
N−アセチル−(R,S)−β−アミノ酸アシラーゼ遺伝子
【課題】 N−アセチル−(R,S)−β−アミノ酸アシラーゼの遺伝子を単離同定し、大腸菌のような宿主を用いて、これら遺伝子を高発現する系を構築するために、これらの酵素を菌体から単離精製し、該酵素をコードする遺伝子の塩基配列を決定すること。
【解決手段】Variovorax sp. AJ 110348が産生するN−アセチル−(R)−β−アミノ酸アシラーゼ、及び、該酵素をコードする遺伝子。
【解決手段】Variovorax sp. AJ 110348が産生するN−アセチル−(R)−β−アミノ酸アシラーゼ、及び、該酵素をコードする遺伝子。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、新規なN−アセチル−(R)−β−アミノ酸アシラーゼ及びN−アセチル−(S)−β−アミノ酸アシラーゼ、及びそれらをコードする遺伝子等に関するものである。
【背景技術】
【0002】
N−アセチル−β−アミノ酸に対してアシラーゼ活性を有する酵素は従来知られていなかったが、本発明者等は、N−アセチル−β−Phe等のN−アセチル−β−アミノ酸を培養菌体によって光学選択的に脱アセチル化する菌株としてVariovorax sp. AJ110348(FERM BP−10367)とBurkholderia sp. AJ110349(FERM BP−10366)を初めて見出し、これらの菌体又は菌体粉砕液上清を用いる光学選択的な脱アセチル化方法は特開2006−42722号公報(特許文献1)に記載されている。尚、これらの菌株は、夫々、平成16年7月22日付けで独立行政法人産業技術総合研究所特許生物寄託センターに寄託されたFERM P−20129及びFERM P−20128から、2005年7月4日付けで特許手続上の微生物の寄託等の国際的承認に関するブダペスト条約に基づく国際寄託に移管され、夫々、受託番号FERM BP−10367及びFERM BP−10366が付されている。
【特許文献1】特開2006−42722号公報
【発明の概要】
【発明が解決しようとする課題】
【0003】
これら選抜された野生株を用いた変換反応も変換の一手法ではあるが、Burkholdelia sp.AJ110349由来N−アセチル−β−アミノ酸アシラーゼは、(R)/(S)双方の光学選択性のアシラーゼを保持しており、野生株を用いて光学選択性の高いβ−アミノ酸を生成することが難しい状況であった。より効果的な変換を達成するためには、反応を触媒する酵素を単離・同定し、更にそのコード遺伝子も同定した上、例えば、大腸菌(Echerichia coli)のような宿主を用いて、その遺伝子を高発現する系を構築することが重要である。
【課題を解決するための手段】
【0004】
従来、ある酵素活性が見出されている菌株から、その活性を担う酵素あるいはその遺伝子を単離・同定する手法に関しては数多く報告されており、具体的には、一つはショットガンクローニングによる遺伝子単離法、もう一つは酵素単離、その部分アミノ酸配列決定、その配列情報に基づく遺伝子単離法が挙げられる。
【0005】
そこで、本発明者は、特許文献1に記載された菌株からN−アセチル−β−アミノ酸を光学選択的に脱アセチル化するN−アセチル−β−アミノ酸アシラーゼの遺伝子を取得すべく、まず第一に、常法であるショットガンクローニングによる方法を試みた。
【0006】
即ち、Burkholdelia sp. AJ110349株のゲノムDNAのライブラリーを作成し、N−アセチル−R/S−β−Pheを単一炭素源とする培地に塗布し37℃で1週間培養を行った。
また、供与検体数は、別途LB培地にライブラリーを塗布し、LB培地に生育した菌体数から算出した。約1万株に対してスクリーニングを行った結果、N−アセチル−R/S−β−Pheを単一炭素源として生育する株は得られなかった。
【0007】
また、Variovorax sp. AJ110348株に関しても同様に、N−アセチル−R/S−β−Pheを単一炭素源とするスクリーニングを約5万1千株に対して行ったが、Burkholdelia sp. AJ110349株のスクリーニングの際と同様に、N−アセチル−R/S−β−Pheを単一炭素源として生育する株は得られなかった。
【0008】
以上のように、非常に多くのクローンを調べたにも関わらず、単一炭素源の資化を指標とする様なスクリーニング手法では、目的とする酵素遺伝子の取得には至らなかった。
【0009】
このため、次に第二の方法である酵素の単離精製からのアプローチを試みることとした。酵素の単離精製の方法は数多く報告されているが、あらゆるタンパク質の単離精製が約束されるような単離精製法の一般則は存在せず、また、精製元の総タンパク質量に占める対象とするタンパク質の存在比によって、単離精製の難易度は大きく異なる。本発明におけるN−アセチル−β−アミノ酸アシラーゼの精製方法については後に詳述するが、Burkholderia sp. AJ110349株からの N−アセチル−(R)−β−アミノ酸アシラーゼおよび N−アセチル−(S)−β−アミノ酸アシラーゼの精製の場合、無細胞抽出液中での目的酵素の比活性に比べ、単離精製酵素の比活性は数百倍となっており、その精製は非常に困難なものであった。この困難な状況の原因の一つに、酵素生産菌であるBurkholderia sp. AJ110349株の性質の内、遠心操作による集菌のし難さが挙げられた。菌体内のタンパク質を単離精製する場合、通常、培地中のタンパク質の混入をさけるため、集菌後、緩衝液を用いて十分に菌体を洗浄する方法が採られるが、本菌株は、集菌手法として通常採用される遠心分離操作においてペレットとして回収されにくく、培地成分由来のタンパク質と菌体由来のタンパク質の分離が困難であった。このため、本株からのタンパク質単離精製は、通常の場合に比べ、多くの夾雑タンパク質が存在する状況から単離を行う必要があり、これが精製の困難さの一因となった。
【0010】
この問題を解決するため、発明者は通常のタンパク質精製の場合に比べ多くの条件等の検討に鋭意努力し、その結果、当業者でも予想し得ない5種という多くのクロマトグラフィーを用いる精製ステップを経ることによって、初めて上記酵素の単離精製に成功し、その結果、該酵素をコードする遺伝子の塩基配列を決定した。
また、Variovorax sp. AJ110348株に関しては、数種類の酵素を別々に取得する必要がないため、手法を改良し更にショットガンクローニングを継続した。上述したように目的の遺伝子を保持する株が選択的に増殖するような簡便なスクリーニングでは酵素遺伝子の取得には至らなかったため、発明者は、後述するように、各クローンの活性を個別に確認するという非常に時間の掛かる作業を実施し、目的の酵素遺伝子の取得に成功した。
上述のように、非常に多くの工夫を重ねながら、本発明を完成させた。
【0011】
即ち、本発明は以下の各態様に係るものである。
[態様1]下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号8で示されるアミノ酸配列からなる蛋白質、
(b)配列番号8で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号8で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様2]下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号7に示される塩基配列からなるDNA、
(b)配列番号7に示される塩基配列の相補鎖を含む核酸とストリンジェントな条件下でハイブリダイズし、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性
を有する蛋白質をコードするDNA、及び
(c)配列番号7に示される塩基配列のDNAと70%以上の配列同一性を示すDNAまたはその部分断片であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
[態様3]下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様4]下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号9に示される塩基配列からなるDNA、
(b)配列番号9に示される塩基配列の相補鎖を含む核酸とストリンジェントな条件下でハイブリダイズし、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性
を有する蛋白質をコードするDNA、及び
(c)配列番号9に示される塩基配列のDNAと70%以上の配列同一性を示すDNAまたはその部分断片であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
[態様5]下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号12で示されるアミノ酸配列からなる蛋白質、
(b)配列番号12で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号12で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様6]下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号11に示される塩基配列からなるDNA、
(b)配列番号11に示される塩基配列の相補鎖を含む核酸とストリンジェントな条件下でハイブリダイズし、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性
を有する蛋白質をコードするDNA、及び
(c)配列番号11に示される塩基配列のDNAと70%以上の配列同一性を示すDNAまたはその部分断片であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
[態様7] β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、態様1〜6のいずれか一項に記載の遺伝子。
[態様8]下記のいずれか一つに示す蛋白質。
(a)配列番号8で示されるアミノ酸配列からなる蛋白質、
(b)配列番号8で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号8で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様9]下記のいずれか一つに示す蛋白質。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様10]下記のいずれか一つに示す蛋白質。
(a)配列番号12で示されるアミノ酸配列からなる蛋白質、
(b)配列番号12で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号12で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様11]β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、態様8から10いずれか一項に記載のβ−アミノ酸アシラーゼ。
[態様12] 態様1から6のいずれか一項記載の遺伝子によって形質転換された微生物。
[態様13] 態様1から6のいずれか一項記載の遺伝子によって形質転換された微生物であるEscherichia coli。
[態様14] 態様12記載の微生物をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
[態様15] 態様12記載の微生物から得られるN−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
【発明の効果】
【0012】
本発明によって、N−アセチル−(R)−β−アミノ酸アシラーゼ及びN−アセチル−(S)−β−アミノ酸アシラーゼが単離同定され、更に、それらの遺伝子がクローニングされ、その塩基配列及びその遺伝子がコードする蛋白質のアミノ酸配列が明らかにされた。
【図面の簡単な説明】
【0013】
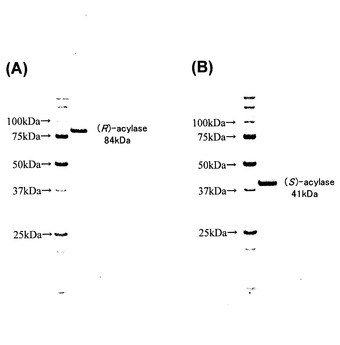
【図1】精製したN−アセチル−(R)−β−アミノ酸アシラーゼ(A)およびN−アセチル−(S)−β−アミノ酸アシラーゼ(B)のSDS−PAGEによる観察結果を示したものである。
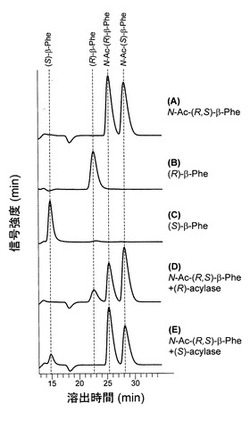
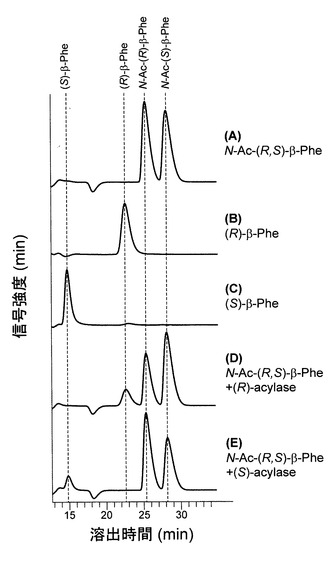
【図2】N−アセチル−(R,S)−β−Pheを基質、精製したN−アセチル−(R)−β−アミノ酸アシラーゼまたはN−アセチル−(S)−β−アミノ酸アシラーゼを酵素源として変換を行った結果をHPLCにて解析した結果である。AはN−アセチル−(R,S)−β−Pheのみで酵素を添加していない場合、BおよびCはそれぞれ(R)−β−Pheおよび(S)−β−Phe標準標品、D及びEはN−アセチル−(R,S)−β−PheをN−アセチル−(R)−β−アミノ酸アシラーゼまたはN−アセチル−(S)−β−アミノ酸アシラーゼで変換した結果をそれぞれ示している。
【発明を実施するための形態】
【0014】
本発明の遺伝子
本発明の遺伝子は、N−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼをコードするものであり、これらの酵素は、夫々、分子量約84kDa及び分子量約41kDaのサブユニット(構成単位)から成るホモダイマー又はホモトライマーの構造を有している。従って、より具体的には、本発明の遺伝子は、これらの各サブユニットであるポリペプチドをコードするものである。これらの酵素の代表例は、Burkholderia sp. AJ 110349(FERM BP−10366)が産生するものであり、その具体的なアミノ酸配列の一例は、夫々、配列番号8及び配列番号10に示されたものである。これらの酵素の別の代表例は、Variovorax sp. AJ 110348(FERM BP−10367)が産生するものであり、その具体的なアミノ酸配列の一例は、配列番号12に示されたものである。尚、本発明において、N−アセチル−(R,S)−β−アミノ酸アシラーゼの代表例として、N−アセチル−(R,S)−β−フェニルアラニン(Phe)アシラーゼを挙げることが出来る。N−アセチル−(R,S)−β−アミノ酸アシラーゼが、R体特異的な活性を有するか、S体特異的な活性を有するかは、N−アセチル−(R,S)−β−フェニルアラニンを基質として決定する。本発明のN−アセチル−(R,S)−β−アミノ酸アシラーゼは、α−アミノ酸のD体とL体を認識するかのごとく、側鎖、アミノ基、カルボキシル基、水素の相対位置を認識する。一方、RS表記では、β位に結合する側鎖によっては、−C−COOHと順位法則の優先性が入れ替わることがある。例えば、後述の実施例に示す、N−アセチル−β−homoLeuとN−アセチル−β−homoPheの場合は、N−アセチル−(R)−β−アミノ酸アシラーゼからS体が、N−アセチル−(S)−β−アミノ酸アシラーゼからR体がそれぞれ生成している。
【0015】
又、本明細書において、「N−アセチル−(R)−β−アミノ酸アシラーゼ活性又はN−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質」とは、ホモダイマー又はホモトライマーの構造を有する各アシラーゼ酵素の構成単位となり得る(構成単位としての機能を有する)蛋白質(ポリペプチド)をも意味する。
【0016】
本発明遺伝子は当業者に公知の方法で調製することが出来る。
例えば、本明細書の実施例に記載された方法に従い、各プローブを用いるコロニーハイブリダイズによる他、Burkholderia sp. AJ 110349(FERM BP−10366)またはVariovorax sp. AJ 110348(FERM BP−10367)のゲノムDNAを鋳型として、本明細書に開示された本発明DNAの塩基配列又はアミノ酸配列の情報に基づき適当な作製されるプライマーを使用した、当業者に公知の任意のPCRにより増幅して調製することも出来る。
【0017】
例えば、94℃で2分の後、94℃で10秒、55℃で20秒、72℃で2分を30サイクル行い、最後に72℃で5分を行う。なお、サーマルサイクラーとしては、Perkin Elmer社製9600など一般のサーマルサイクラーを用いることができる。耐熱性 DNAポリメラーゼとしては、ExTaq DNA Polymerase(宝酒造製)などの一般の市販品を用い、反応液の組成はポリメラーゼに添付の説明書に従って実施する。
【0018】
更に、当業者に周知の化学合成によって、本発明の各遺伝子を調製することも出来る。
【0019】
本明細書において、「ストリンジェントな条件下」とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。一例を示せば、相同性が高いDNA同士、例えば50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上の相同性を有するDNA同士がハイブリダイズし、それより相同性が低いDNA同士がハイブリダイズしない条件が挙げられる。なお、塩基配列間の同一性は、Blast等の当業者に公知のアルゴリズムを用いて決定することができる。より具体的には、通常のサザンハイブリダイゼーションの洗いの条件である60℃、1×SSC、0.1%SDS、好ましくは、0.1×SSC、0.1%SDSに相当する塩濃度でハイブリダイズする条件が挙げられる。このような条件でハイブリダイズするDNAの中には途中にストップコドンが発生したものや、活性中心の変異により活性を失ったものも含まれるが、それらについては、所定のN−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼ活性を後述の方法で測定し取り除くことができる。
【0020】
ハイブリダイゼーションは、例えば、カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current protocols in molecular biology(edited by Frederick M. Ausubel et al., 1987))に記載の方法等、当業界で公知の方法あるいはそれに準じる方法に従って行なうことができる。また、市販のライブ、添付の使用説明書に記載の方法に従って行なうことができる。
