
N−アセチルグルコサミンがαで結合した糖誘導体の調製方法
【課題】 N−アセチルグルコサミン(GlcNAc)がαで結合した糖誘導体を、酵素的に調製する新規な方法を提供する。
【解決手段】 バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来のα−N−アセチルグルコサミニダーゼ1、2または3を用いて、N−アセチルグルコサミンがαで結合したジメトキシトリアゾール (GlcNAc−α−DMT)またはその誘導体と糖受容体を反応させて、N−アセチルグルコサミンがαで結合した糖誘導体を調製する方法。
【解決手段】 バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来のα−N−アセチルグルコサミニダーゼ1、2または3を用いて、N−アセチルグルコサミンがαで結合したジメトキシトリアゾール (GlcNAc−α−DMT)またはその誘導体と糖受容体を反応させて、N−アセチルグルコサミンがαで結合した糖誘導体を調製する方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗ピロリ菌効果等を有する機能性オリゴ糖鎖として有用な、α結合型N−アセチルグルコサミン糖誘導体を、酵素的に調製する新規な方法に関する。
【背景技術】
【0002】
近年高等生物の胃や十二指腸腺粘液中のムチン型糖タンパク質糖鎖の非還元末端にN-アセチルグルコサミン(GlcNAc)がαでガラクトース(Gal)に結合したオリゴ糖鎖、GlcNAcα1→4Gal−Rが存在することが報告されている(非特許文献1)。この糖鎖は近年、胃癌や胃潰瘍の原因とされるヘリコバクターピロリ菌を殺菌もしくは増殖抑制する物質として注目されており(非特許文献2)、最近になって、GlcNAcがαで結合した種々の誘導体の抗ピロリ菌効果が報告されるようになった。
【0003】
また、最近、GlcNAcα1→4Gal−Rに関連する糖鎖に対するいくつかのモノクローナル抗体が商品化されたことから、GlcNAcがαで結合した糖鎖の新たな機能の発見が期待されている。これらの物質のうち、比較的高い抗ピロリ菌効果を有する物質は、少なくとも上記のようにGlcNAcがαでガラクトースに結合した物質であると考えられており、いくつかの化学的合成手法が試みられている(非特許文献3)。しかしながら、一般にGlcNAcがαで結合した化合物の調製において、特にそのグリコシル化における立体制御法が化学的合成法では未だに達成されておらず、多段階の誘導化なしに一度の反応において100%のα選択的グリコシド化が達成された例は報告されていない(非特許文献4)。さらに上記のように、目的のオリゴ糖鎖を化学的に調製するためには、複雑で多段階の合成手法や技術が必要であるために、いまだに大量調製化への目途はたっていない。
【0004】
また、上記の天然型糖鎖は、高等動物が有する糖転移酵素、α1,4−N−アセチルグルコサミニルトランスフェラーゼ(α4GnT)の働きによって生成することが知られていることから、α4GnTによる調製も試みられている(非特許文献5、特許文献1)。しかしながら、この方法は高価な基質である糖ヌクレオチドを使用し、かつα4GnTの調製においても、動物細胞等しか生産媒体にできないために、大量生産には向かない。さらにα4GnTはその糖受容体に対する構造特異性が比較的厳密であるために、GlcNAcをαで結合した新しい糖鎖およびその誘導体を調製するには不向きであると考えられる。
【0005】
一方、糖加水分解酵素によるオリゴ糖鎖の合成手法は、用いる糖供与体が比較的調製しやすく、酵素自体も微生物由来のものが多いために、目的とする物質の大量調製も視野に入れることができる。このような観点から、今までに多くの酵素的合成法が報告されてきた。しかしながら、GlcNAcをαで結合した糖鎖およびその誘導体の酵素的調製方法は今までに報告例がなく、本発明者ら自身も、多糖類(ヘパリン硫酸など)からGlcNAcを遊離する酵素として報告されているヒトのα−N-アセチルグルコサミニダーゼのアミノ酸配列に似た微生物由来の酵素ホモログ(これらのホモログはグリコシルハイドロラーゼファミリー89(GH89)に属する)を見出し、既存の糖供与体(パラニトロフェニル誘導体(GlcNAc−α−pNP)、およびメチルウンベリフェリル誘導体(GlcNAc−α−MU))を糖供与体とする (特許文献2)糖転移を試みてきたが、達成することはできなかった。したがって、新しい糖供与体と糖加水分解酵素の組み合わせによる調製方法が求められていた。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2001−46077号公報
【特許文献2】特開2009−232838号公報
【非特許文献】
【0007】
【非特許文献1】イシハラ ケー(Ishihara K)ら、バイオケミストリー ジャーナル(Biochemstry Journal)、1996年、第318巻、p409−416.
【非特許文献2】カワクボ エム(Kawakubo M)ら、サイエンス(Science)、2004年、第305巻、p1003−1006.
【非特許文献3】マナベ エス(Manabe S)ら、ジャーナル オブ オルガニック ケミストリー(Journal of Organic Chemistry)、2007年、第72巻、p6107−6115.
【非特許文献4】ボルナーニ エフ エル(Bornaghi F L)ら、テトラヘドロン レターズ(Tetrahedron Letters)、2005年、第46巻、p3485−3488.
【非特許文献5】ナカヤマ ジェー(Nakayama J)ら、プロシーディングズ オブ ザ ナショナル アカデミー オブ サイエンスィズ オブ ユーエスエー(Proceedings of the National Academy of Sciences of USA)、1999年、第96巻、p8991−8996.
【非特許文献6】タナカ ティー(Tanaka T)ら、ケミカル コミュニケーション、(Chemical Communication)、2008年、第17巻、p2016−2018.
【非特許文献7】ハンコック エス エム(Hancock S M)ら、カレント オピニオン イン ケミカル バイオロジー(Current Opinion in Chemical Biology)、2006年、第5巻、p509−519.
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は前記の課題を解決するためになされたもので、N−アセチルグルコサミンがαで結合した糖誘導体を、簡便にかつ大量に製造できる、新規な酵素的調製方法を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者らは、上記現状にかんがみ、まずGlcNAcがαで結合した糖鎖からGlcNAcを遊離する酵素の遺伝子と考えられるいくつかのDNA配列情報を基に、いくつかの組換えタンパク質を作製したところ、あるアミノ酸配列を持つタンパク質がGlcNAcα1→4Gal−Rの糖鎖をはじめとするいくつかのGlcNAcがαで結合した糖誘導体からGlcNAcを遊離する活性を持つことを見出していた(特許文献2)。しかしながら、既存の糖供与体(パラニトロフェニル誘導体、GlcNAc−α−pNP、およびメチルウンベリフェリル誘導体、GlcNAc−α−MU)で上記α−N−アセチルグルコサミニダーゼによる糖転移を試みたが、目的とする糖誘導体を得ることはできなかった。
【0010】
そこで、近年、糖加水分解酵素によりすみやかに加水分解されるDMT(ジメトキシトリアゾール)化糖(非特許文献6)に焦点を当て、N−アセチルグルコサミンがαで結合したジメトキシトリアゾール(GlcNAc−α−DMT。4,6-ジメトキシ−1,3,5−トリアジン−2−イル2−アセタミド−2−デオキシ−α−D−グルコピラノシド)に対するα−N−アセチルグルコサミニダーゼの水溶液中における挙動を調査したところ、上記の合成基質よりも、すみやかに加水分解されることをつきとめ、本発明の調製方法を完成するに至った。
【0011】
本発明は、水溶液中一段階で調製できるジメトキシトリアゾールをαで有するN−アセチルグルコサミン(GlcNAc−α−DMT)もしくはその誘導体を糖供与体とし、微生物由来の糖加水分解酵素の作用により、厳密にα選択的に糖受容体へ糖転移するというものである。本発明により、結果として抗ピロリ菌効果を有するオリゴ糖鎖(GlcNAcα1→4Gal−R)を特別な設備を必要とせずに簡単に調製することができる。また、用いる糖加水分解酵素により、位置特異性の異なるオリゴ糖鎖およびその誘導体を得ることもできる。
【0012】
以上のように、本法によって、糖転移酵素を使用せずに、はじめてN−アセチルグルコサミンをα選択的に一段階で糖転移させることができる。また糖加水分解酵素の糖受容体特異性の幅広さにより、糖転移酵素を用いた方法では成しえない多くの種類の化合物をαグルコサミニル化できることになる。
【0013】
すなわち、本発明は、以下の調製方法を提供するものである。
[1] N−アセチルグルコサミン誘導体と、そのアノマー位にグリコシド結合しうる糖受容体とを糖加水分解酵素により反応させて、N−アセチルグルコサミンがαグリコシド結合した糖誘導体を選択的に調製する方法。
[2] N−アセチルグルコサミン誘導体が、(化1)の式で表されるGlcNAc−α−DMTまたはその誘導体である[1]に記載の方法。
【化1】

(式中、XはO、C、S、Nまたはなくてもよく、RはH、置換または非置換アルキル、もしくは置換または非置換ヘテロアルキルを表す。)
[3] 糖加水分解酵素が、グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼである[1]に記載の調製方法。
[4] 糖加水分解酵素が、バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来のα−N−アセチルグルコサミニダーゼ1、2または3である[1]に記載の調製方法。
[5] そのアノマー位にグリコシド結合しうる糖受容体が、アルコール類、ガラクトース誘導体、またはガラクトースを含む糖の誘導体である[1]に記載の調製方法。
【発明の効果】
【0014】
本発明により、GlcNAcがαで結合した糖誘導体および糖鎖を、より簡単かつ大量に調製可能となる。特異的にピロリ菌増殖を抑制する化合物、およびそれを含み安全で、耐性菌を生じさせないピロリ菌増殖抑制剤の調製が可能となる。さらに、従来の方法では調製できなかった多くの種類の糖誘導体が調製可能となることから、より強い抗ピロリ剤を見出す可能性を有する。
【図面の簡単な説明】
【0015】
【図1】実施例1で得た反応溶液を分析した結果を示すHPLCチャート。
【図2】実施例2で得た反応溶液を分析した結果を示すHPLCチャート。
【図3】実施例3で得た反応溶液を分析した結果を示すHPLCチャート。
【図4】実施例4で得た反応溶液を分析した結果を示すHPLCチャート。
【発明を実施するための形態】
【0016】
