
NOGOレセプター結合タンパク質
本発明は、Sp35ポリペプチドおよびその融合タンパク質、Sp35抗体およびその抗原結合フラグメントならびにそれらをコードする核酸を提供する。本発明はまた、このようなSp35抗体、その抗原結合フラグメント、Sp35ポリペプチドおよびその融合タンパク質を含有する組成物ならびにこのようなSp35抗体、その抗原結合フラグメント、Sp35ポリペプチドおよびその融合タンパク質の製造方法および使用方法を提供する。

【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、神経学、神経生物学および分子生物学に関する。より詳細には、本発明は、神経学的疾患、神経学的障害および神経学的損傷(例えば、脊髄損傷)の処置のための分子および方法に関する。
【背景技術】
【0002】
(発明の背景)
軸索および樹状突起は、ニューロンから伸長する。伸長する軸索または神経突起の遠位端は、成長円錐として公知の特定の領域を含む。成長円錐は、局所的環境を感じ取り、ニューロンの標的細胞に向かう軸索成長を誘導する。成長円錐は、環境の合図(例えば、表面への接着、成長因子、神経伝達物質および電場)に応答する。上記成長円錐は、一般的に、一日に1mm〜2mmの速度で成長する。上記成長円錐は、ラメリポディウムおよび糸状足として分類される伸長手段によって、その前方領域およびいずれもの側方領域を探索する。伸長部が不都合な表面に接触する場合、その伸長部は引っ込む。伸長部が、都合のよい成長表面と接触する場合、その伸長部は、伸長を続け、上記成長円錐をその方向に誘導する。上記成長円錐が適切な標的細胞に到達する場合、シナプス結合が生成される。
【0003】
神経細胞機能は、ニューロンとそれらの隣接環境における他の細胞との間の接触により、影響される(Rutishauserら,1988,Physiol.Rev.68:819)。これらの細胞としては、特定の神経膠細胞、中枢神経系(CNS)における希乏突起神経膠細胞ならびに末梢神経系(PNS)におけるシュヴァン細胞が挙げられ、これらの細胞は、神経軸索がミエリンで覆われている(Lemke,1992,An Introduction to Molecular Neurobiology,Z.Hall編,281頁,Sinauer)。
【0004】
CNSニューロンは、損傷後に再生成する固有の潜在能力を有するが、CNSニューロンは、ミエリンに存在する阻害タンパク質によって損傷後に再生成することを阻害される(Brittisら,2001,Neuron 30:11−14;Jonesら,2002,J.Neurosci.22:2792−2803;Grimpeら,2002,J.Neurosci.:22:3144−3160)。
【0005】
希乏突起神経膠細胞において見出されたいくつかのミエリン阻害タンパク質が、特徴付けられている。ミエリン阻害タンパク質の公知の例としては、NogoA(Chenら,Nature,2000,403,434−439;Grandpreら,Nature 2000,403,439−444)、ミエリン関連糖タンパク質(MAG)(McKerracherら,1994,Neuron 13:805−811;Mukhopadhyayら,1994,Neuron 13:757−767)および希乏突起神経膠細胞糖タンパク質(OM−gp)(Mikolら,1998,J.Cell.Biol.106:1273−1279)が挙げられる。これらのタンパク質の各々は、ニューロンのNgR1に対するリガンドであることが、別々に示されている(Wangら,Nature 2002,417,941−944;Grandpreら,Nature 2000,403,439−444;Chenら,Nature,2000,403,434−439;2002年6月28日にインターネット上で公開された,Domeniconiら,Neuron 2002)。
【0006】
Nogoレセプター−1(NgR1)は、8個のロイシンリッチ繰返し配列を含むGPI−固定膜タンパク質である(Fournierら,2001,Nature 409:341−346)。阻害タンパク質(例えば、NogoA、MAGおよびOM−gp)と相互作用すると、上記NgR1複合体は、成長円錐崩壊および神経突起伸長阻害を引き起こすシグナルを伝達する。
【発明の開示】
【発明が解決しようとする課題】
【0007】
NgR1媒介性の成長円錐崩壊およびその結果もたらされる神経突起伸長阻害を阻害するための分子および方法に関する必要性は、未だ満たされていない。
【課題を解決するための手段】
【0008】
(発明の要旨)
発明者らは、「Sp35」と称されるポリペプチド(発明者らが命名)に関する種々の発見を行ってきた。Sp35のための代替的な名称としては、「LINGO」および「LINGO−1」が挙げられる。発明者らの発見は、以下を含む。Sp35は、NgR1に結合する。Sp35は、同型相互作用で、それ自体に結合する。Sp35−Fc融合タンパク質は、顆粒状ニューロンにおいて、線維束性攣縮を誘導または促進する。Sp35−Fc融合タンパク質は、赤核脊髄路半側切除損傷モデルおよび視神経離断モデルの両方において、ニューロンの生存を促進する。Sp35レトロウイルス感染皮質初代細胞は、脊髄損傷ラットに送達される場合、ニューロン生存の促進、軸索のβIIIチューブリン染色の増大およびミエリン含量の上昇をもたらす。
【0009】
これらの発見に部分的に基づいて、本発明は、ポリペプチドをコードするヌクレオチド配列を含む単離された核酸を特徴とし:(a)このポリペプチドは(i)Sp35 LRRドメイン、(ii)このLRRドメインに対してC末端側のSp35塩基性領域および(iii)この塩基性領域に対してC末端側のSp35免疫グロブリン(Ig)ドメイン、を含み;そして(b)このポリペプチドは、膜貫通ドメインを欠く。上記Sp35 LRRドメインは、カルボキシ末端LRR(LRRCT)、アミノ末端LRR(LRRNT)またはその両方を含み得る。本発明のいくつかの実施形態において、上記コードされたSp35ポリペプチドは、細胞質ドメインを欠く。いくつかの実施形態において、上記コードされたSp35ポリペプチドは、配列番号2のアミノ酸残基34〜532を含み、アミノ酸残基533〜614を欠く。
【0010】
本発明はまた、ポリペプチドをコードする核酸を含み、このポリペプチドは、Sp35 Igドメインを含み、そして、Sp35 LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く。
【0011】
本発明はまた、ポリペプチドをコードする核酸を含み、このポリペプチドは、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く。
【0012】
本発明はまた、ポリペプチドをコードする核酸を含み、このポリペプチドは、機能性細胞質ドメインを欠くが、全ての他のSp35ドメインを含む。例えば、上記コードされたポリペプチドは、(シグナル配列のプロセシング前に)配列番号2のアミノ酸1〜576を含み得る。
【0013】
本発明のいくつかの実施形態において、上記コードされたポリペプチドは、非Sp35部分を含む融合タンパク質である。上記非Sp35部分は、例えば、Ig部分、血清アルブミン部分、標的化部分、レポーター部分または精製容易化部分であり得る。好ましい非Sp35部分は、Ig部分(例えば、Fc部分)である。
【0014】
上記ヌクレオチド配列は、例えば、発現ベクターにおいて、発現制御配列に作動可能に結合され得る。本発明はまた、本発明のSp35ポリペプチドを発現するベクターにより形質転換される宿主細胞を含む。
【0015】
本発明はまた、上に記載される核酸のいずれかによりコードされるSp35ポリペプチドを含む。
【0016】
本発明はまた、ポリマー(例えば、ポリアルキレングリコール、糖ポリマーおよびポリペプチド)に結合されたSp35ポリペプチドを含む。好ましいポリマーは、ポリアルキレングリコール(例えば、ポリエチレングリコール(PEG))である。上記ポリペプチドは、1つ、2つ、3つまたは4つのポリマーに結合され得る。好ましくは、上記結合されたポリマーの総分子量は、Sp35ポリペプチド当たり20,000Da〜40,000Daである。
【0017】
本発明はまた、NgR1によるシグナル伝達を阻害する方法を含む。上記方法は、上記NgR1と有効量のSp35ポリペプチドとを接触させる工程を包含する。上記方法における使用のための好ましいポリペプチドとしては、以下が挙げられる:
(a)Sp35ポリペプチドであって:(a)このポリペプチドが(i)Sp35 LRRドメイン、(ii)このLRRドメインに対してC末端側のSp35塩基性領域および(iii)この塩基性領域に対してC末端側のSp35免疫グロブリン(Ig)ドメイン、を含み;そして(b)このポリペプチドが、膜貫通ドメインを欠く、ポリペプチド;ならびに
(b)Sp35 Igドメインを含み、そして、Sp35 LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠くSp35ポリペプチド。
【0018】
本発明はまた、中枢神経系(CNS)ニューロンの軸索成長阻害を減少する方法を含む。上記方法は、上記ニューロンと有効量のポリペプチド(例えば、Sp35ポリペプチド、抗Sp35抗体または抗Sp35抗体の抗原結合フラグメント)とを接触させる工程を包含する。
【0019】
本発明はまた、CNSニューロンの成長円錐崩壊を阻害する方法を含む。上記方法は、上記ニューロンと有効量のポリペプチド(例えば、Sp35ポリペプチド、抗Sp35抗体または抗Sp35抗体の抗原結合フラグメント)とを接触させる工程を包含する。
【0020】
本発明はまた、哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置する方法を含む。上記方法は、治療有効量のポリペプチド(例えば、Sp35ポリペプチド、抗Sp35抗体または抗Sp35抗体の抗原結合フラグメント)を上記哺乳動物に投与する工程を包含する。本発明のいくつかの実施形態において、上記CNS疾患、CNS障害またはCNS損傷は、脊髄損傷である。上記Sp35ポリペプチドは、局所投与され得る。上記方法のいくつかの実施形態において、上記Sp35ポリペプチドは、脊髄損傷の48時間以内に最初に投与される。局所投与のために、上記治療有効量の上記ポリペプチドは、好ましくは、10μg/kg〜10mg/kgである。全身投与のために、上記治療有効量の上記ポリペプチドは、好ましくは、1mg/kg〜20mg/kgである。
【0021】
本発明はまた、哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置するエキソビボ遺伝子治療方法を含む。上記方法は(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)上記CNS疾患、CNS障害またはCNS損傷(例えば、脊髄損傷)の部位で、上記哺乳動物中にこの宿主細胞を導入する工程、を包含する。上記培養宿主細胞は、処置されるべき哺乳動物由来であり得る。このエキソビボ遺伝子治療方法において、上記組換えSp35ポリペプチドは、全長Sp35ポリペプチドであり得る。
【0022】
本発明はまた、上記CNS疾患、CNS障害またはCNS損傷の部位での髄鞘形成を促進する方法を含む。上記方法は、上記CNS疾患、CNS障害またはCNS損傷の部位と有効量のSp35ポリペプチド(例えば、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠くポリペプチド)とを接触させる工程を包含する。
【0023】
本発明はまた、CNS疾患、CNS障害またはCNS損傷をインビボ遺伝子治療により処置するインビボ遺伝子治療方法を含む。上記方法は、上記疾患、障害または損傷の部位あるいはその近傍で、哺乳動物にウイルスベクターを投与する工程を包含し、このウイルスベクターは、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、このSp35ポリペプチドは、この哺乳動物において、損傷の部位またはその近傍のニューロンによる軸索伸長阻害を減少するために十分な量で、このヌクレオチド配列から発現される。上記ウイルスベクターは、例えば、アデノウイルスベクター、レンチウイルスベクター、バキュロウイルスベクター、エプスタイン−バーウイルスベクター、パポバウイルスベクター、ワクシニアウイルスベクターおよび単純疱疹イルスベクターであり得る。上記疾患、障害または損傷は、例えば、脊髄損傷または視覚神経損傷であり得る。上記ウイルスベクターは、例えば、局所投与、眼内投与、非経口投与、くも膜下腔内投与、硬膜下投与および皮下投与のような経路により投与され得る。
【0024】
本発明はまた、死の危険性にあるニューロンの生存を促進する方法を含む。上記方法は、上記ニューロンと有効量のSp35ポリペプチドとを接触させる工程を包含する。上記Sp35ポリペプチドは、Sp35の可溶性形態(例えば、Sp35−Fc融合タンパク質)であり得る。上記ニューロンは、インビトロまたはインビボ(例えば、神経変性疾患、神経変性障害または神経変性損傷(例えば、多発性硬化症、ALS、ハンチントン病、アルツハイマー病、パーキンソン病、糖尿病性ニューロパシー、脳卒中、外傷性脳損傷および外傷性脊髄損傷)を有する哺乳動物中)であり得る。本発明のいくつかの実施形態において、上記Sp35ポリペプチドは、以下により間接的に投与される:(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)上記ニューロンの部位で、上記哺乳動物中にこの宿主細胞を導入する工程。本発明のいくつかの実施形態において、上記ポリペプチドは、インビボ遺伝子治療を介して間接的に投与される。いくつかの実施形態において、上記方法は、上記ニューロンの部位またはその近傍にウイルスベクターを投与する工程を包含し、このウイルスベクターは、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、このSp35ポリペプチドは、上記哺乳動物において、このニューロンの生存を促進するために十分な量で、このヌクレオチド配列から発現される。
【0025】
本明細書中で使用される場合、「全長ヒトSp35ポリペプチド」とは、そのアミノ酸配列が配列番号2のアミノ酸34〜614であるポリペプチドを意味する。
【0026】
本明細書中で使用される場合、「異種部分」とは、全長Sp35ポリペプチドに存在しないアミノ酸配列を意味する。
【0027】
本明細書中で使用される場合、「nogoレセプター−1」とは、その配列がGenbank受託番号AAG53612の下で公的に入手可能であるポリペプチドを意味する。
【0028】
本明細書中で使用される場合、「Sp35アンタゴニストポリペプチド」とは、天然に生じるSp35の生物学的活性を阻止、阻害または妨害するSp35ポリペプチドを意味する。
【0029】
本明細書中で使用される場合、「Sp35塩基性領域」とは、以下のアミノ酸モチーフを意味する:
【0030】
【化1】
最上列のアミノ酸(太字;配列番号4)は、好ましいSp35塩基性領域配列であり、任意の置換を示す変異体を、下に示す(配列番号5、6、7および8)。
【0031】
本明細書中で使用される場合、「Sp35融合タンパク質」とは、異種部分に融合されたSp35部分を含む融合タンパク質を意味する。
【0032】
本明細書中で使用される場合、「Sp35 Igドメイン」とは、配列番号2のアミノ酸433〜493を意味し、ただし、この配列は、5個までの独立したアミノ酸の挿入、欠失または保存的アミノ酸置換を含み得る。以下の置換(配列番号2に基づく番号付け)が、明らかに含まれる:6位のVからM;294位のSからG;348位のVからA;419位のRからH。
【0033】
本明細書中で使用される場合、「Sp35 LRRドメイン」とは、10個〜14個のロイシンリッチ繰返し配列を含むドメインを意味し、このような配列としては、表1に列挙されるLRRNTおよびLRRCTが挙げられるが、ただし、5個までのアミノ酸の挿入、欠失または保存的アミノ酸置換が、10個〜14個のロイシンリッチ繰返し配列の集合体中に現れ得る。
【0034】
本明細書中で使用される場合、「Sp35部分」とは、全長Sp35ポリペプチドの生物学的に活性なフラグメントを意味する。
【0035】
本明細書中で使用される場合、「Sp35ポリペプチド」とは、Sp35部分またはSp35部分を含む融合タンパク質を意味する。
【0036】
別に規定されなければ、本明細書中で使用される全ての技術用語および科学用語は、本発明の属する分野の当業者により一般に理解されるものと同じ意味を有する。争いが生じた場合、定義を含む本明細書の記載に従う。本明細書中で言及される全ての刊行物、特許および他の参考文献が、参考として援用される。
【0037】
本明細書中に記載される方法および物質と類似または等価な方法および物質が、本発明の実施または試験において使用され得るが、好ましい方法および物質は、以下に記載される。上記物質、方法および例は、例示に過ぎず、限定するものとは意図されない。本発明の他の特徴および利点は、詳細な説明および特許請求の範囲から明らかである。
【発明を実施するための最良の形態】
【0038】
(発明の詳細な説明)
天然に生じるヒトSp35は、614アミノ酸を含むグリコシル化CNS特異的タンパク質である(図2;配列番号2)。ヒト全長野生型Sp35ポリペプチドは、14個のロイシンリッチ繰返し配列からなるLRRドメイン(N末端キャップおよびC末端キャップを含む)、Igドメイン、膜貫通領域および細胞質ドメインを含む(図3)。上記細胞質ドメインは、典型的なチロシンリン酸化部位を含む。さらに、上記天然に生じるSp35タンパク質は、シグナル配列、LRRCTとIgドメインとの間の短い塩基性領域およびこのIgドメインと上記細胞質ドメインと間の膜貫通領域を含む(図3)。ヒトSp35遺伝子は、代替的な翻訳開始コドンを含み、それにより、6個のさらなるアミノ酸(すなわち、MQVSKR(配列番号9))が、上記Sp35シグナル配列のN末端に存在していても存在していなくてもよい。表1は、図2の配列(配列番号2)に基づいたアミノ酸残基番号に従って、上記Sp35ドメインおよび他の領域を列挙する。
【0039】
【表1】
Sp35の組織分布および発生における発現は、ヒトおよびラットにおいて研究されている。Sp35生物学は、実験動物モデル(ラット)において研究されている。ラットSP35の発現は、ノザンブロットおよび免疫組織学的染色により決定される場合、CNSニューロンおよび脳の希乏突起神経膠細胞に局在している。ラットSp35 mRNA発現レベルは、発生において調節され、出生直後(すなわち、生後約1日目)がピークである。ラット脊髄離断損傷モデルにおいて、Sp35は、RT−PCRにより決定される場合、上記損傷部位でアップレギュレートされる。
【0040】
発明者らは、全長野生型Sp35が、NgR1に結合することを発見している。Sp35の可溶性誘導体は、NgR1に結合してその機能を阻止、阻害または妨害し、それにより、CNSニューロンにおいて正常に起こる軸索伸長のNgR1媒介性阻害を軽減することにより、Sp35アンタゴニストポリペプチドとして機能する。これは、軸索伸長または神経突起成長が脳または脊髄において必要とされる状態において、有益である。脊髄損傷(部分的または完全な挫傷または切断を含む)は、軸索伸長が必要とされる状態を例示するが、通常、Nogo経路の操作により阻害される。脳における軸索伸長および/もしくは神経突起成長が有益である疾患または障害の例としては、脳卒中、多発性硬化症および他の神経変性疾患または神経変性障害が挙げられる。
【0041】
本発明の方法において、Sp35ポリペプチドまたはSp35ブロッキング抗体(または抗原結合抗体フラグメント)は、事前に形成されたポリペプチドとして直接投与され得るか、または核酸ベクターを介して間接的に投与され得、NgR1機能に拮抗し、有益な軸索伸長を可能にする。
【0042】
本発明のいくつかの実施形態において、可溶性Sp35アンタゴニストポリペプチドは、以下を包含する処置方法において投与される:(1)移植可能な宿主細胞をSp35ポリペプチドを発現する核酸(例えば、ベクター)を用いて形質転換またはトランスフェクションする工程;および(2)形質転換された宿主細胞を、哺乳動物中に、疾患、障害または損傷の部位で移植する工程。例えば、上記形質転換された宿主細胞は、脊髄損傷部位に移植され得る。本発明のいくつかの実施形態において、上記移植可能宿主細胞は、哺乳動物から取り出され、一時的に培養され、可溶性Sp35ポリペプチドをコードする単離された核酸を用いて形質転換またはトランスフェクションされ、そして、それが取り出された同じ哺乳動物に移植して戻される。上記細胞は、移植される同じ部位から取り出されてもよいが、その必要はない。このような実施形態は、時に、エキソビボ遺伝子治療として公知であり、限定された時間、作用部位に局在化したSp35ポリペプチドの連続的供給を提供し得る。
【0043】
本発明は、NgR1とSp35との相互作用およびSp35同種相互作用の調節因子として有用なオリゴペプチドを提供する。上記オリゴペプチドは、以下のアミノ酸モチーフを含む:
【0044】
【化2】
アミノ酸の最上列(太字;配列番号10)が好ましい配列であり、任意の置換を含む変異体を、下に示す(配列番号11、12および13)。
【0045】
種々の例示的なSp35ポリペプチド、抗Sp35抗体および抗体フラグメントならびに本発明の実施のためにこれらの分子を得るための方法および材料を、以下に記載する。
【0046】
(融合タンパク質および結合されたポリペプチド)
本発明のいくつかの実施形態は、Sp35部分が異種ポリペプチド部分に融合されてSp35融合タンパク質を形成するSp35ポリペプチド(例えば、Sp35アンタゴニストポリペプチド)の使用を含む。Sp35融合タンパク質は、種々の目的(例えば、血清半減期の上昇、バイオアベイラビリティの改善、特定の器官または組織のタイプに対するインビボ標的化、組換え発現効率の改善、宿主細胞分泌の改善、精製の容易化および高いアビディティ)を達成するために使用され得る。達成されるべき目的に依存して、上記異種部分は、不活性であっても生物学的に活性であってもよい。また、上記異種部分は、インビボまたはインビトロにおいて、上記Sp35部分に安定に融合されるかまたは切断可能であるように選択され得る。異なる目的を達成するための異種部分は、当該分野において公知である。
【0047】
Sp35融合タンパク質の発現の代替として、選択された異種部分は、事前に形成され、上記Sp35部分に化学的に結合され得る。ほとんどの場合、選択された異種部分は、上記Sp35部分に融合されていても、または結合されていても、同様に機能する。従って、異種アミノ酸配列の以下の議論において、別に示されなければ、その異種配列は、融合タンパク質の形態かまたは化学結合体として上記Sp35部分に結合され得ることが理解されるべきである。
【0048】
薬理学的に活性なポリペプチド(例えば、Sp35)は、しばしば、迅速なインビボでのクリアランスを示し、身体における治療有効濃度を達成するためには多用量を必要とする。さらに、約60kDaよりも小さなポリペプチドは糸球体濾過を経験する可能性があり、これは、時に、腎毒性を引き起こす。比較的小さいポリペプチド(例えば、Sp35フラグメント)の融合または結合は、このような腎毒性の危険性を軽減または回避するために用いられ得る。治療ポリペプチドのインビボ安定性(すなわち、血清半減期)を増加するための種々の異種アミノ酸配列(すなわち、ポリペプチド部分または「キャリア」)が公知である。
【0049】
その長い半減期、広いインビボ分布および酵素的機能または免疫学的機能の欠如に起因して、本質的に全長のヒト血清アルブミン(HSA)またはHSAフラグメントは、好ましい異種部分である。Yehら,1992,Proc.Natl.Acad.Sci.USA,89:1904−1908およびSyedら,1997,Blood 89:3243−3252に教示されているような方法および材料の適用によって、HSAは、上記Sp35部分に起因する薬理学的活性を示し、一方で、有意なインビボ安定性の増加(例えば、10倍〜100倍高い)を示すSp35融合タンパク質またはSp35結合体を形成するために使用され得る。好ましくは、上記HSAのC末端は、上記Sp35部分のN末端に融合される。HSAは、天然に分泌されるタンパク質であるから、このHSAシグナル配列は、上記融合タンパク質が真核生物(例えば、哺乳動物)発現系において生成される場合、上記Sp35融合タンパク質の細胞培養培地への分泌を得るために利用され得る。
【0050】
本発明のいくつかの実施形態は、Sp35部分がFc領域(すなわち、Ig重鎖定常領域のC末端部分)に融合されるSp35ポリペプチドを用いる。Sp35−Fc融合の潜在的な利点としては、可溶性、インビボ安定性および多価性(multivalency)(例えば、二量体化)が挙げられる。使用されるFc領域は、IgA、IgDまたはIgGのFc領域(ヒンジ−CH2−CH3)であり得る。あるいは、使用されるFc領域は、IgEまたはIgMのFc領域(ヒンジ−CH2−CH3−CH4)であり得る。IgGのFc領域(例えば、IgG1のFc領域またはIgG4のFc領域)が好ましい。Fc融合体をコードするDNAを構築し、発現するための材料および方法は、当該分野において公知であり、過度の実験をすることなくSp35融合体を得るために適用され得る。本発明のいくつかの実施形態は、Caponらの米国特許第5,428,130号および同第5,565,335号に記載されるようなSp35融合タンパク質を用いる。
【0051】
上記シグナル配列は、小胞体膜を通るタンパク質輸送を開始するアミノ酸配列をコードする、ポリヌクレオチドである。免疫融合体(immunofusin)を構築するために有用なシグナル配列としては、抗体軽鎖シグナル配列(例えば、抗体14.18(Gilliesら,1989,J.Immunol.Meth.,125:191−202))、抗体重鎖シグナル配列(例えば、MOPC141抗体重鎖シグナル配列(Sakanoら,1980,Nature 286:5774))が挙げられる。あるいは、他のシグナル配列が使用され得る。例えば、Watson,1984,Nucleic Acids Research 12:5145を参照のこと。上記シグナルペプチドは、通常、シグナルペプチダーゼにより上記小胞体の管腔において切断される。この結果、Fc領域および上記Sp35部分を含む免疫融合体タンパク質の分泌がもたらされる。
【0052】
いくつかの実施形態において、DNA配列は、上記分泌カセットと上記Sp35部分との間のタンパク分解性切断部位をコードする。切断部位は、コードされた融合タンパク質のタンパク分解性切断を提供し、従って、上記Fcドメインを上記標的タンパク質から分離する。有用なタンパク分解性切断部位としては、タンパク分解性酵素(例えば、トリプシン、プラスミン、トロンビン、因子XaまたはエンテロキナーゼK)により認識されるアミノ酸配列が挙げられる。
【0053】
上記分泌カセットは、複製可能発現ベクターに組み込まれ得る。有用なベクターとしては、直鎖状核酸、プラスミド、ファージミド、コスミドなどが挙げられる。例示的な発現ベクターはpdCであり、ここで、上記免疫融合体DNAの転写は、ヒトサイトメガロウイルスのエンハンサーおよびプロモーターの制御下で行われる。例えば、Loら,1991,Biochem.Biophys.Acta 1088:712;およびLoら,1998,Protein Engineering 11:495−500を参照のこと。適切な宿主細胞は、Sp35ポリペプチドをコードするDNAを用いて形質転換またはトランスフェクションされ得、Sp35ポリペプチドの発現および分泌のために使用される。好ましい宿主細胞としては、不死化ハイブリドーマ細胞、骨髄腫細胞、293細胞、チャイニーズハムスター卵巣(CHO)細胞、Hela細胞およびCOS細胞が挙げられる。
【0054】
完全にインタクトな野生型Fc領域は、本発明に従うFc融合タンパク質において、通常、不必要であり、そして、所望されないエフェクター機能を示す。従って、特定の結合部位は、好ましくは、上記分泌カセットの構築の間に上記Fc領域から削除される。例えば、上記軽鎖との共発現は不必要であるので、上記重鎖結合タンパク質であるBip(Hendershotら,1987,Immunol.Today 8:111−114)に対する結合部位は、IgEのFc領域のCH2ドメインから削除され、それにより、この部位は、免疫融合体の有効な分泌を妨害しない。同様に、免疫グロブリンの軽鎖への結合に関与するFc領域に存在するシステイン残基は、削除されるかまたは別のアミノ酸に置換され、それにより、これらのシステイン残基が、免疫融合体として生成される場合に、上記Fc領域の適切な折り畳みを妨害しないようにするべきである。膜貫通ドメイン配列(例えば、IgMに存在する膜貫通ドメイン配列)は、削除されるべきである。
【0055】
IgG1のFc領域が好ましい。あるいは、他の免疫グロブリンγサブクラス(γ−2、γ−3およびγ−4)のFc領域が、上記分泌カセットにおいて使用され得る。免疫グロブリンγ−1のIgG1 Fc領域は、好ましくは、ヒンジ領域(少なくとも一部)、CH2領域およびCH3領域を含む分泌カセットにおいて使用される。いくつかの実施形態において、免疫グロブリンγ−1のFc領域は、CH2削除されたFcであり、これは、ヒンジ領域およびCH3領域の一部を含むが、CH2領域は含まない。CH2削除されたFcは、Gilliesら,1990,Hum.Antibod.Hybridomas,1:47により記載されている。いくつかの実施形態において、IgA、IgD、IgEまたはIgMのFc領域が使用される。
【0056】
Sp35−Fc融合タンパク質は、いくつかの異なる構造で構築され得る。1つの構造において、上記Sp35部分のC末端は、上記Fc部分のN末端に直接融合される。わずかに異なる構造において、短いポリペプチド(例えば、2個〜10個のアミノ酸)が、上記Sp35部分のN末端と上記Fc部分のC末端との間の融合中に組み込まれる。このようなリンカーは、いくつかの状態において生物学的活性を改善し得る、コンホメーションの柔軟性を提供する。ヒンジ領域の充分な部分が上記Fc部分において維持される場合、上記Sp35−Fc融合体は、二量体化し、従って、二価の分子を形成する。単量体Fc融合体の同種の集団は、単一の特異性を有する二価の二量体をもたらす。各々が異なる特異性を有する2つの単量体Fc融合体の混合物は、2種の特異性を有する二価の二量体をもたらす。
【0057】
対応するアミノ反応性基およびチオール反応性基を含む任意の数の架橋剤が、Sp35を血清アルブミンに結合するために使用され得る。適切なリンカーの例としては、チオール反応性マレイミドを挿入するアミン反応性架橋剤(例えば、SMCC、AMAS、BMPS、MBS、EMCS、SMPB、SMPH、KMUSおよびGMBS)が挙げられる。他の適切なリンカーは、チオール反応性ハロアセテート基(例えば、SBAP、SIA、SIAB)を挿入する。スルフヒドリル基との反応のための保護チオールまたは非保護チオールを提供し、還元可能な結合を生じるリンカーとしては、SPDP、SMPT、SATAおよびSATPが挙げられる。このような試薬は、市販されている(例えば、Pierce Chemicals)。
【0058】
結合は、Sp35ポリペプチドのN末端または血清アルブミン上のチオール部分を含まなくてもよい。例えば、Sp35−アルブミン融合体は、遺伝子工学的技術を用いて得られ得、上記Sp35部分は、そのN末端、C末端または両方で上記血清アルブミン遺伝子に融合される。
【0059】
Sp35ポリペプチドは、異種ペプチドに融合されて、上記Sp35部分の精製または同定を容易にし得る。例えば、ヒスチジンタグは、Sp35ポリペプチドに融合されて、市販のクロマトグラフィー媒体を用いる精製を容易にし得る。
【0060】
本発明のいくつかの実施形態において、Sp35融合構築体は、細菌におけるSp35部分の生成を促進するために使用される。このような構築体において、通常、高レベルで発現および/または分泌される細菌タンパク質が、Sp35ポリペプチドのN末端融合パートナーとして用いられる。例えば、Smithら,1988 Gene 67:31;Hoppら,1988,Biotechnology 6:1204;La Vallieら,1993,Biotechnology 11:187を参照のこと。
【0061】
本発明のいくつかの実施形態において、融合構築体としては、Sp35部分および第2のヒトNgR1結合部分(例えば、希乏突起神経膠細胞−ミエリン糖タンパク質(OMgp)部分、ミエリン関連糖タンパク質(MAG)部分またはNogo66部分)が挙げられる。このような構築体の利点としては、NgR1結合親和性の上昇が挙げられる。
【0062】
全長OMgpアミノ酸配列は、当該分野において公知である(GenBank受託番号P23515)。Sp35−OMgp融合体の具体例としては、以下が挙げられる:
Sp35(aa 34〜532)+IgG1 Fc+OMgp(アミノ酸残基25〜400);および
Sp35(aa 34〜532)+HSA+OMgp(アミノ酸残基25〜400)。
【0063】
全長MAGアミノ酸配列は、当該分野において公知である(GenBank受託番号A61084)。Sp35−MAG融合体の具体例としては、以下が挙げられる:
Sp35(aa 34〜532)+IgG1 Fc+MAG(アミノ酸残基12〜500);および
Sp35(aa 34〜532)+HSA+MAG(アミノ酸残基12〜500)。
【0064】
全長Nogoアミノ酸配列は、当該分野において公知である(NogoA GenBank受託番号AY102279)。Sp35−Nogo融合体の具体例としては、以下が挙げられる:
Sp35(aa 34〜532)+IgG1 Fc+Nogo66(NogoA アミノ酸残基1056〜1122);
Sp35(aa 34〜532)+HSA+Nogo66(NogoA アミノ酸残基1056〜1122);
Sp35(aa 34〜532)+IgG1 Fc+アミノNogo(NogoA アミノ酸残基1〜949);および
Sp35(aa 34〜532)+HSA+アミノNogo(NogoA アミノ酸残基1〜949)。
【0065】
適切な融合パートナーのアミノ末端およびカルボキシ末端でSp35部分を融合することにより、Sp35ポリペプチドの二価形態または四価形態が得られ得る。例えば、Sp35部分は、Ig部分のアミノ末端およびカルボキシ末端に融合され、2つのSp35部分を含む二価の単量体ポリペプチドを生成し得る。2つのこれらのモノマーが二量体化されると、Ig部分により、Sp35タンパク質の四価形態が得られる。このような多価形態は、標的に対する結合親和性の上昇を達成するために使用され得る。Sp35の多価形態はまた、Sp35部分を縦列に配置し、コンカテマーを形成することによって得られ得、これは、単独で用いられ得るかまたは融合パートナー(例えば、IgまたはHSA)に融合され得る。
【0066】
(結合ポリマー(ポリペプチド以外))
