
P53活性化化合物
本発明は、p53応答を活性化する化合物に関し、および、例えば、癌の治療および潜在的な他の疾患/状態(サーチュイン機能を含む)などの過増殖性疾患において見出された使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、p53応答を活性化する化合物に関し、および、例えば、癌の治療および潜在的な他の疾患/状態(サーチュイン機能を含む)などの過増殖性疾患において見出された使用に関する。
【背景技術】
【0002】
P53腫瘍抑制タンパク質は、細胞ストレス応答の中心的なメディエーターである。P53は、腫瘍の発生を抑制する中心的な役割を担う。保護的なメカニズム、特に細胞周期の停止やアポトーシスを活性化させることにより、潜在的に発癌性ストレスの範囲に応答する。腫瘍抑制剤としての重要性は、50%を超える成人ヒト腫瘍で、TP53遺伝子における変異や欠失を不活性化する、ヒト癌における高頻度の変異に反映される。p53が野生型である多くの癌では、他の発癌性イベントによりp53系が変化しうる。これは、おそらくp53応答が、殆どの癌において欠損していることを意味する。
【0003】
成人の固形腫瘍における知見に反し、ウイルス関連性の悪性腫瘍(例えば、子宮頸癌)、血液の悪性疾患および小児癌におけるp53変異の発生は、非常に低い。これは、成人と比較し小児の癌の予後がより良好であることへの手がかりになりうる。しかし、若年患者の長期間の療法や治療を考慮した場合、現在の多くの療法の変異原性作用を知ることが重要である。さらに、文献には、DNA障害性アルキル化物質によるB−CLL患者の治療が、p53の変異の発生に関連し、著しい不良転帰および薬剤抵抗性を伴うことが示されている。変異によってp53機能が消失していない癌の治療の向上は、新規な、非遺伝毒性的なp53応答活性剤の発見によるだろう。
【発明の概要】
【発明が解決しようとする課題】
【0004】
多くの現在の抗癌療法は、DNA傷害によりp53応答を活性化する。p53系の非遺伝毒性的活性化は、癌の予防的な治療も含め、長期化へ通じる。この要件に合う分子は、変異によるp53機能を消失していない過増殖性状態を伴う患者に適用する治療物質として有用となりうる。
【0005】
本発明の目的は、過増殖性状態や、癌などの異常なP53機能、および関連する状態を含む状態および疾患の治療および/または予防のための分子、およびそれらの分子の使用を提供することである。
【0006】
本発明の更なる目的は、癌、糖尿病、筋分化、心不全、神経変性、老化、HIV感染およびマラリアなどの、サーチュイン発現および/または機能を伴う状態および疾患の治療および/または予防のための分子、およびそれらの分子の使用を提供することである。
【0007】
本発明の第1の局面によれば、式(I)の化合物の医薬としての使用が提供される。
【0008】
【化1】

【0009】
[式中、
【0010】
R1は独立してHであるか;分枝状または非分枝状の、モノ、ジ、もしくはトリ置換または無置換の、アルキル、アルケニルもしくはアルキニルであるか;または、アリールであるか;または、Z−アルキル、Z−アルケニル、Z−アルキニルもしくはZ−アリール(ここでZはO、NH、N、またはSであるか、ラベル、プローブもしくは基を可溶化する溶媒に結合する基を含む)であり;
Yは存在しないか、−C(O)−、−C=S−、または−SO2−であり;
Ar、Ar’はアリールであり;
XはOまたはSであり;
R3およびR4は、独立して、非存在であるか存在し、存在するときは、独立してH、または分枝状または非分枝状の、モノ、ジもしくはトリ置換または無置換の、アルキルもしくはZ−アルキル(ここで、ZがO、NH、NもしくはS)であるか、またはR3およびR4は、共に結合して、分枝状または非分枝状の、置換または無置換の、アルキレンもしくはZ−アルケン(ここで、ZはO、NH、NまたはSである)を形成し;
R5およびR6は、独立して、非存在であるか存在し、存在するときは、独立して分枝状または非分枝状の、置換または無置換のアルキルであり;ここで、
R3が存在するときは、破線bは単結合であり、R3が非存在のときは、破線bは二重結合であり;
R4が存在するときは、破線cおよびdは単結合であり、R4が非存在のときは、破線cまたはdの一方は二重結合であり、他方は単結合であり;そして、
R5およびR6が非存在のとき、破線aおよびeはそれぞれ二重結合であり、R5およびR6が存在するときは、破線aおよびeは、それぞれ単結合である]
で示される、医薬としての使用に供される化合物、または、その生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【0011】
このように、破線a、b、c、dおよびeの単結合または二重結合の要件は、付随する炭素原子の四価、および窒素原子の三価を確保し、維持するために選択される。しかし、窒素原子は、四価となり、例えば、アルキル基などの他の部分の窒素原子への結合を通じて、正電荷を有しうることに留意する。
【0012】
誤解を避けるために、式(I)の種々の化合物は、以下の化合物式(IA−D)により示される。
【0013】
【化2】
【0014】
理論により拘束されることを意図することなしに、構造活性相関の研究から、式(I)におけるR1−Y−NH−の一群は、例えば、NH残基により規定されるように、水素結合供与体基を備えることにより活性を有するように思われる。また、R2は、立体的に嵩高い基が好ましいようである。
【0015】
Yは、存在または非存在となり得るが、好ましくは存在する。Yは、好ましくは−C(O)−基である。
R1は、好ましくは独立してH、または置換または無置換のアルキルまたはアリール基である。
アリール基、Arは、置換または無置換の、単環または多環系アリールもしくはヘテロアリール部分であってもよい。
好ましくは、Arはフェニルである。
Arがフェニルのとき、フェニルにパラ位で結合する基、すなわち、−NH−が−NR3−基に対しパラ位であるのが好ましい。
好ましくはAr’は、1以上の可能な位置に、分枝状または非分枝状の、置換また
は無置換C2−C10アルキルで置換されたフェニルである。好ましくは、R2は二級または三級アルキル基などの、分枝状のアルキル基である。
【0016】
最も好ましくは、R2はiso−プロピルまたは三級のブチル基(すなわち、tert−ブチル)である。
R2基は、結合するフェニル環のパラ位で結合するのが好ましい。
好ましくは、破線aおよびeが二重結合となるようにR5およびR6は非存在である。
好ましくは、R3およびR4は存在し、好ましくは水素、または、例えば、メチルなどのC1−C4アルキルである。
【0017】
アルキル、アルケニルおよびアルキニルという語は、ここでは、これらの基の分枝状または非分枝状で、置換され、または無置換の、線状または環状のものを含む。
アリールという語は、置換または無置換の単環もしくは多環系のアリールまたはヘテロアリールを含む。
【0018】
R1がHでないとき、各存在で、アルキル、アルケニル、アルキニル、アリールまたはヘテロアリール、カルボキシ、アルキルオキシカルボニル ヒドロキシル、アミノ、モルホリノ、ニトロ、アルキルオキシ、アルキルチオ、ホルミル、シアノ、カルバモイル、ハロ(例えば、フルオロ、クロロ、ブロモまたはヨード)、ケトン、−S(O)NR7R8または−S(O)R9(ここで、R7、R8およびR9は、それぞれ独立してアルキル、アルケニル、アルキニル、アリールまたはヘテロアリールから選択される)からなる群から、独立して選択される基で1回以上置換されうる。
【0019】
通常、R1はアルキル基であり、好ましくは直鎖のC3−C6アルキル、例えば、n−ブチルなどのC4アルキルである。
通常、アルキル基は、遊離末端で、フェニル、ヒドロキシル、アミノ、モルホリノ、ニトロ、アルキルオキシ、アルキルチオ、ホルミル、シアノ、カルバモイル、ハロ(例えば、フルオロ、クロロ、ブロモまたはヨード)、ケトン、−S(O)NR7R8または−S(O)R9(ここで、R7、R8およびR9は、それぞれ独立してアルキル、アルケニル、アルキニル、アリールまたはヘテロアリールから選択される)から選択される置換基で置換される。
【0020】
末端置換基がモルホリノ基のとき、好ましくは、その窒素原子によりR1の末端に結合する。
末端置換基は、ハロゲンであってもよく、例えば、ブロモの場合は、式Br(CH2)4−を有するR1基を与える。
末端置換基が、アミノまたはモルホリノ基のとき、その窒素原子は、プロトン化(例えば、酸反応により)して、正電荷されたものとなってもよい。
【0021】
末端R1は、ラベル、親和性樹脂または基質プローブおよび/または水溶性基に結合するリンカー基として有利に考慮されても良い。例えば、リンカーは、活性メカニズムの研究に有用な分子を提供するために、式(I)の化合物(すなわち、活性化合物)の残存部分と、生物化学実験で用いられるラベルまたはプローブに結合してもよい。リンカーは、アルキル鎖、エチレングリコールのポリマー類(すなわち、(OCH2CH2)n1)および6−アミノヘキサン酸のペプチド類(すなわち、(NH(CH2)5CO)n2)r;ここで、n1およびn2は、分子がポリマーであることを示し、通常、1から6または8ぐらいの範囲の整数である)などの本目的に適するどのような部分であってもよい。ラベルやプローブは、ビオチン、ストレプロアバジン(streptavadin)、フルオロフォア類(例えば、bodipy、フルオレセインおよびローダミン)、放射活性ラベル類および固相マトリックス、例えば、ポリマービーズなどのポリマー類(例えばポリスチレン)などの、その目的において知られる物を含む。水溶性化する基は、その目的において適する糖類、アミン類、アミノ酸類、リン酸類等およびそれらの塩を含む。
【0022】
水溶性化する基は、例えば、特に医薬として使用されるとき、化合物の生物学的利用性および/または薬物動態を最適化するために、活性化合物の水溶性を変化させるための修飾に用いられうる。
【0023】
アミノという語は、ここでは、サイクリックアミノ基、すなわち、アミンの窒素原子が環の構成員である基を含む。
アミノという語は、ここでは、プロトン化された種々のアミン類およびその塩についても含む。例えば、アミンは、プロトン化されてもよく、塩酸、硫酸の酸など、カルボン酸類を含む多くの酸類との塩を形成してもよい。例えば、アミンの塩酸塩は、水や水溶性溶媒中で高い水溶性を示す。
【0024】
式(I)において、アリール残基は、例えば、5または6員の単環アリールもしくはヘテロアリール環構造、または他の多環系アリールもしくはヘテロアリール残基となりうる。
フェニルは、6員アリール基の具体例である。
【0025】
通常、ヘテロアリール構造中のヘテロ原子は、酸素または窒素から選択される。
フリルおよびピロリルは、それぞれ酸素および窒素ヘテロ原子を含む5員ヘテロアリール基の具体例である。
ピリジニルおよびピリミジニルは、それぞれ1つの窒素原子、および2つの窒素原子を含む6員ヘテロアリール基の具体例である。
ナフチルは、縮合6員環を形成する10個の炭素原子の骨格を有するアリール基の具体例である。
【0026】
アリール基は、示されるように、1以上の位置で置換されてもよく、適する置換基は、それぞれの置換位置において、R1またはR2で定義された置換基から独立して選択されうる
【0027】
置換基は、アリール基に直接的に、または、−O−、−S−、−N−または−(CR10R11)n−(ここで、nは1〜25の整数であり、例えば、1〜10、1〜4などであり、R10およびR11は、独立してアルキル、アルケニル、アルキニル、アリールまたはヘテロアリール、カルボキシ、アルキルオキシカルボニル ヒドロキシル、アミノ、モルホリノ、ニトロ、アルキルオキシ、アルキルチオ、ホルミル、シアノ、カルバモイル、ハロ(例えば、フルオロ、クロロ、ブロモまたはヨード)、ケトン、−S(O)NR7R8または−S(O)R9(ここで、R7、R8およびR9は前記の定義の通り))から独立して選択される基を介して結合してもよい。
【0028】
医薬としての使用に関し、式(I)の好ましい化合物は、以下の式(II)
【0029】
【化3】
[式中、R1、R2およびYは前記定義の通りである]
または、その生理的に許容可能な塩、溶媒和物、エステルまたは他の生理的機能性誘導体、を有する。
【0030】
好ましくは、R1YNH−および/またはR2−基が、それらが結合するそれぞれのフェニル環上のパラ位にある。
好ましくは、Yは−C(O)−基であり、このため、好ましくは、R1YNH−基はR1C(O)NH−である。
【0031】
好ましい化合物、その薬学的に許容可能な塩、溶媒和物、エステルまたは他の生理的機能性誘導体は、実施例の項においても示される。
本発明は、さらに以下に詳細を説明するように、その薬学的に許容可能な担体と共に式(I)または(II)の化合物を含む医薬製剤にも及ぶ。
本発明は、さらに以下により詳細に記載するように、ここに記載された1以上の式(I)または(II)の化合物を、それを必要とする患者に投与することを含む治療または予防方法にも及ぶ。
本発明は、式(I)および(II)の範囲に含まれる新規な化合物にも及ぶ。
【0032】
このように、更なる局面において、本発明は、下記化合物
【化4】
を除く、ここに示される式(I)または(II)の化合物を提供する。
【0033】
ここに記載された化合物式において、アルキル基は、独立してC1−C22アルキル、好ましくはC1−C10アルキル、好ましくはC1−C4アルキル、例えば、メチル、エチル、プロピル、ブチルであってもよい。
アルケニル基は、独立してC2−C22アルケニル、好ましくはC2−C10アルケニル、好ましくはC2−C4アルケニルであってもよい。
アルキニル基は、独立してC2−C22アルキニル、好ましくはC2−C10アルキニル、好ましくはC2−C4アルキニルであってもよい。
アルキル、アルケニルまたはアルキニル基は、分枝状または非分枝状、置換または無置換であってもよい。例えば典型的な分枝状のアルキル基は、iso−プロピル、iso−ブチル、sec−ブチル、tert−ブチル、3−メチルブチル、3,3−ジメチルブチルおよびそれらの異性体を含む変形体を含む。
【0034】
ここに記載されたように、アルキル、アルケニルまたはアルキニル基は、置換されてもよく、置換基は、ヒドロキシル、置換または無置換のアミン、置換または無置換のアミド、ハライド(フルオロ、クロロ、ブロモ、ヨードなど)、アルコキシ、チオ、ニトロ、カルボキシ、エステル、シアノ、またはアリール(フェニル、ナフチルおよびピリジルなど)などのあらゆる化学分子であってよい。
【0035】
ここに記載された化合物、例えば、アルケニル基、における二重結合の幾何学的配置はcisまたはtransの幾何学的配置であってよい。
【0036】
本発明の化合物の生理的に許容可能な塩の例には、有機カルボン酸類(酢酸、乳酸、酒石酸、マレイン酸、クエン酸、ピルビン酸、シュウ酸、フマル酸、オキザロ酢酸、イセチオン酸、ラクトビオン酸および琥珀酸など);有機スルホン酸類(メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸およびp−トルエンスルホン酸など)および無機酸類(塩酸、硫酸、リン酸およびスルファミン酸等)と形成される酸付加塩を含む。
【0037】
本発明化合物の生理的機能性誘導体は、体内で親化合物に変換される誘導体である。そのような生理的機能性誘導体は、「プロドラッグ」や「バイオプレカーサー」とも言われる。本発明化合物の生理的機能性誘導体は、生体内で加水分解性のエステル類を含む。
【0038】
記載された使用/方法のいずれか1において使用されうる、ここに記載された化合物に対応する溶媒和物を、調製、精製、および/または処理することが、簡便であるか、好ましい。溶媒和物という語は、ここでは、化合物またはその化合物の塩などの溶質と溶媒との複合体を言うのに用いられる。溶媒が水のとき、その溶媒和物を、例えば基質1分子当たりの水分子の数により、一水和物、二水和物、三水和物など、水和物と称してもよい。
【0039】
当然のことながら、本発明の化合物は、種々の立体異性体が存在し得、上記に定義された本発明の化合物は、エナンチオマーおよびラセミ混合物を含む、全ての立体異性体およびその混合物を含む。本発明は、式(I)または(II)の化合物の個々のエナンチオマーやそのようなエナンチオマーの全または部分ラセミ混合物を含む、そのような全ての立体異性体や立体異性体の混合物の使用を、その範囲に含む。
【0040】
本発明の化合物は、後述するように、技術的に容易に利用可能な試薬と技術を用いて調製されうる。本発明の化合物の調製のための合成経路において新規な中間体化合物は、本発明の分子の調製についての一般適用に関し、重要な分子となりうる。従って、本発明はこれらの新規な中間体化合物を含む範囲にまで及ぶ。
【0041】
具体例として、化合物は、公報番号EP 0 136 745 B1の欧州特許、および公開番号EP 0 193 249 A2の欧州特許出願に記載された方法を用いて合成されうる。
【0042】
式(II)に記載の化合物の調製に有用な合成方法には、以下の手法において示されるように、溶媒(例えば、アセトン)中、種々の塩化ベンゾイル類を金属チオシアン酸塩(例えば、チオシアン酸ナトリウム)で処理し、ベンゾイルイソチオシアネート類を得、次いでアニリン類と反応(好ましくはin situで)させ、以下の式(II)の化合物を得る。
【0043】
【化5】
【0044】
上記の手法において、フェニル基は、あらゆるアリール基を表す。更なる機能付与は、アシル化やアルキル化により達成されうる。塩基性水溶性条件下で、化合物は加水分解され、N−アリールチオ尿素類が得られる。
【0045】
上記に示した通り、本発明は、ここに示される化合物を、それを必要とする患者に投与することを含む、ここに挙げられる疾患、病態もしくは状態の治療または予防を提供する。
【0046】
本発明に関する疾患は、P53タンパク質、その機能および/またはP53経路の異常を伴う異常な細胞死に関与するものを含む。
【0047】
特に、異常な細胞増殖が関与する疾患は、ここに挙げられた化合物で治療可能である。そのような疾患の例には、癌、過剰増殖性疾病(疣贅類、乾癬、炎症性腸疾患を含む)、リウマチ/自己免疫状態、鎌状赤血球性貧血、サラセミア等が含まれる。
【0048】
本活性化合物により治療されうる癌の例には、これらに限定されないが、例えば、膀胱癌、乳房癌、大腸癌(例えば、大腸腺癌や大腸腺腫などの結腸直腸の癌)、腎臓癌、表皮性癌、肝癌、肺癌(例えば腺癌、小細胞肺癌や非小細胞肺癌)、食道癌、胆嚢癌、卵巣癌、膵臓癌(例えば、膵外分泌部の癌)、胃癌、頚部癌、甲状腺癌、前立腺癌または皮膚癌(例えば扁平上皮癌);リンパ系の造血性腫瘍(例えば白血病、急性リンパ性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキン病、非ホジキンリンパ腫、毛様細胞リンパ腫、またはバーキットリンパ腫);骨髄疾患系の造血性腫瘍(例えば急性および慢性骨髄性白血病、骨髄異形成症候群、または前骨髄球性白血病);濾胞性甲状腺癌;間葉系起源の腫瘍(例えば線維肉腫または横紋筋肉腫);中枢または末梢神経系の腫瘍(例えば星状細胞腫、神経芽細胞腫、神経膠腫またはシュワン細胞腫);メラノーマ;精上皮腫;奇形癌腫;骨肉腫;色素性乾皮症;角化棘細胞腫;濾胞性甲状腺癌;またはカポジ肉腫が含まれる。
【0049】
本発明者らは、ここに示されるJH164などの本発明の化合物が、SirT1活性の抑制に特に有効であることに注目している。このため、JH164などの本発明の化合物は、SirT1発現/機能に関連する疾患/状態の治療に使用されうる。
【0050】
SirT1または関連するタンパク質は、癌、炎症、免疫反応、肥満、老化、糖尿病、筋分化、心不全、神経変性、HIV感染およびマラリアを含む非常に多くの疾患/状態において標的であると確認されており(例えば、Bordone LおよびGuarente L、Cancer Res.,66(8):4368−77(2006年4月15日);Heltwegら、Trends Pharmacol Sci.,26(2):94−103、2005年2月;Pagansら、PLoS Biology 2005 Vol.3, No.2,e41;Deitsch KW,Cell.,121(1):1−2(2005年4月8日);Freitas−Junior LHら、Cell.,121(1):25−36(2005年4月8日);Nayagam VM、J Biomol Screen.(2006年11月12日)を参照のこと)、従って本発明の化合物は、前記のあらゆる疾患/状態の治療/予防において有用性が見出されうる。
【0051】
式(I)の化合物と、(同時に、または異なる時間間隔を問わず)一緒に投与されうる他の治療物質の例には、これらに限定されないが、シスプラチン、シクロホスファミド、ドキソルビシン、イリノテカン、フルダラビン、5FU、タキサン類、マイトマイシンCなどの、トポイソメラーゼ阻害剤、アルキル化剤、代謝拮抗薬、DNA結合剤および微小管阻害薬(チューブリン標的剤)、または放射線療法などが含まれる。活性化合物を他の療法と組み合わせる場合、2以上の治療剤が、異なる経路により種々の個別の投薬計画で投与されうる。
【0052】
本発明の化合物と上記に挙げられた物質との組み合わせは、通常の一般的な知見と熟練した施術者に公知の投薬計画を用い、投与量を選択する医師の裁量でなされる。
【0053】
式(I)の化合物が、1、2、3、4またはそれ以上、好ましくは1または2、好ましくは1以上の治療剤との組み合わせ療法で投与される場合、化合物を同時にまたは連続して投与できる。連続して投与する場合、狭い間隔(例えば、5〜10分間隔以上)または、より長い間隔(例えば1、2、3、4時間またはそれ以上開けて、または必要な場合はより長い間隔)で、治療の作用と治療剤に見合った的確な投薬計画で投与されうる。
【0054】
本発明の化合物はまた、放射線療法、光力学療法、遺伝子療法;外科手術および栄養制限食などの非化学療法と組み合わせて投与されうる。
患者は、通常、動物であり、例えば、哺乳動物、特にヒトである。
【0055】
本発明による使用に関し、ここに記載された化合物またはその生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体は、該化合物またはその生理的に許容可能な塩、エステルもしくは生理的機能性誘導体と共に、1以上の薬学的に許容可能な担体および任意に他の治療的および/または予防的成分を含む医薬製剤でありうる。担体は、製剤の他の成分に適合するという意味で許容可能でなくてはならず、服用者に有害であってはならない。
【0056】
医薬製剤は、経口、局所(経皮、バッカルおよび舌下を含む)、直腸、または非経口的(皮下、内皮内、筋肉内および静脈内を含む)、経鼻的および肺内投与(例えば、吸入による)に適するものを含む。製剤は、必要に応じて、別個の投与量単位として好都合に存在し得、薬学の当該技術において周知のあらゆる方法により調製されうる。全ての方法には、活性化合物を液体担体や細かく分割された固体担体あるいはその両方と組み合わせる状態にする工程を含み、次いで、所望により、生成物を所望の剤型に形成する。
【0057】
経口投与に適する医薬製剤は、担体が固体であり、最も好ましくは、巨丸剤(ボーラス)、カプセルや錠剤などの、それぞれに活性化合物の所定量を含む単位用量製剤である。錠剤は、場合により、1以上の副成分とともに、圧縮や成型によって製造されてもよい。圧縮錠剤は、粉末や顆粒などの自由流動形態の活性化合物を、場合により、結合剤、滑沢剤、不活性希釈剤、潤滑剤、表面活性化物質または分散剤と混合し、適する機器で圧縮することにより調製されうる。成型錠剤は、活性化合物を不活性な液体希釈剤で成型することにより製造されうる。錠剤は、場合により被覆されてもよく、被覆されていない場合には、場合により割線を入れても良い。カプセル剤は、活性化合物を、単独あるいは1以上の副成分との混合剤として、カプセル殻に充填し、次いで常法により封止することにより調製しうる。サッシェ剤は、カプセル剤に類似し、活性化合物を副成分とともにライスペーパー製の封筒に入れ封止する。活性化合物は、分散可能な顆粒として製剤化してもよく、例えば、投与前に水に懸濁してもよく、食品に散布してもよい。顆粒剤は、例えば、サッシェに包装してもよい。担体が液体である経口投与に適する製剤は、水溶性もしくは非水溶性の溶液または懸濁液として、あるいは水中油型液体エマルジョンとして存在しうる。
【0058】
放出制御投与形態を含む経口投与製剤は、例えば、活性化合物が適した放出制御マトリックスで製剤化され、あるいは、適する放出制御フィルムで被覆された錠剤を含む。そのような製剤は、特に予防的使用に好都合となりうる。
【0059】
直腸投与に適する担体が固体である医薬製剤は、最も好ましくは、単位用量の坐薬である。適する担体は、カカオ脂や技術的に通常用いられる他の物質を含む。坐薬は、活性化合物を柔らかい、あるいは溶けた担体と混合し、次いで冷却し、成型することにより簡便に形成しうる。
【0060】
非経口的投与に適する医薬製剤は、無菌の溶液、または、水溶性もしくは油性の担体中の活性化合物の懸濁液を含む。
【0061】
注射用製剤は、急速注入や連続的な注入に用いられる。そのような製剤は、使用に必要とされるまで製剤を導入後封止した、単位用量または多用量の容器で好都合に存在する。あるいは、活性化合物は、使用前に、無菌の発熱性物質の存在しない水などの適する担体と構成される粉末形態であってもよい。
