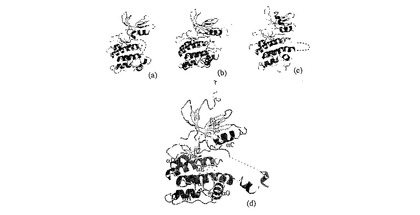
【図3】
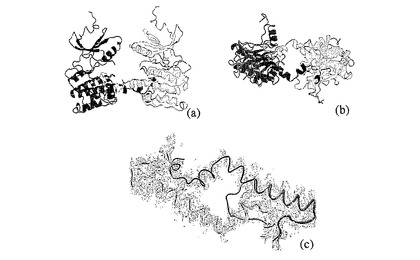
【図4】
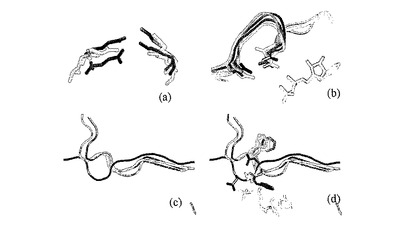
【図5】
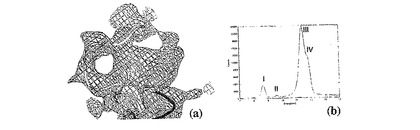
【図6】
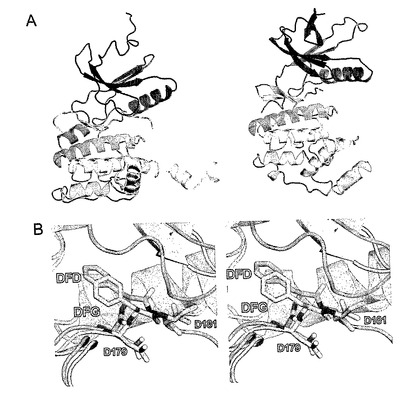
【図7】
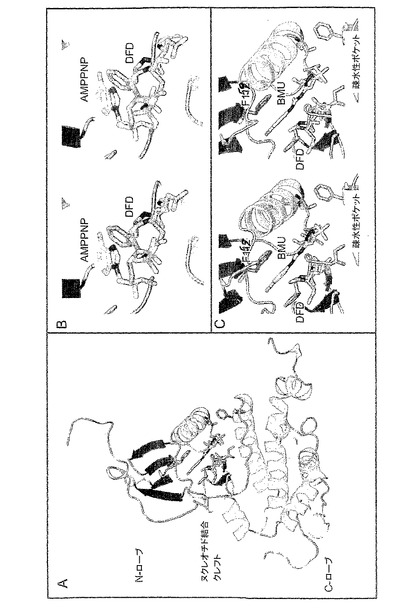
【図8】
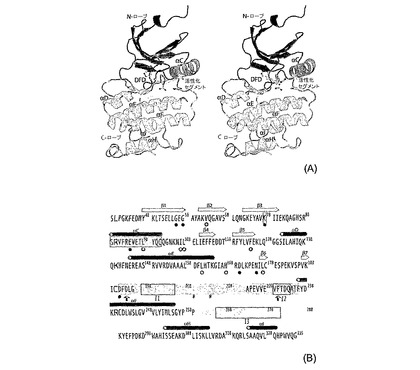
【図9】
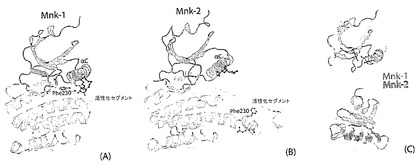
【図10】
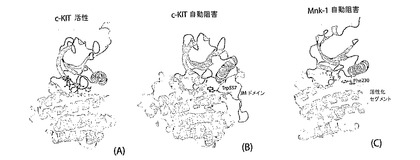
【図11】
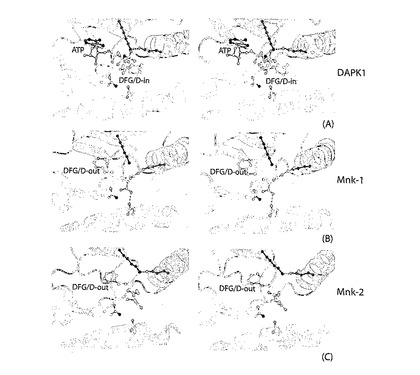
【図12】
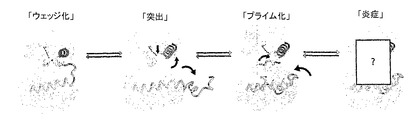
【図13】
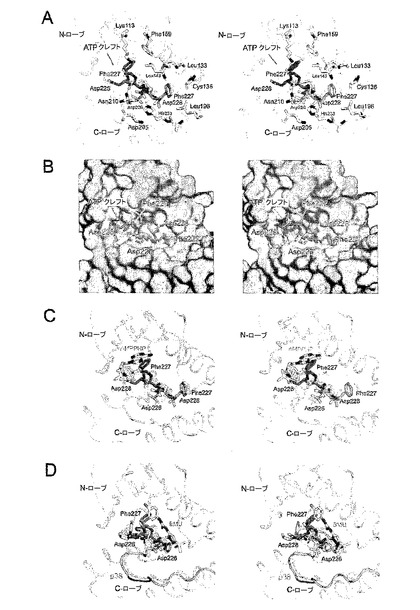
【図14−1】