【0021】
本発明の蛋白質
本発明の遺伝子がコードする蛋白質の中で、N−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼを構成するユニットポリペプチドのアミノ酸配列(配列番号8、配列番号10又は配列番号12)において、1個若しくは数個のアミノ酸残基の置換、欠失、又は付加を含むアミノ酸配列を有する蛋白質であって、上記酵素活性を有するものは、当業者に公知の任意の方法、例えば、部位特異的変異導入法、遺伝子相同組換え法、プライマー伸長法、及びPCR法等の当業者に周知の方法を適宜組み合わせて、容易に作成することが可能である。
【0022】
尚、その際に、実質的に同等の機能を有するためには、当該ポリペプチドを構成するアミノ酸のうち、同族アミノ酸(極性β非極性アミノ酸、疎水性β親水性アミノ酸、陽性β陰性荷電アミノ酸、芳香族アミノ酸など)同士の置換が可能性として考えられる。又、実質的に同等の機能の維持のためには、本発明の各ポリペプチドに含まれる機能ドメイン内のアミノ酸は保持されることが望ましい。
【0023】
更に、本発明の蛋白質として、上記のアミノ酸配列と、全体の平均で約70%、好ましくは約80%以上、より好ましくは約90%以上、更に好ましくは約95%以上であるような高い配列同一性を有するアミノ酸配列を含む蛋白質又はその断片であって、所定のN−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼを有するものを挙げることができる。尚、アミノ酸配列間の同一性も、当業者に公知のアルゴリズム、例えば、実施例で使用されているBlast を用いて決定することができる。又、上記酵素の活性は本明細書の実施例に記載した方法で測定することが出来る。又、このような蛋白質も、当業者に公知の任意の方法、例えば、部位特異的変異導入法、遺伝子相同組換え法、プライマー伸長法、及びPCR法等の当業者に周知の方法を適宜組み合わせて、容易に作成することが可能である。
尚、本発明のβ-アミノ酸は、下記の構造式で示される。
【0024】
【化1】

【0025】
構造式中、Rは、炭素数1〜6のアルキル基、炭素数6〜14のアリール基、炭素数3〜10のシクロアルキル基、炭素数7〜19のアラルキル基、炭素数2〜11のアルコキシアルキル基、これらの炭素骨格中にヘテロ原子を含む基、およびこれらの炭素骨格中に炭素−炭素不飽和結合を含む基からなる群より選ばれ、これらの基は直鎖であっても分岐していてもよく、さらに置換基を有していてもよい。好ましくは、Rは、炭素数1〜4のアルキル基、炭素数6〜7のアリール基、これらの基は直鎖であっても分岐していてもよく、さらに置換基を有していてもよい。好ましくは、β−アミノ酸は、β−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンから選ばれる。
【0026】
本発明の遺伝子の発現
上記で得られた本発明遺伝子を当業者に公知の任意の方法によって組換えベクターに組み込み、本発明の組換え発現ベクターを作成することが出来る。
例えば、(1)本発明の遺伝子を含有するDNA断片を切り出し、(2)該DNA断片を適当な組換えベクター中の制限酵素部位又はマルチクローニングサイトに挿入して該ベクターに連結する挿入することにより製造することができる。組換えベクターに特に制限はなく、例えば、麹菌由来のプラスミド(例えば、pSal23, pTAex3, pNGU113, pRBG1, pGM32, pSE52, pNAGL142等)、大腸菌由来のプラスミド(例、pT7Blue T−Vector、pRSET、pBR322、pBR325、pUC18、pUC118)、枯草菌由来のプラスミド(例、pUB110、pTP5、pC194)、及び、酵母由来プラスミド(例、pSH19、pSH15)、等の組換えベクター等を利用することが出来る。
【0027】
上記組換えベクターには、以上の他に、本発明の転写調節配列の活性を損なわない限り、所望により当該技術分野で公知のプロモーター等の各種転写調節要素、シャイン・ダルガルノ配列、選択マーカー、転写終結シグナル等を付加することができる。また、必要に応じて、本発明の外来遺伝子にコードされた所望の蛋白質を他の蛋白質又はペプチド(例えば、グルタチオンSトランスフェラーゼ、ヒスチジンタグ、カルモデュリンバインディング蛋白質、及びプロテインA等)との融合蛋白質として発現させることも可能である。このような融合蛋白質は、適当なプロテアーゼを使用して切断し、それぞれの蛋白質に分離することが出来る。
【0028】
本発明の遺伝子が有効に発現される限り、本発明の組換え発現ベクターを有する微生物(形質転換体)を調製するために使用する宿主の種類及び由来等に特に制限はなく、例えば、大腸菌のような原核細胞、又はサッカロマイセス・セレビシエ(パン酵母)真核細胞等の当業者に公知の任意の微生物細胞を使用することが可能である。
【0029】
これら宿主細胞の形質転換は、例えば、塩化カルシウム法、パーティクルガン、エレクトロポレーション法等の、当該技術分野で公知の方法に従って行うことが出来る。例えば、以下に記載の文献を参照することが出来る。Proc. Natl. Acad. Sci. USA, 69巻、2110(1972); Gene, 17巻、107(1982);Molecular & General Genetics,168巻, 111(1979);Methods in Enzymology,194巻、182−187(1991);Proc. Natl. Acad. Sci. USA)、75巻、1929(1978);細胞工学別冊8 新細胞工学実験プロトコール。263−267(1995)(秀潤社発行);及びVirology、52巻、456(1973)。
【0030】
このようにして得られた、本発明の形質転換体は、当該技術分野で公知の方法に従って培養することが出来る。
【0031】
本発明における蛋白質の製造に際しては、当業者に公知の方法を適宜選択することができる。例えば、該蛋白質を含む培養液から、例えば、各種クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS−ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等の当業者に公知の方法を適宜選択、組み合わせることによって、実質的に純粋で均一な蛋白質として分離、精製することができる。
【0032】
更に、蛋白質をグルタチオン S−トランスフェラーゼ蛋白質との融合蛋白質、又はヒスチジンを複数付加させた組換え蛋白質として発現させた場合には、発現させた組換え蛋白質はグルタチオンカラムあるいはニッケルカラムを用いて精製することができる。融合蛋白質の精製後、必要に応じて、目的の蛋白質以外の領域を、トロンビンまたはファクターXaなどにより切断し、除去することも可能である。或いは、トリプシン、キモトリプシン、リシルエンドペプチダーゼ、プロテインキナーゼ、グルコシダーゼ等の適当な蛋白質修飾酵素で、精製前又は精製後に蛋白質を処理することにより、任意に修飾を加えたり、部分的にペプチドを除去することもできる。
【0033】
本発明は、N−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をコードする遺伝子で形質転換された微生物をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法を提供する。また、同微生物から得られるN−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法を提供する。
本発明の製造法において、各アシラーゼを作用させる様式、方法、及び条件等は、N−アセチル−β−アミノ酸の種類及び量、各アシラーゼの種類、製造規模等の諸要件に応じて、当業者に公知の任意のものを適宜選択することが出来る。
例えば、本発明の菌体の懸濁液中にN−アセチル−β−アミノ酸を添加したり、又は、該菌体の破砕液上清中にN−アセチル−β−アミノ酸を添加することにより該アシラーゼを作用させることが出来る。
反応温度は、好ましくは10〜60℃、より好ましくは20〜40℃である。また、反応系のpHは、好ましくは4〜10であり、より好ましくは6〜9である。また、反応時間は10分〜120時間、好ましくは、1〜60時間、より好ましくは1〜48時間である。反応溶媒は、水溶液、MeOH、DMF、DMSOなどを用いることもできるし、それらを混合して使用することも可能である。
【実施例】
【0034】
以下の実施例を示し、本発明をより具体的に説明するが、本発明の技術的範囲はこれらによって制限されるものではない。
【0035】
1.試薬
(R,S)−β−Phe(DL−3−amino−3−phenyl−propionic acid)は、Sigma−Aldrich社より購入した。(R)−β−Phe及び(S)−β−Pheは渡辺化学より購入した。
その他β−アミノ酸は下記のメーカーより購入した。
(R,S)−β−Leu(DL−β−Leucine 、Fluka社);
(R)−β−Leu(L−β−Leucine hydrochloride、Fluka社);
(R,S)−β−homoPhe(DL−β−Homophyenylalanine、Fluka社);
(S)−β−homoPhe(L−β−Homophyenylalanine hydrochloride、Fluka社);
(R,S)−β−homoLeu(3−amino−5−methyl−hexanoic acid、Astatech, Inc.社);
(R)−β−homoLeu((R)−3−amino−5−methyl−hexanoic acid、Astatech, Inc.社);
(R,S)−β−Tyr(3−amino−3−(4−hydroxyphenyl)−propanoic acid、Bionet Building Blocks社);
(R)−β−Tyr((R)−3−amino−3−(4−hydroxyphenyl)−propanoic acid、Peptech社);
(R,S)−4−Fluoro−β−Phe(3−amino−3−(4−fluorophenyl)−propanoic acid、Bionet Building Blocks社);
(R)−4−Fluoro−β−Phe((R)−3−amino−3−(4−fluoro−phenyl)−propanoic acid、Peptech社);及び
(R,S)-β−3−Pyr−Ala(3−amino−3−(3−pyridyl)−propanoic acid、Bachem社);
【0036】
2.合成
各種N−アセチル−β−アミノ酸は、ラセミ体はラセミ体のβ−アミノ酸を、光学活性体は各光学活性体のβ−アミノ酸をアセチル化して合成した。合成法は以下に示すとおりである。なお、新規物質に関しては、NMRの解析結果を記載した。
2−1.N−アセチル−(R,S)−β−Pheの合成
(R,S)−β−Phe(50g,303mmol)を水200mlに懸濁させ、氷冷下25% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように25%NaOH水溶液でpHを調節しながら、滴下ロートで無水酢酸(62.8ml,664mmol)を室温で滴下した。一晩撹拌後、反応混合物の分析を行ったところ原料の残存が認められたため、さらに無水酢酸(6ml,63mmol)を加えた。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下60℃で一晩乾燥し、N−アセチル−(R,S)−β−Phe(56.9g,274.3mmol,91%)を得た。
【0037】
2−2.N−アセチル−(S)−β−Pheの合成
(S)−β−Phe(250mg,1.51mmol)を水5mlに懸濁させ、氷冷下20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.33ml,3.49mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(S)−β−Phe(238mg,1.15mmol,76%)を得た。
【0038】
2−3.N−アセチル−(R)−β−Pheの合成
(R)−β−Phe塩酸塩(250mg,1.24mmol)を水5mlに懸濁させ、氷冷下20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.28ml,2.96mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R)−β−Phe(236mg,1.13mmol,91%)を得た。
【0039】
2−4.N−アセチル−(R,S)-β−Leuの合成
(R,S)-β−Leu(995mg、7.58mmol)を水8mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(1.3ml、13.3mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−Leu(382mg、2.21mmol、29.0%)を得た。
ESI−MS [M−H]=172
【0040】
2−5.N−アセチル−(R,S)-β−homoPheの合成
(R,S)-β−homoPhe(198mg、1.12mmol)を水5mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.24ml、2.46mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−homoPhe(64.5mg、0.36mmol,32.1%)を得た。
ESI−MS [M−H]=220
【0041】
2−6.N−アセチル−(R,S)-β−homoLeuの合成
(R,S)-β−homoLeu(1.01g、6.9mmol)を水16mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(1.35ml、13.8mmol)を室温で滴下した。反応混合物をろ過し不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−homoLeu(64.5mg、5.07mmol、73.1%)を得た。この生成物は減圧乾燥により溶解、凝固していたが、NMRにより確認し、求める物質が生成していたのでそのまま反応に用いた。
ESI−MS [M−H]=186
【0042】
2−7.N−アセチル−(R,S)-β−Tyrの合成
(R,S)-β−Tyr(202mg、1.16mmol)を水16mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.33ml、3.39mmol)を室温で滴下した。反応混合物をろ過し不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し、エバポレーターで濃縮し結晶を析出させた。結晶をろ過し水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−Tyr(55.5mg、0.25mmol、21.4%)を得た。
1H NMR(400MHz, D2O); 2.05 (s, 3H), 2.60−2.70(m, 2H), 5.1(dd, 1H, J=7.5 , 7.5 Hz), 6.88(d, 2H, J=8.6 Hz), 7.26 (d, 2H, J=8.6 Hz)
ESI−MS [M−H]=222
【0043】
2−8.N−アセチル−(R,S)-4−Fluoro−β−Pheの合成
(R,S)-4−Fluoro−β−Phe(206mg、1.12mmol)を水16mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.22ml、2.26mmol)を室温で滴下した。反応混合物をろ過し不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し、エバポレーターで濃縮し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-4−Fluoro−β−Phe(143.9mg,0.64mmol,57.1%)を得た。
ESI−MS [M−H]=224
【0044】
以上の生成物は結晶を取得後に、NMRにてチャートを確認し、質量分析を行い目的化合物の生成を確認した。
【0045】
2−9.N−アセチル−(R,S)-β−aminobutylic acidの合成
3−アセトアミド−2−ブテン酸メチルはJ.Am.Chem.Soc. 2002, 124, 14552記載の方法に従い調製した。
3−アセトアミド−2−ブテン酸メチル(1.35g)のメタノール溶液に5%パラジウム−活性炭を加え、1気圧の水素下、30℃で16時間撹拌した。反応終了後、ろ過にて5%パラジウム−活性炭を除き、得られた溶液を濃縮して3−アセトアミド−2−酪酸メチル(1.37g)を得た。
続いて得られた3−アセトアミド−2−ブテン酸メチル(570mg)のエタノール溶液3mLに8M水酸化水溶液(900μL)を加え、35℃で1時間撹拌した。反応終了後、6M塩酸(1.2mL)を加え中和し、濃縮してジクロロメタン(20mL)を加えた。析出した結晶をろ過にて取り除き、ろ液を濃縮して表題化合物265mgを得た。
NMRにて、目的化合物の生成を確認した。
1H NMR(400MHz, MeOH−d4); 1.41 (q, 3H, J=2.6 Hz), 1.99 (s, 3H), 2.55 (t, 2H, J=5.1 Hz), 4.28(m, 1H)
13C NMR(400MHz, MeOH−d4);175.6, 170.88, 42.62, 40.22, 23.60, 20.34
ESI−MS [M−H]=144
【0046】
2−10.N−アセチル−3,4−(−O−CH2−O−)−β−Phenylalanine
US20060035345記載の方法で作成したものを使用した。
【0047】
2−11.N−アセチル−(R,S)-3−Pyr−Alaの合成
β-3-Pyr-Ala(200mg、1.2mmol)を水5mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.257ml、2.46mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調整したが沈殿が析出しなかったため、エバポレーターを用いて総て液体を蒸発させて、乾燥物をNMRにより解析し、反応に用いた。
