
PDE7阻害剤としてのスピロ環式キナゾリン誘導体
本発明は、mが0、1、または2であり、XがO、S、またはN−CNであり、RがF、Cl、またはCNであり、Aが、C1〜4アルキル基で置換されていてもよいC3〜6シクロアルキレン基であり、Bが単結合またはC1〜2アルキレン基である式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグを提供する。この化合物は、PDE7阻害剤であり、特に疼痛、とりわけ神経因性疼痛の治療においていくつかの治療用途を有する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、スピロ環式誘導体、ならびにそのような誘導体の調製方法、その調製で使用する中間体、それを含有する組成物、およびその使用に関する。
【0002】
本発明のスピロ環式誘導体は、PDE7阻害剤であり、特に疼痛、特に神経因性疼痛の治療においていくつかの治療用途を有する。
【背景技術】
【0003】
ホスホジエステラーゼ(PDE)は、セカンドメッセンジャー分子のcAMPおよびcGMPを対応する不活性な5’−一リン酸ヌクレオチドに加水分解し、それによりその生理的なレベルを調節する過程によって、様々な細胞内シグナル伝達過程に影響を及ぼす酵素のファミリーである。二次的なメッセンジャーであるcAMPおよびcGMPは、数多くの細胞内過程の調節を司る。PDEファミリーは、少なくとも11種あり、cAMPに特異的なもの(PDE3、4、7、8)もあれば、cGMPに特異的なもの(PDE5、6、および9)もある。
【0004】
PDE7は、PDEファミリーの一員であり、2種のサブクラスメンバーPDE7AおよびBを含む。PDE7のmRNAは、T細胞関連障害などのいくつかの疾患の病因において重要であることがわかっている様々な組織および細胞型で発現される。特にPDE7Aおよびそのスプライスバリアントは、活性化型T細胞中(L.Li、C.Yee、およびJ.A.Beavo、Science(1999年)、第283巻、848〜851ページ)およびBリンパ球中(R.Lee、S.Wolda、E.Moon、J.Esselstyn、C.Hertel、およびA.Lerner、Cell.Signal(2002年)、第14巻、277〜284ページ)、自己免疫疾患(L.Liら、上記)、ならびに気道疾患(S.J.Smithら、Am.J.Physiol.Lung.Cell.Mol.Physiol.(2003年)、第284巻、L279〜L289ページ)で上方制御される。したがって、PDE7の選択的阻害剤は、免疫抑制剤として、また呼吸器の状態、たとえば慢性閉塞性肺疾患および喘息の治療薬として広範な用途を有することが予想される(N.A.Glavas、C.Ostenson、J.B.Schaefer、V.Vasta、およびJ.A.Beavo.PNAS(2001年)、第98巻、6319〜6324ページ)。
【0005】
ラットでの研究は、PDE7A mRNAが、ラット脳のニューロンおよび非ニューロンの両方の細胞集団中に広く分布しているという知見を示している。最高濃度は嗅球、嗅結節、海馬、小脳、内側手綱核、松果体、最後野、および脈絡叢で認められる。PDE7A mRNAは、他の非脳組織でも広く検出される。これらの結果は、PDE7Aが多くの脳機能においてcAMPシグナル伝達の調節に関与していることと一致し、PDE7Aが、記憶、うつ病、および嘔吐に対して効果を有するかもしれないことを示唆している(X.Miro、S.Perez−Torres、J.M.Palacios、P.Puigdomenech、G.Mengod、Synapse(2001年)、第40巻、201〜214ページ)。アルツハイマー病との関連も示唆されている(S.Perez Torresら、Experimental Neurology(2003年)第182巻、322〜334ページ)。PDE7はさらに、受精障害(WO01/83772)および白血病(R.Leeら、Cell Signalling(2002年)第14巻、277〜284ページ)とも関連付けられている。
【0006】
PDE7Aは、酵母(T.Michaeliら、J.Biol.Chem.(1993年)第268巻、12925〜12932ページ)、ヒト(P.Han、Z.Xiaoyan、およびM.Tamar、J.Biol.Chem.(1997年)第272巻、16152〜16157ページ)、マウス(T.BloomおよびJ.A.Beavo、Proc.Natl.Acad.Sci.USA(1996年)、第93巻、14188〜14192ページ)から単離されており、PDE7Aレベルの上方制御は、ヒトTリンパ球中で見られる(M.IchimuraおよびH.Kase.Biochem.Biophys.Res.Commun.(1993年)、第193巻、985〜990ページ)。
【0007】
PDE7ファミリーの第2のメンバーであるPDE7Bは、C末端の触媒ドメインにおいてPDE7Aと70%のアミノ酸相同性を共有している(N末端ドメインは、PDEファミリー全体に保存されている、リン酸化部位を含有する調節性ドメインである)。PDE7Bは、cAMPに特異的であり、マウス(受託番号−AJ251858)およびヒト(受託番号−AJ251860)供給源からクローン化されている(C.Gardner、N.Robas、D.Cawkill、およびM.Fidock、Biochem.Biophys.Res.Commun.(2000年)、第272巻、186〜192ページ)。PDE7Bは、広範な種類の組織、すなわち脳の尾状核、被殻、および後頭葉、末梢では心臓、卵巣、下垂体、腎臓、肝臓、小腸、および胸腺、さらには骨格筋、大腸、膀胱、子宮、前立腺、胃、副腎、および甲状腺で発現されることが示されている。PDE7Bは、いくつかの一般のPDE阻害剤を識別することも示されている(J.M.Hetman、S.H.Soderling、N.A.Glavas、およびJ.A.Beavo、PNAS(2000年)、第97巻、472〜476ページ)。しかし、ザプリナスト、ロリプラム、ミルリノンなどの多くの標準的PDE阻害剤は、PDE7Bを特異的には阻害しない。
【0008】
PDE7の阻害剤は、様々なPDE7関連疾患の治療で使用されることが知られている。たとえば、WO02/074754は、次式の化合物
【0009】
【化1】
ならびにT細胞関連疾患、自己免疫疾患、骨関節炎、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群、アレルギー、または炎症性腸疾患などのPDE7関連障害の治療におけるその使用を記載している。
【0010】
WO2004/026818は、次式の化合物
【0011】
【化2】
【0012】
およびPDE7関連障害の治療におけるその使用を記載している。
【0013】
WO2006/092691は、神経因性疼痛の治療におけるPDE7阻害剤の使用を記載している。
【発明の開示】
【発明が解決しようとする課題】
【0014】
発明者らは、驚いたことに、WO02/074754の一般の開示内に含まれるが、その中で詳細には開示または例示されていない1クラスの化合物が、WO02/074754で例示されている最も近い化合物と比べて予想外に優れた薬物動態特性を示すことを見出した。これらの化合物は、低い身体からのクリアランスを示すことが予想され、1日1回投与したときに治療効果を実現する可能性を有している。
【課題を解決するための手段】
【0015】
本発明は、式(I)の化合物
【0016】
【化3】
[式中、
mは、0、1、または2であり、
Xは、O、S、またはN−CNであり、
Rは、F、Cl、またはCNであり、
Aは、C1〜4アルキル基で置換されていてもよいC3〜6シクロアルキレン基であり、
Bは、単結合またはC1〜2アルキレン基である]
または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグを提供する。
【発明を実施するための最良の形態】
【0017】
本発明の文脈では、用語「アルキレン」とは、1または2個の炭素原子を有する二価の飽和炭化水素鎖を示す。アルキレン基の例には、メチレン、エチレン、およびメチルメチレンが含まれ、メチレンが好ましい。
【0018】
用語「シクロアルキレン」とは、3〜6個の炭素原子を有する二価の飽和炭素環を示す。シクロアルキレン基の例には、シクロプロピレン(たとえば、1,1−シクロプロピレン、シス−およびトランス−1,2−シクロプロピレン)、シクロブチレン(たとえば、1,1−シクロブチレン、シス−およびトランス−1,2−シクロブチレン、シス−およびトランス−1,3−シクロブチレン)、シクロペンチレン(たとえば、1,1−シクロペンチレン、シス−およびトランス−1,2−シクロペンチレン、シス−およびトランス−1,3−シクロペンチレン)、ならびにシクロヘキシレン(たとえば、1,1−シクロヘキシレン、シス−およびトランス−1,2−シクロヘキシレン、シス−およびトランス−1,3−シクロヘキシレン、シス−およびトランス−1,4−シクロヘキシレン)が含まれる。好ましい例には、シクロブチレンおよびシクロヘキシレン、より好ましくはシクロブチレン、さらにより好ましくは1,3−シクロブチレン、最も好ましくはトランス−1,3−シクロブチレンが含まれる。
【0019】
用語「アルキル」とは、1〜4個の炭素原子を含有している直線状または分枝状の一価の飽和炭化水素鎖を示す。アルキル基の例には、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、s−ブチル、およびt−ブチルが含まれる。好ましい例には、メチルおよびエチル、特にメチルが含まれる。
【0020】
シクロアルキレン基は、C1〜4アルキル基で置換されていてもよい。存在するならば、アルキル置換基は、メチルまたはエチル基、より好ましくはメチル基であることが好ましい。存在するならば、アルキル置換基は、環上のどの位置に存在してもよいが、1位(すなわちカルボン酸基と同位置)に存在することが好ましい。
【0021】
mは、好ましくは1または2、より好ましくは1である。
【0022】
Xは、好ましくはOまたはN−CN、より好ましくはOである。
【0023】
Rは、好ましくはFまたはCl、より好ましくはClである。
【0024】
Aは、好ましくは、メチル基で置換されていてもよいシクロブチレンまたはシクロヘキシレン基である。Aは、より好ましくはシクロブチレン基である。Aは、さらにより好ましくは1,3−シクロブチレン基、特にトランス−1,3−シクロブチレン基である。
【0025】
Bは、好ましくは単結合またはメチレン基である。Bは、より好ましくは単結合である。
【0026】
本発明の特に好ましい化合物には、式(I)中の各可変基が、各可変基について好適かつ/または好ましい基から選択されるものが含まれる。本発明のより一層好ましい化合物には、式(I)中の各可変基が、各可変基についてより好ましいまたは最も好ましい基から選択されるものが含まれる。
【0027】
以下の化合物が特に好ましい。
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−シアノ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
ならびにこれらの薬学的に許容できる塩、溶媒和物、およびプロドラッグ。
【0028】
以下の化合物が特に好ましい。
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
ならびにこれらの薬学的に許容できる塩、溶媒和物、多形体、およびプロドラッグ。
【0029】
特に以下で記載する溶媒和していない結晶形態(A型)として、および以下で記載する酢酸溶媒和物としてのトランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸化合物、ならびに薬学的に許容できるその塩、溶媒和物、多形体、およびプロドラッグが最も好ましい。
【0030】
一実施形態では、本発明は、Cu Kα放射線照射(波長=1.5406Å)を使用して測定したとき以下の粉末X線回折ピーク、すなわち6.3、17.8、21.5、22.1、22.4、26.3(2θ、±0.1°の度数)を特徴とする、溶媒和していない結晶形態(A型)のトランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]−シクロブタンカルボン酸化合物を含む。