【図14−2】

【図14−3】

【図14−4】

【図14−5】

【図14−6】

【図14−7】

【図14−8】
【図14−9】
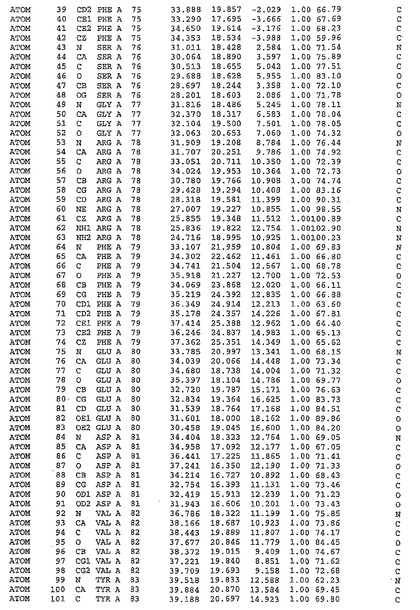
【図14−10】
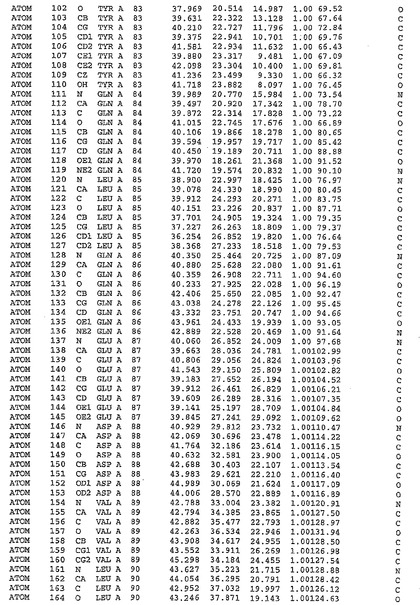
【図14−11】
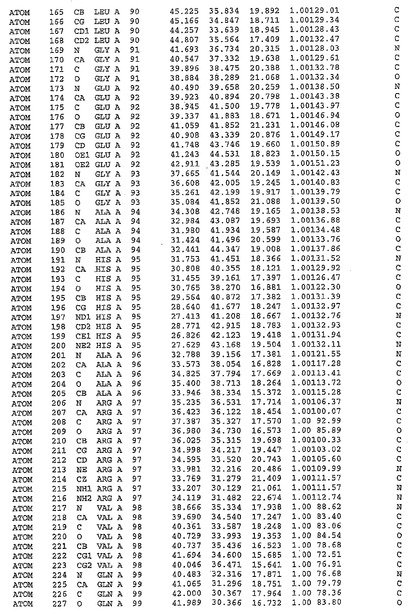
【図14−12】
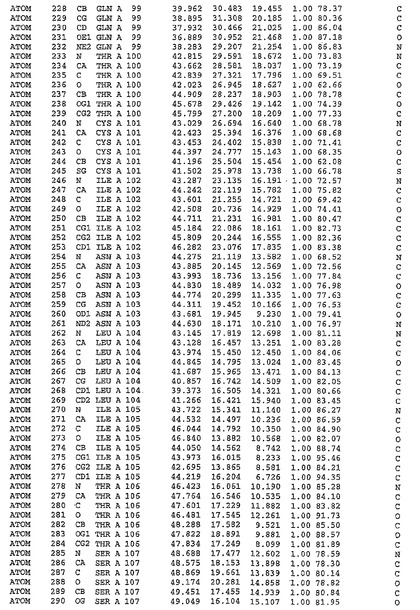
【図14−13】
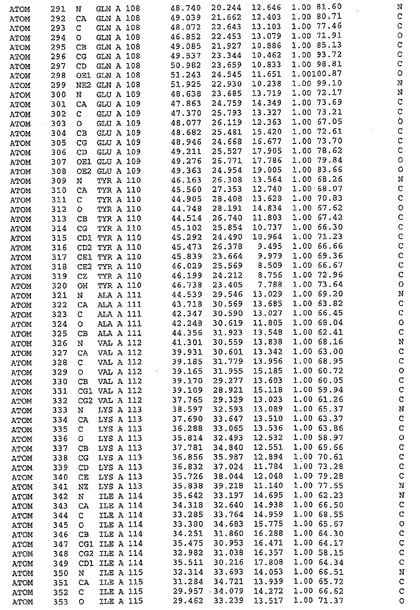
【図14−14】
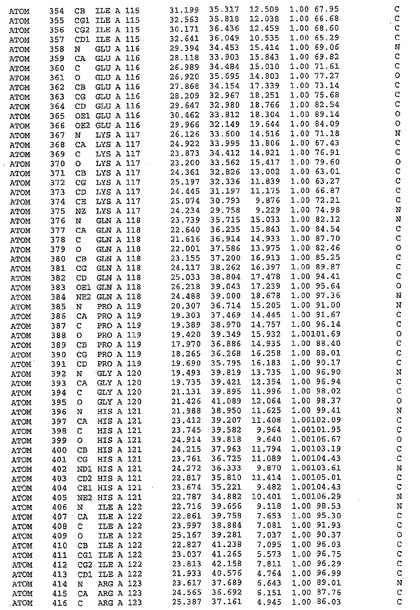
【図14−15】
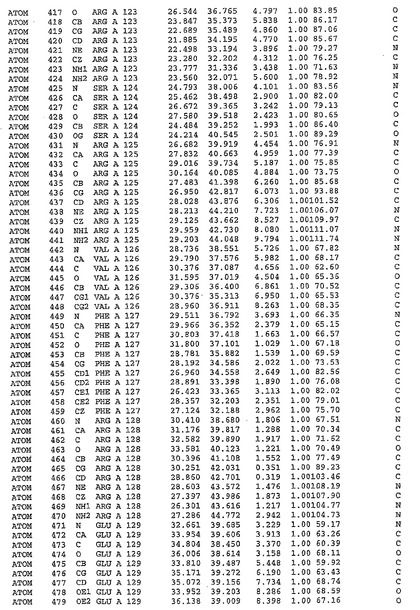
【図14−16】
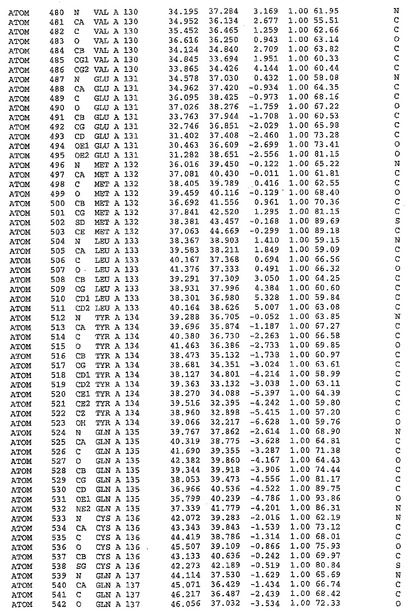
【図14−17】
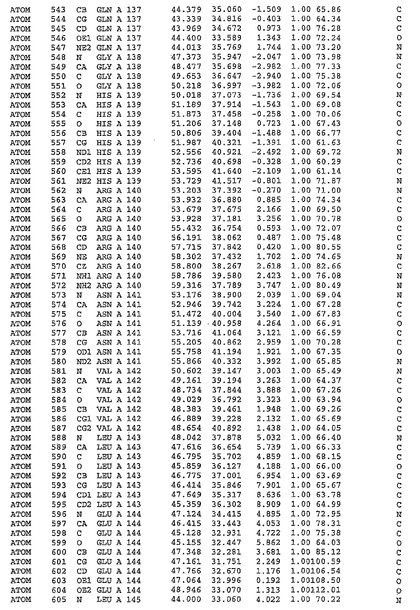
【図14−18】
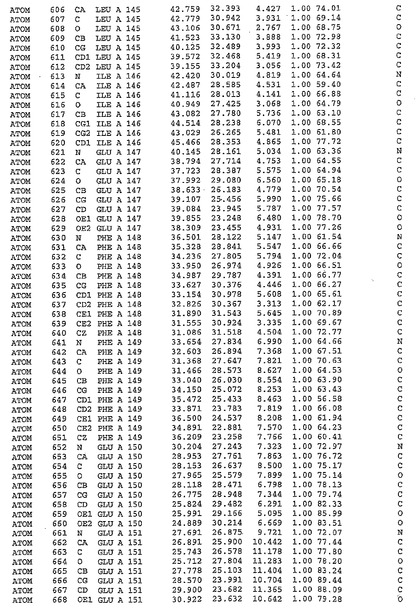
【図14−19】
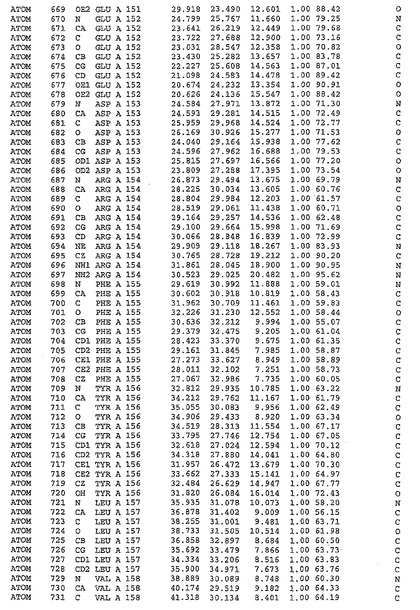
【図14−20】
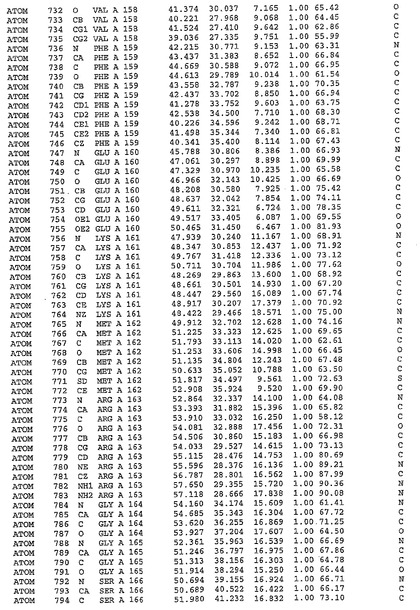
【図14−21】
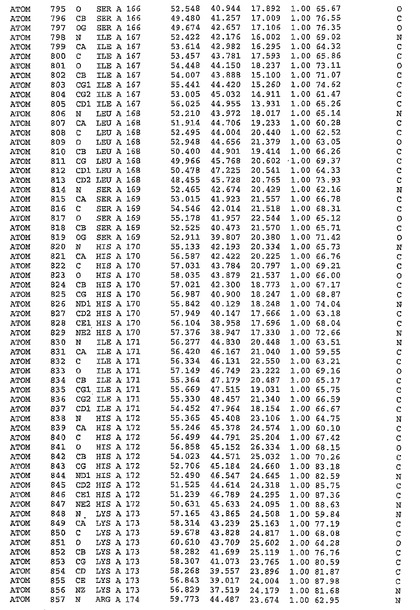
【図14−22】
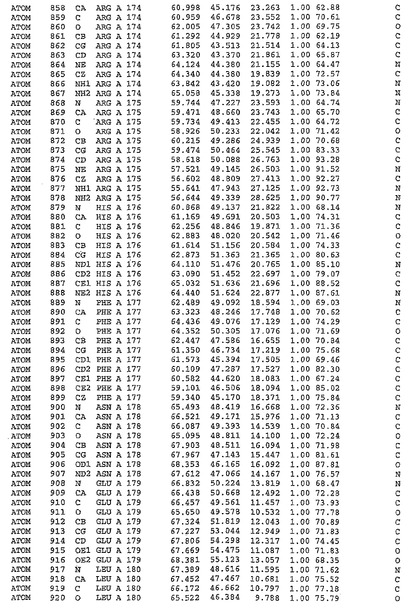
【図14−23】
【図14−24】
【図14−25】
【図14−26】
【図14−27】
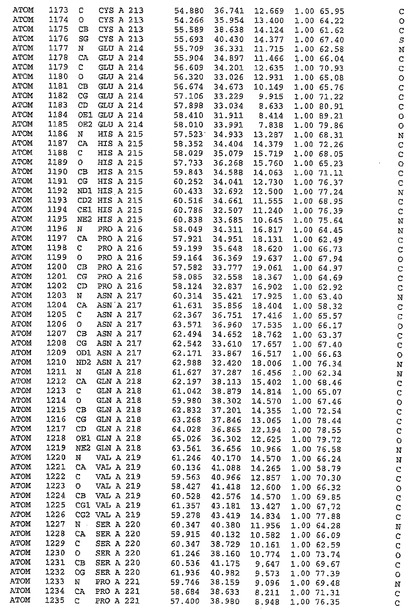
【図14−28】
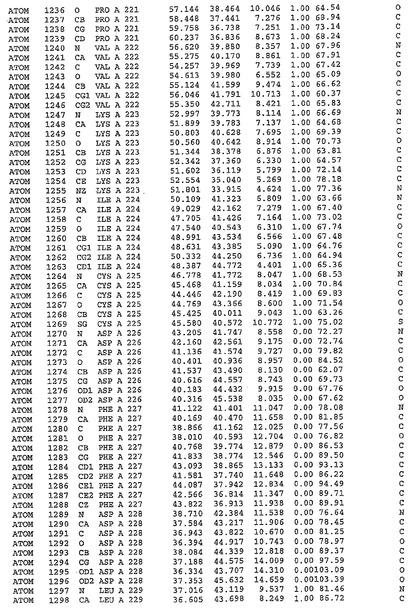
【図14−29】
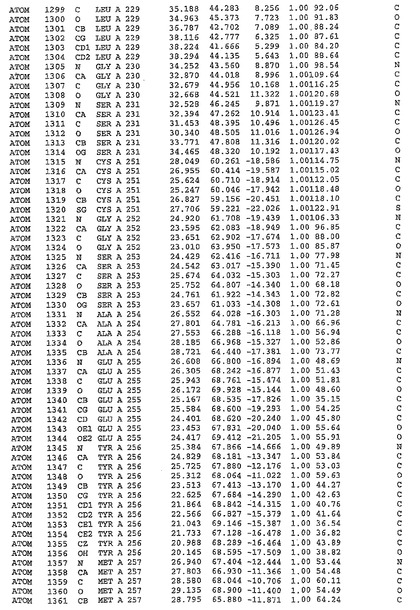
【図14−30】
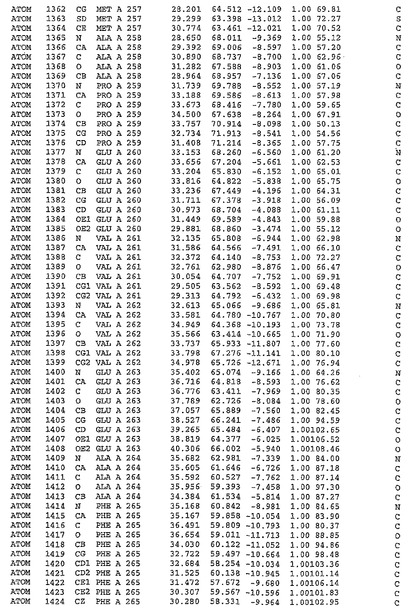
【図14−31】
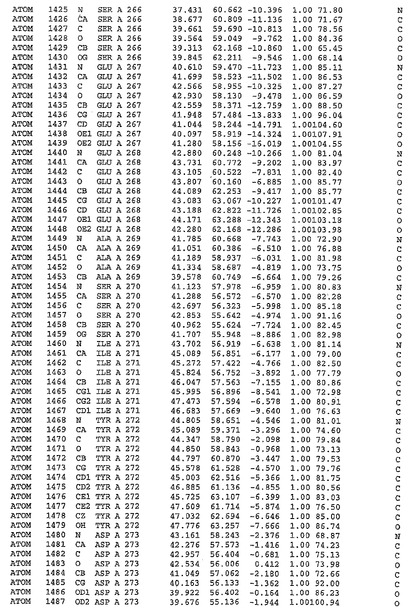
【図14−32】
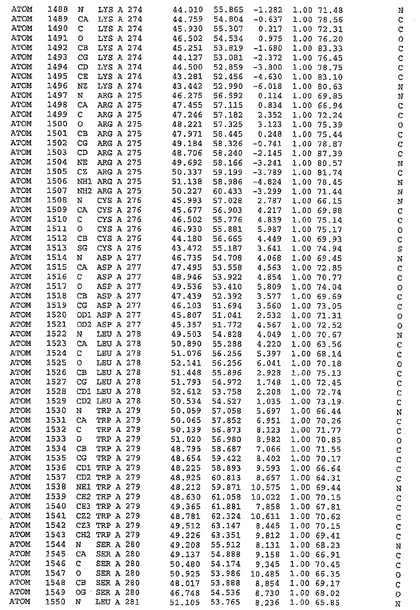
【図14−33】
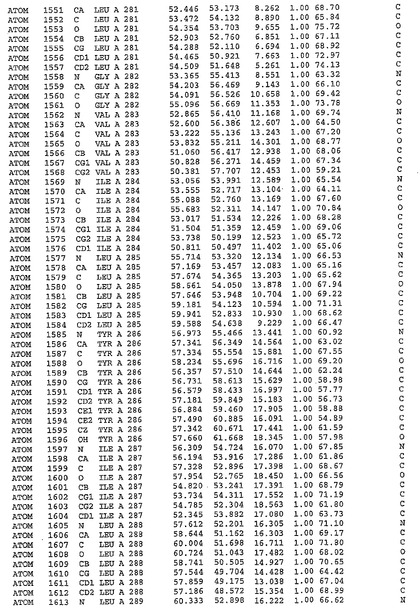
【図14−34】
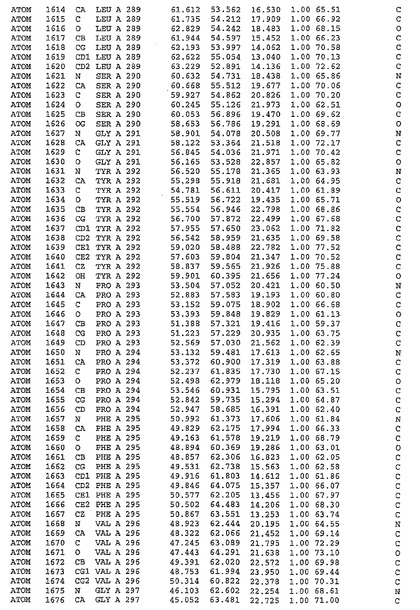
【図14−35】
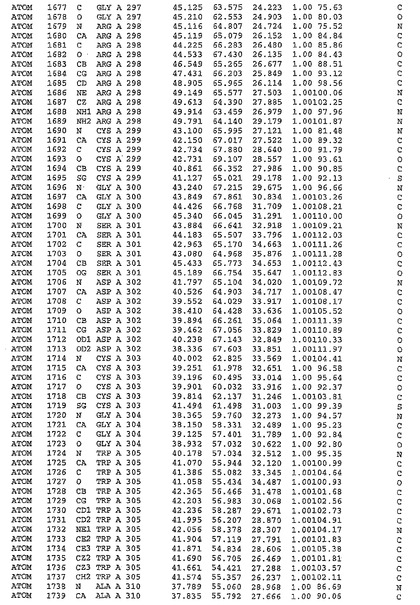
【図14−36】
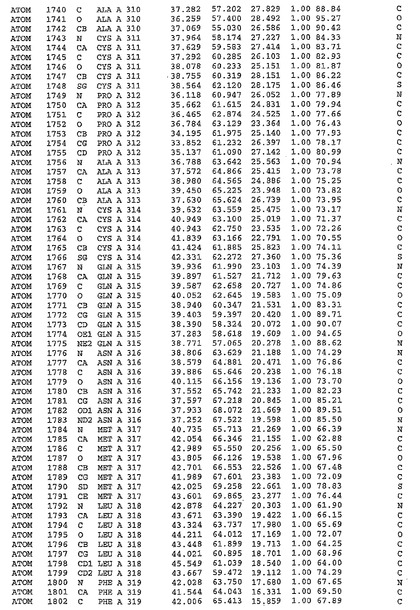
【図14−37】
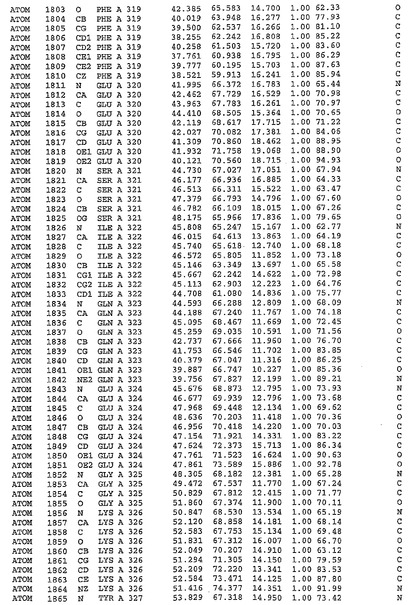
【図14−38】
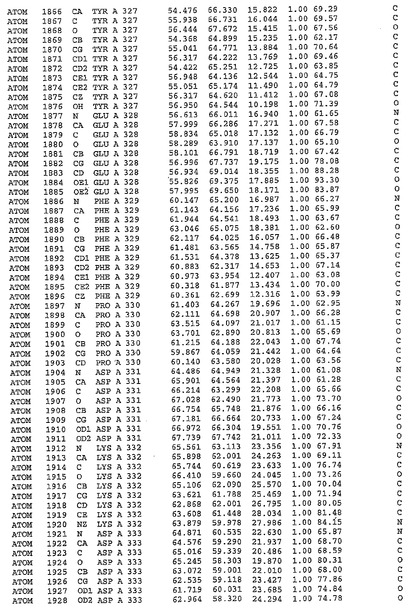
【図14−39】
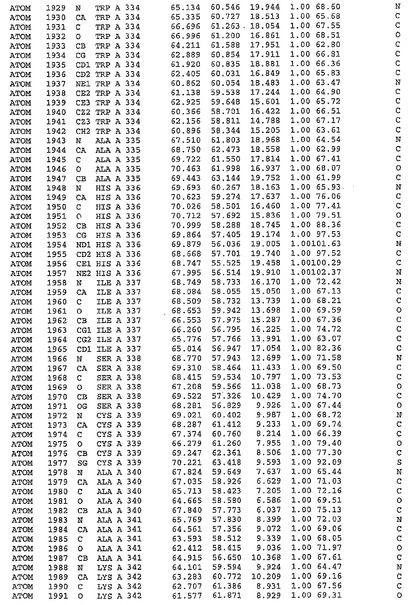
【図14−40】
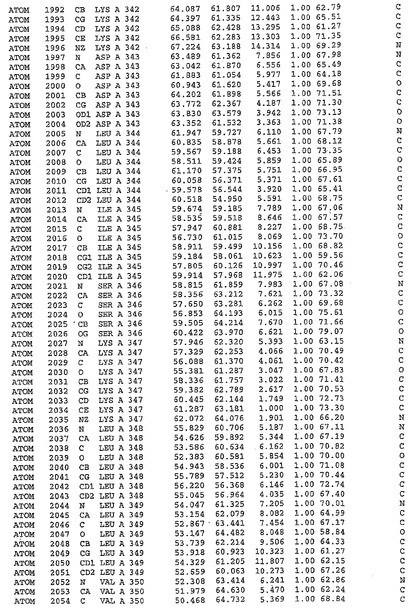
【図14−41】
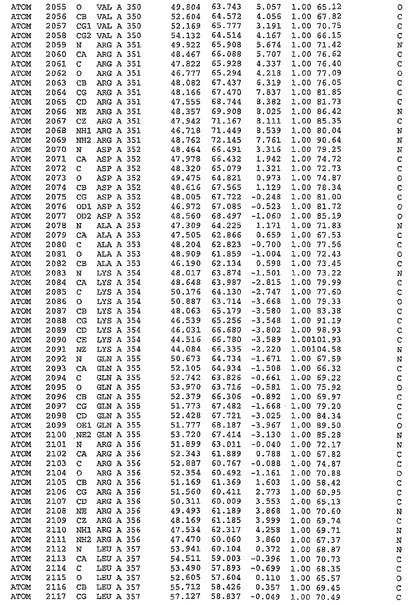
【図14−42】
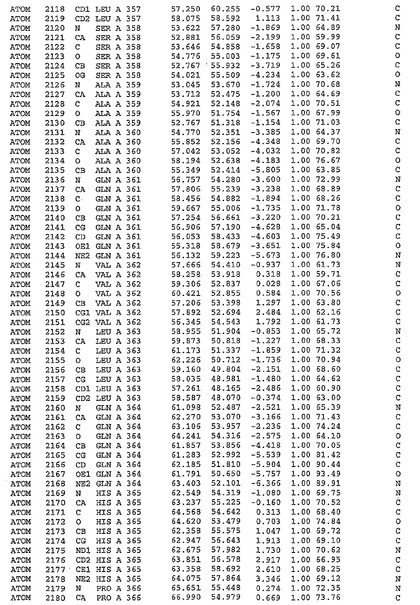
【図14−43】
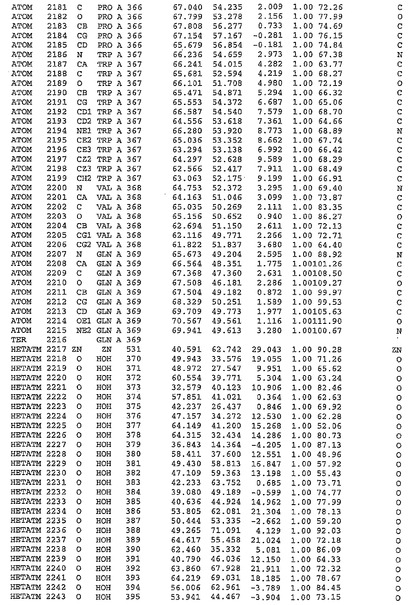
【図14−44】
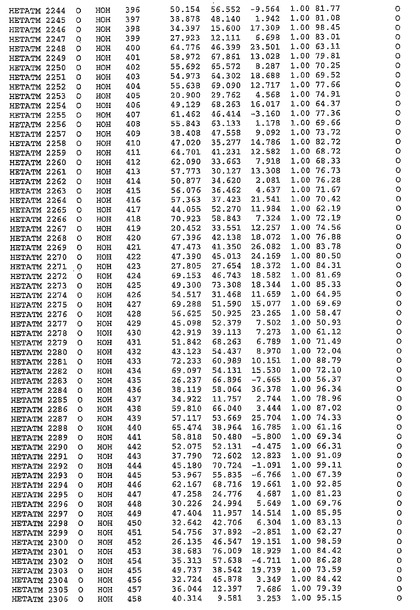
【図14−45】
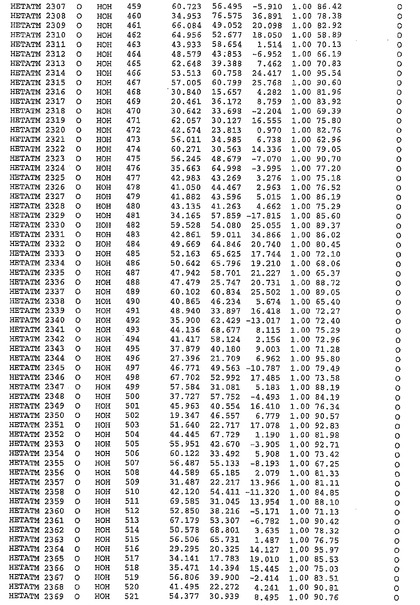
【図14−46】
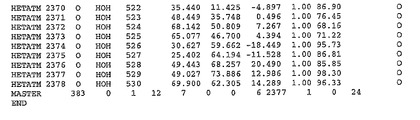
【図14−47】

