
PCR増幅での再現性の改善およびミスプライミングの低減のための試薬および方法
【課題】ポリメラーゼ連鎖反応(PCR)増幅およびアッセイでのミスプライミングを防ぐための添加剤の提供。
【解決手段】6ヌクレオチド長より長いステム二重鎖と安定化されたステム末端とを有するヘアピンオリゴヌクレオチドを含んでなる添加剤に関する。当該添加剤は、初めの増幅反応混合物中に加えた場合、LATE−PCR増幅をはじめとするPCR増幅を改善する。当該添加剤は、PCR増幅およびアッセイのためのオリゴヌクレオチドセットならびにキットに含めることができる。
【解決手段】6ヌクレオチド長より長いステム二重鎖と安定化されたステム末端とを有するヘアピンオリゴヌクレオチドを含んでなる添加剤に関する。当該添加剤は、初めの増幅反応混合物中に加えた場合、LATE−PCR増幅をはじめとするPCR増幅を改善する。当該添加剤は、PCR増幅およびアッセイのためのオリゴヌクレオチドセットならびにキットに含めることができる。
【発明の詳細な説明】
【技術分野】
【0001】
技術分野
本発明は、ポリメラーゼ連鎖反応(PCR)を利用する核酸増幅反応およびアッセイに関し、それにはリアルタイムアッセイおよびエンドポイントアッセイの両者のホモジニアスアッセイが含まれる。
【背景技術】
【0002】
背景
ポリメラーゼ連鎖反応(PCR)を利用する核酸増幅(PCR増幅を含むアッセイなど)は周知である。米国特許第4,683,202号、同第4,683,195号および同第4,965,188号、および、一般には、PCR PROTOCOLS, a guide to Methods and Applications, Innis et al.(編), Academic Press(San Diego, CA (USA) 1990)を参照されたい。未結合検出試薬またはプローブを除去するための洗浄を必要とせず、ゆえに増幅反応容器を開けることなく実施できるホモジニアスPCRアッセイもまた周知である。ホモジニアスPCRアッセイには、増幅反応の終了時点で増幅産物を検出するエンドポイントアッセイ、および反応が進行しているときに一部のまたはすべての熱サイクル中に増幅産物を検出するリアルタイムアッセイの両者が含まれる。米国特許第5,994,056号、同第5,487,972号、同第5,925,517号および同第6,150,097号を参照されたい。
【0003】
PCR増幅反応は、概して、対称的であるように、すなわち、「対応する(matched)」フォワードプライマーおよびリバースプライマーを利用して二本鎖アンプリコンを作り出すように設計され;すなわち、それらは可能な限り近い融解温度を有し、かつそれらは等モル濃度で反応物に添加される。PCR反応において、直接的に一本鎖DNAを作り出すために限られた用途が見出されている技術は、「非対称的PCR」である。Gyllensten and Erlich, "Generation of Single-Stranded DNA by the Polymerase Chain Reaction and Its Application to Direct Sequencing of the HLA-DQA Locus," Proc. Natl. Acad. Sci. (USA) 85: 7652-7656 (1988);および米国特許第5,066,584号。非対称的PCRは、プライマーの一方が制限的な量で添加され、典型的には他方のプライマーの濃度の1〜20%の量で添加される点で対称的PCRと異なっている。
【0004】
さらに最近、本発明者らは、「直線後指数的(Linear-After-The-Exponential)」PCRまたは、略して「LATE−PCR」として知られる非対称的PCR増幅法を開発した。Sanchez et al. (2004) PNAS 101: 1933-1938, Pierce et al. (2005) PNAS 102: 8609-8614、および公開されている国際特許出願WO 03/054233(2003年7月3日)を参照のこと。前記文献は参照によりその全体が本明細書中に組み入れられる。LATE−PCRでは、Tm[0]と称される増幅の出発時点でのPCRプライマーの実際の融解温度を考慮に入れる。Tm[0]は、非天然ヌクレオチドが使用される場合に必要であるように、経験的に決定することができ、あるいは「近接(nearest neighbor)」法(Santa Lucia, J. (1998) PNAS (USA) 95: 1460-1465;およびAllawi, H. T. and Santa Lucia, J. (1997) Biochem. 36: 10581-10594)にしたがって、塩濃度の補正を使用して、算出することができる。本発明者らの研究では、0.07Mの一価塩濃度を利用した。
【0005】
LATE−PCRの場合には低減されるPCR増幅の望ましくない特徴は、反復実験(replicates)間のばらつき(scatter)である。対数期の増幅に続いてリアルタイムで追跡される反復実験増幅はそれぞれ相違し、種々のレベルでプラトーに達する。ばらつきとは、反復実験が同一の反応キネティクスを有さず、精度を低下させることを意味する。このことはPCRアッセイ全般に関する問題であるが、特にエンドポイントアッセイおよび直線期のシグナルの勾配に依存するアッセイに関して問題である。
【0006】
PCR増幅に伴う別の重大な問題はミスプライミングである。本発明者らの考えでは、ミスプライミングは以下の少なくとも3種のタイプで現れる:タイプ1、増幅開始前の反応混合物の調製中に生じるミスプライミング;タイプ2、サイクル温度がプライマーの融解温度よりかなり低いいずれかの温度を含む場合に、増幅中に生じるミスプライミング;およびタイプ3、高濃度のアンプリコンが作り出された後に継続されるPCR増幅の後期段階に生じるミスプライミング。第1のタイプのミスプライミングに対処するためにいくつかのアプローチが使用されている。アプローチの1つは、ポリメラーゼを化学的に修飾して、高温、例えば95℃に加熱されるまで不活性であるようにすることである。米国特許第5,677,152号および同第5,773,258号を参照されたい。別のアプローチは、ポリメラーゼに抗体を結合させ、反応物が高温、例えば95℃に加熱されて該抗体が不可逆的に変性するまで該ポリメラーゼを阻害することである。米国特許第5,338,671号を参照されたい。さらに別のアプローチは、反応混合物中にアプタマーを含ませることである。Doug and Jayasena (1996), J. Mol. Biol. 264: 268-278および米国特許第6,020,130号を参照のこと。アプタマーは約30ヌクレオチド長の一本鎖オリゴヌクレオチドであり、ポリメラーゼに結合して、低温で陥凹3’末端を伸長するその能力を阻害する。アプタマーは、PCRサイクルの典型的な最高温度である95℃で不可逆的に変性しない。Kainz et al. (2000) Biotechniques 28: 278-282では、16〜21ヌクレオチド長を有する、一定量の二本鎖DNA断片をPCR反応混合物に加えると、典型的なPCR伸長温度より低い温度でポリメラーゼが阻害され、非特異的産物の合成が抑制されることが報告された。DNA断片はPCRサイクル処理中に不可逆的に変性しない。Eppendorf-5 Prime, Inc.は、温度依存的様式でTaqポリメラーゼに結合し、約50℃より低い温度で二本鎖DNAに対するその結合を阻害するとされている専売のリガンドを市販している。前述の多数の試みにもかかわらず、ミスプライミングは依然としてPCR増幅に伴う問題である。
【0007】
PCR増幅中のミスプライミングの別の現象はプライマー二量体形成および増幅として知られている。この現象では、一方のプライマーが他方のプライマーまたは自身とハイブリダイズし、次いで3’末端の伸長を受けて、小さい二本鎖アンプリコンが生成される。その後、アンプリコンはさらに増幅されるか、あるいは多量体化されてさらに増幅されうる。プライマー二量体形成は標的の非存在下で生じうる。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第4,683,202号
【特許文献2】米国特許第4,683,195号
【特許文献3】米国特許第4,965,188号
【特許文献4】米国特許第5,994,056号
【特許文献5】米国特許第5,487,972号
【特許文献6】米国特許第5,925,517号
【特許文献7】米国特許第6,150,097号
【特許文献8】米国特許第5,066,584号
【特許文献9】国際特許出願WO 03/054233
【特許文献10】米国特許第5,677,152号
【特許文献11】米国特許第5,773,258号
【特許文献12】米国特許第5,338,671号
【特許文献13】米国特許第6,020,130号
【非特許文献】
【0009】
【非特許文献1】PCR PROTOCOLS, a guide to Methods and Applications, Innis et al.(編), Academic Press(San Diego, CA (USA) 1990)
【非特許文献2】Gyllensten and Erlich, "Generation of Single-Stranded DNA by the Polymerase Chain Reaction and Its Application to Direct Sequencing of the HLA-DQA Locus," Proc. Natl. Acad. Sci. (USA) 85: 7652-7656 (1988)
【非特許文献3】Sanchez et al. (2004) PNAS 101: 1933-1938
【非特許文献4】Pierce et al. (2005) PNAS 102: 8609-8614
【非特許文献5】Santa Lucia, J. (1998) PNAS (USA) 95: 1460-1465
【非特許文献6】Allawi, H. T. and Santa Lucia, J. (1997) Biochem. 36: 10581-10594
【非特許文献7】Doug and Jayasena (1996), J. Mol. Biol. 264: 268-278
【非特許文献8】Kainz et al. (2000) Biotechniques 28: 278-282
【発明の概要】
【発明が解決しようとする課題】
【0010】
リアルタイム検出方法によるPCR増幅の定量的解析が可能になっていて、その場合、反応の閾値サイクルすなわちCTを超えると蛍光シグナルが可視になるPCRサイクルにより出発標的濃度が示唆される。エンドポイント解析は良くても半定量的である。その部分的な原因は、反応が指数的増幅から脱する時の反復実験間のばらつきである。二本鎖アンプリコンの電気泳動による解析は半定量的であり、蛍光標識プライマーを利用する場合がある。蛍光標識プローブ、対立遺伝子識別プローブまたはミスマッチ寛容プローブを利用するエンドポイント解析もまた、良くても半定量的である。ばらつきを低減させ、一本鎖産物を生成することによって、LATE−PCRは、エンドポイント解析における大きな改善を提供するが、反復実験間のばらつきは完全には排除されないことが多く、定量的多重検出は依然として所望の精度より低精度である。
【課題を解決するための手段】
【0011】
本発明の一態様は、PCR増幅反応において生成物の特異性を改善し、かつミスプライミングの影響を排除する、あるクラスの試薬添加剤である。該添加剤はすべてのタイプのPCRにおいて既存の「ホットスタート」方法論より性能が優れていて、その使用により、反応の初期、および多数のサイクル(典型的に60サイクル以上)を有するLATE−PCR反応中のいずれにおいても、プライマー二量体およびミスプライミングされたアンプリコンを含む望ましくない生成物の蓄積を防止することができる。
【0012】
本発明の別の態様は、PCR増幅およびアッセイ方法、対称的PCRまたは非対称的PCRの両者であり、非限定的にLATE−PCRが挙げられ、ならびに前記試薬添加剤を含むキット、部分的キット、およびオリゴヌクレオチドセットである。
【0013】
要旨
本発明の試薬は、少なくとも一部のPCR増幅においてミスプライミングの1種以上の現象を防止することが可能な添加剤である。「現象を防止する(防ぐ)」とは、反応の終了時点で、本明細書中に記載の技術、すなわち蛍光性DNA色素、ゲル電気泳動、DNAシークエンシングおよび融点解析によって、ミスプライミングの生成物または生成物群が検出されないことを意味する。本発明に基づく試薬は、重合活性および5’−3’エキソヌクレアーゼ活性(5' - to - 3' exonuclease activity)の両方を有するポリメラーゼを利用する場合でさえ、増幅開始前に、1マイクロモル濃度(μM)(すなわち1000nM)未満、好ましくは650ナノモル濃度(nM)以下、より好ましくは300nM以下、最も好ましくは50〜250nMの比較的低い濃度でPCR増幅混合物中に含ませてよい。本発明に基づく試薬は修飾された一本鎖オリゴヌクレオチドである。本発明に基づく試薬を構築するために利用してよいオリゴヌクレオチドは広範囲のオリゴヌクレオチドである。それらはDNA、RNAまたは混合DNA−RNAであってよい。それらは、修飾ヌクレオチド、非天然ヌクレオチド、例えば2’O−メチルリボヌクレオチド、非天然ヌクレオチド間結合、非ヌクレオチドリンカー、PNA、LNAおよび付加化学部分、例えばGlenn Researchによって報告されているキャップヌクレオチド(capped nucleotides)を含有してよい。
【0014】
本発明の試薬は、PCR増幅反応混合物中で、一般に「ヘアピン」構造と称されるステム・ループ構造(stem-and-loop structure)を形成する一本鎖オリゴヌクレオチドであるが、それらは、ループがヌクレオチドから構成されていないステム・ループ構造から構成されていてもよい。そのような構造では、分子の中心部分が一本鎖の(ハイブリダイズしていない)ままであり(ループ)、末端は互いにハイブリダイズしてステムを形成する。ステムは平滑末端であってよく、あるいは一方の鎖が他方の末端を超えて伸長していてもよい。ステムは連続する二本鎖領域を含んでよく、あるいはそれは内部ミスマッチを含んでもよい。内部ミスマッチは隆起を生じさせる。オリゴヌクレオチドの末端によって形成されるステムの端部は、DNA−DNAハイブリッドよりも堅固に結合するように安定化される。本発明者らは、本発明の試薬のステムをその融解温度、すなわちTmを参照することによって特徴付ける。融解温度とは、ステムの相補配列の50%がハイブリダイズしておらず(開放型立体配置)、かつ該相補配列の50%が自身にハイブリダイズしているか、あるいは部分的にハイブリダイズしている(閉鎖型立体配置)摂氏温度である。本出願では、ステムの「計算上のTm」とは、M−foldプログラム:Zucker, M. (2003). "Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction." Nucl. Acids Res. 31: 3406-3415を使用して取得される、対応する安定化されていない完全DNAオリゴヌクレオチドのステム部分の計算上の融解温度を意味する。その場合、ナトリウム濃度が70ミリモル濃度(mM)であり、かつマグネシウム濃度が3mMであると仮定する。添加されたクエンチャーペア、好ましくは光を吸収するが、吸収エネルギーを熱として放出する非蛍光クエンチャー、例えばDabcylおよびBlack Holeクエンチャーを有する好ましい実施形態の場合、Tmは、クエンチャーを伴わないDNAヘアピンについての計算上のTmである。末端にキャップヌクレオチドを有する実施形態の場合、Tmは、同等のキャップなしのヌクレオチドを含有するDNAヘアピンについての計算上のTmである。ステム端部の2’−O−メチルリボヌクレオチド含有物の場合、Tmは、2’−O−メチルリボヌクレオチドのデオキシリボヌクレオチドアナログを含有する全DNAヘアピンについての計算上のTmである。本発明者らは、安定化されたヘアピンの実際の融点を取得することが困難であるという実施上の理由のために本方法論を利用するが、安定化用の修飾に起因して実際の融点が計算上のTmより数度高いことを認識している。本発明の試薬は、94℃を超えない計算上のステムTmを有し、好ましくは計算上のステムTmは50〜85℃の範囲である。一部の実施形態では、プライマーアニーリング温度より高い(典型的に増幅反応は55〜72℃であり、すなわち72〜85℃の範囲の)計算上のステムTmが好ましいが、他の実施形態では、プライマーアニーリング温度より低い、すなわち50〜71℃の範囲の計算上のステムTmが好ましい。ステムの相補配列は、PCRサイクル処理中の鎖融解に使用される温度である95℃で開いて、一本鎖になる。さらに、本発明の試薬のステムは、閉鎖型、自己ハイブリダイズ型のコンホメーション(confirmation)の形成のために十分に低い温度で6ヌクレオチドを超える長さを有する。現在、本発明者らの好ましい実施形態では、9〜12塩基対の長さ、好ましくは9〜11塩基対、閉鎖型の場合、より好ましくは9〜12、最も好ましくは9〜11の、内部ミスマッチを含まない連続した相補的塩基対、最も好ましくは9〜12塩基対の長さで、かつ完全に相補的な平滑末端型ステムであるステムを形成する。試薬の3’末端は、増幅反応においてDNAポリメラーゼによって伸長可能ではなく、したがって該反応物はPCRプライマーではない。すべての突出3’末端は、例えばリン酸部分または何らかのブロック用化学部分を付加することによって、伸長を防止するためにブロックされる。
【0015】
ループの長さはかなり変動しうる。ループがヌクレオチドおよびヌクレオチド間結合からなる場合、それは少なくとも3ヌクレオチド長である。さらに、それがちょうど3ヌクレオチド長であれば、チミジン残基を含有する。現在、本発明者らの好ましいヌクレオチドループは3〜22ヌクレオチドの範囲の長さを有する。ループは非ヌクレオチド化学リンカー、例えばアルキレン鎖であってもよい。炭素鎖リンカーに関しては、3〜6炭素原子の長さが好ましく、最も好ましい長さは3炭素原子である。アルキレン炭素鎖において、さらに、炭素鎖の各メンバーの残りの2価電子は共有結合に関与する。そのような結合は、水素原子、短鎖アルキル基または短鎖アルキレン基、試薬を固体表面に連結するための置換基を対象としうる。化学リンカーを含む非オリゴヌクレオチドループは炭化水素鎖に限定されず、ヘテロ(炭素または水素でない)原子を含んでよい。化学リンカーはポリメラーゼに結合しないように電気的に中性であることが好ましい。試薬の活性はステムの閉鎖型コンホメーション(confirmation)に依存するため、5個を超えるヌクレオチドのループを有する試薬に関して、ループの組成は、反応中の、または反応によって生成される、任意の他の配列または配列群に対して広範囲にわたって相補性であるわけではないことが好ましい。本発明に基づく試薬は、増幅反応のいずれの生成物に対するハイブリダイゼーションプローブでもなく、生成物の蓄積をシグナルしない。
【0016】
上記のように、本発明に基づく試薬は、ループの反対側のステムの端部を安定化して、該ステムの端部がDNA−DNAハイブリッドの端部よりも堅固に結合するようにすることを含む。本発明者らの現在の最も好ましい実施形態では、ステムを平滑末端にし、天然DNA−DNAハイブリッドと比較して部分的鎖分離を阻害する様式で該ステムの末端を修飾する。本発明者らは、安定化用の相互作用性化学部分をステムの端部に付加する効果、市販のリンカーを用いてステムの3’および5’ヌクレオチドに共有結合によって付加される1ペアのDabcyl部分および1ペアの市販のBlack HoleTMクエンチャー(Biosearch Technologies, Novato, CA, U.S.A.によって市販されている専売のクエンチャー)の両者の効果を以下で実証する。また本発明者らは、ステムの端部で、強力に結合する非天然ヌクレオチド、2’O−メチルリボヌクレオチドを利用する効果を実証する。試薬が増幅および増幅アッセイにバックグラウンド蛍光を付与しないように、好ましい安定化用部分は非蛍光性である。本発明の試薬の、安定化されていないオリゴヌクレオチドDNAヘアピンを、1μM未満の濃度で加えても、水素結合しているステム末端を安定化するよう上記のように修飾されていない場合はミスプライミングを必ずしも実質的に低減せず、多くの場合でそうならない。ゆえに、本発明の試薬において安定化は必須である。
【0017】
本発明のヘアピン試薬は、6個を超える塩基対からなるステムを形成できるようにつながり合う2つの相補的ヌクレオチドオリゴマーを含有する分子と説明することができ、該ステムの開放端は、該ステムがその開放端でほどけるか、あるいは「ほつれる」傾向が抑制されるように、何らかの手段によって化学的に修飾される。作用の分子的メカニズムの徹底的な理解には、本発明の試薬の存在および非存在下での酵素キネティクスによる追加の分析、ならびに電子顕微鏡観察、核磁気共鳴、およびX線結晶構造解析による前記試薬とポリメラーゼとの相互作用についての構造解析が待たれるが、本明細書および科学文献中に示されている情報に基づいて、メカニズムの部分的な解釈を予想することができる。該文献は、Taqポリメラーゼが、他のDNAポリメラーゼと同様に、合成ドメインおよび5’エキソヌクレオチド分解性(exonucleolytic)ドメインからなることを教示している。5’エキソヌクレオチド分解性ドメインはStoffel断片には存在しない。該文献は、さらに、合成ドメインがまず二本鎖DNAに結合し、それに沿ってスライドすることによってDNA重合を実行することを教示している。合成ドメインは「広げた手」に例えられている形状を有し、dNTPの存在下で、二本鎖DNAと接触すると、ポリペプチドレベルで形状変化を受ける。その結果、「手」の「指」が二本鎖DNA分子を取り囲んで「閉じる」。二本鎖分子内のミスマッチ配列に起因して、ポリメラーゼは数ヌクレオチド後退し、合成ドメインの3’編集機能を使用して正しい塩基で置換する。酵素が鋳型鎖を読んで、相補的プライマーの3’末端を伸長するにつれて新規DNA鎖の合成が生じる。酵素が、鋳型鎖にすでに結合しているオリゴヌクレオチドの5’尾部に遭遇した場合、ポリメラーゼは結合済みオリゴヌクレオチドの領域に1〜2ヌクレオチド侵入し、酵素の5’エキソヌクレアーゼドメインを用いて、得られた5’尾部を切断することができる。
【0018】
この情報に基づいて、本発明の試薬の二本鎖ステムが、少なくとも部分的には、開放型コンホメーションにおいてポリメラーゼの合成ドメインに結合し、それを閉鎖型にすることによって機能すると仮定することができる。ポリメラーゼの合成活性はそれによって阻害される。ゆえに、いかなる理論にも拘束されることを望まないが、第1の作用様式がポリメラーゼの合成活性についての温度依存的インヒビターとしての様式であると仮定することができる。以下に挙げる証拠は、少なくともいくつかのバージョンの試薬が、その二本鎖領域、例えばステムの融解温度で酵素から遊離せず、特に典型的なPCR伸長温度を含めたより高い温度でも結合したままであることを示すものである。しかし、PCRサイクルの変性(すなわち鎖融解)ステップ中は、本発明の試薬はDNAポリメラーゼに結合していない状態になる。典型的に90℃を超えるPCRサイクルの変性ステップ(鎖融解ステップとも称される)中には、本発明のヘアピン試薬のステムは融解して離れ、該二本鎖領域が再形成するために十分に温度が低下した場合にのみ、その時点で、それらは再びポリメラーゼに結合する。したがって、第一の変性までの間だけ二本鎖であり、すなわち増幅前に(一般に室温で)PCR反応混合物に加えた時点では二本鎖であるが、その後、そのステムのTmを超える温度で維持される、本発明の試薬を設計することを選択することができる。そのような実施形態では、すべての増幅サイクルのプライマーアニーリング温度および任意のLATE−PCR低温検出温度は、計算上のステムTmより高く、好ましくは少なくとも5℃高い。あるいは、増幅中の後期に再び二本鎖になる、本発明の試薬を設計することを選んでもよい。その設計は、すべてまたは一部の増幅サイクルで使用されるプライマーアニーリング温度より高いステムTm(記載されているように算出される)を利用することによって達成することができる。あるいは、LATE−PCR増幅において、プライマーアニーリング温度より低いが、低温検出温度より高いステムTmを利用することによって、その設計を達成することができる。前者の設計タイプは、概して、CTの遅延によって示されるように重合を阻害しないが、一方、ステムTmより低いプライマーアニーリング温度を利用すると、概して、1〜3増幅サイクルの中程度のCTの遅延が生じる。
【0019】
さらに、いかなる理論にも拘束されることを望まないが、本発明に基づく試薬の第2の作用様式が、ポリメラーゼの5’エキソヌクレオチド分解活性についての温度依存的インヒビターとしての様式であると仮定することができる。本発明者らは、本発明の試薬が、少なくとも55℃までの温度でポリメラーゼ酵素の5’−3’エキソヌクレアーゼ活性を阻害し、その阻害は、酵素が(PCR増幅においてPCRプライマーが伸長されるにつれて)DNA鎖をその3’末端の重合によって伸長する能力を過度に抑制することなく行われることを以下に示す。
【0020】
さらに、いかなる理論にも拘束されることを望まないが、本発明の試薬の第3の作用様式が、ポリメラーゼに結合し、該ポリメラーゼのポリペプチドの形状を変化させる温度依存的リガンドとしての様式であると仮定することができる。前記リガンドが酵素から遊離すると、該酵素は、いくらかの時間、変化した形状のままであり、その後、その元の形状に戻る。変化した形状であるうちは、酵素は、部分的にミスマッチしたプライマー−鋳型ハイブリッドと比較して、完全に相補的なプライマー−鋳型ハイブリッドに優先的に結合する。
【0021】
このモデルに基づいて、本発明の試薬のステムがポリメラーゼの合成ドメインの形状を変化させる能力が、本発明の試薬がポリメラーゼの合成機能(上記第1の様式)またはポリメラーゼの5’エキソヌクレアーゼ機能(上記第2の様式)を阻害する能力とは異なっていると予想することができる。第1の作用様式および第2の作用様式は両者とも、結合している試薬がポリメラーゼから遊離する速度に実際に依存する。速やかに遊離する本発明の試薬は、第3の様式によって優先的に作用し、高濃度でさえ、ポリメラーゼの合成活性を大きく阻害することはない。対照的に、ポリメラーゼからゆっくり遊離する本発明の試薬は高濃度では阻害性である可能性が高い。一例として、ステムと3つのメチレン(−CH2−)基からなる炭素リンカーループとにより構成される化合物12−C3DDも、ステムと6つのメチレン(−CH2−)基からなる炭素リンカーループとにより構成される化合物12−C3C3DDも、3000nMの濃度でPCR増幅を部分的にしか阻害しない。対照的に、ヌクレオチドにより構成されるループを有する本発明の試薬は、典型的には、実施例1のアッセイにおいて、PCR増幅を<1000nMの濃度で完全に阻害し、時には<500nM未満の濃度で阻害する。
【0022】
タンパク質−核酸相互作用に詳しい当業者が認識するように、本発明の試薬の作用は様式1〜3のうち2つ以上に基づくものでありうる。それらは相互に排他的ではなく、むしろ関連している。以下に記載されるように、ステムを含んでなる2つのオリゴヌクレオチドを連結する特定の分子構造が、化合物全体を(第1の様式または第2の様式によって)酵素インヒビターとして作用させるか、あるいは第3の作用様式で主に酵素特異性のエンハンサーとして作用させる程度を決定するために使用できる定量的試験(実施例14)を本発明者らは考案した。
【0023】
上記モデルに基づいて、さらに、1種以上のポリメラーゼ分子を本発明の試薬の等しい数の分子と結合させ、次いでそれを、より大きい部分、例えばビーズ、粒子、または固体材料からなる材料に共有結合によって結合させることができることを予想できる。それにより、固体物にポリメラーゼが「積載」されていると考えることができる。固体物への本発明の試薬の結合は、一時的であってよく、あるいは当技術分野において公知の種々の手段によって切断可能であってよい。結合が切断されると、試薬または試薬−ポリメラーゼ複合体が溶液中に放出される(その後、該試薬はポリメラーゼから遊離しうる)。
【0024】
本発明の試薬の設計は当分野の技能の範囲内である。ステムの融解温度は、そのGC含量を変動させることによって、かなり調節できることが理解される。例えば、以下に記載される2つの好ましい実施形態のステムは、いずれも、9ヌクレオチドの長さを有するが、それらは25℃異なる計算上のTm(81℃および56℃)を有する。本発明者らは通常、ステムTmの算出に関して、上で引用したM−foldコンピュータソフトウェアプログラムを1種以上のループ配列候補について使用する。ループ配列が、それ以外の点で、本発明のミスプライミング試薬において重要であることは見出されていない。本発明者らの設計では、完全な平滑末端のステム、すなわち内部ミスマッチおよび末端突出部を有しないステムを形成する配列を利用する。そして、簡単な試行によって、内部ミスマッチ(実施例1に示されるように、末端ミスマッチは不安定であり、許容されない)の導入または二本鎖領域を超える1個または最大2個のヌクレオチドの短い伸長の影響を評価することができる。
【0025】
本発明の試薬の構築は当分野の技能の範囲内である。例えば、オリゴヌクレオチド配列をオリゴヌクレオチド合成機で調製することができる。公知の方法を使用して安定化用部分を含ませることができる。例えば、dabcyl化カラム(Glen Research)を用いて出発し、Dabcylで修飾されたヌクレオチドで合成を終えることによって、好都合にDabcylを付加することができる。合成の最初、および最後から2番目、および最後のヌクレオチドとして非天然ヌクレオチドを使用することができる。
【0026】
候補試薬を設計し、構築した後、一部の場合には、蛍光性DNA結合色素を加え、該色素を刺激しながら融解解析を実施することによって、そのステムの融解温度を経験的に概算することができる(色素自体がTmに対して効果を有することを認識して行う)。また、本発明の試薬の安定化されたステムの実際の融解温度に関する実施上有用な情報を、多くの場合、種々のアニーリング温度を利用するリアルタイムPCR増幅を実施することにより推定的に取得することができる。必要であればステムの長さまたはGC含量の調節を施して、所望のTmを達成することができる。そして本発明者らは、ミスプライミングおよび増幅効率の種々の現象に対する候補試薬の効果を、以下の実施例1に記載されるような厳密なPCR増幅におけるその性能を決定することによって評価する。候補試薬は、実施例1の反応混合物に1000nM未満、好ましくは650nM以下のいずれかの濃度で加えられた場合に純粋なアンプリコンが取得されれば、本発明の試薬であると判断される(図1を参照のこと)。本発明者らは、所望の標的および所望のプライマーを用いること以外は実施例1のアッセイを使用して候補アッセイを評価する。しかし、本発明の増幅およびアッセイ(増幅および検出)は実施例1の条件または手順に限定されない。本発明の試薬は、25μLの反応混合物あたり1.25単位のポリメラーゼ濃度(実施例1)と比較して、1000nM未満の濃度で加えられた場合に、ミスプライミングの少なくとも1種の現象を阻害する必要があるが、該試薬はポリメラーゼ濃度に対する任意の有効な濃度;すなわち、ミスプライミングを防止するが、増幅を実質的に妨害しない任意の濃度(すなわち相対濃度)で使用することができる。評価において所望のプライマーを利用すると、意図せぬ結果、例えば試薬の3’末端が遮断されないことまたは相補性の見落としが明らかになる。
【0027】
本発明には、PCR増幅反応およびPCR増幅反応を含むアッセイが含まれ、そのようなものとしては、増幅反応混合物が、重合活性および5’−3’エキソヌクレアーゼ活性のいずれをも有する熱安定性DNAポリメラーゼ、例えばTaqDNAポリメラーゼを含み、かつ本発明の少なくとも1種の試薬が該増幅反応混合物に含まれる反応が挙げられる。本発明の多数の試薬はポリメラーゼの活性を阻害するものであり、熱サイクル処理前または処理中に上記範囲内のポリメラーゼ濃度に対して相対的な濃度、すなわち25μLの反応容量あたり1.25単位のDNAポリメラーゼを含有する反応に関して1000nM以下、好ましくは650nM以下の濃度で加えられる。本発明のいくつかの試薬は、1000nM未満の濃度、好ましくは650nM未満の濃度で有効であるが、ポリメラーゼの活性を低い程度にしか阻害せず、前記ポリメラーゼ濃度に関して1500nMまでの濃度で、あるいは3000nMでさえ加えることができる。本発明の2種以上の試薬を含有するアッセイでは、各試薬は各自のTmを有するステムを有することができ、それを各自の濃度で加えて、増幅プロセス中のステップの種々の部分で種々の試薬が機能するようにすることができる。本発明の試薬が熱サイクル処理中に加えられる場合、反応混合物を作業領域に引き込むことなく加えられるべきであろう。そのような増幅反応としては、対称的PCR増幅、非対称的PCR増幅およびLATE−PCR増幅が挙げられ、それらはいずれも、RNA標的が関与する場合には逆転写をさらに含んでよい。PCR増幅を使用し、任意の目的で、例えばジデオキシシークエンシングの出発材料として増幅産物を調製することができる。アッセイにおいてPCR増幅を増幅産物の検出と組み合わせてよく、そのようなアッセイとしては、特に、標識プライマー、標識プローブまたは蛍光性DNA結合色素、例えばSYBR Greenまたは臭化エチジウムを使用するホモジニアスアッセイが挙げられる。前記アッセイは、検出の読み取り値が複数の増幅サイクル中に取得されるリアルタイムアッセイであってよく、あるいは増幅の完了後に検出が実施されるエンドポイントアッセイであってよい。それは定性的または定量的であってよく、非限定的に、定量的エンドポイントアッセイが含まれる。前記アッセイは、単一の二本鎖産物または一本鎖産物、または2種以上の二本鎖産物を、関連の一本鎖産物を伴わず、あるいは伴って、増幅するように設計することができる。
【0028】
