
N−(4−{[6,7−ビス(メチルオキシ)キノリン−4−イル]オキシ}フェニル)−N’−(4−フルオロフェニル)シクロプロパン−1,1−ジカルボキサミドのリンゴ酸塩およびその結晶質形態
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩を、L-リンゴ酸塩、D-リンゴ酸塩、DL-リンゴ酸塩、ならびにその混合物、およびリンゴ酸塩の結晶質ならびに非晶質の形態を含め、開示する。リンゴ酸塩s of N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の一以上を含む医薬組成物およびN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の一以上を投与することを含む癌治療法も開示する。
【発明の詳細な説明】
【技術分野】
【0001】
関連出願の相互参照
本願は2009年1月16日出願の米国仮特許出願No. 61/145,421号に対する優先権を主張し、その仮特許出願は全体として参照により本明細書に組み込まれる。
【0002】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドリンゴ酸塩およびN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の結晶質形態ならびに非晶質形態に関する。N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩は、(1) L-リンゴ酸塩、(2) D-リンゴ酸塩、(3) D,L-リンゴ酸塩、および (4) それらの混合のいずれかを含む。本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩を含む 医薬組成物にも関する。
【0003】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩の結晶質形態または非晶質形態を含む医薬組成物にも関する。
【0004】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩を投与することを含む癌の治療方法にも関する。
【0005】
本発明はさらに、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩の結晶質形態または非晶質形態を投与することを含む癌の治療方法に関する。
【背景技術】
【0006】
従来、癌治療における劇的な改善は、新規の機序を介して作用する治療薬の同定と関連づけられてきた。プロテインキナーゼ活性を通じて行われるシグナル伝達は、腫瘍細胞の特徴の多くをもたらす原因であり、プロテインキナーゼ活性の調節は、癌治療に利用可能な機序として知られている。プロテインキナーゼによるシグナル伝達は特に、例えば甲状腺癌、胃癌、頭頸部癌、肺癌、乳癌、前立腺癌、結腸直腸癌、および脳腫瘍細胞の拡大ならびに増殖に関連する。
【0007】
プロテインキナーゼは受容体型と非受容体型に分類できる。受容体型チロシンキナーゼは多数の膜貫通受容体で構成され、その生物活性は多岐にわたる。受容体型チロシンキナーゼに関しては、Plowman et al., DN&P 7(6): 334-339, 1994で詳細に論じられている。プロテインキナーゼおよびそのリガンドは、種々の細胞活動においてきわめて重要な役割を果たすため、プロテインキナーゼの酵素活性が脱調節を受けることにより、癌と関連する無制御の細胞増殖を含め、細胞の性質が変性を起こす場合がある。腫瘍学的な適応症に加え、キナーゼによるシグナル伝達の変性は、例えば免疫障害、循環器系疾患、炎症性疾患、変性疾患など、多数の他の疾患にも関与している。従って、プロテインキナーゼは低分子薬の創薬に関する魅力的な標的である。抗血管新生活性および抗増殖活性を対象とする低分子活性調節の特に魅力的な標的が、受容体型チロシンキナーゼのRet、c-Met、VEGFR2である。
【0008】
c-Metキナーゼは、Met、Ron、Seaを含むヘテロ二量体の受容体型チロシンキナーゼ(RTK)サブファミリーのプロトタイプの1種である。c-Metの内因性リガンドは、血管新生の強力な誘導因子である肝細胞増殖因子(HGF)である。HGFがc-Metと結合することにより、自己リン酸化を介して受容体の活性化を誘導し、それにより受容体依存性のシグナル伝達が増加し、細胞の増殖と浸潤が促進される。抗HGF抗体またはHGFアンタゴニストは、in vivoで腫瘍の転移を阻害することが報告されている(参照:Maulik et al Cytokine & Growth Factor Reviews 2002 13, 41-59)。乳腺腫瘍、結腸腫瘍、腎腫瘍、肺腫瘍、扁平上皮癌、骨髄性白血病、血管腫、黒色腫、星細胞腫(神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫を含む)などの多様な腫瘍タイプで、c-Met、VEGFR2、およびRetまたはそのいずれかの過剰発現が実証されている。Retタンパク質はチロシンキナーゼ活性を持つ膜受容体である。家族性甲状腺髄様癌の ほとんどで、Retに変異が起きている。これらの変異がRetのキナーゼ機能を活性化し、それを癌遺伝子産物に変える。
【0009】
EGF、VEGF、エフリンのシグナル伝達の阻害は、腫瘍の増殖および生存に必要な重要な2つの細胞プロセスである細胞増殖および血管新生を防ぐ(Matter A. Drug Disc. Technol. 2001 6, 1005-1024)。キナーゼKDR(キナーゼ挿入ドメイン受容体チロシンキナーゼ)および(fms様チロシンキナーゼ4)はどちらも内皮増殖因子(VEGF)受容体である。EGF、VEGF、エフリンのシグナル伝達の阻害は、腫瘍の増殖および生存に必要な重要な2つの細胞プロセスである細胞増殖および血管新生を防ぐ(Matter A. Drug Disc. Technol. 2001 6, 1005-1024)。EGF受容体およびVEGF受容体は、低分子阻害の望ましい標的である。
【0010】
従って、特に上記のRet、c-Met、VEGFR2を含むキナーゼのシグナル伝達を特異的に阻害し、調節し、かつ修飾するか、またはそのいずれかを行う低分子化合物は、異常な細胞増殖および血管新生を伴う病態の治療または予防手段として特に望ましい。そのような低分子の一つが、下記の化学構造式を持つN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドである。

WO 2005/030140に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの合成(実施例12、37、38、48)を説明し、かつ、キナーゼのシグナル伝達の阻害、調節、修飾のすべて、またはそのいずれかを行う分子の治療活性を開示する(アッセイ、表4、289項)。実施例48はWO 2005/030140の請求項[0353]に記載する。
【0011】
薬剤の開発者は、治療薬としての有効性に加え、薬剤としての加工、製造、保存安定性、有用性の全部または一部に関する性質を備えた適当な形態の治療薬の提供に努める。従って、これらの望ましい性質の一部または全部を備えた形態の発見が、創薬において不可欠である。
【0012】
出願人は、癌などの増殖性疾患の治療を目的とする医薬組成物での使用に適した性質を備えた薬剤N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの塩形態を発見した。本発明の新規塩形態は結晶質および非晶質の形態で存在する。
【発明の概要】
【0013】
本発明は明細書中に記載するN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩、明細書中に記載するその医薬組成物、および明細書中に記載するその使用法に関する。
【0014】
別の局面は、明細書中に記載するN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の結晶質および非晶質の形態、明細書中に記載するその医薬組成物、および明細書中に記載するその使用法に関する。
【図面の簡単な説明】
【0015】
【図1】に結晶質化合物 (I)、形態N-1の25 °CでのXRPD回折実験で得たパターンを示す。
【図2】に結晶質化合物 (I)、形態 N-1の固体13C NMRスペクトルを示す。
【図3】に結晶質化合物 (I)、形態 N-1の固体15N NMRスペクトルを示す。
【図4】に結晶質化合物 (I)、形態 N-1の固体19F NMRスペクトルを示す。
【図5】に結晶質化合物 (I)、形態 N-1の熱重量分析(TGA)結果を示す。
【図6】に結晶質化合物 (I)、形態 N-1の示差走査熱量分析(DSC)結果を示す。
【図7】に結晶質化合物 (I)、形態 N-1の水分吸着を示す。
【図8】に結晶質化合物 (I)、形態N-2の25 °CでのXRPD回折実験で得たパターンを示す。
【図9】に結晶質化合物 (I)、形態 N-2の固体13C NMRスペクトルを示す。
【図10】に結晶質化合物 (I)、形態 N-2の固体15N NMRスペクトルを示す。
【図11】に結晶質化合物 (I)、形態 N-2の固体19F NMRスペクトルを示す。
【図12】に結晶質化合物 (I)、形態 N-2の熱重量分析(TGA)結果を示す。
【図13】に結晶質化合物 (I)、形態 N-2の示差走査熱量分析(DSC)結果を示す。
【図14】に結晶質化合物 (I)、形態 N-2の水分吸着を示す。
【図15】に結晶質化合物 (III)、形態N-1の室温でのXRPD回折実験で得たパターンおよびシミュレーションによるパターンを示す。
【図16】に結晶質化合物 (III)、形態 N-1の固体13C NMRスペクトルを示す。
【図17】に結晶質化合物 (III)、形態 N-1の固体15N NMRスペクトルを示す。
【図18】に結晶質化合物 (III)、形態 N-1の固体19F NMRスペクトルを示す。
【図19】に結晶質化合物 (III)、形態 N-1の熱重量分析(TGA)結果を示す。
【図20】に結晶質化合物 (III)、形態 N-1の示差走査熱量分析(DSC)結果を示す。
【図21】に結晶質化合物 (III)、形態 N-1の水分吸着を示す。
【図22】に非晶質化合物 (I) の室温でのXRPDパターンを示す。
【図23】に非晶質化合物 (I) の固体13C NMRスペクトルを示す。
【図24】に非晶質化合物 (I) の固体15N NMRスペクトルを示す。
【図25】に非晶質化合物 (I) の固体19F NMRスペクトルを示す。
【図26】に非晶質化合物 (I) の示差走査熱量分析(DSC)結果を示す。
【図27】に非晶質化合物 (I) の水分吸着を示す。
【発明を実施するための形態】
【0016】
本発明は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの物理化学的性質を、薬剤開発に適するよう改善することに関する。本明細書で開示するのは、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩である。それらの塩の新規の固体形態も開示する。本明細書で開示するリンゴ酸塩およびその結晶質ならびに非晶質の形態は、各々が本発明の個別の局面を構成する。本明細書にはリンゴ酸塩およびその固体形態を記載するが、本発明は開示する塩および固体形態を含む新規組成にも関する。記載する塩ならびに固体形態およびそれらを含む治療用組成物の治療目的の使用は、本発明の個別の局面を構成する。塩およびその固体形態を特徴づけるために使用した技法は、下記の実施例に記載する。これらの技法は、単独で、または組み合わせて、本明細書に開示する塩およびその固体形態を特徴づけるために使用できる。塩およびその固体形態は、開示する図を参照して特徴づけることもできる。
【0017】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドでは、酵素Retの IC50値が約5.2 nM(ナノモル濃度)、酵素c-MetのIC50値が約1.3 nM(ナノモル濃度)であった。このc-Met活性の測定に使用したアッセイ法はWO2005‐030140の請求項[0468]に記載する。
【0018】
RETの生化学活性は、WO2005‐030140に記載するルシフェラーゼ共役化学発光キナーゼアッセイ(LCCA)法を使い測定した。キナーゼ活性はキナーゼ反応後に残ったATPの割合(%)として測定した。残ったATPはルシフェラーゼ - ルシフェリン共役化学発光により検出した。具体的には、2mM ATP、1mMポリEY、15nM RET(N末端に(His)6タグを導入し、バキュロウイルスで発現させたヒトRETキナーゼドメインM700-D1042)を20uLのアッセイ用緩衝液(20mM Tris-HCL pH 7.5、10mM MgCl2、0.01% Triton X-100、1 mM DTT、3mM MnCl2)中で混合し、反応を開始させた。この混合液を常温で2時間インキュベートした後、ルシフェラーゼ - ルシフェリン混合液を加え、Wallac Victor2プレートリーダーを使い、化学発光を測定した。ルシフェラーゼ - ルシフェリン混合液の組成は50 mM HEPES (pH 7.8)、8.5ug/mLシュウ酸 (pH 7.8)、5mM DTT、0.4% Triton X-100、0.25 mg/mLコエンザイムA、63 mM AMP、28 mg/mLルシフェリン、40,000発光単位/mLルシフェラーゼである。
【0019】
リンゴ酸塩s of N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩に関する。これらのリンゴ酸塩は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドとリンゴ酸を化合し、1:1の比でN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩を形成したものである。
【0020】
リンゴ酸の構造式を下記に示す。
不斉炭素原子により、リンゴ酸にはL-リンゴ酸およびD-リンゴ酸の2種類のエナンチオマーが存在する。
【0021】
L-リンゴ酸の構造式
当業者の間では、L-リンゴ酸に対してさまざまな名称または呼称が使われている。例えば、ブタンジオイック酸、ヒドロキシ-、(2S)- (9CI)、ブタンジオイック酸、ヒドロキシ-、(S)-、リンゴ酸、L- (8CI)、リンゴ酸、l- (3CI)、(-)-(S)-リンゴ酸、(-)-ヒドロキシコハク酸、(-)-(L)-リンゴ酸、(-)-リンゴ酸、(2S)-2-ヒドロキシブタンジオイック酸、(2S)-2-ヒドロキシコハク酸、(S)-リンゴ酸、リンゴ酸、L-(-)-リンゴ酸、(L)-リンゴ酸、NSC 9232、S-(-)-リンゴ酸、S-2-ヒドロキシコハク酸などである。
【0022】
D-リンゴ酸の構造式
当業者の間では、D-リンゴ酸に対してさまざまな名称または呼称が使われている。例えば、ブタンジオイック酸、2-ヒドロキシ-、(2R)-、ブタンジオイック酸、ヒドロキシ-、(2R)- (9CI)、ブタンジオイック酸、ヒドロキシ-、(R)-、(+)-リンゴ酸、(2R)-2-ヒドロキシブタンジオイック酸、(2R)-リンゴ酸、(R)-(+)-リンゴ酸、(R)-リンゴ酸、D-(+)-2-ヒドロキシコハク酸、D-(+)-リンゴ酸、D-リンゴ酸などである。
【0023】
上記のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの化学構造式を示す。
化学構造には不斉炭素原子はない。 N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドは一般にさまざまな名称で呼ばれ、それらの名称または呼称の例としては、1,1-シクロプロパンジカルボキサミド、N'-[4-[(6,7-ジメトキシ-4-キノリニル)オキシ]フェニル]-N-(4-フルオロフェニル)- および1,1-シクロプロパンジカルボキサミド、N-[4-[(6,7-ジメトキシ-4-キノリニル)オキシ]フェニル]-N'-(4-フルオロフェニル)- (9CI)などがある。
【0024】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドは、グラム単位(1 kg未満)またはキロ単位(1 kg以上)のいずれかにより、数種類の方法のいずれかに従い調製できる。グラム単位の方法はWO 2005-030140に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの合成法(実施例25、37、38、48)として記載し、それは参照として本明細書の一部とする。別法として、有効化合物を含むN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドを、下記実施例1に示す手順を使い、キロ単位で調製することができる。
【0025】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩に関する。
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩(化合物 (I))
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのD-リンゴ酸塩(化合物 (II))および
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのDL-リンゴ酸塩(化合物 (III))
上記の各々が、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドおよびその他の塩と比較し、改善された性質を持つ。例えば「N-2」など、特定の形態の特徴を示すために本明細書で用いる名称は、同様または同一の物理的および科学的な特徴を持つ他の物質を除外するための限定的な名称ではなく、むしろそのような名称は単なる識別記号として、本明細書に提示する特徴に従い解釈するものとする。
【0026】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩、特に化合物 (I) は、開発のために好ましい医薬品特性の組み合わせを持つ。25°C/60%相対湿度(RH)の条件と40 °C/60% RHの条件において、化合物 (I) の分析結果、純度、水分、溶解度に変化はなかった。DSC/TGAにより、化合物 (I) は185°Cまで安定であることが示された。溶剤損失は観察されなかった。L-リンゴ酸塩による水分吸収は可逆性であり、ヒステリシスは低かった。水分吸収量は90% RHで約0.60 wt%と算出された。L-リンゴ酸塩の合成は収率が良く、純度は90%を超え、医薬組成物中で使用するために十分な溶解度を備えていた。この塩の水分量はカールフィッシャー分析により約0.5 wt%と算出され、これはTGA分析およびGVS分析と相関する。N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのD-リンゴ酸塩 は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩と同じ性質を持つ。
【0027】
化合物 (I) の塩それ自体、およびそれとは別に、その結晶質および非晶質の形態は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの遊離塩基および他の塩と比較し、有益な性質を示す。例えば、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの塩酸塩は、望ましくない感湿性を示し、高湿(湿度75%)高温(40°C)条件に曝露すると、相変化を起こす。マレイン酸塩は溶解度が低かった。酒石酸塩は結晶性が低く、溶解度が低かった。リン酸塩では、H2Oの吸収により重量が8%増加し、これは試験を行った塩の中で最高であった。
【0028】
さまざまな塩の水溶性は、水1 mLに対して10 mgの固体を使い測定した。塩の調製には、ソルトスクリーンを使い、遊離塩基のアセトン溶液を、ある範囲の酸をテトラヒドロフラン(THF)に溶解した保存溶液と、約1:1のモル比で反応させた。下記の表1は遊離塩基と各塩に関する水溶性などのデータの要約である。
【0029】
本発明の別の局面は、本明細書に記載する化合物 (I) のN-1およびN-2結晶質形態またはそのいずれかを含む化合物 (I) 結晶質形態に関する。化合物 (I) の各形態は本発明の個別の局面である。同様に、本発明の別の局面は、本明細書に記載する化合物 (II) のN-1およびN-2結晶質形態またはそのいずれかを含む化合物 (II) 結晶質形態に関する。その各形態も本発明の個別の局面である。当業者に知られているように、結晶性のD-リンゴ酸塩は、結晶性の化合物 (I) と同じ結晶質形態を形成し、同じ性質を持つ。結晶のエナンチオマーの性質について考察したWO 2008/083319を参照のこと。化合物 (I) および (II) の結晶質形態の混合は、本発明の別の局面である。
【0030】
本明細書に記載する化合物 (I) および (II) の結晶性のN-1形態は、以下の1項目以上の特徴を持つ場合がある。
(i) 固体13C NMRスペクトルのピークが18.1、42.9、44.5、70.4、123.2、156.2、170.8、175.7、および182.1 ppm ± 0.2 ppmにある。
(ii) 固体13C NMRスペクトルが図 2に示すパターンと実質的に一致する。
(iii) 粉末X線回折パターン(CuKα λ=1.5418Å)が、以下の中から選択される4点以上のピークを含む。6.4、9.0、12.0、12.8、13.5、16.9、19.4、21.5、22.8、25.1、および27.6 °2θ ± 0.2 °2θ。結晶質形態の測定は室温で行う。
(iv) 粉末X線回折(XRPD)スペクトルが図1に示したパターンと実質的に一致する。
(v) 固体15N NMRスペクトルのピークが118.6、119.6、120.7、134.8、167.1、176.0、および180 ppm ±0.2 ppmにある。
(vi) 固体15N NMRスペクトルが図3に示すパターンと実質的に一致する。
【0031】
化合物 (I) および (II) の結晶質N-1形態の特徴づけに使用できる他の固体状態特性を図に示し、下記の実施例で考察する。結晶質化合物 (I) に関しては、40°C で1週間、75% RHに曝露後、固体状態と結晶化度に変化はなかった。
【0032】
本明細書に記載する化合物 (I) および (II) の結晶質N-2形態は、以下の1項目以上の特徴を持つ。
(i) 固体13C NMRスペクトルのピークが 23.0、25.9、38.0、54.4、56.11、41.7、69.7、102.0、122.5、177.3、179.3、180.0、および180.3 ± 0.2 ppmにある。
(ii) 固体13C NMRスペクトルが図9に示すパターンと実質的に一致する。
(ii) 粉末X線回折パターン(CuKα λ=1.5418Å)が、以下の中から選択される4点以上のピークを含む。6.4、9.1、12.0、12.8、13.7、17.1、20.9、21.9、22.6、および23.7 °2θ ± 0.2 °2θ。結晶質形態の測定は室温で行う。