本発明において用いる糖加水分解酵素は、バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来の3種のα−N−アセチルグルコサミニダーゼ1、2または3が特に好ましいが、グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼのホモログは上記以外の宿主にも多数見出すことができるので、それらも本法と同様に用いることができる。特に、ヒト、マウス、蛇由来のα−N−アセチルグルコサミニダーゼではGlcNAc−α−pNPだけでなくヘパリン構成糖であるGlcNAc−α1,4−IdoA−RやGlcNAc−α1,4−GlcA−Rにも加水分解活性があるので、同様に糖受容体としてイズロン酸誘導体(IdoA−R)やグルクロン酸誘導体(GlcA−R)も用いることができる。また、本発明において、酵素においては糖加水分解酵素や糖転移酵素が有する活性アミノ酸残基の変異体も用いることができる(非特許文献7)。これにより、生成したαグルコサミン化合物に対する加水分解を、野生型の糖加水分解酵素を用いた時に比べて、顕著に抑えることができ、結果的に目的物の収率向上させることができる。
【0017】
バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来の3種のα−N−アセチルグルコサミニダーゼ1、2または3は、特許文献2を参照してリコンビナント蛋白質として調製できる。グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼも、それぞれの出典の文献(ヒト由来のリコンビナント酵素調製(ウェバー ビー(Webber B)ら、プロテイン エクスプレッション アンド ピュアリフィケーション(Protein Expression and Purification)、2001年、第21巻2号、p251−259)、蛇毒からの調製(アンドリュー ジェー エヌ(Andrew J N)ら、ジャーナル オブ ケミカル アンド モレキュラー トキシコロジー(Journal of Biochemical and Molecular Toxicology)、2001年、第15巻4号、p221−227)を参照してリコンビナント蛋白質としてもしくは天然からの抽出物として調製できる。
【0018】
本発明において糖供与体として用いるN−アセチルグルコサミン誘導体とは、糖のアノマー位に脱離基を有するN−アセチルグルコサミン誘導体であり、上記[1]に記載の調製方法により、非還元末端にN−アセチルグルコサミンがα選択的に結合した化合物を与える。N−アセチルグルコサミン誘導体としてはGlcNAc−α−DMTが好ましいが、この化合物に限られない。用いる糖加水分解酵素によっては(糖加水分解酵素の活性アミノ酸残基等の変異体も含む)その他の脱離基を有するN−アセチルグルコサミン誘導体も糖供与体となりうるからである。
【0019】
前記[2]の(化1)の式に記載のXは、O、C、S、Nであればよく、なくてもトリアジン基として、酵素の糖供与体となりうる。Rはアルキルを示すが、「アルキル」という用語は、それ自体によってまたは別の置換基の一部として、特記しない限り、完全飽和、モノまたはポリ不飽和のいずれであってもよく、指定の数の炭素原子を有する(すなわち、C1−C10は1〜10炭素を意味する)二価または多価ラジカルを含んでいてもよい直鎖または分枝鎖、または環状炭化水素基、またはその組み合わせを意味する。飽和炭化水素基の例としては、限定はされないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、t−ブチル、イソブチル、sec−ブチル、シクロヘキシル、(シクロヘキシル)メチル、シクロプロピルメチルなどの基、たとえばn−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチルなどの同族体および異性体などが挙げられる。不飽和アルキル基は、1つ以上の二重結合または三重結合を有するものである。不飽和アルキル基の例としては、限定されないが、ビニル、2−プロペニル、クロチル、2−イソペンテニル、2−(ブダジエニル)、2,4−ペンタジエニル、3−(1,4−ペンタジエニル)、エチニル、1−および3−プロピニル、3−ブチニル、およびより高次の同族体および異性体が挙げられる。
【0020】
本発明において用いる、そのアノマー位にグリコシド結合しうる糖受容体とは、前記[5]に示すように、水酸基を有する化合物であり糖供与体と反応できるものであれば特に限定しないが、具体的にはメタノール、フェニルアルコールなどのアルコール類、ベンジル β−D−ガラクトピラノシド(Gal−β−OBn)やp−メトキシフェニル−β−D−ガラクトピラノシド(Gal−β−pMP)などのガラクトース誘導体、グルクロン酸やイズロン酸の誘導体などの例を挙げることができる。
【0021】
本発明において酵素を用いる糖転移反応の条件は、公知の条件を用いることができるが、具体的には以下のようになる。
溶液は、リン酸緩衝液、炭酸緩衝液などの例を挙げることができ、特にリン酸緩衝化生理食塩水(phosphate buffered saline,PBS)が好ましい。
溶液のpHは、中性付近が好ましく、pH6.0からpH8.0が特に好ましい。
反応の温度は、25℃から45℃が好ましく、32℃が特に好ましい。
反応の時間は、酵素濃度が数十から数百nMで数時間から24時間が好ましく、さらに酵素濃度800nMとした場合に、4から5時間程度が特に好ましい。しかしながら、酵素濃度が数十nM以下においては、反応時間が24時間以上で、比較的高い糖転移収率を得ることがある。
【0022】
反応容器においては、系内の温度を制御でき、かつ溶解しているタンパク質や糖質化合物を特に吸着させる容器でないかぎりどのようなものを用いてもよい。また、本発明において用いているリコンビナントタンパクや基質はいずれも室温から37℃付近では比較的安定であるために、反応において、振とうや撹拌操作を行えば、出発物質の分解もほとんどなく反応時間を短縮させることができる。
本発明で調製される糖誘導体には、糖鎖、糖蛋白質、糖脂質などが含まれる。
【0023】
糖供与体として用いる、GlcNAc−α−DMTは、次のようにして合成した。
N−アセチル−D−グルコサミン(GlcNAc)221mg(1.0mmol)を水6.25mlに溶解し、塩化4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウム(DMT−MM))553mg(2.0mmol)、次いで2,6−ルチジン0.23ml(2.0mmol)を室温で加え、反応溶液を室温で24時間撹拌した。(化2)式の反応が進行した。薄層クロマトグラフィー(TLC)で反応終了を確認後、減圧下溶媒を除去し、シリカゲルフラッシュカラムクロマトグラフィ(展開溶媒:酢酸エチル/メタノール=7/1)により精製して、4,6−ジメトキシ−1,3,5−トリアジン−2−イル2−アセタミド−2−デオキシ−α−D−グルコピラノシド(GlcNAc−α−DMT)288mg(0.80mmol、80%)を得た。
【化2】
以下に、本発明の調製方法(糖転移反応)を、実施例を挙げて具体的に説明するが、本発明を何ら限定するものではない。
【実施例1】
【0024】
糖供与体としてGlcNAc−α−DMT 1.8mg(5μmol、終濃度50mM)をPBS溶液に溶解し、糖受容体としてメタノール12.3μl(300μM、12.3vol.%)、およびBacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB2(4.95μg、0.495μg/μl、特許文献2参照)のPBS溶液を加え、37℃で22時間インキュベートした。(化3)式の反応が進行した。反応溶液をHPLCで分析(カラム:TSK−GelAmide−80(4.6×250mm,TOSOH), 溶離液:アセトニトリル/水=3/1, 流速:1ml/min, 温度:30℃, 検出:UV(214nm))したところ、メチル 2−アセタミド−2−デオキシ−α−D−グルコピラノシド (GlcNAc−α−OMe)が27%の収率で生成していることを確認した。その結果を図1に示す。
【化3】
【実施例2】
【0025】
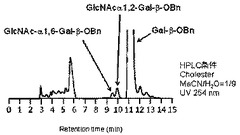
糖供与体としてGlcNAc−αDMT 1.8mg(5μmol、終濃度50mM)、糖受容体としてベンジル β−D−ガラクトピラノシド(Gal−β−OBn)9.5mg(35μM)をPBS溶液に溶解し、Bacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB2(4.95μg、0.495μg/μl、特許文献2参照)のPBS溶液を加え、37℃で6時間インキュベートした。(化4)式の反応が進行した。反応溶液をHPLCで分析(カラム:COSMOSIL Cholester(4.6×250mm,Nacalai Tesque), 溶離液:アセトニトリル/水=9/91, 流速:1ml/min, 温度:30℃, 検出:UV(254nm))したところ、ベンジル 6−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→6−Gal−β−OBn)、ベンジル 2−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→2−Gal−β−OBn)が、各々4%、8%の収率で生成していることを確認した。その結果を図2に示す。
【化4】
【実施例3】
【0026】
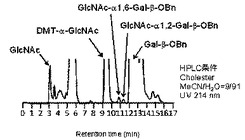
糖供与体としてGlcNAc−αDMT 0.36mg(1μmol、終濃度50mM)、糖受容体としてベンジル β−D−ガラクトピラノシド(Gal−β−OBn)1.9mg(7μmol)をPBS溶液に溶解し、Bacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB3(0.10μg、0.086μg/μl、特許文献2参照)のPBS溶液を加え、37℃で29時間インキュベートした。(化4)式の反応が進行した。反応溶液をHPLCで分析(カラム:COSMOSIL Cholester(4.6×250mm,Nakarai tesque), 溶離液:アセトニトリル/水=9/91, 流速:1ml/min, 温度:30℃, 検出:UV(254nm))したところ、ベンジル 6−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→6−Gal−β−OBn)、ベンジル 2−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→2−Gal−β−OBn)が、各々0.4%、0.3%の収率で生成していることを確認した。その結果を図3に示す。
【0027】
<生成物のNMR法による解析結果>
ベンジル 6−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→6−Gal−β−OBn)
1H NMR(500 MHz, CD3OD) : δ 7.47−7.30(5H, m, Ph), 4.95(1H, d, −CH2−, J = 12.0 Hz) , (1H, d, H−1’), 4.68(1H, d, −CH2−, J = 11.8 Hz), 4.38(1H, d, H−1, J = 7.6 Hz), 2.03(3H, s, −COCH3).