本出願で使用される「LATE−PCR」とは、おおむね十分なPCRサイクル中に使い果たされて蛍光によって検出可能な二本鎖アンプリコンを生じるように、それ自体は200nMまでの低濃度で利用される一方のプライマー(「制限的プライマー(Limiting Primer)」)と比較して少なくとも5倍過剰のもう一方のオリゴヌクレオチドプライマー(「過剰プライマー」)を利用するポリメラーゼ連鎖反応(PCR)プロセスを使用する、非対称的DNA増幅を意味し、その場合、増幅の開始時点での制限的プライマーの、濃度について補正した融解温度Tm[0]は、増幅の開始時点での過剰プライマーの、濃度について補正した融解温度Tm[0]Xより高いか、あるいは最大5℃まで低く、好ましくは3〜10℃高く;ならびに、熱サイクル処理は制限的プライマーが使い果たされた後に複数サイクル継続されて、一本鎖産物、すなわち過剰プライマーの伸長産物を生じる。LATE−PCRアッセイは、少なくとも一部の直線的増幅サイクル中にプライマーアニーリング温度より温度を下げる低温検出ステップを含んでよい。好ましくは、鎖融解前の伸長後に当該ステップを行う。
【0029】
また本発明は、完全PCRキット、部分的キット、およびオリゴヌクレオチドセットを包含する。完全PCR増幅キットは、少なくとも、PCR増幅またはアッセイを実行するためのすべての試薬を含み、少なくとも1ペアのPCRプライマー、dNTP、反応バッファー、熱安定性DNAポリメラーゼ(好ましくは5’−3’エキソヌクレアーゼ活性を有するポリメラーゼ)、および少なくとも1種の本発明の試薬が含まれる。ホモジニアスPCRアッセイのための完全キットは、必要とされる任意の追加検出試薬、例えば蛍光性DNA色素または蛍光標識プローブをさらに含む。いずれのタイプの完全キットも、好ましくはサンプル調製用の試薬を含み、一部の実施形態では、逆転写酵素を含んでよい。本発明の部分的キットは、完全キットの少なくとも一部の構成要素を省略するが、少なくとも熱安定性DNAポリメラーゼ(および、必要であれば、逆転写酵素)および少なくとも1種の本発明の試薬を含む。例えば、商業的に「マスターミックス(master mix)」または「基本キット(basic kit)」として公知の製品は、典型的には、PCRプライマーおよびプローブを省略する。好ましい部分的キットは、サンプル調製のためのものを除いて、増幅またはアッセイに必要とされるすべての試薬を含む。本発明に基づくオリゴヌクレオチドセットは、少なくとも1ペアのPCRプライマーおよび少なくとも1種の本発明の試薬を含む。それらはオリゴヌクレオチドプローブまたはシークエンシングプライマーをさらに含んでよい。
【0030】
本発明の1種以上の実施形態の詳細は添付の図面および以下の説明中に記載される。本発明の他の特徴、目的、および利点は、その説明および図面から明らかになり、さらに特許請求の範囲から明らかになるであろう。
【0031】
図面の説明
種々の図面中の参照記号などは要素などを示す。
【図面の簡単な説明】
【0032】
【図1】本発明に記載の試薬の阻害活性を試験するために設計したミスプライミングエラーに関する厳密な定性的PCRアッセイから得たサンプルのゲル電気泳動解析を示す。
【図2】本発明の試薬によってPCRベースのリアルタイムアッセイおよびエンドポイントアッセイが可能になることを示す。
【図3】本発明に記載の試薬を使用し、ミスプライミングエラーの非存在下で70サイクルのLATE−PCR増幅後に生成された一本鎖DNA産物から取得したDNA配列クロマトグラフを示す。
【図4】五重(pentaplex)LATE−PCR増幅に対する化合物9−3DDの効果を示す。
【図5】本発明の試薬がTaqポリメラーゼに結合すると、PCRサイクル処理温度が変性ステップに達するまでそれらはポリメラーゼから遊離しないことの証拠を示す。
【図6】25℃でのDNAポリメラーゼ活性に対する効果に関する、本発明の試薬の直接比較を示す。
【図7】ポリメラーゼ活性の非存在下で、ポリメラーゼのエキソヌクレアーゼ活性に対する、本発明に基づく試薬の効果を評価するためのアッセイを示す。
【図8A】対称的PCRアッセイにおいてミスプライミングを防止する本発明に基づく試薬の能力を示す。
【図8B】対称的PCRアッセイにおいてミスプライミングを防止する本発明に基づく試薬の能力を示す。
【図9】エキソヌクレアーゼ阻害を定量化するためのアッセイを示す。
【図10】LATE−PCR増幅でのプライマー二量体およびプライマーオリゴマーの形成に対する、本発明の試薬の用量依存的効果を示す。
【図11】2ペアのプライマーを用いる2種の標的配列についての二重LATE−PCR増幅に対する、低濃度の試薬9−22DDの効果を示す。
【図12】2ペアのプライマーを用いた2種の標的配列についての二重LATE−PCR増幅に対する、試薬9−3DDのステム組成を変化させることの効果を示す。
【図13】2ペアのプライマーを用いる2種の標的配列についての二重LATE−PCR増幅に対する、試薬9−3bDDの濃度を変化させることの効果を示す。
【図14】CT値に関して、LATE−PCR増幅の効率に対する試薬9−3DDの濃度上昇の効果を示す。
【図15】蛍光シグナル勾配および最終蛍光に関して、LATE−PCR増幅の効率に対する試薬9−3DDの濃度上昇の効果を示す。
【図16】本発明の化合物の混合物を用いるか用いないで実行された多重反応の産物を示す。
【図17】化合物12−3DDの作用に関する試験の結果を示す。
【図18】化合物12−C3DDの作用に関する試験の結果を示す。
【発明を実施するための形態】
【0033】
詳細な説明
上記のように、本発明者らは、ミスプライミングエラーの3種の異なるタイプおよび原因であると考えられるものを特定した。全3タイプのミスプライミングを評価するために、本発明者らは以下の実施例1に示す厳密なアッセイを開発した。増幅は剪断DNAを用いて開始され、これはミスプライミングを促進する。物理的操作、例えばDNAをピペット内に吸い込むことによって、DNAは剪断されやすい。さらに、「ホットスタート」試薬および方法によってはタイプ1のミスプライミングしか防止できないので、本発明者らは多数の温度サイクル(少なくとも60サイクル)をアッセイに組み入れた。さらに本発明者らは、一部のLATE−PCRアッセイで利用される低温検出ステップを組み入れた。さらにまた、本発明者らは、プライマー二量体の形成およびさらにそれらのオリゴマー形成および増幅を追加のタイプのミスプライミングとして認識している。プライマー二量体形成および増幅は、添加鋳型DNAの存在下および非存在下の両方で生じうるものであり、本発明者らはこれらの現象に関するアッセイをさらに開発した。
【0034】
ホットスタート酵素は、高温、例えば95℃に加熱されると生じる不可逆的な変化のために、増幅の開始前にしか有効でないが、本発明のミスプライミング試薬は不可逆的に変性せず、低温ステップが含まれる場合、反応の後期に、正の効果もしくは負の効果、または両者を有しうる。
【0035】
特定のサイクル温度より高いか、またはそれより低い融解温度Tmを有し、増幅に対して種々の影響を有するステムを設計することができる。ステムの設計では、好ましくは、融解が、一定の温度範囲にわたって起こる動的現象であり、このときTmは、50パーセントの分子が二本鎖形態であり、かつ50パーセントが一本鎖形態である温度を指定するものであることが考慮される。本発明者らは、一部の閉鎖型ステムがポリメラーゼに結合すると、ポリメラーゼに未結合のままである分子間の平衡が、より多くのステムが閉鎖型になる方へシフトすると考えている。特定のサイクル温度、例えばプライマーアニーリング温度で大多数のステムが閉鎖型であることが所望であれば、一般に、その温度より少なくとも5℃高い計算上のTmを有する無修飾のオリゴヌクレオチドステムを用いて出発し、その場合、安定化用のステムの修飾によって実際のTmが数度上昇することを認識している。反対に、特定のサイクル温度で大多数のステムが開放型であることが所望であれば、一般に、該特定のサイクル温度より少なくとも5℃低く、好ましくは少なくとも約10℃低い計算上のTmを有する無修飾のオリゴヌクレオチドステムを用いて出発し、その場合も、安定化用のステムの修飾によって実際のTmが数度上がる可能性が高いことを認識している。
【0036】
以下の実施例5で実証されるように、本発明の特定の試薬を種々のプライマーアニーリング温度とともに利用すると、増幅効率に対する種々の効果を有しうる。以下で考察される、本発明者らが現在意図している実施形態の1つ(化合物9−22DD)では、81℃の計算上のTmを有する。温度が鎖融解温度(例えば95℃)からプライマーアニーリング温度(例えば55℃)に下がるにつれて、前記化合物はすべての典型的なPCR熱サイクル中に閉鎖型になる。そのようなステムを有する試薬は増幅の各サイクル中に効果を有する。別の現在の本発明者らの好ましい実施形態(化合物9−3DD)では、56℃の計算上のTmを有する。PCR増幅混合物が室温で調製される時に、この試薬は閉鎖型のステムを有し、ポリメラーゼに結合する。増幅の開始時点で温度が上がっても、試薬は結合したままである。ポリメラーゼ結合型の9−3DD分子は最初の高温ステップ中に結合がはずれて開放型になり、未結合分子のステムが閉鎖型になるほど十分に温度が下がらない限り、ポリメラーゼに再結合しない。ゆえに、最低サイクル温度を65℃で維持することによって、ステムは再形成しない。これらの条件下では、本実施形態は、該増幅において、増幅の開始前に生じるミスプライミングを防止するホットスタート試薬として機能する。しかし、LATE−PCRサイクルにおける伸長後の、増幅の直線期に蓄積する一本鎖に対する低温分子ビーコンまたは他の標識プローブのハイブリダイゼーションを可能にする低温検出ステップ、例えば、40℃でのインキュベーションを含ませる場合、56℃の計算上の融解温度を有するステムは閉鎖型になり、高温の鎖融解が達成されるまで試薬がポリメラーゼに再結合することが可能になる。
【0037】
以下の実施例1では、本発明の試薬の性能を評価するために本発明者らが利用する厳密なLATE−PCRアッセイを記載する。図1は、「通常の」TaqDNAポリメラーゼ、すなわち5’−3’エキソヌクレアーゼ活性を有するが、ホットスタート修飾を有しない熱安定性DNAポリメラーゼを用いた場合(レーンbおよびレーンc)だけでなく、ホットスタートTaqポリメラーゼを用いた場合(レーンdおよびレーンe)にもミスプライミングエラーが起こることを示す。図1は、さらに、ホットスタートポリメラーゼを用いる増幅(レーンhおよびレーンi)に対してだけでなく、通常のTaqDNAポリメラーゼを用いる増幅(レーンfおよびレーンg)に対しても、300nMの濃度で試薬9−22DDを加えることによって達成される、ミスプライミングエラーにより形成される非特異的産物の排除、および所望の特異的産物しか存在しないことを示す。実施例1に記載のアッセイを利用して、ミスプライミングの抑制に関する化合物9−22DDおよび9−3DDの有効性を評価した。両化合物はタイプ1のミスプライミングを抑制することがわかった。9−3DDのステムの計算上のTmは低い(56℃)ので、すべてのサイクル温度が60℃以上であるPCR増幅プロトコルを利用することによって、それを単に有効なホットスタート試薬として使用することが可能である。ステムの融点より高いままであることによって、増幅が進行してもステムは再形成されない。化合物9−3DDをそのように使用しても重合効率に対して阻害効果を有しない。以下の実施例5はこれに関して有益な情報を提供する。実施例5では、試薬9−3DDならびに65℃および55℃のアニーリング温度を利用する増幅を比較する。65℃が利用される場合、CTが反映するように重合効率に対する影響はないことがわかった。しかし、55℃が利用される場合、CTは遅延した。このことは、ステムが閉鎖型になり、ポリメラーゼが結合するほど十分に温度が下がった場合に、プライマー伸長のために温度が上がっても、プライマー伸長温度がステムのTmより高い場合でも、結合が継続することの証拠である。一方、9−22DDのステムのTmは高い(81℃)ので、毎PCRサイクルのプライマーアニーリングステップ中にステムが再形成されてヘアピンになることが可能である。この化合物は、タイプ1のミスプライミングだけでなく、タイプ2のミスプライミング、タイプ3のミスプライミング、およびプライマー二量体形成の現象をも防止する。しかし、この化合物は重合効率に実際にいくらか影響する。このことは、100nMの濃度で使用された場合に、SYBR Green 1または標的特異的分子ビーコンプローブを用いてリアルタイムで追跡したPCR増幅の閾値サイクル(CT)が1.5サイクル遅延することによって示される。
【0038】
実施例1の表Iは、図1のレーンfおよびgに示されるように、ホットスタート型ではなく通常のTaqDNAポリメラーゼを使用して、一連の添加剤に対する前記厳密なアッセイの適用を報告し、さらに、ミスプライミングの現象の防止、すなわちミスプライミングエラーにより形成される検出可能なレベルの非特異的産物の回避を達成するために、および所望の特異的増幅産物しか存在しないことを達成するために必要な最低濃度を報告する。表Iでは、化合物は本発明者らの用語体系によって特定される。無修飾のヘアピン分子はステムループの長さによって特定される。例えば、化合物6−22は6ヌクレオチド長のステムと22ヌクレオチド長のループとを有する。そのループ識別名は数字で始まるか、あるいは数字のみである(「22」または「3b」)ため、該ループはヌクレオチドである。非ヌクレオチドループは別に指定される。化合物12−C3C3は12ヌクレオチド長のステムと、6炭素鎖、すなわち6つのメチレン(CH2)基の鎖であるループとを有する。基本となるヘアピンの一端または両端に種々の修飾を施した。これらの修飾は接尾辞によって指定され、その場合、3Dは3’末端ヌクレオチドに付加されたDabcyl(5’−ジメトキシトリチルオキシ−5−[(N−4’−カルボキシ−4−(ジメチルアミノ)−アゾベンゼン)−アミノヘキシル−3−アクリルイミド]−2’−デオキシウリジン−3’−[(2−シアノエチル)−(N,N−ジイソプロピル)]−ホスホルアミダイト)であり、5Dは5’末端ヌクレオチドに付加されたDabcylであり、DDは両末端ヌクレオチドに付加されたDabcylであり、BHQBHQは両末端ヌクレオチドに付加されたBlack Holeクエンチャーであり、FFは両末端ヌクレオチドに付加された蛍光団(この場合はFAM)であり、AAは両末端ヌクレオチドに付加されたアデノシン(A)であり、ならびにTTは両末端ヌクレオチドに付加されたチミジン(T)である。
【0039】
付加に加えて、基本となるステムにいくつかの修飾を施した。接尾辞2’OM4とは、4個の末端ヌクレオチド、各ステムオリゴヌクレオチド上の最後の2個、またはアーム、がデオキシリボヌクレオチドから2’−O−メチルヌクレオチドに変更されたことを示す。ステムの融解温度を変更するために、本発明者らはそのGC含量を変更した。ステムの長さではなくステムの配列が変更された化合物を、末端修飾の接尾辞の前の小文字識別子によって示す。例えば、9−3bDDまたは9−3iDDである。表Iの種々の化合物に関するヌクレオチド配列を表IIに挙げる。その配列は、使用された炭素鎖ループを同時に示す。表Iはいくつかの点で有益な情報を提供する。例えば、表Iは、前記アッセイにおいて、無修飾のDNAヘアピンオリゴヌクレオチドが有効ではないか、または1000nM以上の高濃度でしか有効でないことを示す。オリゴマー9−22(9ヌクレオチド長のステム、22ヌクレオチド長のループ)は3000nMの濃度でしか有効でなかった。これは二本鎖DNAによるミスプライミング抑制に関して報告された量である。オリゴマー6−22(6ヌクレオチド長のステム、22ヌクレオチド長のループ)および9−5(9ヌクレオチド長のステム、5ヌクレオチド長のループ)は3000nMの濃度でさえ有効でなかった。しかし、9−22オリゴマーを、そのステムの両端にDabcyl部分を付加することによって修飾して、本発明に基づく好ましい試薬である化合物9−22DDを作成した場合、ミスプライミングの現象を防止するために50nMの濃度で十分であった。9−3オリゴマーを同様に修飾した場合、100nMの濃度で十分であった。
【0040】
表Iはステムの長さの影響を示す。9ヌクレオチドおよび12ヌクレオチドのステムを有するいくつかのオリゴマーは、1ペアの相互作用性Dabcylクエンチャーで修飾した場合に、低濃度でミスプライミングを排除したが、オリゴマー6−22は修飾によりそのような効果を示さなかった:組成物6−22DD(本発明の試薬ではない)は高濃度(3000nM)でしか有効でなかった。本発明の試薬は、6ヌクレオチドより長く、ほとんどの場合で14塩基対より短いステムを有する。現時点では、14塩基対より短い長さが好ましい。
【0041】
また表Iは、ステムの末端での結合強度を増大させる他の修飾の有効性を示す。1ペアの相互作用性Black HoleTMクエンチャーをオリゴマー9−5のステムに付加して試薬9−5BHQBHQを作成したところ、ミスプライミングエラーにより形成される非特異的産物の排除、および所望の特異的増幅産物しか存在しないことを達成するために、該試薬は100nMの濃度で十分であった。オリゴマー9−5の各末端を2個の2’−O−メチルリボヌクレオチドで置換して試薬9−5 2’OM4を作成したところ、該試薬は50nMの濃度で十分であった。
【0042】
表Iはオリゴヌクレオチドループの長さをかなり変動させることが可能であることを示す。3ヌクレオチド(試薬9−3DD)、5ヌクレオチド(試薬9−5DD)、および22ヌクレオチド(試薬9−22DD)のすべてのループ長で、100nM以下の濃度でミスプライミングを防止する本発明の試薬が得られた。同時に、表Iは、最適化のためにいくらかの試行錯誤による調整が必要であることを示す。オリゴマー9−5の安定化用の修飾においてDabcylクエンチャーの代わりに1ペアの2’−O−メチルリボヌクレオチドを用いる場合(試薬9−5 2’OM4と試薬9−5DDの比較)、有効性のレベルは変わらない:50nMの濃度で十分である。しかし、修飾型オリゴマー9−3に同じ変更を施すと(試薬9−3 2’OM4と試薬9−3DDの比較)、有効性のレベルは、依然として良好ではあるが減少した:50nMの濃度ではなく300nMの濃度が必要とされた。同データは、試薬9−3 2’OM4のループ長を2ヌクレオチド増加させて、試薬9−5 2’OM4を作成すると、有効性が改善されたことを示す:300nMの濃度ではなく50nMの濃度で十分であった。実施例1に記載されるアッセイのような厳密なアッセイを用いて、候補薬物をルーチンの方法で評価し、調節することができる。
【0043】
また表Iは、非ヌクレオチドブリッジ(ループ)によって逆の極性でつながり合う2つの相補的オリゴヌクレオチドからなる化合物もまた、そのオリゴヌクレオチドの自由な3’末端および5’末端を本発明にしたがって(試験された実施形態では、Dabcyl部分によって)修飾した場合に、活性であることを示す。表Iは、非ヌクレオチドブリッジのサイズが重要ではないことを示す。表Iに記載のブリッジは3〜6炭素原子の線状鎖からなる化学リンカーであるが、当業者は、非ヌクレオチドブリッジ(ループ)について多数の他のバリエーションを作成することができる。表Iのデータは、同一オリゴヌクレオチドステムに関して3炭素原子鎖のブリッジが6炭素原子鎖のブリッジより良好である(より低い濃度で活性である)ことを示唆し、厳密に試験されてはいないが、その差異がブリッジの相対的な可動性および、その結果として、ステムの融解温度を上げるかまたは下げるそれらの効果もしくは能力を反映すると仮定される。本発明者らは、好ましい化学的に中性のリンカー(無電荷)を利用する。
【0044】
本発明の試薬および方法の種々の適用が以下の実施例2〜4に例示される。実施例2では、リアルタイムアッセイであるLATE−PCRアッセイにおける、試薬9−22DDの利用が記載されるが、これはエンドポイントアッセイでも同様に好適であることが示されている。実施例2のアッセイでは、本発明者らが同時に出願した米国仮特許出願第60/619,654号、表題「Primers, Probes and Methods for Nucleic Acid Amplification」の対象である検出技術を利用する。該文献は参照によりその全体が本明細書中に組み入れられる。該検出技術は、二本鎖DNAに結合すると蛍光を発する蛍光性DNA色素(例えばSYBR Gold)および、制限的プライマーが使い果たされた後のLATE−PCR増幅において生成される一本鎖アンプリコンに相補的な蛍光団標識ハイブリダイゼーションプローブの両者を加えるステップ、その場合、該蛍光団は該色素からの発光によって刺激される;色素を刺激するステップ;色素および蛍光団の両方からの発光を検出するステップ;および色素シグナルに対する蛍光団シグナルの比を算出するステップを含む。ゆえに実施例2は、本発明の試薬と蛍光プローブおよび蛍光色素の適合性、およびそのいずれかまたは両者とともに使用された場合のミスプライミングの効果的な防止を示す。
【0045】
実施例4では、2ペア、3ペア、4ペアおよび5ペアまでものプライマーを利用するいくつかの多重LATE−PCR増幅が記載され、試薬9−3DDを反応混合物に加えた場合の全例で正しいアンプリコンが増幅されることが示される。実施例4の目的ではゲル電気泳動によって増幅産物を分析したが、同様に複数の方法を多重アッセイに適用可能であり、それには他の検出手段(例えば所望のアンプリコンに関する蛍光性ハイブリダイゼーションプローブ)を使用する定性的アッセイおよび定量的アッセイ、例えばリアルタイムアッセイが含まれる。リアルタイムLATE−PCRアッセイでは、プライマー伸長のステップ後の低温検出ステップおよび例えば実施例3で示される低温プローブを利用することが好ましい。
【0046】
実施例13では、2種以上の型の本発明の試薬を含有する増幅反応混合物を用いる増幅反応もまた利用することができることが示される。例えば、増幅混合物は、高い融解温度を有する本発明の化合物および低い融解温度を有する本発明の別の化合物の両者を含有することができ、各化合物はそれぞれの最適濃度で加えられる。81℃の計算上のTmを有する化合物9−22DDのステムは、反応の開始時点および各熱サイクル中に温度が低下した直後は閉鎖型コンホメーションであるが、58℃の計算上のTmを有する化合物12−3DDは相対的に低い温度でしか閉鎖型にならない。したがって、それは「ホットスタート」として作用し、反応混合物が室温で調製される場合にミスプライミングを防止するが、60℃以上のアニーリング温度を有するPCR増幅の以後の熱サイクル中には閉鎖型にならず、ポリメラーゼに結合しない。試薬の混合物は、複数のアンプリコンの増幅のために一定範囲のアニーリング温度にまたがる複数ペアのプライマーが組み合わされている多重反応の構築に関して特に有用である。実施例13で使用される試薬の混合物はいずれかの試薬の単独使用より良好である。
【0047】
実施例3では、十分な量で、ミスプライミングから生じる非特異的産物が十分に排除されている増幅一本鎖産物を調製するための、およびシークエンシング用の出発材料としての使用に好適な、試薬9−3DDを利用するLATE−PCR増幅が記載される。十分な出発材料を保証するために増幅を70サイクルまで延長した。本発明者らは、いくつかのアンプリコンが他のアンプリコンよりタイプ2およびタイプ3のミスプライミングエラーを生じやすいことを発見した。そのようなミスプライミングがより生じやすい増幅では、鎖融解ステップからプライマーアニーリングステップへサイクル処理温度が下がった時に閉鎖型になる本発明の試薬のステムが好ましい。このことはステムTm、アニーリング温度、またはその両者を調節することによって確かに実現することができる。関連する研究で、本発明者らは、本発明者らが同時に出願した上記の米国仮特許出願で開示されているように、生成物を希釈することが、シークエンシングとともに使用するためのシンプルな精製方法であることを発見した。
【0048】
本発明者らは、無修飾のDNAヘアピン形成オリゴマーと比較して、および互いに、本発明の試薬の実施形態の機能性および有効性を調べた。本発明者らは、試験試薬の存在下でのTaqDNAポリメラーゼによる標識プライマーの伸長を測定するためのDNA重合アッセイを考案した。該アッセイでは、本発明者らが同時に出願した上記の米国仮特許出願で開示されているように、蛍光色素からの発光で励起される蛍光団で標識されているプライマーを利用する。該アッセイの混合物は、0.5μMの濃度の合成オリゴヌクレオチドDNA鋳型、1.5μMの濃度のDNAプライマー(鋳型の3’末端に相補的で、その5’末端がCy5で標識されている)、PCRバッファー、MgCl2、TaqDNAポリメラーゼ、1:40,000希釈のSYBR Green蛍光性DNA色素、および試験試薬を含む。反応の制御された開始はdNTPの添加によって達成される。該反応は等温性である:それは所定の温度で所定の時間進行する。DNAポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する試験化合物の効果を評価するために、「ブロッカー」、すなわち鋳型の5’末端に相補的で、その3’末端がリン酸基でブロックされ、その5’末端がROXで標識されているオリゴヌクレオチドを反応混合物に加えた。DNAポリメラーゼの重合活性に対する試験化合物の効果を評価するためには、ブロッカーを使用しない。融解曲線解析による反応産物の分析を容易にするために、鋳型、プライマーおよびブロッカーは、以下のハイブリッドがその融解温度によって容易に識別可能であるように設計する:プライマー−鋳型、ブロッカー−鋳型、完全長伸長産物−鋳型、および部分的伸長産物(ブロッカーまで)−鋳型。リアルタイム解析は、色素を周期的に励起し、該色素および2種の蛍光団からの蛍光発光を周期的にモニタリングすることによって達成される。両蛍光団は色素からの発光によって間接的に励起される。SYBR Green Iの蛍光の増加は重合の指標である。ROXの蛍光の減少はブロッカーの分解の指標である。エンドポイント解析は、12mM EDTAを加えて反応を停止させ、SYBR Green Iの希釈を1:14,200に調節し、試験化合物濃度を800nMに調節し;次いで、色素を刺激し、2種の蛍光団の各々からの蛍光をモニタリングしながら、標準的な融解曲線解析を実施することによって達成される。
【0049】
本発明者らは、55℃で60分間のインキュベーションを行い、化合物9−22、9−22DDおよび9−3DDを評価した。試験化合物を含まない対照も組み入れられた。試験化合物を含まないがブロッカーの存在下でインキュベートされた対照から得られた結果は、これらの条件下で、完全にではないが、TaqDNAポリメラーゼがブロッカー領域を通過してプライマーを伸長し、Cy5蛍光によればほぼ単一の融解ピークを有する産物が生成されたことを示す。ゆえに、酵素は重合活性および5’−3’エキソヌクレアーゼ活性の両方を示した。300nMの濃度または1000nMの濃度で存在する化合物9−22DDを用いて得られた結果は、部分的伸長産物の生成および残留ブロッカーの量に対する用量依存的様式の効果を示した。ゆえに化合物9−22DDは、55℃で、ポリメラーゼの5’−3’エキソヌクレアーゼ活性を阻害した。完全長産物の生成が特に1000nMの濃度で減少した。このことは、この高濃度では、化合物9−22DDが重合を阻害し始めることを示唆する。
【0050】
本発明の試薬の特性を調べるために本発明者らが利用した別のアッセイは以下の実施例6に記載のアッセイである。該アッセイは、25℃でのTaqDNAポリメラーゼによる伸長を阻害する試験試薬の能力を検出する。実施例7に報告される結果は、無修飾のヘアピンオリゴヌクレオチド9−22および9−3は対照と比較して効果を示さなかったが、試薬9−22DDおよび9−3DDはプライマー伸長を用量依存的様式で阻害したことを示す。ゆえに、本発明の試薬が低濃度で重合活性を阻害するのには十分に低い温度でさえ、それらの無修飾のアナログは同一濃度で該特性を示さなかった。
【0051】
本発明者らは、DNAポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する本発明の試薬の阻害効果を調べるために2種のアッセイを利用した。一方のアッセイは以下の実施例7に記載される。該アッセイでは、熱サイクル処理中の5’−3’エキソヌクレアーゼ活性の阻害を測定する。図7は、試薬9−22DDが50nM〜300nMの範囲の低濃度で用量依存的様式で該エキソヌクレアーゼ活性を阻害することを示す。
【0052】
第2のアッセイは以下の実施例9に記載される。該アッセイでは、25℃での5’−3’エキソヌクレアーゼ活性の阻害を定量的に測定する。図9は、50nM〜1000nMの濃度の試薬9−22DDによる用量依存的阻害を示す。
【0053】
以下の実施例6で記載されるさらに別のアッセイでは、TaqDNAポリメラーゼのStoffel断片によって実行されるプライマー伸長の、該試薬による阻害を測定する。インタクトなTaqポリメラーゼと同様に、伸長の阻害には、オリゴヌクレオチドステムでの修飾端部の存在が必要とされ、ステム間のループの長さによる大きな影響はなかった。しかし、インタクトなTaqポリメラーゼとは異なり、ほんの6ヌクレオチドのステムを有する試薬である化合物6−22DDによってStoffel断片は阻害される。本発明者らは、Stoffel断片が、インタクトなポリメラーゼと同一の様式で試薬と相互作用するわけではないと結論づける。
【0054】
本発明の試薬は、以下の実施例8で実証されるように、対称的PCRアッセイでのミスプライミングの防止に有用である。該アッセイでは、非常に近いTm値を有する等モルのペアのプライマーを使用して、嚢胞性線維症遺伝子における配列の増幅を測定した。
【0055】
現在使用されているPCR増幅およびアッセイは、典型的に、室温での増幅反応混合物の調製ステップを含み、ゆえに、タイプ1のミスプライミングを阻害するために最初の混合物に本発明の試薬を含ませることを必要とする。しかし、必ずしもそうではない。特定の自動化された方法、例えばマイクロ流体工学的(microfluidics)方法では、ホットスタート用の高い温度でポリメラーゼを反応混合物に加え、それによってタイプ1のミスプライミングを回避することが可能である。そのような方法では、増幅の開始後に本発明の試薬を加えることができる。
【0056】
プライマー二量体の形成は、完全反応物、すなわち増幅に必要なすべての構成要素と最初の標的配列とを含有する反応物中、ならびに標的配列を含有しない反応物中で生じうるミスプライミングの現象である。プライマー二量体は、反応物中に存在する別のコピーの同じ一本鎖プライマーまたは何らかの他のプライマーに対するハイブリダイゼーションおよびそれらに沿った伸長に基づいて、反応物中の1種以上のプライマーのミスプライミングによって形成される典型的には短い二本鎖DNA配列である。標的配列の存在下でのプライマー二量体の形成は、予測されるアンプリコンの蓄積に伴うプライマー二量体の蓄積として観察される。標的配列の非存在下でのプライマー二量体の形成は、特異的アンプリコンの蓄積を伴わないプライマー二量体の蓄積として観察される。またプライマー二量体は、基本となるプライマー二量体より配列が長いオリゴマーを形成しうる。その理由は、それらが追加の、連結コピーの一方または両方のプライマーを含有するからである。オリゴマー形成のプロセスは十分に理解されていないが、オリゴマー形成は反応産物のゲル電気泳動または融点解析によって容易に検出できる。プライマー二量体の形成により、プライマー二量体の生成および蓄積に一方または両方のプライマーが消費されるにつれ、所定数の熱サイクル中に生成される予測アンプリコンの量が減少する。したがって、プライマー二量体を排除することが望ましい。その理由は、それによって正常産物の特異性および収量がともに増加するからである。プライマー二量体のこれらの特徴、および本発明に記載の試薬を加えることによるその排除は、実施例10で例証される。
【0057】
プライマー二量体の形成確率は、多数の他の要因、例えばプライマー濃度の上昇、マグネシウム濃度の上昇、プライマーの長さの増加、熱サイクルのアニーリング温度の低下、および反応に含まれるプライマーの総数の増加に加えて、一方のプライマーの3’末端と同プライマーの内部配列または異なるプライマーとの間の配列相同性によって上昇する。さらに、プライマー二量体の形成確率が、ホットスタートポリメラーゼを使用する反応と比較して、非ホットスタートポリメラーゼを使用する反応においてずっと高いことが当技術分野において周知である。その理由は、反応の成分の混合中にプライマー−プライマーハイブリダイゼーションおよび伸長が相対的に低い温度で生じうるからである。ホットスタート酵素はプライマー二量体の生成および蓄積を低減するが、完全には排除しない。プライマー二量体のこれらの特徴、ならびに本発明に記載の試薬を加えることによるその排除は、実施例11で例証される。
【0058】
またプライマー二量体の形成は反応のキネティクスに対して微妙な効果を有しうる。本明細書中に記載の試薬を加えることによるそれらの排除を観察し、キネティクスに関して最適化することができる。実施例12では、2ペアのプライマーからなり、2種の一本鎖産物を生成させるLATE−PCR増幅が記載される。それらの一方の産物の動的蓄積を蛍光プローブに対するハイブリダイゼーションに基づいて検出した。結果を図13および図14に示す。該データは、キネティクスの直線性が、試薬のステムの厳密な組成および試薬の濃度の両者によって影響を受けることを実証する。試薬のステムの厳密な組成の影響は融解温度に対するその効果に起因する。さらに、試薬の最適使用および最適に近い使用によってLATE−PCRにおける直線的増幅の速度を変化させ、それによって反応の終了時点でのシグナルの大きさを変化させることができるが、反応のCT値には影響しない。図15および図16は、化合物9−3DDを150および300nMで使用した場合、一定範囲の鋳型出発濃度にわたってCT値によって判断される増幅効率は同一であったが、300nMの9−3DDの存在下で鋳型出発数100および1000から生じたシグナルの大きさは、150nMの9−3DDを使用して得られたシグナルより大きかったことを実証する。おそらく、それはプライマー二量体形成が存在しないせいである。
【0059】
実施例
