
Nogo受容体アンタゴニスト
免疫原性Nogo受容体−1ポリペプチド、Nogo受容体−1抗体、その抗原結合断片、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸が開示される。このようなNogo受容体抗体、その抗原結合断片、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸を含む組成物、ならびに、このようなNogo受容体抗体、その抗原結合断片、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸を作成し、使用するための方法もまた、開示される。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の分野)
本発明は、神経生物学および分子生物学に関する。さらに具体的には、本発明は、免疫原性Nogo受容体−1ポリペプチド、Nogo受容体−1抗体、その抗原結合断片、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸に関する。本発明はさらに、このようなNogo受容体抗体、その抗原結合断片、免疫原性Nogo受容体−1ポリペプチド、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸を含む組成物、さらには、このようなNogo受容体抗体、その抗原結合断片、免疫原性Nogo受容体−1ポリペプチド、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸を作成し、使用するための方法に関する。
【背景技術】
【0002】
(発明の背景)
ニューロンの軸索および樹状突起は、ニューロンからの長い細胞の伸張である。伸張途中の軸索または神経突起の先端部には、成長円錐として知られている特殊な領域が含まれている。成長円錐は、局所環境を感知し、ニューロンの標的細胞に向かっての軸索の成長を導く。成長円錐は、いくつかの環境による合図、例えば、表面接着、成長因子、神経伝達物質、および電場に反応する。円錐での成長の誘導には、種々のクラスの接着分子、細胞内シグナル、さらには、成長円錐を刺激し阻害する因子が関与している。成長途中にある神経突起の成長円錐は種々の速度で前進するが、通常は、1日あたり1から2ミリメートルの速度で前進する。
【0003】
成長円錐は手の形状であり、広がった扁平な伸張(微小突起または糸状仮足)を有しており、これは胚の中の表面に対して別々に接着する。糸状仮足は、絶えず活動しており、一部の糸状仮足は、成長円錐の内部へと引っ込み、一方、他の糸状仮足は、基体を通り抜けて伸張し続ける。種々の糸状仮足の間での伸張により、葉状仮足が形成される。
【0004】
成長円錐は、その葉状仮足と糸状仮足を用いて、その前方と両側の領域を探索する。伸張によって、成長に好ましくない表面と接触すると、それは引っ込む。伸張によって、好ましい成長表面に接触すると、それは伸張し続け、その方向に成長円錐を導く。成長円錐は基体の表面レパートリーにおける小さなバリエーションによって導かれ得る。成長円錐が適切な標的細胞に到達すると、シナプス結合が作成される。
【0005】
神経細胞の機能は、その当面の環境におけるニューロンと他の細胞との間での接触によって大きく影響される(U.Rutishauser,T.M.Jessell,Physiol.Rev.1988,68,p.819)。これらの細胞としては、特定の神経膠細胞、中枢神経系(CNS)の乏突起膠細胞、および末梢神経系(PNS)のシュワン細胞(これは、神経軸索をミエリン鞘で覆う(多層膜の絶縁構造))(G.Lemke,An Introduction to Molecular Neurobiology,Z.Hall,Ed.[Sinauer,Sanderland,Mass.,1992],p.281)が挙げられる。
【0006】
CNSニューロンは、損傷後に再生する能力を有しているが、この能力は、ミエリンの中に存在する阻害タンパク質が原因であり、そして、それらの局所環境に通常見られる他のタイプの分子もおそらく原因で、再生が阻害される(Brittis and Flanagan,Neuron 2001,30,pp.11−14;Jones et al.,J.Neurosci.2002,22,pp.2792−2803;Grimpe et al.,J.Neurosci.2002,22,pp.3144−3160)。
【0007】
乏突起膠細胞上に見られるいくつかのミエリン阻害タンパク質、例えば、NogoA(Chen et al.,Nature,2000,403,434−439;Grandpre et al.,Nature 2000,403,439−444)、ミエリン関連糖タンパク質(MAG,McKerracher et al.,Neuron 1994,13,805−811;Mukhopadhyay et al.,Neuron 1994,13,757−767)、および乏突起膠細胞糖タンパク質(OM−gp、Mikol and Stefansson,J.Cell.Biol.1988,106,1273−1279)の特性が明らかにされている。これらのタンパク質は、それぞれ、ニューロンのNogo受容体−1のリガンドであることが別々に示されている(Wang et al.,Nature 2002,417,941−944;Liu et al.,Science,2002,297,1190−93;Grandpre et al.,Nature 2000,403,439−444;Chen et al.,Nature,2000,403,434−439;Domeniconi et al.,Neuron,2002,35,283−90)。
【0008】
Nogo受容体−1は8個のロイシンを多く含む繰り返しを含む、GPI結合膜タンパク質である(Fournier et al.,Nature 2001,409,341−346)。阻害タンパク質(例えば、NogoA、MAG、およびOM−gp)と相互作用すると、Nogo受容体−1複合体は、成長円錐の崩壊および神経突起成長の阻害を導くシグナルを変換する。
【発明の開示】
【発明が解決しようとする課題】
【0009】
そのリガンドに対するNogo受容体−1の結合を阻害し、ミエリンによって媒介される成長円錐の崩壊と、神経突起成長の阻害を弱める分子が、緊急に必要とされている。
【課題を解決するための手段】
【0010】
(発明の要旨)
本発明は、可溶性Nogo受容体−1ポリペプチドおよびそれを含む融合タンパク質、ならびに、Nogo受容体−1の特異的な免疫原性領域に対する抗体およびその抗原性断片に関する。本発明はまた、本発明の抗体に結合する免疫原性Nogo受容体−1ポリペプチドにも関する。本発明はまた、Nogo受容体−1に結合するモノクローナル抗体によって結合されるNogo受容体−1ポリペプチドにも関する。このようなポリペプチドは、特に、免疫原として、または、本発明の抗体に対して同様の特異性を有している抗体を同定するために抗体をスクリーニングするために使用することができる。本発明はさらに、本発明のポリペプチドをコードする核酸、そのような核酸を含むベクターおよび宿主細胞、ならびにペプチドを作成する方法に関する。本発明の抗体、可溶性受容体、および受容体融合タンパク質は、Nogo受容体−1をアンタゴナイズするかまたはブロックし、そしてそのリガンドに対するNogo受容体−1の結合を阻害すること、ニューロンにおける成長円錐の崩壊を阻害すること、およびニューロンにおける神経突起成長または萌芽(sprouting)の阻害を減少させることについて有用である。
【0011】
いくつかの実施形態においては、本発明により、AAAFGLTLLEQLDLSDNAQLR(配列番号26);LDLSDNAQLR(配列番号27);LDLSDDAELR(配列番号29);LDLASDNAQLR(配列番号30);LDLASDDAELR(配列番号31);LDALSDNAQLR(配列番号32);LDALSDDAELR(配列番号33);LDLSSDNAQLR(配列番号34);LDLSSDEAELR(配列番号35);DNAQLRVVDPTT(配列番号36);DNAQLR(配列番号37);ADLSDNAQLRVVDPTT(配列番号41);LALSDNAQLRVVDPTT(配列番号42);LDLSDNAALRVVDPTT(配列番号43);LDLSDNAQLHVVDPTT(配列番号44);およびLDLSDNAQLAVVDPTT(配列番号45)からなる群より選択されるポリペプチドが提供される。
【0012】
いくつかの実施形態においては、本発明により、本発明のポリペプチドをコードする核酸が提供される。いくつかの実施形態においては、核酸は、発現制御配列に動作可能であるように連結される。いくつかの実施形態においては、本発明により、本発明の核酸を含むベクターが提供される。
【0013】
いくつかの実施形態においては、本発明により、本発明の核酸を含むか、または本発明のベクターを含む宿主細胞が提供される。いくつかの実施形態においては、本発明により、本発明の核酸またはベクターを含む宿主細胞を培養すること、および宿主細胞または培養培地からポリペプチドを回収することを含む、本発明のポリペプチドを産生する方法が提供される。
【0014】
いくつかの実施形態においては、本発明により、宿主を、本発明のポリペプチドまたは本発明の核酸を含むかまたは本発明のベクターを含む宿主細胞で免疫化し、抗体を回収する工程を含む、抗体を産生する方法が提供される。本発明により、また、この方法によって産生される抗体、またはその抗原結合断片も提供される。
【0015】
いくつかの実施形態においては、本発明により、本発明のポリペプチドに特異的に結合する抗体またはその抗原結合断片が提供される。ここでは、抗体はハイブリドーマ細胞株HB 7E11によって産生されるモノクローナル抗体(ATCC(登録商標)登録番号PTA−4587)ではない。
【0016】
本発明のいくつかの実施形態においては、抗体または抗原結合断片は、(a)ニューロンの神経円錐の崩壊を阻害する;(b)ニューロンにおける神経突起成長および萌芽の阻害を減少させる;ならびに(c)リガンドに対するNogo受容体−1の結合を阻害する。いくつかの実施形態においては、神経突起成長および萌芽は、軸索成長である。いくつかの実施形態においては、ニューロンは中枢神経系のニューロンである。
【0017】
いくつかの実施形態においては、本発明の抗体はモノクローナルである。いくつかの実施形態においては、本発明の抗体はマウス抗体である。いくつかの実施形態においては、本発明の抗体は、ヒト化抗体、キメラ抗体、および単鎖抗体からなる群より選択される。
【0018】
いくつかの実施形態においては、本発明により、リガンドへのNogo受容体−1の結合を阻害する方法が提供される。この方法には、Nogo受容体−1を本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、リガンドは、NogoA、NogoB、NogoC、MAG、およびOM−gpからなる群より選択される。
【0019】
いくつかの実施形態においては、本発明により、ニューロンにおける神経円錐の崩壊を阻害するための方法が提供される。この方法には、ニューロンを、本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。
【0020】
いくつかの実施形態においては、本発明により、ニューロンにおける神経突起成長および萌芽の阻害を減少させるための方法が提供される。この方法には、ニューロンを、本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、神経突起成長または萌芽は、軸索成長である。いくつかの実施形態においては、ニューロンは中枢神経系のニューロンである。
【0021】
いくつかの実施形態においては、本発明により、薬学的に許容される担体と、本発明の抗体またはその抗原結合断片を含む組成物が提供される。いくつかの実施形態においては、組成物にはさらに、1つ以上のさらなる治療薬が含まれる。
【0022】
いくつかの実施形態においては、本発明により、死滅の危険性があるニューロンの生存を促進する方法が提供される。この方法には、ニューロンを、有効量の本発明の抗Nogo受容体−1抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、ニューロンは生体外にある。いくつかの実施形態においては、ニューロンは哺乳動物中に存在する。いくつかの実施形態においては、哺乳動物は、多発性硬化症、ALS、ハンチントン病、またはアルツハイマー病、パーキンソン病、糖尿病性神経障害、脳卒中、外傷性脳損傷、または脊髄損傷の兆候または症状を呈する。
【0023】
いくつかの実施形態においては、本発明により、哺乳動物において死滅の危険性があるニューロンの生存を促進する方法が提供される。この方法には、(a)本発明の抗Nogo受容体−1抗体またはその抗原結合断片を発現する培養された宿主細胞を提供すること;および(b)ニューロンの部位またはその付近に哺乳動物中に宿主細胞を導入することが含まれる。
【0024】
いくつかの実施形態においては、本発明により、哺乳動物中にある死滅の危険性があるニューロンの生存を促進する遺伝子治療方法が提供される。この方法には、本発明の抗Nogo受容体−1抗体またはその抗原結合断片をコードするヌクレオチド配列を含むウイルスベクターを、ニューロンの部位またはその付近に投与することが含まれる。ここでは、抗Nogo受容体−1抗体または抗原結合断片は、哺乳動物中のヌクレオチド配列から、ニューロンの生存を促進するために十分な量で発現される。
【発明を実施するための最良の形態】
【0025】
(発明の詳細な説明)
(定義および一般的な技術)
他の場所で定義されない限りは、本明細書中で使用される全ての技術用語および科学用語は、本発明が属する分野の当業者によって通常理解されている意味と同じ意味を有する。矛盾する場合は、本出願が含む定義が支配する。また、他の方法で状況によって必要とされない限りは、単数形の用語に複数形が含まれ、複数形の用語に単数形が含まれる。本明細書中で言及される全ての刊行物、特許、および他の参考文献は、全ての目的についてそれらの全体が引用によって本明細書中に組み入れられる。
【0026】
本明細書中に記載されるものと同様であるかまたはそれらと同等である方法および材料を、本発明の実施または試験において使用することができるが、適切な方法および材料は以下に記載される。材料、方法、および実施例は、例示的なものにすぎず、限定を意図するものではない。本発明の他の特徴および利点は、詳細な説明から、そして特許請求の範囲から明らかであろう。
【0027】
本明細書および特許請求の範囲全体を通じて、語「含まれる(comprise)」、または「含む(comprises)」もしくは「含んでいる(comprising)」のようなバリエーションは、記載される整数、または整数のグループの包含を暗に意味するが、任意の他の整数または整数のグループを排除するようには理解されない。
【0028】
本発明をさらに定義するために、以下の用語と定義が本明細書において提供される。
【0029】
本明細書中で使用される場合は、「抗体」は、完全な免疫グロブリン、またはその抗原結合断片を意味する。本発明の抗体は、任意のイソ型またはクラス(例えば、M、D、G、E、およびA)、あるいは、任意のサブクラス(例えば、G1−4、A1−2)であり得、そしてカッパ(κ)軽鎖またはラムダ(λ)軽鎖のいずれかを有し得る。
【0030】
本明細書中で使用される場合は、「Fc」は、パパインでの消化によって得ることができる抗体の重鎖定常領域の部分を意味する。
【0031】
本明細書中で使用される場合は、「NogoR融合タンパク質」は、異種ポリペプチドに融合させられた可溶性Nogo受容体−1部分を含むタンパク質を意味する。
【0032】
本明細書中で使用される場合は、「ヒト化抗体」は、ヒト以外の配列の少なくとも一部がヒト配列で置き換えられている抗体を意味する。ヒト化抗体を作成するための方法の例は、米国特許第6,054,297号、同第5,886,152号、および同5,877,293号に見ることができる。
【0033】
本明細書中で使用される場合は、「キメラ抗体」は、第1の抗体に由来する1つ以上の領域と、少なくとも1つの他の抗体に由来する1つ以上の領域を含む抗体を意味する。第1の抗体とさらなる抗体は、同じ種に由来するものであっても、異なる種に由来するものであってもよい。
【0034】
本明細書中、および米国特許出願番号第60/402,866において使用される場合は、「Nogo受容体」、「NogoR」、「NogoR−1」、「NgR」、および「NgR−1」はそれぞれ、Nogo受容体−1を意味する。
【0035】
(Nogo受容体−1ポリペプチド)
1つの態様においては、本発明は、免疫原性であるNogo受容体−1ポリペプチドに関する。本発明のいくつかの実施形態においては、免疫原性ポリペプチドは、LDLSDNAQLRVVDPTT(ラット)(配列番号1);LDLSDNAQLRSVDPAT(ヒト)(配列番号2);AVASGPFRPFQTNQLTDEELLGLPKCCQPDAADKA(ラット)(配列番号3);AVATGPYHPIWTGRATDEEPLGLPKCCQPDAADKA(ヒト)(配列番号4);およびCRLGQAGSGA(マウス)(配列番号5)からなる群より選択されるアミノ酸配列から本質的には構成される。
【0036】
いくつかの実施形態においては、本発明は、Nogo受容体−1に結合するモノクローナル抗体によって結合されるNogo受容体−1ポリペプチドに関する。いくつかの実施形態においては、ポリペプチドは、7E11モノクローナル抗体によって認識される。いくつかの実施形態においては、ポリペプチドは、LDLSDNAQLR(配列番号28);LDLSDDAELR(配列番号30);LDLASDNAQLR(配列番号31);LDLASDDAELR(配列番号32);LDALSDNAQLR(配列番号33);LDALSDDAELR(配列番号34);LDLSSDNAQLR(配列番号35);LDLSSDEAELR(配列番号36);DNAQLRVVDPTT(配列番号37);DNAQLR(配列番号38);ADLSDNAQLRVVDPTT(配列番号42);LALSDNAQLRVVDPTT(配列番号43);LDLSDNAALRVVDPTT(配列番号44);LDLSDNAQLHVVDPTT(配列番号45);およびLDLSDNAQLAVVDPTT(配列番号46)からなる群より選択される。
【0037】
いくつかの実施形態においては、本発明は、配列番号1〜5、26〜27、29〜37、および41〜45のポリペプチドをコードする核酸に関する。本発明のいくつかの実施形態においては、核酸分子は、発現制御配列(例えば、pCDNA(I))に連結される。
【0038】
本発明はまた、本発明のポリペプチドをコードする核酸を含むベクターにも関する。本発明のいくつかの実施形態においては、ベクターはクローニングベクターである。本発明のいくつかの実施形態においては、ベクターは発現ベクターである。本発明のいくつかの実施形態においては、ベクターには、少なくとも1つの選択マーカーが含まれる。
【0039】
本発明はまた、上記の核酸またはベクターを含む宿主細胞にも関する。
【0040】
本発明はまた、宿主細胞を培養する工程を含む、本発明の免疫原性ポリペプチドを産生する方法にも関する。いくつかの実施形態においては、宿主細胞は原核生物である。いくつかの実施形態においては、宿主細胞は真核生物である。いくつかの実施形態においては宿主細胞は酵母である。
【0041】
(抗体)
本発明はさらに、本発明のNogo受容体−1ポリペプチドに特異的に結合する抗体またはその抗原結合断片に関する。いくつかの実施形態においては、抗体または抗原結合断片は、配列番号1〜5、26〜27、29〜37、および41〜45からなる群より選択されるアミノ酸配列から本質的には構成されるポリペプチドに結合する。本発明の抗体または抗原結合断片は、生体内または生体外で産生することができる。抗体または抗原結合断片の産生については以下で議論される。
【0042】
本発明の抗体またはその抗原結合断片は、リガンド(例えば、NogoA、NogoB、NogoC、MAG、OM−gp)に対するNogo受容体−1の結合を阻害し、神経突起成長および萌芽、具体的には、軸索成長のミエリンによって媒介される阻害を減少させ、ミエリンによって媒介される成長円錐の崩壊を弱める。
【0043】
いくつかの実施形態においては、抗Nogo受容体−1抗体またはその抗原結合断片は、マウスのものである。いくつかの実施形態においては、Nogo受容体−1はラット由来のものである。他の実施形態においては、Nogo受容体−1はヒトのものである。いくつかの実施形態においては、抗Nogo受容体−1抗体またはその抗原結合断片は組み換えのもの、遺伝子操作されたもの、ヒト化されたもの、および/またはキメラである。
【0044】
いくつかの実施形態においては、抗体は、モノクローナル7E11(ATCC(登録商標)登録番号PTA−4587);モノクローナル1H2(ATCC(登録商標)登録番号PTA−4584);モノクローナル2F7(ATCC(登録商標)登録番号PTA−4585);モノクローナル3G5(ATCC(登録商標)登録番号PTA−4586);およびモノクローナル5B10(ATCC(登録商標)登録番号PTA−4588)からなる群より選択される。いくつかの実施形態においては、抗体はポリクローナル抗体46である。
【0045】
例示的な抗原結合断片は、Fab、Fab’、F(ab’)2、Fv、Fd、dAb、および相補性決定領域(CDR)断片を含む断片、単鎖抗体(scFv)、キメラ抗体、ポリペプチドに特異的抗原結合性(例えば、免疫接着体)を付与するために十分である免疫グロブリンの部分を少なくとも含むダイアボディーおよびポリペプチドである。
【0046】
本明細書中で使用される場合は、FdはVHドメインとCH1ドメインからなる断片を意味し、Fvは、抗体の単一のアームのVLドメインとVHドメインからなる断片を意味し、そしてdAbは、VHドメインからなる断片を意味する(Ward et al.,Nature 341:544−546,1989)。本明細書中で使用される場合は、単鎖抗体(scFv)は、VL領域とVH領域が、それらが1本のタンパク質鎖として作成されることを可能にする合成のリンカーを介して、一価の分子を形成するように対を形成している抗体を意味する(Bird et al.,Science 242:423−426,1988、およびHuston et al.,Proc.Natl.Acad.Sci.USA,85:5879−5883、1988)。本明細書中で使用される場合は、ダイアボディーは、VHとVLドメインが1本のポリペプチド鎖の上で発現されるが、同じ鎖の上にある2つのドメインの間で対を形成することを可能にするためには短すぎるリンカーを使用しており、それによって、これらのドメインが別の鎖の相補ドメインと対を形成するようにさせて、2つの抗原結合部位を作成する、二重特異的抗体を意味する(例えば、Holliger,P.et al.,Proc.Natl.Acad.Sci.USA,90:6444−6448,1993、およびPoljak,R.J.,et al.,Structure 2:1121−1123,1994を参照のこと)。本明細書中で使用される場合は、目的の抗原に特異的に結合する免疫接着体は、1つ以上のCDRが、共有結合によって、または非共有結合によってのいずれかで組み込まれ得る分子を意味する。
【0047】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1抗体のサブユニットポリペプチドが提供される。ここでは、サブユニットポリペプチドは、(a)重鎖またはその可変領域;および(b)軽鎖またはその可変領域からなる群より選択される。
【0048】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1抗体のサブユニットポリペプチドの、重鎖またはその可変領域、あるいは、軽鎖またはその可変領域をコードする核酸が提供される。
【0049】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1抗体の超可変領域(CDR)、またはCDRをコードする核酸が提供される。
【0050】
(免疫化)
本発明の抗体は、適切な宿主(例えば、ヒト、マウス、ラット、ヒツジ、ヤギ、ブタ、ウシ、ウマ、爬虫類、魚類、両生類を含む脊椎動物、ならびに、鳥類、爬虫類、および魚類の卵の中)の免疫化によって作成することができる。
【0051】
いくつかの実施形態においては、宿主は、本発明の免疫原性Nogo受容体−1ポリペプチドで免疫される。他の実施形態においては、宿主は、完全な、または破壊された細胞の細胞膜と会合させられたNogo受容体−1で免疫化され、本発明の抗体が、本発明のNogo受容体−1ポリペプチドに対する結合によって同定される。
【0052】
いくつかの実施形態においては、Nogo受容体−1抗原は、免疫応答を刺激するために、アジュバントとともに投与される。アジュバントは、多くの場合、抗原に対する免疫応答を誘発するために、抗原に加えて投与されることが必要とされる。これらのアジュバントは、通常、非特異的炎症を促進し、免疫化の部位への単核の食細胞の動員を伴う、不溶性または非分解性の物質である。アジュバントの例としては、フロイトのアジュバント、RIBI(ムラミルジペプチダーゼ)、ISCOM(免疫刺激複合体)またはそれらの断片が挙げられるが、これらに限定されない。
【0053】
抗体を作成するための方法の概要については、例えば、Harlow and Lane(1988),Antibodies,A Laboratory Manual,Yelton,D.E.et al.,(1981);Ann.Rev.of Biochem.,50,pp.657−80.,およびAusubel et al.,(1989);Current Protocols in Molecular Biology(New York:John Wiley & Sons)を参照のこと。本発明の免疫原性Nogo受容体−1ポリペプチドとの免疫反応性の決定は、当該分野で周知のいくつかの方法のいずれかによって行うことができ、例えば、免疫ブロットアッセイおよびELISAが含まれる。
【0054】
(抗体の産生および抗体を産生する細胞株)
本発明のモノクローナル抗体は、例えば、Harlow and Lane(1988)前出に記載されているような標準的な手順によって行うことができる。
【0055】
簡単に説明すると、適切な時点で、動物が屠殺され、リンパ節および/または脾臓B細胞が、当該分野で周知であるいくつかの技術のうちのいずれか1つによって不死化させられる。このような技術としては、例えばEBVでの形質転換、または不死化細胞株、例えば、骨髄腫細胞との融合が挙げられるが、これらに限定されない。その後、細胞は、クローンに分けられ、それぞれのクローンの上清が、本発明の免疫原性Nogo受容体−1ポリペプチドに特異的な抗体の産生について試験される。ハイブリドーマの選択、クローニング、および拡大の方法は当該分野で周知である。同様に、免疫グロブリン遺伝子のヌクレオチド配列およびアミノ酸配列を同定するための方法は当該分野で公知である。
【0056】
本発明の抗体を産生するための他の適切な技術には、リンパ球のNogo受容体−1への、もしくは本発明の免疫原性ポリペプチドへの生体外での暴露、あるいは、ファージもしくは同様のベクター中の抗体のライブラリーの選択が含まれる。Huse et al.,Science,246,pp.1275−81(1989)を参照のこと。本発明において有用である抗体は、修飾して使用することができ、また、修飾することなく使用することもできる。
【0057】
抗原(この場合は、本発明のNogo受容体−1または免疫原性ポリペプチド)および抗体は、検出可能なシグナルを提供する物質に共有結合または非共有結合のいずれかによって連結させることによって標識することができる。種々の標識および結合技術が当該分野で知られており、これらを本発明の実施において使用することができる。適切な標識としては、放射性核種、酵素、基質、補因子、阻害因子、蛍光剤、化学発光剤、磁性粒子などが挙げられるが、これらに限定されない。このような標識の使用を教示している特許としては、米国特許第3,817,837号;同第3,850,752号;同第3,939,350号;同第3,996,345号;同第4,277,437号;同第4,275,149号、および同第4,366,241号が挙げられる。また、組み換え免疫グロブリンを産生することもできる(米国特許第4,816,567号を参照のこと)。
【0058】
本発明のいくつかの実施形態においては、抗体は、複数の結合特異性を有する。例えば、二官能性抗体は、当業者に公知である多数の技術のうちのいずれか1つによって調製される。このような技術としては、ハイブリッドハイブリドーマの産生、ジスルフィド交換、化学的架橋、2つのモノクローナル抗体の間へのペプチドリンカーの付加、特定の細胞株への免疫グロブリン重鎖および軽鎖の2つのセットの導入などが挙げられる(さらに詳細な議論については下記を参照のこと)。
【0059】
本発明の抗体はまた、ヒトモノクローナル抗体である場合もあり、例えば、不死化ヒト細胞によって産生されるもの、「ヒト」抗体を産生することができるSCID−huマウスまたは他のヒト以外の動物によって産生されるものであり得る。
【0060】
(ファージディスプレイライブラリー)
本発明の抗Nogo受容体−1抗体は、組み換えの組み合わせ抗体ライブラリーをスクリーニングすることによって単離することができる。例示的な組み合わせライブラリーは、本発明の免疫原性Nogo受容体−1ポリペプチドに対する結合のためのものであり、例えば、本発明の免疫原性Nogo受容体−1ポリペプチドで免疫化した動物に由来するmRNAから調製されたVLおよびVH cDNAを使用して調製されたscFvファージディスプレイライブラリーである。このようなライブラリーを調製しスクリーニングするための方法論は当該分野で公知である。ファージディスプレイライブラリーを作成するための方法および材料は市販されている(例えば、Pharmacia Recombinant Phage Antibody System,カタログ番号27−9400−01;Stratagene SurfZAP(登録商標)ファージディスプレイキット、カタログ番号240612;およびMorphoSysによる他のもの)。抗体ディスプレイライブラリーを作成し、スクリーニングすることにおいて使用することができる方法および試薬は、他にも存在する(例えば、Ladner et al.,米国特許第5,223,409号;Kang et al.,PCT公開番号WO92/18619;Dower et al.,PCT公開番号WO91/17271;Winter et al.,PCT公開番号WO92/20791;Markland et al.,PCT公開番号WO92/15679;Breitling et al.,PCT公開番号WO93/01288;McCafferty et al.,PCT公開番号WO93/01047;Garrard et al.,PCT公開番号WO92/09690;Fuchs et al.,(1991)Bio/Technology 9:1370−1372;Hay et al.,(1992)Hum.Antibod.Hybridomas 3:81−85;Huse et al.,(1989)Science 246:1275−1281;McCafferty et al.,Nature(1990)348:552−554;Griffiths et al.,(1993)EMBO J.12:725−734;Hawkins et al.,(1992)J.Mol.Biol.,226:889−896;Clackson et al.,(1991)Nature 352:624−628;Gram et al.,(1992)Proc.Natl.Acad.Sci.USA,89:3576−3580;Garrad et al.,(1991)Bio/Technology 9:1373−1377;Hoogenboom et al.,(1991)Nucl.Acids Res.19:4133−4137;およびBarbas et al.,(1991)Proc.Natl.Acad.Sci.USA,88:7978−7982を参照のこと)。
【0061】
組み換え免疫グロブリンディスプレイライブラリーから本発明の抗Nogo受容体−1抗体をスクリーニングし単離した後、選択された抗体をコードする核酸をディスプレイパッケージから(例えば、ファージゲノムから)回収することができ、標準的な組み換えDNA技術によって他の発現ベクターにサブクローニングすることができる。所望される場合は、核酸はさらに、本発明の他の抗体形態を生じるように、以下に記載されるように操作され得る。組み合わせライブラリーのスクリーニングによって単離した抗体を発現させるためには、抗体重鎖および軽鎖、またはその可変領域をコードするDNAが、組み換え発現ベクター中にクローニングされ、そして上記のように、哺乳動物宿主細胞に導入される。
【0062】
(クラススイッチ)
本発明の抗Nogo受容体−1抗体は任意のイソ型であり得る。任意の所望されるイソ型の抗体は、クラススイッチによって産生することができる。クラススイッチのためには、CLまたはCHをコードするヌクレオチド配列のいずれをも含まない、VLまたはVHをコードする核酸が、当該分野で周知の方法を使用して単離される。VLまたはVHをコードする核酸は、その後、免疫グロブリン分子の所望されるクラスに由来するCLまたはCHをコードするヌクレオチド配列に対して動作可能であるように連結させられる。これは、ベクターと、上記のようなCLまたはCH鎖を含む核酸を使用して行うことができる。例えば、もともとIgMであった本発明の抗Nogo受容体−1抗体は、IgGへとクラススイッチさせることができる。さらに、クラススイッチを、1つのIgGサブクラスから別のサブクラスへ、例えば、IgG1からIgG2へと変換させるために使用することもできる。
【0063】
(変異させられた抗体)
他の実施形態においては、本発明の抗体または抗原結合断片は、重鎖および/または軽鎖の種々のドメインにおいて変異させることができ、それによって、抗体の結合特性を変化させることができる。例えば、変異は、1つ以上のCDR領域中に作成することができ、これによって、Nogo受容体−1についての抗体のKdを増大または低下させることができ、Koffを増大または低下させることができ、あるいは、抗体の結合特異性を変化させることができる。部位特異的変異誘発における技術は、当該分野で周知である。例えば、Sambrook et al.,およびAusubel et al.,(前出)を参照のこと。好ましい実施形態においては、変異は、本発明の抗Nogo受容体−1抗体の可変領域中の生殖系と比較して異なっていることが知られているアミノ酸残基に作成される。いくつかの実施形態においては、変異は、本発明の抗Nogo受容体−1抗体の可変領域中の生殖系と比較して異なっていることが知られている1つ以上のアミノ酸残基に作成される。別の実施形態においては、抗体の重鎖または軽鎖可変領域をコードする核酸が、1つ以上のフレームワーク領域の中で変異させられる。変異は、半減期が長くなるように、フレームワーク領域または定常ドメインに作成される場合もある。フレームワーク領域または定常ドメイン中の変異はまた、抗体の免疫原性を変化させるように作成される場合もあり、これによって、別の分子に対する共有結合または非共有結合のための部位が提供されるか、あるいは、補体結合のような特性が変化する。変異は、1つの変異抗体中のフレームワーク領域、定常ドメイン、および可変領域のそれぞれに作成することができる。あるいは、変異は、1つの変異抗体中の1つのフレームワーク領域の中だけに、複数の可変領域の中だけに、または定常ドメインの中だけに作成される場合もある。
【0064】
(融合抗体および免疫接着体)
別の実施形態においては、別のポリペプチドに連結させられた本発明の抗Nogo受容体−1抗体の全てまたは一部を含む、融合抗体または免疫接着体が作成される場合もある。いくつかの実施形態においては、抗Nogo受容体−1抗体の可変領域だけが、ポリペプチドに連結させられる。他の実施形態においては、本発明の抗Nogo受容体−1抗体のVHドメインが、第1のポリペプチドに連結させられ、一方では、VHドメインとVLドメインとが抗体結合部位を形成するように互いに相互作用できるような様式で、抗体のVLドメインが、第1のポリペプチドと会合する第2のポリペプチドに対して連結させられる。他の実施形態においては、VHドメインは、VHドメインとVLドメインとが互いに相互作用できるように、リンカーによってVLドメインから間隔を空けられる(下記の単鎖抗体の項を参照のこと)。VH−リンカー−VL抗体は、その後、目的のポリペプチドに連結させられる。融合抗体は、Nogo受容体−1リガンドを発現する細胞または組織に対してポリペプチドを向けるために有用である。目的のポリペプチドは、治療薬、例えば、毒素である場合も、また、診断薬、例えば、西洋ワサビペルオキシダーゼのような、容易に視覚化させることができる酵素である場合もある。さらに、「融合抗体」は、2つ(またはそれ以上)の単鎖抗体が互いに連結されるように作成され得る。これは、1本のポリペプチド鎖の上に二価または多価の抗体を作成することが望まれる場合、あるいは、二重特異的抗体を作成することが望まれる場合に、有用である。
【0065】
(単鎖抗体)
本発明には、本発明のNogo受容体−1ポリペプチドに結合する単鎖抗体(scFv)が含まれる。ScFvを産生するためには、VH−、およびVL−をコードするDNAが、可撓性リンカー、例えば、アミノ酸配列(Gly4−Ser)3(配列番号10)をコードするDNAに対して動作可能であるように連結させられる。その結果、VHおよびVL配列は、可撓性リンカーによって連結させられたVLおよびVH領域を有している連続する1本鎖のタンパク質として発現させられ得る(例えば、Bird et al.,(1988)Science 242:423−426;Huston et al.,(1988)Proc.Natl.Acad.Sci.USA,85:5879−5883;McCafferty et al.,Nature(1990)348:552−554を参照のこと)。単鎖抗体は、VHとVLが1つだけ使用される場合には一価であり、また、VHとVLが2つ使用される場合には二価であり、また、2つ以上のVHとVLが使用される場合には多価である場合もある。
【0066】
(キメラ抗体)
本発明にはさらに、二重特異的抗体またはその抗原結合断片が含まれる。これらにおいては、1つの特異性は本発明のNogo受容体−1ポリペプチドに対する特異性である。1つの実施形態においては、1つの結合ドメインを通じて本発明のNogo受容体−1ポリペプチドに特異的に結合し、第2の結合ドメインを通じて第2の分子に特異的に結合するキメラ抗体が作成され得る。キメラ抗体は、組み換え分子生物学の技術によって産生することができ、また、両方を物理的に結合させることもできる。さらに、本発明のポリペプチドに特異的に結合し、ミエリンによって媒介される成長円錐の崩壊、ならびに神経突起成長および萌芽の阻害を弱めることに関係している別の分子にも特異的に結合する、1つ以上のVHおよびVLを含む単鎖抗体を作成することができる。このような二重特異的抗体は、周知の技術、例えば、Fanger et al.,Immunol Methods 4:72−81(1994)およびWright and Harris(前出)を使用して、そして、(iii)と組み合わせて作成することができる(例えば、Traunecker et al.,Int.J.Cancer(Suppl.)7:51−52(1992)を参照のこと。
【0067】
いくつかの実施形態においては、キメラ抗体は、本発明の抗体に由来する1つ以上の可変領域を使用して調製される。別の実施形態においては、キメラ抗体は、上記の抗体に由来する1つ以上のCDR領域を使用して調製される。
【0068】
(誘導された抗体および標識された抗体)
本発明の抗体または抗原結合断片は、別の分子(例えば、別のペプチドまたはタンパク質)になるように誘導することができ、また、別の分子に連結させることもできる。一般的には、抗体または抗原結合断片は、本発明のポリペプチドに対する結合が、誘導または標識によって悪影響を受けないように誘導される。例えば、本発明の抗体または抗体部分は、1つ以上の他の分子構成要素、例えば、別の抗体(例えば、二重特異的抗体またはダイアボディー)、検出試薬、細胞傷害性因子、薬剤、および/あるいは別の分子との抗体もしくは抗原結合断片の会合を媒介することができるタンパク質またはペプチド(例えば、ストレプトアビジンコア領域またはポリヒスチジンタグ)に対して、機能するように連結させられ得る(化学結合、遺伝子融合、非共有結合などによる)。
【0069】
いくつかの実施形態においては、誘導された抗体は、2つ以上の抗体(同じタイプの抗体、またはたとえば二重特異的抗体を作成するための異なるタイプの抗体)を架橋することによって産生される。適切な架橋基としては、適切なスペーサー(例えば、m−マレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル)によって間隔を隔てられた2つの異なる反応基を有しているヘテロ二官能性の架橋基、またはホモ二官能性の架橋基(例えば、スベリン酸ジスクシンイミジル)が挙げられる。このようなリンカーは、Pierce Chemical Company,Rockford,Illから入手することができる。
【0070】
いくつかの実施形態においては、誘導された抗体は標識された抗体である。例えば、それらを用いて本発明の抗体または抗体部分を誘導することができる検出試薬は、蛍光化合物(フルオレセイン、フルオレセインイソチオシアネート、ローダミン、5−ジメチルアミン−1−ナフタレンスルフォニルクロライド、フィコエリスリン、ランタニドリンなどを含む)である。抗体はまた、検出に有用である酵素、例えば、西洋ワサビペルオキシダーゼ、β−ガラクトシダーゼ、ルシフェラーゼ、アルカリホスファターゼ、グルコースオキシダーゼなどを用いて標識することができる。検出可能な酵素で標識される実施形態においては、抗体は、検出可能な反応産物を生じるために酵素を使用する別の試薬を添加することによって検出される。例えば、西洋ワサビペルオキシダーゼは、過酸化水素およびジアミノベンジジンを用いる。抗体はまた、ビオチンで標識し、アビジンまたはストレプトアビジンの結合の間接的な測定によって検出される場合もある。抗体はまた、二次的なレポーター(例えば、ロイシンジッパー対合配列、二次抗体の結合部位、金属結合ドメイン、エピトープタグ)によって認識される予め決定されたポリペプチドエピトープで標識される場合もある。
【0071】
抗Nogo受容体−1抗体またはその抗原断片もまた、放射標識されたアミノ酸で標識され得る。放射標識は、診断目的と治療目的の両方に使用され得る。放射標識された抗Nogo受容体−1抗体は、診断的に、例えば、被験体においてNogo受容体−1レベルを決定するために使用することができる。さらに、放射標識された抗Nogo受容体−1抗体は、脊髄損傷を処置するために治療的に使用される場合もある。ポリペプチドの標識の例としては、以下の放射性同位元素または放射性核種が挙げられるが、これらに限定されない:3H、14C、15N、35S、90Y、99Tc、111In、125I、131I。
【0072】
