
O6−アルキルグアニン−DNAアルキルトランスフェラーゼのための基質
本発明は、標識を新規基質からO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)及びO6−アルキルグアニン−DNAアルキルトランスフェラーゼ融合タンパク質に転移させる方法、並びに、そのような方法において適切な新規基質に関する。目的とするタンパク質をAGT融合タンパク質に組み入れ、得られたAGT融合タンパク質を標識を有している特定のAGT基質と接触させ、並びに、該標識を認識及び/又は操作できるように設計されているシステム内で該標識を用いて、該AGT融合タンパク質を検出し、及び、場合により、さらに操作する。本発明の方法で使用する特定のAGT基質は、O6−置換グアニン誘導体又は関連する窒素含有ヒドロキシ−ヘテロ環及びそれらの硫黄類似体(ここで、該O6−置換基は、グアニン又は対応するヘテロ環からAGTへ移動するのに適する活性化されたメチル誘導体である)であり、さらに、1つの標識又は複数の同一であるか若しくは異なっている標識を有している。本発明は、さらに、そのような新規AGT基質自体にも関し、また、そのような新規基質の製造方法及びそのような新規AGT基質の合成において有用な中間体にも関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、標識を新規基質からO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)及びO6−アルキルグアニン−DNAアルキルトランスフェラーゼ融合タンパク質に転移させる方法、並びに、そのような方法において適切な新規基質に関する。
【0002】
発明の背景
N−メチル−N−ニトロソウレアのような求電子試薬の突然変異誘発作用及び発癌作用は、主として、DNA内のグアニンのO6−アルキル化に起因している。それらをDNA−アルキル化から保護するために、哺乳動物及び細菌類は、それらの損傷を修復するタンパク質O6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)を有している。AGTは、アルキル化されたグアニン及びグアニン誘導体のO−6位からそれが有している1つのシステインのメルカプト基にアルキル基を転移させ、不可逆的にアルキル化されたAGTを生成する。その基礎的なメカニズムは、SN2型の求核反応であり、それにより、メチル基のみではなくベンジル基も容易に転移される理由が説明される。腫瘍細胞におけるAGTの過剰発現は、プロカルバジン、ダカルバジン、テモゾロマイド及びビス−2−クロロエチル−N−ニトロソウレアなどのアルキル化薬に対する抵抗性の主な原因であるので、化学療法における増感剤として使用するためのAGT阻害薬が求められてきた(Peggら,Prog Nucleic Acid Res Mol Biol 51:167−223,1995)。US5,691,307には、ベンジル基に種々の置換基を有するO6−ベンジルグアニン類が記述されており、また、腫瘍細胞内のAGTレベルを低下させ、それによりアルキル化抗腫瘍薬に対する反応性を増大させるためのO6−ベンジルグアニン類の使用についても記述されている。同様に、WO97/20843には、さらに、O6−ベンジル−ピリミジン誘導体及びO6−ヘテロアリールメチル−ピリミジン誘導体である、AGTを低減させる化合物が開示されている。
【0003】
DE19903895には、AGTのレベルを測定するためのアッセイが開示されており、ここで、該アッセイは、ビオチニル化O6−アルキルグアニン誘導体とAGTを反応させてAGTをビオチニル化することに依拠している。これにより、次に、ストレプトアビジンでコーティングしたプレート上でのAGTの分離と検出(例えば、ELISAアッセイ)が可能となる。このアッセイは、腫瘍組織内のAGTのレベルのモニタリング及びAGT阻害薬のスクリーニングにおける使用に示唆されるアッセイである。
【0004】
WO01/85221では、AGTの検出及びAGTレベルのモニタリングのために、放射能標識フルオロ置換O6−ベンジル−グアニン類又は放射能標識ヨード置換O6−ベンジル−グアニン類の使用が提案されている。
【0005】
Damoiseauxら(ChemBiochem.4:285−287,2001)は、研究及び化学療法において有用な、癌細胞内におけるこの酵素のレベルの検出を容易にする、AGTを標識するための化学プローブとして使用するためのオリゴデオキシリボヌクレオチドに組み入れた修飾されたO6−アルキル化グアニン誘導体を開示している。
【0006】
PCT/GB02/01636(WO02/083937)には、目的とするタンパク質を検出及び/又は操作する方法が開示されており、ここで、該方法では、タンパク質をAGTに融合させ、得られたAGT融合タンパク質を標識を有しているAGT基質と接触させ、そして、該標識を用いて、該AGT融合タンパク質を検出し、そして、場合により、該AGT融合タンパク質をさらに操作する。使用する数種類のAGT融合タンパク質、AGT基質の構造上の一般的な原則、並びに、該方法において有用な広範な種類の標識及び該標識の検出方法が開示されている。
【0007】
発明の要旨
本発明は、目的とするタンパク質を検出及び/又操作する方法に関し、ここで、該方法では、目的とするタンパク質をAGT融合タンパク質に組み入れ、得られたAGT融合タンパク質を標識を有している特定のAGT基質と接触させ、そして、該標識を認識及び/又は処理するように設計されているシステム内で該標識を用いて、該AGT融合タンパク質を検出し、及び、場合により、さらに操作する。
【0008】
本発明の方法で使用する特定のAGT基質は、O6−置換グアニン誘導体又は関連する窒素含有ヒドロキシ−ヘテロ環及びそれらの硫黄類似体(ここで、該O6−置換基は、グアニン又は対応するヘテロ環からAGTへ移動するのに適する活性化されたメチル誘導体である)であり、さらに、標識を有している。該標識は、同一であるか又は異なっている複数の標識からなるものであってよい。活性化されたメチル誘導体は、例えば、アリール環内で適切に置換されているアリールメチル誘導体、ヘテロアリール環内で適切に置換されているヘテロアリールメチル誘導体、及び、二重結合が適切に置換されているアリルタイプの誘導体などである。アリール環、ヘテロアリール環又はアリル二重結合の適切な置換基は、該アリール環、該ヘテロアリール環又は該アリル基に標識を連結するリンカーであり、好ましくは、さらに修飾され得るか又は切断され得るリンカー、及び、AGT基質を二量体化又は環化するリンカーである。本発明は、さらに、そのような新規AGT基質自体にも関し、また、そのような新規基質の製造方法及びそのような新規AGT基質の合成において有用な中間体にも関する。
【0009】
発明の詳細な説明
本発明では、目的とするタンパク質又はペプチドをO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)に融合させる。目的とするタンパク質又はペプチドは、任意の長さを有するものであることができ、また、二次構造、三次構造若しくは四次構造を有していても又は有していなくてもよい。該タンパク質又はペプチドは、好ましくは、少なくとも12個から2000個以下のアミノ酸からなる。そのような目的とするタンパク質又はペプチドの例は以下で与えられており、例えば、酵素、DNA結合タンパク質、転写調節タンパク質、膜タンパク質、核内受容体タンパク質、核局在化シグナルタンパク質、タンパク質補因子、小単量体GTPアーゼ、ATP結合カセットタンパク質、細胞内構造タンパク質、特定の細胞コンパートメントに対するタンパク質の標的に関与する配列を有するタンパク質、標識又はアフィニティータグとして一般に使用されるタンパク質、並びに、前記タンパク質のドメイン及びサブドメインなどがある。目的とするタンパク質又はペプチドは、好ましくは、酵素により切断され得るリンカー、例えば、DNA段階で適切な制限酵素により切断され得るリンカー(例えば、BglIIにより切断され得るAGATCT)、及び/又は、タンパク質段階で適切な酵素(例えば、タバコエッチウイルスNla(TEV)プロテアーゼ)により切断され得るリンカーを介してAGTに融合させる。融合タンパク質は、原核生物の宿主(好ましくは、E.coli)、又は、真核生物の宿主(例えば、酵母細胞、昆虫細胞若しくは哺乳動物細胞)で発現させることができる。
【0010】
O6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)は、基質上に存在している標識を融合タンパク質の中のAGTを形成している部分に含まれているシステイン残基の1つに転移させる性質を有している。好ましい実施形態では、AGTは、公知ヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)である。マウス形態又はラット形態の酵素も、それらがヒトAGTのような基質と反応する上で同様の性質を有しているかぎり、同様に考慮される。本発明においては、1個以上のアミノ酸の置換、欠失又は付加により異なり得るが、基質上に存在している標識を融合タンパク質内のAGT部分に転移させる性質は維持している野生型AGTの変異体も、O6−アルキルグアニン−DNAアルキルトランスフェラーゼに包含される。AGT変異体は、当業者が周知している技術を用いて化学的に修飾することにより得ることができる。AGT変異体は、好ましくは、当業者に公知のタンパク質工学技術を用いて製造することができるか、及び/又は、分子進化を用いて新しいO6−アルキルグアニン−DNAアルキルトランスフェラーゼを生成及び選択することにより製造することができる。そのような技術は、例えば、飽和突然変異誘発、配列中の何れかに変異を導入する変異導入型PCR、飽和突然変異誘発及び/又は変異導入型PCRの後で用いるDNAシャフリング、又は、幾つかの種から得た遺伝子を用いるファミリーシャフリングなどである。
【0011】
目的とするタンパク質及びO6−アルキルグアニン−DNAアルキル−トランスフェラーゼ(AGT)を含んでいる融合タンパク質を、標識を有している特定の基質と接触させる。反応条件は、AGTが基質と反応して、基質の標識を転移させるように選択する。一般的な条件は、約pH7で室温(約25℃)の緩衝液である。しかしながら、AGTがさまざまな別の反応条件下でも反応すること、及び、本明細書において述べられている反応条件により本発明の範囲が限定されることはないことは理解される。
【0012】
AGTは、その基質であるO6−アルキルグアニン−DNAからそのシステイン残基の内の1つに、アルキル基を不可逆的に転移させる。hAGTと急速に反応する基質類似体は、O6−ベンジル−グアニンであり、二次反応速度定数は、約103秒−1M−1である。O6−ベンジルグアニンをベンジル環のC−4位で置換しても、O6−ベンジルグアニン誘導体に対するhAGTの反応性に対して有意な影響は与えない。これまで、この性質を利用して、ベンジル環のC−4位に結合させた標識をAGTに転移させてきた。
【0013】
当業者は、該融合タンパク質について意図している用途に応じて、基質の標識部分を選定することができる。AGT含有融合タンパク質を基質と接触させた後、標識を共有結合的に融合タンパク質に結合させる。次いで、標識されたAGT融合タンパク質を、転移させた標識に基づいて、さらに操作するか、及び/又は、検出する。該標識は、複数の同一であるか又は異なっている標識から成ることができる。基質が2以上の標識を含んでいる場合、標識された対応するAGT融合タンパク質は、同様に、2以上の標識を含有し、これは、標識された融合タンパク質をさらに操作するか、及び/又は、検出するためのより多くの選択肢を与える。
【0014】
特定のAGT基質は、式1:
【0015】
【化9】

【0016】
[R1−R2は、AGTにより基質として認識される基であり;
Xは、酸素又は硫黄であり;
R3は、芳香族基若しくはヘテロ芳香族基であるか、又は、CH2に結合した二重結合を有する、場合により置換されていてもよい不飽和のアルキル基、シクロアルキル基若しくはヘテロシクリル基であり;
R4は、リンカーであり;
そして、
Lは、1つの標識であるか、同一であるか若しくは異なっている複数の標識であるか、R4をR1に連結して環状基質を形成する結合であるか、又は、さらなる基−R3−CH2−X−R1−R2である]
で表される化合物である。
【0017】
基R1−R2において、残基R1は、好ましくは、AGTにより基質として認識されるヘテロ芳香族基(ここで、該ヘテロ芳香族基1〜5の窒素原子を含んでいる)である。
【0018】
ヘテロ芳香族基R1は、単環式又は二環式であり、5〜12個、好ましくは、6又は9又は10個の環原子を有する。ヘテロ芳香族基R1は、置換基R2を有する他に、置換されていなくてもよいし、又は、以下のものからなる群から選択される1以上の置換基、特に、1、2若しくは3個の置換基で置換されていてもよい:低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ若しくはエトキシ、ヒドロキシ、オキソ、アミノ、低級アルキルアミノ、ジ−低級アルキルアミノ、アシルアミノ、ハロゲン、例えば、塩素若しくは臭素、ハロゲン化低級アルキル、例えば、トリフルオロメチル、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、又は、低級アルキルカルボニル。
【0019】
低級アルキルは、好ましくは、1〜7個、好ましくは、1〜4個のC原子を有するアルキルであり、直鎖又は分枝鎖である。好ましくは、低級アルキルは、ブチル、例えば、n−ブチル、s−ブチル、イソブチル若しくはt−ブチル、プロピル、例えば、n−プロピル若しくはイソプロピル、エチル、又は、メチルである。好ましくは、低級アルキルは、メチルである。
【0020】
低級アルコキシにおいて、低級アルキル基は、上記で定義されているとおりである。低級アルコキシは、好ましくは、n−ブトキシ、t−ブトキシ、イソプロポキシ、エトキシ、又は、メトキシを表し、特に、メトキシである。
【0021】
好ましくは、単環式又は二環式のヘテロ芳香族基R1は、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、8−アザプリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、トリアゾリル、テトラゾリル、又は、ベンゾ[d]ピラゾリルである。さらに好ましくは、単環式又は二環式のヘテロ芳香族基R1は、プリニル、8−アザプリニル、ピリジル、ピラジニル、ピリミジニル及びピリダジニルからなる群から選択される。
【0022】
例えば、基R1−R2は、式2:
【0023】
【化10】
【0024】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
R5は、水素、ハロゲン、例えば、クロロ若しくはブロモ、トリフルオロメチル、又は、ヒドロキシであり;
そして、
R6は、水素、ヒドロキシ、又は、置換されていないか若しくは置換されているアミノである]
で表されるプリンラジカルであることができる。
【0025】
R5又はR6がヒドロキシである場合、上記プリンラジカルは、主として、その互変異性形態(ここで、R5又はR6を有している炭素原子に隣接している窒素は水素原子を有し、この窒素原子とR5又はR6を有している炭素原子の間の二重結合は単結合であり、及び、R5又はR6は、それぞれ、二重結合で結合してる酸素である)で存在している。
【0026】
置換されているアミノ基R6は、1〜4個の炭素原子を有する低級アルキルアミノ又はアシルアミノであり、その際、該アシル基は、1〜5個の炭素原子を有する低級アルキルカルボニル、例えば、アセチル、プロピオニル、n−プロピルカルボニル若しくはイソプロピルカルボニル、n−ブチルカルボニル、イソブチルカルボニル若しくはt−ブチルカルボニル、又は、アリールカルボニル、例えば、ベンゾイルである。
【0027】
R6が置換されていないか又は置換されているアミノであり、且つ、残基Xがプリンラジカルの結合に連結している場合、式2で表されるラジカルは、グアニン誘導体である。
【0028】
1〜10個の炭素原子を有するアルキルとしてのR2は、直鎖又は分枝鎖であり、そのようなR2には、1〜4の炭素原子を有する低級アルキル、例えば、メチル、エチル、ブチル、例えば、n−ブチル、s−ブチル、イソブチル又はt−ブチル、及び、プロピル、例えば、n−プロピル又はイソプロピルなどがある。アルキルとしてのR2は、さらにまた、ペンチル、ヘキシル、ヘプチル、オクチル、ノニル、又は、デシル、例えば、n−ヘキシルであってもよい。
【0029】
糖部分R2は、可変的な長さのスペーサーでグアニン塩基の第N9位に結合している糖単量体又はオリゴマーである。これに関連して、スペーサーは、アルキル鎖(好ましくは、1〜15個の炭素原子を有するアルキル鎖)、1〜200個のエチレングリコール単位からなるポリエチレングリコールスペーサー、アミド基−CO−NH−、エステル基−CO−O−、若しくは、アルキレン基−CH=CH−であるか、又は、アルキル鎖、ポリエチレングリコール基、アミド基、エステル基及び/若しくはアルキレン基の組合せである。
【0030】
本発明との関連において、糖部分R2としては、さらに、β−D−2’−デオキシリボシル、又は、2〜99個のヌクレオチドからなる長さを有する一本鎖オリゴデオキシリボヌクレオチドに組み込まれているβ−D−2’−デオキシリボシルなどがあり、その際、グアニン誘導体R1は、オリゴヌクレオチド配列内の任意の位置を占める。
【0031】
特に好ましいのは、基R1−R2が式2で表されるプリンラジカルであり、R2が水素であり、R5が水素であり、R6が置換されていないアミノであり、且つ、Xが酸素である化合物、即ち、置換されていないグアニン誘導体である。
【0032】
本発明の好ましい別の実施形態では、基R1−R2は、式3:
【0033】
【化11】
【0034】
[式中、置換基R2及びR6は、式2におけるR2及びR6に関して定義されている意味を有する]
で表される8−アザプリンラジカルである。
【0035】
本発明の好ましいさらに別の実施形態では、基R1−R2は、式4:
【0036】
【化12】
【0037】
[式中、
置換基R2は、式2において定義されている意味を有し、好ましくは、水素であり;
そして、
R7及びR8は、いずれも、互いに独立して、水素、ハロゲン、例えば、クロロ若しくはブロモ、1〜4の炭素原子を有する低級アルキル、例えば、メチル、アミノ、又は、ニトロである]
で表されるピリミジンラジカルである。
【0038】
Xは、好ましくは、酸素である。
【0039】
芳香族基若しくはヘテロ芳香族基としてのR3、又は、場合により置換されていてもよい不飽和アルキル基、シクロアルキル基若しくはヘテロシクリル基としてのR3は、AGTにより(その反応機構に従って)、立体的及び電子的に受容され、それにより、R3−R4−L単位が融合タンパク質へと共有結合的に転移するのを可能とする基である。R3−R4−L単位において、R4−Lは、さらにまた、同一であるか又は異なっている複数の標識Lを有している同一であるか又は異なっている複数のリンカーR4を意味し得る。
【0040】
芳香族基としてのR3は、好ましくは、フェニル又はナフチル、特に、フェニル、例えば、パラ位又はメタ位がR4で置換されているフェニルである。
【0041】
ヘテロ芳香族基R3は、0、1、2、3若しくは4個の環窒素原子と0若しくは1個の酸素原子と0若しくは1個の硫黄原子を含み、5〜12個、好ましくは、5若しくは6個の環原子を有する単環式若しくは二環式のヘテロアリール基(但し、少なくとも1の環炭素原子は、窒素原子、酸素原子又は硫黄原子で置き換えられている)であり、ここで、該ヘテロ芳香族基R3は、置換基R4を有している他に、置換されていなくてもよいし、又は、以下のものからなる群から選択される1つ以上の置換基、特に、1つのさらなる置換基で置換されていてもよい:低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ若しくはエトキシ、ハロゲン、例えば、塩素、臭素若しくはフッ素、ハロゲン化低級アルキル、例えば、トリフルオロメチル、又は、ヒドロキシ。
【0042】
好ましくは、単環式又は二環式のヘテロアリール基R3は、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、フラザニル、ベンゾ[d]ピラゾリル、チエニル、及び、フラニルから選択される。さらに好ましくは、該単環式又は二環式のヘテロアリール基は、以下のものからなる群から選択される:ピロリル、イミダゾリル、例えば、1H−イミダゾール−1−イル、ベンゾイミダゾリル、例えば、1−ベンゾイミダゾリル、インダゾリル、特に、5−インダゾリル、ピリジル、例えば、2−ピリジル、3−ピリジル若しくは4−ピリジル、ピリミジニル、特に、2−ピリミジニル、ピラジニル、イソキノリニル、特に、3−イソキノリニル、キノリニル、特に、4−キノリニル若しくは8−キノリニル、インドリル、特に、3−インドリル、チアゾリル、トリアゾリル、テトラゾリル、ベンゾ[d]ピラゾリル、チエニル、及び、フラニル。
【0043】
本発明の特に好ましい実施形態では、該ヘテロアリール基R3は、トリアゾリル、特に、4位若しくは5位にさらなる置換基R4を有している1−トリアゾリル、テトラゾリル、特に、4位若しくは5位にさらなる置換基R4を有している1−テトラゾリル若しくは5位にさらなる置換基R4を有している2−テトラゾリル、イソオキサゾリル、特に、5位にさらなる置換基R4を有している3−イソオキサゾリル若しくは3位にさらなる置換基R4を有している5−イソオキサゾリル、又は、チエニル、特に、3位、4位若しくは5位(好ましくは、4位)にさらなる置換基R4を有している2−チエニル若しくは4位にさらなる置換基R4を有している3−チエニルである。
【0044】
最も好ましいのは、4位又は5位に置換基R4を有しているトリアゾリルとしてのヘテロアリール基R3であり、また、同様に、4位又は5位に置換基R4を有している2−チエニルとしてのR3である。
【0045】
場合により置換されていてもよい不飽和アルキル基R3は、1位若しくは2位(好ましくは、2位)にさらなる置換基R4を有している1−アルケニルであるか、又は、1−アルキニルである。1−アルケニルにおいて考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロである。本発明の特に好ましい実施形態では、R3は、1−アルキニルである。
【0046】
場合により置換されていてもよい不飽和シクロアルキル基は、3〜7個の炭素原子を有し、1位が不飽和であるシクロアルキル基、例えば、任意の位置にさらなる置換基R4を有する1−シクロペンチル又は1−シクロヘキシルである。考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロなどである。
【0047】
場合により置換されていてもよい不飽和ヘテロシクリル基は、3〜12個の原子、窒素、酸素及び硫黄から選択される1〜5個のヘテロ原子、並びに、該ヘテロシクリル基をメチレンCH2に連結させている位置における二重結合を有する。考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロなどである。
【0048】
特に、場合により置換されていてもよい不飽和ヘテロシクリル基は、ヘテロ芳香族基R3に関して上記で定義されている部分的に飽和しているヘテロ芳香族基である。そのようなヘテロシクリル基の例は、イソオキサゾリジニル、特に、5位にさらなる置換基を有している3−イソオキサゾリジニル、又は、3位にさらなる置換基を有している5−イソオキサゾリジニルである。
【0049】
リンカー基R4は、好ましくは、1つの標識L又は同一であるか若しくは異なっている複数の標識Lを基質に結合させる可変性リンカーである。リンカー単位は、想定される用途に照らして、即ち、AGTを含有している融合タンパク質への基質の転移に関連して選択される。それらは、さらに、適切な溶媒中における基質の溶解度も増大させる。使用されるリンカーは、実際に適用する条件下において化学的に安定である。そのようなリンカーは、AGTとの反応を妨害せず、また、標識Lの検出も妨害しないが、例えば、式1の化合物とAGT含有融合タンパク質の反応完了後のある時点で切断されるように構築し得る。
【0050】
リンカーR4は、1〜300個の炭素原子を有する直鎖又は分枝鎖のアルキレン基であり、その際、場合により、
(a)1個以上の炭素原子が酸素により置き換えられており、特に、2つおきの炭素原子が酸素で置き換えられており(例えば、1〜100個のエチレンオキシ単位を有するポリエチレンオキシ基);
(b)1個以上の炭素原子が水素原子を有している窒素で置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、アミド官能基−NH−CO−を表し;
(c)1個以上の炭素原子が酸素により置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、エステル官能基−O−CO−を表し;
(d)隣接してる2つの炭素原子の間の結合が二重結合又は三重結合であって、官能基−CH=CH−又は−C≡C−を表し;
(e)1個以上の炭素原子が、フェニレン、飽和若しくは不飽和のシクロアルキレン、飽和若しくは不飽和のビシクロアルキレン、架橋ヘテロ芳香族、又は、飽和若しくは不飽和の架橋ヘテロシクリル基により置き換えられており;
(f)隣接してる2個の炭素原子がジスルフィド結合−S−S−により置き換えられており;
又は、上記(a)〜(f)で定義されている2以上(特に、2又は3)のアルキレン基及び/又は修飾されているアルキレン基の組合せは、場合により、置換基を含んでいてもよい。
【0051】
考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロなどである。
【0052】
考慮されるさらなる置換基は、例えば、α−アミノ酸、特に、天然α−アミノ酸をリンカーR4に組み入れたときに得られる置換基(ここで、炭素原子は、(b)で定義されているアミド官能基−NH−CO−により置き換えられている)などである。そのようなリンカーにおいては、アルキレン基R4の炭素鎖の一部は、基−(NH−CHR−CO)n−(ここで、nは、1〜100であり、Rは、α−アミノ酸の種々の残基を表す)で置き換えられている。
【0053】
さらなる置換基は、光切断可能なリンカーR4、例えば、o−ニトロフェニル基などをもたらす置換基である。特に、この置換基o−ニトロフェニルは、アミド結合に隣接する炭素原子に位置するか(例えば、基−NH−CO−CH2−CH(o−ニトロフェニル)−NH−CO−)、又は、ポリエチレングリコール鎖内の置換基として存在する(例えば、基−O−CH2−CH(o−ニトロフェニル)−O−)。考慮される別の光切断可能なリンカーは、例えば、フェナシル、アルコキシベンゾイン、ベンジルチオエーテル及びピバロイルグリコール誘導体などである。
【0054】
上記(e)で定義されている炭素原子に置き換えられるフェニレン基は、例えば、1,2−フェニレン、1,3−フェニレン、又は、好ましくは、1,4−フェニレンである。。特定の実施形態において、該フェニレン基は、ニトロ基でさらに置換され、また、上記(a)、(b)、(c)、(d)及び(f)において述べられているような別の置き換えと組み合わされ、光切断可能な基を表し、例えば、4−ニトロ−1,3−フェニレン、例えば、−CO−NH−CH2−4−ニトロ−1,3−フェニレン−CH(CH3)−O−CO−、又は、2−メトキシ−5−ニトロ−1,4−フェニレン、例えば、−CH2−O−2−メトキシ−5−ニトロ−1,4−フェニレン−CH(CH3)−O−などである。光切断可能なリンカーを表す特定の別の実施形態は、例えば、−1,4−フェニレン−CO−CH2−O−CO−CH2−(フェナシル基)、−1,4−フェニレン−CH(OR)−CO−1,4−フェニレン−(アルコキシベンゾイン)、又は、−3,5−ジメトキシ−1,4−フェニレン−CH2−O−(ジメトキシベンジル部分)などである。上記(e)で定義されている炭素原子と置き換えられる飽和又は不飽和のシクロアルキレン基は、3〜7個の炭素原子を有するシクロアルキルから、好ましくは、シクロペンチル又はシクロヘキシルから誘導され、例えば、1,2−シクロペンチレン、若しくは、1,3−シクロペンチレン、1,2−シクロヘキシレン、1,3−シクロヘキシレン、若しくは、好ましくは、1,4−シクロヘキシレンなどであるか、又は、さらに、例えば1位若しくは2位が不飽和である1,4−シクロヘキシレンである。上記(e)で定義されている炭素原子と置き換えられる飽和又は不飽和のビシクロアルキレン基は、7又は8個の炭素原子を有するビシクロアルキルから誘導され、例えば、ビシクロ[2.2.1]へプチレン、又は、ビシクロ[2.2.2]オクチレン、好ましくは、場合により2位に不飽和を有するか若しくは2位と5位に二重に不飽和を有していてもよい1,4−ビシクロ[2.2.1]へプチレン、及び、場合により2位に不飽和を有するか若しくは2位と5位に二重に不飽和を有していてもよい1,4−ビシクロ[2.2.2]オクチレンなどである。上記(e)で定義されている炭素原子と置き換えられる架橋ヘテロ芳香族基は、例えば、トリアゾリデン、好ましくは、1,4−トリアゾリデン、又は、イソオキサゾリデン、好ましくは、3,5−イソオキサゾリデンなどである。上記(e)で定義されている炭素原子と置き換えられる飽和若しくは不飽和の架橋ヘテロシクリル基は、例えば、上記R3で定義されている不飽和ヘテロシクリル基から誘導され、例えば、イソオキサゾリジネン、好ましくは、3,5−イソオキサゾリジネンなどであるか、又は、3〜12の原子(そのうちの1〜3は、窒素、酸素及び硫黄から選択されるヘテロ原子である)を有する完全に飽和しているヘテロシクリル基から誘導され、例えば、ピロリジンジイル、ピペリジンジイル、テトラヒドロフランジイル、ジオキサンジイル、モルホリンジイル、若しくは、テトラヒドロチオフェンジイル、好ましくは、2,5−テトラヒドロフランジイル、若しくは、2,5−ジオキサンジイルなどである。考慮される特定のヘテロシクリル基は、糖部分、例えば、α−フラノシル部分若しくはβ−フラノシル部分、又は、α−ピラノシル部分若しくはβ−ピラノシル部分などである。
【0055】
リンカーR4内の環状下部構造は、R4内の回転可能な結合の数で評価した場合に、分子の柔軟性を低下させるが、これにより、良好な膜透過速度が得られる。これは、全てのインビボでの標識化用途にとって重要である。
【0056】
好ましくは、リンカーR4は、場合により−CH=CH−又は−C≡C−基で基R3に結合している、4〜100のエチレンオキシ単位を有する直鎖ポリエチレングリコール基、又は、1〜25個の炭素原子を有する直鎖アルキレン基である。さらに好ましいのは、1〜25個の炭素原子(ここで、炭素原子は場合によりアミド官能基−NH−CO−で置き換えられていてもよい)を有し、光切断可能なサブユニット(例えば、o−ニトロフェニル)を有している直鎖アルキレン基である。さらに好ましいのは、3〜6のエチレングリコール単位を有するポリエチレングリコール基とアルキレン基(炭素原子はアミド結合で置き換えられている)を含み、さらに、置換されているアミノ官能基及びヒドロキシ官能基を有している分枝リンカーである。好ましい別の分枝鎖リンカーは、アルキレン基の炭素原子がアミン、カルボキサミド及び/又はエーテルで置き換えられている樹状構造(木のような構造)を有している。
【0057】
特に好ましいリンカーR4は、3〜12個の炭素原子が酸素で置き換えられており、1又は2の炭素原子がそれぞれ1又は2個の1,4−トリアゾリデン単位で置き換えられており、また、場合により1個の炭素原子が1,4−フェニレン単位で置き換えられている、10〜40個の炭素原子を有する直鎖アルキレン基である。
【0058】
特に好ましい別のリンカーR4は、3〜12個の炭素原子が酸素で置き換えられており、1又は2個の炭素原子が窒素で置き換えられている、場合によりオキソで置換されていてもよい10〜40個の炭素原子を有する直鎖アルキレン基である。
【0059】
特に好ましい別のリンカーR4は、2〜12個の炭素原子が酸素で置き換えられており、2個の隣接する炭素原子の間の1又は2つの結合が二重結合であって、官能基−CH=CH−を表している、6〜40個の炭素原子を有する直鎖アルキレン基である。
【0060】
リンカーR4は、1つ以上の同一であるか又は異なっている標識、例えば、1〜100の同一であるか又は異なっている標識、特に、1〜5つ、好ましくは、1つ、2つ又は3つ、特に、1つ又は2つの同一であるか又は異なっている標識を有し得る。
【0061】
基質の標識部分Lは、意図されている融合タンパク質についての用途に応じて、当業者が選択することができる。標識は、例えば、標識された融合タンパク質が、容易に検出されるか又はその環境から容易に分離されるようなものであり得る。考慮される別の標識は、該標識化融合タンパク質の周囲における変化を探知及び惹起することが可能な標識であるか、及び/又は、該標識によって融合タンパク質に特異的に導入された物理的及び/又は化学的性質により該融合タンパク質を操作するのを助ける標識である。
【0062】
標識Lの例には、分光学的プローブ、例えば、発蛍光団、発色団、磁気プローブ又は造影剤;放射能で標識された分子;結合パートナーに特異的に結合することが可能な特異的結合対の一方である分子;別の生体分子と相互作用すると推測される分子;別の生体分子と相互作用すると推測される分子のライブラリー;別の分子と架橋することが可能な分子;H2O2及びアスコルベートと接触したときにヒドロキシルラジカルを生成することが可能な分子、例えば、テザー金属キレート(tethered metal-chelate);光を照射されたときに反応性ラジカルを生成することが可能な分子、例えば、マラカイトグリーン;固体支持体に共有結合で結合している分子(ここで、支持体は、スライドガラス、マイクロタイタープレート又は当業者に知られている任意のポリマーであり得る);その相補鎖と塩基対を成すことが可能な核酸若しくはその誘導体;膜挿入特性を有する脂質若しくは別の疎水性分子;酵素的、化学的若しくは物理的に望ましい特性を有している生体分子;又は、上記で挙げた特性のいずれかの組合せを有する分子などである。好ましいものは、放射能標識された分子を除く、上記で挙げた標識Lである。標識Lとして最も好ましいのは、分光学的プローブ、及び、結合パートナーに特異的に結合することが可能な特異的結合対の一方である分子(いわゆる、アフィニティー標識)である。
