
NK細胞の増殖
閉鎖型細胞培養システムにおける、NK細胞およびNKT細胞の大規模増殖および同時活性化のための方法であって、該増殖した細胞が増加した細胞傷害性を示す方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
技術分野
本発明は、細胞培養および免疫療法、特に、治療に使用するためのナチュラルキラー細胞(以下、NK細胞と記す)およびナチュラルキラー様T細胞(以下、NKT細胞と記す)細胞の大規模増殖および同時活性化に関する。該細胞を、閉鎖型自動化培養システム、例えばバイオリアクターを用いて増殖させる。
【背景技術】
【0002】
発明の背景
癌に対する細胞免疫療法の使用は、1980年代半ばにリンホカイン活性化キラー(LAK)細胞が導入されて以来、徹底して調査されている(Grimm EA. et al., 1982; Rosenberg S., 1985)。
【0003】
最も研究された手法の1つは、移植片対腫瘍(GvT)効果をもたらし腫瘍細胞を殺す可能性のある、自己(autologous)または同種異系(allogeneic)の細胞傷害性エフェクターの養子移植であった。可能性のある抗腫瘍効果を有する種々のエフェクター集団のうち、NKおよびNKT細胞は、それらの高い細胞傷害能により注目されている(Sutlu T and Alici E., 2009)。
【0004】
NKおよびNKT細胞は通常、末梢血単核球細胞(PBMC)およびLAK細胞のようなエフェクター細胞調製物中に少数しか存在しない。故に、健常なドナー(Carlens S. et al., 2001 および US 10/242,788)ならびにB細胞慢性リンパ性白血病を有する患者(Guven H. et al., 2003)および多発性骨髄腫(MM)を発症した患者(Alici E. et al., 2008)由来のPBMCを用いて細胞培養フラスコ中、ポリクローナルNK細胞およびNKT細胞の増殖を可能とする、現在の良好な製造法であるcGMP適合の成分を含む方法が開発されている。これらの細胞は、臨床の場で評価される可能性を開く、ヒト腫瘍のインビトロモデルおよび実験モデル(Guimaraes F. et al., 2006)において、新鮮なヒト腫瘍細胞に対して特定の細胞傷害活性を発揮することが示されている。しかしながら、従来のフラスコベースの培養は、多大な労力を要し、かつ扱いにくく、故に、実際に扱える細胞数に限りがある。エフェクター細胞調製物を対象とする既報のプロトコール(例えば、Miller JS. et al., 1994;Pierson BA. et al., 1996;Luhm J. et al., 2002;Klingermann HG and Martinson J., 2004)は、培養前のNK前駆体またはCD56の単離、およびフィーダー細胞またはcGMP不適合成分の使用のような工程も含む。これらの不都合は、既報のプロトコールが大規模な臨床研究を支持するのに至適ではなく、かつ実施不能であることを示す。
【0005】
さらに、細胞培養フラスコ中でのNK細胞の増殖は、外部物質への暴露および汚染の固有のリスクを有する。このリスクは、GMP適合の実験室環境で最小化されるが、閉鎖型自動化システムの使用は、それが十分量の細胞を供給する限り間違いなく好ましい。
【0006】
造血細胞はせん断に比較的感受性であるため、高せん断処理は、エクスビボ増殖には好ましくない(Nielsen, 1999)との仮定は当然である。故に、外部フィルターおよび高流速に依存する振盪タンク型バイオリアクター(Pierson. et al., 1996)またはかん流培養システムは、高効率を達成する可能性が低い。
【0007】
癌の処置用の免疫療法に基づくNK細胞およびNKT細胞を用いる多くの有望な方法がある。しかしながら、GMP適合の閉鎖系でのこれらのエフェクター細胞のエクスビボ増殖および活性化は、臨床的な使用機会を増加させるための非常に重要な因子である。
【0008】
NK細胞をエクスボビで増殖および/または活性化するための他の研究者による試みもされており、精製/静止に用いる複数の処理オプションがあり、短時間でまたは高度に精製され、かつ長時間活性化されるNK細胞が研究されている。これらの研究は、NK細胞注入が良好な耐容性を示し、部分的に有効であることを報告する。しかし、エフェクター細胞調製に一般的に用いられるプロトコールは、培養前のNK前駆体またはCD56の単離、およびフィーダー細胞および/またはcGMP不適合成分の使用のようなさらなる工程を含む。これらの不都合は、かかるプロトコールを、GMP適合生産に適当ではなく、大規模な臨床研究を支持することを不可能にする。
【0009】
上記の報告の詳細な評価は、NK細胞の最適化エクスビボ増殖のための自動化法の必要性を示す。常套のフラスコベースの培養の1つの問題は、規模に関し、すなわち細胞数がフラスコ操作の煩雑さのために制限されることに関する。さらに、細菌感染の危険性は、該システムが培地交換や細胞の分配時に周囲環境に曝されるために、極めて高い。解決されるべきさらなる問題には、費用対効果が高く、取り扱いが容易で、明確に定義されたcGMP品質の成分を含む、エフェクター細胞の増殖法の開発が含まれる。好ましくは、培養システムはまた、動物由来成分およびフィーダー細胞を含まない。
【発明の概要】
【0010】
発明の概要
本発明は、細胞治療薬として使用されるべき、NK細胞およびNKT細胞の大規模増殖および同時活性化のための閉鎖型システムに関する。特に、本発明は、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の大規模増殖および同時活性化のための方法であって、該増殖細胞が、同一または類似の条件下で、例えばフラスコのような常套法を用いて培養された細胞と比較して、増加した細胞傷害性を示す方法を開示する。本発明の方法は、発明の詳細な説明、非限定的実施例および特許請求の範囲にさらに開示され得る。
【0011】
本発明者らは、閉鎖型システムを用いる大規模NK細胞増殖の実現可能性を調査した。2つの異なる閉鎖型システム(細胞培養バッグおよび自動化バイオリアクター)を、NK細胞の大規模生産が免疫療法に用いられるのを可能とする自動化GMP適合プロトコールを開発する目的で、健常なドナーおよび多発性骨髄腫(MM)の患者由来のPBMCを用いて、常套の細胞培養フラスコと比較して評価した。
【0012】
同時に、本発明者らは、癌患者の第I相臨床試験での同種異型処置におけるこの細胞薬の安全性評価を成功裏に完了した(Barkholt L. et al., 2009)。
【0013】
本発明の第一の態様によれば、特定の細胞タイプの大規模増殖および同時活性化のための方法を提供し、ここで、閉鎖型細胞培養系を用い、この方法によって得られた増殖した細胞は、インビトロの細胞傷害性試験で決定される通り増加した細胞傷害性を示す。好ましくは、該細胞型は、表現型CD3−CD56+のNK細胞および/または表現型CD3+CD56+のNKT細胞である。
【0014】
好ましい態様によれば、本発明の方法は、該細胞を、血清、インターロイキン−2(IL−2)および抗CD3抗体を補充した増殖培地を含む該閉鎖システムに添加し;該細胞を、増殖した細胞集団の少なくとも35%が活性化NK細胞およびNKT細胞となるまで振盪(agitation)および加熱して該システム内で増殖させ;該増殖した細胞が、インビトロ細胞傷害性試験で決定される通り増加した細胞傷害性を示す工程を含む。
【0015】
記載した方法の一態様によれば、血清は、ヒト血清および自己血清からなる群から選択される。培地には、約50ないし約1500U/mlのIL−2、約1ないし約50ng/mlの抗CD3抗体および約1ないし約40%の血清を補充する。
【0016】
別の態様によれば、振盪および加熱を以下の条件下で行う:約36−40℃の温度;約4.7−5.1%のCO2濃度;および、細胞が閉鎖型細胞システムの表面に付着できる速度および角度での穏やかな振盪。振盪を、約4−8/分の振盪速度、および約4−8°の振盪角度で行う。
【0017】
増殖は、好ましくは、全細胞数が、少なくとも10倍に増すか、または増殖した細胞集団の少なくとも約50%が活性化NK細胞およびNKT細胞それぞれを含むまで行われる。
【0018】
該細胞を増殖させるために用いる細胞サンプルは、好ましくは、末梢血、細胞株またはサイトカインで刺激した末梢血のサンプルである。最も好ましくは、細胞サンプルは、末梢血単核球細胞(PBMC)サンプルである。
【0019】
該方法の一態様によれば、細胞サンプルを健常な対象から集める。
【0020】
別の態様によれば、細胞サンプルを、腫瘍を有する対象、好ましくは血液系腫瘍および固形腫瘍からなる群から選択される腫瘍を有する対象から集める。
【0021】
別の態様によれば、細胞は、主に、表現型CD3−CD56+のNK細胞で構成される。
【0022】
閉鎖型細胞システムに最初に添加される細胞の濃度は、好ましくは、増殖培地1ml当たり、約0.5x106ないし約2x106である。
【0023】
本発明の一態様において、本発明の方法は、1日当たり、全培養量の約50%に相当する量の血清およびIL−2を補充した培地を添加し、該閉鎖型細胞システムから1日当たりおよそ同量の増殖培地を捨てる工程をさらに含み、ここで該工程は、全細胞密度が、最初の細胞密度の少なくとも約1.5倍に増大したときに行われる。
【0024】
好ましくは、該工程は、全細胞密度が、最初の細胞密度から少なくとも約300%増大したときに行われ、その場合は、全培養量の約75%を交換する。
【0025】
別法として、該工程は、全細胞密度が、最初の細胞密度から少なくとも約500%増大したときに行われ、その場合は、全培養量の約100%を交換する。
【0026】
さらに別の態様によれば、上記の態様の1つ以上を自由に組合せて、細胞を少なくとも約10日間インキュベートする。
【0027】
一態様によれば、閉鎖型細胞システムは予め滅菌したバッグである。
【0028】
別の態様によれば、該閉鎖型細胞システムは、バイオリアクターである。
【0029】
本発明の別の態様は、増加した細胞傷害性を示す、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液である。
【0030】
好ましくは、これらの細胞は、フラスコ中で増加させた細胞と比較して、インビトロ細胞傷害性試験で決定される通り、増加した細胞傷害性を示す。
【0031】
本発明の表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液中、増殖した細胞集団の少なくとも35%、好ましくは少なくとも50%は、活性化NK細胞からなる。
【0032】
本明細書中、バイオリアクターは、再利用可能な自動チャンバーシステムであって、生物学的に活性な環境を支持する何らかの装置またはシステムを意味し得る。記載した本発明の範囲内において、用語バイオリアクターは、細胞培養に関して、細胞または組織を増殖させることを意図する装置またはシステムを意味する。バイオリアクターシステムの非限定的例は、GE−Healthcareから提供されるWave bioreactor system 2/10であるが、当業者は、他の供給者から提供される別のバイオリアクターを利用可能であることを理解し得るか、または本発明の方法を実行可能なバイオリアクターを構築可能である。
【0033】
本明細書中、閉鎖型システムは、さらなる処理工程を経ることなくエクスビボで効率的かつ迅速に細胞を増殖させ得る中央の細胞培養バッグからなる細胞増殖チャンバーシステムである。
【0034】
用語“細胞傷害性”は、細胞に対する傷害性を意味する。傷害性物質の例には、化学物質、または細胞傷害性T細胞、NK細胞およびNKT細胞のような細胞傷害性リンパ球のような免疫細胞が含まれる。NK細胞およびNKT細胞は、それらの高い細胞傷害能により注目される。
【0035】
当業者は、利用可能な方法を用いて細胞傷害性を決定できる。細胞が増加した細胞傷害性を示すかどうかを決定する1つの方法は、標準的な4時間の51Cr−放出アッセイを用いて、K562細胞に対する細胞媒介性細胞傷害のインビトロ分析を用いることである。あるいは、脱顆粒アッセイを用い得る。これらの両方法を、以下の実施例に記載する。
【0036】
本明細書中、用語“活性化”および“活性化NK細胞”は、活性化シグナルを受容したNK細胞を意味する。活性化NK細胞は、MHCクラスI発現が低下している細胞を殺し得る。
【0037】
用語「NK細胞およびNKT細胞の“同時活性化”」は、細胞が、実質的に同時に、好ましくは、同じ細胞培養において、活性化されることを意味する。
【0038】
それらの強力な細胞傷害活性および自己反応性の可能性を考慮して、NK細胞活性は、厳密に制御される。MHCクラスI発現を喪失したか、またはそれが異常な細胞を殺すために、NK細胞は活性化される必要がある。NK細胞は、種々の形態をとり得る活性化シグナルを受容しなければならず、中でもサイトカイン、Fc−受容体、活性化受容体および阻害性受容体が最も重要である。
【0039】
本明細書中、用語“細胞増殖”は、一連の細胞分裂過程を経て、故に培養中に存在する細胞数が増大する細胞の培養に関する。故に、用語“NK細胞増殖”は、一連の細胞分裂過程を経て、故に培養中に存在する細胞数が増大するNK細胞の培養に関する。用語“増殖したNK細胞”は、NK細胞増殖を介して得られたNK細胞に関する。より具体的には、一態様において、用語“増殖したNK細胞”は、特定のcGMPグレードの環境およびcGMPグレードの培地中で増殖される、慢性的に活性化したCD3−CD56+細胞ならびに表現型CD3+CD56+のNKT細胞のポリクローナル群に関する。
【0040】