抗Nogo受容体−1抗体またはその抗原断片を、ポリエチレングリコール(PEG)、メチルまたはエチル基、あるいは炭水化物基のような化学基で誘導することができる場合もある。これらの基は、抗体の生物学的特性を改善するため、例えば、血清半減期を長くするため、または組織結合を増大させるために、有用であり得る。
【0073】
(抗Nogo受容体−1抗体の特徴づけ)
(抗Nogo受容体−1抗体のクラスおよびサブクラス)
抗Nogo受容体−1抗体のクラスおよびサブクラスは、当該分野で公知の任意の方法によって決定することができる。一般的には、抗体のクラスおよびサブクラスは、抗体の特定のクラスおよびサブクラスに特異的である抗体を使用して決定することができる。このような抗体は市販によって入手することができる。クラスおよびサブクラスは、ELISA、ウェスタンブロット、さらに他の技術によっても決定することができる。あるいは、クラスおよびサブクラスは、抗体の重鎖および/または軽鎖の定常ドメインの全てまたは一部を配列決定し、免疫グロブリンの種々のクラスおよびサブクラスの既知のアミノ酸配列に対してそれらのアミノ酸配列を比較し、そして抗体のクラスおよびサブクラスを決定することによって決定することもできる。
【0074】
(Nogo受容体−1に対する抗Nogo受容体−1抗体の結合親和性)
本発明のNogo受容体−1ポリペプチドに対する本発明の抗Nogo受容体−1抗体の結合親和性および解離速度は、当該分野で公知の任意の方法によって決定することができる。例えば、結合親和性は、競合ELISA、RIA、BIAcore、またはKinExA技術によって測定することができる。解離速度も、また、BIAcoreまたはKinExA技術によって測定することができる。結合親和性と解離速度は、例えば、BIAcoreを使用して、表面プラスモン共鳴によって測定される。
【0075】
7E11および1H2のKdは、それぞれ、1×10−7Mおよび2×10−8Mであることが決定された。
【0076】
(抗Nogo受容体−1抗体によるNogo受容体−1活性の阻害)
いくつかの実施形態においては、本発明の抗Nogo受容体−1抗体またはその抗原結合断片は、リガンドに対するNogo受容体−1の結合を阻害する。このような阻害のIC50は、当該分野で公知の任意の方法によって、例えば、ELISA、RIA、または機能的拮抗によって、測定することができる。いくつかの実施形態においては、IC50は、0.1から500nMの間である。いくつかの実施形態においては、IC50は10から400nMの間である。さらに他の実施形態においては、抗体またはその一部は、60nMから400nMの間のIC50を有する。7E11および1H2のIC50は、結合アッセイにおいて、それぞれ、400nMおよび60nMであることが決定された。下の表3を参照のこと。
【0077】
つまり、当業者であれば、本発明の教示が提供されると、本発明の抗体の生物学的特性を変化させるために、あるいは、それを特定の用途により適するように任意の他の方法においてこれを変化させるために使用することができる、種々の方法を利用することができる。本発明の抗体の生物学的特性を変化させるための方法には、安定性または半減期、免疫原性、毒性、親和性、あるいは、所定の抗体分子の量を増大または減少させる方法が含まれる。
【0078】
本発明の抗体を含む組成物、およびその使用が以下に記載される。
【0079】
(可溶性Nogo受容体−1ポリペプチド)
(タンパク質)
全長のNogo受容体−1は、シグナル配列、N末端領域(NT)、8個のロイシンを多く含む反復(LRR)、LRRCT領域(8個のロイシンを多く含む反復のC末端のロイシンを多く含む反復ドメイン)、C末端領域(CT)、およびGPIアンカーから構成されている(図1を参照のこと)。
【0080】
本発明のいくつかの実施形態では、可溶性Nogo受容体−1ポリペプチドが提供される。本発明の可溶性Nogo受容体−1ポリペプチドには、NTドメイン;8個のLRR、およびLRRCTドメインが含まれており、シグナル配列と機能的なGPIアンカーは含まれていない(すなわち、GPIアンカーは含まれないか、細胞膜に対して効率的に会合する能力を欠いているGPIアンカーが含まれる)。
【0081】
いくつかの実施形態においては、可溶性Nogo受容体−1ポリペプチドには、異種LRRが含まれる。いくつかの実施形態においては、可溶性Nogo受容体−1ポリペプチドには、2個、3個、4個、5個、6個、7個、または8個の異種LRRが含まれる。異種LRRは、Nogo受容体−1以外のタンパク質から得られるLRRを意味する。異種LRRを得ることができる例示的なタンパク質は、トル様(toll−like)受容体(TLR1.2);T細胞活性化ロイシン反復を多く含むタンパク質;デセオリン;OM−gp;インシュリン様成長因子結合タンパク質酸性不安定サブユニットslitおよびrobo;ならびにトル様受容体4である。
【0082】
いくつかの実施形態においては、本発明により、319アミノ酸の可溶性Nogo受容体−1ポリペプチド(可溶性Nogo受容体−1 344、sNogoR1−344、またはsNogoR344)(配列番号6および8の残基26〜344、または配列番号8の残基27〜344)が提供される。いくつかの実施形態においては、本発明により、285アミノ酸の可溶性Nogo受容体−1ポリペプチド(可溶性Nogo受容体−1 310、sNogoR1−310、またはsNogoR310)(配列番号7および9の残基26〜310、または配列番号9の残基27〜310)が提供される。図1を参照のこと。
【0083】
(表1 ヒトおよびラットのNogo受容体−1ポリペプチドの配列)
【0084】
【表1】

本発明のいくつかの実施形態においては、本発明の可溶性Nogo受容体−1ポリペプチドは、Nogo受容体−1に対するリガンドの結合を阻害し、Nogo受容体−1リガンドのアンタゴニストとしての役割を果たすように使用される。本発明のいくつかの実施形態においては、本発明の可溶性Nogo受容体−1ポリペプチドは、ニューロンにおいて神経突起成長および萌芽、例えば、軸索成長の阻害を減少させるために、そしてニューロンにおけるミエリンによって媒介される成長円錐の崩壊を阻害するために、使用される。いくつかの実施形態においては、ニューロンはCNSニューロンである。
【0085】
sNogoR310およびsNogoR344は、驚くべきことに、NogoA、NogoB、NogoC、MAG、およびOM−gpのNogo受容体−1に対する結合をブロックする。
【0086】
いくつかの実施形態においては、本発明の可溶性Nogo受容体−1ポリペプチドは、異種ポリペプチドをさらに含む融合タンパク質の構成要素である。いくつかの実施形態においては、異種ポリペプチドは、免疫グロブリン定常ドメインである。いくつかの実施形態においては、免疫グロブリン定常ドメインは、重鎖定常ドメインである。いくつかの実施形態においては、異種ポリペプチドはFc断片である。いくつかの実施形態においては、Fcは、本発明の可溶性Nogo受容体−1ポリペプチドのC末端に連結させられる。いくつかの実施形態においては、融合Nogo受容体−1タンパク質は二量体である。
【0087】
(本発明の核酸分子)
本発明により、配列番号1〜9、26〜27、29〜37、および41〜45のいずれか1つのポリペプチドを含む、本発明のポリペプチドをコードする核酸が提供される。いくつかの実施形態においては、核酸は、配列番号6および8に示されるNogo受容体−1のアミノ酸残基26〜344、または配列番号8に示されるNogo受容体−1のアミノ酸残基27〜344からなる群より選択されるポリペプチドをコードする。いくつかの実施形態においては、核酸分子は、配列番号7および9に示されるNogo受容体−1のアミノ酸残基26〜310、または配列番号9に示されるNogo受容体−1のアミノ酸残基27〜310からなる群より選択されるポリペプチドをコードする。本明細書中で使用される場合は、「核酸」は、ゲノムDNA、cDNA、mRNA、およびアンチセンス分子、さらに、自然界の供給源に由来するかまたは合成されたかにはかかわらず、別の骨格をベースとするかまたは別の塩基を含む核酸を意味する。いくつかの実施形態においては、核酸にはさらに、転写プロモーターおよび状況に応じたシグナル配列を含む。これらのそれぞれは、本発明のポリペプチドをコードするヌクレオチド配列に動作可能であるように連結される。
【0088】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1融合タンパク質をコードする核酸が提供される。本発明のNogo受容体−1融合タンパク質には、配列番号6および8に示されるNogo受容体−1のアミノ酸残基26〜344、または配列番号8のアミノ酸残基27〜344、および配列番号7および9に示されるNogo受容体−1のアミノ酸残基26〜310、または配列番号9のアミノ酸残基27〜310からなる群より選択されるポリペプチドを含む融合タンパク質が含まれる。いくつかの実施形態においては、核酸は、配列番号26〜27、29〜37、および41〜45からなる群より選択されるポリペプチドを含むNogo受容体−1融合タンパク質をコードする。いくつかの実施形態においては、Nogo受容体−1融合タンパク質をコードする核酸にはさらに、転写プロモーターおよび状況に応じたシグナル配列が含まれる。いくつかの実施形態においては、ヌクレオチド配列はさらに、免疫グロブリン定常領域をコードする。いくつかの実施形態においては、免疫グロブリン定常領域は重鎖定常領域である。いくつかの実施形態においては、ヌクレオチド配列はさらに、ヒンジ領域に連結させられた免疫グロブリン重鎖定常領域をコードする。いくつかの実施形態においては、核酸はさらにFcをコードする。いくつかの実施形態においては、Nogo受容体−1融合タンパク質にはFc断片が含まれる。
【0089】
本発明のコード核酸はさらに、診断目的およびプローブとしての目的のための検出可能な標識を含むように修飾される場合もある。種々のこのような標識は当該分野で公知であり、本明細書中に記載されるコード分子とともに容易に使用することができる。適切な標識としては、ビオチン、放射標識核種などが挙げられるがこれらに限定されない。当業者は、標識されたコード核酸分子を得るために、当該分野で公知の標識のうちの任意のものを使用することができる。
【0090】
(組成物)
いくつかの実施形態においては、本発明により、配列番号1〜5、26〜27、29〜37、および41〜45からなる群より選択されるポリペプチドを含む組成物が提供される。
【0091】
いくつかの実施形態においては、本発明により、抗Nogo受容体−1抗体またはその抗原結合断片、あるいは可溶性Nogo受容体−1ポリペプチド、または本発明の融合タンパク質を含む組成物が提供される。
【0092】
いくつかの実施形態においては、本発明には、作用部位への投与のために薬学的に使用することができる調製物へと活性のある化合物を加工することを容易にする賦形剤および助剤を含む、適切な薬学的に許容される担体が含まれる場合もある。非経口投与に適している処方物としては、水溶性の形態、例えば、水溶性の塩である活性のある化合物の水溶液が挙げられる。さらに、適切な油状の注射懸濁液としての活性のある化合物の懸濁液が投与される場合もある。適切な親油性溶媒またはビヒクルとして、脂肪油、例えば、ゴマ油、または合成の脂肪酸エステル、例えば、オレイン酸エチルまたはトリグリセリドが挙げられる。水性の注射用懸濁液には、懸濁液の粘度を高める物質、例えば、カルボキシメチルセルロース・ナトリウム、ソルビトール、およびデキストランが含まれる場合もある。状況に応じて、懸濁液には、安定剤も含まれる場合がある。リポソームを、本発明の分子を細胞への送達のためにカプセル化するために使用することもできる。例示的な「薬学的に許容される担体」は、生理学的に適合性である任意の全ての溶媒、分散媒体、コーティング剤、抗菌剤および抗真菌剤、等張性の吸収遅延剤など、水、生理食塩水、リン酸緩衝化生理食塩水、デキストロース、グリセロール、エタノールなど、さらにはそれらの組み合わせである。いくつかの実施形態においては、組成物には、等張化剤、例えば、糖類、多価アルコール、例えば、マンニトール、ソルビトール、または塩化ナトリウムが含まれる。いくつかの実施形態においては、組成物には、湿潤剤のような薬学的に許容される物質、または少量の、湿潤剤、乳化剤、保存剤、または緩衝液のような補助物質が含まれる。補助物質は、本発明の抗体、抗原結合断片、可溶性Nogo受容体、または融合タンパク質の保存期間または有効性を増強させる。
【0093】
本発明の組成物は種々の形態で存在することができ、例えば、液体、半固体、および固体である投与形態、たとえは、液体の溶液(例えば、注射することができる、および注入することができる溶液)、分散液、または懸濁液が挙げられる。好ましい形態は、意図される投与の態様、および治療用途に応じて変化する。1つの実施形態においては、組成物は、注射することができるか、または注入することができる溶液の形態であり、例えば、他の抗体でのヒトの受動免疫法に使用されるものと同様の組成物である。
【0094】
組成物は、溶液、マイクロエマルジョン、分散液、リポソーム、または高い薬剤濃度に適している他の秩序構造として、処方することができる。滅菌の注射することができる溶液は、上記に列挙した成分の1つまたはそれらの組み合わせとともに、適切な溶媒の中に必要とされる量の抗Nogo受容体−1抗体を配合し、必要である場合には、その後に濾過滅菌することによって、調製することができる。一般的には、分散液は、塩基性の分散媒体と、上記に列挙されたものからの必要とされる他の成分を含む滅菌のビヒクルの中に活性のある化合物を配合することによって調製される。滅菌の注射することができる溶液の調製のための滅菌の粉末の場合には、好ましい調製方法は、減圧乾燥および凍結乾燥であり、これにより、活性のある成分と任意のさらなる所望される成分の粉末が、それらの予め滅菌濾過された溶液から得られる。溶液の適切な流動性は、例えば、レシチンのようなコーティングの使用によって、分散液の場合には必要とされる粒子の大きさを維持することによって、および界面活性剤の使用によって、維持することができる。注射することができる組成物の持続的吸収は、吸収を遅らせる物質、例えば、モノステアリン酸塩およびゼラチンを組成物中に含めることによってもたらすことができる。
【0095】
いくつかの実施形態においては、活性のある化合物は、迅速な放出から化合物を保護する担体を用いて調製することができ、例えば、移植物、経皮パッチ、およびマイクロカプセルに入れられた送達システムを含む徐放処方物である。生分解性の生体適合性高分子、例えば、エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸を使用することができる。このような処方物の調製のための多くの方法については特許がとられており、当業者に一般的に知られている。例えば、Sustained and Controlled Release Drug Delivery Systems,J.R.Robinson,ed.,Marcel Dekker,Inc.,New York,1978を参照のこと。
【0096】
補助的な活性のある化合物をもまた、組成物中に配合することもできる。いくつかの実施形態においては、本発明のNogo受容体−1抗体もしくはその抗原結合断片、または可溶性Nogo受容体−1ポリペプチドまたは融合タンパク質は、1つ以上のさらなる治療薬と一緒に処方され、そして/または一緒に投与される。
【0097】
本発明の薬学的組成物には、「治療有効量」または「予防上有効である量」の、本発明の抗体、抗原結合断片、ポリペプチド(単数または複数)、または融合タンパク質が含まれる場合もある。「治療有効量」は、所望される治療結果に到達するために必要な、有効量、投与量、および期間をいう。Nogo受容体−1抗体もしくはその抗原結合断片、可溶性Nogo受容体−1ポリペプチド、またはNogo受容体融合タンパク質の治療有効量は、疾患のステージ、個体の年齢、性別、および体重のような要因によって変化する場合がある。治療有効量はまた、抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、またはNogo受容体融合タンパク質の任意の毒性作用または有害な作用よりも、治療上の利点がある効果が上回るような量である。「予防上有効である量」は、所望される予防的な結果に到達するために必要な、有効量、投与量、またはそのために必要な期間をいう。通常は、予防のための用量は、疾患の前、または疾患の初期段階に被験体において使用されるので、予防上有効である量は、治療有効量よりも少ない。
【0098】
投与レジュメは、最適な所望される反応(例えば、治療応答または予防応答)を提供するように調整することができる。例えば、1回のボーラスが投与される場合があり、数回に分けられた用量が時間をかけて投与される場合もあり、また、治療状況の緊急性によって望まれる場合には、用量が比例的に減少させられる場合も、増大させられる場合もある。投与を容易にするための単位投与量形態に非経口組成物を処方することが特に有効であり、そして、投与量単位が均質であることは、本明細書中で使用される場合には、処置される哺乳動物被験体について単一の投与量として適している物理的に分かれている単位をいう。個々の単位には、必要とされる薬学的担体と組み合わせて、所望される治療効果を生じるように計算された予め決定された量の活性のある化合物が含まれる。本発明の単位投与量形態についての詳細は、(a)抗体、抗原結合断片、および可溶性受容体−1ポリペプチド、またはNogo受容体融合タンパク質の特有の特性、ならびに到達させられる特定の治療または予防効果、さらには(b)個体の過敏症の処置については、そのような抗体、抗原結合断片、および可溶性受容体−1ポリペプチド、またはNogo受容体融合タンパク質を混合することに関する当該分野に特有の限界に影響されるか、またはそれらに直接依存する。いくつかの実施形態においては、Nogo受容体−1抗体またはその抗原結合断片についての治療有効量の範囲は、0.1〜4mg/kg/日である。いくつかの実施形態においては、Nogo受容体−1抗体またはその抗原結合断片についての治療有効量の範囲は、0.2〜4mg/kg/日である。いくつかの実施形態においては、Nogo受容体−1抗体またはその抗原結合断片についての治療有効量の範囲は、0.2mg/kg/日である。
【0099】
(抗体、抗原結合断片、可溶性受容体、および融合タンパク質の使用)
いくつかの実施形態においては、本発明により、抗Nogo受容体−1抗体、そのような抗体の抗原結合断片、可溶性Nogo受容体−1ポリペプチド、またはそのようなポリペプチドを含む融合タンパク質を、それを必要としている哺乳動物に投与することによって、Nogo受容体−1活性を阻害するための方法が提供される。
【0100】
いくつかの実施形態においては、本発明により、リガンドに対するNogo受容体−1の結合を阻害する方法が提供される。この方法には、Nogo受容体−1を、本発明の抗体または抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、リガンドは、NogoA、NogoB、NogoC、MAG、およびOM−gpからなる群より選択される。
【0101】
いくつかの実施形態においては、本発明により、ニューロンにおける成長円錐の崩壊を阻害するための方法が提供される。この方法には、ニューロンを、本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、本発明により、ニューロンにおいて神経突起成長または萌芽の阻害を減少させるための方法が提供される。この方法には、ニューロンを、本発明の抗体または抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、ニューロンは、CNSニューロンである。これらの方法のいくつかにおいては、神経突起成長または萌芽は、軸索成長である。
【0102】
いくつかの実施形態においては、本発明により、哺乳動物において死滅の危険性があるニューロンの生存を促進する方法が提供される。この方法には、(a)(i)抗Nogo受容体−1抗体もしくはその抗原結合断片、または(ii)可溶性Nogo受容体−1ポリペプチドを発現する、培養された宿主細胞を提供すること;および、(b)哺乳動物中に、ニューロンの部位またはその付近に、宿主細胞を導入することが含まれる。Almudena Ramon−Cueto,M Isabel Cordero,Fernando F Santos−Benito and Jesus Avila(2000)Functional recovery of paralegic rats and motor axon regeneraton in their spinal cords by olfactory ensheathing cells.Neuron 25,425−435。
【0103】
いくつかの実施形態においては、本発明により、哺乳動物中にある死滅の危険性があるニューロンの生存を促進する遺伝子治療方法が提供される。この方法には、(a)抗Nogo受容体−1抗体もしくはその抗原結合断片、または(b)可溶性Nogo受容体−1ポリペプチドをコードするヌクレオチド配列を含むウイルスベクターを、ニューロンの部位またはその付近に投与することが含まれる。ここでは、抗Nogo受容体−1抗体、抗原結合断片、または可溶性Nogo受容体−1ポリペプチドは、哺乳動物中のヌクレオチド配列から、ニューロンの生存を促進するために十分な量で発現される。これらの実施形態に有用であるウイルスベクターおよび方法は、例えば、Noel et al.,Human Gene Therapy,13,1483−93(2002)に記載されている。
【0104】
いくつかの実施形態においては、本発明により、リガンドに対するNogo受容体−1の結合を阻害する方法が提供される。この方法には、リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。
【0105】
いくつかの実施形態においては、本発明により、Nogo受容体−1リガンドの活性を調節する方法が提供される。この方法には、Nogo受容体−1リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。
【0106】
いくつかの実施形態においては、本発明により、ニューロンにおける成長円錐の崩壊を阻害するための方法が提供される。この方法には、Nogo受容体−1リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。いくつかの実施形態においては、本発明により、ニューロンにおいて神経突起成長または萌芽の阻害を減少させるための方法が提供される。この方法には、Nogo受容体−1リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。いくつかの実施形態においては、ニューロンはCNSニューロンである。いくつかの実施形態においては、リガンドは、NogoA、NogoB、NogoC,MAG、およびOM−gpからなる群より選択される。いくつかの実施形態においては、神経突起成長および萌芽は、軸索成長である。
【0107】
本明細書中に記載される任意のタイプの抗体または受容体を、治療的に使用することができる。いくつかの実施形態においては、抗Nogo受容体−1抗体はヒト抗体である。いくつかの実施形態においては、哺乳動物はヒト患者である。いくつかの実施形態においては、抗体またはその抗原結合断片は、獣医学的な目的のために、またはヒトの疾患についての動物モデルとして、抗体が交差反応するNogo受容体−1を発現するヒト以外の哺乳動物(例えば、霊長類、カニクイザル、またはアカゲザル)に投与される。このような動物モデルは、本発明の抗体の治療効力を評価するために有用であり得る。
【0108】
いくつかの実施形態においては、抗Nogo受容体−1抗体もしくはその抗原結合断片、または可溶性Nogo受容体−1ポリペプチド、または融合タンパク質の投与は、損傷部位からの軸索成長を促進するように、脊髄損傷を処置するために使用される。
【0109】
本発明の抗Nogo受容体−1抗体もしくは抗原結合断片、または可溶性Nogo受容体−1ポリペプチドもしくは融合タンパク質は、単独で提供することもでき、また、特定の病理学的プロセスを調節する他の物質と組み合わせて、または順次組み合わせて提供することもできる。例えば、抗炎症薬が、ニューロンのさらなる損傷をブロックするため、および軸索の再生の阻害のための手段として、脳梗塞の後に同時に投与される場合がある。本明細書中で使用される場合は、Nogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1、およびNogo受容体融合タンパク質は、2つが同時に、連続して、または別々に投与される場合に、1つ以上のさらなる治療薬と組み合わせて投与されると言われる。
【0110】
本発明の抗Nogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、Nogo受容体−1融合タンパク質は、非経口、皮下、静脈内、筋肉内、腹腔内、経皮、吸入、または口腔経路によって投与することができる。例えば、薬剤は、微量注入によって損傷部位に局所投与される場合がある。典型的な部位としては、怪我によって生じた脊髄の損傷した領域が挙げられるが、これに限定されない。投与される投与量は、レシピエントの年齢、健康状態、および体重、同時に行われている(行われている場合には)処置の種類、処置の頻度、所望される効果の性質に応じて変化する。
【0111】
本発明の化合物は、通常は、ヒト、ヒツジ、ウマ、ウシ、ブタ、イヌ、ネコ、ラット、およびマウスのような哺乳動物に生体内で、あるいは生体外で利用することができる。
【0112】
(本発明のベクター)
いくつかの実施形態においては、本発明により、コード配列を含む組み換えDNA分子(rDNA)が提供される。本明細書中で使用される場合は、rDNA分子は、分子操作が行われたDNA分子である。rDNA分子を作成するための方法は当該分野で周知である。例えば、Sambrook et al.,(1989)Molecular Cloning−A Laboratory Manual,Cold Spring Harbor Laboratory Pressを参照のこと。いくつかのrDNA分子においては、コードDNA配列は、発現制御配列およびベクター配列に動作可能であるように連結される。
【0113】
いくつかの実施形態においては、本発明により、本発明のポリペプチドをコードする核酸を含むベクターが提供される。本発明の核酸が動作可能であるように連結されるベクターおよび発現制御配列の選択は、当該分野で周知であるように、所望される機能的特性(例えば、タンパク質の発現および形質転換される宿主細胞)に直接依存する。本発明のベクターは、少なくとも、複製、または宿主染色体への組み込みを指示することができ、rDNA分子中に含まれる構造遺伝子を発現できることが好ましい。
【0114】
動作可能であるように連結されたタンパク質をコードする配列の発現を調節するために使用される発現制御エレメントは当該分野で公知である。発現制御エレメントとしては、誘導性プロモーター、構成的プロモーター、分泌シグナル、および他の調節エレメントが挙げられるが、これらに限定されない。宿主細胞の培地中の栄養素に反応する誘導性プロモーターは、容易に制御することができるので好ましい。
【0115】
1つの実施形態においては、コード核酸分子を含むベクターには、原核生物のレプリコンが含まれる。すなわち、DNA配列は、形質転換される原核生物宿主細胞、例えば、細菌宿主中で自律複製を指示し、染色体外で組み換えDNA分子を維持する能力を有する。そのようなレプリコンは当該分野で周知である。さらに、原核生物のレプリコンを含むベクターにはまた、薬剤耐性のような、その発現により検出可能な、または選択可能なマーカーを付与する遺伝子が含まれる場合もある。細菌の薬剤耐性遺伝子の典型は、アンピシリンまたはテトラサイクリンに対する耐性を付与する遺伝子である。
【0116】
原核生物のレプリコンを含むベクターには、さらに、大腸菌(E.coli)のような細菌宿主細胞中のコード遺伝子配列の発現(転写および翻訳)を指示することができる原核生物またはバクテリオファージのプロモーターが含まれ得る。プロモーターは、RNAポリメラーゼの結合を可能にし、転写を生じさせることができるDNA配列によって形成される発現制御エレメントである。細菌宿主と適合するプロモーター配列は、通常、本発明のDNAセグメントの挿入のための便利な制限部位を含むプラスミドベクターとして提供される。このようなベクタープラスミドの例は、pUC8、pUC9、pBR322、およびpBR329(Bio−Rad(登録商標)Laboratories)、pPL、およびpKK223(Pharmacia)である。任意の適切な原核生物宿主を、本発明のタンパク質をコードする組み換えDNA分子を発現させるために使用することができる。
【0117】
真核生物細胞と適合する発現ベクター、好ましくは、脊椎動物細胞と適合する発現ベクターもまた、コード配列を含むrDNA分子を形成させるために使用することができる。真核生物細胞発現ベクターは、当該分野で周知であり、いくつかの市販の供給業者から入手することができる。通常は、このようなベクターは、所望されるDNAセグメントの挿入のための便利な制限部位を含んで提供される。このようなベクターの例は、pSVLおよびpKSV−10(Pharmacia)、pBPV−1、pML2d(International Biotechnologies)、pTDT1(ATCC(登録商標)31255)、および他の真核生物発現ベクターである。
【0118】
本発明のrDNA分子を構築するために使用される真核生物発現ベクターには、さらに、真核生物細胞中で有効である選択マーカー、好ましくは、薬剤耐性選択マーカーを含めることができる。好ましい薬剤耐性マーカーは、その発現によりネオマイシン耐性を生じる遺伝子、すなわち、ネオマイシンホスホトランスフェラーゼ(neo)遺伝子である。(Southern et al.,(1982)J.Mol.Anal.Genet.1,327−341)。あるいは、選択マーカーは、別のプラスミド上に存在させることができ、2つのベクターを宿主細胞の同時トランスフェクションによって導入し、トランスフェクタントを、選択マーカーについて適切な薬剤の中で培養することによって選択することができる。
【0119】
本発明の抗体または抗体部分を発現させるためには、遺伝子が、転写および翻訳制御配列に動作可能であるように連結されるように、軽鎖および重鎖の一部または全長をコードするDNAが発現ベクター中に挿入される。発現ベクターとしては、プラスミド、レトロウイルス、コスミド、YAC、EBVに由来するエピソームなどが挙げられる。抗体遺伝子は、ベクター中の転写および翻訳制御配列が、抗体遺伝子の転写および翻訳を調節するそれらの意図される機能を果たすように、ベクター中に連結される。発現ベクターと発現制御配列は、使用される発現宿主細胞と適合するように選択される。抗体の軽鎖遺伝子と、抗体の重鎖遺伝子が、別々のベクターに挿入され得る。いくつかの実施形態においては、両方の遺伝子が同じ発現ベクターに挿入される。抗体遺伝子は、標準的な方法(例えば、抗体遺伝子断片とベクター上の相補的な制限部位の連結、または制限部位が存在しない場合は、平滑末端の連結)によって、発現ベクター中に挿入される。
【0120】
便利なベクターは、機能的に完全なヒトCHまたはCL免疫グロブリン配列をコードし、上記のように任意のVHまたはVL配列を容易に挿入し、発現することができるように操作された適切な制限部位を有しているベクターである。このようなベクター中では、スプライシングは、通常、挿入されたJ領域中のスプライシングドナー部位と、ヒトC領域の前にあるスプライシングアクセプター部位との間で、また、ヒトCHエキソンの中に存在するスプライシング領域でも生じる。ポリアデニル化および転写終結は、コード領域の下流に存在するもともとの染色体部位で生じる。組み換え発現ベクターはまた、宿主細胞からの抗体鎖の分泌を促進するシグナルペプチドをコードすることもできる。抗体鎖遺伝子は、シグナルペプチドが抗体鎖遺伝子のアミノ末端にインフレームで連結されるように、ベクター中にクローニングすることもできる。シグナルペプチドは、免疫グロブリンシグナルペプチドである場合も、また異種シグナルペプチド(すなわち、免疫グロブリン以外のタンパク質に由来するシグナルペプチド)である場合もある。
【0121】
本発明の免疫原性ポリペプチド、Nogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質に加えて、本発明の組み換え発現ベクターは、宿主細胞中でのそれらの発現を制御する調節配列を有する。調節配列の選択を含む発現ベクターの設計が、形質転換される宿主細胞の選択、所望されるタンパク質の発現のレベルなどのような要因によって変化する場合があることは、当業者によって理解されるであろう。哺乳動物宿主細胞での発現に好ましい調節配列としては、哺乳動物細胞中で高レベルのタンパク質の発現を指示するウイルスエレメント、例えば、レトロウイルスLTR由来のプロモーターおよび/またはエンハンサー、サイトメガロウイルス(CMV)由来のプロモーターおよび/またはエンハンサー(例えば、CMVプロモーター/エンハンサー)、シミアンウイルス40(SV40)由来のプロモーターおよび/またはエンハンサー(例えば、SV40プロモーター/エンハンサー)、アデノウイルス由来のプロモーターおよび/またはエンハンサー(例えば、アデノウイルス主要後期プロモーター(AdMLP))、ポリオーマおよび強力哺乳動物プロモーター(例えば、本来の免疫グロブリンおよびアクチンプロモーター)が挙げられる。ウイルス調節エレメントおよびその配列のさらなる詳細については、例えば、Stinskiによる米国特許第5,168,062号、Bell et al.,による米国特許第4,510,245号、およびSchaffner et al.,による米国特許第4,968,615号を参照のこと。
【0122】
異種遺伝子および調節配列に加えて、本発明の組み換え発現ベクターは、宿主細胞中でのベクターの複製を調節する配列(例えば、複製起点)、および選択マーカー遺伝子のような、さらなる配列を有する場合がある。選択マーカー遺伝子は、ベクターが導入された宿主細胞の選択を容易にする(例えば、米国特許第4,399,216号、同第4,634,655号、および同第5,179,017号を参照のこと、全て、Axel et al.,による)。例えば、通常、選択マーカー遺伝子によって、薬剤、例えば、G418、ハイグロマイシン、またはメトトレキセートに対する耐性が、ベクターが導入される宿主に付与される。好ましい選択マーカー遺伝子としては、ジヒドロ葉酸レダクターゼ(DHFR)遺伝子(例えば、dhfr−宿主細胞におけるメトトレキセート選択/増幅の使用について)、およびneo遺伝子(G418選択について)が挙げられる。
【0123】
(本発明のタンパク質を組み換えにより産生する宿主および方法)
本発明の抗Nogo受容体−1抗体、免疫原性ペプチド、可溶性Nogo受容体−1ポリペプチド、可溶性Nogo受容体−1融合タンパク質をコードする核酸分子、ならびにこれらの核酸分子を含むベクターを、適切な宿主細胞の形質転換のために使用することができる。形質転換は、宿主細胞にポリヌクレオチドを導入するための任意の公知の方法によって行うことができる。異種ポリヌクレオチドを哺乳動物細胞に導入するための方法は当該分野で周知である。これには、デキストラン媒介トランスフェクション、リン酸カルシウム沈殿、ポリブレン媒介トランスフェクション、プロトプラスト融合、エレクトロポレーション、リポソーム中へのポリヌクレオチド(単数または複数)のカプセル化、および核へのDNAの直接のマイクロインジェクションが含まれる。さらに、核酸分子を、ウイルスベクターによって哺乳動物細胞に導入することができる場合もある。
【0124】
適切な細胞宿主の本発明のrDNA分子での形質転換は、周知の方法によって行われる。これらは、通常、使用されるベクターおよび使用される宿主システムのタイプに応じて異なる。原核生物宿主細胞の形質転換に関しては、エレクトロポレーションおよび塩処理方法が使用され得る(例えば、Sambrook et al.,(1989)Molecular Cloning−A Laboratory Manual,Cold Spring Harbor Laboratory Press;Cohen et al.,(1972)Proc.Natl.Acad.Sci.USA,69,2110−2114を参照のこと)。脊椎動物細胞のrDNAを含むベクターでの形質転換に関しては、エレクトロポレーション、陽イオン性脂質、または塩処理方法が使用され得る(例えば、Graham et al.,(1973)Virology 52,456−467;Wigler et al.,(1979)Proc.Natl.Acad.Sci.USA,76,1373−1376を参照のこと)。
【0125】
うまく形質転換された細胞、すなわち、本発明のrDNA分子を含む細胞は、選択マーカーについての選択を含む、周知の技術によって同定することができる。例えば、本発明のrDNAの導入によって得られる細胞は、単一コロニーを生じるようにクローニングされ得る。これらのコロニーに由来する細胞を、回収し、溶解させ、そしてそれらのDNA内容物を、Southern,(1975)J.Mol.Biol.,98,503−517によって記載されているような方法を使用してrDNAの存在について試験することができ、また、細胞によって産生されるタンパク質が、免疫学的方法によってアッセイされる場合もある。
【0126】
発現のための宿主として利用することができる哺乳動物細胞株は当該分野で周知であり、これには、アメリカンタイプカルチャーコレクション(ATCC(登録商標))から入手することができる多くの不死化細胞株が含まれる。これらとしては、特に、チャイニーズハムスター卵巣(CHO)細胞、NSO、SP2細胞、HeLa細胞、ベビーハムスター腎臓(BHK)細胞、サル腎臓細胞(COS)、ヒト肝細胞ガン細胞(例えば、Hep G2)、A549細胞、および多数の他の細胞株が挙げられる。特に好ましい細胞株は、どの細胞株が高い発現レベルを有しているかを決定することによって選択される。使用することができる他の細胞株は、昆虫細胞株、例えば、Sf9細胞である。本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質をコードする組み換え発現ベクターが哺乳動物宿主細胞に導入される場合は、これらは、宿主細胞中で抗体、ポリペプチド、および融合ポリペプチドを発現させるために、あるいは、さらに好ましくは、本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質を宿主が増殖させられる培養培地中に分泌させるために十分な時間の間、宿主細胞を培養することによって産生される。本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質は、標準的なタンパク質精製方法を使用して培養培地から回収することができる。
【0127】
さらに、本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質(あるいはそれらに由来する他の部分)の、産生細胞株からの発現は、多数の公知の技術を使用して増強させることができる。例えば、グルタミン合成酵素の遺伝子発現システム(GSシステム)が、特定の条件下で発現を増強させるための一般的なアプローチである。GSシステムは、欧州特許第0,216,846号、同第0,256,055号、および同第0,323,997号、ならびに欧州特許出願番号89303964.4と組み合わせて、完全にまたは一部議論されている。
【0128】
(宿主細胞)
本発明により、さらに、本発明のNogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および/または可溶性Nogo受容体−1融合タンパク質をコードする核酸分子で形質転換された宿主細胞が提供される。宿主細胞は、原核生物である場合も、また真核生物である場合もある。細胞株が細胞培養方法と適合しており、発現ベクターの増幅、および遺伝子産物の発現と適合している限りは、本発明のタンパク質の発現に有用である真核生物細胞は限定されない。好ましい真核生物宿主細胞としては、酵母細胞、昆虫細胞、および哺乳動物細胞、好ましくは、マウス、ラット、サル、またはヒト細胞株のような脊椎動物細胞株が挙げられるが、これらに限定されない。有用な真核生物細胞の例として、CCL61としてATCC(登録商標)から入手することができるチャイニーズハムスター卵巣(CHO)細胞、CRL1658としてATCCから入手することができるNIH Swissマウス胚性細胞NIH−3T3、ベビーハムスター腎臓細胞(BHK)、同様の真核生物組織培養細胞株が挙げられる。
【0129】
(rDNA分子を使用する組み換えタンパク質の産生)
本発明により、さらに、本発明のNogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および/または可溶性Nogo受容体−1融合タンパク質を、本明細書中に記載される核酸分子を使用して産生するための方法が提供される。一般的な用語においては、組み換え形態のタンパク質の産生には、通常、以下の工程が含まれる:
最初に、本発明のタンパク質をコードする核酸分子が得られる。コード配列にイントロンが間に挟まっていない場合は、これはいずれの宿主での発現にも適している。
【0130】
その後、核酸分子は、状況に応じて、上記のような適切な制御配列と動作可能であるように連結させられて、タンパク質のオープンリーディングフレームを含む発現ユニットが形成される。発現ユニットは、適切な宿主を形質転換するために使用され、形質転換された宿主が組み換えタンパク質が産生される条件下で培養される。状況に応じて、組み換えタンパク質は、培地から、または細胞から単離される。タンパク質の回収および精製は、何らかの不純物が寛容であるいくつかの場合においては必要ない場合もある。
【0131】
上記の工程のそれぞれは、種々の方法で行うことができる。例えば、所望されるコード配列が、ゲノム断片から得られ、適切な宿主の中で直接使用される場合もある。種々の宿主において動作可能である発現ベクターの構築は、適切なレプリコン、および制御配列を使用して、上記のように行われる。制御配列、発現ベクター、および形質転換方法は、遺伝子を発現させるために使用される宿主細胞のタイプに応じて変化し、上記で詳細に議論されている。適切な制限部位を、もともと利用できない場合は、摘出可能な遺伝子がこれらのベクターの中に挿入されることを可能にするために、コード配列の末端に付加することができる。当業者であれば、当該分野で公知の任意の宿主/発現システムを、組み換えタンパク質を産生させるように本発明の核酸分子とともに使用するために容易に適応させることができる。
【0132】
本発明がより理解されるように、以下の実施例が示される。これらの実施例は、説明の目的のためのものにすぎず、いかなる様式においても本発明の範囲を限定するとは解釈さらない。
【実施例】
【0133】
(実施例1)
(マウスモノクローナル抗Nogo受容体−1抗体の産生)
本発明の免疫原性Nogo受容体−1ポリペプチドに特異的に結合する抗Nogo受容体−1抗体を、以下の方法および手順を使用して作成した。
【0134】
(免疫化)
2つの免疫化アプローチを使用した:
(1.免疫原としてのNogo受容体−1(NogoR−1)を含むNOS−7細胞または細胞膜)
