
N末端標識膜蛋白質の作製方法、及びN末端標識膜蛋白質を表層に有する細胞
【課題】対象蛋白質に特異的にプローブを修飾することで対象蛋白質のみを特異的に標識する方法、特に、N末端が細胞外に存在する膜蛋白質を特異的に標識する方法、当該方法によって特異的に修飾された膜蛋白質を有する細胞、及び、当該細胞を用いるスクリーニング方法を提供すること。
【解決手段】N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質、ペプチド転移酵素Sortase A、及び、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す)が付加された標識分子プローブを用いることを特徴とする、N末端標識膜蛋白質の作成方法。
【解決手段】N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質、ペプチド転移酵素Sortase A、及び、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す)が付加された標識分子プローブを用いることを特徴とする、N末端標識膜蛋白質の作成方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、分子生物学分野におけるバイオイメージング技術、及び蛋白質工学分野における蛋白質改変技術に関する。詳しくは、細胞が生きたままの状態で特定の蛋白質の機能や動態を直接観察するためのバイオイメージング技術、そのための蛋白質標識技術に関する。
【背景技術】
【0002】
細胞機能の解明や疾病の発症メカニズムの解明といった分子細胞生物学分野の研究開発において、特定の蛋白質の機能、細胞内での局在や活性の変化を知ることが必要不可欠である。そのための解析手法として、観測対象の蛋白質を可視化して直接観察するバイオイメージングの手法が幅広く利用されている。現在のバイオイメージングの代表的な手法としては、以下の3つの手法がある。
【0003】
(1)緑色蛍光蛋白質 (GFP)等の蛍光蛋白質を融合して蛋白質を蛍光標識する手法。
この手法では、遺伝子工学的に蛍光蛋白質と対象蛋白質を融合させるため、対象蛋白質のみを特異的に蛍光標識した状態で発現させることが可能である。
しかしこの手法の問題点として、1)蛍光蛋白質のサイズが非常に大きいため、標識に伴い対象蛋白質の発現・機能が阻害される可能性があること、2)常に対象蛋白質が蛍光標識された状態で発現されるために、発現時間の違いを区別した細胞内局在の変化を解析すること (パルスチェイス標識) が難しいこと等を挙げることができる。
【0004】
(2)有機化学反応により蛍光性合成分子プローブを修飾して蛋白質を蛍光標識する手法。
この手法では、観測したい任意のタイミングで有機化学反応によりサイズの小さな蛍光性合成分子プローブを修飾して、対象蛋白質を蛍光標識することができる。そのため、上述の蛍光蛋白質融合法の問題点を補うものであるといえる。また、蛍光性合成分子プローブのみならず、光反応性プローブ、pH感受性プローブ等の様々な機能性合成分子プローブを対象蛋白質に修飾することが可能である。
しかし、有機化学反応は官能基特異的反応、つまり「アミノ酸」特異的反応であるため、多種多様な蛋白質が存在する細胞内及び細胞表層で、対象蛋白質のみを特異的に蛍光標識することは非常に難しい。
【0005】
(3)蛍光修飾した抗体を用いて蛋白質を蛍光標識する手法。
この手法では、抗原抗体反応の高い特異性を利用し、蛍光修飾した抗体により対象蛋白質のみを特異的に蛍光標識することができる。また、パルスチェイス解析を行うことも可能である。
しかし、この手法にも1)抗体自体のサイズが非常に大きいため、標識に伴い対象蛋白質の機能が阻害される可能性があること、2)標識したい蛋白質それぞれに対し、蛍光修飾した抗体 (非常に高価である) を個別に用意する必要があること、3)抗原−抗体間の相互作用は強固であるものの、非共有結合的な相互作用であるため、条件によって蛍光修飾した抗体が対象蛋白質からはずれる可能性があること等の問題点を挙げることができる。
【0006】
また、蛋白質標識技術においては、目的の蛋白質を特異的に修飾する技術として生物の翻訳系を利用する手法が知られている。例えば、標識物質よりなるラベル部と、タンパク質のC末端に結合する能力を有する化合物よりなるアクセプター部とから構成されるラベル化試薬の存在下、プロモーター領域の制御下にある、終止コドンが削除されたコーディング領域からなるDNAから転写された産物を鋳型として無細胞翻訳系または生細胞中でタンパク質合成を行わせる方法が知られている(特許文献1、2)。
しかしこの手法はC末端に結合する能力を有する化合物を利用するC末端特異的な反応であり、N末端の修飾に適用することはできない。
【0007】
酵素反応の高い基質特異性を利用して対象蛋白質に特異的に蛍光分子プローブを修飾する手法が知られている。発明者らは既に、ペプチド転移酵素Sortase A(SrtA)を用いて細胞表層に存在する対象膜蛋白質のC末端部位に特異的に蛍光分子プローブを修飾し、対象膜蛋白質を蛍光標識する手法を開発している(非特許文献1)。
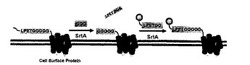
【0008】
SrtAは、認識配列LPXTGのTとGの間を切断し、そこにGG末端をもつ別のペプチドあるいは蛋白質を連結する反応を触媒する酵素である(図1参照)。発明者らの前記手法はこの基質特異性を利用し、C末端領域にLPETG配列を有する膜蛋白質と、GG配列を付加した蛍光分子プローブとをSrtAの酵素反応によって結合することで、特定の膜蛋白質のC末端を蛍光標識するものである。
【0009】
しかし、薬剤開発の主要なターゲットであるG蛋白質共役受容体(GPCR)をはじめとする多くの膜蛋白質は、N末端部位が細胞外、C末端部位が細胞内に位置するものであり、C末端部位が細胞表層に提示される膜蛋白質はごく少数に過ぎない。そのため、C末端部位が細胞表層に提示される膜蛋白質のみにしか適用できない前述の手法は汎用性の点で問題がある。
【0010】
【特許文献1】特開平11−322781号公報
【特許文献2】特開2000−139468号公報
【非特許文献1】T.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明の課題は、従来の各バイオイメージング手法の欠点を補う新たな蛋白質標識法として、酵素反応の高い基質特異性を利用して対象蛋白質に特異的にプローブを修飾することで対象蛋白質のみを特異的に標識する方法、特に、細胞外に存在する膜蛋白質のN末端部位を特異的に標識する方法、当該方法によって特異的に修飾された膜蛋白質を有する細胞、及び、当該細胞を用いるスクリーニング方法を提供することにある。
【課題を解決するための手段】
【0012】
発明者らは、シグナル伝達や物質輸送等生命現象において非常に重要な役割を果たしている細胞表層の膜蛋白質をターゲットとし、ペプチド転移酵素Sortase A(SrtA)を用いて対象膜蛋白質のN末端部位に特異的に蛍光分子プローブを修飾する手法の検討を進める中で、Sortase A認識配列とSortase A転移配列の両方を有する被標識蛋白質と、Sortase A認識配列を付加したプローブとを用いて、目的の膜蛋白質のN末端を特異的に標識することができることを見い出し、本発明を完成するに至った。
【0013】
すなわち本発明は、
[1]N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質、
ペプチド転移酵素Sortase A、及び、
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ、
を用いることを特徴とする、N末端標識膜蛋白質の作製方法や、
[2]次の各工程を含むことを特徴とする、上記[1]に記載のN末端標識膜蛋白質の作製方法に関する。
(i)N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質に、N末端に残基数2以上のポリグリシンを含むぺプチドの存在下にペプチド転移酵素Sortase Aを作用させる工程;
(ii)工程(i)によりN末端がポリグリシン配列となった膜蛋白質に、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブの存在下にペプチド転移酵素Sortase Aを作用させる工程。
【0014】
また本発明は、
[3]N末端が標識分子プローブで標識された膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されていることを特徴とする細胞、
[4]LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を細胞外N末端側領域に有する膜蛋白質を表層に有する細胞、
[5]N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むための、LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、
[6]LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ、
[7]N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むためのLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)を含むペプチドが付加された標識分子プローブ、ペプチド転移酵素Sortase A、及び、N末端に残基数2以上のポリグリシンを含むペプチド、を含むことを特徴とする、細胞表層膜蛋白質の標識キットに関する。
【0015】
さらに本発明は、
[8]N末端標識膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞を用いることを特徴とする、創薬のためのスクリーニング方法に関する。
【発明の効果】
【0016】
本発明によれば、細胞表層に存在する多種多様な膜蛋白質のうち、対象とする特定の膜蛋白質のみを特異的に標識できる。
また、標識に伴い付加される分子サイズが非常に小さいため、標識に伴う対象膜蛋白質の発現阻害・機能阻害を回避することができる。標識物質についても、蛍光性合成分子プローブのみならず、光反応性プローブ、pH感受性プローブ等の様々な機能性合成分子プローブを膜蛋白質に修飾することが可能であるため、膜蛋白質を単に標識するだけでなく、膜蛋白質に様々な機能性合成分子プローブを修飾して膜蛋白質の機能解析を行うことが可能である。本手法で利用する合成分子プローブは大変自由度が高く設計することができ、また、一般的なペプチド固相合成と簡単な化学反応の組み合わせで大変容易に合成することが可能である。
さらに、Sortase Aが触媒するペプチド転移反応は血清を含む培地中において短時間で進行し、ATPなどの補因子を必要としないため、生きた細胞上で機能を保ったまま膜蛋白質を改変する手法として非常に有用であると考えられる。
そして、細胞表層に発現している膜蛋白質に対し、酵素反応により「後付け」でプローブを修飾するため、観測したい任意のタイミングで膜蛋白質を標識し、パルスチェイス解析を行うことができる。
本発明の方法及び本発明の細胞は、創薬や生物基礎研究等の分野において強力なツールとなると考えられる。
