
NP−1アンタゴニストおよびそれらの治療上の使用
式(I):

(式中、Wはアリーレン、ヘテロアリーレンまたは式(a)であり、各Lは独立してアルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、アルキレン−C=Oまたは−C=Oであり、各Xは独立してN含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;YはN含有ヘテロアリール、N含有シクロアルキル、NR2、OR1、CNまたはCO2Rであり、Z1は式(b)である)の化合物、またはその医薬的に許容される塩は治療、特に神経変性および癌の治療に有用である。
(式中、Wはアリーレン、ヘテロアリーレンまたは式(a)であり、各Lは独立してアルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、アルキレン−C=Oまたは−C=Oであり、各Xは独立してN含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;YはN含有ヘテロアリール、N含有シクロアルキル、NR2、OR1、CNまたはCO2Rであり、Z1は式(b)である)の化合物、またはその医薬的に許容される塩は治療、特に神経変性および癌の治療に有用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、NP−1アンタゴニスト活性を有し、故に治療において有用となる化合物に関連する。
【背景技術】
【0002】
非チロシンキナーゼ型膜貫通タンパク質であるニューロピリン−1(NP−1)は、血管新生関連サイトカインのVEGFファミリーのメンバー、特に血管発生に不可欠なVEGF−A165の受容体であり、哺乳類の発生における神経軸索のガイダンスで重要な役割を果たすセマホリンまたはコラプシンと呼ばれる分子のファミリーの受容体でもある。とりわけ、NP−1はセマホリン3Aの成長円錐退縮および化学反発活性を仲介することが知られている。NP−1が一次T細胞免疫応答、ならびにヒトTリンパ好性ウイルス(HTLV−1)の細胞への侵入および感染に関与することが示されている。
【0003】
NP−1が病理において重要な役割を有し得る病態は数多く存在する。かかる病態は、例えば、脳卒中、虚血性癌疾患、癌、特に肺癌、および関節リウマチである。
【発明の概要】
【0004】
VEGFのNP−1への結合のアンタゴナイズにおいて驚くほど強力な活性を有する新たな化合物が発見された。
【0005】
第1の態様によると、本発明は式(I):
【化1】
[式中、
Wはアリーレン、ヘテロアリーレンまたは
【化2】
であり;
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、アルキレン−C=Oまたは−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR2、OR1、CNまたはCO2Rであり;
Z1は
【化3】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは2、3、4または5であり;
mは1、2または3である]
の化合物、またはその医薬的に許容される塩である。
【0006】
第2の態様によると、本発明は式(II):
【化4】
[式中、
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、またはアルキレン−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR、OR1、CNまたはCO2Rであり;
Z1は
【化5】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは0、1、2、3、4または5であり;
mは1、2または3である]
の化合物、またはその医薬的に許容される塩である。
【図面の簡単な説明】
【0007】
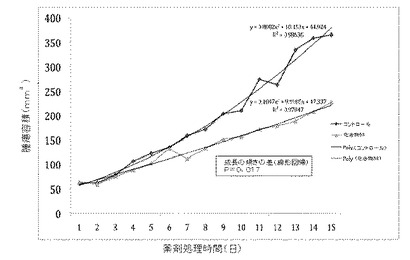
【図1】図1は、本発明の化合物である化合物58の腫瘍増殖における効果を示す。
【発明を実施するための最良の形態】
【0008】
当然のことながら、本発明の化合物は非対称に置換された炭素原子を含有する。特に、アルギニンの側鎖が主骨格に結合している一般式(I)および(II)には不斉中心が存在する。キラル中心の絶対配置はRでもSでもよい。両方のエナンチオマーが本発明の範囲に包含される。
【0009】
本発明の化合物のかかる不斉中心の存在により立体異性体が存在し得、それぞれの場合において、本発明は、エナンチオマーおよびジアステレオマー、ならびにその混合物(ラセミおよび非ラセミ混合物を含む)を含む全ての立体異性体に拡張されると理解されるべきである。
【0010】
本発明の特定の化合物の互変異性体が存在することもまた当然であり、これらの本発明の範囲に包含される。これらの互変異性体は、水素原子のホルマール移動、ならびに単結合と隣接する二重結合の転換後に形成され得る。互変異性化の方法は当業者に周知である。
【0011】
疑義を避けるため、nが1より大きい場合、括弧内の各Xおよび各L基は独立して選択される。例えば、nが2の場合(即ち(XL)−(XL))、各X基はそれぞれ異なっていてもよく、各L基もそれぞれ異なっていてもよい。
【0012】
疑義を避けるため、例えばW−L−XにおいてLが直接結合である場合、該用語はL基が「不存在」であることを意味する。言い換えれば、例えばW−L−Xにおいて、Lが直接結合である場合、W原子は直接X原子に結合する。
【0013】
本明細書中で用いられるように、用語「アルキル」または「アルキレン」は、一価又は二価の直鎖または分枝鎖のアルキル部位を意味し、例えば、メチル、エチル、プロピレン、イソプロピル、ブチル、tert−ブチル、ペンチレン、ヘキシルなどである。好ましくは、アルキルおよびアルキレン基はそれぞれ1から10個の炭素原子を含む。より好ましくは、アルキルおよびアルキレンは、それぞれC1−C6アルキルおよびC1−C6アルキレンを意味する。
【0014】
本明細書中で用いられるように、アルケニルは好ましくはC2−C10アルケニル基を意味する。好ましくは、それはC2−C6アルケニル基である。より好ましくは、それはC2−C4アルケニル基である。アルケニル基は一飽和でも二飽和でもよいが、より好ましくは一飽和である。例は、ビニル、アリル、1−プロペニル、イソプロペニルおよび1−ブテニルである。それは二価でもよく、例えば、プロペニレンである。
【0015】
本明細書中で用いられるように、アルキニルは好ましくはC2−C10アルキニル基であり、直鎖でも分枝鎖でもよい。好ましくは、それはC2−C4アルキニル基または部分である。それは二価でもよい。
【0016】
アルキル、C2−C10アルケニルおよびC2−C10アルキニル基のそれぞれは互いで適宜置換されていてもよく、即ち、C2−C10アルケニルで適宜置換されていてもよいC1−C10アルキルであってもよい。それらはまた、アリール、シクロアルキル(好ましくはC3−C10)、アリールまたはヘテロアリールで適宜置換されていてもよい。
【0017】
用語「アリール」または「アリーレン」または「Ar」は、一価または二価の芳香族炭化水素部分を意味し、例えば、フェニレン、ビフェニルまたはナフチル基である。該環は最大5個までの置換基で置換されていてもよい。考えられる他の置換基は、C1−C6アルキル、ヒドロキシ、C1−C3ヒドロキシアルキル、C1−C3アルコキシ、C1−C3ハロアルコキシ、アミノ、C1−C3モノアルキルアミノ、C1−C3ビスアルキルアミノ、C1−C3アシルアミノ、C1−C3アミノアルキル、モノ(C1−C3アルキル)アミノC1−C3アルキル、ビス(C1−C3アルキル)アミノC1−C3アルキル、C1−C3−アシルアミノ、C1−C3アルキルスルホニルアミノ、ハロ、ニトロ、シアノ、トリフルオロメチル、カルボキシ、C1−C3アルコキシカルボニル、アミノカルボニル、モノC1−C3アルキルアミノカルボニル、ビスC1−C3アルキルアミノカルボニル、−SO3H、C1−C3アルキルスルホニル、アミノスルホニル、モノC1−C3アルキルアミノスルホニルおよびビスC1−C3−アルキルアミノスルホニルである。好ましい実施態様において、Arはベンジルまたはベンジレンである。
【0018】
該アリールまたはアリーレン環は、好ましくは5または6員である。
【0019】
用語「ヘテロアリール」または「ヘテロアリーレン」は、一価または二価の芳香族環系を意味し、少なくとも1つのその環原子はO、N、またはSから選択され、例えば、ベンゾ縮合フラニル、チオフェニレン、チオフェニレン(フェニル)、ピリジル、インドリル、ピリダニジル、ピペラジニル、ピリミジニル、チアゾリレンなどである。該ヘテロアリールまたはヘテロアリーレンは、好ましくは5、6または7員であり、最大5個までの置換基、例えば、アミノ、アルキルまたはカルボン酸基などで置換されていてもよい。考えられる他の置換基は、前記の「アリール」で記載したものである。
【0020】
本明細書中で用いられるように、シクロアルキルまたはシクロアルキレンは一価または二価の飽和環系を意味し、N、OまたはSといったヘテロ原子を含んでいてもよい。「N含有シクロアルキル」は、少なくとも1個のN原子を含んでいなければならない。好ましくは、それは2個のN原子を含む。好ましくは、該環は5または6個の原子を含む。例は、シクロヘキシルまたはシクロペンチレンである。該環は、好ましくは前記「アリール」の定義において考えられる置換基として記載した少なくとも1つの基により、置換されていてもよい。
【0021】
本明細書中で用いられるように、ヘテロ環は、酸素、窒素および硫黄から独立して選択される最大4個のヘテロ原子を含む一価または二価の炭素環基である。
【0022】