ラットNogo受容体−1遺伝子(GenBank(登録商標)No.AF462390)を、CMVプロモーターと薬剤選択のためのゲネチシン耐性遺伝子を含む哺乳動物発現ベクターpEAG1256(Biogen(登録商標))にサブクローニングした。組み換えプラスミドを、Superfect(Qiagen(登録商標))を使用してCOS−7細胞にトランスフェクトした。トランスフェクタントをゲネチシン(Gibco(登録商標)、2mg/mL)を使用して選択し、クローニングし、そしてNogo受容体−1タンパク質の表面での発現についてFACSによって確認した。COS−7膜を、記載されている手順にしたがってこれらの細胞から調製し(Wang et al.,J.Neurochem.,75:1155−1161(2000))、2回洗浄し、1mg/mL(タンパク質濃度)で10%のグリセロール中で−70℃で保存した。
【0135】
8週齢の雌RBFマウス(Jackson Labs,Bar Harbor,ME)を、50μgのラットNogo受容体−1−COS−7膜、または表面上にNogo受容体−1を発現するCOS−7細胞全体のいずれか、および50μlのRIBI MPL−TDM−CWSアジュバント(Sigma(登録商標)Chemical Co.,St.Louis,MO)を含む乳濁物のいずれかで、2週間ごとに1回、腹腔内に免疫した(Lipman et al.,1992)。免疫化したマウスから血清を、最初の免疫化の前、2回目の免疫化の7日後、および3回目の免疫化の7日後、および3回目の免疫化の38日後に採取し、抗Nogo受容体−1抗体力価を、以下に記載するようにELISAによって測定した。
【0136】
(2.免疫原としての特異的Nogo受容体−1ペプチド)
ラットNogo受容体−1遺伝子配列を、Vector NTi(登録商標)ソフトウェアを使用して抗原性分析した(図2)。分析において同定した抗原性ペプチドを、標準的なグルタルアルデヒド手順を使用してキーホールリンペットヘモシアニン(KLH)に結合させた。
【0137】
8週齢の雌RBFマウス(Jackson Labs,Bar Harbor,ME)を、50μgのKLH結合ペプチドと50μlの完全なフロイトのアジュバント(Sigma(登録商標)Chemical Co.,St.Louis,MO)を含む乳濁物で、2週間ごとに1回、腹腔内に免疫した。免疫化したマウスから血清を、最初の免疫化の前、2回目の免疫化の1週間後、3回目の免疫化の1週間後に採取し、抗Nogo受容体−1抗体力価を測定した。追加免疫の用量を、3回目の免疫化の後に投与した。この追加免疫用量での免疫化の3日後に、融合実験を開始した。
【0138】
(ハイブリドーマの産生およびスクリーニング)
抗原性Nogo受容体−1ペプチドで免疫化したマウスに由来する血清をELISAによってスクリーニングし、一方、Nogo受容体−1を発現するCOS−7細胞で免疫化したマウスに由来する血清は、フローサイトメトリーによってスクリーニングした。Nogo受容体−1−COS−7細胞に特異的に結合した抗体についてポジティブであるマウスをフローサイトメトリーによって同定し、屠殺した。脾細胞をマウスから単離し、記載されている(Kennett et al.,1993、Monoclonal Antibodies:A New Dimension in Biological Analysis.Plenum Press,New York)ように、FL653骨髄腫(Ig−/HGPRT−Balb/cマウス骨髄腫のAPRT誘導株、10%のFBS、4500mg/Lのグルコース、4mMのL−グルタミン、および20mg/mLの8−アザグアニンを含むDMEM中で維持した)に融合させた。融合させた細胞を、24ウェルまたは48ウェルプレート(Corning Glass Works,Corning,NY)にプレートし、アデニン、アミノプテリン、およびチミジンを含む培養培地を供給した。AAT耐性培養物を、ELISAまたはフローサイトメトリーによって、Nogo受容体−1−COS−7細胞、またはNogo受容体−1抗原性ペプチドのいずれかに対する結合について、下記のようにスクリーニングした。ポジティブであるウェルの細胞を、限界希釈によってさらにサブクローニングした。
【0139】
Nogo受容体−1抗原性ペプチドに対する抗体の結合についてスクリーニングするために、免疫原として使用したペプチドをBSAに結合させた。50μLの0.1M重炭酸ナトリウム緩衝液(pH9.0)中の0.5μgの結合ペプチドを、96ウェルMaxiSorp(登録商標)プレート(Nunc(登録商標))のそれぞれのウェルに添加した。その後、プレートを、37℃で1時間、または4℃で16時間インキュベートし、非特異的結合部位を、0.1%のBSA、0.1%のオボアルブミン、0.1%のブロット(blotto)、および0.001%のアジドを含む25mMのHEPES(pH7.4)を使用してブロックした。ハイブリドーマ上清を添加し、25℃で1時間インキュベートした。PBSでの3回の洗浄の後、西洋ワサビペルオキシダーゼ結合ヤギ抗マウス二次抗体(Jackson ImmunoResearch Inc.)の50μlの1:10,000希釈液をそれぞれのウェルに添加し、さらに1時間インキュベートした。3回の洗浄の後、TMB(Pierce)によって発色させ、2Mの硫酸で停止させた。色の強さを、分光光度計において450nmでモニターした。
【0140】
抗体を、全長のNogo受容体−1に対する結合について以下のようにスクリーニングした。COS−7細胞を、0.1μMのCellTracker(登録商標)Green CMFDA(Molecular Probes,Eugene,OR)で製造供給元によって記載されているように標識した。等量のCellTracker(登録商標)で標識した対照細胞を、抗Nogo受容体−1試験血清とのインキュベーションの前に、洗浄したNogo受容体−1−COS−7細胞と混合した。50μLの細胞混合物を、96ウェルV底ポリスチレンプレート(Coster(登録商標)3877、Corning,NY)のそれぞれのウェルに分配し、100μLのハイブリドーマ上清または対照抗Nogo受容体−1抗体を添加した。4℃で30分間のインキュベーションの後、細胞を洗浄し、50μLの、PBS中のR−フィコエリスリン結合親和性純粋F(ab’)2断片ヤギ坑マウスIgG Fcγ特異的二次抗体(1:200、Jackson ImmunoResearch Laboratory,West Grove,PA)とともにインキュベートした。インキュベーションの終わりに、細胞をPBSで2回洗浄し、1%のFBSを含む200μLのPBS中に懸濁し、FACS分析を行った。あるいは、Nogo受容体−1−COS−7細胞をハイブリドーマ上清と混合し、その後、R−フィコエリスリン結合ヤギ坑マウス二次抗体で処理し、標準的なFACS分析に直接供した。
【0141】
本発明者らは、種々の免疫原を使用して25個の抗Nogo受容体−1抗体を作成した。本発明者らは、免疫原としてラットNogo受容体−1の残基110〜125に対応するペプチド配列を使用して2つの抗体7E11および5B10を作成した。本発明者らは、免疫原として全長のラットNogo受容体−1でトランスフェクトしたCOS7細胞から調製した膜を使用して、3つの抗体1H2、3G5、および2F7を作成した。本発明者らは、免疫原としてsNogoR310−Fcを使用して13個の抗体(1D9.3、1E4.7、1B4.3、2C4.3、1F10.3、2H1.4、1H3.3、1G4.1、1E4.1、2G7.1、2C4.1、2F11.1、および1H4.1)を作成し、免疫原としてラットNogo受容体−1の残基423−434に対応するペプチド配列を使用して7個の抗体(2E8.1、2G11.2、および1B5.1)を作成した。
【0142】
(モノクローナル抗体7E11および5B10の配列分析)
本発明者らは、Qiagen(登録商標)RNeasy(登録商標)ミニキットを使用して全RNAを抽出し、単離したRNAからcDNAを作成した。本発明者らは、プライマー5’−TGAGGAGACGGTGACCGTGGTCCCTTGGCCCCAG−3’(配列番号12)および5’−AGGTSMARCTGCAGSAGTCWGG−3’(配列番号25)を使用してPCRによって、軽鎖配列を増幅した。本発明者らは、プライマー5’−GGGGATATCCACCATGAAGTTGCCTGTTAGGCTGTTG−3’(配列番号13)および5’−GGGGATATCCACCATGAGGKCCCCWGCTCAGYTYCTKGGA−3’(配列番号14)を使用してPCRによって重鎖配列を増幅した。これらのプライマーには、以下のような縮重ヌクレオチドが含まれている:SはGまたはCを示し;MはAまたはCを示し;RはGまたはAを示し;WはAまたはTを示し;KはGまたはTを示し;そしてYはTまたはCを示す。本発明者らは、PCR断片を配列決定ベクターにクローニングし、配列決定ベクターに特異的なプライマーを使用して、ジデオキシ鎖終結によってCDRのDNA配列を決定した。本発明者らは、DNA配列を概念的に翻訳し、モノクローナル抗体7E11および5B10の重鎖および軽鎖のCDR領域の部分的なアミノ酸配列を表2に示す。mAbの重鎖および軽鎖に由来する3個のCDRには、表2の中で下線を付けて示す。7E11および5B10の軽鎖は、94%のアミノ酸配列同一性を有しており、重鎖は91%のアミノ酸配列同一性を有している。mAb 7E11、5B10、と1H2は、IgG1イソ型であり、mAb 3G5と2F7は、IgG2aイソ型である。これらの5個のmAbのそれぞれは、κイソ型の軽鎖を有している。本発明者らは、このアプローチによって他のモノクローナル抗体の配列を分析した。
【0143】
(表2 mAb 7E11と5B10のアミノ酸配列)
【0144】
【表2】
(モノクローナル抗体7E11のエピトープマッピング)
Mab 7E11は、ラットNgR1およびヒトNgR1の両方に結合する。7E11の結合に関与しているエピトープを決定するために、本発明者らは、ラットNgR1の断片と合成ペプチドを作成し、7E11への結合についてそれらを試験した。
【0145】
全8個のLRRドメインと、N末端およびC末端キャップとを含むラットNgR1の組み換え断片(sNgR310)を、酸または臭化シアン(CNBr)のいずれかで処理し、ゲル電気泳動によって断片を分離した。未処理のsNgR310は、42kDaの見かけの分子量で移動した。sNgR310の酸処理により、15kDa(aa27〜aa122)と30kDa(aa123〜aa310)の2つの主要な切断産物が生じた。CNBr処理によって、3つの断片が生じ、これらは、33/35kDaの二重線(aa27〜229)(これは、異種のグリコシル化を受けた断片をおそらく示している)、10kDaの産物(aa241〜aa310)、および11アミノ酸の断片(aa230〜aa240)(これは、ゲル上には留まらない)である。ゲルのウェスタンブロットを7E11でプローブし、これによって、これが完全なラットNgR1(aa27〜aa310)、15kDaの酸による断片(aa27〜122)、および35kDaのCNBrによる断片(aa27〜aa229)に結合することが明らかになった。7E11は、30kDaの酸による断片(aa123〜aa310)、または10kDaのCNBrによる断片(aa241〜aa310)には結合しなかった。15kDaの酸による断片と35kDaのCNBrによる断片のいずれにも、配列LDLSDNAQLRVVDPTT(配列番号1)が含まれていた。これは、NgR1上にある1つのエプトープに対する7E11の結合と一致した。
【0146】
7E11結合部位を、さらに、sNgR310のトリプシンペプチド消化物を試験することによって分析した。HPLC分析はいくつかの断片を示し、これによって、いくつかのトリプシン反応性であるリジンおよびアルギニン残基がNgR1配列中に存在することが示された。7E11は、トリプシン消化ペプチドの1つだけに結合し、このことは、7E11がNgR1上の1つのエピトープだけに結合することについてのさらなる証拠を提供する。その後の質量スペクトル分析(MS)および配列分析によって、結合されるペプチドがAAAFGLTLLEQLDLSDNAQLR(配列番号26)であることが明らかになった。
【0147】
LDLSDNAQLRVVDPTTペプチド(配列番号1)について、さらなるマッピング分析を行った。ペプチドをトリプシンで消化した。これによって2つの主要な断片LDLSDNAQLR(配列番号27)およびVVDPTT(配列番号28)が生じた。それらに結合する7E11の能力を試験した。MS分析によって、抗体が、ペプチドLDLSDNAQLR(配列番号27)に結合し、したがって、このペプチドに7E11についての結合エピトープが含まれていることが明らかになった。このペプチド断片の中での詳細なMS分析によって、これもまた7E11に結合する、Asn115およびGln117の脱アミノ化、112または113でのアラニンの付加、または114でのセリンの付加を有しているペプチドを含む、いくつかのスクランブルペプチドが同定された(表3)。これらのデータは、このペプチド断片中に存在しているいくつかのアミノ酸残基には、7E11への結合に重要ではないものがある可能性を示している。
【0148】
(表3 7E11によって結合される変異体ペプチド)
【0149】
【表3】
LDLSDNAQLRVVDPTTペプチドを、エンドペプチダーゼAsp−Nでも消化し、7E11の結合を試験した。エンドペプチダーゼAsp−Nは、このペプチドを3つのペプチド断片、L、DLS、およびDNAQLRVVDPTT(配列番号36)に切断した。これらの産物のうち、7E11はDNAQLRVVDPTTペプチドに結合した。まとめると、トリプシンとAsp−Nでの切断のデータによって、さらに、7E11の結合エピトープは、それらの間で共有されている配列DNAQLR(配列番号37)に限定された。
【0150】
7E11はラットNgR1およびヒトNgR1に結合するが、マウスNgR1、ヒトNgR2、またはマウスNgR3には結合しないという観察に基づいて、種々の種に由来するNgR1、NgR2、およびNgR3のアミノ酸配列を、7E11の結合エピトープ中の重要な残基を推定するために分析した。配列アラインメントにより、ラットNgR1のアミノ酸110〜125と、ヒトNgR1の対応する配列が同一であり、マウスNgR1配列は119位の1つのアミノ酸だけが異なることが明らかになった(ラットNgR1およびヒトNgR1においてはArg119、マウスNgR1においてはHis119、表4)。
【0151】
(表4 種々の種に由来するNgRの配列アラインメント)
【0152】
【表4】
NgR1上のArg119は、7E11との結合に関与している。なぜなら、これは、ラットNgR1とヒトNgR1にはしっかりと結合するが、マウスNgR1にはほとんど結合しないからである。同様に、7E11はNgR3には十分には結合しないので、Ala116はエピトープには含まれる。なぜなら、DNAQLR配列(配列番号38)の中において、NgR3は、対応する配列においてアルギニンだけがNgR1とは異なるからである。DNAQLR配列のうち、NgR2の6個の残基のうちの4個は、ラットNgR1と同じである。Ala116とGln117が、アルギニンおよびヒスチジンでそれぞれ置き換えられている。これにより、Ala116が、7E11との結合に寄与している重要なアミノ酸残基ではあるが、Gln117の関与を必ずしも妨げるわけではないことが確認される。
【0153】
これらの接点を確認するために、LDLSDNAQLR配列(配列番号27)の中に点変異を含むいくつかのペプチドを作成し、7E11との結合について試験した。ペプチドを、MaxiSorp(登録商標)プレート(Nunc(登録商標))上に固定し、7E11の段階希釈物を載せた。得られたEC50値を表5に示す。7E11は、変異体Leu110AlaおよびAsp111Alaに結合し、もとのペプチドと同様のEC50値を有していた。Gla117Alaを試験すると、EC50値は30倍に増大し、Arg119Hisを試験すると、EC50値は25倍に増大していた。もっとも顕著なEC50値の変化は、Arg119をアラニンに変異させた場合に観察された。
【0154】
(表5 種々のEC50値を伴った変異体ペプチドに対する7E11の結合)
【0155】
【表5】
7E11結合エピトープの位置を、また、sNgR310についての最近明らかにされた結晶構造において決定した。予想したように、7E11エピトープが分子の表面に露出している構造が示された。残基Arg119、Gln117、Ala116、およびAsp114は、構造から突き出しており、一方、Leu118とAsn115は内部に存在している。エピトープは、構造の凹面にある酸性パッチの先端と、一方の側面に面している塩基性表面にある。
【0156】
(モノクローナル抗Nogo受容体−1抗体による可溶性Nogo受容体−1へのリガンド結合の阻害)
上記のように産生した抗Nogo受容体−1モノクローナル抗体を、これらがNogo受容体−1に対するリガンドの結合を阻害するかどうかを決定するために試験した。
【0157】
0.5μgの、下記のように産生したラットNogo受容体−1のアミノ酸残基26〜344と、ラットIgG1分子のヒンジ領域およびFc領域とを含む可溶性Nogo受容体−1融合タンパク質(sNogoR344−Fc)を、250μgのプロテインA、またはコムギ胚芽アグルチニン結合SPAビーズ(Amersham Pharmacia Biotech)上に、25℃で2時間かけて固定した。50μLのHEPES緩衝化インキュベーション培地(10mMのHEPES(pH7.4)、0.1%のウシ血清アルブミン、0.1%のオボアルブミン、2mMのMgCl2、2mMのCaCl2、およびプロテアーゼ阻害因子)中の、Fc−sNogoR−1に結合させたSPAビーズ、抗Nogo受容体−1 mAb、および1μLの125I−Nogo66(Amersham,2000Ci/mmol、1nM)を、それぞれの試料ウェルに添加した。16時間後、放射活性を、TopCount(登録商標)(Packard)を使用して、4連の試料において測定した。IC50値を、曲線適合分析によって計算した(図3)(PRISM、GraphPad Software,NJ)。いくつかの実験においては、本発明者らは、AP−リガンド結合体(例えば、AP−Nogo66)を使用し、アルカリホスファターゼ活性をモニターすることによって結合を検出した。本発明者らは、Nogo受容体−1に対するリガンドMAG−FcおよびAP−OM−gpの結合をブロックするmAbの能力もまたアッセイした。
【0158】
モノクローナル抗体7E11、5B10、1H2、3G5、および2F7は全て、Nogo66、MAG、およびOM−gpのsNogoR344−Fcに対する結合を阻害した。7E11および1H2についてのNogo66に対する計算したIC50は、それぞれ、400nMおよび60nMであった。Nogo受容体−1に対する3つのリガンドの結合のmAbによって媒介される阻害をモニターするELISAによるデータを、表6にまとめる。
【0159】
(表6 mAbはNogo受容体−1に対する、Nogo66、MAG、およびOM−gpの結合を阻害する)
【0160】
【表6】
置き換えの割合(%)を30nMの抗体について示し、そして特定のmAbについてのEC50を記載した曲線適合分析によって決定した。“−”は、検出できる程度の活性がないことを示し、“ND”は決定しなかったことを示す。
【0161】
(実施例2)
(Fab−ファージ抗Nogo受容体−1抗体の産生)
本発明の免疫原性Nogo受容体−1ポリペプチドに特異的に結合する抗Nogo受容体−1Fabファージ抗体をもまた、以下のように、Fab−ファージライブラリーをスクリーニングすることによって作成した。
【0162】
MorphoSys Fab−ファージライブラリーHuCAL(登録商標)GOLDを、組み換えラット可溶性sNogoR310−Fcタンパク質、およびラットNogo受容体−1を発現するCOS7細胞に対してスクリーニングした。Nogo受容体−1に特異的に結合するFab−ファージを精製し、特徴を決定した。14D5の重鎖は、VH2遺伝子に由来するものであり、軽鎖はVK1遺伝子に由来するモノであった。これらのFab−ファージ14D5の1つについて重鎖および軽鎖のCDRのアミノ酸配列を、表7に示す。
【0163】
(表7 14D5のCDRのアミノ酸配列)
【0164】
【表7】
14D5は、一価および二価のいずれの形態でもラットNogo受容体−1に結合する。さらに、14D5は、マウスNogo受容体−1およびヒトNogo受容体−1、ならびにヒトNogo受容体−2に結合するが、マウスNogo受容体−3には結合しない。
【0165】
(実施例3)
(抗Nogo受容体−1モノクローナル抗体によるNogo受容体−1の免疫沈降)
免疫沈降を行うために、100μLの溶解させた細胞、または50μLのPiPLCで処理した細胞を、400または450μLの抽出緩衝液(10mMのTris−HCl(pH7.2)、0.5%のTween−20(登録商標)、0.2mMのPMSF)、またはRIPA緩衝液と、それぞれ、30μLのプロテインAまたはG、および1〜2μgの抗体の存在下で、混合した。混合物を、4℃で16時間、震盪装置の中でインキュベートした。
【0166】
試料を、穏やかに攪拌してプロテインAまたはGが結合したビーズをペレットとした。ビーズを1mLの洗浄緩衝液(10mMのTris−HCl(pH7.2)、0.1%のTween−20(登録商標))で3回洗浄した。最後の洗浄は、10%のもとの洗浄緩衝液を使用して行った。
【0167】
ビーズを、10%のβ−メルカプトエタノールを含む100μLの2×SDS中に再度懸濁した。試料を室温でインキュベートし、その後、SDS−PAGEのために、4〜20%のTris−グリシンゲル上で泳動した。SDS−PAGEゲル分析によって決定すると、モノクローナル抗体3G5および2F7がNogo受容体−1を免疫沈降させた。
【0168】
(実施例4)
(ELISAによる抗体特異性の決定)
実施例1および2で産生したモノクローナル抗体およびFab−ファージ抗体の特異性を決定するために、本発明者らは、Nogo受容体−1ポリペプチドのパネルを使用してELISAを行った。パネルは、sNogoR310−Fc(ラットNogo受容体−1のアミノ酸26〜310とラットFc断片を含む融合タンパク質)、sNogoR344−Fc(上記を参照のこと)、ポリペプチドp−617(配列番号1)、ポリペプチドp−618(a19〜ラットNogo受容体−1のLRR7領域に由来するアミノ酸ポリペプチド;図2;配列11)、ならびに、ポリペプチドp−4およびp−5(それぞれ、Nogo受容体−1のLRR5およびLRRCT領域に由来するポリペプチド)から構成された。オボアルブミンおよびBSAを対照として使用した。図4に示すように、mAb 1H2、3G5、および2F7は全て、sNogoR344−Fcに特異的に結合した。同様の実験においては、これらの抗体は、ラットNogoレセプター−1のアミノ酸310〜344からなるポリペプチド(配列番号3)にも特異的に結合し、mAb 7E11および5B10はポリペプチドp−617(配列番号1)に特異的に結合した。
【0169】
sNogoR310−Fcでの免疫化による10個の抗体(1D9.3、1E4.7、1B4.3、2C4.3、1F10.3、2H1.4、1H3.3、1G4.1、1E4.1、および2G7.1)は、結合について互いに置換することができ、このことは、これらが、sNogoR310−Fc上の同様であるかまたは重複しているエピトープを認識することを示している。sNogoR310−Fcでの免疫化による他の3個の抗体(2C4.1、2F11.1、および1H4.1)は、アミノ酸残基26〜310に存在する異なるエピトープを認識する。
【0170】
本発明者らは、また、Fab−ファージ14D5を使用してELISA結合アッセイを行った。AP−Nogo66、AP−OM−gp、およびMAG−Fcリガンドを、固定したsNogoR344−Fcに結合させた場合には、1μMの14D5はNogoおよびMAGの結合を完全に阻害した。10μMの14D5が、sNogoR344−Fcに対するOM−gpの結合を完全に阻害するためには必要であった。
【0171】
(実施例5)
(神経突起成長についてのアッセイ)
ニューロンに対するCNSミエリンの阻害作用を弱める、上記で産生したモノクローナル抗体およびFab−ファージ抗体の能力を試験するために、Lab−Tek(登録商標)培養スライド(4ウェル)を、0.1mg/mLのポリ−D−リジン(Sigma(登録商標)))でコーティングした。CNSミエリンまたはPBSを、3μLの液滴としてスポットした。蛍光ミクロスフェア(Polyscience)を、ミエリン/PBSに添加して、後で液滴が同定できるようにした(Grandpre et al.,Nature 403,2000)。その後、Lab−Tek(登録商標)スライドをリンスし、10μg/mLのラミニン(Gibco(登録商標))でコーティングした。P3−4 Sprague Dawleyラットの子供から後根神経節(DRG)を取り出し、1mg/mLのコラゲナーゼ1型(Worthington)で解離させ、炎磨きパスツールピペット(fire−polished Pasteur pipette)で粉末状にし、ニューロン細胞を多く含むように予めプレーティングし、最後に予めコーティングしたLab−Tek(登録商標)培養スライド上に23,000細胞/ウェルでプレートした。培養培地は、5%の熱不活化ドナーウマ血清、5%の熱不活化ウシ胎児血清、および50ng/mLのmNGFを含むF12とし、37℃、5%のCO2で6時間インキュベートした。15μg/mLのmAb 7E11を、プレーティングの直後に添加した。
【0172】
スライドを、20%のスクロースを含む4%のパラホルムアルデヒドで20分間固定し、1:500希釈した、ニューロンマーカーである抗β−III−チューブリン(Covance TUJ1)について染色した。二次抗体である抗マウスAlexa Flour(登録商標)594(Molecular Probes)を1:300希釈し、スライドを、Gel/Mount(登録商標)(Biomedia(登録商標))を用いてカバースライドをかけた。OpenLab(登録商標)ソフトウェアを用いて5倍のデジタル画像を獲得し、MetaMorph(登録商標)ソフトウェアを使用して、神経突起成長の定量のために分析した。
【0173】
Mab 7E11は、神経突起成長のミエリンによって媒介される阻害からDRGニューロンを保護した(図5)。同様の結果が、mAb 1H2、および3G5を用いても観察された。
【0174】
ラットP7 DRGニューロンを、CNSミエリン基質上で培養した場合の神経突起成長の保護アッセイにおいては、二価の14D5もまた、神経突起成長を効率よく促進した。
【0175】
(実施例6)
(Nogo受容体−1でトランスフェクトした細胞についての7E11を用いた免疫組織化学)
実施例1に記載したように産生した抗Nogo受容体−1 mAbの結合特性をさらに特徴付けるために、本発明者らは、ラットNogo受容体−1またはヒトNogo受容体−1を発現する、固定されたおよび生存しているCOS−7または293細胞の両方に対する結合を比較した。
【0176】
(固定された細胞)
Nogo受容体−1でトランスフェクトした細胞と、トランスフェクトしていない細胞を、8ウェルLab−Tek(登録商標)培養スライド中にプレートし、PBS中の、4%のパラホルムアルデヒドで15分間固定し、10%の正常なヤギの血清、0.1%のTriton X−100で1時間ブロックした。Mab 7E11を、ブロッキング溶液中で15μg/mL、および1.5μg/mLで添加し、室温で2時間インキュベートした。Alexa(登録商標)結合二次抗体抗マウス(Molecular Probes)を、ブロッキング溶液中で1:300希釈で1時間インキュベートし、DAPIを全ての核種を標識するために二次抗体に対して5μg/mLで添加した。
【0177】
(生存している細胞)
トランスフェクトした細胞と、トランスフェクトしていない細胞を、8ウェルLab−Tek(登録商標)培養スライド中にプレートし、FACS緩衝液(4%のドナーウマ血清を含む)で、4℃で30分間ブロックし、FACS緩衝液中の15μg/mL、および1.5μg/mLの7E11とともに、4℃で1時間インキュベートし、リンスし、二次抗体抗マウス−Alexa(登録商標)(FACS緩衝液中で1:300希釈)とともに4℃で30分間インキュベートした。
【0178】
免疫組織化学染色実験は、全てのmAbがラットのNogo受容体−1を発現する細胞に結合したことを示した。mAb 7E11、2G7.1、および2C4.1は、ヒトNogo受容体−1を発現する、固定した細胞および生存している細胞の両方に結合した。
【0179】
(実施例7)
(脊髄挫傷のマウスモデル)
生体内でのニューロンに対する実施例1で産生した抗Nogo受容体−1 mAbの効果を試験するために、本発明者らは、マウスの脊髄挫傷モデルを使用した。
【0180】
雌のマウス(18〜22g)を、鎮痛薬および抗生物質で予防的に処置した。マウスに麻酔をかけ定位固定装置に置き、立体顕微鏡下にて脊柱固定を行った。脊髄への外傷を、落錘方法の修正版によって行った(M.Li et al.,Functional role and therapeutic implications of neuronal caspase−1 and −3 in a mouse model of traumatic spinal cord injury.Neuroscience Vol.99,pp.333−342,2000)。
【0181】
簡単に説明すると、T9およびT10椎弓切除術を行い、脊柱を、一対のマウス用の横方向のクランプを使用して安定させ、T9−T10横方向の処理を双方からサポートした。直径1.4mm、重さ2gのステンレス鋼製の衝突棒(impact rod)を、硬膜の上2.5cmに上げ、T10レベルで脊髄に落とした。外科手術の間、マウスは37℃の加温毛布の上に維持し、1mLの暖めた滅菌の生理食塩水を、脱水を避けるために外科手術後、それぞれのマウスに皮下投与した。膀胱を、反射的な膀胱の制御が回復するまで、1日に1回、手で絞った。
【0182】
全ての動物に、外科手術後8〜12時間おきに術後鎮痛剤を投与し、その後7日間にわたって抗生物質での処置を1日に2回行った。動物は、研究期間を通じて食物および水を自由に摂取できるようにした。抗Nogo受容体−1抗体を、以下のラットの脊髄離断モデルにおいて記載するように、28日間にわたり、髄腔内注射によって損傷部位に投与した。
【0183】
(実施例8)
(可溶性Nogo受容体−1融合タンパク質の特徴づけ)
可溶性Nogo受容体−1ポリペプチド(sNogoR−1)および融合タンパク質(Fc−sNogoR−1)を特徴付けるために、本発明者らは以下の実験を行った。
【0184】
3μgの可溶性Nogo受容体−1(sNogoR310−FcおよびsNogoR344−Fc)を、250μgのWGA−SPAビーズ上に固定し、100μLの最終容量の結合緩衝液(20mMのHEPES(pH7.4)、2mMのCa、2mMのMg、0.1%のBSA、0.1%のオボアルブミンおよびプロテアーゼ阻害因子)中の0.5μLの放射活性リガンド(最終濃度0.5nM)を投与した。リガンドには、10μMのNogo66、10μMの125I−Nogo40(NogoAのアミノ酸1〜40)、および10μLの抗Nogo受容体−1抗体上清を、それぞれのリガンドのセットについて含めた。Nogo40上の3個のチロシンを別々にヨウ素化し、Nogo40−A、−B、および−Cと、それぞれ命名した。三連の平均値を、較正した%結合放射活性として示す(図6、7、および8)。エラーバーはSEMを示す。阻害因子が存在しない条件下で結合した放射活性を100%とし、10μMのNogo40の存在下での最も低い結合放射活性を、データを較正するための0%とした。
【0185】
(実施例9)
(可溶性Nogo受容体−1融合タンパク質に対するリガンドの結合の阻害)
実施例8の結合アッセイと同様の結合アッセイを、実施例1で産生した2つのmAbの、sNogoR344−Fcに対する125I−Nogo66の結合を阻害する能力を試験するために使用した。Mab 2F7および3G5は、sNogoR344−Fcに対する125I−Nogo66の結合を阻害した。
【0186】
(実施例10)
(神経突起成長アッセイ)
Lab−Tek(登録商標)培養スライド(4ウェル)を、0.1mg/mLのポリ−D−リジン(Sigma(登録商標)))でコーティングした。CNSミエリンのみ、あるいは、sNogoR310、sNogoR310−Fc融合タンパク質、mAB 5B10、または対照としてのPBSと混合したCNSミエリンを、3μLの液滴として別々にスポットした。蛍光ミクロスフェア(Polyscience)を、ミエリン/PBSに添加して、後で液滴が同定できるようにした(Grandpre et al.,Nature 403,2000)。その後、Lab−Tek(登録商標)スライドをリンスし、10μg/mLのラミニン(Gibco(登録商標))でコーティングした。
【0187】
P3−4 Sprague Dawleyラットの子供から後根神経節(DRG)を取り出し、1mg/mLのコラゲナーゼ1型(Worthington)で解離させ、炎磨きパスツールピペット(fire−polished Pasteur pipette)で粉末状にし、ニューロン細胞を多く含むように予めプレーティングし、最後に予めコーティングしたLab−Tek培養スライド上に23,000細胞/ウェルでプレートした。培養培地は、5%の熱不活化ドナーウマ血清、5%の熱不活化ウシ胎児血清、および50ng/mLのmNGFを含むF12とし、37℃、5%のCO2で6時間インキュベートした。
【0188】
スライドを、20%のスクロースを含む4%のパラホルムアルデヒドで20分間固定し、1:500希釈した、ニューロンマーカーである抗β−III−チューブリン(Covance TUJ1)について染色した。二次抗体である抗マウスAlexa Flour(登録商標)594(Molecular Probes)を1:300希釈し、スライドを、Gel/Mount(登録商標)(Biomeda(登録商標))を用いてカバースライドをかけた。OpenLab(登録商標)ソフトウェアを用いて5倍のデジタル画像を獲得し、MetaMorph(登録商標)ソフトウェアを使用して、神経突起成長の量について分析した。
【0189】
sNogoR310、sNogoR310−Fc、およびmAb 5B10は全て、神経突起の成長のミエリンによって媒介される阻害からDRGニューロンを保護した(図9〜11)。sNogoR310を、ニワトリのニューロンを使用する同様のアッセイにおいて使用し、保護することを見出した。
【0190】
本発明者らは、また、ラミニンの存在下で増殖させた細胞と、ラミニンが存在しない条件下で増殖させた細胞を用いて実験を行うことにより、可溶性Nogo受容体の神経保護効果を試験した。ラミニンを含まない培地中でのニューロン細胞の成長は乏しく、ニューロンのストレス条件のモデルとなる。
【0191】
DRGを、ラットの子供から生後6〜7日で切り出し(P6〜7)、単細胞になるように解離させ、上記のようにポリ−D−リジンを予めコーティングした96ウェルプレート上にプレーティングした。いくつかのウェルには、2μg/mLのラミニンを2〜3時間かけて添加し、細胞をプレーティングする前にリンスした。18〜20時間のインキュベーションの後、プレートを、4%のパラホルムアルデヒドで固定し、1:500希釈した、ウサギ抗β−III−チューブリン抗体(Covance(登録商標))および1:100希釈した抗HuC/D(Molecular Probes)で染色した。蛍光二次抗体(Molecular Probes)を、1:200希釈で添加した。ArrayScan(登録商標)II(Cellomics(登録商標))を使用して、5倍のデジタル画像を獲得し、Neurite outgrowth applicationを使用することによって、ウェルあたりの平均の神経突起成長/ニューロンとして神経突起成長を定量した。1種類の条件について3つのウェルからの9個の5倍の画像を分析した。
【0192】
いくつかの実験においては、PC12細胞のサブクローン(Neuroscreen(登録商標))を使用した(Cellomics(登録商標))。Neuroscreen(登録商標)細胞を、200ng/mLのNGFを用いて7日間の間予め分化させ、分離させ、ポリ−D−リジンをあらかじめコーティングした96ウェルプレート上に再度プレーティングした。いくつかのウェルには、5μg/mLのラミニンを2〜3時間かけて添加し、細胞をプレーティングする前にリンスした。2日間のインキュベーションの後、プレートを、4%のパラホルムアルデヒドで固定し、1:500希釈した、ウサギ抗β−III−チューブリン抗体(Covance(登録商標))およびHoechst(核染色)で染色した。ArrayScan(登録商標)IIを使用して、DRG細胞中のものとして神経突起成長を定量した。
【0193】
sNogoR344−FcまたはラットIgGを、プレーティング時に、P6−7 DRGニューロンおよび分化させたNeuroscreen(登録商標)細胞に、溶液中で添加した。
【0194】
P6 DRGニューロンをラミニンが存在しない条件下で増殖させた場合のsNogoR344−Fcの神経保護作用を、1μMおよび10μMで観察した。神経突起成長の定量は、sNogoR344−Fcの添加に伴う用量依存性の増加を示した。ラミニン基質上でのDRGニューロンの成長に対する同じ濃度のsNogoR344の添加は、何ら特異な効果を生じなかった。このことは、sNogoR344−Fcはストレスを受けた細胞に対してのみ活性があることを示している。ラミニンが存在しない条件下での同じ濃度のsNogoR344−Fcの神経保護効果を、Neuroscreen(登録商標)細胞を用いて観察した。
【0195】
(実施例11)
(Fc−sNogoR−1融合タンパク質の産生および精製)
ラットNogo受容体−1のアミノ酸1〜310をコードするcDNA構築物を、哺乳動物発現ベクター中に含まれるラットIgG1 Fcに融合させ、このベクターをチャイニーズハムスター卵巣(CHO)(DG44)細胞にエレクトロポレーションした。細胞を、10%の透析したウシ胎児血清、2mMのグルタミン、および抗生抗かび剤を補充したα−MEM中で維持した。トランスフェクションの2日後、馴化培地を回収し、還元条件下でのウェスタンブロットによって分析した。約60kDaのタンパク質のバンドを、ポリクローナルウサギ抗Nogo受容体−1抗体を使用して検出した。細胞を増殖させ、R−PE結合ヤギ坑ラットIgG抗体を使用して分類した。2回目の分類の後、細胞を、96ウェルプレートに1細胞/ウェルの密度でプレーティングした。個々のウェルから分泌された可溶性Nogo受容体−1タンパク質のレベルを試験し、サンドイッチELISAを使用して比較した。ELISAプレートを、ヤギ坑ラットIgG Fcκ特異的抗体を用いてコーティングした。馴化培地をアプライした。結合した可溶性Nogo受容体−1タンパク質を、HRP結合ロバ抗ラットIgG Fab、Fc特異的抗体によって検出した。クローン4C12は最も高い分泌レベルを有していた。4C12を、スピナーフラスコの中のCHO−M7培地中で拡大、増殖させた。分泌レベルは、37℃で約10mg/Lであった。
【0196】
sNogoR310−Fc融合タンパク質を発現するCHO細胞を、大スケールで培養した。1.7Lの濃縮した馴化培地を、10Lのバイオリアクターを運用することによって得た。1.0MのTris−HCl(pH8.9)の1/10用量を添加することによって、pHを上昇させた。固形の塩化ナトリウムおよびグリシンをそれぞれ、3.0Mおよび1.5Mになるように添加した。10mMのTris−HCl、3Mの塩化ナトリウム、1.5Mのグリシン(pH8.9)で平衡化させた60mLのプロテインA−Sepharose(登録商標)カラムを準備した。濃縮した馴化培地をカラムに、蠕動ポンプを使用して1.5mL/分でアプライした。カラムを、300mLの10mMのTris−HCl、3Mの塩化ナトリウム、1.5Mのグリシン(pH8.9)で洗浄し、その後、120mLの5mMのTris−HCl、3Mの塩化ナトリウム(pH8.9)で洗浄した。タンパク質を、25mMのリン酸ナトリウム、100mMの塩化ナトリウム(pH2.8)を用いて溶出させた。10mLの画分を、1.0mLの1.0MのHEPES(pH8.5)を含むチューブに回収した。タンパク質画分をプールし、3×2Lの5mMのリン酸ナトリウム、300mMのNaCl(pH7.4)に対して透析した。
【0197】
(実施例12)
(脊髄離断アッセイ)
生体内で機能回復を促進するそれらの能力を試験するために、sNogoR−1融合タンパク質を、ラットの脊髄離断アッセイにおいて試験した。
【0198】
Alzet(登録商標)浸透圧ポンプに、使用する日に新しく作成した試験溶液(PBS中のsNogoR310−Fc)を充填した。充填濃度は、5μMおよび50μMになるように計算した。ポンプを、動物への埋め込みの前に、37℃で>40時間になるように準備した。雌のLong Evansラットに、術前鎮痛剤と鎮静剤を投与し、イソフルラン(O2中3%)を使用して麻酔した。
【0199】
ラットを、定位フレームに入れ、運動皮質を露出させ、左右双方から菅追跡剤(tract tracing agent)BDA(10,000MW)を注入した。その後、ラットに、T5−T6で脊髄の背部片側切断を行い、その後、試験化合物を投与するための髄腔内カテーテルとポンプシステムを挿入した(1つのグループあたりn=11)。
【0200】
ラットを回復させ、ラットは外科手術後28日まで生存していた。BBBシステムを用いた行動採点法により、研究の生存期間が終わる直前である損傷の誘導後28日までを記録した。潅流および固定後、脊髄を採取し、凍結防止し、切片とし、染色し、軸索のカウントを行った。
【0201】
Basso−Beattie−Bresnahan(BBB)運動機能評価尺度(Basso et al.,1996,Neurotrauma 13,343−359)、傾斜板試験、および傾斜格子歩行試験(Li and Strittmatter,2003,J Neurosci.2003,23,4219−27)を、損傷の後、ラットとマウスにおいてモニターした。傾斜板試験のために、本発明者らは、50cm×60cmの板がマウスが滑り落ちることなく5秒間傾いていられる最大角を測定した。傾斜格子歩行のために、マウスに、45度の傾斜のワイヤー格子をよじ登るようにトレーニングした(2.54cm四方の35cmの長さ)。格子面よりも下に後肢が落ちた回数を、下から上までの各歩行について記録した。ラットの行動試験については、BBB運動機能尺度、格子歩行分析および足跡の分析を行った。格子歩行のために、ラットに、ワイヤー格子上を歩くようにトレーニングし(2.54cm四方の70cmの長さ)、格子面よりも下に後肢が落ちた回数を数えた。足跡の分析のために、ラットの後肢の歩行パターンを、90cmの通路を途切れることなく動く間に、インクで記録し、それぞれの側のストライドの長さおよびストライドの幅を計算した(Metz et al.,2000、Brain Res.,883,165−177)。これらの行動試験の全てを、少なくとも2匹の個体によって行った。外科手術、行動試験、および組織学的分析の間中、研究は、ミニポンプの中の化合物の実体について盲検とした。
【0202】
sNogoR310−Fcは機能の回復を促進した(図12)。
【0203】
(実施例13)
(ラットの脊髄挫傷アッセイ)