【図14−48】

【図14−49】

【図14−50】

【図14−51】

【図14−52】

【図14−53】
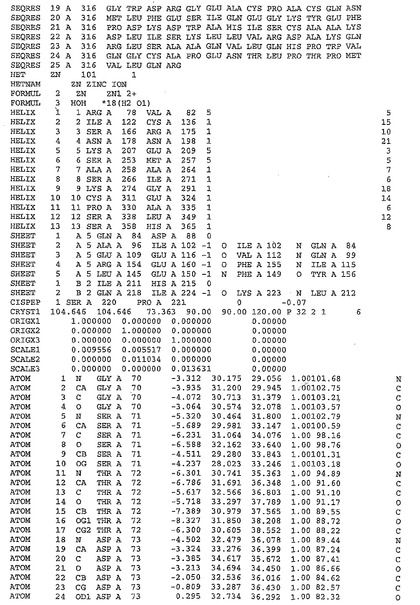
【図14−54】
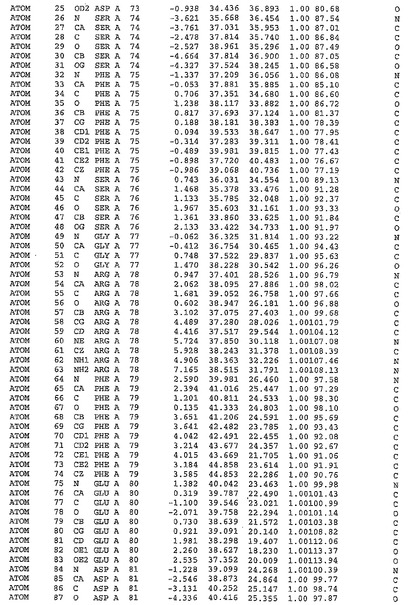
【図14−55】
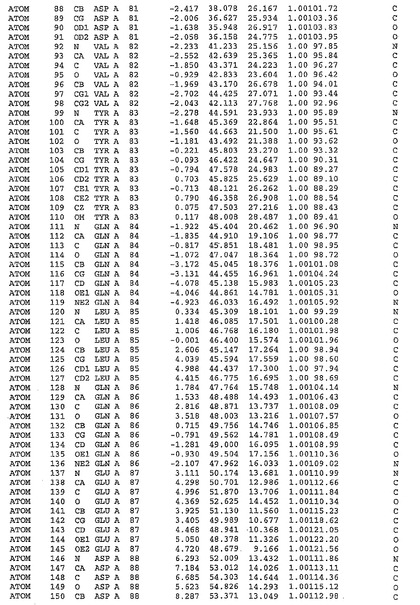
【図14−56】
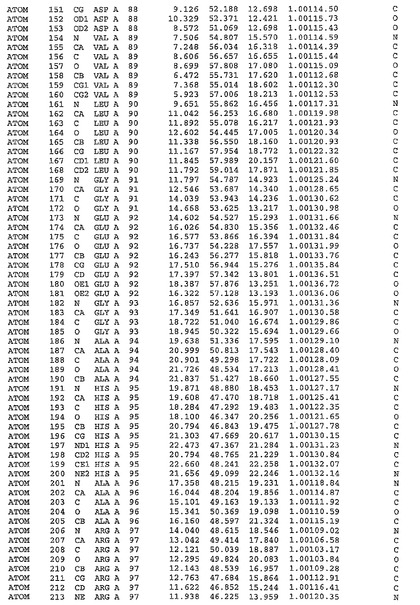
【図14−57】
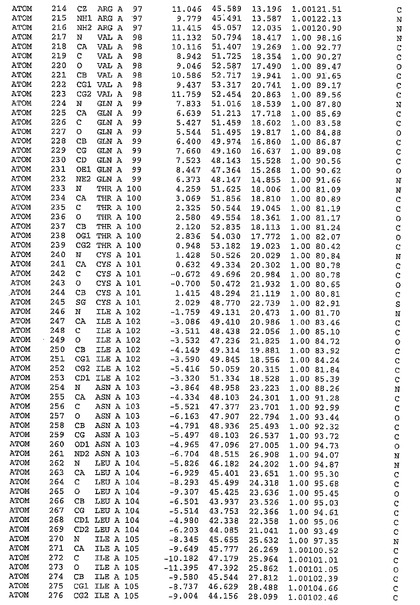
【図14−58】
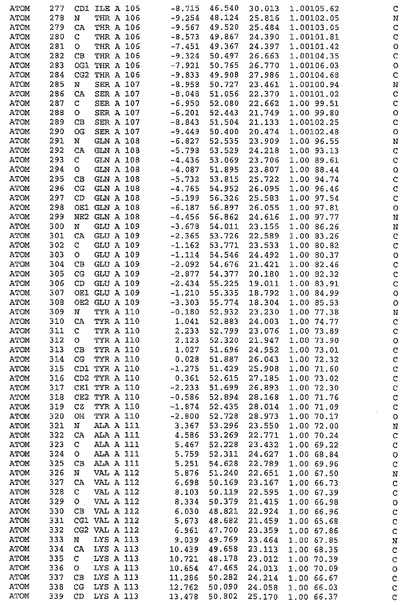
【図14−59】
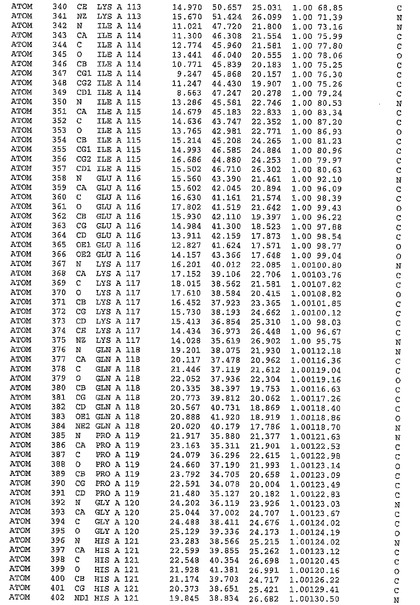
【図14−60】
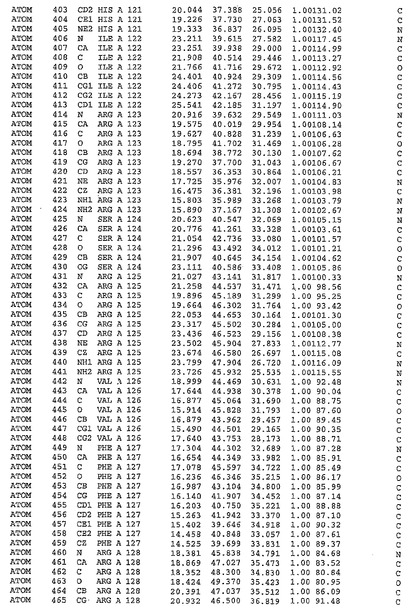
【図14−61】
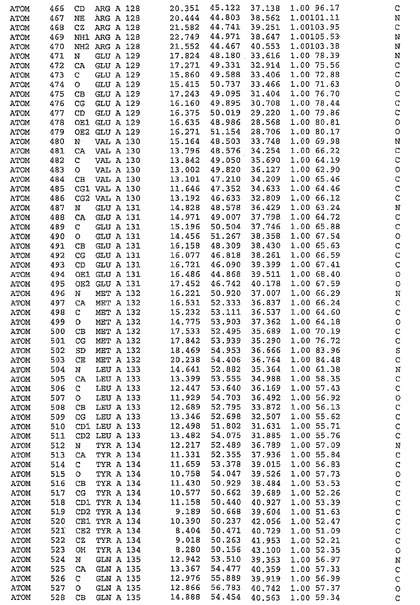
【図14−62】
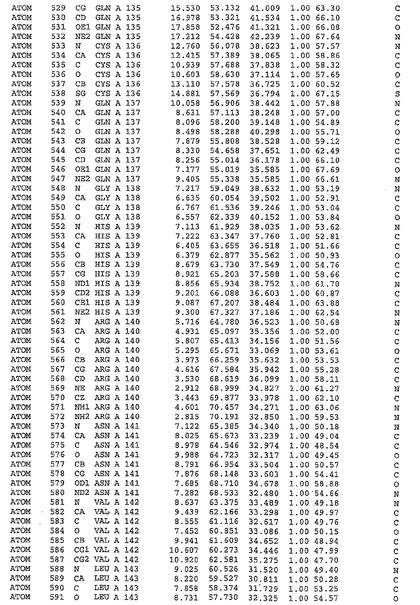
【図14−63】
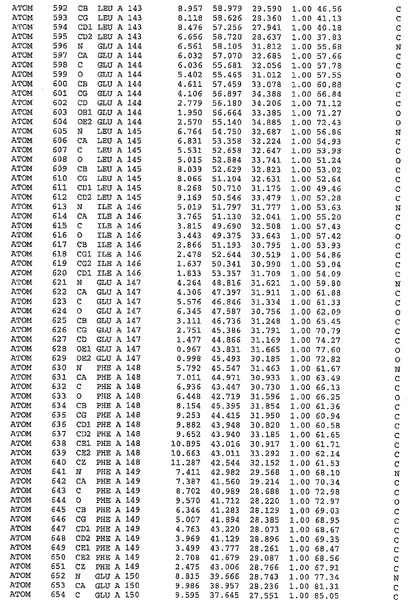
【図14−64】
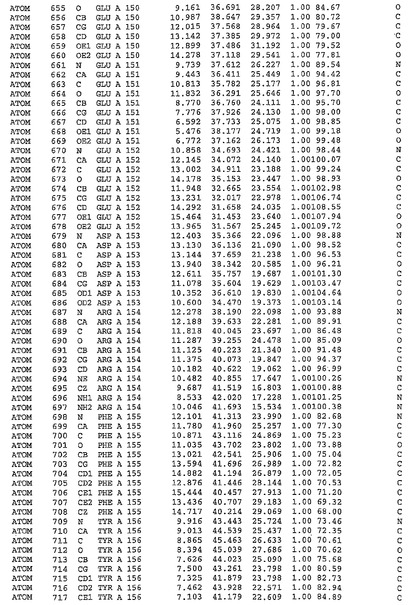
【図14−65】
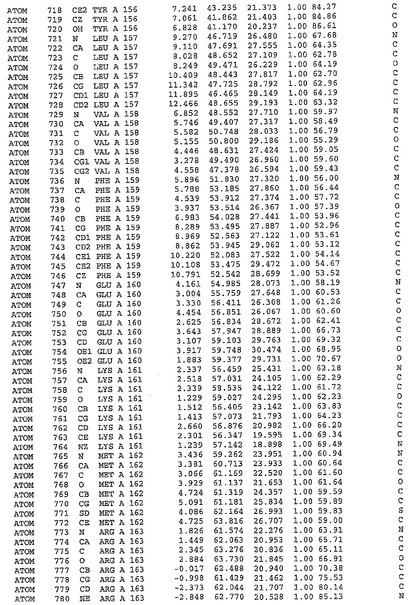
【図14−66】
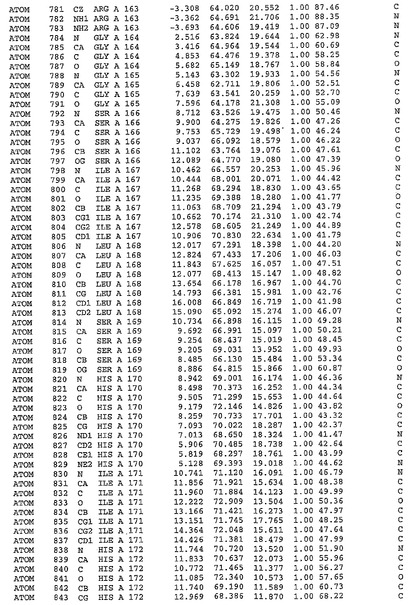
【図14−67】
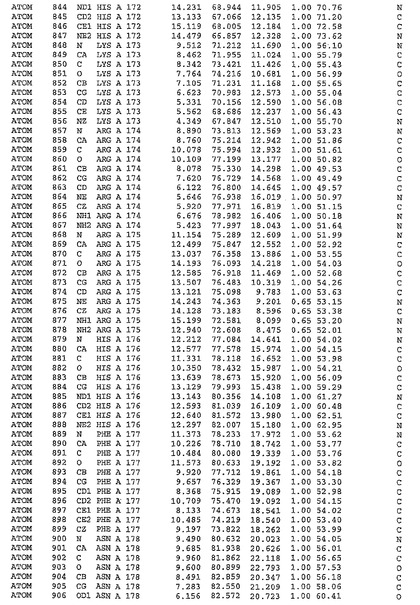
【図14−68】
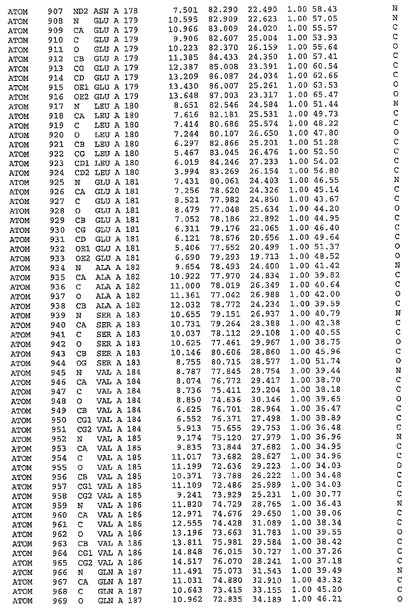
【図14−69】
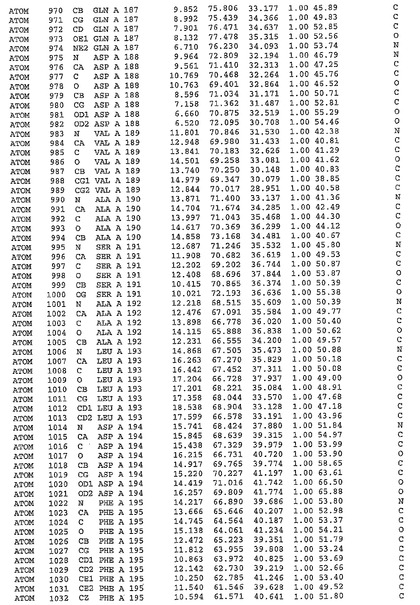
【図14−70】
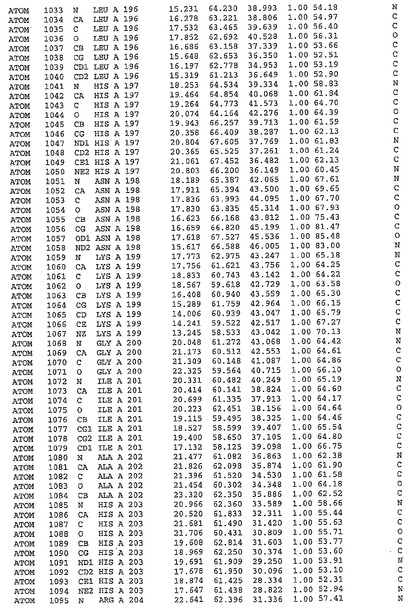
【図14−71】
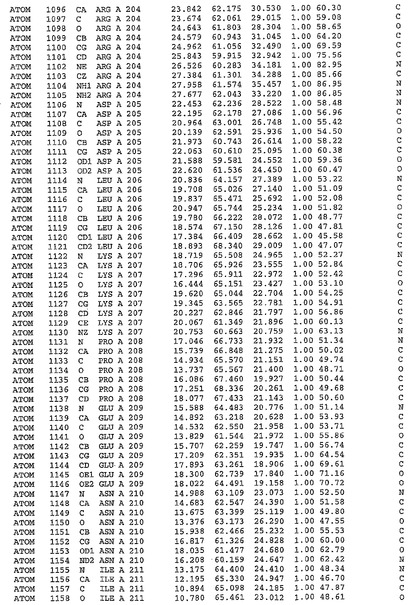
【図14−72】
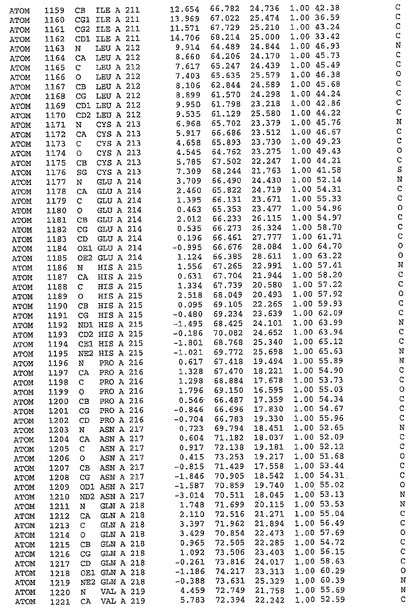
【図14−73】
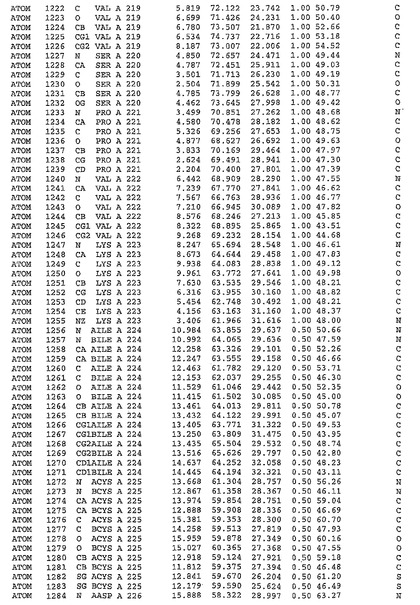
【図14−74】
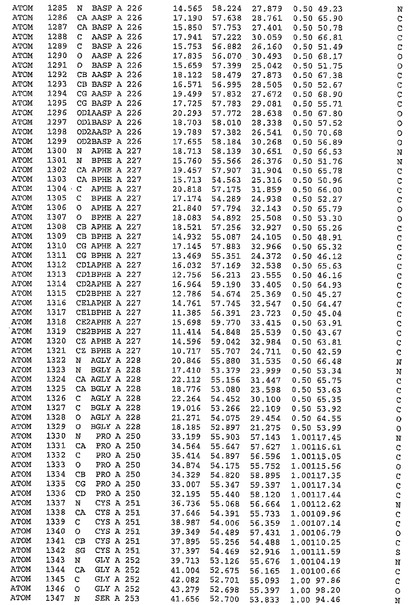
【図14−75】
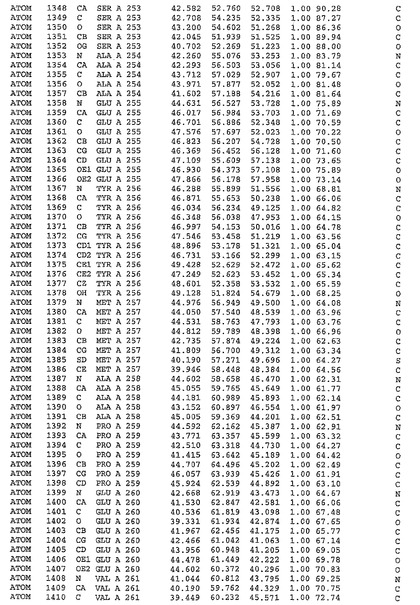
【図14−76】
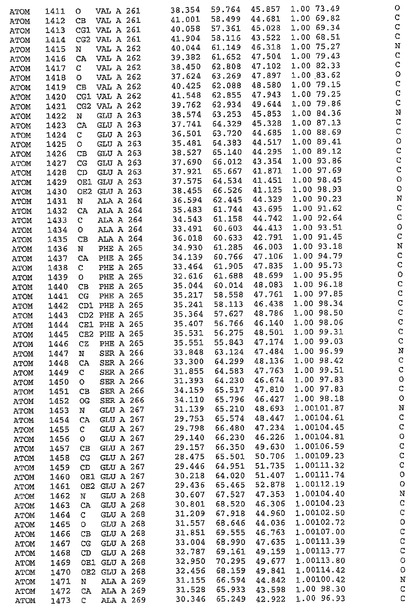
【図14−77】
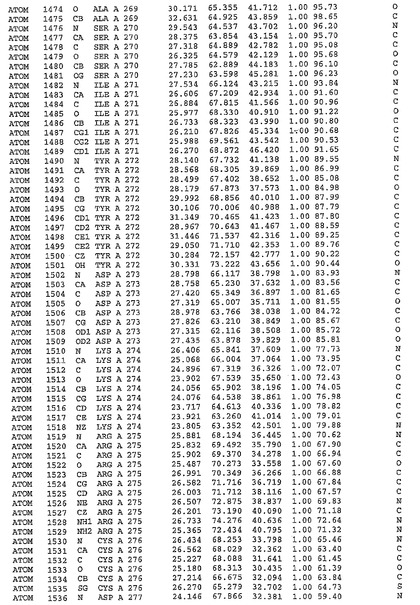
【図14−78】
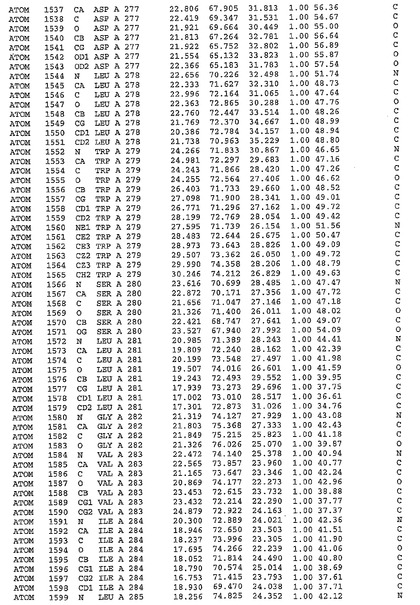
【図14−79】
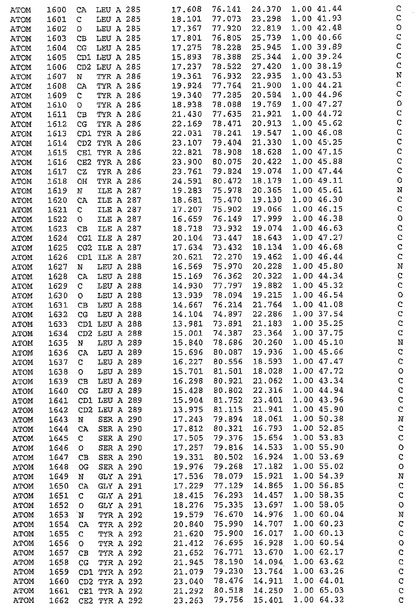
【図14−80】
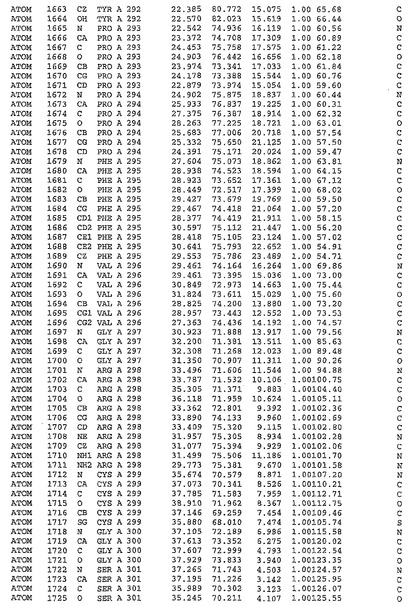
【図14−81】
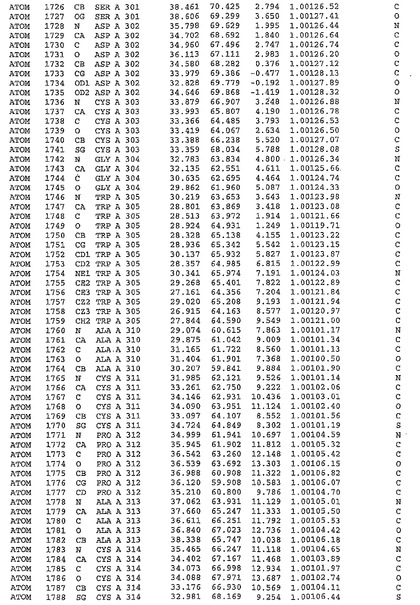
【図14−82】
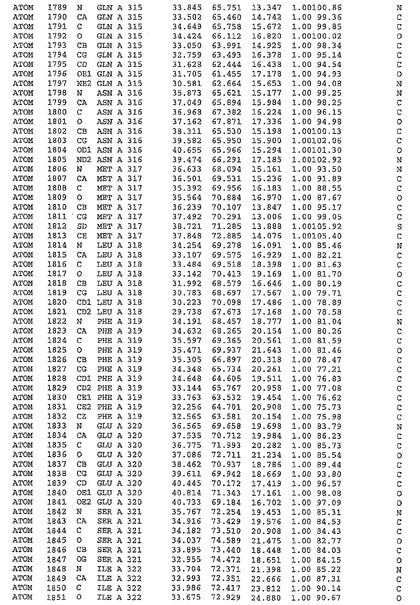
【図14−83】
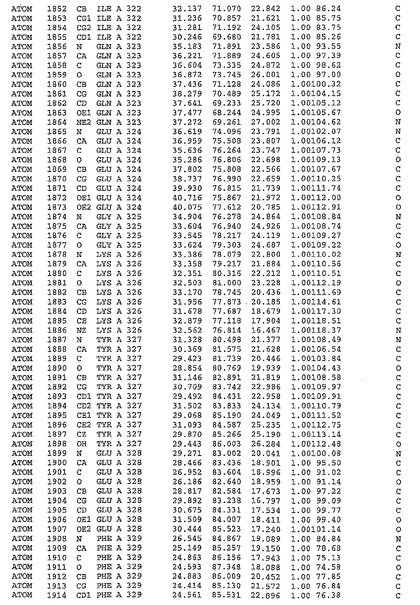
【図14−84】
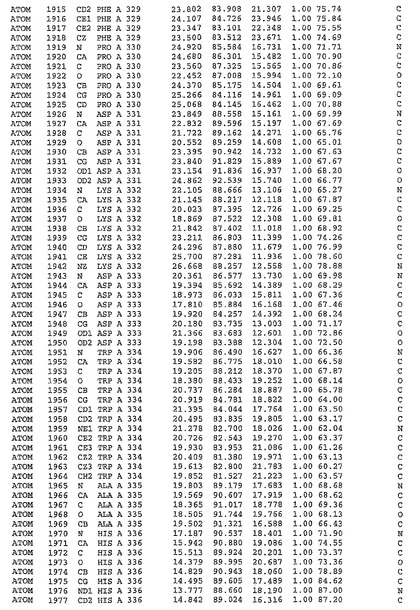
【図14−85】
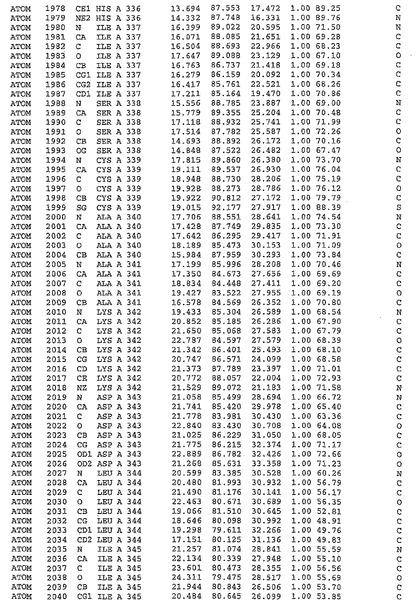
【図14−86】
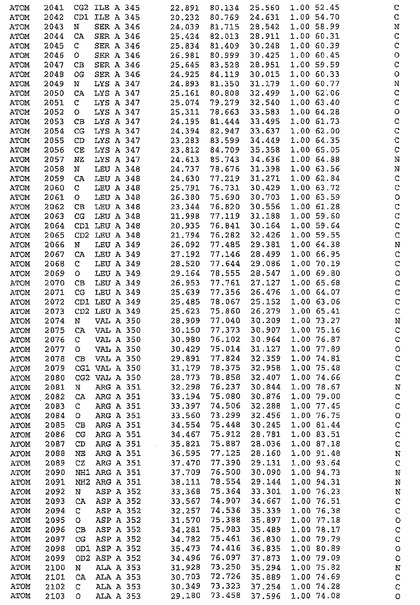
【図14−87】
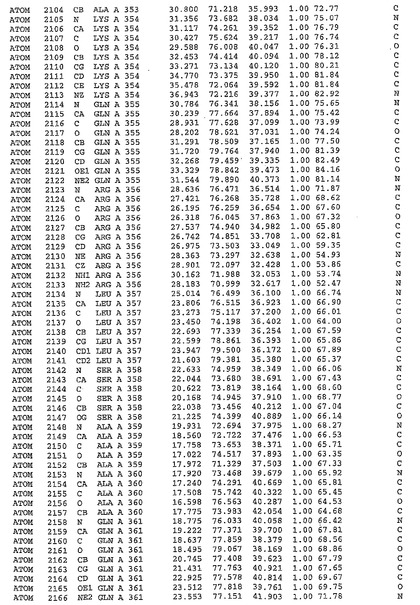
【図14−88】
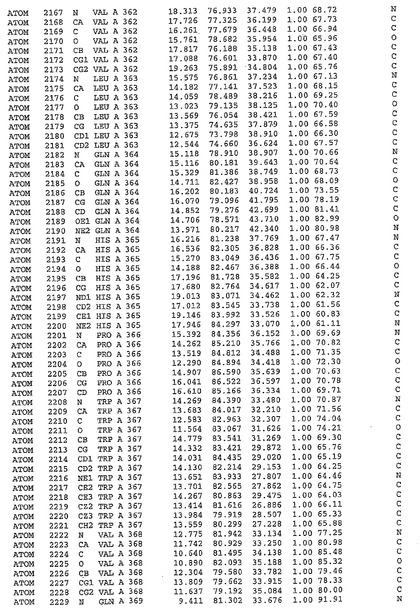
【図14−89】
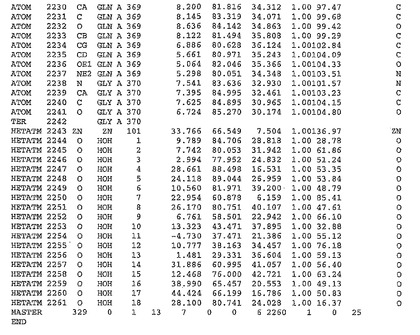
【図14−90】