【発明を実施するための最良の形態】
【0017】
本発明のN末端標識膜蛋白質の作製方法において標識される膜蛋白質は、N末端が細胞外に存在する膜蛋白質であり、例えば、I型サイトカイン受容体ファミリー、II型サイトカイン受容体ファミリー、免疫グロブリンスーパーファミリー(IgSF)、G蛋白質共役型受容体ファミリー(GPCR)等に属する蛋白質を具体的に挙げることができる。
【0018】
I型サイトカイン受容体ファミリーの蛋白質としては、例えば、インターロイキン−2受容体、インターロイキン−3受容体、インターロイキン−4受容体、インターロイキン−5受容体、インターロイキン−6受容体、インターロイキン−7受容体、インターロイキン−9受容体、インターロイキン−11受容体、インターロイキン−12受容体、インターロイキン−13受容体、インターロイキン−15受容体、顆粒球マクロファージコロニー刺激因子受容体、毛様体神経栄養因子受容体、エリスロポエチン受容体、白血病抑制因子受容体、OSM受容体、又は顆粒球コロニー刺激因子受容体等を挙げることができる。
【0019】
II型サイトカイン受容体ファミリーの蛋白質としては、例えば、インターフェロン−α受容体、インターフェロン−β受容体、インターフェロン−γ受容体、又はインターロイキン−10受容体等を挙げることができる。
【0020】
免疫グロブリンスーパーファミリーの蛋白質としては、例えば、インターロイキン−1受容体又はインターロイキン−18受容体等を挙げることができる。
【0021】
G蛋白質共役型受容体ファミリーの蛋白質としては、例えば、ロドプシン受容体、カテコールアミン受容体[β1、β2、β3、α1、α2]、ドーパミン受容体[D1、D5、D2]、ムスカリン受容体[m1、m3、m2、m4、m5]、アデノシン受容体[A1、A3、A2]、PGE受容体[EP2、EP4、EP3、EP1]、ロイコトリエン受容体[BLT、Cys−LT1]、スフィンゴシン1−リン酸受容体[S1P1、S1P2、S1P3]、リゾホスファチジン酸受容体[LPA1、LPA2、LPA3]、ノシセプチン受容体、hGPCR44、hGPCR4、hGPCR10、hGPCR17、hGPCR19、hGPCR39、又はhGPCR48等を挙げることができる。
【0022】
本発明の作製方法において、そのN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する膜蛋白質(以下、「LPX1X2(G)n配列融合膜蛋白質」ということがある。)における「N末端側領域」とは、膜蛋白質のN末端近傍の細胞外領域を意味し、細胞質内で合成された当該蛋白質の翻訳後修飾や輸送等に影響しない部位であることが好ましい。例えば、N末端シグナル配列の下流に、上記アミノ酸配列を有することが好ましい。
【0023】
LPX1X2(G)nにおけるX1は任意のアミノ酸を表し、アラニン(A)、アルギニン(R)、アスパラギン(N)、アスパラギン酸(D)、システイン(C)、グルタミン(Q)、グルタミン酸(E)、グリシン(G)、ヒスチジン(H)、イソロイシン(I)、ロイシン(L)、リジン(K)、メチオニン(M)、フェニルアラニン(F)、プロリン(P)、セリン(S)、トレオニン(T)、トリプトファン(W)、チロシン(Y)、バリン(V)等が挙げられ、これらのうち、グルタミン酸、メチオニン、チロシン、ロイシン、グルタミン、アスパラギン、フェニルアラニンがより好ましい。
X2はトレオニン(T)又はアラニン(A)であり、トレオニンがより好ましい。
nは2〜8のいずれかの整数を表し、4〜6であることがより好ましい。
【0024】
上述のLPX1X2(G)n配列融合膜蛋白質は、任意の細胞の表層に発現した形で得ることができる。このLPX1X2(G)n配列融合膜蛋白質を細胞の表層に発現した細胞の調製方法は公知の方法を含め特に制限されず、例えば、標的とする膜蛋白質をコードするDNAのN末端側に相当する領域に、LPETGGGGG配列(配列番号1)をコードするDNA断片を挿入した、LPETGGGGG配列融合膜蛋白質をコードするDNAを含むプラスミドベクターを構築し、このプラスミドベクターを、公知の遺伝子導入方法、例えば、リン酸カルシウム法、リポフェクション法、電気穿孔法のいずれかの方法を用いて細胞に導入し、LPETGGGGG配列融合膜蛋白質を発現させることができる。また、LPETGGGGG配列等のLPX1X2(G)n配列の上流及び/又は下流にはさらに別の配列、例えば、LPX1X2(G)n配列の挿入を確認するためのタグ配列等を含めることもできる。
【0025】
LPX1X2(G)n配列融合膜蛋白質を発現させる細胞としては、目的に応じて選択でき特に制限されず、例えば、公知の哺乳動物細胞又は昆虫細胞等を用いることができる。公知の哺乳動物細胞としては、例えばHEK293細胞、Vero細胞、Hela細胞、CV1細胞、COS1細胞、CHO細胞、ナマルバ細胞等を挙げることができる。公知の昆虫細胞としては、例えばSf9細胞、MG1細胞等を挙げることができる。
【0026】
本発明で用いられるペプチド転移酵素Sortase Aは、Staphylococcus属菌等に存在する酵素であって、認識配列LPXTGのTとGの間を切断し、そこにGG末端を持つ別のペプチド又は蛋白質を結合する反応を触媒する(図1参照)。Sortase Aは、公知の遺伝子工学的方法(例えば、T.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)に記載の方法)で微生物に発現させ、精製して得たものを用いることができる。
【0027】
本発明の作製方法に用いられる標識分子プローブとしては、LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を介して、膜蛋白質と結合できる限り特に制限されるものではなく、例えば蛍光性合成分子プローブ、発光性プローブ、発色性プローブ、ビオチン標識プローブ、放射性同位元素プローブ、受容体プローブ等の検出プローブを挙げることができる。これらのうち、バイオイメージングにおける検出の簡便性等の観点から、蛍光性合成分子プローブが好ましい。また、検出プローブだけでなく、光反応性プローブ、pH感受性プローブ等の機能性プローブを用いることもできる。
【0028】
上記標識分子プローブには、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加されているが、LPX1X2(G)m配列において、X1及びX2は前記に例示したものと同様であり、mは1又は2であり、2であることがより好ましい。
上記アミノ酸配列LPX1X2(G)mはプローブに化学的方法によって付加されていてもよく、例えばT.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)に記載の方法によって付加することができる。
また、上記アミノ酸配列はプローブに直接付加されていてもよく、LPX1X2(G)m配列とプローブの間に別のペプチド及び/又はポリマー等からなるスペーサーを介して付加されていてもよい。さらに、上記アミノ酸配列の下流に、別のペプチド及び/又はポリマー等を有していてもよい。
【0029】
本発明の方法は、前記に詳説したLPX1X2(G)n配列融合膜蛋白質、ペプチド転移酵素Sortase A、及び標識分子プローブを用いて標的膜蛋白質のN末端を標識するものであるが、そのストラテジーは、
(1)Sortase AによってLPX1X2(G)n配列融合膜蛋白質のSortase認識配列部位が切断され、N末端にGG配列が提示される;
(2)Sortase Aによって、GG配列と標識分子プローブのSortase A認識配列とが結合される;
なる2段階の反応による。
【0030】
Sortase A及び標識分子プローブの添加量は、反応が進行する限り特に制限されないが、例えば、Sortase Aを1〜1000μM、標識プローブを1〜1000μM含有する培養液、好ましくは、Sortase Aを5〜500μM、標識プローブを2〜500μM含有する培養液を用いることができる。
反応温度としては、細胞が生存できる条件であれば特に制限されないが、例えば20℃〜40℃で反応させることができ、好ましくは37℃のインキュベーター中で反応させることができる。
反応時間としては、5分〜10時間、好ましくは5分〜2時間である。
【0031】
反応の開始は、LPX1X2(G)n配列融合膜蛋白質を発現している細胞の培養液に、Sortase A及び標識分子プローブを添加してもよいし、予めSortase A及び標識分子プローブを含む培養液を調製しておき、当該培養液に培地交換してもよい。
反応の終了は、所望の時間経過後、PBS等で細胞を洗浄し、Sortase Aや余分のプローブ、遊離したN末端断片等を除去することで反応を終了させることができる。
【0032】
上記反応によりN末端標識膜蛋白質が得られたことは、公知の方法で確認することができ、例えばプローブが蛍光性のものである場合、共焦点レーザー顕微鏡により確認できる。
以上のように、N末端標識膜蛋白質は、前記標識分子プローブがLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を介してN末端に付加されている膜蛋白質である。
【0033】
また、反応時間・反応効率のさらなる向上のため、前述したLPX1X2(G)n配列融合膜蛋白質、ペプチド転移酵素Sortase A及び標識分子プローブに加えて、N末端に残基数2以上のポリグリシンを含むペプチドを用いる方法も、本発明の方法である。
N末端に残基数2以上のポリグリシンを含むペプチドの残基数は、目的の反応を阻害しない限り特に制限されないが、2〜50が好ましく、2〜20がより好ましい。
N末端のポリグリシンの残基数は2以上であれば特に制限されないが、2〜12であることがより好ましく、3〜8であることがさらに好ましい。
上記ペプチドはポリグリシンのみで構成されていてもよく、ポリグリシン以外に任意のアミノ酸を含んでいてもよいが、ポリグリシンのみで構成されることがより好ましく、具体的には残基数3〜8のポリグリシンを例示できる。ポリグリシン以外の配列としては、ペプチドが反応系に溶解し、反応を阻害しない限りにおいて特に制限されない。
【0034】
N末端に残基数2以上のポリグリシンを含むペプチドを用いる場合、次の工程を含む方法によってN末端標識膜蛋白質を製造する(図3参照)。
(i)N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質に、N末端に残基数2以上のポリグリシンを含むペプチドの存在下にペプチド転移酵素Sortase Aを作用させる工程;
(ii)工程(i)によりN末端がポリグリシン配列となった膜蛋白質に、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブの存在下にペプチド転移酵素Sortase Aを作用させる工程。
【0035】
まず、工程(i)において、Sortase A認識配列を有するLPX1X2(G)n配列融合膜蛋白質にSortase Aを作用させると、LPX1X2(G)n配列融合膜蛋白質は認識配列部分で切断されてN末端部断片が遊離し、残る膜蛋白質のN末端にはポリグリシン配列が提示される。遊離したN末端部断片は、反応系中のフリーのN末端にポリグリシンを含むペプチドと結合する。
【0036】