DEPT−135 NMR (126 MHz, CD3OD): δ 128.0−127.3(Ph), 102.6(C−1), 97.3(C−1’), 70.4(−CH2−), 66.4(C−6), 61.3(C−6’), 21.3(−COCH3).
MALDI−TOF MS ; m/z calcd for C21H31NO11[M+Na]+ : 496.5, Found : 496.8
【0028】
ベンジル 2−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→2−Gal−β−OBn)
1H NMR(500 MHz, CD3OD) : δ 7.47−7.29(5H, m, Ph), 5.32(1H, d, H−1’, J = 3.7 Hz), 5.03(1H, d, −CH2−, J = 11.4 Hz), 4.63(1H, d, −CH2−, J = 11.4 Hz), 4.60(1H, d, H−1, J = 7.8 Hz), 4.15(1H, ddd, H−5’, J = 2.1, 5.2, 10.1 Hz), 4.0(1H, dd, H−2’, J = 3.6, 10.7 Hz), 3.89−3.71(7H, m, H−2, H−4, H−6a, H−6b, H−3’, H−6’a, H−6’b), 3.64−3.53(2H, m, H−3, H−5), 3.42(1H, t, H−4’, J = 9.5 Hz), 1.83(3H, s, −COCH3).
DEPT−135 NMR (126 MHz, CD3OD): δ 128.1−127.3(Ph), 102.8(C−1), 97.5(C−1’), 76.2(C−2), 75.4(C−5), 72.5(C−3), 72.2(C−5’), 71.7(C−3’), 70.9(C−4’), 70.6(−CH2−), 69.3(C−4), 61.3(C−6’), 61.1(C−6), 53.8(C−2’), 21.3(−COCH3).
MALDI−TOF MS ; m/z calcd for C21H31NO11[M+Na]+ : 496.5, Found : 496.6
【実施例4】
【0029】
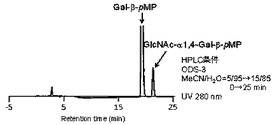
糖供与体としてGlcNAc−αDMT 0.22mg(0.6μmol、終濃度30mM)、糖受容体としてp−メトキシフェニル β−D−ガラクトピラノシド(Gal−β−pMP)0.17mg(0.6μmol)をPBS溶液に溶解し、Bacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB1(14.4μg、14.4μg/μl、特許文献2参照)のPBS溶液を加え、37℃で5時間インキュベートした。(化5)式の反応が進行した。反応溶液をHPLCで分析(カラム:Inertsil ODS−3(4.6×250mm,GL−Science), 溶離液:アセトニトリル/水=5/95→15/85(0→25min), 流速:1ml/min, 温度:30℃, 検出:UV(280nm))したところ、p−メトキシフェニル 4−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→4−Gal−β−pMP)が、10%の収率で生成していることを確認した。その結果を図4に示す。
【化5】
【0030】
<生成物のNMR法による解析結果>
p−メトキシフェニル−4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル−β−D−ガラクトピラノシド
1H NMR(500 MHz, CD3OD) : δ 7.08(2H, d, Ph, J = 9.1 Hz), 6.86(2H, d, Ph, J = 9.1 Hz), 4.95(1H, d, H−1’, J = 3.6 Hz), 4.84(1H, d, H−1, J = 7.6 Hz), 4.29(1H, ddd, H−5’, J = 2.5, 4.0, 9.9 Hz), 4.04(1H, d, H−4, J = 2.8 Hz), 3.97(1H, dd, H−2’, J = 3.6, 10.9 Hz), 3.83(1H, dd, H−6a’, J = 2.2, 11.8 Hz), 3.79−3.71(8H, m, H−2, H−5, H−6a, H−3’, H−6’b, −OCH3), 3.69−3.65(2H, m, H−3, H−6b), 3.46(1H, t, H−4’, J = 9.4, 9.5 Hz), 2.02(3H, s, −COCH3).
13C NMR(126 MHz, CD3OD): δ 173.8(−COCH3), 156.7, 153.0, 119.1, and 115.5(Ph), 104.0(C−1), 100.3(C−1’), 77.7(C−4), 76.8(C−5), 74.3(C−3), 73.7(C−5’), 72.6(C−3’), 72.4(C−2), 72.0(C−4’), 62.3(C−6’), 60.7(C−6), 56.1(−OCH3), 55.5(C−2’), 22.7(−COCH3).
【実施例5】
【0031】
GlcNAc−α−DMTを50mM、pMP−β−Gal 14mgを50mMになるようにpH6.5の100mMリン酸緩衝液に溶解し、37℃で上記と同様にα−N−アセチルグルコサミニダーゼB1のPBS溶液(酵素濃度0.8μM)を加え、反応を8時間行い、HPLCにより測定した。上記と同様に測定し、GlcNAc−α(1→4)−Gal−β−OpMPが収率52%で得られていることがわかった。
【実施例6】
【0032】
GlcNAc−α−DMT 45.0mg(0.13mmol、50mM)、メトキシβ−D−ガラクトピラノシド(Gal−β−O−Me 25.0mg(0.13mmol,50mM))をpH6.5、100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、α−N−アセチルグルコサミニダーゼB1のPBS溶液(2.88mg/ml,134μlを加え、全量2.5mlとして反応を開始した。(化6)式の反応1時間後に溶離液で5倍に希釈し、分取HPLCにより単離した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=4/1、流量を8ml/min、カラム温度を40℃、検出器をUV240nmとした。NMRにより構造決定を行ったところメトキシ−4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル−β−D−ガラクトピラノシド (GlcNAc−α(1→4)−Gal−β−O−Me)を得た。(収量;7.8mg,収率;16.5%)
【化6】
【0033】
<生成物のNMR法による解析結果>
メトキシ−4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル−β−D−ガラクトピラノシド
1H NMR (500 MHz, CD3OD) : δ 4.88(1H, d, H−1’, J = 3.7 Hz), 4.24(1H, ddd, H−5’, J = 2.5 Hz, 4.4 Hz, 10.2 Hz), 4.21(1H, d, H−1, J = 7.4 Hz), 3.97(1H, d, H−4, J = 3.0 Hz), 3.94(1H, dd, H−2’, J = 3.7 Hz, 10.8 Hz), 3.79(1H, dd, H−6’a, J = 2.6 Hz, 11.8 Hz), 3.74−3.58(5H, m, H−6’b, H−6a, H−3’, H−6b, H−5), 3.58(3H, s, −OCH3), 3.55(1H, dd, H−3, J = 3.0 Hz, 10.1 Hz), 3.50(1H, dd, H−2, J = 7.5 Hz, 10.1 Hz),3.43(1H, dd, H−4’, J = 9.0 Hz, 10.1 Hz), 2.03(3H, s, −COCH3).
13C NMR (126 MHz, CD3OD) : δ 173.7(−COCH3), 106.3(C−1), 100.3(C−1’), 77.6(C−4), 76.7(C−5), 74.4(C−3), 73.6(C−5’), 72.7(C−3’), 72.6(C−2), 72.0(C−4’), 62.3(C−6’), 60.8(C−6), 57.8(−OCH3), 55.5(C−2’), 22.7(−COCH3)
【実施例7】
【0034】
終濃度がGlcNAc−α−DMT 75mM、2−ピリジル−1−チオ−2−アセトアミド−2−デオキシ−4−O−(β−D−ガラクトピラノシル)−β−D−グルコピラノシド (LacNAc−β−Spy)25mMになるようpH6.5,100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、α−N−アセチルグルコサミニダーゼB1 PBS溶液(2.75mg/ml)3.6μlを加え、全量100μlとして反応を開始した。この反応を12サンプル用意し、反応3時間後、分取HPLCにより分取した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=3/1、流量を8ml/min、カラム温度を30℃、検出器をUV240nmとした。2−ピリジル−1−チオ−2−アセトアミド−2−デオキシ−4−O−(4−O−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド (GlcNAc−α(1→4)−LacNAc−β−Spy)8.2mg(収率39%)を得た。
【0035】
<生成物のNMR法による解析結果>
2−ピリジル−1−チオ−2−アセトアミド−2−デオキシ−4−O−(4−O−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド
1H NMR (400 MHz, CD3OD): δ 8.38, 7.68, 7,40, and 7.16(4H, Pyridyl), 5.45(1H, d, H−1, J = 10.7 Hz), 4.90(1H, d, H−1”, J = 3.7 Hz), 4.48(1H, d, H−1’, J = 7.4 Hz), 4.20(1H, ddd, H−5” , J = 2.4, 4.6, 7.8 Hz), 3.99(1H, dd, H−2, J = 9.6, 10.5 Hz), 3.95(1H, d, H−4’, J = 2.2 Hz), 3.93−3.89(3H, m, H−6a, H−6b, H−2”), 3.81(1H, dd, H−6”a, J = 2.5, 11.9 Hz), 3.78−3.54(10H, m, H−3, H−4, H−5, H−2’, H−3’, H−5’, H−6’a, H−6’b, H−3”, H−6”b), 3.42(1H, dd, H−4”, J = 8.9, 10.1 Hz), 2.01(3H, s, −COCH3), 1.95(3H, s, −COCH3).
13C NMR (101 MHz, CD3OD): δ 173.7 and 173.5(−COCH3), 158.9, 150.3, 138.5, 124.4, and 122.0(Pyridyl), 105.4(C−1’), 100.1(C−1”), 84.8(C−1), 80.9(C−5), 80.6(C−4), 78.1(C−4’), 77.2(C−5’), 75.7(C−3), 74.5(C−3’), 73.8(C−5”), 72.5(C−2’, C−3”), 72.1(C−4”), 62.4(C−6”), 61.8(C−6), 61.3(C−6’), 55.6(C−2”), 55.5(C−2), 22.9 and 22.7(−COCH3).