【0063】
標識Lが、発蛍光団、発色団、磁気標識又は放射能標識などである場合、検出は、当該方法をインビトロ又はインビボで使用するかにより、該標識に適合させた標準的な方法による。該方法は、目的とするタンパク質に遺伝的に融合していて生細胞内でのタンパク質の研究を可能とするグリーン蛍光タンパク質(GFP)の適用と比較することができる。さらにまた、標識Lの特定の例は、非線形光学的性質を示すホウ素化合物、又は、標識された基質とAGT融合タンパク質が反応する際にその分光学的性質を変化させるFRET対のメンバーである。
【0064】
標識Lの性質に応じて、目的とするタンパク質及びAGTを含んでいる融合タンパク質を固体支持体に結合させ得る。AGT含有融合タンパク質と反応する基質の標識は、AGTとの反応に入るときには既に固体支持体に結合させておいてもよいし、又は、その後で、即ち、AGTへ転移した後で、AGT含有融合タンパク質と反応する基質の標識を用いてAGT融合タンパク質を固体支持体に結合させてもよい。該標識は、特異的結合対の一方のメンバーであってもよく、その際、該特異的結合対の他方のメンバーは、共有結合又は別の任意の手段により、固体支持体に結合しているか又は結合可能である。考慮される特異的結合対は、例えば、ビオチンとアビジン又はストレプトアビジンなどである。結合対のいずれのメンバーも基質の標識Lとすることが可能であり、その際、他方のメンバーは、固体支持体に結合されていてもよい。固体支持体に都合よく結合することを可能にする標識のさらなる例は、例えば、マルトース結合タンパク質、糖タンパク質、FLAGタグ、又は、固体支持体の表面上に存在している相補的な官能基との間で化学選択的に反応することが可能な反応性置換基である。そのような反応性置換基と相補的な官能基の対の例は、例えば、アミドを形成するアミンと活性化カルボキシ基、1,3−双極子付加環化反応を受けるアジドとプロピオル酸誘導体、活性化ビス−ジカルボン酸誘導体のタイプの添加された二官能性リンカー試薬と反応して2つのアミド結合を生成させるアミンと別のアミン官能基、又は、当業界で知られている別の組合せなどである。
【0065】
都合のよい固体支持体の例は、例えば、ガラス表面、例えば、スライドガラス、マイクロタイタープレート、及び、適切なセンサーエレメント、特に、官能化ポリマー(例えば、ビーズ形態にある官能化ポリマー)、化学的に修飾された酸化物表面、例えば、二酸化ケイ素、五酸化タンタル若しくは二酸化チタン、又は、化学的に修飾された金属表面、例えば、貴金属表面、例えば、金若しくは銀の表面などである。不可逆的に結合及び/又はスポットするAGT基質を用いて、AGT融合タンパク質を、空間的に分離したやり方で、特に、スポットにより、固体支持体上に結合させることができ、これらは、タンパク質マイクロアレイ、DNAマイクロアレイ又は小分子のアレイに相当する。
【0066】
標識Lが、外部刺激に晒されたときに反応性ラジカル(例えば、ヒドロキシルラジカル)を生成し得る場合、生成されたラジカルは、AGT融合タンパク質及びAGT融合タンパク質に近接しているタンパク質を不活性化することができ、これは、これらのタンパク質の役割についての研究を可能にする。そのような標識の例は、H2O2及びアスコルベートと接触したときにヒドロキシルラジカルを生成するテザー金属キレート錯体(tethered metal-chelate complex)、及び、レーザーを照射されたときにヒドロキシルラジカルを生成する発色団、例えば、マラカイトグリーンなどである。ヒドロキシルラジカルを生成させるために発色団とレーザーを使用することは、当技術分野では、発色団アシストレーザー分子機能不活化(chromophore assisted laser induced inactivation)(CALI)として知られている。本発明では、発色団(例えば、マラカイトグリーン)を用いてAGT融合タンパク質を標識した後、レーザーを照射して、時間を制御し且つ空間的に分離したやり方で、AGT融合タンパク質を不活性化すると共にAGT融合タンパク質と相互作用するタンパク質を不活性化する。この方法は、インビボ又はインビトロの両方に適用可能である。さらに、AGT融合タンパク質に近接しているタンパク質は、該タンパク質のフラグメントを特異的抗体により検出することにより、又は、高分解2D−電気泳動ゲル上における該タンパク質の消失により、又は、分離及び配列決定法(例えば、質量分析法又はN−末端分解によるタンパク質配列決定法など)による切断されたタンパク質フラグメントを同定することにより、同定することができる。
【0067】
標識Lが、別のタンパク質と架橋可能な分子、例えば、官能基(例えば、マレイミド、活性エステル又はアジド及び当業者に知られている別のものなど)を含んでいる分子である場合、そのような標識されたAGT基質を別のタンパク質と相互作用するAGT融合タンパク質と接触させる(インビボ又はインビトロ)ことにより、AGT融合タンパク質をそれが相互作用するタンパク質と該標識を介して共有結合的に架橋することができる。これにより、AGT融合タンパク質と相互作用するタンパク質を同定することが可能となる。光活性化架橋のための標識Lは、例えば、ベンゾフェノン類である。架橋の特定の態様において、標識Lは、それ自体がAGT基質である分子であり、それにより、AGT融合タンパク質が二量体化される。そのような二量体の化学的構造は、対称(ホモ二量体)又は非対照(ヘテロ二量体)であり得る。
【0068】
考慮される別の標識Lは、例えば、フラーレン類、中性子捕獲処理のためのボラン類、例えばセルフアドレッシングチップ(self-addressing chip)用のヌクレオチド類若しくはオリゴヌクレオチド類、ペプチド核酸類、及び、金属キレート類、例えば、DNAに特異的に結合する白金キレートなどである。
【0069】
望ましい酵素的性質、化学的性質又は物理的性質を有している特定の生体分子は、メトトレキサートである。メトトレキサートは、酵素であるジヒドロ葉酸レダクターゼ(DHFR)の強結合阻害剤である。式中のLがメトトレキサートである式1の化合物は、いわゆる「二量体化誘発物質(chemical inducers of dimerization)」(CID)と称されるよく知られている種類に属する。hAGTとDNA結合ドメインLexAの融合タンパク質を使用し、式1(式中、Lはメトトレキサートである)の化合物によるhAGT融合タンパク質のインビボ標識化に、転写活性化ドメインB42を有するDHFRを添加することにより、hAGT−LexA融合タンパク質とDHFR−B42融合タンパク質のカップリング(「二量体化」)が誘発され、その結果、図4に示されているように、LexAとB42が空間的に近接し、その後、転写が刺激される。
【0070】
基質が2つ以上の標識を有している場合、これら標識は、同一でもよく、又は、異なっていてもよい。特に好ましい組合せは、2つの異なったアフィニティー標識、又は、1つのアフィニティー標識と1つの発色団標識、特に、1つのアフィニティー標識と1つの発蛍光団標識である。
【0071】
本発明は、さらに、インビボ及びインビトロの両方において、AGT融合タンパク質を標識する方法を提供する。AGT融合タンパク質のインビボ標識化という用語には、細胞の全てのコンパートメントにおける標識化、及び、細胞外空間に面しているAGT融合タンパク質の標識化が包含される。AGT融合タンパク質の標識化がインビボで行われ且つAGTに融合しているタンパク質が膜タンパク質(より特定的には、細胞膜タンパク質)である場合、該融合タンパク質内のAGT部分は、該膜のいずれの面にも結合させることが可能であり、例えば、細胞質面又は細胞膜の細胞外の面に結合させることが可能である。
【0072】
標識化をインビトロで行う場合、融合タンパク質の標識化は、細胞抽出物中で、又は、AGT融合タンパク質の精製された形態若しくは濃縮された形態で行うことができる。
【0073】
標識化をインビボ又は細胞抽出物中で行う場合、宿主の内因性AGTの標識化を考慮に入れるのが有利である。宿主の内因性AGTが、O6−アルキルグアニン誘導体又はその関連する化合物を基質として受容しない場合、該融合タンパク質の標識化は、特異的である。哺乳類細胞、例えば、ヒト、マウス又はラットの細胞においては、内因性AGTの標識化が可能である。内因性AGTとAGT融合タンパク質を同時に標識化すると問題が生じるような実験においては、公知のAGT欠失細胞系を使用することができる。
【0074】
特定の態様において、本発明は、候補化合物又は候補化合物のライブラリーと標的タンパク質又は標的タンパク質のライブラリーの相互作用を測定する方法を提供する。候補化合物と標的タンパク質の例には、リガンドとタンパク質、薬物と該薬物の標的、又は、小分子とタンパク質などがある。本発明のこの特定の方法において、AGTに融合させる目的とするタンパク質は、転写因子のDNA結合ドメイン又は転写因子の活性化ドメインを含んでいる。物質又はタンパク質のライブラリーの推定されるタンパク質標的を、機能的転写因子が形成され得るような方法で、転写因子のDNA結合ドメイン又は活性化ドメインのいずれかに結合させ、本発明によるAGT基質の標識Lは、1種又は複数の標的物質と相互作用すると推定される候補化合物又は候補化合物のライブラリーである。基質の一部分である候補化合物又は候補化合物のライブラリーを、次いで、AGT融合タンパク質に転移させる。転移後は、1種又は複数の標的物質を含んでいる1種又は複数のAGT融合タンパク質は、1種又は複数の候補化合物で標識されている。AGT融合タンパク質に連結させた候補化合物とDNA結合ドメイン又は活性化ドメインのいずれかに融合させた標的タンパク質を相互作用させることにより、機能的転写因子が形成される。活性化された転写因子は、次に、レポーターを発現させることができる。このレポーターは、該方法を細胞内で行う場合、レポーターの発現が細胞に選択有利性をもたらす場合には検出される。
【0075】
特定の実施形態では、該方法は、1以上のさらなるステップ、例えば、検出ステップ、単離ステップ、1種若しくは複数の候補化合物を同定若しくは特性付けするステップ、又は、1種若しくは複数の標的物質を同定若しくは特性付けするステップなどを含み得る。
【0076】
特定の例では、標識Lは、薬物であるか又は未だ同定されていないタンパク質Yに結合する生物学的に活性な小分子である。未知の標的タンパク質Yを発現することが期待される生物のcDNAライブラリーを転写因子の活性化ドメインに融合させ、AGTを転写因子のDNA結合ドメインに融合させる。上記のような標識Lを含んでいる本発明のAGT基質を加えることにより、機能的転写因子が形成され、この小分子が、cDNAライブラリー内に存在し且つ活性化ドメインに融合したその標的タンパク質Yに結合したときにのみ、遺伝子が発現する。遺伝子発現を選択有利性に結びつけると、該薬物又は生理活性分子の標的タンパク質Yをコードする遺伝子を有しているプラスミドを保持している対応する宿主を同定することができる。
【0077】
さらに別の特定の例では、標識Lは、化学分子のライブラリーである。該ライブラリーは、インビボ条件下で既知の薬物標的タンパク質Yに結合する未だ同定されていない化合物を含んでいることが期待される。標的タンパク質Yを転写因子の活性化ドメインに融合させ、AGTを転写因子のDNA結合ドメインに融合させる。化学的化合物のライブラリーを含んでいる基質を添加することにより、機能的転写因子が形成され、該標識(即ち、該化学的ライブラリー内の化合物)が活性化ドメインに融合したその標的タンパク質Yに結合したときにのみ、遺伝子が発現する。遺伝子発現を選択有利性に結びつけると、宿主の成長をもたらすライブラリーの分子を同定することができる。
【0078】
Lが、R4とR1を連結して環状基質を形成させる結合である場合、好ましい化合物は、R4からR1への結合が、リンカーR4を式2で定義されているアミノ基R6に連結させる結合である環状基質である。そのような好ましい環状基質において、R2は、好ましくは、オリゴヌクレオチド、即ち、上記で詳述した2〜99のヌクレオチドの長さを有する一本鎖オリゴデオキシリボヌクレオチドに組み入れられているβ−D−2’−デオキシリボシルである。このオリゴヌクレオチドは、検出され得ることで標識として機能するように、さらに化学的に修飾することができる。置換基のそのような化学的修飾は、標識Lについて上記で述べたのと同じ種類であり得る。
【0079】
Lが、さらなる基−R3−CH2−X−R1−R2である場合、該基質は二量体化合物であり、その結果、AGT含有融合タンパク質と反応させると、二量体化融合タンパク質が得られる。残基−R3−CH2−X−R1−R2としてのサブユニットLにおいて、R1、R2、R3及びXの意味は、別の基R2−R1−X−CH2−R3−内の対応する意味と同一であり得る(ホモ二量体)か、又は、異なり得る(ヘテロ二量体)。
【0080】
最も好ましいのは、スキームにおいて示され、及び、実施例において記載されている式1の化合物である。
【0081】
新規基質及び中間体を製造する方法も、同様に、本発明の目的である。これらの方法は、概して当技術分野において知られており、また、本発明の好ましい基質が最もよく生成されるように選択され、また、下記において例示されている。特に、本発明は、式:R2−R1−Y(ここで、Yは、反応性脱離基、例えば、アンモニウム塩などである)で表される化合物を、式:HX−CH2−R3−R4−L又はその前駆物質で処理し、得られた化合物を、例えばリンカーR4を構築すること及び/又は標識Lを導入することにより、さらに操作する方法に関する。あるいは、式:R2−R1−XHで表される化合物を、式:Y−CH2−R3−R4−L(ここで、Yは、脱離基、例えば、ハロゲンである)で表される化合物又はその前駆物質でアルキル化し、得られた化合物を、例えば、残基R3及び/若しくはリンカーR4を構築することにより、並びに/又は、標識Lを導入することにより、さらに操作する。
【0082】
特に、本発明は、1以上の標識を有する新規基質を製造する方法に関する。該合成は、式:R2−R1−X−CH2−R3−R4(ここで、R4は、2以上の反応性求核基又は求電子基を有する多官能性残基である)で表されるコア化合物を介して進行させる。そのような基の例には、ヒドロキシ、アミノ、スルフヒドリル、ハロ又はカルボキシなどがある。反応性基のさらなる例は、マイケルアクセプター(Michael acceptor)、例えば、マレイミド又はビニルスルホンなどである。さらに、付加環化反応に関与し得る官能基も挙げることができ、そのような官能基は、例えば、ジエン類又はジエノフィル類、アジド類、ニトロン類、ニトリルオキシド類、アセチレン類及びアルケン類などである。
【0083】
適切な標識を結合させるために、残基R4上に、特に、分枝鎖残基の外圏に配置し得る特定の官能基は、それらが互いに反応し得るか又はそれらが本発明の基質を構築するのに使用される任意の化合物若しくは試薬と反応し得るということが想定されないという要件によってのみ制限される。従って、それらの選定は、望ましい基質を構築するために選択される特定の試薬に依存する。分枝鎖残基R4の外圏に配置することが可能な官能基の例には、フルオロ、クロロ、ブロモ、シアノ、ニトロ、アミノ、アルキルカルボニルアミノ、アリールカルボニルアミノ、アルコキシカルボニルアミノ、アリールオキシカルボニルアミノ、カルボキシ、カルバモイル、アルコキシカルボニル、アリールオキシカルボニル、カルバルデヒド、ヒドロキシ、アルコキシ、アリールオキシ、アルキルカルボニルオキシ、アリールカルボニルオキシ、炭素−炭素二重結合、及び、炭素−炭素三重結合などである。最も好ましい例としては、アミノ、ヒドロキシ、シアノ(潜在的なカルボキシ官能基)、カルバモイル、カルバルデヒド、又は、炭素−炭素二重結合などを挙げることができる。
【0084】
特に、複数の標識Lを有している分枝鎖R4の好ましい合成は、オルソゴナルに保護されている(orthogonally protected)官能基を用いる。保護基をそのように選択することにより、別々に脱保護することが可能となり、それによって、遊離された各官能基を次に化学的にさらに操作して、それに標識を結合させることができるか、又は、リンカーR4のさらなる拡大が可能となる。当業者は、想定される官能性についての適切な保護基を選定することが可能であり、そのような保護基は、例えば、以下の文献に要約されている:T.W.Greene 及び P.G.M.Wuts “Protective Groups in Organic Synthesis”,John Wiley & Sons,New York 1991。
【0085】
本発明による好ましい中間体は、式中のR1−R2が式2(ここで、R2は水素であり、R5は水素であり、R6は置換されていないアミノである)で表されるラジカルであり;Xが酸素であり;R3がトリアゾリル、テトラゾリル、イソオキサゾリル、チエニル、イソオキサゾリジニル又はアルキニルであり;R4がリンカーであり;及び、Lがアミノ又はアジド(特に、アミノ)である式1で表される化合物である。
【0086】
本発明による好ましい別の中間体は、式中のR1−R2が式2(ここで、R2は水素であり、R5は水素であり、R6は置換されていないアミノである)で表されるラジカルであり;Xが酸素であり;R3は1,4−フェニレンであり;R4は場合によりオキソで置換されていてもよい10〜40の炭素原子を有する直鎖アルキレン基(ここで、12以下の炭素原子は酸素で置き換えられており、0、1又は2の炭素原子は窒素で置き換えられている)であり;及び、Lがアミノ又はアジド(特に、アミノ)である式1で表される化合物である。
【0087】
最も好ましい中間体は、スキーム及び実施例に記載されている中間体である。
【0088】
式中のR3がテトラゾリル基、イソオキサゾリル基又はイソオキサゾリジニル基である式1の化合物の合成において有用な中間体の合成については、スキーム1及びスキーム2に要約してある。
【0089】
アジド化合物7は、市販されているテトラエチレングリコール5から、メシル化(メタンスルホニルクロリド、トリエチルアミン)した後、エタノール中でアジ化ナトリウムと反応させることにより調製する。7を再度メシル化した後、Gabrielアミン合成に付すことにより、アジド−アミン9を得る(Carolayら,J.Org.Chem.56:4326−4329,1991)。アジド9とアセチレン誘導体10の間でCu(I)を触媒として用いる1,3−双極付加環化(Griffinら,J.Med.Chem.43:4071−4083,2000)を行うことにより、1,4−置換トリアゾール11を得る。あるいは、アジド9とシアノ誘導体12をルイス酸(ZnBr2)の触媒作用下で反応させて、テトラゾール13を形成させる(スキーム1)。
【0090】
【化13】
【0091】
アジド7をSwerns酸化(塩化オキサリル、DMSO、トリエチルアミン)により中心的なビルディングブロック(アルデヒド14)に変換する。14をヒドロキシルアミン誘導体と反応させることにより、ニトロン17が生成され、これをアセチレン誘導体10と反応させることにより、イソオキサゾリジン類18が形成される。アルデヒド14から、オキシム15が異性体の等モル混合物として形成される。次亜塩素酸ナトリウムで酸化することにより対応するニトリルオキシドがインシトゥで形成され、次いで、10と反応させることにより、イソオキサゾール16が生成する(スキーム2)。
【0092】
【化14】
【0093】
式中のR3がチエニル基である式1の化合物の合成において有用な中間体の合成については、スキーム3に要約してある。
【0094】
【化15】
【0095】
市販されているテトラエチレングリコール5を、強塩基条件下で1当量のヨウ化アリルと反応させることにより単官能化して22を生成させ、これを、さらに、ジメトキシトリチル(DMT)で保護して23とする。この中間体は、パラジウムを触媒として使用するチオフェン誘導体21とのスズキカップリングに付すことができ、それにより、完全に保護された化合物25が得られる。
【0096】
DMT基をモノ脱保護した後、メシル化し(メタンスルホニルクロリド、トリエチルアミン)、その後、エタノール中でアジ化ナトリウムと反応させることにより、保護されたアジドを得る。これをHF/ピリジンで脱保護することにより26を得る。遊離ヒドロキシ基を活性化グアニン−カチオン27とカップリングさせることにより、アジド−中間体が得られるが、これは、別の官能化方策のための前駆物質として使用することができる。最後に、前記アジドを還元してアミン28とすることにより、標識単位Lを導入することが可能であるか、又は、別の表面に結合させることができる。
【0097】
式中のR3がフェニレン基である式1の化合物の合成において有用な中間体の合成については、スキーム4に要約してある。
【0098】
【化16】
【0099】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4が2つのトリアゾリル−4−メトキシ単位とテトラエチレンオキシ単位から構成される単位であり、及び、Lが−R3−CH2−X−R1−R2である式1の化合物は、スキーム5に示してあるようにして調製する。
【0100】
【化17】
【0101】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4がトリアゾリル−4−メトキシキをさらに有しているテトラエチレンオキシ単位であり、及び、Lが−R3−CH2−X−R1−R2である式1の化合物は、スキーム6、スキーム7及びスキーム8に示してあるようにして調製する。
【0102】
【化18】
【0103】
【化19】
【0104】
【化20】
【0105】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4が6−アミノカプロイル基をさらに有するテトラエチレンオキシ単位であり、及び、Lが2種類の異なったアフィニティー標識の組合せを表している式1の化合物の合成については、スキーム9〜スキーム11に例示してある。
【0106】
トリ−オルソゴナル保護基戦略(tri-orthogonal protecting group strategy)のために、ジアゾ転移法(diazo transfer method)により、ε−N−Fmoc−リシン47を対応するα−アジド−ε−N−Fmoc−リシン48に変換する(Lundquistら,J.Org.Lett.3:781−783,2001)。α−アジド−ε−N−Fmoc−リシンを、標準的なペプチドカップリング条件下で、適切な官能化アミノ−ベンジルグアニン誘導体(例えば、49)と縮合させることにより、完全に保護された分子骨格50が得られる。水性1,4−ジオキサン中で、中性条件下、α−アジド基をトリメチルホスフィンで還元して対応するアミン51とすることにより、種々の異なったビルディングブロック(例えばアフィニティー標識を含んでいるもの)と縮合させるのに適する化合物が得られる。例えば、N−(+)−ビオチン−6−アミノ−カプロン酸N−スクシンイミジルエステルとカップリングさせることにより、52が得られ、その後、ジエチルアミンでFmocを除去することにより、遊離アミノ基を有する中間体53が生成される。これを用いて、さらに別の標識を中心の単位にカップリングさせることができる。53の前記遊離アミン基にジゴキシゲニン−3−O−メチルカルボニル−ε−アミノカプロン酸−N−ヒドロキシ−スクシンイミドエステルをカップリングさせることにより、二重に標識された基質54が得られる(スキーム9)。
【0107】
【化21】
【0108】
【化22】
【0109】
【化23】
【0110】
二重に標識された基質54の別法による合成に関し、別のオルソゴナル保護戦略(orthogonal protection strategy)についてスキーム10で説明する。α−N−Fmoc−ε−N−Dde−リシン55を、標準的なペプチドカップリング条件下、適切な官能化アミノ−ベンジルグアニン誘導体(例えば、49)と縮合させることにより、完全に保護された分子骨格56が得られる。ジエチルアミンでFmocを除去した後、N−(+)−ビオチン−6−アミノカプロン酸N−スクシンイミジルエステルとカップリングさせることにより、中間体58が得られる。4%ヒドラジン溶液を用いて残留しているDde保護基を切断することで第二のアミノ基を遊離させ、それにより、化合物53が得られる。これは、さらに、誘導体化することができる。
【0111】
【化24】
【0112】
スキーム11は、標識の異なった組合せを有しているベンジルグアニン誘導体のさらに2つの例の合成を示している(59における2つのアフィニティー標識ビオチン及びジニトロフェニル、又は、60における発蛍光団と組み合わせたアフィニティー標識)。53から出発して(スキーム9を参照されたい)、2,4−ジニトロフルオロベンゼンを遊離アミノ基にカップリングさせることにより、59が得られる。53内の遊離アミノ基にフルオレセイン−5(6)−カルボキサミドカプロン酸N−スクシンイミジルエステルをカップリングさせることにより、60が得られる。
【0113】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4が尿素官能基を介して結合している二量体化テトラエチレンオキシ単位であり、及び、Lが−R3−CH2−X−R1−R2である式1のさらに別の化合物は、スキーム12に示されているように調製する。
【0114】
【化25】
【0115】
式中のR1がグアニンであり、R2が水素であり、R3がプロパルギルであり、R4がさらに6−アミノカプロイル基を含んでいるテトラエチレンオキシ単位であり、及び、Lがビオチンである式1の化合物の合成は、スキーム13及びスキーム14に示されている。
【0116】
【化26】
【0117】
【化27】
【0118】
別のタンパク質と相互作用可能な合成リガンドでの融合タンパク質の共有結合的な標識化に基づいて生細胞内でタンパク質の二量体化を誘導する小分子として作用する標識Lとしての酵素阻害薬を用いたベンジルグアニン誘導体の合成は、スキーム15及びスキーム16に示されている。特に、式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4がω−アミノ−ドデカノイルアミノ−メチル残基であるか又はグルタミン酸単位をさらに含んでいるテトラエチレンオキシ単位であり、及び、Lがメトトレキサートである式1の化合物の合成に関して記載されている。メトトレキサートは、酵素ジヒドロ葉酸レダクターゼの強結合阻害剤であり、これらのヘテロ二量体構造は、いわゆる「二量体化誘発物質」(CID)と称されるよく知られている種類の化合物に属する。
【0119】
【化28】
【0120】
【化29】
【0121】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4がブト−2−エン−1,4−ジオール誘導体であり、及び、Lが−R3−CH2−X−R1−R2である式1の化合物は、スキーム17及びスキーム18に示されているように調製する。
【0122】
【化30】
【0123】
【化31】
【0124】
O6−ベンジルグアニン誘導体を溶解させるのに必要な極性溶媒は、オレフィン複分解触媒ベンジリデン−ビス(トリシクロヘキシルホスフィン)−ジクロロルテニウムとは適合しないと考えられる。従って、最初に、保護された対称性コア単位81及び85を構築し、脱保護した後、最終的なビス−ベンジルグアニン82及び86を合成する。
【0125】
本発明の特に好ましい化合物は、スキーム1〜スキーム18に示されている化合物及び実施例に記載されている化合物である。
【0126】
実施例
実施例1:AGT基質を共有結合により結合させるためのスライドガラスの調製、及び、それに続く、タンパク質マイクロアレイを調製するためのAGT融合タンパク質の共有結合による固定化
市販されている顕微鏡スライドガラス(SiO2)を、超音波浴内で、ジクロロメタン、アセトン、H2O2/H2SO4を用いて充分に清浄化し、さらに、2回蒸留させた蒸留水で充分に洗浄した。それを、公表されている手順に従い、エタノール/水(95:5)の溶媒混合物中で3−アミノプロピルトリエトキシシランを用いて、1時間、アミノシリル化した後、ジスクシンイミジルグルタレート(10mM)をジクロロメタン/ジイソプロピルエチルアミン(100:1)に溶解させた溶液で、アルゴン下、室温で2時間処理すした。該表面を、ジクロロメタンで数回洗浄した。活性化カルボキシ官能基を保持しているガラス表面を、標識Lの代わりに遊離アミノ基を有している式1の化合物(例えば、化合物11、化合物13、化合物16、化合物18、化合物28、化合物34、化合物49、化合物68又は化合物73)を10mMの濃度でメタノールに溶解させトリエチルアミンを添加した溶液と一緒に、4時間インキュベーションした。スライドガラスをメタノールで3回以上洗浄することにより、対応するAGT−基質がアミド結合により共有結合的に結合した表面が得られた。得られたスライドガラスをさらに使用する際に副反応が起こるのを避けるために、全ての未反応スクシンイミジル基を、6−アミノヘキサノール(DMF中100mM)を添加することによりクエンチした。
【0127】
実施例2:11−アジド−3,6,9−トリオキサウンデカノール(7)及び1,11−ジアジド−3,6,9−トリオキサウンデカン(36)
50.0g(260mM)のテトラエチレングリコールと50mLのトリエチルアミンを200mLの乾燥ジエチルエーテルに溶解させた溶液を、アルゴン雰囲気下、0℃に冷却し、15.0g(130mM)のメタンスルホニルクロリドを3時間かけて添加して室温で20分間撹拌した。減圧下に溶媒を除去し、300mLの95%エタノール及び18.0g(280mM)のアジ化ナトリウムを添加した。得られた混合物を24時間加熱還流し、室温まで冷却し、減圧下に濃縮した。残留した混合物を400mLのジクロロメタンで希釈し、ブラインで洗浄し、MgSO4で脱水した。減圧下に濃縮した後、モノアジドとジアジドの粗混合物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 3:1)で精製することにより、15.03g(68.5mmol,26%)のモノアジド及び3.46g(14.18mmol,5.5%)のジアジドを得た。
【0128】
実施例3:1−アジド−11−フタルイミド−3,6,9−トリオキサウンデカン(8)
1.17g(5.35mmol)の11−アジド−3,6,9−トリオキサウンデカノール(7)と1.2mLのトリエチルアミンを35mLの塩化メチレンに溶解させた溶液を0℃に冷却し、シリンジを用いて0.5mL(6.45mmol)のメタンスルホニルクロリドを20分間かけて滴下して加えた。得られた混合物を室温まで昇温させ、1.5時間撹拌した。次いで、その混合物を、10mLの飽和水性NaHCO3で2回洗浄し、5mLの水で3回洗浄した。有機層を減圧下に乾燥濃縮して、1.5gの8を黄色の油状物として得た。これは、それ以上精製することなく、使用した。
【0129】
実施例4:4−ブロモテニル(bromothenyl)アルコール(20)
5.0g(26.17mmol)の4−ブロモチオフェン−2−カルボキシアルデヒド(19)を75mLのイソプロパノールに溶解させ、1.11g(29.31mmol)のNaBH4を一度に添加し、得られた混合物を2時間撹拌した。20mLの飽和水性NH4Clを添加し、濾過により固体を除去し、得られた混合物を減圧下に濃縮した。生成物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 10:1)により精製して、4.64gの20(24.07mmol,92%)を無色の固体として得た。
【0130】
実施例5:4,7,10,13−テトラオキサ−1−ペンタデセン−15−オール(22)
2.3g(19.5mmol)のカリウムt−ブトキシドを500mLの乾燥THFに溶解させ、7.18g(37mmol)のテトラエチレングリコールを滴下して加えた。30分間撹拌した後、3.31g(19.7mmol)のヨウ化アリルを60mLの乾燥THFに溶解させた溶液を1時間かけて添加し、撹拌を24時間継続した。得られた粗混合物をシリカゲルで濾過し、減圧下に溶媒を除去した。生成物をシリカゲルクロマトグラフィー(勾配:石油エーテル/酢酸エチル 10:1 → 酢酸エチル)により精製して、2.41gの22(10.3mmol,27%)を無色の液体として得た。
【0131】
実施例6:1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27)
1.0g(5.9mmol)の6−クロログアニンを40℃で40mLのDMFに溶解させた。室温まで冷却した後、1.4mLの1−メチルピロリジン(13.2mmol)を添加し、得られた反応混合物を18時間撹拌した。2mLのアセトンを添加して、沈澱を完了させた。固体を濾過し、エーテルで洗浄し、減圧下に乾燥させて、1.03gの27(3.9mmol,66%)を得た。
【0132】
実施例7:O6−(4−アミノメチル−ベンジル)グアニン(32)
(a)4−アミノメチル−ベンジルアルコール: 2.83gのLiAlH4(74.5mmol)を150mLの乾燥エーテルに懸濁させ、冷却しながら、1.9mLのH2SO4(100%,37.2mmol)を滴下して加えた。得られた混合物を室温で1時間撹拌した後、12mLのエーテル中の2.0g(12.4mmol)の4−シアノベンゾエートを滴下して加えた。2時間還流した後、20mLの水とそれに続く60mLの水中の7.4gのNaOHにより反応物をクエンチした。有機層をデカントし、水層をエーテルと酢酸エチルで抽出した。有機層をMgSO4で脱水し、溶媒を除去し、生成物を減圧下に乾燥させて、0.92g(6.7mmol,54%)を得た。