【0062】
活性化合物は、長時間作用性デポ製剤として製剤化されてもよく、例えば、皮下投与や筋肉内投与などの、筋肉内注射や埋め込みにより投与されうる。デポ製剤は、例えば、適する重合性や疎水性の物質、またはイオン交換樹脂を含んでもよい。そのような長時間作用性の製剤は、特に予防的使用に好都合となりうる。
【0063】
口腔を介した肺内投与に適する製剤は、活性化合物を含み、好ましくは0.5〜7ミクロンの範囲の直径を有し、服用者の気管支樹に到達するような粒子である。
そのような製剤の1つの可能性としては、吸入器で使用される穴の開けられるカプセル(好ましくは、例えば、ゼラチン)で、あるいは、活性化合物と、適する液体または気体噴射剤、および、場合により界面活性剤および/または固体希釈剤などの他の成分を含む自己噴射性製剤として用いられる、細かく粉砕した粉末の形態である。適する液体噴射剤は、プロパンおよびクロロフルオロカーボン類を含み、適する気体噴射剤は、二酸化炭素を含む。活性化合物が、溶液または懸濁液の溶滴の形態で分散している自己噴射性製剤も、使用されうる。
【0064】
そのような自己噴射性製剤は、当該技術において公知のものの類似物であり、確立された手順により調製しうる。好ましくは、所望のスプレー特性を有する手動で操作可能な機能性バルブ、または、有利には、各操作において、例えば、25〜100マイクロリットルの固定容量を送達する計量型のバルブ、を備える容器を用いる。
【0065】
更なる可能性として、吸入の微滴霧を作り出すのに、加速気流や超音波撹拌が用いられる噴霧器やネブライザーでの使用に、活性化合物を溶液や懸濁液の状態で用いることができる。
【0066】
経鼻投与に適する製剤は、一般的に上記に記載された肺内投与と類似の調製法を含む。分散する場合、そのような製剤は、鼻腔中に保持可能に、好ましくは、10〜200ミクロンの範囲の粒子直径を有するべきであり、必要に応じて、適する粒径の粉末の使用や適切なバルブの選択により、達成されうる。他の適する製剤は、鼻および鼻腔に近づけ保持された容器から、鼻腔を通じて、水溶性もしくは油性溶液または懸濁液中に0.2〜5%w/vの活性化合物を含む滴を、高速吸入により投与するために、20〜500ミクロンの範囲の粒子径を有する粗粉末である。
【0067】
前述の担体成分に加え、上記に記載された医薬製剤は、希釈剤、緩衝剤、香味剤、結合剤、界面活性剤、増粘剤、滑沢剤、保存料(抗酸化剤を含む)等の、適当な1以上の追加的な担体成分、および製剤を対象とする服用者の血液と等張にする目的で加えられる物質を含んでもよい。
【0068】
薬学的に許容可能な担体は、当業者に周知であり、これらに限定されないが、0.1M、好ましくは0.05Mリン酸バッファまたは0.8%生理食塩水を含む。また、そのような薬学的に許容可能な担体は、水溶性もしくは非水溶性溶液、懸濁液、およびエマルジョンであってもよい。非水溶性溶媒の例には、プロピレングリコール、ポリエチレングリコール、オリーブ油などの植物油、およびエチルオレイン酸塩などの注射可能な有機エステル類がある。水溶性担体は、水、アルコール/水溶性溶液、生理食塩水や緩衝培地を含むエマルジョンまたは懸濁液を含む。非経口的担体は、塩化ナトリウム溶液、リンゲルデキストロース液、デキストロースおよび塩化ナトリウム、乳酸化されたリンゲル液または固定油を含む。例えば、抗菌剤、抗酸化剤、キレート化剤、不活性ガスなどの、保存料および他の添加物も存在してもよい。
【0069】
局所製剤に適する製剤は、例えば、ゲル、クリームまたは軟膏として供されうる。そのような製剤は、例えば、創傷や潰瘍に適用でき、創傷や潰瘍の表面上に直接塗布したり、治療する箇所および部分一面に適用可能な、包帯、ガーゼ、メッシュなどの適するサポートに保持してもよい。
例えば、創傷や潰瘍に、治療する箇所に直接スプレーや散布しうる液体や粉末製剤も提供されうる。また、製剤を包帯、ガーゼ、メッシュなどの担体にスプレーでき、あるいは散布し、治療する箇所に適用することもできる。
【0070】
動物用の治療用製剤は、粉末や液体の濃縮形態が好都合であろう。動物用製剤の手技標準により、ラクトースやスクロースなどの従来の水溶性添加剤は、その物性を向上させるために加えられうる。このように、特に適する本発明の粉末は、活性成分を50〜100%w/w、好ましくは60〜80%w/w含み、従来の動物用添加剤を0〜50%w/w、好ましくは20〜40%w/w含む。これらの粉末は、例えば、予め混ぜた中間体を経由し、動物用飼料に加えたり、動物用飲料水で希釈してもよい。
【0071】
本発明の液体濃縮物は、化合物や誘導体またはその塩を好適に含み、場合により、獣医学的に許容可能な水相溶性溶媒、例えばポリエチレングリコール、プロピレングリコール、グリセロール、グリセロールホルマールや30%v/vまでのエタノールと混合したような溶媒などを含んでも良い。液体濃縮物は、動物用飲料水に投与してもよい。
【0072】
本発明を、以下の図面、限定されない実施例を参照して詳細に説明する。
以下の図面を参照して本発明を詳細に説明する。
【図面の簡単な説明】
【0073】
【図1】化合物JJ91を種々の量で処理したT22 RGC−Fos−lacZ細胞におけるp53−依存性転写誘導の量を示すグラフである。
【図2】ウェスタンブロット画像を示す。
【図3】FACS分析グラフである。
【図4】(A)および(B)は、ウェスタンブロット画像を示す。
【図5】マウスへの化合物投与の結果を示すグラフである。
【図6】BL2バーキットリンパ腫細胞の生存に対する種々の濃度のJJ91の効果を示すグラフである。
【図7】SCIDマウスにおけるBL2バーキットリンパ腫異種移植腫瘍に対するJJ91の効果を示すグラフである。
【図8】SirT1活性に対するJH164の抑制効果を示すグラフである。
【発明を実施するための形態】
【0074】
詳細な説明
化合物合成
ここに記載された化合物は、手法1および2を参照し、以下の方法に従って準備された。
【0075】
手法1を参照し、本発明に記載されたN−ベンゾイルチオ尿素類は、種々のベンゾイルクロライド類をアセトン溶媒中、チオシアン酸ナトリウムで処理し、そして所望の化合物を得るためのアニリン類とin situで反応させ、ベンゾイルイソチオシアネート類を得ることにより、合成した。
【0076】
【化6】
【0077】
さらに、アシル化またはアルキル化によって機能付与を行った。
【0078】
具体例として、化合物(JH129)の合成を以下のように行った。
アセトン(20mL)中4−tert−ブチルベンゾイルクロライド(10mmol、1.97g)の撹拌溶液に、アルゴン雰囲気下で、チオシアン酸ナトリウム(10mol、0.81g)を加えた。2時間後、この混合物を、0℃に冷却されたアルゴン下で、アセトン(50mL)中1,4−フェニレンジアミン(20mmol、2.16g)溶液に滴下した。周囲温度まで加温後、反応混合物を36時間攪拌した。混合物を減圧下濃縮し、残渣をジクロロメタン中にとり、濾過し、濾液を濃縮し、シリカゲルカラムでクロマトグラフィにかけ、酢酸エチル−石油エーテル混液で溶出した。生じた固体をジエチルエーテルで粉砕し、分析的に純粋な物質2.45g(75%)を得た。分析により以下のデータを得た:mpt 189−191℃;1H−NMR(CDCl3)δ1.36(s,9H),6.81(m,2H),7.47(m,2H),7.54(m,2H),7.81(m,2H),9.05(s,1H),12.41(s,1H);MS(ES+)m/z350[M+Na]+;C18H21N3ONaSの計算値350.1300,実測値350.1303.
【0079】
さらなる具体例として、以下のようにアシル化によって、化合物JH129をさらに機能付与した。
ジクロロメタン(1mL)中、JH129(0.2mmol、65mg)の撹拌溶液に、アルゴン雰囲気下で、5−ブロモプロパノイルクロライド(ジクロロメタン0.2mL中0.2mmol)の溶液を加えた。生じた懸濁液に、トリエチルアミン(0.2mmol、27μL)を加えた。反応混合物を90分間攪拌し、ジクロロメタン(5mL)で希釈し、1MのHCl、2MのNaOHおよび飽和NaCl溶液で洗浄した。有機層を乾燥(MgSO4)し、灰白色固体になるまで濃縮した。酢酸エチルから再結晶し、分析的に純粋な物質64mg(65%)を得た。分析により以下のデータを得た:mpt 152−153℃;1H−NMR(CDCl3)δ1.36(s,9H),1.92(m,4H),2.42(t,2H),3.46(t,2H),7.22(s,1H),7.57(m,4H),7.68(m,2H),7.82(m,2H),9.04(s,1H),12.60(s,1H);MS(ES+)m/z512,514[M+Na]+;C23H2879BrN3O2NaSの計算値512.0983,実測値512.0995.
【0080】
更なる具体例として、化合物JH164HClは、式(I)において、R1基が、水溶性基に活性化合物を結合するためのリンカー基であると考えられ得る具体的化合物である。JH164の合成およびその塩酸塩の形成は、次のようになされた。
ジクロロメタン(10mL)中、JH140(0.1mmol、50mg)の溶液に、ジメチルアミン水溶液(40重量%の2mL)を加えた。二相性の混合物を20時間攪拌した。有機層を分離し、乾燥(MgSO4)し、エバポレートして、乾固するまで濃縮した。残渣をアセトンに溶解し、この溶液をHCl蒸気に曝した。生じたHCl塩を濾過し、純白色固体、33mg(67%)を単離した。分析により以下のデータを得た:mpt 205−206℃;1H−NMR(D6−DMSO)δ1.32(s,9H),1.64(brs,4H),2.39(t,2H),2.75(s,6H),3.06(t,2H),7.60(m,6H),7.94(m,2H),9.64(brs,<1H),10.11(s,1H),11.45(s,1H),12.60(s,1H);MS(ES+)m/z 455[M−Cl]+;C25H35N4O2Sの計算値455.2481,実測値455.2477;分析 C25H35ClN4O2Sの計算値:C,61.14;H,7.18;N,11.41%.実測値:C,60.75;H,7.46;N,11.30%.
【0081】
生物学的評価
in vitro実験:
材料と方法:
ケムブリッジケミカルス(ChemBridge Corporation、16981 Via Tazon、Suite G、サンディエゴ、CA 92127)から得た30,000個の化合物ライブラリー(DIVERSetTM)を、p53腫瘍抑制剤機能の活性化剤についてスクリーニングした。
1次および2次スクリーニングを、以下の細胞ベースのアッセイを用いて行った。
【0082】
1.1次スクリーニング
Lu X、Burbidge SA、Griffin SおよびSmith HMによりOncogene.,13(2):413−8(1996年7月18日)に記載された、p53−依存性プロモーター支配下でベータ−ガラクトシダーゼを発現するT22 RGC−ΔFos−lacZ細胞を用いた。
・セレクションなしのDMEM90μl、10%FCSおよびゲンタマイシン1mg/mlを有する96ウェル組織培養プレートに1ウェル当たり、1x104個の低継代T22細胞を播種する。
・細胞播種48時間後に化合物を加える。DMSOは、培地中最終濃度1:100を超えない。非処理の対照と、アクチノマイシンD5ng/mlで処理したポジティブコントロールを使用する。総量=100μl
・18時間後、96ウェルプレートから培地を除き、1ウェルあたり50μlの1倍溶解バッファ(プロメガ)を加える。
・室温で1時間振盪する(すぐに使えるようになるまでプレートを−80℃で冷凍できる)
・1ウェルあたり150μlのCPRG反応混合物を加える。
CPRG反応混合物15mlの調製
[15mlの0.1Mのリン酸バッファ、pH7.5
300μlのCPRG 4mg/ml(ベーリンガー・マンハイム)
80μl(0.1MのMgCl2/0.1M β−メルカプトエタノール)]
・37℃で4時間湿室中でインキュベートする。色が黄色から桃色に変わると、p53活性を示す。
・各ウェルから新たな96ウェルプレートに100μlを移す。これは、細胞壊死組織片が、吸光度の読み取りに干渉するのを防止する。プレートリーダーを用いて570nMでの吸光度を測定する。
・可溶化液を一晩4℃で放置し、次いで、再び吸光度を測定する。
【0083】
2.FACS分析
Oncogene.,18(51):7378−86(1999年12月2日)に、Smart P,Lane EB,Lane DP,Midgley C,Vojtesek B,Lain Sにより開示されている神経芽細胞腫細胞株SKNSH−CMVNeo(p53機能あり)およびSKNSH−DDp53(不活化p53)を用いた。
1日目
・6ウェルプレートの1ウェルあたり50,000個の細胞をDMEM−10% FCSで播種する。
2日目
・薬剤を細胞に加える。
4日目
・ブロモデオキシウリジン(BrdU)を30μMになるまで加え、20分間細胞をインキュベートする。
・細胞から培地を除去し、13mlのファルコンチューブに移す。PBSで細胞をすすぎ、これも移す。細胞をトリプシン処理し、チューブに移し、次いで最後にPBSで再度すすぐ。一旦チューブに全てを移し、1500rpmで5分、細胞をペレット化する。
・1mlのPBSに細胞を再懸濁し、ボルテックスする間、3mlのエタノールに流滴を加える。4℃で最短1〜2時間(上限なし)インキュベートする。
5日目
・2,500rpmで5分間の遠心分離によりペレット化し、上澄みを捨てる。
30mMのHCl(pH1.5)中1mg/mlの濃度で、1チューブあたりペプシン溶液2mlを新たに調製し、37℃に予め温める。
・予熱したペプシン溶液2mlを各チューブに加え、37℃で30分間混合する。
・2,500rpmで5分間の遠心分離によりペレット化し、上澄みを捨てる(ペレットは、透明になろう)。
・室温で15−20分間1mlの2M HClを加える(貯蔵瓶は、11.6M)。タイミングが重要であり、長時間のインキュベートは、ブロードなDNAピークとなる。PBSを追加し、前記と同様にペレット化する。
・PBSで再度次いで抗体緩衝液で一度洗浄し、毎回細胞をペレット化する。
抗体緩衝剤:PBS、0.5%のBSA、0.5%のTween−20
・抗体緩衝剤中1:50に希釈されたベクトン・ディッキンソン抗BrdU抗体の200μL中にペレットを再懸濁する。室温で1時間インキュベートする。
・PBSで洗浄し、細胞をペレット化する。
・抗体緩衝剤中1:64に希釈されたシグマFITC抗体(#3008)の200μL中にペレットを再懸濁する。抗体の消失を予防するために暗所下、室温で30分間インキュベートする。
・PBSで洗浄し、細胞をペレット化する。
・25μg/mlのヨウ化プロピジウム対比染色液を含む500μLのPBS中に最終的なペレットを再懸濁する。FACScanで分析にかけるまで暗所下氷冷する。
・FACScanによるDNA含量の測定(ヨウ化プロピジウム蛍光)およびDNA合成(BrdU取り込み)
【0084】
3.ウエスタンブロット法
・6ウェルプレートの1ウェルあたり、2x105個のMCF−7細胞を播種する。
・24〜36時間インキュベーション後、細胞に薬剤を加え、必要な時間インキュベートする。
・プレートの培地を捨て、PBSで洗浄する。PBSの残りを吸引し100μlの1倍LDS添加緩衝剤(インビトロジェン)をプレートに直接加える。プレートの表面を片隅へかき取り、細胞/LDSをピペットでチューブに取る。
・試料を90℃まで5分間加熱し、次いで各15秒間を2回超音波処理する。最大速で5分間遠心分離し、必要時まで氷冷する。全ての試料のタンパク質濃度を測定(ピアスBCAキット)し、濃度を均等化する。各試料に1:10DTTを加える。
・試料を4〜12%ノベックス・ゲルに添加し、MOPSバッファで1度流し、メーカーの使用説明書(インビトロジェン)に従い、PVDF膜に移す。
・膜をブロックし、標準手順を用い1次、次いで2次抗体でインキュベートし、検出にアマシャムECLを用いた。
・関連する1次抗体は、抗p53DO1マウスモノクローナル抗体、抗p53ホスホセリン−15(サンタクルズ)、抗p21 118マウスモノクローナル抗体を含む。負荷対照としてアクチン検出を用いた。
【0085】
水溶性分析の一般手順
化合物の相対的水溶性は、UVスペクトルに基づく方法を使用して求めた。各化合物の吸光係数の生成は、分光学的グレードのアセトニトリル(100mL)中に1mgの化合物(一部の化合物は、最初に最小量のDMSOに溶解することを要する)を溶解することにより行われる。次いで6x2倍の連続希釈を行い、合計7溶液を得る。これらの溶液をUVスペクトルで分析し、適当な波長における吸光度をモル濃度に対しプロットした。吸光係数を最適な線勾配から算出した。次いで吸光係数を、40mMのDMSOストックと100μMの化合物の水溶液の実際の濃度を計算するのに用いた。
40mMのDMSOストックと100μMの化合物の水溶液の実際の濃度は、次のように計算した。1mgの化合物を必要量のDMSOに溶解し、40mMストックを得た。次いで、不溶の化合物を遠心分離でペレット化した。上澄み(2μL)をアセトニトリルで4mLに希釈し、理論上2x10-5Mの溶液を作った。次いで溶液をUVスペクトルで分析した。
100μM水溶液の実際の濃度は、40mMDMSO溶液のアリコート(4.2μL)を1mLになるまで水で希釈することにより求めた。この溶液のアリコート(60μL)を水(40μL)に加え、不溶の化合物を遠心分離によりペレット化した。上澄み(80μL)をアセトニトリルで4mLになるまで希釈し、理論上2x10-6Mの溶液を作った。次いでこの溶液をUVスペクトルで分析した。
次いで水における実験的に測定した水溶性を、理論値(パーセンテージとして)と比較し、テノビン1に対する相対的水溶性値を得た。
【0086】
in vitro脱アセチル化アッセイ
酵素的SirTlアッセイを、Fluor de Lys蛍光アッセイシステム(バイオモルキットAK555)で精製成分を用い、以下のメーカーの使用説明書に従い行った。FdL基質を7*MおよびNAD+1mMで用いた。化合物をDMSO中、反応において0.25%より小さい最終DSMO濃度で可溶化した。1反応あたり酵素1単位を用いた。反応は、37℃で1時間行った。
【0087】
結果
図1に示される結果は、JJ91はp53転写因子機能を活性化することを示す。T22 RGC−ΔFos−lacZ細胞をJJ91の指示量で16時間処理した。p53−依存性転写の誘導率を測定した
図2に示される結果は、JJ91がp53を活性化して、選択的に神経芽細胞腫細胞を死滅させることを示す。SKNSH−CMVNeoおよびSKNSH−DDp53細胞は、非処理にしたまま、あるいは10μMのJJ91で48時間処理した。細胞をFACS分析で分析した。JJ91は、明らかにDNA合成と細胞死例を低下させた(sub−G1/G0細胞の数を増加)。これらの効果は、非活性化p53を伴うSKNSH細胞においては、見られない。
図3に示される結果は、JJ91がp53レベルを増加させることを示す。MCF−7細胞を、DNA傷害物質であるマイトマイシン C(10μM)、非遺伝毒性物質nutlin−3(6μM)やJJ91(10μM)で指示時間処理した。JJ91は、nutlin−3と同様の効果を有する。P53レベルは、急速に増加する。p53ホスホセリン−15のレベルは、遺伝毒性物質であるマイトマイシンCで見られたほど高くはない。アクチンを増加させるp53の下流の標的p21のレベルは、負荷対照で分析された。
図4Aおよび4Bに示される結果は、p53レベルにおけるJJ91類縁物質の効果を示す。
図4A:MCF−7細胞を4時間(レーン2から9)または6時間(レーン11から15)、10μMのマイトマイシンC(レーン2)、JJ91(レーン3および11)、JH118(レーン4)、JH129(レーン5)、JH132(レーン6)、JH140(レーン7および12)、JH141(レーン8)および4−アミノアセトアニリド(レーン9)、JH151(レーン13)、JH156(レーン14)および5406085(レーン15)で処理した。レーン2とレーン10において、細胞を非処理とした。細胞抽出液をp53、ホスホセリン−15 p53、p21およびアクチンに対する抗体で、ウエスタンブロット法により分析した。
図4B:MCF−7細胞を、4時間(レーン2から6)6μMのnutlin−3(レーン2)、10μMのJJ91(レーン3)、10μMの7322366(レーン4)、40nMのレプトマイシンB(レーン5)および20μMのMG132 JH129で処理した。細胞抽出液を、p53、アクチン、p21、病原性毒素およびmdm2に対する抗体でウエスタンブロット法により分析した。
【0088】
表1は、活性化合物の構造、および化合物JJ91活性を100%とし、示された化合物によるT22 RGC−ΔFos−lacZ細胞におけるp53−依存性転写に関する活性レベルを示す。
【0089】
【表1】
【0090】
【表2】
【0091】
in vitroおよびin vivo実験
図5に関し、JJ91をマウスに5mg/kgの用量で投与した。◆と□は、それぞれi.p.とp.o.の投与ルートに対応する(挿入図は、データの対数プロットを示す。)。血中濃度は、上記のLC−MS/MSで示される時点において測定し、示される値は、3つの測定値の±SDを意味する。結果は、化合物の腹膜内注射が、血中でマイクロモル濃度に達し、顕著な体重や行動変化を起こさず、約1.3時間の半減期を有することを示す。
図6に関し、BL2バーキットリンパ腫細胞を、1μM〜10μMの範囲の指示濃度のJJ91(70%シクロデキストリンに溶解)で2時間処理した。このとき、生存細胞の数をカウントした。この短い暴露のあと、細胞を洗浄し、化合物を除去した。この処理は、6日間毎日繰り返した。実験を三回実施し、標準偏差を示した。図6に示されるように化合物への6回の短い暴露のあと、BL2細胞の生存は、大きく減少した。
図7に関し、SCIDマウスにおけるBL2バーキットリンパ腫異種移植腫瘍を腫瘍が触知できるまで7日間樹立した。このとき、ビヒクル(70%シクロデキストリン)(上段パネル)およびJJ91(92mg/kg)(下段パネル)を毎日腹膜内注射で投与した。腫瘍サイズを、注射した日(1日目)および注射後4、8および11日目で測定した。示されるように、JJ91は、SCIDマウスにおけるBL2異種移植腫瘍の成長を低下させる。
SirT1活性を、メーカーにより特定されるバイオモルのFluor de Lys SirT1蛍光活性アッセイ(カタログ番号AK−555)を用いて評価した。反応は、1ミリモルのNAD+、7マイクロモルのFluor de Lys基質および過量のJH164を含んだ。脱アセチル化と展開液反応を37℃で1時間行った。JH164のIC50は、23.5マイクロモルである。図8はSirT1に対するJH164の抑制効果を示す。
上記態様は、本発明の代表的なものであり、請求の範囲に定義された発明の範囲を制限すると解釈されるものではない。
【0092】
更なる化合物は、初期の化合物JJ91(以後、テノビン1と称する)に基づき、合成される。
【0093】
【化7】
【0094】
調製された追加の化合物は、4つのカテゴリーに分類される。
カテゴリー1:水溶性類縁物質
a)テノビン6(JH164):中間体JHl40から調製
JH140は、可逆的にp53を活性化するこれまでに調製された他の化合物全てとは異なり、非可逆的p53活性化を示すことから、潜在的に興味深い。その合成は、テノビン6の手順中、以下に説明される。
化合物1 上記
化合物5〜8 上記
【0095】
下記手法1で示されるこれらの化合物の実験的合成
【化8】
【0096】
b
手法1:a テノビン6および類縁物質1、5〜7の合成
試薬と条件:(i)SOCl2、CH2Cl2、室温16時間(96%)。(ii)NaSCN、アセトン、室温、8時間、次いで1,4−フェニレンジアミン、16時間(58%)。(iii)酸塩化物、NEt3、DCM、室温。(iv)40% HNMe2水溶液、DCM/H2O、室温、24時間。(v)2M HCl(ジエチルエーテル中)。
【0097】
テノビン1(JJ91):N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アセトアミドの合成
アセトン(50mL)中、3(J.Am.Chem.Soc.1985、207、898に記載の2から調製)の溶液(6.53g、33mmol、1当量)に、チオシアン酸ナトリウム(2.69g、33mmol、1当量)を加えた。クリーム色の懸濁液を室温で16時間攪拌し、次いで、アセトン(50mL)中、p−アミノアセトアニリドの溶液(4.95g、33mmol、1当量)を加えた。黄色懸濁液を室温で16時間攪拌し、溶媒を減圧下濃縮し、淡橙色残渣を得、それをCH2Cl2中に再懸濁し、不溶性固体を濾過した。濾液を減圧下濃縮し、淡橙色固体を得、それを酢酸エチルから再結晶することにより精製し、テノビン1(7.49g、61%)を淡黄色固体として得た。
1H−NMR(アセトン−d6,300MHz)δ1.36(s,9H,(CH3)3),2.09(s,3H,CH3),7.63(d,2H,J=8.7Hz,ArH),7.71(s,6H,ArH),8.03(d,2H,J=8.7Hz,ArH),9.26(s(br),1H,NH),10.12(s(br),1H,NH),12.80(s(br),1H,NH).
