
O6−アルキルグアニンDNAアルキルトランスフェラーゼ特異的基質
本発明は、式R1−A−X−CH2−R3−R4−L1[式中、Aは基質としてAGTによって認識される基であり、Xは酸素または硫黄であり、R1は基−R2−L2または基R5であり、R2およびR4は互いに独立したリンカーであり、R3は芳香族基もしくは複素環式芳香族基;または場合により置換されている不飽和アルキル、シクロアルキル、もしくはCH2に結合する二重結合を有するヘテロシクリル基であり、R5はアリールメチルもしくはヘテロアリールメチル、または場合により置換されているシクロアルキル、シクロアルケニル、もしくはヘテロシクリル基であり、L1は1個の標識、複数の同じもしくは異なる標識、R4とAを結合して環状基質を形成する結合、またはさらなる基−R3CH2−X−A−R1であり、かつL2は、1個の標識、または複数の同じもしくは異なる標識である]のO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)基質に関する。本発明は、さらに、これらの基質からO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)に標識を移動させる方法、およびAGT融合蛋白質に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、基質からO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)に標識を移動させる方法、O6−アルキルグアニンDNAアルキルトランスフェラーゼ融合蛋白質、およびそのような方法に適した新規な特異的基質に関する。
【背景技術】
【0002】
N−メチル−N−ニトロソウレアなどの求電子試剤の変異原性および発癌性作用は、主としてDNAのグアニンのO6−アルキル化によるものである。DNAアルキル化からそれ自体を守るために、哺乳動物および細菌は、これらの病変部を修復する蛋白質O6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)を保持している。AGTは、アルキル化したグアニンおよびグアニン誘導体のO6位からアルキル基を自身のシステインの一つであるメルカプト基に移動させ、不可逆的にアルキル化AGTを生じさせる。根底にある機序は、SN2型の求核反応であり、この反応によってメチル基のみならずベンジル基までも容易に移動する理由が説明される。腫瘍細胞でAGTが過剰発現することは、プロカルバジン、ダカルバジン、テモゾロマイド、ビス−2−クロロエチル−N−ニトロソウレアなどのアルキル化薬物に対する耐性の主たる理由であり、その結果、化学療法においてAGT阻害薬を感作物質として使用することが提案されきた(Peggら, Prog Nucleic Acid Res Mol Biol 51:167-223, 1995)。米国特許第5,691,307号は、ベンジル基に様々な置換基を有するO6−ベンジルグアニン、および腫瘍細胞でAGT濃度を枯渇させ、それによってアルキル化抗腫瘍薬物に対する応答性を増大するためのその使用について記載している。同様に、国際公開公報第97/20843号は、O6−ベンジル−およびO6−ヘテロアリールメチル−ピリミジン誘導体を表す、さらに別のAGT枯渇化合物を開示している。
【0003】
ドイツ国特許第19903895号は、AGTレベルを測定するためのアッセイを開示し、このアッセイはビオチン化されたO6−アルキルグアニン誘導体とAGTの間の反応を頼りに、AGTをビオチン標識化する。これにより、順次、ストレプトアビジン被膜プレート上のAGTを分離し、例えば、ELISAアッセイでAGTを検出できるようにする。このアッセイは、腫瘍組織でAGT濃度をモニタリングすること、およびAGT阻害薬スクリーニングでの使用に向けて提案される。
【0004】
国際公開公報第01/85221号は、AGTの検出に放射性標識したフルオロまたはヨード置換O6−ベンジルグアニンの使用およびAGTレベルのモニタリングを提案している。
【0005】
Damoiseaux ら, ChemBiochem. 4:285-287, 2001は、AGT標識化の化学プローブとして使用し、再度、癌細胞におけるこの酵素のレベルの検出を容易にして研究および化学療法に役立てるために、オリゴデオキシリボヌクレオチドに組み込まれた改変O6−アルキル化グアニン誘導体を開示している。
【0006】
国際公開公報第02/083937号は、対象となる蛋白質を検出し、かつ/または操作する方法であって、蛋白質とAGTを融合し、標識を有するAGT基質にAGT融合蛋白質を接触させ、そして標識を使用しAGT融合蛋白質を検出し、場合によりさらに操作する方法を開示した。使用する数種のAGT融合蛋白質、AGT基質の一般構造原理、ならびに多種多様な標識、およびその方法で使用可能な標識を検出するための方法が記載されている。
【0007】
PCT/EP03/10859(国際公開公報第2004/031404号)は、対象となる蛋白質を検出しかつ/または操作する上記の方法で使用される特定のAGT融合蛋白質、この方法によって得られる標識した融合蛋白質、および特定のAGT融合蛋白質の使用方法について記載している。
【0008】
PCT/EP03/10889(国際公開公報第2004/031405号)は、対象となる蛋白質を検出しかつ/または操作する上記の方法に特に適した標識を有する追加のAGT基質、およびそのような特に標識した基質の適用を開示している。この特許出願はまた、これらの追加のAGT基質の製造方法について記載している。
【0009】
発明の概要
本発明は、式(1)
【0010】
【化6】
【0011】
[式中、Aは基質としてAGTによって認識される基であり;
Xは、酸素または硫黄であり;
R1は、基−R2−L2または基R5であり;
R2およびR4は、互いに独立したリンカーであり;
R3は、芳香族基もしくは複素環式芳香族基;または場合により置換されている不飽和アルキル、シクロアルキル、もしくはCH2に結合する二重結合を有するヘテロシクリル基であり;
R5は、アリールメチルもしくはヘテロアリールメチル、または場合により置換されているシクロアルキル、シクロアルケニル、もしくはヘテロシクリル基であり;
L1は、1個の標識、複数の同じもしくは異なる標識、R4とAを結合して環状基質を形成する結合、またはさらに基−R3−CH2−X−A−R1であり;かつ
L2は、1個の標識、または複数の同じもしくは異なる標識である]
のO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)基質に関する。
【0012】
本発明は、さらに、これらの基質からO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)に標識を移動させる方法およびAGT融合蛋白質に関する。
【0013】
本発明の詳細な説明
本発明の特定のAGT基質は、式(1)
【0014】
【化7】
【0015】
[式中、
Aは基質としてAGTによって認識される基であり;
Xは、酸素または硫黄であり;
R1は、基−R2−L2または基R5であり;
R2およびR4は、互いに独立したリンカーであり;
R3は、芳香族基もしくは複素環式芳香族基;または場合により置換されている不飽和アルキル、シクロアルキル、もしくはCH2に結合する二重結合も有するヘテロシクリル基であり;
R5は、アリールメチルもしくはヘテロアリールメチル、または場合により置換されているシクロアルキル、シクロアルケニル、もしくはヘテロシクリル基であり;
L1は、1個の標識、複数の同じもしくは異なる標識、R4とAを結合して環状基質を形成する結合、またはさらに基−R3−CH2−X−A−R1であり;かつ
L2は、1個の標識、または複数の同じもしくは異なる標識である]
の化合物である。
【0016】
基R1−Aでは、残基Aは、1〜5個の窒素原子を含み、基質としてAGTによって認識される複素環式芳香族基であることが好ましい。
【0017】
複素環式芳香族基Aは、単環式もしくは2環式であり、5〜12個の、好ましくは6個の、または9個もしくは10個の環原子を有し;そしてこの基Aは、置換基R1を有することに加えて、メチルなどの低級アルキル、メトキシやエトキシなどの低級アルコキシ、ヒドロキシ、オキソ、アミノ、低級アルキルアミノ、ジ低級アルキルアミノ、アシルアミノ、塩素や臭素などのハロゲン、トリフルオロメチルなどのハロゲン化した低級アルキル、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、または低級アルキルカルボニルからなる群から選択される一個以上の、特に1、2、もしくは3個のさらなる置換基によって置換されていなくても置換されていてもよい。
【0018】
低級アルキルは、1〜7個の、好ましくは1〜4個のC原子を有するアルキルであり、直鎖もしくは分枝鎖であることが好ましく;低級アルキルはn−ブチル、sec−ブチル、iso−ブチル、tert−ブチルなどのブチル、n−プロピルやイソプロピルなどのプロピル、エチルまたはメチルが好ましい。低級アルキルはメチルが最も好ましい。
【0019】
低級アルコキシでは、低級アルキル基は先に定義した通りである。低級アルコキシは、好ましくはn−ブトキシ、tert−ブトキシ、iso−プロポキシ、エトキシ、またはメトキシ、特にメトキシを示す。
【0020】
好ましい単環式もしくは2環式複素環式芳香族基Aは、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、8−アザプリニル、7−デアザプリニル、8−アザ−7−デアザプリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、トリアジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、トリアゾリル、テトラゾリル、またはベンゾ[d]ピラゾリルから選択される。より好ましくは、単環式もしくは2環式複素環式芳香族基Aは、プリニル、8−アザプリニル、7−デアザプリニル、8−アザ−7−デアザプリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、およびトリアジニルからなる群から選択される。
【0021】
例えば、基R1−Aは、式(2)
【0022】
【化8】
【0023】
「式中、R6は、水素、ヒドロキシ、または非置換もしくは置換アミノであり、R7およびR8の一方はR1であり他方は水素である]のプリンラジカルでよい。
【0024】
R6がヒドロキシである場合、プリンラジカルは主に互変異性体で存在し、ここで、それぞれ、R6を有する炭素原子に隣接する窒素は水素原子を有し、この窒素原子とR6を有する炭素原子の間の二重結合は単結合であり、そしてR6は二重結合酸素である。
【0025】
置換されたアミノ基R6は、1〜4個の炭素原子の低級アルキルアミノまたはアシルアミノであり、ここで、アシル基は、1〜5個の炭素原子を有する低級アルキルカルボニル、例えばアセチル、プロピオニル、n−もしくはiso−プロピルカルボニル、またはn−、iso−、もしくはtert−ブチルカルボニルもしくはアリ−ルカルボニルであり、例えばベンゾイルである。
【0026】
R6が非置換もしくは置換アミノであり、プリンラジカルの結合に接続する残基Xが酸素である場合、式(2)の残基はグアニン誘導体である。
【0027】
特に好ましいものは、基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7がR1であり、R8が水素であり、かつXが酸素である化合物、すなわち、N9位にさらに置換基を有するグアニン誘導体である。
【0028】
本発明の別の好ましい態様では、基R1−Aが式(3)
【0029】
【化9】
【0030】
[式中、置換基R6は、式(2)下でR6について定義した意味を有する]
の8−アザプリンラジカルである。
【0031】
本発明のさらに好ましい態様では、基R1−Aは、式(4a)または(4b)
【0032】
【化10】
【0033】
[式中、R9は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、またはアミノであり、アミノが好ましく、かつR10は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、ニトロ、またはニトロソである]のピリミジンラジカルである。ハロゲンR9またはR10は、例えばフルオロ、クロロ、ブロモ、またはヨードである。
【0034】
Xは、酸素であることが好ましい。
【0035】
本発明のさらに好ましい態様では、基R1−Aが式(4c)
【0036】
【化11】
【0037】
[式中、R6は、非置換もしくは置換アミノであり、R7およびR8の一方はR1であり他方は水素である]
のプテリジンラジカルである。
【0038】
リンカー基R2またはR4は、それぞれ、一個の標識L2またはL1、あるいは複数の同じもしくは異なる標識L2またはL1を基質に結合する柔軟なリンカーであることが好ましい。リンカー単位は、想定した適用と関連して、すなわち、AGTを含む融合蛋白質への基質の移動に際して選択される。リンカー単位はまた、適当な溶媒への基質溶解性も増大させる。使用するリンカーは、実際の適用条件下で化学的に安定している。リンカーは、AGTによる反応にも、標識L1および/またはL2の検出にも干渉しないが、例えば、式(1)の化合物とAGTを含む融合蛋白質が反応した後のある時点で切断されるように構築しうる。
【0039】
リンカーR2またはR4は、1〜300個の炭素原子を有する直鎖もしくは分枝鎖アルキレン基であって、場合により
(a)一個以上の炭素原子が酸素によって、特に、全ての第3炭素原子が酸素によって置き換えられている、例えば、1〜100個のエチレンオキシ単位を有するポリエチレンオキシ基である;
(b)一個以上の炭素原子が、水素原子を有する窒素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、アミド官能基−NH−CO−を示す;
(c)一個以上の炭素原子が酸素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、エステル官能基−O−CO−を示す;
(d)2個の隣接する炭素原子間の結合が二重または三重結合であり、官能基−CH=CH−または−C≡C−を表す;
(e)一個以上の炭素原子が、フェニレン、飽和もしくは不飽和シクロアルキレン、飽和もしくは不飽和ビシクロアルキレン、架橋ヘテロ芳香族、または架橋飽和もしくは不飽和ヘテロシクリル基によって置き換えられている;
(f)2個の隣接する炭素原子が、ジスルフィド結合−S−S−によって置き換えられている;
または場合により置換基を含み、先に(a)〜(f)下で定義したような2個以上の、特に、2個もしくは3個のアルキレン基および/または修飾アルキレン基の組合せ
である。
【0040】
考えられる置換基は、メチルなどの例えば低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0041】
考えられるさらなる置換基は、例えば、αアミノ酸、特に自然に存在するαアミノ酸がリンカーR2またはR4に組み込まれたときに得られる置換基であり、ここで、炭素原子は、(b)下で定義したアミド官能基−NH−CO−によって置き換えられている。そのようなリンカーでは、アルキレン基R2またはR4の炭素鎖の一部が、基−(NH−CHR−CO)n−[式中、nは1〜100であり、Rはαアミノ酸の様々な残基を表す]によって置き換えられている。
【0042】
さらなる置換基は、光切断リンカーR2またはR4をもたらす置換基であり、例えばo−ニトロフェニル基である。特に、この置換基o−ニトロフェニルは、アミド結合に隣接する炭素原子に位置し、例えば基−NH−CO−CH2−CH(o−ニトロフェニル)−NH−CO−に位置し、またはポリエチレングリコール鎖中の、例えば、基−O−CH2−CH(o−ニトロフェニル)−O−中の置換基として位置する。考えられる他の光切断リンカーは、例えば、フェナシル、アルコキシベンゾイン、ベンジルチオエーテル、ピバロイルグリコール誘導体である。
【0043】
先に(e)下で定義した炭素原子に置き換わるフェニレン基は、例えば、1,2−、1,3−、であり、1,4−フェニレンが好ましい。特定の態様では、フェニレン基はニトロ基によってさらに置換され、そして(a)、(b)、(c)、(d)、および(f)下で上記した他の置き換えと組み合わされて光切断基を表し、例えば−CO−NH−CH2−4−ニトロ−1,3−フェニレン−CH(CH3)−O−CO−中の4−ニトロ−1,3−フェニレン、または例えば−CH2−O−2−メトキシ−5−ニトロ−1,4−フェニレン−CH(CH3)−O−中の2−メトキシ−5−ニトロ−1,4−フェニレンなどがある。光切断リンカーを表す他の特定の態様は、例えば、−1,4−フェニレン−CO−CH2−O−CO−CH2−(フェナシル基)、−1,4−フェニレン−CH(OR)−CO−1,4−フェニレン−(アルコキシベンゾイン)、又は−3,5−ジメトキシ−1,4−フェニレン−CH2−O−(ジメトキシベンジル部分)である。先に(e)下で定義した炭素原子に置き換わる飽和もしくは不飽和シクロアルキレン基は、3〜7個の炭素原子を有するシクロアルキルに由来し、シクロペンチルまたはシクロヘキシルに由来するのが好ましく、例えば、1,2−もしくは1,3−シクロペンチレン、1,2−、1,3−、好ましくは1,4−シクロヘキシレン、または例えば1位もしくは2位で不飽和である1,4−シクロヘキシレンがある。先に(e)下で定義した炭素原子に置き換わる飽和もしくは不飽和ビシクロアルキレン基は、7もしくは8個の炭素原子を有するビシクロアルキルに由来し、例えば、ビシクロ[2.2.1]ヘプチレンやビシクロ[2.2.2]オクチレンであり、場合により、2位で不飽和であり、または2位と5位で二重に不飽和である1,4−ビシクロ[2.2.1]ヘプチレン、ならびに場合により、2位で不飽和であり、または2位と5位で二重に不飽和である1,4−ビシクロ[2.2.2]オクチレンが好ましい。先に(e)下で定義した炭素原子に置き換わる架橋複素環式芳香族基は、例えばトリアゾリデン、好ましくは1,4−トリアゾリデン、またはイソオキサゾリデン、好ましくは3,5−イソオキサゾリデンである。先に(e)下で定義した炭素原子に置き換わる架橋飽和もしくは不飽和ヘテロシクリル基は、例えば、先にR3下で定義した不飽和ヘテロシクリル基に由来し、例えばイソオキサゾリジネン、好ましくは3,5−イソオキサゾリジネン、または3〜12個の原子を有し、その1〜3個が窒素、酸素、硫黄から選択したヘテロ原子である完全飽和ヘテロシクリル基、例えばピロリジンジイル、ピペリジンジイル、テトラヒドロフランジイル、ジオキサンジイル、モルホリンジイル、またはテトラヒドロチオフェンジイルであり、2,5−テトラヒドロフランジイル、または2,5−ジオキサンジイルが好ましい。考えられる特定のヘテロシクリル基は、糖類部分であり、例えばα−もしくはβ−フラノシル部分、またはα−もしくはβ−ピラノシル部分である。
【0044】
リンカーR2またはR4の環状下部構造は、R2またはR4内で回転可能な結合数によって測定される分子柔軟性を減少させ、それによりインビボでの標識適用例全てにおいて重要である膜透過速度が上昇する。
【0045】
リンカーR2またはR4は、それぞれ、−CH=CH−基または−C≡C−基によって基AまたはR3に場合により結合している、1〜25個の炭素原子を有する直鎖アルキレン基、または4〜100個のエチレンオキシ単位を有する直鎖ポリエチレングリコール基であることが好ましい。さらに好ましいものは、1〜25個の炭素原子を有する直鎖アルキレン基であり、ここで、炭素原子はアミド官能基−NH−CO−によって場合により置き換えられており、光切断サブユニット、例えば、o−ニトロフェニルを有している。さらに好ましいものは、3〜6個のエチレングリコール単位であるポリエチレングリコール基と、炭素原子がアミド結合によって置き換えられているアルキレン基を含み、さらに置換されたアミノ官能基とヒドロキシ官能基を有する分枝リンカーである。他の好ましい分枝リンカーは、アミン、カルボキサミド、および/またはエーテル官能基がアルキレン基の炭素原子に置き換わっているデンドリティック(樹状)構造を有している。
【0046】
特に好ましいリンカーR2またはR4は、10〜40個の炭素原子の直鎖アルキレン基であり、ここで、3〜12個の炭素原子は酸素によって置き換えられ、1個もしくは2個の炭素原子は、それぞれ、1個もしくは2個の1,4−トリアゾリデン単位によって置き換えられ、かつ場合により1個の炭素原子は1,4−フェニレン単位によって置き換えられている。
【0047】
別の特に好ましいリンカーR2またはR4は、オキソによって場合により置換されている、10〜40個の炭素原子の直鎖アルキレン基であり、ここで、3〜12個の炭素原子は、酸素によって置き換えられ、1個もしくは2個の炭素原子は、窒素によって置き換えられている。
【0048】
別の特に好ましいリンカーR2またはR4は、6〜40個の炭素原子の直鎖アルキレン基であり、ここで、2〜12個の炭素原子は、酸素によって置き換えられ、2個の隣接する炭素原子間の1個もしくは2個の結合は官能基−CH=CH−を表す二重結合である。
【0049】
別の特に好ましいリンカーR2またはR4は、1〜15個の炭素原子の直鎖アルキレン基であり、N−メチルイソオキサゾリジン−3,5−ジメチル基である。
【0050】
リンカーR2またはR4は、一個もしくは複数の同じもしくは異なる標識、例えば1〜100個の同じもしくは異なる標識、特に1〜5個、好ましくは1、2、もしくは3個の、特に1個もしくは2個の同じもしくは異なる標識を有しうる。
【0051】
芳香族基もしくは複素環式芳香族基としてR3、または場合により置換されている不飽和アルキル、シクロアルキル、もしくはヘテロシクリル基としてR3は、(その反応機序に従い)AGTによって立体的に、そして電子的に受け入れられる基であり、それによってR3−R4−L1単位は融合蛋白質に共有結合的に移動する。R3−R4−L1単位では、R4−L1は、複数の同じもしくは異なる標識L1を有する複数の同じもしくは異なるリンカーR4である意味を含みうる。
【0052】
芳香族基としてR3は、フェニルまたはナフチル、特にフェニルが好ましく、例えばパラ位もしくはメタ位でR4によって置換されているフェニルである。
【0053】
複素環式芳香族基R3は、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)であり、そして、5〜12個の、好ましくは5または6個の環原子を有し;そして置換基R4を有することに加えて、置換されていなくても、あるいはメチルなどの低級アルキル、メトキシやエトキシなどの低級アルコキシ、例えば塩素、臭素、フッ素などのハロゲン、トリフルオロメチルなどのハロゲン化した低級アルキル、またはヒドロキシからなる群から選択される、一つ以上、特に一つの、さらなる置換基によって置換されていてもよい。
【0054】
単環式もしくは2環式ヘテロアリール基R3は、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、フラザニル、ベンゾ[d]ピラゾリル、チエニル、およびフラニルから選択されることが好ましい。より好ましくは単環式もしくは2環式ヘテロアリール基は、ピロリル、1H−イミダゾール−1−イルなどのイミダゾリル、1−ベンゾイミダゾリルなどのベンゾイミダゾリル;インダゾリル、特に5−インダゾリル:例えば2−、3−、または4−ピリジルなどのピリジル;ピリミジニル、特に2−ピリミジニル;ピラジニル;イソキノリニル、特に3−イソキノリニル;キノリニル、特に4−、または8−キノリニル;インドリル、特に3−インドリル;チアゾリル、トリアゾリル、テトラゾリル、ベンゾ[d]ピラゾリル、チエニル、ならびにフラニルからなる群からを選択される。
【0055】
特に好ましい本発明の態様では、ヘテロアリール基R3は、トリアゾリル、特に4位もしくは5位にさらに置換基R4を有する1−トリアゾリル;テトラゾリル、特に4位もしくは5位にさらに置換基R4を有する1−テトラゾリル、または5位にさらに置換基R4を有する2−テトラゾリル;イソオキサゾリル、特に5位にさらに置換基R4を有する3−イソオキサゾリル、または3位にさらに置換基R4を有する5−イソオキサゾリル;あるいはチエニル、特に3位、4位、もしくは5位、好ましくは4位にさらに置換基R4を有する2−チエニル、または4位にさらに置換基R4を有する3−チエニルである。
【0056】
最も好ましいものは、4位もしくは5位に置換基R4を有するトリアゾリルとしてのヘテロアリール基R3であり、また4位もしくは5位に置換基R4を有する2−チエニルとしてのR3である。
【0057】
場合により置換されている不飽和アルキル基R3は、1位もしくは2位に、好ましくは2位にさらに置換基R4を有する1−アルケニル;または1−アルキニルである。1−アルケニルで考慮される置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。特に好ましい本発明の態様では、R3は1−アルキニルである。
【0058】
場合により置換されている不飽和シクロアルキル基は、1位に不飽和で5〜7個の炭素原子を有するシクロアルケニル基であり、例えば任意の位置にさらに置換基R4を有する、1−シクロペンテニルまたは1−シクロヘキセニルである。考えられる置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0059】
場合により置換されている不飽和ヘテロシクリル基は、3〜12個の原子;窒素、酸素、硫黄から選択した1〜5個のヘテロ原子;およびヘテロシクリル基をメチレンCH2に結合する位置にある二重結合を有する。考えられる置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0060】
特に、場合により置換されている不飽和ヘテロシクリル基は、複素環式芳香族基R3について先に定義した部分的に飽和した複素環式芳香族基である。そのようなヘテロシクリル基の一例は、イソオキサゾリジニル、特に5位にさらに置換基を有する3−イソオキサゾリジニル、または3位にさらに置換基を有する5−イソオキサゾリジニルである。
【0061】
アリールメチルを意味するR5では、アリールは、フェニルまたはナフチルであることが好ましく、特に、フェニルまたは置換されたフェニル、例えば、メチルやエチルなどの低級アルキル、メトキシなどの低級アルコキシ、フッ素や塩素などのハロゲン、アミノ、またはアシルアミノによってパラ位もしくはメタ位で置換されたフェニルである。
【0062】
ヘテロアリールメチルを意味するR5では、ヘテロアリールは、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)であり、そして5〜12個の、好ましくは5または6個の環原子を有し;そして置換されていなくても、あるいはメチルなどの低級アルキル、メトキシもしくはエトキシなどの低級アルコキシ、例えば塩素、臭素、フッ素などのハロゲン、トリフルオロメチルもしくはヒドロキシなどのハロゲン化した低級アルキルからなる群から選択される、一つもしくは複数の、特に一つの、さらなる置換基によって置換されていてもよい。ヘテロアリールメチルR5のヘテロアリールとして好ましいものは、ヘテロアリールR3下で好ましいものとして記載したヘテロアリール、例えばトリアゾリルや2−チエニルである。
【0063】
場合により置換されているシクロアルキルとしてR5は、3〜7個の炭素原子を有するシクロアルキル基であり、例えば、任意の位置に場合により置換基を有する、シクロプロピル、シクロペンチル、またはシクロヘキシルである。考えられる置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0064】
場合により置換されているシクロアルケニルとしてR5は、任意の位置に、例えば1位に不飽和で5〜7個の炭素原子を有するシクロアルケニル基であり、例えば、任意の位置に場合により置換基を有する1−シクロペンテニルや1−シクロヘキセニルである。考えられる置換基は、シクロアルキルR5下で挙げた置換基である。
【0065】
場合により置換されているヘテロシクリル基としてR5は、飽和もしくは不飽和であり、3〜12個の原子、および窒素、酸素、硫黄から選択した1〜5個のヘテロ原子を有する。考えられる置換基は、例えば、メチルなどの低級アルキル、ビニルやアリルなどの低級アルケニル、アセチレニルなどのアルキニル、フェニルなどのアリール、トリフルオロメチルなどのハロ低級アルキル、ヒドロキシメチルなどのヒドロキシアルキル、ヒドロキシ、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、カルボキシ、カルバモイル、メトキシカルボニルなどの低級アルコキシカルボニル、アミノ、アセチルアミノなどのアシルアミノ、ニトロ、アジド、シアノ、低級アルキル−またはアミノ低級アルキル−スルフェニル、−スルフィニル、または−スルホニル、あるいはクロロなどのハロゲニルがある。好ましいヘテロシクリルは、例えば、2−テトラヒドロフラニルなどのテトラヒドロフラニルである。
【0066】
先に定義したR5は、例えば、2−テトラヒドロフラニル中に1個以上のキラル中心を有し、野生型hAGTによって唯一の鏡像異性体(またはジアステレオマー)を選択的に認識するかもしれない。
【0067】
好ましいR5は、シクロペンチル、シクロヘキシル、ベンジル;ヒドロキシ置換したシクロペンチル、シクロヘキシル、もしくはベンジル;およびヒドロキシ、もしくはヒドロキシ低級アルキル置換したテトラヒドロフラニルである。
【0068】
基質の標識L1およびL2は、融合蛋白質が意図される適用に依存して、当業者が選択することができる。標識は、例えば、標識L1を有する標識融合蛋白質を容易に検出し、または標識L1をその環境から分離するようなものであってよい。考えられる他の標識は、標識した融合蛋白質および/または基質環境において変化を感知し誘発することができる標識、あるいは基質の物理的および/または化学的特性によって融合蛋白質を操作することに役立つ標識で、融合蛋白質に特異的に導入されたものである。
【0069】
標識L1およびL2の例には、フルオロフォアや発色団などの分光プローブ、磁気プローブ、造影剤;放射性に標識した分子;パートナーに特異的に結合することができる特異的結合対の片方である分子;他の生体分子と相互に作用すると推測される分子;他の生体分子と相互に作用すると推測される分子ライブラリ;他の分子に架橋結合ことができる分子;繋留した金属キレートなど、H2O2およびアスコルバートに曝露されるとヒドロキシルラジカルを発生することができる分子;マラカイトグリーンなど、光を照射すると反応性ラジカルを発生することができる分子;固体支持体に共有結合している分子であって、支持体がガラススライド、マイクロタイタープレート、または当業者に公知の任意のポリマーであってよい分子;その相補鎖と塩基対を形成することができる核酸もしくはその誘導体;膜挿入特性を有する脂質もしくは他の疎水性分子;望ましい酵素的、化学的、もしくは物理的性を有する生体分子;あるいは上で挙げた特性の任意の組合せを有する分子が含まれる。
【0070】
さらに、標識L1およびL2は、結合させた分子が生細胞原形質膜を越えて移動するのを促進することが知られている正電荷の直鎖もしくは分枝鎖ポリマーである。これは、細胞膜透過性が低いか、そうでなければ実質的に生細胞の細胞膜不透過性である物質では特に重要である。細胞不透過性AGT基質は、そのような基L1またはL2にコンジュゲートすることによって細胞膜透過性になる。そのような細胞膜輸送エンハンサ基L1およびL2には、例えば、6〜15個のアルギニン残基を有するD−および/またはL−アルギニンの直鎖ポリ(アルギニン)、それぞれグアニジウム基を有する6〜15個のサブ単位の直鎖ポリマー、その一部がグアニジウム基に結合する6〜50サブユニットのオリゴマーもしくは短鎖ポリマー、および/またはHIV−tat蛋白質配列、特にサブユニットTat49−Tat57(一文字アミノ酸コードでRKKRRQRRR)配列の一部が含まれる。AGT基質は、先に定義したリンカーR2またはR4によってこのL1またはL2基に共有結合し、このリンカーは生細胞内側で不安定であるのが好ましく、例えば、細胞内エステル分解酵素によるエステル基R2もしくはR4の切断によって分解し、直接的に、またはエステル官能基の切断によって誘発されたさらに別の反応で、AGT基質と、細胞膜透過性を亢進する単位L1およびL2を分離しうる。
【0071】
好ましいものは、放射性標識分子を除く先に記載した標識L1およびL2である。本発明の範囲から除外されるものは、核酸を意味する標識L2である。標識L1として最も好ましいものは、分光プローブ、およびパートナーに特異的に結合することができる特異的結合対の片方である分子、いわゆる親和性標識である。標識L2として最も好ましいものは、パートナーに特異的に結合することができる特異的結合対の片方を表す分子、および固体支持体に共有結合している分子である。
【0072】
標識L1またはL2が、フルオロフォア、発色団、磁気標識、放射性標識などである場合、検出は、その方法がインビトロもしくはインビボで使用されようが、標識に適合させた標準的手段によって行われる。L1がフルオロフォアである場合、その方法を緑色蛍光蛋白質(GFP)の適用例と比較することができ、この緑色蛍光蛋白質は対象となる蛋白質に遺伝子的に融合され、生細胞において蛋白質研究を可能にするものである。標識L1およびL2の特定の例には、非線形光学特性を示すホウ素化合物もある。特に好ましいものは、2個の相互作用する分光プローブL1/L2の、L1が一構成要素であり、L2が他方の構成要素であって、ドナーとアクセプターが密接(10nmの距離未満)している場合、動的または静的消光によって、エネルギーをドナーとアクセプター(クエンチャー)間で無放射に移動できるようにする標識である。そのような1対の標識L1/L2は、標識基質とAGT融合蛋白質の反応においてその分光学的特性を変化させる。そのような1対の標識L1/L2の一例は、以下にさらに詳細説明するFRET対である。
【0073】
