
N−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミドの合成方法
本発明は0.2%未満の不純物を含有する式(IV)のN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミドに関するものであり、これは治療用途に適する。更に本発明は0.2%未満の不純物を含有する式(IV)の治療上適用しうる化合物の合成方法に関するものであり、この方法は式(II)の新規な中間体N−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチルアセタミド・ヒドロクロライドと式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基とを酸フリーの媒体にて反応させる。更に本発明は式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチルアセタミド・ヒドロクロライドにも関するものである。更に本発明は99.5%より大の純度を有する式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの合成方法にも関するものである。本発明は更に、式(IV)の化合物の合成に際し0.07%の収率で生成される新規な単離不純物である式(V)のN−{3−(3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドにも関するものである。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
本発明は、治療用途に適する0.2%未満の不純物を含有する式(IV)のN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミドに関するものである。
【0002】
更に本発明は、0.2%未満の不純物を含有する式(IV)の治療上適用しうる化合物の合成方法にも関するものであり、この方法は式(II)の新規な中間体N−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]−フェニル}−N−エチル−アセタミド・ヒドロクロライドを酸フリーの媒体中で式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と反応させる。
【0003】
更に本発明は式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドにも関するものである。
【0004】
さらに本発明は、式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドに関するものであり、また本発明は、99.5%より大の純度を有する。
【0005】
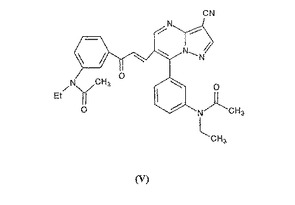
更に本発明は、式(IV)の化合物の合成に際し0.07%収率にて形成される新規な単離不純物である式(V)のN−{3−(3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドにも関するものである。
【0006】
式(IV)の化合物(ザレプロン)は、GABAAリセプタ作用剤として作用する抗不安剤、抗てんかん薬、鎮静薬、催眠薬および骨格筋弛緩剤である。
【0007】
式(IV)の化合物は米国特許第4,626,538号に表8における例14で記載されている。最終生成物は、式(I)のN−[3−[3−(ジメチルアミノ)−オキソ−2−プロペニル]フェニル]−N−エチル−アセタミド塩基を式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と氷酢酸中で8時間にわたり還流させることにより得られる。化合物の正確な物理化学的性質、純度および収率はこの米国特許には記載されておらず、186〜187℃である融点のみが示されている。
【0008】
米国特許第4,626,538号の方法は、欧州特許第776,898号にて改良される。式(IV)の化合物の改良合成方法は次のプロセスに従う:式(I)のN−[3−[3−(ジメチルアミノ)−オキソ−2−プロペニル]フェニル]−N−エチル−アセタミド塩基を式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と酢酸および水(10容量%〜85容量%)の混液にて、好ましくは1:2v/vのこれらの混合物にて反応させる。得られる生成物の純度はHPLCにより98.86〜99.40%である。最も純度の高い(99.4%)の化合物の収率は例1に記載されたように83.5%である。出発材料は各例の全てにおいて塩基性である。式(IV)の化合物の合成方法は中間体に基づくだけでなく、その塩からも出発して請求項に記載されている。出発材料として塩を使用する上記欧州特許に記載されたプロセスは存在せず、更に適用しうる中間塩またはその合成についても記載されていない。
【0009】
更に、開示された国際公開第2002/100828号も式(IV)の化合物の合成方法を記載しており、この方法は式(I)のN−[3−[3−(ジメチルアミノ)−オキソ−2−プロペニル]フェニル]−N−エチル−アセタミド塩基を式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と水および水混和性有機溶剤の混合液中で酸性条件下に反応させることからなっている。酸性条件は、有機酸もしくは無機酸(特に塩酸)を反応混合物に添加して達成される。酸の量は出発塩基の1種と等モル量とすることができ、或いは大過剰で用いることもできる。請求項12〜15において、出発材料としての式(I)の塩基の酸付加塩の適用および酸の添加が別々に記載されているが一方では請求項6および1にて塩基のみが出発材料として使用され、他方では出発材料としての塩の使用を支持する記載はない。国際公開第2002/100828号(欧州特許第776,898号と同様)は、式(I)もしくは(III)の塩基の塩につき作成、単離および好適具体例の所定の例もしくは記載を示していない。例12においては、1当量の塩酸が使用され、これは請求項21に記載されているように適用した酸の量の下限値である。この例によれば生成物は24時間の反応時間後に82%収率にて得られ、純度は僅か98.95%である。本発明者等の実験(上記例12による合成を行うと共に1当量の塩酸を用いる)において、測定されたpHは全反応時間に際し約1.5であった。例1〜21に記載されたように用いた酸の量が1当量より多いこれら反応において、測定pHは1.0未満であり、すなわち強酸性反応条件が用いられる。
【0010】
国際公開第2003/011228号には、式(IV)の化合物の新規なレジオアイソマーN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−5−イル)−フェニル]−N−エチル−アセタミドが記載されており、これは式(IV)の化合物の合成に際し生成される。このレジオアイソマーの相当量(0.2〜0.5%)が、たとえば米国特許第2003/0040522号に記載された式(IV)の化合物の合成方法に際し生成される。
【0011】
上記化学的純度の数値は現在の医薬工業にて使用されるよりも低いので、本発明の目的は、高純度(最も厳密な品質要件を満たす)の生成物を良好な収率にて作成するのに適する新規な式(IV)の化合物の合成方法を見出すことであった。
【0012】