ヘテロ環式環は一飽和でも二飽和でもよい。該基は、C1−C6アルキル、ヒドロキシ、C1−C3ヒドロキシアルキル、C1−C3アルコキシ、C1−C3ハロアルコキシ、アミノ、C1−C3モノアルキルアミノ、C1−C3ビスアルキルアミノ、C1−C3アシルアミノ、C1−C3アミノアルキル、モノ(C1−C3アルキル)アミノC1−C3アルキル、ビス(C1−C3アルキル)アミノC1−C3アルキル、C1−C3−アシルアミノ、C1−C3アルキルスルホニルアミノ、ハロ(例えば、F)、ニトロ、シアノ、トリフルオロメチル、カルボキシ、C1−C3アルコキシカルボニル、アミノカルボニル、モノC1−C3アルキルアミノカルボニル、ビスC1−C3アルキルアミノカルボニル、−SO3H、C1−C3アルキルスルホニル、アミノスルホニル、モノC1−C3アルキルアミノスルホニルおよびビスC1−C3−アルキルアミノスルホニルから独立して選択される最大3個の置換基で適宜置換されていてもよい。
【0023】
本明細書中で用いられるように、前記の基に接頭辞エンが続いてもよい。これは、該基が二価、つまり連結基であることを意味する。
【0024】
好ましくは、少なくとも1つのLはアルキレンである。好ましくは、それはCH2である。より好ましくは、少なくとも1つのLは結合である。さらにより好ましくは、少なくとも1つのLはアリーレンである。
【0025】
好ましくは、Wはベンジレンである。
【0026】
好ましくは、XはNR(ここで、Rは上と同義である)である。より好ましくは、Xは少なくとも1つのN原子を含有する6員のシクロアルキレンである。
【0027】
好ましくは、Yは少なくとも1つのN原子を含有する6員のシクロアルキルである。より好ましくは、Yは少なくとも1つのN原子、ならびにOもしくはSおよびNから選択される別の1つの原子を含有する置換または非置換の5員のヘテロアリールである。さらにより好ましくは、Yはピリジンであるか、YはC6H4CNである。
【0028】
好ましくは、構造IIにおけるnは1から5である。好ましくは、構造IおよびIIにおけるnは2である。より好ましくは、nは3である。
【0029】
好ましくは、構造IおよびIIにおけるArはベンジレンである。
【0030】
好ましい実施態様において、Z1は:
【化6】
である。
【0031】
好ましい実施態様において、本発明の化合物は本明細書中で58と命名される化合物である。
【0032】
本発明の化合物の活性とは、NP−1が病理において重要な役割を有し得る疾患の治療にそれらが有用であり得ることを意味する。本発明の化合物は神経修復の促進、神経変性の治療、および例えば肺癌に対する抗癌療法に有用であり得る。本発明の化合物はまた、例えば、移植手術後、免疫系の調節が要求される疾患の治療にも有用であり得る。本発明の化合物を用いて治療することが可能であり得る別の疾患は、例えば、乾癬などの皮膚病、免疫調節が必要な疾患、眼球における血管新生、糖尿病、黄斑変性、緑内障、心不全およびアルツハイマー病である。本発明の化合物はまた、血小板凝集阻害、ならびに白血病およびリンパ腫およびHTLV1感染により引き起こされる他の疾患の治療にも有用であり得る。
【0033】
本発明の化合物はまた、肝疾患、多発性硬化症の治療およびNRP−1発現腫瘍において、獣医学的適用の有用性を有し得る。
【0034】
本発明の化合物は、アバスチンといった別の1つの抗癌剤と組み合わされてもよい。本発明の化合物はまた、抗血管新生薬と組み合わされてもよい。組み合わせは別々、時間差、または同時に治療に用いることができる。治療は上と同義のものである。
【0035】
治療的使用のために、本発明の化合物は当業者に周知の製造方法により、当業者に周知の成分を用いて製剤化され、投与されてもよい。該化合物の適当な投与量は、治療対象の病態、化合物の薬力、投与経路などといった通例の因子により当業者により選択されてもよい。適切な投与経路は、例えば、経口、静脈内、筋肉内、腹腔内、鼻腔内および皮下である。
【0036】
特定の理論に制約されないが、NP−1アンタゴニストは、NP−1との結合においてセマホリン−3Aと競合し、それによりセマホリン−3Aの軸索伸張および神経細胞移動に対する阻害効果をアンタゴナイズする可能性がある。これの有力な適用は、神経突起伸張の促進、神経修復の刺激または神経変性の治療におけるものである。さらに、NP−1アンタゴニストはセマホリン−3A感受性神経細胞の生存を促進する可能性があるが、この効果は上記の適用におけるその有用性を裏付ける、または高めるものであり、これにより、これらの適用を、例えば、脳卒中やいくつかの癌疾患における虚血の発現による神経細胞死の治療に広げることができる。
【0037】
最近の証拠によりNP−1が血管新生に関与することが示唆された。それによると、NP−1は、癌、癌疾患、関節リウマチおよび他の疾患におけるVEGF誘導性血管新生に不可欠である可能性がある。故に、NP−1アンタゴニストは、疾患におけるVEGF依存性血管新生の阻害の適用を有し得る。
【0038】
NP−1アンタゴニストはまた、免疫系の調節にも関与し得る。故に、移植前、移植中または移植後に本発明の化合物を投与することは有用であり得る。
【0039】
加えて、NP−1アンタゴニストは腫瘍細胞におけるNP−1との結合においてVEGFと競合し、NP−1発現腫瘍細胞の細胞死を促進する可能性がある。これの有力な適用は抗癌療法におけるものである。さらに、NP−1アンタゴニストは、カルシノーマ細胞の細胞外マトリックスへの接着および細胞遊走を効果的に阻害するため、抗転移能を有する。
【0040】
好ましい実施態様において、本発明の化合物は核種、または常磁性核種(例えば、ガドリニウム、金属と錯体形成するための当業者に周知の適当なタイプのキレートと共に)と共に、放射線イメージングに、または磁気共鳴画像法の造影剤として用いられてもよい。
【0041】
以下の例は本発明を説明するものである。本発明の化合物の一般的な製造方法が提供される。例示される化合物は一覧表にされ、LC−MSによりキャラクタリゼーションされる。NP−1結合データもまた、いくつかの化合物について提供される。
【0042】
定義および最終化合物のキャラクタリゼーション
略語
Arg,アルギニン;eq,当量;Boc,tert−ブトキシカルボニル;tBu,tert−ブチル;DIPEA,N,N−ジイソプロピルエチルアミン;HPLC,高速液体クロマトグラフィー;LC−MS,液体クロマトグラフ質量分析;Pbf,2,2,4,6,7‐ペンタメチルジヒドロベンゾフラン−5−スルホニル;PG,保護基;py,ピリジン;PyBrOP,ブロモ−トリス−ピロリジノ−ホスホニウム ヘキサフルオロホスフェート;SCX−2,ISOLUTE SCX−2 強陽イオン交換樹脂;TLC,薄層クロマトグラフィー
【0043】
プレパラティブLC−MS:プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μm)を用いた質量分析連結精製プレパラティブLC−MS。
中間体化合物、即ち、所定の番号で示されていない化合物は、逆相LC−MS(分析用C−18カラム、 Phenomenex Luna C18 (2), 50 x 3.0 mm, 3 μm、B:5−95%のABグラジエント、6.5分間、流速1.1mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリルまたはメタノール)で分析した。
全ての最終化合物、即ち、所定の番号を付与されている化合物は、逆相LC−MS(分析用C−18カラム、Phenomenex Luna C18 (2), 150 x 4.6 mm, 5 μm、B:5−95%のABグラジエント、13分間、流速1.5mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリルまたはメタノール)で分析した。
【0044】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸 メチルエステル
【化7】
窒素下(バルーン)、20℃の撹拌した5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルクロリド(1.25当量、2g、6.72mmol)のピリジン(無水、10mL)溶液に、メチル−3−アミノチオフェン−2−カルボキシレート(1当量、845mg、5.38mmol)/ピリジン(無水、5mL)を120分間かけて滴下して加えた。反応混合物を20℃で18時間撹拌し、その後、反応混合物を冷却(約0℃)し、水(5mL)を滴下して加えた。沈殿物が生じ、混合物にさらに水(20mL)を加え、目的生成物を濾取し、氷冷した水(2x10mL)で洗浄し、減圧乾燥し、灰白色の固形物(2.2g、98%)を得、さらに精製することなく用いた。
LC-MS 保持時間:4.42分;純度:98 %; MS m/z - 416/418 [M - 1]−.
【0045】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸
【化8】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸 メチルエステル(1当量、2.17g、5.2mmol)をテトラヒドロフラン(20mL)およびメタノール(12mL)に溶解した。1M 水酸化リチウム(5当量、26mL、26mmol)を一度に加えた。混合物を45℃で20時間撹拌し、その後、有機溶媒を減圧除去し、(水性の)残渣を水(30mL)で希釈し、次いで、6M 塩酸でpH1に酸性化すると、沈殿物が生じた。灰白色の固形物を吸引濾過により回収し、水(2x20mL)で洗浄し、減圧乾燥し、灰白色の固形物(1.8g、86%)を得た。
LC-MS 保持時間:4.54分;純度:95%; MS m/z - 402/404 [M - 1]−.
