
PCRにより増幅させた伸長DNA鎖の検出方法、および、標的配列の測定方法
【課題】定量性に優れ且つ同一反応液中の複数の増幅産物を各々検出可能な、新たなPCR増幅産物の検出方法を提供する。
【解決手段】標的配列を鋳型とし、これにプライマーと標識化プローブとをアニーリングさせる。DNAポリメラーゼのポリメラーゼ活性により、標的配列にアニーリングしたプライマーから標的配列に相補的なDNA鎖を5’→3’方向に伸長させ、且つ、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性により、標的配列にアニーリングした標識化プローブを分解し、標識化物質が結合した断片を遊離させる。この伸長反応(それに伴う分解反応)を繰り返し、標識配列に相補的な伸長DNA鎖を増幅する。この反応液について質量分析を行い、遊離した標識化物質結合断片の有無や量を検出して、標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する。
【解決手段】標的配列を鋳型とし、これにプライマーと標識化プローブとをアニーリングさせる。DNAポリメラーゼのポリメラーゼ活性により、標的配列にアニーリングしたプライマーから標的配列に相補的なDNA鎖を5’→3’方向に伸長させ、且つ、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性により、標的配列にアニーリングした標識化プローブを分解し、標識化物質が結合した断片を遊離させる。この伸長反応(それに伴う分解反応)を繰り返し、標識配列に相補的な伸長DNA鎖を増幅する。この反応液について質量分析を行い、遊離した標識化物質結合断片の有無や量を検出して、標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、PCRにより増幅させた伸長DNA鎖の検出方法、および、PCRの鋳型となる標的配列の測定方法に関する。
【背景技術】
【0002】
病理学や臨床学の様々な分野において、遺伝子の発現解析、機能解析、診断等を目的としてポリメラーゼチェーンリアクション(PCR)法が広く利用されている。PCR法は、一般に、(1)二本鎖DNAから鋳型となる一本鎖DNAへの解離、(2)鋳型一本鎖DNAへのプライマーのアニーリング、(3)DNAポリメラーゼを用いた前記プライマーの伸長、という3ステップを1サイクルとし、このサイクルを繰り返すことによって、試料中の標的核酸に相補的なDNAを増幅する方法である。
【0003】
近年では、PCRにおける増幅産物の生成過程を経時的にモニタリングする、いわゆるリアルタイムPCRが、様々な用途に利用されている。リアルタイムPCRは、一般的に、PCR産物の増幅に伴って発光強度が増大する蛍光物質(もしくは発光強度が減少する蛍光物質)を共存させてPCRを行い、増加する発光強度(もしくは減少する蛍光強度)を、PCR機器と光学的な検出器とを組み合わせた装置で、経時的にモニタリングする方法である。このようなリアルタイムPCRによれば、例えば、1サイクルごとの増幅産物の定量や、増幅産物が所定量(閾値)に達する際のサイクル数をカウントすることが可能であり、さらに、それらの情報に基づいて、試料に含まれていた標的配列を定量することも可能である。特に、RNAの発現解析においては、RNAの発現量が低い場合でも、逆転写PCR法によってRNAからcDNAを合成し、目的のcDNAをリアルタイムPCRで増幅することによって、従来よりも正確な定量が可能となっている。
【0004】
リアルタイムPCRにおける蛍光検出法としては、インターカレーター法、Taq
Man(商標)プローブ法、ハイブリダイゼーション法等が広く採用されている。「インターカレーター法」は、二本鎖DNAにインターカレートし、励起光を照射すると蛍光を発するインターカレーター(例えば、商品名SYBER Green I)を使用する方法である。この方法では、相補鎖が伸長されれば、鋳型と相補鎖との二本鎖にインーカレーターが入り込み、励起光照射によって蛍光を発するため、増幅による蛍光強度の増加をモニタリングすればよい。また、「Taq Man(商標)プローブ法」は、5’末端に蛍光物質を、3’末端にクエンチャーを結合させたプローブを使用する方法である。前記プローブは、鋳型にアニールしたのみでは、励起光を照射しても前記クエンチャーにより蛍光物質の蛍光は抑制されるが、相補鎖が伸長されると、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性により前記プローブが分解され、蛍光物質がクエンチャーから離れることにより蛍光を発する。このため、この方法では、増幅による蛍光強度の増加をモニタリングすればよい。「ハイブリダイゼーション法」は、3’末端がアクセプター蛍光物質で標識化されたプローブと、5’末端がドナー蛍光物質で標識化されたプローブとを用いる方法である。前記二種類のプローブは、鋳型にアニールすると、アクセプター蛍光物質とドナー蛍光物質とが隣接して蛍光が発生するが、相補鎖が伸長されると、アクセプター蛍光物質で標識されたプローブが分解され、アクセプター蛍光物質がドナー蛍光物質から離れることにより蛍光を発生しなくなる。したがって、この方法では、増幅による蛍光強度の減少を測定すればよい。
【0005】
しかしながら、前述のようなリアルタイムPCRは、スループットや定量性に優れるものの、蛍光検出が必須であることから、クロストーク(漏話)等が問題視されている。特に、1つの反応液中で二種類以上の標的核酸についてPCRを行った場合、それぞれの標的核酸に対する相補的伸長鎖を検出するには、各プローブについて異なる検出波長の蛍光物質を結合させる必要がある。しかし、各蛍光物質の最大吸収波長が異なっていても、例えば、スペクトルの端部が重複するおそれがあるため、互いの影響を受けることなく、それぞれの発現量を正確に測定することが困難である。また、スペクトルが重複しない蛍光物質を選択する方法も考えられるが、そのような蛍光物質の組み合わせには限りがあるため、一つの反応液中で所望の複数種類の相補的伸長鎖を分析することは現実的ではない。
【非特許文献1】BioTechniques, 27, 342−349.(1999) “Seven-color, homogeneous detection of six PCR products.” Lee,L.G., Livak,K.J., Mullah,B., Graham,R.J., Vinayak,R.S. and Woudenberg,T.M.
【非特許文献2】Nucleic Acids Research, 2006, Vol.34, No.1 “TaqMan probe array for quantitative detection of DNA targets” Heping Liu1, Hong Wang, Zhiyang Shi, Hua Wang, Chaoyong Yang, Spering Silke, Weihong Tan and Zuhong Lu
【発明の開示】
【発明が解決しようとする課題】
【0006】
そこで、本発明は、PCRの増幅産物を検出する新たな方法の提供を目的とする。より詳細には、定量性に優れ、且つ、同一反応液中の複数の増幅産物をそれぞれ検出することが可能な、新たなPCR増幅産物の検出方法の提供を目的とする。
【課題を解決するための手段】
【0007】
前記目的を達成するために、本発明の検出方法は、PCRによって増幅した、標的配列に相補的な伸長DNA鎖を検出する方法であって、下記(A)〜(F)工程を含むことを特徴とする。
(A) 標的配列、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有するDNAポリメラーゼ、プライマー、ならびに、オリゴヌクレオチドプローブに標識化物質が結合した標識化プローブを含む反応液を準備する工程
(B) 前記標的配列を鋳型として、これに前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(C) DNAポリメラーゼのポリメラーゼ活性によって、前記標的配列にアニーリングしたプライマーから前記標的配列に相補的なDNA鎖を5’→3’方向に向かって伸長させ、且つ、前記DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質が結合した断片(以下、「標識化物質結合断片」という)を遊離させる工程
(D) 前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(E) 前記工程(C)および工程(D)を繰り返し行うことによって、前記標識配列に相補的な伸長DNA鎖を増幅する工程
(F) 前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質が結合した断片の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する工程
【0008】
また、本発明の測定方法は、PCRによって増幅した、標識配列に相補的な伸長DNA鎖の検出により、標的配列を測定する方法であって、下記(G)および(H)工程を含むことを特徴とする。
(G) 本発明の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出する工程
(H) 前記(G)工程の検出結果に基づいて、前記反応液における標的配列の定性または定量を行う工程
【発明の効果】
【0009】
本発明の検出方法では、質量分析によって、PCR反応において遊離する標識化物質結合断片の有無や量を検出し、間接的に、標識配列に相補的な伸長DNA鎖を検出する方法である。質量分析は、非常に定量性に優れることから、これを利用した本発明は、定量性に優れる新たな増幅産物の検出方法といえる。また、蛍光物質を使用した蛍光強度の測定とは異なり、質量分析においては、例えば、クロストーク等の問題がない。このため、同一反応液中の複数の伸長DNA鎖を検出する場合でも、各標的配列に対応する標識化プローブから遊離する標識化物質結合断片のそれぞれが、例えば、イオン化によって、異なる分子イオンのピークを示したり、異なるフラグメントイオンピークを示せば、質量分析によって各々を検出することが可能である。したがって、本発明の検出方法によれば、分析対象試料について、複数の標的配列の定性・定量を行う場合であっても、同一反応液において複数の標的配列に対するPCRを行うことができ、且つ、互いの影響を受けることなく、各々の伸長DNA鎖を高精度で分析することができる。このため、本発明は、病理学や臨床学、分子生物学等の様々な分野において、極めて有用な方法といえる。なお、PCRにより増幅した伸長DNA鎖自体を質量分析により検出することは試みられているが、本発明のように、PCR反応によって標識化プローブより遊離する標識化物質結合断片を質量分析により検出することは、本発明者らが初めて見出した手法である。
【発明を実施するための最良の形態】
【0010】
本発明の検出方法は、前述のように、PCRによって増幅した、標的配列に相補的な伸長DNA鎖を検出する方法であって、下記(A)〜(F)工程を含むことを特徴とする。
(A) 標的配列、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有するDNAポリメラーゼ、プライマー、ならびに、オリゴヌクレオチドプローブに標識化物質が結合した標識化プローブを含む反応液を準備する工程
(B) 前記標的配列を鋳型として、これに前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(C) DNAポリメラーゼのポリメラーゼ活性によって、前記標的配列にアニーリングしたプライマーから前記標的配列に相補的なDNA鎖を5’→3’方向に向かって伸長させ、且つ、前記DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質結合断片を遊離させる工程
(D) 前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(E) 前記工程(C)および工程(D)を繰り返し行うことによって、前記標識配列に相補的な伸長DNA鎖を増幅する工程
(F) 前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質結合断片の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する工程
【0011】
PCRにおける標的配列は、特に制限されないが、例えば、DNAやRNA(mRNA、Total RNA)の他に、RNAから逆転写により合成したcDNA等があげられる。特に、RNAから合成したcDNAを標的配列とすれば、例えば、低発現のRNAの有無を間接的に検出することが可能である。
【0012】
前記標識化物質としては、何ら制限されず、あらゆる物質が使用できる。例えば、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、標識化プローブから標識化物質のみが切断されるものでないことが好ましい。前記標識化物質としては、例えば、イオン化可能な有機化合物が好ましく、より好ましくはイオン化可能な芳香族有機化合物である。具体例としては、例えば、6−カルボキシフルオレセイン(FAM)等のフルオレセイン、ローダミン、ビオチン等があげられるが、これらは本発明における一例であり、何ら制限されない。
【0013】
オリゴヌクレオチドプローブにおいて、標識化物質を結合させる基は、特に制限されず、例えば、塩基、糖、リン酸のいずれの基でもよい。また、前記(F)工程においては、標識化プローブの分解物のうち標識化物質が結合した断片を、質量分析によって検出することから、前記標識化物質は、少なくとも前記(C)工程においてDNAポリメラーゼにより遊離されることとなる部位に結合していればよい。中でも、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によれば、通常、プローブの5’側から断片が遊離するため、例えば、前記オリゴヌクレオチドプローブの5’末端に結合させることが好ましい。
【0014】
前記(C)工程において、標識化プローブの分解により遊離する標識化物質結合断片(以下、質量分析の対象となることから、「ターゲット物質」ともいう)としては、例えば、前記標識化物質とヌクレオチド1分子とが結合した結合体であることが好ましい。このヌクレオチドは、前記標識化プローブにおいて、前記標識化物質が結合した部位のヌクレオチドである。遊離する断片が、前記標識化物質とヌクレオチド1分子との結合体であることは、特に、1つのPCR反応液で、二種類以上の伸長DNA鎖をそれぞれ検出する際に好ましい。この理由については後述する。遊離する断片としては、この他に、前記標識化物質がオリゴヌクレオチドに結合した結合体であってもよい。なお、この結合体は標識化プローブの分解物であるため、前記結合体を構成するオリゴヌクレオチドは、少なくとも前記オリゴヌクレオチドプローブよりも短い配列となる。
【0015】
なお、本発明においては、5’→3’エキソヌクレアーゼ活性により遊離したヌクレオチドを「遊離ヌクレオチド」、遊離したオリゴヌクレオチドを「遊離オリゴヌクレオチド」、標識化物質を結合させるプローブを「オリゴヌクレオチドプローブ」、前記プローブの構成成分であるヌクレオチドを「構成ヌクレオチド」という。