本発明の基礎は次の知見にある:反応混合物のpHは反応に際し3.0〜3.5の範囲に保って、化学反応の最高の選択率に到達すべきである。本発明による研究の結果、式(IV)の高純度の化合物を得るのに要する混合物の最適pHは自動調節的に式(II)の高純度塩酸塩の適用により維持される。
【0013】
本発明の研究に際し、高純度の中間体(特に最後のもの)が最終生成物の高純度の前提であることが判明した。式(III)の高品質の3−アミノ−4−ピラゾール−カルボニトリル塩基は市販入手することができる。式(I)の塩基は多段階合成にて得ることができ、適する純度の化合物の基本形態は多量の材料を損失した後にのみ単離することができる。驚くことに、特に式(I)の塩基の塩類から塩酸塩(従来特性化されていない)を式(I)の塩基から高純度および極めて良好な収率にて容易に得られることが判明した。
【0014】
式(IV)の化合物の合成を取り扱った従来技術の特許および特許出願の例において、反応は高酸性条件下で行われ、或る場合には1当量より多い酸が使用される。先ず最初に、本発明者等はこれら公知方法を反復した。これら高酸性条件下にて全ての場合、式(IV)の得られる化合物は0.2%より多いレジオアイソマーN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−5−イル)−フェニル]−N−エチル−アセタミドを含有することを突き止めた。次いで、本発明者等は高純度の式(IV)の化合物の新規な合成方法につき検討した。
【0015】
驚くことに式(IV)の化合物は、式(I)の高純度塩基のうち式(II)の新規な塩酸塩(これは本発明の方法により良好な収率で得られる)を式(III)の塩基と反応混合物に酸を添加することなく反応させることにより合成しうること、更に生成物の収率および純度は従来技術に記載された数値よりもずっと高いことが突き止められた。
【0016】
0.2%未満の不純物を含有する式(IV)の高純度化合物の合成は刊行物に記載されておらず、更に反応混合物へ酸を添加することのない式(IV)の化合物の合成に関する本発明による方法も記載されていない。
【0017】
本発明は、0.2%未満の不純物を含有し治療用途に適する式(IV)の化合物に関するものである。
【0018】
更に本発明は0.2%未満の不純物を含有する式(IV)の治療上適用しうる化合物の合成方法にも関するものであり、この方法は式(II)の新規な塩を酸フリーの媒体にて式(III)の塩基と反応させる。
【0019】
また本発明は、式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドにも関するものである。
【0020】
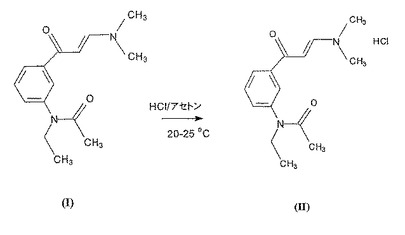
更にまた本発明は99.5%より大の純度を有する式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの合成方法にも関し、この方法は式(I)の化合物をアセトン中に20〜25℃にて窒素下で懸濁させると共に等モル量の濃塩酸を添加することを特徴とする。
【0021】
更に本発明は式(V)のN−{3−[3−シアノピラゾロ(1,5−a)−ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドにも関し、この化合物は式(IV)の化合物の合成に際し形成される新規な単離不純物である。
【0022】
本発明の詳細な説明は次の通りである:
【0023】
「室温」の意味は20〜25℃である。
【0024】
「粗製最終生成物」の意味は、従来技術の式(IV)の化合物の合成手順〜反復して得られる最終生成物である。
【0025】
「高酸性条件」の意味は、反応をpH1.0未満で行うことを意味する。
【0026】
本発明によれば式(II)の新規な塩は、式(I)の塩基をアセトン中に室温にて窒素下で懸濁させると共に等モル量の濃塩酸を添加することにより得られる。攪拌後沈殿した結晶を濾過し、洗浄し、次いで乾燥させる。このように得られた生成物の純度は99.5%より大である。
【0027】
本発明によれば、0.2%未満の不純物を含有する式(IV)の治療上適用しうる化合物の合成は次の通りである:
【0028】
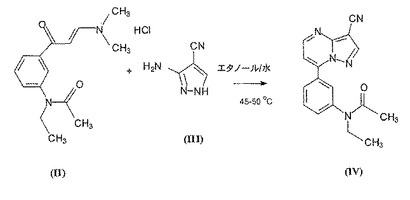
式(III)の塩基を水とアルコールとの混液に溶解させると共に、式(II)の固体塩を少量ずつ加温溶液に添加する。添加に際し、激しい攪拌を持続する。他の具体例によれば、式(II)の塩を溶剤に溶解させると共に、溶液を反応混合物に添加する。先ず最初に、溶液(後に得られる懸濁液)を連続的に攪拌し、次いで生成物を単離する。通常、懸濁物を冷却すると共に攪拌を持続する。沈殿した結晶を濾別し、洗浄し、次いで乾燥させる。得られた結晶物質を温メタノールに窒素下で溶解させ、溶液を濾過し、冷却し、沈殿生成物を濾別し、洗浄し、次いで乾燥させる。
【0029】
本発明による方法は、好ましくは有機溶剤中で或いは水と水混和性有機溶剤との混液中で行われる。有機溶剤としては単鎖脂肪族アルコール(好ましくはメタノール、エタノール、イソプロパノール、n−プロパノール)を使用することができる。本発明による方法は好ましくは15〜50℃にて0.5〜1時間にわたり行われる。式(III)の3−アミノ−4−シアノ−ピラゾール塩基を好ましくは溶剤の適する混液に溶解させ、式(II)の塩酸塩を少量ずつ溶液に添加する。冷却後、沈殿生成物を濾別すると共に乾燥させる。このように得られた式(IV)の生成物の純度は、HPLCによれば99.8%より大である。
【0030】
本発明による方法の適用(式(II)の新規な高純度の塩酸塩と式(III)の化合物との反応)は、従来技術により得られるもの(すなわち反応を水性酢酸にて行うことにより得られるもの)よりもずっと高品質(99.4%より大の純度)の式(IV)の化合物をもたらす。
【0031】
本発明の方法により得られる式(IV)の最終生成物の高純度(これは医薬工業にて特に有利である)は、式(II)の新規な塩酸塩の高純度に部分的に基づき、更に酸フリーの反応混合物に部分的に基づき、また少量ずつの式(II)の新規な塩酸塩の添加に部分的に基づき、更にまた短い反応時間に部分的に基づいている。
【0032】
式(IV)の化合物の合成にて本発明により最初に作成されると共に使用される式(II)の新規な塩酸塩の適用に関する大きい利点は、高純度の式(IV)の化合物の合成にて高純度の出発材料が使用される(これは医薬工業にて必要とされる)点である。HPLCによれば、式(I)の塩基の純度は一般に90〜95%である一方、この塩基から得られる式(II)の塩酸塩の純度は98.5%より大である。
【0033】
式(IV)の化合物の本発明による合成方法に際し、反応のpHは3.0〜3.5であることを強調せねばならない。本発明による方法の大きな利点は、追加量の酸を反応に添加する必要がないことである。何故なら、出発材料として使用する式(II)の新規な塩酸塩は等モル量の酸を含有し、更に反応混合物のpHは全反応時間にわたり式(II)の塩酸塩の反応混合物に対する少量ずつの添加に基づき3.0〜3.5の範囲に留まり、その結果式(IV)の化合物の本発明による合成方法の反応条件は反応をpH1.0未満または最高pH1.5にて行う従来技術の手法と比較してずっと緩和であるからである。
【0034】
式(IV)の化合物の合成に際し本発明により最初に作成されかつ使用された式(II)の新規な塩酸塩の適用に関する大きな利点は、式(IV)の化合物のレジオアイソマーが0.1%未満の収率でのみ生成される点である。従来技術の手法により作成される式(IV)の最終生成物(明細書に開示した通り)は或る場合には0.5%の不純物としてレジオアイソマーを含有する。