【0046】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7−ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸メチルエステル
【化9】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸(1当量、2.11g、5.2mmol)およびブロモ−トリス−ピロリジノ−ホスホニウム ヘキサフルオロホスフェート(PyBrOP;1.1当量、2.66g、5.7mmol)をジクロロメタン(45mL)に懸濁し、混合物を20℃で10分間撹拌した。N,N−ジイソプロピルエチルアミン(7当量、6.34mL、36.4mmol)を混合物に加え、さらに15分間撹拌した。H−L−アルギニン(Pbf)−OMe(塩酸塩;1.1当量、7g、14.8mmol)を一度に加え、反応混合物(いくらかの白色の沈殿物を含む)を20℃で18時間撹拌した。次いで溶媒を減圧除去し、得られた残渣を酢酸エチル(60mL)に溶解し、1M 塩酸(40 mL)で分液処理した。水層を分離し、有機層をさらに1M 塩酸のアリコート(3x40L)で洗浄した。有機層をブライン(飽和、水溶液;50mL)で洗浄し、硫酸マグネシウムで乾燥し、濾過し、溶媒を減圧除去した。粗生成物(灰白色の泡状物質;約4.5g)をシリカゲルフラッシュカラムクロマトグラフィー(溶出液:酢酸エチル/イソ−ヘキサン;50:50、酢酸エチルのみ増加)で精製し、目的生成物を灰白色の固形物として得た(3.38g、79%)。
LC-MS 保持時間:4.77分;純度:95 %; MS m/z - 826/828 [M + 1]+.
【0047】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7−ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸
【化10】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7‐ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸 メチルエステル(1当量、2.45g、2.96mmol)を1M 水酸化リチウム(5当量、14.82mL、14.82mmol)/テトラヒドロフラン(29mL)と共に20℃で3時間撹拌した。次いで有機溶媒を減圧除去し、有機溶媒を減圧除去し、(水性の)残渣を水(30mL)で希釈し、6M 塩酸でpH1に酸性化した。酢酸エチル(200mL)を得られた懸濁液に加え、撹拌後、有機層を分離した。水層をさらに酢酸エチル(150mL)で抽出し、有機抽出物を合わせ、ブライン(飽和、水溶液;3x75mL)で洗浄し、硫酸マグネシウムで乾燥し、濾過し、減圧除去した。生成物(淡黄色の泡状物質、2.42g、100%)をさらに精製することなく用いた。
LC-MS 保持時間:4.89分;純度:90 %; MS m/z - 812/814 [M + 1]+.
【0048】
合成ボロン酸の一般的製造方法
【化11】
適当なホルミル−フェニルボロン酸(1.2当量)およびアミン(1当量)を合わせ、ジクロロメタン(15mL)に溶解した。酢酸(0.2mL)を加え、反応物を周囲温度で2時間撹拌した。次いで、ナトリウム シアノボロヒドリド(2当量)を一度に加え、反応物を20℃でさらに20時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製されたボロン酸を溶媒のエバポレートにより単離し、さらに精製することなく用いた。
【0049】
液相鈴木カップリング(カルボン酸)の一般的方法
【化12】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7‐ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸(約1g、1.0当量)、対応するボロン酸(1.5当量)およびテトラキス(トリフェニルホスフィン)パラジウム(0)(0.05当量)を脱気した1,2−ジメトキシエタン(3mL)に懸濁した。脱気したリン酸化リウム(三塩基、2M水溶液、4当量)をさらに加え、マイクロ波(100ワット、90℃、照射時間=10分間)を用いて反応混合物を加熱した。次いで、溶媒を減圧除去し、得られた残渣を酢酸エチル(200mL)および塩酸(1M水溶液;150mL)で分液処理した。相を分離し、水相をさらに酢酸エチル(200mL)で抽出した。有機抽出物を合わせ、ブライン(飽和、水溶液;2x100mL)で洗浄し、硫酸マグネシウムで乾燥し、濾過し、溶媒を減圧除去した。粗生成物(通常は黄色の固形物;約1.5g)をシリカゲルフラッシュカラムクロマトグラフィー(溶出液:ジクロロメタンからジクロロメタン/メタノール 75:25に漸増)で精製し、表1にまとめられるような生成物を得た。
【表1−1】
【表1−2】
【0050】
メチルエステル加水分解およびPbf除去の一般的方法
【化13】
撹拌したメチルエステル(1当量)の1,4−ジオキサン(1.2mL)懸濁液に1M 水酸化リチウム(水溶液、4当量)および水(1.2mL)を加えた。反応物を20℃で24時間撹拌し、反応物をエバポレートして乾燥して白色の固形物を得、さらに精製することなく用いた。
残渣をジクロロメタン/トリフルオロ酢酸(1:1、5mL)に溶解し、室温で1時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1%ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法を用いて作製した最終化合物を表2にまとめる。
【表2】
【0051】
Pbf除去の一般的方法
【化14】
残渣をジクロロメタン/トリフルオロ酢酸(1:1、5mL)に溶解し、室温で1時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製されたペプチドミメティックを溶媒のエバポレートにより単離した。
【表3】
【0052】
還元的アミノ化およびPbf除去の一般的方法
【化15】
アルデヒド(1当量)のテトラヒドロフラン/メタノール(1:1、1.5mL)溶液にアミン(市販;1.1当量)、次いで酢酸(1−2滴、〜pH6)を加えた。反応物を20℃で2時間撹拌し、ナトリウム シアノボロヒドリド(2当量)/メタノール(0.1mL)を一度に加えた。反応物を20℃でさらに16時間撹拌した。反応物を前処理したSCX−2(1g)カートリッジに通して濾過し、生成物を2M アンモニア/メタノールで溶出した。溶媒をエバポレートして生成物を黄色の油状物として得、それをジクロロメタン/トリフルオロ酢酸(1:1、8mL)に溶解し、20℃で1時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、i.溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール、またはii.溶出液A:10mM 炭酸水素アンモニウム(pH9)、溶出液B:100% メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法を用いて作製した最終化合物を表4にまとめる。
【表4−1】
【表4−2】
【表4−3】
【表4−4】
【表4−5】
【表4−6】
【表4−7】
【表4−8】
【表4−9】
【表4−10】
【表4−11】
【表4−12】
【表4−13】
【0053】
アミノ−チアゾールアルデヒド形成の一般的方法
【化16】
ブロモ−チアゾール(1当量)、アミン(市販;1.1当量)および水酸化リチウム(1.15当量)を合わせ、テトラヒドロフラン(2mL)/水(0.1mL)に溶解した。マイクロ波で反応混合物を75℃で15分間加熱した。次いで、水(5mL)を反応混合物に加え、塩酸(1M、水溶液)を用いてpHを約7に調整した。溶媒を減圧除去し、ジメチルスルホキシドに再溶解し、さらに精製することなく用いるか、あるいはプレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1%)を用いた(質量分析連結)プレパラティブLC−MSで精製した。これらの方法を用いて作製されたチアゾールアルデヒドを表5にまとめる。
【表5−1】
【表5−2】
【表5−3】
【0054】
液相還元的アミノ化の一般的方法
【化17】
アニリン(1当量)、アルデヒド(市販または前記のようにして製造;0.5−6.0当量)およびナトリウム シアノボロヒドリド(0.6−2.0当量)を合わせ、メタノール(2mL)に溶解した。塩酸(1M、水溶液)または酢酸をpHが5−6の間になるまで加え、反応混合物を20℃で16時間撹拌した。溶媒を減圧除去し、得られた残渣をジメチルスルホキシドに再溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法により作製した化合物を表6にまとめる。
【表6−1】
【表6−2】
【表6−3】
【表6−4】
【0055】
アミノエチル−ピラゾールアルデヒド形成およびそれに次ぐ還元的アミノ化の一般的方法
【化18】