【0133】
(b)2,2,2−トリフルオロ−N−(4−ヒドロキシメチル−ベンジル)−アセトアミド: 866mg(6.3mmol)の4−アミノメチル−ベンジルアルコールと880μL(6.3mmol)のトリエチルアミンを10mLの乾燥メタノールに溶解させた溶液に、980μL(8.2mmol)のトリフルオロ酢酸エチルエステルを滴下して加えた。得られた反応混合物を45分間撹拌し、10mLの酢酸エチルと10mLの水で希釈した。水層を酢酸エチルで希釈し、有機層を合して飽和NaClで洗浄し、Na2SO4で脱水した。減圧下に溶媒を除去した後、粗生成物をフラッシュカラムクロマトグラフィー(酢酸エチル/シクロヘキサン 1:2)で精製した。収量:1.32g(5.7mmol,90%)。
【0134】
(c)N−[4−(2−アミノ−7H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロアセトアミド: 592mg(2.54mmol)の2,2,2−トリフルオロ−N−(4−ヒドロキシメチル−ベンジル)アセトアミドを、アルゴン雰囲気下、乾燥DMFに溶解させ、599mg(5.33mmol)のカリウムt−ブトキシドを添加した。次いで、300mg(1.18mmol)の1−(2−アミノ−7H−プリン−6−イル)−1−メチルピロリジニウムクロリド(27)を添加し、この溶液を3時間撹拌した。減圧下に溶媒を除去した後、粗生成物をフラッシュカラムクロマトグラフィー(300mL メタノール/ジクロロメタン 1:50,500mL メタノール/ジクロロメタン 1:10)で精製した。収量:382mg(1.04mmol,88%)。
【0135】
(d)O6−(4−アミノメチル−ベンジル)グアニン(32): 335mg(0.91mmol)のN−[4−(2−アミノ−7H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロアセトアミドを34mLのメタノールと2mLの水に懸濁させた。656mg(4.75mmol)のK2CO3を添加した後、得られた反応混合物を2時間還流した。減圧下に溶媒を除去し、生成物をフラッシュカラムクロマトグラフィー(メタノール/トリエチルアミン/ジクロロメタン 1:0.05:5)で精製した。収量:209mgの32(0.77mmol,85%)。
【0136】
実施例8:O6−(4−[プロプ−2−イニルオキシメチル]−ベンジル)グアニン(35)
662mg(3.8mmol)の4−(プロプ−2−イニルオキシメチル)−ベンジルアルコール(39)を3mLの乾燥DMSOに溶解させ、61mgのNaHを5分間かけて少量ずつ添加した。300mg(1.27mmol)の1−(2−アミノ−7H−プリン−6−イル)−1−メチルピロリジニウムクロリド(27)を添加し、得られた混合物をさらに4時間撹拌した。反応物を0.2mLの酢酸でクエンチし、蒸発乾固させ、フラッシュカラムクロマトグラフィー(勾配:ジクロロメタン/メタノール 50:1 → 10:1)により精製して、188mgの35(0.61mmol,53%)を得た。
【0137】
実施例9:ホモ−O6−ベンジルグアニン−二量体37
50.0mg(0.162mmol)のO6−(4−[プロプ−2−イニルオキシメチル]−ベンジル)グアニン(35)と19.7mg(0.081mmol)の1,11−ジアジド−3,6,9−トリオキサウンデカン(36)を0.5mLのDMFに溶解させた溶液に、15.43mg(0.081mmol)のCu2Cl2を0.15mLの水に懸濁させた懸濁液を添加した。得られた混合物を室温で24時間撹拌した。
【0138】
実施例10:4−(プロプ−2−イニルオキシメチル)ベンジルアルコール(39)及び1,4−ビス−(プロプ−2−イニルオキシ−メチル)ベンゼン(40)
2.5g(18.1mmol)の4−ヒドロキシメチルベンジルアルコール(38)の溶液に、477.5mg(19.9mmol)のNaHを20分間かけて少量ずつ添加した。2.15mLの臭化プロパルギル溶液(トルエン中80%)を滴下して加え、15時間撹拌した。得られた混合物に100mLの水を添加し、生成物をジエチルエーテルで抽出した。相を合して乾燥させ、減圧下に溶媒を除去した。生成物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 4:1)により分離して、1.08gの39(6.17mmol,34%)及び1.05gの40(4.94mmol,27%)を得た。
【0139】
実施例11:4−[(t−ブチルジメチルシリルオキシ)メチル]ベンジルアルコール(44)
810mg(33.77mmol)のNaHを室温で90mLの乾燥THFに懸濁させ、4.2g(30.39mmol)の固形4−ヒドロキシメチル−ベンジルアルコール(38)を3回に分けて5分間かけて添加した。得られた反応混合物を45分間撹拌した。4.83g(32.08mmol)のt−ブチルジメチルシリルクロリドを3回に分けて5分間かけて添加し、さらに1.5時間撹拌した後、その混合物を水でクエンチし、次いで、100mLの水と100mLのジエチルエーテルで希釈した。有機相を分離し、水相をジエチルエーテルで抽出した。有機相を合してブラインで洗浄し、MgSO4で脱水し、濾過し、減圧下に濃縮した。生成物をフラッシュクロマトグラフィー(石油エーテル/酢酸エチル 10:1)で精製して、3.0gの44(11.88mmol,40%)を得た。
【0140】
実施例12:1−[(t−ブチルジメチルシリルオキシ)メチル]−4−(ヨードメチル)ベンゼン(45)
9.15g(34.88mmol)のトリフェニルホスフィン及び3.2g(44.5mmol)のイミダゾールをジエチルエーテル/アセトニトリルの3:1混合物(30mL)に溶解させた。黄色の懸濁液が形成されるまで、激しく撹拌しながら、8.85g(34.9mmol)のヨウ素を添加した。6.1g(23.25mmol)のモノ保護ベンジルアルコール44を20mLの上記と同一の溶媒混合物に溶解させた溶液を添加し、得られた混合物を2時間撹拌した。濾過により固体を除去し、濾液を100mLのジエチルエーテルで希釈し、100mLの重亜硫酸ナトリウム飽和溶液で洗浄した。この水溶液をジエチルエーテルで逆抽出し、有機相を合してMgSO4で脱水し、減圧下に濃縮した。フラッシュクロマトグラフィー(石油エーテル/酢酸エチル 95:5)に付すことにより、4.8gの45(13.25mmol,57%)を得た。
【0141】
実施例13:4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジルアルコール(46)
4.8g(13.25mmol)の1−[(t−ブチルジメチルシリルオキシ)メチル]−4−(ヨードメチル)ベンゼン(45)をアルゴン下、70mLの乾燥THFに溶解させた。0.954g(39.75mmol)のNaHを10分間かけて少量ずつ添加した。3.2g(14.58mmol)の11−アジド−3,6,9−トリオキサウンデカノール(7)を20mLの乾燥THFに溶解させた溶液を滴下して加え、得られた反応混合物を室温で15時間撹拌した。2mLの水を添加して反応物をクエンチし、この混合物を減圧下に約50%になるまで濃縮した。70mLの水を添加し、反応混合物をジエチルエーテルで抽出した。有機相をMgSO4で脱水し、溶媒を除去した。シリカゲルクロマトグラフィー(勾配:石油エーテル/酢酸エチル 10:1 → 3:1)で精製して、3.8g(8.38mmol,63%)のTBDMSで保護された生成物を得た。これを、プラスチック製チューブ内で80mLの乾燥THFに溶解させ、0℃に冷却し、8mLのピリジン/HF(70:30)溶液を添加し、室温で3時間撹拌した。100mLの水性飽和NaHCO3を添加し、有機相を分離し、ブラインで洗浄し、MgSO4で脱水した。溶媒を除去した後、生成物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 1:1)で精製して、1.27gの46(2.87mmol,74%)を得た。
【0142】
実施例14:O6−[4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジル]グアニン(41)
0.974g(2.87mmol)の4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジルアルコール(46)を5mLの乾燥DMFに溶解させ、1.3g(11.5mmol)のカリウムt−ブトキシドを添加した。0.731g(2.87mmol)の1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリドを添加し、この溶液を22時間撹拌した。減圧下に溶媒を除去した後、粗生成物をフラッシュカラムクロマトグラフィー(メタノール/ジクロロメタン 5:95)で精製した。収量:0.675g(50%)。
【0143】
実施例15:ヘテロ−O6−ベンジルグアニン−二量体42
45mg(0.09mmol)のアジド41と29.5mg(0.09mmol)のO6−(4−[プロプ−2−イニルオキシメチル]−ベンジル)グアニン(35)を1mLの水/アセトニトリル(1:1)に溶解させた溶液に、9.8mgのCuCl2を0.1mLの水に懸濁させた懸濁液を添加し、得られた反応混合物を室温で24時間撹拌した。全ての不溶性物質を濾過し、減圧下に溶媒を蒸発させ、生成物をフラッシュカラムクロマトグラフィー(メタノール/ジクロロメタン 5:1)で精製して、3.5mg(0.004mmol,10%)を得た。ESI/MS:781.44(M+)。
【0144】
実施例16:ホモ−O6−ベンジルグアニン−二量体43
アジド41(100mg,0.21mmol)及び1,4−(ジプロプ−2−イニルオキシメチル)ベンゼン(40)(22.6mg,0.10mmol)を、300μLのエタノールと200μLの水と500μLのt−ブタノールからなる溶媒混合物に溶解させた。10μLのCuSO4溶液(40mM)、5mgの銅線及び3mgのCuI2を添加し、得られた反応混合物を室温で48時間撹拌した。全ての不溶性物質を濾過し、減圧下に溶媒を蒸発させ、生成物をフラッシュカラムクロマトグラフィー(クロロホルム/メタノール 20:1)で精製して、34mg(28%)の二量体43を得た。ESI/MS:1159.37(M+)。
【0145】
実施例17:11−アミノ−1−アジド−3,6,9−トリオキサウンデカン(9)
1−アジド−11−フタルイミド−3,6,9−トリオキサウンデカン(8,実施例3,2.0g,5.74mmol)を、50mLの無水メタノール中で、1.0mLの55%ヒドラジン水和物で処理し、2時間加熱還流した。室温まで冷却した後、得られた混合物を減圧下に濃縮し、50mLの水で希釈し、5mLの濃HClで処理した。全ての沈澱物を濾過し、水性濾液をNaOHで中和し、減圧下に溶媒を除去した。残渣をジクロロメタンに溶解させ、3N NaOH及び水で洗浄した。有機相をMgSO4で脱水し、減圧下に濃縮して、0.63g(2.9mmol,51%)の無色の油状物を得た。これは、それ以上精製することなく次のステップで使用した。
【0146】
実施例18:O6−(1−[11−アミノ−3,6,9−トリオキサウンデシル]−1,2,3−トリアゾリル−4−メチル)−グアニン(11)
O6−プロパルギル−グアニン10(75mg,0.4mmol)及びアジド9(44mg,2mmol)を、400μLのN,N−ジメチルアセトアミドと100μLのアセトニトリルと100μLの水からなる溶媒混合物に溶解させた。10μLのCuSO4溶液(40mM)、銅線(5mg)及びCuI2(2mg)を添加し、得られた反応混合物を室温で48時間撹拌した。全ての不溶性物質を濾過し、減圧下に溶媒を蒸発させた。フラッシュクロマトグラフィー(ジクロロメタン/メタノール 20:1)に付すことにより、35mgの11(21%)を得た。
【0147】
実施例19:α−アジド−ε−N−Fmoc−リシン48
18mLのトリフリルアジド(6.3mmol,ジクロロメタン中2.3当量)を、27mLの溶媒混合物(水/メタノール 1:2)中のCuSO4(8mg)及びε−N−Fmoc−リシン47(1.10g,2.98mmol)に添加した。ジイソプロピルエチルアミン(971μL,5.58mmol)を添加し、得られた反応混合物を室温で一晩撹拌した。減圧下に有機溶媒を除去し、100mLの水を添加した。6N HClでpHを2.5に調節した。酢酸エチルで抽出した後、有機フラクションを合して水で洗浄し、脱水した(MgSO4)。溶媒を蒸発させることにより、粗生成物を黄色の油状物として得た。フラッシュクロマトグラフィー(ジクロロメタン/メタノール 97:3)に付すことにより、670mg(59%)の白色の固体を得た。Rf=0.32(ジクロロメタン/メタノール 95:5)。ESI−MS(m/z)395.23[M+H]+(MW計算値=394.3)。
【0148】
実施例20:O6−[4−(13−アミノ−2,5,8,11−テトラオキサトリデシル)−ベンジル]グアニン(49)
O6−[4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジル]グアニン(41,0.21g,0.44mmol)を加熱することにより8mLの乾燥メタノールに溶解させた。トリエチルアミン(410μL,2.2mmol)及び1,3−プロパンジチオール(225μL,2.2mmol)をアルゴン雰囲気下に添加した。得られた反応混合物を40℃で加熱し、48時間撹拌した。この溶液をデカントし、固体をメタノールで洗浄し、減圧下に溶媒を蒸発させた。残渣をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 95:5)で精製し、標題化合物を淡黄色の油状物として得た(120mg,0.26mmol,61%)。Rf=0.01(ジクロロメタン/メタノール 9:1)。
【0149】
【表1】
【0150】
実施例21:O6−ベンジルグアニン−PEG−アミノ−α−アジド−ε−N−Fmoc−リシン50
O6−ベンジルグアニン−PEG−アミン49(76mg,0.17mmol)及びα−アジド−ε−N−Fmoc−リシン48(67.2mg,0.17mmol)を0.8mLの乾燥DMFに溶解させ、1−ヒドロキシベンゾトリアゾール(HOBT)(380μL,0.38mmol,1−メチル−2−ピロリドン(NMP)中1M)を添加した。得られた混合物を40℃で2時間撹拌し、25mLのジエチルエーテル中に移し、粗生成物を沈澱させた。残渣をメタノールに溶解させ、SiO2(0.2g)に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、O6−ベンジルグアニン−PEG−アミノ−α−アジド−ε−N−Fmoc−リシン50を淡黄色の油状物として得た(81mg,0.098mmol,57%)。ESI−MS(m/z)823.3[M+H]+(MW計算値=822.9)。
【0151】
実施例22:O6−ベンジルグアニン−PEG−アミノ−ε−N−Fmoc−リシン51
O6−ベンジルグアニン−PEG−アミノ−α−アジド−ε−N−Fmoc−リシン50(81mg,0.098mmol)を400μLの1,4−ジオキサン/100μLの水に溶解させ、590μLのトリメチルホスフィン(THF中1M)を添加した。室温で2時間撹拌した後、生成物を20mLのジエチルエーテル中で沈澱させ、減圧下で一晩乾燥させた。ESI−MS(m/z)797.35[M+H]+(MW計算値=796.9)。この生成物は、それ以上精製することなく使用した。
【0152】
実施例23:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Fmoc−リシン52
O6−ベンジルグアニン−PEG−アミノ−ε−N−Fmoc−リシン51(78.4mg,0.098mmol)及び30μLのトリエチルアミンを0.5mLのDMFに溶解させた。N−(+)−ビオチン−6−アミノカプロン酸N−スクシンイミジルエステル(49.2mg,0.1mmol)を0.5mLのDMFに溶解させた。両方の溶液を合し、40℃で2時間撹拌した。得られた反応混合物を30mLのジエチルエーテル中に移して粗生成物を沈澱させた。残渣をメタノールに溶解させ、SiO2(0.2g)に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、80mg(0.07mmol,71%)の無色の固体を得た。ESI−MS(m/z)1136.38[M+H]+(MW計算値=1135.38)。
【0153】
実施例24:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Fmoc−リシン52(16mg,0.014mmol)及び50μLのジエチルアミンを0.4mLのDMFに溶解させ、室温で1時間撹拌した。20mLのジエチルエーテルで生成物を沈澱させ、 減圧下に乾燥させた。
ESI−MS(m/z)914.38[M+H]+(MW計算値=914.13)。この生成物は、それ以上精製することなく次のステップで使用した。
【0154】
実施例25:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−[ジゴキシゲニン−3−オキシメチルカルボニル]−6−アミノカプロリル)−リシン54
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53(6.93mg,0.0075mmol)及び10μLのトリエチルアミンを0.4mLの乾燥DMFに溶解させた溶液に、(ジゴキシゲニン−3−オキシメチル−カルボニル)−6−アミノカプロン酸N−ヒドロキシスクシンイミドエステル(5mg,0.0075mmol)を添加し、得られた反応混合物を40℃で2時間撹拌した。20mLのジエチルエーテルで生成物を沈澱させ、減圧下に乾燥させ、クロロホルム中の5%メタノールから15%メタノールまでの勾配を用いるフラッシュカラムクロマトグラフィーで精製して、5mg(0.0034mmol,45%)の標題生成物を得た。ESI−MS(m/z)1458.78[M+H]+(MW計算値=1457.82)。
【0155】
実施例26:O6−ベンジルグアニン−PEG−アミノ−α−N−Fmoc−ε−N−Dde−リシン56
O6−ベンジルグアニン−PEG−アミン49(50mg,0.11mmol)及びα−N−Fmoc−ε−N−Dde−リシン55(58mg,0.11mmol)をDMF(600μL)に溶解させた。HOBT(NMP中の1M溶液の280μL,0.28mmol)及び1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(EDC,54mg,0.28mmol)を添加し、得られた反応物を室温で1時間撹拌した。この溶液にエチルエーテルを添加して生成物を沈澱させ、上清をピペットで除去した。残留している溶媒を減圧下に蒸発させた。生成物を、ジクロロメタン中の2%メタノールから5%メタノールまでの勾配を用いるシリカゲルカラムクロマトグラフィーで精製した(収率:64%)。
【0156】
実施例27:O6−ベンジルグアニン−PEG−アミノ−ε−N−Dde−リシン57
O6−ベンジルグアニン−PEG−アミノ−α−N−Fmoc−ε−N−Dde−リシン56(66mg,68.6μmol)をDMF(600μL)に溶解させ、ジエチルアミン(71μL,686μmol)を添加した。得られた反応混合物を室温で1時間撹拌し、減圧下に溶媒を除去した。この生成物は、それ以上精製することなく次のステップで使用した。
【0157】
実施例28:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Dde−リシン58
O6−ベンジルグアニン−PEG−アミノ−ε−N−Dde−リシン57及びN−(+)−ビオチニル−6−アミノカプロン酸N−スクシンイミジルエステル(31mg,68.6μmol)をDMF(700μL)に溶解させた。トリエチルアミン(30μL)を添加し、得られた反応物を2時間撹拌した。この溶液にジエチルエーテルを添加して生成物を沈澱させ、上清をピペットで除去した。残留している溶媒を減圧下に蒸発させた。生成物をシリカゲルカラムクロマトグラフィーで精製した(クロロホルム中の2%メタノールから3%メタノールまでの勾配;収率:2ステップで37%)。
【0158】
実施例29:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53
DMF(250μL)中の2%ヒドラジンの溶液中で、O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Dde−リシン58(27mg,25.0μmol)を20分間撹拌した。得られた反応混合物にアセトンを添加し、その混合物をさらに3分間撹拌した。この溶液にエチルエーテルを添加して生成物を沈澱させ、上清をピペットで除去した。残留している溶媒を減圧下に蒸発させた。この生成物をそれ以上精製することなく実施例24に準じて使用して、ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−([ジゴキシゲニン−3−オキシメチル−カルボニル]−6−アミノカプロイル)−リシン54を調製した(収率:2ステップで40%)。
【0159】
実施例30:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−(2,4−ジニトロベンゾイル)−リシン59
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53(16.9mg,18.5 gmol)をDMF(400μL)に溶解させ、2,4−ジニトロフルオロベンゼン(4.1mg,22μmol)及びトリエチルアミン(40μL)を添加した。得られた反応混合物を45℃で2時間撹拌した。残留している溶媒を減圧下に蒸発させた。生成物を、クロロホルム中の0%メタノールから15%メタノールまでの段階的な勾配を用いるシリカゲルフラッシュカラムクロマトグラフィーで精製した(収量:15mg,75%)。
【0160】
実施例31:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−(6−[フルオレセイン−5−カルボニル]−アミノカプロイル)−リシン60
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53(8.5mg,0.93 gmol)及び6−(フルオレセイン−5−カルボニル)−アミノカプロン酸N−ヒドロキシスクシンイミジルエステルをトリエチルアミン(10μL)と一緒に、200μLのジメチルアセトアミド中で、40℃で4時間撹拌した。8mLのジエチルエーテル中で生成物を沈澱させ、減圧下に乾燥させた。ESI−MS(m/z)1385.38[M+H]+(MW計算値=1385.13)。
【0161】
実施例32:ビス−N,N’−(O6−ベンジルグアニン−PEG)−尿素61
O6−[4−(13−アミノ−2,5,8,11−テトラオキサトリデシル)−ベンジル]−グアニン(49,30mg,67μL)とN,N’−ジスクシンイミジルカルボネート(8.6mg,33μL)を0.8mLの乾燥DMFに溶解させた溶液にトリエチルアミン(15μL)を添加し、得られた反応混合物を室温で24時間撹拌した。粗生成物をジエチルエーテル(10mL)で沈澱させ、SiO2に吸着させ、フラッシュカラムクロマトグラフィー(メタノール/ジクロロメタン 5:1)で精製した。収量:8.5mg(9.2μmol,27%)。ESI/MS:919.78[M+H]+,(MW計算値=918.99)。
【0162】
実施例33:1−(t−ブチルジメチルシリルオキシ)ブト−2−イン−4−オール(63)
t−ブチルジメチルシリルクロリド(8.0g,53mmol)を乾燥ジクロロメタンに溶解させ、2−ブチン−1,4−ジオール(13.71g,159.2mmol,3当量)と4−ジメチル−アミノピリジン(1.29g,10.6mmol,0.2当量)とトリエチルアミン(6.9mL)をジクロロメタン(200mL)とTHF(100mL)の溶媒混合物に溶解させた溶液に、滴下して加えた。得られた反応混合物を室温で15時間撹拌し、水及び飽和塩化アンモニウムで洗浄し、MgSO4で脱水した。フラッシュカラムクロマトグラフィー(石油エーテル/酢酸エチル 1:1)に付すことにより、7.2g(67%)を得た。
【0163】
実施例34:1−(t−ブチルジメチルシリルオキシ)−4−ヨードブト−2−イン(64)
J.Robertsonら(J.Chem.Soc.,Perkin Trans.1,3389−3369,2000)に従い、100mLのジクロロメタン中で、1−(t−ブチルジメチルシリルオキシ)−ブト−2−イン−4−オール(63,4.0g,19.96mmol)、イミダゾール(1.80g,26.45mmol)、ヨウ素(6.58g,25.95mmol)及びトリフェニルホスフィン(6.80g,25.95mmol)を処理した。後処理に付すことにより、4.42g(71%)の無色の油状物を得た。この生成物は、直接、次のステップで使用した。
【0164】
実施例35:4−(11−アジド−3,6,9−トリオキサウンデシルオキシ)−1−(t−ブチルジメチルシリルオキシ)−2−ブチン(65)
11−アジド−3,6,9−トリオキサウンデカノール(7,3.79g,17.02mmol)を乾燥THFに溶解させ、水素化ナトリウム(1.02g,42.45mmol)を少量ずつ添加した。室温で40分間撹拌した後、1−(t−ブチルジメチルシリルオキシ)−4−ヨードブト−2−イン(64,4.4g,14.18mmol)を添加し、得られた反応混合物をさらに15時間撹拌した。この反応物を水でクエンチし、減圧下に溶媒の量を約30%に低減させた。ジエチルエーテル(50mL)を添加し、この混合物を水で洗浄し、有機相をMgSO4で脱水した。溶媒を蒸発させた後、生成物をフラッシュカラムクロマトグラフィー(石油エーテル/酢酸エチル 4:1)で精製して、3.12g(54%)を得た。
【0165】
実施例36:(13−アジド−2,5,8,11−テトラオキサトリデシル)−プロパルギルアルコール(66)
t−ブチルジメチルシリルエーテル65(1.5g,3.37mmol)を、0.25mLの酢酸を含んでいる25mLのTHFに溶解させ、テトラブチルアンモニウムフルオリド(4.1mL,THF中1M)で処理した。得られた反応混合物を2時間撹拌し、減圧下に溶媒の量を約30%に低減させた。酢酸エチル(50mL)を添加した後、有機相を飽和重炭酸ナトリウム(20mL)及びブライン(30mL)で洗浄し、MgSO4で脱水した。この生成物は、それ以上精製することなく次のステップで使用した。収量:0.774g(80%)。
【0166】
実施例37:O6−[(13−アジド−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン(67)
プロパルギルアルコール66(724mg,2.5mmol)を乾燥DMF(1mL)に溶解させ、水素化ナトリウム(181mg,7.55mmol)を2回に分けて添加した。室温で30分間撹拌した後、1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27,641mg,2.5mmol)及び4−ジメチルアミノピリジン(100mg,0.81mmol)を添加した。撹拌を15時間継続し、30mLのジエチルエーテルを添加して生成物を沈澱させた。フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 95:5)に付すことにより、470mg(44%)の僅かに黄色い油状物を得た。ESI/MS:421.29[M+H]+。
【0167】
実施例38:O6−[(13−アミノ−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン(68)
アジド67(400mg,0.951mmol)及びトリフェニルホスフィン(748mg,2.85mmol)を、溶媒混合物(6mLの1,4−ジオキサン/400μLの水)中で、室温で15時間撹拌した。減圧下に溶媒を除去し、残渣をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、312mg(83%)を得た。
【0168】
実施例39:O6−[(13−(ビオチニル−6−アミノカプロイル−アミノ)−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン(69)
プロパルギルグアニン68(26.9mg,88μmol)及び(+)−ビオチニル−6−アミノヘキサン酸N−ヒドロキシ−スクシンイミドエステル(20.0mg,44 gmol)を15μLのトリエチルアミンと一緒に、1mLのDMF中で、40℃で3時間撹拌した。減圧下に溶媒を除去し、残渣をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、20mg(68%)を得た。
【0169】
実施例40:O6−[(13−(ジゴキシゲニン−3−オキシメチルカルボニル−6−アミノカプロイル−アミノ)−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン70
プロパルギルグアニン68(11.6mg,37μmol)、ジゴキシゲニン−3−オキシメチルカルボニル−6−アミノカプロン酸N−ヒドロキシスクシンイミドエステル(5mg,7.5μmol)及び10μLのトリエチルアミンを40℃で2時間撹拌した。20mLのジエチルエーテルで生成物を沈澱させ、減圧下に乾燥させ、クロロホルム中の5%メタノールから15%メタノールまでの勾配を用いるフラッシュカラムクロマトグラフィーで精製して、3.2mgの標題化合物を得た(0.0034mmol,45%)。ESI−MS(m/z)939.78[M+H]+(MW計算値=938.12)。
【0170】
実施例41:O6−[4−(12−Fmoc−アミノ−ドデカノイル)アミノメチル]ベンジルグアニン72
12−Fmoc−アミノドデカン酸(147mg,0.33mmol)及び1−ベンゾトリアゾリルオキシ−トリス−ピロリジノ−ホスホニウムヘキサフルオロホスフェート(PyBOP,193mg,0.37mmol)を11mLのDMFに溶解させた。室温で45分間撹拌した後、5mLのDMF中のO6−4−アミノメチル−ベンジルグアニン(71,100mg,0.37mmol)を添加した。得られた反応混合物を50℃で5分間撹拌し、次いで、室温で1時間撹拌した。減圧下に溶媒を蒸発させた後、生成物をフラッシュカラムクロマトグラフィー(勾配:メタノール/ジクロロメタン 1:50から1:10まで)で精製して、185mg(0.27mmol,79%)の白色の固体生成物を得た。Rf=0.33(メタノール/ジクロロメタン 1:10)。
【0171】
【表2】
【0172】
実施例42:O6−[4−12−アミノドデカノイル)−アミノメチル]−ベンジルグアニン(73)
Fmocで保護された誘導体72(180mg,0.26mmol)を、20%(v/v,6.5mmol)のピペリジンを含んでいる3.2mLのDMFに溶解させた。室温で30分間撹拌した後、減圧下に溶媒を除去し、生成物をフラッシュカラムクロマトグラフィー(勾配:メタノール/ジクロロメタン 1:20から1:5まで(1%トリエチルアミン含有))で精製して、95mg(0.20mmol,78%)の淡黄色の固体を得た。Rf=0.05(メタノール/トリエチルアミン/ジクロロメタン 20:1:100)。
【0173】
【表3】
【0174】
実施例43:L−2−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ}−ペンタン二酸1−ベンジルエステル(76)
L−グルタミン酸1−ベンジルエステル(75,114mg,0.48mmol)及びK2CO3(66.4mg,0.48mmol)を3mLのDMSOに添加し、60℃で1時間超音波処理に付した。この混合物を、4−(N−[2,4−ジアミノ−6−プテリジニルメチル]−メチルアミノ)−安息香酸74(150mg,0.46mmol)とジイソプロピルエチルアミン(112μL,0.65mmol)とPyBOP(250mg,0.48mmol)を1.3mLの1時間撹拌しておいたDMSOに溶解させた溶液に添加した。この反応混合物を50℃で10分間撹拌し、次いで、室温で2.5時間撹拌した。0.5mLの1Mトリエチルアミンでクエンチした後、粗生成物を直線勾配(水/0.1%トリフルオロ酢酸(TFA):アセトニトリル/0.08%TFA 100:0から20:80まで)を用いるHPLCで精製した。この溶液をpH7に調節し、減圧下に溶媒を除去して、50mg(0.09mmol,20%)の橙色の固体を得た。
【0175】
【表4】
【0176】
実施例44:O6−4−([12−N−(4−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ)−4−ベンジルオキシカルボニルブタノイル)−アミノドデカノイル]−アミノメチル)−ベンジルグアニン(77)
(ベンゾトリアゾール−1−イルオキシ)トリピロリジノ−ホスホニウムヘキサフルオロホスフェート(PyBOP,152mg,0.29mmol)を1−ヒドロキシベンゾトリアゾール(HOBT)の溶液(1−メチル−2−ピロリドン(NMP)中の1M溶液の146μL,0.15mmol)に溶解させ、ベンジルエステル76(NMP中の0.154M溶液の380μL,0.06mmol)に添加した。得られた混合物を30分間振盪し、次いで、ジイソプロピルエチルアミン(30μL,0.18mmol)及び200μLのNMP中のアミノ−グアニン73(27.2mg,0.06mmol)に添加した。この反応混合物を室温で2時間撹拌し、次いで、200μLの1MのTFAでクエンチした。減圧下に溶媒を除去した後、生成物をフラッシュカラムクロマトグラフィー(勾配:メタノール/ジクロロメタン 1:50から1:5まで(2%酢酸含有))で精製した。19mg(0.2mmol,33%)の橙色の固体が得られた。Rf=0.08(メタノール/酢酸/ジクロロメタン 5:1:50)。ESI−MS(m/z)994.9[M+H]+(MW計算値=994.2)。