【0031】
別の実施形態では、本発明は、Cu Kα放射線照射(波長=1.5406Å)を使用して測定したとき以下の粉末X線回折ピーク、すなわち8.3、10.8、16.6、17.1、19.5、20.5、23.7(2θ、±0.1°の度数)を特徴とする、酢酸溶媒和物としてのトランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]−シクロブタンカルボン酸化合物を含む。
【0032】
本発明はさらに、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグと、薬学的に許容できる担体または希釈剤とを含む医薬組成物を含む。
【0033】
本発明はさらに、医薬として使用するための、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグを含む。
【0034】
本発明はさらに、PDE7阻害剤による治療が適切である疾患または状態を治療する医薬の製造における、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグの使用を含む。
【0035】
本発明はさらに、PDE7阻害剤による治療が適切である疾患または状態の治療方法であって、その最も広い態様もしくは好ましい態様の有効量の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグの投与を含む方法を含む。
【0036】
PDE7阻害剤である式(I)の化合物は、ある範囲の障害の治療において潜在的に有用である。疼痛、詳細には神経因性疼痛の治療が好ましい使用である。
【0037】
生理的な痛みは、外部環境からの有害な可能性のある刺激の危険を警告するように設計された重要な防御機構である。このシステムは、特定のセットの一次感覚ニューロンを通して作動し、侵害刺激によって、末梢の変換機構を介して活性化される(総説については、Millan、Prog.Neurobiol.、(1999年)、第57巻、1〜164ページを参照されたい)。これらの感覚線維は、侵害受容器として知られており、伝導速度の緩徐な直径の小さい軸索を特徴とする。侵害受容器は、侵害刺激の強度、持続時間、および質、さらにはその組織分布的に系統立てられた脊髄への投射によって刺激の位置をコードする。侵害受容器は、Aδ線維(有髄)およびC線維(無随)を主要な2タイプとする侵害受容性神経線維上に見られる。侵害受容器による入力によって生成された活性は、後角での複雑な処理の後、直接にまたは脳幹中継核を介して、視床基底複側、次いで皮質へと伝達され、皮質で痛みの感覚が生じる。
【0038】
疼痛は、一般に急性または慢性として分類することができる。急性痛は、突如始まり、短命(通常は12週間以下)である。急性痛は、通常は特定の損傷などの特定の原因に関連し、しばしば鋭く激しい。急性痛は、手術、歯科作業、挫傷、または捻挫の結果として生じる特定の損傷の後に起こり得る種類の疼痛である。急性痛は一般に、どんな持続的な心理的応答ももたらさない。対照的に、慢性痛は、通常は3ヶ月間より長い間持続し、著しい心理的および情緒的問題をもたらす長期間の疼痛である。慢性痛の一般的な例は、神経因性疼痛(たとえば、有痛性の糖尿病性ニューロパチー、ヘルペス後神経痛)、手根管症候群、背痛、頭痛、癌性疼痛、関節炎疼痛、および慢性の術後疼痛である。
【0039】
疾患または外傷によって体組織に相当な損傷が起こると、侵害受容器活性化の特性は変更され、感作は、末梢で、損傷周囲で局所的に、また侵害受容器が終わる中枢で起こる。これらの影響は、疼痛の感覚を強める。急性痛では、これらの機序は、修復過程が起こるのを一層可能にし得る保護的な挙動を促進する点で有用な場合がある。通常の予想では、損傷が治癒してしまえば、その敏感性が正常に戻るということになる。しかし、多くの慢性疼痛状態では、過敏性は、治癒過程よりはるかに長く残り、しばしば神経系損傷を原因とする。この損傷はしばしば、適応不良および異常な活動に関連する感覚神経繊維の異常をもたらす(Woolf&Salter、Science、(2000年)、第288巻、1765〜1768ページ)。
【0040】
患者の症状の中で不快感および異常な敏感性が特徴となっているとき、臨床上の痛みが存在する。患者は、全く均一でない傾向があり、様々な痛み症状で診察を受けにくる場合がある。そのような症状には、1)鈍い、焼けるような、または刺すようであるといえる自発痛、2)侵害刺激に対する疼痛反応の悪化(痛覚過敏)、および3)通常は非侵害性の刺激によって生じる痛み(異痛症−Meyerら、1994、Textbook of Pain、13〜44ページ)が含まれる。様々な形態の急性痛および慢性痛に罹患している患者が同様の症状を有することがあるとしても、根底にある機序は異なる場合があり、したがって異なる治療戦略が必要となり得る。したがって、痛みは、異なる病態生理に従って、侵害受容性、炎症性、および神経因性の疼痛を含むいくつかの異なるサブタイプに分けることができる。
【0041】
侵害受容性疼痛は、組織損傷、または損傷を引き起こす潜在性のある激しい刺激によって誘発される。痛みの求心性は、損傷部位での侵害受容器による刺激の伝達によって活性化され、それが終結するレベルにある脊髄のニューロンを活性化する。次いで、これが脊髄路を上って脳へと中継され、そこで痛みが知覚される(Meyerら、1994年、Textbook of Pain、13〜44ページ)。侵害受容器の活性化は、2タイプの求心性神経線維を活性化する。有髄のAδ線維は、急速な伝達を行い、鋭く刺すような痛覚を司るのに対し、無髄のC線維は、より緩慢な速度で伝達を行い、鈍いまたはうずくような痛みを伝える。中程度から重度の急性侵害受容性疼痛は、中枢神経系外傷、挫傷/捻挫、火傷、心筋梗塞、および急性膵炎の疼痛、術後疼痛(任意のタイプの手術手順後の疼痛)、外傷後疼痛、腎疝痛、癌性疼痛、および背痛からくる疼痛の顕著な特色である。癌性疼痛は、腫瘍関連疼痛(たとえば、骨痛、頭痛、顔面痛、または内臓痛)や、癌治療に関連する疼痛(たとえば、化学療法後症候群、慢性術後疼痛症候群、または放射線照射後症候群)などの慢性痛であるといえる。癌性疼痛は、化学療法、免疫療法、ホルモン療法、または放射線療法に応答して起こる場合もある。背痛は、椎間板ヘルニアもしくは椎間板破裂、または腰椎椎間関節、仙腸関節、傍脊椎筋、もしくは後縦方向靱帯の異常によるものであるといえる。背痛は、自然に消散することもあるが、これが12週間を超えて持続する一部の患者では、特に消耗性となり得る慢性状態になる。
【0042】
神経因性疼痛は現在、神経系の一次病巣または機能不全によって始まる、または引き起こされる疼痛であると定義されている。神経損傷は、外傷および疾患によって引き起こされる場合があり、したがって用語「神経因性疼痛」は、病因の多様な多くの障害を包含する。これらには、末梢性神経障害、糖尿病性ニューロパチー、ヘルペス後神経痛、三叉神経痛、背痛、癌性ニューロパチー、HIVニューロパチー、幻肢痛、手根管症候群、中心性卒中後痛、ならびに慢性アルコール中毒、甲状腺機能低下、尿毒症、多発性硬化症、脊椎損傷、パーキンソン病、てんかん、およびビタミン欠乏に関連する疼痛が含まれるがこの限りでない。保護的な役割をもたない神経因性疼痛は病的である。神経因性疼痛はしばしば、もとの原因が消失した後も多分に存在し、患者の生活の質を著しく低下させながら、一般的に何年間も持続する(Woolf and Mannion、Lancet、(1999年)第353巻、1959〜1964ページ)。神経因性疼痛の症状は、同じ疾患の患者間でさえもしばしば均一でないので、治療が難しい(Woolf&Decosterd、Pain Supp.(1999年)、第6巻、S141〜S147ページ;WoolfおよびMannion、上記)。神経因性疼痛の症状には、持続的になることのある自発痛、ならびに痛覚過敏(侵害刺激に対する敏感性の増大)や異痛症(通常は非侵害性の刺激に対する敏感性)などの発作性または異常性の誘発痛が含まれる。
【0043】
炎症の過程は、組織損傷または異物の存在に応答して活性化される、腫れおよび痛みをもたらす一連の複雑な生化学的および細胞性の事象である(LevineおよびTaiwo、1994年、Textbook of Pain、45〜56ページ)。関節炎疼痛は、最も一般的な炎症性疼痛である。リウマチ様疾患は、先進諸国において最も一般的な慢性炎症状態の1つであり、関節リウマチは、身体障害の一般的な原因である。関節リウマチの正確な病因は不明であるが、現在の仮説は、遺伝的要因と微生物学的要因の両方が重要である可能性があることを示唆している(Grennan&Jayson、1994年、Textbook of Pain、397〜407ページ)。ほぼ1600万人のアメリカ人が症候性の骨関節炎(OA)または変形性関節疾患に罹患し、その大部分が60才を超えていることが推定されており、集団の年齢が増すにつれてこれが4000万人に増えることが想定され、そのためこれが重要性の非常に大きい公衆衛生問題になっている(Houge & Mersfelder、Ann Pharmacother.(2002年)、第36巻、679〜686ページ;McCarthyら、1994年、Textbook of Pain、387〜395ページ)。ほとんどの変形性関節炎患者は、それに伴う疼痛のために医学的な対応処置を捜し求める。関節炎は、心理社会的および身体的な機能に著しい影響を及ぼし、後の人生で身体障害の主な原因となることが知られている。強直性脊椎炎も、脊椎および仙腸関節の関節炎を引き起こすリウマチ性疾患である。強直性脊椎炎は、生涯にわたって起こる背痛の断続的なエピソードから、脊椎、末梢の関節、および他の身体臓器を襲う重度の慢性疾患に至るまで様々である。
【0044】
別のタイプの炎症性疼痛は内臓痛であり、これには、炎症性腸疾患(IBD)に関連する疼痛が含まれる。内臓痛は、腹腔の臓器を包含する内臓に関連する疼痛である。これらの臓器には、生殖器、脾臓、および消化器系の一部が含まれる。内臓に関連する疼痛は、消化系の内臓痛および非消化系の内臓痛に分けることができる。一般的に遭遇する、疼痛を引き起こす胃腸(GI)障害には、機能性腸障害(FBD)および炎症性腸疾患(IBD)が含まれる。これらのGI障害には、FBDに関しては胃食道逆流症、消化不良、過敏性大腸症候群(IBS)、および機能性腹痛症候群(FAPS)、IBDに関してはクローン病、回腸炎、および潰瘍性大腸炎を含めて、すべてが定期的に内臓痛を生じる、現在中等度にしかコントロールされない広範囲な疾患状態が含まれる。他のタイプの内臓痛には、月経困難症、膀胱炎、および膵炎に関連する疼痛、ならびに骨盤痛が含まれる。
【0045】
一部のタイプの疼痛は、複数の病因を有し、すなわち複数の領域に分類される場合があり、たとえば背痛および癌性疼痛は、侵害受容性および神経障害性の両方の構成要素を有することに留意されたい。
【0046】
他のタイプの疼痛には、以下のものが含まれる。
・筋肉痛、線維筋痛、椎骨炎、血清反応陰性(非リウマチ様)関節症、非関節性リウマチ、ジストロフィノパチー、グリコーゲン分解、多発性筋炎、および化膿性筋炎を含む筋骨格障害の結果として生じる疼痛、
・狭心症、心筋梗塞、僧帽弁狭窄、心外膜炎、レイノー現象、浮腫性硬化症、および骨格筋虚血によって引き起こされる疼痛を含む心臓および血管の疼痛、
・偏頭痛(前兆を伴う偏頭痛および前兆を伴わない偏頭痛を含む)、群発性頭痛、筋緊張型頭痛、混合型頭痛、および血管性障害に関連する頭痛などの頭痛、ならびに
・歯痛、耳痛、口腔内灼熱症候群、および側頭下顎の筋筋膜痛を含む口腔顔面痛。
【0047】
本発明の式(I)の化合物は、疼痛以外の状態の治療においても有用である。特に、本発明の式(I)の化合物は、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、および炎症性腸疾患の治療において有用である。
【0048】