【0016】
前記遊離する断片を、標識化物質が結合した「遊離ヌクレオチド」とするか、標識化物質が結合した「遊離オリゴヌクレオチド」とするかは、通常、使用するDNAポリメラーゼの種類に応じて決定できる。DNAポリメラーゼとしては、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有していれば、その種類は特に制限されず、例えば、従来公知の耐熱性細菌由来のDNAポリメラーゼがあげられる。DNAポリメラーゼは、例えば、自家調製してもよいし、市販のものを使用してもよい。具体例としては、テルムス・アクアティカス(Thermus aquaticus)由来DNAポリメラーゼ(米国特許第4,889,818号および同第5,079,352号)(商品名Taq DNAポリメラーゼ)、テルムス・テルモフィラス(Thermus thermophilus)由来DNAポリメラーゼ(WO91/09950)(rTth DNA polymerase)、ピロコッカス・フリオサス(Pyrococcus furiosus)由来DNAポリメラーゼ(WO92/9688)(Pfu DNA polymerase:Stratagenes社製)、テルモコッカス・リトラリス(Thermococcus litoralis)由来DNAポリメラーゼ(EP−A455430)(商標Vent:Biolab New England社製)等が商業的に入手可能である。中でも、5’末端からヌクレオチドを1分子ずつ遊離させる場合は、テルムス・アクアティカス由来DNAポリメラーゼ(商品名Taq DNAポリメラーゼ)等を使用することが好ましい。なお、DNAポリメラーゼが、ヌクレオチドを遊離するかオリゴヌクレオチドを遊離するかが不明の場合でも、例えば、予め確認すれば足り、当業者であれば容易に行うことができる。
【0017】
本発明の検出方法によれば、一種類の伸長DNA鎖を検出するのはもちろんのこと、一つの反応液において二種類の伸長DNA鎖を増幅させた場合であっても、それぞれの伸長DNA鎖を検出することができる。以下に、一種類の伸長DNA鎖の検出方法ならびに二種類以上の伸長DNA鎖の検出方法のそれぞれについて、例をあげて説明する。なお、本発明は、これらには限定されない。
【0018】
まず、一種類の伸長DNA鎖の検出方法の例について説明する。
【0019】
実施形態1
(A)PCR反応液の準備
標的配列を含むDNA、DNAポリメラーゼ、dNTP(ヌクレオシド三リン酸)、前記標的配列に特異的なプライマー、ならびに、標識化プローブを含む反応液を準備する。
【0020】
前記標識化プローブとしては、特に制限されないが、前述のようにオリゴヌクレオチドプローブの5’末端に標識化物質を結合させたものが好ましい。このように5’末端に標識化物質が結合した標識化プローブであれば、例えば、DNAポリメラーゼがヌクレオチドを1分子ずつ遊離する場合であっても、2mer以上のオリゴヌクレオチドを遊離させる場合であっても、確実に、はじめに遊離した断片が標識化物質を有することとなる。本発明においては、この標識化物質が結合した遊離断片の有無または量が、伸長DNA鎖の増幅の有無または増幅量に相当するため、はじめに遊離した断片を検出できれば、分析精度を十分に確保できる。
【0021】
標識化プローブならびにプライマーの配列ならびに長さは、特に制限されず、標的配列に応じて、従来公知の方法により適宜決定できる。また、本発明では、後述する(C)工程において、DNAポリメラーゼによって、プライマーからの伸長反応を行うとともに、標的配列にアニーリングした標識化プローブを5’末端から分解するが、この反応自体は、例えば、Taq Man(商標)Probe法等において公知であることから、プライマーならびにプローブの長さや配列は、当業者であれば技術常識に基づいて設計可能である。通常、標識化プローブは、プライマーのアニーリング部位よりも5’側(鋳型の5’側)にアニーリングするように設計することが好ましい。なお、プライマーとしては、フォワードプライマーのみでもよいが、フォワードプライマーとリバースプライマーとを含むプライマーセットを用いることが好ましい。また、プライマーの長さは、一般的に10〜50mer程度であり、プローブの長さは、一般的に20〜30merであるが、これらには制限されない。
【0022】
前記PCR反応液中のプライマーの添加割合は、特に制限されないが、例えば、フォワードプライマーとリバースプライマーとが、それぞれ0.01〜5μmol/Lであり、好ましくは0.1〜3μmol/Lであり、より好ましくは0.1〜1μmol/Lである。また、前記PCR反応液中の標識化プローブの添加割合は、特に制限されないが、例えば、0.01〜5μmol/Lであり、好ましくは0.1〜1μmol/Lであり、より好ましくは0.2〜0.5μmol/Lである
【0023】
また、DNAポリメラーゼとしては、例えば、5’→3’エキソヌクレアーゼ活性により、ヌクレオチドを1分子ずつ遊離するものでも、2mer以上のオリゴヌクレオチドを遊離させるものでもよいが、前者が好ましい。このようなDNAポリメラーゼとしては、例えば、前述のようなテルムス・アクアティカス由来DNAポリメラーゼ(商品名Taq DNAポリメラーゼ)があげられる。
【0024】
前記PCR反応液中のDNAポリメラーゼの添加割合は、特に制限されないが、例えば、5〜50U/mlであり、好ましくは1〜100U/mlであり、より好ましくは20〜30U/mlである。なお、DNAポリメラーゼの活性単位(U)は、一般に、活性化サケ精子DNAを鋳型プライマーとして、活性測定用反応液(25mM TAPS buffer(pH9.3、25℃)、50mM KCl、2mM MgCl2、1mMメルカプトエタノール、200μM dATP、200μM dGTP、200μM dTTP、100μM「α−32P」dCTP、0.25mg/ml活性化サケ精子DNA)中、74℃で、30分間に10nmolの全ヌクレオチドを酸不溶性沈殿物に取り込む活性が1Uである。
【0025】
前記dNTPとしては、通常、dATP、dCTP、dTTPの混合物があげられる。前記PCR反応液中のdNTPの添加割合(合計添加割合)は、特に制限されないが、例えば、0.01〜1mmol/Lであり、好ましくは0.05〜0.5mmol/Lであり、より好ましくは0.1〜0.3mmol/Lである。
【0026】
前記PCR反応液の溶媒としては、特に制限されないが、例えば、Tricine、MES、MOPS、HEPES、CAPS等の緩衝液、市販のPCR用緩衝液や市販のPCRキットの緩衝液等が使用できる。反応液のpHは、例えば、7〜9である。
【0027】
(B)〜(E)PCR工程
つぎに、標的配列を鋳型としてPCR反応を行う。この反応自体は、特に制限されず、従来公知の方法に基づいて行うことができる。
【0028】
すなわち、まず、(B):鋳型である前記標的配列に、プライマーおよび標識化プローブをアニーリングさせる。鋳型である前記標的配列が二本鎖DNAを形成している場合には、この(B)工程に先立って、例えば、熱処理等によって一本鎖DNAへの解離を行うことが好ましい。そして、(C):DNAポリメラーゼによって、前記プライマーから前記標的配列に相補的なDNA鎖を伸長させ、且つ、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質結合断片を遊離させる。さらに、(D):前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび標識化プローブをアニーリングさせてから、前記(C)工程および(D)工程を繰り返す。このように、一本鎖DNAへの解離、アニーリングおよびDNAポリメラーゼ反応の3ステップを1サイクルとして、繰り返し行うことによって、標的配列に相補的な伸長DNA鎖を増幅することができる。前記(C)工程および(D)工程のサイクル数は、特に制限されないが、例えば、30サイクル以上であれば、標的配列に相補的な伸長DNA鎖の増幅を十分に確認することができ、好ましくは35サイクル以上、より好ましくは40サイクル以上である。なお、本発明においては、最終サイクルの(D)工程は割愛することができる。
【0029】
二本鎖DNAから一本鎖DNAへの解離の処理条件は、特に制限されないが、例えば、温度93〜95℃、時間15〜60秒であり、好ましくは温度94℃、時間30秒であり、アニーリングの処理条件は、特に制限されないが、例えば、温度(Tm値−5℃)〜(Tm値−2℃)、時間20〜60秒であり、好ましくは温度(Tm値−4℃、時間45秒であり、DNAポリメラーゼ反応は、特に制限されないが、例えば、温度60〜80℃、時間1〜45秒であり、好ましくは温度65〜75℃、時間30秒である。なお、各ステップにおける処理温度や処理時間は、例えば、サーマルサイクラー等の装置を用いて自動的に制御することができる。
【0030】
(F)標識化物質が結合した断片の質量分析による検出
そして、前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質結合断片の有無または量を検出することによって、標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する。
【0031】
前記(B)工程において、標識化プローブは、プライマーのアニーリング部位よりも5’側(鋳型の5’側)にアニーリングする。そして、前記(C)工程において、DNAポリメラーゼメラーゼは、プライマーを開始部位として5’→3’方向に伸長反応を進めると同時に、鋳型の3’→5’方向に向かって移動する。そして、移動したDNAポリメラーゼが、プライマーのアニーリング位置より5’側の標識化プローブのアニーリング部位に近づくと、5’→3’エキソヌクレアーゼ活性により、標識化プローブを5’側から切断していく。他方、ポリメラーゼ活性による伸長反応が起こらなかった場合には、DNAポリメラーゼも標識化プローブのアニーリング部位に近づかないため、エキソヌクレアーゼ活性による標識化プローブの切断は生じない。したがって、5’→3’エキソヌクレアーゼ活性により切断された断片の有無を検出すれば、それは、ポリメラーゼ活性により相補的な伸長DNA鎖が合成されたことを間接的に検出することとなる。このため、遊離する前記標識化物質結合断片の有無や量を検出すれば、伸長鎖DNAの有無や量を検出することができる。
【0032】
遊離する断片の長さは、前述のようにDNAポリメラーゼの種類によって異なるが、5’末端からヌクレオチドを1分子ずつ遊離するDNAポリメラーゼを使用した場合、前記(C)工程において、標識化プローブの5’末端ヌクレオチドの3’側が切断され、標識化物質が結合した前記5’末端ヌクレオチドが遊離する。具体例としては、標識化プローブの5’末端ヌクレオチドがデオキシアデノシンリン酸であれば、デオキシアデノシンリン酸に標識化物質が結合した結合体が遊離される。同様に、5’末端ヌクレオチドにおける塩基の種類によって、デオキシグアノシンリン酸、デオキシシチジンリン酸またはデオキシチミジンリン酸に標識化物質が結合した結合体が遊離される。
【0033】
前記反応液は、例えば、質量分析に供する前に脱塩処理等の不純物除去処理を施すことが好ましい。これによって分析精度をさらに向上することができる。
【0034】
質量分析の方法は、何ら制限されず、従来公知の方法が採用できる。質量分析におけるイオン化法としては、例えば、ESI(Electro Spray Ionization)法、LD(Laser Desorption Ionization)法、MALDI(Matrix Assisted Laser Desorption Ionization)法、EI(Electron Impact)法、CI(Chemical Ionization)法、API(atmospheric pressure Ionization)法、FI(Field Ionization)法、FD(Field Desorption)法、SIMS(Secondary Ion Mass Spectroscopy)法、FAB(Fast Atom Bonbardment)法、TSP(thermospray Ionization)法等があげられ、特に制限されないが、中でもESI法が好ましい。
【0035】
質量分析において、前述のようなイオン化法により生成したイオンは、通常、磁場や電場により分離され、分析される。分析方法の原理としては、特に制限されないが、例えば、磁場単収束型、電場磁場二重収束型、四重極型、三次元四重極型、飛行時間(time of flight、TOF)型、イオンサイクロトロン共鳴(Ion cyclotron resonance、ICR)型、イオントラップ(Ion Trap、IT)型、フーリエ変換(FT)型等があげられる。
【0036】
質量分析は、通常、質量分析装置(MS)により行うことができる。前記MSは、通常、イオン化室、質量分析部およびイオン検出器を備え、一般に、磁場型質量分析装置、四重極質量分析装置等に大別されるが、本発明においては、従来公知のあらゆる装置を使用することができる。質量分析装置の種類は、例えば、必要とされる分解能に応じて選択可能であり、例えば、高分解能が必要な場合には、MS/MS法を採用したり、二重収束質量分析装置を使用することも好ましい。また、本発明における分析対象試料はPCR反応液であって、例えば、未反応のdNTPやタンパク質、他の分解物等が含まれるため、例えば、液体クロマトグラフィー(LC)を質量分析装置(MS)に連結させたLC−MSや、LC−MS/MS等を使用することが好ましい。これにより、分析精度をさらに向上することができる。
【0037】
質量分析は、例えば、PCRの反応終了時のみ、または、PCRの反応途中のみに行ってもよいし、経時的に行ってもよい。このように経時的に質量分析を行えば、増幅産物(増幅した伸長DNA鎖)の生成過程を経時的にモニタリングすることができ、いわゆる従来のリアルタイムPCRと同様の解析が可能となる。経時的に分析を行う場合、例えば、断続的な分析でもよいし、連続的な分析でもよい。このため、本発明において、(F)工程の実施時期は、何ら制限されず、例えば、(C)工程と共に行ってもよいし、(C)工程後に行ってもよく、また、(C)工程を繰り返し行う間、連続的または断続的に行ってもよい。
【0038】
なお、標的核酸がRNA(mRNA、total RNA)の場合には、通常、前記RNAから逆転写反応によりcDNAを生成させ、このcDNAを標的配列とすればよい。そして、これをPCRの鋳型として使用する以外は、前述と同様にしてPCRを行えばよい(いわゆる、Reverse Transcription PCR)。
【0039】
逆転写反応は、例えば、従来公知の逆転写酵素、前記RNAに対するプライマーおよびヌクレオシド三リン酸(dNTP)を含む逆転写反応液を使用することができる。逆転写反応の反応条件は、特に制限されないが、例えば、反応温度30〜70℃、反応時間1〜120分であり、好ましくは反応温度40〜60℃、反応時間10〜60分であり、より好ましくは反応温度45〜50℃、反応時間20〜40分である。
【0040】