工程(i)の後には洗浄工程を設けて、遊離したN末端部断片とポリグリシンとが結合したポリペプチド、余分なSortase AやN末端にポリグリシンを含むぺプチド等を除去することが望ましい。洗浄はPBS洗浄等の一般的な方法でおこなうことができる。
【0037】
次に、工程(ii)において、Sortase A及びSortase A認識配列を有する標識分子プローブを反応系中に添加し、Sortase Aの作用によって、N末端にポリグリシン配列を提示している膜蛋白質と標識分子プローブとをポリグリシン配列を介して結合することができる。
【0038】
上記の反応において、反応の濃度及び温度は、前述のN末端にポリグリシンを含むペプチドを使用しない方法と同様の条件を用いることができる。
反応時間は、工程(i)は5分〜10時間、好ましくは10分〜2時間とすることができる。工程(ii)は5分〜4時間、好ましくは5分〜1時間であり、5分程度の短時間でも標識プローブは膜蛋白質に結合し、短時間での標識が可能である。
【0039】
本発明の細胞としては、N末端が標識分子プローブで標識された膜蛋白質を表層に有する細胞であって、標識分子プローブがLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞や、LPX1X2(G)n配列融合膜蛋白質を表層に有する細胞であれば特に制限されず、これら細胞のうち前者は、創薬のためのスクリーニング方法において有用であり、後者は前者の作製に有利に用いることができる。
【0040】
本発明はまた細胞表層膜蛋白質の標識キットに関し、本発明のキットは、少なくとも、N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むためのLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)を含むペプチドが付加された標識分子プローブ、ペプチド転移酵素Sortase A、及び、N末端の残基数2以上のポリグリシンを含むペプチド、を含む。
プラスミドベクター、標識分子プローブ、ペプチド転移酵素Sortase A、及びN末端にポリグリシンを含むペプチドは前記に詳述したものと同様であり、本発明のキットにはこれらの他に必要と目的に応じて、緩衝液、遺伝子導入用試薬、エネルギー供給液、pH調整剤、反応容器等が含まれていてもよい。
【0041】
本発明はまた、N末端標識膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞を用いることを特徴とする、創薬のためのスクリーニング方法に関し、スクリーニング対象としては膜蛋白質と作用してその機能を活性化する物質(例えばアゴニスト等)又は不活性化する物質(例えばアンタゴニスト等)等を挙げることができ、物質としては例えば、低分子有機化合物、蛋白質、天然ペプチド、人工ペプチド、ポリマー、ホルモン、糖質、脂質、核酸、ウイルス、細胞等を挙げることができる(以下「スクリーニング対象物質」ということがある。)。
【0042】
スクリーニング方法としては例えば、蛍光観察を利用する方法、FRET(蛍光共鳴エネルギー移動)を利用する方法等を挙げることができる。
【0043】
蛍光観察を利用する方法としては、本発明の方法でターゲットとする膜蛋白質(例えばGPCR等の受容体)を蛍光標識し、リガンド候補物質を添加した際の脱感作現象の有無などを蛍光から判定し、リガンド物質を特定するリガンドのスクリーニング方法を挙げることができる。
【0044】
FRETを利用する方法としては例えば、本発明の標識方法によってターゲットの膜蛋白質を標識した上で、蛍光標識したリガンド候補の物質を添加し、細胞表面でのFRETの有無を観察することで当該物質の膜蛋白質への結合の有無を判定し、リガンド物質を特定するリガンドのスクリーニング方法を挙げることができる。
或いはまた、ターゲットの膜蛋白質に結合することが知られている蛍光標識リガンドを添加して予めFRETを起こしておき、さらに、蛍光標識していない別種の物質を添加したときのFRETの変化の有無を確認する方法、例えば、膜蛋白質である受容体とそのアゴニストが知られている場合、本発明の方法で標識した膜蛋白質と標識したアゴニストとでFRETを起こし、様々なアンタゴニスト候補物質を添加したときのFRETの変化によってアンタゴニストを特定する、アンタゴニストのスクリーニング方法を挙げることができる。
【0045】
FRETを利用した薬剤スクリーニングの別の例として、ターゲットの膜蛋白質の一部を本発明の方法で蛍光アクセプター標識、残りを蛍光ドナー標識し、ここにオリゴマー化を誘発するリガンド候補物質を添加し、或いは、何らかの刺激を与えた場合のFRETの有無でオリゴマー形成の有無を確認し、リガンド物質や刺激感受性を特定するスクリーニング方法を挙げることができる。
或いはまた、ターゲットとする膜蛋白質のN末端を本発明の方法で蛍光アクセプター或いはドナー標識し、さらに当該膜蛋白質の別の部位を別法によって蛍光ドナー或いはアクセプター標識して、様々な物質の存在下或いは非存在下でのFRETの変化を観察することにより、ターゲット膜蛋白質の構造変化を観察し、リガンド物質を特定するリガンドのスクリーニング方法を挙げることができる。
【0046】
また、本発明の標識方法は、創薬目的に限らない薬剤のスクリーニング、膜蛋白質の機能・動態の観察に使用され得る。
【0047】
以下、実施例により本発明をより具体的に説明するが、本発明の技術的範囲はこれらの例示に限定されるものではない。
【実施例】
【0048】
[実施例1]
(1)AF647-LPETGGの合成及びLPETGGGGG配列を付加したECFP−TMの構築
標識分子としてAlexa Fluor 647-LPETGG (AF647-LPETGG) を用いた。Fmoc固相合成法によりペプチドH−LPETGG−NH2(配列番号2)を合成した後、Alexa Fluor 647サクシニミジルエステル (Invitrogen社製) を用いてN末端のアミノ基にAlexa Fluor 647を修飾することで、標識分子AF647-LPETGGを合成した。具体的には、DMSO(脱水) に溶解させたペプチドH−LPETGG−NH2及びDMSO(脱水)に溶解させたAlexa Fluor 647 サクシニミジルエステル (Invitrogen社製) を2:1のモル比で添加し、3時間反応を行った。塩基として、DIEAを4等量添加した。反応後は高速液体クロマトグラフィー (HPLC)により精製し、同定はMALDI−TOF−MSにより行った。
また、N末端近傍にLPETGGGGG配列(配列番号1) (以下LPETG5配列と表記する) を付加した、シアン蛍光蛋白質(ECFP) と血小板由来増殖因子受容体の膜貫通ドメイン(TM)との融合体 (以下LPETG5-ECFP-TMと表記する) の発現プラスミドベクター (図4) を構築した。まず、プライマー5’-EcoRI-BglII-LPETGGGGG-ECFP (塩基配列GCC GAA TTC GAC AGA TCT CTG CCG GAA ACT GGT GGC GGT GGC GGT TCT GGA GTG AGC AAG GGC GAG GAG CTG TTC ACC) (配列番号3)及び3’-ECFP-PstI-BamHI (塩基配列CGT GGA TCC GAA CTG CAG GCC CTT GTA CAG CTC GTC CAT GCC GAG AGT GAT CCC GGC) (配列番号4)を用いてECFP−C1(Clontech社製) から該当する遺伝子領域をクローニングし、EcoRI / BamHIサイトでpBlueScriptII SK(-) (Stratagene社製) に組み込んでシークエンスを確認した。その後、BglII/PstIサイトでpDisplay (Invitrogen社製) に組み替え、LPETG5-ECFP-TM発現プラスミドベクター (図4) を得た。
【0049】
(2)His6−SrtAの作成
His6-SrtA発現ベクター (T.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)に記載) をBL21 STAR (DE3) (Novagen社製) へ形質転換した。TB培地で培養し、O.D.=0.6の時点で0.1mM IPTGを添加して蛋白質発現を誘導した後、さらに27℃で18時間振とう培養を行った。そして、集菌して超音波破砕を行い、遠心して上清を回収した。次に、上清についてNi Sepharose High Performance (GE Healthcare社製) を用いてアフィニティー精製を行った。精製後、20mM Tris,150mM NaCl, pH8.0のバッファーに透析した。Sortase Aの蛋白質濃度はBCA Protein Assay Kit (PIERCE社製) を用いて求めた。
【0050】
(3)トリグリシン及びSrtAの添加によるLPETG5配列の切断
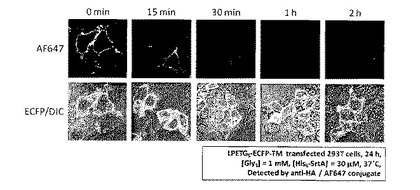
LPETG5-ECFP-TMを発現させたHEK 293T細胞(培養時間:24時間)にトリグリシン (Sigma社製)1mM及びHis6−SrtA30μMを添加し、37℃のCO2インキュベータ内で、15分、30分、1時間、2時間の各時間、切断反応を行った後、Alexa Fluor 647修飾抗HA抗体を用いた免疫蛍光染色を行った。
LPETG5配列の上流に位置するHAタグ (図4参照) の細胞表層における残存を、共焦点レーザー顕微鏡観察により測定することで、LPETG5配列の切断を評価した。図5に測定結果を示す。
【0051】
図5より、切断反応時間を長くとるにつれて観察されるAlexa Fluor 647由来の紫色蛍光強度は減少した。切断反応時間30分で細胞表層のHAタグのほとんどがなくなり、細胞表層に存在するLPETG5配列の多くが切断されたことが確認された。
【0052】
(4)細胞表層におけるECFP-TMへのAlexa Fluor 647の標識
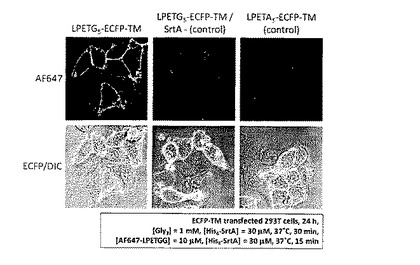
LPETG5-ECFP-TMを発現させたHEK 293T細胞(培養時間:24時間)に、トリグリシン 1mM及びHis6−SrtA 30μMを血清含有DMEM培地とともに添加し、37℃のCO2インキュベータ内で30分間切断反応を行った。His6−SrtA、余分なトリグリシン等を除去した後、標識分子であるAF647-LPETGG 10μM及びHis6−SrtA 30μMを血清含有DMEM培地とともに添加し、37℃のCO2インキュベータ内で15分間標識反応を行った。
比較例として、標識反応の際にHis6−SrtAを添加せずに同様に反応させた。またLPETG5配列をLPETA5配列に置換したLPETA5-ECFP-TMを発現させたHEK 293T細胞についても同様に反応させた。
SrtA及び余分なAF647-LPETGGを除去した後、共焦点レーザー顕微鏡を用いて蛍光観察を行った。結果を図6に示す。
【0053】
図6より、LPETG5-ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察された(左図)。一方、SrtAを加えない場合 (中図) 及びLPETA5-ECFP-TMを発現させたHEK 293T細胞を用いた場合 (右図) には細胞の輪郭に紫色蛍光が観察されなかった。
以上より、SrtAが触媒するペプチド転移反応により、部位特異的にECFP-TMにAlexa Fluor 647を標識することに成功した。