本発明のいくつかの実施形態は、Sp35ポリペプチドを含み、1つ以上のポリマーが、上記Sp35ポリペプチドに結合(共有結合)される。このような結合のために適切なポリマーの例としては、(上で議論された)ポリペプチド、糖ポリマーおよびポリアルキレングリコール鎖が挙げられる。代表的に、ポリマーは、以下のうちの1つ以上を改善する目的のために上記Sp35ポリペプチドに結合されるが、必ずしもそうでなくてもよい:可溶性、安定性またはバイオアベイラビリティ。
【0067】
Sp35ポリペプチドに対する結合のための好ましいポリマーのクラスは、ポリアルキレングリコールである。ポリエチレングリコール(PEG)が、特に好ましい。PEG部分(例えば、1PEGポリマー、2PEGポリマー、3PEGポリマー、4PEGポリマーまたは5PEGポリマー)は、各々のSp35ポリペプチドに結合されて、上記Sp35ポリペプチド単独と比較して、血清半減期を上昇し得る。PEG部分は非抗原性であり、本質的に、生物学的に不活性である。本発明の実施において使用されるPEG部分は、分枝鎖でも、非分枝鎖でもよい。
【0068】
上記Sp35ポリペプチドに結合されるPEG部分の数および個々のPEG鎖の分子量は、変化し得る。一般的に、上記ポリマーの分子量が大きくなるほど、上記ポリペプチドに結合されるポリマー鎖は少なくなる。好ましくは、上記Sp35ポリペプチドに結合される総ポリマー質量は、20kDa〜40kDaである。従って、1つのポリマー鎖が結合される場合、その鎖の好ましい分子量は、20kDa〜40kDaである。2つの鎖が結合される場合、各々の鎖の好ましい分子量は、10kDa〜20kDaである。3つの鎖が結合される場合、好ましい分子量は、7kDa〜14kDaである。
【0069】
上記ポリマー(例えば、PEG)は、上記ポリペプチド上の任意の適切な露出した反応性基を介して、そのSp35ポリペプチドに結合され得る。上記露出した反応性基は、例えば、N末端アミノ基もしくは内部リジン残基のεアミノ基または両方であり得る。活性化されたポリマーは、上記Sp35ポリペプチド上の任意のフリーのアミノ基において、反応して、共有結合し得る。上記Sp35ポリペプチドのフリーのカルボキシル基、適切に活性化されたカルボニル基、ヒドロキシル、グアニジル、イミダゾール、酸化された炭水化物部分およびメルカプト基はまた(利用可能である場合)、ポリマー結合のための反応性基として使用され得る。
【0070】
好ましくは、結合反応において、ポリペプチド濃度に依存して、1モルのポリペプチドあたり約1.0モル〜約10モルの活性化されたポリマーが用いられる。通常、選択される比は、上記反応の最大化と、上記Sp35部分の所望される薬理学的活性を損ない得る(しばしば、非特異的な)副反応の最小化との間の均衡を表す。好ましくは、上記Sp35ポリペプチドの生物学的活性の少なくとも50%(例えば、本明細書中で記載されるかまたは当該分野で公知のあらゆるアッセイにおいて実証される場合)が維持され、最も好ましくは、約100%が維持される。
【0071】
上記ポリマーは、従来の化学を用いて上記Sp35ポリペプチドに結合され得る。例えば、ポリアルキレングリコール部分は、上記Sp35ポリペプチドのリジンεアミノ基に結合され得る。リジン側鎖に対する結合は、N−ヒドロキシルスクシニミド(NHS)活性エステル(例えば、PEGスクシンイミジルサクシネート(SS−PEG)およびスクシンイミジルプロピオネート(SPA−PEG))を用いて実施され得る。適切なポリアルキレングリコール部分としては、例えば、カルボキシメチル−NHS、ノルロイシン−NHS、SC−PEG、トレシレート(tresylate)、アルデヒド、エポキシド、カルボニルイミダゾールおよびPNPカーボネートが挙げられる。これらの試薬は、市販されている。さらなるアミン反応性PEGリンカーは、スクシンイミジル部分に置換され得る。これらとしては、例えば、イソチオシアネート、ニトロフェニルカーボネート、エポキシドおよびベンゾトリアゾールカーボネートが挙げられる。好ましくは、選択性および反応程度(extent or reaction)を最大化するように、条件が選択される。このような反応条件の最適化は、当該分野における通常の技術内である。
【0072】
PEG化は、当該分野において公知の任意のPEG化反応により実施され得る。例えば、Focus on Growth Factors,3:4−10,1992;公開された欧州特許出願EP 0 154 316およびEP 0 401 384を参照のこと。PEG化は、反応性ポリエチレングリコール分子(または類似の反応性水溶性ポリマー)とのアシル化反応またはアルキル化反応を用いて、実施され得る。
【0073】
アシル化によるPEG化は、一般的に、ポリエチレングリコールの活性エステル誘導体の反応を含む。任意の反応性PEG分子が、上記PEG化において用いられ得る。好ましい活性化PEGエステルは、N−ヒドロキシスクシニミド(NHS)に対してエステル化されたPEGである。本明細書中で使用される場合、「アシル化」は、治療タンパク質と水溶性ポリマー(例えば、PEG)との間の以下のタイプの結合を含むが、これらに限定されない:アミド、カーボネート、ウレタンなど。Bioconjugate Chem.5:133−140,1994を参照のこと。反応パラメータは、上記Sp35ポリペプチドに損傷を与えるかまたは不活性化する温度条件、溶媒条件およびpH条件を避けるように選択されるべきである。
【0074】
好ましくは、上記連結する結合は、アミドである。好ましくは、結果としてもたらされる生成物のうちの少なくとも95%が、モノPEG化、ジPEG化またはトリPEG化される。しかし、より高い程度のPEG化を有するいくつかの種が、使用される具体的反応条件に依存した量で形成され得る。必要に応じて、精製されたPEG化種は、従来の精製方法によってその混合物(特に、未反応種)から分離され、これら従来の精製方法としては、例えば、透析、塩析、限外濾過、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーおよび電気泳動が挙げられる。
【0075】
アルキル化によるPEG化は、一般的に、還元剤存在下でのPEGの末端アルデヒド誘導体とSp35との反応を含む。さらに、実質的にSp35のN末端アミノ基においてのみのPEG化(すなわち、モノPEG化タンパク質)に都合のよいように上記反応条件を操作し得る。モノPEG化またはポリPEG化のいずれかにおいて、上記PEG基は、好ましくは、−CH2−NH−基を介して上記タンパク質に結合される。特に−CH2−基に関して、このタイプの結合は、「アルキル」結合として公知である。
【0076】
モノPEG化生成物を生成するための還元的アルキル化を介した誘導体化は、誘導体化のために利用可能な一級アミノ基の異なるタイプの異なる反応性(リジン対N末端)を利用する。上記反応は、リジン残基のεアミノ基とタンパク質のN末端アミノ基との間のpKaの差を利用可能なpHにおいて、実施される。このような選択的誘導体化により、反応性基(例えば、アルデヒド)を含む水溶性ポリマーとタンパク質との結合は、制御される:上記ポリマーとの結合は、上記タンパク質のN末端で主に起こり、他の反応性基(例えば、上記リジン側鎖アミノ基)の有意な改変は、起こらない。
【0077】
上記アシル化方法およびアルキル化方法の両方において使用されるポリマー分子は、水溶性ポリマーの中から選択される。選択されるポリマーは、単一の反応性基(例えば、アシル化のための活性エステルまたはアルキル化のためのアルデヒド)を有するように、好ましくは、重合度が本発明の方法のために提供されるように制御され得るように、改変されるべきである。例示的な反応性PEGアルデヒドは、水溶性であるポリエチレングリコールプロピオンアルデヒドまたはそのモノC1〜C10アルコキシ誘導体もしくはそのモノC1〜C10アリールオキシ誘導体である(米国特許第5,252,714号を参照のこと)。上記ポリマーは、分枝鎖であっても、非分枝鎖であってもよい。上記アシル化反応のために、選択されるポリマーは、単一の反応性エステル基を有する。還元的アルキル化のために、選択されるポリマーは、単一の反応性アルデヒド基を有する。一般的に、上記水溶性ポリマーは、天然に生じるグリコシル残基から選択される。なぜなら、これらは、通常、哺乳動物組換え発現系によって、より都合よく作製されるからである。
【0078】
PEG化Sp35を調製するための方法は、一般的に、(a)Sp35タンパク質またはSp35ポリペプチドと、ポリエチレングリコール(例えば、PEGの反応性エステル誘導体または反応性アルデヒド誘導体)とを、それによってその分子が1つ以上のPEG基に結合されるようになる条件下で反応させる工程、および(b)その反応生成物を得る工程、を包含する。一般的に、上記アシル化反応のための最適反応条件は、公知のパラメータおよび所望される結果に基づいて、場合に応じて決定される。例えば、PEG:タンパク質の比が大きくなるほど、ポリPEG化生成物の百分率は大きくなる。
【0079】
モノ−ポリマー/Sp35の実質的に均一な集団を生成するための還元的アルキル化は、一般的に、以下の工程:(a)Sp35のN末端アミノ基の選択的改変を可能にする(pen−nit)ために適切なpHの還元的アルキル化条件下、Sp35タンパク質またはSp35ポリペプチドと、反応性PEG分子とを反応させる工程;および(b)その反応性生物を得る工程、を包含する。
【0080】
モノ−ポリマー/Sp35の実質的に均一な集団のために、上記還元的アルキル化反応条件は、上記水溶性ポリマー部分と上記Sp35のN末端との選択的結合を可能にする反応条件である。このような反応条件は、一般的に、リジン側鎖アミノ基とN末端アミノ基との間のpKaの差を考慮に入れる。本発明の目的のために、好ましいpHは、3〜9の範囲内、好ましくは、3〜6の範囲内である。
【0081】
Sp35ポリペプチドは、タグ(例えば、その後にタンパク分解により放出され得る部分)を含み得る。従って、上記リジン部分は、His−タグ改変体と低分子量リンカー(例えば、Traut’s試薬(Pierce))とが最初に反応することにより、選択的に改変され得、この低分子量リンカーは、そのリジンおよびN末端の両方と反応し、次いでhisタグを放出する。次いで、上記ポリペプチドは、フリーのSH基を含み、このフリーのSH基は、チオール反応性末端基(例えば、マレイミド基、ビニルスルホン基、ハロアセテート基またはフリーもしくは保護されたSH)を含むPEGにより選択的に改変され得る。
【0082】
Traut’s試薬は、PEG結合のための特異的部位を設定する任意のリンカーにより置換され得る。例えば、Traut’s試薬は、SPDP、SMPT、SATAまたはSATP(Pierce)により置換され得る。同様に、上記タンパク質と、マレイミドを挿入するアミン反応性リンカー(例えば、SMCC、AMAS、BMPS、MBS、EMCS、SMPB、SMPH、KMUSまたはGMBS)、ハロアセテート基を挿入するアミン反応性リンカー(SBAP、SIA、SIAB)またはビニルスルホン基を挿入するアミン反応性リンカーとが反応し得、結果として生じた生成物と、フリーのSHを含むPEGとが反応し得る。
【0083】
いくつかの実施形態において、上記ポリアルキレングリコール部分は、上記Sp35ポリペプチドのシステイン基に結合される。結合は、例えば、マレイミド基、ビニルスルホン基、ハロアセテート基またはチオール基を用いてもたらされ得る。
【0084】
必要に応じて、上記Sp35ポリペプチドは、不安定な結合を介して、上記ポリエチレングリコール部分に結合される。上記不安定な結合は、例えば、生物化学的加水分解、タンパク分解またはスルフヒドリル切断で切断され得る。例えば、上記結合はインビボ(生理的)条件下で切断され得る。
【0085】
上記反応は、上記反応性基がN末端のαアミノ基上である場合、生物学的に活性な物質と不活性ポリマーとを反応させるために使用される任意の適切な方法により、好ましくは、約pH5〜8(例えば、pH5、6、7または8)で行われ得る。一般的に、上記プロセスは、活性化されたポリマーを調製する工程およびその後の上記タンパク質とその活性化されたポリマーとを反応させ、処方物のための適切な可溶性タンパク質を生成する工程を包含する。
【0086】
(ベクター)
本発明は、Sp35ポリペプチドをコードする核酸を含むベクターを提供する。ベクターおよび本発明の核酸が作動可能に連結する発現制御配列の選択は、所望される機能特性(例えば、タンパク質発現)および形質転換されるべき宿主細胞に依存する。
【0087】
作動可能に連結されたコード配列の発現調節のために有用な発現制御エレメントは、当該分野において公知である。例としては、誘導性プロモーター、構成性プロモーター、分泌シグナルおよび他の調節エレメントが挙げられるが、これらに限定されない。誘導性プロモーターが使用される場合、それは、例えば、上記宿主細胞培地における栄養状態変化または温度変化によって制御され得る。
【0088】
上記ベクターとしては、原核生物レプリコン(すなわち、細菌宿主細胞において、染色体外の組換えDNA分子の自律的な複製および維持を行う能力を有するDNA配列)が挙げられ得る。このようなレプリコンは、当該分野において周知である。さらに、原核生物レプリコンを含むベクターはまた、その発現が検出可能マーカー(例えば、薬物耐性)を与える遺伝子を含み得る。細菌薬物耐性遺伝子の例は、アンピシリンまたはテトラサイクリンに対する耐性を与える遺伝子である。
【0089】
原核生物レプリコンを含むベクターはまた、細菌宿主細胞において、上記コード遺伝子配列の発現を行うための原核生物プロモーターまたはバクテリオファージプロモーターを含み得る。細菌宿主に適合性のプロモーター配列は、代表的に、発現されるべきDNAセグメントの挿入のために都合のよい制限酵素部位を含むプラスミドベクター中に、提供される。このようなプラスミドベクターの例は、pUC8、pUC9、pBR322およびpBR329(BioRad)、pPLおよびpKK223(Pharmacia)である。任意の適切な原核生物宿主が、本発明のタンパク質をコードする組換えDNA分子を発現するために、使用され得る。
【0090】
真核生物細胞発現ベクターは、当該分野において公知であり、市販されている。代表的に、このようなベクターは、所望されるDNAセグメントの挿入のために都合のよい制限酵素部位を含む。例示的なベクターとしては、pSVLおよびpKSV−10(Pharmacia)、pBPV−1、pML2d(International Biotechnologies)、pTDT1(ATCC 31255)、レトロウイルス発現ベクターpMIG、アデノウイルスシャトルベクターpDC315およびAAVベクターが挙げられる。
【0091】
真核生物細胞発現ベクターは、選択可能マーカー(例えば、薬物耐性遺伝子)を含み得る。ネオマイシンホスホトランスフェラーゼ(neo)遺伝子(Southernら,1982,J.Mol.Anal.Genet.1:327−341)は、このような遺伝子の例である。
【0092】
抗体または抗体フラグメントを発現するために、一部または全長の軽鎖および重鎖をコードするDNAが、発現ベクター(例えば、プラスミド、レトロウイルス、コスミド、YAC、EBV由来エピソームなど)に挿入される。上記発現ベクターおよび発現制御配列は、使用される発現宿主細胞に適合性であるように選択される。上記抗体軽鎖遺伝子および上記抗体重鎖遺伝子は、別個のベクターに挿入され得る。いくつかの実施形態において、両方の遺伝子は、同じ発現ベクターに挿入される。
【0093】
都合のよいベクターは、機能的に完全なヒトCH免疫グロブリン配列またはヒトCL免疫グロブリン配列をコードするベクターである。好ましくは、制限酵素部位が作製され、それにより、任意のVH配列またはVL配列が、容易に挿入され、発現され得る。このようなベクターにおいて、スプライシングは、通常、挿入されたJ領域のスプライスドナー部位とヒトC領域の前のスプライスアクセプター部位との間で、そしてまた、ヒトCHエキソン内に生じるスプライス領域で、生じる。ポリアデニル化および転写終結は、上記コード領域の下流の天然の染色体部位で生じる。上記組換え発現ベクターはまた、宿主細胞からの上記抗体鎖の分泌を促進するシグナルペプチドをコードし得る。
【0094】
哺乳動物宿主細胞発現のための好ましい調節配列としては、哺乳動物細胞における高レベルのタンパク質発現を導くウイルスエレメント(例えば、レトロウイルスLTR由来のプロモーターおよびエンハンサー、サイトメガロウイルス(CMV)(例えば、CMVプロモーター/エンハンサー)、シミアンウイルス40(SV40)(例えば、SV40プロモーター/エンハンサー)、アデノウイルス(例えば、アデノウイルス主要後期プロモーター(AdMLP))、ポリオーマ)ならびに強力な哺乳動物プロモーター(例えば、天然の免疫グロブリンプロモーターおよびアクチンプロモーター)が挙げられる。ウイルス調節エレメントおよびその配列のさらなる記載については、例えば、Stinskiの米国特許第5,168,062号;Bellの同第4,510,245号;およびSchaffnerの同第4,968,615号を参照のこと。
【0095】
組換え発現ベクターは、宿主細胞においてそのベクターの複製を調節する配列(例えば、複製起点)および選択マーカー遺伝子を有してもよい。上記選択マーカー遺伝子は、そのベクターが導入された宿主細胞の選択を容易にする(例えば、Axelの米国特許第4,399,216号;同第4,634,665号および同第5,179,017号を参照のこと)。例えば、代表的に、上記選択マーカー遺伝子は、上記ベクターが導入された宿主細胞に薬物(例えば、G418、ハイグロマイシンまたはメトトレキサート)に対する耐性を与える。好ましい選択マーカー遺伝子としては、ジヒドロ葉酸還元酵素(DHFR)遺伝子(メトトレキサート選択/増殖を用いるdhfr−宿主細胞における使用のため)およびneo遺伝子(G418選択のため)が挙げられる。
【0096】
Sp35ポリペプチドおよび抗Sp35抗体をコードする核酸分子ならびにこれらの核酸分子を含むベクターは、適切な宿主細胞の形質転換のために使用され得る。任意の適切な方法により、形質転換され得る。哺乳動物細胞への外因性DNAの導入のための方法は、当該分野において周知であり、これらの方法としては、デキストラン媒介性トランスフェクション、リン酸カルシウム沈殿法、ポリブレン媒介性トランスフェクション、プロトプラスト融合、エレクトロポレーション、リポソーム中へのポリヌクレオチドの封入および核へのDNAの直接的なマイクロインジェクションが挙げられる。さらに、核酸分子は、ウイルスベクターにより哺乳動物細胞に導入され得る。
【0097】
宿主細胞の形質転換は、用いられるベクターおよび宿主細胞にとって適切な従来の方法により達成され得る。原核生物宿主細胞の形質転換のために、エレクトロポレーション法および塩処理法が用いられ得る(Cohenら,1972,Proc.Natl.Acad.Sci.USA 69:2110−2114)。脊椎動物細胞の形質転換のために、エレクトロポレーション法、カチオン性脂質法または塩処置法が用いられ得る。例えば、Grahamら,1973,Virology 52:456−467;Wiglerら,1979,Proc.Natl.Acad.Sci.USA 76:1373−1376fを参照のこと。
【0098】
発現のための宿主として利用可能な哺乳動物細胞株は、当該分野において公知であり、これらの細胞株としては、American Type Culture Collection(ATCC)から入手可能な多くの不死化細胞株が挙げられる。これらとしては、特に、チャイニーズハムスター卵巣(CHO)細胞、NSO、SP2細胞、HeLa細胞、乳仔ハムスター腎臓(BHK)細胞、サル腎臓細胞(COS)、ヒト肝細胞癌細胞(例えば、Hep G2)、A549細胞および多くの他の細胞株が挙げられる。
【0099】
産生細胞株からのポリペプチドの発現は、公知の技術を用いて促進され得る。例えば、グルタミンシンセターゼ(GS)系は、特定条件下で発現を促進するために、一般的に使用される。例えば、欧州特許第0216846号、同第0256055号および同第0323997号ならびに欧州特許出願番号89303964.4を参照のこと。
【0100】
(宿主細胞)
宿主細胞は、原核生物起源または真核生物起源であり得る。好ましい真核生物の宿主細胞としては、限定されないが、酵母細胞および哺乳動物細胞(例えば、チャイニーズハムスター卵巣細胞(CHO)(ATCC登録番号CCL61)、NIHのスイスマウス胚細胞NIH−3T3(ATCC登録番号CRL1658)、および新生児のハムスター腎臓細胞(BHK))が挙げられる。他の有用な真核生物の宿主細胞としては、昆虫細胞および植物細胞が挙げられる。例示的な原核生物の宿主細胞は、E.coliおよびStreptomycesである。
【0101】
(処方物)
Sp35ポリペプチド、抗Sp35抗体、または抗Sp35抗体の抗原結合フラングメントを含む組成物は、適切な薬学的に受容可能なキャリアを含み得る。例えば、それらは、作用部位への送達のために設計された調製物への、活性化合物の処理を促進する賦形剤および/または補助剤を含み得る。非経口投与のための適切な処方物としては、水溶性形態(例えば、水溶性塩)の活性化合物の水溶液が挙げられる。さらに、適切な油状の注射懸濁液のような活性化合物の懸濁液が、投与され得る。適切な新油性溶媒またはビヒクルとしては、脂肪油(例えば、ゴマ油、または合成脂肪酸エステル(例えば、オレイン酸エチルまたはトリグリセリド))が挙げられる。水溶性の注射懸濁液は、懸濁液の粘性を増加させる物質(例えば、カルボキシメチルセルロースナトリウム、ソルビトールおよびデキストランが挙げられる)を含み得る。必要に応じて、懸濁液はまた、安定剤を含み得る。リポソームもまた、細胞または間隙の空間の中への送達のために本発明の分子をカプセル化するために使用され得る。例示的な薬学的に受容可能なキャリアは、生理学的に適合性のある溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤、水、生理食塩水、リン酸緩衝化生理食塩水、デキストロース、グリセロール、エタノールなどである。いくつかの実施形態において、組成物は、等張剤(例えば、糖、ポリアルコール(例えば、マンニトール)、ソルビトール、または塩化ナトリウム)を含む。いくつかの実施形態において、組成物は、薬学的に受容可能な物質(例えば、湿潤剤または少量の補助的な物質(例えば、湿潤剤もしくは乳化剤、防腐剤または緩衝液))を含み、これらは、活性成分の貯蔵寿命または有効性を高める。
【0102】
本発明の組成物は、例えば、液体(例えば、注射剤および不溶解性溶液)、分散剤、懸濁液、半固形ならびに固形投薬形態を含む、様々な形態であり得る。好ましい形態は、投与および治療の用途の様式に依存する。
【0103】
組成物は、溶液、ミクロエマルジョン、分散、リポソーム、または高い薬物濃度に適する他の指示される構造として処方され得る。滅菌注射溶液は、上に列挙した成分の1つまたは組み合わせと共に、適切な溶媒中に、必要とされる量の活性成分を組み込むことによって調製され得、必要な場合、続いて、濾過滅菌される。概して、分散剤は、塩基性分散媒体および上に列挙したものからの必要とされる他の成分を含む滅菌ビヒクルの中に活性成分を組み込むことによって調製される。滅菌注射溶液の調製のための滅菌粉末の場合、好ましい調製方法は、活性成分の粉末に加えて、前以て滅菌濾過した溶液から任意のさらなる所望の成分を生じる真空乾燥および凍結乾燥である。溶液の適切な流動性が、例えば、コーティング(例えば、レシチン)の使用、分散剤の場合は、必要とされる粒子サイズの維持、および界面活性剤の使用によって維持され得る。注射用組成物の長期の吸収は、吸収を遅らせる薬剤(例えば、モノステアリン酸塩およびゼラチン)を組成物中に含むことによってもたらされ得る。
【0104】
活性成分は、制御放出性の処方物またはデバイスを用いて処方され得る。そのような処方物およびデバイスの例としては、インプラント、経皮パッチ、およびマイクロカプセルに入れられた送達系が挙げられる。生分解性で、生体適合性のポリマー(例えば、酢酸エチレンビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸)が、使用され得る。そのような処方物およびデバイスの調製のための方法は、当該分野において公知である。例えば、Sustained and Controlled Release Drug Delivery Systems,J.R.Robinson,ed.,Marcel Dekker,Inc.,New York,1978を参照のこと。
【0105】
注射用の貯蔵処方物は、生分解性ポリマー(例えば、ポリラクチド−ポリグリコリド)中でマイクロカプセルに入れられた薬物のマトリックスを形成することによって作製され得る。ポリマーに対する薬物の割合、および使用されるポリマーの性質に依存して、薬物放出の速度が、制御され得る。他の例示的な生分解性ポリマーは、ポリオルトエステルおよびポリ無水物である。貯蔵注射用処方物はまた、リポソームまたはマイクロエマルジョン中に薬物を包括することによって調製され得る。
【0106】
補助的な活性化合物は、組成物の中に組み込まれ得る。いくつかの実施形態において、Sp35ポリペプチド、抗Sp35抗体またはそのフラグメントは、抗NgR1抗体、もしくはその抗原結合断片、または溶解性のNgR1ポリペプチドもしくはNgR1融合タンパク質と一緒に同時投与される。
【0107】
投薬レジメンは、最適な所望の反応を提供するように調整され得る。例えば、単一のボーラスが投与され得、いくつかの分割用量が、時間をかけて投与され得るか、または用量は、治療状態の緊急性によって示されるにつれて、比例して減少もしくは増加され得る。投与の容易さおよび投薬量の均一性のための投薬単位形態で、非経口組成物を処方することが有利である。例えば、Remington’s Pharmaceutical Sciences(Mack Pub.Co.,Easton,PA1980)を参照のこと。
【0108】
活性化合物に加えて、液体投薬形態は、不活性成分(例えば、水、エチルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、ジメチルホルムアミド、油、グリセロール、テトラヒドロフルフリルアルコール、ポリエチレングリコール、およびソルビタンの脂肪酸エステル)を含み得る。
【0109】
(遺伝子治療)
Sp35ポリペプチドは、軸策の伸長の阻害を減少させることが治療的に有益であるCNS疾患、CNS障害またはCNS損傷の処置のための遺伝子治療アプローチを使用して、哺乳動物(例えば、ヒト患者)においてインビボで生成され得る。これは、適切な発現制御配列に作動可能に連結される適切なSp35ポリペプチドをコードする核酸の投与を含む。好ましくは、これらの配列は、ウイルスベクターの中へ組み込まれる。このような遺伝子治療のための適切なウイルスベクターとしては、アデノウイルスベクター、レンチウイルスベクター、バキュロウイルスベクター、Epstein Barrウイルスベクター、パポバウイルスベクター、ワクシニアウイルスベクター、単純ヘルペスベクター、およびアデノ関連ウイルス(AAV)ベクターが挙げられる。ウイルスベクターは、複製欠損のウイルスベクターであり得る。好ましいアデノウイルスベクターは、そのE1遺伝子またはE3遺伝子に欠損を有し得る。アデノウイルスベクターが使用される場合、好ましくは、哺乳動物は、選択マーカー遺伝子をコードする核酸に曝露されない。
【実施例】
【0110】
本発明はさらに、以下の実験の実施例によって例示される。実施例は、例示の目的のためだけに提供され、あらゆる方法においても本発明の範囲または内容を制限すると解釈されるべきではない。
【0111】
(実施例1:Sp35発現パターン)
ヒトの組織におけるSp35の発現をノーザンブロット解析によって調べた。12個のヒトの主要な組織または14個のヒトのCNS組織を含む複数の組織ブロットを、P32で標識したSp35プローブ(Sp35cDNA配列のヌクレオチド150〜450)を用いて68℃で一晩ハイブリダイズした。そのブロットを、2×SSC、0.5%SDSで3回洗浄し、次いで、0.5×SSC、0.1%SDSで3回洗浄した。次いで、そのブロットをX線フィルムに露光し、mRNAレベルをオートラジオグラフィーによって可視化した。
【0112】
Sp35は、ヒトの脳においては心臓、高度に発現したが、骨格筋、結腸、胸腺、脾臓、腎臓、肝臓、小腸、胎盤、肺および末梢血白血球では発現しなかった。Sp35は、前頭皮質、後頭皮質、嗅内皮質、海馬、嗅球、線条、視床、小脳、中脳、脳橋、髄質および脊髄から単離された組織を含む、試験した全ての脳組織において発現した。吻/索軸に沿った遺伝子発現の勾配を、Sp35について観察し、皮質において最もレベルが高く、そして脊髄において最もレベルが低かった。
【0113】
Sp35が特定の脳細胞で発現するかどうかを、免疫組織化学(IHC)染色を使用して決定した。4%パラホルムアルデヒドで固定したラットの脳、脊髄の切片、または初代顆粒状ニューロン培養物を、示されるようにSp35に対する一次抗体と一緒にインキュベートし、その後、Alexa480またはAlexa590(Molecular Probes Inc.)に結合した二次抗体と一緒にインキュベートした。次いで、その切片を、Vectashieldに乗せて、蛍光顕微鏡検査によって可視化した。IHCのために使用される抗Sp35特異的抗体を、MorPhosys技術を使用してFabファージディスプレイライブラリーから作製した。
【0114】
Sp35は、ニューロンおよび希突起膠細胞において特異的に発現したが、星状細胞では発現しなかった。このことを、抗星状細胞マーカーGFAP、希突起膠細胞マーカー(O4)に対する抗体、およびニューロンマーカーβIIIチューブリンに対する抗体(全て抗Sp35抗体を用いて対比染色を行う)を含む種々の薬剤で、ラットの脳組織切片を染色し、実験において測定した。希突起膠細胞およびニューロンは、抗Sp35抗体で強く染色された。星状細胞の染色は、観測されなかった。
【0115】
Sp35の発現パターンの非依存性の確認として、本発明者らは、ラットの精製した星状細胞、希突起膠細胞、および小脳顆粒状ニューロンの初代細胞培養物から抽出したmRNA(Ambion kit)を使用して半定量的なRT−PCRを実施した。
【0116】
【数1】
26サイクルの後、強いバンドをニューロン由来のmRNAにおいて観察し、識別可能であるが、弱いシグナルを希突起膠細胞mRNAにおいて検出し、星状細胞においてはバンドが観察されなかった。
【0117】
(実施例2:Sp35−Fc融合タンパク質)
Sp35の生物学的機能を研究するために、ヒトSp35の細胞外部分(残基1〜531)をヒトIgG1のヒンジ領域およびFc領域と融合して、構築物を作製した。ヒトSp35を部分的にコードする配列を、
【0118】
【数2】
を使用してクローン227.2(Incyte)からPCRによって得た。
【0119】
平滑末端PCR産物を、PCR SCRIPT AMP−sp35を作製するためにPCR SCRIPT AMPベクター(Stratagene)のSrfI部位の中へサブクローニングした。SalIフラグメントを、PCR SCRIPT AMP−sp35から単離し、PCRCAMP Igベクター(StratageneベクターPCR SCRIPT AMPの誘導体で、このベクターでは、Fcγ配列がSalI(5’)〜NotI(3’)フラグメントとしてサブクローンされている)の中へサブクローニングし、インフレームでSp35シグナル配列および外部ドメイン配列(コドン1〜531)をヒトIg1のヒンジ領域およびFc領域をコードする配列と融合した。正しい単離物を同定し、Sp35 Fcフラグメントを包含するNotIフラグメントを、293E発現ベクター(市販の発現ベクターREP4(Invitrogen)の誘導体である、CH274)の単一のNot Iクローニング部位の中へサブクローニングした。新しいベクターである、CH274/sp35−FcによってコードされるSp35−Fc融合を、プラスミドGT123としてDNA配列決定によって確認した。
【0120】
Sp35−Fc融合タンパク質を発現する安定な細胞株を、プラスミドGT123を用いてCHO宿主細胞DG44のエレクトロポレーションによって作製した。トランスフェクトされたCHO細胞を、ヌクレオシド非依存性増殖について選択するために、10%の透析血清および4mMグルタミンの存在下のα−MEM中で培養した。トランスフェクションの14日後、細胞に新鮮な培地を与えた。Sp35−Fcを発現する細胞をスクリーニングするために、CHO細胞をフィコエリトリン(PE)標識したヤギ抗ヒトIgG(Jackson Labs)で標識し、FACS Mo−Flo(Cytomation)において高速フローサイトメトリーソーティングに供した。最も高いレベルのSp35−Igを発現した細胞を選択した。これらの細胞を7日間培養液で拡張させて、次いで、再び標識し、再びソートした。最も高いレベルのSp35−Igを発現する細胞を、96−ウェルプレート中の個々のクローンとして単離した。これらのクローンを2週間増殖させ、次いで、発現レベルを検査するために、FACS解析の1日前に新鮮な培地を与えた。最も高いレベルのSp35−Fcを発現したクローンを拡張し、凍結して、セルバンクを確立した。その細胞株を、無血清のBCM16培地の懸濁培養物中で増殖させるために適合した。これらのクローンによって生成されたSp35−Fcの力価を、4〜5継代の間、37℃での増殖細胞株によって測定し、次いで、50%最大細胞密度まで細胞を増殖させ、生存可能な細胞密度が75%に落ちるまで、28℃で10〜15日間培養した。この時点で、培地を収集し、遠心分離によって細胞および残屑を除き、プローブとして抗ヒトIg抗体(Jackson Lab)を使用して、ウェスタンブロット解析によってSp35−Fcレベルについての培養上清を力価測定した。
【0121】
Sp35−Fc融合タンパク質を、以下のような浄化した培養培地から精製した:9mlの1M HEPES pH7.5を900mlの馴化培地に添加した。この培地を3mlのプロテインAセファロース(Pharmacia)に対して4℃で3時間、負荷して一括処理した。樹脂を1.5cm(I.D.)のカラムで回収し、3ml PBSで4回、800mM NaClを含む4mlのPBSで2回洗浄し、次いで、3mLのPBSで再び洗浄した。Sp35−Fcを、1.5mL画分において25mM NaH2PO4 pH2.8、100mM NaClを用いてカラムから溶出し、75μLの0.5M NaH2PO4 pH8.6を添加することによって中和した。ピークタンパク質含有画分を、280nmでの吸光度によって同定して、プールし、さらに1mLプロテインAカラム上での精製に供した。負荷するより前に、NaClを600mMまで、そしてHEPES pH7.5を50mMまで添加した。カラムを、600μLの10mM HEPES pH7.5、1M NaClで2回洗浄し、次いで、1mL PBSで洗浄した。Sp35−Fcを、25mM NaH2PO4 pH2.8、100mM NaClを用いてカラムから溶出し、0.5mL画分を回収して、25μLの0.5M NaH2PO4 pH8.6を添加することによって中和した。ピークタンパク質含有画分を、280nmでの吸光度によって同定して、プールした。SDS−PAGEを還元することによって、Sp35−Igを90kDaの見かけの質量を有するシングルバンド(95%より高い純粋)として移動した。還元していない条件の下で、タンパク質をおよそ180kDaの質量を有する二量体として、電気泳動した。精製したSp35−Fcを等分して、−70℃で保存した。Sp35アミノ酸1〜531およびヒトIgG1 Fcを含むGT123のNotIフラグメントを、DB002を作製するためにPV90ベクターのNotI部位の中にサブクローニングした。