【実施例1】
【0060】
実施例1. ミスプライミングエラーを評価するための厳密なアッセイ
PCR増幅反応においてタイプ1、タイプ2およびタイプ3のミスプライミングを評価するために、本発明者らは、剪断ゲノムDNAを利用するLATE−PCR増幅を実施し、融点解析およびゲル電気泳動によってその産物を調べる。使用した具体的な増幅は以下の通りである。
【0061】
A. 基質:10〜10,000ゲノムに相当する剪断ゲノムDNA。市販のDNAを購入し、ゲノムDNAを複数回凍結・解凍するか、またはDNAを剪断するための当業者に公知の任意の他の同様の方法によって処理した。
【0062】
B. PCR増幅混合物ベース(Sanchez et al. (1994) PNAS 101:1933-1938を参照のこと):
基質:2000ゲノムの剪断ゲノムDNA
1×PCRバッファー
Mg+2:3ミリモル濃度(mM)
dNTP:250マイクロモル濃度(μM)の4種の各dNTP
過剰プライマー:1000ナノモル濃度(nM)の配列5’ CTTTGATGACGCTTCTGTATCTA 3’(配列番号13)
制限的プライマー:50nMの配列5’ CCTGGATTATGCCTGGCACCAT 3’(配列番号14)
DNAポリメラーゼ:25マイクロリットル(μL)の反応混合物あたり1.25単位。
【0063】
C. 増幅プロトコル:
低温インキュベーション:室温で35分
PCR増幅プロトコル:95℃で15分間の高温浸漬;95℃で10秒間、55℃で30秒間、および70℃で30秒間の10サイクル;次いで95℃で10秒間、50℃で30秒間、および70℃で30秒間の70サイクル。
【0064】
いずれのホットスタート方法論をも使用せずに2種の熱安定性DNAポリメラーゼ(Promega TaqおよびInvitrogen Taq)を利用する増幅によって、ミスプライミングに関するベースラインの事例を確立した。2種の異なる市販のホットスタートDNAポリメラーゼ(Qiagen Hot Start TaqおよびPlatinum Taq(Invitrogen))を利用して追加の増幅を実施した。最後に、300nMの現在の好ましい実施形態の本発明に基づく試薬を加えて、追加の増幅を実施した。本実施例で使用される一実施形態は、組成物9−22DDと称される試薬である(本発明者らの用語体系において、「9」はステム長であり、「22」はループ長であり、「DD」は安定化用の修飾を意味し、この場合は1ペアのDabcylクエンチャーである)。化合物9−22DDは81℃の計算上のTmを有する。該化合物は、表IIに示される配列を有し、それは5’末端および3’末端のDabcyl部分の付加によって修飾されている。
【0065】
臭化エチジウム染色を施した種々の増幅のゲル電気泳動の結果を図1に示す。図1は、ラベルがない両端のカラムにサイズマーカー(100塩基対ずつ異なるサイズ)を含む。重複実験を行ったために、各LATE−PCR増幅の産物に関して2レーンずつが存在する。レーンa,DNAが省略されたベースラインの事例;レーンb,DNAポリメラーゼがPromega Taqであったベースラインの事例;レーンc,DNAポリメラーゼがInvitrogen Taqであったベースラインの事例;レーンd,ホットスタート酵素であるQiagen Hot Star Taqを代用;レーンe,ホットスタート酵素であるPlatinum Taqを代用;レーンf,化合物9−22DDを加えるとともにPromega Taqポリメラーゼを利用する増幅;レーンg,Invitrogen Taqポリメラーゼおよび化合物9−22DDを利用する増幅;レーンh,Qiagen Hot Star Taq DNAポリメラーゼおよび化合物9−22DDを利用する増幅;レーンi,Platinum Taqポリメラーゼおよび化合物9−22DDを利用する増幅。
【0066】
図1から、重合活性および5’−3’エキソヌクレアーゼ活性の両者を有する標準のTaqDNAポリメラーゼ(レーンbおよびレーンc)が一定範囲の産物サイズをもたらしたが、そのうちのほとんどが、レーンf〜iに見られる産物である、プライマーペアによって規定される所望のアンプリコンではなかったことがわかる。修飾型「ホットスタート」ポリメラーゼ(レーンd〜e)への切り替えは有益であったが、依然として、ミスプライミングエラーにより形成される非特異的産物は排除されなかった。しかし、化合物9−22DDが300nMの濃度で存在した場合は、酵素が標準のTaqDNAポリメラーゼであった場合(レーンf〜g)および酵素がホットスタートTaqDNAポリメラーゼであった場合(レーンh〜i)でいずれも、所望のアンプリコンが得られ、ミスプライミングエラーにより形成される非特異的産物は含まれなかった。
【0067】
本実施例の厳密なアッセイを使用して、いくつかの無修飾および修飾DNAヘアピン分子を、末端安定化を伴わずに;各アームの端部に同一のヌクレオチドを付加することによって達成される末端不安定化を伴って;本発明に基づく末端安定化を伴って;ならびに安定化用ではない末端付加を伴って比較した。定量的比較を行うために、標準のTaqDNAポリメラーゼを用いて図1に示されるように達成される、ミスプライミングエラーにより形成される非特異的産物の排除、および所望の特異的産物しか存在しないことをもたらすために必要とされる最低濃度を決定するために、ミスプライミング抑制活性を示した各化合物に関して前記アッセイを使用する用量応答試験を実施した。その結果は表Iに示される通りであり、表には試験対象の各試薬に関する計算上のステムTmが含まれる。表Iに報告される種々の化合物の配列は表IIに記載される通りである。
【表1】

【0068】
【表2】
【0069】
表Iでは、修飾および無修飾のいくつかのDNAヘアピンが比較され、本発明の試薬(1000nM未満の濃度でミスプライミングの産物を伴わない純粋なアンプリコンが得られる)であるか、または本発明の好ましい試薬(650nM以下の濃度でミスプライミングの産物を伴わない純粋なアンプリコンが得られる)であるいくつかの末端安定化ヘアピン分子が特定される。無修飾のヘアピンは2つの数字によって特定される。第一の数字はステム長であり、第二の数字はループ長である。表Iに挙げられる本発明に基づく化合物は以下のような数種類の安定化用の修飾を有する:市販のリンカーを用いて末端ヌクレオチドに共有結合している5’Dabcylクエンチャーおよび3’Dabcylクエンチャー(接尾辞「DD」によって示される)、同様に結合している3’Black HoleTMクエンチャーおよび5’Black HoleTMクエンチャー(接尾辞「BHQBHQ」によって示される)、およびステム末端でデオキシリボヌクレオチドの代わりに用いられる2個の3’2’−O−メチルリボヌクレオチドおよび2個の5’2’−O−メチルリボヌクレオチド(「2’OM4」によって示される)。ステム末端に対する不安定化用の修飾には、分子の3’および5’末端を2個のAまたはTで置換することが含まれた。ステムを安定化する様式で互いに相互作用しないと考えられる1ペアのFAM蛍光団を付加すると、単一の5’Dabcylの付加と同様に不安定であった。しかし、単一の3’Dabcylの付加は9−22ヘアピンをわずかに安定化した。それは必要とされる濃度の低下によって示される通りである。ヘアピン9−22は高濃度(3000nM)でしかミスプライミングを抑制しなかったが、9−22DDは低濃度(50nM)でミスプライミングを抑制した。
【0070】
現在の本発明者らの好ましい別のヘアピンである化合物9−3は高濃度(1000nM)でしかミスプライミングを抑制しなかったが、9−3DDおよび9−3 2’OM4はずっと低い濃度(100〜300nM)でミスプライミングを抑制した。さらに別の実施形態である9−5DD、9−5BHQBHQおよび9−5 2’OM4もまた低濃度(50〜100nM)でミスプライミングを抑制した。オリゴヌクレオチド9−5はオリゴヌクレオチド9−22と同一のステムを有するが、ヌクレオチドがAおよびTである短いループを有する。オリゴヌクレオチド9−5の計算上のステムTmは93℃であり、ループの影響によりオリゴヌクレオチド9−22の計算上のステムTmより約12℃高い。
【0071】
さらに長いステムを用いても同様の結果が得られた。化合物12−3はミスプライミングを抑制しなかったが、化合物12−3DDはほんの50nMの濃度で有効であった。
【0072】
表Iは、安定化ステムを修飾して、計算上のステム融解温度(Tm)を変化させることができることを示す。例えば、Tm56℃の化合物9−3DDは100nMの濃度で有効であった。ステムのGC含量を変えることによって、化合物9−3bDD(Tm62℃)および9−3iDD(Tm68℃)を作成した。両化合物もまた100nMの濃度で有効であった。Tm58℃の化合物12−3DDは50nMの濃度で有効であった。改変されたステムを有する化合物、すなわちTm63℃の化合物12−3bDDおよびTm68℃の化合物12−3cDDは100nMの濃度で有効であり、依然として非常に良好であった。
【実施例2】
【0073】
実施例2. LATE−PCRエンドポイントアッセイ
反復実験サンプル間の反応キネティクスの確率的変動によってハイブリダイゼーションプローブ由来のシグナルのばらつきが生じ、エンドポイント解析が妨害される。LATE−PCRはその問題を著しく軽減する。本発明の試薬を利用するとエンドポイント検出を利用する能力がさらに改善される。図2は、テイ・サックス病に関与するヘキソサミニダーゼA遺伝子のG269対立遺伝子を含むアンプリコンを増幅する際に得られる結果を示す。反応混合物は0.6μMのハイブリダイゼーションプローブおよび1000ゲノムのホモ接合性野生型DNA標的(+/+)またはヘテロ接合性DNA標的(G269/+)を含んでいた。各タイプの反復サンプルを以下のように増幅した:25μLの反応物中、100nMの実施形態9−22DDの存在または非存在下での、1×PCRバッファー、3mMのMgCl2、250μMの各dNTP、配列5’ CGAGGTCATTGAATACGCACGGCTCC 3’(配列番号15)を有する25nMの制限的プライマー、配列5’ TAACAAGCAGAGTCCCTCTGGT 3’(配列番号16)を有する1000nMの過剰プライマー、1.25単位のPlatinum(ホットスタート) Taqポリメラーゼ、配列5’ Cy5−GGGACCAGGTAAGAA−リン酸 3’(配列番号17)を有する0.6μMのCy5標識ハイブリダイゼーションプローブ、および1:40,000希釈のSYBR Gold I。PCRサイクルのパラメータは、95℃で3分間;95℃で10秒間、65℃で20秒間、および72℃で20秒間の25サイクル;および、95℃で10秒間、65℃で20秒間、72℃で20秒間、55℃で20秒間、および45℃で20秒間の30サイクルであり、72℃でSYBR Goldに関する蛍光を取得し、55℃および45℃でCy5に関する蛍光を取得した。反応キネティクスの試験管ごとの変動を補正するために、72℃でのSYBR Goldシグナルに対する55℃でのCy5シグナルの比によってハイブリダイゼーションシグナルを標準化した。そのような標準化の影響の1つはすべてのCTの遅延がマスクされることである。
【0074】
図2の、サークル21によって特定されるライン群およびサークル22によって特定されるライン群は、本発明の試薬を加えないが、ホットスタートポリメラーゼを用いた場合の複数回の反復実験の結果を示す。ホモ接合性の反復実験(サークル21)およびヘテロ接合性の反復(サークル22)はそれらの直線プロットの勾配によって識別することができる。約40サイクル後のハイブリダイゼーションプローブシグナルの低下(「Hook効果(hook effect)」)によって立証される、これらのサンプルにおける反復実験間のばらつきおよびミスプライミングエラーは、これらのサンプルのエンドポイント識別を不可能にする。図2のサークル23〜24によって特定されるライン群は、本発明の試薬、すなわち100nMの9−22DDを加えた場合の結果を示す。これらの条件下では、ミスプライミングエラーが生じず、アッセイの経過中に直線的キネティクスが維持され、ばらつきが低減され、Hook効果が回避され、50サイクルの時点でホモ接合性およびヘテロ接合性サンプルのエンドポイント識別が可能である。
【実施例3】
【0075】
実施例3. ジデオキシシークエンシング用のDNAサンプルの調製
LATE−PCRは、増幅反応が60以上の熱サイクルにわたって実行される場合に、DNAシークエンシングに好適な大量の一本鎖DNAを生産しうる非対称増幅法である。しかし、そのような多数の増幅サイクルはミスプライミングの産物の出現を助長し、それにはタイプ2およびタイプ3のミスプライミングエラーの形成が含まれると考えられる。該ミスプライミングエラーは検出可能な非特異的産物の蓄積として現れ、それは、最終的に、反応において最も多数を占める分子種になる。本発明の試薬は、70以上のサイクルのLATE−PCR増幅を使用する場合に、非特異的産物を生じずに、DNAシークエンシングに好適な大量の一本鎖DNAの合成を可能にする。キャピラリーゲル電気泳動によるジデオキシシークエンシングに好適なサンプルを以下のように調製した。まずLATE−PCR反応混合物を、1×PCRバッファー、3mMのMgCl2、配列5’ GCCAGGGGTTCCACTACGTAGA 3’(配列番号18)を有する1000nMの過剰プライマー、配列5’ CCGCCCTTCTCTCTGCCCCCTGGT 3’(配列番号19)を有する25nMの制限的プライマー、1.25単位のPlatinum Taqポリメラーゼ、600nMの試薬9−3DDおよび250μMの各dNTPを含有する25μLで調製した。化合物9−3DDは、オリゴヌクレオチド9−3の各末端ヌクレオチドに付加されたDabcylクエンチャーを有する。増幅は、95℃で3分間;95℃で10秒間、65℃で20秒間、および72℃で20秒間の10サイクル;および、95℃で10秒間、65℃で20秒間、72℃で20秒間;および45℃で20秒間の60サイクル実行した。二本鎖および一本鎖DNA合成を別々にモニタリングするために、1:40,000希釈のSYBR Greenまたは配列5’ FAM−CGTGCGCTCTGGTAAGGGTTTGCACG−Dabcyl 3’(配列番号20)を有する2.4μMの低Tm分子ビーコンを用いて並行して反応を実施した。各サンプルは6ng(約1000ゲノム)のヒトDNAを含有した。さらに、化合物9−3DDを加えずに対照反応を実施した。
【0076】
増幅反応の結果、ホットスタートTaqDNAポリメラーゼを用いるが、化合物9−3DDを用いない対照は顕著なミスプライミングを示した。制限的プライマーが使い果たされた後の増幅の後期段階中に、SYBR Greenシグナルが二次的に上昇することによってそれは立証される。該産物はそのような混合物であるため、該対照をシークエンシングすることは不可能であった。一方、化合物9−3DDを加えた場合の増幅の結果では、所望の特異的産物しか存在せず、70サイクルを通してSYBR Greenシグナルの遅延された上昇はないことが示された。制限的プライマーの濃度および50%の直線的増幅の見かけの効率から、反応によって250フェムトモル(fmoles)/μLの純粋な産物が生産されると見積もられる。該産物をBrandeis Universityのジデオキシシークエンシング施設に送り、シークエンシングを行った。
【0077】
シークエンシング反応は、Beckman CEQ 2000 DNAシーケンサーを利用するキャピラリー電気泳動などの標準プロトコルを使用して実施した。増幅産物の1/5アリコート、すなわちわずかに50フェムトモルの産物を用いてシークエンシング反応を実施した。機械により作成された配列を図3に挙げる。図3では、チャートの上部に沿って配列を示し、クリアで不明瞭なところがない産物ピーク(これから配列を導き出した)を示し、底部では非常に低レベルのバックグラウンドシグナルを示す。本発明者らは、機械により作成された配列をGenBank登録番号NT 010235から入手可能な該アンプリコンの公知の配列と比較することによって、その正確さを確認した。
【実施例4】
【0078】
実施例4. 多重化PCR増幅
ミスプライミングエラーの抑制は、複数の産物が同時に増幅される多重化反応において特に重要である。複数のプライマーペアおよび増幅産物が存在すると、これらの反応種の2者間および3者以上の間のミスプライミング相互作用の確率が高くなる。増加した数のアンプリコンからなる多重化反応を化合物9−3DDの不存在または存在下で実行した。以下に示すプライマーペアの種々の組み合わせに関して、300nMの試薬9−3DDの非存在下または存在下で、サンプル、1×PCRバッファー、3mMのMgCl2、100μMの各dNTP、1000nMの過剰プライマー、50nMの制限的プライマー、および1.25単位の非ホットスタートPromega Taqポリメラーゼからなる25μL容量のLATE−PCR反応物を調製した。サンプルの増幅は、95℃で3分間;95℃で10秒間,65℃で30秒間,および70℃で30秒間の10サイクル;および、95℃で10秒間,50℃で30秒間,70℃で30秒間の40サイクルの熱サイクルプロファイルを使用して行った。反応の終了時点で、0.5×TBE中の3.5%アガロースゲルでのゲル電気泳動によってサンプルを分析した。試験対象の所望のアンプリコンおよびプライマーペアは以下の通りであった。
【0079】
反応A:テイ・サックス病に関連するHex−A遺伝子の2領域TSD1278+TSD1421
TSD1278過剰プライマー:5’ GCCAGGGGTTCCACTACGTAGA 3’(配列番号21)
TSD1278制限的プライマー:5’ CCGCCCTTCTCTCTGCCCCCTGGT 3’(配列番号22)
TSD1421過剰プライマー:5’ CCGGGTCTCTAAGGGAGAACTCCT 3’(配列番号23)
TSD1421制限的プライマー:5’ CCGGCCGACAACACAAACCTGGTCC 3’(配列番号24)。
【0080】
反応B:TSD1278アンプリコン、TSD1421アンプリコン、およびCFTR遺伝子の1領域、CFエキソン10アンプリコン
本反応は反応Aと同じプライマーペアに加えて以下のプライマーペアを含有した:
CF ex10過剰プライマー:5’ GCTTTGATGACGCTTCTGTATCTA 3’(配列番号25)
CF ex10制限的プライマー:5’ CAGTTTTCCTGGATTATGCCTGGCACCAT 3’(配列番号26)。
【0081】
反応C:TSD1278アンプリコン、TSD1421アンプリコン、CFエキソン10アンプリコン、およびCFTR遺伝子の別の領域、CFエキソン11アンプリコン
本反応は反応Bと同じプライマーペアに加えて以下のプライマーペアを含有した:
CF ex11過剰プライマー:5’ TCGAAGTTTGCAGAGAAAGACAAT 3’(配列番号27)
CF ex11制限的プライマー:5’ TGACGTTTACAGCGAATGCTTGCTAGACCAAT 3’(配列番号28)。
【0082】
反応D:TSD1278アンプリコン、TSD1421アンプリコン、CFエキソン10アンプリコン、CF ex11アンプリコン、およびヒトβグロビン遺伝子の1領域
本反応は反応Cと同じプライマーペアに加えて以下のプライマーペアを含有した:
βグロビン過剰プライマー:5’ TGGGTTTCTGATACGCACTGACTCTCTC 3’(配列番号29)
βグロビン制限的プライマー:5’ GGCCATCACTAAAGGCACCGAGCACT 3’(配列番号30)。
【0083】
電気泳動ゲルを臭化エチジウムで染色し、LATE−PCR反応の対数期に生成された二本鎖産物を検出した。図4は、多重化反応A〜Dにより得られた産物についての該当部分のゲルを示す。矢印は特異的二本鎖増幅産物を指し示し、無数のバンドは二次構造を有する特異的一本鎖DNA産物に相当し、アスタリスクは非特異的産物を特定する。ゲルのペアA〜Dはそれぞれ反応A〜Dに相当する。「+」でラベルされている各ゲルは化合物9−3DDを含む増幅により得られた産物である。「−」でラベルされている各ゲルは化合物9−3DDを含まない増幅により得られた産物である。
【0084】
ゲルのペアAは、9−3DD化合物の非存在下で、領域TSD1278(矢印41)は増幅されなかったが、領域TSD1421(矢印42)は、主にまたは完全に二本鎖の多数の非特異的産物とともに増幅されたことを示す。該非特異的産物はゲルではスメアとして見られる。9−3DD化合物を加えると、非特異的産物のバックグラウンドが減少し、TSD1278(矢印41)およびTSD1421(矢印42)の両者の増幅が可能になった。それらはよりきれいに増幅されていて、ミスプライミングのいくつかの現象が防止されたことが示される。
【0085】
ゲルのペアBは、9−3DD化合物の非存在下で、領域TSD1278、TSD1421、およびCFエキソン10(矢印43)が、ゲルではスメアとして見られる多数の非特異的産物とともに増幅されたことを示す。9−3DD化合物を加えると、バックグラウンドの非特異的産物が排除され、3種の予測されるアンプリコンのすべてが得られた。
【0086】
ゲルのペアCは、9−3DD化合物の非存在下で、領域TSD1278、TSD1421、CFエキソン10、およびCFエキソン11(矢印44)が、アスタリスクによって示される非特異的産物とともに増幅されたことを示す。9−3DD化合物を加えると、非特異的産物の合成が排除され、4種の予測されるアンプリコンのすべてが得られた。
【0087】
ゲルのペアDは、9−3DD化合物の非存在下で、βグロビン遺伝子の選択された領域(矢印45)は増幅されなかったが、TSD1278、TSD1421、CFエキソン10、およびCFエキソン11は、アスタリスクによって示される非特異的産物とともに増幅されたことを示す。9−3DD化合物を加えると、非特異的産物の合成が排除され、5種の予測されるアンプリコンのすべての増幅が可能になった。
【0088】
また本発明者らは、感染性病原体に関してスクリーニングする場合に生じうるものなどの、任意の所定の試験サンプル中に1つの標的しか存在しない場合の多重増幅および検出反応における本発明の化合物の用途を実証した。そのような場合、複数ペアのプライマー(本発明者らは複数ペアの過剰プライマーおよび制限的プライマーをLATE−PCR増幅に利用した)が存在するが、特定のサンプルに関して1ペアのプライマーしか有効ではない。SYBR green蛍光および増幅後の電気泳動による解析によって示されるように、それは実際に生じたことであり、したがって、すべての基質より少数の基質しか存在しない場合の多重反応において本発明の化合物が効果的にミスプライミングを阻害することが実証される。
【実施例5】
【0089】
実施例5. ステムTmとアニーリング温度の関係
標的を含む同じ増幅混合物を利用して2種の異なるLATE−PCR増幅プロトコルを比較した。600nMの化合物9−3DDを含む場合および化合物9−3DDを含まない場合の両方で各増幅を実施した。上記のように、化合物9−3DDのステムTm(すなわち無修飾のオリゴヌクレオチド9−3の計算上の融解温度)は56℃である。ホットスタートPlatinum Taq DNAポリメラーゼを使用した。2種の増幅は、主として、使用されるプライマーアニーリング温度が異なり、65℃(ステムTmより高い)または55℃(ステムTmより低い)であった。増幅サイクルのパラメータは以下の通りであった:
プロファイルA:95℃で3分間;95℃で10秒間,65℃で20秒間,72℃で20秒間の10サイクル;95℃で15秒間,55℃で20秒間,72℃で20秒間,50℃で20秒間の60サイクル
プロファイルB:最後の60サイクルが95℃で10秒間,65℃で20秒間,72℃で20秒間,45℃で20秒間であったことを除いて、プロファイルAと同一。
【0090】
増幅反応をリアルタイムでモニタリングした。その場合、二本鎖産物の合成をモニタリングするためにSYBR Green I蛍光色素を使用した。ミスプライミングによって増幅の後期部分がばらつかない限り、二本鎖産物の合成は、制限的プライマーが使い果たされた後のLATE−PCR反応においてプラトーに達した。低温検出ステップ中に読み取りを行った。
【0091】
結果を図5に挙げる。パネルAは、化合物9−3DDを加えた場合(サークル52)および加えなかった場合(サークル51)の両方において、10サイクルの伸長後に55℃のアニーリング温度が利用された場合のプロファイルAの反復実験サンプルから得られた蛍光読み取り値を示す。パネルBは、化合物9−3DDを加えた場合(サークル54)および加えなかった場合(サークル53)の両方において、10サイクルの伸長後に65℃のアニーリング温度が使用された場合のプロファイルBの反復実験サンプルから得られた蛍光読み取り値を示す。
【0092】
図5は、本発明の試薬のステムTmより低いプライマーアニーリング温度が使用された場合(パネルA)に、閾値サイクルCTの数サイクルの遅延が観察されたことを示す。しかし、指数的PCR相の初めから終わりまでステムTmよりかなり高いプライマーアニーリング温度が維持された場合(パネルB)には、CTの遅延は、たとえあったとしても、ほとんど観察されなかった。本増幅で利用された特定のアンプリコンに関しては、ホットスタートTaqが使用された場合に、化合物9−3DDを用いても用いなくても、タイプ2/タイプ3のミスプライミングが観察されなかったことがわかった。このことは、そのようなミスプライミングエラーの出現が予測不能であることを示す。本実施例は、反応の温度が55℃に下がると試薬9−3DDのステムが閉鎖型になること、および反応の高温伸長ステップ中は該試薬がポリメラーゼに結合したままであることを実証する。
【実施例6】
【0093】
実施例6. 25℃でのDNAポリメラーゼ活性に対する効果
本発明の試薬およびそれらの無修飾のオリゴヌクレオチドアナログの効果を評価するために、一連のプライマー伸長実験を25℃で実行した。各反応混合物には、鋳型、プライマーおよびTaqDNAポリメラーゼを含めた。各場合において2時間、反応を実行した。プライマーをCy5で蛍光標識した。SYBR Green蛍光色素を反応混合物に加えた。伸長反応後に、融解曲線を作成した。その場合、本発明者らが同時に出願した米国仮特許出願、表題「Primers, Probes and Methods for Nucleic Acid amplification」に開示されるように、色素を刺激し、該色素からのFRET転移による蛍光団の発光を読み取った。結果を図6に挙げる。
【0094】
図6のパネルAは、反応混合物からdNTPが省略されることによって伸長が妨げられた場合に得られた融解曲線61を含む。ゆえに曲線61はプライマー・標的ハイブリッドの融解曲線であり、58℃のTmを示す。またパネルAは、dNTPを、単独で、あるいは無修飾のオリゴヌクレオチド9−3(300nM、1000nM)または9−22(50nM、100nM、300nM)とともに含有するいくつかの対照を含む。サークル62によって特定される、前記対照に関する曲線は、すべての場合で伸長を示し、すなわち58℃のプライマー融解ピークが消失した。図6のパネルBは、反応混合物中に試薬9−22DDを加えることの用量依存的効果を示す:曲線63、50nM;曲線64、100nM;曲線65、300nM。58℃のピークが大きいほど、等温性試験条件下で重合による伸長を防止する効果が大きい。図6のパネルCは、反応混合物中に試薬9−3DDを加えることの用量依存的効果を示す:曲線66、300nM;曲線67、1000nM。この場合もやはり、58℃のピークが大きいほど、試験条件下で重合による伸長を防止する効果が大きい。
【0095】
また本発明者らは、5’−3’エキソヌクレアーゼ活性を欠いているDNAポリメラーゼであるStoffel断片(Lawyer et al. (1993) PCR Methods and Applications 2: 275-287)に対する効果を調べた。鋳型、蛍光標識プライマー、Stoffel断片およびdNTPしか含有しない陰性対照と、表1に列挙される種々の化合物を比較した。この一連の試験では、40℃で伸長を実施し、SYBR Green読み取り値を30分間以上にわたって20秒毎に取得した。目視によって曲線を比較したが、その比較は信頼できるものであった。その理由は、得られた曲線が対照をほぼ正確にたどるか、あるいは得られた曲線が対照とは著しく異なっていたからである。50nM、100nMおよび300nMの濃度で試験化合物を加えた。無修飾のヘアピン6−22、9−3、9−5および9−22は、いかなる濃度においてもStoffel断片による伸長を抑制しなかった。化合物6−22DDは、化合物9−3DD、9−5DDおよび9−22DDと同様に、すべての3種の濃度で伸長を抑制した。以下の化合物は、50Mまたは100nMの濃度では伸長を抑制しなかったが、300nMの濃度では伸長を抑制した:9−22−5D、9−22−3D、9−5 2’OM4、9−5BHQBHQ、および9−32’OM4。
【実施例7】
【0096】
実施例7. ポリメラーゼ活性の非存在下でのTaqエキソヌクレアーゼ活性に対する効果
本発明者らは、DNA合成の非存在下での5’−3’エキソヌクレアーゼ活性の阻害を実証するアッセイを開発した。本アッセイでは、一方の鎖の5’末端がFAM蛍光団で標識され、他方の鎖の3’末端がDabcylクエンチャーで標識されている特異的二本鎖DNA分子を基質として使用する。蛍光団はクエンチャーと近接していて、蛍光団が刺激された場合に蛍光は生じない。加熱および冷却による鎖変性およびアニーリングによって、FAM標識鎖の切断および蛍光団の遊離が起こる。何らかのサイクル処理によってTaqポリメラーゼの5’−3’エキソヌクレアーゼ活性に関する基質が生成されると考えられる。したがって、蛍光の増大はTaqポリメラーゼの5’−3’エキソヌクレアーゼ活性の尺度を提供する。
【0097】
25μLの反応混合物は、適切な濃度の9−22DD化合物の存在または非存在下で、1×PCRバッファー中の300nMの二本鎖DNA鋳型、3mMのMgCl2、および1.25単位(U)のTaqポリメラーゼを含有する。該反応物は、いかなるdNTPをも含有せず、前記二本鎖DNA以外のいかなる他の核酸標的をも含有しない。前記二本鎖DNA鋳型の相補鎖の配列は、5’ FAM−AGTGTGATGATGGTGAGG −リン酸基3’(配列番号31)および5’ ACTTTCAACTCTGTCT 3’−Dabcyl(配列番号32)である。サンプルを95℃で3分間変性させ、次いで95℃で10秒間,67℃で30秒間,72℃で30秒間,および45℃で20秒間のサイクルに付する。各サイクル中に45℃で蛍光を取得する。
【0098】
複数のサンプルに対するアッセイの実施から得られた蛍光読み取り値を図7に報告する。図7では、読み取り値を標準化して、同じバックグラウンド蛍光で出発するようにしてある。曲線76は、Taqポリメラーゼが存在しない場合の蛍光を示す対照である。残りのサンプルにはTaqポリメラーゼを加えた。曲線71は、試薬9−22DDを加えなかった場合に得られた蛍光の増大を示す。30サイクルにわたって蛍光は安定的に増大した。曲線72〜75は、それぞれ50nM、100nM、200nMおよび300nMの濃度で試薬9−22DDを加えた場合の蛍光の増大を示す。上記反応物に9−22DD化合物を加えると、観察される蛍光の増大が用量依存的様式で減少した。
【実施例8】
【0099】
実施例8. 対称的PCR増幅
本発明者らは、剪断ゲノムDNAおよび等モル濃度で近似のTmを有する1ペアのプライマーを利用してリアルタイムPCR増幅アッセイを実施した。400nMの試薬9−3DDを加えない場合および加えた場合で反復アッセイを実施した。DNA全体としての蓄積のキネティクスに関して、SYBR Greenでの染色によって、ならびに臭化エチジウム染色を用いるゲル電気泳動によって試薬の効果を評価した。
【0100】
増幅に関する所望の標的はCFTR遺伝子のイントロン19に一塩基転位として存在する対立遺伝子であった(GenBank登録番号AC000061)。25μLの反応混合物は、400nMの試薬9−3DDの不存在または存在下で、120ピコグラム(pg)の剪断ヒトゲノムDNAの基質、1×PCRバッファー、5mMのMgCl2、250μMの各dNTP、1:40,000希釈のSYBR Green、1000nMのフォワードプライマー:配列5’ TAATTACAAGAGTCTTCCAT 3’(配列番号33)(Tm56.6℃)、および1000nMのリバースプライマー:配列5’ CATGAATAGAACATTTCCTT 3’(配列番号34)(Tm56.3℃)、および1.25単位の非ホットスタートInvitrogen Taqポリメラーゼを含有した。以下の熱サイクル処理プロファイルを使用してサンプルを増幅した:95℃で3分間;95℃で10秒間,55℃で30秒間,および72℃で30秒間の60サイクル(SYBR Green I蛍光を72℃でモニタリング)、最後に、30秒間に1℃刻みで増加する54℃〜96℃の温度勾配を用いる。反応の終了時点で、各組の反応から得られたランダムに選択された複数のサンプルを、0.5×Tris−ホウ酸−EDTA溶液(TBE)中、3.0%アガロースゲルでのゲル電気泳動に付することよって分析した。
【0101】
図8は得られた結果を示す。図8のパネルAの増幅プロットは60サイクルにわたるSYBR Green I蛍光を示し、サークル81によって特定される曲線群は試薬9−3DDを含有しない反復実験サンプルであり、サークル82によって特定される曲線群は400nMの9−3DDを含む反復実験サンプルである。2組のサンプルから生じた蛍光シグナルのキネティクスによれば、二本鎖DNA全体は、試薬9−3DDの非存在下で、該試薬の存在下よりいくらか速やかに蓄積したが、60サイクルによって蓄積された二本鎖DNAの総量は2組の反応において実質的に同一であることが実証される。
【0102】
図8のパネルBは、臭化エチジウム染色を施した電気泳動ゲルを示す。左側の図では、レーンaはサイズマーカー(50塩基対ずつ異なるサイズ)であり、レーンb〜iは試薬9−3DDを含まないサンプルである。右側の図では、レーンj〜qは400nMの9−3DDを含むサンプルであり、レーンrはサイズマーカーである。この結果は、本発明の試薬を含有しない反応では、適正な産物ならびに高分子量の多数の非特異的産物が生成されたが、試薬9−3DDを含有する反応では、適正な産物しか生成されなかったことを示す。さらに、ゲル中の適正な産物のバンドの相対強度によって判断すると、試薬9−3DDを含有する反応では、該試薬を含有しない反応の約2倍の適正な産物が生成された。