生体内でのニューロンに対する可溶性Nogo受容体−1ポリペプチドおよび融合タンパク質の作用を、ラットの脊髄挫傷アッセイにおいて試験した。
【0204】
雌のフードをかぶせたLong Evansラット(170〜190g)を、鎮痛剤と抗生物質で予防的に処置した。外科手術の10分前に、動物を2.5mg/kgのミダゾラムを用いてi.p.で鎮静し、O2中の2〜3%のイソフルランで麻酔した。その後、ラットの毛を剃り、アルコールとベタジンで拭き、眼に眼用滑剤を点眼した。次に、中線切開を行い、T7からT12脊椎を露出させた。
【0205】
背部椎弓切除術を、T9 1/2とT10で行い、脊髄を露出させた。ラットを衝撃部材の上に載せた。T7およびT8セグメントを最初にクランプし、その後、T11およびT12セグメントを尾側のクランプに取り付けた。やわらかい物質をラットの胸部の下に入れた。衝撃部材の棒をゼロ位置にセットし、電気設置クリップを傷の端に取り付けた。その後、衝撃部材の棒を25.0mmまで上げ、露出させた脊髄の真上の位置になるように適切に調節した。次に、衝撃部材の棒を、露出させた脊髄に当たるように放し、衝撃部材の棒をすぐに拾い上げた。
【0206】
その後、ラットを取り外し、Gelform(登録商標)を傷の上に置いた。傷を覆う筋肉を縫合し、切開部を外科用ホッチキスで留めた。動物を、麻酔から回復するまでインキュベーターに入れた。ラットには、必要に応じて、抗生物質、鎮痛剤、および生理食塩水を投与した。膀胱を、機能が回復するまで毎朝晩に絞った。
【0207】
可溶性Nogo受容体−1融合タンパク質(例えば、sNogoR310−Fc)を、上記のラットの脊髄離断モデルにおいて記載したように、髄腔内投与した。BBB採点法を、外科手術の1日後、その後4週間から6週間まで毎週行った。
【0208】
(実施例14)
(トランスジェニックマウスにおけるsNogoR310の発現)
本発明者らは、生体内で発現させた場合のその効果を試験するために、可溶性Nogo受容体−1タンパク質を発現するトランスジェニックマウスを作成した。
【0209】
本発明者らは、マウスsNogoR310 cDNA(Nogo受容体−1のアミノ酸1〜310に対応する)を、C−3123ベクターのNotI部位にクローニングした。このベクターの中では、sNogoR310の発現はグリア細胞繊維性酸性タンパク質(gfap)遺伝子調節エレメントの制御下にある。グリア細胞繊維性酸性タンパク質遺伝子調節エレメントは、損傷の部位の反応性星状細胞からの分泌の増強を伴う、高レベルの発現を可能にする。本発明者らは、得られたベクターをAatIIとSfiIで連続して消化し、gfap::sNogoR310構築物を3.4kbの断片上に単離した。本発明者らは、この断片を胚にマイクロインジェクションして、トランスジェニックマウスを作成した。本発明者らは、PCRによって、トランス遺伝子が組み込まれていることを確認し、5つの創始株を同定した。本発明者らは、最も高い発現レベルを有している2つの創始株のヘテロ接合型雄を、雌のC57BL/6Jマウスと交配させた。本発明者らは、ヘテロ接合型トランスジェニックマウスにおいてGFAP陽性細胞がsNogoR310を発現し、分泌していることを、Nogo受容体−1に対して惹起させた抗体を使用したウェスタンブロット分析によって確認した。
【0210】
本発明者らは、プロテアーゼ阻害因子(Roche)を補充したTris緩衝化生理食塩水中で皮質および脊髄をホモジナイズし、ホモジネートを40,000rpmで4℃で20分間遠心分離した。本発明者らは、抗体特異性を高めるために上清を4%のパラホルムアルデヒドで20分間処理し、免疫ブロッティングの前に透析した。本発明者らは、RIPA緩衝液(PBS中の1%のTriton(登録商標)X−100、0.5%のデオキシコール酸ナトリウム、0.1%のSDS)中での超音波処理によって微粒子画分をホモジナイズし、得られたホモジネートを遠心分離し、この上清(界面活性剤可溶性微粒子画分)を上記のように処理した。本発明者らは、20μgの脳または脊髄タンパク質を、1:2000希釈のNogo受容体−1に対して惹起させられたウサギ抗血清を使用する免疫ブロットによって分析した。本発明者らは、AP結合抗ウサギIgGおよびNBT/BCIP AP基質とのインキュベーションによって、免疫反応性を視覚化した。
【0211】
本発明者らは、2つのトランスジェニック株Tg08およびTg01に由来する皮質および脊髄の界面活性物質を含まない可溶性抽出物中に、分泌された37kDaのsNogoR310を検出した。しかし、37kDaまたは81kDaのいずれの可溶性Nogo受容体−1タンパク質は、同腹子の野生型(WT)マウス中に存在するとしても、ごくわずかであった。微粒子画分の試験により、WTおよびトランスジェニックマウスの両方に相当のレベルの内因性Nogo受容体−1が存在することが明らかになった。
【0212】
(実施例15)
(損傷の後のトランスジェニックマウスにおけるsNogoR310の発現)
本発明者らは、背部の全片側切断(over−hemisection)損傷を行うことにより、トランスジェニックマウスにおけるsNogoR310の発現に対するCNS損傷の影響を試験した。本発明者らは、実施例14に記載したようにヘテロ接合型の雄をC57/BL6雌と交配させることによって、sNogoR310トランスジェニックマウスと、トランスジェニックではない対照動物を得た。
【0213】
本発明者らは、成体である雌のヘテロ接合型トランスジェニックマウスまたは同腹子であるWTマウス(10〜16週齢)に深く麻酔をかけ、完全な椎弓切除術を行って、T6およびT7のレベルで脊髄の背部を完全に露出させた。本発明者らは、30ゲージ針と一対のマイクロシザーズでT6に背部全片側切断(over−hemisection)を行い、背部と背側部の皮質脊髄路(CST)を完全に切断した。本発明者らは、記号のついた針を脊髄の背部を数回貫通させて、病変が確実に1.0mmの深さになるようにした。本発明者らは、椎弓切除術を行った筋肉層を縫合し、外科用ホッチキスで背面で皮膚を閉じた。皮質脊髄路を追跡するために、本発明者らは、脊髄損傷の14日後に、頭蓋骨内において右側に大脳皮質を覆うバーホール(burr hole)を作成した。本発明者らは、トレーサーBDA(MW 10,000、PBS中で10%)(Molecular Probes,Eugene,OR)を、皮質表面から0.7mmの深さで4つの注射部位に注入した。損傷の4週間後、マウスにPBSを噴門から潅流させ、その後、4%のパラホルムアルデヒドを潅流させた。sNogoR310の発現実験に使用したマウスには、トレーサーの注射を全く行わなかった。
【0214】
ウェスタンブロット分析に使用したマウスについては、T3とL3の間のレベルで脊髄を採取し、損傷の14日後の潅流は行わなかった。Nogo受容体−1免疫組織化学的染色に使用したマウスには、片側切断の10日後に4%のパラホルムアルデヒドを潅流させ、損傷をうけた脊髄を切片を作成するために取り出した。トランスジェニックマウスとWTマウスの損傷した脳におけるsNogoR310の発現を試験するために、皮質の刺し傷を、定位固定装置に取り付けた11番手術用メスを用いて作成した(David Kopf,Tujunga,CA)。ブレグマ(Bregma)の0.5mm後方、中線から1.5mm側方、3.5mmの深さに、4mmの傍矢状の切開を行った。
【0215】
本発明者らは、トランスジェニックマウスにおいては、損傷の10日後に、脊髄の可溶性抽出物中においてsNogoR310のレベルの増大を観察したが、WTマウスにおいては見られなかった。このことは、病変の周辺でのGFAPの損傷後のアップレギュレーションと一致する。この原因が、Nogo−Aの代償性アップレギュレーションではないことを確認するために、本発明者らはその発現を試験し、WTマウスとトランスジェニックマウスの両方に由来する、完全な、または損傷を受けた皮質および脊髄のいずれにおいても同様であったことを見出した。
【0216】
本発明者らは、病変の領域を含む損傷を受けた脳および脊髄の、Nogo受容体−1およびGFAPに対する抗体での免疫染色によって、損傷を受けたCNS中でのsNogoR310の細胞性発現を試験した。反応性の星状細胞神経膠の一般的な形態は、WTマウスとトランスジェニックマウスの間では差はなかったが、細胞内空間および細胞外空間の両方において、Nogo受容体−1について染色された密度は、WTマウスよりも、gfap::sNogoR310トランスジェニックマウスにおいて顕著に高かった。このことは、トランスジェニックマウスの病変の周辺での高いsNogoR310の発現を示している。Nogo受容体−1タンパク質は、トランスジェニックマウスだけにおいて、星状細胞マーカーであるGFAPと一緒に存在している。トランスジェニックの試料においては、大幅に強い拡散した非細胞性染色もまた存在し、これは、細胞外空間にあるsNogoR310と一致する。神経細胞体Nogo受容体−1染色は、WTマウスとトランスジェニックマウスの両方において検出された。
【0217】
(実施例16)
(分泌されたsNogoR310はトランスジェニックマウスにおいてCSTの萌芽を誘導する)
本発明者らは、トランスジェニックマウスにおける病変の周辺でのsNogoR310の発現の増大が損傷を受けた軸索の再生を生じるかどうかを試験した。
【0218】
本発明者らは、Li and Strittmatter,2003,J.Neurosci.,23,4219−27に記載されているように、順行性トレーサーであるビオチンデキストランアミン(BDA)を右運動皮質に注入することによって、下行皮質脊髄路(CST)の完全性を調べた。同腹子のWTマウスにおいては、突出した背部CST(dCST)は、病変に対して吻側にしっかりとまとまっており、いくつかの背側部のCST線維は同側上に見ることができる。少数のBDA標識された短い付随する萌芽が、灰白質、具体的には、前索に突き出しているが、萌芽は、トレーサーの注入に対して対側性の脊髄の側に、大部分が確認できた。しかし、損傷を受けたsNogoR310トランスジェニックマウスに由来する背部片側切断に対して吻側の切片は、極めて異なるBDA標識パターンを示した。高密度のBDA標識されたCST線維が、株Tg08または株Tg01に由来する全てのトランスジェニックマウスにおいて突出したdCSTの外側に観察された。異所性の線維は、灰白質の領域を貫通して伸び、いくつかの線維は外側白質および背側部の白質にまで達していた。いくつかの線維(横断切片あたり4〜12個の萌芽)が、脊髄の反対側(トレーサーの注入部位に対して同側)に見られた。付随する萌芽のマイクロデンシトメトリーによる測定は、sNogoR310トランスジェニックマウスにおいて萌芽の密度がおよそ10倍増大したことを示していた。病変に対して1から4mm吻側の傍矢状の縦断面の試験により、dCST線維がsNogoR310トランスジェニックマウスにおいては腹部の灰白質の領域に入るような多数の分岐する萌芽を延ばし、これが同腹子であるWT動物とは対照的であることが明らかになった。一般的には、トランスジェニックマウスの病変に対して吻側の萌芽のパターンおよび広がりは、Nogo受容体−1アンタゴニストペプチドNEP1−40で全身的に処置したマウスにおいて観察されるものと似ている(Li and Strittmatter,2003)。
【0219】
これらの結果は、分泌されたsNogoR310がトランスジェニックマウスにおいてCST萌芽を誘導することを示している。
【0220】
(実施例17)
(sNogoR310トランスジェニックマウスにおいては、CST軸索の再生により病変部位を回避して脊髄の先端へとバイパスする)
本発明者らは、トランスジェニックマウスから、病変部位に対して吻側4mmから尾側4mmの脊髄(合計8mm長)を単離した。これを、グルタルアルデヒド重合アルブミンマトリックスに包埋し、ビブラトーム(vibratome)(30μmの厚み)の上で傍矢状に切断した。本発明者らは、損傷部位に対して吻側5〜7mm、尾側5〜7mmの脊髄から、横断切片(50μm)を回収した。ラットにおけるsNogoR310−Fcの注射実験のために、病変部位から、吻側10mmから尾側10mmまでまたがる脊髄を、バイブレーションミクロトーム上で傍矢状(50μm)に切断した。横断切片を、損傷部位に対して吻側11〜16mmから尾側11〜16mmの脊髄から回収した。本発明者らは、これらの切片を、アビジン−ビオチン−ペルオキシダーゼ複合体とともにインキュベートし、ニッケル増強ジアミノベンジジンHRP反応によってBDAトレーサーを視覚化した(Grandpre,2002,Nature,417,547−551)。本発明者らは、間接的な免疫蛍光によって、セロトニン免疫組織化学(抗−5−HT抗体)のためにいくつかの切片を処理した。病変の領域を視覚化するために、本発明者らは、いくつかの切片を、GFAPに対する抗体(Sigma(登録商標)St.Louis,MO)で二重染色した。本発明者らは、封入剤で切片を封入し、脱水し、保護した。
【0221】
本発明者らは、損傷後にトランスジェニックマウスにおいて発現されたsNogoR310によって誘導された線維 (実施例16)が、病変の領域を抜けて尾側の脊髄に達し、機能回復をもたらすかどうかを試験した。
【0222】
カメラルシーダディスプレイに取り込んだ損傷部位の全域を越える連続する傍矢状切片は、病変から数ミリメートルの再生しつつあるCST線維の全体的な分布パターンを示す。WTマウスに由来する切片は、損傷部位を越えて伸びるCST繊維は示さなかった。sNogoR310トランスジェニックマウスに由来する同様の切片は、離断領域を超える多数のCST線維と、先端部の灰白質および白質の領域に伸びる突起を、高度に分岐したパターンで示した。片側切断に対してすぐ吻側には、高密度のBDA標識されたCST萌芽が、突出したdCST突起から病変の領域の方向に存在するが、ほとんどのCST萌芽は、傷跡の形成および組織の空孔が顕著である離段領域を超えることはできなかった。少しではあるが極めて顕著な、再生しつつある軸索の画分が、背部および背側部の灰白質および白質の残っている組織の細胞間橋を通って病変部位を回避する。さらに、ごく少数のCST線維は、離断領域自体を通過して、病変の背部および背側部の脊髄を通って先端領域へと伸びるようである。病変の近傍では、再生しつつある線維の進路は通常、蛇行性であり、吻側のCST中の正常なまっすぐの線維とは極めて異なる。側枝および樹枝状の線維は、脊髄の先端部の灰白質において最も頻繁に見られる。再構築により、それぞれのトランスジェニックマウスにおいて病変に対して尾側1〜4mmのいずれかのレベルで、吻−尾軸において、5〜15本のBDA標識された再生しつつある線維の追跡が明らかになった。背部片側切断に対して5〜7mm尾側の横断面については、BDA標識されたCST軸索が、それぞれのトランスジェニックマウスにおいて灰白質と白質の両方の領域において見られた。トランスジェニックマウスについての繊維の数は、矢状断面の最も近いレベルとほぼ同数のBDA標識されたCST線維を示した。
【0223】
CST線維に加えて、他の下行路、例えば、縫線核脊髄線維もまた、マウスの運動機能に関与している。このマウスの背部全片側切断(over−hemisection)モデルにおいては、離断によりセロトニン作動性線維の大部分が損傷を受け、前角においてはこれらの線維の密度がおよそ80%減少した。尾側の脊髄の前角におけるセロトニン線維の全長の分析は、トランスジェニックマウスにおいてはWTグループよりもはるかに多い数のこれらの線維を示し、このことは、トランスジェニックマウスにおけるsNogoR310の成長促進作用が、1つの軸索の下行路には限定されないことを示している。
【0224】
(実施例18)
(sNogoR310のトランスジェニックでの発現は運動機能の回復を改善する)
CST軸策の追跡およびセロトニン作動性線維の分析は、トランスジェニックマウスにおいて星状細胞から分泌されたsNogoR310が、脊髄の損傷をうけた下行軸索の広範囲の解剖学的再生を刺激することを示している。本発明者らは、これらの再生した線維が機能の回復に利点をもたらすかどうかを決定するために、実施例12に記載したいくつかの行動試験を行った。
【0225】
BBB試験によって評価した場合には、WTマウスは、4週間の生存期間の間に運動機能を一部回復した。損傷後4週間では、ほとんどのWTマウスが、しっかりと体重を支えながら、しっかりと足底で踏みしめることによって特徴付けられるレベルに回復したが、これらは、時折、前肢−後肢の同調を生じるだけで、主に、最初に表面に接触させた足の位置で旋回していた。対照的に、株Tg08およびTg01の両方に由来するsNogoR310トランスジェニックマウスのBBBスコアは、7〜28日の観察期間を通じて、対照のグループよりも有意に高かった(図13Aおよび13B)。損傷の28日後には、ほとんどのトランスジェニックマウスが一貫して前肢−後肢の同調を示し、主に、足の位置は体に対して並行であった。
【0226】
本発明者らは、sNogoR310トランスジェニックマウスの能力をさらに特徴付けるために、2つのさらなる行動試験を使用した。最初に、本発明者らは、5秒以内にマウスがその格子を落ちることなく傾けられた板につかまっていられる最大角を測定した。背部片側切断による損傷の前には、トランスジェニックマウスおよびWTマウスの両方が、55度に傾けられた板の上でその姿勢を維持できた。損傷の7〜28日後に、維持できる角度は、全てのマウスにおいて低くなったが、トランスジェニックマウスが維持できる角度は、対照のグループよりも有意に大きかった(図13C)。別の行動試験においては、マウスに、垂直に対して45度の角度に置いた格子に上らせ、格子面から後肢が脱落した回数を数えた(Metz et al.,2000)。損傷前のトレーニングの間のこの試験においては、マウスはミスすることはなかった。損傷後2〜6週間の期間の間には、WTマウスは多数回の足を踏み外すミスをし、最少程度の改善しか見られなかった。対照的に、sNogoR310トランスジェニックマウスは、この期間の間に、格子をよじ登ることにおいて進行性の改善を示し、ほとんどの改善は損傷後1〜3週間の間に生じた(図13D)。したがって、星状細胞からsNogoR310を分泌するトランスジェニックマウスは、胸部脊髄の片側切断の後、CSTの再生、縫線核脊髄萌芽、および運動機能の改善を示した。
【0227】
(実施例19)
(sNogoR310−Fcタンパク質の髄腔内投与によりCSTの萌芽が誘導される)
脊髄の外傷後の可溶性Nogo受容体−1の成長促進の利点についての第2の試験として、本発明者らは、精製したタンパク質を髄腔内投与した。
【0228】
本発明者らは、安定性を増大させ精製を促進するために、ラットのNogo受容体−1のリガンド結合ドメイン(27〜310)をラットIgG1 Fcドメインに融合させた。本発明者らは、安定にトランスフェクトされたCHO細胞からタンパク質を精製した。このタンパク質は、マウスsNogoR310−Myc Hisについて以前に示されたとおり(Fournier et al.,2002、J Neurosci.,22,8876−8883;Liu et al.,2002、Science,297,1190−1193)、Nogo−66、MAG、およびミエリンの作用を生体外でブロックする。本発明者らは、胸中部の背部片側切断損傷を行ったラットに、浸透圧ミニポンプによりsNogoR310−Fcタンパク質を髄腔内投与した。損傷後の4週間の生存期間の間に、1.2mgのsNogoR310−Fcタンパク質を、それぞれのラットに局所投与した。ビヒクル(1.2mgのラットIgG)での処置を受けたラットにおいては、片側切断部位に対して吻側の切片は、しっかりとまとまった突出した背部CSTを示し、病変部位の上部にはごく少数の異所性BDA標識CST線維を示した。sNogoR310−Fcタンパク質を投与した損傷を受けたラットに由来する、病変に対して吻側の切片は、極めて異なる標識パターンを示した。BDA標識されたCSTからの多数の異所性線維の萌芽が、横断切片および傍矢状切片から観察された。いくつかの場合には、突起が、中線付近のdCST領域から脊髄の境界線に向かってのび、背側部CSTと混ざりあっていた。芽がでかかった軸索は灰白質を通り、白質よりもさらに伸びていた。dCSTに隣接する異所性の芽がでかかった線維の測定(横断切片において≧100μm、矢状断片においては≧200μm)により、sNogoR310−Fcで処置したラットにおけるさらなる増大が明らかになった。
【0229】
(実施例20)
(sNogoR310で処置したラットにおいて、CST軸索は脊髄の先端へと再生する)
本発明者らは、雌のSprague−Dawleyラット(190〜250g)に深く麻酔をかけ、T6〜7の脊髄レベルで椎弓切除術を行い、脊髄を露出させた。本発明者らは、脊髄の背部の半分を、30ゲージ針と一対のマイクロシザーズで切断し、CSGT経路の背部を完全に切断し、11番手術用メスの鋭い部分を脊髄の背部の半分を横断させることにより、病変の深さ(1.8mm)を確保した(Grandpre et al.,2002,Nature,417,547−551)。PBS中の1.2mgのラットIgG、またはPBS中の1.2mgのsNogoR310−Fc融合タンパク質を充填した浸透圧ミニポンプ(Alzet(登録商標)、2ML4、2mLの容量、2.5μL/h、28日間の投与)を、動物の背中の皮膚の下の筋肉に埋め、縫合した。ミニポンプの出口に接続したカテーテルを、硬膜の中の小さな穴を通じてT7〜8のレベルで脊髄の髄腔内空間に挿入した。
【0230】
Nogo受容体−1アンタゴニストタンパク質の注入により、ラットの片側切断に対して吻側で広範囲の萌芽を誘導したが、より重要な問題は、芽を出しつつあるCST線維が脊髄の先端に突き出すかどうか、そして運動機能の回復に関係しているかどうかである。ビヒクルで処置したラットに由来する病変を横切る縦断面は、病変のレベルより下では、検出できる程度のBDA標識された腹部CST線維は示さなかったか、またはごく少数のBDA標識された腹部CST線維しか示さなかった(GrandPre et al.,2002;Weidner et al.,2001、Proc.Natl.Acad.Sci.USA,98,3513−3518)。sNogoR310−Fcで処置したラットに由来する同様の切片は、多くのBDA標識線維が離断部位をバイパスし、主に腹部および腹側両面脊髄の組織の細胞間橋を通って尾側の脊髄に向かって突出することを示した。星状細胞マーカーGFAPについての免疫染色は、離断の程度が脊髄中心菅の領域よりも深くまで到達していることを示した。突出した背部CST中の吻側の線維の直線的なプロフィールとは異なり、再生したCST線維は、通常、脊髄先端、特に、灰白質の領域においては、高度に分岐している軌跡を残す。これらの線維は、脊髄の多くの領域で検出されたが、これらは、脊髄の中心部分、および脊髄の背部の半分から脊髄の隅々にわたって容易に見ることができた。矢状断面によるCST線維の数は、sNogoR310−Fcで処置したラットのそれぞれに由来する病変に対して1〜2mm尾側ではおよそ20個のBDA標識された軸索を、病変に対して7〜8mm遠位では15個の追跡された軸索を示した。
【0231】
一般的には、これらの線維の分岐パターンは、NEP 1−40ペプチドで局所的に処置された動物から観察されるパターンと類似しているが、それぞれの萌芽にさらに付随する分岐が、sNogoR310−Fcタンパク質で処置した切片から見られた。脊髄の先端からの萌芽の測定は、sNogoR310−Fcで処置したラットにおいてそれぞれの萌芽に付随する全ての長さが、NEP 1−40で処置した動物よりも2倍長いことを示した。Nogo受容体−1アンタゴニストで処置したグループの両方における脊髄に対して1〜10mm尾側の萌芽の数(長さに関して≧200μm)は、対照のグループよりもおよそ20〜40倍多かった。萌芽も、NEP 1−40で局所的に処置したラットよりもsNogoR310−Fcで処置したラットにおいて多く見られたが(25個の萌芽/ラットに対しておよそ50個の萌芽/ラット)、この差は、統計学的に有意ではなかった(p=0.1713、t検定)。
【0232】
再生しつつあるCST軸索を、sNogoR310−Fc処置を受けさせたラットにおいて、片側切断に対して11〜15mm尾側の脊髄の横断切片において観察した。これらの線維は、脊髄の灰白質と白質の両方において検出された。灰白質で検出された繊維は、多くの場合には、白質の領域で見られるものよりも多くの分岐の付随を示した。対照的に、ビヒクルで処置したグループに由来する横断切片においては、腹部の白質の領域において、ごくたまにBDA標識が見られ、これは、損傷を受けていない腹部のCST軸索と一致した。脊髄の先端のこのレベルでは、Nogo受容体−1アンタゴニスト(sNogoR310−FcおよびNEP 1−40)で処置されたグループの両方に由来するBDA標識されたCST線維の平均の数は、ビヒクルで処置されたラットよりもおよそ20倍多かった。まとめると、いずれのNogo受容体アンタゴニスト(sNogoR310−Fcタンパク質およびNEP 1−40ペプチド)も、脊髄の先端において劇的なCST軸索の再生を生じるが、前者によって誘導される萌芽は、より高度に分岐したパターンを示す。
【0233】
(実施例21)
(局所sNogoR310−Fcは、損傷を受けたラットの脊髄において赤核脊髄およびセロトニン作動性軸索の萌芽を誘導する)
片側切断の14日後、バーホールを、CST線維の軌跡をたどるために、下肢の感覚運動皮質に重なる頭蓋骨のそれぞれの側に空けた。順行性のニューロンのトレーサーであるBDA(PBS中に10%、3.5μL/皮質)を、それぞれの側の硬膜から1.5mmの深さの7個の注射部位に注射した(Grandpre,2002)。ラットにおいて赤核脊髄路の軌跡をたどるために、トレーサーBDA(1μL:MW 10,000;PBS中10%
)を、左側の赤核(ブレグマの前方5.8mm、0.7mm側方、頭蓋骨表面に対して7.0mm腹部側)に注射した。BDAの注射の2週間後、これらの動物にBPSを潅流させ、その後、4%のパラホルムアルデヒドを潅流させ、組織を組織学のために採取した。
【0234】
損傷を受けた赤核脊髄路(RST)繊維の修復は、脊髄損傷後の機能の改善に関与している(Liu et al.,1999,J.Neurosci.,19,4370−4387)。CNSニューロンにおけるNogo受容体−1の広範囲の分布(Wang et al.,2002,J.Neurosci.,22,5505−5515)は、Nogo受容体−1のそのアンタゴニストでの阻害により、損傷後のRST軸索の再成長を生じさせることができることを可能にする。損傷を受けたRSTに対するsNogoR310−Fcの作用を試験するために、この経路の完全性を、左の赤核にBDAを注射することによって軌跡をたどった。脊髄のレベルでは、RST線維は通常は、脊髄の背側部の白質の領域に局在しており、これを、この研究の背部片側切断によって離断した。対照ラットに由来する病変から11〜15mm吻側の横断切片においては、少数の短いBDA標識線維が、突出したRSTと背部の角状灰白質の間に見られた。sNogoR310−Fcで処置した同じレベルの切片は、メインのRSTと背部の角状灰白質との間に多くの連結している線維を示した。SCIに対して11〜15mm遠位の横断切片については、ビヒクルで処置されたラットにおいてはBDA標識されたRST線維は見られなかった。対照的に、sNogoR310−Fcでの処置を受けた同じレベルの切片は、トレーサーの注射部位に対して対側にある灰白質および白質の両方において多くのBDA標識されたRST線維を示した。分岐パターンを伴ういくつかの萌芽が、BDA注射部位に対して同側の灰白質に見られた。
【0235】
赤核脊髄線維もまた、sNogoR310−Fcで処置された脊髄損傷を受けたラットにおいて試験した。免疫染色は、病変に対して11〜15mm吻側のセロトニン作動性線維の密度を示し、これは、ビヒクルで処置されたグループとsNogoR310−Fcで処置されたグループの間で類似していた。病変の11〜15mm下の切片においては、sNogoR310−Fcで処置されたラットにおけるセロトン線維は、対照のグループの線維の2倍多く存在した。これらの結果は、sNogoR310−Fcタンパク質によるNogo受容体−1の阻害に対する反応性が、CST線維に限定されておらず、他の下方路、例えば、赤核脊髄およびセロトニン作動性軸索もまた、Nogo受容体−1アンタゴニズムに反応することを示している。
【0236】
(実施例22)
(sNogoR310−Fcでの局所処置によりラットにおける機能回復が改善する)
sNogoR310−Fcタンパク質の髄腔内投与により、外傷性脊髄損傷後にいくつかの下方路において軸索の再生が刺激される。本発明者らは、このタンパク質により、損傷を受けた脊髄における機能回復もまた改善されるかどうかを試験した。
【0237】
片側切断の2週間後に、ビヒクルで処置したラットの運動機能BBBスコアは、12の安定なレベルに達した(図14A)。外傷の4週間後、対照のほとんど(7匹のちの6匹)は、頻度が一定である体重を支える足底での踏み、および頻度が一定である前肢−後肢の同調を有していたが、これらは、主に、最初に表面に接触させた足の位置で旋回していた。対照的に、sNogoR310−Fcタンパク質での処置を受けさせたラットにおいては、運動機能スコアは、外傷後2〜4週間の間改善し続けた。損傷の4週間後、sNogoR310−Fcで処置された9匹の動物全てが、一貫した前肢−後肢の同調、および、最初に試験表面と接触させた足の位置と並行な足の位置を有していた。
【0238】
格子の歩行は、脊髄損傷後の下行する細かい運動機能の制御の欠損を評価するために使用されている(Metz et al.,2000)。この能力には、前肢−後肢の同調、ならびに、腹側両面、皮質脊髄、および赤核脊髄線維によって媒介される随意運動の制御が必要である。損傷の前のトレーニングの間は、全てのラットが格子の棒の上に彼らの後肢を正確に置いていた。損傷後2〜4週間は、対照のラットは、1回のセッションについて8〜9回のミスをし、時間とともに最少程度の改善しか見られなかった。対照的に、sNogoR310−Fcで処置したラットは、格子歩行について進行性の改善を示し、有意に少ないミスしかしなかった(平均で4〜7回/セッション)。改善のほとんどは、損傷後2〜3週間で生じた。対照のグループにおける後肢の足跡の分析は、損傷を受けていないラットまたはsNogoR310−Fcでの処置を受けた損傷を受けたラットと比較して、片側切断後4週間で、ストライドの長さが顕著に小さく、立脚の幅が大きかったことを示した(図14C)。したがって、これらの複数の行動試験は、Nogo受容体−1機能の、アンタゴニストタンパク質の局所注射での遮断により、損傷後の運動機能の回復に改善が生じたことを示している。
【0239】
(生物学的寄託物)
ハイブリドーマHB 7E11(ATCC(登録商標)登録番号PTA−4587)、HB 1H2(ATCC(登録商標)登録番号PTA−4584)、HB 3G5(ATCC(登録商標)登録番号PTA−4586)、HB 5B10(ATCC(登録商標)登録番号PTA−4588)、およびHB 2F7(ATCC(登録商標)登録番号PTA−4585)を、アメリカンタイプカルチャーコレクション(「ATCC(登録商標)」、10801 University Boulevard,Manassas,VA 20110−2209,USAに、2002年8月9日に寄託した。
【0240】
当業者に明らかであるように、多数の変更および修飾を、本発明の精神を逸脱することなく本発明の好ましい実施形態に対して行うことができる。全てのこのようなバリエーションは、本発明の範囲に含まれるように意図される。
【図面の簡単な説明】
【0241】
【図1】図1は、Nogo受容体−1の構造の模式図である。ヒトsNogoR310には、残基26〜310が含まれており、sNogoR344には残基26〜344が含まれている。ラットsNogoR310には残基27〜310が含まれており、sNogoR344には残基27〜344が含まれている。
【図2】図2は、Vector Nti(登録商標)ソフトウェアを使用したNogo受容体−1タンパク質についての抗原性プロットを示す。ラットP−617は配列番号10であり、ラットP−618は配列番号11である。
【図3】図3Aは、抗Nogo受容体−1抗体7E11の結合活性を示すグラフである。このグラフは、Nogo受容体−1に対するNogo66の結合に対する7E11の濃度の効果を示す。図3Bは、抗Nogo受容体−1抗体1H2の結合活性を示す。グラフは、sNogoR344−Fc(本明細書中と、米国特許出願番号60/402,866においては、Fc−sNogoR344またはIg−sNogoR344とも呼ばれる)に対するNogo66の結合に対する1H2の濃度の効果を示す。Fc−MAGはsNogoR344−Fcへの結合についてNogo66と競合しなかった。
【図4】図4は、抗Nogo−R−1抗体1H2、3G5、および2F7についてのELISAの結果を示す。固定された抗原の存在下でのOD450に対する抗体の効果が決定された。固定された抗原はsNogoR310−Fc(本明細書中と、米国特許出願番号60/402,866においては、Fc−sNogoR310またはIg−sNogoR310とも呼ばれる)、sNogoR344−Fc、p−617、p−618、p−4、p−5、オボアルブミンおよびBSAである。
【図5】図5は、種々の量のミエリンの存在下でのラットDRG神経突起成長に対する、モノクローナル抗体7E11の効果を示すグラフである。
【図6】図6Aは、以下の競合因子の存在下での125I−Nogo66および125I−Nogo40に対するsNogoR310の結合の効果を示すグラフである:Nogo66、Nogo40、および抗Nogo受容体−1モノクローナル抗体上清。図6Bは、sNogoR310に対する125I−Nogo66の結合活性を示す。
【図7】図7は、sNogoR310への125I−Nogo40の結合に対するsNogoR310−Fcの効果を示すグラフである。
【図8】図8は、125I−Nogo40に対するsNogoR310−Fcの結合活性を示すグラフである。
【図9A】図9Aは、ミエリンの存在下、またはミエリンが存在しない条件下での、神経突起成長/細胞に対するsNogoR310の効果のグラフである。
【図9B】図9Bは、ミエリンの存在下、またはミエリンが存在しない条件下での、神経突起成長に対するsNogoR310の効果のグラフである。
【図10A】図10Aは、漸増量のミエリンの存在下、またはミエリンが存在しない条件下での、P4ラットDRG神経突起成長に対するsNogoR310−Fcの効果を示すグラフである。
【図10B】図10Bは、PBS、PBS+sNogoR310−Fc、20ngのミエリン、およびミエリン+sNogoR310−Fcでの処置後の、神経突起の数/細胞を示す。
【図11】図11は、漸増量のミエリンの存在下での、DRG神経突起成長/細胞に対するモノクローナル抗体5B10の効果を示すグラフである。
【図12】図12は、ラットの脊髄離断モデルにおける損傷の誘導後30日までのBBBスコアに対するsNogoR310−Fcの効果を示すグラフである。
【図13】図13Aおよび13Bは、株08または株01によるWTまたはトランスジェニックマウスにおける背部片側切断後の時間の関数として、運動機能BBBスコアを報告する。図13Cは、WTおよびトランスジェニックマウスについて、損傷後の時間の関数として、最大の耐え得る斜平面角を示す。図13Dは、損傷後の時間の関数として、斜めの格子をよじ登る間の後肢のミスを示す。全てのグラフにおいて、それぞれのグループにおいて7〜9匹のマウスによる平均±s.e.m.が報告される。トランスジェニックのグループによる値は、WTのマウスの値とは統計学的に異なる(二重のアスタリスク、P<0.01;スチューデントt検定)。
【図14】図14Aは、ビヒクルで処置した動物またはsNogoR310−Fcで処置した動物における背部片側切断後の時間の関数として、運動機能BBBスコアを示す。図14Bは、損傷後の時間の関数として、格子を歩行する間の後肢のミスを示す。図14Cは、足跡の分析を示し、これにより、損傷を受けさせていないラット、または損傷を受けさせたsNogoR310−Fcラットよりも、対照マウスにおいて、ストライドがより短く、ストライド幅がより大きいことが明らかである。全てのグラフにおいて、それぞれのグループにおいて7〜9匹のラットによる平均±s.e.m.が報告される。sNogoR310−Fcグループの値は、対照とは統計学的に異なる(図14A〜B)。対照の値は、図14Cにおいては、SCIなしのラット、またはSCI+sNogoR310−Fcラットとは統計学的に異なる(アスタリスク、P<0.05;二重のアスタリスク、p<0.01;スチューデントt検定)。
【技術分野】
【0001】
(発明の分野)
本発明は、神経生物学および分子生物学に関する。さらに具体的には、本発明は、免疫原性Nogo受容体−1ポリペプチド、Nogo受容体−1抗体、その抗原結合断片、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸に関する。本発明はさらに、このようなNogo受容体抗体、その抗原結合断片、免疫原性Nogo受容体−1ポリペプチド、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸を含む組成物、さらには、このようなNogo受容体抗体、その抗原結合断片、免疫原性Nogo受容体−1ポリペプチド、可溶性Nogo受容体およびその融合タンパク質、ならびにそれらをコードする核酸を作成し、使用するための方法に関する。
【背景技術】
【0002】
(発明の背景)
ニューロンの軸索および樹状突起は、ニューロンからの長い細胞の伸張である。伸張途中の軸索または神経突起の先端部には、成長円錐として知られている特殊な領域が含まれている。成長円錐は、局所環境を感知し、ニューロンの標的細胞に向かっての軸索の成長を導く。成長円錐は、いくつかの環境による合図、例えば、表面接着、成長因子、神経伝達物質、および電場に反応する。円錐での成長の誘導には、種々のクラスの接着分子、細胞内シグナル、さらには、成長円錐を刺激し阻害する因子が関与している。成長途中にある神経突起の成長円錐は種々の速度で前進するが、通常は、1日あたり1から2ミリメートルの速度で前進する。
【0003】
成長円錐は手の形状であり、広がった扁平な伸張(微小突起または糸状仮足)を有しており、これは胚の中の表面に対して別々に接着する。糸状仮足は、絶えず活動しており、一部の糸状仮足は、成長円錐の内部へと引っ込み、一方、他の糸状仮足は、基体を通り抜けて伸張し続ける。種々の糸状仮足の間での伸張により、葉状仮足が形成される。
【0004】
成長円錐は、その葉状仮足と糸状仮足を用いて、その前方と両側の領域を探索する。伸張によって、成長に好ましくない表面と接触すると、それは引っ込む。伸張によって、好ましい成長表面に接触すると、それは伸張し続け、その方向に成長円錐を導く。成長円錐は基体の表面レパートリーにおける小さなバリエーションによって導かれ得る。成長円錐が適切な標的細胞に到達すると、シナプス結合が作成される。
【0005】
神経細胞の機能は、その当面の環境におけるニューロンと他の細胞との間での接触によって大きく影響される(U.Rutishauser,T.M.Jessell,Physiol.Rev.1988,68,p.819)。これらの細胞としては、特定の神経膠細胞、中枢神経系(CNS)の乏突起膠細胞、および末梢神経系(PNS)のシュワン細胞(これは、神経軸索をミエリン鞘で覆う(多層膜の絶縁構造))(G.Lemke,An Introduction to Molecular Neurobiology,Z.Hall,Ed.[Sinauer,Sanderland,Mass.,1992],p.281)が挙げられる。
【0006】
CNSニューロンは、損傷後に再生する能力を有しているが、この能力は、ミエリンの中に存在する阻害タンパク質が原因であり、そして、それらの局所環境に通常見られる他のタイプの分子もおそらく原因で、再生が阻害される(Brittis and Flanagan,Neuron 2001,30,pp.11−14;Jones et al.,J.Neurosci.2002,22,pp.2792−2803;Grimpe et al.,J.Neurosci.2002,22,pp.3144−3160)。
【0007】
乏突起膠細胞上に見られるいくつかのミエリン阻害タンパク質、例えば、NogoA(Chen et al.,Nature,2000,403,434−439;Grandpre et al.,Nature 2000,403,439−444)、ミエリン関連糖タンパク質(MAG,McKerracher et al.,Neuron 1994,13,805−811;Mukhopadhyay et al.,Neuron 1994,13,757−767)、および乏突起膠細胞糖タンパク質(OM−gp、Mikol and Stefansson,J.Cell.Biol.1988,106,1273−1279)の特性が明らかにされている。これらのタンパク質は、それぞれ、ニューロンのNogo受容体−1のリガンドであることが別々に示されている(Wang et al.,Nature 2002,417,941−944;Liu et al.,Science,2002,297,1190−93;Grandpre et al.,Nature 2000,403,439−444;Chen et al.,Nature,2000,403,434−439;Domeniconi et al.,Neuron,2002,35,283−90)。
【0008】
Nogo受容体−1は8個のロイシンを多く含む繰り返しを含む、GPI結合膜タンパク質である(Fournier et al.,Nature 2001,409,341−346)。阻害タンパク質(例えば、NogoA、MAG、およびOM−gp)と相互作用すると、Nogo受容体−1複合体は、成長円錐の崩壊および神経突起成長の阻害を導くシグナルを変換する。
【発明の開示】
【発明が解決しようとする課題】
【0009】
そのリガンドに対するNogo受容体−1の結合を阻害し、ミエリンによって媒介される成長円錐の崩壊と、神経突起成長の阻害を弱める分子が、緊急に必要とされている。
【課題を解決するための手段】
【0010】
(発明の要旨)
本発明は、可溶性Nogo受容体−1ポリペプチドおよびそれを含む融合タンパク質、ならびに、Nogo受容体−1の特異的な免疫原性領域に対する抗体およびその抗原性断片に関する。本発明はまた、本発明の抗体に結合する免疫原性Nogo受容体−1ポリペプチドにも関する。本発明はまた、Nogo受容体−1に結合するモノクローナル抗体によって結合されるNogo受容体−1ポリペプチドにも関する。このようなポリペプチドは、特に、免疫原として、または、本発明の抗体に対して同様の特異性を有している抗体を同定するために抗体をスクリーニングするために使用することができる。本発明はさらに、本発明のポリペプチドをコードする核酸、そのような核酸を含むベクターおよび宿主細胞、ならびにペプチドを作成する方法に関する。本発明の抗体、可溶性受容体、および受容体融合タンパク質は、Nogo受容体−1をアンタゴナイズするかまたはブロックし、そしてそのリガンドに対するNogo受容体−1の結合を阻害すること、ニューロンにおける成長円錐の崩壊を阻害すること、およびニューロンにおける神経突起成長または萌芽(sprouting)の阻害を減少させることについて有用である。
【0011】