本発明はさらに、疼痛(特に神経因性疼痛)、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、および炎症性腸疾患から選択される状態または障害を治療するための医薬の製造における、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、もしくはプロドラッグの使用を含む。
【0049】
本発明はさらに、疼痛(特に神経因性疼痛)、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、または炎症性腸疾患から選択される疾患または状態の治療方法であって、その最も広い態様もしくは好ましい態様の有効量の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、もしくはプロドラッグを投与することを含む方法を含む。
【0050】
式(I)の化合物の薬学的に許容できる塩には、その酸付加塩および塩基の塩が含まれる。
【0051】
好適な酸付加塩は、非毒性の塩を形成する酸から生成されるものである。例には、酢酸塩、アジピン酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、炭酸水素塩/炭酸塩、重硫酸塩/硫酸塩、ホウ酸塩、カンシル酸塩、クエン酸塩、シクラミン酸塩、エジシル酸塩、エシル酸塩、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンズ酸塩、塩酸塩/塩化物、臭化水素酸塩/臭化物、ヨウ化水素酸塩/ヨウ化物、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシル酸塩、メチル硫酸塩、ナフチル酸塩、2−ナプシル酸塩、ニコチン酸塩、硝酸塩、オロト酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素塩/リン酸二水素塩、ピログルタミン酸塩、糖酸塩、ステアリン酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、トシル酸塩、トリフルオロ酢酸塩、およびキシノホ酸塩(xinofoate)が含まれる。
【0052】
好適な塩基の塩は、非毒性の塩を形成する塩基から生成されるものである。例には、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオールアミン、グリシン、リジン、マグネシウム、メグルミン、オールアミン、カリウム、ナトリウム、トロメタミン、および亜鉛の塩が含まれる。
【0053】
酸および塩基の半塩、たとえば半硫酸塩および半カルシウム塩を生成してもよい。
【0054】
好適な塩に関する総説については、StahlおよびWermuthによる「Handbook of Pharmaceutical Salts:Properties,Selection,and Use」(Wiley−VCH、2002年)を参照されたい。
【0055】
式(I)の化合物の薬学的に許容できる塩は、次の3つの方法の1つまたは複数によって調製することができる。
(i)式(I)の化合物を所望の酸または塩基と反応させることによる方法、
(ii)式(I)の化合物の好適な前駆体から、酸もしくは塩基に不安定な保護基を除去する、または所望の酸もしくは塩基を使用して好適な環状前駆体、たとえばラクトンもしくはラクタムを開環することによる方法、または
(iii)式(I)の化合物の塩を、適切な酸もしくは塩基との反応によって、または好適なイオン交換カラムによって別の塩に変換することによる方法。
【0056】
3つの反応はすべて、通常は溶液中で実施する。生じる塩が析出することもあり、濾過によって収集してもよいし、または溶媒を蒸発させて回収してもよい。得られる塩のイオン化の程度は、完全にイオン化したものからほとんどイオン化していないものまで様々となり得る。
【0057】
本発明の化合物は、完全な非晶質から完全な結晶性に至る範囲の一連の固体状態で存在し得る。用語「非晶質」とは、材料が分子レベルで長いきちんとした序列を欠き、温度に応じて固体または液体の物理的性質を示し得る状態を指す。通常、そのような材料は、特有のX線回折パターンを与えず、固体の性質を示しながらもより正式には液体であると記述される。加熱すると、固体の性質から液体の性質への変化が起こるが、これは通常、二次の状態変化(「ガラス転移」)を特徴とする。用語「結晶性」とは、材料が、分子レベルで規則的な整った内部構造を有し、明確なピークを伴う特有のX線回折パターンを与える固相を指す。十分に加熱したとき、そのような材料も液体の性質を示すが、固体から液体への変化は、通常は一次の相変化(「融点」)を特徴とする。
【0058】
本発明の化合物はまた、溶媒和していない形態および溶媒和した形態で存在する場合がある。用語「溶媒和物」は、本明細書では、本発明の化合物と、薬学的に許容できる1つまたは複数の溶媒分子、たとえばエタノールとを含む分子複合体について述べるのに使用する。用語「水和物」は、前記溶媒が水であるときに使用する。本発明は、溶媒和していない形態とすべての溶媒和形態の両方を含む。
【0059】
現在受け入れられている有機水和物の分類系統は、隔離部位水和物、チャネル水和物、または金属イオン配位水和物を規定するものである。K.R.Morrisによる「Polymorphism in Pharmaceutical Solids」(H.G.Brittain編、Marcel Dekker、1995年)を参照されたい。隔離部位水和物は、水分子が介在する有機分子によって互いの直接の接触から隔離されているものである。チャネル水和物では、水分子は格子チャネルにあり、そこで他の水分子と隣り合っている。金属イオン配位水和物では、水分子は金属イオンに結合している。
【0060】
溶媒または水が堅く結合しているとき、複合体は、湿度に関係なく明確な化学量論性を有する。しかし、チャネル溶媒和物および吸湿性化合物でのように溶媒または水が弱く結合しているとき、水/溶媒含有量は湿度および乾燥条件に左右される。そのような場合では、非化学量論性が標準となる。
【0061】
以下では、式(I)の化合物へのすべての言及は、その塩および溶媒和物への言及ならびにその塩の溶媒和物を含む。
【0062】
本発明の化合物には、そのすべての多形体および晶癖、以下で定義するそのプロドラッグおよび異性体(光学異性体、幾何異性体、および互変異性体を含む)、ならびに同位体標識された式(I)の化合物を含めて、上で規定した式(I)の化合物が含まれる。
【0063】
指摘したように、式(I)の化合物のいわゆる「プロドラッグ」も、本発明の範囲内にある。したがって、それ自体は薬理活性をほとんどまたは全くもたなくてもよい式(I)の化合物の特定の誘導体は、身体中または身体上に投与されたとき、たとえば加水分解による切断によって、所望の活性を有する式(I)の化合物に変換され得る。そのような誘導体を「プロドラッグ」と呼ぶ。プロドラッグの使用についてのそれ以上の情報は、「Pro−drugs as Novel Delivery Systems」、第14巻、ACS Symposium Series(T.HiguchiおよびW.Stella)および「Bioreversible Carriers in Drug Design」、Pergamon Press、1987年(E.B.Roche編、米国薬剤師会)で見ることができる。
【0064】
本発明によるプロドラッグは、たとえば、たとえばH.Bundgaardによる「Design of Prodrugs」(Elsevier、1985年)に記載されているように、式(I)の化合物中に存在する適切な官能基を、当業者に「pro−部分」として知られている特定の部分と交換することによって生成することができる。
【0065】
本発明の式(I)の化合物は、カルボン酸官能基(−COOH)を含有する。したがって、好適なプロドラッグは、式(I)の化合物のカルボン酸官能基の水素がエステル残基によって置換されている、そのエステルを含む。用語「エステル残基」とは、加水分解などの生物学的方法によってin vivoで切断することができ、遊離のカルボン酸基を有する式(I)の化合物またはその塩を生成するエステル基を意味する。
【0066】
化合物がそのようなプロドラッグであるか否かは、たとえば、それをラットやマウスなどの実験動物に静脈内注射によって投与し、次いで動物の体液を調査して、式(I)の化合物または薬学的に許容できるその塩が検出できるか否かを判定することによって決定することができる。
【0067】
エステル残基の好ましい例には、以下のものが含まれる。
メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、s−ブチル、t−ブチル、n−ペンチル、イソペンチル、ネオペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、トリデシル、テトラデシル、ペンタデシル、ヘキサデシル、ヘプタデシル、オクタデシル、ノナデシル、イコサニルなどの直鎖または分枝鎖のアルキル基でよい、C1〜20アルキル基、特にC1〜12アルキル基、好ましくはC1〜8アルキル基、より好ましくはC1〜6アルキル基、最も好ましくは上で定義し例示したものなどのC1〜4アルキル基、
C1〜10ハロアルキル基(1個または複数のハロゲン原子、好ましくはフッ素または塩素原子、より好ましくはフッ素原子で置換されたアルキル基であると定義される)、好ましくはC1〜8ハロアルキル基、より好ましくはC1〜6ハロアルキル基、最も好ましくはC1〜4ハロアルキル基、たとえば、モノ−、ジ−、またはトリフルオロメチル、モノ−、ジ−、またはトリクロロメチル、ブロモメチル、2−フルオロエチル、2,2−ジフルオロエチル、2,2,2−トリフルオロエチル、2−クロロエチル、2,2−ジクロロエチル、2,2,2−トリクロロエチル、ペルフルオロエチル、ペルフルオロプロピル、ペルフルオロブチルなど、
C1〜10ヒドロキシアルキル基(ヒドロキシ(−OH)基で置換されたアルキル基であると定義される)、好ましくはC1〜8ヒドロキシアルキル基、より好ましくはC1〜6ヒドロキシアルキル基、最も好ましくはC1〜4ヒドロキシアルキル基、たとえば、ヒドロキシメチル、1−または2−ヒドロキシエチル、1−、2−、または3−ヒドロキシプロピル、1−、2−、3−、または4−ヒドロキシブチルなど、
(C1〜10アルコキシ)C1〜10アルキル基(アルコキシ基で置換されたアルキル基であると定義される)、好ましくは(C1〜6アルコキシ)C1〜6アルキル基、より好ましくは(C1〜4アルコキシ)C1〜4アルキル基、最も好ましくは(C1〜4アルコキシ)メチル基、たとえば、メトキシメチル、1,1−ジメチル−1−メトキシメチル、エトキシメチル、プロポキシメチル、イソプロポキシメチル、ブトキシメチル、t−ブトキシメチル基など、
C1〜6アルコキシル化された(C1〜6アルコキシ)メチル基、たとえば、2−メトキシエトキシメチル基など、
ハロ(C1〜6アルコキシ)メチル基、たとえば、2,2,2−トリクロロエトキシメチルやビス(2−クロロエトキシ)メチル基など、
C3〜8シクロアルキル基、たとえば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル基など、
アラルキル基、たとえば、1〜3個のC6〜14アリール基で置換されたC1〜6アルキル基(アリール部分は、フェニル、ナフチル、アントリル、およびフェナントリルから選択される)、たとえば、ベンジル、α−ナフチルメチル、β−ナフチルメチル、ジフェニルメチル、トリフェニルメチル、α−ナフチルジフェニルメチル、9−アントリルメチル基など;ならびに1〜3個の置換C6〜14アリール基で置換されたC1〜6アルキル基(アリール基の1個または複数は、1個または複数(好ましくは1〜3個、より好ましくは1個のみ)のC1〜6アルキル、C1〜6アルコキシ、ニトロ、ハロゲン、またはシアノ置換基で置換されている)、たとえば、4−メチルベンジル、2,4,6−トリメチルベンジル、3,4,5−トリメチルベンジル、4−メトキシベンジル、4−メトキシフェニルジフェニルメチル、2−ニトロベンジル、4−ニトロベンジル、4−クロロベンジル、4−ブロモベンジル、4−シアノベンジル基など;特にベンジル基、