【0177】
実施例45:O6−4−([12−N−(4−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ}−4−カルボキシブタノイル)−アミノドデカノイル]−アミノメチル)−ベンジルグアニン(78)
ベンジルエステル77(13mg,0.01mmol)を2mLのメタノール/水(1:1)に溶解させ、K2CO3(785μL メタノール/水(1:1)中の1M,0.79mmol)を添加した。得られた混合物を室温で2.5時間振盪した後、785μL の1MのTFAでクエンチした。粗生成物を、直線勾配(水/0.1%TFA:アセトニトリル/0.08%TFA 100:0から20:80まで)を用いるHPLCで精製した。10mg(0.01mmol,85%)の生成物が得られた。ESI−MS(m/z)904.7[M+H]+(MW計算値=904.0)。
【0178】
実施例46:O6−4−[13−N−(4−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ}−4−カルボキシブタノイル)−アミノ−2,5,8,11−テトラオキサトリデシル]−ベンジルグアニン(79)
PyBOP(201mg,0.39mmol)及びHOBT(NMP中の1M溶液の194μL、0.19mmol)をベンジルエステル76(21mg,0.04mmol)に添加し、得られた混合物を室温で30分間振盪した。O6−[4−(13−アミノ−2,5,8,11−テトラオキサトリデシル)−ベンジルグアニン(49,21mg,0.05mmol)及びジイソプロピルエチルアミン(24μL,0.14mmol)を490μLのNMPに溶解させ、活性化させた76の溶液に添加した。室温で3時間振盪した後、この反応混合物を1mLのアセトニトリル/2mLの1MのTFAでクエンチした。粗生成物を、直線勾配(水/0.1%TFA:アセトニトリル/0.08%TFA 100:0から20:80まで)を用いるHPLCで精製した。1.5MのNaOHを用いてこの溶液のpHを10に調節することにより、カルボキシ基を脱保護した。先のステップと同じ勾配を用いるHPLCで粗生成物を精製した後、14mg(0.02mmol,40%)の標題化合物を得た。ESI−MS(m/z)883.4[M+H]+(MW計算値=882.9)。
【0179】
実施例47:4−アリルオキシメチル−1−(t−ブチルジメチルシリルオキシメチル)−ベンゼン(80)
4−(t−ブチルジメチルシリルオキシメチル)−ベンジルアルコール44(2.85g,11.31mmol)を15mLの乾燥THFに溶解させた溶液に、アルゴン雰囲気下、水素化ナトリウム(326mg,13.57mmol)を少量ずつ添加し、この反応混合物を室温で30分間撹拌した。ヨウ化アリル(3.80g,22.62mmol)を5mLの乾燥THFに溶解させた溶液を添加し、得られた混合物を17時間撹拌した。この反応物を水でクエンチし、溶媒の容積を約30%に低減させ、残渣を酢酸エチル(20mLで3回)で抽出した。有機相をMgSO4で脱水し、減圧下に溶媒を除去し、生成物をフラッシュカラムクロマトグラフィー(石油エーテル/酢酸エチル 100:1)で精製して、1.83g(6.25mmol,59%)を得た。
【0180】
実施例48:1,4−ジ−[4−(t−ブチルジメチルシリルオキシメチル)−ベンジルオキシ]−2−ブテン(81)
4−アリルオキシメチル−1−(t−ブチルジメチル−シリルオキシメチル)−ベンゼン(80,1.0g,3.24mmol)を70mLの乾燥ジクロロメタンに溶解させた溶液に、アルゴン雰囲気下、ベンジリデン−ビス(トリシクロヘキシルホスフィン)−ジクロロルテニウム(420mg,0.513mmol,15mol%)を添加した。得られた反応混合物を室温で15時間撹拌し、触媒(200mg)を追加し、その反応混合物を5時間加熱還流した。減圧下に溶媒を除去し、生成物をカラムクロマトグラフィー(石油エーテル/酢酸エチル 50:1)で精製して、520mg(0.93mmol,55%)の標題化合物を得た。
【0181】
実施例49:ビス−1,4−(O6−ベンジルグアニン−メトキシ)−2−ブテン82
TBDMS−二量体81(351mg,0.63mmol)を5mLの乾燥THFに溶解させ、テトラブチルアンモニウムフルオリド(3.78mmol)及び酢酸(3.78mmol)を添加した。得られた反応混合物を室温で15時間撹拌した。溶媒を蒸発させた後、生成物を溶媒の勾配(石油エーテル/酢酸エチル 1:1から1:3まで)を用いるカラムクロマトグラフィーで精製して、150mg(0.45mmol,73%)の無色の固体を得た。この脱保護した二量体(100mg,0.30mmol)を0.8mLの乾燥DMFに溶解させた溶液に、水素化ナトリウム(44mg,1.82mmol)を添加し、この反応混合物を30分間撹拌した。1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27,186mg,0.73mmol)及びDMAP(11mg,0.09mmol)を添加し、この反応混合物を室温でさらに3時間撹拌した。この反応物を水でクエンチし、減圧下に溶媒を除去した。残渣をメタノールに溶解させ、SiO2に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、二量体82を無色の固体として得た(30mg,0.05mmol,16%)。
【0182】
実施例50:11−アリルオキシ−3,6,9−トリオキサウンデカノール(83)
カリウムt−ブトキシド(2.3g,19.5mmol)をTHF(500mL)に溶解させた無水溶液に、室温で、テトラエチレングリコール(7.18g,37.0mmol)を添加した。得られた混合物を30分間撹拌し、ヨウ化アリル(3.31g,19.7mmol)を乾燥THF(60mL)に溶解させた溶液を、1時間かけて滴下して加えた。この反応混合物を30時間撹拌し、酢酸エチル(800mL)を用いてシリカプラグ(10g)を通して濾過して生成物を溶出させた。減圧下に溶媒を蒸発させた後、生成物を溶媒の勾配(石油エーテル/酢酸エチル 10:1から、酢酸エチル 100%まで)を用いるカラムクロマトグラフィー で精製して、無色の油状物を得た(2.41g,10.3mmol,27%)。
【0183】
実施例51:1−(t−ブチルジメチルシリルオキシメチル)−4−(13−アリルオキシ−2,5,8,11−テトラオキサトリデシル−ベンゼン(84)
11−アリルオキシ−3,6,9−トリオキサウンデカノール(83,2.33g,9.94mmol)を乾燥THF(40mL)に溶解させた溶液に、アルゴン雰囲気下、水素化ナトリウム(0.95g,39.8mmol)を数回に分けて添加した。得られた混合物を30分間撹拌した。ヨード誘導体45(3.0g,8.28mmol)を乾燥THF(10mL)に溶解させた溶液を滴下して加え、この混合物を15時間撹拌した。2mLの水を添加することにより反応物をクエンチし、減圧下に70%の量のTHFを蒸発させた。70mLの水を添加してこの混合物を希釈し、得られた溶液を50mLの酢酸エチルで5回抽出した。有機抽出物を合してMgSO4で脱水し、減圧下に濃縮した。得られた黄色の油状物をフラッシュカラムクロマトグラフィー(勾配:石油エーテル/酢酸エチル 10:1から5:1まで)で精製して、標題生成物84を淡黄色の油状物として得た(1.96g,4.2mmol,51%)。
【0184】
実施例52:TBDMSで保護されたビス−1,4−(ベンジルアルコール−PEG)−2−ブテン85
TBDMSで保護されたアリルオキシ誘導体84(1.0g,2.13mmol)を40mLの乾燥ジクロロメタンに溶解させた溶液に、アルゴン雰囲気下、ベンジリデン−ビス(トリシクロヘキシルホスフィン)−ジクロロルテニウム(263mg,0.32mmol,15mol%)を添加した。得られた反応混合物を5時間加熱還流した。触媒(200mg)を追加し、加熱を15時間継続した。減圧下に溶媒を除去し、生成物を、溶媒の勾配(石油エーテル/酢酸エチル 1:1から1:5まで)を用いるカラムクロマトグラフィーで精製して、530mg(0.58mmol,54%)の二量体85を得た。
【0185】
実施例53:ビス−1,4−(O6−ベンジルグアニン−PEG)−2−ブテン86
TBDMSで保護された二量体85(200mg,0.22mmol)を3mLの乾燥THFに溶解させ、テトラブチルアンモニウムフルオリド(2.5mmol)及び酢酸(2.5mmol)を添加した。得られた反応混合物を室温で15時間撹拌した。溶媒を蒸発させた後、生成物を、溶媒の勾配(石油エーテル/酢酸エチル 1:1から、酢酸エチル 100%まで)を用いるカラムクロマトグラフィーで精製して、80mg(0.11mmol,50%)の無色の固体を得た。この脱保護された二量体(61mg,0.089mmol)と1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27,54mg,0.22mmol)とDMAP(3.0mg,0.027mmol)を1.0mLの乾燥DMFに溶解させた溶液に、水素化ナトリウム(13mg,0.54mmol)を添加し、得られた反応混合物を2時間撹拌した。この反応物を水でクエンチし、減圧下に溶媒を除去した。残渣をメタノールに溶解させ、SiO2に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、二量体86を無色の固体として得た(35mg,0.036mmol,41%)。
【0186】
実施例54:式1の化合物とヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)融合タンパク質の反応性の実証
(a) O6−(1−[11−アミノ−3,6,9−トリオキサウンデシル]−1,2,3−トリアゾリル−4−メチル)−グアニン(11)の反応性
総容積15μLの50mMのHEPES(pH7.5)中で、18pmolのPGEAhAGT−GST(A.Juilleratら,Chem.Biol.10:313−317,2003)をDMSO中の11の5mM溶液(0.2μL)と一緒に1時間インキュベーションした。10pmolのビオチニル化O6−ベンジルグアニン−オリゴヌクレオチド(R.Damoiseauxら,ChemBiochem 4:285−287,2001)を添加し、さらに15分間、反応を継続させた。対照として、総容積15μLの50mMのHEPES(pH7.5)中で、基質を含まない状態で、18pmolのPGEAhAGT−GSTを10pmolの同一のビオチニル化オリゴヌクレオチドと一緒に15分間インキュベーションした。hAGT−GSTとビオチニル化オリゴヌクレオチドの間のビオチニル化反応生成物は、HRP(Pierce)に結合させた抗ビオチンコンジュゲートを用いるウエスタンブロット法により検出した。図1Aのレーン「C」は、PGEAhAGT−GSTとビオチニル化オリゴヌクレオチドの反応生成物を示していた。PGEAhAGT−GSTが最初に11と反応する場合は、ビオチニル化オリゴヌクレオチドとのさらなる反応は観察されなかった(レーン1及びレーン2、図1A)。これにより、O6−トリアゾリルメチルグアニンがhAGTに対する効果的な基質であることが証明された。
【0187】
(b) O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−(6−[フルオレセイン−5−カルボニル]−アミノカプロイル)−リシン60の反応性
総容積15μLの50mMのHEPES(pH7.5)中で、18pmolのPGEAhAGT−GSTを0.5nmolの60と一緒にインキュベーションした。反応生成物は、ヤギ抗フルオレセイン抗体(Rockland)及びHRPコンジュゲート抗ヤギ抗体(Sigma)を用いるウエスタンブロット法により検出した。これにより、2つの異なった標識を含んでいる化合物60がhAGTに対する効果的な基質であることが証明された。
【0188】
実施例55:固相上におけるヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)融合タンパク質の固定化
80μLのPGEAhAGT−GST融合タンパク質溶液(HBSバッファー中200μg/mL)を、O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノ−カプロイル)−ε−N−([ジゴキシゲニン−3−オキシメチルカルボニル]−6−アミノカプロイル)−リシン54の0.4mM溶液(20μL)と一緒に、室温で10分間インキュベーションした。この溶液を、Neutravidin Biacore チップ上に、1μL/分の流速で35分間通過させた。次いで、このチップを、同じ流速のバッファー(HBST)で40分間パージした。次に、GST抗体(1980μg/mL)を注入して該融合タンパク質の先の結合を明らかにし、次いで、再度、HBSTで洗浄した。固定化実験は、表面プラズモン共鳴により評価した。表面プラズモン共鳴分析は、研究グレードのSAセンサーチップを備えたBiacore 1000 光学バイオセンサー(Biacore)を用いて行った。EDTAを含まないHBS(10mM HEPES,pH7.4,150mM NaCl,0.005% Tween 20)は、新たに調製し、22μmの膜を通して濾過した。得られたセンソグラムは、図2に示してある。
【0189】
実施例56:ヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)の二量体化
(a) PGEGhAGTと二量体化基質43の反応
二量体化実験は、HEPESバッファー(HEPES 50mM,pH7.2,1mM DTT,200μg/mL BSA)中の0.4μMの一定の濃度のPGEGhAGT−GST(A.Juilleratら,Chem.Biol.10:313−317,2003)で、総容積40μLで行った。0〜100:1の基質:タンパク質の比率をカバーするように、化合物43の濃度を変化させた。インキュベーション時間は、すべての実験に関して、40分間とした。ウェスタンブロットは、抗hAGT抗体(PBS中1:2000,Chemicon)と第二の抗体としての抗マウス−HRPコンジュゲート(PBS中1:2000,Sigma)を用いて得た。結果は、図3Aに示してある。
【0190】
(b) E.coli抽出物中の二量体化基質61とHA−W160hAGTとの反応
実験は、hAGT変異体Gly160Trp(W160hAGT)(M.Xu−Welliverら,Biochemical Pharmacology 58:1279−1285,1999)を用いて行った。HA−W160hAGTは、LB培地中、24℃で、IPTG(1mM)と種々の濃度(0μM、10μM、20μM、及び、50μM)の基質61を指数関数的に増殖する培養に添加した後、E.coli BL21で2時間発現させた。遠心分離により細胞を回収した。各標識化実験について、抽出物の総容積は、約1mL(容積は、OD600が一定となるように調節する)であった。得られたペレットを50μLの1×SDSに再度懸濁させた後、O6−ベンジルグアニンを1mMの濃度となるまで添加した。95℃に5分間加熱した後、サンプルを12%SDS−PAGEにロードした。ウェスタンブロットは、ヤギ抗MGMT(TBST中1:100,Chemicon)と第二の抗体としての抗ヤギIgG(TBST中1:1000,Santa Cruz Biotechnology)を用いて得た。結果は、図3Bに示してある。
【0191】
実施例57:GST−3HYhAGTに対する78と79の反応性
3HYhAGTが78及び79に対して活性を有していることを実証するために、3HYhAGTを、E.coliにおいて、グルタチオンS−トランスフェラーゼとの融合タンパク質(GST−3HYhAGT)として発現させ、その融合タンパク質を精製し、及び、その活性をインビトロアッセイで測定した。GST−3HYhAGTを過剰量の78又は79と一緒にインキュベーションして反応させた後、抗メトトレキサート抗体を用いるウェスタンブロット法に付した。このアッセイにおいて、GST−3HYhAGTは、78との反応については約1500秒−1M−1の二次反応速度定数を示し、79との反応については約900秒−1M−1の二次反応速度定数を示した。従って、78の濃度1μMで、及び、3HYhAGTに対して過剰な78(擬一次条件)で、該タンパク質の50%が10分以内に標識された。
【0192】
実施例58:hAGT変異体
hAGT融合タンパク質を構築するために、好ましくは、変異体Asn157Gly Ser159Gluを使用した。この変異体は、野生型hAGTと比較して、1型のベンジルグアニン誘導体に対して、約20倍の増大した活性を示した。以下の文献を参照されたい:A.Juilleratら,Chem.Biol.10:313−317,2003。以下に示すhAGTのさらなる変異体は、hAGTのDNAへの結合を分断するが、ベンジルグアニン誘導体に対する活性は有意には阻害しないことが示されている:Lys125Ala,Ala127Thr,及び,Arg128Ala。以下の文献を参照されたい:A.Limら,EMBO J.15:4050−4060,1996,及び,D.S.Danielsら,EMBO J.19:1719−1730,2000。これらの変異体を導入する理論的根拠は、3ハイブリッド系におけるhAGT融合タンパク質とDNAの相互作用を最少にすることである。結果として得られた、上記の5つの変位を有しているhAGT変異体は、3HYhAGTと略記する。
【0193】
実施例59:酵母L40株の増殖アッセイ
LexA−3HYhAGT及びB42−DHFR融合タンパク質をコード化するプラスミドの対を酵母株L40内に形質転換し、機能的LexA−B42転写因子を再構築することにより、レポーター遺伝子HIS3及びlacZが転写された。78又は79のいずれかの存在下において融合タンパク質の異なった組合せを発現させることにより酵母L40のヒスチジン要求性が補完されるか否かについて試験した(表1)。LexA−3HYhAGTとB42−DHFRを同時に発現させることにより、酵母L40はヒスチジンは含んでいないが78又は79のいずれかを含んでいるプレート上で増殖することが可能となった。このことは、HIS3が転写されたことを示している。78又は79の非存在下では、増殖は観察されなかった。
【0194】
【表5】
【0195】
原則として、ベンジルグアニン78及び79を介した機能的転写因子の再構築は、どのリガンド結合性タンパク質がLexAに融合するかとは関係がなく、また、どのリガンド結合性タンパク質がB42に融合するかとは関係がない。このことを実証するために、LexA−DHFRとB42−3HYhAGTを同時に発現する酵母L40の、ヒスチジンは含んでいないが78又は79のいずれかを含んでいるプレート上での増殖について調べた。予想されたとおり、化合物78又は79に依存する増殖が観察されるが、その増殖速度は、LexA−3HYhAGTとB42−DHFRを同時に発現する酵母で観察される増殖速度を下回った(表1)。
【0196】
実施例60:ONPGを用いる定量的β−ガラクトシダーゼアッセイ
LexA融合タンパク質とB42融合タンパク質の別の組合せを発現する酵母株L40における、ベンジルグアニンをベースとする化合物78及び79によるlacZレポーター遺伝子の転写の活性化について調べた(表2)。このアッセイにおいて、液体培養の細胞抽出物中の色素原基質o−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)の加水分解速度を測定することにより、lacZ遺伝子の産物であるβ−ガラクトシダーゼの活性を決定した。
【0197】
【表6】
【図面の簡単な説明】
【0198】
【図1A】HRP(Pierce)に結合した抗ビオチンコンジュゲートのウェスタンブロット。レーン1及び2:化合物11(式中のR3がトリアゾリルメチルである式1の化合物)と反応させた後、AGT基質として知られているビオチニル化O6−ベンジル−グアニンオリゴヌクレオチドと反応させた後のPGEAhAGT−GST(AGT融合タンパク質)。「C」の印が付いているレーン(対照):PGEAhAGT−GSTとビオチニル化O6−ベンジルグアニンオリゴヌクレオチドの反応生成物。
【図1B】ヤギ抗フルオレセイン抗体(Rockland)及びHRPコンジュゲート抗ヤギ抗体(Sigma)のウェスタンブロット。レーン1及び2(2種類の異なった濃度)は、フルオレセイン標識とビオチン標識を有している化合物60(式中のLが複数の標識である式1の化合物)とPGEAhAGT−GSTの反応生成物を示している。左側のレーンは、47kDa及び79kDaの分子量マーカーを示している。
【図2】ストレプトアビジンBiacoreチップに適用した化合物54(式中のLがビオチンである式1の化合物)とPGEAhAGT−GST(AGT融合タンパク質)の反応生成物の表面プラズモン共鳴のセンソグラム。該チップを、αGST抗体の溶液でさらに処理する。A:化合物54と一緒にプレインキュベーションしたPGEAhAGT−GSTを添加。B:緩衝液(HBST)を添加。C:αGST抗体を添加。
【図3A】抗hAGT抗体及び第二抗体としての抗マウスHRPコンジュゲートを用いたウェスタンブロット。レーンの下に、化合物43(式中のLがさらなる−R3−CH2−X−R1−R2残基である式1の化合物、即ち、二量体化化合物)とPGEGhAGT−GSTの比率を示してある。
【図3B】抗ヤギhAGT、及びE.coli中における二量体化化合物61と融合タンパク質HA−W160hAGTのインビボ反応生成物の抗ヤギIgGを用いたウェスタンブロット。レーンの下に、61(式中のLが−R3−CH2−X−R1−R2残基である式1の化合物)の種々の濃度を示してある。
【図4】hAGTをベースとする3ハイブリッド系の略図。融合タンパク質DHFR−B42の存在下におけるhAGT−LexA融合タンパク質と式1(式中、Lはメトトレキサート(M)である)の化合物の反応において、hAGT−LexAとDHFR−B42をカップリングさせ、転写を開始させる。
【図1】
【技術分野】
【0001】
本発明は、標識を新規基質からO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)及びO6−アルキルグアニン−DNAアルキルトランスフェラーゼ融合タンパク質に転移させる方法、並びに、そのような方法において適切な新規基質に関する。
【0002】
発明の背景
N−メチル−N−ニトロソウレアのような求電子試薬の突然変異誘発作用及び発癌作用は、主として、DNA内のグアニンのO6−アルキル化に起因している。それらをDNA−アルキル化から保護するために、哺乳動物及び細菌類は、それらの損傷を修復するタンパク質O6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)を有している。AGTは、アルキル化されたグアニン及びグアニン誘導体のO−6位からそれが有している1つのシステインのメルカプト基にアルキル基を転移させ、不可逆的にアルキル化されたAGTを生成する。その基礎的なメカニズムは、SN2型の求核反応であり、それにより、メチル基のみではなくベンジル基も容易に転移される理由が説明される。腫瘍細胞におけるAGTの過剰発現は、プロカルバジン、ダカルバジン、テモゾロマイド及びビス−2−クロロエチル−N−ニトロソウレアなどのアルキル化薬に対する抵抗性の主な原因であるので、化学療法における増感剤として使用するためのAGT阻害薬が求められてきた(Peggら,Prog Nucleic Acid Res Mol Biol 51:167−223,1995)。US5,691,307には、ベンジル基に種々の置換基を有するO6−ベンジルグアニン類が記述されており、また、腫瘍細胞内のAGTレベルを低下させ、それによりアルキル化抗腫瘍薬に対する反応性を増大させるためのO6−ベンジルグアニン類の使用についても記述されている。同様に、WO97/20843には、さらに、O6−ベンジル−ピリミジン誘導体及びO6−ヘテロアリールメチル−ピリミジン誘導体である、AGTを低減させる化合物が開示されている。
【0003】
DE19903895には、AGTのレベルを測定するためのアッセイが開示されており、ここで、該アッセイは、ビオチニル化O6−アルキルグアニン誘導体とAGTを反応させてAGTをビオチニル化することに依拠している。これにより、次に、ストレプトアビジンでコーティングしたプレート上でのAGTの分離と検出(例えば、ELISAアッセイ)が可能となる。このアッセイは、腫瘍組織内のAGTのレベルのモニタリング及びAGT阻害薬のスクリーニングにおける使用に示唆されるアッセイである。
【0004】
WO01/85221では、AGTの検出及びAGTレベルのモニタリングのために、放射能標識フルオロ置換O6−ベンジル−グアニン類又は放射能標識ヨード置換O6−ベンジル−グアニン類の使用が提案されている。
【0005】
Damoiseauxら(ChemBiochem.4:285−287,2001)は、研究及び化学療法において有用な、癌細胞内におけるこの酵素のレベルの検出を容易にする、AGTを標識するための化学プローブとして使用するためのオリゴデオキシリボヌクレオチドに組み入れた修飾されたO6−アルキル化グアニン誘導体を開示している。
【0006】
PCT/GB02/01636(WO02/083937)には、目的とするタンパク質を検出及び/又は操作する方法が開示されており、ここで、該方法では、タンパク質をAGTに融合させ、得られたAGT融合タンパク質を標識を有しているAGT基質と接触させ、そして、該標識を用いて、該AGT融合タンパク質を検出し、そして、場合により、該AGT融合タンパク質をさらに操作する。使用する数種類のAGT融合タンパク質、AGT基質の構造上の一般的な原則、並びに、該方法において有用な広範な種類の標識及び該標識の検出方法が開示されている。
【0007】
発明の要旨
本発明は、目的とするタンパク質を検出及び/又操作する方法に関し、ここで、該方法では、目的とするタンパク質をAGT融合タンパク質に組み入れ、得られたAGT融合タンパク質を標識を有している特定のAGT基質と接触させ、そして、該標識を認識及び/又は処理するように設計されているシステム内で該標識を用いて、該AGT融合タンパク質を検出し、及び、場合により、さらに操作する。
【0008】
本発明の方法で使用する特定のAGT基質は、O6−置換グアニン誘導体又は関連する窒素含有ヒドロキシ−ヘテロ環及びそれらの硫黄類似体(ここで、該O6−置換基は、グアニン又は対応するヘテロ環からAGTへ移動するのに適する活性化されたメチル誘導体である)であり、さらに、標識を有している。該標識は、同一であるか又は異なっている複数の標識からなるものであってよい。活性化されたメチル誘導体は、例えば、アリール環内で適切に置換されているアリールメチル誘導体、ヘテロアリール環内で適切に置換されているヘテロアリールメチル誘導体、及び、二重結合が適切に置換されているアリルタイプの誘導体などである。アリール環、ヘテロアリール環又はアリル二重結合の適切な置換基は、該アリール環、該ヘテロアリール環又は該アリル基に標識を連結するリンカーであり、好ましくは、さらに修飾され得るか又は切断され得るリンカー、及び、AGT基質を二量体化又は環化するリンカーである。本発明は、さらに、そのような新規AGT基質自体にも関し、また、そのような新規基質の製造方法及びそのような新規AGT基質の合成において有用な中間体にも関する。
【0009】
発明の詳細な説明
本発明では、目的とするタンパク質又はペプチドをO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)に融合させる。目的とするタンパク質又はペプチドは、任意の長さを有するものであることができ、また、二次構造、三次構造若しくは四次構造を有していても又は有していなくてもよい。該タンパク質又はペプチドは、好ましくは、少なくとも12個から2000個以下のアミノ酸からなる。そのような目的とするタンパク質又はペプチドの例は以下で与えられており、例えば、酵素、DNA結合タンパク質、転写調節タンパク質、膜タンパク質、核内受容体タンパク質、核局在化シグナルタンパク質、タンパク質補因子、小単量体GTPアーゼ、ATP結合カセットタンパク質、細胞内構造タンパク質、特定の細胞コンパートメントに対するタンパク質の標的に関与する配列を有するタンパク質、標識又はアフィニティータグとして一般に使用されるタンパク質、並びに、前記タンパク質のドメイン及びサブドメインなどがある。目的とするタンパク質又はペプチドは、好ましくは、酵素により切断され得るリンカー、例えば、DNA段階で適切な制限酵素により切断され得るリンカー(例えば、BglIIにより切断され得るAGATCT)、及び/又は、タンパク質段階で適切な酵素(例えば、タバコエッチウイルスNla(TEV)プロテアーゼ)により切断され得るリンカーを介してAGTに融合させる。融合タンパク質は、原核生物の宿主(好ましくは、E.coli)、又は、真核生物の宿主(例えば、酵母細胞、昆虫細胞若しくは哺乳動物細胞)で発現させることができる。
【0010】
O6−アルキルグアニン−DNAアルキルトランスフェラーゼ(AGT)は、基質上に存在している標識を融合タンパク質の中のAGTを形成している部分に含まれているシステイン残基の1つに転移させる性質を有している。好ましい実施形態では、AGTは、公知ヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)である。マウス形態又はラット形態の酵素も、それらがヒトAGTのような基質と反応する上で同様の性質を有しているかぎり、同様に考慮される。本発明においては、1個以上のアミノ酸の置換、欠失又は付加により異なり得るが、基質上に存在している標識を融合タンパク質内のAGT部分に転移させる性質は維持している野生型AGTの変異体も、O6−アルキルグアニン−DNAアルキルトランスフェラーゼに包含される。AGT変異体は、当業者が周知している技術を用いて化学的に修飾することにより得ることができる。AGT変異体は、好ましくは、当業者に公知のタンパク質工学技術を用いて製造することができるか、及び/又は、分子進化を用いて新しいO6−アルキルグアニン−DNAアルキルトランスフェラーゼを生成及び選択することにより製造することができる。そのような技術は、例えば、飽和突然変異誘発、配列中の何れかに変異を導入する変異導入型PCR、飽和突然変異誘発及び/又は変異導入型PCRの後で用いるDNAシャフリング、又は、幾つかの種から得た遺伝子を用いるファミリーシャフリングなどである。
【0011】
目的とするタンパク質及びO6−アルキルグアニン−DNAアルキル−トランスフェラーゼ(AGT)を含んでいる融合タンパク質を、標識を有している特定の基質と接触させる。反応条件は、AGTが基質と反応して、基質の標識を転移させるように選択する。一般的な条件は、約pH7で室温(約25℃)の緩衝液である。しかしながら、AGTがさまざまな別の反応条件下でも反応すること、及び、本明細書において述べられている反応条件により本発明の範囲が限定されることはないことは理解される。
【0012】
AGTは、その基質であるO6−アルキルグアニン−DNAからそのシステイン残基の内の1つに、アルキル基を不可逆的に転移させる。hAGTと急速に反応する基質類似体は、O6−ベンジル−グアニンであり、二次反応速度定数は、約103秒−1M−1である。O6−ベンジルグアニンをベンジル環のC−4位で置換しても、O6−ベンジルグアニン誘導体に対するhAGTの反応性に対して有意な影響は与えない。これまで、この性質を利用して、ベンジル環のC−4位に結合させた標識をAGTに転移させてきた。
【0013】
当業者は、該融合タンパク質について意図している用途に応じて、基質の標識部分を選定することができる。AGT含有融合タンパク質を基質と接触させた後、標識を共有結合的に融合タンパク質に結合させる。次いで、標識されたAGT融合タンパク質を、転移させた標識に基づいて、さらに操作するか、及び/又は、検出する。該標識は、複数の同一であるか又は異なっている標識から成ることができる。基質が2以上の標識を含んでいる場合、標識された対応するAGT融合タンパク質は、同様に、2以上の標識を含有し、これは、標識された融合タンパク質をさらに操作するか、及び/又は、検出するためのより多くの選択肢を与える。
【0014】
特定のAGT基質は、式1:
【0015】
【化9】
【0016】
[R1−R2は、AGTにより基質として認識される基であり;
Xは、酸素又は硫黄であり;
R3は、芳香族基若しくはヘテロ芳香族基であるか、又は、CH2に結合した二重結合を有する、場合により置換されていてもよい不飽和のアルキル基、シクロアルキル基若しくはヘテロシクリル基であり;
R4は、リンカーであり;
そして、
Lは、1つの標識であるか、同一であるか若しくは異なっている複数の標識であるか、R4をR1に連結して環状基質を形成する結合であるか、又は、さらなる基−R3−CH2−X−R1−R2である]
で表される化合物である。
【0017】
基R1−R2において、残基R1は、好ましくは、AGTにより基質として認識されるヘテロ芳香族基(ここで、該ヘテロ芳香族基1〜5の窒素原子を含んでいる)である。
【0018】