標識L1の特性に応じて、対象となる蛋白質とAGTを含む融合蛋白質は、基質と反応すると固体支持体に結合しうる。AGTを含む融合蛋白質と反応する基質の標識L1は、予め固体支持体に結合させておいてよく、AGTと反応し始めたとき、あるいは続いて、すなわちAGTに移動後、標識したAGT融合蛋白質を固体支持体に結合させるために使用しうる。あるいは、基質の標識L2は、固体支持体であっても、または固体支持体に結合してあっても、結合することもでき、この固体支持体によって、反応後に標識L1を有する標識融合蛋白質が基質残余部分から分離しL2を含むようになる。標識は、特異的結合対の一構成要素でよく、特異的結合対の他方の構成要素は、共有結合によって、または他の任意の手段によって固体支持体に結合し、または結合可能である。考えられる特異的結合対は、例えば、ビオチン、およびアビジンもしくはストレプトアビジンである。結合対のいずれかの構成要素は、基質の標識L1および/またはL2でよく、他方は固体支持体に結合されている。固体支持体へ好都合に結合できるようにする標識のさらに別の例には、例えば、マルトース結合蛋白質、糖蛋白、FLAGタグ、あるいはそのような置換基と、固体支持体の表面上の相補的官能基の間の化学選択的反応を可能にする反応性置換基がある。反応性置換基および相補的官能基のそのような対の例には、例えば、アミド、アジドを形成するアミンおよび活性化したカルボキシ基;および1,3−双極子環化付加反応を起こすプロピオル酸誘導体;2個のアミド結合を生じる活性化したビスジカルボン酸誘導体型の付加されたニ官能基リンカー試薬と反応する、アミンおよび別のアミン官能基、あるいは当技術分野で公知の他の組合せがある。
【0074】
好都合な固体支持体の例は、例えば、ガラススライドなどのガラス表面、マイクロタイタープレート、および適当なセンサー素子、特に機能性を付与したポリマー(例えばビーズ形)、化学的に改変した酸化物表面、例えば、二酸化ケイ素、タンタル五酸化物、もしくは二酸化チタン、または化学的に改変した金属表面、例えば金や銀表面などの貴金属表面である。次いで、不可逆的に結合させ、かつ/またはスポットしたAGT基質を使用してAGT融合蛋白質を空間的に離して、特に、固体支持体にスポットすることによって結合させ、蛋白質マイクロアレイ、DNAマイクロアレイ、または小分子のアレイを表してよい。
【0075】
標識L1またはL2が、外部刺激への曝露によってヒドロキシルラジカルなどの反応性ラジカルを生成できる場合、生成したラジカルは次いでAGT融合蛋白質、およびAGT融合蛋白質に密接しているような蛋白質を不活化することができ、これらの蛋白質の役割を研究できるようになる。そのような標識の例には、H2O2およびアスコルビン酸塩に曝露されるとヒドロキシルラジカルを発生する繋留した金属キレート錯体、およびレーザー照射によってヒドロキシルラジカルを発生するマラカイトグリーンなどの発色団がある。ヒドロキシルラジカルを発生させるために、発色団およびレーザーを使用することもまた、クロモホア援助レーザー誘起不活化法(CALI)として当技術分野で公知である。本発明では、標識L1としてマラカイトグリーンなどの発色団を有する基質でAGT融合蛋白質を標識し、続いてレーザー照射することによって、時間を制御し空間的に離して、標識したAGT融合蛋白質、およびAGT融合蛋白質と相互に作用するような蛋白質を失活させる。この方法は、インビボ、またはインビトロで適用することができる。さらに、AGT融合蛋白質に密接する蛋白質は、特異的抗体による蛋白質フラグメントの検出、高解像度2D電気泳動ゲル上でそれらの蛋白質の消失、あるいは分離による切断した蛋白質フラグメントの同定、および質量分析やN末端分解による蛋白質配列決定などの配列決定技術によって同定することができる。
【0076】
標識L1が、他の蛋白質、例えば、マレイミド、活性エステル、アジド、および当業者に公知の他のものなど、官能基を含む分子に架橋できる分子の場合、他の蛋白質と(インビボもしくはインビトロで)相互に作用するAGT融合蛋白質にそのような標識したAGT基質を接触させると、標識を介してAGT融合蛋白質とその相互作用する蛋白質の共有結合性架橋がもたらされる。これによりAGT融合蛋白質と相互に作用する蛋白質を同定できるようになる。光架橋用の標識L1(およびL2)は、例えば、ベンゾフェノンである。架橋の特殊な局面では、標識L1は、それ自体、AGT融合蛋白質を二量体化するAGT基質の分子である。そのような二量体の化学構造は、対称(ホモ二量体)でも、非対称(ヘテロ二量体)でもよい。
【0077】
考えられる他の標識L1は、例えば、フラーレン、中性子捕獲治療用ボラン、ヌクレオチド、オリゴヌクレオチド、例えば、自己アドレス指定(self-addressing)チップ用のもの、ペプチド核酸、および金属キレート、例えば、DNAに特異的に結合する白金キレートである。
【0078】
望ましい酵素的、化学的、もしくは物理的性を有する特定の生体分子はメトトレキサートである。メトトレキサートは、酵素、ジヒドロ葉酸還元酵素(DHFR)に強固に結合する阻害薬である。L1がメトトレキサートである式(1)の化合物は、いわゆる「二量体化化学誘導物質」(CID)の周知のクラスに属する。DNA結合ドメインLexAを有する融合蛋白質hAGTを使用し、L1がメトトレキサートである式(1)の化合物を有するhAGT融合蛋白質のインビボ標識に転写活性化ドメインB42を有するDHFRを加えることによって、hAGT−LexA融合蛋白質とDHFR−B42融合蛋白質のカップリング(「二量体化」)が誘発され、LexAとB42が空間的に近接し、続いて転写が刺激される。
【0079】
基質が2個以上の標識を有する場合、これらの標識は、同一でもまたは異なってもよい。特定の好ましい組合せは、異なる2個の親和性標識、または1個の親和性標識と1個の発色団標識、特に、1個の親和性標識と1個のフルオロフォア標識、または1対の分光学的に相互作用する標識L1/L2、例えばFRET対である。
【0080】
好ましいものは、Xが酸素である式(1)の化合物である。さらに好ましいものは、Xが酸素であり、R3がフェニルである化合物であり、特に、パラ置換フェニルもしくはチエニル、特に2,4−2置換チエニルである。
【0081】
同様に好ましいものは、基R1−Aが式(2)[式中、R6は、水素、ヒドロキシ、または非置換もしくは置換アミノであり、非置換アミノが好ましく、R7およびR8の一方はR1であり他方は水素である]のプリンラジカルである式(1)の化合物である。R7がR1である場合、R1は基−R2−L2もしくは残基R5であってよい。特に好ましいものは、R8がR1であり、R1が基−R2−L2である化合物に対応する。R1が残基R5である場合、好ましいR5の意味はシクロペンチルである。
【0082】
他の好ましい化合物は、基R1−Aが
式(3)の8−アザプリンラジカル[式中、置換基R6は水素、ヒドロキシ、または非置換もしくは置換アミノであり、非置換アミノが好ましい]、
式(4a)または(4b)のピリミジンラジカル[式中、R9は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、またはアミノであり、かつR10は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、ニトロ、またはニトロソである]、および
式(4c)プテリジンラジカル[式中、R6は、非置換もしくは置換アミノであり、非置換アミノが好ましく、かつR7およびR8の一方はR1であり他方は水素である。好ましくは、R7が水素であり、R8が基−R2−L2である]
である式(1)の化合物である。
【0083】
好ましいものは、L2が分光プローブである化合物、特にL1およびL2が分光プローブである化合物、例えば、FRET対を表す。同様に好ましいものは、L2が特異的結合対の片方を表す分子である化合物、L2が固体支持体に共有結合している分子である化合物、およびL2が細胞膜輸送エンハンサ基である化合物である。
【0084】
最も好ましいものは、実施例の化合物である。
【0085】
本発明は、さらに、対象となる蛋白質を検出し、かつ/または操作する方法であって、対象となる蛋白質をAGT融合蛋白質中に組み込み、該AGT融合蛋白質を先に記載した標識を有する特定のAGT基質に接触させ、そして、標識を認識しかつ/または取り扱うために設計した系で標識を使用して、該AGT融合蛋白質を検出し、場合によりさらに操作する方法に関する。
【0086】
本発明の方法では、対象となる蛋白質またはペプチドをO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)に融合する。対象となる蛋白質またはペプチドは、任意の長さでよく、どちらも二次、三次、または四次構造を有していても有していなくてもよく、かつ少なくとも12アミノ酸〜2000アミノ酸からなることが好ましい。そのような対象となる蛋白質またはペプチドの例には、例えば、酵素、DNA結合蛋白質、転写調節蛋白質、膜蛋白質、核内受容体蛋白質、核移行シグナル蛋白質、蛋白質補因子、小モノマーGTP加水分解酵素、ATP結合カセット蛋白質、細胞内構造蛋白質、特定の細胞区画に蛋白質を向かわせる配列を有する蛋白質、一般的に標識もしくは親和性タグとして使用される蛋白質、および前述の蛋白質ドメインもしくはサブドメインがある。対象となる蛋白質またはペプチドは、酵素によって、例えば、DNA期に適当な制限酵素によって切断されうるリンカー、および/または蛋白質期に適当な酵素によって切断可能なリンカーを経てAGTに融合されていることが好ましい。
【0087】
O6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)は、基質上に存在する標識をAGTのシステイン残基の一つに移動し、融合蛋白質の一部を形成する特性を有する。好ましい態様では、AGTは、野生型ヒトO6−アルキルグアニンDNAアルキルトランスフェラーゼ、hAGT、またはその変異体、例えば、Juilleratら., Chem Biol 10:313-317, 2003もしくは後の実施例に記載されている変異体である。本発明で使用される変異体AGTは、一個以上のアミノ酸置換、欠失、または付加という点で野生型hAGTと異なってよいが、依然として基質上に存在する標識を融合蛋白質のAGT部分に移動する特性を保持している。変異体AGTは、当業者に公知の蛋白質設計技術、例えば、飽和変異誘発、配列の任意の場所に変異を導入するためのエラープローン(error prone)PCR、あるいは飽和変異誘発および/またはエラープローンPCR後に使用されるDNAシャフリングを使用し生成されることが好ましいであろう。
【0088】
対象となる蛋白質およびO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)を含む融合蛋白質は、標識を有する特定の基質と接触させる。反応条件は、AGTが基質と反応し、基質の標識を移動させるように選択する。通常の条件は、pH7付近、室温、例えば、およそ25℃の緩衝液である。しかし、AGTは様々な他の条件下でも反応し、ここに述べたような条件が本発明の範囲を制限することはないことは理解されよう。
【0089】
反応速度は、基質の構造に非常に大きく依存する。本発明に記載した化合物は、反応性に違いがあり、これらの差は、異なるO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)、例えば異なる種に由来する野生型AGT、すなわち特に実施例で示す、野生型AGTの幾つかのアミノ酸が異なるアミノ酸によって置き換えられているAGT変異体と、本発明の化合物とを選択的に反応させるために使用しうる。
【0090】
反応性の差は、例えば、式(1){式中、Xは酸素であり、R3はパラ置換フェニルであり、基R1−Aは式(2)[式中、R6は非置換アミノであり、R7およびR8の一つは基−R2−L2であり、他方の一つは水素である]のプリンラジカルである}の化合物に見られる。R7が水素であり、R8が基−R2−L2であるような化合物は、R7が基−R2−L2(例えば同じ基−R2−L2)でありR8が水素である、対応する化合物よりも少なくとも3倍迅速に反応し、さらにR7とR8が水素である化合物よりも迅速に反応する。
【0091】
AGTは、その天然基質O6−アルキルグアニンDNAからアルキル基をそのシステイン残基の一つへ不可逆的移動する。同様に、AGTは、本発明の基質が関連する位置にあるグアニン型O6置換基またはその対応する置換基をそのシステイン残基の一つに移動する。AGTのこの特性を本発明の方法に使用して、式(1)の化合物の残基CH2−R3−R4−に結合した標識L1をAGTに移動する。
【0092】
この基質の標識L1は、融合蛋白質を意図したその適用に依存して当業者が選択することができる。AGTを含む融合蛋白質を基質と接触させた後、標識L1を融合蛋白質に共有結合させる。次いで、移動した標識によって、標識したAGT融合蛋白質をさらに操作し、かつ/または検出する。標識L1は、複数の同じもしくは異なる標識からなってよい。基質が、一つを超える標識L1を含む場合、その対応する標識したAGT融合蛋白質は、標識した融合蛋白質をさらに操作し、かつ/または検出するための選択肢をさらに提供する標識を一つを超えてさらに含む。
【0093】
特定の局面では、本発明は、標識L1をAGT融合蛋白質に移動させ、全ての未反応AGT基質を除去するのに特に好都合な方法を提供する。R2を介して基質としてAGTによって認識される複素環基Aに結合された標識L2が、固体支持体を意味し、または固体支持体に結合可能な反応基、例えば、固体支持体に結合するパートナーに特異的に結合することができる特異的結合対の片方を意味する標識L2である場合、これは簡単に行われる。次いで、L1で標識されたAGT融合蛋白質を含む、固体支持体および蛋白質溶液は、例えば、ろ過によって、さらに別の操作をせずに容易に分離される。より具体的には、分子過剰な式(1)[式中、R1はR2−L2であり、L2は固体支持体または固体支持体に結合された基である]の基質をAGT融合蛋白質と反応させ、得られた標識L1を有する標識AGT融合蛋白を固体支持体から分離するが、この固体支持体には反応した基質の残りの部分、すなわち残基R2−Aが結合し、さらにこの固体支持体には式(1)の過剰な未反応基質が依然として結合している。他の点では、AGT融合蛋白質は、基質と反応すると固体支持体から標識L1を離し、引き継ぎ、固体支持体に結合している全ての未反応標識L1はそのまま残される。
【0094】
この特定の局面では、標識L1は、先に記載した任意の標識、例えば、分光プローブまたはフルオロフォアでよい。固体支持体すなわちL2、またはL2が結合する支持体は、任意の固体支持体、例えば、ビーズ(例えば磁気ビーズ)、ポリマー支持体、金属表面でよい。未反応基質から標識したAGT融合蛋白質を分離することは、固体支持体の特性に応じて、ろ過、遠心分離、または他の適当な方法によってよく、例えば、固体支持体が磁気ビーズの場合は磁場をかけてよい。
【0095】
本発明の別の特定の局面では、AGT融合蛋白質による反応において標識L1およびL2の空間的分離を利用する方法を提供する。本発明の特定の基質は、相互作用性分光学的標識として、例えば、一端(例えばL1)にドナー(レポーター)を、他端(L2)にアクセプター(クエンチャー)を共有結合標識し、逆も同様であるプロ蛍光プローブとして設計される。そのようなプローブでの消光は、蛍光共鳴エネルギー移動(FRET)、またはドナーアクセプター対が密接することによる静的消光により生じうることが一般的に知られている。ドナーとアクセプターは、そのスペクトルの重なりが最大になるように選択すべきである。蛍光エネルギー転移は、典型的には、20〜100Åまでの距離で生じる。
【0096】
L1がドナー(レポーター)であり、L2がアクセプター(クエンチャー)であり、またはL1がクエンチャーであり、L2がレポーターである特定の本発明の方法では、AGT融合蛋白質と基質の反応によって蛍光が変化する。二重に標識した基質内のリポーター−クエンチャー間距離が、AGT融合蛋白質との反応によって変化し、レポーターとクエンチャーを空間的に分離し、それによって蛍光が出現する。それぞれ、標識L1またはL2として例えば赤外放射フルオロフォアを含む幅広い選択肢のレポーター基を使用してよく、唯一のフルオロフォア標識L1を有する基質と比較して、方法の感受性および特異性がかなり改善する。レポーターとクエンチャーを含む基質は、AGT融合蛋白質と反応するまで暗いままであり、反応混合物が「点灯」されると、フルオロフォアの発光スイッチが入る。なぜならば、今度は、レポーター標識とクエンチャー標識が、空間的に分離されるからである。蛍光消光およびエネルギー転移は、2個の標識、消光した標識またはエネルギードナー標識の一つだけが発光することによって測定できる。エネルギー転移が生じ、エネルギー受容標識も蛍光である場合、アクセプター標識蛍光も測定することができる。これらの2個の相互作用性標識のドナー標識は、励起ランプの必要性を取り除き、アクセプターバックグラウンド蛍光を低減する化学発光ドナープローブから選択することができる。そのような二重標識した基質を使用する記載した特定の方法は、蛍光時間測定に基づく反応速度の定量に有用であり、インビボおよびインビトロで利用してよい。
【0097】
インビトロでは、一般的に、AGT融合蛋白質と本発明の基質の反応は、細胞抽出物中で、またはAGT融合蛋白質の精製体もしくは濃縮体を用いて実施することができる。
【0098】
本発明の基質を用いる実験をインビボでまたは細胞抽出物中で行う場合、宿主の内在性AGTの反応が有利に考慮される。宿主の内在性AGTが、O6−アルキルグアニン誘導体、または基質として関連する化合物を受け入れない場合、(外来性)AGT融合蛋白質の反応は特異的である。ヒト、マウス、ラット細胞などの哺乳動物細胞中で内在性AGTを用いた非特異的反応が可能である。内在性AGTと(外来性)AGT融合蛋白質の同時反応に問題があるような実験では、公知のAGT欠乏細胞系を使用することができる。
【0099】
記載した特定の本発明の態様では、レポーターとクエンチャーを有する二重に標識した基質によって、インビトロもしくはインビボでAGT融合蛋白質の濃度を定量できるようになる。インビボでの適用については、レポーターは、近赤外(NIR)領域のエミッターであることが好ましい。この領域が生物蛍光に干渉しないからである。これらの要件に合致する公知のシアニンNIR色素を本発明の基質に組み込むことが好ましい。
【0100】
適当なレポーターとクエンチャー対は、当業者が選択することができる。典型的には、レポーターとクエンチャーは、スペクトルが大きく重なる蛍光色素であり、例えば、レポーターとしてフルオレセイン、およびクエンチャーとしてローダミンである。他のクエンチャーは、金クラスタや金属クリプテートである。
【0101】
本発明で使用される第2クラスのクエンチャーは、「ダーククエンチャー」(Johnasson, M. K.ら, Chem. Eur. J. 9:3466-3471, 2003)であり、すなわち、天然蛍光を持たず、一般的なレポーター色素の発光スペクトルと重複し、最大のFRET消光をもたらす吸収スペクトルを有する色素である。一般的なレポーター色素の発光スペクトルと重複し最大のFRET消光をもたらす吸収スペクトルを有する天然蛍光を持たない色素である。ずさらに、基底状態の複合体内で共鳴双極子−双極子相互作用機序(静的消光)を促進するために、その吸収帯が重なるように色素対を選択することができる。
【0102】
考えられる特定のフルオロフォアおよびクエンチャーは以下のものである。Alexa350、Alexa488、Alexa532、Alexa546、Alexa555、Alexa635、Alexa647を含むAlexa色素(PanchukVoloshina, N.ら, J. Histochem. & Cytochem. 47:1179-1188, 1999)、ジメチルアミノクマリン(7−ジメチルアミノクマリン−4−酢酸スクシンイミジルエステル、Moelcular Probes社製、商品名D374)、クエンチャーQSY35、QSY9、およびQSY21(Molecular Probes社製、Eugene OR 97402、米国)、シアニン−3(Cy3)、シアニン5(Cy5)、およびシアニン5.5(Cy5.5)(Amersham - GE Healthcare, Solingen, Germany);BHQ−1、BHQ−2、およびBHQ−3(Biosearch Technologies, Inc.のBlack Hole Quencher(商標), Novato, CA 94949, USA)、フルオロフォアATTO488、ATTO532、ATTO600、およびATTO655、およびクエンチャーATTO540Q、およびATTO612Q(Atto-Tec, D57076 Siegen, Germany)、フルオロフォアDY−505、DY−547、DY−632、およびDY−647(Dyomics, Jena, Germany)、5/6−カルボキシフルオレセイン、テトラメチルローダミン、4−ジメチルアミノアゾベンゼン−4'−スルホニル誘導体(Dabcyl)、および4−ジメチルアミノアゾベンゼン−4'−カルボニル誘導体(Dabcyl)。これらは、以下の組合せで有利に併用される。
フルオロフォア クエンチャー
・Alexa350、ジメチルアミノクマリン・Dabsyl、Dabcyl
5/6−カルボキシ−フルオレセイン、 BHQ1、QSY35
Alexa488、ATTO488、
DY−505
・5/6−カルボキシフルオレセイン、 ・BHQ2、QSY9、
Alexa488、Alexa532、 ATTO540Q
Alexa546、Alexa555、
ATTO488、ATTO532、
テトラメチルローダミン、Cy3、
DY−505、DY−547、
・Alexa635、Alexa647、 ・BHQ3、ATTO612Q、
ATTO600、ATTO655、 QSY21
DY−632、Cy5、DY−647
Cy5.5
【0103】
特定の態様では、この方法には、L2が固体支持体であり、もしくはレポーター/クエンチャー対の一構成要素をさらに有する固体支持体に結合しており、またはL2が固体支持体とレポーター/クエンチャー対の一構成要素の組合せであり、かつL1がこの対の他方の構成要素である基質が関与する。このように、AGT基質であるダーク固体支持体は、AGT融合蛋白質と反応すると蛍光性になる。
【0104】
レポーター/クエンチャー対、例えば、FRET対に基づく蛍光の定量が関与する記載の特定の方法は、相当の蛋白質調製物、および非特異的シグナルを除去するための条件操作を必要とする、SDS/PAGE、ウェスタンブロッティングなどに基づく濃度または反応速度を測定用の他の方法よりもはるかに好都合である。エネルギー移動法は、反応および未反応のAGT基質を分離する必要がない。従って、この方法は、化合物ライブラリの高処理スクリーニング、活性AGT変異体の同定、またはAGT阻害薬の同定に特に好都合である。
【0105】
さらに別の特定の本発明の方法では、R1が、基R5、特に、シクロアルキル、シクロアルケニル、シクロアルキルメチル、アリールメチル、またはヘテロアリールメチル基である基質を使用する。好ましい基R5の例は、シクロペンチルおよびベンジルである。AGT融合蛋白質では、変異体AGTは融合パートナーとして選択され、このパートナーは、R5が記載した特定の、または好ましい置換基の一つである基質に対して反応性が低い。そのような特定の対象となる蛋白質を検出しかつ/または操作する方法では、対象となる蛋白質は、変異体AGTと融合され、変異体AGT融合蛋白質を(a)基質であって、R1は基R5であり、変異体AGTによって認識されない基質、および(b)別のAGT基質であって、変異体AGT融合蛋白質によって認識され、先に記載した標識を有する基質の混合物に接触させ、標識を認識しかつ/または取り扱うために設計した系で標識を使用し、変異体AGT融合蛋白質を検出し、場合によりさらに操作する。そのような方法は、反応が内在性AGTを含む系においてインビボで実施され、または内在性AGTを含む細胞抽出物中で実施される場合の選択方法である。内在性AGTが飽和し、必要な場合は、R1が基R5である本発明の基質の標識L1を使用し分離する。並列反応では、変異体AGT融合蛋白質は、先に記載した一つの標識もしくは複数の標識を有する別のAGT基質と反応し、適宜検出し、かつ/または操作する。
【0106】
製造方法
本発明の基質は、一般的に、当技術分野で公知の標準的方法によって調製される。特定の方法は、例えば、特許出願PCT/EP03/10889に説明される。式(2)の基質、すなわち、Aがプリン物質である化合物の合成では、公知の方法を使用してO6、C8、およびN9の位置を改変することができる。本発明は、以下に記載する新規な方法、および使用し得られた新規な中間体にも関する。
【0107】
特に、複数の標識(例えばL1およびL2)を有するAGT基質の好ましい合成は、直交的に保護された官能基を利用する。保護基をそのように選択することによって、脱保護の分離が可能になり、その結果、各遊離された官能基を順次さらに化学的に操作して、標識をそれに結合させ、あるいはリンカーR2および/またはR4のそれ以上の伸展を導入することができる。想定される官能基に適した保護基は、当業者が選択することができ、例えば、T.W. GreeneおよびP.G.M. Wuts in "Protective Groups in Organic Synthesis", John Wiley & Sons, New York 1991に要約されている。
【0108】
グアニン誘導体の調製には、アンモニウム塩中間体を介した対応する6−クロログアニン誘導体と、三級アミン例えばメチルピロリジンとの反応に、基−CH2−R3−R4−L1もしくはその前駆体を導入する。アルコラート−O−CH2−R3−R4−L1もしくはその前駆体によりアンモニウム基を置き換えることによって、塩素を直接置き換えるのに比べて全体収量が多くなり、アンモニウム塩との反応を室温で実施することができる。
【0109】
式(2)のグアニンのN9位への基R1の導入は、ハロゲン誘導体R1−Halを使用するSN2反応による直接アルキル化によって実施しうる。しかし、位置選択性は、一般的に不十分であり、N7およびN9のアルキル化したグアニン生成混合物が得られる。アルキル化した生成物のN9/N7割合は、LiHで前処置し、またはN9置換に好都合な高温で得られた生成物を平衡化ことによってイミダゾール環を活性化すると高くなる。式(5)の好都合な中間体の調製をスキーム1に示す。
【0110】
【化12】
【0111】
式(5)の中間体は、N9のリンカーおよびO6位のベンジル官能基で直交的に保護されている。次いで、この中間体をさらに操作して残基−R4−L1をO6のベンジル官能基で、残基−R2−L2をN9位で完成させてよい。あるいはMitsunobuの条件(アルコール誘導体R1−OH、トリフェニルホスフィン、ジエチルアゾジカルボキシラート)を、N9位に置換基R1(すなわちR5、または−R2−L2)を導入するために使用してよい。
【0112】
中間体(5)に密接に関係するが、N9位にアジドプロピル基を有する化合物の合成を実施例8〜10の実験パートに例示する。
【0113】
【化13】
【0114】
スキーム2は、N9位に置換基を位置選択的に導入する閉環反応を示す。式(6)の中間体は、既に、標識L1を含み、アジド官能基を手直しすることによって適当な標識L2を導入できるようになる。
【0115】
トリフェニルホスフィンによって式(6)の化合物のアジド官能基を還元し、QSY−9−NHS(Molecular Probes)によってアシル化すると、式(7)[式中、L1(カルボキシ−フルオレセイン)およびL2(QSY−9)は、FRET対(スキーム3)を表す]のプロ蛍光AGT基質が得られる。
【0116】
【化14】
【0117】
標識L1およびL2を、それぞれ、O6位およびN9位に有し、FRET対を表す、さらに別の(7)型化合物を以下の実施例14、15、および19に記載する。
【0118】
N9の位置選択的アルキル化の別の方法は、スキーム4に示すパラジウム触媒アリル位アルキル化である。
【0119】
リンカー部分R2のレベルで構造を手直しするために、適当な官能性を持たせたホモ−N,O−ヌクレオシドの合成も同様にスキーム4に示すが、このヌクレオシドのグアノシンの糖部分はイソオキサゾリジン環によって置き換えられている(TBDPS=tert−ブチル−ジフェニル−シリル)。そのような手直しによって、一般的ヌクレオシドの比較的反応性アミン結合と比較して加水分解切断または酵素切断への耐性の増大、および高次構造の柔軟性の上昇が導入される。反応配列は可変であり、アリル二重結合に異なる適当な官能性を持たせたニトロンを結合できるようになる。
【0120】
あるいは、水素化ホウ素試薬9−ボラビシクロ[3.3.1]ノナン(9−BBN)によって、N9のアリル基を一級ヒドロキシ官能基に転換し、次いで適当な基R5または−R2−L2を得るためにさらに操作するすることができる。
【0121】
【化15】
【0122】
【化16】
【0123】
L1がフルオロフォア(Alexa594、Molecular Probes)であり、L2が固体支持体(例えばビーズB、またはアビジンもしくはストレプトアビジンを有するビーズに結合可能なビオチン残基B)である、式(9)の本発明の基質の合成をスキーム5に要約する。
【0124】
異なる2種の標識がプリン塩基のO6位およびC8位に結合している式(1)の化合物の合成に使用可能な中間体の合成をスキーム6および7に要約する。
【0125】
【化17】
【0126】
【化18】
【0127】
式(11)の化合物の代替合成法を実施例20〜23に例示する。この代替合成法では、p−(トリフルオロアセトアミドメチル)ベンジル置換基は初期に導入される。次いで、二重に保護された式(11)の化合物は、標識L1およびL2を、それぞれ、O6位およびC8に有する化合物、例えば、実施例24〜42に記載されている化合物にさらに作り上げることができる。
【0128】
R1−Aが式(4a)のピリミジン残基であり、R9がアミノであり、R10がニトロソである式(1)の化合物の調製については、スキーム8または9に示す反応順序を実施する。4−クロロ−2,6−ジアミノピリミジンを適当なナトリウムアルコキシドと反応させ、次いで亜硝酸ナトリウムの30%酢酸溶液で処理してニトロソ官能基を導入する。N6−置換誘導体は、適当な無水物または塩化アシルで処理することによって調製される。
【0129】
【化19】
【0130】
化合物(12)の直前の前駆体2,6−ジアミノ−5−ニトロソピリミジンをその対応する2,5,6−トリアミノピリミジンに還元する場合、式(4c)のプテリジンはジヒドロキシアセトンによる縮合、および実施例43〜46に記載した側鎖をさらに作成することによって得られる。
【0131】
【化20】
【0132】
N6位へのアミノアルキル置換基の導入をスキーム9に示す。11−アジド−3,6,9−トリオキサ−ウンデカ−アミンと2,4,6−トリクロロピリミジンの反応によって、クロマトグラフィーによって分離することができる2,6−二置換生成物と共に、6−置換クロロピリミジンが得られる。マスクをかけたアンモニア同等物として、2位をビス−p−メトキシ−ベンジルアミン(PMB)で置換することによって2置換ピリミジンが生じ、この2置換ピリミジンは、続いて、適当なアルコキシドとの反応によりO4誘導体へ転換される。60℃でトリフルオロ酢酸により脱保護すると2−アミノピリミジンが遊離される。5−ニトロソ化合物は、酢酸に含まれる亜硝酸ナトリウムによって調製される。
【0133】
実施例
略語:
DMF=ジメチルホルムアミド
DMSO=ジメチルスルホキシド
DTT=ジチオトレイトール
equiv.=当量
MPLC=中圧液体クロマトグラフィー
sat.=飽和
THF=テトラヒドロフラン
TEA=トリエチルアミン
TLC=薄層クロマトグラフィー
【実施例1】
【0134】
(4−ブロモチオフェン−2−イル)−メタノール(14)
【0135】
NaBH4(1.11g、29.31mmol)を4−ブロモチオフェン−2−カルボアルデヒド(5g、26.17mmol)を含むイソプロパノール(70mL)溶液に加える。反応混合物を室温で2時間攪拌する。NH4Cl(15mL)の飽和水溶液を溶液に加える。懸濁液をろ過し、ろ液を減圧濃縮し、CH2Cl2(70mL)に溶解し、MgSO4で乾燥し、再度減圧濃縮する。残留物をフラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:10)により精製する。収量4.55g(23.55mmol、90%)。TLC:Rf=0.75(酢酸エチル/石油エーテル1:1)。
【0136】
【表1】
【実施例2】
【0137】
2−シクロペンテニルメチルカルボナート(15)
【0138】
シクロペンタ−2−エン−1−オール(1.0g、11.9mmol、J.-L. Luche, J. Am. Chem. Soc., 1978, 100:7, 2226-2227)を含むCH2Cl2(30mL)およびピリジン(10mL)の0℃の攪拌溶液にクロロぎ酸メチル(2.94mL、38mmol)を30分かけて滴下する。4時間後、TLCは完全反応を示し、反応混合物を飽和NH4Cl(50mL)に注ぎ、Et2O(3×50mL)で抽出する。有機相を洗浄液が酸性になるまで1M HClで洗浄し、水洗(50mL)し、塩水(50mL)洗浄し、MgSO4で乾燥する。粗生成物をフラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:100)によって精製する。収量1.777g(12.5mmol、53%)。TLC Rf=0.70(酢酸エチル/石油エーテル1:12)。
【0139】
【表2】
【実施例3】
【0140】
N9−(シクロペンタ−2−エニル)−6−クロログアニン(16)
【0141】
この反応は、無水条件下(アルゴン雰囲気下、かつ溶媒は分子篩にかける)で実施する。6−クロログアニン(596mg、3.52mmol)を含むDMSO(10mL)溶液に、Pd(PPh3)4(404mg、0.35mmol)を加え、続いて2−シクロペンテニルメチルカルボナート(13)(500mg、3.52mmol)を含むTHF(10mL)溶液を加える。反応混合物を室温で1時間攪拌する。それをH2O(60mL)に注ぎ、酢酸エチル(3×50mL)で抽出する。混合有機層を塩水(60mL)で洗浄し、MgSO4で乾燥し、減圧濃縮する。残留物をフラッシュカラムクロマトグラフィー(酢酸エチル/石油エーテル6 4)によって精製する。収量:381mg(1.62mmol、61%)。TLC Rf=0.33(酢酸エチル/石油エーテル6:4)。
【0142】
【表3】
【実施例4】
【0143】
N9−シクロペンチル−6−クロログアニン(17)
【0144】
パラジウム担持活性炭(100mg)をN9−(シクロペンタ−2−エニル)−6−クロログアニン(16)(200mg、0.85mmol)を含むメタノール(33mL)溶液に加える。水素ガスを30分間溶液に通す。粗生成物をシリカ(500mg)に吸着させ、フラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:4、3:7、および2:3)によって精製する。収量:111mg(0.47mmol、55%)。TLC Rf=0.34(酢酸エチル/石油エーテル1:1)。
【0145】
【表4】
【0146】