【0103】
キネティクス解析および電気泳動による解析の組み合わせでは、非特異的産物を生成する反応81が、適正産物しか生成しない反応82より早く蛍光シグナルを生じたことが示される。SYBR Green色素は配列特異性とは無関係に二本鎖DNA内に挿入される。ゆえに、反応81の直線的なキネティクスと比較して、よりS字状である反応82のキネティクスは、対称的反応が適正な産物のみを蓄積しているか否かを判断するために使用することができる。
【実施例9】
【0104】
実施例9. エキソヌクレアーゼ阻害の定量化
本実施例は、TaqDNAポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する本発明の試薬の阻害効果を測定するための厳密なアッセイを記載する。本アッセイでは、M. W. Kaiser, N. Lyamicheva, W. Ma, C. Miller, B. Neri, L. Fors, and V. I. Lyamicheva in J. Bio. Chem., 274, pp. 21387-21394(1999)に記載の系と同様のプライマー−鋳型系を使用した。ただし、鋳型の5’末端をCy5で標識し、二本鎖DNAに結合すると蛍光を発するDNA色素であるSYBR Green Iの存在下でアッセイを実行した点で異なる。本アッセイ用の鋳型は、ヌクレオチド配列:5’−ACGAGCGTCTTTC−3’(配列番号36)を有するプライマーにアニーリングするオリゴヌクレオチド配列5’−Cy5−AAAACGCTGTCTCGCTGAAAGCGAGACAGCGAAAGACGCTCGT−3’(配列番号35)からなる。長い方のオリゴヌクレオチドは、異なる長さの2個の一本鎖尾部を有するヘアピン構造を形成する。鋳型の短い5’尾部はCy5蛍光団を含有し、4アデノシン残基からなり;鋳型の長い方の3’尾部は、プライマーオリゴヌクレオチドに対する標的となり、これにより、該プライマーが1塩基対の重複を有して5’の鋳型尾部の直前に位置する。この鋳型−プライマー複合体にSYBR Green Iを加えると、該複合体の二本鎖DNA領域に結合したSYBR Green Iが生じる。480nmレーザーでSYBR Green Iが励起されると、結合したDNA色素からの蛍光エネルギーがCy5によって完全に吸収され、それは蛍光共鳴エネルギー転移によって生じると考えられ、最大SYBR Green発光波長(最大発光波長:521nm)で検出可能な蛍光は存在しない。Taqポリメラーゼの5’−3’エキソヌクレアーゼ活性によって5’尾部が加水分解されると、鋳型−プライマー複合体からCy5部分が除去され、検出可能なSYBR Green I蛍光が回復する。TaqDNAポリメラーゼの存在下でのSYBR Green I蛍光の回復についてのキネティクスは、5’−3’エキソヌクレアーゼ活性の定量的尺度を提供する。本発明の試薬はTaqポリメラーゼの5’−3’エキソヌクレアーゼ活性を阻害し、SYBR Green I蛍光の増大を減速させる。
【0105】
本アッセイは、25マイクロリットル(μL)の容量中、50nM、100nM、300nM、または1000nMの9−22DD化合物の不存在または存在下で、1×PCRバッファー中で混合された0.5μMの上記鋳型および1.5μMの上記プライマー、3mMのMgCl2、1:40000希釈の市販のSYBR Green Iストック溶液(Molecular Probes, Eugene, OR)、1.25UのTaqポリメラーゼ(Invitrogen, Calrsbad, CA)から構成されるものとした。該反応混合物にはdNTPを加えなかった。その理由は、本アッセイがDNAポリメラーゼ活性に依拠しないからである。鋳型およびプライマーを含まない反応混合物を25℃で調製して、9−22DD化合物およびTaqDNAポリメラーゼ間の相互作用を促進させた。次いで、サンプルを氷上に置き、鋳型およびプライマーを加え、反応の開始まで氷上で置いた。陰性対照にはTaqDNAポリメラーゼを加えなかった。ABI Prism Sequence Detector 7700にて25℃で60分間サンプルをインキュベートすることによって反応を開始し、SYBR Green Iチャネルにおける蛍光収集を30秒毎に行った。各対照および各濃度の試薬9−22DDに関して3回の異なる試行を平均した。
【0106】
図9は本アッセイの結果を示す。検出可能なSYBR Green I蛍光は、5’−3’エキソヌクレアーゼ活性の程度についての尺度を提供する。「Taqなし」の対照はSYBR蛍光シグナルを示さず、Cy5によるSYBR Green I蛍光の完全吸収および5’−3’エキソヌクレアーゼ活性の完全な不存在と矛盾しない(ライン96)。Taqポリメラーゼを加えると、SYBR Green I蛍光が回復し、当該アッセイ条件下での5’−3’エキソヌクレアーゼ活性の最大レベルに関するベースラインが提供される(ライン91)。反応混合物に9−22DD化合物を加えると、Taqポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する9−22DDの阻害効果に起因して、SYBR Green I蛍光の回復が用量依存的様式で減速される(ライン92、50nM;ライン93、100nM;ライン94、300nM;ライン95、1000nM)。以下の表IIIでは、25℃で60分間のインキュベーション後に達成される相対的SYBR Green I蛍光のレベルに基づいて、種々の濃度の9−22DD化合物による5’−3’エキソヌクレアーゼ活性の阻害割合を定量化する。
【表3】
【実施例10】
【0107】
実施例10. 標的DNAの存在下および非存在下でのプライマー二量体形成およびオリゴマー形成の防止
最終容量25μL中の100ゲノムのヒト胎盤DNA(Sigma, St Louis, MO)の存在下または非存在下で、一連のLATE−PCR増幅反応を準備した。この実験では、LATE−PCR増幅反応物は、1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.25mMのdNTP、1000nMの過剰プライマー、50nMの制限的プライマー、0.25μMのFAM標識プローブ鎖、0.3μMのDabcyl標識リバース相補鎖、1.25単位のPlatinum TaqDNAポリメラーゼ(Invitrogen, Carlsbad, CA)から構成するものとした。プライマーおよびプローブの配列は以下の通りであった:
過剰プライマー:5’ GTTTCTTTGCTGCCGTGTTC 3’(配列番号37)
制限的プライマー:5’ CCCCAGAGACCCCAGTTGCTAACCAGAC 3’(配列番号38)
FAM標識プローブ鎖:5’ [TET]AGACAGAGTTGAAAGTCAGG[Phos] 3’(配列番号39)
Dabcyl標識リバース相補鎖:5’ ACTTTCAACTCTGTCT[Dabcyl] 3’(配列番号40)。
【0108】
前記プライマーおよびプローブは、ヒトp53遺伝子のエキソン5および6を包含する488塩基対(bp)のアンプリコンを増幅および検出する。
【0109】
ABI Prism 7700 Sequence Detector(Applied Biosystems, CA)で増幅を実行した。その場合、95℃で3分間の1サイクル;95℃で10秒間,64℃で30秒間,75℃で30秒間の25サイクル;および95℃で10秒間,64℃で30秒間,75℃で30秒間,45℃で20秒間の35サイクルからなる熱プロファイルを用い、45℃ステップ中にTETチャネルにおいて蛍光を検出した。
【0110】
得られた増幅産物を、0.5×TBEバッファー中で、2時間、3%アガロースゲルでのゲル電気泳動に付することよって分析し、臭化エチジウムで染色した。結果を図10に示す。レーン3、5、7、9、11のサンプルはゲノムDNAを加えずに調製されたものである。レーン2、4、6、8、10のサンプルはゲノムDNAを加えて調製されたものである。レーン1には100塩基対ラダーの電気泳動用サイズマーカーを含めた。試薬9−22DDを以下のように出発反応物に加えた:レーン2および3、0nM;レーン4および5、50nM;レーン6および7、100nM;レーン8および9、200nM;レーン10および11、300nM。
【0111】
ゲルの右側に隣接して、適正産物のサイズ、ならびにプライマー二量体およびプライマーオリゴマーであると推測されるサイズをマークした。図10に示される結果は、ゲノムDNAを加えて開始された反応において、試薬9−22DDの濃度を増加させると、プライマー二量体およびプライマーオリゴマーを含めた非特異的産物の出現を防止するように用量依存的様式で作用し、それによって適正産物の特異性および収量の両者が高まることを実証する。また増加濃度の試薬9−22DDは、ゲノムDNAを含有しない反応においても、プライマー二量体およびプライマーオリゴマーの出現を防止する。
【実施例11】
【0112】
実施例11. 二重反応におけるプライマー二量体形成の防止
最終容量25μL中の100ゲノムのヒト胎盤DNA(Sigma, St Louis, MO)の存在下または非存在下で、一連のLATE−PCR増幅反応を準備した。この実験では、LATE−PCR増幅反応物は、1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.20mMのdNTPから構成するものとした。サンプルごとに1.25単位のTaqポリメラーゼを使用した。第一の増幅標的配列(産物1)は嚢胞性線維症遺伝子のエキソン11の一部分であり、100nMの制限的プライマー:5’ GACGTTTACAGCGAATGCTTGCTAGACCAAT 3’(配列番号41)および2,000nMの過剰プライマー:5’ TCCAAGTTTGCAGAGAAAGACAAT 3’(配列番号42)を用いて増幅した。第二の増幅標的配列(産物2)は嚢胞性線維症遺伝子のエキソン10の一部分であり、50nMの制限的プライマー:5’ CAGTTTTCCTGGATTATGCCTGGCACCAT 3’(配列番号43)および1000nMの過剰プライマー:5’ GCTTTGATGACGCTTCTGTATCTA 3’(配列番号44)を用いて増幅した。
【0113】
ABI Prism 7700 Sequence Detector(Applied Biosystems, CA)において、95℃で2分間、次いで95℃で10秒間,56℃で15秒間,70℃で20秒間の25サイクル、次いで95℃で10秒間,56℃で15秒間,70℃で20秒間,および45℃で30秒間(蛍光の取得を伴う)の50サイクルで増幅を実行した。
【0114】
得られた増幅産物を、0.5×TBEバッファー中の3%アガロースゲルでの2時間のゲル電気泳動に付すことによって分析し、臭化エチジウムで染色した。結果を図11に示す。レーン1〜6で分析された反応では、非ホットスタートTaqポリメラーゼを利用し、レーン7〜12で分析された反応では、Taqポリメラーゼに加えてホットスタート抗体を利用した。レーン4〜6および10〜12で分析された反応では、100nMの試薬9−22DDを加えた。
【0115】
ゲルの右側に隣接して、産物1および産物2の一本鎖アンプリコン(ssDNA)および二本鎖アンプリコン(dsDNA)、ならびにプライマー二量体であると推測される短い産物を含めた増幅産物の特定についての解釈を付した。他の非特異的産物はアスタリスクでマークされる。非ホットスタートポリメラーゼを用い、本発明の試薬を用いない増幅(レーン1〜3)では、プライマー二量体および他の非特異的産物が生成された。ホットスタートポリメラーゼを用い、本発明の試薬を用いない増幅(レーン7〜9)では、概して、いくらか不純物が減少したが、依然としてミスプライミングの現象が示された。本発明に基づく試薬をホットスタート増幅に加えたレーン10〜12では、目的の産物のみが生成された。本発明の試薬を非ホットスタート増幅に加えたレーン4〜6では、プライマー二量体が排除され、3回の反復実験のうちの2回(レーン4およびレーン6)では、すべての他の非特異的産物も同様に排除されたが、1回の反復実験(レーン5)のみでは非特異的産物が出現した。図11に示される結果は、非ホットスタートTaqポリメラーゼまたはホットスタートTaqポリメラーゼが使用された場合、低濃度(100nM)の試薬9−22DDが、プライマー二量体の形成を防止するために十分であったこと、さらに、その低濃度の試薬9−22DDが、2ペアのプライマーを用いる2種の標的の増幅において非ホットスタートTaqポリメラーゼとともに使用された場合でさえ、ミスプライミングのすべての現象を防止するためにほぼ十分であったことを実証する。
【実施例12】
【0116】
実施例12. プライマー二量体形成の防止による二重リアルタイムPCRおよびリアルタイムPCRのキネティクスの最適化
マウスOct4およびXist遺伝子のエキソン内の配列(それぞれGenBank登録番号NM_013633およびL04961)を同時に増幅するための二重リアルタイムLATE−PCRアッセイを設計した。各反応は最終容量50μL中で実行し、以下の試薬を含有した:20mMのTris−HCl,pH8.4、および50mMのKClによって構成される1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.4mMの各dNTP、配列5’ TGGCTGGACACCTGGCTTCAGACT 3’(配列番号45)を有する50nMのOct4制限的プライマー、配列5’ CAACTTGGGGGACTAGGC 3’(配列番号46)を有する2μMのOct4過剰プライマー、配列5’ GGTCGTACAGGAAAAGATGGCGGCTCAA 3’(配列番号47)を有する100nMのXist制限的プライマー、配列5’ TGAAAGAAACCACTAGAGGGCA 3’(配列番号48)を有する2μMのXist過剰プライマー、配列5’ TET−CCG CCT GGG ATG GCA TAC TGT GGA AGG CGG−Dabcyl 3’(配列番号49)を有する1μMの低TmのOct4分子ビーコンおよび2単位の抗体複合体化Platinum(登録商標)Taq DNAポリメラーゼ(Invitrogen, Carlsbad, CA)。さらに化合物9−3DDまたは化合物9−3bDDを、以下に指定される濃度でPCR混合物に加えた。本実施例では、Xistアンプリコンを検出するための分子ビーコンを加えなかった。各アッセイは、PurAmpプロトコル(Hartshorn et al. (2005) BMC Biotechnol 5: 2を参照のこと)にしたがって、2.5μL容量中に細胞溶解および逆転写に必要な試薬をさらに含有した。この二重LATE−PCRでは、前記試薬の最終濃度は以下の通りであった:2.5mMのTris酢酸,pH8.4、3.75mMの酢酸カリウムおよび0.4mMの酢酸マグネシウム(希釈したcDNA合成バッファー, ThermoScriptTM RT-PCR System, Invitrogen, Carlsbad, CA)、追加の50μMの各dNTP、0.13mMのイソチオシアン酸グアニジン、6.7μMのβ−メルカプトエタノール、0.7μMのクエン酸ナトリウム,pH7.0、0.7×10−4%(vol/vol)のジメチルスルホキシドおよび0.2×10−4%のサルコシル(sarcosyl)。
【0117】
さらにマウスゲノムDNA(Sigma, St Louis, MO)を各アッセイに加え、PCR増幅用の鋳型を提供した。各試験管に加えるゲノム数は6pg/ゲノムサイズに基づいて算出した(Vendrely and Vendrely (1949) Experientia 5: 327-329を参照のこと)。
【0118】
ABI Prism 7700 Sequence Detector(Applied Biosystems, CA)において増幅を実行した。その場合、95℃で5分間の1サイクル;95℃で10秒間,63℃で20秒間,および72℃で30秒間の15サイクル;および95℃で15秒間,55℃で25秒間,72℃で35秒間,および45℃で30秒間の40サイクルから構成される熱プロファイルを用い、TETチャネルにおいて45℃で蛍光を取得した。
【0119】
PCRの終了時点で、増幅産物を、0.5×TBEバッファー中の3%アガロースゲルでの2時間のゲル電気泳動に付すことによって分析し、臭化エチジウムで染色した。2種の同時に増幅された遺伝子に関して、予測されたサイズの二本鎖および一本鎖産物がともに可視であった。このことは二重LATE−PCRが有効であることを示す。
【0120】
図12は、Oct4およびXist遺伝子内の2種の非相同配列を増幅するための2組のプライマーを含有する上記二重LATE−PCRのキネティクスに対する、試薬9−3DDのステム組成を変化させることの効果を示す。該図は、TET−Oct4分子ビーコンとのハイブリダイゼーションを介してOct4アンプリコンの蓄積によって生じる蛍光シグナルを示す。その結果は、分析された各ゲノム濃度(10ゲノム、サークル122;100ゲノム、サークル123;1000ゲノム、サークル124)で、300nMの9−3DD(三角形付きの線)または同濃度のその修飾型である9−3bDD(三角形が付いていない太線)の存在下でCT値が非常に近いことを実証する。そうではなくむしろ、リアルタイム蛍光シグナルのキネティクスは試薬のステムの組成によって影響を受ける。化合物9−3bDDは、9−3DDと比較して高いTmのステムを有し、ゆえに、プライマー二量体形成を最適に防止すると本発明者らは考え、その結果、すべての試験ゲノム濃度で非常に直線的で平行なシグナルが生じる。一方、低ストリンジェントの化合物9−3DDの存在下では、蛍光シグナルの一部は急な勾配を有するが、他のものは反応の早期にプラトーに達し始め、それはプライマー二量体のランダムな形成を示す。種々の鋳型濃度で化合物9−3DDを用いて生じるシグナルは、化合物9−3bDD(改変型ステムを有する)の存在下で得られるシグナルより平行ではない。理想的には、完全に平行なシグナル(一定勾配を有する)を生じる直線的増幅が望ましく、エンドポイントタイプの解析に関しては特に妥当である。
【0121】
9−3DDを含有するサンプルのアガロースゲル解析では、特に多数の鋳型数で可視の、プライマー二量体であろうバンドが示され、少数の鋳型数での非常に小さいバンドはプライマーと合致する。そのようなバンドは9−3bDDを含有するサンプルの分析では現れなかった。
【0122】
図13は、本実施例に記載の、図12に関しても使用された二重LATE−PCRにおいて、試薬9−3bDDの濃度を変化させることの効果を示す。この場合、試薬9−3bDDの濃度を300nM(三角形が付いていない太線)から200nM(三角形付きの線)に低下させると、アガロースゲル解析によって確認されるように、やはり、プライマー二量体形成に起因して直線的増幅の勾配に影響する。結果として、少数の当初鋳型数(10ゲノム、サークル132)を含有するサンプルの一部は、最後のサイクル(「エンドポイント」)で、多数の当初鋳型数(100ゲノム、サークル133および1000ゲノム、サークル134)を含有するサンプルより高い蛍光を有する。
【0123】
PCR効率に対する、本出願に記載される試薬の濃度の効果を、1ペアのプライマーで1鋳型を増幅するLATE−PCRにおいてさらに試験した。増幅対象の鋳型は、図12および図13に関して記載された二重反応で使用されたのと同じマウスOct4配列であり、プライマーおよび分子ビーコン配列も該反応と同一であった。
【0124】
各反応は最終容量100μL中で実行し、以下の試薬を含有した:20mMのTris−HCl,pH8.4、および50mMのKClによって構成される1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.25mMの各dNTP、50nMのOct4制限的プライマー、2μMのOct4過剰プライマー、1μMの低TmのTET−Oct4分子ビーコン、および2単位の抗体複合体化Platinum(登録商標)Taq DNAポリメラーゼ(Invitrogen, Carlsbad, CA)。さらに化合物9−3DDを、150、または300または450nMの濃度でPCR混合物に加えた。上述の二重反応の場合(図12および図13を参照のこと)と同様に、各アッセイは、PurAmpプロトコルにしたがって、10.5μL容量中に細胞溶解および逆転写に必要な試薬をさらに含有した。このLATE−PCRでは、前記試薬の最終濃度は以下の通りであった:5mMのTris酢酸,pH8.4、7.5mMの酢酸カリウムおよび0.8mMの酢酸マグネシウム(希釈したcDNA合成バッファー)、1ng/μLのランダムヘキサマーおよび追加の100μMの各dNTP(ThermoScriptTM RT-PCR System, Invitrogen, Carlsbad, CAの全構成要素)、0.4mMのイソチオシアン酸グアニジン、20μMのβ−メルカプトエタノール、2μMのクエン酸ナトリウム,pH7.0、2×10−4%(vol/vol)のジメチルスルホキシドおよび0.5×10−4%のサルコシル。
【0125】
さらにマウスゲノムDNAを各アッセイに加え、本実施例に記載の二重LATE−PCRに関して指定される、PCR増幅用の鋳型を提供した。ABI Prism 7700 Sequence Detectorにおいて、二重反応に関して詳述されるプロファイルと同じ熱プロファイルを用いて、増幅を実行した。PCRの終了時点で、増幅産物を、0.5×TBEバッファー中の3%アガロースゲルでの2時間のゲル電気泳動に付すことによって分析し、臭化エチジウムで染色した。ゲル上では二本鎖および一本鎖産物がともに可視であり、予測されるサイズを有した。このことはLATE−PCRによる増幅が有効であることを示す。
【0126】
さらにOct4鋳型の増幅効率に対する増加濃度の化合物9−3DDの効果を試験した。150nM、300nM、450nMの9−3DDの各試験濃度で実施された5ゲノム(9−3DDの各試験濃度に関して2回の反復実験の平均)、20ゲノム(9−3DDの各試験濃度に関して2回の反復実験の平均)、100ゲノム、および1000ゲノムを含有するリアルタイムPCRアッセイからCT値を取得した。各濃度での各CT値のシリーズに関する直線回帰を図14に示す。図14では、三角は450nMの濃度であり、白抜きの四角は300nMであり、黒い四角は150nMである。このプロットからすぐにわかることは、450nMの9−3DDが閾値を超える蛍光シグナルの出現を大きく遅延させ、さらにPCR増幅の用量依存性を変更することである(回帰直線から離れている10遺伝子コピーおよび40遺伝子コピーの点を参照のこと)。一方、150nMまたは300nMの9−3DDを使用して得られたCT値は非常に類似し、鋳型コピー数および蛍光シグナルの最初の出現の間の直線的関連性を実証する。
【0127】
しかし、蛍光のリアルタイムパターンの分析では、図15に示されるように2つの条件(濃度)間の差異が強調される。100ゲノム、サークル151、または1000ゲノム、サークル152によって作成される曲線の勾配は、300nMの9−3DDが使用された場合(三角形が付いていない太線)に、150nMの9−3DD(三角形付きの線)が使用された場合より急勾配であり、よりストリンジェントな300nMの条件がプライマー二量体を排除し、ゆえに増幅効率を高めることが示唆される。
【実施例13】
【0128】
実施例13. 本発明に基づく複数の試薬の使用
ヒトゲノムDNA出発材料の5種の異なる標的配列を個別に増幅するために、および多重反応においてすべての5配列を一緒に増幅するために、LATE−PCR反応混合物を調製した。各標的配列はその独自のペアの制限的および過剰プライマーを必要とした。前記標的配列は、(1)191塩基対のグロビン対立遺伝子、(2)312塩基対のテイ・サックスG269対立遺伝子、(3)452塩基対のテイ・サックス1278および1421対立遺伝子、(4)ミトコンドリア超可変領域1の549塩基対セグメント、および(5)p53遺伝子エキソン7および8を含む611塩基対セグメントであった。25μLの反応混合物は、1×PCRバッファー(Invitrogen)、0.4mMのdNTP、3mMのMg++、0.24×SYBR Greenおよび1.5単位のTaqDNAポリメラーゼ(Invitrogen)を含有した。制限的プライマーに関しては50nMおよび過剰プライマーに関しては1000nMの濃度でプライマーを加えた。すべての5ペアのプライマーを多重反応に加えた。熱サイクル処理は、95℃で10秒間,64℃で20秒間,および72℃で1分間の45サイクルであった。
【0129】
個別のプライマーペアおよび個別の標的を含む反応混合物、ならびにすべての5ペアのプライマーおよびすべての5種の標的を含む反応混合物を、25nMの化合物9−22DDおよび100nMの化合物12−3DDの組み合わせの存在下で増幅した。さらに、本発明に基づくいかなる試薬をも加えずに五重反応混合物を増幅した。反応産物をゲル電気泳動によって調べた。種々の増幅のゲル電気泳動の結果を図16に示す。図16は、レーンrにサイズマーカー(100塩基対ずつ異なるサイズ)を含む。中央のレーンg〜kはそれぞれ標的配列(1)〜(5)の個別の増幅の産物である。レーンl〜qは、化合物9−22DDおよび化合物12−3DDの混合物を加えた6回反復の五重増幅の産物である。レーンa〜fは、本発明の試薬を加えなかった6回反復の五重増幅の産物である。図16は、いずれの化合物も加えない多重反応と比較して、化合物の混合物が増幅を増強した(より多量で純粋な所望のアンプリコンが得られた)ことを示し、本発明のキット、増幅およびアッセイにおいて試薬の混合物を使用しうることを実証する。
【実施例14】
【0130】
実施例14. 作用様式の試験
本発明者らは、本発明の試薬のミスプライミングの低減(特異性改善)およびポリメラーゼ阻害効果を定量的に研究する試験を考案した。該試験は第1の作用様式と第3の作用様式を識別すると考えられる。該試験は、実施例1に記載のアッセイと本質的に同様のPCR増幅アッセイであるが、以下の例外を有する:増幅反応混合物は半数(1000ゲノム)の剪断ゲノムDNAを含有し、熱サイクル処理の最終部分は70サイクルから40サイクルに減少される。種々の量、典型的には25nM、50nM、100nM、300nM、1500nMおよび3000nMの本発明の試薬を加えて増幅を実施する。試験は2種の分析を含む:第一は、特異性に関する、増幅産物の融解曲線の分析であり;第二は、阻害に関する、合成二本鎖産物のリアルタイム蛍光曲線の分析である。
【0131】
図17〜18は、本発明に基づく2種の試薬(それぞれ化合物12−3DDおよび化合物12−C3DD)に関する分析結果の一部分を示す。各図のカラムIは、表示通りの数種の選択された濃度の試薬に関する融解曲線を示す。各図のカラムIIは、表示通りの同濃度に関するリアルタイム蛍光曲線を示す。図17では、カラムIの融解曲線は、化合物12−3DDが100nM以上の濃度で高い特異性(ミスプライミングの回避)を達成したことを示した(最低濃度100nMは、実施例1に記載の反応に関して見出された最低濃度よりわずかに高い)。図17では、カラムIIのリアルタイム蛍光曲線は、化合物12−3DDの濃度が100nMを超えて増加するにつれて、1000nMを超える濃度で反応が本質的に停止されるまで、ポリメラーゼ阻害が進行的に増大したことを示した。阻害は、プラトーの蛍光レベル、およびCT値の遅延(100nMでは19.6、300nMでは21.1、1500nMおよび3000nMでは存在せず、すなわち少なくとも40)によって示される、生成二本鎖産物(群)全体(所望の特異的産物およびミスプライミングの非特異的産物を含む)の量が少ないことに反映される。図18では、カラムIの融解曲線は、化合物12−3DDが300nM以上の濃度で高い特異性を達成したことを示した(最低濃度300nMは、実施例1に記載の反応に関して見出された最低濃度と同一である)。図18では、カラムIIのリアルタイム蛍光曲線は、化合物12−C3DDの濃度が300nMを超えて増加するにつれて、ポリメラーゼ阻害が進行的に増大した(CT値は100nMでは18.8、300nMでは18.8、1500nMでは21.1および3000nMでは24.5であった)が、該反応は3000nMの濃度でさえ停止されなかったことを示した。
【0132】
図17と図18を比較すると、化合物12−C3DDの総合的な性能では、化合物12−3DDの総合的な性能より濃度依存性の程度が低いことがわかる。ゆえに、この特定の増幅反応では、化合物12−C3DDの濃度が300nMから1500nMになると生じるCT遅延およびプラトー蛍光の減少は、化合物12−3DDの濃度が100nMから300nMになると生じる結果とおおむね同一である。
【0133】
本発明の多数の実施形態を記載してきた。しかしながら、本発明の精神および範囲から逸脱することなく種々の修飾を施してよいことが理解されよう。したがって、他の実施形態は特許請求の範囲内である。
【技術分野】
【0001】
技術分野
本発明は、ポリメラーゼ連鎖反応(PCR)を利用する核酸増幅反応およびアッセイに関し、それにはリアルタイムアッセイおよびエンドポイントアッセイの両者のホモジニアスアッセイが含まれる。
【背景技術】
【0002】
背景
ポリメラーゼ連鎖反応(PCR)を利用する核酸増幅(PCR増幅を含むアッセイなど)は周知である。米国特許第4,683,202号、同第4,683,195号および同第4,965,188号、および、一般には、PCR PROTOCOLS, a guide to Methods and Applications, Innis et al.(編), Academic Press(San Diego, CA (USA) 1990)を参照されたい。未結合検出試薬またはプローブを除去するための洗浄を必要とせず、ゆえに増幅反応容器を開けることなく実施できるホモジニアスPCRアッセイもまた周知である。ホモジニアスPCRアッセイには、増幅反応の終了時点で増幅産物を検出するエンドポイントアッセイ、および反応が進行しているときに一部のまたはすべての熱サイクル中に増幅産物を検出するリアルタイムアッセイの両者が含まれる。米国特許第5,994,056号、同第5,487,972号、同第5,925,517号および同第6,150,097号を参照されたい。
【0003】
PCR増幅反応は、概して、対称的であるように、すなわち、「対応する(matched)」フォワードプライマーおよびリバースプライマーを利用して二本鎖アンプリコンを作り出すように設計され;すなわち、それらは可能な限り近い融解温度を有し、かつそれらは等モル濃度で反応物に添加される。PCR反応において、直接的に一本鎖DNAを作り出すために限られた用途が見出されている技術は、「非対称的PCR」である。Gyllensten and Erlich, "Generation of Single-Stranded DNA by the Polymerase Chain Reaction and Its Application to Direct Sequencing of the HLA-DQA Locus," Proc. Natl. Acad. Sci. (USA) 85: 7652-7656 (1988);および米国特許第5,066,584号。非対称的PCRは、プライマーの一方が制限的な量で添加され、典型的には他方のプライマーの濃度の1〜20%の量で添加される点で対称的PCRと異なっている。
【0004】
さらに最近、本発明者らは、「直線後指数的(Linear-After-The-Exponential)」PCRまたは、略して「LATE−PCR」として知られる非対称的PCR増幅法を開発した。Sanchez et al. (2004) PNAS 101: 1933-1938, Pierce et al. (2005) PNAS 102: 8609-8614、および公開されている国際特許出願WO 03/054233(2003年7月3日)を参照のこと。前記文献は参照によりその全体が本明細書中に組み入れられる。LATE−PCRでは、Tm[0]と称される増幅の出発時点でのPCRプライマーの実際の融解温度を考慮に入れる。Tm[0]は、非天然ヌクレオチドが使用される場合に必要であるように、経験的に決定することができ、あるいは「近接(nearest neighbor)」法(Santa Lucia, J. (1998) PNAS (USA) 95: 1460-1465;およびAllawi, H. T. and Santa Lucia, J. (1997) Biochem. 36: 10581-10594)にしたがって、塩濃度の補正を使用して、算出することができる。本発明者らの研究では、0.07Mの一価塩濃度を利用した。
【0005】
LATE−PCRの場合には低減されるPCR増幅の望ましくない特徴は、反復実験(replicates)間のばらつき(scatter)である。対数期の増幅に続いてリアルタイムで追跡される反復実験増幅はそれぞれ相違し、種々のレベルでプラトーに達する。ばらつきとは、反復実験が同一の反応キネティクスを有さず、精度を低下させることを意味する。このことはPCRアッセイ全般に関する問題であるが、特にエンドポイントアッセイおよび直線期のシグナルの勾配に依存するアッセイに関して問題である。
【0006】