【0054】
[実施例2]
標識分子としてLPETGG配列を付加したビオチン分子を用い、切断反応時間を1時間、標識反応時間を5分とした他は実施例1と同様にして、細胞表層に存在するECFP-TMへのビオチン分子の標識を行った。標識の確認は、ストレプトアビジン−HRP(Invitrogen社製) を用いたウエスタンブロッティングにより行った。結果を図7に示す。
【0055】
図7より、LPETG5-ECFP-TMに挿入されたc-mycタグ (図4参照) に対する抗c-myc抗体 (Bethyl Laboratories社製) と同じ分子量付近 (36kDa) にストレプトアビジン−HRPのバンドが確認され、LPETG5-ECFP-TMにビオチン分子が標識されたことを確認した(最左レーン)。また、他の位置にはバンドが確認されず、細胞表層には多種多様な膜蛋白質が存在する中で、LPETG5-ECFP-TMのみに特異的にビオチン分子が標識されたことを確認した。一方で、SrtAを用いなかった場合 (左から2番目のレーン) 及びLPETA5-ECFP-TMを発現させたHEK 293T細胞を用いた場合 (右側2レーン) には同じ位置にバンドが確認されず、ビオチン分子が標識されなかったことを確認した。
【0056】
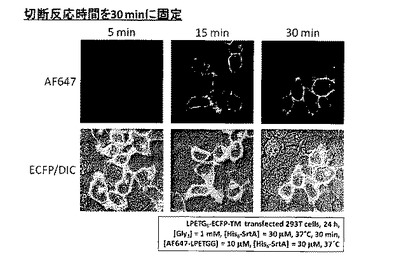
[実施例3]
実施例1と同様にしてLPETG5-ECFP-TMを発現させたHEK 293T細胞に、トリグリシン1mM及びHis6−SrtA30μMを血清含有DMEM培地とともに添加し、37℃のCO2インキュベータ内で1時間切断反応を行った。SrtA、余分なトリグリシン等を除去した後、標識分子であるAF647-LPETGG及びSrtAを血清含有DMEM培地とともに添加し、CO2インキュベータ内で5分、15分、30分間の各時間、標識反応を行った。SrtA及び余分なAF647-LPETGGを除去した後、共焦点レーザー顕微鏡を用いて蛍光観察を行った。結果を図8に示す。
【0057】
図8より、標識反応時間を5分としても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間を長くとるに従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0058】
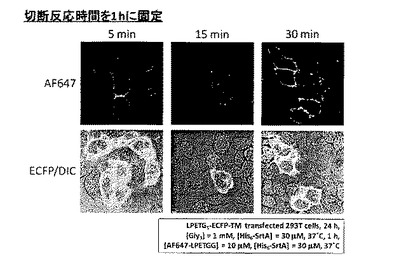
[実施例4]
切断反応の反応時間を30分とした以外は実施例3と同様にして標識化を行った。結果を図9に示す。
【0059】
図9より、標識反応を5分としても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間に従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0060】
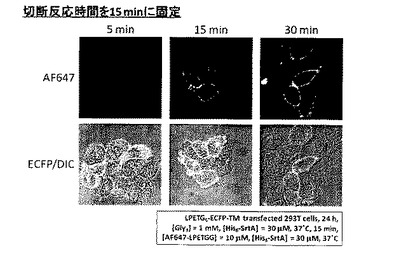
[実施例5]
切断反応の反応時間を15分とした以外は実施例3と同様にして標識化を行った。結果を図10に示す。
【0061】
図10より、標識反応を5分としても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間に従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0062】
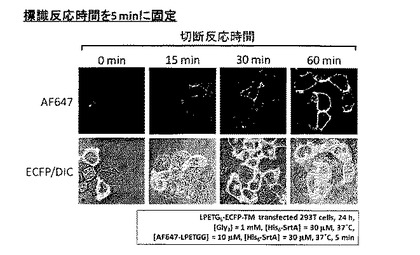
[実施例6]
標識反応時間を5分と固定し、切断反応の反応時間を0分、15分、30分、1時間とした上で、実施例1と同様の手順でAlexa Fluor 647の標識を行った。結果を図11に示す。
【0063】
図11の通り、切断反応時間を長くとるにつれてECFP-TM発現細胞の輪郭に観察されるAlexa Fluor 647由来の紫色蛍光強度は増大した。トリグリシン及びSrtAを添加して予め切断反応を行うことで、その後の標識反応効率が上昇することが示唆された。
【0064】
[実施例7]
実施例1と同様にしてLPETG5-ECFP-TMを発現させたHEK 293T細胞を得た。トリグリシンは用いず、標識分子であるAF647-LPETGG10μM、及びHis6−SrtA30μMを加え、37℃において、30分間標識反応を行った。結果を図12に示す。
【0065】
図12より、ECFP-TMへのAlexa Fluor 647の標識が確認された(右図)。トリグリシン存在下で切断反応を15分行った例(左図)には及ばないものの、トリグリシン非存在下でも標識が可能であることが確認された。
【0066】
[実施例8]
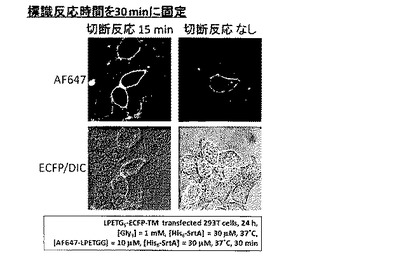
AF647-LPETGGの濃度を30μMとした以外は実施例3と同様にして標識化を行った。結果を図13に示す。
【0067】
図13より、AF647-LPETGGの濃度を30μMとしても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間に従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0068】
[参考例1]
実施例1と同様にしてLPETG5-ECFP-TMを発現させたHEK 293T細胞を得た。
LPETG5-ECFP-TMを発現させたHEK 293T細胞(培養24時間)に、トリグリシン100μMを添加して37℃のCO2インキュベータ内で、0分、15分、30分、1時間、2時間の各時間、切断反応を行った後、Alexa Fluor 647標識抗HA抗体を用いた免疫蛍光染色を行った。
LPETG5配列の上流に位置するHAタグの細胞表層における残存を、共焦点レーザー顕微鏡観察により測定することで、LPETG5配列の切断効率を評価した。図14に測定結果を示す。
【0069】
図14の通り、トリグリシン濃度が1mMの場合 (図5) と比較して切断反応効率は低下するものの、トリグリシン濃度が100μMの場合にも、Alexa Fluor 647由来の紫色蛍光強度は経時的に消失し、LPETG5配列が確かに切断されていることが確認された。
【図面の簡単な説明】
【0070】
【図1】本発明に用いるペプチド転移酵素Sortase Aが触媒する反応を模式的に示した図である。
【図2】本発明に用いるプラスミドベクターを概念的に示した図である。
【図3】本発明のN末端標識方法を模式的に示した図である。
【図4】実施例で用いたLPETG5-ECFP-TM発現プラスミドベクターのコンストラクトを模式的に示した図である。
【図5】トリグリシン及びSrtAの添加によるLPETG5配列の切断効率を免疫蛍光染色で評価した図である。
【図6】細胞表層におけるECFP-TMへのAlexa Fluor 647の標識を、蛍光観察により確認した図である。
【図7】ECFP-TMへのビオチン分子の標識を、ウエスタンブロッティングにより確認した図である。
【図8】切断反応を1時間とし、ECFP-TMへのAlexa Fluor 647の標識の経時変化を、蛍光観察により確認した図である。
【図9】切断反応を30分とし、ECFP-TMへのAlexa Fluor 647の標識の経時変化を、蛍光観察により確認した図である。
【図10】切断反応を15分とし、ECFP-TMへのAlexa Fluor 647の標識の経時変化を、蛍光観察により確認した図である。
【図11】標識反応時間を5分と固定し、切断反応時間を0分、15分、30分、60分とした際のECFP-TMへのAlexa Fluor 647の標識反応効率を蛍光観察により評価した図である。
【図12】トリグリシンを用いず、切断反応と標識反応を一段階で行った場合の、細胞表層におけるECFP-TMへのAlexa Fluor 647の標識を、蛍光観察により確認した図である。
【図13】Alexa Fluor 647の濃度を30μMとした場合の、標識反応効率を蛍光観察により評価した図である。
【図14】切断反応においてトリグリシンを100μMとした場合の、LPETG5配列の切断を免疫蛍光染色で確認した図である。
【技術分野】
【0001】
本発明は、分子生物学分野におけるバイオイメージング技術、及び蛋白質工学分野における蛋白質改変技術に関する。詳しくは、細胞が生きたままの状態で特定の蛋白質の機能や動態を直接観察するためのバイオイメージング技術、そのための蛋白質標識技術に関する。
【背景技術】
【0002】
細胞機能の解明や疾病の発症メカニズムの解明といった分子細胞生物学分野の研究開発において、特定の蛋白質の機能、細胞内での局在や活性の変化を知ることが必要不可欠である。そのための解析手法として、観測対象の蛋白質を可視化して直接観察するバイオイメージングの手法が幅広く利用されている。現在のバイオイメージングの代表的な手法としては、以下の3つの手法がある。
【0003】
(1)緑色蛍光蛋白質 (GFP)等の蛍光蛋白質を融合して蛋白質を蛍光標識する手法。
この手法では、遺伝子工学的に蛍光蛋白質と対象蛋白質を融合させるため、対象蛋白質のみを特異的に蛍光標識した状態で発現させることが可能である。
しかしこの手法の問題点として、1)蛍光蛋白質のサイズが非常に大きいため、標識に伴い対象蛋白質の発現・機能が阻害される可能性があること、2)常に対象蛋白質が蛍光標識された状態で発現されるために、発現時間の違いを区別した細胞内局在の変化を解析すること (パルスチェイス標識) が難しいこと等を挙げることができる。
【0004】
(2)有機化学反応により蛍光性合成分子プローブを修飾して蛋白質を蛍光標識する手法。
この手法では、観測したい任意のタイミングで有機化学反応によりサイズの小さな蛍光性合成分子プローブを修飾して、対象蛋白質を蛍光標識することができる。そのため、上述の蛍光蛋白質融合法の問題点を補うものであるといえる。また、蛍光性合成分子プローブのみならず、光反応性プローブ、pH感受性プローブ等の様々な機能性合成分子プローブを対象蛋白質に修飾することが可能である。
しかし、有機化学反応は官能基特異的反応、つまり「アミノ酸」特異的反応であるため、多種多様な蛋白質が存在する細胞内及び細胞表層で、対象蛋白質のみを特異的に蛍光標識することは非常に難しい。
【0005】
(3)蛍光修飾した抗体を用いて蛋白質を蛍光標識する手法。
この手法では、抗原抗体反応の高い特異性を利用し、蛍光修飾した抗体により対象蛋白質のみを特異的に蛍光標識することができる。また、パルスチェイス解析を行うことも可能である。
しかし、この手法にも1)抗体自体のサイズが非常に大きいため、標識に伴い対象蛋白質の機能が阻害される可能性があること、2)標識したい蛋白質それぞれに対し、蛍光修飾した抗体 (非常に高価である) を個別に用意する必要があること、3)抗原−抗体間の相互作用は強固であるものの、非共有結合的な相互作用であるため、条件によって蛍光修飾した抗体が対象蛋白質からはずれる可能性があること等の問題点を挙げることができる。
【0006】
また、蛋白質標識技術においては、目的の蛋白質を特異的に修飾する技術として生物の翻訳系を利用する手法が知られている。例えば、標識物質よりなるラベル部と、タンパク質のC末端に結合する能力を有する化合物よりなるアクセプター部とから構成されるラベル化試薬の存在下、プロモーター領域の制御下にある、終止コドンが削除されたコーディング領域からなるDNAから転写された産物を鋳型として無細胞翻訳系または生細胞中でタンパク質合成を行わせる方法が知られている(特許文献1、2)。