または、臭化シクロペンチル(200mg、1.34mmol)を6−クロログアニンを含むジメチルアセトアミド懸濁液に加え、続いてNaOMe(144mg、2.67mmol)を加える。溶液を100℃で終夜攪拌する。溶媒を減圧蒸発させる。残留物をシリカ(1g)に吸着させ、フラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:1)によって精製する。収量:140mg(0.59mmol、44%)。
【実施例5】
【0147】
N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(CPTG、18)
【0148】
N9−シクロペンチル−6−クロログアニン(17、50mg、0.21mmol)を含むDMF(1.3mL)溶液に1,4−ジアザビシクロ[2.2.2]オクタン(DABCO、71mg、0.63mmol)を加える。反応混合物を室温で3時間攪拌する。TLCは、完全反応を示す。次いで、(4−ブロモチオフェン−2−イル)−メタノール(14、49mg、0.25mmol)および1,8−ジアザビシクロ[5.4.0]−ウンデカ−7−エン(DBU)(96mg、0.094mL、0.63mmol)を含むDMF(0.7mL)溶液を反応混合物に加える。溶液を終夜室温で攪拌する。粗生成物をフラッシュクロマトグラフィーによって精製する(酢酸エチル/石油エーテル1:9→3:7)。収量25mg(0.063mmol、30%)。TLC Rf=0.23(酢酸エチル/石油エーテル1:1)。
【0149】
【表5】
【実施例6】
【0150】
N9−ベンジル−6−クロログアニン(19)
【0151】
フラスコに6−クロログアニン、PPh3、およびベンジルアルコールを装入する。混合物を3時間減圧乾燥し、続いて活性化分子篩(4Å)を加えた無水THFに溶解する。15分間攪拌した後、ジイソプロピルアゾジカルボキシラート(DIAD)を加える。反応を終夜攪拌する。溶媒を減圧下で除去し、生成物をカラムクロマトグラフィー(石油エーテル/酢酸エチル3:1)によって精製する。
【0152】
【表6】
【実施例7】
【0153】
N9−ベンジル−O6−(4−ブロモチオフェン−2−イル)−グアニン(20)
【0154】
N9−ベンジル−6−クロログアニン(19、50mg、0.19mmol)を含むDMF(1.5mL)溶液に、1,4−ジアザビシクロ[2.2.2]オクタン(DABCO、65mg、0.57mmol)を加える。反応混合物を室温で3時間攪拌する。TLCは、完全反応を示す。次いで、(4−ブロモ−チオフェン−2−イル)−メタノール(14,44mg、0.23mmol)および1,8−ジアザビシクロ[5.4.0]−ウンデカ−7−エン(DBU)(88mg、0.086mL、0.57mmol)を含むDMF(0.7mL)溶液を反応混合物に加える。溶液を終夜室温で攪拌する。粗生成物をフラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:4→3:7)によって精製する。収量27mg(0.065mmol、34%)。TLC Rf=0.20(酢酸エチル/石油エーテル1:1)。
【0155】
【表7】
【実施例8】
【0156】
N9−(3−クロロプロピル)−2−アミノ−6−クロルプリン(21)
【0157】
6−クロロプリン(5.0g、29.48mmol)、およびK2CO3(4.48g、32.42mmol)を含む無水DMFの攪拌懸濁液に、1−ブロモ−3−クロロプロパン(13.92g、88.45mmol)を加える。反応混合物を50℃で1分間加熱し、室温でさらに3時間攪拌する。反応混合物を120mLの水に注ぎ、CH2Cl2で抽出する。混合有機相を水洗し、MgSO4で乾燥し、減圧蒸発させる。残留物をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール50:1〜10:1)によって精製すると、2.6g(10.65mmol、35%)の表題化合物が無色固体として得られる。
【0158】
【表8】
【実施例9】
【0159】
N9−(3−アジドプロピル)−2−アミノ−6−クロルプリン(22)
【0160】
N9−(3−クロロプロピル)−2−アミノ−6−クロルプリン(21、2.56g、10.40mmol)、およびアジ化ナトリウム(811mg、12.48mmol)を25mLのDMSOに溶解し、80℃で16時間攪拌する。室温に冷却後、反応混合物を150mLの水に注ぎ、CH2Cl2で抽出する。混合有機相を水洗し、MgSO4で乾燥し、減圧蒸発させる。残留物をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール50:1〜10:1)によって精製すると表題化合物が無色固体(650mg、2,57mmol、24%)として得られる。
【0161】
【表9】
【実施例10】
【0162】
N−[4−(2−アミノ−9−(3−アジドプロピル)−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(23)
【0163】
【化21】
【0164】
2,2,2−トリフルオロ−N−(4−ヒドロキシメチル−ベンジル)−アセトアミド(138mg、0.59mmol)をアルゴン雰囲気下で3mLの無水ジメチルアセトアミドに溶解し、NaH(31mg、1.31mmol)を加える。N9−(3−アジドプロピル)−2−アミノ−6−クロロプリン(22、100mg、0.39mmol)を加え、溶液を90℃で16時間加熱する。室温に冷却後、混合物を80mLの水に注ぎ、トリフルオロ酢酸でpHを6に調整する。水相をCH2Cl2および酢酸エチルで抽出する。有機相を水と塩水で洗浄し、MgSO4で乾燥し、溶媒を蒸発させる。粗生成物をメタノールに溶解し、SiO2(150mg)に吸着させ、ジクロロメタン:メタノール(95:5)のフラッシュカラムクロマトグラフィーによって精製する。収量:151mg(0.0336mmol、85%)。
【0165】
【表10】
【実施例11】
【0166】
N−[4−(2−アミノ−9−(3−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(24)
【0167】
アジド23(実施例10、100mg、0.23mmol)を0.9mLの1,4−ジオキサン/H2O(8:1)に溶解し、0.3mLのトリメチルホスピン溶液(THF中1M)を加える。反応を室温で3時間攪拌し、次いで全揮発性物質を減圧除去する。粗生成物をジクロロメタン/メタノール(95:5〜10:1)のフラッシュカラムクロマトグラフィーによって精製する。収量:87mg(0.021mmol、91%)。
【実施例12】
【0168】
N−[4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(25)
【0169】
4−([4−ジメチルアミノ−フェニル]−アゾ)−安息香酸スクシンイミジルエステル(14.3mg、0.0039mmol)およびトリフルオロアセタミド24(実施例11、16.6mg、0.0039mmol)をTEA10μL含有DMF0.8mLに溶解する。反応混合物を室温で終夜攪拌し、生成物を逆相MPLCによって精製する。
【実施例13】
【0170】
4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)プリン−6−イルオキシメチル)−ベンジルアミン(26)
【0171】
トリフルオロアセタミド25(実施例12、50mg)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を終夜室温で攪拌し、全揮発性物質を減圧除去する。生成物を次のステップでさらに精製することなく使用する。MS(ESI)m/z 579.1[M+H]+。
【実施例14】
【0172】
N−[4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(27)
【0173】
【化22】
【0174】
アミン26(実施例13、2mg)、およびATTO488−スクシンイミジルエステル(28、2mg、Atto-Tech, Siegen, Germany, Cat.-No. AD488-3)をTEA1μL含有DMF150μLに溶解し、室温で24時間放置する。生成物(27)を逆相MPLCによって精製する。
【実施例15】
【0175】
N−[4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(29)
【0176】
【化23】
【0177】
アミン26(実施例13、2mg、0.0034mmol)および5(6)−カルボキシフルオレセインスクシンイミジルエステル(30、2.0mg、0.0042mmol)をTEA1μL含有DMF150μLに溶解し、室温で24時間放置する。生成物(29)を逆相MPLCによって精製する。
【実施例16】
【0178】
4−(2−アミノ−9−(3−アジドプロピル)−プリン−6−イルオキシメチル)−ベンジルアミン(31)
【0179】
アジド23(実施例10、50mg、0.11mmol)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を50℃で終夜攪拌し、全揮発性物質を減圧除去する。生成物をフラッシュカラムクロマトグラフィー(CH2Cl2/MeOH10:1)によって精製し33.5mgを得た。
【0180】
【表11】
【実施例17】
【0181】
N−[4−(2−アミノ−9−(3−アジドプロピル)−プリン−6−イルオキシメチル)−ベンジル]−テトラメチル−ローダミン−5(6)−カルボキサミド(32)
【0182】
アジド31(実施例16、10mg、0.08mmol)および5(6)−カルボキシテトラメチルローダミンスクシンイミジルエステル(33、7.47mg、0.014mmol)をTEA20μL含有DMF850μLに溶解し、室温で24時間放置する。生成物を逆相MPLCによって精製し9.0mg(0.012mmol、79%)を得た。MS(ESI)m/z 766.0[M]+。
【0183】
【化24】
【実施例18】
【0184】
N−[4−(2−アミノ−9−(3−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−テトラメチルローダミン−5(6)−カルボキサミド(34)
【0185】
アジド32(実施例17、9.0mg、0.012mmol)を600μLの1,4−ジオキサンおよび80μLの水の混合物に溶解する。この貯蔵液の200μLを14μLのトリメチルホスフィン(THF中1M)溶液で3時間処理する。全揮発性物質を減圧除去し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製する。収量3.47mg(0.0047mmol,40%)。MS(ESI)m/z 740.1[M+]。
【実施例19】
【0186】
N−[4−(2−アミノ−9−(3−QSY9−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]テトラメチルローダミン−5(6)−カルボキサミド(35)
【0187】
【化25】
【0188】
アミン34(実施例18、100μL、DMF中4.7mM)溶液にQSY−9−スクシンイミジルエステル(36、Molecular Probes、Eugene OR、USA、Cat.No.Q20131)(0.8mg)および1μLのTEAを加え、混合物をシェーカーに乗せ室温で終夜放置する。水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製する。MS(ESI)m/z 1539.9[M+H]+。
【実施例20】
【0189】
2,6−ジアミノ−4−[4−(2,2,2−トリフルオロ−アセトアミドメチル)−ベンジルオキシ]−ピリミジン37
【0190】
N−(4−ヒドロキシメチル−ベンジル)−2,2,2−トリフルオロ−アセトアミド(12.0g、52mmol)を80mLの無水DMSOに溶解し、NaH(4.08g、102mmol、鉱油中60%懸濁液)をアルゴン雰囲気下、30分かけて少しずつ加える。室温で1時間攪拌後、2,6−ジアミノ−4−クロロピリミジン(7.52g、52mmol)を加え、反応混合物を60℃まで終夜加熱する。室温に冷却後、混合物を1Lの1N HClに注ぎ、生成物を酢酸エチル(500mL)で抽出する。混合有機相を水と塩水で洗浄し、MgSO4で乾燥し、溶媒を蒸発させる。残留物をフラッシュカラムクロマトグラフィー(酢酸エチル/メタノール100:0〜80:20)によって精製すると、表題化合物が無色固体(4.7g、13.78mmol、27%)として得られる。
【0191】
【表12】
【実施例21】
【0192】
2,6−ジアミノ−5−ニトロソ−4−[4−(2,2,2−トリフルオロ−アセトアミドメチル)−ベンジルオキシ]ピリミジン(38)
【0193】
ピリミジン37(実施例20、4.72g、13.7mmol)を60mLの酢酸(30%)に溶解し、70℃に加熱する。5mLの水に溶解したNaNO2(1.4g、20.03mmol)をKJ澱粉紙が黒色に留まっているまで滴下する。氷浴で0℃に冷却後、紫色沈殿物をろ過によって収集し、アセトンから再結晶化すると、3.9g(10.54mmol、77%)の紫色固体が得られる。
【0194】
【表13】
【実施例22】
【0195】
2−アミノ−6−(4−(tert−ブトキシカルボニルアミノ)−ブタノイルアミノ)−5−ニトロソ−4−[4−(2,2,2−トリフルオロ−アセトアミドメチル)−ベンジルオキシ]−ピリミジン(39)
【0196】
アルゴン雰囲気下、4−(tert−ブトキシカルボニルアミノ)−酪酸(1.58g、8mmol)を20mLの乾燥THFに溶解し、0℃に冷却する。ジイソプロピルエチルアミン(1.18g、9mmol)を加え、混合物を−20℃に冷却し、クロロギ酸イソブチルエステル(1.15g、8mmol)を加える。混合物をこの温度で5分間攪拌し、20mLの乾燥THFに溶解させたニトロソピリミジン38(実施例21、2.64g、7mmol)を注射器によって加える。反応混合物を放置して室温まで暖め、55℃まで加熱し、この温度で終夜攪拌する。室温に冷却後、混合物を100mLの1N HClに注ぎ、生成物を酢酸エチル(300mL)で抽出する。混合有機相を水と塩水で洗浄し、続いてMgSO4で乾燥し、溶媒を蒸発させる。残留物をSiO2に吸着させ、フラッシュカラムクロマトグラフィー(シクロヘキサン/酢酸エチル1:2〜1:10)によって精製すると、表題化合物が青色固体(1.96g、3.53mmol、50.4%)として得られる。この粗製物質より、分析用試料を酢酸エチルから再結晶化する。
【実施例23】
【0197】
N−[4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(11)
【化26】
【0198】
ニトロソ−ピリミジン39(実施例22、1.84g、3.3mmol)およびトリフェニルホスフィン(1.89g、7mmol)を含む20mLのo−キシレンを10時間加熱還流する。室温に冷却後、溶媒を減圧除去し、残存する残留物を酢酸エチルに再溶解する。残留物をSiO2に吸着させ、フラッシュカラムクロマトグラフィー(酢酸エチル/エタノール1:0〜10:1)によって精製すると、表題化合物が無色固体(1.04g、3.53mmol、50.4%)としてが得られる。
【0199】
【表14】
【実施例24】
【0200】
N−[4−(2−アミノ−8−(3−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(41)
【0201】
tert−ブトキシカルボニル誘導体11(実施例23、100mg、0.19mmol)を10mLのジクロロメタンに懸濁し、2mLのトリフルオロ酢酸を加える。混合物を室温で30分間攪拌し、70mLのジエチルエーテルに注ぐ。沈殿物をろ過によって収集し、フラッシュカラムクロマトグラフィー(CH2Cl2/MeOH5:1)によって精製すると、74mg(0.17mmol、92%)の表題化合物が得られる。MS(ESI)m/z 424.1[M+H]+。
【実施例25】
【0202】
N−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(42)
【0203】
アミン41(実施例24、20mg、0.047mmol)および4−([4−ジメチルアミノ−フェニル]−アゾ)安息香酸スクシンイミジルエステル(17.3mg、0.047mmol)をTEA20μL含有DMF0.6mLに溶解する。反応混合物を終夜室温で攪拌し、溶媒を減圧除去する。生成物をフラッシュカラムクロマトグラフィー(CH2Cl2/MeOH95:5〜10:1)によって精製すると23mg(0.035mmol、75%)が得られる。MS(ESI)m/z 675.1[M+H]+。
【実施例26】
【0204】
4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)9H−プリン−6−イルオキシメチル)−ベンジル−アミン(43)
【0205】
トリフルオロアセタミド42(実施例25、23mg、0.035mmol)を2mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を50℃で終夜攪拌し、全揮発性物質を減圧除去する。生成物をさらに精製することなく使用する。MS(ESI)m/z 579.1[M+H]+。
【実施例27】
【0206】
N−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(44)
【0207】
【化27】
【0208】
アミン43(実施例26、2.0mg、0.0035mmol)および5−(6)−カルボキシフルオレセインスクシンイミジルエステル(1.63mg、0.0035mmol)をTEA5μL含有DMF200μLに溶解する。反応混合物を終夜室温で放置し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって粗生成物を精製する。MS(ESI)m/z 937.0[M+H]+。
【実施例28】
【0209】
N−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(45)
【0210】
【化28】
【0211】
アミン43(実施例26、1.0mg、0.0017mmol)およびATTO488−スクシンイミジルエステル(28、1.0mg、0.0015mmol)をTEA1μL含有DMF100μLに溶解する。反応混合物を終夜室温で放置し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって粗生成物を精製する。MS(ESI)m/z 1150.0[M+]。
【実施例29】
【0212】
N−[4−(2−アミノ−8−(3−QSY9−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]2,2,2−トリフルオロ−アセトアミド(46)
【0213】
アミン41(実施例24、1.6mg、0.038mmol)およびQSY−9−スクシンイミジルエステル(36、3.7mg、0.038mmol)をTEA2μL含有DMF0.25mLに溶解する。反応混合物を終夜室温で攪拌する。反応混合物を1mLの水/アセトニトリル(80:20)で希釈し、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって生成物を精製すると2.1mg(0.0017mmol、44%)の表題化合物が得られる。
【実施例30】
【0214】
4−(2−アミノ−8−(3−QSY9−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジルアミン(47)
【0215】
トリフルオロアセタミド46(実施例29、2.1mg、1.72μmol)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を終夜室温で攪拌し、全揮発性物質を減圧除去する。生成物をさらに精製することなく使用する。
【実施例31】
【0216】
N−[4−(2−アミノ−8−(3−QSY9−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−テトラメチルローダミン−5(6)−カルボキサミド(48)
【0217】
【化29】
【0218】
アミン47(実施例30、1mg、0.886μmol)、5(6)−カルボキシテトラメチルローダミンスクシンイミジルエステル(33、0.5mg、0.886μmol)、および1μLのTEAを100μLのDMFに溶解し、混合物をシェーカーに乗せ終夜室温で放置する。水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製する。
【実施例32】
【0219】
N−[4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(49)
【0220】
アミン41(実施例24、10mg、0.023mmol)およびN−(+)−ビオチニル−6−アミノカプロン酸スクシンイミジルエステル(50、10mg、0.023mmol)をTEA5μL含有DMF650μLに溶解し、終夜室温で放置する。水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製すると、15.1mg(0.019mmol、86%)の表題化合物が得られる。MS(ESI)m/z 763.0[M+H]+。
【0221】
【化30】
【実施例33】
【0222】
4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル−アミン(51)
【0223】
トリフルオロアセタミド49(実施例32、15.1mg、0.019mmol)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物室温で48時間を攪拌し、全揮発性物質を減圧除去する。生成物をさらに精製することなく使用する。MS(ESI)m/z 667.5[M+H]+。
【実施例34】
【0224】
N−[4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(52)
【0225】
【化31】
【0226】
アミン51(実施例33、10mg、0.015mmol)および5(6)−カルボキシフルオレセインスクシンイミジルエステル(30、7.0mg、0.015mmol)をTEA10μL含有DMF 500μLに溶解する。反応を24時間室温で放置し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製すると、8.2mg(0.008mmol、53%)の表題化合物が得られる。MS(ESI)m/z 1025.0[M+H]+。
【実施例35】
【0227】
4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル−アミン(53)
【0228】
トリフルオロアセタミド11(実施例23、100mg、0.19mmol)を2mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を終夜50℃で攪拌し、全揮発性物質を減圧除去する。生成物をフラッシュカラムクロマトグラフィー(CH2Cl2/MeOH95:5〜10:1)によって精製すると、77mg(0.017mmol、94%)の表題化合物が得られる。
【実施例36】
【0229】
N−[4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(54)
【0230】
アミン53(実施例35、10mg、0.023mmol)および5(6)−カルボキシフルオレセインスクシンイミジルエステル(30、11.4mg、0.023mmol)をTEA10μL含有DMF 500μLに溶解し、終夜室温で攪拌し、続いて水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって生成物を精製すると、13.4mg(0.017mmol、73%)の表題化合物が得られる。MS(ESI)m/z 766.0[M+H]+。
【実施例37】
【0231】
N−[4−(2−アミノ−8−(3−アミノ−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]フルオレセイン−5(6)−カルボキサミド(55)
【0232】
tert−ブトキシカルボニルアミド54(実施例36、5mg、0.0062mmol)を2mLのCH2Cl2/MeOH(1:1)に溶解し、1mLのTFAを加える。混合物を室温で4時間攪拌し、全揮発性物質を減圧除去する。粗生成物を0.5mLのMeOHに再溶解し、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって精製すると、0.5mg(0.0006mmol、10%)の表題化合物が得られる。MS(ESI)m/z 686.2[M+H]+。
【実施例38】
【0233】
O6−フルオレセイン−C8−ビーズコンジュゲート(56)
【0234】
【化32】
【0235】
NHS活性化セファロース(商標)をポリプロピレンカラム(HiTrap(商標)、カラム容量1mL、Amersham Biosciences)に充填し、アミン55(実施例37、8mM)を含む0.5M NaCl溶液pH8.5と共に30分間インキュベートする。カラムを最初に6mLの緩衝液A(0.5M エタノールアミン、0.5M NaCl、pH8.3)で、続いて6mLの緩衝液B(0.1M 酢酸、0.5M NaCl、pH4)で洗浄する。残存する全てのNHSエステルをクエンチするために、続いてカラムを緩衝液Aと共に30分間インキュベートし、次いで再度6mLの緩衝液Aで、続いて6mLの緩衝液Bで、次いで6mLの緩衝液Aで再度洗浄する。式56のビーズがhAGT用基質として使用するために今準備される。
【実施例39】
【0236】
N−[4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(57)
【0237】
アミン53(実施例35、2.5mg、0.08mmol)およびATTO488−スクシンイミジルエステル(28,4mg、0.08mmol)をTEA10μL含有DMF 500μLに溶解し、終夜室温で攪拌し、続いて水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって生成物を精製すると、13.4mg(0.017mmol、73%)の表題化合物が得られる。
【実施例40】
【0238】
N−[4−(2−アミノ−8−(3−アミノ−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]ATTO488−アミド(58)
【0239】
tert−ブトキシカルボニルアミド57(実施例39、2mg、0.002mmol)を1mLのCH2Cl2/MeOH(1:1)に溶解し、0.1mLのTFAを加える。混合物を室温で30分間攪拌し、全揮発性物質を減圧除去する。粗生成物を0.5mLのMeOHに再溶解し、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって精製する。
【実施例41】
【0240】
N−[4−(2−アミノ−8−(3−(マレイミドアセチル−アミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(59)
【0241】
アミン58(実施例40、1mg、0.0011mmol)およびマレイミド酢酸スクシンイミジルエステル(1mg、0.0039mmol)をTEA1μL含有DMF100μLに溶解し、終夜室温で放置する。水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって粗生成物を精製する。
【実施例42】
【0242】
O6−ATTO488−C8−TyrArg9Cysコンジュゲート(60)
【0243】
【化33】
【0244】
マレイミドフルオロフォア59(実施例41、4当量)を含む50mLのアセトニトリルを、TyrArg9Cysを含む0.5mLの脱酸素化MeOHに加え、次いでN2下、2〜4時間室温で振盪する。エチルエーテル(10倍過剰な)を加えて生成物を沈殿させる。精製は、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し、その対応する波長で検出しながら、逆相MPLCによって実施する。生成物は、MALDI−TOFMSによって特徴付ける。
【実施例43】
【0245】
2,5,6−トリアミノ−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]ピリミジン(61)
【0246】
2,6−ジアミノ−5−ニトロソ−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]−ピリミジン(1.22g、3.3mmol)およびトリフェニルホスフィン(1.89g、7mmol)を含む20mLのo−キシレンを1時間加熱還流する。室温に冷却後、溶媒を減圧除去し、残存する残留物をメタノールに再溶解する。残留物をSiO2に吸着させ、フラッシュカラムクロマトグラフィー(CH2Cl2/メタノール95:5〜5:1)によって精製すると表題化合物が得られる。
【実施例44】
【0247】
2−アミノ−6−ヒドロキシメチル−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]−プテリジン(62)
【0248】
ピリミジン61(実施例43、5.0g、14.1mmol)をアスコルビン酸ナトリウム(2.85g、14.4mmol)と共にジメチルアセトアミド/水(1:1)に溶解する。ジヒドロキシアセトン二量体(2.57g、14.3mmol)を加え、エアを反応混合物に吹き入れながら反応混合物を40℃に加熱する。4時間後、反応混合物を250mLのH2Oに注ぎ、形成した固体をろ過によって収集する。この固体をCH2Cl2/MeOH(3:1、500mL)に再溶解し、MgSO4で乾燥する。粗生成物をSiO2に吸着し、CH2Cl2/MeOH(20:1)を使用するフラッシュカラムクロマトグラフィーによって精製する。
【実施例45】
【0249】
2−アミノ−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]−プテリジン−6−カルボン酸(63)
【0250】
ヒドロキシメチル化合物62(実施例44、0.342g、0.84mmol)をアセトン/0.5M リン酸緩衝液、pH7(1:1、20mL)に懸濁し、過マンガン酸カリウム(0.34g、2.18mmol)を2時間かけて少しずつ加える。反応混合物を室温で3時間攪拌し、H2O(50mL)で希釈し、過マンガン酸が消費するまで亜硫酸ナトリウムを加えると、黒褐色沈殿物が生じ、これをろ過除去する。pHを2M HClを加えて2.5に調整すると黄色固体が生じ、これをろ過収集する。固体をH2O(50mL)に溶解しpHが30分間一定になるまで0.1M NaOHを加えてpHを7.0に調整する。混合物をろ過し、再度酸性化し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって粗生成物を精製する。
【実施例46】
【0251】
2−アミノ−6−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−4−[4−(2,2,2−トリフルオロアセチルアミノ−メチル)−ベンジルオキシ]−プテリジン(64)
【0252】
カルボン酸63(実施例45、0.10g、0.24mmol)およびPyBOP((ベンゾトリアゾール−1−イルオキシ)−トリピロリジノホスホニウムヘキサフルオロホスファート、0.12g、0.24mmol)を1mLの無水DMFに溶解し、室温で30分間攪拌する。TEA(50μL)およびtert−ブチル2−アミノエチルカルバマート(48mg、0.3mmol)を加える。反応混合物をさらに3時間攪拌し50mLの水に注入する。生成物をCH2Cl2で抽出し、MgSO4で乾燥し、CH2Cl2/MeOH(20:1)を使用するフラッシュカラムクロマトグラフィーによって精製する。
【実施例47】
【0253】
N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(18、CPTG)を有するAGT変異体の反応速度
【0254】
最初にAGT変異体Gly131Lys、Gly132Thr、Met134Leu、Arg135Ser、Asn157Gly、Ser159Gluを指向進化法によって調製する。PGEG−hAGT遺伝子の部分的に重複する2領域、変異Asn157Gly、Ser159Gluを含むAGT(Juillerat et al., Chem Biol 10:313-317, 2003)を別々の反応において適当なプライマーで増幅する。プライマーには、hAGT遺伝子のコドン131、132、134、135に対応する位置にヌクレオチド混合物NNK(N=A、C、G、もしくはT;K=GもしくはT)が含まれる。その部分的相補性に関して、さらなるPCR反応でこれらの2つのPCRフラグメントを合わせ、増幅してコドン131、132、134、135でランダム化された完全長遺伝子を得る。これらを、Sfil制限部位によりベクターpAK100の繊維状ファージのg3蛋白質に融合してクローン化する。得られた遺伝子ライブラリをファージディスプレイに使用する。
【0255】
このライブラリのファージは、大腸菌JM101細胞で産生される。指数関数的培養をヘルパーファージに重感染させ、終夜24℃で増殖する。この培養上清を1μMジゴキシゲニン化O6−ベンジルグアニン(Juillerat et al., Chem Biol 10:313-317, 2003の物質2)と共に6分間インキュベートする。次の選択ラウンドでは、反応時間をそれぞれ90秒および45秒に減少し、基質濃度を10nMに減少して選択圧力を増大させる。4%PEG/3%NaClで沈殿させることによって、この反応からファージを精製する。今回共有結合によりジゴキシゲニンで標識した変異体AGTを有するファージは、抗ジゴキシゲニン抗体(Roche Diagnostics)でコートした磁気ビーズと共にインキュベートすることによって単離し、細菌の再感染に使用される。
【0256】
選択したAGT変異体を増幅し、続いて発現ベクターpGEX−2T(Amersham)のBamH1とEcoR1間部位にクローン化する。これによりGST蛋白質C末端融合体として挿入遺伝子の発現が可能になり、ベクターによってこの遺伝子を提供する。
【0257】
このベクターからの蛋白質発現は大腸菌株BL21で行う。指数関数的に増殖する培養を0.5mM IPTGによって誘発し、24℃で3.5時間発現させる。
【0258】