ハロおよびC1〜6アルコキシから選択される置換基で置換されていてもよいテトラヒドロピラニルまたはテトラヒドロチオピラニル基、たとえば、テトラヒドロピラン−2−イル、3−ブロモテトラヒドロピラン−2−イル、4−メトキシ−テトラヒドロピラン−4−イル、テトラヒドロチオピラン−2−イル、4−メトキシ−テトラヒドロチオピラン−4−イル基など、
ハロおよびC1〜6アルコキシから選択される置換基で置換されていてもよいテトラヒドロフラニルまたはテトラヒドロチオフラニル基、たとえば、テトラヒドロフラン−2−イル基やテトラヒドロチオフラン−2−イル基など、
C2〜10アルケニル基、たとえば、ビニル、プロペニル、ブテニル、ペンテニル、ヘキセニル、ヘプテニル、オクテニル、ノネニル、デセニル基など、ならびに
C2〜10アルキニル基、たとえば、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、ヘプチニル、オクチニル、ノニニル、デシニル基など。
【0068】
前述の例に従う置換基のこれ以上の例、および他のプロドラッグタイプの例は、上述の参考文献で見ることができる。
【0069】
また、特定の式(I)の化合物は、それ自体が他の式(I)の化合物のプロドラッグとして働く場合もある。
【0070】
1個または複数の不斉炭素原子を含有している式(I)の化合物は、2種以上の立体異性体として存在する場合がある。式(I)の化合物は、シクロアルキレン基を含有しているので、CO2H基およびB基が同じ炭素上にないとき、シス/トランス異性体が考えられる。構造異性体が低いエネルギー障壁で相互変換可能である場合、互変異性体異性(「互変異性」)が起こり得る。互変異性は、環状尿素、チオ尿素、またはシアノグアニジン基を含有している式(I)の化合物ではプロトン互変異性の形態、または芳香族部分を含有している化合物ではいわゆる原子価互変異性の形態を取り得る。これは、単一の化合物が1種類に留まらない異性を示す場合もあるということである。
【0071】
本発明の範囲内には、1種類を超える異性を示す化合物を含めて、式(I)の化合物のすべての立体異性体、ジアステレオ異性体(特にシス/トランス異性体)、および互変異性体形態、ならびにこれらの1種または複数の混合物が含まれる。また、酸付加塩または塩基の塩において対イオンが光学活性を有するもの、たとえばd−乳酸塩もしくはl−リジン、またはラセミ体であるもの、たとえばdl−酒石酸塩もしくはdl−アルギニンも含まれる。
【0072】
シス/トランス異性体は、当業者によく知られている従来の技術、たとえば、クロマトグラフィーおよび分別結晶によって分離することができる。
【0073】
個々の鏡像異性体を調製/単離するための従来の技術には、光学的に純粋な好適な前駆体からのキラル合成、またはたとえば、キラルな高圧液体クロマトグラフィー(HPLC)を使用するラセミ体(または塩もしくは誘導体のラセミ体)の分割が含まれる。
【0074】
別法として、ラセミ体(またはラセミ前駆体)を、光学活性を有する好適な化合物、たとえばアルコールと、または式(I)の化合物が酸性もしくは塩基性の部分を含有している場合には1−フェニルエチルアミンや酒石酸などの塩基もしくは酸と反応させることができる。得られるジアステレオ異性体混合物は、クロマトグラフィーおよび/または分別結晶によって分離し、当業者によく知られる方法によってジアステレオ異性体の一方または両方を対応する純粋な鏡像異性体に変換することができる。
【0075】
本発明のキラルな化合物(およびそのキラルな前駆体)は、0〜50体積%、通常は2%〜20%のイソプロパノール、および0〜5体積%のアルキルアミン、通常は0.1%のジエチルアミンを含有する炭化水素、通常はヘプタンまたはヘキサンからなる移動相を用いる不斉樹脂でのクロマトグラフィー、通常はHPLCを使用して、鏡像異性体を豊富に含む形で得ることができる。溶出液を濃縮すると、濃縮された混合物が得られる。
【0076】
ラセミ体が結晶化するとき、異なる2タイプの結晶が考えられる。第1のタイプは、両方の鏡像異性体を等モル量で含有する1つの均質な形態の結晶が生成する、上で言及したラセミ化合物(真のラセミ体)である。第2のタイプは、それぞれ単一の鏡像異性体を含む2形態の結晶が等モル量で生成する、ラセミ混合物または集晶体である。
【0077】
ラセミ混合物中に存在する結晶形は両方とも同一の物理的性質を有するが、これらは真のラセミ体と比較すると異なる物理的性質を有する場合もある。ラセミ混合物は、当業者に知られている従来の技術によって分離することができる。たとえば、E.L.ElielおよびS.H.Wilenによる「Stereochemistry of Organic Compounds」(Wiley、1994年)を参照されたい。
【0078】
本発明は、1個または複数の原子が、原子番号が同じであるが、原子質量または質量数が自然界で優位を占める原子質量または質量数と異なっている原子によって置換されている、薬学的に許容できるすべての同位体標識された式(I)の化合物を含む。
【0079】
本発明の化合物中に含めるのに好適な同位体の例には、2Hや3Hなどの水素、11C、13C、14Cなどの炭素、36Clなどの塩素、18Fなどのフッ素、123Iや125Iなどのヨウ素、13Nや15Nなどの窒素、15O、17O、18Oなどの酸素、32Pなどのリン、および35Sなどの硫黄の同位体が含まれる。
【0080】
特定の同位体標識された式(I)の化合物、たとえば放射性同位体が組み込まれているものは、薬物および/または基質の組織分布調査において有用である。放射性同位体トリチウム、すなわち3H、およびカーボン14、すなわち14Cは、組込みが容易であり、検出手段が整っていることを考えて、この目的に特に有用である。
【0081】
ジュウテリウム、すなわち2Hなどのより重い同位体で置換すると、より高い代謝安定性の結果として生じる特定の治療上の利点、たとえば、in vivo半減期の延長または投与必要量の減少がもたらされる場合もあり、したがってある状況で好ましいといえる。
【0082】
11C、18F、15O、13Nなどの陽電子放射同位体での置換は、基質受容体占有率を調べる陽電子放射断層撮影(PET)研究において有用となり得る。
【0083】
同位体標識された式(I)の化合物は一般に、以前から用いられる標識されていない試薬の代わりに同位体標識された適切な試薬を使用して、当業者に知られている従来の技術によって、または添付の実施例および調製例に記載のものと類似の方法によって調製することができる。
【0084】
本発明による薬学的に許容できる溶媒和物には、結晶化の溶媒が同位体によって置換されているもの、たとえばD2O、d6−アセトン、d6−DMSOでよいものが含まれる。
【0085】
上で規定した式(I)の中間体化合物、上で式(I)の化合物について定義したようなそのすべての塩、溶媒和物、および複合体、ならびにその塩のすべての溶媒和物および複合体も、本発明の範囲内にある。本発明は、上述の化学種およびその晶癖のすべての多形体を含む。
【0086】
本発明に従って式(I)の化合物を調製するとき、当業者は、この目的のために最高の特徴の組合せをもたらす式(I)の化合物の形態を常法どおりに選択することができる。そのような特徴には、融点、溶解性、加工性、および中間体形態の収率、ならびにその結果としての、生成物を単離時に容易に精製できることが含まれる。
【0087】
式(I)の化合物は、提案された適応症を治療するのに最も適切な剤形および投与経路を選択するために、(全pHでの)溶解性および溶液安定性、透過性などのその生物薬剤学的な性質について評価すべきである。
【0088】
医薬としての使用を目的とする本発明の化合物は、結晶性または非晶質の生成物として投与することができる。そのような本発明の化合物は、沈殿、結晶化、凍結乾燥、噴霧乾燥、または蒸発乾燥などの方法によって、たとえば、固体充填物、粉末、またはフィルムとして得ることができる。マイクロ波または高周波乾燥をこの目的のために使用してもよい。
【0089】
化合物は、単独で、または1種または複数の他の本発明化合物と組み合わせて、または1種または複数の他の薬物と組み合わせて(またはこれらの任意の組合せとして)投与することができる。一般に、化合物は、1種または複数の薬学的に許容できる賦形剤と合同で製剤として投与される。用語「賦形剤」は、本明細書では、本発明の化合物以外の任意の成分について述べるのに使用する。賦形剤の選択は、大部分は、特定の投与方式、賦形剤が溶解性および安定性に及ぼす影響、剤形の性質などの要素に応じて決まる。
【0090】
本発明の化合物の送達に好適な医薬組成物およびその調製方法は、当業者には言うまでもない。そのような組成物およびその調製方法は、たとえば、「Remington’s Pharmaceutical Sciences」第19版(Mack Publishing Company、1995年)で見ることができる。
【0091】
経口投与
本発明の化合物は、経口投与することができる。経口投与は、化合物がGIに入るように飲み込むものでもよいし、および/または化合物がそれによって口から直接血流に入る頬側、舌側、もしくは舌下投与を含むものでもよい。
【0092】
経口投与に好適な製剤には、錠剤;多粒子もしくはナノ粒子、液体、または粉末を含有する軟または硬カプセル剤;ロゼンジ(液体充填型を含む);咀嚼剤;ゲル;急速分散型剤形;フィルム;膣坐剤;スプレー;ならびに頬側/粘膜付着性パッチなどの、固体、半固体、および液体の系が含まれる。
【0093】
液体製剤には、懸濁液、溶液、シロップ、およびエリキシルが含まれる。このような製剤は、(たとえばゼラチンまたはヒドロキシプロピルメチルセルロース製の)軟もしくは硬カプセル中に充填剤として用いることもでき、通常は、担体、たとえば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロース、または好適な油と、1種または複数の乳化剤および/または懸濁化剤とを含む。液体製剤は、たとえば小袋から出した固体を再構成して、調製することもできる。
【0094】
本発明の化合物は、LiangおよびChenによるExpert Opinion in Therapeutic Patents、第11巻(6)、981〜986ページ(2001年)に記載のものなどの急速溶解急速崩壊型の剤形にして使用してもよい。
【0095】
錠剤剤形では、薬物は、用量に応じて、剤形の1重量%〜80重量%、より典型的な例では剤形の5重量%〜60重量%を占めてよい。錠剤は一般に、薬物に加えて崩壊剤を含有する。崩壊剤の例には、ナトリウムデンプングリコラート、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶セルロース、低級アルキル置換されたヒドロキシプロピルセルロース、デンプン、α化デンプン、およびアルギン酸ナトリウムが含まれる。一般に、崩壊剤は、剤形の1重量%〜25重量%、好ましくは5重量%〜20重量%を占める。
【0096】
結合剤は一般に、錠剤製剤に粘着性の性質を付与するために使用する。好適な結合剤には、微結晶セルロース、ゼラチン、糖、ポリエチレングリコール、天然および合成のゴム、ポリビニルピロリドン、α化デンプン、ヒドロキシプロピルセルロース、およびヒドロキシプロピルメチルセルロースが含まれる。錠剤は、ラクトース(一水和物、噴霧乾燥一水和物、無水物など)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶セルロース、デンプン、第二リン酸カルシウム二水和物などの希釈剤も含有する場合がある。
【0097】
錠剤は、ラウリル硫酸ナトリウムやポリソルベート80などの界面活性剤、および二酸化ケイ素やタルクなどの滑剤を場合により含むこともある。存在するとき、界面活性剤は錠剤の0.2重量%〜5重量%を占めてよく、滑剤は錠剤の0.2重量%〜1重量%を占めてよい。
【0098】
錠剤は一般に、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、フマル酸ステアリルナトリウム、ステアリン酸マグネシウムとラウリル硫酸ナトリウムの混合物などの滑沢剤も含有する。滑沢剤は一般に、錠剤の0.25重量%〜10重量%、好ましくは0.5重量%〜3重量%を占める。
【0099】
他の考えられる成分には、抗酸化剤、着色剤、着香剤、保存剤、および矯味剤が含まれる。
【0100】