【0035】
式(IV)の化合物の合成に際し本発明により最初に作成されかつ使用された式(II)の新規な塩酸塩の使用に関する更なる利点は、式(IV)のレジオアイソマーの生成される少量の他に、刊行物または開示された特許には記載されていない式(V)の不純物が微量(0.07%)でのみ生成される点である。
【0036】
式(V)の不純度の分析的特性化を実施例2に示す。本発明による分析測定は式(V)の化合物の構造を証明し、その化学名はN−{3−[3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドである。
【0037】
式(V)の上記不純物は、式(IV)の化合物の合成につき文献に記載された方法により得られる粗製最終生成物から単離することができる。何故なら、これら粗製生成物はより多量(0.1%)の式(V)の不純物を含有するからである。式(V)の不純物を、作成HPLCにより再結晶化の母液から単離した。驚くことに、式(V)の不純物は式(IV)の化合物の本発明による合成方法にて0.07%の収率でのみ生成することが判明した。
【0038】
本発明者等は、式(V)の不純物の生成を式(IV)の化合物の合成に関する本発明の方法に際し反応混合物へ少量ずつ式(II)の塩酸塩を添加すると共に適するpH値、温度、溶剤の割合および反応時間を保つことにより、減少させうることを認識した。これは医薬工業において特に有利である。
【0039】
要約すれば、公知方法と比較した式(IV)の化合物の本発明による合成方法の利点は次の通りである:
【0040】
−式(IV)の最終生成物は、0.2%未満の不純物(医薬品製造のGMP要件に従う)を含有して良好な収率で作成される;
−式(II)の塩酸塩の純度は極めて高い;
−式(IV)の最終生成物は酸の添加なしに作成される;
−式(IV)の最終生成物は、従来技術の手法により得られるものと比較してずっと少ない(最大0.1%)のレジオアイソマーを不純物として含有する;
−式(IV)の最終生成物は、従来技術の手法により得られるものと比較してずっと少ない(0.07%)の本発明者等により単離された式(V)の不純物を含有する;
−薬物の高純度は式(IV)の最終生成物の製造に際し安全に制御することができ、これは式(IV)の最終生成物の高純度だけでなく式(II)の最終中間体の高純度も実現しうる。
【0041】
以下、実施例を例示するが本発明の範囲を決して限定するものでない。
【0042】
実施例においては、式(II)の新規な化合物、式(IV)の最終生成物、および本発明の方法により得られる式(V)の不純物を以下の分析法により特性化する。
【0043】
本発明の方法により得られる生成物の純度はHPLCにより測定した。HPLC装置:熱分離品、ポンプ:スペクトルシステムP2000、検出器:スペクトルシステムUV2000。制御、データ収集およびデータ処理:クロムクエスト・プログラム、Ver.2.51。クロマトグラフィーの条件:カラム:YMC−パック・プロC18、150x4.6mm、ID5μm。溶出剤A:140mlのアセトニトリル+30mlのメタノール+30mlのテトラヒドロフラン+800mlの0.1M酢酸アンモニウムの混液。溶出剤B:400mlのアセトニトリル+100mlのメタノール+100mlのテトラヒドロフラン+400mlの0.1M酢酸アンモニウムの混液。これらから表1に示す線状勾配の溶剤システムを使用した。
【0044】
流速:1ml/min;カラムの温度:室温;検出の波長:235nm。分析試料の溶解:77%の溶出剤A+23%の溶出剤Bの混液。注入量:10μl。
【0045】
【表1】

【0046】
1H−NMRおよび13C−NMRスペクトルをバリアンINOVA(1H−NMRにつき500MHz)スペクトロメータにて記録した。各測定はデューテロ−ピリジンまたはデューテロ−ジクロルメタンにて30℃で行った。化学シフト(δ)は、テトラメチルシラン(TMS)を内部標準(δTMS=0.00ppm)として使用しppmにて示す。NMRスペクトルにおいては次の記号を使用する:s=シングレット、d=デュープレット、t=トリプレット、q=クワルテット、m=マルチプレット、dd=ダブルデュープレット、br=ブロードマルチプレット、dm=デュープレット・マルチプレット。
【0047】
各化合物の高分解MSスペクトルはFAB−MS法により測定した。装置:フィニガンMAT95SQ。FABイオン化法:20kV Cs+イオンガン、3−ニトロ−ベンジルアルコール・マトリックス溶剤。スペクトルの評価:m/z。FAB−娘イオンスペクトルはタンデムMS法により得た。
【0048】
IRスペクトルは臭化カリウムペレットの4cm−1スペクトル分解を用いてパーキン−エルマー1000スペクトロメータで記録した。
【0049】
実施例1
式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライド
窒素下で31.5gの式(I)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミドを300mlのアセトン中に室温にて懸濁させると共に、10.4mlの濃塩酸を滴加した。0.5mlの濃塩酸を添加した後、均質溶液が得られ、結晶の分離を3−4mlの濃塩酸の添加の後に開始した。全量の濃塩酸を添加した後、反応混合物を室温にて30分間および0〜5℃にて更に1時間にわたり攪拌した。沈殿した結晶を濾別し、10mlの冷(3〜5℃)アセトンで2回洗浄すると共に、50℃未満で乾燥させて31.15g(87%)の標記化合物を得た。
【0050】
融点:136〜137℃。
【0051】
得られた生成物の分析特性化:
【0052】
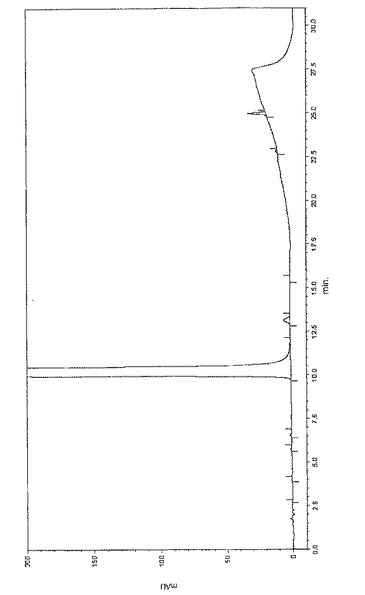
HPLCにより得られた式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの溶出プロフィルを図1に示す。横軸:溶出時間(0〜31分間にわたるmin);縦軸:吸収率(mAU)試料の濃度は0.5mg/mlであった。式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの反応時間は4.0minである。評価は面積基準化により行った。
【0053】
HPLCにより生成物の純度は98.5%であった。
【0054】
デューテロ−ピリジンにおける式(II)の化合物の1H−NMRスペクトルを図2に示し、ここには化学シフト(δ)を内部標準としてテトラメチルシラン(TMS)(δTMS=0.00ppm)を使用してppmにて示す。
【0055】
特徴的化学シフトは次の通りである:
【0056】
【数1】
【0057】
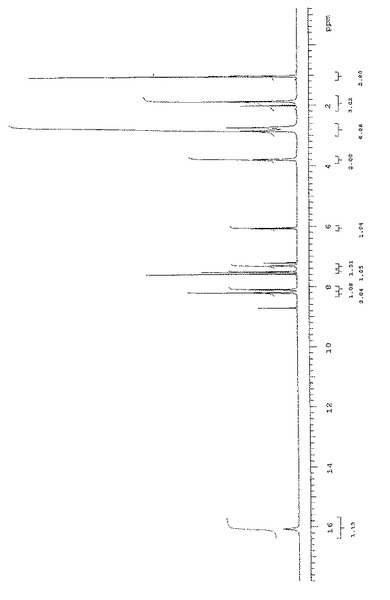
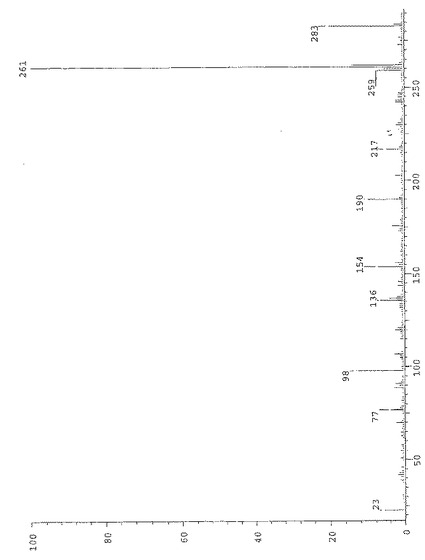
式(II)の化合物のFAB−MSスペクトルを図3に示す。特徴的なプロトン化分子イオンはm/z=261である一方、ナトリウム陽イオンを有する分子イオンはm/z=283である。他の断片的ピーク:3−(ジメチルアミノ)−1−オキソ−2−プロペニルイオンの形成(m/z=98)、2−ジメチルアミノ−エチレン側鎖の開裂(m/z=190)、アセチル基の開裂(m/z=217)、プロトン化分子イオンからのエタンの排除(m/z=231)。M/z=77、136および154の各ピークをマトリックス溶剤から得ることができる。