1−(2−クロロエチル)−1H−ピラゾール−4−カルボアルデヒド(4.2当量)、アミン(5当量)およびトリエチルアミン(8.4当量)をN−メチル−2−ピロリドン(1mL)と共に85℃で16時間加熱した。溶媒を減圧除去し、アニリン(1当量)/メタノール(1mL)、ナトリウム シアノボロヒドリド(2.0当量)を加えた。次いで、塩酸(1M、水溶液)をpHが5−6の間になるまで加え、反応混合物を20℃で16時間撹拌した。メタノールを減圧除去し、混合物をジメチルスルホキシドで希釈し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離し、さらに精製することなく用いた。これらの方法により作製された化合物を表7にまとめる。
【表7】
【0056】
ブロモ−チアゾールによる置換反応の一般的方法
【化19】
ブロモ−チアゾール(1当量)および1−(2−ヒドロキシエチル)ピペラジン(10当量)を合わせ、トリエチルアミン(10当量)およびN−メチル−2−ピロリドン(2mL)に溶解し、24時間加熱還流した。混合物を周囲温度に冷却し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法により作製した化合物を表8にまとめる。
【表8】
【0057】
液相アルキル化の一般的方法
【化20】
アニリン(1当量)をメタノール(2mL)またはジメチルスルホキシド(2mL)に溶解した。アルキルハライド(1当量)、次いで、トリエチルアミン(2当量)を加え、反応混合物を50−100℃で16時間撹拌した。メタノールを溶媒をとして用いた場合、メタノールを減圧除去し、得られた残渣をジメチルスルホキシドに再溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)、および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離し、さらに精製することなく用いた。この方法を用いて作製した化合物を表9にまとめる。
【表9】
【0058】
メチルエステル加水分解およびPbf/Bocの除去の一般的方法
【化21】
完全に保護した出発物質(1当量)を水酸化リチウム(5当量)/テトラヒドロフラン/水(4:1;2.5mL)と共に、必要に応じ、20−60℃で1−3時間撹拌した。次いで溶媒を減圧除去し、残渣をトリフルオロ酢酸(2mL)および水(0.1mL)で処理した。反応混合物を20℃でさらに3−16時間撹拌した。溶媒を減圧除去し、得られた残渣をジメチルスルホキシドに再溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(2−95% B、12分間、流速20mL/分、i.溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル;またはii.溶出液A:10mM 炭酸水素アンモニウム(pH9)、溶出液B:メタノールのみ)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。この方法で作製した化合物を表10にまとめる。
【表10−1】
【表10−2】
【表10−3】
【表10−4】
【表10−5】
【表10−6】
【0059】
インビトロにおけるNP−1の結合の評価およびインビボ腫瘍モデルにおける研究
いくつかの化合物についてNP−1の結合を評価した。1個の化合物(化合物58)について、ヒト肺癌細胞の異種移植片を負荷されたマウスモデルにおいて抗癌活性を評価した。実験方法および結果を以下に示す。
【0060】
一般的実験方法
細胞培養およびアデノウィルスを介したNP−1トランスフェクション
ヒト前立腺癌 DU145細胞を成長培地(10% FBSおよびL−グルタミン含有RPMI 1640)中で培養した。DU145細胞を2x104細胞/ウェル(96ウェルプレート)の密度において0.1mlの成長培地を用いて播種し、ヒトNP−1の翻訳領域の全長を含むアデノウィルスベクターをトランスフェクションした。Ad.NP−1−トランスフェクション細胞を結合アッセイに先立って2日間培養した。
【0061】
細胞におけるビオチン化VEGF−A165の結合
96ウェルプレート内のコンフルーエントなAd.NP−1−トランスフェクション細胞をリン酸緩衝生理食塩水(PBS)で2回洗浄した。結合培地(ダルベッコ改変イーグル培地、25mM HEPES、pH7.3、0.1% BSA含有)で希釈した異なる濃度の化合物を加え、次いで2nMのbt−VEGF−A165を加えた。室温で2時間インキュベーション後、培地を吸引し、PBSで3回洗浄した。NP−1に結合したbt−VEGF−A165を、ストレプトアビジン−西洋ワサビペルオキシダーゼ複合体および酵素基質により検出し、Tecan Geniosプレートリーダー(A450nm、リファレンス波長:A595nm)で測定した。非特異的結合は、100倍過剰量の非標識VEGF−A165の存在下で決定した。
【0062】
無細胞系におけるビオチン化VEGF−A165の結合
96ウェルプレートを前もってNP−1タンパク(3μg/ml)により終夜4℃でコーティングした。翌日、プレートをブロッキングバッファー(1% BSA含有PBS)で処理し、洗浄バッファー(0.1% Tween−20含有PBS)で3回洗浄した。1% DMSO含有PBSで希釈した異なる濃度の化合物を加え、次いで、0.25nMのbt−VEGF−A165を加えた。室温で2時間インキュベーション後、プレートを洗浄バッファーで3回洗浄した。NP−1に結合したbt−VEGF−A165をストレプトアビジン−西洋ワサビペルオキシダーゼ複合体および酵素基質で検出し、Tecan Geniosプレートリーダー(A450nm、リファレンス波長:A595nm)で測定した。非特異的結合は、プレートのNP−1コーティングウェルの非存在下で決定した。
【0063】
結合実験の結果を下の表11に示す。
【表11−1】
【表11−2】
【0064】
肺癌の生物学的研究(インビボ)
化合物58もまた、肺癌の前臨床モデルにおいて、研究概念の実証(proof of principle study)に成功した。化合物58は腫瘍増殖の速度を有意に低下させ、毒性の証拠は示さなかった。
肺癌マウスモデルにおけるこの前臨床の研究概念の実証では、1日1回投与において2週間投与された化合物58が、腫瘍増殖速度を52%(p=0.017)低下させることが示された。先に行われた高用量の毒性実験の結果と一致して、この研究では毒性の証拠は見られなかった。
【0065】
方法
有効性研究用に、ヒト肺非小細胞癌A549細胞を成長培地RPMI 1640中で培養した。90%コンフルーエントの細胞を剥し、細胞数をカウントし、PBSに懸濁し、接種用に最終細胞濃度を5x107/mlとした。
【0066】
動物研究
A549細胞をメスBalb/cヌードマウスに接種してから2週間後、化合物の投与を開始した。化合物58を1日当たり80mg/kgの投与量において2週間投与した。2週間の期間中、デジタルノギス(Fisher Scientific)を用いて腫瘍の長さおよび幅を毎日測定することにより、腫瘍容積をモニターした。腫瘍容積は式(長さx幅2/2)を用いて算出した。実験の終了時、腫瘍を解剖し、重量を測定した。
【0067】
結果
インビボ研究の結果を図1に示す。
図1のデータにより、化合物58処理群において腫瘍増殖速度が50%低下した(p=0.017、直線回帰分析)。さらに、腫瘍重量は27%減少した(p=0.04、マン・ホイットニー検定)。
【技術分野】
【0001】
本発明は、NP−1アンタゴニスト活性を有し、故に治療において有用となる化合物に関連する。
【背景技術】
【0002】
非チロシンキナーゼ型膜貫通タンパク質であるニューロピリン−1(NP−1)は、血管新生関連サイトカインのVEGFファミリーのメンバー、特に血管発生に不可欠なVEGF−A165の受容体であり、哺乳類の発生における神経軸索のガイダンスで重要な役割を果たすセマホリンまたはコラプシンと呼ばれる分子のファミリーの受容体でもある。とりわけ、NP−1はセマホリン3Aの成長円錐退縮および化学反発活性を仲介することが知られている。NP−1が一次T細胞免疫応答、ならびにヒトTリンパ好性ウイルス(HTLV−1)の細胞への侵入および感染に関与することが示されている。
【0003】
NP−1が病理において重要な役割を有し得る病態は数多く存在する。かかる病態は、例えば、脳卒中、虚血性癌疾患、癌、特に肺癌、および関節リウマチである。
【発明の概要】
【0004】
VEGFのNP−1への結合のアンタゴナイズにおいて驚くほど強力な活性を有する新たな化合物が発見された。
【0005】
第1の態様によると、本発明は式(I):
【化1】
[式中、
Wはアリーレン、ヘテロアリーレンまたは
【化2】
であり;
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、アルキレン−C=Oまたは−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR2、OR1、CNまたはCO2Rであり;
Z1は
【化3】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは2、3、4または5であり;
mは1、2または3である]
の化合物、またはその医薬的に許容される塩である。
【0006】
第2の態様によると、本発明は式(II):
【化4】
[式中、
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、またはアルキレン−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR、OR1、CNまたはCO2Rであり;