しかしこの手法はC末端に結合する能力を有する化合物を利用するC末端特異的な反応であり、N末端の修飾に適用することはできない。
【0007】
酵素反応の高い基質特異性を利用して対象蛋白質に特異的に蛍光分子プローブを修飾する手法が知られている。発明者らは既に、ペプチド転移酵素Sortase A(SrtA)を用いて細胞表層に存在する対象膜蛋白質のC末端部位に特異的に蛍光分子プローブを修飾し、対象膜蛋白質を蛍光標識する手法を開発している(非特許文献1)。
【0008】
SrtAは、認識配列LPXTGのTとGの間を切断し、そこにGG末端をもつ別のペプチドあるいは蛋白質を連結する反応を触媒する酵素である(図1参照)。発明者らの前記手法はこの基質特異性を利用し、C末端領域にLPETG配列を有する膜蛋白質と、GG配列を付加した蛍光分子プローブとをSrtAの酵素反応によって結合することで、特定の膜蛋白質のC末端を蛍光標識するものである。
【0009】
しかし、薬剤開発の主要なターゲットであるG蛋白質共役受容体(GPCR)をはじめとする多くの膜蛋白質は、N末端部位が細胞外、C末端部位が細胞内に位置するものであり、C末端部位が細胞表層に提示される膜蛋白質はごく少数に過ぎない。そのため、C末端部位が細胞表層に提示される膜蛋白質のみにしか適用できない前述の手法は汎用性の点で問題がある。
【0010】
【特許文献1】特開平11−322781号公報
【特許文献2】特開2000−139468号公報
【非特許文献1】T.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)
【発明の開示】
【発明が解決しようとする課題】
【0011】
本発明の課題は、従来の各バイオイメージング手法の欠点を補う新たな蛋白質標識法として、酵素反応の高い基質特異性を利用して対象蛋白質に特異的にプローブを修飾することで対象蛋白質のみを特異的に標識する方法、特に、細胞外に存在する膜蛋白質のN末端部位を特異的に標識する方法、当該方法によって特異的に修飾された膜蛋白質を有する細胞、及び、当該細胞を用いるスクリーニング方法を提供することにある。
【課題を解決するための手段】
【0012】
発明者らは、シグナル伝達や物質輸送等生命現象において非常に重要な役割を果たしている細胞表層の膜蛋白質をターゲットとし、ペプチド転移酵素Sortase A(SrtA)を用いて対象膜蛋白質のN末端部位に特異的に蛍光分子プローブを修飾する手法の検討を進める中で、Sortase A認識配列とSortase A転移配列の両方を有する被標識蛋白質と、Sortase A認識配列を付加したプローブとを用いて、目的の膜蛋白質のN末端を特異的に標識することができることを見い出し、本発明を完成するに至った。
【0013】
すなわち本発明は、
[1]N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質、
ペプチド転移酵素Sortase A、及び、
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ、
を用いることを特徴とする、N末端標識膜蛋白質の作製方法や、
[2]次の各工程を含むことを特徴とする、上記[1]に記載のN末端標識膜蛋白質の作製方法に関する。
(i)N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質に、N末端に残基数2以上のポリグリシンを含むぺプチドの存在下にペプチド転移酵素Sortase Aを作用させる工程;
(ii)工程(i)によりN末端がポリグリシン配列となった膜蛋白質に、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブの存在下にペプチド転移酵素Sortase Aを作用させる工程。
【0014】
また本発明は、
[3]N末端が標識分子プローブで標識された膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されていることを特徴とする細胞、
[4]LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を細胞外N末端側領域に有する膜蛋白質を表層に有する細胞、
[5]N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むための、LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、
[6]LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ、
[7]N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むためのLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)を含むペプチドが付加された標識分子プローブ、ペプチド転移酵素Sortase A、及び、N末端に残基数2以上のポリグリシンを含むペプチド、を含むことを特徴とする、細胞表層膜蛋白質の標識キットに関する。
【0015】
さらに本発明は、
[8]N末端標識膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞を用いることを特徴とする、創薬のためのスクリーニング方法に関する。
【発明の効果】
【0016】
本発明によれば、細胞表層に存在する多種多様な膜蛋白質のうち、対象とする特定の膜蛋白質のみを特異的に標識できる。
また、標識に伴い付加される分子サイズが非常に小さいため、標識に伴う対象膜蛋白質の発現阻害・機能阻害を回避することができる。標識物質についても、蛍光性合成分子プローブのみならず、光反応性プローブ、pH感受性プローブ等の様々な機能性合成分子プローブを膜蛋白質に修飾することが可能であるため、膜蛋白質を単に標識するだけでなく、膜蛋白質に様々な機能性合成分子プローブを修飾して膜蛋白質の機能解析を行うことが可能である。本手法で利用する合成分子プローブは大変自由度が高く設計することができ、また、一般的なペプチド固相合成と簡単な化学反応の組み合わせで大変容易に合成することが可能である。
さらに、Sortase Aが触媒するペプチド転移反応は血清を含む培地中において短時間で進行し、ATPなどの補因子を必要としないため、生きた細胞上で機能を保ったまま膜蛋白質を改変する手法として非常に有用であると考えられる。
そして、細胞表層に発現している膜蛋白質に対し、酵素反応により「後付け」でプローブを修飾するため、観測したい任意のタイミングで膜蛋白質を標識し、パルスチェイス解析を行うことができる。
本発明の方法及び本発明の細胞は、創薬や生物基礎研究等の分野において強力なツールとなると考えられる。
【発明を実施するための最良の形態】
【0017】
本発明のN末端標識膜蛋白質の作製方法において標識される膜蛋白質は、N末端が細胞外に存在する膜蛋白質であり、例えば、I型サイトカイン受容体ファミリー、II型サイトカイン受容体ファミリー、免疫グロブリンスーパーファミリー(IgSF)、G蛋白質共役型受容体ファミリー(GPCR)等に属する蛋白質を具体的に挙げることができる。
【0018】
I型サイトカイン受容体ファミリーの蛋白質としては、例えば、インターロイキン−2受容体、インターロイキン−3受容体、インターロイキン−4受容体、インターロイキン−5受容体、インターロイキン−6受容体、インターロイキン−7受容体、インターロイキン−9受容体、インターロイキン−11受容体、インターロイキン−12受容体、インターロイキン−13受容体、インターロイキン−15受容体、顆粒球マクロファージコロニー刺激因子受容体、毛様体神経栄養因子受容体、エリスロポエチン受容体、白血病抑制因子受容体、OSM受容体、又は顆粒球コロニー刺激因子受容体等を挙げることができる。
【0019】
II型サイトカイン受容体ファミリーの蛋白質としては、例えば、インターフェロン−α受容体、インターフェロン−β受容体、インターフェロン−γ受容体、又はインターロイキン−10受容体等を挙げることができる。
【0020】
免疫グロブリンスーパーファミリーの蛋白質としては、例えば、インターロイキン−1受容体又はインターロイキン−18受容体等を挙げることができる。
【0021】
G蛋白質共役型受容体ファミリーの蛋白質としては、例えば、ロドプシン受容体、カテコールアミン受容体[β1、β2、β3、α1、α2]、ドーパミン受容体[D1、D5、D2]、ムスカリン受容体[m1、m3、m2、m4、m5]、アデノシン受容体[A1、A3、A2]、PGE受容体[EP2、EP4、EP3、EP1]、ロイコトリエン受容体[BLT、Cys−LT1]、スフィンゴシン1−リン酸受容体[S1P1、S1P2、S1P3]、リゾホスファチジン酸受容体[LPA1、LPA2、LPA3]、ノシセプチン受容体、hGPCR44、hGPCR4、hGPCR10、hGPCR17、hGPCR19、hGPCR39、又はhGPCR48等を挙げることができる。
【0022】
本発明の作製方法において、そのN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する膜蛋白質(以下、「LPX1X2(G)n配列融合膜蛋白質」ということがある。)における「N末端側領域」とは、膜蛋白質のN末端近傍の細胞外領域を意味し、細胞質内で合成された当該蛋白質の翻訳後修飾や輸送等に影響しない部位であることが好ましい。例えば、N末端シグナル配列の下流に、上記アミノ酸配列を有することが好ましい。
【0023】
LPX1X2(G)nにおけるX1は任意のアミノ酸を表し、アラニン(A)、アルギニン(R)、アスパラギン(N)、アスパラギン酸(D)、システイン(C)、グルタミン(Q)、グルタミン酸(E)、グリシン(G)、ヒスチジン(H)、イソロイシン(I)、ロイシン(L)、リジン(K)、メチオニン(M)、フェニルアラニン(F)、プロリン(P)、セリン(S)、トレオニン(T)、トリプトファン(W)、チロシン(Y)、バリン(V)等が挙げられ、これらのうち、グルタミン酸、メチオニン、チロシン、ロイシン、グルタミン、アスパラギン、フェニルアラニンがより好ましい。
X2はトレオニン(T)又はアラニン(A)であり、トレオニンがより好ましい。
nは2〜8のいずれかの整数を表し、4〜6であることがより好ましい。
【0024】
上述のLPX1X2(G)n配列融合膜蛋白質は、任意の細胞の表層に発現した形で得ることができる。このLPX1X2(G)n配列融合膜蛋白質を細胞の表層に発現した細胞の調製方法は公知の方法を含め特に制限されず、例えば、標的とする膜蛋白質をコードするDNAのN末端側に相当する領域に、LPETGGGGG配列(配列番号1)をコードするDNA断片を挿入した、LPETGGGGG配列融合膜蛋白質をコードするDNAを含むプラスミドベクターを構築し、このプラスミドベクターを、公知の遺伝子導入方法、例えば、リン酸カルシウム法、リポフェクション法、電気穿孔法のいずれかの方法を用いて細胞に導入し、LPETGGGGG配列融合膜蛋白質を発現させることができる。また、LPETGGGGG配列等のLPX1X2(G)n配列の上流及び/又は下流にはさらに別の配列、例えば、LPX1X2(G)n配列の挿入を確認するためのタグ配列等を含めることもできる。
【0025】
