
PBRリガンドとしての三環系インドール誘導体
本発明は、PBRと高い親和性で結合し、投与後に脳に良好に取り込まれ、PBRとの良好な選択的結合を有するインドール系インビボ造影剤を提供する。本発明はまた、本発明のインビボ造影剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の使用を含む前記インビボ造影剤の合成方法、及び前記方法を実施するためのキットを含む。また、インビボ造影剤の自動合成のためのカセットを提供する。本発明のさらなる態様は、本発明のインビボ造影剤を含む放射性医薬組成物、及び前記インビボ造影剤の使用方法を含む。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、インビボイメージング、特に末梢性ベンゾジアゼピン受容体(PBR)のインビボイメージングに関する。PBRと高い親和性で結合し、投与後に脳に良好に取り込まれ、PBRとの良好な選択的結合を有するインドール系インビボ造影剤を提供する。本発明はまた、本発明のインビボ造影剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の合成方法を提供する。本発明の他の態様は、本発明の前駆体化合物の使用を含む本発明のインビボ造影剤の合成方法、前記方法を実施するためのキット、及び自動化された前記方法を実施するためのカセットを含む。さらに本発明は、本発明のインビボ造影剤を含む放射性医薬組成物、並びに前記インビボ造影剤の使用方法を提供する。
【背景技術】
【0002】
末梢性ベンゾジアゼピン受容体(PBR)は、主に末梢組織及びグリア細胞内に局在することが知られているが、その生理的機能は未だ解明されていない。PBRは、細胞内ではミトコンドリア外膜上に局在することが知られており、ミトコンドリア機能の調節及び免疫系において潜在的な役割を示す。さらにPBRは、細胞増殖、ステロイド産生、カルシウム流及び細胞呼吸に関与すると想定されている。
【0003】
異常なPBR発現は、多発性硬化症(Banati et al,2001 Neuroreport;12(16):3439−42;Debruyne et al,2002 Acta Neurol Belg;102(3):127−35)、ラスムッセン脳炎(Banati et al,1999 Neurology;53(9):2199−203)、脳血管炎(Goerres et al,2001 Am J Roentgenol;176(4):1016−8)、ヘルペス脳炎(Cagnin et al,2001 Brain;124(Pt 10):2014−27)、及びAIDS関連認知症(Hammoud et al,2005 J Neurovirol;11(4):346−55)を含む、中枢神経系(CNS)の炎症性病状に関連するとされている。
【0004】
またCNSでは、パーキンソン病(Gerhard et al,2006 Neurobiol Dis;21(2):404−12;Ouchi et al,2005 Ann Neurol;57(2):161−2)、大脳皮質基底核変性症(Gerhard et al,2004 Mov Disord;19(10):1221−6)、進行性核上性麻痺(Gerhard et al,2006 Neurobiol Dis;21(2):404−12)、多系統萎縮症(Gerhard et al,2003 Neurology;61(5):686−9)、ハンチントン病(Pavese et al,2006 Neurology;66(11):1638−43;Tai et al,2007 Brain Res Bull;72(2−3):148−51)、筋萎縮性側索硬化症(Turner et al,2004 Neurobiol Dis;15(3):601−9)、及びアルツハイマー病(Cagnin et al,2001 Lancet;358(9283):766;Yasuno et al,2008 Biol Psychiatry;64(10):835−41)などの変性疾患において、PBRとの関係が報告されている。
【0005】
いくつかのCNS虚血状態は、虚血性脳卒中(Gerhard et al,2005 Neuroimage;24(2):591−5)、末梢神経損傷(Banati et al,2001 Neuroreport;12(16):3439−42)、てんかん(Sauvageau 2002 Metab Brain Dis;17(1):3−11;Kumar et al,2008 Pediatr Neurol;38(6))を含む異常なPBR発現に関係することが示されている。PBRは、外傷性脳損傷における損傷の程度を決定するためのバイオマーカーと想定されており(Toyama et al,2008 Ann Nucl Med;22(5):417−24)、外傷性脳損傷の動物モデルにおいて、PBR発現の増大が報告された(Venneti et al,2007 Exp Neurol;207(1):118−27)。興味深いことに、急性ストレスは、脳内のPBR発現の増大に相関するとされており、慢性ストレスは、PBRの下方制御と相関するとされている(Lehmann et al,1999 Brain Res;851(1−2):141−7)。PBRを結像するために[11C]PK11195を使用して、神経膠腫の境界を描写できることが報告されている(Junck et al,1989 Ann Neurol;26(6):752−8)。神経障害性疼痛を有する対象において活性化したミクログリアを観測したTsudaらによれば、PBRは神経障害性疼痛にも関連し得る(2005 TINS 28(2) pp101−7)。
【0006】
末梢では、PBR発現は、肺炎症(Branley et al,2008 Nucl. Med. Biol;35(8):901−9)、慢性閉塞性肺疾患及び喘息(Jones et al,2003 Eur Respir J;21(4):567−73)、炎症性腸疾患(Ostuni et al,Inflamm Bowel Dis;2010 online publication)、関節リウマチ(van der Laken et al,2008 Arthritis Rheum;58(11):3350−5)、原発性線維筋痛症(Faggioli et al,2004 Rheumatology;43(10):1224−1225)、神経損傷(Durrenberger et al,2004 J Peripher Nerv Syst;9(1):15−25)、アテローム性動脈硬化症(Fujimura et al,2008 Atherosclerosis;201(1):108−111)、結腸、前立腺及び乳癌(Deane et al,2007 Mol Cancer Res;5(4):341−9;Miettinen et al,1995 Cancer Res;55(12):2691−5;Han et al,2003 J Recept Signal Transduct Res;23(2−3):225−38)、腎炎(Tam et al,1999 Nephrol Dial Transplant;14(7):1658−66;Cook et al,1999 Kidney Int;55(4):1319−26)、並びに虚血再灌流障害(Zhang et al,2006 J Am Coll Surg;203(3):353−64)に関係するとされている。
【0007】
PBR選択的リガンドである(R)−[11C]PK11195を使用する陽電子放射断層撮影(PET)イメージングは、中枢神経系(CNS)炎症の一般的な指標を提供する。しかし、(R)−[11C]PK11195は、タンパク質結合性が高く、特異性の低い結合を有するものから非特異的な結合を有するものがあることが知られている。さらに、放射性標識したその代謝産物の役割は知られておらず、結合の定量化には、複雑なモデリングを必要とする。
【0008】
三環系インドール化合物は、当技術分野で公知である。Davies et al(J. Med. Chem. 1998;41(4):451−67)には、ある種の三環系インドール化合物が教示されており、それらをメラトニン作動薬及び拮抗薬として特徴づけている。Napper et al(J. Med. Chem. 2005;48:8045−54)には、ヒストン及び他のタンパク質のリシン残基からアセチル基を除去する酵素ファミリーの一員である酵素SIRT1の選択的阻害に関連して、ある種の三環系インドール化合物の構造と活性の関係について教示・議論されている。米国特許第6451795号には、別の種類の三環系インドール化合物が開示されており、この化合物はPBR関連病状の治療に有用なものとして論じられている。米国特許第6451795号には、最も活性な化合物について0.2nM〜5.0nMのIC50値を開示しており、その化合物が末梢神経障害の予防又は治療、及び中枢神経変性疾患の治療にとって有用であると記載されている。
【0009】
Okubu et al,(Bioorganic & Medicinal Chemistry 2004 12 3569−80)には、四環系インドール化合物群の設計、合成及び構造、並びにPBRに対するそれらの親和性(IC50値は約0.4nMに過ぎない)を記載している。本発明の出願人に譲渡された国際公開第2007/057705号には、様々なインビボイメージング部分で標識した四環系インドール誘導体が開示されている。国際公開第2007/057705号に開示された好ましいインビボイメージング部分は、陽電子放射断層撮影(PET)又は単一光子放出断層撮影(SPECT)イメージング、最も好ましくはPETに適した部分である。
【0010】
さらに、同時係属注の国際出願PCT/EP2009/062827には、国際公開第2007/057705号の造影剤に類似の四環系インドール由来のインビボ造影剤が記載されている。
【0011】
国際公開第2007/057705号及び同時係属の国際出願PCT/EP2009/062827に記載の四環系インドール誘導体は、PBR受容体に対して良好な親和性を有し、注入の60分後における脳内の高い割合の放射活性は、親インビボ造影剤の活性を表している。これらの四環系インドール誘導体はまた、体内分布研究においてラットの脳内で妥当な初期濃度を達成するが、その取込みはやはり相対的に低く、改善の余地がある。本発明者らは、これらの従来技術の四環系インドール誘導体の嗅球(最高濃度のPBR受容体を有する脳領域)における相対的な保持が、インビボイメージングにとって望ましいものに満たないことも見出した。したがって、上述の従来技術の四環系インドールインビボ造影剤の有利な特性を保持するPBRインビボ造影剤にも、脳内取込みが改善され、PBR受容体との特異的結合が改善される余地がある。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】国際公開第00/44751号
【発明の概要】
【0013】
本発明は、インビボ造影剤としての使用に適した新規な三環系インドール化合物を提供する。本発明はまた、本発明のインビボ造影剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の合成方法を提供する。本発明の前駆体化合物の使用を含む、インビボ造影剤の調製方法も提供する。さらに、本発明のインビボ造影剤を含む医薬組成物に加えて、医薬組成物の容易な調製に適したキットを提供する。さらなる一態様では、異常なPBR発現に関連する状態のインビボイメージングのためのインビボ造影剤の使用を提供する。本発明のインビボ造影剤は、末梢性ベンゾジアゼピン受容体に対する改善された脳内取込み及び特異性と共に、公知の四環系インビボ造影剤の有利な特性を保持する。
【図面の簡単な説明】
【0014】
【図1】造影剤5と非放射性造影剤5の共溶出を示す図。
【図2】造影剤6と非放射性造影剤6の共溶出を示す図。
【図3】造影剤7と非放射性造影剤7の共溶出を示す図。
【図4】造影剤9と非放射性造影剤9の共溶出を示す図。
【図5】造影剤10(上)及び7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(中間)及び7−フルオロ−9−(2−[19F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(下)を示す図。
【図6】造影剤11と非放射性造影剤11の共溶出を示す図。
【図7】四環系造影剤の脳内での体内分布プロファイルを示す図。
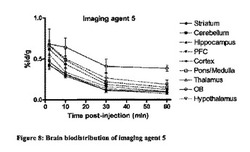
【図8】造影剤5の脳内での体内分布プロファイルを示す図。
【図9】造影剤6の脳内での体内分布プロファイルを示す図。
【図10】造影剤7の脳内での体内分布プロファイルを示す図。
【図11】造影剤9の脳内での体内分布プロファイルを示す図。
【図12】造影剤10の脳内での体内分布プロファイルを示す図。
【図13】造影剤11の脳内での体内分布プロファイルを示す図。
【発明を実施するための形態】
【0015】
造影剤
一態様では、本発明は、次の式Iのインビボ造影剤を提供する。
【0016】
【化1】

式中、
R1はC1〜3アルキル又はC1〜3フルオロアルキルであり、
R2は水素、ヒドロキシル、ハロ、シアノ、C1〜3アルキル、C1〜3アルコキシ、C1〜3フルオロアルキル又はC1〜3フルオロアルコキシであり、
R3及びR4は独立にC1〜3アルキル、C7〜10アラルキルであるか、或いはR3及びR4はそれらと結合した窒素と共に含窒素C4〜6脂肪族環を形成するもので、適宜、窒素、酸素及び硫黄から選択される追加の1個のヘテロ原子を含んでいてもよく、
Y1はO、S、SO、SO2又はCH2であり、
Y2はCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
上記の式Iは、インビボイメージングに適した放射性同位体である原子を含む。
【0017】
「インビボ造影剤」は、本発明の文脈では、インビボイメージングに適した放射性標識合物である。用語「インビボイメージング」は、本明細書で使用される場合、対象の内面のすべて又は一部の画像を非侵襲的に作成する技術を指す。
【0018】
別途記載しない限り、単独又は組合せにおける用語「アルキル」は、好ましくは1〜3個の炭素原子を含有する直鎖又は枝分れアルキル基を意味する。かかる基の例としては、メチル、エチル及びプロピルが挙げられる。
【0019】
別途記載しない限り、用語「アルコキシ」は、エーテル結合を含む上記で定義したアルキル基を意味し、用語「エーテル結合」は、基−C−O−C−を指す。適切なアルキルエーテル基の例としては、メトキシ、エトキシ及びプロポキシが挙げられる。
【0020】
用語「ハロゲン」又は「ハロ−」は、フッ素、塩素、臭素又はヨウ素から選択される置換基を意味する。「ハロアルキル」及び「ハロアルコキシ」は、それぞれ1つ以上のハロゲンで置換されている上記で定義したアルキル及びアルコキシ基である。ハロアルキル及びハロアルコキシ置換基の場合、適切には、ハロゲンは、基の末端の水素を置き換え、すなわち−アルキレン−ハロゲン又は−アルコキシレン−ハロゲンとなる。用語「アルキレン」は、nが1〜3である二価の基−(CH2)n−を指し、用語「アルコキシレン」は、エーテル結合を含むアルキレン基を指し、エーテル結合は上記で定義した通りである。
【0021】
用語「シアノ」は、基−CNを指す。
【0022】
用語「ヒドロキシル」は、基−OHを指す。
【0023】
用語「アラルキル」は、基−アルキレン−フェニルを指し、アルキレンは上記で定義した通りである。
【0024】
「含窒素C4〜6脂肪族環」は、窒素ヘテロ原子を含む飽和C4〜6アルキル環である。その例としては、ピロリジニル、ピペリジニル及びモルホリニル環が挙げられる。
【0025】
用語「インビボイメージングに適した放射性同位体である原子を含む」とは、上記で定義した式Iにおいて、原子の1つの同位体が、インビボイメージングに適した放射性同位体であることを意味する。インビボイメージングに適したものとなるには、放射性同位体が、前記対象への投与後に外部から検出できることが必要である。
【0026】
キラル中心又は別の形態の異性体中心が、本発明のインビボ造影剤に存在する場合、鏡像異性体及びジアステレオ異性体を含むかかる異性体のすべての形態が本発明に包含される。キラル中心を含有する本発明のインビボ造影剤は、ラセミ混合物若しくは鏡像異性体に富んだ混合物として使用することができ、又は周知の技術を使用してラセミ混合物を分離することができ、個々の鏡像異性体を単独で使用することもできる。
【0027】
好ましい造影剤
R1は、好ましくはメチル又はC2〜3フルオロアルキルであり、最も好ましくは−エチレン−F(すなわち−CH2−CH2−F)である。
【0028】
R2は、好ましくは水素、ハロ、C1〜3アルコキシ又はC1〜3フルオロアルコキシである。R2は、最も好ましくは水素、ハロ又はC1〜3アルコキシであり、特に最も好ましくは水素、フルオロ又はメトキシである。R2が置換基である場合、5位又は6位にあるのが好ましく、最も好ましくは5−メトキシ、6−メトキシ、5−フルオロ及び6−フルオロから選択される。
【0029】
R3及びR4は、好ましくは独立に、メチル、エチル又はベンジルであり、最も好ましくは共にエチルである。
【0030】
或いは好ましくは、R3及びR4は、それらに結合した窒素と共に含窒素C5〜6脂肪族環を形成する。
【0031】
Y1は、好ましくはCH2である。
【0032】
本発明の最も好ましいインビボ造影剤では、Y2はCH2−CH2である。
【0033】
本発明の好ましいインビボ造影剤は、単光子放射断層撮影(SPECT)又は陽電子放射断層撮影(PET)を使用するイメージングに適している。SPECTでは、適切な放射性同位体は、γ線を放射する放射性ハロゲンである。本発明における使用に適したγ線を放射する放射性ハロゲンの例は、123I、131I及び77Brである。γ線を放射する好ましい放射性ハロゲンは123Iである。インビボ造影剤の放射性同位体が123Iである場合、R2が123Iであることが好ましい。PETでは、適切な放射性同位体は、陽電子を放出する放射性非金属である。本発明における使用に適した陽電子を放出する放射性非金属の例は、11C、18F及び124Iである。陽電子を放出する好ましい放射性非金属は、11C及び18Fである。11Cの場合、R1が11Cメチルであることが好ましい。放射性同位体が18Fである場合、R1がC2〜3[18F]フルオロアルキルであることが好ましく、最も好ましくは−エチレン−18Fである。
【0034】
本発明のインビボ造影剤がPETイメージングに適していることが好ましく、18Fは、PETイメージングに適した好ましい放射性同位体である。本発明の方法においてPETが好ましいのは、その優れた感受性及び分解能に起因するものであり、したがって病変の相対的に小さい変化も、経時的に観測することができる。PETスキャナーは、ピコモル範囲の放射能濃度を定期的に測定する。マイクロPETスキャナーは、現在約1mmの空間分解及び約4〜5mmの臨床的スキャナーにアプローチするものである。
【0035】
式Iの好ましいインビボ造影剤は、次の式Iaのものである。
【0036】
【化2】
式中、
R2aは水素、ハロ又はC1〜3アルコキシであり、
R3a及びR4aは独立にメチル、エチル若しくはベンジルであるか、或いはそれらに結合した窒素と共にピロリジニル、ピペリジニル、アゼパニル若しくはモルホリニル環を形成するものであり、
Y2aはCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
nは1、2又は3である。
【0037】
式Iaでは、R3a及びR4aは、好ましくは共にエチルであるか、或いはR3aはメチルであってR4aはベンジルであるか、或いはそれらに結合した窒素と共にアゼパニル環を形成するものである。
【0038】
R2aは、好ましくは水素、メトキシ又はフルオロである。
【0039】
Y2aは、好ましくはCH2−CH2又はCH(CH3)−CH2である。
【0040】
nは好ましくは2である。
【0041】
式Iaの好ましいインビボ造影剤では、
R3a及びR4aは共にエチルであるか、或いはR3aがメチルであってR4aがベンジルであるか、或いはそれらに結合した窒素と共にアゼパニル環を形成するものであり、
R2aは水素、メトキシ又はフルオロであり、
Y2aはCH2−CH2又はCH(CH3)−CH2であり、
nは2である。
【0042】
式Iaのインビボ造影剤の非限定的な例は、以下の通りである。
【0043】
【化3】
上記のインビボ造影剤1〜11の中でも、インビボ造影剤5、6、7、9、10及び11が好ましく、インビボ造影剤5及び10が最も好ましく、インビボ造影剤5が特に好ましい。本発明の任意のインビボ造影剤では、鏡像異性的に純粋な形態が特に好ましい。
【0044】
前駆体化合物
別の態様では、本発明は、本発明のインビボ造影剤の調製のための前駆体化合物を提供し、前駆体化合物は、次の式IIの化合物である。
【0045】
【化4】
式中、R11及びR12の一方は、本発明のインビボ造影剤について上記で定義した放射性同位体の適切な供給源と反応して、前駆体化合物と放射性同位体の適切な供給源との反応によって本発明のインビボ造影剤を形成する化学基であり、R11及びR12の他方は、それぞれR1及びR2について本明細書に定義した通りであり、適宜保護基を含んでおり、
R13〜14及びY11〜12は、それぞれR3〜4及びY1〜2について本明細書に定義した通りであり、適宜保護基を含んでいる。
【0046】
「前駆体化合物」は、好都合な化学的形態の検出可能な標識との化学反応が部位特異的に生じ、最小数の段階(理想的には単一段階)で著しい精製の必要なしに(理想的にはさらなる精製せずに)実施されて、所望のインビボ造影剤を生成し得るように設計された、放射性標識合物の非放射性誘導体を含む。かかる前駆体化合物は、合成することができ、好都合には良好な化学的純度で得ることができる。
【0047】
用語「保護基」は、望ましくない化学反応を阻害又は抑制するが、分子の残りを修飾しない穏やかな条件下で、問題の官能基から開裂して所望の生成物をもたらし得るのに十分な反応性を有するように設計されている基を意味する。保護基は、当業者に周知であり、’Protective Groups in Organic Synthesis’,Theorodora W. Greene and Peter G. M. Wuts,(Third Edition,John Wiley & Sons,1999)に記載されている。
【0048】
用語「放射性同位体の適切な供給源」は、前駆体化合物の置換基と反応性であり、したがって放射性同位体が前駆体化合物と共有結合するような化学的形態の放射性同位体を意味する。以下の部分に提示したそれぞれ特定の放射性同位体では、放射性同位体の1つ以上の適切な供給源を論じる。インビボ造影剤の当業者には、本発明における適用に適した放射性同位体のこれら及び他の供給源が周知であろう。
【0049】
以下のスキーム1は、前駆体化合物としてそれ自体使用することができ、又は少数のさらなる段階を用いて前駆体化合物に変換することができる化合物を得る方法を示す一般的な反応スキームである。スキーム1のR11〜14及びY11〜12は、式IIについて上記で定義した通りである。
【0050】
【化5】
或いは、前駆体化合物のR12が環の一番上の位置にある場合、以下のスキームIaに示した一般的な合成経路を使用することができる。
【0051】
【化6】
上記のスキーム1aでは、−R11a−PGは、保護されたR11基を表し、Rは、適切には本明細書に定義したものであることが好ましい。R11がヒドロキシである場合、−R11a−PGは、例えば−O−ベンジルであってよい。R12〜14及びY11〜12は、適切には上記の式IIについて提示したものであることが好ましく、但しR12はクロロではない。この合成経路では、環の一番下の位置の塩素によって、唯一の異性体が生成されるような唯一の方法で、強制的に環化が生じる。類似の方法は、国際公開第2003/014082号に開示されている。しかし、本発明者らが本発明の前駆体化合物を得るために国際公開第2003/014082号の教示を適用すると、その収率は低かった(実施例2(d)参照)。この問題は、環化段階で使用する溶媒系を変更することによって克服した。国際公開第2003/014082号では、環化段階をトルエン中で実施しているが、本発明者らは、トルエンの代わりにジエチルエーテルを使用すると最適な収率が得られることを見出した。環化段階の生成物はジエチルエーテルに溶解するが、非環化出発化合物は溶解しない。したがって、非環化出発化合物は反応容器の底部にZnCl2と共に残存し、環化生成物は反応容器の上部のジエチルエーテル中に移動する。
【0052】
したがって別の一態様では、本発明は、次の式IIbの前駆体化合物の調製方法であって、
【0053】
【化7】
(式中、
R11bは、スキームIaでR11aについて定義した通りであり、
R12b〜14bは、式IIのR12〜14について定義した通りであり、但しR12bはクロロではなく、
Y11b〜12bは、式IIのY11〜12について定義した通りである。)
下記の式IIcの化合物とZnCl2との反応によって、下記の式IIdの化合物を形成する段階を含んでいて、上記の反応を、ジエチルエーテルを含む溶媒系中で実施する方法を提供する。
【0054】
【化8】
(式中、R12c、Y11c及びY12cは、適切にはそれぞれR12、Y11及びY12について本明細書に定義した通りであることが好ましく、PGcは保護基である。)
【0055】
【化9】
(式中、R12d、Y11d、Y12d及びPGdは、それぞれR12c、Y11c、Y12c及びPGcについて定義した通りである。)
好ましくは、前記保護基のPGc、PGdは、−ベンジルである。式IIbの前駆体化合物は、式IIの好ましい前駆体化合物を表す。
【0056】
インビボ造影剤の放射性同位体が18Fである場合、18Fによる標識は、前駆体化合物の脱離基の求核置換によって達成され得る。
【0057】
適切な脱離基にはCl、Br、I、トシレート(OTs)、メシレート(OMs)及びトリフラート(OTf)が挙げられる。もう1つの戦略は、前駆体化合物に存在するアルキルアミド基上の所定位置に適切な脱離基を有することである。いずれの場合も前駆体化合物は、普通は核反応18O(p,n)18Fから水溶液として得られ、カチオン性対イオンの付加及びその後の水の除去によって反応性になる[18F]フッ化物イオン(18F-)の適切な供給源と反応することによって、一段階で標識することができる。18Fは、18F(CH2)3−LG(LGは上記で定義した脱離基を表す)を有する前駆体化合物のヒドロキシル基をO−アルキル化することによって導入することもできる。或いは放射性フッ素原子は、ベンゼン環などの芳香族環との直接共有結合を介して結合することができる。アリール系では、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第4級アンモニウム塩の18F−フッ化物求核置換が、アリール−18F誘導体に適した経路である。
【0058】
以下のスキーム2に示す通り、上記のスキーム1又はスキーム1aのいずれかを継続して、本発明の18Fインビボ造影剤を得るのに適した前駆体化合物に達することができる。
【0059】
【化10】
出発化合物及び中間体は、市販されており、又は刊行された学術論文、例えばNapper et al,J Med Chem 2005;48:8045−54;Davies et al,J Med Chem 1998;41:451−467から公知である。
【0060】
18Fを含むインビボ造影剤を得るための、式IIの好ましい前駆体化合物では、R11はC1〜3アルキレン−LGであり、LGは脱離基を表す。最も好ましいかかる前駆体化合物は、式IIa
【0061】
【化11】
(式中、
LGは、メシレート、トシレート及びトリフラートから選択され、
R12a〜14a、Y12a及びmは、適切にはそれぞれ式IaのR2a〜4a、Y2a及びnについて上記で定義した通りであることが好ましい]
の化合物である。
【0062】
式IIaの好ましい前駆体化合物の非限定的な例は、以下の通りである。
【0063】
【化12】
上記の前駆体化合物1〜11の中でも、前駆体化合物5、6、7、9、10及び11が好ましく、前駆体化合物5及び10が最も好ましく、前駆体化合物5が特に好ましい。
【0064】
11Cで標識したPETトレーサー化合物は、前駆体化合物を11Cヨウ化メチルと反応させることによって合成することができる。11Cの半減期はわずか20.4分なので、中間体11Cヨウ化メチルが高い特異的活性を有し、その結果、可能な限り急速な反応過程を使用して中間体11Cヨウ化メチルを生成することが重要である。かかる11Cによる標識技術の詳しい総説は、Antoni et al,“Aspects on the Synthesis of 11C−Labelled Compounds” in Handbook of Radiopharmaceuticals,Ed. M.J. Welch and C.S. Redvanly(2003,John Wiley and Sons)に見ることができる。
【0065】
本発明の11Cで標識したインビボ造影剤は、以下のスキーム3に示す通り上記のスキーム1を継続することによって得ることができる。
【0066】
【化13】
イメージング部分が放射性ヨウ素である場合、好ましい前駆体化合物は、求電子性ヨウ素化を受ける誘導体を含む化合物である。この例は、トリアルキルスタンナン(例えば、トリメチルスタンニル又はトリブチルスタンニル)などの有機金属誘導体、又はトリアルキルシラン(例えば、トリメチルシリル)、又は有機ホウ素化合物(例えば、ボロン酸エステル又は有機三フッ化ホウ素)である。
【0067】
求電子性の放射性ヨウ素標識では、前駆体化合物は、好ましくは活性化有機金属前駆体化合物(例えば、トリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)を含む。放射性ヨウ素を有機分子に導入する前駆体化合物及び方法は、Bolton(J. Lab. Comp. Radiopharm. 2002;45:485−528)に記載されている。適切なボロン酸エステルである有機ホウ素化合物及びそれらの調製は、Kabalaka et al(Nucl. Med. Biol.,2002;29:841−843 and 2003;30:369−373)に記載されている。適切な有機三フッ化ホウ素及びそれらの調製は、Kabalaka et al(Nucl. Med. Biol.,2004;31:935−938)に記載されている。放射性ヨウ素標識にとって好ましい前駆体化合物は、有機金属前駆体化合物、最も好ましくはトリアルキルスズを含む。
【0068】
放射性ヨウ素で標識した本発明のインビボ造影剤は、以下のスキーム4に示す通り上記のスキーム1を継続することによって得ることができる。
【0069】
【化14】
放射性臭素標識は、放射性ヨウ素標識について前述の方法と類似の方法によって達成することができる。Kabalka及びVarmaは、放射性臭素標識化合物を含む放射性ハロゲン標識化合物の合成のための様々な方法を総説した(Tetrahedron 1989年;45(21):6601〜21頁)。
【0070】
本発明の前駆体化合物は、理想的には非発熱性の滅菌形態で提供される。したがって前駆体化合物は、インビボ造影剤を哺乳動物への投与に適した生体適合性の担体と一緒に含む医薬組成物の調製のために使用することができる。前駆体化合物はまた、かかる医薬組成物の調製のためのキット又はカセットの成分として含まれるのに適している。これらの態様を、以下により詳細に論じる。
【0071】
別の好ましい実施形態では、前駆体化合物は固相と結合している。前駆体化合物は、好ましくは、固体支持マトリックスと共有結合した状態で供給される。この方法では、所望の生成物は溶液の形態であり、出発材料及び不純物は固相に結合したままである。かかる系の一例として、18F−フッ化物による固相求電子性フッ素化のための前駆体化合物は、国際公開第03/002489号に記載されており、18F−フッ化物による固相求核フッ素化のための前駆体化合物は、国際公開第03/002157号に記載されている。
【0072】
調製方法
さらなる一態様では、本発明は、
(i)本発明の前駆体化合物を提供する段階と、
(ii)本明細書に定義の前記放射性同位体の適切な供給源を提供する段階と、
(iii)段階(i)の前駆体化合物を、段階(ii)の放射性同位体と反応させて、本発明のインビボ造影剤を得る段階と
を含む、本発明のインビボ造影剤を調製する方法を提供する。
【0073】
段階(i)では、前駆体化合物は、前駆体化合物の説明で前述した通り、自動合成装置との併用に適したキット又はカセット内の溶液として、或いは固体支持体に結合した状態で提供することができる。キット及びカセットは、本発明のさらなる態様を形成するものであり、これらを以下により詳細に論じることにする。
【0074】
前駆体化合物を放射性同位体と「反応させる」段階は、可能な限り高い放射化学的収率(RCY)で所望のインビボ造影剤を形成するのに適した反応条件下で、2つの反応物を一緒にすることを含む。本発明のインビボ造影剤を得るためのいくつかの特定の合成経路を、以下の実験部分に提示する。
【0075】
本発明の調製方法について、インビボ造影剤、前駆体化合物及び放射性同位体の適切な好ましい実施形態は、既に本明細書に提示した通りである。
【0076】
キット及びカセット
またさらなる一態様では、本発明は、本発明のインビボ造影剤を調製するためのキットを提供し、前記キットは、本発明の前駆体化合物を含み、したがって放射性同位体の滅菌供給源との反応によって、最小限の操作数で所望のインビボ造影剤を得るようにされている。かかる考察は、放射性同位体が相対的に短い半減期を有する場合に特に重要であり、放射線技師(radiopharmacist)にとっての取扱いを容易にし、したがって放射線量を低減するのに重要である。前駆体化合物は、好ましくは凍結乾燥形態でキット内に存在し、かかるキットの再構成のための反応媒体は、好ましくは生体適合性の担体である。
【0077】
「生体適合性の担体」は、インビボ造影剤を懸濁又は溶解することによって組成物が生理的に容認できるものとなる、すなわち毒性又は過度の不快感なしに哺乳動物の身体に投与することができる流体、特に液体である。生体適合性の担体は、適切には、発熱物質を含まない注入用の滅菌水;生理食塩水(注入用の最終生成物が、等張性であるように、又は低張性にならないように平衡化できることが有利である)などの水溶液;1つ以上の浸透圧調節物質(例えば、血漿カチオンと生体適合性の対イオンの塩)、糖類(例えば、グルコース又はスクロース)、糖アルコール(例えば、ソルビトール又はマンニトール)、グリコール(例えば、グリセロール)又は他の非イオン性ポリオール材料(例えば、ポリエチレングリコール、プロピレングリコール等)の水溶液などの注入可能な担体液体である。生体適合性の担体は、エタノールなどの生体適合性の有機溶媒を含むこともできる。かかる有機溶媒は、より親油性が高い化合物又は配合物を可溶化するのに有用である。好ましくは、生体適合性の担体は、発熱物質を含まない注入用の水、等張食塩水又はエタノール水溶液である。静脈内注入のための生体適合性の担体のpHは、適切には4.0〜10.5の範囲である。
【0078】
本発明のキットでは、前駆体化合物は、好ましくは滅菌上の完全性及び/又は放射性の安全性と、任意選択により不活性なヘッドスペースガス(例えば、窒素又はアルゴン)の維持を可能にすると同時に、シリンジによる溶液の添加及び除去を可能にする封止容器中に存在する。好ましい封止容器は、セプタムで封止したバイアルであり、その場合気密にする蓋は、オーバーシール(一般にアルミニウム製)で圧着される。かかる封止容器は、その蓋が所望に応じて真空に耐えて、例えばヘッドスペースガスを取り替え、又は溶液を脱気することができるというさらなる利点を有する。
【0079】
キットで使用される場合、前駆体化合物の好ましい実施形態は、既に本明細書に記載した通りである。
【0080】
キットで使用するための前駆体化合物は、所望の非発熱性の滅菌材料を得るために、無菌の製造条件下で使用することができる。或いは、前駆体化合物を非滅菌条件下で使用し、その後、例えばγ照射、高圧蒸気殺菌法、乾熱又は化学的処理(例えば、エチレンオキシドを用いる)を使用して最終滅菌にかけることができる。好ましくは、前駆体化合物は、非発熱性の滅菌形態で提供される。最も好ましくは、非発熱性の滅菌前駆体化合物は、前述の封止容器で提供される。
【0081】
キットのすべての成分は、実施と実施の間の汚染の可能性を最小限に抑え、無菌性及び品質の保持を確実にするために使い捨てできることが好ましい。
【0082】
現在、特に[18F]放射性トレーサーは、好都合には自動化放射合成(radiosynthesis)装置で調製されることが多い。Tracerlab(商標)及びFastlab(商標)(GE Healthcare Ltd)を含む、かかる装置の市販で利用可能ないくつかの例がある。かかる装置は一般に、放射合成を実施するための装置に備え付けられる、しばしば使い捨ての「カセット」を含み、そのカセットの中では放射化学が行われる。カセットは、普通は、流体経路、反応容器、及び試薬のバイアルを入れるポート、並びに放射合成後の除去段階で使用する任意の固相抽出カートリッジを含む。
【0083】
したがって本発明は、別の態様では、
(i)本明細書に定義の前駆体化合物を含有する容器と、
(ii)その容器を、本明細書に定義のインビボイメージングに適した前記放射性同位体の適切な供給源を用いて溶出するための手段と
を含む、本明細書に定義のインビボ造影剤の自動合成のためのカセットを提供する。
【0084】
本発明のカセットについて、前駆体化合物及び放射性同位体の適切な供給源の適切な好ましい実施形態は、既に本明細書に定義した通りである。
【0085】
カセットは、
(iii)過剰の放射性同位体を除去するためのイオン交換カートリッジと、任意選択により
(iv)前駆体化合物が1つ以上の保護基を含む場合、得られた放射性標識生成物を脱保護して本明細書に定義のインビボ造影剤を形成するためのカートリッジと
をさらに含むことができる。
【0086】
放射性医薬組成物
別のさらなる一態様では、本発明は、哺乳動物への投与に適した形態の生体適合性の担体と一緒に本発明のインビボ造影剤を含む、「放射性医薬組成物」を提供する。生体適合性の担体は、本発明のキットに関して上記で定義した通りである。本発明の放射性医薬組成物について、インビボ造影剤の適切な好ましい実施形態は、既に本明細書に定義した通りである。
【0087】
放射性医薬組成物は、非経口によって、すなわち注入によって投与することができ、最も好ましくは水溶液である。かかる組成物は、任意選択により緩衝液;薬学的に許容される可溶化剤(例えば、シクロデキストリン又はPluronic、Tween若しくはリン脂質などの界面活性剤);薬学的に許容される安定剤又は抗酸化剤(アスコルビン酸、ゲンチシン酸又はパラアミノ安息香酸など)などのさらなる成分を含有することができる。本発明のインビボ造影剤が放射性医薬組成物として提供される場合、前記インビボ造影剤の調製方法は、放射性医薬組成物を得るために必要な段階、例えば有機溶媒の除去、生体適合性の緩衝液の添加、及び任意の場合によるさらなる成分の添加をさらに含むことができる。非経口投与では、放射性医薬組成物が滅菌され、非発熱性であることを確実にする段階を含む必要もある。
【0088】
使用方法
またさらなる一態様では、本発明は、
(i)前記対象に、本発明のインビボ造影剤を投与する段階と、
(ii)前記インビボ造影剤を前記対象のPBRと結合させる段階と、
(iii)インビボイメージング手順によって、前記インビボ造影剤の放射性同位体から放出されたシグナルを検出する段階と、
(iv)前記シグナルの位置及び/又は量の代表的な画像を作成する段階と、
(v)前記インビボ造影剤によって放出された前記シグナルと直接相関する、前記対象におけるPBR発現の分布及び程度を決定する段階と
を含む、対象におけるPBR発現の分布及び/又は程度を決定するためのインビボイメージング法を提供する。
【0089】
本発明のインビボイメージング法について、インビボ造影剤の適切な好ましい実施形態は、既に本明細書に定義した通りである。
【0090】
インビボ造影剤の「投与」は、好ましくは非経口により、最も好ましくは静脈内により実施される。静脈内経路は、インビボ造影剤を対象の身体にわたって送達し、したがって血液脳関門(BBB)にも通過させ、前記対象の中枢神経系(CNS)に発現したPBRと接触させるための最も効率的な方法である。さらに、静脈内投与は、実質的な身体的介入又は実質的な健康上の危険性を示さない。本発明のインビボ造影剤は、好ましくは本明細書に定義の本発明の医薬組成物として投与される。本発明のインビボイメージング法は、本発明のインビボ造影剤を予め投与した対象に対して実施される上記で定義した段階(ii)〜(v)を含むと理解することもできる。
【0091】
投与段階の後、検出段階の前に、インビボ造影剤をPBRと結合させる。例えば、対象が無傷哺乳動物である場合、インビボ造影剤は、哺乳動物の身体中を動的に移動し、その様々な組織と接触することになる。インビボ造影剤がPBRと接触すると特異的な相互反応が生じ、その結果、PBRを有する組織からのインビボ造影剤のクリアランスは、PBRのない、又はPBRの少ない組織からのクリアランスよりも長くかかる。PBRを有する組織と結合したインビボ造影剤と、PBRのない、又はPBRの少ない組織において結合したインビボ造影剤との間の比の結果、PBRに特異的に結合したインビボ造影剤の検出が可能となるある時点に達することになる。かかる理想的な比は、約2:1である。
【0092】
本発明の方法の「検出」段階は、前記シグナルに感受性のある検出器を用いて放射性同位体が放出するシグナルを検出することを含む。この検出段階は、シグナルデータの取得と理解することもできる。単光子放射型断層撮影(SPECT)及び陽電子放射断層撮影(PET)は、本発明の方法における使用に最も適したインビボイメージング手順である。PETは、本発明の方法における使用に好ましいインビボイメージング手順である。
【0093】
本発明の方法の「作成」段階は、取得したシグナルデータに再構成アルゴリズムを適用してデータセットを得るコンピューターによって実施される。次いで、このデータセットを操作して、前記放射性同位体が放出するシグナルの位置及び/又は量を示す画像を作成する。放出されたシグナルは、PBRの発現と直接相関し、したがって、作成した画像を評価することによって「決定」段階を行うことができる。
【0094】
本発明の「対象」は、任意のヒト又は動物対象であってよい。好ましくは、本発明の対象は哺乳動物である。最も好ましくは、前記対象は、インビボの無傷哺乳動物の身体である。特に好ましい一実施形態では、本発明の対象はヒトである。インビボイメージング法は、健康な対象において、又はPBRの異常発現に関連する病理学的状態(以下「PBR状態」)を有することが公知の、若しくはその疑いのある対象においてPBRを試験するために使用することができる。好ましくは、前記方法は、PBR状態を有することが公知の、又はその疑いのある対象におけるインビボイメージングに関し、したがって前記状態の診断方法において有用性がある。