本明細書中、用語“エフェクター細胞”は、免疫系の細胞のような、刺激に応答して特定の機能を果たす細胞に関する。一態様において、エフェクター細胞は、細胞傷害能(すなわち、他の細胞に細胞死を誘導する能力)を有するリンパ球の一種である。別の態様は、分泌型抗体に積極的に関与するリンパ球である。エフェクター細胞の非限定的例は、NK細胞、T細胞およびNKT細胞である。
【図面の簡単な説明】
【0041】
図面の簡単な説明
【図1】図1A−Dは、フラスコおよびバッグ(1A)ならびにフラスコおよびバイオリアクター(1B)における増殖倍の比較、ならびにフラスコおよびバッグ(1C)ならびにフラスコおよびバイオリアクター(1D)における最終産物の純度を示す図である。
【図2】図2A−Cは、異なる増殖プロトコールで増殖させた細胞の表現型(AおよびB)および細胞傷害活性(C)の比較を示す図である。
【図3】図3A−Dは、フラスコと比較して、バイオリアクターで開始した細胞培養の増殖を示す図である。
【図4】図4A−Bは、バイオリアクターおよびフラスコそれぞれで直接増殖させた細胞の機能的比較を示す図である。
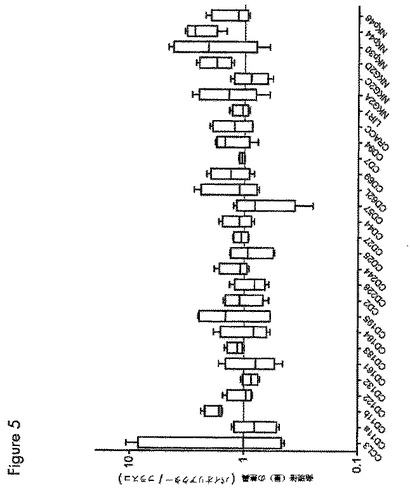
【図5】図5は、バイオリアクターおよびフラスコそれぞれで直接増殖させたNK細胞の表現型の比較を示す。
【発明を実施するための形態】
【0042】
発明の詳細な説明
本発明を記載する前に、本発明の範囲は、添付の特許請求の範囲およびその均等物でのみ限定され得るから、本明細書で用いる専門用語は、特定の態様を記載する目的でのみ用いられ、限定すべきことを意図しないことが、理解されるべきである。
【0043】
他に特に記載がなければ、本明細書で用いる用語および技術用語は、本発明が属する分野の当業者に通常理解される意味を有することを意図する。
【0044】
本明細書および添付の特許請求の範囲に用いられる、単数形“a”、“an”および“the”は、文中に他に明記されない限り、複数形も含むことに留意すべきである。
【0045】
また、用語“約”は、適用されるとき、所定の値の+/−2%、好ましくは、+/−5%、最も好ましくは、+/−10%の数値の偏差を示すために用いる。
【0046】
悪性疾患に罹患した患者に対するNK細胞のエクスビボ増殖および再注入は、かかる疾患に対抗する新規かつ可能性のある興味深い治療方法を提供する。このための必要条件は、臨床用途の要求を満たすNK細胞の増殖および使用の可能性である。本発明者らは、驚くべきことに、臨床適用のために大規模でNK細胞を増殖させることができた。従って、臨床で適用可能なエフェクター集団に富むNKおよびNKT細胞の大規模製造のためのGMP適合自動化閉鎖型培養システムを提供する。
【0047】
以下の実施例に示す通り、本発明の方法は、満足できる細胞傷害能(すなわち、他の細胞の細胞死を誘導する能力)を有するNK細胞を増殖および活性化させる。他の細胞に対するNK細胞の細胞傷害性は、例えば、活性化NK細胞に暴露する前後で細胞を計数する従来の方法により、容易に測定可能である。かかる方法は、当業者によく知られている。
【実施例】
【0048】
実施例
1.末梢血由来のNK細胞およびNKT細胞のエクスビボ増殖
材料および方法
末梢血単核球細胞(PBMC)のサンプリングおよび単離
バフィーコート、末梢血および血液成分(apheresis product)を、Karolinska University Hospital, Huddingeの血液バンクに登録された健常なドナーまたはMM患者から得た。実験プロトコールは、倫理委員会の承認を得た。
【0049】
末梢血単核球細胞(PBMC)を、Lymphoprep (Nyegaard, Oslo, Norway)を用いる勾配遠心分離により単離した。PBMCを、リン酸緩衝生理食塩水(PBS)(Gibco, Grand Island, NY, USA)で2回洗浄し、細胞の生存を、トリパンブルー色素排除試験により評価した。
【0050】
増殖培地および細胞計数
全てのシステムに関して、5%ヒト血清(Biowhittaker−Cambrex, Walkersville, MD, USA)および500U/ml rhIL−2(Proleukin(商標), Novartis Pharmaceuticals, East Hanover, NJ, USA)を添加したCellGro SCGM血清不含有培地(CellGenix, Freiburg, Germany)を増殖培地として用いた。培養開始時、培地に、モノクローナル抗CD3抗体(Orthoclone OKT−3, Ortho Biotech, Raritan, NJ, USA)を終濃度10ng/mlでさらに添加した。全細胞数を、培養0日、5−6日、9−10日、14−15日および21日目にトリパンブルー色素を用いて細胞を染色して評価した。最終製品を、安全性、純度および同一性(細胞生存能および表現型)について評価した。細胞の絶対数を、全細胞数とフローサイトメトリー(BD FacsCalibur; BD Biosciences, San Jose, CA, USA)により決定されたこれらの亜集団の割合を乗ずることにより計算した。
【0051】
細胞培養フラスコでの増殖
細胞培養フラスコ中での細胞傷害性細胞の増殖のための培養条件は、健常な個体由来のPBMC(Carlens S. et al., 2001)に対して予め最適化した。簡単には、PBMCを、T25フラスコ(TPP, Trasadingen, Switzerland)中で、初め0.5x106細胞/mlの濃度で培養した。5日後、培養を、培養が終了するまでの間、5%ヒト血清およびIL−2(500U/ml)を含むがOKT−3を含まない新鮮な培地を2−3日毎に補充して行った。細胞増殖の接触阻止を阻止するため(Heiskala M. et al., 1987)、細胞を、より大きなフラスコ(T75 または T150: TPP, Trasadingen, Switzerland)に移すか、または必要なとき、複数のフラスコに移した。培地の補充において、細胞濃度を、10日目までは0.5x105細胞/mlに、10日目以降は1x106細胞/mlに合わせた。時には、細胞の一部を、扱いやすいフラスコ数に維持するために凍結した。
【0052】
Waveのバイオリアクターシステムでの増殖
Waveのバイオリアクターは、温度およびCO2が制御された、振とう加熱プラットホーム上に置く使い捨ての滅菌バッグ内で細胞を増殖させる細胞培養システムである。発明者らは、Waveのバイオリアクターシステム 2/10 (GE Healthcare, Somerset, NJ, USA)を用いた。このシステムでの本発明者らの以前の結果は、低容量および少数の細胞で培養を開始したとき不十分な効率を示した。しかし、ドナーの末梢血サンプル中の細胞数は、該バイオリアクターで直接培養を開始するのを可能にしない。故に、本発明者らは、最初の最適化実験においてフラスコ中で培養を開始し、十分な細胞数に達した時点のおよそ5日目に該細胞をバイオリアクターに移した。この日のバイオリアクター培養は、800ml中に、2x106細胞/mlで開始した。
【0053】
最後の立証実験において、ドナー由来の末梢血全血または血液成分を得て、培養を0日目からバイオリアクターで直接開始した。バイオリアクターの設定は、全ての時点で以下の通りであった:温度:37℃、CO2:5%、気流:0.1、振とう速度:6/分、振とう角度:6°。細胞を1日おきにサンプリングし、計数し、さらなる培地供給を、細胞密度が3x106細胞/mlに達するまで行わなかった。以降、培養物に1日当たり500mlの培地を供給した(50ml/回)。細胞が6x106細胞/mlの密度に達したとき、培地供給を750ml/日に増し、1x107細胞/mlに達した後に1000ml/日に増した。
【0054】
Vuelife(商標)バッグでの増殖
Vuelife(商標)(American Fluoroseal Corporation, MD, USA)は、生物学的、免疫学的および化学的に不活性なフッ素化エチレン−プロピレン製の滅菌細胞培養バッグである。それは、高度にガス透過性であり、光学的に透明である。Vuelife バッグ中での培養を、72mlのVuelife バッグを用いて、培地60ml中に5x105細胞/mlで開始した。該バッグを、37℃および5%CO2で加湿インキュベーター中でインキュベートした。新鮮な培地を2−3日毎に添加して、10日目まで1x106細胞/mlの濃度に調節し、10日目以降、2x106細胞/mlの濃度に調節した。必要なとき、細胞をより大きなバッグに分割した。
【0055】
2.フローサイトメトリーによるリンパ球サブセットおよび
表現型の分析
材料および方法
細胞の表現型および亜集団の割合を、CD3、CD14、CD19、CD45およびCD56に対するmAbsと結合した蛍光色素を用いて、標準法により、培養0日目、5−6日目、9−10日目、14−15日目および20日目にフローサイトメトリーによって分析した。
【0056】
最初の最適化実験において、各増殖条件からの0日目および20日目の細胞を、より詳細な免疫表現型分析に付した。細胞収集中の変動を避けるために、全ての凍結サンプルを、フローサイトメトリーによりNK細胞サブセットの詳細な表現型特性化を行うために同時に解凍した。このパネルは、以下の表面抗原:CD11a(HI111)、CD3(UCHT−1)、CD7(M−T701)、CD14(MOP9)、CD16(3G8)、CD19(HIB19)、CD25(M−A251)、CD27(M−T271)、CD56(B159)、CD57(NK−1)、CD226(DX11)、NKB1(DX9)およびCD62L(DREG56)(BD Biosciences, San Jose, CA, USAから購入);CD244(2B4)(C1.7)、NKG2D(ON71)、NKp30(Z25)、NKp44(Z231)、NKp46(BAB281)(Beckman Coulter Inc., Fullerton, CA, USAから購入);NKG2A(131411)、NKG2C(134591)、KIR2DL1(143211)、KIR2DL3(180701)(R&D Systems, Minneapolis, MN, USAから購入)に対するmAbsと結合した蛍光色素を含んだ。
【0057】
フローサイトメトリー用の全ての抗体染色は、以下のプロトコールに従って行った。細胞をPBSで1回洗浄し、適当量の抗体と共に4℃で30分間インキュベートした。その後、データ収集前に、標識した細胞をPBSで洗浄し、4%PFAで固定化した。データ収集を、FACSCalibur(BD)またはCyFlow ML(Partec GmbH, Munster, Germany)で行い、データをCellQuestまたはFloMaxソフトウェアで分析した。分析に関して、CD14−CD19−リンパ球集団付近の適当なSSC/FSCゲートを用いた。NK細胞は、CD3−CD56+集団としてゲートを通した。NKT細胞およびT細胞は、CD3+CD56+およびCD3+CD56−集団としてそれぞれゲートを通した。
【0058】
各細胞表面受容体を分析するために、平均蛍光強度(MFI)値を、0日目および20日目のサンプルで計測した。受容体発現の変化を概算するために、本発明者らは、各受容体についてMFI比(MFIday20/MFIday0)を計測した。20日目のサンプルのMFIが0日目のものよりも高かったとき、MFI比は1より大きく、それは、その受容体の上方制御の相対的程度を示す。同様に、MFI比が1以下のときは、その受容体の発現が下方制御されたと解釈した。
【0059】
3.細胞媒介性細胞傷害の評価
材料および方法
最終産物の細胞傷害能を、K562細胞に対する標準的な4時間の51Cr−放出アッセイを用いてインビトロで評価した(Aktas E. et al., 2009; Alter, G. et al., 2004)。簡単には、K562標的細胞を、37℃で1時間かけて100μCiの51Crで標識し、PBSで2回洗浄し、RPMI培地に再懸濁した。100μlのRPMI培地中、全量3x104の標的細胞を、V底型の96ウェルプレートにトリプリケートで播種し、エフェクター:標的比が1:3ないし10:1となるよう適当な濃度で100μlのエフェクター細胞と共に4時間インキュベートした。上清のアリコートを、Packard Cobra Auto−Gamma 5000 Series Counting System (Meridien, CT, USA)を用いてカウントした。特異的な51Cr放出の割合を、式に当てはめて計算した:特異的放出割合=[(実験による放出−非特異的放出)/(最大放出−非特異的放出)]x100。
【0060】
NK細胞脱顆粒の分析
増殖産物を、丸底96ウェルプレート中で、37℃および5%CO2で6時間、終量200μl中に1:1比でK562標的細胞と共にインキュベートした。蛍光色素結合抗CD107a mAbまたは対応するIgG1イソ型対照を、アッセイの開始時に添加した。1時間、共にインキュベーション後、Monensin (GolgiStop, Becton Dickinson)を、1:100希釈して添加した。表面染色を、抗CD3および抗CD56 mAbsと共に氷上で30分間インキュベートして行った。その後、細胞を洗浄し、PBSに再懸濁し、フローサイトメトリーで直ちに分析した。
【0061】
4.統計的分析
データ分析、グラフの作成および統計比較を、GraphPad Prism ソフトウェア(GraphPad Software Inc. CA, USA)を用いて行った。
【0062】
結果
細胞培養バッグおよびバイオリアクターのNK細胞増殖に関する評価