【実施例8】
【0036】
終濃度がGlcNAc−α−DMT 75mM、2−ピリジル−1−チオ−4−O−(4−O−β−D−ガラクトピラノシル)−β−D−グルコピラノシド (Lac−β−Spy)25mMになるようpH6.5,100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、α−N−アセチルグルコサミニダーゼB1 PBS溶液(2.75mg/ml)7.3μlを加え、全量100μlとして反応を行った。(化7)式の反応24時間後、分取HPLCにより分取した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=3/1、流量を8ml/min、カラム温度を30℃、検出器をUV240nmとした。2−ピリジル−1−チオ−4−O−(4−O−(2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド (GlcNAc−α(1→4)−Lac−β−Spy)4.3mg(収率45%)を得た。
【化7】
【0037】
<生成物のNMR法による解析結果>
2−ピリジル−1−チオ−4−O−(4−O−(2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド
1H NMR (500 MHz, CD3OD): δ 8.42, 7.73, 7,49, and 7.21(4H, Pyridyl), 5.29(1H, d, H−1, J = 10.7 Hz), 4.93(1H, d, H−1”, J = 3.7 Hz), 4.49(1H, d, H−1’, J = 7.4 Hz), 4.24(1H, m, H−5”), 3.98(1H, d, H−4, J = 1.9 Hz), 3.94(1H, dd, H−2”, J = 3.4, 11.2 Hz), 3.92(2H, d, H−6a, H−6b, J = 2.7 Hz), 3.84(1H, dd, H−6”a, J = 1.9, 11.8 Hz), 3.77−3.59(10H, m, H−3, H−4, H−5, H−2’, H−3’, H−5’, H−6’a, H−6’b, H−3”, H−6”b), 3.49−3.44(2H, m, H−2, H−4”), 2.05(3H, s, −COCH3).
13C NMR (126 MHz, CD3OD): δ 172.4(−COCH3), 157.8, 148.9, 137.3, 123.2, and 120.7(Pyridyl), 104.0(C−1’), 98.6(C−1”), 84.6(C−1), 79.3(C−5), 78.7(C−4), 76.8(C−3), 76.6(C−4’), 75.8(C−5’), 73.1(C−3’), 72.3(C−2, C−5”), 71.1(C−2’, C−3”), 70.1(C−4”), 61.0(C−6”), 60.4(C−6), 59.9(C−6’), 54.3(C−2”), 21.3(−COCH3).
FAB−MS ; m/z calcd for C25H38N2O15S[M+H]+ : 639.2071, Found : 639.2075
【実施例9】
【0038】
終濃度がGlcNAc−α−DMT 50mM、GNB−α−DMT 25mMになるようpH6.5、100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、GlcNAcase B1 PBS溶液(2.75mg/ml)5.5μlを加え、全量100μlとして反応を開始した。(化8)式の反応を16サンプル用意し、反応4時間後、分取HPLCにより分取した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=3/1、流量を8ml/min、カラム温度を30℃、検出器をUV214nmとした。4,6ジメトキシ−1,3,5−トリアジン−2−イル2−アセトアミド−2−デオキシ−3−O−(4−O−(2−アセトアミド−2デオキシα−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α(1→4)−GNB−α−O−DMT)12.7mg(収率44%)を得た。
【0039】
<生成物のNMR法による解析結果>
4,6ジメトキシ−1,3,5−トリアジン−2−イル2−アセトアミド−2−デオキシ−3−O−(4−O−(2−アセトアミド−2デオキシα−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−ガラクトピラノシド
1H NMR (400 MHz, CD3OD): δ 6.63(1H, d, H−1, J = 3.72 Hz), 4.93(1H, d, H−1”, J = 3.7 Hz), 4.66(1H, dd, H−2, J = 3.7, 11.3 Hz), 4.56(1H, d, H−1’, J = 7.1 Hz), 4.27(1H, d, H−4, J = 2.1 Hz), 4.20−4.16(2H, m, H−3, H−5”), 4.02−4.00(7H, m, −OCH3, H−5), 3.97(1H, d, H−4’, J = 2.8 Hz), 3.94(1H, dd, H−2”, J = 3.7, 10.9 Hz), 3.81(1H, d, H−6”a, J = 2.3 11.8 Hz), 3.76−3.54(9H, m, H−6a, H−6b, H−2’, H−3’, H−5’, H−6’a, H−6’b, H−3”, H−6”b), 3.42(1H, dd, H−4”, J = 8.9, 10.0 Hz), 2.02(3H, s, −COCH3), 1.92(3H, s, −COCH3).
13C NMR (101 MHz, CD3OD): δ 175.1, 174.3, 173.8, and 173.7(triazine, −COCH3), 106.1(C−1’), 100.1(C−1”), 96.4(C−1), 78.4(C−3), 78.1(C−4’), 76.8(C−5’), 74.8(C−5), 74.5(C−3’), 73.8(C−5”), 72.6(C−3”), 72.5(C−2’), 72.1(C−4”), 69.5(C−4), 62.5(C−6), 62.4(C−6”), 61.3(C−6’), 56.1(−OCH3), 55.6(C−2”), 49.6−48.4(C−2 in peak of CHD2OD), 22.7 and 22.6(−COCH3).
【化8】
【実施例10】
【0040】
GlcNAc−α−DMT 39.6mg(0.11mmol,50mM)、イソプロピル−1−チオ−β−Gal−β−D−ガラクトピラノシド (Gal−β−SiPr)79.2mg(0.33mmol、150mM)をpH6.5、100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、GlcNAcase B1 PBS溶液(2.88mg/ml)118μlを加え、全量2.2mlとして反応を開始した。(化9)式の反応1時間後に溶離液で5倍に希釈し、分取HPLCにより単離した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=4/1、流量を8ml/min、カラム温度を40℃とし、UV214nmにおける紫外吸収を測定した。NMRにより構造決定を行ったところイソプロピル−1−チオ−β−(4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシドを得た。(収量;14mg、収率;28.6%)
【化9】
【0041】
<生成物のNMR法による解析結果>
イソプロピル−1−チオ―β―4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド
1H NMR (500 MHz, CD3OD) : δ 4.89(1H, d, H−1’, J = 3.7 Hz), 4.51(1H, d, H−1, J = 9.6 Hz), 4.22(1H, ddd, H−5’, J = 2.5, 4.4, 10.1 Hz), 4.01(1H, d, H−4, J = 3.0 Hz), 3.95(1H, dd, H−2’, J = 3.7, 10.8 Hz), 3.79(1H, dd, H−6’a, J = 2.5, 11.8 Hz), 3.73(1H, dd, H−6’b, J = 4.5, 11.8 Hz), 3.70−3.59(4H, m, H−3’, H−5, H−6a, H−6b), 3.56(1H, dd, H−3, J = 3.0, 9.6 Hz), 3.48(1H, t, H−2, J = 9.6 Hz), 3.43(1H, dd, H−4’, J = 9.0, 10.1 Hz), 3.25(1H, sept, isopropyl, J = 6.8 Hz), 2.03(3H, s, −COCH3) 1.34(3H, d, isopropyl, J = 6.8 Hz), 1.33(3H, d, isopropyl, J = 6.8 Hz),.
13C NMR (126 MHz, CD3OD) : δ 173.7(−COCH3), 100.53(C−1’), 87.3(C−1), 80.5(C−5), 78.3(C−4), 75.9(C−3), 73.6(C−5’), 72.7(C−3’), 72.0(C−4’), 71.7(C−2), 62.3(C−6’), 60.8(C−6), 55.5(C−2’), 36.2(isopropyl), 24.4, 24.1(isopropyl), 22.7(−COCH3).
【産業上の利用可能性】
【0042】
N−アセチルグルコサミン(GlcNAc)がαでガラクトース(Gal)に結合したオリゴ糖鎖(αGlcNAc含有オリゴ糖鎖)は、従来の抗生物質とは全く異なり、あらゆるピロリ菌の生育に必須の増殖活動を抑制するという機序でピロリ菌に対する抗菌作用を示すから、抗ピロリ菌剤として有用である。また、本発明によりαGlcNAc含有オリゴ糖鎖を、人工高分子担体だけでなく、クラゲや卵白などの容易に入手可能な天然のムチン型糖蛋白質糖鎖へも導入可能であることが明らかである。
【0043】
したがって、調製した物質は、ピロリ菌増殖抑制剤として、サプリメントや飲食品添加物として有用であると考えられる。またそのピロリ菌増殖抑制剤を含有する飲食品は、機能性飲食品や健康飲食品として有用である。そのピロリ菌増殖抑制剤を含有する医薬製剤は、ピロリ菌に起因する消化器系疾患、特に胃炎、胃潰瘍、十二指腸潰瘍のような胃疾患を軽減したり治癒したり予防したりする医薬品として、有用である。
【技術分野】
【0001】
本発明は、抗ピロリ菌効果等を有する機能性オリゴ糖鎖として有用な、α結合型N−アセチルグルコサミン糖誘導体を、酵素的に調製する新規な方法に関する。
【背景技術】
【0002】
近年高等生物の胃や十二指腸腺粘液中のムチン型糖タンパク質糖鎖の非還元末端にN-アセチルグルコサミン(GlcNAc)がαでガラクトース(Gal)に結合したオリゴ糖鎖、GlcNAcα1→4Gal−Rが存在することが報告されている(非特許文献1)。この糖鎖は近年、胃癌や胃潰瘍の原因とされるヘリコバクターピロリ菌を殺菌もしくは増殖抑制する物質として注目されており(非特許文献2)、最近になって、GlcNAcがαで結合した種々の誘導体の抗ピロリ菌効果が報告されるようになった。
【0003】
また、最近、GlcNAcα1→4Gal−Rに関連する糖鎖に対するいくつかのモノクローナル抗体が商品化されたことから、GlcNAcがαで結合した糖鎖の新たな機能の発見が期待されている。これらの物質のうち、比較的高い抗ピロリ菌効果を有する物質は、少なくとも上記のようにGlcNAcがαでガラクトースに結合した物質であると考えられており、いくつかの化学的合成手法が試みられている(非特許文献3)。しかしながら、一般にGlcNAcがαで結合した化合物の調製において、特にそのグリコシル化における立体制御法が化学的合成法では未だに達成されておらず、多段階の誘導化なしに一度の反応において100%のα選択的グリコシド化が達成された例は報告されていない(非特許文献4)。さらに上記のように、目的のオリゴ糖鎖を化学的に調製するためには、複雑で多段階の合成手法や技術が必要であるために、いまだに大量調製化への目途はたっていない。
【0004】
また、上記の天然型糖鎖は、高等動物が有する糖転移酵素、α1,4−N−アセチルグルコサミニルトランスフェラーゼ(α4GnT)の働きによって生成することが知られていることから、α4GnTによる調製も試みられている(非特許文献5、特許文献1)。しかしながら、この方法は高価な基質である糖ヌクレオチドを使用し、かつα4GnTの調製においても、動物細胞等しか生産媒体にできないために、大量生産には向かない。さらにα4GnTはその糖受容体に対する構造特異性が比較的厳密であるために、GlcNAcをαで結合した新しい糖鎖およびその誘導体を調製するには不向きであると考えられる。
【0005】
一方、糖加水分解酵素によるオリゴ糖鎖の合成手法は、用いる糖供与体が比較的調製しやすく、酵素自体も微生物由来のものが多いために、目的とする物質の大量調製も視野に入れることができる。このような観点から、今までに多くの酵素的合成法が報告されてきた。しかしながら、GlcNAcをαで結合した糖鎖およびその誘導体の酵素的調製方法は今までに報告例がなく、本発明者ら自身も、多糖類(ヘパリン硫酸など)からGlcNAcを遊離する酵素として報告されているヒトのα−N-アセチルグルコサミニダーゼのアミノ酸配列に似た微生物由来の酵素ホモログ(これらのホモログはグリコシルハイドロラーゼファミリー89(GH89)に属する)を見出し、既存の糖供与体(パラニトロフェニル誘導体(GlcNAc−α−pNP)、およびメチルウンベリフェリル誘導体(GlcNAc−α−MU))を糖供与体とする (特許文献2)糖転移を試みてきたが、達成することはできなかった。したがって、新しい糖供与体と糖加水分解酵素の組み合わせによる調製方法が求められていた。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】特開2001−46077号公報
【特許文献2】特開2009−232838号公報
【非特許文献】
【0007】
【非特許文献1】イシハラ ケー(Ishihara K)ら、バイオケミストリー ジャーナル(Biochemstry Journal)、1996年、第318巻、p409−416.