13C−NMR(アセトン−d6,100MHz)δ23.36,29.70,30.40,34.83,118.95,124.14,125.71,128.15,129.36,133.31,137.82,157.00,167.97,178.73.
【0098】
中間体化合物1:(4−アミノ−フェニル)−3−(4−tert−ブチル−ベンゾイル)−チオ尿素(4)の合成
アセトン(30mL)中、3(J.Am.Chem.Soc.1985、207、898に記載の2から調製)の溶液(4g、20.4mmol、1当量)に、チオシアン酸ナトリウム(1.68g、20.8mmol、1.02当量)を加えた。生じた淡黄色懸濁液を室温で8時間攪拌し、次いで、0℃でアセトン(30mL)中1,4−フェニレンジアミン溶液(4.41g、40.8mmol、2当量)を加えた。
褐色懸濁液を室温まで加温し、16時間撹拌した。溶媒を減圧下濃縮し、残った残渣をCH2Cl2に再懸濁し、不溶性固体を濾過した。濾液を減圧下濃縮し、褐色固体を得、シリカのカラムクロマトグラフィにより精製(50%酢酸エチル/石油エーテル)し、続いて酢酸エチルから再結晶により4(4.42g、58%)を淡黄色固体として得た。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3,3.75(s(br),2H,NH2),6.71(d,2H,J=11.8Hz,ArH),7.49(dd,2H,J=8.6,33.4Hz,ArH),7.81(d,2H,J=10.8Hz,ArH),7.68(dd,4H,J=8.6,81.3Hz,ArH),9.02(s(br),1H,NH),12.37(s(br),1H,NH).
13C−NMR(CDCl3,100MHz)δ31.1,35.0,115.1,125.8,126.2,127.4,128.7,128.8,145.4,157.7,166.8,178.5.
【0099】
中間体JHl40からのテノビン6(JHl64):5−ジメチルアミノ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩の合成
CH2Cl2(110mL)中、チオ尿素4(6g、18.3mmol、1当量)をN2雰囲気下で攪拌した。5−ブロムワレリルクロライド(3.65g、18.3mmol、1当量)、続いてトリエチルアミン(2.51mL、18.3mmol、1当量)を加えた。ベージュ色の懸濁液を室温で4時間攪拌し、CH2Cl2(50mL)で希釈した。有機溶液を1Mの塩酸水溶液(1x50mL)、10%水酸化ナトリウム水溶液(1x50mL)、および飽和塩水(1x50mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた褐色泡をシリカのフラッシュクロマトグラフィで精製(50%酢酸エチル/石油エーテル)し、5−ブロモ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(中間体JH140)(6.76g、77%)を黄色粉末として得た。
【0100】
【化9】
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),1.93(m,4H,(CH2)2),2.42(t,1H,J=6.9Hz,COCH2),1H−NMR(300MHz)ppm 3.45(t,1H,J=6.3Hz,CH2Br),7.22(s(br),1H,NH),7.57(m,4H,ArH),7.68(d,2H,J=8.9Hz),7.82(m,2H,ArH),9.05(s(br),1H,NH),12.60(s(br),1H,NH).
13C−NMR(CDCl3,100MHz)δ24.6,27.7,31.1,32.5,33.6,37.4,119.9,124.9,126.3,127.4,128.6,133.6,157.9,166.9,170.9,178.4.
【0101】
上記ブロモ化合物(中間体JH140)(2.36g、4.80mmol)のH2O(75mL)中のジメチルアミン(30mL)との処理により、5−ジメチルアミノ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを含む粗混合物を得た。この混合物を最小量のアセトンに溶解し、黄色沈殿が生じるまで、エーテル溶液中、2M塩酸を滴下した。固体を濾過し、粗混合物を得、エタノールから再結晶することにより精製し、テノビン6(1.4g、59%)を黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.33(s,9H,(CH3)3),1.66(m,4H,(CH2)2),2.40(t,2H,J=6.1Hz,COCH2),2.75(s,6H,N(CH3)2),3.06(m,2H,CH2NH(CH3)2),7.59(m,6H,ArH),7.95(d,2H,J=8.4Hz,ArH),9.77(s(br),1H,NH(CH3)2),10.13(s(br),1H,NH),11.43(s(br),1H,NH),12.61(s(br),1H,NH).
13C−NMR(DMSO−d6,75MHz)δ22.4,23.8,31.3,35.3,35.9,42.5,56.7,119.5,125.3,125.8,129.1,129.7,133.3,137.9,156.8,168.5,171.2,179.4.
HRMS実測値455.2477(C17H39N6O4S2の計算値455.2474).
【0102】
化合物1:5−モルホリン−4−イル−ペンタン酸 {4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩(1)の合成
アセトニトリル(4mL)中、5−ブロモ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(中間体JH140、テノビン6に記載の通り調製)(80mg、0.16mmol)の溶液に、モルホリン(0.4mL)およびヨウ化カリウム(触媒量)を加えた。反応混合物を40分間加熱還流し、室温まで冷却し、減圧下濃縮した。残った残渣を、75%飽和炭酸水素ナトリウム水溶液とCH2Cl2間で分配した。有機層をMgSO4で乾燥し、減圧下濃縮し、ベージュ色の粗固体を得た。EtOAcから再結晶により精製し、5−モルホリン−4−イル−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを得、蒸気拡散により塩酸塩に変換した。25%CDCl3/ジエチルエーテル中ジメチルアミンの溶液を、塩化水素に曝し、1を白色沈殿として得、濾過により集めた。
HRMS実測値497.2585(C27H37N4O3Sの計算値.497.2586).
【0103】
化合物5:N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−4−ジメチルアミノ−ブチルアミド塩酸塩(5)の合成
チオ尿素4(200mg、0.61mmol、1当量)をCH2Cl2(10mL)中、N2雰囲気下で攪拌した。4−ブロモブチリルクロライド(169mg、0.92mmol、1.5当量)、続いてトリエチルアミン(85μL、0.61mmol、1当量)を加えた。橙色溶液を室温で1.5時間攪拌し、CH2Cl2(10mL)で希釈した。有機溶液を1M塩酸水溶液(1x10mL)、10%水酸化ナトリウム水溶液(1x50mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた淡黄色泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィにより精製し、4−ブロモ−N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−ブチルアミド(138mg、48%)を黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.35(s,9H,(CH3)3),2.26(m,2H,CH2CH2Br),2.57(t,2H,J=7.0Hz,CH2),3.52(t,3H,J=6.2Hz,CH2),7.60(m,6H,ArH),7.82(d,2H,J=8.7Hz,ArH),9.08(s(br),1H,NH),12.59(s(br),1H,NH).
【0104】
前記ブロモ化合物(138mg、0.29mmol)を、H2O(7.5mL)中、ジメチルアミン(3mL)で処理し、N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−4−ジメチルアミノ−ブチルアミドを含有する粗混合物を得た。この混合物を最小量のアセトンに溶解し、エーテル溶液中、2M塩酸を沈殿が生じるまで滴下した。固体を濾過し、濾液を減圧下濃縮した。生じた黄色オイルをヘキサンで研和し、黄色固体を得、イソプロパノールから再結晶し、5(75mg、55%)を粘性黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.32(s,9H,(CH3)3),1.95(m,2H,CH2),2.44(t,2H,J=7.3Hz,CH2),2.79(d,6H,J=4.8Hz,N(CH3)2),3.09(m,2H,CH2),7.62(m,6H,ArH),7.95(d,2H,J=8.5Hz,ArH),9.64(s(br),1H,NH(CH3)2),10.17(s(br),1H,NH),11.44(s(br),1H,NH),12.61(s(br),1H,NH).
【0105】
化合物6:6−ジメチルアミノ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩(6)の合成
チオ尿素4(200mg、0.61mmol、1当量)をCH2Cl2(10mL)中、N2雰囲気下で攪拌した。6−ブロモヘキサノイルクロリド(195.8mg、0.92mmol、1.5当量)、続いてトリエチルアミン(85μL、0.61mmol、1当量)を加えた。
ベージュ色の懸濁液を室温で1.5時間攪拌し、CH2Cl2(10mL)で希釈した。有機溶液を1M塩酸水溶液(1x10mL)、10%水酸化ナトリウム水溶液(1x50mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた褐色の泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィにより精製し、6−ブロモ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(166mg、55%)を黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.35(s,9H,(CH3)3),1.51(m,2H,CH2),1.73(m,2H,CH2),1.89(m,2H,CH2),2.36(t,2H,J=7.4Hz,COCH2),3.40(t,2H,J=6.7Hz,CH2Br),7.57(m,7H,ArH,NH),9.10(s(br),1H,NH),12.58(s(br),1H,NH).
【0106】
前記ブロモ化合物(136mg、0.27mmol)をH2O(6.25mL)中、ジメチルアミン(2.5mL)で処理することにより、6−ジメチルアミノ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを含有する粗混合物を得た。この混合物を最小量のCH2Cl2に溶解し、エーテル溶液中、2M塩酸を、クリーム色の沈殿が生じるまで滴下した。固体を濾過し、濾液を減圧下濃縮した。生じた黄色オイルをヘキサンで研和し、黄色固体を得、イソプロパノールから再結晶し、6(80mg、59%)を粘性黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.32(s,9H,(CH3)3),1.64(m,4H,(CH2)2),2.35(t,2H,J=7.1Hz,CH2),2.73(d,6H,J=4.8Hz,N(CH3)2),3.01(m,2H,CH2),7.59(m,6H,ArH),7.94(d,2H,J=8.4Hz,ArH),9.83(s(br),1H,NH(CH3)2),10.08(s,1H,NH),11.42(s,1H,NH),12.59(s,1H,NH).
【0107】
化合物7:8−ジメチルアミノ−オクタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩(7)の合成
8−ブロモオクタン酸(2g、8.90mmol、1当量)をCH2Cl2(15mL)中、N2雰囲気で攪拌し、塩化チオニル(784μL、10.68mmol、1.2当量)、続いて、DMF(数滴)を加えた。溶液を16時間攪拌し、減圧下濃縮して8−ブロモオクタノイルクロライド(2.04g、95%)を黄色オイルとして得、さらなる精製をせずに用いた。チオ尿素4(600mg、1.84mmol、1当量)を、CH2Cl2(15mL)中、N2雰囲気下で攪拌した。8−ブロモオクタノイルクロライド(532mg、2.02mmol、1.2当量)、続いてトリエチルアミン(256μL、1.84mmol、1当量)を加えた。ベージュ色の懸濁液を室温で16時間攪拌し、CH2Cl2(20mL)で希釈した。有機溶液を1M塩酸水溶液(1x20mL)、10%水酸化ナトリウム水溶液(1x20mL)、および飽和塩水(1x20mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた黄色の泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィで精製し、8−ブロモ−オクタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(579mg、59%)を黄色オイルとして得た。
1H−NMR(CDCl3,400MHz)δ1.36(s,9H,(CH3)3),1.44(m,6H,(CH2)3),1.74(m,2H,CH2),1.86(m,2H,CH2),2.37(t,2H,J=7.4Hz,COCH2),3.41(t,2H,J=6.8Hz,CH2Br),7.18(s,1H,NH),7.56(m,6H,ArH),7.63(d,2H,J=8.9Hz,ArH),7.82(d,2H,J=8.6Hz,ArH),9.04(s,1H,NH),12.59(1H,NH).
13C−NMR(CDCl3,75MHz)δ25.5,28.0,28.6,29.1,31.1,32.7,34.0,37.6,120.1,124.8,126.2,127.5,128.6,133.4,136.8,157.8,167.0,171.6,178.5.
【0108】
前記ブロモ化合物(487mg、0.92mmol)をH2O(25mL)中、ジメチルアミン(10mL)で処理することにより、8−ジメチルアミノ−オクタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを含有する粗混合物を得た。この混合物を最小量のCH2Cl2に溶解し、エーテル溶液中、2M塩酸をクリーム色の沈殿が生じるまで滴下した。固体を濾過し、濾液を減圧下濃縮した。生じた黄色オイルをヘキサンで研和し、黄色固体を得、イソプロパノールから再結晶し、7(301mg、62%)を粘性黄色固体として得た。
1H−NMR(DMSO−d6,400MHz)δ1.32(s,9H,(CH3)3),1.32(m,6H,CH2)3),1.62(m,4H,(CH2)2),2.33(m,2H,CH2),2.72(s(br),6H,N(CH3)2),2.99(m,2H,CH2),7.62(m,6H,ArH),7.97(dd,2H,J=8.5,16.5Hz,ArH),9.95(s,1H,NH),10.08(s(br),1H,NH),11.44(s,1H,NH),12.60(s,1H,NH).
【0109】
化合物8:N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−2−[2−(2−{2−[2−(2−ジメチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−アセトアミド塩酸塩(8)の合成
【0110】
【化10】
【0111】
類縁物質8の合成
試薬と条件:(i)JOC 2004、69、639に記載された文献反応により調製。(87%)(ii)CrO3/H2SO4、アセトン、0℃から室温、16時間(65%)。(iii)SOCl2、CH2Cl2、室温、16時間。(iv)4、NEt3、CH2Cl2、室温、16時間、(46%)。(v)LiBr、アセトン、還流、24時間(81%)。(vi)40%HNMe2水溶液、CH2Cl2/H2O、室温、24時間(91%)。(vii)ジエチルエーテル中2MのHCl、CH2Cl2、(73%)。
【0112】
ベンゼンスルホン酸 2−[2−(2−{2−[2−(2−ヒドロキシ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エチルエステル(12)
ヘキサエチレングリコール(11)(2.82g、10mmol、1当量)を、N2雰囲気下、CH2Cl2(100mL)に溶解し、酸化銀(3.48g、15mmol、1.5当量)、続いて塩化トシル(2.10g、11mmol、1.1当量)およびヨウ化カリウム(332mg、2mmol、0.2mmol)を加えた。溶液を室温で1時間攪拌し、次いで、濃縮し、セライトを通じて濾過し、酢酸エチルで溶出した。濾液を減圧下濃縮し、淡黄色オイルを得、シリカ(酢酸エチル)のカラムクロマトグラフィで精製し、12(3.79g、87%)を淡黄色オイルとして得た。
1H−NMR(DMSO−d6,300MHz)δ1.80(s(br),1H,OH),2.44(s,3H,CH3),3.64(m,22H,(CH2)11),4.15(m,2H,CH2),7.33(d,2H,J=8.0Hz,ArH),7.79(d,2H,J=8.4Hz,ArH).
【0113】
[2−(2−{2−[2−(2−ベンゼンスルホニルオキシ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−酢酸(13)
1.5M硫酸水溶液(10mL)を0℃に調製し、三酸化クロムを加えて撹拌し、橙色溶液を得た。アルコール12(1g、2.29mmol)をアセトン(15mL)に溶解し、その溶液を硫酸に0℃で滴下した。混合物を室温まで温め、16時間撹拌した。生じた緑色の溶液をセライトで濾過し、減圧下濃縮した。生じた水溶性の残渣をH2Oで希釈し、CH2Cl2(3x50mL)で抽出した。有機相をMgSO4で乾燥し、減圧下濃縮し、13(672mg、65%)を淡黄色オイルとして得、更なる精製は行わなかった。
1H−NMR(アセトン−d6,300MHz)δ2.46(s,3H,CH3),3.59(m,18H,(CH2)9),4.13(m,4H,(CH2)2),7.49(d,2H,J=8.0Hz,ArH),7.82(d,2H,J=8.0Hz).
13C−NMR(CDCl3,100MHz)δ21.6,68.5,68.6,68.8,69.1,69.3,70.3,70.4,70.4,70.5,70.6,70.6,70.7,70.8,71.2,71.3,76.8,77.1,77.4,127.9,129.9,132.9,144.9,172.7.