(iv) 粉末X線回折(XRPD)スペクトルが図8に示すパターンと実質的に一致する。
(v) 固体15N NMRスペクトルのピークが118.5、120.8、135.1、167.3、および180.1 ppmにある。
(vi) 固体15N NMRスペクトルが図10に示すパターンと実質的に一致する。
化合物 (I) および (II) の結晶質N-2形態の特徴づけに使用できる他の固体状態特性を図に示し、下記の実施例で考察する。
【0033】
別の実施形態では、本発明は化合物 (I) の結晶質形態に関し、それは本発明のいかなる局面および実施形態に関して記載される場合も、実質的に純粋なN-1形態である。
【0034】
別の実施形態では、本発明は化合物 (I) の結晶質形態に関し、それは本発明のいかなる局面および実施形態に関して記載される場合も、実質的に純粋なN-2形態である。
【0035】
本発明は化合物 (I) および (II) の非晶質形態にも関する。化合物 (I) の非晶質形態の調製法および固体の性質ならびに特徴は、下記の実施例に記載する。化合物 (I) および (II) の非晶質形態は、本発明の別の局面を表す。
【0036】
本発明のさらに別の局面は、化合物 (I) と化合物 (II) の混合物に関する。混合物は、化合物 (I) と化合物 (II) の合計重量に基づき、ゼロ重量%を超え、100重量%未満の化合物 (I) および100重量%未満でゼロ重量%を超える化合物 (II) を含む。他の実施形態では、混合物はその混合物中の化合物 (I) と化合物 (II) の合計重量に基づき、約1重量%から約99重量%の化合物 (I) および約99重量%から約1重量%の化合物 (II) を含む。さらに別の実施形態では、混合物は化合物 (I) と化合物 (II) の合計重量に基づき、約90重量%から100重量%未満の化合物 (I) およびゼロ重量%を超え、約10重量%までの化合物 (II) を含む。従って、混合物は1〜10重量%の化合物 (I)、11〜20重量%の化合物 (I)、21〜30重量%の化合物 (I)、31〜40重量%の化合物 (I)、41〜50重量%の化合物 (I)、51〜60重量%の化合物 (I)、61〜70重量%の化合物 (I)、71〜80重量%の化合物 (I)、81〜90重量%の化合物 (I)、91〜99重量%の化合物 (I) のいずれかを含み、リンゴ酸塩の残りの重量%は化合物 (II) の重量%である。
【0037】
本発明の別の局面は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのDL-リンゴ酸塩の結晶質形態、化合物 (III) に関する。DL-リンゴ酸塩はラセミ体のリンゴ酸から調製する。本明細書に記載する化合物 (III) の結晶性のN-1形態は、以下の1項目以上の特徴を持つ場合がある。
(i) 固体13C NMRスペクトルが、20.8、26.2、44.8、55.7、70.7、100.4、101.0、114.7、115.2、116.0、119.7、120.4、121.6、124.4、136.9、138.9、141.1、145.7、150.3、156.5、157.6、159.6、165.2、167.4、171.2、176.3、182.1 ppm ± 0.2 ppmから選択される4点以上のピークを含む。
(ii) 固体13C NMRスペクトルが図16に示すパターンと実質的に一致する。
(iii) 粉末X線回折パターン(CuKα λ=1.5418Å)が、以下の中から選択される4点以上の2θ値を含む。12.8、13.5、16.9、19.4、21.5、22.8、25.1、および27.6 ±0.2 °2θ。結晶質形態の測定は室温で行う。
(iv) 粉末X線回折(XRPD)スペクトルが図15に示すパターンと実質的に一致する。
(v) 固体15N NMRスペクトルのピークが119.6、134,7、および175.5 ppm ± 0.2 ppmにある。
(vi) 固体15N NMRスペクトルが図17に示すパターンと実質的に一致する。
化合物 (III) の結晶質N-1形態の特徴づけに使用できる他の固体状態特性を図に示し、下記の実施例で考察する。1つの実施形態では、化合物 (III) のN-1形態が、ほぼ以下の値に等しい単位格子パラメータを示す。
結晶の寸法 a = 14.60 Å
b = 5.20 Å
c = 39.09 Å
α = 90.0°
β = 90.4°
γ = 90.0°
空間群: P21/n
単位格子あたりの化合物 (I) の分子数:4
体積 = 2969 Å3
密度(計算値)= 1.422 g/cm3
化合物 (III) のN-1形態の単位格子パラメータは、例えば外気温または室温などに相当する約25°Cで測定した。
【0038】
化合物 (I) ならびに (II) の結晶質形態N-1ならびにN-2、および化合物 (III) の結晶質形態N-1の各々が、互いに識別可能な固有の特徴を持つ。これらの特徴は、下記の実施例で示す固体形態の物性を比較することにより理解できる。例えば表2は、結晶質化合物 (III) の形態 N-1および結晶質化合物 (I) の形態N-1ならびにN-2に特徴的なXRPDのピークの位置(°2θ±0.2 °2θ)を示したものである。非晶質形態はXRPDのパターンで回折ピークを示さない。
結晶質化合物 (II) の形態N-1とN-2の間で固有な反射にマーク(*)を付けた。前述のように、化合物 (II) は化合物 (I) のエナンチオマーであるため、化合物 (II) の形態 N-1は、化合物 (I) の形態 N-1に関して表2に掲げたものと同じ特徴の回折パターンおよび固有のピークを持つ。同様に、化合物 (II) の形態 N-2は、化合物 (I) の形態 N-2に関して表2に掲げたものと同じ特徴の回折パターンおよび固有のピークを持つ。化合物 (I) と (II) はその絶対立体化学、すなわちL-リンゴ酸塩とD-リンゴ酸塩の違いにより、その間の識別が可能である。結晶質化合物 (III) の形態 N-1は、D,L-リンゴ酸塩として識別される。
【0039】
固体NMRでの特徴的なピークも、本明細書で開示する結晶質形態と非晶質形態を識別する役割を果たす。例えば表3は、結晶質化合物 (III) の形態 N-1、結晶質化合物 (I) の形態N-1ならびにN-2、および化合物 (I) の非晶質形態に特徴的な固体13C NMRピークを示したものである。
後述する固体19Fおよび15N NMRスペクトルは、同様の比較および特徴づけのためのデータを提供する。前述のように、化合物 (I) のエナンチオマーであるため、結晶質形態 N-1ならびにN-2と化合物 (II) の非晶質形態は、結晶質化合物 (I) の形態N-1およびN-2に関して表3に示したように、同じ固体NMR共鳴を持ち、それらの間で異なる固有のピークを持つ。
【0040】
医薬組成物および処理方法
本発明の別の局面は、化合物 (I)、化合物 (II)、化合物 (III) またはその組み合わせの一以上、および薬学的に許容される賦形剤を含む医薬組成物に関する。医薬組成物中の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの量は、治療上有効な量とすることができる。化合物 (I)、化合物 (II)、または化合物 (III) は医薬組成物中で個別に、上記のいずれかの固体形態またはその組み合わせとして存在することができる。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、結晶質形態の治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上、および薬学的に許容される賦形剤を含む固体または分散の医薬組成物に関する。
【0041】
本発明の別の局面は、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、それを必要とする患者に投与することを含む癌治療法に関する。投与する化合物 (I)、化合物 (II)、またはその組み合わせは、治療上有効な量とすることができる。 化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれか、またはその組み合わせとして個別に投与できる。結晶質形態が好ましい固体形態であり、結晶質化合物 (I)、形態 N-1またはN-2が好ましい。従って、本発明の別の局面は、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、それを必要とする患者に投与することを含む癌治療法に関し、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。
【0042】
本発明の別の局面は、上記のように癌を治療する方法に関し、治療される癌は胃癌、食道癌、腎癌、肝癌、卵巣癌、子宮頸癌、大腸癌、小腸癌、脳腫瘍(神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫などの星細胞腫を含む)、肺癌(非小細胞肺癌を含む)、骨癌、前立腺癌、膵癌、皮膚癌、骨癌、リンパ腫、固形腫瘍、ホジキン病、非ホジキンリンパ腫、または甲状腺癌(甲状腺髄様癌を含む)である。
【0043】
チロシンキナーゼ阻害剤は、非小細胞肺癌(NSCLC)の治療にも使われてきた。ゲフィチニブおよびエルロチニブは、チロシンキナーゼと呼ばれる上皮増殖因子の受容体を標的とする血管新生阻害剤である。エルロチニブおよびゲフィチニブは現在、NSCLCの治療に使われている。本発明の別の局面は、患者の小細胞肺癌(NSCLC)を治療する方法に関し、その方法は、治療上有効な量のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド、またはその薬学的に許容される塩を、場合によってはエルロチニブまたはゲフィチニブと組み合わせ、治療を必要とする患者に投与することを含む。別の実施形態では、エルロチニブと組み合わせる。
【0044】
本発明の別の局面は、患者の小細胞肺癌(NSCLC)を治療する方法に関し、その方法は、治療上有効な量のエルロチニブまたはゲフィチニブを、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上と組み合わせ、治療を必要とする患者に投与することを含む。化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれかとして個別に投与してもよく、またはそれらを組み合わせて投与してもよい。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、患者の小細胞肺癌(NSCLC)を治療する方法に関し、その方法は、治療上有効な量のエルロチニブまたはゲフィチニブを、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上と組み合わせ、治療を必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。別の実施形態では、この方法で投与される組み合わせは、エルロチニブと化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上である。
【0045】
本発明の別の局面は、患者における星細胞腫(患者における神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫を含む)を治療する方法に関し、その方法は、治療上有効な量のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドを、治療を必要とする患者に投与することを含む。
【0046】
本発明の別の局面は、患者における星細胞腫(患者における神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫を含む)を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含む。化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれかとして個別に投与してもよく、またはそれらを組み合わせて投与してもよい。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、星細胞腫を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。
【0047】
本発明の別の局面は、患者における甲状腺癌(甲状腺髄様癌を含む)を治療する方法に関し、その方法は、治療上有効な量のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド、またはその薬学的に許容される塩を、治療を必要とする患者に投与することを含む。投与量は治療上有効な量とすることができる。
【0048】
本発明の別の局面は、患者における甲状腺癌(甲状腺髄様癌を含む)を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含む。化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれかとして個別に投与してもよく、またはそれらを組み合わせて投与してもよい。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、甲状腺癌を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。
【0049】
本発明の別の局面は、制御されず、異常であり、望ましくないか、またはそのいずれかである細胞活性と関連する疾患または障害を治療する方法に関する。この方法では、化合物 (I)、化合物 (II)、化合物 (III) またはその組み合わせの一以上を、それを必要とする患者に投与する。投与する化合物 (I)、化合物 (II)、またはその組み合わせは、治療上有効な量とすることができる。 化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれか、またはその組み合わせとして個別に投与できる。結晶質形態が好ましい固体形態である。
【0050】
従って、本発明の別の局面は、制御されず、異常であり、望ましくないか、またはそのいずれかである細胞活性と関連する疾患または障害を治療する方法に関し、それは化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、それを必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。本発明の別の局面は、制御されず、異常であり、望ましくないか、またはそのいずれかである細胞活性と関連する疾患または障害を治療する方法に関する。この方法では、結晶質形態の化合物 (I)、化合物 (II)、または化合物 (I) および (II) の組み合わせを、それを必要とする患者に投与する。投与する化合物 (I)、化合物 (II)、または化合物 (I) および (II) の組み合わせの量は、治療上有効な量とすることができる。
【0051】
本発明の別の局面は、上記の疾患または障害を治療する薬剤を製造するために、上記実施形態のいずれかに従い、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩を使用することに関する。溶解すると、本発明に従う結晶質形態または非晶質形態は固体構造を失い、このため、それを例えば化合物 (I) の溶液などと呼ぶ。本発明に従う一以上の結晶質形態が溶解および懸濁された(またはそのいずれか)液体製剤を調製するために、本発明中の結晶質形態の一以上を使うことができる。
【0052】
上記の医薬組成物は、固体形態を含め、活性のある化合物 (I)、化合物 (II)、および化合物 (III)、またはそのいずれか(以降、活性化合物と呼ぶ)を含む任意の医薬形態である。例えば、医薬組成物は錠剤、カプセル剤、懸濁液、注射液、局所薬、経皮薬の場合がある。医薬組成物は一般に、約1重量%から約99重量%の活性化合物または活性化合物の結晶質形態と、99重量%から1重量%の好適な医薬賦形剤を含む。一例では、組成物は約5重量%と約75重量%の間の活性化合物を含み、残りは下記の好適な医薬賦形剤または他のアジュバントである。
【0053】
本発明に従い、キナーゼのシグナル伝達を阻害し、調節し、かつ修飾するか、またはそのいずれかを行うための、活性化合物、または活性化合物の結晶質形態または非晶質形態(ここでは医薬組成物に関して論じる)の「治療上有効な量」とは、異常な細胞増殖および血管新生と関連する多種の癌のいずれかに罹患した患者を治療するために十分な量を意味する。本発明に従う治療上有効な量は、本明細書で論じる疾患状態および障害の治療または予防に対して治療上有用な量である。化合物 (I)、(II)、および (III) またはそのいずれか(それらの固体形態を含む)は、WO2005‐030140に記載するキナーゼのシグナル伝達を阻害し、調節し、かつ修飾するか、またはそのいずれかによる治療作用を持つ。N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド。
【0054】
特定の患者の治療に必要な実質量は多様な要因に依存し、例えば、治療する疾患の状態およびその重症度、使用する特定の医薬組成物、患者の年齢、体重、健康状態、性別、食生活、投与方法、投与回数、投与経路、本発明に従う活性化合物または活性化合物の結晶質形態の排出速度、治療期間、使用する特定化合物と併用する薬剤または同時に服薬される薬剤、および医学分野で周知の他の要因がある。これらの要因については、Goodman and Gilman’s “The Pharmacological Basis of Therapeutics”, Tenth Edition, A. Gilman, J.Hardman and L. Limbird, eds., McGraw-Hill Press, 155-173, 2001で論じられており、それは参照により本明細書の一部とする。本発明に従う活性化合物、または活性化合物の結晶質形態、およびそれらを含む医薬組成物は、癌の治療を受ける患者に一般に投与される抗癌剤その他の薬剤と併用してもよい。単一の医薬組成物内で、一以上のそれらの薬剤と合わせて製剤してもよい。
【0055】
医薬組成物のタイプに従い、当業者の間で知られるいずれかの担体またはその組み合わせの中から、薬学的に許容される担体を選択できる。薬学的に許容される担体の選択は部分的に、使用する望ましい投与方法に依存する。本発明の医薬組成物、すなわち本発明の活性化合物、または活性化合物の結晶質形態に関しては、結晶質であるか否かに関わらず、担体は活性化合物の特定の形態を実質的に維持するよう選択すること。言い換えれば、担体は活性化合物の形態を実質的に変えてはならない。それ以外にも、担体は、望ましくない生物学的影響を与えることや、それ以外に、医薬組成物の他の構成要素と有害な形で相互作用することなどにより、活性化合物の形態と不適合ではないものとする。
【0056】
本発明の医薬組成物は、例えばRemington's Pharmaceutical Sciences, 18th Ed., (Mack Publishing Company, Easton, Pa., 1990) に掲載されている、製薬分野で周知の方法により調整できる。固形剤形では化合物 (I) に、一以上のクエン酸ナトリウムまたはリン酸水素カルシウムなどの薬学的に許容される賦形剤、または (a) 例えばデンプン、乳糖、ショ糖、ブドウ糖、マンニトール、ケイ酸などの充填剤または増量剤、(b) 例えばセルロース誘導体、デンプン、アルギン酸、ゼラチン、ポリビニルピロリドン、ショ糖、アラビアゴムなどの結合剤、(c) 例えばグリセロールなどの保湿剤、(d) 例えば寒天、炭酸カルシウム、ジャガイモまたはタピオカのデンプン、アルギニン酸、クロスカルメロースナトリウム、複合ケイ酸塩、炭酸ナトリウムなどの崩壊剤、(e) 例えばパラフィンなどの溶解遅延剤、(f) 例えば四級アンモニウム化合物などの吸収促進剤、(g) 例えばセチルアルコール、モノステアリン酸グリセロール、ステアリン酸マグネシウムなどの湿潤剤、(h) 例えばカオリン、ベントナイトなどの吸湿剤、(i) 例えばタルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、またはその混合などの潤滑剤を混合できる。カプセル剤、錠剤、丸剤の場合、投与剤形には緩衝剤を含めてもよい。
【0057】
製薬分野で周知の薬学的に許容されるアジュバントも、本発明の医薬組成物で使用できる。これは保存剤、湿潤剤、懸濁剤、甘味剤、香味剤、芳香剤、乳化剤、分散剤を含むが、それらに限定されない。微生物の作用は、例えばパラベン、クロロブタノール、フェノール、ソルビン酸、およびその類似物など、さまざまな抗菌剤および抗真菌剤により、確実に防止できる。例えば糖類、塩化ナトリウム、およびその類似物などの等張剤を含めることが望ましい場合もある。望ましい場合は、本発明の医薬組成物に、例えばクエン酸、ソルビタンモノラウレート、オレイン酸トリエタノールアミン、ブチル化ヒドロキシトルエンなど、湿潤剤または懸濁剤、pH緩衝剤、抗酸化剤などの少量の添加剤を含めてもよい。
【0058】
上記の固形剤形は、腸溶コーティングおよび当業者の間で周知の他のコーティングなど、コーティングおよびシェルを加えて調製できる。安定剤を含めてもよく、活性化合物を腸管の特定箇所で遅延して放出するような組成にすることもできる。使用可能な埋め込み組成の例としては、高分子物質およびワックスがある。活性化合物は、適切であれば、一以上の上記賦形剤と共に、マイクロカプセル化することもできる。
【0059】
懸濁液には活性化合物に加え、例えばエトキシル化イソステアリルアルコール、ポリオキシエチレンソルビトルおよびソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム、ベントナイト、寒天およびトラガカント、またはこれらの物質の混合、およびその類似物などの懸濁剤を含むことができる。
【0060】
直腸投与用の組成物は、例えば活性化合物または活性化合物の結晶質形態を、例えばココアバター、ポリエチレングリコールまたは坐剤ワックスなど、常温では固体であるが、体温では液体であり、従って、好適な体腔内で溶融し、そこで活性化合物を放出する好適な非刺激性の賦形剤または担体と混合することにより調製できる坐剤である。
【0061】
活性化合物または活性化合物の結晶質形態は、調製中に維持されるため、本発明の医薬組成物には、固体投与剤形が好ましい。カプセル剤、錠剤、丸剤、粉剤、顆粒剤を含む経口投与用固体投与剤形は、特に好ましい。そのような固体投与剤形において、活性化合物は一以上の不活性な薬学的に許容される賦形剤(薬学的に許容される担体とも称する)と混合される。純粋な形態または適当な医薬組成物としての活性化合物または活性化合物の結晶質形態の投与は、同様の用途のために許容される投与方法または薬剤を介して実行できる。従って、投与は例えば経口、経鼻、非経口(静脈内、筋肉内、または皮下)、局所的、経皮、膣内、膀胱内、槽内、経直腸で行うことができ、剤形は例えば錠剤、坐剤、丸剤、軟質弾性ゼラチンカプセル剤ならびに硬質ゼラチンカプセル剤、粉剤、液剤、懸濁剤、噴霧剤、または類似物などの固体、半固体、凍結乾燥粉剤、または液体投与剤形であり、正確な用量を簡単に投与するために適した単位用量ごとに投与できる剤形が好ましい。一つの好ましい投与経路は経口投与であり、治療する疾患状態の重症度に従い調整できる便利な投与計画に従い投与する。