【0058】
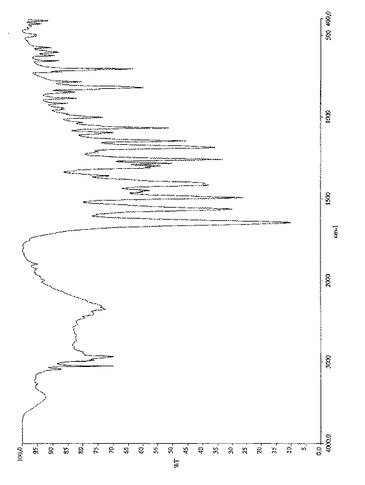
式(II)の化合物のIRスペクトルを図4に示す。特徴的IR吸収バンド(cm−1)は次の通りである:
N+−H 2700−2000
C−O 1645
Ar 820,706
【0059】
更なる特徴的吸収バンド(cm−1):3050、2932、1562、1492、1259、1185、1146、1067。
【0060】
実施例2
式(IV)のN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミド
窒素を150mlの精製水と50mlのエタノールとの混液中にバブリングさせると共に、窒素下で10.8gの式(III)の3−アミノ−4−ピラゾール−カルボニトリルをこの混合物に添加した。溶解後、反応混合物を45〜50℃まで加温すると共に、式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの29.6gを3つの等しい部分にて全て15分間で添加した。最初に溶液であると共に後に懸濁液となった反応混合物を、この温度にて1時間にわたり窒素下で攪拌した。次いで得られた懸濁物を0〜5℃まで冷却すると共に、この温度にて2時間にわたり攪拌した。沈殿した結晶を濾別し、25mlの氷冷エタノールおよび2x25mlの氷冷水で洗浄した。生成物を50℃未満で減圧下に乾燥させた。得られた生成物を280mlのメタノール中に窒素下で65〜67℃にて溶解させて機械的不純物を除去した。この溶液を濾過すると共に濾液を0〜5℃まで冷却した。混合物をこの温度に2時間にわたり保ち、次いで沈殿結晶を濾別し、2x10mlの氷冷メタノールで洗浄し、更に50℃未満で乾燥させて28.2g(92.5%)の標記化合物を得た。
【0061】
得られた生成物の分析特性化:
【0062】
HPLCにより得られた式(IV)の化合物の溶出プロフィルを図5に示す。横軸:溶出時間(0〜31分間にわたるmin);縦軸:吸収率(mAU)。試料の濃度は1.0mg/mlであった。式(IV)の化合物の滞留時間は10.2minである。評価は、標準試料溶液の1000倍希釈の外部標準法により行った。
【0063】
HPLCにより式(IV)の生成物の純度は99.8%であった。
【0064】
0.07%収率にて形成されると共に本発明により特性化された不純物である式(V)のN−{3−(3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドの滞留時間は、図5に見られるように24.9minである。
【0065】
この不純物を更に式(IV)の化合物の合成に関する公知手順により得られた粗製最終生成物から単離し、これら手順の最終生成物は多量(0.1%)の式(V)の不純物を含有した。
【0066】
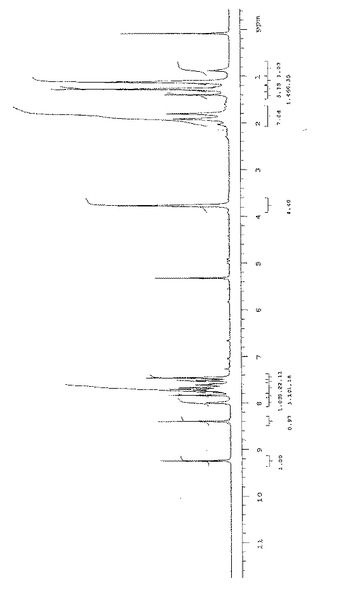
デューテロジクロルメタンにおける式(V)の化合物の1H−NMRおよび13C−NMRスペクトルを図6および図7に示し、ここで化学シフト(δ)はテトラメチルシラン(TMS)を内部標準(δTMS=0.00ppm)として使用しppmにて示す。
【0067】
式(V)の特徴的化学シフトは次の通りである:
【0068】
【数2】
【0069】
【数3】
【0070】
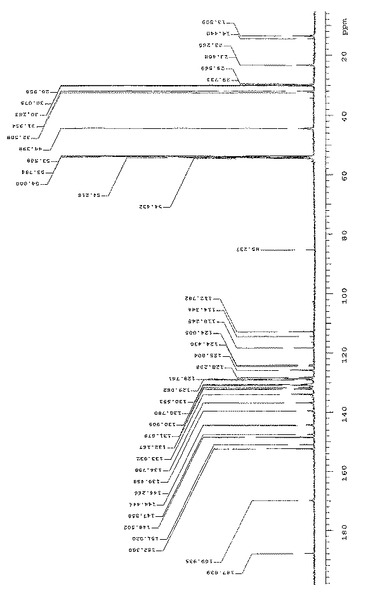
式(V)の化合物のFAB−MSスペクトルを図8に示す。特徴的プロトン化分子イオンはm/z=521である。M/z=107、136、137、154、289および307のピークはマトリックス溶剤から得ることができる。
【0071】
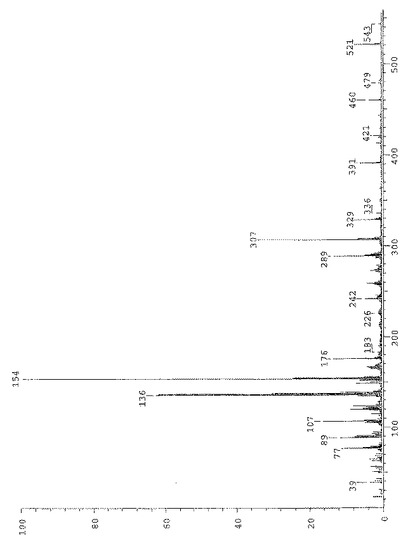
式(V)の化合物のFAB−娘イオンスペクトルを図9に示す。断片的ピーク(m/z=479および437)は、それぞれ1個もしくは2個のケテン基の排除によりプロトン化分子イオンから得ることができる。
【0072】
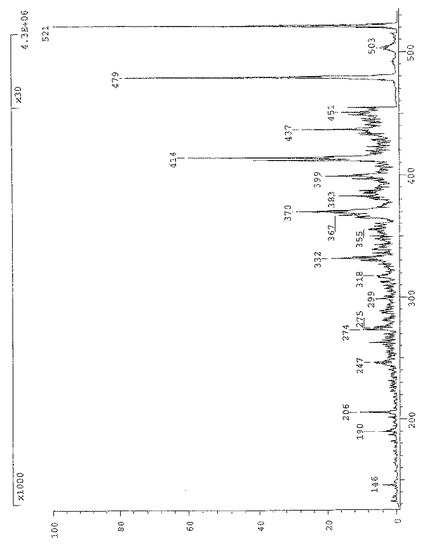
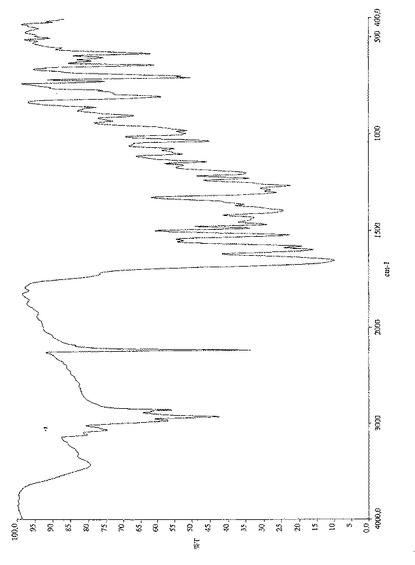
式(V)の化合物のIRスペクトルを図10に示す。特徴的IR吸収バンド(cm−1)は次の通りである:
C=N 2230
C=O 1652
Ar 1597、1579、807、707
【0073】
更なる特徴的吸収バンド(cm−1):2926、2854、1520、1394、1265、1199、1035、641、583。
【図面の簡単な説明】
【0074】
【図1】HPLCにより得られる式(II)の最終生成物の溶出プロフィルを示す。横軸:溶出時間(min)、縦軸:吸光率(mAU)である。
【図2】デューテロピリジンにおける式(II)の化合物の1H−NMRスペクトル、化学シフトをppmで示す。
【図3】式(II)の化合物のFAB−MSスペクトルを示す。横軸:m/z、縦軸:イオン強度(%)である。
【図4】式(II)の化合物のIRスペクトルを示す。横軸:波長(cm−1)、縦軸:透過率(%)である。
【図5】HPLCにより得られた式(IV)の最終生成物の溶出プロフィルを示す。横軸:溶出時間(min)、縦軸:吸光率(mAU)である。
【図6】デューテロ−ジクロルメタンにおける式(V)の化合物の1H−NMRスペクトル、化学シフトをppmで示す。
【図7】デューテロ−ジクロルメタンにおける式(V)の化合物の13C−NMR、化学シフトをppmで示す。
【図8】式(V)の化合物のFAB−MSスペクトルを示す。横軸:m/z、縦軸:イオン強度(%)である。
【図9】式(V)の化合物のFAB−娘イオンスペクトルを示す。横軸:m/z、縦軸:イオン強度(%)である。
【図10】式(V)の化合物のIRスペクトルを示す。横軸:波長(cm−1)、縦軸:透過率(%)である。
【発明の詳細な説明】
【0001】
本発明は、治療用途に適する0.2%未満の不純物を含有する式(IV)のN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミドに関するものである。
【0002】
更に本発明は、0.2%未満の不純物を含有する式(IV)の治療上適用しうる化合物の合成方法にも関するものであり、この方法は式(II)の新規な中間体N−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]−フェニル}−N−エチル−アセタミド・ヒドロクロライドを酸フリーの媒体中で式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と反応させる。
【0003】
更に本発明は式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドにも関するものである。
【0004】
さらに本発明は、式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドに関するものであり、また本発明は、99.5%より大の純度を有する。
【0005】
更に本発明は、式(IV)の化合物の合成に際し0.07%収率にて形成される新規な単離不純物である式(V)のN−{3−(3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドにも関するものである。
【0006】
式(IV)の化合物(ザレプロン)は、GABAAリセプタ作用剤として作用する抗不安剤、抗てんかん薬、鎮静薬、催眠薬および骨格筋弛緩剤である。
【0007】