1H NMR(400MHz, D2O); 2.03 (s, 3H), 2.78−2.90(m, 2H), 5.33(dd, 1H, J=7.3, 7.3 Hz),7.97(dd, 1H , J=5.7, 8.2 Hz), 8.49 (d, 1H, J=8.2 Hz), 8.67(d, 1H, J=5.7 Hz), 8.75(s, 1H,)
ESI−MS [M−H]=207
【0048】
3.酵素活性測定
3−1.アッセイ条件
10 mM N−アセチル−(R,S)−β−Phe, 50mM Tris−HCl (pH 7.6)および適宜酵素サンプルを含む反応液を30℃にて15minから2h静置した後、5minの煮沸処理にて反応を停止した。反応液を遠心し、その上清を適宜希釈して、HPLCにて分析した。この標準反応条件にて、1min間に1μmolの(R)−β−Pheまたは(S)−β−Pheを生成するアシラーゼ活性を1 Unitと定義した。以後N−アセチル−(R,S)−β−Pheから(R)−β−Pheを生成する活性をN−アセチル−(R)−β−Pheアシラーゼ活性、(S)−β−Pheを生成する活性をN−アセチル−(S)−β−Pheアシラーゼ活性と記述する。
3−2.HPLC
【0049】
3−2−1.(R),(S)−β−Phe光学分割条件
(R)−β−Phe、(S)−β−Phe、N−アセチル−(R)−β−PheおよびN−アセチル−(S)−β−PheはHPLCによって定量分析した。カラムはジーエルサイエンス社製Inertsil ODS3(0.46cmΦ, 5cm)とダイセル化学社製Chiralpak WH(0.46cmΦ, 25cm)をこの順に直列につないで用いた。移動相は0.25mM CuSO4, 2%(v/v)アセトニトリルとし、流速1.5ml/min、カラム温度50℃、検出はUV detector 210nmとした。この分析で上記4種化合物は異なる溶出時間に溶出し、溶出順は早いほうから(S)−β−Phe、(R)−β−Phe、N−アセチル−(R)−β−Phe、N−アセチル−(S)−β−Pheの順であった。定量は、各標準標品を対照とし、ピークエリア値から算出した。
【0050】
3−2−2. N−アセチル−β−Pheとβ−Pheの分離条件
カラムはGLサイエンス社製Inertsil Ph−3(0.46cmΦ、25cm)を用いた。移動相に10%アセトニトリル(リン酸にてpH3.0に調整)を用い、温度40℃、流速1.0mL/min、検出UV210nmの条件で各ラセミ体標準品を対照とし、ピークエリア値から算出し定量した。
【0051】
3−2−3. (R),(S)−β−Leu、(R),(S)−β−homoLeu、(R),(S)−β−homoPhe分割条件
カラムはastec社製CHIROBIOTEC T(0.46cmΦ、25cm)を用いた。移動相に90% MeOHを用い、温度40℃、流速0.4mL/min、検出UV205nmの条件で、(R),(S)体標準品と比較し、光学選択性を決定した。
【0052】
3−2−4.(R),(S)−β−Tyr、(R),(S)−β−4−Fluoro−Phe分離条件
カラムはジーエルサイエンス社製Inertsil ODS3(0.46cmΦ, 5cm)とダイセル化学社製Chiralpak WH(0.46cmΦ, 25cm)をこの順に直列につないで用いた。移動相に1mM CuSO4、10% MeOHを用い、温度 50℃、流速1.0ml/min、検出UV210nmの条件で(R),(S)体標準品と比較し光学選択性を決定した。
【0053】
4.Burkholderia sp. AJ110349由来N−アセチル−(R)/(S)−β−アミノ酸アシラーゼの精製
4−1.使用菌株と培養条件
Burkholderia sp. AJ110349を用いた。保存菌株を5g/l D−glucose, 10g/l yeast extract, 10g/l peptone, 5g/l NaCl, 20g/l agar (pH 7.0)から成るCM2G寒天培地上で、30℃で48h静置培養することによりリフレッシュした。これを、10g/l D−glucose, 10g/l (NH4)2SO4, 10g/l (R,S)−β−Phe, 2g/l casamino acid, 1g/l KH2PO4, 0.4g/l MgSO4・7H2O,1g/l NaCl, 19.5g/l 2−(N−morpholino) ethanesufonic acid (MES), 5mg/l nicotinamide, 0.2mg/l thiamin, 10mg/l FeSO4・7H2O, 10mg/l MnSO4・4〜5H2Oからなる酵素生産培地100 mlに接種し、500ml用の坂口フラスコを用いて30℃、120rpmで66h、振盪培養した。
【0054】
4−2.無細胞抽出液の調製
培養液およそ2,000mlから、6,800g,10minの遠心操作により菌体を集菌した。この際、菌体の沈降が十分に見られなかったため、およそ1,600 mlの上清を除去し、残りの部分をピペッティングにより均一化した。この濃縮した培養液をバッファー等で洗浄することなく、200W, 20minの超音波処理に供し菌体を破砕した。得られた破砕液を200,000g, 30min遠心操作し、得られた遠心上清およそ200mlを無細胞抽出液とした。
【0055】
4−3.硫安分画
得られた無細胞抽出液に(NH4)2SO4を終濃度40%飽和となるよう加え、氷上1h攪拌した後、9,200g, 15min遠心した。得られた沈殿を少量の25mM Tris−HCl(pH 7.6)にて溶解後、25mM Tris−HCl(pH7.6)に対して透析した。この透析後の溶液を、以後クロマトグラフィーに供するサンプルとした。
【0056】
4−4.クロマトグラフィー
4−4−1)Phenyl Sepharose 26/10 (Amersham Pharmacia)
上記で得られた硫安分画画分を25mM Tris−HCl (pH7.6), 0.6M (NH4)2SO4から成る緩衝液に対して透析後、同緩衝液で平衡化したPhenyl Sepharose 26/10に供した。非吸着タンパク質溶出後、緩衝液中の(NH4)2SO4濃度を直線的に0.6 Mから0 Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、(NH4)2SO4濃度およそ0.2Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性は(NH4)2SO4濃度およそ0.1Mの付近にそれぞれ検出された。活性の存在する画分をN−アセチル−(R)−β−Pheアシラーゼ溶出画分、N−アセチル−(S)−β−Pheアシラーゼ活性溶出画分に分けて、それぞれ回収した。
【0057】
4−4−2)Q−Sepharose 16/10 (Amersham Pharmacia)
得られたPhenyl−Sepharose分画画分を濃縮し、25mM Tris−HCl (pH7.6)に対して透析後、同緩衝液で平衡化したQ−Sepharose 16/10に供した。非吸着タンパク質溶出後、緩衝液中のNaCl濃度を直線的に0 Mから0.5Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、NaCl濃度およそ0.22Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性はNaCl濃度およそ0.3Mの付近にそれぞれ検出された。活性の存在する画分をそれぞれ回収した。
【0058】
4−4−3)Superdex 200 16/60 (Amersham Pharmacia)
得られたQ−Sepharose分画画分を濃縮し、25mM Tris−HCl (pH7.6)で平衡化したSuperdex 200 16/20に供した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は分子量206kDaと見積もられる溶出位置に、N−アセチル−(S)−β−Pheアシラーゼ活性は分子量101kDaと見積もられる溶出位置でそれぞれ検出された。活性の存在する画分をそれぞれ回収した。
【0059】
4−4−4)Resource phenyl (Amersham Pharmacia)
得られたSuperdex分画画分を濃縮し、25mM Tris−HCl (pH7.6), 0.6M (NH4)2SO4から成る緩衝液に対して透析後、同緩衝液で平衡化したResource phenylに供した。非吸着タンパク質溶出後、緩衝液中の(NH4)2SO4濃度を直線的に0.6Mから0 Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、(NH4)2SO4濃度およそ0.35Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性は(NH4)2SO4濃度およそ0.45Mの付近にそれぞれ検出された。活性の存在する画分をそれぞれ回収した。
【0060】
4−4−5)Mono Q 5/5 (Amersham Pharmacia)
得られたResource phenyl分画画分を濃縮し、25 mM Tris−HCl (pH 7.6)に対して透析後、同緩衝液で平衡化したMono Q 5/5に供した。非吸着タンパク質溶出後、緩衝液中のNaCl濃度を直線的に0 Mから0.5Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、NaCl濃度およそ0.2Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性はNaCl濃度およそ0.28Mの付近にそれぞれ検出された。活性の存在する画分をそれぞれ回収し、それぞれ精製N−アセチル−(R)−β−アミノ酸アシラーゼ、精製N−アセチル−(S)−β−アミノ酸アシラーゼ標品とした。
【0061】
以上の酵素精製の結果を表1(N−アセチル−(R)−β−アミノ酸アシラーゼ)、表2(N−アセチル−(S)−β−アミノ酸アシラーゼ)にそれぞれまとめた。この精製操作により、N−アセチル−(R)−β−アミノ酸アシラーゼは比活性262倍に、N−アセチル−(S)−β−アミノ酸アシラーゼは比活性809倍にそれぞれ精製された。
【0062】
【表1】
【0063】
【表2】
【0064】
4−5.SDS−PAGE
上記で得られた精製N−アセチル−(R)−β−アミノ酸アシラーゼおよび精製N−アセチル−(S)−β−アミノ酸アシラーゼをSDS−PAGEに供した(Fig.1)。結果、N−アセチル−(R)−β−アミノ酸アシラーゼは分子量約84kDaと見積もられる単一のバンドとして(Fig.1A)、N−アセチル−(S)−β−アミノ酸アシラーゼは分子量約41kDaと見積もられる単一のバンドとして(Fig.1B)、それぞれ観察された。上記ゲルろ過クロマトグラフィーにおいて、両酵素の未変性状態の分子量はそれぞれ206kDa(N−アセチル−(R)−β−アミノ酸アシラーゼ)、101kDa(N−アセチル−(S)−β−アミノ酸アシラーゼ)と見積もられたため、SDS−PAGEで見積もられた分子量と併せて考えると、何れの酵素もホモダイマーあるいはホモトライマー構造を有している酵素であると考えられた。
【0065】
4−6.光学特異性
精製酵素を酵素源として、その光学選択性を測定した。上記酵素活性条件において反応時間30 min、酵素添加量4.6μg/ml(N−アセチル−(R)−β−アミノ酸アシラーゼ)または3.1μg/ml(N−アセチル−(S)−β−アミノ酸アシラーゼ)として反応を行った。結果、N−アセチル−(R)−β−アミノ酸アシラーゼを酵素源とした場合には(R)−β−Pheの生成のみが観察され、(S)−β−Pheの生成は検出限界以下であった(Fig.2D)。一方、N−アセチル−(S)−β−アミノ酸アシラーゼを酵素源とした場合には(S)−β−Pheの生成のみが観察され、(R)−β−Pheの生成は検出限界以下であった(Fig.2E)。これらの結果から、両酵素はいずれも高い光学選択性を有していることが示された。
【0066】
5.N末端アミノ酸配列解析
精製酵素標品を材料に、そのN末端アミノ酸配列を決定した。方法は、酵素標品をSDS−PAGEに供した後、PVDF膜に転写し、クマシーブルーで染色後、バンドの見られる位置を切り出し、これをプロテインシーケンサーに供することにより行った。結果、N−アセチル−(R)−β−アミノ酸アシラーゼの場合は配列番号1に示す20残基の配列が、N−アセチル−(S)−β−アミノ酸アシラーゼの場合は配列番号2に示す26残基のアミノ酸配列が、それぞれ決定された。
【0067】
6.遺伝子クローニングと塩基配列決定
決定されたN末端アミノ酸配列に基づいて合成プライマーを設計し、Takara社製LA PCR in vitro Cloning Kitを用いて、アシラーゼをコードする遺伝子部分断片の取得を行った。結果、N−アセチル−(R)−β−アミノ酸アシラーゼN末端近傍をコードする遺伝子領域を含む、5’−PstI認識配列、3’−EcoRI認識配列を両端に有する794baseのDNA断片、およびN−アセチル−(S)−β−アミノ酸アシラーゼN末端近傍をコードする遺伝子領域を含む、5’−PstI認識配列、3’−SalI認識配列を両端に有する393baseのDNA断片をそれぞれ得て、その塩基配列を決定した。
【0068】
塩基配列の決定により得られた情報より、プライマーR7F、R8R(配列番号3、4)/プライマーS3F、S4R(配列番号5、6)を用いてBurkholdelia sp. AJ110349のゲノムDNAを鋳型としてPCRを行いN−アセチル−(R)−β−アミノ酸アシラーゼ/ N−アセチル−(S)−β−アミノ酸アシラーゼのN末端近傍をコードする遺伝子領域含む0.3kb/0.4kbのDNA断片を増幅し、DIGラベリングを行ってそれぞれをR−プローブ/S−プローブとした。
【0069】
Burkholderia sp. AJ110349染色体DNA5μgをBamHI/HindIII(100/50U)にて切断した後、R−プローブを用いてサザン解析を行った。ハイブリダイゼーションの条件は、42℃、16時間で、ハイブリダイゼーションの溶液にはDIG Easy Hyb(Roche社)を用いた。その結果、約5kbにポジティブシグナルを確認した。
【0070】
次いで、Burkholderia sp. AJ110349染色体DNA5μg をBamHI/HindIII(100/50U)処理後、アガロース電気泳動し、5kb付近の断片を精製し、pUC118のBamHI/HindIIIサイトにライゲートした。この反応液をもちいてEscherichia coli JM109を形質転換し、ライブラリーを作製した。上記プローブをもちいてコロニーハイブリダイズを行い、ポジティブコロニーを取得し、プラスミドを抽出した。得られたプラスミドをpBRACY_A3として、挿入配列について塩基配列を決定したところ、760アミノ酸(配列番号8)をコードするORF(配列番号7)が存在した。
【0071】
Burkholderia sp. AJ110349染色体DNA5μgをPstI/HindIII(各50U)にて切断した後、S−プローブを用いてサザン解析を行った。ハイブリダイゼーションの条件は、42℃、16時間で、ハイブリダイゼーションの溶液にはDIG Easy Hyb(Roche社)を用いた。その結果、約1.5kbにポジティブシグナルを確認した。
【0072】
次いで、Burkholderia sp. AJ110349染色体DNA5μgをPstI/HindIII(各50U)処理後アガロース電気泳動し、1.5kb付近の断片を精製し、pUC118のPstI/HindIIIサイトにライゲートした。この反応液をもちいてEscherichia coli JM109を形質転換し、ライブラリーを作製した。上記プローブをもちいてコロニーハイブリダイズを行い、ポジティブコロニーを取得し、プラスミドを抽出した。得られたプラスミドをpBSACY_PHとして、挿入配列について塩基配列を決定したところ、352アミノ酸(配列番号10)をコードするORF(配列番号9)が存在した。
【0073】
7. Variovorax sp. AJ110348株由来N−アセチル−(R)-β−アミノ酸アシラーゼ遺伝子のショットガンクローニングによる取得
Variovorax sp.AJ110348株よりゲノムDNAを抽出し、制限酵素Sau3AIにて部分分解した後、約3〜8kb付近の断片を回収しpUC118へと連結した。この反応液をもちいてEscherichia coli JM109を形質転換し、ライブラリーを作製した。
得られた形質転換体をブルーホワイトセレクションにより選別し、単コロニーとして得られた3500株に対してマスタープレートを作成した。これらの菌株をN−アセチル−(R,S)−β−Phe液体培地[硫酸アンモニウム10.0g/l、KH2PO4 1.0g/l、MgSO4・7H2O 0.4g/l、FeSO4・7H2O 10mg/l、MnSO4・5H2O 10mg/l、ビタミンB1・HCl 0.2mg/l、N−アセチル−(R,S)−β−Phe 1.0g/l, pH8.0、IPTG 100μM、Amp 100μg/ml] に10株ずつ接種し、37℃、48時間振とう培養した。
得られた培養上清をSiliagel 60F254プレート(Merck)、ブタノール:酢酸:水=4:1:2にて分離し、UV254nm及びニンヒドリン発色にてβ−Pheの生成を確認した。
その結果、混合した培養上清からβ−Pheが検出されたので、さらに、1株ずつ同様の手法で培養し、TLCにて培養上清を確認したところ、K83株と命名した株からβ−Pheが検出された。
K83株のプラスミドDNAを抽出し挿入配列を確認したところ、約3kbの挿入配列が存在した。塩基配列を決定した結果、779アミノ酸(配列番号12)をコードするORF(配列番号11)が存在した。
【0074】
8.K83株の活性測定及び光学選択性の測定
得られたK83株を100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリン、1mM IPTGを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、50mM Tris−HCl(pH7.6)、0.2% N−アセチル−β−Phe、37℃、10分間反応し、N−アセチル−β−Pheとβ−Pheの分離条件にてN−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、2.3U/mgであった。