【0062】
結晶質形態の一般的な調製法
結晶質形態は、例えば好適な溶媒混合液からの結晶化または再結晶化、昇華、融液からの成長、別の相からの固相変換、超臨界液流体からの結晶化、およびジェット噴霧などの多様な方法により調整することができ、それらに限定されない。溶媒混合液からの結晶質形態の結晶化または再結晶化の技法は、例えば溶媒の蒸発、溶媒混合液の冷却、化合物およびその塩またはそのいずれかの超飽和溶媒混合液への結晶の接種、化合物およびその塩またはそのいずれかの超飽和溶媒混合への結晶の接種、溶媒混合液の凍結乾燥、および溶媒混合液への逆溶媒(逆溶剤)の添加などを含み、それらに限定されない。多形体を含む結晶質形態を調製するために、高処理能力結晶化技法を採用してもよい。
【0063】
多形体を含む薬剤の結晶、調製法、薬剤結晶の特徴づけは、Solid-State Chemistry of Drugs, S.R. Byrn, R.R. Pfeiffer, and J.G. Stowell, 2nd Edition, SSCI, West Lafayette, Indiana (1999) で考察されている。
【0064】
溶媒を採用する結晶化技法において、典型的には溶媒は一以上の要素に基づき選択され、それには例えば化合物の溶解度、使用する結晶化技法、および溶媒の蒸気圧が含まれ、それらに限定されない。複数の溶媒を組み合わせて使用してもよい。例えば、最初の溶媒に化合物を溶解させて溶液を作り、次に、それに逆溶媒を加えて溶液中の化合物 (I) の溶解度を下げ、結晶を析出させることができる。逆溶媒は、化合物の溶解度が低い溶媒である。
【0065】
結晶の調製に使用できる一方法では、化合物 (I)、化合物 (II) および化合物 (III)、またはそのいずれかを好適な溶媒中に懸濁し、攪拌して得たスラリーを熱し、溶解を促進する。本明細書で使用する限り、「スラリー」という用語は、当該化合物の飽和溶液を意味し、その溶液に化合物が添加され、任意の温度で化合物と溶媒の不均質な混合状態が生じる場合があることを意味する。
【0066】
結晶化混合液に種結晶を添加し、結晶化を促進することができる。特定の多形体の成長を制御するため、および結晶の粒子サイズの分散を制御するため、またはそのいずれかを目的として、接種を利用することができる。従って、必要な種結晶の量の計算は、例えば “Programmed Cooling Batch Crystallizers,” J.W. Mullin and J. Nyvlt, Chemical Engineering Science, 1971, 26, 3690377に記載されているように、入手可能な種結晶の大きさおよび結晶産物について所望する平均粒子サイズに依存する。一般に、バッチ中の結晶の成長を有効に制御するには、小さい種結晶が必要とされる。小さい種結晶は、大きい結晶のふるい分け、微粉砕、もしくは超微粉砕、または溶液の微結晶化により生成できる。結晶の微粉砕または超微粉砕においては、所望する結晶質形態から結晶化度が変化しないよう(すなわち非晶質または他の多形体への変化)、注意が必要である。
【0067】
冷却した結晶化混合液を減圧濾過し、分離された固体産物を、例えば低温の再結晶溶媒などの好適な溶媒で洗浄することができる。洗浄後、当該産物を窒素パージ下で乾燥させることにより、所望する結晶質形態が得られる。化合物の結晶質形態が形成されたことを確認するために、当該産物は好適な分光学的または分析的技法により分析することができ、それには例えば示差走査熱量計(DSC)、粉末X線回折(XRPD)、および熱重量分析(TGA)が含まれ、それらに限定されない。得られる結晶質形態の分離収率は、結晶化手順に最初に投入した化合物の重量に基づき、約70重量%であり、好ましくは約90重量%以上である。場合によっては、共粉砕するか、またはメッシュスクリーンを通すことにより、産物の塊を崩してもよい。
【0068】
当業者は以下の詳細な説明を読むことにより、本発明の特徴および優位性を、より容易に理解できるであろう。理解すべき点として、本発明の特徴は、明確にするために、上記および下記で個別の実施形態を背景として説明したが、それらの特定の特徴を、一つの実施形態で組み合わせることも可能である。逆に、簡潔にするために、一つの実施形態を背景として説明した種々の特徴を、それらよりも下位の項で組み合わせて使うこともできる。本発明は以下の実施例により、さらに詳しく例示されるが、そこに記載された特定の手順に本発明の範囲または主旨を限定するものと解釈すべきではない。
【0069】
本明細書に明記する定義は、参照により本明細書の一部とするいかなる特許、特許出願、特許出願公報に明記された定義よりも優先する。すべての測定値において実験上の誤差が生じる可能性があるが、本発明の主旨から逸脱しない。
【0070】
本明細書で使用する限り、「非晶質」とは、結晶質ではない分子およびイオンの固体形態を意味する。非晶質固体は、シャープな最大値を持つ確定的なX線回折パターンを示さない。
【0071】
本明細書で使用する限り、「実質的に純粋」とは、言及する化合物 (I) の結晶質形態が、結晶質形態の重量に基づき約90重量%以上を含むことを意味する。「約90重量%以上」は、請求範囲に対する等価性理論の適用の制限を意図しないが、しかし、言及する結晶質形態の重量に基づき、例えば約90、約91、約92、約93、約94、約95、約96、約97、約98、約99、および約100重量%を含み、それらに限定されない。化合物 (I) の結晶質形態の残りは、化合物 (I) の他の形態、および、例えば結晶質形態を調製するときに生じる反応不純物および処理不純物、またはそのいずれかを含む場合がある。反応不純物および処理不純物、またはそのいずれかの存在は、例えばクロマトグラフィー、核磁気共鳴分光法、質量分光分析法、および赤外分光法、またはそのいずれかなど、当業者の間で周知の分析法により決定できる。
【0072】
調製実施例
実施例1:N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドおよびそのL-リンゴ酸塩 (化合物 (I))の調製
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドおよびそのL-リンゴ酸塩の調製に使用する合成経路を反応式1に示す。
反応式1に示した過程を以下に詳しく説明する。
【0073】
1.1 4‐クロロ‐6,7‐ジメトキシキノリンの調製
反応器に、最初に6,7‐ジメトキシキノリン‐4‐オル(1 L、10.0 kg)、次にアセトニトリル(64.0 L)を順に入れた。その混合物を約65°Cに熱し、オキシ塩化リン(POCl3、50.0 kg)を加えた。POCl3を加えた後、反応混合物の温度を約80°Cに上げた。開始物質の2%未満が残る(反応進行を確認する高速液体クロマトグラフィー[HPLC]分析による)時点で、反応は完了と見なした(約9.0時間)。反応混合物を約10°Cに冷却した後、クエンチするために、ジクロロメタン(DCM、238.0 kg)、30% NH4OH(135.0 kg)、および氷(440.0 kg)の冷却溶液に投入した。混合物を約14°Cに加熱し、相を分離させた。有機相を水(40.0 kg)で洗浄し、減圧蒸留で溶媒を除去して濃縮した(約190.0 kg)。メチル-t-ブチルエーテル(MTBE、50.0 kg)をバッチに加え、混合物を約10°Cまで冷却する間に、結晶が析出した。固体を遠心分離により回収し、n-ヘプタン(20.0 kg)で洗浄し、約40°Cで乾燥し、表題化合物を得た(8.0 kg)。
【0074】
1.2 6,7‐ジメチル‐4‐(4-ニトロフェノキシ)‐キノリンの調製
反応器に、4‐クロロ‐6,7‐ジメトキシキノリン(8.0 kg)、4-ニトロフェノール(7.0 kg)、4-ジメチルアミノピリジン(0.9 kg)、2,6-ルチジン(40.0 kg)を順に入れた。反応器の内容物を約147°Cに熱した。反応完了後(反応中のHPLC分析により、残った反応物が5%未満であることを確認。約20時間)、約25°Cに冷却するまで、反応器の内容物を放置した。メタノール(26.0 kg)を加え、次に、水(50.0 kg)に溶解した炭酸カリウム(3.0 kg)を加えた。反応器の内容物を約2時間、攪拌した。沈殿した固体を濾過し、水(67.0 kg)で洗浄し、25°Cで約12時間乾燥させ、表題化合物を得た(4.0 kg)。
【0075】
1.3 4‐(6,7 ‐ジメトキシキノリン‐4‐イロキシ)‐フェニルアミンの調製
ギ酸カリウム(5.0 kg)、ギ酸(3.0 kg)、および水(16.0 kg)を、テトラヒドロフラン(40.0 kg)中に6,7-ジメトキシ‐4‐(4-ニトロフェノキシ)‐キノリン(4.0 kg)および10%パラジウム炭素(50%水湿潤、0.4 kg)を混合し、約60°Cに熱したものに加えた。反応混合物の温度が約60°Cに保たれるような方法で、それらを加えた。反応中のHPLC分析により反応が完了したと見なされた時点で(反応物の2%未満が残る状態。通常1 5時間)、反応器の内容物を濾過した。濾過液を約 35°Cで減圧蒸留し、最初の容積の半分まで濃縮し、反応産物を沈殿させた。反応産物を濾過により回収し、水(12.0 kg)で洗浄し、約50°Cで減圧乾燥し、表題化合物を得た(3.0 kg。97% AUC)。
【0076】
1.4 1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボン酸の調製
THF(63.0 kg)に市販のシクロプロパン‐1,1‐ジカルボン酸(2 1、10.0 kg)を溶解して冷却した(約4°C)溶液に、トリエチルアミン(8.0 kg)を、バッチ温度が10°Cを超えない速度で加えた。溶液を約30分間攪拌した後、バッチ温度を10°C以下に保ちつつ、塩化チオニル(9.0 kg)を加えた。添加完了後、THF(25.0 kg)に4-フルオロアラニン(9.0 kg)を溶解した溶液を、バッチ温度が10°Cを超えない速度で加えた。混合物を約4時間攪拌した後、酢酸イソプロピル(87.0 kg)で希釈した。この溶液を、順に水酸化ナトリウム水溶液(50.0 Lの水に2.0 kgを溶解)、水(40.0 L)、塩化ナトリウム水溶液(40.0 Lの水に10.0 kgを溶解)で洗浄した。この有機溶液を減圧蒸留により濃縮した後、ヘプタンを加え、固体の沈殿物を得た。遠心分離により固体を回収し、約35°Cで減圧乾燥し、表題化合物を得た(10.0 kg)。
【0077】
1.5 1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボニルクロリドの調製
THF(11 kg)とN, N-ジメチルホルムアミド(DMF。0.02 kg)の混合液に1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボン酸(2.0 kg)を溶解した溶液に、塩化オキサリル(1.0 kg)を、バッチ温度が30°Cを超えない速度で加えた。それ以上処理せずに、この溶液を次の段階で使用した。
【0078】
1.6 N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの調製
前段階の1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボニルクロリドを含む溶液を、THF(27.0 kg)と水(13.0 kg)に4-(6,7-ジメトキシキノリン-4-イロキシ)-フェニルアミン(3.0 kg)と炭酸カリウム(4.0 kg)を溶解した混合物に、バッチ温度が30°Cを超えない速度で加えた。反応完了後(通常、10分間)、水(74.0 kg)を加えた。混合物を15〜30°Cで約10時間攪拌すると、反応産物が沈殿した。反応産物を濾過により回収し、事前に調整したTHF(11.0 kg)と水(24.0 kg)の溶液で洗浄し、約65°Cで約12時間、減圧乾燥し、表題化合物(遊離塩基、5.0 kg)を得た。1H NMR (400 MHz、d6-DMSO): d 10.2 (s、1H)、10.05 (s、1H)、8.4 (s、1H)、7.8 (m、2H)、7.65 (m、2H)、7.5 (s、1H)、7.35 (s、1H)、7.25 (m、2H)、7.15(m、2H)、6.4 (s、1H)、4.0 (d、6H)、1.5 (s、4H). LC/MS: M+H= 502.
【0079】
1.7 N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩(化合物 (I))の調製
L-リンゴ酸(2.0 kg)を水(2.0 kg)に溶解した溶液を、エタノールにシクロプロパン‐1,1‐ジカルボン酸[4‐(6,7‐ジメトキシキノリン‐4‐イロキシ)‐フェニル]‐アミド(4‐フルオロフェニル)‐アミド遊離塩基(1 5、5.0 kg)を溶解した溶液に、バッチ温度を約25°Cに維持しつつ加えた。次に、炭素(0.5 kg)とチオールシリカ(0.1 kg)を加え、その混合物を約78°Cまで加熱し、その時点で水(6.0 kg)を加えた。反応混合物を濾過し、イソプロパノール(38.0 kg)を加え、温度が約25°Cに下がるまで放置した。その産物を濾過により回収し、イソプロパノール(20.0 kg)で洗浄し、約65°Cで乾燥させ、化合物 (I)(5.0 kg)を得た。
【0080】
実施例2:結晶質化合物 (I)、形態 N-1の調製
1 Lの反応器にテトラヒドロフラン(12 mL/gバルクLR(限定試薬)、1.20 L)およびN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド(100 g、1.00当量、100.00 g)およびL-リンゴ酸(1.2当量(モル濃度)、32.08 g)を投入し、溶液を作った。水(0.5317 mL/gバルクLR、53.17 mL)を加え、溶液を60 °Cに熱し、固体が完全に溶解するまで1時間、その温度に保った。溶液をポリッシュフィルタに通した。
【0081】
60 °Cで8時間をかけてアセトニトリル(12 mL/gバルクLR、1.20 L)を加えた。溶液を60 °Cに維持し、10時間放置した。その後、容器を20 °Cに冷却し、その温度で1時間放置した。固体を濾過し、アセトニトリル(12 mL/gバルクLR、1.20 L)で洗浄した。固体を60 °C(25 mm Hg)で6時間乾燥させ、白色の結晶質固体として化合物 (I)、形態 N-1を得た(108 g、0.85当量、108.00 g、収量85.22%)。
【0082】
実施例3:結晶質化合物 (I)、形態 N-1の別の調製
190 mLのテトラヒドロフラン(110 mL)、メチルイソブチルケトン、29 mLの水で溶液を作った。次に、20 mLの溶液を琥珀色の遮光ビンに移し、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩を、混濁したスラリーが形成されるまで加えて飽和させ、室温で2時間以上攪拌し、寝かせる。ブフナー漏斗で濾過して固体を除去し、透明な飽和溶液状態にした。
【0083】
それとは別に、化合物 (I) の2回分のバッチから次の量を取り、粉末ブレンドを調製した。(1) バッチ1から300 mg。ラマン分光分析で約41%の化合物 (I)、形態 N-1および約59%の化合物 (I)、形態 N-2を含む。 (2) バッチ2から200 mg。XPRDパターンが化合物 (I)、形態 N-2と類似。
【0084】
この化合物 (I)、形態 N-1と化合物 (I)、形態 N-2の粉末ブレンドを飽和溶液に加え、室温でマグネチックスタラーを使い攪拌し、スラリーを25日間寝かせた。その後、スラリーをサンプリングし、ブフナー漏斗で濾過し、162 mgのウェットケーキを得た。ウェットケーキを真空オーブン中で45°Cで乾燥させ、128 mg のN-1形態の結晶質化合物 (I) を得た。
【0085】
実施例4:結晶質化合物 (I)、形態 N-2の調製
4.1 結晶質化合物 (I)、形態 N-2の種 結晶の調製
25 mLねじ口バイアルで、20 mLのアセトンと300 mgの遊離塩基N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドを混合し、溶液を調製した。次に、マグネチックスタラーで攪拌しながら、0.79MのL-リンゴ酸の保存溶液0.658 mLを加えた。この溶液を室温で24時間、攪拌した。その後、試料を0.45μm PTFE フィルターカートリッジを通して吸引濾過し、室温で1夜、減圧乾燥させた。
【0086】
4.2 結晶質化合物 (I)、形態 N-2の調製
反応器に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド(48 g、1.00当量、48.00 g)およびテトラヒドロフラン(16.5 mL/gバルクLR、792.00 mL)を加えた。水分含有量を1重量%に調整した。溶液を60°Cに熱した。溶解後、溶液をポリッシュフィルタで濾過し、第1の溶液を得た。
【0087】
別の反応器で、メチルイソブチルケトン(10 mL/gバルクLR、480.00 mL)およびテトラヒドロフラン(1 mL/gバルクLR、48.00 mL)にL-リンゴ酸(1.2当量(モル濃度)、15.40 g)を溶解した。次に、50°Cで第1の溶液に50 mLのL-リンゴ酸溶液を加えた。種結晶を加え(1%、480 mg)、50°Cでリンゴ酸溶液を添加漏斗で滴下して加えた(1.3 mL/分(3時間))。そのスラリーを50°Cで18時間放置した後、30分間で25°Cに冷却した。固体を濾過し、20%テトラヒドロフラン/メチルイソブチルケトン(10V、480 mL)で洗浄した。固体を60 °Cで5時間、減圧乾燥させ、オフホワイト色の結晶質固体の化合物 (I)(55.7 g、0.92当量、55.70 g、収率91.56%)を得た。
【0088】
実施例5:結晶質化合物 (III)、形態 N-1の調整
テトラヒドロフラン(THF)中でスラリー状にしたN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのDL-リンゴ酸塩を1 mL分取し、ハーフドラムのバイアルに入れ、ホットプレート上で60 °Cに熱した。次に、ほぼ透明な溶液が得られるまで、テトラヒドロフランを滴下した。バイアルに蓋をし、ホットプレートから降ろし、室温で振動を加えずに放置し、平衡化した。数時間後に結晶が析出し、溶液はそのまま1夜放置し、析出を完了させた。生じたスラリーを数滴、ガラススライドに落とし、顕微鏡で分析した。結晶質の物質は、最長60ミクロンまでの多数の細長い板状の物質で構成されていた。
【0089】
結晶質化合物 (III)、形態 N-1の別の調製
反応器に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド(15 g、1.00当量、15.00 g)とテトラヒドロフラン(16.5 mL/gバルクLR、792.00 mL)を入れた。水分含有量は1重量%に調整した。溶液を60 °Cに熱した。溶解後、溶液をポリッシュフィルタで濾過し、第1の溶液を得た。
【0090】
別の反応器で、DL-リンゴ酸(1.2当量(モル濃度)、4.53 g)を、メチルイソブチルケトン(8 mL/gバルクLR、120.00 mL)およびテトラヒドロフラン(1 mL/gバルクLR、15.00 mL)に溶解した。次に、この溶液20 mLを、50 °Cで第1の溶液に加えた。50°Cでリンゴ酸溶液を添加漏斗で滴下して加えた(1.3 mL/分(3時間))。そのスラリーを50°Cで18時間放置した後、30分間で25°Cに冷却した。固体を濾過し、20% THF/MIBK(10V、150 mL)で洗浄した。固体を60 °Cで5時間減圧乾燥し、オフホワイト色の固体の化合物 (III)(15.52 g、収率86.68%)を得た。
【0091】
実施例6:非晶質化合物 (I) の調製
5 gのN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩およびメタノールとジクロロメタンの1:1(v:v)混合液250 mLで溶液を調製した。この不透明な溶液を0.45ミクロンのフィルターで濾過し、透明な黄色を帯びた溶液を得た。溶液を12.9 cc/分の速度でスプレードライヤーのノズルを通し、10.9 L/分の速度で吹き込まれる窒素ガスで微粒化した。サイクロンのインレットの温度を65 °Cに設定し、液滴を乾燥させた。乾燥した非晶質粉末(1.5 g)を回収した(収率= 30%)。
【0092】
特徴づけの例
I. ジメチルスルホキシド溶液中でのNMRスペクトル
I.1 化合物 (I)、形態 N-1
1H NMR (400 MHz、d6-DMSO): d 1.48 (s、1 H)、2.42-2.48 (m、1 H)、2.60-2.65 (m、1 H)、3.93-3.96 (m、6 H)、4.25-4.30 (dd、1 H、J = 5、8 Hz)、6.44 (d、1H、J = 5 Hz、1 H)、7.12-7.19 (m、2 H)、7.22-7.26 (m、2 H)、7.40 (s、1 H)、7.51 (s、1 H)、7.63-7.68 (m、2 H)、7.76-7.80 (m、2 H)、8.46-8.49 (m、1 H)、10.08 (s、1 H)、10.21 (s、1 H).
【0093】
13C NMR(d6-DMSO):15.36、31.55、55.64、55.67、66.91、99.03、102.95、107.66、114.89、115.07、115.11、121.17、122.11、122.32、122.39、135.15、136.41、146.25、148.7、149.28、149.38、152.54、157.03、159.42、160.02、168.07、171.83、174.68。
【0094】
I.2 化合物 (I)、形態 N-2
1H NMR (400 MHz、d6-DMSO):d 1.48 (s、1 H)、2.42-2.48 (m、1 H)、2.60-2.65 (m、1 H)、3.93-3.96 (m、6 H)、4.25-4.30 (dd、1 H、J = 5、8 Hz)、6.44 (d、J = 5 Hz、1 H)、7.12-7.19 (m、2 H)、7.22-7.26 (m、2 H)、7.40 (s、1 H)、7.51 (s、1 H)、7.63-7.68 (m、2 H)、7.76-7.80 (m、2 H)、8.46-8.49 (m、1 H)、10.08 (s、1 H)、10.21 (s、1 H)。
【0095】
13C NMR(d6-DMSO):15.36、31.55、55.64、55.67、66.91、99.03、102.95、107.66、114.89、115.07、115.11、121.17、122.11、122.32、122.39、135.15、136.41、146.25、148.7、149.28、149.38、152.54、157.03、159.42、160.02、168.07、171.83、174.68.