PCR増幅に伴う別の重大な問題はミスプライミングである。本発明者らの考えでは、ミスプライミングは以下の少なくとも3種のタイプで現れる:タイプ1、増幅開始前の反応混合物の調製中に生じるミスプライミング;タイプ2、サイクル温度がプライマーの融解温度よりかなり低いいずれかの温度を含む場合に、増幅中に生じるミスプライミング;およびタイプ3、高濃度のアンプリコンが作り出された後に継続されるPCR増幅の後期段階に生じるミスプライミング。第1のタイプのミスプライミングに対処するためにいくつかのアプローチが使用されている。アプローチの1つは、ポリメラーゼを化学的に修飾して、高温、例えば95℃に加熱されるまで不活性であるようにすることである。米国特許第5,677,152号および同第5,773,258号を参照されたい。別のアプローチは、ポリメラーゼに抗体を結合させ、反応物が高温、例えば95℃に加熱されて該抗体が不可逆的に変性するまで該ポリメラーゼを阻害することである。米国特許第5,338,671号を参照されたい。さらに別のアプローチは、反応混合物中にアプタマーを含ませることである。Doug and Jayasena (1996), J. Mol. Biol. 264: 268-278および米国特許第6,020,130号を参照のこと。アプタマーは約30ヌクレオチド長の一本鎖オリゴヌクレオチドであり、ポリメラーゼに結合して、低温で陥凹3’末端を伸長するその能力を阻害する。アプタマーは、PCRサイクルの典型的な最高温度である95℃で不可逆的に変性しない。Kainz et al. (2000) Biotechniques 28: 278-282では、16〜21ヌクレオチド長を有する、一定量の二本鎖DNA断片をPCR反応混合物に加えると、典型的なPCR伸長温度より低い温度でポリメラーゼが阻害され、非特異的産物の合成が抑制されることが報告された。DNA断片はPCRサイクル処理中に不可逆的に変性しない。Eppendorf-5 Prime, Inc.は、温度依存的様式でTaqポリメラーゼに結合し、約50℃より低い温度で二本鎖DNAに対するその結合を阻害するとされている専売のリガンドを市販している。前述の多数の試みにもかかわらず、ミスプライミングは依然としてPCR増幅に伴う問題である。
【0007】
PCR増幅中のミスプライミングの別の現象はプライマー二量体形成および増幅として知られている。この現象では、一方のプライマーが他方のプライマーまたは自身とハイブリダイズし、次いで3’末端の伸長を受けて、小さい二本鎖アンプリコンが生成される。その後、アンプリコンはさらに増幅されるか、あるいは多量体化されてさらに増幅されうる。プライマー二量体形成は標的の非存在下で生じうる。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】米国特許第4,683,202号
【特許文献2】米国特許第4,683,195号
【特許文献3】米国特許第4,965,188号
【特許文献4】米国特許第5,994,056号
【特許文献5】米国特許第5,487,972号
【特許文献6】米国特許第5,925,517号
【特許文献7】米国特許第6,150,097号
【特許文献8】米国特許第5,066,584号
【特許文献9】国際特許出願WO 03/054233
【特許文献10】米国特許第5,677,152号
【特許文献11】米国特許第5,773,258号
【特許文献12】米国特許第5,338,671号
【特許文献13】米国特許第6,020,130号
【非特許文献】
【0009】
【非特許文献1】PCR PROTOCOLS, a guide to Methods and Applications, Innis et al.(編), Academic Press(San Diego, CA (USA) 1990)
【非特許文献2】Gyllensten and Erlich, "Generation of Single-Stranded DNA by the Polymerase Chain Reaction and Its Application to Direct Sequencing of the HLA-DQA Locus," Proc. Natl. Acad. Sci. (USA) 85: 7652-7656 (1988)
【非特許文献3】Sanchez et al. (2004) PNAS 101: 1933-1938
【非特許文献4】Pierce et al. (2005) PNAS 102: 8609-8614
【非特許文献5】Santa Lucia, J. (1998) PNAS (USA) 95: 1460-1465
【非特許文献6】Allawi, H. T. and Santa Lucia, J. (1997) Biochem. 36: 10581-10594
【非特許文献7】Doug and Jayasena (1996), J. Mol. Biol. 264: 268-278
【非特許文献8】Kainz et al. (2000) Biotechniques 28: 278-282
【発明の概要】
【発明が解決しようとする課題】
【0010】
リアルタイム検出方法によるPCR増幅の定量的解析が可能になっていて、その場合、反応の閾値サイクルすなわちCTを超えると蛍光シグナルが可視になるPCRサイクルにより出発標的濃度が示唆される。エンドポイント解析は良くても半定量的である。その部分的な原因は、反応が指数的増幅から脱する時の反復実験間のばらつきである。二本鎖アンプリコンの電気泳動による解析は半定量的であり、蛍光標識プライマーを利用する場合がある。蛍光標識プローブ、対立遺伝子識別プローブまたはミスマッチ寛容プローブを利用するエンドポイント解析もまた、良くても半定量的である。ばらつきを低減させ、一本鎖産物を生成することによって、LATE−PCRは、エンドポイント解析における大きな改善を提供するが、反復実験間のばらつきは完全には排除されないことが多く、定量的多重検出は依然として所望の精度より低精度である。
【課題を解決するための手段】
【0011】
本発明の一態様は、PCR増幅反応において生成物の特異性を改善し、かつミスプライミングの影響を排除する、あるクラスの試薬添加剤である。該添加剤はすべてのタイプのPCRにおいて既存の「ホットスタート」方法論より性能が優れていて、その使用により、反応の初期、および多数のサイクル(典型的に60サイクル以上)を有するLATE−PCR反応中のいずれにおいても、プライマー二量体およびミスプライミングされたアンプリコンを含む望ましくない生成物の蓄積を防止することができる。
【0012】
本発明の別の態様は、PCR増幅およびアッセイ方法、対称的PCRまたは非対称的PCRの両者であり、非限定的にLATE−PCRが挙げられ、ならびに前記試薬添加剤を含むキット、部分的キット、およびオリゴヌクレオチドセットである。
【0013】
要旨
本発明の試薬は、少なくとも一部のPCR増幅においてミスプライミングの1種以上の現象を防止することが可能な添加剤である。「現象を防止する(防ぐ)」とは、反応の終了時点で、本明細書中に記載の技術、すなわち蛍光性DNA色素、ゲル電気泳動、DNAシークエンシングおよび融点解析によって、ミスプライミングの生成物または生成物群が検出されないことを意味する。本発明に基づく試薬は、重合活性および5’−3’エキソヌクレアーゼ活性(5' - to - 3' exonuclease activity)の両方を有するポリメラーゼを利用する場合でさえ、増幅開始前に、1マイクロモル濃度(μM)(すなわち1000nM)未満、好ましくは650ナノモル濃度(nM)以下、より好ましくは300nM以下、最も好ましくは50〜250nMの比較的低い濃度でPCR増幅混合物中に含ませてよい。本発明に基づく試薬は修飾された一本鎖オリゴヌクレオチドである。本発明に基づく試薬を構築するために利用してよいオリゴヌクレオチドは広範囲のオリゴヌクレオチドである。それらはDNA、RNAまたは混合DNA−RNAであってよい。それらは、修飾ヌクレオチド、非天然ヌクレオチド、例えば2’O−メチルリボヌクレオチド、非天然ヌクレオチド間結合、非ヌクレオチドリンカー、PNA、LNAおよび付加化学部分、例えばGlenn Researchによって報告されているキャップヌクレオチド(capped nucleotides)を含有してよい。
【0014】
本発明の試薬は、PCR増幅反応混合物中で、一般に「ヘアピン」構造と称されるステム・ループ構造(stem-and-loop structure)を形成する一本鎖オリゴヌクレオチドであるが、それらは、ループがヌクレオチドから構成されていないステム・ループ構造から構成されていてもよい。そのような構造では、分子の中心部分が一本鎖の(ハイブリダイズしていない)ままであり(ループ)、末端は互いにハイブリダイズしてステムを形成する。ステムは平滑末端であってよく、あるいは一方の鎖が他方の末端を超えて伸長していてもよい。ステムは連続する二本鎖領域を含んでよく、あるいはそれは内部ミスマッチを含んでもよい。内部ミスマッチは隆起を生じさせる。オリゴヌクレオチドの末端によって形成されるステムの端部は、DNA−DNAハイブリッドよりも堅固に結合するように安定化される。本発明者らは、本発明の試薬のステムをその融解温度、すなわちTmを参照することによって特徴付ける。融解温度とは、ステムの相補配列の50%がハイブリダイズしておらず(開放型立体配置)、かつ該相補配列の50%が自身にハイブリダイズしているか、あるいは部分的にハイブリダイズしている(閉鎖型立体配置)摂氏温度である。本出願では、ステムの「計算上のTm」とは、M−foldプログラム:Zucker, M. (2003). "Mfold Web Server for Nucleic Acid Folding and Hybridization Prediction." Nucl. Acids Res. 31: 3406-3415を使用して取得される、対応する安定化されていない完全DNAオリゴヌクレオチドのステム部分の計算上の融解温度を意味する。その場合、ナトリウム濃度が70ミリモル濃度(mM)であり、かつマグネシウム濃度が3mMであると仮定する。添加されたクエンチャーペア、好ましくは光を吸収するが、吸収エネルギーを熱として放出する非蛍光クエンチャー、例えばDabcylおよびBlack Holeクエンチャーを有する好ましい実施形態の場合、Tmは、クエンチャーを伴わないDNAヘアピンについての計算上のTmである。末端にキャップヌクレオチドを有する実施形態の場合、Tmは、同等のキャップなしのヌクレオチドを含有するDNAヘアピンについての計算上のTmである。ステム端部の2’−O−メチルリボヌクレオチド含有物の場合、Tmは、2’−O−メチルリボヌクレオチドのデオキシリボヌクレオチドアナログを含有する全DNAヘアピンについての計算上のTmである。本発明者らは、安定化されたヘアピンの実際の融点を取得することが困難であるという実施上の理由のために本方法論を利用するが、安定化用の修飾に起因して実際の融点が計算上のTmより数度高いことを認識している。本発明の試薬は、94℃を超えない計算上のステムTmを有し、好ましくは計算上のステムTmは50〜85℃の範囲である。一部の実施形態では、プライマーアニーリング温度より高い(典型的に増幅反応は55〜72℃であり、すなわち72〜85℃の範囲の)計算上のステムTmが好ましいが、他の実施形態では、プライマーアニーリング温度より低い、すなわち50〜71℃の範囲の計算上のステムTmが好ましい。ステムの相補配列は、PCRサイクル処理中の鎖融解に使用される温度である95℃で開いて、一本鎖になる。さらに、本発明の試薬のステムは、閉鎖型、自己ハイブリダイズ型のコンホメーション(confirmation)の形成のために十分に低い温度で6ヌクレオチドを超える長さを有する。現在、本発明者らの好ましい実施形態では、9〜12塩基対の長さ、好ましくは9〜11塩基対、閉鎖型の場合、より好ましくは9〜12、最も好ましくは9〜11の、内部ミスマッチを含まない連続した相補的塩基対、最も好ましくは9〜12塩基対の長さで、かつ完全に相補的な平滑末端型ステムであるステムを形成する。試薬の3’末端は、増幅反応においてDNAポリメラーゼによって伸長可能ではなく、したがって該反応物はPCRプライマーではない。すべての突出3’末端は、例えばリン酸部分または何らかのブロック用化学部分を付加することによって、伸長を防止するためにブロックされる。
【0015】
ループの長さはかなり変動しうる。ループがヌクレオチドおよびヌクレオチド間結合からなる場合、それは少なくとも3ヌクレオチド長である。さらに、それがちょうど3ヌクレオチド長であれば、チミジン残基を含有する。現在、本発明者らの好ましいヌクレオチドループは3〜22ヌクレオチドの範囲の長さを有する。ループは非ヌクレオチド化学リンカー、例えばアルキレン鎖であってもよい。炭素鎖リンカーに関しては、3〜6炭素原子の長さが好ましく、最も好ましい長さは3炭素原子である。アルキレン炭素鎖において、さらに、炭素鎖の各メンバーの残りの2価電子は共有結合に関与する。そのような結合は、水素原子、短鎖アルキル基または短鎖アルキレン基、試薬を固体表面に連結するための置換基を対象としうる。化学リンカーを含む非オリゴヌクレオチドループは炭化水素鎖に限定されず、ヘテロ(炭素または水素でない)原子を含んでよい。化学リンカーはポリメラーゼに結合しないように電気的に中性であることが好ましい。試薬の活性はステムの閉鎖型コンホメーション(confirmation)に依存するため、5個を超えるヌクレオチドのループを有する試薬に関して、ループの組成は、反応中の、または反応によって生成される、任意の他の配列または配列群に対して広範囲にわたって相補性であるわけではないことが好ましい。本発明に基づく試薬は、増幅反応のいずれの生成物に対するハイブリダイゼーションプローブでもなく、生成物の蓄積をシグナルしない。
【0016】
上記のように、本発明に基づく試薬は、ループの反対側のステムの端部を安定化して、該ステムの端部がDNA−DNAハイブリッドの端部よりも堅固に結合するようにすることを含む。本発明者らの現在の最も好ましい実施形態では、ステムを平滑末端にし、天然DNA−DNAハイブリッドと比較して部分的鎖分離を阻害する様式で該ステムの末端を修飾する。本発明者らは、安定化用の相互作用性化学部分をステムの端部に付加する効果、市販のリンカーを用いてステムの3’および5’ヌクレオチドに共有結合によって付加される1ペアのDabcyl部分および1ペアの市販のBlack HoleTMクエンチャー(Biosearch Technologies, Novato, CA, U.S.A.によって市販されている専売のクエンチャー)の両者の効果を以下で実証する。また本発明者らは、ステムの端部で、強力に結合する非天然ヌクレオチド、2’O−メチルリボヌクレオチドを利用する効果を実証する。試薬が増幅および増幅アッセイにバックグラウンド蛍光を付与しないように、好ましい安定化用部分は非蛍光性である。本発明の試薬の、安定化されていないオリゴヌクレオチドDNAヘアピンを、1μM未満の濃度で加えても、水素結合しているステム末端を安定化するよう上記のように修飾されていない場合はミスプライミングを必ずしも実質的に低減せず、多くの場合でそうならない。ゆえに、本発明の試薬において安定化は必須である。
【0017】
本発明のヘアピン試薬は、6個を超える塩基対からなるステムを形成できるようにつながり合う2つの相補的ヌクレオチドオリゴマーを含有する分子と説明することができ、該ステムの開放端は、該ステムがその開放端でほどけるか、あるいは「ほつれる」傾向が抑制されるように、何らかの手段によって化学的に修飾される。作用の分子的メカニズムの徹底的な理解には、本発明の試薬の存在および非存在下での酵素キネティクスによる追加の分析、ならびに電子顕微鏡観察、核磁気共鳴、およびX線結晶構造解析による前記試薬とポリメラーゼとの相互作用についての構造解析が待たれるが、本明細書および科学文献中に示されている情報に基づいて、メカニズムの部分的な解釈を予想することができる。該文献は、Taqポリメラーゼが、他のDNAポリメラーゼと同様に、合成ドメインおよび5’エキソヌクレオチド分解性(exonucleolytic)ドメインからなることを教示している。5’エキソヌクレオチド分解性ドメインはStoffel断片には存在しない。該文献は、さらに、合成ドメインがまず二本鎖DNAに結合し、それに沿ってスライドすることによってDNA重合を実行することを教示している。合成ドメインは「広げた手」に例えられている形状を有し、dNTPの存在下で、二本鎖DNAと接触すると、ポリペプチドレベルで形状変化を受ける。その結果、「手」の「指」が二本鎖DNA分子を取り囲んで「閉じる」。二本鎖分子内のミスマッチ配列に起因して、ポリメラーゼは数ヌクレオチド後退し、合成ドメインの3’編集機能を使用して正しい塩基で置換する。酵素が鋳型鎖を読んで、相補的プライマーの3’末端を伸長するにつれて新規DNA鎖の合成が生じる。酵素が、鋳型鎖にすでに結合しているオリゴヌクレオチドの5’尾部に遭遇した場合、ポリメラーゼは結合済みオリゴヌクレオチドの領域に1〜2ヌクレオチド侵入し、酵素の5’エキソヌクレアーゼドメインを用いて、得られた5’尾部を切断することができる。
【0018】
この情報に基づいて、本発明の試薬の二本鎖ステムが、少なくとも部分的には、開放型コンホメーションにおいてポリメラーゼの合成ドメインに結合し、それを閉鎖型にすることによって機能すると仮定することができる。ポリメラーゼの合成活性はそれによって阻害される。ゆえに、いかなる理論にも拘束されることを望まないが、第1の作用様式がポリメラーゼの合成活性についての温度依存的インヒビターとしての様式であると仮定することができる。以下に挙げる証拠は、少なくともいくつかのバージョンの試薬が、その二本鎖領域、例えばステムの融解温度で酵素から遊離せず、特に典型的なPCR伸長温度を含めたより高い温度でも結合したままであることを示すものである。しかし、PCRサイクルの変性(すなわち鎖融解)ステップ中は、本発明の試薬はDNAポリメラーゼに結合していない状態になる。典型的に90℃を超えるPCRサイクルの変性ステップ(鎖融解ステップとも称される)中には、本発明のヘアピン試薬のステムは融解して離れ、該二本鎖領域が再形成するために十分に温度が低下した場合にのみ、その時点で、それらは再びポリメラーゼに結合する。したがって、第一の変性までの間だけ二本鎖であり、すなわち増幅前に(一般に室温で)PCR反応混合物に加えた時点では二本鎖であるが、その後、そのステムのTmを超える温度で維持される、本発明の試薬を設計することを選択することができる。そのような実施形態では、すべての増幅サイクルのプライマーアニーリング温度および任意のLATE−PCR低温検出温度は、計算上のステムTmより高く、好ましくは少なくとも5℃高い。あるいは、増幅中の後期に再び二本鎖になる、本発明の試薬を設計することを選んでもよい。その設計は、すべてまたは一部の増幅サイクルで使用されるプライマーアニーリング温度より高いステムTm(記載されているように算出される)を利用することによって達成することができる。あるいは、LATE−PCR増幅において、プライマーアニーリング温度より低いが、低温検出温度より高いステムTmを利用することによって、その設計を達成することができる。前者の設計タイプは、概して、CTの遅延によって示されるように重合を阻害しないが、一方、ステムTmより低いプライマーアニーリング温度を利用すると、概して、1〜3増幅サイクルの中程度のCTの遅延が生じる。
【0019】
さらに、いかなる理論にも拘束されることを望まないが、本発明に基づく試薬の第2の作用様式が、ポリメラーゼの5’エキソヌクレオチド分解活性についての温度依存的インヒビターとしての様式であると仮定することができる。本発明者らは、本発明の試薬が、少なくとも55℃までの温度でポリメラーゼ酵素の5’−3’エキソヌクレアーゼ活性を阻害し、その阻害は、酵素が(PCR増幅においてPCRプライマーが伸長されるにつれて)DNA鎖をその3’末端の重合によって伸長する能力を過度に抑制することなく行われることを以下に示す。
【0020】
さらに、いかなる理論にも拘束されることを望まないが、本発明の試薬の第3の作用様式が、ポリメラーゼに結合し、該ポリメラーゼのポリペプチドの形状を変化させる温度依存的リガンドとしての様式であると仮定することができる。前記リガンドが酵素から遊離すると、該酵素は、いくらかの時間、変化した形状のままであり、その後、その元の形状に戻る。変化した形状であるうちは、酵素は、部分的にミスマッチしたプライマー−鋳型ハイブリッドと比較して、完全に相補的なプライマー−鋳型ハイブリッドに優先的に結合する。
【0021】
このモデルに基づいて、本発明の試薬のステムがポリメラーゼの合成ドメインの形状を変化させる能力が、本発明の試薬がポリメラーゼの合成機能(上記第1の様式)またはポリメラーゼの5’エキソヌクレアーゼ機能(上記第2の様式)を阻害する能力とは異なっていると予想することができる。第1の作用様式および第2の作用様式は両者とも、結合している試薬がポリメラーゼから遊離する速度に実際に依存する。速やかに遊離する本発明の試薬は、第3の様式によって優先的に作用し、高濃度でさえ、ポリメラーゼの合成活性を大きく阻害することはない。対照的に、ポリメラーゼからゆっくり遊離する本発明の試薬は高濃度では阻害性である可能性が高い。一例として、ステムと3つのメチレン(−CH2−)基からなる炭素リンカーループとにより構成される化合物12−C3DDも、ステムと6つのメチレン(−CH2−)基からなる炭素リンカーループとにより構成される化合物12−C3C3DDも、3000nMの濃度でPCR増幅を部分的にしか阻害しない。対照的に、ヌクレオチドにより構成されるループを有する本発明の試薬は、典型的には、実施例1のアッセイにおいて、PCR増幅を<1000nMの濃度で完全に阻害し、時には<500nM未満の濃度で阻害する。
【0022】
タンパク質−核酸相互作用に詳しい当業者が認識するように、本発明の試薬の作用は様式1〜3のうち2つ以上に基づくものでありうる。それらは相互に排他的ではなく、むしろ関連している。以下に記載されるように、ステムを含んでなる2つのオリゴヌクレオチドを連結する特定の分子構造が、化合物全体を(第1の様式または第2の様式によって)酵素インヒビターとして作用させるか、あるいは第3の作用様式で主に酵素特異性のエンハンサーとして作用させる程度を決定するために使用できる定量的試験(実施例14)を本発明者らは考案した。
【0023】
上記モデルに基づいて、さらに、1種以上のポリメラーゼ分子を本発明の試薬の等しい数の分子と結合させ、次いでそれを、より大きい部分、例えばビーズ、粒子、または固体材料からなる材料に共有結合によって結合させることができることを予想できる。それにより、固体物にポリメラーゼが「積載」されていると考えることができる。固体物への本発明の試薬の結合は、一時的であってよく、あるいは当技術分野において公知の種々の手段によって切断可能であってよい。結合が切断されると、試薬または試薬−ポリメラーゼ複合体が溶液中に放出される(その後、該試薬はポリメラーゼから遊離しうる)。
【0024】
本発明の試薬の設計は当分野の技能の範囲内である。ステムの融解温度は、そのGC含量を変動させることによって、かなり調節できることが理解される。例えば、以下に記載される2つの好ましい実施形態のステムは、いずれも、9ヌクレオチドの長さを有するが、それらは25℃異なる計算上のTm(81℃および56℃)を有する。本発明者らは通常、ステムTmの算出に関して、上で引用したM−foldコンピュータソフトウェアプログラムを1種以上のループ配列候補について使用する。ループ配列が、それ以外の点で、本発明のミスプライミング試薬において重要であることは見出されていない。本発明者らの設計では、完全な平滑末端のステム、すなわち内部ミスマッチおよび末端突出部を有しないステムを形成する配列を利用する。そして、簡単な試行によって、内部ミスマッチ(実施例1に示されるように、末端ミスマッチは不安定であり、許容されない)の導入または二本鎖領域を超える1個または最大2個のヌクレオチドの短い伸長の影響を評価することができる。
【0025】
本発明の試薬の構築は当分野の技能の範囲内である。例えば、オリゴヌクレオチド配列をオリゴヌクレオチド合成機で調製することができる。公知の方法を使用して安定化用部分を含ませることができる。例えば、dabcyl化カラム(Glen Research)を用いて出発し、Dabcylで修飾されたヌクレオチドで合成を終えることによって、好都合にDabcylを付加することができる。合成の最初、および最後から2番目、および最後のヌクレオチドとして非天然ヌクレオチドを使用することができる。
【0026】
候補試薬を設計し、構築した後、一部の場合には、蛍光性DNA結合色素を加え、該色素を刺激しながら融解解析を実施することによって、そのステムの融解温度を経験的に概算することができる(色素自体がTmに対して効果を有することを認識して行う)。また、本発明の試薬の安定化されたステムの実際の融解温度に関する実施上有用な情報を、多くの場合、種々のアニーリング温度を利用するリアルタイムPCR増幅を実施することにより推定的に取得することができる。必要であればステムの長さまたはGC含量の調節を施して、所望のTmを達成することができる。そして本発明者らは、ミスプライミングおよび増幅効率の種々の現象に対する候補試薬の効果を、以下の実施例1に記載されるような厳密なPCR増幅におけるその性能を決定することによって評価する。候補試薬は、実施例1の反応混合物に1000nM未満、好ましくは650nM以下のいずれかの濃度で加えられた場合に純粋なアンプリコンが取得されれば、本発明の試薬であると判断される(図1を参照のこと)。本発明者らは、所望の標的および所望のプライマーを用いること以外は実施例1のアッセイを使用して候補アッセイを評価する。しかし、本発明の増幅およびアッセイ(増幅および検出)は実施例1の条件または手順に限定されない。本発明の試薬は、25μLの反応混合物あたり1.25単位のポリメラーゼ濃度(実施例1)と比較して、1000nM未満の濃度で加えられた場合に、ミスプライミングの少なくとも1種の現象を阻害する必要があるが、該試薬はポリメラーゼ濃度に対する任意の有効な濃度;すなわち、ミスプライミングを防止するが、増幅を実質的に妨害しない任意の濃度(すなわち相対濃度)で使用することができる。評価において所望のプライマーを利用すると、意図せぬ結果、例えば試薬の3’末端が遮断されないことまたは相補性の見落としが明らかになる。
【0027】
本発明には、PCR増幅反応およびPCR増幅反応を含むアッセイが含まれ、そのようなものとしては、増幅反応混合物が、重合活性および5’−3’エキソヌクレアーゼ活性のいずれをも有する熱安定性DNAポリメラーゼ、例えばTaqDNAポリメラーゼを含み、かつ本発明の少なくとも1種の試薬が該増幅反応混合物に含まれる反応が挙げられる。本発明の多数の試薬はポリメラーゼの活性を阻害するものであり、熱サイクル処理前または処理中に上記範囲内のポリメラーゼ濃度に対して相対的な濃度、すなわち25μLの反応容量あたり1.25単位のDNAポリメラーゼを含有する反応に関して1000nM以下、好ましくは650nM以下の濃度で加えられる。本発明のいくつかの試薬は、1000nM未満の濃度、好ましくは650nM未満の濃度で有効であるが、ポリメラーゼの活性を低い程度にしか阻害せず、前記ポリメラーゼ濃度に関して1500nMまでの濃度で、あるいは3000nMでさえ加えることができる。本発明の2種以上の試薬を含有するアッセイでは、各試薬は各自のTmを有するステムを有することができ、それを各自の濃度で加えて、増幅プロセス中のステップの種々の部分で種々の試薬が機能するようにすることができる。本発明の試薬が熱サイクル処理中に加えられる場合、反応混合物を作業領域に引き込むことなく加えられるべきであろう。そのような増幅反応としては、対称的PCR増幅、非対称的PCR増幅およびLATE−PCR増幅が挙げられ、それらはいずれも、RNA標的が関与する場合には逆転写をさらに含んでよい。PCR増幅を使用し、任意の目的で、例えばジデオキシシークエンシングの出発材料として増幅産物を調製することができる。アッセイにおいてPCR増幅を増幅産物の検出と組み合わせてよく、そのようなアッセイとしては、特に、標識プライマー、標識プローブまたは蛍光性DNA結合色素、例えばSYBR Greenまたは臭化エチジウムを使用するホモジニアスアッセイが挙げられる。前記アッセイは、検出の読み取り値が複数の増幅サイクル中に取得されるリアルタイムアッセイであってよく、あるいは増幅の完了後に検出が実施されるエンドポイントアッセイであってよい。それは定性的または定量的であってよく、非限定的に、定量的エンドポイントアッセイが含まれる。前記アッセイは、単一の二本鎖産物または一本鎖産物、または2種以上の二本鎖産物を、関連の一本鎖産物を伴わず、あるいは伴って、増幅するように設計することができる。
【0028】
本出願で使用される「LATE−PCR」とは、おおむね十分なPCRサイクル中に使い果たされて蛍光によって検出可能な二本鎖アンプリコンを生じるように、それ自体は200nMまでの低濃度で利用される一方のプライマー(「制限的プライマー(Limiting Primer)」)と比較して少なくとも5倍過剰のもう一方のオリゴヌクレオチドプライマー(「過剰プライマー」)を利用するポリメラーゼ連鎖反応(PCR)プロセスを使用する、非対称的DNA増幅を意味し、その場合、増幅の開始時点での制限的プライマーの、濃度について補正した融解温度Tm[0]は、増幅の開始時点での過剰プライマーの、濃度について補正した融解温度Tm[0]Xより高いか、あるいは最大5℃まで低く、好ましくは3〜10℃高く;ならびに、熱サイクル処理は制限的プライマーが使い果たされた後に複数サイクル継続されて、一本鎖産物、すなわち過剰プライマーの伸長産物を生じる。LATE−PCRアッセイは、少なくとも一部の直線的増幅サイクル中にプライマーアニーリング温度より温度を下げる低温検出ステップを含んでよい。好ましくは、鎖融解前の伸長後に当該ステップを行う。
【0029】
また本発明は、完全PCRキット、部分的キット、およびオリゴヌクレオチドセットを包含する。完全PCR増幅キットは、少なくとも、PCR増幅またはアッセイを実行するためのすべての試薬を含み、少なくとも1ペアのPCRプライマー、dNTP、反応バッファー、熱安定性DNAポリメラーゼ(好ましくは5’−3’エキソヌクレアーゼ活性を有するポリメラーゼ)、および少なくとも1種の本発明の試薬が含まれる。ホモジニアスPCRアッセイのための完全キットは、必要とされる任意の追加検出試薬、例えば蛍光性DNA色素または蛍光標識プローブをさらに含む。いずれのタイプの完全キットも、好ましくはサンプル調製用の試薬を含み、一部の実施形態では、逆転写酵素を含んでよい。本発明の部分的キットは、完全キットの少なくとも一部の構成要素を省略するが、少なくとも熱安定性DNAポリメラーゼ(および、必要であれば、逆転写酵素)および少なくとも1種の本発明の試薬を含む。例えば、商業的に「マスターミックス(master mix)」または「基本キット(basic kit)」として公知の製品は、典型的には、PCRプライマーおよびプローブを省略する。好ましい部分的キットは、サンプル調製のためのものを除いて、増幅またはアッセイに必要とされるすべての試薬を含む。本発明に基づくオリゴヌクレオチドセットは、少なくとも1ペアのPCRプライマーおよび少なくとも1種の本発明の試薬を含む。それらはオリゴヌクレオチドプローブまたはシークエンシングプライマーをさらに含んでよい。
【0030】
本発明の1種以上の実施形態の詳細は添付の図面および以下の説明中に記載される。本発明の他の特徴、目的、および利点は、その説明および図面から明らかになり、さらに特許請求の範囲から明らかになるであろう。
【0031】
図面の説明
種々の図面中の参照記号などは要素などを示す。
【図面の簡単な説明】
【0032】
【図1】本発明に記載の試薬の阻害活性を試験するために設計したミスプライミングエラーに関する厳密な定性的PCRアッセイから得たサンプルのゲル電気泳動解析を示す。
【図2】本発明の試薬によってPCRベースのリアルタイムアッセイおよびエンドポイントアッセイが可能になることを示す。
【図3】本発明に記載の試薬を使用し、ミスプライミングエラーの非存在下で70サイクルのLATE−PCR増幅後に生成された一本鎖DNA産物から取得したDNA配列クロマトグラフを示す。
【図4】五重(pentaplex)LATE−PCR増幅に対する化合物9−3DDの効果を示す。
【図5】本発明の試薬がTaqポリメラーゼに結合すると、PCRサイクル処理温度が変性ステップに達するまでそれらはポリメラーゼから遊離しないことの証拠を示す。
【図6】25℃でのDNAポリメラーゼ活性に対する効果に関する、本発明の試薬の直接比較を示す。
【図7】ポリメラーゼ活性の非存在下で、ポリメラーゼのエキソヌクレアーゼ活性に対する、本発明に基づく試薬の効果を評価するためのアッセイを示す。
【図8A】対称的PCRアッセイにおいてミスプライミングを防止する本発明に基づく試薬の能力を示す。
【図8B】対称的PCRアッセイにおいてミスプライミングを防止する本発明に基づく試薬の能力を示す。
【図9】エキソヌクレアーゼ阻害を定量化するためのアッセイを示す。
【図10】LATE−PCR増幅でのプライマー二量体およびプライマーオリゴマーの形成に対する、本発明の試薬の用量依存的効果を示す。
【図11】2ペアのプライマーを用いる2種の標的配列についての二重LATE−PCR増幅に対する、低濃度の試薬9−22DDの効果を示す。
【図12】2ペアのプライマーを用いた2種の標的配列についての二重LATE−PCR増幅に対する、試薬9−3DDのステム組成を変化させることの効果を示す。
【図13】2ペアのプライマーを用いる2種の標的配列についての二重LATE−PCR増幅に対する、試薬9−3bDDの濃度を変化させることの効果を示す。
【図14】CT値に関して、LATE−PCR増幅の効率に対する試薬9−3DDの濃度上昇の効果を示す。
【図15】蛍光シグナル勾配および最終蛍光に関して、LATE−PCR増幅の効率に対する試薬9−3DDの濃度上昇の効果を示す。
【図16】本発明の化合物の混合物を用いるか用いないで実行された多重反応の産物を示す。
【図17】化合物12−3DDの作用に関する試験の結果を示す。
【図18】化合物12−C3DDの作用に関する試験の結果を示す。
【発明を実施するための形態】
【0033】
詳細な説明