【0122】
(実施例3:His−Ap−Sp35融合タンパク質)
Sp35についてのレセプターを研究および単離するために、Hisタグ化アルカリホスファターゼ(His−Ap)融合タンパク質として、COS7細胞およびCHO細胞においてタンパク質を発現させた。プラスミドを以下のように構築した:Sp35の細胞外ドメイン(a.a.34〜532)を、Eco RI切断部位(下線)を含むプライマー(フォワード)
【0123】
【数3−1】
およびXba I切断部位(下線)を含むプライマー(リバース)
【0124】
【数3−2】
を使用してPCR増幅した。PCR産物を、Xba Iで切断し、その結果生じる粘着末端を、T4DNAポリメラーゼで満たし、次いで、Eco RIで消化し、そしてゲル精製した。消化した生成物を、His−AP−pcDNA1.1ベクター(Invitrogen)由来のEco RI His−APフラグメント中に埋め込まれたHindIIIに連結した。His−AP−Sp35フラグメントを、Hind IIIおよびEco RIで消化し、満たし、次いで、ベクターpV90のNot Iを満たした部位の中へ連結した。挿入物のDNA配列を、DNA配列決定によって確認した。
【0125】
COS7細胞をトランスフェクション前日に分けた。His−AP−Sp35
ベクターDNA(8μg)を、リポフェクタミン(Invitrogen)を使用して、5×106個の細胞をトランスフェクトするために使用した。馴化培地を、トランスフェクションの48時間後に、収集した。
【0126】
本発明者らは、pV90プラスミドを使用してHis−AP−Sp35融合タンパク質を発現するCHO細胞株を開発した。CHO宿主細胞DG44(2×106個の細胞)を、エレクトロポレーションによって100μgのプラスミドを用いてトランスフェクトした。細胞を、ヌクレオシド非依存性増殖について選択するために、10%の透析血清および4mMグルタミンの存在下のα−MEM中で培養した。トランスフェクションの14日後、FACS Mo−Flo(Cytomation)ソーティングによるスクリーニング前に、新鮮な培地を細胞に与えた。トランスフェクトしたCHO細胞を、ヒト胎盤アルカリホスファターゼ(Sigma)に対するマウスモノクローナル抗体8B6で標識した。二次抗体である、PE標識したヤギ抗マウスIgGを使用して、トランスフェクトした細胞に特異的なシグナルを生成させた。PE標識後、細胞を高速フローサイトメトリーソーティングに供し、上位5%を選択した。
【0127】
His−AP−Sp35を含む馴化した培地を生成するために、最も高いレベルのHISApSp35を発現した細胞を選択した。細胞株を、無血清の培地(BCM16)の懸濁培養液中で増殖させるために適合させた。これらのクローンによって生成したHis−AP−Sp35の力価を、4〜5継代の間、37℃で細胞株を増殖させることによって測定し、次いで、細胞を50%の最大細胞密度まで増殖させ、これらを生存可能な細胞密度が75%に落ちるまで、28℃で10〜15日間、培養した。培地を収集し、遠心分離によって、細胞および残屑を除き、培養上清を、プローブとして抗ヒトAP抗体(Jackson Labs)を使用するウェスタンブロット解析によってHis−AP−Sp35レベルについて力価測定した。
【0128】
His−AP−Sp35を、以下のような馴化培地から精製した:His−AP−Sp35を発現するCHO細胞由来の400mLの馴化培地を400mLの水で希釈した。トリエタノールアミンpH8.5を、0.5Mストックからの25mMに添加し、サンプルを、6mlのFractogel TMAE(EM Industries)アニオン交換樹脂の上に4℃で2時間、負荷して一括処理した。樹脂を、1.5cm(I.D.)カラム中で回収し、6mLの10mM HEPES pH7.5、50mM NaClで2回洗浄した。AP−Sp35を、2mL画分において10mM HEPES pH7.5、200mM NaClを用いてカラムから溶出した。ピーク画分を、AP活性のモニタリング、およびSDS−PAGEによって同定した。TMAEカラムからの貫流画分をさらに、300mlの水で希釈し、6mlのTMAE樹脂の上で4℃で一晩、一括処理で負荷した。樹脂を回収し、上記のように洗浄して、10mM HEPES pH7.5、150mM NaClで溶出した。ピーク画分を再び、AP活性のモニタリング、およびSDS−PAGEによって同定した。第1のカラムからのHis−AP−Sp35は、50%純粋で、第2のカラムからのAP−Sp35は、90%純粋であった。還元条件の下で、His−Ap−Sp35は、130kDaの見かけの質量でSDS−PAGEゲル上を移動した。90%純粋な物質は、ほとんどの調査について適切であったが、いくつかの研究については、His−AP−Sp35を、Ni−NTAアガロース樹脂(Qiagen)でさらに精製した。NaClを、TMAEカラムからの溶出画分に800mM添加し、0.5MトリエタノールアミンpH8.5および1MイミダゾールpH7.0をそれぞれ、25mMおよび15mMまで添加した。4.5mlのサンプルを、400μL NiNTAカラムの上に負荷した。そのカラムを、25mMトリエタノールアミン pH8.5、800mM NaCl、15mMイミダゾールで3回洗浄し、His−AP−Sp35を、200mMイミダゾールpH7.0、350mM NaClを用いてカラムから溶出し、200μLの画分を回収した。ピークAP含有画分をプールし、250容量の10mM HEPES pH7.5、200mM NaClに対して一晩、透析した。MgCl2およびZnCl2をそれぞれ2mMおよび0.25mMまで、保有物に添加した。最終生成物は、SDS−PAGEによって95%純粋より大きく、還元条件の下でおよそ140kDaの質量を有するシングルバンドとして電気泳動した。
【0129】
Sp35構築物をまた、Fc融合体として加工した。Sp−35LRR−Fc構築物を、プライマー(フォワード)
【0130】
【数4】
を使用するPCRによって作製した。このPCR産物を、PV90ベクターのNotI部位の中へ挿入した。Sp35IG−Fc構築物を、プライマー(フォワード)
【0131】
【数5】
を使用するPCRによって作製した。このPCR産物を、PV90ベクターのNotI部位の中へ挿入した。タンパク質をCHO細胞において発現させ、プロテインAセファロースカラムを使用して精製した。
【0132】
(実施例4:NgR1発現細胞へのSp35結合)
NgR1へのSp35結合を示すために、4つの異なる方法を使用した。第1に、本発明者らは、アルカリホスファターゼSp35結合体(AP−Sp35)を、NgR1発現細胞と一緒にインキュベートし、色素形成のAP検出試薬を使用して結合を評価する、直接的な結合アッセイにおいて相互作用を検出した。90%コンフルエントのCOS7細胞を、100mm組織培養皿上で増殖させ、Fugene6試薬(Roche)を使用して、NgR1発現プラスミドを用いてトランスフェクトした。48時間後、トランスフェクトした細胞を、HBH(Hankの平衡化塩緩衝液、1mg/ml BSA、20mM HEPES、pH7.0)で1回洗浄し、次いで、HBH中の4μg/mlのAP−Sp35融合タンパク質と一緒に、23℃で1.5時間インキュベートした。細胞を、氷冷のHBH緩衝液で、各々3分間で3回洗浄し、次いで、15分間、20mM HEPES、pH7.0、150mM NaCl中の3.7%ホルムアルデヒドで固定し、HBH緩衝液の中へ移して戻した。内因性の熱に不安定なAPを、67℃で2時間、熱不活性化した。結合したAP−Sp35を、ニトロブルーテトラゾリウムNBT(Roche)と共にインキュベーションすることによって検出した。Ap−Sp35は、ヒトNgR1レセプターを発現するCOS7細胞に結合したが、ベクター単独でトランスフェクトしたコントロールCOS7細胞には結合しなかった。NgR1についての点状の染色パターンが観測され、このことは、1つの画分のみ、おそらく50%の細胞がNgR1でトランスフェクトされたことを反映する。
【0133】
結合をよりよく定量化するために、本発明者らは同じ実験を行ったが、並行細胞サンプルとして、8μg/ml、4μg/ml、2μg/ml、1μg/ml、0.5μg/ml、0.125μg/ml、0.06μg/mlのAP−Sp35を用いて処理した。結合したAPを、4−ニトロフェニルホスフェートと一緒にインキュベートし、96−ウェルプレートリーダー(Molecular Devices)においてAP活性を測定した。これらのデータから、本発明者らは、ヒトNgR1へのAp−Sp35の結合についてのEC50が、およそ6nMであると推定した。
【0134】
第2に、本発明者らは、ELISAアプローチにおけるNgR1へのSp35の結合を検出した。ELISAプレート(Costar)を、37℃で1時間、0.1M NaHCO3、pH9.0において10μg/ml可溶性NgR1−Fcレセプター(ラットIgG1のヒンジおよびFcと融合したラットNgR1ペプチド35〜310を含むsNgR310−FcならびにラットIgG1と融合したラットNgR1ペプチド35〜344を含むsNgR344−Fc)でコーティングした。そのプレートをブロッキングして、25mM Hepes、pH7.0、0.1%BSA、0.1%オボアルブミン、0.1%脂肪を含まないドライミルクおよび0.001%NaN3で洗浄した。AP−Sp35タンパク質4μg/mlをプレートに添加して、4℃で一晩インキュベートした。次いで、プレートを、10mM Tris pH7.5、150mM NaClで洗浄し、結合したAPを、0.1Mグリシン、1mM MgCl2、1mM ZnCl2 pH10.5で希釈した10μg/mlの色素形成の基質であるリン酸4−ニトロフェニルを使用して検出した。OD410を、Softmaxプログラムを装着するELISAリーダー(Molecular Devices)において測定した。AP−Sp35は、固定化したsNgR−344−Fcに結合したが、sNgR−310−Fcタンパク質には結合せず、より長い種類のNgR1が、Sp35結合に必要とされることを示した。本発明者らは、100倍過剰のsNgR344−Fcと一緒にAP Sp35をプレインキュベートすることによってsNgR344−Fc NgR1へのAP−Sp35の結合を80%まで達成し得た。ラットIg融合コントロールタンパク質としてハリネズミIg1融合タンパク質を使用すると、結合の競合は見られなかった。
【0135】
第3に、本発明者らは、NgR1とSp35を同時に免疫沈降することによってNgR1へのSp35の結合を検出した。この研究のために、100mmの組織培養皿上で増殖した80%コンフルエントのCOS7細胞を、Fugene6試薬(Roche)を使用してSp35血球凝集素(Sp35−HA)およびNgR−FLAGをコードするプラスミドを用いてトランスフェクトし、トランスフェクションの48時間後、細胞を収集して、4℃で30分間、1mlの溶解緩衝液(50mM HEPES、pH7.5、150mM NaCl、1.5mM MgCl2、1mM EGTA、1% Triton X−100および10%グリセロール)中で溶解した。次いで、溶解物を、15分間14,000×gで遠心分離し、上清を回収して、抗HA親和性マトリックス(Roche)を使用して、攪拌しながら4℃で一晩インキュベートした。サンプルを、1mlの溶解緩衝液で3回洗浄し、次いで、Laemmliサンプル緩衝液中で3分間ボイルし、4〜15%SDS−PAGEに供して、Anti−FLAG M2抗体(Sigma)を用いる免疫ブロッティングによって解析した。FLAGの存在によって明らかなように、抗HAタグ親和性樹脂から、Sp35−HAおよびFLAG−NgRの両方を含む複合体を回収した。この複合体は、細胞をSp35−HAプラスミドまたはFLAG−NgR1プラスミド単独で処理したか、またはFlag−NgR1およびNgR1に結合しないHAタグ化コントロールタンパク質で同時トランスフェクションした、コントロールトランスフェクション由来の溶解物中に見られなかった。
【0136】
Sp35−HAを以下のように作製した。Sp35シグナル配列および細胞外ドメイン(アミノ酸1〜531)を、XbaI部位およびSpeI部位(下線)を含むプライマー
【0137】
【数6】
を使用してPCR増幅した。PCR産物を、XbaIおよびSpeIによって消化し、XbaI部位とSpeI部位との間のベクターpCGCHAの中へ挿入した。配列の挿入をDNA配列決定によって確認した。FLAG NgR1構築物はDr.Zhigang He(2002年、11月7日、Nature,Vol420)のご好意により提供された。
【0138】
第4に、本発明者らは、Ap−Sp35が、NgR1を発現するラットの小脳顆粒ニューロン(CGN)に結合することを示した。この実験のために、90%コンフルエントの出後8日のCGN細胞を、100mm組織培養皿上で増殖した。48時間後、細胞を、HBH緩衝液で一度洗浄し、次いで、23℃で1.5時間、HBH緩衝液中で4μg/mlのAP−Sp35と一緒にインキュベートした。次いで、細胞を、各々3分間、氷冷のHBHで3回洗浄し、次いで、15分間、20mM HEPES、pH7.0、および150mM NaCl中で3.7%のホルムアルデヒドで固定し、HBHに移して戻した。内因性の熱に不安定なAPを、67℃で2時間、熱不活性化した。結合したAP−Sp35を、ニトロブルーテトラゾリウムNBT(Roche)と共にインキュベーションすることによって検出した。AP−Sp35は、NgR1を発現する出後8日の小脳顆粒状ニューロンに結合した。ニューロンへのAP−Sp35の結合を、ほとんどの膜表面由来のGPIアンカータンパク質を開裂するPIPLC(5単位/ml)を用いてCGNを処理することによって阻害した。NGR1は、GPI結合タンパク質であるので、この結果は、Sp35が、CGN細胞上のNgR1に結合するという考えをさらに支持する。
【0139】
(実施例5:NgR1とSp35の共存)
Sp35およびNgR1が、同じニューロンにおいて発現するかどうかを決定するために、本発明者らは、共存の研究を行った。4%パラホルムアルデヒドで固体したラットのp8初代顆粒ニューロン培養物を、Sp35およびNgR1に対する抗体(Santa Cruz)と一緒にインキュベートし、次いで、適切なAlexa標識した第2の抗体(Molecular Probes Inc.)と一緒にインキュベートした。細胞を共焦点蛍光顕微鏡によって可視化した。ニューロンは、Sp35およびNgR1抗体によって強く染色した。両方のタンパク質が、細胞体およびニューロンの軸策において発現した。共存解析を補助するために、2つの型の抗体について異なる有色のプローブを使用した。染色(NgR陽性細胞については赤色そしてSp35陽性細胞については緑色)を組み合わせると、本発明者らは、2つのタンパク質がニューロン内で共存していることを示す細胞全体にわたる黄色が見えた。
【0140】
(実施例6:Sp35内のNgR1結合部位)
本発明者らは、NgR1相互作用に関係するSp35の特異的ドメインを規定するために欠損マッピングを使用した。以下の欠損構築物を、Stratagene Quikchange Mutagenesisキットを使用して作製した。本発明者らは、改変した挿入物のDNA配列決定によって全てのベクター構築物を確認した。
【0141】
Sp35のロイシンリッチ反復ドメイン、さらに塩基性領域(a.a34〜432)を含むHis−AP−Sp35bを、PCRによってHis−AP−Sp35(a.a.34〜532)からクローニングした。使用したプライマーは、
【0142】
【数7】
であった。
【0143】
Sp35のIgドメイン、さらに塩基性領域(a.a.417〜531)をコードするHis−AP−Sp35dを、PCRによってHis−AP−Sp35a(a.a.37〜531)ベクターからクローニングした。使用したプライマーは、
【0144】
【数8】
であった。
【0145】
Igドメイン(a.a425〜531)のみをコードするHis−AP−Sp35eを、PCRによってHis−AP−Sp35(aa34〜532)ベクターからクローニングした。使用したプライマーは、
【0146】
【数9】
であった。
【0147】
市販の変異誘発キットおよびプロトコル(Stratagene Quikchange)を、ベクターHis−AP−Sp35(34〜532)におけるSp35のアミノ酸456(アルギニンからグルタミン酸に)およびアミノ酸458(ヒスチジンからバリンに)を変異するために使用した。使用したプライマーは、
【0148】
【数10】
であった。
【0149】
His−AP−Sp35欠損構築物(図3)を、pV90発現ベクターにおいて加工し、293細胞において発現させた。馴化培地を回収し、AP付加物を、Fractogel TMAE樹脂およびNiNTAアガロースでの一連のクロマトグラフィー工程によって精製した。精製したタンパク質を、COS7細胞上で発現したNgR1への結合について試験した。3つの構築物は、すべてSp35に弱く結合した。これらの結果は、Sp35 LRR反復1〜14(アミノ酸34〜417)およびSp35のIgドメイン(アミノ酸425〜531)の両方は、NgR1へのSp35結合に寄与することを示した。Igドメインは、LRRドメインより高い親和性を示した。
【0150】
Sp35のIgドメインについての構造モデルを、フレームワークとしてNCAM結晶構造を使用して作製した(Rasmussenら、2000、Nat.Struct.Biol.7:389−393)。このモデルから、本発明者らは、結合に関与し得るループ(残基数454〜458、アミノ酸:SPRKH;配列番号:34)を観察した。この仮説を試験するために、本発明者らは、456での残基Rおよび458での残基Hを、それぞれEおよびVに変更したSp35構築物を作製した。この構築物を、NgR1結合について試験すると、本発明者らはシグナルにおいて10倍より少ない減少を観察した。結合におけるこのループ領域の寄与を試験するための代わりのアプローチとして、本発明者らは、ペプチドのN末端およびC末端にシステインを付加することによって環化する配列LSPRKH(配列番号:10)に対応するペプチドを合成した。NgR1への結合の際に、このペプチドは、NgR1の機能をブロック、阻害、または妨害する。
【0151】
(実施例7:Sp35はp8 CGN束形成(fasciculation)を誘発する)
ニューロンにおけるSp35の生物学的機能を決定するために、本発明者らは、Sp35−Fcを、出生後8日目の顆粒状ニューロンと共にインキュベートし、Sp35が神経突起の外向きの成長を調節し得るかどうかを観察した。Sp35−Fcタンパク質(16μg/ウェルのタンパク質)をスポットする前に、Labtek培養スライド(8ウェル)を、0.1mg/mlのポリ−D−リジン(Sigma)でコーティングした。このスライドを一晩乾燥させ、次いでリンスし、そして10μg/mlのラミニン(Gibco)でコーティングした。出生後8日目由来の小脳顆粒状ニューロンを解離し、事前コーティングしたスライド上に播種した。このスライド培養物を5%CO2中37℃で24時間インキュベートした。次いで、このスライドを20%スクロースを含有する4%パラホルムアルデヒド中で固定し、そして抗βIIIチューブリン(Covance TUJ1)で染色した。24時間後、CGNはニューロンの束状化(bundling)で明らかなように、はっきりした束形成形態を示した。この束形成は、未処理細胞コントロールまたはFcタンパク質コーティング化サンプルコントロールでは見られなかった。
【0152】
(実施例8:RhoA活性化/不活性化についてのSp35の効果)
Sp35−Fcは、束形成を引き起こす出生後の小脳顆粒状ニューロンを誘発した。シグナル伝達分子RhoAが束形成に関与することは公知であるので、本発明者らは、Sp35−FcがニューロンにおいてRhoA機能を調節し得るかどうかを決定した。本発明者らは、以下のようにRhoA活性化実験を行った:293細胞またはCOS7細胞を、Fugene 6試薬(Roche)を使用して、RhoA、Sp35またはNgR1の組み合わせを含む発現ベクターでトランスフェクトした。トランスフェクションの48時間後、細胞を一晩血清飢餓にし、次いで50mM Tris(pH7.5)、1% Triton X−100、0.5%デオキシコール酸ナトリウム、0.1% SDS、500mM NaCl、10mM MgCl2、さらにプロテアーゼインヒビターカクテルでリンスした。細胞溶解物を、13,000×g、4℃、5分間の遠心分離によって清澄化し、そして95%の上清を、4℃で45分間、20μgの固定化GST−Rho結合ドメイン親和性マトリックス(Rhotekinビーズ、Upstate Biotechnology)と共にインキュベートした。このビーズを、洗浄用緩衝液(50mM Tris(pH7.5)、1% Triton X−100、150mM NaCl、10mM MgCl2、およびプロテアーゼインヒビター)で3回洗浄した。GTP結合Rhoを、SDS−PAGEサンプル緩衝液中95℃で5分間加熱することによって、上記ビーズから溶出させた。結合Rhoタンパク質、およびRhoタンパク質の全体を、RhoAに対するモノクローナル抗体(Santa Cruz)を使用してウェスタンブロッティングによって検出した。Sp35遺伝子でのトランスフェクション後のブロットで検出されたRhoA−GTPの量の増加によって明らかなように、Sp35でトランスフェクトされたCOS7細胞およびHEK293細胞は、RhoA活性化を誘発した。RhoA−GTPのさらなる増加は、Sp35−Fcでの処理後に観察された。Sp35単独でのトランスフェクト後のRhoA−GTPの増加と対照的に、細胞をSp35およびNgR1でトランスフェクトした場合、RhoAは部分的に不活性化された。Sp35−Fcでのこれらの細胞の処理は、RhoAのさらなる不活性化をもたらした。
【0153】
本発明者らは、Ca++流入へのSp35処理の影響を決定するために、FLIPRアッセイ(Molecular Devices)を使用してSp35によるシグナル伝達応答を確認した。本発明者らは、Sp35−Fcの処理でSp35を発現する細胞において有意なCa++流入を観察したが、Sp35−Fcで処理したコントロール細胞においては有意なCa++流入を観察しなかった。NgR1およびSp35で同時トランスフェクトされた細胞がSp35−Fc融合タンパク質で処理された場合、このCa++流入は減少した。
【0154】
(実施例9:Sp35タンパク質とSp35タンパク自体との相互作用)
LRRドメインはホモタイプな相互作用にしばしば関与し、そして、本発明者らはSp35でトランスフェクトされた細胞への可溶性Sp35の添加がSp35トランスフェクション単独で観察されたRhoA−GTPの増加を越えるRhoA−GTPの増加を引き起こすことを観察したので、本発明者らは、Sp35自体に結合するSp35について試験した。この試験を行うために、本発明者らは、免疫共沈降を使用した。COS7細胞の80%コンフルエント(100mm組織培養皿上で増殖された)を、Fugene 6試薬(Roche)を使用して、プラスミドSp35 HAまたはプラスミドSp35−FLAG、あるいはその両方でトランスフェクトした。トランスフェクションの48時間後、細胞を収集し、そして4℃で30分間、1mlの溶解緩衝液(50mM HEPES(pH7.5)、150mM NaCl、1.5mM MgCl2、1mM EGTA、1% Triton X−100、および10%グリセロール)中に溶解した。次いで、この溶解物を14,000×gで15分間遠心分離し、そして上清を収集し、4℃で一晩撹拌しながら抗HA親和性マトリックス(Roche)と共にインキュベートした。次いで、このサンプルを1mlの溶解緩衝液で3回洗浄し、Laemmliサンプル緩衝液中で煮沸し、4〜15%SDS−PAGEに供し、そして抗FLAG抗体での免疫ブロッティングによって分析した。ウェスタンブロッティングによって決定されたように、この抗HA抗体樹脂は、Sp35−FLAGを含んだ複合体を捕獲した。これは、Sp35とSp35自体との直接的な相互作用を示した。本研究者らはまた、Sp35−Fcと一緒にHA−Sp35でトランスフェクトされた細胞を処理し、そして類似の免疫沈降アプローチを使用して、HA−Sp35がSp35−Fcに結合することを示した。
【0155】
Sp35−FLAGを、以下のように作製した。Sp35遺伝子細胞外ドメイン(a.a.1〜531)を、NotI部位(下線が引かれている)を含むプライマー:
【0156】
【数11−1】
およびプライマー:
【0157】
【数11−2】
を使用してPCR増幅した。このPCR産物をNotIによって消化し、そしてベクターpV90のNotI部位に挿入した。この挿入物のDNA配列を、DNA配列決定によって確認した。
【0158】
(実施例10:Sp35で形質転換された細胞のインビトロ移植)
脊髄損傷ラットにおけるSp35の生物学的機能を決定するために、本発明者らは、皮質性初代培養細胞(混合培養物)を、(ラット脊髄の損傷中心への送達のため)全長Sp35を発現するレトロウイルスまたはレトロウイルスコントロールで感染させた。2×106個の細胞を導入し、そしてラットを10日目に屠殺した。この脊髄を4%パラホルムアルデヒド中で一晩固定し、次いで、70%ETOH中で脱水し、続いて95%ETOH中で脱水した。組織サンプルをパラフィン中に包埋した。切片(10ミクロン厚)を、免疫組織化学的染色のために使用した。コントロールと比較して、Sp35発現細胞を受け取ったラットは、より少ない軸索退縮を示し、そして上記中心近くに染まったより多くのβチューブリンを示す。Sp35を受け取る損傷ラットにおいて、上昇したニューロンの生存が観察された。
【0159】
Sp35レトロウイルス構築物を、以下のように作製した。Sp35遺伝子を、XhoI部位(下線が引かれている)を含むプライマー:
【0160】
【数12−1】
およびEcoRI部位(下線が引かれている)を含むプライマー:
【0161】
【数12−2】
を使用してPCR増幅した。このPCR産物をXhoIおよびEcoRIで消化し、次いで、レトロウイルスベクターpMIG(これは、IPES−GFPを含む)に結合し、これをXhoIおよびEcoRIで事前に切断した。この新たなベクターを、pMMC078と命名した。pMMC078の単離物の全てが不注意な点突然変異を含んだので、pMMC078のうちの2つの単離物を一緒に結合した。pMMC078.6をXhoIおよびAccIで切断し、そしてpMMC078.7をXhoIおよびAccIで切断した。これらの2つのフラグメントを一緒に結合し、最終修正(correct)プラスミドpMMC089を作製した。この挿入物のDNA配列を、DNA配列決定によって確認した。Sp35レトロウイルスを、説明したように作製した。293G細胞は、トランスフェクションの前日に分裂された。8μgのSp35−レトロウイルスDNAを、リポフェクタミン(Invitrogen)によって5×106個の細胞をトランスフェクトするために使用した。この馴化培地を、トランスフェクションの92時間後に収集した。この馴化培地を、5000gで10分間遠心分離し、そして上清を、Sp35レトロウイルスストックとして使用した。このストックを4℃で1週間、または−80℃で6ヶ月間保存した。
【0162】
(実施例11:脊髄損傷の動物モデル)
外科的手順の全てを、無菌技術を使用して行う。任意の外科的処置前の一週間、動物を取り扱う。アンピシリン100mg/kgを、SCに手術の前および後に予防的に投与し、膀胱感染損傷の発生を減少させた。
【0163】
動物を、O2中イソフルラン2〜3%と共にミダゾラム2.5mg/kgIPを使用して、つま先を挟むことによって測定して深い麻酔にまで麻酔する。動物を、手術および回復の期間、循環水ヒーティングパッド(heating pad)上で維持する。角膜乾燥を予防するために眼潤滑剤を使用し、そして過剰の唾液分泌を減少させるためにアトロピン0.05mg/kgSCを与える。小さな切開を皮膚に作製し、そして筋肉を引っ張って椎骨を露出させる。脊髄レベルL6の背側椎弓切除(および鞘内カテーテルの置換が必要な場合はL7、以下を参照のこと)を行い、L6/L7および隣接した棘突起を脊髄フレーム(David Kopf Instruments)に強固に固定した。背部片側切断を、主な背内側皮質脊髄(coticospinal)路(CST)構成要素および少数の背外側皮質脊髄(coticospinal)路(CST)構成要素を完全に遮るよく切れる虹彩切除鋏を使用して、L6で行った。手術後、露出した脊柱を保護するために椎弓切除部位を保護材料(例えば、Durafilm)で覆い、そして上にある筋肉を4.0クロミックガット(chromic gut)で縫合した。この皮膚を縫合し、ベータダイン溶液でふき取った。
【0164】
動物の機能回復ISを、脊髄損傷後のラットを評価するために一般的に使用されるBasso Beattie and Bresnehan(BBB)スコアリング法を使用して評価した。この方法は、関節運動能力および体重付加能力の詳細な分析によってラットの後肢機能を定量化する。ラットを、脊髄損傷の翌日に評価し、次いでその後一週間評価する。
【0165】
CST切除の直後に、Sp35もしくはGFPを発現するアデノウイルスまたはコントロールウイルス(1010粒子)を、切除の部位に、ならびに損傷部位のすぐ尾側およびすぐ吻側の領域に注入する。合計で10μlのAdvを、5箇所の異なる部位に注入する(4μl/部位)。Sp35タンパク質の鞘内投与については、小さな穴を外傷(lesion)L7より2mm尾側の脊髄の硬膜に開け、そして鞘内カテーテルをL7のクモ膜下腔に挿入する。このカテーテルを、外傷より約1mm尾側の脊髄の向こう側に、ゆっくりかつ徐々に滑らせる。鞘内空間の外側にあるカテーテルの一部を、所定の周辺組織にしっかりと縫合した。試験物質(Sp35タンパク質またはコントロールタンパク質)を含むプライムされた小型浸透ポンプ(Alza corp.)をガイドカニューレの露出された端部に連結し、そしてクモ膜下腔に挿入する。手術後、露出した脊柱を保護するために椎弓切除部位を保護材料(例えば、Durafilm)で覆い、そして上にある筋肉を4.0クロミックガットで縫合した。この皮膚を縫合し、そしてベータダイン溶液でふき取った。
【0166】
組織学的分析:路追跡手術は、脊髄損傷を誘発する手術時に起こる。頭部の皮膚を薄く削り、そしてベータダインおよび70%アルコールでふき取る。この動物を定位固定フレームに配置する。この頭皮を縦方向に切開し、そして骨膜を頭蓋冠からすくい取る。頭蓋骨に直径約1〜2mmの穴をドリルで開け、そしてガラスマイクロリットル針を、運動皮質の8つの位置に垂直に挿入する(座標は、Paxixnos and Wastonのラット脳図譜(1997)に従って決定される)。約5μlの路追跡物質(例えば、ビオチンデキストランアミン、10,000M.Wt)を注入し、そしてさらに5分間にわたって上記針を定位置に置いたままにして、溶液を拡散させる。針の除去後、スクールキャップ(scull cap)の穴をゲルフォームでふさぎ、そして頭皮を損傷部位をこえてステープル(staple)で閉じる。動物を回復させ、そして手術後の治療を受けさせる(以下に記載される)。4〜10週後、動物を深く麻酔し(ipでイナクチン100〜110mg/kg)、そして以下に記載されるような組織学のために還流する。この路追跡物が、脊髄の尾側端部へと向かう皮質性脊髄路を順行性輸送機序を下がることによって行われ、そして皮質脊髄路内の解剖学的結合性を定量するための手段を提供する。
【0167】
免疫組織化学的実験については、損傷を誘発する手術の2〜8週間後に動物をイナクチン(IPにおいて100〜110mg/kg)で深く麻酔する。胸腔を開き、そして心臓を露出し、灌流させる。100ccの氷冷PBSをゆっくりと押出すことによって(流体逃避を可能にするために、穴が右心室に切り込まれる)、カニューレを左心室に挿入する。これの後、眼/耳/つま先の固定が明らかになるまで、4%パラホルムアルデヒド(50〜100ml)のゆっくりとした規則的な点滴を続ける。損傷部位の改変を最小にすることに注意して脊髄を取り出し、OCTで凍結させ、切片にして、そして免疫組織化学について処置する。必要に応じて、他の組織もまた、後者の分析のために採取する。アデノウイルスSp35を受け取る動物は、ニューロン軸索を染色するβIIIチューブリンによって決定されるように、上昇した軸索出芽を示した。
【0168】
(実施例12:Sp−35ウイルスベクター構築物)
pMIG由来のSp−35ウイルスベクターを、以下のように作製した。全長Sp35コード配列を、XhoI部位を含むプライマー:
【0169】
【数13−1】
およびEcoRI部位を含むプライマー:
【0170】
【数13−2】
を使用してPCR増幅した。このPCR産物を、XhoIおよびEcoRIで切断し、次いで、レトロウイルスベクターpMIG(Chengら、1996、Nat.Biotechnol.145:576)に結合し、これをXhoIおよびEcoRIで切断した。このベクターをpMMC078と命名した。pMMC078の単離物全てが点突然変異を含んだので、pMMC078のうちの2つの単離物を一緒に結合した。ベクターpMMC078.6をXhoIおよびAccIで切断し、そしてpMMC078.7をXhoIおよびAccIで切断した。これらの2つのフラグメントを結合し、プラスミドpMMC089を作製した。
【0171】
pMIG由来のSp35−HAウイルスベクターを以下のように作製した。HA配列を有するフレーム中のSp35アミノ酸326〜614をコードするフラグメントを、SacII部位を含むプライマー:
【0172】
【数14−1】
および鋳型として機能するpMMC089を含むプライマー:
【0173】
【数14−2】
でのPCRを使用することによって得た。このより長いプライマーは、Sp35コドン614の後およびEcoRI部位の前のHAコード配列(イタリック)を含む。次いで、このPCR産物を、Sac IIおよびEcoR Iで切断し、そしてpMIG由来のレトロウイルスベクター中に野生型Sp35コドン326〜614を含むSac II−EcoR Iフラグメントを置換するために使用する。
【0174】
Sp35−バキュロウイルスHAベクターを以下のように作製した。Sp35−HAレトロウイルスベクター由来のSp35−HAコード配列を、Xho IおよびEcoR Iで切り出し、平滑末端化し、そしてバキュロウイルスシャトルベクターpBV−CZPG(米国特許第6,190,887号;および同第6,338,953号)の、Bgl2−フィルインサイト(Bgl2−fill in site)にクローン化し、CMVプロモーターの下でLacZ遺伝子を置換した。
【0175】