ヘテロ芳香族基R1は、単環式又は二環式であり、5〜12個、好ましくは、6又は9又は10個の環原子を有する。ヘテロ芳香族基R1は、置換基R2を有する他に、置換されていなくてもよいし、又は、以下のものからなる群から選択される1以上の置換基、特に、1、2若しくは3個の置換基で置換されていてもよい:低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ若しくはエトキシ、ヒドロキシ、オキソ、アミノ、低級アルキルアミノ、ジ−低級アルキルアミノ、アシルアミノ、ハロゲン、例えば、塩素若しくは臭素、ハロゲン化低級アルキル、例えば、トリフルオロメチル、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、又は、低級アルキルカルボニル。
【0019】
低級アルキルは、好ましくは、1〜7個、好ましくは、1〜4個のC原子を有するアルキルであり、直鎖又は分枝鎖である。好ましくは、低級アルキルは、ブチル、例えば、n−ブチル、s−ブチル、イソブチル若しくはt−ブチル、プロピル、例えば、n−プロピル若しくはイソプロピル、エチル、又は、メチルである。好ましくは、低級アルキルは、メチルである。
【0020】
低級アルコキシにおいて、低級アルキル基は、上記で定義されているとおりである。低級アルコキシは、好ましくは、n−ブトキシ、t−ブトキシ、イソプロポキシ、エトキシ、又は、メトキシを表し、特に、メトキシである。
【0021】
好ましくは、単環式又は二環式のヘテロ芳香族基R1は、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、8−アザプリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、トリアゾリル、テトラゾリル、又は、ベンゾ[d]ピラゾリルである。さらに好ましくは、単環式又は二環式のヘテロ芳香族基R1は、プリニル、8−アザプリニル、ピリジル、ピラジニル、ピリミジニル及びピリダジニルからなる群から選択される。
【0022】
例えば、基R1−R2は、式2:
【0023】
【化10】
【0024】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
R5は、水素、ハロゲン、例えば、クロロ若しくはブロモ、トリフルオロメチル、又は、ヒドロキシであり;
そして、
R6は、水素、ヒドロキシ、又は、置換されていないか若しくは置換されているアミノである]
で表されるプリンラジカルであることができる。
【0025】
R5又はR6がヒドロキシである場合、上記プリンラジカルは、主として、その互変異性形態(ここで、R5又はR6を有している炭素原子に隣接している窒素は水素原子を有し、この窒素原子とR5又はR6を有している炭素原子の間の二重結合は単結合であり、及び、R5又はR6は、それぞれ、二重結合で結合してる酸素である)で存在している。
【0026】
置換されているアミノ基R6は、1〜4個の炭素原子を有する低級アルキルアミノ又はアシルアミノであり、その際、該アシル基は、1〜5個の炭素原子を有する低級アルキルカルボニル、例えば、アセチル、プロピオニル、n−プロピルカルボニル若しくはイソプロピルカルボニル、n−ブチルカルボニル、イソブチルカルボニル若しくはt−ブチルカルボニル、又は、アリールカルボニル、例えば、ベンゾイルである。
【0027】
R6が置換されていないか又は置換されているアミノであり、且つ、残基Xがプリンラジカルの結合に連結している場合、式2で表されるラジカルは、グアニン誘導体である。
【0028】
1〜10個の炭素原子を有するアルキルとしてのR2は、直鎖又は分枝鎖であり、そのようなR2には、1〜4の炭素原子を有する低級アルキル、例えば、メチル、エチル、ブチル、例えば、n−ブチル、s−ブチル、イソブチル又はt−ブチル、及び、プロピル、例えば、n−プロピル又はイソプロピルなどがある。アルキルとしてのR2は、さらにまた、ペンチル、ヘキシル、ヘプチル、オクチル、ノニル、又は、デシル、例えば、n−ヘキシルであってもよい。
【0029】
糖部分R2は、可変的な長さのスペーサーでグアニン塩基の第N9位に結合している糖単量体又はオリゴマーである。これに関連して、スペーサーは、アルキル鎖(好ましくは、1〜15個の炭素原子を有するアルキル鎖)、1〜200個のエチレングリコール単位からなるポリエチレングリコールスペーサー、アミド基−CO−NH−、エステル基−CO−O−、若しくは、アルキレン基−CH=CH−であるか、又は、アルキル鎖、ポリエチレングリコール基、アミド基、エステル基及び/若しくはアルキレン基の組合せである。
【0030】
本発明との関連において、糖部分R2としては、さらに、β−D−2’−デオキシリボシル、又は、2〜99個のヌクレオチドからなる長さを有する一本鎖オリゴデオキシリボヌクレオチドに組み込まれているβ−D−2’−デオキシリボシルなどがあり、その際、グアニン誘導体R1は、オリゴヌクレオチド配列内の任意の位置を占める。
【0031】
特に好ましいのは、基R1−R2が式2で表されるプリンラジカルであり、R2が水素であり、R5が水素であり、R6が置換されていないアミノであり、且つ、Xが酸素である化合物、即ち、置換されていないグアニン誘導体である。
【0032】
本発明の好ましい別の実施形態では、基R1−R2は、式3:
【0033】
【化11】
【0034】
[式中、置換基R2及びR6は、式2におけるR2及びR6に関して定義されている意味を有する]
で表される8−アザプリンラジカルである。
【0035】
本発明の好ましいさらに別の実施形態では、基R1−R2は、式4:
【0036】
【化12】
【0037】
[式中、
置換基R2は、式2において定義されている意味を有し、好ましくは、水素であり;
そして、
R7及びR8は、いずれも、互いに独立して、水素、ハロゲン、例えば、クロロ若しくはブロモ、1〜4の炭素原子を有する低級アルキル、例えば、メチル、アミノ、又は、ニトロである]
で表されるピリミジンラジカルである。
【0038】
Xは、好ましくは、酸素である。
【0039】
芳香族基若しくはヘテロ芳香族基としてのR3、又は、場合により置換されていてもよい不飽和アルキル基、シクロアルキル基若しくはヘテロシクリル基としてのR3は、AGTにより(その反応機構に従って)、立体的及び電子的に受容され、それにより、R3−R4−L単位が融合タンパク質へと共有結合的に転移するのを可能とする基である。R3−R4−L単位において、R4−Lは、さらにまた、同一であるか又は異なっている複数の標識Lを有している同一であるか又は異なっている複数のリンカーR4を意味し得る。
【0040】
芳香族基としてのR3は、好ましくは、フェニル又はナフチル、特に、フェニル、例えば、パラ位又はメタ位がR4で置換されているフェニルである。
【0041】
ヘテロ芳香族基R3は、0、1、2、3若しくは4個の環窒素原子と0若しくは1個の酸素原子と0若しくは1個の硫黄原子を含み、5〜12個、好ましくは、5若しくは6個の環原子を有する単環式若しくは二環式のヘテロアリール基(但し、少なくとも1の環炭素原子は、窒素原子、酸素原子又は硫黄原子で置き換えられている)であり、ここで、該ヘテロ芳香族基R3は、置換基R4を有している他に、置換されていなくてもよいし、又は、以下のものからなる群から選択される1つ以上の置換基、特に、1つのさらなる置換基で置換されていてもよい:低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ若しくはエトキシ、ハロゲン、例えば、塩素、臭素若しくはフッ素、ハロゲン化低級アルキル、例えば、トリフルオロメチル、又は、ヒドロキシ。
【0042】
好ましくは、単環式又は二環式のヘテロアリール基R3は、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、フラザニル、ベンゾ[d]ピラゾリル、チエニル、及び、フラニルから選択される。さらに好ましくは、該単環式又は二環式のヘテロアリール基は、以下のものからなる群から選択される:ピロリル、イミダゾリル、例えば、1H−イミダゾール−1−イル、ベンゾイミダゾリル、例えば、1−ベンゾイミダゾリル、インダゾリル、特に、5−インダゾリル、ピリジル、例えば、2−ピリジル、3−ピリジル若しくは4−ピリジル、ピリミジニル、特に、2−ピリミジニル、ピラジニル、イソキノリニル、特に、3−イソキノリニル、キノリニル、特に、4−キノリニル若しくは8−キノリニル、インドリル、特に、3−インドリル、チアゾリル、トリアゾリル、テトラゾリル、ベンゾ[d]ピラゾリル、チエニル、及び、フラニル。
【0043】
本発明の特に好ましい実施形態では、該ヘテロアリール基R3は、トリアゾリル、特に、4位若しくは5位にさらなる置換基R4を有している1−トリアゾリル、テトラゾリル、特に、4位若しくは5位にさらなる置換基R4を有している1−テトラゾリル若しくは5位にさらなる置換基R4を有している2−テトラゾリル、イソオキサゾリル、特に、5位にさらなる置換基R4を有している3−イソオキサゾリル若しくは3位にさらなる置換基R4を有している5−イソオキサゾリル、又は、チエニル、特に、3位、4位若しくは5位(好ましくは、4位)にさらなる置換基R4を有している2−チエニル若しくは4位にさらなる置換基R4を有している3−チエニルである。
【0044】
最も好ましいのは、4位又は5位に置換基R4を有しているトリアゾリルとしてのヘテロアリール基R3であり、また、同様に、4位又は5位に置換基R4を有している2−チエニルとしてのR3である。
【0045】
場合により置換されていてもよい不飽和アルキル基R3は、1位若しくは2位(好ましくは、2位)にさらなる置換基R4を有している1−アルケニルであるか、又は、1−アルキニルである。1−アルケニルにおいて考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロである。本発明の特に好ましい実施形態では、R3は、1−アルキニルである。
【0046】
場合により置換されていてもよい不飽和シクロアルキル基は、3〜7個の炭素原子を有し、1位が不飽和であるシクロアルキル基、例えば、任意の位置にさらなる置換基R4を有する1−シクロペンチル又は1−シクロヘキシルである。考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロなどである。
【0047】
場合により置換されていてもよい不飽和ヘテロシクリル基は、3〜12個の原子、窒素、酸素及び硫黄から選択される1〜5個のヘテロ原子、並びに、該ヘテロシクリル基をメチレンCH2に連結させている位置における二重結合を有する。考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロなどである。
【0048】
特に、場合により置換されていてもよい不飽和ヘテロシクリル基は、ヘテロ芳香族基R3に関して上記で定義されている部分的に飽和しているヘテロ芳香族基である。そのようなヘテロシクリル基の例は、イソオキサゾリジニル、特に、5位にさらなる置換基を有している3−イソオキサゾリジニル、又は、3位にさらなる置換基を有している5−イソオキサゾリジニルである。
【0049】
リンカー基R4は、好ましくは、1つの標識L又は同一であるか若しくは異なっている複数の標識Lを基質に結合させる可変性リンカーである。リンカー単位は、想定される用途に照らして、即ち、AGTを含有している融合タンパク質への基質の転移に関連して選択される。それらは、さらに、適切な溶媒中における基質の溶解度も増大させる。使用されるリンカーは、実際に適用する条件下において化学的に安定である。そのようなリンカーは、AGTとの反応を妨害せず、また、標識Lの検出も妨害しないが、例えば、式1の化合物とAGT含有融合タンパク質の反応完了後のある時点で切断されるように構築し得る。
【0050】
リンカーR4は、1〜300個の炭素原子を有する直鎖又は分枝鎖のアルキレン基であり、その際、場合により、
(a)1個以上の炭素原子が酸素により置き換えられており、特に、2つおきの炭素原子が酸素で置き換えられており(例えば、1〜100個のエチレンオキシ単位を有するポリエチレンオキシ基);
(b)1個以上の炭素原子が水素原子を有している窒素で置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、アミド官能基−NH−CO−を表し;
(c)1個以上の炭素原子が酸素により置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、エステル官能基−O−CO−を表し;
(d)隣接してる2つの炭素原子の間の結合が二重結合又は三重結合であって、官能基−CH=CH−又は−C≡C−を表し;
(e)1個以上の炭素原子が、フェニレン、飽和若しくは不飽和のシクロアルキレン、飽和若しくは不飽和のビシクロアルキレン、架橋ヘテロ芳香族、又は、飽和若しくは不飽和の架橋ヘテロシクリル基により置き換えられており;
(f)隣接してる2個の炭素原子がジスルフィド結合−S−S−により置き換えられており;
又は、上記(a)〜(f)で定義されている2以上(特に、2又は3)のアルキレン基及び/又は修飾されているアルキレン基の組合せは、場合により、置換基を含んでいてもよい。
【0051】
考慮される置換基は、例えば、低級アルキル、例えば、メチル、低級アルコキシ、例えば、メトキシ、低級アシルオキシ、例えば、アセトキシ、又は、ハロゲニル、例えば、クロロなどである。
【0052】
考慮されるさらなる置換基は、例えば、α−アミノ酸、特に、天然α−アミノ酸をリンカーR4に組み入れたときに得られる置換基(ここで、炭素原子は、(b)で定義されているアミド官能基−NH−CO−により置き換えられている)などである。そのようなリンカーにおいては、アルキレン基R4の炭素鎖の一部は、基−(NH−CHR−CO)n−(ここで、nは、1〜100であり、Rは、α−アミノ酸の種々の残基を表す)で置き換えられている。
【0053】
さらなる置換基は、光切断可能なリンカーR4、例えば、o−ニトロフェニル基などをもたらす置換基である。特に、この置換基o−ニトロフェニルは、アミド結合に隣接する炭素原子に位置するか(例えば、基−NH−CO−CH2−CH(o−ニトロフェニル)−NH−CO−)、又は、ポリエチレングリコール鎖内の置換基として存在する(例えば、基−O−CH2−CH(o−ニトロフェニル)−O−)。考慮される別の光切断可能なリンカーは、例えば、フェナシル、アルコキシベンゾイン、ベンジルチオエーテル及びピバロイルグリコール誘導体などである。
【0054】
上記(e)で定義されている炭素原子に置き換えられるフェニレン基は、例えば、1,2−フェニレン、1,3−フェニレン、又は、好ましくは、1,4−フェニレンである。。特定の実施形態において、該フェニレン基は、ニトロ基でさらに置換され、また、上記(a)、(b)、(c)、(d)及び(f)において述べられているような別の置き換えと組み合わされ、光切断可能な基を表し、例えば、4−ニトロ−1,3−フェニレン、例えば、−CO−NH−CH2−4−ニトロ−1,3−フェニレン−CH(CH3)−O−CO−、又は、2−メトキシ−5−ニトロ−1,4−フェニレン、例えば、−CH2−O−2−メトキシ−5−ニトロ−1,4−フェニレン−CH(CH3)−O−などである。光切断可能なリンカーを表す特定の別の実施形態は、例えば、−1,4−フェニレン−CO−CH2−O−CO−CH2−(フェナシル基)、−1,4−フェニレン−CH(OR)−CO−1,4−フェニレン−(アルコキシベンゾイン)、又は、−3,5−ジメトキシ−1,4−フェニレン−CH2−O−(ジメトキシベンジル部分)などである。上記(e)で定義されている炭素原子と置き換えられる飽和又は不飽和のシクロアルキレン基は、3〜7個の炭素原子を有するシクロアルキルから、好ましくは、シクロペンチル又はシクロヘキシルから誘導され、例えば、1,2−シクロペンチレン、若しくは、1,3−シクロペンチレン、1,2−シクロヘキシレン、1,3−シクロヘキシレン、若しくは、好ましくは、1,4−シクロヘキシレンなどであるか、又は、さらに、例えば1位若しくは2位が不飽和である1,4−シクロヘキシレンである。上記(e)で定義されている炭素原子と置き換えられる飽和又は不飽和のビシクロアルキレン基は、7又は8個の炭素原子を有するビシクロアルキルから誘導され、例えば、ビシクロ[2.2.1]へプチレン、又は、ビシクロ[2.2.2]オクチレン、好ましくは、場合により2位に不飽和を有するか若しくは2位と5位に二重に不飽和を有していてもよい1,4−ビシクロ[2.2.1]へプチレン、及び、場合により2位に不飽和を有するか若しくは2位と5位に二重に不飽和を有していてもよい1,4−ビシクロ[2.2.2]オクチレンなどである。上記(e)で定義されている炭素原子と置き換えられる架橋ヘテロ芳香族基は、例えば、トリアゾリデン、好ましくは、1,4−トリアゾリデン、又は、イソオキサゾリデン、好ましくは、3,5−イソオキサゾリデンなどである。上記(e)で定義されている炭素原子と置き換えられる飽和若しくは不飽和の架橋ヘテロシクリル基は、例えば、上記R3で定義されている不飽和ヘテロシクリル基から誘導され、例えば、イソオキサゾリジネン、好ましくは、3,5−イソオキサゾリジネンなどであるか、又は、3〜12の原子(そのうちの1〜3は、窒素、酸素及び硫黄から選択されるヘテロ原子である)を有する完全に飽和しているヘテロシクリル基から誘導され、例えば、ピロリジンジイル、ピペリジンジイル、テトラヒドロフランジイル、ジオキサンジイル、モルホリンジイル、若しくは、テトラヒドロチオフェンジイル、好ましくは、2,5−テトラヒドロフランジイル、若しくは、2,5−ジオキサンジイルなどである。考慮される特定のヘテロシクリル基は、糖部分、例えば、α−フラノシル部分若しくはβ−フラノシル部分、又は、α−ピラノシル部分若しくはβ−ピラノシル部分などである。
【0055】
リンカーR4内の環状下部構造は、R4内の回転可能な結合の数で評価した場合に、分子の柔軟性を低下させるが、これにより、良好な膜透過速度が得られる。これは、全てのインビボでの標識化用途にとって重要である。
【0056】
好ましくは、リンカーR4は、場合により−CH=CH−又は−C≡C−基で基R3に結合している、4〜100のエチレンオキシ単位を有する直鎖ポリエチレングリコール基、又は、1〜25個の炭素原子を有する直鎖アルキレン基である。さらに好ましいのは、1〜25個の炭素原子(ここで、炭素原子は場合によりアミド官能基−NH−CO−で置き換えられていてもよい)を有し、光切断可能なサブユニット(例えば、o−ニトロフェニル)を有している直鎖アルキレン基である。さらに好ましいのは、3〜6のエチレングリコール単位を有するポリエチレングリコール基とアルキレン基(炭素原子はアミド結合で置き換えられている)を含み、さらに、置換されているアミノ官能基及びヒドロキシ官能基を有している分枝リンカーである。好ましい別の分枝鎖リンカーは、アルキレン基の炭素原子がアミン、カルボキサミド及び/又はエーテルで置き換えられている樹状構造(木のような構造)を有している。
【0057】
特に好ましいリンカーR4は、3〜12個の炭素原子が酸素で置き換えられており、1又は2の炭素原子がそれぞれ1又は2個の1,4−トリアゾリデン単位で置き換えられており、また、場合により1個の炭素原子が1,4−フェニレン単位で置き換えられている、10〜40個の炭素原子を有する直鎖アルキレン基である。
【0058】
特に好ましい別のリンカーR4は、3〜12個の炭素原子が酸素で置き換えられており、1又は2個の炭素原子が窒素で置き換えられている、場合によりオキソで置換されていてもよい10〜40個の炭素原子を有する直鎖アルキレン基である。
【0059】
特に好ましい別のリンカーR4は、2〜12個の炭素原子が酸素で置き換えられており、2個の隣接する炭素原子の間の1又は2つの結合が二重結合であって、官能基−CH=CH−を表している、6〜40個の炭素原子を有する直鎖アルキレン基である。
【0060】
リンカーR4は、1つ以上の同一であるか又は異なっている標識、例えば、1〜100の同一であるか又は異なっている標識、特に、1〜5つ、好ましくは、1つ、2つ又は3つ、特に、1つ又は2つの同一であるか又は異なっている標識を有し得る。
【0061】
基質の標識部分Lは、意図されている融合タンパク質についての用途に応じて、当業者が選択することができる。標識は、例えば、標識された融合タンパク質が、容易に検出されるか又はその環境から容易に分離されるようなものであり得る。考慮される別の標識は、該標識化融合タンパク質の周囲における変化を探知及び惹起することが可能な標識であるか、及び/又は、該標識によって融合タンパク質に特異的に導入された物理的及び/又は化学的性質により該融合タンパク質を操作するのを助ける標識である。
【0062】
標識Lの例には、分光学的プローブ、例えば、発蛍光団、発色団、磁気プローブ又は造影剤;放射能で標識された分子;結合パートナーに特異的に結合することが可能な特異的結合対の一方である分子;別の生体分子と相互作用すると推測される分子;別の生体分子と相互作用すると推測される分子のライブラリー;別の分子と架橋することが可能な分子;H2O2及びアスコルベートと接触したときにヒドロキシルラジカルを生成することが可能な分子、例えば、テザー金属キレート(tethered metal-chelate);光を照射されたときに反応性ラジカルを生成することが可能な分子、例えば、マラカイトグリーン;固体支持体に共有結合で結合している分子(ここで、支持体は、スライドガラス、マイクロタイタープレート又は当業者に知られている任意のポリマーであり得る);その相補鎖と塩基対を成すことが可能な核酸若しくはその誘導体;膜挿入特性を有する脂質若しくは別の疎水性分子;酵素的、化学的若しくは物理的に望ましい特性を有している生体分子;又は、上記で挙げた特性のいずれかの組合せを有する分子などである。好ましいものは、放射能標識された分子を除く、上記で挙げた標識Lである。標識Lとして最も好ましいのは、分光学的プローブ、及び、結合パートナーに特異的に結合することが可能な特異的結合対の一方である分子(いわゆる、アフィニティー標識)である。
【0063】
標識Lが、発蛍光団、発色団、磁気標識又は放射能標識などである場合、検出は、当該方法をインビトロ又はインビボで使用するかにより、該標識に適合させた標準的な方法による。該方法は、目的とするタンパク質に遺伝的に融合していて生細胞内でのタンパク質の研究を可能とするグリーン蛍光タンパク質(GFP)の適用と比較することができる。さらにまた、標識Lの特定の例は、非線形光学的性質を示すホウ素化合物、又は、標識された基質とAGT融合タンパク質が反応する際にその分光学的性質を変化させるFRET対のメンバーである。
【0064】
標識Lの性質に応じて、目的とするタンパク質及びAGTを含んでいる融合タンパク質を固体支持体に結合させ得る。AGT含有融合タンパク質と反応する基質の標識は、AGTとの反応に入るときには既に固体支持体に結合させておいてもよいし、又は、その後で、即ち、AGTへ転移した後で、AGT含有融合タンパク質と反応する基質の標識を用いてAGT融合タンパク質を固体支持体に結合させてもよい。該標識は、特異的結合対の一方のメンバーであってもよく、その際、該特異的結合対の他方のメンバーは、共有結合又は別の任意の手段により、固体支持体に結合しているか又は結合可能である。考慮される特異的結合対は、例えば、ビオチンとアビジン又はストレプトアビジンなどである。結合対のいずれのメンバーも基質の標識Lとすることが可能であり、その際、他方のメンバーは、固体支持体に結合されていてもよい。固体支持体に都合よく結合することを可能にする標識のさらなる例は、例えば、マルトース結合タンパク質、糖タンパク質、FLAGタグ、又は、固体支持体の表面上に存在している相補的な官能基との間で化学選択的に反応することが可能な反応性置換基である。そのような反応性置換基と相補的な官能基の対の例は、例えば、アミドを形成するアミンと活性化カルボキシ基、1,3−双極子付加環化反応を受けるアジドとプロピオル酸誘導体、活性化ビス−ジカルボン酸誘導体のタイプの添加された二官能性リンカー試薬と反応して2つのアミド結合を生成させるアミンと別のアミン官能基、又は、当業界で知られている別の組合せなどである。
【0065】
都合のよい固体支持体の例は、例えば、ガラス表面、例えば、スライドガラス、マイクロタイタープレート、及び、適切なセンサーエレメント、特に、官能化ポリマー(例えば、ビーズ形態にある官能化ポリマー)、化学的に修飾された酸化物表面、例えば、二酸化ケイ素、五酸化タンタル若しくは二酸化チタン、又は、化学的に修飾された金属表面、例えば、貴金属表面、例えば、金若しくは銀の表面などである。不可逆的に結合及び/又はスポットするAGT基質を用いて、AGT融合タンパク質を、空間的に分離したやり方で、特に、スポットにより、固体支持体上に結合させることができ、これらは、タンパク質マイクロアレイ、DNAマイクロアレイ又は小分子のアレイに相当する。
【0066】
標識Lが、外部刺激に晒されたときに反応性ラジカル(例えば、ヒドロキシルラジカル)を生成し得る場合、生成されたラジカルは、AGT融合タンパク質及びAGT融合タンパク質に近接しているタンパク質を不活性化することができ、これは、これらのタンパク質の役割についての研究を可能にする。そのような標識の例は、H2O2及びアスコルベートと接触したときにヒドロキシルラジカルを生成するテザー金属キレート錯体(tethered metal-chelate complex)、及び、レーザーを照射されたときにヒドロキシルラジカルを生成する発色団、例えば、マラカイトグリーンなどである。ヒドロキシルラジカルを生成させるために発色団とレーザーを使用することは、当技術分野では、発色団アシストレーザー分子機能不活化(chromophore assisted laser induced inactivation)(CALI)として知られている。本発明では、発色団(例えば、マラカイトグリーン)を用いてAGT融合タンパク質を標識した後、レーザーを照射して、時間を制御し且つ空間的に分離したやり方で、AGT融合タンパク質を不活性化すると共にAGT融合タンパク質と相互作用するタンパク質を不活性化する。この方法は、インビボ又はインビトロの両方に適用可能である。さらに、AGT融合タンパク質に近接しているタンパク質は、該タンパク質のフラグメントを特異的抗体により検出することにより、又は、高分解2D−電気泳動ゲル上における該タンパク質の消失により、又は、分離及び配列決定法(例えば、質量分析法又はN−末端分解によるタンパク質配列決定法など)による切断されたタンパク質フラグメントを同定することにより、同定することができる。
【0067】
標識Lが、別のタンパク質と架橋可能な分子、例えば、官能基(例えば、マレイミド、活性エステル又はアジド及び当業者に知られている別のものなど)を含んでいる分子である場合、そのような標識されたAGT基質を別のタンパク質と相互作用するAGT融合タンパク質と接触させる(インビボ又はインビトロ)ことにより、AGT融合タンパク質をそれが相互作用するタンパク質と該標識を介して共有結合的に架橋することができる。これにより、AGT融合タンパク質と相互作用するタンパク質を同定することが可能となる。光活性化架橋のための標識Lは、例えば、ベンゾフェノン類である。架橋の特定の態様において、標識Lは、それ自体がAGT基質である分子であり、それにより、AGT融合タンパク質が二量体化される。そのような二量体の化学的構造は、対称(ホモ二量体)又は非対照(ヘテロ二量体)であり得る。
【0068】
考慮される別の標識Lは、例えば、フラーレン類、中性子捕獲処理のためのボラン類、例えばセルフアドレッシングチップ(self-addressing chip)用のヌクレオチド類若しくはオリゴヌクレオチド類、ペプチド核酸類、及び、金属キレート類、例えば、DNAに特異的に結合する白金キレートなどである。
【0069】
望ましい酵素的性質、化学的性質又は物理的性質を有している特定の生体分子は、メトトレキサートである。メトトレキサートは、酵素であるジヒドロ葉酸レダクターゼ(DHFR)の強結合阻害剤である。式中のLがメトトレキサートである式1の化合物は、いわゆる「二量体化誘発物質(chemical inducers of dimerization)」(CID)と称されるよく知られている種類に属する。hAGTとDNA結合ドメインLexAの融合タンパク質を使用し、式1(式中、Lはメトトレキサートである)の化合物によるhAGT融合タンパク質のインビボ標識化に、転写活性化ドメインB42を有するDHFRを添加することにより、hAGT−LexA融合タンパク質とDHFR−B42融合タンパク質のカップリング(「二量体化」)が誘発され、その結果、図4に示されているように、LexAとB42が空間的に近接し、その後、転写が刺激される。
【0070】
基質が2つ以上の標識を有している場合、これら標識は、同一でもよく、又は、異なっていてもよい。特に好ましい組合せは、2つの異なったアフィニティー標識、又は、1つのアフィニティー標識と1つの発色団標識、特に、1つのアフィニティー標識と1つの発蛍光団標識である。
【0071】
本発明は、さらに、インビボ及びインビトロの両方において、AGT融合タンパク質を標識する方法を提供する。AGT融合タンパク質のインビボ標識化という用語には、細胞の全てのコンパートメントにおける標識化、及び、細胞外空間に面しているAGT融合タンパク質の標識化が包含される。AGT融合タンパク質の標識化がインビボで行われ且つAGTに融合しているタンパク質が膜タンパク質(より特定的には、細胞膜タンパク質)である場合、該融合タンパク質内のAGT部分は、該膜のいずれの面にも結合させることが可能であり、例えば、細胞質面又は細胞膜の細胞外の面に結合させることが可能である。
【0072】
標識化をインビトロで行う場合、融合タンパク質の標識化は、細胞抽出物中で、又は、AGT融合タンパク質の精製された形態若しくは濃縮された形態で行うことができる。
【0073】
標識化をインビボ又は細胞抽出物中で行う場合、宿主の内因性AGTの標識化を考慮に入れるのが有利である。宿主の内因性AGTが、O6−アルキルグアニン誘導体又はその関連する化合物を基質として受容しない場合、該融合タンパク質の標識化は、特異的である。哺乳類細胞、例えば、ヒト、マウス又はラットの細胞においては、内因性AGTの標識化が可能である。内因性AGTとAGT融合タンパク質を同時に標識化すると問題が生じるような実験においては、公知のAGT欠失細胞系を使用することができる。
【0074】
特定の態様において、本発明は、候補化合物又は候補化合物のライブラリーと標的タンパク質又は標的タンパク質のライブラリーの相互作用を測定する方法を提供する。候補化合物と標的タンパク質の例には、リガンドとタンパク質、薬物と該薬物の標的、又は、小分子とタンパク質などがある。本発明のこの特定の方法において、AGTに融合させる目的とするタンパク質は、転写因子のDNA結合ドメイン又は転写因子の活性化ドメインを含んでいる。物質又はタンパク質のライブラリーの推定されるタンパク質標的を、機能的転写因子が形成され得るような方法で、転写因子のDNA結合ドメイン又は活性化ドメインのいずれかに結合させ、本発明によるAGT基質の標識Lは、1種又は複数の標的物質と相互作用すると推定される候補化合物又は候補化合物のライブラリーである。基質の一部分である候補化合物又は候補化合物のライブラリーを、次いで、AGT融合タンパク質に転移させる。転移後は、1種又は複数の標的物質を含んでいる1種又は複数のAGT融合タンパク質は、1種又は複数の候補化合物で標識されている。AGT融合タンパク質に連結させた候補化合物とDNA結合ドメイン又は活性化ドメインのいずれかに融合させた標的タンパク質を相互作用させることにより、機能的転写因子が形成される。活性化された転写因子は、次に、レポーターを発現させることができる。このレポーターは、該方法を細胞内で行う場合、レポーターの発現が細胞に選択有利性をもたらす場合には検出される。
【0075】
特定の実施形態では、該方法は、1以上のさらなるステップ、例えば、検出ステップ、単離ステップ、1種若しくは複数の候補化合物を同定若しくは特性付けするステップ、又は、1種若しくは複数の標的物質を同定若しくは特性付けするステップなどを含み得る。
【0076】
特定の例では、標識Lは、薬物であるか又は未だ同定されていないタンパク質Yに結合する生物学的に活性な小分子である。未知の標的タンパク質Yを発現することが期待される生物のcDNAライブラリーを転写因子の活性化ドメインに融合させ、AGTを転写因子のDNA結合ドメインに融合させる。上記のような標識Lを含んでいる本発明のAGT基質を加えることにより、機能的転写因子が形成され、この小分子が、cDNAライブラリー内に存在し且つ活性化ドメインに融合したその標的タンパク質Yに結合したときにのみ、遺伝子が発現する。遺伝子発現を選択有利性に結びつけると、該薬物又は生理活性分子の標的タンパク質Yをコードする遺伝子を有しているプラスミドを保持している対応する宿主を同定することができる。
【0077】
さらに別の特定の例では、標識Lは、化学分子のライブラリーである。該ライブラリーは、インビボ条件下で既知の薬物標的タンパク質Yに結合する未だ同定されていない化合物を含んでいることが期待される。標的タンパク質Yを転写因子の活性化ドメインに融合させ、AGTを転写因子のDNA結合ドメインに融合させる。化学的化合物のライブラリーを含んでいる基質を添加することにより、機能的転写因子が形成され、該標識(即ち、該化学的ライブラリー内の化合物)が活性化ドメインに融合したその標的タンパク質Yに結合したときにのみ、遺伝子が発現する。遺伝子発現を選択有利性に結びつけると、宿主の成長をもたらすライブラリーの分子を同定することができる。
【0078】
Lが、R4とR1を連結して環状基質を形成させる結合である場合、好ましい化合物は、R4からR1への結合が、リンカーR4を式2で定義されているアミノ基R6に連結させる結合である環状基質である。そのような好ましい環状基質において、R2は、好ましくは、オリゴヌクレオチド、即ち、上記で詳述した2〜99のヌクレオチドの長さを有する一本鎖オリゴデオキシリボヌクレオチドに組み入れられているβ−D−2’−デオキシリボシルである。このオリゴヌクレオチドは、検出され得ることで標識として機能するように、さらに化学的に修飾することができる。置換基のそのような化学的修飾は、標識Lについて上記で述べたのと同じ種類であり得る。