式(IV)の化合物は米国特許第4,626,538号に表8における例14で記載されている。最終生成物は、式(I)のN−[3−[3−(ジメチルアミノ)−オキソ−2−プロペニル]フェニル]−N−エチル−アセタミド塩基を式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と氷酢酸中で8時間にわたり還流させることにより得られる。化合物の正確な物理化学的性質、純度および収率はこの米国特許には記載されておらず、186〜187℃である融点のみが示されている。
【0008】
米国特許第4,626,538号の方法は、欧州特許第776,898号にて改良される。式(IV)の化合物の改良合成方法は次のプロセスに従う:式(I)のN−[3−[3−(ジメチルアミノ)−オキソ−2−プロペニル]フェニル]−N−エチル−アセタミド塩基を式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と酢酸および水(10容量%〜85容量%)の混液にて、好ましくは1:2v/vのこれらの混合物にて反応させる。得られる生成物の純度はHPLCにより98.86〜99.40%である。最も純度の高い(99.4%)の化合物の収率は例1に記載されたように83.5%である。出発材料は各例の全てにおいて塩基性である。式(IV)の化合物の合成方法は中間体に基づくだけでなく、その塩からも出発して請求項に記載されている。出発材料として塩を使用する上記欧州特許に記載されたプロセスは存在せず、更に適用しうる中間塩またはその合成についても記載されていない。
【0009】
更に、開示された国際公開第2002/100828号も式(IV)の化合物の合成方法を記載しており、この方法は式(I)のN−[3−[3−(ジメチルアミノ)−オキソ−2−プロペニル]フェニル]−N−エチル−アセタミド塩基を式(III)の3−アミノ−4−ピラゾール−カルボニトリル塩基と水および水混和性有機溶剤の混合液中で酸性条件下に反応させることからなっている。酸性条件は、有機酸もしくは無機酸(特に塩酸)を反応混合物に添加して達成される。酸の量は出発塩基の1種と等モル量とすることができ、或いは大過剰で用いることもできる。請求項12〜15において、出発材料としての式(I)の塩基の酸付加塩の適用および酸の添加が別々に記載されているが一方では請求項6および1にて塩基のみが出発材料として使用され、他方では出発材料としての塩の使用を支持する記載はない。国際公開第2002/100828号(欧州特許第776,898号と同様)は、式(I)もしくは(III)の塩基の塩につき作成、単離および好適具体例の所定の例もしくは記載を示していない。例12においては、1当量の塩酸が使用され、これは請求項21に記載されているように適用した酸の量の下限値である。この例によれば生成物は24時間の反応時間後に82%収率にて得られ、純度は僅か98.95%である。本発明者等の実験(上記例12による合成を行うと共に1当量の塩酸を用いる)において、測定されたpHは全反応時間に際し約1.5であった。例1〜21に記載されたように用いた酸の量が1当量より多いこれら反応において、測定pHは1.0未満であり、すなわち強酸性反応条件が用いられる。
【0010】
国際公開第2003/011228号には、式(IV)の化合物の新規なレジオアイソマーN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−5−イル)−フェニル]−N−エチル−アセタミドが記載されており、これは式(IV)の化合物の合成に際し生成される。このレジオアイソマーの相当量(0.2〜0.5%)が、たとえば米国特許第2003/0040522号に記載された式(IV)の化合物の合成方法に際し生成される。
【0011】
上記化学的純度の数値は現在の医薬工業にて使用されるよりも低いので、本発明の目的は、高純度(最も厳密な品質要件を満たす)の生成物を良好な収率にて作成するのに適する新規な式(IV)の化合物の合成方法を見出すことであった。
【0012】
本発明の基礎は次の知見にある:反応混合物のpHは反応に際し3.0〜3.5の範囲に保って、化学反応の最高の選択率に到達すべきである。本発明による研究の結果、式(IV)の高純度の化合物を得るのに要する混合物の最適pHは自動調節的に式(II)の高純度塩酸塩の適用により維持される。
【0013】
本発明の研究に際し、高純度の中間体(特に最後のもの)が最終生成物の高純度の前提であることが判明した。式(III)の高品質の3−アミノ−4−ピラゾール−カルボニトリル塩基は市販入手することができる。式(I)の塩基は多段階合成にて得ることができ、適する純度の化合物の基本形態は多量の材料を損失した後にのみ単離することができる。驚くことに、特に式(I)の塩基の塩類から塩酸塩(従来特性化されていない)を式(I)の塩基から高純度および極めて良好な収率にて容易に得られることが判明した。
【0014】
式(IV)の化合物の合成を取り扱った従来技術の特許および特許出願の例において、反応は高酸性条件下で行われ、或る場合には1当量より多い酸が使用される。先ず最初に、本発明者等はこれら公知方法を反復した。これら高酸性条件下にて全ての場合、式(IV)の得られる化合物は0.2%より多いレジオアイソマーN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−5−イル)−フェニル]−N−エチル−アセタミドを含有することを突き止めた。次いで、本発明者等は高純度の式(IV)の化合物の新規な合成方法につき検討した。
【0015】
驚くことに式(IV)の化合物は、式(I)の高純度塩基のうち式(II)の新規な塩酸塩(これは本発明の方法により良好な収率で得られる)を式(III)の塩基と反応混合物に酸を添加することなく反応させることにより合成しうること、更に生成物の収率および純度は従来技術に記載された数値よりもずっと高いことが突き止められた。
【0016】
0.2%未満の不純物を含有する式(IV)の高純度化合物の合成は刊行物に記載されておらず、更に反応混合物へ酸を添加することのない式(IV)の化合物の合成に関する本発明による方法も記載されていない。
【0017】
本発明は、0.2%未満の不純物を含有し治療用途に適する式(IV)の化合物に関するものである。
【0018】
更に本発明は0.2%未満の不純物を含有する式(IV)の治療上適用しうる化合物の合成方法にも関するものであり、この方法は式(II)の新規な塩を酸フリーの媒体にて式(III)の塩基と反応させる。
【0019】
また本発明は、式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドにも関するものである。
【0020】
更にまた本発明は99.5%より大の純度を有する式(II)の新規なN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの合成方法にも関し、この方法は式(I)の化合物をアセトン中に20〜25℃にて窒素下で懸濁させると共に等モル量の濃塩酸を添加することを特徴とする。
【0021】
更に本発明は式(V)のN−{3−[3−シアノピラゾロ(1,5−a)−ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドにも関し、この化合物は式(IV)の化合物の合成に際し形成される新規な単離不純物である。
【0022】
本発明の詳細な説明は次の通りである:
【0023】
「室温」の意味は20〜25℃である。
【0024】