【図14−91】
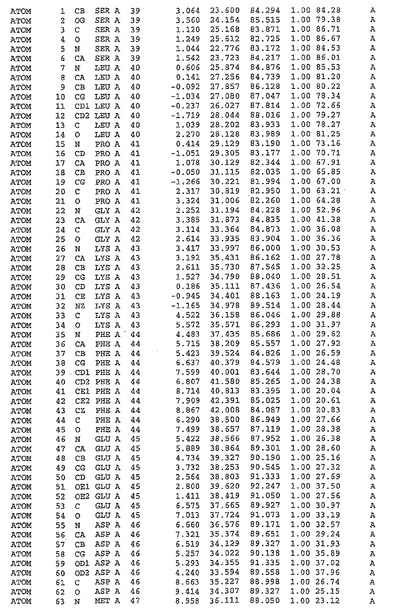
【図14−92】
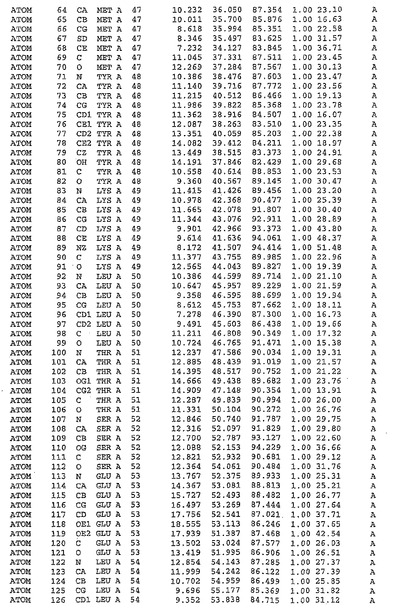
【図14−93】
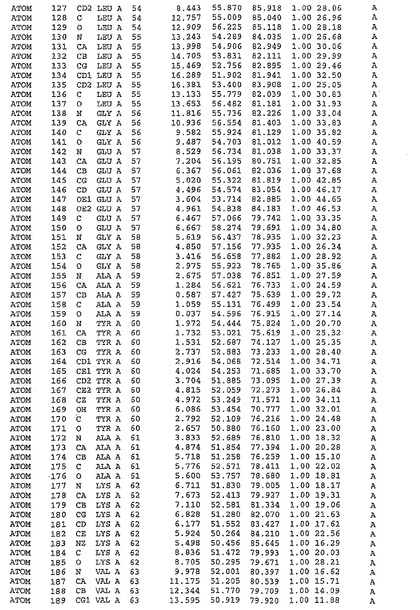
【図14−94】
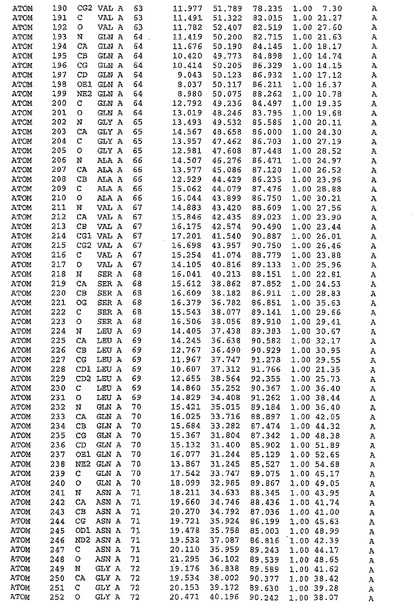
【図14−95】
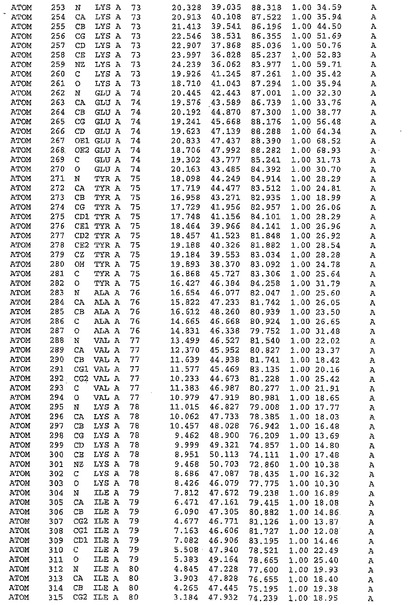
【図14−96】
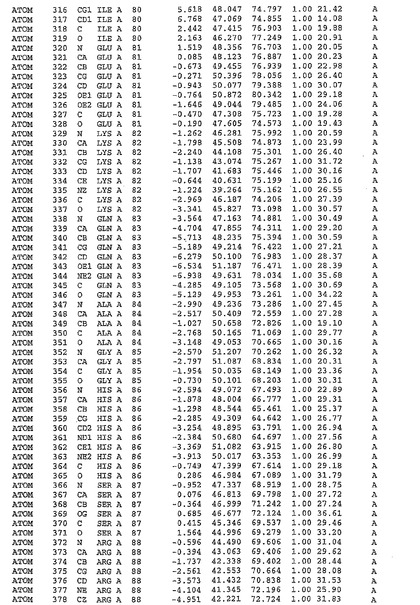
【図14−97】
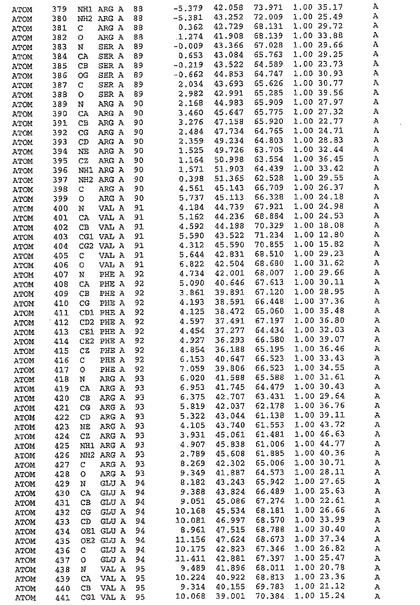
【図14−98】
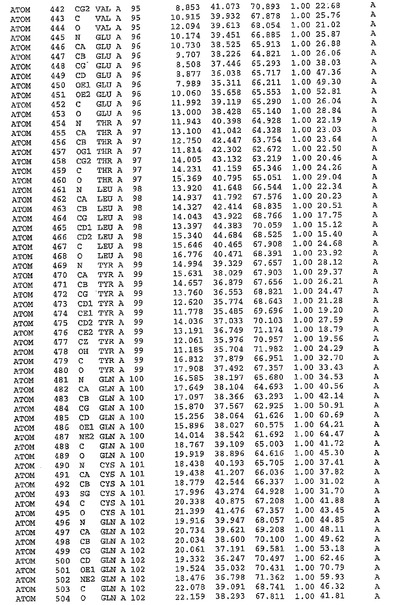
【図14−99】
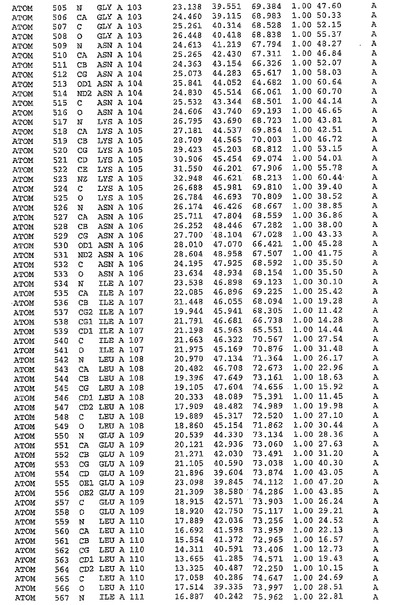
【図14−100】
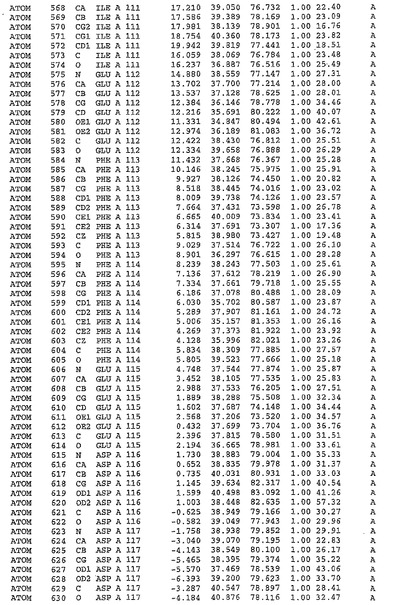
【図14−101】
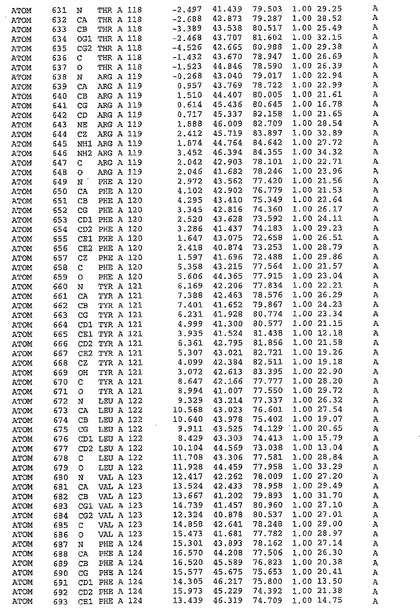
【図14−102】
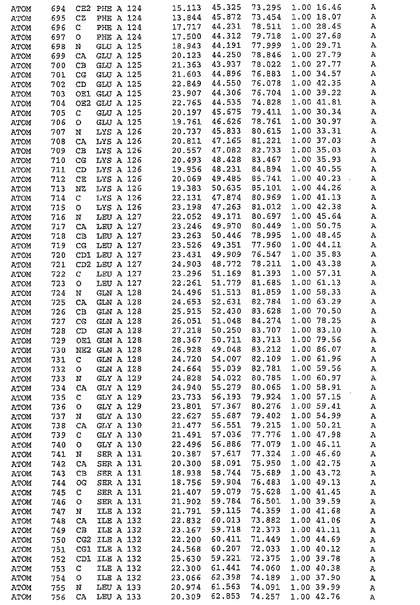
【図14−103】
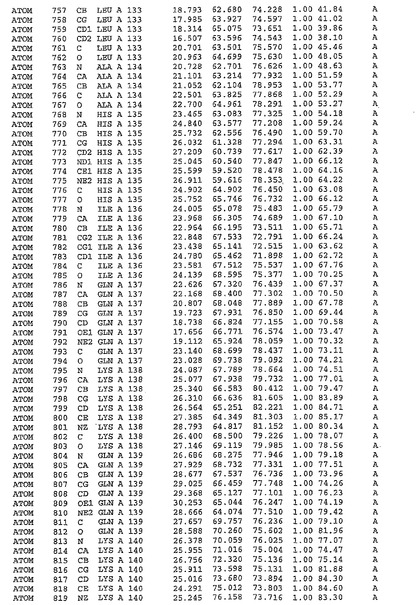
【図14−104】
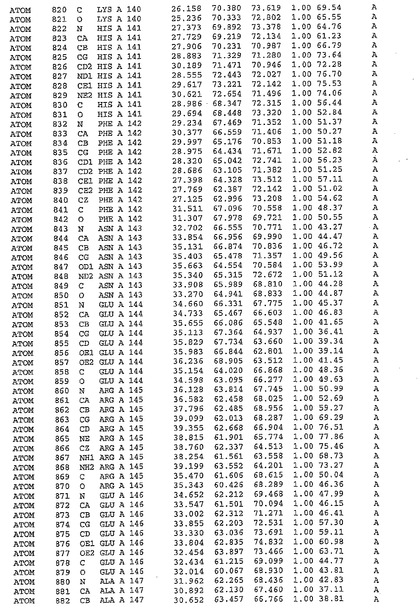
【図14−105】
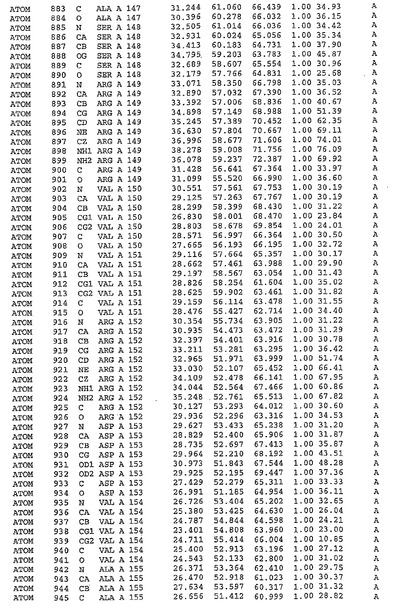
【図14−106】
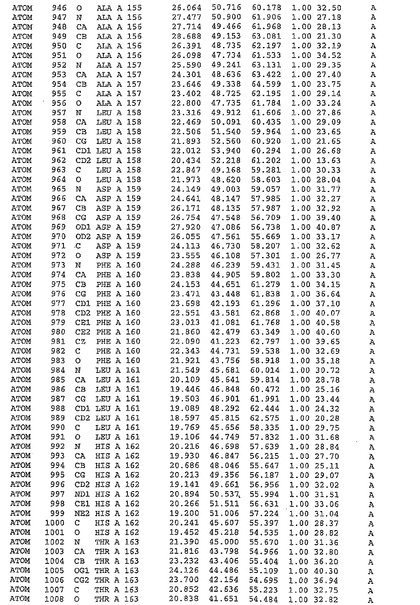
【図14−107】
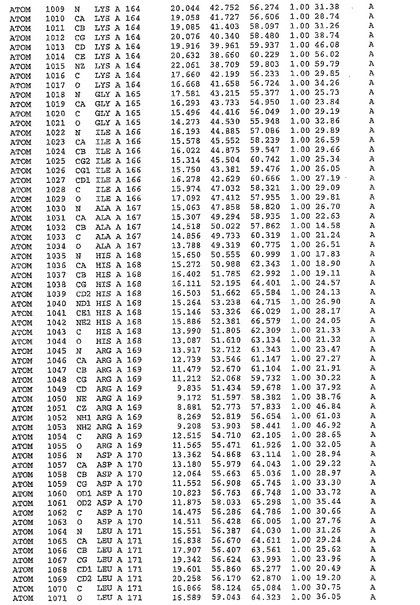
【図14−108】
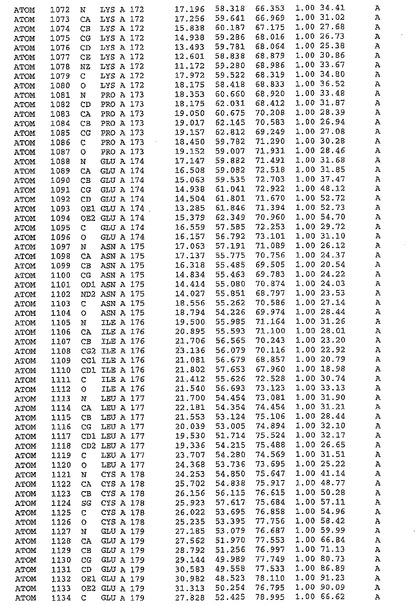
【図14−109】
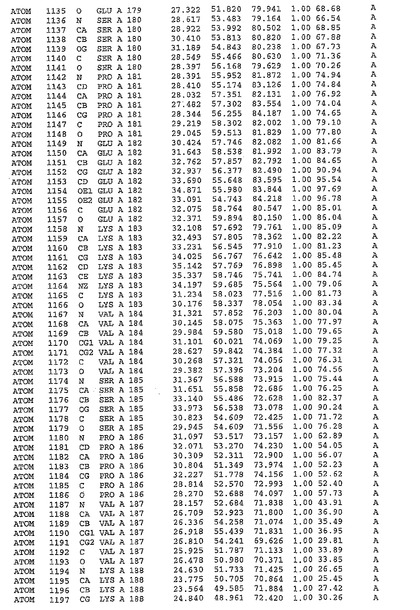
【図14−110】
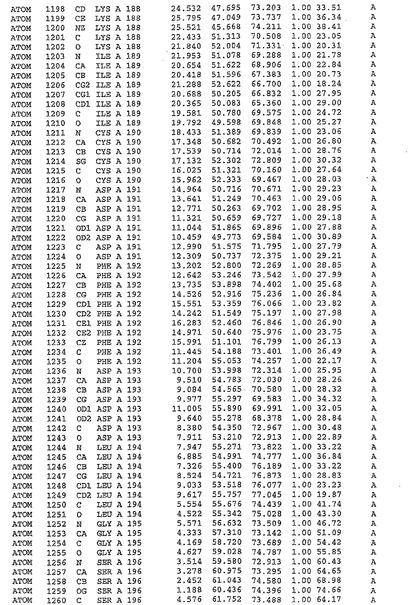
【図14−111】
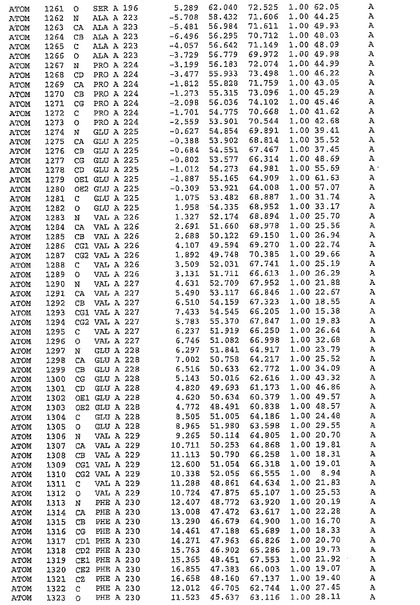
【図14−112】
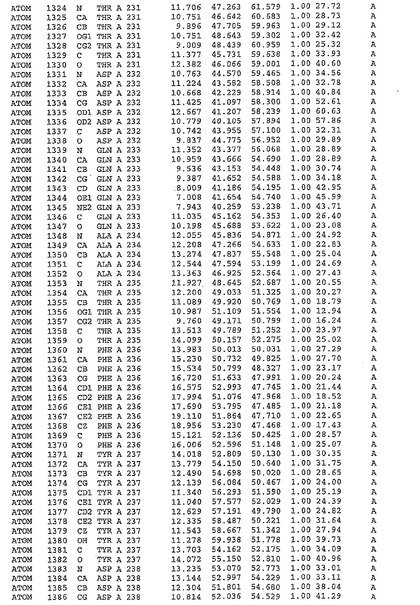
【図14−113】
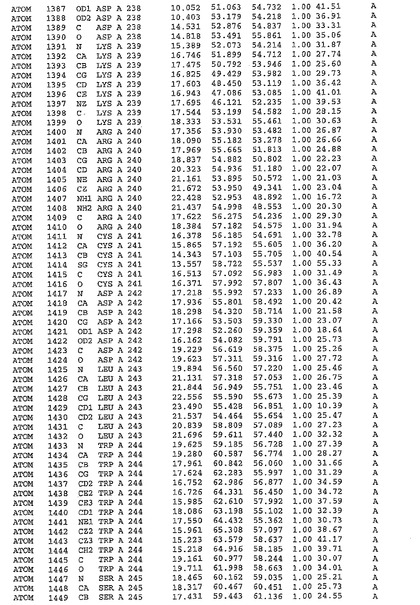
【図14−114】
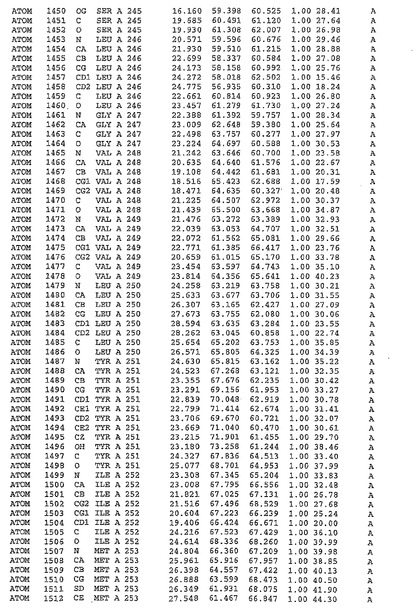