NK細胞の増殖に関して閉鎖型培養システムを利用する試みにおいて、本発明者らは、初めに、5名の健常なドナー由来のPBMCを用いて、細胞培養バッグとフラスコを比較した。図1Aは、増殖期の最後での各ドナー由来のバルク細胞ならびにNKおよびNKT細胞の増殖倍率を示す。平均バルク細胞増殖は、バッグにおいて530倍であり、一方、フラスコにおいて平均1100倍の増殖が得られた。バッグ中でのNK細胞増殖は、とりわけ、5名中3名のドナーで、フラスコでの培養と比較したとき有利に見えた。しかしながら、最終産物中のNK細胞の割合を考慮したとき(図1C)、バッグ中での増殖は、フラスコ中での増殖と相関せず、培養終了時点でより低いNK細胞純度となり得ることが分かった。バッグ中の最終産物は、NK細胞を平均31%含み、一方で、フラスコの最終産物中のNK細胞割合は53%であった。
【0063】
フラスコと同程度かつ対応する収率をもたらす閉鎖型増殖システムの探索において、発明者らは、5名の健常なドナー由来のPBMCを用いて、フラスコでの増殖と比較した自動化バイオリアクターシステムの使用を評価した。発明者らは、バルク細胞の増殖を観察し(平均増殖:フラスコ:770倍、バイオリアクター:77倍)、一方で、NK細胞が優先的に増殖し、両条件下で全体に占めるNK細胞の割合を増加させた。NK細胞の増加倍率(図1B)は、5名中4名のドナーでフラスコでの増加よりも低かったが、最終産物中のこれらの亜集団の割合は、フラスコ増殖とより同程度かつ対応していた(図1D)。増殖プロトコールの最終産物は、バイオリアクター中で平均38%のNK細胞を有し、一方、フラスコ中で44%のNK細胞および16%が存在した。
【0064】
バッグについての上記の結果と比較したとき、バイオリアクターでのNK細胞の割合は、このドナー集団が、フラスコ増殖でのNK細胞の純度が悪かったという事実にもかかわらず、バッグでのNK細胞の割合よりも高かった。まとめると、上記の結果は、NK細胞の増殖はバッグでの方がより増加し、一方、その純度は、バイオリアクターでの増殖がわずかに高いことを示唆する。
【0065】
異なるドナー群の使用およびNK細胞増殖の個人差の高さ故に、バッグとバイオリアクターシステムの相対的効率を直接見いだすことは不可能であった。これらの異なる群中の5名のドナーのうち2名が、実際に同一の個人であった(ドナー1および2)という事実によって、発明者らは、増殖効率を直接的に比較する機会を得た。増殖倍率が低かったが、バイオリアクターは、フラスコ(74%)と比較して、最終産物中に同程度の割合のNK細胞を有し(64%)、バッグ(47%)での増殖よりも高かった。NKT細胞の場合には、3つ全てのシステムで5%にかなり近い割合であった。
【0066】
これらの2名のドナー由来の増殖産物を、受容体発現の変化のパターンを観察するために、さらなる表現型分析に付した。通常通り個人差があったが、異なる最終産物中の受容体発現レベルの変化は、非常に似ていた(図2Aおよび2B)。所定の受容体の上方または下方制御の程度は、培養プロトコールが用いられるかどうかに関わらず同じであることは、明確に理解され得る。
【0067】
異なるシステムの最終産物中のNK細胞が、同様の活性状態を保持し、同程度の細胞傷害性を示すかどうかを明らかにするために、発明者らは、NK感受性細胞株K562に対する最終産物の細胞傷害能を評価した(図2C)。本発明者らは、異なる条件下で増殖させた細胞調製物の細胞傷害活性の顕著な相違を見いださなかった。これらの結果は、検討した方法が、増加した細胞傷害能を有する細胞調製物を産生するのに十分であることを保証する。
【0068】
バイオリアクターでのNK細胞増殖法の検討
増殖法へのバイオリアクターの使用可能性を明らかにした後、本発明者らは、0日目からバイオリアクターで直接培養を開始するために、血液成分または末梢血全体を用いるcGMP条適合件下で、バイオリアクターでの増殖法の検討を継続した。2名の健常なドナーおよび2名のMM患者由来のPBMCを、この検討で用いた。比較のために、出発物質であるPBMCを、フラスコを並行して用いて増殖させた。図3は、バイオリアクターおよびフラスコ中の全てのドナーについてのバルク細胞およびリンパ球サブ集団の増殖曲線を示す。達した全細胞数は、バイオリアクター増殖で非常に多く、平均37.5%純度であるが、フラスコでは43%純度であった。NK細胞純度はバイオリアクターでわずかに低かったが、達したNK細胞の最終的な数は、癌の免疫療法設定における増殖したNK細胞の臨床使用を容易にするのに十分である。
【0069】
バイオリアクター中で増殖したNK細胞は、より高い細胞傷害能を示す
上記の実験において、増殖をフラスコで開始し、その後バイオリアクターに移したとき、本発明者らは、フラスコでの増殖と比較して、最終産物の細胞傷害能および表現型に相違を検出しなかった。興味深いことに、発明者らは、バイオリアクターで直接増殖を開始したとき、K562細胞に対する最終産物の細胞傷害活性が、4名中3名のドナーで、フラスコで増殖させた最終産物と比較して顕著に高かったことを見いだした(図4A)。この現象をよく調べるために、本発明者らは、K562細胞に対する脱顆粒アッセイを行い、各リンパ球サブ集団中の脱顆粒細胞の割合を測定した(図4B)。驚くことに、本発明者らは、バイオリアクター増殖からのNK細胞画分にて観察される脱顆粒の程度が、4名全てのドナーにおいてフラスコ増殖からのNK細胞よりも顕著に高いことを見いだした。同様に、NKT細胞画分の脱顆粒は、4名中3名のドナーで顕著に高かった。纏めると、これらの結果は、バイオリアクターで行われる増殖法が、NK細胞の細胞傷害能を上げる点で、より良いことを示唆する。
【0070】
バイオリアクターおよびフラスコ増殖からの最終産物の細胞傷害能の相違を説明するために、発明者らは、マルチカラーフローサイトメトリーを用いてNK細胞の詳細な表現型特性化を行った。図5は、バイオリアクターおよびフラスコ増殖からのNK細胞上の種々の表面受容体の発現を示し、全てのドナーの結果をまとめて、バイオリアクター増殖とフラスコ増殖の表現型の比較全体図を示す。概して、最終産物中のNK細SSS胞は、何れの増殖システムを用いるかに関わらず同様であって、CD11b、NKG2DおよびNKp44の発現レベルがわずかであるが、顕著に異なる。NKp44の上方制御は、上昇した細胞傷害能の鍵となる要因の1つである。
【0071】
この研究は、閉鎖型自動化システムでGMP適合品質のエフェクター細胞を生産し得る可能性を検討する試みにおいて、ヒトドナーのバルクPBMCに由来するNKおよびNKT細胞のエクスビボ増殖のための細胞培養フラスコ、バッグおよびバイオリアクターの使用を比較評価する。
【0072】
発明者らは、細胞培養フラスコ中で活性化NK細胞の選択的富化を促進するGMP適合培養培地を以前に報告した(Carlens S. et al., 2001)。そこで、本発明者らは、臨床適用可能な、エフェクター集団に富む大規模NK細胞の調製のためのGMP適合品質の自動化閉鎖型培養システムを最適化する最終工程を示し(Miller JS. et al., 1994;Luhm J. et al., 2002;Klingemann HG and Martinson J., 2004;Koehl U. et al., 2005)、および/またはフィーダー細胞株の使用(Ishikawa E. et al., 2004;Torelli GF, et al., 2005)が、既報の通り、NK細胞増殖に広く用いられていたことを示した。この研究において、本発明者らは、NK細胞含有量および抗腫瘍活性(Bordignon C. et al., 1999)の両方に関して、如何なる単離工程も用いないが、LAK(Ramsdell FJ and Golub SH., 1987)およびサイトカイン誘導性キラー(CIK)細胞(Chan JK. et al., 2006;Lu PH and Negrin RS., 1994)とは別のNK細胞に富む細胞集団をもたらすバルクPBMCを用いた。
【0073】
本発明者らは、バッグおよびバイオリアクターシステムの両方が、NK細胞を増殖させることを証明した。この研究で利用される2つの閉鎖型システムの増殖率比較および最終製品純度の全体的な比較は、バッグと比較したとき、バイオリアクターシステムが、より高い純度を有する十分量のNK細胞を提供し、さらに最終産物中のT細胞をより少なくすることを明らかにする。
【0074】
驚くことに、本発明者らは、NK細胞活性が、フラスコからの増殖産物と比較して、バイオリアクターからの増殖産物で顕著に高いことを見いだした。受容体発現レベルとK562に対するNK細胞の反応との相関関係は、CD132、CD25、CD57およびNKG2C発現レベルが、その反応と逆相関しており、NKp30、NKp44およびNKp46の発現レベルが、直接的に相関することを明らかにした。活性化NK細胞受容体NKG2Cの発現で逆相関を観察することは珍しくないが、標的K562細胞が、そのリガンドHLA−Eの発現を欠失することが公知の通りであり、この場合にはあまり意味がない(Khalil−Daher I. et al., 1999)。NK細胞応答と相関する受容体の統計的分析により、NKp44は、反応に正に相関し、フラスコ増殖と比較したとき、バイオリアクター製品で顕著に高レベルで発現されることが明らかとなった。このことは、少なくとも1つには、バイオリアクター産物の高い細胞毒性能の観察を説明し得る。他のNCRとは異なり、NKp44(Vitale M. et al., 1998)は、活性化NK細胞でのみ発現され、インビトロでのIL−2刺激後に上方制御される(Biassoni R. et al., 2002)。故に、この場合、培養中にIL−2がどのくらいの頻度で用いられているか、およびNK細胞集団の活性化がどの程度かを、代理マーカーとして示し得る。従って、NKp44の上昇した発現は、常套の細胞培養フラスコよりもバイオリアクターで行われる増殖法に機能的意義を提供する。
【0075】
実用性に関して、本発明者らは、当該全てのシステムには有利点と不利点があることを明らかにした。細胞培養フラスコでの増殖は、外部物質および汚染物質への暴露という固有の危険性がある。この危険性は、GMP適合の実験室環境で最小化されるが、閉鎖型自動化システムの使用は、それが十分量の細胞を供給する限り好まれることは明らかである。フラスコでの培養開始は、多くの細胞を必要としないが、細胞は、取り扱い不可能な数のフラスコ数になるまで、増殖中、新しいフラスコに分けられる間、ある程度の濃度内に維持されなければならない(Heiskala M. et al., 1987)。培養はまた、小さなバッグ中で少量の細胞で開始可能であり、良好な増殖をもたらすが、NK細胞の純度は、他のシステムよりも低く、細胞は、2個以上のバッグに分けられる必要がある。しかし、バッグでの増殖は、さらなる装置に投資を必要とすることなく、標準的な細胞培養実験室で容易に最適化され得る。2種以上の増殖が、インキュベーターのスペースが限界でない限り、バッグを用いて同時に行われ得て、バイオリアクターは、機械の購入のために追加の投資を必要とするが、一度に一回の増殖に用いられ得る。
【0076】
バイオリアクターは、要する時間が最小限であるために、最も実用的な方法である。しかしながら、このシステムの開始には多くの細胞が必要とされ、増殖率が低い。バイオリアクターの連続した振とう動作は、例えば均一な培養条件、サンプリングの容易さならびにpHおよび溶存酸素の測定のような良好な制御処理品質のような多くの利点を提供する、同時かつ同種の培養環境を確実にする。そのような同時培養条件の利用は、バイオリアクター中でより濃縮される細胞増殖が実行可能かどうかに関与する主な要因であるはずである。このことは、培地またはさらなる成分の無駄遣いを避け、処理全体の費用を劇的に減少させる。プロトコール毎に平均増殖曲線を用いて、本発明者らは、同程度のNK細胞数を得るために、バイオリアクターシステムは、バッグに用いる培地成分のおよそ1/10倍、およびフラスコに用いる培地成分の1/25倍を用いることを概算した。培地およびサイトカインの消費は、フラスコでよりも多い(バッグのおよそ2.5倍)。
【0077】
当然、エクスビボ培養のための同時バイオリアクターシステムの使用は、幾つかの要因が慎重に評価されることを要する。造血細胞培養は、せん断に比較的感受性であり、高せん断処理がエクスビボ増殖に不適当であるという仮定は合理的である(Nielsen LK., 1999)。従って、振盪タンク型バイオリアクター(Pierson BA. et al., 1996)または外部フィルターおよび高流速に依存するかん流培養システムは、高い効率を提供しない。バイオリアクターの最大限の利点を達成するために、培地を除去するための内部かん流フィルターを備える低せん断力生成システムの使用が望ましく(Nielsen LK., 1999)、この実験で用いるバイオリアクターシステムは、これらの予想に合致し得る。
【0078】
慎重に考慮する別の要因は、培養環境で用いられる材料である。わずかな材料のみが、効率的な造血細胞の増殖を支持し得て、洗浄、滅菌および再利用のような要因が、それらのパフォーマンスにかなり影響を及ぼす(LaIuppa JA. et al., 1997)。従って、使い捨てで、かつ予め滅菌された適当な材料の使用が好ましい。この実験で用いるバッグおよびバイオリアクターシステムの両方が、この点で生産に好適である。
【0079】
フラスコと比較するとき、観察される1つの違いは、バイオリアクターおよびバッグ増殖の最終産物中の細胞のわずかに低い生存率であった。このことは、主に、死んだ細胞が、フラスコ培養において2−3日毎に継続的に流されているという事実のためであり、培地を交換するとき、閉鎖型システムではかかる操作を伴わない。しかし、投与前に、培養終了時のGMP適合品質の洗浄工程を常に利用可能である。
【0080】
問題とする全てのシステムは、特定の実施上の有利点および不利点がある。細胞培養フラスコ中のNK細胞の増殖は、外部物質および汚染物質への暴露の固有の危険性を有する。この危険性は、GMP適合実験室環境で最小化されるが、閉鎖型自動化システムの使用は、それが十分量の細胞を供給する限り、間違いなく好ましい。フラスコでの培養は、非常に少ない数の細胞で開始できるが、細胞は、取り扱い不可能なフラスコ数になるまで、増殖中、新しいフラスコに分けられる間、ある程度の濃度内に維持されなければならない。