Z1は
【化5】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは0、1、2、3、4または5であり;
mは1、2または3である]
の化合物、またはその医薬的に許容される塩である。
【図面の簡単な説明】
【0007】
【図1】図1は、本発明の化合物である化合物58の腫瘍増殖における効果を示す。
【発明を実施するための最良の形態】
【0008】
当然のことながら、本発明の化合物は非対称に置換された炭素原子を含有する。特に、アルギニンの側鎖が主骨格に結合している一般式(I)および(II)には不斉中心が存在する。キラル中心の絶対配置はRでもSでもよい。両方のエナンチオマーが本発明の範囲に包含される。
【0009】
本発明の化合物のかかる不斉中心の存在により立体異性体が存在し得、それぞれの場合において、本発明は、エナンチオマーおよびジアステレオマー、ならびにその混合物(ラセミおよび非ラセミ混合物を含む)を含む全ての立体異性体に拡張されると理解されるべきである。
【0010】
本発明の特定の化合物の互変異性体が存在することもまた当然であり、これらの本発明の範囲に包含される。これらの互変異性体は、水素原子のホルマール移動、ならびに単結合と隣接する二重結合の転換後に形成され得る。互変異性化の方法は当業者に周知である。
【0011】
疑義を避けるため、nが1より大きい場合、括弧内の各Xおよび各L基は独立して選択される。例えば、nが2の場合(即ち(XL)−(XL))、各X基はそれぞれ異なっていてもよく、各L基もそれぞれ異なっていてもよい。
【0012】
疑義を避けるため、例えばW−L−XにおいてLが直接結合である場合、該用語はL基が「不存在」であることを意味する。言い換えれば、例えばW−L−Xにおいて、Lが直接結合である場合、W原子は直接X原子に結合する。
【0013】
本明細書中で用いられるように、用語「アルキル」または「アルキレン」は、一価又は二価の直鎖または分枝鎖のアルキル部位を意味し、例えば、メチル、エチル、プロピレン、イソプロピル、ブチル、tert−ブチル、ペンチレン、ヘキシルなどである。好ましくは、アルキルおよびアルキレン基はそれぞれ1から10個の炭素原子を含む。より好ましくは、アルキルおよびアルキレンは、それぞれC1−C6アルキルおよびC1−C6アルキレンを意味する。
【0014】
本明細書中で用いられるように、アルケニルは好ましくはC2−C10アルケニル基を意味する。好ましくは、それはC2−C6アルケニル基である。より好ましくは、それはC2−C4アルケニル基である。アルケニル基は一飽和でも二飽和でもよいが、より好ましくは一飽和である。例は、ビニル、アリル、1−プロペニル、イソプロペニルおよび1−ブテニルである。それは二価でもよく、例えば、プロペニレンである。
【0015】
本明細書中で用いられるように、アルキニルは好ましくはC2−C10アルキニル基であり、直鎖でも分枝鎖でもよい。好ましくは、それはC2−C4アルキニル基または部分である。それは二価でもよい。
【0016】
アルキル、C2−C10アルケニルおよびC2−C10アルキニル基のそれぞれは互いで適宜置換されていてもよく、即ち、C2−C10アルケニルで適宜置換されていてもよいC1−C10アルキルであってもよい。それらはまた、アリール、シクロアルキル(好ましくはC3−C10)、アリールまたはヘテロアリールで適宜置換されていてもよい。
【0017】
用語「アリール」または「アリーレン」または「Ar」は、一価または二価の芳香族炭化水素部分を意味し、例えば、フェニレン、ビフェニルまたはナフチル基である。該環は最大5個までの置換基で置換されていてもよい。考えられる他の置換基は、C1−C6アルキル、ヒドロキシ、C1−C3ヒドロキシアルキル、C1−C3アルコキシ、C1−C3ハロアルコキシ、アミノ、C1−C3モノアルキルアミノ、C1−C3ビスアルキルアミノ、C1−C3アシルアミノ、C1−C3アミノアルキル、モノ(C1−C3アルキル)アミノC1−C3アルキル、ビス(C1−C3アルキル)アミノC1−C3アルキル、C1−C3−アシルアミノ、C1−C3アルキルスルホニルアミノ、ハロ、ニトロ、シアノ、トリフルオロメチル、カルボキシ、C1−C3アルコキシカルボニル、アミノカルボニル、モノC1−C3アルキルアミノカルボニル、ビスC1−C3アルキルアミノカルボニル、−SO3H、C1−C3アルキルスルホニル、アミノスルホニル、モノC1−C3アルキルアミノスルホニルおよびビスC1−C3−アルキルアミノスルホニルである。好ましい実施態様において、Arはベンジルまたはベンジレンである。
【0018】
該アリールまたはアリーレン環は、好ましくは5または6員である。
【0019】
用語「ヘテロアリール」または「ヘテロアリーレン」は、一価または二価の芳香族環系を意味し、少なくとも1つのその環原子はO、N、またはSから選択され、例えば、ベンゾ縮合フラニル、チオフェニレン、チオフェニレン(フェニル)、ピリジル、インドリル、ピリダニジル、ピペラジニル、ピリミジニル、チアゾリレンなどである。該ヘテロアリールまたはヘテロアリーレンは、好ましくは5、6または7員であり、最大5個までの置換基、例えば、アミノ、アルキルまたはカルボン酸基などで置換されていてもよい。考えられる他の置換基は、前記の「アリール」で記載したものである。
【0020】
本明細書中で用いられるように、シクロアルキルまたはシクロアルキレンは一価または二価の飽和環系を意味し、N、OまたはSといったヘテロ原子を含んでいてもよい。「N含有シクロアルキル」は、少なくとも1個のN原子を含んでいなければならない。好ましくは、それは2個のN原子を含む。好ましくは、該環は5または6個の原子を含む。例は、シクロヘキシルまたはシクロペンチレンである。該環は、好ましくは前記「アリール」の定義において考えられる置換基として記載した少なくとも1つの基により、置換されていてもよい。
【0021】
本明細書中で用いられるように、ヘテロ環は、酸素、窒素および硫黄から独立して選択される最大4個のヘテロ原子を含む一価または二価の炭素環基である。
【0022】
ヘテロ環式環は一飽和でも二飽和でもよい。該基は、C1−C6アルキル、ヒドロキシ、C1−C3ヒドロキシアルキル、C1−C3アルコキシ、C1−C3ハロアルコキシ、アミノ、C1−C3モノアルキルアミノ、C1−C3ビスアルキルアミノ、C1−C3アシルアミノ、C1−C3アミノアルキル、モノ(C1−C3アルキル)アミノC1−C3アルキル、ビス(C1−C3アルキル)アミノC1−C3アルキル、C1−C3−アシルアミノ、C1−C3アルキルスルホニルアミノ、ハロ(例えば、F)、ニトロ、シアノ、トリフルオロメチル、カルボキシ、C1−C3アルコキシカルボニル、アミノカルボニル、モノC1−C3アルキルアミノカルボニル、ビスC1−C3アルキルアミノカルボニル、−SO3H、C1−C3アルキルスルホニル、アミノスルホニル、モノC1−C3アルキルアミノスルホニルおよびビスC1−C3−アルキルアミノスルホニルから独立して選択される最大3個の置換基で適宜置換されていてもよい。
【0023】
本明細書中で用いられるように、前記の基に接頭辞エンが続いてもよい。これは、該基が二価、つまり連結基であることを意味する。
【0024】
好ましくは、少なくとも1つのLはアルキレンである。好ましくは、それはCH2である。より好ましくは、少なくとも1つのLは結合である。さらにより好ましくは、少なくとも1つのLはアリーレンである。
【0025】
好ましくは、Wはベンジレンである。
【0026】
好ましくは、XはNR(ここで、Rは上と同義である)である。より好ましくは、Xは少なくとも1つのN原子を含有する6員のシクロアルキレンである。
【0027】
好ましくは、Yは少なくとも1つのN原子を含有する6員のシクロアルキルである。より好ましくは、Yは少なくとも1つのN原子、ならびにOもしくはSおよびNから選択される別の1つの原子を含有する置換または非置換の5員のヘテロアリールである。さらにより好ましくは、Yはピリジンであるか、YはC6H4CNである。
【0028】
好ましくは、構造IIにおけるnは1から5である。好ましくは、構造IおよびIIにおけるnは2である。より好ましくは、nは3である。
【0029】
好ましくは、構造IおよびIIにおけるArはベンジレンである。
【0030】
好ましい実施態様において、Z1は:
【化6】
である。
【0031】
好ましい実施態様において、本発明の化合物は本明細書中で58と命名される化合物である。
【0032】
本発明の化合物の活性とは、NP−1が病理において重要な役割を有し得る疾患の治療にそれらが有用であり得ることを意味する。本発明の化合物は神経修復の促進、神経変性の治療、および例えば肺癌に対する抗癌療法に有用であり得る。本発明の化合物はまた、例えば、移植手術後、免疫系の調節が要求される疾患の治療にも有用であり得る。本発明の化合物を用いて治療することが可能であり得る別の疾患は、例えば、乾癬などの皮膚病、免疫調節が必要な疾患、眼球における血管新生、糖尿病、黄斑変性、緑内障、心不全およびアルツハイマー病である。本発明の化合物はまた、血小板凝集阻害、ならびに白血病およびリンパ腫およびHTLV1感染により引き起こされる他の疾患の治療にも有用であり得る。
【0033】
本発明の化合物はまた、肝疾患、多発性硬化症の治療およびNRP−1発現腫瘍において、獣医学的適用の有用性を有し得る。
【0034】