【非特許文献2】カワクボ エム(Kawakubo M)ら、サイエンス(Science)、2004年、第305巻、p1003−1006.
【非特許文献3】マナベ エス(Manabe S)ら、ジャーナル オブ オルガニック ケミストリー(Journal of Organic Chemistry)、2007年、第72巻、p6107−6115.
【非特許文献4】ボルナーニ エフ エル(Bornaghi F L)ら、テトラヘドロン レターズ(Tetrahedron Letters)、2005年、第46巻、p3485−3488.
【非特許文献5】ナカヤマ ジェー(Nakayama J)ら、プロシーディングズ オブ ザ ナショナル アカデミー オブ サイエンスィズ オブ ユーエスエー(Proceedings of the National Academy of Sciences of USA)、1999年、第96巻、p8991−8996.
【非特許文献6】タナカ ティー(Tanaka T)ら、ケミカル コミュニケーション、(Chemical Communication)、2008年、第17巻、p2016−2018.
【非特許文献7】ハンコック エス エム(Hancock S M)ら、カレント オピニオン イン ケミカル バイオロジー(Current Opinion in Chemical Biology)、2006年、第5巻、p509−519.
【発明の概要】
【発明が解決しようとする課題】
【0008】
本発明は前記の課題を解決するためになされたもので、N−アセチルグルコサミンがαで結合した糖誘導体を、簡便にかつ大量に製造できる、新規な酵素的調製方法を提供することを目的とする。
【課題を解決するための手段】
【0009】
本発明者らは、上記現状にかんがみ、まずGlcNAcがαで結合した糖鎖からGlcNAcを遊離する酵素の遺伝子と考えられるいくつかのDNA配列情報を基に、いくつかの組換えタンパク質を作製したところ、あるアミノ酸配列を持つタンパク質がGlcNAcα1→4Gal−Rの糖鎖をはじめとするいくつかのGlcNAcがαで結合した糖誘導体からGlcNAcを遊離する活性を持つことを見出していた(特許文献2)。しかしながら、既存の糖供与体(パラニトロフェニル誘導体、GlcNAc−α−pNP、およびメチルウンベリフェリル誘導体、GlcNAc−α−MU)で上記α−N−アセチルグルコサミニダーゼによる糖転移を試みたが、目的とする糖誘導体を得ることはできなかった。
【0010】
そこで、近年、糖加水分解酵素によりすみやかに加水分解されるDMT(ジメトキシトリアゾール)化糖(非特許文献6)に焦点を当て、N−アセチルグルコサミンがαで結合したジメトキシトリアゾール(GlcNAc−α−DMT。4,6-ジメトキシ−1,3,5−トリアジン−2−イル2−アセタミド−2−デオキシ−α−D−グルコピラノシド)に対するα−N−アセチルグルコサミニダーゼの水溶液中における挙動を調査したところ、上記の合成基質よりも、すみやかに加水分解されることをつきとめ、本発明の調製方法を完成するに至った。
【0011】
本発明は、水溶液中一段階で調製できるジメトキシトリアゾールをαで有するN−アセチルグルコサミン(GlcNAc−α−DMT)もしくはその誘導体を糖供与体とし、微生物由来の糖加水分解酵素の作用により、厳密にα選択的に糖受容体へ糖転移するというものである。本発明により、結果として抗ピロリ菌効果を有するオリゴ糖鎖(GlcNAcα1→4Gal−R)を特別な設備を必要とせずに簡単に調製することができる。また、用いる糖加水分解酵素により、位置特異性の異なるオリゴ糖鎖およびその誘導体を得ることもできる。
【0012】
以上のように、本法によって、糖転移酵素を使用せずに、はじめてN−アセチルグルコサミンをα選択的に一段階で糖転移させることができる。また糖加水分解酵素の糖受容体特異性の幅広さにより、糖転移酵素を用いた方法では成しえない多くの種類の化合物をαグルコサミニル化できることになる。
【0013】
すなわち、本発明は、以下の調製方法を提供するものである。
[1] N−アセチルグルコサミン誘導体と、そのアノマー位にグリコシド結合しうる糖受容体とを糖加水分解酵素により反応させて、N−アセチルグルコサミンがαグリコシド結合した糖誘導体を選択的に調製する方法。
[2] N−アセチルグルコサミン誘導体が、(化1)の式で表されるGlcNAc−α−DMTまたはその誘導体である[1]に記載の方法。
【化1】
(式中、XはO、C、S、Nまたはなくてもよく、RはH、置換または非置換アルキル、もしくは置換または非置換ヘテロアルキルを表す。)
[3] 糖加水分解酵素が、グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼである[1]に記載の調製方法。
[4] 糖加水分解酵素が、バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来のα−N−アセチルグルコサミニダーゼ1、2または3である[1]に記載の調製方法。
[5] そのアノマー位にグリコシド結合しうる糖受容体が、アルコール類、ガラクトース誘導体、またはガラクトースを含む糖の誘導体である[1]に記載の調製方法。
【発明の効果】
【0014】
本発明により、GlcNAcがαで結合した糖誘導体および糖鎖を、より簡単かつ大量に調製可能となる。特異的にピロリ菌増殖を抑制する化合物、およびそれを含み安全で、耐性菌を生じさせないピロリ菌増殖抑制剤の調製が可能となる。さらに、従来の方法では調製できなかった多くの種類の糖誘導体が調製可能となることから、より強い抗ピロリ剤を見出す可能性を有する。
【図面の簡単な説明】
【0015】
【図1】実施例1で得た反応溶液を分析した結果を示すHPLCチャート。
【図2】実施例2で得た反応溶液を分析した結果を示すHPLCチャート。
【図3】実施例3で得た反応溶液を分析した結果を示すHPLCチャート。
【図4】実施例4で得た反応溶液を分析した結果を示すHPLCチャート。
【発明を実施するための形態】
【0016】
本発明において用いる糖加水分解酵素は、バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来の3種のα−N−アセチルグルコサミニダーゼ1、2または3が特に好ましいが、グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼのホモログは上記以外の宿主にも多数見出すことができるので、それらも本法と同様に用いることができる。特に、ヒト、マウス、蛇由来のα−N−アセチルグルコサミニダーゼではGlcNAc−α−pNPだけでなくヘパリン構成糖であるGlcNAc−α1,4−IdoA−RやGlcNAc−α1,4−GlcA−Rにも加水分解活性があるので、同様に糖受容体としてイズロン酸誘導体(IdoA−R)やグルクロン酸誘導体(GlcA−R)も用いることができる。また、本発明において、酵素においては糖加水分解酵素や糖転移酵素が有する活性アミノ酸残基の変異体も用いることができる(非特許文献7)。これにより、生成したαグルコサミン化合物に対する加水分解を、野生型の糖加水分解酵素を用いた時に比べて、顕著に抑えることができ、結果的に目的物の収率向上させることができる。
【0017】
バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来の3種のα−N−アセチルグルコサミニダーゼ1、2または3は、特許文献2を参照してリコンビナント蛋白質として調製できる。グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼも、それぞれの出典の文献(ヒト由来のリコンビナント酵素調製(ウェバー ビー(Webber B)ら、プロテイン エクスプレッション アンド ピュアリフィケーション(Protein Expression and Purification)、2001年、第21巻2号、p251−259)、蛇毒からの調製(アンドリュー ジェー エヌ(Andrew J N)ら、ジャーナル オブ ケミカル アンド モレキュラー トキシコロジー(Journal of Biochemical and Molecular Toxicology)、2001年、第15巻4号、p221−227)を参照してリコンビナント蛋白質としてもしくは天然からの抽出物として調製できる。
【0018】
本発明において糖供与体として用いるN−アセチルグルコサミン誘導体とは、糖のアノマー位に脱離基を有するN−アセチルグルコサミン誘導体であり、上記[1]に記載の調製方法により、非還元末端にN−アセチルグルコサミンがα選択的に結合した化合物を与える。N−アセチルグルコサミン誘導体としてはGlcNAc−α−DMTが好ましいが、この化合物に限られない。用いる糖加水分解酵素によっては(糖加水分解酵素の活性アミノ酸残基等の変異体も含む)その他の脱離基を有するN−アセチルグルコサミン誘導体も糖供与体となりうるからである。
【0019】
前記[2]の(化1)の式に記載のXは、O、C、S、Nであればよく、なくてもトリアジン基として、酵素の糖供与体となりうる。Rはアルキルを示すが、「アルキル」という用語は、それ自体によってまたは別の置換基の一部として、特記しない限り、完全飽和、モノまたはポリ不飽和のいずれであってもよく、指定の数の炭素原子を有する(すなわち、C1−C10は1〜10炭素を意味する)二価または多価ラジカルを含んでいてもよい直鎖または分枝鎖、または環状炭化水素基、またはその組み合わせを意味する。飽和炭化水素基の例としては、限定はされないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、t−ブチル、イソブチル、sec−ブチル、シクロヘキシル、(シクロヘキシル)メチル、シクロプロピルメチルなどの基、たとえばn−ペンチル、n−ヘキシル、n−ヘプチル、n−オクチルなどの同族体および異性体などが挙げられる。不飽和アルキル基は、1つ以上の二重結合または三重結合を有するものである。不飽和アルキル基の例としては、限定されないが、ビニル、2−プロペニル、クロチル、2−イソペンテニル、2−(ブダジエニル)、2,4−ペンタジエニル、3−(1,4−ペンタジエニル)、エチニル、1−および3−プロピニル、3−ブチニル、およびより高次の同族体および異性体が挙げられる。