【0081】
培養はまた、小さなバッグ中で少量の細胞で開始可能で良好な増殖をもたらすが、NK細胞およびNKT細胞の純度は、他のシステムよりも低く、細胞は、2個以上のバッグに分けられる必要がある。バッグでの増殖は、さらなる装置に投資を必要とすることなく、標準的な細胞培養実験室で容易に最適化され得る。2個以上の増殖が、インキュベーターのスペースが限界でない限り、バッグを用いて同時に行われ得て、バイオリアクターは、一度に一回の増殖に用いられ得る。
【0082】
結論として、本明細書に記載の結果は、適用可能な免疫療法適用に予想される使用のための大量の高度に活性化されたエフェクター細胞が、GMP適合条件下で、閉鎖型培養システムで生産され得ることを、明確に証明する。
【0083】
特定の態様が明細書中に詳細に記載されるが、これは説明のみを目的とし、例示として記載されたもので、添付の特許請求の範囲を限定することを意図しない。特に、種々の置換、変更および修正が、特許請求の範囲によって定義される本発明の精神および範囲から逸脱することなく本発明にされ得ることは、本発明者らにより考慮されている。
【0084】
参考文献
Aktas E. et al. Relationship between CD107α expression and cytotoxic activity. Cell Immunol. 2009, 254 pp.149−54.
Alici E. et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP−compliant components. Blood 2008, 111 pp. 3155−62.
Alter G. et al. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004, 294 pp.15−22.
Barkholt L. et al. Safety analysis of ex vivo expanded NK and NK−like T cells administered to cancer patients: a Phase I clinical study. Immunotherapy. 2009, 1 pp. 753−764.
Biassoni R. et al. Human natural killer receptors and their ligands. Curr Protoc Immunol. 2002; Chapter 14: Unit 14 10.
Bordignon C. et al. Cell therapy: achievements and perspectives. Haematologica. 1999, 84 pp.1110−1149.
Carlens S. et al. A new method for in vitro expansion of cytotoxic human CD3−CD56+ natural killer cells. Hum Immunol. 2001, 62 pp.1092−8.
Chan JK. et al. Enhanced killing of primary ovarian cancer by retargeting autologous cytokine−induced killer cells with bispecific antibodies: a preclinical study. Clin Cancer Res. 2006, 12 pp.1859−1867.
Grimm EA. et al. Lymphokine−activated killer cell phenomenon. Lysis of natural killer−resistant fresh solid tumor cells by interleukin 2−activated autologous human peripheral blood lymphocytes. J Exp Med. 1982,155 pp.1823−1841.
Guimaraes F. et al. Evaluation of ex vivo expanded human NK cells on antileukemia activity in SCID−beige mice. Leukemia. 2006, 20 pp. 833−839.
Guven H. et al. Expansion of natural killer (NK) and natural killer−like T (NKT)−cell populations derived from patients with B−chronic lymphocytic leukemia (B−CLL): a potential source for cellular immunotherapy. Leukemia 2003, 17 pp.1973−80.
Heiskala M. et al. Mechanism of cell contact−mediated inhibition of natural killer activity. J Immunol. 1987, 139 pp.1414−1418.
Ishikawa E. et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004, 24 pp.1861−1871.
Khalil−Daher I. et al. Role of HLA−G versus HLA−E on NK function: HLA−G is able to inhibit NK cytolysis by itself. J Reprod Immunol. 1999, 43 pp.175−182.
Klingemann HG and Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy 2004, 6 pp. 15−22.
Koehl U. et al. Ex vivo expansion of highly purified NK cells for immunotherapy after haploidentical stem cell transplantation in children. Klin Padiatr. 2005, 217 pp. 345−350.
LaIuppa JA. et al. Culture materials affect ex vivo expansion of hematopoietic progenitor cells. J Biomed Mater Res. 1997, 36 pp. 347−359.
Lu PH and Negrin RS. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J Immunol. 1994, 153 pp.1687−1696.
Luhm J. et al. Large−scale generation of natural killer lymphocytes for clinical application. J Hematother Stem Cell Res. 2002, 11 pp. 651−7.
Miller JS. et al. Large scale ex vivo expansion and activation of human natural killer cells for autologous therapy. Bone Marrow Transplant 1994, 14 pp. 555−62.
Nielsen LK. Bioreactors for hematopoietic cell culture. Annu Rev Biomed Eng. 1999, 1 pp. 129−152.
Pierson BA. et al. Production of human natural killer cells for adoptive immunotherapy using a computer−controlled stirred−tank bioreactor. J Hematother 1996, 5 pp. 475−83.
Ramsdell FJ and Golub SH. Generation of lymphokine−activated killer cell activity from human thymocytes. J Immunol. 1987,139 pp.1446−1453.
Rosenberg S. Lymphokine−activated killer cells: a new approach to immunotherapy of cancer. J Natl Cancer Inst. 1985, 75 pp. 595−603.
Sutlu T and Alici E. Natural killer cell−based immunotherapy in cancer: current insights and future prospects. J Intern Med. 2009, 266 pp.154−181.
Torelli GF. et al. Expansion of natural killer cells with lytic activity against autologous blasts from adult and pediatric acute lymphoid leukemia patients in complete hematologic remission. Haematologica 2005, 90 pp. 785−792.
Vitale M. et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non−major histocompatibility complex−restricted tumor cell lysis. J Exp Med. 1998, 187 pp. 2065−2072.
US 10/242,7881
【図1A】

【図1B】
【図1C】
【図1D】
【図2A】
【図2B】
【図2C】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【技術分野】
【0001】
技術分野
本発明は、細胞培養および免疫療法、特に、治療に使用するためのナチュラルキラー細胞(以下、NK細胞と記す)およびナチュラルキラー様T細胞(以下、NKT細胞と記す)細胞の大規模増殖および同時活性化に関する。該細胞を、閉鎖型自動化培養システム、例えばバイオリアクターを用いて増殖させる。
【背景技術】
【0002】
発明の背景
癌に対する細胞免疫療法の使用は、1980年代半ばにリンホカイン活性化キラー(LAK)細胞が導入されて以来、徹底して調査されている(Grimm EA. et al., 1982; Rosenberg S., 1985)。
【0003】
最も研究された手法の1つは、移植片対腫瘍(GvT)効果をもたらし腫瘍細胞を殺す可能性のある、自己(autologous)または同種異系(allogeneic)の細胞傷害性エフェクターの養子移植であった。可能性のある抗腫瘍効果を有する種々のエフェクター集団のうち、NKおよびNKT細胞は、それらの高い細胞傷害能により注目されている(Sutlu T and Alici E., 2009)。
【0004】
NKおよびNKT細胞は通常、末梢血単核球細胞(PBMC)およびLAK細胞のようなエフェクター細胞調製物中に少数しか存在しない。故に、健常なドナー(Carlens S. et al., 2001 および US 10/242,788)ならびにB細胞慢性リンパ性白血病を有する患者(Guven H. et al., 2003)および多発性骨髄腫(MM)を発症した患者(Alici E. et al., 2008)由来のPBMCを用いて細胞培養フラスコ中、ポリクローナルNK細胞およびNKT細胞の増殖を可能とする、現在の良好な製造法であるcGMP適合の成分を含む方法が開発されている。これらの細胞は、臨床の場で評価される可能性を開く、ヒト腫瘍のインビトロモデルおよび実験モデル(Guimaraes F. et al., 2006)において、新鮮なヒト腫瘍細胞に対して特定の細胞傷害活性を発揮することが示されている。しかしながら、従来のフラスコベースの培養は、多大な労力を要し、かつ扱いにくく、故に、実際に扱える細胞数に限りがある。エフェクター細胞調製物を対象とする既報のプロトコール(例えば、Miller JS. et al., 1994;Pierson BA. et al., 1996;Luhm J. et al., 2002;Klingermann HG and Martinson J., 2004)は、培養前のNK前駆体またはCD56の単離、およびフィーダー細胞またはcGMP不適合成分の使用のような工程も含む。これらの不都合は、既報のプロトコールが大規模な臨床研究を支持するのに至適ではなく、かつ実施不能であることを示す。
【0005】
さらに、細胞培養フラスコ中でのNK細胞の増殖は、外部物質への暴露および汚染の固有のリスクを有する。このリスクは、GMP適合の実験室環境で最小化されるが、閉鎖型自動化システムの使用は、それが十分量の細胞を供給する限り間違いなく好ましい。
【0006】
造血細胞はせん断に比較的感受性であるため、高せん断処理は、エクスビボ増殖には好ましくない(Nielsen, 1999)との仮定は当然である。故に、外部フィルターおよび高流速に依存する振盪タンク型バイオリアクター(Pierson. et al., 1996)またはかん流培養システムは、高効率を達成する可能性が低い。
【0007】