Sp35アデノウイルスベクターを、以下のように作製した。Sp35レトロウイルス由来のSp35−IRES−GFPコード配列を、XhoI−フィルインサイトおよびNhe Iで切り出し、次いで、最小限のCMVプロモータ−の下、アデノウイルスシャトルベクターpDC315の、EcoR I−フィルインサイト/Nhe I部位にクローン化した。
【0176】
(実施例13:再髄鞘化の動物モデル)
雌性のLong Evansラットを、全ての研究に使用する。ラットを、イソフルランを使用して麻酔し、T3/T4露出をし、そして背側半椎弓切除を行う。次いで、化学的脱髄剤であるリソレクチン(0.9%生理食塩水中1%リソレクチン3μl)を、索表面より0.5〜1mm下の脊髄の脊柱の右側に注入する。適切な鎮痛処置を手術前後に施す。
【0177】
3日後、この注入部位を再び露出させ(イソフルラン麻酔下、適切な鎮痛処置を使用して)、以下の治療を損傷した脊髄に投入し、そしてタンパク質Sp35/コントロールタンパク質をコードするアデノウイルスベクターを損傷部位に注入する。Sp35をコードするアデノウイルスの1010個の粒子、または10μl体積のGFPコントロールを、リソレシチン誘発脱髄の部位またはその周囲の5箇所までの異なる部位における損傷されたラット脊髄に注入する。2μl以下の容量を、5箇所の注入部位の各々に注入する。脊髄の脱髄/再髄鞘化の組織学的分析ついては、手術の2週間後、3週間後、4週間後または6週間後、動物をイナクチン(ipにおいて100〜110mg/kg)で深く麻酔し、固定液で心臓を介して還流する。次いで、この脊髄を取り出し、分析のために処理する。Sp35処置を受ける動物は、抗MBPタンパク質抗体またはルクソールファーストブルー(luxol fast blue)を使用するIHCによって決定されるように、上昇した軸索髄鞘形成を示した。
【0178】
(実施例14:Sp35 RNAi)
脳機能におけるSp35の役割に取り組むために、本発明者らは、レンチウイルスSp35 RNAiを出生後8日目のCGN細胞に導入した。Sp35 RNAi感染細胞は、コントロール細胞よりも短い神経突起を有し、そしてコントロール細胞よりも高い増殖速度を有した。これらの結果は、RhoA活性化を調節することにおけるSp35についての役割を示す。
【0179】
マウスSp35 DNA配列とラットSp35 DNA配列とを比較して相同領域を見出し、候補shRNAに使用した。CH324を、オリゴヌクレオチドLV1−035およびオリゴヌクレオチドLV1−036をアニーリングさせ、そしてHpa1およびXho1で消化したpLL3.7に結合することによって構築した。このオリゴヌクレオチドを、MWGから購入した。この配列は、以下である:
【0180】
【数15】
ウイルスを産生する前に、pLL3.7由来のDNAまたはpLL3.7中の候補shRNAを、6ウェル型中のCHO細胞に5対1の割合で、マウスSP35−HAタグ化プラスミドで共トランスフェクトした。トランスフェクトされたCHO細胞溶解物由来のSP35−HAタグのウエスタンブロット検出によって、および2連のウェルから調製されたRNA全体のノーザンブロットによって、ノックダウンを分析した。このブロットを、mSP35の0.7kbフラグメントでプローブした。アッセイを、トランスフェクションの48時間後に行った(データは示していない)。ウイルスを、ラットニューロンの培養物における使用について最善の候補物から産生した。ベクター、さらなる方法論およびウイルス産生は、Rubinsonら「A lentivirus−based system to functionally silence genes in primary mammalian cells,stem cells and transgenic mice by RNA interference.」Nat.Genet.33,401−6(2003)に記載されるようなものであった。
【0181】
(実施例15:RhoA活性化)
NgR1およびSP35を共発現するCOS7細胞は、OMgpに応答してRhoA/GTPレベルの変化を示さなかった。これは、SP35/NgR1複合体がミエリンインヒビターによるシグナル伝達を媒介するのに重要でないことを示唆した。
【0182】
本発明者らは、SP35/NgR1/p75の三つ組複合体がシグナル伝達を媒介するという可能性を検討した。2つのアプローチを、SP35とNgR1とp75との間の相互作用を評価するために使用した。1つ目は、AP−SP35結合体を使用する直接的な結合アッセイにおいて、結合を評価した。このAP−SP35結合体は、p75発現細胞に弱く結合した。AP−P75は、NgR1発現細胞に結合した。NgR1およびp75へのAP−SP35の結合を、ELISAによって測定した(図4)。2つ目は、NgR1およびp75へのSP35の結合を、SP35、NgR1およびp75を共発現するCOS7細胞からの免疫共沈降によって評価した。抗NgR1抗体は、SP35およびp75を含む複合体を免疫沈降させた。抗SP35抗体はまた、p75を含む複合体を免疫沈降させた。この相互作用データおよび免疫共沈降データは、SP35とNgR1とp75との間の直接的な相互作用についての証拠を提供した。本発明者らは、共焦点顕微鏡法ならびにSP35、p75およびNgR1に対する抗体を使用し、SP35、NgR1およびp75が、ラット由来のp7 CGニューロンの細胞体および軸索に共に局在することを示した。
【0183】
次に、本発明者らは、SP35、NgR1およびp75の組み合わせが、ミエリンインヒビターの活性に充分であることを示した。非ニューロン性COS7細胞を、3つの構成要素全てを発現するように操作した。これらの細胞を使用して、本発明者らは、RhoA/GTPレベルがOMgpによってアップレギュレーションされることを示した。OMgp−Fc処理は、これらの3つの構成要素の他の組み合わせと比較して、SP35/p75/NgR1を共発現する細胞におけるRhoA/GTPレベルを上昇させた。本発明者らは、COS7細胞溶解物のウエスタンブロッティングによってタンパク質の発現を確認した。NgR1に結合するミエリンインヒビターの親和力は、p75の存在によっても、p75およびSP35の存在によっても影響を受けなかった。この組み合わせた結果は、NgR1、SP35およびp75の三つ組複合体が、NgR1リガンドの存在下でのRhoA調節のために必要とされるというモデルを支持する(図5)。
【0184】
SP35は、シグナル伝達への潜在的な直接関与または潜在的な間接関与を有する細胞質ドメインを含む。細胞質ドメインの役割を決定するために、本発明者らは、シグナル伝達ができない、非産生的な三つ組複合体を形成することにより、ドミナントネガティブ様式で機能する、SP35の細胞質ドメイン切断物(truncation)(配列番号2のアミノ酸34〜576)を産生した。本発明者らは、細胞質ドメイン切断を有するこの分子を「DN−SP35」(dominant negative SP35)と命名した。本発明者らは、出生後7日目の(p7)CGニューロンを、全長SP35またはDN−SP35でトランスフェクトし、次いで、阻害性ミエリン成分(Omgp、ミエリンおよびNogo66)への応答についてアッセイした。図6に示されるように、DN−SP35トランスフェクト細胞は、阻害性ミエリン成分に応答せず、そしてコントロールよりも長い神経突起を示した。対照的に、全長SP35構築物でトランスフェクトされた細胞は、阻害性基質への高められた応答を示し、そしてコントロールと比較してより短い神経突起を有した。これは、DN−SP35が、ミエリン成分によって引き起こされる神経突起の伸長阻害を弱めるような競合物として作用することを示した。本発明者らは、外因性の可溶性SP35−Fcはまた、NgR1と結合し、そして阻害性基質の作用を遮断すると予測した。図7に示されるように、SP35−Fcは、Omgp、Nogo66およびMAGによる神経突起伸長阻害を低減させた。
【0185】
(実施例16:神経保護活性)
等数のラットp6小脳顆粒ニューロンを、50nMのsp35−Fcタンパク質の存在下または非存在下で、12ウェルの細胞培養プレートの各ウェルにプレートした。これらのポリ−D−リジンプレートを、10μgのCNSミエリンまたは200ngのNogo66、MAGおよびOMgpまたはコントロール−Fcで事前コーティングした(乾燥させた)。このニューロン培養物を、1〜7日間、37℃で、および5%CO2で維持した。ニューロンは、3日後に検査した(ニューロンの特異性マーカー IIIチューブリンによって決定した)ところ、十分な神経突起伸長をして、sp35−Fc処理とは無関係に、PBSコントロールウェルにおいて健常であり、そして良好に成長した。sp35−Fcの非存在下では、ニューロンは、ミエリン、Nogo66、MAGおよびOMgpでコーティングされたウェルにおいて良好に成長しなかった。最小限の神経突起出芽(短く、歪曲した)が存在し、そしてこのニューロンは健常には見えず、丸い細胞体および縮合された核物質を有した。DAPI染色は、これらのウェルで検出されたニューロンの数が、PBSコントロールウェル中で検出されたニューロンの数よりも少なく、ニューロンの損失を示唆していることを示した。sp35−Fcの存在下では、長い神経突起が存在し、そしてこのニューロンは健常に見えた。DAPI染色は、sp35−Fcを受け取らなかったウェルよりも、これらのウェル中でニューロンの数がより多いことを実証した。このデータを、以下の表2に要約する。
【0186】
【表2】
これらのデータは、Sp35の可溶性形態(例えば、Sp35−Fc)が、神経保護活性を保有すると示した。
【0187】
脊髄を半分切除された(T9、SCT)ラットにおいて、脊髄切片のβ−IIIチューブリン染色は、損傷部位におけるニューロンの実質的な損失を示した。sp35を発現する組換えウイルスは、SCT動物を損傷部位において感染させるために使用した。これらの脊髄の組織学的染色は、ベクターウイルスで感染されたコントロールグループと比較して、損傷部位周辺のニューロンの数の増加を示した。これは、上記のインビトロ実験の知見と一致し、そしてSp35に関連する神経保護特性をさらに示す。
【0188】
(実施例17:脊髄損傷の動物モデルにおけるSp35)
Sp35−Fcは、インビトロでOMgp、Nogo−66およびMAGによって引き起こされる神経突起成長阻害を低減させたので、本発明者らは、この分子がインビボでCNS損傷の機能回復を促進することを予測した。これを確かめるために、本発明者らは、脊髄を半分切除したラット(すなわち、急性CNS外傷の動物モデル)にSp35−Fcを投与した。図8および図9に示されるように、Sp35−Fc処置ラットは、IgGで処置されたコントロールラットと比較して、有意に向上した機能回復を示した。
【0189】
脊髄損傷および行動分析を、以下のように行った。外科的手順の全てを、Biogen Institutional Animal Use and Care Committeeの指針に従って行った。雌のLong Evansラット(190〜210g、Charles River、Wilmington、MA)を、2.5mg/kgのミダゾラム(I.P.において)およびO2中2〜3%のフルオタンを使用して麻酔した。背部椎弓切除を、脊髄レベルT6および脊髄レベルT7で行った。背側の片側切除を行い、主な背内側皮質脊髄路(CST)構成要素および少数の背外側皮質脊髄路(CST)構成要素を完全に遮断した。CST切除の直後に、鞘内カテーテルをT7のクモ膜下腔に挿入し、そしてクモ膜下腔に挿入されたプライムされた小型浸透ポンプ(Alzet model 2004)に連結した。小型浸透ポンプは、0.25μl/時間の速度で、Hu IgGアイソタイプコントロールタンパク質(5mg/ml、n=5、Pharmingen)、PBS(n=3)可溶性Hu Sp35−Ig融合タンパク質(4.3mg/ml、n=8)を送達した。手術後、椎弓切除部位を縫合し、そして皮膚の傷をステープルで閉じた。術後処置は、手術後3日間の鎮痛処置(s.c.においてブプレノルフィン0.05mg/kg)および手術後7日間の抗生物質処置(s.c.においてアンピシリン100mg/kgを1日に2回)を包含した。膀胱を、研究の期間(28日)または機能回復まで(その時間を記述した)、1日2回、手動で圧搾した。動物全てを、オープンフィールドBBBスコアリングシステム(Bassoら,1995,J.Neurotrauma 12:1−21;Onoら,2003,J.Neurosci.23:5887−5896)を使用して盲目的に採点した。ラットを、CST切除の翌日(2日目)およびその後四週間にわたって毎週、Basso−Beattie−Bresnahan (BBB)歩行運動評価尺度を使用して評価した。調査者を、研究の期間にわたって、処置群に対して盲目的にした。
【0190】
(実施例18:赤核脊髄路(RST)片側切除損傷モデルにおけるニューロンの生存および軸索再生)
本研究者らはまた、歩行運動に直接的に寄与する赤核脊髄路におけるニューロンの再生に対するSp35処置の効果を調査した。
【0191】
成体9週齢のSprague−Dawleyラット(200〜250g)を、ケタミン(80mg/kg)およびキシラジン(8mg/kg)の腹腔内注入によって麻酔した。顕微鏡の操作下で、背部椎弓切除を行い、そして7番目の胸椎(C7)を同定した。硬膜を開いた後、右側切除を、1つのスプリング鋏を使用して脊髄レベルC7において行った。脊髄を半分切除した後、動物に、Sp35−Fcの2μg/ml溶液(10μl)、またはヒトIgの2μg/ml溶液(10μl)、またはPBS(10μl)のいずれかで浸漬された1つのゲルフォームを与えた(損傷部位の上に配置した)。手術後、各群の動物を、軸索追跡および行動分析のために細分した。軸索追跡についての動物(各群についてn=5)および行動分析についての動物(各群についてn=7)を、1ヶ月間生存させた。
【0192】
フルオロゴールド(FG、6%w/v、Fluorochrome)を使用して、損傷瘢痕にわたって軸索を再生したRSTを標識し、そして、尾側の脊髄に戻した。損傷後の生存期間(1ヶ月)の末日より2日前、動物を、ケタミン(80mg/kg)およびキシラジン(8mg/kg)の腹腔内注入によって麻酔した。背部の椎弓切除を行い、そしてT2脊髄断片を同定した。0.5ml体積のFGを、ハミルトンシリンジを使用して右側のT2脊髄に手動で注入した。2日後、この動物を、致死用量のケタミン(150mg/kg)およびキシラジン(8mg/kg)で麻酔して屠殺し、そしてこの動物を、正常食塩水で心臓内に還流し、続いて0.1×PBS中4%パラホルムアルデヒドを含む400mlの固定液で心臓内に還流した。脳および脊髄を除去し、一晩パラホルムアルデヒド中で事後固定し、次いで、30%リン酸緩衝化スクロース中に配置した。脳組織および脊髄組織を、クリオスタットにおいて30mmの切片に切り出し、ゼラチンがコーティングされたスライド上に取り付けた。損傷部位上のFG標識RSTニューロンの数を、対側のインタクトな側のFG標識ニューロンの合計数の百分率として表した。この群間の百分率を、一方向ANOVAを使用して、続いてTukey−Kramer多重比較検定を使用して、統計学的に比較した。表3に示されるように、2μg/mlのSp35−Fcは、赤核脊髄路(RST)ニューロンの生存を促進した。
【0193】
【表3】
行動分析については、自発性垂直探索(exploration)の間の前肢の使用を、軽度の改変を含む記載されるような(Liuら、1999)種々の処置の1ヵ月後に試験した。ラットを、5分間の垂直探索について前肢の使用を促進する透明なPlexiglasシリンダー(直径15cmおよび高さ30cm)中に配置した。以下の行動を記録した:(1)シリンダーの壁と接触するための左肢(非障害性)または右肢(障害性)の独立的な使用;および(2)シリンダーの壁と接触する前肢両方の同時使用。以下の点に関して、垂直探索行動が発現された:(1)障害性の肢の使用、非障害性の肢の使用、および両方の肢の使用の合計数に相対する左肢(非障害性)の使用の百分率;(2)障害性の肢の使用、非障害性の肢の使用、および両方の肢の使用の合計数に対する右肢(障害性)の使用の百分率;ならびに(3)障害性の肢の使用、非障害性の肢の使用、および両方の肢の使用の合計数に対する両肢の使用の百分率。群間の差異を、一方向ANOVAによって、続いてBonferroniポストホック分析によって検定した。Sp35−Fc処置動物は、有意に向上した前肢運動を示した:Sp35−1−Fc処置動物においては両肢について30%の使用 対 コントロール−Fc処置動物またはPBS処置動物においては両肢について10%の使用;Sp35−1−Fc処置動物においては55%の左肢(非障害性)の使用 対 コントロール−Fc処置動物またはPBS処置動物においては80%の使用;および、Sp35−1−Fc処置動物においては29%の右肢(障害性)の使用 対 コントロール−Fc処置動物またはPBS処置動物においては約15%の使用。
【0194】
(実施例19:Sp35−Fcは、視神経切除モデルにおける網膜神経節細胞(RGC)生存を促進する)
本発明者らは、視神経切除モデルを使用してSp35の活性をさらに確認した。これは、ニューロンの機能に影響を与える因子を調査する。若年成体の雌のSprague Dawley(SD)ラットを、この研究で使用した。各動物の右側の視神経を、眼窩内で、視神経円板から1.5mm切除した。6%フルオロゴールド(FG)で浸漬した1つのゲルフォームを、視神経円板の真後ろの新たに切除した部位に適用し、生存網膜神経節細胞(RGC)を標識した。この動物を、6つの群(各群においてn=6)に分割しSp35−Fc、ヒトIgG1、またはPBSのみのいずれかを硝子体内注入によって与えた。各硝子体内注入の体積は、4mlであった。他方、各注入の投与量は2mgであった。この硝子体内注入を、視神経切除の直後に行った。
【0195】
全ての動物を1週間生存させた。動物を屠殺する2日前、各動物の左側の視神経を切除し、6%FGを使用して、内部コントロールとして機能する生存RGCを標識した。動物を、過量のネンブタールで屠殺し、網膜を、4%パラホルムアルデヒド中で解剖した。4つのラジアルカットを、網膜を4つの四分円(上方、下方、鼻側、および頭側)に分割するように作製した。次いで、この網膜を封入剤(Dako)でフラットに取り付ける前に、この網膜を同じ固定液中で1時間事後固定した。このスライドを、蛍光顕微鏡の下、紫外線フィルター(励起波長=330〜380nm)を使用して試験した。標識RGCを、200×200mmの接眼グリッドの下で、視神経平板から出発して500mm間を置いた網膜の末梢境界まで、各四分円の中線に沿って数えた。各処置がもたらした生存RGCの百分率を、損傷眼における生存RGCの数と反対側の側眼における生存RGCの数とを比較することによって表した。全てのデータを、平均±SEMとして表した。統計学的有意性を、一方向ANOVAによって評価し、続いてTukey−Kramerポストホック検定によって評価した。差異は、p<0.05が有意であると考えられた。Sp35−Fc処置動物は、コントロール−Fc処置動物またはPBS処置動物と比較した場合に有意なニューロンの生存(83%)を示した。コントロールFc処置動物またはPBS処置動物は各々、約50%のニューロンの生存を示しただけだった。
【0196】
(他の実施形態)
他の実施形態は、添付の特許請求の範囲内である。
【図面の簡単な説明】
【0197】
【図1−1】図1は、全長ヒトSp35 cDNAのヌクレオチド配列である(配列番号1)。
【図1−2】図1は、全長ヒトSp35 cDNAのヌクレオチド配列である(配列番号1)。
【図2】図2は、全長ヒトSp35ポリペプチドのアミノ酸配列である(配列番号2)。
【図3】図3は、Sp35ドメイン構造およびNgR1に結合するSp35配列を同定するための欠失マッピングの概略図である。
【図4】図4は、ラットp75をコードする発現ベクターまたはベクターコントロールをトランスフェクションされたCOS7細胞に結合するSP35に関するデータを要約するヒストグラムである。48時間後、AP−SP35またはAPを、上記細胞とともにインキュベートした。結合したAPを、発色AP検出試薬を用いて検出した。
【図5】図5は、NgR1;NgR1およびp75;NgR1、p75およびSP35をコードする発現ベクターまたはベクターコントロールをトランスフェクションされたCOS7細胞に対する、AP−OmgpおよびAP−Nogo−66の結合に関するデータを要約するヒストグラムである。48時間後、AP−Omgp、AP−Nogo−66またはAPを、上記細胞とともにインキュベートした。結合したAPを、発色AP検出試薬を用いて検出した。
【図6】図6は、インビトロでの神経突起伸長に対するミエリンインヒビターの阻害活性の軽減に関するデータを要約するヒストグラムである。神経突起長を、固定化した基質Omgp、ミエリンおよびNogo−66上で培養された、DN−Sp35、全長Sp35を発現する生後7日ラット小脳顆粒状ニューロン、またはコントロール生後7日ラット小脳顆粒状ニューロンについて測定した。DN−Sp35を用いてトランスフェクションした細胞は、阻害基質に対する応答の減少を示した。神経突起長を、2回の独立した実験からの処置群あたり1000ニューロンから定量した(p<0.01)。
【図7】図7は、SP35−Fcによるミエリンインヒビターの阻害活性の逆転に関するデータを要約するヒストグラムである。生後7日ラット小脳顆粒状ニューロン(1000ニューロン)の神経突起長を、SP35−Fcの存在下または非存在下、固定化した基質OMgp、ミエリンまたはNogo−66上で培養した。SP35−Fcは、Omgp、Nogo−66およびMAGにより引き起こされる神経突起伸長阻害を減少した。神経突起長を、2回の独立した実験からの処置群あたり1000ニューロンから定量した(p<0.01)。
【図8】図8は、くも膜下腔内投与されたSp35−Fcがラットにおける脊椎半側切除後の機能的回復を改善したことを示す実験からのデータを要約するグラフである。運動性BBBスコアを、コントロール(IgG)ラットまたはSp35−Fc処置ラットにおける脊椎半側切除後の時間の関数として測定した(群あたり動物8匹)。脊髄損傷時に、処置を開始した。
【図9】図9は、図8で要約される実験における、4週目の個々の動物のBBBスコアを示すグラフである。
【図1】
【技術分野】
【0001】
(発明の分野)
本発明は、神経学、神経生物学および分子生物学に関する。より詳細には、本発明は、神経学的疾患、神経学的障害および神経学的損傷(例えば、脊髄損傷)の処置のための分子および方法に関する。
【背景技術】
【0002】
(発明の背景)
軸索および樹状突起は、ニューロンから伸長する。伸長する軸索または神経突起の遠位端は、成長円錐として公知の特定の領域を含む。成長円錐は、局所的環境を感じ取り、ニューロンの標的細胞に向かう軸索成長を誘導する。成長円錐は、環境の合図(例えば、表面への接着、成長因子、神経伝達物質および電場)に応答する。上記成長円錐は、一般的に、一日に1mm〜2mmの速度で成長する。上記成長円錐は、ラメリポディウムおよび糸状足として分類される伸長手段によって、その前方領域およびいずれもの側方領域を探索する。伸長部が不都合な表面に接触する場合、その伸長部は引っ込む。伸長部が、都合のよい成長表面と接触する場合、その伸長部は、伸長を続け、上記成長円錐をその方向に誘導する。上記成長円錐が適切な標的細胞に到達する場合、シナプス結合が生成される。
【0003】
神経細胞機能は、ニューロンとそれらの隣接環境における他の細胞との間の接触により、影響される(Rutishauserら,1988,Physiol.Rev.68:819)。これらの細胞としては、特定の神経膠細胞、中枢神経系(CNS)における希乏突起神経膠細胞ならびに末梢神経系(PNS)におけるシュヴァン細胞が挙げられ、これらの細胞は、神経軸索がミエリンで覆われている(Lemke,1992,An Introduction to Molecular Neurobiology,Z.Hall編,281頁,Sinauer)。
【0004】
CNSニューロンは、損傷後に再生成する固有の潜在能力を有するが、CNSニューロンは、ミエリンに存在する阻害タンパク質によって損傷後に再生成することを阻害される(Brittisら,2001,Neuron 30:11−14;Jonesら,2002,J.Neurosci.22:2792−2803;Grimpeら,2002,J.Neurosci.:22:3144−3160)。
【0005】
希乏突起神経膠細胞において見出されたいくつかのミエリン阻害タンパク質が、特徴付けられている。ミエリン阻害タンパク質の公知の例としては、NogoA(Chenら,Nature,2000,403,434−439;Grandpreら,Nature 2000,403,439−444)、ミエリン関連糖タンパク質(MAG)(McKerracherら,1994,Neuron 13:805−811;Mukhopadhyayら,1994,Neuron 13:757−767)および希乏突起神経膠細胞糖タンパク質(OM−gp)(Mikolら,1998,J.Cell.Biol.106:1273−1279)が挙げられる。これらのタンパク質の各々は、ニューロンのNgR1に対するリガンドであることが、別々に示されている(Wangら,Nature 2002,417,941−944;Grandpreら,Nature 2000,403,439−444;Chenら,Nature,2000,403,434−439;2002年6月28日にインターネット上で公開された,Domeniconiら,Neuron 2002)。
【0006】
Nogoレセプター−1(NgR1)は、8個のロイシンリッチ繰返し配列を含むGPI−固定膜タンパク質である(Fournierら,2001,Nature 409:341−346)。阻害タンパク質(例えば、NogoA、MAGおよびOM−gp)と相互作用すると、上記NgR1複合体は、成長円錐崩壊および神経突起伸長阻害を引き起こすシグナルを伝達する。
【発明の開示】
【発明が解決しようとする課題】
【0007】
NgR1媒介性の成長円錐崩壊およびその結果もたらされる神経突起伸長阻害を阻害するための分子および方法に関する必要性は、未だ満たされていない。
【課題を解決するための手段】
【0008】
(発明の要旨)
発明者らは、「Sp35」と称されるポリペプチド(発明者らが命名)に関する種々の発見を行ってきた。Sp35のための代替的な名称としては、「LINGO」および「LINGO−1」が挙げられる。発明者らの発見は、以下を含む。Sp35は、NgR1に結合する。Sp35は、同型相互作用で、それ自体に結合する。Sp35−Fc融合タンパク質は、顆粒状ニューロンにおいて、線維束性攣縮を誘導または促進する。Sp35−Fc融合タンパク質は、赤核脊髄路半側切除損傷モデルおよび視神経離断モデルの両方において、ニューロンの生存を促進する。Sp35レトロウイルス感染皮質初代細胞は、脊髄損傷ラットに送達される場合、ニューロン生存の促進、軸索のβIIIチューブリン染色の増大およびミエリン含量の上昇をもたらす。
【0009】
これらの発見に部分的に基づいて、本発明は、ポリペプチドをコードするヌクレオチド配列を含む単離された核酸を特徴とし:(a)このポリペプチドは(i)Sp35 LRRドメイン、(ii)このLRRドメインに対してC末端側のSp35塩基性領域および(iii)この塩基性領域に対してC末端側のSp35免疫グロブリン(Ig)ドメイン、を含み;そして(b)このポリペプチドは、膜貫通ドメインを欠く。上記Sp35 LRRドメインは、カルボキシ末端LRR(LRRCT)、アミノ末端LRR(LRRNT)またはその両方を含み得る。本発明のいくつかの実施形態において、上記コードされたSp35ポリペプチドは、細胞質ドメインを欠く。いくつかの実施形態において、上記コードされたSp35ポリペプチドは、配列番号2のアミノ酸残基34〜532を含み、アミノ酸残基533〜614を欠く。
【0010】
本発明はまた、ポリペプチドをコードする核酸を含み、このポリペプチドは、Sp35 Igドメインを含み、そして、Sp35 LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く。
【0011】
本発明はまた、ポリペプチドをコードする核酸を含み、このポリペプチドは、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く。
【0012】
本発明はまた、ポリペプチドをコードする核酸を含み、このポリペプチドは、機能性細胞質ドメインを欠くが、全ての他のSp35ドメインを含む。例えば、上記コードされたポリペプチドは、(シグナル配列のプロセシング前に)配列番号2のアミノ酸1〜576を含み得る。
【0013】
本発明のいくつかの実施形態において、上記コードされたポリペプチドは、非Sp35部分を含む融合タンパク質である。上記非Sp35部分は、例えば、Ig部分、血清アルブミン部分、標的化部分、レポーター部分または精製容易化部分であり得る。好ましい非Sp35部分は、Ig部分(例えば、Fc部分)である。
【0014】
上記ヌクレオチド配列は、例えば、発現ベクターにおいて、発現制御配列に作動可能に結合され得る。本発明はまた、本発明のSp35ポリペプチドを発現するベクターにより形質転換される宿主細胞を含む。
【0015】
本発明はまた、上に記載される核酸のいずれかによりコードされるSp35ポリペプチドを含む。
【0016】
本発明はまた、ポリマー(例えば、ポリアルキレングリコール、糖ポリマーおよびポリペプチド)に結合されたSp35ポリペプチドを含む。好ましいポリマーは、ポリアルキレングリコール(例えば、ポリエチレングリコール(PEG))である。上記ポリペプチドは、1つ、2つ、3つまたは4つのポリマーに結合され得る。好ましくは、上記結合されたポリマーの総分子量は、Sp35ポリペプチド当たり20,000Da〜40,000Daである。
【0017】
本発明はまた、NgR1によるシグナル伝達を阻害する方法を含む。上記方法は、上記NgR1と有効量のSp35ポリペプチドとを接触させる工程を包含する。上記方法における使用のための好ましいポリペプチドとしては、以下が挙げられる:
(a)Sp35ポリペプチドであって:(a)このポリペプチドが(i)Sp35 LRRドメイン、(ii)このLRRドメインに対してC末端側のSp35塩基性領域および(iii)この塩基性領域に対してC末端側のSp35免疫グロブリン(Ig)ドメイン、を含み;そして(b)このポリペプチドが、膜貫通ドメインを欠く、ポリペプチド;ならびに
(b)Sp35 Igドメインを含み、そして、Sp35 LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠くSp35ポリペプチド。
【0018】
本発明はまた、中枢神経系(CNS)ニューロンの軸索成長阻害を減少する方法を含む。上記方法は、上記ニューロンと有効量のポリペプチド(例えば、Sp35ポリペプチド、抗Sp35抗体または抗Sp35抗体の抗原結合フラグメント)とを接触させる工程を包含する。
【0019】
本発明はまた、CNSニューロンの成長円錐崩壊を阻害する方法を含む。上記方法は、上記ニューロンと有効量のポリペプチド(例えば、Sp35ポリペプチド、抗Sp35抗体または抗Sp35抗体の抗原結合フラグメント)とを接触させる工程を包含する。
【0020】
本発明はまた、哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置する方法を含む。上記方法は、治療有効量のポリペプチド(例えば、Sp35ポリペプチド、抗Sp35抗体または抗Sp35抗体の抗原結合フラグメント)を上記哺乳動物に投与する工程を包含する。本発明のいくつかの実施形態において、上記CNS疾患、CNS障害またはCNS損傷は、脊髄損傷である。上記Sp35ポリペプチドは、局所投与され得る。上記方法のいくつかの実施形態において、上記Sp35ポリペプチドは、脊髄損傷の48時間以内に最初に投与される。局所投与のために、上記治療有効量の上記ポリペプチドは、好ましくは、10μg/kg〜10mg/kgである。全身投与のために、上記治療有効量の上記ポリペプチドは、好ましくは、1mg/kg〜20mg/kgである。
【0021】
本発明はまた、哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置するエキソビボ遺伝子治療方法を含む。上記方法は(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)上記CNS疾患、CNS障害またはCNS損傷(例えば、脊髄損傷)の部位で、上記哺乳動物中にこの宿主細胞を導入する工程、を包含する。上記培養宿主細胞は、処置されるべき哺乳動物由来であり得る。このエキソビボ遺伝子治療方法において、上記組換えSp35ポリペプチドは、全長Sp35ポリペプチドであり得る。
【0022】
本発明はまた、上記CNS疾患、CNS障害またはCNS損傷の部位での髄鞘形成を促進する方法を含む。上記方法は、上記CNS疾患、CNS障害またはCNS損傷の部位と有効量のSp35ポリペプチド(例えば、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠くポリペプチド)とを接触させる工程を包含する。
【0023】
本発明はまた、CNS疾患、CNS障害またはCNS損傷をインビボ遺伝子治療により処置するインビボ遺伝子治療方法を含む。上記方法は、上記疾患、障害または損傷の部位あるいはその近傍で、哺乳動物にウイルスベクターを投与する工程を包含し、このウイルスベクターは、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、このSp35ポリペプチドは、この哺乳動物において、損傷の部位またはその近傍のニューロンによる軸索伸長阻害を減少するために十分な量で、このヌクレオチド配列から発現される。上記ウイルスベクターは、例えば、アデノウイルスベクター、レンチウイルスベクター、バキュロウイルスベクター、エプスタイン−バーウイルスベクター、パポバウイルスベクター、ワクシニアウイルスベクターおよび単純疱疹イルスベクターであり得る。上記疾患、障害または損傷は、例えば、脊髄損傷または視覚神経損傷であり得る。上記ウイルスベクターは、例えば、局所投与、眼内投与、非経口投与、くも膜下腔内投与、硬膜下投与および皮下投与のような経路により投与され得る。
【0024】
本発明はまた、死の危険性にあるニューロンの生存を促進する方法を含む。上記方法は、上記ニューロンと有効量のSp35ポリペプチドとを接触させる工程を包含する。上記Sp35ポリペプチドは、Sp35の可溶性形態(例えば、Sp35−Fc融合タンパク質)であり得る。上記ニューロンは、インビトロまたはインビボ(例えば、神経変性疾患、神経変性障害または神経変性損傷(例えば、多発性硬化症、ALS、ハンチントン病、アルツハイマー病、パーキンソン病、糖尿病性ニューロパシー、脳卒中、外傷性脳損傷および外傷性脊髄損傷)を有する哺乳動物中)であり得る。本発明のいくつかの実施形態において、上記Sp35ポリペプチドは、以下により間接的に投与される:(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)上記ニューロンの部位で、上記哺乳動物中にこの宿主細胞を導入する工程。本発明のいくつかの実施形態において、上記ポリペプチドは、インビボ遺伝子治療を介して間接的に投与される。いくつかの実施形態において、上記方法は、上記ニューロンの部位またはその近傍にウイルスベクターを投与する工程を包含し、このウイルスベクターは、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、このSp35ポリペプチドは、上記哺乳動物において、このニューロンの生存を促進するために十分な量で、このヌクレオチド配列から発現される。