【図14−115】
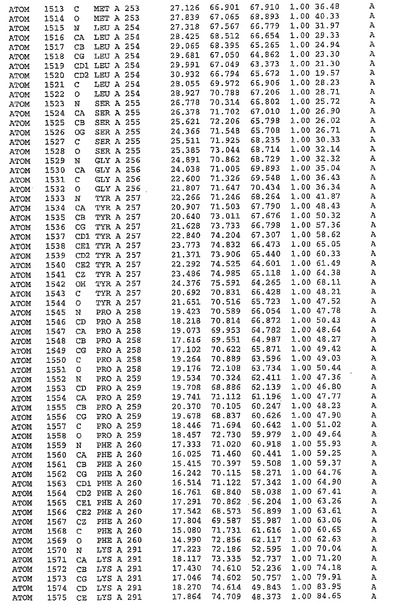
【図14−116】
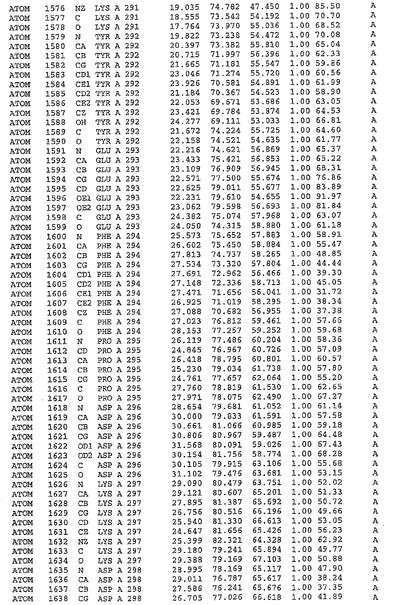
【図14−117】
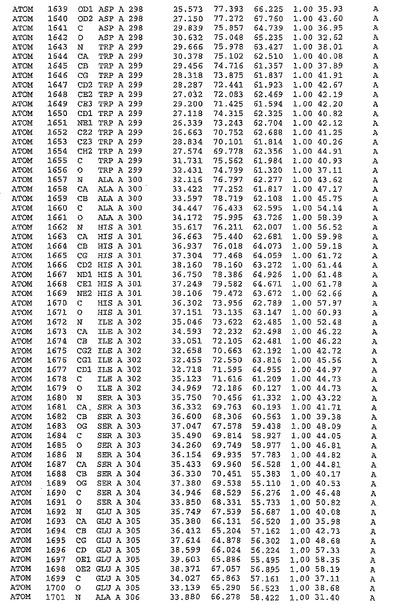
【図14−118】
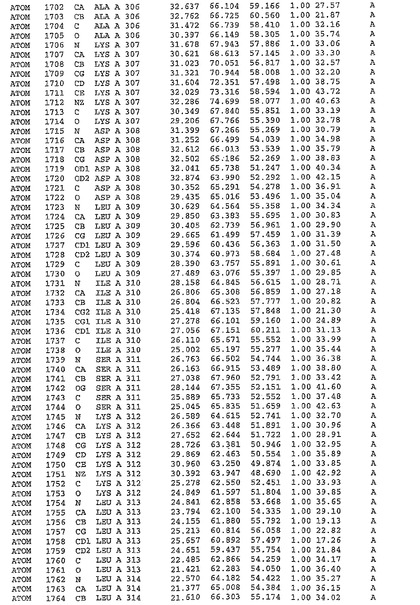
【図14−119】
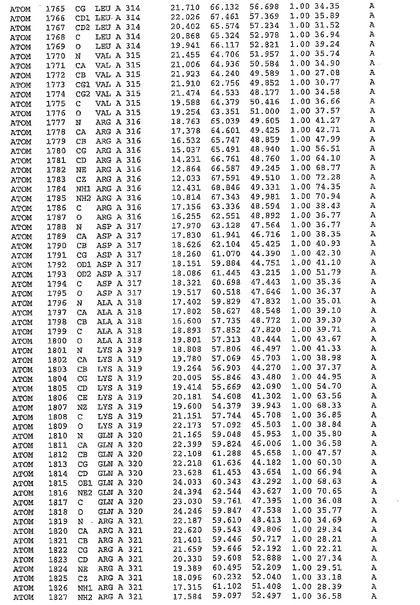
【図14−120】
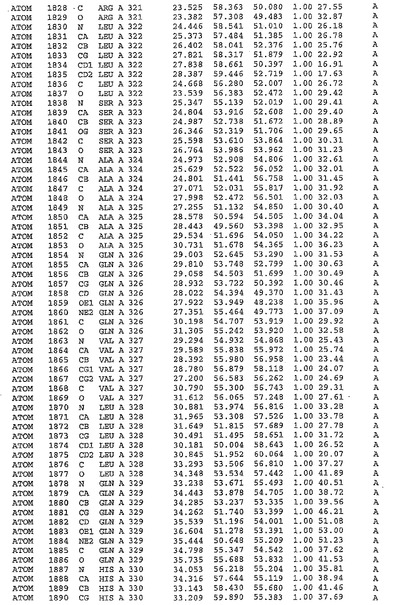
【図14−121】
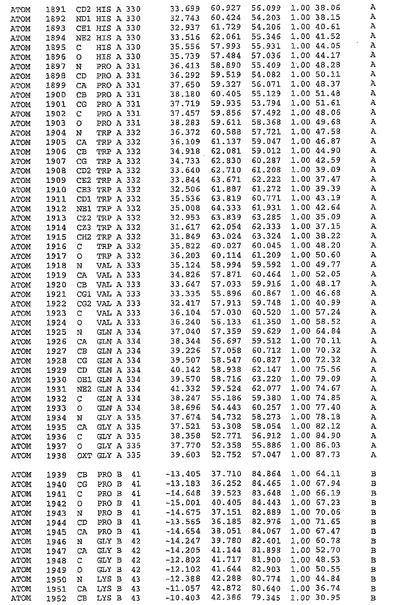
【図14−122】
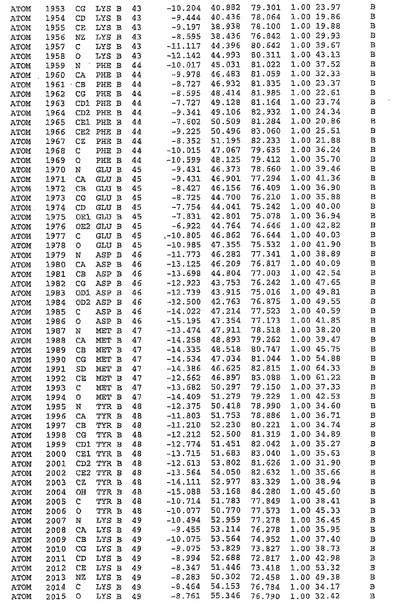
【図14−123】
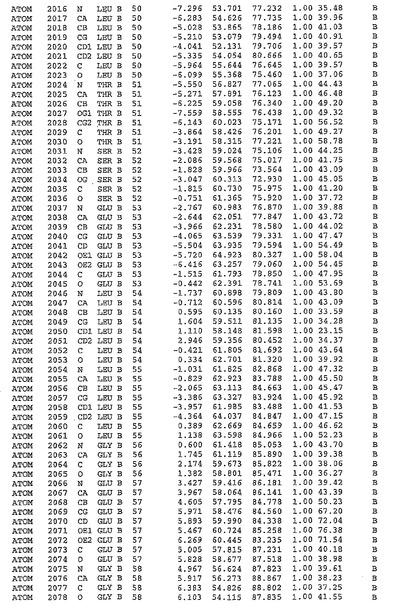
【図14−124】
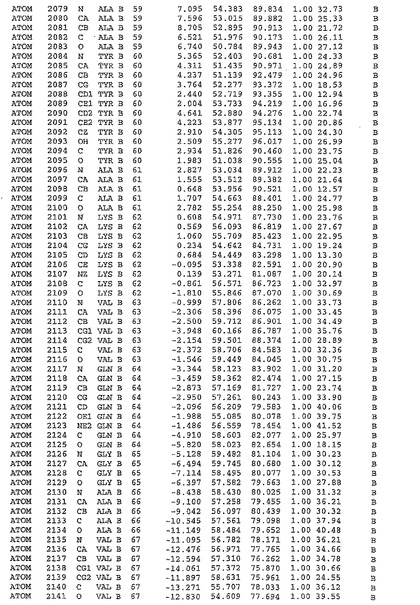
【図14−125】
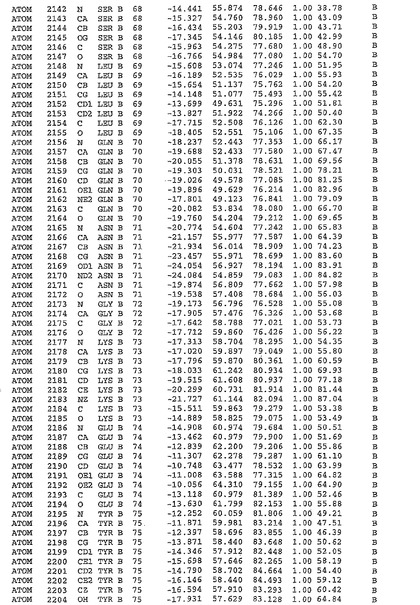
【図14−126】
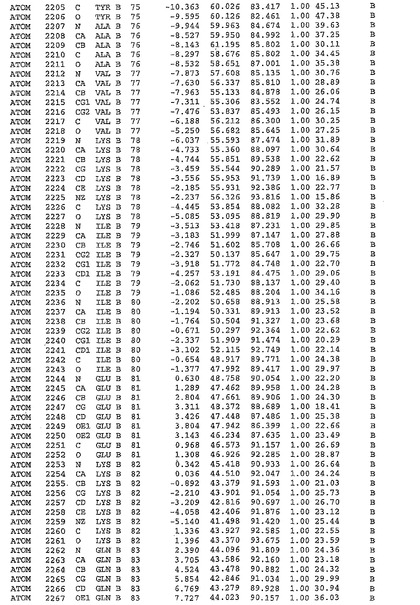
【図14−127】
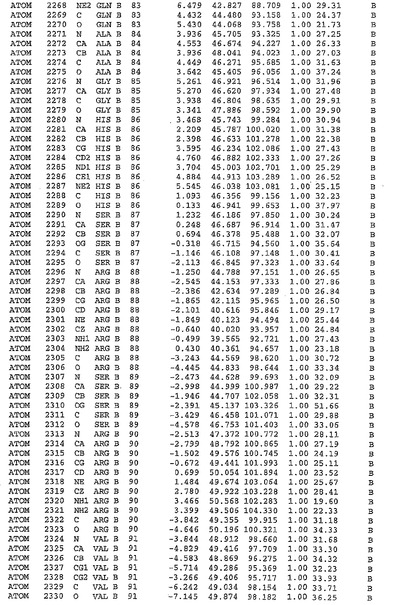
【図14−128】
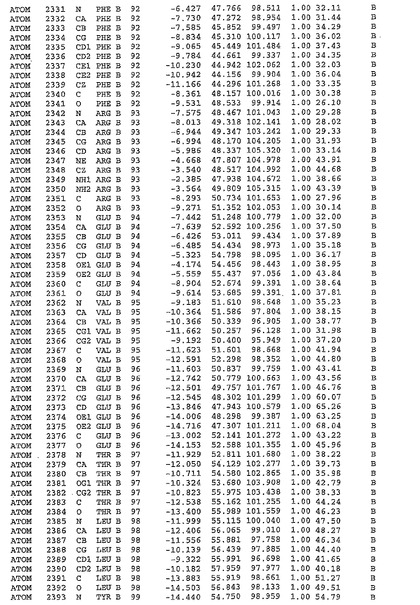
【図14−129】
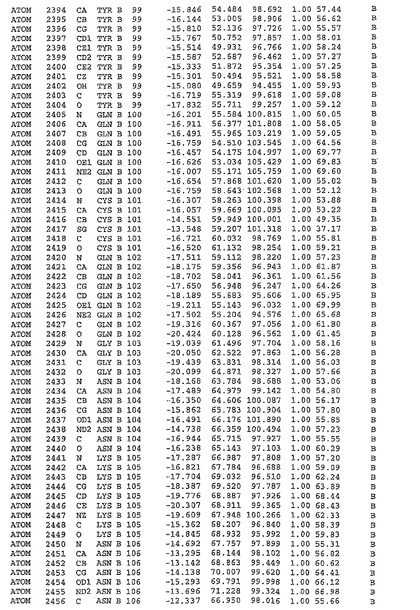
【図14−130】
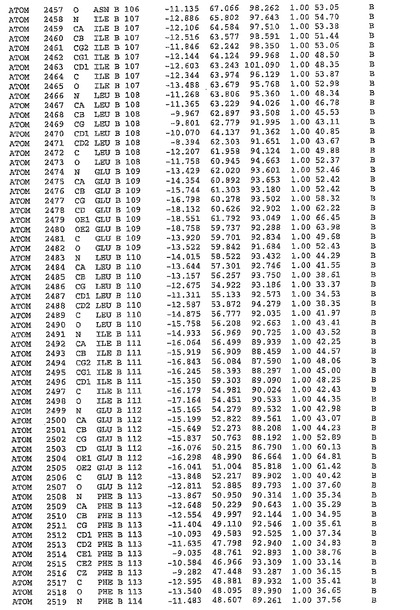
【図14−131】
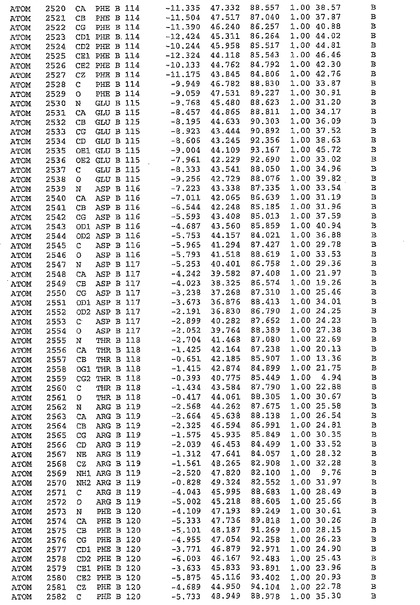
【図14−132】
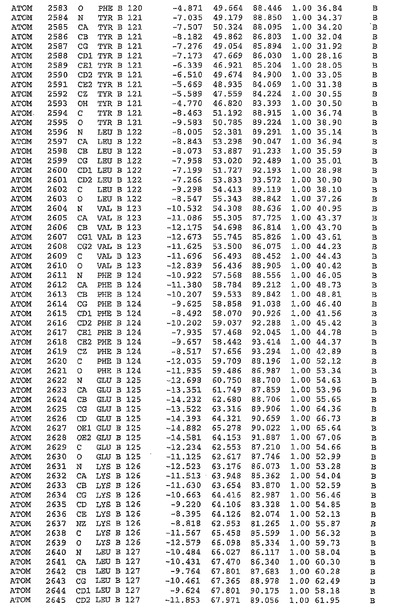
【図14−133】
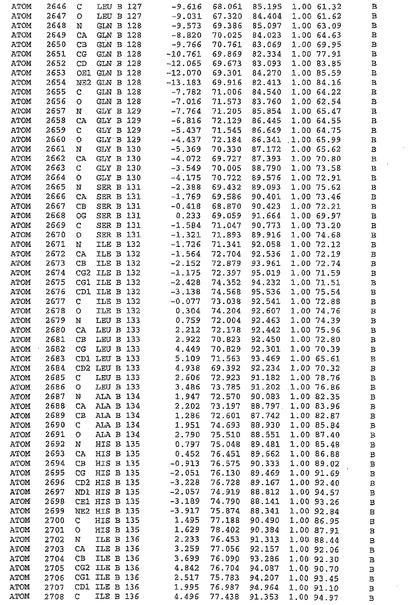
【図14−134】
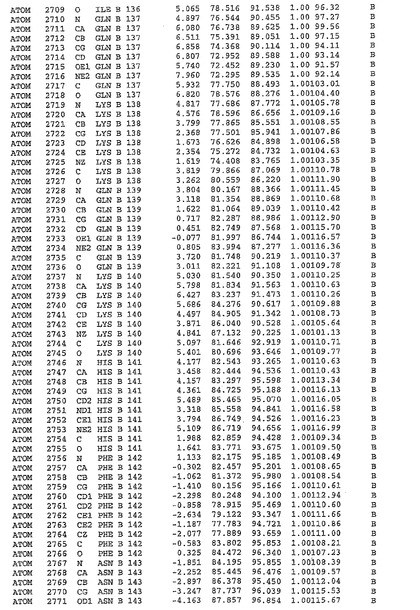
【図14−135】
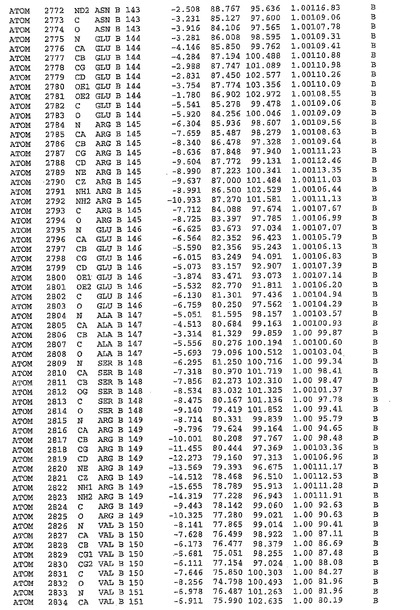
【図14−136】
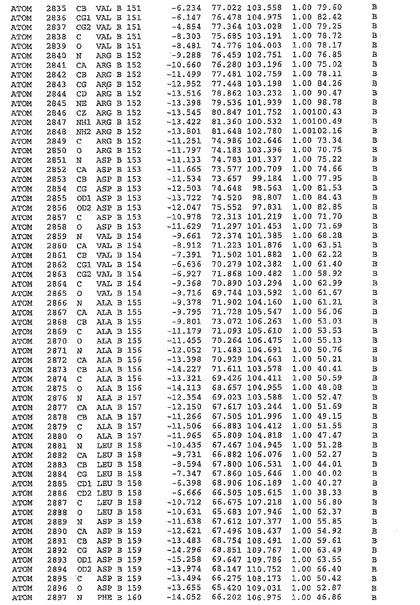
【図14−137】
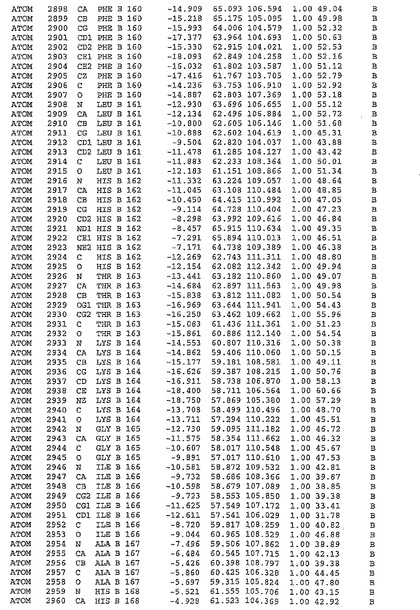
【図14−138】