つぎに、同一反応液中における二種類以上の伸長DNA鎖の検出方法の例について説明する。
【0041】
同一反応液において、二種類以上の標的配列に相補的なDNA鎖をそれぞれ伸長させた場合、各伸長DNA鎖を検出することが必要となる。本発明は、遊離した標識化物質結合断片を質量分析で検出することによって、間接的に伸長DNA鎖を検出できる。したがって、本発明において二種類以上の伸長DNA鎖を検出する場合には、各標識化プローブから遊離される各標識化物質結合断片が、例えば、イオン化によって、異なる分子イオンピークを示したり、異なるフラグメントピークを示すもの等であれば、ターゲット物質とすることができる。なお、各標識化物質結合断片の質量が同じであっても、例えば、各フラグメントイオンが異なるm/z値にピークを示せば、それぞれを検出することが可能である。
【0042】
具体例としては、前記(C)工程において、二種類以上の標的配列に対応する各標識化プローブの分解により、前記標識化物質が結合した断片がそれぞれ遊離するが、これらの各断片が、それぞれ異なる質量であることが好ましい。各標識化物質結合断片の質量を変化させる方法は、特に制限されないが、例えば、異なる標識化物質を使用する方法、標識化物質結合断片として、異なる塩基のヌクレオチドと標識化物質との結合体を遊離させる方法、さらに、これらを組み合わせた方法等があげられる。これらの方法について、以下に具体例をあげるが、本発明は、これらには制限されない。
【0043】
実施形態2−1
この実施形態は、各標的配列に対応する標識化プローブについて、それぞれ質量が異なる標識化物質を使用する形態である。なお、本実施形態においては、オリゴヌクレオチドの5’末端に標識化物質が結合された標識化プローブであり、5’→3’エキソヌクレアーゼ活性によって、5’末端ヌクレオチド1分子と標識化物質との結合体が遊離する例をあげて説明する。
【0044】
本実施形態においては、前記(A)工程における反応液として、二種類以上の標的配列、および、前記各標的配列に対応する二種類以上のプライマーならびに二種類以上の標識化プローブを含む反応液を使用する。なお、各標的配列に対応するプライマーの配列や長さは、特に制限されず、例えば、標的配列に応じて、従来公知の方法によって適宜決定できる。
【0045】
本実施形態のように、異なる質量の標識化物質を使用する場合、例えば、各標識化プローブにおける5’末端のヌクレオチドは、同じ塩基のヌクレオチドであってもよい。例えば、各標識化プローブの5’末端ヌクレオチドが同じデオキシアデノシンリン酸であっても、5’末端に結合した標識化物質が質量の異なる物質であれば、遊離する標識化物質結合断片の全体の質量は、それぞれ異なる。したがって、各標的配列について相補的なDNA鎖を伸長する場合、各標識化プローブの標識化物質を異なる質量とすることで、各伸長DNA鎖を質量分析によって検出することが可能となる。
【0046】
質量分析では、例えば、1原子質量単位が異なるのみであっても、別の物質として検出することができる。このため、本実施形態においては、質量が異なれば、どのような標識化物質でも使用することができるため、同一反応液において検出できる伸長DNA鎖の数は、何ら制限されない。なお、検出できる伸長DNA鎖の数としては、特に制限されないが、下限は、例えば、2以上であり、上限は、例えば、100以下、好ましくは20以下、より好ましくは10以下である。
【0047】
実施形態2−2
この実施形態は、各標的配列に対応する標識化プローブにおいて、各標識化物質が同じ質量であり、各標識化物質結合断片として、異なる塩基のヌクレオチドと標識化物質との結合体を遊離させる形態である。なお、本実施形態においては、オリゴヌクレオチドの5’末端に標識化物質が結合された標識化プローブであり、5’→3’エキソヌクレアーゼ活性によって、5’末端ヌクレオチド1分子と標識化物質との結合体が遊離する例をあげて説明する。
【0048】
本実施形態においては、前記実施形態2−1と同様に、前記(A)工程における反応液として、二種類以上の標的配列、および、前記各標的配列に対応する二種類以上のプライマーならびに二種類以上の標識化プローブを含む反応液を使用する。なお、各標的配列に対応するプライマーの配列や長さは、特に制限されず、標的配列に応じて、従来公知の方法によって適宜決定できる。
【0049】
本実施形態では、前述のように、前記標識化物質結合断片におけるヌクレオチドの塩基の違いによって、各断片を検出する。したがって、各標識化プローブにおいて、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基であればよい。例えば、各標識化プローブの標識化物質が同じであっても、5’末端ヌクレオチドの塩基が異なれば、遊離する標識化物質結合断片の全体質量はそれぞれ異なる。したがって、各標的配列について、相補的なDNA鎖を伸長する場合、各標識化プローブにおいて、標識化物質を結合するヌクレオチドの塩基を異なる塩基に設定することで、それぞれを質量分析により検出することが可能となる。具体例としては、オリゴヌクレオチドプローブを、その5’末端ヌクレオチドが、デオキシアデノシンリン酸、デオキシグアノシンリン酸、デオキシシチジンリン酸またはデオキシチミジンリン酸となるように設計し、これに標識化物質を結合して標識化プローブを調製すればよい。このように5’末端ヌクレオチドの塩基を異なる塩基とすれば、各標識化プローブから遊離する断片は、質量が異なるため、同一反応液であっても、少なくとも4種類の伸長DNA鎖の検出を行うことが可能となる。
【0050】
実施形態2−3
この実施形態は、二種類以上の標的配列に対応する各標識化プローブとして、それぞれの標識化物質が異なる質量の物質であり、且つ、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基であるプローブを使用する形態である。このような形態であれば、標識化物質の組み合わせと塩基の組み合わせが使用できるため、同一反応液であっても、極めて多くの種類塩基の種類の数×標識化物質の種類の数)の伸長DNA鎖を検出できる。
【0051】
つぎに、本発明の測定方法は、PCRによって増幅した、標識配列に相補的な伸長DNA鎖の検出により、標的配列を測定する方法であって、下記(G)および(H)工程を含むことを特徴とする。
(G) 本発明の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出する工程
(H) 前記(G)工程の検出結果に基づいて、前記反応液における標的配列の定性または定量を行う工程
【0052】
本発明の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出すれば、その結果から、前記反応液における標的配列の有無(定性)または量(定量)を測定することができる。具体的には、前記(G)工程の検出結果に基づき、増幅した伸長DNA鎖を定量し、前記伸長DNA鎖が所定量となった際のPCRサイクル数をカウントすることによって、前記反応液における標的配列を定量することができる。
【0053】
また、反応液における標的配列がRNAから逆転写反応により生成させたcDNAであれば、cDNAを定性または定量することによって、その鋳型であるRNAの有無または量を定性もしくは定量することもできる。
【0054】
PCRでは、通常、増幅産物がある一定の量(閾値)に達するとそれ以上の増幅は起こらないため、最終的に生成した増幅産物の量を測定するのみでは、反応液に含まれる標的核酸の量を定量することは困難である。しかし、このようなリアルタイムPCR様の方法によれば、サイクルごとの増幅産物の生成をモニタリングし、設定した閾値に達するサイクル数(Ct値)をカウントするため、例えば、閾値とCt値とに基づいて、反応液に含まれる標的配列を定量することも可能である。
【0055】
つぎに、本発明の実施例について、比較例と併せて説明する。ただし、本発明は以下の実施例および比較例により制限されない。
【実施例1】
【0056】
LC/MS分析によって、1分子のヌクレオチドと標識化物質とが結合したターゲット物質を定量的に検出できるか否かを確認した。
【0057】
1.サンプルの調製
FAMにアデニンを有するヌクレオチド(デオキシアデノシンリン酸)を付加したターゲット物質(SIGMA GENOSYS社製、以下、「FAM+A」ともいう)を準備し、これを水で希釈した希釈系列をサンプルとした。なお、「FAM+A」の構造を下記式に表すが、後述する他のターゲット物質(FAM+ヌクレオチド)は、下記式における塩基が異なるのみで、その他は同様の構造である。
【化1】

【0058】
2.LC/MS分析
capillary LCシステムは、Ultimate、SwithosII、Famos(LC packings、日本ダイオネクス社製)を、質量分析計は、Esquire3000plus(Bruker Daltonics社製)をそれぞれ用いた。分析カラムはInertsil ODS−3 0.300mm i.d.×150mm(GL sciences社製)、プレカラムはμ−Precolumn(商標)Cartridge C18 0.300mm i.d.×5mm(LC packings社製)を使用した。前記「FAM+A」の各サンプル5μlをインジェクトし、0.1体積%ギ酸を用いて流速10μl/minで5分間プレカラムに送液することによって、前記サンプルを前記プレカラムに吸着させた。その後、バルブを切り替え、前記プレカラムに吸着したサンプルを分析カラムに導入した。この際の溶離液は、A液として、H2O(和光純薬社製)/アセトニトリル(キシダ化学、以下同じ)/ギ酸(体積比95/5/0.1)、B液として、H2O/アセトニトリル/ギ酸(体積比5/95/0.1)をそれぞれ使用し、A液とB液との混合液におけるB液の割合(体積%)が下記条件となるグラジェントで溶離を行った。前記溶離液の流量は、4μl/minとした。
【0059】
B液体積% 溶離時間
40% 0min〜10min
90% 10min〜25min
40% 25min〜35min
【0060】
(質量分析計の分析パラメーター)
polarity:negative mode
target mass:787m/z
scan range:100−1200m/z
capillary voltage:3500V
nebulizar gas:15.0psi
dry gas flow rate:8.0 l/min
dry gas temp.:300℃
ICC target:20000
average:7
【0061】
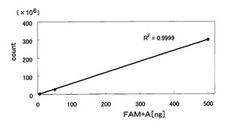
このLC/MS分析の結果を図1に示す。同図は、各サンプルにおけるターゲット物質の量とカウント(イオン強度)との相関関係を示すグラフである。同図に示すように、ターゲット物質とカウントとの関係を示す相関係数は0.9999と非常に高く、ターゲット物質の量が数百pg〜数百ngの間で優れた相関性を示した。このことから、「FAM+A」は、イオン化効率に優れ、ターゲット物質として本発明の方法に非常に適していると言える。
【実施例2】
【0062】
塩基のみが異なる4種類のターゲット物質を含む混合液についてLC/MS分析を行い、各ターゲット物質の検出が可能であるか否かを確認した。
【0063】
ターゲット物質として、FAMに異なる塩基(A,T、C、G)を有するヌクレオチド(デオキシアデノシンリン酸、デオキシチミジンリン酸、デオキシグアノシンリン酸、デオキシシチジンリン酸)を付加した4種類の物質を使用した。そして、4種類のターゲット物質をそれぞれ1ppmずつ含有する混合液を調製し、この混合液をサンプルとして、前記実施例1と同様にしてLC/MS分析を行い、Data Analysis(Bruker Daltonics社製)で解析した。なお、前記ターゲット物質は、塩基に応じてそれぞれ「FAM+A」、「FAM+T」、「FAM+C」および「FAM+G」という(以下、同様)。
【0064】
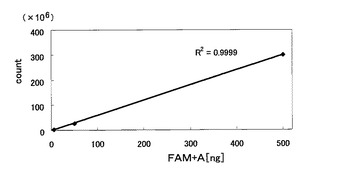
これらのLC/MS分析の結果を図2に示す。同図において、(A)はFAM+Cのクロマトグラム、(B)はFAM+Aのクロマトグラム、(C)はFAM+Gのクロマトグラム、(D)はFAM+Tのクロマトグラムである。これらのターゲット物質は、それぞれFAMに付加したヌクレオチドにおける塩基が異なるのみであるが、図2に示すように、それぞれ異なるリテンションタイムにピークが見られた。このことから、1つの反応液に塩基が異なる複数のターゲット物質(例えば、2〜4種類)が含まれる場合であっても、検出可能であることがわかった。
【実施例3】
【0065】
亜熱帯性植物Torenia hybrida(以下、「トレニア」という)の葉由来のmRNAよりcDNAを調製し、CHS(chalcon synthase)遺伝子およびGAPDH(glyceraldehyde−3−phosphate dehydrogenase)遺伝子を標的配列としてPCRを行い、増幅産物についてLC/MS分析を行った。
【0066】
1. RT−PCR
(1)cDNAの調製
トレニアの葉から、RNeasy(登録商標)Plant Mini Kit(QIAGEN社製)を用いて、TotalRNAを調製し、さらにDNaseIで処理した後、同キットを用いてTotalRNAを精製した。
【0067】
次いで、SuperScript(商標)First−Strand Synthesis System for RT−PCRキット(Invitrogen社)を用いて、以下に示す逆転写反応を行い、さらにRNaseHで処理して一本鎖cDNAを調製した。
【0068】
まず、精製した前記TotalRNAを用いて、全量5μgのTotal RNA溶液を調製した。これに、10mM dNTP mix(1μl)および0.5μg/μl Oligo(dT)12−18(1μl)を添加し、全量10μlとなるようにDEPC−treated waterを添加して、65℃で5分インキュベートした後、氷上で1分静置した。前記混合液に、さらに、10×RT buffer(2μl)、25mM MgCl2(4μl)、0.1M DTT(2μl)およびRNaseOUT(商標)Recombinant RNase Inhibitor(Invitrogen社製)(1μl)を順に添加し、42℃で2分インキュベートした。インキュベート後の混合液に、SuperScript(商標)II RT(1μl)を添加し、42℃で50分、70℃で15分の条件で順次インキュベートを行ってから、RNaseH(1μl)を添加した。これらの処理によって一本鎖cDNAを調製した。
【0069】
(2)PCR
調製した一本鎖cDNAを鋳型とし、下記プライマー、プローブおよびHotStarTaq(登録商標)PCR Master Mixキット(QIAGEN社製)を用いて、以下に示す条件で、CHS遺伝子とGAPDH遺伝子のそれぞれについてPCRを行った。なお、下記PCR反応液AおよびPCR条件Aは、CHS遺伝子およびGAPDH遺伝子の一方を検出する際の組成と条件である。
【0070】
CHS用プライマー
フォワードプライマー(配列番号1)
5’−ATACTGGACGAGATGAGGAA−3’
リバースプライマー (配列番号2)
5’−ATATCACCAGAATCATGCCA−3’