いくつかの実施形態においては、本発明により、AAAFGLTLLEQLDLSDNAQLR(配列番号26);LDLSDNAQLR(配列番号27);LDLSDDAELR(配列番号29);LDLASDNAQLR(配列番号30);LDLASDDAELR(配列番号31);LDALSDNAQLR(配列番号32);LDALSDDAELR(配列番号33);LDLSSDNAQLR(配列番号34);LDLSSDEAELR(配列番号35);DNAQLRVVDPTT(配列番号36);DNAQLR(配列番号37);ADLSDNAQLRVVDPTT(配列番号41);LALSDNAQLRVVDPTT(配列番号42);LDLSDNAALRVVDPTT(配列番号43);LDLSDNAQLHVVDPTT(配列番号44);およびLDLSDNAQLAVVDPTT(配列番号45)からなる群より選択されるポリペプチドが提供される。
【0012】
いくつかの実施形態においては、本発明により、本発明のポリペプチドをコードする核酸が提供される。いくつかの実施形態においては、核酸は、発現制御配列に動作可能であるように連結される。いくつかの実施形態においては、本発明により、本発明の核酸を含むベクターが提供される。
【0013】
いくつかの実施形態においては、本発明により、本発明の核酸を含むか、または本発明のベクターを含む宿主細胞が提供される。いくつかの実施形態においては、本発明により、本発明の核酸またはベクターを含む宿主細胞を培養すること、および宿主細胞または培養培地からポリペプチドを回収することを含む、本発明のポリペプチドを産生する方法が提供される。
【0014】
いくつかの実施形態においては、本発明により、宿主を、本発明のポリペプチドまたは本発明の核酸を含むかまたは本発明のベクターを含む宿主細胞で免疫化し、抗体を回収する工程を含む、抗体を産生する方法が提供される。本発明により、また、この方法によって産生される抗体、またはその抗原結合断片も提供される。
【0015】
いくつかの実施形態においては、本発明により、本発明のポリペプチドに特異的に結合する抗体またはその抗原結合断片が提供される。ここでは、抗体はハイブリドーマ細胞株HB 7E11によって産生されるモノクローナル抗体(ATCC(登録商標)登録番号PTA−4587)ではない。
【0016】
本発明のいくつかの実施形態においては、抗体または抗原結合断片は、(a)ニューロンの神経円錐の崩壊を阻害する;(b)ニューロンにおける神経突起成長および萌芽の阻害を減少させる;ならびに(c)リガンドに対するNogo受容体−1の結合を阻害する。いくつかの実施形態においては、神経突起成長および萌芽は、軸索成長である。いくつかの実施形態においては、ニューロンは中枢神経系のニューロンである。
【0017】
いくつかの実施形態においては、本発明の抗体はモノクローナルである。いくつかの実施形態においては、本発明の抗体はマウス抗体である。いくつかの実施形態においては、本発明の抗体は、ヒト化抗体、キメラ抗体、および単鎖抗体からなる群より選択される。
【0018】
いくつかの実施形態においては、本発明により、リガンドへのNogo受容体−1の結合を阻害する方法が提供される。この方法には、Nogo受容体−1を本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、リガンドは、NogoA、NogoB、NogoC、MAG、およびOM−gpからなる群より選択される。
【0019】
いくつかの実施形態においては、本発明により、ニューロンにおける神経円錐の崩壊を阻害するための方法が提供される。この方法には、ニューロンを、本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。
【0020】
いくつかの実施形態においては、本発明により、ニューロンにおける神経突起成長および萌芽の阻害を減少させるための方法が提供される。この方法には、ニューロンを、本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、神経突起成長または萌芽は、軸索成長である。いくつかの実施形態においては、ニューロンは中枢神経系のニューロンである。
【0021】
いくつかの実施形態においては、本発明により、薬学的に許容される担体と、本発明の抗体またはその抗原結合断片を含む組成物が提供される。いくつかの実施形態においては、組成物にはさらに、1つ以上のさらなる治療薬が含まれる。
【0022】
いくつかの実施形態においては、本発明により、死滅の危険性があるニューロンの生存を促進する方法が提供される。この方法には、ニューロンを、有効量の本発明の抗Nogo受容体−1抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、ニューロンは生体外にある。いくつかの実施形態においては、ニューロンは哺乳動物中に存在する。いくつかの実施形態においては、哺乳動物は、多発性硬化症、ALS、ハンチントン病、またはアルツハイマー病、パーキンソン病、糖尿病性神経障害、脳卒中、外傷性脳損傷、または脊髄損傷の兆候または症状を呈する。
【0023】
いくつかの実施形態においては、本発明により、哺乳動物において死滅の危険性があるニューロンの生存を促進する方法が提供される。この方法には、(a)本発明の抗Nogo受容体−1抗体またはその抗原結合断片を発現する培養された宿主細胞を提供すること;および(b)ニューロンの部位またはその付近に哺乳動物中に宿主細胞を導入することが含まれる。
【0024】
いくつかの実施形態においては、本発明により、哺乳動物中にある死滅の危険性があるニューロンの生存を促進する遺伝子治療方法が提供される。この方法には、本発明の抗Nogo受容体−1抗体またはその抗原結合断片をコードするヌクレオチド配列を含むウイルスベクターを、ニューロンの部位またはその付近に投与することが含まれる。ここでは、抗Nogo受容体−1抗体または抗原結合断片は、哺乳動物中のヌクレオチド配列から、ニューロンの生存を促進するために十分な量で発現される。
【発明を実施するための最良の形態】
【0025】
(発明の詳細な説明)
(定義および一般的な技術)
他の場所で定義されない限りは、本明細書中で使用される全ての技術用語および科学用語は、本発明が属する分野の当業者によって通常理解されている意味と同じ意味を有する。矛盾する場合は、本出願が含む定義が支配する。また、他の方法で状況によって必要とされない限りは、単数形の用語に複数形が含まれ、複数形の用語に単数形が含まれる。本明細書中で言及される全ての刊行物、特許、および他の参考文献は、全ての目的についてそれらの全体が引用によって本明細書中に組み入れられる。
【0026】
本明細書中に記載されるものと同様であるかまたはそれらと同等である方法および材料を、本発明の実施または試験において使用することができるが、適切な方法および材料は以下に記載される。材料、方法、および実施例は、例示的なものにすぎず、限定を意図するものではない。本発明の他の特徴および利点は、詳細な説明から、そして特許請求の範囲から明らかであろう。
【0027】
本明細書および特許請求の範囲全体を通じて、語「含まれる(comprise)」、または「含む(comprises)」もしくは「含んでいる(comprising)」のようなバリエーションは、記載される整数、または整数のグループの包含を暗に意味するが、任意の他の整数または整数のグループを排除するようには理解されない。
【0028】
本発明をさらに定義するために、以下の用語と定義が本明細書において提供される。
【0029】
本明細書中で使用される場合は、「抗体」は、完全な免疫グロブリン、またはその抗原結合断片を意味する。本発明の抗体は、任意のイソ型またはクラス(例えば、M、D、G、E、およびA)、あるいは、任意のサブクラス(例えば、G1−4、A1−2)であり得、そしてカッパ(κ)軽鎖またはラムダ(λ)軽鎖のいずれかを有し得る。
【0030】
本明細書中で使用される場合は、「Fc」は、パパインでの消化によって得ることができる抗体の重鎖定常領域の部分を意味する。
【0031】
本明細書中で使用される場合は、「NogoR融合タンパク質」は、異種ポリペプチドに融合させられた可溶性Nogo受容体−1部分を含むタンパク質を意味する。
【0032】
本明細書中で使用される場合は、「ヒト化抗体」は、ヒト以外の配列の少なくとも一部がヒト配列で置き換えられている抗体を意味する。ヒト化抗体を作成するための方法の例は、米国特許第6,054,297号、同第5,886,152号、および同5,877,293号に見ることができる。
【0033】
本明細書中で使用される場合は、「キメラ抗体」は、第1の抗体に由来する1つ以上の領域と、少なくとも1つの他の抗体に由来する1つ以上の領域を含む抗体を意味する。第1の抗体とさらなる抗体は、同じ種に由来するものであっても、異なる種に由来するものであってもよい。
【0034】
本明細書中、および米国特許出願番号第60/402,866において使用される場合は、「Nogo受容体」、「NogoR」、「NogoR−1」、「NgR」、および「NgR−1」はそれぞれ、Nogo受容体−1を意味する。
【0035】
(Nogo受容体−1ポリペプチド)
1つの態様においては、本発明は、免疫原性であるNogo受容体−1ポリペプチドに関する。本発明のいくつかの実施形態においては、免疫原性ポリペプチドは、LDLSDNAQLRVVDPTT(ラット)(配列番号1);LDLSDNAQLRSVDPAT(ヒト)(配列番号2);AVASGPFRPFQTNQLTDEELLGLPKCCQPDAADKA(ラット)(配列番号3);AVATGPYHPIWTGRATDEEPLGLPKCCQPDAADKA(ヒト)(配列番号4);およびCRLGQAGSGA(マウス)(配列番号5)からなる群より選択されるアミノ酸配列から本質的には構成される。
【0036】
いくつかの実施形態においては、本発明は、Nogo受容体−1に結合するモノクローナル抗体によって結合されるNogo受容体−1ポリペプチドに関する。いくつかの実施形態においては、ポリペプチドは、7E11モノクローナル抗体によって認識される。いくつかの実施形態においては、ポリペプチドは、LDLSDNAQLR(配列番号28);LDLSDDAELR(配列番号30);LDLASDNAQLR(配列番号31);LDLASDDAELR(配列番号32);LDALSDNAQLR(配列番号33);LDALSDDAELR(配列番号34);LDLSSDNAQLR(配列番号35);LDLSSDEAELR(配列番号36);DNAQLRVVDPTT(配列番号37);DNAQLR(配列番号38);ADLSDNAQLRVVDPTT(配列番号42);LALSDNAQLRVVDPTT(配列番号43);LDLSDNAALRVVDPTT(配列番号44);LDLSDNAQLHVVDPTT(配列番号45);およびLDLSDNAQLAVVDPTT(配列番号46)からなる群より選択される。
【0037】
いくつかの実施形態においては、本発明は、配列番号1〜5、26〜27、29〜37、および41〜45のポリペプチドをコードする核酸に関する。本発明のいくつかの実施形態においては、核酸分子は、発現制御配列(例えば、pCDNA(I))に連結される。
【0038】
本発明はまた、本発明のポリペプチドをコードする核酸を含むベクターにも関する。本発明のいくつかの実施形態においては、ベクターはクローニングベクターである。本発明のいくつかの実施形態においては、ベクターは発現ベクターである。本発明のいくつかの実施形態においては、ベクターには、少なくとも1つの選択マーカーが含まれる。
【0039】
本発明はまた、上記の核酸またはベクターを含む宿主細胞にも関する。
【0040】
本発明はまた、宿主細胞を培養する工程を含む、本発明の免疫原性ポリペプチドを産生する方法にも関する。いくつかの実施形態においては、宿主細胞は原核生物である。いくつかの実施形態においては、宿主細胞は真核生物である。いくつかの実施形態においては宿主細胞は酵母である。
【0041】
(抗体)
本発明はさらに、本発明のNogo受容体−1ポリペプチドに特異的に結合する抗体またはその抗原結合断片に関する。いくつかの実施形態においては、抗体または抗原結合断片は、配列番号1〜5、26〜27、29〜37、および41〜45からなる群より選択されるアミノ酸配列から本質的には構成されるポリペプチドに結合する。本発明の抗体または抗原結合断片は、生体内または生体外で産生することができる。抗体または抗原結合断片の産生については以下で議論される。
【0042】
本発明の抗体またはその抗原結合断片は、リガンド(例えば、NogoA、NogoB、NogoC、MAG、OM−gp)に対するNogo受容体−1の結合を阻害し、神経突起成長および萌芽、具体的には、軸索成長のミエリンによって媒介される阻害を減少させ、ミエリンによって媒介される成長円錐の崩壊を弱める。
【0043】
いくつかの実施形態においては、抗Nogo受容体−1抗体またはその抗原結合断片は、マウスのものである。いくつかの実施形態においては、Nogo受容体−1はラット由来のものである。他の実施形態においては、Nogo受容体−1はヒトのものである。いくつかの実施形態においては、抗Nogo受容体−1抗体またはその抗原結合断片は組み換えのもの、遺伝子操作されたもの、ヒト化されたもの、および/またはキメラである。
【0044】
いくつかの実施形態においては、抗体は、モノクローナル7E11(ATCC(登録商標)登録番号PTA−4587);モノクローナル1H2(ATCC(登録商標)登録番号PTA−4584);モノクローナル2F7(ATCC(登録商標)登録番号PTA−4585);モノクローナル3G5(ATCC(登録商標)登録番号PTA−4586);およびモノクローナル5B10(ATCC(登録商標)登録番号PTA−4588)からなる群より選択される。いくつかの実施形態においては、抗体はポリクローナル抗体46である。
【0045】
例示的な抗原結合断片は、Fab、Fab’、F(ab’)2、Fv、Fd、dAb、および相補性決定領域(CDR)断片を含む断片、単鎖抗体(scFv)、キメラ抗体、ポリペプチドに特異的抗原結合性(例えば、免疫接着体)を付与するために十分である免疫グロブリンの部分を少なくとも含むダイアボディーおよびポリペプチドである。
【0046】
本明細書中で使用される場合は、FdはVHドメインとCH1ドメインからなる断片を意味し、Fvは、抗体の単一のアームのVLドメインとVHドメインからなる断片を意味し、そしてdAbは、VHドメインからなる断片を意味する(Ward et al.,Nature 341:544−546,1989)。本明細書中で使用される場合は、単鎖抗体(scFv)は、VL領域とVH領域が、それらが1本のタンパク質鎖として作成されることを可能にする合成のリンカーを介して、一価の分子を形成するように対を形成している抗体を意味する(Bird et al.,Science 242:423−426,1988、およびHuston et al.,Proc.Natl.Acad.Sci.USA,85:5879−5883、1988)。本明細書中で使用される場合は、ダイアボディーは、VHとVLドメインが1本のポリペプチド鎖の上で発現されるが、同じ鎖の上にある2つのドメインの間で対を形成することを可能にするためには短すぎるリンカーを使用しており、それによって、これらのドメインが別の鎖の相補ドメインと対を形成するようにさせて、2つの抗原結合部位を作成する、二重特異的抗体を意味する(例えば、Holliger,P.et al.,Proc.Natl.Acad.Sci.USA,90:6444−6448,1993、およびPoljak,R.J.,et al.,Structure 2:1121−1123,1994を参照のこと)。本明細書中で使用される場合は、目的の抗原に特異的に結合する免疫接着体は、1つ以上のCDRが、共有結合によって、または非共有結合によってのいずれかで組み込まれ得る分子を意味する。
【0047】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1抗体のサブユニットポリペプチドが提供される。ここでは、サブユニットポリペプチドは、(a)重鎖またはその可変領域;および(b)軽鎖またはその可変領域からなる群より選択される。
【0048】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1抗体のサブユニットポリペプチドの、重鎖またはその可変領域、あるいは、軽鎖またはその可変領域をコードする核酸が提供される。
【0049】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1抗体の超可変領域(CDR)、またはCDRをコードする核酸が提供される。
【0050】
(免疫化)
本発明の抗体は、適切な宿主(例えば、ヒト、マウス、ラット、ヒツジ、ヤギ、ブタ、ウシ、ウマ、爬虫類、魚類、両生類を含む脊椎動物、ならびに、鳥類、爬虫類、および魚類の卵の中)の免疫化によって作成することができる。
【0051】
いくつかの実施形態においては、宿主は、本発明の免疫原性Nogo受容体−1ポリペプチドで免疫される。他の実施形態においては、宿主は、完全な、または破壊された細胞の細胞膜と会合させられたNogo受容体−1で免疫化され、本発明の抗体が、本発明のNogo受容体−1ポリペプチドに対する結合によって同定される。
【0052】
いくつかの実施形態においては、Nogo受容体−1抗原は、免疫応答を刺激するために、アジュバントとともに投与される。アジュバントは、多くの場合、抗原に対する免疫応答を誘発するために、抗原に加えて投与されることが必要とされる。これらのアジュバントは、通常、非特異的炎症を促進し、免疫化の部位への単核の食細胞の動員を伴う、不溶性または非分解性の物質である。アジュバントの例としては、フロイトのアジュバント、RIBI(ムラミルジペプチダーゼ)、ISCOM(免疫刺激複合体)またはそれらの断片が挙げられるが、これらに限定されない。
【0053】
抗体を作成するための方法の概要については、例えば、Harlow and Lane(1988),Antibodies,A Laboratory Manual,Yelton,D.E.et al.,(1981);Ann.Rev.of Biochem.,50,pp.657−80.,およびAusubel et al.,(1989);Current Protocols in Molecular Biology(New York:John Wiley & Sons)を参照のこと。本発明の免疫原性Nogo受容体−1ポリペプチドとの免疫反応性の決定は、当該分野で周知のいくつかの方法のいずれかによって行うことができ、例えば、免疫ブロットアッセイおよびELISAが含まれる。
【0054】
(抗体の産生および抗体を産生する細胞株)
本発明のモノクローナル抗体は、例えば、Harlow and Lane(1988)前出に記載されているような標準的な手順によって行うことができる。
【0055】
簡単に説明すると、適切な時点で、動物が屠殺され、リンパ節および/または脾臓B細胞が、当該分野で周知であるいくつかの技術のうちのいずれか1つによって不死化させられる。このような技術としては、例えばEBVでの形質転換、または不死化細胞株、例えば、骨髄腫細胞との融合が挙げられるが、これらに限定されない。その後、細胞は、クローンに分けられ、それぞれのクローンの上清が、本発明の免疫原性Nogo受容体−1ポリペプチドに特異的な抗体の産生について試験される。ハイブリドーマの選択、クローニング、および拡大の方法は当該分野で周知である。同様に、免疫グロブリン遺伝子のヌクレオチド配列およびアミノ酸配列を同定するための方法は当該分野で公知である。
【0056】
本発明の抗体を産生するための他の適切な技術には、リンパ球のNogo受容体−1への、もしくは本発明の免疫原性ポリペプチドへの生体外での暴露、あるいは、ファージもしくは同様のベクター中の抗体のライブラリーの選択が含まれる。Huse et al.,Science,246,pp.1275−81(1989)を参照のこと。本発明において有用である抗体は、修飾して使用することができ、また、修飾することなく使用することもできる。
【0057】
抗原(この場合は、本発明のNogo受容体−1または免疫原性ポリペプチド)および抗体は、検出可能なシグナルを提供する物質に共有結合または非共有結合のいずれかによって連結させることによって標識することができる。種々の標識および結合技術が当該分野で知られており、これらを本発明の実施において使用することができる。適切な標識としては、放射性核種、酵素、基質、補因子、阻害因子、蛍光剤、化学発光剤、磁性粒子などが挙げられるが、これらに限定されない。このような標識の使用を教示している特許としては、米国特許第3,817,837号;同第3,850,752号;同第3,939,350号;同第3,996,345号;同第4,277,437号;同第4,275,149号、および同第4,366,241号が挙げられる。また、組み換え免疫グロブリンを産生することもできる(米国特許第4,816,567号を参照のこと)。
【0058】
本発明のいくつかの実施形態においては、抗体は、複数の結合特異性を有する。例えば、二官能性抗体は、当業者に公知である多数の技術のうちのいずれか1つによって調製される。このような技術としては、ハイブリッドハイブリドーマの産生、ジスルフィド交換、化学的架橋、2つのモノクローナル抗体の間へのペプチドリンカーの付加、特定の細胞株への免疫グロブリン重鎖および軽鎖の2つのセットの導入などが挙げられる(さらに詳細な議論については下記を参照のこと)。
【0059】
本発明の抗体はまた、ヒトモノクローナル抗体である場合もあり、例えば、不死化ヒト細胞によって産生されるもの、「ヒト」抗体を産生することができるSCID−huマウスまたは他のヒト以外の動物によって産生されるものであり得る。
【0060】
(ファージディスプレイライブラリー)
本発明の抗Nogo受容体−1抗体は、組み換えの組み合わせ抗体ライブラリーをスクリーニングすることによって単離することができる。例示的な組み合わせライブラリーは、本発明の免疫原性Nogo受容体−1ポリペプチドに対する結合のためのものであり、例えば、本発明の免疫原性Nogo受容体−1ポリペプチドで免疫化した動物に由来するmRNAから調製されたVLおよびVH cDNAを使用して調製されたscFvファージディスプレイライブラリーである。このようなライブラリーを調製しスクリーニングするための方法論は当該分野で公知である。ファージディスプレイライブラリーを作成するための方法および材料は市販されている(例えば、Pharmacia Recombinant Phage Antibody System,カタログ番号27−9400−01;Stratagene SurfZAP(登録商標)ファージディスプレイキット、カタログ番号240612;およびMorphoSysによる他のもの)。抗体ディスプレイライブラリーを作成し、スクリーニングすることにおいて使用することができる方法および試薬は、他にも存在する(例えば、Ladner et al.,米国特許第5,223,409号;Kang et al.,PCT公開番号WO92/18619;Dower et al.,PCT公開番号WO91/17271;Winter et al.,PCT公開番号WO92/20791;Markland et al.,PCT公開番号WO92/15679;Breitling et al.,PCT公開番号WO93/01288;McCafferty et al.,PCT公開番号WO93/01047;Garrard et al.,PCT公開番号WO92/09690;Fuchs et al.,(1991)Bio/Technology 9:1370−1372;Hay et al.,(1992)Hum.Antibod.Hybridomas 3:81−85;Huse et al.,(1989)Science 246:1275−1281;McCafferty et al.,Nature(1990)348:552−554;Griffiths et al.,(1993)EMBO J.12:725−734;Hawkins et al.,(1992)J.Mol.Biol.,226:889−896;Clackson et al.,(1991)Nature 352:624−628;Gram et al.,(1992)Proc.Natl.Acad.Sci.USA,89:3576−3580;Garrad et al.,(1991)Bio/Technology 9:1373−1377;Hoogenboom et al.,(1991)Nucl.Acids Res.19:4133−4137;およびBarbas et al.,(1991)Proc.Natl.Acad.Sci.USA,88:7978−7982を参照のこと)。
【0061】
組み換え免疫グロブリンディスプレイライブラリーから本発明の抗Nogo受容体−1抗体をスクリーニングし単離した後、選択された抗体をコードする核酸をディスプレイパッケージから(例えば、ファージゲノムから)回収することができ、標準的な組み換えDNA技術によって他の発現ベクターにサブクローニングすることができる。所望される場合は、核酸はさらに、本発明の他の抗体形態を生じるように、以下に記載されるように操作され得る。組み合わせライブラリーのスクリーニングによって単離した抗体を発現させるためには、抗体重鎖および軽鎖、またはその可変領域をコードするDNAが、組み換え発現ベクター中にクローニングされ、そして上記のように、哺乳動物宿主細胞に導入される。
【0062】
(クラススイッチ)
本発明の抗Nogo受容体−1抗体は任意のイソ型であり得る。任意の所望されるイソ型の抗体は、クラススイッチによって産生することができる。クラススイッチのためには、CLまたはCHをコードするヌクレオチド配列のいずれをも含まない、VLまたはVHをコードする核酸が、当該分野で周知の方法を使用して単離される。VLまたはVHをコードする核酸は、その後、免疫グロブリン分子の所望されるクラスに由来するCLまたはCHをコードするヌクレオチド配列に対して動作可能であるように連結させられる。これは、ベクターと、上記のようなCLまたはCH鎖を含む核酸を使用して行うことができる。例えば、もともとIgMであった本発明の抗Nogo受容体−1抗体は、IgGへとクラススイッチさせることができる。さらに、クラススイッチを、1つのIgGサブクラスから別のサブクラスへ、例えば、IgG1からIgG2へと変換させるために使用することもできる。
【0063】
(変異させられた抗体)
他の実施形態においては、本発明の抗体または抗原結合断片は、重鎖および/または軽鎖の種々のドメインにおいて変異させることができ、それによって、抗体の結合特性を変化させることができる。例えば、変異は、1つ以上のCDR領域中に作成することができ、これによって、Nogo受容体−1についての抗体のKdを増大または低下させることができ、Koffを増大または低下させることができ、あるいは、抗体の結合特異性を変化させることができる。部位特異的変異誘発における技術は、当該分野で周知である。例えば、Sambrook et al.,およびAusubel et al.,(前出)を参照のこと。好ましい実施形態においては、変異は、本発明の抗Nogo受容体−1抗体の可変領域中の生殖系と比較して異なっていることが知られているアミノ酸残基に作成される。いくつかの実施形態においては、変異は、本発明の抗Nogo受容体−1抗体の可変領域中の生殖系と比較して異なっていることが知られている1つ以上のアミノ酸残基に作成される。別の実施形態においては、抗体の重鎖または軽鎖可変領域をコードする核酸が、1つ以上のフレームワーク領域の中で変異させられる。変異は、半減期が長くなるように、フレームワーク領域または定常ドメインに作成される場合もある。フレームワーク領域または定常ドメイン中の変異はまた、抗体の免疫原性を変化させるように作成される場合もあり、これによって、別の分子に対する共有結合または非共有結合のための部位が提供されるか、あるいは、補体結合のような特性が変化する。変異は、1つの変異抗体中のフレームワーク領域、定常ドメイン、および可変領域のそれぞれに作成することができる。あるいは、変異は、1つの変異抗体中の1つのフレームワーク領域の中だけに、複数の可変領域の中だけに、または定常ドメインの中だけに作成される場合もある。
【0064】
(融合抗体および免疫接着体)
別の実施形態においては、別のポリペプチドに連結させられた本発明の抗Nogo受容体−1抗体の全てまたは一部を含む、融合抗体または免疫接着体が作成される場合もある。いくつかの実施形態においては、抗Nogo受容体−1抗体の可変領域だけが、ポリペプチドに連結させられる。他の実施形態においては、本発明の抗Nogo受容体−1抗体のVHドメインが、第1のポリペプチドに連結させられ、一方では、VHドメインとVLドメインとが抗体結合部位を形成するように互いに相互作用できるような様式で、抗体のVLドメインが、第1のポリペプチドと会合する第2のポリペプチドに対して連結させられる。他の実施形態においては、VHドメインは、VHドメインとVLドメインとが互いに相互作用できるように、リンカーによってVLドメインから間隔を空けられる(下記の単鎖抗体の項を参照のこと)。VH−リンカー−VL抗体は、その後、目的のポリペプチドに連結させられる。融合抗体は、Nogo受容体−1リガンドを発現する細胞または組織に対してポリペプチドを向けるために有用である。目的のポリペプチドは、治療薬、例えば、毒素である場合も、また、診断薬、例えば、西洋ワサビペルオキシダーゼのような、容易に視覚化させることができる酵素である場合もある。さらに、「融合抗体」は、2つ(またはそれ以上)の単鎖抗体が互いに連結されるように作成され得る。これは、1本のポリペプチド鎖の上に二価または多価の抗体を作成することが望まれる場合、あるいは、二重特異的抗体を作成することが望まれる場合に、有用である。
【0065】
(単鎖抗体)
本発明には、本発明のNogo受容体−1ポリペプチドに結合する単鎖抗体(scFv)が含まれる。ScFvを産生するためには、VH−、およびVL−をコードするDNAが、可撓性リンカー、例えば、アミノ酸配列(Gly4−Ser)3(配列番号10)をコードするDNAに対して動作可能であるように連結させられる。その結果、VHおよびVL配列は、可撓性リンカーによって連結させられたVLおよびVH領域を有している連続する1本鎖のタンパク質として発現させられ得る(例えば、Bird et al.,(1988)Science 242:423−426;Huston et al.,(1988)Proc.Natl.Acad.Sci.USA,85:5879−5883;McCafferty et al.,Nature(1990)348:552−554を参照のこと)。単鎖抗体は、VHとVLが1つだけ使用される場合には一価であり、また、VHとVLが2つ使用される場合には二価であり、また、2つ以上のVHとVLが使用される場合には多価である場合もある。
【0066】
(キメラ抗体)
本発明にはさらに、二重特異的抗体またはその抗原結合断片が含まれる。これらにおいては、1つの特異性は本発明のNogo受容体−1ポリペプチドに対する特異性である。1つの実施形態においては、1つの結合ドメインを通じて本発明のNogo受容体−1ポリペプチドに特異的に結合し、第2の結合ドメインを通じて第2の分子に特異的に結合するキメラ抗体が作成され得る。キメラ抗体は、組み換え分子生物学の技術によって産生することができ、また、両方を物理的に結合させることもできる。さらに、本発明のポリペプチドに特異的に結合し、ミエリンによって媒介される成長円錐の崩壊、ならびに神経突起成長および萌芽の阻害を弱めることに関係している別の分子にも特異的に結合する、1つ以上のVHおよびVLを含む単鎖抗体を作成することができる。このような二重特異的抗体は、周知の技術、例えば、Fanger et al.,Immunol Methods 4:72−81(1994)およびWright and Harris(前出)を使用して、そして、(iii)と組み合わせて作成することができる(例えば、Traunecker et al.,Int.J.Cancer(Suppl.)7:51−52(1992)を参照のこと。
【0067】
いくつかの実施形態においては、キメラ抗体は、本発明の抗体に由来する1つ以上の可変領域を使用して調製される。別の実施形態においては、キメラ抗体は、上記の抗体に由来する1つ以上のCDR領域を使用して調製される。
【0068】
(誘導された抗体および標識された抗体)
本発明の抗体または抗原結合断片は、別の分子(例えば、別のペプチドまたはタンパク質)になるように誘導することができ、また、別の分子に連結させることもできる。一般的には、抗体または抗原結合断片は、本発明のポリペプチドに対する結合が、誘導または標識によって悪影響を受けないように誘導される。例えば、本発明の抗体または抗体部分は、1つ以上の他の分子構成要素、例えば、別の抗体(例えば、二重特異的抗体またはダイアボディー)、検出試薬、細胞傷害性因子、薬剤、および/あるいは別の分子との抗体もしくは抗原結合断片の会合を媒介することができるタンパク質またはペプチド(例えば、ストレプトアビジンコア領域またはポリヒスチジンタグ)に対して、機能するように連結させられ得る(化学結合、遺伝子融合、非共有結合などによる)。
【0069】
いくつかの実施形態においては、誘導された抗体は、2つ以上の抗体(同じタイプの抗体、またはたとえば二重特異的抗体を作成するための異なるタイプの抗体)を架橋することによって産生される。適切な架橋基としては、適切なスペーサー(例えば、m−マレイミドベンゾイル−N−ヒドロキシスクシンイミドエステル)によって間隔を隔てられた2つの異なる反応基を有しているヘテロ二官能性の架橋基、またはホモ二官能性の架橋基(例えば、スベリン酸ジスクシンイミジル)が挙げられる。このようなリンカーは、Pierce Chemical Company,Rockford,Illから入手することができる。
【0070】
いくつかの実施形態においては、誘導された抗体は標識された抗体である。例えば、それらを用いて本発明の抗体または抗体部分を誘導することができる検出試薬は、蛍光化合物(フルオレセイン、フルオレセインイソチオシアネート、ローダミン、5−ジメチルアミン−1−ナフタレンスルフォニルクロライド、フィコエリスリン、ランタニドリンなどを含む)である。抗体はまた、検出に有用である酵素、例えば、西洋ワサビペルオキシダーゼ、β−ガラクトシダーゼ、ルシフェラーゼ、アルカリホスファターゼ、グルコースオキシダーゼなどを用いて標識することができる。検出可能な酵素で標識される実施形態においては、抗体は、検出可能な反応産物を生じるために酵素を使用する別の試薬を添加することによって検出される。例えば、西洋ワサビペルオキシダーゼは、過酸化水素およびジアミノベンジジンを用いる。抗体はまた、ビオチンで標識し、アビジンまたはストレプトアビジンの結合の間接的な測定によって検出される場合もある。抗体はまた、二次的なレポーター(例えば、ロイシンジッパー対合配列、二次抗体の結合部位、金属結合ドメイン、エピトープタグ)によって認識される予め決定されたポリペプチドエピトープで標識される場合もある。
【0071】
抗Nogo受容体−1抗体またはその抗原断片もまた、放射標識されたアミノ酸で標識され得る。放射標識は、診断目的と治療目的の両方に使用され得る。放射標識された抗Nogo受容体−1抗体は、診断的に、例えば、被験体においてNogo受容体−1レベルを決定するために使用することができる。さらに、放射標識された抗Nogo受容体−1抗体は、脊髄損傷を処置するために治療的に使用される場合もある。ポリペプチドの標識の例としては、以下の放射性同位元素または放射性核種が挙げられるが、これらに限定されない:3H、14C、15N、35S、90Y、99Tc、111In、125I、131I。
【0072】
抗Nogo受容体−1抗体またはその抗原断片を、ポリエチレングリコール(PEG)、メチルまたはエチル基、あるいは炭水化物基のような化学基で誘導することができる場合もある。これらの基は、抗体の生物学的特性を改善するため、例えば、血清半減期を長くするため、または組織結合を増大させるために、有用であり得る。
【0073】
(抗Nogo受容体−1抗体の特徴づけ)
(抗Nogo受容体−1抗体のクラスおよびサブクラス)
抗Nogo受容体−1抗体のクラスおよびサブクラスは、当該分野で公知の任意の方法によって決定することができる。一般的には、抗体のクラスおよびサブクラスは、抗体の特定のクラスおよびサブクラスに特異的である抗体を使用して決定することができる。このような抗体は市販によって入手することができる。クラスおよびサブクラスは、ELISA、ウェスタンブロット、さらに他の技術によっても決定することができる。あるいは、クラスおよびサブクラスは、抗体の重鎖および/または軽鎖の定常ドメインの全てまたは一部を配列決定し、免疫グロブリンの種々のクラスおよびサブクラスの既知のアミノ酸配列に対してそれらのアミノ酸配列を比較し、そして抗体のクラスおよびサブクラスを決定することによって決定することもできる。
【0074】
(Nogo受容体−1に対する抗Nogo受容体−1抗体の結合親和性)
本発明のNogo受容体−1ポリペプチドに対する本発明の抗Nogo受容体−1抗体の結合親和性および解離速度は、当該分野で公知の任意の方法によって決定することができる。例えば、結合親和性は、競合ELISA、RIA、BIAcore、またはKinExA技術によって測定することができる。解離速度も、また、BIAcoreまたはKinExA技術によって測定することができる。結合親和性と解離速度は、例えば、BIAcoreを使用して、表面プラスモン共鳴によって測定される。
【0075】
7E11および1H2のKdは、それぞれ、1×10−7Mおよび2×10−8Mであることが決定された。
【0076】
(抗Nogo受容体−1抗体によるNogo受容体−1活性の阻害)
いくつかの実施形態においては、本発明の抗Nogo受容体−1抗体またはその抗原結合断片は、リガンドに対するNogo受容体−1の結合を阻害する。このような阻害のIC50は、当該分野で公知の任意の方法によって、例えば、ELISA、RIA、または機能的拮抗によって、測定することができる。いくつかの実施形態においては、IC50は、0.1から500nMの間である。いくつかの実施形態においては、IC50は10から400nMの間である。さらに他の実施形態においては、抗体またはその一部は、60nMから400nMの間のIC50を有する。7E11および1H2のIC50は、結合アッセイにおいて、それぞれ、400nMおよび60nMであることが決定された。下の表3を参照のこと。
【0077】
つまり、当業者であれば、本発明の教示が提供されると、本発明の抗体の生物学的特性を変化させるために、あるいは、それを特定の用途により適するように任意の他の方法においてこれを変化させるために使用することができる、種々の方法を利用することができる。本発明の抗体の生物学的特性を変化させるための方法には、安定性または半減期、免疫原性、毒性、親和性、あるいは、所定の抗体分子の量を増大または減少させる方法が含まれる。
【0078】
本発明の抗体を含む組成物、およびその使用が以下に記載される。
【0079】
(可溶性Nogo受容体−1ポリペプチド)
(タンパク質)
全長のNogo受容体−1は、シグナル配列、N末端領域(NT)、8個のロイシンを多く含む反復(LRR)、LRRCT領域(8個のロイシンを多く含む反復のC末端のロイシンを多く含む反復ドメイン)、C末端領域(CT)、およびGPIアンカーから構成されている(図1を参照のこと)。
【0080】
本発明のいくつかの実施形態では、可溶性Nogo受容体−1ポリペプチドが提供される。本発明の可溶性Nogo受容体−1ポリペプチドには、NTドメイン;8個のLRR、およびLRRCTドメインが含まれており、シグナル配列と機能的なGPIアンカーは含まれていない(すなわち、GPIアンカーは含まれないか、細胞膜に対して効率的に会合する能力を欠いているGPIアンカーが含まれる)。
【0081】
いくつかの実施形態においては、可溶性Nogo受容体−1ポリペプチドには、異種LRRが含まれる。いくつかの実施形態においては、可溶性Nogo受容体−1ポリペプチドには、2個、3個、4個、5個、6個、7個、または8個の異種LRRが含まれる。異種LRRは、Nogo受容体−1以外のタンパク質から得られるLRRを意味する。異種LRRを得ることができる例示的なタンパク質は、トル様(toll−like)受容体(TLR1.2);T細胞活性化ロイシン反復を多く含むタンパク質;デセオリン;OM−gp;インシュリン様成長因子結合タンパク質酸性不安定サブユニットslitおよびrobo;ならびにトル様受容体4である。
【0082】
いくつかの実施形態においては、本発明により、319アミノ酸の可溶性Nogo受容体−1ポリペプチド(可溶性Nogo受容体−1 344、sNogoR1−344、またはsNogoR344)(配列番号6および8の残基26〜344、または配列番号8の残基27〜344)が提供される。いくつかの実施形態においては、本発明により、285アミノ酸の可溶性Nogo受容体−1ポリペプチド(可溶性Nogo受容体−1 310、sNogoR1−310、またはsNogoR310)(配列番号7および9の残基26〜310、または配列番号9の残基27〜310)が提供される。図1を参照のこと。
【0083】
(表1 ヒトおよびラットのNogo受容体−1ポリペプチドの配列)
【0084】
【表1】
本発明のいくつかの実施形態においては、本発明の可溶性Nogo受容体−1ポリペプチドは、Nogo受容体−1に対するリガンドの結合を阻害し、Nogo受容体−1リガンドのアンタゴニストとしての役割を果たすように使用される。本発明のいくつかの実施形態においては、本発明の可溶性Nogo受容体−1ポリペプチドは、ニューロンにおいて神経突起成長および萌芽、例えば、軸索成長の阻害を減少させるために、そしてニューロンにおけるミエリンによって媒介される成長円錐の崩壊を阻害するために、使用される。いくつかの実施形態においては、ニューロンはCNSニューロンである。
【0085】
sNogoR310およびsNogoR344は、驚くべきことに、NogoA、NogoB、NogoC、MAG、およびOM−gpのNogo受容体−1に対する結合をブロックする。
【0086】
いくつかの実施形態においては、本発明の可溶性Nogo受容体−1ポリペプチドは、異種ポリペプチドをさらに含む融合タンパク質の構成要素である。いくつかの実施形態においては、異種ポリペプチドは、免疫グロブリン定常ドメインである。いくつかの実施形態においては、免疫グロブリン定常ドメインは、重鎖定常ドメインである。いくつかの実施形態においては、異種ポリペプチドはFc断片である。いくつかの実施形態においては、Fcは、本発明の可溶性Nogo受容体−1ポリペプチドのC末端に連結させられる。いくつかの実施形態においては、融合Nogo受容体−1タンパク質は二量体である。
【0087】
(本発明の核酸分子)
本発明により、配列番号1〜9、26〜27、29〜37、および41〜45のいずれか1つのポリペプチドを含む、本発明のポリペプチドをコードする核酸が提供される。いくつかの実施形態においては、核酸は、配列番号6および8に示されるNogo受容体−1のアミノ酸残基26〜344、または配列番号8に示されるNogo受容体−1のアミノ酸残基27〜344からなる群より選択されるポリペプチドをコードする。いくつかの実施形態においては、核酸分子は、配列番号7および9に示されるNogo受容体−1のアミノ酸残基26〜310、または配列番号9に示されるNogo受容体−1のアミノ酸残基27〜310からなる群より選択されるポリペプチドをコードする。本明細書中で使用される場合は、「核酸」は、ゲノムDNA、cDNA、mRNA、およびアンチセンス分子、さらに、自然界の供給源に由来するかまたは合成されたかにはかかわらず、別の骨格をベースとするかまたは別の塩基を含む核酸を意味する。いくつかの実施形態においては、核酸にはさらに、転写プロモーターおよび状況に応じたシグナル配列を含む。これらのそれぞれは、本発明のポリペプチドをコードするヌクレオチド配列に動作可能であるように連結される。