【0095】
インビボイメージングが役立つかかるPBR状態の例としては、多発性硬化症、ラスムッセン脳炎、脳血管炎、ヘルペス脳炎、AIDS関連認知症、パーキンソン病、大脳皮質基底核変性症、進行性核上性麻痺、多系統萎縮症、ハンチントン病、筋萎縮性側索硬化症、アルツハイマー病、虚血性脳卒中、末梢神経損傷、てんかん、外傷性脳損傷、急性ストレス、慢性ストレス、神経障害性疼痛、肺炎症、慢性閉塞性肺疾患、喘息、炎症性腸疾患、関節リウマチ、原発性線維筋痛症、神経損傷、アテローム性動脈硬化症、腎炎、虚血再灌流障害、及び癌、特に結腸、前立腺又は乳房の癌が挙げられる。本発明のインビボ造影剤は、特に、脳へのそれらの良好な取込みに起因して、CNSのインビボイメージングに適している。
【0096】
代替の一実施形態では、本発明のインビボイメージング法は、前記対象のための治療投与計画の過程中、反復して実施することができ、前記投与計画は、PBR状態に対抗するための薬物を投与する段階を含む。例えば、本発明のインビボイメージング法は、PBR状態に対抗するための薬物を用いる治療の前、その最中、及びその後に実施することができる。この方法では、前記治療の作用を経時的にモニタすることができる。好ましくはこの実施形態では、インビボイメージング手順はPETである。PETは、優れた感受性及び分解能を有し、したがって病変における相対的に小さい変化も経時的に観察することができ、このことは治療モニタリングにとって特に有利である。
【0097】
さらなる一態様では、本発明は、PBR状態の診断方法を提供する。本発明の診断方法は、上記で定義したインビボイメージング法を、PBR発現の分布及び程度が特定の臨床像に帰するさらなる段階(vi)、すなわち演繹的な医学的決定段階と一緒に含む。
【0098】
別の態様では、本発明は、本明細書に定義の診断方法において使用するための、本明細書に定義のインビボ造影剤を提供する。
【0099】
またさらなる一態様では、本発明は、本明細書に定義の診断方法において使用する本明細書に定義の放射性医薬組成物の製造に使用するための、本明細書に定義のインビボ造影剤を提供する。
【0100】
本発明を、ここで一連の非限定的な例によって例示する。
【実施例】
【0101】
実施例の簡単な説明
実施例1は、前駆体化合物5及び造影剤5の合成を記載する。
【0102】
実施例2は、造影剤5の非放射性類似体の合成を記載する。
【0103】
実施例3は、前駆体化合物6及び造影剤6の合成を記載する。
【0104】
実施例4は、造影剤6の非放射性類似体の合成を記載する。
【0105】
実施例5は、前駆体化合物7及び造影剤7の合成を記載する。
【0106】
実施例6は、造影剤7の非放射性類似体の合成を記載する。
【0107】
実施例7は、前駆体化合物9及び造影剤9の合成を記載する。
【0108】
実施例8は、造影剤9の非放射性類似体の合成を記載する。
【0109】
実施例9は、前駆体化合物10及び造影剤10の合成を記載する。
【0110】
実施例10は、造影剤10の非放射性類似体の合成を記載する。
【0111】
実施例11は、前駆体化合物11及び造影剤11の合成を記載する。
【0112】
実施例12は、造影剤11の非放射性類似体の合成を記載する。
【0113】
実施例13は、前駆体化合物5の鏡像異性体分離を記載する。
【0114】
実施例14は、非放射性造影剤5の鏡像異性体分離を記載する。
【0115】
実施例15は、PBRに対する親和性を試験するために使用したインビトロ効力アッセイを記載する。
【0116】
実施例16は、インビボでの本発明の造影剤の性能を試験するために使用した体内分布法を記載する。
【0117】
実施例17は、過去の四環系インドール造影剤の非放射性類似体の合成を記載する。
【0118】
実施例18は、過去の四環系インドール造影剤の合成を記載する。
【0119】
実施例で使用した略語一覧
aq 水性
DCM ジクロロメタン
DMAP 4−ジメチルアミノピリジン
DMF ジメチルホルムアミド
EDC 1−エチル−3−[3−ジメチルアミノプロピル]カルボジイミド塩酸塩
EOS 合成の終了
EtOAc 酢酸エチル
IPA イソプロピルアルコール
LC−MS 液体クロマトグラフィー−質量分析
NMR 核磁気共鳴
OBn ベンジルオキシ
OMs メシレート
OTs トシレート
RT 室温
TLC 薄層クロマトグラフィー
Tol トルエン
実施例
実施例1:メタンスルホン酸2−(4−ジエチルカルバミル−5−メトキシ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)エチルエステル(前駆体化合物5)及び9−(2−[18F]フルオロ−エチル)−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤5)の合成
実施例1(a):塩化ベンジルオキシアセチル(1)
ジクロロメタン(50mL)中ベンジルオキシ酢酸(10.0g、60.0mmol、8.6mL)に、塩化オキサリル(9.1g、72.0mmol、6.0mL)及びDMF(30.0mg、0.4mmol、32.0μL)を添加し、RTで3時間撹拌した。最初、反応が進行するにつれてガスが急速に発生したが、反応が完了すると発生は停止した。ジクロロメタン溶液を真空中で濃縮してガムを得た。このガムを、さらなる塩化オキサリル(4.5g、35.7mmol、3.0mL)、ジクロロメタン(50mL)及び一滴のDMFで処理した。ガスが急速に発生し、反応物をさらに2時間撹拌した。次いで、反応物を真空中で濃縮して、塩化ベンジルオキシアセチル(1)11.0g(定量的)をガムとして得た。構造を、13C NMR(75MHz,CDCl3)δC73.6,74.8,128.1,128.4,128.6,130.0及び171.9によって確認した。
【0120】
実施例1(b):2−ベンジルオキシ−N−(2−クロロ−5−メトキシ−フェニル)アセトアミド(2)
ジクロロメタン(100mL)中塩化ベンジルオキシアセチル(1)(11.0g、60.0mmol)及び2−クロロ−5−メトキシアニリン塩酸塩(11.7g、60.2mmol)を0℃で撹拌し、トリエチルアミン(13.0g、126.0mmol、18.0mL)を15分かけてゆっくり添加した。撹拌した反応物を、RTにして18時間かけて温めた。トリエチルアミン塩酸塩の大量の沈殿が生じた。ジクロロメタン溶液を10%炭酸カリウム水溶液(50mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、2−ベンジルオキシ−N−(2−クロロ−5−メトキシ−フェニル)アセトアミド(2)18.9g(定量的)をガムとして得た。構造を、13C NMR(75MHz,CDCl3):δC55.6,69.6,73.6,106.2,111.1,114.1,127.7,128.3,128.6,129.2,134.6,136.5,158.9及び167.7によって確認した。
【0121】
実施例1(c):(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシフェニル)アミン(3)
THF(100mL)中2−ベンジルオキシ−N−(2−クロロ−5−メトキシ−フェニル)アセトアミド(2)(18.9g、62.0mmol)を撹拌し、水素化アルミニウムリチウム(4.9g、130.0mmol)を15分かけてゆっくり添加した。最初に水酸化アルミニウムリチウムを添加すると、水素ガスが急速に発生した。次いで、反応物を4時間加熱還流し、週末にかけてRTで静置した。次いで、撹拌した溶液に水(50mL)を滴下添加することによって、反応物をクエンチした。水素が強烈に発生して、反応混合物が還流した。次いで、反応物を真空中で濃縮してスラリーを得た。水(200mL)及び酢酸エチル(200mL)を添加し、混合物を激しく振とうした。次いで、セライトを介して反応物を濾過して、沈殿した水酸化アルミニウムを除去し、酢酸エチル溶液を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシフェニル)アミン(3)18.4g(定量的)をガムとして得た。構造を、13C NMR(75MHz,CDCl3)δC43.3,55.3,68.2,73.0,98.1,101.8,111.6,127.6,127.7,128.4,129.3,137.9,144.8及び159.5によって確認した。
【0122】
実施例1(d):3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4)
2−オキソシクロヘキサンカルボン酸エチル(30g、176mmol、28mL)をジエチルエーテル(30mL)に溶解し、窒素下で0℃に冷却した。臭素(28g、176mmol、9.0mL)を15分かけて滴下添加し、反応混合物をRTにして90分かけて温めた。混合物を、氷冷した飽和炭酸カリウム水溶液(250mL)にゆっくり注ぎ、酢酸エチル(3×200mL)で抽出した。混合有機層を硫酸マグネシウムで乾燥させ、濾過し、真空中で濃縮し、真空ラインで18時間乾燥させて、3−ブロモ−2−ヒドロキシ−1−エンカルボン酸エチルエステル(4)41.4g(94%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3):δC14.1,17.7,21.8,32.0,60.0,60.8,99.7,166.3及び172.8によって確認した。
【0123】
実施例1(e):3[(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシ−フェニル)−アミノ]−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(5)
(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシフェニル)アミン(3)(10.0g、34.2mmol)を、乾燥THF(100mL)中、窒素下において−40℃で撹拌し、カリウムビス(トリメチルシリル)アミド(トルエン中0.5M溶液143.0mL、72.0mmol)を30分かけて添加した。次いで、乾燥THF(10mL)中3−ブロモ−2−ヒドロキシシクロヘキセ−1−エンカルボン酸エチルエステル(4)(8.5g、34.2mmol)を添加し、RTにして1.5時間かけて温めた。酢酸(10.0g、166mmol、10.0mL)を添加し、真空中で濃縮してTHFを除去した。酢酸エチル(200mL)及び10%炭酸カリウム水溶液(100mL)を添加し、混合物を激しく振とうした。酢酸エチル溶液を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、3[(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシ−フェニル)−アミノ]−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(5)16.5g(定量的)をガムとして得、それを次の段階で粗生成物として使用した。粗反応混合物のHPLC(Gemini 150×4.6mm、20分間50〜95%のメタノール/水)、18.9分(38%)、19.2分(25%)、23.1分(28%)。
【0124】
反応物の一成分を単離した13C NMR(75MHz,CDCl3)δC14.3,20.6,21.8,26.4,38.6,43.0,55.8,60.5,68.7,73.3,93,4,106.3,108.2,119.3,121.5,127.5,127.6,128.3,135.7,137.0,137.9,155.7及び175.0。
【0125】
実施例1(f):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)
塩化亜鉛(7.1g、52.0mmol)を、乾燥ジエチルエーテル(150mL)中3[(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシ−フェニル)−アミノ]−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(5)(8.0g、17.0mmol)に窒素下で添加し、5.5時間加熱還流した。反応物を還流すると、反応物中に高密度の褐色油が形成した。次いで反応物を冷却し、上清であるジエチルエーテルをデカントで除去し、酢酸エチル(100mL)を添加し、2NのHCl(50mL)及び10%炭酸カリウム水溶液(50mL)で洗浄した。ジエチルエーテル層を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮して油(2.0g)を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(10〜40%の(B)、340g、22CV、150mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)1.8gを得た。高密度の褐色層を、酢酸エチル(100mL)及び2NのHCl(50mL)で処理した。酢酸エチル溶液を分離し、10%炭酸カリウム水溶液(50mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して油(5.2g)を得た。ジエチルエーテル(100mL)及び無水塩化亜鉛(7.0g)を添加した。混合物を、さらに5日間加熱還流した。エーテル層を暗色ガムからデカントで除去し、2NのHCl(50mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガム(2.8g)を得た。このガムを、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(5〜35%の(B)、340g、150mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)2.1gを得た。得られたすべての材料は、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)4.1g(50%)であった。構造を、13C NMR(75MHz,CDCl3):δC14.4,20.5,22.3,27.5,40.2,43.9,55.0,60.2,70.7,73.3,100.2,107.5,108.4,120.1,122.8,127.4,127.5,128.2,132.0,137.4,138.1,152.6及び175.8によって確認した。
【0126】
実施例1(g):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(7)
エタノール(50mL)中9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)(2.0g、4.1mmol)に、水酸化ナトリウム(1.1g、27.1mmol)及び水(5mL)を添加し、80℃で18時間加熱した。次いで、エタノールを真空中で蒸発することによって除去し、残渣をジエチルエーテル(50mL)と水(50mL)に分離した。ジエチルエーテル層を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガム(71.0mg)を得た。水層を、2NのHCl(20mL)でpH1の酸性にし、ジクロロメタン(2×100mL)で抽出した。ジクロロメタン層を硫酸マグネシウムで乾燥させ、真空中で濃縮して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(7)1.6g(87%)を気泡として得た。構造を、13C NMR(75MHz;CDCl3):δC20.2,22.2,27.1,39.7,44.0,55.1,70.7,73.3,100.6,106.3,108.9,123.0,127.4,127.5,128.3,132.0,138.0及び152.0によって確認した。
【0127】
実施例1(h):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(8)
9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(7)(1.5g、3.7mmol)をジクロロメタン(50mL)に溶解し、塩化オキサリル(700mg、5.5mmol、470μL)及びDMF(1滴)を添加し、反応物を20℃で2時間撹拌した。反応が進行するにつれて、約30分間ガスが穏やかに発生した。次いで、反応物を真空中で濃縮して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(8)をガムとして得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz;CDCl3):δC 20.8,22.1,26.4,44.2,51.8,55.1,70.7,73.3,100.7,106.0,108.6,119.5,123.4,127.3,127.7,128.3,131.9,138.0,138.2,152.0.及び176.3によって確認した。
【0128】
実施例1(i):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(9)
9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(8)(1.6g、3.7mmol)を、次いでジクロロメタン(50mL)に溶解し、0℃に冷却し、撹拌し、ジエチルアミン(810mg、11.0mmol、1.1mL)を滴下添加した。反応物を、室温にして18時間かけて温めた。次いで、反応混合物を10%炭酸カリウム水溶液(50mL)で洗浄し、分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガムを得た。粗材料を、ジエチルエーテルから結晶化させて、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(9)1.2g(71%)を白色結晶固体として得た。構造を、13C NMR(75MHz;CDCl3):δC 13.0,14.5,19.8,22.2,27.9,36.4,40.4,41.9,43.8,55.0,70.8,73.3,100.2,108.5,108.6,119.9,122.5,127.4,127.5,128.3,131.5,137.8,138.2,152.4及び174.5によって確認した。
【0129】
実施例1(j):9−(2−ベンジルオキシ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(10)
メタノール(100ml)中9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(9)(1.0g、2.1mmol)を、木炭上10%パラジウム(1.0g)、トリエチルアミン(2.9mg、2.9mmol、4μL)と共に、水素ガス雰囲気下において55℃で18時間振とうした。次いで、セライトパッドを介して反応物を濾過し、濾液を真空中で濃縮してガム(908mg)を得た。次いで、ガムをジクロロメタン(100ml)に溶解し、5%炭酸カリウム水溶液(50ml)で洗浄した。次いで、ジクロロメタン溶液を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガムを得た。次いで、ガムをジエチルエーテル(50ml)から結晶化させ、結晶を濾過によって収集して、9−(2−ベンジルオキシ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(10)523mg(57%)を得た。構造を、13C NMR(75MHz;CDCl3):δC13.1,14.6,20.1,22.0,28.1,36.4,40.5,42.0,43.0,54.7,68.8,73.3,99.4,102.4,107.8,116.4,121.2,127.6,127.6,128.3,135.6,137.8,138.0 153.6及び175.0によって確認した。
【0130】
実施例1(k):9−(2−ヒドロキシエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(11)
メタノール(50ml)中9−(2−ベンジルオキシ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(10)(1.0g、2.1mmol)を、木炭上10%パラジウム(300mg)及び過剰の水素ガスを用いて55℃で18時間振とうした。次いで、セライトパッドを介して反応物を濾過し、濾液を真空中で濃縮して、9−(2−ヒドロキシエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(11)578mg(100%)を気泡として得た。構造を、13C NMR(75MHz;CDCl3):δC13.0,14.4,20.0,22.0,28.0,36.4,40.6,42.0,54.7,60.6,99.2,102.6,107.0,116.7,121.1,136.1,137.5,138.0 153.5及び175.7によって確認した。
【0131】
実施例1(l):メタンスルホン酸2−(4−ジエチルカルバミル−5−メトキシ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)エチルエステル(前駆体化合物5)
ジクロロメタン(30ml)中9−(2−ヒドロキシエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(11)(478mg、1.4mmol)を0℃に冷却し、塩化メタンスルホニル(477mg、4.2mmol、324μL)及びトリエチルアミン(420mg、4.2mmol、578μL)を添加し、RTにして終夜温めた。反応物を5%炭酸カリウム水溶液で洗浄した。層を分離した。混合有機物を硫酸マグネシウムで乾燥させ、真空中で濃縮してガム(696mg)を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(75〜100%のB、22CV、120g、85mL/分)で溶出して精製して、メタンスルホン酸2−(4−ジエチルカルバミル−5−メトキシ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)エチルエステル(前駆体化合物5)をガムとして得、それをジエチルエーテルから結晶化させて、無色固体346mg(59%)を得た。構造を、13C NMR(75MHz;CDCl3):δC13.1,14.5,20.0,21.9,28.0,36.3,36.7,40.3,41.8,41.9,54.7,68.1,100.0,102.0,109.0,116.4,122.0 135.1,137.3,153.8及び174.6によって確認した。
【0132】
実施例1(m):9−(2−[18F]フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤5)
GE HealthcareからGE PETraceサイクロトロンにより[18F]フッ化物が供給された。Kryptofix 2.2.2(2mg、5μmol)、重炭酸カリウム(0.1mol dm-3、0.1ml、5mg、5μmol)及びアセトニトリル(0.5ml)を、COC反応容器中[18F]F-/H2O(約400MBq、0.1〜0.3ml)に添加した。混合物を、100℃で20〜25分間、窒素流の下で加熱することによって乾燥させた。乾燥後、冷却なしにアセトニトリル(1ml)中前駆体化合物5(0.5〜1mg、1.2〜2.4μmol)をCOC反応容器に添加し、100℃で10分間加熱した。冷却後、反応混合物を除去し、COC反応容器を水(1.5ml)ですすぎ、主な粗反応物に添加した。
【0133】
この後、粗生成物に、半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配(flow gradient):3ml/分;0〜1分、40%B;1〜20分、40〜95%B;波長254nm;tR造影剤5、16分を適用した。HPLCの精製ピークである造影剤5を、水で体積10mlに希釈し、tC18 Sep−Pak(lite)カートリッジに吸着させた。カートリッジを水(2ml)で洗浄し、無水エタノール(0.5ml)で溶出し、その後ダルベッコリン酸緩衝食塩水(4.5ml)で溶出した。放射化学的収率30±7%(n=4)減衰補正なし、時間90〜120分、放射化学的純度≧99%。
【0134】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、40%B;1〜20分、40〜95%B;波長230nm;tR造影剤5、16分。図1は、造影剤5と非放射性造影剤5の共溶出を示す。
【0135】
実施例2:9−(2−フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤5)の合成
実施例2(a):フルオロエチルトシレート(12)
2−フルオロエタノール(640mg、10mmol、0.6mL)を、窒素下でピリジン(10mL)に溶解した。溶液を0℃で撹拌し、塩化トシル(4.2g、21.8mmol)を、5℃未満の温度を維持しながら30分かけて溶液に少しずつ添加した。反応物を0℃で3時間撹拌した。氷をゆっくり添加し、その後水(20mL)を添加した。反応混合物を酢酸エチルに抽出し、水で洗浄した。水層が酸性になるまで1NのHCl溶液で洗浄することによって、過剰のピリジンを除去した。過剰の塩化トシルを、1Mの炭酸ナトリウム水溶液で洗浄することによって除去した。有機層をブラインで洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、フルオロエチルトシレート(12)2.1g(98%)を無色油として得た。構造を、13C NMR(75MHz,CDCl3):δC21.6(CCH3),68.5(d,JCF=173Hz,OCH2CH2F),80.6(d,JCF=173Hz,OCH2CH2F),128.0,129.9,132.6及び145.1によって確認した。
【0136】
実施例2(b):2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13)
2−クロロ−5−メトキシアニリン塩酸塩(5.0g、26.0mmol)をDMF(50mL)に溶解し、水素化ナトリウム(2.3g、油中60%、57.0mmol)を添加した。反応物を窒素下においてRTで30分間撹拌した。DMF(5mL)中フルオロエチルトシレート(12)(6.7g、31.0mmol)を滴下添加し、反応物をRTで2時間撹拌した。次いで、反応物を100℃で18時間加熱した。反応物を冷却し、溶媒を減圧下で除去した。残渣を酢酸エチル(100mL)に溶解し、水(2×100mL)で洗浄した。有機物を収集し、硫酸マグネシウムで乾燥させ、真空中で濃縮して褐色油を得、それをシリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(5〜30%(B)、330g、18.1CV、120mL/分)で溶出して精製して、2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13)1.3g(25%)を黄色油として得た。構造を、13C NMR(75MHz;CDCl3):δC43.8(d,JCF=23Hz),55.3,82.0(d,JCF=165Hz),98.1,102.2,111.6,129.5,144.1及び159.5によって確認した。
【0137】
実施例2(c):3−[(2−クロロ−5−メトキシ−フェニル)−(2−フルオロエチル)アミノ]−2−ヒドロキシ−シクロヘキシル−1−エンカルボン酸エチルエステル(14)
2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13)(6.1g、30.0mmol)のTHF(170mL)溶液を−40℃に冷却した。カリウムビス(トリメチルシリル)アミド(トルエン中0.5M溶液126.0mL、63.0mmol)を滴下添加し、反応物を−40℃で30分間撹拌した。THF(30mL)中3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4;実施例1(d)に従って調製した)(7.4g、30.0mmol)を、−40℃で滴下添加した。冷却浴を除去し、反応物をRTで4時間撹拌した。反応物をブライン(300mL)でクエンチし、酢酸エチル(2×400mL)で抽出し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、3−[(2−クロロ−5−メトキシ−フェニル)−(2−フルオロエチル)アミノ]−2−ヒドロキシ−シクロヘキシル−1−エンカルボン酸エチルエステル(14)12.0g(定量的)を褐色油として得、それを次の段階で粗生成物として使用した。異性体混合物としての構造を、1H NMR(300MHz,CDCl3):δH1.08(0.8H,t,J=9Hz,CO2CH2CH3),1.22−1.33(2.2 H,m,CO2CH2CH3),1.40−2.60(7H,m,4−,5−及び6−CH2,CHN),3.20−4.50(10H,m,NCH2CH2F,NCH2CH2F,OCH3,CHCO2CH2CH3),6.50−6.70(1H,m,CHC(OCH3)CHCH),6.95(0.5H,dd,J=3及び6Hz,CHC(OCH3)CHCH),7.08(0.5H,d,J=3Hz,CHC(OCH3)CHCH)及び7.20−7.30(1H,m,CHC(OCH3)CHCH)によって確認した。
【0138】
実施例2(d):8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)
8−クロロ−9−(2−フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)の合成を、まず国際公開第2003/014082号に記載の条件を使用して試みた。2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13;実施例2(b)に従って調製した)(600mg、3.8mmol)の乾燥THF(20mL)溶液を氷浴で冷却し、カリウムビス(トリメチルシリル)アミド(トルエン中0.5M溶液16mL、8.0mmol)で処理した。30分後、THF(4mL)中3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4;実施例1(d)に従って調製した)(1.04g、4.2mmol)を添加し、反応物をRTにして2時間かけて温めた。反応物を飽和塩化アンモニウム溶液でクエンチし、エーテルで2回抽出した。抽出物を、水、ブラインで洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(2.5〜50%のB、50g、25CV、40mL/分)で溶出して精製した。主な点は、3つの化合物の混合物であった。この混合物を、トルエン(20mL)中、乾燥塩化亜鉛(1.7g、12.6mmol)と共に終夜還流した。反応物を真空中で濃縮し、残渣を1NのHCL(25mL)と酢酸エチル(25mL)に分離し、次いで酢酸エチルで再度抽出した。有機層を水及びブラインで洗浄し、乾燥させ、真空中で濃縮して褐色油を得た。1H NMRは、これがいくつかの化合物の混合物であったことを示した。様々な溶媒におけるシリカによるTLCでは、この混合物を別個の点に分離することができなかった。混合物の1H NMRと標準品の1H NMRの比較によって、混合物が推定25%の8−クロロ−9−(2−フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)を含有していることが示された。
【0139】
次いで、修正を加えた方法を実施した。3−[(2−クロロ−5−メトキシ−フェニル)−(2−フルオロエチル)アミノ]−2−ヒドロキシ−シクロヘキシル−1−エンカルボン酸エチルエステル(14)(12.2g、30.0mmol)をジエチルエーテル(250mL)に溶解し、塩化亜鉛(16.4g、120.0mmol)を添加した。反応物を16時間加熱還流した。酢酸エチル(500mL)を添加してすべてを溶解し、2NのHCl(200mL)、水(200mL)、10%炭酸カリウム水溶液(200mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(5〜20%のB、12CV、10g、100mL/分)で溶出して精製して、8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)5.3g(2段階にわたって50%)を黄色固体として得た。構造を、13C NMR(75MHz,CDCl3):δC14.4,20.4,22.2,27.4,40.1,44.2(d,JCF=23Hz),55.1,60.2,83.9(d,JCF=173Hz),100.6,107.9,108.2,119.8,123.1,131.9,137.2,152.7及び175.7によって確認した。
【0140】
実施例2(e):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(16)
8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)(5.3g、15.0mmol)をメタノール(180mL)に溶解し、トリエチルアミン(1.8g、18.0mmol、2.5mL)及び10%Pd/C(メタノール(20mL)中2g)を添加した。混合物をParr水素化装置(hydrogenator)に入れ、水素雰囲気下で18時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、溶媒を真空中で除去した。残渣を酢酸エチル(300mL)に溶解し、10%炭酸カリウム水溶液(200mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(16)4.2g(88%)を淡褐色固体として得た。構造を、13C NMR(75MHz,CDCl3):δC14.3,20.6,21.8,27.6,40.3,43.3(d,JCF=23Hz),54.9,60.1,82.0(d,JCF=165Hz),99.8,102.1,107.3,117.2,121.8,134.9,137.6,153.8及び176.0によって確認した。
【0141】
HPLC(Gemini 150×4.6mm、20分間50〜95%のメタノール/水)13.6分(94%)。
【0142】
実施例2(f):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(17)
8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(16)(380mg、1.2mmol)をエタノール(4mL)に溶解した。水6mLに溶解した水酸化ナトリウム溶液(580mg、14.5mmol)を添加した。反応混合物を、終夜加熱還流した。溶媒を真空中で除去し、粗混合物を水で希釈し、酸性になるまで2NのHClで酸性にし、ジクロロメタンで洗浄した。有機物を混合し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(17)347mg(定量的)をオフホワイト色の固体として得、それを次の段階で粗生成物として使用した。構造を、13C NMR(75MHz;CDCl3):δC20.4,21.9,27.2,39.9,43.3(d,JCF=23Hz),55.1,81.9(d,JCF=173Hz),100.3,102.8,106.2,117.1,122.2,135.6,137.8,153.3及び180.8によって確認した。
【0143】
実施例2(g):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(18)
9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(17)(347mg、1.2mmol)の乾燥ジクロロメタン(2mL)溶液を窒素下で撹拌した。塩化オキサリル(453mg、3.6mmol、300μL)を添加し、その後一滴のDMFを添加し、反応混合物を窒素下においてRTで2時間撹拌し、次いで真空中で蒸発させて、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物371mg(定量的)をガムとして得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz,CDCl3):δC20.2,21.7,26.4,43.3(d,JCF=23Hz),54.9,80.5,83.1,100.2,102.2,105.8,116.7,122.4,135.5,137.4,153.5及び176.6によって確認した。
【0144】
実施例2(h):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤5)
9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(18)(371mg、1.2mmol)をジクロロメタン(2mL)に溶解し、0℃に冷却した。次いでジエチルアミン(177mg、2.4mmol、250μL)を添加し、反応物を終夜RTで撹拌した。10%炭酸カリウム水溶液(2mL)を用いて反応物をクエンチした。ジクロロメタン層を相分離機によって収集し、次いで真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(50〜100%の(B)、50g、35.2CV、40mL/分)で溶出して精製して、薄黄色固体を得た。次に、固体を最小量のジエチルエーテルで倍散して、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤5)240mg(58%)を得た。構造を、13C NMR(75MHz,CDCl3):δC13.0,14.6,19.9,21.9,28.0,36.3,40.5,41.9,43.1(d,JCF=23Hz),54.7,82.0(d,JCF=173Hz),99.7,102.1,108.3,117.0,121.5,135.3,137.4,153.3及び174.8によって確認した。
【0145】
実施例3:メタンスルホン酸2−[4−(ピペリジン−1−カルボニル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物6)及び[9−(2−[18F]フルオロエチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(造影剤6)の合成
実施例3(a):2−(ピペリジン−1−カルボニル)−シクロヘキサノン(19)
トルエン(100mL)中2−オキソシクロヘキサン−カルボン酸エチル(5.3g、31mmol、5.0mL)、DMAP(1.05g、9.4mmol)及びピペリジン(5.3g、63mmol、6.2mL)を4日間加熱還流した。反応物を冷却し、反応物を真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜80%(B)、100g、8CV、85mL/分)で溶出して精製して、2−(ピペリジン−1−カルボニル)−シクロヘキサノン(19)6.26g(96%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC23.5,24.5,25.5,26.2,27.1,30.4,41.9,42.9,46.8,54.2,167.6,207.6によって確認した。
【0146】
実施例3(b):2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20)
2−(ピペリジン−1−カルボニル)−シクロヘキサノン(19)(4.0g、19mmol)をジエチルエーテル(5mL)に溶解し、N2下で0℃に冷却した。臭素(5.9g、19mmol、1.0mL)を15分かけて滴下添加し、反応混合物を室温にして90分かけて温めた。固体を濾過によって収集して、2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20)5.86g(定量的)を白色固体として得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz,DMSO−d6)δC17.3,24.2,25.3,25.8,32.5,44.0,51.6,108.3,145.5,167.8によって確認した。
【0147】
実施例3(c):(2−ベンジルオキシ−エチル)−フェニル−アミン(21)
丸底フラスコ中、アニリン(2.0g、21.5mmol、2.0mL)、2,6−ルチジン(2.30g、21.5mmol)及びベンジル2−ブロモエチルエーテル(4.6g、21.5mmol、3.4mL)を、DMF(10mL)中で混合し、100℃で終夜撹拌した。反応物を冷却し、次いで酢酸エチル(50mL)で希釈した。これを水(3×20mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜50%のB、100g、19.5CV、85mL/分)で溶出して精製して、(2−ベンジルオキシ−エチル)−フェニル−アミン(21)2.22g(37%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3)δC43.6,68.6,73.2,113.1,117.5,127.5,127.7,128.4,129.1,138.2,148.1によって確認した。
【0148】
実施例3(d):[9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(22)
2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20)(1.5g、5.2mmol)及び(2−ベンジルオキシ−エチル)−フェニル−アミン(21)(3.2g、10.4mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(5mL)に溶解し、乾燥塩化亜鉛(2.13g、15.6mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(100mL)に溶解し、2NのHCl(30mL)、水(2×30mL)及び炭酸カリウム水溶液(2×30mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、SCXカートリッジ及び次いでシリカゲルクロマトグラフィーによって、石油(A)及び酢酸エチル(B)(30〜100%のB、12g、41CV、30mL/分)で溶出して精製して、[9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(22)600mg(27%)を油として得た。構造を、13C NMR(75MHz,CDCl3)δC21.5,21.7,24.5,25..7,26.3,273,37.7,42.8,43.1,46.7,60.2,68.7,73.1,108.2,108.7,117.8,118.9,120.5,126.4,127.3,127.4,128.1,136.2,137.8,172.9によって確認した。