癌の処置用の免疫療法に基づくNK細胞およびNKT細胞を用いる多くの有望な方法がある。しかしながら、GMP適合の閉鎖系でのこれらのエフェクター細胞のエクスビボ増殖および活性化は、臨床的な使用機会を増加させるための非常に重要な因子である。
【0008】
NK細胞をエクスボビで増殖および/または活性化するための他の研究者による試みもされており、精製/静止に用いる複数の処理オプションがあり、短時間でまたは高度に精製され、かつ長時間活性化されるNK細胞が研究されている。これらの研究は、NK細胞注入が良好な耐容性を示し、部分的に有効であることを報告する。しかし、エフェクター細胞調製に一般的に用いられるプロトコールは、培養前のNK前駆体またはCD56の単離、およびフィーダー細胞および/またはcGMP不適合成分の使用のようなさらなる工程を含む。これらの不都合は、かかるプロトコールを、GMP適合生産に適当ではなく、大規模な臨床研究を支持することを不可能にする。
【0009】
上記の報告の詳細な評価は、NK細胞の最適化エクスビボ増殖のための自動化法の必要性を示す。常套のフラスコベースの培養の1つの問題は、規模に関し、すなわち細胞数がフラスコ操作の煩雑さのために制限されることに関する。さらに、細菌感染の危険性は、該システムが培地交換や細胞の分配時に周囲環境に曝されるために、極めて高い。解決されるべきさらなる問題には、費用対効果が高く、取り扱いが容易で、明確に定義されたcGMP品質の成分を含む、エフェクター細胞の増殖法の開発が含まれる。好ましくは、培養システムはまた、動物由来成分およびフィーダー細胞を含まない。
【発明の概要】
【0010】
発明の概要
本発明は、細胞治療薬として使用されるべき、NK細胞およびNKT細胞の大規模増殖および同時活性化のための閉鎖型システムに関する。特に、本発明は、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の大規模増殖および同時活性化のための方法であって、該増殖細胞が、同一または類似の条件下で、例えばフラスコのような常套法を用いて培養された細胞と比較して、増加した細胞傷害性を示す方法を開示する。本発明の方法は、発明の詳細な説明、非限定的実施例および特許請求の範囲にさらに開示され得る。
【0011】
本発明者らは、閉鎖型システムを用いる大規模NK細胞増殖の実現可能性を調査した。2つの異なる閉鎖型システム(細胞培養バッグおよび自動化バイオリアクター)を、NK細胞の大規模生産が免疫療法に用いられるのを可能とする自動化GMP適合プロトコールを開発する目的で、健常なドナーおよび多発性骨髄腫(MM)の患者由来のPBMCを用いて、常套の細胞培養フラスコと比較して評価した。
【0012】
同時に、本発明者らは、癌患者の第I相臨床試験での同種異型処置におけるこの細胞薬の安全性評価を成功裏に完了した(Barkholt L. et al., 2009)。
【0013】
本発明の第一の態様によれば、特定の細胞タイプの大規模増殖および同時活性化のための方法を提供し、ここで、閉鎖型細胞培養系を用い、この方法によって得られた増殖した細胞は、インビトロの細胞傷害性試験で決定される通り増加した細胞傷害性を示す。好ましくは、該細胞型は、表現型CD3−CD56+のNK細胞および/または表現型CD3+CD56+のNKT細胞である。
【0014】
好ましい態様によれば、本発明の方法は、該細胞を、血清、インターロイキン−2(IL−2)および抗CD3抗体を補充した増殖培地を含む該閉鎖システムに添加し;該細胞を、増殖した細胞集団の少なくとも35%が活性化NK細胞およびNKT細胞となるまで振盪(agitation)および加熱して該システム内で増殖させ;該増殖した細胞が、インビトロ細胞傷害性試験で決定される通り増加した細胞傷害性を示す工程を含む。
【0015】
記載した方法の一態様によれば、血清は、ヒト血清および自己血清からなる群から選択される。培地には、約50ないし約1500U/mlのIL−2、約1ないし約50ng/mlの抗CD3抗体および約1ないし約40%の血清を補充する。
【0016】
別の態様によれば、振盪および加熱を以下の条件下で行う:約36−40℃の温度;約4.7−5.1%のCO2濃度;および、細胞が閉鎖型細胞システムの表面に付着できる速度および角度での穏やかな振盪。振盪を、約4−8/分の振盪速度、および約4−8°の振盪角度で行う。
【0017】
増殖は、好ましくは、全細胞数が、少なくとも10倍に増すか、または増殖した細胞集団の少なくとも約50%が活性化NK細胞およびNKT細胞それぞれを含むまで行われる。
【0018】
該細胞を増殖させるために用いる細胞サンプルは、好ましくは、末梢血、細胞株またはサイトカインで刺激した末梢血のサンプルである。最も好ましくは、細胞サンプルは、末梢血単核球細胞(PBMC)サンプルである。
【0019】
該方法の一態様によれば、細胞サンプルを健常な対象から集める。
【0020】
別の態様によれば、細胞サンプルを、腫瘍を有する対象、好ましくは血液系腫瘍および固形腫瘍からなる群から選択される腫瘍を有する対象から集める。
【0021】
別の態様によれば、細胞は、主に、表現型CD3−CD56+のNK細胞で構成される。
【0022】
閉鎖型細胞システムに最初に添加される細胞の濃度は、好ましくは、増殖培地1ml当たり、約0.5x106ないし約2x106である。
【0023】
本発明の一態様において、本発明の方法は、1日当たり、全培養量の約50%に相当する量の血清およびIL−2を補充した培地を添加し、該閉鎖型細胞システムから1日当たりおよそ同量の増殖培地を捨てる工程をさらに含み、ここで該工程は、全細胞密度が、最初の細胞密度の少なくとも約1.5倍に増大したときに行われる。
【0024】
好ましくは、該工程は、全細胞密度が、最初の細胞密度から少なくとも約300%増大したときに行われ、その場合は、全培養量の約75%を交換する。
【0025】
別法として、該工程は、全細胞密度が、最初の細胞密度から少なくとも約500%増大したときに行われ、その場合は、全培養量の約100%を交換する。
【0026】
さらに別の態様によれば、上記の態様の1つ以上を自由に組合せて、細胞を少なくとも約10日間インキュベートする。
【0027】
一態様によれば、閉鎖型細胞システムは予め滅菌したバッグである。
【0028】
別の態様によれば、該閉鎖型細胞システムは、バイオリアクターである。
【0029】
本発明の別の態様は、増加した細胞傷害性を示す、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液である。
【0030】
好ましくは、これらの細胞は、フラスコ中で増加させた細胞と比較して、インビトロ細胞傷害性試験で決定される通り、増加した細胞傷害性を示す。
【0031】
本発明の表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液中、増殖した細胞集団の少なくとも35%、好ましくは少なくとも50%は、活性化NK細胞からなる。
【0032】
本明細書中、バイオリアクターは、再利用可能な自動チャンバーシステムであって、生物学的に活性な環境を支持する何らかの装置またはシステムを意味し得る。記載した本発明の範囲内において、用語バイオリアクターは、細胞培養に関して、細胞または組織を増殖させることを意図する装置またはシステムを意味する。バイオリアクターシステムの非限定的例は、GE−Healthcareから提供されるWave bioreactor system 2/10であるが、当業者は、他の供給者から提供される別のバイオリアクターを利用可能であることを理解し得るか、または本発明の方法を実行可能なバイオリアクターを構築可能である。
【0033】
本明細書中、閉鎖型システムは、さらなる処理工程を経ることなくエクスビボで効率的かつ迅速に細胞を増殖させ得る中央の細胞培養バッグからなる細胞増殖チャンバーシステムである。
【0034】
用語“細胞傷害性”は、細胞に対する傷害性を意味する。傷害性物質の例には、化学物質、または細胞傷害性T細胞、NK細胞およびNKT細胞のような細胞傷害性リンパ球のような免疫細胞が含まれる。NK細胞およびNKT細胞は、それらの高い細胞傷害能により注目される。
【0035】
当業者は、利用可能な方法を用いて細胞傷害性を決定できる。細胞が増加した細胞傷害性を示すかどうかを決定する1つの方法は、標準的な4時間の51Cr−放出アッセイを用いて、K562細胞に対する細胞媒介性細胞傷害のインビトロ分析を用いることである。あるいは、脱顆粒アッセイを用い得る。これらの両方法を、以下の実施例に記載する。
【0036】
本明細書中、用語“活性化”および“活性化NK細胞”は、活性化シグナルを受容したNK細胞を意味する。活性化NK細胞は、MHCクラスI発現が低下している細胞を殺し得る。
【0037】
用語「NK細胞およびNKT細胞の“同時活性化”」は、細胞が、実質的に同時に、好ましくは、同じ細胞培養において、活性化されることを意味する。
【0038】
それらの強力な細胞傷害活性および自己反応性の可能性を考慮して、NK細胞活性は、厳密に制御される。MHCクラスI発現を喪失したか、またはそれが異常な細胞を殺すために、NK細胞は活性化される必要がある。NK細胞は、種々の形態をとり得る活性化シグナルを受容しなければならず、中でもサイトカイン、Fc−受容体、活性化受容体および阻害性受容体が最も重要である。
【0039】
本明細書中、用語“細胞増殖”は、一連の細胞分裂過程を経て、故に培養中に存在する細胞数が増大する細胞の培養に関する。故に、用語“NK細胞増殖”は、一連の細胞分裂過程を経て、故に培養中に存在する細胞数が増大するNK細胞の培養に関する。用語“増殖したNK細胞”は、NK細胞増殖を介して得られたNK細胞に関する。より具体的には、一態様において、用語“増殖したNK細胞”は、特定のcGMPグレードの環境およびcGMPグレードの培地中で増殖される、慢性的に活性化したCD3−CD56+細胞ならびに表現型CD3+CD56+のNKT細胞のポリクローナル群に関する。
【0040】
本明細書中、用語“エフェクター細胞”は、免疫系の細胞のような、刺激に応答して特定の機能を果たす細胞に関する。一態様において、エフェクター細胞は、細胞傷害能(すなわち、他の細胞に細胞死を誘導する能力)を有するリンパ球の一種である。別の態様は、分泌型抗体に積極的に関与するリンパ球である。エフェクター細胞の非限定的例は、NK細胞、T細胞およびNKT細胞である。
【図面の簡単な説明】
【0041】
図面の簡単な説明
【図1】図1A−Dは、フラスコおよびバッグ(1A)ならびにフラスコおよびバイオリアクター(1B)における増殖倍の比較、ならびにフラスコおよびバッグ(1C)ならびにフラスコおよびバイオリアクター(1D)における最終産物の純度を示す図である。
【図2】図2A−Cは、異なる増殖プロトコールで増殖させた細胞の表現型(AおよびB)および細胞傷害活性(C)の比較を示す図である。
【図3】図3A−Dは、フラスコと比較して、バイオリアクターで開始した細胞培養の増殖を示す図である。
【図4】図4A−Bは、バイオリアクターおよびフラスコそれぞれで直接増殖させた細胞の機能的比較を示す図である。
【図5】図5は、バイオリアクターおよびフラスコそれぞれで直接増殖させたNK細胞の表現型の比較を示す。
【発明を実施するための形態】
【0042】
発明の詳細な説明
本発明を記載する前に、本発明の範囲は、添付の特許請求の範囲およびその均等物でのみ限定され得るから、本明細書で用いる専門用語は、特定の態様を記載する目的でのみ用いられ、限定すべきことを意図しないことが、理解されるべきである。
【0043】
他に特に記載がなければ、本明細書で用いる用語および技術用語は、本発明が属する分野の当業者に通常理解される意味を有することを意図する。
【0044】
本明細書および添付の特許請求の範囲に用いられる、単数形“a”、“an”および“the”は、文中に他に明記されない限り、複数形も含むことに留意すべきである。
【0045】
また、用語“約”は、適用されるとき、所定の値の+/−2%、好ましくは、+/−5%、最も好ましくは、+/−10%の数値の偏差を示すために用いる。
【0046】
悪性疾患に罹患した患者に対するNK細胞のエクスビボ増殖および再注入は、かかる疾患に対抗する新規かつ可能性のある興味深い治療方法を提供する。このための必要条件は、臨床用途の要求を満たすNK細胞の増殖および使用の可能性である。本発明者らは、驚くべきことに、臨床適用のために大規模でNK細胞を増殖させることができた。従って、臨床で適用可能なエフェクター集団に富むNKおよびNKT細胞の大規模製造のためのGMP適合自動化閉鎖型培養システムを提供する。
【0047】
以下の実施例に示す通り、本発明の方法は、満足できる細胞傷害能(すなわち、他の細胞の細胞死を誘導する能力)を有するNK細胞を増殖および活性化させる。他の細胞に対するNK細胞の細胞傷害性は、例えば、活性化NK細胞に暴露する前後で細胞を計数する従来の方法により、容易に測定可能である。かかる方法は、当業者によく知られている。
【実施例】
【0048】
実施例
1.末梢血由来のNK細胞およびNKT細胞のエクスビボ増殖
材料および方法
末梢血単核球細胞(PBMC)のサンプリングおよび単離
バフィーコート、末梢血および血液成分(apheresis product)を、Karolinska University Hospital, Huddingeの血液バンクに登録された健常なドナーまたはMM患者から得た。実験プロトコールは、倫理委員会の承認を得た。
【0049】
末梢血単核球細胞(PBMC)を、Lymphoprep (Nyegaard, Oslo, Norway)を用いる勾配遠心分離により単離した。PBMCを、リン酸緩衝生理食塩水(PBS)(Gibco, Grand Island, NY, USA)で2回洗浄し、細胞の生存を、トリパンブルー色素排除試験により評価した。
【0050】
増殖培地および細胞計数