【0079】
Lが、さらなる基−R3−CH2−X−R1−R2である場合、該基質は二量体化合物であり、その結果、AGT含有融合タンパク質と反応させると、二量体化融合タンパク質が得られる。残基−R3−CH2−X−R1−R2としてのサブユニットLにおいて、R1、R2、R3及びXの意味は、別の基R2−R1−X−CH2−R3−内の対応する意味と同一であり得る(ホモ二量体)か、又は、異なり得る(ヘテロ二量体)。
【0080】
最も好ましいのは、スキームにおいて示され、及び、実施例において記載されている式1の化合物である。
【0081】
新規基質及び中間体を製造する方法も、同様に、本発明の目的である。これらの方法は、概して当技術分野において知られており、また、本発明の好ましい基質が最もよく生成されるように選択され、また、下記において例示されている。特に、本発明は、式:R2−R1−Y(ここで、Yは、反応性脱離基、例えば、アンモニウム塩などである)で表される化合物を、式:HX−CH2−R3−R4−L又はその前駆物質で処理し、得られた化合物を、例えばリンカーR4を構築すること及び/又は標識Lを導入することにより、さらに操作する方法に関する。あるいは、式:R2−R1−XHで表される化合物を、式:Y−CH2−R3−R4−L(ここで、Yは、脱離基、例えば、ハロゲンである)で表される化合物又はその前駆物質でアルキル化し、得られた化合物を、例えば、残基R3及び/若しくはリンカーR4を構築することにより、並びに/又は、標識Lを導入することにより、さらに操作する。
【0082】
特に、本発明は、1以上の標識を有する新規基質を製造する方法に関する。該合成は、式:R2−R1−X−CH2−R3−R4(ここで、R4は、2以上の反応性求核基又は求電子基を有する多官能性残基である)で表されるコア化合物を介して進行させる。そのような基の例には、ヒドロキシ、アミノ、スルフヒドリル、ハロ又はカルボキシなどがある。反応性基のさらなる例は、マイケルアクセプター(Michael acceptor)、例えば、マレイミド又はビニルスルホンなどである。さらに、付加環化反応に関与し得る官能基も挙げることができ、そのような官能基は、例えば、ジエン類又はジエノフィル類、アジド類、ニトロン類、ニトリルオキシド類、アセチレン類及びアルケン類などである。
【0083】
適切な標識を結合させるために、残基R4上に、特に、分枝鎖残基の外圏に配置し得る特定の官能基は、それらが互いに反応し得るか又はそれらが本発明の基質を構築するのに使用される任意の化合物若しくは試薬と反応し得るということが想定されないという要件によってのみ制限される。従って、それらの選定は、望ましい基質を構築するために選択される特定の試薬に依存する。分枝鎖残基R4の外圏に配置することが可能な官能基の例には、フルオロ、クロロ、ブロモ、シアノ、ニトロ、アミノ、アルキルカルボニルアミノ、アリールカルボニルアミノ、アルコキシカルボニルアミノ、アリールオキシカルボニルアミノ、カルボキシ、カルバモイル、アルコキシカルボニル、アリールオキシカルボニル、カルバルデヒド、ヒドロキシ、アルコキシ、アリールオキシ、アルキルカルボニルオキシ、アリールカルボニルオキシ、炭素−炭素二重結合、及び、炭素−炭素三重結合などである。最も好ましい例としては、アミノ、ヒドロキシ、シアノ(潜在的なカルボキシ官能基)、カルバモイル、カルバルデヒド、又は、炭素−炭素二重結合などを挙げることができる。
【0084】
特に、複数の標識Lを有している分枝鎖R4の好ましい合成は、オルソゴナルに保護されている(orthogonally protected)官能基を用いる。保護基をそのように選択することにより、別々に脱保護することが可能となり、それによって、遊離された各官能基を次に化学的にさらに操作して、それに標識を結合させることができるか、又は、リンカーR4のさらなる拡大が可能となる。当業者は、想定される官能性についての適切な保護基を選定することが可能であり、そのような保護基は、例えば、以下の文献に要約されている:T.W.Greene 及び P.G.M.Wuts “Protective Groups in Organic Synthesis”,John Wiley & Sons,New York 1991。
【0085】
本発明による好ましい中間体は、式中のR1−R2が式2(ここで、R2は水素であり、R5は水素であり、R6は置換されていないアミノである)で表されるラジカルであり;Xが酸素であり;R3がトリアゾリル、テトラゾリル、イソオキサゾリル、チエニル、イソオキサゾリジニル又はアルキニルであり;R4がリンカーであり;及び、Lがアミノ又はアジド(特に、アミノ)である式1で表される化合物である。
【0086】
本発明による好ましい別の中間体は、式中のR1−R2が式2(ここで、R2は水素であり、R5は水素であり、R6は置換されていないアミノである)で表されるラジカルであり;Xが酸素であり;R3は1,4−フェニレンであり;R4は場合によりオキソで置換されていてもよい10〜40の炭素原子を有する直鎖アルキレン基(ここで、12以下の炭素原子は酸素で置き換えられており、0、1又は2の炭素原子は窒素で置き換えられている)であり;及び、Lがアミノ又はアジド(特に、アミノ)である式1で表される化合物である。
【0087】
最も好ましい中間体は、スキーム及び実施例に記載されている中間体である。
【0088】
式中のR3がテトラゾリル基、イソオキサゾリル基又はイソオキサゾリジニル基である式1の化合物の合成において有用な中間体の合成については、スキーム1及びスキーム2に要約してある。
【0089】
アジド化合物7は、市販されているテトラエチレングリコール5から、メシル化(メタンスルホニルクロリド、トリエチルアミン)した後、エタノール中でアジ化ナトリウムと反応させることにより調製する。7を再度メシル化した後、Gabrielアミン合成に付すことにより、アジド−アミン9を得る(Carolayら,J.Org.Chem.56:4326−4329,1991)。アジド9とアセチレン誘導体10の間でCu(I)を触媒として用いる1,3−双極付加環化(Griffinら,J.Med.Chem.43:4071−4083,2000)を行うことにより、1,4−置換トリアゾール11を得る。あるいは、アジド9とシアノ誘導体12をルイス酸(ZnBr2)の触媒作用下で反応させて、テトラゾール13を形成させる(スキーム1)。
【0090】
【化13】
【0091】
アジド7をSwerns酸化(塩化オキサリル、DMSO、トリエチルアミン)により中心的なビルディングブロック(アルデヒド14)に変換する。14をヒドロキシルアミン誘導体と反応させることにより、ニトロン17が生成され、これをアセチレン誘導体10と反応させることにより、イソオキサゾリジン類18が形成される。アルデヒド14から、オキシム15が異性体の等モル混合物として形成される。次亜塩素酸ナトリウムで酸化することにより対応するニトリルオキシドがインシトゥで形成され、次いで、10と反応させることにより、イソオキサゾール16が生成する(スキーム2)。
【0092】
【化14】
【0093】
式中のR3がチエニル基である式1の化合物の合成において有用な中間体の合成については、スキーム3に要約してある。
【0094】
【化15】
【0095】
市販されているテトラエチレングリコール5を、強塩基条件下で1当量のヨウ化アリルと反応させることにより単官能化して22を生成させ、これを、さらに、ジメトキシトリチル(DMT)で保護して23とする。この中間体は、パラジウムを触媒として使用するチオフェン誘導体21とのスズキカップリングに付すことができ、それにより、完全に保護された化合物25が得られる。
【0096】
DMT基をモノ脱保護した後、メシル化し(メタンスルホニルクロリド、トリエチルアミン)、その後、エタノール中でアジ化ナトリウムと反応させることにより、保護されたアジドを得る。これをHF/ピリジンで脱保護することにより26を得る。遊離ヒドロキシ基を活性化グアニン−カチオン27とカップリングさせることにより、アジド−中間体が得られるが、これは、別の官能化方策のための前駆物質として使用することができる。最後に、前記アジドを還元してアミン28とすることにより、標識単位Lを導入することが可能であるか、又は、別の表面に結合させることができる。
【0097】
式中のR3がフェニレン基である式1の化合物の合成において有用な中間体の合成については、スキーム4に要約してある。
【0098】
【化16】
【0099】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4が2つのトリアゾリル−4−メトキシ単位とテトラエチレンオキシ単位から構成される単位であり、及び、Lが−R3−CH2−X−R1−R2である式1の化合物は、スキーム5に示してあるようにして調製する。
【0100】
【化17】
【0101】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4がトリアゾリル−4−メトキシキをさらに有しているテトラエチレンオキシ単位であり、及び、Lが−R3−CH2−X−R1−R2である式1の化合物は、スキーム6、スキーム7及びスキーム8に示してあるようにして調製する。
【0102】
【化18】
【0103】
【化19】
【0104】
【化20】
【0105】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4が6−アミノカプロイル基をさらに有するテトラエチレンオキシ単位であり、及び、Lが2種類の異なったアフィニティー標識の組合せを表している式1の化合物の合成については、スキーム9〜スキーム11に例示してある。
【0106】
トリ−オルソゴナル保護基戦略(tri-orthogonal protecting group strategy)のために、ジアゾ転移法(diazo transfer method)により、ε−N−Fmoc−リシン47を対応するα−アジド−ε−N−Fmoc−リシン48に変換する(Lundquistら,J.Org.Lett.3:781−783,2001)。α−アジド−ε−N−Fmoc−リシンを、標準的なペプチドカップリング条件下で、適切な官能化アミノ−ベンジルグアニン誘導体(例えば、49)と縮合させることにより、完全に保護された分子骨格50が得られる。水性1,4−ジオキサン中で、中性条件下、α−アジド基をトリメチルホスフィンで還元して対応するアミン51とすることにより、種々の異なったビルディングブロック(例えばアフィニティー標識を含んでいるもの)と縮合させるのに適する化合物が得られる。例えば、N−(+)−ビオチン−6−アミノ−カプロン酸N−スクシンイミジルエステルとカップリングさせることにより、52が得られ、その後、ジエチルアミンでFmocを除去することにより、遊離アミノ基を有する中間体53が生成される。これを用いて、さらに別の標識を中心の単位にカップリングさせることができる。53の前記遊離アミン基にジゴキシゲニン−3−O−メチルカルボニル−ε−アミノカプロン酸−N−ヒドロキシ−スクシンイミドエステルをカップリングさせることにより、二重に標識された基質54が得られる(スキーム9)。
【0107】
【化21】
【0108】
【化22】
【0109】
【化23】
【0110】
二重に標識された基質54の別法による合成に関し、別のオルソゴナル保護戦略(orthogonal protection strategy)についてスキーム10で説明する。α−N−Fmoc−ε−N−Dde−リシン55を、標準的なペプチドカップリング条件下、適切な官能化アミノ−ベンジルグアニン誘導体(例えば、49)と縮合させることにより、完全に保護された分子骨格56が得られる。ジエチルアミンでFmocを除去した後、N−(+)−ビオチン−6−アミノカプロン酸N−スクシンイミジルエステルとカップリングさせることにより、中間体58が得られる。4%ヒドラジン溶液を用いて残留しているDde保護基を切断することで第二のアミノ基を遊離させ、それにより、化合物53が得られる。これは、さらに、誘導体化することができる。
【0111】
【化24】
【0112】
スキーム11は、標識の異なった組合せを有しているベンジルグアニン誘導体のさらに2つの例の合成を示している(59における2つのアフィニティー標識ビオチン及びジニトロフェニル、又は、60における発蛍光団と組み合わせたアフィニティー標識)。53から出発して(スキーム9を参照されたい)、2,4−ジニトロフルオロベンゼンを遊離アミノ基にカップリングさせることにより、59が得られる。53内の遊離アミノ基にフルオレセイン−5(6)−カルボキサミドカプロン酸N−スクシンイミジルエステルをカップリングさせることにより、60が得られる。
【0113】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4が尿素官能基を介して結合している二量体化テトラエチレンオキシ単位であり、及び、Lが−R3−CH2−X−R1−R2である式1のさらに別の化合物は、スキーム12に示されているように調製する。
【0114】
【化25】
【0115】
式中のR1がグアニンであり、R2が水素であり、R3がプロパルギルであり、R4がさらに6−アミノカプロイル基を含んでいるテトラエチレンオキシ単位であり、及び、Lがビオチンである式1の化合物の合成は、スキーム13及びスキーム14に示されている。
【0116】
【化26】
【0117】
【化27】
【0118】
別のタンパク質と相互作用可能な合成リガンドでの融合タンパク質の共有結合的な標識化に基づいて生細胞内でタンパク質の二量体化を誘導する小分子として作用する標識Lとしての酵素阻害薬を用いたベンジルグアニン誘導体の合成は、スキーム15及びスキーム16に示されている。特に、式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4がω−アミノ−ドデカノイルアミノ−メチル残基であるか又はグルタミン酸単位をさらに含んでいるテトラエチレンオキシ単位であり、及び、Lがメトトレキサートである式1の化合物の合成に関して記載されている。メトトレキサートは、酵素ジヒドロ葉酸レダクターゼの強結合阻害剤であり、これらのヘテロ二量体構造は、いわゆる「二量体化誘発物質」(CID)と称されるよく知られている種類の化合物に属する。
【0119】
【化28】
【0120】
【化29】
【0121】
式中のR1がグアニンであり、R2が水素であり、R3が1,4−フェニレンであり、R4がブト−2−エン−1,4−ジオール誘導体であり、及び、Lが−R3−CH2−X−R1−R2である式1の化合物は、スキーム17及びスキーム18に示されているように調製する。
【0122】
【化30】
【0123】
【化31】
【0124】
O6−ベンジルグアニン誘導体を溶解させるのに必要な極性溶媒は、オレフィン複分解触媒ベンジリデン−ビス(トリシクロヘキシルホスフィン)−ジクロロルテニウムとは適合しないと考えられる。従って、最初に、保護された対称性コア単位81及び85を構築し、脱保護した後、最終的なビス−ベンジルグアニン82及び86を合成する。
【0125】
本発明の特に好ましい化合物は、スキーム1〜スキーム18に示されている化合物及び実施例に記載されている化合物である。
【0126】
実施例
実施例1:AGT基質を共有結合により結合させるためのスライドガラスの調製、及び、それに続く、タンパク質マイクロアレイを調製するためのAGT融合タンパク質の共有結合による固定化
市販されている顕微鏡スライドガラス(SiO2)を、超音波浴内で、ジクロロメタン、アセトン、H2O2/H2SO4を用いて充分に清浄化し、さらに、2回蒸留させた蒸留水で充分に洗浄した。それを、公表されている手順に従い、エタノール/水(95:5)の溶媒混合物中で3−アミノプロピルトリエトキシシランを用いて、1時間、アミノシリル化した後、ジスクシンイミジルグルタレート(10mM)をジクロロメタン/ジイソプロピルエチルアミン(100:1)に溶解させた溶液で、アルゴン下、室温で2時間処理すした。該表面を、ジクロロメタンで数回洗浄した。活性化カルボキシ官能基を保持しているガラス表面を、標識Lの代わりに遊離アミノ基を有している式1の化合物(例えば、化合物11、化合物13、化合物16、化合物18、化合物28、化合物34、化合物49、化合物68又は化合物73)を10mMの濃度でメタノールに溶解させトリエチルアミンを添加した溶液と一緒に、4時間インキュベーションした。スライドガラスをメタノールで3回以上洗浄することにより、対応するAGT−基質がアミド結合により共有結合的に結合した表面が得られた。得られたスライドガラスをさらに使用する際に副反応が起こるのを避けるために、全ての未反応スクシンイミジル基を、6−アミノヘキサノール(DMF中100mM)を添加することによりクエンチした。
【0127】
実施例2:11−アジド−3,6,9−トリオキサウンデカノール(7)及び1,11−ジアジド−3,6,9−トリオキサウンデカン(36)
50.0g(260mM)のテトラエチレングリコールと50mLのトリエチルアミンを200mLの乾燥ジエチルエーテルに溶解させた溶液を、アルゴン雰囲気下、0℃に冷却し、15.0g(130mM)のメタンスルホニルクロリドを3時間かけて添加して室温で20分間撹拌した。減圧下に溶媒を除去し、300mLの95%エタノール及び18.0g(280mM)のアジ化ナトリウムを添加した。得られた混合物を24時間加熱還流し、室温まで冷却し、減圧下に濃縮した。残留した混合物を400mLのジクロロメタンで希釈し、ブラインで洗浄し、MgSO4で脱水した。減圧下に濃縮した後、モノアジドとジアジドの粗混合物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 3:1)で精製することにより、15.03g(68.5mmol,26%)のモノアジド及び3.46g(14.18mmol,5.5%)のジアジドを得た。
【0128】
実施例3:1−アジド−11−フタルイミド−3,6,9−トリオキサウンデカン(8)
1.17g(5.35mmol)の11−アジド−3,6,9−トリオキサウンデカノール(7)と1.2mLのトリエチルアミンを35mLの塩化メチレンに溶解させた溶液を0℃に冷却し、シリンジを用いて0.5mL(6.45mmol)のメタンスルホニルクロリドを20分間かけて滴下して加えた。得られた混合物を室温まで昇温させ、1.5時間撹拌した。次いで、その混合物を、10mLの飽和水性NaHCO3で2回洗浄し、5mLの水で3回洗浄した。有機層を減圧下に乾燥濃縮して、1.5gの8を黄色の油状物として得た。これは、それ以上精製することなく、使用した。
【0129】
実施例4:4−ブロモテニル(bromothenyl)アルコール(20)
5.0g(26.17mmol)の4−ブロモチオフェン−2−カルボキシアルデヒド(19)を75mLのイソプロパノールに溶解させ、1.11g(29.31mmol)のNaBH4を一度に添加し、得られた混合物を2時間撹拌した。20mLの飽和水性NH4Clを添加し、濾過により固体を除去し、得られた混合物を減圧下に濃縮した。生成物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 10:1)により精製して、4.64gの20(24.07mmol,92%)を無色の固体として得た。
【0130】
実施例5:4,7,10,13−テトラオキサ−1−ペンタデセン−15−オール(22)
2.3g(19.5mmol)のカリウムt−ブトキシドを500mLの乾燥THFに溶解させ、7.18g(37mmol)のテトラエチレングリコールを滴下して加えた。30分間撹拌した後、3.31g(19.7mmol)のヨウ化アリルを60mLの乾燥THFに溶解させた溶液を1時間かけて添加し、撹拌を24時間継続した。得られた粗混合物をシリカゲルで濾過し、減圧下に溶媒を除去した。生成物をシリカゲルクロマトグラフィー(勾配:石油エーテル/酢酸エチル 10:1 → 酢酸エチル)により精製して、2.41gの22(10.3mmol,27%)を無色の液体として得た。
【0131】
実施例6:1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27)
1.0g(5.9mmol)の6−クロログアニンを40℃で40mLのDMFに溶解させた。室温まで冷却した後、1.4mLの1−メチルピロリジン(13.2mmol)を添加し、得られた反応混合物を18時間撹拌した。2mLのアセトンを添加して、沈澱を完了させた。固体を濾過し、エーテルで洗浄し、減圧下に乾燥させて、1.03gの27(3.9mmol,66%)を得た。
【0132】
実施例7:O6−(4−アミノメチル−ベンジル)グアニン(32)
(a)4−アミノメチル−ベンジルアルコール: 2.83gのLiAlH4(74.5mmol)を150mLの乾燥エーテルに懸濁させ、冷却しながら、1.9mLのH2SO4(100%,37.2mmol)を滴下して加えた。得られた混合物を室温で1時間撹拌した後、12mLのエーテル中の2.0g(12.4mmol)の4−シアノベンゾエートを滴下して加えた。2時間還流した後、20mLの水とそれに続く60mLの水中の7.4gのNaOHにより反応物をクエンチした。有機層をデカントし、水層をエーテルと酢酸エチルで抽出した。有機層をMgSO4で脱水し、溶媒を除去し、生成物を減圧下に乾燥させて、0.92g(6.7mmol,54%)を得た。
【0133】
(b)2,2,2−トリフルオロ−N−(4−ヒドロキシメチル−ベンジル)−アセトアミド: 866mg(6.3mmol)の4−アミノメチル−ベンジルアルコールと880μL(6.3mmol)のトリエチルアミンを10mLの乾燥メタノールに溶解させた溶液に、980μL(8.2mmol)のトリフルオロ酢酸エチルエステルを滴下して加えた。得られた反応混合物を45分間撹拌し、10mLの酢酸エチルと10mLの水で希釈した。水層を酢酸エチルで希釈し、有機層を合して飽和NaClで洗浄し、Na2SO4で脱水した。減圧下に溶媒を除去した後、粗生成物をフラッシュカラムクロマトグラフィー(酢酸エチル/シクロヘキサン 1:2)で精製した。収量:1.32g(5.7mmol,90%)。
【0134】
(c)N−[4−(2−アミノ−7H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロアセトアミド: 592mg(2.54mmol)の2,2,2−トリフルオロ−N−(4−ヒドロキシメチル−ベンジル)アセトアミドを、アルゴン雰囲気下、乾燥DMFに溶解させ、599mg(5.33mmol)のカリウムt−ブトキシドを添加した。次いで、300mg(1.18mmol)の1−(2−アミノ−7H−プリン−6−イル)−1−メチルピロリジニウムクロリド(27)を添加し、この溶液を3時間撹拌した。減圧下に溶媒を除去した後、粗生成物をフラッシュカラムクロマトグラフィー(300mL メタノール/ジクロロメタン 1:50,500mL メタノール/ジクロロメタン 1:10)で精製した。収量:382mg(1.04mmol,88%)。
【0135】
(d)O6−(4−アミノメチル−ベンジル)グアニン(32): 335mg(0.91mmol)のN−[4−(2−アミノ−7H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロアセトアミドを34mLのメタノールと2mLの水に懸濁させた。656mg(4.75mmol)のK2CO3を添加した後、得られた反応混合物を2時間還流した。減圧下に溶媒を除去し、生成物をフラッシュカラムクロマトグラフィー(メタノール/トリエチルアミン/ジクロロメタン 1:0.05:5)で精製した。収量:209mgの32(0.77mmol,85%)。
【0136】
実施例8:O6−(4−[プロプ−2−イニルオキシメチル]−ベンジル)グアニン(35)
662mg(3.8mmol)の4−(プロプ−2−イニルオキシメチル)−ベンジルアルコール(39)を3mLの乾燥DMSOに溶解させ、61mgのNaHを5分間かけて少量ずつ添加した。300mg(1.27mmol)の1−(2−アミノ−7H−プリン−6−イル)−1−メチルピロリジニウムクロリド(27)を添加し、得られた混合物をさらに4時間撹拌した。反応物を0.2mLの酢酸でクエンチし、蒸発乾固させ、フラッシュカラムクロマトグラフィー(勾配:ジクロロメタン/メタノール 50:1 → 10:1)により精製して、188mgの35(0.61mmol,53%)を得た。
【0137】
実施例9:ホモ−O6−ベンジルグアニン−二量体37
50.0mg(0.162mmol)のO6−(4−[プロプ−2−イニルオキシメチル]−ベンジル)グアニン(35)と19.7mg(0.081mmol)の1,11−ジアジド−3,6,9−トリオキサウンデカン(36)を0.5mLのDMFに溶解させた溶液に、15.43mg(0.081mmol)のCu2Cl2を0.15mLの水に懸濁させた懸濁液を添加した。得られた混合物を室温で24時間撹拌した。
【0138】
実施例10:4−(プロプ−2−イニルオキシメチル)ベンジルアルコール(39)及び1,4−ビス−(プロプ−2−イニルオキシ−メチル)ベンゼン(40)
2.5g(18.1mmol)の4−ヒドロキシメチルベンジルアルコール(38)の溶液に、477.5mg(19.9mmol)のNaHを20分間かけて少量ずつ添加した。2.15mLの臭化プロパルギル溶液(トルエン中80%)を滴下して加え、15時間撹拌した。得られた混合物に100mLの水を添加し、生成物をジエチルエーテルで抽出した。相を合して乾燥させ、減圧下に溶媒を除去した。生成物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 4:1)により分離して、1.08gの39(6.17mmol,34%)及び1.05gの40(4.94mmol,27%)を得た。
【0139】
実施例11:4−[(t−ブチルジメチルシリルオキシ)メチル]ベンジルアルコール(44)
810mg(33.77mmol)のNaHを室温で90mLの乾燥THFに懸濁させ、4.2g(30.39mmol)の固形4−ヒドロキシメチル−ベンジルアルコール(38)を3回に分けて5分間かけて添加した。得られた反応混合物を45分間撹拌した。4.83g(32.08mmol)のt−ブチルジメチルシリルクロリドを3回に分けて5分間かけて添加し、さらに1.5時間撹拌した後、その混合物を水でクエンチし、次いで、100mLの水と100mLのジエチルエーテルで希釈した。有機相を分離し、水相をジエチルエーテルで抽出した。有機相を合してブラインで洗浄し、MgSO4で脱水し、濾過し、減圧下に濃縮した。生成物をフラッシュクロマトグラフィー(石油エーテル/酢酸エチル 10:1)で精製して、3.0gの44(11.88mmol,40%)を得た。
【0140】
実施例12:1−[(t−ブチルジメチルシリルオキシ)メチル]−4−(ヨードメチル)ベンゼン(45)
9.15g(34.88mmol)のトリフェニルホスフィン及び3.2g(44.5mmol)のイミダゾールをジエチルエーテル/アセトニトリルの3:1混合物(30mL)に溶解させた。黄色の懸濁液が形成されるまで、激しく撹拌しながら、8.85g(34.9mmol)のヨウ素を添加した。6.1g(23.25mmol)のモノ保護ベンジルアルコール44を20mLの上記と同一の溶媒混合物に溶解させた溶液を添加し、得られた混合物を2時間撹拌した。濾過により固体を除去し、濾液を100mLのジエチルエーテルで希釈し、100mLの重亜硫酸ナトリウム飽和溶液で洗浄した。この水溶液をジエチルエーテルで逆抽出し、有機相を合してMgSO4で脱水し、減圧下に濃縮した。フラッシュクロマトグラフィー(石油エーテル/酢酸エチル 95:5)に付すことにより、4.8gの45(13.25mmol,57%)を得た。
【0141】
実施例13:4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジルアルコール(46)
4.8g(13.25mmol)の1−[(t−ブチルジメチルシリルオキシ)メチル]−4−(ヨードメチル)ベンゼン(45)をアルゴン下、70mLの乾燥THFに溶解させた。0.954g(39.75mmol)のNaHを10分間かけて少量ずつ添加した。3.2g(14.58mmol)の11−アジド−3,6,9−トリオキサウンデカノール(7)を20mLの乾燥THFに溶解させた溶液を滴下して加え、得られた反応混合物を室温で15時間撹拌した。2mLの水を添加して反応物をクエンチし、この混合物を減圧下に約50%になるまで濃縮した。70mLの水を添加し、反応混合物をジエチルエーテルで抽出した。有機相をMgSO4で脱水し、溶媒を除去した。シリカゲルクロマトグラフィー(勾配:石油エーテル/酢酸エチル 10:1 → 3:1)で精製して、3.8g(8.38mmol,63%)のTBDMSで保護された生成物を得た。これを、プラスチック製チューブ内で80mLの乾燥THFに溶解させ、0℃に冷却し、8mLのピリジン/HF(70:30)溶液を添加し、室温で3時間撹拌した。100mLの水性飽和NaHCO3を添加し、有機相を分離し、ブラインで洗浄し、MgSO4で脱水した。溶媒を除去した後、生成物をシリカゲルクロマトグラフィー(石油エーテル/酢酸エチル 1:1)で精製して、1.27gの46(2.87mmol,74%)を得た。
【0142】
実施例14:O6−[4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジル]グアニン(41)
0.974g(2.87mmol)の4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジルアルコール(46)を5mLの乾燥DMFに溶解させ、1.3g(11.5mmol)のカリウムt−ブトキシドを添加した。0.731g(2.87mmol)の1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリドを添加し、この溶液を22時間撹拌した。減圧下に溶媒を除去した後、粗生成物をフラッシュカラムクロマトグラフィー(メタノール/ジクロロメタン 5:95)で精製した。収量:0.675g(50%)。
【0143】
実施例15:ヘテロ−O6−ベンジルグアニン−二量体42
45mg(0.09mmol)のアジド41と29.5mg(0.09mmol)のO6−(4−[プロプ−2−イニルオキシメチル]−ベンジル)グアニン(35)を1mLの水/アセトニトリル(1:1)に溶解させた溶液に、9.8mgのCuCl2を0.1mLの水に懸濁させた懸濁液を添加し、得られた反応混合物を室温で24時間撹拌した。全ての不溶性物質を濾過し、減圧下に溶媒を蒸発させ、生成物をフラッシュカラムクロマトグラフィー(メタノール/ジクロロメタン 5:1)で精製して、3.5mg(0.004mmol,10%)を得た。ESI/MS:781.44(M+)。
【0144】
実施例16:ホモ−O6−ベンジルグアニン−二量体43
アジド41(100mg,0.21mmol)及び1,4−(ジプロプ−2−イニルオキシメチル)ベンゼン(40)(22.6mg,0.10mmol)を、300μLのエタノールと200μLの水と500μLのt−ブタノールからなる溶媒混合物に溶解させた。10μLのCuSO4溶液(40mM)、5mgの銅線及び3mgのCuI2を添加し、得られた反応混合物を室温で48時間撹拌した。全ての不溶性物質を濾過し、減圧下に溶媒を蒸発させ、生成物をフラッシュカラムクロマトグラフィー(クロロホルム/メタノール 20:1)で精製して、34mg(28%)の二量体43を得た。ESI/MS:1159.37(M+)。
【0145】
実施例17:11−アミノ−1−アジド−3,6,9−トリオキサウンデカン(9)
1−アジド−11−フタルイミド−3,6,9−トリオキサウンデカン(8,実施例3,2.0g,5.74mmol)を、50mLの無水メタノール中で、1.0mLの55%ヒドラジン水和物で処理し、2時間加熱還流した。室温まで冷却した後、得られた混合物を減圧下に濃縮し、50mLの水で希釈し、5mLの濃HClで処理した。全ての沈澱物を濾過し、水性濾液をNaOHで中和し、減圧下に溶媒を除去した。残渣をジクロロメタンに溶解させ、3N NaOH及び水で洗浄した。有機相をMgSO4で脱水し、減圧下に濃縮して、0.63g(2.9mmol,51%)の無色の油状物を得た。これは、それ以上精製することなく次のステップで使用した。
【0146】
実施例18:O6−(1−[11−アミノ−3,6,9−トリオキサウンデシル]−1,2,3−トリアゾリル−4−メチル)−グアニン(11)
O6−プロパルギル−グアニン10(75mg,0.4mmol)及びアジド9(44mg,2mmol)を、400μLのN,N−ジメチルアセトアミドと100μLのアセトニトリルと100μLの水からなる溶媒混合物に溶解させた。10μLのCuSO4溶液(40mM)、銅線(5mg)及びCuI2(2mg)を添加し、得られた反応混合物を室温で48時間撹拌した。全ての不溶性物質を濾過し、減圧下に溶媒を蒸発させた。フラッシュクロマトグラフィー(ジクロロメタン/メタノール 20:1)に付すことにより、35mgの11(21%)を得た。
【0147】
実施例19:α−アジド−ε−N−Fmoc−リシン48
18mLのトリフリルアジド(6.3mmol,ジクロロメタン中2.3当量)を、27mLの溶媒混合物(水/メタノール 1:2)中のCuSO4(8mg)及びε−N−Fmoc−リシン47(1.10g,2.98mmol)に添加した。ジイソプロピルエチルアミン(971μL,5.58mmol)を添加し、得られた反応混合物を室温で一晩撹拌した。減圧下に有機溶媒を除去し、100mLの水を添加した。6N HClでpHを2.5に調節した。酢酸エチルで抽出した後、有機フラクションを合して水で洗浄し、脱水した(MgSO4)。溶媒を蒸発させることにより、粗生成物を黄色の油状物として得た。フラッシュクロマトグラフィー(ジクロロメタン/メタノール 97:3)に付すことにより、670mg(59%)の白色の固体を得た。Rf=0.32(ジクロロメタン/メタノール 95:5)。ESI−MS(m/z)395.23[M+H]+(MW計算値=394.3)。