【0149】
実施例3(e):[9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(23)
[9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(22)(600mg、1.4mmol)のメタノール(15mL)溶液に、メタノール(10mL)中Pd/C(200mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮した。粗材料を研和して、[9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(23)332mg(71%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3):δC21.2,21.9,24.7,27.4,36.4,43.4,45.0,47.0,60.9,107.8,109.0,117.7,119.0,120.7,126.6,136.2,137.2,173.5によって確認した。
【0150】
実施例3(f):メタンスルホン酸2−[4−(ピペリジン−1−カルボニル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物6)
[9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(23)(260mg、0.8mmol)のジクロロメタン(15mL)溶液に、ピリジン(633mg、8.0mmol、0.65mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(458mg、4.0mmol、0.31mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×50mL)及び水(2×50mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料をジエチルエーテルで倍散して、メタンスルホン酸2−[4−(ピペリジン−1−カルボニル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物6)263mg(82%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC21.4,21.8,24.7,25.9,26.9,27.4,36.6,36.8,41.7,43.3,47.0,67.9,108.5,109.5,118.4,119.7,121.3,126.9,136.2,172.7によって確認した。
【0151】
実施例3(g):[9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(造影剤6)
18Fによる前駆体化合物6の標識を、実施例1(f)に記載の通り実施した。
【0152】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長254nm;tR造影剤6、17分。
【0153】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤6、16分。放射化学的収率23±2%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図2は、造影剤6と非放射性造影剤6の共溶出を示す。
【0154】
実施例4:[9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(造影剤6の非放射性類似体)の合成
実施例4(a):(2−フルオロ−エチル)−フェニル−アミン(24)
丸底フラスコ中、アニリン(0.5g、5.4mmol)、2,6−ルチジン(0.58g、5.4mmol)及び2−フルオロエチルトシレート(12;実施例2(a)に従って調製した)(1.17g、5.4mmol)を、DMF(2.5mL)中で混合し、100℃で終夜撹拌した。反応物を冷却し、次いで酢酸エチル(50mL)で希釈した。これを水(3×20mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(100g、0〜100%のB、18CV、85mL/分)で溶出して精製して、(2−フルオロ−エチル)−フェニル−アミン(24)435mg(60%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH3.41(1H,t,J=3Hz,NCH2CH2F),3.50(1H,t,J=3Hz,NCH2CH2F),3.93(1H,s,br),4.54(1H,t,J=3Hz,NCH2CH2F),4.71(1H,t,J=3Hz,NCH2CH2F),6.65−6.82(3H,m,2×NCCH,NCCHCHCH),7.14−7.28(2H,m,2×NCCHCHCH)によって確認した。
【0155】
実施例4(b):[9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(非放射性造影剤6)
2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20;実施例3(b)に従って調製した)(500mg、1.7mmol)及び(2−フルオロ−エチル)−フェニル−アミン(24)(890mg、3.5mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(2mL)に溶解し、乾燥塩化亜鉛(682mg、5mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(50mL)に溶解し、2NのHCl(20mL)、水(2×20mL)及び炭酸カリウム水溶液(2×20mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料をジエチルエーテルで倍散して、[9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(非放射性造影剤6)151mg(27%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC21.6,21.8,24.7,26.5,26.9,27.4,37.3,43.1(d,JCF=45Hz),47.0,82.1(d,JCF=173Hz),108.5,108.9,118.6,119.4,121.0,126.8,136.2,172.7によって確認した。
【0156】
実施例5:メタンスルホン酸2−[4−(ベンジル−メチル−カルバモイル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物7)及び9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(造影剤7)の合成
実施例5(a):9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(25)
(2−ベンジルオキシ−エチル)−フェニル−アミン(21;実施例3(c)に従って調製した)(8.0g、26mmol)及び3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4;実施例1(d)に従って調製した)(3.2g、13mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(30mL)に溶解し、乾燥塩化亜鉛(10.6g、78mmol)を添加した。混合物をN2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(300mL)に溶解し、2NのHCl(100mL)、水(2×100mL)及び炭酸カリウム水溶液(2×100mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(2.5〜40%のB、17CV、330g、100mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(25)3.49g(72%)を油として得た。構造を、13C NMR(75MHz,CDCl3)δC14.2,20.5,21.8,26.5,38.6,42.9,60.4,68.7,73.2,106.4,108.8,118.7,120.7,127.4,127.5,128.3,136.2,136.9,137.8,175.0によって確認した。
【0157】
実施例5(b):9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(26)
9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(25)(35g、9.3mmol)をエタノール(9mL)に溶解し、次いで水(15mL)中NaOH(1.56g)を添加した。反応物を、2時間加熱還流した。反応物を真空中で濃縮し、残渣を水で希釈し、ジクロロメタン(2×150mL)で洗浄した。水層を2NのHCl(150mL)に滴下添加し、次いでジクロロメタン(3×150mL)に抽出した。有機物を乾燥させ、真空中で濃縮して、9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(26)2.48g(92%)を黄色固体として得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz,CDCl3)δC20.4,21.8,26.4,38.3,42.9,68.7,73.3,105.7,108.8,118.7,119.3,102.9,127.4,127.6,128.3,136.2,137.1,137.8,108.9によって確認した。
【0158】
実施例5(c):9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(27)
9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(26)(600mg、1.7mmol)を、窒素下で乾燥DCM(8mL)に溶解し、塩化オキサリル(393mg、3.1mmol、0.26mL)を添加した。反応物を室温で3時間撹拌すると、ガスが激しく発生した。反応物を真空中で濃縮し、次いでジクロロメタン(8mL)に再溶解し、0℃に冷却し、N−ベンジルメチルアミン(412mg、3.4mmol、0.44mL)を添加した。反応物を室温にして終夜温めた。反応物を5%炭酸カリウム水溶液で洗浄し、乾燥させ、真空中で濃縮して褐色油を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(30%のB、10g)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(27)246mg(64%)を黄色油として得た。構造を、1H NMR(CDCl3)δH1.60−2.30(4H,m,CHCH2CH2CH2),2.70−2.90(2H,m,CHCH2CH2CH2),3.10(1.5H,s,N(CH3)CH2Ph),3.13(1.5H,s,N(CH3)CH2Ph),3.73(2H,t,J=6Hz,NCH2CH2O),4.10−4.30(3H,m,NCH2CH2O,CHCH2CH2CH2),4.42(1H,s,OCH2Ph),4.44(1H,s,OCH2Ph),4.80(1H,s,N(CH3)CH2Ph),4.81(1H,s,N(CH3)CH2Ph),6.90−7.50(14H,m)によって確認した。
【0159】
実施例5(d):9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(28)
9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(27)(246mg、0.5mmol)のメタノール(15mL)溶液に、メタノール(10mL)中Pd/C(200mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、9−(2ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(28)36mg(20%)を緑色油として得、それを精製せずに次の段階で使用した。構造を、1H NMR(CDCl3) δH 1.80-2.20 (4H,m), 2.70-3.00 (2H,m), 3.20-4.30 (10H,m), 6.90-7.50 (9H,m)によって確認した。
【0160】
実施例5(e):メタンスルホン酸2−[4−(ベンジル−メチル−カルバモイル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物7)
9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(28)(36mg、0.1mmol)のジクロロメタン(2mL)溶液に、ピリジン(7.91g、1.0mmol、8.1mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(57mg、0.5mmol、0.04mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×10mL)及び水(2×10mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜80%のB、4g、45CV、18mL/分)で溶出して精製して、メタンスルホン酸2−[4−(ベンジル−メチル−カルバモイル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物7)14mg(32%)を黄色油として得た。構造を、1H NMR(CDCl3)δH1.10−2.40(5H,m),2.51(1.5H,s,OSO2CH3),2.54(1.5H,s,OSO2CH3),2.70−2.90(2H,m),3.08(1.5H,s,NCH3),3.15(1.5H,s,NCH3),3.40−3.70(1H,m),4.10−4.80(4H,m),7.00−7.50(9H,m)によって確認した。
【0161】
実施例5(f):9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(造影剤7)
18Fによる前駆体化合物7の標識を、実施例1(f)に記載の通り実施した。半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長254nm;tR造影剤7、17分。
【0162】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤7、16分。放射化学的収率23±2%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図3は、造影剤7と非放射性造影剤7の共溶出を示す。
【0163】
実施例6:9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(非放射性造影剤7)
実施例6(a):3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(29)
2−オキソシクロヘキサンカルボン酸エチル(5.0g、29mmol、4.7mL)をジエチルエーテル(5mL)に溶解し、N2下で0℃に冷却した。臭素(4.6g、29mmol、4.2mL)を15分かけて滴下添加し、反応混合物を室温にして90分かけて温めた。混合物を、氷冷した飽和炭酸ナトリウム水溶液(40mL)にゆっくり注ぎ、酢酸エチル(3×40mL)で抽出した。混合有機層を乾燥させ、真空中で濃縮して、3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(29)5.96g(81%)を薄黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,DMSO−d6)δC14.14,17.65,21.77,32.02,59.95,60.83,99.70,166.33,172.81によって確認した。
【0164】
実施例6(b):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(30)
(2−フルオロ−エチル)−フェニル−アミン(24;実施例4(a)に従って調製した)(560mg、4.0mmol)及び3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(29)(500mg、2.0mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(4mL)に溶解し、乾燥塩化亜鉛(820mg、6mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。生成物を酢酸エチル/エーテル(30mL/150mL)に溶解し、2NのHCl(40mL)、水(2×100mL)及び炭酸カリウム水溶液(2×100mL)で洗浄し、次いで乾燥させ、濃縮して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(30)447mg(91%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC14.3,20.4,21.7,26.4,38.5,43.1(d,JCF=15Hz),60.6,76.6,77.0,77.4,82.1(d,JCF=173Hz),106.9,108.5,118.9,119.4,121.1,127.1,136.2,136.7,174.9によって確認した。
【0165】
実施例6(c):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(31)
9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(30)(380mg、1.3mmol)をエタノール(3mL)に溶解し、次いで水(5mL)中NaOH(520mg)を添加した。反応物を、2時間加熱還流した。反応物を真空中で濃縮し、残渣を水で希釈し、ジクロロメタン(2×50mL)で洗浄した。水層を2NのHCl(50mL)に滴下添加し、次いでジクロロメタン(3×50mL)に抽出した。有機物を乾燥させ、真空中で濃縮して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(31)130mg(37%)を黄色固体として得、それを次の段階で精製せずに使用した。構造を、1H NMR(300MHz,CDCl3)δ 1.90−2.42(4H,m,2−及び3−CH2),2.60−2.91(2H,m,1−CH2),3.94(1H,t,J=6Hz,4−CH),4.30(1H,t,J=6Hz,NCH2CH2F),4.37(1H,t,J=6Hz,NCH2CH2F),4.59(1H,t,J=6Hz,NCH2CH2F),4.74(1H,t,J=6Hz,NCH2CH2F),7.05−7.26(3H,m,ArH),7.59(1H,d,J=9Hz,ArH)によって確認した。
【0166】
実施例6(d):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(32)
乾燥ジクロロメタン(6mL)中9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(31)(0.5g、1.91mmol)を、窒素雰囲気下で、塩化オキサリル(490mg、3.8mmol、0.34mL)及び一滴のDMFと共に室温で撹拌した。反応物を真空中で濃縮して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(32)545mg(定量的)を得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC20.2,21.6,26.7,43.1,43.4,50.6,80.9,83.1,105.3,108.8,118.3,120.0,121.6,126.5,136.2,137.5,176.1によって確認した。
【0167】
実施例6(e):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(非放射性造影剤7)
9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(32)(110mg、0.4mmol)をジクロロメタン(1mL)に溶解し、0℃に冷却した。次いで、N−ベンジルメチルアミン(92mg、0.8mmol、98μL)を添加し、反応物を終夜RTで撹拌した。反応物を10%炭酸カリウム水溶液(2mL)でクエンチした。ジクロロメタン層を相分離機によって収集し、次いで真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜100%のB、12g、30CV、30mL/分)で溶出して精製して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(非放射性造影剤7)39mg(28%)を得た。構造を、1H NMR(300MHz,CDCl3)δH1.75−2.32,(4H,m,2−及び3−CH2),2.68−2.86(2H,m,1−CH2),3.10(1H,s,NCH3),3.14(2H,s,NCH3),4.17−4.39(3H,m,NCH2CH2F及び4−CH2),4.52−4.87(4H,m,NCH2Ph及びNCH2CH2F),6.96−7.42(9H,m,ArH)によって確認した。
【0168】
実施例7:メタンスルホン酸2−(4−ジエチルカルバモイル−6−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物9)及び6−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤9)の合成
実施例7(a):2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)
ベンジルオキシ酢酸(4.6g、28.0mmol、4.0mL)のDCM(52mL)溶液に、塩化オキサリル(7.7g、61mmol、5.3mL)及び一滴のDMFを添加した。反応混合物を室温で4時間撹拌した。過剰の塩化オキサリルを真空中で除去して、ベンジルオキシ−塩化アセチルを得た。粗塩化アシルをDCM(100mL)で希釈し、トリエチルアミン(5.3mL、41.6mmol、4.2g)を添加し、その後4−フルオロアニリン(3.5g、32mmol、3.0mL)を添加した。反応混合物を終夜RTで撹拌した。次いで、反応物を1MのHCl水溶液(100mL)でクエンチし、乾燥させ、真空中で濃縮して、2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)7.1g(95%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC69.2,73.5,115.4(d,JCF=22Hz),121.4(d,JCF=7Hz),127.9,128.2,128.5,132.5(d,JCF=3Hz),136.3,157.6,160.8及び167.5によって確認した。
【0169】
実施例7(b):(2−ベンジルオキシ−エチル)−(4−フルオロ−フェニル)−アミン(34)
LAH(1.25g、27mmol)の乾燥ジエチルエーテル(100mL)懸濁液に、2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)(6.9g、27mmol)の乾燥ジエチルエーテル(100mL)溶液を滴下添加した。還流が維持されるように添加を実施した。添加が終了したら、反応混合物を4時間加熱還流し、次いで氷水に注ぎ、DCMを添加した。アルミニウム塩を分解するために、強塩基のpHが得られるまで2M水酸化ナトリウム水溶液を添加した。層を分離し、水層をDCMで洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(5〜50%のB、100g、12CV、60mL/分)で溶出して精製して、(2−ベンジルオキシ−エチル)−(4−フルオロ−フェニル)−アミン(34)5.5g(84%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3)δC44.0,68.3,72.8,113.7(d,JCF=7Hz),115.3(d,JCF=22Hz),127.5,127.6(d,JCF=3Hz),128.3,137.8,144.5,154.1及び157.2によって確認した。
【0170】
実施例7(c):3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35)
2−シクロヘキサン(cyclohexone)−カルボン酸エチル(7.50mL、47.0mmol)、DMAP(1.72g、14.1mmol)及びジエチルアミン(9.77mL、94.0mmol)を、トルエン(100mL)中で72時間加熱還流した。反応物を冷却し、トルエンを減圧下で除去した。粗油を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(1:1、100g、SiO2)で溶出して精製して、2−オキソ−シクロヘキサンカルボン酸ジエチルアミン6.8g(73%)を橙色油として得た。構造を、13C NMR(CDCl3)δ11.1,12.7,21.3,24.9,28.5,39.4,39.6,51.7,166.5,205.9によって確認した。
【0171】
2−オキソ−シクロヘキサンカルボン酸ジエチルアミン(3.56mL、19.3mmol)をジエチルエーテル(5mL)に溶解し、N2下で撹拌しながら0℃に冷却した。臭素(0.99mL、19.3mmol)を15分かけて滴下添加し、反応混合物を室温にして3時間かけて温めた。反応物から固体が沈殿した。それを濾過によって収集し、エーテルで洗浄して、3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35)5.85g(109%)を薄黄色固体として得た。構造を、13C NMR(CDCl3)δ11.2,12.8,22.7,28.8,37.6,37.9,39.4,51.0,55.7,165.5,197.2によって確認した。
【0172】
実施例7(d):9−(2−ベンジルオキシ−エチル)−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(36)
2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)(5.3g、22mmol)及び3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35)(3.0g、13mmol))の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(30mL)に溶解し、乾燥塩化亜鉛(9.0g、66mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(300mL)に溶解し、2NのHCl(100mL)、水(2×100mL)及び炭酸カリウム水溶液(2×100mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10〜50%のB、100g)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(36)196mg(11%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.14(3H,t,J=7Hz,N(CH2CH3)2),1.30(3H,t,J=7Hz,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びNCH2CH2OBn),3.66−3.75(1H,m,4−CH),4.00−4.25(2H,m,NCH2CH2OBn),4.41(2H,s,OCH2Ph),6.75−6.95(2H,m,NCCHCHCFCH),7.05−7.15(1H,m,NCCHCHCFCH)及び7.16−7.25(5H,m,Ph)によって確認した。
【0173】
実施例7(d):6−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(37)
9−(2−ベンジルオキシ−エチル)−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(36)(600mg、1.4mmol)のメタノール(40mL)溶液に、メタノール(5mL)中Pd/C(100mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、6−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(37)460mg(80%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、1H NMR(300MHz,MeOD−d3)δH1.18(3H,t,J=9Hz,N(CH2CH3)2),1.35(3H,t,J=9Hz,N(CH2CH3)2),1.80−2.20(4H,m,2−及び3−CH2),2.69−3.88(2H,m,1−CH2),3.40−3.86(6H,m,N(CH2CH3)2及びNCH2CH2OH),4.03−4.22(3H,m,NCH2CH2OH及び4−CH),6.75−6.95(2H,m,NCCHCHCFCH)及び7.05−7.15(1H,m,NCCHCHCFCHによって確認した。
【0174】
実施例7(e):メタンスルホン酸2−(4−ジエチルカルバモイル−6−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物9)
6−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(37)(460mg、1.4mmol)のジクロロメタン(20mL)溶液に、ピリジン(1.11g、14.0mmol、1.1mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(722mg、6.3mmol、0.5mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×30mL)及び水(2×30mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜100%(B)、10g、45CV、30mL/分)で溶出して精製し、次いでジエチルエーテルで倍散して、メタンスルホン酸2−(4−ジエチルカルバモイル−6−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物9)166mg(30%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC12.9,15.0,21.1,27.7,36.1,36.7,40.6,41.7,67.8,103.3(d,JCF=23Hz),108.7,109.0,109.1,109.4(d,JCF=5Hz),126.9(d,JCF=10Hz),132.4,138.4,156.1,159.2及び173.3によって確認した。
【0175】
実施例7(f):6−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤9)
18Fによる前駆体化合物9の標識を、実施例1(f)に記載の通り実施した。
【0176】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、40%のB;1〜20分、40〜95%のB;波長254nm;tR造影剤9、15分。
【0177】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤9、14分。放射化学的収率26±8%(n=4)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図4は、造影剤9と非放射性造影剤9の共溶出を示す。
【0178】
実施例8:6−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤9)の合成
実施例8(a):(2−フルオロ−エチル)−(4−フルオロ−フェニル)−アミン(38)
丸底フラスコ中、4−フルオロアニリン(1.3g、11.6mmol、1.6mL)、2,6−ルチジン(1.24g、11.6mmol)及び2−フルオロエチルトシレート(12;実施例2(a)に従って調製した)(2.5g、11.6mmol)を、DMF(5mL)中で混合し、100℃で終夜撹拌した。反応物を冷却し、次いで酢酸エチル(100mL)で希釈した。これを水(3×40mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10%のB、100g、12CV、60mL/分)で溶出して精製して、(2−フルオロ−エチル)−(4−フルオロ−フェニル)−アミン(38)383mg(20%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH3.30−3.35(1H,m,NCH2CH2F),3.40−3.45(1H,m,NCH2CH2F),3.90(1H,s,br,NH),4.53(1H,t,J=3Hz,NCH2CH2F),4.69(1H,t,J=3Hz,NCH2CH2F),6.51−6.72(2H,m,2 x NCCH),6.85−7.05(2H,m,2 x NCCHCH)によって確認した。
【0179】
実施例8(b):6−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤9)
3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35;実施例7(c)に従って調製した)(336mg、1.2mmol)及び(2−フルオロ−エチル)−(4−フルオロ−フェニル)−アミン(38)(383mg、2.4mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(2mL)に溶解し、乾燥塩化亜鉛(491mg、3.6mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(20mL)に溶解し、2NのHCl(10mL)、水(2×10mL)及び炭酸カリウム水溶液(2×5mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料をジエチルエーテルで倍散して、6−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤9)40mg(10%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.13(3H,t,J=9Hz,N(CH2CH3)2),1.30(3H,t,J=9Hz,N(CH2CH3)2),1.55−2.14(4H,m,2−及び3−CH2),2.78−2.86(2H,m,1−CH2),3.36−3.67(4H,m,N(CH2CH3)2),4.00−4.10(1H,m,4−CH),4.30(2H,dm,J=21Hz,NCH2CH2F),4.60(2H,dm,J=41Hz,NCH2CH2F),6.75−6.95(2H,m,NCCHCHCFCH)及び7.05−7.15(1H,m,NCCHCHCFCHによって確認した。
【0180】
実施例9:メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)及び5−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤10)の合成
実施例9(a):2−ベンジルオキシ−N−(3−フルオロ−フェニル)−アセトアミド(39)
ベンジルオキシ酢酸(4.65g、28mmol、4.0mL)のDCM(52mL)溶液に、塩化オキサリル(7.7g、61mmol、5.3mL)及び一滴のDMFを添加した。反応混合物を室温で4時間撹拌した。過剰の塩化オキサリルを真空中で除去し、粗塩化アシルをDCM(100mL)で希釈し、トリエチルアミン(5.3mL、41.6mmol、4.2g)を添加し、その後3−フルオロアニリン(3.5g、32mmol、3.0mL)を添加した。反応混合物を終夜RTで撹拌した。次いで、反応物を1MのHCl水溶液(100mL)でクエンチし、乾燥させ、真空中で濃縮して、2−ベンジルオキシ−N−(3−フルオロ−フェニル)−アセトアミド(39)7.10g(95%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC69.2,73.5,106.9,107.2,111.0(d,JCF=24Hz),114.9(d,JCF=3Hz),127.8,128.2,128.5,129.7(d,JCF=9Hz),136.2及び167.6によって確認した。
【0181】
実施例9(b):(2−ベンジルオキシ−エチル)−(3−フルオロ−フェニル)−アミン(40)
LAH(1.25g、27mmol)の乾燥ジエチルエーテル(100mL)懸濁液に、2−ベンジルオキシ−N−(3−フルオロ−フェニル)−アセトアミド(39)(7.0g、27mmol)の乾燥ジエチルエーテル(100mL)溶液を滴下添加した。還流が維持されるように添加を実施した。添加が終了したら、反応混合物を4時間加熱還流し、次いで氷水に注ぎ、DCMを添加した。アルミニウム塩を分解するために、強塩基のpHが得られるまで2M水酸化ナトリウム水溶液を添加した。層を分離し、水層をDCMで洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(5〜50%のB、100g、12CV、60mL/分)で溶出して精製して、(2−ベンジルオキシ−エチル)−(3−フルオロ−フェニル)−アミン(40)4.1g(84%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3):δC43.3,68.2,73.0,99.4(d,JCF=24Hz),103.5,103.8,108.8,127.4(d,JCF=3Hz),127.6,128.4,130.0(d,JCF=9Hz)及び138.8によって確認した。
【0182】
実施例9(c):9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)
3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35;実施例7(c)に従って調製した)(2.3g、10mmol)及び(2−ベンジルオキシ−エチル)−(3−フルオロ−フェニル)−アミン(40)(4.1g、17mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(10mL)に溶解し、乾燥塩化亜鉛(4.09g、30mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(200mL)に溶解し、2NのHCl(50mL)、水(2×50mL)及び炭酸カリウム水溶液(2×50mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(5〜100%のB、100g、28CV、60mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)1.3g(30%)を、異性体9−(2−ベンジルオキシ−エチル)−7−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドと一緒に混合物として得、それを次の段階で精製せずに使用した。9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)の構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びCH2CH2OBn),4.00−4.30(3H,m,CH2CH2OBn及び4−CH),4.43(2H,s,OCH2Ph),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF),7.05−7.15(1H,m,NCCHCHCHCF)及び7.16−7.25(5H,m,Ph)によって確認した。
【0183】
9−(2−ベンジルオキシ−エチル)−7−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びNCH2CH2OBn),4.00−4.30(3H,m,NCH2CH2Obn及び4−CH),4.55(2H,s,OCH2Ph),6.70−6.80(1H,m,NCCHCFCHCH)及び7.00−7.40(7H,m,NCCHCFCHCH及びPh)によって確認した。
【0184】
実施例9(d):5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(42)
9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)及び9−(2−ベンジルオキシ−エチル)−7−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(1.3g、3.0mmol)の混合物のメタノール(75mL)溶液に、メタノール(10mL)中Pa/C(200mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(42)及び7−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの混合物743mg(80%)を黄色油として得、それを次の段階で精製せずに使用した。5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(55)の構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びCH2CH2OH),4.00−4.30(3H,m,CH2CH2OH,4−CH),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF)及び7.05−7.15(1H,m,NCCHCHCHCF)によって確認した。
【0185】
7−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びCH2CH2OH),4.00−4.30(3H,m,NCH2CH2OH,4−CH),6.70−6.80(1H,m,NCCHCFCHCH)及び7.00−7.40(2H,m,NCCHCFCHCH)によって確認した。