本発明の化合物は、アバスチンといった別の1つの抗癌剤と組み合わされてもよい。本発明の化合物はまた、抗血管新生薬と組み合わされてもよい。組み合わせは別々、時間差、または同時に治療に用いることができる。治療は上と同義のものである。
【0035】
治療的使用のために、本発明の化合物は当業者に周知の製造方法により、当業者に周知の成分を用いて製剤化され、投与されてもよい。該化合物の適当な投与量は、治療対象の病態、化合物の薬力、投与経路などといった通例の因子により当業者により選択されてもよい。適切な投与経路は、例えば、経口、静脈内、筋肉内、腹腔内、鼻腔内および皮下である。
【0036】
特定の理論に制約されないが、NP−1アンタゴニストは、NP−1との結合においてセマホリン−3Aと競合し、それによりセマホリン−3Aの軸索伸張および神経細胞移動に対する阻害効果をアンタゴナイズする可能性がある。これの有力な適用は、神経突起伸張の促進、神経修復の刺激または神経変性の治療におけるものである。さらに、NP−1アンタゴニストはセマホリン−3A感受性神経細胞の生存を促進する可能性があるが、この効果は上記の適用におけるその有用性を裏付ける、または高めるものであり、これにより、これらの適用を、例えば、脳卒中やいくつかの癌疾患における虚血の発現による神経細胞死の治療に広げることができる。
【0037】
最近の証拠によりNP−1が血管新生に関与することが示唆された。それによると、NP−1は、癌、癌疾患、関節リウマチおよび他の疾患におけるVEGF誘導性血管新生に不可欠である可能性がある。故に、NP−1アンタゴニストは、疾患におけるVEGF依存性血管新生の阻害の適用を有し得る。
【0038】
NP−1アンタゴニストはまた、免疫系の調節にも関与し得る。故に、移植前、移植中または移植後に本発明の化合物を投与することは有用であり得る。
【0039】
加えて、NP−1アンタゴニストは腫瘍細胞におけるNP−1との結合においてVEGFと競合し、NP−1発現腫瘍細胞の細胞死を促進する可能性がある。これの有力な適用は抗癌療法におけるものである。さらに、NP−1アンタゴニストは、カルシノーマ細胞の細胞外マトリックスへの接着および細胞遊走を効果的に阻害するため、抗転移能を有する。
【0040】
好ましい実施態様において、本発明の化合物は核種、または常磁性核種(例えば、ガドリニウム、金属と錯体形成するための当業者に周知の適当なタイプのキレートと共に)と共に、放射線イメージングに、または磁気共鳴画像法の造影剤として用いられてもよい。
【0041】
以下の例は本発明を説明するものである。本発明の化合物の一般的な製造方法が提供される。例示される化合物は一覧表にされ、LC−MSによりキャラクタリゼーションされる。NP−1結合データもまた、いくつかの化合物について提供される。
【0042】
定義および最終化合物のキャラクタリゼーション
略語
Arg,アルギニン;eq,当量;Boc,tert−ブトキシカルボニル;tBu,tert−ブチル;DIPEA,N,N−ジイソプロピルエチルアミン;HPLC,高速液体クロマトグラフィー;LC−MS,液体クロマトグラフ質量分析;Pbf,2,2,4,6,7‐ペンタメチルジヒドロベンゾフラン−5−スルホニル;PG,保護基;py,ピリジン;PyBrOP,ブロモ−トリス−ピロリジノ−ホスホニウム ヘキサフルオロホスフェート;SCX−2,ISOLUTE SCX−2 強陽イオン交換樹脂;TLC,薄層クロマトグラフィー
【0043】
プレパラティブLC−MS:プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μm)を用いた質量分析連結精製プレパラティブLC−MS。
中間体化合物、即ち、所定の番号で示されていない化合物は、逆相LC−MS(分析用C−18カラム、 Phenomenex Luna C18 (2), 50 x 3.0 mm, 3 μm、B:5−95%のABグラジエント、6.5分間、流速1.1mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリルまたはメタノール)で分析した。
全ての最終化合物、即ち、所定の番号を付与されている化合物は、逆相LC−MS(分析用C−18カラム、Phenomenex Luna C18 (2), 150 x 4.6 mm, 5 μm、B:5−95%のABグラジエント、13分間、流速1.5mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリルまたはメタノール)で分析した。
【0044】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸 メチルエステル
【化7】
窒素下(バルーン)、20℃の撹拌した5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルクロリド(1.25当量、2g、6.72mmol)のピリジン(無水、10mL)溶液に、メチル−3−アミノチオフェン−2−カルボキシレート(1当量、845mg、5.38mmol)/ピリジン(無水、5mL)を120分間かけて滴下して加えた。反応混合物を20℃で18時間撹拌し、その後、反応混合物を冷却(約0℃)し、水(5mL)を滴下して加えた。沈殿物が生じ、混合物にさらに水(20mL)を加え、目的生成物を濾取し、氷冷した水(2x10mL)で洗浄し、減圧乾燥し、灰白色の固形物(2.2g、98%)を得、さらに精製することなく用いた。
LC-MS 保持時間:4.42分;純度:98 %; MS m/z - 416/418 [M - 1]−.
【0045】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸
【化8】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸 メチルエステル(1当量、2.17g、5.2mmol)をテトラヒドロフラン(20mL)およびメタノール(12mL)に溶解した。1M 水酸化リチウム(5当量、26mL、26mmol)を一度に加えた。混合物を45℃で20時間撹拌し、その後、有機溶媒を減圧除去し、(水性の)残渣を水(30mL)で希釈し、次いで、6M 塩酸でpH1に酸性化すると、沈殿物が生じた。灰白色の固形物を吸引濾過により回収し、水(2x20mL)で洗浄し、減圧乾燥し、灰白色の固形物(1.8g、86%)を得た。
LC-MS 保持時間:4.54分;純度:95%; MS m/z - 402/404 [M - 1]−.
【0046】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7−ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸メチルエステル
【化9】
3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボン酸(1当量、2.11g、5.2mmol)およびブロモ−トリス−ピロリジノ−ホスホニウム ヘキサフルオロホスフェート(PyBrOP;1.1当量、2.66g、5.7mmol)をジクロロメタン(45mL)に懸濁し、混合物を20℃で10分間撹拌した。N,N−ジイソプロピルエチルアミン(7当量、6.34mL、36.4mmol)を混合物に加え、さらに15分間撹拌した。H−L−アルギニン(Pbf)−OMe(塩酸塩;1.1当量、7g、14.8mmol)を一度に加え、反応混合物(いくらかの白色の沈殿物を含む)を20℃で18時間撹拌した。次いで溶媒を減圧除去し、得られた残渣を酢酸エチル(60mL)に溶解し、1M 塩酸(40 mL)で分液処理した。水層を分離し、有機層をさらに1M 塩酸のアリコート(3x40L)で洗浄した。有機層をブライン(飽和、水溶液;50mL)で洗浄し、硫酸マグネシウムで乾燥し、濾過し、溶媒を減圧除去した。粗生成物(灰白色の泡状物質;約4.5g)をシリカゲルフラッシュカラムクロマトグラフィー(溶出液:酢酸エチル/イソ−ヘキサン;50:50、酢酸エチルのみ増加)で精製し、目的生成物を灰白色の固形物として得た(3.38g、79%)。
LC-MS 保持時間:4.77分;純度:95 %; MS m/z - 826/828 [M + 1]+.
【0047】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7−ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸
【化10】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7‐ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸 メチルエステル(1当量、2.45g、2.96mmol)を1M 水酸化リチウム(5当量、14.82mL、14.82mmol)/テトラヒドロフラン(29mL)と共に20℃で3時間撹拌した。次いで有機溶媒を減圧除去し、有機溶媒を減圧除去し、(水性の)残渣を水(30mL)で希釈し、6M 塩酸でpH1に酸性化した。酢酸エチル(200mL)を得られた懸濁液に加え、撹拌後、有機層を分離した。水層をさらに酢酸エチル(150mL)で抽出し、有機抽出物を合わせ、ブライン(飽和、水溶液;3x75mL)で洗浄し、硫酸マグネシウムで乾燥し、濾過し、減圧除去した。生成物(淡黄色の泡状物質、2.42g、100%)をさらに精製することなく用いた。
LC-MS 保持時間:4.89分;純度:90 %; MS m/z - 812/814 [M + 1]+.