好例となる錠剤は、約80%までの薬物、約10重量%〜約90重量%の結合剤、約0重量%〜約85重量%の希釈剤、約2重量%〜約10重量%の崩壊剤、および約0.25重量%〜約10重量%の滑沢剤を含有する。
【0101】
錠剤ブレンドを直接にまたはローラーによって圧縮すると、錠剤を生成することができる。別法として、錠剤ブレンドまたはブレンドの一部分を、湿式、乾式、もしくは溶融造粒、溶融凝固、または押出し成形処理した後に打錠してもよい。最終製剤は、1重または複数の層を含むこともあり、コーティングされていてもされていなくてもよく、カプセル封入されていてもよい。
【0102】
錠剤の製剤については、H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets」、第1巻(Marcel Dekker、ニューヨーク、1980年)で論述されている。
【0103】
ヒトまたは獣医学で使用する摂取可能な経口フィルムは通常、急速溶解型または粘膜付着性でよい可撓性の水溶性または水膨張性薄膜剤形であり、通常は式(I)の化合物、フィルム形成ポリマー、結合剤、溶媒、湿潤剤、可塑剤、安定剤または乳化剤、粘度調整剤、ならびに溶媒を含む。製剤の一部の構成要素が複数の機能を果たす場合もある。
【0104】
式(I)の化合物は、水溶性である場合もあれば不溶性である場合もある。水溶性の化合物は通常、溶質の1重量%〜80重量%、より典型的な例では20重量%〜50重量%を占める。それほど可溶性でない化合物は、組成物のより高い割合、通常は溶質の88重量%までを占めてよい。別法として、式(I)の化合物を多粒子ビーズの形にしてもよい。
【0105】
フィルム形成ポリマーは、天然の多糖類、タンパク質、または合成の親水コロイドから選択されるものでよく、通常は0.01〜99重量%の範囲、より典型的な例では30〜80重量%の範囲で存在する。
【0106】
他の考えられる成分には、抗酸化剤、着色剤、着香剤、および香味剤、保存剤、唾液腺刺激剤、冷却剤、共溶媒(油を含む)、緩和剤、増量剤、消泡剤、界面活性剤、および矯味剤が含まれる。
【0107】
本発明によるフィルムは通常、可剥性の支持担体または紙上にコーティングした薄い水性フィルムを蒸発乾燥して調製する。これは、乾燥オーブンもしくは乾燥トンネル、通常は複合塗工乾燥機で、または凍結乾燥もしくは減圧によって行うことができる。
【0108】
経口投与用の固体製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0109】
本発明の目的に好適な変更型放出製剤は、米国特許第6106864号に記載されている。高エネルギー分散や浸透性粒子および被覆粒子などの他の好適な放出技術の詳細は、VermaらによるPharmaceutical Technology On−line、第25巻(2)、1〜14ページ(2001年)で見られる。制御放出を実現するためのチューインガムの使用は、WO00/35298に記載されている。
【0110】
非経口投与
本発明の化合物は、血流中、筋肉中、または内臓中に直接投与することもできる。非経口投与に好適な手段には、静脈内、動脈内、腹腔内、くも膜下腔内、側脳室内、尿道内、胸骨内、脳内、筋肉内、滑液包内、および皮下が含まれる。非経口投与に好適な装置には、(微細針を含む)針注射器、無針注射器、および注入技術が含まれる。
【0111】
非経口製剤は通常、塩、炭水化物、(好ましくはpH3〜9にする)緩衝剤などの賦形剤を含有することもある水溶液であるが、一部の適用例では、無菌の非水性溶液として、または発熱物質を含まない無菌水などの好適な媒体と共に使用する乾燥形態としてより適切に製剤してもよい。
【0112】
たとえば凍結乾燥による無菌条件下での非経口製剤の調製は、当業者によく知られている標準の製薬技術を使用して容易に実現することができる。
【0113】
非経口溶液の調製で使用する式(I)の化合物の溶解性は、溶解性改善剤を混ぜるなどの適切な製剤技術を使用して増大させることができる。
【0114】
非経口投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。したがって、本発明の化合物は、懸濁液として、または活性化合物の変更型放出をもたらす移植デポー剤として投与するための固体、半固体、もしくは揺変性液体として製剤することができる。そのような製剤の例には、薬物でコーティングされたステント、ならびに薬物を載せたdl−乳酸−グリコール酸共重合体(PGLA)ミクロスフェアを含む半固体および懸濁液が含まれる。
【0115】
局所投与
本発明の化合物は、皮膚または粘膜に局所的に、(皮内)皮膚上に、または経皮的に投与することもできる。この目的のための典型的な製剤には、ゲル、ヒドロゲル、ローション、溶液、クリーム、軟膏、散粉剤、包帯剤、フォーム、フィルム、皮膚パッチ、ウェーハ、植込錠、スポンジ、線維、絆創膏、およびマイクロエマルジョンが含まれる。リポソームを使用してもよい。典型的な担体には、アルコール、水、鉱油、流動パラフィン、白色ワセリン、グリセリン、ポリエチレングリコール、およびプロピレングリコールが含まれる。浸透性改善剤を混ぜてもよい。たとえば、FinninおよびMorganによるJ.Pharm.Sci.、第88巻(10)、955〜958ページ(1999年10月)を参照されたい。
【0116】
他の局所投与手段には、電気穿孔法、イオン導入法、音波泳動法、超音波導入法、ならびに微細針または無針(たとえばPowderject(商標)、Bioject(商標)など)注射による送達が含まれる。
【0117】
局所投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0118】
吸入投与/鼻腔内投与
本発明の化合物は通常、乾燥粉末吸入器から(単独で、たとえばラクトースとの乾燥ブレンドにした混合物として、またはたとえばホスファチジルコリンなどのリン脂質と混合した混合型成分粒子としての)乾燥粉末の形で、加圧容器、ポンプ、スプレー、アトマイザー(好ましくは電気水力学を使用して微細な霧を生成するアトマイザー)、もしくはネブライザーから、1,1,1,2−テトラフルオロエタンや1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの好適な噴射剤を使用しまたは使用せずにエアロゾルスプレーとして、または点鼻液として、鼻腔内にまたは吸入によって投与することもできる。鼻腔内の使用では、粉末は、生体接着剤、たとえばキトサンまたはシクロデキストリンを含んでもよい。
【0119】
加圧容器、ポンプ、スプレー、アトマイザー、またはネブライザーは、たとえばエタノール、エタノール水溶液、または活性物を分散させ、可溶化し、もしくはその放出を延長するのに好適な別の薬剤を含む本発明の化合物の溶液または懸濁液、溶媒としての噴射剤、ならびにトリオレイン酸ソルビタン、オレイン酸、オリゴ乳酸などのオプションの界面活性剤を含有する。
【0120】
薬物製品は、乾燥粉末または懸濁液製剤中に使用する前に、吸入による送達に好適なサイズ(通常は5ミクロン未満)に微粒子化する。これは、スパイラルジェット粉砕、流動層ジェット粉砕、ナノ粒子を生成するための超臨界流体処理、高圧ホモジナイズ、噴霧乾燥などの任意の適切な微粉砕法によって実現することができる。
【0121】
吸入器または注入器に入れて使用する(たとえばゼラチンまたはヒドロキシプロピルメチルセルロース製の)カプセル、ブリスター、およびカートリッジは、本発明の化合物、ラクトースやデンプンなどの好適な粉末基剤、およびl−ロイシン、マンニトール、ステアリン酸マグネシウムなどの性能調節剤の粉末混合物を含有するように製剤することができる。ラクトースは、無水でも一水和物の形でもよいが、後者が好ましい。他の好適な賦形剤には、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロース、およびトレハロースが含まれる。
【0122】
電気水力学を使用して微細な霧を生成するアトマイザーに入れて使用するのに好適な溶液製剤は、1作動あたり1μg〜20mgの本発明の化合物を含有するものでよく、作動体積は1μl〜100μlと様々でよい。典型的な製剤は、式(I)の化合物、プロピレングリコール、滅菌水、エタノール、および塩化ナトリウムを含む。プロピレングリコールの代わりに使用することのできる別の溶媒には、グリセロールおよびポリエチレングリコールが含まれる。
【0123】
メントールやl−メントールなどの好適な着香剤、またはサッカリンやサッカリンナトリウムなどの甘味剤を、吸入投与/鼻腔内投与を目的とする本発明の製剤に加えてもよい。
【0124】
吸入投与/鼻腔内投与用の製剤は、たとえばPGLAを使用して、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0125】
直腸/膣内投与
本発明の化合物は、たとえば坐剤、膣坐剤、または浣腸の形で直腸にまたは経膣的に投与することができる。カカオ脂が伝統的な坐剤基剤であるが、様々な代替品を適宜使用することができる。
【0126】
直腸/経膣投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0127】
眼/耳への投与
本発明の化合物は、通常は、等張性のpH調整された無菌食塩水中の微粒子化された懸濁液または溶液の液滴の形で眼または耳に直接投与することもできる。眼および耳への投与に好適な他の製剤には、軟膏、ゲル、生分解性(たとえば吸収性のゲルスポンジ、コラーゲン)および非生分解性(たとえばケイ素樹脂)の植込錠、ウェーハ、レンズ、ならびにニオソームやリポソームなどの微粒子系またはベシクル系が含まれる。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸などのポリマー;セルロース系ポリマー、たとえば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、もしくはメチルセルロース;またはヘテロ多糖ポリマー、たとえばゲランガムを、塩化ベンザルコニウムなどの保存剤と共に混ぜることができる。このような製剤は、イオン導入法によって送達してもよい。
【0128】
眼/耳への投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、またはプログラム放出が含まれる。
【0129】
他の技術
本発明の化合物は、上述の投与様式のいずれかで使用するため、その溶解性、溶出速度、矯味、バイオアベイラビリティおよび/または安定性を改善するためにシクロデキストリンおよびその好適な誘導体やポリエチレングリコール含有ポリマーなどの可溶性の高分子実在物と混ぜ合わせてもよい。
【0130】
たとえば、薬物−シクロデキストリン複合体は、一般にほとんどの剤形および投与経路に有用であることがわかっている。包接複合体および非包接複合体の両方を使用することができる。シクロデキストリンは、薬物との直接の複合体形成に代わるものとして、補助添加剤、すなわち担体、希釈剤、または可溶化剤として使用してもよい。これらの目的のために最も一般に使用されるのは、α、β、およびγシクロデキストリンであり、その例は、WO91/11172、WO94/02518、およびWO98/55148で見ることができる。
【0131】
成分キット
たとえばある特定の疾患または状態を治療する目的のために活性化合物の組合せを投与することが望ましい場合もあるので、その少なくとも1種が本発明による化合物を含有する2種以上の医薬組成物を、組成物の共投与に好適なキットの形で好都合に組み合わせてよいことは、本発明の範囲内である。
【0132】
したがって、本発明のキットは、その少なくとも1種が本発明による式(I)の化合物を含有する2種以上の別個の医薬組成物と、容器、分割されたボトル、または分割されたホイル製袋などの、前記組成物を別々に保持するための手段とを含む。そのようなキットの例は、錠剤、カプセルなどの包装に使用される見慣れたブリスターパックである。
【0133】
本発明のキットは、たとえば経口と非経口の異なる剤形を投与する、別個の組成物を異なる投与間隔で投与する、または別個の組成物を互いに対して滴定するのに特に好適である。服薬遵守を援助するために、キットは通常、投与の説明書を含み、いわゆるメモリーエイドを添えて提供することができる。