【0186】
実施例9(e):メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)
5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(42)及び7−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(743mg、2.2mmol)の混合物のジクロロメタン(30mL)溶液に、ピリジン(1.74g、22.0mmol、1.8mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(1.01g、8.8mmol、0.7mL)を添加した。反応物を室温にして終夜温めた。混合物を2NのHCl(2×50mL)及び水(2×50mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、半分取HPLCによって水(A)及びメタノール(B)(Gemini 5u、C18、110A、150×21mm、20分間50〜95%のB、21mL/分)で溶出して精製して、メタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル10mg(1%)を白色固体として得、メタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル及びメタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)の混合物30mg(9%)を白色固体として得た。これらの精製条件を使用すると、メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)を、単一成分として単離することができなかった。メタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステルの構造を、1H NMR(300MHz,CDCl3)δH1.18(3H,t,J=7Hz,N(CH2CH3)2),1.39(3H,t,J=7Hz,N(CH2CH3)2) 1.70−2.30(4H,m,2−及び3−CH2),2.58(3H,s,OSO2CH3),2.60−2.80(2H,m,1−CH2),3.40−3.65(4H,m,N(CH2CH3)2),4.02(1H,t,J=6Hz,4−CH),4.20(2H,t,J=7Hz,NCH2CH2OMs),4.35(2H,t,J=7Hz,NCH2CH2OMs),6.70−6.85(1H,m,NCCHCFCHCH),6.90−7.00(1H,m,NCCHCFCHCH)及び7.05−7.15(2H,m,NCCHCFCHCH)によって確認した。
【0187】
メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)の構造を、1H NMR(300MHz,CDCl3)δH1.18(3H,t,J=7Hz,N(CH2CH3)2),1.39(3H,t,J=7Hz,N(CH2CH3)2) 1.70−2.30(4H,m,2−及び3−CH2),2.58(3H,s,OSO2CH3),2.60−2.80(2H,m,1−CH2),3.40−3.65(4H,m,N(CH2CH3)2),4.15(1H,m,4−CH),4.20(2H,t,J=7Hz,NCH2CH2OMs),4.35(2H,t,J=7Hz,NCH2CH2OMs),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF)及び7.05−7.15(1H,m,NCCHCHCHCF)によって確認した。
【0188】
実施例9(f):5−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤10)
前駆体化合物10及びメタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステルの混合物を、反応物の放射性標識に使用した。18Fによる標識を、実施例1(f)に記載の通り実施した。7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの造影剤10を得た。
【0189】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長254nm;tR造影剤10、15分;tR7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド、14分。
【0190】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤10、16分;tR7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド、14分。造影剤10の放射化学的収率8.7±1%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図5は、造影剤10(上)及び7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(中間)及び7−フルオロ−9−(2−[19F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(下)を示す。
【0191】
実施例10:5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸−ジエチルアミド(非放射性造影剤10)の合成
実施例10(a):(2−フルオロ−エチル)−(3−フルオロ−フェニル)−アミン(43)
3−フルオロアニリン(1.4g、11.6mmol、1.2mL)及び2−フルオロエチルトシレート(12;実施例2(a)に従って調製した)(2.5g、11.6mmol)及びルチジン(1.24g、11.6mmol)を撹拌し、DMF(5mL)中、100℃で終夜加熱した。反応物を冷却し、次いで酢酸エチル(100mL)で希釈した。これを水(3×40mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10%のB、100g、12CV、60mL/分)で溶出して精製して、(2−フルオロ−エチル)−(3−フルオロ−フェニル)−アミン(43)184mg(10%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH3.37(1H,q,J=6Hz,NCH2CH2F),3.46(1H,q,J=6Hz,NCH2CH2F),4.12(1H,s,br,NH),4.54(1H,t,J=3Hz,NCH2CH2F),4.69(1H,t,J=3Hz,NCH2CH2F),6.3 1−6.50(3H,m,NCCHCHCH),7.10−7.25(1H,m,NCCHCF)によって確認した。
【0192】
実施例10(b):5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤10)
3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35;実施例7(c)に従って調製した)(161mg、0.6mmol)及び(2−フルオロ−エチル)−(3−フルオロ−フェニル)−アミン(43)(184mg、1.2mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(1mL)に溶解し、乾燥塩化亜鉛(245mg、1.8mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(10mL)に溶解し、2NのHCl(5mL)、水(2×5mL)及び炭酸カリウム水溶液(2×5mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、半分取HPLCによって水(A)及びメタノール(B)(Gemini 5u、C18、110A、150×21mm、20分間50〜95%のB、21mL/分)で溶出して精製して、7−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド20mg(6%)を白色固体として得、5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤10)10mg(3%)を白色固体として得た。7−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの構造を、1H NMR(300MHz,CDCl3)δH1.14(3H,t,J=7Hz,N(CH2CH3)2),1.33(3H,t,J=7Hz,N(CH2CH3)2),1.80−2.15(4H,m,2−及び3−CH2),2.70−2.80(2H,m,1−CH2),3.50−3.80(4H,m,N(CH2CH3)2),4.20−4.35(1H,m,4−CH),4.40(2H,dm,J=21Hz,NCH2CH2F),4.60(2H,dm,J=41Hz,NCH2CH2F),6.70−6.80(1H,m,NCCHCFCHCH)及び7.00−7.10(2H,m,NCCHCFCHCH)によって確認した。
【0193】
5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤10)の構造を、1H NMR(300MHz,CDCl3)δH1.14(3H,t,J=7Hz,N(CH2CH3)2),1.33(3H,t,J=7Hz,N(CH2CH3)2),1.80−2.15(4H,m,2−及び3−CH2),2.70−2.80(2H,m,1−CH2),3.50−3.80(4H,m,N(CH2CH3)2),4.20−4.35(1H,m,4−CH),4.40(2H,dm,J=21Hz,NCH2CH2F),4.60(2H,dm,J=41Hz,NCH2CH2F),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF)及び7.05−7.15(1H,m,NCCHCHCHCF)によって確認した。
【0194】
実施例11:メタンスルホン酸2−(4−ジエチルカルバモイル−2−メチル−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物11)及び9−(2−[18F]フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤11)
実施例11(a):4−(4−メチル−シクロヘキセ−1−エニル)−モルホリン(44)
ディーンスタークを備えたフラスコ中、4−メチルシクロヘキサノン(20.1g、179.3mmol、22mL)及びモルホリン(31.3g、359.0mmol、31.4mL)の溶液を、ベンゼン(55mL)中で26時間還流した。ベンゼンを真空下で除去し、粗生成物を減圧下で蒸留によって精製して、4−(4−メチル−シクロヘキセ−1−エニル)−モルホリン(44)23g(70%)を油として得た(10mmHgにおいてb.p.120℃)。構造を、1H NMR(300MHz,CDCl3):δH0.94(3H,d,J=6.0Hz,CH3),1.15−1.35(1H,m,CH2CH=CN),1.50−1.80(3H,m,CH2CH2CHCH3),2.00−2.25(4H,m,CH2CH=CN及びCH2CH2CHCH3),2.65−2.95(4H,m,OCH2NCH2),3.73(4H,t,J=6.0Hz,OCH2NCH2)及び4.60−4.65(1H,m,CH2CH=CN)によって確認した。
【0195】
実施例11(b):5−メチル−2−オキソ−シクロヘキサンカルボン酸エチルエステル(45)
4−(4−メチル−シクロヘキセ−1−エニル)−モルホリン(44)(23g、127.0mmol)のベンゼン(55mL)溶液に、クロロギ酸エチル(7.5g、69.0mmol、6.6mL)を窒素下で添加すると同時に、エナミン溶液を急速に撹拌した。18時間還流した後、溶液を冷却し、濾過した。エナミン塩酸塩の沈殿物を乾燥エーテルで洗浄した。濾液及び洗浄物を反応フラスコに戻し、10%HCl水溶液(40mL)を添加した。混合物を、15〜30分間激しく撹拌した。層を分離し、水層を酢酸エチル(2×100mL)で抽出し、混合有機層を真空中で濃縮した。粗材料を、減圧下で蒸留によって精製して、5−メチル−2−オキソ−シクロヘキサンカルボン酸エチルエステル(45)12.5g(53%)を油として得た(10mmHgにおいてb.p.85℃〜90℃)。構造を、1H NMR(300MHz,CDCl3):δH0.85−0.95(3H,m,CH3),1.17(3H,t,J=7Hz,OCH2CH3),1.25−2.00(5H,m,5−CH,4−及び6−CH2),2.15−2.40(3H,m,1−CH及び3−CH2)及び4.00−4.20(2H,m,OCH2CH3)によって確認した。
【0196】
実施例11(c):5−メチル−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(46)
トルエン(90mL)中5−メチル−2−オキソ−シクロヘキサンカルボン酸エチルエステル(45)(5.9g、32mmol)、DMAP(1.12g、10mmol)及びジエチルアミン(4.7g、65mmol、6.7mL)を、4日間加熱還流した。反応物を冷却し、トルエンを減圧下で除去して、黄色油を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜50%のB、80g)で溶出して精製して、5−メチル−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(46)4.4g(65%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH0.8−1.05(9H,m,CH3及びN(CH2CH3)2),1.05−2.10(5H,m,5−CH及び4−及び6−CH2),2.15−2.80(2H,m,3−CH2),2.95−3.55(5H,m,1−CH及びN(CH2CH3)2)によって確認した。
【0197】
実施例11(d):3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47)
5−メチル−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(46)(4.4g、21mmol)をジエチルエーテル(5mL)に溶解し、N2下で0℃に冷却した。臭素(3.32g、21mmol、1.1mL)を15分かけて滴下添加し、反応混合物を室温にして90分かけて温めた。混合物を、氷冷した飽和炭酸ナトリウム水溶液(40mL)にゆっくり注ぎ、酢酸エチル(3×40mL)で抽出した。混合有機層を乾燥させ、真空中で濃縮して、3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47)6.1g(定量的)をオフホワイト色の固体として得た。構造を、1H NMR(300MHz,CDCl3)δH0.8−1.20(9H,m,CH3及びN(CH2CH3)2),1.80−2.40(5H,m,CH2CH(CH3)CH2),3.15−3.55(4H,m,N(CH2CH3)2),4.65−4.74(1H,m,CHBr)及び12.04(1H,s,OH)によって確認した。
【0198】
実施例11(e):9−(2−ベンジルオキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(48)
3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47)(4.0g、14mmol)及び(2−ベンジルオキシ−エチル)−フェニル−アミン(21;実施例3(c)に従って調製した)(6.3g、28mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(14mL)に溶解し、乾燥塩化亜鉛(5.72g、42mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(200mL)に溶解し、2NのHCl(50mL)、水(2×50mL)及び炭酸カリウム水溶液(2×50mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗混合物を、SCXカートリッジ(40mL)及び次いでシリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10〜50%のB、100g、12CV、85mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(48)467mg(8%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.20−1.40(9H,m,CH3及びN(CH2CH3)2),1.90−2.20(3H,m,2−CH及び3−CH2),2.35−2.45(1H,m,1−CH2),2.85−2.95(1H,m,1−CH2),3.40−3.70(4H,m,N(CH2CH3)2),3.70−3.80(1H,m,4−CH),4.10−4.30(4H,m,NCH2CH2OBn),4.43(2H,s,OCH2Ph)及び7.00−7.30(9H,m,CHCHCHCH及びPh)によって確認した。
【0199】
実施例11(f):9−(2−ヒドロキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(49)
9−(2−ベンジルオキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(48)(460mg、1.1mmol)のメタノール(25mL)溶液に、メタノール(5mL)中Pd/C(100mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、9−(2−ヒドロキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(49)250mg(79%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、1H NMR(300MHz,CDCl3)δH1.20−1.40(9H,m,CH3及びN(CH2CH3)2),1.90−2.20(3H,m,2−CH及び3−CH2),2.35−2.45(1H,m,1−CH2),2.85−2.95(1H,m,1−CH2),3.40−3.70(4H,m,N(CH2CH3)2),3.70−3.80(1H,m,4−CH),4.10−4.30(4H,m,NCH2CH2OH),6.91(1H,t,J=7Hz,NCCHCHCHCH),7.00(1H,t,J=7Hz,NCCHCHCHCH),7.12(1H,d,J=7Hz,NCCHCHCHCH)及び7.15(1H,d,J=7Hz,NCCHCHCHCH)によって確認した。
【0200】
実施例11(g):メタンスルホン酸2−(4−ジエチルカルバモイル−2−メチル−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物11)
9−(2−ヒドロキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(49)(250mg、0.8mmol)のジクロロメタン(10mL)溶液に、ピリジン(633mg、8.0mmol、0.6mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(367mg、3.2mmol、0.2mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×20mL)及び水(2×20mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜100%のB、10g、34CV、30mL/分)で溶出して精製し、次いでジエチルエーテルで倍散して、メタンスルホン酸2−(4−ジエチルカルバモイル−2−メチル−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物11)250mg(80%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC12.9,13.0,15.2,22.0,29.7,30.2,36.7,36.8,40.8,41.6,42.0,67.8,108.6,109.5,118.6,119.6,121.2,126.4,136.2,136.4,173.7によって確認した。
【0201】
実施例11(h):9−(2−[18F]フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤11)
18Fによる前駆体化合物11の標識を、実施例1(f)に記載の通り実施した。
【0202】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜26分、50%のB;波長254nm;tR造影剤11、15分。
【0203】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、40%のB;1〜20分、40〜95%のB;波長230nm;tR造影剤11、17分。放射化学的収率14±13%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図6は、造影剤11と非放射性造影剤11の共溶出を示す。
【0204】
実施例12:9−(2−フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤11)の合成
3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47;実施例11(d)に従って調製した)(2.0g、7mmol)及び(2−フルオロ−エチル)−フェニル−アミン(24;実施例4(a)に従って調製した)(1.9g、14mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(7mL)に溶解し、乾燥塩化亜鉛(2.86g、21mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(100mL)に溶解し、2NのHCl(30mL)、水(2×30mL)及び炭酸カリウム水溶液(2×30mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗混合物を、SCXカートリッジ(40mL)及び次いでシリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜100%のB、100g、12CV、85mL/分)で溶出して精製して、9−(2−フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤11)400mg(17%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.10−1.35(9H,m,CH3及びN(CH2CH3)2),1.95−2.10(2H,m,3−CH2),2.30−2.50(1H,m,2−CH),2.70−2.80(2H,m,1−CH2),3.40−3.70(4H,m,N(CH2CH3)2),4.05−4.15(1H,m,4−CH),4.30(2H,dm,J=21Hz,NCH2CH2F),4.65(2H,dm,J=41Hz,NCH2CH2F)及び7.00−7.30(4H,m,NCCHCHCHCHによって確認した。
【0205】
実施例13:前駆体化合物5の鏡像異性体分離
【0206】
【化15】
前駆体化合物5(実施例1に記載の通り得た)を、超臨界流体(CO2)キラルクロマトグラフィーを使用して、Kromasil Amycoat、250×10mm、5μm、100Åカラムにて40℃において30%IPAを用いて、毎分13mlの実施時間6分でその鏡像異性体に分離した。前駆体化合物5(60mg)を1,4−ジオキサン(2ml)に溶解し、実施ごとに注入する場合、1度に200μlまでとした。2つの鏡像異性体間のベースライン分離を達成した。Chiral Technologies製のIC、250×4.6mm、5μm、均一溶媒による実施(run isocratic)、80:20−MeOH:IPA、0.5ml/分及び室温で、分離した2つの鏡像異性体の鏡像異性体純度を分析的HPLCによって測定すると、鏡像異性体のそれぞれについて99.5%の鏡像異性体純度を示した。
【0207】
実施例14:非放射性造影剤5の鏡像異性体分離
【0208】
【化16】
非放射性造影剤5(実施例2に記載の通り得た)を、超臨界流体(CO2)キラルクロマトグラフィーを使用して、Kromasil Amycoat、250×10mm、5μm、100Åカラムにて40℃において20%IPAを用いて、毎分14mlの実施時間6分でその鏡像異性体に分離した。化合物5(100mg)を1,4−ジオキサン(2.5ml)に溶解し、実施ごとに注入する場合、1度に200μlまでとした。時間ごとに画分をカットして、混合画分が収集されないようにした。Chiral Technologies製のIC、250×4.6mm、5μm、均一溶媒による実施、80:20−MeOH:IPA、0.5ml/分及び室温で、分離した2つの鏡像異性体の鏡像異性体純度を分析的HPLCによって測定すると、鏡像異性体のそれぞれについて99.5%の鏡像異性体純度を示した。
【0209】
実施例15:インビトロ効力アッセイ
PBRに対する親和性を、Le Fur et al,(Life Sci. 1983;USA 33:449−57)に記載された方法を使用してスクリーニングした。本発明のインビボ造影剤の非放射性類似体を、過去の四環系インドール造影剤の非放射性類似体(同時係属の特許出願PCT/EP2009/062827のもの;以下の実施例17に記載の合成)と一緒に試験した。
【0210】
各試験化合物(50mMトリス−HCl、pH7.4、1%DMSOを含有する10mMのMgCl2に溶解した)は、Wistarラット心臓のPBRとの結合について、0.3nMの[3H]PK−11195と競合した。反応を、50mMトリス−HCl、pH7.4、10mMのMgCl2中で25℃において15分間実施した。各試験化合物を、6つの異なる濃度で、推定Ki値周辺の300倍の範囲濃度にわたってスクリーニングした。以下のデータが観測された。
【0211】
【表1】
実施例16:インビボ体内分布法
本発明の造影剤を、過去の四環系造影剤(同時係属の特許出願PCT/EP2009/062827のもの;以下の実施例18に記載の合成)と一緒に、インビボ体内分布モデルで試験した。
【0212】
成体雄性Wistarラット(200〜300g)に、側尾静脈を介して試験化合物1〜3MBqを注入した。注入の2、10、30又は60分後(n=3)、ラットを安楽死させ、組織又は体液を、γ計数器で放射性を測定するために試料採取した。
【0213】
以下の記録データが観測された。
【0214】
【表2】
図7〜13は、それぞれ四環系造影剤並びに造影剤5〜7及び9〜11の脳内の体内分布プロファイルを示す。本発明のインビボ造影剤は、四環系造影剤と比較して、脳に良好に取り込まれ、PBR発現組織における特異的取込みが改善していることがわかる。
【0215】
実施例17:(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性四環系インドール造影剤)の調製
17(a):(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(50)
乾燥DCM(100ml)中、T. Okubo et al,(Bioorg. Med. Chem. 2004;12:3569−3580)に記載の通り調製した(±)−4−オキソ−チオクロマン−2−カルボン酸(10.4g、50mmol)を、塩化オキサリル(12.6g、100mmol)及び一滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。次いで、反応物を真空中で蒸発させてガムを得、次いでDCM(100ml)に再溶解し、氷浴で0℃に冷却し、撹拌し、DCM(20ml)中ジエチルアミン(8.03g、110mmol)を用いて1時間かけて滴下処理した。反応物を室温にして1時間かけて温め、10%炭酸カリウム水溶液(100ml)を添加し、反応混合物を激しく撹拌した。DCM溶液を分離した。DCMの2つのさらなるバッチ(100ml)で水溶液を抽出し、混合抽出物を硫酸マグネシウムで乾燥させた。DCM溶液を真空中で濃縮して暗緑色油を得、それを静置して結晶化させた。結晶固体をジエチルエーテル(50ml)で倍散し、濾過して、標記化合物(50)(8.57g、65%)を薄緑色固体として得た。構造を、1H NMR(300MHz,CDCl3)δ 1.06(t,J=7.1Hz,3H),1.23(t,J=7.1Hz,3H),3.0−3.5(m,6H),4.25(m,1H),7.15−7.21(m,2H),7.32−7.39(m,1H),8.10−8.14(m,1H)によって確認した。
【0216】
17(b):(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(51)
(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(50)(1.32g、5.0mmol)及び4−メトキシフェニルヒドラジン塩酸塩(0.87g、5.0mmol)のエタノール(10ml)溶液に、濃硫酸(0.73ml、1.35g、13.8mmol)を窒素下で添加した。反応混合物を還流下で24時間加熱した。冷却後、反応混合物を濾過し、固体をエタノールで洗浄し、真空中で乾燥させて(45℃)、標記化合物(51)(1.05g、57%)を薄黄色固体として得た。構造を、13C NMR(75MHz,DMSO−d6)δ 10.5,12.7,32.7,37.9,39.5,53.0,97.6,103.3,109.87,109.92,120.3,123.5,123.8,124.3,124.7,124.9,127.8,129.4,131.8,151.3,166.2によって確認した。
【0217】
17(c):(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(過去の四環系インドール造影剤の非放射性類似体)
(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(51)(150mg、0.41mmol;実施例17(b)に従って調製した)の無水DMF(4ml)溶液に、L.Croninら(J.Org.Chem.2004年;69:5934〜5946頁)に記載の通り調製した2−フルオロエチルトシレート(166mg、0.82mmol)を添加し、その後鉱油(34mg、0.82mmol)中水素化ナトリウム60%分散液を窒素下で添加した。反応混合物を80℃で1時間加熱した。冷却後、溶媒を真空中で除去し、残渣を水(30ml)でクエンチし、DCM(2×30ml)で抽出し、乾燥させ(MgSO4)、溶媒を真空中で除去した。残渣を、シリカカラムクロマトグラフィーによって5〜10%のEtOAc/CH2Cl2で溶出して精製した。粗固体を、エーテル/ミネラルスピリットでクエンチし、濾過し、真空中で乾燥させて(45℃)、標記化合物(非放射性四環系インドール造影剤)(77mg、46%)を薄褐色固体として得た。構造を、1H NMR(300MHz,CDCl3)δ 1.12(t,J=7.0Hz,3H),1.36(t,J=7.0Hz,3H),3.25−3.70(m,4H),3.83(s,3H),4.45−4.70(m,2H),4.80(t,J=5.2Hz,1H),4.96(t,J=5.2Hz,1H),5.09(s,1H),6.84−6.93(m,2H),7.13−7.32(m,3H),7.46(m,1H),7.58(d,J=8.0Hz,1H)によって確認した。
【0218】
実施例18:(±)−11−(2−[18F]フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(四環系インドール造影剤)の合成
18F-/水を、反応容器中、K222(4mg)、K2CO3水溶液(0.1モル溶液50μl)及びアセトニトリル(500μl)に添加し、窒素流の下で100℃において20〜30分間乾燥させた。アセトニトリル(1000ul)中エチル−1,2−ジトシレート(4mg)を添加し、100℃で10分間加熱した。反応混合物を冷却し、半分取(preperative)HPLCによって精製し、18F−フルオロエチルトシレートを含有する画分を収集した。この画分を、H2Oで体積約20mlに希釈し、コンディショニングしたlight t−C18 sep pakに搭載し、H2O(1×2ml)でフラッシュした。sep pakを大流量のN2ラインで20分間乾燥させた。次いで、18FフルオロエチルトシレートをDMF(500μl)で溶出した。
【0219】
DMF(250ul)中前駆体化合物(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(51;実施例17(b)に従って調製した)(13mg)を、第2の反応容器に添加し、N2で5分間パージした。次いで、DMF(2×250ul)中NaH(1.3mg)を窒素下で添加し、反応容器を45℃で0.5〜1時間加熱した。次いでこれに、上記で調製したDMF中18Fフルオロエチルトシレートを添加し、N2でパージした反応容器中、100℃で10分間加熱した。反応物を冷却し、水(1ml)で反応容器から洗浄した。シリンジフィルターによって溶液を濾過し、分取HPLCで精製した。主な放射性ピークを含有する画分を収集した。これを、H2Oで体積約10mlに希釈し、コンディショニングしたlight t−C18 sep pakに搭載し、H2O(1×2ml)でフラッシュし、EtOH(0.5ml)でP6バイアルに溶出し、リン酸緩衝食塩水(5ml)を添加した。
【技術分野】
【0001】
本発明は、インビボイメージング、特に末梢性ベンゾジアゼピン受容体(PBR)のインビボイメージングに関する。PBRと高い親和性で結合し、投与後に脳に良好に取り込まれ、PBRとの良好な選択的結合を有するインドール系インビボ造影剤を提供する。本発明はまた、本発明のインビボ造影剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の合成方法を提供する。本発明の他の態様は、本発明の前駆体化合物の使用を含む本発明のインビボ造影剤の合成方法、前記方法を実施するためのキット、及び自動化された前記方法を実施するためのカセットを含む。さらに本発明は、本発明のインビボ造影剤を含む放射性医薬組成物、並びに前記インビボ造影剤の使用方法を提供する。
【背景技術】
【0002】
末梢性ベンゾジアゼピン受容体(PBR)は、主に末梢組織及びグリア細胞内に局在することが知られているが、その生理的機能は未だ解明されていない。PBRは、細胞内ではミトコンドリア外膜上に局在することが知られており、ミトコンドリア機能の調節及び免疫系において潜在的な役割を示す。さらにPBRは、細胞増殖、ステロイド産生、カルシウム流及び細胞呼吸に関与すると想定されている。
【0003】
異常なPBR発現は、多発性硬化症(Banati et al,2001 Neuroreport;12(16):3439−42;Debruyne et al,2002 Acta Neurol Belg;102(3):127−35)、ラスムッセン脳炎(Banati et al,1999 Neurology;53(9):2199−203)、脳血管炎(Goerres et al,2001 Am J Roentgenol;176(4):1016−8)、ヘルペス脳炎(Cagnin et al,2001 Brain;124(Pt 10):2014−27)、及びAIDS関連認知症(Hammoud et al,2005 J Neurovirol;11(4):346−55)を含む、中枢神経系(CNS)の炎症性病状に関連するとされている。
【0004】
またCNSでは、パーキンソン病(Gerhard et al,2006 Neurobiol Dis;21(2):404−12;Ouchi et al,2005 Ann Neurol;57(2):161−2)、大脳皮質基底核変性症(Gerhard et al,2004 Mov Disord;19(10):1221−6)、進行性核上性麻痺(Gerhard et al,2006 Neurobiol Dis;21(2):404−12)、多系統萎縮症(Gerhard et al,2003 Neurology;61(5):686−9)、ハンチントン病(Pavese et al,2006 Neurology;66(11):1638−43;Tai et al,2007 Brain Res Bull;72(2−3):148−51)、筋萎縮性側索硬化症(Turner et al,2004 Neurobiol Dis;15(3):601−9)、及びアルツハイマー病(Cagnin et al,2001 Lancet;358(9283):766;Yasuno et al,2008 Biol Psychiatry;64(10):835−41)などの変性疾患において、PBRとの関係が報告されている。
【0005】
いくつかのCNS虚血状態は、虚血性脳卒中(Gerhard et al,2005 Neuroimage;24(2):591−5)、末梢神経損傷(Banati et al,2001 Neuroreport;12(16):3439−42)、てんかん(Sauvageau 2002 Metab Brain Dis;17(1):3−11;Kumar et al,2008 Pediatr Neurol;38(6))を含む異常なPBR発現に関係することが示されている。PBRは、外傷性脳損傷における損傷の程度を決定するためのバイオマーカーと想定されており(Toyama et al,2008 Ann Nucl Med;22(5):417−24)、外傷性脳損傷の動物モデルにおいて、PBR発現の増大が報告された(Venneti et al,2007 Exp Neurol;207(1):118−27)。興味深いことに、急性ストレスは、脳内のPBR発現の増大に相関するとされており、慢性ストレスは、PBRの下方制御と相関するとされている(Lehmann et al,1999 Brain Res;851(1−2):141−7)。PBRを結像するために[11C]PK11195を使用して、神経膠腫の境界を描写できることが報告されている(Junck et al,1989 Ann Neurol;26(6):752−8)。神経障害性疼痛を有する対象において活性化したミクログリアを観測したTsudaらによれば、PBRは神経障害性疼痛にも関連し得る(2005 TINS 28(2) pp101−7)。
【0006】
末梢では、PBR発現は、肺炎症(Branley et al,2008 Nucl. Med. Biol;35(8):901−9)、慢性閉塞性肺疾患及び喘息(Jones et al,2003 Eur Respir J;21(4):567−73)、炎症性腸疾患(Ostuni et al,Inflamm Bowel Dis;2010 online publication)、関節リウマチ(van der Laken et al,2008 Arthritis Rheum;58(11):3350−5)、原発性線維筋痛症(Faggioli et al,2004 Rheumatology;43(10):1224−1225)、神経損傷(Durrenberger et al,2004 J Peripher Nerv Syst;9(1):15−25)、アテローム性動脈硬化症(Fujimura et al,2008 Atherosclerosis;201(1):108−111)、結腸、前立腺及び乳癌(Deane et al,2007 Mol Cancer Res;5(4):341−9;Miettinen et al,1995 Cancer Res;55(12):2691−5;Han et al,2003 J Recept Signal Transduct Res;23(2−3):225−38)、腎炎(Tam et al,1999 Nephrol Dial Transplant;14(7):1658−66;Cook et al,1999 Kidney Int;55(4):1319−26)、並びに虚血再灌流障害(Zhang et al,2006 J Am Coll Surg;203(3):353−64)に関係するとされている。
【0007】
PBR選択的リガンドである(R)−[11C]PK11195を使用する陽電子放射断層撮影(PET)イメージングは、中枢神経系(CNS)炎症の一般的な指標を提供する。しかし、(R)−[11C]PK11195は、タンパク質結合性が高く、特異性の低い結合を有するものから非特異的な結合を有するものがあることが知られている。さらに、放射性標識したその代謝産物の役割は知られておらず、結合の定量化には、複雑なモデリングを必要とする。
【0008】
三環系インドール化合物は、当技術分野で公知である。Davies et al(J. Med. Chem. 1998;41(4):451−67)には、ある種の三環系インドール化合物が教示されており、それらをメラトニン作動薬及び拮抗薬として特徴づけている。Napper et al(J. Med. Chem. 2005;48:8045−54)には、ヒストン及び他のタンパク質のリシン残基からアセチル基を除去する酵素ファミリーの一員である酵素SIRT1の選択的阻害に関連して、ある種の三環系インドール化合物の構造と活性の関係について教示・議論されている。米国特許第6451795号には、別の種類の三環系インドール化合物が開示されており、この化合物はPBR関連病状の治療に有用なものとして論じられている。米国特許第6451795号には、最も活性な化合物について0.2nM〜5.0nMのIC50値を開示しており、その化合物が末梢神経障害の予防又は治療、及び中枢神経変性疾患の治療にとって有用であると記載されている。
【0009】
Okubu et al,(Bioorganic & Medicinal Chemistry 2004 12 3569−80)には、四環系インドール化合物群の設計、合成及び構造、並びにPBRに対するそれらの親和性(IC50値は約0.4nMに過ぎない)を記載している。本発明の出願人に譲渡された国際公開第2007/057705号には、様々なインビボイメージング部分で標識した四環系インドール誘導体が開示されている。国際公開第2007/057705号に開示された好ましいインビボイメージング部分は、陽電子放射断層撮影(PET)又は単一光子放出断層撮影(SPECT)イメージング、最も好ましくはPETに適した部分である。
【0010】
さらに、同時係属注の国際出願PCT/EP2009/062827には、国際公開第2007/057705号の造影剤に類似の四環系インドール由来のインビボ造影剤が記載されている。