また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(R)−β−Phe特異的に生成しており、(S)−β−Pheは検出限界以下であった。
【0075】
9.Burkholderia sp. AJ110349由来N−アセチル−(R)−β−アミノ酸アシラーゼ高発現株の作成
Burkholderia sp.AJ110349染色体DNAを鋳型として、プライマー R_7F(配列番号13)、及び R_R_HindIII(配列番号14)によりPCR反応により、得られた2.3kbの増幅断片をBamHI/HindIII処理し、ptrp4(参考文献Journal of Molecular Catalysis B: Enzymatic 32 (2005)205−211)のBamHI/HindIIIサイトに挿入し、ptrp4_3BRとした。このプラスミドによりEscherichia coli JM109を形質転換した。この形質転換体をJM109/ptrp4_3BRと命名した。
JM109/ptrp4_3BRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むM9カザミノ酸培地50mLに植菌し、30℃、18時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、50mM Tris−HCl(pH7.6)、0.2% N−アセチル−(R,S)−β−Phe、37℃、30分間反応し、生成したβ−Pheの量をHPLCにて測定した。N−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、0.29U/mgであった。また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(R)−β−Phe特異的に生成しており、(S)−β−Pheは検出限界以下であった。
【0076】
10.Burkholderia sp. AJ110349由来N−アセチル−(S)−β−アミノ酸アシラーゼ高発現株の作成
Burkholderia sp.AJ110349染色体DNAを鋳型として、プライマー S_F_NdeI_2(配列番号15)、及び S_R_HindIII(配列番号16)によりPCR反応により、得られた1.1kbの増幅断片をNdeI/BamHIで処理した。これとは別に、pSFN_Sm_Aet(参考文献WO2006075486)をNdeI/HindIIIで切断し、3kb付近のDNAを切り出し精製した。このpSFNのNdeI/HindIIIサイトに1.1kbPCR産物を制限酵素にて処理したDNAを挿入し、pSFN_2BSとした。このプラスミドによりEscherichi a coli JM109を形質転換した。この形質転換体をJM109/pSFN_2BSと命名した。
なお、このプラスミドの挿入配列は、配列10の6残基目のアミノ酸にあたるメチオニンを開始コドンとしているが、発明者らは1残基目からのアミノ酸を開始コドンとするプラスミドとともに作成した。しかしながら、特に活性の違いはなかった。
JM109/pSFN_2BSを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、上記の方法と同様にN−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、0.33U/mgであった。また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(S)−β−Phe特異的に生成しており、(R)−β−Pheは検出限界以下であった。
【0077】
11.Variovorax sp. AJ110348由来N−アセチル−(R)−β−アミノ酸アシラーゼ高発現株の作成
Variovorax sp. AJ110348染色体DNAを鋳型として、プライマー VRACY_1F_NdeI(配列番号17)、及びVRACY_R_HindIII(配列番号18)によりPCR反応により、得られた2.4kbの増幅断片をNdeI/HindIIIで処理した。このDNA産物を実施例9で作成したpSFN NdeI/HindIII精製産物のNdeI/HindIIIサイトに挿入し、pSFN_1VRとした。このプラスミドによりEscherichia coli JM109を形質転換した。この形質転換体をJM109/pSFN_1VRと命名した。
JM109/pSFN_1VRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、上記の方法と同様にN−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、1.5U/mgであった。また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(R)−β−Phe特異的に生成しており、(S)−β−Pheは検出限界以下であった。
【0078】
12.基質特異性の調査
12−1.各種N−アセチル−(R,S)-β−アミノ酸への基質特異性調査
JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、実施例2−1〜9に記した各種N−アセチル−(R,S)−β−アミノ酸への基質特異性を調査した。
酵素は上記の方法で調整後、活性を測定し、N−アセチル−(R,S)−β−Pheから1分間に1μmolのβ−Pheを生成する活性を1Uとし、各種基質特異性の調査には、50mUの無細胞抽出液を用いた。ネガティブコントロールとして、上記のように調整した無細胞中質液の代わりに、50mM Tris緩衝液(pH7.6)を用いて各基質と反応を行ったものを「enzyme-」の実験区とし、各基質の代わりに50mM Tris緩衝液(pH7.6)を用いて各無細胞抽出液と反応させたものを「基質-」の実験区とした。
50mM Tris−HCl(pH7.6)、0.2% 各N−アセチル−(R,S)−β−アミノ酸、37℃、1時間、または24時間反応し、96℃、10分間処理し、反応を停止した。アセチル基の分解により生成した酢酸の量を酢酸キット(Roche)のプロトコールに従って定量した。
1時間後の酢酸生成量を表3に、24時間後の酢酸生成量を表4に示した。その結果、N−アセチル−(R,S)-β−aminobutylic acid(N−アセチル−β−Aba)、N−アセチル−(R,S)-β−Leu、N−アセチル−(R,S)-β−homoLeu、N−アセチル−(R,S)-β−Tyr、N−アセチル−(R,S)-homoPhe、N−アセチル−(R,S)-β−4−Fluoro−Pheを基質とした実験区から酢酸が検出された。
【0079】
【表3】
【0080】
【表4】
【0081】
12−2.光学特異性の決定
反応24時間後のサンプルを各種キラル分割カラムにて解析し、光学選択性を決定した。
12−2−1.(R),(S)−β−Leuの分離
3−2−3.の分割条件で、標準品は、N−アセチル-(R),(S)−β−Leu、(S)−β−Leu、(R)−β−Leuの順に溶出された。
JM109/ptrp4_3BRを用いたN−アセチル−β−Leu反応用液では(R)−β−Leuが検出され(S)−β−Leuは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−Leu反応用液では(S)−β−Leuが検出され(R)−β−Leuは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−Leu反応用液では(R)−β−Leuが検出され(S)−β−Leuは検出限界以下であり、> 99 % eeであった。
【0082】
12−2−2.(R),(S)−β−homoLeuの分離
3−2−3.の分割条件で、標準品は、N-アセチル-(R,S)−β−homoLeu、(R)−β−homoLeu、(S)−β−homoLeuの順に溶出された。
JM109/ptrp4_3BRを用いたN−アセチル−β−homoLeu反応用液では(S)−β−homoLeuが検出され、少量の(R)−β−Leuは検出され69% eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−homoLeu反応用液では(R)−β−homoLeuが検出され(S)−β−Leuは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−homoLeu反応用液では(S)−β−homoLeuが検出され、(R)−β−homoLeuは検出限界以下であり、> 99 % eeであった。
【0083】
12−2−3.(R),(S)−β−homoPheの分離
3−2−3.の分割条件で、標準品は、N−アセチル−(R,S) −β−homoPhe、(R)−β−homoPhe、(S)−β−homoPheの順に溶出された。
JM109/ptrp4_3BRを用いたN−アセチル−β−homoPhe反応用液では(S)−β−homoPheが検出され、微量の(R)−β−homoPheも検出され、98% eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−homoPhe反応用液では(R)−β−homoPheが検出され(S)−β−は検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−homoPhe反応用液では(S)−β−homoPheが検出され(R)−β−homoPheは検出限界以下であり、> 99 % eeであった。
【0084】
12−2−4.(R),(S)−β−Tyrの分離
3−2−4.の分離条件で、標準品は、(S)−β−Tyr、(R)−β−Tyr、N−アセチル−(R)/(S)−β−Tyrの順に溶出された。(N-アセチル-β-Tyrの標準品はラセミ体であるので、分離はされるものの溶出順は未決)
JM109/ptrp4_3BRを用いたN−アセチル−β−Tyr反応用液では(R)−β−Tyrが検出され、少量の(S)−β−Tyrも検出され61 % eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−Tyr反応用液では(S)−β−Tyrが検出され(R)−β−Tyrは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−Tyr反応用液では(R)−β−Tyrが検出され(S)−β−Tyrは検出限界以下であり、> 99 % eeであった。
【0085】
12−2−5.(R),(S)−4−Fluoro−β−Pheの分離
3−2−4.の分離条件で、標準品は、(S)−4−Fluoro−β−Phe、(R)−4−Fluoro−β−Phe、N−アセチル−(R)/(S)−4−Fluoro−β−Pheの順に溶出された。(N-アセチル-4-Fluoro-β-Pheの標準品はラセミ体であるので、分離はされるものの溶出順は未決)
JM109/ptrp4_3BRを用いたN−アセチル−4−Fluoro−β−Phe反応用液では(R)−4−Fluoro−β−Pheが検出され、少量の(S)−4−Fluoro−β−Pheが検出され、94% eeであった。
JM109/pSFN_2BSを用いたN−アセチル−4−Fluoro−β−Phe反応用液では(S)−4−Fluoro−β−Pheが検出され(R)−4−Fluoro−β−Pheは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−4−Fluoro−β−Phe反応用液では(R)−4−Fluoro−β−Pheが検出され(S)−4−Fluoro−β−Pheは検出限界以下であり、> 99 % eeであった。
【0086】
12−3.N−アセチル−3,4−(−O−CH2−O−)−β−Phenylalanineへの基質特異性調査
JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを上記のような方法で調整し、各種N−アセチル−3,4−(−O−CH2−O−)−β−Phenylalanineへの基質特異性を調査した。
培養液300μLを50mM Tris−HCl(pH7.6)で洗浄し、300μLの50mM Tris−HCl(pH7.6)に懸濁し、超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として反応に用いた。
50mM Tris−HCl(pH7.6)、0.2% N−アセチル−(R)/(S)−3,4−(−O−CH2−O−)−β−Phenylalanine、37℃、10分間反応した。N−アセチル−β−Pheとβ−Pheの分離条件で、3,4−(−O−CH2−O−)−β−Phenylalanineを定量したところ、無細胞抽出液1mLあたりの活性は、JM109/ptrp4_3BRは0.2U、JM109/pSFN_2BSは3.9U、JM109/pSFN_1VRは27.8Uであった。
(R),(S)−β−Phe光学分割条件にて標準品を分離したところ、(S)−3,4−(−O−CH2−O−)−β−Phenylalanine、(R)−3,4−(−O−CH2−O−)−β−Phenylalanine、N−アセチル−(R)/(S)− 3,4−(−O−CH2−O−)−β−Phenylalanineの順に溶出された。(N−アセチル−(R)/(S)−3,4−(−O−CH2−O−)−β−Phenylalanineの溶出順は未決)
この条件で、反応溶液を解析したところ、JM109/ptrp4_3BRの反応液からは(R)-3,4−(−O−CH2−O−)−β−Phenylalanineが、JM109/pSFN_2BSの反応液からは(S)−3,4−(−O−CH2−O−)−β−Phenylalanineが、JM109/pSFN_1VRの反応液からは(R)−3,4−(−O−CH2−O−)−β−Phenylalanineが検出された。
【0087】
12−4.N−アセチル−3−Pyr−Alaへの基質特異性調査
12−1と同様の菌体を用いて、同様の方法で各酵素を調整し、50mUの無細胞抽出液を反応に用いた。50mM Tris−HCl(pH7.6)、1% N−アセチル−(R,S)−β−3−Pyr−Ala、37℃、1時間反応し、70℃、10分間処理し、反応を停止した。アセチル基の分解により生成した酢酸の量を酢酸キット(Roche)のプロトコールに従って定量した。
その結果、JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを用いた反応液では、それぞれ3.5mM、8.4mM、0.8mMの酢酸が検出され、「基質−」及び「enzyme−」の実験区からは酢酸は検出されなかった。
【0088】
13.蓄積量の評価
JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて10倍に濃縮した菌体懸濁液を調製した。
この菌体懸濁液を用いて、100mM Tris−HCl(pH 7.6)、5% N−アセチル−(R,S)−β−Phe、37℃、24時間、全量2.5mLで反応した。その後、3−2−1の分析法で反応液を解析した。
JM109/ptrp4_3BRの10倍濃縮の菌体懸濁液を500μL加え反応したところ、(R)−β−Pheの収率21.6%、>99% eeで、(S)−β−Pheは検出限界以下であった。
JM109/pSFN_2BSの10倍濃縮の菌体懸濁液を250μL加え反応したところ、(S)−β−Pheの収率49.5%、>99% eeで、(R)−β−Pheは検出限界以下であった。
JM109/pSFN_1VRの10倍濃縮の菌体懸濁液を125μL加え反応したところ、(R)−β−Pheの収率45.4%、>99% eeで、(S)−β−Pheは検出限界以下であった。
【産業上の利用可能性】
【0089】
本発明で同定された遺伝子を用いた大腸菌(Echerichia coli)のような宿主を組換えることによって、N−アセチル−(R)−β−アミノ酸アシラーゼ及びN−アセチル−(S)−β−アミノ酸アシラーゼを高発現する系を構築することができる。その結果、該酵素を利用してR体又はS体のβアミノ酸を選択的にする系を簡便かつ安価に提供することが可能となる。
【技術分野】
【0001】
本発明は、新規なN−アセチル−(R)−β−アミノ酸アシラーゼ及びN−アセチル−(S)−β−アミノ酸アシラーゼ、及びそれらをコードする遺伝子等に関するものである。
【背景技術】
【0002】
N−アセチル−β−アミノ酸に対してアシラーゼ活性を有する酵素は従来知られていなかったが、本発明者等は、N−アセチル−β−Phe等のN−アセチル−β−アミノ酸を培養菌体によって光学選択的に脱アセチル化する菌株としてVariovorax sp. AJ110348(FERM BP−10367)とBurkholderia sp. AJ110349(FERM BP−10366)を初めて見出し、これらの菌体又は菌体粉砕液上清を用いる光学選択的な脱アセチル化方法は特開2006−42722号公報(特許文献1)に記載されている。尚、これらの菌株は、夫々、平成16年7月22日付けで独立行政法人産業技術総合研究所特許生物寄託センターに寄託されたFERM P−20129及びFERM P−20128から、2005年7月4日付けで特許手続上の微生物の寄託等の国際的承認に関するブダペスト条約に基づく国際寄託に移管され、夫々、受託番号FERM BP−10367及びFERM BP−10366が付されている。
【特許文献1】特開2006−42722号公報
【発明の概要】
【発明が解決しようとする課題】
【0003】
これら選抜された野生株を用いた変換反応も変換の一手法ではあるが、Burkholdelia sp.AJ110349由来N−アセチル−β−アミノ酸アシラーゼは、(R)/(S)双方の光学選択性のアシラーゼを保持しており、野生株を用いて光学選択性の高いβ−アミノ酸を生成することが難しい状況であった。より効果的な変換を達成するためには、反応を触媒する酵素を単離・同定し、更にそのコード遺伝子も同定した上、例えば、大腸菌(Echerichia coli)のような宿主を用いて、その遺伝子を高発現する系を構築することが重要である。
【課題を解決するための手段】
【0004】
従来、ある酵素活性が見出されている菌株から、その活性を担う酵素あるいはその遺伝子を単離・同定する手法に関しては数多く報告されており、具体的には、一つはショットガンクローニングによる遺伝子単離法、もう一つは酵素単離、その部分アミノ酸配列決定、その配列情報に基づく遺伝子単離法が挙げられる。
【0005】