LPX1X2(G)n配列融合膜蛋白質を発現させる細胞としては、目的に応じて選択でき特に制限されず、例えば、公知の哺乳動物細胞又は昆虫細胞等を用いることができる。公知の哺乳動物細胞としては、例えばHEK293細胞、Vero細胞、Hela細胞、CV1細胞、COS1細胞、CHO細胞、ナマルバ細胞等を挙げることができる。公知の昆虫細胞としては、例えばSf9細胞、MG1細胞等を挙げることができる。
【0026】
本発明で用いられるペプチド転移酵素Sortase Aは、Staphylococcus属菌等に存在する酵素であって、認識配列LPXTGのTとGの間を切断し、そこにGG末端を持つ別のペプチド又は蛋白質を結合する反応を触媒する(図1参照)。Sortase Aは、公知の遺伝子工学的方法(例えば、T.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)に記載の方法)で微生物に発現させ、精製して得たものを用いることができる。
【0027】
本発明の作製方法に用いられる標識分子プローブとしては、LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を介して、膜蛋白質と結合できる限り特に制限されるものではなく、例えば蛍光性合成分子プローブ、発光性プローブ、発色性プローブ、ビオチン標識プローブ、放射性同位元素プローブ、受容体プローブ等の検出プローブを挙げることができる。これらのうち、バイオイメージングにおける検出の簡便性等の観点から、蛍光性合成分子プローブが好ましい。また、検出プローブだけでなく、光反応性プローブ、pH感受性プローブ等の機能性プローブを用いることもできる。
【0028】
上記標識分子プローブには、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加されているが、LPX1X2(G)m配列において、X1及びX2は前記に例示したものと同様であり、mは1又は2であり、2であることがより好ましい。
上記アミノ酸配列LPX1X2(G)mはプローブに化学的方法によって付加されていてもよく、例えばT.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)に記載の方法によって付加することができる。
また、上記アミノ酸配列はプローブに直接付加されていてもよく、LPX1X2(G)m配列とプローブの間に別のペプチド及び/又はポリマー等からなるスペーサーを介して付加されていてもよい。さらに、上記アミノ酸配列の下流に、別のペプチド及び/又はポリマー等を有していてもよい。
【0029】
本発明の方法は、前記に詳説したLPX1X2(G)n配列融合膜蛋白質、ペプチド転移酵素Sortase A、及び標識分子プローブを用いて標的膜蛋白質のN末端を標識するものであるが、そのストラテジーは、
(1)Sortase AによってLPX1X2(G)n配列融合膜蛋白質のSortase認識配列部位が切断され、N末端にGG配列が提示される;
(2)Sortase Aによって、GG配列と標識分子プローブのSortase A認識配列とが結合される;
なる2段階の反応による。
【0030】
Sortase A及び標識分子プローブの添加量は、反応が進行する限り特に制限されないが、例えば、Sortase Aを1〜1000μM、標識プローブを1〜1000μM含有する培養液、好ましくは、Sortase Aを5〜500μM、標識プローブを2〜500μM含有する培養液を用いることができる。
反応温度としては、細胞が生存できる条件であれば特に制限されないが、例えば20℃〜40℃で反応させることができ、好ましくは37℃のインキュベーター中で反応させることができる。
反応時間としては、5分〜10時間、好ましくは5分〜2時間である。
【0031】
反応の開始は、LPX1X2(G)n配列融合膜蛋白質を発現している細胞の培養液に、Sortase A及び標識分子プローブを添加してもよいし、予めSortase A及び標識分子プローブを含む培養液を調製しておき、当該培養液に培地交換してもよい。
反応の終了は、所望の時間経過後、PBS等で細胞を洗浄し、Sortase Aや余分のプローブ、遊離したN末端断片等を除去することで反応を終了させることができる。
【0032】
上記反応によりN末端標識膜蛋白質が得られたことは、公知の方法で確認することができ、例えばプローブが蛍光性のものである場合、共焦点レーザー顕微鏡により確認できる。
以上のように、N末端標識膜蛋白質は、前記標識分子プローブがLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を介してN末端に付加されている膜蛋白質である。
【0033】
また、反応時間・反応効率のさらなる向上のため、前述したLPX1X2(G)n配列融合膜蛋白質、ペプチド転移酵素Sortase A及び標識分子プローブに加えて、N末端に残基数2以上のポリグリシンを含むペプチドを用いる方法も、本発明の方法である。
N末端に残基数2以上のポリグリシンを含むペプチドの残基数は、目的の反応を阻害しない限り特に制限されないが、2〜50が好ましく、2〜20がより好ましい。
N末端のポリグリシンの残基数は2以上であれば特に制限されないが、2〜12であることがより好ましく、3〜8であることがさらに好ましい。
上記ペプチドはポリグリシンのみで構成されていてもよく、ポリグリシン以外に任意のアミノ酸を含んでいてもよいが、ポリグリシンのみで構成されることがより好ましく、具体的には残基数3〜8のポリグリシンを例示できる。ポリグリシン以外の配列としては、ペプチドが反応系に溶解し、反応を阻害しない限りにおいて特に制限されない。
【0034】
N末端に残基数2以上のポリグリシンを含むペプチドを用いる場合、次の工程を含む方法によってN末端標識膜蛋白質を製造する(図3参照)。
(i)N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質に、N末端に残基数2以上のポリグリシンを含むペプチドの存在下にペプチド転移酵素Sortase Aを作用させる工程;
(ii)工程(i)によりN末端がポリグリシン配列となった膜蛋白質に、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブの存在下にペプチド転移酵素Sortase Aを作用させる工程。
【0035】
まず、工程(i)において、Sortase A認識配列を有するLPX1X2(G)n配列融合膜蛋白質にSortase Aを作用させると、LPX1X2(G)n配列融合膜蛋白質は認識配列部分で切断されてN末端部断片が遊離し、残る膜蛋白質のN末端にはポリグリシン配列が提示される。遊離したN末端部断片は、反応系中のフリーのN末端にポリグリシンを含むペプチドと結合する。
【0036】
工程(i)の後には洗浄工程を設けて、遊離したN末端部断片とポリグリシンとが結合したポリペプチド、余分なSortase AやN末端にポリグリシンを含むぺプチド等を除去することが望ましい。洗浄はPBS洗浄等の一般的な方法でおこなうことができる。
【0037】
次に、工程(ii)において、Sortase A及びSortase A認識配列を有する標識分子プローブを反応系中に添加し、Sortase Aの作用によって、N末端にポリグリシン配列を提示している膜蛋白質と標識分子プローブとをポリグリシン配列を介して結合することができる。
【0038】
上記の反応において、反応の濃度及び温度は、前述のN末端にポリグリシンを含むペプチドを使用しない方法と同様の条件を用いることができる。
反応時間は、工程(i)は5分〜10時間、好ましくは10分〜2時間とすることができる。工程(ii)は5分〜4時間、好ましくは5分〜1時間であり、5分程度の短時間でも標識プローブは膜蛋白質に結合し、短時間での標識が可能である。
【0039】
本発明の細胞としては、N末端が標識分子プローブで標識された膜蛋白質を表層に有する細胞であって、標識分子プローブがLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞や、LPX1X2(G)n配列融合膜蛋白質を表層に有する細胞であれば特に制限されず、これら細胞のうち前者は、創薬のためのスクリーニング方法において有用であり、後者は前者の作製に有利に用いることができる。
【0040】
本発明はまた細胞表層膜蛋白質の標識キットに関し、本発明のキットは、少なくとも、N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むためのLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)を含むペプチドが付加された標識分子プローブ、ペプチド転移酵素Sortase A、及び、N末端の残基数2以上のポリグリシンを含むペプチド、を含む。
プラスミドベクター、標識分子プローブ、ペプチド転移酵素Sortase A、及びN末端にポリグリシンを含むペプチドは前記に詳述したものと同様であり、本発明のキットにはこれらの他に必要と目的に応じて、緩衝液、遺伝子導入用試薬、エネルギー供給液、pH調整剤、反応容器等が含まれていてもよい。
【0041】
本発明はまた、N末端標識膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞を用いることを特徴とする、創薬のためのスクリーニング方法に関し、スクリーニング対象としては膜蛋白質と作用してその機能を活性化する物質(例えばアゴニスト等)又は不活性化する物質(例えばアンタゴニスト等)等を挙げることができ、物質としては例えば、低分子有機化合物、蛋白質、天然ペプチド、人工ペプチド、ポリマー、ホルモン、糖質、脂質、核酸、ウイルス、細胞等を挙げることができる(以下「スクリーニング対象物質」ということがある。)。
【0042】
スクリーニング方法としては例えば、蛍光観察を利用する方法、FRET(蛍光共鳴エネルギー移動)を利用する方法等を挙げることができる。
【0043】
蛍光観察を利用する方法としては、本発明の方法でターゲットとする膜蛋白質(例えばGPCR等の受容体)を蛍光標識し、リガンド候補物質を添加した際の脱感作現象の有無などを蛍光から判定し、リガンド物質を特定するリガンドのスクリーニング方法を挙げることができる。
【0044】
FRETを利用する方法としては例えば、本発明の標識方法によってターゲットの膜蛋白質を標識した上で、蛍光標識したリガンド候補の物質を添加し、細胞表面でのFRETの有無を観察することで当該物質の膜蛋白質への結合の有無を判定し、リガンド物質を特定するリガンドのスクリーニング方法を挙げることができる。
或いはまた、ターゲットの膜蛋白質に結合することが知られている蛍光標識リガンドを添加して予めFRETを起こしておき、さらに、蛍光標識していない別種の物質を添加したときのFRETの変化の有無を確認する方法、例えば、膜蛋白質である受容体とそのアゴニストが知られている場合、本発明の方法で標識した膜蛋白質と標識したアゴニストとでFRETを起こし、様々なアンタゴニスト候補物質を添加したときのFRETの変化によってアンタゴニストを特定する、アンタゴニストのスクリーニング方法を挙げることができる。
【0045】
FRETを利用した薬剤スクリーニングの別の例として、ターゲットの膜蛋白質の一部を本発明の方法で蛍光アクセプター標識、残りを蛍光ドナー標識し、ここにオリゴマー化を誘発するリガンド候補物質を添加し、或いは、何らかの刺激を与えた場合のFRETの有無でオリゴマー形成の有無を確認し、リガンド物質や刺激感受性を特定するスクリーニング方法を挙げることができる。