【0011】
国際公開第2007/057705号及び同時係属の国際出願PCT/EP2009/062827に記載の四環系インドール誘導体は、PBR受容体に対して良好な親和性を有し、注入の60分後における脳内の高い割合の放射活性は、親インビボ造影剤の活性を表している。これらの四環系インドール誘導体はまた、体内分布研究においてラットの脳内で妥当な初期濃度を達成するが、その取込みはやはり相対的に低く、改善の余地がある。本発明者らは、これらの従来技術の四環系インドール誘導体の嗅球(最高濃度のPBR受容体を有する脳領域)における相対的な保持が、インビボイメージングにとって望ましいものに満たないことも見出した。したがって、上述の従来技術の四環系インドールインビボ造影剤の有利な特性を保持するPBRインビボ造影剤にも、脳内取込みが改善され、PBR受容体との特異的結合が改善される余地がある。
【先行技術文献】
【特許文献】
【0012】
【特許文献1】国際公開第00/44751号
【発明の概要】
【0013】
本発明は、インビボ造影剤としての使用に適した新規な三環系インドール化合物を提供する。本発明はまた、本発明のインビボ造影剤の合成において有用な前駆体化合物、並びに前記前駆体化合物の合成方法を提供する。本発明の前駆体化合物の使用を含む、インビボ造影剤の調製方法も提供する。さらに、本発明のインビボ造影剤を含む医薬組成物に加えて、医薬組成物の容易な調製に適したキットを提供する。さらなる一態様では、異常なPBR発現に関連する状態のインビボイメージングのためのインビボ造影剤の使用を提供する。本発明のインビボ造影剤は、末梢性ベンゾジアゼピン受容体に対する改善された脳内取込み及び特異性と共に、公知の四環系インビボ造影剤の有利な特性を保持する。
【図面の簡単な説明】
【0014】
【図1】造影剤5と非放射性造影剤5の共溶出を示す図。
【図2】造影剤6と非放射性造影剤6の共溶出を示す図。
【図3】造影剤7と非放射性造影剤7の共溶出を示す図。
【図4】造影剤9と非放射性造影剤9の共溶出を示す図。
【図5】造影剤10(上)及び7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(中間)及び7−フルオロ−9−(2−[19F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(下)を示す図。
【図6】造影剤11と非放射性造影剤11の共溶出を示す図。
【図7】四環系造影剤の脳内での体内分布プロファイルを示す図。
【図8】造影剤5の脳内での体内分布プロファイルを示す図。
【図9】造影剤6の脳内での体内分布プロファイルを示す図。
【図10】造影剤7の脳内での体内分布プロファイルを示す図。
【図11】造影剤9の脳内での体内分布プロファイルを示す図。
【図12】造影剤10の脳内での体内分布プロファイルを示す図。
【図13】造影剤11の脳内での体内分布プロファイルを示す図。
【発明を実施するための形態】
【0015】
造影剤
一態様では、本発明は、次の式Iのインビボ造影剤を提供する。
【0016】
【化1】
式中、
R1はC1〜3アルキル又はC1〜3フルオロアルキルであり、
R2は水素、ヒドロキシル、ハロ、シアノ、C1〜3アルキル、C1〜3アルコキシ、C1〜3フルオロアルキル又はC1〜3フルオロアルコキシであり、
R3及びR4は独立にC1〜3アルキル、C7〜10アラルキルであるか、或いはR3及びR4はそれらと結合した窒素と共に含窒素C4〜6脂肪族環を形成するもので、適宜、窒素、酸素及び硫黄から選択される追加の1個のヘテロ原子を含んでいてもよく、
Y1はO、S、SO、SO2又はCH2であり、
Y2はCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
上記の式Iは、インビボイメージングに適した放射性同位体である原子を含む。
【0017】
「インビボ造影剤」は、本発明の文脈では、インビボイメージングに適した放射性標識合物である。用語「インビボイメージング」は、本明細書で使用される場合、対象の内面のすべて又は一部の画像を非侵襲的に作成する技術を指す。
【0018】
別途記載しない限り、単独又は組合せにおける用語「アルキル」は、好ましくは1〜3個の炭素原子を含有する直鎖又は枝分れアルキル基を意味する。かかる基の例としては、メチル、エチル及びプロピルが挙げられる。
【0019】
別途記載しない限り、用語「アルコキシ」は、エーテル結合を含む上記で定義したアルキル基を意味し、用語「エーテル結合」は、基−C−O−C−を指す。適切なアルキルエーテル基の例としては、メトキシ、エトキシ及びプロポキシが挙げられる。
【0020】
用語「ハロゲン」又は「ハロ−」は、フッ素、塩素、臭素又はヨウ素から選択される置換基を意味する。「ハロアルキル」及び「ハロアルコキシ」は、それぞれ1つ以上のハロゲンで置換されている上記で定義したアルキル及びアルコキシ基である。ハロアルキル及びハロアルコキシ置換基の場合、適切には、ハロゲンは、基の末端の水素を置き換え、すなわち−アルキレン−ハロゲン又は−アルコキシレン−ハロゲンとなる。用語「アルキレン」は、nが1〜3である二価の基−(CH2)n−を指し、用語「アルコキシレン」は、エーテル結合を含むアルキレン基を指し、エーテル結合は上記で定義した通りである。
【0021】
用語「シアノ」は、基−CNを指す。
【0022】
用語「ヒドロキシル」は、基−OHを指す。
【0023】
用語「アラルキル」は、基−アルキレン−フェニルを指し、アルキレンは上記で定義した通りである。
【0024】
「含窒素C4〜6脂肪族環」は、窒素ヘテロ原子を含む飽和C4〜6アルキル環である。その例としては、ピロリジニル、ピペリジニル及びモルホリニル環が挙げられる。
【0025】
用語「インビボイメージングに適した放射性同位体である原子を含む」とは、上記で定義した式Iにおいて、原子の1つの同位体が、インビボイメージングに適した放射性同位体であることを意味する。インビボイメージングに適したものとなるには、放射性同位体が、前記対象への投与後に外部から検出できることが必要である。
【0026】
キラル中心又は別の形態の異性体中心が、本発明のインビボ造影剤に存在する場合、鏡像異性体及びジアステレオ異性体を含むかかる異性体のすべての形態が本発明に包含される。キラル中心を含有する本発明のインビボ造影剤は、ラセミ混合物若しくは鏡像異性体に富んだ混合物として使用することができ、又は周知の技術を使用してラセミ混合物を分離することができ、個々の鏡像異性体を単独で使用することもできる。
【0027】
好ましい造影剤
R1は、好ましくはメチル又はC2〜3フルオロアルキルであり、最も好ましくは−エチレン−F(すなわち−CH2−CH2−F)である。
【0028】
R2は、好ましくは水素、ハロ、C1〜3アルコキシ又はC1〜3フルオロアルコキシである。R2は、最も好ましくは水素、ハロ又はC1〜3アルコキシであり、特に最も好ましくは水素、フルオロ又はメトキシである。R2が置換基である場合、5位又は6位にあるのが好ましく、最も好ましくは5−メトキシ、6−メトキシ、5−フルオロ及び6−フルオロから選択される。
【0029】
R3及びR4は、好ましくは独立に、メチル、エチル又はベンジルであり、最も好ましくは共にエチルである。
【0030】
或いは好ましくは、R3及びR4は、それらに結合した窒素と共に含窒素C5〜6脂肪族環を形成する。
【0031】
Y1は、好ましくはCH2である。
【0032】
本発明の最も好ましいインビボ造影剤では、Y2はCH2−CH2である。
【0033】
本発明の好ましいインビボ造影剤は、単光子放射断層撮影(SPECT)又は陽電子放射断層撮影(PET)を使用するイメージングに適している。SPECTでは、適切な放射性同位体は、γ線を放射する放射性ハロゲンである。本発明における使用に適したγ線を放射する放射性ハロゲンの例は、123I、131I及び77Brである。γ線を放射する好ましい放射性ハロゲンは123Iである。インビボ造影剤の放射性同位体が123Iである場合、R2が123Iであることが好ましい。PETでは、適切な放射性同位体は、陽電子を放出する放射性非金属である。本発明における使用に適した陽電子を放出する放射性非金属の例は、11C、18F及び124Iである。陽電子を放出する好ましい放射性非金属は、11C及び18Fである。11Cの場合、R1が11Cメチルであることが好ましい。放射性同位体が18Fである場合、R1がC2〜3[18F]フルオロアルキルであることが好ましく、最も好ましくは−エチレン−18Fである。
【0034】
本発明のインビボ造影剤がPETイメージングに適していることが好ましく、18Fは、PETイメージングに適した好ましい放射性同位体である。本発明の方法においてPETが好ましいのは、その優れた感受性及び分解能に起因するものであり、したがって病変の相対的に小さい変化も、経時的に観測することができる。PETスキャナーは、ピコモル範囲の放射能濃度を定期的に測定する。マイクロPETスキャナーは、現在約1mmの空間分解及び約4〜5mmの臨床的スキャナーにアプローチするものである。
【0035】
式Iの好ましいインビボ造影剤は、次の式Iaのものである。
【0036】
【化2】
式中、
R2aは水素、ハロ又はC1〜3アルコキシであり、
R3a及びR4aは独立にメチル、エチル若しくはベンジルであるか、或いはそれらに結合した窒素と共にピロリジニル、ピペリジニル、アゼパニル若しくはモルホリニル環を形成するものであり、
Y2aはCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
nは1、2又は3である。
【0037】
式Iaでは、R3a及びR4aは、好ましくは共にエチルであるか、或いはR3aはメチルであってR4aはベンジルであるか、或いはそれらに結合した窒素と共にアゼパニル環を形成するものである。
【0038】
R2aは、好ましくは水素、メトキシ又はフルオロである。
【0039】
Y2aは、好ましくはCH2−CH2又はCH(CH3)−CH2である。
【0040】
nは好ましくは2である。
【0041】
式Iaの好ましいインビボ造影剤では、
R3a及びR4aは共にエチルであるか、或いはR3aがメチルであってR4aがベンジルであるか、或いはそれらに結合した窒素と共にアゼパニル環を形成するものであり、
R2aは水素、メトキシ又はフルオロであり、
Y2aはCH2−CH2又はCH(CH3)−CH2であり、
nは2である。
【0042】
式Iaのインビボ造影剤の非限定的な例は、以下の通りである。
【0043】
【化3】
上記のインビボ造影剤1〜11の中でも、インビボ造影剤5、6、7、9、10及び11が好ましく、インビボ造影剤5及び10が最も好ましく、インビボ造影剤5が特に好ましい。本発明の任意のインビボ造影剤では、鏡像異性的に純粋な形態が特に好ましい。
【0044】
前駆体化合物
別の態様では、本発明は、本発明のインビボ造影剤の調製のための前駆体化合物を提供し、前駆体化合物は、次の式IIの化合物である。
【0045】
【化4】
式中、R11及びR12の一方は、本発明のインビボ造影剤について上記で定義した放射性同位体の適切な供給源と反応して、前駆体化合物と放射性同位体の適切な供給源との反応によって本発明のインビボ造影剤を形成する化学基であり、R11及びR12の他方は、それぞれR1及びR2について本明細書に定義した通りであり、適宜保護基を含んでおり、
R13〜14及びY11〜12は、それぞれR3〜4及びY1〜2について本明細書に定義した通りであり、適宜保護基を含んでいる。
【0046】
「前駆体化合物」は、好都合な化学的形態の検出可能な標識との化学反応が部位特異的に生じ、最小数の段階(理想的には単一段階)で著しい精製の必要なしに(理想的にはさらなる精製せずに)実施されて、所望のインビボ造影剤を生成し得るように設計された、放射性標識合物の非放射性誘導体を含む。かかる前駆体化合物は、合成することができ、好都合には良好な化学的純度で得ることができる。
【0047】
用語「保護基」は、望ましくない化学反応を阻害又は抑制するが、分子の残りを修飾しない穏やかな条件下で、問題の官能基から開裂して所望の生成物をもたらし得るのに十分な反応性を有するように設計されている基を意味する。保護基は、当業者に周知であり、’Protective Groups in Organic Synthesis’,Theorodora W. Greene and Peter G. M. Wuts,(Third Edition,John Wiley & Sons,1999)に記載されている。
【0048】
用語「放射性同位体の適切な供給源」は、前駆体化合物の置換基と反応性であり、したがって放射性同位体が前駆体化合物と共有結合するような化学的形態の放射性同位体を意味する。以下の部分に提示したそれぞれ特定の放射性同位体では、放射性同位体の1つ以上の適切な供給源を論じる。インビボ造影剤の当業者には、本発明における適用に適した放射性同位体のこれら及び他の供給源が周知であろう。
【0049】
以下のスキーム1は、前駆体化合物としてそれ自体使用することができ、又は少数のさらなる段階を用いて前駆体化合物に変換することができる化合物を得る方法を示す一般的な反応スキームである。スキーム1のR11〜14及びY11〜12は、式IIについて上記で定義した通りである。
【0050】
【化5】
或いは、前駆体化合物のR12が環の一番上の位置にある場合、以下のスキームIaに示した一般的な合成経路を使用することができる。
【0051】
【化6】
上記のスキーム1aでは、−R11a−PGは、保護されたR11基を表し、Rは、適切には本明細書に定義したものであることが好ましい。R11がヒドロキシである場合、−R11a−PGは、例えば−O−ベンジルであってよい。R12〜14及びY11〜12は、適切には上記の式IIについて提示したものであることが好ましく、但しR12はクロロではない。この合成経路では、環の一番下の位置の塩素によって、唯一の異性体が生成されるような唯一の方法で、強制的に環化が生じる。類似の方法は、国際公開第2003/014082号に開示されている。しかし、本発明者らが本発明の前駆体化合物を得るために国際公開第2003/014082号の教示を適用すると、その収率は低かった(実施例2(d)参照)。この問題は、環化段階で使用する溶媒系を変更することによって克服した。国際公開第2003/014082号では、環化段階をトルエン中で実施しているが、本発明者らは、トルエンの代わりにジエチルエーテルを使用すると最適な収率が得られることを見出した。環化段階の生成物はジエチルエーテルに溶解するが、非環化出発化合物は溶解しない。したがって、非環化出発化合物は反応容器の底部にZnCl2と共に残存し、環化生成物は反応容器の上部のジエチルエーテル中に移動する。
【0052】
したがって別の一態様では、本発明は、次の式IIbの前駆体化合物の調製方法であって、
【0053】
【化7】
(式中、
R11bは、スキームIaでR11aについて定義した通りであり、
R12b〜14bは、式IIのR12〜14について定義した通りであり、但しR12bはクロロではなく、
Y11b〜12bは、式IIのY11〜12について定義した通りである。)
下記の式IIcの化合物とZnCl2との反応によって、下記の式IIdの化合物を形成する段階を含んでいて、上記の反応を、ジエチルエーテルを含む溶媒系中で実施する方法を提供する。
【0054】
【化8】
(式中、R12c、Y11c及びY12cは、適切にはそれぞれR12、Y11及びY12について本明細書に定義した通りであることが好ましく、PGcは保護基である。)
【0055】
【化9】
(式中、R12d、Y11d、Y12d及びPGdは、それぞれR12c、Y11c、Y12c及びPGcについて定義した通りである。)
好ましくは、前記保護基のPGc、PGdは、−ベンジルである。式IIbの前駆体化合物は、式IIの好ましい前駆体化合物を表す。
【0056】
インビボ造影剤の放射性同位体が18Fである場合、18Fによる標識は、前駆体化合物の脱離基の求核置換によって達成され得る。
【0057】
適切な脱離基にはCl、Br、I、トシレート(OTs)、メシレート(OMs)及びトリフラート(OTf)が挙げられる。もう1つの戦略は、前駆体化合物に存在するアルキルアミド基上の所定位置に適切な脱離基を有することである。いずれの場合も前駆体化合物は、普通は核反応18O(p,n)18Fから水溶液として得られ、カチオン性対イオンの付加及びその後の水の除去によって反応性になる[18F]フッ化物イオン(18F-)の適切な供給源と反応することによって、一段階で標識することができる。18Fは、18F(CH2)3−LG(LGは上記で定義した脱離基を表す)を有する前駆体化合物のヒドロキシル基をO−アルキル化することによって導入することもできる。或いは放射性フッ素原子は、ベンゼン環などの芳香族環との直接共有結合を介して結合することができる。アリール系では、アリールジアゾニウム塩、アリールニトロ化合物又はアリール第4級アンモニウム塩の18F−フッ化物求核置換が、アリール−18F誘導体に適した経路である。
【0058】
以下のスキーム2に示す通り、上記のスキーム1又はスキーム1aのいずれかを継続して、本発明の18Fインビボ造影剤を得るのに適した前駆体化合物に達することができる。
【0059】
【化10】
出発化合物及び中間体は、市販されており、又は刊行された学術論文、例えばNapper et al,J Med Chem 2005;48:8045−54;Davies et al,J Med Chem 1998;41:451−467から公知である。
【0060】
18Fを含むインビボ造影剤を得るための、式IIの好ましい前駆体化合物では、R11はC1〜3アルキレン−LGであり、LGは脱離基を表す。最も好ましいかかる前駆体化合物は、式IIa
【0061】
【化11】
(式中、
LGは、メシレート、トシレート及びトリフラートから選択され、
R12a〜14a、Y12a及びmは、適切にはそれぞれ式IaのR2a〜4a、Y2a及びnについて上記で定義した通りであることが好ましい]
の化合物である。
【0062】
式IIaの好ましい前駆体化合物の非限定的な例は、以下の通りである。
【0063】
【化12】
上記の前駆体化合物1〜11の中でも、前駆体化合物5、6、7、9、10及び11が好ましく、前駆体化合物5及び10が最も好ましく、前駆体化合物5が特に好ましい。
【0064】
11Cで標識したPETトレーサー化合物は、前駆体化合物を11Cヨウ化メチルと反応させることによって合成することができる。11Cの半減期はわずか20.4分なので、中間体11Cヨウ化メチルが高い特異的活性を有し、その結果、可能な限り急速な反応過程を使用して中間体11Cヨウ化メチルを生成することが重要である。かかる11Cによる標識技術の詳しい総説は、Antoni et al,“Aspects on the Synthesis of 11C−Labelled Compounds” in Handbook of Radiopharmaceuticals,Ed. M.J. Welch and C.S. Redvanly(2003,John Wiley and Sons)に見ることができる。
【0065】
本発明の11Cで標識したインビボ造影剤は、以下のスキーム3に示す通り上記のスキーム1を継続することによって得ることができる。
【0066】
【化13】
イメージング部分が放射性ヨウ素である場合、好ましい前駆体化合物は、求電子性ヨウ素化を受ける誘導体を含む化合物である。この例は、トリアルキルスタンナン(例えば、トリメチルスタンニル又はトリブチルスタンニル)などの有機金属誘導体、又はトリアルキルシラン(例えば、トリメチルシリル)、又は有機ホウ素化合物(例えば、ボロン酸エステル又は有機三フッ化ホウ素)である。
【0067】
求電子性の放射性ヨウ素標識では、前駆体化合物は、好ましくは活性化有機金属前駆体化合物(例えば、トリアルキルスズ、トリアルキルシリル又は有機ホウ素化合物)を含む。放射性ヨウ素を有機分子に導入する前駆体化合物及び方法は、Bolton(J. Lab. Comp. Radiopharm. 2002;45:485−528)に記載されている。適切なボロン酸エステルである有機ホウ素化合物及びそれらの調製は、Kabalaka et al(Nucl. Med. Biol.,2002;29:841−843 and 2003;30:369−373)に記載されている。適切な有機三フッ化ホウ素及びそれらの調製は、Kabalaka et al(Nucl. Med. Biol.,2004;31:935−938)に記載されている。放射性ヨウ素標識にとって好ましい前駆体化合物は、有機金属前駆体化合物、最も好ましくはトリアルキルスズを含む。
【0068】
放射性ヨウ素で標識した本発明のインビボ造影剤は、以下のスキーム4に示す通り上記のスキーム1を継続することによって得ることができる。
【0069】
【化14】
放射性臭素標識は、放射性ヨウ素標識について前述の方法と類似の方法によって達成することができる。Kabalka及びVarmaは、放射性臭素標識化合物を含む放射性ハロゲン標識化合物の合成のための様々な方法を総説した(Tetrahedron 1989年;45(21):6601〜21頁)。
【0070】
本発明の前駆体化合物は、理想的には非発熱性の滅菌形態で提供される。したがって前駆体化合物は、インビボ造影剤を哺乳動物への投与に適した生体適合性の担体と一緒に含む医薬組成物の調製のために使用することができる。前駆体化合物はまた、かかる医薬組成物の調製のためのキット又はカセットの成分として含まれるのに適している。これらの態様を、以下により詳細に論じる。
【0071】
別の好ましい実施形態では、前駆体化合物は固相と結合している。前駆体化合物は、好ましくは、固体支持マトリックスと共有結合した状態で供給される。この方法では、所望の生成物は溶液の形態であり、出発材料及び不純物は固相に結合したままである。かかる系の一例として、18F−フッ化物による固相求電子性フッ素化のための前駆体化合物は、国際公開第03/002489号に記載されており、18F−フッ化物による固相求核フッ素化のための前駆体化合物は、国際公開第03/002157号に記載されている。
【0072】
調製方法
さらなる一態様では、本発明は、
(i)本発明の前駆体化合物を提供する段階と、
(ii)本明細書に定義の前記放射性同位体の適切な供給源を提供する段階と、
(iii)段階(i)の前駆体化合物を、段階(ii)の放射性同位体と反応させて、本発明のインビボ造影剤を得る段階と
を含む、本発明のインビボ造影剤を調製する方法を提供する。
【0073】
段階(i)では、前駆体化合物は、前駆体化合物の説明で前述した通り、自動合成装置との併用に適したキット又はカセット内の溶液として、或いは固体支持体に結合した状態で提供することができる。キット及びカセットは、本発明のさらなる態様を形成するものであり、これらを以下により詳細に論じることにする。
【0074】
前駆体化合物を放射性同位体と「反応させる」段階は、可能な限り高い放射化学的収率(RCY)で所望のインビボ造影剤を形成するのに適した反応条件下で、2つの反応物を一緒にすることを含む。本発明のインビボ造影剤を得るためのいくつかの特定の合成経路を、以下の実験部分に提示する。
【0075】
本発明の調製方法について、インビボ造影剤、前駆体化合物及び放射性同位体の適切な好ましい実施形態は、既に本明細書に提示した通りである。
【0076】
キット及びカセット
またさらなる一態様では、本発明は、本発明のインビボ造影剤を調製するためのキットを提供し、前記キットは、本発明の前駆体化合物を含み、したがって放射性同位体の滅菌供給源との反応によって、最小限の操作数で所望のインビボ造影剤を得るようにされている。かかる考察は、放射性同位体が相対的に短い半減期を有する場合に特に重要であり、放射線技師(radiopharmacist)にとっての取扱いを容易にし、したがって放射線量を低減するのに重要である。前駆体化合物は、好ましくは凍結乾燥形態でキット内に存在し、かかるキットの再構成のための反応媒体は、好ましくは生体適合性の担体である。
【0077】
「生体適合性の担体」は、インビボ造影剤を懸濁又は溶解することによって組成物が生理的に容認できるものとなる、すなわち毒性又は過度の不快感なしに哺乳動物の身体に投与することができる流体、特に液体である。生体適合性の担体は、適切には、発熱物質を含まない注入用の滅菌水;生理食塩水(注入用の最終生成物が、等張性であるように、又は低張性にならないように平衡化できることが有利である)などの水溶液;1つ以上の浸透圧調節物質(例えば、血漿カチオンと生体適合性の対イオンの塩)、糖類(例えば、グルコース又はスクロース)、糖アルコール(例えば、ソルビトール又はマンニトール)、グリコール(例えば、グリセロール)又は他の非イオン性ポリオール材料(例えば、ポリエチレングリコール、プロピレングリコール等)の水溶液などの注入可能な担体液体である。生体適合性の担体は、エタノールなどの生体適合性の有機溶媒を含むこともできる。かかる有機溶媒は、より親油性が高い化合物又は配合物を可溶化するのに有用である。好ましくは、生体適合性の担体は、発熱物質を含まない注入用の水、等張食塩水又はエタノール水溶液である。静脈内注入のための生体適合性の担体のpHは、適切には4.0〜10.5の範囲である。
【0078】
本発明のキットでは、前駆体化合物は、好ましくは滅菌上の完全性及び/又は放射性の安全性と、任意選択により不活性なヘッドスペースガス(例えば、窒素又はアルゴン)の維持を可能にすると同時に、シリンジによる溶液の添加及び除去を可能にする封止容器中に存在する。好ましい封止容器は、セプタムで封止したバイアルであり、その場合気密にする蓋は、オーバーシール(一般にアルミニウム製)で圧着される。かかる封止容器は、その蓋が所望に応じて真空に耐えて、例えばヘッドスペースガスを取り替え、又は溶液を脱気することができるというさらなる利点を有する。
【0079】
キットで使用される場合、前駆体化合物の好ましい実施形態は、既に本明細書に記載した通りである。
【0080】
キットで使用するための前駆体化合物は、所望の非発熱性の滅菌材料を得るために、無菌の製造条件下で使用することができる。或いは、前駆体化合物を非滅菌条件下で使用し、その後、例えばγ照射、高圧蒸気殺菌法、乾熱又は化学的処理(例えば、エチレンオキシドを用いる)を使用して最終滅菌にかけることができる。好ましくは、前駆体化合物は、非発熱性の滅菌形態で提供される。最も好ましくは、非発熱性の滅菌前駆体化合物は、前述の封止容器で提供される。
【0081】
キットのすべての成分は、実施と実施の間の汚染の可能性を最小限に抑え、無菌性及び品質の保持を確実にするために使い捨てできることが好ましい。
【0082】
現在、特に[18F]放射性トレーサーは、好都合には自動化放射合成(radiosynthesis)装置で調製されることが多い。Tracerlab(商標)及びFastlab(商標)(GE Healthcare Ltd)を含む、かかる装置の市販で利用可能ないくつかの例がある。かかる装置は一般に、放射合成を実施するための装置に備え付けられる、しばしば使い捨ての「カセット」を含み、そのカセットの中では放射化学が行われる。カセットは、普通は、流体経路、反応容器、及び試薬のバイアルを入れるポート、並びに放射合成後の除去段階で使用する任意の固相抽出カートリッジを含む。
【0083】
したがって本発明は、別の態様では、
(i)本明細書に定義の前駆体化合物を含有する容器と、
(ii)その容器を、本明細書に定義のインビボイメージングに適した前記放射性同位体の適切な供給源を用いて溶出するための手段と
を含む、本明細書に定義のインビボ造影剤の自動合成のためのカセットを提供する。
【0084】
本発明のカセットについて、前駆体化合物及び放射性同位体の適切な供給源の適切な好ましい実施形態は、既に本明細書に定義した通りである。
【0085】
カセットは、
(iii)過剰の放射性同位体を除去するためのイオン交換カートリッジと、任意選択により
(iv)前駆体化合物が1つ以上の保護基を含む場合、得られた放射性標識生成物を脱保護して本明細書に定義のインビボ造影剤を形成するためのカートリッジと
をさらに含むことができる。
【0086】
放射性医薬組成物
別のさらなる一態様では、本発明は、哺乳動物への投与に適した形態の生体適合性の担体と一緒に本発明のインビボ造影剤を含む、「放射性医薬組成物」を提供する。生体適合性の担体は、本発明のキットに関して上記で定義した通りである。本発明の放射性医薬組成物について、インビボ造影剤の適切な好ましい実施形態は、既に本明細書に定義した通りである。
【0087】
放射性医薬組成物は、非経口によって、すなわち注入によって投与することができ、最も好ましくは水溶液である。かかる組成物は、任意選択により緩衝液;薬学的に許容される可溶化剤(例えば、シクロデキストリン又はPluronic、Tween若しくはリン脂質などの界面活性剤);薬学的に許容される安定剤又は抗酸化剤(アスコルビン酸、ゲンチシン酸又はパラアミノ安息香酸など)などのさらなる成分を含有することができる。本発明のインビボ造影剤が放射性医薬組成物として提供される場合、前記インビボ造影剤の調製方法は、放射性医薬組成物を得るために必要な段階、例えば有機溶媒の除去、生体適合性の緩衝液の添加、及び任意の場合によるさらなる成分の添加をさらに含むことができる。非経口投与では、放射性医薬組成物が滅菌され、非発熱性であることを確実にする段階を含む必要もある。
【0088】
使用方法
またさらなる一態様では、本発明は、
(i)前記対象に、本発明のインビボ造影剤を投与する段階と、
(ii)前記インビボ造影剤を前記対象のPBRと結合させる段階と、
(iii)インビボイメージング手順によって、前記インビボ造影剤の放射性同位体から放出されたシグナルを検出する段階と、
(iv)前記シグナルの位置及び/又は量の代表的な画像を作成する段階と、
(v)前記インビボ造影剤によって放出された前記シグナルと直接相関する、前記対象におけるPBR発現の分布及び程度を決定する段階と
を含む、対象におけるPBR発現の分布及び/又は程度を決定するためのインビボイメージング法を提供する。
【0089】
本発明のインビボイメージング法について、インビボ造影剤の適切な好ましい実施形態は、既に本明細書に定義した通りである。
【0090】
インビボ造影剤の「投与」は、好ましくは非経口により、最も好ましくは静脈内により実施される。静脈内経路は、インビボ造影剤を対象の身体にわたって送達し、したがって血液脳関門(BBB)にも通過させ、前記対象の中枢神経系(CNS)に発現したPBRと接触させるための最も効率的な方法である。さらに、静脈内投与は、実質的な身体的介入又は実質的な健康上の危険性を示さない。本発明のインビボ造影剤は、好ましくは本明細書に定義の本発明の医薬組成物として投与される。本発明のインビボイメージング法は、本発明のインビボ造影剤を予め投与した対象に対して実施される上記で定義した段階(ii)〜(v)を含むと理解することもできる。
【0091】
投与段階の後、検出段階の前に、インビボ造影剤をPBRと結合させる。例えば、対象が無傷哺乳動物である場合、インビボ造影剤は、哺乳動物の身体中を動的に移動し、その様々な組織と接触することになる。インビボ造影剤がPBRと接触すると特異的な相互反応が生じ、その結果、PBRを有する組織からのインビボ造影剤のクリアランスは、PBRのない、又はPBRの少ない組織からのクリアランスよりも長くかかる。PBRを有する組織と結合したインビボ造影剤と、PBRのない、又はPBRの少ない組織において結合したインビボ造影剤との間の比の結果、PBRに特異的に結合したインビボ造影剤の検出が可能となるある時点に達することになる。かかる理想的な比は、約2:1である。
【0092】
本発明の方法の「検出」段階は、前記シグナルに感受性のある検出器を用いて放射性同位体が放出するシグナルを検出することを含む。この検出段階は、シグナルデータの取得と理解することもできる。単光子放射型断層撮影(SPECT)及び陽電子放射断層撮影(PET)は、本発明の方法における使用に最も適したインビボイメージング手順である。PETは、本発明の方法における使用に好ましいインビボイメージング手順である。
【0093】
本発明の方法の「作成」段階は、取得したシグナルデータに再構成アルゴリズムを適用してデータセットを得るコンピューターによって実施される。次いで、このデータセットを操作して、前記放射性同位体が放出するシグナルの位置及び/又は量を示す画像を作成する。放出されたシグナルは、PBRの発現と直接相関し、したがって、作成した画像を評価することによって「決定」段階を行うことができる。
【0094】
本発明の「対象」は、任意のヒト又は動物対象であってよい。好ましくは、本発明の対象は哺乳動物である。最も好ましくは、前記対象は、インビボの無傷哺乳動物の身体である。特に好ましい一実施形態では、本発明の対象はヒトである。インビボイメージング法は、健康な対象において、又はPBRの異常発現に関連する病理学的状態(以下「PBR状態」)を有することが公知の、若しくはその疑いのある対象においてPBRを試験するために使用することができる。好ましくは、前記方法は、PBR状態を有することが公知の、又はその疑いのある対象におけるインビボイメージングに関し、したがって前記状態の診断方法において有用性がある。
【0095】
インビボイメージングが役立つかかるPBR状態の例としては、多発性硬化症、ラスムッセン脳炎、脳血管炎、ヘルペス脳炎、AIDS関連認知症、パーキンソン病、大脳皮質基底核変性症、進行性核上性麻痺、多系統萎縮症、ハンチントン病、筋萎縮性側索硬化症、アルツハイマー病、虚血性脳卒中、末梢神経損傷、てんかん、外傷性脳損傷、急性ストレス、慢性ストレス、神経障害性疼痛、肺炎症、慢性閉塞性肺疾患、喘息、炎症性腸疾患、関節リウマチ、原発性線維筋痛症、神経損傷、アテローム性動脈硬化症、腎炎、虚血再灌流障害、及び癌、特に結腸、前立腺又は乳房の癌が挙げられる。本発明のインビボ造影剤は、特に、脳へのそれらの良好な取込みに起因して、CNSのインビボイメージングに適している。
【0096】
代替の一実施形態では、本発明のインビボイメージング法は、前記対象のための治療投与計画の過程中、反復して実施することができ、前記投与計画は、PBR状態に対抗するための薬物を投与する段階を含む。例えば、本発明のインビボイメージング法は、PBR状態に対抗するための薬物を用いる治療の前、その最中、及びその後に実施することができる。この方法では、前記治療の作用を経時的にモニタすることができる。好ましくはこの実施形態では、インビボイメージング手順はPETである。PETは、優れた感受性及び分解能を有し、したがって病変における相対的に小さい変化も経時的に観察することができ、このことは治療モニタリングにとって特に有利である。
【0097】
さらなる一態様では、本発明は、PBR状態の診断方法を提供する。本発明の診断方法は、上記で定義したインビボイメージング法を、PBR発現の分布及び程度が特定の臨床像に帰するさらなる段階(vi)、すなわち演繹的な医学的決定段階と一緒に含む。
【0098】
別の態様では、本発明は、本明細書に定義の診断方法において使用するための、本明細書に定義のインビボ造影剤を提供する。
【0099】
またさらなる一態様では、本発明は、本明細書に定義の診断方法において使用する本明細書に定義の放射性医薬組成物の製造に使用するための、本明細書に定義のインビボ造影剤を提供する。
【0100】
本発明を、ここで一連の非限定的な例によって例示する。
【実施例】
【0101】
実施例の簡単な説明
実施例1は、前駆体化合物5及び造影剤5の合成を記載する。
【0102】
実施例2は、造影剤5の非放射性類似体の合成を記載する。
【0103】
実施例3は、前駆体化合物6及び造影剤6の合成を記載する。
【0104】
実施例4は、造影剤6の非放射性類似体の合成を記載する。
【0105】
実施例5は、前駆体化合物7及び造影剤7の合成を記載する。
【0106】
実施例6は、造影剤7の非放射性類似体の合成を記載する。
【0107】
実施例7は、前駆体化合物9及び造影剤9の合成を記載する。
【0108】
実施例8は、造影剤9の非放射性類似体の合成を記載する。
【0109】
実施例9は、前駆体化合物10及び造影剤10の合成を記載する。
【0110】
実施例10は、造影剤10の非放射性類似体の合成を記載する。
【0111】
実施例11は、前駆体化合物11及び造影剤11の合成を記載する。
【0112】
実施例12は、造影剤11の非放射性類似体の合成を記載する。
【0113】
実施例13は、前駆体化合物5の鏡像異性体分離を記載する。
【0114】
実施例14は、非放射性造影剤5の鏡像異性体分離を記載する。
【0115】
実施例15は、PBRに対する親和性を試験するために使用したインビトロ効力アッセイを記載する。
【0116】
実施例16は、インビボでの本発明の造影剤の性能を試験するために使用した体内分布法を記載する。
【0117】
実施例17は、過去の四環系インドール造影剤の非放射性類似体の合成を記載する。
【0118】
実施例18は、過去の四環系インドール造影剤の合成を記載する。
【0119】
実施例で使用した略語一覧
aq 水性
DCM ジクロロメタン
DMAP 4−ジメチルアミノピリジン
DMF ジメチルホルムアミド
EDC 1−エチル−3−[3−ジメチルアミノプロピル]カルボジイミド塩酸塩
EOS 合成の終了
EtOAc 酢酸エチル
IPA イソプロピルアルコール
LC−MS 液体クロマトグラフィー−質量分析
NMR 核磁気共鳴
OBn ベンジルオキシ
OMs メシレート
OTs トシレート
RT 室温
TLC 薄層クロマトグラフィー
Tol トルエン
実施例
実施例1:メタンスルホン酸2−(4−ジエチルカルバミル−5−メトキシ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)エチルエステル(前駆体化合物5)及び9−(2−[18F]フルオロ−エチル)−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤5)の合成
実施例1(a):塩化ベンジルオキシアセチル(1)
ジクロロメタン(50mL)中ベンジルオキシ酢酸(10.0g、60.0mmol、8.6mL)に、塩化オキサリル(9.1g、72.0mmol、6.0mL)及びDMF(30.0mg、0.4mmol、32.0μL)を添加し、RTで3時間撹拌した。最初、反応が進行するにつれてガスが急速に発生したが、反応が完了すると発生は停止した。ジクロロメタン溶液を真空中で濃縮してガムを得た。このガムを、さらなる塩化オキサリル(4.5g、35.7mmol、3.0mL)、ジクロロメタン(50mL)及び一滴のDMFで処理した。ガスが急速に発生し、反応物をさらに2時間撹拌した。次いで、反応物を真空中で濃縮して、塩化ベンジルオキシアセチル(1)11.0g(定量的)をガムとして得た。構造を、13C NMR(75MHz,CDCl3)δC73.6,74.8,128.1,128.4,128.6,130.0及び171.9によって確認した。
【0120】
実施例1(b):2−ベンジルオキシ−N−(2−クロロ−5−メトキシ−フェニル)アセトアミド(2)
ジクロロメタン(100mL)中塩化ベンジルオキシアセチル(1)(11.0g、60.0mmol)及び2−クロロ−5−メトキシアニリン塩酸塩(11.7g、60.2mmol)を0℃で撹拌し、トリエチルアミン(13.0g、126.0mmol、18.0mL)を15分かけてゆっくり添加した。撹拌した反応物を、RTにして18時間かけて温めた。トリエチルアミン塩酸塩の大量の沈殿が生じた。ジクロロメタン溶液を10%炭酸カリウム水溶液(50mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、2−ベンジルオキシ−N−(2−クロロ−5−メトキシ−フェニル)アセトアミド(2)18.9g(定量的)をガムとして得た。構造を、13C NMR(75MHz,CDCl3):δC55.6,69.6,73.6,106.2,111.1,114.1,127.7,128.3,128.6,129.2,134.6,136.5,158.9及び167.7によって確認した。
【0121】
実施例1(c):(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシフェニル)アミン(3)
THF(100mL)中2−ベンジルオキシ−N−(2−クロロ−5−メトキシ−フェニル)アセトアミド(2)(18.9g、62.0mmol)を撹拌し、水素化アルミニウムリチウム(4.9g、130.0mmol)を15分かけてゆっくり添加した。最初に水酸化アルミニウムリチウムを添加すると、水素ガスが急速に発生した。次いで、反応物を4時間加熱還流し、週末にかけてRTで静置した。次いで、撹拌した溶液に水(50mL)を滴下添加することによって、反応物をクエンチした。水素が強烈に発生して、反応混合物が還流した。次いで、反応物を真空中で濃縮してスラリーを得た。水(200mL)及び酢酸エチル(200mL)を添加し、混合物を激しく振とうした。次いで、セライトを介して反応物を濾過して、沈殿した水酸化アルミニウムを除去し、酢酸エチル溶液を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシフェニル)アミン(3)18.4g(定量的)をガムとして得た。構造を、13C NMR(75MHz,CDCl3)δC43.3,55.3,68.2,73.0,98.1,101.8,111.6,127.6,127.7,128.4,129.3,137.9,144.8及び159.5によって確認した。
【0122】
実施例1(d):3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4)
2−オキソシクロヘキサンカルボン酸エチル(30g、176mmol、28mL)をジエチルエーテル(30mL)に溶解し、窒素下で0℃に冷却した。臭素(28g、176mmol、9.0mL)を15分かけて滴下添加し、反応混合物をRTにして90分かけて温めた。混合物を、氷冷した飽和炭酸カリウム水溶液(250mL)にゆっくり注ぎ、酢酸エチル(3×200mL)で抽出した。混合有機層を硫酸マグネシウムで乾燥させ、濾過し、真空中で濃縮し、真空ラインで18時間乾燥させて、3−ブロモ−2−ヒドロキシ−1−エンカルボン酸エチルエステル(4)41.4g(94%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3):δC14.1,17.7,21.8,32.0,60.0,60.8,99.7,166.3及び172.8によって確認した。
【0123】
実施例1(e):3[(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシ−フェニル)−アミノ]−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(5)