【0020】
本発明において用いる、そのアノマー位にグリコシド結合しうる糖受容体とは、前記[5]に示すように、水酸基を有する化合物であり糖供与体と反応できるものであれば特に限定しないが、具体的にはメタノール、フェニルアルコールなどのアルコール類、ベンジル β−D−ガラクトピラノシド(Gal−β−OBn)やp−メトキシフェニル−β−D−ガラクトピラノシド(Gal−β−pMP)などのガラクトース誘導体、グルクロン酸やイズロン酸の誘導体などの例を挙げることができる。
【0021】
本発明において酵素を用いる糖転移反応の条件は、公知の条件を用いることができるが、具体的には以下のようになる。
溶液は、リン酸緩衝液、炭酸緩衝液などの例を挙げることができ、特にリン酸緩衝化生理食塩水(phosphate buffered saline,PBS)が好ましい。
溶液のpHは、中性付近が好ましく、pH6.0からpH8.0が特に好ましい。
反応の温度は、25℃から45℃が好ましく、32℃が特に好ましい。
反応の時間は、酵素濃度が数十から数百nMで数時間から24時間が好ましく、さらに酵素濃度800nMとした場合に、4から5時間程度が特に好ましい。しかしながら、酵素濃度が数十nM以下においては、反応時間が24時間以上で、比較的高い糖転移収率を得ることがある。
【0022】
反応容器においては、系内の温度を制御でき、かつ溶解しているタンパク質や糖質化合物を特に吸着させる容器でないかぎりどのようなものを用いてもよい。また、本発明において用いているリコンビナントタンパクや基質はいずれも室温から37℃付近では比較的安定であるために、反応において、振とうや撹拌操作を行えば、出発物質の分解もほとんどなく反応時間を短縮させることができる。
本発明で調製される糖誘導体には、糖鎖、糖蛋白質、糖脂質などが含まれる。
【0023】
糖供与体として用いる、GlcNAc−α−DMTは、次のようにして合成した。
N−アセチル−D−グルコサミン(GlcNAc)221mg(1.0mmol)を水6.25mlに溶解し、塩化4−(4,6−ジメトキシ−1,3,5−トリアジン−2−イル)−4−メチルモルホリニウム(DMT−MM))553mg(2.0mmol)、次いで2,6−ルチジン0.23ml(2.0mmol)を室温で加え、反応溶液を室温で24時間撹拌した。(化2)式の反応が進行した。薄層クロマトグラフィー(TLC)で反応終了を確認後、減圧下溶媒を除去し、シリカゲルフラッシュカラムクロマトグラフィ(展開溶媒:酢酸エチル/メタノール=7/1)により精製して、4,6−ジメトキシ−1,3,5−トリアジン−2−イル2−アセタミド−2−デオキシ−α−D−グルコピラノシド(GlcNAc−α−DMT)288mg(0.80mmol、80%)を得た。
【化2】
以下に、本発明の調製方法(糖転移反応)を、実施例を挙げて具体的に説明するが、本発明を何ら限定するものではない。
【実施例1】
【0024】
糖供与体としてGlcNAc−α−DMT 1.8mg(5μmol、終濃度50mM)をPBS溶液に溶解し、糖受容体としてメタノール12.3μl(300μM、12.3vol.%)、およびBacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB2(4.95μg、0.495μg/μl、特許文献2参照)のPBS溶液を加え、37℃で22時間インキュベートした。(化3)式の反応が進行した。反応溶液をHPLCで分析(カラム:TSK−GelAmide−80(4.6×250mm,TOSOH), 溶離液:アセトニトリル/水=3/1, 流速:1ml/min, 温度:30℃, 検出:UV(214nm))したところ、メチル 2−アセタミド−2−デオキシ−α−D−グルコピラノシド (GlcNAc−α−OMe)が27%の収率で生成していることを確認した。その結果を図1に示す。
【化3】
【実施例2】
【0025】
糖供与体としてGlcNAc−αDMT 1.8mg(5μmol、終濃度50mM)、糖受容体としてベンジル β−D−ガラクトピラノシド(Gal−β−OBn)9.5mg(35μM)をPBS溶液に溶解し、Bacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB2(4.95μg、0.495μg/μl、特許文献2参照)のPBS溶液を加え、37℃で6時間インキュベートした。(化4)式の反応が進行した。反応溶液をHPLCで分析(カラム:COSMOSIL Cholester(4.6×250mm,Nacalai Tesque), 溶離液:アセトニトリル/水=9/91, 流速:1ml/min, 温度:30℃, 検出:UV(254nm))したところ、ベンジル 6−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→6−Gal−β−OBn)、ベンジル 2−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→2−Gal−β−OBn)が、各々4%、8%の収率で生成していることを確認した。その結果を図2に示す。
【化4】
【実施例3】
【0026】
糖供与体としてGlcNAc−αDMT 0.36mg(1μmol、終濃度50mM)、糖受容体としてベンジル β−D−ガラクトピラノシド(Gal−β−OBn)1.9mg(7μmol)をPBS溶液に溶解し、Bacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB3(0.10μg、0.086μg/μl、特許文献2参照)のPBS溶液を加え、37℃で29時間インキュベートした。(化4)式の反応が進行した。反応溶液をHPLCで分析(カラム:COSMOSIL Cholester(4.6×250mm,Nakarai tesque), 溶離液:アセトニトリル/水=9/91, 流速:1ml/min, 温度:30℃, 検出:UV(254nm))したところ、ベンジル 6−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→6−Gal−β−OBn)、ベンジル 2−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→2−Gal−β−OBn)が、各々0.4%、0.3%の収率で生成していることを確認した。その結果を図3に示す。
【0027】
<生成物のNMR法による解析結果>
ベンジル 6−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→6−Gal−β−OBn)
1H NMR(500 MHz, CD3OD) : δ 7.47−7.30(5H, m, Ph), 4.95(1H, d, −CH2−, J = 12.0 Hz) , (1H, d, H−1’), 4.68(1H, d, −CH2−, J = 11.8 Hz), 4.38(1H, d, H−1, J = 7.6 Hz), 2.03(3H, s, −COCH3).
DEPT−135 NMR (126 MHz, CD3OD): δ 128.0−127.3(Ph), 102.6(C−1), 97.3(C−1’), 70.4(−CH2−), 66.4(C−6), 61.3(C−6’), 21.3(−COCH3).
MALDI−TOF MS ; m/z calcd for C21H31NO11[M+Na]+ : 496.5, Found : 496.8
【0028】
ベンジル 2−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→2−Gal−β−OBn)
1H NMR(500 MHz, CD3OD) : δ 7.47−7.29(5H, m, Ph), 5.32(1H, d, H−1’, J = 3.7 Hz), 5.03(1H, d, −CH2−, J = 11.4 Hz), 4.63(1H, d, −CH2−, J = 11.4 Hz), 4.60(1H, d, H−1, J = 7.8 Hz), 4.15(1H, ddd, H−5’, J = 2.1, 5.2, 10.1 Hz), 4.0(1H, dd, H−2’, J = 3.6, 10.7 Hz), 3.89−3.71(7H, m, H−2, H−4, H−6a, H−6b, H−3’, H−6’a, H−6’b), 3.64−3.53(2H, m, H−3, H−5), 3.42(1H, t, H−4’, J = 9.5 Hz), 1.83(3H, s, −COCH3).
DEPT−135 NMR (126 MHz, CD3OD): δ 128.1−127.3(Ph), 102.8(C−1), 97.5(C−1’), 76.2(C−2), 75.4(C−5), 72.5(C−3), 72.2(C−5’), 71.7(C−3’), 70.9(C−4’), 70.6(−CH2−), 69.3(C−4), 61.3(C−6’), 61.1(C−6), 53.8(C−2’), 21.3(−COCH3).