そこで、本発明者は、特許文献1に記載された菌株からN−アセチル−β−アミノ酸を光学選択的に脱アセチル化するN−アセチル−β−アミノ酸アシラーゼの遺伝子を取得すべく、まず第一に、常法であるショットガンクローニングによる方法を試みた。
【0006】
即ち、Burkholdelia sp. AJ110349株のゲノムDNAのライブラリーを作成し、N−アセチル−R/S−β−Pheを単一炭素源とする培地に塗布し37℃で1週間培養を行った。
また、供与検体数は、別途LB培地にライブラリーを塗布し、LB培地に生育した菌体数から算出した。約1万株に対してスクリーニングを行った結果、N−アセチル−R/S−β−Pheを単一炭素源として生育する株は得られなかった。
【0007】
また、Variovorax sp. AJ110348株に関しても同様に、N−アセチル−R/S−β−Pheを単一炭素源とするスクリーニングを約5万1千株に対して行ったが、Burkholdelia sp. AJ110349株のスクリーニングの際と同様に、N−アセチル−R/S−β−Pheを単一炭素源として生育する株は得られなかった。
【0008】
以上のように、非常に多くのクローンを調べたにも関わらず、単一炭素源の資化を指標とする様なスクリーニング手法では、目的とする酵素遺伝子の取得には至らなかった。
【0009】
このため、次に第二の方法である酵素の単離精製からのアプローチを試みることとした。酵素の単離精製の方法は数多く報告されているが、あらゆるタンパク質の単離精製が約束されるような単離精製法の一般則は存在せず、また、精製元の総タンパク質量に占める対象とするタンパク質の存在比によって、単離精製の難易度は大きく異なる。本発明におけるN−アセチル−β−アミノ酸アシラーゼの精製方法については後に詳述するが、Burkholderia sp. AJ110349株からの N−アセチル−(R)−β−アミノ酸アシラーゼおよび N−アセチル−(S)−β−アミノ酸アシラーゼの精製の場合、無細胞抽出液中での目的酵素の比活性に比べ、単離精製酵素の比活性は数百倍となっており、その精製は非常に困難なものであった。この困難な状況の原因の一つに、酵素生産菌であるBurkholderia sp. AJ110349株の性質の内、遠心操作による集菌のし難さが挙げられた。菌体内のタンパク質を単離精製する場合、通常、培地中のタンパク質の混入をさけるため、集菌後、緩衝液を用いて十分に菌体を洗浄する方法が採られるが、本菌株は、集菌手法として通常採用される遠心分離操作においてペレットとして回収されにくく、培地成分由来のタンパク質と菌体由来のタンパク質の分離が困難であった。このため、本株からのタンパク質単離精製は、通常の場合に比べ、多くの夾雑タンパク質が存在する状況から単離を行う必要があり、これが精製の困難さの一因となった。
【0010】
この問題を解決するため、発明者は通常のタンパク質精製の場合に比べ多くの条件等の検討に鋭意努力し、その結果、当業者でも予想し得ない5種という多くのクロマトグラフィーを用いる精製ステップを経ることによって、初めて上記酵素の単離精製に成功し、その結果、該酵素をコードする遺伝子の塩基配列を決定した。
また、Variovorax sp. AJ110348株に関しては、数種類の酵素を別々に取得する必要がないため、手法を改良し更にショットガンクローニングを継続した。上述したように目的の遺伝子を保持する株が選択的に増殖するような簡便なスクリーニングでは酵素遺伝子の取得には至らなかったため、発明者は、後述するように、各クローンの活性を個別に確認するという非常に時間の掛かる作業を実施し、目的の酵素遺伝子の取得に成功した。
上述のように、非常に多くの工夫を重ねながら、本発明を完成させた。
【0011】
即ち、本発明は以下の各態様に係るものである。
[態様1]下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号8で示されるアミノ酸配列からなる蛋白質、
(b)配列番号8で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号8で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様2]下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号7に示される塩基配列からなるDNA、
(b)配列番号7に示される塩基配列の相補鎖を含む核酸とストリンジェントな条件下でハイブリダイズし、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性
を有する蛋白質をコードするDNA、及び
(c)配列番号7に示される塩基配列のDNAと70%以上の配列同一性を示すDNAまたはその部分断片であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
[態様3]下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様4]下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号9に示される塩基配列からなるDNA、
(b)配列番号9に示される塩基配列の相補鎖を含む核酸とストリンジェントな条件下でハイブリダイズし、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性
を有する蛋白質をコードするDNA、及び
(c)配列番号9に示される塩基配列のDNAと70%以上の配列同一性を示すDNAまたはその部分断片であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
[態様5]下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号12で示されるアミノ酸配列からなる蛋白質、
(b)配列番号12で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号12で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様6]下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号11に示される塩基配列からなるDNA、
(b)配列番号11に示される塩基配列の相補鎖を含む核酸とストリンジェントな条件下でハイブリダイズし、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性
を有する蛋白質をコードするDNA、及び
(c)配列番号11に示される塩基配列のDNAと70%以上の配列同一性を示すDNAまたはその部分断片であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
[態様7] β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、態様1〜6のいずれか一項に記載の遺伝子。
[態様8]下記のいずれか一つに示す蛋白質。
(a)配列番号8で示されるアミノ酸配列からなる蛋白質、
(b)配列番号8で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号8で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様9]下記のいずれか一つに示す蛋白質。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様10]下記のいずれか一つに示す蛋白質。
(a)配列番号12で示されるアミノ酸配列からなる蛋白質、
(b)配列番号12で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号12で示されるアミノ酸配列と70%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(R)−β−アミノ酸アシラーゼ活性を有する蛋白質。
[態様11]β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、態様8から10いずれか一項に記載のβ−アミノ酸アシラーゼ。
[態様12] 態様1から6のいずれか一項記載の遺伝子によって形質転換された微生物。
[態様13] 態様1から6のいずれか一項記載の遺伝子によって形質転換された微生物であるEscherichia coli。
[態様14] 態様12記載の微生物をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
[態様15] 態様12記載の微生物から得られるN−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
【発明の効果】
【0012】
本発明によって、N−アセチル−(R)−β−アミノ酸アシラーゼ及びN−アセチル−(S)−β−アミノ酸アシラーゼが単離同定され、更に、それらの遺伝子がクローニングされ、その塩基配列及びその遺伝子がコードする蛋白質のアミノ酸配列が明らかにされた。
【図面の簡単な説明】
【0013】
【図1】精製したN−アセチル−(R)−β−アミノ酸アシラーゼ(A)およびN−アセチル−(S)−β−アミノ酸アシラーゼ(B)のSDS−PAGEによる観察結果を示したものである。
【図2】N−アセチル−(R,S)−β−Pheを基質、精製したN−アセチル−(R)−β−アミノ酸アシラーゼまたはN−アセチル−(S)−β−アミノ酸アシラーゼを酵素源として変換を行った結果をHPLCにて解析した結果である。AはN−アセチル−(R,S)−β−Pheのみで酵素を添加していない場合、BおよびCはそれぞれ(R)−β−Pheおよび(S)−β−Phe標準標品、D及びEはN−アセチル−(R,S)−β−PheをN−アセチル−(R)−β−アミノ酸アシラーゼまたはN−アセチル−(S)−β−アミノ酸アシラーゼで変換した結果をそれぞれ示している。
【発明を実施するための形態】
【0014】
本発明の遺伝子
本発明の遺伝子は、N−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼをコードするものであり、これらの酵素は、夫々、分子量約84kDa及び分子量約41kDaのサブユニット(構成単位)から成るホモダイマー又はホモトライマーの構造を有している。従って、より具体的には、本発明の遺伝子は、これらの各サブユニットであるポリペプチドをコードするものである。これらの酵素の代表例は、Burkholderia sp. AJ 110349(FERM BP−10366)が産生するものであり、その具体的なアミノ酸配列の一例は、夫々、配列番号8及び配列番号10に示されたものである。これらの酵素の別の代表例は、Variovorax sp. AJ 110348(FERM BP−10367)が産生するものであり、その具体的なアミノ酸配列の一例は、配列番号12に示されたものである。尚、本発明において、N−アセチル−(R,S)−β−アミノ酸アシラーゼの代表例として、N−アセチル−(R,S)−β−フェニルアラニン(Phe)アシラーゼを挙げることが出来る。N−アセチル−(R,S)−β−アミノ酸アシラーゼが、R体特異的な活性を有するか、S体特異的な活性を有するかは、N−アセチル−(R,S)−β−フェニルアラニンを基質として決定する。本発明のN−アセチル−(R,S)−β−アミノ酸アシラーゼは、α−アミノ酸のD体とL体を認識するかのごとく、側鎖、アミノ基、カルボキシル基、水素の相対位置を認識する。一方、RS表記では、β位に結合する側鎖によっては、−C−COOHと順位法則の優先性が入れ替わることがある。例えば、後述の実施例に示す、N−アセチル−β−homoLeuとN−アセチル−β−homoPheの場合は、N−アセチル−(R)−β−アミノ酸アシラーゼからS体が、N−アセチル−(S)−β−アミノ酸アシラーゼからR体がそれぞれ生成している。
【0015】
又、本明細書において、「N−アセチル−(R)−β−アミノ酸アシラーゼ活性又はN−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質」とは、ホモダイマー又はホモトライマーの構造を有する各アシラーゼ酵素の構成単位となり得る(構成単位としての機能を有する)蛋白質(ポリペプチド)をも意味する。
【0016】
本発明遺伝子は当業者に公知の方法で調製することが出来る。
例えば、本明細書の実施例に記載された方法に従い、各プローブを用いるコロニーハイブリダイズによる他、Burkholderia sp. AJ 110349(FERM BP−10366)またはVariovorax sp. AJ 110348(FERM BP−10367)のゲノムDNAを鋳型として、本明細書に開示された本発明DNAの塩基配列又はアミノ酸配列の情報に基づき適当な作製されるプライマーを使用した、当業者に公知の任意のPCRにより増幅して調製することも出来る。
【0017】
例えば、94℃で2分の後、94℃で10秒、55℃で20秒、72℃で2分を30サイクル行い、最後に72℃で5分を行う。なお、サーマルサイクラーとしては、Perkin Elmer社製9600など一般のサーマルサイクラーを用いることができる。耐熱性 DNAポリメラーゼとしては、ExTaq DNA Polymerase(宝酒造製)などの一般の市販品を用い、反応液の組成はポリメラーゼに添付の説明書に従って実施する。
【0018】
更に、当業者に周知の化学合成によって、本発明の各遺伝子を調製することも出来る。
【0019】
本明細書において、「ストリンジェントな条件下」とは、いわゆる特異的なハイブリッドが形成され、非特異的なハイブリッドが形成されない条件をいう。一例を示せば、相同性が高いDNA同士、例えば50%以上、好ましくは70%以上、より好ましくは80%以上、さらに好ましくは90%以上、特に好ましくは95%以上の相同性を有するDNA同士がハイブリダイズし、それより相同性が低いDNA同士がハイブリダイズしない条件が挙げられる。なお、塩基配列間の同一性は、Blast等の当業者に公知のアルゴリズムを用いて決定することができる。より具体的には、通常のサザンハイブリダイゼーションの洗いの条件である60℃、1×SSC、0.1%SDS、好ましくは、0.1×SSC、0.1%SDSに相当する塩濃度でハイブリダイズする条件が挙げられる。このような条件でハイブリダイズするDNAの中には途中にストップコドンが発生したものや、活性中心の変異により活性を失ったものも含まれるが、それらについては、所定のN−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼ活性を後述の方法で測定し取り除くことができる。
【0020】
ハイブリダイゼーションは、例えば、カレント・プロトコールズ・イン・モレキュラー・バイオロジー(Current protocols in molecular biology(edited by Frederick M. Ausubel et al., 1987))に記載の方法等、当業界で公知の方法あるいはそれに準じる方法に従って行なうことができる。また、市販のライブ、添付の使用説明書に記載の方法に従って行なうことができる。
【0021】
本発明の蛋白質
本発明の遺伝子がコードする蛋白質の中で、N−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼを構成するユニットポリペプチドのアミノ酸配列(配列番号8、配列番号10又は配列番号12)において、1個若しくは数個のアミノ酸残基の置換、欠失、又は付加を含むアミノ酸配列を有する蛋白質であって、上記酵素活性を有するものは、当業者に公知の任意の方法、例えば、部位特異的変異導入法、遺伝子相同組換え法、プライマー伸長法、及びPCR法等の当業者に周知の方法を適宜組み合わせて、容易に作成することが可能である。
【0022】
尚、その際に、実質的に同等の機能を有するためには、当該ポリペプチドを構成するアミノ酸のうち、同族アミノ酸(極性β非極性アミノ酸、疎水性β親水性アミノ酸、陽性β陰性荷電アミノ酸、芳香族アミノ酸など)同士の置換が可能性として考えられる。又、実質的に同等の機能の維持のためには、本発明の各ポリペプチドに含まれる機能ドメイン内のアミノ酸は保持されることが望ましい。
【0023】
更に、本発明の蛋白質として、上記のアミノ酸配列と、全体の平均で約70%、好ましくは約80%以上、より好ましくは約90%以上、更に好ましくは約95%以上であるような高い配列同一性を有するアミノ酸配列を含む蛋白質又はその断片であって、所定のN−アセチル−(R)−β−アミノ酸アシラーゼ又はN−アセチル−(S)−β−アミノ酸アシラーゼを有するものを挙げることができる。尚、アミノ酸配列間の同一性も、当業者に公知のアルゴリズム、例えば、実施例で使用されているBlast を用いて決定することができる。又、上記酵素の活性は本明細書の実施例に記載した方法で測定することが出来る。又、このような蛋白質も、当業者に公知の任意の方法、例えば、部位特異的変異導入法、遺伝子相同組換え法、プライマー伸長法、及びPCR法等の当業者に周知の方法を適宜組み合わせて、容易に作成することが可能である。
尚、本発明のβ-アミノ酸は、下記の構造式で示される。
【0024】
【化1】
【0025】
構造式中、Rは、炭素数1〜6のアルキル基、炭素数6〜14のアリール基、炭素数3〜10のシクロアルキル基、炭素数7〜19のアラルキル基、炭素数2〜11のアルコキシアルキル基、これらの炭素骨格中にヘテロ原子を含む基、およびこれらの炭素骨格中に炭素−炭素不飽和結合を含む基からなる群より選ばれ、これらの基は直鎖であっても分岐していてもよく、さらに置換基を有していてもよい。好ましくは、Rは、炭素数1〜4のアルキル基、炭素数6〜7のアリール基、これらの基は直鎖であっても分岐していてもよく、さらに置換基を有していてもよい。好ましくは、β−アミノ酸は、β−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンから選ばれる。
【0026】
本発明の遺伝子の発現
上記で得られた本発明遺伝子を当業者に公知の任意の方法によって組換えベクターに組み込み、本発明の組換え発現ベクターを作成することが出来る。