(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシフェニル)アミン(3)(10.0g、34.2mmol)を、乾燥THF(100mL)中、窒素下において−40℃で撹拌し、カリウムビス(トリメチルシリル)アミド(トルエン中0.5M溶液143.0mL、72.0mmol)を30分かけて添加した。次いで、乾燥THF(10mL)中3−ブロモ−2−ヒドロキシシクロヘキセ−1−エンカルボン酸エチルエステル(4)(8.5g、34.2mmol)を添加し、RTにして1.5時間かけて温めた。酢酸(10.0g、166mmol、10.0mL)を添加し、真空中で濃縮してTHFを除去した。酢酸エチル(200mL)及び10%炭酸カリウム水溶液(100mL)を添加し、混合物を激しく振とうした。酢酸エチル溶液を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、3[(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシ−フェニル)−アミノ]−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(5)16.5g(定量的)をガムとして得、それを次の段階で粗生成物として使用した。粗反応混合物のHPLC(Gemini 150×4.6mm、20分間50〜95%のメタノール/水)、18.9分(38%)、19.2分(25%)、23.1分(28%)。
【0124】
反応物の一成分を単離した13C NMR(75MHz,CDCl3)δC14.3,20.6,21.8,26.4,38.6,43.0,55.8,60.5,68.7,73.3,93,4,106.3,108.2,119.3,121.5,127.5,127.6,128.3,135.7,137.0,137.9,155.7及び175.0。
【0125】
実施例1(f):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)
塩化亜鉛(7.1g、52.0mmol)を、乾燥ジエチルエーテル(150mL)中3[(2−ベンジルオキシ−エチル)−(2−クロロ−5−メトキシ−フェニル)−アミノ]−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(5)(8.0g、17.0mmol)に窒素下で添加し、5.5時間加熱還流した。反応物を還流すると、反応物中に高密度の褐色油が形成した。次いで反応物を冷却し、上清であるジエチルエーテルをデカントで除去し、酢酸エチル(100mL)を添加し、2NのHCl(50mL)及び10%炭酸カリウム水溶液(50mL)で洗浄した。ジエチルエーテル層を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮して油(2.0g)を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(10〜40%の(B)、340g、22CV、150mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)1.8gを得た。高密度の褐色層を、酢酸エチル(100mL)及び2NのHCl(50mL)で処理した。酢酸エチル溶液を分離し、10%炭酸カリウム水溶液(50mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して油(5.2g)を得た。ジエチルエーテル(100mL)及び無水塩化亜鉛(7.0g)を添加した。混合物を、さらに5日間加熱還流した。エーテル層を暗色ガムからデカントで除去し、2NのHCl(50mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガム(2.8g)を得た。このガムを、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(5〜35%の(B)、340g、150mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)2.1gを得た。得られたすべての材料は、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)4.1g(50%)であった。構造を、13C NMR(75MHz,CDCl3):δC14.4,20.5,22.3,27.5,40.2,43.9,55.0,60.2,70.7,73.3,100.2,107.5,108.4,120.1,122.8,127.4,127.5,128.2,132.0,137.4,138.1,152.6及び175.8によって確認した。
【0126】
実施例1(g):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(7)
エタノール(50mL)中9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(6)(2.0g、4.1mmol)に、水酸化ナトリウム(1.1g、27.1mmol)及び水(5mL)を添加し、80℃で18時間加熱した。次いで、エタノールを真空中で蒸発することによって除去し、残渣をジエチルエーテル(50mL)と水(50mL)に分離した。ジエチルエーテル層を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガム(71.0mg)を得た。水層を、2NのHCl(20mL)でpH1の酸性にし、ジクロロメタン(2×100mL)で抽出した。ジクロロメタン層を硫酸マグネシウムで乾燥させ、真空中で濃縮して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(7)1.6g(87%)を気泡として得た。構造を、13C NMR(75MHz;CDCl3):δC20.2,22.2,27.1,39.7,44.0,55.1,70.7,73.3,100.6,106.3,108.9,123.0,127.4,127.5,128.3,132.0,138.0及び152.0によって確認した。
【0127】
実施例1(h):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(8)
9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(7)(1.5g、3.7mmol)をジクロロメタン(50mL)に溶解し、塩化オキサリル(700mg、5.5mmol、470μL)及びDMF(1滴)を添加し、反応物を20℃で2時間撹拌した。反応が進行するにつれて、約30分間ガスが穏やかに発生した。次いで、反応物を真空中で濃縮して、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(8)をガムとして得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz;CDCl3):δC 20.8,22.1,26.4,44.2,51.8,55.1,70.7,73.3,100.7,106.0,108.6,119.5,123.4,127.3,127.7,128.3,131.9,138.0,138.2,152.0.及び176.3によって確認した。
【0128】
実施例1(i):9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(9)
9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(8)(1.6g、3.7mmol)を、次いでジクロロメタン(50mL)に溶解し、0℃に冷却し、撹拌し、ジエチルアミン(810mg、11.0mmol、1.1mL)を滴下添加した。反応物を、室温にして18時間かけて温めた。次いで、反応混合物を10%炭酸カリウム水溶液(50mL)で洗浄し、分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガムを得た。粗材料を、ジエチルエーテルから結晶化させて、9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(9)1.2g(71%)を白色結晶固体として得た。構造を、13C NMR(75MHz;CDCl3):δC 13.0,14.5,19.8,22.2,27.9,36.4,40.4,41.9,43.8,55.0,70.8,73.3,100.2,108.5,108.6,119.9,122.5,127.4,127.5,128.3,131.5,137.8,138.2,152.4及び174.5によって確認した。
【0129】
実施例1(j):9−(2−ベンジルオキシ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(10)
メタノール(100ml)中9−(2−ベンジルオキシ−エチル)−8−クロロ−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(9)(1.0g、2.1mmol)を、木炭上10%パラジウム(1.0g)、トリエチルアミン(2.9mg、2.9mmol、4μL)と共に、水素ガス雰囲気下において55℃で18時間振とうした。次いで、セライトパッドを介して反応物を濾過し、濾液を真空中で濃縮してガム(908mg)を得た。次いで、ガムをジクロロメタン(100ml)に溶解し、5%炭酸カリウム水溶液(50ml)で洗浄した。次いで、ジクロロメタン溶液を分離し、硫酸マグネシウムで乾燥させ、真空中で濃縮してガムを得た。次いで、ガムをジエチルエーテル(50ml)から結晶化させ、結晶を濾過によって収集して、9−(2−ベンジルオキシ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(10)523mg(57%)を得た。構造を、13C NMR(75MHz;CDCl3):δC13.1,14.6,20.1,22.0,28.1,36.4,40.5,42.0,43.0,54.7,68.8,73.3,99.4,102.4,107.8,116.4,121.2,127.6,127.6,128.3,135.6,137.8,138.0 153.6及び175.0によって確認した。
【0130】
実施例1(k):9−(2−ヒドロキシエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(11)
メタノール(50ml)中9−(2−ベンジルオキシ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(10)(1.0g、2.1mmol)を、木炭上10%パラジウム(300mg)及び過剰の水素ガスを用いて55℃で18時間振とうした。次いで、セライトパッドを介して反応物を濾過し、濾液を真空中で濃縮して、9−(2−ヒドロキシエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(11)578mg(100%)を気泡として得た。構造を、13C NMR(75MHz;CDCl3):δC13.0,14.4,20.0,22.0,28.0,36.4,40.6,42.0,54.7,60.6,99.2,102.6,107.0,116.7,121.1,136.1,137.5,138.0 153.5及び175.7によって確認した。
【0131】
実施例1(l):メタンスルホン酸2−(4−ジエチルカルバミル−5−メトキシ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)エチルエステル(前駆体化合物5)
ジクロロメタン(30ml)中9−(2−ヒドロキシエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミン(11)(478mg、1.4mmol)を0℃に冷却し、塩化メタンスルホニル(477mg、4.2mmol、324μL)及びトリエチルアミン(420mg、4.2mmol、578μL)を添加し、RTにして終夜温めた。反応物を5%炭酸カリウム水溶液で洗浄した。層を分離した。混合有機物を硫酸マグネシウムで乾燥させ、真空中で濃縮してガム(696mg)を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(75〜100%のB、22CV、120g、85mL/分)で溶出して精製して、メタンスルホン酸2−(4−ジエチルカルバミル−5−メトキシ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)エチルエステル(前駆体化合物5)をガムとして得、それをジエチルエーテルから結晶化させて、無色固体346mg(59%)を得た。構造を、13C NMR(75MHz;CDCl3):δC13.1,14.5,20.0,21.9,28.0,36.3,36.7,40.3,41.8,41.9,54.7,68.1,100.0,102.0,109.0,116.4,122.0 135.1,137.3,153.8及び174.6によって確認した。
【0132】
実施例1(m):9−(2−[18F]フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤5)
GE HealthcareからGE PETraceサイクロトロンにより[18F]フッ化物が供給された。Kryptofix 2.2.2(2mg、5μmol)、重炭酸カリウム(0.1mol dm-3、0.1ml、5mg、5μmol)及びアセトニトリル(0.5ml)を、COC反応容器中[18F]F-/H2O(約400MBq、0.1〜0.3ml)に添加した。混合物を、100℃で20〜25分間、窒素流の下で加熱することによって乾燥させた。乾燥後、冷却なしにアセトニトリル(1ml)中前駆体化合物5(0.5〜1mg、1.2〜2.4μmol)をCOC反応容器に添加し、100℃で10分間加熱した。冷却後、反応混合物を除去し、COC反応容器を水(1.5ml)ですすぎ、主な粗反応物に添加した。
【0133】
この後、粗生成物に、半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配(flow gradient):3ml/分;0〜1分、40%B;1〜20分、40〜95%B;波長254nm;tR造影剤5、16分を適用した。HPLCの精製ピークである造影剤5を、水で体積10mlに希釈し、tC18 Sep−Pak(lite)カートリッジに吸着させた。カートリッジを水(2ml)で洗浄し、無水エタノール(0.5ml)で溶出し、その後ダルベッコリン酸緩衝食塩水(4.5ml)で溶出した。放射化学的収率30±7%(n=4)減衰補正なし、時間90〜120分、放射化学的純度≧99%。
【0134】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、40%B;1〜20分、40〜95%B;波長230nm;tR造影剤5、16分。図1は、造影剤5と非放射性造影剤5の共溶出を示す。
【0135】
実施例2:9−(2−フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤5)の合成
実施例2(a):フルオロエチルトシレート(12)
2−フルオロエタノール(640mg、10mmol、0.6mL)を、窒素下でピリジン(10mL)に溶解した。溶液を0℃で撹拌し、塩化トシル(4.2g、21.8mmol)を、5℃未満の温度を維持しながら30分かけて溶液に少しずつ添加した。反応物を0℃で3時間撹拌した。氷をゆっくり添加し、その後水(20mL)を添加した。反応混合物を酢酸エチルに抽出し、水で洗浄した。水層が酸性になるまで1NのHCl溶液で洗浄することによって、過剰のピリジンを除去した。過剰の塩化トシルを、1Mの炭酸ナトリウム水溶液で洗浄することによって除去した。有機層をブラインで洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、フルオロエチルトシレート(12)2.1g(98%)を無色油として得た。構造を、13C NMR(75MHz,CDCl3):δC21.6(CCH3),68.5(d,JCF=173Hz,OCH2CH2F),80.6(d,JCF=173Hz,OCH2CH2F),128.0,129.9,132.6及び145.1によって確認した。
【0136】
実施例2(b):2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13)
2−クロロ−5−メトキシアニリン塩酸塩(5.0g、26.0mmol)をDMF(50mL)に溶解し、水素化ナトリウム(2.3g、油中60%、57.0mmol)を添加した。反応物を窒素下においてRTで30分間撹拌した。DMF(5mL)中フルオロエチルトシレート(12)(6.7g、31.0mmol)を滴下添加し、反応物をRTで2時間撹拌した。次いで、反応物を100℃で18時間加熱した。反応物を冷却し、溶媒を減圧下で除去した。残渣を酢酸エチル(100mL)に溶解し、水(2×100mL)で洗浄した。有機物を収集し、硫酸マグネシウムで乾燥させ、真空中で濃縮して褐色油を得、それをシリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(5〜30%(B)、330g、18.1CV、120mL/分)で溶出して精製して、2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13)1.3g(25%)を黄色油として得た。構造を、13C NMR(75MHz;CDCl3):δC43.8(d,JCF=23Hz),55.3,82.0(d,JCF=165Hz),98.1,102.2,111.6,129.5,144.1及び159.5によって確認した。
【0137】
実施例2(c):3−[(2−クロロ−5−メトキシ−フェニル)−(2−フルオロエチル)アミノ]−2−ヒドロキシ−シクロヘキシル−1−エンカルボン酸エチルエステル(14)
2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13)(6.1g、30.0mmol)のTHF(170mL)溶液を−40℃に冷却した。カリウムビス(トリメチルシリル)アミド(トルエン中0.5M溶液126.0mL、63.0mmol)を滴下添加し、反応物を−40℃で30分間撹拌した。THF(30mL)中3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4;実施例1(d)に従って調製した)(7.4g、30.0mmol)を、−40℃で滴下添加した。冷却浴を除去し、反応物をRTで4時間撹拌した。反応物をブライン(300mL)でクエンチし、酢酸エチル(2×400mL)で抽出し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、3−[(2−クロロ−5−メトキシ−フェニル)−(2−フルオロエチル)アミノ]−2−ヒドロキシ−シクロヘキシル−1−エンカルボン酸エチルエステル(14)12.0g(定量的)を褐色油として得、それを次の段階で粗生成物として使用した。異性体混合物としての構造を、1H NMR(300MHz,CDCl3):δH1.08(0.8H,t,J=9Hz,CO2CH2CH3),1.22−1.33(2.2 H,m,CO2CH2CH3),1.40−2.60(7H,m,4−,5−及び6−CH2,CHN),3.20−4.50(10H,m,NCH2CH2F,NCH2CH2F,OCH3,CHCO2CH2CH3),6.50−6.70(1H,m,CHC(OCH3)CHCH),6.95(0.5H,dd,J=3及び6Hz,CHC(OCH3)CHCH),7.08(0.5H,d,J=3Hz,CHC(OCH3)CHCH)及び7.20−7.30(1H,m,CHC(OCH3)CHCH)によって確認した。
【0138】
実施例2(d):8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)
8−クロロ−9−(2−フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)の合成を、まず国際公開第2003/014082号に記載の条件を使用して試みた。2−クロロ−5−メトキシ−フェニル)(2−フルオロエチル)アミン(13;実施例2(b)に従って調製した)(600mg、3.8mmol)の乾燥THF(20mL)溶液を氷浴で冷却し、カリウムビス(トリメチルシリル)アミド(トルエン中0.5M溶液16mL、8.0mmol)で処理した。30分後、THF(4mL)中3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4;実施例1(d)に従って調製した)(1.04g、4.2mmol)を添加し、反応物をRTにして2時間かけて温めた。反応物を飽和塩化アンモニウム溶液でクエンチし、エーテルで2回抽出した。抽出物を、水、ブラインで洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(2.5〜50%のB、50g、25CV、40mL/分)で溶出して精製した。主な点は、3つの化合物の混合物であった。この混合物を、トルエン(20mL)中、乾燥塩化亜鉛(1.7g、12.6mmol)と共に終夜還流した。反応物を真空中で濃縮し、残渣を1NのHCL(25mL)と酢酸エチル(25mL)に分離し、次いで酢酸エチルで再度抽出した。有機層を水及びブラインで洗浄し、乾燥させ、真空中で濃縮して褐色油を得た。1H NMRは、これがいくつかの化合物の混合物であったことを示した。様々な溶媒におけるシリカによるTLCでは、この混合物を別個の点に分離することができなかった。混合物の1H NMRと標準品の1H NMRの比較によって、混合物が推定25%の8−クロロ−9−(2−フルオロ−エチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)を含有していることが示された。
【0139】
次いで、修正を加えた方法を実施した。3−[(2−クロロ−5−メトキシ−フェニル)−(2−フルオロエチル)アミノ]−2−ヒドロキシ−シクロヘキシル−1−エンカルボン酸エチルエステル(14)(12.2g、30.0mmol)をジエチルエーテル(250mL)に溶解し、塩化亜鉛(16.4g、120.0mmol)を添加した。反応物を16時間加熱還流した。酢酸エチル(500mL)を添加してすべてを溶解し、2NのHCl(200mL)、水(200mL)、10%炭酸カリウム水溶液(200mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(5〜20%のB、12CV、10g、100mL/分)で溶出して精製して、8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)5.3g(2段階にわたって50%)を黄色固体として得た。構造を、13C NMR(75MHz,CDCl3):δC14.4,20.4,22.2,27.4,40.1,44.2(d,JCF=23Hz),55.1,60.2,83.9(d,JCF=173Hz),100.6,107.9,108.2,119.8,123.1,131.9,137.2,152.7及び175.7によって確認した。
【0140】
実施例2(e):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(16)
8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(15)(5.3g、15.0mmol)をメタノール(180mL)に溶解し、トリエチルアミン(1.8g、18.0mmol、2.5mL)及び10%Pd/C(メタノール(20mL)中2g)を添加した。混合物をParr水素化装置(hydrogenator)に入れ、水素雰囲気下で18時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、溶媒を真空中で除去した。残渣を酢酸エチル(300mL)に溶解し、10%炭酸カリウム水溶液(200mL)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(16)4.2g(88%)を淡褐色固体として得た。構造を、13C NMR(75MHz,CDCl3):δC14.3,20.6,21.8,27.6,40.3,43.3(d,JCF=23Hz),54.9,60.1,82.0(d,JCF=165Hz),99.8,102.1,107.3,117.2,121.8,134.9,137.6,153.8及び176.0によって確認した。
【0141】
HPLC(Gemini 150×4.6mm、20分間50〜95%のメタノール/水)13.6分(94%)。
【0142】
実施例2(f):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(17)
8−クロロ−9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(16)(380mg、1.2mmol)をエタノール(4mL)に溶解した。水6mLに溶解した水酸化ナトリウム溶液(580mg、14.5mmol)を添加した。反応混合物を、終夜加熱還流した。溶媒を真空中で除去し、粗混合物を水で希釈し、酸性になるまで2NのHClで酸性にし、ジクロロメタンで洗浄した。有機物を混合し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(17)347mg(定量的)をオフホワイト色の固体として得、それを次の段階で粗生成物として使用した。構造を、13C NMR(75MHz;CDCl3):δC20.4,21.9,27.2,39.9,43.3(d,JCF=23Hz),55.1,81.9(d,JCF=173Hz),100.3,102.8,106.2,117.1,122.2,135.6,137.8,153.3及び180.8によって確認した。
【0143】
実施例2(g):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(18)
9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(17)(347mg、1.2mmol)の乾燥ジクロロメタン(2mL)溶液を窒素下で撹拌した。塩化オキサリル(453mg、3.6mmol、300μL)を添加し、その後一滴のDMFを添加し、反応混合物を窒素下においてRTで2時間撹拌し、次いで真空中で蒸発させて、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物371mg(定量的)をガムとして得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz,CDCl3):δC20.2,21.7,26.4,43.3(d,JCF=23Hz),54.9,80.5,83.1,100.2,102.2,105.8,116.7,122.4,135.5,137.4,153.5及び176.6によって確認した。
【0144】
実施例2(h):9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤5)
9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(18)(371mg、1.2mmol)をジクロロメタン(2mL)に溶解し、0℃に冷却した。次いでジエチルアミン(177mg、2.4mmol、250μL)を添加し、反応物を終夜RTで撹拌した。10%炭酸カリウム水溶液(2mL)を用いて反応物をクエンチした。ジクロロメタン層を相分離機によって収集し、次いで真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A):酢酸エチル(B)(50〜100%の(B)、50g、35.2CV、40mL/分)で溶出して精製して、薄黄色固体を得た。次に、固体を最小量のジエチルエーテルで倍散して、9−(2−フルオロエチル)−5−メトキシ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤5)240mg(58%)を得た。構造を、13C NMR(75MHz,CDCl3):δC13.0,14.6,19.9,21.9,28.0,36.3,40.5,41.9,43.1(d,JCF=23Hz),54.7,82.0(d,JCF=173Hz),99.7,102.1,108.3,117.0,121.5,135.3,137.4,153.3及び174.8によって確認した。
【0145】
実施例3:メタンスルホン酸2−[4−(ピペリジン−1−カルボニル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物6)及び[9−(2−[18F]フルオロエチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(造影剤6)の合成
実施例3(a):2−(ピペリジン−1−カルボニル)−シクロヘキサノン(19)
トルエン(100mL)中2−オキソシクロヘキサン−カルボン酸エチル(5.3g、31mmol、5.0mL)、DMAP(1.05g、9.4mmol)及びピペリジン(5.3g、63mmol、6.2mL)を4日間加熱還流した。反応物を冷却し、反応物を真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜80%(B)、100g、8CV、85mL/分)で溶出して精製して、2−(ピペリジン−1−カルボニル)−シクロヘキサノン(19)6.26g(96%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC23.5,24.5,25.5,26.2,27.1,30.4,41.9,42.9,46.8,54.2,167.6,207.6によって確認した。
【0146】
実施例3(b):2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20)
2−(ピペリジン−1−カルボニル)−シクロヘキサノン(19)(4.0g、19mmol)をジエチルエーテル(5mL)に溶解し、N2下で0℃に冷却した。臭素(5.9g、19mmol、1.0mL)を15分かけて滴下添加し、反応混合物を室温にして90分かけて温めた。固体を濾過によって収集して、2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20)5.86g(定量的)を白色固体として得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz,DMSO−d6)δC17.3,24.2,25.3,25.8,32.5,44.0,51.6,108.3,145.5,167.8によって確認した。
【0147】
実施例3(c):(2−ベンジルオキシ−エチル)−フェニル−アミン(21)
丸底フラスコ中、アニリン(2.0g、21.5mmol、2.0mL)、2,6−ルチジン(2.30g、21.5mmol)及びベンジル2−ブロモエチルエーテル(4.6g、21.5mmol、3.4mL)を、DMF(10mL)中で混合し、100℃で終夜撹拌した。反応物を冷却し、次いで酢酸エチル(50mL)で希釈した。これを水(3×20mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜50%のB、100g、19.5CV、85mL/分)で溶出して精製して、(2−ベンジルオキシ−エチル)−フェニル−アミン(21)2.22g(37%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3)δC43.6,68.6,73.2,113.1,117.5,127.5,127.7,128.4,129.1,138.2,148.1によって確認した。
【0148】
実施例3(d):[9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(22)
2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20)(1.5g、5.2mmol)及び(2−ベンジルオキシ−エチル)−フェニル−アミン(21)(3.2g、10.4mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(5mL)に溶解し、乾燥塩化亜鉛(2.13g、15.6mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(100mL)に溶解し、2NのHCl(30mL)、水(2×30mL)及び炭酸カリウム水溶液(2×30mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、SCXカートリッジ及び次いでシリカゲルクロマトグラフィーによって、石油(A)及び酢酸エチル(B)(30〜100%のB、12g、41CV、30mL/分)で溶出して精製して、[9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(22)600mg(27%)を油として得た。構造を、13C NMR(75MHz,CDCl3)δC21.5,21.7,24.5,25..7,26.3,273,37.7,42.8,43.1,46.7,60.2,68.7,73.1,108.2,108.7,117.8,118.9,120.5,126.4,127.3,127.4,128.1,136.2,137.8,172.9によって確認した。
【0149】
実施例3(e):[9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(23)
[9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(22)(600mg、1.4mmol)のメタノール(15mL)溶液に、メタノール(10mL)中Pd/C(200mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮した。粗材料を研和して、[9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(23)332mg(71%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3):δC21.2,21.9,24.7,27.4,36.4,43.4,45.0,47.0,60.9,107.8,109.0,117.7,119.0,120.7,126.6,136.2,137.2,173.5によって確認した。
【0150】
実施例3(f):メタンスルホン酸2−[4−(ピペリジン−1−カルボニル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物6)
[9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(23)(260mg、0.8mmol)のジクロロメタン(15mL)溶液に、ピリジン(633mg、8.0mmol、0.65mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(458mg、4.0mmol、0.31mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×50mL)及び水(2×50mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料をジエチルエーテルで倍散して、メタンスルホン酸2−[4−(ピペリジン−1−カルボニル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物6)263mg(82%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC21.4,21.8,24.7,25.9,26.9,27.4,36.6,36.8,41.7,43.3,47.0,67.9,108.5,109.5,118.4,119.7,121.3,126.9,136.2,172.7によって確認した。
【0151】
実施例3(g):[9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(造影剤6)
18Fによる前駆体化合物6の標識を、実施例1(f)に記載の通り実施した。
【0152】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長254nm;tR造影剤6、17分。
【0153】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤6、16分。放射化学的収率23±2%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図2は、造影剤6と非放射性造影剤6の共溶出を示す。
【0154】
実施例4:[9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(造影剤6の非放射性類似体)の合成
実施例4(a):(2−フルオロ−エチル)−フェニル−アミン(24)
丸底フラスコ中、アニリン(0.5g、5.4mmol)、2,6−ルチジン(0.58g、5.4mmol)及び2−フルオロエチルトシレート(12;実施例2(a)に従って調製した)(1.17g、5.4mmol)を、DMF(2.5mL)中で混合し、100℃で終夜撹拌した。反応物を冷却し、次いで酢酸エチル(50mL)で希釈した。これを水(3×20mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(100g、0〜100%のB、18CV、85mL/分)で溶出して精製して、(2−フルオロ−エチル)−フェニル−アミン(24)435mg(60%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH3.41(1H,t,J=3Hz,NCH2CH2F),3.50(1H,t,J=3Hz,NCH2CH2F),3.93(1H,s,br),4.54(1H,t,J=3Hz,NCH2CH2F),4.71(1H,t,J=3Hz,NCH2CH2F),6.65−6.82(3H,m,2×NCCH,NCCHCHCH),7.14−7.28(2H,m,2×NCCHCHCH)によって確認した。
【0155】
実施例4(b):[9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(非放射性造影剤6)
2−ブロモ−6−(ピペリジン−1−カルボニル)−シクロヘキサノン(20;実施例3(b)に従って調製した)(500mg、1.7mmol)及び(2−フルオロ−エチル)−フェニル−アミン(24)(890mg、3.5mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(2mL)に溶解し、乾燥塩化亜鉛(682mg、5mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(50mL)に溶解し、2NのHCl(20mL)、水(2×20mL)及び炭酸カリウム水溶液(2×20mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料をジエチルエーテルで倍散して、[9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−イル]−ピペリジン−1−イル−メタノン(非放射性造影剤6)151mg(27%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC21.6,21.8,24.7,26.5,26.9,27.4,37.3,43.1(d,JCF=45Hz),47.0,82.1(d,JCF=173Hz),108.5,108.9,118.6,119.4,121.0,126.8,136.2,172.7によって確認した。
【0156】
実施例5:メタンスルホン酸2−[4−(ベンジル−メチル−カルバモイル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物7)及び9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(造影剤7)の合成
実施例5(a):9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(25)
(2−ベンジルオキシ−エチル)−フェニル−アミン(21;実施例3(c)に従って調製した)(8.0g、26mmol)及び3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(4;実施例1(d)に従って調製した)(3.2g、13mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(30mL)に溶解し、乾燥塩化亜鉛(10.6g、78mmol)を添加した。混合物をN2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(300mL)に溶解し、2NのHCl(100mL)、水(2×100mL)及び炭酸カリウム水溶液(2×100mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(2.5〜40%のB、17CV、330g、100mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(25)3.49g(72%)を油として得た。構造を、13C NMR(75MHz,CDCl3)δC14.2,20.5,21.8,26.5,38.6,42.9,60.4,68.7,73.2,106.4,108.8,118.7,120.7,127.4,127.5,128.3,136.2,136.9,137.8,175.0によって確認した。
【0157】
実施例5(b):9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(26)
9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(25)(35g、9.3mmol)をエタノール(9mL)に溶解し、次いで水(15mL)中NaOH(1.56g)を添加した。反応物を、2時間加熱還流した。反応物を真空中で濃縮し、残渣を水で希釈し、ジクロロメタン(2×150mL)で洗浄した。水層を2NのHCl(150mL)に滴下添加し、次いでジクロロメタン(3×150mL)に抽出した。有機物を乾燥させ、真空中で濃縮して、9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(26)2.48g(92%)を黄色固体として得、それを精製せずに次の段階で使用した。構造を、13C NMR(75MHz,CDCl3)δC20.4,21.8,26.4,38.3,42.9,68.7,73.3,105.7,108.8,118.7,119.3,102.9,127.4,127.6,128.3,136.2,137.1,137.8,108.9によって確認した。
【0158】
実施例5(c):9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(27)