【0088】
いくつかの実施形態においては、本発明により、本発明のNogo受容体−1融合タンパク質をコードする核酸が提供される。本発明のNogo受容体−1融合タンパク質には、配列番号6および8に示されるNogo受容体−1のアミノ酸残基26〜344、または配列番号8のアミノ酸残基27〜344、および配列番号7および9に示されるNogo受容体−1のアミノ酸残基26〜310、または配列番号9のアミノ酸残基27〜310からなる群より選択されるポリペプチドを含む融合タンパク質が含まれる。いくつかの実施形態においては、核酸は、配列番号26〜27、29〜37、および41〜45からなる群より選択されるポリペプチドを含むNogo受容体−1融合タンパク質をコードする。いくつかの実施形態においては、Nogo受容体−1融合タンパク質をコードする核酸にはさらに、転写プロモーターおよび状況に応じたシグナル配列が含まれる。いくつかの実施形態においては、ヌクレオチド配列はさらに、免疫グロブリン定常領域をコードする。いくつかの実施形態においては、免疫グロブリン定常領域は重鎖定常領域である。いくつかの実施形態においては、ヌクレオチド配列はさらに、ヒンジ領域に連結させられた免疫グロブリン重鎖定常領域をコードする。いくつかの実施形態においては、核酸はさらにFcをコードする。いくつかの実施形態においては、Nogo受容体−1融合タンパク質にはFc断片が含まれる。
【0089】
本発明のコード核酸はさらに、診断目的およびプローブとしての目的のための検出可能な標識を含むように修飾される場合もある。種々のこのような標識は当該分野で公知であり、本明細書中に記載されるコード分子とともに容易に使用することができる。適切な標識としては、ビオチン、放射標識核種などが挙げられるがこれらに限定されない。当業者は、標識されたコード核酸分子を得るために、当該分野で公知の標識のうちの任意のものを使用することができる。
【0090】
(組成物)
いくつかの実施形態においては、本発明により、配列番号1〜5、26〜27、29〜37、および41〜45からなる群より選択されるポリペプチドを含む組成物が提供される。
【0091】
いくつかの実施形態においては、本発明により、抗Nogo受容体−1抗体またはその抗原結合断片、あるいは可溶性Nogo受容体−1ポリペプチド、または本発明の融合タンパク質を含む組成物が提供される。
【0092】
いくつかの実施形態においては、本発明には、作用部位への投与のために薬学的に使用することができる調製物へと活性のある化合物を加工することを容易にする賦形剤および助剤を含む、適切な薬学的に許容される担体が含まれる場合もある。非経口投与に適している処方物としては、水溶性の形態、例えば、水溶性の塩である活性のある化合物の水溶液が挙げられる。さらに、適切な油状の注射懸濁液としての活性のある化合物の懸濁液が投与される場合もある。適切な親油性溶媒またはビヒクルとして、脂肪油、例えば、ゴマ油、または合成の脂肪酸エステル、例えば、オレイン酸エチルまたはトリグリセリドが挙げられる。水性の注射用懸濁液には、懸濁液の粘度を高める物質、例えば、カルボキシメチルセルロース・ナトリウム、ソルビトール、およびデキストランが含まれる場合もある。状況に応じて、懸濁液には、安定剤も含まれる場合がある。リポソームを、本発明の分子を細胞への送達のためにカプセル化するために使用することもできる。例示的な「薬学的に許容される担体」は、生理学的に適合性である任意の全ての溶媒、分散媒体、コーティング剤、抗菌剤および抗真菌剤、等張性の吸収遅延剤など、水、生理食塩水、リン酸緩衝化生理食塩水、デキストロース、グリセロール、エタノールなど、さらにはそれらの組み合わせである。いくつかの実施形態においては、組成物には、等張化剤、例えば、糖類、多価アルコール、例えば、マンニトール、ソルビトール、または塩化ナトリウムが含まれる。いくつかの実施形態においては、組成物には、湿潤剤のような薬学的に許容される物質、または少量の、湿潤剤、乳化剤、保存剤、または緩衝液のような補助物質が含まれる。補助物質は、本発明の抗体、抗原結合断片、可溶性Nogo受容体、または融合タンパク質の保存期間または有効性を増強させる。
【0093】
本発明の組成物は種々の形態で存在することができ、例えば、液体、半固体、および固体である投与形態、たとえは、液体の溶液(例えば、注射することができる、および注入することができる溶液)、分散液、または懸濁液が挙げられる。好ましい形態は、意図される投与の態様、および治療用途に応じて変化する。1つの実施形態においては、組成物は、注射することができるか、または注入することができる溶液の形態であり、例えば、他の抗体でのヒトの受動免疫法に使用されるものと同様の組成物である。
【0094】
組成物は、溶液、マイクロエマルジョン、分散液、リポソーム、または高い薬剤濃度に適している他の秩序構造として、処方することができる。滅菌の注射することができる溶液は、上記に列挙した成分の1つまたはそれらの組み合わせとともに、適切な溶媒の中に必要とされる量の抗Nogo受容体−1抗体を配合し、必要である場合には、その後に濾過滅菌することによって、調製することができる。一般的には、分散液は、塩基性の分散媒体と、上記に列挙されたものからの必要とされる他の成分を含む滅菌のビヒクルの中に活性のある化合物を配合することによって調製される。滅菌の注射することができる溶液の調製のための滅菌の粉末の場合には、好ましい調製方法は、減圧乾燥および凍結乾燥であり、これにより、活性のある成分と任意のさらなる所望される成分の粉末が、それらの予め滅菌濾過された溶液から得られる。溶液の適切な流動性は、例えば、レシチンのようなコーティングの使用によって、分散液の場合には必要とされる粒子の大きさを維持することによって、および界面活性剤の使用によって、維持することができる。注射することができる組成物の持続的吸収は、吸収を遅らせる物質、例えば、モノステアリン酸塩およびゼラチンを組成物中に含めることによってもたらすことができる。
【0095】
いくつかの実施形態においては、活性のある化合物は、迅速な放出から化合物を保護する担体を用いて調製することができ、例えば、移植物、経皮パッチ、およびマイクロカプセルに入れられた送達システムを含む徐放処方物である。生分解性の生体適合性高分子、例えば、エチレン酢酸ビニル、ポリ無水物、ポリグリコール酸、コラーゲン、ポリオルトエステル、およびポリ乳酸を使用することができる。このような処方物の調製のための多くの方法については特許がとられており、当業者に一般的に知られている。例えば、Sustained and Controlled Release Drug Delivery Systems,J.R.Robinson,ed.,Marcel Dekker,Inc.,New York,1978を参照のこと。
【0096】
補助的な活性のある化合物をもまた、組成物中に配合することもできる。いくつかの実施形態においては、本発明のNogo受容体−1抗体もしくはその抗原結合断片、または可溶性Nogo受容体−1ポリペプチドまたは融合タンパク質は、1つ以上のさらなる治療薬と一緒に処方され、そして/または一緒に投与される。
【0097】
本発明の薬学的組成物には、「治療有効量」または「予防上有効である量」の、本発明の抗体、抗原結合断片、ポリペプチド(単数または複数)、または融合タンパク質が含まれる場合もある。「治療有効量」は、所望される治療結果に到達するために必要な、有効量、投与量、および期間をいう。Nogo受容体−1抗体もしくはその抗原結合断片、可溶性Nogo受容体−1ポリペプチド、またはNogo受容体融合タンパク質の治療有効量は、疾患のステージ、個体の年齢、性別、および体重のような要因によって変化する場合がある。治療有効量はまた、抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、またはNogo受容体融合タンパク質の任意の毒性作用または有害な作用よりも、治療上の利点がある効果が上回るような量である。「予防上有効である量」は、所望される予防的な結果に到達するために必要な、有効量、投与量、またはそのために必要な期間をいう。通常は、予防のための用量は、疾患の前、または疾患の初期段階に被験体において使用されるので、予防上有効である量は、治療有効量よりも少ない。
【0098】
投与レジュメは、最適な所望される反応(例えば、治療応答または予防応答)を提供するように調整することができる。例えば、1回のボーラスが投与される場合があり、数回に分けられた用量が時間をかけて投与される場合もあり、また、治療状況の緊急性によって望まれる場合には、用量が比例的に減少させられる場合も、増大させられる場合もある。投与を容易にするための単位投与量形態に非経口組成物を処方することが特に有効であり、そして、投与量単位が均質であることは、本明細書中で使用される場合には、処置される哺乳動物被験体について単一の投与量として適している物理的に分かれている単位をいう。個々の単位には、必要とされる薬学的担体と組み合わせて、所望される治療効果を生じるように計算された予め決定された量の活性のある化合物が含まれる。本発明の単位投与量形態についての詳細は、(a)抗体、抗原結合断片、および可溶性受容体−1ポリペプチド、またはNogo受容体融合タンパク質の特有の特性、ならびに到達させられる特定の治療または予防効果、さらには(b)個体の過敏症の処置については、そのような抗体、抗原結合断片、および可溶性受容体−1ポリペプチド、またはNogo受容体融合タンパク質を混合することに関する当該分野に特有の限界に影響されるか、またはそれらに直接依存する。いくつかの実施形態においては、Nogo受容体−1抗体またはその抗原結合断片についての治療有効量の範囲は、0.1〜4mg/kg/日である。いくつかの実施形態においては、Nogo受容体−1抗体またはその抗原結合断片についての治療有効量の範囲は、0.2〜4mg/kg/日である。いくつかの実施形態においては、Nogo受容体−1抗体またはその抗原結合断片についての治療有効量の範囲は、0.2mg/kg/日である。
【0099】
(抗体、抗原結合断片、可溶性受容体、および融合タンパク質の使用)
いくつかの実施形態においては、本発明により、抗Nogo受容体−1抗体、そのような抗体の抗原結合断片、可溶性Nogo受容体−1ポリペプチド、またはそのようなポリペプチドを含む融合タンパク質を、それを必要としている哺乳動物に投与することによって、Nogo受容体−1活性を阻害するための方法が提供される。
【0100】
いくつかの実施形態においては、本発明により、リガンドに対するNogo受容体−1の結合を阻害する方法が提供される。この方法には、Nogo受容体−1を、本発明の抗体または抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、リガンドは、NogoA、NogoB、NogoC、MAG、およびOM−gpからなる群より選択される。
【0101】
いくつかの実施形態においては、本発明により、ニューロンにおける成長円錐の崩壊を阻害するための方法が提供される。この方法には、ニューロンを、本発明の抗体またはその抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、本発明により、ニューロンにおいて神経突起成長または萌芽の阻害を減少させるための方法が提供される。この方法には、ニューロンを、本発明の抗体または抗原結合断片と接触させる工程が含まれる。いくつかの実施形態においては、ニューロンは、CNSニューロンである。これらの方法のいくつかにおいては、神経突起成長または萌芽は、軸索成長である。
【0102】
いくつかの実施形態においては、本発明により、哺乳動物において死滅の危険性があるニューロンの生存を促進する方法が提供される。この方法には、(a)(i)抗Nogo受容体−1抗体もしくはその抗原結合断片、または(ii)可溶性Nogo受容体−1ポリペプチドを発現する、培養された宿主細胞を提供すること;および、(b)哺乳動物中に、ニューロンの部位またはその付近に、宿主細胞を導入することが含まれる。Almudena Ramon−Cueto,M Isabel Cordero,Fernando F Santos−Benito and Jesus Avila(2000)Functional recovery of paralegic rats and motor axon regeneraton in their spinal cords by olfactory ensheathing cells.Neuron 25,425−435。
【0103】
いくつかの実施形態においては、本発明により、哺乳動物中にある死滅の危険性があるニューロンの生存を促進する遺伝子治療方法が提供される。この方法には、(a)抗Nogo受容体−1抗体もしくはその抗原結合断片、または(b)可溶性Nogo受容体−1ポリペプチドをコードするヌクレオチド配列を含むウイルスベクターを、ニューロンの部位またはその付近に投与することが含まれる。ここでは、抗Nogo受容体−1抗体、抗原結合断片、または可溶性Nogo受容体−1ポリペプチドは、哺乳動物中のヌクレオチド配列から、ニューロンの生存を促進するために十分な量で発現される。これらの実施形態に有用であるウイルスベクターおよび方法は、例えば、Noel et al.,Human Gene Therapy,13,1483−93(2002)に記載されている。
【0104】
いくつかの実施形態においては、本発明により、リガンドに対するNogo受容体−1の結合を阻害する方法が提供される。この方法には、リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。
【0105】
いくつかの実施形態においては、本発明により、Nogo受容体−1リガンドの活性を調節する方法が提供される。この方法には、Nogo受容体−1リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。
【0106】
いくつかの実施形態においては、本発明により、ニューロンにおける成長円錐の崩壊を阻害するための方法が提供される。この方法には、Nogo受容体−1リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。いくつかの実施形態においては、本発明により、ニューロンにおいて神経突起成長または萌芽の阻害を減少させるための方法が提供される。この方法には、Nogo受容体−1リガンドを、本発明の可溶性Nogo受容体−1ポリペプチドまたはNogo受容体−1融合タンパク質と接触させる工程が含まれる。いくつかの実施形態においては、ニューロンはCNSニューロンである。いくつかの実施形態においては、リガンドは、NogoA、NogoB、NogoC,MAG、およびOM−gpからなる群より選択される。いくつかの実施形態においては、神経突起成長および萌芽は、軸索成長である。
【0107】
本明細書中に記載される任意のタイプの抗体または受容体を、治療的に使用することができる。いくつかの実施形態においては、抗Nogo受容体−1抗体はヒト抗体である。いくつかの実施形態においては、哺乳動物はヒト患者である。いくつかの実施形態においては、抗体またはその抗原結合断片は、獣医学的な目的のために、またはヒトの疾患についての動物モデルとして、抗体が交差反応するNogo受容体−1を発現するヒト以外の哺乳動物(例えば、霊長類、カニクイザル、またはアカゲザル)に投与される。このような動物モデルは、本発明の抗体の治療効力を評価するために有用であり得る。
【0108】
いくつかの実施形態においては、抗Nogo受容体−1抗体もしくはその抗原結合断片、または可溶性Nogo受容体−1ポリペプチド、または融合タンパク質の投与は、損傷部位からの軸索成長を促進するように、脊髄損傷を処置するために使用される。
【0109】
本発明の抗Nogo受容体−1抗体もしくは抗原結合断片、または可溶性Nogo受容体−1ポリペプチドもしくは融合タンパク質は、単独で提供することもでき、また、特定の病理学的プロセスを調節する他の物質と組み合わせて、または順次組み合わせて提供することもできる。例えば、抗炎症薬が、ニューロンのさらなる損傷をブロックするため、および軸索の再生の阻害のための手段として、脳梗塞の後に同時に投与される場合がある。本明細書中で使用される場合は、Nogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1、およびNogo受容体融合タンパク質は、2つが同時に、連続して、または別々に投与される場合に、1つ以上のさらなる治療薬と組み合わせて投与されると言われる。
【0110】
本発明の抗Nogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、Nogo受容体−1融合タンパク質は、非経口、皮下、静脈内、筋肉内、腹腔内、経皮、吸入、または口腔経路によって投与することができる。例えば、薬剤は、微量注入によって損傷部位に局所投与される場合がある。典型的な部位としては、怪我によって生じた脊髄の損傷した領域が挙げられるが、これに限定されない。投与される投与量は、レシピエントの年齢、健康状態、および体重、同時に行われている(行われている場合には)処置の種類、処置の頻度、所望される効果の性質に応じて変化する。
【0111】
本発明の化合物は、通常は、ヒト、ヒツジ、ウマ、ウシ、ブタ、イヌ、ネコ、ラット、およびマウスのような哺乳動物に生体内で、あるいは生体外で利用することができる。
【0112】
(本発明のベクター)
いくつかの実施形態においては、本発明により、コード配列を含む組み換えDNA分子(rDNA)が提供される。本明細書中で使用される場合は、rDNA分子は、分子操作が行われたDNA分子である。rDNA分子を作成するための方法は当該分野で周知である。例えば、Sambrook et al.,(1989)Molecular Cloning−A Laboratory Manual,Cold Spring Harbor Laboratory Pressを参照のこと。いくつかのrDNA分子においては、コードDNA配列は、発現制御配列およびベクター配列に動作可能であるように連結される。
【0113】
いくつかの実施形態においては、本発明により、本発明のポリペプチドをコードする核酸を含むベクターが提供される。本発明の核酸が動作可能であるように連結されるベクターおよび発現制御配列の選択は、当該分野で周知であるように、所望される機能的特性(例えば、タンパク質の発現および形質転換される宿主細胞)に直接依存する。本発明のベクターは、少なくとも、複製、または宿主染色体への組み込みを指示することができ、rDNA分子中に含まれる構造遺伝子を発現できることが好ましい。
【0114】
動作可能であるように連結されたタンパク質をコードする配列の発現を調節するために使用される発現制御エレメントは当該分野で公知である。発現制御エレメントとしては、誘導性プロモーター、構成的プロモーター、分泌シグナル、および他の調節エレメントが挙げられるが、これらに限定されない。宿主細胞の培地中の栄養素に反応する誘導性プロモーターは、容易に制御することができるので好ましい。
【0115】
1つの実施形態においては、コード核酸分子を含むベクターには、原核生物のレプリコンが含まれる。すなわち、DNA配列は、形質転換される原核生物宿主細胞、例えば、細菌宿主中で自律複製を指示し、染色体外で組み換えDNA分子を維持する能力を有する。そのようなレプリコンは当該分野で周知である。さらに、原核生物のレプリコンを含むベクターにはまた、薬剤耐性のような、その発現により検出可能な、または選択可能なマーカーを付与する遺伝子が含まれる場合もある。細菌の薬剤耐性遺伝子の典型は、アンピシリンまたはテトラサイクリンに対する耐性を付与する遺伝子である。
【0116】
原核生物のレプリコンを含むベクターには、さらに、大腸菌(E.coli)のような細菌宿主細胞中のコード遺伝子配列の発現(転写および翻訳)を指示することができる原核生物またはバクテリオファージのプロモーターが含まれ得る。プロモーターは、RNAポリメラーゼの結合を可能にし、転写を生じさせることができるDNA配列によって形成される発現制御エレメントである。細菌宿主と適合するプロモーター配列は、通常、本発明のDNAセグメントの挿入のための便利な制限部位を含むプラスミドベクターとして提供される。このようなベクタープラスミドの例は、pUC8、pUC9、pBR322、およびpBR329(Bio−Rad(登録商標)Laboratories)、pPL、およびpKK223(Pharmacia)である。任意の適切な原核生物宿主を、本発明のタンパク質をコードする組み換えDNA分子を発現させるために使用することができる。
【0117】
真核生物細胞と適合する発現ベクター、好ましくは、脊椎動物細胞と適合する発現ベクターもまた、コード配列を含むrDNA分子を形成させるために使用することができる。真核生物細胞発現ベクターは、当該分野で周知であり、いくつかの市販の供給業者から入手することができる。通常は、このようなベクターは、所望されるDNAセグメントの挿入のための便利な制限部位を含んで提供される。このようなベクターの例は、pSVLおよびpKSV−10(Pharmacia)、pBPV−1、pML2d(International Biotechnologies)、pTDT1(ATCC(登録商標)31255)、および他の真核生物発現ベクターである。
【0118】
本発明のrDNA分子を構築するために使用される真核生物発現ベクターには、さらに、真核生物細胞中で有効である選択マーカー、好ましくは、薬剤耐性選択マーカーを含めることができる。好ましい薬剤耐性マーカーは、その発現によりネオマイシン耐性を生じる遺伝子、すなわち、ネオマイシンホスホトランスフェラーゼ(neo)遺伝子である。(Southern et al.,(1982)J.Mol.Anal.Genet.1,327−341)。あるいは、選択マーカーは、別のプラスミド上に存在させることができ、2つのベクターを宿主細胞の同時トランスフェクションによって導入し、トランスフェクタントを、選択マーカーについて適切な薬剤の中で培養することによって選択することができる。
【0119】
本発明の抗体または抗体部分を発現させるためには、遺伝子が、転写および翻訳制御配列に動作可能であるように連結されるように、軽鎖および重鎖の一部または全長をコードするDNAが発現ベクター中に挿入される。発現ベクターとしては、プラスミド、レトロウイルス、コスミド、YAC、EBVに由来するエピソームなどが挙げられる。抗体遺伝子は、ベクター中の転写および翻訳制御配列が、抗体遺伝子の転写および翻訳を調節するそれらの意図される機能を果たすように、ベクター中に連結される。発現ベクターと発現制御配列は、使用される発現宿主細胞と適合するように選択される。抗体の軽鎖遺伝子と、抗体の重鎖遺伝子が、別々のベクターに挿入され得る。いくつかの実施形態においては、両方の遺伝子が同じ発現ベクターに挿入される。抗体遺伝子は、標準的な方法(例えば、抗体遺伝子断片とベクター上の相補的な制限部位の連結、または制限部位が存在しない場合は、平滑末端の連結)によって、発現ベクター中に挿入される。
【0120】
便利なベクターは、機能的に完全なヒトCHまたはCL免疫グロブリン配列をコードし、上記のように任意のVHまたはVL配列を容易に挿入し、発現することができるように操作された適切な制限部位を有しているベクターである。このようなベクター中では、スプライシングは、通常、挿入されたJ領域中のスプライシングドナー部位と、ヒトC領域の前にあるスプライシングアクセプター部位との間で、また、ヒトCHエキソンの中に存在するスプライシング領域でも生じる。ポリアデニル化および転写終結は、コード領域の下流に存在するもともとの染色体部位で生じる。組み換え発現ベクターはまた、宿主細胞からの抗体鎖の分泌を促進するシグナルペプチドをコードすることもできる。抗体鎖遺伝子は、シグナルペプチドが抗体鎖遺伝子のアミノ末端にインフレームで連結されるように、ベクター中にクローニングすることもできる。シグナルペプチドは、免疫グロブリンシグナルペプチドである場合も、また異種シグナルペプチド(すなわち、免疫グロブリン以外のタンパク質に由来するシグナルペプチド)である場合もある。
【0121】
本発明の免疫原性ポリペプチド、Nogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質に加えて、本発明の組み換え発現ベクターは、宿主細胞中でのそれらの発現を制御する調節配列を有する。調節配列の選択を含む発現ベクターの設計が、形質転換される宿主細胞の選択、所望されるタンパク質の発現のレベルなどのような要因によって変化する場合があることは、当業者によって理解されるであろう。哺乳動物宿主細胞での発現に好ましい調節配列としては、哺乳動物細胞中で高レベルのタンパク質の発現を指示するウイルスエレメント、例えば、レトロウイルスLTR由来のプロモーターおよび/またはエンハンサー、サイトメガロウイルス(CMV)由来のプロモーターおよび/またはエンハンサー(例えば、CMVプロモーター/エンハンサー)、シミアンウイルス40(SV40)由来のプロモーターおよび/またはエンハンサー(例えば、SV40プロモーター/エンハンサー)、アデノウイルス由来のプロモーターおよび/またはエンハンサー(例えば、アデノウイルス主要後期プロモーター(AdMLP))、ポリオーマおよび強力哺乳動物プロモーター(例えば、本来の免疫グロブリンおよびアクチンプロモーター)が挙げられる。ウイルス調節エレメントおよびその配列のさらなる詳細については、例えば、Stinskiによる米国特許第5,168,062号、Bell et al.,による米国特許第4,510,245号、およびSchaffner et al.,による米国特許第4,968,615号を参照のこと。
【0122】
異種遺伝子および調節配列に加えて、本発明の組み換え発現ベクターは、宿主細胞中でのベクターの複製を調節する配列(例えば、複製起点)、および選択マーカー遺伝子のような、さらなる配列を有する場合がある。選択マーカー遺伝子は、ベクターが導入された宿主細胞の選択を容易にする(例えば、米国特許第4,399,216号、同第4,634,655号、および同第5,179,017号を参照のこと、全て、Axel et al.,による)。例えば、通常、選択マーカー遺伝子によって、薬剤、例えば、G418、ハイグロマイシン、またはメトトレキセートに対する耐性が、ベクターが導入される宿主に付与される。好ましい選択マーカー遺伝子としては、ジヒドロ葉酸レダクターゼ(DHFR)遺伝子(例えば、dhfr−宿主細胞におけるメトトレキセート選択/増幅の使用について)、およびneo遺伝子(G418選択について)が挙げられる。
【0123】
(本発明のタンパク質を組み換えにより産生する宿主および方法)
本発明の抗Nogo受容体−1抗体、免疫原性ペプチド、可溶性Nogo受容体−1ポリペプチド、可溶性Nogo受容体−1融合タンパク質をコードする核酸分子、ならびにこれらの核酸分子を含むベクターを、適切な宿主細胞の形質転換のために使用することができる。形質転換は、宿主細胞にポリヌクレオチドを導入するための任意の公知の方法によって行うことができる。異種ポリヌクレオチドを哺乳動物細胞に導入するための方法は当該分野で周知である。これには、デキストラン媒介トランスフェクション、リン酸カルシウム沈殿、ポリブレン媒介トランスフェクション、プロトプラスト融合、エレクトロポレーション、リポソーム中へのポリヌクレオチド(単数または複数)のカプセル化、および核へのDNAの直接のマイクロインジェクションが含まれる。さらに、核酸分子を、ウイルスベクターによって哺乳動物細胞に導入することができる場合もある。
【0124】
適切な細胞宿主の本発明のrDNA分子での形質転換は、周知の方法によって行われる。これらは、通常、使用されるベクターおよび使用される宿主システムのタイプに応じて異なる。原核生物宿主細胞の形質転換に関しては、エレクトロポレーションおよび塩処理方法が使用され得る(例えば、Sambrook et al.,(1989)Molecular Cloning−A Laboratory Manual,Cold Spring Harbor Laboratory Press;Cohen et al.,(1972)Proc.Natl.Acad.Sci.USA,69,2110−2114を参照のこと)。脊椎動物細胞のrDNAを含むベクターでの形質転換に関しては、エレクトロポレーション、陽イオン性脂質、または塩処理方法が使用され得る(例えば、Graham et al.,(1973)Virology 52,456−467;Wigler et al.,(1979)Proc.Natl.Acad.Sci.USA,76,1373−1376を参照のこと)。
【0125】
うまく形質転換された細胞、すなわち、本発明のrDNA分子を含む細胞は、選択マーカーについての選択を含む、周知の技術によって同定することができる。例えば、本発明のrDNAの導入によって得られる細胞は、単一コロニーを生じるようにクローニングされ得る。これらのコロニーに由来する細胞を、回収し、溶解させ、そしてそれらのDNA内容物を、Southern,(1975)J.Mol.Biol.,98,503−517によって記載されているような方法を使用してrDNAの存在について試験することができ、また、細胞によって産生されるタンパク質が、免疫学的方法によってアッセイされる場合もある。
【0126】
発現のための宿主として利用することができる哺乳動物細胞株は当該分野で周知であり、これには、アメリカンタイプカルチャーコレクション(ATCC(登録商標))から入手することができる多くの不死化細胞株が含まれる。これらとしては、特に、チャイニーズハムスター卵巣(CHO)細胞、NSO、SP2細胞、HeLa細胞、ベビーハムスター腎臓(BHK)細胞、サル腎臓細胞(COS)、ヒト肝細胞ガン細胞(例えば、Hep G2)、A549細胞、および多数の他の細胞株が挙げられる。特に好ましい細胞株は、どの細胞株が高い発現レベルを有しているかを決定することによって選択される。使用することができる他の細胞株は、昆虫細胞株、例えば、Sf9細胞である。本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質をコードする組み換え発現ベクターが哺乳動物宿主細胞に導入される場合は、これらは、宿主細胞中で抗体、ポリペプチド、および融合ポリペプチドを発現させるために、あるいは、さらに好ましくは、本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質を宿主が増殖させられる培養培地中に分泌させるために十分な時間の間、宿主細胞を培養することによって産生される。本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質は、標準的なタンパク質精製方法を使用して培養培地から回収することができる。
【0127】
さらに、本発明の免疫原性ポリペプチド、Nogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および可溶性Nogo受容体−1融合タンパク質(あるいはそれらに由来する他の部分)の、産生細胞株からの発現は、多数の公知の技術を使用して増強させることができる。例えば、グルタミン合成酵素の遺伝子発現システム(GSシステム)が、特定の条件下で発現を増強させるための一般的なアプローチである。GSシステムは、欧州特許第0,216,846号、同第0,256,055号、および同第0,323,997号、ならびに欧州特許出願番号89303964.4と組み合わせて、完全にまたは一部議論されている。
【0128】
(宿主細胞)
本発明により、さらに、本発明のNogo受容体−1抗体、抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および/または可溶性Nogo受容体−1融合タンパク質をコードする核酸分子で形質転換された宿主細胞が提供される。宿主細胞は、原核生物である場合も、また真核生物である場合もある。細胞株が細胞培養方法と適合しており、発現ベクターの増幅、および遺伝子産物の発現と適合している限りは、本発明のタンパク質の発現に有用である真核生物細胞は限定されない。好ましい真核生物宿主細胞としては、酵母細胞、昆虫細胞、および哺乳動物細胞、好ましくは、マウス、ラット、サル、またはヒト細胞株のような脊椎動物細胞株が挙げられるが、これらに限定されない。有用な真核生物細胞の例として、CCL61としてATCC(登録商標)から入手することができるチャイニーズハムスター卵巣(CHO)細胞、CRL1658としてATCCから入手することができるNIH Swissマウス胚性細胞NIH−3T3、ベビーハムスター腎臓細胞(BHK)、同様の真核生物組織培養細胞株が挙げられる。
【0129】
(rDNA分子を使用する組み換えタンパク質の産生)
本発明により、さらに、本発明のNogo受容体−1抗体もしくは抗原結合断片、可溶性Nogo受容体−1ポリペプチド、および/または可溶性Nogo受容体−1融合タンパク質を、本明細書中に記載される核酸分子を使用して産生するための方法が提供される。一般的な用語においては、組み換え形態のタンパク質の産生には、通常、以下の工程が含まれる:
最初に、本発明のタンパク質をコードする核酸分子が得られる。コード配列にイントロンが間に挟まっていない場合は、これはいずれの宿主での発現にも適している。
【0130】
その後、核酸分子は、状況に応じて、上記のような適切な制御配列と動作可能であるように連結させられて、タンパク質のオープンリーディングフレームを含む発現ユニットが形成される。発現ユニットは、適切な宿主を形質転換するために使用され、形質転換された宿主が組み換えタンパク質が産生される条件下で培養される。状況に応じて、組み換えタンパク質は、培地から、または細胞から単離される。タンパク質の回収および精製は、何らかの不純物が寛容であるいくつかの場合においては必要ない場合もある。
【0131】
上記の工程のそれぞれは、種々の方法で行うことができる。例えば、所望されるコード配列が、ゲノム断片から得られ、適切な宿主の中で直接使用される場合もある。種々の宿主において動作可能である発現ベクターの構築は、適切なレプリコン、および制御配列を使用して、上記のように行われる。制御配列、発現ベクター、および形質転換方法は、遺伝子を発現させるために使用される宿主細胞のタイプに応じて変化し、上記で詳細に議論されている。適切な制限部位を、もともと利用できない場合は、摘出可能な遺伝子がこれらのベクターの中に挿入されることを可能にするために、コード配列の末端に付加することができる。当業者であれば、当該分野で公知の任意の宿主/発現システムを、組み換えタンパク質を産生させるように本発明の核酸分子とともに使用するために容易に適応させることができる。
【0132】
本発明がより理解されるように、以下の実施例が示される。これらの実施例は、説明の目的のためのものにすぎず、いかなる様式においても本発明の範囲を限定するとは解釈さらない。
【実施例】
【0133】
(実施例1)
(マウスモノクローナル抗Nogo受容体−1抗体の産生)
本発明の免疫原性Nogo受容体−1ポリペプチドに特異的に結合する抗Nogo受容体−1抗体を、以下の方法および手順を使用して作成した。
【0134】
(免疫化)
2つの免疫化アプローチを使用した:
(1.免疫原としてのNogo受容体−1(NogoR−1)を含むNOS−7細胞または細胞膜)
ラットNogo受容体−1遺伝子(GenBank(登録商標)No.AF462390)を、CMVプロモーターと薬剤選択のためのゲネチシン耐性遺伝子を含む哺乳動物発現ベクターpEAG1256(Biogen(登録商標))にサブクローニングした。組み換えプラスミドを、Superfect(Qiagen(登録商標))を使用してCOS−7細胞にトランスフェクトした。トランスフェクタントをゲネチシン(Gibco(登録商標)、2mg/mL)を使用して選択し、クローニングし、そしてNogo受容体−1タンパク質の表面での発現についてFACSによって確認した。COS−7膜を、記載されている手順にしたがってこれらの細胞から調製し(Wang et al.,J.Neurochem.,75:1155−1161(2000))、2回洗浄し、1mg/mL(タンパク質濃度)で10%のグリセロール中で−70℃で保存した。
【0135】
8週齢の雌RBFマウス(Jackson Labs,Bar Harbor,ME)を、50μgのラットNogo受容体−1−COS−7膜、または表面上にNogo受容体−1を発現するCOS−7細胞全体のいずれか、および50μlのRIBI MPL−TDM−CWSアジュバント(Sigma(登録商標)Chemical Co.,St.Louis,MO)を含む乳濁物のいずれかで、2週間ごとに1回、腹腔内に免疫した(Lipman et al.,1992)。免疫化したマウスから血清を、最初の免疫化の前、2回目の免疫化の7日後、および3回目の免疫化の7日後、および3回目の免疫化の38日後に採取し、抗Nogo受容体−1抗体力価を、以下に記載するようにELISAによって測定した。
【0136】
(2.免疫原としての特異的Nogo受容体−1ペプチド)
ラットNogo受容体−1遺伝子配列を、Vector NTi(登録商標)ソフトウェアを使用して抗原性分析した(図2)。分析において同定した抗原性ペプチドを、標準的なグルタルアルデヒド手順を使用してキーホールリンペットヘモシアニン(KLH)に結合させた。
【0137】
8週齢の雌RBFマウス(Jackson Labs,Bar Harbor,ME)を、50μgのKLH結合ペプチドと50μlの完全なフロイトのアジュバント(Sigma(登録商標)Chemical Co.,St.Louis,MO)を含む乳濁物で、2週間ごとに1回、腹腔内に免疫した。免疫化したマウスから血清を、最初の免疫化の前、2回目の免疫化の1週間後、3回目の免疫化の1週間後に採取し、抗Nogo受容体−1抗体力価を測定した。追加免疫の用量を、3回目の免疫化の後に投与した。この追加免疫用量での免疫化の3日後に、融合実験を開始した。
【0138】
(ハイブリドーマの産生およびスクリーニング)
抗原性Nogo受容体−1ペプチドで免疫化したマウスに由来する血清をELISAによってスクリーニングし、一方、Nogo受容体−1を発現するCOS−7細胞で免疫化したマウスに由来する血清は、フローサイトメトリーによってスクリーニングした。Nogo受容体−1−COS−7細胞に特異的に結合した抗体についてポジティブであるマウスをフローサイトメトリーによって同定し、屠殺した。脾細胞をマウスから単離し、記載されている(Kennett et al.,1993、Monoclonal Antibodies:A New Dimension in Biological Analysis.Plenum Press,New York)ように、FL653骨髄腫(Ig−/HGPRT−Balb/cマウス骨髄腫のAPRT誘導株、10%のFBS、4500mg/Lのグルコース、4mMのL−グルタミン、および20mg/mLの8−アザグアニンを含むDMEM中で維持した)に融合させた。融合させた細胞を、24ウェルまたは48ウェルプレート(Corning Glass Works,Corning,NY)にプレートし、アデニン、アミノプテリン、およびチミジンを含む培養培地を供給した。AAT耐性培養物を、ELISAまたはフローサイトメトリーによって、Nogo受容体−1−COS−7細胞、またはNogo受容体−1抗原性ペプチドのいずれかに対する結合について、下記のようにスクリーニングした。ポジティブであるウェルの細胞を、限界希釈によってさらにサブクローニングした。
【0139】
Nogo受容体−1抗原性ペプチドに対する抗体の結合についてスクリーニングするために、免疫原として使用したペプチドをBSAに結合させた。50μLの0.1M重炭酸ナトリウム緩衝液(pH9.0)中の0.5μgの結合ペプチドを、96ウェルMaxiSorp(登録商標)プレート(Nunc(登録商標))のそれぞれのウェルに添加した。その後、プレートを、37℃で1時間、または4℃で16時間インキュベートし、非特異的結合部位を、0.1%のBSA、0.1%のオボアルブミン、0.1%のブロット(blotto)、および0.001%のアジドを含む25mMのHEPES(pH7.4)を使用してブロックした。ハイブリドーマ上清を添加し、25℃で1時間インキュベートした。PBSでの3回の洗浄の後、西洋ワサビペルオキシダーゼ結合ヤギ抗マウス二次抗体(Jackson ImmunoResearch Inc.)の50μlの1:10,000希釈液をそれぞれのウェルに添加し、さらに1時間インキュベートした。3回の洗浄の後、TMB(Pierce)によって発色させ、2Mの硫酸で停止させた。色の強さを、分光光度計において450nmでモニターした。
【0140】
抗体を、全長のNogo受容体−1に対する結合について以下のようにスクリーニングした。COS−7細胞を、0.1μMのCellTracker(登録商標)Green CMFDA(Molecular Probes,Eugene,OR)で製造供給元によって記載されているように標識した。等量のCellTracker(登録商標)で標識した対照細胞を、抗Nogo受容体−1試験血清とのインキュベーションの前に、洗浄したNogo受容体−1−COS−7細胞と混合した。50μLの細胞混合物を、96ウェルV底ポリスチレンプレート(Coster(登録商標)3877、Corning,NY)のそれぞれのウェルに分配し、100μLのハイブリドーマ上清または対照抗Nogo受容体−1抗体を添加した。4℃で30分間のインキュベーションの後、細胞を洗浄し、50μLの、PBS中のR−フィコエリスリン結合親和性純粋F(ab’)2断片ヤギ坑マウスIgG Fcγ特異的二次抗体(1:200、Jackson ImmunoResearch Laboratory,West Grove,PA)とともにインキュベートした。インキュベーションの終わりに、細胞をPBSで2回洗浄し、1%のFBSを含む200μLのPBS中に懸濁し、FACS分析を行った。あるいは、Nogo受容体−1−COS−7細胞をハイブリドーマ上清と混合し、その後、R−フィコエリスリン結合ヤギ坑マウス二次抗体で処理し、標準的なFACS分析に直接供した。
【0141】
本発明者らは、種々の免疫原を使用して25個の抗Nogo受容体−1抗体を作成した。本発明者らは、免疫原としてラットNogo受容体−1の残基110〜125に対応するペプチド配列を使用して2つの抗体7E11および5B10を作成した。本発明者らは、免疫原として全長のラットNogo受容体−1でトランスフェクトしたCOS7細胞から調製した膜を使用して、3つの抗体1H2、3G5、および2F7を作成した。本発明者らは、免疫原としてsNogoR310−Fcを使用して13個の抗体(1D9.3、1E4.7、1B4.3、2C4.3、1F10.3、2H1.4、1H3.3、1G4.1、1E4.1、2G7.1、2C4.1、2F11.1、および1H4.1)を作成し、免疫原としてラットNogo受容体−1の残基423−434に対応するペプチド配列を使用して7個の抗体(2E8.1、2G11.2、および1B5.1)を作成した。
【0142】