【0096】
I.3 化合物 (III)、形態 N-1
1H NMR (400 MHz、d6-DMSO):d 1.48 (s、1 H)、2.42-2.48 (m、1 H)、2.60-2.65 (m、1 H)、3.93-3.96 (m、6 H)、4.25-4.30 (dd、1 H、J = 5、8 Hz)、6.44 (d、J = 5 Hz、1 H)、7.12-7.19 (m、2 H)、7.22-7.26 (m、2 H)、7.40 (s、1 H)、7.51 (s、1 H)、7.63-7.68 (m、2 H)、7.76-7.80 (m、2 H)、8.46-8.49 (m、1 H)、10.08 (s、1 H)、10.21 (s、1 H)。
【0097】
13C NMR(d6-DMSO):15.36、31.55、55.64、55.67、66.91、99.03、102.95、107.66、114.89、115.07、115.11、121.17、122.11、122.32、122.39、135.15、136.41、146.25、148.7、149.28、149.38、152.54、157.03、159.42、160.02、168.07、171.83、174.68。
【0098】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の固体形態の特徴づけ
II. 粉末X線回折(XRPD)実験
粉末X線回折(XRPD)パターンは、自動化されたXYZステージ、自動試料位置調整用のレーザービデオ顕微鏡、HiStar 2次元エリア検出器を装備したBruker AXS C2 GADDS回折装置を使い検出した。放射線源には銅を使用し(Cu Kα = 1.5406 Å)、電圧を40 kV、電流を40 mAに設定し、X線光学系はシングル・ゲーベル多層膜ミラーと0.3 mmのピンホール・コリメータを組み合わせた構成である。ビームの発散、すなわち試料面上でのX線ビームの有効サイズは約4 mmであった。θ-θ連続スキャンモードを採用し、試料と検出器の距離は20 cmとし、2θの有効範囲は3.2 °〜29.8 °であった。試料は室温で(約18 °Cから約25 °C)、受け取った状態の粉のまま、粉砕せずに使い、平板状の標本として調製された。約1〜2 mgの試料をガラススライド上で軽く押し、表面を平らにした。典型的には120秒間、試料にX線ビームを照射した。ビームの発散(すなわち、X線スポットの有効サイズ)は約4mmである。別の方法として、粉末試料を直径1 mm以下の密封ガラスキャピラリーに入れた。試料と検出器の距離を15 cmとし、キャピラリーを回転させながらデータを収集した。データは3≦2θ≦35°の範囲で収集し、試料への照射時間は2,000秒以上であった。結果として得た2次元回折アークを積分することにより、範囲3〜35 °2θ± 0.2 °2θおよびステップサイズ0.02 °2θの従来の1次元XRPDパターンを求めた。データ収集用のソフトウェアとしてWNT 4.1.16用GADDSを使用し、データの分析と表示にはDiffrac Plus EVA v 9.0.0.2またはv 13.0.0.2を使用した。
【0099】
II.1 化合物 (I)、形態 N-1
図 1に、結晶質化合物 (I)、形態 N-1の室温(約25 °C)での回折実験によるXRPDパターンを示す。ピークのリストは前掲の表2に示した。2θの値19.4、21.5、22.8、25.1、および27.6(± 0.2 °2θ)が、結晶質化合物 (I)、形態 N-1の特徴づけに有用である。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-1の特徴づけに十分な場合がある。
【0100】
II.2 化合物 (I)、形態 N-2
図 8に、結晶質化合物 (I)、形態 N-2の室温(約25 °C)での回折実験によるXRPDパターンを示す。ピークのリストは前掲の表2に示した。2θの値20.9および21.9(± 0.2 °2θ)結晶質化合物 (I)、形態 N-2の特徴づけに有用である。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0101】
II.3 化合物 (III)、形態 N-1
図 15に、実験とシミュレーションにより、25°Cで回転式キャピラリー標本を使って求めた結晶質化合物 (III)、形態 N-1のXRPDパターンを示す。ピークのリストは前掲の表2に示した。ピークのリスト全体またはその一部が、結晶質化合物 (III)、形態 N-1の特徴づけに十分な場合がある。
【0102】
II.4 非晶質化合物 (I)
図 22に、室温(約25 °C)での回折実験で求めた非晶質化合物 (I) のXRPDパターンを示す。スペクトルの特徴として、ピークの幅が広く、鋭いピークがない。これは非晶質物質と一致する。
【0103】
III.化合物 (III)、形態 N-1の単位格子X線回折実験
Bruker-Nonius CAD4連続回折装置でデータを収集した。単位格子パラメータは、回折実験で高角反射データを25個に設定し、最小二乗法で解析して求めた。回折強度はCu Ka放射線(l = 1.5418 Å)を使い、定温でθ-2θ可変スキャン法により求め、ローレンツ偏光因子に関してのみ補正を加えた。バックグラウンド計数値は、スキャンの半分に時間に関するスキャンの極値で収集した。別の方法として、Bruker-Nonius Kappa CCD 2000システムでCu Ka放射線(l = 1.5418 Å)を使い、単結晶データを収集した。測定した回折強度データの指数付けと処理は、HKL2000ソフトウェア・パッケージ(Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W.C. Jr & Sweet, R.M. (Academic, NY), Vol. 276, pp.307-326)を使い、Collectプログラム・スイート( Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B.V., 1998)で行った。別の方法として、Bruker-AXS APEX2 CCDシステムでCu Ka放射線(l = 1.5418 Å)を使い、単結晶データを収集した。測定した回折強度データの指数付けと処理は、APEX2ソフトウェア・パッケージ/プログラム・スイート( Data collection and processing user interface: APEX2 User Manual, v1.27)で行った。指示がある場合は、データ収集中に、結晶をOxfordクライオ・システム( Cryostream cooler: J. Cosier and A.M. Glazer, J. Appl. Cryst., 1986, 19, 105)の低温流中で冷却した。
【0104】
構造の解析は直接法で行い、SDPソフトウェア・パッケージ(SDP, Structure Determination Package, Enraf-Nonius, Bohemia NY 11716。SDPソフトウェアで、f'およびf''を含む散乱因子には、“International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1の値を使用した)にわずかな局所的変更を加えるか、または結晶学用パッケージのMAXUS(maXus solution and refinement software suite: S. Mackay, C.J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus:回折データによる結晶構造の解析および精密化を行うためのコンピュータ・プログラム)もしくはSHELXTL(Data collection and processing user interface: APEX2 User Manual, v1.27)を使い、観測された反射に基づく精密化を行った。
【0105】
求めた原子パラメータ(座標と温度因子)に対し、フルマトリックス最小二乗法を使い、精密化を行った。精密化で値を最小にする関数はSw(|Fo| - |Fc|)2である。RをS ||Fo| - |Fc||/S |Fo|と定義され、Rw = [Sw( |Fo| - |Fc|)2/Sw |Fo|2]1/2において、wは観測された回折強度の誤差に基づく適当な重み関数である。差フーリエ合成図の検討を精密化の全段階で行った。等方性温度因子と仮定した理想位置に水素原子を導入した計算では、水素のパラメータに変化はなかった。
【0106】
参考文献の説明に従い、シミュレーションによる「ハイブリッド」の粉末X線回折パターンを作成した(Yin. S.; Scaringe, R. P.; DiMarco, J.; Galella, M. and Gougoutas, J. Z., American Pharmaceutical Review, 2003, 6,2, 80)。CellRefine.xlsプログラムを使い、結晶格子の精密化を行うことにより、室温での結晶格子パラメータを求めた。プログラムに入力した数値は、室温での粉末X線回折パターンの実験結果から求めた約10個の反射の2θ位置を含む。低温で収集した単結晶データに基づき、それに対応するミラー指数hklを割り当てた。この手順の最初に求めた室温での結晶格子に、低温で決定された分子構造を挿入することにより、新たな(ハイブリッド)XRPDを計算した(Alexまたは LatticeViewのいずれかのソフトウェア・プログラムを用いた)。分子のサイズならびに形状および結晶格子の原点に対する分子の位置が変わらず、しかも分子間の距離が結晶格子と共に拡大できるよう、分子を挿入した。
【0107】
単結晶回折解析のために、実施例5に記載した結晶のスラリーから、40 x 30 x 10ミクロンの単結晶を選択した。選択した結晶を少量の軽いグリースで細いガラス繊維に接着し、回転する銅陽極管を備えたBruker ApexII単結晶回折装置に、室温で装着した。
【0108】
結晶質化合物 (III)、形態N-1は、表4に報告する値とほぼ等しい単位格子パラメータを特徴とする。単位格子パラメータは約25°Cで測定した。
【0109】
単位格子中に4個の化学式単位を持つ単斜晶系の空間群P21/nにおける構造の解析および精密化は、通常の方法で行った。この構造はキノリン窒素原子がプロトン化されたN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの陽イオンと、単独でイオン化されたリンゴ酸の陰イオンを、1:1の比率で含んでいた。さらに、この結晶は、L-リンゴ酸イオンおよびD-リンゴ酸イオンを1:1の比率で含んでいた。表5に、約25°Cで計算した化合物 (III)、形態 N-1に関する分率原子座標を示す。
【0110】
単結晶X線回折データに基づき、図15に示すシミュレートしたパターンと実質的に一致するシミュレートした粉末X線回折(XRPD)パターン、および図15に示す実験結果のパターンと実質的に一致する観測XRPDパターン、またはそのいずれかにより、結晶質化合物 (III)、形態 N-1の特徴を決定できる。
【0111】
IV. 固体核磁気共鳴(SSNMR)
すべての固体C-13 NMR測定をBruker DSX-400, 400 MHz NMR回折装置で行った。高出力プロトンデカップリングならびにTPPMパルスシーケンス、および傾斜振幅交差分極法(ramp amplitude cross-polarization)(RAMP-CP)を、約12 kHzでのマジック角回転法(MAS)と併用し、高分解能スペクトルを得た(A.E. Bennett et al, J. Chem. Phys.,1995, 103, 6951),(G. Metz, X. Wu and S.O. Smith, J. Magn. Reson. A,. 1994, 110, 219-227)。円筒型のジルコニア製ローターに、約70 mgの試料を詰め、各実験で使用した。化学シフト(d)の指標として外部のアダマンタンを使い、高周波共鳴を38.56 ppmに設定した(W.L. Earl and D.L. VanderHart, J. Magn. Reson., 1982, 48, 35-54)。
【0112】
IV.1 化合物 (I)、形態 N-1
図2に結晶質化合物 (I)、形態 N-1の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-1の特徴づけに十分な場合がある。
【0113】
SS 13C NMRのピーク:18.1、20.6、26.0、42.9、44.5、54.4、55.4、56.1、70.4、99.4、100.1、100.6、114.4、114.9、 115.8、119.6、120.1、121.6、123.2、124.1、136.4、138.6、140.6、145.4、150.1、150.9、156.2、157.4、159.4、164.9、167.1、170.8、175.7、および182.1 ppm、± 0.2 ppm。
【0114】
図3に結晶質化合物 (I)、形態 N-1の固体15N NMRスペクトルを示す。スペクトルのピークは118.6、119.6、120.7、134.8、167.1、176.0、および180 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-1の特徴づけに十分な場合がある。
【0115】
図 4に結晶質化合物 (I)、形態 N-1の固体19F NMRスペクトルを示す。スペクトルのピークは-121.6、-120.8、および-118.0 ppm、± 0.2 ppmである。
【0116】
IV.2 化合物 (I), 形態 N-2
図9に結晶質化合物 (I)、形態 N-2の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0117】
SS 13C NMRのピーク:20.5、21.8、23.0、25.9、26.4、38.0、41.7、54.7、55.8、56.2、56.6、69.7、99.4、100.0、100.4、100.8、102.3、114.5、115.5、116.7、119.0、120.2、121.1、121.2、122.1、122.9、124.5、136.0、137.3、138.1、138.9、139.5、140.2、144.9、145.7、146.1、150.7、156.7、157.7、159.6、159.7、165.1、167.0、168.0、171.5、177.3、179.3、180.0、および180.3 ppm、± 0.2 ppm。
【0118】
図 10 に結晶質化合物 (I)、形態 N-2の固体15N NMRスペクトルを示す。スペクトルのピークは118.5、120.8、135.1、167.3、および180.1 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0119】
図 11に結晶質化合物 (I)、形態 N-2の固体19F NMRスペクトルを示す。スペクトルのピークは-121.0および-119.1 ppm、± 0.2 ppmである。それらのピークは個別に、または両方で、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0120】
IV.3 化合物 (III)、形態 N-1
図16に結晶質化合物 (III)、形態 N-1の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、結晶質化合物 (III)、形態 N-1の特徴づけに十分な場合がある。
【0121】
SS 13C NMRのピーク:20.8、26.2、44.8、55.7、70.7、100.4、101.0、114.7、115.2、116.0、119.7、120.4、121.6、124.4、136.9、138.9、141.1、145.7、150.3、156.5、157.6、159.6、165.2、167.4、171.2、176.3、および182.1 ppm、± 0.2 ppm。
【0122】
図 17に結晶質化合物 (III)、形態 N-1の固体15N NMRスペクトルを示す。スペクトルのピークは119.6、134.7、および175.5 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、結晶質化合物 (III)、形態 N-1の特徴づけに十分な場合がある。
【0123】
図 18に結晶質化合物 (III)、形態 N-1の固体19F NMRスペクトルを示す。スペクトルのピークは-120.5 ppm、± 0.2 ppmである。
【0124】
IV.4 化合物 (I)、非晶質
図 23に非晶質化合物 (I) の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、非晶質化合物 (I) の特徴づけに十分な場合がある。
【0125】
SS 13C NMRのピーク(ppm):12.2、17.8、20.3、21.8、27.2、33.8、41.7、56.9、69.9、99.9、102.2、115.6、122.2、134.4、137.8、142.9、149.1、150.9、157.3、159.7、167.0、171.7、173.1、177.4、および179.5 ppm、± 0.2 ppm。
【0126】
図 24に非晶質化合物 (I) の固体15N NMRスペクトルを示す。スペクトルのピークは120.8、131.8、174.7、および178.3 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、非晶質化合物 (I) の特徴づけに十分な場合がある。
【0127】
図 25に非晶質化合物 (I) の固体19F NMRスペクトルを示す。スペクトルのピークは-118.9 ppm、± 0.2 ppmである。
【0128】
V. 熱特性測定
熱重量分析(TGA)
TGA測定は、オープンパンを装備したTA Instruments(TM)モデルQ500または2950で実施した。試料(約10-30 mg)を、事前に風袋調節したプラチナパンに乗せた。試料の重量はこの装置により、1000分の1ミリグラム単位まで正確に測定・記録された。加熱炉は100 mL/分の窒素ガスでパージした。10°C/分の加熱速度で、室温から300°Cの間のデータを収集した。
【0129】
示差走査熱量(DSC)分析
DSC測定は、オープンパンを装備したTA Instruments(TM)モデルQ2000、Q1000または2920で実施した。試料(約2-6 mg)をアルミニウムパンの上で計量し、100分の1ミリグラム単位まで正確に記録し、DSCに移した。装置は50 mL/分の窒素ガスでパージした。10°C/分の加熱速度で、室温から300°Cの間のデータを収集した。データをプロットし、下向きの吸熱ピークを求めた。
【0130】
V.1 化合物 (I), 形態 N-1
図 5に示す結晶質化合物 (I)、形態 N-1のTGAサーモグラムで、170°Cにおける重量減少は約0.4重量%である。
【0131】
図 6に示す結晶質化合物 (I)、形態 N-1のDSCサーモグラムで、融点は約187°Cである。
【0132】
V.2 化合物 (I)、形態 N-2
図 12に示す結晶質化合物 (I)、形態 N-2のTGAサーモグラムで、170°Cにおける重量減少は約0.1重量%である。
【0133】
図 13に示す結晶質化合物 (I)、形態 N-2のDSCサーモグラムで、融点は約186°Cである。
【0134】
V.3 化合物 (III)、形態 N-1
図 19に示す結晶質化合物 (III)、形態 N-1のTGAサーモグラムで、170°Cにおける重量減少は約0.2重量%である。
【0135】
図 20に示す結晶質化合物 (III)、形態 N-1のDSCサーモグラムで、融点は約186°Cである。
【0136】
V.2 化合物 (I)、非晶質
図 26に非晶質化合物 (I) のDSCを示す。
【0137】
VI. 水蒸気吸着等温線測定
水蒸気吸着等温線は、約10 mgの試料を使い、VTI SGA-100 Symmetric Vapor Analyzerで求めた。試料を60°C で、10分間の減少速度が0.0005 重量%/分になるまで乾燥させた。試料を25°Cで、3または4、5、15、25、35、45、50、65、75、85、および95% RHで測定した。35分間の減少速度が0.0003重量%/分に達した時、または最長600分間で、各RHにおける平衡に達した。
【0138】
VI.1 化合物 (I)、形態 N-1
図 7に結晶質化合物 (I)、形態 N-1の水蒸気吸着等温線を示す。
【0139】
VI.2 化合物 (I)、形態 N-2
図 14に結晶質化合物 (I)、形態 N-2の水蒸気吸着等温線を示す。
【0140】
VI.3 化合物 (III)、形態 N-1
図 21に結晶質化合物 (III)、形態 N-1の水蒸気吸着等温線を示す。
【0141】
VI.4 化合物 (I)、非晶質
図 27に非晶質化合物 (I) の水蒸気吸着等温線を示す。
【0142】
上記の開示内容は、明確性と理解の促進を目的として、図面および例を使い、ある程度詳細に記載されている。本発明は、さまざまな特定および好ましい実施形態および技法に参照して記載されている。しかし、本発明の主旨および範囲内にとどまりつつ、多数の変形および修正を加えることができると理解するものとする。添付した請求項の範囲内で、変更および修正を実施できることは、当業者には明白なはずである。従って、上記の説明は、例示を意図したものであり、限定的ではないと理解するものとする。従って、本発明の範囲は、上記の説明を参照して決定すべきではなく、以下に添付する請求項、および係る請求項に関して認められる同等物の全範囲を参照して決定すべきである。
【技術分野】
【0001】
関連出願の相互参照
本願は2009年1月16日出願の米国仮特許出願No. 61/145,421号に対する優先権を主張し、その仮特許出願は全体として参照により本明細書に組み込まれる。
【0002】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドリンゴ酸塩およびN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の結晶質形態ならびに非晶質形態に関する。N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩は、(1) L-リンゴ酸塩、(2) D-リンゴ酸塩、(3) D,L-リンゴ酸塩、および (4) それらの混合のいずれかを含む。本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩を含む 医薬組成物にも関する。
【0003】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩の結晶質形態または非晶質形態を含む医薬組成物にも関する。
【0004】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩を投与することを含む癌の治療方法にも関する。
【0005】
本発明はさらに、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの一以上のリンゴ酸塩の結晶質形態または非晶質形態を投与することを含む癌の治療方法に関する。
【背景技術】
【0006】
従来、癌治療における劇的な改善は、新規の機序を介して作用する治療薬の同定と関連づけられてきた。プロテインキナーゼ活性を通じて行われるシグナル伝達は、腫瘍細胞の特徴の多くをもたらす原因であり、プロテインキナーゼ活性の調節は、癌治療に利用可能な機序として知られている。プロテインキナーゼによるシグナル伝達は特に、例えば甲状腺癌、胃癌、頭頸部癌、肺癌、乳癌、前立腺癌、結腸直腸癌、および脳腫瘍細胞の拡大ならびに増殖に関連する。
【0007】
プロテインキナーゼは受容体型と非受容体型に分類できる。受容体型チロシンキナーゼは多数の膜貫通受容体で構成され、その生物活性は多岐にわたる。受容体型チロシンキナーゼに関しては、Plowman et al., DN&P 7(6): 334-339, 1994で詳細に論じられている。プロテインキナーゼおよびそのリガンドは、種々の細胞活動においてきわめて重要な役割を果たすため、プロテインキナーゼの酵素活性が脱調節を受けることにより、癌と関連する無制御の細胞増殖を含め、細胞の性質が変性を起こす場合がある。腫瘍学的な適応症に加え、キナーゼによるシグナル伝達の変性は、例えば免疫障害、循環器系疾患、炎症性疾患、変性疾患など、多数の他の疾患にも関与している。従って、プロテインキナーゼは低分子薬の創薬に関する魅力的な標的である。抗血管新生活性および抗増殖活性を対象とする低分子活性調節の特に魅力的な標的が、受容体型チロシンキナーゼのRet、c-Met、VEGFR2である。
【0008】
c-Metキナーゼは、Met、Ron、Seaを含むヘテロ二量体の受容体型チロシンキナーゼ(RTK)サブファミリーのプロトタイプの1種である。c-Metの内因性リガンドは、血管新生の強力な誘導因子である肝細胞増殖因子(HGF)である。HGFがc-Metと結合することにより、自己リン酸化を介して受容体の活性化を誘導し、それにより受容体依存性のシグナル伝達が増加し、細胞の増殖と浸潤が促進される。抗HGF抗体またはHGFアンタゴニストは、in vivoで腫瘍の転移を阻害することが報告されている(参照:Maulik et al Cytokine & Growth Factor Reviews 2002 13, 41-59)。乳腺腫瘍、結腸腫瘍、腎腫瘍、肺腫瘍、扁平上皮癌、骨髄性白血病、血管腫、黒色腫、星細胞腫(神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫を含む)などの多様な腫瘍タイプで、c-Met、VEGFR2、およびRetまたはそのいずれかの過剰発現が実証されている。Retタンパク質はチロシンキナーゼ活性を持つ膜受容体である。家族性甲状腺髄様癌の ほとんどで、Retに変異が起きている。これらの変異がRetのキナーゼ機能を活性化し、それを癌遺伝子産物に変える。
【0009】
EGF、VEGF、エフリンのシグナル伝達の阻害は、腫瘍の増殖および生存に必要な重要な2つの細胞プロセスである細胞増殖および血管新生を防ぐ(Matter A. Drug Disc. Technol. 2001 6, 1005-1024)。キナーゼKDR(キナーゼ挿入ドメイン受容体チロシンキナーゼ)および(fms様チロシンキナーゼ4)はどちらも内皮増殖因子(VEGF)受容体である。EGF、VEGF、エフリンのシグナル伝達の阻害は、腫瘍の増殖および生存に必要な重要な2つの細胞プロセスである細胞増殖および血管新生を防ぐ(Matter A. Drug Disc. Technol. 2001 6, 1005-1024)。EGF受容体およびVEGF受容体は、低分子阻害の望ましい標的である。