9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(26)(600mg、1.7mmol)を、窒素下で乾燥DCM(8mL)に溶解し、塩化オキサリル(393mg、3.1mmol、0.26mL)を添加した。反応物を室温で3時間撹拌すると、ガスが激しく発生した。反応物を真空中で濃縮し、次いでジクロロメタン(8mL)に再溶解し、0℃に冷却し、N−ベンジルメチルアミン(412mg、3.4mmol、0.44mL)を添加した。反応物を室温にして終夜温めた。反応物を5%炭酸カリウム水溶液で洗浄し、乾燥させ、真空中で濃縮して褐色油を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(30%のB、10g)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(27)246mg(64%)を黄色油として得た。構造を、1H NMR(CDCl3)δH1.60−2.30(4H,m,CHCH2CH2CH2),2.70−2.90(2H,m,CHCH2CH2CH2),3.10(1.5H,s,N(CH3)CH2Ph),3.13(1.5H,s,N(CH3)CH2Ph),3.73(2H,t,J=6Hz,NCH2CH2O),4.10−4.30(3H,m,NCH2CH2O,CHCH2CH2CH2),4.42(1H,s,OCH2Ph),4.44(1H,s,OCH2Ph),4.80(1H,s,N(CH3)CH2Ph),4.81(1H,s,N(CH3)CH2Ph),6.90−7.50(14H,m)によって確認した。
【0159】
実施例5(d):9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(28)
9−(2−ベンジルオキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(27)(246mg、0.5mmol)のメタノール(15mL)溶液に、メタノール(10mL)中Pd/C(200mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、9−(2ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(28)36mg(20%)を緑色油として得、それを精製せずに次の段階で使用した。構造を、1H NMR(CDCl3) δH 1.80-2.20 (4H,m), 2.70-3.00 (2H,m), 3.20-4.30 (10H,m), 6.90-7.50 (9H,m)によって確認した。
【0160】
実施例5(e):メタンスルホン酸2−[4−(ベンジル−メチル−カルバモイル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物7)
9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(28)(36mg、0.1mmol)のジクロロメタン(2mL)溶液に、ピリジン(7.91g、1.0mmol、8.1mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(57mg、0.5mmol、0.04mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×10mL)及び水(2×10mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜80%のB、4g、45CV、18mL/分)で溶出して精製して、メタンスルホン酸2−[4−(ベンジル−メチル−カルバモイル)−1,2,3,4−テトラヒドロ−カルバゾール−9−イル]−エチルエステル(前駆体化合物7)14mg(32%)を黄色油として得た。構造を、1H NMR(CDCl3)δH1.10−2.40(5H,m),2.51(1.5H,s,OSO2CH3),2.54(1.5H,s,OSO2CH3),2.70−2.90(2H,m),3.08(1.5H,s,NCH3),3.15(1.5H,s,NCH3),3.40−3.70(1H,m),4.10−4.80(4H,m),7.00−7.50(9H,m)によって確認した。
【0161】
実施例5(f):9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(造影剤7)
18Fによる前駆体化合物7の標識を、実施例1(f)に記載の通り実施した。半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長254nm;tR造影剤7、17分。
【0162】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤7、16分。放射化学的収率23±2%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図3は、造影剤7と非放射性造影剤7の共溶出を示す。
【0163】
実施例6:9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(非放射性造影剤7)
実施例6(a):3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(29)
2−オキソシクロヘキサンカルボン酸エチル(5.0g、29mmol、4.7mL)をジエチルエーテル(5mL)に溶解し、N2下で0℃に冷却した。臭素(4.6g、29mmol、4.2mL)を15分かけて滴下添加し、反応混合物を室温にして90分かけて温めた。混合物を、氷冷した飽和炭酸ナトリウム水溶液(40mL)にゆっくり注ぎ、酢酸エチル(3×40mL)で抽出した。混合有機層を乾燥させ、真空中で濃縮して、3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(29)5.96g(81%)を薄黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,DMSO−d6)δC14.14,17.65,21.77,32.02,59.95,60.83,99.70,166.33,172.81によって確認した。
【0164】
実施例6(b):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(30)
(2−フルオロ−エチル)−フェニル−アミン(24;実施例4(a)に従って調製した)(560mg、4.0mmol)及び3−ブロモ−2−ヒドロキシ−シクロヘキセ−1−エンカルボン酸エチルエステル(29)(500mg、2.0mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(4mL)に溶解し、乾燥塩化亜鉛(820mg、6mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。生成物を酢酸エチル/エーテル(30mL/150mL)に溶解し、2NのHCl(40mL)、水(2×100mL)及び炭酸カリウム水溶液(2×100mL)で洗浄し、次いで乾燥させ、濃縮して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(30)447mg(91%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC14.3,20.4,21.7,26.4,38.5,43.1(d,JCF=15Hz),60.6,76.6,77.0,77.4,82.1(d,JCF=173Hz),106.9,108.5,118.9,119.4,121.1,127.1,136.2,136.7,174.9によって確認した。
【0165】
実施例6(c):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(31)
9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸エチルエステル(30)(380mg、1.3mmol)をエタノール(3mL)に溶解し、次いで水(5mL)中NaOH(520mg)を添加した。反応物を、2時間加熱還流した。反応物を真空中で濃縮し、残渣を水で希釈し、ジクロロメタン(2×50mL)で洗浄した。水層を2NのHCl(50mL)に滴下添加し、次いでジクロロメタン(3×50mL)に抽出した。有機物を乾燥させ、真空中で濃縮して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(31)130mg(37%)を黄色固体として得、それを次の段階で精製せずに使用した。構造を、1H NMR(300MHz,CDCl3)δ 1.90−2.42(4H,m,2−及び3−CH2),2.60−2.91(2H,m,1−CH2),3.94(1H,t,J=6Hz,4−CH),4.30(1H,t,J=6Hz,NCH2CH2F),4.37(1H,t,J=6Hz,NCH2CH2F),4.59(1H,t,J=6Hz,NCH2CH2F),4.74(1H,t,J=6Hz,NCH2CH2F),7.05−7.26(3H,m,ArH),7.59(1H,d,J=9Hz,ArH)によって確認した。
【0166】
実施例6(d):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(32)
乾燥ジクロロメタン(6mL)中9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸(31)(0.5g、1.91mmol)を、窒素雰囲気下で、塩化オキサリル(490mg、3.8mmol、0.34mL)及び一滴のDMFと共に室温で撹拌した。反応物を真空中で濃縮して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(32)545mg(定量的)を得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC20.2,21.6,26.7,43.1,43.4,50.6,80.9,83.1,105.3,108.8,118.3,120.0,121.6,126.5,136.2,137.5,176.1によって確認した。
【0167】
実施例6(e):9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(非放射性造影剤7)
9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボニル塩化物(32)(110mg、0.4mmol)をジクロロメタン(1mL)に溶解し、0℃に冷却した。次いで、N−ベンジルメチルアミン(92mg、0.8mmol、98μL)を添加し、反応物を終夜RTで撹拌した。反応物を10%炭酸カリウム水溶液(2mL)でクエンチした。ジクロロメタン層を相分離機によって収集し、次いで真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜100%のB、12g、30CV、30mL/分)で溶出して精製して、9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ベンジル−メチル−アミド(非放射性造影剤7)39mg(28%)を得た。構造を、1H NMR(300MHz,CDCl3)δH1.75−2.32,(4H,m,2−及び3−CH2),2.68−2.86(2H,m,1−CH2),3.10(1H,s,NCH3),3.14(2H,s,NCH3),4.17−4.39(3H,m,NCH2CH2F及び4−CH2),4.52−4.87(4H,m,NCH2Ph及びNCH2CH2F),6.96−7.42(9H,m,ArH)によって確認した。
【0168】
実施例7:メタンスルホン酸2−(4−ジエチルカルバモイル−6−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物9)及び6−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤9)の合成
実施例7(a):2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)
ベンジルオキシ酢酸(4.6g、28.0mmol、4.0mL)のDCM(52mL)溶液に、塩化オキサリル(7.7g、61mmol、5.3mL)及び一滴のDMFを添加した。反応混合物を室温で4時間撹拌した。過剰の塩化オキサリルを真空中で除去して、ベンジルオキシ−塩化アセチルを得た。粗塩化アシルをDCM(100mL)で希釈し、トリエチルアミン(5.3mL、41.6mmol、4.2g)を添加し、その後4−フルオロアニリン(3.5g、32mmol、3.0mL)を添加した。反応混合物を終夜RTで撹拌した。次いで、反応物を1MのHCl水溶液(100mL)でクエンチし、乾燥させ、真空中で濃縮して、2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)7.1g(95%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC69.2,73.5,115.4(d,JCF=22Hz),121.4(d,JCF=7Hz),127.9,128.2,128.5,132.5(d,JCF=3Hz),136.3,157.6,160.8及び167.5によって確認した。
【0169】
実施例7(b):(2−ベンジルオキシ−エチル)−(4−フルオロ−フェニル)−アミン(34)
LAH(1.25g、27mmol)の乾燥ジエチルエーテル(100mL)懸濁液に、2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)(6.9g、27mmol)の乾燥ジエチルエーテル(100mL)溶液を滴下添加した。還流が維持されるように添加を実施した。添加が終了したら、反応混合物を4時間加熱還流し、次いで氷水に注ぎ、DCMを添加した。アルミニウム塩を分解するために、強塩基のpHが得られるまで2M水酸化ナトリウム水溶液を添加した。層を分離し、水層をDCMで洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(5〜50%のB、100g、12CV、60mL/分)で溶出して精製して、(2−ベンジルオキシ−エチル)−(4−フルオロ−フェニル)−アミン(34)5.5g(84%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3)δC44.0,68.3,72.8,113.7(d,JCF=7Hz),115.3(d,JCF=22Hz),127.5,127.6(d,JCF=3Hz),128.3,137.8,144.5,154.1及び157.2によって確認した。
【0170】
実施例7(c):3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35)
2−シクロヘキサン(cyclohexone)−カルボン酸エチル(7.50mL、47.0mmol)、DMAP(1.72g、14.1mmol)及びジエチルアミン(9.77mL、94.0mmol)を、トルエン(100mL)中で72時間加熱還流した。反応物を冷却し、トルエンを減圧下で除去した。粗油を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(1:1、100g、SiO2)で溶出して精製して、2−オキソ−シクロヘキサンカルボン酸ジエチルアミン6.8g(73%)を橙色油として得た。構造を、13C NMR(CDCl3)δ11.1,12.7,21.3,24.9,28.5,39.4,39.6,51.7,166.5,205.9によって確認した。
【0171】
2−オキソ−シクロヘキサンカルボン酸ジエチルアミン(3.56mL、19.3mmol)をジエチルエーテル(5mL)に溶解し、N2下で撹拌しながら0℃に冷却した。臭素(0.99mL、19.3mmol)を15分かけて滴下添加し、反応混合物を室温にして3時間かけて温めた。反応物から固体が沈殿した。それを濾過によって収集し、エーテルで洗浄して、3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35)5.85g(109%)を薄黄色固体として得た。構造を、13C NMR(CDCl3)δ11.2,12.8,22.7,28.8,37.6,37.9,39.4,51.0,55.7,165.5,197.2によって確認した。
【0172】
実施例7(d):9−(2−ベンジルオキシ−エチル)−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(36)
2−ベンジルオキシ−N−(4−フルオロ−フェニル)−アセトアミド(33)(5.3g、22mmol)及び3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35)(3.0g、13mmol))の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(30mL)に溶解し、乾燥塩化亜鉛(9.0g、66mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(300mL)に溶解し、2NのHCl(100mL)、水(2×100mL)及び炭酸カリウム水溶液(2×100mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10〜50%のB、100g)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(36)196mg(11%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.14(3H,t,J=7Hz,N(CH2CH3)2),1.30(3H,t,J=7Hz,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びNCH2CH2OBn),3.66−3.75(1H,m,4−CH),4.00−4.25(2H,m,NCH2CH2OBn),4.41(2H,s,OCH2Ph),6.75−6.95(2H,m,NCCHCHCFCH),7.05−7.15(1H,m,NCCHCHCFCH)及び7.16−7.25(5H,m,Ph)によって確認した。
【0173】
実施例7(d):6−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(37)
9−(2−ベンジルオキシ−エチル)−6−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(36)(600mg、1.4mmol)のメタノール(40mL)溶液に、メタノール(5mL)中Pd/C(100mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、6−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(37)460mg(80%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、1H NMR(300MHz,MeOD−d3)δH1.18(3H,t,J=9Hz,N(CH2CH3)2),1.35(3H,t,J=9Hz,N(CH2CH3)2),1.80−2.20(4H,m,2−及び3−CH2),2.69−3.88(2H,m,1−CH2),3.40−3.86(6H,m,N(CH2CH3)2及びNCH2CH2OH),4.03−4.22(3H,m,NCH2CH2OH及び4−CH),6.75−6.95(2H,m,NCCHCHCFCH)及び7.05−7.15(1H,m,NCCHCHCFCHによって確認した。
【0174】
実施例7(e):メタンスルホン酸2−(4−ジエチルカルバモイル−6−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物9)
6−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(37)(460mg、1.4mmol)のジクロロメタン(20mL)溶液に、ピリジン(1.11g、14.0mmol、1.1mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(722mg、6.3mmol、0.5mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×30mL)及び水(2×30mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜100%(B)、10g、45CV、30mL/分)で溶出して精製し、次いでジエチルエーテルで倍散して、メタンスルホン酸2−(4−ジエチルカルバモイル−6−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物9)166mg(30%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC12.9,15.0,21.1,27.7,36.1,36.7,40.6,41.7,67.8,103.3(d,JCF=23Hz),108.7,109.0,109.1,109.4(d,JCF=5Hz),126.9(d,JCF=10Hz),132.4,138.4,156.1,159.2及び173.3によって確認した。
【0175】
実施例7(f):6−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤9)
18Fによる前駆体化合物9の標識を、実施例1(f)に記載の通り実施した。
【0176】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、40%のB;1〜20分、40〜95%のB;波長254nm;tR造影剤9、15分。
【0177】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤9、14分。放射化学的収率26±8%(n=4)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図4は、造影剤9と非放射性造影剤9の共溶出を示す。
【0178】
実施例8:6−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤9)の合成
実施例8(a):(2−フルオロ−エチル)−(4−フルオロ−フェニル)−アミン(38)
丸底フラスコ中、4−フルオロアニリン(1.3g、11.6mmol、1.6mL)、2,6−ルチジン(1.24g、11.6mmol)及び2−フルオロエチルトシレート(12;実施例2(a)に従って調製した)(2.5g、11.6mmol)を、DMF(5mL)中で混合し、100℃で終夜撹拌した。反応物を冷却し、次いで酢酸エチル(100mL)で希釈した。これを水(3×40mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10%のB、100g、12CV、60mL/分)で溶出して精製して、(2−フルオロ−エチル)−(4−フルオロ−フェニル)−アミン(38)383mg(20%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH3.30−3.35(1H,m,NCH2CH2F),3.40−3.45(1H,m,NCH2CH2F),3.90(1H,s,br,NH),4.53(1H,t,J=3Hz,NCH2CH2F),4.69(1H,t,J=3Hz,NCH2CH2F),6.51−6.72(2H,m,2 x NCCH),6.85−7.05(2H,m,2 x NCCHCH)によって確認した。
【0179】
実施例8(b):6−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤9)
3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35;実施例7(c)に従って調製した)(336mg、1.2mmol)及び(2−フルオロ−エチル)−(4−フルオロ−フェニル)−アミン(38)(383mg、2.4mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(2mL)に溶解し、乾燥塩化亜鉛(491mg、3.6mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(20mL)に溶解し、2NのHCl(10mL)、水(2×10mL)及び炭酸カリウム水溶液(2×5mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料をジエチルエーテルで倍散して、6−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤9)40mg(10%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.13(3H,t,J=9Hz,N(CH2CH3)2),1.30(3H,t,J=9Hz,N(CH2CH3)2),1.55−2.14(4H,m,2−及び3−CH2),2.78−2.86(2H,m,1−CH2),3.36−3.67(4H,m,N(CH2CH3)2),4.00−4.10(1H,m,4−CH),4.30(2H,dm,J=21Hz,NCH2CH2F),4.60(2H,dm,J=41Hz,NCH2CH2F),6.75−6.95(2H,m,NCCHCHCFCH)及び7.05−7.15(1H,m,NCCHCHCFCHによって確認した。
【0180】
実施例9:メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)及び5−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤10)の合成
実施例9(a):2−ベンジルオキシ−N−(3−フルオロ−フェニル)−アセトアミド(39)
ベンジルオキシ酢酸(4.65g、28mmol、4.0mL)のDCM(52mL)溶液に、塩化オキサリル(7.7g、61mmol、5.3mL)及び一滴のDMFを添加した。反応混合物を室温で4時間撹拌した。過剰の塩化オキサリルを真空中で除去し、粗塩化アシルをDCM(100mL)で希釈し、トリエチルアミン(5.3mL、41.6mmol、4.2g)を添加し、その後3−フルオロアニリン(3.5g、32mmol、3.0mL)を添加した。反応混合物を終夜RTで撹拌した。次いで、反応物を1MのHCl水溶液(100mL)でクエンチし、乾燥させ、真空中で濃縮して、2−ベンジルオキシ−N−(3−フルオロ−フェニル)−アセトアミド(39)7.10g(95%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、13C NMR(75MHz,CDCl3)δC69.2,73.5,106.9,107.2,111.0(d,JCF=24Hz),114.9(d,JCF=3Hz),127.8,128.2,128.5,129.7(d,JCF=9Hz),136.2及び167.6によって確認した。
【0181】
実施例9(b):(2−ベンジルオキシ−エチル)−(3−フルオロ−フェニル)−アミン(40)
LAH(1.25g、27mmol)の乾燥ジエチルエーテル(100mL)懸濁液に、2−ベンジルオキシ−N−(3−フルオロ−フェニル)−アセトアミド(39)(7.0g、27mmol)の乾燥ジエチルエーテル(100mL)溶液を滴下添加した。還流が維持されるように添加を実施した。添加が終了したら、反応混合物を4時間加熱還流し、次いで氷水に注ぎ、DCMを添加した。アルミニウム塩を分解するために、強塩基のpHが得られるまで2M水酸化ナトリウム水溶液を添加した。層を分離し、水層をDCMで洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(5〜50%のB、100g、12CV、60mL/分)で溶出して精製して、(2−ベンジルオキシ−エチル)−(3−フルオロ−フェニル)−アミン(40)4.1g(84%)を黄色油として得た。構造を、13C NMR(75MHz,CDCl3):δC43.3,68.2,73.0,99.4(d,JCF=24Hz),103.5,103.8,108.8,127.4(d,JCF=3Hz),127.6,128.4,130.0(d,JCF=9Hz)及び138.8によって確認した。
【0182】
実施例9(c):9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)
3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35;実施例7(c)に従って調製した)(2.3g、10mmol)及び(2−ベンジルオキシ−エチル)−(3−フルオロ−フェニル)−アミン(40)(4.1g、17mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(10mL)に溶解し、乾燥塩化亜鉛(4.09g、30mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(200mL)に溶解し、2NのHCl(50mL)、水(2×50mL)及び炭酸カリウム水溶液(2×50mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(5〜100%のB、100g、28CV、60mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)1.3g(30%)を、異性体9−(2−ベンジルオキシ−エチル)−7−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドと一緒に混合物として得、それを次の段階で精製せずに使用した。9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)の構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びCH2CH2OBn),4.00−4.30(3H,m,CH2CH2OBn及び4−CH),4.43(2H,s,OCH2Ph),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF),7.05−7.15(1H,m,NCCHCHCHCF)及び7.16−7.25(5H,m,Ph)によって確認した。
【0183】
9−(2−ベンジルオキシ−エチル)−7−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びNCH2CH2OBn),4.00−4.30(3H,m,NCH2CH2Obn及び4−CH),4.55(2H,s,OCH2Ph),6.70−6.80(1H,m,NCCHCFCHCH)及び7.00−7.40(7H,m,NCCHCFCHCH及びPh)によって確認した。
【0184】
実施例9(d):5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(42)
9−(2−ベンジルオキシ−エチル)−5−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(41)及び9−(2−ベンジルオキシ−エチル)−7−フルオロ−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(1.3g、3.0mmol)の混合物のメタノール(75mL)溶液に、メタノール(10mL)中Pa/C(200mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(42)及び7−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの混合物743mg(80%)を黄色油として得、それを次の段階で精製せずに使用した。5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(55)の構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びCH2CH2OH),4.00−4.30(3H,m,CH2CH2OH,4−CH),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF)及び7.05−7.15(1H,m,NCCHCHCHCF)によって確認した。
【0185】
7−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの構造を、1H NMR(300MHz,CDCl3)δH1.10−1.40(6H,m,N(CH2CH3)2),1.60−2.60(4H,m,2−及び3−CH2),2.70−2.85(2H,m,1−CH2),3.10−3.65(4H,m,N(CH2CH3)2及びCH2CH2OH),4.00−4.30(3H,m,NCH2CH2OH,4−CH),6.70−6.80(1H,m,NCCHCFCHCH)及び7.00−7.40(2H,m,NCCHCFCHCH)によって確認した。
【0186】
実施例9(e):メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)
5−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(42)及び7−フルオロ−9−(2−ヒドロキシ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(743mg、2.2mmol)の混合物のジクロロメタン(30mL)溶液に、ピリジン(1.74g、22.0mmol、1.8mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(1.01g、8.8mmol、0.7mL)を添加した。反応物を室温にして終夜温めた。混合物を2NのHCl(2×50mL)及び水(2×50mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、半分取HPLCによって水(A)及びメタノール(B)(Gemini 5u、C18、110A、150×21mm、20分間50〜95%のB、21mL/分)で溶出して精製して、メタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル10mg(1%)を白色固体として得、メタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル及びメタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)の混合物30mg(9%)を白色固体として得た。これらの精製条件を使用すると、メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)を、単一成分として単離することができなかった。メタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステルの構造を、1H NMR(300MHz,CDCl3)δH1.18(3H,t,J=7Hz,N(CH2CH3)2),1.39(3H,t,J=7Hz,N(CH2CH3)2) 1.70−2.30(4H,m,2−及び3−CH2),2.58(3H,s,OSO2CH3),2.60−2.80(2H,m,1−CH2),3.40−3.65(4H,m,N(CH2CH3)2),4.02(1H,t,J=6Hz,4−CH),4.20(2H,t,J=7Hz,NCH2CH2OMs),4.35(2H,t,J=7Hz,NCH2CH2OMs),6.70−6.85(1H,m,NCCHCFCHCH),6.90−7.00(1H,m,NCCHCFCHCH)及び7.05−7.15(2H,m,NCCHCFCHCH)によって確認した。
【0187】
メタンスルホン酸2−(4−ジエチルカルバモイル−5−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物10)の構造を、1H NMR(300MHz,CDCl3)δH1.18(3H,t,J=7Hz,N(CH2CH3)2),1.39(3H,t,J=7Hz,N(CH2CH3)2) 1.70−2.30(4H,m,2−及び3−CH2),2.58(3H,s,OSO2CH3),2.60−2.80(2H,m,1−CH2),3.40−3.65(4H,m,N(CH2CH3)2),4.15(1H,m,4−CH),4.20(2H,t,J=7Hz,NCH2CH2OMs),4.35(2H,t,J=7Hz,NCH2CH2OMs),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF)及び7.05−7.15(1H,m,NCCHCHCHCF)によって確認した。
【0188】
実施例9(f):5−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤10)
前駆体化合物10及びメタンスルホン酸2−(4−ジエチルカルバモイル−7−フルオロ−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステルの混合物を、反応物の放射性標識に使用した。18Fによる標識を、実施例1(f)に記載の通り実施した。7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの造影剤10を得た。
【0189】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長254nm;tR造影剤10、15分;tR7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド、14分。
【0190】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、50%のB;1〜20分、50〜95%のB;波長230nm;tR造影剤10、16分;tR7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド、14分。造影剤10の放射化学的収率8.7±1%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図5は、造影剤10(上)及び7−フルオロ−9−(2−[18F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(中間)及び7−フルオロ−9−(2−[19F]フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(下)を示す。
【0191】
実施例10:5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸−ジエチルアミド(非放射性造影剤10)の合成
実施例10(a):(2−フルオロ−エチル)−(3−フルオロ−フェニル)−アミン(43)
3−フルオロアニリン(1.4g、11.6mmol、1.2mL)及び2−フルオロエチルトシレート(12;実施例2(a)に従って調製した)(2.5g、11.6mmol)及びルチジン(1.24g、11.6mmol)を撹拌し、DMF(5mL)中、100℃で終夜加熱した。反応物を冷却し、次いで酢酸エチル(100mL)で希釈した。これを水(3×40mL)で洗浄し、有機物を乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10%のB、100g、12CV、60mL/分)で溶出して精製して、(2−フルオロ−エチル)−(3−フルオロ−フェニル)−アミン(43)184mg(10%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH3.37(1H,q,J=6Hz,NCH2CH2F),3.46(1H,q,J=6Hz,NCH2CH2F),4.12(1H,s,br,NH),4.54(1H,t,J=3Hz,NCH2CH2F),4.69(1H,t,J=3Hz,NCH2CH2F),6.3 1−6.50(3H,m,NCCHCHCH),7.10−7.25(1H,m,NCCHCF)によって確認した。
【0192】
実施例10(b):5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤10)
3−ブロモ−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(35;実施例7(c)に従って調製した)(161mg、0.6mmol)及び(2−フルオロ−エチル)−(3−フルオロ−フェニル)−アミン(43)(184mg、1.2mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(1mL)に溶解し、乾燥塩化亜鉛(245mg、1.8mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(10mL)に溶解し、2NのHCl(5mL)、水(2×5mL)及び炭酸カリウム水溶液(2×5mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗材料を、半分取HPLCによって水(A)及びメタノール(B)(Gemini 5u、C18、110A、150×21mm、20分間50〜95%のB、21mL/分)で溶出して精製して、7−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド20mg(6%)を白色固体として得、5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤10)10mg(3%)を白色固体として得た。7−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミドの構造を、1H NMR(300MHz,CDCl3)δH1.14(3H,t,J=7Hz,N(CH2CH3)2),1.33(3H,t,J=7Hz,N(CH2CH3)2),1.80−2.15(4H,m,2−及び3−CH2),2.70−2.80(2H,m,1−CH2),3.50−3.80(4H,m,N(CH2CH3)2),4.20−4.35(1H,m,4−CH),4.40(2H,dm,J=21Hz,NCH2CH2F),4.60(2H,dm,J=41Hz,NCH2CH2F),6.70−6.80(1H,m,NCCHCFCHCH)及び7.00−7.10(2H,m,NCCHCFCHCH)によって確認した。
【0193】