上記のように、本発明者らは、ミスプライミングエラーの3種の異なるタイプおよび原因であると考えられるものを特定した。全3タイプのミスプライミングを評価するために、本発明者らは以下の実施例1に示す厳密なアッセイを開発した。増幅は剪断DNAを用いて開始され、これはミスプライミングを促進する。物理的操作、例えばDNAをピペット内に吸い込むことによって、DNAは剪断されやすい。さらに、「ホットスタート」試薬および方法によってはタイプ1のミスプライミングしか防止できないので、本発明者らは多数の温度サイクル(少なくとも60サイクル)をアッセイに組み入れた。さらに本発明者らは、一部のLATE−PCRアッセイで利用される低温検出ステップを組み入れた。さらにまた、本発明者らは、プライマー二量体の形成およびさらにそれらのオリゴマー形成および増幅を追加のタイプのミスプライミングとして認識している。プライマー二量体形成および増幅は、添加鋳型DNAの存在下および非存在下の両方で生じうるものであり、本発明者らはこれらの現象に関するアッセイをさらに開発した。
【0034】
ホットスタート酵素は、高温、例えば95℃に加熱されると生じる不可逆的な変化のために、増幅の開始前にしか有効でないが、本発明のミスプライミング試薬は不可逆的に変性せず、低温ステップが含まれる場合、反応の後期に、正の効果もしくは負の効果、または両者を有しうる。
【0035】
特定のサイクル温度より高いか、またはそれより低い融解温度Tmを有し、増幅に対して種々の影響を有するステムを設計することができる。ステムの設計では、好ましくは、融解が、一定の温度範囲にわたって起こる動的現象であり、このときTmは、50パーセントの分子が二本鎖形態であり、かつ50パーセントが一本鎖形態である温度を指定するものであることが考慮される。本発明者らは、一部の閉鎖型ステムがポリメラーゼに結合すると、ポリメラーゼに未結合のままである分子間の平衡が、より多くのステムが閉鎖型になる方へシフトすると考えている。特定のサイクル温度、例えばプライマーアニーリング温度で大多数のステムが閉鎖型であることが所望であれば、一般に、その温度より少なくとも5℃高い計算上のTmを有する無修飾のオリゴヌクレオチドステムを用いて出発し、その場合、安定化用のステムの修飾によって実際のTmが数度上昇することを認識している。反対に、特定のサイクル温度で大多数のステムが開放型であることが所望であれば、一般に、該特定のサイクル温度より少なくとも5℃低く、好ましくは少なくとも約10℃低い計算上のTmを有する無修飾のオリゴヌクレオチドステムを用いて出発し、その場合も、安定化用のステムの修飾によって実際のTmが数度上がる可能性が高いことを認識している。
【0036】
以下の実施例5で実証されるように、本発明の特定の試薬を種々のプライマーアニーリング温度とともに利用すると、増幅効率に対する種々の効果を有しうる。以下で考察される、本発明者らが現在意図している実施形態の1つ(化合物9−22DD)では、81℃の計算上のTmを有する。温度が鎖融解温度(例えば95℃)からプライマーアニーリング温度(例えば55℃)に下がるにつれて、前記化合物はすべての典型的なPCR熱サイクル中に閉鎖型になる。そのようなステムを有する試薬は増幅の各サイクル中に効果を有する。別の現在の本発明者らの好ましい実施形態(化合物9−3DD)では、56℃の計算上のTmを有する。PCR増幅混合物が室温で調製される時に、この試薬は閉鎖型のステムを有し、ポリメラーゼに結合する。増幅の開始時点で温度が上がっても、試薬は結合したままである。ポリメラーゼ結合型の9−3DD分子は最初の高温ステップ中に結合がはずれて開放型になり、未結合分子のステムが閉鎖型になるほど十分に温度が下がらない限り、ポリメラーゼに再結合しない。ゆえに、最低サイクル温度を65℃で維持することによって、ステムは再形成しない。これらの条件下では、本実施形態は、該増幅において、増幅の開始前に生じるミスプライミングを防止するホットスタート試薬として機能する。しかし、LATE−PCRサイクルにおける伸長後の、増幅の直線期に蓄積する一本鎖に対する低温分子ビーコンまたは他の標識プローブのハイブリダイゼーションを可能にする低温検出ステップ、例えば、40℃でのインキュベーションを含ませる場合、56℃の計算上の融解温度を有するステムは閉鎖型になり、高温の鎖融解が達成されるまで試薬がポリメラーゼに再結合することが可能になる。
【0037】
以下の実施例1では、本発明の試薬の性能を評価するために本発明者らが利用する厳密なLATE−PCRアッセイを記載する。図1は、「通常の」TaqDNAポリメラーゼ、すなわち5’−3’エキソヌクレアーゼ活性を有するが、ホットスタート修飾を有しない熱安定性DNAポリメラーゼを用いた場合(レーンbおよびレーンc)だけでなく、ホットスタートTaqポリメラーゼを用いた場合(レーンdおよびレーンe)にもミスプライミングエラーが起こることを示す。図1は、さらに、ホットスタートポリメラーゼを用いる増幅(レーンhおよびレーンi)に対してだけでなく、通常のTaqDNAポリメラーゼを用いる増幅(レーンfおよびレーンg)に対しても、300nMの濃度で試薬9−22DDを加えることによって達成される、ミスプライミングエラーにより形成される非特異的産物の排除、および所望の特異的産物しか存在しないことを示す。実施例1に記載のアッセイを利用して、ミスプライミングの抑制に関する化合物9−22DDおよび9−3DDの有効性を評価した。両化合物はタイプ1のミスプライミングを抑制することがわかった。9−3DDのステムの計算上のTmは低い(56℃)ので、すべてのサイクル温度が60℃以上であるPCR増幅プロトコルを利用することによって、それを単に有効なホットスタート試薬として使用することが可能である。ステムの融点より高いままであることによって、増幅が進行してもステムは再形成されない。化合物9−3DDをそのように使用しても重合効率に対して阻害効果を有しない。以下の実施例5はこれに関して有益な情報を提供する。実施例5では、試薬9−3DDならびに65℃および55℃のアニーリング温度を利用する増幅を比較する。65℃が利用される場合、CTが反映するように重合効率に対する影響はないことがわかった。しかし、55℃が利用される場合、CTは遅延した。このことは、ステムが閉鎖型になり、ポリメラーゼが結合するほど十分に温度が下がった場合に、プライマー伸長のために温度が上がっても、プライマー伸長温度がステムのTmより高い場合でも、結合が継続することの証拠である。一方、9−22DDのステムのTmは高い(81℃)ので、毎PCRサイクルのプライマーアニーリングステップ中にステムが再形成されてヘアピンになることが可能である。この化合物は、タイプ1のミスプライミングだけでなく、タイプ2のミスプライミング、タイプ3のミスプライミング、およびプライマー二量体形成の現象をも防止する。しかし、この化合物は重合効率に実際にいくらか影響する。このことは、100nMの濃度で使用された場合に、SYBR Green 1または標的特異的分子ビーコンプローブを用いてリアルタイムで追跡したPCR増幅の閾値サイクル(CT)が1.5サイクル遅延することによって示される。
【0038】
実施例1の表Iは、図1のレーンfおよびgに示されるように、ホットスタート型ではなく通常のTaqDNAポリメラーゼを使用して、一連の添加剤に対する前記厳密なアッセイの適用を報告し、さらに、ミスプライミングの現象の防止、すなわちミスプライミングエラーにより形成される検出可能なレベルの非特異的産物の回避を達成するために、および所望の特異的増幅産物しか存在しないことを達成するために必要な最低濃度を報告する。表Iでは、化合物は本発明者らの用語体系によって特定される。無修飾のヘアピン分子はステムループの長さによって特定される。例えば、化合物6−22は6ヌクレオチド長のステムと22ヌクレオチド長のループとを有する。そのループ識別名は数字で始まるか、あるいは数字のみである(「22」または「3b」)ため、該ループはヌクレオチドである。非ヌクレオチドループは別に指定される。化合物12−C3C3は12ヌクレオチド長のステムと、6炭素鎖、すなわち6つのメチレン(CH2)基の鎖であるループとを有する。基本となるヘアピンの一端または両端に種々の修飾を施した。これらの修飾は接尾辞によって指定され、その場合、3Dは3’末端ヌクレオチドに付加されたDabcyl(5’−ジメトキシトリチルオキシ−5−[(N−4’−カルボキシ−4−(ジメチルアミノ)−アゾベンゼン)−アミノヘキシル−3−アクリルイミド]−2’−デオキシウリジン−3’−[(2−シアノエチル)−(N,N−ジイソプロピル)]−ホスホルアミダイト)であり、5Dは5’末端ヌクレオチドに付加されたDabcylであり、DDは両末端ヌクレオチドに付加されたDabcylであり、BHQBHQは両末端ヌクレオチドに付加されたBlack Holeクエンチャーであり、FFは両末端ヌクレオチドに付加された蛍光団(この場合はFAM)であり、AAは両末端ヌクレオチドに付加されたアデノシン(A)であり、ならびにTTは両末端ヌクレオチドに付加されたチミジン(T)である。
【0039】
付加に加えて、基本となるステムにいくつかの修飾を施した。接尾辞2’OM4とは、4個の末端ヌクレオチド、各ステムオリゴヌクレオチド上の最後の2個、またはアーム、がデオキシリボヌクレオチドから2’−O−メチルヌクレオチドに変更されたことを示す。ステムの融解温度を変更するために、本発明者らはそのGC含量を変更した。ステムの長さではなくステムの配列が変更された化合物を、末端修飾の接尾辞の前の小文字識別子によって示す。例えば、9−3bDDまたは9−3iDDである。表Iの種々の化合物に関するヌクレオチド配列を表IIに挙げる。その配列は、使用された炭素鎖ループを同時に示す。表Iはいくつかの点で有益な情報を提供する。例えば、表Iは、前記アッセイにおいて、無修飾のDNAヘアピンオリゴヌクレオチドが有効ではないか、または1000nM以上の高濃度でしか有効でないことを示す。オリゴマー9−22(9ヌクレオチド長のステム、22ヌクレオチド長のループ)は3000nMの濃度でしか有効でなかった。これは二本鎖DNAによるミスプライミング抑制に関して報告された量である。オリゴマー6−22(6ヌクレオチド長のステム、22ヌクレオチド長のループ)および9−5(9ヌクレオチド長のステム、5ヌクレオチド長のループ)は3000nMの濃度でさえ有効でなかった。しかし、9−22オリゴマーを、そのステムの両端にDabcyl部分を付加することによって修飾して、本発明に基づく好ましい試薬である化合物9−22DDを作成した場合、ミスプライミングの現象を防止するために50nMの濃度で十分であった。9−3オリゴマーを同様に修飾した場合、100nMの濃度で十分であった。
【0040】
表Iはステムの長さの影響を示す。9ヌクレオチドおよび12ヌクレオチドのステムを有するいくつかのオリゴマーは、1ペアの相互作用性Dabcylクエンチャーで修飾した場合に、低濃度でミスプライミングを排除したが、オリゴマー6−22は修飾によりそのような効果を示さなかった:組成物6−22DD(本発明の試薬ではない)は高濃度(3000nM)でしか有効でなかった。本発明の試薬は、6ヌクレオチドより長く、ほとんどの場合で14塩基対より短いステムを有する。現時点では、14塩基対より短い長さが好ましい。
【0041】
また表Iは、ステムの末端での結合強度を増大させる他の修飾の有効性を示す。1ペアの相互作用性Black HoleTMクエンチャーをオリゴマー9−5のステムに付加して試薬9−5BHQBHQを作成したところ、ミスプライミングエラーにより形成される非特異的産物の排除、および所望の特異的増幅産物しか存在しないことを達成するために、該試薬は100nMの濃度で十分であった。オリゴマー9−5の各末端を2個の2’−O−メチルリボヌクレオチドで置換して試薬9−5 2’OM4を作成したところ、該試薬は50nMの濃度で十分であった。
【0042】
表Iはオリゴヌクレオチドループの長さをかなり変動させることが可能であることを示す。3ヌクレオチド(試薬9−3DD)、5ヌクレオチド(試薬9−5DD)、および22ヌクレオチド(試薬9−22DD)のすべてのループ長で、100nM以下の濃度でミスプライミングを防止する本発明の試薬が得られた。同時に、表Iは、最適化のためにいくらかの試行錯誤による調整が必要であることを示す。オリゴマー9−5の安定化用の修飾においてDabcylクエンチャーの代わりに1ペアの2’−O−メチルリボヌクレオチドを用いる場合(試薬9−5 2’OM4と試薬9−5DDの比較)、有効性のレベルは変わらない:50nMの濃度で十分である。しかし、修飾型オリゴマー9−3に同じ変更を施すと(試薬9−3 2’OM4と試薬9−3DDの比較)、有効性のレベルは、依然として良好ではあるが減少した:50nMの濃度ではなく300nMの濃度が必要とされた。同データは、試薬9−3 2’OM4のループ長を2ヌクレオチド増加させて、試薬9−5 2’OM4を作成すると、有効性が改善されたことを示す:300nMの濃度ではなく50nMの濃度で十分であった。実施例1に記載されるアッセイのような厳密なアッセイを用いて、候補薬物をルーチンの方法で評価し、調節することができる。
【0043】
また表Iは、非ヌクレオチドブリッジ(ループ)によって逆の極性でつながり合う2つの相補的オリゴヌクレオチドからなる化合物もまた、そのオリゴヌクレオチドの自由な3’末端および5’末端を本発明にしたがって(試験された実施形態では、Dabcyl部分によって)修飾した場合に、活性であることを示す。表Iは、非ヌクレオチドブリッジのサイズが重要ではないことを示す。表Iに記載のブリッジは3〜6炭素原子の線状鎖からなる化学リンカーであるが、当業者は、非ヌクレオチドブリッジ(ループ)について多数の他のバリエーションを作成することができる。表Iのデータは、同一オリゴヌクレオチドステムに関して3炭素原子鎖のブリッジが6炭素原子鎖のブリッジより良好である(より低い濃度で活性である)ことを示唆し、厳密に試験されてはいないが、その差異がブリッジの相対的な可動性および、その結果として、ステムの融解温度を上げるかまたは下げるそれらの効果もしくは能力を反映すると仮定される。本発明者らは、好ましい化学的に中性のリンカー(無電荷)を利用する。
【0044】
本発明の試薬および方法の種々の適用が以下の実施例2〜4に例示される。実施例2では、リアルタイムアッセイであるLATE−PCRアッセイにおける、試薬9−22DDの利用が記載されるが、これはエンドポイントアッセイでも同様に好適であることが示されている。実施例2のアッセイでは、本発明者らが同時に出願した米国仮特許出願第60/619,654号、表題「Primers, Probes and Methods for Nucleic Acid Amplification」の対象である検出技術を利用する。該文献は参照によりその全体が本明細書中に組み入れられる。該検出技術は、二本鎖DNAに結合すると蛍光を発する蛍光性DNA色素(例えばSYBR Gold)および、制限的プライマーが使い果たされた後のLATE−PCR増幅において生成される一本鎖アンプリコンに相補的な蛍光団標識ハイブリダイゼーションプローブの両者を加えるステップ、その場合、該蛍光団は該色素からの発光によって刺激される;色素を刺激するステップ;色素および蛍光団の両方からの発光を検出するステップ;および色素シグナルに対する蛍光団シグナルの比を算出するステップを含む。ゆえに実施例2は、本発明の試薬と蛍光プローブおよび蛍光色素の適合性、およびそのいずれかまたは両者とともに使用された場合のミスプライミングの効果的な防止を示す。
【0045】
実施例4では、2ペア、3ペア、4ペアおよび5ペアまでものプライマーを利用するいくつかの多重LATE−PCR増幅が記載され、試薬9−3DDを反応混合物に加えた場合の全例で正しいアンプリコンが増幅されることが示される。実施例4の目的ではゲル電気泳動によって増幅産物を分析したが、同様に複数の方法を多重アッセイに適用可能であり、それには他の検出手段(例えば所望のアンプリコンに関する蛍光性ハイブリダイゼーションプローブ)を使用する定性的アッセイおよび定量的アッセイ、例えばリアルタイムアッセイが含まれる。リアルタイムLATE−PCRアッセイでは、プライマー伸長のステップ後の低温検出ステップおよび例えば実施例3で示される低温プローブを利用することが好ましい。
【0046】
実施例13では、2種以上の型の本発明の試薬を含有する増幅反応混合物を用いる増幅反応もまた利用することができることが示される。例えば、増幅混合物は、高い融解温度を有する本発明の化合物および低い融解温度を有する本発明の別の化合物の両者を含有することができ、各化合物はそれぞれの最適濃度で加えられる。81℃の計算上のTmを有する化合物9−22DDのステムは、反応の開始時点および各熱サイクル中に温度が低下した直後は閉鎖型コンホメーションであるが、58℃の計算上のTmを有する化合物12−3DDは相対的に低い温度でしか閉鎖型にならない。したがって、それは「ホットスタート」として作用し、反応混合物が室温で調製される場合にミスプライミングを防止するが、60℃以上のアニーリング温度を有するPCR増幅の以後の熱サイクル中には閉鎖型にならず、ポリメラーゼに結合しない。試薬の混合物は、複数のアンプリコンの増幅のために一定範囲のアニーリング温度にまたがる複数ペアのプライマーが組み合わされている多重反応の構築に関して特に有用である。実施例13で使用される試薬の混合物はいずれかの試薬の単独使用より良好である。
【0047】
実施例3では、十分な量で、ミスプライミングから生じる非特異的産物が十分に排除されている増幅一本鎖産物を調製するための、およびシークエンシング用の出発材料としての使用に好適な、試薬9−3DDを利用するLATE−PCR増幅が記載される。十分な出発材料を保証するために増幅を70サイクルまで延長した。本発明者らは、いくつかのアンプリコンが他のアンプリコンよりタイプ2およびタイプ3のミスプライミングエラーを生じやすいことを発見した。そのようなミスプライミングがより生じやすい増幅では、鎖融解ステップからプライマーアニーリングステップへサイクル処理温度が下がった時に閉鎖型になる本発明の試薬のステムが好ましい。このことはステムTm、アニーリング温度、またはその両者を調節することによって確かに実現することができる。関連する研究で、本発明者らは、本発明者らが同時に出願した上記の米国仮特許出願で開示されているように、生成物を希釈することが、シークエンシングとともに使用するためのシンプルな精製方法であることを発見した。
【0048】
本発明者らは、無修飾のDNAヘアピン形成オリゴマーと比較して、および互いに、本発明の試薬の実施形態の機能性および有効性を調べた。本発明者らは、試験試薬の存在下でのTaqDNAポリメラーゼによる標識プライマーの伸長を測定するためのDNA重合アッセイを考案した。該アッセイでは、本発明者らが同時に出願した上記の米国仮特許出願で開示されているように、蛍光色素からの発光で励起される蛍光団で標識されているプライマーを利用する。該アッセイの混合物は、0.5μMの濃度の合成オリゴヌクレオチドDNA鋳型、1.5μMの濃度のDNAプライマー(鋳型の3’末端に相補的で、その5’末端がCy5で標識されている)、PCRバッファー、MgCl2、TaqDNAポリメラーゼ、1:40,000希釈のSYBR Green蛍光性DNA色素、および試験試薬を含む。反応の制御された開始はdNTPの添加によって達成される。該反応は等温性である:それは所定の温度で所定の時間進行する。DNAポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する試験化合物の効果を評価するために、「ブロッカー」、すなわち鋳型の5’末端に相補的で、その3’末端がリン酸基でブロックされ、その5’末端がROXで標識されているオリゴヌクレオチドを反応混合物に加えた。DNAポリメラーゼの重合活性に対する試験化合物の効果を評価するためには、ブロッカーを使用しない。融解曲線解析による反応産物の分析を容易にするために、鋳型、プライマーおよびブロッカーは、以下のハイブリッドがその融解温度によって容易に識別可能であるように設計する:プライマー−鋳型、ブロッカー−鋳型、完全長伸長産物−鋳型、および部分的伸長産物(ブロッカーまで)−鋳型。リアルタイム解析は、色素を周期的に励起し、該色素および2種の蛍光団からの蛍光発光を周期的にモニタリングすることによって達成される。両蛍光団は色素からの発光によって間接的に励起される。SYBR Green Iの蛍光の増加は重合の指標である。ROXの蛍光の減少はブロッカーの分解の指標である。エンドポイント解析は、12mM EDTAを加えて反応を停止させ、SYBR Green Iの希釈を1:14,200に調節し、試験化合物濃度を800nMに調節し;次いで、色素を刺激し、2種の蛍光団の各々からの蛍光をモニタリングしながら、標準的な融解曲線解析を実施することによって達成される。
【0049】
本発明者らは、55℃で60分間のインキュベーションを行い、化合物9−22、9−22DDおよび9−3DDを評価した。試験化合物を含まない対照も組み入れられた。試験化合物を含まないがブロッカーの存在下でインキュベートされた対照から得られた結果は、これらの条件下で、完全にではないが、TaqDNAポリメラーゼがブロッカー領域を通過してプライマーを伸長し、Cy5蛍光によればほぼ単一の融解ピークを有する産物が生成されたことを示す。ゆえに、酵素は重合活性および5’−3’エキソヌクレアーゼ活性の両方を示した。300nMの濃度または1000nMの濃度で存在する化合物9−22DDを用いて得られた結果は、部分的伸長産物の生成および残留ブロッカーの量に対する用量依存的様式の効果を示した。ゆえに化合物9−22DDは、55℃で、ポリメラーゼの5’−3’エキソヌクレアーゼ活性を阻害した。完全長産物の生成が特に1000nMの濃度で減少した。このことは、この高濃度では、化合物9−22DDが重合を阻害し始めることを示唆する。
【0050】
本発明の試薬の特性を調べるために本発明者らが利用した別のアッセイは以下の実施例6に記載のアッセイである。該アッセイは、25℃でのTaqDNAポリメラーゼによる伸長を阻害する試験試薬の能力を検出する。実施例7に報告される結果は、無修飾のヘアピンオリゴヌクレオチド9−22および9−3は対照と比較して効果を示さなかったが、試薬9−22DDおよび9−3DDはプライマー伸長を用量依存的様式で阻害したことを示す。ゆえに、本発明の試薬が低濃度で重合活性を阻害するのには十分に低い温度でさえ、それらの無修飾のアナログは同一濃度で該特性を示さなかった。
【0051】
本発明者らは、DNAポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する本発明の試薬の阻害効果を調べるために2種のアッセイを利用した。一方のアッセイは以下の実施例7に記載される。該アッセイでは、熱サイクル処理中の5’−3’エキソヌクレアーゼ活性の阻害を測定する。図7は、試薬9−22DDが50nM〜300nMの範囲の低濃度で用量依存的様式で該エキソヌクレアーゼ活性を阻害することを示す。
【0052】
第2のアッセイは以下の実施例9に記載される。該アッセイでは、25℃での5’−3’エキソヌクレアーゼ活性の阻害を定量的に測定する。図9は、50nM〜1000nMの濃度の試薬9−22DDによる用量依存的阻害を示す。
【0053】
以下の実施例6で記載されるさらに別のアッセイでは、TaqDNAポリメラーゼのStoffel断片によって実行されるプライマー伸長の、該試薬による阻害を測定する。インタクトなTaqポリメラーゼと同様に、伸長の阻害には、オリゴヌクレオチドステムでの修飾端部の存在が必要とされ、ステム間のループの長さによる大きな影響はなかった。しかし、インタクトなTaqポリメラーゼとは異なり、ほんの6ヌクレオチドのステムを有する試薬である化合物6−22DDによってStoffel断片は阻害される。本発明者らは、Stoffel断片が、インタクトなポリメラーゼと同一の様式で試薬と相互作用するわけではないと結論づける。
【0054】
本発明の試薬は、以下の実施例8で実証されるように、対称的PCRアッセイでのミスプライミングの防止に有用である。該アッセイでは、非常に近いTm値を有する等モルのペアのプライマーを使用して、嚢胞性線維症遺伝子における配列の増幅を測定した。
【0055】
現在使用されているPCR増幅およびアッセイは、典型的に、室温での増幅反応混合物の調製ステップを含み、ゆえに、タイプ1のミスプライミングを阻害するために最初の混合物に本発明の試薬を含ませることを必要とする。しかし、必ずしもそうではない。特定の自動化された方法、例えばマイクロ流体工学的(microfluidics)方法では、ホットスタート用の高い温度でポリメラーゼを反応混合物に加え、それによってタイプ1のミスプライミングを回避することが可能である。そのような方法では、増幅の開始後に本発明の試薬を加えることができる。
【0056】
プライマー二量体の形成は、完全反応物、すなわち増幅に必要なすべての構成要素と最初の標的配列とを含有する反応物中、ならびに標的配列を含有しない反応物中で生じうるミスプライミングの現象である。プライマー二量体は、反応物中に存在する別のコピーの同じ一本鎖プライマーまたは何らかの他のプライマーに対するハイブリダイゼーションおよびそれらに沿った伸長に基づいて、反応物中の1種以上のプライマーのミスプライミングによって形成される典型的には短い二本鎖DNA配列である。標的配列の存在下でのプライマー二量体の形成は、予測されるアンプリコンの蓄積に伴うプライマー二量体の蓄積として観察される。標的配列の非存在下でのプライマー二量体の形成は、特異的アンプリコンの蓄積を伴わないプライマー二量体の蓄積として観察される。またプライマー二量体は、基本となるプライマー二量体より配列が長いオリゴマーを形成しうる。その理由は、それらが追加の、連結コピーの一方または両方のプライマーを含有するからである。オリゴマー形成のプロセスは十分に理解されていないが、オリゴマー形成は反応産物のゲル電気泳動または融点解析によって容易に検出できる。プライマー二量体の形成により、プライマー二量体の生成および蓄積に一方または両方のプライマーが消費されるにつれ、所定数の熱サイクル中に生成される予測アンプリコンの量が減少する。したがって、プライマー二量体を排除することが望ましい。その理由は、それによって正常産物の特異性および収量がともに増加するからである。プライマー二量体のこれらの特徴、および本発明に記載の試薬を加えることによるその排除は、実施例10で例証される。
【0057】
プライマー二量体の形成確率は、多数の他の要因、例えばプライマー濃度の上昇、マグネシウム濃度の上昇、プライマーの長さの増加、熱サイクルのアニーリング温度の低下、および反応に含まれるプライマーの総数の増加に加えて、一方のプライマーの3’末端と同プライマーの内部配列または異なるプライマーとの間の配列相同性によって上昇する。さらに、プライマー二量体の形成確率が、ホットスタートポリメラーゼを使用する反応と比較して、非ホットスタートポリメラーゼを使用する反応においてずっと高いことが当技術分野において周知である。その理由は、反応の成分の混合中にプライマー−プライマーハイブリダイゼーションおよび伸長が相対的に低い温度で生じうるからである。ホットスタート酵素はプライマー二量体の生成および蓄積を低減するが、完全には排除しない。プライマー二量体のこれらの特徴、ならびに本発明に記載の試薬を加えることによるその排除は、実施例11で例証される。
【0058】
またプライマー二量体の形成は反応のキネティクスに対して微妙な効果を有しうる。本明細書中に記載の試薬を加えることによるそれらの排除を観察し、キネティクスに関して最適化することができる。実施例12では、2ペアのプライマーからなり、2種の一本鎖産物を生成させるLATE−PCR増幅が記載される。それらの一方の産物の動的蓄積を蛍光プローブに対するハイブリダイゼーションに基づいて検出した。結果を図13および図14に示す。該データは、キネティクスの直線性が、試薬のステムの厳密な組成および試薬の濃度の両者によって影響を受けることを実証する。試薬のステムの厳密な組成の影響は融解温度に対するその効果に起因する。さらに、試薬の最適使用および最適に近い使用によってLATE−PCRにおける直線的増幅の速度を変化させ、それによって反応の終了時点でのシグナルの大きさを変化させることができるが、反応のCT値には影響しない。図15および図16は、化合物9−3DDを150および300nMで使用した場合、一定範囲の鋳型出発濃度にわたってCT値によって判断される増幅効率は同一であったが、300nMの9−3DDの存在下で鋳型出発数100および1000から生じたシグナルの大きさは、150nMの9−3DDを使用して得られたシグナルより大きかったことを実証する。おそらく、それはプライマー二量体形成が存在しないせいである。
【0059】
実施例
【実施例1】
【0060】
実施例1. ミスプライミングエラーを評価するための厳密なアッセイ
PCR増幅反応においてタイプ1、タイプ2およびタイプ3のミスプライミングを評価するために、本発明者らは、剪断ゲノムDNAを利用するLATE−PCR増幅を実施し、融点解析およびゲル電気泳動によってその産物を調べる。使用した具体的な増幅は以下の通りである。
【0061】
A. 基質:10〜10,000ゲノムに相当する剪断ゲノムDNA。市販のDNAを購入し、ゲノムDNAを複数回凍結・解凍するか、またはDNAを剪断するための当業者に公知の任意の他の同様の方法によって処理した。
【0062】
B. PCR増幅混合物ベース(Sanchez et al. (1994) PNAS 101:1933-1938を参照のこと):
基質:2000ゲノムの剪断ゲノムDNA
1×PCRバッファー
Mg+2:3ミリモル濃度(mM)
dNTP:250マイクロモル濃度(μM)の4種の各dNTP
過剰プライマー:1000ナノモル濃度(nM)の配列5’ CTTTGATGACGCTTCTGTATCTA 3’(配列番号13)
制限的プライマー:50nMの配列5’ CCTGGATTATGCCTGGCACCAT 3’(配列番号14)
DNAポリメラーゼ:25マイクロリットル(μL)の反応混合物あたり1.25単位。
【0063】
C. 増幅プロトコル:
低温インキュベーション:室温で35分
PCR増幅プロトコル:95℃で15分間の高温浸漬;95℃で10秒間、55℃で30秒間、および70℃で30秒間の10サイクル;次いで95℃で10秒間、50℃で30秒間、および70℃で30秒間の70サイクル。
【0064】
いずれのホットスタート方法論をも使用せずに2種の熱安定性DNAポリメラーゼ(Promega TaqおよびInvitrogen Taq)を利用する増幅によって、ミスプライミングに関するベースラインの事例を確立した。2種の異なる市販のホットスタートDNAポリメラーゼ(Qiagen Hot Start TaqおよびPlatinum Taq(Invitrogen))を利用して追加の増幅を実施した。最後に、300nMの現在の好ましい実施形態の本発明に基づく試薬を加えて、追加の増幅を実施した。本実施例で使用される一実施形態は、組成物9−22DDと称される試薬である(本発明者らの用語体系において、「9」はステム長であり、「22」はループ長であり、「DD」は安定化用の修飾を意味し、この場合は1ペアのDabcylクエンチャーである)。化合物9−22DDは81℃の計算上のTmを有する。該化合物は、表IIに示される配列を有し、それは5’末端および3’末端のDabcyl部分の付加によって修飾されている。
【0065】
臭化エチジウム染色を施した種々の増幅のゲル電気泳動の結果を図1に示す。図1は、ラベルがない両端のカラムにサイズマーカー(100塩基対ずつ異なるサイズ)を含む。重複実験を行ったために、各LATE−PCR増幅の産物に関して2レーンずつが存在する。レーンa,DNAが省略されたベースラインの事例;レーンb,DNAポリメラーゼがPromega Taqであったベースラインの事例;レーンc,DNAポリメラーゼがInvitrogen Taqであったベースラインの事例;レーンd,ホットスタート酵素であるQiagen Hot Star Taqを代用;レーンe,ホットスタート酵素であるPlatinum Taqを代用;レーンf,化合物9−22DDを加えるとともにPromega Taqポリメラーゼを利用する増幅;レーンg,Invitrogen Taqポリメラーゼおよび化合物9−22DDを利用する増幅;レーンh,Qiagen Hot Star Taq DNAポリメラーゼおよび化合物9−22DDを利用する増幅;レーンi,Platinum Taqポリメラーゼおよび化合物9−22DDを利用する増幅。
【0066】
図1から、重合活性および5’−3’エキソヌクレアーゼ活性の両者を有する標準のTaqDNAポリメラーゼ(レーンbおよびレーンc)が一定範囲の産物サイズをもたらしたが、そのうちのほとんどが、レーンf〜iに見られる産物である、プライマーペアによって規定される所望のアンプリコンではなかったことがわかる。修飾型「ホットスタート」ポリメラーゼ(レーンd〜e)への切り替えは有益であったが、依然として、ミスプライミングエラーにより形成される非特異的産物は排除されなかった。しかし、化合物9−22DDが300nMの濃度で存在した場合は、酵素が標準のTaqDNAポリメラーゼであった場合(レーンf〜g)および酵素がホットスタートTaqDNAポリメラーゼであった場合(レーンh〜i)でいずれも、所望のアンプリコンが得られ、ミスプライミングエラーにより形成される非特異的産物は含まれなかった。
【0067】
本実施例の厳密なアッセイを使用して、いくつかの無修飾および修飾DNAヘアピン分子を、末端安定化を伴わずに;各アームの端部に同一のヌクレオチドを付加することによって達成される末端不安定化を伴って;本発明に基づく末端安定化を伴って;ならびに安定化用ではない末端付加を伴って比較した。定量的比較を行うために、標準のTaqDNAポリメラーゼを用いて図1に示されるように達成される、ミスプライミングエラーにより形成される非特異的産物の排除、および所望の特異的産物しか存在しないことをもたらすために必要とされる最低濃度を決定するために、ミスプライミング抑制活性を示した各化合物に関して前記アッセイを使用する用量応答試験を実施した。その結果は表Iに示される通りであり、表には試験対象の各試薬に関する計算上のステムTmが含まれる。表Iに報告される種々の化合物の配列は表IIに記載される通りである。
【表1】
【0068】
【表2】
【0069】