或いはまた、ターゲットとする膜蛋白質のN末端を本発明の方法で蛍光アクセプター或いはドナー標識し、さらに当該膜蛋白質の別の部位を別法によって蛍光ドナー或いはアクセプター標識して、様々な物質の存在下或いは非存在下でのFRETの変化を観察することにより、ターゲット膜蛋白質の構造変化を観察し、リガンド物質を特定するリガンドのスクリーニング方法を挙げることができる。
【0046】
また、本発明の標識方法は、創薬目的に限らない薬剤のスクリーニング、膜蛋白質の機能・動態の観察に使用され得る。
【0047】
以下、実施例により本発明をより具体的に説明するが、本発明の技術的範囲はこれらの例示に限定されるものではない。
【実施例】
【0048】
[実施例1]
(1)AF647-LPETGGの合成及びLPETGGGGG配列を付加したECFP−TMの構築
標識分子としてAlexa Fluor 647-LPETGG (AF647-LPETGG) を用いた。Fmoc固相合成法によりペプチドH−LPETGG−NH2(配列番号2)を合成した後、Alexa Fluor 647サクシニミジルエステル (Invitrogen社製) を用いてN末端のアミノ基にAlexa Fluor 647を修飾することで、標識分子AF647-LPETGGを合成した。具体的には、DMSO(脱水) に溶解させたペプチドH−LPETGG−NH2及びDMSO(脱水)に溶解させたAlexa Fluor 647 サクシニミジルエステル (Invitrogen社製) を2:1のモル比で添加し、3時間反応を行った。塩基として、DIEAを4等量添加した。反応後は高速液体クロマトグラフィー (HPLC)により精製し、同定はMALDI−TOF−MSにより行った。
また、N末端近傍にLPETGGGGG配列(配列番号1) (以下LPETG5配列と表記する) を付加した、シアン蛍光蛋白質(ECFP) と血小板由来増殖因子受容体の膜貫通ドメイン(TM)との融合体 (以下LPETG5-ECFP-TMと表記する) の発現プラスミドベクター (図4) を構築した。まず、プライマー5’-EcoRI-BglII-LPETGGGGG-ECFP (塩基配列GCC GAA TTC GAC AGA TCT CTG CCG GAA ACT GGT GGC GGT GGC GGT TCT GGA GTG AGC AAG GGC GAG GAG CTG TTC ACC) (配列番号3)及び3’-ECFP-PstI-BamHI (塩基配列CGT GGA TCC GAA CTG CAG GCC CTT GTA CAG CTC GTC CAT GCC GAG AGT GAT CCC GGC) (配列番号4)を用いてECFP−C1(Clontech社製) から該当する遺伝子領域をクローニングし、EcoRI / BamHIサイトでpBlueScriptII SK(-) (Stratagene社製) に組み込んでシークエンスを確認した。その後、BglII/PstIサイトでpDisplay (Invitrogen社製) に組み替え、LPETG5-ECFP-TM発現プラスミドベクター (図4) を得た。
【0049】
(2)His6−SrtAの作成
His6-SrtA発現ベクター (T.Tanaka et al. ChemBioChem, 9 (2008) pp.802-807 (February 22,2008)に記載) をBL21 STAR (DE3) (Novagen社製) へ形質転換した。TB培地で培養し、O.D.=0.6の時点で0.1mM IPTGを添加して蛋白質発現を誘導した後、さらに27℃で18時間振とう培養を行った。そして、集菌して超音波破砕を行い、遠心して上清を回収した。次に、上清についてNi Sepharose High Performance (GE Healthcare社製) を用いてアフィニティー精製を行った。精製後、20mM Tris,150mM NaCl, pH8.0のバッファーに透析した。Sortase Aの蛋白質濃度はBCA Protein Assay Kit (PIERCE社製) を用いて求めた。
【0050】
(3)トリグリシン及びSrtAの添加によるLPETG5配列の切断
LPETG5-ECFP-TMを発現させたHEK 293T細胞(培養時間:24時間)にトリグリシン (Sigma社製)1mM及びHis6−SrtA30μMを添加し、37℃のCO2インキュベータ内で、15分、30分、1時間、2時間の各時間、切断反応を行った後、Alexa Fluor 647修飾抗HA抗体を用いた免疫蛍光染色を行った。
LPETG5配列の上流に位置するHAタグ (図4参照) の細胞表層における残存を、共焦点レーザー顕微鏡観察により測定することで、LPETG5配列の切断を評価した。図5に測定結果を示す。
【0051】
図5より、切断反応時間を長くとるにつれて観察されるAlexa Fluor 647由来の紫色蛍光強度は減少した。切断反応時間30分で細胞表層のHAタグのほとんどがなくなり、細胞表層に存在するLPETG5配列の多くが切断されたことが確認された。
【0052】
(4)細胞表層におけるECFP-TMへのAlexa Fluor 647の標識
LPETG5-ECFP-TMを発現させたHEK 293T細胞(培養時間:24時間)に、トリグリシン 1mM及びHis6−SrtA 30μMを血清含有DMEM培地とともに添加し、37℃のCO2インキュベータ内で30分間切断反応を行った。His6−SrtA、余分なトリグリシン等を除去した後、標識分子であるAF647-LPETGG 10μM及びHis6−SrtA 30μMを血清含有DMEM培地とともに添加し、37℃のCO2インキュベータ内で15分間標識反応を行った。
比較例として、標識反応の際にHis6−SrtAを添加せずに同様に反応させた。またLPETG5配列をLPETA5配列に置換したLPETA5-ECFP-TMを発現させたHEK 293T細胞についても同様に反応させた。
SrtA及び余分なAF647-LPETGGを除去した後、共焦点レーザー顕微鏡を用いて蛍光観察を行った。結果を図6に示す。
【0053】
図6より、LPETG5-ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察された(左図)。一方、SrtAを加えない場合 (中図) 及びLPETA5-ECFP-TMを発現させたHEK 293T細胞を用いた場合 (右図) には細胞の輪郭に紫色蛍光が観察されなかった。
以上より、SrtAが触媒するペプチド転移反応により、部位特異的にECFP-TMにAlexa Fluor 647を標識することに成功した。
【0054】
[実施例2]
標識分子としてLPETGG配列を付加したビオチン分子を用い、切断反応時間を1時間、標識反応時間を5分とした他は実施例1と同様にして、細胞表層に存在するECFP-TMへのビオチン分子の標識を行った。標識の確認は、ストレプトアビジン−HRP(Invitrogen社製) を用いたウエスタンブロッティングにより行った。結果を図7に示す。
【0055】
図7より、LPETG5-ECFP-TMに挿入されたc-mycタグ (図4参照) に対する抗c-myc抗体 (Bethyl Laboratories社製) と同じ分子量付近 (36kDa) にストレプトアビジン−HRPのバンドが確認され、LPETG5-ECFP-TMにビオチン分子が標識されたことを確認した(最左レーン)。また、他の位置にはバンドが確認されず、細胞表層には多種多様な膜蛋白質が存在する中で、LPETG5-ECFP-TMのみに特異的にビオチン分子が標識されたことを確認した。一方で、SrtAを用いなかった場合 (左から2番目のレーン) 及びLPETA5-ECFP-TMを発現させたHEK 293T細胞を用いた場合 (右側2レーン) には同じ位置にバンドが確認されず、ビオチン分子が標識されなかったことを確認した。
【0056】
[実施例3]
実施例1と同様にしてLPETG5-ECFP-TMを発現させたHEK 293T細胞に、トリグリシン1mM及びHis6−SrtA30μMを血清含有DMEM培地とともに添加し、37℃のCO2インキュベータ内で1時間切断反応を行った。SrtA、余分なトリグリシン等を除去した後、標識分子であるAF647-LPETGG及びSrtAを血清含有DMEM培地とともに添加し、CO2インキュベータ内で5分、15分、30分間の各時間、標識反応を行った。SrtA及び余分なAF647-LPETGGを除去した後、共焦点レーザー顕微鏡を用いて蛍光観察を行った。結果を図8に示す。
【0057】
図8より、標識反応時間を5分としても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間を長くとるに従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0058】
[実施例4]
切断反応の反応時間を30分とした以外は実施例3と同様にして標識化を行った。結果を図9に示す。
【0059】
図9より、標識反応を5分としても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間に従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0060】
[実施例5]
切断反応の反応時間を15分とした以外は実施例3と同様にして標識化を行った。結果を図10に示す。
【0061】
図10より、標識反応を5分としても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間に従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0062】
[実施例6]
標識反応時間を5分と固定し、切断反応の反応時間を0分、15分、30分、1時間とした上で、実施例1と同様の手順でAlexa Fluor 647の標識を行った。結果を図11に示す。
【0063】
図11の通り、切断反応時間を長くとるにつれてECFP-TM発現細胞の輪郭に観察されるAlexa Fluor 647由来の紫色蛍光強度は増大した。トリグリシン及びSrtAを添加して予め切断反応を行うことで、その後の標識反応効率が上昇することが示唆された。
【0064】
[実施例7]
実施例1と同様にしてLPETG5-ECFP-TMを発現させたHEK 293T細胞を得た。トリグリシンは用いず、標識分子であるAF647-LPETGG10μM、及びHis6−SrtA30μMを加え、37℃において、30分間標識反応を行った。結果を図12に示す。
【0065】
図12より、ECFP-TMへのAlexa Fluor 647の標識が確認された(右図)。トリグリシン存在下で切断反応を15分行った例(左図)には及ばないものの、トリグリシン非存在下でも標識が可能であることが確認された。
【0066】
[実施例8]
AF647-LPETGGの濃度を30μMとした以外は実施例3と同様にして標識化を行った。結果を図13に示す。
【0067】
図13より、AF647-LPETGGの濃度を30μMとしても、ECFP-TM発現細胞の輪郭にのみAlexa Fluor 647由来の紫色蛍光が観察され、ECFP-TMへのAlexa Fluor 647の標識が確認された。反応時間に従って観察されるAlexa Fluor 647由来の紫色蛍光強度は増大し、徐々に飽和していくことが確認された。