例えば、(1)本発明の遺伝子を含有するDNA断片を切り出し、(2)該DNA断片を適当な組換えベクター中の制限酵素部位又はマルチクローニングサイトに挿入して該ベクターに連結する挿入することにより製造することができる。組換えベクターに特に制限はなく、例えば、麹菌由来のプラスミド(例えば、pSal23, pTAex3, pNGU113, pRBG1, pGM32, pSE52, pNAGL142等)、大腸菌由来のプラスミド(例、pT7Blue T−Vector、pRSET、pBR322、pBR325、pUC18、pUC118)、枯草菌由来のプラスミド(例、pUB110、pTP5、pC194)、及び、酵母由来プラスミド(例、pSH19、pSH15)、等の組換えベクター等を利用することが出来る。
【0027】
上記組換えベクターには、以上の他に、本発明の転写調節配列の活性を損なわない限り、所望により当該技術分野で公知のプロモーター等の各種転写調節要素、シャイン・ダルガルノ配列、選択マーカー、転写終結シグナル等を付加することができる。また、必要に応じて、本発明の外来遺伝子にコードされた所望の蛋白質を他の蛋白質又はペプチド(例えば、グルタチオンSトランスフェラーゼ、ヒスチジンタグ、カルモデュリンバインディング蛋白質、及びプロテインA等)との融合蛋白質として発現させることも可能である。このような融合蛋白質は、適当なプロテアーゼを使用して切断し、それぞれの蛋白質に分離することが出来る。
【0028】
本発明の遺伝子が有効に発現される限り、本発明の組換え発現ベクターを有する微生物(形質転換体)を調製するために使用する宿主の種類及び由来等に特に制限はなく、例えば、大腸菌のような原核細胞、又はサッカロマイセス・セレビシエ(パン酵母)真核細胞等の当業者に公知の任意の微生物細胞を使用することが可能である。
【0029】
これら宿主細胞の形質転換は、例えば、塩化カルシウム法、パーティクルガン、エレクトロポレーション法等の、当該技術分野で公知の方法に従って行うことが出来る。例えば、以下に記載の文献を参照することが出来る。Proc. Natl. Acad. Sci. USA, 69巻、2110(1972); Gene, 17巻、107(1982);Molecular & General Genetics,168巻, 111(1979);Methods in Enzymology,194巻、182−187(1991);Proc. Natl. Acad. Sci. USA)、75巻、1929(1978);細胞工学別冊8 新細胞工学実験プロトコール。263−267(1995)(秀潤社発行);及びVirology、52巻、456(1973)。
【0030】
このようにして得られた、本発明の形質転換体は、当該技術分野で公知の方法に従って培養することが出来る。
【0031】
本発明における蛋白質の製造に際しては、当業者に公知の方法を適宜選択することができる。例えば、該蛋白質を含む培養液から、例えば、各種クロマトグラフィーカラム、フィルター、限外濾過、塩析、溶媒沈殿、溶媒抽出、蒸留、免疫沈降、SDS−ポリアクリルアミドゲル電気泳動、等電点電気泳動法、透析、再結晶等の当業者に公知の方法を適宜選択、組み合わせることによって、実質的に純粋で均一な蛋白質として分離、精製することができる。
【0032】
更に、蛋白質をグルタチオン S−トランスフェラーゼ蛋白質との融合蛋白質、又はヒスチジンを複数付加させた組換え蛋白質として発現させた場合には、発現させた組換え蛋白質はグルタチオンカラムあるいはニッケルカラムを用いて精製することができる。融合蛋白質の精製後、必要に応じて、目的の蛋白質以外の領域を、トロンビンまたはファクターXaなどにより切断し、除去することも可能である。或いは、トリプシン、キモトリプシン、リシルエンドペプチダーゼ、プロテインキナーゼ、グルコシダーゼ等の適当な蛋白質修飾酵素で、精製前又は精製後に蛋白質を処理することにより、任意に修飾を加えたり、部分的にペプチドを除去することもできる。
【0033】
本発明は、N−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をコードする遺伝子で形質転換された微生物をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法を提供する。また、同微生物から得られるN−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法を提供する。
本発明の製造法において、各アシラーゼを作用させる様式、方法、及び条件等は、N−アセチル−β−アミノ酸の種類及び量、各アシラーゼの種類、製造規模等の諸要件に応じて、当業者に公知の任意のものを適宜選択することが出来る。
例えば、本発明の菌体の懸濁液中にN−アセチル−β−アミノ酸を添加したり、又は、該菌体の破砕液上清中にN−アセチル−β−アミノ酸を添加することにより該アシラーゼを作用させることが出来る。
反応温度は、好ましくは10〜60℃、より好ましくは20〜40℃である。また、反応系のpHは、好ましくは4〜10であり、より好ましくは6〜9である。また、反応時間は10分〜120時間、好ましくは、1〜60時間、より好ましくは1〜48時間である。反応溶媒は、水溶液、MeOH、DMF、DMSOなどを用いることもできるし、それらを混合して使用することも可能である。
【実施例】
【0034】
以下の実施例を示し、本発明をより具体的に説明するが、本発明の技術的範囲はこれらによって制限されるものではない。
【0035】
1.試薬
(R,S)−β−Phe(DL−3−amino−3−phenyl−propionic acid)は、Sigma−Aldrich社より購入した。(R)−β−Phe及び(S)−β−Pheは渡辺化学より購入した。
その他β−アミノ酸は下記のメーカーより購入した。
(R,S)−β−Leu(DL−β−Leucine 、Fluka社);
(R)−β−Leu(L−β−Leucine hydrochloride、Fluka社);
(R,S)−β−homoPhe(DL−β−Homophyenylalanine、Fluka社);
(S)−β−homoPhe(L−β−Homophyenylalanine hydrochloride、Fluka社);
(R,S)−β−homoLeu(3−amino−5−methyl−hexanoic acid、Astatech, Inc.社);
(R)−β−homoLeu((R)−3−amino−5−methyl−hexanoic acid、Astatech, Inc.社);
(R,S)−β−Tyr(3−amino−3−(4−hydroxyphenyl)−propanoic acid、Bionet Building Blocks社);
(R)−β−Tyr((R)−3−amino−3−(4−hydroxyphenyl)−propanoic acid、Peptech社);
(R,S)−4−Fluoro−β−Phe(3−amino−3−(4−fluorophenyl)−propanoic acid、Bionet Building Blocks社);
(R)−4−Fluoro−β−Phe((R)−3−amino−3−(4−fluoro−phenyl)−propanoic acid、Peptech社);及び
(R,S)-β−3−Pyr−Ala(3−amino−3−(3−pyridyl)−propanoic acid、Bachem社);
【0036】
2.合成
各種N−アセチル−β−アミノ酸は、ラセミ体はラセミ体のβ−アミノ酸を、光学活性体は各光学活性体のβ−アミノ酸をアセチル化して合成した。合成法は以下に示すとおりである。なお、新規物質に関しては、NMRの解析結果を記載した。
2−1.N−アセチル−(R,S)−β−Pheの合成
(R,S)−β−Phe(50g,303mmol)を水200mlに懸濁させ、氷冷下25% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように25%NaOH水溶液でpHを調節しながら、滴下ロートで無水酢酸(62.8ml,664mmol)を室温で滴下した。一晩撹拌後、反応混合物の分析を行ったところ原料の残存が認められたため、さらに無水酢酸(6ml,63mmol)を加えた。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下60℃で一晩乾燥し、N−アセチル−(R,S)−β−Phe(56.9g,274.3mmol,91%)を得た。
【0037】
2−2.N−アセチル−(S)−β−Pheの合成
(S)−β−Phe(250mg,1.51mmol)を水5mlに懸濁させ、氷冷下20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.33ml,3.49mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(S)−β−Phe(238mg,1.15mmol,76%)を得た。
【0038】
2−3.N−アセチル−(R)−β−Pheの合成
(R)−β−Phe塩酸塩(250mg,1.24mmol)を水5mlに懸濁させ、氷冷下20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.28ml,2.96mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R)−β−Phe(236mg,1.13mmol,91%)を得た。
【0039】
2−4.N−アセチル−(R,S)-β−Leuの合成
(R,S)-β−Leu(995mg、7.58mmol)を水8mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(1.3ml、13.3mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−Leu(382mg、2.21mmol、29.0%)を得た。
ESI−MS [M−H]=172
【0040】
2−5.N−アセチル−(R,S)-β−homoPheの合成
(R,S)-β−homoPhe(198mg、1.12mmol)を水5mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.24ml、2.46mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−homoPhe(64.5mg、0.36mmol,32.1%)を得た。
ESI−MS [M−H]=220
【0041】
2−6.N−アセチル−(R,S)-β−homoLeuの合成
(R,S)-β−homoLeu(1.01g、6.9mmol)を水16mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(1.35ml、13.8mmol)を室温で滴下した。反応混合物をろ過し不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−homoLeu(64.5mg、5.07mmol、73.1%)を得た。この生成物は減圧乾燥により溶解、凝固していたが、NMRにより確認し、求める物質が生成していたのでそのまま反応に用いた。
ESI−MS [M−H]=186
【0042】
2−7.N−アセチル−(R,S)-β−Tyrの合成
(R,S)-β−Tyr(202mg、1.16mmol)を水16mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.33ml、3.39mmol)を室温で滴下した。反応混合物をろ過し不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し、エバポレーターで濃縮し結晶を析出させた。結晶をろ過し水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-β−Tyr(55.5mg、0.25mmol、21.4%)を得た。
1H NMR(400MHz, D2O); 2.05 (s, 3H), 2.60−2.70(m, 2H), 5.1(dd, 1H, J=7.5 , 7.5 Hz), 6.88(d, 2H, J=8.6 Hz), 7.26 (d, 2H, J=8.6 Hz)
ESI−MS [M−H]=222
【0043】
2−8.N−アセチル−(R,S)-4−Fluoro−β−Pheの合成
(R,S)-4−Fluoro−β−Phe(206mg、1.12mmol)を水16mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.22ml、2.26mmol)を室温で滴下した。反応混合物をろ過し不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調節し、エバポレーターで濃縮し結晶を析出させた。結晶をろ過し、水で洗浄し、減圧下50℃で一晩乾燥し、N−アセチル−(R,S)-4−Fluoro−β−Phe(143.9mg,0.64mmol,57.1%)を得た。
ESI−MS [M−H]=224
【0044】
以上の生成物は結晶を取得後に、NMRにてチャートを確認し、質量分析を行い目的化合物の生成を確認した。
【0045】
2−9.N−アセチル−(R,S)-β−aminobutylic acidの合成
3−アセトアミド−2−ブテン酸メチルはJ.Am.Chem.Soc. 2002, 124, 14552記載の方法に従い調製した。
3−アセトアミド−2−ブテン酸メチル(1.35g)のメタノール溶液に5%パラジウム−活性炭を加え、1気圧の水素下、30℃で16時間撹拌した。反応終了後、ろ過にて5%パラジウム−活性炭を除き、得られた溶液を濃縮して3−アセトアミド−2−酪酸メチル(1.37g)を得た。
続いて得られた3−アセトアミド−2−ブテン酸メチル(570mg)のエタノール溶液3mLに8M水酸化水溶液(900μL)を加え、35℃で1時間撹拌した。反応終了後、6M塩酸(1.2mL)を加え中和し、濃縮してジクロロメタン(20mL)を加えた。析出した結晶をろ過にて取り除き、ろ液を濃縮して表題化合物265mgを得た。
NMRにて、目的化合物の生成を確認した。
1H NMR(400MHz, MeOH−d4); 1.41 (q, 3H, J=2.6 Hz), 1.99 (s, 3H), 2.55 (t, 2H, J=5.1 Hz), 4.28(m, 1H)
13C NMR(400MHz, MeOH−d4);175.6, 170.88, 42.62, 40.22, 23.60, 20.34
ESI−MS [M−H]=144
【0046】
2−10.N−アセチル−3,4−(−O−CH2−O−)−β−Phenylalanine
US20060035345記載の方法で作成したものを使用した。
【0047】
2−11.N−アセチル−(R,S)-3−Pyr−Alaの合成
β-3-Pyr-Ala(200mg、1.2mmol)を水5mlに懸濁させ、20% NaOH水溶液を加え、pH11−12に調節した。得られた水溶液のpHが11−12の範囲をできるだけ保つように20%NaOH水溶液でpHを調節しながら、無水酢酸(0.257ml、2.46mmol)を室温で滴下した。反応混合物をろ過し、不溶物を除去した後、得られた溶液に濃塩酸を加え、pHを2に調整したが沈殿が析出しなかったため、エバポレーターを用いて総て液体を蒸発させて、乾燥物をNMRにより解析し、反応に用いた。
1H NMR(400MHz, D2O); 2.03 (s, 3H), 2.78−2.90(m, 2H), 5.33(dd, 1H, J=7.3, 7.3 Hz),7.97(dd, 1H , J=5.7, 8.2 Hz), 8.49 (d, 1H, J=8.2 Hz), 8.67(d, 1H, J=5.7 Hz), 8.75(s, 1H,)
ESI−MS [M−H]=207
【0048】
3.酵素活性測定
3−1.アッセイ条件
10 mM N−アセチル−(R,S)−β−Phe, 50mM Tris−HCl (pH 7.6)および適宜酵素サンプルを含む反応液を30℃にて15minから2h静置した後、5minの煮沸処理にて反応を停止した。反応液を遠心し、その上清を適宜希釈して、HPLCにて分析した。この標準反応条件にて、1min間に1μmolの(R)−β−Pheまたは(S)−β−Pheを生成するアシラーゼ活性を1 Unitと定義した。以後N−アセチル−(R,S)−β−Pheから(R)−β−Pheを生成する活性をN−アセチル−(R)−β−Pheアシラーゼ活性、(S)−β−Pheを生成する活性をN−アセチル−(S)−β−Pheアシラーゼ活性と記述する。
3−2.HPLC
【0049】
3−2−1.(R),(S)−β−Phe光学分割条件
(R)−β−Phe、(S)−β−Phe、N−アセチル−(R)−β−PheおよびN−アセチル−(S)−β−PheはHPLCによって定量分析した。カラムはジーエルサイエンス社製Inertsil ODS3(0.46cmΦ, 5cm)とダイセル化学社製Chiralpak WH(0.46cmΦ, 25cm)をこの順に直列につないで用いた。移動相は0.25mM CuSO4, 2%(v/v)アセトニトリルとし、流速1.5ml/min、カラム温度50℃、検出はUV detector 210nmとした。この分析で上記4種化合物は異なる溶出時間に溶出し、溶出順は早いほうから(S)−β−Phe、(R)−β−Phe、N−アセチル−(R)−β−Phe、N−アセチル−(S)−β−Pheの順であった。定量は、各標準標品を対照とし、ピークエリア値から算出した。
【0050】
3−2−2. N−アセチル−β−Pheとβ−Pheの分離条件
カラムはGLサイエンス社製Inertsil Ph−3(0.46cmΦ、25cm)を用いた。移動相に10%アセトニトリル(リン酸にてpH3.0に調整)を用い、温度40℃、流速1.0mL/min、検出UV210nmの条件で各ラセミ体標準品を対照とし、ピークエリア値から算出し定量した。
【0051】
3−2−3. (R),(S)−β−Leu、(R),(S)−β−homoLeu、(R),(S)−β−homoPhe分割条件
カラムはastec社製CHIROBIOTEC T(0.46cmΦ、25cm)を用いた。移動相に90% MeOHを用い、温度40℃、流速0.4mL/min、検出UV205nmの条件で、(R),(S)体標準品と比較し、光学選択性を決定した。
【0052】
3−2−4.(R),(S)−β−Tyr、(R),(S)−β−4−Fluoro−Phe分離条件
カラムはジーエルサイエンス社製Inertsil ODS3(0.46cmΦ, 5cm)とダイセル化学社製Chiralpak WH(0.46cmΦ, 25cm)をこの順に直列につないで用いた。移動相に1mM CuSO4、10% MeOHを用い、温度 50℃、流速1.0ml/min、検出UV210nmの条件で(R),(S)体標準品と比較し光学選択性を決定した。