LRMS(M+Na)実測値473.16.
【0114】
N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−2−[2−(2−{2−[2−(2−ジメチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−アセトアミド塩酸塩(8)
酸13(117mg、0.26mmol)を、触媒量のDMFを含有する塩化オキサリル中で攪拌した。溶液を1時間加熱還流し、次いで、室温に冷却し、溶媒を減圧下濃縮し、対応する酸塩化物を得、更なる精製を行わずに用いた。酸塩化物(138mg、0.29、1.3当量)を乾燥CH2Cl2(10mL)中、N2雰囲気下で攪拌し、チオ尿素4(74mg、0.23mmol、1当量)を加え、明黄色溶液を得た。トリエチルアミン(41μL、0.23mmol、1当量)を加え、混合物を室温で16時間撹拌した。溶液をCH2Cl2(1x10mL)で希釈し、1M塩酸水溶液(1x10mL)、1M水酸化ナトリウム水溶液、続いて飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、溶媒を減圧下濃縮した。生じた黄色の泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィで精製し、トルエン−4−スルホン酸 2−[2−(2−{2−[2−({4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニルカルバモイル}−メトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エチルエステル(79mg、46%)を黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.34(s,9H,(CH3)3),2.41(s,3H,CH3),3.63(m,18H,(CH2)9),4.10(m,4H,(CH2)2),7.31(d,2H,J=8.0Hz,ArH),7.53(d,2H,J=8.6Hz,ArH),7.67(m,4H,ArH),7.79(m,m,4H,ArH),8.99(s,1H,NH),9.15(s,1H,NH),12.61(s,1H,NH).
13C−NMR(CDCl3,100MHz)δ21.7,31.1,35.3,68.7,69.3,70.1,70.4,70.5,70.6,70.7,71.2,120.2,124.6,126.1,126.2,127.5,127.9,128.6,129.8,133.7,136.1,144.8,157.8,166.89,168.4,178.3.
HRMS(M+Na)実測値782.2756(C37H49N3O10NaS2の計算値782.2757).
【0115】
上記に記載されたトシレート(76mg、0.1mmol、1当量)をアセトン(3mL)中で攪拌し、臭化リチウム(44mg、0.5mmol、5当量)を加えた。混合物を50℃で24時間加熱した。溶媒を減圧下濃縮し、生じた残渣を酢酸エチルに再懸濁し、固体を濾過した。濾液を減圧下濃縮し、2−[2−(2−{2−[2−(2−ブロモ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アセトアミド(54mg、81%)を淡黄色オイルとして得、更なる精製を行わずに用いた。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),3.45(t,2H,J=6.3Hz,CH2Br),3.71(m,16H,(CH2)8),4.18(s,2H,NHCOCH2),7.55(d,2H,J=8.6Hz,ArH),7.70(q,4H,J=9.0Hz,ArH),7.82(d,2H,J=8.6Hz,ArH),9.06(s,1H,NH),9.16(s(br),1H,NH),12.61(s,1H,NH).
【0116】
続いてこの臭化物(30mg、0.05mmol)を、40%ジメチルアミン水溶液(0.75mL)、H2O(1.8mL)およびCH2Cl2(2mL)中、室温で24時間撹拌することによりジメチルアミン誘導体に変換した。反応混合物をCH2Cl2 (20mL)で希釈し、有機相をH2O(1x10mL)、1M水酸化ナトリウム水溶液(1x10mL)および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、溶媒を減圧下濃縮し、ジメチルアミン体を得た。淡黄色オイルを最少量のCH2Cl2にとり、ジエチルエーテル中2M塩酸溶液を、わずかな沈殿が生じるまで滴下した。混合物を減圧下濃縮し、8(20mg、73%)を黄色の泡として得た。
1H−NMR(CDCl3,300MHz)δ1.32(s,9H,(CH3)3),2.77(d(br),6H,J=4.0Hz,NH(CH3)2),3.61(m,18H,(CH2)9),4.10(m,4H,(CH2)2),7.63(m,6H,ArH),7.95(d,2H,J=8.4Hz,ArH),9.55(s,1H,NH),9.77(s,1H,NH),11.45(s,1H,NH),12.62(s,1H,NH).
【0117】
カテゴリー2:オキソ−テノビン類縁体、化合物24
以下の手法に従い24を調製した。
【0118】
【化11】
【0119】
{4−[3−(4−tert−ブチル−ベンゾイル)−ウレイド]−フェニル}−カルバミン酸 tert−ブチルエステル(28)
上記に記載されたとおり、ベンズアミド26(1g、5.64mmol、1当量)を塩化オキサリル(834μL、9.59mmol、1.7当量)と反応させた。生じたイソシアネート(27)を、アニリン18と共に乾燥アセトニトリル中で素早く撹拌し、3時間加熱還流した。生じたベージュ色の懸濁液を室温まで冷却し、沈殿を濾過により集め、28(400mg、17%)をベージュ色の固体として得、さらに精製しなかった。
1H−NMR(DMSO−d6,300MHz)δ1.31(s,9H,(CH3)3),1.47(s,9H,OC(CH3)3),7.43(m,4H,ArH),7.55(d,2H,J=8.6Hz,ArH),7.97(d,2H,J=8.6Hz,ArH),9.31(s,1H,NH),10.77(s,1H,NH),10.91(s,1H,NH).
【0120】
5−ブロモ−ペンタン酸 {4−[3−(4−tert−ブチル−ベンゾイル)−ウレイド]−フェニル}−アミド(29)
N−アシル尿素28(300mg、0.73mmol)をトリフルオロ酢酸(1.2mL)中、40分間攪拌し、減圧下濃縮し、1−(4−アミノ−フェニル)−3−(4−tert−ブチル−ベンゾイル)−ウレア(トリフルオロ酢酸塩として)(383mg、定量的)を褐色な綿状の固体として得、更なる精製をしなかった。
1H−NMR(DMSO−d6,300MHz)δ1.30(s,9H,(CH3)3),7.32(d,2H,J=8.8Hz,ArH),7.55(d,2H,J=8.6Hz,ArH),7.70(d,2H,J=8.9Hz,ArH),7.98(d,2H,J=8.6Hz,ArH),10.96(s,1H,NH),11.02(s,1H,NH).
【0121】
脱保護したアミン(368mg、0.70mmol)を乾燥CH2Cl2(10mL)中、N2雰囲気下で攪拌し、NEt3(98μL、0.70mmol)を加え、清明な褐色溶液を得た。5−ブロムワレリルクロライド(140μL、1.05mmol、1.5.当量)、続いてトリエチルアミン(98μL、0.70mmol)を加えた。反応混合物を室温で16時間攪拌し、次いでCH2Cl2(1x20mL)で希釈し、1M塩酸水溶液(1x20mL)、1M水酸化ナトリウム水溶液(1x20mL)および飽和塩水(1x20mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮し、粗黄色チョーク質の固体を得、シリカ(50%酢酸エチル/石油エーテル)のカラムクロマトグラフィで精製し、29(153mg、48%)を黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.31(s,9H,(CH3)3),1.80(m,4H,(CH2)2),2.33(t,2H,J=7.2Hz,COCH2),3.57(t,2H,J=6.6Hz,CH2Br),7.54(m,6H,ArH),7.98(d,2H,J=8.5Hz,ArH),9.90(s,1H,NH),10.82(s,1H,NH),10.93(s,1H,NH).
【0122】
5−ジメチルアミノ−ペンタン酸 {4−[3−(4−tert−ブチル−ベンゾイル)−ウレイド]−フェニル}−アミド塩酸塩(24)
ブロモ化合物29(123mg、0.27mmol)をCH2Cl2(10mL)、40%HNMe2水溶液(3mL)およびH2O(7.5mL)中、室温で20時間攪拌した。反応混合物をCH2Cl2(20mL)で希釈し、H2O(1x10mL)、10%NaOH水溶液(1x10mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮し、クリーム色の固体を得、アセトンに溶解し、ジエチルエーテル中2M塩酸で処理した。白色沈殿を得、濾過により単離し、24(44mg、36%)をクリーム色の粉末として得た。
1H−NMR(DMSO−d6,400MHz)δ1.31(s,9H,(CH3)3),1.64(m,4H,(CH2)2),2.36(t,2H,J=6.7Hz,COCH2),2.75(s,6H,NH(CH3)2),3.05(m,2H,CH2Br),7.53(m,6H,ArH),7.98(d,2H,J=8.6Hz,ArH),9.63(s(br),1H,NH),9.98(s,1H,NH),10.82(s,1H,NH),10.93(s,1H,NH).
【0123】
カテゴリー3:Boc−テノビン類縁体、化合物21
この類縁物質は、以下に示すp53活性化アッセイにおいて、非常に良好な活性を示したので、重要な化合物である。
【0124】
{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−カルバミン酸 tert−ブチルエステル(21)
アセトン(15mL)中3(869μL、4.79mmol、1当量)の撹拌溶液に、N2雰囲気下、チオシアン酸ナトリウム(389mg、4.79mmol、1当量)を加え、反応混合物を室温で4時間撹拌した。アニリン18(1g、4.79mmol、1当量)をアセトン(10mL)中に溶液として加えた。生じたベージュ色の懸濁液を室温で16時間攪拌し、溶媒を減圧下濃縮した。残った残渣を、CH2Cl2に再懸濁し、不溶の固体を濾過した。濾液を減圧下濃縮し、クリーム色の粗固体を得、50%酢酸エチル/石油エーテルから再結晶することにより精製し、{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−カルバミン酸 tert−ブチルエステル(1.4g、66%)を金色微粉末として得た。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),1.63(s,9H,OC(CH3)3),6.52(s,1H,NH),7.42(d,2H,J=8.7Hz,ArH),7.55(d,2H,J=8.5Hz,ArH),7.64(d,2H,J=8.8Hz,ArH),8.82(d,2H,J=8.7Hz,ArH),9.03(s,1H,NH),12.56(s,1H,NH).
【0125】
カテゴリー4:テノビン類に基づく親和性マトリックス
このタイプの試薬として、テノビン類に基づく親和性マトリックスの調製が含まれ、テノビン類の可能性のあるタンパク質標的や、サーチュインタンパク質ファミリーやその他の範囲の抑制剤の選択性のアッセイにおいて、可能性のある関連物質(例えばMeijer,Laurent;Skaltsounis,Alexios−Leandros;Magiatis,Prokopios;Polychronopoulos,Panagiotis;Knockaert,Marie;Leost,Maryse;Ryan,Xiaozhou P.;Vonica,Claudia Alin;Brivanlou,Ali;Dajani,Rana;Crovace,Claudia;Tarricone,Cataldo;Musacchio,Andrea;Roe,S.Mark;Pearl,Laurence;Greengard,Paul.「古代紫インディルビン類から誘導されるGSK−3−選択的な抑制剤」Chemistry&Biology(2003),10(12),1255−1266により報告される以下の類縁物質プロトコール)を同定するのに重要である。親和性マトリックスを調製する手順は次のとおりである。
【0126】
親和性樹脂の使用
【0127】
【化12】
【0128】
6−tert−ブトキシカルボニルアミノ−ヘキサン酸(31)
6−アミノヘキサン酸(3.1g、23.63mmol、1当量)を、ジオキサン/H20(1:1、75mL)中、0℃で水酸化ナトリウム(1.05g、23.63mmol)と共に攪拌した。この溶液に、Boc無水物(5.2g、21.27mmol、0.9当量)を加え、反応を16時間室温まで温めた。反応混合物を1M塩酸水溶液でpH5まで酸性にした。混合物をCH2Cl2(3x50mL)で抽出し、合わせた有機相をMgSO4で乾燥し、減圧下濃縮し、31を無色のオイル(4.5g、89%)として得、更なる精製を行わなかった。
1H−NMR(CDCl3,300MHz)δ1.42(m,4H,(CH2)2),1.44(s,9H,(CH3)3),1.65(m,2H,CH2),2.35(t,2H,J=7.4Hz,CH2),3.10(m,2H,CH2),4.55(s,(br),1H,NH).
【0129】
(5−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニルカルバモイル}−ペンチル)−カルバミン酸 tert−ブチルエステル(32)
チオ尿素4(100mg、0.31mmol、1当量)を乾燥CH2Cl2(5mL)中、N2雰囲気下で攪拌し、酸31(106mg、0.46mmol、1.5当量)、続いてブロモ−トリス−ピロリジノホスホニウムヘキサフルオロホスファート(214mg、0.46mmol、1.5当量)、およびN,N−ジイソプロピルエチルアミン(53μL、0.31mmol、1当量)を加えた。反応混合物を室温で16時間攪拌し、次いで、CH2Cl2(20mL)で希釈し、1M塩酸水溶液(1x10mL)、1M水酸化ナトリウム水溶液(1x10mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮し、粗黄色オイルを得、シリカ(25〜50%酢酸エチル/石油エーテル)のカラムクロマトグラフィで精製し、32(86mg、54%)を無色のオイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),1.40(s,9H,OC(CH3)3),1.40(m,2H,CH2),1.52(m,2H,CH2),1.75(m,2H,CH2),2.36(t,2H,J=7.5Hz,CH2),3.12(m,2H,CH2),4.61(s(br),1H,NHBoc),7.54(d,2H,J=8.7Hz,ArH),7.62(m,4H,ArH),7.82(d,2H,J=8.6Hz,ArH),9.09(s,1H,NH),12.59(s,1H,NH).
LRMS(M+Na)実測値563.15.
【0130】
6−アミノ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドトリフルオロ酢酸塩(33)
Boc保護されたアミン32(20mg、0.04mmol)をCH2Cl2(1mL)およびトリフルオロ酢酸(166μL)中、室温で30分間攪拌した。減圧下濃縮し、33を淡黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.29(m,2H,CH2)1.30(s,9H,(CH3)3),1.59(m,4H,(CH2)2),2.28(m,2H,CH2),2.90(m,2H,CH2),7.60(m,6H,ArH),7.75(d,2H,J=8.2Hz,ArH),8.75(s(br),1H,NH),9.21(s,1H,NH),12.50(s,1H,NH).
【0131】
親和性樹脂(Affigel−15)への33の固定化
Affigel−15(BioRad)(1mL)を冷イソプロパノールで洗浄し、濾過した。Affigel(210mg)に、ジオキサン(1mL)、33(2mg)および過剰のトリエチルアミンを加えた。さらにジオキサンを加え、総量を1.5mLに調節した。混合物を4℃で24時間撹拌し、次いで3000rpmで3分間遠心分離した。母液をデカントし、新鮮なジオキサンを加え、混合物を振盪した。遠心分離し、2回デカントした。
【0132】
上記に記載された化合物の活性および水溶性を試験し、結果を表2に示す。
【0133】
【表3】
表2活性データ:a細胞ベースのアッセイにおいて類縁物質がp53を活性化する活性化能の順序.この形式は、定量化が困難なp53活性化アッセイに選択的に用いられている。n.d.=測定せず
【技術分野】
【0001】
本発明は、p53応答を活性化する化合物に関し、および、例えば、癌の治療および潜在的な他の疾患/状態(サーチュイン機能を含む)などの過増殖性疾患において見出された使用に関する。
【背景技術】
【0002】
P53腫瘍抑制タンパク質は、細胞ストレス応答の中心的なメディエーターである。P53は、腫瘍の発生を抑制する中心的な役割を担う。保護的なメカニズム、特に細胞周期の停止やアポトーシスを活性化させることにより、潜在的に発癌性ストレスの範囲に応答する。腫瘍抑制剤としての重要性は、50%を超える成人ヒト腫瘍で、TP53遺伝子における変異や欠失を不活性化する、ヒト癌における高頻度の変異に反映される。p53が野生型である多くの癌では、他の発癌性イベントによりp53系が変化しうる。これは、おそらくp53応答が、殆どの癌において欠損していることを意味する。
【0003】
成人の固形腫瘍における知見に反し、ウイルス関連性の悪性腫瘍(例えば、子宮頸癌)、血液の悪性疾患および小児癌におけるp53変異の発生は、非常に低い。これは、成人と比較し小児の癌の予後がより良好であることへの手がかりになりうる。しかし、若年患者の長期間の療法や治療を考慮した場合、現在の多くの療法の変異原性作用を知ることが重要である。さらに、文献には、DNA障害性アルキル化物質によるB−CLL患者の治療が、p53の変異の発生に関連し、著しい不良転帰および薬剤抵抗性を伴うことが示されている。変異によってp53機能が消失していない癌の治療の向上は、新規な、非遺伝毒性的なp53応答活性剤の発見によるだろう。
【発明の概要】
【発明が解決しようとする課題】
【0004】
多くの現在の抗癌療法は、DNA傷害によりp53応答を活性化する。p53系の非遺伝毒性的活性化は、癌の予防的な治療も含め、長期化へ通じる。この要件に合う分子は、変異によるp53機能を消失していない過増殖性状態を伴う患者に適用する治療物質として有用となりうる。
【0005】
本発明の目的は、過増殖性状態や、癌などの異常なP53機能、および関連する状態を含む状態および疾患の治療および/または予防のための分子、およびそれらの分子の使用を提供することである。
【0006】
本発明の更なる目的は、癌、糖尿病、筋分化、心不全、神経変性、老化、HIV感染およびマラリアなどの、サーチュイン発現および/または機能を伴う状態および疾患の治療および/または予防のための分子、およびそれらの分子の使用を提供することである。
【0007】
本発明の第1の局面によれば、式(I)の化合物の医薬としての使用が提供される。
【0008】
【化1】
【0009】
[式中、
【0010】
R1は独立してHであるか;分枝状または非分枝状の、モノ、ジ、もしくはトリ置換または無置換の、アルキル、アルケニルもしくはアルキニルであるか;または、アリールであるか;または、Z−アルキル、Z−アルケニル、Z−アルキニルもしくはZ−アリール(ここでZはO、NH、N、またはSであるか、ラベル、プローブもしくは基を可溶化する溶媒に結合する基を含む)であり;
Yは存在しないか、−C(O)−、−C=S−、または−SO2−であり;
Ar、Ar’はアリールであり;
XはOまたはSであり;
R3およびR4は、独立して、非存在であるか存在し、存在するときは、独立してH、または分枝状または非分枝状の、モノ、ジもしくはトリ置換または無置換の、アルキルもしくはZ−アルキル(ここで、ZがO、NH、NもしくはS)であるか、またはR3およびR4は、共に結合して、分枝状または非分枝状の、置換または無置換の、アルキレンもしくはZ−アルケン(ここで、ZはO、NH、NまたはSである)を形成し;
R5およびR6は、独立して、非存在であるか存在し、存在するときは、独立して分枝状または非分枝状の、置換または無置換のアルキルであり;ここで、
R3が存在するときは、破線bは単結合であり、R3が非存在のときは、破線bは二重結合であり;
R4が存在するときは、破線cおよびdは単結合であり、R4が非存在のときは、破線cまたはdの一方は二重結合であり、他方は単結合であり;そして、
R5およびR6が非存在のとき、破線aおよびeはそれぞれ二重結合であり、R5およびR6が存在するときは、破線aおよびeは、それぞれ単結合である]
で示される、医薬としての使用に供される化合物、または、その生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【0011】
このように、破線a、b、c、dおよびeの単結合または二重結合の要件は、付随する炭素原子の四価、および窒素原子の三価を確保し、維持するために選択される。しかし、窒素原子は、四価となり、例えば、アルキル基などの他の部分の窒素原子への結合を通じて、正電荷を有しうることに留意する。
【0012】
誤解を避けるために、式(I)の種々の化合物は、以下の化合物式(IA−D)により示される。
【0013】
【化2】
【0014】
理論により拘束されることを意図することなしに、構造活性相関の研究から、式(I)におけるR1−Y−NH−の一群は、例えば、NH残基により規定されるように、水素結合供与体基を備えることにより活性を有するように思われる。また、R2は、立体的に嵩高い基が好ましいようである。
【0015】
Yは、存在または非存在となり得るが、好ましくは存在する。Yは、好ましくは−C(O)−基である。
R1は、好ましくは独立してH、または置換または無置換のアルキルまたはアリール基である。
アリール基、Arは、置換または無置換の、単環または多環系アリールもしくはヘテロアリール部分であってもよい。
好ましくは、Arはフェニルである。
Arがフェニルのとき、フェニルにパラ位で結合する基、すなわち、−NH−が−NR3−基に対しパラ位であるのが好ましい。
好ましくはAr’は、1以上の可能な位置に、分枝状または非分枝状の、置換また
は無置換C2−C10アルキルで置換されたフェニルである。好ましくは、R2は二級または三級アルキル基などの、分枝状のアルキル基である。
【0016】
最も好ましくは、R2はiso−プロピルまたは三級のブチル基(すなわち、tert−ブチル)である。
R2基は、結合するフェニル環のパラ位で結合するのが好ましい。
好ましくは、破線aおよびeが二重結合となるようにR5およびR6は非存在である。
好ましくは、R3およびR4は存在し、好ましくは水素、または、例えば、メチルなどのC1−C4アルキルである。
【0017】
アルキル、アルケニルおよびアルキニルという語は、ここでは、これらの基の分枝状または非分枝状で、置換され、または無置換の、線状または環状のものを含む。
アリールという語は、置換または無置換の単環もしくは多環系のアリールまたはヘテロアリールを含む。
【0018】
R1がHでないとき、各存在で、アルキル、アルケニル、アルキニル、アリールまたはヘテロアリール、カルボキシ、アルキルオキシカルボニル ヒドロキシル、アミノ、モルホリノ、ニトロ、アルキルオキシ、アルキルチオ、ホルミル、シアノ、カルバモイル、ハロ(例えば、フルオロ、クロロ、ブロモまたはヨード)、ケトン、−S(O)NR7R8または−S(O)R9(ここで、R7、R8およびR9は、それぞれ独立してアルキル、アルケニル、アルキニル、アリールまたはヘテロアリールから選択される)からなる群から、独立して選択される基で1回以上置換されうる。
【0019】
通常、R1はアルキル基であり、好ましくは直鎖のC3−C6アルキル、例えば、n−ブチルなどのC4アルキルである。
通常、アルキル基は、遊離末端で、フェニル、ヒドロキシル、アミノ、モルホリノ、ニトロ、アルキルオキシ、アルキルチオ、ホルミル、シアノ、カルバモイル、ハロ(例えば、フルオロ、クロロ、ブロモまたはヨード)、ケトン、−S(O)NR7R8または−S(O)R9(ここで、R7、R8およびR9は、それぞれ独立してアルキル、アルケニル、アルキニル、アリールまたはヘテロアリールから選択される)から選択される置換基で置換される。