【0025】
本明細書中で使用される場合、「全長ヒトSp35ポリペプチド」とは、そのアミノ酸配列が配列番号2のアミノ酸34〜614であるポリペプチドを意味する。
【0026】
本明細書中で使用される場合、「異種部分」とは、全長Sp35ポリペプチドに存在しないアミノ酸配列を意味する。
【0027】
本明細書中で使用される場合、「nogoレセプター−1」とは、その配列がGenbank受託番号AAG53612の下で公的に入手可能であるポリペプチドを意味する。
【0028】
本明細書中で使用される場合、「Sp35アンタゴニストポリペプチド」とは、天然に生じるSp35の生物学的活性を阻止、阻害または妨害するSp35ポリペプチドを意味する。
【0029】
本明細書中で使用される場合、「Sp35塩基性領域」とは、以下のアミノ酸モチーフを意味する:
【0030】
【化1】
最上列のアミノ酸(太字;配列番号4)は、好ましいSp35塩基性領域配列であり、任意の置換を示す変異体を、下に示す(配列番号5、6、7および8)。
【0031】
本明細書中で使用される場合、「Sp35融合タンパク質」とは、異種部分に融合されたSp35部分を含む融合タンパク質を意味する。
【0032】
本明細書中で使用される場合、「Sp35 Igドメイン」とは、配列番号2のアミノ酸433〜493を意味し、ただし、この配列は、5個までの独立したアミノ酸の挿入、欠失または保存的アミノ酸置換を含み得る。以下の置換(配列番号2に基づく番号付け)が、明らかに含まれる:6位のVからM;294位のSからG;348位のVからA;419位のRからH。
【0033】
本明細書中で使用される場合、「Sp35 LRRドメイン」とは、10個〜14個のロイシンリッチ繰返し配列を含むドメインを意味し、このような配列としては、表1に列挙されるLRRNTおよびLRRCTが挙げられるが、ただし、5個までのアミノ酸の挿入、欠失または保存的アミノ酸置換が、10個〜14個のロイシンリッチ繰返し配列の集合体中に現れ得る。
【0034】
本明細書中で使用される場合、「Sp35部分」とは、全長Sp35ポリペプチドの生物学的に活性なフラグメントを意味する。
【0035】
本明細書中で使用される場合、「Sp35ポリペプチド」とは、Sp35部分またはSp35部分を含む融合タンパク質を意味する。
【0036】
別に規定されなければ、本明細書中で使用される全ての技術用語および科学用語は、本発明の属する分野の当業者により一般に理解されるものと同じ意味を有する。争いが生じた場合、定義を含む本明細書の記載に従う。本明細書中で言及される全ての刊行物、特許および他の参考文献が、参考として援用される。
【0037】
本明細書中に記載される方法および物質と類似または等価な方法および物質が、本発明の実施または試験において使用され得るが、好ましい方法および物質は、以下に記載される。上記物質、方法および例は、例示に過ぎず、限定するものとは意図されない。本発明の他の特徴および利点は、詳細な説明および特許請求の範囲から明らかである。
【発明を実施するための最良の形態】
【0038】
(発明の詳細な説明)
天然に生じるヒトSp35は、614アミノ酸を含むグリコシル化CNS特異的タンパク質である(図2;配列番号2)。ヒト全長野生型Sp35ポリペプチドは、14個のロイシンリッチ繰返し配列からなるLRRドメイン(N末端キャップおよびC末端キャップを含む)、Igドメイン、膜貫通領域および細胞質ドメインを含む(図3)。上記細胞質ドメインは、典型的なチロシンリン酸化部位を含む。さらに、上記天然に生じるSp35タンパク質は、シグナル配列、LRRCTとIgドメインとの間の短い塩基性領域およびこのIgドメインと上記細胞質ドメインと間の膜貫通領域を含む(図3)。ヒトSp35遺伝子は、代替的な翻訳開始コドンを含み、それにより、6個のさらなるアミノ酸(すなわち、MQVSKR(配列番号9))が、上記Sp35シグナル配列のN末端に存在していても存在していなくてもよい。表1は、図2の配列(配列番号2)に基づいたアミノ酸残基番号に従って、上記Sp35ドメインおよび他の領域を列挙する。
【0039】
【表1】
Sp35の組織分布および発生における発現は、ヒトおよびラットにおいて研究されている。Sp35生物学は、実験動物モデル(ラット)において研究されている。ラットSP35の発現は、ノザンブロットおよび免疫組織学的染色により決定される場合、CNSニューロンおよび脳の希乏突起神経膠細胞に局在している。ラットSp35 mRNA発現レベルは、発生において調節され、出生直後(すなわち、生後約1日目)がピークである。ラット脊髄離断損傷モデルにおいて、Sp35は、RT−PCRにより決定される場合、上記損傷部位でアップレギュレートされる。
【0040】
発明者らは、全長野生型Sp35が、NgR1に結合することを発見している。Sp35の可溶性誘導体は、NgR1に結合してその機能を阻止、阻害または妨害し、それにより、CNSニューロンにおいて正常に起こる軸索伸長のNgR1媒介性阻害を軽減することにより、Sp35アンタゴニストポリペプチドとして機能する。これは、軸索伸長または神経突起成長が脳または脊髄において必要とされる状態において、有益である。脊髄損傷(部分的または完全な挫傷または切断を含む)は、軸索伸長が必要とされる状態を例示するが、通常、Nogo経路の操作により阻害される。脳における軸索伸長および/もしくは神経突起成長が有益である疾患または障害の例としては、脳卒中、多発性硬化症および他の神経変性疾患または神経変性障害が挙げられる。
【0041】
本発明の方法において、Sp35ポリペプチドまたはSp35ブロッキング抗体(または抗原結合抗体フラグメント)は、事前に形成されたポリペプチドとして直接投与され得るか、または核酸ベクターを介して間接的に投与され得、NgR1機能に拮抗し、有益な軸索伸長を可能にする。
【0042】
本発明のいくつかの実施形態において、可溶性Sp35アンタゴニストポリペプチドは、以下を包含する処置方法において投与される:(1)移植可能な宿主細胞をSp35ポリペプチドを発現する核酸(例えば、ベクター)を用いて形質転換またはトランスフェクションする工程;および(2)形質転換された宿主細胞を、哺乳動物中に、疾患、障害または損傷の部位で移植する工程。例えば、上記形質転換された宿主細胞は、脊髄損傷部位に移植され得る。本発明のいくつかの実施形態において、上記移植可能宿主細胞は、哺乳動物から取り出され、一時的に培養され、可溶性Sp35ポリペプチドをコードする単離された核酸を用いて形質転換またはトランスフェクションされ、そして、それが取り出された同じ哺乳動物に移植して戻される。上記細胞は、移植される同じ部位から取り出されてもよいが、その必要はない。このような実施形態は、時に、エキソビボ遺伝子治療として公知であり、限定された時間、作用部位に局在化したSp35ポリペプチドの連続的供給を提供し得る。
【0043】
本発明は、NgR1とSp35との相互作用およびSp35同種相互作用の調節因子として有用なオリゴペプチドを提供する。上記オリゴペプチドは、以下のアミノ酸モチーフを含む:
【0044】
【化2】
アミノ酸の最上列(太字;配列番号10)が好ましい配列であり、任意の置換を含む変異体を、下に示す(配列番号11、12および13)。
【0045】
種々の例示的なSp35ポリペプチド、抗Sp35抗体および抗体フラグメントならびに本発明の実施のためにこれらの分子を得るための方法および材料を、以下に記載する。
【0046】
(融合タンパク質および結合されたポリペプチド)
本発明のいくつかの実施形態は、Sp35部分が異種ポリペプチド部分に融合されてSp35融合タンパク質を形成するSp35ポリペプチド(例えば、Sp35アンタゴニストポリペプチド)の使用を含む。Sp35融合タンパク質は、種々の目的(例えば、血清半減期の上昇、バイオアベイラビリティの改善、特定の器官または組織のタイプに対するインビボ標的化、組換え発現効率の改善、宿主細胞分泌の改善、精製の容易化および高いアビディティ)を達成するために使用され得る。達成されるべき目的に依存して、上記異種部分は、不活性であっても生物学的に活性であってもよい。また、上記異種部分は、インビボまたはインビトロにおいて、上記Sp35部分に安定に融合されるかまたは切断可能であるように選択され得る。異なる目的を達成するための異種部分は、当該分野において公知である。
【0047】
Sp35融合タンパク質の発現の代替として、選択された異種部分は、事前に形成され、上記Sp35部分に化学的に結合され得る。ほとんどの場合、選択された異種部分は、上記Sp35部分に融合されていても、または結合されていても、同様に機能する。従って、異種アミノ酸配列の以下の議論において、別に示されなければ、その異種配列は、融合タンパク質の形態かまたは化学結合体として上記Sp35部分に結合され得ることが理解されるべきである。
【0048】
薬理学的に活性なポリペプチド(例えば、Sp35)は、しばしば、迅速なインビボでのクリアランスを示し、身体における治療有効濃度を達成するためには多用量を必要とする。さらに、約60kDaよりも小さなポリペプチドは糸球体濾過を経験する可能性があり、これは、時に、腎毒性を引き起こす。比較的小さいポリペプチド(例えば、Sp35フラグメント)の融合または結合は、このような腎毒性の危険性を軽減または回避するために用いられ得る。治療ポリペプチドのインビボ安定性(すなわち、血清半減期)を増加するための種々の異種アミノ酸配列(すなわち、ポリペプチド部分または「キャリア」)が公知である。
【0049】
その長い半減期、広いインビボ分布および酵素的機能または免疫学的機能の欠如に起因して、本質的に全長のヒト血清アルブミン(HSA)またはHSAフラグメントは、好ましい異種部分である。Yehら,1992,Proc.Natl.Acad.Sci.USA,89:1904−1908およびSyedら,1997,Blood 89:3243−3252に教示されているような方法および材料の適用によって、HSAは、上記Sp35部分に起因する薬理学的活性を示し、一方で、有意なインビボ安定性の増加(例えば、10倍〜100倍高い)を示すSp35融合タンパク質またはSp35結合体を形成するために使用され得る。好ましくは、上記HSAのC末端は、上記Sp35部分のN末端に融合される。HSAは、天然に分泌されるタンパク質であるから、このHSAシグナル配列は、上記融合タンパク質が真核生物(例えば、哺乳動物)発現系において生成される場合、上記Sp35融合タンパク質の細胞培養培地への分泌を得るために利用され得る。
【0050】
本発明のいくつかの実施形態は、Sp35部分がFc領域(すなわち、Ig重鎖定常領域のC末端部分)に融合されるSp35ポリペプチドを用いる。Sp35−Fc融合の潜在的な利点としては、可溶性、インビボ安定性および多価性(multivalency)(例えば、二量体化)が挙げられる。使用されるFc領域は、IgA、IgDまたはIgGのFc領域(ヒンジ−CH2−CH3)であり得る。あるいは、使用されるFc領域は、IgEまたはIgMのFc領域(ヒンジ−CH2−CH3−CH4)であり得る。IgGのFc領域(例えば、IgG1のFc領域またはIgG4のFc領域)が好ましい。Fc融合体をコードするDNAを構築し、発現するための材料および方法は、当該分野において公知であり、過度の実験をすることなくSp35融合体を得るために適用され得る。本発明のいくつかの実施形態は、Caponらの米国特許第5,428,130号および同第5,565,335号に記載されるようなSp35融合タンパク質を用いる。
【0051】
上記シグナル配列は、小胞体膜を通るタンパク質輸送を開始するアミノ酸配列をコードする、ポリヌクレオチドである。免疫融合体(immunofusin)を構築するために有用なシグナル配列としては、抗体軽鎖シグナル配列(例えば、抗体14.18(Gilliesら,1989,J.Immunol.Meth.,125:191−202))、抗体重鎖シグナル配列(例えば、MOPC141抗体重鎖シグナル配列(Sakanoら,1980,Nature 286:5774))が挙げられる。あるいは、他のシグナル配列が使用され得る。例えば、Watson,1984,Nucleic Acids Research 12:5145を参照のこと。上記シグナルペプチドは、通常、シグナルペプチダーゼにより上記小胞体の管腔において切断される。この結果、Fc領域および上記Sp35部分を含む免疫融合体タンパク質の分泌がもたらされる。
【0052】
いくつかの実施形態において、DNA配列は、上記分泌カセットと上記Sp35部分との間のタンパク分解性切断部位をコードする。切断部位は、コードされた融合タンパク質のタンパク分解性切断を提供し、従って、上記Fcドメインを上記標的タンパク質から分離する。有用なタンパク分解性切断部位としては、タンパク分解性酵素(例えば、トリプシン、プラスミン、トロンビン、因子XaまたはエンテロキナーゼK)により認識されるアミノ酸配列が挙げられる。
【0053】
上記分泌カセットは、複製可能発現ベクターに組み込まれ得る。有用なベクターとしては、直鎖状核酸、プラスミド、ファージミド、コスミドなどが挙げられる。例示的な発現ベクターはpdCであり、ここで、上記免疫融合体DNAの転写は、ヒトサイトメガロウイルスのエンハンサーおよびプロモーターの制御下で行われる。例えば、Loら,1991,Biochem.Biophys.Acta 1088:712;およびLoら,1998,Protein Engineering 11:495−500を参照のこと。適切な宿主細胞は、Sp35ポリペプチドをコードするDNAを用いて形質転換またはトランスフェクションされ得、Sp35ポリペプチドの発現および分泌のために使用される。好ましい宿主細胞としては、不死化ハイブリドーマ細胞、骨髄腫細胞、293細胞、チャイニーズハムスター卵巣(CHO)細胞、Hela細胞およびCOS細胞が挙げられる。
【0054】
完全にインタクトな野生型Fc領域は、本発明に従うFc融合タンパク質において、通常、不必要であり、そして、所望されないエフェクター機能を示す。従って、特定の結合部位は、好ましくは、上記分泌カセットの構築の間に上記Fc領域から削除される。例えば、上記軽鎖との共発現は不必要であるので、上記重鎖結合タンパク質であるBip(Hendershotら,1987,Immunol.Today 8:111−114)に対する結合部位は、IgEのFc領域のCH2ドメインから削除され、それにより、この部位は、免疫融合体の有効な分泌を妨害しない。同様に、免疫グロブリンの軽鎖への結合に関与するFc領域に存在するシステイン残基は、削除されるかまたは別のアミノ酸に置換され、それにより、これらのシステイン残基が、免疫融合体として生成される場合に、上記Fc領域の適切な折り畳みを妨害しないようにするべきである。膜貫通ドメイン配列(例えば、IgMに存在する膜貫通ドメイン配列)は、削除されるべきである。
【0055】
IgG1のFc領域が好ましい。あるいは、他の免疫グロブリンγサブクラス(γ−2、γ−3およびγ−4)のFc領域が、上記分泌カセットにおいて使用され得る。免疫グロブリンγ−1のIgG1 Fc領域は、好ましくは、ヒンジ領域(少なくとも一部)、CH2領域およびCH3領域を含む分泌カセットにおいて使用される。いくつかの実施形態において、免疫グロブリンγ−1のFc領域は、CH2削除されたFcであり、これは、ヒンジ領域およびCH3領域の一部を含むが、CH2領域は含まない。CH2削除されたFcは、Gilliesら,1990,Hum.Antibod.Hybridomas,1:47により記載されている。いくつかの実施形態において、IgA、IgD、IgEまたはIgMのFc領域が使用される。
【0056】
Sp35−Fc融合タンパク質は、いくつかの異なる構造で構築され得る。1つの構造において、上記Sp35部分のC末端は、上記Fc部分のN末端に直接融合される。わずかに異なる構造において、短いポリペプチド(例えば、2個〜10個のアミノ酸)が、上記Sp35部分のN末端と上記Fc部分のC末端との間の融合中に組み込まれる。このようなリンカーは、いくつかの状態において生物学的活性を改善し得る、コンホメーションの柔軟性を提供する。ヒンジ領域の充分な部分が上記Fc部分において維持される場合、上記Sp35−Fc融合体は、二量体化し、従って、二価の分子を形成する。単量体Fc融合体の同種の集団は、単一の特異性を有する二価の二量体をもたらす。各々が異なる特異性を有する2つの単量体Fc融合体の混合物は、2種の特異性を有する二価の二量体をもたらす。
【0057】
対応するアミノ反応性基およびチオール反応性基を含む任意の数の架橋剤が、Sp35を血清アルブミンに結合するために使用され得る。適切なリンカーの例としては、チオール反応性マレイミドを挿入するアミン反応性架橋剤(例えば、SMCC、AMAS、BMPS、MBS、EMCS、SMPB、SMPH、KMUSおよびGMBS)が挙げられる。他の適切なリンカーは、チオール反応性ハロアセテート基(例えば、SBAP、SIA、SIAB)を挿入する。スルフヒドリル基との反応のための保護チオールまたは非保護チオールを提供し、還元可能な結合を生じるリンカーとしては、SPDP、SMPT、SATAおよびSATPが挙げられる。このような試薬は、市販されている(例えば、Pierce Chemicals)。
【0058】
結合は、Sp35ポリペプチドのN末端または血清アルブミン上のチオール部分を含まなくてもよい。例えば、Sp35−アルブミン融合体は、遺伝子工学的技術を用いて得られ得、上記Sp35部分は、そのN末端、C末端または両方で上記血清アルブミン遺伝子に融合される。
【0059】
Sp35ポリペプチドは、異種ペプチドに融合されて、上記Sp35部分の精製または同定を容易にし得る。例えば、ヒスチジンタグは、Sp35ポリペプチドに融合されて、市販のクロマトグラフィー媒体を用いる精製を容易にし得る。
【0060】
本発明のいくつかの実施形態において、Sp35融合構築体は、細菌におけるSp35部分の生成を促進するために使用される。このような構築体において、通常、高レベルで発現および/または分泌される細菌タンパク質が、Sp35ポリペプチドのN末端融合パートナーとして用いられる。例えば、Smithら,1988 Gene 67:31;Hoppら,1988,Biotechnology 6:1204;La Vallieら,1993,Biotechnology 11:187を参照のこと。
【0061】
本発明のいくつかの実施形態において、融合構築体としては、Sp35部分および第2のヒトNgR1結合部分(例えば、希乏突起神経膠細胞−ミエリン糖タンパク質(OMgp)部分、ミエリン関連糖タンパク質(MAG)部分またはNogo66部分)が挙げられる。このような構築体の利点としては、NgR1結合親和性の上昇が挙げられる。
【0062】
全長OMgpアミノ酸配列は、当該分野において公知である(GenBank受託番号P23515)。Sp35−OMgp融合体の具体例としては、以下が挙げられる:
Sp35(aa 34〜532)+IgG1 Fc+OMgp(アミノ酸残基25〜400);および
Sp35(aa 34〜532)+HSA+OMgp(アミノ酸残基25〜400)。
【0063】
全長MAGアミノ酸配列は、当該分野において公知である(GenBank受託番号A61084)。Sp35−MAG融合体の具体例としては、以下が挙げられる:
Sp35(aa 34〜532)+IgG1 Fc+MAG(アミノ酸残基12〜500);および
Sp35(aa 34〜532)+HSA+MAG(アミノ酸残基12〜500)。
【0064】
全長Nogoアミノ酸配列は、当該分野において公知である(NogoA GenBank受託番号AY102279)。Sp35−Nogo融合体の具体例としては、以下が挙げられる:
Sp35(aa 34〜532)+IgG1 Fc+Nogo66(NogoA アミノ酸残基1056〜1122);
Sp35(aa 34〜532)+HSA+Nogo66(NogoA アミノ酸残基1056〜1122);
Sp35(aa 34〜532)+IgG1 Fc+アミノNogo(NogoA アミノ酸残基1〜949);および
Sp35(aa 34〜532)+HSA+アミノNogo(NogoA アミノ酸残基1〜949)。
【0065】
適切な融合パートナーのアミノ末端およびカルボキシ末端でSp35部分を融合することにより、Sp35ポリペプチドの二価形態または四価形態が得られ得る。例えば、Sp35部分は、Ig部分のアミノ末端およびカルボキシ末端に融合され、2つのSp35部分を含む二価の単量体ポリペプチドを生成し得る。2つのこれらのモノマーが二量体化されると、Ig部分により、Sp35タンパク質の四価形態が得られる。このような多価形態は、標的に対する結合親和性の上昇を達成するために使用され得る。Sp35の多価形態はまた、Sp35部分を縦列に配置し、コンカテマーを形成することによって得られ得、これは、単独で用いられ得るかまたは融合パートナー(例えば、IgまたはHSA)に融合され得る。
【0066】
(結合ポリマー(ポリペプチド以外))
本発明のいくつかの実施形態は、Sp35ポリペプチドを含み、1つ以上のポリマーが、上記Sp35ポリペプチドに結合(共有結合)される。このような結合のために適切なポリマーの例としては、(上で議論された)ポリペプチド、糖ポリマーおよびポリアルキレングリコール鎖が挙げられる。代表的に、ポリマーは、以下のうちの1つ以上を改善する目的のために上記Sp35ポリペプチドに結合されるが、必ずしもそうでなくてもよい:可溶性、安定性またはバイオアベイラビリティ。
【0067】
Sp35ポリペプチドに対する結合のための好ましいポリマーのクラスは、ポリアルキレングリコールである。ポリエチレングリコール(PEG)が、特に好ましい。PEG部分(例えば、1PEGポリマー、2PEGポリマー、3PEGポリマー、4PEGポリマーまたは5PEGポリマー)は、各々のSp35ポリペプチドに結合されて、上記Sp35ポリペプチド単独と比較して、血清半減期を上昇し得る。PEG部分は非抗原性であり、本質的に、生物学的に不活性である。本発明の実施において使用されるPEG部分は、分枝鎖でも、非分枝鎖でもよい。
【0068】
上記Sp35ポリペプチドに結合されるPEG部分の数および個々のPEG鎖の分子量は、変化し得る。一般的に、上記ポリマーの分子量が大きくなるほど、上記ポリペプチドに結合されるポリマー鎖は少なくなる。好ましくは、上記Sp35ポリペプチドに結合される総ポリマー質量は、20kDa〜40kDaである。従って、1つのポリマー鎖が結合される場合、その鎖の好ましい分子量は、20kDa〜40kDaである。2つの鎖が結合される場合、各々の鎖の好ましい分子量は、10kDa〜20kDaである。3つの鎖が結合される場合、好ましい分子量は、7kDa〜14kDaである。
【0069】
上記ポリマー(例えば、PEG)は、上記ポリペプチド上の任意の適切な露出した反応性基を介して、そのSp35ポリペプチドに結合され得る。上記露出した反応性基は、例えば、N末端アミノ基もしくは内部リジン残基のεアミノ基または両方であり得る。活性化されたポリマーは、上記Sp35ポリペプチド上の任意のフリーのアミノ基において、反応して、共有結合し得る。上記Sp35ポリペプチドのフリーのカルボキシル基、適切に活性化されたカルボニル基、ヒドロキシル、グアニジル、イミダゾール、酸化された炭水化物部分およびメルカプト基はまた(利用可能である場合)、ポリマー結合のための反応性基として使用され得る。
【0070】
好ましくは、結合反応において、ポリペプチド濃度に依存して、1モルのポリペプチドあたり約1.0モル〜約10モルの活性化されたポリマーが用いられる。通常、選択される比は、上記反応の最大化と、上記Sp35部分の所望される薬理学的活性を損ない得る(しばしば、非特異的な)副反応の最小化との間の均衡を表す。好ましくは、上記Sp35ポリペプチドの生物学的活性の少なくとも50%(例えば、本明細書中で記載されるかまたは当該分野で公知のあらゆるアッセイにおいて実証される場合)が維持され、最も好ましくは、約100%が維持される。
【0071】
上記ポリマーは、従来の化学を用いて上記Sp35ポリペプチドに結合され得る。例えば、ポリアルキレングリコール部分は、上記Sp35ポリペプチドのリジンεアミノ基に結合され得る。リジン側鎖に対する結合は、N−ヒドロキシルスクシニミド(NHS)活性エステル(例えば、PEGスクシンイミジルサクシネート(SS−PEG)およびスクシンイミジルプロピオネート(SPA−PEG))を用いて実施され得る。適切なポリアルキレングリコール部分としては、例えば、カルボキシメチル−NHS、ノルロイシン−NHS、SC−PEG、トレシレート(tresylate)、アルデヒド、エポキシド、カルボニルイミダゾールおよびPNPカーボネートが挙げられる。これらの試薬は、市販されている。さらなるアミン反応性PEGリンカーは、スクシンイミジル部分に置換され得る。これらとしては、例えば、イソチオシアネート、ニトロフェニルカーボネート、エポキシドおよびベンゾトリアゾールカーボネートが挙げられる。好ましくは、選択性および反応程度(extent or reaction)を最大化するように、条件が選択される。このような反応条件の最適化は、当該分野における通常の技術内である。
【0072】
PEG化は、当該分野において公知の任意のPEG化反応により実施され得る。例えば、Focus on Growth Factors,3:4−10,1992;公開された欧州特許出願EP 0 154 316およびEP 0 401 384を参照のこと。PEG化は、反応性ポリエチレングリコール分子(または類似の反応性水溶性ポリマー)とのアシル化反応またはアルキル化反応を用いて、実施され得る。
【0073】
アシル化によるPEG化は、一般的に、ポリエチレングリコールの活性エステル誘導体の反応を含む。任意の反応性PEG分子が、上記PEG化において用いられ得る。好ましい活性化PEGエステルは、N−ヒドロキシスクシニミド(NHS)に対してエステル化されたPEGである。本明細書中で使用される場合、「アシル化」は、治療タンパク質と水溶性ポリマー(例えば、PEG)との間の以下のタイプの結合を含むが、これらに限定されない:アミド、カーボネート、ウレタンなど。Bioconjugate Chem.5:133−140,1994を参照のこと。反応パラメータは、上記Sp35ポリペプチドに損傷を与えるかまたは不活性化する温度条件、溶媒条件およびpH条件を避けるように選択されるべきである。
【0074】
好ましくは、上記連結する結合は、アミドである。好ましくは、結果としてもたらされる生成物のうちの少なくとも95%が、モノPEG化、ジPEG化またはトリPEG化される。しかし、より高い程度のPEG化を有するいくつかの種が、使用される具体的反応条件に依存した量で形成され得る。必要に応じて、精製されたPEG化種は、従来の精製方法によってその混合物(特に、未反応種)から分離され、これら従来の精製方法としては、例えば、透析、塩析、限外濾過、イオン交換クロマトグラフィー、ゲル濾過クロマトグラフィーおよび電気泳動が挙げられる。
【0075】
アルキル化によるPEG化は、一般的に、還元剤存在下でのPEGの末端アルデヒド誘導体とSp35との反応を含む。さらに、実質的にSp35のN末端アミノ基においてのみのPEG化(すなわち、モノPEG化タンパク質)に都合のよいように上記反応条件を操作し得る。モノPEG化またはポリPEG化のいずれかにおいて、上記PEG基は、好ましくは、−CH2−NH−基を介して上記タンパク質に結合される。特に−CH2−基に関して、このタイプの結合は、「アルキル」結合として公知である。
【0076】
モノPEG化生成物を生成するための還元的アルキル化を介した誘導体化は、誘導体化のために利用可能な一級アミノ基の異なるタイプの異なる反応性(リジン対N末端)を利用する。上記反応は、リジン残基のεアミノ基とタンパク質のN末端アミノ基との間のpKaの差を利用可能なpHにおいて、実施される。このような選択的誘導体化により、反応性基(例えば、アルデヒド)を含む水溶性ポリマーとタンパク質との結合は、制御される:上記ポリマーとの結合は、上記タンパク質のN末端で主に起こり、他の反応性基(例えば、上記リジン側鎖アミノ基)の有意な改変は、起こらない。
【0077】
上記アシル化方法およびアルキル化方法の両方において使用されるポリマー分子は、水溶性ポリマーの中から選択される。選択されるポリマーは、単一の反応性基(例えば、アシル化のための活性エステルまたはアルキル化のためのアルデヒド)を有するように、好ましくは、重合度が本発明の方法のために提供されるように制御され得るように、改変されるべきである。例示的な反応性PEGアルデヒドは、水溶性であるポリエチレングリコールプロピオンアルデヒドまたはそのモノC1〜C10アルコキシ誘導体もしくはそのモノC1〜C10アリールオキシ誘導体である(米国特許第5,252,714号を参照のこと)。上記ポリマーは、分枝鎖であっても、非分枝鎖であってもよい。上記アシル化反応のために、選択されるポリマーは、単一の反応性エステル基を有する。還元的アルキル化のために、選択されるポリマーは、単一の反応性アルデヒド基を有する。一般的に、上記水溶性ポリマーは、天然に生じるグリコシル残基から選択される。なぜなら、これらは、通常、哺乳動物組換え発現系によって、より都合よく作製されるからである。
【0078】
PEG化Sp35を調製するための方法は、一般的に、(a)Sp35タンパク質またはSp35ポリペプチドと、ポリエチレングリコール(例えば、PEGの反応性エステル誘導体または反応性アルデヒド誘導体)とを、それによってその分子が1つ以上のPEG基に結合されるようになる条件下で反応させる工程、および(b)その反応生成物を得る工程、を包含する。一般的に、上記アシル化反応のための最適反応条件は、公知のパラメータおよび所望される結果に基づいて、場合に応じて決定される。例えば、PEG:タンパク質の比が大きくなるほど、ポリPEG化生成物の百分率は大きくなる。
【0079】
モノ−ポリマー/Sp35の実質的に均一な集団を生成するための還元的アルキル化は、一般的に、以下の工程:(a)Sp35のN末端アミノ基の選択的改変を可能にする(pen−nit)ために適切なpHの還元的アルキル化条件下、Sp35タンパク質またはSp35ポリペプチドと、反応性PEG分子とを反応させる工程;および(b)その反応性生物を得る工程、を包含する。
【0080】
モノ−ポリマー/Sp35の実質的に均一な集団のために、上記還元的アルキル化反応条件は、上記水溶性ポリマー部分と上記Sp35のN末端との選択的結合を可能にする反応条件である。このような反応条件は、一般的に、リジン側鎖アミノ基とN末端アミノ基との間のpKaの差を考慮に入れる。本発明の目的のために、好ましいpHは、3〜9の範囲内、好ましくは、3〜6の範囲内である。
【0081】
Sp35ポリペプチドは、タグ(例えば、その後にタンパク分解により放出され得る部分)を含み得る。従って、上記リジン部分は、His−タグ改変体と低分子量リンカー(例えば、Traut’s試薬(Pierce))とが最初に反応することにより、選択的に改変され得、この低分子量リンカーは、そのリジンおよびN末端の両方と反応し、次いでhisタグを放出する。次いで、上記ポリペプチドは、フリーのSH基を含み、このフリーのSH基は、チオール反応性末端基(例えば、マレイミド基、ビニルスルホン基、ハロアセテート基またはフリーもしくは保護されたSH)を含むPEGにより選択的に改変され得る。
【0082】
Traut’s試薬は、PEG結合のための特異的部位を設定する任意のリンカーにより置換され得る。例えば、Traut’s試薬は、SPDP、SMPT、SATAまたはSATP(Pierce)により置換され得る。同様に、上記タンパク質と、マレイミドを挿入するアミン反応性リンカー(例えば、SMCC、AMAS、BMPS、MBS、EMCS、SMPB、SMPH、KMUSまたはGMBS)、ハロアセテート基を挿入するアミン反応性リンカー(SBAP、SIA、SIAB)またはビニルスルホン基を挿入するアミン反応性リンカーとが反応し得、結果として生じた生成物と、フリーのSHを含むPEGとが反応し得る。
【0083】
いくつかの実施形態において、上記ポリアルキレングリコール部分は、上記Sp35ポリペプチドのシステイン基に結合される。結合は、例えば、マレイミド基、ビニルスルホン基、ハロアセテート基またはチオール基を用いてもたらされ得る。
【0084】
必要に応じて、上記Sp35ポリペプチドは、不安定な結合を介して、上記ポリエチレングリコール部分に結合される。上記不安定な結合は、例えば、生物化学的加水分解、タンパク分解またはスルフヒドリル切断で切断され得る。例えば、上記結合はインビボ(生理的)条件下で切断され得る。
【0085】
上記反応は、上記反応性基がN末端のαアミノ基上である場合、生物学的に活性な物質と不活性ポリマーとを反応させるために使用される任意の適切な方法により、好ましくは、約pH5〜8(例えば、pH5、6、7または8)で行われ得る。一般的に、上記プロセスは、活性化されたポリマーを調製する工程およびその後の上記タンパク質とその活性化されたポリマーとを反応させ、処方物のための適切な可溶性タンパク質を生成する工程を包含する。
【0086】
(ベクター)
本発明は、Sp35ポリペプチドをコードする核酸を含むベクターを提供する。ベクターおよび本発明の核酸が作動可能に連結する発現制御配列の選択は、所望される機能特性(例えば、タンパク質発現)および形質転換されるべき宿主細胞に依存する。
【0087】
作動可能に連結されたコード配列の発現調節のために有用な発現制御エレメントは、当該分野において公知である。例としては、誘導性プロモーター、構成性プロモーター、分泌シグナルおよび他の調節エレメントが挙げられるが、これらに限定されない。誘導性プロモーターが使用される場合、それは、例えば、上記宿主細胞培地における栄養状態変化または温度変化によって制御され得る。
【0088】
上記ベクターとしては、原核生物レプリコン(すなわち、細菌宿主細胞において、染色体外の組換えDNA分子の自律的な複製および維持を行う能力を有するDNA配列)が挙げられ得る。このようなレプリコンは、当該分野において周知である。さらに、原核生物レプリコンを含むベクターはまた、その発現が検出可能マーカー(例えば、薬物耐性)を与える遺伝子を含み得る。細菌薬物耐性遺伝子の例は、アンピシリンまたはテトラサイクリンに対する耐性を与える遺伝子である。
【0089】
原核生物レプリコンを含むベクターはまた、細菌宿主細胞において、上記コード遺伝子配列の発現を行うための原核生物プロモーターまたはバクテリオファージプロモーターを含み得る。細菌宿主に適合性のプロモーター配列は、代表的に、発現されるべきDNAセグメントの挿入のために都合のよい制限酵素部位を含むプラスミドベクター中に、提供される。このようなプラスミドベクターの例は、pUC8、pUC9、pBR322およびpBR329(BioRad)、pPLおよびpKK223(Pharmacia)である。任意の適切な原核生物宿主が、本発明のタンパク質をコードする組換えDNA分子を発現するために、使用され得る。