GAPDH用プライマー
フォワードプライマー(配列番号3)
5’−TGTCGATGTCTCCGTAGTGG−3’
リバースプライマー (配列番号4)
5’−CATCGTCTTCGGTATAGCCC−3’
【0071】
CHS用プローブ (配列番号5)
5’−FAM−AGACCGTTGTGCTG−MGB−3’
GAPDH用プローブ(配列番号6)
5’−FAM−CTGCTATCAAGGAG−MGB−3’
【0072】
【表1】
【0073】
【表2】
【0074】
2. LC/MS分析
得られた二種類のPCR増幅産物(CHS遺伝子の増幅産物およびGAPDH遺伝子の増幅産物)のそれぞれについて、LC/MS分析前、ziptip(商品名、SIGMA MILLIPORE社製)を用いて脱塩処理を行った。
【0075】
まず、ziptipをアセトニトリルで2回ピペッティング(ピペッティングは全て10μl、以下同様)してから、精製水で2回ピペッティングすることにより平衡化した。つぎに、前記PCR増幅産物(50μl)を5回ピペッティングし、PCR反応により遊離したターゲット物質(FAM+ヌクレオチド)をトラップさせてから、精製水で2回ピペッティングして平衡化した。続いて、精製水12.5μlとアセトニトリル12.5μlの混合液中で5回ピペッティングを行い、トラップした前記ターゲット物質を溶出させた(計25μl)。この溶出液に、内部標準物質として、1ppmの「FAM+G」(標準品、SIGMA
GENOSYS社製)5μlを添加して(計30μl)、分析用サンプルとした。
【0076】
この分析用サンプルを、前記実施例1と同様の条件でLC/MS分析に供し、その結果をData Analysis(Bruker Daltonics社製)で解析した。
【0077】
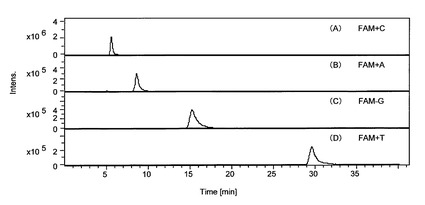
LC/MS分析の結果を図3に示す。同図において、(A)は、CHS遺伝子のPCR増幅産物の分析に、5’末端のAがFAMで標識されたプローブ(配列番号5)を使用した結果であり、(B)は、GAPDH遺伝子のPCR増幅産物の分析に、5’末端のCがFAMで標識されたプローブ(配列番号6)を使用した結果である。同図(A)および(B)に示すように、それぞれのPCR増幅産物について、異なるリテンションタイムでピークが検出された。同図(A)で検出されたピークは、「FAM+A」、同図(B)で検出されたピークは、「FAM+C」である。これらの結果から、LC/MS分析によって、PCR反応で遊離されたヌクレオチドが付加したターゲット物質を検出でき、この検出により、PCRにおける増幅の有無や増幅量を確認(定性、定量)できることがわかった。また、この結果から、サイクル数40で、十分にPCR増幅産物を検出できることが確認できた。
【実施例4】
【0078】
1つのPCR反応液内でCHS遺伝子およびGAPDH遺伝子のPCRを同時に行い、それぞれのPCR増幅産物をLC/MSにより分析した。
【0079】
1. RT−PCR
前記実施例3で調製した一本鎖DNAを鋳型として、下記PCR反応液Bを用いて、下記PCR条件BによりPCRを行った。なお、GAPDH用プローブとしては、以下に示す2種類のプローブを使用し、各プローブを用いて別個にPCRを行った。なお、下記PCR反応液BおよびPCR条件Bは、それぞれ1つの反応液でCHSおよびGAPDHの両方を検出するマルチプレックスPCRの組成ならびに条件である。
【0080】
【表3】
【0081】
GAPDH用プローブ
5’−FAM−CTGCTATCAAGGAG−MGB−3’(配列番号6)
5’−FAM−TCAAGGAGGAATCC−MGB−3’(配列番号7)
【0082】
【表4】
【0083】
そして、得られたPCR増幅産物について、前記実施例3と同様に脱塩処理を施した後、前記実施例1と同様にLC/MS分析を行い、その結果をData Analysis(Bruker Daltonics社製)で解析した。
【0084】
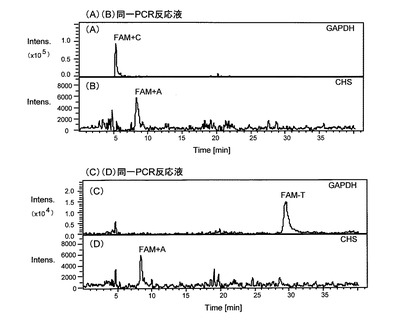
これらのLC/MS分析の結果を図4に示す。同図において、(A)と(B)は、同じPCR反応液についての結果、(A)は、GAPDH遺伝子のPCR増幅産物の分析に、5’末端のCがFAMで標識されたプローブ(配列番号6)を使用した結果、(B)は、CHS遺伝子のPCR増幅産物の分析に、5’末端のAがFAMで標識されたプローブ(配列番号5)を使用した結果である。また、(C)と(D)は、同じPCR反応液についての結果であり、(C)は、GAPDH遺伝子のPCR増幅産物の分析に、5’末端のTがFAMで標識されたプローブ(配列番号7)を使用した結果、(D)は、CHS遺伝子のPCR増幅産物の分析に5’末端のAがFAMで標識されたプローブ(配列番号5)を使用した結果である。同図(A)および(B)、ならびに、(C)および(D)に示すように、一つのPCR反応液において2つの遺伝子の増幅を行った場合でも、両方のターゲット物質をそれぞれ検出することができた。なお、同図(A)で検出されたピークは「FAM+C」、同図(B)および(D)で検出されたピークは「FAM+A」、同図(C)で検出されたピークは「FAM+T」である。このため、本発明によれば、同一反応液において複数の遺伝子を分析することが可能であるといえる。
【実施例5】
【0085】
本発明の測定方法による定量性を確認するために、トレニアの葉と花弁におけるCHS発現量について、TaqMan(登録商標)プローブを用いたリアルタイムPCRとの比較を行った。
【0086】
(LC/MS分析)
まず、前記実施例3と同様の方法により、トレニアの葉と花弁とからTotal RNAを抽出し、それぞれの一本鎖cDNAを調製した。そして、トレニア葉由来cDNAおよびトレニア花弁cDNAを鋳型としてPCRを行い、前記実施例4と同様にしてLC/MS分析を行った。なお、PCR反応液BにおけるGAPDH用プローブとCHS用プローブの組み合わせも、前記実施例4と同様とした。
【0087】
(TaqManプローブを用いたリアルタイムPCR)
前述のLC/MS分析用に調製したトレニア葉由来cDNAおよびトレニア花弁由来cDNAを鋳型として、下記PCR反応液Cを用いて、下記条件CでPCRを行い、Gene Amp(登録商標)5700 Sequence Detection System(Applied Biosystems社製)により検出を行った。なお、標準化のため、前記LC/MS分析と同じプライマーセットおよび下記GAPDH用プローブを用いて、GAPDH遺伝子についてもリアルタイムPCRを行った。これらのデータ解析は、ABI PRISM7700Sequencing Detection System(Applied Biosystems社製)により行った。この結果を参考例とした。
【0088】
CHS用プローブ
5’−FAM−AGACCGTTGTGCTG−MGB−3’(配列番号5)
GAPDH用プローブ
5’−FAM−CTGCTATCAAGGAG−MGB−3’(配列番号6)
【0089】
【表5】
【0090】
【表6】
【0091】
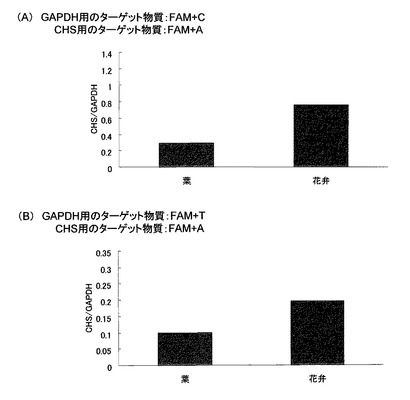
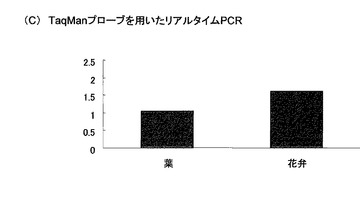
これらの結果を、図5および図6に示す。図5は、葉および花弁由来cDNAを鋳型とするPCR増幅産物についてLC/MS分析を行った実施例5の結果であり、同図(A)は、配列番号5(CHS用)および配列番号6(GAPDH用)のプローブを用いたCSH遺伝子/GAPDH遺伝子の結果であり、同図(B)は、配列番号5(CHS用)および配列番号7(GAPDH用)のプローブを用いたCSH遺伝子/GAPDH遺伝子の結果である。また、図6(C)は、リアルタイムPCRによるCHS遺伝子/GAPDH遺伝子の結果(参考例)である。
【0092】
図5(A)および(B)に示すように、LC/MS分析による実施例5の結果は、いずれのプローブを用いた場合も、同様の傾向、すなわち、葉と比較して花弁の方がGAPDH遺伝子よりもCHS遺伝子が多く発現しているという傾向を示した。さらに、図6(C)に示す、TaqManプローブを用いたリアルタイムPCRの結果(参考例)とも同じ挙動を示した。これらの結果より、本発明は、従来のリアルタイムPCRの代替法となり得ることが確認できた。さらに、本発明によれば、同一PCR反応液内で複数の標的遺伝子を増幅させた場合であっても、それぞれの増幅産物を検出可能であることがわかった。
【産業上の利用可能性】
【0093】
以上のように、本発明の検出方法では、質量分析によって、PCR反応において遊離する標識化物質結合断片の有無や量を検出し、間接的に、標識配列に相補的な伸長DNA鎖を検出する。質量分析は、非常に定量性に優れることから、本発明は、PCRの増幅産物の新たな検出方法となる。また、蛍光物質を使用した蛍光強度の測定とは異なり、質量分析においては、例えば、クロストーク等の問題がない。このため、同一反応液中の複数の伸長DNA鎖を検出する場合でも、各標的配列に対応する標識化プローブから遊離する標識化物質結合断片が、例えば、異なる質量や異なるm/z値等であれば、質量分析によって各々を検出することが可能である。したがって、本発明の検出方法によれば、分析対象試料について、複数の標的配列の定性・定量を行う場合であっても、同一反応液において複数の標的配列に対するPCRを行うことができ、且つ、互いの影響をうけることなく、各々を高精度で分析することができる。このため、本発明は、病理学や臨床学、分子生物学等の様々な分野において、極めて有用な方法といえる。
【図面の簡単な説明】
【0094】
【図1】図1は、本発明の一実施例であり、標識化物質とヌクレオチド1分子との結合体(ターゲット物質)に関するLC/MS分析の結果を示すグラフである。
【図2】図1は、本発明のその他の実施例であり、ヌクレオチドの塩基が異なる4種類のターゲット物質に関するLC/MS分析の結果を示すクロマトグラムである。
【図3】図3は、本発明のさらにその他の実施例であり、トレニア由来cDNAを鋳型とするPCR産物のLC/MS分析の結果を示すクロマトグラムである。
【図4】図4は、本発明のさらにその他の実施例であり、トレニア由来cDNAを鋳型とするPCR産物のLC/MS分析の結果を示すクロマトグラムである。
【図5】図5は、本発明のさらにその他の実施例であり、トレニア由来cDNAを鋳型とするPCR産物のLC/MS分析の結果を示すグラフである。
【図6】図6は、参考例であり、トレニア由来cDNAを鋳型とするリアルタイムPCRの結果を示すグラフである。
【技術分野】
【0001】
本発明は、PCRにより増幅させた伸長DNA鎖の検出方法、および、PCRの鋳型となる標的配列の測定方法に関する。
【背景技術】
【0002】
病理学や臨床学の様々な分野において、遺伝子の発現解析、機能解析、診断等を目的としてポリメラーゼチェーンリアクション(PCR)法が広く利用されている。PCR法は、一般に、(1)二本鎖DNAから鋳型となる一本鎖DNAへの解離、(2)鋳型一本鎖DNAへのプライマーのアニーリング、(3)DNAポリメラーゼを用いた前記プライマーの伸長、という3ステップを1サイクルとし、このサイクルを繰り返すことによって、試料中の標的核酸に相補的なDNAを増幅する方法である。
【0003】
近年では、PCRにおける増幅産物の生成過程を経時的にモニタリングする、いわゆるリアルタイムPCRが、様々な用途に利用されている。リアルタイムPCRは、一般的に、PCR産物の増幅に伴って発光強度が増大する蛍光物質(もしくは発光強度が減少する蛍光物質)を共存させてPCRを行い、増加する発光強度(もしくは減少する蛍光強度)を、PCR機器と光学的な検出器とを組み合わせた装置で、経時的にモニタリングする方法である。このようなリアルタイムPCRによれば、例えば、1サイクルごとの増幅産物の定量や、増幅産物が所定量(閾値)に達する際のサイクル数をカウントすることが可能であり、さらに、それらの情報に基づいて、試料に含まれていた標的配列を定量することも可能である。特に、RNAの発現解析においては、RNAの発現量が低い場合でも、逆転写PCR法によってRNAからcDNAを合成し、目的のcDNAをリアルタイムPCRで増幅することによって、従来よりも正確な定量が可能となっている。
【0004】
リアルタイムPCRにおける蛍光検出法としては、インターカレーター法、Taq
Man(商標)プローブ法、ハイブリダイゼーション法等が広く採用されている。「インターカレーター法」は、二本鎖DNAにインターカレートし、励起光を照射すると蛍光を発するインターカレーター(例えば、商品名SYBER Green I)を使用する方法である。この方法では、相補鎖が伸長されれば、鋳型と相補鎖との二本鎖にインーカレーターが入り込み、励起光照射によって蛍光を発するため、増幅による蛍光強度の増加をモニタリングすればよい。また、「Taq Man(商標)プローブ法」は、5’末端に蛍光物質を、3’末端にクエンチャーを結合させたプローブを使用する方法である。前記プローブは、鋳型にアニールしたのみでは、励起光を照射しても前記クエンチャーにより蛍光物質の蛍光は抑制されるが、相補鎖が伸長されると、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性により前記プローブが分解され、蛍光物質がクエンチャーから離れることにより蛍光を発する。このため、この方法では、増幅による蛍光強度の増加をモニタリングすればよい。「ハイブリダイゼーション法」は、3’末端がアクセプター蛍光物質で標識化されたプローブと、5’末端がドナー蛍光物質で標識化されたプローブとを用いる方法である。前記二種類のプローブは、鋳型にアニールすると、アクセプター蛍光物質とドナー蛍光物質とが隣接して蛍光が発生するが、相補鎖が伸長されると、アクセプター蛍光物質で標識されたプローブが分解され、アクセプター蛍光物質がドナー蛍光物質から離れることにより蛍光を発生しなくなる。したがって、この方法では、増幅による蛍光強度の減少を測定すればよい。
【0005】
しかしながら、前述のようなリアルタイムPCRは、スループットや定量性に優れるものの、蛍光検出が必須であることから、クロストーク(漏話)等が問題視されている。特に、1つの反応液中で二種類以上の標的核酸についてPCRを行った場合、それぞれの標的核酸に対する相補的伸長鎖を検出するには、各プローブについて異なる検出波長の蛍光物質を結合させる必要がある。しかし、各蛍光物質の最大吸収波長が異なっていても、例えば、スペクトルの端部が重複するおそれがあるため、互いの影響を受けることなく、それぞれの発現量を正確に測定することが困難である。また、スペクトルが重複しない蛍光物質を選択する方法も考えられるが、そのような蛍光物質の組み合わせには限りがあるため、一つの反応液中で所望の複数種類の相補的伸長鎖を分析することは現実的ではない。
【非特許文献1】BioTechniques, 27, 342−349.(1999) “Seven-color, homogeneous detection of six PCR products.” Lee,L.G., Livak,K.J., Mullah,B., Graham,R.J., Vinayak,R.S. and Woudenberg,T.M.