【0020】
末端置換基がモルホリノ基のとき、好ましくは、その窒素原子によりR1の末端に結合する。
末端置換基は、ハロゲンであってもよく、例えば、ブロモの場合は、式Br(CH2)4−を有するR1基を与える。
末端置換基が、アミノまたはモルホリノ基のとき、その窒素原子は、プロトン化(例えば、酸反応により)して、正電荷されたものとなってもよい。
【0021】
末端R1は、ラベル、親和性樹脂または基質プローブおよび/または水溶性基に結合するリンカー基として有利に考慮されても良い。例えば、リンカーは、活性メカニズムの研究に有用な分子を提供するために、式(I)の化合物(すなわち、活性化合物)の残存部分と、生物化学実験で用いられるラベルまたはプローブに結合してもよい。リンカーは、アルキル鎖、エチレングリコールのポリマー類(すなわち、(OCH2CH2)n1)および6−アミノヘキサン酸のペプチド類(すなわち、(NH(CH2)5CO)n2)r;ここで、n1およびn2は、分子がポリマーであることを示し、通常、1から6または8ぐらいの範囲の整数である)などの本目的に適するどのような部分であってもよい。ラベルやプローブは、ビオチン、ストレプロアバジン(streptavadin)、フルオロフォア類(例えば、bodipy、フルオレセインおよびローダミン)、放射活性ラベル類および固相マトリックス、例えば、ポリマービーズなどのポリマー類(例えばポリスチレン)などの、その目的において知られる物を含む。水溶性化する基は、その目的において適する糖類、アミン類、アミノ酸類、リン酸類等およびそれらの塩を含む。
【0022】
水溶性化する基は、例えば、特に医薬として使用されるとき、化合物の生物学的利用性および/または薬物動態を最適化するために、活性化合物の水溶性を変化させるための修飾に用いられうる。
【0023】
アミノという語は、ここでは、サイクリックアミノ基、すなわち、アミンの窒素原子が環の構成員である基を含む。
アミノという語は、ここでは、プロトン化された種々のアミン類およびその塩についても含む。例えば、アミンは、プロトン化されてもよく、塩酸、硫酸の酸など、カルボン酸類を含む多くの酸類との塩を形成してもよい。例えば、アミンの塩酸塩は、水や水溶性溶媒中で高い水溶性を示す。
【0024】
式(I)において、アリール残基は、例えば、5または6員の単環アリールもしくはヘテロアリール環構造、または他の多環系アリールもしくはヘテロアリール残基となりうる。
フェニルは、6員アリール基の具体例である。
【0025】
通常、ヘテロアリール構造中のヘテロ原子は、酸素または窒素から選択される。
フリルおよびピロリルは、それぞれ酸素および窒素ヘテロ原子を含む5員ヘテロアリール基の具体例である。
ピリジニルおよびピリミジニルは、それぞれ1つの窒素原子、および2つの窒素原子を含む6員ヘテロアリール基の具体例である。
ナフチルは、縮合6員環を形成する10個の炭素原子の骨格を有するアリール基の具体例である。
【0026】
アリール基は、示されるように、1以上の位置で置換されてもよく、適する置換基は、それぞれの置換位置において、R1またはR2で定義された置換基から独立して選択されうる
【0027】
置換基は、アリール基に直接的に、または、−O−、−S−、−N−または−(CR10R11)n−(ここで、nは1〜25の整数であり、例えば、1〜10、1〜4などであり、R10およびR11は、独立してアルキル、アルケニル、アルキニル、アリールまたはヘテロアリール、カルボキシ、アルキルオキシカルボニル ヒドロキシル、アミノ、モルホリノ、ニトロ、アルキルオキシ、アルキルチオ、ホルミル、シアノ、カルバモイル、ハロ(例えば、フルオロ、クロロ、ブロモまたはヨード)、ケトン、−S(O)NR7R8または−S(O)R9(ここで、R7、R8およびR9は前記の定義の通り))から独立して選択される基を介して結合してもよい。
【0028】
医薬としての使用に関し、式(I)の好ましい化合物は、以下の式(II)
【0029】
【化3】
[式中、R1、R2およびYは前記定義の通りである]
または、その生理的に許容可能な塩、溶媒和物、エステルまたは他の生理的機能性誘導体、を有する。
【0030】
好ましくは、R1YNH−および/またはR2−基が、それらが結合するそれぞれのフェニル環上のパラ位にある。
好ましくは、Yは−C(O)−基であり、このため、好ましくは、R1YNH−基はR1C(O)NH−である。
【0031】
好ましい化合物、その薬学的に許容可能な塩、溶媒和物、エステルまたは他の生理的機能性誘導体は、実施例の項においても示される。
本発明は、さらに以下に詳細を説明するように、その薬学的に許容可能な担体と共に式(I)または(II)の化合物を含む医薬製剤にも及ぶ。
本発明は、さらに以下により詳細に記載するように、ここに記載された1以上の式(I)または(II)の化合物を、それを必要とする患者に投与することを含む治療または予防方法にも及ぶ。
本発明は、式(I)および(II)の範囲に含まれる新規な化合物にも及ぶ。
【0032】
このように、更なる局面において、本発明は、下記化合物
【化4】
を除く、ここに示される式(I)または(II)の化合物を提供する。
【0033】
ここに記載された化合物式において、アルキル基は、独立してC1−C22アルキル、好ましくはC1−C10アルキル、好ましくはC1−C4アルキル、例えば、メチル、エチル、プロピル、ブチルであってもよい。
アルケニル基は、独立してC2−C22アルケニル、好ましくはC2−C10アルケニル、好ましくはC2−C4アルケニルであってもよい。
アルキニル基は、独立してC2−C22アルキニル、好ましくはC2−C10アルキニル、好ましくはC2−C4アルキニルであってもよい。
アルキル、アルケニルまたはアルキニル基は、分枝状または非分枝状、置換または無置換であってもよい。例えば典型的な分枝状のアルキル基は、iso−プロピル、iso−ブチル、sec−ブチル、tert−ブチル、3−メチルブチル、3,3−ジメチルブチルおよびそれらの異性体を含む変形体を含む。
【0034】
ここに記載されたように、アルキル、アルケニルまたはアルキニル基は、置換されてもよく、置換基は、ヒドロキシル、置換または無置換のアミン、置換または無置換のアミド、ハライド(フルオロ、クロロ、ブロモ、ヨードなど)、アルコキシ、チオ、ニトロ、カルボキシ、エステル、シアノ、またはアリール(フェニル、ナフチルおよびピリジルなど)などのあらゆる化学分子であってよい。
【0035】
ここに記載された化合物、例えば、アルケニル基、における二重結合の幾何学的配置はcisまたはtransの幾何学的配置であってよい。
【0036】
本発明の化合物の生理的に許容可能な塩の例には、有機カルボン酸類(酢酸、乳酸、酒石酸、マレイン酸、クエン酸、ピルビン酸、シュウ酸、フマル酸、オキザロ酢酸、イセチオン酸、ラクトビオン酸および琥珀酸など);有機スルホン酸類(メタンスルホン酸、エタンスルホン酸、ベンゼンスルホン酸およびp−トルエンスルホン酸など)および無機酸類(塩酸、硫酸、リン酸およびスルファミン酸等)と形成される酸付加塩を含む。
【0037】
本発明化合物の生理的機能性誘導体は、体内で親化合物に変換される誘導体である。そのような生理的機能性誘導体は、「プロドラッグ」や「バイオプレカーサー」とも言われる。本発明化合物の生理的機能性誘導体は、生体内で加水分解性のエステル類を含む。
【0038】
記載された使用/方法のいずれか1において使用されうる、ここに記載された化合物に対応する溶媒和物を、調製、精製、および/または処理することが、簡便であるか、好ましい。溶媒和物という語は、ここでは、化合物またはその化合物の塩などの溶質と溶媒との複合体を言うのに用いられる。溶媒が水のとき、その溶媒和物を、例えば基質1分子当たりの水分子の数により、一水和物、二水和物、三水和物など、水和物と称してもよい。
【0039】
当然のことながら、本発明の化合物は、種々の立体異性体が存在し得、上記に定義された本発明の化合物は、エナンチオマーおよびラセミ混合物を含む、全ての立体異性体およびその混合物を含む。本発明は、式(I)または(II)の化合物の個々のエナンチオマーやそのようなエナンチオマーの全または部分ラセミ混合物を含む、そのような全ての立体異性体や立体異性体の混合物の使用を、その範囲に含む。
【0040】
本発明の化合物は、後述するように、技術的に容易に利用可能な試薬と技術を用いて調製されうる。本発明の化合物の調製のための合成経路において新規な中間体化合物は、本発明の分子の調製についての一般適用に関し、重要な分子となりうる。従って、本発明はこれらの新規な中間体化合物を含む範囲にまで及ぶ。
【0041】
具体例として、化合物は、公報番号EP 0 136 745 B1の欧州特許、および公開番号EP 0 193 249 A2の欧州特許出願に記載された方法を用いて合成されうる。
【0042】
式(II)に記載の化合物の調製に有用な合成方法には、以下の手法において示されるように、溶媒(例えば、アセトン)中、種々の塩化ベンゾイル類を金属チオシアン酸塩(例えば、チオシアン酸ナトリウム)で処理し、ベンゾイルイソチオシアネート類を得、次いでアニリン類と反応(好ましくはin situで)させ、以下の式(II)の化合物を得る。
【0043】
【化5】
【0044】
上記の手法において、フェニル基は、あらゆるアリール基を表す。更なる機能付与は、アシル化やアルキル化により達成されうる。塩基性水溶性条件下で、化合物は加水分解され、N−アリールチオ尿素類が得られる。
【0045】
上記に示した通り、本発明は、ここに示される化合物を、それを必要とする患者に投与することを含む、ここに挙げられる疾患、病態もしくは状態の治療または予防を提供する。
【0046】
本発明に関する疾患は、P53タンパク質、その機能および/またはP53経路の異常を伴う異常な細胞死に関与するものを含む。
【0047】
特に、異常な細胞増殖が関与する疾患は、ここに挙げられた化合物で治療可能である。そのような疾患の例には、癌、過剰増殖性疾病(疣贅類、乾癬、炎症性腸疾患を含む)、リウマチ/自己免疫状態、鎌状赤血球性貧血、サラセミア等が含まれる。
【0048】
本活性化合物により治療されうる癌の例には、これらに限定されないが、例えば、膀胱癌、乳房癌、大腸癌(例えば、大腸腺癌や大腸腺腫などの結腸直腸の癌)、腎臓癌、表皮性癌、肝癌、肺癌(例えば腺癌、小細胞肺癌や非小細胞肺癌)、食道癌、胆嚢癌、卵巣癌、膵臓癌(例えば、膵外分泌部の癌)、胃癌、頚部癌、甲状腺癌、前立腺癌または皮膚癌(例えば扁平上皮癌);リンパ系の造血性腫瘍(例えば白血病、急性リンパ性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキン病、非ホジキンリンパ腫、毛様細胞リンパ腫、またはバーキットリンパ腫);骨髄疾患系の造血性腫瘍(例えば急性および慢性骨髄性白血病、骨髄異形成症候群、または前骨髄球性白血病);濾胞性甲状腺癌;間葉系起源の腫瘍(例えば線維肉腫または横紋筋肉腫);中枢または末梢神経系の腫瘍(例えば星状細胞腫、神経芽細胞腫、神経膠腫またはシュワン細胞腫);メラノーマ;精上皮腫;奇形癌腫;骨肉腫;色素性乾皮症;角化棘細胞腫;濾胞性甲状腺癌;またはカポジ肉腫が含まれる。
【0049】
本発明者らは、ここに示されるJH164などの本発明の化合物が、SirT1活性の抑制に特に有効であることに注目している。このため、JH164などの本発明の化合物は、SirT1発現/機能に関連する疾患/状態の治療に使用されうる。
【0050】
SirT1または関連するタンパク質は、癌、炎症、免疫反応、肥満、老化、糖尿病、筋分化、心不全、神経変性、HIV感染およびマラリアを含む非常に多くの疾患/状態において標的であると確認されており(例えば、Bordone LおよびGuarente L、Cancer Res.,66(8):4368−77(2006年4月15日);Heltwegら、Trends Pharmacol Sci.,26(2):94−103、2005年2月;Pagansら、PLoS Biology 2005 Vol.3, No.2,e41;Deitsch KW,Cell.,121(1):1−2(2005年4月8日);Freitas−Junior LHら、Cell.,121(1):25−36(2005年4月8日);Nayagam VM、J Biomol Screen.(2006年11月12日)を参照のこと)、従って本発明の化合物は、前記のあらゆる疾患/状態の治療/予防において有用性が見出されうる。
【0051】
式(I)の化合物と、(同時に、または異なる時間間隔を問わず)一緒に投与されうる他の治療物質の例には、これらに限定されないが、シスプラチン、シクロホスファミド、ドキソルビシン、イリノテカン、フルダラビン、5FU、タキサン類、マイトマイシンCなどの、トポイソメラーゼ阻害剤、アルキル化剤、代謝拮抗薬、DNA結合剤および微小管阻害薬(チューブリン標的剤)、または放射線療法などが含まれる。活性化合物を他の療法と組み合わせる場合、2以上の治療剤が、異なる経路により種々の個別の投薬計画で投与されうる。
【0052】
本発明の化合物と上記に挙げられた物質との組み合わせは、通常の一般的な知見と熟練した施術者に公知の投薬計画を用い、投与量を選択する医師の裁量でなされる。
【0053】
式(I)の化合物が、1、2、3、4またはそれ以上、好ましくは1または2、好ましくは1以上の治療剤との組み合わせ療法で投与される場合、化合物を同時にまたは連続して投与できる。連続して投与する場合、狭い間隔(例えば、5〜10分間隔以上)または、より長い間隔(例えば1、2、3、4時間またはそれ以上開けて、または必要な場合はより長い間隔)で、治療の作用と治療剤に見合った的確な投薬計画で投与されうる。
【0054】
本発明の化合物はまた、放射線療法、光力学療法、遺伝子療法;外科手術および栄養制限食などの非化学療法と組み合わせて投与されうる。
患者は、通常、動物であり、例えば、哺乳動物、特にヒトである。
【0055】
本発明による使用に関し、ここに記載された化合物またはその生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体は、該化合物またはその生理的に許容可能な塩、エステルもしくは生理的機能性誘導体と共に、1以上の薬学的に許容可能な担体および任意に他の治療的および/または予防的成分を含む医薬製剤でありうる。担体は、製剤の他の成分に適合するという意味で許容可能でなくてはならず、服用者に有害であってはならない。
【0056】
医薬製剤は、経口、局所(経皮、バッカルおよび舌下を含む)、直腸、または非経口的(皮下、内皮内、筋肉内および静脈内を含む)、経鼻的および肺内投与(例えば、吸入による)に適するものを含む。製剤は、必要に応じて、別個の投与量単位として好都合に存在し得、薬学の当該技術において周知のあらゆる方法により調製されうる。全ての方法には、活性化合物を液体担体や細かく分割された固体担体あるいはその両方と組み合わせる状態にする工程を含み、次いで、所望により、生成物を所望の剤型に形成する。
【0057】
経口投与に適する医薬製剤は、担体が固体であり、最も好ましくは、巨丸剤(ボーラス)、カプセルや錠剤などの、それぞれに活性化合物の所定量を含む単位用量製剤である。錠剤は、場合により、1以上の副成分とともに、圧縮や成型によって製造されてもよい。圧縮錠剤は、粉末や顆粒などの自由流動形態の活性化合物を、場合により、結合剤、滑沢剤、不活性希釈剤、潤滑剤、表面活性化物質または分散剤と混合し、適する機器で圧縮することにより調製されうる。成型錠剤は、活性化合物を不活性な液体希釈剤で成型することにより製造されうる。錠剤は、場合により被覆されてもよく、被覆されていない場合には、場合により割線を入れても良い。カプセル剤は、活性化合物を、単独あるいは1以上の副成分との混合剤として、カプセル殻に充填し、次いで常法により封止することにより調製しうる。サッシェ剤は、カプセル剤に類似し、活性化合物を副成分とともにライスペーパー製の封筒に入れ封止する。活性化合物は、分散可能な顆粒として製剤化してもよく、例えば、投与前に水に懸濁してもよく、食品に散布してもよい。顆粒剤は、例えば、サッシェに包装してもよい。担体が液体である経口投与に適する製剤は、水溶性もしくは非水溶性の溶液または懸濁液として、あるいは水中油型液体エマルジョンとして存在しうる。
【0058】
放出制御投与形態を含む経口投与製剤は、例えば、活性化合物が適した放出制御マトリックスで製剤化され、あるいは、適する放出制御フィルムで被覆された錠剤を含む。そのような製剤は、特に予防的使用に好都合となりうる。
【0059】
直腸投与に適する担体が固体である医薬製剤は、最も好ましくは、単位用量の坐薬である。適する担体は、カカオ脂や技術的に通常用いられる他の物質を含む。坐薬は、活性化合物を柔らかい、あるいは溶けた担体と混合し、次いで冷却し、成型することにより簡便に形成しうる。
【0060】
非経口的投与に適する医薬製剤は、無菌の溶液、または、水溶性もしくは油性の担体中の活性化合物の懸濁液を含む。
【0061】
注射用製剤は、急速注入や連続的な注入に用いられる。そのような製剤は、使用に必要とされるまで製剤を導入後封止した、単位用量または多用量の容器で好都合に存在する。あるいは、活性化合物は、使用前に、無菌の発熱性物質の存在しない水などの適する担体と構成される粉末形態であってもよい。
【0062】
活性化合物は、長時間作用性デポ製剤として製剤化されてもよく、例えば、皮下投与や筋肉内投与などの、筋肉内注射や埋め込みにより投与されうる。デポ製剤は、例えば、適する重合性や疎水性の物質、またはイオン交換樹脂を含んでもよい。そのような長時間作用性の製剤は、特に予防的使用に好都合となりうる。
【0063】
口腔を介した肺内投与に適する製剤は、活性化合物を含み、好ましくは0.5〜7ミクロンの範囲の直径を有し、服用者の気管支樹に到達するような粒子である。
そのような製剤の1つの可能性としては、吸入器で使用される穴の開けられるカプセル(好ましくは、例えば、ゼラチン)で、あるいは、活性化合物と、適する液体または気体噴射剤、および、場合により界面活性剤および/または固体希釈剤などの他の成分を含む自己噴射性製剤として用いられる、細かく粉砕した粉末の形態である。適する液体噴射剤は、プロパンおよびクロロフルオロカーボン類を含み、適する気体噴射剤は、二酸化炭素を含む。活性化合物が、溶液または懸濁液の溶滴の形態で分散している自己噴射性製剤も、使用されうる。
【0064】
そのような自己噴射性製剤は、当該技術において公知のものの類似物であり、確立された手順により調製しうる。好ましくは、所望のスプレー特性を有する手動で操作可能な機能性バルブ、または、有利には、各操作において、例えば、25〜100マイクロリットルの固定容量を送達する計量型のバルブ、を備える容器を用いる。
【0065】
更なる可能性として、吸入の微滴霧を作り出すのに、加速気流や超音波撹拌が用いられる噴霧器やネブライザーでの使用に、活性化合物を溶液や懸濁液の状態で用いることができる。
【0066】
経鼻投与に適する製剤は、一般的に上記に記載された肺内投与と類似の調製法を含む。分散する場合、そのような製剤は、鼻腔中に保持可能に、好ましくは、10〜200ミクロンの範囲の粒子直径を有するべきであり、必要に応じて、適する粒径の粉末の使用や適切なバルブの選択により、達成されうる。他の適する製剤は、鼻および鼻腔に近づけ保持された容器から、鼻腔を通じて、水溶性もしくは油性溶液または懸濁液中に0.2〜5%w/vの活性化合物を含む滴を、高速吸入により投与するために、20〜500ミクロンの範囲の粒子径を有する粗粉末である。
【0067】
前述の担体成分に加え、上記に記載された医薬製剤は、希釈剤、緩衝剤、香味剤、結合剤、界面活性剤、増粘剤、滑沢剤、保存料(抗酸化剤を含む)等の、適当な1以上の追加的な担体成分、および製剤を対象とする服用者の血液と等張にする目的で加えられる物質を含んでもよい。
【0068】
薬学的に許容可能な担体は、当業者に周知であり、これらに限定されないが、0.1M、好ましくは0.05Mリン酸バッファまたは0.8%生理食塩水を含む。また、そのような薬学的に許容可能な担体は、水溶性もしくは非水溶性溶液、懸濁液、およびエマルジョンであってもよい。非水溶性溶媒の例には、プロピレングリコール、ポリエチレングリコール、オリーブ油などの植物油、およびエチルオレイン酸塩などの注射可能な有機エステル類がある。水溶性担体は、水、アルコール/水溶性溶液、生理食塩水や緩衝培地を含むエマルジョンまたは懸濁液を含む。非経口的担体は、塩化ナトリウム溶液、リンゲルデキストロース液、デキストロースおよび塩化ナトリウム、乳酸化されたリンゲル液または固定油を含む。例えば、抗菌剤、抗酸化剤、キレート化剤、不活性ガスなどの、保存料および他の添加物も存在してもよい。
【0069】
局所製剤に適する製剤は、例えば、ゲル、クリームまたは軟膏として供されうる。そのような製剤は、例えば、創傷や潰瘍に適用でき、創傷や潰瘍の表面上に直接塗布したり、治療する箇所および部分一面に適用可能な、包帯、ガーゼ、メッシュなどの適するサポートに保持してもよい。
例えば、創傷や潰瘍に、治療する箇所に直接スプレーや散布しうる液体や粉末製剤も提供されうる。また、製剤を包帯、ガーゼ、メッシュなどの担体にスプレーでき、あるいは散布し、治療する箇所に適用することもできる。
【0070】
動物用の治療用製剤は、粉末や液体の濃縮形態が好都合であろう。動物用製剤の手技標準により、ラクトースやスクロースなどの従来の水溶性添加剤は、その物性を向上させるために加えられうる。このように、特に適する本発明の粉末は、活性成分を50〜100%w/w、好ましくは60〜80%w/w含み、従来の動物用添加剤を0〜50%w/w、好ましくは20〜40%w/w含む。これらの粉末は、例えば、予め混ぜた中間体を経由し、動物用飼料に加えたり、動物用飲料水で希釈してもよい。
【0071】
本発明の液体濃縮物は、化合物や誘導体またはその塩を好適に含み、場合により、獣医学的に許容可能な水相溶性溶媒、例えばポリエチレングリコール、プロピレングリコール、グリセロール、グリセロールホルマールや30%v/vまでのエタノールと混合したような溶媒などを含んでも良い。液体濃縮物は、動物用飲料水に投与してもよい。
【0072】
本発明を、以下の図面、限定されない実施例を参照して詳細に説明する。
以下の図面を参照して本発明を詳細に説明する。
【図面の簡単な説明】
【0073】
【図1】化合物JJ91を種々の量で処理したT22 RGC−Fos−lacZ細胞におけるp53−依存性転写誘導の量を示すグラフである。
【図2】ウェスタンブロット画像を示す。
【図3】FACS分析グラフである。
【図4】(A)および(B)は、ウェスタンブロット画像を示す。
【図5】マウスへの化合物投与の結果を示すグラフである。
【図6】BL2バーキットリンパ腫細胞の生存に対する種々の濃度のJJ91の効果を示すグラフである。
【図7】SCIDマウスにおけるBL2バーキットリンパ腫異種移植腫瘍に対するJJ91の効果を示すグラフである。
【図8】SirT1活性に対するJH164の抑制効果を示すグラフである。
【発明を実施するための形態】
【0074】
詳細な説明
化合物合成
ここに記載された化合物は、手法1および2を参照し、以下の方法に従って準備された。
【0075】
手法1を参照し、本発明に記載されたN−ベンゾイルチオ尿素類は、種々のベンゾイルクロライド類をアセトン溶媒中、チオシアン酸ナトリウムで処理し、そして所望の化合物を得るためのアニリン類とin situで反応させ、ベンゾイルイソチオシアネート類を得ることにより、合成した。
【0076】
【化6】
【0077】
さらに、アシル化またはアルキル化によって機能付与を行った。
【0078】
具体例として、化合物(JH129)の合成を以下のように行った。
アセトン(20mL)中4−tert−ブチルベンゾイルクロライド(10mmol、1.97g)の撹拌溶液に、アルゴン雰囲気下で、チオシアン酸ナトリウム(10mol、0.81g)を加えた。2時間後、この混合物を、0℃に冷却されたアルゴン下で、アセトン(50mL)中1,4−フェニレンジアミン(20mmol、2.16g)溶液に滴下した。周囲温度まで加温後、反応混合物を36時間攪拌した。混合物を減圧下濃縮し、残渣をジクロロメタン中にとり、濾過し、濾液を濃縮し、シリカゲルカラムでクロマトグラフィにかけ、酢酸エチル−石油エーテル混液で溶出した。生じた固体をジエチルエーテルで粉砕し、分析的に純粋な物質2.45g(75%)を得た。分析により以下のデータを得た:mpt 189−191℃;1H−NMR(CDCl3)δ1.36(s,9H),6.81(m,2H),7.47(m,2H),7.54(m,2H),7.81(m,2H),9.05(s,1H),12.41(s,1H);MS(ES+)m/z350[M+Na]+;C18H21N3ONaSの計算値350.1300,実測値350.1303.