【0090】
真核生物細胞発現ベクターは、当該分野において公知であり、市販されている。代表的に、このようなベクターは、所望されるDNAセグメントの挿入のために都合のよい制限酵素部位を含む。例示的なベクターとしては、pSVLおよびpKSV−10(Pharmacia)、pBPV−1、pML2d(International Biotechnologies)、pTDT1(ATCC 31255)、レトロウイルス発現ベクターpMIG、アデノウイルスシャトルベクターpDC315およびAAVベクターが挙げられる。
【0091】
真核生物細胞発現ベクターは、選択可能マーカー(例えば、薬物耐性遺伝子)を含み得る。ネオマイシンホスホトランスフェラーゼ(neo)遺伝子(Southernら,1982,J.Mol.Anal.Genet.1:327−341)は、このような遺伝子の例である。
【0092】
抗体または抗体フラグメントを発現するために、一部または全長の軽鎖および重鎖をコードするDNAが、発現ベクター(例えば、プラスミド、レトロウイルス、コスミド、YAC、EBV由来エピソームなど)に挿入される。上記発現ベクターおよび発現制御配列は、使用される発現宿主細胞に適合性であるように選択される。上記抗体軽鎖遺伝子および上記抗体重鎖遺伝子は、別個のベクターに挿入され得る。いくつかの実施形態において、両方の遺伝子は、同じ発現ベクターに挿入される。
【0093】
都合のよいベクターは、機能的に完全なヒトCH免疫グロブリン配列またはヒトCL免疫グロブリン配列をコードするベクターである。好ましくは、制限酵素部位が作製され、それにより、任意のVH配列またはVL配列が、容易に挿入され、発現され得る。このようなベクターにおいて、スプライシングは、通常、挿入されたJ領域のスプライスドナー部位とヒトC領域の前のスプライスアクセプター部位との間で、そしてまた、ヒトCHエキソン内に生じるスプライス領域で、生じる。ポリアデニル化および転写終結は、上記コード領域の下流の天然の染色体部位で生じる。上記組換え発現ベクターはまた、宿主細胞からの上記抗体鎖の分泌を促進するシグナルペプチドをコードし得る。
【0094】
哺乳動物宿主細胞発現のための好ましい調節配列としては、哺乳動物細胞における高レベルのタンパク質発現を導くウイルスエレメント(例えば、レトロウイルスLTR由来のプロモーターおよびエンハンサー、サイトメガロウイルス(CMV)(例えば、CMVプロモーター/エンハンサー)、シミアンウイルス40(SV40)(例えば、SV40プロモーター/エンハンサー)、アデノウイルス(例えば、アデノウイルス主要後期プロモーター(AdMLP))、ポリオーマ)ならびに強力な哺乳動物プロモーター(例えば、天然の免疫グロブリンプロモーターおよびアクチンプロモーター)が挙げられる。ウイルス調節エレメントおよびその配列のさらなる記載については、例えば、Stinskiの米国特許第5,168,062号;Bellの同第4,510,245号;およびSchaffnerの同第4,968,615号を参照のこと。
【0095】
組換え発現ベクターは、宿主細胞においてそのベクターの複製を調節する配列(例えば、複製起点)および選択マーカー遺伝子を有してもよい。上記選択マーカー遺伝子は、そのベクターが導入された宿主細胞の選択を容易にする(例えば、Axelの米国特許第4,399,216号;同第4,634,665号および同第5,179,017号を参照のこと)。例えば、代表的に、上記選択マーカー遺伝子は、上記ベクターが導入された宿主細胞に薬物(例えば、G418、ハイグロマイシンまたはメトトレキサート)に対する耐性を与える。好ましい選択マーカー遺伝子としては、ジヒドロ葉酸還元酵素(DHFR)遺伝子(メトトレキサート選択/増殖を用いるdhfr−宿主細胞における使用のため)およびneo遺伝子(G418選択のため)が挙げられる。
【0096】
Sp35ポリペプチドおよび抗Sp35抗体をコードする核酸分子ならびにこれらの核酸分子を含むベクターは、適切な宿主細胞の形質転換のために使用され得る。任意の適切な方法により、形質転換され得る。哺乳動物細胞への外因性DNAの導入のための方法は、当該分野において周知であり、これらの方法としては、デキストラン媒介性トランスフェクション、リン酸カルシウム沈殿法、ポリブレン媒介性トランスフェクション、プロトプラスト融合、エレクトロポレーション、リポソーム中へのポリヌクレオチドの封入および核へのDNAの直接的なマイクロインジェクションが挙げられる。さらに、核酸分子は、ウイルスベクターにより哺乳動物細胞に導入され得る。
【0097】
宿主細胞の形質転換は、用いられるベクターおよび宿主細胞にとって適切な従来の方法により達成され得る。原核生物宿主細胞の形質転換のために、エレクトロポレーション法および塩処理法が用いられ得る(Cohenら,1972,Proc.Natl.Acad.Sci.USA 69:2110−2114)。脊椎動物細胞の形質転換のために、エレクトロポレーション法、カチオン性脂質法または塩処置法が用いられ得る。例えば、Grahamら,1973,Virology 52:456−467;Wiglerら,1979,Proc.Natl.Acad.Sci.USA 76:1373−1376fを参照のこと。
【0098】
発現のための宿主として利用可能な哺乳動物細胞株は、当該分野において公知であり、これらの細胞株としては、American Type Culture Collection(ATCC)から入手可能な多くの不死化細胞株が挙げられる。これらとしては、特に、チャイニーズハムスター卵巣(CHO)細胞、NSO、SP2細胞、HeLa細胞、乳仔ハムスター腎臓(BHK)細胞、サル腎臓細胞(COS)、ヒト肝細胞癌細胞(例えば、Hep G2)、A549細胞および多くの他の細胞株が挙げられる。
【0099】
産生細胞株からのポリペプチドの発現は、公知の技術を用いて促進され得る。例えば、グルタミンシンセターゼ(GS)系は、特定条件下で発現を促進するために、一般的に使用される。例えば、欧州特許第0216846号、同第0256055号および同第0323997号ならびに欧州特許出願番号89303964.4を参照のこと。
【0100】
(宿主細胞)
宿主細胞は、原核生物起源または真核生物起源であり得る。好ましい真核生物の宿主細胞としては、限定されないが、酵母細胞および哺乳動物細胞(例えば、チャイニーズハムスター卵巣細胞(CHO)(ATCC登録番号CCL61)、NIHのスイスマウス胚細胞NIH−3T3(ATCC登録番号CRL1658)、および新生児のハムスター腎臓細胞(BHK))が挙げられる。他の有用な真核生物の宿主細胞としては、昆虫細胞および植物細胞が挙げられる。例示的な原核生物の宿主細胞は、E.coliおよびStreptomycesである。
【0101】
(処方物)
Sp35ポリペプチド、抗Sp35抗体、または抗Sp35抗体の抗原結合フラングメントを含む組成物は、適切な薬学的に受容可能なキャリアを含み得る。例えば、それらは、作用部位への送達のために設計された調製物への、活性化合物の処理を促進する賦形剤および/または補助剤を含み得る。非経口投与のための適切な処方物としては、水溶性形態(例えば、水溶性塩)の活性化合物の水溶液が挙げられる。さらに、適切な油状の注射懸濁液のような活性化合物の懸濁液が、投与され得る。適切な新油性溶媒またはビヒクルとしては、脂肪油(例えば、ゴマ油、または合成脂肪酸エステル(例えば、オレイン酸エチルまたはトリグリセリド))が挙げられる。水溶性の注射懸濁液は、懸濁液の粘性を増加させる物質(例えば、カルボキシメチルセルロースナトリウム、ソルビトールおよびデキストランが挙げられる)を含み得る。必要に応じて、懸濁液はまた、安定剤を含み得る。リポソームもまた、細胞または間隙の空間の中への送達のために本発明の分子をカプセル化するために使用され得る。例示的な薬学的に受容可能なキャリアは、生理学的に適合性のある溶媒、分散媒、コーティング、抗菌剤および抗真菌剤、等張剤および吸収遅延剤、水、生理食塩水、リン酸緩衝化生理食塩水、デキストロース、グリセロール、エタノールなどである。いくつかの実施形態において、組成物は、等張剤(例えば、糖、ポリアルコール(例えば、マンニトール)、ソルビトール、または塩化ナトリウム)を含む。いくつかの実施形態において、組成物は、薬学的に受容可能な物質(例えば、湿潤剤または少量の補助的な物質(例えば、湿潤剤もしくは乳化剤、防腐剤または緩衝液))を含み、これらは、活性成分の貯蔵寿命または有効性を高める。
【0102】
本発明の組成物は、例えば、液体(例えば、注射剤および不溶解性溶液)、分散剤、懸濁液、半固形ならびに固形投薬形態を含む、様々な形態であり得る。好ましい形態は、投与および治療の用途の様式に依存する。
【0103】
組成物は、溶液、ミクロエマルジョン、分散、リポソーム、または高い薬物濃度に適する他の指示される構造として処方され得る。滅菌注射溶液は、上に列挙した成分の1つまたは組み合わせと共に、適切な溶媒中に、必要とされる量の活性成分を組み込むことによって調製され得、必要な場合、続いて、濾過滅菌される。概して、分散剤は、塩基性分散媒体および上に列挙したものからの必要とされる他の成分を含む滅菌ビヒクルの中に活性成分を組み込むことによって調製される。滅菌注射溶液の調製のための滅菌粉末の場合、好ましい調製方法は、活性成分の粉末に加えて、前以て滅菌濾過した溶液から任意のさらなる所望の成分を生じる真空乾燥および凍結乾燥である。溶液の適切な流動性が、例えば、コーティング(例えば、レシチン)の使用、分散剤の場合は、必要とされる粒子サイズの維持、および界面活性剤の使用によって維持され得る。注射用組成物の長期の吸収は、吸収を遅らせる薬剤(例えば、モノステアリン酸塩およびゼラチン)を組成物中に含むことによってもたらされ得る。
【0104】
活性成分は、制御放出性の処方物またはデバイスを用いて処方され得る。そのような処方物およびデバイスの例としては、インプラント、経皮パッチ、およびマイクロカプセルに入れられた送達系が挙げられる。生分解性で、生体適合性のポリマー(例えば、酢酸エチレンビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸)が、使用され得る。そのような処方物およびデバイスの調製のための方法は、当該分野において公知である。例えば、Sustained and Controlled Release Drug Delivery Systems,J.R.Robinson,ed.,Marcel Dekker,Inc.,New York,1978を参照のこと。
【0105】
注射用の貯蔵処方物は、生分解性ポリマー(例えば、ポリラクチド−ポリグリコリド)中でマイクロカプセルに入れられた薬物のマトリックスを形成することによって作製され得る。ポリマーに対する薬物の割合、および使用されるポリマーの性質に依存して、薬物放出の速度が、制御され得る。他の例示的な生分解性ポリマーは、ポリオルトエステルおよびポリ無水物である。貯蔵注射用処方物はまた、リポソームまたはマイクロエマルジョン中に薬物を包括することによって調製され得る。
【0106】
補助的な活性化合物は、組成物の中に組み込まれ得る。いくつかの実施形態において、Sp35ポリペプチド、抗Sp35抗体またはそのフラグメントは、抗NgR1抗体、もしくはその抗原結合断片、または溶解性のNgR1ポリペプチドもしくはNgR1融合タンパク質と一緒に同時投与される。
【0107】
投薬レジメンは、最適な所望の反応を提供するように調整され得る。例えば、単一のボーラスが投与され得、いくつかの分割用量が、時間をかけて投与され得るか、または用量は、治療状態の緊急性によって示されるにつれて、比例して減少もしくは増加され得る。投与の容易さおよび投薬量の均一性のための投薬単位形態で、非経口組成物を処方することが有利である。例えば、Remington’s Pharmaceutical Sciences(Mack Pub.Co.,Easton,PA1980)を参照のこと。
【0108】
活性化合物に加えて、液体投薬形態は、不活性成分(例えば、水、エチルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香酸ベンジル、プロピレングリコール、1,3−ブチレングリコール、ジメチルホルムアミド、油、グリセロール、テトラヒドロフルフリルアルコール、ポリエチレングリコール、およびソルビタンの脂肪酸エステル)を含み得る。
【0109】
(遺伝子治療)
Sp35ポリペプチドは、軸策の伸長の阻害を減少させることが治療的に有益であるCNS疾患、CNS障害またはCNS損傷の処置のための遺伝子治療アプローチを使用して、哺乳動物(例えば、ヒト患者)においてインビボで生成され得る。これは、適切な発現制御配列に作動可能に連結される適切なSp35ポリペプチドをコードする核酸の投与を含む。好ましくは、これらの配列は、ウイルスベクターの中へ組み込まれる。このような遺伝子治療のための適切なウイルスベクターとしては、アデノウイルスベクター、レンチウイルスベクター、バキュロウイルスベクター、Epstein Barrウイルスベクター、パポバウイルスベクター、ワクシニアウイルスベクター、単純ヘルペスベクター、およびアデノ関連ウイルス(AAV)ベクターが挙げられる。ウイルスベクターは、複製欠損のウイルスベクターであり得る。好ましいアデノウイルスベクターは、そのE1遺伝子またはE3遺伝子に欠損を有し得る。アデノウイルスベクターが使用される場合、好ましくは、哺乳動物は、選択マーカー遺伝子をコードする核酸に曝露されない。
【実施例】
【0110】
本発明はさらに、以下の実験の実施例によって例示される。実施例は、例示の目的のためだけに提供され、あらゆる方法においても本発明の範囲または内容を制限すると解釈されるべきではない。
【0111】
(実施例1:Sp35発現パターン)
ヒトの組織におけるSp35の発現をノーザンブロット解析によって調べた。12個のヒトの主要な組織または14個のヒトのCNS組織を含む複数の組織ブロットを、P32で標識したSp35プローブ(Sp35cDNA配列のヌクレオチド150〜450)を用いて68℃で一晩ハイブリダイズした。そのブロットを、2×SSC、0.5%SDSで3回洗浄し、次いで、0.5×SSC、0.1%SDSで3回洗浄した。次いで、そのブロットをX線フィルムに露光し、mRNAレベルをオートラジオグラフィーによって可視化した。
【0112】
Sp35は、ヒトの脳においては心臓、高度に発現したが、骨格筋、結腸、胸腺、脾臓、腎臓、肝臓、小腸、胎盤、肺および末梢血白血球では発現しなかった。Sp35は、前頭皮質、後頭皮質、嗅内皮質、海馬、嗅球、線条、視床、小脳、中脳、脳橋、髄質および脊髄から単離された組織を含む、試験した全ての脳組織において発現した。吻/索軸に沿った遺伝子発現の勾配を、Sp35について観察し、皮質において最もレベルが高く、そして脊髄において最もレベルが低かった。
【0113】
Sp35が特定の脳細胞で発現するかどうかを、免疫組織化学(IHC)染色を使用して決定した。4%パラホルムアルデヒドで固定したラットの脳、脊髄の切片、または初代顆粒状ニューロン培養物を、示されるようにSp35に対する一次抗体と一緒にインキュベートし、その後、Alexa480またはAlexa590(Molecular Probes Inc.)に結合した二次抗体と一緒にインキュベートした。次いで、その切片を、Vectashieldに乗せて、蛍光顕微鏡検査によって可視化した。IHCのために使用される抗Sp35特異的抗体を、MorPhosys技術を使用してFabファージディスプレイライブラリーから作製した。
【0114】
Sp35は、ニューロンおよび希突起膠細胞において特異的に発現したが、星状細胞では発現しなかった。このことを、抗星状細胞マーカーGFAP、希突起膠細胞マーカー(O4)に対する抗体、およびニューロンマーカーβIIIチューブリンに対する抗体(全て抗Sp35抗体を用いて対比染色を行う)を含む種々の薬剤で、ラットの脳組織切片を染色し、実験において測定した。希突起膠細胞およびニューロンは、抗Sp35抗体で強く染色された。星状細胞の染色は、観測されなかった。
【0115】
Sp35の発現パターンの非依存性の確認として、本発明者らは、ラットの精製した星状細胞、希突起膠細胞、および小脳顆粒状ニューロンの初代細胞培養物から抽出したmRNA(Ambion kit)を使用して半定量的なRT−PCRを実施した。
【0116】
【数1】
26サイクルの後、強いバンドをニューロン由来のmRNAにおいて観察し、識別可能であるが、弱いシグナルを希突起膠細胞mRNAにおいて検出し、星状細胞においてはバンドが観察されなかった。
【0117】
(実施例2:Sp35−Fc融合タンパク質)
Sp35の生物学的機能を研究するために、ヒトSp35の細胞外部分(残基1〜531)をヒトIgG1のヒンジ領域およびFc領域と融合して、構築物を作製した。ヒトSp35を部分的にコードする配列を、
【0118】
【数2】
を使用してクローン227.2(Incyte)からPCRによって得た。
【0119】
平滑末端PCR産物を、PCR SCRIPT AMP−sp35を作製するためにPCR SCRIPT AMPベクター(Stratagene)のSrfI部位の中へサブクローニングした。SalIフラグメントを、PCR SCRIPT AMP−sp35から単離し、PCRCAMP Igベクター(StratageneベクターPCR SCRIPT AMPの誘導体で、このベクターでは、Fcγ配列がSalI(5’)〜NotI(3’)フラグメントとしてサブクローンされている)の中へサブクローニングし、インフレームでSp35シグナル配列および外部ドメイン配列(コドン1〜531)をヒトIg1のヒンジ領域およびFc領域をコードする配列と融合した。正しい単離物を同定し、Sp35 Fcフラグメントを包含するNotIフラグメントを、293E発現ベクター(市販の発現ベクターREP4(Invitrogen)の誘導体である、CH274)の単一のNot Iクローニング部位の中へサブクローニングした。新しいベクターである、CH274/sp35−FcによってコードされるSp35−Fc融合を、プラスミドGT123としてDNA配列決定によって確認した。
【0120】
Sp35−Fc融合タンパク質を発現する安定な細胞株を、プラスミドGT123を用いてCHO宿主細胞DG44のエレクトロポレーションによって作製した。トランスフェクトされたCHO細胞を、ヌクレオシド非依存性増殖について選択するために、10%の透析血清および4mMグルタミンの存在下のα−MEM中で培養した。トランスフェクションの14日後、細胞に新鮮な培地を与えた。Sp35−Fcを発現する細胞をスクリーニングするために、CHO細胞をフィコエリトリン(PE)標識したヤギ抗ヒトIgG(Jackson Labs)で標識し、FACS Mo−Flo(Cytomation)において高速フローサイトメトリーソーティングに供した。最も高いレベルのSp35−Igを発現した細胞を選択した。これらの細胞を7日間培養液で拡張させて、次いで、再び標識し、再びソートした。最も高いレベルのSp35−Igを発現する細胞を、96−ウェルプレート中の個々のクローンとして単離した。これらのクローンを2週間増殖させ、次いで、発現レベルを検査するために、FACS解析の1日前に新鮮な培地を与えた。最も高いレベルのSp35−Fcを発現したクローンを拡張し、凍結して、セルバンクを確立した。その細胞株を、無血清のBCM16培地の懸濁培養物中で増殖させるために適合した。これらのクローンによって生成されたSp35−Fcの力価を、4〜5継代の間、37℃での増殖細胞株によって測定し、次いで、50%最大細胞密度まで細胞を増殖させ、生存可能な細胞密度が75%に落ちるまで、28℃で10〜15日間培養した。この時点で、培地を収集し、遠心分離によって細胞および残屑を除き、プローブとして抗ヒトIg抗体(Jackson Lab)を使用して、ウェスタンブロット解析によってSp35−Fcレベルについての培養上清を力価測定した。
【0121】
Sp35−Fc融合タンパク質を、以下のような浄化した培養培地から精製した:9mlの1M HEPES pH7.5を900mlの馴化培地に添加した。この培地を3mlのプロテインAセファロース(Pharmacia)に対して4℃で3時間、負荷して一括処理した。樹脂を1.5cm(I.D.)のカラムで回収し、3ml PBSで4回、800mM NaClを含む4mlのPBSで2回洗浄し、次いで、3mLのPBSで再び洗浄した。Sp35−Fcを、1.5mL画分において25mM NaH2PO4 pH2.8、100mM NaClを用いてカラムから溶出し、75μLの0.5M NaH2PO4 pH8.6を添加することによって中和した。ピークタンパク質含有画分を、280nmでの吸光度によって同定して、プールし、さらに1mLプロテインAカラム上での精製に供した。負荷するより前に、NaClを600mMまで、そしてHEPES pH7.5を50mMまで添加した。カラムを、600μLの10mM HEPES pH7.5、1M NaClで2回洗浄し、次いで、1mL PBSで洗浄した。Sp35−Fcを、25mM NaH2PO4 pH2.8、100mM NaClを用いてカラムから溶出し、0.5mL画分を回収して、25μLの0.5M NaH2PO4 pH8.6を添加することによって中和した。ピークタンパク質含有画分を、280nmでの吸光度によって同定して、プールした。SDS−PAGEを還元することによって、Sp35−Igを90kDaの見かけの質量を有するシングルバンド(95%より高い純粋)として移動した。還元していない条件の下で、タンパク質をおよそ180kDaの質量を有する二量体として、電気泳動した。精製したSp35−Fcを等分して、−70℃で保存した。Sp35アミノ酸1〜531およびヒトIgG1 Fcを含むGT123のNotIフラグメントを、DB002を作製するためにPV90ベクターのNotI部位の中にサブクローニングした。
【0122】
(実施例3:His−Ap−Sp35融合タンパク質)
Sp35についてのレセプターを研究および単離するために、Hisタグ化アルカリホスファターゼ(His−Ap)融合タンパク質として、COS7細胞およびCHO細胞においてタンパク質を発現させた。プラスミドを以下のように構築した:Sp35の細胞外ドメイン(a.a.34〜532)を、Eco RI切断部位(下線)を含むプライマー(フォワード)
【0123】
【数3−1】
およびXba I切断部位(下線)を含むプライマー(リバース)
【0124】
【数3−2】
を使用してPCR増幅した。PCR産物を、Xba Iで切断し、その結果生じる粘着末端を、T4DNAポリメラーゼで満たし、次いで、Eco RIで消化し、そしてゲル精製した。消化した生成物を、His−AP−pcDNA1.1ベクター(Invitrogen)由来のEco RI His−APフラグメント中に埋め込まれたHindIIIに連結した。His−AP−Sp35フラグメントを、Hind IIIおよびEco RIで消化し、満たし、次いで、ベクターpV90のNot Iを満たした部位の中へ連結した。挿入物のDNA配列を、DNA配列決定によって確認した。
【0125】
COS7細胞をトランスフェクション前日に分けた。His−AP−Sp35
ベクターDNA(8μg)を、リポフェクタミン(Invitrogen)を使用して、5×106個の細胞をトランスフェクトするために使用した。馴化培地を、トランスフェクションの48時間後に、収集した。
【0126】
本発明者らは、pV90プラスミドを使用してHis−AP−Sp35融合タンパク質を発現するCHO細胞株を開発した。CHO宿主細胞DG44(2×106個の細胞)を、エレクトロポレーションによって100μgのプラスミドを用いてトランスフェクトした。細胞を、ヌクレオシド非依存性増殖について選択するために、10%の透析血清および4mMグルタミンの存在下のα−MEM中で培養した。トランスフェクションの14日後、FACS Mo−Flo(Cytomation)ソーティングによるスクリーニング前に、新鮮な培地を細胞に与えた。トランスフェクトしたCHO細胞を、ヒト胎盤アルカリホスファターゼ(Sigma)に対するマウスモノクローナル抗体8B6で標識した。二次抗体である、PE標識したヤギ抗マウスIgGを使用して、トランスフェクトした細胞に特異的なシグナルを生成させた。PE標識後、細胞を高速フローサイトメトリーソーティングに供し、上位5%を選択した。
【0127】
His−AP−Sp35を含む馴化した培地を生成するために、最も高いレベルのHISApSp35を発現した細胞を選択した。細胞株を、無血清の培地(BCM16)の懸濁培養液中で増殖させるために適合させた。これらのクローンによって生成したHis−AP−Sp35の力価を、4〜5継代の間、37℃で細胞株を増殖させることによって測定し、次いで、細胞を50%の最大細胞密度まで増殖させ、これらを生存可能な細胞密度が75%に落ちるまで、28℃で10〜15日間、培養した。培地を収集し、遠心分離によって、細胞および残屑を除き、培養上清を、プローブとして抗ヒトAP抗体(Jackson Labs)を使用するウェスタンブロット解析によってHis−AP−Sp35レベルについて力価測定した。
【0128】
His−AP−Sp35を、以下のような馴化培地から精製した:His−AP−Sp35を発現するCHO細胞由来の400mLの馴化培地を400mLの水で希釈した。トリエタノールアミンpH8.5を、0.5Mストックからの25mMに添加し、サンプルを、6mlのFractogel TMAE(EM Industries)アニオン交換樹脂の上に4℃で2時間、負荷して一括処理した。樹脂を、1.5cm(I.D.)カラム中で回収し、6mLの10mM HEPES pH7.5、50mM NaClで2回洗浄した。AP−Sp35を、2mL画分において10mM HEPES pH7.5、200mM NaClを用いてカラムから溶出した。ピーク画分を、AP活性のモニタリング、およびSDS−PAGEによって同定した。TMAEカラムからの貫流画分をさらに、300mlの水で希釈し、6mlのTMAE樹脂の上で4℃で一晩、一括処理で負荷した。樹脂を回収し、上記のように洗浄して、10mM HEPES pH7.5、150mM NaClで溶出した。ピーク画分を再び、AP活性のモニタリング、およびSDS−PAGEによって同定した。第1のカラムからのHis−AP−Sp35は、50%純粋で、第2のカラムからのAP−Sp35は、90%純粋であった。還元条件の下で、His−Ap−Sp35は、130kDaの見かけの質量でSDS−PAGEゲル上を移動した。90%純粋な物質は、ほとんどの調査について適切であったが、いくつかの研究については、His−AP−Sp35を、Ni−NTAアガロース樹脂(Qiagen)でさらに精製した。NaClを、TMAEカラムからの溶出画分に800mM添加し、0.5MトリエタノールアミンpH8.5および1MイミダゾールpH7.0をそれぞれ、25mMおよび15mMまで添加した。4.5mlのサンプルを、400μL NiNTAカラムの上に負荷した。そのカラムを、25mMトリエタノールアミン pH8.5、800mM NaCl、15mMイミダゾールで3回洗浄し、His−AP−Sp35を、200mMイミダゾールpH7.0、350mM NaClを用いてカラムから溶出し、200μLの画分を回収した。ピークAP含有画分をプールし、250容量の10mM HEPES pH7.5、200mM NaClに対して一晩、透析した。MgCl2およびZnCl2をそれぞれ2mMおよび0.25mMまで、保有物に添加した。最終生成物は、SDS−PAGEによって95%純粋より大きく、還元条件の下でおよそ140kDaの質量を有するシングルバンドとして電気泳動した。
【0129】
Sp35構築物をまた、Fc融合体として加工した。Sp−35LRR−Fc構築物を、プライマー(フォワード)
【0130】
【数4】
を使用するPCRによって作製した。このPCR産物を、PV90ベクターのNotI部位の中へ挿入した。Sp35IG−Fc構築物を、プライマー(フォワード)
【0131】
【数5】
を使用するPCRによって作製した。このPCR産物を、PV90ベクターのNotI部位の中へ挿入した。タンパク質をCHO細胞において発現させ、プロテインAセファロースカラムを使用して精製した。
【0132】
(実施例4:NgR1発現細胞へのSp35結合)
NgR1へのSp35結合を示すために、4つの異なる方法を使用した。第1に、本発明者らは、アルカリホスファターゼSp35結合体(AP−Sp35)を、NgR1発現細胞と一緒にインキュベートし、色素形成のAP検出試薬を使用して結合を評価する、直接的な結合アッセイにおいて相互作用を検出した。90%コンフルエントのCOS7細胞を、100mm組織培養皿上で増殖させ、Fugene6試薬(Roche)を使用して、NgR1発現プラスミドを用いてトランスフェクトした。48時間後、トランスフェクトした細胞を、HBH(Hankの平衡化塩緩衝液、1mg/ml BSA、20mM HEPES、pH7.0)で1回洗浄し、次いで、HBH中の4μg/mlのAP−Sp35融合タンパク質と一緒に、23℃で1.5時間インキュベートした。細胞を、氷冷のHBH緩衝液で、各々3分間で3回洗浄し、次いで、15分間、20mM HEPES、pH7.0、150mM NaCl中の3.7%ホルムアルデヒドで固定し、HBH緩衝液の中へ移して戻した。内因性の熱に不安定なAPを、67℃で2時間、熱不活性化した。結合したAP−Sp35を、ニトロブルーテトラゾリウムNBT(Roche)と共にインキュベーションすることによって検出した。Ap−Sp35は、ヒトNgR1レセプターを発現するCOS7細胞に結合したが、ベクター単独でトランスフェクトしたコントロールCOS7細胞には結合しなかった。NgR1についての点状の染色パターンが観測され、このことは、1つの画分のみ、おそらく50%の細胞がNgR1でトランスフェクトされたことを反映する。
【0133】
結合をよりよく定量化するために、本発明者らは同じ実験を行ったが、並行細胞サンプルとして、8μg/ml、4μg/ml、2μg/ml、1μg/ml、0.5μg/ml、0.125μg/ml、0.06μg/mlのAP−Sp35を用いて処理した。結合したAPを、4−ニトロフェニルホスフェートと一緒にインキュベートし、96−ウェルプレートリーダー(Molecular Devices)においてAP活性を測定した。これらのデータから、本発明者らは、ヒトNgR1へのAp−Sp35の結合についてのEC50が、およそ6nMであると推定した。
【0134】
第2に、本発明者らは、ELISAアプローチにおけるNgR1へのSp35の結合を検出した。ELISAプレート(Costar)を、37℃で1時間、0.1M NaHCO3、pH9.0において10μg/ml可溶性NgR1−Fcレセプター(ラットIgG1のヒンジおよびFcと融合したラットNgR1ペプチド35〜310を含むsNgR310−FcならびにラットIgG1と融合したラットNgR1ペプチド35〜344を含むsNgR344−Fc)でコーティングした。そのプレートをブロッキングして、25mM Hepes、pH7.0、0.1%BSA、0.1%オボアルブミン、0.1%脂肪を含まないドライミルクおよび0.001%NaN3で洗浄した。AP−Sp35タンパク質4μg/mlをプレートに添加して、4℃で一晩インキュベートした。次いで、プレートを、10mM Tris pH7.5、150mM NaClで洗浄し、結合したAPを、0.1Mグリシン、1mM MgCl2、1mM ZnCl2 pH10.5で希釈した10μg/mlの色素形成の基質であるリン酸4−ニトロフェニルを使用して検出した。OD410を、Softmaxプログラムを装着するELISAリーダー(Molecular Devices)において測定した。AP−Sp35は、固定化したsNgR−344−Fcに結合したが、sNgR−310−Fcタンパク質には結合せず、より長い種類のNgR1が、Sp35結合に必要とされることを示した。本発明者らは、100倍過剰のsNgR344−Fcと一緒にAP Sp35をプレインキュベートすることによってsNgR344−Fc NgR1へのAP−Sp35の結合を80%まで達成し得た。ラットIg融合コントロールタンパク質としてハリネズミIg1融合タンパク質を使用すると、結合の競合は見られなかった。
【0135】
第3に、本発明者らは、NgR1とSp35を同時に免疫沈降することによってNgR1へのSp35の結合を検出した。この研究のために、100mmの組織培養皿上で増殖した80%コンフルエントのCOS7細胞を、Fugene6試薬(Roche)を使用してSp35血球凝集素(Sp35−HA)およびNgR−FLAGをコードするプラスミドを用いてトランスフェクトし、トランスフェクションの48時間後、細胞を収集して、4℃で30分間、1mlの溶解緩衝液(50mM HEPES、pH7.5、150mM NaCl、1.5mM MgCl2、1mM EGTA、1% Triton X−100および10%グリセロール)中で溶解した。次いで、溶解物を、15分間14,000×gで遠心分離し、上清を回収して、抗HA親和性マトリックス(Roche)を使用して、攪拌しながら4℃で一晩インキュベートした。サンプルを、1mlの溶解緩衝液で3回洗浄し、次いで、Laemmliサンプル緩衝液中で3分間ボイルし、4〜15%SDS−PAGEに供して、Anti−FLAG M2抗体(Sigma)を用いる免疫ブロッティングによって解析した。FLAGの存在によって明らかなように、抗HAタグ親和性樹脂から、Sp35−HAおよびFLAG−NgRの両方を含む複合体を回収した。この複合体は、細胞をSp35−HAプラスミドまたはFLAG−NgR1プラスミド単独で処理したか、またはFlag−NgR1およびNgR1に結合しないHAタグ化コントロールタンパク質で同時トランスフェクションした、コントロールトランスフェクション由来の溶解物中に見られなかった。
【0136】
Sp35−HAを以下のように作製した。Sp35シグナル配列および細胞外ドメイン(アミノ酸1〜531)を、XbaI部位およびSpeI部位(下線)を含むプライマー
【0137】
【数6】
を使用してPCR増幅した。PCR産物を、XbaIおよびSpeIによって消化し、XbaI部位とSpeI部位との間のベクターpCGCHAの中へ挿入した。配列の挿入をDNA配列決定によって確認した。FLAG NgR1構築物はDr.Zhigang He(2002年、11月7日、Nature,Vol420)のご好意により提供された。
【0138】