【0048】
合成ボロン酸の一般的製造方法
【化11】
適当なホルミル−フェニルボロン酸(1.2当量)およびアミン(1当量)を合わせ、ジクロロメタン(15mL)に溶解した。酢酸(0.2mL)を加え、反応物を周囲温度で2時間撹拌した。次いで、ナトリウム シアノボロヒドリド(2当量)を一度に加え、反応物を20℃でさらに20時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製されたボロン酸を溶媒のエバポレートにより単離し、さらに精製することなく用いた。
【0049】
液相鈴木カップリング(カルボン酸)の一般的方法
【化12】
(S)−2−{[3−(5−ブロモ−2,3−ジヒドロ−ベンゾフラン−7−スルホニルアミノ)−チオフェン−2−カルボニル]−アミノ}−5−(2,2,4,6,7‐ペンタメチル−2,3−ジヒドロ−ベンゾフラン−5−スルホニル−グアニジノ)−ペンタン酸(約1g、1.0当量)、対応するボロン酸(1.5当量)およびテトラキス(トリフェニルホスフィン)パラジウム(0)(0.05当量)を脱気した1,2−ジメトキシエタン(3mL)に懸濁した。脱気したリン酸化リウム(三塩基、2M水溶液、4当量)をさらに加え、マイクロ波(100ワット、90℃、照射時間=10分間)を用いて反応混合物を加熱した。次いで、溶媒を減圧除去し、得られた残渣を酢酸エチル(200mL)および塩酸(1M水溶液;150mL)で分液処理した。相を分離し、水相をさらに酢酸エチル(200mL)で抽出した。有機抽出物を合わせ、ブライン(飽和、水溶液;2x100mL)で洗浄し、硫酸マグネシウムで乾燥し、濾過し、溶媒を減圧除去した。粗生成物(通常は黄色の固形物;約1.5g)をシリカゲルフラッシュカラムクロマトグラフィー(溶出液:ジクロロメタンからジクロロメタン/メタノール 75:25に漸増)で精製し、表1にまとめられるような生成物を得た。
【表1−1】
【表1−2】
【0050】
メチルエステル加水分解およびPbf除去の一般的方法
【化13】
撹拌したメチルエステル(1当量)の1,4−ジオキサン(1.2mL)懸濁液に1M 水酸化リチウム(水溶液、4当量)および水(1.2mL)を加えた。反応物を20℃で24時間撹拌し、反応物をエバポレートして乾燥して白色の固形物を得、さらに精製することなく用いた。
残渣をジクロロメタン/トリフルオロ酢酸(1:1、5mL)に溶解し、室温で1時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1%ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法を用いて作製した最終化合物を表2にまとめる。
【表2】
【0051】
Pbf除去の一般的方法
【化14】
残渣をジクロロメタン/トリフルオロ酢酸(1:1、5mL)に溶解し、室温で1時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製されたペプチドミメティックを溶媒のエバポレートにより単離した。
【表3】
【0052】
還元的アミノ化およびPbf除去の一般的方法
【化15】
アルデヒド(1当量)のテトラヒドロフラン/メタノール(1:1、1.5mL)溶液にアミン(市販;1.1当量)、次いで酢酸(1−2滴、〜pH6)を加えた。反応物を20℃で2時間撹拌し、ナトリウム シアノボロヒドリド(2当量)/メタノール(0.1mL)を一度に加えた。反応物を20℃でさらに16時間撹拌した。反応物を前処理したSCX−2(1g)カートリッジに通して濾過し、生成物を2M アンモニア/メタノールで溶出した。溶媒をエバポレートして生成物を黄色の油状物として得、それをジクロロメタン/トリフルオロ酢酸(1:1、8mL)に溶解し、20℃で1時間撹拌した。溶媒を減圧除去し、粗残渣をジメチルスルホキシドに溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、i.溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール、またはii.溶出液A:10mM 炭酸水素アンモニウム(pH9)、溶出液B:100% メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法を用いて作製した最終化合物を表4にまとめる。
【表4−1】
【表4−2】
【表4−3】
【表4−4】
【表4−5】
【表4−6】
【表4−7】
【表4−8】
【表4−9】
【表4−10】
【表4−11】
【表4−12】
【表4−13】
【0053】
アミノ−チアゾールアルデヒド形成の一般的方法
【化16】
ブロモ−チアゾール(1当量)、アミン(市販;1.1当量)および水酸化リチウム(1.15当量)を合わせ、テトラヒドロフラン(2mL)/水(0.1mL)に溶解した。マイクロ波で反応混合物を75℃で15分間加熱した。次いで、水(5mL)を反応混合物に加え、塩酸(1M、水溶液)を用いてpHを約7に調整した。溶媒を減圧除去し、ジメチルスルホキシドに再溶解し、さらに精製することなく用いるか、あるいはプレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1%)を用いた(質量分析連結)プレパラティブLC−MSで精製した。これらの方法を用いて作製されたチアゾールアルデヒドを表5にまとめる。
【表5−1】
【表5−2】
【表5−3】
【0054】
液相還元的アミノ化の一般的方法
【化17】
アニリン(1当量)、アルデヒド(市販または前記のようにして製造;0.5−6.0当量)およびナトリウム シアノボロヒドリド(0.6−2.0当量)を合わせ、メタノール(2mL)に溶解した。塩酸(1M、水溶液)または酢酸をpHが5−6の間になるまで加え、反応混合物を20℃で16時間撹拌した。溶媒を減圧除去し、得られた残渣をジメチルスルホキシドに再溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法により作製した化合物を表6にまとめる。
【表6−1】
【表6−2】
【表6−3】
【表6−4】
【0055】
アミノエチル−ピラゾールアルデヒド形成およびそれに次ぐ還元的アミノ化の一般的方法
【化18】
1−(2−クロロエチル)−1H−ピラゾール−4−カルボアルデヒド(4.2当量)、アミン(5当量)およびトリエチルアミン(8.4当量)をN−メチル−2−ピロリドン(1mL)と共に85℃で16時間加熱した。溶媒を減圧除去し、アニリン(1当量)/メタノール(1mL)、ナトリウム シアノボロヒドリド(2.0当量)を加えた。次いで、塩酸(1M、水溶液)をpHが5−6の間になるまで加え、反応混合物を20℃で16時間撹拌した。メタノールを減圧除去し、混合物をジメチルスルホキシドで希釈し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離し、さらに精製することなく用いた。これらの方法により作製された化合物を表7にまとめる。
【表7】
【0056】
ブロモ−チアゾールによる置換反応の一般的方法
【化19】
ブロモ−チアゾール(1当量)および1−(2−ヒドロキシエチル)ピペラジン(10当量)を合わせ、トリエチルアミン(10当量)およびN−メチル−2−ピロリドン(2mL)に溶解し、24時間加熱還流した。混合物を周囲温度に冷却し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。これらの方法により作製した化合物を表8にまとめる。
【表8】
【0057】
液相アルキル化の一般的方法
【化20】
アニリン(1当量)をメタノール(2mL)またはジメチルスルホキシド(2mL)に溶解した。アルキルハライド(1当量)、次いで、トリエチルアミン(2当量)を加え、反応混合物を50−100℃で16時間撹拌した。メタノールを溶媒をとして用いた場合、メタノールを減圧除去し、得られた残渣をジメチルスルホキシドに再溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)、および直線ABグラジエント(5−95% B、12分間、流速20mL/分、溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/メタノール)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離し、さらに精製することなく用いた。この方法を用いて作製した化合物を表9にまとめる。
【表9】
【0058】
メチルエステル加水分解およびPbf/Bocの除去の一般的方法
【化21】
完全に保護した出発物質(1当量)を水酸化リチウム(5当量)/テトラヒドロフラン/水(4:1;2.5mL)と共に、必要に応じ、20−60℃で1−3時間撹拌した。次いで溶媒を減圧除去し、残渣をトリフルオロ酢酸(2mL)および水(0.1mL)で処理した。反応混合物を20℃でさらに3−16時間撹拌した。溶媒を減圧除去し、得られた残渣をジメチルスルホキシドに再溶解し、プレパラティブC−18カラム(Phenomenex Luna C18 (2), 100 x 21.2 mm, 5 μM)および直線ABグラジエント(2−95% B、12分間、流速20mL/分、i.溶出液A:0.1% ギ酸/水、溶出液B:0.1% ギ酸/アセトニトリル;またはii.溶出液A:10mM 炭酸水素アンモニウム(pH9)、溶出液B:メタノールのみ)を用いた(質量分析連結)プレパラティブLC−MSで精製した。精製したペプチドミメティックを溶媒のエバポレートにより単離した。この方法で作製した化合物を表10にまとめる。
【表10−1】
【表10−2】
【表10−3】
【表10−4】
【表10−5】
【表10−6】
【0059】
インビトロにおけるNP−1の結合の評価およびインビボ腫瘍モデルにおける研究
いくつかの化合物についてNP−1の結合を評価した。1個の化合物(化合物58)について、ヒト肺癌細胞の異種移植片を負荷されたマウスモデルにおいて抗癌活性を評価した。実験方法および結果を以下に示す。
【0060】
一般的実験方法
細胞培養およびアデノウィルスを介したNP−1トランスフェクション
ヒト前立腺癌 DU145細胞を成長培地(10% FBSおよびL−グルタミン含有RPMI 1640)中で培養した。DU145細胞を2x104細胞/ウェル(96ウェルプレート)の密度において0.1mlの成長培地を用いて播種し、ヒトNP−1の翻訳領域の全長を含むアデノウィルスベクターをトランスフェクションした。Ad.NP−1−トランスフェクション細胞を結合アッセイに先立って2日間培養した。
【0061】
細胞におけるビオチン化VEGF−A165の結合
96ウェルプレート内のコンフルーエントなAd.NP−1−トランスフェクション細胞をリン酸緩衝生理食塩水(PBS)で2回洗浄した。結合培地(ダルベッコ改変イーグル培地、25mM HEPES、pH7.3、0.1% BSA含有)で希釈した異なる濃度の化合物を加え、次いで2nMのbt−VEGF−A165を加えた。室温で2時間インキュベーション後、培地を吸引し、PBSで3回洗浄した。NP−1に結合したbt−VEGF−A165を、ストレプトアビジン−西洋ワサビペルオキシダーゼ複合体および酵素基質により検出し、Tecan Geniosプレートリーダー(A450nm、リファレンス波長:A595nm)で測定した。非特異的結合は、100倍過剰量の非標識VEGF−A165の存在下で決定した。
【0062】
無細胞系におけるビオチン化VEGF−A165の結合
96ウェルプレートを前もってNP−1タンパク(3μg/ml)により終夜4℃でコーティングした。翌日、プレートをブロッキングバッファー(1% BSA含有PBS)で処理し、洗浄バッファー(0.1% Tween−20含有PBS)で3回洗浄した。1% DMSO含有PBSで希釈した異なる濃度の化合物を加え、次いで、0.25nMのbt−VEGF−A165を加えた。室温で2時間インキュベーション後、プレートを洗浄バッファーで3回洗浄した。NP−1に結合したbt−VEGF−A165をストレプトアビジン−西洋ワサビペルオキシダーゼ複合体および酵素基質で検出し、Tecan Geniosプレートリーダー(A450nm、リファレンス波長:A595nm)で測定した。非特異的結合は、プレートのNP−1コーティングウェルの非存在下で決定した。