【非特許文献2】Nucleic Acids Research, 2006, Vol.34, No.1 “TaqMan probe array for quantitative detection of DNA targets” Heping Liu1, Hong Wang, Zhiyang Shi, Hua Wang, Chaoyong Yang, Spering Silke, Weihong Tan and Zuhong Lu
【発明の開示】
【発明が解決しようとする課題】
【0006】
そこで、本発明は、PCRの増幅産物を検出する新たな方法の提供を目的とする。より詳細には、定量性に優れ、且つ、同一反応液中の複数の増幅産物をそれぞれ検出することが可能な、新たなPCR増幅産物の検出方法の提供を目的とする。
【課題を解決するための手段】
【0007】
前記目的を達成するために、本発明の検出方法は、PCRによって増幅した、標的配列に相補的な伸長DNA鎖を検出する方法であって、下記(A)〜(F)工程を含むことを特徴とする。
(A) 標的配列、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有するDNAポリメラーゼ、プライマー、ならびに、オリゴヌクレオチドプローブに標識化物質が結合した標識化プローブを含む反応液を準備する工程
(B) 前記標的配列を鋳型として、これに前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(C) DNAポリメラーゼのポリメラーゼ活性によって、前記標的配列にアニーリングしたプライマーから前記標的配列に相補的なDNA鎖を5’→3’方向に向かって伸長させ、且つ、前記DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質が結合した断片(以下、「標識化物質結合断片」という)を遊離させる工程
(D) 前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(E) 前記工程(C)および工程(D)を繰り返し行うことによって、前記標識配列に相補的な伸長DNA鎖を増幅する工程
(F) 前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質が結合した断片の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する工程
【0008】
また、本発明の測定方法は、PCRによって増幅した、標識配列に相補的な伸長DNA鎖の検出により、標的配列を測定する方法であって、下記(G)および(H)工程を含むことを特徴とする。
(G) 本発明の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出する工程
(H) 前記(G)工程の検出結果に基づいて、前記反応液における標的配列の定性または定量を行う工程
【発明の効果】
【0009】
本発明の検出方法では、質量分析によって、PCR反応において遊離する標識化物質結合断片の有無や量を検出し、間接的に、標識配列に相補的な伸長DNA鎖を検出する方法である。質量分析は、非常に定量性に優れることから、これを利用した本発明は、定量性に優れる新たな増幅産物の検出方法といえる。また、蛍光物質を使用した蛍光強度の測定とは異なり、質量分析においては、例えば、クロストーク等の問題がない。このため、同一反応液中の複数の伸長DNA鎖を検出する場合でも、各標的配列に対応する標識化プローブから遊離する標識化物質結合断片のそれぞれが、例えば、イオン化によって、異なる分子イオンのピークを示したり、異なるフラグメントイオンピークを示せば、質量分析によって各々を検出することが可能である。したがって、本発明の検出方法によれば、分析対象試料について、複数の標的配列の定性・定量を行う場合であっても、同一反応液において複数の標的配列に対するPCRを行うことができ、且つ、互いの影響を受けることなく、各々の伸長DNA鎖を高精度で分析することができる。このため、本発明は、病理学や臨床学、分子生物学等の様々な分野において、極めて有用な方法といえる。なお、PCRにより増幅した伸長DNA鎖自体を質量分析により検出することは試みられているが、本発明のように、PCR反応によって標識化プローブより遊離する標識化物質結合断片を質量分析により検出することは、本発明者らが初めて見出した手法である。
【発明を実施するための最良の形態】
【0010】
本発明の検出方法は、前述のように、PCRによって増幅した、標的配列に相補的な伸長DNA鎖を検出する方法であって、下記(A)〜(F)工程を含むことを特徴とする。
(A) 標的配列、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有するDNAポリメラーゼ、プライマー、ならびに、オリゴヌクレオチドプローブに標識化物質が結合した標識化プローブを含む反応液を準備する工程
(B) 前記標的配列を鋳型として、これに前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(C) DNAポリメラーゼのポリメラーゼ活性によって、前記標的配列にアニーリングしたプライマーから前記標的配列に相補的なDNA鎖を5’→3’方向に向かって伸長させ、且つ、前記DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質結合断片を遊離させる工程
(D) 前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(E) 前記工程(C)および工程(D)を繰り返し行うことによって、前記標識配列に相補的な伸長DNA鎖を増幅する工程
(F) 前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質結合断片の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する工程
【0011】
PCRにおける標的配列は、特に制限されないが、例えば、DNAやRNA(mRNA、Total RNA)の他に、RNAから逆転写により合成したcDNA等があげられる。特に、RNAから合成したcDNAを標的配列とすれば、例えば、低発現のRNAの有無を間接的に検出することが可能である。
【0012】
前記標識化物質としては、何ら制限されず、あらゆる物質が使用できる。例えば、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、標識化プローブから標識化物質のみが切断されるものでないことが好ましい。前記標識化物質としては、例えば、イオン化可能な有機化合物が好ましく、より好ましくはイオン化可能な芳香族有機化合物である。具体例としては、例えば、6−カルボキシフルオレセイン(FAM)等のフルオレセイン、ローダミン、ビオチン等があげられるが、これらは本発明における一例であり、何ら制限されない。
【0013】
オリゴヌクレオチドプローブにおいて、標識化物質を結合させる基は、特に制限されず、例えば、塩基、糖、リン酸のいずれの基でもよい。また、前記(F)工程においては、標識化プローブの分解物のうち標識化物質が結合した断片を、質量分析によって検出することから、前記標識化物質は、少なくとも前記(C)工程においてDNAポリメラーゼにより遊離されることとなる部位に結合していればよい。中でも、DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によれば、通常、プローブの5’側から断片が遊離するため、例えば、前記オリゴヌクレオチドプローブの5’末端に結合させることが好ましい。
【0014】
前記(C)工程において、標識化プローブの分解により遊離する標識化物質結合断片(以下、質量分析の対象となることから、「ターゲット物質」ともいう)としては、例えば、前記標識化物質とヌクレオチド1分子とが結合した結合体であることが好ましい。このヌクレオチドは、前記標識化プローブにおいて、前記標識化物質が結合した部位のヌクレオチドである。遊離する断片が、前記標識化物質とヌクレオチド1分子との結合体であることは、特に、1つのPCR反応液で、二種類以上の伸長DNA鎖をそれぞれ検出する際に好ましい。この理由については後述する。遊離する断片としては、この他に、前記標識化物質がオリゴヌクレオチドに結合した結合体であってもよい。なお、この結合体は標識化プローブの分解物であるため、前記結合体を構成するオリゴヌクレオチドは、少なくとも前記オリゴヌクレオチドプローブよりも短い配列となる。
【0015】
なお、本発明においては、5’→3’エキソヌクレアーゼ活性により遊離したヌクレオチドを「遊離ヌクレオチド」、遊離したオリゴヌクレオチドを「遊離オリゴヌクレオチド」、標識化物質を結合させるプローブを「オリゴヌクレオチドプローブ」、前記プローブの構成成分であるヌクレオチドを「構成ヌクレオチド」という。
【0016】
前記遊離する断片を、標識化物質が結合した「遊離ヌクレオチド」とするか、標識化物質が結合した「遊離オリゴヌクレオチド」とするかは、通常、使用するDNAポリメラーゼの種類に応じて決定できる。DNAポリメラーゼとしては、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有していれば、その種類は特に制限されず、例えば、従来公知の耐熱性細菌由来のDNAポリメラーゼがあげられる。DNAポリメラーゼは、例えば、自家調製してもよいし、市販のものを使用してもよい。具体例としては、テルムス・アクアティカス(Thermus aquaticus)由来DNAポリメラーゼ(米国特許第4,889,818号および同第5,079,352号)(商品名Taq DNAポリメラーゼ)、テルムス・テルモフィラス(Thermus thermophilus)由来DNAポリメラーゼ(WO91/09950)(rTth DNA polymerase)、ピロコッカス・フリオサス(Pyrococcus furiosus)由来DNAポリメラーゼ(WO92/9688)(Pfu DNA polymerase:Stratagenes社製)、テルモコッカス・リトラリス(Thermococcus litoralis)由来DNAポリメラーゼ(EP−A455430)(商標Vent:Biolab New England社製)等が商業的に入手可能である。中でも、5’末端からヌクレオチドを1分子ずつ遊離させる場合は、テルムス・アクアティカス由来DNAポリメラーゼ(商品名Taq DNAポリメラーゼ)等を使用することが好ましい。なお、DNAポリメラーゼが、ヌクレオチドを遊離するかオリゴヌクレオチドを遊離するかが不明の場合でも、例えば、予め確認すれば足り、当業者であれば容易に行うことができる。
【0017】
本発明の検出方法によれば、一種類の伸長DNA鎖を検出するのはもちろんのこと、一つの反応液において二種類の伸長DNA鎖を増幅させた場合であっても、それぞれの伸長DNA鎖を検出することができる。以下に、一種類の伸長DNA鎖の検出方法ならびに二種類以上の伸長DNA鎖の検出方法のそれぞれについて、例をあげて説明する。なお、本発明は、これらには限定されない。
【0018】
まず、一種類の伸長DNA鎖の検出方法の例について説明する。
【0019】
実施形態1
(A)PCR反応液の準備
標的配列を含むDNA、DNAポリメラーゼ、dNTP(ヌクレオシド三リン酸)、前記標的配列に特異的なプライマー、ならびに、標識化プローブを含む反応液を準備する。
【0020】
前記標識化プローブとしては、特に制限されないが、前述のようにオリゴヌクレオチドプローブの5’末端に標識化物質を結合させたものが好ましい。このように5’末端に標識化物質が結合した標識化プローブであれば、例えば、DNAポリメラーゼがヌクレオチドを1分子ずつ遊離する場合であっても、2mer以上のオリゴヌクレオチドを遊離させる場合であっても、確実に、はじめに遊離した断片が標識化物質を有することとなる。本発明においては、この標識化物質が結合した遊離断片の有無または量が、伸長DNA鎖の増幅の有無または増幅量に相当するため、はじめに遊離した断片を検出できれば、分析精度を十分に確保できる。
【0021】
標識化プローブならびにプライマーの配列ならびに長さは、特に制限されず、標的配列に応じて、従来公知の方法により適宜決定できる。また、本発明では、後述する(C)工程において、DNAポリメラーゼによって、プライマーからの伸長反応を行うとともに、標的配列にアニーリングした標識化プローブを5’末端から分解するが、この反応自体は、例えば、Taq Man(商標)Probe法等において公知であることから、プライマーならびにプローブの長さや配列は、当業者であれば技術常識に基づいて設計可能である。通常、標識化プローブは、プライマーのアニーリング部位よりも5’側(鋳型の5’側)にアニーリングするように設計することが好ましい。なお、プライマーとしては、フォワードプライマーのみでもよいが、フォワードプライマーとリバースプライマーとを含むプライマーセットを用いることが好ましい。また、プライマーの長さは、一般的に10〜50mer程度であり、プローブの長さは、一般的に20〜30merであるが、これらには制限されない。
【0022】
前記PCR反応液中のプライマーの添加割合は、特に制限されないが、例えば、フォワードプライマーとリバースプライマーとが、それぞれ0.01〜5μmol/Lであり、好ましくは0.1〜3μmol/Lであり、より好ましくは0.1〜1μmol/Lである。また、前記PCR反応液中の標識化プローブの添加割合は、特に制限されないが、例えば、0.01〜5μmol/Lであり、好ましくは0.1〜1μmol/Lであり、より好ましくは0.2〜0.5μmol/Lである
【0023】
また、DNAポリメラーゼとしては、例えば、5’→3’エキソヌクレアーゼ活性により、ヌクレオチドを1分子ずつ遊離するものでも、2mer以上のオリゴヌクレオチドを遊離させるものでもよいが、前者が好ましい。このようなDNAポリメラーゼとしては、例えば、前述のようなテルムス・アクアティカス由来DNAポリメラーゼ(商品名Taq DNAポリメラーゼ)があげられる。
【0024】
前記PCR反応液中のDNAポリメラーゼの添加割合は、特に制限されないが、例えば、5〜50U/mlであり、好ましくは1〜100U/mlであり、より好ましくは20〜30U/mlである。なお、DNAポリメラーゼの活性単位(U)は、一般に、活性化サケ精子DNAを鋳型プライマーとして、活性測定用反応液(25mM TAPS buffer(pH9.3、25℃)、50mM KCl、2mM MgCl2、1mMメルカプトエタノール、200μM dATP、200μM dGTP、200μM dTTP、100μM「α−32P」dCTP、0.25mg/ml活性化サケ精子DNA)中、74℃で、30分間に10nmolの全ヌクレオチドを酸不溶性沈殿物に取り込む活性が1Uである。
【0025】
前記dNTPとしては、通常、dATP、dCTP、dTTPの混合物があげられる。前記PCR反応液中のdNTPの添加割合(合計添加割合)は、特に制限されないが、例えば、0.01〜1mmol/Lであり、好ましくは0.05〜0.5mmol/Lであり、より好ましくは0.1〜0.3mmol/Lである。
【0026】
前記PCR反応液の溶媒としては、特に制限されないが、例えば、Tricine、MES、MOPS、HEPES、CAPS等の緩衝液、市販のPCR用緩衝液や市販のPCRキットの緩衝液等が使用できる。反応液のpHは、例えば、7〜9である。
【0027】
(B)〜(E)PCR工程
つぎに、標的配列を鋳型としてPCR反応を行う。この反応自体は、特に制限されず、従来公知の方法に基づいて行うことができる。
【0028】