【図14−139】
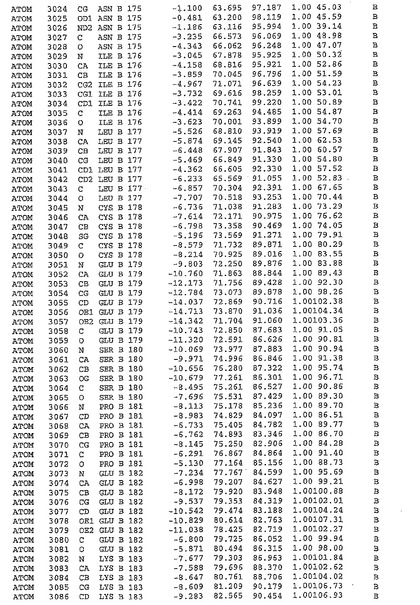
【図14−140】
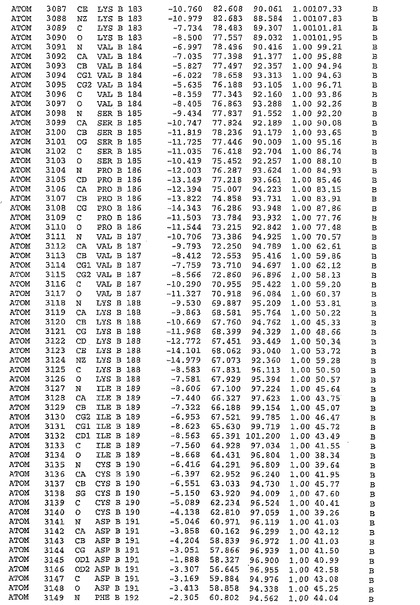
【図14−141】
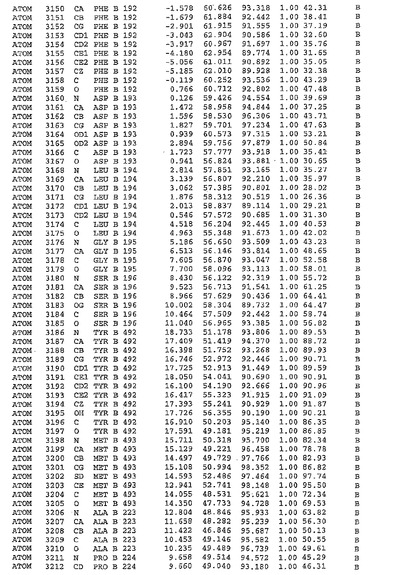
【図14−142】
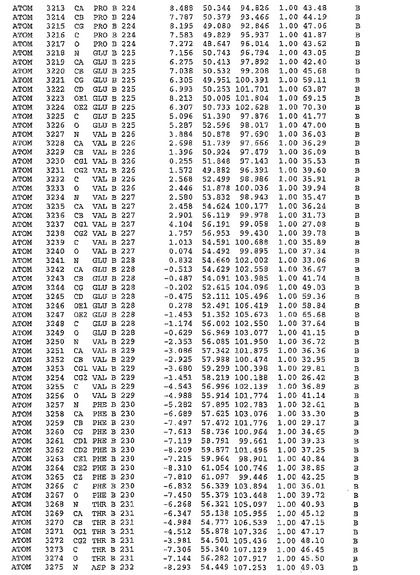
【図14−143】
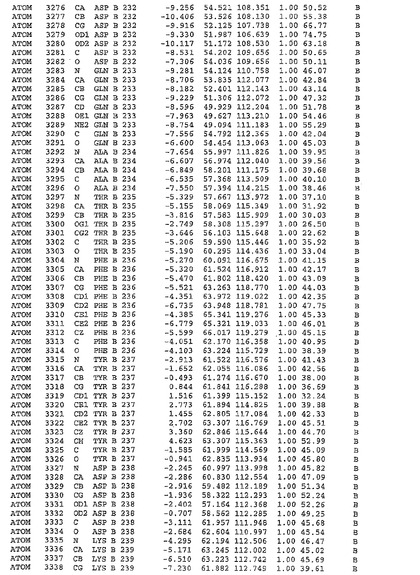
【図14−144】
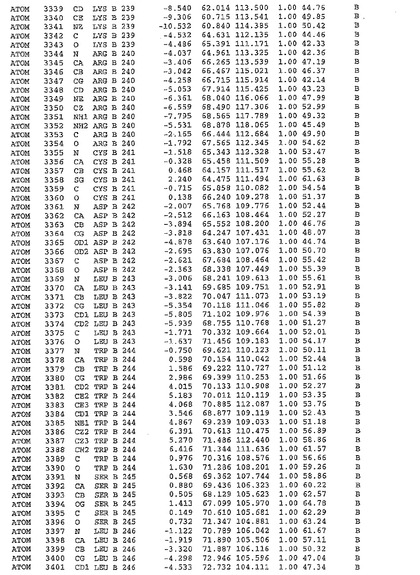
【図14−145】