全てのシステムに関して、5%ヒト血清(Biowhittaker−Cambrex, Walkersville, MD, USA)および500U/ml rhIL−2(Proleukin(商標), Novartis Pharmaceuticals, East Hanover, NJ, USA)を添加したCellGro SCGM血清不含有培地(CellGenix, Freiburg, Germany)を増殖培地として用いた。培養開始時、培地に、モノクローナル抗CD3抗体(Orthoclone OKT−3, Ortho Biotech, Raritan, NJ, USA)を終濃度10ng/mlでさらに添加した。全細胞数を、培養0日、5−6日、9−10日、14−15日および21日目にトリパンブルー色素を用いて細胞を染色して評価した。最終製品を、安全性、純度および同一性(細胞生存能および表現型)について評価した。細胞の絶対数を、全細胞数とフローサイトメトリー(BD FacsCalibur; BD Biosciences, San Jose, CA, USA)により決定されたこれらの亜集団の割合を乗ずることにより計算した。
【0051】
細胞培養フラスコでの増殖
細胞培養フラスコ中での細胞傷害性細胞の増殖のための培養条件は、健常な個体由来のPBMC(Carlens S. et al., 2001)に対して予め最適化した。簡単には、PBMCを、T25フラスコ(TPP, Trasadingen, Switzerland)中で、初め0.5x106細胞/mlの濃度で培養した。5日後、培養を、培養が終了するまでの間、5%ヒト血清およびIL−2(500U/ml)を含むがOKT−3を含まない新鮮な培地を2−3日毎に補充して行った。細胞増殖の接触阻止を阻止するため(Heiskala M. et al., 1987)、細胞を、より大きなフラスコ(T75 または T150: TPP, Trasadingen, Switzerland)に移すか、または必要なとき、複数のフラスコに移した。培地の補充において、細胞濃度を、10日目までは0.5x105細胞/mlに、10日目以降は1x106細胞/mlに合わせた。時には、細胞の一部を、扱いやすいフラスコ数に維持するために凍結した。
【0052】
Waveのバイオリアクターシステムでの増殖
Waveのバイオリアクターは、温度およびCO2が制御された、振とう加熱プラットホーム上に置く使い捨ての滅菌バッグ内で細胞を増殖させる細胞培養システムである。発明者らは、Waveのバイオリアクターシステム 2/10 (GE Healthcare, Somerset, NJ, USA)を用いた。このシステムでの本発明者らの以前の結果は、低容量および少数の細胞で培養を開始したとき不十分な効率を示した。しかし、ドナーの末梢血サンプル中の細胞数は、該バイオリアクターで直接培養を開始するのを可能にしない。故に、本発明者らは、最初の最適化実験においてフラスコ中で培養を開始し、十分な細胞数に達した時点のおよそ5日目に該細胞をバイオリアクターに移した。この日のバイオリアクター培養は、800ml中に、2x106細胞/mlで開始した。
【0053】
最後の立証実験において、ドナー由来の末梢血全血または血液成分を得て、培養を0日目からバイオリアクターで直接開始した。バイオリアクターの設定は、全ての時点で以下の通りであった:温度:37℃、CO2:5%、気流:0.1、振とう速度:6/分、振とう角度:6°。細胞を1日おきにサンプリングし、計数し、さらなる培地供給を、細胞密度が3x106細胞/mlに達するまで行わなかった。以降、培養物に1日当たり500mlの培地を供給した(50ml/回)。細胞が6x106細胞/mlの密度に達したとき、培地供給を750ml/日に増し、1x107細胞/mlに達した後に1000ml/日に増した。
【0054】
Vuelife(商標)バッグでの増殖
Vuelife(商標)(American Fluoroseal Corporation, MD, USA)は、生物学的、免疫学的および化学的に不活性なフッ素化エチレン−プロピレン製の滅菌細胞培養バッグである。それは、高度にガス透過性であり、光学的に透明である。Vuelife バッグ中での培養を、72mlのVuelife バッグを用いて、培地60ml中に5x105細胞/mlで開始した。該バッグを、37℃および5%CO2で加湿インキュベーター中でインキュベートした。新鮮な培地を2−3日毎に添加して、10日目まで1x106細胞/mlの濃度に調節し、10日目以降、2x106細胞/mlの濃度に調節した。必要なとき、細胞をより大きなバッグに分割した。
【0055】
2.フローサイトメトリーによるリンパ球サブセットおよび
表現型の分析
材料および方法
細胞の表現型および亜集団の割合を、CD3、CD14、CD19、CD45およびCD56に対するmAbsと結合した蛍光色素を用いて、標準法により、培養0日目、5−6日目、9−10日目、14−15日目および20日目にフローサイトメトリーによって分析した。
【0056】
最初の最適化実験において、各増殖条件からの0日目および20日目の細胞を、より詳細な免疫表現型分析に付した。細胞収集中の変動を避けるために、全ての凍結サンプルを、フローサイトメトリーによりNK細胞サブセットの詳細な表現型特性化を行うために同時に解凍した。このパネルは、以下の表面抗原:CD11a(HI111)、CD3(UCHT−1)、CD7(M−T701)、CD14(MOP9)、CD16(3G8)、CD19(HIB19)、CD25(M−A251)、CD27(M−T271)、CD56(B159)、CD57(NK−1)、CD226(DX11)、NKB1(DX9)およびCD62L(DREG56)(BD Biosciences, San Jose, CA, USAから購入);CD244(2B4)(C1.7)、NKG2D(ON71)、NKp30(Z25)、NKp44(Z231)、NKp46(BAB281)(Beckman Coulter Inc., Fullerton, CA, USAから購入);NKG2A(131411)、NKG2C(134591)、KIR2DL1(143211)、KIR2DL3(180701)(R&D Systems, Minneapolis, MN, USAから購入)に対するmAbsと結合した蛍光色素を含んだ。
【0057】
フローサイトメトリー用の全ての抗体染色は、以下のプロトコールに従って行った。細胞をPBSで1回洗浄し、適当量の抗体と共に4℃で30分間インキュベートした。その後、データ収集前に、標識した細胞をPBSで洗浄し、4%PFAで固定化した。データ収集を、FACSCalibur(BD)またはCyFlow ML(Partec GmbH, Munster, Germany)で行い、データをCellQuestまたはFloMaxソフトウェアで分析した。分析に関して、CD14−CD19−リンパ球集団付近の適当なSSC/FSCゲートを用いた。NK細胞は、CD3−CD56+集団としてゲートを通した。NKT細胞およびT細胞は、CD3+CD56+およびCD3+CD56−集団としてそれぞれゲートを通した。
【0058】
各細胞表面受容体を分析するために、平均蛍光強度(MFI)値を、0日目および20日目のサンプルで計測した。受容体発現の変化を概算するために、本発明者らは、各受容体についてMFI比(MFIday20/MFIday0)を計測した。20日目のサンプルのMFIが0日目のものよりも高かったとき、MFI比は1より大きく、それは、その受容体の上方制御の相対的程度を示す。同様に、MFI比が1以下のときは、その受容体の発現が下方制御されたと解釈した。
【0059】
3.細胞媒介性細胞傷害の評価
材料および方法
最終産物の細胞傷害能を、K562細胞に対する標準的な4時間の51Cr−放出アッセイを用いてインビトロで評価した(Aktas E. et al., 2009; Alter, G. et al., 2004)。簡単には、K562標的細胞を、37℃で1時間かけて100μCiの51Crで標識し、PBSで2回洗浄し、RPMI培地に再懸濁した。100μlのRPMI培地中、全量3x104の標的細胞を、V底型の96ウェルプレートにトリプリケートで播種し、エフェクター:標的比が1:3ないし10:1となるよう適当な濃度で100μlのエフェクター細胞と共に4時間インキュベートした。上清のアリコートを、Packard Cobra Auto−Gamma 5000 Series Counting System (Meridien, CT, USA)を用いてカウントした。特異的な51Cr放出の割合を、式に当てはめて計算した:特異的放出割合=[(実験による放出−非特異的放出)/(最大放出−非特異的放出)]x100。
【0060】
NK細胞脱顆粒の分析
増殖産物を、丸底96ウェルプレート中で、37℃および5%CO2で6時間、終量200μl中に1:1比でK562標的細胞と共にインキュベートした。蛍光色素結合抗CD107a mAbまたは対応するIgG1イソ型対照を、アッセイの開始時に添加した。1時間、共にインキュベーション後、Monensin (GolgiStop, Becton Dickinson)を、1:100希釈して添加した。表面染色を、抗CD3および抗CD56 mAbsと共に氷上で30分間インキュベートして行った。その後、細胞を洗浄し、PBSに再懸濁し、フローサイトメトリーで直ちに分析した。
【0061】
4.統計的分析
データ分析、グラフの作成および統計比較を、GraphPad Prism ソフトウェア(GraphPad Software Inc. CA, USA)を用いて行った。
【0062】
結果
細胞培養バッグおよびバイオリアクターのNK細胞増殖に関する評価
NK細胞の増殖に関して閉鎖型培養システムを利用する試みにおいて、本発明者らは、初めに、5名の健常なドナー由来のPBMCを用いて、細胞培養バッグとフラスコを比較した。図1Aは、増殖期の最後での各ドナー由来のバルク細胞ならびにNKおよびNKT細胞の増殖倍率を示す。平均バルク細胞増殖は、バッグにおいて530倍であり、一方、フラスコにおいて平均1100倍の増殖が得られた。バッグ中でのNK細胞増殖は、とりわけ、5名中3名のドナーで、フラスコでの培養と比較したとき有利に見えた。しかしながら、最終産物中のNK細胞の割合を考慮したとき(図1C)、バッグ中での増殖は、フラスコ中での増殖と相関せず、培養終了時点でより低いNK細胞純度となり得ることが分かった。バッグ中の最終産物は、NK細胞を平均31%含み、一方で、フラスコの最終産物中のNK細胞割合は53%であった。
【0063】
フラスコと同程度かつ対応する収率をもたらす閉鎖型増殖システムの探索において、発明者らは、5名の健常なドナー由来のPBMCを用いて、フラスコでの増殖と比較した自動化バイオリアクターシステムの使用を評価した。発明者らは、バルク細胞の増殖を観察し(平均増殖:フラスコ:770倍、バイオリアクター:77倍)、一方で、NK細胞が優先的に増殖し、両条件下で全体に占めるNK細胞の割合を増加させた。NK細胞の増加倍率(図1B)は、5名中4名のドナーでフラスコでの増加よりも低かったが、最終産物中のこれらの亜集団の割合は、フラスコ増殖とより同程度かつ対応していた(図1D)。増殖プロトコールの最終産物は、バイオリアクター中で平均38%のNK細胞を有し、一方、フラスコ中で44%のNK細胞および16%が存在した。
【0064】
バッグについての上記の結果と比較したとき、バイオリアクターでのNK細胞の割合は、このドナー集団が、フラスコ増殖でのNK細胞の純度が悪かったという事実にもかかわらず、バッグでのNK細胞の割合よりも高かった。まとめると、上記の結果は、NK細胞の増殖はバッグでの方がより増加し、一方、その純度は、バイオリアクターでの増殖がわずかに高いことを示唆する。
【0065】
異なるドナー群の使用およびNK細胞増殖の個人差の高さ故に、バッグとバイオリアクターシステムの相対的効率を直接見いだすことは不可能であった。これらの異なる群中の5名のドナーのうち2名が、実際に同一の個人であった(ドナー1および2)という事実によって、発明者らは、増殖効率を直接的に比較する機会を得た。増殖倍率が低かったが、バイオリアクターは、フラスコ(74%)と比較して、最終産物中に同程度の割合のNK細胞を有し(64%)、バッグ(47%)での増殖よりも高かった。NKT細胞の場合には、3つ全てのシステムで5%にかなり近い割合であった。
【0066】
これらの2名のドナー由来の増殖産物を、受容体発現の変化のパターンを観察するために、さらなる表現型分析に付した。通常通り個人差があったが、異なる最終産物中の受容体発現レベルの変化は、非常に似ていた(図2Aおよび2B)。所定の受容体の上方または下方制御の程度は、培養プロトコールが用いられるかどうかに関わらず同じであることは、明確に理解され得る。
【0067】
異なるシステムの最終産物中のNK細胞が、同様の活性状態を保持し、同程度の細胞傷害性を示すかどうかを明らかにするために、発明者らは、NK感受性細胞株K562に対する最終産物の細胞傷害能を評価した(図2C)。本発明者らは、異なる条件下で増殖させた細胞調製物の細胞傷害活性の顕著な相違を見いださなかった。これらの結果は、検討した方法が、増加した細胞傷害能を有する細胞調製物を産生するのに十分であることを保証する。
【0068】
バイオリアクターでのNK細胞増殖法の検討
増殖法へのバイオリアクターの使用可能性を明らかにした後、本発明者らは、0日目からバイオリアクターで直接培養を開始するために、血液成分または末梢血全体を用いるcGMP条適合件下で、バイオリアクターでの増殖法の検討を継続した。2名の健常なドナーおよび2名のMM患者由来のPBMCを、この検討で用いた。比較のために、出発物質であるPBMCを、フラスコを並行して用いて増殖させた。図3は、バイオリアクターおよびフラスコ中の全てのドナーについてのバルク細胞およびリンパ球サブ集団の増殖曲線を示す。達した全細胞数は、バイオリアクター増殖で非常に多く、平均37.5%純度であるが、フラスコでは43%純度であった。NK細胞純度はバイオリアクターでわずかに低かったが、達したNK細胞の最終的な数は、癌の免疫療法設定における増殖したNK細胞の臨床使用を容易にするのに十分である。