【0068】
[参考例1]
実施例1と同様にしてLPETG5-ECFP-TMを発現させたHEK 293T細胞を得た。
LPETG5-ECFP-TMを発現させたHEK 293T細胞(培養24時間)に、トリグリシン100μMを添加して37℃のCO2インキュベータ内で、0分、15分、30分、1時間、2時間の各時間、切断反応を行った後、Alexa Fluor 647標識抗HA抗体を用いた免疫蛍光染色を行った。
LPETG5配列の上流に位置するHAタグの細胞表層における残存を、共焦点レーザー顕微鏡観察により測定することで、LPETG5配列の切断効率を評価した。図14に測定結果を示す。
【0069】
図14の通り、トリグリシン濃度が1mMの場合 (図5) と比較して切断反応効率は低下するものの、トリグリシン濃度が100μMの場合にも、Alexa Fluor 647由来の紫色蛍光強度は経時的に消失し、LPETG5配列が確かに切断されていることが確認された。
【図面の簡単な説明】
【0070】
【図1】本発明に用いるペプチド転移酵素Sortase Aが触媒する反応を模式的に示した図である。
【図2】本発明に用いるプラスミドベクターを概念的に示した図である。
【図3】本発明のN末端標識方法を模式的に示した図である。
【図4】実施例で用いたLPETG5-ECFP-TM発現プラスミドベクターのコンストラクトを模式的に示した図である。
【図5】トリグリシン及びSrtAの添加によるLPETG5配列の切断効率を免疫蛍光染色で評価した図である。
【図6】細胞表層におけるECFP-TMへのAlexa Fluor 647の標識を、蛍光観察により確認した図である。
【図7】ECFP-TMへのビオチン分子の標識を、ウエスタンブロッティングにより確認した図である。
【図8】切断反応を1時間とし、ECFP-TMへのAlexa Fluor 647の標識の経時変化を、蛍光観察により確認した図である。
【図9】切断反応を30分とし、ECFP-TMへのAlexa Fluor 647の標識の経時変化を、蛍光観察により確認した図である。
【図10】切断反応を15分とし、ECFP-TMへのAlexa Fluor 647の標識の経時変化を、蛍光観察により確認した図である。
【図11】標識反応時間を5分と固定し、切断反応時間を0分、15分、30分、60分とした際のECFP-TMへのAlexa Fluor 647の標識反応効率を蛍光観察により評価した図である。
【図12】トリグリシンを用いず、切断反応と標識反応を一段階で行った場合の、細胞表層におけるECFP-TMへのAlexa Fluor 647の標識を、蛍光観察により確認した図である。
【図13】Alexa Fluor 647の濃度を30μMとした場合の、標識反応効率を蛍光観察により評価した図である。
【図14】切断反応においてトリグリシンを100μMとした場合の、LPETG5配列の切断を免疫蛍光染色で確認した図である。
【特許請求の範囲】
【請求項1】
N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質、
ペプチド転移酵素Sortase A、及び、
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ、
を用いることを特徴とする、N末端標識膜蛋白質の作製方法。
【請求項2】
次の各工程を含むことを特徴とする、請求項1に記載のN末端標識膜蛋白質の作製方法。
(i)N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質に、N末端に残基数2以上のポリグリシンを含むペプチドの存在下にペプチド転移酵素Sortase Aを作用させる工程;
(ii)工程(i)によりN末端がポリグリシン配列となった膜蛋白質に、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブの存在下にペプチド転移酵素Sortase Aを作用させる工程。
【請求項3】
N末端が標識分子プローブで標識された膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されていることを特徴とする細胞。
【請求項4】
LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を細胞外N末端側領域に有する膜蛋白質を表層に有する細胞。
【請求項5】
N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むための、LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター。
【請求項6】
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ。
【請求項7】
N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むためのLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)を含むペプチドが付加された標識分子プローブ、
ペプチド転移酵素Sortase A、及び
N末端に残基数2以上のポリグリシンを含むペプチド、
を含むことを特徴とする、細胞表層膜蛋白質の標識キット。
【請求項8】
N末端標識膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞を用いることを特徴とする、創薬のためのスクリーニング方法。
【請求項1】
N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質、
ペプチド転移酵素Sortase A、及び、
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ、
を用いることを特徴とする、N末端標識膜蛋白質の作製方法。
【請求項2】
次の各工程を含むことを特徴とする、請求項1に記載のN末端標識膜蛋白質の作製方法。
(i)N末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を有する、N末端が細胞外に存在する膜蛋白質に、N末端に残基数2以上のポリグリシンを含むペプチドの存在下にペプチド転移酵素Sortase Aを作用させる工程;
(ii)工程(i)によりN末端がポリグリシン配列となった膜蛋白質に、LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブの存在下にペプチド転移酵素Sortase Aを作用させる工程。
【請求項3】
N末端が標識分子プローブで標識された膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されていることを特徴とする細胞。
【請求項4】
LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を細胞外N末端側領域に有する膜蛋白質を表層に有する細胞。
【請求項5】
N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むための、LPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター。
【請求項6】
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)が付加された標識分子プローブ。
【請求項7】
N末端が細胞外に存在する膜蛋白質のN末端側領域にLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を組み込むためのLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)をコードするDNA断片を含むプラスミドベクター、
LPX1X2(G)m配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。mは1又は2を表す。)を含むペプチドが付加された標識分子プローブ、
ペプチド転移酵素Sortase A、及び
N末端に残基数2以上のポリグリシンを含むペプチド、
を含むことを特徴とする、細胞表層膜蛋白質の標識キット。
【請求項8】
N末端標識膜蛋白質を表層に有する細胞であって、標識がLPX1X2(G)n配列(X1は任意のアミノ酸を表し、X2はトレオニン又はアラニンを表す。nは2〜8のいずれかの整数を表す。)を含むポリペプチドを介して膜蛋白質に付加されている細胞を用いることを特徴とする、創薬のためのスクリーニング方法。
【図1】



【図2】

【図3】

【図4】
【図5】
【図6】
【図7】

【図8】
【図9】
【図10】
【図11】
【図12】
【図13】

【図14】

【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公開番号】特開2010−115136(P2010−115136A)
【公開日】平成22年5月27日(2010.5.27)
【国際特許分類】
【出願番号】特願2008−290026(P2008−290026)
【出願日】平成20年11月12日(2008.11.12)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年8月24日 社団法人化学工学会発行の「化学工学会第40回秋季大会研究発表講演要旨集」に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年9月8日 日本化学会 生体機能関連化学部会、日本化学会 バイオテクノロジー部会、日本化学会 フロンティア生命化学研究会、日本化学会 ホスト−ゲスト・超分子化学研究会発行の「第3回バイオ関連化学合同シンポジウム講演要旨集」に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年9月23日 東京大学生命科学研究ネットワーク発行の「東京大学生命科学研究ネットワークシンポジウム2008抄録集」に発表
【出願人】(501163761)
【Fターム(参考)】
【公開日】平成22年5月27日(2010.5.27)
【国際特許分類】
【出願日】平成20年11月12日(2008.11.12)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年8月24日 社団法人化学工学会発行の「化学工学会第40回秋季大会研究発表講演要旨集」に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年9月8日 日本化学会 生体機能関連化学部会、日本化学会 バイオテクノロジー部会、日本化学会 フロンティア生命化学研究会、日本化学会 ホスト−ゲスト・超分子化学研究会発行の「第3回バイオ関連化学合同シンポジウム講演要旨集」に発表
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り 平成20年9月23日 東京大学生命科学研究ネットワーク発行の「東京大学生命科学研究ネットワークシンポジウム2008抄録集」に発表
【出願人】(501163761)
【Fターム(参考)】
[ Back to top ]