すなわち、まず、(B):鋳型である前記標的配列に、プライマーおよび標識化プローブをアニーリングさせる。鋳型である前記標的配列が二本鎖DNAを形成している場合には、この(B)工程に先立って、例えば、熱処理等によって一本鎖DNAへの解離を行うことが好ましい。そして、(C):DNAポリメラーゼによって、前記プライマーから前記標的配列に相補的なDNA鎖を伸長させ、且つ、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質結合断片を遊離させる。さらに、(D):前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび標識化プローブをアニーリングさせてから、前記(C)工程および(D)工程を繰り返す。このように、一本鎖DNAへの解離、アニーリングおよびDNAポリメラーゼ反応の3ステップを1サイクルとして、繰り返し行うことによって、標的配列に相補的な伸長DNA鎖を増幅することができる。前記(C)工程および(D)工程のサイクル数は、特に制限されないが、例えば、30サイクル以上であれば、標的配列に相補的な伸長DNA鎖の増幅を十分に確認することができ、好ましくは35サイクル以上、より好ましくは40サイクル以上である。なお、本発明においては、最終サイクルの(D)工程は割愛することができる。
【0029】
二本鎖DNAから一本鎖DNAへの解離の処理条件は、特に制限されないが、例えば、温度93〜95℃、時間15〜60秒であり、好ましくは温度94℃、時間30秒であり、アニーリングの処理条件は、特に制限されないが、例えば、温度(Tm値−5℃)〜(Tm値−2℃)、時間20〜60秒であり、好ましくは温度(Tm値−4℃、時間45秒であり、DNAポリメラーゼ反応は、特に制限されないが、例えば、温度60〜80℃、時間1〜45秒であり、好ましくは温度65〜75℃、時間30秒である。なお、各ステップにおける処理温度や処理時間は、例えば、サーマルサイクラー等の装置を用いて自動的に制御することができる。
【0030】
(F)標識化物質が結合した断片の質量分析による検出
そして、前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質結合断片の有無または量を検出することによって、標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する。
【0031】
前記(B)工程において、標識化プローブは、プライマーのアニーリング部位よりも5’側(鋳型の5’側)にアニーリングする。そして、前記(C)工程において、DNAポリメラーゼメラーゼは、プライマーを開始部位として5’→3’方向に伸長反応を進めると同時に、鋳型の3’→5’方向に向かって移動する。そして、移動したDNAポリメラーゼが、プライマーのアニーリング位置より5’側の標識化プローブのアニーリング部位に近づくと、5’→3’エキソヌクレアーゼ活性により、標識化プローブを5’側から切断していく。他方、ポリメラーゼ活性による伸長反応が起こらなかった場合には、DNAポリメラーゼも標識化プローブのアニーリング部位に近づかないため、エキソヌクレアーゼ活性による標識化プローブの切断は生じない。したがって、5’→3’エキソヌクレアーゼ活性により切断された断片の有無を検出すれば、それは、ポリメラーゼ活性により相補的な伸長DNA鎖が合成されたことを間接的に検出することとなる。このため、遊離する前記標識化物質結合断片の有無や量を検出すれば、伸長鎖DNAの有無や量を検出することができる。
【0032】
遊離する断片の長さは、前述のようにDNAポリメラーゼの種類によって異なるが、5’末端からヌクレオチドを1分子ずつ遊離するDNAポリメラーゼを使用した場合、前記(C)工程において、標識化プローブの5’末端ヌクレオチドの3’側が切断され、標識化物質が結合した前記5’末端ヌクレオチドが遊離する。具体例としては、標識化プローブの5’末端ヌクレオチドがデオキシアデノシンリン酸であれば、デオキシアデノシンリン酸に標識化物質が結合した結合体が遊離される。同様に、5’末端ヌクレオチドにおける塩基の種類によって、デオキシグアノシンリン酸、デオキシシチジンリン酸またはデオキシチミジンリン酸に標識化物質が結合した結合体が遊離される。
【0033】
前記反応液は、例えば、質量分析に供する前に脱塩処理等の不純物除去処理を施すことが好ましい。これによって分析精度をさらに向上することができる。
【0034】
質量分析の方法は、何ら制限されず、従来公知の方法が採用できる。質量分析におけるイオン化法としては、例えば、ESI(Electro Spray Ionization)法、LD(Laser Desorption Ionization)法、MALDI(Matrix Assisted Laser Desorption Ionization)法、EI(Electron Impact)法、CI(Chemical Ionization)法、API(atmospheric pressure Ionization)法、FI(Field Ionization)法、FD(Field Desorption)法、SIMS(Secondary Ion Mass Spectroscopy)法、FAB(Fast Atom Bonbardment)法、TSP(thermospray Ionization)法等があげられ、特に制限されないが、中でもESI法が好ましい。
【0035】
質量分析において、前述のようなイオン化法により生成したイオンは、通常、磁場や電場により分離され、分析される。分析方法の原理としては、特に制限されないが、例えば、磁場単収束型、電場磁場二重収束型、四重極型、三次元四重極型、飛行時間(time of flight、TOF)型、イオンサイクロトロン共鳴(Ion cyclotron resonance、ICR)型、イオントラップ(Ion Trap、IT)型、フーリエ変換(FT)型等があげられる。
【0036】
質量分析は、通常、質量分析装置(MS)により行うことができる。前記MSは、通常、イオン化室、質量分析部およびイオン検出器を備え、一般に、磁場型質量分析装置、四重極質量分析装置等に大別されるが、本発明においては、従来公知のあらゆる装置を使用することができる。質量分析装置の種類は、例えば、必要とされる分解能に応じて選択可能であり、例えば、高分解能が必要な場合には、MS/MS法を採用したり、二重収束質量分析装置を使用することも好ましい。また、本発明における分析対象試料はPCR反応液であって、例えば、未反応のdNTPやタンパク質、他の分解物等が含まれるため、例えば、液体クロマトグラフィー(LC)を質量分析装置(MS)に連結させたLC−MSや、LC−MS/MS等を使用することが好ましい。これにより、分析精度をさらに向上することができる。
【0037】
質量分析は、例えば、PCRの反応終了時のみ、または、PCRの反応途中のみに行ってもよいし、経時的に行ってもよい。このように経時的に質量分析を行えば、増幅産物(増幅した伸長DNA鎖)の生成過程を経時的にモニタリングすることができ、いわゆる従来のリアルタイムPCRと同様の解析が可能となる。経時的に分析を行う場合、例えば、断続的な分析でもよいし、連続的な分析でもよい。このため、本発明において、(F)工程の実施時期は、何ら制限されず、例えば、(C)工程と共に行ってもよいし、(C)工程後に行ってもよく、また、(C)工程を繰り返し行う間、連続的または断続的に行ってもよい。
【0038】
なお、標的核酸がRNA(mRNA、total RNA)の場合には、通常、前記RNAから逆転写反応によりcDNAを生成させ、このcDNAを標的配列とすればよい。そして、これをPCRの鋳型として使用する以外は、前述と同様にしてPCRを行えばよい(いわゆる、Reverse Transcription PCR)。
【0039】
逆転写反応は、例えば、従来公知の逆転写酵素、前記RNAに対するプライマーおよびヌクレオシド三リン酸(dNTP)を含む逆転写反応液を使用することができる。逆転写反応の反応条件は、特に制限されないが、例えば、反応温度30〜70℃、反応時間1〜120分であり、好ましくは反応温度40〜60℃、反応時間10〜60分であり、より好ましくは反応温度45〜50℃、反応時間20〜40分である。
【0040】
つぎに、同一反応液中における二種類以上の伸長DNA鎖の検出方法の例について説明する。
【0041】
同一反応液において、二種類以上の標的配列に相補的なDNA鎖をそれぞれ伸長させた場合、各伸長DNA鎖を検出することが必要となる。本発明は、遊離した標識化物質結合断片を質量分析で検出することによって、間接的に伸長DNA鎖を検出できる。したがって、本発明において二種類以上の伸長DNA鎖を検出する場合には、各標識化プローブから遊離される各標識化物質結合断片が、例えば、イオン化によって、異なる分子イオンピークを示したり、異なるフラグメントピークを示すもの等であれば、ターゲット物質とすることができる。なお、各標識化物質結合断片の質量が同じであっても、例えば、各フラグメントイオンが異なるm/z値にピークを示せば、それぞれを検出することが可能である。
【0042】
具体例としては、前記(C)工程において、二種類以上の標的配列に対応する各標識化プローブの分解により、前記標識化物質が結合した断片がそれぞれ遊離するが、これらの各断片が、それぞれ異なる質量であることが好ましい。各標識化物質結合断片の質量を変化させる方法は、特に制限されないが、例えば、異なる標識化物質を使用する方法、標識化物質結合断片として、異なる塩基のヌクレオチドと標識化物質との結合体を遊離させる方法、さらに、これらを組み合わせた方法等があげられる。これらの方法について、以下に具体例をあげるが、本発明は、これらには制限されない。
【0043】
実施形態2−1
この実施形態は、各標的配列に対応する標識化プローブについて、それぞれ質量が異なる標識化物質を使用する形態である。なお、本実施形態においては、オリゴヌクレオチドの5’末端に標識化物質が結合された標識化プローブであり、5’→3’エキソヌクレアーゼ活性によって、5’末端ヌクレオチド1分子と標識化物質との結合体が遊離する例をあげて説明する。
【0044】
本実施形態においては、前記(A)工程における反応液として、二種類以上の標的配列、および、前記各標的配列に対応する二種類以上のプライマーならびに二種類以上の標識化プローブを含む反応液を使用する。なお、各標的配列に対応するプライマーの配列や長さは、特に制限されず、例えば、標的配列に応じて、従来公知の方法によって適宜決定できる。
【0045】
本実施形態のように、異なる質量の標識化物質を使用する場合、例えば、各標識化プローブにおける5’末端のヌクレオチドは、同じ塩基のヌクレオチドであってもよい。例えば、各標識化プローブの5’末端ヌクレオチドが同じデオキシアデノシンリン酸であっても、5’末端に結合した標識化物質が質量の異なる物質であれば、遊離する標識化物質結合断片の全体の質量は、それぞれ異なる。したがって、各標的配列について相補的なDNA鎖を伸長する場合、各標識化プローブの標識化物質を異なる質量とすることで、各伸長DNA鎖を質量分析によって検出することが可能となる。
【0046】
質量分析では、例えば、1原子質量単位が異なるのみであっても、別の物質として検出することができる。このため、本実施形態においては、質量が異なれば、どのような標識化物質でも使用することができるため、同一反応液において検出できる伸長DNA鎖の数は、何ら制限されない。なお、検出できる伸長DNA鎖の数としては、特に制限されないが、下限は、例えば、2以上であり、上限は、例えば、100以下、好ましくは20以下、より好ましくは10以下である。
【0047】
実施形態2−2
この実施形態は、各標的配列に対応する標識化プローブにおいて、各標識化物質が同じ質量であり、各標識化物質結合断片として、異なる塩基のヌクレオチドと標識化物質との結合体を遊離させる形態である。なお、本実施形態においては、オリゴヌクレオチドの5’末端に標識化物質が結合された標識化プローブであり、5’→3’エキソヌクレアーゼ活性によって、5’末端ヌクレオチド1分子と標識化物質との結合体が遊離する例をあげて説明する。
【0048】
本実施形態においては、前記実施形態2−1と同様に、前記(A)工程における反応液として、二種類以上の標的配列、および、前記各標的配列に対応する二種類以上のプライマーならびに二種類以上の標識化プローブを含む反応液を使用する。なお、各標的配列に対応するプライマーの配列や長さは、特に制限されず、標的配列に応じて、従来公知の方法によって適宜決定できる。
【0049】
本実施形態では、前述のように、前記標識化物質結合断片におけるヌクレオチドの塩基の違いによって、各断片を検出する。したがって、各標識化プローブにおいて、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基であればよい。例えば、各標識化プローブの標識化物質が同じであっても、5’末端ヌクレオチドの塩基が異なれば、遊離する標識化物質結合断片の全体質量はそれぞれ異なる。したがって、各標的配列について、相補的なDNA鎖を伸長する場合、各標識化プローブにおいて、標識化物質を結合するヌクレオチドの塩基を異なる塩基に設定することで、それぞれを質量分析により検出することが可能となる。具体例としては、オリゴヌクレオチドプローブを、その5’末端ヌクレオチドが、デオキシアデノシンリン酸、デオキシグアノシンリン酸、デオキシシチジンリン酸またはデオキシチミジンリン酸となるように設計し、これに標識化物質を結合して標識化プローブを調製すればよい。このように5’末端ヌクレオチドの塩基を異なる塩基とすれば、各標識化プローブから遊離する断片は、質量が異なるため、同一反応液であっても、少なくとも4種類の伸長DNA鎖の検出を行うことが可能となる。
【0050】
実施形態2−3
この実施形態は、二種類以上の標的配列に対応する各標識化プローブとして、それぞれの標識化物質が異なる質量の物質であり、且つ、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基であるプローブを使用する形態である。このような形態であれば、標識化物質の組み合わせと塩基の組み合わせが使用できるため、同一反応液であっても、極めて多くの種類塩基の種類の数×標識化物質の種類の数)の伸長DNA鎖を検出できる。
【0051】
つぎに、本発明の測定方法は、PCRによって増幅した、標識配列に相補的な伸長DNA鎖の検出により、標的配列を測定する方法であって、下記(G)および(H)工程を含むことを特徴とする。
(G) 本発明の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出する工程
(H) 前記(G)工程の検出結果に基づいて、前記反応液における標的配列の定性または定量を行う工程
【0052】
本発明の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出すれば、その結果から、前記反応液における標的配列の有無(定性)または量(定量)を測定することができる。具体的には、前記(G)工程の検出結果に基づき、増幅した伸長DNA鎖を定量し、前記伸長DNA鎖が所定量となった際のPCRサイクル数をカウントすることによって、前記反応液における標的配列を定量することができる。
【0053】
また、反応液における標的配列がRNAから逆転写反応により生成させたcDNAであれば、cDNAを定性または定量することによって、その鋳型であるRNAの有無または量を定性もしくは定量することもできる。
【0054】
PCRでは、通常、増幅産物がある一定の量(閾値)に達するとそれ以上の増幅は起こらないため、最終的に生成した増幅産物の量を測定するのみでは、反応液に含まれる標的核酸の量を定量することは困難である。しかし、このようなリアルタイムPCR様の方法によれば、サイクルごとの増幅産物の生成をモニタリングし、設定した閾値に達するサイクル数(Ct値)をカウントするため、例えば、閾値とCt値とに基づいて、反応液に含まれる標的配列を定量することも可能である。
【0055】
つぎに、本発明の実施例について、比較例と併せて説明する。ただし、本発明は以下の実施例および比較例により制限されない。
【実施例1】
【0056】
LC/MS分析によって、1分子のヌクレオチドと標識化物質とが結合したターゲット物質を定量的に検出できるか否かを確認した。