【0010】
従って、特に上記のRet、c-Met、VEGFR2を含むキナーゼのシグナル伝達を特異的に阻害し、調節し、かつ修飾するか、またはそのいずれかを行う低分子化合物は、異常な細胞増殖および血管新生を伴う病態の治療または予防手段として特に望ましい。そのような低分子の一つが、下記の化学構造式を持つN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドである。
WO 2005/030140に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの合成(実施例12、37、38、48)を説明し、かつ、キナーゼのシグナル伝達の阻害、調節、修飾のすべて、またはそのいずれかを行う分子の治療活性を開示する(アッセイ、表4、289項)。実施例48はWO 2005/030140の請求項[0353]に記載する。
【0011】
薬剤の開発者は、治療薬としての有効性に加え、薬剤としての加工、製造、保存安定性、有用性の全部または一部に関する性質を備えた適当な形態の治療薬の提供に努める。従って、これらの望ましい性質の一部または全部を備えた形態の発見が、創薬において不可欠である。
【0012】
出願人は、癌などの増殖性疾患の治療を目的とする医薬組成物での使用に適した性質を備えた薬剤N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの塩形態を発見した。本発明の新規塩形態は結晶質および非晶質の形態で存在する。
【発明の概要】
【0013】
本発明は明細書中に記載するN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩、明細書中に記載するその医薬組成物、および明細書中に記載するその使用法に関する。
【0014】
別の局面は、明細書中に記載するN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の結晶質および非晶質の形態、明細書中に記載するその医薬組成物、および明細書中に記載するその使用法に関する。
【図面の簡単な説明】
【0015】
【図1】に結晶質化合物 (I)、形態N-1の25 °CでのXRPD回折実験で得たパターンを示す。
【図2】に結晶質化合物 (I)、形態 N-1の固体13C NMRスペクトルを示す。
【図3】に結晶質化合物 (I)、形態 N-1の固体15N NMRスペクトルを示す。
【図4】に結晶質化合物 (I)、形態 N-1の固体19F NMRスペクトルを示す。
【図5】に結晶質化合物 (I)、形態 N-1の熱重量分析(TGA)結果を示す。
【図6】に結晶質化合物 (I)、形態 N-1の示差走査熱量分析(DSC)結果を示す。
【図7】に結晶質化合物 (I)、形態 N-1の水分吸着を示す。
【図8】に結晶質化合物 (I)、形態N-2の25 °CでのXRPD回折実験で得たパターンを示す。
【図9】に結晶質化合物 (I)、形態 N-2の固体13C NMRスペクトルを示す。
【図10】に結晶質化合物 (I)、形態 N-2の固体15N NMRスペクトルを示す。
【図11】に結晶質化合物 (I)、形態 N-2の固体19F NMRスペクトルを示す。
【図12】に結晶質化合物 (I)、形態 N-2の熱重量分析(TGA)結果を示す。
【図13】に結晶質化合物 (I)、形態 N-2の示差走査熱量分析(DSC)結果を示す。
【図14】に結晶質化合物 (I)、形態 N-2の水分吸着を示す。
【図15】に結晶質化合物 (III)、形態N-1の室温でのXRPD回折実験で得たパターンおよびシミュレーションによるパターンを示す。
【図16】に結晶質化合物 (III)、形態 N-1の固体13C NMRスペクトルを示す。
【図17】に結晶質化合物 (III)、形態 N-1の固体15N NMRスペクトルを示す。
【図18】に結晶質化合物 (III)、形態 N-1の固体19F NMRスペクトルを示す。
【図19】に結晶質化合物 (III)、形態 N-1の熱重量分析(TGA)結果を示す。
【図20】に結晶質化合物 (III)、形態 N-1の示差走査熱量分析(DSC)結果を示す。
【図21】に結晶質化合物 (III)、形態 N-1の水分吸着を示す。
【図22】に非晶質化合物 (I) の室温でのXRPDパターンを示す。
【図23】に非晶質化合物 (I) の固体13C NMRスペクトルを示す。
【図24】に非晶質化合物 (I) の固体15N NMRスペクトルを示す。
【図25】に非晶質化合物 (I) の固体19F NMRスペクトルを示す。
【図26】に非晶質化合物 (I) の示差走査熱量分析(DSC)結果を示す。
【図27】に非晶質化合物 (I) の水分吸着を示す。
【発明を実施するための形態】
【0016】
本発明は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの物理化学的性質を、薬剤開発に適するよう改善することに関する。本明細書で開示するのは、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩である。それらの塩の新規の固体形態も開示する。本明細書で開示するリンゴ酸塩およびその結晶質ならびに非晶質の形態は、各々が本発明の個別の局面を構成する。本明細書にはリンゴ酸塩およびその固体形態を記載するが、本発明は開示する塩および固体形態を含む新規組成にも関する。記載する塩ならびに固体形態およびそれらを含む治療用組成物の治療目的の使用は、本発明の個別の局面を構成する。塩およびその固体形態を特徴づけるために使用した技法は、下記の実施例に記載する。これらの技法は、単独で、または組み合わせて、本明細書に開示する塩およびその固体形態を特徴づけるために使用できる。塩およびその固体形態は、開示する図を参照して特徴づけることもできる。
【0017】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドでは、酵素Retの IC50値が約5.2 nM(ナノモル濃度)、酵素c-MetのIC50値が約1.3 nM(ナノモル濃度)であった。このc-Met活性の測定に使用したアッセイ法はWO2005‐030140の請求項[0468]に記載する。
【0018】
RETの生化学活性は、WO2005‐030140に記載するルシフェラーゼ共役化学発光キナーゼアッセイ(LCCA)法を使い測定した。キナーゼ活性はキナーゼ反応後に残ったATPの割合(%)として測定した。残ったATPはルシフェラーゼ - ルシフェリン共役化学発光により検出した。具体的には、2mM ATP、1mMポリEY、15nM RET(N末端に(His)6タグを導入し、バキュロウイルスで発現させたヒトRETキナーゼドメインM700-D1042)を20uLのアッセイ用緩衝液(20mM Tris-HCL pH 7.5、10mM MgCl2、0.01% Triton X-100、1 mM DTT、3mM MnCl2)中で混合し、反応を開始させた。この混合液を常温で2時間インキュベートした後、ルシフェラーゼ - ルシフェリン混合液を加え、Wallac Victor2プレートリーダーを使い、化学発光を測定した。ルシフェラーゼ - ルシフェリン混合液の組成は50 mM HEPES (pH 7.8)、8.5ug/mLシュウ酸 (pH 7.8)、5mM DTT、0.4% Triton X-100、0.25 mg/mLコエンザイムA、63 mM AMP、28 mg/mLルシフェリン、40,000発光単位/mLルシフェラーゼである。
【0019】
リンゴ酸塩s of N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩に関する。これらのリンゴ酸塩は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドとリンゴ酸を化合し、1:1の比でN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩を形成したものである。
【0020】
リンゴ酸の構造式を下記に示す。
不斉炭素原子により、リンゴ酸にはL-リンゴ酸およびD-リンゴ酸の2種類のエナンチオマーが存在する。
【0021】
L-リンゴ酸の構造式
当業者の間では、L-リンゴ酸に対してさまざまな名称または呼称が使われている。例えば、ブタンジオイック酸、ヒドロキシ-、(2S)- (9CI)、ブタンジオイック酸、ヒドロキシ-、(S)-、リンゴ酸、L- (8CI)、リンゴ酸、l- (3CI)、(-)-(S)-リンゴ酸、(-)-ヒドロキシコハク酸、(-)-(L)-リンゴ酸、(-)-リンゴ酸、(2S)-2-ヒドロキシブタンジオイック酸、(2S)-2-ヒドロキシコハク酸、(S)-リンゴ酸、リンゴ酸、L-(-)-リンゴ酸、(L)-リンゴ酸、NSC 9232、S-(-)-リンゴ酸、S-2-ヒドロキシコハク酸などである。
【0022】
D-リンゴ酸の構造式
当業者の間では、D-リンゴ酸に対してさまざまな名称または呼称が使われている。例えば、ブタンジオイック酸、2-ヒドロキシ-、(2R)-、ブタンジオイック酸、ヒドロキシ-、(2R)- (9CI)、ブタンジオイック酸、ヒドロキシ-、(R)-、(+)-リンゴ酸、(2R)-2-ヒドロキシブタンジオイック酸、(2R)-リンゴ酸、(R)-(+)-リンゴ酸、(R)-リンゴ酸、D-(+)-2-ヒドロキシコハク酸、D-(+)-リンゴ酸、D-リンゴ酸などである。
【0023】
上記のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの化学構造式を示す。
化学構造には不斉炭素原子はない。 N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドは一般にさまざまな名称で呼ばれ、それらの名称または呼称の例としては、1,1-シクロプロパンジカルボキサミド、N'-[4-[(6,7-ジメトキシ-4-キノリニル)オキシ]フェニル]-N-(4-フルオロフェニル)- および1,1-シクロプロパンジカルボキサミド、N-[4-[(6,7-ジメトキシ-4-キノリニル)オキシ]フェニル]-N'-(4-フルオロフェニル)- (9CI)などがある。
【0024】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドは、グラム単位(1 kg未満)またはキロ単位(1 kg以上)のいずれかにより、数種類の方法のいずれかに従い調製できる。グラム単位の方法はWO 2005-030140に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの合成法(実施例25、37、38、48)として記載し、それは参照として本明細書の一部とする。別法として、有効化合物を含むN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドを、下記実施例1に示す手順を使い、キロ単位で調製することができる。
【0025】
本発明はN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩に関する。
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩(化合物 (I))
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのD-リンゴ酸塩(化合物 (II))および
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのDL-リンゴ酸塩(化合物 (III))
上記の各々が、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドおよびその他の塩と比較し、改善された性質を持つ。例えば「N-2」など、特定の形態の特徴を示すために本明細書で用いる名称は、同様または同一の物理的および科学的な特徴を持つ他の物質を除外するための限定的な名称ではなく、むしろそのような名称は単なる識別記号として、本明細書に提示する特徴に従い解釈するものとする。
【0026】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩、特に化合物 (I) は、開発のために好ましい医薬品特性の組み合わせを持つ。25°C/60%相対湿度(RH)の条件と40 °C/60% RHの条件において、化合物 (I) の分析結果、純度、水分、溶解度に変化はなかった。DSC/TGAにより、化合物 (I) は185°Cまで安定であることが示された。溶剤損失は観察されなかった。L-リンゴ酸塩による水分吸収は可逆性であり、ヒステリシスは低かった。水分吸収量は90% RHで約0.60 wt%と算出された。L-リンゴ酸塩の合成は収率が良く、純度は90%を超え、医薬組成物中で使用するために十分な溶解度を備えていた。この塩の水分量はカールフィッシャー分析により約0.5 wt%と算出され、これはTGA分析およびGVS分析と相関する。N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのD-リンゴ酸塩 は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩と同じ性質を持つ。
【0027】
化合物 (I) の塩それ自体、およびそれとは別に、その結晶質および非晶質の形態は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの遊離塩基および他の塩と比較し、有益な性質を示す。例えば、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの塩酸塩は、望ましくない感湿性を示し、高湿(湿度75%)高温(40°C)条件に曝露すると、相変化を起こす。マレイン酸塩は溶解度が低かった。酒石酸塩は結晶性が低く、溶解度が低かった。リン酸塩では、H2Oの吸収により重量が8%増加し、これは試験を行った塩の中で最高であった。
【0028】
さまざまな塩の水溶性は、水1 mLに対して10 mgの固体を使い測定した。塩の調製には、ソルトスクリーンを使い、遊離塩基のアセトン溶液を、ある範囲の酸をテトラヒドロフラン(THF)に溶解した保存溶液と、約1:1のモル比で反応させた。下記の表1は遊離塩基と各塩に関する水溶性などのデータの要約である。
【0029】
本発明の別の局面は、本明細書に記載する化合物 (I) のN-1およびN-2結晶質形態またはそのいずれかを含む化合物 (I) 結晶質形態に関する。化合物 (I) の各形態は本発明の個別の局面である。同様に、本発明の別の局面は、本明細書に記載する化合物 (II) のN-1およびN-2結晶質形態またはそのいずれかを含む化合物 (II) 結晶質形態に関する。その各形態も本発明の個別の局面である。当業者に知られているように、結晶性のD-リンゴ酸塩は、結晶性の化合物 (I) と同じ結晶質形態を形成し、同じ性質を持つ。結晶のエナンチオマーの性質について考察したWO 2008/083319を参照のこと。化合物 (I) および (II) の結晶質形態の混合は、本発明の別の局面である。
【0030】
本明細書に記載する化合物 (I) および (II) の結晶性のN-1形態は、以下の1項目以上の特徴を持つ場合がある。
(i) 固体13C NMRスペクトルのピークが18.1、42.9、44.5、70.4、123.2、156.2、170.8、175.7、および182.1 ppm ± 0.2 ppmにある。
(ii) 固体13C NMRスペクトルが図 2に示すパターンと実質的に一致する。
(iii) 粉末X線回折パターン(CuKα λ=1.5418Å)が、以下の中から選択される4点以上のピークを含む。6.4、9.0、12.0、12.8、13.5、16.9、19.4、21.5、22.8、25.1、および27.6 °2θ ± 0.2 °2θ。結晶質形態の測定は室温で行う。
(iv) 粉末X線回折(XRPD)スペクトルが図1に示したパターンと実質的に一致する。
(v) 固体15N NMRスペクトルのピークが118.6、119.6、120.7、134.8、167.1、176.0、および180 ppm ±0.2 ppmにある。
(vi) 固体15N NMRスペクトルが図3に示すパターンと実質的に一致する。
【0031】
化合物 (I) および (II) の結晶質N-1形態の特徴づけに使用できる他の固体状態特性を図に示し、下記の実施例で考察する。結晶質化合物 (I) に関しては、40°C で1週間、75% RHに曝露後、固体状態と結晶化度に変化はなかった。
【0032】
本明細書に記載する化合物 (I) および (II) の結晶質N-2形態は、以下の1項目以上の特徴を持つ。
(i) 固体13C NMRスペクトルのピークが 23.0、25.9、38.0、54.4、56.11、41.7、69.7、102.0、122.5、177.3、179.3、180.0、および180.3 ± 0.2 ppmにある。
(ii) 固体13C NMRスペクトルが図9に示すパターンと実質的に一致する。
(ii) 粉末X線回折パターン(CuKα λ=1.5418Å)が、以下の中から選択される4点以上のピークを含む。6.4、9.1、12.0、12.8、13.7、17.1、20.9、21.9、22.6、および23.7 °2θ ± 0.2 °2θ。結晶質形態の測定は室温で行う。
(iv) 粉末X線回折(XRPD)スペクトルが図8に示すパターンと実質的に一致する。
(v) 固体15N NMRスペクトルのピークが118.5、120.8、135.1、167.3、および180.1 ppmにある。
(vi) 固体15N NMRスペクトルが図10に示すパターンと実質的に一致する。
化合物 (I) および (II) の結晶質N-2形態の特徴づけに使用できる他の固体状態特性を図に示し、下記の実施例で考察する。
【0033】
別の実施形態では、本発明は化合物 (I) の結晶質形態に関し、それは本発明のいかなる局面および実施形態に関して記載される場合も、実質的に純粋なN-1形態である。
【0034】
別の実施形態では、本発明は化合物 (I) の結晶質形態に関し、それは本発明のいかなる局面および実施形態に関して記載される場合も、実質的に純粋なN-2形態である。
【0035】
本発明は化合物 (I) および (II) の非晶質形態にも関する。化合物 (I) の非晶質形態の調製法および固体の性質ならびに特徴は、下記の実施例に記載する。化合物 (I) および (II) の非晶質形態は、本発明の別の局面を表す。
【0036】
本発明のさらに別の局面は、化合物 (I) と化合物 (II) の混合物に関する。混合物は、化合物 (I) と化合物 (II) の合計重量に基づき、ゼロ重量%を超え、100重量%未満の化合物 (I) および100重量%未満でゼロ重量%を超える化合物 (II) を含む。他の実施形態では、混合物はその混合物中の化合物 (I) と化合物 (II) の合計重量に基づき、約1重量%から約99重量%の化合物 (I) および約99重量%から約1重量%の化合物 (II) を含む。さらに別の実施形態では、混合物は化合物 (I) と化合物 (II) の合計重量に基づき、約90重量%から100重量%未満の化合物 (I) およびゼロ重量%を超え、約10重量%までの化合物 (II) を含む。従って、混合物は1〜10重量%の化合物 (I)、11〜20重量%の化合物 (I)、21〜30重量%の化合物 (I)、31〜40重量%の化合物 (I)、41〜50重量%の化合物 (I)、51〜60重量%の化合物 (I)、61〜70重量%の化合物 (I)、71〜80重量%の化合物 (I)、81〜90重量%の化合物 (I)、91〜99重量%の化合物 (I) のいずれかを含み、リンゴ酸塩の残りの重量%は化合物 (II) の重量%である。
【0037】
本発明の別の局面は、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのDL-リンゴ酸塩の結晶質形態、化合物 (III) に関する。DL-リンゴ酸塩はラセミ体のリンゴ酸から調製する。本明細書に記載する化合物 (III) の結晶性のN-1形態は、以下の1項目以上の特徴を持つ場合がある。
(i) 固体13C NMRスペクトルが、20.8、26.2、44.8、55.7、70.7、100.4、101.0、114.7、115.2、116.0、119.7、120.4、121.6、124.4、136.9、138.9、141.1、145.7、150.3、156.5、157.6、159.6、165.2、167.4、171.2、176.3、182.1 ppm ± 0.2 ppmから選択される4点以上のピークを含む。
(ii) 固体13C NMRスペクトルが図16に示すパターンと実質的に一致する。
(iii) 粉末X線回折パターン(CuKα λ=1.5418Å)が、以下の中から選択される4点以上の2θ値を含む。12.8、13.5、16.9、19.4、21.5、22.8、25.1、および27.6 ±0.2 °2θ。結晶質形態の測定は室温で行う。
(iv) 粉末X線回折(XRPD)スペクトルが図15に示すパターンと実質的に一致する。
(v) 固体15N NMRスペクトルのピークが119.6、134,7、および175.5 ppm ± 0.2 ppmにある。
(vi) 固体15N NMRスペクトルが図17に示すパターンと実質的に一致する。
化合物 (III) の結晶質N-1形態の特徴づけに使用できる他の固体状態特性を図に示し、下記の実施例で考察する。1つの実施形態では、化合物 (III) のN-1形態が、ほぼ以下の値に等しい単位格子パラメータを示す。
結晶の寸法 a = 14.60 Å
b = 5.20 Å
c = 39.09 Å
α = 90.0°
β = 90.4°
γ = 90.0°
空間群: P21/n
単位格子あたりの化合物 (I) の分子数:4
体積 = 2969 Å3
密度(計算値)= 1.422 g/cm3
化合物 (III) のN-1形態の単位格子パラメータは、例えば外気温または室温などに相当する約25°Cで測定した。
【0038】
化合物 (I) ならびに (II) の結晶質形態N-1ならびにN-2、および化合物 (III) の結晶質形態N-1の各々が、互いに識別可能な固有の特徴を持つ。これらの特徴は、下記の実施例で示す固体形態の物性を比較することにより理解できる。例えば表2は、結晶質化合物 (III) の形態 N-1および結晶質化合物 (I) の形態N-1ならびにN-2に特徴的なXRPDのピークの位置(°2θ±0.2 °2θ)を示したものである。非晶質形態はXRPDのパターンで回折ピークを示さない。
結晶質化合物 (II) の形態N-1とN-2の間で固有な反射にマーク(*)を付けた。前述のように、化合物 (II) は化合物 (I) のエナンチオマーであるため、化合物 (II) の形態 N-1は、化合物 (I) の形態 N-1に関して表2に掲げたものと同じ特徴の回折パターンおよび固有のピークを持つ。同様に、化合物 (II) の形態 N-2は、化合物 (I) の形態 N-2に関して表2に掲げたものと同じ特徴の回折パターンおよび固有のピークを持つ。化合物 (I) と (II) はその絶対立体化学、すなわちL-リンゴ酸塩とD-リンゴ酸塩の違いにより、その間の識別が可能である。結晶質化合物 (III) の形態 N-1は、D,L-リンゴ酸塩として識別される。
【0039】
固体NMRでの特徴的なピークも、本明細書で開示する結晶質形態と非晶質形態を識別する役割を果たす。例えば表3は、結晶質化合物 (III) の形態 N-1、結晶質化合物 (I) の形態N-1ならびにN-2、および化合物 (I) の非晶質形態に特徴的な固体13C NMRピークを示したものである。
後述する固体19Fおよび15N NMRスペクトルは、同様の比較および特徴づけのためのデータを提供する。前述のように、化合物 (I) のエナンチオマーであるため、結晶質形態 N-1ならびにN-2と化合物 (II) の非晶質形態は、結晶質化合物 (I) の形態N-1およびN-2に関して表3に示したように、同じ固体NMR共鳴を持ち、それらの間で異なる固有のピークを持つ。
【0040】
医薬組成物および処理方法
本発明の別の局面は、化合物 (I)、化合物 (II)、化合物 (III) またはその組み合わせの一以上、および薬学的に許容される賦形剤を含む医薬組成物に関する。医薬組成物中の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの量は、治療上有効な量とすることができる。化合物 (I)、化合物 (II)、または化合物 (III) は医薬組成物中で個別に、上記のいずれかの固体形態またはその組み合わせとして存在することができる。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、結晶質形態の治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上、および薬学的に許容される賦形剤を含む固体または分散の医薬組成物に関する。
【0041】
本発明の別の局面は、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、それを必要とする患者に投与することを含む癌治療法に関する。投与する化合物 (I)、化合物 (II)、またはその組み合わせは、治療上有効な量とすることができる。 化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれか、またはその組み合わせとして個別に投与できる。結晶質形態が好ましい固体形態であり、結晶質化合物 (I)、形態 N-1またはN-2が好ましい。従って、本発明の別の局面は、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、それを必要とする患者に投与することを含む癌治療法に関し、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。
【0042】
本発明の別の局面は、上記のように癌を治療する方法に関し、治療される癌は胃癌、食道癌、腎癌、肝癌、卵巣癌、子宮頸癌、大腸癌、小腸癌、脳腫瘍(神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫などの星細胞腫を含む)、肺癌(非小細胞肺癌を含む)、骨癌、前立腺癌、膵癌、皮膚癌、骨癌、リンパ腫、固形腫瘍、ホジキン病、非ホジキンリンパ腫、または甲状腺癌(甲状腺髄様癌を含む)である。
【0043】