表Iでは、修飾および無修飾のいくつかのDNAヘアピンが比較され、本発明の試薬(1000nM未満の濃度でミスプライミングの産物を伴わない純粋なアンプリコンが得られる)であるか、または本発明の好ましい試薬(650nM以下の濃度でミスプライミングの産物を伴わない純粋なアンプリコンが得られる)であるいくつかの末端安定化ヘアピン分子が特定される。無修飾のヘアピンは2つの数字によって特定される。第一の数字はステム長であり、第二の数字はループ長である。表Iに挙げられる本発明に基づく化合物は以下のような数種類の安定化用の修飾を有する:市販のリンカーを用いて末端ヌクレオチドに共有結合している5’Dabcylクエンチャーおよび3’Dabcylクエンチャー(接尾辞「DD」によって示される)、同様に結合している3’Black HoleTMクエンチャーおよび5’Black HoleTMクエンチャー(接尾辞「BHQBHQ」によって示される)、およびステム末端でデオキシリボヌクレオチドの代わりに用いられる2個の3’2’−O−メチルリボヌクレオチドおよび2個の5’2’−O−メチルリボヌクレオチド(「2’OM4」によって示される)。ステム末端に対する不安定化用の修飾には、分子の3’および5’末端を2個のAまたはTで置換することが含まれた。ステムを安定化する様式で互いに相互作用しないと考えられる1ペアのFAM蛍光団を付加すると、単一の5’Dabcylの付加と同様に不安定であった。しかし、単一の3’Dabcylの付加は9−22ヘアピンをわずかに安定化した。それは必要とされる濃度の低下によって示される通りである。ヘアピン9−22は高濃度(3000nM)でしかミスプライミングを抑制しなかったが、9−22DDは低濃度(50nM)でミスプライミングを抑制した。
【0070】
現在の本発明者らの好ましい別のヘアピンである化合物9−3は高濃度(1000nM)でしかミスプライミングを抑制しなかったが、9−3DDおよび9−3 2’OM4はずっと低い濃度(100〜300nM)でミスプライミングを抑制した。さらに別の実施形態である9−5DD、9−5BHQBHQおよび9−5 2’OM4もまた低濃度(50〜100nM)でミスプライミングを抑制した。オリゴヌクレオチド9−5はオリゴヌクレオチド9−22と同一のステムを有するが、ヌクレオチドがAおよびTである短いループを有する。オリゴヌクレオチド9−5の計算上のステムTmは93℃であり、ループの影響によりオリゴヌクレオチド9−22の計算上のステムTmより約12℃高い。
【0071】
さらに長いステムを用いても同様の結果が得られた。化合物12−3はミスプライミングを抑制しなかったが、化合物12−3DDはほんの50nMの濃度で有効であった。
【0072】
表Iは、安定化ステムを修飾して、計算上のステム融解温度(Tm)を変化させることができることを示す。例えば、Tm56℃の化合物9−3DDは100nMの濃度で有効であった。ステムのGC含量を変えることによって、化合物9−3bDD(Tm62℃)および9−3iDD(Tm68℃)を作成した。両化合物もまた100nMの濃度で有効であった。Tm58℃の化合物12−3DDは50nMの濃度で有効であった。改変されたステムを有する化合物、すなわちTm63℃の化合物12−3bDDおよびTm68℃の化合物12−3cDDは100nMの濃度で有効であり、依然として非常に良好であった。
【実施例2】
【0073】
実施例2. LATE−PCRエンドポイントアッセイ
反復実験サンプル間の反応キネティクスの確率的変動によってハイブリダイゼーションプローブ由来のシグナルのばらつきが生じ、エンドポイント解析が妨害される。LATE−PCRはその問題を著しく軽減する。本発明の試薬を利用するとエンドポイント検出を利用する能力がさらに改善される。図2は、テイ・サックス病に関与するヘキソサミニダーゼA遺伝子のG269対立遺伝子を含むアンプリコンを増幅する際に得られる結果を示す。反応混合物は0.6μMのハイブリダイゼーションプローブおよび1000ゲノムのホモ接合性野生型DNA標的(+/+)またはヘテロ接合性DNA標的(G269/+)を含んでいた。各タイプの反復サンプルを以下のように増幅した:25μLの反応物中、100nMの実施形態9−22DDの存在または非存在下での、1×PCRバッファー、3mMのMgCl2、250μMの各dNTP、配列5’ CGAGGTCATTGAATACGCACGGCTCC 3’(配列番号15)を有する25nMの制限的プライマー、配列5’ TAACAAGCAGAGTCCCTCTGGT 3’(配列番号16)を有する1000nMの過剰プライマー、1.25単位のPlatinum(ホットスタート) Taqポリメラーゼ、配列5’ Cy5−GGGACCAGGTAAGAA−リン酸 3’(配列番号17)を有する0.6μMのCy5標識ハイブリダイゼーションプローブ、および1:40,000希釈のSYBR Gold I。PCRサイクルのパラメータは、95℃で3分間;95℃で10秒間、65℃で20秒間、および72℃で20秒間の25サイクル;および、95℃で10秒間、65℃で20秒間、72℃で20秒間、55℃で20秒間、および45℃で20秒間の30サイクルであり、72℃でSYBR Goldに関する蛍光を取得し、55℃および45℃でCy5に関する蛍光を取得した。反応キネティクスの試験管ごとの変動を補正するために、72℃でのSYBR Goldシグナルに対する55℃でのCy5シグナルの比によってハイブリダイゼーションシグナルを標準化した。そのような標準化の影響の1つはすべてのCTの遅延がマスクされることである。
【0074】
図2の、サークル21によって特定されるライン群およびサークル22によって特定されるライン群は、本発明の試薬を加えないが、ホットスタートポリメラーゼを用いた場合の複数回の反復実験の結果を示す。ホモ接合性の反復実験(サークル21)およびヘテロ接合性の反復(サークル22)はそれらの直線プロットの勾配によって識別することができる。約40サイクル後のハイブリダイゼーションプローブシグナルの低下(「Hook効果(hook effect)」)によって立証される、これらのサンプルにおける反復実験間のばらつきおよびミスプライミングエラーは、これらのサンプルのエンドポイント識別を不可能にする。図2のサークル23〜24によって特定されるライン群は、本発明の試薬、すなわち100nMの9−22DDを加えた場合の結果を示す。これらの条件下では、ミスプライミングエラーが生じず、アッセイの経過中に直線的キネティクスが維持され、ばらつきが低減され、Hook効果が回避され、50サイクルの時点でホモ接合性およびヘテロ接合性サンプルのエンドポイント識別が可能である。
【実施例3】
【0075】
実施例3. ジデオキシシークエンシング用のDNAサンプルの調製
LATE−PCRは、増幅反応が60以上の熱サイクルにわたって実行される場合に、DNAシークエンシングに好適な大量の一本鎖DNAを生産しうる非対称増幅法である。しかし、そのような多数の増幅サイクルはミスプライミングの産物の出現を助長し、それにはタイプ2およびタイプ3のミスプライミングエラーの形成が含まれると考えられる。該ミスプライミングエラーは検出可能な非特異的産物の蓄積として現れ、それは、最終的に、反応において最も多数を占める分子種になる。本発明の試薬は、70以上のサイクルのLATE−PCR増幅を使用する場合に、非特異的産物を生じずに、DNAシークエンシングに好適な大量の一本鎖DNAの合成を可能にする。キャピラリーゲル電気泳動によるジデオキシシークエンシングに好適なサンプルを以下のように調製した。まずLATE−PCR反応混合物を、1×PCRバッファー、3mMのMgCl2、配列5’ GCCAGGGGTTCCACTACGTAGA 3’(配列番号18)を有する1000nMの過剰プライマー、配列5’ CCGCCCTTCTCTCTGCCCCCTGGT 3’(配列番号19)を有する25nMの制限的プライマー、1.25単位のPlatinum Taqポリメラーゼ、600nMの試薬9−3DDおよび250μMの各dNTPを含有する25μLで調製した。化合物9−3DDは、オリゴヌクレオチド9−3の各末端ヌクレオチドに付加されたDabcylクエンチャーを有する。増幅は、95℃で3分間;95℃で10秒間、65℃で20秒間、および72℃で20秒間の10サイクル;および、95℃で10秒間、65℃で20秒間、72℃で20秒間;および45℃で20秒間の60サイクル実行した。二本鎖および一本鎖DNA合成を別々にモニタリングするために、1:40,000希釈のSYBR Greenまたは配列5’ FAM−CGTGCGCTCTGGTAAGGGTTTGCACG−Dabcyl 3’(配列番号20)を有する2.4μMの低Tm分子ビーコンを用いて並行して反応を実施した。各サンプルは6ng(約1000ゲノム)のヒトDNAを含有した。さらに、化合物9−3DDを加えずに対照反応を実施した。
【0076】
増幅反応の結果、ホットスタートTaqDNAポリメラーゼを用いるが、化合物9−3DDを用いない対照は顕著なミスプライミングを示した。制限的プライマーが使い果たされた後の増幅の後期段階中に、SYBR Greenシグナルが二次的に上昇することによってそれは立証される。該産物はそのような混合物であるため、該対照をシークエンシングすることは不可能であった。一方、化合物9−3DDを加えた場合の増幅の結果では、所望の特異的産物しか存在せず、70サイクルを通してSYBR Greenシグナルの遅延された上昇はないことが示された。制限的プライマーの濃度および50%の直線的増幅の見かけの効率から、反応によって250フェムトモル(fmoles)/μLの純粋な産物が生産されると見積もられる。該産物をBrandeis Universityのジデオキシシークエンシング施設に送り、シークエンシングを行った。
【0077】
シークエンシング反応は、Beckman CEQ 2000 DNAシーケンサーを利用するキャピラリー電気泳動などの標準プロトコルを使用して実施した。増幅産物の1/5アリコート、すなわちわずかに50フェムトモルの産物を用いてシークエンシング反応を実施した。機械により作成された配列を図3に挙げる。図3では、チャートの上部に沿って配列を示し、クリアで不明瞭なところがない産物ピーク(これから配列を導き出した)を示し、底部では非常に低レベルのバックグラウンドシグナルを示す。本発明者らは、機械により作成された配列をGenBank登録番号NT 010235から入手可能な該アンプリコンの公知の配列と比較することによって、その正確さを確認した。
【実施例4】
【0078】
実施例4. 多重化PCR増幅
ミスプライミングエラーの抑制は、複数の産物が同時に増幅される多重化反応において特に重要である。複数のプライマーペアおよび増幅産物が存在すると、これらの反応種の2者間および3者以上の間のミスプライミング相互作用の確率が高くなる。増加した数のアンプリコンからなる多重化反応を化合物9−3DDの不存在または存在下で実行した。以下に示すプライマーペアの種々の組み合わせに関して、300nMの試薬9−3DDの非存在下または存在下で、サンプル、1×PCRバッファー、3mMのMgCl2、100μMの各dNTP、1000nMの過剰プライマー、50nMの制限的プライマー、および1.25単位の非ホットスタートPromega Taqポリメラーゼからなる25μL容量のLATE−PCR反応物を調製した。サンプルの増幅は、95℃で3分間;95℃で10秒間,65℃で30秒間,および70℃で30秒間の10サイクル;および、95℃で10秒間,50℃で30秒間,70℃で30秒間の40サイクルの熱サイクルプロファイルを使用して行った。反応の終了時点で、0.5×TBE中の3.5%アガロースゲルでのゲル電気泳動によってサンプルを分析した。試験対象の所望のアンプリコンおよびプライマーペアは以下の通りであった。
【0079】
反応A:テイ・サックス病に関連するHex−A遺伝子の2領域TSD1278+TSD1421
TSD1278過剰プライマー:5’ GCCAGGGGTTCCACTACGTAGA 3’(配列番号21)
TSD1278制限的プライマー:5’ CCGCCCTTCTCTCTGCCCCCTGGT 3’(配列番号22)
TSD1421過剰プライマー:5’ CCGGGTCTCTAAGGGAGAACTCCT 3’(配列番号23)
TSD1421制限的プライマー:5’ CCGGCCGACAACACAAACCTGGTCC 3’(配列番号24)。
【0080】
反応B:TSD1278アンプリコン、TSD1421アンプリコン、およびCFTR遺伝子の1領域、CFエキソン10アンプリコン
本反応は反応Aと同じプライマーペアに加えて以下のプライマーペアを含有した:
CF ex10過剰プライマー:5’ GCTTTGATGACGCTTCTGTATCTA 3’(配列番号25)
CF ex10制限的プライマー:5’ CAGTTTTCCTGGATTATGCCTGGCACCAT 3’(配列番号26)。
【0081】
反応C:TSD1278アンプリコン、TSD1421アンプリコン、CFエキソン10アンプリコン、およびCFTR遺伝子の別の領域、CFエキソン11アンプリコン
本反応は反応Bと同じプライマーペアに加えて以下のプライマーペアを含有した:
CF ex11過剰プライマー:5’ TCGAAGTTTGCAGAGAAAGACAAT 3’(配列番号27)
CF ex11制限的プライマー:5’ TGACGTTTACAGCGAATGCTTGCTAGACCAAT 3’(配列番号28)。
【0082】
反応D:TSD1278アンプリコン、TSD1421アンプリコン、CFエキソン10アンプリコン、CF ex11アンプリコン、およびヒトβグロビン遺伝子の1領域
本反応は反応Cと同じプライマーペアに加えて以下のプライマーペアを含有した:
βグロビン過剰プライマー:5’ TGGGTTTCTGATACGCACTGACTCTCTC 3’(配列番号29)
βグロビン制限的プライマー:5’ GGCCATCACTAAAGGCACCGAGCACT 3’(配列番号30)。
【0083】
電気泳動ゲルを臭化エチジウムで染色し、LATE−PCR反応の対数期に生成された二本鎖産物を検出した。図4は、多重化反応A〜Dにより得られた産物についての該当部分のゲルを示す。矢印は特異的二本鎖増幅産物を指し示し、無数のバンドは二次構造を有する特異的一本鎖DNA産物に相当し、アスタリスクは非特異的産物を特定する。ゲルのペアA〜Dはそれぞれ反応A〜Dに相当する。「+」でラベルされている各ゲルは化合物9−3DDを含む増幅により得られた産物である。「−」でラベルされている各ゲルは化合物9−3DDを含まない増幅により得られた産物である。
【0084】
ゲルのペアAは、9−3DD化合物の非存在下で、領域TSD1278(矢印41)は増幅されなかったが、領域TSD1421(矢印42)は、主にまたは完全に二本鎖の多数の非特異的産物とともに増幅されたことを示す。該非特異的産物はゲルではスメアとして見られる。9−3DD化合物を加えると、非特異的産物のバックグラウンドが減少し、TSD1278(矢印41)およびTSD1421(矢印42)の両者の増幅が可能になった。それらはよりきれいに増幅されていて、ミスプライミングのいくつかの現象が防止されたことが示される。
【0085】
ゲルのペアBは、9−3DD化合物の非存在下で、領域TSD1278、TSD1421、およびCFエキソン10(矢印43)が、ゲルではスメアとして見られる多数の非特異的産物とともに増幅されたことを示す。9−3DD化合物を加えると、バックグラウンドの非特異的産物が排除され、3種の予測されるアンプリコンのすべてが得られた。
【0086】
ゲルのペアCは、9−3DD化合物の非存在下で、領域TSD1278、TSD1421、CFエキソン10、およびCFエキソン11(矢印44)が、アスタリスクによって示される非特異的産物とともに増幅されたことを示す。9−3DD化合物を加えると、非特異的産物の合成が排除され、4種の予測されるアンプリコンのすべてが得られた。
【0087】
ゲルのペアDは、9−3DD化合物の非存在下で、βグロビン遺伝子の選択された領域(矢印45)は増幅されなかったが、TSD1278、TSD1421、CFエキソン10、およびCFエキソン11は、アスタリスクによって示される非特異的産物とともに増幅されたことを示す。9−3DD化合物を加えると、非特異的産物の合成が排除され、5種の予測されるアンプリコンのすべての増幅が可能になった。
【0088】
また本発明者らは、感染性病原体に関してスクリーニングする場合に生じうるものなどの、任意の所定の試験サンプル中に1つの標的しか存在しない場合の多重増幅および検出反応における本発明の化合物の用途を実証した。そのような場合、複数ペアのプライマー(本発明者らは複数ペアの過剰プライマーおよび制限的プライマーをLATE−PCR増幅に利用した)が存在するが、特定のサンプルに関して1ペアのプライマーしか有効ではない。SYBR green蛍光および増幅後の電気泳動による解析によって示されるように、それは実際に生じたことであり、したがって、すべての基質より少数の基質しか存在しない場合の多重反応において本発明の化合物が効果的にミスプライミングを阻害することが実証される。
【実施例5】
【0089】
実施例5. ステムTmとアニーリング温度の関係
標的を含む同じ増幅混合物を利用して2種の異なるLATE−PCR増幅プロトコルを比較した。600nMの化合物9−3DDを含む場合および化合物9−3DDを含まない場合の両方で各増幅を実施した。上記のように、化合物9−3DDのステムTm(すなわち無修飾のオリゴヌクレオチド9−3の計算上の融解温度)は56℃である。ホットスタートPlatinum Taq DNAポリメラーゼを使用した。2種の増幅は、主として、使用されるプライマーアニーリング温度が異なり、65℃(ステムTmより高い)または55℃(ステムTmより低い)であった。増幅サイクルのパラメータは以下の通りであった:
プロファイルA:95℃で3分間;95℃で10秒間,65℃で20秒間,72℃で20秒間の10サイクル;95℃で15秒間,55℃で20秒間,72℃で20秒間,50℃で20秒間の60サイクル
プロファイルB:最後の60サイクルが95℃で10秒間,65℃で20秒間,72℃で20秒間,45℃で20秒間であったことを除いて、プロファイルAと同一。
【0090】
増幅反応をリアルタイムでモニタリングした。その場合、二本鎖産物の合成をモニタリングするためにSYBR Green I蛍光色素を使用した。ミスプライミングによって増幅の後期部分がばらつかない限り、二本鎖産物の合成は、制限的プライマーが使い果たされた後のLATE−PCR反応においてプラトーに達した。低温検出ステップ中に読み取りを行った。
【0091】
結果を図5に挙げる。パネルAは、化合物9−3DDを加えた場合(サークル52)および加えなかった場合(サークル51)の両方において、10サイクルの伸長後に55℃のアニーリング温度が利用された場合のプロファイルAの反復実験サンプルから得られた蛍光読み取り値を示す。パネルBは、化合物9−3DDを加えた場合(サークル54)および加えなかった場合(サークル53)の両方において、10サイクルの伸長後に65℃のアニーリング温度が使用された場合のプロファイルBの反復実験サンプルから得られた蛍光読み取り値を示す。
【0092】
図5は、本発明の試薬のステムTmより低いプライマーアニーリング温度が使用された場合(パネルA)に、閾値サイクルCTの数サイクルの遅延が観察されたことを示す。しかし、指数的PCR相の初めから終わりまでステムTmよりかなり高いプライマーアニーリング温度が維持された場合(パネルB)には、CTの遅延は、たとえあったとしても、ほとんど観察されなかった。本増幅で利用された特定のアンプリコンに関しては、ホットスタートTaqが使用された場合に、化合物9−3DDを用いても用いなくても、タイプ2/タイプ3のミスプライミングが観察されなかったことがわかった。このことは、そのようなミスプライミングエラーの出現が予測不能であることを示す。本実施例は、反応の温度が55℃に下がると試薬9−3DDのステムが閉鎖型になること、および反応の高温伸長ステップ中は該試薬がポリメラーゼに結合したままであることを実証する。
【実施例6】
【0093】
実施例6. 25℃でのDNAポリメラーゼ活性に対する効果
本発明の試薬およびそれらの無修飾のオリゴヌクレオチドアナログの効果を評価するために、一連のプライマー伸長実験を25℃で実行した。各反応混合物には、鋳型、プライマーおよびTaqDNAポリメラーゼを含めた。各場合において2時間、反応を実行した。プライマーをCy5で蛍光標識した。SYBR Green蛍光色素を反応混合物に加えた。伸長反応後に、融解曲線を作成した。その場合、本発明者らが同時に出願した米国仮特許出願、表題「Primers, Probes and Methods for Nucleic Acid amplification」に開示されるように、色素を刺激し、該色素からのFRET転移による蛍光団の発光を読み取った。結果を図6に挙げる。
【0094】
図6のパネルAは、反応混合物からdNTPが省略されることによって伸長が妨げられた場合に得られた融解曲線61を含む。ゆえに曲線61はプライマー・標的ハイブリッドの融解曲線であり、58℃のTmを示す。またパネルAは、dNTPを、単独で、あるいは無修飾のオリゴヌクレオチド9−3(300nM、1000nM)または9−22(50nM、100nM、300nM)とともに含有するいくつかの対照を含む。サークル62によって特定される、前記対照に関する曲線は、すべての場合で伸長を示し、すなわち58℃のプライマー融解ピークが消失した。図6のパネルBは、反応混合物中に試薬9−22DDを加えることの用量依存的効果を示す:曲線63、50nM;曲線64、100nM;曲線65、300nM。58℃のピークが大きいほど、等温性試験条件下で重合による伸長を防止する効果が大きい。図6のパネルCは、反応混合物中に試薬9−3DDを加えることの用量依存的効果を示す:曲線66、300nM;曲線67、1000nM。この場合もやはり、58℃のピークが大きいほど、試験条件下で重合による伸長を防止する効果が大きい。
【0095】
また本発明者らは、5’−3’エキソヌクレアーゼ活性を欠いているDNAポリメラーゼであるStoffel断片(Lawyer et al. (1993) PCR Methods and Applications 2: 275-287)に対する効果を調べた。鋳型、蛍光標識プライマー、Stoffel断片およびdNTPしか含有しない陰性対照と、表1に列挙される種々の化合物を比較した。この一連の試験では、40℃で伸長を実施し、SYBR Green読み取り値を30分間以上にわたって20秒毎に取得した。目視によって曲線を比較したが、その比較は信頼できるものであった。その理由は、得られた曲線が対照をほぼ正確にたどるか、あるいは得られた曲線が対照とは著しく異なっていたからである。50nM、100nMおよび300nMの濃度で試験化合物を加えた。無修飾のヘアピン6−22、9−3、9−5および9−22は、いかなる濃度においてもStoffel断片による伸長を抑制しなかった。化合物6−22DDは、化合物9−3DD、9−5DDおよび9−22DDと同様に、すべての3種の濃度で伸長を抑制した。以下の化合物は、50Mまたは100nMの濃度では伸長を抑制しなかったが、300nMの濃度では伸長を抑制した:9−22−5D、9−22−3D、9−5 2’OM4、9−5BHQBHQ、および9−32’OM4。
【実施例7】
【0096】
実施例7. ポリメラーゼ活性の非存在下でのTaqエキソヌクレアーゼ活性に対する効果
本発明者らは、DNA合成の非存在下での5’−3’エキソヌクレアーゼ活性の阻害を実証するアッセイを開発した。本アッセイでは、一方の鎖の5’末端がFAM蛍光団で標識され、他方の鎖の3’末端がDabcylクエンチャーで標識されている特異的二本鎖DNA分子を基質として使用する。蛍光団はクエンチャーと近接していて、蛍光団が刺激された場合に蛍光は生じない。加熱および冷却による鎖変性およびアニーリングによって、FAM標識鎖の切断および蛍光団の遊離が起こる。何らかのサイクル処理によってTaqポリメラーゼの5’−3’エキソヌクレアーゼ活性に関する基質が生成されると考えられる。したがって、蛍光の増大はTaqポリメラーゼの5’−3’エキソヌクレアーゼ活性の尺度を提供する。
【0097】
25μLの反応混合物は、適切な濃度の9−22DD化合物の存在または非存在下で、1×PCRバッファー中の300nMの二本鎖DNA鋳型、3mMのMgCl2、および1.25単位(U)のTaqポリメラーゼを含有する。該反応物は、いかなるdNTPをも含有せず、前記二本鎖DNA以外のいかなる他の核酸標的をも含有しない。前記二本鎖DNA鋳型の相補鎖の配列は、5’ FAM−AGTGTGATGATGGTGAGG −リン酸基3’(配列番号31)および5’ ACTTTCAACTCTGTCT 3’−Dabcyl(配列番号32)である。サンプルを95℃で3分間変性させ、次いで95℃で10秒間,67℃で30秒間,72℃で30秒間,および45℃で20秒間のサイクルに付する。各サイクル中に45℃で蛍光を取得する。
【0098】
複数のサンプルに対するアッセイの実施から得られた蛍光読み取り値を図7に報告する。図7では、読み取り値を標準化して、同じバックグラウンド蛍光で出発するようにしてある。曲線76は、Taqポリメラーゼが存在しない場合の蛍光を示す対照である。残りのサンプルにはTaqポリメラーゼを加えた。曲線71は、試薬9−22DDを加えなかった場合に得られた蛍光の増大を示す。30サイクルにわたって蛍光は安定的に増大した。曲線72〜75は、それぞれ50nM、100nM、200nMおよび300nMの濃度で試薬9−22DDを加えた場合の蛍光の増大を示す。上記反応物に9−22DD化合物を加えると、観察される蛍光の増大が用量依存的様式で減少した。
【実施例8】
【0099】
実施例8. 対称的PCR増幅
本発明者らは、剪断ゲノムDNAおよび等モル濃度で近似のTmを有する1ペアのプライマーを利用してリアルタイムPCR増幅アッセイを実施した。400nMの試薬9−3DDを加えない場合および加えた場合で反復アッセイを実施した。DNA全体としての蓄積のキネティクスに関して、SYBR Greenでの染色によって、ならびに臭化エチジウム染色を用いるゲル電気泳動によって試薬の効果を評価した。
【0100】
増幅に関する所望の標的はCFTR遺伝子のイントロン19に一塩基転位として存在する対立遺伝子であった(GenBank登録番号AC000061)。25μLの反応混合物は、400nMの試薬9−3DDの不存在または存在下で、120ピコグラム(pg)の剪断ヒトゲノムDNAの基質、1×PCRバッファー、5mMのMgCl2、250μMの各dNTP、1:40,000希釈のSYBR Green、1000nMのフォワードプライマー:配列5’ TAATTACAAGAGTCTTCCAT 3’(配列番号33)(Tm56.6℃)、および1000nMのリバースプライマー:配列5’ CATGAATAGAACATTTCCTT 3’(配列番号34)(Tm56.3℃)、および1.25単位の非ホットスタートInvitrogen Taqポリメラーゼを含有した。以下の熱サイクル処理プロファイルを使用してサンプルを増幅した:95℃で3分間;95℃で10秒間,55℃で30秒間,および72℃で30秒間の60サイクル(SYBR Green I蛍光を72℃でモニタリング)、最後に、30秒間に1℃刻みで増加する54℃〜96℃の温度勾配を用いる。反応の終了時点で、各組の反応から得られたランダムに選択された複数のサンプルを、0.5×Tris−ホウ酸−EDTA溶液(TBE)中、3.0%アガロースゲルでのゲル電気泳動に付することよって分析した。
【0101】
図8は得られた結果を示す。図8のパネルAの増幅プロットは60サイクルにわたるSYBR Green I蛍光を示し、サークル81によって特定される曲線群は試薬9−3DDを含有しない反復実験サンプルであり、サークル82によって特定される曲線群は400nMの9−3DDを含む反復実験サンプルである。2組のサンプルから生じた蛍光シグナルのキネティクスによれば、二本鎖DNA全体は、試薬9−3DDの非存在下で、該試薬の存在下よりいくらか速やかに蓄積したが、60サイクルによって蓄積された二本鎖DNAの総量は2組の反応において実質的に同一であることが実証される。
【0102】
図8のパネルBは、臭化エチジウム染色を施した電気泳動ゲルを示す。左側の図では、レーンaはサイズマーカー(50塩基対ずつ異なるサイズ)であり、レーンb〜iは試薬9−3DDを含まないサンプルである。右側の図では、レーンj〜qは400nMの9−3DDを含むサンプルであり、レーンrはサイズマーカーである。この結果は、本発明の試薬を含有しない反応では、適正な産物ならびに高分子量の多数の非特異的産物が生成されたが、試薬9−3DDを含有する反応では、適正な産物しか生成されなかったことを示す。さらに、ゲル中の適正な産物のバンドの相対強度によって判断すると、試薬9−3DDを含有する反応では、該試薬を含有しない反応の約2倍の適正な産物が生成された。
【0103】
キネティクス解析および電気泳動による解析の組み合わせでは、非特異的産物を生成する反応81が、適正産物しか生成しない反応82より早く蛍光シグナルを生じたことが示される。SYBR Green色素は配列特異性とは無関係に二本鎖DNA内に挿入される。ゆえに、反応81の直線的なキネティクスと比較して、よりS字状である反応82のキネティクスは、対称的反応が適正な産物のみを蓄積しているか否かを判断するために使用することができる。
【実施例9】
【0104】
実施例9. エキソヌクレアーゼ阻害の定量化
本実施例は、TaqDNAポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する本発明の試薬の阻害効果を測定するための厳密なアッセイを記載する。本アッセイでは、M. W. Kaiser, N. Lyamicheva, W. Ma, C. Miller, B. Neri, L. Fors, and V. I. Lyamicheva in J. Bio. Chem., 274, pp. 21387-21394(1999)に記載の系と同様のプライマー−鋳型系を使用した。ただし、鋳型の5’末端をCy5で標識し、二本鎖DNAに結合すると蛍光を発するDNA色素であるSYBR Green Iの存在下でアッセイを実行した点で異なる。本アッセイ用の鋳型は、ヌクレオチド配列:5’−ACGAGCGTCTTTC−3’(配列番号36)を有するプライマーにアニーリングするオリゴヌクレオチド配列5’−Cy5−AAAACGCTGTCTCGCTGAAAGCGAGACAGCGAAAGACGCTCGT−3’(配列番号35)からなる。長い方のオリゴヌクレオチドは、異なる長さの2個の一本鎖尾部を有するヘアピン構造を形成する。鋳型の短い5’尾部はCy5蛍光団を含有し、4アデノシン残基からなり;鋳型の長い方の3’尾部は、プライマーオリゴヌクレオチドに対する標的となり、これにより、該プライマーが1塩基対の重複を有して5’の鋳型尾部の直前に位置する。この鋳型−プライマー複合体にSYBR Green Iを加えると、該複合体の二本鎖DNA領域に結合したSYBR Green Iが生じる。480nmレーザーでSYBR Green Iが励起されると、結合したDNA色素からの蛍光エネルギーがCy5によって完全に吸収され、それは蛍光共鳴エネルギー転移によって生じると考えられ、最大SYBR Green発光波長(最大発光波長:521nm)で検出可能な蛍光は存在しない。Taqポリメラーゼの5’−3’エキソヌクレアーゼ活性によって5’尾部が加水分解されると、鋳型−プライマー複合体からCy5部分が除去され、検出可能なSYBR Green I蛍光が回復する。TaqDNAポリメラーゼの存在下でのSYBR Green I蛍光の回復についてのキネティクスは、5’−3’エキソヌクレアーゼ活性の定量的尺度を提供する。本発明の試薬はTaqポリメラーゼの5’−3’エキソヌクレアーゼ活性を阻害し、SYBR Green I蛍光の増大を減速させる。
【0105】
本アッセイは、25マイクロリットル(μL)の容量中、50nM、100nM、300nM、または1000nMの9−22DD化合物の不存在または存在下で、1×PCRバッファー中で混合された0.5μMの上記鋳型および1.5μMの上記プライマー、3mMのMgCl2、1:40000希釈の市販のSYBR Green Iストック溶液(Molecular Probes, Eugene, OR)、1.25UのTaqポリメラーゼ(Invitrogen, Calrsbad, CA)から構成されるものとした。該反応混合物にはdNTPを加えなかった。その理由は、本アッセイがDNAポリメラーゼ活性に依拠しないからである。鋳型およびプライマーを含まない反応混合物を25℃で調製して、9−22DD化合物およびTaqDNAポリメラーゼ間の相互作用を促進させた。次いで、サンプルを氷上に置き、鋳型およびプライマーを加え、反応の開始まで氷上で置いた。陰性対照にはTaqDNAポリメラーゼを加えなかった。ABI Prism Sequence Detector 7700にて25℃で60分間サンプルをインキュベートすることによって反応を開始し、SYBR Green Iチャネルにおける蛍光収集を30秒毎に行った。各対照および各濃度の試薬9−22DDに関して3回の異なる試行を平均した。
【0106】
図9は本アッセイの結果を示す。検出可能なSYBR Green I蛍光は、5’−3’エキソヌクレアーゼ活性の程度についての尺度を提供する。「Taqなし」の対照はSYBR蛍光シグナルを示さず、Cy5によるSYBR Green I蛍光の完全吸収および5’−3’エキソヌクレアーゼ活性の完全な不存在と矛盾しない(ライン96)。Taqポリメラーゼを加えると、SYBR Green I蛍光が回復し、当該アッセイ条件下での5’−3’エキソヌクレアーゼ活性の最大レベルに関するベースラインが提供される(ライン91)。反応混合物に9−22DD化合物を加えると、Taqポリメラーゼの5’−3’エキソヌクレアーゼ活性に対する9−22DDの阻害効果に起因して、SYBR Green I蛍光の回復が用量依存的様式で減速される(ライン92、50nM;ライン93、100nM;ライン94、300nM;ライン95、1000nM)。以下の表IIIでは、25℃で60分間のインキュベーション後に達成される相対的SYBR Green I蛍光のレベルに基づいて、種々の濃度の9−22DD化合物による5’−3’エキソヌクレアーゼ活性の阻害割合を定量化する。
【表3】
【実施例10】
【0107】
実施例10. 標的DNAの存在下および非存在下でのプライマー二量体形成およびオリゴマー形成の防止