【0148】
実施例20:O6−[4−(13−アミノ−2,5,8,11−テトラオキサトリデシル)−ベンジル]グアニン(49)
O6−[4−(13−アジド−2,5,8,11−テトラオキサトリデシル)−ベンジル]グアニン(41,0.21g,0.44mmol)を加熱することにより8mLの乾燥メタノールに溶解させた。トリエチルアミン(410μL,2.2mmol)及び1,3−プロパンジチオール(225μL,2.2mmol)をアルゴン雰囲気下に添加した。得られた反応混合物を40℃で加熱し、48時間撹拌した。この溶液をデカントし、固体をメタノールで洗浄し、減圧下に溶媒を蒸発させた。残渣をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 95:5)で精製し、標題化合物を淡黄色の油状物として得た(120mg,0.26mmol,61%)。Rf=0.01(ジクロロメタン/メタノール 9:1)。
【0149】
【表1】
【0150】
実施例21:O6−ベンジルグアニン−PEG−アミノ−α−アジド−ε−N−Fmoc−リシン50
O6−ベンジルグアニン−PEG−アミン49(76mg,0.17mmol)及びα−アジド−ε−N−Fmoc−リシン48(67.2mg,0.17mmol)を0.8mLの乾燥DMFに溶解させ、1−ヒドロキシベンゾトリアゾール(HOBT)(380μL,0.38mmol,1−メチル−2−ピロリドン(NMP)中1M)を添加した。得られた混合物を40℃で2時間撹拌し、25mLのジエチルエーテル中に移し、粗生成物を沈澱させた。残渣をメタノールに溶解させ、SiO2(0.2g)に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、O6−ベンジルグアニン−PEG−アミノ−α−アジド−ε−N−Fmoc−リシン50を淡黄色の油状物として得た(81mg,0.098mmol,57%)。ESI−MS(m/z)823.3[M+H]+(MW計算値=822.9)。
【0151】
実施例22:O6−ベンジルグアニン−PEG−アミノ−ε−N−Fmoc−リシン51
O6−ベンジルグアニン−PEG−アミノ−α−アジド−ε−N−Fmoc−リシン50(81mg,0.098mmol)を400μLの1,4−ジオキサン/100μLの水に溶解させ、590μLのトリメチルホスフィン(THF中1M)を添加した。室温で2時間撹拌した後、生成物を20mLのジエチルエーテル中で沈澱させ、減圧下で一晩乾燥させた。ESI−MS(m/z)797.35[M+H]+(MW計算値=796.9)。この生成物は、それ以上精製することなく使用した。
【0152】
実施例23:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Fmoc−リシン52
O6−ベンジルグアニン−PEG−アミノ−ε−N−Fmoc−リシン51(78.4mg,0.098mmol)及び30μLのトリエチルアミンを0.5mLのDMFに溶解させた。N−(+)−ビオチン−6−アミノカプロン酸N−スクシンイミジルエステル(49.2mg,0.1mmol)を0.5mLのDMFに溶解させた。両方の溶液を合し、40℃で2時間撹拌した。得られた反応混合物を30mLのジエチルエーテル中に移して粗生成物を沈澱させた。残渣をメタノールに溶解させ、SiO2(0.2g)に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、80mg(0.07mmol,71%)の無色の固体を得た。ESI−MS(m/z)1136.38[M+H]+(MW計算値=1135.38)。
【0153】
実施例24:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Fmoc−リシン52(16mg,0.014mmol)及び50μLのジエチルアミンを0.4mLのDMFに溶解させ、室温で1時間撹拌した。20mLのジエチルエーテルで生成物を沈澱させ、 減圧下に乾燥させた。
ESI−MS(m/z)914.38[M+H]+(MW計算値=914.13)。この生成物は、それ以上精製することなく次のステップで使用した。
【0154】
実施例25:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−[ジゴキシゲニン−3−オキシメチルカルボニル]−6−アミノカプロリル)−リシン54
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53(6.93mg,0.0075mmol)及び10μLのトリエチルアミンを0.4mLの乾燥DMFに溶解させた溶液に、(ジゴキシゲニン−3−オキシメチル−カルボニル)−6−アミノカプロン酸N−ヒドロキシスクシンイミドエステル(5mg,0.0075mmol)を添加し、得られた反応混合物を40℃で2時間撹拌した。20mLのジエチルエーテルで生成物を沈澱させ、減圧下に乾燥させ、クロロホルム中の5%メタノールから15%メタノールまでの勾配を用いるフラッシュカラムクロマトグラフィーで精製して、5mg(0.0034mmol,45%)の標題生成物を得た。ESI−MS(m/z)1458.78[M+H]+(MW計算値=1457.82)。
【0155】
実施例26:O6−ベンジルグアニン−PEG−アミノ−α−N−Fmoc−ε−N−Dde−リシン56
O6−ベンジルグアニン−PEG−アミン49(50mg,0.11mmol)及びα−N−Fmoc−ε−N−Dde−リシン55(58mg,0.11mmol)をDMF(600μL)に溶解させた。HOBT(NMP中の1M溶液の280μL,0.28mmol)及び1−エチル−3−(3−ジメチルアミノプロピル)カルボジイミド(EDC,54mg,0.28mmol)を添加し、得られた反応物を室温で1時間撹拌した。この溶液にエチルエーテルを添加して生成物を沈澱させ、上清をピペットで除去した。残留している溶媒を減圧下に蒸発させた。生成物を、ジクロロメタン中の2%メタノールから5%メタノールまでの勾配を用いるシリカゲルカラムクロマトグラフィーで精製した(収率:64%)。
【0156】
実施例27:O6−ベンジルグアニン−PEG−アミノ−ε−N−Dde−リシン57
O6−ベンジルグアニン−PEG−アミノ−α−N−Fmoc−ε−N−Dde−リシン56(66mg,68.6μmol)をDMF(600μL)に溶解させ、ジエチルアミン(71μL,686μmol)を添加した。得られた反応混合物を室温で1時間撹拌し、減圧下に溶媒を除去した。この生成物は、それ以上精製することなく次のステップで使用した。
【0157】
実施例28:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Dde−リシン58
O6−ベンジルグアニン−PEG−アミノ−ε−N−Dde−リシン57及びN−(+)−ビオチニル−6−アミノカプロン酸N−スクシンイミジルエステル(31mg,68.6μmol)をDMF(700μL)に溶解させた。トリエチルアミン(30μL)を添加し、得られた反応物を2時間撹拌した。この溶液にジエチルエーテルを添加して生成物を沈澱させ、上清をピペットで除去した。残留している溶媒を減圧下に蒸発させた。生成物をシリカゲルカラムクロマトグラフィーで精製した(クロロホルム中の2%メタノールから3%メタノールまでの勾配;収率:2ステップで37%)。
【0158】
実施例29:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53
DMF(250μL)中の2%ヒドラジンの溶液中で、O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−Dde−リシン58(27mg,25.0μmol)を20分間撹拌した。得られた反応混合物にアセトンを添加し、その混合物をさらに3分間撹拌した。この溶液にエチルエーテルを添加して生成物を沈澱させ、上清をピペットで除去した。残留している溶媒を減圧下に蒸発させた。この生成物をそれ以上精製することなく実施例24に準じて使用して、ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−([ジゴキシゲニン−3−オキシメチル−カルボニル]−6−アミノカプロイル)−リシン54を調製した(収率:2ステップで40%)。
【0159】
実施例30:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−(2,4−ジニトロベンゾイル)−リシン59
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53(16.9mg,18.5 gmol)をDMF(400μL)に溶解させ、2,4−ジニトロフルオロベンゼン(4.1mg,22μmol)及びトリエチルアミン(40μL)を添加した。得られた反応混合物を45℃で2時間撹拌した。残留している溶媒を減圧下に蒸発させた。生成物を、クロロホルム中の0%メタノールから15%メタノールまでの段階的な勾配を用いるシリカゲルフラッシュカラムクロマトグラフィーで精製した(収量:15mg,75%)。
【0160】
実施例31:O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−(6−[フルオレセイン−5−カルボニル]−アミノカプロイル)−リシン60
O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−リシン53(8.5mg,0.93 gmol)及び6−(フルオレセイン−5−カルボニル)−アミノカプロン酸N−ヒドロキシスクシンイミジルエステルをトリエチルアミン(10μL)と一緒に、200μLのジメチルアセトアミド中で、40℃で4時間撹拌した。8mLのジエチルエーテル中で生成物を沈澱させ、減圧下に乾燥させた。ESI−MS(m/z)1385.38[M+H]+(MW計算値=1385.13)。
【0161】
実施例32:ビス−N,N’−(O6−ベンジルグアニン−PEG)−尿素61
O6−[4−(13−アミノ−2,5,8,11−テトラオキサトリデシル)−ベンジル]−グアニン(49,30mg,67μL)とN,N’−ジスクシンイミジルカルボネート(8.6mg,33μL)を0.8mLの乾燥DMFに溶解させた溶液にトリエチルアミン(15μL)を添加し、得られた反応混合物を室温で24時間撹拌した。粗生成物をジエチルエーテル(10mL)で沈澱させ、SiO2に吸着させ、フラッシュカラムクロマトグラフィー(メタノール/ジクロロメタン 5:1)で精製した。収量:8.5mg(9.2μmol,27%)。ESI/MS:919.78[M+H]+,(MW計算値=918.99)。
【0162】
実施例33:1−(t−ブチルジメチルシリルオキシ)ブト−2−イン−4−オール(63)
t−ブチルジメチルシリルクロリド(8.0g,53mmol)を乾燥ジクロロメタンに溶解させ、2−ブチン−1,4−ジオール(13.71g,159.2mmol,3当量)と4−ジメチル−アミノピリジン(1.29g,10.6mmol,0.2当量)とトリエチルアミン(6.9mL)をジクロロメタン(200mL)とTHF(100mL)の溶媒混合物に溶解させた溶液に、滴下して加えた。得られた反応混合物を室温で15時間撹拌し、水及び飽和塩化アンモニウムで洗浄し、MgSO4で脱水した。フラッシュカラムクロマトグラフィー(石油エーテル/酢酸エチル 1:1)に付すことにより、7.2g(67%)を得た。
【0163】
実施例34:1−(t−ブチルジメチルシリルオキシ)−4−ヨードブト−2−イン(64)
J.Robertsonら(J.Chem.Soc.,Perkin Trans.1,3389−3369,2000)に従い、100mLのジクロロメタン中で、1−(t−ブチルジメチルシリルオキシ)−ブト−2−イン−4−オール(63,4.0g,19.96mmol)、イミダゾール(1.80g,26.45mmol)、ヨウ素(6.58g,25.95mmol)及びトリフェニルホスフィン(6.80g,25.95mmol)を処理した。後処理に付すことにより、4.42g(71%)の無色の油状物を得た。この生成物は、直接、次のステップで使用した。
【0164】
実施例35:4−(11−アジド−3,6,9−トリオキサウンデシルオキシ)−1−(t−ブチルジメチルシリルオキシ)−2−ブチン(65)
11−アジド−3,6,9−トリオキサウンデカノール(7,3.79g,17.02mmol)を乾燥THFに溶解させ、水素化ナトリウム(1.02g,42.45mmol)を少量ずつ添加した。室温で40分間撹拌した後、1−(t−ブチルジメチルシリルオキシ)−4−ヨードブト−2−イン(64,4.4g,14.18mmol)を添加し、得られた反応混合物をさらに15時間撹拌した。この反応物を水でクエンチし、減圧下に溶媒の量を約30%に低減させた。ジエチルエーテル(50mL)を添加し、この混合物を水で洗浄し、有機相をMgSO4で脱水した。溶媒を蒸発させた後、生成物をフラッシュカラムクロマトグラフィー(石油エーテル/酢酸エチル 4:1)で精製して、3.12g(54%)を得た。
【0165】
実施例36:(13−アジド−2,5,8,11−テトラオキサトリデシル)−プロパルギルアルコール(66)
t−ブチルジメチルシリルエーテル65(1.5g,3.37mmol)を、0.25mLの酢酸を含んでいる25mLのTHFに溶解させ、テトラブチルアンモニウムフルオリド(4.1mL,THF中1M)で処理した。得られた反応混合物を2時間撹拌し、減圧下に溶媒の量を約30%に低減させた。酢酸エチル(50mL)を添加した後、有機相を飽和重炭酸ナトリウム(20mL)及びブライン(30mL)で洗浄し、MgSO4で脱水した。この生成物は、それ以上精製することなく次のステップで使用した。収量:0.774g(80%)。
【0166】
実施例37:O6−[(13−アジド−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン(67)
プロパルギルアルコール66(724mg,2.5mmol)を乾燥DMF(1mL)に溶解させ、水素化ナトリウム(181mg,7.55mmol)を2回に分けて添加した。室温で30分間撹拌した後、1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27,641mg,2.5mmol)及び4−ジメチルアミノピリジン(100mg,0.81mmol)を添加した。撹拌を15時間継続し、30mLのジエチルエーテルを添加して生成物を沈澱させた。フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 95:5)に付すことにより、470mg(44%)の僅かに黄色い油状物を得た。ESI/MS:421.29[M+H]+。
【0167】
実施例38:O6−[(13−アミノ−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン(68)
アジド67(400mg,0.951mmol)及びトリフェニルホスフィン(748mg,2.85mmol)を、溶媒混合物(6mLの1,4−ジオキサン/400μLの水)中で、室温で15時間撹拌した。減圧下に溶媒を除去し、残渣をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、312mg(83%)を得た。
【0168】
実施例39:O6−[(13−(ビオチニル−6−アミノカプロイル−アミノ)−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン(69)
プロパルギルグアニン68(26.9mg,88μmol)及び(+)−ビオチニル−6−アミノヘキサン酸N−ヒドロキシ−スクシンイミドエステル(20.0mg,44 gmol)を15μLのトリエチルアミンと一緒に、1mLのDMF中で、40℃で3時間撹拌した。減圧下に溶媒を除去し、残渣をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、20mg(68%)を得た。
【0169】
実施例40:O6−[(13−(ジゴキシゲニン−3−オキシメチルカルボニル−6−アミノカプロイル−アミノ)−2,5,8,11−テトラオキサトリデシル)−プロパルギル]−グアニン70
プロパルギルグアニン68(11.6mg,37μmol)、ジゴキシゲニン−3−オキシメチルカルボニル−6−アミノカプロン酸N−ヒドロキシスクシンイミドエステル(5mg,7.5μmol)及び10μLのトリエチルアミンを40℃で2時間撹拌した。20mLのジエチルエーテルで生成物を沈澱させ、減圧下に乾燥させ、クロロホルム中の5%メタノールから15%メタノールまでの勾配を用いるフラッシュカラムクロマトグラフィーで精製して、3.2mgの標題化合物を得た(0.0034mmol,45%)。ESI−MS(m/z)939.78[M+H]+(MW計算値=938.12)。
【0170】
実施例41:O6−[4−(12−Fmoc−アミノ−ドデカノイル)アミノメチル]ベンジルグアニン72
12−Fmoc−アミノドデカン酸(147mg,0.33mmol)及び1−ベンゾトリアゾリルオキシ−トリス−ピロリジノ−ホスホニウムヘキサフルオロホスフェート(PyBOP,193mg,0.37mmol)を11mLのDMFに溶解させた。室温で45分間撹拌した後、5mLのDMF中のO6−4−アミノメチル−ベンジルグアニン(71,100mg,0.37mmol)を添加した。得られた反応混合物を50℃で5分間撹拌し、次いで、室温で1時間撹拌した。減圧下に溶媒を蒸発させた後、生成物をフラッシュカラムクロマトグラフィー(勾配:メタノール/ジクロロメタン 1:50から1:10まで)で精製して、185mg(0.27mmol,79%)の白色の固体生成物を得た。Rf=0.33(メタノール/ジクロロメタン 1:10)。
【0171】
【表2】
【0172】
実施例42:O6−[4−12−アミノドデカノイル)−アミノメチル]−ベンジルグアニン(73)
Fmocで保護された誘導体72(180mg,0.26mmol)を、20%(v/v,6.5mmol)のピペリジンを含んでいる3.2mLのDMFに溶解させた。室温で30分間撹拌した後、減圧下に溶媒を除去し、生成物をフラッシュカラムクロマトグラフィー(勾配:メタノール/ジクロロメタン 1:20から1:5まで(1%トリエチルアミン含有))で精製して、95mg(0.20mmol,78%)の淡黄色の固体を得た。Rf=0.05(メタノール/トリエチルアミン/ジクロロメタン 20:1:100)。
【0173】
【表3】
【0174】
実施例43:L−2−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ}−ペンタン二酸1−ベンジルエステル(76)
L−グルタミン酸1−ベンジルエステル(75,114mg,0.48mmol)及びK2CO3(66.4mg,0.48mmol)を3mLのDMSOに添加し、60℃で1時間超音波処理に付した。この混合物を、4−(N−[2,4−ジアミノ−6−プテリジニルメチル]−メチルアミノ)−安息香酸74(150mg,0.46mmol)とジイソプロピルエチルアミン(112μL,0.65mmol)とPyBOP(250mg,0.48mmol)を1.3mLの1時間撹拌しておいたDMSOに溶解させた溶液に添加した。この反応混合物を50℃で10分間撹拌し、次いで、室温で2.5時間撹拌した。0.5mLの1Mトリエチルアミンでクエンチした後、粗生成物を直線勾配(水/0.1%トリフルオロ酢酸(TFA):アセトニトリル/0.08%TFA 100:0から20:80まで)を用いるHPLCで精製した。この溶液をpH7に調節し、減圧下に溶媒を除去して、50mg(0.09mmol,20%)の橙色の固体を得た。
【0175】
【表4】
【0176】
実施例44:O6−4−([12−N−(4−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ)−4−ベンジルオキシカルボニルブタノイル)−アミノドデカノイル]−アミノメチル)−ベンジルグアニン(77)
(ベンゾトリアゾール−1−イルオキシ)トリピロリジノ−ホスホニウムヘキサフルオロホスフェート(PyBOP,152mg,0.29mmol)を1−ヒドロキシベンゾトリアゾール(HOBT)の溶液(1−メチル−2−ピロリドン(NMP)中の1M溶液の146μL,0.15mmol)に溶解させ、ベンジルエステル76(NMP中の0.154M溶液の380μL,0.06mmol)に添加した。得られた混合物を30分間振盪し、次いで、ジイソプロピルエチルアミン(30μL,0.18mmol)及び200μLのNMP中のアミノ−グアニン73(27.2mg,0.06mmol)に添加した。この反応混合物を室温で2時間撹拌し、次いで、200μLの1MのTFAでクエンチした。減圧下に溶媒を除去した後、生成物をフラッシュカラムクロマトグラフィー(勾配:メタノール/ジクロロメタン 1:50から1:5まで(2%酢酸含有))で精製した。19mg(0.2mmol,33%)の橙色の固体が得られた。Rf=0.08(メタノール/酢酸/ジクロロメタン 5:1:50)。ESI−MS(m/z)994.9[M+H]+(MW計算値=994.2)。
【0177】
実施例45:O6−4−([12−N−(4−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ}−4−カルボキシブタノイル)−アミノドデカノイル]−アミノメチル)−ベンジルグアニン(78)
ベンジルエステル77(13mg,0.01mmol)を2mLのメタノール/水(1:1)に溶解させ、K2CO3(785μL メタノール/水(1:1)中の1M,0.79mmol)を添加した。得られた混合物を室温で2.5時間振盪した後、785μL の1MのTFAでクエンチした。粗生成物を、直線勾配(水/0.1%TFA:アセトニトリル/0.08%TFA 100:0から20:80まで)を用いるHPLCで精製した。10mg(0.01mmol,85%)の生成物が得られた。ESI−MS(m/z)904.7[M+H]+(MW計算値=904.0)。
【0178】
実施例46:O6−4−[13−N−(4−{4−[N−(2,4−ジアミノ−6−プテリジニルメチル)−メチルアミノ]−ベンゾイルアミノ}−4−カルボキシブタノイル)−アミノ−2,5,8,11−テトラオキサトリデシル]−ベンジルグアニン(79)
PyBOP(201mg,0.39mmol)及びHOBT(NMP中の1M溶液の194μL、0.19mmol)をベンジルエステル76(21mg,0.04mmol)に添加し、得られた混合物を室温で30分間振盪した。O6−[4−(13−アミノ−2,5,8,11−テトラオキサトリデシル)−ベンジルグアニン(49,21mg,0.05mmol)及びジイソプロピルエチルアミン(24μL,0.14mmol)を490μLのNMPに溶解させ、活性化させた76の溶液に添加した。室温で3時間振盪した後、この反応混合物を1mLのアセトニトリル/2mLの1MのTFAでクエンチした。粗生成物を、直線勾配(水/0.1%TFA:アセトニトリル/0.08%TFA 100:0から20:80まで)を用いるHPLCで精製した。1.5MのNaOHを用いてこの溶液のpHを10に調節することにより、カルボキシ基を脱保護した。先のステップと同じ勾配を用いるHPLCで粗生成物を精製した後、14mg(0.02mmol,40%)の標題化合物を得た。ESI−MS(m/z)883.4[M+H]+(MW計算値=882.9)。
【0179】
実施例47:4−アリルオキシメチル−1−(t−ブチルジメチルシリルオキシメチル)−ベンゼン(80)
4−(t−ブチルジメチルシリルオキシメチル)−ベンジルアルコール44(2.85g,11.31mmol)を15mLの乾燥THFに溶解させた溶液に、アルゴン雰囲気下、水素化ナトリウム(326mg,13.57mmol)を少量ずつ添加し、この反応混合物を室温で30分間撹拌した。ヨウ化アリル(3.80g,22.62mmol)を5mLの乾燥THFに溶解させた溶液を添加し、得られた混合物を17時間撹拌した。この反応物を水でクエンチし、溶媒の容積を約30%に低減させ、残渣を酢酸エチル(20mLで3回)で抽出した。有機相をMgSO4で脱水し、減圧下に溶媒を除去し、生成物をフラッシュカラムクロマトグラフィー(石油エーテル/酢酸エチル 100:1)で精製して、1.83g(6.25mmol,59%)を得た。
【0180】
実施例48:1,4−ジ−[4−(t−ブチルジメチルシリルオキシメチル)−ベンジルオキシ]−2−ブテン(81)
4−アリルオキシメチル−1−(t−ブチルジメチル−シリルオキシメチル)−ベンゼン(80,1.0g,3.24mmol)を70mLの乾燥ジクロロメタンに溶解させた溶液に、アルゴン雰囲気下、ベンジリデン−ビス(トリシクロヘキシルホスフィン)−ジクロロルテニウム(420mg,0.513mmol,15mol%)を添加した。得られた反応混合物を室温で15時間撹拌し、触媒(200mg)を追加し、その反応混合物を5時間加熱還流した。減圧下に溶媒を除去し、生成物をカラムクロマトグラフィー(石油エーテル/酢酸エチル 50:1)で精製して、520mg(0.93mmol,55%)の標題化合物を得た。
【0181】
実施例49:ビス−1,4−(O6−ベンジルグアニン−メトキシ)−2−ブテン82
TBDMS−二量体81(351mg,0.63mmol)を5mLの乾燥THFに溶解させ、テトラブチルアンモニウムフルオリド(3.78mmol)及び酢酸(3.78mmol)を添加した。得られた反応混合物を室温で15時間撹拌した。溶媒を蒸発させた後、生成物を溶媒の勾配(石油エーテル/酢酸エチル 1:1から1:3まで)を用いるカラムクロマトグラフィーで精製して、150mg(0.45mmol,73%)の無色の固体を得た。この脱保護した二量体(100mg,0.30mmol)を0.8mLの乾燥DMFに溶解させた溶液に、水素化ナトリウム(44mg,1.82mmol)を添加し、この反応混合物を30分間撹拌した。1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27,186mg,0.73mmol)及びDMAP(11mg,0.09mmol)を添加し、この反応混合物を室温でさらに3時間撹拌した。この反応物を水でクエンチし、減圧下に溶媒を除去した。残渣をメタノールに溶解させ、SiO2に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、二量体82を無色の固体として得た(30mg,0.05mmol,16%)。
【0182】
実施例50:11−アリルオキシ−3,6,9−トリオキサウンデカノール(83)
カリウムt−ブトキシド(2.3g,19.5mmol)をTHF(500mL)に溶解させた無水溶液に、室温で、テトラエチレングリコール(7.18g,37.0mmol)を添加した。得られた混合物を30分間撹拌し、ヨウ化アリル(3.31g,19.7mmol)を乾燥THF(60mL)に溶解させた溶液を、1時間かけて滴下して加えた。この反応混合物を30時間撹拌し、酢酸エチル(800mL)を用いてシリカプラグ(10g)を通して濾過して生成物を溶出させた。減圧下に溶媒を蒸発させた後、生成物を溶媒の勾配(石油エーテル/酢酸エチル 10:1から、酢酸エチル 100%まで)を用いるカラムクロマトグラフィー で精製して、無色の油状物を得た(2.41g,10.3mmol,27%)。
【0183】
実施例51:1−(t−ブチルジメチルシリルオキシメチル)−4−(13−アリルオキシ−2,5,8,11−テトラオキサトリデシル−ベンゼン(84)
11−アリルオキシ−3,6,9−トリオキサウンデカノール(83,2.33g,9.94mmol)を乾燥THF(40mL)に溶解させた溶液に、アルゴン雰囲気下、水素化ナトリウム(0.95g,39.8mmol)を数回に分けて添加した。得られた混合物を30分間撹拌した。ヨード誘導体45(3.0g,8.28mmol)を乾燥THF(10mL)に溶解させた溶液を滴下して加え、この混合物を15時間撹拌した。2mLの水を添加することにより反応物をクエンチし、減圧下に70%の量のTHFを蒸発させた。70mLの水を添加してこの混合物を希釈し、得られた溶液を50mLの酢酸エチルで5回抽出した。有機抽出物を合してMgSO4で脱水し、減圧下に濃縮した。得られた黄色の油状物をフラッシュカラムクロマトグラフィー(勾配:石油エーテル/酢酸エチル 10:1から5:1まで)で精製して、標題生成物84を淡黄色の油状物として得た(1.96g,4.2mmol,51%)。
【0184】
実施例52:TBDMSで保護されたビス−1,4−(ベンジルアルコール−PEG)−2−ブテン85
TBDMSで保護されたアリルオキシ誘導体84(1.0g,2.13mmol)を40mLの乾燥ジクロロメタンに溶解させた溶液に、アルゴン雰囲気下、ベンジリデン−ビス(トリシクロヘキシルホスフィン)−ジクロロルテニウム(263mg,0.32mmol,15mol%)を添加した。得られた反応混合物を5時間加熱還流した。触媒(200mg)を追加し、加熱を15時間継続した。減圧下に溶媒を除去し、生成物を、溶媒の勾配(石油エーテル/酢酸エチル 1:1から1:5まで)を用いるカラムクロマトグラフィーで精製して、530mg(0.58mmol,54%)の二量体85を得た。
【0185】
実施例53:ビス−1,4−(O6−ベンジルグアニン−PEG)−2−ブテン86
TBDMSで保護された二量体85(200mg,0.22mmol)を3mLの乾燥THFに溶解させ、テトラブチルアンモニウムフルオリド(2.5mmol)及び酢酸(2.5mmol)を添加した。得られた反応混合物を室温で15時間撹拌した。溶媒を蒸発させた後、生成物を、溶媒の勾配(石油エーテル/酢酸エチル 1:1から、酢酸エチル 100%まで)を用いるカラムクロマトグラフィーで精製して、80mg(0.11mmol,50%)の無色の固体を得た。この脱保護された二量体(61mg,0.089mmol)と1−(2−アミノ−7H−プリン−6−イル)−1−メチル−ピロリジニウムクロリド(27,54mg,0.22mmol)とDMAP(3.0mg,0.027mmol)を1.0mLの乾燥DMFに溶解させた溶液に、水素化ナトリウム(13mg,0.54mmol)を添加し、得られた反応混合物を2時間撹拌した。この反応物を水でクエンチし、減圧下に溶媒を除去した。残渣をメタノールに溶解させ、SiO2に吸着させ、フラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール 10:1)で精製して、二量体86を無色の固体として得た(35mg,0.036mmol,41%)。
【0186】
実施例54:式1の化合物とヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)融合タンパク質の反応性の実証
(a) O6−(1−[11−アミノ−3,6,9−トリオキサウンデシル]−1,2,3−トリアゾリル−4−メチル)−グアニン(11)の反応性
総容積15μLの50mMのHEPES(pH7.5)中で、18pmolのPGEAhAGT−GST(A.Juilleratら,Chem.Biol.10:313−317,2003)をDMSO中の11の5mM溶液(0.2μL)と一緒に1時間インキュベーションした。10pmolのビオチニル化O6−ベンジルグアニン−オリゴヌクレオチド(R.Damoiseauxら,ChemBiochem 4:285−287,2001)を添加し、さらに15分間、反応を継続させた。対照として、総容積15μLの50mMのHEPES(pH7.5)中で、基質を含まない状態で、18pmolのPGEAhAGT−GSTを10pmolの同一のビオチニル化オリゴヌクレオチドと一緒に15分間インキュベーションした。hAGT−GSTとビオチニル化オリゴヌクレオチドの間のビオチニル化反応生成物は、HRP(Pierce)に結合させた抗ビオチンコンジュゲートを用いるウエスタンブロット法により検出した。図1Aのレーン「C」は、PGEAhAGT−GSTとビオチニル化オリゴヌクレオチドの反応生成物を示していた。PGEAhAGT−GSTが最初に11と反応する場合は、ビオチニル化オリゴヌクレオチドとのさらなる反応は観察されなかった(レーン1及びレーン2、図1A)。これにより、O6−トリアゾリルメチルグアニンがhAGTに対する効果的な基質であることが証明された。
【0187】
(b) O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノカプロイル)−ε−N−(6−[フルオレセイン−5−カルボニル]−アミノカプロイル)−リシン60の反応性
総容積15μLの50mMのHEPES(pH7.5)中で、18pmolのPGEAhAGT−GSTを0.5nmolの60と一緒にインキュベーションした。反応生成物は、ヤギ抗フルオレセイン抗体(Rockland)及びHRPコンジュゲート抗ヤギ抗体(Sigma)を用いるウエスタンブロット法により検出した。これにより、2つの異なった標識を含んでいる化合物60がhAGTに対する効果的な基質であることが証明された。
【0188】
実施例55:固相上におけるヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)融合タンパク質の固定化