【図14−146】
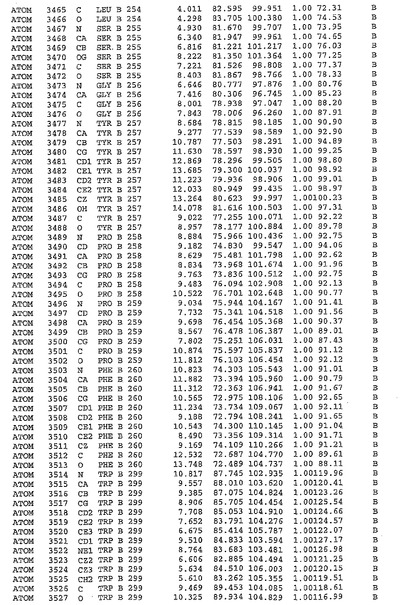
【図14−147】
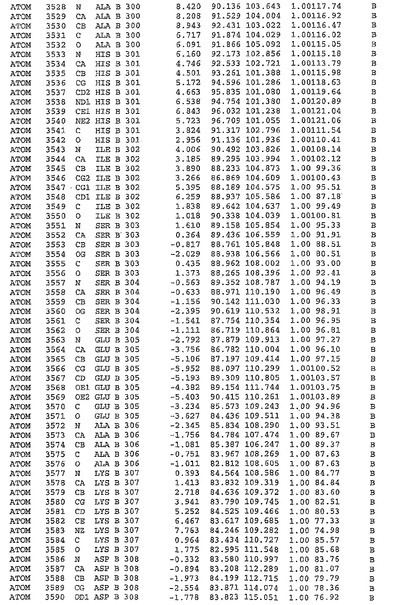
【図14−148】
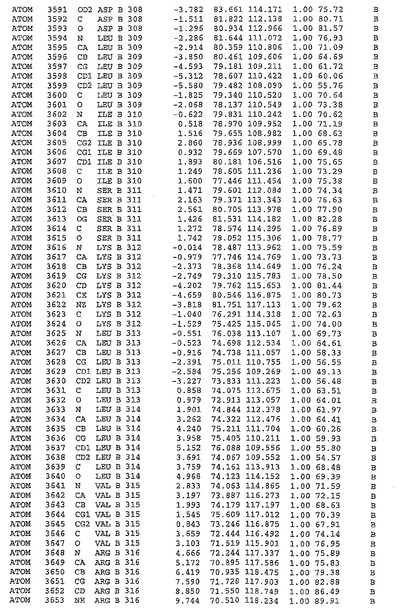
【図14−149】
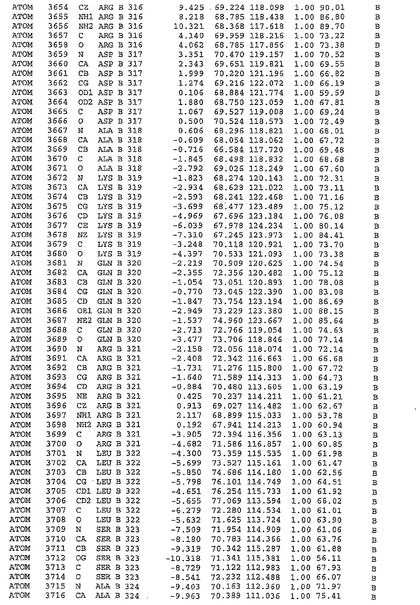
【図14−150】
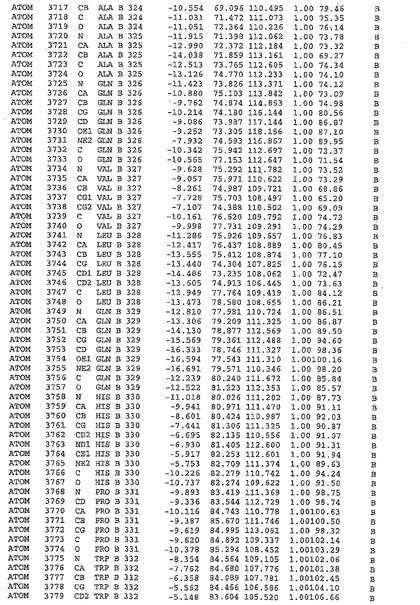
【図14−151】
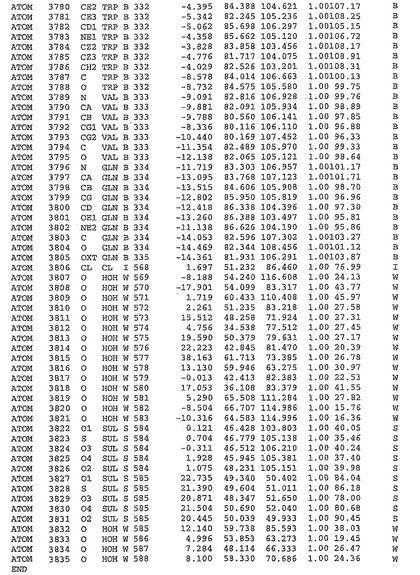
【図14−152】