5−フルオロ−9−(2−フルオロ−エチル)−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤10)の構造を、1H NMR(300MHz,CDCl3)δH1.14(3H,t,J=7Hz,N(CH2CH3)2),1.33(3H,t,J=7Hz,N(CH2CH3)2),1.80−2.15(4H,m,2−及び3−CH2),2.70−2.80(2H,m,1−CH2),3.50−3.80(4H,m,N(CH2CH3)2),4.20−4.35(1H,m,4−CH),4.40(2H,dm,J=21Hz,NCH2CH2F),4.60(2H,dm,J=41Hz,NCH2CH2F),6.55−6.65(1H,m,NCCHCHCHCF),6.90−7.05(1H,m,NCCHCHCHCF)及び7.05−7.15(1H,m,NCCHCHCHCF)によって確認した。
【0194】
実施例11:メタンスルホン酸2−(4−ジエチルカルバモイル−2−メチル−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物11)及び9−(2−[18F]フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤11)
実施例11(a):4−(4−メチル−シクロヘキセ−1−エニル)−モルホリン(44)
ディーンスタークを備えたフラスコ中、4−メチルシクロヘキサノン(20.1g、179.3mmol、22mL)及びモルホリン(31.3g、359.0mmol、31.4mL)の溶液を、ベンゼン(55mL)中で26時間還流した。ベンゼンを真空下で除去し、粗生成物を減圧下で蒸留によって精製して、4−(4−メチル−シクロヘキセ−1−エニル)−モルホリン(44)23g(70%)を油として得た(10mmHgにおいてb.p.120℃)。構造を、1H NMR(300MHz,CDCl3):δH0.94(3H,d,J=6.0Hz,CH3),1.15−1.35(1H,m,CH2CH=CN),1.50−1.80(3H,m,CH2CH2CHCH3),2.00−2.25(4H,m,CH2CH=CN及びCH2CH2CHCH3),2.65−2.95(4H,m,OCH2NCH2),3.73(4H,t,J=6.0Hz,OCH2NCH2)及び4.60−4.65(1H,m,CH2CH=CN)によって確認した。
【0195】
実施例11(b):5−メチル−2−オキソ−シクロヘキサンカルボン酸エチルエステル(45)
4−(4−メチル−シクロヘキセ−1−エニル)−モルホリン(44)(23g、127.0mmol)のベンゼン(55mL)溶液に、クロロギ酸エチル(7.5g、69.0mmol、6.6mL)を窒素下で添加すると同時に、エナミン溶液を急速に撹拌した。18時間還流した後、溶液を冷却し、濾過した。エナミン塩酸塩の沈殿物を乾燥エーテルで洗浄した。濾液及び洗浄物を反応フラスコに戻し、10%HCl水溶液(40mL)を添加した。混合物を、15〜30分間激しく撹拌した。層を分離し、水層を酢酸エチル(2×100mL)で抽出し、混合有機層を真空中で濃縮した。粗材料を、減圧下で蒸留によって精製して、5−メチル−2−オキソ−シクロヘキサンカルボン酸エチルエステル(45)12.5g(53%)を油として得た(10mmHgにおいてb.p.85℃〜90℃)。構造を、1H NMR(300MHz,CDCl3):δH0.85−0.95(3H,m,CH3),1.17(3H,t,J=7Hz,OCH2CH3),1.25−2.00(5H,m,5−CH,4−及び6−CH2),2.15−2.40(3H,m,1−CH及び3−CH2)及び4.00−4.20(2H,m,OCH2CH3)によって確認した。
【0196】
実施例11(c):5−メチル−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(46)
トルエン(90mL)中5−メチル−2−オキソ−シクロヘキサンカルボン酸エチルエステル(45)(5.9g、32mmol)、DMAP(1.12g、10mmol)及びジエチルアミン(4.7g、65mmol、6.7mL)を、4日間加熱還流した。反応物を冷却し、トルエンを減圧下で除去して、黄色油を得た。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(20〜50%のB、80g)で溶出して精製して、5−メチル−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(46)4.4g(65%)を黄色油として得た。構造を、1H NMR(300MHz,CDCl3)δH0.8−1.05(9H,m,CH3及びN(CH2CH3)2),1.05−2.10(5H,m,5−CH及び4−及び6−CH2),2.15−2.80(2H,m,3−CH2),2.95−3.55(5H,m,1−CH及びN(CH2CH3)2)によって確認した。
【0197】
実施例11(d):3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47)
5−メチル−2−オキソ−シクロヘキサンカルボン酸ジエチルアミド(46)(4.4g、21mmol)をジエチルエーテル(5mL)に溶解し、N2下で0℃に冷却した。臭素(3.32g、21mmol、1.1mL)を15分かけて滴下添加し、反応混合物を室温にして90分かけて温めた。混合物を、氷冷した飽和炭酸ナトリウム水溶液(40mL)にゆっくり注ぎ、酢酸エチル(3×40mL)で抽出した。混合有機層を乾燥させ、真空中で濃縮して、3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47)6.1g(定量的)をオフホワイト色の固体として得た。構造を、1H NMR(300MHz,CDCl3)δH0.8−1.20(9H,m,CH3及びN(CH2CH3)2),1.80−2.40(5H,m,CH2CH(CH3)CH2),3.15−3.55(4H,m,N(CH2CH3)2),4.65−4.74(1H,m,CHBr)及び12.04(1H,s,OH)によって確認した。
【0198】
実施例11(e):9−(2−ベンジルオキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(48)
3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47)(4.0g、14mmol)及び(2−ベンジルオキシ−エチル)−フェニル−アミン(21;実施例3(c)に従って調製した)(6.3g、28mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(14mL)に溶解し、乾燥塩化亜鉛(5.72g、42mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(200mL)に溶解し、2NのHCl(50mL)、水(2×50mL)及び炭酸カリウム水溶液(2×50mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗混合物を、SCXカートリッジ(40mL)及び次いでシリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(10〜50%のB、100g、12CV、85mL/分)で溶出して精製して、9−(2−ベンジルオキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(48)467mg(8%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.20−1.40(9H,m,CH3及びN(CH2CH3)2),1.90−2.20(3H,m,2−CH及び3−CH2),2.35−2.45(1H,m,1−CH2),2.85−2.95(1H,m,1−CH2),3.40−3.70(4H,m,N(CH2CH3)2),3.70−3.80(1H,m,4−CH),4.10−4.30(4H,m,NCH2CH2OBn),4.43(2H,s,OCH2Ph)及び7.00−7.30(9H,m,CHCHCHCH及びPh)によって確認した。
【0199】
実施例11(f):9−(2−ヒドロキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(49)
9−(2−ベンジルオキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(48)(460mg、1.1mmol)のメタノール(25mL)溶液に、メタノール(5mL)中Pd/C(100mg)のスラリーを添加した。混合物をParr水素化装置に入れ、水素雰囲気下で24時間振とうした。反応物を、セライトパッドを介して濾過し、メタノールで洗浄し、真空中で濃縮して、9−(2−ヒドロキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(49)250mg(79%)を黄色油として得、それを次の段階で精製せずに使用した。構造を、1H NMR(300MHz,CDCl3)δH1.20−1.40(9H,m,CH3及びN(CH2CH3)2),1.90−2.20(3H,m,2−CH及び3−CH2),2.35−2.45(1H,m,1−CH2),2.85−2.95(1H,m,1−CH2),3.40−3.70(4H,m,N(CH2CH3)2),3.70−3.80(1H,m,4−CH),4.10−4.30(4H,m,NCH2CH2OH),6.91(1H,t,J=7Hz,NCCHCHCHCH),7.00(1H,t,J=7Hz,NCCHCHCHCH),7.12(1H,d,J=7Hz,NCCHCHCHCH)及び7.15(1H,d,J=7Hz,NCCHCHCHCH)によって確認した。
【0200】
実施例11(g):メタンスルホン酸2−(4−ジエチルカルバモイル−2−メチル−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物11)
9−(2−ヒドロキシ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(49)(250mg、0.8mmol)のジクロロメタン(10mL)溶液に、ピリジン(633mg、8.0mmol、0.6mL)を添加した。反応物を0℃に冷却し、塩化メタンスルホニル(367mg、3.2mmol、0.2mL)を添加した。反応物を室温にして終夜温めた。混合物を、2NのHCl(2×20mL)及び水(2×20mL)で洗浄し、乾燥させ、真空中で濃縮した。粗材料を、シリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜100%のB、10g、34CV、30mL/分)で溶出して精製し、次いでジエチルエーテルで倍散して、メタンスルホン酸2−(4−ジエチルカルバモイル−2−メチル−1,2,3,4−テトラヒドロ−カルバゾール−9−イル)−エチルエステル(前駆体化合物11)250mg(80%)を白色固体として得た。構造を、13C NMR(75MHz,CDCl3)δC12.9,13.0,15.2,22.0,29.7,30.2,36.7,36.8,40.8,41.6,42.0,67.8,108.6,109.5,118.6,119.6,121.2,126.4,136.2,136.4,173.7によって確認した。
【0201】
実施例11(h):9−(2−[18F]フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(造影剤11)
18Fによる前駆体化合物11の標識を、実施例1(f)に記載の通り実施した。
【0202】
半分取HPLC:HICHROM ACE5 C18カラム(100×10mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:3ml/分;0〜26分、50%のB;波長254nm;tR造影剤11、15分。
【0203】
分析的HPLC:Phenomenex Luna C18カラム(150×4.6mm i.d.)、粒径5μm;移動相A:水、移動相B:メタノール;流速勾配:1ml/分;0〜1分、40%のB;1〜20分、40〜95%のB;波長230nm;tR造影剤11、17分。放射化学的収率14±13%(n=3)減衰補正なし、時間90〜120分、放射化学的純度≧99%。図6は、造影剤11と非放射性造影剤11の共溶出を示す。
【0204】
実施例12:9−(2−フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤11)の合成
3−ブロモ−2−ヒドロキシ−5−メチル−シクロヘキセ−1−エンカルボン酸ジエチルアミド(47;実施例11(d)に従って調製した)(2.0g、7mmol)及び(2−フルオロ−エチル)−フェニル−アミン(24;実施例4(a)に従って調製した)(1.9g、14mmol)の混合物を、N2下において50℃で3時間撹拌すると、反応物が褐色になった。得られた混合物をプロパン−2−オール(7mL)に溶解し、乾燥塩化亜鉛(2.86g、21mmol)を添加した。混合物を、N2下で16時間加熱還流し、次いで真空中で濃縮した。残渣を酢酸エチル(100mL)に溶解し、2NのHCl(30mL)、水(2×30mL)及び炭酸カリウム水溶液(2×30mL)で洗浄し、次いで乾燥させ、真空中で濃縮した。粗混合物を、SCXカートリッジ(40mL)及び次いでシリカゲルクロマトグラフィーによって石油(A)及び酢酸エチル(B)(0〜100%のB、100g、12CV、85mL/分)で溶出して精製して、9−(2−フルオロ−エチル)−2−メチル−2,3,4,9−テトラヒドロ−1H−カルバゾール−4−カルボン酸ジエチルアミド(非放射性造影剤11)400mg(17%)を白色固体として得た。構造を、1H NMR(300MHz,CDCl3)δH1.10−1.35(9H,m,CH3及びN(CH2CH3)2),1.95−2.10(2H,m,3−CH2),2.30−2.50(1H,m,2−CH),2.70−2.80(2H,m,1−CH2),3.40−3.70(4H,m,N(CH2CH3)2),4.05−4.15(1H,m,4−CH),4.30(2H,dm,J=21Hz,NCH2CH2F),4.65(2H,dm,J=41Hz,NCH2CH2F)及び7.00−7.30(4H,m,NCCHCHCHCHによって確認した。
【0205】
実施例13:前駆体化合物5の鏡像異性体分離
【0206】
【化15】
前駆体化合物5(実施例1に記載の通り得た)を、超臨界流体(CO2)キラルクロマトグラフィーを使用して、Kromasil Amycoat、250×10mm、5μm、100Åカラムにて40℃において30%IPAを用いて、毎分13mlの実施時間6分でその鏡像異性体に分離した。前駆体化合物5(60mg)を1,4−ジオキサン(2ml)に溶解し、実施ごとに注入する場合、1度に200μlまでとした。2つの鏡像異性体間のベースライン分離を達成した。Chiral Technologies製のIC、250×4.6mm、5μm、均一溶媒による実施(run isocratic)、80:20−MeOH:IPA、0.5ml/分及び室温で、分離した2つの鏡像異性体の鏡像異性体純度を分析的HPLCによって測定すると、鏡像異性体のそれぞれについて99.5%の鏡像異性体純度を示した。
【0207】
実施例14:非放射性造影剤5の鏡像異性体分離
【0208】
【化16】
非放射性造影剤5(実施例2に記載の通り得た)を、超臨界流体(CO2)キラルクロマトグラフィーを使用して、Kromasil Amycoat、250×10mm、5μm、100Åカラムにて40℃において20%IPAを用いて、毎分14mlの実施時間6分でその鏡像異性体に分離した。化合物5(100mg)を1,4−ジオキサン(2.5ml)に溶解し、実施ごとに注入する場合、1度に200μlまでとした。時間ごとに画分をカットして、混合画分が収集されないようにした。Chiral Technologies製のIC、250×4.6mm、5μm、均一溶媒による実施、80:20−MeOH:IPA、0.5ml/分及び室温で、分離した2つの鏡像異性体の鏡像異性体純度を分析的HPLCによって測定すると、鏡像異性体のそれぞれについて99.5%の鏡像異性体純度を示した。
【0209】
実施例15:インビトロ効力アッセイ
PBRに対する親和性を、Le Fur et al,(Life Sci. 1983;USA 33:449−57)に記載された方法を使用してスクリーニングした。本発明のインビボ造影剤の非放射性類似体を、過去の四環系インドール造影剤の非放射性類似体(同時係属の特許出願PCT/EP2009/062827のもの;以下の実施例17に記載の合成)と一緒に試験した。
【0210】
各試験化合物(50mMトリス−HCl、pH7.4、1%DMSOを含有する10mMのMgCl2に溶解した)は、Wistarラット心臓のPBRとの結合について、0.3nMの[3H]PK−11195と競合した。反応を、50mMトリス−HCl、pH7.4、10mMのMgCl2中で25℃において15分間実施した。各試験化合物を、6つの異なる濃度で、推定Ki値周辺の300倍の範囲濃度にわたってスクリーニングした。以下のデータが観測された。
【0211】
【表1】
実施例16:インビボ体内分布法
本発明の造影剤を、過去の四環系造影剤(同時係属の特許出願PCT/EP2009/062827のもの;以下の実施例18に記載の合成)と一緒に、インビボ体内分布モデルで試験した。
【0212】
成体雄性Wistarラット(200〜300g)に、側尾静脈を介して試験化合物1〜3MBqを注入した。注入の2、10、30又は60分後(n=3)、ラットを安楽死させ、組織又は体液を、γ計数器で放射性を測定するために試料採取した。
【0213】
以下の記録データが観測された。
【0214】
【表2】
図7〜13は、それぞれ四環系造影剤並びに造影剤5〜7及び9〜11の脳内の体内分布プロファイルを示す。本発明のインビボ造影剤は、四環系造影剤と比較して、脳に良好に取り込まれ、PBR発現組織における特異的取込みが改善していることがわかる。
【0215】
実施例17:(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(非放射性四環系インドール造影剤)の調製
17(a):(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(50)
乾燥DCM(100ml)中、T. Okubo et al,(Bioorg. Med. Chem. 2004;12:3569−3580)に記載の通り調製した(±)−4−オキソ−チオクロマン−2−カルボン酸(10.4g、50mmol)を、塩化オキサリル(12.6g、100mmol)及び一滴のDMFと共に窒素雰囲気下において室温で18時間撹拌した。次いで、反応物を真空中で蒸発させてガムを得、次いでDCM(100ml)に再溶解し、氷浴で0℃に冷却し、撹拌し、DCM(20ml)中ジエチルアミン(8.03g、110mmol)を用いて1時間かけて滴下処理した。反応物を室温にして1時間かけて温め、10%炭酸カリウム水溶液(100ml)を添加し、反応混合物を激しく撹拌した。DCM溶液を分離した。DCMの2つのさらなるバッチ(100ml)で水溶液を抽出し、混合抽出物を硫酸マグネシウムで乾燥させた。DCM溶液を真空中で濃縮して暗緑色油を得、それを静置して結晶化させた。結晶固体をジエチルエーテル(50ml)で倍散し、濾過して、標記化合物(50)(8.57g、65%)を薄緑色固体として得た。構造を、1H NMR(300MHz,CDCl3)δ 1.06(t,J=7.1Hz,3H),1.23(t,J=7.1Hz,3H),3.0−3.5(m,6H),4.25(m,1H),7.15−7.21(m,2H),7.32−7.39(m,1H),8.10−8.14(m,1H)によって確認した。
【0216】
17(b):(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(51)
(±)−4−オキソ−チオクロマン−2−カルボン酸ジエチルアミド(50)(1.32g、5.0mmol)及び4−メトキシフェニルヒドラジン塩酸塩(0.87g、5.0mmol)のエタノール(10ml)溶液に、濃硫酸(0.73ml、1.35g、13.8mmol)を窒素下で添加した。反応混合物を還流下で24時間加熱した。冷却後、反応混合物を濾過し、固体をエタノールで洗浄し、真空中で乾燥させて(45℃)、標記化合物(51)(1.05g、57%)を薄黄色固体として得た。構造を、13C NMR(75MHz,DMSO−d6)δ 10.5,12.7,32.7,37.9,39.5,53.0,97.6,103.3,109.87,109.92,120.3,123.5,123.8,124.3,124.7,124.9,127.8,129.4,131.8,151.3,166.2によって確認した。
【0217】
17(c):(±)−11−(2−フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(過去の四環系インドール造影剤の非放射性類似体)
(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(51)(150mg、0.41mmol;実施例17(b)に従って調製した)の無水DMF(4ml)溶液に、L.Croninら(J.Org.Chem.2004年;69:5934〜5946頁)に記載の通り調製した2−フルオロエチルトシレート(166mg、0.82mmol)を添加し、その後鉱油(34mg、0.82mmol)中水素化ナトリウム60%分散液を窒素下で添加した。反応混合物を80℃で1時間加熱した。冷却後、溶媒を真空中で除去し、残渣を水(30ml)でクエンチし、DCM(2×30ml)で抽出し、乾燥させ(MgSO4)、溶媒を真空中で除去した。残渣を、シリカカラムクロマトグラフィーによって5〜10%のEtOAc/CH2Cl2で溶出して精製した。粗固体を、エーテル/ミネラルスピリットでクエンチし、濾過し、真空中で乾燥させて(45℃)、標記化合物(非放射性四環系インドール造影剤)(77mg、46%)を薄褐色固体として得た。構造を、1H NMR(300MHz,CDCl3)δ 1.12(t,J=7.0Hz,3H),1.36(t,J=7.0Hz,3H),3.25−3.70(m,4H),3.83(s,3H),4.45−4.70(m,2H),4.80(t,J=5.2Hz,1H),4.96(t,J=5.2Hz,1H),5.09(s,1H),6.84−6.93(m,2H),7.13−7.32(m,3H),7.46(m,1H),7.58(d,J=8.0Hz,1H)によって確認した。
【0218】
実施例18:(±)−11−(2−[18F]フルオロエチル)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(四環系インドール造影剤)の合成
18F-/水を、反応容器中、K222(4mg)、K2CO3水溶液(0.1モル溶液50μl)及びアセトニトリル(500μl)に添加し、窒素流の下で100℃において20〜30分間乾燥させた。アセトニトリル(1000ul)中エチル−1,2−ジトシレート(4mg)を添加し、100℃で10分間加熱した。反応混合物を冷却し、半分取(preperative)HPLCによって精製し、18F−フルオロエチルトシレートを含有する画分を収集した。この画分を、H2Oで体積約20mlに希釈し、コンディショニングしたlight t−C18 sep pakに搭載し、H2O(1×2ml)でフラッシュした。sep pakを大流量のN2ラインで20分間乾燥させた。次いで、18FフルオロエチルトシレートをDMF(500μl)で溶出した。
【0219】
DMF(250ul)中前駆体化合物(±)−8−メトキシ−6,11−ジヒドロ−5−チア−11−アザ−ベンゾ[a]フルオレン−6−カルボン酸ジエチルアミド(51;実施例17(b)に従って調製した)(13mg)を、第2の反応容器に添加し、N2で5分間パージした。次いで、DMF(2×250ul)中NaH(1.3mg)を窒素下で添加し、反応容器を45℃で0.5〜1時間加熱した。次いでこれに、上記で調製したDMF中18Fフルオロエチルトシレートを添加し、N2でパージした反応容器中、100℃で10分間加熱した。反応物を冷却し、水(1ml)で反応容器から洗浄した。シリンジフィルターによって溶液を濾過し、分取HPLCで精製した。主な放射性ピークを含有する画分を収集した。これを、H2Oで体積約10mlに希釈し、コンディショニングしたlight t−C18 sep pakに搭載し、H2O(1×2ml)でフラッシュし、EtOH(0.5ml)でP6バイアルに溶出し、リン酸緩衝食塩水(5ml)を添加した。
【特許請求の範囲】
【請求項1】
次の式Iのインビボ造影剤。
【化1】
式中、
R1はC1〜3アルキル又はC1〜3フルオロアルキルであり、
R2は水素、ヒドロキシル、ハロ、シアノ、C1〜3アルキル、C1〜3アルコキシ、C1〜3フルオロアルキル又はC1〜3フルオロアルコキシであり、
R3及びR4は独立にC1〜3アルキル、C7〜10アラルキルであるか、或いはR3及びR4はそれらと結合した窒素と共に含窒素C4〜6脂肪族環を形成するもので、適宜、窒素、酸素及び硫黄から選択される追加の1個のヘテロ原子を含んでいてもよく、
Y1はO、S、SO、SO2又はCH2であり、
Y2はCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
上記の式Iは、インビボイメージングに適した放射性同位体である原子を含む。
【請求項2】
前記インビボイメージングに適した放射性同位体が、11C、18F及び123Iから選択される、請求項1記載のインビボ造影剤。
【請求項3】
R1がメチル又はC2〜3フルオロアルキルである、請求項1又は請求項2記載のインビボ造影剤。
【請求項4】
R2が水素、ハロ、C1〜3アルコキシ又はC1〜3フルオロアルコキシである、請求項1乃至請求項3記載のインビボ造影剤。
【請求項5】
R3及びR4が独立にメチル、エチル又はベンジルである、請求項1乃至請求項4のいずれか1項記載のインビボ造影剤。
【請求項6】
R3及びR4がそれらに結合した窒素と共に含窒素C5〜6脂肪族環を形成する、請求項1乃至請求項4のいずれか1項記載のインビボ造影剤。
【請求項7】
Y1がCH2である、請求項1乃至請求項6のいずれか1項記載のインビボ造影剤。
【請求項8】
Y2がCH2−CH2である、請求項1乃至請求項6のいずれか1項記載のインビボ造影剤。
【請求項9】
次の式Iaのものである、請求項1記載のインビボ造影剤。
【化2】
式中、
R2aは水素、ハロ又はC1〜3アルコキシであり、
R3a及びR4aは独立にメチル、エチル又はベンジルであるか、或いはそれらに結合した窒素と共にピロリジニル、ピペリジニル、アゼパニル若しくはモルホリニル環を形成するものであり、
Y2aはCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
nは1、2又は3である。
【請求項10】
R3a及びR4aが共にエチルであるか、或いはR3aがメチルであってR4aがベンジルであるか、或いはそれらに結合した窒素と共にアゼパニル環を形成するものであり、
R2aが水素、メトキシ又はフルオロであり、
Y2aがCH2−CH2又はCH(CH3)−CH2であり、
nが2である、請求項9記載のインビボ造影剤。
【請求項11】
以下の化学構造を有する、請求項10記載のインビボ造影剤。
【化3】
【請求項12】
請求項1乃至請求項11のいずれか1項記載のインビボ造影剤の調製のための前駆体化合物であって、次の式IIの前駆体化合物。
【化4】
式中、R11及びR12の一方は、請求項1又は請求項2記載の放射性同位体の適切な供給源と反応して、当該前駆体化合物と放射性同位体の適切な供給源との反応によって前記インビボ造影剤を形成する化学基であり、R11及びR12の他方は、それぞれR1及びR2について請求項1記載の通りであって、適宜保護基を含んでおり、
R13〜14及びY11〜12は、請求項1乃至請求項8のいずれか1項において、それぞれR3〜4及びY1〜2について記載の通りであり、適宜保護基を含んでいる。
【請求項13】
R11がC1〜3アルキレン−LGであり、LGが、メシレート、トシレート及びトリフラートから選択される脱離基である、請求項12記載の前駆体化合物。
【請求項14】
次の式IIaの化合物である、請求項13記載の前駆体化合物。
【化5】
式中、
LGは、請求項13記載の通りであり、
R12a〜14a、Y12a及びmは、それぞれR2a〜4a、Y2a及びnについて請求項9及び請求項10記載の通りである。
【請求項15】
R12がトリメチルスズである、請求項12記載の前駆体化合物。
【請求項16】
次の式IIbの化合物である、請求項12記載の前駆体化合物。
【化6】
式中、
R11bは、請求項1又は請求項2記載の放射性同位体の適切な供給源と反応する化学基であり、
R12b〜14bは、R12〜14について請求項12記載の通りであり、但しR12bはクロロではなく、
Y11b〜12bは、Y11〜12について請求項12記載の通りである。
【請求項17】
請求項16記載の式IIbの前駆体化合物の調製方法であって、下記の式IIcの化合物とZnCl2との反応によって、下記の式IIdの化合物を形成する段階を含んでいて、上記反応がジエチルエーテルを含む溶媒系中で実施される、方法。
【化7】
(式中、R12c、Y11c及びY12cは、それぞれR12、Y11及びY12について請求項12記載の通りであり、PGcは保護基である。)
【化8】
(式中、R12d、Y11d、Y12d及びPGdは、それぞれR12c、Y11c、Y12c及びPGcについて記載の通りである。)
【請求項18】
(i)請求項12乃至請求項16のいずれか1項記載の前駆体化合物を提供する段階と、
(ii)請求項1又は請求項2記載の前記放射性同位体の適切な供給源を提供する段階と、
(iii)前記段階(i)の前駆体化合物を、前記段階(ii)の放射性同位体と反応させて、前記インビボ造影剤を得る段階と
を含む、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤を調製する方法。
【請求項19】
自動化された、請求項18記載の方法。
【請求項20】
請求項12乃至請求項16のいずれか1項記載の前駆体を含む、請求項18記載の方法を実施するためのキット。
【請求項21】
(i)請求項12乃至請求項16のいずれか1項記載の前駆体化合物を含有する容器と、
(ii)前記段階(i)の容器を、請求項1又は請求項2記載のインビボイメージングに適した放射性同位体の適切な供給源を用いて溶出するための手段と
を含む、請求項19記載の方法を実施するためのカセット。
【請求項22】
(iii)過剰の放射性同位体を除去するためのイオン交換カートリッジと、任意選択により
(iv)前記前駆体化合物が1つ以上の保護基を含む場合、得られた放射性標識生成物を脱保護して本明細書記載のインビボ造影剤を形成するためのカートリッジと
をさらに含む、請求項21記載のカセット。
【請求項23】
哺乳動物への投与に適した形態の生体適合性の担体と一緒に、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤を含む、放射性医薬組成物。
【請求項24】
対象におけるPBR発現の分布及び/又は程度を決定するためのインビボイメージング法であって、
(i)前記対象に、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤を投与する段階と、
(ii)前記インビボ造影剤を前記対象のPBRと結合させる段階と、
(iii)インビボイメージング手順によって、前記インビボ造影剤の放射性同位体から放出されたシグナルを検出する段階と、
(iv)前記シグナルの位置及び/又は量の代表的な画像を作成する段階と、
(v)前記インビボ造影剤によって放出された前記シグナルと直接相関する、前記対象におけるPBR発現の分布及び程度を決定する段階と
を含む方法。
【請求項25】
前記対象のための治療投与計画の過程中、反復して実施される請求項24記載のインビボイメージング法であって、前記投与計画がPBR状態に対抗するための薬物を投与する段階を含む方法。
【請求項26】
請求項24記載のインビボイメージング法を、PBR発現の分布及び程度が特定の臨床像に帰するさらなる段階(vi)と一緒に含む、PBRが上方制御されている状態を診断する方法。
【請求項27】
請求項26記載の診断方法において使用するための、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤。
【請求項28】
請求項26記載の診断方法において使用する請求項21記載の放射性医薬組成物の製造に使用するための、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤。
【請求項1】
次の式Iのインビボ造影剤。
【化1】
式中、
R1はC1〜3アルキル又はC1〜3フルオロアルキルであり、
R2は水素、ヒドロキシル、ハロ、シアノ、C1〜3アルキル、C1〜3アルコキシ、C1〜3フルオロアルキル又はC1〜3フルオロアルコキシであり、
R3及びR4は独立にC1〜3アルキル、C7〜10アラルキルであるか、或いはR3及びR4はそれらと結合した窒素と共に含窒素C4〜6脂肪族環を形成するもので、適宜、窒素、酸素及び硫黄から選択される追加の1個のヘテロ原子を含んでいてもよく、
Y1はO、S、SO、SO2又はCH2であり、
Y2はCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
上記の式Iは、インビボイメージングに適した放射性同位体である原子を含む。
【請求項2】
前記インビボイメージングに適した放射性同位体が、11C、18F及び123Iから選択される、請求項1記載のインビボ造影剤。
【請求項3】
R1がメチル又はC2〜3フルオロアルキルである、請求項1又は請求項2記載のインビボ造影剤。
【請求項4】
R2が水素、ハロ、C1〜3アルコキシ又はC1〜3フルオロアルコキシである、請求項1乃至請求項3記載のインビボ造影剤。
【請求項5】
R3及びR4が独立にメチル、エチル又はベンジルである、請求項1乃至請求項4のいずれか1項記載のインビボ造影剤。
【請求項6】
R3及びR4がそれらに結合した窒素と共に含窒素C5〜6脂肪族環を形成する、請求項1乃至請求項4のいずれか1項記載のインビボ造影剤。
【請求項7】
Y1がCH2である、請求項1乃至請求項6のいずれか1項記載のインビボ造影剤。
【請求項8】
Y2がCH2−CH2である、請求項1乃至請求項6のいずれか1項記載のインビボ造影剤。
【請求項9】
次の式Iaのものである、請求項1記載のインビボ造影剤。
【化2】
式中、
R2aは水素、ハロ又はC1〜3アルコキシであり、
R3a及びR4aは独立にメチル、エチル又はベンジルであるか、或いはそれらに結合した窒素と共にピロリジニル、ピペリジニル、アゼパニル若しくはモルホリニル環を形成するものであり、
Y2aはCH2、CH2−CH2、CH(CH3)−CH2又はCH2−CH2−CH2であり、
nは1、2又は3である。
【請求項10】
R3a及びR4aが共にエチルであるか、或いはR3aがメチルであってR4aがベンジルであるか、或いはそれらに結合した窒素と共にアゼパニル環を形成するものであり、
R2aが水素、メトキシ又はフルオロであり、
Y2aがCH2−CH2又はCH(CH3)−CH2であり、
nが2である、請求項9記載のインビボ造影剤。
【請求項11】
以下の化学構造を有する、請求項10記載のインビボ造影剤。
【化3】
【請求項12】
請求項1乃至請求項11のいずれか1項記載のインビボ造影剤の調製のための前駆体化合物であって、次の式IIの前駆体化合物。
【化4】
式中、R11及びR12の一方は、請求項1又は請求項2記載の放射性同位体の適切な供給源と反応して、当該前駆体化合物と放射性同位体の適切な供給源との反応によって前記インビボ造影剤を形成する化学基であり、R11及びR12の他方は、それぞれR1及びR2について請求項1記載の通りであって、適宜保護基を含んでおり、
R13〜14及びY11〜12は、請求項1乃至請求項8のいずれか1項において、それぞれR3〜4及びY1〜2について記載の通りであり、適宜保護基を含んでいる。
【請求項13】
R11がC1〜3アルキレン−LGであり、LGが、メシレート、トシレート及びトリフラートから選択される脱離基である、請求項12記載の前駆体化合物。
【請求項14】
次の式IIaの化合物である、請求項13記載の前駆体化合物。
【化5】
式中、
LGは、請求項13記載の通りであり、
R12a〜14a、Y12a及びmは、それぞれR2a〜4a、Y2a及びnについて請求項9及び請求項10記載の通りである。
【請求項15】
R12がトリメチルスズである、請求項12記載の前駆体化合物。
【請求項16】
次の式IIbの化合物である、請求項12記載の前駆体化合物。
【化6】
式中、
R11bは、請求項1又は請求項2記載の放射性同位体の適切な供給源と反応する化学基であり、
R12b〜14bは、R12〜14について請求項12記載の通りであり、但しR12bはクロロではなく、
Y11b〜12bは、Y11〜12について請求項12記載の通りである。
【請求項17】
請求項16記載の式IIbの前駆体化合物の調製方法であって、下記の式IIcの化合物とZnCl2との反応によって、下記の式IIdの化合物を形成する段階を含んでいて、上記反応がジエチルエーテルを含む溶媒系中で実施される、方法。
【化7】
(式中、R12c、Y11c及びY12cは、それぞれR12、Y11及びY12について請求項12記載の通りであり、PGcは保護基である。)
【化8】
(式中、R12d、Y11d、Y12d及びPGdは、それぞれR12c、Y11c、Y12c及びPGcについて記載の通りである。)
【請求項18】
(i)請求項12乃至請求項16のいずれか1項記載の前駆体化合物を提供する段階と、
(ii)請求項1又は請求項2記載の前記放射性同位体の適切な供給源を提供する段階と、
(iii)前記段階(i)の前駆体化合物を、前記段階(ii)の放射性同位体と反応させて、前記インビボ造影剤を得る段階と
を含む、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤を調製する方法。
【請求項19】
自動化された、請求項18記載の方法。
【請求項20】
請求項12乃至請求項16のいずれか1項記載の前駆体を含む、請求項18記載の方法を実施するためのキット。
【請求項21】
(i)請求項12乃至請求項16のいずれか1項記載の前駆体化合物を含有する容器と、
(ii)前記段階(i)の容器を、請求項1又は請求項2記載のインビボイメージングに適した放射性同位体の適切な供給源を用いて溶出するための手段と
を含む、請求項19記載の方法を実施するためのカセット。
【請求項22】
(iii)過剰の放射性同位体を除去するためのイオン交換カートリッジと、任意選択により
(iv)前記前駆体化合物が1つ以上の保護基を含む場合、得られた放射性標識生成物を脱保護して本明細書記載のインビボ造影剤を形成するためのカートリッジと
をさらに含む、請求項21記載のカセット。
【請求項23】
哺乳動物への投与に適した形態の生体適合性の担体と一緒に、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤を含む、放射性医薬組成物。
【請求項24】
対象におけるPBR発現の分布及び/又は程度を決定するためのインビボイメージング法であって、
(i)前記対象に、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤を投与する段階と、
(ii)前記インビボ造影剤を前記対象のPBRと結合させる段階と、
(iii)インビボイメージング手順によって、前記インビボ造影剤の放射性同位体から放出されたシグナルを検出する段階と、
(iv)前記シグナルの位置及び/又は量の代表的な画像を作成する段階と、
(v)前記インビボ造影剤によって放出された前記シグナルと直接相関する、前記対象におけるPBR発現の分布及び程度を決定する段階と
を含む方法。
【請求項25】
前記対象のための治療投与計画の過程中、反復して実施される請求項24記載のインビボイメージング法であって、前記投与計画がPBR状態に対抗するための薬物を投与する段階を含む方法。
【請求項26】
請求項24記載のインビボイメージング法を、PBR発現の分布及び程度が特定の臨床像に帰するさらなる段階(vi)と一緒に含む、PBRが上方制御されている状態を診断する方法。
【請求項27】
請求項26記載の診断方法において使用するための、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤。
【請求項28】
請求項26記載の診断方法において使用する請求項21記載の放射性医薬組成物の製造に使用するための、請求項1乃至請求項11のいずれか1項記載のインビボ造影剤。
【図1】

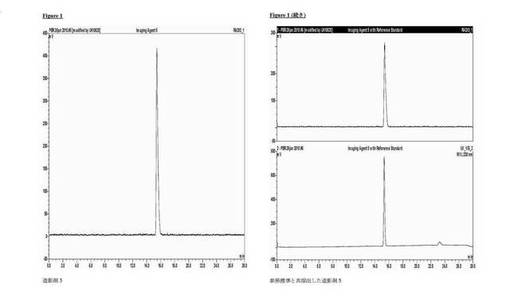
【図2】
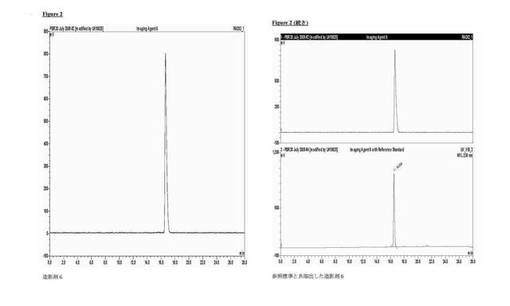
【図3】
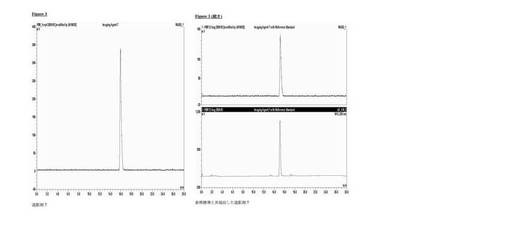
【図4】

【図5】
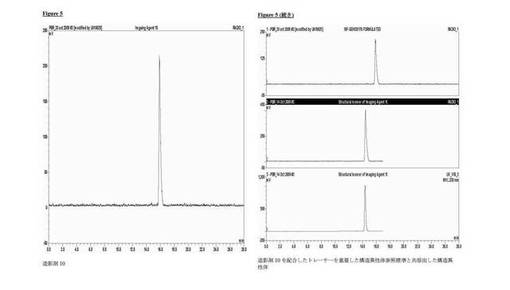
【図6】

【図7】
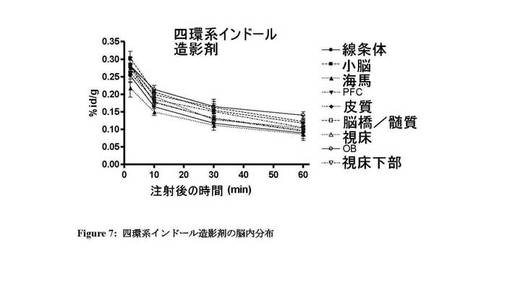
【図8】
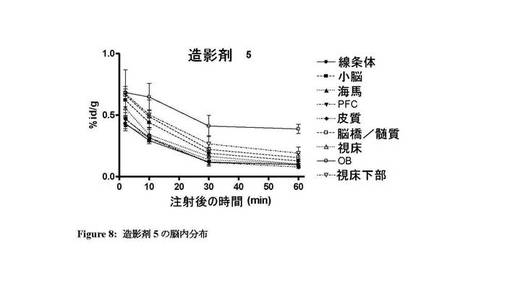
【図9】
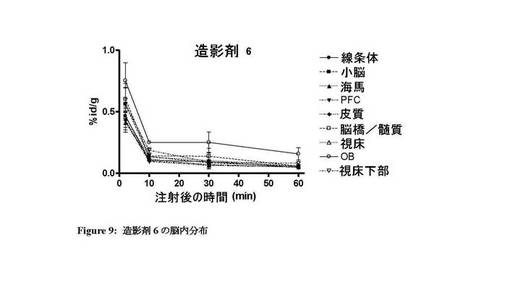
【図10】
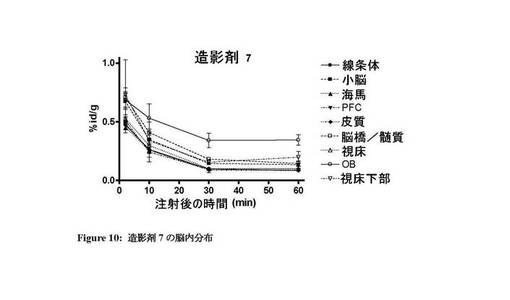
【図11】
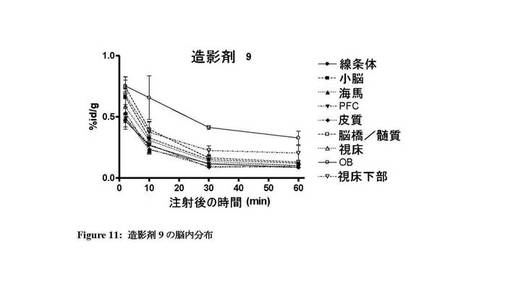
【図12】
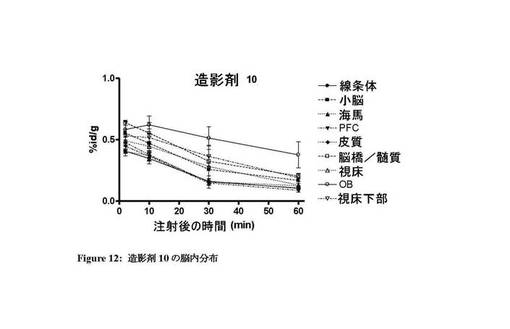
【図13】
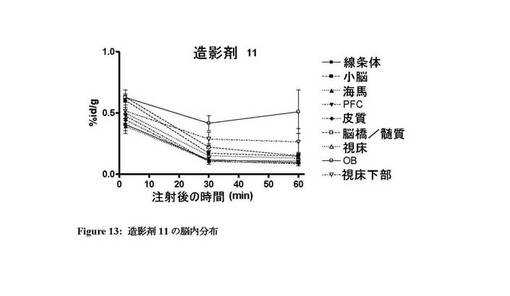
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【公表番号】特表2012−521973(P2012−521973A)
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願番号】特願2012−501315(P2012−501315)
【出願日】平成22年3月26日(2010.3.26)
【国際出願番号】PCT/EP2010/053998
【国際公開番号】WO2010/109007
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
【公表日】平成24年9月20日(2012.9.20)
【国際特許分類】
【出願日】平成22年3月26日(2010.3.26)
【国際出願番号】PCT/EP2010/053998
【国際公開番号】WO2010/109007
【国際公開日】平成22年9月30日(2010.9.30)
【出願人】(305040710)ジーイー・ヘルスケア・リミテッド (99)
【Fターム(参考)】
[ Back to top ]