MALDI−TOF MS ; m/z calcd for C21H31NO11[M+Na]+ : 496.5, Found : 496.6
【実施例4】
【0029】
糖供与体としてGlcNAc−αDMT 0.22mg(0.6μmol、終濃度30mM)、糖受容体としてp−メトキシフェニル β−D−ガラクトピラノシド(Gal−β−pMP)0.17mg(0.6μmol)をPBS溶液に溶解し、Bacteroides thetaiotaomicron VPI5482由来α−N−アセチルグルコサミニダーゼB1(14.4μg、14.4μg/μl、特許文献2参照)のPBS溶液を加え、37℃で5時間インキュベートした。(化5)式の反応が進行した。反応溶液をHPLCで分析(カラム:Inertsil ODS−3(4.6×250mm,GL−Science), 溶離液:アセトニトリル/水=5/95→15/85(0→25min), 流速:1ml/min, 温度:30℃, 検出:UV(280nm))したところ、p−メトキシフェニル 4−O−(2−アセタミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α1→4−Gal−β−pMP)が、10%の収率で生成していることを確認した。その結果を図4に示す。
【化5】
【0030】
<生成物のNMR法による解析結果>
p−メトキシフェニル−4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル−β−D−ガラクトピラノシド
1H NMR(500 MHz, CD3OD) : δ 7.08(2H, d, Ph, J = 9.1 Hz), 6.86(2H, d, Ph, J = 9.1 Hz), 4.95(1H, d, H−1’, J = 3.6 Hz), 4.84(1H, d, H−1, J = 7.6 Hz), 4.29(1H, ddd, H−5’, J = 2.5, 4.0, 9.9 Hz), 4.04(1H, d, H−4, J = 2.8 Hz), 3.97(1H, dd, H−2’, J = 3.6, 10.9 Hz), 3.83(1H, dd, H−6a’, J = 2.2, 11.8 Hz), 3.79−3.71(8H, m, H−2, H−5, H−6a, H−3’, H−6’b, −OCH3), 3.69−3.65(2H, m, H−3, H−6b), 3.46(1H, t, H−4’, J = 9.4, 9.5 Hz), 2.02(3H, s, −COCH3).
13C NMR(126 MHz, CD3OD): δ 173.8(−COCH3), 156.7, 153.0, 119.1, and 115.5(Ph), 104.0(C−1), 100.3(C−1’), 77.7(C−4), 76.8(C−5), 74.3(C−3), 73.7(C−5’), 72.6(C−3’), 72.4(C−2), 72.0(C−4’), 62.3(C−6’), 60.7(C−6), 56.1(−OCH3), 55.5(C−2’), 22.7(−COCH3).
【実施例5】
【0031】
GlcNAc−α−DMTを50mM、pMP−β−Gal 14mgを50mMになるようにpH6.5の100mMリン酸緩衝液に溶解し、37℃で上記と同様にα−N−アセチルグルコサミニダーゼB1のPBS溶液(酵素濃度0.8μM)を加え、反応を8時間行い、HPLCにより測定した。上記と同様に測定し、GlcNAc−α(1→4)−Gal−β−OpMPが収率52%で得られていることがわかった。
【実施例6】
【0032】
GlcNAc−α−DMT 45.0mg(0.13mmol、50mM)、メトキシβ−D−ガラクトピラノシド(Gal−β−O−Me 25.0mg(0.13mmol,50mM))をpH6.5、100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、α−N−アセチルグルコサミニダーゼB1のPBS溶液(2.88mg/ml,134μlを加え、全量2.5mlとして反応を開始した。(化6)式の反応1時間後に溶離液で5倍に希釈し、分取HPLCにより単離した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=4/1、流量を8ml/min、カラム温度を40℃、検出器をUV240nmとした。NMRにより構造決定を行ったところメトキシ−4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル−β−D−ガラクトピラノシド (GlcNAc−α(1→4)−Gal−β−O−Me)を得た。(収量;7.8mg,収率;16.5%)
【化6】
【0033】
<生成物のNMR法による解析結果>
メトキシ−4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル−β−D−ガラクトピラノシド
1H NMR (500 MHz, CD3OD) : δ 4.88(1H, d, H−1’, J = 3.7 Hz), 4.24(1H, ddd, H−5’, J = 2.5 Hz, 4.4 Hz, 10.2 Hz), 4.21(1H, d, H−1, J = 7.4 Hz), 3.97(1H, d, H−4, J = 3.0 Hz), 3.94(1H, dd, H−2’, J = 3.7 Hz, 10.8 Hz), 3.79(1H, dd, H−6’a, J = 2.6 Hz, 11.8 Hz), 3.74−3.58(5H, m, H−6’b, H−6a, H−3’, H−6b, H−5), 3.58(3H, s, −OCH3), 3.55(1H, dd, H−3, J = 3.0 Hz, 10.1 Hz), 3.50(1H, dd, H−2, J = 7.5 Hz, 10.1 Hz),3.43(1H, dd, H−4’, J = 9.0 Hz, 10.1 Hz), 2.03(3H, s, −COCH3).
13C NMR (126 MHz, CD3OD) : δ 173.7(−COCH3), 106.3(C−1), 100.3(C−1’), 77.6(C−4), 76.7(C−5), 74.4(C−3), 73.6(C−5’), 72.7(C−3’), 72.6(C−2), 72.0(C−4’), 62.3(C−6’), 60.8(C−6), 57.8(−OCH3), 55.5(C−2’), 22.7(−COCH3)
【実施例7】
【0034】
終濃度がGlcNAc−α−DMT 75mM、2−ピリジル−1−チオ−2−アセトアミド−2−デオキシ−4−O−(β−D−ガラクトピラノシル)−β−D−グルコピラノシド (LacNAc−β−Spy)25mMになるようpH6.5,100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、α−N−アセチルグルコサミニダーゼB1 PBS溶液(2.75mg/ml)3.6μlを加え、全量100μlとして反応を開始した。この反応を12サンプル用意し、反応3時間後、分取HPLCにより分取した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=3/1、流量を8ml/min、カラム温度を30℃、検出器をUV240nmとした。2−ピリジル−1−チオ−2−アセトアミド−2−デオキシ−4−O−(4−O−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド (GlcNAc−α(1→4)−LacNAc−β−Spy)8.2mg(収率39%)を得た。
【0035】
<生成物のNMR法による解析結果>
2−ピリジル−1−チオ−2−アセトアミド−2−デオキシ−4−O−(4−O−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド
1H NMR (400 MHz, CD3OD): δ 8.38, 7.68, 7,40, and 7.16(4H, Pyridyl), 5.45(1H, d, H−1, J = 10.7 Hz), 4.90(1H, d, H−1”, J = 3.7 Hz), 4.48(1H, d, H−1’, J = 7.4 Hz), 4.20(1H, ddd, H−5” , J = 2.4, 4.6, 7.8 Hz), 3.99(1H, dd, H−2, J = 9.6, 10.5 Hz), 3.95(1H, d, H−4’, J = 2.2 Hz), 3.93−3.89(3H, m, H−6a, H−6b, H−2”), 3.81(1H, dd, H−6”a, J = 2.5, 11.9 Hz), 3.78−3.54(10H, m, H−3, H−4, H−5, H−2’, H−3’, H−5’, H−6’a, H−6’b, H−3”, H−6”b), 3.42(1H, dd, H−4”, J = 8.9, 10.1 Hz), 2.01(3H, s, −COCH3), 1.95(3H, s, −COCH3).
13C NMR (101 MHz, CD3OD): δ 173.7 and 173.5(−COCH3), 158.9, 150.3, 138.5, 124.4, and 122.0(Pyridyl), 105.4(C−1’), 100.1(C−1”), 84.8(C−1), 80.9(C−5), 80.6(C−4), 78.1(C−4’), 77.2(C−5’), 75.7(C−3), 74.5(C−3’), 73.8(C−5”), 72.5(C−2’, C−3”), 72.1(C−4”), 62.4(C−6”), 61.8(C−6), 61.3(C−6’), 55.6(C−2”), 55.5(C−2), 22.9 and 22.7(−COCH3).
【実施例8】
【0036】
終濃度がGlcNAc−α−DMT 75mM、2−ピリジル−1−チオ−4−O−(4−O−β−D−ガラクトピラノシル)−β−D−グルコピラノシド (Lac−β−Spy)25mMになるようpH6.5,100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、α−N−アセチルグルコサミニダーゼB1 PBS溶液(2.75mg/ml)7.3μlを加え、全量100μlとして反応を行った。(化7)式の反応24時間後、分取HPLCにより分取した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=3/1、流量を8ml/min、カラム温度を30℃、検出器をUV240nmとした。2−ピリジル−1−チオ−4−O−(4−O−(2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド (GlcNAc−α(1→4)−Lac−β−Spy)4.3mg(収率45%)を得た。
【化7】
【0037】
<生成物のNMR法による解析結果>
2−ピリジル−1−チオ−4−O−(4−O−(2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−グルコピラノシド
1H NMR (500 MHz, CD3OD): δ 8.42, 7.73, 7,49, and 7.21(4H, Pyridyl), 5.29(1H, d, H−1, J = 10.7 Hz), 4.93(1H, d, H−1”, J = 3.7 Hz), 4.49(1H, d, H−1’, J = 7.4 Hz), 4.24(1H, m, H−5”), 3.98(1H, d, H−4, J = 1.9 Hz), 3.94(1H, dd, H−2”, J = 3.4, 11.2 Hz), 3.92(2H, d, H−6a, H−6b, J = 2.7 Hz), 3.84(1H, dd, H−6”a, J = 1.9, 11.8 Hz), 3.77−3.59(10H, m, H−3, H−4, H−5, H−2’, H−3’, H−5’, H−6’a, H−6’b, H−3”, H−6”b), 3.49−3.44(2H, m, H−2, H−4”), 2.05(3H, s, −COCH3).
13C NMR (126 MHz, CD3OD): δ 172.4(−COCH3), 157.8, 148.9, 137.3, 123.2, and 120.7(Pyridyl), 104.0(C−1’), 98.6(C−1”), 84.6(C−1), 79.3(C−5), 78.7(C−4), 76.8(C−3), 76.6(C−4’), 75.8(C−5’), 73.1(C−3’), 72.3(C−2, C−5”), 71.1(C−2’, C−3”), 70.1(C−4”), 61.0(C−6”), 60.4(C−6), 59.9(C−6’), 54.3(C−2”), 21.3(−COCH3).