精製:1mM PMSFおよび2μg/mLアプロチニンを補充し、50mMリン酸塩、0.5M NaCl、1mM DTTを含む緩衝液に回収した細胞を再懸濁し、リゾチームおよび超音波処理により破壊する。細胞残屑は、40000×gでの遠心分離によって分離する。抽出物を予め平衡化したグルタチオンセファロース(Amersham)にアプライし、次いでこれを20ベッド容量(50mMリン酸塩、0.5M NaCl、1mM DTT)で洗浄する。変異GST−AGT融合蛋白質を、10mM還元グルタチオンを含む50mM Tris−HClpH7.9で溶出する。精製した蛋白質を50mM HEPES pH7.2、1mM DTT、30%グリセロールに対して透析し、次いで−80℃で貯蔵する。
【0259】
変異Cys62Ala、Gln115Ser、Gln116His、Lys125Ala、Ala127Thr、Arg128Ala、Gly131Lys、Gly132Thr、Met134Leu、Arg135Ser、Cys150Asn、Ser151lle、Ser152Asn、Asn157Gly、Ser159Glu、および182での切り詰めを含む第2のAGT変異体(「AGTM」)は上記AGT変異体に由来するが、さらなる変異およびコドン182後の切り詰めを変異体AGT遺伝子に続いて導入することによる。続くPCR増幅によって、第1の変異体について記載したpGEX2T中にサブクローン化された、さらなる変異遺伝子が生じる。GST融合蛋白質が発現し同様に精製する。
【0260】
AGTMに関係し、変異Cys62Ala、Gln115Ser、Gln116His、Lys125Ala、Ala127Thr、Arg128Ala、Arg135Ser、Cys150Asn、Ser151lle、Ser152Asn、Asn157Gly、Ser159Glu、および182での切り詰めを含む第3のAGT変異体(「AGT21」)を同じ方法によって調製する。
【0261】
AGTMに関係し、変異Cys62Ala、Lys125Ala、Ala127Thr、Arg128Ala、Gly131Lys、Gly132Thr、Met134Leu、Arg135Ser、Cys150Ser、Asn157Gly、Ser159Glu、182での切り詰めを含む第4のAGT変異体(「AGT26」)も同じ方法によって調製する。
【0262】
AGTM(1μM終濃度)をN9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(18、CPTG)(1%DMSO中の終濃度10μM)を含む反応緩衝液(50mM HEPES、1mM DTT、200mg/mL BSA、pH7.3)と共に24℃でインキュベートする。試料を所定の時間(5、10、15、20、30、45、60、90、120分)に採取し、直接、ビオチン化O6−ベンジルグアニン(BGBT;Juillerat et al., Chem Biol 10:313−317, 2003の物質3a、10μM終濃度)と共に4分間インキュベートする。SDS−Laemmli緩衝液を加えることによって反応をクエンチし95℃で2分間加熱する。試料を12%アクリルアミドゲル上で泳動させ、ウェスタンブロッティングによって分析する。結果を図2に示す。
【0263】
2次速度定数:k2=92.5[S−1M−1]。この値は、BGBTによるAGTMの活性と比較して非常に遅い(k2=〜3,000[S−1M−1])。2種類の基質BGBT/CPTGの活性比は少なくとも25である。
【実施例48】
【0264】
N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)グアニン(18、CPTG)とビオチン化O6−ベンジルグアニン(BGBT)の間のインビトロでの競合
【0265】
変異体AGT(AGTM)および野生型ヒトAGT(0.5μM終濃度)を、別々に、BGBT(0.5μM終濃度)および異なる濃度のCPTG(終濃度0、0.5、1、5、10μM)を含む反応緩衝液(50mM HEPES、1mM DTT、200mg/mL BSA、pH7.3)と共に45分間インキュベートする。反応を2×SDS緩衝液を加えることによってクエンチし95℃で2分間放置する。試料を12%アクリルアミドゲル上で泳動させ、ウェスタンブロッティングによって分析する。
【0266】
AGTMおよび野生型hAGTは、化合物CPTGおよびBGBTの存在下、CPTGに対して著しく異なる反応性を示す。BGBTの20倍の濃度のCPTGでは、95%の野生型AGTが45分後にCPTGによってクエンチされるが、87%の変異体AGTMは依然として活性である。BGBTを有する野生型hAGTの反応速度定数は400S−1M−1であることが知られているので、CPTGを有する野生型hAGTの速度定数は、立体的に匹敵する、340S−1M−1の値(Lodewijkら、欧州特許第704445号)を示すN9−デスオキシリボシル−O6−ベンジルグアニンを有するhAGTの既知の反応性に基づいて著しく影響を受けないとみなすことができる。
【0267】
AGTM−6×His融合蛋白質(0.2μM終濃度)および野生型hAGT−GST融合蛋白質(1.2μM終濃度)を含む反応緩衝液(50mM HEPES、1mM DTT、200mg/mL BSA、pH7.3)溶液をBGBT(5μM終濃度)および異なる濃度のCPTG(0、5、10μM終濃度)と共に30分間インキュベートする。反応を2×SDS緩衝液を加えることによってクエンチし95℃で2分間放置する。試料を12%アクリルアミドゲル上で泳動させ、ウェスタンブロッティングによって分析する。結果を図3に示す。
【0268】
AGTMおよび野生型hAGTの反応を比較すると、CPTGのhAGTに対する高い特異性が観察される。同じ濃度(両基質、終濃度5μM)では、AGTMのわずか5%に対して95%の野生型AGTがCPTGと反応した。
【実施例49】
【0269】
異なる2種の基質を有する一試料における、異なる2つのAGT変異体の細胞内競合
【0270】
変異体AGTM(実施例47)を適当なプライマーによって増幅し、ベクターpEGFP−NucのNheIとBglII部位の間にクローン化する。酵母β−ガラクトシダーゼ遺伝子を同様に増幅し、BglII/BamHIによりこのベクター中にサブクローン化して、AGT−β−Gal融合遺伝子を有するベクターを得る。野生型hAGT遺伝子を増幅し、NheI/BglIIによってpEGFP−Nuc中にクローン化して、変異Gly160TrpをコードするAGT−NLS3融合遺伝子を得る。CHO細胞中で並行して両AGT変異体を一時的に発現させた後、細胞を10分間5μM CPTG(18)と共にインキュベートし、続いてJuillerat et al., Chem Biol 10:313-317, 2003、(O6−(4−(ジアセチルフルオレセイン−カルボニルアミノメチル)−ベンジル)−グアニン(BG−AcFL)の物質4の5μM共に20分間インキュベートする。細胞を洗浄し標準手順によって画像化する。
【0271】
CHO細胞では、蛍光基質BG−AcFLと反応させることによって、細胞質内に局在化するAGT−β−ガラクトシダーゼ融合蛋白質を選択的に蛍光標識する。核内にはその他のAGT融合蛋白質の有意な標識は観察されない。AGT(Gly160Trp)は、細胞内でCPTG(18)と効率よく反応し、従ってその後蛍光基質で標識されることは有り得ないと結論付けらる。変異体AGTMは、細胞内でCPTGとのプレインキュベーション後も依然として蛍光基質に反応性であり、従って第1ステップでCPTGと反応しない。
【実施例50】
【0272】
蛍光出現アッセイ
100μLのhAGT−GST融合蛋白質を含む反応緩衝液(50mM Tris、1mM DTT、1mg/mL BSA、pH8.0、hAGT−GSTの終濃度:1μM)の2個のアリコットを黒色平底マイクロタイタープレートに入れる。一つのアリコットを対照として使用し、これを40分間ベンジルグアニン(終濃度は100μM)でクエンチする。化合物(7)を含むDMSOをhAGT−GST(終濃度10nM)の両試料に加え、25℃で1時間インキュベートする。フルオレセイン−プロトコルを備えたVictor2Vプレートリーダー(Perkin-Elmer)で蛍光を読み取る。試料の蛍光読取り値は、対照の読取り値の少なくとも2倍である。
【0273】
さらに別のアッセイでは、蛋白質濃度10μMのAGT変異体AGTMおよびAGT21(実施例47)の精製蛋白質を、100mM NaCl、50mM Tris、pH8.0、0.1%Tween、1mM DTTを含む緩衝液に含まれた100nM N−[4−(2−アミノ−9−(3−QSY9−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−テトラメチルローダミン−5(6)−カルボキサミド(35、N9−QSY9−BG−TMR、実施例19)と共に室温でインキュベートする。蛍光強度(531nm励起/595nm発光)をVictor2マイクロプレートリーダー(Perkin Elmer)を使用し追跡する。変異体AGT21は、N9−QSY9−BG−TMR(化合物35)に対して有意な反応性を示し、25℃で1時間にわたってテトラメチルローダミン蛍光を約80%増大させる。それに反して、変異体AGTMでは、N9−QSY9−BG−TMRにより25℃で1時間におよぶインキュベーションすることで5%未満のテトラメチルローダミンの蛍光が増大する。
【0274】
同様に、AGTMおよびAGT21をN−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノフェニル]−アゾ−ベンゾイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(45、C8−Dabcyl−BG−Atto488、実施例28)、およびO6−(4−(ATT0488−アミドメチル)−ベンジル)グアニン(BG−Atto488、Atto-Tec Siegen, D, Prod Nr.: AD 488-2)と共にインキュベートし、蛍光強度を490nm(励起)および535nm(発光)で測定する。変異体AGTMは、BG−Atto488と比較してC8−Dabcyl−BG−Atto488に対して3倍の反応性を示す。変異体AGT21は、BG−Atto488と比較してC8−Dabcyl−BG−Atto488に対して二倍の反応性を示す。
【0275】
さらなる反応比較により以下の結果が得られた。
AGT21は、化合物48(C8−QSY9−BG−TMR、実施例31)と、C8/N9非置換化合物(BG−TMR、Molecular Probes製品C300)との場合より2倍速く、化合物35(N9−QSY9−BG−TMR、実施例19)との場合より少なくとも4倍速く反応する。AGT21は、化合物45(C8−Dabcyl−BG−Atto488、実施例28)と、C8/N9非置換化合物(BG−Atto488、Atto-Tec Siegen, D, Prod. No. AD 488-2)との場合より2倍速く、化合物27(N9−Dabcyl−BG−Atto488、実施例14)との場合より少なくとも4倍速く反応する。
【0276】
AGT21は、化合物44(C8−Dabcyl−BG−FL、実施例27)と、C8/N9非置換化合物(BG−FL、Juillerat et al., Chem Biol 10:313-317, 2003の物質5)との場合より2倍速く、化合物29(N9−Dabcyl−BG−FL、実施例15)との場合より少なくとも4倍速く反応する。
【0277】
AGTMは、化合物48(C8−QSY9−BG−TMR、実施例31)と、C8/N9非置換化合物(BG−TMR)との場合より2倍速く、化合物35(N9−QSY9−BG−TMR、実施例19)との場合より少なくとも10倍速く反応する。
【0278】
AGTMは、化合物45(C8−Dabcyl−BG−Atto488、実施例28)と、C8/N9非置換化合物(BG−Atto488)との場合より3倍速く、化合物27(N9−Dabcyl−BG−Atto488、実施例14)との場合より少なくとも15倍速く反応する。
【0279】
AGTMは、化合物44(C8−Dabcyl−BG−FL、実施例27)と、C8/N9非置換化合物(BG−FL)との場合より2倍速く、化合物29(N9−Dabcyl−BG−FL、実施例15)との場合より少なくとも10倍速く反応する。
【実施例51】
【0280】
ワンポット標識および分離アッセイ
50μLのhAGT−GFP融合蛋白質を含む反応緩衝液(50mM Tris、1mM DTT、1mg/mL BSA、pH7.3、hAGT−GFPの終濃度:5μM)を基質(9)(B=ビオチン、終濃度10μM)と共に1時間インキュベートする。150μLのストレプトアビジン被膜ビーズ(Dynabeads M−280)を加え、インキュベーション時間および上清からの磁気分離を製造業者の使用説明書に従って行う。488nmでのGFP吸収を測定することによって、磁気ビーズによる処理前後の溶液中の融合蛋白質量を定量する。融合蛋白質の損失は30%未満である。未反応基質を除去後、上清中のALEXA色素の吸収を594nmで測定によって標識効率を定量する。標識効率は少なくとも20%である。
【0281】
他の実験では、蛋白質濃度が5μMである精製AGTMまたはAGT21(実施例47)を、100mM NaCl、50mM Tris、pH8.0、0.1%Tween、1mM DTTを含む緩衝液50μlに含まれる10μMのN−[4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(52、実施例34)と共に室温でインキュベートする。1時間後、少なくとも1nmolビオチンを結合する能力を有するストレプトアビジン被膜磁気ビーズ(Magnabindストレプトアビジン、Pierce)を加え、インキュベーション時間、およびフルオレセイン標識蛋白質を含む上清からの磁気分離を製造業者の使用説明書に従って行う。磁気ビーズによる処理前後の溶液中の蛋白質量をBradfordアッセイによって決定する。蛋白質の損失は30%未満である。未反応基質およびビーズへの結合および磁気分離によって遊離した生成物を除去後の上清中の蛍光強度を、490nm励起および535nm発光のVictor2マイクロプレートリーダー(Perkin Elmer)を使用して測定することによって標識効率を定量する。標識効率は少なくとも20%である。
【0282】
基質が予め固定されるさらに別の実験では、1ml当たり2μgのビオチンに結合する能力を有するストレプトアビジン被膜磁気ビーズ(Magnabindストレプトアビジン、Pierce)を、100mM NaCl、50mM Tris、pH8.0、0.1%Tweenを含む緩衝液に含まれた2倍過剰の化合物52(上のように)と共に室温でインキュベートして基質をビーズに固定する。インキュベーション時間、および上清からの磁気分離を製造業者の使用説明書に従って行う。未結合基質は、続く3回の洗浄ステップで緩衝液により除去する。蛋白質濃度が50μMのAGTMを、100mM NaCl、50mM Tris、pH8.0、0.1%Tween、1mM DTTを含む緩衝液50μlに含まれたこれらの基質改変ビーズと共に室温でインキュベートする。20時間後、フルオレセイン標識蛋白質を含む上清からビーズを分離する。磁気ビーズによる処理前後の溶液中の蛋白質量をBradfordアッセイによって決定する。蛋白質の損失は30%未満である。上のように、蛍光強度を測定することによって標識効率を定量するが、少なくとも20%である。
【実施例52】
【0283】
標識ビーズ
機能付与したセファロースビーズ56(実施例38)を5mLの緩衝液C(10mM HEPES、pH7.4、150mM NaCl、0.05%Tween20)で洗浄することによって平衡化する。この懸濁液に100μLのhAGT−GFP(終濃度50μM)を加え、25℃で1時間インキュベートする。セファロースビーズを遠心分離によって除去し、488nmで上清中のフルオレセインの吸収を測定することによって標識効率を定量する。標識効率は少なくとも20%である。488nmでのGFP吸収を測定することによって、改変セファロースビーズ処理前後の溶液中の融合蛋白質量を定量する。融合蛋白質の損失は30%未満である。
【0284】
別の実験では、製造業者により提供された使用説明書に従って、化合物56をセファロース4FastFlow(NHS−エステルとして活性化させたカルボキシル基、Amersham Prod No. 17-0906-01)によって調製する。洗浄液中に蛍光が検出されなくなるまで樹脂物質を洗浄し、暗所において4℃で貯蔵する。以下の全ステップで使用する緩衝液は、150mM NaClと50mM Tris、pH8.0に1mM DTTを加えたものである。各実験には、製造業者の使用説明書に従い、200μLの改変樹脂をBio-Rad Micro Biospinスピンカラムに充填し、2分間1000×gで遠心分離する。素通りは廃棄する。その後50μM濃度の非標識AGTM100μLを加え、25℃の暗所で1時間放置する。
【0285】
対照実験として、100μMのO6−ベンジルグアニンで予め30分間ブロックした50μM AGTMを100μL使用する。その後、カラムを1000×gで2分間遠心分離し、その度に200μLの緩衝液を加えることによって4回洗浄する。最終容量を1mLに調整する。蛋白質含有量をBCAアッセイ(Pierce BCA蛋白質アッセイキット−Prod. Nr. 23225, Perbio Science, Switzerland)によって決定する。見出された値は、予想された5μM濃度の少なくとも80%である。pH9に調整した100mM Na2CO3緩衝液で1:50に希釈した後、全溶液の蛍光を定量する。黒色96ウェルプレート(Costar)には各試料の200μLが備わる。試験試料のフルオレセイン範囲の蛍光をVictor2プレートリーダー(フルオレセインフィルタセット)によって読み取る。試験試料で見られた蛍光は、対照実験で見られた蛍光の少なくとも5倍である。
【実施例53】
【0286】
コンジュゲートO6ATTO488−C8−TyrArg9Cys(60)の細胞透過性
AGT26およびCaaX−ファルネシル化シグナル融合用ベクターを、内在性野生型AGTを持たないCHO細胞中に形質移入する。安定した形質移入体をジェネティシン(geneticine)処理下で選択する。その目的のために、AGT26遺伝子をpCMV−Script(Stratagene)中にクローン化し、そのC末端にpECP−F(Clontech)中にコードされているCaaX−box配列を続けた。最終蛋白質配列(位置690〜1271)によって、194アミノ酸を有する単一ポリペプチドが得られる。安定した形質移入細胞系統では、このポリペプチドによって、細胞膜内面に局在化するAGT融合蛋白質がもたらされる。これらの細胞を5μM O6−テトラメチルローダミン−ベンジルグアニン(BG−TMR、TMR−NHS−エステルから合成、Molecular Probes製品C300)で30分間標識し、続いて培地を2回交換し、次に30分間休止し、さらに培地を交換した場合、このAGT融合蛋白質は細胞膜に明白なシグナルをもたらす。テトラメチルローダミン用およびフルオレセイン用のフィルタセットを備えた逆位Zeiss蛍光顕微鏡Axiovert40CFLで全画像化を行う。同ステップ順序に続いて、5μM O6−Atto488−ベンジルグアニン(BG−ATTO488、Atto-Tec Siegen, D, Prod. No. AD 488-2)でインキュベーションしても、有意な細胞標識は観察されない。これは、ビス−スルホン化Atto488誘導体が非常に低い細胞膜透過性しか有していないことを示している。コンジュゲートO6−ATTO488−C8−TyrArg9Cys(60、実施例42)が同じ条件下、5μM濃度で使用される場合、細胞膜の主要標識によって有意な細胞内染色が観察される。これは、ベンジルグアニン誘導体が細胞透過性でない場合でも、ポリ−アルギニンリンカーの導入が透過性を増大させることを示している。
【図面の簡単な説明】
【0287】
【図1】対象となる蛋白質(P)およびAGTを含む融合蛋白質と、式R1−A−X−CH2−R3−R4−L1の基質との反応を示す模式図である。
【図2】AGTM−CPTGの動力学的測定:発光ペルオキシダーゼ基質により検出したAGTM−BGBT複合体の強度のウエスタンブロットを示す図である。実施例8を参照。変異体AGTM−6×Hisのアリコットを、緩衝化した水溶液中で、10倍過剰のN9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(CPTG)によりインキュベートする。所定の時間を経た後、ビオチン化O6−ベンジルグアニンによって反応が終了(BGBT、Juilleratら., Chem Biol 10: 313-317, 2003の基質3a)するが、これは蛋白質と非常に素早く反応し、それによって未反応AGTMを捕獲すると考えられる。次いで、蛋白質をSDSおよび熱で変性させる。試料をSDS−PAGEとウエスタンブロット分析にかける。対応するバンド強度は、化学発光染色によって検出する。
【図3】2種の蛋白質(〜25kDaのAGTM−6×His、0.2μM、および〜50kDaの野生型hAGT−GST融合蛋白質、1.2μM)と、2種の基質N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(CPTG、0、5、10μM)、およびビオチン化O6−ベンジルグアニン(BGBT、5μM)の反応のウエスタンブロットを示す図である。実施例9参照。発光ペルオキシダーゼ基質によるAGTM−BGBT複合体の検出は、図2に記載されている通りである。
【技術分野】
【0001】
本発明は、基質からO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)に標識を移動させる方法、O6−アルキルグアニンDNAアルキルトランスフェラーゼ融合蛋白質、およびそのような方法に適した新規な特異的基質に関する。
【背景技術】
【0002】
N−メチル−N−ニトロソウレアなどの求電子試剤の変異原性および発癌性作用は、主としてDNAのグアニンのO6−アルキル化によるものである。DNAアルキル化からそれ自体を守るために、哺乳動物および細菌は、これらの病変部を修復する蛋白質O6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)を保持している。AGTは、アルキル化したグアニンおよびグアニン誘導体のO6位からアルキル基を自身のシステインの一つであるメルカプト基に移動させ、不可逆的にアルキル化AGTを生じさせる。根底にある機序は、SN2型の求核反応であり、この反応によってメチル基のみならずベンジル基までも容易に移動する理由が説明される。腫瘍細胞でAGTが過剰発現することは、プロカルバジン、ダカルバジン、テモゾロマイド、ビス−2−クロロエチル−N−ニトロソウレアなどのアルキル化薬物に対する耐性の主たる理由であり、その結果、化学療法においてAGT阻害薬を感作物質として使用することが提案されきた(Peggら, Prog Nucleic Acid Res Mol Biol 51:167-223, 1995)。米国特許第5,691,307号は、ベンジル基に様々な置換基を有するO6−ベンジルグアニン、および腫瘍細胞でAGT濃度を枯渇させ、それによってアルキル化抗腫瘍薬物に対する応答性を増大するためのその使用について記載している。同様に、国際公開公報第97/20843号は、O6−ベンジル−およびO6−ヘテロアリールメチル−ピリミジン誘導体を表す、さらに別のAGT枯渇化合物を開示している。
【0003】
ドイツ国特許第19903895号は、AGTレベルを測定するためのアッセイを開示し、このアッセイはビオチン化されたO6−アルキルグアニン誘導体とAGTの間の反応を頼りに、AGTをビオチン標識化する。これにより、順次、ストレプトアビジン被膜プレート上のAGTを分離し、例えば、ELISAアッセイでAGTを検出できるようにする。このアッセイは、腫瘍組織でAGT濃度をモニタリングすること、およびAGT阻害薬スクリーニングでの使用に向けて提案される。
【0004】
国際公開公報第01/85221号は、AGTの検出に放射性標識したフルオロまたはヨード置換O6−ベンジルグアニンの使用およびAGTレベルのモニタリングを提案している。
【0005】
Damoiseaux ら, ChemBiochem. 4:285-287, 2001は、AGT標識化の化学プローブとして使用し、再度、癌細胞におけるこの酵素のレベルの検出を容易にして研究および化学療法に役立てるために、オリゴデオキシリボヌクレオチドに組み込まれた改変O6−アルキル化グアニン誘導体を開示している。
【0006】
国際公開公報第02/083937号は、対象となる蛋白質を検出し、かつ/または操作する方法であって、蛋白質とAGTを融合し、標識を有するAGT基質にAGT融合蛋白質を接触させ、そして標識を使用しAGT融合蛋白質を検出し、場合によりさらに操作する方法を開示した。使用する数種のAGT融合蛋白質、AGT基質の一般構造原理、ならびに多種多様な標識、およびその方法で使用可能な標識を検出するための方法が記載されている。
【0007】
PCT/EP03/10859(国際公開公報第2004/031404号)は、対象となる蛋白質を検出しかつ/または操作する上記の方法で使用される特定のAGT融合蛋白質、この方法によって得られる標識した融合蛋白質、および特定のAGT融合蛋白質の使用方法について記載している。
【0008】
PCT/EP03/10889(国際公開公報第2004/031405号)は、対象となる蛋白質を検出しかつ/または操作する上記の方法に特に適した標識を有する追加のAGT基質、およびそのような特に標識した基質の適用を開示している。この特許出願はまた、これらの追加のAGT基質の製造方法について記載している。
【0009】
発明の概要
本発明は、式(1)
【0010】
【化6】
【0011】
[式中、Aは基質としてAGTによって認識される基であり;
Xは、酸素または硫黄であり;
R1は、基−R2−L2または基R5であり;
R2およびR4は、互いに独立したリンカーであり;
R3は、芳香族基もしくは複素環式芳香族基;または場合により置換されている不飽和アルキル、シクロアルキル、もしくはCH2に結合する二重結合を有するヘテロシクリル基であり;
R5は、アリールメチルもしくはヘテロアリールメチル、または場合により置換されているシクロアルキル、シクロアルケニル、もしくはヘテロシクリル基であり;
L1は、1個の標識、複数の同じもしくは異なる標識、R4とAを結合して環状基質を形成する結合、またはさらに基−R3−CH2−X−A−R1であり;かつ
L2は、1個の標識、または複数の同じもしくは異なる標識である]
のO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)基質に関する。
【0012】
本発明は、さらに、これらの基質からO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)に標識を移動させる方法およびAGT融合蛋白質に関する。
【0013】
本発明の詳細な説明
本発明の特定のAGT基質は、式(1)
【0014】
【化7】
【0015】
[式中、
Aは基質としてAGTによって認識される基であり;
Xは、酸素または硫黄であり;
R1は、基−R2−L2または基R5であり;
R2およびR4は、互いに独立したリンカーであり;
R3は、芳香族基もしくは複素環式芳香族基;または場合により置換されている不飽和アルキル、シクロアルキル、もしくはCH2に結合する二重結合も有するヘテロシクリル基であり;
R5は、アリールメチルもしくはヘテロアリールメチル、または場合により置換されているシクロアルキル、シクロアルケニル、もしくはヘテロシクリル基であり;
L1は、1個の標識、複数の同じもしくは異なる標識、R4とAを結合して環状基質を形成する結合、またはさらに基−R3−CH2−X−A−R1であり;かつ
L2は、1個の標識、または複数の同じもしくは異なる標識である]
の化合物である。
【0016】
基R1−Aでは、残基Aは、1〜5個の窒素原子を含み、基質としてAGTによって認識される複素環式芳香族基であることが好ましい。
【0017】
複素環式芳香族基Aは、単環式もしくは2環式であり、5〜12個の、好ましくは6個の、または9個もしくは10個の環原子を有し;そしてこの基Aは、置換基R1を有することに加えて、メチルなどの低級アルキル、メトキシやエトキシなどの低級アルコキシ、ヒドロキシ、オキソ、アミノ、低級アルキルアミノ、ジ低級アルキルアミノ、アシルアミノ、塩素や臭素などのハロゲン、トリフルオロメチルなどのハロゲン化した低級アルキル、カルボキシ、低級アルコキシカルボニル、カルバモイル、低級アルキルカルバモイル、または低級アルキルカルボニルからなる群から選択される一個以上の、特に1、2、もしくは3個のさらなる置換基によって置換されていなくても置換されていてもよい。
【0018】
低級アルキルは、1〜7個の、好ましくは1〜4個のC原子を有するアルキルであり、直鎖もしくは分枝鎖であることが好ましく;低級アルキルはn−ブチル、sec−ブチル、iso−ブチル、tert−ブチルなどのブチル、n−プロピルやイソプロピルなどのプロピル、エチルまたはメチルが好ましい。低級アルキルはメチルが最も好ましい。
【0019】
低級アルコキシでは、低級アルキル基は先に定義した通りである。低級アルコキシは、好ましくはn−ブトキシ、tert−ブトキシ、iso−プロポキシ、エトキシ、またはメトキシ、特にメトキシを示す。
【0020】
好ましい単環式もしくは2環式複素環式芳香族基Aは、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、8−アザプリニル、7−デアザプリニル、8−アザ−7−デアザプリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、トリアジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、トリアゾリル、テトラゾリル、またはベンゾ[d]ピラゾリルから選択される。より好ましくは、単環式もしくは2環式複素環式芳香族基Aは、プリニル、8−アザプリニル、7−デアザプリニル、8−アザ−7−デアザプリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、およびトリアジニルからなる群から選択される。
【0021】
例えば、基R1−Aは、式(2)
【0022】
【化8】
【0023】
「式中、R6は、水素、ヒドロキシ、または非置換もしくは置換アミノであり、R7およびR8の一方はR1であり他方は水素である]のプリンラジカルでよい。
【0024】
R6がヒドロキシである場合、プリンラジカルは主に互変異性体で存在し、ここで、それぞれ、R6を有する炭素原子に隣接する窒素は水素原子を有し、この窒素原子とR6を有する炭素原子の間の二重結合は単結合であり、そしてR6は二重結合酸素である。
【0025】
置換されたアミノ基R6は、1〜4個の炭素原子の低級アルキルアミノまたはアシルアミノであり、ここで、アシル基は、1〜5個の炭素原子を有する低級アルキルカルボニル、例えばアセチル、プロピオニル、n−もしくはiso−プロピルカルボニル、またはn−、iso−、もしくはtert−ブチルカルボニルもしくはアリ−ルカルボニルであり、例えばベンゾイルである。
【0026】
R6が非置換もしくは置換アミノであり、プリンラジカルの結合に接続する残基Xが酸素である場合、式(2)の残基はグアニン誘導体である。
【0027】
特に好ましいものは、基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7がR1であり、R8が水素であり、かつXが酸素である化合物、すなわち、N9位にさらに置換基を有するグアニン誘導体である。
【0028】
本発明の別の好ましい態様では、基R1−Aが式(3)
【0029】
【化9】
【0030】
[式中、置換基R6は、式(2)下でR6について定義した意味を有する]
の8−アザプリンラジカルである。
【0031】
本発明のさらに好ましい態様では、基R1−Aは、式(4a)または(4b)
【0032】
【化10】
【0033】
[式中、R9は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、またはアミノであり、アミノが好ましく、かつR10は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、ニトロ、またはニトロソである]のピリミジンラジカルである。ハロゲンR9またはR10は、例えばフルオロ、クロロ、ブロモ、またはヨードである。
【0034】
Xは、酸素であることが好ましい。
【0035】
本発明のさらに好ましい態様では、基R1−Aが式(4c)
【0036】
【化11】
【0037】
[式中、R6は、非置換もしくは置換アミノであり、R7およびR8の一方はR1であり他方は水素である]
のプテリジンラジカルである。
【0038】
リンカー基R2またはR4は、それぞれ、一個の標識L2またはL1、あるいは複数の同じもしくは異なる標識L2またはL1を基質に結合する柔軟なリンカーであることが好ましい。リンカー単位は、想定した適用と関連して、すなわち、AGTを含む融合蛋白質への基質の移動に際して選択される。リンカー単位はまた、適当な溶媒への基質溶解性も増大させる。使用するリンカーは、実際の適用条件下で化学的に安定している。リンカーは、AGTによる反応にも、標識L1および/またはL2の検出にも干渉しないが、例えば、式(1)の化合物とAGTを含む融合蛋白質が反応した後のある時点で切断されるように構築しうる。
【0039】
リンカーR2またはR4は、1〜300個の炭素原子を有する直鎖もしくは分枝鎖アルキレン基であって、場合により
(a)一個以上の炭素原子が酸素によって、特に、全ての第3炭素原子が酸素によって置き換えられている、例えば、1〜100個のエチレンオキシ単位を有するポリエチレンオキシ基である;
(b)一個以上の炭素原子が、水素原子を有する窒素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、アミド官能基−NH−CO−を示す;
(c)一個以上の炭素原子が酸素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、エステル官能基−O−CO−を示す;
(d)2個の隣接する炭素原子間の結合が二重または三重結合であり、官能基−CH=CH−または−C≡C−を表す;
(e)一個以上の炭素原子が、フェニレン、飽和もしくは不飽和シクロアルキレン、飽和もしくは不飽和ビシクロアルキレン、架橋ヘテロ芳香族、または架橋飽和もしくは不飽和ヘテロシクリル基によって置き換えられている;
(f)2個の隣接する炭素原子が、ジスルフィド結合−S−S−によって置き換えられている;