80μLのPGEAhAGT−GST融合タンパク質溶液(HBSバッファー中200μg/mL)を、O6−ベンジルグアニン−PEG−アミノ−α−N−(ビオチニル−6−アミノ−カプロイル)−ε−N−([ジゴキシゲニン−3−オキシメチルカルボニル]−6−アミノカプロイル)−リシン54の0.4mM溶液(20μL)と一緒に、室温で10分間インキュベーションした。この溶液を、Neutravidin Biacore チップ上に、1μL/分の流速で35分間通過させた。次いで、このチップを、同じ流速のバッファー(HBST)で40分間パージした。次に、GST抗体(1980μg/mL)を注入して該融合タンパク質の先の結合を明らかにし、次いで、再度、HBSTで洗浄した。固定化実験は、表面プラズモン共鳴により評価した。表面プラズモン共鳴分析は、研究グレードのSAセンサーチップを備えたBiacore 1000 光学バイオセンサー(Biacore)を用いて行った。EDTAを含まないHBS(10mM HEPES,pH7.4,150mM NaCl,0.005% Tween 20)は、新たに調製し、22μmの膜を通して濾過した。得られたセンソグラムは、図2に示してある。
【0189】
実施例56:ヒトO6−アルキルグアニン−DNAアルキルトランスフェラーゼ(hAGT)の二量体化
(a) PGEGhAGTと二量体化基質43の反応
二量体化実験は、HEPESバッファー(HEPES 50mM,pH7.2,1mM DTT,200μg/mL BSA)中の0.4μMの一定の濃度のPGEGhAGT−GST(A.Juilleratら,Chem.Biol.10:313−317,2003)で、総容積40μLで行った。0〜100:1の基質:タンパク質の比率をカバーするように、化合物43の濃度を変化させた。インキュベーション時間は、すべての実験に関して、40分間とした。ウェスタンブロットは、抗hAGT抗体(PBS中1:2000,Chemicon)と第二の抗体としての抗マウス−HRPコンジュゲート(PBS中1:2000,Sigma)を用いて得た。結果は、図3Aに示してある。
【0190】
(b) E.coli抽出物中の二量体化基質61とHA−W160hAGTとの反応
実験は、hAGT変異体Gly160Trp(W160hAGT)(M.Xu−Welliverら,Biochemical Pharmacology 58:1279−1285,1999)を用いて行った。HA−W160hAGTは、LB培地中、24℃で、IPTG(1mM)と種々の濃度(0μM、10μM、20μM、及び、50μM)の基質61を指数関数的に増殖する培養に添加した後、E.coli BL21で2時間発現させた。遠心分離により細胞を回収した。各標識化実験について、抽出物の総容積は、約1mL(容積は、OD600が一定となるように調節する)であった。得られたペレットを50μLの1×SDSに再度懸濁させた後、O6−ベンジルグアニンを1mMの濃度となるまで添加した。95℃に5分間加熱した後、サンプルを12%SDS−PAGEにロードした。ウェスタンブロットは、ヤギ抗MGMT(TBST中1:100,Chemicon)と第二の抗体としての抗ヤギIgG(TBST中1:1000,Santa Cruz Biotechnology)を用いて得た。結果は、図3Bに示してある。
【0191】
実施例57:GST−3HYhAGTに対する78と79の反応性
3HYhAGTが78及び79に対して活性を有していることを実証するために、3HYhAGTを、E.coliにおいて、グルタチオンS−トランスフェラーゼとの融合タンパク質(GST−3HYhAGT)として発現させ、その融合タンパク質を精製し、及び、その活性をインビトロアッセイで測定した。GST−3HYhAGTを過剰量の78又は79と一緒にインキュベーションして反応させた後、抗メトトレキサート抗体を用いるウェスタンブロット法に付した。このアッセイにおいて、GST−3HYhAGTは、78との反応については約1500秒−1M−1の二次反応速度定数を示し、79との反応については約900秒−1M−1の二次反応速度定数を示した。従って、78の濃度1μMで、及び、3HYhAGTに対して過剰な78(擬一次条件)で、該タンパク質の50%が10分以内に標識された。
【0192】
実施例58:hAGT変異体
hAGT融合タンパク質を構築するために、好ましくは、変異体Asn157Gly Ser159Gluを使用した。この変異体は、野生型hAGTと比較して、1型のベンジルグアニン誘導体に対して、約20倍の増大した活性を示した。以下の文献を参照されたい:A.Juilleratら,Chem.Biol.10:313−317,2003。以下に示すhAGTのさらなる変異体は、hAGTのDNAへの結合を分断するが、ベンジルグアニン誘導体に対する活性は有意には阻害しないことが示されている:Lys125Ala,Ala127Thr,及び,Arg128Ala。以下の文献を参照されたい:A.Limら,EMBO J.15:4050−4060,1996,及び,D.S.Danielsら,EMBO J.19:1719−1730,2000。これらの変異体を導入する理論的根拠は、3ハイブリッド系におけるhAGT融合タンパク質とDNAの相互作用を最少にすることである。結果として得られた、上記の5つの変位を有しているhAGT変異体は、3HYhAGTと略記する。
【0193】
実施例59:酵母L40株の増殖アッセイ
LexA−3HYhAGT及びB42−DHFR融合タンパク質をコード化するプラスミドの対を酵母株L40内に形質転換し、機能的LexA−B42転写因子を再構築することにより、レポーター遺伝子HIS3及びlacZが転写された。78又は79のいずれかの存在下において融合タンパク質の異なった組合せを発現させることにより酵母L40のヒスチジン要求性が補完されるか否かについて試験した(表1)。LexA−3HYhAGTとB42−DHFRを同時に発現させることにより、酵母L40はヒスチジンは含んでいないが78又は79のいずれかを含んでいるプレート上で増殖することが可能となった。このことは、HIS3が転写されたことを示している。78又は79の非存在下では、増殖は観察されなかった。
【0194】
【表5】
【0195】
原則として、ベンジルグアニン78及び79を介した機能的転写因子の再構築は、どのリガンド結合性タンパク質がLexAに融合するかとは関係がなく、また、どのリガンド結合性タンパク質がB42に融合するかとは関係がない。このことを実証するために、LexA−DHFRとB42−3HYhAGTを同時に発現する酵母L40の、ヒスチジンは含んでいないが78又は79のいずれかを含んでいるプレート上での増殖について調べた。予想されたとおり、化合物78又は79に依存する増殖が観察されるが、その増殖速度は、LexA−3HYhAGTとB42−DHFRを同時に発現する酵母で観察される増殖速度を下回った(表1)。
【0196】
実施例60:ONPGを用いる定量的β−ガラクトシダーゼアッセイ
LexA融合タンパク質とB42融合タンパク質の別の組合せを発現する酵母株L40における、ベンジルグアニンをベースとする化合物78及び79によるlacZレポーター遺伝子の転写の活性化について調べた(表2)。このアッセイにおいて、液体培養の細胞抽出物中の色素原基質o−ニトロフェニル−β−D−ガラクトピラノシド(ONPG)の加水分解速度を測定することにより、lacZ遺伝子の産物であるβ−ガラクトシダーゼの活性を決定した。
【0197】
【表6】
【図面の簡単な説明】
【0198】
【図1A】HRP(Pierce)に結合した抗ビオチンコンジュゲートのウェスタンブロット。レーン1及び2:化合物11(式中のR3がトリアゾリルメチルである式1の化合物)と反応させた後、AGT基質として知られているビオチニル化O6−ベンジル−グアニンオリゴヌクレオチドと反応させた後のPGEAhAGT−GST(AGT融合タンパク質)。「C」の印が付いているレーン(対照):PGEAhAGT−GSTとビオチニル化O6−ベンジルグアニンオリゴヌクレオチドの反応生成物。
【図1B】ヤギ抗フルオレセイン抗体(Rockland)及びHRPコンジュゲート抗ヤギ抗体(Sigma)のウェスタンブロット。レーン1及び2(2種類の異なった濃度)は、フルオレセイン標識とビオチン標識を有している化合物60(式中のLが複数の標識である式1の化合物)とPGEAhAGT−GSTの反応生成物を示している。左側のレーンは、47kDa及び79kDaの分子量マーカーを示している。
【図2】ストレプトアビジンBiacoreチップに適用した化合物54(式中のLがビオチンである式1の化合物)とPGEAhAGT−GST(AGT融合タンパク質)の反応生成物の表面プラズモン共鳴のセンソグラム。該チップを、αGST抗体の溶液でさらに処理する。A:化合物54と一緒にプレインキュベーションしたPGEAhAGT−GSTを添加。B:緩衝液(HBST)を添加。C:αGST抗体を添加。
【図3A】抗hAGT抗体及び第二抗体としての抗マウスHRPコンジュゲートを用いたウェスタンブロット。レーンの下に、化合物43(式中のLがさらなる−R3−CH2−X−R1−R2残基である式1の化合物、即ち、二量体化化合物)とPGEGhAGT−GSTの比率を示してある。
【図3B】抗ヤギhAGT、及びE.coli中における二量体化化合物61と融合タンパク質HA−W160hAGTのインビボ反応生成物の抗ヤギIgGを用いたウェスタンブロット。レーンの下に、61(式中のLが−R3−CH2−X−R1−R2残基である式1の化合物)の種々の濃度を示してある。
【図4】hAGTをベースとする3ハイブリッド系の略図。融合タンパク質DHFR−B42の存在下におけるhAGT−LexA融合タンパク質と式1(式中、Lはメトトレキサート(M)である)の化合物の反応において、hAGT−LexAとDHFR−B42をカップリングさせ、転写を開始させる。
【図1】
【特許請求の範囲】
【請求項1】
式1:
【化1】
[式中、
R1−R2は、AGTにより基質として認識される基であり;
Xは、酸素又は硫黄であり;
R3は、芳香族基若しくはヘテロ芳香族基であるか、又は、CH2に結合した二重結合を有する、場合により置換されていてもよい不飽和のアルキル基、シクロアルキル基若しくはヘテロシクリル基であり;
R4は、リンカーであり;
そして、
Lは、1つの標識であるか、同一であるか若しくは異なっている複数の標識であるか、R4をR1に連結して環状基質を形成する結合であるか、又は、さらなる基−R3−CH2−X−R1−R2である]
で表される化合物。
【請求項2】
R1が、1〜5個の窒素原子を含んでいるヘテロ芳香族基であり;
R2が、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
Xが、酸素であり;
R3が、フェニルであるか、又は、0、1、2、3若しくは4個の環窒素原子と0若しくは1個の酸素原子と0若しくは1個の硫黄原子を含み5若しくは6個の環原子を有する置換されていないか若しくは置換されている単環式若しくは二環式のヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素原子、酸素原子又は硫黄原子で置き換えられている)であるか、又は、1−アルケニル、1−アルキニル、若しくは、3〜7個の炭素原子を有する1−シクロヘキセニルであるか、又は、3〜12個の原子と窒素、酸素及び硫黄から選択される1〜5個のヘテロ原子を含んでいる、場合により置換されていてもよい不飽和ヘテロシクリル基(ここで、ヘテロシクリル基は、該ヘテロシクリル基をメチレンCH2に連結する位置に二重結合を有する);
R4が、1〜300個の炭素原子を有する、場合により置換されていてもよい直鎖又は分枝鎖のアルキレン基であり、その際、場合により、
(a)1個以上の炭素原子が酸素により置き換えられており;
(b)1個以上の炭素原子が水素原子を有している窒素で置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、アミド官能基−NH−CO−を表し;
(c)1個以上の炭素原子が酸素により置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、エステル官能基−O−CO−を表し;
(d)隣接してる2つの炭素原子の間の結合が二重結合又は三重結合であって、官能基−CH=CH−又は−C≡C−を表し;
(e)1個以上の炭素原子が、フェニレン、飽和若しくは不飽和のシクロアルキレン、飽和若しくは不飽和のビシクロアルキレン、架橋ヘテロ芳香族、又は、飽和若しくは不飽和の架橋ヘテロシクリル基により置き換えられており;
及び/又は、
(f)隣接してる2個の炭素原子がジスルフィド結合−S−S−により置き換えられており;
並びに、
Lが、分光学的プローブ、磁気プローブ、造影剤、結合パートナーに特異的に結合可能な特異的結合対の一方である分子、別の生体分子と相互作用すると推測される分子、別の生体分子と相互作用すると推測される分子のライブラリー、別の分子と架橋することが可能な分子、H2O2及びアスコルベートと接触したときにヒドロキシルラジカルを生成することが可能な分子、光を照射されたときに反応性ラジカルを生成することが可能な分子、固体支持体に共有結合で結合している分子、その相補鎖と塩基対を成すことが可能な核酸若しくはその誘導体、膜挿入特性を有する脂質若しくは別の疎水性分子、酵素的、化学的若しくは物理的に望ましい特性を有している生体分子、R4をR1に連結して環状基質を形成する結合、及び、さらなる基−R3−CH2−X−R1−R2から選択される1つの標識であるか又は複数の同一であるか若しくは異なっている標識である、請求項1に記載の式1で表される化合物。
【請求項3】
R1−R2が、式2:
【化2】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
R5は、水素、ハロゲン、トリフルオロメチル、又は、ヒドロキシであり;
及び、
R6は、水素、ヒドロキシ、又は、置換されていないか若しくは置換されているアミノである]
で表される基及びその互変異性体形態である、請求項1に記載の式1で表される化合物。
【請求項4】
前記糖部分R2が、β−D−2’−デオキシリボシルであるか、又は、2〜99のヌクレオチドの長さを有する一本鎖オリゴデオキシリボヌクレオチドに組み入れられているβ−D−2’−デオキシリボシルであり、ここで、グアニン誘導体R1は、該オリゴヌクレオチド配列内の任意の位置を占めている、請求項3に記載の式1で表される化合物。
【請求項5】
R2が水素であり、R5が水素であり、R6が置換されていないアミノであり、及び、Xが酸素である、請求項3に記載の式1で表される化合物。
【請求項6】
R1−R2が、式3:
【化3】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
及び、
R6は、水素、ヒドロキシ、又は、置換されていないか若しくは置換されているアミノである]
で表される基及びその互変異性体形態である、請求項1に記載の式1で表される化合物。
【請求項7】
R1−R2が、式4:
【化4】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
並びに、
R7及びR8は、互いに独立して、いずれも、水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、又は、ニトロである]
で表される基である、請求項1に記載の式1で表される化合物。
【請求項8】
R3が、トリアゾリル、テトラゾリル、イソオキサゾリル、チエニル、又は、イソオキサゾリジニルである、請求項1に記載の式1で表される化合物。
【請求項9】
R3がトリアゾリルである、請求項8に記載の式1で表される化合物。
【請求項10】
R3がテトラゾリルである、請求項8に記載の式1で表される化合物。
【請求項11】
R3がイソオキサゾリルである、請求項8に記載の式1で表される化合物。
【請求項12】
R3がチエニルである、請求項8に記載の式1で表される化合物。
【請求項13】
R3がイソオキサゾリジニルである、請求項8に記載の式1で表される化合物。
【請求項14】
R3が1−アルキニルである、請求項1に記載の式1で表される化合物。
【請求項15】
R4が、場合により−CH=CH−又は−C≡C−を介して基R3に結合している、2〜25個の炭素原子を有する直鎖アルキレン基、又は4〜100のエチレンオキシ単位を有する直鎖ポリエチレングリコール基、又は2個以上の炭素原子がアミド官能基−NH−COで置き換えられている2〜25個の炭素原子を有する直鎖アルキレン基である、請求項1に記載の式1で表される化合物。
【請求項16】
R4が、分枝鎖アルキレン基(ここで、該分枝鎖アルキレン基は、3〜6のエチレングリコール単位を有するポリエチレングリコール基及び炭素原子がアミド結合で置き換えられているアルキレン基を含んでおり、さらに、置換されているアミノ官能基とヒドロキシ官能基を有している)である、請求項1に記載の式1で表される化合物。
【請求項17】
R4が、アルキレン基の炭素原子がアミン官能基、カルボキサミド官能基及びエーテル官能基で置き換えられている樹枝状構造を有する分枝鎖アルキレン基である、請求項1に記載の式1で表される化合物。
【請求項18】
R3がフェニレンであり、そして、Lがさらなる基−R3−CH2−X−R1−R2である、請求項3に記載の式1で表される化合物。
【請求項19】
R3が1,4−フェニレンであり、そして、リンカーR4が、10〜40個の炭素原子を有する直鎖アルキレン基(ここで、3〜12個の炭素原子は、酸素で置き換えられており、1又は2個の炭素原子は、1,4−トリアゾリデン単位で置き換えられており、及び、場合により、1の炭素原子は、1,4−フェニレン単位で置き換えられている)である、請求項3又は18に記載の式1で表される化合物。
【請求項20】
R3が1,4−フェニレンであり、そして、リンカーR4が、場合によりオキソで置換されていてもよい10〜40個の炭素原子を有する直鎖アルキレン基(ここで、3〜12個の炭素原子は、酸素で置き換えられており、及び、1若しくは2個の炭素原子は窒素で置き換えられている )である、請求項3又は18に記載の式1で表される化合物。
【請求項21】
R3が1,4−フェニレンであり、そして、リンカーR4が、6〜40の炭素原子を有する直鎖アルキレン基(ここで、2〜12個の炭素原子は、酸素で置き換えられており、及び、隣接する炭素原子の間の1若しくは2つの結合は、二重結合である)である、請求項3又は18に記載の式1で表される化合物。
【請求項22】
R3がフェニレンであり、R6がアミノであり、そして、Lが、R4をR6に連結する結合である、請求項3に記載の式1で表される化合物。
【請求項23】
R3がフェニレンであり、そして、Lがメトトレキサートである、請求項3に記載の式1で表される化合物。
【請求項24】
R3がフェニレンであり、そして、Lが、同一であるか又は異なっている複数の標識である、請求項3に記載の式1で表される化合物。
【請求項25】
R3がフェニレンであり、そして、Lが、異なっている2つの標識である、請求項24に記載の式1で表される化合物。
【請求項26】
請求項1〜25のいずれか1項に記載の式1で表される化合物を合成する方法。
【請求項27】
式R2−R1−X−CH2−R3−R4[式中、R4は、2つ以上の反応性求核基又は反応性求電子基を有する多官能性残基である]で表される化合物を、1以上の標識Lを導入する適切な試薬と反応させることを特徴とする、請求項26に記載の方法。
【請求項28】
反応性求核基又は求電子基が、オルソゴナルに保護されている官能基であり、そして、反応条件が、遊離された各官能基を次に化学的にさらに操作して、それに標識を結合させることができるように、又は、リンカーR4のさらなる拡大の導入が可能となるように、別々の脱保護を可能とするように選択される、請求項27に記載の方法。
【請求項29】
式1:
【化5】
[式中、
R1−R2は、
【化6】
で表されるラジカルであり、その際、R2は水素であり、R5は水素であり、そして、R6は置換されていないアミノであり;
Xは酸素であり;
R3は、トリアゾリル、テトラゾリル、イソオキサゾリル、チエニル、イソオキサゾリジニル又はアルキニル;
R4はリンカーであり;
そして、
Lは、アミノ又はアジドである]
で表される化合物。
【請求項30】
式1:
【化7】
[式中、
R1−R2は、
【化8】
で表されるラジカルであり、その際、R2は水素であり、R5は水素であり、そして、R6は置換されていないアミノであり;
Xは酸素であり;
R3は1,4−フェニレンであり;
R4は、場合によりオキソで置換されていてもよい10〜40個の炭素原子を有する直鎖アルキレン基(ここで、12個以下の炭素原子は、酸素で置き換えられており、及び、0、1若しくは2個の炭素原子は窒素で置き換えられている)であり;
及び、
Lは、アミノ又はアジドである]
で表される化合物。
【請求項31】
目的とするタンパク質を検出及び操作する方法であって、AGT融合タンパク質に組み入れた目的とする該タンパク質を標識を有しているAGT基質と接触させ、そして、該標識を認識又は処理するように設計されているシステム内で該標識を用いて、該AGT融合タンパク質を検出し、そして、場合により、さらに操作することを特徴とし、ここで、標識を有しているAGT基質は、請求項1〜25のいずれか1項に記載の式1で表される化合物である、前記方法。
【請求項1】
式1:
【化1】
[式中、
R1−R2は、AGTにより基質として認識される基であり;
Xは、酸素又は硫黄であり;
R3は、芳香族基若しくはヘテロ芳香族基であるか、又は、CH2に結合した二重結合を有する、場合により置換されていてもよい不飽和のアルキル基、シクロアルキル基若しくはヘテロシクリル基であり;
R4は、リンカーであり;
そして、
Lは、1つの標識であるか、同一であるか若しくは異なっている複数の標識であるか、R4をR1に連結して環状基質を形成する結合であるか、又は、さらなる基−R3−CH2−X−R1−R2である]
で表される化合物。
【請求項2】
R1が、1〜5個の窒素原子を含んでいるヘテロ芳香族基であり;
R2が、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
Xが、酸素であり;
R3が、フェニルであるか、又は、0、1、2、3若しくは4個の環窒素原子と0若しくは1個の酸素原子と0若しくは1個の硫黄原子を含み5若しくは6個の環原子を有する置換されていないか若しくは置換されている単環式若しくは二環式のヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素原子、酸素原子又は硫黄原子で置き換えられている)であるか、又は、1−アルケニル、1−アルキニル、若しくは、3〜7個の炭素原子を有する1−シクロヘキセニルであるか、又は、3〜12個の原子と窒素、酸素及び硫黄から選択される1〜5個のヘテロ原子を含んでいる、場合により置換されていてもよい不飽和ヘテロシクリル基(ここで、ヘテロシクリル基は、該ヘテロシクリル基をメチレンCH2に連結する位置に二重結合を有する);
R4が、1〜300個の炭素原子を有する、場合により置換されていてもよい直鎖又は分枝鎖のアルキレン基であり、その際、場合により、
(a)1個以上の炭素原子が酸素により置き換えられており;
(b)1個以上の炭素原子が水素原子を有している窒素で置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、アミド官能基−NH−CO−を表し;
(c)1個以上の炭素原子が酸素により置き換えられており、そして、それに隣接する炭素原子がオキソにより置換されていて、エステル官能基−O−CO−を表し;
(d)隣接してる2つの炭素原子の間の結合が二重結合又は三重結合であって、官能基−CH=CH−又は−C≡C−を表し;
(e)1個以上の炭素原子が、フェニレン、飽和若しくは不飽和のシクロアルキレン、飽和若しくは不飽和のビシクロアルキレン、架橋ヘテロ芳香族、又は、飽和若しくは不飽和の架橋ヘテロシクリル基により置き換えられており;
及び/又は、
(f)隣接してる2個の炭素原子がジスルフィド結合−S−S−により置き換えられており;
並びに、
Lが、分光学的プローブ、磁気プローブ、造影剤、結合パートナーに特異的に結合可能な特異的結合対の一方である分子、別の生体分子と相互作用すると推測される分子、別の生体分子と相互作用すると推測される分子のライブラリー、別の分子と架橋することが可能な分子、H2O2及びアスコルベートと接触したときにヒドロキシルラジカルを生成することが可能な分子、光を照射されたときに反応性ラジカルを生成することが可能な分子、固体支持体に共有結合で結合している分子、その相補鎖と塩基対を成すことが可能な核酸若しくはその誘導体、膜挿入特性を有する脂質若しくは別の疎水性分子、酵素的、化学的若しくは物理的に望ましい特性を有している生体分子、R4をR1に連結して環状基質を形成する結合、及び、さらなる基−R3−CH2−X−R1−R2から選択される1つの標識であるか又は複数の同一であるか若しくは異なっている標識である、請求項1に記載の式1で表される化合物。
【請求項3】
R1−R2が、式2:
【化2】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
R5は、水素、ハロゲン、トリフルオロメチル、又は、ヒドロキシであり;
及び、
R6は、水素、ヒドロキシ、又は、置換されていないか若しくは置換されているアミノである]
で表される基及びその互変異性体形態である、請求項1に記載の式1で表される化合物。
【請求項4】
前記糖部分R2が、β−D−2’−デオキシリボシルであるか、又は、2〜99のヌクレオチドの長さを有する一本鎖オリゴデオキシリボヌクレオチドに組み入れられているβ−D−2’−デオキシリボシルであり、ここで、グアニン誘導体R1は、該オリゴヌクレオチド配列内の任意の位置を占めている、請求項3に記載の式1で表される化合物。
【請求項5】
R2が水素であり、R5が水素であり、R6が置換されていないアミノであり、及び、Xが酸素である、請求項3に記載の式1で表される化合物。
【請求項6】
R1−R2が、式3:
【化3】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
及び、
R6は、水素、ヒドロキシ、又は、置換されていないか若しくは置換されているアミノである]
で表される基及びその互変異性体形態である、請求項1に記載の式1で表される化合物。
【請求項7】
R1−R2が、式4:
【化4】
[式中、
R2は、水素、1〜10個の炭素原子を有するアルキル、又は、糖部分であり;
並びに、
R7及びR8は、互いに独立して、いずれも、水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、又は、ニトロである]
で表される基である、請求項1に記載の式1で表される化合物。
【請求項8】
R3が、トリアゾリル、テトラゾリル、イソオキサゾリル、チエニル、又は、イソオキサゾリジニルである、請求項1に記載の式1で表される化合物。
【請求項9】
R3がトリアゾリルである、請求項8に記載の式1で表される化合物。
【請求項10】
R3がテトラゾリルである、請求項8に記載の式1で表される化合物。
【請求項11】
R3がイソオキサゾリルである、請求項8に記載の式1で表される化合物。
【請求項12】
R3がチエニルである、請求項8に記載の式1で表される化合物。
【請求項13】
R3がイソオキサゾリジニルである、請求項8に記載の式1で表される化合物。
【請求項14】
R3が1−アルキニルである、請求項1に記載の式1で表される化合物。
【請求項15】
R4が、場合により−CH=CH−又は−C≡C−を介して基R3に結合している、2〜25個の炭素原子を有する直鎖アルキレン基、又は4〜100のエチレンオキシ単位を有する直鎖ポリエチレングリコール基、又は2個以上の炭素原子がアミド官能基−NH−COで置き換えられている2〜25個の炭素原子を有する直鎖アルキレン基である、請求項1に記載の式1で表される化合物。
【請求項16】
R4が、分枝鎖アルキレン基(ここで、該分枝鎖アルキレン基は、3〜6のエチレングリコール単位を有するポリエチレングリコール基及び炭素原子がアミド結合で置き換えられているアルキレン基を含んでおり、さらに、置換されているアミノ官能基とヒドロキシ官能基を有している)である、請求項1に記載の式1で表される化合物。
【請求項17】
R4が、アルキレン基の炭素原子がアミン官能基、カルボキサミド官能基及びエーテル官能基で置き換えられている樹枝状構造を有する分枝鎖アルキレン基である、請求項1に記載の式1で表される化合物。
【請求項18】
R3がフェニレンであり、そして、Lがさらなる基−R3−CH2−X−R1−R2である、請求項3に記載の式1で表される化合物。
【請求項19】
R3が1,4−フェニレンであり、そして、リンカーR4が、10〜40個の炭素原子を有する直鎖アルキレン基(ここで、3〜12個の炭素原子は、酸素で置き換えられており、1又は2個の炭素原子は、1,4−トリアゾリデン単位で置き換えられており、及び、場合により、1の炭素原子は、1,4−フェニレン単位で置き換えられている)である、請求項3又は18に記載の式1で表される化合物。
【請求項20】
R3が1,4−フェニレンであり、そして、リンカーR4が、場合によりオキソで置換されていてもよい10〜40個の炭素原子を有する直鎖アルキレン基(ここで、3〜12個の炭素原子は、酸素で置き換えられており、及び、1若しくは2個の炭素原子は窒素で置き換えられている )である、請求項3又は18に記載の式1で表される化合物。
【請求項21】
R3が1,4−フェニレンであり、そして、リンカーR4が、6〜40の炭素原子を有する直鎖アルキレン基(ここで、2〜12個の炭素原子は、酸素で置き換えられており、及び、隣接する炭素原子の間の1若しくは2つの結合は、二重結合である)である、請求項3又は18に記載の式1で表される化合物。
【請求項22】
R3がフェニレンであり、R6がアミノであり、そして、Lが、R4をR6に連結する結合である、請求項3に記載の式1で表される化合物。
【請求項23】
R3がフェニレンであり、そして、Lがメトトレキサートである、請求項3に記載の式1で表される化合物。
【請求項24】
R3がフェニレンであり、そして、Lが、同一であるか又は異なっている複数の標識である、請求項3に記載の式1で表される化合物。
【請求項25】
R3がフェニレンであり、そして、Lが、異なっている2つの標識である、請求項24に記載の式1で表される化合物。
【請求項26】
請求項1〜25のいずれか1項に記載の式1で表される化合物を合成する方法。
【請求項27】
式R2−R1−X−CH2−R3−R4[式中、R4は、2つ以上の反応性求核基又は反応性求電子基を有する多官能性残基である]で表される化合物を、1以上の標識Lを導入する適切な試薬と反応させることを特徴とする、請求項26に記載の方法。
【請求項28】
反応性求核基又は求電子基が、オルソゴナルに保護されている官能基であり、そして、反応条件が、遊離された各官能基を次に化学的にさらに操作して、それに標識を結合させることができるように、又は、リンカーR4のさらなる拡大の導入が可能となるように、別々の脱保護を可能とするように選択される、請求項27に記載の方法。
【請求項29】
式1:
【化5】
[式中、
R1−R2は、
【化6】
で表されるラジカルであり、その際、R2は水素であり、R5は水素であり、そして、R6は置換されていないアミノであり;
Xは酸素であり;
R3は、トリアゾリル、テトラゾリル、イソオキサゾリル、チエニル、イソオキサゾリジニル又はアルキニル;
R4はリンカーであり;
そして、
Lは、アミノ又はアジドである]
で表される化合物。
【請求項30】
式1:
【化7】
[式中、
R1−R2は、
【化8】
で表されるラジカルであり、その際、R2は水素であり、R5は水素であり、そして、R6は置換されていないアミノであり;
Xは酸素であり;
R3は1,4−フェニレンであり;
R4は、場合によりオキソで置換されていてもよい10〜40個の炭素原子を有する直鎖アルキレン基(ここで、12個以下の炭素原子は、酸素で置き換えられており、及び、0、1若しくは2個の炭素原子は窒素で置き換えられている)であり;
及び、
Lは、アミノ又はアジドである]
で表される化合物。
【請求項31】
目的とするタンパク質を検出及び操作する方法であって、AGT融合タンパク質に組み入れた目的とする該タンパク質を標識を有しているAGT基質と接触させ、そして、該標識を認識又は処理するように設計されているシステム内で該標識を用いて、該AGT融合タンパク質を検出し、そして、場合により、さらに操作することを特徴とし、ここで、標識を有しているAGT基質は、請求項1〜25のいずれか1項に記載の式1で表される化合物である、前記方法。
【図2】

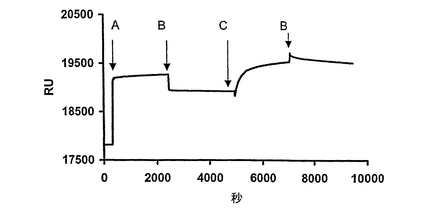
【図3】
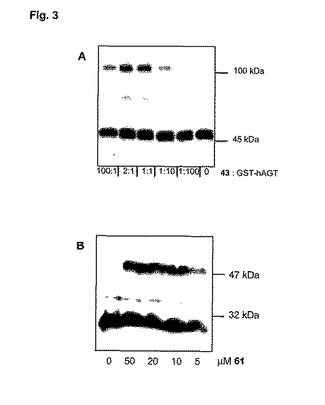
【図4】
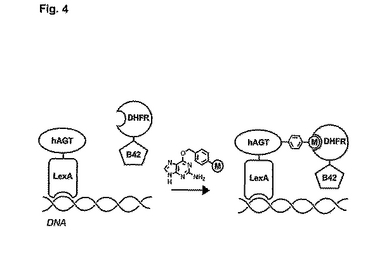
【図3】
【図4】
【公表番号】特表2006−501289(P2006−501289A)
【公表日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願番号】特願2004−540766(P2004−540766)
【出願日】平成15年10月1日(2003.10.1)
【国際出願番号】PCT/EP2003/010889
【国際公開番号】WO2004/031405
【国際公開日】平成16年4月15日(2004.4.15)
【出願人】(503183293)エコル・ポリテクニック・フェデラル・ドゥ・ローザンヌ(エーペーエフエル) (11)
【氏名又は名称原語表記】ECOLE POLYTECHNIQUE FEDERALE DE LAUSANNE(EPFL)
【Fターム(参考)】
【公表日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願日】平成15年10月1日(2003.10.1)
【国際出願番号】PCT/EP2003/010889
【国際公開番号】WO2004/031405
【国際公開日】平成16年4月15日(2004.4.15)
【出願人】(503183293)エコル・ポリテクニック・フェデラル・ドゥ・ローザンヌ(エーペーエフエル) (11)
【氏名又は名称原語表記】ECOLE POLYTECHNIQUE FEDERALE DE LAUSANNE(EPFL)
【Fターム(参考)】
[ Back to top ]