(モノクローナル抗体7E11および5B10の配列分析)
本発明者らは、Qiagen(登録商標)RNeasy(登録商標)ミニキットを使用して全RNAを抽出し、単離したRNAからcDNAを作成した。本発明者らは、プライマー5’−TGAGGAGACGGTGACCGTGGTCCCTTGGCCCCAG−3’(配列番号12)および5’−AGGTSMARCTGCAGSAGTCWGG−3’(配列番号25)を使用してPCRによって、軽鎖配列を増幅した。本発明者らは、プライマー5’−GGGGATATCCACCATGAAGTTGCCTGTTAGGCTGTTG−3’(配列番号13)および5’−GGGGATATCCACCATGAGGKCCCCWGCTCAGYTYCTKGGA−3’(配列番号14)を使用してPCRによって重鎖配列を増幅した。これらのプライマーには、以下のような縮重ヌクレオチドが含まれている:SはGまたはCを示し;MはAまたはCを示し;RはGまたはAを示し;WはAまたはTを示し;KはGまたはTを示し;そしてYはTまたはCを示す。本発明者らは、PCR断片を配列決定ベクターにクローニングし、配列決定ベクターに特異的なプライマーを使用して、ジデオキシ鎖終結によってCDRのDNA配列を決定した。本発明者らは、DNA配列を概念的に翻訳し、モノクローナル抗体7E11および5B10の重鎖および軽鎖のCDR領域の部分的なアミノ酸配列を表2に示す。mAbの重鎖および軽鎖に由来する3個のCDRには、表2の中で下線を付けて示す。7E11および5B10の軽鎖は、94%のアミノ酸配列同一性を有しており、重鎖は91%のアミノ酸配列同一性を有している。mAb 7E11、5B10、と1H2は、IgG1イソ型であり、mAb 3G5と2F7は、IgG2aイソ型である。これらの5個のmAbのそれぞれは、κイソ型の軽鎖を有している。本発明者らは、このアプローチによって他のモノクローナル抗体の配列を分析した。
【0143】
(表2 mAb 7E11と5B10のアミノ酸配列)
【0144】
【表2】
(モノクローナル抗体7E11のエピトープマッピング)
Mab 7E11は、ラットNgR1およびヒトNgR1の両方に結合する。7E11の結合に関与しているエピトープを決定するために、本発明者らは、ラットNgR1の断片と合成ペプチドを作成し、7E11への結合についてそれらを試験した。
【0145】
全8個のLRRドメインと、N末端およびC末端キャップとを含むラットNgR1の組み換え断片(sNgR310)を、酸または臭化シアン(CNBr)のいずれかで処理し、ゲル電気泳動によって断片を分離した。未処理のsNgR310は、42kDaの見かけの分子量で移動した。sNgR310の酸処理により、15kDa(aa27〜aa122)と30kDa(aa123〜aa310)の2つの主要な切断産物が生じた。CNBr処理によって、3つの断片が生じ、これらは、33/35kDaの二重線(aa27〜229)(これは、異種のグリコシル化を受けた断片をおそらく示している)、10kDaの産物(aa241〜aa310)、および11アミノ酸の断片(aa230〜aa240)(これは、ゲル上には留まらない)である。ゲルのウェスタンブロットを7E11でプローブし、これによって、これが完全なラットNgR1(aa27〜aa310)、15kDaの酸による断片(aa27〜122)、および35kDaのCNBrによる断片(aa27〜aa229)に結合することが明らかになった。7E11は、30kDaの酸による断片(aa123〜aa310)、または10kDaのCNBrによる断片(aa241〜aa310)には結合しなかった。15kDaの酸による断片と35kDaのCNBrによる断片のいずれにも、配列LDLSDNAQLRVVDPTT(配列番号1)が含まれていた。これは、NgR1上にある1つのエプトープに対する7E11の結合と一致した。
【0146】
7E11結合部位を、さらに、sNgR310のトリプシンペプチド消化物を試験することによって分析した。HPLC分析はいくつかの断片を示し、これによって、いくつかのトリプシン反応性であるリジンおよびアルギニン残基がNgR1配列中に存在することが示された。7E11は、トリプシン消化ペプチドの1つだけに結合し、このことは、7E11がNgR1上の1つのエピトープだけに結合することについてのさらなる証拠を提供する。その後の質量スペクトル分析(MS)および配列分析によって、結合されるペプチドがAAAFGLTLLEQLDLSDNAQLR(配列番号26)であることが明らかになった。
【0147】
LDLSDNAQLRVVDPTTペプチド(配列番号1)について、さらなるマッピング分析を行った。ペプチドをトリプシンで消化した。これによって2つの主要な断片LDLSDNAQLR(配列番号27)およびVVDPTT(配列番号28)が生じた。それらに結合する7E11の能力を試験した。MS分析によって、抗体が、ペプチドLDLSDNAQLR(配列番号27)に結合し、したがって、このペプチドに7E11についての結合エピトープが含まれていることが明らかになった。このペプチド断片の中での詳細なMS分析によって、これもまた7E11に結合する、Asn115およびGln117の脱アミノ化、112または113でのアラニンの付加、または114でのセリンの付加を有しているペプチドを含む、いくつかのスクランブルペプチドが同定された(表3)。これらのデータは、このペプチド断片中に存在しているいくつかのアミノ酸残基には、7E11への結合に重要ではないものがある可能性を示している。
【0148】
(表3 7E11によって結合される変異体ペプチド)
【0149】
【表3】
LDLSDNAQLRVVDPTTペプチドを、エンドペプチダーゼAsp−Nでも消化し、7E11の結合を試験した。エンドペプチダーゼAsp−Nは、このペプチドを3つのペプチド断片、L、DLS、およびDNAQLRVVDPTT(配列番号36)に切断した。これらの産物のうち、7E11はDNAQLRVVDPTTペプチドに結合した。まとめると、トリプシンとAsp−Nでの切断のデータによって、さらに、7E11の結合エピトープは、それらの間で共有されている配列DNAQLR(配列番号37)に限定された。
【0150】
7E11はラットNgR1およびヒトNgR1に結合するが、マウスNgR1、ヒトNgR2、またはマウスNgR3には結合しないという観察に基づいて、種々の種に由来するNgR1、NgR2、およびNgR3のアミノ酸配列を、7E11の結合エピトープ中の重要な残基を推定するために分析した。配列アラインメントにより、ラットNgR1のアミノ酸110〜125と、ヒトNgR1の対応する配列が同一であり、マウスNgR1配列は119位の1つのアミノ酸だけが異なることが明らかになった(ラットNgR1およびヒトNgR1においてはArg119、マウスNgR1においてはHis119、表4)。
【0151】
(表4 種々の種に由来するNgRの配列アラインメント)
【0152】
【表4】
NgR1上のArg119は、7E11との結合に関与している。なぜなら、これは、ラットNgR1とヒトNgR1にはしっかりと結合するが、マウスNgR1にはほとんど結合しないからである。同様に、7E11はNgR3には十分には結合しないので、Ala116はエピトープには含まれる。なぜなら、DNAQLR配列(配列番号38)の中において、NgR3は、対応する配列においてアルギニンだけがNgR1とは異なるからである。DNAQLR配列のうち、NgR2の6個の残基のうちの4個は、ラットNgR1と同じである。Ala116とGln117が、アルギニンおよびヒスチジンでそれぞれ置き換えられている。これにより、Ala116が、7E11との結合に寄与している重要なアミノ酸残基ではあるが、Gln117の関与を必ずしも妨げるわけではないことが確認される。
【0153】
これらの接点を確認するために、LDLSDNAQLR配列(配列番号27)の中に点変異を含むいくつかのペプチドを作成し、7E11との結合について試験した。ペプチドを、MaxiSorp(登録商標)プレート(Nunc(登録商標))上に固定し、7E11の段階希釈物を載せた。得られたEC50値を表5に示す。7E11は、変異体Leu110AlaおよびAsp111Alaに結合し、もとのペプチドと同様のEC50値を有していた。Gla117Alaを試験すると、EC50値は30倍に増大し、Arg119Hisを試験すると、EC50値は25倍に増大していた。もっとも顕著なEC50値の変化は、Arg119をアラニンに変異させた場合に観察された。
【0154】
(表5 種々のEC50値を伴った変異体ペプチドに対する7E11の結合)
【0155】
【表5】
7E11結合エピトープの位置を、また、sNgR310についての最近明らかにされた結晶構造において決定した。予想したように、7E11エピトープが分子の表面に露出している構造が示された。残基Arg119、Gln117、Ala116、およびAsp114は、構造から突き出しており、一方、Leu118とAsn115は内部に存在している。エピトープは、構造の凹面にある酸性パッチの先端と、一方の側面に面している塩基性表面にある。
【0156】
(モノクローナル抗Nogo受容体−1抗体による可溶性Nogo受容体−1へのリガンド結合の阻害)
上記のように産生した抗Nogo受容体−1モノクローナル抗体を、これらがNogo受容体−1に対するリガンドの結合を阻害するかどうかを決定するために試験した。
【0157】
0.5μgの、下記のように産生したラットNogo受容体−1のアミノ酸残基26〜344と、ラットIgG1分子のヒンジ領域およびFc領域とを含む可溶性Nogo受容体−1融合タンパク質(sNogoR344−Fc)を、250μgのプロテインA、またはコムギ胚芽アグルチニン結合SPAビーズ(Amersham Pharmacia Biotech)上に、25℃で2時間かけて固定した。50μLのHEPES緩衝化インキュベーション培地(10mMのHEPES(pH7.4)、0.1%のウシ血清アルブミン、0.1%のオボアルブミン、2mMのMgCl2、2mMのCaCl2、およびプロテアーゼ阻害因子)中の、Fc−sNogoR−1に結合させたSPAビーズ、抗Nogo受容体−1 mAb、および1μLの125I−Nogo66(Amersham,2000Ci/mmol、1nM)を、それぞれの試料ウェルに添加した。16時間後、放射活性を、TopCount(登録商標)(Packard)を使用して、4連の試料において測定した。IC50値を、曲線適合分析によって計算した(図3)(PRISM、GraphPad Software,NJ)。いくつかの実験においては、本発明者らは、AP−リガンド結合体(例えば、AP−Nogo66)を使用し、アルカリホスファターゼ活性をモニターすることによって結合を検出した。本発明者らは、Nogo受容体−1に対するリガンドMAG−FcおよびAP−OM−gpの結合をブロックするmAbの能力もまたアッセイした。
【0158】
モノクローナル抗体7E11、5B10、1H2、3G5、および2F7は全て、Nogo66、MAG、およびOM−gpのsNogoR344−Fcに対する結合を阻害した。7E11および1H2についてのNogo66に対する計算したIC50は、それぞれ、400nMおよび60nMであった。Nogo受容体−1に対する3つのリガンドの結合のmAbによって媒介される阻害をモニターするELISAによるデータを、表6にまとめる。
【0159】
(表6 mAbはNogo受容体−1に対する、Nogo66、MAG、およびOM−gpの結合を阻害する)
【0160】
【表6】
置き換えの割合(%)を30nMの抗体について示し、そして特定のmAbについてのEC50を記載した曲線適合分析によって決定した。“−”は、検出できる程度の活性がないことを示し、“ND”は決定しなかったことを示す。
【0161】
(実施例2)
(Fab−ファージ抗Nogo受容体−1抗体の産生)
本発明の免疫原性Nogo受容体−1ポリペプチドに特異的に結合する抗Nogo受容体−1Fabファージ抗体をもまた、以下のように、Fab−ファージライブラリーをスクリーニングすることによって作成した。
【0162】
MorphoSys Fab−ファージライブラリーHuCAL(登録商標)GOLDを、組み換えラット可溶性sNogoR310−Fcタンパク質、およびラットNogo受容体−1を発現するCOS7細胞に対してスクリーニングした。Nogo受容体−1に特異的に結合するFab−ファージを精製し、特徴を決定した。14D5の重鎖は、VH2遺伝子に由来するものであり、軽鎖はVK1遺伝子に由来するモノであった。これらのFab−ファージ14D5の1つについて重鎖および軽鎖のCDRのアミノ酸配列を、表7に示す。
【0163】
(表7 14D5のCDRのアミノ酸配列)
【0164】
【表7】
14D5は、一価および二価のいずれの形態でもラットNogo受容体−1に結合する。さらに、14D5は、マウスNogo受容体−1およびヒトNogo受容体−1、ならびにヒトNogo受容体−2に結合するが、マウスNogo受容体−3には結合しない。
【0165】
(実施例3)
(抗Nogo受容体−1モノクローナル抗体によるNogo受容体−1の免疫沈降)
免疫沈降を行うために、100μLの溶解させた細胞、または50μLのPiPLCで処理した細胞を、400または450μLの抽出緩衝液(10mMのTris−HCl(pH7.2)、0.5%のTween−20(登録商標)、0.2mMのPMSF)、またはRIPA緩衝液と、それぞれ、30μLのプロテインAまたはG、および1〜2μgの抗体の存在下で、混合した。混合物を、4℃で16時間、震盪装置の中でインキュベートした。
【0166】
試料を、穏やかに攪拌してプロテインAまたはGが結合したビーズをペレットとした。ビーズを1mLの洗浄緩衝液(10mMのTris−HCl(pH7.2)、0.1%のTween−20(登録商標))で3回洗浄した。最後の洗浄は、10%のもとの洗浄緩衝液を使用して行った。
【0167】
ビーズを、10%のβ−メルカプトエタノールを含む100μLの2×SDS中に再度懸濁した。試料を室温でインキュベートし、その後、SDS−PAGEのために、4〜20%のTris−グリシンゲル上で泳動した。SDS−PAGEゲル分析によって決定すると、モノクローナル抗体3G5および2F7がNogo受容体−1を免疫沈降させた。
【0168】
(実施例4)
(ELISAによる抗体特異性の決定)
実施例1および2で産生したモノクローナル抗体およびFab−ファージ抗体の特異性を決定するために、本発明者らは、Nogo受容体−1ポリペプチドのパネルを使用してELISAを行った。パネルは、sNogoR310−Fc(ラットNogo受容体−1のアミノ酸26〜310とラットFc断片を含む融合タンパク質)、sNogoR344−Fc(上記を参照のこと)、ポリペプチドp−617(配列番号1)、ポリペプチドp−618(a19〜ラットNogo受容体−1のLRR7領域に由来するアミノ酸ポリペプチド;図2;配列11)、ならびに、ポリペプチドp−4およびp−5(それぞれ、Nogo受容体−1のLRR5およびLRRCT領域に由来するポリペプチド)から構成された。オボアルブミンおよびBSAを対照として使用した。図4に示すように、mAb 1H2、3G5、および2F7は全て、sNogoR344−Fcに特異的に結合した。同様の実験においては、これらの抗体は、ラットNogoレセプター−1のアミノ酸310〜344からなるポリペプチド(配列番号3)にも特異的に結合し、mAb 7E11および5B10はポリペプチドp−617(配列番号1)に特異的に結合した。
【0169】
sNogoR310−Fcでの免疫化による10個の抗体(1D9.3、1E4.7、1B4.3、2C4.3、1F10.3、2H1.4、1H3.3、1G4.1、1E4.1、および2G7.1)は、結合について互いに置換することができ、このことは、これらが、sNogoR310−Fc上の同様であるかまたは重複しているエピトープを認識することを示している。sNogoR310−Fcでの免疫化による他の3個の抗体(2C4.1、2F11.1、および1H4.1)は、アミノ酸残基26〜310に存在する異なるエピトープを認識する。
【0170】
本発明者らは、また、Fab−ファージ14D5を使用してELISA結合アッセイを行った。AP−Nogo66、AP−OM−gp、およびMAG−Fcリガンドを、固定したsNogoR344−Fcに結合させた場合には、1μMの14D5はNogoおよびMAGの結合を完全に阻害した。10μMの14D5が、sNogoR344−Fcに対するOM−gpの結合を完全に阻害するためには必要であった。
【0171】
(実施例5)
(神経突起成長についてのアッセイ)
ニューロンに対するCNSミエリンの阻害作用を弱める、上記で産生したモノクローナル抗体およびFab−ファージ抗体の能力を試験するために、Lab−Tek(登録商標)培養スライド(4ウェル)を、0.1mg/mLのポリ−D−リジン(Sigma(登録商標)))でコーティングした。CNSミエリンまたはPBSを、3μLの液滴としてスポットした。蛍光ミクロスフェア(Polyscience)を、ミエリン/PBSに添加して、後で液滴が同定できるようにした(Grandpre et al.,Nature 403,2000)。その後、Lab−Tek(登録商標)スライドをリンスし、10μg/mLのラミニン(Gibco(登録商標))でコーティングした。P3−4 Sprague Dawleyラットの子供から後根神経節(DRG)を取り出し、1mg/mLのコラゲナーゼ1型(Worthington)で解離させ、炎磨きパスツールピペット(fire−polished Pasteur pipette)で粉末状にし、ニューロン細胞を多く含むように予めプレーティングし、最後に予めコーティングしたLab−Tek(登録商標)培養スライド上に23,000細胞/ウェルでプレートした。培養培地は、5%の熱不活化ドナーウマ血清、5%の熱不活化ウシ胎児血清、および50ng/mLのmNGFを含むF12とし、37℃、5%のCO2で6時間インキュベートした。15μg/mLのmAb 7E11を、プレーティングの直後に添加した。
【0172】
スライドを、20%のスクロースを含む4%のパラホルムアルデヒドで20分間固定し、1:500希釈した、ニューロンマーカーである抗β−III−チューブリン(Covance TUJ1)について染色した。二次抗体である抗マウスAlexa Flour(登録商標)594(Molecular Probes)を1:300希釈し、スライドを、Gel/Mount(登録商標)(Biomedia(登録商標))を用いてカバースライドをかけた。OpenLab(登録商標)ソフトウェアを用いて5倍のデジタル画像を獲得し、MetaMorph(登録商標)ソフトウェアを使用して、神経突起成長の定量のために分析した。
【0173】
Mab 7E11は、神経突起成長のミエリンによって媒介される阻害からDRGニューロンを保護した(図5)。同様の結果が、mAb 1H2、および3G5を用いても観察された。
【0174】
ラットP7 DRGニューロンを、CNSミエリン基質上で培養した場合の神経突起成長の保護アッセイにおいては、二価の14D5もまた、神経突起成長を効率よく促進した。
【0175】
(実施例6)
(Nogo受容体−1でトランスフェクトした細胞についての7E11を用いた免疫組織化学)
実施例1に記載したように産生した抗Nogo受容体−1 mAbの結合特性をさらに特徴付けるために、本発明者らは、ラットNogo受容体−1またはヒトNogo受容体−1を発現する、固定されたおよび生存しているCOS−7または293細胞の両方に対する結合を比較した。
【0176】
(固定された細胞)
Nogo受容体−1でトランスフェクトした細胞と、トランスフェクトしていない細胞を、8ウェルLab−Tek(登録商標)培養スライド中にプレートし、PBS中の、4%のパラホルムアルデヒドで15分間固定し、10%の正常なヤギの血清、0.1%のTriton X−100で1時間ブロックした。Mab 7E11を、ブロッキング溶液中で15μg/mL、および1.5μg/mLで添加し、室温で2時間インキュベートした。Alexa(登録商標)結合二次抗体抗マウス(Molecular Probes)を、ブロッキング溶液中で1:300希釈で1時間インキュベートし、DAPIを全ての核種を標識するために二次抗体に対して5μg/mLで添加した。
【0177】
(生存している細胞)
トランスフェクトした細胞と、トランスフェクトしていない細胞を、8ウェルLab−Tek(登録商標)培養スライド中にプレートし、FACS緩衝液(4%のドナーウマ血清を含む)で、4℃で30分間ブロックし、FACS緩衝液中の15μg/mL、および1.5μg/mLの7E11とともに、4℃で1時間インキュベートし、リンスし、二次抗体抗マウス−Alexa(登録商標)(FACS緩衝液中で1:300希釈)とともに4℃で30分間インキュベートした。
【0178】
免疫組織化学染色実験は、全てのmAbがラットのNogo受容体−1を発現する細胞に結合したことを示した。mAb 7E11、2G7.1、および2C4.1は、ヒトNogo受容体−1を発現する、固定した細胞および生存している細胞の両方に結合した。
【0179】
(実施例7)
(脊髄挫傷のマウスモデル)
生体内でのニューロンに対する実施例1で産生した抗Nogo受容体−1 mAbの効果を試験するために、本発明者らは、マウスの脊髄挫傷モデルを使用した。
【0180】
雌のマウス(18〜22g)を、鎮痛薬および抗生物質で予防的に処置した。マウスに麻酔をかけ定位固定装置に置き、立体顕微鏡下にて脊柱固定を行った。脊髄への外傷を、落錘方法の修正版によって行った(M.Li et al.,Functional role and therapeutic implications of neuronal caspase−1 and −3 in a mouse model of traumatic spinal cord injury.Neuroscience Vol.99,pp.333−342,2000)。
【0181】
簡単に説明すると、T9およびT10椎弓切除術を行い、脊柱を、一対のマウス用の横方向のクランプを使用して安定させ、T9−T10横方向の処理を双方からサポートした。直径1.4mm、重さ2gのステンレス鋼製の衝突棒(impact rod)を、硬膜の上2.5cmに上げ、T10レベルで脊髄に落とした。外科手術の間、マウスは37℃の加温毛布の上に維持し、1mLの暖めた滅菌の生理食塩水を、脱水を避けるために外科手術後、それぞれのマウスに皮下投与した。膀胱を、反射的な膀胱の制御が回復するまで、1日に1回、手で絞った。
【0182】
全ての動物に、外科手術後8〜12時間おきに術後鎮痛剤を投与し、その後7日間にわたって抗生物質での処置を1日に2回行った。動物は、研究期間を通じて食物および水を自由に摂取できるようにした。抗Nogo受容体−1抗体を、以下のラットの脊髄離断モデルにおいて記載するように、28日間にわたり、髄腔内注射によって損傷部位に投与した。
【0183】
(実施例8)
(可溶性Nogo受容体−1融合タンパク質の特徴づけ)
可溶性Nogo受容体−1ポリペプチド(sNogoR−1)および融合タンパク質(Fc−sNogoR−1)を特徴付けるために、本発明者らは以下の実験を行った。
【0184】
3μgの可溶性Nogo受容体−1(sNogoR310−FcおよびsNogoR344−Fc)を、250μgのWGA−SPAビーズ上に固定し、100μLの最終容量の結合緩衝液(20mMのHEPES(pH7.4)、2mMのCa、2mMのMg、0.1%のBSA、0.1%のオボアルブミンおよびプロテアーゼ阻害因子)中の0.5μLの放射活性リガンド(最終濃度0.5nM)を投与した。リガンドには、10μMのNogo66、10μMの125I−Nogo40(NogoAのアミノ酸1〜40)、および10μLの抗Nogo受容体−1抗体上清を、それぞれのリガンドのセットについて含めた。Nogo40上の3個のチロシンを別々にヨウ素化し、Nogo40−A、−B、および−Cと、それぞれ命名した。三連の平均値を、較正した%結合放射活性として示す(図6、7、および8)。エラーバーはSEMを示す。阻害因子が存在しない条件下で結合した放射活性を100%とし、10μMのNogo40の存在下での最も低い結合放射活性を、データを較正するための0%とした。
【0185】
(実施例9)
(可溶性Nogo受容体−1融合タンパク質に対するリガンドの結合の阻害)
実施例8の結合アッセイと同様の結合アッセイを、実施例1で産生した2つのmAbの、sNogoR344−Fcに対する125I−Nogo66の結合を阻害する能力を試験するために使用した。Mab 2F7および3G5は、sNogoR344−Fcに対する125I−Nogo66の結合を阻害した。
【0186】
(実施例10)
(神経突起成長アッセイ)
Lab−Tek(登録商標)培養スライド(4ウェル)を、0.1mg/mLのポリ−D−リジン(Sigma(登録商標)))でコーティングした。CNSミエリンのみ、あるいは、sNogoR310、sNogoR310−Fc融合タンパク質、mAB 5B10、または対照としてのPBSと混合したCNSミエリンを、3μLの液滴として別々にスポットした。蛍光ミクロスフェア(Polyscience)を、ミエリン/PBSに添加して、後で液滴が同定できるようにした(Grandpre et al.,Nature 403,2000)。その後、Lab−Tek(登録商標)スライドをリンスし、10μg/mLのラミニン(Gibco(登録商標))でコーティングした。
【0187】
P3−4 Sprague Dawleyラットの子供から後根神経節(DRG)を取り出し、1mg/mLのコラゲナーゼ1型(Worthington)で解離させ、炎磨きパスツールピペット(fire−polished Pasteur pipette)で粉末状にし、ニューロン細胞を多く含むように予めプレーティングし、最後に予めコーティングしたLab−Tek培養スライド上に23,000細胞/ウェルでプレートした。培養培地は、5%の熱不活化ドナーウマ血清、5%の熱不活化ウシ胎児血清、および50ng/mLのmNGFを含むF12とし、37℃、5%のCO2で6時間インキュベートした。
【0188】
スライドを、20%のスクロースを含む4%のパラホルムアルデヒドで20分間固定し、1:500希釈した、ニューロンマーカーである抗β−III−チューブリン(Covance TUJ1)について染色した。二次抗体である抗マウスAlexa Flour(登録商標)594(Molecular Probes)を1:300希釈し、スライドを、Gel/Mount(登録商標)(Biomeda(登録商標))を用いてカバースライドをかけた。OpenLab(登録商標)ソフトウェアを用いて5倍のデジタル画像を獲得し、MetaMorph(登録商標)ソフトウェアを使用して、神経突起成長の量について分析した。
【0189】
sNogoR310、sNogoR310−Fc、およびmAb 5B10は全て、神経突起の成長のミエリンによって媒介される阻害からDRGニューロンを保護した(図9〜11)。sNogoR310を、ニワトリのニューロンを使用する同様のアッセイにおいて使用し、保護することを見出した。
【0190】
本発明者らは、また、ラミニンの存在下で増殖させた細胞と、ラミニンが存在しない条件下で増殖させた細胞を用いて実験を行うことにより、可溶性Nogo受容体の神経保護効果を試験した。ラミニンを含まない培地中でのニューロン細胞の成長は乏しく、ニューロンのストレス条件のモデルとなる。
【0191】
DRGを、ラットの子供から生後6〜7日で切り出し(P6〜7)、単細胞になるように解離させ、上記のようにポリ−D−リジンを予めコーティングした96ウェルプレート上にプレーティングした。いくつかのウェルには、2μg/mLのラミニンを2〜3時間かけて添加し、細胞をプレーティングする前にリンスした。18〜20時間のインキュベーションの後、プレートを、4%のパラホルムアルデヒドで固定し、1:500希釈した、ウサギ抗β−III−チューブリン抗体(Covance(登録商標))および1:100希釈した抗HuC/D(Molecular Probes)で染色した。蛍光二次抗体(Molecular Probes)を、1:200希釈で添加した。ArrayScan(登録商標)II(Cellomics(登録商標))を使用して、5倍のデジタル画像を獲得し、Neurite outgrowth applicationを使用することによって、ウェルあたりの平均の神経突起成長/ニューロンとして神経突起成長を定量した。1種類の条件について3つのウェルからの9個の5倍の画像を分析した。
【0192】
いくつかの実験においては、PC12細胞のサブクローン(Neuroscreen(登録商標))を使用した(Cellomics(登録商標))。Neuroscreen(登録商標)細胞を、200ng/mLのNGFを用いて7日間の間予め分化させ、分離させ、ポリ−D−リジンをあらかじめコーティングした96ウェルプレート上に再度プレーティングした。いくつかのウェルには、5μg/mLのラミニンを2〜3時間かけて添加し、細胞をプレーティングする前にリンスした。2日間のインキュベーションの後、プレートを、4%のパラホルムアルデヒドで固定し、1:500希釈した、ウサギ抗β−III−チューブリン抗体(Covance(登録商標))およびHoechst(核染色)で染色した。ArrayScan(登録商標)IIを使用して、DRG細胞中のものとして神経突起成長を定量した。
【0193】
sNogoR344−FcまたはラットIgGを、プレーティング時に、P6−7 DRGニューロンおよび分化させたNeuroscreen(登録商標)細胞に、溶液中で添加した。
【0194】
P6 DRGニューロンをラミニンが存在しない条件下で増殖させた場合のsNogoR344−Fcの神経保護作用を、1μMおよび10μMで観察した。神経突起成長の定量は、sNogoR344−Fcの添加に伴う用量依存性の増加を示した。ラミニン基質上でのDRGニューロンの成長に対する同じ濃度のsNogoR344の添加は、何ら特異な効果を生じなかった。このことは、sNogoR344−Fcはストレスを受けた細胞に対してのみ活性があることを示している。ラミニンが存在しない条件下での同じ濃度のsNogoR344−Fcの神経保護効果を、Neuroscreen(登録商標)細胞を用いて観察した。
【0195】
(実施例11)
(Fc−sNogoR−1融合タンパク質の産生および精製)
ラットNogo受容体−1のアミノ酸1〜310をコードするcDNA構築物を、哺乳動物発現ベクター中に含まれるラットIgG1 Fcに融合させ、このベクターをチャイニーズハムスター卵巣(CHO)(DG44)細胞にエレクトロポレーションした。細胞を、10%の透析したウシ胎児血清、2mMのグルタミン、および抗生抗かび剤を補充したα−MEM中で維持した。トランスフェクションの2日後、馴化培地を回収し、還元条件下でのウェスタンブロットによって分析した。約60kDaのタンパク質のバンドを、ポリクローナルウサギ抗Nogo受容体−1抗体を使用して検出した。細胞を増殖させ、R−PE結合ヤギ坑ラットIgG抗体を使用して分類した。2回目の分類の後、細胞を、96ウェルプレートに1細胞/ウェルの密度でプレーティングした。個々のウェルから分泌された可溶性Nogo受容体−1タンパク質のレベルを試験し、サンドイッチELISAを使用して比較した。ELISAプレートを、ヤギ坑ラットIgG Fcκ特異的抗体を用いてコーティングした。馴化培地をアプライした。結合した可溶性Nogo受容体−1タンパク質を、HRP結合ロバ抗ラットIgG Fab、Fc特異的抗体によって検出した。クローン4C12は最も高い分泌レベルを有していた。4C12を、スピナーフラスコの中のCHO−M7培地中で拡大、増殖させた。分泌レベルは、37℃で約10mg/Lであった。
【0196】
sNogoR310−Fc融合タンパク質を発現するCHO細胞を、大スケールで培養した。1.7Lの濃縮した馴化培地を、10Lのバイオリアクターを運用することによって得た。1.0MのTris−HCl(pH8.9)の1/10用量を添加することによって、pHを上昇させた。固形の塩化ナトリウムおよびグリシンをそれぞれ、3.0Mおよび1.5Mになるように添加した。10mMのTris−HCl、3Mの塩化ナトリウム、1.5Mのグリシン(pH8.9)で平衡化させた60mLのプロテインA−Sepharose(登録商標)カラムを準備した。濃縮した馴化培地をカラムに、蠕動ポンプを使用して1.5mL/分でアプライした。カラムを、300mLの10mMのTris−HCl、3Mの塩化ナトリウム、1.5Mのグリシン(pH8.9)で洗浄し、その後、120mLの5mMのTris−HCl、3Mの塩化ナトリウム(pH8.9)で洗浄した。タンパク質を、25mMのリン酸ナトリウム、100mMの塩化ナトリウム(pH2.8)を用いて溶出させた。10mLの画分を、1.0mLの1.0MのHEPES(pH8.5)を含むチューブに回収した。タンパク質画分をプールし、3×2Lの5mMのリン酸ナトリウム、300mMのNaCl(pH7.4)に対して透析した。
【0197】
(実施例12)
(脊髄離断アッセイ)
生体内で機能回復を促進するそれらの能力を試験するために、sNogoR−1融合タンパク質を、ラットの脊髄離断アッセイにおいて試験した。
【0198】
Alzet(登録商標)浸透圧ポンプに、使用する日に新しく作成した試験溶液(PBS中のsNogoR310−Fc)を充填した。充填濃度は、5μMおよび50μMになるように計算した。ポンプを、動物への埋め込みの前に、37℃で>40時間になるように準備した。雌のLong Evansラットに、術前鎮痛剤と鎮静剤を投与し、イソフルラン(O2中3%)を使用して麻酔した。
【0199】
ラットを、定位フレームに入れ、運動皮質を露出させ、左右双方から菅追跡剤(tract tracing agent)BDA(10,000MW)を注入した。その後、ラットに、T5−T6で脊髄の背部片側切断を行い、その後、試験化合物を投与するための髄腔内カテーテルとポンプシステムを挿入した(1つのグループあたりn=11)。
【0200】
ラットを回復させ、ラットは外科手術後28日まで生存していた。BBBシステムを用いた行動採点法により、研究の生存期間が終わる直前である損傷の誘導後28日までを記録した。潅流および固定後、脊髄を採取し、凍結防止し、切片とし、染色し、軸索のカウントを行った。
【0201】
Basso−Beattie−Bresnahan(BBB)運動機能評価尺度(Basso et al.,1996,Neurotrauma 13,343−359)、傾斜板試験、および傾斜格子歩行試験(Li and Strittmatter,2003,J Neurosci.2003,23,4219−27)を、損傷の後、ラットとマウスにおいてモニターした。傾斜板試験のために、本発明者らは、50cm×60cmの板がマウスが滑り落ちることなく5秒間傾いていられる最大角を測定した。傾斜格子歩行のために、マウスに、45度の傾斜のワイヤー格子をよじ登るようにトレーニングした(2.54cm四方の35cmの長さ)。格子面よりも下に後肢が落ちた回数を、下から上までの各歩行について記録した。ラットの行動試験については、BBB運動機能尺度、格子歩行分析および足跡の分析を行った。格子歩行のために、ラットに、ワイヤー格子上を歩くようにトレーニングし(2.54cm四方の70cmの長さ)、格子面よりも下に後肢が落ちた回数を数えた。足跡の分析のために、ラットの後肢の歩行パターンを、90cmの通路を途切れることなく動く間に、インクで記録し、それぞれの側のストライドの長さおよびストライドの幅を計算した(Metz et al.,2000、Brain Res.,883,165−177)。これらの行動試験の全てを、少なくとも2匹の個体によって行った。外科手術、行動試験、および組織学的分析の間中、研究は、ミニポンプの中の化合物の実体について盲検とした。
【0202】
sNogoR310−Fcは機能の回復を促進した(図12)。
【0203】
(実施例13)
(ラットの脊髄挫傷アッセイ)
生体内でのニューロンに対する可溶性Nogo受容体−1ポリペプチドおよび融合タンパク質の作用を、ラットの脊髄挫傷アッセイにおいて試験した。
【0204】
雌のフードをかぶせたLong Evansラット(170〜190g)を、鎮痛剤と抗生物質で予防的に処置した。外科手術の10分前に、動物を2.5mg/kgのミダゾラムを用いてi.p.で鎮静し、O2中の2〜3%のイソフルランで麻酔した。その後、ラットの毛を剃り、アルコールとベタジンで拭き、眼に眼用滑剤を点眼した。次に、中線切開を行い、T7からT12脊椎を露出させた。
【0205】
背部椎弓切除術を、T9 1/2とT10で行い、脊髄を露出させた。ラットを衝撃部材の上に載せた。T7およびT8セグメントを最初にクランプし、その後、T11およびT12セグメントを尾側のクランプに取り付けた。やわらかい物質をラットの胸部の下に入れた。衝撃部材の棒をゼロ位置にセットし、電気設置クリップを傷の端に取り付けた。その後、衝撃部材の棒を25.0mmまで上げ、露出させた脊髄の真上の位置になるように適切に調節した。次に、衝撃部材の棒を、露出させた脊髄に当たるように放し、衝撃部材の棒をすぐに拾い上げた。
【0206】
その後、ラットを取り外し、Gelform(登録商標)を傷の上に置いた。傷を覆う筋肉を縫合し、切開部を外科用ホッチキスで留めた。動物を、麻酔から回復するまでインキュベーターに入れた。ラットには、必要に応じて、抗生物質、鎮痛剤、および生理食塩水を投与した。膀胱を、機能が回復するまで毎朝晩に絞った。
【0207】
可溶性Nogo受容体−1融合タンパク質(例えば、sNogoR310−Fc)を、上記のラットの脊髄離断モデルにおいて記載したように、髄腔内投与した。BBB採点法を、外科手術の1日後、その後4週間から6週間まで毎週行った。
【0208】
(実施例14)
(トランスジェニックマウスにおけるsNogoR310の発現)
本発明者らは、生体内で発現させた場合のその効果を試験するために、可溶性Nogo受容体−1タンパク質を発現するトランスジェニックマウスを作成した。
【0209】
本発明者らは、マウスsNogoR310 cDNA(Nogo受容体−1のアミノ酸1〜310に対応する)を、C−3123ベクターのNotI部位にクローニングした。このベクターの中では、sNogoR310の発現はグリア細胞繊維性酸性タンパク質(gfap)遺伝子調節エレメントの制御下にある。グリア細胞繊維性酸性タンパク質遺伝子調節エレメントは、損傷の部位の反応性星状細胞からの分泌の増強を伴う、高レベルの発現を可能にする。本発明者らは、得られたベクターをAatIIとSfiIで連続して消化し、gfap::sNogoR310構築物を3.4kbの断片上に単離した。本発明者らは、この断片を胚にマイクロインジェクションして、トランスジェニックマウスを作成した。本発明者らは、PCRによって、トランス遺伝子が組み込まれていることを確認し、5つの創始株を同定した。本発明者らは、最も高い発現レベルを有している2つの創始株のヘテロ接合型雄を、雌のC57BL/6Jマウスと交配させた。本発明者らは、ヘテロ接合型トランスジェニックマウスにおいてGFAP陽性細胞がsNogoR310を発現し、分泌していることを、Nogo受容体−1に対して惹起させた抗体を使用したウェスタンブロット分析によって確認した。
【0210】
本発明者らは、プロテアーゼ阻害因子(Roche)を補充したTris緩衝化生理食塩水中で皮質および脊髄をホモジナイズし、ホモジネートを40,000rpmで4℃で20分間遠心分離した。本発明者らは、抗体特異性を高めるために上清を4%のパラホルムアルデヒドで20分間処理し、免疫ブロッティングの前に透析した。本発明者らは、RIPA緩衝液(PBS中の1%のTriton(登録商標)X−100、0.5%のデオキシコール酸ナトリウム、0.1%のSDS)中での超音波処理によって微粒子画分をホモジナイズし、得られたホモジネートを遠心分離し、この上清(界面活性剤可溶性微粒子画分)を上記のように処理した。本発明者らは、20μgの脳または脊髄タンパク質を、1:2000希釈のNogo受容体−1に対して惹起させられたウサギ抗血清を使用する免疫ブロットによって分析した。本発明者らは、AP結合抗ウサギIgGおよびNBT/BCIP AP基質とのインキュベーションによって、免疫反応性を視覚化した。
【0211】
本発明者らは、2つのトランスジェニック株Tg08およびTg01に由来する皮質および脊髄の界面活性物質を含まない可溶性抽出物中に、分泌された37kDaのsNogoR310を検出した。しかし、37kDaまたは81kDaのいずれの可溶性Nogo受容体−1タンパク質は、同腹子の野生型(WT)マウス中に存在するとしても、ごくわずかであった。微粒子画分の試験により、WTおよびトランスジェニックマウスの両方に相当のレベルの内因性Nogo受容体−1が存在することが明らかになった。
【0212】
(実施例15)
(損傷の後のトランスジェニックマウスにおけるsNogoR310の発現)
本発明者らは、背部の全片側切断(over−hemisection)損傷を行うことにより、トランスジェニックマウスにおけるsNogoR310の発現に対するCNS損傷の影響を試験した。本発明者らは、実施例14に記載したようにヘテロ接合型の雄をC57/BL6雌と交配させることによって、sNogoR310トランスジェニックマウスと、トランスジェニックではない対照動物を得た。
【0213】
本発明者らは、成体である雌のヘテロ接合型トランスジェニックマウスまたは同腹子であるWTマウス(10〜16週齢)に深く麻酔をかけ、完全な椎弓切除術を行って、T6およびT7のレベルで脊髄の背部を完全に露出させた。本発明者らは、30ゲージ針と一対のマイクロシザーズでT6に背部全片側切断(over−hemisection)を行い、背部と背側部の皮質脊髄路(CST)を完全に切断した。本発明者らは、記号のついた針を脊髄の背部を数回貫通させて、病変が確実に1.0mmの深さになるようにした。本発明者らは、椎弓切除術を行った筋肉層を縫合し、外科用ホッチキスで背面で皮膚を閉じた。皮質脊髄路を追跡するために、本発明者らは、脊髄損傷の14日後に、頭蓋骨内において右側に大脳皮質を覆うバーホール(burr hole)を作成した。本発明者らは、トレーサーBDA(MW 10,000、PBS中で10%)(Molecular Probes,Eugene,OR)を、皮質表面から0.7mmの深さで4つの注射部位に注入した。損傷の4週間後、マウスにPBSを噴門から潅流させ、その後、4%のパラホルムアルデヒドを潅流させた。sNogoR310の発現実験に使用したマウスには、トレーサーの注射を全く行わなかった。
【0214】
ウェスタンブロット分析に使用したマウスについては、T3とL3の間のレベルで脊髄を採取し、損傷の14日後の潅流は行わなかった。Nogo受容体−1免疫組織化学的染色に使用したマウスには、片側切断の10日後に4%のパラホルムアルデヒドを潅流させ、損傷をうけた脊髄を切片を作成するために取り出した。トランスジェニックマウスとWTマウスの損傷した脳におけるsNogoR310の発現を試験するために、皮質の刺し傷を、定位固定装置に取り付けた11番手術用メスを用いて作成した(David Kopf,Tujunga,CA)。ブレグマ(Bregma)の0.5mm後方、中線から1.5mm側方、3.5mmの深さに、4mmの傍矢状の切開を行った。
【0215】
本発明者らは、トランスジェニックマウスにおいては、損傷の10日後に、脊髄の可溶性抽出物中においてsNogoR310のレベルの増大を観察したが、WTマウスにおいては見られなかった。このことは、病変の周辺でのGFAPの損傷後のアップレギュレーションと一致する。この原因が、Nogo−Aの代償性アップレギュレーションではないことを確認するために、本発明者らはその発現を試験し、WTマウスとトランスジェニックマウスの両方に由来する、完全な、または損傷を受けた皮質および脊髄のいずれにおいても同様であったことを見出した。