第4に、本発明者らは、Ap−Sp35が、NgR1を発現するラットの小脳顆粒ニューロン(CGN)に結合することを示した。この実験のために、90%コンフルエントの出後8日のCGN細胞を、100mm組織培養皿上で増殖した。48時間後、細胞を、HBH緩衝液で一度洗浄し、次いで、23℃で1.5時間、HBH緩衝液中で4μg/mlのAP−Sp35と一緒にインキュベートした。次いで、細胞を、各々3分間、氷冷のHBHで3回洗浄し、次いで、15分間、20mM HEPES、pH7.0、および150mM NaCl中で3.7%のホルムアルデヒドで固定し、HBHに移して戻した。内因性の熱に不安定なAPを、67℃で2時間、熱不活性化した。結合したAP−Sp35を、ニトロブルーテトラゾリウムNBT(Roche)と共にインキュベーションすることによって検出した。AP−Sp35は、NgR1を発現する出後8日の小脳顆粒状ニューロンに結合した。ニューロンへのAP−Sp35の結合を、ほとんどの膜表面由来のGPIアンカータンパク質を開裂するPIPLC(5単位/ml)を用いてCGNを処理することによって阻害した。NGR1は、GPI結合タンパク質であるので、この結果は、Sp35が、CGN細胞上のNgR1に結合するという考えをさらに支持する。
【0139】
(実施例5:NgR1とSp35の共存)
Sp35およびNgR1が、同じニューロンにおいて発現するかどうかを決定するために、本発明者らは、共存の研究を行った。4%パラホルムアルデヒドで固体したラットのp8初代顆粒ニューロン培養物を、Sp35およびNgR1に対する抗体(Santa Cruz)と一緒にインキュベートし、次いで、適切なAlexa標識した第2の抗体(Molecular Probes Inc.)と一緒にインキュベートした。細胞を共焦点蛍光顕微鏡によって可視化した。ニューロンは、Sp35およびNgR1抗体によって強く染色した。両方のタンパク質が、細胞体およびニューロンの軸策において発現した。共存解析を補助するために、2つの型の抗体について異なる有色のプローブを使用した。染色(NgR陽性細胞については赤色そしてSp35陽性細胞については緑色)を組み合わせると、本発明者らは、2つのタンパク質がニューロン内で共存していることを示す細胞全体にわたる黄色が見えた。
【0140】
(実施例6:Sp35内のNgR1結合部位)
本発明者らは、NgR1相互作用に関係するSp35の特異的ドメインを規定するために欠損マッピングを使用した。以下の欠損構築物を、Stratagene Quikchange Mutagenesisキットを使用して作製した。本発明者らは、改変した挿入物のDNA配列決定によって全てのベクター構築物を確認した。
【0141】
Sp35のロイシンリッチ反復ドメイン、さらに塩基性領域(a.a34〜432)を含むHis−AP−Sp35bを、PCRによってHis−AP−Sp35(a.a.34〜532)からクローニングした。使用したプライマーは、
【0142】
【数7】
であった。
【0143】
Sp35のIgドメイン、さらに塩基性領域(a.a.417〜531)をコードするHis−AP−Sp35dを、PCRによってHis−AP−Sp35a(a.a.37〜531)ベクターからクローニングした。使用したプライマーは、
【0144】
【数8】
であった。
【0145】
Igドメイン(a.a425〜531)のみをコードするHis−AP−Sp35eを、PCRによってHis−AP−Sp35(aa34〜532)ベクターからクローニングした。使用したプライマーは、
【0146】
【数9】
であった。
【0147】
市販の変異誘発キットおよびプロトコル(Stratagene Quikchange)を、ベクターHis−AP−Sp35(34〜532)におけるSp35のアミノ酸456(アルギニンからグルタミン酸に)およびアミノ酸458(ヒスチジンからバリンに)を変異するために使用した。使用したプライマーは、
【0148】
【数10】
であった。
【0149】
His−AP−Sp35欠損構築物(図3)を、pV90発現ベクターにおいて加工し、293細胞において発現させた。馴化培地を回収し、AP付加物を、Fractogel TMAE樹脂およびNiNTAアガロースでの一連のクロマトグラフィー工程によって精製した。精製したタンパク質を、COS7細胞上で発現したNgR1への結合について試験した。3つの構築物は、すべてSp35に弱く結合した。これらの結果は、Sp35 LRR反復1〜14(アミノ酸34〜417)およびSp35のIgドメイン(アミノ酸425〜531)の両方は、NgR1へのSp35結合に寄与することを示した。Igドメインは、LRRドメインより高い親和性を示した。
【0150】
Sp35のIgドメインについての構造モデルを、フレームワークとしてNCAM結晶構造を使用して作製した(Rasmussenら、2000、Nat.Struct.Biol.7:389−393)。このモデルから、本発明者らは、結合に関与し得るループ(残基数454〜458、アミノ酸:SPRKH;配列番号:34)を観察した。この仮説を試験するために、本発明者らは、456での残基Rおよび458での残基Hを、それぞれEおよびVに変更したSp35構築物を作製した。この構築物を、NgR1結合について試験すると、本発明者らはシグナルにおいて10倍より少ない減少を観察した。結合におけるこのループ領域の寄与を試験するための代わりのアプローチとして、本発明者らは、ペプチドのN末端およびC末端にシステインを付加することによって環化する配列LSPRKH(配列番号:10)に対応するペプチドを合成した。NgR1への結合の際に、このペプチドは、NgR1の機能をブロック、阻害、または妨害する。
【0151】
(実施例7:Sp35はp8 CGN束形成(fasciculation)を誘発する)
ニューロンにおけるSp35の生物学的機能を決定するために、本発明者らは、Sp35−Fcを、出生後8日目の顆粒状ニューロンと共にインキュベートし、Sp35が神経突起の外向きの成長を調節し得るかどうかを観察した。Sp35−Fcタンパク質(16μg/ウェルのタンパク質)をスポットする前に、Labtek培養スライド(8ウェル)を、0.1mg/mlのポリ−D−リジン(Sigma)でコーティングした。このスライドを一晩乾燥させ、次いでリンスし、そして10μg/mlのラミニン(Gibco)でコーティングした。出生後8日目由来の小脳顆粒状ニューロンを解離し、事前コーティングしたスライド上に播種した。このスライド培養物を5%CO2中37℃で24時間インキュベートした。次いで、このスライドを20%スクロースを含有する4%パラホルムアルデヒド中で固定し、そして抗βIIIチューブリン(Covance TUJ1)で染色した。24時間後、CGNはニューロンの束状化(bundling)で明らかなように、はっきりした束形成形態を示した。この束形成は、未処理細胞コントロールまたはFcタンパク質コーティング化サンプルコントロールでは見られなかった。
【0152】
(実施例8:RhoA活性化/不活性化についてのSp35の効果)
Sp35−Fcは、束形成を引き起こす出生後の小脳顆粒状ニューロンを誘発した。シグナル伝達分子RhoAが束形成に関与することは公知であるので、本発明者らは、Sp35−FcがニューロンにおいてRhoA機能を調節し得るかどうかを決定した。本発明者らは、以下のようにRhoA活性化実験を行った:293細胞またはCOS7細胞を、Fugene 6試薬(Roche)を使用して、RhoA、Sp35またはNgR1の組み合わせを含む発現ベクターでトランスフェクトした。トランスフェクションの48時間後、細胞を一晩血清飢餓にし、次いで50mM Tris(pH7.5)、1% Triton X−100、0.5%デオキシコール酸ナトリウム、0.1% SDS、500mM NaCl、10mM MgCl2、さらにプロテアーゼインヒビターカクテルでリンスした。細胞溶解物を、13,000×g、4℃、5分間の遠心分離によって清澄化し、そして95%の上清を、4℃で45分間、20μgの固定化GST−Rho結合ドメイン親和性マトリックス(Rhotekinビーズ、Upstate Biotechnology)と共にインキュベートした。このビーズを、洗浄用緩衝液(50mM Tris(pH7.5)、1% Triton X−100、150mM NaCl、10mM MgCl2、およびプロテアーゼインヒビター)で3回洗浄した。GTP結合Rhoを、SDS−PAGEサンプル緩衝液中95℃で5分間加熱することによって、上記ビーズから溶出させた。結合Rhoタンパク質、およびRhoタンパク質の全体を、RhoAに対するモノクローナル抗体(Santa Cruz)を使用してウェスタンブロッティングによって検出した。Sp35遺伝子でのトランスフェクション後のブロットで検出されたRhoA−GTPの量の増加によって明らかなように、Sp35でトランスフェクトされたCOS7細胞およびHEK293細胞は、RhoA活性化を誘発した。RhoA−GTPのさらなる増加は、Sp35−Fcでの処理後に観察された。Sp35単独でのトランスフェクト後のRhoA−GTPの増加と対照的に、細胞をSp35およびNgR1でトランスフェクトした場合、RhoAは部分的に不活性化された。Sp35−Fcでのこれらの細胞の処理は、RhoAのさらなる不活性化をもたらした。
【0153】
本発明者らは、Ca++流入へのSp35処理の影響を決定するために、FLIPRアッセイ(Molecular Devices)を使用してSp35によるシグナル伝達応答を確認した。本発明者らは、Sp35−Fcの処理でSp35を発現する細胞において有意なCa++流入を観察したが、Sp35−Fcで処理したコントロール細胞においては有意なCa++流入を観察しなかった。NgR1およびSp35で同時トランスフェクトされた細胞がSp35−Fc融合タンパク質で処理された場合、このCa++流入は減少した。
【0154】
(実施例9:Sp35タンパク質とSp35タンパク自体との相互作用)
LRRドメインはホモタイプな相互作用にしばしば関与し、そして、本発明者らはSp35でトランスフェクトされた細胞への可溶性Sp35の添加がSp35トランスフェクション単独で観察されたRhoA−GTPの増加を越えるRhoA−GTPの増加を引き起こすことを観察したので、本発明者らは、Sp35自体に結合するSp35について試験した。この試験を行うために、本発明者らは、免疫共沈降を使用した。COS7細胞の80%コンフルエント(100mm組織培養皿上で増殖された)を、Fugene 6試薬(Roche)を使用して、プラスミドSp35 HAまたはプラスミドSp35−FLAG、あるいはその両方でトランスフェクトした。トランスフェクションの48時間後、細胞を収集し、そして4℃で30分間、1mlの溶解緩衝液(50mM HEPES(pH7.5)、150mM NaCl、1.5mM MgCl2、1mM EGTA、1% Triton X−100、および10%グリセロール)中に溶解した。次いで、この溶解物を14,000×gで15分間遠心分離し、そして上清を収集し、4℃で一晩撹拌しながら抗HA親和性マトリックス(Roche)と共にインキュベートした。次いで、このサンプルを1mlの溶解緩衝液で3回洗浄し、Laemmliサンプル緩衝液中で煮沸し、4〜15%SDS−PAGEに供し、そして抗FLAG抗体での免疫ブロッティングによって分析した。ウェスタンブロッティングによって決定されたように、この抗HA抗体樹脂は、Sp35−FLAGを含んだ複合体を捕獲した。これは、Sp35とSp35自体との直接的な相互作用を示した。本研究者らはまた、Sp35−Fcと一緒にHA−Sp35でトランスフェクトされた細胞を処理し、そして類似の免疫沈降アプローチを使用して、HA−Sp35がSp35−Fcに結合することを示した。
【0155】
Sp35−FLAGを、以下のように作製した。Sp35遺伝子細胞外ドメイン(a.a.1〜531)を、NotI部位(下線が引かれている)を含むプライマー:
【0156】
【数11−1】
およびプライマー:
【0157】
【数11−2】
を使用してPCR増幅した。このPCR産物をNotIによって消化し、そしてベクターpV90のNotI部位に挿入した。この挿入物のDNA配列を、DNA配列決定によって確認した。
【0158】
(実施例10:Sp35で形質転換された細胞のインビトロ移植)
脊髄損傷ラットにおけるSp35の生物学的機能を決定するために、本発明者らは、皮質性初代培養細胞(混合培養物)を、(ラット脊髄の損傷中心への送達のため)全長Sp35を発現するレトロウイルスまたはレトロウイルスコントロールで感染させた。2×106個の細胞を導入し、そしてラットを10日目に屠殺した。この脊髄を4%パラホルムアルデヒド中で一晩固定し、次いで、70%ETOH中で脱水し、続いて95%ETOH中で脱水した。組織サンプルをパラフィン中に包埋した。切片(10ミクロン厚)を、免疫組織化学的染色のために使用した。コントロールと比較して、Sp35発現細胞を受け取ったラットは、より少ない軸索退縮を示し、そして上記中心近くに染まったより多くのβチューブリンを示す。Sp35を受け取る損傷ラットにおいて、上昇したニューロンの生存が観察された。
【0159】
Sp35レトロウイルス構築物を、以下のように作製した。Sp35遺伝子を、XhoI部位(下線が引かれている)を含むプライマー:
【0160】
【数12−1】
およびEcoRI部位(下線が引かれている)を含むプライマー:
【0161】
【数12−2】
を使用してPCR増幅した。このPCR産物をXhoIおよびEcoRIで消化し、次いで、レトロウイルスベクターpMIG(これは、IPES−GFPを含む)に結合し、これをXhoIおよびEcoRIで事前に切断した。この新たなベクターを、pMMC078と命名した。pMMC078の単離物の全てが不注意な点突然変異を含んだので、pMMC078のうちの2つの単離物を一緒に結合した。pMMC078.6をXhoIおよびAccIで切断し、そしてpMMC078.7をXhoIおよびAccIで切断した。これらの2つのフラグメントを一緒に結合し、最終修正(correct)プラスミドpMMC089を作製した。この挿入物のDNA配列を、DNA配列決定によって確認した。Sp35レトロウイルスを、説明したように作製した。293G細胞は、トランスフェクションの前日に分裂された。8μgのSp35−レトロウイルスDNAを、リポフェクタミン(Invitrogen)によって5×106個の細胞をトランスフェクトするために使用した。この馴化培地を、トランスフェクションの92時間後に収集した。この馴化培地を、5000gで10分間遠心分離し、そして上清を、Sp35レトロウイルスストックとして使用した。このストックを4℃で1週間、または−80℃で6ヶ月間保存した。
【0162】
(実施例11:脊髄損傷の動物モデル)
外科的手順の全てを、無菌技術を使用して行う。任意の外科的処置前の一週間、動物を取り扱う。アンピシリン100mg/kgを、SCに手術の前および後に予防的に投与し、膀胱感染損傷の発生を減少させた。
【0163】
動物を、O2中イソフルラン2〜3%と共にミダゾラム2.5mg/kgIPを使用して、つま先を挟むことによって測定して深い麻酔にまで麻酔する。動物を、手術および回復の期間、循環水ヒーティングパッド(heating pad)上で維持する。角膜乾燥を予防するために眼潤滑剤を使用し、そして過剰の唾液分泌を減少させるためにアトロピン0.05mg/kgSCを与える。小さな切開を皮膚に作製し、そして筋肉を引っ張って椎骨を露出させる。脊髄レベルL6の背側椎弓切除(および鞘内カテーテルの置換が必要な場合はL7、以下を参照のこと)を行い、L6/L7および隣接した棘突起を脊髄フレーム(David Kopf Instruments)に強固に固定した。背部片側切断を、主な背内側皮質脊髄(coticospinal)路(CST)構成要素および少数の背外側皮質脊髄(coticospinal)路(CST)構成要素を完全に遮るよく切れる虹彩切除鋏を使用して、L6で行った。手術後、露出した脊柱を保護するために椎弓切除部位を保護材料(例えば、Durafilm)で覆い、そして上にある筋肉を4.0クロミックガット(chromic gut)で縫合した。この皮膚を縫合し、ベータダイン溶液でふき取った。
【0164】
動物の機能回復ISを、脊髄損傷後のラットを評価するために一般的に使用されるBasso Beattie and Bresnehan(BBB)スコアリング法を使用して評価した。この方法は、関節運動能力および体重付加能力の詳細な分析によってラットの後肢機能を定量化する。ラットを、脊髄損傷の翌日に評価し、次いでその後一週間評価する。
【0165】
CST切除の直後に、Sp35もしくはGFPを発現するアデノウイルスまたはコントロールウイルス(1010粒子)を、切除の部位に、ならびに損傷部位のすぐ尾側およびすぐ吻側の領域に注入する。合計で10μlのAdvを、5箇所の異なる部位に注入する(4μl/部位)。Sp35タンパク質の鞘内投与については、小さな穴を外傷(lesion)L7より2mm尾側の脊髄の硬膜に開け、そして鞘内カテーテルをL7のクモ膜下腔に挿入する。このカテーテルを、外傷より約1mm尾側の脊髄の向こう側に、ゆっくりかつ徐々に滑らせる。鞘内空間の外側にあるカテーテルの一部を、所定の周辺組織にしっかりと縫合した。試験物質(Sp35タンパク質またはコントロールタンパク質)を含むプライムされた小型浸透ポンプ(Alza corp.)をガイドカニューレの露出された端部に連結し、そしてクモ膜下腔に挿入する。手術後、露出した脊柱を保護するために椎弓切除部位を保護材料(例えば、Durafilm)で覆い、そして上にある筋肉を4.0クロミックガットで縫合した。この皮膚を縫合し、そしてベータダイン溶液でふき取った。
【0166】
組織学的分析:路追跡手術は、脊髄損傷を誘発する手術時に起こる。頭部の皮膚を薄く削り、そしてベータダインおよび70%アルコールでふき取る。この動物を定位固定フレームに配置する。この頭皮を縦方向に切開し、そして骨膜を頭蓋冠からすくい取る。頭蓋骨に直径約1〜2mmの穴をドリルで開け、そしてガラスマイクロリットル針を、運動皮質の8つの位置に垂直に挿入する(座標は、Paxixnos and Wastonのラット脳図譜(1997)に従って決定される)。約5μlの路追跡物質(例えば、ビオチンデキストランアミン、10,000M.Wt)を注入し、そしてさらに5分間にわたって上記針を定位置に置いたままにして、溶液を拡散させる。針の除去後、スクールキャップ(scull cap)の穴をゲルフォームでふさぎ、そして頭皮を損傷部位をこえてステープル(staple)で閉じる。動物を回復させ、そして手術後の治療を受けさせる(以下に記載される)。4〜10週後、動物を深く麻酔し(ipでイナクチン100〜110mg/kg)、そして以下に記載されるような組織学のために還流する。この路追跡物が、脊髄の尾側端部へと向かう皮質性脊髄路を順行性輸送機序を下がることによって行われ、そして皮質脊髄路内の解剖学的結合性を定量するための手段を提供する。
【0167】
免疫組織化学的実験については、損傷を誘発する手術の2〜8週間後に動物をイナクチン(IPにおいて100〜110mg/kg)で深く麻酔する。胸腔を開き、そして心臓を露出し、灌流させる。100ccの氷冷PBSをゆっくりと押出すことによって(流体逃避を可能にするために、穴が右心室に切り込まれる)、カニューレを左心室に挿入する。これの後、眼/耳/つま先の固定が明らかになるまで、4%パラホルムアルデヒド(50〜100ml)のゆっくりとした規則的な点滴を続ける。損傷部位の改変を最小にすることに注意して脊髄を取り出し、OCTで凍結させ、切片にして、そして免疫組織化学について処置する。必要に応じて、他の組織もまた、後者の分析のために採取する。アデノウイルスSp35を受け取る動物は、ニューロン軸索を染色するβIIIチューブリンによって決定されるように、上昇した軸索出芽を示した。
【0168】
(実施例12:Sp−35ウイルスベクター構築物)
pMIG由来のSp−35ウイルスベクターを、以下のように作製した。全長Sp35コード配列を、XhoI部位を含むプライマー:
【0169】
【数13−1】
およびEcoRI部位を含むプライマー:
【0170】
【数13−2】
を使用してPCR増幅した。このPCR産物を、XhoIおよびEcoRIで切断し、次いで、レトロウイルスベクターpMIG(Chengら、1996、Nat.Biotechnol.145:576)に結合し、これをXhoIおよびEcoRIで切断した。このベクターをpMMC078と命名した。pMMC078の単離物全てが点突然変異を含んだので、pMMC078のうちの2つの単離物を一緒に結合した。ベクターpMMC078.6をXhoIおよびAccIで切断し、そしてpMMC078.7をXhoIおよびAccIで切断した。これらの2つのフラグメントを結合し、プラスミドpMMC089を作製した。
【0171】
pMIG由来のSp35−HAウイルスベクターを以下のように作製した。HA配列を有するフレーム中のSp35アミノ酸326〜614をコードするフラグメントを、SacII部位を含むプライマー:
【0172】
【数14−1】
および鋳型として機能するpMMC089を含むプライマー:
【0173】
【数14−2】
でのPCRを使用することによって得た。このより長いプライマーは、Sp35コドン614の後およびEcoRI部位の前のHAコード配列(イタリック)を含む。次いで、このPCR産物を、Sac IIおよびEcoR Iで切断し、そしてpMIG由来のレトロウイルスベクター中に野生型Sp35コドン326〜614を含むSac II−EcoR Iフラグメントを置換するために使用する。
【0174】
Sp35−バキュロウイルスHAベクターを以下のように作製した。Sp35−HAレトロウイルスベクター由来のSp35−HAコード配列を、Xho IおよびEcoR Iで切り出し、平滑末端化し、そしてバキュロウイルスシャトルベクターpBV−CZPG(米国特許第6,190,887号;および同第6,338,953号)の、Bgl2−フィルインサイト(Bgl2−fill in site)にクローン化し、CMVプロモーターの下でLacZ遺伝子を置換した。
【0175】
Sp35アデノウイルスベクターを、以下のように作製した。Sp35レトロウイルス由来のSp35−IRES−GFPコード配列を、XhoI−フィルインサイトおよびNhe Iで切り出し、次いで、最小限のCMVプロモータ−の下、アデノウイルスシャトルベクターpDC315の、EcoR I−フィルインサイト/Nhe I部位にクローン化した。
【0176】
(実施例13:再髄鞘化の動物モデル)
雌性のLong Evansラットを、全ての研究に使用する。ラットを、イソフルランを使用して麻酔し、T3/T4露出をし、そして背側半椎弓切除を行う。次いで、化学的脱髄剤であるリソレクチン(0.9%生理食塩水中1%リソレクチン3μl)を、索表面より0.5〜1mm下の脊髄の脊柱の右側に注入する。適切な鎮痛処置を手術前後に施す。
【0177】
3日後、この注入部位を再び露出させ(イソフルラン麻酔下、適切な鎮痛処置を使用して)、以下の治療を損傷した脊髄に投入し、そしてタンパク質Sp35/コントロールタンパク質をコードするアデノウイルスベクターを損傷部位に注入する。Sp35をコードするアデノウイルスの1010個の粒子、または10μl体積のGFPコントロールを、リソレシチン誘発脱髄の部位またはその周囲の5箇所までの異なる部位における損傷されたラット脊髄に注入する。2μl以下の容量を、5箇所の注入部位の各々に注入する。脊髄の脱髄/再髄鞘化の組織学的分析ついては、手術の2週間後、3週間後、4週間後または6週間後、動物をイナクチン(ipにおいて100〜110mg/kg)で深く麻酔し、固定液で心臓を介して還流する。次いで、この脊髄を取り出し、分析のために処理する。Sp35処置を受ける動物は、抗MBPタンパク質抗体またはルクソールファーストブルー(luxol fast blue)を使用するIHCによって決定されるように、上昇した軸索髄鞘形成を示した。
【0178】
(実施例14:Sp35 RNAi)
脳機能におけるSp35の役割に取り組むために、本発明者らは、レンチウイルスSp35 RNAiを出生後8日目のCGN細胞に導入した。Sp35 RNAi感染細胞は、コントロール細胞よりも短い神経突起を有し、そしてコントロール細胞よりも高い増殖速度を有した。これらの結果は、RhoA活性化を調節することにおけるSp35についての役割を示す。
【0179】
マウスSp35 DNA配列とラットSp35 DNA配列とを比較して相同領域を見出し、候補shRNAに使用した。CH324を、オリゴヌクレオチドLV1−035およびオリゴヌクレオチドLV1−036をアニーリングさせ、そしてHpa1およびXho1で消化したpLL3.7に結合することによって構築した。このオリゴヌクレオチドを、MWGから購入した。この配列は、以下である:
【0180】
【数15】
ウイルスを産生する前に、pLL3.7由来のDNAまたはpLL3.7中の候補shRNAを、6ウェル型中のCHO細胞に5対1の割合で、マウスSP35−HAタグ化プラスミドで共トランスフェクトした。トランスフェクトされたCHO細胞溶解物由来のSP35−HAタグのウエスタンブロット検出によって、および2連のウェルから調製されたRNA全体のノーザンブロットによって、ノックダウンを分析した。このブロットを、mSP35の0.7kbフラグメントでプローブした。アッセイを、トランスフェクションの48時間後に行った(データは示していない)。ウイルスを、ラットニューロンの培養物における使用について最善の候補物から産生した。ベクター、さらなる方法論およびウイルス産生は、Rubinsonら「A lentivirus−based system to functionally silence genes in primary mammalian cells,stem cells and transgenic mice by RNA interference.」Nat.Genet.33,401−6(2003)に記載されるようなものであった。
【0181】
(実施例15:RhoA活性化)
NgR1およびSP35を共発現するCOS7細胞は、OMgpに応答してRhoA/GTPレベルの変化を示さなかった。これは、SP35/NgR1複合体がミエリンインヒビターによるシグナル伝達を媒介するのに重要でないことを示唆した。
【0182】
本発明者らは、SP35/NgR1/p75の三つ組複合体がシグナル伝達を媒介するという可能性を検討した。2つのアプローチを、SP35とNgR1とp75との間の相互作用を評価するために使用した。1つ目は、AP−SP35結合体を使用する直接的な結合アッセイにおいて、結合を評価した。このAP−SP35結合体は、p75発現細胞に弱く結合した。AP−P75は、NgR1発現細胞に結合した。NgR1およびp75へのAP−SP35の結合を、ELISAによって測定した(図4)。2つ目は、NgR1およびp75へのSP35の結合を、SP35、NgR1およびp75を共発現するCOS7細胞からの免疫共沈降によって評価した。抗NgR1抗体は、SP35およびp75を含む複合体を免疫沈降させた。抗SP35抗体はまた、p75を含む複合体を免疫沈降させた。この相互作用データおよび免疫共沈降データは、SP35とNgR1とp75との間の直接的な相互作用についての証拠を提供した。本発明者らは、共焦点顕微鏡法ならびにSP35、p75およびNgR1に対する抗体を使用し、SP35、NgR1およびp75が、ラット由来のp7 CGニューロンの細胞体および軸索に共に局在することを示した。
【0183】
次に、本発明者らは、SP35、NgR1およびp75の組み合わせが、ミエリンインヒビターの活性に充分であることを示した。非ニューロン性COS7細胞を、3つの構成要素全てを発現するように操作した。これらの細胞を使用して、本発明者らは、RhoA/GTPレベルがOMgpによってアップレギュレーションされることを示した。OMgp−Fc処理は、これらの3つの構成要素の他の組み合わせと比較して、SP35/p75/NgR1を共発現する細胞におけるRhoA/GTPレベルを上昇させた。本発明者らは、COS7細胞溶解物のウエスタンブロッティングによってタンパク質の発現を確認した。NgR1に結合するミエリンインヒビターの親和力は、p75の存在によっても、p75およびSP35の存在によっても影響を受けなかった。この組み合わせた結果は、NgR1、SP35およびp75の三つ組複合体が、NgR1リガンドの存在下でのRhoA調節のために必要とされるというモデルを支持する(図5)。
【0184】
SP35は、シグナル伝達への潜在的な直接関与または潜在的な間接関与を有する細胞質ドメインを含む。細胞質ドメインの役割を決定するために、本発明者らは、シグナル伝達ができない、非産生的な三つ組複合体を形成することにより、ドミナントネガティブ様式で機能する、SP35の細胞質ドメイン切断物(truncation)(配列番号2のアミノ酸34〜576)を産生した。本発明者らは、細胞質ドメイン切断を有するこの分子を「DN−SP35」(dominant negative SP35)と命名した。本発明者らは、出生後7日目の(p7)CGニューロンを、全長SP35またはDN−SP35でトランスフェクトし、次いで、阻害性ミエリン成分(Omgp、ミエリンおよびNogo66)への応答についてアッセイした。図6に示されるように、DN−SP35トランスフェクト細胞は、阻害性ミエリン成分に応答せず、そしてコントロールよりも長い神経突起を示した。対照的に、全長SP35構築物でトランスフェクトされた細胞は、阻害性基質への高められた応答を示し、そしてコントロールと比較してより短い神経突起を有した。これは、DN−SP35が、ミエリン成分によって引き起こされる神経突起の伸長阻害を弱めるような競合物として作用することを示した。本発明者らは、外因性の可溶性SP35−Fcはまた、NgR1と結合し、そして阻害性基質の作用を遮断すると予測した。図7に示されるように、SP35−Fcは、Omgp、Nogo66およびMAGによる神経突起伸長阻害を低減させた。
【0185】
(実施例16:神経保護活性)
等数のラットp6小脳顆粒ニューロンを、50nMのsp35−Fcタンパク質の存在下または非存在下で、12ウェルの細胞培養プレートの各ウェルにプレートした。これらのポリ−D−リジンプレートを、10μgのCNSミエリンまたは200ngのNogo66、MAGおよびOMgpまたはコントロール−Fcで事前コーティングした(乾燥させた)。このニューロン培養物を、1〜7日間、37℃で、および5%CO2で維持した。ニューロンは、3日後に検査した(ニューロンの特異性マーカー IIIチューブリンによって決定した)ところ、十分な神経突起伸長をして、sp35−Fc処理とは無関係に、PBSコントロールウェルにおいて健常であり、そして良好に成長した。sp35−Fcの非存在下では、ニューロンは、ミエリン、Nogo66、MAGおよびOMgpでコーティングされたウェルにおいて良好に成長しなかった。最小限の神経突起出芽(短く、歪曲した)が存在し、そしてこのニューロンは健常には見えず、丸い細胞体および縮合された核物質を有した。DAPI染色は、これらのウェルで検出されたニューロンの数が、PBSコントロールウェル中で検出されたニューロンの数よりも少なく、ニューロンの損失を示唆していることを示した。sp35−Fcの存在下では、長い神経突起が存在し、そしてこのニューロンは健常に見えた。DAPI染色は、sp35−Fcを受け取らなかったウェルよりも、これらのウェル中でニューロンの数がより多いことを実証した。このデータを、以下の表2に要約する。
【0186】
【表2】
これらのデータは、Sp35の可溶性形態(例えば、Sp35−Fc)が、神経保護活性を保有すると示した。
【0187】
脊髄を半分切除された(T9、SCT)ラットにおいて、脊髄切片のβ−IIIチューブリン染色は、損傷部位におけるニューロンの実質的な損失を示した。sp35を発現する組換えウイルスは、SCT動物を損傷部位において感染させるために使用した。これらの脊髄の組織学的染色は、ベクターウイルスで感染されたコントロールグループと比較して、損傷部位周辺のニューロンの数の増加を示した。これは、上記のインビトロ実験の知見と一致し、そしてSp35に関連する神経保護特性をさらに示す。
【0188】
(実施例17:脊髄損傷の動物モデルにおけるSp35)
Sp35−Fcは、インビトロでOMgp、Nogo−66およびMAGによって引き起こされる神経突起成長阻害を低減させたので、本発明者らは、この分子がインビボでCNS損傷の機能回復を促進することを予測した。これを確かめるために、本発明者らは、脊髄を半分切除したラット(すなわち、急性CNS外傷の動物モデル)にSp35−Fcを投与した。図8および図9に示されるように、Sp35−Fc処置ラットは、IgGで処置されたコントロールラットと比較して、有意に向上した機能回復を示した。
【0189】
脊髄損傷および行動分析を、以下のように行った。外科的手順の全てを、Biogen Institutional Animal Use and Care Committeeの指針に従って行った。雌のLong Evansラット(190〜210g、Charles River、Wilmington、MA)を、2.5mg/kgのミダゾラム(I.P.において)およびO2中2〜3%のフルオタンを使用して麻酔した。背部椎弓切除を、脊髄レベルT6および脊髄レベルT7で行った。背側の片側切除を行い、主な背内側皮質脊髄路(CST)構成要素および少数の背外側皮質脊髄路(CST)構成要素を完全に遮断した。CST切除の直後に、鞘内カテーテルをT7のクモ膜下腔に挿入し、そしてクモ膜下腔に挿入されたプライムされた小型浸透ポンプ(Alzet model 2004)に連結した。小型浸透ポンプは、0.25μl/時間の速度で、Hu IgGアイソタイプコントロールタンパク質(5mg/ml、n=5、Pharmingen)、PBS(n=3)可溶性Hu Sp35−Ig融合タンパク質(4.3mg/ml、n=8)を送達した。手術後、椎弓切除部位を縫合し、そして皮膚の傷をステープルで閉じた。術後処置は、手術後3日間の鎮痛処置(s.c.においてブプレノルフィン0.05mg/kg)および手術後7日間の抗生物質処置(s.c.においてアンピシリン100mg/kgを1日に2回)を包含した。膀胱を、研究の期間(28日)または機能回復まで(その時間を記述した)、1日2回、手動で圧搾した。動物全てを、オープンフィールドBBBスコアリングシステム(Bassoら,1995,J.Neurotrauma 12:1−21;Onoら,2003,J.Neurosci.23:5887−5896)を使用して盲目的に採点した。ラットを、CST切除の翌日(2日目)およびその後四週間にわたって毎週、Basso−Beattie−Bresnahan (BBB)歩行運動評価尺度を使用して評価した。調査者を、研究の期間にわたって、処置群に対して盲目的にした。
【0190】
(実施例18:赤核脊髄路(RST)片側切除損傷モデルにおけるニューロンの生存および軸索再生)
本研究者らはまた、歩行運動に直接的に寄与する赤核脊髄路におけるニューロンの再生に対するSp35処置の効果を調査した。
【0191】
成体9週齢のSprague−Dawleyラット(200〜250g)を、ケタミン(80mg/kg)およびキシラジン(8mg/kg)の腹腔内注入によって麻酔した。顕微鏡の操作下で、背部椎弓切除を行い、そして7番目の胸椎(C7)を同定した。硬膜を開いた後、右側切除を、1つのスプリング鋏を使用して脊髄レベルC7において行った。脊髄を半分切除した後、動物に、Sp35−Fcの2μg/ml溶液(10μl)、またはヒトIgの2μg/ml溶液(10μl)、またはPBS(10μl)のいずれかで浸漬された1つのゲルフォームを与えた(損傷部位の上に配置した)。手術後、各群の動物を、軸索追跡および行動分析のために細分した。軸索追跡についての動物(各群についてn=5)および行動分析についての動物(各群についてn=7)を、1ヶ月間生存させた。