または場合により置換基を含み、先に(a)〜(f)下で定義したような2個以上の、特に、2個もしくは3個のアルキレン基および/または修飾アルキレン基の組合せ
である。
【0040】
考えられる置換基は、メチルなどの例えば低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0041】
考えられるさらなる置換基は、例えば、αアミノ酸、特に自然に存在するαアミノ酸がリンカーR2またはR4に組み込まれたときに得られる置換基であり、ここで、炭素原子は、(b)下で定義したアミド官能基−NH−CO−によって置き換えられている。そのようなリンカーでは、アルキレン基R2またはR4の炭素鎖の一部が、基−(NH−CHR−CO)n−[式中、nは1〜100であり、Rはαアミノ酸の様々な残基を表す]によって置き換えられている。
【0042】
さらなる置換基は、光切断リンカーR2またはR4をもたらす置換基であり、例えばo−ニトロフェニル基である。特に、この置換基o−ニトロフェニルは、アミド結合に隣接する炭素原子に位置し、例えば基−NH−CO−CH2−CH(o−ニトロフェニル)−NH−CO−に位置し、またはポリエチレングリコール鎖中の、例えば、基−O−CH2−CH(o−ニトロフェニル)−O−中の置換基として位置する。考えられる他の光切断リンカーは、例えば、フェナシル、アルコキシベンゾイン、ベンジルチオエーテル、ピバロイルグリコール誘導体である。
【0043】
先に(e)下で定義した炭素原子に置き換わるフェニレン基は、例えば、1,2−、1,3−、であり、1,4−フェニレンが好ましい。特定の態様では、フェニレン基はニトロ基によってさらに置換され、そして(a)、(b)、(c)、(d)、および(f)下で上記した他の置き換えと組み合わされて光切断基を表し、例えば−CO−NH−CH2−4−ニトロ−1,3−フェニレン−CH(CH3)−O−CO−中の4−ニトロ−1,3−フェニレン、または例えば−CH2−O−2−メトキシ−5−ニトロ−1,4−フェニレン−CH(CH3)−O−中の2−メトキシ−5−ニトロ−1,4−フェニレンなどがある。光切断リンカーを表す他の特定の態様は、例えば、−1,4−フェニレン−CO−CH2−O−CO−CH2−(フェナシル基)、−1,4−フェニレン−CH(OR)−CO−1,4−フェニレン−(アルコキシベンゾイン)、又は−3,5−ジメトキシ−1,4−フェニレン−CH2−O−(ジメトキシベンジル部分)である。先に(e)下で定義した炭素原子に置き換わる飽和もしくは不飽和シクロアルキレン基は、3〜7個の炭素原子を有するシクロアルキルに由来し、シクロペンチルまたはシクロヘキシルに由来するのが好ましく、例えば、1,2−もしくは1,3−シクロペンチレン、1,2−、1,3−、好ましくは1,4−シクロヘキシレン、または例えば1位もしくは2位で不飽和である1,4−シクロヘキシレンがある。先に(e)下で定義した炭素原子に置き換わる飽和もしくは不飽和ビシクロアルキレン基は、7もしくは8個の炭素原子を有するビシクロアルキルに由来し、例えば、ビシクロ[2.2.1]ヘプチレンやビシクロ[2.2.2]オクチレンであり、場合により、2位で不飽和であり、または2位と5位で二重に不飽和である1,4−ビシクロ[2.2.1]ヘプチレン、ならびに場合により、2位で不飽和であり、または2位と5位で二重に不飽和である1,4−ビシクロ[2.2.2]オクチレンが好ましい。先に(e)下で定義した炭素原子に置き換わる架橋複素環式芳香族基は、例えばトリアゾリデン、好ましくは1,4−トリアゾリデン、またはイソオキサゾリデン、好ましくは3,5−イソオキサゾリデンである。先に(e)下で定義した炭素原子に置き換わる架橋飽和もしくは不飽和ヘテロシクリル基は、例えば、先にR3下で定義した不飽和ヘテロシクリル基に由来し、例えばイソオキサゾリジネン、好ましくは3,5−イソオキサゾリジネン、または3〜12個の原子を有し、その1〜3個が窒素、酸素、硫黄から選択したヘテロ原子である完全飽和ヘテロシクリル基、例えばピロリジンジイル、ピペリジンジイル、テトラヒドロフランジイル、ジオキサンジイル、モルホリンジイル、またはテトラヒドロチオフェンジイルであり、2,5−テトラヒドロフランジイル、または2,5−ジオキサンジイルが好ましい。考えられる特定のヘテロシクリル基は、糖類部分であり、例えばα−もしくはβ−フラノシル部分、またはα−もしくはβ−ピラノシル部分である。
【0044】
リンカーR2またはR4の環状下部構造は、R2またはR4内で回転可能な結合数によって測定される分子柔軟性を減少させ、それによりインビボでの標識適用例全てにおいて重要である膜透過速度が上昇する。
【0045】
リンカーR2またはR4は、それぞれ、−CH=CH−基または−C≡C−基によって基AまたはR3に場合により結合している、1〜25個の炭素原子を有する直鎖アルキレン基、または4〜100個のエチレンオキシ単位を有する直鎖ポリエチレングリコール基であることが好ましい。さらに好ましいものは、1〜25個の炭素原子を有する直鎖アルキレン基であり、ここで、炭素原子はアミド官能基−NH−CO−によって場合により置き換えられており、光切断サブユニット、例えば、o−ニトロフェニルを有している。さらに好ましいものは、3〜6個のエチレングリコール単位であるポリエチレングリコール基と、炭素原子がアミド結合によって置き換えられているアルキレン基を含み、さらに置換されたアミノ官能基とヒドロキシ官能基を有する分枝リンカーである。他の好ましい分枝リンカーは、アミン、カルボキサミド、および/またはエーテル官能基がアルキレン基の炭素原子に置き換わっているデンドリティック(樹状)構造を有している。
【0046】
特に好ましいリンカーR2またはR4は、10〜40個の炭素原子の直鎖アルキレン基であり、ここで、3〜12個の炭素原子は酸素によって置き換えられ、1個もしくは2個の炭素原子は、それぞれ、1個もしくは2個の1,4−トリアゾリデン単位によって置き換えられ、かつ場合により1個の炭素原子は1,4−フェニレン単位によって置き換えられている。
【0047】
別の特に好ましいリンカーR2またはR4は、オキソによって場合により置換されている、10〜40個の炭素原子の直鎖アルキレン基であり、ここで、3〜12個の炭素原子は、酸素によって置き換えられ、1個もしくは2個の炭素原子は、窒素によって置き換えられている。
【0048】
別の特に好ましいリンカーR2またはR4は、6〜40個の炭素原子の直鎖アルキレン基であり、ここで、2〜12個の炭素原子は、酸素によって置き換えられ、2個の隣接する炭素原子間の1個もしくは2個の結合は官能基−CH=CH−を表す二重結合である。
【0049】
別の特に好ましいリンカーR2またはR4は、1〜15個の炭素原子の直鎖アルキレン基であり、N−メチルイソオキサゾリジン−3,5−ジメチル基である。
【0050】
リンカーR2またはR4は、一個もしくは複数の同じもしくは異なる標識、例えば1〜100個の同じもしくは異なる標識、特に1〜5個、好ましくは1、2、もしくは3個の、特に1個もしくは2個の同じもしくは異なる標識を有しうる。
【0051】
芳香族基もしくは複素環式芳香族基としてR3、または場合により置換されている不飽和アルキル、シクロアルキル、もしくはヘテロシクリル基としてR3は、(その反応機序に従い)AGTによって立体的に、そして電子的に受け入れられる基であり、それによってR3−R4−L1単位は融合蛋白質に共有結合的に移動する。R3−R4−L1単位では、R4−L1は、複数の同じもしくは異なる標識L1を有する複数の同じもしくは異なるリンカーR4である意味を含みうる。
【0052】
芳香族基としてR3は、フェニルまたはナフチル、特にフェニルが好ましく、例えばパラ位もしくはメタ位でR4によって置換されているフェニルである。
【0053】
複素環式芳香族基R3は、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)であり、そして、5〜12個の、好ましくは5または6個の環原子を有し;そして置換基R4を有することに加えて、置換されていなくても、あるいはメチルなどの低級アルキル、メトキシやエトキシなどの低級アルコキシ、例えば塩素、臭素、フッ素などのハロゲン、トリフルオロメチルなどのハロゲン化した低級アルキル、またはヒドロキシからなる群から選択される、一つ以上、特に一つの、さらなる置換基によって置換されていてもよい。
【0054】
単環式もしくは2環式ヘテロアリール基R3は、2H−ピロリル、ピロリル、イミダゾリル、ベンゾイミダゾリル、ピラゾリル、インダゾリル、プリニル、ピリジル、ピラジニル、ピリミジニル、ピリダジニル、4H−キノリジニル、イソキノリル、キノリル、フタラジニル、ナフチリジニル、キノキサリル、キナゾリニル、キノリニル、プテリジニル、インドリジニル、3H−インドリル、インドリル、イソインドリル、オキサゾリル、イソオキサゾリル、チアゾリル、イソチアゾリル、トリアゾリル、テトラゾリル、フラザニル、ベンゾ[d]ピラゾリル、チエニル、およびフラニルから選択されることが好ましい。より好ましくは単環式もしくは2環式ヘテロアリール基は、ピロリル、1H−イミダゾール−1−イルなどのイミダゾリル、1−ベンゾイミダゾリルなどのベンゾイミダゾリル;インダゾリル、特に5−インダゾリル:例えば2−、3−、または4−ピリジルなどのピリジル;ピリミジニル、特に2−ピリミジニル;ピラジニル;イソキノリニル、特に3−イソキノリニル;キノリニル、特に4−、または8−キノリニル;インドリル、特に3−インドリル;チアゾリル、トリアゾリル、テトラゾリル、ベンゾ[d]ピラゾリル、チエニル、ならびにフラニルからなる群からを選択される。
【0055】
特に好ましい本発明の態様では、ヘテロアリール基R3は、トリアゾリル、特に4位もしくは5位にさらに置換基R4を有する1−トリアゾリル;テトラゾリル、特に4位もしくは5位にさらに置換基R4を有する1−テトラゾリル、または5位にさらに置換基R4を有する2−テトラゾリル;イソオキサゾリル、特に5位にさらに置換基R4を有する3−イソオキサゾリル、または3位にさらに置換基R4を有する5−イソオキサゾリル;あるいはチエニル、特に3位、4位、もしくは5位、好ましくは4位にさらに置換基R4を有する2−チエニル、または4位にさらに置換基R4を有する3−チエニルである。
【0056】
最も好ましいものは、4位もしくは5位に置換基R4を有するトリアゾリルとしてのヘテロアリール基R3であり、また4位もしくは5位に置換基R4を有する2−チエニルとしてのR3である。
【0057】
場合により置換されている不飽和アルキル基R3は、1位もしくは2位に、好ましくは2位にさらに置換基R4を有する1−アルケニル;または1−アルキニルである。1−アルケニルで考慮される置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。特に好ましい本発明の態様では、R3は1−アルキニルである。
【0058】
場合により置換されている不飽和シクロアルキル基は、1位に不飽和で5〜7個の炭素原子を有するシクロアルケニル基であり、例えば任意の位置にさらに置換基R4を有する、1−シクロペンテニルまたは1−シクロヘキセニルである。考えられる置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0059】
場合により置換されている不飽和ヘテロシクリル基は、3〜12個の原子;窒素、酸素、硫黄から選択した1〜5個のヘテロ原子;およびヘテロシクリル基をメチレンCH2に結合する位置にある二重結合を有する。考えられる置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0060】
特に、場合により置換されている不飽和ヘテロシクリル基は、複素環式芳香族基R3について先に定義した部分的に飽和した複素環式芳香族基である。そのようなヘテロシクリル基の一例は、イソオキサゾリジニル、特に5位にさらに置換基を有する3−イソオキサゾリジニル、または3位にさらに置換基を有する5−イソオキサゾリジニルである。
【0061】
アリールメチルを意味するR5では、アリールは、フェニルまたはナフチルであることが好ましく、特に、フェニルまたは置換されたフェニル、例えば、メチルやエチルなどの低級アルキル、メトキシなどの低級アルコキシ、フッ素や塩素などのハロゲン、アミノ、またはアシルアミノによってパラ位もしくはメタ位で置換されたフェニルである。
【0062】
ヘテロアリールメチルを意味するR5では、ヘテロアリールは、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)であり、そして5〜12個の、好ましくは5または6個の環原子を有し;そして置換されていなくても、あるいはメチルなどの低級アルキル、メトキシもしくはエトキシなどの低級アルコキシ、例えば塩素、臭素、フッ素などのハロゲン、トリフルオロメチルもしくはヒドロキシなどのハロゲン化した低級アルキルからなる群から選択される、一つもしくは複数の、特に一つの、さらなる置換基によって置換されていてもよい。ヘテロアリールメチルR5のヘテロアリールとして好ましいものは、ヘテロアリールR3下で好ましいものとして記載したヘテロアリール、例えばトリアゾリルや2−チエニルである。
【0063】
場合により置換されているシクロアルキルとしてR5は、3〜7個の炭素原子を有するシクロアルキル基であり、例えば、任意の位置に場合により置換基を有する、シクロプロピル、シクロペンチル、またはシクロヘキシルである。考えられる置換基は、例えば、メチルなどの低級アルキル、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、またはクロロなどのハロゲニルである。
【0064】
場合により置換されているシクロアルケニルとしてR5は、任意の位置に、例えば1位に不飽和で5〜7個の炭素原子を有するシクロアルケニル基であり、例えば、任意の位置に場合により置換基を有する1−シクロペンテニルや1−シクロヘキセニルである。考えられる置換基は、シクロアルキルR5下で挙げた置換基である。
【0065】
場合により置換されているヘテロシクリル基としてR5は、飽和もしくは不飽和であり、3〜12個の原子、および窒素、酸素、硫黄から選択した1〜5個のヘテロ原子を有する。考えられる置換基は、例えば、メチルなどの低級アルキル、ビニルやアリルなどの低級アルケニル、アセチレニルなどのアルキニル、フェニルなどのアリール、トリフルオロメチルなどのハロ低級アルキル、ヒドロキシメチルなどのヒドロキシアルキル、ヒドロキシ、メトキシなどの低級アルコキシ、アセトキシなどの低級アシルオキシ、カルボキシ、カルバモイル、メトキシカルボニルなどの低級アルコキシカルボニル、アミノ、アセチルアミノなどのアシルアミノ、ニトロ、アジド、シアノ、低級アルキル−またはアミノ低級アルキル−スルフェニル、−スルフィニル、または−スルホニル、あるいはクロロなどのハロゲニルがある。好ましいヘテロシクリルは、例えば、2−テトラヒドロフラニルなどのテトラヒドロフラニルである。
【0066】
先に定義したR5は、例えば、2−テトラヒドロフラニル中に1個以上のキラル中心を有し、野生型hAGTによって唯一の鏡像異性体(またはジアステレオマー)を選択的に認識するかもしれない。
【0067】
好ましいR5は、シクロペンチル、シクロヘキシル、ベンジル;ヒドロキシ置換したシクロペンチル、シクロヘキシル、もしくはベンジル;およびヒドロキシ、もしくはヒドロキシ低級アルキル置換したテトラヒドロフラニルである。
【0068】
基質の標識L1およびL2は、融合蛋白質が意図される適用に依存して、当業者が選択することができる。標識は、例えば、標識L1を有する標識融合蛋白質を容易に検出し、または標識L1をその環境から分離するようなものであってよい。考えられる他の標識は、標識した融合蛋白質および/または基質環境において変化を感知し誘発することができる標識、あるいは基質の物理的および/または化学的特性によって融合蛋白質を操作することに役立つ標識で、融合蛋白質に特異的に導入されたものである。
【0069】
標識L1およびL2の例には、フルオロフォアや発色団などの分光プローブ、磁気プローブ、造影剤;放射性に標識した分子;パートナーに特異的に結合することができる特異的結合対の片方である分子;他の生体分子と相互に作用すると推測される分子;他の生体分子と相互に作用すると推測される分子ライブラリ;他の分子に架橋結合ことができる分子;繋留した金属キレートなど、H2O2およびアスコルバートに曝露されるとヒドロキシルラジカルを発生することができる分子;マラカイトグリーンなど、光を照射すると反応性ラジカルを発生することができる分子;固体支持体に共有結合している分子であって、支持体がガラススライド、マイクロタイタープレート、または当業者に公知の任意のポリマーであってよい分子;その相補鎖と塩基対を形成することができる核酸もしくはその誘導体;膜挿入特性を有する脂質もしくは他の疎水性分子;望ましい酵素的、化学的、もしくは物理的性を有する生体分子;あるいは上で挙げた特性の任意の組合せを有する分子が含まれる。
【0070】
さらに、標識L1およびL2は、結合させた分子が生細胞原形質膜を越えて移動するのを促進することが知られている正電荷の直鎖もしくは分枝鎖ポリマーである。これは、細胞膜透過性が低いか、そうでなければ実質的に生細胞の細胞膜不透過性である物質では特に重要である。細胞不透過性AGT基質は、そのような基L1またはL2にコンジュゲートすることによって細胞膜透過性になる。そのような細胞膜輸送エンハンサ基L1およびL2には、例えば、6〜15個のアルギニン残基を有するD−および/またはL−アルギニンの直鎖ポリ(アルギニン)、それぞれグアニジウム基を有する6〜15個のサブ単位の直鎖ポリマー、その一部がグアニジウム基に結合する6〜50サブユニットのオリゴマーもしくは短鎖ポリマー、および/またはHIV−tat蛋白質配列、特にサブユニットTat49−Tat57(一文字アミノ酸コードでRKKRRQRRR)配列の一部が含まれる。AGT基質は、先に定義したリンカーR2またはR4によってこのL1またはL2基に共有結合し、このリンカーは生細胞内側で不安定であるのが好ましく、例えば、細胞内エステル分解酵素によるエステル基R2もしくはR4の切断によって分解し、直接的に、またはエステル官能基の切断によって誘発されたさらに別の反応で、AGT基質と、細胞膜透過性を亢進する単位L1およびL2を分離しうる。
【0071】
好ましいものは、放射性標識分子を除く先に記載した標識L1およびL2である。本発明の範囲から除外されるものは、核酸を意味する標識L2である。標識L1として最も好ましいものは、分光プローブ、およびパートナーに特異的に結合することができる特異的結合対の片方である分子、いわゆる親和性標識である。標識L2として最も好ましいものは、パートナーに特異的に結合することができる特異的結合対の片方を表す分子、および固体支持体に共有結合している分子である。
【0072】
標識L1またはL2が、フルオロフォア、発色団、磁気標識、放射性標識などである場合、検出は、その方法がインビトロもしくはインビボで使用されようが、標識に適合させた標準的手段によって行われる。L1がフルオロフォアである場合、その方法を緑色蛍光蛋白質(GFP)の適用例と比較することができ、この緑色蛍光蛋白質は対象となる蛋白質に遺伝子的に融合され、生細胞において蛋白質研究を可能にするものである。標識L1およびL2の特定の例には、非線形光学特性を示すホウ素化合物もある。特に好ましいものは、2個の相互作用する分光プローブL1/L2の、L1が一構成要素であり、L2が他方の構成要素であって、ドナーとアクセプターが密接(10nmの距離未満)している場合、動的または静的消光によって、エネルギーをドナーとアクセプター(クエンチャー)間で無放射に移動できるようにする標識である。そのような1対の標識L1/L2は、標識基質とAGT融合蛋白質の反応においてその分光学的特性を変化させる。そのような1対の標識L1/L2の一例は、以下にさらに詳細説明するFRET対である。
【0073】
標識L1の特性に応じて、対象となる蛋白質とAGTを含む融合蛋白質は、基質と反応すると固体支持体に結合しうる。AGTを含む融合蛋白質と反応する基質の標識L1は、予め固体支持体に結合させておいてよく、AGTと反応し始めたとき、あるいは続いて、すなわちAGTに移動後、標識したAGT融合蛋白質を固体支持体に結合させるために使用しうる。あるいは、基質の標識L2は、固体支持体であっても、または固体支持体に結合してあっても、結合することもでき、この固体支持体によって、反応後に標識L1を有する標識融合蛋白質が基質残余部分から分離しL2を含むようになる。標識は、特異的結合対の一構成要素でよく、特異的結合対の他方の構成要素は、共有結合によって、または他の任意の手段によって固体支持体に結合し、または結合可能である。考えられる特異的結合対は、例えば、ビオチン、およびアビジンもしくはストレプトアビジンである。結合対のいずれかの構成要素は、基質の標識L1および/またはL2でよく、他方は固体支持体に結合されている。固体支持体へ好都合に結合できるようにする標識のさらに別の例には、例えば、マルトース結合蛋白質、糖蛋白、FLAGタグ、あるいはそのような置換基と、固体支持体の表面上の相補的官能基の間の化学選択的反応を可能にする反応性置換基がある。反応性置換基および相補的官能基のそのような対の例には、例えば、アミド、アジドを形成するアミンおよび活性化したカルボキシ基;および1,3−双極子環化付加反応を起こすプロピオル酸誘導体;2個のアミド結合を生じる活性化したビスジカルボン酸誘導体型の付加されたニ官能基リンカー試薬と反応する、アミンおよび別のアミン官能基、あるいは当技術分野で公知の他の組合せがある。
【0074】
好都合な固体支持体の例は、例えば、ガラススライドなどのガラス表面、マイクロタイタープレート、および適当なセンサー素子、特に機能性を付与したポリマー(例えばビーズ形)、化学的に改変した酸化物表面、例えば、二酸化ケイ素、タンタル五酸化物、もしくは二酸化チタン、または化学的に改変した金属表面、例えば金や銀表面などの貴金属表面である。次いで、不可逆的に結合させ、かつ/またはスポットしたAGT基質を使用してAGT融合蛋白質を空間的に離して、特に、固体支持体にスポットすることによって結合させ、蛋白質マイクロアレイ、DNAマイクロアレイ、または小分子のアレイを表してよい。
【0075】
標識L1またはL2が、外部刺激への曝露によってヒドロキシルラジカルなどの反応性ラジカルを生成できる場合、生成したラジカルは次いでAGT融合蛋白質、およびAGT融合蛋白質に密接しているような蛋白質を不活化することができ、これらの蛋白質の役割を研究できるようになる。そのような標識の例には、H2O2およびアスコルビン酸塩に曝露されるとヒドロキシルラジカルを発生する繋留した金属キレート錯体、およびレーザー照射によってヒドロキシルラジカルを発生するマラカイトグリーンなどの発色団がある。ヒドロキシルラジカルを発生させるために、発色団およびレーザーを使用することもまた、クロモホア援助レーザー誘起不活化法(CALI)として当技術分野で公知である。本発明では、標識L1としてマラカイトグリーンなどの発色団を有する基質でAGT融合蛋白質を標識し、続いてレーザー照射することによって、時間を制御し空間的に離して、標識したAGT融合蛋白質、およびAGT融合蛋白質と相互に作用するような蛋白質を失活させる。この方法は、インビボ、またはインビトロで適用することができる。さらに、AGT融合蛋白質に密接する蛋白質は、特異的抗体による蛋白質フラグメントの検出、高解像度2D電気泳動ゲル上でそれらの蛋白質の消失、あるいは分離による切断した蛋白質フラグメントの同定、および質量分析やN末端分解による蛋白質配列決定などの配列決定技術によって同定することができる。
【0076】
標識L1が、他の蛋白質、例えば、マレイミド、活性エステル、アジド、および当業者に公知の他のものなど、官能基を含む分子に架橋できる分子の場合、他の蛋白質と(インビボもしくはインビトロで)相互に作用するAGT融合蛋白質にそのような標識したAGT基質を接触させると、標識を介してAGT融合蛋白質とその相互作用する蛋白質の共有結合性架橋がもたらされる。これによりAGT融合蛋白質と相互に作用する蛋白質を同定できるようになる。光架橋用の標識L1(およびL2)は、例えば、ベンゾフェノンである。架橋の特殊な局面では、標識L1は、それ自体、AGT融合蛋白質を二量体化するAGT基質の分子である。そのような二量体の化学構造は、対称(ホモ二量体)でも、非対称(ヘテロ二量体)でもよい。
【0077】
考えられる他の標識L1は、例えば、フラーレン、中性子捕獲治療用ボラン、ヌクレオチド、オリゴヌクレオチド、例えば、自己アドレス指定(self-addressing)チップ用のもの、ペプチド核酸、および金属キレート、例えば、DNAに特異的に結合する白金キレートである。
【0078】
望ましい酵素的、化学的、もしくは物理的性を有する特定の生体分子はメトトレキサートである。メトトレキサートは、酵素、ジヒドロ葉酸還元酵素(DHFR)に強固に結合する阻害薬である。L1がメトトレキサートである式(1)の化合物は、いわゆる「二量体化化学誘導物質」(CID)の周知のクラスに属する。DNA結合ドメインLexAを有する融合蛋白質hAGTを使用し、L1がメトトレキサートである式(1)の化合物を有するhAGT融合蛋白質のインビボ標識に転写活性化ドメインB42を有するDHFRを加えることによって、hAGT−LexA融合蛋白質とDHFR−B42融合蛋白質のカップリング(「二量体化」)が誘発され、LexAとB42が空間的に近接し、続いて転写が刺激される。
【0079】
基質が2個以上の標識を有する場合、これらの標識は、同一でもまたは異なってもよい。特定の好ましい組合せは、異なる2個の親和性標識、または1個の親和性標識と1個の発色団標識、特に、1個の親和性標識と1個のフルオロフォア標識、または1対の分光学的に相互作用する標識L1/L2、例えばFRET対である。
【0080】
好ましいものは、Xが酸素である式(1)の化合物である。さらに好ましいものは、Xが酸素であり、R3がフェニルである化合物であり、特に、パラ置換フェニルもしくはチエニル、特に2,4−2置換チエニルである。
【0081】
同様に好ましいものは、基R1−Aが式(2)[式中、R6は、水素、ヒドロキシ、または非置換もしくは置換アミノであり、非置換アミノが好ましく、R7およびR8の一方はR1であり他方は水素である]のプリンラジカルである式(1)の化合物である。R7がR1である場合、R1は基−R2−L2もしくは残基R5であってよい。特に好ましいものは、R8がR1であり、R1が基−R2−L2である化合物に対応する。R1が残基R5である場合、好ましいR5の意味はシクロペンチルである。
【0082】
他の好ましい化合物は、基R1−Aが
式(3)の8−アザプリンラジカル[式中、置換基R6は水素、ヒドロキシ、または非置換もしくは置換アミノであり、非置換アミノが好ましい]、
式(4a)または(4b)のピリミジンラジカル[式中、R9は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、またはアミノであり、かつR10は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、ニトロ、またはニトロソである]、および
式(4c)プテリジンラジカル[式中、R6は、非置換もしくは置換アミノであり、非置換アミノが好ましく、かつR7およびR8の一方はR1であり他方は水素である。好ましくは、R7が水素であり、R8が基−R2−L2である]
である式(1)の化合物である。
【0083】
好ましいものは、L2が分光プローブである化合物、特にL1およびL2が分光プローブである化合物、例えば、FRET対を表す。同様に好ましいものは、L2が特異的結合対の片方を表す分子である化合物、L2が固体支持体に共有結合している分子である化合物、およびL2が細胞膜輸送エンハンサ基である化合物である。
【0084】
最も好ましいものは、実施例の化合物である。
【0085】
本発明は、さらに、対象となる蛋白質を検出し、かつ/または操作する方法であって、対象となる蛋白質をAGT融合蛋白質中に組み込み、該AGT融合蛋白質を先に記載した標識を有する特定のAGT基質に接触させ、そして、標識を認識しかつ/または取り扱うために設計した系で標識を使用して、該AGT融合蛋白質を検出し、場合によりさらに操作する方法に関する。
【0086】
本発明の方法では、対象となる蛋白質またはペプチドをO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)に融合する。対象となる蛋白質またはペプチドは、任意の長さでよく、どちらも二次、三次、または四次構造を有していても有していなくてもよく、かつ少なくとも12アミノ酸〜2000アミノ酸からなることが好ましい。そのような対象となる蛋白質またはペプチドの例には、例えば、酵素、DNA結合蛋白質、転写調節蛋白質、膜蛋白質、核内受容体蛋白質、核移行シグナル蛋白質、蛋白質補因子、小モノマーGTP加水分解酵素、ATP結合カセット蛋白質、細胞内構造蛋白質、特定の細胞区画に蛋白質を向かわせる配列を有する蛋白質、一般的に標識もしくは親和性タグとして使用される蛋白質、および前述の蛋白質ドメインもしくはサブドメインがある。対象となる蛋白質またはペプチドは、酵素によって、例えば、DNA期に適当な制限酵素によって切断されうるリンカー、および/または蛋白質期に適当な酵素によって切断可能なリンカーを経てAGTに融合されていることが好ましい。
【0087】
O6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)は、基質上に存在する標識をAGTのシステイン残基の一つに移動し、融合蛋白質の一部を形成する特性を有する。好ましい態様では、AGTは、野生型ヒトO6−アルキルグアニンDNAアルキルトランスフェラーゼ、hAGT、またはその変異体、例えば、Juilleratら., Chem Biol 10:313-317, 2003もしくは後の実施例に記載されている変異体である。本発明で使用される変異体AGTは、一個以上のアミノ酸置換、欠失、または付加という点で野生型hAGTと異なってよいが、依然として基質上に存在する標識を融合蛋白質のAGT部分に移動する特性を保持している。変異体AGTは、当業者に公知の蛋白質設計技術、例えば、飽和変異誘発、配列の任意の場所に変異を導入するためのエラープローン(error prone)PCR、あるいは飽和変異誘発および/またはエラープローンPCR後に使用されるDNAシャフリングを使用し生成されることが好ましいであろう。
【0088】
対象となる蛋白質およびO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)を含む融合蛋白質は、標識を有する特定の基質と接触させる。反応条件は、AGTが基質と反応し、基質の標識を移動させるように選択する。通常の条件は、pH7付近、室温、例えば、およそ25℃の緩衝液である。しかし、AGTは様々な他の条件下でも反応し、ここに述べたような条件が本発明の範囲を制限することはないことは理解されよう。
【0089】
反応速度は、基質の構造に非常に大きく依存する。本発明に記載した化合物は、反応性に違いがあり、これらの差は、異なるO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)、例えば異なる種に由来する野生型AGT、すなわち特に実施例で示す、野生型AGTの幾つかのアミノ酸が異なるアミノ酸によって置き換えられているAGT変異体と、本発明の化合物とを選択的に反応させるために使用しうる。
【0090】
反応性の差は、例えば、式(1){式中、Xは酸素であり、R3はパラ置換フェニルであり、基R1−Aは式(2)[式中、R6は非置換アミノであり、R7およびR8の一つは基−R2−L2であり、他方の一つは水素である]のプリンラジカルである}の化合物に見られる。R7が水素であり、R8が基−R2−L2であるような化合物は、R7が基−R2−L2(例えば同じ基−R2−L2)でありR8が水素である、対応する化合物よりも少なくとも3倍迅速に反応し、さらにR7とR8が水素である化合物よりも迅速に反応する。
【0091】
AGTは、その天然基質O6−アルキルグアニンDNAからアルキル基をそのシステイン残基の一つへ不可逆的移動する。同様に、AGTは、本発明の基質が関連する位置にあるグアニン型O6置換基またはその対応する置換基をそのシステイン残基の一つに移動する。AGTのこの特性を本発明の方法に使用して、式(1)の化合物の残基CH2−R3−R4−に結合した標識L1をAGTに移動する。
【0092】
この基質の標識L1は、融合蛋白質を意図したその適用に依存して当業者が選択することができる。AGTを含む融合蛋白質を基質と接触させた後、標識L1を融合蛋白質に共有結合させる。次いで、移動した標識によって、標識したAGT融合蛋白質をさらに操作し、かつ/または検出する。標識L1は、複数の同じもしくは異なる標識からなってよい。基質が、一つを超える標識L1を含む場合、その対応する標識したAGT融合蛋白質は、標識した融合蛋白質をさらに操作し、かつ/または検出するための選択肢をさらに提供する標識を一つを超えてさらに含む。
【0093】
特定の局面では、本発明は、標識L1をAGT融合蛋白質に移動させ、全ての未反応AGT基質を除去するのに特に好都合な方法を提供する。R2を介して基質としてAGTによって認識される複素環基Aに結合された標識L2が、固体支持体を意味し、または固体支持体に結合可能な反応基、例えば、固体支持体に結合するパートナーに特異的に結合することができる特異的結合対の片方を意味する標識L2である場合、これは簡単に行われる。次いで、L1で標識されたAGT融合蛋白質を含む、固体支持体および蛋白質溶液は、例えば、ろ過によって、さらに別の操作をせずに容易に分離される。より具体的には、分子過剰な式(1)[式中、R1はR2−L2であり、L2は固体支持体または固体支持体に結合された基である]の基質をAGT融合蛋白質と反応させ、得られた標識L1を有する標識AGT融合蛋白を固体支持体から分離するが、この固体支持体には反応した基質の残りの部分、すなわち残基R2−Aが結合し、さらにこの固体支持体には式(1)の過剰な未反応基質が依然として結合している。他の点では、AGT融合蛋白質は、基質と反応すると固体支持体から標識L1を離し、引き継ぎ、固体支持体に結合している全ての未反応標識L1はそのまま残される。
【0094】
この特定の局面では、標識L1は、先に記載した任意の標識、例えば、分光プローブまたはフルオロフォアでよい。固体支持体すなわちL2、またはL2が結合する支持体は、任意の固体支持体、例えば、ビーズ(例えば磁気ビーズ)、ポリマー支持体、金属表面でよい。未反応基質から標識したAGT融合蛋白質を分離することは、固体支持体の特性に応じて、ろ過、遠心分離、または他の適当な方法によってよく、例えば、固体支持体が磁気ビーズの場合は磁場をかけてよい。
【0095】