【0057】
1.サンプルの調製
FAMにアデニンを有するヌクレオチド(デオキシアデノシンリン酸)を付加したターゲット物質(SIGMA GENOSYS社製、以下、「FAM+A」ともいう)を準備し、これを水で希釈した希釈系列をサンプルとした。なお、「FAM+A」の構造を下記式に表すが、後述する他のターゲット物質(FAM+ヌクレオチド)は、下記式における塩基が異なるのみで、その他は同様の構造である。
【化1】
【0058】
2.LC/MS分析
capillary LCシステムは、Ultimate、SwithosII、Famos(LC packings、日本ダイオネクス社製)を、質量分析計は、Esquire3000plus(Bruker Daltonics社製)をそれぞれ用いた。分析カラムはInertsil ODS−3 0.300mm i.d.×150mm(GL sciences社製)、プレカラムはμ−Precolumn(商標)Cartridge C18 0.300mm i.d.×5mm(LC packings社製)を使用した。前記「FAM+A」の各サンプル5μlをインジェクトし、0.1体積%ギ酸を用いて流速10μl/minで5分間プレカラムに送液することによって、前記サンプルを前記プレカラムに吸着させた。その後、バルブを切り替え、前記プレカラムに吸着したサンプルを分析カラムに導入した。この際の溶離液は、A液として、H2O(和光純薬社製)/アセトニトリル(キシダ化学、以下同じ)/ギ酸(体積比95/5/0.1)、B液として、H2O/アセトニトリル/ギ酸(体積比5/95/0.1)をそれぞれ使用し、A液とB液との混合液におけるB液の割合(体積%)が下記条件となるグラジェントで溶離を行った。前記溶離液の流量は、4μl/minとした。
【0059】
B液体積% 溶離時間
40% 0min〜10min
90% 10min〜25min
40% 25min〜35min
【0060】
(質量分析計の分析パラメーター)
polarity:negative mode
target mass:787m/z
scan range:100−1200m/z
capillary voltage:3500V
nebulizar gas:15.0psi
dry gas flow rate:8.0 l/min
dry gas temp.:300℃
ICC target:20000
average:7
【0061】
このLC/MS分析の結果を図1に示す。同図は、各サンプルにおけるターゲット物質の量とカウント(イオン強度)との相関関係を示すグラフである。同図に示すように、ターゲット物質とカウントとの関係を示す相関係数は0.9999と非常に高く、ターゲット物質の量が数百pg〜数百ngの間で優れた相関性を示した。このことから、「FAM+A」は、イオン化効率に優れ、ターゲット物質として本発明の方法に非常に適していると言える。
【実施例2】
【0062】
塩基のみが異なる4種類のターゲット物質を含む混合液についてLC/MS分析を行い、各ターゲット物質の検出が可能であるか否かを確認した。
【0063】
ターゲット物質として、FAMに異なる塩基(A,T、C、G)を有するヌクレオチド(デオキシアデノシンリン酸、デオキシチミジンリン酸、デオキシグアノシンリン酸、デオキシシチジンリン酸)を付加した4種類の物質を使用した。そして、4種類のターゲット物質をそれぞれ1ppmずつ含有する混合液を調製し、この混合液をサンプルとして、前記実施例1と同様にしてLC/MS分析を行い、Data Analysis(Bruker Daltonics社製)で解析した。なお、前記ターゲット物質は、塩基に応じてそれぞれ「FAM+A」、「FAM+T」、「FAM+C」および「FAM+G」という(以下、同様)。
【0064】
これらのLC/MS分析の結果を図2に示す。同図において、(A)はFAM+Cのクロマトグラム、(B)はFAM+Aのクロマトグラム、(C)はFAM+Gのクロマトグラム、(D)はFAM+Tのクロマトグラムである。これらのターゲット物質は、それぞれFAMに付加したヌクレオチドにおける塩基が異なるのみであるが、図2に示すように、それぞれ異なるリテンションタイムにピークが見られた。このことから、1つの反応液に塩基が異なる複数のターゲット物質(例えば、2〜4種類)が含まれる場合であっても、検出可能であることがわかった。
【実施例3】
【0065】
亜熱帯性植物Torenia hybrida(以下、「トレニア」という)の葉由来のmRNAよりcDNAを調製し、CHS(chalcon synthase)遺伝子およびGAPDH(glyceraldehyde−3−phosphate dehydrogenase)遺伝子を標的配列としてPCRを行い、増幅産物についてLC/MS分析を行った。
【0066】
1. RT−PCR
(1)cDNAの調製
トレニアの葉から、RNeasy(登録商標)Plant Mini Kit(QIAGEN社製)を用いて、TotalRNAを調製し、さらにDNaseIで処理した後、同キットを用いてTotalRNAを精製した。
【0067】
次いで、SuperScript(商標)First−Strand Synthesis System for RT−PCRキット(Invitrogen社)を用いて、以下に示す逆転写反応を行い、さらにRNaseHで処理して一本鎖cDNAを調製した。
【0068】
まず、精製した前記TotalRNAを用いて、全量5μgのTotal RNA溶液を調製した。これに、10mM dNTP mix(1μl)および0.5μg/μl Oligo(dT)12−18(1μl)を添加し、全量10μlとなるようにDEPC−treated waterを添加して、65℃で5分インキュベートした後、氷上で1分静置した。前記混合液に、さらに、10×RT buffer(2μl)、25mM MgCl2(4μl)、0.1M DTT(2μl)およびRNaseOUT(商標)Recombinant RNase Inhibitor(Invitrogen社製)(1μl)を順に添加し、42℃で2分インキュベートした。インキュベート後の混合液に、SuperScript(商標)II RT(1μl)を添加し、42℃で50分、70℃で15分の条件で順次インキュベートを行ってから、RNaseH(1μl)を添加した。これらの処理によって一本鎖cDNAを調製した。
【0069】
(2)PCR
調製した一本鎖cDNAを鋳型とし、下記プライマー、プローブおよびHotStarTaq(登録商標)PCR Master Mixキット(QIAGEN社製)を用いて、以下に示す条件で、CHS遺伝子とGAPDH遺伝子のそれぞれについてPCRを行った。なお、下記PCR反応液AおよびPCR条件Aは、CHS遺伝子およびGAPDH遺伝子の一方を検出する際の組成と条件である。
【0070】
CHS用プライマー
フォワードプライマー(配列番号1)
5’−ATACTGGACGAGATGAGGAA−3’
リバースプライマー (配列番号2)
5’−ATATCACCAGAATCATGCCA−3’
GAPDH用プライマー
フォワードプライマー(配列番号3)
5’−TGTCGATGTCTCCGTAGTGG−3’
リバースプライマー (配列番号4)
5’−CATCGTCTTCGGTATAGCCC−3’
【0071】
CHS用プローブ (配列番号5)
5’−FAM−AGACCGTTGTGCTG−MGB−3’
GAPDH用プローブ(配列番号6)
5’−FAM−CTGCTATCAAGGAG−MGB−3’
【0072】
【表1】
【0073】
【表2】
【0074】
2. LC/MS分析
得られた二種類のPCR増幅産物(CHS遺伝子の増幅産物およびGAPDH遺伝子の増幅産物)のそれぞれについて、LC/MS分析前、ziptip(商品名、SIGMA MILLIPORE社製)を用いて脱塩処理を行った。
【0075】
まず、ziptipをアセトニトリルで2回ピペッティング(ピペッティングは全て10μl、以下同様)してから、精製水で2回ピペッティングすることにより平衡化した。つぎに、前記PCR増幅産物(50μl)を5回ピペッティングし、PCR反応により遊離したターゲット物質(FAM+ヌクレオチド)をトラップさせてから、精製水で2回ピペッティングして平衡化した。続いて、精製水12.5μlとアセトニトリル12.5μlの混合液中で5回ピペッティングを行い、トラップした前記ターゲット物質を溶出させた(計25μl)。この溶出液に、内部標準物質として、1ppmの「FAM+G」(標準品、SIGMA
GENOSYS社製)5μlを添加して(計30μl)、分析用サンプルとした。
【0076】
この分析用サンプルを、前記実施例1と同様の条件でLC/MS分析に供し、その結果をData Analysis(Bruker Daltonics社製)で解析した。
【0077】
LC/MS分析の結果を図3に示す。同図において、(A)は、CHS遺伝子のPCR増幅産物の分析に、5’末端のAがFAMで標識されたプローブ(配列番号5)を使用した結果であり、(B)は、GAPDH遺伝子のPCR増幅産物の分析に、5’末端のCがFAMで標識されたプローブ(配列番号6)を使用した結果である。同図(A)および(B)に示すように、それぞれのPCR増幅産物について、異なるリテンションタイムでピークが検出された。同図(A)で検出されたピークは、「FAM+A」、同図(B)で検出されたピークは、「FAM+C」である。これらの結果から、LC/MS分析によって、PCR反応で遊離されたヌクレオチドが付加したターゲット物質を検出でき、この検出により、PCRにおける増幅の有無や増幅量を確認(定性、定量)できることがわかった。また、この結果から、サイクル数40で、十分にPCR増幅産物を検出できることが確認できた。
【実施例4】
【0078】
1つのPCR反応液内でCHS遺伝子およびGAPDH遺伝子のPCRを同時に行い、それぞれのPCR増幅産物をLC/MSにより分析した。
【0079】
1. RT−PCR
前記実施例3で調製した一本鎖DNAを鋳型として、下記PCR反応液Bを用いて、下記PCR条件BによりPCRを行った。なお、GAPDH用プローブとしては、以下に示す2種類のプローブを使用し、各プローブを用いて別個にPCRを行った。なお、下記PCR反応液BおよびPCR条件Bは、それぞれ1つの反応液でCHSおよびGAPDHの両方を検出するマルチプレックスPCRの組成ならびに条件である。
【0080】
【表3】
【0081】
GAPDH用プローブ
5’−FAM−CTGCTATCAAGGAG−MGB−3’(配列番号6)
5’−FAM−TCAAGGAGGAATCC−MGB−3’(配列番号7)
【0082】
【表4】
【0083】
そして、得られたPCR増幅産物について、前記実施例3と同様に脱塩処理を施した後、前記実施例1と同様にLC/MS分析を行い、その結果をData Analysis(Bruker Daltonics社製)で解析した。
【0084】
これらのLC/MS分析の結果を図4に示す。同図において、(A)と(B)は、同じPCR反応液についての結果、(A)は、GAPDH遺伝子のPCR増幅産物の分析に、5’末端のCがFAMで標識されたプローブ(配列番号6)を使用した結果、(B)は、CHS遺伝子のPCR増幅産物の分析に、5’末端のAがFAMで標識されたプローブ(配列番号5)を使用した結果である。また、(C)と(D)は、同じPCR反応液についての結果であり、(C)は、GAPDH遺伝子のPCR増幅産物の分析に、5’末端のTがFAMで標識されたプローブ(配列番号7)を使用した結果、(D)は、CHS遺伝子のPCR増幅産物の分析に5’末端のAがFAMで標識されたプローブ(配列番号5)を使用した結果である。同図(A)および(B)、ならびに、(C)および(D)に示すように、一つのPCR反応液において2つの遺伝子の増幅を行った場合でも、両方のターゲット物質をそれぞれ検出することができた。なお、同図(A)で検出されたピークは「FAM+C」、同図(B)および(D)で検出されたピークは「FAM+A」、同図(C)で検出されたピークは「FAM+T」である。このため、本発明によれば、同一反応液において複数の遺伝子を分析することが可能であるといえる。
【実施例5】
【0085】
本発明の測定方法による定量性を確認するために、トレニアの葉と花弁におけるCHS発現量について、TaqMan(登録商標)プローブを用いたリアルタイムPCRとの比較を行った。
【0086】
(LC/MS分析)
まず、前記実施例3と同様の方法により、トレニアの葉と花弁とからTotal RNAを抽出し、それぞれの一本鎖cDNAを調製した。そして、トレニア葉由来cDNAおよびトレニア花弁cDNAを鋳型としてPCRを行い、前記実施例4と同様にしてLC/MS分析を行った。なお、PCR反応液BにおけるGAPDH用プローブとCHS用プローブの組み合わせも、前記実施例4と同様とした。
【0087】
(TaqManプローブを用いたリアルタイムPCR)
前述のLC/MS分析用に調製したトレニア葉由来cDNAおよびトレニア花弁由来cDNAを鋳型として、下記PCR反応液Cを用いて、下記条件CでPCRを行い、Gene Amp(登録商標)5700 Sequence Detection System(Applied Biosystems社製)により検出を行った。なお、標準化のため、前記LC/MS分析と同じプライマーセットおよび下記GAPDH用プローブを用いて、GAPDH遺伝子についてもリアルタイムPCRを行った。これらのデータ解析は、ABI PRISM7700Sequencing Detection System(Applied Biosystems社製)により行った。この結果を参考例とした。
【0088】
CHS用プローブ
5’−FAM−AGACCGTTGTGCTG−MGB−3’(配列番号5)
GAPDH用プローブ
5’−FAM−CTGCTATCAAGGAG−MGB−3’(配列番号6)
【0089】
【表5】
【0090】
【表6】
【0091】
これらの結果を、図5および図6に示す。図5は、葉および花弁由来cDNAを鋳型とするPCR増幅産物についてLC/MS分析を行った実施例5の結果であり、同図(A)は、配列番号5(CHS用)および配列番号6(GAPDH用)のプローブを用いたCSH遺伝子/GAPDH遺伝子の結果であり、同図(B)は、配列番号5(CHS用)および配列番号7(GAPDH用)のプローブを用いたCSH遺伝子/GAPDH遺伝子の結果である。また、図6(C)は、リアルタイムPCRによるCHS遺伝子/GAPDH遺伝子の結果(参考例)である。
【0092】
図5(A)および(B)に示すように、LC/MS分析による実施例5の結果は、いずれのプローブを用いた場合も、同様の傾向、すなわち、葉と比較して花弁の方がGAPDH遺伝子よりもCHS遺伝子が多く発現しているという傾向を示した。さらに、図6(C)に示す、TaqManプローブを用いたリアルタイムPCRの結果(参考例)とも同じ挙動を示した。これらの結果より、本発明は、従来のリアルタイムPCRの代替法となり得ることが確認できた。さらに、本発明によれば、同一PCR反応液内で複数の標的遺伝子を増幅させた場合であっても、それぞれの増幅産物を検出可能であることがわかった。
【産業上の利用可能性】
【0093】
以上のように、本発明の検出方法では、質量分析によって、PCR反応において遊離する標識化物質結合断片の有無や量を検出し、間接的に、標識配列に相補的な伸長DNA鎖を検出する。質量分析は、非常に定量性に優れることから、本発明は、PCRの増幅産物の新たな検出方法となる。また、蛍光物質を使用した蛍光強度の測定とは異なり、質量分析においては、例えば、クロストーク等の問題がない。このため、同一反応液中の複数の伸長DNA鎖を検出する場合でも、各標的配列に対応する標識化プローブから遊離する標識化物質結合断片が、例えば、異なる質量や異なるm/z値等であれば、質量分析によって各々を検出することが可能である。したがって、本発明の検出方法によれば、分析対象試料について、複数の標的配列の定性・定量を行う場合であっても、同一反応液において複数の標的配列に対するPCRを行うことができ、且つ、互いの影響をうけることなく、各々を高精度で分析することができる。このため、本発明は、病理学や臨床学、分子生物学等の様々な分野において、極めて有用な方法といえる。