【0134】
投与量
ヒト患者への投与について、本発明の化合物の合計1日量は通常、当然ながら投与方式に応じて10mg〜1000mgの範囲にある。たとえば、経口投与には10mg〜1000mgの合計1日量が必要である場合があり、静脈内用量には10mg〜1000mgが必要となる場合がある。合計1日量は、1回で、または数回に分けて投与することができ、医師の裁量で、本明細書で示す典型的な範囲の範囲外になることもある。
【0135】
これらの投与量は、体重が約60kg〜70kgである平均的なヒト対象に基づく。医師は、小児や高齢者などの、体重がこの範囲外にある対象のための用量を容易に決定することができよう。
【0136】
疑義を避けるために、本明細書における「治療」への言及は、治療的、姑息的、および予防的な治療への言及を含む。
【0137】
式(I)の化合物はすべて、以下で述べる一般法に記載の手順によって、または実施例の項および調製例の項に記載の特定の方法によって、またはその常法どおりの変更形態によって調製することができる。本発明はまた、式(I)の化合物の調製方法のいずれか1つまたは複数に加えて、その中で使用する任意の新規な中間体も包含する。
【0138】
一般法
以下の略語を使用する。
DMF=ジメチルホルムアミド
DMSO=ジメチルスルホキシド
TEMPO=2,2,6,6−テトラメチルピペリジン−N−オキシド
THF=テトラヒドロフラン
DCM=ジクロロメタン
【0139】
式(I)の化合物は、以下のスキーム1に示すとおりに調製することができる。
【0140】
【化4】
【0141】
スキーム1において、Pはヒドロキシ保護基を表し、その好適な例は、T.W.GreeneおよびP.Wutsによる「Protective Groups in Organic Synthesis」、Wiley and Sons、1991年に記載されており、LGは、ハロゲン、(C1〜6アルキル)スルホニルオキシ(たとえばメタン−スルホニルオキシ)、(C1〜6ハロアルキル)スルホニルオキシ(たとえばトリフルオロメタンスルホニルオキシ)、またはベンゼンもしくはトルエンスルホニルオキシ(たとえばp−トルエンスルホニルオキシ)などの好適な脱離基を表す。Pがベンジルであり、LGがp−トルエンスルホニルオキシであることが好ましい。
【0142】
ステップ(a):式(III)の化合物は、化合物(II)と、ヒドロキシ基を脱離基に変換することのできる適切な薬剤、通常はスルホニル化試薬(たとえば塩化メタンスルホニルまたは塩化p−トルエンスルホニル)とから、塩基(たとえばトリエチルアミンまたはピリジン)の存在下、好適な溶媒(たとえばピリジンまたはジクロロメタン)中にて、0℃から室温で15分間〜24時間かけて調製することができる。
【0143】
好ましい条件は次のとおりである。ジクロロメタン中の1当量の化合物(II)、1.2当量の塩化p−トルエンスルホニル、2当量のピリジン、室温で18時間。
【0144】
ステップ(b):式(IV)の化合物は、化合物(III)と式(VI)のヒドロキシ化合物とから、好適な溶媒(たとえばDMF、DMSO)中にて、好適な塩基(たとえばCs2CO3、K2CO3)の存在下、場合によりクラウンエーテル(たとえば18−クラウン−6)の存在下、50〜120℃で一晩かけて調製することができる。
【0145】
好ましい条件は次のとおりである。1当量の化合物(VI)、1.1当量の化合物(III)、1.2当量のCs2CO3、DMF中にて80℃で24時間。
【0146】
式(VI)の化合物は、WO02/074754に一般に記載されている。XがOであり、mが1であり、RがClである特定の式(VI)の化合物は、Bioorg.Med.Chem.Lett.(2004年)、第14巻(18)、4627〜32ページに記載のとおり、または以下のスキーム5で概略を述べるとおりに調製することができる。
【0147】
ステップ(c):式(IV)の化合物を好適な溶媒中で脱保護剤と反応させて脱保護すると、式(V)の化合物を得ることができる。好適な試薬および方法は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。Pがベンジルであるとき、好適な試薬の例には、三塩化ホウ素または塩化鉄(III)が含まれる。
【0148】
好ましい条件は次のとおりである。ジクロロメタン中の1当量の化合物(IV)、4当量のBCl3、室温で18時間。
【0149】
ステップ(d):好適な溶媒中で酸化剤を使用して式(V)の化合物を酸化することにより、式(I)の化合物を調製することができる。典型的な試薬および条件としては、三酸化クロムおよび過ヨウ素酸(H5IO6)を触媒とし、アセトニトリルなどの溶媒中にて室温から50℃で18〜36時間、または別法としてNaOClおよびNaClO2、触媒のTEMPOの存在下、アセトニトリルなどの溶媒中にて0℃〜室温で18〜36時間が挙げられる。
【0150】
好ましい条件は次のとおりである。1当量の化合物(V)、2.5当量の過ヨウ素酸、0.02当量のCrO3、0.75%アセトニトリル水溶液中にて40℃で24時間。
【0151】
別法として、式(I)の化合物は、スキーム2に示すように、式(V)の化合物を、式(VII)のアルデヒドを介する二段階の手順で酸化して調製することができる。
【0152】
【化5】
【0153】
ステップ(a):アルコール(V)をアルデヒド(VII)にする酸化は通常、触媒としてのTEMPOと共にNaOClを使用し、好適な溶媒、たとえばアセトニトリル、アセトン中にて0℃から室温で2〜18時間、または別法として、DMSOと共に三酸化物硫黄−ピリジン錯体を使用し、THFなどの溶媒中にて0℃から室温で2〜18時間実施する。
【0154】
ステップ(b):アルデヒド(VII)を酸(I)にするさらなる酸化は通常、NaClO2を使用し、リン酸カリウムの存在下、t−ブタノール水溶液などの溶媒中にて0℃から室温で2〜18時間、または別法として、触媒としてのTEMPOと共にトリクロロイソシアニル酸を使用し、好適な溶媒、たとえばアセトンまたはアセトニトリル中にて0℃から室温で2〜18時間実施する。
【0155】
式(II)の化合物は文献で知られている。たとえば、Aがシス−1,3−シクロブチレン基であり、Bが単結合である式(II)の化合物は、J.Chem.Soc.、Perkin Trans.1(1995年)、第18巻、2281〜7ページに記載のとおりに調製することができる。
【0156】
別法として、Aがシス−またはトランス−1,3−シクロブチレン基であり、Bが単結合である式(I)の化合物である式(Ib)の化合物は、化合物(VIII)または化合物(IX)から、スキーム3に示すような標準の方法によって調製することができる。トランス化合物(II)および(X)は、それぞれシス化合物(II)および(X)から、Synthesis(1981年)、第1巻に記載のものと類似の光延化学を使用する反転によって得ることができる。
【0157】
【化6】
【0158】
スキーム3において、Raはエステル残基であり、その好適な例は、上でプロドラッグに関して述べ、また「Protective Groups in Organic Synthesis」(前掲書)に記載されており(たとえば、(C1〜6)アルキル、ベンジル、または(+)もしくは(−)−メンチル)、LGは、ハロゲン、(C1〜6アルキル)スルホニルオキシ(たとえばメタンスルホニルオキシ)、(C1〜6ハロアルキル)スルホニルオキシ(たとえばトリフルオロメタンスルホニルオキシ)、またはベンゼン−もしくはトルエンスルホニルオキシ(たとえばp−トルエンスルホニルオキシ)などの脱離基である。
【0159】
ステップ(a):式(IX)の化合物は、化合物(VIII)と式RaOHの好適なアルコール(たとえばメタノール、t−ブタノール、ベンジルアルコール、または(−)メントール)とを、様々な条件下で反応させて調製することができ、その好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。
【0160】
好ましい条件は次のとおりである。1当量の化合物(VIII)、1.1当量の1,1’−カルボニルジイミダゾール、酢酸エチル中にて還流温度で1時間、その後1当量のRaOH、室温で4時間。
【0161】
ステップ(b):化合物(IX)をアルコール(X)にする還元は、THFなどの好適な溶媒中で、好適な還元剤、たとえば水素化ホウ素ナトリウムまたはL−Selectride(登録商標)を使用して実施することができる。
【0162】
好ましい条件は次のとおりである。1当量の化合物(IX)、0.5当量のNaBH4、20:1のTHF:メタノール中にて0℃で20分間。
【0163】
ステップ(c):式(XI)の化合物は、化合物(X)から、スキーム1ステップ(a)に記載のものと同様の試薬および条件を使用して調製することができる。
【0164】
好ましい条件は次のとおりである。1当量の化合物(X)、1.05当量の塩化p−トルエンスルホニル、ピリジン中、0℃から室温。
【0165】
ステップ(d):式(Ia)の化合物は、化合物(XI)と式(VI)のヒドロキシ化合物とから、スキーム1ステップ(b)に記載のものと同様の試薬および条件を使用して調製することができる。
【0166】
好ましい条件は次のとおりである。1.2当量の化合物(XI)、1.0当量の化合物(VI)、1.5当量のCs2CO3、DMF中にて80℃で18時間。
【0167】
ステップ(e):式(Ia)の化合物を加水分解すると、式(Ib)の化合物を得ることができる。この反応は、様々な条件下で実現することができ、その好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。好ましい条件は次のとおりである。化合物(Ia)、2当量のNaOH、1:1のエタノール:水中にて60℃で2時間。
【0168】
化合物(VIII)は、J.Org.Chem.、(1981年)、第53巻、3841〜43ページに記載されており、Raがメチル基である化合物(IX)は、J.Org.Chem.、(1994年)、第59巻、2132〜34ページに記載されている。
【0169】
【化7】
【0170】
Bがメチレン基である式(I)の化合物である式(Id)の化合物は、スキーム4に示すとおりに調製することができる。
【0171】
【化8】
【0172】
スキーム4において、Raはエステル残基であり、その好適な例は、上でプロドラッグに関して述べており、また「Protective Groups in Organic Synthesis」(前掲書)に記載されており(たとえば(C1〜6)アルキルまたはベンジル)、LGは、ハロゲン、(C1〜6アルキル)スルホニルオキシ(たとえばメタンスルホニルオキシ)、(C1〜6ハロアルキル)スルホニルオキシ(たとえばトリフルオロメタンスルホニルオキシ)、またはベンゼン−もしくはトルエンスルホニルオキシ(たとえばp−トルエンスルホニルオキシ)などの脱離基である。Raがベンジルであり、LGがp−トルエンスルホニルオキシであることが好ましい。式(XII)の化合物は、市販品として得ることができる。
【0173】
ステップ(a):式(XIII)の化合物は、酸性または塩基性条件下、たとえば、メタノール、エタノール、1,4−ジオキサンなどを好適な共溶媒とする水酸化ナトリウム水溶液中で、またはエタノールや1,4−ジオキサンなどを場合により好適な共溶媒とする塩酸もしくは硫酸水溶液中で、式(XII)の化合物を加水分解することにより調製できる。
【0174】
好ましい条件は次のとおりである。1当量の化合物(XII)、4当量のNaOH、1:1のエタノール:水中にて還流温度で2.5時間。
【0175】
ステップ(b):式(XIV)の化合物は、式(XIII)の化合物と式RaOHの好適なアルコール(たとえばメタノール、t−ブタノール、ベンジルアルコール)とを、様々な条件下で反応させて調製することができ、その条件の好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。好ましい条件は次のとおりである。1当量の化合物(XIII)、1.1当量の1,1’−カルボニルジイミダゾール、酢酸エチル中、約1時間、その後1.2当量のベンジルアルコール、室温で18時間。
【0176】