チロシンキナーゼ阻害剤は、非小細胞肺癌(NSCLC)の治療にも使われてきた。ゲフィチニブおよびエルロチニブは、チロシンキナーゼと呼ばれる上皮増殖因子の受容体を標的とする血管新生阻害剤である。エルロチニブおよびゲフィチニブは現在、NSCLCの治療に使われている。本発明の別の局面は、患者の小細胞肺癌(NSCLC)を治療する方法に関し、その方法は、治療上有効な量のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド、またはその薬学的に許容される塩を、場合によってはエルロチニブまたはゲフィチニブと組み合わせ、治療を必要とする患者に投与することを含む。別の実施形態では、エルロチニブと組み合わせる。
【0044】
本発明の別の局面は、患者の小細胞肺癌(NSCLC)を治療する方法に関し、その方法は、治療上有効な量のエルロチニブまたはゲフィチニブを、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上と組み合わせ、治療を必要とする患者に投与することを含む。化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれかとして個別に投与してもよく、またはそれらを組み合わせて投与してもよい。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、患者の小細胞肺癌(NSCLC)を治療する方法に関し、その方法は、治療上有効な量のエルロチニブまたはゲフィチニブを、化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上と組み合わせ、治療を必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。別の実施形態では、この方法で投与される組み合わせは、エルロチニブと化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上である。
【0045】
本発明の別の局面は、患者における星細胞腫(患者における神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫を含む)を治療する方法に関し、その方法は、治療上有効な量のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドを、治療を必要とする患者に投与することを含む。
【0046】
本発明の別の局面は、患者における星細胞腫(患者における神経膠芽細胞腫、巨細胞神経膠芽腫、膠肉腫、オリゴデンドログリア細胞がある神経膠芽細胞腫を含む)を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含む。化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれかとして個別に投与してもよく、またはそれらを組み合わせて投与してもよい。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、星細胞腫を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。
【0047】
本発明の別の局面は、患者における甲状腺癌(甲状腺髄様癌を含む)を治療する方法に関し、その方法は、治療上有効な量のN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド、またはその薬学的に許容される塩を、治療を必要とする患者に投与することを含む。投与量は治療上有効な量とすることができる。
【0048】
本発明の別の局面は、患者における甲状腺癌(甲状腺髄様癌を含む)を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含む。化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれかとして個別に投与してもよく、またはそれらを組み合わせて投与してもよい。結晶質形態が好ましい固体形態である。従って、本発明の別の局面は、甲状腺癌を治療する方法に関し、その方法は、治療上有効な量の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、治療を必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。
【0049】
本発明の別の局面は、制御されず、異常であり、望ましくないか、またはそのいずれかである細胞活性と関連する疾患または障害を治療する方法に関する。この方法では、化合物 (I)、化合物 (II)、化合物 (III) またはその組み合わせの一以上を、それを必要とする患者に投与する。投与する化合物 (I)、化合物 (II)、またはその組み合わせは、治療上有効な量とすることができる。 化合物 (I)、化合物 (II)、または化合物 (III) は、上記の固体形態のいずれか、またはその組み合わせとして個別に投与できる。結晶質形態が好ましい固体形態である。
【0050】
従って、本発明の別の局面は、制御されず、異常であり、望ましくないか、またはそのいずれかである細胞活性と関連する疾患または障害を治療する方法に関し、それは化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上を、それを必要とする患者に投与することを含み、その場合に化合物 (I)、化合物 (II)、または化合物 (III) は結晶質形態で存在する。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。本発明の別の局面では、上記の化合物 (I)、化合物 (II)、化合物 (III)、またはその組み合わせの一以上の医薬組成物を投与することにより、この治療法を実行できる。本発明の別の局面は、制御されず、異常であり、望ましくないか、またはそのいずれかである細胞活性と関連する疾患または障害を治療する方法に関する。この方法では、結晶質形態の化合物 (I)、化合物 (II)、または化合物 (I) および (II) の組み合わせを、それを必要とする患者に投与する。投与する化合物 (I)、化合物 (II)、または化合物 (I) および (II) の組み合わせの量は、治療上有効な量とすることができる。
【0051】
本発明の別の局面は、上記の疾患または障害を治療する薬剤を製造するために、上記実施形態のいずれかに従い、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩を使用することに関する。溶解すると、本発明に従う結晶質形態または非晶質形態は固体構造を失い、このため、それを例えば化合物 (I) の溶液などと呼ぶ。本発明に従う一以上の結晶質形態が溶解および懸濁された(またはそのいずれか)液体製剤を調製するために、本発明中の結晶質形態の一以上を使うことができる。
【0052】
上記の医薬組成物は、固体形態を含め、活性のある化合物 (I)、化合物 (II)、および化合物 (III)、またはそのいずれか(以降、活性化合物と呼ぶ)を含む任意の医薬形態である。例えば、医薬組成物は錠剤、カプセル剤、懸濁液、注射液、局所薬、経皮薬の場合がある。医薬組成物は一般に、約1重量%から約99重量%の活性化合物または活性化合物の結晶質形態と、99重量%から1重量%の好適な医薬賦形剤を含む。一例では、組成物は約5重量%と約75重量%の間の活性化合物を含み、残りは下記の好適な医薬賦形剤または他のアジュバントである。
【0053】
本発明に従い、キナーゼのシグナル伝達を阻害し、調節し、かつ修飾するか、またはそのいずれかを行うための、活性化合物、または活性化合物の結晶質形態または非晶質形態(ここでは医薬組成物に関して論じる)の「治療上有効な量」とは、異常な細胞増殖および血管新生と関連する多種の癌のいずれかに罹患した患者を治療するために十分な量を意味する。本発明に従う治療上有効な量は、本明細書で論じる疾患状態および障害の治療または予防に対して治療上有用な量である。化合物 (I)、(II)、および (III) またはそのいずれか(それらの固体形態を含む)は、WO2005‐030140に記載するキナーゼのシグナル伝達を阻害し、調節し、かつ修飾するか、またはそのいずれかによる治療作用を持つ。N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド。
【0054】
特定の患者の治療に必要な実質量は多様な要因に依存し、例えば、治療する疾患の状態およびその重症度、使用する特定の医薬組成物、患者の年齢、体重、健康状態、性別、食生活、投与方法、投与回数、投与経路、本発明に従う活性化合物または活性化合物の結晶質形態の排出速度、治療期間、使用する特定化合物と併用する薬剤または同時に服薬される薬剤、および医学分野で周知の他の要因がある。これらの要因については、Goodman and Gilman’s “The Pharmacological Basis of Therapeutics”, Tenth Edition, A. Gilman, J.Hardman and L. Limbird, eds., McGraw-Hill Press, 155-173, 2001で論じられており、それは参照により本明細書の一部とする。本発明に従う活性化合物、または活性化合物の結晶質形態、およびそれらを含む医薬組成物は、癌の治療を受ける患者に一般に投与される抗癌剤その他の薬剤と併用してもよい。単一の医薬組成物内で、一以上のそれらの薬剤と合わせて製剤してもよい。
【0055】
医薬組成物のタイプに従い、当業者の間で知られるいずれかの担体またはその組み合わせの中から、薬学的に許容される担体を選択できる。薬学的に許容される担体の選択は部分的に、使用する望ましい投与方法に依存する。本発明の医薬組成物、すなわち本発明の活性化合物、または活性化合物の結晶質形態に関しては、結晶質であるか否かに関わらず、担体は活性化合物の特定の形態を実質的に維持するよう選択すること。言い換えれば、担体は活性化合物の形態を実質的に変えてはならない。それ以外にも、担体は、望ましくない生物学的影響を与えることや、それ以外に、医薬組成物の他の構成要素と有害な形で相互作用することなどにより、活性化合物の形態と不適合ではないものとする。
【0056】
本発明の医薬組成物は、例えばRemington's Pharmaceutical Sciences, 18th Ed., (Mack Publishing Company, Easton, Pa., 1990) に掲載されている、製薬分野で周知の方法により調整できる。固形剤形では化合物 (I) に、一以上のクエン酸ナトリウムまたはリン酸水素カルシウムなどの薬学的に許容される賦形剤、または (a) 例えばデンプン、乳糖、ショ糖、ブドウ糖、マンニトール、ケイ酸などの充填剤または増量剤、(b) 例えばセルロース誘導体、デンプン、アルギン酸、ゼラチン、ポリビニルピロリドン、ショ糖、アラビアゴムなどの結合剤、(c) 例えばグリセロールなどの保湿剤、(d) 例えば寒天、炭酸カルシウム、ジャガイモまたはタピオカのデンプン、アルギニン酸、クロスカルメロースナトリウム、複合ケイ酸塩、炭酸ナトリウムなどの崩壊剤、(e) 例えばパラフィンなどの溶解遅延剤、(f) 例えば四級アンモニウム化合物などの吸収促進剤、(g) 例えばセチルアルコール、モノステアリン酸グリセロール、ステアリン酸マグネシウムなどの湿潤剤、(h) 例えばカオリン、ベントナイトなどの吸湿剤、(i) 例えばタルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、固体ポリエチレングリコール、ラウリル硫酸ナトリウム、またはその混合などの潤滑剤を混合できる。カプセル剤、錠剤、丸剤の場合、投与剤形には緩衝剤を含めてもよい。
【0057】
製薬分野で周知の薬学的に許容されるアジュバントも、本発明の医薬組成物で使用できる。これは保存剤、湿潤剤、懸濁剤、甘味剤、香味剤、芳香剤、乳化剤、分散剤を含むが、それらに限定されない。微生物の作用は、例えばパラベン、クロロブタノール、フェノール、ソルビン酸、およびその類似物など、さまざまな抗菌剤および抗真菌剤により、確実に防止できる。例えば糖類、塩化ナトリウム、およびその類似物などの等張剤を含めることが望ましい場合もある。望ましい場合は、本発明の医薬組成物に、例えばクエン酸、ソルビタンモノラウレート、オレイン酸トリエタノールアミン、ブチル化ヒドロキシトルエンなど、湿潤剤または懸濁剤、pH緩衝剤、抗酸化剤などの少量の添加剤を含めてもよい。
【0058】
上記の固形剤形は、腸溶コーティングおよび当業者の間で周知の他のコーティングなど、コーティングおよびシェルを加えて調製できる。安定剤を含めてもよく、活性化合物を腸管の特定箇所で遅延して放出するような組成にすることもできる。使用可能な埋め込み組成の例としては、高分子物質およびワックスがある。活性化合物は、適切であれば、一以上の上記賦形剤と共に、マイクロカプセル化することもできる。
【0059】
懸濁液には活性化合物に加え、例えばエトキシル化イソステアリルアルコール、ポリオキシエチレンソルビトルおよびソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム、ベントナイト、寒天およびトラガカント、またはこれらの物質の混合、およびその類似物などの懸濁剤を含むことができる。
【0060】
直腸投与用の組成物は、例えば活性化合物または活性化合物の結晶質形態を、例えばココアバター、ポリエチレングリコールまたは坐剤ワックスなど、常温では固体であるが、体温では液体であり、従って、好適な体腔内で溶融し、そこで活性化合物を放出する好適な非刺激性の賦形剤または担体と混合することにより調製できる坐剤である。
【0061】
活性化合物または活性化合物の結晶質形態は、調製中に維持されるため、本発明の医薬組成物には、固体投与剤形が好ましい。カプセル剤、錠剤、丸剤、粉剤、顆粒剤を含む経口投与用固体投与剤形は、特に好ましい。そのような固体投与剤形において、活性化合物は一以上の不活性な薬学的に許容される賦形剤(薬学的に許容される担体とも称する)と混合される。純粋な形態または適当な医薬組成物としての活性化合物または活性化合物の結晶質形態の投与は、同様の用途のために許容される投与方法または薬剤を介して実行できる。従って、投与は例えば経口、経鼻、非経口(静脈内、筋肉内、または皮下)、局所的、経皮、膣内、膀胱内、槽内、経直腸で行うことができ、剤形は例えば錠剤、坐剤、丸剤、軟質弾性ゼラチンカプセル剤ならびに硬質ゼラチンカプセル剤、粉剤、液剤、懸濁剤、噴霧剤、または類似物などの固体、半固体、凍結乾燥粉剤、または液体投与剤形であり、正確な用量を簡単に投与するために適した単位用量ごとに投与できる剤形が好ましい。一つの好ましい投与経路は経口投与であり、治療する疾患状態の重症度に従い調整できる便利な投与計画に従い投与する。
【0062】
結晶質形態の一般的な調製法
結晶質形態は、例えば好適な溶媒混合液からの結晶化または再結晶化、昇華、融液からの成長、別の相からの固相変換、超臨界液流体からの結晶化、およびジェット噴霧などの多様な方法により調整することができ、それらに限定されない。溶媒混合液からの結晶質形態の結晶化または再結晶化の技法は、例えば溶媒の蒸発、溶媒混合液の冷却、化合物およびその塩またはそのいずれかの超飽和溶媒混合液への結晶の接種、化合物およびその塩またはそのいずれかの超飽和溶媒混合への結晶の接種、溶媒混合液の凍結乾燥、および溶媒混合液への逆溶媒(逆溶剤)の添加などを含み、それらに限定されない。多形体を含む結晶質形態を調製するために、高処理能力結晶化技法を採用してもよい。
【0063】
多形体を含む薬剤の結晶、調製法、薬剤結晶の特徴づけは、Solid-State Chemistry of Drugs, S.R. Byrn, R.R. Pfeiffer, and J.G. Stowell, 2nd Edition, SSCI, West Lafayette, Indiana (1999) で考察されている。
【0064】
溶媒を採用する結晶化技法において、典型的には溶媒は一以上の要素に基づき選択され、それには例えば化合物の溶解度、使用する結晶化技法、および溶媒の蒸気圧が含まれ、それらに限定されない。複数の溶媒を組み合わせて使用してもよい。例えば、最初の溶媒に化合物を溶解させて溶液を作り、次に、それに逆溶媒を加えて溶液中の化合物 (I) の溶解度を下げ、結晶を析出させることができる。逆溶媒は、化合物の溶解度が低い溶媒である。
【0065】
結晶の調製に使用できる一方法では、化合物 (I)、化合物 (II) および化合物 (III)、またはそのいずれかを好適な溶媒中に懸濁し、攪拌して得たスラリーを熱し、溶解を促進する。本明細書で使用する限り、「スラリー」という用語は、当該化合物の飽和溶液を意味し、その溶液に化合物が添加され、任意の温度で化合物と溶媒の不均質な混合状態が生じる場合があることを意味する。
【0066】
結晶化混合液に種結晶を添加し、結晶化を促進することができる。特定の多形体の成長を制御するため、および結晶の粒子サイズの分散を制御するため、またはそのいずれかを目的として、接種を利用することができる。従って、必要な種結晶の量の計算は、例えば “Programmed Cooling Batch Crystallizers,” J.W. Mullin and J. Nyvlt, Chemical Engineering Science, 1971, 26, 3690377に記載されているように、入手可能な種結晶の大きさおよび結晶産物について所望する平均粒子サイズに依存する。一般に、バッチ中の結晶の成長を有効に制御するには、小さい種結晶が必要とされる。小さい種結晶は、大きい結晶のふるい分け、微粉砕、もしくは超微粉砕、または溶液の微結晶化により生成できる。結晶の微粉砕または超微粉砕においては、所望する結晶質形態から結晶化度が変化しないよう(すなわち非晶質または他の多形体への変化)、注意が必要である。
【0067】
冷却した結晶化混合液を減圧濾過し、分離された固体産物を、例えば低温の再結晶溶媒などの好適な溶媒で洗浄することができる。洗浄後、当該産物を窒素パージ下で乾燥させることにより、所望する結晶質形態が得られる。化合物の結晶質形態が形成されたことを確認するために、当該産物は好適な分光学的または分析的技法により分析することができ、それには例えば示差走査熱量計(DSC)、粉末X線回折(XRPD)、および熱重量分析(TGA)が含まれ、それらに限定されない。得られる結晶質形態の分離収率は、結晶化手順に最初に投入した化合物の重量に基づき、約70重量%であり、好ましくは約90重量%以上である。場合によっては、共粉砕するか、またはメッシュスクリーンを通すことにより、産物の塊を崩してもよい。
【0068】
当業者は以下の詳細な説明を読むことにより、本発明の特徴および優位性を、より容易に理解できるであろう。理解すべき点として、本発明の特徴は、明確にするために、上記および下記で個別の実施形態を背景として説明したが、それらの特定の特徴を、一つの実施形態で組み合わせることも可能である。逆に、簡潔にするために、一つの実施形態を背景として説明した種々の特徴を、それらよりも下位の項で組み合わせて使うこともできる。本発明は以下の実施例により、さらに詳しく例示されるが、そこに記載された特定の手順に本発明の範囲または主旨を限定するものと解釈すべきではない。
【0069】
本明細書に明記する定義は、参照により本明細書の一部とするいかなる特許、特許出願、特許出願公報に明記された定義よりも優先する。すべての測定値において実験上の誤差が生じる可能性があるが、本発明の主旨から逸脱しない。
【0070】
本明細書で使用する限り、「非晶質」とは、結晶質ではない分子およびイオンの固体形態を意味する。非晶質固体は、シャープな最大値を持つ確定的なX線回折パターンを示さない。
【0071】
本明細書で使用する限り、「実質的に純粋」とは、言及する化合物 (I) の結晶質形態が、結晶質形態の重量に基づき約90重量%以上を含むことを意味する。「約90重量%以上」は、請求範囲に対する等価性理論の適用の制限を意図しないが、しかし、言及する結晶質形態の重量に基づき、例えば約90、約91、約92、約93、約94、約95、約96、約97、約98、約99、および約100重量%を含み、それらに限定されない。化合物 (I) の結晶質形態の残りは、化合物 (I) の他の形態、および、例えば結晶質形態を調製するときに生じる反応不純物および処理不純物、またはそのいずれかを含む場合がある。反応不純物および処理不純物、またはそのいずれかの存在は、例えばクロマトグラフィー、核磁気共鳴分光法、質量分光分析法、および赤外分光法、またはそのいずれかなど、当業者の間で周知の分析法により決定できる。
【0072】
調製実施例
実施例1:N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドおよびそのL-リンゴ酸塩 (化合物 (I))の調製
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドおよびそのL-リンゴ酸塩の調製に使用する合成経路を反応式1に示す。
反応式1に示した過程を以下に詳しく説明する。
【0073】
1.1 4‐クロロ‐6,7‐ジメトキシキノリンの調製
反応器に、最初に6,7‐ジメトキシキノリン‐4‐オル(1 L、10.0 kg)、次にアセトニトリル(64.0 L)を順に入れた。その混合物を約65°Cに熱し、オキシ塩化リン(POCl3、50.0 kg)を加えた。POCl3を加えた後、反応混合物の温度を約80°Cに上げた。開始物質の2%未満が残る(反応進行を確認する高速液体クロマトグラフィー[HPLC]分析による)時点で、反応は完了と見なした(約9.0時間)。反応混合物を約10°Cに冷却した後、クエンチするために、ジクロロメタン(DCM、238.0 kg)、30% NH4OH(135.0 kg)、および氷(440.0 kg)の冷却溶液に投入した。混合物を約14°Cに加熱し、相を分離させた。有機相を水(40.0 kg)で洗浄し、減圧蒸留で溶媒を除去して濃縮した(約190.0 kg)。メチル-t-ブチルエーテル(MTBE、50.0 kg)をバッチに加え、混合物を約10°Cまで冷却する間に、結晶が析出した。固体を遠心分離により回収し、n-ヘプタン(20.0 kg)で洗浄し、約40°Cで乾燥し、表題化合物を得た(8.0 kg)。
【0074】
1.2 6,7‐ジメチル‐4‐(4-ニトロフェノキシ)‐キノリンの調製
反応器に、4‐クロロ‐6,7‐ジメトキシキノリン(8.0 kg)、4-ニトロフェノール(7.0 kg)、4-ジメチルアミノピリジン(0.9 kg)、2,6-ルチジン(40.0 kg)を順に入れた。反応器の内容物を約147°Cに熱した。反応完了後(反応中のHPLC分析により、残った反応物が5%未満であることを確認。約20時間)、約25°Cに冷却するまで、反応器の内容物を放置した。メタノール(26.0 kg)を加え、次に、水(50.0 kg)に溶解した炭酸カリウム(3.0 kg)を加えた。反応器の内容物を約2時間、攪拌した。沈殿した固体を濾過し、水(67.0 kg)で洗浄し、25°Cで約12時間乾燥させ、表題化合物を得た(4.0 kg)。
【0075】
1.3 4‐(6,7 ‐ジメトキシキノリン‐4‐イロキシ)‐フェニルアミンの調製
ギ酸カリウム(5.0 kg)、ギ酸(3.0 kg)、および水(16.0 kg)を、テトラヒドロフラン(40.0 kg)中に6,7-ジメトキシ‐4‐(4-ニトロフェノキシ)‐キノリン(4.0 kg)および10%パラジウム炭素(50%水湿潤、0.4 kg)を混合し、約60°Cに熱したものに加えた。反応混合物の温度が約60°Cに保たれるような方法で、それらを加えた。反応中のHPLC分析により反応が完了したと見なされた時点で(反応物の2%未満が残る状態。通常1 5時間)、反応器の内容物を濾過した。濾過液を約 35°Cで減圧蒸留し、最初の容積の半分まで濃縮し、反応産物を沈殿させた。反応産物を濾過により回収し、水(12.0 kg)で洗浄し、約50°Cで減圧乾燥し、表題化合物を得た(3.0 kg。97% AUC)。
【0076】
1.4 1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボン酸の調製
THF(63.0 kg)に市販のシクロプロパン‐1,1‐ジカルボン酸(2 1、10.0 kg)を溶解して冷却した(約4°C)溶液に、トリエチルアミン(8.0 kg)を、バッチ温度が10°Cを超えない速度で加えた。溶液を約30分間攪拌した後、バッチ温度を10°C以下に保ちつつ、塩化チオニル(9.0 kg)を加えた。添加完了後、THF(25.0 kg)に4-フルオロアラニン(9.0 kg)を溶解した溶液を、バッチ温度が10°Cを超えない速度で加えた。混合物を約4時間攪拌した後、酢酸イソプロピル(87.0 kg)で希釈した。この溶液を、順に水酸化ナトリウム水溶液(50.0 Lの水に2.0 kgを溶解)、水(40.0 L)、塩化ナトリウム水溶液(40.0 Lの水に10.0 kgを溶解)で洗浄した。この有機溶液を減圧蒸留により濃縮した後、ヘプタンを加え、固体の沈殿物を得た。遠心分離により固体を回収し、約35°Cで減圧乾燥し、表題化合物を得た(10.0 kg)。
【0077】
1.5 1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボニルクロリドの調製
THF(11 kg)とN, N-ジメチルホルムアミド(DMF。0.02 kg)の混合液に1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボン酸(2.0 kg)を溶解した溶液に、塩化オキサリル(1.0 kg)を、バッチ温度が30°Cを超えない速度で加えた。それ以上処理せずに、この溶液を次の段階で使用した。
【0078】
1.6 N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの調製
前段階の1‐(4‐フルオロフェニルカルバモイル)‐シクロプロパンカルボニルクロリドを含む溶液を、THF(27.0 kg)と水(13.0 kg)に4-(6,7-ジメトキシキノリン-4-イロキシ)-フェニルアミン(3.0 kg)と炭酸カリウム(4.0 kg)を溶解した混合物に、バッチ温度が30°Cを超えない速度で加えた。反応完了後(通常、10分間)、水(74.0 kg)を加えた。混合物を15〜30°Cで約10時間攪拌すると、反応産物が沈殿した。反応産物を濾過により回収し、事前に調整したTHF(11.0 kg)と水(24.0 kg)の溶液で洗浄し、約65°Cで約12時間、減圧乾燥し、表題化合物(遊離塩基、5.0 kg)を得た。1H NMR (400 MHz、d6-DMSO): d 10.2 (s、1H)、10.05 (s、1H)、8.4 (s、1H)、7.8 (m、2H)、7.65 (m、2H)、7.5 (s、1H)、7.35 (s、1H)、7.25 (m、2H)、7.15(m、2H)、6.4 (s、1H)、4.0 (d、6H)、1.5 (s、4H). LC/MS: M+H= 502.