【0192】
フルオロゴールド(FG、6%w/v、Fluorochrome)を使用して、損傷瘢痕にわたって軸索を再生したRSTを標識し、そして、尾側の脊髄に戻した。損傷後の生存期間(1ヶ月)の末日より2日前、動物を、ケタミン(80mg/kg)およびキシラジン(8mg/kg)の腹腔内注入によって麻酔した。背部の椎弓切除を行い、そしてT2脊髄断片を同定した。0.5ml体積のFGを、ハミルトンシリンジを使用して右側のT2脊髄に手動で注入した。2日後、この動物を、致死用量のケタミン(150mg/kg)およびキシラジン(8mg/kg)で麻酔して屠殺し、そしてこの動物を、正常食塩水で心臓内に還流し、続いて0.1×PBS中4%パラホルムアルデヒドを含む400mlの固定液で心臓内に還流した。脳および脊髄を除去し、一晩パラホルムアルデヒド中で事後固定し、次いで、30%リン酸緩衝化スクロース中に配置した。脳組織および脊髄組織を、クリオスタットにおいて30mmの切片に切り出し、ゼラチンがコーティングされたスライド上に取り付けた。損傷部位上のFG標識RSTニューロンの数を、対側のインタクトな側のFG標識ニューロンの合計数の百分率として表した。この群間の百分率を、一方向ANOVAを使用して、続いてTukey−Kramer多重比較検定を使用して、統計学的に比較した。表3に示されるように、2μg/mlのSp35−Fcは、赤核脊髄路(RST)ニューロンの生存を促進した。
【0193】
【表3】
行動分析については、自発性垂直探索(exploration)の間の前肢の使用を、軽度の改変を含む記載されるような(Liuら、1999)種々の処置の1ヵ月後に試験した。ラットを、5分間の垂直探索について前肢の使用を促進する透明なPlexiglasシリンダー(直径15cmおよび高さ30cm)中に配置した。以下の行動を記録した:(1)シリンダーの壁と接触するための左肢(非障害性)または右肢(障害性)の独立的な使用;および(2)シリンダーの壁と接触する前肢両方の同時使用。以下の点に関して、垂直探索行動が発現された:(1)障害性の肢の使用、非障害性の肢の使用、および両方の肢の使用の合計数に相対する左肢(非障害性)の使用の百分率;(2)障害性の肢の使用、非障害性の肢の使用、および両方の肢の使用の合計数に対する右肢(障害性)の使用の百分率;ならびに(3)障害性の肢の使用、非障害性の肢の使用、および両方の肢の使用の合計数に対する両肢の使用の百分率。群間の差異を、一方向ANOVAによって、続いてBonferroniポストホック分析によって検定した。Sp35−Fc処置動物は、有意に向上した前肢運動を示した:Sp35−1−Fc処置動物においては両肢について30%の使用 対 コントロール−Fc処置動物またはPBS処置動物においては両肢について10%の使用;Sp35−1−Fc処置動物においては55%の左肢(非障害性)の使用 対 コントロール−Fc処置動物またはPBS処置動物においては80%の使用;および、Sp35−1−Fc処置動物においては29%の右肢(障害性)の使用 対 コントロール−Fc処置動物またはPBS処置動物においては約15%の使用。
【0194】
(実施例19:Sp35−Fcは、視神経切除モデルにおける網膜神経節細胞(RGC)生存を促進する)
本発明者らは、視神経切除モデルを使用してSp35の活性をさらに確認した。これは、ニューロンの機能に影響を与える因子を調査する。若年成体の雌のSprague Dawley(SD)ラットを、この研究で使用した。各動物の右側の視神経を、眼窩内で、視神経円板から1.5mm切除した。6%フルオロゴールド(FG)で浸漬した1つのゲルフォームを、視神経円板の真後ろの新たに切除した部位に適用し、生存網膜神経節細胞(RGC)を標識した。この動物を、6つの群(各群においてn=6)に分割しSp35−Fc、ヒトIgG1、またはPBSのみのいずれかを硝子体内注入によって与えた。各硝子体内注入の体積は、4mlであった。他方、各注入の投与量は2mgであった。この硝子体内注入を、視神経切除の直後に行った。
【0195】
全ての動物を1週間生存させた。動物を屠殺する2日前、各動物の左側の視神経を切除し、6%FGを使用して、内部コントロールとして機能する生存RGCを標識した。動物を、過量のネンブタールで屠殺し、網膜を、4%パラホルムアルデヒド中で解剖した。4つのラジアルカットを、網膜を4つの四分円(上方、下方、鼻側、および頭側)に分割するように作製した。次いで、この網膜を封入剤(Dako)でフラットに取り付ける前に、この網膜を同じ固定液中で1時間事後固定した。このスライドを、蛍光顕微鏡の下、紫外線フィルター(励起波長=330〜380nm)を使用して試験した。標識RGCを、200×200mmの接眼グリッドの下で、視神経平板から出発して500mm間を置いた網膜の末梢境界まで、各四分円の中線に沿って数えた。各処置がもたらした生存RGCの百分率を、損傷眼における生存RGCの数と反対側の側眼における生存RGCの数とを比較することによって表した。全てのデータを、平均±SEMとして表した。統計学的有意性を、一方向ANOVAによって評価し、続いてTukey−Kramerポストホック検定によって評価した。差異は、p<0.05が有意であると考えられた。Sp35−Fc処置動物は、コントロール−Fc処置動物またはPBS処置動物と比較した場合に有意なニューロンの生存(83%)を示した。コントロールFc処置動物またはPBS処置動物は各々、約50%のニューロンの生存を示しただけだった。
【0196】
(他の実施形態)
他の実施形態は、添付の特許請求の範囲内である。
【図面の簡単な説明】
【0197】
【図1−1】図1は、全長ヒトSp35 cDNAのヌクレオチド配列である(配列番号1)。
【図1−2】図1は、全長ヒトSp35 cDNAのヌクレオチド配列である(配列番号1)。
【図2】図2は、全長ヒトSp35ポリペプチドのアミノ酸配列である(配列番号2)。
【図3】図3は、Sp35ドメイン構造およびNgR1に結合するSp35配列を同定するための欠失マッピングの概略図である。
【図4】図4は、ラットp75をコードする発現ベクターまたはベクターコントロールをトランスフェクションされたCOS7細胞に結合するSP35に関するデータを要約するヒストグラムである。48時間後、AP−SP35またはAPを、上記細胞とともにインキュベートした。結合したAPを、発色AP検出試薬を用いて検出した。
【図5】図5は、NgR1;NgR1およびp75;NgR1、p75およびSP35をコードする発現ベクターまたはベクターコントロールをトランスフェクションされたCOS7細胞に対する、AP−OmgpおよびAP−Nogo−66の結合に関するデータを要約するヒストグラムである。48時間後、AP−Omgp、AP−Nogo−66またはAPを、上記細胞とともにインキュベートした。結合したAPを、発色AP検出試薬を用いて検出した。
【図6】図6は、インビトロでの神経突起伸長に対するミエリンインヒビターの阻害活性の軽減に関するデータを要約するヒストグラムである。神経突起長を、固定化した基質Omgp、ミエリンおよびNogo−66上で培養された、DN−Sp35、全長Sp35を発現する生後7日ラット小脳顆粒状ニューロン、またはコントロール生後7日ラット小脳顆粒状ニューロンについて測定した。DN−Sp35を用いてトランスフェクションした細胞は、阻害基質に対する応答の減少を示した。神経突起長を、2回の独立した実験からの処置群あたり1000ニューロンから定量した(p<0.01)。
【図7】図7は、SP35−Fcによるミエリンインヒビターの阻害活性の逆転に関するデータを要約するヒストグラムである。生後7日ラット小脳顆粒状ニューロン(1000ニューロン)の神経突起長を、SP35−Fcの存在下または非存在下、固定化した基質OMgp、ミエリンまたはNogo−66上で培養した。SP35−Fcは、Omgp、Nogo−66およびMAGにより引き起こされる神経突起伸長阻害を減少した。神経突起長を、2回の独立した実験からの処置群あたり1000ニューロンから定量した(p<0.01)。
【図8】図8は、くも膜下腔内投与されたSp35−Fcがラットにおける脊椎半側切除後の機能的回復を改善したことを示す実験からのデータを要約するグラフである。運動性BBBスコアを、コントロール(IgG)ラットまたはSp35−Fc処置ラットにおける脊椎半側切除後の時間の関数として測定した(群あたり動物8匹)。脊髄損傷時に、処置を開始した。
【図9】図9は、図8で要約される実験における、4週目の個々の動物のBBBスコアを示すグラフである。
【図1】
【特許請求の範囲】
【請求項1】
単離された核酸であって、該核酸が、ポリペプチドをコードするヌクレオチド配列を含み、:(a)該ポリペプチドが(i)Sp35 LRRドメイン、(ii)該LRRドメインに対してC末端側のSp35塩基性領域および(iii)該塩基性領域に対してC末端側のSp35免疫グロブリン(Ig)ドメイン、を含み;そして(b)該ポリペプチドが、膜貫通ドメインを欠く、核酸。
【請求項2】
単離された核酸であって、該核酸が、ポリペプチドをコードするヌクレオチド配列を含み、該ポリペプチドが、Sp35 Igドメインを含み、そして、Sp35 LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、核酸。
【請求項3】
単離された核酸であって、該核酸が、ポリペプチドをコードするヌクレオチド配列を含み、該ポリペプチドが、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、核酸。
【請求項4】
請求項1に記載の核酸であって、前記Sp35ポリペプチドが、細胞質ドメインを欠く、核酸。
【請求項5】
請求項1に記載の核酸であって、前記ポリペプチドが、配列番号2のアミノ酸残基34〜532を含む、核酸。
【請求項6】
請求項1〜3のいずれか1項に記載の核酸であって、前記ポリペプチドが、非Sp35部分を含む融合ポリペプチドである、核酸。
【請求項7】
請求項6に記載の核酸であって、前記非Sp35部分が、Ig部分、血清アルブミン部分、標的化部分、レポーター部分および精製容易化部分からなる群より選択される、核酸。
【請求項8】
請求項7に記載の核酸であって、前記非Sp35部分がIg部分である、核酸。
【請求項9】
請求項8に記載の核酸であって、前記Ig部分がFc部分である、核酸。
【請求項10】
請求項1〜3のいずれか1項に記載の核酸であって、前記ヌクレオチド配列が、発現制御配列に作動可能に結合される、核酸。
【請求項11】
請求項10に記載の核酸を含む、ベクター。
【請求項12】
請求項11に記載のベクターを含む、宿主細胞。
【請求項13】
単離されたポリペプチドであって:(a)該ポリペプチドが(i)Sp35 LRRドメイン、(ii)該LRRドメインに対してC末端側のSp35塩基性領域および(iii)該塩基性領域に対してC末端側の免疫グロブリン(Ig)ドメイン、を含み;そして(b)該ポリペプチドが、膜貫通ドメインを欠く、ポリペプチド。
【請求項14】
単離されたポリペプチドであって、該ポリペプチドが、Sp35 Igドメインを含み、そして、LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、ポリペプチド。
【請求項15】
単離されたポリペプチドであって、該ポリペプチドが、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、ポリペプチド。
【請求項16】
請求項13に記載のポリペプチドであって、ロイシンリッチ繰返し配列のうちの1つが、カルボキシ末端ロイシンリッチ繰返し配列(LRRCT)である、ポリペプチド。
【請求項17】
請求項13に記載のポリペプチドであって、ロイシンリッチ繰返し配列のうちの1つが、アミノ末端ロイシンリッチ繰返し配列(LRRNT)である、ポリペプチド。
【請求項18】
請求項13に記載のポリペプチドであって、前記Sp35ポリペプチドが、細胞質ドメインを欠く、ポリペプチド。
【請求項19】
請求項13に記載のポリペプチドであって、該ポリペプチドが、配列番号2のアミノ酸残基34〜532を含む、ポリペプチド。
【請求項20】
請求項13、14または15に記載のポリペプチドであって、該ポリペプチドが、非Sp35部分を含む融合ポリペプチドである、ポリペプチド。
【請求項21】
請求項20に記載のポリペプチドであって、前記非Sp35部分が、Ig部分、血清アルブミン部分、標的化部分、レポーター部分および精製容易化部分からなる群より選択される、ポリペプチド。
【請求項22】
請求項21に記載のポリペプチドであって、前記非Sp35部分がIg部分である、ポリペプチド。
【請求項23】
請求項22に記載のポリペプチドであって、前記Ig部分がFc部分である、ポリペプチド。
【請求項24】
請求項13、14または15に記載のポリペプチドであって、該ポリペプチドが、ポリマーに結合される、ポリペプチド。
【請求項25】
請求項24に記載のポリペプチドであって、前記ポリマーが、ポリアルキレングリコール、糖ポリマーおよびポリペプチドからなる群より選択される、ポリペプチド。
【請求項26】
請求項25に記載のポリペプチドであって、前記ポリマーがポリアルキレングリコールである、ポリペプチド。
【請求項27】
請求項26に記載のポリペプチドであって、前記ポリアルキレングリコーがポリエチレングリコール(PEG)である、ポリペプチド。
【請求項28】
請求項24に記載のポリペプチドであって、該ポリペプチドが、1つ、2つ、3つまたは4つのポリマーに結合される、ポリペプチド。
【請求項29】
請求項28に記載のポリペプチドであって、前記ポリマーの総分子量が、20,000Da〜40,000Daである、ポリペプチド。
【請求項30】
NgR1によるシグナル伝達を阻害する方法であって、該方法が、該NgR1と有効量のSp35ポリペプチドとを接触させる工程を包含する、方法。
【請求項31】
中枢神経系(CNS)ニューロンの軸索成長阻害を減少する方法であって、該方法が、該ニューロンと有効量のポリペプチドとを接触させる工程を包含し、該ポリペプチドが、Sp35ポリペプチド、抗Sp35抗体および抗Sp35抗体の抗原結合フラグメントからなる群より選択される、方法。
【請求項32】
CNSニューロンの成長円錐崩壊を阻害する方法であって、該方法が、該ニューロンと有効量のポリペプチドとを接触させる工程を包含し、該ポリペプチドが、Sp35ポリペプチド、抗Sp35抗体および抗Sp35抗体の抗原結合フラグメントからなる群より選択される、方法。
【請求項33】
哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置する方法であって、該方法が、治療有効量のポリペプチドを該哺乳動物に投与する工程を包含し、該ポリペプチドが、Sp35ポリペプチド、抗Sp35抗体および抗Sp35抗体の抗原結合フラグメントからなる群より選択される、方法。
【請求項34】
請求項30〜33のいずれか1項に記載の方法であって、前記Sp35ポリペプチドが、以下、
ポリペプチドであって:(a)該ポリペプチドが(i)12個〜14個のSp35ロイシンリッチ繰返し配列を含むLRRドメイン、(ii)該LRRドメインに対してC末端側のSp35塩基性領域および(iii)該塩基性領域に対してC末端側の免疫グロブリン(Ig)ドメイン、を含み;そして(b)該ポリペプチドが、膜貫通ドメインを欠く、ポリペプチド;ならびに
ポリペプチドであって、該ポリペプチドが、Sp35 Igドメインを含み、そして、LRRドメイン、塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、ポリペプチド
からなる群より選択される、方法。
【請求項35】
請求項33に記載の方法であって、前記CNS疾患、CNS障害またはCNS損傷が、脊髄損傷または視覚神経損傷である、方法。
【請求項36】
請求項33に記載の方法であって、前記ポリペプチドが局所投与される、方法。
【請求項37】
請求項33に記載の方法であって、前記ポリペプチドが、脊髄損傷の48時間以内に最初に投与される、方法。
【請求項38】
請求項36に記載の方法であって、前記ポリペプチドの治療有効量が、10μg〜10mgである、方法。
【請求項39】
哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置する方法であって、該方法が、(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)該CNS疾患、CNS障害またはCNS損傷の部位またはその近傍で、該哺乳動物中に該宿主細胞を導入する工程、を包含する、方法。
【請求項40】
請求項39に記載の方法であって、前記疾患、障害または損傷が脊髄損傷である、方法。
【請求項41】
請求項39に記載の方法であって、前記培養宿主細胞が、前記処置されるべき哺乳動物由来である、方法。
【請求項42】
請求項39に記載の方法であって、前記組換えSp35ポリペプチドが、全長Sp35ポリペプチドである、方法。
【請求項43】
CNS疾患、CNS障害またはCNS損傷の部位での髄鞘形成を促進する方法であって、該方法が、該CNS疾患、CNS障害またはCNS損傷の部位を有効量のSp35ポリペプチドと接触させる工程を包含する、方法。
【請求項44】
請求項43に記載の方法であって、前記Sp35ポリペプチドが、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、方法。
【請求項45】
インビボ遺伝子治療によってCNS疾患、CNS障害またはCNS損傷を処置する方法であって、該方法が、該疾患、障害または損傷の部位あるいはその近傍で、哺乳動物にウイルスベクターを投与する工程を包含し、該ウイルスベクターが、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、該Sp35ポリペプチドが、該哺乳動物において、該損傷の部位またはその近傍のニューロンによる軸索伸長阻害を減少するために十分な量で、該ヌクレオチド配列から発現される、方法。
【請求項46】
請求項36に記載の方法であって、前記ウイルスベクターが、アデノウイルスベクター、レンチウイルスベクター、バキュロウイルスベクター、エプスタイン−バーウイルスベクター、パポバウイルスベクター、ワクシニアウイルスベクターおよび単純疱疹ウイルスベクターからなる群より選択される、方法。
【請求項47】
請求項45に記載の方法であって、前記疾患、障害または損傷が、脊髄損傷および視覚神経損傷からなる群より選択される、方法。
【請求項48】
請求項45に記載の方法であって、前記ウイルスベクターが、局所投与、眼内投与、非経口投与、くも膜下腔内投与、硬膜下投与および皮下投与からなる群より選択される経路によって投与される、方法。
【請求項49】
ポリペプチドをコードする核酸であって、該ポリペプチドが、Sp35 LRRドメイン、塩基性領域、Igドメイン、連結配列および膜貫通ドメインを含み:そして、機能性細胞質ドメインを欠く、核酸。
【請求項50】
請求項49に記載の核酸であって、該核酸が、本質的に、配列番号2のアミノ酸1〜576からなるポリペプチドをコードする、核酸。
【請求項51】
死の危険性にあるニューロンの生存を促進する方法であって、該方法が、該ニューロンと有効量のSp35ポリペプチドを接触させる工程を包含する、方法。
【請求項52】
請求項51に記載の方法であって、前記ニューロンがインビトロである、方法。
【請求項53】
請求項51に記載の方法であって、前記ニューロンが、神経変性疾患、神経変性障害または神経変性損傷を有する哺乳動物中に存在する、方法。
【請求項54】
請求項53に記載の方法であって、前記神経変性疾患、神経変性障害または神経変性損傷が、多発性硬化症、ALS、ハンチントン病、アルツハイマー病、パーキンソン病、糖尿病性ニューロパシー、脳卒中、外傷性脳損傷および外傷性脊髄損傷からなる群より選択される、方法。
【請求項55】
神経変性疾患、神経変性障害または神経変性損傷を有する哺乳動物において、死の危険性にあるニューロンの生存を促進する方法であって、該方法が、(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)該ニューロン部位で、該哺乳動物中に該宿主細胞を導入する工程、を包含する、方法。
【請求項56】
死の危険性にあるニューロンの生存を促進するインビボ遺伝子治療方法であって、該方法が、該ニューロン部位またはその近傍で、哺乳動物にウイルスベクターを投与する工程を包含し、該ウイルスベクターが、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、該Sp35ポリペプチドが、該哺乳動物において、該ニューロンの生存を促進するために十分な量で該ヌクレオチド配列から発現される、方法。
【請求項57】
請求項51に記載の方法であって、前記Sp35ポリペプチドが可溶性である、方法。
【請求項1】
単離された核酸であって、該核酸が、ポリペプチドをコードするヌクレオチド配列を含み、:(a)該ポリペプチドが(i)Sp35 LRRドメイン、(ii)該LRRドメインに対してC末端側のSp35塩基性領域および(iii)該塩基性領域に対してC末端側のSp35免疫グロブリン(Ig)ドメイン、を含み;そして(b)該ポリペプチドが、膜貫通ドメインを欠く、核酸。
【請求項2】
単離された核酸であって、該核酸が、ポリペプチドをコードするヌクレオチド配列を含み、該ポリペプチドが、Sp35 Igドメインを含み、そして、Sp35 LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、核酸。
【請求項3】
単離された核酸であって、該核酸が、ポリペプチドをコードするヌクレオチド配列を含み、該ポリペプチドが、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、核酸。
【請求項4】
請求項1に記載の核酸であって、前記Sp35ポリペプチドが、細胞質ドメインを欠く、核酸。
【請求項5】
請求項1に記載の核酸であって、前記ポリペプチドが、配列番号2のアミノ酸残基34〜532を含む、核酸。
【請求項6】
請求項1〜3のいずれか1項に記載の核酸であって、前記ポリペプチドが、非Sp35部分を含む融合ポリペプチドである、核酸。
【請求項7】
請求項6に記載の核酸であって、前記非Sp35部分が、Ig部分、血清アルブミン部分、標的化部分、レポーター部分および精製容易化部分からなる群より選択される、核酸。
【請求項8】
請求項7に記載の核酸であって、前記非Sp35部分がIg部分である、核酸。
【請求項9】
請求項8に記載の核酸であって、前記Ig部分がFc部分である、核酸。
【請求項10】
請求項1〜3のいずれか1項に記載の核酸であって、前記ヌクレオチド配列が、発現制御配列に作動可能に結合される、核酸。
【請求項11】
請求項10に記載の核酸を含む、ベクター。
【請求項12】
請求項11に記載のベクターを含む、宿主細胞。
【請求項13】
単離されたポリペプチドであって:(a)該ポリペプチドが(i)Sp35 LRRドメイン、(ii)該LRRドメインに対してC末端側のSp35塩基性領域および(iii)該塩基性領域に対してC末端側の免疫グロブリン(Ig)ドメイン、を含み;そして(b)該ポリペプチドが、膜貫通ドメインを欠く、ポリペプチド。
【請求項14】
単離されたポリペプチドであって、該ポリペプチドが、Sp35 Igドメインを含み、そして、LRRドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、ポリペプチド。
【請求項15】
単離されたポリペプチドであって、該ポリペプチドが、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、ポリペプチド。
【請求項16】
請求項13に記載のポリペプチドであって、ロイシンリッチ繰返し配列のうちの1つが、カルボキシ末端ロイシンリッチ繰返し配列(LRRCT)である、ポリペプチド。
【請求項17】
請求項13に記載のポリペプチドであって、ロイシンリッチ繰返し配列のうちの1つが、アミノ末端ロイシンリッチ繰返し配列(LRRNT)である、ポリペプチド。
【請求項18】
請求項13に記載のポリペプチドであって、前記Sp35ポリペプチドが、細胞質ドメインを欠く、ポリペプチド。
【請求項19】
請求項13に記載のポリペプチドであって、該ポリペプチドが、配列番号2のアミノ酸残基34〜532を含む、ポリペプチド。
【請求項20】
請求項13、14または15に記載のポリペプチドであって、該ポリペプチドが、非Sp35部分を含む融合ポリペプチドである、ポリペプチド。
【請求項21】
請求項20に記載のポリペプチドであって、前記非Sp35部分が、Ig部分、血清アルブミン部分、標的化部分、レポーター部分および精製容易化部分からなる群より選択される、ポリペプチド。
【請求項22】
請求項21に記載のポリペプチドであって、前記非Sp35部分がIg部分である、ポリペプチド。
【請求項23】
請求項22に記載のポリペプチドであって、前記Ig部分がFc部分である、ポリペプチド。
【請求項24】
請求項13、14または15に記載のポリペプチドであって、該ポリペプチドが、ポリマーに結合される、ポリペプチド。
【請求項25】
請求項24に記載のポリペプチドであって、前記ポリマーが、ポリアルキレングリコール、糖ポリマーおよびポリペプチドからなる群より選択される、ポリペプチド。
【請求項26】
請求項25に記載のポリペプチドであって、前記ポリマーがポリアルキレングリコールである、ポリペプチド。
【請求項27】
請求項26に記載のポリペプチドであって、前記ポリアルキレングリコーがポリエチレングリコール(PEG)である、ポリペプチド。
【請求項28】
請求項24に記載のポリペプチドであって、該ポリペプチドが、1つ、2つ、3つまたは4つのポリマーに結合される、ポリペプチド。
【請求項29】
請求項28に記載のポリペプチドであって、前記ポリマーの総分子量が、20,000Da〜40,000Daである、ポリペプチド。
【請求項30】
NgR1によるシグナル伝達を阻害する方法であって、該方法が、該NgR1と有効量のSp35ポリペプチドとを接触させる工程を包含する、方法。
【請求項31】
中枢神経系(CNS)ニューロンの軸索成長阻害を減少する方法であって、該方法が、該ニューロンと有効量のポリペプチドとを接触させる工程を包含し、該ポリペプチドが、Sp35ポリペプチド、抗Sp35抗体および抗Sp35抗体の抗原結合フラグメントからなる群より選択される、方法。
【請求項32】
CNSニューロンの成長円錐崩壊を阻害する方法であって、該方法が、該ニューロンと有効量のポリペプチドとを接触させる工程を包含し、該ポリペプチドが、Sp35ポリペプチド、抗Sp35抗体および抗Sp35抗体の抗原結合フラグメントからなる群より選択される、方法。
【請求項33】
哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置する方法であって、該方法が、治療有効量のポリペプチドを該哺乳動物に投与する工程を包含し、該ポリペプチドが、Sp35ポリペプチド、抗Sp35抗体および抗Sp35抗体の抗原結合フラグメントからなる群より選択される、方法。
【請求項34】
請求項30〜33のいずれか1項に記載の方法であって、前記Sp35ポリペプチドが、以下、
ポリペプチドであって:(a)該ポリペプチドが(i)12個〜14個のSp35ロイシンリッチ繰返し配列を含むLRRドメイン、(ii)該LRRドメインに対してC末端側のSp35塩基性領域および(iii)該塩基性領域に対してC末端側の免疫グロブリン(Ig)ドメイン、を含み;そして(b)該ポリペプチドが、膜貫通ドメインを欠く、ポリペプチド;ならびに
ポリペプチドであって、該ポリペプチドが、Sp35 Igドメインを含み、そして、LRRドメイン、塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、ポリペプチド
からなる群より選択される、方法。
【請求項35】
請求項33に記載の方法であって、前記CNS疾患、CNS障害またはCNS損傷が、脊髄損傷または視覚神経損傷である、方法。
【請求項36】
請求項33に記載の方法であって、前記ポリペプチドが局所投与される、方法。
【請求項37】
請求項33に記載の方法であって、前記ポリペプチドが、脊髄損傷の48時間以内に最初に投与される、方法。
【請求項38】
請求項36に記載の方法であって、前記ポリペプチドの治療有効量が、10μg〜10mgである、方法。
【請求項39】
哺乳動物におけるCNS疾患、CNS障害またはCNS損傷を処置する方法であって、該方法が、(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)該CNS疾患、CNS障害またはCNS損傷の部位またはその近傍で、該哺乳動物中に該宿主細胞を導入する工程、を包含する、方法。
【請求項40】
請求項39に記載の方法であって、前記疾患、障害または損傷が脊髄損傷である、方法。
【請求項41】
請求項39に記載の方法であって、前記培養宿主細胞が、前記処置されるべき哺乳動物由来である、方法。
【請求項42】
請求項39に記載の方法であって、前記組換えSp35ポリペプチドが、全長Sp35ポリペプチドである、方法。
【請求項43】
CNS疾患、CNS障害またはCNS損傷の部位での髄鞘形成を促進する方法であって、該方法が、該CNS疾患、CNS障害またはCNS損傷の部位を有効量のSp35ポリペプチドと接触させる工程を包含する、方法。
【請求項44】
請求項43に記載の方法であって、前記Sp35ポリペプチドが、Sp35 LRRドメインを含み、そして、Sp35 Igドメイン、Sp35塩基性領域、膜貫通ドメインおよび細胞質ドメインを欠く、方法。
【請求項45】
インビボ遺伝子治療によってCNS疾患、CNS障害またはCNS損傷を処置する方法であって、該方法が、該疾患、障害または損傷の部位あるいはその近傍で、哺乳動物にウイルスベクターを投与する工程を包含し、該ウイルスベクターが、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、該Sp35ポリペプチドが、該哺乳動物において、該損傷の部位またはその近傍のニューロンによる軸索伸長阻害を減少するために十分な量で、該ヌクレオチド配列から発現される、方法。
【請求項46】
請求項36に記載の方法であって、前記ウイルスベクターが、アデノウイルスベクター、レンチウイルスベクター、バキュロウイルスベクター、エプスタイン−バーウイルスベクター、パポバウイルスベクター、ワクシニアウイルスベクターおよび単純疱疹ウイルスベクターからなる群より選択される、方法。
【請求項47】
請求項45に記載の方法であって、前記疾患、障害または損傷が、脊髄損傷および視覚神経損傷からなる群より選択される、方法。
【請求項48】
請求項45に記載の方法であって、前記ウイルスベクターが、局所投与、眼内投与、非経口投与、くも膜下腔内投与、硬膜下投与および皮下投与からなる群より選択される経路によって投与される、方法。
【請求項49】
ポリペプチドをコードする核酸であって、該ポリペプチドが、Sp35 LRRドメイン、塩基性領域、Igドメイン、連結配列および膜貫通ドメインを含み:そして、機能性細胞質ドメインを欠く、核酸。
【請求項50】
請求項49に記載の核酸であって、該核酸が、本質的に、配列番号2のアミノ酸1〜576からなるポリペプチドをコードする、核酸。
【請求項51】
死の危険性にあるニューロンの生存を促進する方法であって、該方法が、該ニューロンと有効量のSp35ポリペプチドを接触させる工程を包含する、方法。
【請求項52】
請求項51に記載の方法であって、前記ニューロンがインビトロである、方法。
【請求項53】
請求項51に記載の方法であって、前記ニューロンが、神経変性疾患、神経変性障害または神経変性損傷を有する哺乳動物中に存在する、方法。
【請求項54】
請求項53に記載の方法であって、前記神経変性疾患、神経変性障害または神経変性損傷が、多発性硬化症、ALS、ハンチントン病、アルツハイマー病、パーキンソン病、糖尿病性ニューロパシー、脳卒中、外傷性脳損傷および外傷性脊髄損傷からなる群より選択される、方法。
【請求項55】
神経変性疾患、神経変性障害または神経変性損傷を有する哺乳動物において、死の危険性にあるニューロンの生存を促進する方法であって、該方法が、(a)組換えSp35ポリペプチドを発現する培養宿主細胞を提供する工程;および(b)該ニューロン部位で、該哺乳動物中に該宿主細胞を導入する工程、を包含する、方法。
【請求項56】
死の危険性にあるニューロンの生存を促進するインビボ遺伝子治療方法であって、該方法が、該ニューロン部位またはその近傍で、哺乳動物にウイルスベクターを投与する工程を包含し、該ウイルスベクターが、Sp35ポリペプチドをコードするヌクレオチド配列を含み、それにより、該Sp35ポリペプチドが、該哺乳動物において、該ニューロンの生存を促進するために十分な量で該ヌクレオチド配列から発現される、方法。
【請求項57】
請求項51に記載の方法であって、前記Sp35ポリペプチドが可溶性である、方法。
【図2】


【図3】
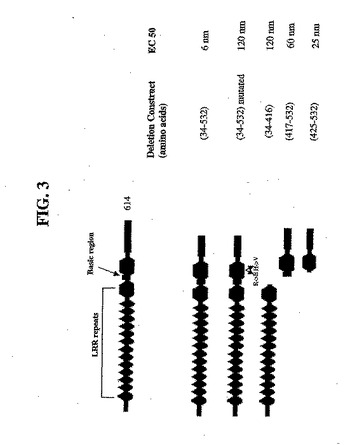
【図4】
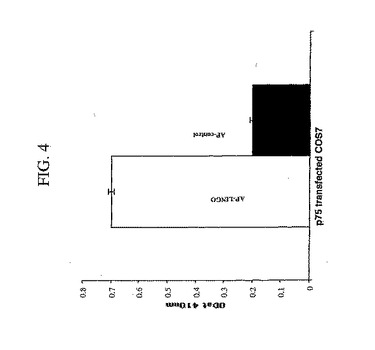
【図5】
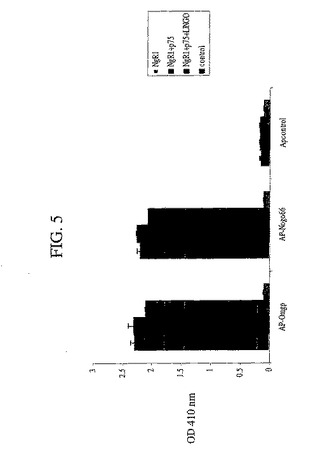
【図6】
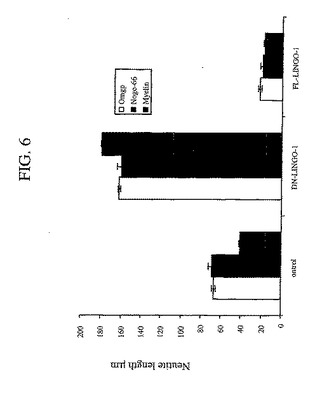
【図7】
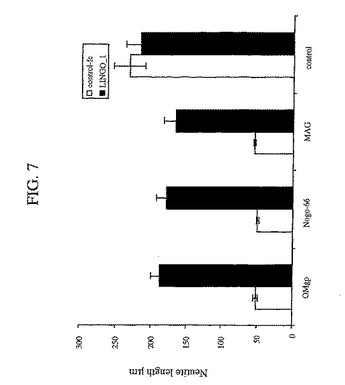
【図8】
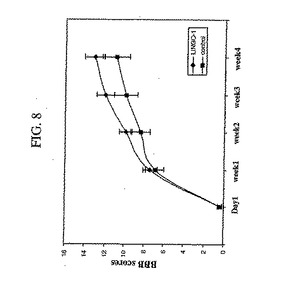
【図9】
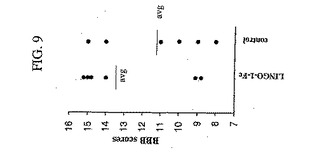
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公表番号】特表2007−524370(P2007−524370A)
【公表日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願番号】特願2006−507330(P2006−507330)
【出願日】平成16年3月17日(2004.3.17)
【国際出願番号】PCT/US2004/008323
【国際公開番号】WO2004/085648
【国際公開日】平成16年10月7日(2004.10.7)
【出願人】(592221528)バイオジェン・アイデック・エムエイ・インコーポレイテッド (224)
【Fターム(参考)】
【公表日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願日】平成16年3月17日(2004.3.17)
【国際出願番号】PCT/US2004/008323
【国際公開番号】WO2004/085648
【国際公開日】平成16年10月7日(2004.10.7)
【出願人】(592221528)バイオジェン・アイデック・エムエイ・インコーポレイテッド (224)
【Fターム(参考)】
[ Back to top ]