【0079】
さらなる具体例として、以下のようにアシル化によって、化合物JH129をさらに機能付与した。
ジクロロメタン(1mL)中、JH129(0.2mmol、65mg)の撹拌溶液に、アルゴン雰囲気下で、5−ブロモプロパノイルクロライド(ジクロロメタン0.2mL中0.2mmol)の溶液を加えた。生じた懸濁液に、トリエチルアミン(0.2mmol、27μL)を加えた。反応混合物を90分間攪拌し、ジクロロメタン(5mL)で希釈し、1MのHCl、2MのNaOHおよび飽和NaCl溶液で洗浄した。有機層を乾燥(MgSO4)し、灰白色固体になるまで濃縮した。酢酸エチルから再結晶し、分析的に純粋な物質64mg(65%)を得た。分析により以下のデータを得た:mpt 152−153℃;1H−NMR(CDCl3)δ1.36(s,9H),1.92(m,4H),2.42(t,2H),3.46(t,2H),7.22(s,1H),7.57(m,4H),7.68(m,2H),7.82(m,2H),9.04(s,1H),12.60(s,1H);MS(ES+)m/z512,514[M+Na]+;C23H2879BrN3O2NaSの計算値512.0983,実測値512.0995.
【0080】
更なる具体例として、化合物JH164HClは、式(I)において、R1基が、水溶性基に活性化合物を結合するためのリンカー基であると考えられ得る具体的化合物である。JH164の合成およびその塩酸塩の形成は、次のようになされた。
ジクロロメタン(10mL)中、JH140(0.1mmol、50mg)の溶液に、ジメチルアミン水溶液(40重量%の2mL)を加えた。二相性の混合物を20時間攪拌した。有機層を分離し、乾燥(MgSO4)し、エバポレートして、乾固するまで濃縮した。残渣をアセトンに溶解し、この溶液をHCl蒸気に曝した。生じたHCl塩を濾過し、純白色固体、33mg(67%)を単離した。分析により以下のデータを得た:mpt 205−206℃;1H−NMR(D6−DMSO)δ1.32(s,9H),1.64(brs,4H),2.39(t,2H),2.75(s,6H),3.06(t,2H),7.60(m,6H),7.94(m,2H),9.64(brs,<1H),10.11(s,1H),11.45(s,1H),12.60(s,1H);MS(ES+)m/z 455[M−Cl]+;C25H35N4O2Sの計算値455.2481,実測値455.2477;分析 C25H35ClN4O2Sの計算値:C,61.14;H,7.18;N,11.41%.実測値:C,60.75;H,7.46;N,11.30%.
【0081】
生物学的評価
in vitro実験:
材料と方法:
ケムブリッジケミカルス(ChemBridge Corporation、16981 Via Tazon、Suite G、サンディエゴ、CA 92127)から得た30,000個の化合物ライブラリー(DIVERSetTM)を、p53腫瘍抑制剤機能の活性化剤についてスクリーニングした。
1次および2次スクリーニングを、以下の細胞ベースのアッセイを用いて行った。
【0082】
1.1次スクリーニング
Lu X、Burbidge SA、Griffin SおよびSmith HMによりOncogene.,13(2):413−8(1996年7月18日)に記載された、p53−依存性プロモーター支配下でベータ−ガラクトシダーゼを発現するT22 RGC−ΔFos−lacZ細胞を用いた。
・セレクションなしのDMEM90μl、10%FCSおよびゲンタマイシン1mg/mlを有する96ウェル組織培養プレートに1ウェル当たり、1x104個の低継代T22細胞を播種する。
・細胞播種48時間後に化合物を加える。DMSOは、培地中最終濃度1:100を超えない。非処理の対照と、アクチノマイシンD5ng/mlで処理したポジティブコントロールを使用する。総量=100μl
・18時間後、96ウェルプレートから培地を除き、1ウェルあたり50μlの1倍溶解バッファ(プロメガ)を加える。
・室温で1時間振盪する(すぐに使えるようになるまでプレートを−80℃で冷凍できる)
・1ウェルあたり150μlのCPRG反応混合物を加える。
CPRG反応混合物15mlの調製
[15mlの0.1Mのリン酸バッファ、pH7.5
300μlのCPRG 4mg/ml(ベーリンガー・マンハイム)
80μl(0.1MのMgCl2/0.1M β−メルカプトエタノール)]
・37℃で4時間湿室中でインキュベートする。色が黄色から桃色に変わると、p53活性を示す。
・各ウェルから新たな96ウェルプレートに100μlを移す。これは、細胞壊死組織片が、吸光度の読み取りに干渉するのを防止する。プレートリーダーを用いて570nMでの吸光度を測定する。
・可溶化液を一晩4℃で放置し、次いで、再び吸光度を測定する。
【0083】
2.FACS分析
Oncogene.,18(51):7378−86(1999年12月2日)に、Smart P,Lane EB,Lane DP,Midgley C,Vojtesek B,Lain Sにより開示されている神経芽細胞腫細胞株SKNSH−CMVNeo(p53機能あり)およびSKNSH−DDp53(不活化p53)を用いた。
1日目
・6ウェルプレートの1ウェルあたり50,000個の細胞をDMEM−10% FCSで播種する。
2日目
・薬剤を細胞に加える。
4日目
・ブロモデオキシウリジン(BrdU)を30μMになるまで加え、20分間細胞をインキュベートする。
・細胞から培地を除去し、13mlのファルコンチューブに移す。PBSで細胞をすすぎ、これも移す。細胞をトリプシン処理し、チューブに移し、次いで最後にPBSで再度すすぐ。一旦チューブに全てを移し、1500rpmで5分、細胞をペレット化する。
・1mlのPBSに細胞を再懸濁し、ボルテックスする間、3mlのエタノールに流滴を加える。4℃で最短1〜2時間(上限なし)インキュベートする。
5日目
・2,500rpmで5分間の遠心分離によりペレット化し、上澄みを捨てる。
30mMのHCl(pH1.5)中1mg/mlの濃度で、1チューブあたりペプシン溶液2mlを新たに調製し、37℃に予め温める。
・予熱したペプシン溶液2mlを各チューブに加え、37℃で30分間混合する。
・2,500rpmで5分間の遠心分離によりペレット化し、上澄みを捨てる(ペレットは、透明になろう)。
・室温で15−20分間1mlの2M HClを加える(貯蔵瓶は、11.6M)。タイミングが重要であり、長時間のインキュベートは、ブロードなDNAピークとなる。PBSを追加し、前記と同様にペレット化する。
・PBSで再度次いで抗体緩衝液で一度洗浄し、毎回細胞をペレット化する。
抗体緩衝剤:PBS、0.5%のBSA、0.5%のTween−20
・抗体緩衝剤中1:50に希釈されたベクトン・ディッキンソン抗BrdU抗体の200μL中にペレットを再懸濁する。室温で1時間インキュベートする。
・PBSで洗浄し、細胞をペレット化する。
・抗体緩衝剤中1:64に希釈されたシグマFITC抗体(#3008)の200μL中にペレットを再懸濁する。抗体の消失を予防するために暗所下、室温で30分間インキュベートする。
・PBSで洗浄し、細胞をペレット化する。
・25μg/mlのヨウ化プロピジウム対比染色液を含む500μLのPBS中に最終的なペレットを再懸濁する。FACScanで分析にかけるまで暗所下氷冷する。
・FACScanによるDNA含量の測定(ヨウ化プロピジウム蛍光)およびDNA合成(BrdU取り込み)
【0084】
3.ウエスタンブロット法
・6ウェルプレートの1ウェルあたり、2x105個のMCF−7細胞を播種する。
・24〜36時間インキュベーション後、細胞に薬剤を加え、必要な時間インキュベートする。
・プレートの培地を捨て、PBSで洗浄する。PBSの残りを吸引し100μlの1倍LDS添加緩衝剤(インビトロジェン)をプレートに直接加える。プレートの表面を片隅へかき取り、細胞/LDSをピペットでチューブに取る。
・試料を90℃まで5分間加熱し、次いで各15秒間を2回超音波処理する。最大速で5分間遠心分離し、必要時まで氷冷する。全ての試料のタンパク質濃度を測定(ピアスBCAキット)し、濃度を均等化する。各試料に1:10DTTを加える。
・試料を4〜12%ノベックス・ゲルに添加し、MOPSバッファで1度流し、メーカーの使用説明書(インビトロジェン)に従い、PVDF膜に移す。
・膜をブロックし、標準手順を用い1次、次いで2次抗体でインキュベートし、検出にアマシャムECLを用いた。
・関連する1次抗体は、抗p53DO1マウスモノクローナル抗体、抗p53ホスホセリン−15(サンタクルズ)、抗p21 118マウスモノクローナル抗体を含む。負荷対照としてアクチン検出を用いた。
【0085】
水溶性分析の一般手順
化合物の相対的水溶性は、UVスペクトルに基づく方法を使用して求めた。各化合物の吸光係数の生成は、分光学的グレードのアセトニトリル(100mL)中に1mgの化合物(一部の化合物は、最初に最小量のDMSOに溶解することを要する)を溶解することにより行われる。次いで6x2倍の連続希釈を行い、合計7溶液を得る。これらの溶液をUVスペクトルで分析し、適当な波長における吸光度をモル濃度に対しプロットした。吸光係数を最適な線勾配から算出した。次いで吸光係数を、40mMのDMSOストックと100μMの化合物の水溶液の実際の濃度を計算するのに用いた。
40mMのDMSOストックと100μMの化合物の水溶液の実際の濃度は、次のように計算した。1mgの化合物を必要量のDMSOに溶解し、40mMストックを得た。次いで、不溶の化合物を遠心分離でペレット化した。上澄み(2μL)をアセトニトリルで4mLに希釈し、理論上2x10-5Mの溶液を作った。次いで溶液をUVスペクトルで分析した。
100μM水溶液の実際の濃度は、40mMDMSO溶液のアリコート(4.2μL)を1mLになるまで水で希釈することにより求めた。この溶液のアリコート(60μL)を水(40μL)に加え、不溶の化合物を遠心分離によりペレット化した。上澄み(80μL)をアセトニトリルで4mLになるまで希釈し、理論上2x10-6Mの溶液を作った。次いでこの溶液をUVスペクトルで分析した。
次いで水における実験的に測定した水溶性を、理論値(パーセンテージとして)と比較し、テノビン1に対する相対的水溶性値を得た。
【0086】
in vitro脱アセチル化アッセイ
酵素的SirTlアッセイを、Fluor de Lys蛍光アッセイシステム(バイオモルキットAK555)で精製成分を用い、以下のメーカーの使用説明書に従い行った。FdL基質を7*MおよびNAD+1mMで用いた。化合物をDMSO中、反応において0.25%より小さい最終DSMO濃度で可溶化した。1反応あたり酵素1単位を用いた。反応は、37℃で1時間行った。
【0087】
結果
図1に示される結果は、JJ91はp53転写因子機能を活性化することを示す。T22 RGC−ΔFos−lacZ細胞をJJ91の指示量で16時間処理した。p53−依存性転写の誘導率を測定した
図2に示される結果は、JJ91がp53を活性化して、選択的に神経芽細胞腫細胞を死滅させることを示す。SKNSH−CMVNeoおよびSKNSH−DDp53細胞は、非処理にしたまま、あるいは10μMのJJ91で48時間処理した。細胞をFACS分析で分析した。JJ91は、明らかにDNA合成と細胞死例を低下させた(sub−G1/G0細胞の数を増加)。これらの効果は、非活性化p53を伴うSKNSH細胞においては、見られない。
図3に示される結果は、JJ91がp53レベルを増加させることを示す。MCF−7細胞を、DNA傷害物質であるマイトマイシン C(10μM)、非遺伝毒性物質nutlin−3(6μM)やJJ91(10μM)で指示時間処理した。JJ91は、nutlin−3と同様の効果を有する。P53レベルは、急速に増加する。p53ホスホセリン−15のレベルは、遺伝毒性物質であるマイトマイシンCで見られたほど高くはない。アクチンを増加させるp53の下流の標的p21のレベルは、負荷対照で分析された。
図4Aおよび4Bに示される結果は、p53レベルにおけるJJ91類縁物質の効果を示す。
図4A:MCF−7細胞を4時間(レーン2から9)または6時間(レーン11から15)、10μMのマイトマイシンC(レーン2)、JJ91(レーン3および11)、JH118(レーン4)、JH129(レーン5)、JH132(レーン6)、JH140(レーン7および12)、JH141(レーン8)および4−アミノアセトアニリド(レーン9)、JH151(レーン13)、JH156(レーン14)および5406085(レーン15)で処理した。レーン2とレーン10において、細胞を非処理とした。細胞抽出液をp53、ホスホセリン−15 p53、p21およびアクチンに対する抗体で、ウエスタンブロット法により分析した。
図4B:MCF−7細胞を、4時間(レーン2から6)6μMのnutlin−3(レーン2)、10μMのJJ91(レーン3)、10μMの7322366(レーン4)、40nMのレプトマイシンB(レーン5)および20μMのMG132 JH129で処理した。細胞抽出液を、p53、アクチン、p21、病原性毒素およびmdm2に対する抗体でウエスタンブロット法により分析した。
【0088】
表1は、活性化合物の構造、および化合物JJ91活性を100%とし、示された化合物によるT22 RGC−ΔFos−lacZ細胞におけるp53−依存性転写に関する活性レベルを示す。
【0089】
【表1】
【0090】
【表2】
【0091】
in vitroおよびin vivo実験
図5に関し、JJ91をマウスに5mg/kgの用量で投与した。◆と□は、それぞれi.p.とp.o.の投与ルートに対応する(挿入図は、データの対数プロットを示す。)。血中濃度は、上記のLC−MS/MSで示される時点において測定し、示される値は、3つの測定値の±SDを意味する。結果は、化合物の腹膜内注射が、血中でマイクロモル濃度に達し、顕著な体重や行動変化を起こさず、約1.3時間の半減期を有することを示す。
図6に関し、BL2バーキットリンパ腫細胞を、1μM〜10μMの範囲の指示濃度のJJ91(70%シクロデキストリンに溶解)で2時間処理した。このとき、生存細胞の数をカウントした。この短い暴露のあと、細胞を洗浄し、化合物を除去した。この処理は、6日間毎日繰り返した。実験を三回実施し、標準偏差を示した。図6に示されるように化合物への6回の短い暴露のあと、BL2細胞の生存は、大きく減少した。
図7に関し、SCIDマウスにおけるBL2バーキットリンパ腫異種移植腫瘍を腫瘍が触知できるまで7日間樹立した。このとき、ビヒクル(70%シクロデキストリン)(上段パネル)およびJJ91(92mg/kg)(下段パネル)を毎日腹膜内注射で投与した。腫瘍サイズを、注射した日(1日目)および注射後4、8および11日目で測定した。示されるように、JJ91は、SCIDマウスにおけるBL2異種移植腫瘍の成長を低下させる。
SirT1活性を、メーカーにより特定されるバイオモルのFluor de Lys SirT1蛍光活性アッセイ(カタログ番号AK−555)を用いて評価した。反応は、1ミリモルのNAD+、7マイクロモルのFluor de Lys基質および過量のJH164を含んだ。脱アセチル化と展開液反応を37℃で1時間行った。JH164のIC50は、23.5マイクロモルである。図8はSirT1に対するJH164の抑制効果を示す。
上記態様は、本発明の代表的なものであり、請求の範囲に定義された発明の範囲を制限すると解釈されるものではない。
【0092】
更なる化合物は、初期の化合物JJ91(以後、テノビン1と称する)に基づき、合成される。
【0093】
【化7】
【0094】
調製された追加の化合物は、4つのカテゴリーに分類される。
カテゴリー1:水溶性類縁物質
a)テノビン6(JH164):中間体JHl40から調製
JH140は、可逆的にp53を活性化するこれまでに調製された他の化合物全てとは異なり、非可逆的p53活性化を示すことから、潜在的に興味深い。その合成は、テノビン6の手順中、以下に説明される。
化合物1 上記
化合物5〜8 上記
【0095】
下記手法1で示されるこれらの化合物の実験的合成
【化8】
【0096】
b
手法1:a テノビン6および類縁物質1、5〜7の合成
試薬と条件:(i)SOCl2、CH2Cl2、室温16時間(96%)。(ii)NaSCN、アセトン、室温、8時間、次いで1,4−フェニレンジアミン、16時間(58%)。(iii)酸塩化物、NEt3、DCM、室温。(iv)40% HNMe2水溶液、DCM/H2O、室温、24時間。(v)2M HCl(ジエチルエーテル中)。
【0097】
テノビン1(JJ91):N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アセトアミドの合成
アセトン(50mL)中、3(J.Am.Chem.Soc.1985、207、898に記載の2から調製)の溶液(6.53g、33mmol、1当量)に、チオシアン酸ナトリウム(2.69g、33mmol、1当量)を加えた。クリーム色の懸濁液を室温で16時間攪拌し、次いで、アセトン(50mL)中、p−アミノアセトアニリドの溶液(4.95g、33mmol、1当量)を加えた。黄色懸濁液を室温で16時間攪拌し、溶媒を減圧下濃縮し、淡橙色残渣を得、それをCH2Cl2中に再懸濁し、不溶性固体を濾過した。濾液を減圧下濃縮し、淡橙色固体を得、それを酢酸エチルから再結晶することにより精製し、テノビン1(7.49g、61%)を淡黄色固体として得た。
1H−NMR(アセトン−d6,300MHz)δ1.36(s,9H,(CH3)3),2.09(s,3H,CH3),7.63(d,2H,J=8.7Hz,ArH),7.71(s,6H,ArH),8.03(d,2H,J=8.7Hz,ArH),9.26(s(br),1H,NH),10.12(s(br),1H,NH),12.80(s(br),1H,NH).
13C−NMR(アセトン−d6,100MHz)δ23.36,29.70,30.40,34.83,118.95,124.14,125.71,128.15,129.36,133.31,137.82,157.00,167.97,178.73.