【0079】
1.7 N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩(化合物 (I))の調製
L-リンゴ酸(2.0 kg)を水(2.0 kg)に溶解した溶液を、エタノールにシクロプロパン‐1,1‐ジカルボン酸[4‐(6,7‐ジメトキシキノリン‐4‐イロキシ)‐フェニル]‐アミド(4‐フルオロフェニル)‐アミド遊離塩基(1 5、5.0 kg)を溶解した溶液に、バッチ温度を約25°Cに維持しつつ加えた。次に、炭素(0.5 kg)とチオールシリカ(0.1 kg)を加え、その混合物を約78°Cまで加熱し、その時点で水(6.0 kg)を加えた。反応混合物を濾過し、イソプロパノール(38.0 kg)を加え、温度が約25°Cに下がるまで放置した。その産物を濾過により回収し、イソプロパノール(20.0 kg)で洗浄し、約65°Cで乾燥させ、化合物 (I)(5.0 kg)を得た。
【0080】
実施例2:結晶質化合物 (I)、形態 N-1の調製
1 Lの反応器にテトラヒドロフラン(12 mL/gバルクLR(限定試薬)、1.20 L)およびN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド(100 g、1.00当量、100.00 g)およびL-リンゴ酸(1.2当量(モル濃度)、32.08 g)を投入し、溶液を作った。水(0.5317 mL/gバルクLR、53.17 mL)を加え、溶液を60 °Cに熱し、固体が完全に溶解するまで1時間、その温度に保った。溶液をポリッシュフィルタに通した。
【0081】
60 °Cで8時間をかけてアセトニトリル(12 mL/gバルクLR、1.20 L)を加えた。溶液を60 °Cに維持し、10時間放置した。その後、容器を20 °Cに冷却し、その温度で1時間放置した。固体を濾過し、アセトニトリル(12 mL/gバルクLR、1.20 L)で洗浄した。固体を60 °C(25 mm Hg)で6時間乾燥させ、白色の結晶質固体として化合物 (I)、形態 N-1を得た(108 g、0.85当量、108.00 g、収量85.22%)。
【0082】
実施例3:結晶質化合物 (I)、形態 N-1の別の調製
190 mLのテトラヒドロフラン(110 mL)、メチルイソブチルケトン、29 mLの水で溶液を作った。次に、20 mLの溶液を琥珀色の遮光ビンに移し、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩を、混濁したスラリーが形成されるまで加えて飽和させ、室温で2時間以上攪拌し、寝かせる。ブフナー漏斗で濾過して固体を除去し、透明な飽和溶液状態にした。
【0083】
それとは別に、化合物 (I) の2回分のバッチから次の量を取り、粉末ブレンドを調製した。(1) バッチ1から300 mg。ラマン分光分析で約41%の化合物 (I)、形態 N-1および約59%の化合物 (I)、形態 N-2を含む。 (2) バッチ2から200 mg。XPRDパターンが化合物 (I)、形態 N-2と類似。
【0084】
この化合物 (I)、形態 N-1と化合物 (I)、形態 N-2の粉末ブレンドを飽和溶液に加え、室温でマグネチックスタラーを使い攪拌し、スラリーを25日間寝かせた。その後、スラリーをサンプリングし、ブフナー漏斗で濾過し、162 mgのウェットケーキを得た。ウェットケーキを真空オーブン中で45°Cで乾燥させ、128 mg のN-1形態の結晶質化合物 (I) を得た。
【0085】
実施例4:結晶質化合物 (I)、形態 N-2の調製
4.1 結晶質化合物 (I)、形態 N-2の種 結晶の調製
25 mLねじ口バイアルで、20 mLのアセトンと300 mgの遊離塩基N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドを混合し、溶液を調製した。次に、マグネチックスタラーで攪拌しながら、0.79MのL-リンゴ酸の保存溶液0.658 mLを加えた。この溶液を室温で24時間、攪拌した。その後、試料を0.45μm PTFE フィルターカートリッジを通して吸引濾過し、室温で1夜、減圧乾燥させた。
【0086】
4.2 結晶質化合物 (I)、形態 N-2の調製
反応器に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド(48 g、1.00当量、48.00 g)およびテトラヒドロフラン(16.5 mL/gバルクLR、792.00 mL)を加えた。水分含有量を1重量%に調整した。溶液を60°Cに熱した。溶解後、溶液をポリッシュフィルタで濾過し、第1の溶液を得た。
【0087】
別の反応器で、メチルイソブチルケトン(10 mL/gバルクLR、480.00 mL)およびテトラヒドロフラン(1 mL/gバルクLR、48.00 mL)にL-リンゴ酸(1.2当量(モル濃度)、15.40 g)を溶解した。次に、50°Cで第1の溶液に50 mLのL-リンゴ酸溶液を加えた。種結晶を加え(1%、480 mg)、50°Cでリンゴ酸溶液を添加漏斗で滴下して加えた(1.3 mL/分(3時間))。そのスラリーを50°Cで18時間放置した後、30分間で25°Cに冷却した。固体を濾過し、20%テトラヒドロフラン/メチルイソブチルケトン(10V、480 mL)で洗浄した。固体を60 °Cで5時間、減圧乾燥させ、オフホワイト色の結晶質固体の化合物 (I)(55.7 g、0.92当量、55.70 g、収率91.56%)を得た。
【0088】
実施例5:結晶質化合物 (III)、形態 N-1の調整
テトラヒドロフラン(THF)中でスラリー状にしたN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのDL-リンゴ酸塩を1 mL分取し、ハーフドラムのバイアルに入れ、ホットプレート上で60 °Cに熱した。次に、ほぼ透明な溶液が得られるまで、テトラヒドロフランを滴下した。バイアルに蓋をし、ホットプレートから降ろし、室温で振動を加えずに放置し、平衡化した。数時間後に結晶が析出し、溶液はそのまま1夜放置し、析出を完了させた。生じたスラリーを数滴、ガラススライドに落とし、顕微鏡で分析した。結晶質の物質は、最長60ミクロンまでの多数の細長い板状の物質で構成されていた。
【0089】
結晶質化合物 (III)、形態 N-1の別の調製
反応器に、N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミド(15 g、1.00当量、15.00 g)とテトラヒドロフラン(16.5 mL/gバルクLR、792.00 mL)を入れた。水分含有量は1重量%に調整した。溶液を60 °Cに熱した。溶解後、溶液をポリッシュフィルタで濾過し、第1の溶液を得た。
【0090】
別の反応器で、DL-リンゴ酸(1.2当量(モル濃度)、4.53 g)を、メチルイソブチルケトン(8 mL/gバルクLR、120.00 mL)およびテトラヒドロフラン(1 mL/gバルクLR、15.00 mL)に溶解した。次に、この溶液20 mLを、50 °Cで第1の溶液に加えた。50°Cでリンゴ酸溶液を添加漏斗で滴下して加えた(1.3 mL/分(3時間))。そのスラリーを50°Cで18時間放置した後、30分間で25°Cに冷却した。固体を濾過し、20% THF/MIBK(10V、150 mL)で洗浄した。固体を60 °Cで5時間減圧乾燥し、オフホワイト色の固体の化合物 (III)(15.52 g、収率86.68%)を得た。
【0091】
実施例6:非晶質化合物 (I) の調製
5 gのN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩およびメタノールとジクロロメタンの1:1(v:v)混合液250 mLで溶液を調製した。この不透明な溶液を0.45ミクロンのフィルターで濾過し、透明な黄色を帯びた溶液を得た。溶液を12.9 cc/分の速度でスプレードライヤーのノズルを通し、10.9 L/分の速度で吹き込まれる窒素ガスで微粒化した。サイクロンのインレットの温度を65 °Cに設定し、液滴を乾燥させた。乾燥した非晶質粉末(1.5 g)を回収した(収率= 30%)。
【0092】
特徴づけの例
I. ジメチルスルホキシド溶液中でのNMRスペクトル
I.1 化合物 (I)、形態 N-1
1H NMR (400 MHz、d6-DMSO): d 1.48 (s、1 H)、2.42-2.48 (m、1 H)、2.60-2.65 (m、1 H)、3.93-3.96 (m、6 H)、4.25-4.30 (dd、1 H、J = 5、8 Hz)、6.44 (d、1H、J = 5 Hz、1 H)、7.12-7.19 (m、2 H)、7.22-7.26 (m、2 H)、7.40 (s、1 H)、7.51 (s、1 H)、7.63-7.68 (m、2 H)、7.76-7.80 (m、2 H)、8.46-8.49 (m、1 H)、10.08 (s、1 H)、10.21 (s、1 H).
【0093】
13C NMR(d6-DMSO):15.36、31.55、55.64、55.67、66.91、99.03、102.95、107.66、114.89、115.07、115.11、121.17、122.11、122.32、122.39、135.15、136.41、146.25、148.7、149.28、149.38、152.54、157.03、159.42、160.02、168.07、171.83、174.68。
【0094】
I.2 化合物 (I)、形態 N-2
1H NMR (400 MHz、d6-DMSO):d 1.48 (s、1 H)、2.42-2.48 (m、1 H)、2.60-2.65 (m、1 H)、3.93-3.96 (m、6 H)、4.25-4.30 (dd、1 H、J = 5、8 Hz)、6.44 (d、J = 5 Hz、1 H)、7.12-7.19 (m、2 H)、7.22-7.26 (m、2 H)、7.40 (s、1 H)、7.51 (s、1 H)、7.63-7.68 (m、2 H)、7.76-7.80 (m、2 H)、8.46-8.49 (m、1 H)、10.08 (s、1 H)、10.21 (s、1 H)。
【0095】
13C NMR(d6-DMSO):15.36、31.55、55.64、55.67、66.91、99.03、102.95、107.66、114.89、115.07、115.11、121.17、122.11、122.32、122.39、135.15、136.41、146.25、148.7、149.28、149.38、152.54、157.03、159.42、160.02、168.07、171.83、174.68.
【0096】
I.3 化合物 (III)、形態 N-1
1H NMR (400 MHz、d6-DMSO):d 1.48 (s、1 H)、2.42-2.48 (m、1 H)、2.60-2.65 (m、1 H)、3.93-3.96 (m、6 H)、4.25-4.30 (dd、1 H、J = 5、8 Hz)、6.44 (d、J = 5 Hz、1 H)、7.12-7.19 (m、2 H)、7.22-7.26 (m、2 H)、7.40 (s、1 H)、7.51 (s、1 H)、7.63-7.68 (m、2 H)、7.76-7.80 (m、2 H)、8.46-8.49 (m、1 H)、10.08 (s、1 H)、10.21 (s、1 H)。
【0097】
13C NMR(d6-DMSO):15.36、31.55、55.64、55.67、66.91、99.03、102.95、107.66、114.89、115.07、115.11、121.17、122.11、122.32、122.39、135.15、136.41、146.25、148.7、149.28、149.38、152.54、157.03、159.42、160.02、168.07、171.83、174.68。
【0098】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の固体形態の特徴づけ
II. 粉末X線回折(XRPD)実験
粉末X線回折(XRPD)パターンは、自動化されたXYZステージ、自動試料位置調整用のレーザービデオ顕微鏡、HiStar 2次元エリア検出器を装備したBruker AXS C2 GADDS回折装置を使い検出した。放射線源には銅を使用し(Cu Kα = 1.5406 Å)、電圧を40 kV、電流を40 mAに設定し、X線光学系はシングル・ゲーベル多層膜ミラーと0.3 mmのピンホール・コリメータを組み合わせた構成である。ビームの発散、すなわち試料面上でのX線ビームの有効サイズは約4 mmであった。θ-θ連続スキャンモードを採用し、試料と検出器の距離は20 cmとし、2θの有効範囲は3.2 °〜29.8 °であった。試料は室温で(約18 °Cから約25 °C)、受け取った状態の粉のまま、粉砕せずに使い、平板状の標本として調製された。約1〜2 mgの試料をガラススライド上で軽く押し、表面を平らにした。典型的には120秒間、試料にX線ビームを照射した。ビームの発散(すなわち、X線スポットの有効サイズ)は約4mmである。別の方法として、粉末試料を直径1 mm以下の密封ガラスキャピラリーに入れた。試料と検出器の距離を15 cmとし、キャピラリーを回転させながらデータを収集した。データは3≦2θ≦35°の範囲で収集し、試料への照射時間は2,000秒以上であった。結果として得た2次元回折アークを積分することにより、範囲3〜35 °2θ± 0.2 °2θおよびステップサイズ0.02 °2θの従来の1次元XRPDパターンを求めた。データ収集用のソフトウェアとしてWNT 4.1.16用GADDSを使用し、データの分析と表示にはDiffrac Plus EVA v 9.0.0.2またはv 13.0.0.2を使用した。
【0099】
II.1 化合物 (I)、形態 N-1
図 1に、結晶質化合物 (I)、形態 N-1の室温(約25 °C)での回折実験によるXRPDパターンを示す。ピークのリストは前掲の表2に示した。2θの値19.4、21.5、22.8、25.1、および27.6(± 0.2 °2θ)が、結晶質化合物 (I)、形態 N-1の特徴づけに有用である。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-1の特徴づけに十分な場合がある。
【0100】
II.2 化合物 (I)、形態 N-2
図 8に、結晶質化合物 (I)、形態 N-2の室温(約25 °C)での回折実験によるXRPDパターンを示す。ピークのリストは前掲の表2に示した。2θの値20.9および21.9(± 0.2 °2θ)結晶質化合物 (I)、形態 N-2の特徴づけに有用である。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0101】
II.3 化合物 (III)、形態 N-1
図 15に、実験とシミュレーションにより、25°Cで回転式キャピラリー標本を使って求めた結晶質化合物 (III)、形態 N-1のXRPDパターンを示す。ピークのリストは前掲の表2に示した。ピークのリスト全体またはその一部が、結晶質化合物 (III)、形態 N-1の特徴づけに十分な場合がある。
【0102】
II.4 非晶質化合物 (I)
図 22に、室温(約25 °C)での回折実験で求めた非晶質化合物 (I) のXRPDパターンを示す。スペクトルの特徴として、ピークの幅が広く、鋭いピークがない。これは非晶質物質と一致する。
【0103】
III.化合物 (III)、形態 N-1の単位格子X線回折実験
Bruker-Nonius CAD4連続回折装置でデータを収集した。単位格子パラメータは、回折実験で高角反射データを25個に設定し、最小二乗法で解析して求めた。回折強度はCu Ka放射線(l = 1.5418 Å)を使い、定温でθ-2θ可変スキャン法により求め、ローレンツ偏光因子に関してのみ補正を加えた。バックグラウンド計数値は、スキャンの半分に時間に関するスキャンの極値で収集した。別の方法として、Bruker-Nonius Kappa CCD 2000システムでCu Ka放射線(l = 1.5418 Å)を使い、単結晶データを収集した。測定した回折強度データの指数付けと処理は、HKL2000ソフトウェア・パッケージ(Otwinowski, Z. & Minor, W. (1997) in Macromolecular Crystallography, eds. Carter, W.C. Jr & Sweet, R.M. (Academic, NY), Vol. 276, pp.307-326)を使い、Collectプログラム・スイート( Data collection and processing user interface: Collect: Data collection software, R. Hooft, Nonius B.V., 1998)で行った。別の方法として、Bruker-AXS APEX2 CCDシステムでCu Ka放射線(l = 1.5418 Å)を使い、単結晶データを収集した。測定した回折強度データの指数付けと処理は、APEX2ソフトウェア・パッケージ/プログラム・スイート( Data collection and processing user interface: APEX2 User Manual, v1.27)で行った。指示がある場合は、データ収集中に、結晶をOxfordクライオ・システム( Cryostream cooler: J. Cosier and A.M. Glazer, J. Appl. Cryst., 1986, 19, 105)の低温流中で冷却した。
【0104】
構造の解析は直接法で行い、SDPソフトウェア・パッケージ(SDP, Structure Determination Package, Enraf-Nonius, Bohemia NY 11716。SDPソフトウェアで、f'およびf''を含む散乱因子には、“International Tables for Crystallography”, Kynoch Press, Birmingham, England, 1974; Vol IV, Tables 2.2A and 2.3.1の値を使用した)にわずかな局所的変更を加えるか、または結晶学用パッケージのMAXUS(maXus solution and refinement software suite: S. Mackay, C.J. Gilmore, C. Edwards, M. Tremayne, N. Stewart, K. Shankland. maXus:回折データによる結晶構造の解析および精密化を行うためのコンピュータ・プログラム)もしくはSHELXTL(Data collection and processing user interface: APEX2 User Manual, v1.27)を使い、観測された反射に基づく精密化を行った。
【0105】
求めた原子パラメータ(座標と温度因子)に対し、フルマトリックス最小二乗法を使い、精密化を行った。精密化で値を最小にする関数はSw(|Fo| - |Fc|)2である。RをS ||Fo| - |Fc||/S |Fo|と定義され、Rw = [Sw( |Fo| - |Fc|)2/Sw |Fo|2]1/2において、wは観測された回折強度の誤差に基づく適当な重み関数である。差フーリエ合成図の検討を精密化の全段階で行った。等方性温度因子と仮定した理想位置に水素原子を導入した計算では、水素のパラメータに変化はなかった。
【0106】
参考文献の説明に従い、シミュレーションによる「ハイブリッド」の粉末X線回折パターンを作成した(Yin. S.; Scaringe, R. P.; DiMarco, J.; Galella, M. and Gougoutas, J. Z., American Pharmaceutical Review, 2003, 6,2, 80)。CellRefine.xlsプログラムを使い、結晶格子の精密化を行うことにより、室温での結晶格子パラメータを求めた。プログラムに入力した数値は、室温での粉末X線回折パターンの実験結果から求めた約10個の反射の2θ位置を含む。低温で収集した単結晶データに基づき、それに対応するミラー指数hklを割り当てた。この手順の最初に求めた室温での結晶格子に、低温で決定された分子構造を挿入することにより、新たな(ハイブリッド)XRPDを計算した(Alexまたは LatticeViewのいずれかのソフトウェア・プログラムを用いた)。分子のサイズならびに形状および結晶格子の原点に対する分子の位置が変わらず、しかも分子間の距離が結晶格子と共に拡大できるよう、分子を挿入した。
【0107】
単結晶回折解析のために、実施例5に記載した結晶のスラリーから、40 x 30 x 10ミクロンの単結晶を選択した。選択した結晶を少量の軽いグリースで細いガラス繊維に接着し、回転する銅陽極管を備えたBruker ApexII単結晶回折装置に、室温で装着した。
【0108】
結晶質化合物 (III)、形態N-1は、表4に報告する値とほぼ等しい単位格子パラメータを特徴とする。単位格子パラメータは約25°Cで測定した。
【0109】
単位格子中に4個の化学式単位を持つ単斜晶系の空間群P21/nにおける構造の解析および精密化は、通常の方法で行った。この構造はキノリン窒素原子がプロトン化されたN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの陽イオンと、単独でイオン化されたリンゴ酸の陰イオンを、1:1の比率で含んでいた。さらに、この結晶は、L-リンゴ酸イオンおよびD-リンゴ酸イオンを1:1の比率で含んでいた。表5に、約25°Cで計算した化合物 (III)、形態 N-1に関する分率原子座標を示す。
【0110】
単結晶X線回折データに基づき、図15に示すシミュレートしたパターンと実質的に一致するシミュレートした粉末X線回折(XRPD)パターン、および図15に示す実験結果のパターンと実質的に一致する観測XRPDパターン、またはそのいずれかにより、結晶質化合物 (III)、形態 N-1の特徴を決定できる。
【0111】
IV. 固体核磁気共鳴(SSNMR)
すべての固体C-13 NMR測定をBruker DSX-400, 400 MHz NMR回折装置で行った。高出力プロトンデカップリングならびにTPPMパルスシーケンス、および傾斜振幅交差分極法(ramp amplitude cross-polarization)(RAMP-CP)を、約12 kHzでのマジック角回転法(MAS)と併用し、高分解能スペクトルを得た(A.E. Bennett et al, J. Chem. Phys.,1995, 103, 6951),(G. Metz, X. Wu and S.O. Smith, J. Magn. Reson. A,. 1994, 110, 219-227)。円筒型のジルコニア製ローターに、約70 mgの試料を詰め、各実験で使用した。化学シフト(d)の指標として外部のアダマンタンを使い、高周波共鳴を38.56 ppmに設定した(W.L. Earl and D.L. VanderHart, J. Magn. Reson., 1982, 48, 35-54)。