本発明の別の特定の局面では、AGT融合蛋白質による反応において標識L1およびL2の空間的分離を利用する方法を提供する。本発明の特定の基質は、相互作用性分光学的標識として、例えば、一端(例えばL1)にドナー(レポーター)を、他端(L2)にアクセプター(クエンチャー)を共有結合標識し、逆も同様であるプロ蛍光プローブとして設計される。そのようなプローブでの消光は、蛍光共鳴エネルギー移動(FRET)、またはドナーアクセプター対が密接することによる静的消光により生じうることが一般的に知られている。ドナーとアクセプターは、そのスペクトルの重なりが最大になるように選択すべきである。蛍光エネルギー転移は、典型的には、20〜100Åまでの距離で生じる。
【0096】
L1がドナー(レポーター)であり、L2がアクセプター(クエンチャー)であり、またはL1がクエンチャーであり、L2がレポーターである特定の本発明の方法では、AGT融合蛋白質と基質の反応によって蛍光が変化する。二重に標識した基質内のリポーター−クエンチャー間距離が、AGT融合蛋白質との反応によって変化し、レポーターとクエンチャーを空間的に分離し、それによって蛍光が出現する。それぞれ、標識L1またはL2として例えば赤外放射フルオロフォアを含む幅広い選択肢のレポーター基を使用してよく、唯一のフルオロフォア標識L1を有する基質と比較して、方法の感受性および特異性がかなり改善する。レポーターとクエンチャーを含む基質は、AGT融合蛋白質と反応するまで暗いままであり、反応混合物が「点灯」されると、フルオロフォアの発光スイッチが入る。なぜならば、今度は、レポーター標識とクエンチャー標識が、空間的に分離されるからである。蛍光消光およびエネルギー転移は、2個の標識、消光した標識またはエネルギードナー標識の一つだけが発光することによって測定できる。エネルギー転移が生じ、エネルギー受容標識も蛍光である場合、アクセプター標識蛍光も測定することができる。これらの2個の相互作用性標識のドナー標識は、励起ランプの必要性を取り除き、アクセプターバックグラウンド蛍光を低減する化学発光ドナープローブから選択することができる。そのような二重標識した基質を使用する記載した特定の方法は、蛍光時間測定に基づく反応速度の定量に有用であり、インビボおよびインビトロで利用してよい。
【0097】
インビトロでは、一般的に、AGT融合蛋白質と本発明の基質の反応は、細胞抽出物中で、またはAGT融合蛋白質の精製体もしくは濃縮体を用いて実施することができる。
【0098】
本発明の基質を用いる実験をインビボでまたは細胞抽出物中で行う場合、宿主の内在性AGTの反応が有利に考慮される。宿主の内在性AGTが、O6−アルキルグアニン誘導体、または基質として関連する化合物を受け入れない場合、(外来性)AGT融合蛋白質の反応は特異的である。ヒト、マウス、ラット細胞などの哺乳動物細胞中で内在性AGTを用いた非特異的反応が可能である。内在性AGTと(外来性)AGT融合蛋白質の同時反応に問題があるような実験では、公知のAGT欠乏細胞系を使用することができる。
【0099】
記載した特定の本発明の態様では、レポーターとクエンチャーを有する二重に標識した基質によって、インビトロもしくはインビボでAGT融合蛋白質の濃度を定量できるようになる。インビボでの適用については、レポーターは、近赤外(NIR)領域のエミッターであることが好ましい。この領域が生物蛍光に干渉しないからである。これらの要件に合致する公知のシアニンNIR色素を本発明の基質に組み込むことが好ましい。
【0100】
適当なレポーターとクエンチャー対は、当業者が選択することができる。典型的には、レポーターとクエンチャーは、スペクトルが大きく重なる蛍光色素であり、例えば、レポーターとしてフルオレセイン、およびクエンチャーとしてローダミンである。他のクエンチャーは、金クラスタや金属クリプテートである。
【0101】
本発明で使用される第2クラスのクエンチャーは、「ダーククエンチャー」(Johnasson, M. K.ら, Chem. Eur. J. 9:3466-3471, 2003)であり、すなわち、天然蛍光を持たず、一般的なレポーター色素の発光スペクトルと重複し、最大のFRET消光をもたらす吸収スペクトルを有する色素である。一般的なレポーター色素の発光スペクトルと重複し最大のFRET消光をもたらす吸収スペクトルを有する天然蛍光を持たない色素である。ずさらに、基底状態の複合体内で共鳴双極子−双極子相互作用機序(静的消光)を促進するために、その吸収帯が重なるように色素対を選択することができる。
【0102】
考えられる特定のフルオロフォアおよびクエンチャーは以下のものである。Alexa350、Alexa488、Alexa532、Alexa546、Alexa555、Alexa635、Alexa647を含むAlexa色素(PanchukVoloshina, N.ら, J. Histochem. & Cytochem. 47:1179-1188, 1999)、ジメチルアミノクマリン(7−ジメチルアミノクマリン−4−酢酸スクシンイミジルエステル、Moelcular Probes社製、商品名D374)、クエンチャーQSY35、QSY9、およびQSY21(Molecular Probes社製、Eugene OR 97402、米国)、シアニン−3(Cy3)、シアニン5(Cy5)、およびシアニン5.5(Cy5.5)(Amersham - GE Healthcare, Solingen, Germany);BHQ−1、BHQ−2、およびBHQ−3(Biosearch Technologies, Inc.のBlack Hole Quencher(商標), Novato, CA 94949, USA)、フルオロフォアATTO488、ATTO532、ATTO600、およびATTO655、およびクエンチャーATTO540Q、およびATTO612Q(Atto-Tec, D57076 Siegen, Germany)、フルオロフォアDY−505、DY−547、DY−632、およびDY−647(Dyomics, Jena, Germany)、5/6−カルボキシフルオレセイン、テトラメチルローダミン、4−ジメチルアミノアゾベンゼン−4'−スルホニル誘導体(Dabcyl)、および4−ジメチルアミノアゾベンゼン−4'−カルボニル誘導体(Dabcyl)。これらは、以下の組合せで有利に併用される。
フルオロフォア クエンチャー
・Alexa350、ジメチルアミノクマリン・Dabsyl、Dabcyl
5/6−カルボキシ−フルオレセイン、 BHQ1、QSY35
Alexa488、ATTO488、
DY−505
・5/6−カルボキシフルオレセイン、 ・BHQ2、QSY9、
Alexa488、Alexa532、 ATTO540Q
Alexa546、Alexa555、
ATTO488、ATTO532、
テトラメチルローダミン、Cy3、
DY−505、DY−547、
・Alexa635、Alexa647、 ・BHQ3、ATTO612Q、
ATTO600、ATTO655、 QSY21
DY−632、Cy5、DY−647
Cy5.5
【0103】
特定の態様では、この方法には、L2が固体支持体であり、もしくはレポーター/クエンチャー対の一構成要素をさらに有する固体支持体に結合しており、またはL2が固体支持体とレポーター/クエンチャー対の一構成要素の組合せであり、かつL1がこの対の他方の構成要素である基質が関与する。このように、AGT基質であるダーク固体支持体は、AGT融合蛋白質と反応すると蛍光性になる。
【0104】
レポーター/クエンチャー対、例えば、FRET対に基づく蛍光の定量が関与する記載の特定の方法は、相当の蛋白質調製物、および非特異的シグナルを除去するための条件操作を必要とする、SDS/PAGE、ウェスタンブロッティングなどに基づく濃度または反応速度を測定用の他の方法よりもはるかに好都合である。エネルギー移動法は、反応および未反応のAGT基質を分離する必要がない。従って、この方法は、化合物ライブラリの高処理スクリーニング、活性AGT変異体の同定、またはAGT阻害薬の同定に特に好都合である。
【0105】
さらに別の特定の本発明の方法では、R1が、基R5、特に、シクロアルキル、シクロアルケニル、シクロアルキルメチル、アリールメチル、またはヘテロアリールメチル基である基質を使用する。好ましい基R5の例は、シクロペンチルおよびベンジルである。AGT融合蛋白質では、変異体AGTは融合パートナーとして選択され、このパートナーは、R5が記載した特定の、または好ましい置換基の一つである基質に対して反応性が低い。そのような特定の対象となる蛋白質を検出しかつ/または操作する方法では、対象となる蛋白質は、変異体AGTと融合され、変異体AGT融合蛋白質を(a)基質であって、R1は基R5であり、変異体AGTによって認識されない基質、および(b)別のAGT基質であって、変異体AGT融合蛋白質によって認識され、先に記載した標識を有する基質の混合物に接触させ、標識を認識しかつ/または取り扱うために設計した系で標識を使用し、変異体AGT融合蛋白質を検出し、場合によりさらに操作する。そのような方法は、反応が内在性AGTを含む系においてインビボで実施され、または内在性AGTを含む細胞抽出物中で実施される場合の選択方法である。内在性AGTが飽和し、必要な場合は、R1が基R5である本発明の基質の標識L1を使用し分離する。並列反応では、変異体AGT融合蛋白質は、先に記載した一つの標識もしくは複数の標識を有する別のAGT基質と反応し、適宜検出し、かつ/または操作する。
【0106】
製造方法
本発明の基質は、一般的に、当技術分野で公知の標準的方法によって調製される。特定の方法は、例えば、特許出願PCT/EP03/10889に説明される。式(2)の基質、すなわち、Aがプリン物質である化合物の合成では、公知の方法を使用してO6、C8、およびN9の位置を改変することができる。本発明は、以下に記載する新規な方法、および使用し得られた新規な中間体にも関する。
【0107】
特に、複数の標識(例えばL1およびL2)を有するAGT基質の好ましい合成は、直交的に保護された官能基を利用する。保護基をそのように選択することによって、脱保護の分離が可能になり、その結果、各遊離された官能基を順次さらに化学的に操作して、標識をそれに結合させ、あるいはリンカーR2および/またはR4のそれ以上の伸展を導入することができる。想定される官能基に適した保護基は、当業者が選択することができ、例えば、T.W. GreeneおよびP.G.M. Wuts in "Protective Groups in Organic Synthesis", John Wiley & Sons, New York 1991に要約されている。
【0108】
グアニン誘導体の調製には、アンモニウム塩中間体を介した対応する6−クロログアニン誘導体と、三級アミン例えばメチルピロリジンとの反応に、基−CH2−R3−R4−L1もしくはその前駆体を導入する。アルコラート−O−CH2−R3−R4−L1もしくはその前駆体によりアンモニウム基を置き換えることによって、塩素を直接置き換えるのに比べて全体収量が多くなり、アンモニウム塩との反応を室温で実施することができる。
【0109】
式(2)のグアニンのN9位への基R1の導入は、ハロゲン誘導体R1−Halを使用するSN2反応による直接アルキル化によって実施しうる。しかし、位置選択性は、一般的に不十分であり、N7およびN9のアルキル化したグアニン生成混合物が得られる。アルキル化した生成物のN9/N7割合は、LiHで前処置し、またはN9置換に好都合な高温で得られた生成物を平衡化ことによってイミダゾール環を活性化すると高くなる。式(5)の好都合な中間体の調製をスキーム1に示す。
【0110】
【化12】
【0111】
式(5)の中間体は、N9のリンカーおよびO6位のベンジル官能基で直交的に保護されている。次いで、この中間体をさらに操作して残基−R4−L1をO6のベンジル官能基で、残基−R2−L2をN9位で完成させてよい。あるいはMitsunobuの条件(アルコール誘導体R1−OH、トリフェニルホスフィン、ジエチルアゾジカルボキシラート)を、N9位に置換基R1(すなわちR5、または−R2−L2)を導入するために使用してよい。
【0112】
中間体(5)に密接に関係するが、N9位にアジドプロピル基を有する化合物の合成を実施例8〜10の実験パートに例示する。
【0113】
【化13】
【0114】
スキーム2は、N9位に置換基を位置選択的に導入する閉環反応を示す。式(6)の中間体は、既に、標識L1を含み、アジド官能基を手直しすることによって適当な標識L2を導入できるようになる。
【0115】
トリフェニルホスフィンによって式(6)の化合物のアジド官能基を還元し、QSY−9−NHS(Molecular Probes)によってアシル化すると、式(7)[式中、L1(カルボキシ−フルオレセイン)およびL2(QSY−9)は、FRET対(スキーム3)を表す]のプロ蛍光AGT基質が得られる。
【0116】
【化14】
【0117】
標識L1およびL2を、それぞれ、O6位およびN9位に有し、FRET対を表す、さらに別の(7)型化合物を以下の実施例14、15、および19に記載する。
【0118】
N9の位置選択的アルキル化の別の方法は、スキーム4に示すパラジウム触媒アリル位アルキル化である。
【0119】
リンカー部分R2のレベルで構造を手直しするために、適当な官能性を持たせたホモ−N,O−ヌクレオシドの合成も同様にスキーム4に示すが、このヌクレオシドのグアノシンの糖部分はイソオキサゾリジン環によって置き換えられている(TBDPS=tert−ブチル−ジフェニル−シリル)。そのような手直しによって、一般的ヌクレオシドの比較的反応性アミン結合と比較して加水分解切断または酵素切断への耐性の増大、および高次構造の柔軟性の上昇が導入される。反応配列は可変であり、アリル二重結合に異なる適当な官能性を持たせたニトロンを結合できるようになる。
【0120】
あるいは、水素化ホウ素試薬9−ボラビシクロ[3.3.1]ノナン(9−BBN)によって、N9のアリル基を一級ヒドロキシ官能基に転換し、次いで適当な基R5または−R2−L2を得るためにさらに操作するすることができる。
【0121】
【化15】
【0122】
【化16】
【0123】
L1がフルオロフォア(Alexa594、Molecular Probes)であり、L2が固体支持体(例えばビーズB、またはアビジンもしくはストレプトアビジンを有するビーズに結合可能なビオチン残基B)である、式(9)の本発明の基質の合成をスキーム5に要約する。
【0124】
異なる2種の標識がプリン塩基のO6位およびC8位に結合している式(1)の化合物の合成に使用可能な中間体の合成をスキーム6および7に要約する。
【0125】
【化17】
【0126】
【化18】
【0127】
式(11)の化合物の代替合成法を実施例20〜23に例示する。この代替合成法では、p−(トリフルオロアセトアミドメチル)ベンジル置換基は初期に導入される。次いで、二重に保護された式(11)の化合物は、標識L1およびL2を、それぞれ、O6位およびC8に有する化合物、例えば、実施例24〜42に記載されている化合物にさらに作り上げることができる。
【0128】
R1−Aが式(4a)のピリミジン残基であり、R9がアミノであり、R10がニトロソである式(1)の化合物の調製については、スキーム8または9に示す反応順序を実施する。4−クロロ−2,6−ジアミノピリミジンを適当なナトリウムアルコキシドと反応させ、次いで亜硝酸ナトリウムの30%酢酸溶液で処理してニトロソ官能基を導入する。N6−置換誘導体は、適当な無水物または塩化アシルで処理することによって調製される。
【0129】
【化19】
【0130】
化合物(12)の直前の前駆体2,6−ジアミノ−5−ニトロソピリミジンをその対応する2,5,6−トリアミノピリミジンに還元する場合、式(4c)のプテリジンはジヒドロキシアセトンによる縮合、および実施例43〜46に記載した側鎖をさらに作成することによって得られる。
【0131】
【化20】
【0132】
N6位へのアミノアルキル置換基の導入をスキーム9に示す。11−アジド−3,6,9−トリオキサ−ウンデカ−アミンと2,4,6−トリクロロピリミジンの反応によって、クロマトグラフィーによって分離することができる2,6−二置換生成物と共に、6−置換クロロピリミジンが得られる。マスクをかけたアンモニア同等物として、2位をビス−p−メトキシ−ベンジルアミン(PMB)で置換することによって2置換ピリミジンが生じ、この2置換ピリミジンは、続いて、適当なアルコキシドとの反応によりO4誘導体へ転換される。60℃でトリフルオロ酢酸により脱保護すると2−アミノピリミジンが遊離される。5−ニトロソ化合物は、酢酸に含まれる亜硝酸ナトリウムによって調製される。
【0133】
実施例
略語:
DMF=ジメチルホルムアミド
DMSO=ジメチルスルホキシド
DTT=ジチオトレイトール
equiv.=当量
MPLC=中圧液体クロマトグラフィー
sat.=飽和
THF=テトラヒドロフラン
TEA=トリエチルアミン
TLC=薄層クロマトグラフィー
【実施例1】
【0134】
(4−ブロモチオフェン−2−イル)−メタノール(14)
【0135】
NaBH4(1.11g、29.31mmol)を4−ブロモチオフェン−2−カルボアルデヒド(5g、26.17mmol)を含むイソプロパノール(70mL)溶液に加える。反応混合物を室温で2時間攪拌する。NH4Cl(15mL)の飽和水溶液を溶液に加える。懸濁液をろ過し、ろ液を減圧濃縮し、CH2Cl2(70mL)に溶解し、MgSO4で乾燥し、再度減圧濃縮する。残留物をフラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:10)により精製する。収量4.55g(23.55mmol、90%)。TLC:Rf=0.75(酢酸エチル/石油エーテル1:1)。
【0136】
【表1】
【実施例2】
【0137】
2−シクロペンテニルメチルカルボナート(15)
【0138】
シクロペンタ−2−エン−1−オール(1.0g、11.9mmol、J.-L. Luche, J. Am. Chem. Soc., 1978, 100:7, 2226-2227)を含むCH2Cl2(30mL)およびピリジン(10mL)の0℃の攪拌溶液にクロロぎ酸メチル(2.94mL、38mmol)を30分かけて滴下する。4時間後、TLCは完全反応を示し、反応混合物を飽和NH4Cl(50mL)に注ぎ、Et2O(3×50mL)で抽出する。有機相を洗浄液が酸性になるまで1M HClで洗浄し、水洗(50mL)し、塩水(50mL)洗浄し、MgSO4で乾燥する。粗生成物をフラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:100)によって精製する。収量1.777g(12.5mmol、53%)。TLC Rf=0.70(酢酸エチル/石油エーテル1:12)。
【0139】
【表2】
【実施例3】
【0140】
N9−(シクロペンタ−2−エニル)−6−クロログアニン(16)
【0141】
この反応は、無水条件下(アルゴン雰囲気下、かつ溶媒は分子篩にかける)で実施する。6−クロログアニン(596mg、3.52mmol)を含むDMSO(10mL)溶液に、Pd(PPh3)4(404mg、0.35mmol)を加え、続いて2−シクロペンテニルメチルカルボナート(13)(500mg、3.52mmol)を含むTHF(10mL)溶液を加える。反応混合物を室温で1時間攪拌する。それをH2O(60mL)に注ぎ、酢酸エチル(3×50mL)で抽出する。混合有機層を塩水(60mL)で洗浄し、MgSO4で乾燥し、減圧濃縮する。残留物をフラッシュカラムクロマトグラフィー(酢酸エチル/石油エーテル6 4)によって精製する。収量:381mg(1.62mmol、61%)。TLC Rf=0.33(酢酸エチル/石油エーテル6:4)。
【0142】
【表3】
【実施例4】
【0143】
N9−シクロペンチル−6−クロログアニン(17)
【0144】
パラジウム担持活性炭(100mg)をN9−(シクロペンタ−2−エニル)−6−クロログアニン(16)(200mg、0.85mmol)を含むメタノール(33mL)溶液に加える。水素ガスを30分間溶液に通す。粗生成物をシリカ(500mg)に吸着させ、フラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:4、3:7、および2:3)によって精製する。収量:111mg(0.47mmol、55%)。TLC Rf=0.34(酢酸エチル/石油エーテル1:1)。
【0145】
【表4】
【0146】
または、臭化シクロペンチル(200mg、1.34mmol)を6−クロログアニンを含むジメチルアセトアミド懸濁液に加え、続いてNaOMe(144mg、2.67mmol)を加える。溶液を100℃で終夜攪拌する。溶媒を減圧蒸発させる。残留物をシリカ(1g)に吸着させ、フラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:1)によって精製する。収量:140mg(0.59mmol、44%)。
【実施例5】
【0147】
N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(CPTG、18)
【0148】
N9−シクロペンチル−6−クロログアニン(17、50mg、0.21mmol)を含むDMF(1.3mL)溶液に1,4−ジアザビシクロ[2.2.2]オクタン(DABCO、71mg、0.63mmol)を加える。反応混合物を室温で3時間攪拌する。TLCは、完全反応を示す。次いで、(4−ブロモチオフェン−2−イル)−メタノール(14、49mg、0.25mmol)および1,8−ジアザビシクロ[5.4.0]−ウンデカ−7−エン(DBU)(96mg、0.094mL、0.63mmol)を含むDMF(0.7mL)溶液を反応混合物に加える。溶液を終夜室温で攪拌する。粗生成物をフラッシュクロマトグラフィーによって精製する(酢酸エチル/石油エーテル1:9→3:7)。収量25mg(0.063mmol、30%)。TLC Rf=0.23(酢酸エチル/石油エーテル1:1)。
【0149】
【表5】
【実施例6】
【0150】
N9−ベンジル−6−クロログアニン(19)
【0151】
フラスコに6−クロログアニン、PPh3、およびベンジルアルコールを装入する。混合物を3時間減圧乾燥し、続いて活性化分子篩(4Å)を加えた無水THFに溶解する。15分間攪拌した後、ジイソプロピルアゾジカルボキシラート(DIAD)を加える。反応を終夜攪拌する。溶媒を減圧下で除去し、生成物をカラムクロマトグラフィー(石油エーテル/酢酸エチル3:1)によって精製する。
【0152】
【表6】
【実施例7】
【0153】
N9−ベンジル−O6−(4−ブロモチオフェン−2−イル)−グアニン(20)
【0154】
N9−ベンジル−6−クロログアニン(19、50mg、0.19mmol)を含むDMF(1.5mL)溶液に、1,4−ジアザビシクロ[2.2.2]オクタン(DABCO、65mg、0.57mmol)を加える。反応混合物を室温で3時間攪拌する。TLCは、完全反応を示す。次いで、(4−ブロモ−チオフェン−2−イル)−メタノール(14,44mg、0.23mmol)および1,8−ジアザビシクロ[5.4.0]−ウンデカ−7−エン(DBU)(88mg、0.086mL、0.57mmol)を含むDMF(0.7mL)溶液を反応混合物に加える。溶液を終夜室温で攪拌する。粗生成物をフラッシュクロマトグラフィー(酢酸エチル/石油エーテル1:4→3:7)によって精製する。収量27mg(0.065mmol、34%)。TLC Rf=0.20(酢酸エチル/石油エーテル1:1)。
【0155】
【表7】
【実施例8】
【0156】
N9−(3−クロロプロピル)−2−アミノ−6−クロルプリン(21)
【0157】
6−クロロプリン(5.0g、29.48mmol)、およびK2CO3(4.48g、32.42mmol)を含む無水DMFの攪拌懸濁液に、1−ブロモ−3−クロロプロパン(13.92g、88.45mmol)を加える。反応混合物を50℃で1分間加熱し、室温でさらに3時間攪拌する。反応混合物を120mLの水に注ぎ、CH2Cl2で抽出する。混合有機相を水洗し、MgSO4で乾燥し、減圧蒸発させる。残留物をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール50:1〜10:1)によって精製すると、2.6g(10.65mmol、35%)の表題化合物が無色固体として得られる。
【0158】
【表8】
【実施例9】
【0159】
N9−(3−アジドプロピル)−2−アミノ−6−クロルプリン(22)
【0160】
N9−(3−クロロプロピル)−2−アミノ−6−クロルプリン(21、2.56g、10.40mmol)、およびアジ化ナトリウム(811mg、12.48mmol)を25mLのDMSOに溶解し、80℃で16時間攪拌する。室温に冷却後、反応混合物を150mLの水に注ぎ、CH2Cl2で抽出する。混合有機相を水洗し、MgSO4で乾燥し、減圧蒸発させる。残留物をフラッシュカラムクロマトグラフィー(ジクロロメタン/メタノール50:1〜10:1)によって精製すると表題化合物が無色固体(650mg、2,57mmol、24%)として得られる。
【0161】
【表9】
【実施例10】
【0162】
N−[4−(2−アミノ−9−(3−アジドプロピル)−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(23)
【0163】
【化21】
【0164】
2,2,2−トリフルオロ−N−(4−ヒドロキシメチル−ベンジル)−アセトアミド(138mg、0.59mmol)をアルゴン雰囲気下で3mLの無水ジメチルアセトアミドに溶解し、NaH(31mg、1.31mmol)を加える。N9−(3−アジドプロピル)−2−アミノ−6−クロロプリン(22、100mg、0.39mmol)を加え、溶液を90℃で16時間加熱する。室温に冷却後、混合物を80mLの水に注ぎ、トリフルオロ酢酸でpHを6に調整する。水相をCH2Cl2および酢酸エチルで抽出する。有機相を水と塩水で洗浄し、MgSO4で乾燥し、溶媒を蒸発させる。粗生成物をメタノールに溶解し、SiO2(150mg)に吸着させ、ジクロロメタン:メタノール(95:5)のフラッシュカラムクロマトグラフィーによって精製する。収量:151mg(0.0336mmol、85%)。
【0165】
【表10】
【実施例11】
【0166】
N−[4−(2−アミノ−9−(3−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(24)
【0167】
アジド23(実施例10、100mg、0.23mmol)を0.9mLの1,4−ジオキサン/H2O(8:1)に溶解し、0.3mLのトリメチルホスピン溶液(THF中1M)を加える。反応を室温で3時間攪拌し、次いで全揮発性物質を減圧除去する。粗生成物をジクロロメタン/メタノール(95:5〜10:1)のフラッシュカラムクロマトグラフィーによって精製する。収量:87mg(0.021mmol、91%)。
【実施例12】
【0168】
N−[4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(25)
【0169】
4−([4−ジメチルアミノ−フェニル]−アゾ)−安息香酸スクシンイミジルエステル(14.3mg、0.0039mmol)およびトリフルオロアセタミド24(実施例11、16.6mg、0.0039mmol)をTEA10μL含有DMF0.8mLに溶解する。反応混合物を室温で終夜攪拌し、生成物を逆相MPLCによって精製する。
【実施例13】
【0170】
4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)プリン−6−イルオキシメチル)−ベンジルアミン(26)
【0171】
トリフルオロアセタミド25(実施例12、50mg)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を終夜室温で攪拌し、全揮発性物質を減圧除去する。生成物を次のステップでさらに精製することなく使用する。MS(ESI)m/z 579.1[M+H]+。
【実施例14】
【0172】
N−[4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(27)
【0173】
【化22】
【0174】
アミン26(実施例13、2mg)、およびATTO488−スクシンイミジルエステル(28、2mg、Atto-Tech, Siegen, Germany, Cat.-No. AD488-3)をTEA1μL含有DMF150μLに溶解し、室温で24時間放置する。生成物(27)を逆相MPLCによって精製する。
【実施例15】
【0175】
N−[4−(2−アミノ−9−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(29)
【0176】
【化23】
【0177】
アミン26(実施例13、2mg、0.0034mmol)および5(6)−カルボキシフルオレセインスクシンイミジルエステル(30、2.0mg、0.0042mmol)をTEA1μL含有DMF150μLに溶解し、室温で24時間放置する。生成物(29)を逆相MPLCによって精製する。
【実施例16】
【0178】
4−(2−アミノ−9−(3−アジドプロピル)−プリン−6−イルオキシメチル)−ベンジルアミン(31)
【0179】
アジド23(実施例10、50mg、0.11mmol)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を50℃で終夜攪拌し、全揮発性物質を減圧除去する。生成物をフラッシュカラムクロマトグラフィー(CH2Cl2/MeOH10:1)によって精製し33.5mgを得た。
【0180】
【表11】
【実施例17】
【0181】
N−[4−(2−アミノ−9−(3−アジドプロピル)−プリン−6−イルオキシメチル)−ベンジル]−テトラメチル−ローダミン−5(6)−カルボキサミド(32)
【0182】
アジド31(実施例16、10mg、0.08mmol)および5(6)−カルボキシテトラメチルローダミンスクシンイミジルエステル(33、7.47mg、0.014mmol)をTEA20μL含有DMF850μLに溶解し、室温で24時間放置する。生成物を逆相MPLCによって精製し9.0mg(0.012mmol、79%)を得た。MS(ESI)m/z 766.0[M]+。
【0183】
【化24】
【実施例18】
【0184】
N−[4−(2−アミノ−9−(3−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−テトラメチルローダミン−5(6)−カルボキサミド(34)
【0185】
アジド32(実施例17、9.0mg、0.012mmol)を600μLの1,4−ジオキサンおよび80μLの水の混合物に溶解する。この貯蔵液の200μLを14μLのトリメチルホスフィン(THF中1M)溶液で3時間処理する。全揮発性物質を減圧除去し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製する。収量3.47mg(0.0047mmol,40%)。MS(ESI)m/z 740.1[M+]。
【実施例19】
【0186】
N−[4−(2−アミノ−9−(3−QSY9−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]テトラメチルローダミン−5(6)−カルボキサミド(35)
【0187】
【化25】
【0188】
アミン34(実施例18、100μL、DMF中4.7mM)溶液にQSY−9−スクシンイミジルエステル(36、Molecular Probes、Eugene OR、USA、Cat.No.Q20131)(0.8mg)および1μLのTEAを加え、混合物をシェーカーに乗せ室温で終夜放置する。水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製する。MS(ESI)m/z 1539.9[M+H]+。
【実施例20】
【0189】
2,6−ジアミノ−4−[4−(2,2,2−トリフルオロ−アセトアミドメチル)−ベンジルオキシ]−ピリミジン37
【0190】
N−(4−ヒドロキシメチル−ベンジル)−2,2,2−トリフルオロ−アセトアミド(12.0g、52mmol)を80mLの無水DMSOに溶解し、NaH(4.08g、102mmol、鉱油中60%懸濁液)をアルゴン雰囲気下、30分かけて少しずつ加える。室温で1時間攪拌後、2,6−ジアミノ−4−クロロピリミジン(7.52g、52mmol)を加え、反応混合物を60℃まで終夜加熱する。室温に冷却後、混合物を1Lの1N HClに注ぎ、生成物を酢酸エチル(500mL)で抽出する。混合有機相を水と塩水で洗浄し、MgSO4で乾燥し、溶媒を蒸発させる。残留物をフラッシュカラムクロマトグラフィー(酢酸エチル/メタノール100:0〜80:20)によって精製すると、表題化合物が無色固体(4.7g、13.78mmol、27%)として得られる。
【0191】
【表12】
【実施例21】
【0192】
2,6−ジアミノ−5−ニトロソ−4−[4−(2,2,2−トリフルオロ−アセトアミドメチル)−ベンジルオキシ]ピリミジン(38)
【0193】
ピリミジン37(実施例20、4.72g、13.7mmol)を60mLの酢酸(30%)に溶解し、70℃に加熱する。5mLの水に溶解したNaNO2(1.4g、20.03mmol)をKJ澱粉紙が黒色に留まっているまで滴下する。氷浴で0℃に冷却後、紫色沈殿物をろ過によって収集し、アセトンから再結晶化すると、3.9g(10.54mmol、77%)の紫色固体が得られる。
【0194】
【表13】
【実施例22】
【0195】
2−アミノ−6−(4−(tert−ブトキシカルボニルアミノ)−ブタノイルアミノ)−5−ニトロソ−4−[4−(2,2,2−トリフルオロ−アセトアミドメチル)−ベンジルオキシ]−ピリミジン(39)
【0196】
アルゴン雰囲気下、4−(tert−ブトキシカルボニルアミノ)−酪酸(1.58g、8mmol)を20mLの乾燥THFに溶解し、0℃に冷却する。ジイソプロピルエチルアミン(1.18g、9mmol)を加え、混合物を−20℃に冷却し、クロロギ酸イソブチルエステル(1.15g、8mmol)を加える。混合物をこの温度で5分間攪拌し、20mLの乾燥THFに溶解させたニトロソピリミジン38(実施例21、2.64g、7mmol)を注射器によって加える。反応混合物を放置して室温まで暖め、55℃まで加熱し、この温度で終夜攪拌する。室温に冷却後、混合物を100mLの1N HClに注ぎ、生成物を酢酸エチル(300mL)で抽出する。混合有機相を水と塩水で洗浄し、続いてMgSO4で乾燥し、溶媒を蒸発させる。残留物をSiO2に吸着させ、フラッシュカラムクロマトグラフィー(シクロヘキサン/酢酸エチル1:2〜1:10)によって精製すると、表題化合物が青色固体(1.96g、3.53mmol、50.4%)として得られる。この粗製物質より、分析用試料を酢酸エチルから再結晶化する。