【0069】
バイオリアクター中で増殖したNK細胞は、より高い細胞傷害能を示す
上記の実験において、増殖をフラスコで開始し、その後バイオリアクターに移したとき、本発明者らは、フラスコでの増殖と比較して、最終産物の細胞傷害能および表現型に相違を検出しなかった。興味深いことに、発明者らは、バイオリアクターで直接増殖を開始したとき、K562細胞に対する最終産物の細胞傷害活性が、4名中3名のドナーで、フラスコで増殖させた最終産物と比較して顕著に高かったことを見いだした(図4A)。この現象をよく調べるために、本発明者らは、K562細胞に対する脱顆粒アッセイを行い、各リンパ球サブ集団中の脱顆粒細胞の割合を測定した(図4B)。驚くことに、本発明者らは、バイオリアクター増殖からのNK細胞画分にて観察される脱顆粒の程度が、4名全てのドナーにおいてフラスコ増殖からのNK細胞よりも顕著に高いことを見いだした。同様に、NKT細胞画分の脱顆粒は、4名中3名のドナーで顕著に高かった。纏めると、これらの結果は、バイオリアクターで行われる増殖法が、NK細胞の細胞傷害能を上げる点で、より良いことを示唆する。
【0070】
バイオリアクターおよびフラスコ増殖からの最終産物の細胞傷害能の相違を説明するために、発明者らは、マルチカラーフローサイトメトリーを用いてNK細胞の詳細な表現型特性化を行った。図5は、バイオリアクターおよびフラスコ増殖からのNK細胞上の種々の表面受容体の発現を示し、全てのドナーの結果をまとめて、バイオリアクター増殖とフラスコ増殖の表現型の比較全体図を示す。概して、最終産物中のNK細SSS胞は、何れの増殖システムを用いるかに関わらず同様であって、CD11b、NKG2DおよびNKp44の発現レベルがわずかであるが、顕著に異なる。NKp44の上方制御は、上昇した細胞傷害能の鍵となる要因の1つである。
【0071】
この研究は、閉鎖型自動化システムでGMP適合品質のエフェクター細胞を生産し得る可能性を検討する試みにおいて、ヒトドナーのバルクPBMCに由来するNKおよびNKT細胞のエクスビボ増殖のための細胞培養フラスコ、バッグおよびバイオリアクターの使用を比較評価する。
【0072】
発明者らは、細胞培養フラスコ中で活性化NK細胞の選択的富化を促進するGMP適合培養培地を以前に報告した(Carlens S. et al., 2001)。そこで、本発明者らは、臨床適用可能な、エフェクター集団に富む大規模NK細胞の調製のためのGMP適合品質の自動化閉鎖型培養システムを最適化する最終工程を示し(Miller JS. et al., 1994;Luhm J. et al., 2002;Klingemann HG and Martinson J., 2004;Koehl U. et al., 2005)、および/またはフィーダー細胞株の使用(Ishikawa E. et al., 2004;Torelli GF, et al., 2005)が、既報の通り、NK細胞増殖に広く用いられていたことを示した。この研究において、本発明者らは、NK細胞含有量および抗腫瘍活性(Bordignon C. et al., 1999)の両方に関して、如何なる単離工程も用いないが、LAK(Ramsdell FJ and Golub SH., 1987)およびサイトカイン誘導性キラー(CIK)細胞(Chan JK. et al., 2006;Lu PH and Negrin RS., 1994)とは別のNK細胞に富む細胞集団をもたらすバルクPBMCを用いた。
【0073】
本発明者らは、バッグおよびバイオリアクターシステムの両方が、NK細胞を増殖させることを証明した。この研究で利用される2つの閉鎖型システムの増殖率比較および最終製品純度の全体的な比較は、バッグと比較したとき、バイオリアクターシステムが、より高い純度を有する十分量のNK細胞を提供し、さらに最終産物中のT細胞をより少なくすることを明らかにする。
【0074】
驚くことに、本発明者らは、NK細胞活性が、フラスコからの増殖産物と比較して、バイオリアクターからの増殖産物で顕著に高いことを見いだした。受容体発現レベルとK562に対するNK細胞の反応との相関関係は、CD132、CD25、CD57およびNKG2C発現レベルが、その反応と逆相関しており、NKp30、NKp44およびNKp46の発現レベルが、直接的に相関することを明らかにした。活性化NK細胞受容体NKG2Cの発現で逆相関を観察することは珍しくないが、標的K562細胞が、そのリガンドHLA−Eの発現を欠失することが公知の通りであり、この場合にはあまり意味がない(Khalil−Daher I. et al., 1999)。NK細胞応答と相関する受容体の統計的分析により、NKp44は、反応に正に相関し、フラスコ増殖と比較したとき、バイオリアクター製品で顕著に高レベルで発現されることが明らかとなった。このことは、少なくとも1つには、バイオリアクター産物の高い細胞毒性能の観察を説明し得る。他のNCRとは異なり、NKp44(Vitale M. et al., 1998)は、活性化NK細胞でのみ発現され、インビトロでのIL−2刺激後に上方制御される(Biassoni R. et al., 2002)。故に、この場合、培養中にIL−2がどのくらいの頻度で用いられているか、およびNK細胞集団の活性化がどの程度かを、代理マーカーとして示し得る。従って、NKp44の上昇した発現は、常套の細胞培養フラスコよりもバイオリアクターで行われる増殖法に機能的意義を提供する。
【0075】
実用性に関して、本発明者らは、当該全てのシステムには有利点と不利点があることを明らかにした。細胞培養フラスコでの増殖は、外部物質および汚染物質への暴露という固有の危険性がある。この危険性は、GMP適合の実験室環境で最小化されるが、閉鎖型自動化システムの使用は、それが十分量の細胞を供給する限り好まれることは明らかである。フラスコでの培養開始は、多くの細胞を必要としないが、細胞は、取り扱い不可能な数のフラスコ数になるまで、増殖中、新しいフラスコに分けられる間、ある程度の濃度内に維持されなければならない(Heiskala M. et al., 1987)。培養はまた、小さなバッグ中で少量の細胞で開始可能であり、良好な増殖をもたらすが、NK細胞の純度は、他のシステムよりも低く、細胞は、2個以上のバッグに分けられる必要がある。しかし、バッグでの増殖は、さらなる装置に投資を必要とすることなく、標準的な細胞培養実験室で容易に最適化され得る。2種以上の増殖が、インキュベーターのスペースが限界でない限り、バッグを用いて同時に行われ得て、バイオリアクターは、機械の購入のために追加の投資を必要とするが、一度に一回の増殖に用いられ得る。
【0076】
バイオリアクターは、要する時間が最小限であるために、最も実用的な方法である。しかしながら、このシステムの開始には多くの細胞が必要とされ、増殖率が低い。バイオリアクターの連続した振とう動作は、例えば均一な培養条件、サンプリングの容易さならびにpHおよび溶存酸素の測定のような良好な制御処理品質のような多くの利点を提供する、同時かつ同種の培養環境を確実にする。そのような同時培養条件の利用は、バイオリアクター中でより濃縮される細胞増殖が実行可能かどうかに関与する主な要因であるはずである。このことは、培地またはさらなる成分の無駄遣いを避け、処理全体の費用を劇的に減少させる。プロトコール毎に平均増殖曲線を用いて、本発明者らは、同程度のNK細胞数を得るために、バイオリアクターシステムは、バッグに用いる培地成分のおよそ1/10倍、およびフラスコに用いる培地成分の1/25倍を用いることを概算した。培地およびサイトカインの消費は、フラスコでよりも多い(バッグのおよそ2.5倍)。
【0077】
当然、エクスビボ培養のための同時バイオリアクターシステムの使用は、幾つかの要因が慎重に評価されることを要する。造血細胞培養は、せん断に比較的感受性であり、高せん断処理がエクスビボ増殖に不適当であるという仮定は合理的である(Nielsen LK., 1999)。従って、振盪タンク型バイオリアクター(Pierson BA. et al., 1996)または外部フィルターおよび高流速に依存するかん流培養システムは、高い効率を提供しない。バイオリアクターの最大限の利点を達成するために、培地を除去するための内部かん流フィルターを備える低せん断力生成システムの使用が望ましく(Nielsen LK., 1999)、この実験で用いるバイオリアクターシステムは、これらの予想に合致し得る。
【0078】
慎重に考慮する別の要因は、培養環境で用いられる材料である。わずかな材料のみが、効率的な造血細胞の増殖を支持し得て、洗浄、滅菌および再利用のような要因が、それらのパフォーマンスにかなり影響を及ぼす(LaIuppa JA. et al., 1997)。従って、使い捨てで、かつ予め滅菌された適当な材料の使用が好ましい。この実験で用いるバッグおよびバイオリアクターシステムの両方が、この点で生産に好適である。
【0079】
フラスコと比較するとき、観察される1つの違いは、バイオリアクターおよびバッグ増殖の最終産物中の細胞のわずかに低い生存率であった。このことは、主に、死んだ細胞が、フラスコ培養において2−3日毎に継続的に流されているという事実のためであり、培地を交換するとき、閉鎖型システムではかかる操作を伴わない。しかし、投与前に、培養終了時のGMP適合品質の洗浄工程を常に利用可能である。
【0080】
問題とする全てのシステムは、特定の実施上の有利点および不利点がある。細胞培養フラスコ中のNK細胞の増殖は、外部物質および汚染物質への暴露の固有の危険性を有する。この危険性は、GMP適合実験室環境で最小化されるが、閉鎖型自動化システムの使用は、それが十分量の細胞を供給する限り、間違いなく好ましい。フラスコでの培養は、非常に少ない数の細胞で開始できるが、細胞は、取り扱い不可能なフラスコ数になるまで、増殖中、新しいフラスコに分けられる間、ある程度の濃度内に維持されなければならない。
【0081】
培養はまた、小さなバッグ中で少量の細胞で開始可能で良好な増殖をもたらすが、NK細胞およびNKT細胞の純度は、他のシステムよりも低く、細胞は、2個以上のバッグに分けられる必要がある。バッグでの増殖は、さらなる装置に投資を必要とすることなく、標準的な細胞培養実験室で容易に最適化され得る。2個以上の増殖が、インキュベーターのスペースが限界でない限り、バッグを用いて同時に行われ得て、バイオリアクターは、一度に一回の増殖に用いられ得る。
【0082】
結論として、本明細書に記載の結果は、適用可能な免疫療法適用に予想される使用のための大量の高度に活性化されたエフェクター細胞が、GMP適合条件下で、閉鎖型培養システムで生産され得ることを、明確に証明する。
【0083】
特定の態様が明細書中に詳細に記載されるが、これは説明のみを目的とし、例示として記載されたもので、添付の特許請求の範囲を限定することを意図しない。特に、種々の置換、変更および修正が、特許請求の範囲によって定義される本発明の精神および範囲から逸脱することなく本発明にされ得ることは、本発明者らにより考慮されている。
【0084】
参考文献
Aktas E. et al. Relationship between CD107α expression and cytotoxic activity. Cell Immunol. 2009, 254 pp.149−54.
Alici E. et al. Autologous antitumor activity by NK cells expanded from myeloma patients using GMP−compliant components. Blood 2008, 111 pp. 3155−62.
Alter G. et al. CD107a as a functional marker for the identification of natural killer cell activity. J Immunol Methods. 2004, 294 pp.15−22.
Barkholt L. et al. Safety analysis of ex vivo expanded NK and NK−like T cells administered to cancer patients: a Phase I clinical study. Immunotherapy. 2009, 1 pp. 753−764.
Biassoni R. et al. Human natural killer receptors and their ligands. Curr Protoc Immunol. 2002; Chapter 14: Unit 14 10.
Bordignon C. et al. Cell therapy: achievements and perspectives. Haematologica. 1999, 84 pp.1110−1149.
Carlens S. et al. A new method for in vitro expansion of cytotoxic human CD3−CD56+ natural killer cells. Hum Immunol. 2001, 62 pp.1092−8.
Chan JK. et al. Enhanced killing of primary ovarian cancer by retargeting autologous cytokine−induced killer cells with bispecific antibodies: a preclinical study. Clin Cancer Res. 2006, 12 pp.1859−1867.