ステップ(c):式(XV)の化合物は、THFなどの好適な溶媒中にて、0℃から室温で、式(XIV)の化合物をボラン−ジメチルスルフィド、カテコールボラン、9−ボラビシクロ[3.3.1]ノナン(9−BBN)などのヒドロホウ素化剤で処理した後、in situで、室温から60℃にて、過酸化水素、過ホウ酸ナトリウム、トリメチルアミン−N−オキシドなどの酸化剤で酸化して調製することができる。
【0177】
好ましい条件は次のとおりである。1当量の化合物(XIV)、0.5当量のボラン−ジメチルスルフィド、THF中にて室温で1時間、その後1.2当量の過ホウ酸ナトリウム、60℃で1時間の加熱。
【0178】
ステップ(d):式(XVI)の化合物は、式(XV)の化合物から、スキーム1ステップ(a)に記載のものと同様の試薬および条件を使用して調製することができる。
【0179】
好ましい条件は次のとおりである。1当量の化合物(XV)、1.3当量の塩化p−トルエンスルホニル、2.6当量のピリジン、DCM中、0℃から室温。
【0180】
ステップ(e):式(Ic)の化合物は、式(XVI)の化合物と式(VI)のヒドロキシ化合物とから、スキーム1ステップ(b)に記載のものと同様の試薬および条件を使用して調製することができる。
【0181】
好ましい条件は次のとおりである。1.2当量の化合物(XVI)、1.0当量の化合物(VI)、1.5当量のCs2CO3、DMF中にて80℃で18時間。
【0182】
ステップ(f):式(Ic)の化合物を加水分解すると、式(Id)の化合物を得ることができる。この反応は、様々な条件下で実現することができ、その好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。
【0183】
好ましい条件は次のとおりである。化合物(Ic)、過剰のNaOH、1:1のエタノール:水中にて60℃で2時間。
【0184】
式(VI)の化合物は、WO02/074754に一般に記載されている。XがOまたはSである式(VI)の化合物である特定の式(VIa)の化合物は、Bioorg.Med.Chem.Lett.、(2004年)、第14巻(18)、4627〜32ページに記載のとおりに、または以下のスキーム5で概略を述べるとおりに調製することができる。
【0185】
【化9】
【0186】
スキーム5において、Rbは(C1〜6)アルキルまたはベンジルである。
【0187】
ステップ(a):式(XVIII)の化合物は、アニリン(XVII)とナトリウムまたはカリウムのシアン酸塩またはチオシアン酸塩とを、マレイン酸や酢酸などの酸の存在下、好適な溶媒または溶媒混合物中、たとえばジクロロメタンまたは酢酸:水中で反応させて調製することができる。別法として、式(XVIII)の化合物は、アニリン(XVII)とイソシアン酸またはチオシアン酸塩トリメチルシリルとを、ジクロロメタンなどの溶媒中で反応させた後、in situで水によって加水分解して調製することができる。
【0188】
XがOであるときの好ましい条件は次のとおりである。1当量の化合物(XVII)、酢酸:水(9:1)中、その後1.2当量のシアン酸カリウム、水中、滴下、40℃で1時間保つ。
【0189】
ステップ(b):式(XIX)の化合物は、式(XVIII)の尿素と適切なケトンとを、ポリリン酸やEaton試薬(メタンスルホン酸中7.5%P2O5)などの脱水剤の存在下、50℃〜100℃の間で反応させて調製することができる。
【0190】
好ましい条件は次のとおりである。1当量の化合物(XVIII)、Eaton試薬(30g/g)、60℃、その後2当量のケトン、80℃で1時間の加熱。
【0191】
ステップ(c):式(VIa)の化合物は、式(XIX)の化合物を、ジクロロメタンなどの好適な溶媒中にて室温で三臭化ホウ素などのルイス酸と反応させ、または高温で強酸と、たとえば110℃で臭化水素酸と反応させることにより調製できる。
【0192】
好ましい条件は次のとおりである。1当量の化合物(XIX)、20当量の48%臭化水素水溶液、酢酸中にて110℃で4日間。
【0193】
式(I)のPDE7阻害剤は、特に疼痛の治療において、もう1種の薬理活性のある化合物、または2種以上の他の薬理活性のある化合物と有益に組み合わせることができる。たとえば、上で定義した、式(I)のPDE7阻害剤、または薬学的に許容できるその塩、溶媒和物、もしくはプロドラッグは、以下のものから選択される1種または複数の薬剤と組み合わせて、同時、逐次、または別々に投与することができる。
・オピオイド鎮痛薬、たとえば、モルヒネ、ヘロイン、ヒドロモルフォン、オキシモルフォン、レボルファノール、レバロルファン、メサドン、メペリジン、フェンタニル、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ハイドロコドン、プロポキシフェン、ナルメフェン、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフィン、またはペンタゾシン、
・非ステロイド性抗炎症薬(NSAID)、たとえば、アスピリン、ジクロフェナク、ジフルシナル(diflusinal)、エトドラク、フェンブフェン、フェノプロフェン、フルフェニサール(flufenisal)、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラック、メクロフェナム酸、メフェナム酸、メロキシカム、ナブメトン、ナプロキセン、ニメスリド、ニトロフルルビプロフェン、オルサラジン、オキサプロジン、フェニルブタゾン、ピロキシカム、スルファサラジン、スリンダク、トルメチン、またはゾメピラク、
・バルビツール酸系鎮静薬、たとえば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール(butabital)、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルチタール(phenobartital)、セコバルビタール、タルブタール、テアミラル(theamylal)、またはチオペンタール、
・鎮静作用を有するベンゾジアゼピン、たとえば、クロルジアゼポキシド、クロラゼプ酸、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパム、またはトリアゾラム、
・鎮静作用を有するH1拮抗薬、たとえば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミン、またはクロルシクリジン、
・鎮静薬、たとえば、グルテチミド、メプロバメート、メタカロン、またはジクロラールフェナゾンなど、
・骨格筋弛緩薬、たとえば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモール、またはオルフレナジン(orphrenadine)、
・NMDA受容体拮抗薬、たとえば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルヒナン)もしくはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルヒナン)、ケタミン、メマンチン、ピロロキノリンキニン、シス−4−(ホスホノメチル)−2−ピペリジンカルボン酸、ブジピン、EN−3231(MorphiDex(登録商標)、モルヒネとデキストロメトルファンの合剤)、トピラメート、ネラメキサン(neramexane)、またはペルジンフォテル(perzinfotel)(NR2B拮抗薬、たとえば、イフェンプロジル、トラキソプロジル(traxoprodil)、または(−)−(R)−6−{2−[4−(3−フルオロフェニル)−4−ヒドロキシ−1−ピペリジニル]−1−ヒドロキシエチル−3,4−ジヒドロ−2(1H)−キノリノンを含む)、
・α−アドレナリン作動薬、たとえば、ドキサゾシン、タムスロシン、クロニジン、グァンファシン、デクスメタトミジン(dexmetatomidine)、モダフィニル、または4−アミノ−6,7−ジメトキシ−2−(5−メタン−スルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン、
・三環系抗うつ薬、たとえば、デシプラミン、イミプラミン、アミトリプチリン、またはノルトリプチリン、
・抗痙攣薬、たとえば、カルバマゼピン、ラモトリジン、トピラトメート(topiratmate)、またはバルプロエート、
・タキキニン(NK)拮抗薬、特にNK−3、NK−2、またはNK−1拮抗薬、たとえば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]−ナフチリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]−メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、アプレピタント、ラネピタント、ダピタント、または3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]−メチルアミノ]−2−フェニルピペリジン(2S,3S)、
・ムスカリン性拮抗薬、たとえば、オキシブチニン、トルテロジン、プロピベリン、塩化トロプシウム(tropsium chloride)、ダリフェナシン、ソリフェナシン、テミベリン、およびイプラトロピウム、
・COX−2選択的阻害剤、たとえば、セレコキシブ、ロフェコキシブ、パレコキシブ、バルデコキシブ、デラコキシブ(deracoxib)、エトリコキシブ、またはルミラコキシブ、
・コールタール鎮痛薬、特にパラセタモール、
・神経弛緩薬、たとえば、ドロペリドール、クロルプロマジン、ハロペリドール、ペルフェナジン、チオリダジン、メソリダジン、トリフルオペラジン、フルフェナジン、クロザピン、オランザピン、リスペリドン、ジプラシドン、クエチアピン、セルチンドール、アリピプラゾール、ソネピプラゾール、ブロナンセリン、イロペリドン、ペロスピロン、ラクロプライド、ゾテピン、ビフェプルノックス、アセナピン、ルラシドン、アミスルプリド、バラペリドン、パリンドーレ(palindore)、エプリバンセリン、オサネタント、リモナバント、メクリネルタント(meclinertant)、Miraxion(登録商標)、サリゾタン(sarizotan)など、
・バニロイド受容体作動薬(たとえばレシンフェラトキシン(resinferatoxin))または拮抗薬(たとえばカプサゼピン)、
・プロプラノロールなどのβ−アドレナリン作動薬、
・メキシレチンなどの局所麻酔薬、
・デキサメタゾンなどの副腎皮質ステロイド、
・5−HT受容体作動薬または拮抗薬、特に、エレトリプタン、スマトリプタン、ナラトリプタン、ゾルミトリプタン、リザトリプタンなどの5−HT1B/1D作動薬、
・R(+)−α−(2,3−ジメトキシ−フェニル)−1−[2−(4−フルオロフェニルエチル)]−4−ピペリジンメタノール(MDL−100907)などの5−HT2A受容体拮抗薬、
・コリン作用性(ニコチン性)鎮痛薬、たとえば、イスプロニクリン(TC−1734)、(E)−N−メチル−4−(3−ピリジニル)−3−ブテン−1−アミン(RJR−2403)、(R)−5−(2−アゼチジニルメトキシ)−2−クロロピリジン(ABT−594)、ニコチンなど、
・Tramadol(登録商標)、