【0063】
結合実験の結果を下の表11に示す。
【表11−1】
【表11−2】
【0064】
肺癌の生物学的研究(インビボ)
化合物58もまた、肺癌の前臨床モデルにおいて、研究概念の実証(proof of principle study)に成功した。化合物58は腫瘍増殖の速度を有意に低下させ、毒性の証拠は示さなかった。
肺癌マウスモデルにおけるこの前臨床の研究概念の実証では、1日1回投与において2週間投与された化合物58が、腫瘍増殖速度を52%(p=0.017)低下させることが示された。先に行われた高用量の毒性実験の結果と一致して、この研究では毒性の証拠は見られなかった。
【0065】
方法
有効性研究用に、ヒト肺非小細胞癌A549細胞を成長培地RPMI 1640中で培養した。90%コンフルーエントの細胞を剥し、細胞数をカウントし、PBSに懸濁し、接種用に最終細胞濃度を5x107/mlとした。
【0066】
動物研究
A549細胞をメスBalb/cヌードマウスに接種してから2週間後、化合物の投与を開始した。化合物58を1日当たり80mg/kgの投与量において2週間投与した。2週間の期間中、デジタルノギス(Fisher Scientific)を用いて腫瘍の長さおよび幅を毎日測定することにより、腫瘍容積をモニターした。腫瘍容積は式(長さx幅2/2)を用いて算出した。実験の終了時、腫瘍を解剖し、重量を測定した。
【0067】
結果
インビボ研究の結果を図1に示す。
図1のデータにより、化合物58処理群において腫瘍増殖速度が50%低下した(p=0.017、直線回帰分析)。さらに、腫瘍重量は27%減少した(p=0.04、マン・ホイットニー検定)。
【特許請求の範囲】
【請求項1】
式(I):
【化1】
[式中:
Wはアリーレン、ヘテロアリーレン、または
【化2】
であり;
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、アルキレン−C=Oまたは−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR2、OR1、CNまたはCO2Rであり;
Z1は、
【化3】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは2から5であり;
mは1から3である]
の化合物、またはその医薬的に許容される塩。
【請求項2】
Wがアリーレンである、請求項1に記載される化合物。
【請求項3】
式(II):
【化4】
[式中:
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、またはアルキレン−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR、OR1、CNまたはCO2Rであり;
Z1は、
【化5】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは0から5であり;
mは1から3である]
の化合物、またはその医薬的に許容される塩。
【請求項4】
少なくとも1つのLがアルキレンである、請求項1から3のいずれかに記載される化合物。
【請求項5】
少なくとも1つのLが直接結合である、請求項1から4のいずれかに記載される化合物。
【請求項6】
少なくとも1つのLがアリーレンである、請求項1から5のいずれかに記載される化合物。
【請求項7】
少なくとも1つのXがNRである、請求項1から6のいずれかに記載される化合物。
【請求項8】
少なくとも1つのXが少なくとも1個のN原子を含む6員のシクロアルキレンである、請求項1から7のいずれかに記載される化合物。
【請求項9】
Yが少なくとも1個のN原子を含む6員のシクロアルキルである、請求項1から8のいずれかに記載される化合物。
【請求項10】
Yが、少なくとも1個のN原子、ならびに好ましくはOまたはSおよびNから選択される1個の別の原子を含む5員のヘテロアリールである、請求項1から8のいずれかに記載される化合物。
【請求項11】
Yがピリジンである、請求項1から8のいずれかに記載される化合物。
【請求項12】
YがC6H4CNである、請求項1から8のいずれかに記載される化合物。
【請求項13】
nが3、4または5である、請求項1から12のいずれかに記載される化合物。
【請求項14】
mが2または3である、請求項1から13のいずれかに記載される化合物。
【請求項15】
mが1である、請求項1から13のいずれかに記載される化合物。
【請求項16】
請求項1から15のいずれかに記載される化合物、および明細書中に例示され、化合物1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76または77と呼ばれる化合物。
【請求項17】
請求項1から16のいずれかに記載される化合物、ならびに医薬的に許容される賦形剤を含む医薬組成物。
【請求項18】
治療に用いるための、請求項1から17のいずれかに記載される化合物または組成物。
【請求項19】
神経修復促進または神経変性治療に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項20】
血小板凝集阻害に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項21】
癌の治療に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項22】
免疫系の調節に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項23】
HTLVIの治療に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項24】
放射線イメージングに用いるための、または磁気共鳴画像法における造影剤として用いるための、請求項1から16のいずれかに記載される化合物ならびにi)放射性核種;またはii)常磁性核種および該常磁性核種と錯体形成するためのキレートを含む組成物。
【請求項1】
式(I):
【化1】
[式中:
Wはアリーレン、ヘテロアリーレン、または
【化2】
であり;
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、アルキレン−C=Oまたは−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR2、OR1、CNまたはCO2Rであり;
Z1は、
【化3】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは2から5であり;
mは1から3である]
の化合物、またはその医薬的に許容される塩。
【請求項2】
Wがアリーレンである、請求項1に記載される化合物。
【請求項3】
式(II):
【化4】
[式中:
各Lは独立して、アルキレン、アルケニレン、アルキニレン、直接結合、アリーレン、シクロアルキレン、アルキレン−アリーレン、またはアルキレン−C=Oであり;
各Xは独立して、N含有ヘテロアリーレン、N含有シクロアルキレンまたはNRであり;
YはN含有ヘテロアリール、N含有シクロアルキル、NR、OR1、CNまたはCO2Rであり;
Z1は、
【化5】
であり;
RはHまたはC1−C6アルキルであり;
R1はH、C1−C6アルキルまたはアミノ酸であり;
nは0から5であり;
mは1から3である]
の化合物、またはその医薬的に許容される塩。
【請求項4】
少なくとも1つのLがアルキレンである、請求項1から3のいずれかに記載される化合物。
【請求項5】
少なくとも1つのLが直接結合である、請求項1から4のいずれかに記載される化合物。
【請求項6】
少なくとも1つのLがアリーレンである、請求項1から5のいずれかに記載される化合物。
【請求項7】
少なくとも1つのXがNRである、請求項1から6のいずれかに記載される化合物。
【請求項8】
少なくとも1つのXが少なくとも1個のN原子を含む6員のシクロアルキレンである、請求項1から7のいずれかに記載される化合物。
【請求項9】
Yが少なくとも1個のN原子を含む6員のシクロアルキルである、請求項1から8のいずれかに記載される化合物。
【請求項10】
Yが、少なくとも1個のN原子、ならびに好ましくはOまたはSおよびNから選択される1個の別の原子を含む5員のヘテロアリールである、請求項1から8のいずれかに記載される化合物。
【請求項11】
Yがピリジンである、請求項1から8のいずれかに記載される化合物。
【請求項12】
YがC6H4CNである、請求項1から8のいずれかに記載される化合物。
【請求項13】
nが3、4または5である、請求項1から12のいずれかに記載される化合物。
【請求項14】
mが2または3である、請求項1から13のいずれかに記載される化合物。
【請求項15】
mが1である、請求項1から13のいずれかに記載される化合物。
【請求項16】
請求項1から15のいずれかに記載される化合物、および明細書中に例示され、化合物1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、31、32、33、34、35、36、37、38、39、40、41、42、43、44、45、46、47、48、49、50、51、52、53、54、55、56、57、58、59、60、61、62、63、64、65、66、67、68、69、70、71、72、73、74、75、76または77と呼ばれる化合物。
【請求項17】
請求項1から16のいずれかに記載される化合物、ならびに医薬的に許容される賦形剤を含む医薬組成物。
【請求項18】
治療に用いるための、請求項1から17のいずれかに記載される化合物または組成物。
【請求項19】
神経修復促進または神経変性治療に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項20】
血小板凝集阻害に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項21】
癌の治療に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項22】
免疫系の調節に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項23】
HTLVIの治療に用いるための、請求項1から18のいずれかに記載される化合物または組成物。
【請求項24】
放射線イメージングに用いるための、または磁気共鳴画像法における造影剤として用いるための、請求項1から16のいずれかに記載される化合物ならびにi)放射性核種;またはii)常磁性核種および該常磁性核種と錯体形成するためのキレートを含む組成物。
【図1】

【公表番号】特表2013−503146(P2013−503146A)
【公表日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願番号】特願2012−526129(P2012−526129)
【出願日】平成22年8月25日(2010.8.25)
【国際出願番号】PCT/GB2010/051413
【国際公開番号】WO2011/024001
【国際公開日】平成23年3月3日(2011.3.3)
【出願人】(500175668)アーク・セラピューティックス・リミテッド (13)
【氏名又は名称原語表記】Ark Therapeutics Limited
【Fターム(参考)】
【公表日】平成25年1月31日(2013.1.31)
【国際特許分類】
【出願日】平成22年8月25日(2010.8.25)
【国際出願番号】PCT/GB2010/051413
【国際公開番号】WO2011/024001
【国際公開日】平成23年3月3日(2011.3.3)
【出願人】(500175668)アーク・セラピューティックス・リミテッド (13)
【氏名又は名称原語表記】Ark Therapeutics Limited
【Fターム(参考)】
[ Back to top ]