「粗製最終生成物」の意味は、従来技術の式(IV)の化合物の合成手順〜反復して得られる最終生成物である。
【0025】
「高酸性条件」の意味は、反応をpH1.0未満で行うことを意味する。
【0026】
本発明によれば式(II)の新規な塩は、式(I)の塩基をアセトン中に室温にて窒素下で懸濁させると共に等モル量の濃塩酸を添加することにより得られる。攪拌後沈殿した結晶を濾過し、洗浄し、次いで乾燥させる。このように得られた生成物の純度は99.5%より大である。
【0027】
本発明によれば、0.2%未満の不純物を含有する式(IV)の治療上適用しうる化合物の合成は次の通りである:
【0028】
式(III)の塩基を水とアルコールとの混液に溶解させると共に、式(II)の固体塩を少量ずつ加温溶液に添加する。添加に際し、激しい攪拌を持続する。他の具体例によれば、式(II)の塩を溶剤に溶解させると共に、溶液を反応混合物に添加する。先ず最初に、溶液(後に得られる懸濁液)を連続的に攪拌し、次いで生成物を単離する。通常、懸濁物を冷却すると共に攪拌を持続する。沈殿した結晶を濾別し、洗浄し、次いで乾燥させる。得られた結晶物質を温メタノールに窒素下で溶解させ、溶液を濾過し、冷却し、沈殿生成物を濾別し、洗浄し、次いで乾燥させる。
【0029】
本発明による方法は、好ましくは有機溶剤中で或いは水と水混和性有機溶剤との混液中で行われる。有機溶剤としては単鎖脂肪族アルコール(好ましくはメタノール、エタノール、イソプロパノール、n−プロパノール)を使用することができる。本発明による方法は好ましくは15〜50℃にて0.5〜1時間にわたり行われる。式(III)の3−アミノ−4−シアノ−ピラゾール塩基を好ましくは溶剤の適する混液に溶解させ、式(II)の塩酸塩を少量ずつ溶液に添加する。冷却後、沈殿生成物を濾別すると共に乾燥させる。このように得られた式(IV)の生成物の純度は、HPLCによれば99.8%より大である。
【0030】
本発明による方法の適用(式(II)の新規な高純度の塩酸塩と式(III)の化合物との反応)は、従来技術により得られるもの(すなわち反応を水性酢酸にて行うことにより得られるもの)よりもずっと高品質(99.4%より大の純度)の式(IV)の化合物をもたらす。
【0031】
本発明の方法により得られる式(IV)の最終生成物の高純度(これは医薬工業にて特に有利である)は、式(II)の新規な塩酸塩の高純度に部分的に基づき、更に酸フリーの反応混合物に部分的に基づき、また少量ずつの式(II)の新規な塩酸塩の添加に部分的に基づき、更にまた短い反応時間に部分的に基づいている。
【0032】
式(IV)の化合物の合成にて本発明により最初に作成されると共に使用される式(II)の新規な塩酸塩の適用に関する大きい利点は、高純度の式(IV)の化合物の合成にて高純度の出発材料が使用される(これは医薬工業にて必要とされる)点である。HPLCによれば、式(I)の塩基の純度は一般に90〜95%である一方、この塩基から得られる式(II)の塩酸塩の純度は98.5%より大である。
【0033】
式(IV)の化合物の本発明による合成方法に際し、反応のpHは3.0〜3.5であることを強調せねばならない。本発明による方法の大きな利点は、追加量の酸を反応に添加する必要がないことである。何故なら、出発材料として使用する式(II)の新規な塩酸塩は等モル量の酸を含有し、更に反応混合物のpHは全反応時間にわたり式(II)の塩酸塩の反応混合物に対する少量ずつの添加に基づき3.0〜3.5の範囲に留まり、その結果式(IV)の化合物の本発明による合成方法の反応条件は反応をpH1.0未満または最高pH1.5にて行う従来技術の手法と比較してずっと緩和であるからである。
【0034】
式(IV)の化合物の合成に際し本発明により最初に作成されかつ使用された式(II)の新規な塩酸塩の適用に関する大きな利点は、式(IV)の化合物のレジオアイソマーが0.1%未満の収率でのみ生成される点である。従来技術の手法により作成される式(IV)の最終生成物(明細書に開示した通り)は或る場合には0.5%の不純物としてレジオアイソマーを含有する。
【0035】
式(IV)の化合物の合成に際し本発明により最初に作成されかつ使用された式(II)の新規な塩酸塩の使用に関する更なる利点は、式(IV)のレジオアイソマーの生成される少量の他に、刊行物または開示された特許には記載されていない式(V)の不純物が微量(0.07%)でのみ生成される点である。
【0036】
式(V)の不純度の分析的特性化を実施例2に示す。本発明による分析測定は式(V)の化合物の構造を証明し、その化学名はN−{3−[3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドである。
【0037】
式(V)の上記不純物は、式(IV)の化合物の合成につき文献に記載された方法により得られる粗製最終生成物から単離することができる。何故なら、これら粗製生成物はより多量(0.1%)の式(V)の不純物を含有するからである。式(V)の不純物を、作成HPLCにより再結晶化の母液から単離した。驚くことに、式(V)の不純物は式(IV)の化合物の本発明による合成方法にて0.07%の収率でのみ生成することが判明した。
【0038】
本発明者等は、式(V)の不純物の生成を式(IV)の化合物の合成に関する本発明の方法に際し反応混合物へ少量ずつ式(II)の塩酸塩を添加すると共に適するpH値、温度、溶剤の割合および反応時間を保つことにより、減少させうることを認識した。これは医薬工業において特に有利である。
【0039】
要約すれば、公知方法と比較した式(IV)の化合物の本発明による合成方法の利点は次の通りである:
【0040】
−式(IV)の最終生成物は、0.2%未満の不純物(医薬品製造のGMP要件に従う)を含有して良好な収率で作成される;
−式(II)の塩酸塩の純度は極めて高い;
−式(IV)の最終生成物は酸の添加なしに作成される;
−式(IV)の最終生成物は、従来技術の手法により得られるものと比較してずっと少ない(最大0.1%)のレジオアイソマーを不純物として含有する;
−式(IV)の最終生成物は、従来技術の手法により得られるものと比較してずっと少ない(0.07%)の本発明者等により単離された式(V)の不純物を含有する;
−薬物の高純度は式(IV)の最終生成物の製造に際し安全に制御することができ、これは式(IV)の最終生成物の高純度だけでなく式(II)の最終中間体の高純度も実現しうる。
【0041】
以下、実施例を例示するが本発明の範囲を決して限定するものでない。
【0042】
実施例においては、式(II)の新規な化合物、式(IV)の最終生成物、および本発明の方法により得られる式(V)の不純物を以下の分析法により特性化する。
【0043】
本発明の方法により得られる生成物の純度はHPLCにより測定した。HPLC装置:熱分離品、ポンプ:スペクトルシステムP2000、検出器:スペクトルシステムUV2000。制御、データ収集およびデータ処理:クロムクエスト・プログラム、Ver.2.51。クロマトグラフィーの条件:カラム:YMC−パック・プロC18、150x4.6mm、ID5μm。溶出剤A:140mlのアセトニトリル+30mlのメタノール+30mlのテトラヒドロフラン+800mlの0.1M酢酸アンモニウムの混液。溶出剤B:400mlのアセトニトリル+100mlのメタノール+100mlのテトラヒドロフラン+400mlの0.1M酢酸アンモニウムの混液。これらから表1に示す線状勾配の溶剤システムを使用した。
【0044】
流速:1ml/min;カラムの温度:室温;検出の波長:235nm。分析試料の溶解:77%の溶出剤A+23%の溶出剤Bの混液。注入量:10μl。