Grimm EA. et al. Lymphokine−activated killer cell phenomenon. Lysis of natural killer−resistant fresh solid tumor cells by interleukin 2−activated autologous human peripheral blood lymphocytes. J Exp Med. 1982,155 pp.1823−1841.
Guimaraes F. et al. Evaluation of ex vivo expanded human NK cells on antileukemia activity in SCID−beige mice. Leukemia. 2006, 20 pp. 833−839.
Guven H. et al. Expansion of natural killer (NK) and natural killer−like T (NKT)−cell populations derived from patients with B−chronic lymphocytic leukemia (B−CLL): a potential source for cellular immunotherapy. Leukemia 2003, 17 pp.1973−80.
Heiskala M. et al. Mechanism of cell contact−mediated inhibition of natural killer activity. J Immunol. 1987, 139 pp.1414−1418.
Ishikawa E. et al. Autologous natural killer cell therapy for human recurrent malignant glioma. Anticancer Res. 2004, 24 pp.1861−1871.
Khalil−Daher I. et al. Role of HLA−G versus HLA−E on NK function: HLA−G is able to inhibit NK cytolysis by itself. J Reprod Immunol. 1999, 43 pp.175−182.
Klingemann HG and Martinson J. Ex vivo expansion of natural killer cells for clinical applications. Cytotherapy 2004, 6 pp. 15−22.
Koehl U. et al. Ex vivo expansion of highly purified NK cells for immunotherapy after haploidentical stem cell transplantation in children. Klin Padiatr. 2005, 217 pp. 345−350.
LaIuppa JA. et al. Culture materials affect ex vivo expansion of hematopoietic progenitor cells. J Biomed Mater Res. 1997, 36 pp. 347−359.
Lu PH and Negrin RS. A novel population of expanded human CD3+CD56+ cells derived from T cells with potent in vivo antitumor activity in mice with severe combined immunodeficiency. J Immunol. 1994, 153 pp.1687−1696.
Luhm J. et al. Large−scale generation of natural killer lymphocytes for clinical application. J Hematother Stem Cell Res. 2002, 11 pp. 651−7.
Miller JS. et al. Large scale ex vivo expansion and activation of human natural killer cells for autologous therapy. Bone Marrow Transplant 1994, 14 pp. 555−62.
Nielsen LK. Bioreactors for hematopoietic cell culture. Annu Rev Biomed Eng. 1999, 1 pp. 129−152.
Pierson BA. et al. Production of human natural killer cells for adoptive immunotherapy using a computer−controlled stirred−tank bioreactor. J Hematother 1996, 5 pp. 475−83.
Ramsdell FJ and Golub SH. Generation of lymphokine−activated killer cell activity from human thymocytes. J Immunol. 1987,139 pp.1446−1453.
Rosenberg S. Lymphokine−activated killer cells: a new approach to immunotherapy of cancer. J Natl Cancer Inst. 1985, 75 pp. 595−603.
Sutlu T and Alici E. Natural killer cell−based immunotherapy in cancer: current insights and future prospects. J Intern Med. 2009, 266 pp.154−181.
Torelli GF. et al. Expansion of natural killer cells with lytic activity against autologous blasts from adult and pediatric acute lymphoid leukemia patients in complete hematologic remission. Haematologica 2005, 90 pp. 785−792.
Vitale M. et al. NKp44, a novel triggering surface molecule specifically expressed by activated natural killer cells, is involved in non−major histocompatibility complex−restricted tumor cell lysis. J Exp Med. 1998, 187 pp. 2065−2072.
US 10/242,7881
【図1A】
【図1B】
【図1C】
【図1D】
【図2A】
【図2B】
【図2C】
【図3A】
【図3B】
【図3C】
【図3D】
【図4A】
【図4B】
【特許請求の範囲】
【請求項1】
表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の大規模増殖および同時活性化のための方法であって、該増殖および活性化を閉鎖型システムで行い、得られた増殖細胞が、インビトロ細胞傷害性試験により増加した細胞傷害性を示す、方法。
【請求項2】
−細胞を、血清、インターロイキン−2(IL−2)および抗CD3抗体を添加した増殖培地を含む閉鎖型システムに加え;
−該システム内の細胞を、増殖した細胞集団の少なくとも35%が活性化NK細胞およびNKT細胞を含むまで振盪および加熱しながら増殖させる(ここで、該増殖した細胞が、インビトロ細胞傷害性試験により増加した細胞傷害性を示す。)、
手順を含む、請求項1記載の方法。
【請求項3】
振盪および加熱を、約36℃ないし約40℃の温度;約4.7ないし約5.1%のCO2濃度;および、細胞が閉鎖型細胞システムの表面に接着する速度および角度でゆっくりと振盪するという条件下で行う、請求項2記載の方法。
【請求項4】
振盪を、約4−8/分の振盪速度で行う、請求項2記載の方法。
【請求項5】
振盪を、約4−8°の振盪角度で行う、請求項2記載の方法。
【請求項6】
増殖を、全細胞数が、少なくとも約10倍に増殖するまで行う、請求項2記載の方法。
【請求項7】
増殖を、増殖細胞集団の少なくとも約50%が、活性化NK細胞およびNKT細胞それぞれを含むまで行う、請求項2記載の方法。
【請求項8】
細胞サンプルが、末梢血、細胞株、またはサイトカインで刺激した末梢血サンプルである、請求項1−7のいずれか一項記載の方法。
【請求項9】
細胞サンプルが、末梢血単核球細胞(PBMC)である、請求項8記載の方法。
【請求項10】
閉鎖型細胞システムに最初に添加される細胞濃度が、約0.5x106ないし約2x106/ml増殖培地である、請求項2記載の方法。
【請求項11】
1日当たり、全培養量の約50%に相当する量の血清およびIL−2を含む培地を添加し、閉鎖型細胞システムからほぼ同量の増殖培地を捨てる工程を含み、該工程を、全細胞密度が、初期細胞密度の少なくとも約50%増加したときに行う、請求項1−10のいずれか一項記載の方法。
【請求項12】
細胞密度が、初期細胞密度から少なくとも約300%増加したときに請求項11記載の工程を繰り返すが、全培地量の約75%を添加および破棄することを含む、請求項11記載の方法。
【請求項13】
細胞密度が、初期細胞密度から少なくとも約500%増加したときに請求項11記載の工程を繰り返すが、全培地量の約100%を添加および破棄することを含む、請求項11記載の方法。
【請求項14】
細胞を少なくとも約10日間インキュベートする、請求項1−13のいずれか一項記載の方法。
【請求項15】
閉鎖型細胞システムが予め滅菌したバッグである、請求項1−14のいずれか一項記載の方法。
【請求項16】
閉鎖型細胞システムがバイオリアクターである、請求項1ないし15のいずれか一項記載の方法。
【請求項17】
血清が、ヒト血清および自家血清からなる群から選択される、請求項2記載の方法。
【請求項18】
培地に、約50ないし約1500U/mlのIL−2、約1ないし約50ng/mlの抗CD3抗体および約1ないし約40%血清を添加する、請求項2記載の方法。
【請求項19】
細胞サンプルを、健常な対象から採取する、請求項1ないし19のいずれか一項記載の方法。
【請求項20】
細胞サンプルを、腫瘍を有する対象から採取する、請求項1ないし19のいずれか一項記載の方法。
【請求項21】
腫瘍が、血液系腫瘍および固形腫瘍からなる群から選択される、請求項20記載の方法。
【請求項22】
細胞が、主に、表現型CD3−CD56+のNK細胞である、請求項1−21のいずれか一項記載の方法。
【請求項23】
請求項1−21のいずれか一項記載の方法により得られる、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液。
【請求項24】
インビトロ細胞傷害性試験により決定される通り、フラスコ中で増殖した細胞と比較して増加した細胞傷害性を示す、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液。
【請求項25】
増殖した細胞集団の少なくとも35%、好ましくは少なくとも50%が活性化NK細胞を含む、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液。
【請求項1】
表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の大規模増殖および同時活性化のための方法であって、該増殖および活性化を閉鎖型システムで行い、得られた増殖細胞が、インビトロ細胞傷害性試験により増加した細胞傷害性を示す、方法。
【請求項2】
−細胞を、血清、インターロイキン−2(IL−2)および抗CD3抗体を添加した増殖培地を含む閉鎖型システムに加え;
−該システム内の細胞を、増殖した細胞集団の少なくとも35%が活性化NK細胞およびNKT細胞を含むまで振盪および加熱しながら増殖させる(ここで、該増殖した細胞が、インビトロ細胞傷害性試験により増加した細胞傷害性を示す。)、
手順を含む、請求項1記載の方法。
【請求項3】
振盪および加熱を、約36℃ないし約40℃の温度;約4.7ないし約5.1%のCO2濃度;および、細胞が閉鎖型細胞システムの表面に接着する速度および角度でゆっくりと振盪するという条件下で行う、請求項2記載の方法。
【請求項4】
振盪を、約4−8/分の振盪速度で行う、請求項2記載の方法。
【請求項5】
振盪を、約4−8°の振盪角度で行う、請求項2記載の方法。
【請求項6】
増殖を、全細胞数が、少なくとも約10倍に増殖するまで行う、請求項2記載の方法。
【請求項7】
増殖を、増殖細胞集団の少なくとも約50%が、活性化NK細胞およびNKT細胞それぞれを含むまで行う、請求項2記載の方法。
【請求項8】
細胞サンプルが、末梢血、細胞株、またはサイトカインで刺激した末梢血サンプルである、請求項1−7のいずれか一項記載の方法。
【請求項9】
細胞サンプルが、末梢血単核球細胞(PBMC)である、請求項8記載の方法。
【請求項10】
閉鎖型細胞システムに最初に添加される細胞濃度が、約0.5x106ないし約2x106/ml増殖培地である、請求項2記載の方法。
【請求項11】
1日当たり、全培養量の約50%に相当する量の血清およびIL−2を含む培地を添加し、閉鎖型細胞システムからほぼ同量の増殖培地を捨てる工程を含み、該工程を、全細胞密度が、初期細胞密度の少なくとも約50%増加したときに行う、請求項1−10のいずれか一項記載の方法。
【請求項12】
細胞密度が、初期細胞密度から少なくとも約300%増加したときに請求項11記載の工程を繰り返すが、全培地量の約75%を添加および破棄することを含む、請求項11記載の方法。
【請求項13】
細胞密度が、初期細胞密度から少なくとも約500%増加したときに請求項11記載の工程を繰り返すが、全培地量の約100%を添加および破棄することを含む、請求項11記載の方法。
【請求項14】
細胞を少なくとも約10日間インキュベートする、請求項1−13のいずれか一項記載の方法。
【請求項15】
閉鎖型細胞システムが予め滅菌したバッグである、請求項1−14のいずれか一項記載の方法。
【請求項16】
閉鎖型細胞システムがバイオリアクターである、請求項1ないし15のいずれか一項記載の方法。
【請求項17】
血清が、ヒト血清および自家血清からなる群から選択される、請求項2記載の方法。
【請求項18】
培地に、約50ないし約1500U/mlのIL−2、約1ないし約50ng/mlの抗CD3抗体および約1ないし約40%血清を添加する、請求項2記載の方法。
【請求項19】
細胞サンプルを、健常な対象から採取する、請求項1ないし19のいずれか一項記載の方法。
【請求項20】
細胞サンプルを、腫瘍を有する対象から採取する、請求項1ないし19のいずれか一項記載の方法。
【請求項21】
腫瘍が、血液系腫瘍および固形腫瘍からなる群から選択される、請求項20記載の方法。
【請求項22】
細胞が、主に、表現型CD3−CD56+のNK細胞である、請求項1−21のいずれか一項記載の方法。
【請求項23】
請求項1−21のいずれか一項記載の方法により得られる、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液。
【請求項24】
インビトロ細胞傷害性試験により決定される通り、フラスコ中で増殖した細胞と比較して増加した細胞傷害性を示す、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液。
【請求項25】
増殖した細胞集団の少なくとも35%、好ましくは少なくとも50%が活性化NK細胞を含む、表現型CD3−CD56+のNK細胞および表現型CD3+CD56+のNKT細胞の懸濁液。
【図5】

【公表番号】特表2012−521215(P2012−521215A)
【公表日】平成24年9月13日(2012.9.13)
【国際特許分類】
【出願番号】特願2012−501965(P2012−501965)
【出願日】平成22年3月25日(2010.3.25)
【国際出願番号】PCT/SE2010/050333
【国際公開番号】WO2010/110734
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(505305927)アヴァリス・アクチエボラーグ (5)
【Fターム(参考)】
【公表日】平成24年9月13日(2012.9.13)
【国際特許分類】
【出願日】平成22年3月25日(2010.3.25)
【国際出願番号】PCT/SE2010/050333
【国際公開番号】WO2010/110734
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(505305927)アヴァリス・アクチエボラーグ (5)
【Fターム(参考)】
[ Back to top ]