【0216】
本発明者らは、病変の領域を含む損傷を受けた脳および脊髄の、Nogo受容体−1およびGFAPに対する抗体での免疫染色によって、損傷を受けたCNS中でのsNogoR310の細胞性発現を試験した。反応性の星状細胞神経膠の一般的な形態は、WTマウスとトランスジェニックマウスの間では差はなかったが、細胞内空間および細胞外空間の両方において、Nogo受容体−1について染色された密度は、WTマウスよりも、gfap::sNogoR310トランスジェニックマウスにおいて顕著に高かった。このことは、トランスジェニックマウスの病変の周辺での高いsNogoR310の発現を示している。Nogo受容体−1タンパク質は、トランスジェニックマウスだけにおいて、星状細胞マーカーであるGFAPと一緒に存在している。トランスジェニックの試料においては、大幅に強い拡散した非細胞性染色もまた存在し、これは、細胞外空間にあるsNogoR310と一致する。神経細胞体Nogo受容体−1染色は、WTマウスとトランスジェニックマウスの両方において検出された。
【0217】
(実施例16)
(分泌されたsNogoR310はトランスジェニックマウスにおいてCSTの萌芽を誘導する)
本発明者らは、トランスジェニックマウスにおける病変の周辺でのsNogoR310の発現の増大が損傷を受けた軸索の再生を生じるかどうかを試験した。
【0218】
本発明者らは、Li and Strittmatter,2003,J.Neurosci.,23,4219−27に記載されているように、順行性トレーサーであるビオチンデキストランアミン(BDA)を右運動皮質に注入することによって、下行皮質脊髄路(CST)の完全性を調べた。同腹子のWTマウスにおいては、突出した背部CST(dCST)は、病変に対して吻側にしっかりとまとまっており、いくつかの背側部のCST線維は同側上に見ることができる。少数のBDA標識された短い付随する萌芽が、灰白質、具体的には、前索に突き出しているが、萌芽は、トレーサーの注入に対して対側性の脊髄の側に、大部分が確認できた。しかし、損傷を受けたsNogoR310トランスジェニックマウスに由来する背部片側切断に対して吻側の切片は、極めて異なるBDA標識パターンを示した。高密度のBDA標識されたCST線維が、株Tg08または株Tg01に由来する全てのトランスジェニックマウスにおいて突出したdCSTの外側に観察された。異所性の線維は、灰白質の領域を貫通して伸び、いくつかの線維は外側白質および背側部の白質にまで達していた。いくつかの線維(横断切片あたり4〜12個の萌芽)が、脊髄の反対側(トレーサーの注入部位に対して同側)に見られた。付随する萌芽のマイクロデンシトメトリーによる測定は、sNogoR310トランスジェニックマウスにおいて萌芽の密度がおよそ10倍増大したことを示していた。病変に対して1から4mm吻側の傍矢状の縦断面の試験により、dCST線維がsNogoR310トランスジェニックマウスにおいては腹部の灰白質の領域に入るような多数の分岐する萌芽を延ばし、これが同腹子であるWT動物とは対照的であることが明らかになった。一般的には、トランスジェニックマウスの病変に対して吻側の萌芽のパターンおよび広がりは、Nogo受容体−1アンタゴニストペプチドNEP1−40で全身的に処置したマウスにおいて観察されるものと似ている(Li and Strittmatter,2003)。
【0219】
これらの結果は、分泌されたsNogoR310がトランスジェニックマウスにおいてCST萌芽を誘導することを示している。
【0220】
(実施例17)
(sNogoR310トランスジェニックマウスにおいては、CST軸索の再生により病変部位を回避して脊髄の先端へとバイパスする)
本発明者らは、トランスジェニックマウスから、病変部位に対して吻側4mmから尾側4mmの脊髄(合計8mm長)を単離した。これを、グルタルアルデヒド重合アルブミンマトリックスに包埋し、ビブラトーム(vibratome)(30μmの厚み)の上で傍矢状に切断した。本発明者らは、損傷部位に対して吻側5〜7mm、尾側5〜7mmの脊髄から、横断切片(50μm)を回収した。ラットにおけるsNogoR310−Fcの注射実験のために、病変部位から、吻側10mmから尾側10mmまでまたがる脊髄を、バイブレーションミクロトーム上で傍矢状(50μm)に切断した。横断切片を、損傷部位に対して吻側11〜16mmから尾側11〜16mmの脊髄から回収した。本発明者らは、これらの切片を、アビジン−ビオチン−ペルオキシダーゼ複合体とともにインキュベートし、ニッケル増強ジアミノベンジジンHRP反応によってBDAトレーサーを視覚化した(Grandpre,2002,Nature,417,547−551)。本発明者らは、間接的な免疫蛍光によって、セロトニン免疫組織化学(抗−5−HT抗体)のためにいくつかの切片を処理した。病変の領域を視覚化するために、本発明者らは、いくつかの切片を、GFAPに対する抗体(Sigma(登録商標)St.Louis,MO)で二重染色した。本発明者らは、封入剤で切片を封入し、脱水し、保護した。
【0221】
本発明者らは、損傷後にトランスジェニックマウスにおいて発現されたsNogoR310によって誘導された線維 (実施例16)が、病変の領域を抜けて尾側の脊髄に達し、機能回復をもたらすかどうかを試験した。
【0222】
カメラルシーダディスプレイに取り込んだ損傷部位の全域を越える連続する傍矢状切片は、病変から数ミリメートルの再生しつつあるCST線維の全体的な分布パターンを示す。WTマウスに由来する切片は、損傷部位を越えて伸びるCST繊維は示さなかった。sNogoR310トランスジェニックマウスに由来する同様の切片は、離断領域を超える多数のCST線維と、先端部の灰白質および白質の領域に伸びる突起を、高度に分岐したパターンで示した。片側切断に対してすぐ吻側には、高密度のBDA標識されたCST萌芽が、突出したdCST突起から病変の領域の方向に存在するが、ほとんどのCST萌芽は、傷跡の形成および組織の空孔が顕著である離段領域を超えることはできなかった。少しではあるが極めて顕著な、再生しつつある軸索の画分が、背部および背側部の灰白質および白質の残っている組織の細胞間橋を通って病変部位を回避する。さらに、ごく少数のCST線維は、離断領域自体を通過して、病変の背部および背側部の脊髄を通って先端領域へと伸びるようである。病変の近傍では、再生しつつある線維の進路は通常、蛇行性であり、吻側のCST中の正常なまっすぐの線維とは極めて異なる。側枝および樹枝状の線維は、脊髄の先端部の灰白質において最も頻繁に見られる。再構築により、それぞれのトランスジェニックマウスにおいて病変に対して尾側1〜4mmのいずれかのレベルで、吻−尾軸において、5〜15本のBDA標識された再生しつつある線維の追跡が明らかになった。背部片側切断に対して5〜7mm尾側の横断面については、BDA標識されたCST軸索が、それぞれのトランスジェニックマウスにおいて灰白質と白質の両方の領域において見られた。トランスジェニックマウスについての繊維の数は、矢状断面の最も近いレベルとほぼ同数のBDA標識されたCST線維を示した。
【0223】
CST線維に加えて、他の下行路、例えば、縫線核脊髄線維もまた、マウスの運動機能に関与している。このマウスの背部全片側切断(over−hemisection)モデルにおいては、離断によりセロトニン作動性線維の大部分が損傷を受け、前角においてはこれらの線維の密度がおよそ80%減少した。尾側の脊髄の前角におけるセロトニン線維の全長の分析は、トランスジェニックマウスにおいてはWTグループよりもはるかに多い数のこれらの線維を示し、このことは、トランスジェニックマウスにおけるsNogoR310の成長促進作用が、1つの軸索の下行路には限定されないことを示している。
【0224】
(実施例18)
(sNogoR310のトランスジェニックでの発現は運動機能の回復を改善する)
CST軸策の追跡およびセロトニン作動性線維の分析は、トランスジェニックマウスにおいて星状細胞から分泌されたsNogoR310が、脊髄の損傷をうけた下行軸索の広範囲の解剖学的再生を刺激することを示している。本発明者らは、これらの再生した線維が機能の回復に利点をもたらすかどうかを決定するために、実施例12に記載したいくつかの行動試験を行った。
【0225】
BBB試験によって評価した場合には、WTマウスは、4週間の生存期間の間に運動機能を一部回復した。損傷後4週間では、ほとんどのWTマウスが、しっかりと体重を支えながら、しっかりと足底で踏みしめることによって特徴付けられるレベルに回復したが、これらは、時折、前肢−後肢の同調を生じるだけで、主に、最初に表面に接触させた足の位置で旋回していた。対照的に、株Tg08およびTg01の両方に由来するsNogoR310トランスジェニックマウスのBBBスコアは、7〜28日の観察期間を通じて、対照のグループよりも有意に高かった(図13Aおよび13B)。損傷の28日後には、ほとんどのトランスジェニックマウスが一貫して前肢−後肢の同調を示し、主に、足の位置は体に対して並行であった。
【0226】
本発明者らは、sNogoR310トランスジェニックマウスの能力をさらに特徴付けるために、2つのさらなる行動試験を使用した。最初に、本発明者らは、5秒以内にマウスがその格子を落ちることなく傾けられた板につかまっていられる最大角を測定した。背部片側切断による損傷の前には、トランスジェニックマウスおよびWTマウスの両方が、55度に傾けられた板の上でその姿勢を維持できた。損傷の7〜28日後に、維持できる角度は、全てのマウスにおいて低くなったが、トランスジェニックマウスが維持できる角度は、対照のグループよりも有意に大きかった(図13C)。別の行動試験においては、マウスに、垂直に対して45度の角度に置いた格子に上らせ、格子面から後肢が脱落した回数を数えた(Metz et al.,2000)。損傷前のトレーニングの間のこの試験においては、マウスはミスすることはなかった。損傷後2〜6週間の期間の間には、WTマウスは多数回の足を踏み外すミスをし、最少程度の改善しか見られなかった。対照的に、sNogoR310トランスジェニックマウスは、この期間の間に、格子をよじ登ることにおいて進行性の改善を示し、ほとんどの改善は損傷後1〜3週間の間に生じた(図13D)。したがって、星状細胞からsNogoR310を分泌するトランスジェニックマウスは、胸部脊髄の片側切断の後、CSTの再生、縫線核脊髄萌芽、および運動機能の改善を示した。
【0227】
(実施例19)
(sNogoR310−Fcタンパク質の髄腔内投与によりCSTの萌芽が誘導される)
脊髄の外傷後の可溶性Nogo受容体−1の成長促進の利点についての第2の試験として、本発明者らは、精製したタンパク質を髄腔内投与した。
【0228】
本発明者らは、安定性を増大させ精製を促進するために、ラットのNogo受容体−1のリガンド結合ドメイン(27〜310)をラットIgG1 Fcドメインに融合させた。本発明者らは、安定にトランスフェクトされたCHO細胞からタンパク質を精製した。このタンパク質は、マウスsNogoR310−Myc Hisについて以前に示されたとおり(Fournier et al.,2002、J Neurosci.,22,8876−8883;Liu et al.,2002、Science,297,1190−1193)、Nogo−66、MAG、およびミエリンの作用を生体外でブロックする。本発明者らは、胸中部の背部片側切断損傷を行ったラットに、浸透圧ミニポンプによりsNogoR310−Fcタンパク質を髄腔内投与した。損傷後の4週間の生存期間の間に、1.2mgのsNogoR310−Fcタンパク質を、それぞれのラットに局所投与した。ビヒクル(1.2mgのラットIgG)での処置を受けたラットにおいては、片側切断部位に対して吻側の切片は、しっかりとまとまった突出した背部CSTを示し、病変部位の上部にはごく少数の異所性BDA標識CST線維を示した。sNogoR310−Fcタンパク質を投与した損傷を受けたラットに由来する、病変に対して吻側の切片は、極めて異なる標識パターンを示した。BDA標識されたCSTからの多数の異所性線維の萌芽が、横断切片および傍矢状切片から観察された。いくつかの場合には、突起が、中線付近のdCST領域から脊髄の境界線に向かってのび、背側部CSTと混ざりあっていた。芽がでかかった軸索は灰白質を通り、白質よりもさらに伸びていた。dCSTに隣接する異所性の芽がでかかった線維の測定(横断切片において≧100μm、矢状断片においては≧200μm)により、sNogoR310−Fcで処置したラットにおけるさらなる増大が明らかになった。
【0229】
(実施例20)
(sNogoR310で処置したラットにおいて、CST軸索は脊髄の先端へと再生する)
本発明者らは、雌のSprague−Dawleyラット(190〜250g)に深く麻酔をかけ、T6〜7の脊髄レベルで椎弓切除術を行い、脊髄を露出させた。本発明者らは、脊髄の背部の半分を、30ゲージ針と一対のマイクロシザーズで切断し、CSGT経路の背部を完全に切断し、11番手術用メスの鋭い部分を脊髄の背部の半分を横断させることにより、病変の深さ(1.8mm)を確保した(Grandpre et al.,2002,Nature,417,547−551)。PBS中の1.2mgのラットIgG、またはPBS中の1.2mgのsNogoR310−Fc融合タンパク質を充填した浸透圧ミニポンプ(Alzet(登録商標)、2ML4、2mLの容量、2.5μL/h、28日間の投与)を、動物の背中の皮膚の下の筋肉に埋め、縫合した。ミニポンプの出口に接続したカテーテルを、硬膜の中の小さな穴を通じてT7〜8のレベルで脊髄の髄腔内空間に挿入した。
【0230】
Nogo受容体−1アンタゴニストタンパク質の注入により、ラットの片側切断に対して吻側で広範囲の萌芽を誘導したが、より重要な問題は、芽を出しつつあるCST線維が脊髄の先端に突き出すかどうか、そして運動機能の回復に関係しているかどうかである。ビヒクルで処置したラットに由来する病変を横切る縦断面は、病変のレベルより下では、検出できる程度のBDA標識された腹部CST線維は示さなかったか、またはごく少数のBDA標識された腹部CST線維しか示さなかった(GrandPre et al.,2002;Weidner et al.,2001、Proc.Natl.Acad.Sci.USA,98,3513−3518)。sNogoR310−Fcで処置したラットに由来する同様の切片は、多くのBDA標識線維が離断部位をバイパスし、主に腹部および腹側両面脊髄の組織の細胞間橋を通って尾側の脊髄に向かって突出することを示した。星状細胞マーカーGFAPについての免疫染色は、離断の程度が脊髄中心菅の領域よりも深くまで到達していることを示した。突出した背部CST中の吻側の線維の直線的なプロフィールとは異なり、再生したCST線維は、通常、脊髄先端、特に、灰白質の領域においては、高度に分岐している軌跡を残す。これらの線維は、脊髄の多くの領域で検出されたが、これらは、脊髄の中心部分、および脊髄の背部の半分から脊髄の隅々にわたって容易に見ることができた。矢状断面によるCST線維の数は、sNogoR310−Fcで処置したラットのそれぞれに由来する病変に対して1〜2mm尾側ではおよそ20個のBDA標識された軸索を、病変に対して7〜8mm遠位では15個の追跡された軸索を示した。
【0231】
一般的には、これらの線維の分岐パターンは、NEP 1−40ペプチドで局所的に処置された動物から観察されるパターンと類似しているが、それぞれの萌芽にさらに付随する分岐が、sNogoR310−Fcタンパク質で処置した切片から見られた。脊髄の先端からの萌芽の測定は、sNogoR310−Fcで処置したラットにおいてそれぞれの萌芽に付随する全ての長さが、NEP 1−40で処置した動物よりも2倍長いことを示した。Nogo受容体−1アンタゴニストで処置したグループの両方における脊髄に対して1〜10mm尾側の萌芽の数(長さに関して≧200μm)は、対照のグループよりもおよそ20〜40倍多かった。萌芽も、NEP 1−40で局所的に処置したラットよりもsNogoR310−Fcで処置したラットにおいて多く見られたが(25個の萌芽/ラットに対しておよそ50個の萌芽/ラット)、この差は、統計学的に有意ではなかった(p=0.1713、t検定)。
【0232】
再生しつつあるCST軸索を、sNogoR310−Fc処置を受けさせたラットにおいて、片側切断に対して11〜15mm尾側の脊髄の横断切片において観察した。これらの線維は、脊髄の灰白質と白質の両方において検出された。灰白質で検出された繊維は、多くの場合には、白質の領域で見られるものよりも多くの分岐の付随を示した。対照的に、ビヒクルで処置したグループに由来する横断切片においては、腹部の白質の領域において、ごくたまにBDA標識が見られ、これは、損傷を受けていない腹部のCST軸索と一致した。脊髄の先端のこのレベルでは、Nogo受容体−1アンタゴニスト(sNogoR310−FcおよびNEP 1−40)で処置されたグループの両方に由来するBDA標識されたCST線維の平均の数は、ビヒクルで処置されたラットよりもおよそ20倍多かった。まとめると、いずれのNogo受容体アンタゴニスト(sNogoR310−Fcタンパク質およびNEP 1−40ペプチド)も、脊髄の先端において劇的なCST軸索の再生を生じるが、前者によって誘導される萌芽は、より高度に分岐したパターンを示す。
【0233】
(実施例21)
(局所sNogoR310−Fcは、損傷を受けたラットの脊髄において赤核脊髄およびセロトニン作動性軸索の萌芽を誘導する)
片側切断の14日後、バーホールを、CST線維の軌跡をたどるために、下肢の感覚運動皮質に重なる頭蓋骨のそれぞれの側に空けた。順行性のニューロンのトレーサーであるBDA(PBS中に10%、3.5μL/皮質)を、それぞれの側の硬膜から1.5mmの深さの7個の注射部位に注射した(Grandpre,2002)。ラットにおいて赤核脊髄路の軌跡をたどるために、トレーサーBDA(1μL:MW 10,000;PBS中10%
)を、左側の赤核(ブレグマの前方5.8mm、0.7mm側方、頭蓋骨表面に対して7.0mm腹部側)に注射した。BDAの注射の2週間後、これらの動物にBPSを潅流させ、その後、4%のパラホルムアルデヒドを潅流させ、組織を組織学のために採取した。
【0234】
損傷を受けた赤核脊髄路(RST)繊維の修復は、脊髄損傷後の機能の改善に関与している(Liu et al.,1999,J.Neurosci.,19,4370−4387)。CNSニューロンにおけるNogo受容体−1の広範囲の分布(Wang et al.,2002,J.Neurosci.,22,5505−5515)は、Nogo受容体−1のそのアンタゴニストでの阻害により、損傷後のRST軸索の再成長を生じさせることができることを可能にする。損傷を受けたRSTに対するsNogoR310−Fcの作用を試験するために、この経路の完全性を、左の赤核にBDAを注射することによって軌跡をたどった。脊髄のレベルでは、RST線維は通常は、脊髄の背側部の白質の領域に局在しており、これを、この研究の背部片側切断によって離断した。対照ラットに由来する病変から11〜15mm吻側の横断切片においては、少数の短いBDA標識線維が、突出したRSTと背部の角状灰白質の間に見られた。sNogoR310−Fcで処置した同じレベルの切片は、メインのRSTと背部の角状灰白質との間に多くの連結している線維を示した。SCIに対して11〜15mm遠位の横断切片については、ビヒクルで処置されたラットにおいてはBDA標識されたRST線維は見られなかった。対照的に、sNogoR310−Fcでの処置を受けた同じレベルの切片は、トレーサーの注射部位に対して対側にある灰白質および白質の両方において多くのBDA標識されたRST線維を示した。分岐パターンを伴ういくつかの萌芽が、BDA注射部位に対して同側の灰白質に見られた。
【0235】
赤核脊髄線維もまた、sNogoR310−Fcで処置された脊髄損傷を受けたラットにおいて試験した。免疫染色は、病変に対して11〜15mm吻側のセロトニン作動性線維の密度を示し、これは、ビヒクルで処置されたグループとsNogoR310−Fcで処置されたグループの間で類似していた。病変の11〜15mm下の切片においては、sNogoR310−Fcで処置されたラットにおけるセロトン線維は、対照のグループの線維の2倍多く存在した。これらの結果は、sNogoR310−Fcタンパク質によるNogo受容体−1の阻害に対する反応性が、CST線維に限定されておらず、他の下方路、例えば、赤核脊髄およびセロトニン作動性軸索もまた、Nogo受容体−1アンタゴニズムに反応することを示している。
【0236】
(実施例22)
(sNogoR310−Fcでの局所処置によりラットにおける機能回復が改善する)
sNogoR310−Fcタンパク質の髄腔内投与により、外傷性脊髄損傷後にいくつかの下方路において軸索の再生が刺激される。本発明者らは、このタンパク質により、損傷を受けた脊髄における機能回復もまた改善されるかどうかを試験した。
【0237】
片側切断の2週間後に、ビヒクルで処置したラットの運動機能BBBスコアは、12の安定なレベルに達した(図14A)。外傷の4週間後、対照のほとんど(7匹のちの6匹)は、頻度が一定である体重を支える足底での踏み、および頻度が一定である前肢−後肢の同調を有していたが、これらは、主に、最初に表面に接触させた足の位置で旋回していた。対照的に、sNogoR310−Fcタンパク質での処置を受けさせたラットにおいては、運動機能スコアは、外傷後2〜4週間の間改善し続けた。損傷の4週間後、sNogoR310−Fcで処置された9匹の動物全てが、一貫した前肢−後肢の同調、および、最初に試験表面と接触させた足の位置と並行な足の位置を有していた。
【0238】
格子の歩行は、脊髄損傷後の下行する細かい運動機能の制御の欠損を評価するために使用されている(Metz et al.,2000)。この能力には、前肢−後肢の同調、ならびに、腹側両面、皮質脊髄、および赤核脊髄線維によって媒介される随意運動の制御が必要である。損傷の前のトレーニングの間は、全てのラットが格子の棒の上に彼らの後肢を正確に置いていた。損傷後2〜4週間は、対照のラットは、1回のセッションについて8〜9回のミスをし、時間とともに最少程度の改善しか見られなかった。対照的に、sNogoR310−Fcで処置したラットは、格子歩行について進行性の改善を示し、有意に少ないミスしかしなかった(平均で4〜7回/セッション)。改善のほとんどは、損傷後2〜3週間で生じた。対照のグループにおける後肢の足跡の分析は、損傷を受けていないラットまたはsNogoR310−Fcでの処置を受けた損傷を受けたラットと比較して、片側切断後4週間で、ストライドの長さが顕著に小さく、立脚の幅が大きかったことを示した(図14C)。したがって、これらの複数の行動試験は、Nogo受容体−1機能の、アンタゴニストタンパク質の局所注射での遮断により、損傷後の運動機能の回復に改善が生じたことを示している。
【0239】
(生物学的寄託物)
ハイブリドーマHB 7E11(ATCC(登録商標)登録番号PTA−4587)、HB 1H2(ATCC(登録商標)登録番号PTA−4584)、HB 3G5(ATCC(登録商標)登録番号PTA−4586)、HB 5B10(ATCC(登録商標)登録番号PTA−4588)、およびHB 2F7(ATCC(登録商標)登録番号PTA−4585)を、アメリカンタイプカルチャーコレクション(「ATCC(登録商標)」、10801 University Boulevard,Manassas,VA 20110−2209,USAに、2002年8月9日に寄託した。
【0240】
当業者に明らかであるように、多数の変更および修飾を、本発明の精神を逸脱することなく本発明の好ましい実施形態に対して行うことができる。全てのこのようなバリエーションは、本発明の範囲に含まれるように意図される。
【図面の簡単な説明】
【0241】
【図1】図1は、Nogo受容体−1の構造の模式図である。ヒトsNogoR310には、残基26〜310が含まれており、sNogoR344には残基26〜344が含まれている。ラットsNogoR310には残基27〜310が含まれており、sNogoR344には残基27〜344が含まれている。
【図2】図2は、Vector Nti(登録商標)ソフトウェアを使用したNogo受容体−1タンパク質についての抗原性プロットを示す。ラットP−617は配列番号10であり、ラットP−618は配列番号11である。
【図3】図3Aは、抗Nogo受容体−1抗体7E11の結合活性を示すグラフである。このグラフは、Nogo受容体−1に対するNogo66の結合に対する7E11の濃度の効果を示す。図3Bは、抗Nogo受容体−1抗体1H2の結合活性を示す。グラフは、sNogoR344−Fc(本明細書中と、米国特許出願番号60/402,866においては、Fc−sNogoR344またはIg−sNogoR344とも呼ばれる)に対するNogo66の結合に対する1H2の濃度の効果を示す。Fc−MAGはsNogoR344−Fcへの結合についてNogo66と競合しなかった。
【図4】図4は、抗Nogo−R−1抗体1H2、3G5、および2F7についてのELISAの結果を示す。固定された抗原の存在下でのOD450に対する抗体の効果が決定された。固定された抗原はsNogoR310−Fc(本明細書中と、米国特許出願番号60/402,866においては、Fc−sNogoR310またはIg−sNogoR310とも呼ばれる)、sNogoR344−Fc、p−617、p−618、p−4、p−5、オボアルブミンおよびBSAである。
【図5】図5は、種々の量のミエリンの存在下でのラットDRG神経突起成長に対する、モノクローナル抗体7E11の効果を示すグラフである。
【図6】図6Aは、以下の競合因子の存在下での125I−Nogo66および125I−Nogo40に対するsNogoR310の結合の効果を示すグラフである:Nogo66、Nogo40、および抗Nogo受容体−1モノクローナル抗体上清。図6Bは、sNogoR310に対する125I−Nogo66の結合活性を示す。
【図7】図7は、sNogoR310への125I−Nogo40の結合に対するsNogoR310−Fcの効果を示すグラフである。
【図8】図8は、125I−Nogo40に対するsNogoR310−Fcの結合活性を示すグラフである。
【図9A】図9Aは、ミエリンの存在下、またはミエリンが存在しない条件下での、神経突起成長/細胞に対するsNogoR310の効果のグラフである。
【図9B】図9Bは、ミエリンの存在下、またはミエリンが存在しない条件下での、神経突起成長に対するsNogoR310の効果のグラフである。
【図10A】図10Aは、漸増量のミエリンの存在下、またはミエリンが存在しない条件下での、P4ラットDRG神経突起成長に対するsNogoR310−Fcの効果を示すグラフである。
【図10B】図10Bは、PBS、PBS+sNogoR310−Fc、20ngのミエリン、およびミエリン+sNogoR310−Fcでの処置後の、神経突起の数/細胞を示す。
【図11】図11は、漸増量のミエリンの存在下での、DRG神経突起成長/細胞に対するモノクローナル抗体5B10の効果を示すグラフである。
【図12】図12は、ラットの脊髄離断モデルにおける損傷の誘導後30日までのBBBスコアに対するsNogoR310−Fcの効果を示すグラフである。
【図13】図13Aおよび13Bは、株08または株01によるWTまたはトランスジェニックマウスにおける背部片側切断後の時間の関数として、運動機能BBBスコアを報告する。図13Cは、WTおよびトランスジェニックマウスについて、損傷後の時間の関数として、最大の耐え得る斜平面角を示す。図13Dは、損傷後の時間の関数として、斜めの格子をよじ登る間の後肢のミスを示す。全てのグラフにおいて、それぞれのグループにおいて7〜9匹のマウスによる平均±s.e.m.が報告される。トランスジェニックのグループによる値は、WTのマウスの値とは統計学的に異なる(二重のアスタリスク、P<0.01;スチューデントt検定)。
【図14】図14Aは、ビヒクルで処置した動物またはsNogoR310−Fcで処置した動物における背部片側切断後の時間の関数として、運動機能BBBスコアを示す。図14Bは、損傷後の時間の関数として、格子を歩行する間の後肢のミスを示す。図14Cは、足跡の分析を示し、これにより、損傷を受けさせていないラット、または損傷を受けさせたsNogoR310−Fcラットよりも、対照マウスにおいて、ストライドがより短く、ストライド幅がより大きいことが明らかである。全てのグラフにおいて、それぞれのグループにおいて7〜9匹のラットによる平均±s.e.m.が報告される。sNogoR310−Fcグループの値は、対照とは統計学的に異なる(図14A〜B)。対照の値は、図14Cにおいては、SCIなしのラット、またはSCI+sNogoR310−Fcラットとは統計学的に異なる(アスタリスク、P<0.05;二重のアスタリスク、p<0.01;スチューデントt検定)。
【特許請求の範囲】
【請求項1】
AAAFGLTLLEQLDLSDNAQLR(配列番号26);LDLSDNAQLR(配列番号27);LDLSDDAELR(配列番号29);LDLASDNAQLR(配列番号30);LDLASDDAELR(配列番号31);LDALSDNAQLR(配列番号32);LDALSDDAELR(配列番号33);LDLSSDNAQLR(配列番号34);LDLSSDEAELR(配列番号35);DNAQLRVVDPTT(配列番号36);DNAQLR(配列番号37);ADLSDNAQLRVVDPTT(配列番号41);LALSDNAQLRVVDPTT(配列番号42);LDLSDNAALRVVDPTT(配列番号43);LDLSDNAQLHVVDPTT(配列番号44);およびLDLSDNAQLAVVDPTT(配列番号45)からなる群より選択される、ポリペプチド。
【請求項2】
請求項1に記載のポリペプチドをコードする、核酸。
【請求項3】
発現制御配列に動作可能に連結された、請求項2に記載の核酸。
【請求項4】
請求項2または3に記載の核酸を含む、ベクター。
【請求項5】
請求項2または3に記載の核酸を含むか、あるいは、請求項4に記載のベクターを含む、宿主細胞。
【請求項6】
請求項1に記載のポリペプチドを産生する方法であって、以下の工程:
(a)請求項5に記載の宿主細胞を培養する工程;および
(b)該宿主細胞または培養培地から該ポリペプチドを回収する工程
を含む、方法。
【請求項7】
抗体を産生する方法であって、以下の工程:
(a)請求項1に記載のポリペプチドまたは請求項5に記載の宿主細胞で、宿主を免疫化する工程;および
(b)該抗体を回収する工程
を含む、方法。
【請求項8】
請求項7に記載の方法によって産生された抗体、または、該抗体の抗原結合断片。
【請求項9】
請求項1に記載のポリペプチドに特異的に結合する抗体またはその抗原結合断片であって、該抗体は、ハイブリドーマ細胞株HB 7E11(ATCC(登録商標)登録番号PTA−4587)によって産生されるモノクローナル抗体ではない、抗体またはその抗原結合断片。
【請求項10】
請求項8または9に記載の抗体または抗原結合断片であって、該抗体は、
(a)ニューロンの成長円錐の崩壊を阻害する;
(b)ニューロンにおける神経突起成長および萌芽の阻害を減少させる;ならびに
(c)リガンドに対するNogo受容体−1の結合を阻害する、
抗体または抗原結合断片。
【請求項11】
前記神経突起成長および萌芽が軸索成長である、請求項10に記載の抗体または抗原結合断片。
【請求項12】
前記ニューロンが中枢神経系ニューロンである、請求項10に記載の抗体または抗原結合断片。
【請求項13】
前記抗体がモノクローナル抗体である、請求項8または9に記載の抗体または抗原結合断片。
【請求項14】
前記抗体がマウス抗体である、請求項8または9に記載の抗体または抗原結合断片。
【請求項15】
前記抗体が、ヒト化抗体、キメラ抗体、および単鎖抗体からなる群より選択される、請求項8または9に記載の抗体。
【請求項16】
リガンドへのNogo受容体−1の結合を阻害する方法であって、Nogo受容体−1を請求項10に記載の抗体または抗原結合断片と接触させる工程を含む、方法。
【請求項17】
前記リガンドが、NogoA、NogoB、NogoC、MAG、およびOM−gpからなる群より選択される、請求項16に記載の方法。
【請求項18】
ニューロンにおける成長円錐の崩壊を阻害するための方法であって、該ニューロンを請求項10に記載の抗体またはその抗原結合断片と接触させる工程を含む、方法。
【請求項19】
ニューロンにおける神経突起成長および萌芽の阻害を減少させるための方法であって、該ニューロンを請求項10に記載の抗体またはその抗原結合断片と接触させる工程を含む、方法。
【請求項20】
前記神経突起成長または萌芽が軸索成長である、請求項19に記載の方法。
【請求項21】
前記ニューロンが中枢神経ニューロンである、請求項18または19に記載の方法。
【請求項22】
薬学的に許容される担体と、請求項8または9に記載の抗体または抗原結合断片とを含む、組成物。
【請求項23】
1つ以上のさらなる治療剤をさらに含む、請求項22に記載の組成物。
【請求項24】
死滅の危険性があるニューロンの生存を促進する方法であって、該ニューロンを、有効量の請求項8または9に記載の抗Nogo受容体−1抗体または抗原結合断片と接触させる工程を含む、方法。
【請求項25】
前記ニューロンがインビトロにある、請求項24に記載の方法。
【請求項26】
前記ニューロンが哺乳動物中にある、請求項24に記載の方法。
【請求項27】
前記哺乳動物が、多発性硬化症、ALS、ハンチントン病、アルツハイマー病、パーキンソン病、糖尿病性神経障害、脳卒中、外傷性脳損傷、または脊髄損傷の兆候または症状を呈する、請求項26に記載の方法。
【請求項28】
哺乳動物において死滅の危険性があるニューロンの生存を促進する方法であって、
(a)請求項8または9に記載の抗Nogo受容体−1抗体またはその抗原結合断片を発現する培養された宿主細胞を提供する工程;および
(b)該ニューロンの部位またはその付近において、哺乳動物中に宿主細胞を導入する工程
を含む、方法。
【請求項29】
哺乳動物中にある死滅の危険性があるニューロンの生存を促進する遺伝子治療方法であって、請求項8または9に記載の抗Nogo受容体−1抗体またはその抗原結合断片をコードするヌクレオチド配列を含むウイルスベクターを、該ニューロンの部位またはその付近に投与する工程を含み、該抗Nogo受容体−1抗体または抗原結合断片は、該哺乳動物中のヌクレオチド配列から、該ニューロンの生存を促進するために十分な量で発現される、方法。
【請求項1】
AAAFGLTLLEQLDLSDNAQLR(配列番号26);LDLSDNAQLR(配列番号27);LDLSDDAELR(配列番号29);LDLASDNAQLR(配列番号30);LDLASDDAELR(配列番号31);LDALSDNAQLR(配列番号32);LDALSDDAELR(配列番号33);LDLSSDNAQLR(配列番号34);LDLSSDEAELR(配列番号35);DNAQLRVVDPTT(配列番号36);DNAQLR(配列番号37);ADLSDNAQLRVVDPTT(配列番号41);LALSDNAQLRVVDPTT(配列番号42);LDLSDNAALRVVDPTT(配列番号43);LDLSDNAQLHVVDPTT(配列番号44);およびLDLSDNAQLAVVDPTT(配列番号45)からなる群より選択される、ポリペプチド。
【請求項2】
請求項1に記載のポリペプチドをコードする、核酸。
【請求項3】
発現制御配列に動作可能に連結された、請求項2に記載の核酸。
【請求項4】
請求項2または3に記載の核酸を含む、ベクター。
【請求項5】
請求項2または3に記載の核酸を含むか、あるいは、請求項4に記載のベクターを含む、宿主細胞。
【請求項6】
請求項1に記載のポリペプチドを産生する方法であって、以下の工程:
(a)請求項5に記載の宿主細胞を培養する工程;および
(b)該宿主細胞または培養培地から該ポリペプチドを回収する工程
を含む、方法。
【請求項7】
抗体を産生する方法であって、以下の工程:
(a)請求項1に記載のポリペプチドまたは請求項5に記載の宿主細胞で、宿主を免疫化する工程;および
(b)該抗体を回収する工程
を含む、方法。
【請求項8】
請求項7に記載の方法によって産生された抗体、または、該抗体の抗原結合断片。
【請求項9】
請求項1に記載のポリペプチドに特異的に結合する抗体またはその抗原結合断片であって、該抗体は、ハイブリドーマ細胞株HB 7E11(ATCC(登録商標)登録番号PTA−4587)によって産生されるモノクローナル抗体ではない、抗体またはその抗原結合断片。
【請求項10】
請求項8または9に記載の抗体または抗原結合断片であって、該抗体は、
(a)ニューロンの成長円錐の崩壊を阻害する;
(b)ニューロンにおける神経突起成長および萌芽の阻害を減少させる;ならびに
(c)リガンドに対するNogo受容体−1の結合を阻害する、
抗体または抗原結合断片。
【請求項11】
前記神経突起成長および萌芽が軸索成長である、請求項10に記載の抗体または抗原結合断片。
【請求項12】
前記ニューロンが中枢神経系ニューロンである、請求項10に記載の抗体または抗原結合断片。
【請求項13】
前記抗体がモノクローナル抗体である、請求項8または9に記載の抗体または抗原結合断片。
【請求項14】
前記抗体がマウス抗体である、請求項8または9に記載の抗体または抗原結合断片。
【請求項15】
前記抗体が、ヒト化抗体、キメラ抗体、および単鎖抗体からなる群より選択される、請求項8または9に記載の抗体。
【請求項16】
リガンドへのNogo受容体−1の結合を阻害する方法であって、Nogo受容体−1を請求項10に記載の抗体または抗原結合断片と接触させる工程を含む、方法。
【請求項17】
前記リガンドが、NogoA、NogoB、NogoC、MAG、およびOM−gpからなる群より選択される、請求項16に記載の方法。
【請求項18】
ニューロンにおける成長円錐の崩壊を阻害するための方法であって、該ニューロンを請求項10に記載の抗体またはその抗原結合断片と接触させる工程を含む、方法。
【請求項19】
ニューロンにおける神経突起成長および萌芽の阻害を減少させるための方法であって、該ニューロンを請求項10に記載の抗体またはその抗原結合断片と接触させる工程を含む、方法。
【請求項20】
前記神経突起成長または萌芽が軸索成長である、請求項19に記載の方法。
【請求項21】
前記ニューロンが中枢神経ニューロンである、請求項18または19に記載の方法。
【請求項22】
薬学的に許容される担体と、請求項8または9に記載の抗体または抗原結合断片とを含む、組成物。
【請求項23】
1つ以上のさらなる治療剤をさらに含む、請求項22に記載の組成物。
【請求項24】
死滅の危険性があるニューロンの生存を促進する方法であって、該ニューロンを、有効量の請求項8または9に記載の抗Nogo受容体−1抗体または抗原結合断片と接触させる工程を含む、方法。
【請求項25】
前記ニューロンがインビトロにある、請求項24に記載の方法。
【請求項26】
前記ニューロンが哺乳動物中にある、請求項24に記載の方法。
【請求項27】
前記哺乳動物が、多発性硬化症、ALS、ハンチントン病、アルツハイマー病、パーキンソン病、糖尿病性神経障害、脳卒中、外傷性脳損傷、または脊髄損傷の兆候または症状を呈する、請求項26に記載の方法。
【請求項28】
哺乳動物において死滅の危険性があるニューロンの生存を促進する方法であって、
(a)請求項8または9に記載の抗Nogo受容体−1抗体またはその抗原結合断片を発現する培養された宿主細胞を提供する工程;および
(b)該ニューロンの部位またはその付近において、哺乳動物中に宿主細胞を導入する工程
を含む、方法。
【請求項29】
哺乳動物中にある死滅の危険性があるニューロンの生存を促進する遺伝子治療方法であって、請求項8または9に記載の抗Nogo受容体−1抗体またはその抗原結合断片をコードするヌクレオチド配列を含むウイルスベクターを、該ニューロンの部位またはその付近に投与する工程を含み、該抗Nogo受容体−1抗体または抗原結合断片は、該哺乳動物中のヌクレオチド配列から、該ニューロンの生存を促進するために十分な量で発現される、方法。
【図1】

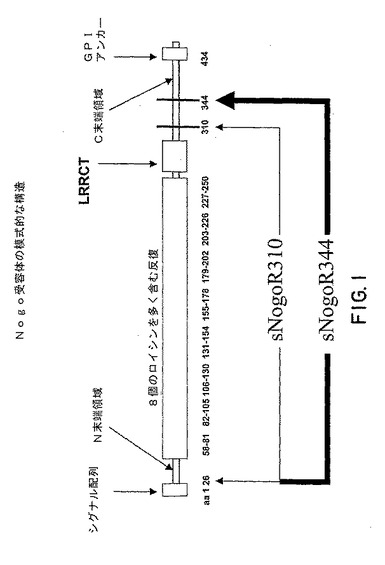
【図2】
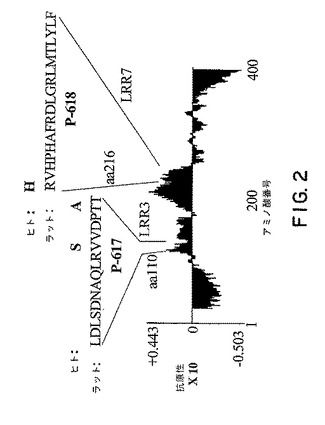
【図3】
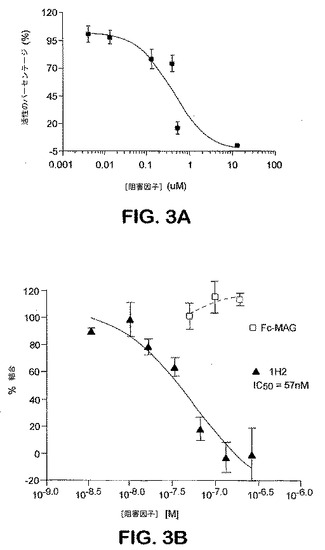
【図4】
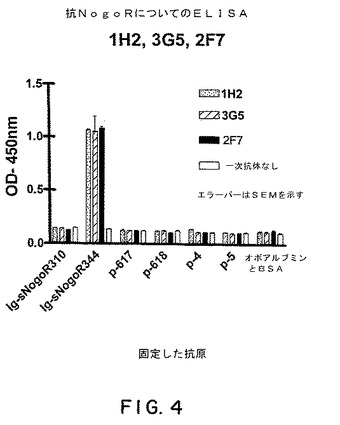
【図5】
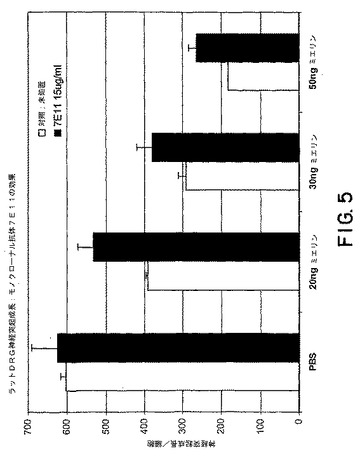
【図6】
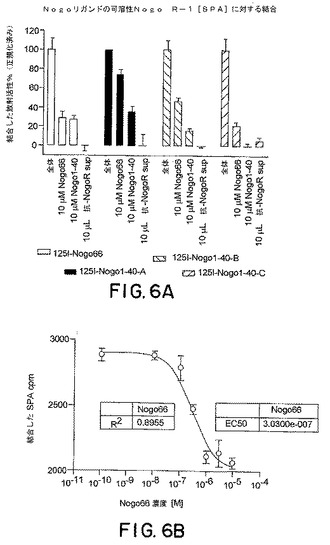
【図7】
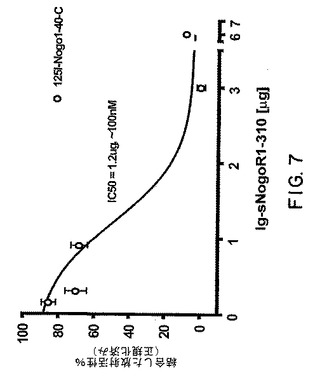
【図8】
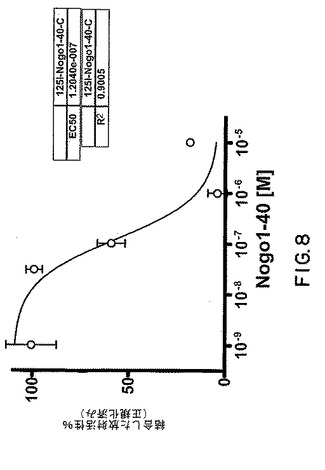
【図9A】
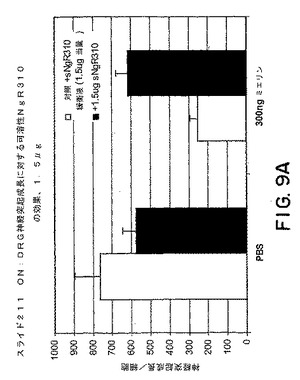
【図9B】
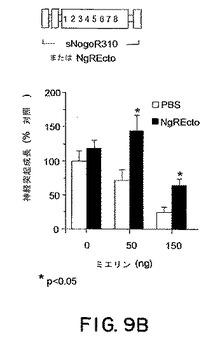
【図10A】
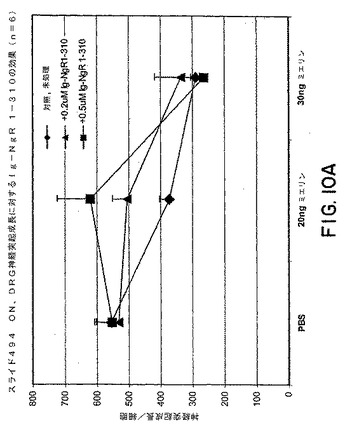
【図10B】
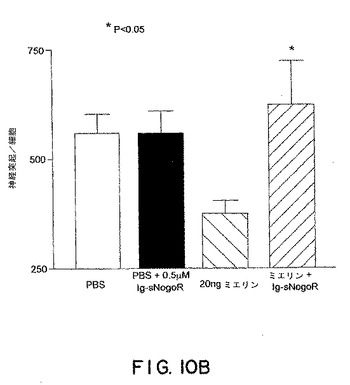
【図11】
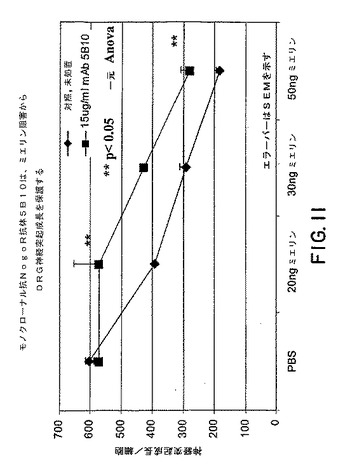
【図12】
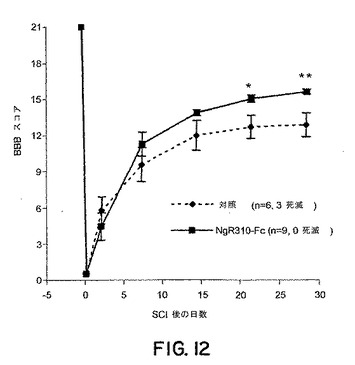
【図13】
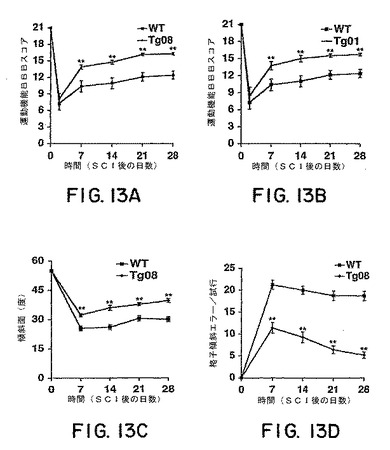
【図14】
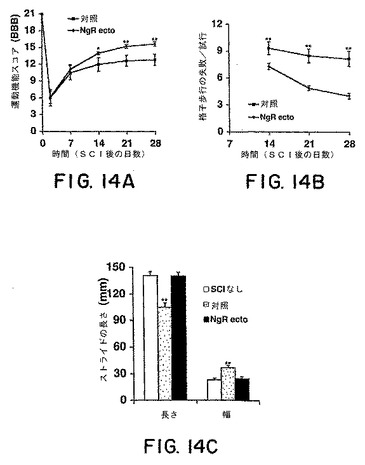
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9A】
【図9B】
【図10A】
【図10B】
【図11】
【図12】
【図13】
【図14】
【公表番号】特表2007−501612(P2007−501612A)
【公表日】平成19年2月1日(2007.2.1)
【国際特許分類】
【出願番号】特願2006−522535(P2006−522535)
【出願日】平成16年1月30日(2004.1.30)
【国際出願番号】PCT/US2004/002702
【国際公開番号】WO2005/016955
【国際公開日】平成17年2月24日(2005.2.24)
【出願人】(592221528)バイオジェン・アイデック・エムエイ・インコーポレイテッド (224)
【出願人】(392019352)イェール ユニバーシティ (38)
【氏名又は名称原語表記】YALE UNIVERSITY
【Fターム(参考)】
【公表日】平成19年2月1日(2007.2.1)
【国際特許分類】
【出願日】平成16年1月30日(2004.1.30)
【国際出願番号】PCT/US2004/002702
【国際公開番号】WO2005/016955
【国際公開日】平成17年2月24日(2005.2.24)
【出願人】(592221528)バイオジェン・アイデック・エムエイ・インコーポレイテッド (224)
【出願人】(392019352)イェール ユニバーシティ (38)
【氏名又は名称原語表記】YALE UNIVERSITY
【Fターム(参考)】
[ Back to top ]