最終容量25μL中の100ゲノムのヒト胎盤DNA(Sigma, St Louis, MO)の存在下または非存在下で、一連のLATE−PCR増幅反応を準備した。この実験では、LATE−PCR増幅反応物は、1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.25mMのdNTP、1000nMの過剰プライマー、50nMの制限的プライマー、0.25μMのFAM標識プローブ鎖、0.3μMのDabcyl標識リバース相補鎖、1.25単位のPlatinum TaqDNAポリメラーゼ(Invitrogen, Carlsbad, CA)から構成するものとした。プライマーおよびプローブの配列は以下の通りであった:
過剰プライマー:5’ GTTTCTTTGCTGCCGTGTTC 3’(配列番号37)
制限的プライマー:5’ CCCCAGAGACCCCAGTTGCTAACCAGAC 3’(配列番号38)
FAM標識プローブ鎖:5’ [TET]AGACAGAGTTGAAAGTCAGG[Phos] 3’(配列番号39)
Dabcyl標識リバース相補鎖:5’ ACTTTCAACTCTGTCT[Dabcyl] 3’(配列番号40)。
【0108】
前記プライマーおよびプローブは、ヒトp53遺伝子のエキソン5および6を包含する488塩基対(bp)のアンプリコンを増幅および検出する。
【0109】
ABI Prism 7700 Sequence Detector(Applied Biosystems, CA)で増幅を実行した。その場合、95℃で3分間の1サイクル;95℃で10秒間,64℃で30秒間,75℃で30秒間の25サイクル;および95℃で10秒間,64℃で30秒間,75℃で30秒間,45℃で20秒間の35サイクルからなる熱プロファイルを用い、45℃ステップ中にTETチャネルにおいて蛍光を検出した。
【0110】
得られた増幅産物を、0.5×TBEバッファー中で、2時間、3%アガロースゲルでのゲル電気泳動に付することよって分析し、臭化エチジウムで染色した。結果を図10に示す。レーン3、5、7、9、11のサンプルはゲノムDNAを加えずに調製されたものである。レーン2、4、6、8、10のサンプルはゲノムDNAを加えて調製されたものである。レーン1には100塩基対ラダーの電気泳動用サイズマーカーを含めた。試薬9−22DDを以下のように出発反応物に加えた:レーン2および3、0nM;レーン4および5、50nM;レーン6および7、100nM;レーン8および9、200nM;レーン10および11、300nM。
【0111】
ゲルの右側に隣接して、適正産物のサイズ、ならびにプライマー二量体およびプライマーオリゴマーであると推測されるサイズをマークした。図10に示される結果は、ゲノムDNAを加えて開始された反応において、試薬9−22DDの濃度を増加させると、プライマー二量体およびプライマーオリゴマーを含めた非特異的産物の出現を防止するように用量依存的様式で作用し、それによって適正産物の特異性および収量の両者が高まることを実証する。また増加濃度の試薬9−22DDは、ゲノムDNAを含有しない反応においても、プライマー二量体およびプライマーオリゴマーの出現を防止する。
【実施例11】
【0112】
実施例11. 二重反応におけるプライマー二量体形成の防止
最終容量25μL中の100ゲノムのヒト胎盤DNA(Sigma, St Louis, MO)の存在下または非存在下で、一連のLATE−PCR増幅反応を準備した。この実験では、LATE−PCR増幅反応物は、1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.20mMのdNTPから構成するものとした。サンプルごとに1.25単位のTaqポリメラーゼを使用した。第一の増幅標的配列(産物1)は嚢胞性線維症遺伝子のエキソン11の一部分であり、100nMの制限的プライマー:5’ GACGTTTACAGCGAATGCTTGCTAGACCAAT 3’(配列番号41)および2,000nMの過剰プライマー:5’ TCCAAGTTTGCAGAGAAAGACAAT 3’(配列番号42)を用いて増幅した。第二の増幅標的配列(産物2)は嚢胞性線維症遺伝子のエキソン10の一部分であり、50nMの制限的プライマー:5’ CAGTTTTCCTGGATTATGCCTGGCACCAT 3’(配列番号43)および1000nMの過剰プライマー:5’ GCTTTGATGACGCTTCTGTATCTA 3’(配列番号44)を用いて増幅した。
【0113】
ABI Prism 7700 Sequence Detector(Applied Biosystems, CA)において、95℃で2分間、次いで95℃で10秒間,56℃で15秒間,70℃で20秒間の25サイクル、次いで95℃で10秒間,56℃で15秒間,70℃で20秒間,および45℃で30秒間(蛍光の取得を伴う)の50サイクルで増幅を実行した。
【0114】
得られた増幅産物を、0.5×TBEバッファー中の3%アガロースゲルでの2時間のゲル電気泳動に付すことによって分析し、臭化エチジウムで染色した。結果を図11に示す。レーン1〜6で分析された反応では、非ホットスタートTaqポリメラーゼを利用し、レーン7〜12で分析された反応では、Taqポリメラーゼに加えてホットスタート抗体を利用した。レーン4〜6および10〜12で分析された反応では、100nMの試薬9−22DDを加えた。
【0115】
ゲルの右側に隣接して、産物1および産物2の一本鎖アンプリコン(ssDNA)および二本鎖アンプリコン(dsDNA)、ならびにプライマー二量体であると推測される短い産物を含めた増幅産物の特定についての解釈を付した。他の非特異的産物はアスタリスクでマークされる。非ホットスタートポリメラーゼを用い、本発明の試薬を用いない増幅(レーン1〜3)では、プライマー二量体および他の非特異的産物が生成された。ホットスタートポリメラーゼを用い、本発明の試薬を用いない増幅(レーン7〜9)では、概して、いくらか不純物が減少したが、依然としてミスプライミングの現象が示された。本発明に基づく試薬をホットスタート増幅に加えたレーン10〜12では、目的の産物のみが生成された。本発明の試薬を非ホットスタート増幅に加えたレーン4〜6では、プライマー二量体が排除され、3回の反復実験のうちの2回(レーン4およびレーン6)では、すべての他の非特異的産物も同様に排除されたが、1回の反復実験(レーン5)のみでは非特異的産物が出現した。図11に示される結果は、非ホットスタートTaqポリメラーゼまたはホットスタートTaqポリメラーゼが使用された場合、低濃度(100nM)の試薬9−22DDが、プライマー二量体の形成を防止するために十分であったこと、さらに、その低濃度の試薬9−22DDが、2ペアのプライマーを用いる2種の標的の増幅において非ホットスタートTaqポリメラーゼとともに使用された場合でさえ、ミスプライミングのすべての現象を防止するためにほぼ十分であったことを実証する。
【実施例12】
【0116】
実施例12. プライマー二量体形成の防止による二重リアルタイムPCRおよびリアルタイムPCRのキネティクスの最適化
マウスOct4およびXist遺伝子のエキソン内の配列(それぞれGenBank登録番号NM_013633およびL04961)を同時に増幅するための二重リアルタイムLATE−PCRアッセイを設計した。各反応は最終容量50μL中で実行し、以下の試薬を含有した:20mMのTris−HCl,pH8.4、および50mMのKClによって構成される1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.4mMの各dNTP、配列5’ TGGCTGGACACCTGGCTTCAGACT 3’(配列番号45)を有する50nMのOct4制限的プライマー、配列5’ CAACTTGGGGGACTAGGC 3’(配列番号46)を有する2μMのOct4過剰プライマー、配列5’ GGTCGTACAGGAAAAGATGGCGGCTCAA 3’(配列番号47)を有する100nMのXist制限的プライマー、配列5’ TGAAAGAAACCACTAGAGGGCA 3’(配列番号48)を有する2μMのXist過剰プライマー、配列5’ TET−CCG CCT GGG ATG GCA TAC TGT GGA AGG CGG−Dabcyl 3’(配列番号49)を有する1μMの低TmのOct4分子ビーコンおよび2単位の抗体複合体化Platinum(登録商標)Taq DNAポリメラーゼ(Invitrogen, Carlsbad, CA)。さらに化合物9−3DDまたは化合物9−3bDDを、以下に指定される濃度でPCR混合物に加えた。本実施例では、Xistアンプリコンを検出するための分子ビーコンを加えなかった。各アッセイは、PurAmpプロトコル(Hartshorn et al. (2005) BMC Biotechnol 5: 2を参照のこと)にしたがって、2.5μL容量中に細胞溶解および逆転写に必要な試薬をさらに含有した。この二重LATE−PCRでは、前記試薬の最終濃度は以下の通りであった:2.5mMのTris酢酸,pH8.4、3.75mMの酢酸カリウムおよび0.4mMの酢酸マグネシウム(希釈したcDNA合成バッファー, ThermoScriptTM RT-PCR System, Invitrogen, Carlsbad, CA)、追加の50μMの各dNTP、0.13mMのイソチオシアン酸グアニジン、6.7μMのβ−メルカプトエタノール、0.7μMのクエン酸ナトリウム,pH7.0、0.7×10−4%(vol/vol)のジメチルスルホキシドおよび0.2×10−4%のサルコシル(sarcosyl)。
【0117】
さらにマウスゲノムDNA(Sigma, St Louis, MO)を各アッセイに加え、PCR増幅用の鋳型を提供した。各試験管に加えるゲノム数は6pg/ゲノムサイズに基づいて算出した(Vendrely and Vendrely (1949) Experientia 5: 327-329を参照のこと)。
【0118】
ABI Prism 7700 Sequence Detector(Applied Biosystems, CA)において増幅を実行した。その場合、95℃で5分間の1サイクル;95℃で10秒間,63℃で20秒間,および72℃で30秒間の15サイクル;および95℃で15秒間,55℃で25秒間,72℃で35秒間,および45℃で30秒間の40サイクルから構成される熱プロファイルを用い、TETチャネルにおいて45℃で蛍光を取得した。
【0119】
PCRの終了時点で、増幅産物を、0.5×TBEバッファー中の3%アガロースゲルでの2時間のゲル電気泳動に付すことによって分析し、臭化エチジウムで染色した。2種の同時に増幅された遺伝子に関して、予測されたサイズの二本鎖および一本鎖産物がともに可視であった。このことは二重LATE−PCRが有効であることを示す。
【0120】
図12は、Oct4およびXist遺伝子内の2種の非相同配列を増幅するための2組のプライマーを含有する上記二重LATE−PCRのキネティクスに対する、試薬9−3DDのステム組成を変化させることの効果を示す。該図は、TET−Oct4分子ビーコンとのハイブリダイゼーションを介してOct4アンプリコンの蓄積によって生じる蛍光シグナルを示す。その結果は、分析された各ゲノム濃度(10ゲノム、サークル122;100ゲノム、サークル123;1000ゲノム、サークル124)で、300nMの9−3DD(三角形付きの線)または同濃度のその修飾型である9−3bDD(三角形が付いていない太線)の存在下でCT値が非常に近いことを実証する。そうではなくむしろ、リアルタイム蛍光シグナルのキネティクスは試薬のステムの組成によって影響を受ける。化合物9−3bDDは、9−3DDと比較して高いTmのステムを有し、ゆえに、プライマー二量体形成を最適に防止すると本発明者らは考え、その結果、すべての試験ゲノム濃度で非常に直線的で平行なシグナルが生じる。一方、低ストリンジェントの化合物9−3DDの存在下では、蛍光シグナルの一部は急な勾配を有するが、他のものは反応の早期にプラトーに達し始め、それはプライマー二量体のランダムな形成を示す。種々の鋳型濃度で化合物9−3DDを用いて生じるシグナルは、化合物9−3bDD(改変型ステムを有する)の存在下で得られるシグナルより平行ではない。理想的には、完全に平行なシグナル(一定勾配を有する)を生じる直線的増幅が望ましく、エンドポイントタイプの解析に関しては特に妥当である。
【0121】
9−3DDを含有するサンプルのアガロースゲル解析では、特に多数の鋳型数で可視の、プライマー二量体であろうバンドが示され、少数の鋳型数での非常に小さいバンドはプライマーと合致する。そのようなバンドは9−3bDDを含有するサンプルの分析では現れなかった。
【0122】
図13は、本実施例に記載の、図12に関しても使用された二重LATE−PCRにおいて、試薬9−3bDDの濃度を変化させることの効果を示す。この場合、試薬9−3bDDの濃度を300nM(三角形が付いていない太線)から200nM(三角形付きの線)に低下させると、アガロースゲル解析によって確認されるように、やはり、プライマー二量体形成に起因して直線的増幅の勾配に影響する。結果として、少数の当初鋳型数(10ゲノム、サークル132)を含有するサンプルの一部は、最後のサイクル(「エンドポイント」)で、多数の当初鋳型数(100ゲノム、サークル133および1000ゲノム、サークル134)を含有するサンプルより高い蛍光を有する。
【0123】
PCR効率に対する、本出願に記載される試薬の濃度の効果を、1ペアのプライマーで1鋳型を増幅するLATE−PCRにおいてさらに試験した。増幅対象の鋳型は、図12および図13に関して記載された二重反応で使用されたのと同じマウスOct4配列であり、プライマーおよび分子ビーコン配列も該反応と同一であった。
【0124】
各反応は最終容量100μL中で実行し、以下の試薬を含有した:20mMのTris−HCl,pH8.4、および50mMのKClによって構成される1×PCRバッファー(Invitrogen, Carlsbad, CA)、3mMのMgCl2、0.25mMの各dNTP、50nMのOct4制限的プライマー、2μMのOct4過剰プライマー、1μMの低TmのTET−Oct4分子ビーコン、および2単位の抗体複合体化Platinum(登録商標)Taq DNAポリメラーゼ(Invitrogen, Carlsbad, CA)。さらに化合物9−3DDを、150、または300または450nMの濃度でPCR混合物に加えた。上述の二重反応の場合(図12および図13を参照のこと)と同様に、各アッセイは、PurAmpプロトコルにしたがって、10.5μL容量中に細胞溶解および逆転写に必要な試薬をさらに含有した。このLATE−PCRでは、前記試薬の最終濃度は以下の通りであった:5mMのTris酢酸,pH8.4、7.5mMの酢酸カリウムおよび0.8mMの酢酸マグネシウム(希釈したcDNA合成バッファー)、1ng/μLのランダムヘキサマーおよび追加の100μMの各dNTP(ThermoScriptTM RT-PCR System, Invitrogen, Carlsbad, CAの全構成要素)、0.4mMのイソチオシアン酸グアニジン、20μMのβ−メルカプトエタノール、2μMのクエン酸ナトリウム,pH7.0、2×10−4%(vol/vol)のジメチルスルホキシドおよび0.5×10−4%のサルコシル。
【0125】
さらにマウスゲノムDNAを各アッセイに加え、本実施例に記載の二重LATE−PCRに関して指定される、PCR増幅用の鋳型を提供した。ABI Prism 7700 Sequence Detectorにおいて、二重反応に関して詳述されるプロファイルと同じ熱プロファイルを用いて、増幅を実行した。PCRの終了時点で、増幅産物を、0.5×TBEバッファー中の3%アガロースゲルでの2時間のゲル電気泳動に付すことによって分析し、臭化エチジウムで染色した。ゲル上では二本鎖および一本鎖産物がともに可視であり、予測されるサイズを有した。このことはLATE−PCRによる増幅が有効であることを示す。
【0126】
さらにOct4鋳型の増幅効率に対する増加濃度の化合物9−3DDの効果を試験した。150nM、300nM、450nMの9−3DDの各試験濃度で実施された5ゲノム(9−3DDの各試験濃度に関して2回の反復実験の平均)、20ゲノム(9−3DDの各試験濃度に関して2回の反復実験の平均)、100ゲノム、および1000ゲノムを含有するリアルタイムPCRアッセイからCT値を取得した。各濃度での各CT値のシリーズに関する直線回帰を図14に示す。図14では、三角は450nMの濃度であり、白抜きの四角は300nMであり、黒い四角は150nMである。このプロットからすぐにわかることは、450nMの9−3DDが閾値を超える蛍光シグナルの出現を大きく遅延させ、さらにPCR増幅の用量依存性を変更することである(回帰直線から離れている10遺伝子コピーおよび40遺伝子コピーの点を参照のこと)。一方、150nMまたは300nMの9−3DDを使用して得られたCT値は非常に類似し、鋳型コピー数および蛍光シグナルの最初の出現の間の直線的関連性を実証する。
【0127】
しかし、蛍光のリアルタイムパターンの分析では、図15に示されるように2つの条件(濃度)間の差異が強調される。100ゲノム、サークル151、または1000ゲノム、サークル152によって作成される曲線の勾配は、300nMの9−3DDが使用された場合(三角形が付いていない太線)に、150nMの9−3DD(三角形付きの線)が使用された場合より急勾配であり、よりストリンジェントな300nMの条件がプライマー二量体を排除し、ゆえに増幅効率を高めることが示唆される。
【実施例13】
【0128】
実施例13. 本発明に基づく複数の試薬の使用
ヒトゲノムDNA出発材料の5種の異なる標的配列を個別に増幅するために、および多重反応においてすべての5配列を一緒に増幅するために、LATE−PCR反応混合物を調製した。各標的配列はその独自のペアの制限的および過剰プライマーを必要とした。前記標的配列は、(1)191塩基対のグロビン対立遺伝子、(2)312塩基対のテイ・サックスG269対立遺伝子、(3)452塩基対のテイ・サックス1278および1421対立遺伝子、(4)ミトコンドリア超可変領域1の549塩基対セグメント、および(5)p53遺伝子エキソン7および8を含む611塩基対セグメントであった。25μLの反応混合物は、1×PCRバッファー(Invitrogen)、0.4mMのdNTP、3mMのMg++、0.24×SYBR Greenおよび1.5単位のTaqDNAポリメラーゼ(Invitrogen)を含有した。制限的プライマーに関しては50nMおよび過剰プライマーに関しては1000nMの濃度でプライマーを加えた。すべての5ペアのプライマーを多重反応に加えた。熱サイクル処理は、95℃で10秒間,64℃で20秒間,および72℃で1分間の45サイクルであった。
【0129】
個別のプライマーペアおよび個別の標的を含む反応混合物、ならびにすべての5ペアのプライマーおよびすべての5種の標的を含む反応混合物を、25nMの化合物9−22DDおよび100nMの化合物12−3DDの組み合わせの存在下で増幅した。さらに、本発明に基づくいかなる試薬をも加えずに五重反応混合物を増幅した。反応産物をゲル電気泳動によって調べた。種々の増幅のゲル電気泳動の結果を図16に示す。図16は、レーンrにサイズマーカー(100塩基対ずつ異なるサイズ)を含む。中央のレーンg〜kはそれぞれ標的配列(1)〜(5)の個別の増幅の産物である。レーンl〜qは、化合物9−22DDおよび化合物12−3DDの混合物を加えた6回反復の五重増幅の産物である。レーンa〜fは、本発明の試薬を加えなかった6回反復の五重増幅の産物である。図16は、いずれの化合物も加えない多重反応と比較して、化合物の混合物が増幅を増強した(より多量で純粋な所望のアンプリコンが得られた)ことを示し、本発明のキット、増幅およびアッセイにおいて試薬の混合物を使用しうることを実証する。
【実施例14】
【0130】
実施例14. 作用様式の試験
本発明者らは、本発明の試薬のミスプライミングの低減(特異性改善)およびポリメラーゼ阻害効果を定量的に研究する試験を考案した。該試験は第1の作用様式と第3の作用様式を識別すると考えられる。該試験は、実施例1に記載のアッセイと本質的に同様のPCR増幅アッセイであるが、以下の例外を有する:増幅反応混合物は半数(1000ゲノム)の剪断ゲノムDNAを含有し、熱サイクル処理の最終部分は70サイクルから40サイクルに減少される。種々の量、典型的には25nM、50nM、100nM、300nM、1500nMおよび3000nMの本発明の試薬を加えて増幅を実施する。試験は2種の分析を含む:第一は、特異性に関する、増幅産物の融解曲線の分析であり;第二は、阻害に関する、合成二本鎖産物のリアルタイム蛍光曲線の分析である。
【0131】
図17〜18は、本発明に基づく2種の試薬(それぞれ化合物12−3DDおよび化合物12−C3DD)に関する分析結果の一部分を示す。各図のカラムIは、表示通りの数種の選択された濃度の試薬に関する融解曲線を示す。各図のカラムIIは、表示通りの同濃度に関するリアルタイム蛍光曲線を示す。図17では、カラムIの融解曲線は、化合物12−3DDが100nM以上の濃度で高い特異性(ミスプライミングの回避)を達成したことを示した(最低濃度100nMは、実施例1に記載の反応に関して見出された最低濃度よりわずかに高い)。図17では、カラムIIのリアルタイム蛍光曲線は、化合物12−3DDの濃度が100nMを超えて増加するにつれて、1000nMを超える濃度で反応が本質的に停止されるまで、ポリメラーゼ阻害が進行的に増大したことを示した。阻害は、プラトーの蛍光レベル、およびCT値の遅延(100nMでは19.6、300nMでは21.1、1500nMおよび3000nMでは存在せず、すなわち少なくとも40)によって示される、生成二本鎖産物(群)全体(所望の特異的産物およびミスプライミングの非特異的産物を含む)の量が少ないことに反映される。図18では、カラムIの融解曲線は、化合物12−3DDが300nM以上の濃度で高い特異性を達成したことを示した(最低濃度300nMは、実施例1に記載の反応に関して見出された最低濃度と同一である)。図18では、カラムIIのリアルタイム蛍光曲線は、化合物12−C3DDの濃度が300nMを超えて増加するにつれて、ポリメラーゼ阻害が進行的に増大した(CT値は100nMでは18.8、300nMでは18.8、1500nMでは21.1および3000nMでは24.5であった)が、該反応は3000nMの濃度でさえ停止されなかったことを示した。
【0132】
図17と図18を比較すると、化合物12−C3DDの総合的な性能では、化合物12−3DDの総合的な性能より濃度依存性の程度が低いことがわかる。ゆえに、この特定の増幅反応では、化合物12−C3DDの濃度が300nMから1500nMになると生じるCT遅延およびプラトー蛍光の減少は、化合物12−3DDの濃度が100nMから300nMになると生じる結果とおおむね同一である。
【0133】
本発明の多数の実施形態を記載してきた。しかしながら、本発明の精神および範囲から逸脱することなく種々の修飾を施してよいことが理解されよう。したがって、他の実施形態は特許請求の範囲内である。
【特許請求の範囲】
【請求項1】
少なくとも1種の増幅DNA産物を生成するためのポリメラーゼ連鎖反応(PCR)増幅において、25μLの反応混合物あたり1.25単位の熱安定性DNAポリメラーゼを含有するPCR増幅混合物に650nM以下の濃度で添加した場合に、少なくとも1種のミスプライミング現象を防ぐことができる試薬であって、該試薬は伸長不可能なオリゴヌクレオチドであり、該オリゴヌクレオチドはステムループ構造を有し、前記少なくとも1種の増幅DNA産物に対するハイブリダイゼーションプローブでなく、6ヌクレオチド長より長いステムを有し、該ループの反対側のステムの末端は安定化されており、かつ計算上のステムの融解温度(Tm)が94℃より低い、上記試薬。
【請求項2】
前記ループが少なくとも3個のヌクレオチドを含むオリゴヌクレオチドである、請求項1に記載の試薬。
【請求項3】
前記ループが非ヌクレオチド化学リンカーである、請求項1に記載の試薬。
【請求項4】
前記ステムが、該ステムの3’および5’ヌクレオチドに共有結合した結合性化学部分により末端で安定化されている、請求項1〜3のいずれか1項に記載の試薬。
【請求項5】
前記結合性化学部分がいずれも蛍光性でない、請求項4に記載の試薬。
【請求項6】
Tm値より低い温度では二本鎖となる前記ステムの一部分が、9〜12塩基対の長さを有する、請求項1〜5のいずれか1項に記載の試薬。
【請求項7】
前記計算上のステムの融解温度が72〜85℃の範囲内である、請求項1〜6のいずれか1項に記載の試薬。
【請求項8】
前記計算上のステムの融解温度が50〜71℃の範囲内である、請求項1〜6のいずれか1項に記載の試薬。
【請求項9】
デオキシヌクレオチドリン酸(dNTP)および熱安定性DNAポリメラーゼを含有する増幅反応混合物から少なくとも1種の増幅DNA産物を生成するためのポリメラーゼ連鎖反応(PCR)増幅での少なくとも1種のミスプライミング現象を防ぐ方法であって、該反応混合物に請求項1〜8のいずれか1項に記載の少なくとも1種の試薬を添加することを含む、上記方法。
【請求項10】
前記試薬がオリゴヌクレオチドループを有し、該試薬の濃度が300nM以下である、請求項9に記載の方法。
【請求項11】
前記試薬が非ヌクレオチド化学リンカーであるループを有し、かつ該試薬を前記反応混合物に1000nM以下の濃度で添加する、請求項9に記載の方法。
【請求項12】
前記増幅が、前記Tmより低い温度で行われる低温検出ステップを含む直線後指数的PCR(LATE−PCR)アッセイである、請求項9〜11のいずれか1項に記載の方法。
【請求項13】
前記増幅が対称的PCRアッセイである、請求項9〜12のいずれか1項に記載の方法。
【請求項14】
前記増幅に続いてエンドポイント検出を行う、請求項9〜13のいずれか1項に記載の方法。
【請求項15】
ポリメラーゼ連鎖反応(PCR)増幅を行うための試薬キットであって、熱安定性DNAポリメラーゼ、少なくとも1ペアのPCRプライマー、dNTP、および請求項1〜8のいずれか1項に記載の少なくとも1種の試薬を含んでなる、上記キット。
【請求項16】
核酸単離のための試薬をさらに含んでなる、請求項15に記載のキット。
【請求項17】
ポリメラーゼ連鎖反応(PCR)増幅を行うためのオリゴヌクレオチドセットであって、少なくとも1ペアのPCRプライマーおよび請求項1〜8のいずれか1項に記載の少なくとも1種の試薬を含んでなる、上記セット。
【請求項18】
前記少なくとも1ペアのPCRプライマーにより規定される増幅産物にハイブリダイズする少なくとも1種の蛍光標識プローブをさらに含有する、請求項17に記載のセット。
【請求項19】
前記少なくとも1ペアのプライマーが、直線後指数的PCR(LATE−PCR)増幅を行うのに適した比率で制限的プライマーと過剰プライマーとを含む、請求項17または18に記載のセット。
【請求項20】
前記増幅産物が一本鎖である、請求項17〜19のいずれか1項に記載のセット。
【請求項1】
少なくとも1種の増幅DNA産物を生成するためのポリメラーゼ連鎖反応(PCR)増幅において、25μLの反応混合物あたり1.25単位の熱安定性DNAポリメラーゼを含有するPCR増幅混合物に650nM以下の濃度で添加した場合に、少なくとも1種のミスプライミング現象を防ぐことができる試薬であって、該試薬は伸長不可能なオリゴヌクレオチドであり、該オリゴヌクレオチドはステムループ構造を有し、前記少なくとも1種の増幅DNA産物に対するハイブリダイゼーションプローブでなく、6ヌクレオチド長より長いステムを有し、該ループの反対側のステムの末端は安定化されており、かつ計算上のステムの融解温度(Tm)が94℃より低い、上記試薬。
【請求項2】
前記ループが少なくとも3個のヌクレオチドを含むオリゴヌクレオチドである、請求項1に記載の試薬。
【請求項3】
前記ループが非ヌクレオチド化学リンカーである、請求項1に記載の試薬。
【請求項4】
前記ステムが、該ステムの3’および5’ヌクレオチドに共有結合した結合性化学部分により末端で安定化されている、請求項1〜3のいずれか1項に記載の試薬。
【請求項5】
前記結合性化学部分がいずれも蛍光性でない、請求項4に記載の試薬。
【請求項6】
Tm値より低い温度では二本鎖となる前記ステムの一部分が、9〜12塩基対の長さを有する、請求項1〜5のいずれか1項に記載の試薬。
【請求項7】
前記計算上のステムの融解温度が72〜85℃の範囲内である、請求項1〜6のいずれか1項に記載の試薬。
【請求項8】
前記計算上のステムの融解温度が50〜71℃の範囲内である、請求項1〜6のいずれか1項に記載の試薬。
【請求項9】
デオキシヌクレオチドリン酸(dNTP)および熱安定性DNAポリメラーゼを含有する増幅反応混合物から少なくとも1種の増幅DNA産物を生成するためのポリメラーゼ連鎖反応(PCR)増幅での少なくとも1種のミスプライミング現象を防ぐ方法であって、該反応混合物に請求項1〜8のいずれか1項に記載の少なくとも1種の試薬を添加することを含む、上記方法。
【請求項10】
前記試薬がオリゴヌクレオチドループを有し、該試薬の濃度が300nM以下である、請求項9に記載の方法。
【請求項11】
前記試薬が非ヌクレオチド化学リンカーであるループを有し、かつ該試薬を前記反応混合物に1000nM以下の濃度で添加する、請求項9に記載の方法。
【請求項12】
前記増幅が、前記Tmより低い温度で行われる低温検出ステップを含む直線後指数的PCR(LATE−PCR)アッセイである、請求項9〜11のいずれか1項に記載の方法。
【請求項13】
前記増幅が対称的PCRアッセイである、請求項9〜12のいずれか1項に記載の方法。
【請求項14】
前記増幅に続いてエンドポイント検出を行う、請求項9〜13のいずれか1項に記載の方法。
【請求項15】
ポリメラーゼ連鎖反応(PCR)増幅を行うための試薬キットであって、熱安定性DNAポリメラーゼ、少なくとも1ペアのPCRプライマー、dNTP、および請求項1〜8のいずれか1項に記載の少なくとも1種の試薬を含んでなる、上記キット。
【請求項16】
核酸単離のための試薬をさらに含んでなる、請求項15に記載のキット。
【請求項17】
ポリメラーゼ連鎖反応(PCR)増幅を行うためのオリゴヌクレオチドセットであって、少なくとも1ペアのPCRプライマーおよび請求項1〜8のいずれか1項に記載の少なくとも1種の試薬を含んでなる、上記セット。
【請求項18】
前記少なくとも1ペアのPCRプライマーにより規定される増幅産物にハイブリダイズする少なくとも1種の蛍光標識プローブをさらに含有する、請求項17に記載のセット。
【請求項19】
前記少なくとも1ペアのプライマーが、直線後指数的PCR(LATE−PCR)増幅を行うのに適した比率で制限的プライマーと過剰プライマーとを含む、請求項17または18に記載のセット。
【請求項20】
前記増幅産物が一本鎖である、請求項17〜19のいずれか1項に記載のセット。
【図1】

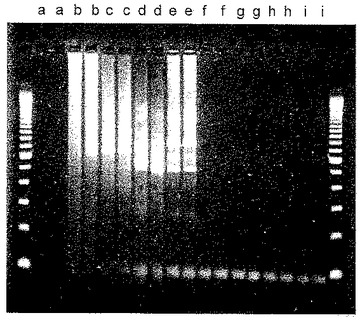
【図2】
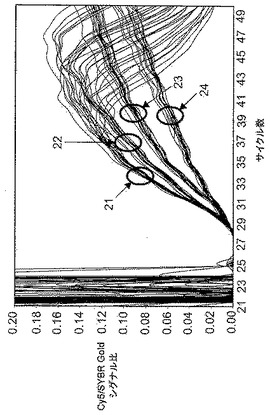
【図3】
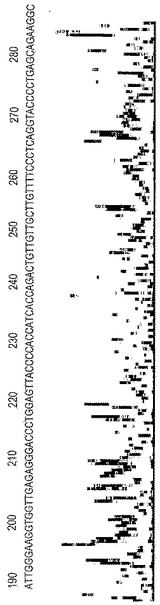
【図4】
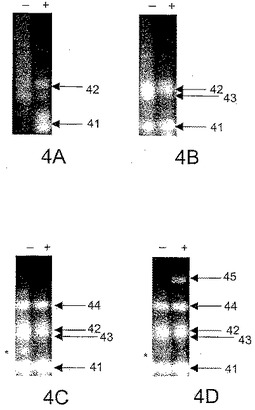
【図5】
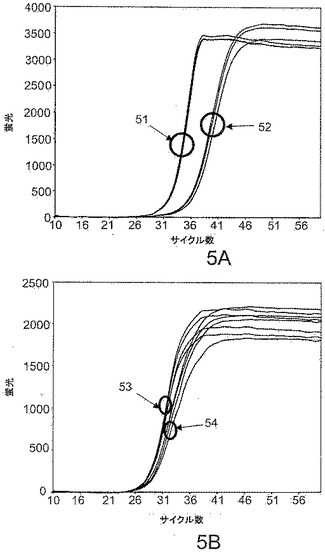
【図6】
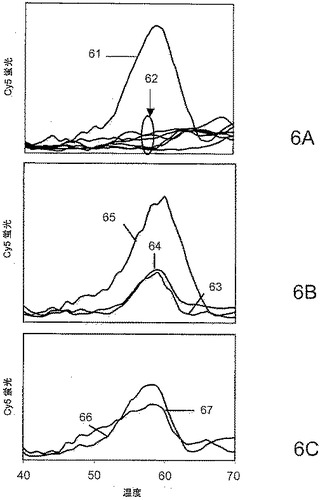
【図7】
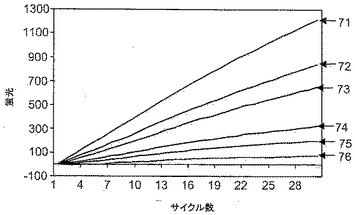
【図8A】
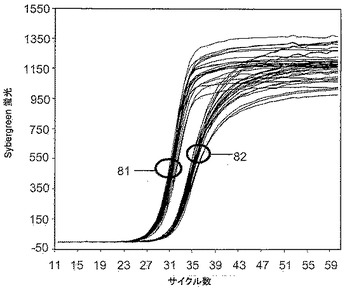
【図8B】
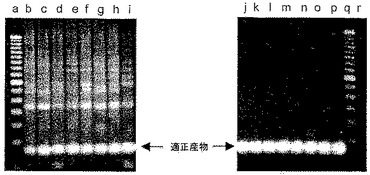
【図9】
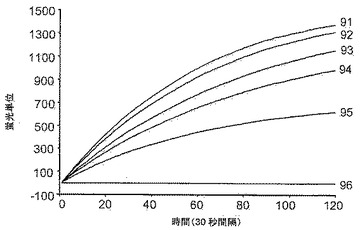
【図10】
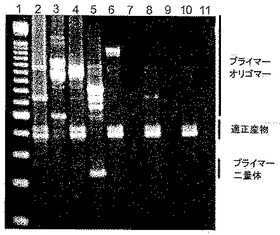
【図11】
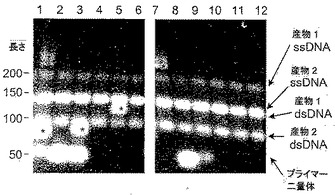
【図12】
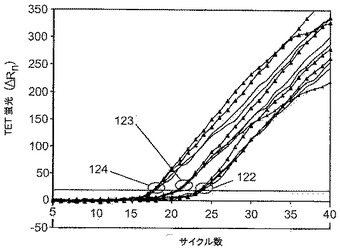
【図13】
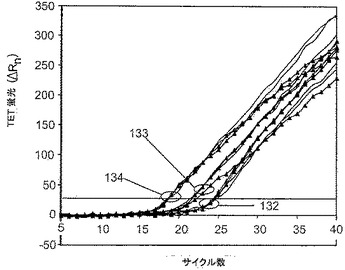
【図14】
【図15】
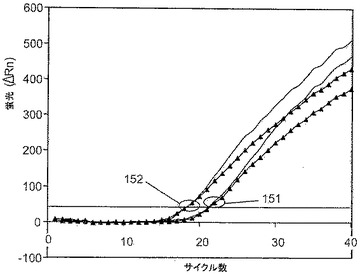
【図16】
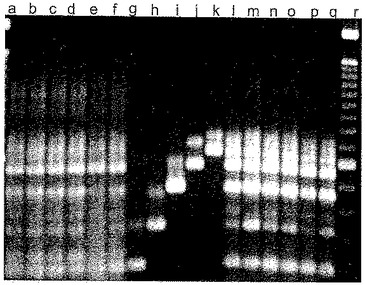
【図17】
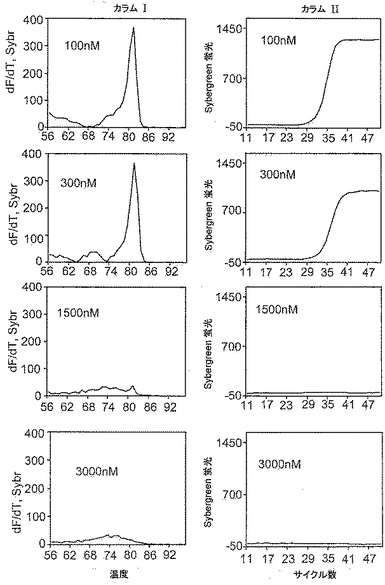
【図18】
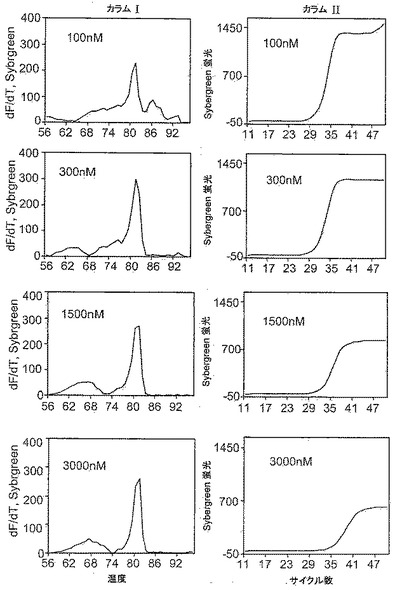
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8A】
【図8B】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【公開番号】特開2013−63070(P2013−63070A)
【公開日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願番号】特願2012−234683(P2012−234683)
【出願日】平成24年10月24日(2012.10.24)
【分割の表示】特願2007−537035(P2007−537035)の分割
【原出願日】平成17年10月17日(2005.10.17)
【出願人】(504238057)ブランデイズ ユニバーシティー (4)
【Fターム(参考)】
【公開日】平成25年4月11日(2013.4.11)
【国際特許分類】
【出願日】平成24年10月24日(2012.10.24)
【分割の表示】特願2007−537035(P2007−537035)の分割
【原出願日】平成17年10月17日(2005.10.17)
【出願人】(504238057)ブランデイズ ユニバーシティー (4)
【Fターム(参考)】
[ Back to top ]