【0098】
中間体化合物1:(4−アミノ−フェニル)−3−(4−tert−ブチル−ベンゾイル)−チオ尿素(4)の合成
アセトン(30mL)中、3(J.Am.Chem.Soc.1985、207、898に記載の2から調製)の溶液(4g、20.4mmol、1当量)に、チオシアン酸ナトリウム(1.68g、20.8mmol、1.02当量)を加えた。生じた淡黄色懸濁液を室温で8時間攪拌し、次いで、0℃でアセトン(30mL)中1,4−フェニレンジアミン溶液(4.41g、40.8mmol、2当量)を加えた。
褐色懸濁液を室温まで加温し、16時間撹拌した。溶媒を減圧下濃縮し、残った残渣をCH2Cl2に再懸濁し、不溶性固体を濾過した。濾液を減圧下濃縮し、褐色固体を得、シリカのカラムクロマトグラフィにより精製(50%酢酸エチル/石油エーテル)し、続いて酢酸エチルから再結晶により4(4.42g、58%)を淡黄色固体として得た。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3,3.75(s(br),2H,NH2),6.71(d,2H,J=11.8Hz,ArH),7.49(dd,2H,J=8.6,33.4Hz,ArH),7.81(d,2H,J=10.8Hz,ArH),7.68(dd,4H,J=8.6,81.3Hz,ArH),9.02(s(br),1H,NH),12.37(s(br),1H,NH).
13C−NMR(CDCl3,100MHz)δ31.1,35.0,115.1,125.8,126.2,127.4,128.7,128.8,145.4,157.7,166.8,178.5.
【0099】
中間体JHl40からのテノビン6(JHl64):5−ジメチルアミノ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩の合成
CH2Cl2(110mL)中、チオ尿素4(6g、18.3mmol、1当量)をN2雰囲気下で攪拌した。5−ブロムワレリルクロライド(3.65g、18.3mmol、1当量)、続いてトリエチルアミン(2.51mL、18.3mmol、1当量)を加えた。ベージュ色の懸濁液を室温で4時間攪拌し、CH2Cl2(50mL)で希釈した。有機溶液を1Mの塩酸水溶液(1x50mL)、10%水酸化ナトリウム水溶液(1x50mL)、および飽和塩水(1x50mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた褐色泡をシリカのフラッシュクロマトグラフィで精製(50%酢酸エチル/石油エーテル)し、5−ブロモ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(中間体JH140)(6.76g、77%)を黄色粉末として得た。
【0100】
【化9】
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),1.93(m,4H,(CH2)2),2.42(t,1H,J=6.9Hz,COCH2),1H−NMR(300MHz)ppm 3.45(t,1H,J=6.3Hz,CH2Br),7.22(s(br),1H,NH),7.57(m,4H,ArH),7.68(d,2H,J=8.9Hz),7.82(m,2H,ArH),9.05(s(br),1H,NH),12.60(s(br),1H,NH).
13C−NMR(CDCl3,100MHz)δ24.6,27.7,31.1,32.5,33.6,37.4,119.9,124.9,126.3,127.4,128.6,133.6,157.9,166.9,170.9,178.4.
【0101】
上記ブロモ化合物(中間体JH140)(2.36g、4.80mmol)のH2O(75mL)中のジメチルアミン(30mL)との処理により、5−ジメチルアミノ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを含む粗混合物を得た。この混合物を最小量のアセトンに溶解し、黄色沈殿が生じるまで、エーテル溶液中、2M塩酸を滴下した。固体を濾過し、粗混合物を得、エタノールから再結晶することにより精製し、テノビン6(1.4g、59%)を黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.33(s,9H,(CH3)3),1.66(m,4H,(CH2)2),2.40(t,2H,J=6.1Hz,COCH2),2.75(s,6H,N(CH3)2),3.06(m,2H,CH2NH(CH3)2),7.59(m,6H,ArH),7.95(d,2H,J=8.4Hz,ArH),9.77(s(br),1H,NH(CH3)2),10.13(s(br),1H,NH),11.43(s(br),1H,NH),12.61(s(br),1H,NH).
13C−NMR(DMSO−d6,75MHz)δ22.4,23.8,31.3,35.3,35.9,42.5,56.7,119.5,125.3,125.8,129.1,129.7,133.3,137.9,156.8,168.5,171.2,179.4.
HRMS実測値455.2477(C17H39N6O4S2の計算値455.2474).
【0102】
化合物1:5−モルホリン−4−イル−ペンタン酸 {4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩(1)の合成
アセトニトリル(4mL)中、5−ブロモ−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(中間体JH140、テノビン6に記載の通り調製)(80mg、0.16mmol)の溶液に、モルホリン(0.4mL)およびヨウ化カリウム(触媒量)を加えた。反応混合物を40分間加熱還流し、室温まで冷却し、減圧下濃縮した。残った残渣を、75%飽和炭酸水素ナトリウム水溶液とCH2Cl2間で分配した。有機層をMgSO4で乾燥し、減圧下濃縮し、ベージュ色の粗固体を得た。EtOAcから再結晶により精製し、5−モルホリン−4−イル−ペンタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを得、蒸気拡散により塩酸塩に変換した。25%CDCl3/ジエチルエーテル中ジメチルアミンの溶液を、塩化水素に曝し、1を白色沈殿として得、濾過により集めた。
HRMS実測値497.2585(C27H37N4O3Sの計算値.497.2586).
【0103】
化合物5:N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−4−ジメチルアミノ−ブチルアミド塩酸塩(5)の合成
チオ尿素4(200mg、0.61mmol、1当量)をCH2Cl2(10mL)中、N2雰囲気下で攪拌した。4−ブロモブチリルクロライド(169mg、0.92mmol、1.5当量)、続いてトリエチルアミン(85μL、0.61mmol、1当量)を加えた。橙色溶液を室温で1.5時間攪拌し、CH2Cl2(10mL)で希釈した。有機溶液を1M塩酸水溶液(1x10mL)、10%水酸化ナトリウム水溶液(1x50mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた淡黄色泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィにより精製し、4−ブロモ−N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−ブチルアミド(138mg、48%)を黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.35(s,9H,(CH3)3),2.26(m,2H,CH2CH2Br),2.57(t,2H,J=7.0Hz,CH2),3.52(t,3H,J=6.2Hz,CH2),7.60(m,6H,ArH),7.82(d,2H,J=8.7Hz,ArH),9.08(s(br),1H,NH),12.59(s(br),1H,NH).
【0104】
前記ブロモ化合物(138mg、0.29mmol)を、H2O(7.5mL)中、ジメチルアミン(3mL)で処理し、N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−4−ジメチルアミノ−ブチルアミドを含有する粗混合物を得た。この混合物を最小量のアセトンに溶解し、エーテル溶液中、2M塩酸を沈殿が生じるまで滴下した。固体を濾過し、濾液を減圧下濃縮した。生じた黄色オイルをヘキサンで研和し、黄色固体を得、イソプロパノールから再結晶し、5(75mg、55%)を粘性黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.32(s,9H,(CH3)3),1.95(m,2H,CH2),2.44(t,2H,J=7.3Hz,CH2),2.79(d,6H,J=4.8Hz,N(CH3)2),3.09(m,2H,CH2),7.62(m,6H,ArH),7.95(d,2H,J=8.5Hz,ArH),9.64(s(br),1H,NH(CH3)2),10.17(s(br),1H,NH),11.44(s(br),1H,NH),12.61(s(br),1H,NH).
【0105】
化合物6:6−ジメチルアミノ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩(6)の合成
チオ尿素4(200mg、0.61mmol、1当量)をCH2Cl2(10mL)中、N2雰囲気下で攪拌した。6−ブロモヘキサノイルクロリド(195.8mg、0.92mmol、1.5当量)、続いてトリエチルアミン(85μL、0.61mmol、1当量)を加えた。
ベージュ色の懸濁液を室温で1.5時間攪拌し、CH2Cl2(10mL)で希釈した。有機溶液を1M塩酸水溶液(1x10mL)、10%水酸化ナトリウム水溶液(1x50mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた褐色の泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィにより精製し、6−ブロモ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(166mg、55%)を黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.35(s,9H,(CH3)3),1.51(m,2H,CH2),1.73(m,2H,CH2),1.89(m,2H,CH2),2.36(t,2H,J=7.4Hz,COCH2),3.40(t,2H,J=6.7Hz,CH2Br),7.57(m,7H,ArH,NH),9.10(s(br),1H,NH),12.58(s(br),1H,NH).
【0106】
前記ブロモ化合物(136mg、0.27mmol)をH2O(6.25mL)中、ジメチルアミン(2.5mL)で処理することにより、6−ジメチルアミノ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを含有する粗混合物を得た。この混合物を最小量のCH2Cl2に溶解し、エーテル溶液中、2M塩酸を、クリーム色の沈殿が生じるまで滴下した。固体を濾過し、濾液を減圧下濃縮した。生じた黄色オイルをヘキサンで研和し、黄色固体を得、イソプロパノールから再結晶し、6(80mg、59%)を粘性黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.32(s,9H,(CH3)3),1.64(m,4H,(CH2)2),2.35(t,2H,J=7.1Hz,CH2),2.73(d,6H,J=4.8Hz,N(CH3)2),3.01(m,2H,CH2),7.59(m,6H,ArH),7.94(d,2H,J=8.4Hz,ArH),9.83(s(br),1H,NH(CH3)2),10.08(s,1H,NH),11.42(s,1H,NH),12.59(s,1H,NH).
【0107】
化合物7:8−ジメチルアミノ−オクタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド塩酸塩(7)の合成
8−ブロモオクタン酸(2g、8.90mmol、1当量)をCH2Cl2(15mL)中、N2雰囲気で攪拌し、塩化チオニル(784μL、10.68mmol、1.2当量)、続いて、DMF(数滴)を加えた。溶液を16時間攪拌し、減圧下濃縮して8−ブロモオクタノイルクロライド(2.04g、95%)を黄色オイルとして得、さらなる精製をせずに用いた。チオ尿素4(600mg、1.84mmol、1当量)を、CH2Cl2(15mL)中、N2雰囲気下で攪拌した。8−ブロモオクタノイルクロライド(532mg、2.02mmol、1.2当量)、続いてトリエチルアミン(256μL、1.84mmol、1当量)を加えた。ベージュ色の懸濁液を室温で16時間攪拌し、CH2Cl2(20mL)で希釈した。有機溶液を1M塩酸水溶液(1x20mL)、10%水酸化ナトリウム水溶液(1x20mL)、および飽和塩水(1x20mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮した。生じた黄色の泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィで精製し、8−ブロモ−オクタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミド(579mg、59%)を黄色オイルとして得た。
1H−NMR(CDCl3,400MHz)δ1.36(s,9H,(CH3)3),1.44(m,6H,(CH2)3),1.74(m,2H,CH2),1.86(m,2H,CH2),2.37(t,2H,J=7.4Hz,COCH2),3.41(t,2H,J=6.8Hz,CH2Br),7.18(s,1H,NH),7.56(m,6H,ArH),7.63(d,2H,J=8.9Hz,ArH),7.82(d,2H,J=8.6Hz,ArH),9.04(s,1H,NH),12.59(1H,NH).
13C−NMR(CDCl3,75MHz)δ25.5,28.0,28.6,29.1,31.1,32.7,34.0,37.6,120.1,124.8,126.2,127.5,128.6,133.4,136.8,157.8,167.0,171.6,178.5.
【0108】
前記ブロモ化合物(487mg、0.92mmol)をH2O(25mL)中、ジメチルアミン(10mL)で処理することにより、8−ジメチルアミノ−オクタン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドを含有する粗混合物を得た。この混合物を最小量のCH2Cl2に溶解し、エーテル溶液中、2M塩酸をクリーム色の沈殿が生じるまで滴下した。固体を濾過し、濾液を減圧下濃縮した。生じた黄色オイルをヘキサンで研和し、黄色固体を得、イソプロパノールから再結晶し、7(301mg、62%)を粘性黄色固体として得た。
1H−NMR(DMSO−d6,400MHz)δ1.32(s,9H,(CH3)3),1.32(m,6H,CH2)3),1.62(m,4H,(CH2)2),2.33(m,2H,CH2),2.72(s(br),6H,N(CH3)2),2.99(m,2H,CH2),7.62(m,6H,ArH),7.97(dd,2H,J=8.5,16.5Hz,ArH),9.95(s,1H,NH),10.08(s(br),1H,NH),11.44(s,1H,NH),12.60(s,1H,NH).
【0109】
化合物8:N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−2−[2−(2−{2−[2−(2−ジメチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−アセトアミド塩酸塩(8)の合成
【0110】
【化10】
【0111】
類縁物質8の合成
試薬と条件:(i)JOC 2004、69、639に記載された文献反応により調製。(87%)(ii)CrO3/H2SO4、アセトン、0℃から室温、16時間(65%)。(iii)SOCl2、CH2Cl2、室温、16時間。(iv)4、NEt3、CH2Cl2、室温、16時間、(46%)。(v)LiBr、アセトン、還流、24時間(81%)。(vi)40%HNMe2水溶液、CH2Cl2/H2O、室温、24時間(91%)。(vii)ジエチルエーテル中2MのHCl、CH2Cl2、(73%)。
【0112】
ベンゼンスルホン酸 2−[2−(2−{2−[2−(2−ヒドロキシ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エチルエステル(12)
ヘキサエチレングリコール(11)(2.82g、10mmol、1当量)を、N2雰囲気下、CH2Cl2(100mL)に溶解し、酸化銀(3.48g、15mmol、1.5当量)、続いて塩化トシル(2.10g、11mmol、1.1当量)およびヨウ化カリウム(332mg、2mmol、0.2mmol)を加えた。溶液を室温で1時間攪拌し、次いで、濃縮し、セライトを通じて濾過し、酢酸エチルで溶出した。濾液を減圧下濃縮し、淡黄色オイルを得、シリカ(酢酸エチル)のカラムクロマトグラフィで精製し、12(3.79g、87%)を淡黄色オイルとして得た。
1H−NMR(DMSO−d6,300MHz)δ1.80(s(br),1H,OH),2.44(s,3H,CH3),3.64(m,22H,(CH2)11),4.15(m,2H,CH2),7.33(d,2H,J=8.0Hz,ArH),7.79(d,2H,J=8.4Hz,ArH).
【0113】
[2−(2−{2−[2−(2−ベンゼンスルホニルオキシ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−酢酸(13)
1.5M硫酸水溶液(10mL)を0℃に調製し、三酸化クロムを加えて撹拌し、橙色溶液を得た。アルコール12(1g、2.29mmol)をアセトン(15mL)に溶解し、その溶液を硫酸に0℃で滴下した。混合物を室温まで温め、16時間撹拌した。生じた緑色の溶液をセライトで濾過し、減圧下濃縮した。生じた水溶性の残渣をH2Oで希釈し、CH2Cl2(3x50mL)で抽出した。有機相をMgSO4で乾燥し、減圧下濃縮し、13(672mg、65%)を淡黄色オイルとして得、更なる精製は行わなかった。
1H−NMR(アセトン−d6,300MHz)δ2.46(s,3H,CH3),3.59(m,18H,(CH2)9),4.13(m,4H,(CH2)2),7.49(d,2H,J=8.0Hz,ArH),7.82(d,2H,J=8.0Hz).
13C−NMR(CDCl3,100MHz)δ21.6,68.5,68.6,68.8,69.1,69.3,70.3,70.4,70.4,70.5,70.6,70.6,70.7,70.8,71.2,71.3,76.8,77.1,77.4,127.9,129.9,132.9,144.9,172.7.
LRMS(M+Na)実測値473.16.
【0114】
N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−2−[2−(2−{2−[2−(2−ジメチルアミノ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−アセトアミド塩酸塩(8)
酸13(117mg、0.26mmol)を、触媒量のDMFを含有する塩化オキサリル中で攪拌した。溶液を1時間加熱還流し、次いで、室温に冷却し、溶媒を減圧下濃縮し、対応する酸塩化物を得、更なる精製を行わずに用いた。酸塩化物(138mg、0.29、1.3当量)を乾燥CH2Cl2(10mL)中、N2雰囲気下で攪拌し、チオ尿素4(74mg、0.23mmol、1当量)を加え、明黄色溶液を得た。トリエチルアミン(41μL、0.23mmol、1当量)を加え、混合物を室温で16時間撹拌した。溶液をCH2Cl2(1x10mL)で希釈し、1M塩酸水溶液(1x10mL)、1M水酸化ナトリウム水溶液、続いて飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、溶媒を減圧下濃縮した。生じた黄色の泡をシリカ(50%酢酸エチル/石油エーテル)のフラッシュクロマトグラフィで精製し、トルエン−4−スルホン酸 2−[2−(2−{2−[2−({4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニルカルバモイル}−メトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−エチルエステル(79mg、46%)を黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.34(s,9H,(CH3)3),2.41(s,3H,CH3),3.63(m,18H,(CH2)9),4.10(m,4H,(CH2)2),7.31(d,2H,J=8.0Hz,ArH),7.53(d,2H,J=8.6Hz,ArH),7.67(m,4H,ArH),7.79(m,m,4H,ArH),8.99(s,1H,NH),9.15(s,1H,NH),12.61(s,1H,NH).
13C−NMR(CDCl3,100MHz)δ21.7,31.1,35.3,68.7,69.3,70.1,70.4,70.5,70.6,70.7,71.2,120.2,124.6,126.1,126.2,127.5,127.9,128.6,129.8,133.7,136.1,144.8,157.8,166.89,168.4,178.3.
HRMS(M+Na)実測値782.2756(C37H49N3O10NaS2の計算値782.2757).
【0115】
上記に記載されたトシレート(76mg、0.1mmol、1当量)をアセトン(3mL)中で攪拌し、臭化リチウム(44mg、0.5mmol、5当量)を加えた。混合物を50℃で24時間加熱した。溶媒を減圧下濃縮し、生じた残渣を酢酸エチルに再懸濁し、固体を濾過した。濾液を減圧下濃縮し、2−[2−(2−{2−[2−(2−ブロモ−エトキシ)−エトキシ]−エトキシ}−エトキシ)−エトキシ]−N−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アセトアミド(54mg、81%)を淡黄色オイルとして得、更なる精製を行わずに用いた。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),3.45(t,2H,J=6.3Hz,CH2Br),3.71(m,16H,(CH2)8),4.18(s,2H,NHCOCH2),7.55(d,2H,J=8.6Hz,ArH),7.70(q,4H,J=9.0Hz,ArH),7.82(d,2H,J=8.6Hz,ArH),9.06(s,1H,NH),9.16(s(br),1H,NH),12.61(s,1H,NH).
【0116】
続いてこの臭化物(30mg、0.05mmol)を、40%ジメチルアミン水溶液(0.75mL)、H2O(1.8mL)およびCH2Cl2(2mL)中、室温で24時間撹拌することによりジメチルアミン誘導体に変換した。反応混合物をCH2Cl2 (20mL)で希釈し、有機相をH2O(1x10mL)、1M水酸化ナトリウム水溶液(1x10mL)および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、溶媒を減圧下濃縮し、ジメチルアミン体を得た。淡黄色オイルを最少量のCH2Cl2にとり、ジエチルエーテル中2M塩酸溶液を、わずかな沈殿が生じるまで滴下した。混合物を減圧下濃縮し、8(20mg、73%)を黄色の泡として得た。
1H−NMR(CDCl3,300MHz)δ1.32(s,9H,(CH3)3),2.77(d(br),6H,J=4.0Hz,NH(CH3)2),3.61(m,18H,(CH2)9),4.10(m,4H,(CH2)2),7.63(m,6H,ArH),7.95(d,2H,J=8.4Hz,ArH),9.55(s,1H,NH),9.77(s,1H,NH),11.45(s,1H,NH),12.62(s,1H,NH).
【0117】
カテゴリー2:オキソ−テノビン類縁体、化合物24
以下の手法に従い24を調製した。
【0118】
【化11】
【0119】
{4−[3−(4−tert−ブチル−ベンゾイル)−ウレイド]−フェニル}−カルバミン酸 tert−ブチルエステル(28)
上記に記載されたとおり、ベンズアミド26(1g、5.64mmol、1当量)を塩化オキサリル(834μL、9.59mmol、1.7当量)と反応させた。生じたイソシアネート(27)を、アニリン18と共に乾燥アセトニトリル中で素早く撹拌し、3時間加熱還流した。生じたベージュ色の懸濁液を室温まで冷却し、沈殿を濾過により集め、28(400mg、17%)をベージュ色の固体として得、さらに精製しなかった。
1H−NMR(DMSO−d6,300MHz)δ1.31(s,9H,(CH3)3),1.47(s,9H,OC(CH3)3),7.43(m,4H,ArH),7.55(d,2H,J=8.6Hz,ArH),7.97(d,2H,J=8.6Hz,ArH),9.31(s,1H,NH),10.77(s,1H,NH),10.91(s,1H,NH).
【0120】
5−ブロモ−ペンタン酸 {4−[3−(4−tert−ブチル−ベンゾイル)−ウレイド]−フェニル}−アミド(29)
N−アシル尿素28(300mg、0.73mmol)をトリフルオロ酢酸(1.2mL)中、40分間攪拌し、減圧下濃縮し、1−(4−アミノ−フェニル)−3−(4−tert−ブチル−ベンゾイル)−ウレア(トリフルオロ酢酸塩として)(383mg、定量的)を褐色な綿状の固体として得、更なる精製をしなかった。
1H−NMR(DMSO−d6,300MHz)δ1.30(s,9H,(CH3)3),7.32(d,2H,J=8.8Hz,ArH),7.55(d,2H,J=8.6Hz,ArH),7.70(d,2H,J=8.9Hz,ArH),7.98(d,2H,J=8.6Hz,ArH),10.96(s,1H,NH),11.02(s,1H,NH).
【0121】
脱保護したアミン(368mg、0.70mmol)を乾燥CH2Cl2(10mL)中、N2雰囲気下で攪拌し、NEt3(98μL、0.70mmol)を加え、清明な褐色溶液を得た。5−ブロムワレリルクロライド(140μL、1.05mmol、1.5.当量)、続いてトリエチルアミン(98μL、0.70mmol)を加えた。反応混合物を室温で16時間攪拌し、次いでCH2Cl2(1x20mL)で希釈し、1M塩酸水溶液(1x20mL)、1M水酸化ナトリウム水溶液(1x20mL)および飽和塩水(1x20mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮し、粗黄色チョーク質の固体を得、シリカ(50%酢酸エチル/石油エーテル)のカラムクロマトグラフィで精製し、29(153mg、48%)を黄色固体として得た。
1H−NMR(DMSO−d6,300MHz)δ1.31(s,9H,(CH3)3),1.80(m,4H,(CH2)2),2.33(t,2H,J=7.2Hz,COCH2),3.57(t,2H,J=6.6Hz,CH2Br),7.54(m,6H,ArH),7.98(d,2H,J=8.5Hz,ArH),9.90(s,1H,NH),10.82(s,1H,NH),10.93(s,1H,NH).