【実施例23】
【0197】
N−[4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(11)
【化26】
【0198】
ニトロソ−ピリミジン39(実施例22、1.84g、3.3mmol)およびトリフェニルホスフィン(1.89g、7mmol)を含む20mLのo−キシレンを10時間加熱還流する。室温に冷却後、溶媒を減圧除去し、残存する残留物を酢酸エチルに再溶解する。残留物をSiO2に吸着させ、フラッシュカラムクロマトグラフィー(酢酸エチル/エタノール1:0〜10:1)によって精製すると、表題化合物が無色固体(1.04g、3.53mmol、50.4%)としてが得られる。
【0199】
【表14】
【実施例24】
【0200】
N−[4−(2−アミノ−8−(3−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(41)
【0201】
tert−ブトキシカルボニル誘導体11(実施例23、100mg、0.19mmol)を10mLのジクロロメタンに懸濁し、2mLのトリフルオロ酢酸を加える。混合物を室温で30分間攪拌し、70mLのジエチルエーテルに注ぐ。沈殿物をろ過によって収集し、フラッシュカラムクロマトグラフィー(CH2Cl2/MeOH5:1)によって精製すると、74mg(0.17mmol、92%)の表題化合物が得られる。MS(ESI)m/z 424.1[M+H]+。
【実施例25】
【0202】
N−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(42)
【0203】
アミン41(実施例24、20mg、0.047mmol)および4−([4−ジメチルアミノ−フェニル]−アゾ)安息香酸スクシンイミジルエステル(17.3mg、0.047mmol)をTEA20μL含有DMF0.6mLに溶解する。反応混合物を終夜室温で攪拌し、溶媒を減圧除去する。生成物をフラッシュカラムクロマトグラフィー(CH2Cl2/MeOH95:5〜10:1)によって精製すると23mg(0.035mmol、75%)が得られる。MS(ESI)m/z 675.1[M+H]+。
【実施例26】
【0204】
4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)9H−プリン−6−イルオキシメチル)−ベンジル−アミン(43)
【0205】
トリフルオロアセタミド42(実施例25、23mg、0.035mmol)を2mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を50℃で終夜攪拌し、全揮発性物質を減圧除去する。生成物をさらに精製することなく使用する。MS(ESI)m/z 579.1[M+H]+。
【実施例27】
【0206】
N−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(44)
【0207】
【化27】
【0208】
アミン43(実施例26、2.0mg、0.0035mmol)および5−(6)−カルボキシフルオレセインスクシンイミジルエステル(1.63mg、0.0035mmol)をTEA5μL含有DMF200μLに溶解する。反応混合物を終夜室温で放置し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって粗生成物を精製する。MS(ESI)m/z 937.0[M+H]+。
【実施例28】
【0209】
N−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノ−フェニル]−アゾ−ベンゾイル)アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(45)
【0210】
【化28】
【0211】
アミン43(実施例26、1.0mg、0.0017mmol)およびATTO488−スクシンイミジルエステル(28、1.0mg、0.0015mmol)をTEA1μL含有DMF100μLに溶解する。反応混合物を終夜室温で放置し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって粗生成物を精製する。MS(ESI)m/z 1150.0[M+]。
【実施例29】
【0212】
N−[4−(2−アミノ−8−(3−QSY9−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]2,2,2−トリフルオロ−アセトアミド(46)
【0213】
アミン41(実施例24、1.6mg、0.038mmol)およびQSY−9−スクシンイミジルエステル(36、3.7mg、0.038mmol)をTEA2μL含有DMF0.25mLに溶解する。反応混合物を終夜室温で攪拌する。反応混合物を1mLの水/アセトニトリル(80:20)で希釈し、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって生成物を精製すると2.1mg(0.0017mmol、44%)の表題化合物が得られる。
【実施例30】
【0214】
4−(2−アミノ−8−(3−QSY9−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジルアミン(47)
【0215】
トリフルオロアセタミド46(実施例29、2.1mg、1.72μmol)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を終夜室温で攪拌し、全揮発性物質を減圧除去する。生成物をさらに精製することなく使用する。
【実施例31】
【0216】
N−[4−(2−アミノ−8−(3−QSY9−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−テトラメチルローダミン−5(6)−カルボキサミド(48)
【0217】
【化29】
【0218】
アミン47(実施例30、1mg、0.886μmol)、5(6)−カルボキシテトラメチルローダミンスクシンイミジルエステル(33、0.5mg、0.886μmol)、および1μLのTEAを100μLのDMFに溶解し、混合物をシェーカーに乗せ終夜室温で放置する。水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製する。
【実施例32】
【0219】
N−[4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−2,2,2−トリフルオロ−アセトアミド(49)
【0220】
アミン41(実施例24、10mg、0.023mmol)およびN−(+)−ビオチニル−6−アミノカプロン酸スクシンイミジルエステル(50、10mg、0.023mmol)をTEA5μL含有DMF650μLに溶解し、終夜室温で放置する。水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製すると、15.1mg(0.019mmol、86%)の表題化合物が得られる。MS(ESI)m/z 763.0[M+H]+。
【0221】
【化30】
【実施例33】
【0222】
4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル−アミン(51)
【0223】
トリフルオロアセタミド49(実施例32、15.1mg、0.019mmol)を1mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物室温で48時間を攪拌し、全揮発性物質を減圧除去する。生成物をさらに精製することなく使用する。MS(ESI)m/z 667.5[M+H]+。
【実施例34】
【0224】
N−[4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(52)
【0225】
【化31】
【0226】
アミン51(実施例33、10mg、0.015mmol)および5(6)−カルボキシフルオレセインスクシンイミジルエステル(30、7.0mg、0.015mmol)をTEA10μL含有DMF 500μLに溶解する。反応を24時間室温で放置し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって生成物を精製すると、8.2mg(0.008mmol、53%)の表題化合物が得られる。MS(ESI)m/z 1025.0[M+H]+。
【実施例35】
【0227】
4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル−アミン(53)
【0228】
トリフルオロアセタミド11(実施例23、100mg、0.19mmol)を2mLのメタノールに溶解し、2mLのメチルアミン(エタノール中33%)を加える。反応混合物を終夜50℃で攪拌し、全揮発性物質を減圧除去する。生成物をフラッシュカラムクロマトグラフィー(CH2Cl2/MeOH95:5〜10:1)によって精製すると、77mg(0.017mmol、94%)の表題化合物が得られる。
【実施例36】
【0229】
N−[4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(54)
【0230】
アミン53(実施例35、10mg、0.023mmol)および5(6)−カルボキシフルオレセインスクシンイミジルエステル(30、11.4mg、0.023mmol)をTEA10μL含有DMF 500μLに溶解し、終夜室温で攪拌し、続いて水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって生成物を精製すると、13.4mg(0.017mmol、73%)の表題化合物が得られる。MS(ESI)m/z 766.0[M+H]+。
【実施例37】
【0231】
N−[4−(2−アミノ−8−(3−アミノ−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]フルオレセイン−5(6)−カルボキサミド(55)
【0232】
tert−ブトキシカルボニルアミド54(実施例36、5mg、0.0062mmol)を2mLのCH2Cl2/MeOH(1:1)に溶解し、1mLのTFAを加える。混合物を室温で4時間攪拌し、全揮発性物質を減圧除去する。粗生成物を0.5mLのMeOHに再溶解し、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって精製すると、0.5mg(0.0006mmol、10%)の表題化合物が得られる。MS(ESI)m/z 686.2[M+H]+。
【実施例38】
【0233】
O6−フルオレセイン−C8−ビーズコンジュゲート(56)
【0234】
【化32】
【0235】
NHS活性化セファロース(商標)をポリプロピレンカラム(HiTrap(商標)、カラム容量1mL、Amersham Biosciences)に充填し、アミン55(実施例37、8mM)を含む0.5M NaCl溶液pH8.5と共に30分間インキュベートする。カラムを最初に6mLの緩衝液A(0.5M エタノールアミン、0.5M NaCl、pH8.3)で、続いて6mLの緩衝液B(0.1M 酢酸、0.5M NaCl、pH4)で洗浄する。残存する全てのNHSエステルをクエンチするために、続いてカラムを緩衝液Aと共に30分間インキュベートし、次いで再度6mLの緩衝液Aで、続いて6mLの緩衝液Bで、次いで6mLの緩衝液Aで再度洗浄する。式56のビーズがhAGT用基質として使用するために今準備される。
【実施例39】
【0236】
N−[4−(2−アミノ−8−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(57)
【0237】
アミン53(実施例35、2.5mg、0.08mmol)およびATTO488−スクシンイミジルエステル(28,4mg、0.08mmol)をTEA10μL含有DMF 500μLに溶解し、終夜室温で攪拌し、続いて水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって生成物を精製すると、13.4mg(0.017mmol、73%)の表題化合物が得られる。
【実施例40】
【0238】
N−[4−(2−アミノ−8−(3−アミノ−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]ATTO488−アミド(58)
【0239】
tert−ブトキシカルボニルアミド57(実施例39、2mg、0.002mmol)を1mLのCH2Cl2/MeOH(1:1)に溶解し、0.1mLのTFAを加える。混合物を室温で30分間攪拌し、全揮発性物質を減圧除去する。粗生成物を0.5mLのMeOHに再溶解し、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって精製する。
【実施例41】
【0240】
N−[4−(2−アミノ−8−(3−(マレイミドアセチル−アミノ)−プロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(59)
【0241】
アミン58(実施例40、1mg、0.0011mmol)およびマレイミド酢酸スクシンイミジルエステル(1mg、0.0039mmol)をTEA1μL含有DMF100μLに溶解し、終夜室温で放置する。水からアセトニトリル(0.08%TFA)までの直線勾配を使用し逆相MPLCによって粗生成物を精製する。
【実施例42】
【0242】
O6−ATTO488−C8−TyrArg9Cysコンジュゲート(60)
【0243】
【化33】
【0244】
マレイミドフルオロフォア59(実施例41、4当量)を含む50mLのアセトニトリルを、TyrArg9Cysを含む0.5mLの脱酸素化MeOHに加え、次いでN2下、2〜4時間室温で振盪する。エチルエーテル(10倍過剰な)を加えて生成物を沈殿させる。精製は、水からアセトニトリル(0.08%TFA)までの直線勾配を使用し、その対応する波長で検出しながら、逆相MPLCによって実施する。生成物は、MALDI−TOFMSによって特徴付ける。
【実施例43】
【0245】
2,5,6−トリアミノ−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]ピリミジン(61)
【0246】
2,6−ジアミノ−5−ニトロソ−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]−ピリミジン(1.22g、3.3mmol)およびトリフェニルホスフィン(1.89g、7mmol)を含む20mLのo−キシレンを1時間加熱還流する。室温に冷却後、溶媒を減圧除去し、残存する残留物をメタノールに再溶解する。残留物をSiO2に吸着させ、フラッシュカラムクロマトグラフィー(CH2Cl2/メタノール95:5〜5:1)によって精製すると表題化合物が得られる。
【実施例44】
【0247】
2−アミノ−6−ヒドロキシメチル−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]−プテリジン(62)
【0248】
ピリミジン61(実施例43、5.0g、14.1mmol)をアスコルビン酸ナトリウム(2.85g、14.4mmol)と共にジメチルアセトアミド/水(1:1)に溶解する。ジヒドロキシアセトン二量体(2.57g、14.3mmol)を加え、エアを反応混合物に吹き入れながら反応混合物を40℃に加熱する。4時間後、反応混合物を250mLのH2Oに注ぎ、形成した固体をろ過によって収集する。この固体をCH2Cl2/MeOH(3:1、500mL)に再溶解し、MgSO4で乾燥する。粗生成物をSiO2に吸着し、CH2Cl2/MeOH(20:1)を使用するフラッシュカラムクロマトグラフィーによって精製する。
【実施例45】
【0249】
2−アミノ−4−[4−(2,2,2−トリフルオロ−アセチルアミノ−メチル)−ベンジルオキシ]−プテリジン−6−カルボン酸(63)
【0250】
ヒドロキシメチル化合物62(実施例44、0.342g、0.84mmol)をアセトン/0.5M リン酸緩衝液、pH7(1:1、20mL)に懸濁し、過マンガン酸カリウム(0.34g、2.18mmol)を2時間かけて少しずつ加える。反応混合物を室温で3時間攪拌し、H2O(50mL)で希釈し、過マンガン酸が消費するまで亜硫酸ナトリウムを加えると、黒褐色沈殿物が生じ、これをろ過除去する。pHを2M HClを加えて2.5に調整すると黄色固体が生じ、これをろ過収集する。固体をH2O(50mL)に溶解しpHが30分間一定になるまで0.1M NaOHを加えてpHを7.0に調整する。混合物をろ過し、再度酸性化し、水からアセトニトリル(0.08%TFA)までの直線勾配の使用による逆相MPLCによって粗生成物を精製する。
【実施例46】
【0251】
2−アミノ−6−(3−(tert−ブトキシカルボニルアミノ)−プロピル)−4−[4−(2,2,2−トリフルオロアセチルアミノ−メチル)−ベンジルオキシ]−プテリジン(64)
【0252】
カルボン酸63(実施例45、0.10g、0.24mmol)およびPyBOP((ベンゾトリアゾール−1−イルオキシ)−トリピロリジノホスホニウムヘキサフルオロホスファート、0.12g、0.24mmol)を1mLの無水DMFに溶解し、室温で30分間攪拌する。TEA(50μL)およびtert−ブチル2−アミノエチルカルバマート(48mg、0.3mmol)を加える。反応混合物をさらに3時間攪拌し50mLの水に注入する。生成物をCH2Cl2で抽出し、MgSO4で乾燥し、CH2Cl2/MeOH(20:1)を使用するフラッシュカラムクロマトグラフィーによって精製する。
【実施例47】
【0253】
N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(18、CPTG)を有するAGT変異体の反応速度
【0254】
最初にAGT変異体Gly131Lys、Gly132Thr、Met134Leu、Arg135Ser、Asn157Gly、Ser159Gluを指向進化法によって調製する。PGEG−hAGT遺伝子の部分的に重複する2領域、変異Asn157Gly、Ser159Gluを含むAGT(Juillerat et al., Chem Biol 10:313-317, 2003)を別々の反応において適当なプライマーで増幅する。プライマーには、hAGT遺伝子のコドン131、132、134、135に対応する位置にヌクレオチド混合物NNK(N=A、C、G、もしくはT;K=GもしくはT)が含まれる。その部分的相補性に関して、さらなるPCR反応でこれらの2つのPCRフラグメントを合わせ、増幅してコドン131、132、134、135でランダム化された完全長遺伝子を得る。これらを、Sfil制限部位によりベクターpAK100の繊維状ファージのg3蛋白質に融合してクローン化する。得られた遺伝子ライブラリをファージディスプレイに使用する。
【0255】
このライブラリのファージは、大腸菌JM101細胞で産生される。指数関数的培養をヘルパーファージに重感染させ、終夜24℃で増殖する。この培養上清を1μMジゴキシゲニン化O6−ベンジルグアニン(Juillerat et al., Chem Biol 10:313-317, 2003の物質2)と共に6分間インキュベートする。次の選択ラウンドでは、反応時間をそれぞれ90秒および45秒に減少し、基質濃度を10nMに減少して選択圧力を増大させる。4%PEG/3%NaClで沈殿させることによって、この反応からファージを精製する。今回共有結合によりジゴキシゲニンで標識した変異体AGTを有するファージは、抗ジゴキシゲニン抗体(Roche Diagnostics)でコートした磁気ビーズと共にインキュベートすることによって単離し、細菌の再感染に使用される。
【0256】
選択したAGT変異体を増幅し、続いて発現ベクターpGEX−2T(Amersham)のBamH1とEcoR1間部位にクローン化する。これによりGST蛋白質C末端融合体として挿入遺伝子の発現が可能になり、ベクターによってこの遺伝子を提供する。
【0257】
このベクターからの蛋白質発現は大腸菌株BL21で行う。指数関数的に増殖する培養を0.5mM IPTGによって誘発し、24℃で3.5時間発現させる。
【0258】
精製:1mM PMSFおよび2μg/mLアプロチニンを補充し、50mMリン酸塩、0.5M NaCl、1mM DTTを含む緩衝液に回収した細胞を再懸濁し、リゾチームおよび超音波処理により破壊する。細胞残屑は、40000×gでの遠心分離によって分離する。抽出物を予め平衡化したグルタチオンセファロース(Amersham)にアプライし、次いでこれを20ベッド容量(50mMリン酸塩、0.5M NaCl、1mM DTT)で洗浄する。変異GST−AGT融合蛋白質を、10mM還元グルタチオンを含む50mM Tris−HClpH7.9で溶出する。精製した蛋白質を50mM HEPES pH7.2、1mM DTT、30%グリセロールに対して透析し、次いで−80℃で貯蔵する。
【0259】
変異Cys62Ala、Gln115Ser、Gln116His、Lys125Ala、Ala127Thr、Arg128Ala、Gly131Lys、Gly132Thr、Met134Leu、Arg135Ser、Cys150Asn、Ser151lle、Ser152Asn、Asn157Gly、Ser159Glu、および182での切り詰めを含む第2のAGT変異体(「AGTM」)は上記AGT変異体に由来するが、さらなる変異およびコドン182後の切り詰めを変異体AGT遺伝子に続いて導入することによる。続くPCR増幅によって、第1の変異体について記載したpGEX2T中にサブクローン化された、さらなる変異遺伝子が生じる。GST融合蛋白質が発現し同様に精製する。
【0260】
AGTMに関係し、変異Cys62Ala、Gln115Ser、Gln116His、Lys125Ala、Ala127Thr、Arg128Ala、Arg135Ser、Cys150Asn、Ser151lle、Ser152Asn、Asn157Gly、Ser159Glu、および182での切り詰めを含む第3のAGT変異体(「AGT21」)を同じ方法によって調製する。
【0261】
AGTMに関係し、変異Cys62Ala、Lys125Ala、Ala127Thr、Arg128Ala、Gly131Lys、Gly132Thr、Met134Leu、Arg135Ser、Cys150Ser、Asn157Gly、Ser159Glu、182での切り詰めを含む第4のAGT変異体(「AGT26」)も同じ方法によって調製する。
【0262】
AGTM(1μM終濃度)をN9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(18、CPTG)(1%DMSO中の終濃度10μM)を含む反応緩衝液(50mM HEPES、1mM DTT、200mg/mL BSA、pH7.3)と共に24℃でインキュベートする。試料を所定の時間(5、10、15、20、30、45、60、90、120分)に採取し、直接、ビオチン化O6−ベンジルグアニン(BGBT;Juillerat et al., Chem Biol 10:313−317, 2003の物質3a、10μM終濃度)と共に4分間インキュベートする。SDS−Laemmli緩衝液を加えることによって反応をクエンチし95℃で2分間加熱する。試料を12%アクリルアミドゲル上で泳動させ、ウェスタンブロッティングによって分析する。結果を図2に示す。
【0263】
2次速度定数:k2=92.5[S−1M−1]。この値は、BGBTによるAGTMの活性と比較して非常に遅い(k2=〜3,000[S−1M−1])。2種類の基質BGBT/CPTGの活性比は少なくとも25である。
【実施例48】
【0264】
N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)グアニン(18、CPTG)とビオチン化O6−ベンジルグアニン(BGBT)の間のインビトロでの競合
【0265】
変異体AGT(AGTM)および野生型ヒトAGT(0.5μM終濃度)を、別々に、BGBT(0.5μM終濃度)および異なる濃度のCPTG(終濃度0、0.5、1、5、10μM)を含む反応緩衝液(50mM HEPES、1mM DTT、200mg/mL BSA、pH7.3)と共に45分間インキュベートする。反応を2×SDS緩衝液を加えることによってクエンチし95℃で2分間放置する。試料を12%アクリルアミドゲル上で泳動させ、ウェスタンブロッティングによって分析する。
【0266】
AGTMおよび野生型hAGTは、化合物CPTGおよびBGBTの存在下、CPTGに対して著しく異なる反応性を示す。BGBTの20倍の濃度のCPTGでは、95%の野生型AGTが45分後にCPTGによってクエンチされるが、87%の変異体AGTMは依然として活性である。BGBTを有する野生型hAGTの反応速度定数は400S−1M−1であることが知られているので、CPTGを有する野生型hAGTの速度定数は、立体的に匹敵する、340S−1M−1の値(Lodewijkら、欧州特許第704445号)を示すN9−デスオキシリボシル−O6−ベンジルグアニンを有するhAGTの既知の反応性に基づいて著しく影響を受けないとみなすことができる。
【0267】
AGTM−6×His融合蛋白質(0.2μM終濃度)および野生型hAGT−GST融合蛋白質(1.2μM終濃度)を含む反応緩衝液(50mM HEPES、1mM DTT、200mg/mL BSA、pH7.3)溶液をBGBT(5μM終濃度)および異なる濃度のCPTG(0、5、10μM終濃度)と共に30分間インキュベートする。反応を2×SDS緩衝液を加えることによってクエンチし95℃で2分間放置する。試料を12%アクリルアミドゲル上で泳動させ、ウェスタンブロッティングによって分析する。結果を図3に示す。
【0268】
AGTMおよび野生型hAGTの反応を比較すると、CPTGのhAGTに対する高い特異性が観察される。同じ濃度(両基質、終濃度5μM)では、AGTMのわずか5%に対して95%の野生型AGTがCPTGと反応した。
【実施例49】
【0269】
異なる2種の基質を有する一試料における、異なる2つのAGT変異体の細胞内競合
【0270】
変異体AGTM(実施例47)を適当なプライマーによって増幅し、ベクターpEGFP−NucのNheIとBglII部位の間にクローン化する。酵母β−ガラクトシダーゼ遺伝子を同様に増幅し、BglII/BamHIによりこのベクター中にサブクローン化して、AGT−β−Gal融合遺伝子を有するベクターを得る。野生型hAGT遺伝子を増幅し、NheI/BglIIによってpEGFP−Nuc中にクローン化して、変異Gly160TrpをコードするAGT−NLS3融合遺伝子を得る。CHO細胞中で並行して両AGT変異体を一時的に発現させた後、細胞を10分間5μM CPTG(18)と共にインキュベートし、続いてJuillerat et al., Chem Biol 10:313-317, 2003、(O6−(4−(ジアセチルフルオレセイン−カルボニルアミノメチル)−ベンジル)−グアニン(BG−AcFL)の物質4の5μM共に20分間インキュベートする。細胞を洗浄し標準手順によって画像化する。
【0271】
CHO細胞では、蛍光基質BG−AcFLと反応させることによって、細胞質内に局在化するAGT−β−ガラクトシダーゼ融合蛋白質を選択的に蛍光標識する。核内にはその他のAGT融合蛋白質の有意な標識は観察されない。AGT(Gly160Trp)は、細胞内でCPTG(18)と効率よく反応し、従ってその後蛍光基質で標識されることは有り得ないと結論付けらる。変異体AGTMは、細胞内でCPTGとのプレインキュベーション後も依然として蛍光基質に反応性であり、従って第1ステップでCPTGと反応しない。
【実施例50】
【0272】
蛍光出現アッセイ
100μLのhAGT−GST融合蛋白質を含む反応緩衝液(50mM Tris、1mM DTT、1mg/mL BSA、pH8.0、hAGT−GSTの終濃度:1μM)の2個のアリコットを黒色平底マイクロタイタープレートに入れる。一つのアリコットを対照として使用し、これを40分間ベンジルグアニン(終濃度は100μM)でクエンチする。化合物(7)を含むDMSOをhAGT−GST(終濃度10nM)の両試料に加え、25℃で1時間インキュベートする。フルオレセイン−プロトコルを備えたVictor2Vプレートリーダー(Perkin-Elmer)で蛍光を読み取る。試料の蛍光読取り値は、対照の読取り値の少なくとも2倍である。
【0273】
さらに別のアッセイでは、蛋白質濃度10μMのAGT変異体AGTMおよびAGT21(実施例47)の精製蛋白質を、100mM NaCl、50mM Tris、pH8.0、0.1%Tween、1mM DTTを含む緩衝液に含まれた100nM N−[4−(2−アミノ−9−(3−QSY9−アミノプロピル)−プリン−6−イルオキシメチル)−ベンジル]−テトラメチルローダミン−5(6)−カルボキサミド(35、N9−QSY9−BG−TMR、実施例19)と共に室温でインキュベートする。蛍光強度(531nm励起/595nm発光)をVictor2マイクロプレートリーダー(Perkin Elmer)を使用し追跡する。変異体AGT21は、N9−QSY9−BG−TMR(化合物35)に対して有意な反応性を示し、25℃で1時間にわたってテトラメチルローダミン蛍光を約80%増大させる。それに反して、変異体AGTMでは、N9−QSY9−BG−TMRにより25℃で1時間におよぶインキュベーションすることで5%未満のテトラメチルローダミンの蛍光が増大する。
【0274】
同様に、AGTMおよびAGT21をN−[4−(2−アミノ−8−(3−(4−[4−ジメチルアミノフェニル]−アゾ−ベンゾイル)−アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−ATTO488−アミド(45、C8−Dabcyl−BG−Atto488、実施例28)、およびO6−(4−(ATT0488−アミドメチル)−ベンジル)グアニン(BG−Atto488、Atto-Tec Siegen, D, Prod Nr.: AD 488-2)と共にインキュベートし、蛍光強度を490nm(励起)および535nm(発光)で測定する。変異体AGTMは、BG−Atto488と比較してC8−Dabcyl−BG−Atto488に対して3倍の反応性を示す。変異体AGT21は、BG−Atto488と比較してC8−Dabcyl−BG−Atto488に対して二倍の反応性を示す。
【0275】
さらなる反応比較により以下の結果が得られた。
AGT21は、化合物48(C8−QSY9−BG−TMR、実施例31)と、C8/N9非置換化合物(BG−TMR、Molecular Probes製品C300)との場合より2倍速く、化合物35(N9−QSY9−BG−TMR、実施例19)との場合より少なくとも4倍速く反応する。AGT21は、化合物45(C8−Dabcyl−BG−Atto488、実施例28)と、C8/N9非置換化合物(BG−Atto488、Atto-Tec Siegen, D, Prod. No. AD 488-2)との場合より2倍速く、化合物27(N9−Dabcyl−BG−Atto488、実施例14)との場合より少なくとも4倍速く反応する。
【0276】
AGT21は、化合物44(C8−Dabcyl−BG−FL、実施例27)と、C8/N9非置換化合物(BG−FL、Juillerat et al., Chem Biol 10:313-317, 2003の物質5)との場合より2倍速く、化合物29(N9−Dabcyl−BG−FL、実施例15)との場合より少なくとも4倍速く反応する。
【0277】
AGTMは、化合物48(C8−QSY9−BG−TMR、実施例31)と、C8/N9非置換化合物(BG−TMR)との場合より2倍速く、化合物35(N9−QSY9−BG−TMR、実施例19)との場合より少なくとも10倍速く反応する。
【0278】
AGTMは、化合物45(C8−Dabcyl−BG−Atto488、実施例28)と、C8/N9非置換化合物(BG−Atto488)との場合より3倍速く、化合物27(N9−Dabcyl−BG−Atto488、実施例14)との場合より少なくとも15倍速く反応する。
【0279】
AGTMは、化合物44(C8−Dabcyl−BG−FL、実施例27)と、C8/N9非置換化合物(BG−FL)との場合より2倍速く、化合物29(N9−Dabcyl−BG−FL、実施例15)との場合より少なくとも10倍速く反応する。
【実施例51】
【0280】
ワンポット標識および分離アッセイ
50μLのhAGT−GFP融合蛋白質を含む反応緩衝液(50mM Tris、1mM DTT、1mg/mL BSA、pH7.3、hAGT−GFPの終濃度:5μM)を基質(9)(B=ビオチン、終濃度10μM)と共に1時間インキュベートする。150μLのストレプトアビジン被膜ビーズ(Dynabeads M−280)を加え、インキュベーション時間および上清からの磁気分離を製造業者の使用説明書に従って行う。488nmでのGFP吸収を測定することによって、磁気ビーズによる処理前後の溶液中の融合蛋白質量を定量する。融合蛋白質の損失は30%未満である。未反応基質を除去後、上清中のALEXA色素の吸収を594nmで測定によって標識効率を定量する。標識効率は少なくとも20%である。
【0281】
他の実験では、蛋白質濃度が5μMである精製AGTMまたはAGT21(実施例47)を、100mM NaCl、50mM Tris、pH8.0、0.1%Tween、1mM DTTを含む緩衝液50μlに含まれる10μMのN−[4−(2−アミノ−8−(3−(N−(+)−ビオチニル−6−アミノカプロイル)アミノプロピル)−9H−プリン−6−イルオキシメチル)−ベンジル]−フルオレセイン−5(6)−カルボキサミド(52、実施例34)と共に室温でインキュベートする。1時間後、少なくとも1nmolビオチンを結合する能力を有するストレプトアビジン被膜磁気ビーズ(Magnabindストレプトアビジン、Pierce)を加え、インキュベーション時間、およびフルオレセイン標識蛋白質を含む上清からの磁気分離を製造業者の使用説明書に従って行う。磁気ビーズによる処理前後の溶液中の蛋白質量をBradfordアッセイによって決定する。蛋白質の損失は30%未満である。未反応基質およびビーズへの結合および磁気分離によって遊離した生成物を除去後の上清中の蛍光強度を、490nm励起および535nm発光のVictor2マイクロプレートリーダー(Perkin Elmer)を使用して測定することによって標識効率を定量する。標識効率は少なくとも20%である。
【0282】