【0045】
【表1】
【0046】
1H−NMRおよび13C−NMRスペクトルをバリアンINOVA(1H−NMRにつき500MHz)スペクトロメータにて記録した。各測定はデューテロ−ピリジンまたはデューテロ−ジクロルメタンにて30℃で行った。化学シフト(δ)は、テトラメチルシラン(TMS)を内部標準(δTMS=0.00ppm)として使用しppmにて示す。NMRスペクトルにおいては次の記号を使用する:s=シングレット、d=デュープレット、t=トリプレット、q=クワルテット、m=マルチプレット、dd=ダブルデュープレット、br=ブロードマルチプレット、dm=デュープレット・マルチプレット。
【0047】
各化合物の高分解MSスペクトルはFAB−MS法により測定した。装置:フィニガンMAT95SQ。FABイオン化法:20kV Cs+イオンガン、3−ニトロ−ベンジルアルコール・マトリックス溶剤。スペクトルの評価:m/z。FAB−娘イオンスペクトルはタンデムMS法により得た。
【0048】
IRスペクトルは臭化カリウムペレットの4cm−1スペクトル分解を用いてパーキン−エルマー1000スペクトロメータで記録した。
【0049】
実施例1
式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライド
窒素下で31.5gの式(I)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミドを300mlのアセトン中に室温にて懸濁させると共に、10.4mlの濃塩酸を滴加した。0.5mlの濃塩酸を添加した後、均質溶液が得られ、結晶の分離を3−4mlの濃塩酸の添加の後に開始した。全量の濃塩酸を添加した後、反応混合物を室温にて30分間および0〜5℃にて更に1時間にわたり攪拌した。沈殿した結晶を濾別し、10mlの冷(3〜5℃)アセトンで2回洗浄すると共に、50℃未満で乾燥させて31.15g(87%)の標記化合物を得た。
【0050】
融点:136〜137℃。
【0051】
得られた生成物の分析特性化:
【0052】
HPLCにより得られた式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの溶出プロフィルを図1に示す。横軸:溶出時間(0〜31分間にわたるmin);縦軸:吸収率(mAU)試料の濃度は0.5mg/mlであった。式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの反応時間は4.0minである。評価は面積基準化により行った。
【0053】
HPLCにより生成物の純度は98.5%であった。
【0054】
デューテロ−ピリジンにおける式(II)の化合物の1H−NMRスペクトルを図2に示し、ここには化学シフト(δ)を内部標準としてテトラメチルシラン(TMS)(δTMS=0.00ppm)を使用してppmにて示す。
【0055】
特徴的化学シフトは次の通りである:
【0056】
【数1】
【0057】
式(II)の化合物のFAB−MSスペクトルを図3に示す。特徴的なプロトン化分子イオンはm/z=261である一方、ナトリウム陽イオンを有する分子イオンはm/z=283である。他の断片的ピーク:3−(ジメチルアミノ)−1−オキソ−2−プロペニルイオンの形成(m/z=98)、2−ジメチルアミノ−エチレン側鎖の開裂(m/z=190)、アセチル基の開裂(m/z=217)、プロトン化分子イオンからのエタンの排除(m/z=231)。M/z=77、136および154の各ピークをマトリックス溶剤から得ることができる。
【0058】
式(II)の化合物のIRスペクトルを図4に示す。特徴的IR吸収バンド(cm−1)は次の通りである:
N+−H 2700−2000
C−O 1645
Ar 820,706
【0059】
更なる特徴的吸収バンド(cm−1):3050、2932、1562、1492、1259、1185、1146、1067。
【0060】
実施例2
式(IV)のN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミド
窒素を150mlの精製水と50mlのエタノールとの混液中にバブリングさせると共に、窒素下で10.8gの式(III)の3−アミノ−4−ピラゾール−カルボニトリルをこの混合物に添加した。溶解後、反応混合物を45〜50℃まで加温すると共に、式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの29.6gを3つの等しい部分にて全て15分間で添加した。最初に溶液であると共に後に懸濁液となった反応混合物を、この温度にて1時間にわたり窒素下で攪拌した。次いで得られた懸濁物を0〜5℃まで冷却すると共に、この温度にて2時間にわたり攪拌した。沈殿した結晶を濾別し、25mlの氷冷エタノールおよび2x25mlの氷冷水で洗浄した。生成物を50℃未満で減圧下に乾燥させた。得られた生成物を280mlのメタノール中に窒素下で65〜67℃にて溶解させて機械的不純物を除去した。この溶液を濾過すると共に濾液を0〜5℃まで冷却した。混合物をこの温度に2時間にわたり保ち、次いで沈殿結晶を濾別し、2x10mlの氷冷メタノールで洗浄し、更に50℃未満で乾燥させて28.2g(92.5%)の標記化合物を得た。
【0061】
得られた生成物の分析特性化:
【0062】
HPLCにより得られた式(IV)の化合物の溶出プロフィルを図5に示す。横軸:溶出時間(0〜31分間にわたるmin);縦軸:吸収率(mAU)。試料の濃度は1.0mg/mlであった。式(IV)の化合物の滞留時間は10.2minである。評価は、標準試料溶液の1000倍希釈の外部標準法により行った。
【0063】
HPLCにより式(IV)の生成物の純度は99.8%であった。
【0064】
0.07%収率にて形成されると共に本発明により特性化された不純物である式(V)のN−{3−(3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミドの滞留時間は、図5に見られるように24.9minである。
【0065】
この不純物を更に式(IV)の化合物の合成に関する公知手順により得られた粗製最終生成物から単離し、これら手順の最終生成物は多量(0.1%)の式(V)の不純物を含有した。
【0066】
デューテロジクロルメタンにおける式(V)の化合物の1H−NMRおよび13C−NMRスペクトルを図6および図7に示し、ここで化学シフト(δ)はテトラメチルシラン(TMS)を内部標準(δTMS=0.00ppm)として使用しppmにて示す。
【0067】
式(V)の特徴的化学シフトは次の通りである:
【0068】
【数2】
【0069】
【数3】
【0070】
式(V)の化合物のFAB−MSスペクトルを図8に示す。特徴的プロトン化分子イオンはm/z=521である。M/z=107、136、137、154、289および307のピークはマトリックス溶剤から得ることができる。
【0071】
式(V)の化合物のFAB−娘イオンスペクトルを図9に示す。断片的ピーク(m/z=479および437)は、それぞれ1個もしくは2個のケテン基の排除によりプロトン化分子イオンから得ることができる。
【0072】
式(V)の化合物のIRスペクトルを図10に示す。特徴的IR吸収バンド(cm−1)は次の通りである:
C=N 2230
C=O 1652
Ar 1597、1579、807、707
【0073】
更なる特徴的吸収バンド(cm−1):2926、2854、1520、1394、1265、1199、1035、641、583。