【図面の簡単な説明】
【0094】
【図1】図1は、本発明の一実施例であり、標識化物質とヌクレオチド1分子との結合体(ターゲット物質)に関するLC/MS分析の結果を示すグラフである。
【図2】図1は、本発明のその他の実施例であり、ヌクレオチドの塩基が異なる4種類のターゲット物質に関するLC/MS分析の結果を示すクロマトグラムである。
【図3】図3は、本発明のさらにその他の実施例であり、トレニア由来cDNAを鋳型とするPCR産物のLC/MS分析の結果を示すクロマトグラムである。
【図4】図4は、本発明のさらにその他の実施例であり、トレニア由来cDNAを鋳型とするPCR産物のLC/MS分析の結果を示すクロマトグラムである。
【図5】図5は、本発明のさらにその他の実施例であり、トレニア由来cDNAを鋳型とするPCR産物のLC/MS分析の結果を示すグラフである。
【図6】図6は、参考例であり、トレニア由来cDNAを鋳型とするリアルタイムPCRの結果を示すグラフである。
【特許請求の範囲】
【請求項1】
PCRによって増幅した、標的配列に相補的な伸長DNA鎖を検出する方法であって、下記(A)〜(F)工程を含むことを特徴とする伸長DNA鎖の検出方法。
(A) 標的配列、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有するDNAポリメラーゼ、プライマー、ならびに、オリゴヌクレオチドプローブに標識化物質が結合した標識化プローブを含む反応液を準備する工程
(B) 前記標的配列を鋳型として、これに前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(C) DNAポリメラーゼのポリメラーゼ活性によって、前記標的配列にアニーリングしたプライマーから前記標的配列に相補的なDNA鎖を5’→3’方向に向かって伸長させ、且つ、前記DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質が結合した断片を遊離させる工程
(D) 前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(E) 前記工程(C)および工程(D)を繰り返し行うことによって、前記標識配列に相補的な伸長DNA鎖を増幅する工程
(F) 前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質が結合した断片の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する工程
【請求項2】
前記標識化プローブが、オリゴヌクレオチドプローブの5’末端に標識化物質が結合したプローブである、請求項1記載の検出方法。
【請求項3】
前記(C)工程において遊離する前記標識化物質が結合した断片が、前記標識化物質とヌクレオチド1分子との結合体であり、
前記(F)工程において、質量分析により前記結合体の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または量を検出する、請求項1または2記載の伸長DNA鎖の検出方法。
【請求項4】
前記(A)工程において、前記反応液が、二種類以上の標的配列、および、前記各標的配列に対応する二種類以上のプライマーならびに二種類以上の標識化プローブを含む、請求項1から3のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項5】
前記(C)工程において、二種類以上の各標識化プローブから、前記標識化物質が結合した断片であり且つ異なる質量の断片を、それぞれ遊離させる、請求項4記載の伸長DNA鎖の検出方法。
【請求項6】
前記二種類以上の各標識化プローブにおいて、それぞれの標識化物質が、異なる質量の物質である、請求項4または5記載の伸長DNA鎖の検出方法。
【請求項7】
前記二種類以上の各標識化プローブにおいて、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基である、請求項4から6のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項8】
前記二種類以上の各標識化プローブにおいて、それぞれの標識化物質が異なる質量の物質であり、且つ、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基である、請求項4から7のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項9】
前記標識化物質が、フルオレセイン、ローダミンおよびビオチンからなる群から選択された少なくとも一つである、請求項1から8のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項10】
フルオレセインが、6−カルボキシフルオレセインである、請求項9記載の伸長DNA鎖の検出方法。
【請求項11】
質量分析におけるイオン化法が、ESI(Electro Spray Ionization)法、LD(Laser Desorption Ionization)法、MALDI(Matrix Assisted Laser Desorption Ionization)法、EI(Electron Impact)法、CI(Chemical Ionization)法、API(atmospheric pressure Ionization)法、FI(Field Ionization)法、FD(Field Desorption)法、SIMS(Secondary Ion Mass Spectroscopy)法、FAB(Fast Atom Bonbardment)法、および、TSP(thermospray Ionization)法からなる群から選択された少なくとも一つの方法である、請求項1から10のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項12】
前記(F)工程において、前記反応液を液体クロマトグラフィーに供した後に質量分析を行う、請求項1から11のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項13】
前記標的配列が、RNAを鋳型として逆転写反応により合成したcDNAである、請求項1から12のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項14】
前記(E)工程において、前記(C)工程のサイクル数が、5以上である、請求項1から13のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項15】
前記(E)工程において、経時的に、前記(F)工程における質量分析を行う、請求項1から14のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項16】
PCRによって増幅した、標識配列に相補的な伸長DNA鎖の検出により、標的配列を測定する方法であって、下記(G)および(H)工程を含むことを特徴とする測定方法。
(G) 請求項1から15のいずれか一項に記載の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出する工程
(H) 前記(G)工程の検出結果に基づいて、前記反応液における標的配列の定性または定量を行う工程
【請求項17】
前記(H)工程において、前記(G)工程の検出結果に基づき、増幅した伸長DNA鎖を定量し、前記伸長DNA鎖が所定量となった際のPCRサイクル数をカウントすることによって、前記反応液における標的配列を定量する、請求項16記載の測定方法。
【請求項18】
前記標的配列がRNAから逆転写反応により合成したcDNAである、請求項16または17記載の測定方法。
【請求項19】
さらに、下記(I)工程を含む、請求項18記載の測定方法。
(I) 前記標的配列であるcDNAを定量し、この結果に基づいて、前記逆転写反応においてcDNAの鋳型となったRNAを定量する工程
【請求項1】
PCRによって増幅した、標的配列に相補的な伸長DNA鎖を検出する方法であって、下記(A)〜(F)工程を含むことを特徴とする伸長DNA鎖の検出方法。
(A) 標的配列、ポリメラーゼ活性および5’→3’エキソヌクレアーゼ活性を有するDNAポリメラーゼ、プライマー、ならびに、オリゴヌクレオチドプローブに標識化物質が結合した標識化プローブを含む反応液を準備する工程
(B) 前記標的配列を鋳型として、これに前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(C) DNAポリメラーゼのポリメラーゼ活性によって、前記標的配列にアニーリングしたプライマーから前記標的配列に相補的なDNA鎖を5’→3’方向に向かって伸長させ、且つ、前記DNAポリメラーゼの5’→3’エキソヌクレアーゼ活性によって、前記標的配列にアニーリングした標識化プローブを分解して、前記標識化物質が結合した断片を遊離させる工程
(D) 前記(C)工程で生成した、前記標的配列とそれに相補的な伸長DNA鎖とからなる二本鎖を解離し、両者を鋳型として、前記プライマーおよび前記標識化プローブをアニーリングさせる工程
(E) 前記工程(C)および工程(D)を繰り返し行うことによって、前記標識配列に相補的な伸長DNA鎖を増幅する工程
(F) 前記反応液について質量分析を行い、前記(C)工程において遊離する前記標識化物質が結合した断片の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または増幅量を検出する工程
【請求項2】
前記標識化プローブが、オリゴヌクレオチドプローブの5’末端に標識化物質が結合したプローブである、請求項1記載の検出方法。
【請求項3】
前記(C)工程において遊離する前記標識化物質が結合した断片が、前記標識化物質とヌクレオチド1分子との結合体であり、
前記(F)工程において、質量分析により前記結合体の有無または量を検出することによって、前記標識配列に相補的な伸長DNA鎖の有無または量を検出する、請求項1または2記載の伸長DNA鎖の検出方法。
【請求項4】
前記(A)工程において、前記反応液が、二種類以上の標的配列、および、前記各標的配列に対応する二種類以上のプライマーならびに二種類以上の標識化プローブを含む、請求項1から3のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項5】
前記(C)工程において、二種類以上の各標識化プローブから、前記標識化物質が結合した断片であり且つ異なる質量の断片を、それぞれ遊離させる、請求項4記載の伸長DNA鎖の検出方法。
【請求項6】
前記二種類以上の各標識化プローブにおいて、それぞれの標識化物質が、異なる質量の物質である、請求項4または5記載の伸長DNA鎖の検出方法。
【請求項7】
前記二種類以上の各標識化プローブにおいて、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基である、請求項4から6のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項8】
前記二種類以上の各標識化プローブにおいて、それぞれの標識化物質が異なる質量の物質であり、且つ、標識化物質が結合するヌクレオチドの塩基がそれぞれ異なる塩基である、請求項4から7のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項9】
前記標識化物質が、フルオレセイン、ローダミンおよびビオチンからなる群から選択された少なくとも一つである、請求項1から8のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項10】
フルオレセインが、6−カルボキシフルオレセインである、請求項9記載の伸長DNA鎖の検出方法。
【請求項11】
質量分析におけるイオン化法が、ESI(Electro Spray Ionization)法、LD(Laser Desorption Ionization)法、MALDI(Matrix Assisted Laser Desorption Ionization)法、EI(Electron Impact)法、CI(Chemical Ionization)法、API(atmospheric pressure Ionization)法、FI(Field Ionization)法、FD(Field Desorption)法、SIMS(Secondary Ion Mass Spectroscopy)法、FAB(Fast Atom Bonbardment)法、および、TSP(thermospray Ionization)法からなる群から選択された少なくとも一つの方法である、請求項1から10のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項12】
前記(F)工程において、前記反応液を液体クロマトグラフィーに供した後に質量分析を行う、請求項1から11のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項13】
前記標的配列が、RNAを鋳型として逆転写反応により合成したcDNAである、請求項1から12のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項14】
前記(E)工程において、前記(C)工程のサイクル数が、5以上である、請求項1から13のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項15】
前記(E)工程において、経時的に、前記(F)工程における質量分析を行う、請求項1から14のいずれか一項に記載の伸長DNA鎖の検出方法。
【請求項16】
PCRによって増幅した、標識配列に相補的な伸長DNA鎖の検出により、標的配列を測定する方法であって、下記(G)および(H)工程を含むことを特徴とする測定方法。
(G) 請求項1から15のいずれか一項に記載の検出方法により、反応液に含まれる増幅した前記伸長DNA鎖を検出する工程
(H) 前記(G)工程の検出結果に基づいて、前記反応液における標的配列の定性または定量を行う工程
【請求項17】
前記(H)工程において、前記(G)工程の検出結果に基づき、増幅した伸長DNA鎖を定量し、前記伸長DNA鎖が所定量となった際のPCRサイクル数をカウントすることによって、前記反応液における標的配列を定量する、請求項16記載の測定方法。
【請求項18】
前記標的配列がRNAから逆転写反応により合成したcDNAである、請求項16または17記載の測定方法。
【請求項19】
さらに、下記(I)工程を含む、請求項18記載の測定方法。
(I) 前記標的配列であるcDNAを定量し、この結果に基づいて、前記逆転写反応においてcDNAの鋳型となったRNAを定量する工程
【図1】

【図2】
【図3】

【図4】
【図5】
【図6】
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2008−154498(P2008−154498A)
【公開日】平成20年7月10日(2008.7.10)
【国際特許分類】
【出願番号】特願2006−346582(P2006−346582)
【出願日】平成18年12月22日(2006.12.22)
【出願人】(801000061)財団法人大阪産業振興機構 (168)
【出願人】(504176911)国立大学法人大阪大学 (1,536)
【Fターム(参考)】
【公開日】平成20年7月10日(2008.7.10)
【国際特許分類】
【出願日】平成18年12月22日(2006.12.22)
【出願人】(801000061)財団法人大阪産業振興機構 (168)
【出願人】(504176911)国立大学法人大阪大学 (1,536)
【Fターム(参考)】
[ Back to top ]