基質が予め固定されるさらに別の実験では、1ml当たり2μgのビオチンに結合する能力を有するストレプトアビジン被膜磁気ビーズ(Magnabindストレプトアビジン、Pierce)を、100mM NaCl、50mM Tris、pH8.0、0.1%Tweenを含む緩衝液に含まれた2倍過剰の化合物52(上のように)と共に室温でインキュベートして基質をビーズに固定する。インキュベーション時間、および上清からの磁気分離を製造業者の使用説明書に従って行う。未結合基質は、続く3回の洗浄ステップで緩衝液により除去する。蛋白質濃度が50μMのAGTMを、100mM NaCl、50mM Tris、pH8.0、0.1%Tween、1mM DTTを含む緩衝液50μlに含まれたこれらの基質改変ビーズと共に室温でインキュベートする。20時間後、フルオレセイン標識蛋白質を含む上清からビーズを分離する。磁気ビーズによる処理前後の溶液中の蛋白質量をBradfordアッセイによって決定する。蛋白質の損失は30%未満である。上のように、蛍光強度を測定することによって標識効率を定量するが、少なくとも20%である。
【実施例52】
【0283】
標識ビーズ
機能付与したセファロースビーズ56(実施例38)を5mLの緩衝液C(10mM HEPES、pH7.4、150mM NaCl、0.05%Tween20)で洗浄することによって平衡化する。この懸濁液に100μLのhAGT−GFP(終濃度50μM)を加え、25℃で1時間インキュベートする。セファロースビーズを遠心分離によって除去し、488nmで上清中のフルオレセインの吸収を測定することによって標識効率を定量する。標識効率は少なくとも20%である。488nmでのGFP吸収を測定することによって、改変セファロースビーズ処理前後の溶液中の融合蛋白質量を定量する。融合蛋白質の損失は30%未満である。
【0284】
別の実験では、製造業者により提供された使用説明書に従って、化合物56をセファロース4FastFlow(NHS−エステルとして活性化させたカルボキシル基、Amersham Prod No. 17-0906-01)によって調製する。洗浄液中に蛍光が検出されなくなるまで樹脂物質を洗浄し、暗所において4℃で貯蔵する。以下の全ステップで使用する緩衝液は、150mM NaClと50mM Tris、pH8.0に1mM DTTを加えたものである。各実験には、製造業者の使用説明書に従い、200μLの改変樹脂をBio-Rad Micro Biospinスピンカラムに充填し、2分間1000×gで遠心分離する。素通りは廃棄する。その後50μM濃度の非標識AGTM100μLを加え、25℃の暗所で1時間放置する。
【0285】
対照実験として、100μMのO6−ベンジルグアニンで予め30分間ブロックした50μM AGTMを100μL使用する。その後、カラムを1000×gで2分間遠心分離し、その度に200μLの緩衝液を加えることによって4回洗浄する。最終容量を1mLに調整する。蛋白質含有量をBCAアッセイ(Pierce BCA蛋白質アッセイキット−Prod. Nr. 23225, Perbio Science, Switzerland)によって決定する。見出された値は、予想された5μM濃度の少なくとも80%である。pH9に調整した100mM Na2CO3緩衝液で1:50に希釈した後、全溶液の蛍光を定量する。黒色96ウェルプレート(Costar)には各試料の200μLが備わる。試験試料のフルオレセイン範囲の蛍光をVictor2プレートリーダー(フルオレセインフィルタセット)によって読み取る。試験試料で見られた蛍光は、対照実験で見られた蛍光の少なくとも5倍である。
【実施例53】
【0286】
コンジュゲートO6ATTO488−C8−TyrArg9Cys(60)の細胞透過性
AGT26およびCaaX−ファルネシル化シグナル融合用ベクターを、内在性野生型AGTを持たないCHO細胞中に形質移入する。安定した形質移入体をジェネティシン(geneticine)処理下で選択する。その目的のために、AGT26遺伝子をpCMV−Script(Stratagene)中にクローン化し、そのC末端にpECP−F(Clontech)中にコードされているCaaX−box配列を続けた。最終蛋白質配列(位置690〜1271)によって、194アミノ酸を有する単一ポリペプチドが得られる。安定した形質移入細胞系統では、このポリペプチドによって、細胞膜内面に局在化するAGT融合蛋白質がもたらされる。これらの細胞を5μM O6−テトラメチルローダミン−ベンジルグアニン(BG−TMR、TMR−NHS−エステルから合成、Molecular Probes製品C300)で30分間標識し、続いて培地を2回交換し、次に30分間休止し、さらに培地を交換した場合、このAGT融合蛋白質は細胞膜に明白なシグナルをもたらす。テトラメチルローダミン用およびフルオレセイン用のフィルタセットを備えた逆位Zeiss蛍光顕微鏡Axiovert40CFLで全画像化を行う。同ステップ順序に続いて、5μM O6−Atto488−ベンジルグアニン(BG−ATTO488、Atto-Tec Siegen, D, Prod. No. AD 488-2)でインキュベーションしても、有意な細胞標識は観察されない。これは、ビス−スルホン化Atto488誘導体が非常に低い細胞膜透過性しか有していないことを示している。コンジュゲートO6−ATTO488−C8−TyrArg9Cys(60、実施例42)が同じ条件下、5μM濃度で使用される場合、細胞膜の主要標識によって有意な細胞内染色が観察される。これは、ベンジルグアニン誘導体が細胞透過性でない場合でも、ポリ−アルギニンリンカーの導入が透過性を増大させることを示している。
【図面の簡単な説明】
【0287】
【図1】対象となる蛋白質(P)およびAGTを含む融合蛋白質と、式R1−A−X−CH2−R3−R4−L1の基質との反応を示す模式図である。
【図2】AGTM−CPTGの動力学的測定:発光ペルオキシダーゼ基質により検出したAGTM−BGBT複合体の強度のウエスタンブロットを示す図である。実施例8を参照。変異体AGTM−6×Hisのアリコットを、緩衝化した水溶液中で、10倍過剰のN9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(CPTG)によりインキュベートする。所定の時間を経た後、ビオチン化O6−ベンジルグアニンによって反応が終了(BGBT、Juilleratら., Chem Biol 10: 313-317, 2003の基質3a)するが、これは蛋白質と非常に素早く反応し、それによって未反応AGTMを捕獲すると考えられる。次いで、蛋白質をSDSおよび熱で変性させる。試料をSDS−PAGEとウエスタンブロット分析にかける。対応するバンド強度は、化学発光染色によって検出する。
【図3】2種の蛋白質(〜25kDaのAGTM−6×His、0.2μM、および〜50kDaの野生型hAGT−GST融合蛋白質、1.2μM)と、2種の基質N9−シクロペンチル−O6−(4−ブロモチオフェン−2−イル)−グアニン(CPTG、0、5、10μM)、およびビオチン化O6−ベンジルグアニン(BGBT、5μM)の反応のウエスタンブロットを示す図である。実施例9参照。発光ペルオキシダーゼ基質によるAGTM−BGBT複合体の検出は、図2に記載されている通りである。
【特許請求の範囲】
【請求項1】
式(1)
【化1】
[式中、
Aは、基質としてO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)によって認識される基であり;
Xは、酸素または硫黄であり;
R1は、基−R2−L2または基R5であり;
R2およびR4は、互いに独立したリンカーであり;
R3は、芳香族基もしくは複素環式芳香族基、または場合により置換されている不飽和アルキル、シクロアルキル、もしくはCH2に結合する二重結合を有するヘテロシクリル基であり;
R5は、アリールメチルもしくはヘテロアリールメチル、または場合により置換されているシクロアルキル、シクロアルケニル、もしくはヘテロシクリル基であり;
L1は、1個の標識、複数の同じもしくは異なる標識、R4とAを結合して環状基質を形成する結合、またはさらに基−R3−CH2−X−A−R1であり;かつ
L2は、1個の標識、または複数の同じもしくは異なる標識である]
の化合物。
【請求項2】
請求項1に記載の式(1)の化合物であって、
Aが、1〜5個の窒素原子を含む複素環式芳香族基であり、
Xが、酸素であり、
R1が、基−R2−L2または基R5であり、
R2およびR4が、互いに独立した、1〜300個の炭素原子を有する直鎖もしくは分枝鎖アルキレン基であって、場合により
(a)一個以上の炭素原子が酸素によって、特に、全ての第3炭素原子が酸素によって置き換えられている、例えば、1〜100個のエチレンオキシ単位を有するポリエチレンオキシ基である;
(b)一個以上の炭素原子が、水素原子を有する窒素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、アミド官能基−NH−CO−を示す;
(c)一個以上の炭素原子が酸素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、エステル官能基−O−CO−を示す;
(d)2個の隣接する炭素原子間の結合が二重または三重結合であり、官能基−CH=CH−または−C≡C−を表す;
(e)一個以上の炭素原子が、フェニレン、飽和もしくは不飽和シクロアルキレン、飽和もしくは不飽和ビシクロアルキレン、架橋ヘテロ芳香族、または架橋飽和もしくは不飽和ヘテロシクリル基によって置き換えられている;
(f)2個の隣接する炭素原子が、ジスルフィド結合−S−S−によって置き換えられている;
または場合により置換基を含み、先の(a)〜(f)下で定義したような2個以上、特に、2個もしくは3個のアルキレン基および/または修飾アルキレン基の組合せであり、
R3が、フェニル、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む、5環もしくは6環原子の非置換もしくは置換、単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)、1−アルケニル、1−アルキニル、3〜7個の炭素原子を有する1−シクロヘキセニル、あるいは3〜12個の原子と、窒素、酸素、硫黄から選択した1〜5個のヘテロ原子と、ヘテロシクリル基とメチレンCH2を結合する位置にある二重結合を有する、場合により置換されている不飽和ヘテロシクリル基であり、
R5が、場合により置換されているフェニルメチルまたはナフチルメチル;場合により置換されているヘテロアリールメチル[ここで、ヘテロアリールは、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)であり、かつ5〜12個の環原子を有する];3〜7個の炭素原子を有する、場合により置換されているシクロアルキル;5〜7個の炭素原子を有する、場合により置換されているシクロアルケニル;3〜12個の原子と、窒素、酸素、硫黄から選択した1〜5個のヘテロ原子とを有する、場合により置換されている飽和もしくは不飽和ヘテロシクリルであり、
L1が、分光プローブ、磁気プローブ、造影剤、パートナーに特異的に結合することができる特異的結合対の片方である分子、他の生体分子と相互に作用すると推測される分子、他の生体分子と相互に作用すると推測される分子ライブラリ、他の分子に架橋結合することができる分子、H2O2およびアスコルバートに曝露されるとヒドロキシルラジカルを発生することができる分子、光を照射すると反応性ラジカルを発生することができる分子、固体支持体に共有結合している分子、その相補鎖と塩基対を形成することができる核酸もしくはその誘導体、膜挿入特性を有する脂質もしくは他の疎水性分子、望ましい酵素的、化学的、もしくは物理的性を有する生体分子、R4とAを結合して環状基質を形成する結合、およびさらに基−R3−CH2−X−A−R1から選択される一つもしくは複数の同じもしくは異なる標識であり、かつ
L2が、分光プローブ、磁気プローブ、造影剤、パートナーに特異的に結合することができる特異的結合対の片方である分子、他の生体分子と相互に作用すると推測される分子、他の生体分子と相互に作用すると推測される分子ライブラリ、他の分子に架橋結合することができる分子、H2O2およびアスコルバートに曝露されるとヒドロキシルラジカルを発生することができる分子、光を照射すると反応性ラジカルを発生することができる分子、固体支持体に共有結合している分子、膜挿入特性を有する脂質もしくは他の疎水性分子、および望ましい酵素的、化学的、もしくは物理的性を有する生体分子から選択される一つもしくは複数の同じもしくは異なる標識である、
化合物。
【請求項3】
基R1−Aが、式(2)
【化2】
「式中、R6は、水素、ヒドロキシ、または非置換もしくは置換アミノであり;R7およびR8の一方はR1であり他方は水素である]
のプリンラジカルである、請求項1に記載の式(1)の化合物。
【請求項4】
Xが酸素であり、R3がフェニルである、請求項3に記載の式(1)の化合物。
【請求項5】
Xが酸素であり、R3がチエニルである、請求項3に記載の式(1)の化合物。
【請求項6】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7がR1であり、R8が水素であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項7】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が基−R2−L2であり、R8が水素であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項8】
L2が分光プローブである、請求項7に記載の化合物。
【請求項9】
L1およびL2が分光プローブである、請求項7に記載の化合物。
【請求項10】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項9に記載の化合物。
【請求項11】
L1およびL2がFRET対を表す、請求項10に記載の化合物。
【請求項12】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が基R5であり、R8が水素であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項13】
R5がシクロペンチルである、請求項12に記載の化合物。
【請求項14】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が水素であり、R8がR1であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項15】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が水素であり、R8が基−R2−L2であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項16】
L2が分光プローブである、請求項15に記載の化合物。
【請求項17】
L1およびL2が分光プローブである、請求項15に記載の化合物。
【請求項18】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項17に記載の化合物。
【請求項19】
L1およびL2がFRET対を表す、請求項18に記載の化合物。
【請求項20】
L2が、特異的結合対の片方を表す分子である、請求項15に記載の化合物。
【請求項21】
L2が、固体支持体に共有結合している分子である、請求項15に記載の化合物。
【請求項22】
L2が、細胞膜輸送エンハンサ基である、請求項15に記載の化合物。
【請求項23】
基R1−Aが、式(3)
【化3】
[式中、置換基R6は、水素、ヒドロキシ、または非置換もしくは置換アミノである]
の8−アザプリンラジカルである、請求項1に記載の式(1)の化合物。
【請求項24】
Xが酸素であり、R3がフェニルである、請求項23に記載の式(1)の化合物。
【請求項25】
基R1−Aが、式(3)の8−アザプリンラジカルであり、R6が非置換アミノであり、R1が基−R2−L2であり、Xが酸素である、請求項23に記載の式(1)の化合物。
【請求項26】
L2が分光プローブである、請求項25に記載の化合物。
【請求項27】
L1およびL2が分光プローブである、請求項25に記載の化合物。
【請求項28】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項27に記載の化合物。
【請求項29】
L1およびL2がFRET対を表す、請求項28に記載の化合物。
【請求項30】
基R1−Aが、式(4a)または(4b)
【化4】
[式中、R9は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、またはアミノであり、かつR10は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、ニトロ、またはニトロソである]
のピリミジンラジカルである、請求項1に記載の式(1)の化合物。
【請求項31】
Xが酸素であり、R3がフェニルである、請求項30に記載の式(1)の化合物。
【請求項32】
基R1−Aが、式(4a)または(4b)のピリミジンラジカルであり、R1が基−R2−L2であり、Xが酸素である、請求項30に記載の式(1)の化合物。
【請求項33】
L2が分光プローブである、請求項32に記載の化合物。
【請求項34】
L1およびL2が分光プローブである、請求項32に記載の化合物。
【請求項35】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項34に記載の化合物。
【請求項36】
L1およびL2がFRET対を表す、請求項35に記載の化合物。
【請求項37】
基R1−Aが式(4c)
【化5】
[式中、R6は、非置換もしくは置換アミノであり、R7およびR8の一方はR1であり他方は水素である]
のプテリジンラジカルである、請求項1に記載の式(1)の化合物。
【請求項38】
Xが酸素であり、R3がフェニルである、請求項37に記載の式(1)の化合物。
【請求項39】
基R1−Aが式(4c)のプテリジンラジカルであり、R6が非置換アミノであり、R7が水素であり、R8がR1であり、R1が基−R2−L2であり、Xが酸素である、請求項37に記載の式(1)の化合物。
【請求項40】
L2が分光プローブである、請求項39に記載の化合物。
【請求項41】
L1およびL2が分光プローブである、請求項39に記載の化合物。
【請求項42】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項41に記載の化合物。
【請求項43】
L1およびL2がFRET対を表す、請求項42に記載の化合物。
【請求項44】
対象となる蛋白質を検出し、かつ/または操作する方法であって、対象となる蛋白質をAGT融合蛋白質中に組み込み、該AGT融合蛋白質を、請求項1から38のいずれか一項記載の式(1)の化合物に接触させ、標識を認識しかつ/または取り扱うために設計した系で標識L1を使用して該AGT融合蛋白質を検出し、場合によりさらに操作する方法。
【請求項45】
式(1)の化合物で、標識L2が固体支持体であり、かつ式(1)の化合物に接触させたAGT融合蛋白質を、ろ過、遠心分離、または磁気ビーズ分離によって式(1)の化合物から分離する、請求項44に記載の方法。
【請求項46】
式(1)の化合物で、2個の相互作用する分光プローブL1/L2の、標識L1が一構成要素であり、標識L2が他方の構成要素であり、AGT融合蛋白質が蛍光によって検出される、請求項44に記載の方法。
【請求項47】
対象となる蛋白質を検出しかつ/または操作するための請求項44に記載の方法であって、対象となる蛋白質を変異体AGTと融合し、変異体AGT融合蛋白質を
(a)R1が基R5であり、変異体AGTによって認識されない式(1)の化合物、および
(b)変異体AGT融合蛋白質によって認識される式(1)の別の化合物、
の混合物に接触させ、そして標識を認識しかつ/または取り扱うために設計した系で標識を使用して、変異体AGT融合蛋白質を検出し、場合によりさらに操作する方法。
【請求項1】
式(1)
【化1】
[式中、
Aは、基質としてO6−アルキルグアニンDNAアルキルトランスフェラーゼ(AGT)によって認識される基であり;
Xは、酸素または硫黄であり;
R1は、基−R2−L2または基R5であり;
R2およびR4は、互いに独立したリンカーであり;
R3は、芳香族基もしくは複素環式芳香族基、または場合により置換されている不飽和アルキル、シクロアルキル、もしくはCH2に結合する二重結合を有するヘテロシクリル基であり;
R5は、アリールメチルもしくはヘテロアリールメチル、または場合により置換されているシクロアルキル、シクロアルケニル、もしくはヘテロシクリル基であり;
L1は、1個の標識、複数の同じもしくは異なる標識、R4とAを結合して環状基質を形成する結合、またはさらに基−R3−CH2−X−A−R1であり;かつ
L2は、1個の標識、または複数の同じもしくは異なる標識である]
の化合物。
【請求項2】
請求項1に記載の式(1)の化合物であって、
Aが、1〜5個の窒素原子を含む複素環式芳香族基であり、
Xが、酸素であり、
R1が、基−R2−L2または基R5であり、
R2およびR4が、互いに独立した、1〜300個の炭素原子を有する直鎖もしくは分枝鎖アルキレン基であって、場合により
(a)一個以上の炭素原子が酸素によって、特に、全ての第3炭素原子が酸素によって置き換えられている、例えば、1〜100個のエチレンオキシ単位を有するポリエチレンオキシ基である;
(b)一個以上の炭素原子が、水素原子を有する窒素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、アミド官能基−NH−CO−を示す;
(c)一個以上の炭素原子が酸素によって置き換えられ、隣接する炭素原子がオキソによって置換されている、エステル官能基−O−CO−を示す;
(d)2個の隣接する炭素原子間の結合が二重または三重結合であり、官能基−CH=CH−または−C≡C−を表す;
(e)一個以上の炭素原子が、フェニレン、飽和もしくは不飽和シクロアルキレン、飽和もしくは不飽和ビシクロアルキレン、架橋ヘテロ芳香族、または架橋飽和もしくは不飽和ヘテロシクリル基によって置き換えられている;
(f)2個の隣接する炭素原子が、ジスルフィド結合−S−S−によって置き換えられている;
または場合により置換基を含み、先の(a)〜(f)下で定義したような2個以上、特に、2個もしくは3個のアルキレン基および/または修飾アルキレン基の組合せであり、
R3が、フェニル、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む、5環もしくは6環原子の非置換もしくは置換、単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)、1−アルケニル、1−アルキニル、3〜7個の炭素原子を有する1−シクロヘキセニル、あるいは3〜12個の原子と、窒素、酸素、硫黄から選択した1〜5個のヘテロ原子と、ヘテロシクリル基とメチレンCH2を結合する位置にある二重結合を有する、場合により置換されている不飽和ヘテロシクリル基であり、
R5が、場合により置換されているフェニルメチルまたはナフチルメチル;場合により置換されているヘテロアリールメチル[ここで、ヘテロアリールは、0、1、2、3、もしくは4個の環窒素原子、0もしくは1個の酸素原子、および0もしくは1個の硫黄原子を含む単環式もしくは2環式ヘテロアリール基(但し、少なくとも1個の環炭素原子は、窒素、酸素、もしくは硫黄原子によって置き換えられている)であり、かつ5〜12個の環原子を有する];3〜7個の炭素原子を有する、場合により置換されているシクロアルキル;5〜7個の炭素原子を有する、場合により置換されているシクロアルケニル;3〜12個の原子と、窒素、酸素、硫黄から選択した1〜5個のヘテロ原子とを有する、場合により置換されている飽和もしくは不飽和ヘテロシクリルであり、
L1が、分光プローブ、磁気プローブ、造影剤、パートナーに特異的に結合することができる特異的結合対の片方である分子、他の生体分子と相互に作用すると推測される分子、他の生体分子と相互に作用すると推測される分子ライブラリ、他の分子に架橋結合することができる分子、H2O2およびアスコルバートに曝露されるとヒドロキシルラジカルを発生することができる分子、光を照射すると反応性ラジカルを発生することができる分子、固体支持体に共有結合している分子、その相補鎖と塩基対を形成することができる核酸もしくはその誘導体、膜挿入特性を有する脂質もしくは他の疎水性分子、望ましい酵素的、化学的、もしくは物理的性を有する生体分子、R4とAを結合して環状基質を形成する結合、およびさらに基−R3−CH2−X−A−R1から選択される一つもしくは複数の同じもしくは異なる標識であり、かつ
L2が、分光プローブ、磁気プローブ、造影剤、パートナーに特異的に結合することができる特異的結合対の片方である分子、他の生体分子と相互に作用すると推測される分子、他の生体分子と相互に作用すると推測される分子ライブラリ、他の分子に架橋結合することができる分子、H2O2およびアスコルバートに曝露されるとヒドロキシルラジカルを発生することができる分子、光を照射すると反応性ラジカルを発生することができる分子、固体支持体に共有結合している分子、膜挿入特性を有する脂質もしくは他の疎水性分子、および望ましい酵素的、化学的、もしくは物理的性を有する生体分子から選択される一つもしくは複数の同じもしくは異なる標識である、
化合物。
【請求項3】
基R1−Aが、式(2)
【化2】
「式中、R6は、水素、ヒドロキシ、または非置換もしくは置換アミノであり;R7およびR8の一方はR1であり他方は水素である]
のプリンラジカルである、請求項1に記載の式(1)の化合物。
【請求項4】
Xが酸素であり、R3がフェニルである、請求項3に記載の式(1)の化合物。
【請求項5】
Xが酸素であり、R3がチエニルである、請求項3に記載の式(1)の化合物。
【請求項6】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7がR1であり、R8が水素であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項7】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が基−R2−L2であり、R8が水素であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項8】
L2が分光プローブである、請求項7に記載の化合物。
【請求項9】
L1およびL2が分光プローブである、請求項7に記載の化合物。
【請求項10】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項9に記載の化合物。
【請求項11】
L1およびL2がFRET対を表す、請求項10に記載の化合物。
【請求項12】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が基R5であり、R8が水素であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項13】
R5がシクロペンチルである、請求項12に記載の化合物。
【請求項14】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が水素であり、R8がR1であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項15】
基R1−Aが式(2)のプリンラジカルであり、R6が非置換アミノであり、R7が水素であり、R8が基−R2−L2であり、Xが酸素である、請求項3に記載の式(1)の化合物。
【請求項16】
L2が分光プローブである、請求項15に記載の化合物。
【請求項17】
L1およびL2が分光プローブである、請求項15に記載の化合物。
【請求項18】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項17に記載の化合物。
【請求項19】
L1およびL2がFRET対を表す、請求項18に記載の化合物。
【請求項20】
L2が、特異的結合対の片方を表す分子である、請求項15に記載の化合物。
【請求項21】
L2が、固体支持体に共有結合している分子である、請求項15に記載の化合物。
【請求項22】
L2が、細胞膜輸送エンハンサ基である、請求項15に記載の化合物。
【請求項23】
基R1−Aが、式(3)
【化3】
[式中、置換基R6は、水素、ヒドロキシ、または非置換もしくは置換アミノである]
の8−アザプリンラジカルである、請求項1に記載の式(1)の化合物。
【請求項24】
Xが酸素であり、R3がフェニルである、請求項23に記載の式(1)の化合物。
【請求項25】
基R1−Aが、式(3)の8−アザプリンラジカルであり、R6が非置換アミノであり、R1が基−R2−L2であり、Xが酸素である、請求項23に記載の式(1)の化合物。
【請求項26】
L2が分光プローブである、請求項25に記載の化合物。
【請求項27】
L1およびL2が分光プローブである、請求項25に記載の化合物。
【請求項28】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項27に記載の化合物。
【請求項29】
L1およびL2がFRET対を表す、請求項28に記載の化合物。
【請求項30】
基R1−Aが、式(4a)または(4b)
【化4】
[式中、R9は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、またはアミノであり、かつR10は水素、ハロゲン、1〜4個の炭素原子を有する低級アルキル、アミノ、ニトロ、またはニトロソである]
のピリミジンラジカルである、請求項1に記載の式(1)の化合物。
【請求項31】
Xが酸素であり、R3がフェニルである、請求項30に記載の式(1)の化合物。
【請求項32】
基R1−Aが、式(4a)または(4b)のピリミジンラジカルであり、R1が基−R2−L2であり、Xが酸素である、請求項30に記載の式(1)の化合物。
【請求項33】
L2が分光プローブである、請求項32に記載の化合物。
【請求項34】
L1およびL2が分光プローブである、請求項32に記載の化合物。
【請求項35】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項34に記載の化合物。
【請求項36】
L1およびL2がFRET対を表す、請求項35に記載の化合物。
【請求項37】
基R1−Aが式(4c)
【化5】
[式中、R6は、非置換もしくは置換アミノであり、R7およびR8の一方はR1であり他方は水素である]
のプテリジンラジカルである、請求項1に記載の式(1)の化合物。
【請求項38】
Xが酸素であり、R3がフェニルである、請求項37に記載の式(1)の化合物。
【請求項39】
基R1−Aが式(4c)のプテリジンラジカルであり、R6が非置換アミノであり、R7が水素であり、R8がR1であり、R1が基−R2−L2であり、Xが酸素である、請求項37に記載の式(1)の化合物。
【請求項40】
L2が分光プローブである、請求項39に記載の化合物。
【請求項41】
L1およびL2が分光プローブである、請求項39に記載の化合物。
【請求項42】
L1およびL2が、蛍光ドナー/蛍光クエンチャー対を表す、請求項41に記載の化合物。
【請求項43】
L1およびL2がFRET対を表す、請求項42に記載の化合物。
【請求項44】
対象となる蛋白質を検出し、かつ/または操作する方法であって、対象となる蛋白質をAGT融合蛋白質中に組み込み、該AGT融合蛋白質を、請求項1から38のいずれか一項記載の式(1)の化合物に接触させ、標識を認識しかつ/または取り扱うために設計した系で標識L1を使用して該AGT融合蛋白質を検出し、場合によりさらに操作する方法。
【請求項45】
式(1)の化合物で、標識L2が固体支持体であり、かつ式(1)の化合物に接触させたAGT融合蛋白質を、ろ過、遠心分離、または磁気ビーズ分離によって式(1)の化合物から分離する、請求項44に記載の方法。
【請求項46】
式(1)の化合物で、2個の相互作用する分光プローブL1/L2の、標識L1が一構成要素であり、標識L2が他方の構成要素であり、AGT融合蛋白質が蛍光によって検出される、請求項44に記載の方法。
【請求項47】
対象となる蛋白質を検出しかつ/または操作するための請求項44に記載の方法であって、対象となる蛋白質を変異体AGTと融合し、変異体AGT融合蛋白質を
(a)R1が基R5であり、変異体AGTによって認識されない式(1)の化合物、および
(b)変異体AGT融合蛋白質によって認識される式(1)の別の化合物、
の混合物に接触させ、そして標識を認識しかつ/または取り扱うために設計した系で標識を使用して、変異体AGT融合蛋白質を検出し、場合によりさらに操作する方法。
【図1】

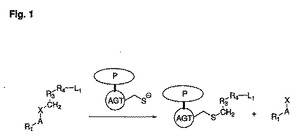
【図2】
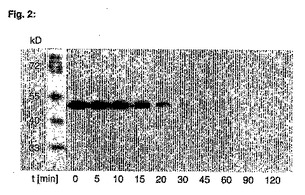
【図3】
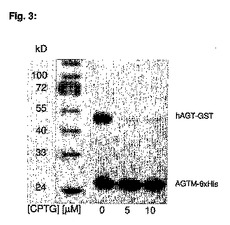
【図2】
【図3】
【公表番号】特表2007−526282(P2007−526282A)
【公表日】平成19年9月13日(2007.9.13)
【国際特許分類】
【出願番号】特願2007−501285(P2007−501285)
【出願日】平成17年3月1日(2005.3.1)
【国際出願番号】PCT/EP2005/050900
【国際公開番号】WO2005/085470
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(503183293)ウペエフエル・エコル・ポリテクニック・フェデラル・ドゥ・ローザンヌ (11)
【氏名又は名称原語表記】EPFL ECOLE POLYTECHNIQUEFEDERALE DE LAUSANNE
【Fターム(参考)】
【公表日】平成19年9月13日(2007.9.13)
【国際特許分類】
【出願日】平成17年3月1日(2005.3.1)
【国際出願番号】PCT/EP2005/050900
【国際公開番号】WO2005/085470
【国際公開日】平成17年9月15日(2005.9.15)
【出願人】(503183293)ウペエフエル・エコル・ポリテクニック・フェデラル・ドゥ・ローザンヌ (11)
【氏名又は名称原語表記】EPFL ECOLE POLYTECHNIQUEFEDERALE DE LAUSANNE
【Fターム(参考)】
[ Back to top ]