【図面の簡単な説明】
【0074】
【図1】HPLCにより得られる式(II)の最終生成物の溶出プロフィルを示す。横軸:溶出時間(min)、縦軸:吸光率(mAU)である。
【図2】デューテロピリジンにおける式(II)の化合物の1H−NMRスペクトル、化学シフトをppmで示す。
【図3】式(II)の化合物のFAB−MSスペクトルを示す。横軸:m/z、縦軸:イオン強度(%)である。
【図4】式(II)の化合物のIRスペクトルを示す。横軸:波長(cm−1)、縦軸:透過率(%)である。
【図5】HPLCにより得られた式(IV)の最終生成物の溶出プロフィルを示す。横軸:溶出時間(min)、縦軸:吸光率(mAU)である。
【図6】デューテロ−ジクロルメタンにおける式(V)の化合物の1H−NMRスペクトル、化学シフトをppmで示す。
【図7】デューテロ−ジクロルメタンにおける式(V)の化合物の13C−NMR、化学シフトをppmで示す。
【図8】式(V)の化合物のFAB−MSスペクトルを示す。横軸:m/z、縦軸:イオン強度(%)である。
【図9】式(V)の化合物のFAB−娘イオンスペクトルを示す。横軸:m/z、縦軸:イオン強度(%)である。
【図10】式(V)の化合物のIRスペクトルを示す。横軸:波長(cm−1)、縦軸:透過率(%)である。
【特許請求の範囲】
【請求項1】
0.2%未満の不純物を含有する式(IV)の治療上適用しうるN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミド。
【請求項2】
0.2%未満の不純物を含有する式(IV)の治療上適用しうるN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミドの合成方法において、式(III)の3−アミノ−4−ピラゾール−カルボニトリルを等モル量の式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドと有機溶剤中または水と有機溶剤との混液中にて3.0〜3.5のpHで反応させ、次いで生成物を単離すると共に所定の場合には生成物を再結晶化させることを特徴とする合成方法。
【請求項3】
有機溶剤として単鎖脂肪族アルコールを使用することを特徴とする請求項2に記載の方法。
【請求項4】
単鎖脂肪族アルコールとしてメタノール、エタノール、イソプロパノールもしくはn−プロパノールを使用することを特徴とする請求項3に記載の方法。
【請求項5】
式(II)の化合物を少量ずつ反応混合物に添加することを特徴とする請求項2〜4のいずれか一項に記載の方法。
【請求項6】
反応を15〜50℃にて行うことを特徴とする請求項2〜5のいずれか一項に記載の方法。
【請求項7】
反応を45〜50℃にて行うことを特徴とする請求項2〜5のいずれか一項に記載の方法。
【請求項8】
反応を0.5〜1時間にわたり行うことを特徴とする請求項2〜7のいずれか一項に記載の方法。
【請求項9】
99.5%より大の純度を有する式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライド。
【請求項10】
99.5%より大の純度を有する式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの合成方法において、式(I)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミドをアセトン中に窒素下で20〜25℃にて懸濁させると共に、等モル量の濃塩酸を懸濁物に添加することを特徴とする合成方法。
【請求項11】
式(V)のN−{3−(3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミド。
【請求項1】
0.2%未満の不純物を含有する式(IV)の治療上適用しうるN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミド。
【請求項2】
0.2%未満の不純物を含有する式(IV)の治療上適用しうるN−[3−(3−シアノピラゾロ[1,5−a]ピリミジン−7−イル)−フェニル]−N−エチル−アセタミドの合成方法において、式(III)の3−アミノ−4−ピラゾール−カルボニトリルを等モル量の式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドと有機溶剤中または水と有機溶剤との混液中にて3.0〜3.5のpHで反応させ、次いで生成物を単離すると共に所定の場合には生成物を再結晶化させることを特徴とする合成方法。
【請求項3】
有機溶剤として単鎖脂肪族アルコールを使用することを特徴とする請求項2に記載の方法。
【請求項4】
単鎖脂肪族アルコールとしてメタノール、エタノール、イソプロパノールもしくはn−プロパノールを使用することを特徴とする請求項3に記載の方法。
【請求項5】
式(II)の化合物を少量ずつ反応混合物に添加することを特徴とする請求項2〜4のいずれか一項に記載の方法。
【請求項6】
反応を15〜50℃にて行うことを特徴とする請求項2〜5のいずれか一項に記載の方法。
【請求項7】
反応を45〜50℃にて行うことを特徴とする請求項2〜5のいずれか一項に記載の方法。
【請求項8】
反応を0.5〜1時間にわたり行うことを特徴とする請求項2〜7のいずれか一項に記載の方法。
【請求項9】
99.5%より大の純度を有する式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライド。
【請求項10】
99.5%より大の純度を有する式(II)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミド・ヒドロクロライドの合成方法において、式(I)のN−{3−[3−(ジメチルアミノ)−1−オキソ−2−プロペニル]フェニル}−N−エチル−アセタミドをアセトン中に窒素下で20〜25℃にて懸濁させると共に、等モル量の濃塩酸を懸濁物に添加することを特徴とする合成方法。
【請求項11】
式(V)のN−{3−(3−シアノピラゾロ(1,5−a)ピリミジン]−6−イル−3−[(3−N−エチル−アセタミド−フェニル)−3−オキソ−プロペン−1−イル]−(ピリミジン−7−イル)−フェニル}−N−エチル−アセタミド。
【図1】


【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公表番号】特表2007−519700(P2007−519700A)
【公表日】平成19年7月19日(2007.7.19)
【国際特許分類】
【出願番号】特願2006−550312(P2006−550312)
【出願日】平成17年1月31日(2005.1.31)
【国際出願番号】PCT/HU2005/000008
【国際公開番号】WO2005/073235
【国際公開日】平成17年8月11日(2005.8.11)
【出願人】(591180314)リヒター ゲデオン ベジェセティ ジャール アール.テー. (33)
【Fターム(参考)】
【公表日】平成19年7月19日(2007.7.19)
【国際特許分類】
【出願日】平成17年1月31日(2005.1.31)
【国際出願番号】PCT/HU2005/000008
【国際公開番号】WO2005/073235
【国際公開日】平成17年8月11日(2005.8.11)
【出願人】(591180314)リヒター ゲデオン ベジェセティ ジャール アール.テー. (33)
【Fターム(参考)】
[ Back to top ]