【0053】
4.Burkholderia sp. AJ110349由来N−アセチル−(R)/(S)−β−アミノ酸アシラーゼの精製
4−1.使用菌株と培養条件
Burkholderia sp. AJ110349を用いた。保存菌株を5g/l D−glucose, 10g/l yeast extract, 10g/l peptone, 5g/l NaCl, 20g/l agar (pH 7.0)から成るCM2G寒天培地上で、30℃で48h静置培養することによりリフレッシュした。これを、10g/l D−glucose, 10g/l (NH4)2SO4, 10g/l (R,S)−β−Phe, 2g/l casamino acid, 1g/l KH2PO4, 0.4g/l MgSO4・7H2O,1g/l NaCl, 19.5g/l 2−(N−morpholino) ethanesufonic acid (MES), 5mg/l nicotinamide, 0.2mg/l thiamin, 10mg/l FeSO4・7H2O, 10mg/l MnSO4・4〜5H2Oからなる酵素生産培地100 mlに接種し、500ml用の坂口フラスコを用いて30℃、120rpmで66h、振盪培養した。
【0054】
4−2.無細胞抽出液の調製
培養液およそ2,000mlから、6,800g,10minの遠心操作により菌体を集菌した。この際、菌体の沈降が十分に見られなかったため、およそ1,600 mlの上清を除去し、残りの部分をピペッティングにより均一化した。この濃縮した培養液をバッファー等で洗浄することなく、200W, 20minの超音波処理に供し菌体を破砕した。得られた破砕液を200,000g, 30min遠心操作し、得られた遠心上清およそ200mlを無細胞抽出液とした。
【0055】
4−3.硫安分画
得られた無細胞抽出液に(NH4)2SO4を終濃度40%飽和となるよう加え、氷上1h攪拌した後、9,200g, 15min遠心した。得られた沈殿を少量の25mM Tris−HCl(pH 7.6)にて溶解後、25mM Tris−HCl(pH7.6)に対して透析した。この透析後の溶液を、以後クロマトグラフィーに供するサンプルとした。
【0056】
4−4.クロマトグラフィー
4−4−1)Phenyl Sepharose 26/10 (Amersham Pharmacia)
上記で得られた硫安分画画分を25mM Tris−HCl (pH7.6), 0.6M (NH4)2SO4から成る緩衝液に対して透析後、同緩衝液で平衡化したPhenyl Sepharose 26/10に供した。非吸着タンパク質溶出後、緩衝液中の(NH4)2SO4濃度を直線的に0.6 Mから0 Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、(NH4)2SO4濃度およそ0.2Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性は(NH4)2SO4濃度およそ0.1Mの付近にそれぞれ検出された。活性の存在する画分をN−アセチル−(R)−β−Pheアシラーゼ溶出画分、N−アセチル−(S)−β−Pheアシラーゼ活性溶出画分に分けて、それぞれ回収した。
【0057】
4−4−2)Q−Sepharose 16/10 (Amersham Pharmacia)
得られたPhenyl−Sepharose分画画分を濃縮し、25mM Tris−HCl (pH7.6)に対して透析後、同緩衝液で平衡化したQ−Sepharose 16/10に供した。非吸着タンパク質溶出後、緩衝液中のNaCl濃度を直線的に0 Mから0.5Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、NaCl濃度およそ0.22Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性はNaCl濃度およそ0.3Mの付近にそれぞれ検出された。活性の存在する画分をそれぞれ回収した。
【0058】
4−4−3)Superdex 200 16/60 (Amersham Pharmacia)
得られたQ−Sepharose分画画分を濃縮し、25mM Tris−HCl (pH7.6)で平衡化したSuperdex 200 16/20に供した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は分子量206kDaと見積もられる溶出位置に、N−アセチル−(S)−β−Pheアシラーゼ活性は分子量101kDaと見積もられる溶出位置でそれぞれ検出された。活性の存在する画分をそれぞれ回収した。
【0059】
4−4−4)Resource phenyl (Amersham Pharmacia)
得られたSuperdex分画画分を濃縮し、25mM Tris−HCl (pH7.6), 0.6M (NH4)2SO4から成る緩衝液に対して透析後、同緩衝液で平衡化したResource phenylに供した。非吸着タンパク質溶出後、緩衝液中の(NH4)2SO4濃度を直線的に0.6Mから0 Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、(NH4)2SO4濃度およそ0.35Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性は(NH4)2SO4濃度およそ0.45Mの付近にそれぞれ検出された。活性の存在する画分をそれぞれ回収した。
【0060】
4−4−5)Mono Q 5/5 (Amersham Pharmacia)
得られたResource phenyl分画画分を濃縮し、25 mM Tris−HCl (pH 7.6)に対して透析後、同緩衝液で平衡化したMono Q 5/5に供した。非吸着タンパク質溶出後、緩衝液中のNaCl濃度を直線的に0 Mから0.5Mまで変化させることにより吸着タンパク質を溶出した。この操作によりN−アセチル−(R)−β−Pheアシラーゼ活性は、NaCl濃度およそ0.2Mの付近に、N−アセチル−(S)−β−Pheアシラーゼ活性はNaCl濃度およそ0.28Mの付近にそれぞれ検出された。活性の存在する画分をそれぞれ回収し、それぞれ精製N−アセチル−(R)−β−アミノ酸アシラーゼ、精製N−アセチル−(S)−β−アミノ酸アシラーゼ標品とした。
【0061】
以上の酵素精製の結果を表1(N−アセチル−(R)−β−アミノ酸アシラーゼ)、表2(N−アセチル−(S)−β−アミノ酸アシラーゼ)にそれぞれまとめた。この精製操作により、N−アセチル−(R)−β−アミノ酸アシラーゼは比活性262倍に、N−アセチル−(S)−β−アミノ酸アシラーゼは比活性809倍にそれぞれ精製された。
【0062】
【表1】
【0063】
【表2】
【0064】
4−5.SDS−PAGE
上記で得られた精製N−アセチル−(R)−β−アミノ酸アシラーゼおよび精製N−アセチル−(S)−β−アミノ酸アシラーゼをSDS−PAGEに供した(Fig.1)。結果、N−アセチル−(R)−β−アミノ酸アシラーゼは分子量約84kDaと見積もられる単一のバンドとして(Fig.1A)、N−アセチル−(S)−β−アミノ酸アシラーゼは分子量約41kDaと見積もられる単一のバンドとして(Fig.1B)、それぞれ観察された。上記ゲルろ過クロマトグラフィーにおいて、両酵素の未変性状態の分子量はそれぞれ206kDa(N−アセチル−(R)−β−アミノ酸アシラーゼ)、101kDa(N−アセチル−(S)−β−アミノ酸アシラーゼ)と見積もられたため、SDS−PAGEで見積もられた分子量と併せて考えると、何れの酵素もホモダイマーあるいはホモトライマー構造を有している酵素であると考えられた。
【0065】
4−6.光学特異性
精製酵素を酵素源として、その光学選択性を測定した。上記酵素活性条件において反応時間30 min、酵素添加量4.6μg/ml(N−アセチル−(R)−β−アミノ酸アシラーゼ)または3.1μg/ml(N−アセチル−(S)−β−アミノ酸アシラーゼ)として反応を行った。結果、N−アセチル−(R)−β−アミノ酸アシラーゼを酵素源とした場合には(R)−β−Pheの生成のみが観察され、(S)−β−Pheの生成は検出限界以下であった(Fig.2D)。一方、N−アセチル−(S)−β−アミノ酸アシラーゼを酵素源とした場合には(S)−β−Pheの生成のみが観察され、(R)−β−Pheの生成は検出限界以下であった(Fig.2E)。これらの結果から、両酵素はいずれも高い光学選択性を有していることが示された。
【0066】
5.N末端アミノ酸配列解析
精製酵素標品を材料に、そのN末端アミノ酸配列を決定した。方法は、酵素標品をSDS−PAGEに供した後、PVDF膜に転写し、クマシーブルーで染色後、バンドの見られる位置を切り出し、これをプロテインシーケンサーに供することにより行った。結果、N−アセチル−(R)−β−アミノ酸アシラーゼの場合は配列番号1に示す20残基の配列が、N−アセチル−(S)−β−アミノ酸アシラーゼの場合は配列番号2に示す26残基のアミノ酸配列が、それぞれ決定された。
【0067】
6.遺伝子クローニングと塩基配列決定
決定されたN末端アミノ酸配列に基づいて合成プライマーを設計し、Takara社製LA PCR in vitro Cloning Kitを用いて、アシラーゼをコードする遺伝子部分断片の取得を行った。結果、N−アセチル−(R)−β−アミノ酸アシラーゼN末端近傍をコードする遺伝子領域を含む、5’−PstI認識配列、3’−EcoRI認識配列を両端に有する794baseのDNA断片、およびN−アセチル−(S)−β−アミノ酸アシラーゼN末端近傍をコードする遺伝子領域を含む、5’−PstI認識配列、3’−SalI認識配列を両端に有する393baseのDNA断片をそれぞれ得て、その塩基配列を決定した。
【0068】
塩基配列の決定により得られた情報より、プライマーR7F、R8R(配列番号3、4)/プライマーS3F、S4R(配列番号5、6)を用いてBurkholdelia sp. AJ110349のゲノムDNAを鋳型としてPCRを行いN−アセチル−(R)−β−アミノ酸アシラーゼ/ N−アセチル−(S)−β−アミノ酸アシラーゼのN末端近傍をコードする遺伝子領域含む0.3kb/0.4kbのDNA断片を増幅し、DIGラベリングを行ってそれぞれをR−プローブ/S−プローブとした。
【0069】
Burkholderia sp. AJ110349染色体DNA5μgをBamHI/HindIII(100/50U)にて切断した後、R−プローブを用いてサザン解析を行った。ハイブリダイゼーションの条件は、42℃、16時間で、ハイブリダイゼーションの溶液にはDIG Easy Hyb(Roche社)を用いた。その結果、約5kbにポジティブシグナルを確認した。
【0070】
次いで、Burkholderia sp. AJ110349染色体DNA5μg をBamHI/HindIII(100/50U)処理後、アガロース電気泳動し、5kb付近の断片を精製し、pUC118のBamHI/HindIIIサイトにライゲートした。この反応液をもちいてEscherichia coli JM109を形質転換し、ライブラリーを作製した。上記プローブをもちいてコロニーハイブリダイズを行い、ポジティブコロニーを取得し、プラスミドを抽出した。得られたプラスミドをpBRACY_A3として、挿入配列について塩基配列を決定したところ、760アミノ酸(配列番号8)をコードするORF(配列番号7)が存在した。
【0071】
Burkholderia sp. AJ110349染色体DNA5μgをPstI/HindIII(各50U)にて切断した後、S−プローブを用いてサザン解析を行った。ハイブリダイゼーションの条件は、42℃、16時間で、ハイブリダイゼーションの溶液にはDIG Easy Hyb(Roche社)を用いた。その結果、約1.5kbにポジティブシグナルを確認した。
【0072】
次いで、Burkholderia sp. AJ110349染色体DNA5μgをPstI/HindIII(各50U)処理後アガロース電気泳動し、1.5kb付近の断片を精製し、pUC118のPstI/HindIIIサイトにライゲートした。この反応液をもちいてEscherichia coli JM109を形質転換し、ライブラリーを作製した。上記プローブをもちいてコロニーハイブリダイズを行い、ポジティブコロニーを取得し、プラスミドを抽出した。得られたプラスミドをpBSACY_PHとして、挿入配列について塩基配列を決定したところ、352アミノ酸(配列番号10)をコードするORF(配列番号9)が存在した。
【0073】
7. Variovorax sp. AJ110348株由来N−アセチル−(R)-β−アミノ酸アシラーゼ遺伝子のショットガンクローニングによる取得
Variovorax sp.AJ110348株よりゲノムDNAを抽出し、制限酵素Sau3AIにて部分分解した後、約3〜8kb付近の断片を回収しpUC118へと連結した。この反応液をもちいてEscherichia coli JM109を形質転換し、ライブラリーを作製した。
得られた形質転換体をブルーホワイトセレクションにより選別し、単コロニーとして得られた3500株に対してマスタープレートを作成した。これらの菌株をN−アセチル−(R,S)−β−Phe液体培地[硫酸アンモニウム10.0g/l、KH2PO4 1.0g/l、MgSO4・7H2O 0.4g/l、FeSO4・7H2O 10mg/l、MnSO4・5H2O 10mg/l、ビタミンB1・HCl 0.2mg/l、N−アセチル−(R,S)−β−Phe 1.0g/l, pH8.0、IPTG 100μM、Amp 100μg/ml] に10株ずつ接種し、37℃、48時間振とう培養した。
得られた培養上清をSiliagel 60F254プレート(Merck)、ブタノール:酢酸:水=4:1:2にて分離し、UV254nm及びニンヒドリン発色にてβ−Pheの生成を確認した。
その結果、混合した培養上清からβ−Pheが検出されたので、さらに、1株ずつ同様の手法で培養し、TLCにて培養上清を確認したところ、K83株と命名した株からβ−Pheが検出された。
K83株のプラスミドDNAを抽出し挿入配列を確認したところ、約3kbの挿入配列が存在した。塩基配列を決定した結果、779アミノ酸(配列番号12)をコードするORF(配列番号11)が存在した。
【0074】
8.K83株の活性測定及び光学選択性の測定
得られたK83株を100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリン、1mM IPTGを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、50mM Tris−HCl(pH7.6)、0.2% N−アセチル−β−Phe、37℃、10分間反応し、N−アセチル−β−Pheとβ−Pheの分離条件にてN−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、2.3U/mgであった。
また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(R)−β−Phe特異的に生成しており、(S)−β−Pheは検出限界以下であった。
【0075】
9.Burkholderia sp. AJ110349由来N−アセチル−(R)−β−アミノ酸アシラーゼ高発現株の作成
Burkholderia sp.AJ110349染色体DNAを鋳型として、プライマー R_7F(配列番号13)、及び R_R_HindIII(配列番号14)によりPCR反応により、得られた2.3kbの増幅断片をBamHI/HindIII処理し、ptrp4(参考文献Journal of Molecular Catalysis B: Enzymatic 32 (2005)205−211)のBamHI/HindIIIサイトに挿入し、ptrp4_3BRとした。このプラスミドによりEscherichia coli JM109を形質転換した。この形質転換体をJM109/ptrp4_3BRと命名した。
JM109/ptrp4_3BRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むM9カザミノ酸培地50mLに植菌し、30℃、18時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、50mM Tris−HCl(pH7.6)、0.2% N−アセチル−(R,S)−β−Phe、37℃、30分間反応し、生成したβ−Pheの量をHPLCにて測定した。N−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、0.29U/mgであった。また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(R)−β−Phe特異的に生成しており、(S)−β−Pheは検出限界以下であった。
【0076】
10.Burkholderia sp. AJ110349由来N−アセチル−(S)−β−アミノ酸アシラーゼ高発現株の作成
Burkholderia sp.AJ110349染色体DNAを鋳型として、プライマー S_F_NdeI_2(配列番号15)、及び S_R_HindIII(配列番号16)によりPCR反応により、得られた1.1kbの増幅断片をNdeI/BamHIで処理した。これとは別に、pSFN_Sm_Aet(参考文献WO2006075486)をNdeI/HindIIIで切断し、3kb付近のDNAを切り出し精製した。このpSFNのNdeI/HindIIIサイトに1.1kbPCR産物を制限酵素にて処理したDNAを挿入し、pSFN_2BSとした。このプラスミドによりEscherichi a coli JM109を形質転換した。この形質転換体をJM109/pSFN_2BSと命名した。
なお、このプラスミドの挿入配列は、配列10の6残基目のアミノ酸にあたるメチオニンを開始コドンとしているが、発明者らは1残基目からのアミノ酸を開始コドンとするプラスミドとともに作成した。しかしながら、特に活性の違いはなかった。
JM109/pSFN_2BSを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、上記の方法と同様にN−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、0.33U/mgであった。また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(S)−β−Phe特異的に生成しており、(R)−β−Pheは検出限界以下であった。