FAB−MS ; m/z calcd for C25H38N2O15S[M+H]+ : 639.2071, Found : 639.2075
【実施例9】
【0038】
終濃度がGlcNAc−α−DMT 50mM、GNB−α−DMT 25mMになるようpH6.5、100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、GlcNAcase B1 PBS溶液(2.75mg/ml)5.5μlを加え、全量100μlとして反応を開始した。(化8)式の反応を16サンプル用意し、反応4時間後、分取HPLCにより分取した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=3/1、流量を8ml/min、カラム温度を30℃、検出器をUV214nmとした。4,6ジメトキシ−1,3,5−トリアジン−2−イル2−アセトアミド−2−デオキシ−3−O−(4−O−(2−アセトアミド−2デオキシα−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−ガラクトピラノシド (GlcNAc−α(1→4)−GNB−α−O−DMT)12.7mg(収率44%)を得た。
【0039】
<生成物のNMR法による解析結果>
4,6ジメトキシ−1,3,5−トリアジン−2−イル2−アセトアミド−2−デオキシ−3−O−(4−O−(2−アセトアミド−2デオキシα−D−グルコピラノシル)−β−D−ガラクトピラノシル)−β−D−ガラクトピラノシド
1H NMR (400 MHz, CD3OD): δ 6.63(1H, d, H−1, J = 3.72 Hz), 4.93(1H, d, H−1”, J = 3.7 Hz), 4.66(1H, dd, H−2, J = 3.7, 11.3 Hz), 4.56(1H, d, H−1’, J = 7.1 Hz), 4.27(1H, d, H−4, J = 2.1 Hz), 4.20−4.16(2H, m, H−3, H−5”), 4.02−4.00(7H, m, −OCH3, H−5), 3.97(1H, d, H−4’, J = 2.8 Hz), 3.94(1H, dd, H−2”, J = 3.7, 10.9 Hz), 3.81(1H, d, H−6”a, J = 2.3 11.8 Hz), 3.76−3.54(9H, m, H−6a, H−6b, H−2’, H−3’, H−5’, H−6’a, H−6’b, H−3”, H−6”b), 3.42(1H, dd, H−4”, J = 8.9, 10.0 Hz), 2.02(3H, s, −COCH3), 1.92(3H, s, −COCH3).
13C NMR (101 MHz, CD3OD): δ 175.1, 174.3, 173.8, and 173.7(triazine, −COCH3), 106.1(C−1’), 100.1(C−1”), 96.4(C−1), 78.4(C−3), 78.1(C−4’), 76.8(C−5’), 74.8(C−5), 74.5(C−3’), 73.8(C−5”), 72.6(C−3”), 72.5(C−2’), 72.1(C−4”), 69.5(C−4), 62.5(C−6), 62.4(C−6”), 61.3(C−6’), 56.1(−OCH3), 55.6(C−2”), 49.6−48.4(C−2 in peak of CHD2OD), 22.7 and 22.6(−COCH3).
【化8】
【実施例10】
【0040】
GlcNAc−α−DMT 39.6mg(0.11mmol,50mM)、イソプロピル−1−チオ−β−Gal−β−D−ガラクトピラノシド (Gal−β−SiPr)79.2mg(0.33mmol、150mM)をpH6.5、100mMリン酸緩衝液に溶解し、37℃で10分間プレインキュベート後、GlcNAcase B1 PBS溶液(2.88mg/ml)118μlを加え、全量2.2mlとして反応を開始した。(化9)式の反応1時間後に溶離液で5倍に希釈し、分取HPLCにより単離した。カラムをAmide−80(Φ21.5×300mm)、溶離液をアセトニトリル/水=4/1、流量を8ml/min、カラム温度を40℃とし、UV214nmにおける紫外吸収を測定した。NMRにより構造決定を行ったところイソプロピル−1−チオ−β−(4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシドを得た。(収量;14mg、収率;28.6%)
【化9】
【0041】
<生成物のNMR法による解析結果>
イソプロピル−1−チオ―β―4−O−2−アセトアミド−2−デオキシ−α−D−グルコピラノシル)−β−D−ガラクトピラノシド
1H NMR (500 MHz, CD3OD) : δ 4.89(1H, d, H−1’, J = 3.7 Hz), 4.51(1H, d, H−1, J = 9.6 Hz), 4.22(1H, ddd, H−5’, J = 2.5, 4.4, 10.1 Hz), 4.01(1H, d, H−4, J = 3.0 Hz), 3.95(1H, dd, H−2’, J = 3.7, 10.8 Hz), 3.79(1H, dd, H−6’a, J = 2.5, 11.8 Hz), 3.73(1H, dd, H−6’b, J = 4.5, 11.8 Hz), 3.70−3.59(4H, m, H−3’, H−5, H−6a, H−6b), 3.56(1H, dd, H−3, J = 3.0, 9.6 Hz), 3.48(1H, t, H−2, J = 9.6 Hz), 3.43(1H, dd, H−4’, J = 9.0, 10.1 Hz), 3.25(1H, sept, isopropyl, J = 6.8 Hz), 2.03(3H, s, −COCH3) 1.34(3H, d, isopropyl, J = 6.8 Hz), 1.33(3H, d, isopropyl, J = 6.8 Hz),.
13C NMR (126 MHz, CD3OD) : δ 173.7(−COCH3), 100.53(C−1’), 87.3(C−1), 80.5(C−5), 78.3(C−4), 75.9(C−3), 73.6(C−5’), 72.7(C−3’), 72.0(C−4’), 71.7(C−2), 62.3(C−6’), 60.8(C−6), 55.5(C−2’), 36.2(isopropyl), 24.4, 24.1(isopropyl), 22.7(−COCH3).
【産業上の利用可能性】
【0042】
N−アセチルグルコサミン(GlcNAc)がαでガラクトース(Gal)に結合したオリゴ糖鎖(αGlcNAc含有オリゴ糖鎖)は、従来の抗生物質とは全く異なり、あらゆるピロリ菌の生育に必須の増殖活動を抑制するという機序でピロリ菌に対する抗菌作用を示すから、抗ピロリ菌剤として有用である。また、本発明によりαGlcNAc含有オリゴ糖鎖を、人工高分子担体だけでなく、クラゲや卵白などの容易に入手可能な天然のムチン型糖蛋白質糖鎖へも導入可能であることが明らかである。
【0043】
したがって、調製した物質は、ピロリ菌増殖抑制剤として、サプリメントや飲食品添加物として有用であると考えられる。またそのピロリ菌増殖抑制剤を含有する飲食品は、機能性飲食品や健康飲食品として有用である。そのピロリ菌増殖抑制剤を含有する医薬製剤は、ピロリ菌に起因する消化器系疾患、特に胃炎、胃潰瘍、十二指腸潰瘍のような胃疾患を軽減したり治癒したり予防したりする医薬品として、有用である。
【特許請求の範囲】
【請求項1】
N−アセチルグルコサミン誘導体と、そのアノマー位にグリコシド結合しうる糖受容体とを糖加水分解酵素により反応させて、N−アセチルグルコサミンがαグリコシド結合した糖誘導体を選択的に調製する方法。
【請求項2】
N−アセチルグルコサミン誘導体が、(化10)の式で表されるGlcNAc−α−DMTまたはその誘導体である請求項1に記載の方法。
【化10】
(式中、XはO、C、S、Nまたはなくてもよく、RはH、置換または非置換アルキル、もしくは置換または非置換ヘテロアルキルを表す。)
【請求項3】
糖加水分解酵素が、グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼである請求項1に記載の調製方法。
【請求項4】
糖加水分解酵素が、バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来のα−N−アセチルグルコサミニダーゼ1、2または3である請求項1に記載の調製方法。
【請求項5】
そのアノマー位にグリコシド結合しうる糖受容体が、アルコール類、ガラクトース誘導体、またはガラクトースを含む糖の誘導体である請求項1に記載の調製方法。
【請求項1】
N−アセチルグルコサミン誘導体と、そのアノマー位にグリコシド結合しうる糖受容体とを糖加水分解酵素により反応させて、N−アセチルグルコサミンがαグリコシド結合した糖誘導体を選択的に調製する方法。
【請求項2】
N−アセチルグルコサミン誘導体が、(化10)の式で表されるGlcNAc−α−DMTまたはその誘導体である請求項1に記載の方法。
【化10】
(式中、XはO、C、S、Nまたはなくてもよく、RはH、置換または非置換アルキル、もしくは置換または非置換ヘテロアルキルを表す。)
【請求項3】
糖加水分解酵素が、グリコシルハイドロラーゼファミリー89(GH89)に属するα−N−アセチルグルコサミニダーゼである請求項1に記載の調製方法。
【請求項4】
糖加水分解酵素が、バクテロイデス セタイオタオミクロン(Bacteroides thetaiotaomicron VPI5482)由来のα−N−アセチルグルコサミニダーゼ1、2または3である請求項1に記載の調製方法。
【請求項5】
そのアノマー位にグリコシド結合しうる糖受容体が、アルコール類、ガラクトース誘導体、またはガラクトースを含む糖の誘導体である請求項1に記載の調製方法。
【図1】


【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2011−244832(P2011−244832A)
【公開日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願番号】特願2011−190079(P2011−190079)
【出願日】平成23年8月31日(2011.8.31)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成23年2月8日に平成22年度 修士論文本審査(研究発表)要旨集にて発表 平成23年2月17日に平成22年度 修士論文本審査(研究発表)にて文書をもって発表
【出願人】(000173924)公益財団法人野口研究所 (108)
【Fターム(参考)】
【公開日】平成23年12月8日(2011.12.8)
【国際特許分類】
【出願日】平成23年8月31日(2011.8.31)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成23年2月8日に平成22年度 修士論文本審査(研究発表)要旨集にて発表 平成23年2月17日に平成22年度 修士論文本審査(研究発表)にて文書をもって発表
【出願人】(000173924)公益財団法人野口研究所 (108)
【Fターム(参考)】
[ Back to top ]