【0112】
IV.1 化合物 (I)、形態 N-1
図2に結晶質化合物 (I)、形態 N-1の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-1の特徴づけに十分な場合がある。
【0113】
SS 13C NMRのピーク:18.1、20.6、26.0、42.9、44.5、54.4、55.4、56.1、70.4、99.4、100.1、100.6、114.4、114.9、 115.8、119.6、120.1、121.6、123.2、124.1、136.4、138.6、140.6、145.4、150.1、150.9、156.2、157.4、159.4、164.9、167.1、170.8、175.7、および182.1 ppm、± 0.2 ppm。
【0114】
図3に結晶質化合物 (I)、形態 N-1の固体15N NMRスペクトルを示す。スペクトルのピークは118.6、119.6、120.7、134.8、167.1、176.0、および180 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-1の特徴づけに十分な場合がある。
【0115】
図 4に結晶質化合物 (I)、形態 N-1の固体19F NMRスペクトルを示す。スペクトルのピークは-121.6、-120.8、および-118.0 ppm、± 0.2 ppmである。
【0116】
IV.2 化合物 (I), 形態 N-2
図9に結晶質化合物 (I)、形態 N-2の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0117】
SS 13C NMRのピーク:20.5、21.8、23.0、25.9、26.4、38.0、41.7、54.7、55.8、56.2、56.6、69.7、99.4、100.0、100.4、100.8、102.3、114.5、115.5、116.7、119.0、120.2、121.1、121.2、122.1、122.9、124.5、136.0、137.3、138.1、138.9、139.5、140.2、144.9、145.7、146.1、150.7、156.7、157.7、159.6、159.7、165.1、167.0、168.0、171.5、177.3、179.3、180.0、および180.3 ppm、± 0.2 ppm。
【0118】
図 10 に結晶質化合物 (I)、形態 N-2の固体15N NMRスペクトルを示す。スペクトルのピークは118.5、120.8、135.1、167.3、および180.1 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0119】
図 11に結晶質化合物 (I)、形態 N-2の固体19F NMRスペクトルを示す。スペクトルのピークは-121.0および-119.1 ppm、± 0.2 ppmである。それらのピークは個別に、または両方で、結晶質化合物 (I)、形態 N-2の特徴づけに十分な場合がある。
【0120】
IV.3 化合物 (III)、形態 N-1
図16に結晶質化合物 (III)、形態 N-1の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、結晶質化合物 (III)、形態 N-1の特徴づけに十分な場合がある。
【0121】
SS 13C NMRのピーク:20.8、26.2、44.8、55.7、70.7、100.4、101.0、114.7、115.2、116.0、119.7、120.4、121.6、124.4、136.9、138.9、141.1、145.7、150.3、156.5、157.6、159.6、165.2、167.4、171.2、176.3、および182.1 ppm、± 0.2 ppm。
【0122】
図 17に結晶質化合物 (III)、形態 N-1の固体15N NMRスペクトルを示す。スペクトルのピークは119.6、134.7、および175.5 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、結晶質化合物 (III)、形態 N-1の特徴づけに十分な場合がある。
【0123】
図 18に結晶質化合物 (III)、形態 N-1の固体19F NMRスペクトルを示す。スペクトルのピークは-120.5 ppm、± 0.2 ppmである。
【0124】
IV.4 化合物 (I)、非晶質
図 23に非晶質化合物 (I) の固体13C NMRスペクトルを示す。ピークのリスト全体またはその一部が、非晶質化合物 (I) の特徴づけに十分な場合がある。
【0125】
SS 13C NMRのピーク(ppm):12.2、17.8、20.3、21.8、27.2、33.8、41.7、56.9、69.9、99.9、102.2、115.6、122.2、134.4、137.8、142.9、149.1、150.9、157.3、159.7、167.0、171.7、173.1、177.4、および179.5 ppm、± 0.2 ppm。
【0126】
図 24に非晶質化合物 (I) の固体15N NMRスペクトルを示す。スペクトルのピークは120.8、131.8、174.7、および178.3 ppm、± 0.2 ppmである。ピークのリスト全体またはその一部が、非晶質化合物 (I) の特徴づけに十分な場合がある。
【0127】
図 25に非晶質化合物 (I) の固体19F NMRスペクトルを示す。スペクトルのピークは-118.9 ppm、± 0.2 ppmである。
【0128】
V. 熱特性測定
熱重量分析(TGA)
TGA測定は、オープンパンを装備したTA Instruments(TM)モデルQ500または2950で実施した。試料(約10-30 mg)を、事前に風袋調節したプラチナパンに乗せた。試料の重量はこの装置により、1000分の1ミリグラム単位まで正確に測定・記録された。加熱炉は100 mL/分の窒素ガスでパージした。10°C/分の加熱速度で、室温から300°Cの間のデータを収集した。
【0129】
示差走査熱量(DSC)分析
DSC測定は、オープンパンを装備したTA Instruments(TM)モデルQ2000、Q1000または2920で実施した。試料(約2-6 mg)をアルミニウムパンの上で計量し、100分の1ミリグラム単位まで正確に記録し、DSCに移した。装置は50 mL/分の窒素ガスでパージした。10°C/分の加熱速度で、室温から300°Cの間のデータを収集した。データをプロットし、下向きの吸熱ピークを求めた。
【0130】
V.1 化合物 (I), 形態 N-1
図 5に示す結晶質化合物 (I)、形態 N-1のTGAサーモグラムで、170°Cにおける重量減少は約0.4重量%である。
【0131】
図 6に示す結晶質化合物 (I)、形態 N-1のDSCサーモグラムで、融点は約187°Cである。
【0132】
V.2 化合物 (I)、形態 N-2
図 12に示す結晶質化合物 (I)、形態 N-2のTGAサーモグラムで、170°Cにおける重量減少は約0.1重量%である。
【0133】
図 13に示す結晶質化合物 (I)、形態 N-2のDSCサーモグラムで、融点は約186°Cである。
【0134】
V.3 化合物 (III)、形態 N-1
図 19に示す結晶質化合物 (III)、形態 N-1のTGAサーモグラムで、170°Cにおける重量減少は約0.2重量%である。
【0135】
図 20に示す結晶質化合物 (III)、形態 N-1のDSCサーモグラムで、融点は約186°Cである。
【0136】
V.2 化合物 (I)、非晶質
図 26に非晶質化合物 (I) のDSCを示す。
【0137】
VI. 水蒸気吸着等温線測定
水蒸気吸着等温線は、約10 mgの試料を使い、VTI SGA-100 Symmetric Vapor Analyzerで求めた。試料を60°C で、10分間の減少速度が0.0005 重量%/分になるまで乾燥させた。試料を25°Cで、3または4、5、15、25、35、45、50、65、75、85、および95% RHで測定した。35分間の減少速度が0.0003重量%/分に達した時、または最長600分間で、各RHにおける平衡に達した。
【0138】
VI.1 化合物 (I)、形態 N-1
図 7に結晶質化合物 (I)、形態 N-1の水蒸気吸着等温線を示す。
【0139】
VI.2 化合物 (I)、形態 N-2
図 14に結晶質化合物 (I)、形態 N-2の水蒸気吸着等温線を示す。
【0140】
VI.3 化合物 (III)、形態 N-1
図 21に結晶質化合物 (III)、形態 N-1の水蒸気吸着等温線を示す。
【0141】
VI.4 化合物 (I)、非晶質
図 27に非晶質化合物 (I) の水蒸気吸着等温線を示す。
【0142】
上記の開示内容は、明確性と理解の促進を目的として、図面および例を使い、ある程度詳細に記載されている。本発明は、さまざまな特定および好ましい実施形態および技法に参照して記載されている。しかし、本発明の主旨および範囲内にとどまりつつ、多数の変形および修正を加えることができると理解するものとする。添付した請求項の範囲内で、変更および修正を実施できることは、当業者には明白なはずである。従って、上記の説明は、例示を意図したものであり、限定的ではないと理解するものとする。従って、本発明の範囲は、上記の説明を参照して決定すべきではなく、以下に添付する請求項、および係る請求項に関して認められる同等物の全範囲を参照して決定すべきである。
【特許請求の範囲】
【請求項1】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩。
【請求項2】
請求項1に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がDL-リンゴ酸塩である。
【請求項3】
請求項1に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がL-リンゴ酸塩またはD-リンゴ酸塩である。
【請求項4】
請求項3に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がL-リンゴ酸塩である。
【請求項5】
請求項3に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がD-リンゴ酸塩である。
【請求項6】
請求項3から5のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩が結晶質である。
【請求項7】
請求項6に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩が結晶質の形態 N-1であり、係る形態 N-1は以下の一以上の特徴を持つ。
(i) 固体13C NMRスペクトルに、18.1、42.9、44.5、70.4、123.2、156.2、170.8、175.7、および182.1 ± 0.2 ppmの中から選択される4個以上のピークがある。
(ii) 室温で結晶質形態を測定した粉末X線回折パターン(CuKα λ=1.5418Å)に、以下から選択される4個以上の2θ値が含まれる。12.8±0.2 °2θ、13.5±0.2 °2θ、16.9±0.2 °2θ、19.4±0.2 °2θ、21.5±0.2 °2θ、22.8±0.2 °2θ、25.1±0.2 °2θ、27.6±0.2 °2θ。
(iii) 粉末X線回折(XRPD)パターンが、図 1に示すパターンと実質的に一致する。
【請求項8】
請求項7に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩の重量に基づき、係る塩が90重量%以上の形態N-1を含む。
【請求項9】
請求項6に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩が結晶質の形態N-2であり、係る形態 N-2は以下の一以上の特徴を持つ。
(i) 固体13C NMRスペクトルに、23.0、25.9、38.0、 41.7、69.7、102.0、122.5、177.3、179.3、180.0、および180.3 ± 0.2 の中から選択される4個以上のピークがある。
(ii) 室温で結晶質形態を測定した粉末X線回折パターン(CuKα λ=1.5418Å)に、20.9±0.2 °2θおよび21.9±0.2 °2θの2θ値が含まれ、さらに以下の中から選択される2個以上の2θ値が含まれる。6.4±0.2 °2θ、9.1±0.2 °2θ、12.0±0.2 °2θ、12.8±0.2、13.7±0.2、17.1±0.2、22.6±0.2、23.7±0.2。
(iii) 粉末X線回折(XRPD)パターンが、図 8に示すパターンと実質的に一致する。
【請求項10】
請求項9に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩の重量に基づき、係る塩が90重量%以上の形態N-2を含む。
【請求項11】
請求項4から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩を含む医薬組成物、および薬学的に許容される賦形剤。
【請求項12】
請求項3から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の癌治療薬剤製造を目的とする使用。
【請求項13】
請求項3から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の癌治療における治療法での使用。
【請求項14】
請求項6から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩の結晶質形態の、甲状腺癌治療を目的とする薬剤としての患者での使用。
【請求項15】
請求項6から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの L-リンゴ酸塩の結晶質形態の、神経膠芽細胞腫治療を目的とする薬剤としての患者での使用。
【請求項1】
N-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩。
【請求項2】
請求項1に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がDL-リンゴ酸塩である。
【請求項3】
請求項1に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がL-リンゴ酸塩またはD-リンゴ酸塩である。
【請求項4】
請求項3に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がL-リンゴ酸塩である。
【請求項5】
請求項3に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩がD-リンゴ酸塩である。
【請求項6】
請求項3から5のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩が結晶質である。
【請求項7】
請求項6に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩が結晶質の形態 N-1であり、係る形態 N-1は以下の一以上の特徴を持つ。
(i) 固体13C NMRスペクトルに、18.1、42.9、44.5、70.4、123.2、156.2、170.8、175.7、および182.1 ± 0.2 ppmの中から選択される4個以上のピークがある。
(ii) 室温で結晶質形態を測定した粉末X線回折パターン(CuKα λ=1.5418Å)に、以下から選択される4個以上の2θ値が含まれる。12.8±0.2 °2θ、13.5±0.2 °2θ、16.9±0.2 °2θ、19.4±0.2 °2θ、21.5±0.2 °2θ、22.8±0.2 °2θ、25.1±0.2 °2θ、27.6±0.2 °2θ。
(iii) 粉末X線回折(XRPD)パターンが、図 1に示すパターンと実質的に一致する。
【請求項8】
請求項7に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩の重量に基づき、係る塩が90重量%以上の形態N-1を含む。
【請求項9】
請求項6に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩が結晶質の形態N-2であり、係る形態 N-2は以下の一以上の特徴を持つ。
(i) 固体13C NMRスペクトルに、23.0、25.9、38.0、 41.7、69.7、102.0、122.5、177.3、179.3、180.0、および180.3 ± 0.2 の中から選択される4個以上のピークがある。
(ii) 室温で結晶質形態を測定した粉末X線回折パターン(CuKα λ=1.5418Å)に、20.9±0.2 °2θおよび21.9±0.2 °2θの2θ値が含まれ、さらに以下の中から選択される2個以上の2θ値が含まれる。6.4±0.2 °2θ、9.1±0.2 °2θ、12.0±0.2 °2θ、12.8±0.2、13.7±0.2、17.1±0.2、22.6±0.2、23.7±0.2。
(iii) 粉末X線回折(XRPD)パターンが、図 8に示すパターンと実質的に一致する。
【請求項10】
請求項9に従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩であって、係る塩の重量に基づき、係る塩が90重量%以上の形態N-2を含む。
【請求項11】
請求項4から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩を含む医薬組成物、および薬学的に許容される賦形剤。
【請求項12】
請求項3から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の癌治療薬剤製造を目的とする使用。
【請求項13】
請求項3から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのリンゴ酸塩の癌治療における治療法での使用。
【請求項14】
請求項6から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドのL-リンゴ酸塩の結晶質形態の、甲状腺癌治療を目的とする薬剤としての患者での使用。
【請求項15】
請求項6から10のいずれかに従うN-(4-{[6,7-ビス(メチルオキシ)-キノリン-4-イル]オキシ}フェニル)-N’-(4-フルオロフェニル)シクロプロパン-1,1-ジカルボキサミドの L-リンゴ酸塩の結晶質形態の、神経膠芽細胞腫治療を目的とする薬剤としての患者での使用。
【図1】

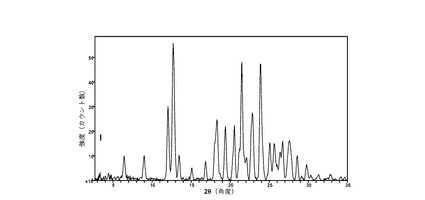
【図2】
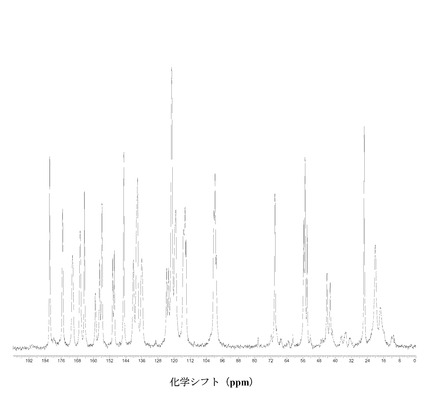
【図3】
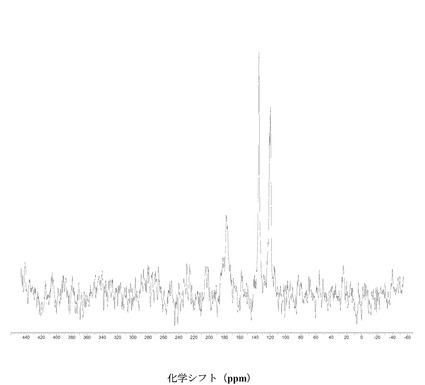
【図4】
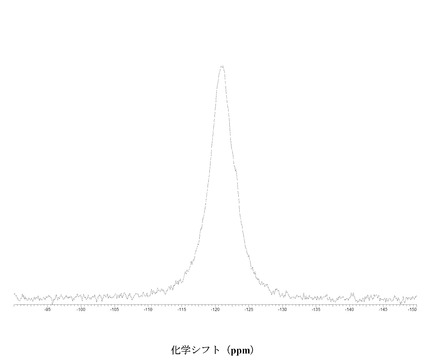
【図5】
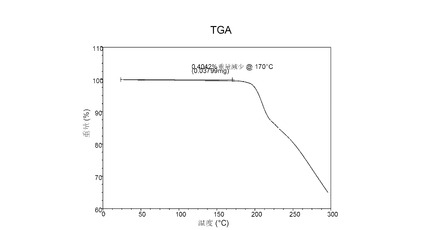
【図6】
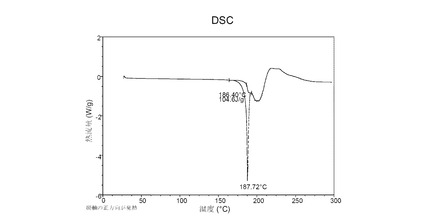
【図7】
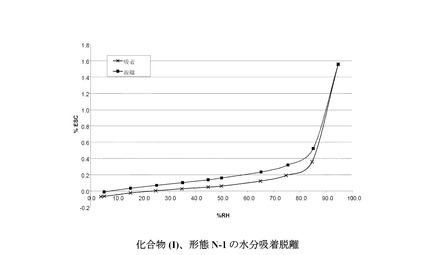
【図8】
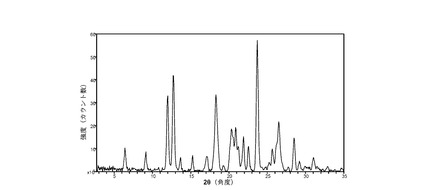
【図9】
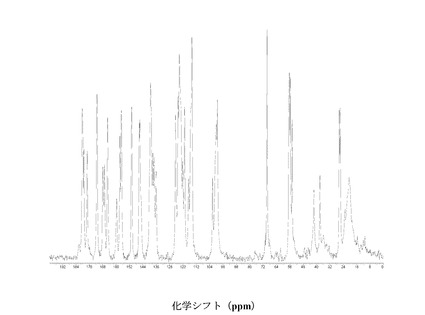
【図10】
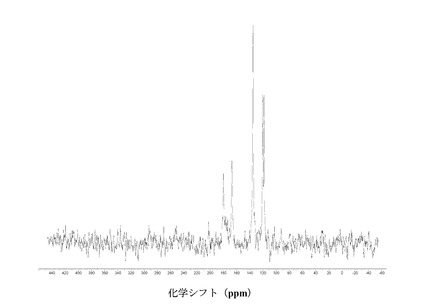
【図11】
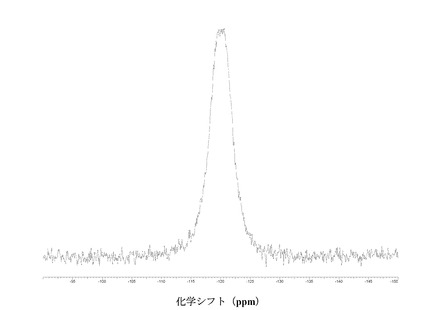
【図12】
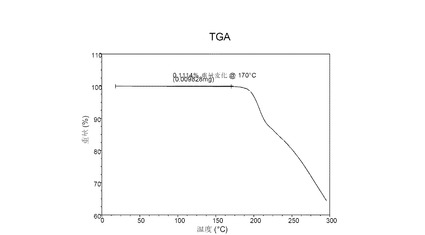
【図13】
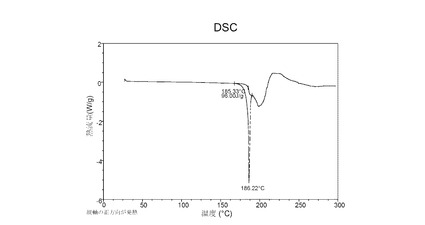
【図14】
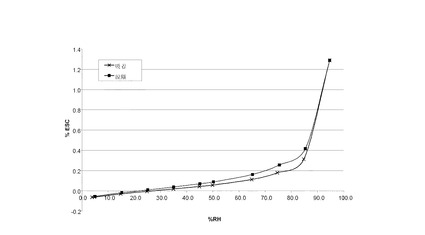
【図15】
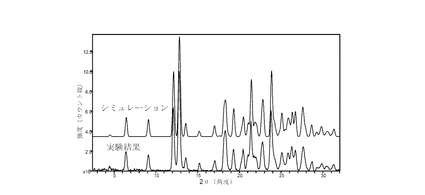
【図16】
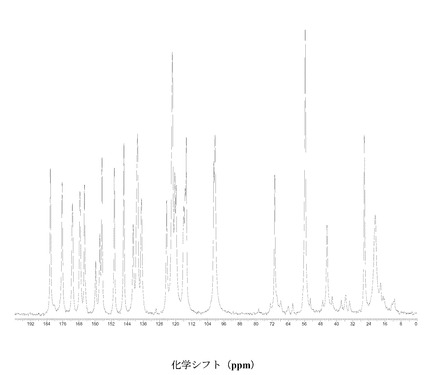
【図17】
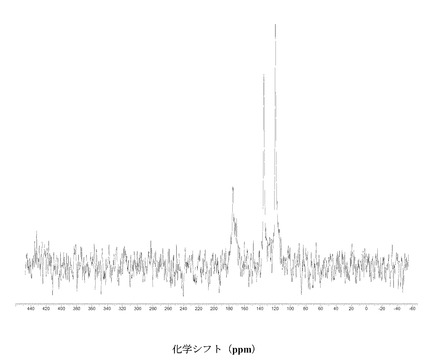
【図18】
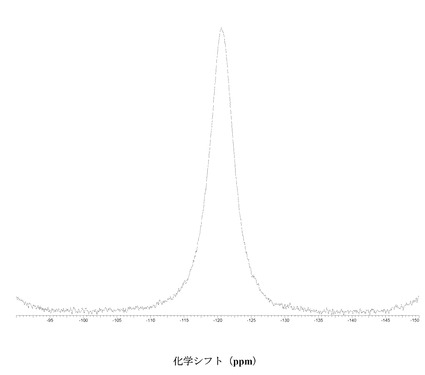
【図19】
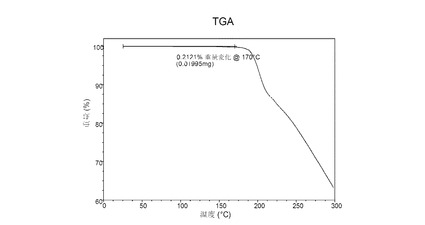
【図20】
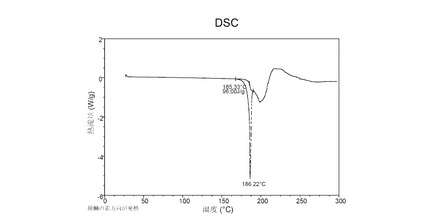
【図21】
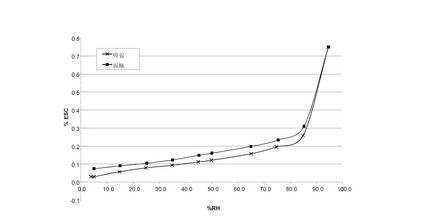
【図22】
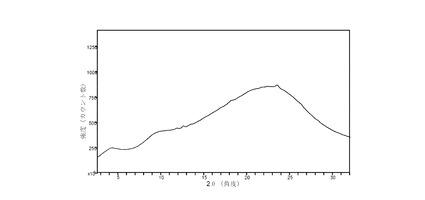
【図23】
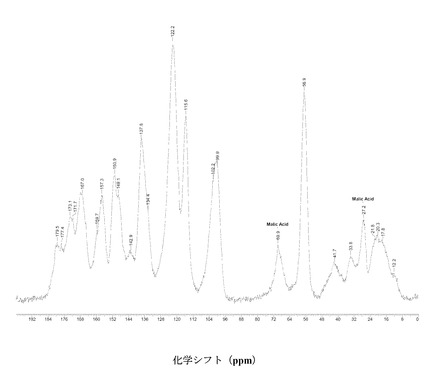
【図24】
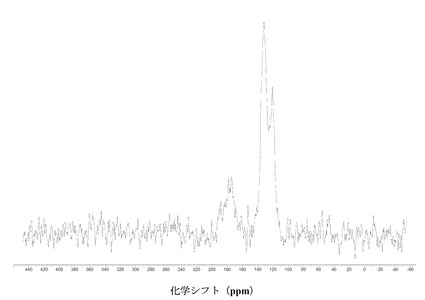
【図25】
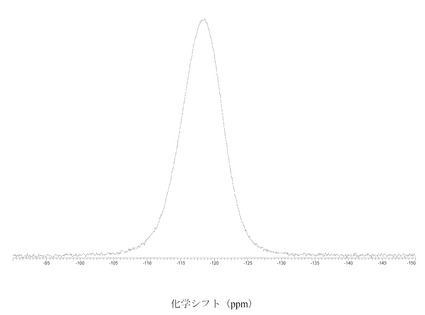
【図26】
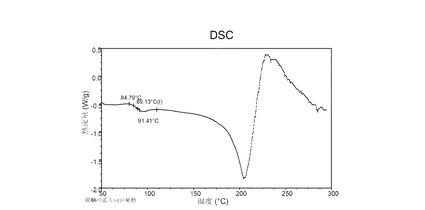
【図27】
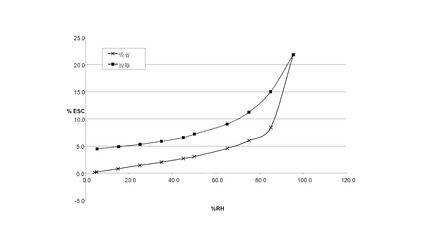
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図23】
【図24】
【図25】
【図26】
【図27】
【公表番号】特表2012−515220(P2012−515220A)
【公表日】平成24年7月5日(2012.7.5)
【国際特許分類】
【出願番号】特願2011−546390(P2011−546390)
【出願日】平成22年1月15日(2010.1.15)
【国際出願番号】PCT/US2010/021194
【国際公開番号】WO2010/083414
【国際公開日】平成22年7月22日(2010.7.22)
【出願人】(504408797)エクセリクシス, インク. (65)
【出願人】(511173147)ブリストル−マイヤーズ スクイブ カンパニー (1)
【Fターム(参考)】
【公表日】平成24年7月5日(2012.7.5)
【国際特許分類】
【出願日】平成22年1月15日(2010.1.15)
【国際出願番号】PCT/US2010/021194
【国際公開番号】WO2010/083414
【国際公開日】平成22年7月22日(2010.7.22)
【出願人】(504408797)エクセリクシス, インク. (65)
【出願人】(511173147)ブリストル−マイヤーズ スクイブ カンパニー (1)
【Fターム(参考)】
[ Back to top ]