【0077】
11.Variovorax sp. AJ110348由来N−アセチル−(R)−β−アミノ酸アシラーゼ高発現株の作成
Variovorax sp. AJ110348染色体DNAを鋳型として、プライマー VRACY_1F_NdeI(配列番号17)、及びVRACY_R_HindIII(配列番号18)によりPCR反応により、得られた2.4kbの増幅断片をNdeI/HindIIIで処理した。このDNA産物を実施例9で作成したpSFN NdeI/HindIII精製産物のNdeI/HindIIIサイトに挿入し、pSFN_1VRとした。このプラスミドによりEscherichia coli JM109を形質転換した。この形質転換体をJM109/pSFN_1VRと命名した。
JM109/pSFN_1VRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、上記の方法と同様にN−アセチル−β−アミノ酸アシラーゼ活性を測定したところ、1.5U/mgであった。また、β−Phe光学分割条件で、HPLCにより光学選択性を決定したところ、(R)−β−Phe特異的に生成しており、(S)−β−Pheは検出限界以下であった。
【0078】
12.基質特異性の調査
12−1.各種N−アセチル−(R,S)-β−アミノ酸への基質特異性調査
JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて菌体懸濁液を調製した。超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として、実施例2−1〜9に記した各種N−アセチル−(R,S)−β−アミノ酸への基質特異性を調査した。
酵素は上記の方法で調整後、活性を測定し、N−アセチル−(R,S)−β−Pheから1分間に1μmolのβ−Pheを生成する活性を1Uとし、各種基質特異性の調査には、50mUの無細胞抽出液を用いた。ネガティブコントロールとして、上記のように調整した無細胞中質液の代わりに、50mM Tris緩衝液(pH7.6)を用いて各基質と反応を行ったものを「enzyme-」の実験区とし、各基質の代わりに50mM Tris緩衝液(pH7.6)を用いて各無細胞抽出液と反応させたものを「基質-」の実験区とした。
50mM Tris−HCl(pH7.6)、0.2% 各N−アセチル−(R,S)−β−アミノ酸、37℃、1時間、または24時間反応し、96℃、10分間処理し、反応を停止した。アセチル基の分解により生成した酢酸の量を酢酸キット(Roche)のプロトコールに従って定量した。
1時間後の酢酸生成量を表3に、24時間後の酢酸生成量を表4に示した。その結果、N−アセチル−(R,S)-β−aminobutylic acid(N−アセチル−β−Aba)、N−アセチル−(R,S)-β−Leu、N−アセチル−(R,S)-β−homoLeu、N−アセチル−(R,S)-β−Tyr、N−アセチル−(R,S)-homoPhe、N−アセチル−(R,S)-β−4−Fluoro−Pheを基質とした実験区から酢酸が検出された。
【0079】
【表3】
【0080】
【表4】
【0081】
12−2.光学特異性の決定
反応24時間後のサンプルを各種キラル分割カラムにて解析し、光学選択性を決定した。
12−2−1.(R),(S)−β−Leuの分離
3−2−3.の分割条件で、標準品は、N−アセチル-(R),(S)−β−Leu、(S)−β−Leu、(R)−β−Leuの順に溶出された。
JM109/ptrp4_3BRを用いたN−アセチル−β−Leu反応用液では(R)−β−Leuが検出され(S)−β−Leuは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−Leu反応用液では(S)−β−Leuが検出され(R)−β−Leuは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−Leu反応用液では(R)−β−Leuが検出され(S)−β−Leuは検出限界以下であり、> 99 % eeであった。
【0082】
12−2−2.(R),(S)−β−homoLeuの分離
3−2−3.の分割条件で、標準品は、N-アセチル-(R,S)−β−homoLeu、(R)−β−homoLeu、(S)−β−homoLeuの順に溶出された。
JM109/ptrp4_3BRを用いたN−アセチル−β−homoLeu反応用液では(S)−β−homoLeuが検出され、少量の(R)−β−Leuは検出され69% eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−homoLeu反応用液では(R)−β−homoLeuが検出され(S)−β−Leuは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−homoLeu反応用液では(S)−β−homoLeuが検出され、(R)−β−homoLeuは検出限界以下であり、> 99 % eeであった。
【0083】
12−2−3.(R),(S)−β−homoPheの分離
3−2−3.の分割条件で、標準品は、N−アセチル−(R,S) −β−homoPhe、(R)−β−homoPhe、(S)−β−homoPheの順に溶出された。
JM109/ptrp4_3BRを用いたN−アセチル−β−homoPhe反応用液では(S)−β−homoPheが検出され、微量の(R)−β−homoPheも検出され、98% eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−homoPhe反応用液では(R)−β−homoPheが検出され(S)−β−は検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−homoPhe反応用液では(S)−β−homoPheが検出され(R)−β−homoPheは検出限界以下であり、> 99 % eeであった。
【0084】
12−2−4.(R),(S)−β−Tyrの分離
3−2−4.の分離条件で、標準品は、(S)−β−Tyr、(R)−β−Tyr、N−アセチル−(R)/(S)−β−Tyrの順に溶出された。(N-アセチル-β-Tyrの標準品はラセミ体であるので、分離はされるものの溶出順は未決)
JM109/ptrp4_3BRを用いたN−アセチル−β−Tyr反応用液では(R)−β−Tyrが検出され、少量の(S)−β−Tyrも検出され61 % eeであった。
JM109/pSFN_2BSを用いたN−アセチル−β−Tyr反応用液では(S)−β−Tyrが検出され(R)−β−Tyrは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−β−Tyr反応用液では(R)−β−Tyrが検出され(S)−β−Tyrは検出限界以下であり、> 99 % eeであった。
【0085】
12−2−5.(R),(S)−4−Fluoro−β−Pheの分離
3−2−4.の分離条件で、標準品は、(S)−4−Fluoro−β−Phe、(R)−4−Fluoro−β−Phe、N−アセチル−(R)/(S)−4−Fluoro−β−Pheの順に溶出された。(N-アセチル-4-Fluoro-β-Pheの標準品はラセミ体であるので、分離はされるものの溶出順は未決)
JM109/ptrp4_3BRを用いたN−アセチル−4−Fluoro−β−Phe反応用液では(R)−4−Fluoro−β−Pheが検出され、少量の(S)−4−Fluoro−β−Pheが検出され、94% eeであった。
JM109/pSFN_2BSを用いたN−アセチル−4−Fluoro−β−Phe反応用液では(S)−4−Fluoro−β−Pheが検出され(R)−4−Fluoro−β−Pheは検出限界以下であり、> 99 % eeであった。
JM109/pSFN_1VRを用いたN−アセチル−4−Fluoro−β−Phe反応用液では(R)−4−Fluoro−β−Pheが検出され(S)−4−Fluoro−β−Pheは検出限界以下であり、> 99 % eeであった。
【0086】
12−3.N−アセチル−3,4−(−O−CH2−O−)−β−Phenylalanineへの基質特異性調査
JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを上記のような方法で調整し、各種N−アセチル−3,4−(−O−CH2−O−)−β−Phenylalanineへの基質特異性を調査した。
培養液300μLを50mM Tris−HCl(pH7.6)で洗浄し、300μLの50mM Tris−HCl(pH7.6)に懸濁し、超音波破砕処理によって、菌体を破砕し、遠心分離(15,000g、10分、4℃)により得られる上澄み液を無細胞抽出液として反応に用いた。
50mM Tris−HCl(pH7.6)、0.2% N−アセチル−(R)/(S)−3,4−(−O−CH2−O−)−β−Phenylalanine、37℃、10分間反応した。N−アセチル−β−Pheとβ−Pheの分離条件で、3,4−(−O−CH2−O−)−β−Phenylalanineを定量したところ、無細胞抽出液1mLあたりの活性は、JM109/ptrp4_3BRは0.2U、JM109/pSFN_2BSは3.9U、JM109/pSFN_1VRは27.8Uであった。
(R),(S)−β−Phe光学分割条件にて標準品を分離したところ、(S)−3,4−(−O−CH2−O−)−β−Phenylalanine、(R)−3,4−(−O−CH2−O−)−β−Phenylalanine、N−アセチル−(R)/(S)− 3,4−(−O−CH2−O−)−β−Phenylalanineの順に溶出された。(N−アセチル−(R)/(S)−3,4−(−O−CH2−O−)−β−Phenylalanineの溶出順は未決)
この条件で、反応溶液を解析したところ、JM109/ptrp4_3BRの反応液からは(R)-3,4−(−O−CH2−O−)−β−Phenylalanineが、JM109/pSFN_2BSの反応液からは(S)−3,4−(−O−CH2−O−)−β−Phenylalanineが、JM109/pSFN_1VRの反応液からは(R)−3,4−(−O−CH2−O−)−β−Phenylalanineが検出された。
【0087】
12−4.N−アセチル−3−Pyr−Alaへの基質特異性調査
12−1と同様の菌体を用いて、同様の方法で各酵素を調整し、50mUの無細胞抽出液を反応に用いた。50mM Tris−HCl(pH7.6)、1% N−アセチル−(R,S)−β−3−Pyr−Ala、37℃、1時間反応し、70℃、10分間処理し、反応を停止した。アセチル基の分解により生成した酢酸の量を酢酸キット(Roche)のプロトコールに従って定量した。
その結果、JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを用いた反応液では、それぞれ3.5mM、8.4mM、0.8mMの酢酸が検出され、「基質−」及び「enzyme−」の実験区からは酢酸は検出されなかった。
【0088】
13.蓄積量の評価
JM109/ptrp4_3BR、JM109/pSFN_2BS、JM109/pSFN_1VRを100mg/lのアンピシリンを含むLB培地で37℃、16時間前培養した。この培養菌体1mLを、100mg/lのアンピシリンを含むTB培地50mLに植菌し、30℃、16時間培養し得られた菌体を遠心分離にて集菌した。この菌体を50mM Tris緩衝液(pH7.6)を用いて洗菌し、同緩衝液を用いて10倍に濃縮した菌体懸濁液を調製した。
この菌体懸濁液を用いて、100mM Tris−HCl(pH 7.6)、5% N−アセチル−(R,S)−β−Phe、37℃、24時間、全量2.5mLで反応した。その後、3−2−1の分析法で反応液を解析した。
JM109/ptrp4_3BRの10倍濃縮の菌体懸濁液を500μL加え反応したところ、(R)−β−Pheの収率21.6%、>99% eeで、(S)−β−Pheは検出限界以下であった。
JM109/pSFN_2BSの10倍濃縮の菌体懸濁液を250μL加え反応したところ、(S)−β−Pheの収率49.5%、>99% eeで、(R)−β−Pheは検出限界以下であった。
JM109/pSFN_1VRの10倍濃縮の菌体懸濁液を125μL加え反応したところ、(R)−β−Pheの収率45.4%、>99% eeで、(S)−β−Pheは検出限界以下であった。
【産業上の利用可能性】
【0089】
本発明で同定された遺伝子を用いた大腸菌(Echerichia coli)のような宿主を組換えることによって、N−アセチル−(R)−β−アミノ酸アシラーゼ及びN−アセチル−(S)−β−アミノ酸アシラーゼを高発現する系を構築することができる。その結果、該酵素を利用してR体又はS体のβアミノ酸を選択的にする系を簡便かつ安価に提供することが可能となる。
【特許請求の範囲】
【請求項1】
下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と90%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
【請求項2】
下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号9に示される塩基配列からなるDNA、及び
(b)配列番号9に示される塩基配列のDNAと90%以上の配列同一性を示すDNAであり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
【請求項3】
β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、請求項1又は2に記載の遺伝子。
【請求項4】
下記のいずれか一つに示す蛋白質。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と90%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
【請求項5】
β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、請求項4記載のβ−アミノ酸アシラーゼ。
【請求項6】
請求項1から3のいずれか一項記載の遺伝子によって形質転換された微生物。
【請求項7】
請求項1から3のいずれか一項記載の遺伝子によって形質転換された微生物であるEscherichia coli。
【請求項8】
請求項6又は7記載の微生物をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
【請求項9】
請求項6又は7記載の微生物から得られるN−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
【請求項1】
下記のいずれか一つに示す蛋白質をコードする遺伝子。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と90%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
【請求項2】
下記のいずれか一つに示すDNAを含む遺伝子。
(a)配列番号9に示される塩基配列からなるDNA、及び
(b)配列番号9に示される塩基配列のDNAと90%以上の配列同一性を示すDNAであり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質をコードするDNA。
【請求項3】
β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、請求項1又は2に記載の遺伝子。
【請求項4】
下記のいずれか一つに示す蛋白質。
(a)配列番号10で示されるアミノ酸配列からなる蛋白質、
(b)配列番号10で示されるアミノ酸配列において、1個若しくは数個のアミノ酸残基が置換、欠失、又は付加されたアミノ酸配列からなり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質、及び
(c)配列番号10で示されるアミノ酸配列と90%以上の配列同一性を示すアミノ酸配列を含む蛋白質であり、かつ、N−アセチル−(S)−β−アミノ酸アシラーゼ活性を有する蛋白質。
【請求項5】
β−アミノ酸がβ−フェニルアラニン、β−ロイシン、β−ホモロイシン、β−ホモフェニルアラニン、β−チロシン、β−4−フルオロフェニルアラニン、β−アミノブチル酸、3,4-(-O-CH2-O-)-β−フェニルアラニン、及びβ−3−ピリジル−アラニンからなる群から選ばれるいずれかである、請求項4記載のβ−アミノ酸アシラーゼ。
【請求項6】
請求項1から3のいずれか一項記載の遺伝子によって形質転換された微生物。
【請求項7】
請求項1から3のいずれか一項記載の遺伝子によって形質転換された微生物であるEscherichia coli。
【請求項8】
請求項6又は7記載の微生物をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
【請求項9】
請求項6又は7記載の微生物から得られるN−アセチル−β−アミノ酸アシラーゼ活性を有する蛋白質をN−アセチル−β−アミノ酸に接触させ、β−アミノ酸を生成させ、これを回収することを特徴とするβ−アミノ酸の製造法。
【図1】

【図2】
【図2】
【公開番号】特開2012−196231(P2012−196231A)
【公開日】平成24年10月18日(2012.10.18)
【国際特許分類】
【出願番号】特願2012−158252(P2012−158252)
【出願日】平成24年7月17日(2012.7.17)
【分割の表示】特願2007−192722(P2007−192722)の分割
【原出願日】平成19年7月25日(2007.7.25)
【出願人】(000000066)味の素株式会社 (887)
【Fターム(参考)】
【公開日】平成24年10月18日(2012.10.18)
【国際特許分類】
【出願日】平成24年7月17日(2012.7.17)
【分割の表示】特願2007−192722(P2007−192722)の分割
【原出願日】平成19年7月25日(2007.7.25)
【出願人】(000000066)味の素株式会社 (887)
【Fターム(参考)】
[ Back to top ]