【図14−153】

【図14−154】

【図14−155】

【図14−156】
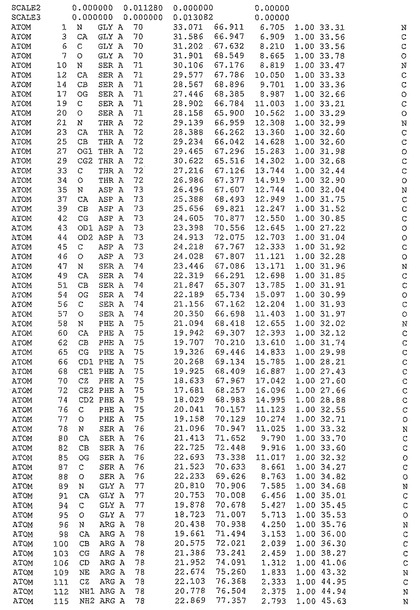
【図14−157】
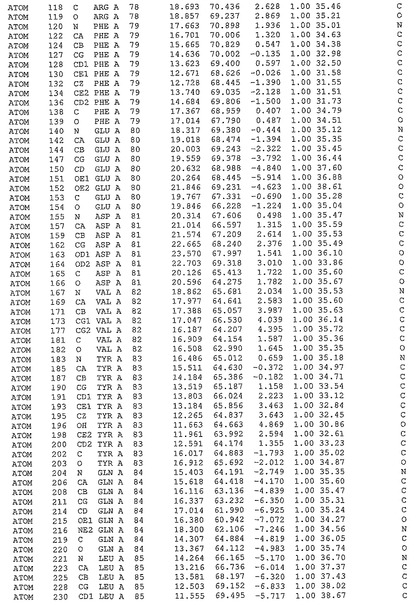
【図14−158】
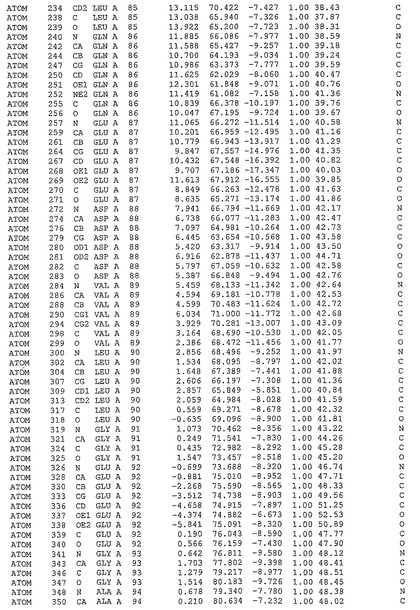
【図14−159】
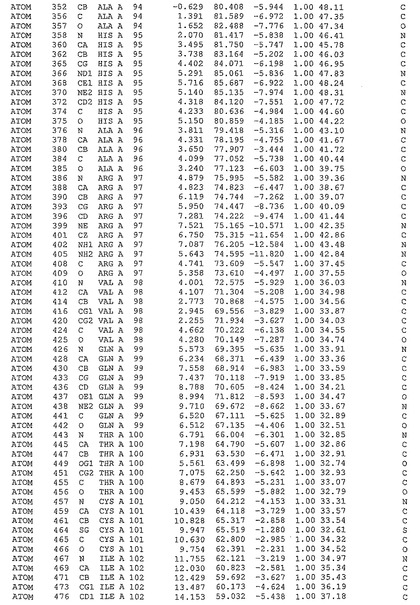
【図14−160】
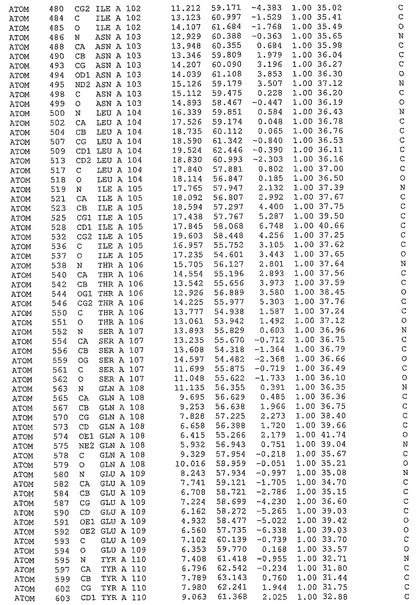
【図14−161】
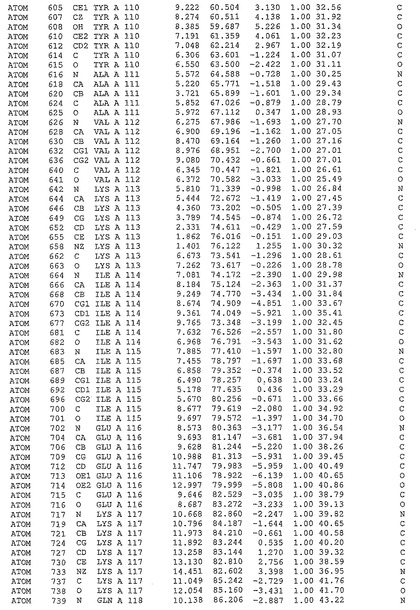
【図14−162】
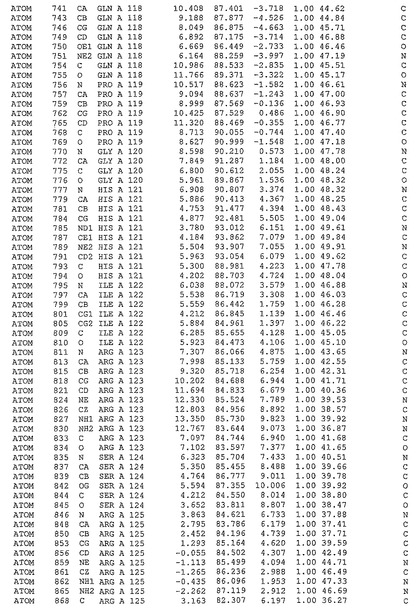
【図14−163】
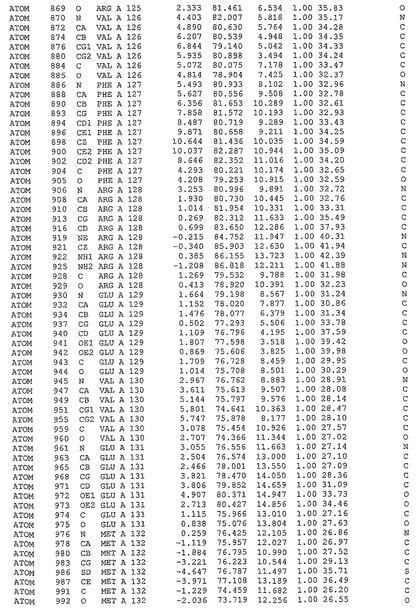
【図14−164】
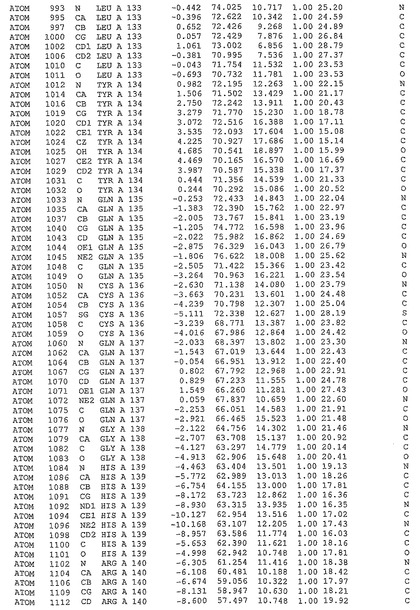
【図14−165】
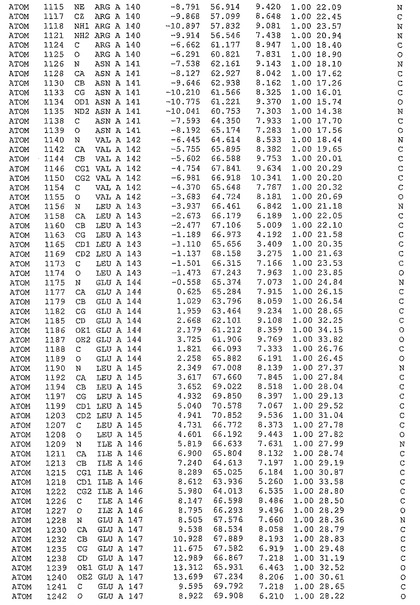
【図14−166】
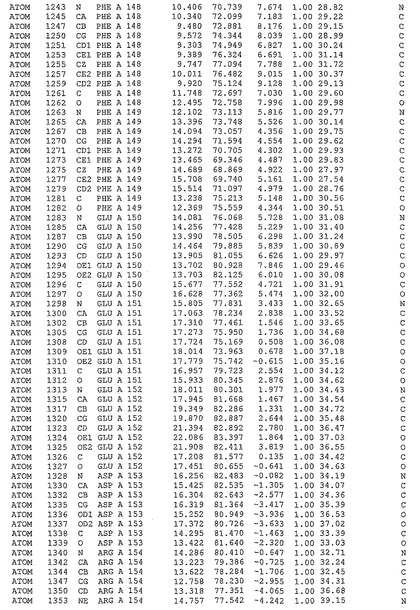
【図14−167】
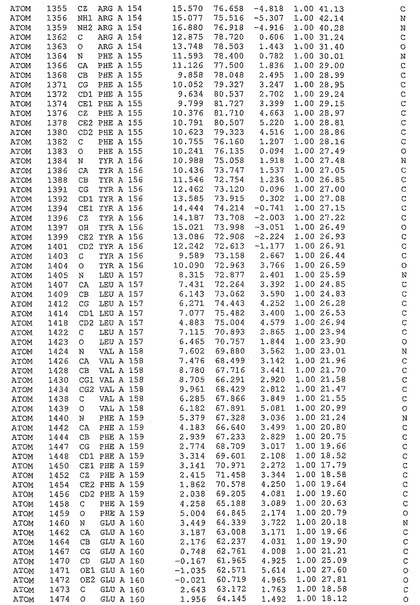
【図14−168】
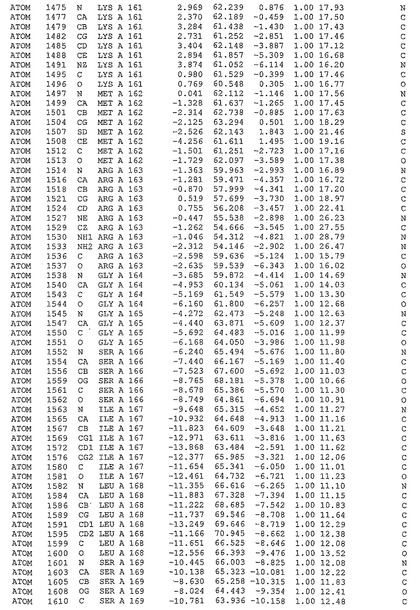
【図14−169】
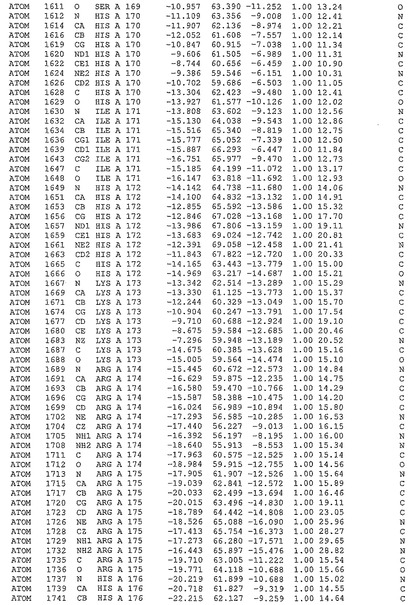
【図14−170】
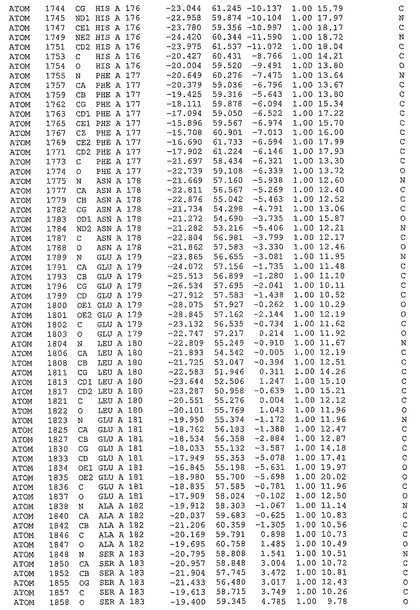
【図14−171】
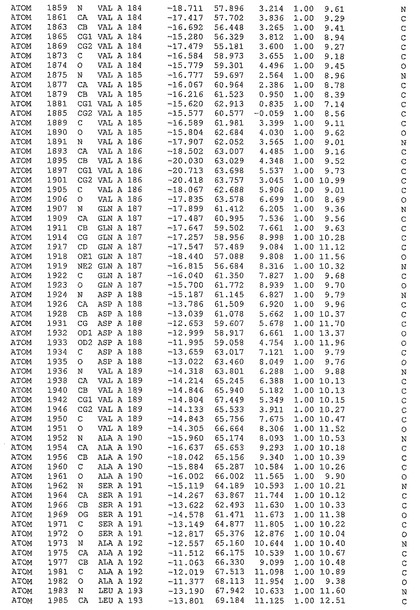
【図14−172】
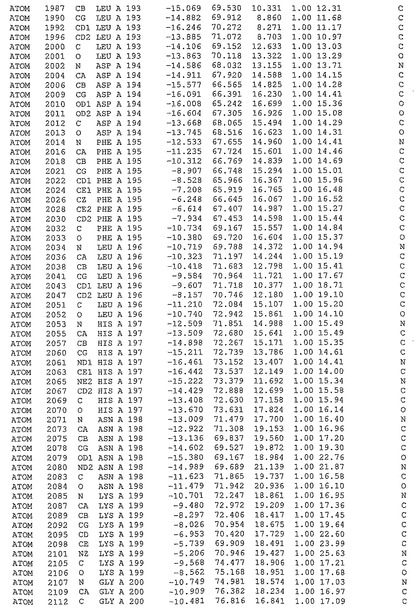
【図14−173】
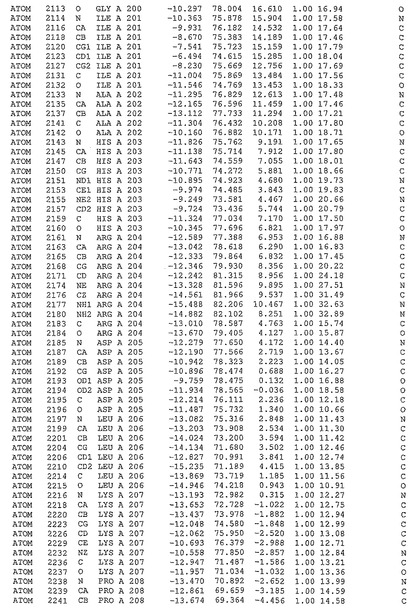
【図14−174】
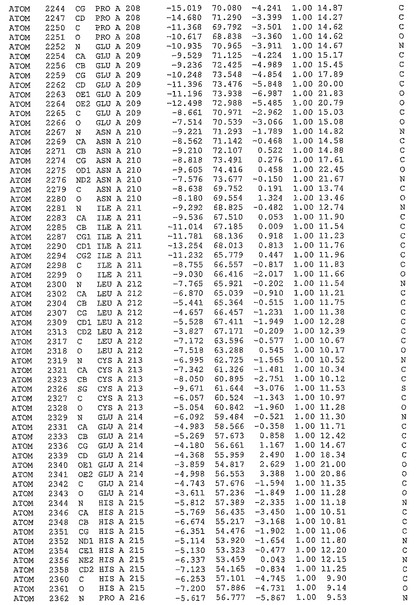
【図14−175】
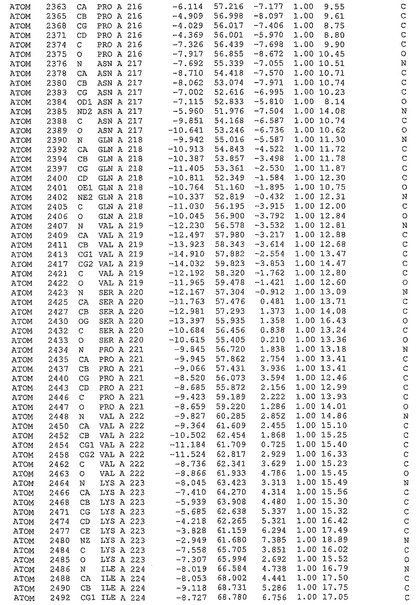
【図14−176】
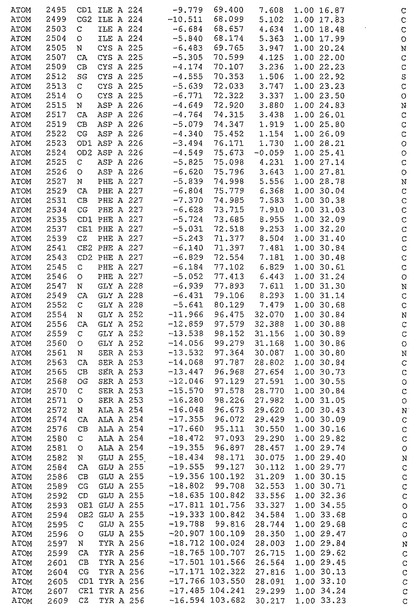
【図14−177】
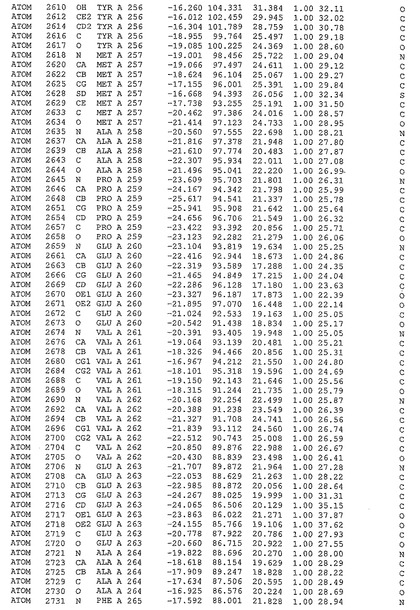
【図14−178】
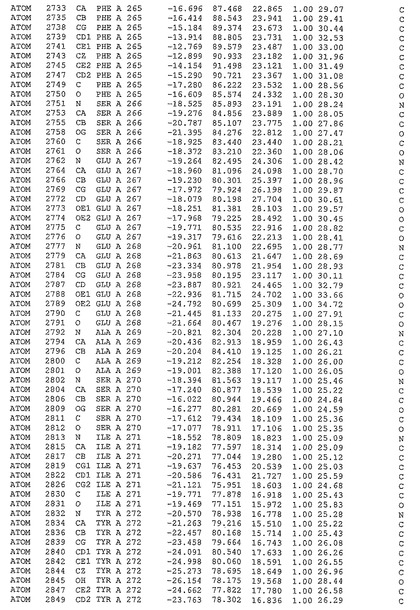
【図14−179】
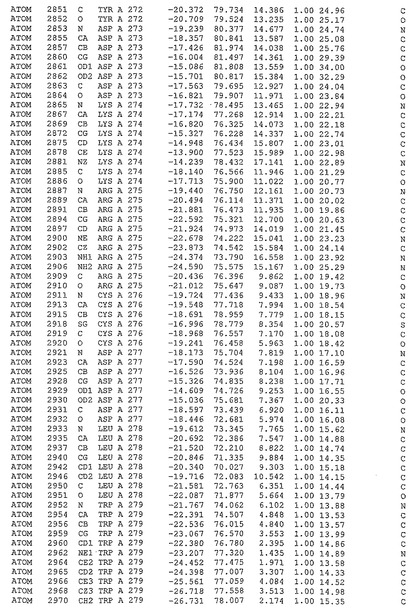
【図14−180】
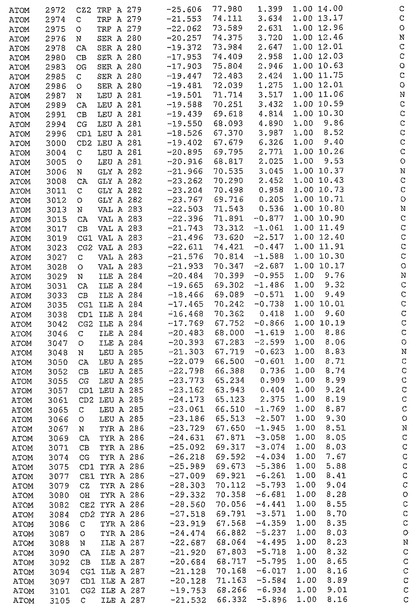
【図14−181】
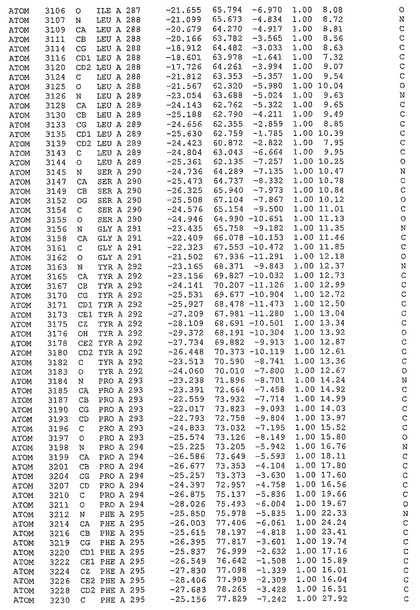
【図14−182】
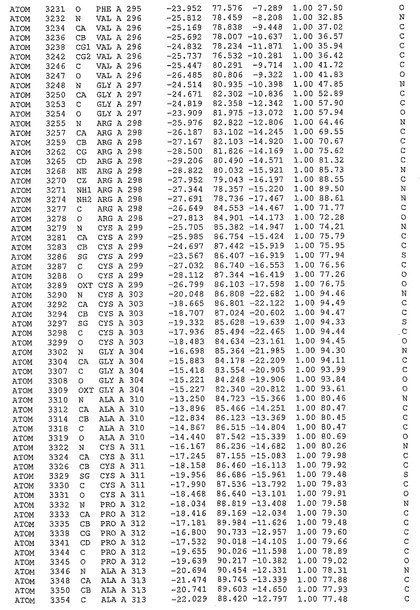
【図14−183】
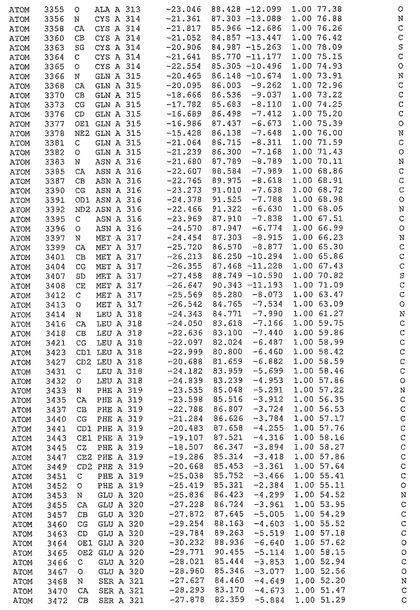
【図14−184】
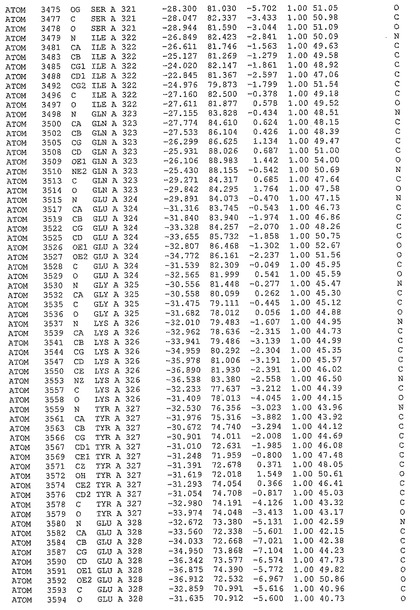
【図14−185】
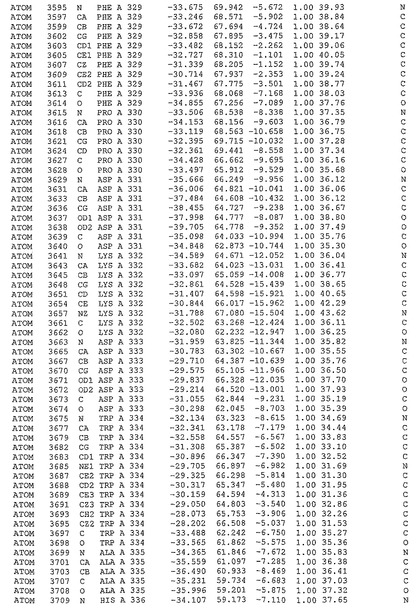
【図14−186】
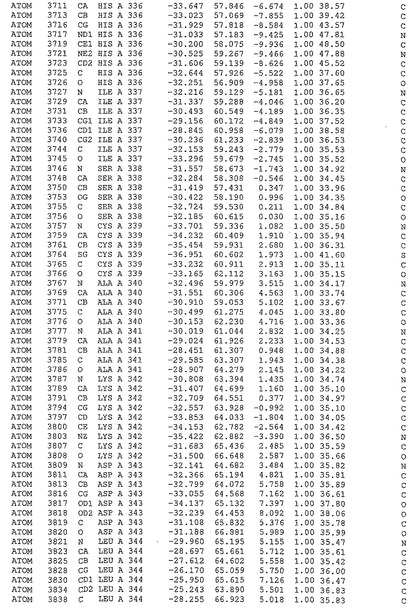
【図14−187】
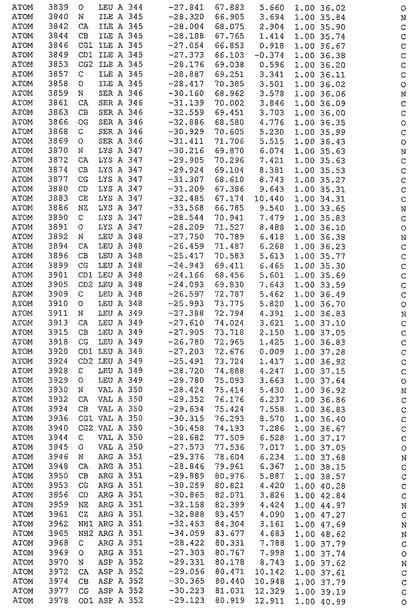
【図14−188】
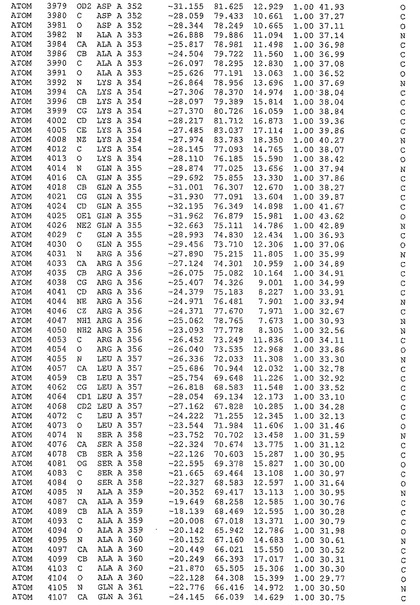
【図14−189】
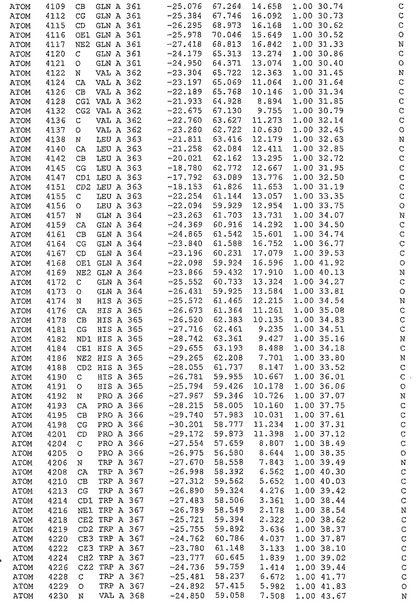
【図14−190】
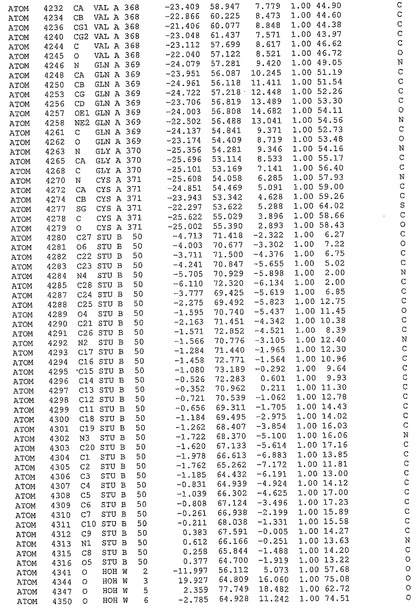
【図14−191】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14−1】
【図14−2】
【図14−3】
【図14−4】
【図14−5】
【図14−6】
【図14−7】
【図14−8】
【図14−9】
【図14−10】
【図14−11】
【図14−12】
【図14−13】
【図14−14】
【図14−15】
【図14−16】
【図14−17】
【図14−18】
【図14−19】
【図14−20】
【図14−21】
【図14−22】
【図14−23】
【図14−24】
【図14−25】
【図14−26】
【図14−27】
【図14−28】
【図14−29】
【図14−30】
【図14−31】
【図14−32】
【図14−33】
【図14−34】
【図14−35】
【図14−36】
【図14−37】
【図14−38】
【図14−39】
【図14−40】
【図14−41】
【図14−42】
【図14−43】
【図14−44】
【図14−45】
【図14−46】
【図14−47】
【図14−48】
【図14−49】
【図14−50】
【図14−51】
【図14−52】
【図14−53】
【図14−54】
【図14−55】
【図14−56】
【図14−57】
【図14−58】
【図14−59】
【図14−60】
【図14−61】
【図14−62】
【図14−63】
【図14−64】
【図14−65】
【図14−66】
【図14−67】
【図14−68】
【図14−69】
【図14−70】
【図14−71】
【図14−72】
【図14−73】
【図14−74】
【図14−75】
【図14−76】
【図14−77】
【図14−78】
【図14−79】
【図14−80】
【図14−81】
【図14−82】
【図14−83】
【図14−84】
【図14−85】
【図14−86】
【図14−87】
【図14−88】
【図14−89】
【図14−90】
【図14−91】
【図14−92】
【図14−93】
【図14−94】
【図14−95】
【図14−96】
【図14−97】
【図14−98】
【図14−99】
【図14−100】
【図14−101】
【図14−102】
【図14−103】
【図14−104】
【図14−105】
【図14−106】
【図14−107】
【図14−108】
【図14−109】
【図14−110】
【図14−111】
【図14−112】
【図14−113】
【図14−114】
【図14−115】
【図14−116】
【図14−117】
【図14−118】
【図14−119】
【図14−120】
【図14−121】
【図14−122】
【図14−123】
【図14−124】
【図14−125】
【図14−126】
【図14−127】
【図14−128】
【図14−129】
【図14−130】
【図14−131】
【図14−132】
【図14−133】
【図14−134】
【図14−135】
【図14−136】
【図14−137】
【図14−138】
【図14−139】
【図14−140】
【図14−141】
【図14−142】
【図14−143】
【図14−144】
【図14−145】
【図14−146】
【図14−147】
【図14−148】
【図14−149】
【図14−150】
【図14−151】
【図14−152】
【図14−153】
【図14−154】
【図14−155】
【図14−156】
【図14−157】
【図14−158】
【図14−159】
【図14−160】
【図14−161】
【図14−162】
【図14−163】
【図14−164】
【図14−165】
【図14−166】
【図14−167】
【図14−168】
【図14−169】
【図14−170】
【図14−171】
【図14−172】
【図14−173】
【図14−174】
【図14−175】
【図14−176】
【図14−177】
【図14−178】
【図14−179】
【図14−180】
【図14−181】
【図14−182】
【図14−183】
【図14−184】
【図14−185】
【図14−186】
【図14−187】
【図14−188】
【図14−189】
【図14−190】
【図14−191】
【公開番号】特開2012−135312(P2012−135312A)
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−22249(P2012−22249)
【出願日】平成24年2月3日(2012.2.3)
【分割の表示】特願2008−500120(P2008−500120)の分割
【原出願日】平成18年3月8日(2006.3.8)
【出願人】(510053400)ベーリンガー インゲルハイム インテルナツィオナール ゲゼルシャフト ミット ベシュレンクテル ハフツング (3)
【氏名又は名称原語表記】Boehringer Ingelheim International GmbH
【住所又は居所原語表記】Binger Strasse 173, D−55216 Ingelheim am Rhein, Germany
【出願人】(390040420)マックス−プランク−ゲゼルシャフト・ツア・フェルデルング・デア・ヴィッセンシャフテン・エー・ファオ (54)
【氏名又は名称原語表記】Max−Planck−Gesellschaft zur Foerderung der Wissenschaften e.V.
【Fターム(参考)】
【公開日】平成24年7月19日(2012.7.19)
【国際特許分類】
【出願番号】特願2012−22249(P2012−22249)
【出願日】平成24年2月3日(2012.2.3)
【分割の表示】特願2008−500120(P2008−500120)の分割
【原出願日】平成18年3月8日(2006.3.8)
【出願人】(510053400)ベーリンガー インゲルハイム インテルナツィオナール ゲゼルシャフト ミット ベシュレンクテル ハフツング (3)
【氏名又は名称原語表記】Boehringer Ingelheim International GmbH
【住所又は居所原語表記】Binger Strasse 173, D−55216 Ingelheim am Rhein, Germany
【出願人】(390040420)マックス−プランク−ゲゼルシャフト・ツア・フェルデルング・デア・ヴィッセンシャフテン・エー・ファオ (54)
【氏名又は名称原語表記】Max−Planck−Gesellschaft zur Foerderung der Wissenschaften e.V.
【Fターム(参考)】
[ Back to top ]