【0122】
5−ジメチルアミノ−ペンタン酸 {4−[3−(4−tert−ブチル−ベンゾイル)−ウレイド]−フェニル}−アミド塩酸塩(24)
ブロモ化合物29(123mg、0.27mmol)をCH2Cl2(10mL)、40%HNMe2水溶液(3mL)およびH2O(7.5mL)中、室温で20時間攪拌した。反応混合物をCH2Cl2(20mL)で希釈し、H2O(1x10mL)、10%NaOH水溶液(1x10mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮し、クリーム色の固体を得、アセトンに溶解し、ジエチルエーテル中2M塩酸で処理した。白色沈殿を得、濾過により単離し、24(44mg、36%)をクリーム色の粉末として得た。
1H−NMR(DMSO−d6,400MHz)δ1.31(s,9H,(CH3)3),1.64(m,4H,(CH2)2),2.36(t,2H,J=6.7Hz,COCH2),2.75(s,6H,NH(CH3)2),3.05(m,2H,CH2Br),7.53(m,6H,ArH),7.98(d,2H,J=8.6Hz,ArH),9.63(s(br),1H,NH),9.98(s,1H,NH),10.82(s,1H,NH),10.93(s,1H,NH).
【0123】
カテゴリー3:Boc−テノビン類縁体、化合物21
この類縁物質は、以下に示すp53活性化アッセイにおいて、非常に良好な活性を示したので、重要な化合物である。
【0124】
{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−カルバミン酸 tert−ブチルエステル(21)
アセトン(15mL)中3(869μL、4.79mmol、1当量)の撹拌溶液に、N2雰囲気下、チオシアン酸ナトリウム(389mg、4.79mmol、1当量)を加え、反応混合物を室温で4時間撹拌した。アニリン18(1g、4.79mmol、1当量)をアセトン(10mL)中に溶液として加えた。生じたベージュ色の懸濁液を室温で16時間攪拌し、溶媒を減圧下濃縮した。残った残渣を、CH2Cl2に再懸濁し、不溶の固体を濾過した。濾液を減圧下濃縮し、クリーム色の粗固体を得、50%酢酸エチル/石油エーテルから再結晶することにより精製し、{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−カルバミン酸 tert−ブチルエステル(1.4g、66%)を金色微粉末として得た。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),1.63(s,9H,OC(CH3)3),6.52(s,1H,NH),7.42(d,2H,J=8.7Hz,ArH),7.55(d,2H,J=8.5Hz,ArH),7.64(d,2H,J=8.8Hz,ArH),8.82(d,2H,J=8.7Hz,ArH),9.03(s,1H,NH),12.56(s,1H,NH).
【0125】
カテゴリー4:テノビン類に基づく親和性マトリックス
このタイプの試薬として、テノビン類に基づく親和性マトリックスの調製が含まれ、テノビン類の可能性のあるタンパク質標的や、サーチュインタンパク質ファミリーやその他の範囲の抑制剤の選択性のアッセイにおいて、可能性のある関連物質(例えばMeijer,Laurent;Skaltsounis,Alexios−Leandros;Magiatis,Prokopios;Polychronopoulos,Panagiotis;Knockaert,Marie;Leost,Maryse;Ryan,Xiaozhou P.;Vonica,Claudia Alin;Brivanlou,Ali;Dajani,Rana;Crovace,Claudia;Tarricone,Cataldo;Musacchio,Andrea;Roe,S.Mark;Pearl,Laurence;Greengard,Paul.「古代紫インディルビン類から誘導されるGSK−3−選択的な抑制剤」Chemistry&Biology(2003),10(12),1255−1266により報告される以下の類縁物質プロトコール)を同定するのに重要である。親和性マトリックスを調製する手順は次のとおりである。
【0126】
親和性樹脂の使用
【0127】
【化12】
【0128】
6−tert−ブトキシカルボニルアミノ−ヘキサン酸(31)
6−アミノヘキサン酸(3.1g、23.63mmol、1当量)を、ジオキサン/H20(1:1、75mL)中、0℃で水酸化ナトリウム(1.05g、23.63mmol)と共に攪拌した。この溶液に、Boc無水物(5.2g、21.27mmol、0.9当量)を加え、反応を16時間室温まで温めた。反応混合物を1M塩酸水溶液でpH5まで酸性にした。混合物をCH2Cl2(3x50mL)で抽出し、合わせた有機相をMgSO4で乾燥し、減圧下濃縮し、31を無色のオイル(4.5g、89%)として得、更なる精製を行わなかった。
1H−NMR(CDCl3,300MHz)δ1.42(m,4H,(CH2)2),1.44(s,9H,(CH3)3),1.65(m,2H,CH2),2.35(t,2H,J=7.4Hz,CH2),3.10(m,2H,CH2),4.55(s,(br),1H,NH).
【0129】
(5−{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニルカルバモイル}−ペンチル)−カルバミン酸 tert−ブチルエステル(32)
チオ尿素4(100mg、0.31mmol、1当量)を乾燥CH2Cl2(5mL)中、N2雰囲気下で攪拌し、酸31(106mg、0.46mmol、1.5当量)、続いてブロモ−トリス−ピロリジノホスホニウムヘキサフルオロホスファート(214mg、0.46mmol、1.5当量)、およびN,N−ジイソプロピルエチルアミン(53μL、0.31mmol、1当量)を加えた。反応混合物を室温で16時間攪拌し、次いで、CH2Cl2(20mL)で希釈し、1M塩酸水溶液(1x10mL)、1M水酸化ナトリウム水溶液(1x10mL)、および飽和塩水(1x10mL)で洗浄した。有機相をMgSO4で乾燥し、減圧下濃縮し、粗黄色オイルを得、シリカ(25〜50%酢酸エチル/石油エーテル)のカラムクロマトグラフィで精製し、32(86mg、54%)を無色のオイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.36(s,9H,(CH3)3),1.40(s,9H,OC(CH3)3),1.40(m,2H,CH2),1.52(m,2H,CH2),1.75(m,2H,CH2),2.36(t,2H,J=7.5Hz,CH2),3.12(m,2H,CH2),4.61(s(br),1H,NHBoc),7.54(d,2H,J=8.7Hz,ArH),7.62(m,4H,ArH),7.82(d,2H,J=8.6Hz,ArH),9.09(s,1H,NH),12.59(s,1H,NH).
LRMS(M+Na)実測値563.15.
【0130】
6−アミノ−ヘキサン酸{4−[3−(4−tert−ブチル−ベンゾイル)−チオウレイド]−フェニル}−アミドトリフルオロ酢酸塩(33)
Boc保護されたアミン32(20mg、0.04mmol)をCH2Cl2(1mL)およびトリフルオロ酢酸(166μL)中、室温で30分間攪拌した。減圧下濃縮し、33を淡黄色オイルとして得た。
1H−NMR(CDCl3,300MHz)δ1.29(m,2H,CH2)1.30(s,9H,(CH3)3),1.59(m,4H,(CH2)2),2.28(m,2H,CH2),2.90(m,2H,CH2),7.60(m,6H,ArH),7.75(d,2H,J=8.2Hz,ArH),8.75(s(br),1H,NH),9.21(s,1H,NH),12.50(s,1H,NH).
【0131】
親和性樹脂(Affigel−15)への33の固定化
Affigel−15(BioRad)(1mL)を冷イソプロパノールで洗浄し、濾過した。Affigel(210mg)に、ジオキサン(1mL)、33(2mg)および過剰のトリエチルアミンを加えた。さらにジオキサンを加え、総量を1.5mLに調節した。混合物を4℃で24時間撹拌し、次いで3000rpmで3分間遠心分離した。母液をデカントし、新鮮なジオキサンを加え、混合物を振盪した。遠心分離し、2回デカントした。
【0132】
上記に記載された化合物の活性および水溶性を試験し、結果を表2に示す。
【0133】
【表3】
表2活性データ:a細胞ベースのアッセイにおいて類縁物質がp53を活性化する活性化能の順序.この形式は、定量化が困難なp53活性化アッセイに選択的に用いられている。n.d.=測定せず
【特許請求の範囲】
【請求項1】
式(I)
【化13】
[式中、
R1は独立してHであるか;分枝状または非分枝の、モノ、ジ、もしくはトリ置換または無置換の、アルキル、アルケニルもしくはアルキニルであるか;または、アリールであるか;または、Z−アルキル、Z−アルケニル、Z−アルキニルもしくはZ−アリール(ここでZはO、NH、N、またはSであるか、ラベル、プローブもしくは基を可溶化する溶媒に結合する基を含む)であり;
Yは存在しないか、−C(O)−、−C=S−、または−SO2−であり;
Ar、Ar’はアリールであり;
XはOまたはSであり;
R3およびR4は、独立して、非存在であるか存在し、存在するときは、独立してH、または、分枝状または非分枝の、モノ、ジもしくはトリ置換または無置換の、アルキルもしくはZ−アルキル(ここで、ZがO、NH、NもしくはS)であるか、またはR3およびR4は、共に結合して、分枝状または非分枝の、置換または無置換のアルキレンもしくはZ−アルケン(ここで、ZはO、NH、NまたはSである)を形成し;
R5およびR6は、独立して、非存在であるか存在し、存在するときは、独立して分枝状または非分枝の、置換または無置換のアルキルであり;ここで、
R3が存在するときは、破線bは単結合であり、R3が非存在のときは、破線bは二重結合であり;
R4が存在するときは、破線cおよびdは単結合であり、R4が非存在のときは、破線cまたはdの一方は二重結合であり、他方は単結合であり;そして、
R5およびR6が非存在のとき、破線aおよびeはそれぞれ二重結合であり、R5およびR6が存在するときは、破線aおよびeは、それぞれ単結合である]
で示される、医薬としての使用に供される化合物、または、その生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体;ただし、下記化合物:
【化14】
を除く。
【請求項2】
式(II)
【化15】
[式中、R1、R2およびYは請求項1に定義された通りである]
で示される請求項1に記載の化合物、または、その生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【請求項3】
R1YNH−および/またはR2−基が、それらが結合するそれぞれのフェニル環上のパラ位にある請求項1または請求項2に記載の化合物、またはその生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【請求項4】
Yが−C(O)−基である、前記請求項のいずれかに記載の化合物、またはその生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【請求項5】
医薬として使用する請求項1〜4のいずれか1項に記載の但し書きのない化合物。
【請求項6】
その薬学的に許容可能な担体と共に請求項1〜4のいずれか1項に記載の但し書きのない化合物を含む医薬組成物
【請求項7】
細胞増殖、特に癌に関連する疾患の治療または予防用医薬の調製のための請求項1〜4のいずれか1項に記載の但し書きのない化合物の使用。
【請求項8】
SirT1に関連する発現および/または機能を伴う疾患/状態の治療または予防用医薬の調製のための請求項1〜4のいずれか1項に記載の但し書きのない化合物の使用。
【請求項9】
糖尿病、筋分化、炎症、異常なまたは望ましくない免疫反応、肥満、心不全、神経変性、老化、HIV感染およびマラリアから選択される疾患/状態の治療または予防用医薬の調製のための請求項1〜4のいずれか1項に記載の但し書きのない化合物の使用。
【請求項10】
請求項1〜4のいずれか1項に記載の但し書きのない化合物の治療学的または予防学的に有用な量を、それを必要とする対象に投与することを含む細胞増殖、特に癌に関連する疾患の治療または予防の方法。
【請求項11】
請求項1〜4のいずれか1項に記載の但し書きのない化合物の治療学的または予防学的に有用な量を、それを必要とする対象に投与することを含むSirT1に関連する発現および/または機能を伴う疾患/状態の治療または予防の方法。
【請求項12】
請求項1〜4のいずれか1項に記載の但し書きのない化合物の治療学的または予防学的に有用な量を、それを必要とする対象に投与することを含む、糖尿病、筋分化、心不全、神経変性、老化、HIV感染およびマラリアからなる群より選択される疾患/状態の治療または予防の方法。
【請求項13】
末端R’基がラベル、親和性樹脂、基質、プローブおよび/または
水溶性基に結合するためのリンカー基である請求項1〜4のいずれか1項に記載の但し書きのない化合物。
【請求項14】
ラベル、親和性樹脂、基質、プローブおよび/または水溶性基に結合する請求項13に記載の化合物。
【請求項15】
サーチュインタンパク質ファミリーの抑制剤の選択性評価において化合物の潜在的タンパク質標的、または潜在的類縁体を同定するための請求項14に記載の化合物の使用。
【請求項1】
式(I)
【化13】
[式中、
R1は独立してHであるか;分枝状または非分枝の、モノ、ジ、もしくはトリ置換または無置換の、アルキル、アルケニルもしくはアルキニルであるか;または、アリールであるか;または、Z−アルキル、Z−アルケニル、Z−アルキニルもしくはZ−アリール(ここでZはO、NH、N、またはSであるか、ラベル、プローブもしくは基を可溶化する溶媒に結合する基を含む)であり;
Yは存在しないか、−C(O)−、−C=S−、または−SO2−であり;
Ar、Ar’はアリールであり;
XはOまたはSであり;
R3およびR4は、独立して、非存在であるか存在し、存在するときは、独立してH、または、分枝状または非分枝の、モノ、ジもしくはトリ置換または無置換の、アルキルもしくはZ−アルキル(ここで、ZがO、NH、NもしくはS)であるか、またはR3およびR4は、共に結合して、分枝状または非分枝の、置換または無置換のアルキレンもしくはZ−アルケン(ここで、ZはO、NH、NまたはSである)を形成し;
R5およびR6は、独立して、非存在であるか存在し、存在するときは、独立して分枝状または非分枝の、置換または無置換のアルキルであり;ここで、
R3が存在するときは、破線bは単結合であり、R3が非存在のときは、破線bは二重結合であり;
R4が存在するときは、破線cおよびdは単結合であり、R4が非存在のときは、破線cまたはdの一方は二重結合であり、他方は単結合であり;そして、
R5およびR6が非存在のとき、破線aおよびeはそれぞれ二重結合であり、R5およびR6が存在するときは、破線aおよびeは、それぞれ単結合である]
で示される、医薬としての使用に供される化合物、または、その生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体;ただし、下記化合物:
【化14】
を除く。
【請求項2】
式(II)
【化15】
[式中、R1、R2およびYは請求項1に定義された通りである]
で示される請求項1に記載の化合物、または、その生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【請求項3】
R1YNH−および/またはR2−基が、それらが結合するそれぞれのフェニル環上のパラ位にある請求項1または請求項2に記載の化合物、またはその生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【請求項4】
Yが−C(O)−基である、前記請求項のいずれかに記載の化合物、またはその生理的に許容可能な塩、溶媒和物、エステルもしくは他の生理的機能性誘導体。
【請求項5】
医薬として使用する請求項1〜4のいずれか1項に記載の但し書きのない化合物。
【請求項6】
その薬学的に許容可能な担体と共に請求項1〜4のいずれか1項に記載の但し書きのない化合物を含む医薬組成物
【請求項7】
細胞増殖、特に癌に関連する疾患の治療または予防用医薬の調製のための請求項1〜4のいずれか1項に記載の但し書きのない化合物の使用。
【請求項8】
SirT1に関連する発現および/または機能を伴う疾患/状態の治療または予防用医薬の調製のための請求項1〜4のいずれか1項に記載の但し書きのない化合物の使用。
【請求項9】
糖尿病、筋分化、炎症、異常なまたは望ましくない免疫反応、肥満、心不全、神経変性、老化、HIV感染およびマラリアから選択される疾患/状態の治療または予防用医薬の調製のための請求項1〜4のいずれか1項に記載の但し書きのない化合物の使用。
【請求項10】
請求項1〜4のいずれか1項に記載の但し書きのない化合物の治療学的または予防学的に有用な量を、それを必要とする対象に投与することを含む細胞増殖、特に癌に関連する疾患の治療または予防の方法。
【請求項11】
請求項1〜4のいずれか1項に記載の但し書きのない化合物の治療学的または予防学的に有用な量を、それを必要とする対象に投与することを含むSirT1に関連する発現および/または機能を伴う疾患/状態の治療または予防の方法。
【請求項12】
請求項1〜4のいずれか1項に記載の但し書きのない化合物の治療学的または予防学的に有用な量を、それを必要とする対象に投与することを含む、糖尿病、筋分化、心不全、神経変性、老化、HIV感染およびマラリアからなる群より選択される疾患/状態の治療または予防の方法。
【請求項13】
末端R’基がラベル、親和性樹脂、基質、プローブおよび/または
水溶性基に結合するためのリンカー基である請求項1〜4のいずれか1項に記載の但し書きのない化合物。
【請求項14】
ラベル、親和性樹脂、基質、プローブおよび/または水溶性基に結合する請求項13に記載の化合物。
【請求項15】
サーチュインタンパク質ファミリーの抑制剤の選択性評価において化合物の潜在的タンパク質標的、または潜在的類縁体を同定するための請求項14に記載の化合物の使用。
【図1】

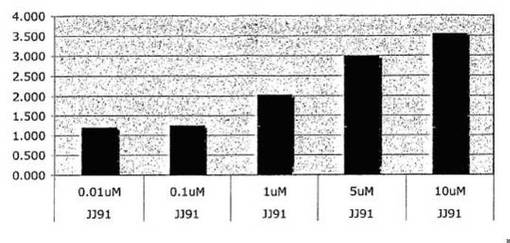
【図2】
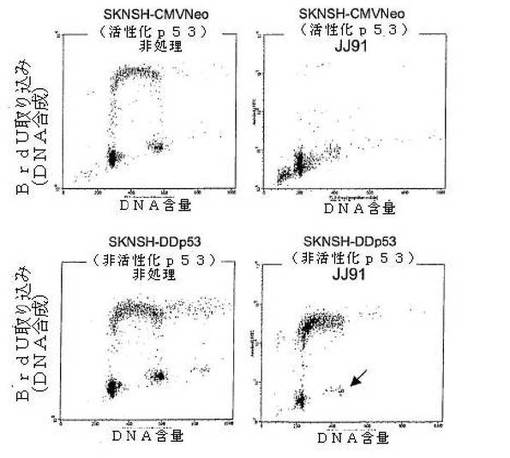
【図3】
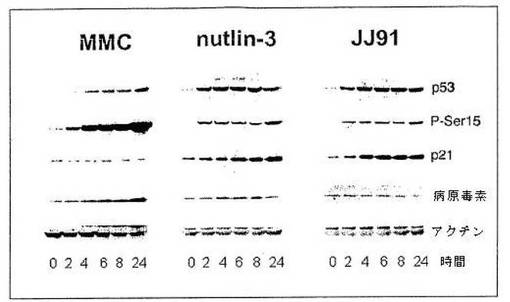
【図4】
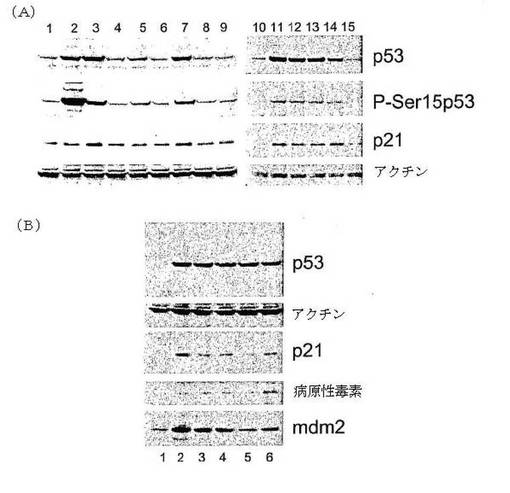
【図5】

【図6】
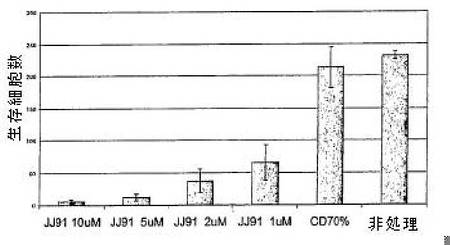
【図7】
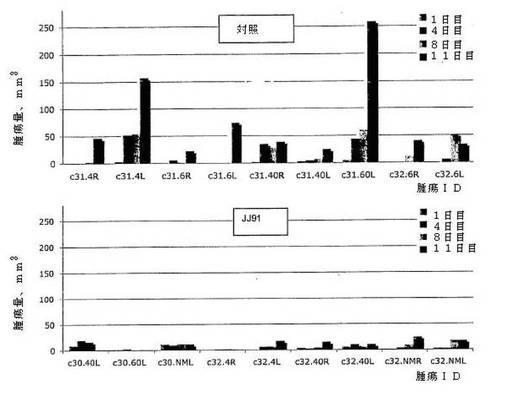
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【公表番号】特表2010−502582(P2010−502582A)
【公表日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願番号】特願2009−526177(P2009−526177)
【出願日】平成19年9月4日(2007.9.4)
【国際出願番号】PCT/GB2007/003302
【国際公開番号】WO2008/029096
【国際公開日】平成20年3月13日(2008.3.13)
【出願人】(501490346)ユニバーシティー・コート・オブ・ザ・ユニバーシティー・オブ・ダンディー (3)
【出願人】(509061760)ユニバーシティー コート オブ ザ ユニバーシティー オブ セイント.アンドリューズ (2)
【Fターム(参考)】
【公表日】平成22年1月28日(2010.1.28)
【国際特許分類】
【出願日】平成19年9月4日(2007.9.4)
【国際出願番号】PCT/GB2007/003302
【国際公開番号】WO2008/029096
【国際公開日】平成20年3月13日(2008.3.13)
【出願人】(501490346)ユニバーシティー・コート・オブ・ザ・ユニバーシティー・オブ・ダンディー (3)
【出願人】(509061760)ユニバーシティー コート オブ ザ ユニバーシティー オブ セイント.アンドリューズ (2)
【Fターム(参考)】
[ Back to top ]