・PDEV阻害剤、たとえば、5−[2−エトキシ−5−(4−メチル−1−ピペラジニル−スルホニル)フェニル]−1−メチル−3−n−プロピル−1,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン(シルデナフィル)、(6R,12aR)−2,3,6,7,12,12a−ヘキサヒドロ−2−メチル−6−(3,4−メチレンジオキシフェニル)−ピラジノ[2’,1’:6,1]−ピリド[3,4−b]インドール−1,4−ジオン(IC−351またはタダラフィル)、2−[2−エトキシ−5−(4−エチル−ピペラジン−1−イル−1−スルホニル)−フェニル]−5−メチル−7−プロピル−3H−イミダゾ[5,1−f][1,2,4]トリアジン−4−オン(バルデナフィル)、5−(5−アセチル−2−ブトキシ−3−ピリジニル)−3−エチル−2−(1−エチル−3−アゼチジニル)−2,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン、5−(5−アセチル−2−プロポキシ−3−ピリジニル)−3−エチル−2−(1−イソプロピル−3−アゼチジニル)−2,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン、5−[2−エトキシ−5−(4−エチルピペラジン−1−イルスルホニル)ピリジン−3−イル]−3−エチル−2−[2−メトキシエチル]−2,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン、4−[(3−クロロ−4−メトキシベンジル)アミノ]−2−[(2S)−2−(ヒドロキシメチル)ピロリジン−1−イル]−N−(ピリミジン−2−イルメチル)ピリミジン−5−カルボキサミド、3−(1−メチル−7−オキソ−3−プロピル−6,7−ジヒドロ−1H−ピラゾロ[4,3−d]ピリミジン−5−イル)−N−[2−(1−メチルピロリジン−2−イル)エチル]−4−プロポキシベンゼンスルホンアミドなど、
・α−2−δリガンド、たとえば、ギャバペンチン、プレガバリン、3−メチルギャバペンチン、(1α,3α,5α)(3−アミノ−メチル−ビシクロ[3.2.0]ヘプタ−3−イル)−酢酸、(3S,5R)−3−アミノメチル−5−メチル−ヘプタン酸、(3S,5R)−3−アミノ−5−メチル−ヘプタン酸、(3S,5R)−3−アミノ−5−メチル−オクタン酸、(2S,4S)−4−(3−クロロフェノキシ)プロリン、(2S,4S)−4−(3−フルオロベンジル)−プロリン、[(1R,5R,6S)−6−(アミノメチル)ビシクロ[3.2.0]ヘプタ−6−イル]酢酸、3−(1−アミノメチル−シクロヘキシルメチル)−4H−[1,2,4]オキサジアゾール−5−オン、C−[1−(1H−テトラゾール−5−イルメチル)−シクロヘプチル]−メチルアミン、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸、(3S,5R)−3−アミノメチル−5−メチル−オクタン酸、(3S,5R)−3−アミノ−5−メチル−ノナン酸、(3S,5R)−3−アミノ−5−メチル−オクタン酸、(3R,4R,5R)−3−アミノ−4,5−ジメチル−ヘプタン酸、(3R,4R,5R)−3−アミノ−4,5−ジメチル−オクタン酸など、
・カンナビノイド、
・代謝調節型グルタミン酸サブタイプ1受容体(mGluR1)拮抗薬、
・セロトニン再取込み阻害剤、たとえばセルトラリン、セルトラリン代謝産物のデメチルセルトラリン、フルオキセチン、ノルフルオキセチン(フルオキセチンデスメチル代謝産物)、フルボキサミン、パロキセチン、シタロプラム、シタロプラム代謝産物のデスメチルシタロプラム、エスシタロプラム、d,l−フェンフルラミン、フェモキセチン、イフォキセチン(ifoxetine)、シアノドチエピン(cyanodothiepin)、リトキセチン、ダポキセチン、ネファゾドン、セリクラミン、トラゾドンなど、
・ノルアドレナリン(ノルエピネフリン)再取込み阻害剤、たとえば、マプロチリン、ロフェプラミン、ミルタゼピン(mirtazepine)、オキサプロチリン、フェゾラミン、トモキセチン、ミアンセリン、ブプロプリオン、ブプロプリオン代謝産物のヒドロキシブプロプリオン、ノミフェンシン、ビロキサジン(Vivalan(登録商標))など、特に選択的ノルアドレナリン再取込み阻害剤、たとえばレボキセチン、特に(S,S)−レボキセチンなど、
・セロトニン−ノルアドレナリン再取込み二重阻害剤、たとえば、ベンラフェキシン、ベンラフェキシン代謝産物のO−デスメチルベンラフェキシン、クロミプラミン、クロミプラミン代謝産物のデスメチルクロミプラミン、デュロキセチン、ミルナシプラン、イミプラミンなど、
・誘導型一酸化窒素合成酵素(iNOS)阻害剤、たとえば、S−[2−[(1−イミノエチル)アミノ]エチル]−L−ホモシステイン、S−[2−[(1−イミノエチル)−アミノ]エチル]−4,4−ジオキソ−L−システイン、S−[2−[(1−イミノエチル)アミノ]エチル]−2−メチル−L−システイン、(2S,5Z)−2−アミノ−2−メチル−7−[(1−イミノエチル)アミノ]−5−ヘプテン酸、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)−ブチル]チオ]−5−クロロ−3−ピリジンカルボニトリル、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)ブチル]チオ]−4−クロロベンゾニトリル、(2S,4R)−2−アミノ−4−[[2−クロロ−5−(トリフルオロメチル)フェニル]チオ]−5−チアゾールブタノール、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)ブチル]チオ]−6−(トリフルオロメチル)−3ピリジンカルボニトリル、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)ブチル]チオ]−5−クロロベンゾニトリル、N−[4−[2−(3−クロロベンジルアミノ)エチル]フェニル]チオフェン−2−カルボキサミジン、グアニジノエチルジスルフィドなど、
・ドネペジルなどのアセチルコリンエステラーゼ阻害剤、
・プロスタグランジンE2サブタイプ4(EP4)拮抗薬、たとえば、N−[({2−[4−(2−エチル−4,6−ジメチル−1H−イミダゾ[4,5−c]ピリジン−1−イル)フェニル]エチル}アミノ)−カルボニル]−4−メチルベンゼンスルホンアミドや4−[(1S)−1−({[5−クロロ−2−(3−フルオロフェノキシ)ピリジン−3−イル]カルボニル}アミノ)エチル]安息香酸など、
・ロイコトリエンB4拮抗薬、たとえば、1−(3−ビフェニル−4−イルメチル−4−ヒドロキシ−クロマン−7−イル)−シクロペンタンカルボン酸(CP−105696)、5−[2−(2−カルボキシエチル)−3−[6−(4−メトキシフェニル)−5E−ヘキセニル]オキシフェノキシ]−吉草酸(ONO−4057)、DPC−11870など、
・5−リポキシゲナーゼ阻害剤、たとえば、ジロートン、6−[(3−フルオロ−5−[4−メトキシ−3,4,5,6−テトラヒドロ−2H−ピラン−4−イル])フェノキシ−メチル]−1−メチル−2−キノロン(ZD−2138)、2,3,5−トリメチル−6−(3−ピリジルメチル),1,4−ベンゾキノン(CV−6504)など、
・リドカインなどのナトリウムチャネル遮断薬、
・オンダンセトロンなどの5−HT3拮抗薬、
ならびにこれらの薬学的に許容できる塩および溶媒和物。
【0194】
式(I)の化合物のPDE7阻害能は、以下のアッセイプロトコルを使用して測定することができる。
【0195】
PDE7AおよびPDE7B酵素は、3’,5’−環状アデノシン一リン酸(cAMP)を5’アデノシン一リン酸酸、すなわち5’AMPにする加水分解を触媒する。マルチウェルプレートにおいて、PDE酵素、[3H]−cAMP、および試験化合物を室温でインキュベートする。硫酸亜鉛を含有する市販のケイ酸イットリウムシンチレーション近接アッセイ(SPA)ビーズを加えて、インキュベートを終了する。ケイ酸イットリウムビーズは、直鎖状のヌクレオチドを優先的に結合し、したがって酵素反応の産物である[3H]−5’AMPがこのビーズに結合して、光シグナルを生成し、これがシンチレーション計数器によって検出される。生成されたシグナルの量は、生成された産物の量、すなわち酵素の活性と直接に相関する。酵素および基質を単独でインキュベートする場合に最大のシグナルが得られる。酵素を含有しないウェル、または最大上濃度の既知のPDE7A/B阻害剤を含有するウェルからのバックグラウンドシグナルを測定する。精製した各酵素バッチを品質管理下に置き、そのKm、Vmax、および比活性を動力学的研究から求めた後、化合物阻害研究で使用する。試験化合物による酵素の阻害を、最大反応およびバックグラウンド反応と比較して算出する。これらのデータを使用し、得られた最大値および最小値と比較して%阻害値を算出する。
【0196】
作用溶液の調製
以下の表1に示す成分から、1000mlの保存緩衝液を調製した。
【0197】
【表1】
【0198】
保存緩衝液を室温でpH7.4に調整し、次いで0.2μmフィルターで濾過した。保存緩衝液は、4℃で調製日から1ヶ月間安定である。
【0199】
実験日に、ウシ血清アルブミン(BSA、Sigmaから入手可能)を必要な体積の緩衝液に加えて、0.00625%のBSA最終溶液を作った。これは、保存10%BSA溶液を次のように調製することにより実現した。
【0200】
保存10%BSA溶液の調製
1gのBSAを10mlの精製水に溶解させ、反転させて混合し、確実に均質になるようにし、適切にラベルを貼った管に100μl体積で等分した。10%BSA溶液は、−20℃で最長6ヶ月間安定である。
【0201】
一定分量の保存10%BSA保存液を保管所から取り出し、使用する前に室温で解凍して、以下の表2に示すようにBSA作用溶液を作った。
【0202】
10mlの作用BSAアッセイ緩衝液の調製
【0203】
【表2】
【0204】
標準化合物および対照の調製
WO02/074754の実施例75の化合物、すなわち5’−カルボキシプロポキシ−8’−クロロ−スピロ[シクロヘキサン−1−4’−(3’,4’−ジヒドロ)キナゾリン]−2’(1’H)−オン(以下では「化合物A」)を標準物質として使用した。
【0205】
100%DMSO中に調製した4mMの保存液は、4℃で保存することができる。DMSOの体積は、次のように算出することができる。
【0206】
【数1】
【0207】
30倍最大対照は、100%DMSOの溶液である。酵素活性をもたらさない30倍最小対照は、30μMの100%DMSO中化合物Aを使用して実現する。100%DMSO4.962mlを4mMの化合物A37.5μlに加えて、化合物Aの30μM溶液5mlを調製することができる。
【0208】
方法
アッセイ日に、前もって詳述したとおりに1倍の最終アッセイ緩衝液を調製し、必要になるまで氷上で保存した。
【0209】
動力学的研究
それぞれの新たな酵素バッチについて、Kmを求め、反応進行曲線の線形部分に残る間、45分間で約1000cpmのシグナルを得るのに必要な酵素の量を評価した。アッセイの過程の間に<10%の利用可能な[3H]−cAMPが加水分解されることが理想的である。
【0210】
酵素溶液
このアッセイの最適化は、全長PDE7AおよびPDE7B酵素を含有する細胞溶解液を使用して実施している。この細胞溶解物サンプル中の酵素の濃度は不明であるので、細胞溶解物の比活性を尺度として用いて、濃度/活性のバッチごとのどんな差異にもかかわらず、確実にウェルあたり同一の活性が使用されるようにする。
【0211】
PDE7A/B酵素の調製
PDE7保存酵素を調製し、凍結/解凍サイクルの数を減らすために適切にサイズ分けされた一定分量にして−20℃で保存した。以下の表3は、9mlのPDE7A/B酵素溶液を生成するのに必要な体積を示す。PDE7Aは1/8000に、PDE7Bは1/10000に希釈する。
【0212】
【表3】
【0213】
酵素溶液を調製したならば、それを使用前に氷上で保存した。
【0214】
50nMのアデノシン3’,5’環状リン酸(cAMP)基質溶液の調製
基質は、未標識cAMPとトリチウムで放射標識したcAMP([3H]−cAMP)の混合物から構成されるものである。[3H]−cAMP保存物の規格によって、使用する体積が決まる。
【0215】
1mCi/mlおよび24Ci/mmol(したがって41.66μM)である[3H]−cAMP保存物を使用する9mlの基質溶液の調製について以下で述べる。
【0216】
酵素バッチのKmは、これまでのところ以下のとおりである。
PDE7A−20nM PDE7B−100nM
【0217】
アッセイでは、15μlの基質溶液が、分注されて30μlの合計アッセイ体積になる、すなわちアッセイプレートで2倍希釈が行われる必要がある。
【0218】
約25nMの最終アッセイ[cAMP]が必要となるので、約50nMの[3H]−cAMPを調製した。
【0219】
10.8μlの[3H]−cAMP(Amershamから入手可能)と8975μlのアッセイ緩衝液とを混合して、9mlの基質溶液を調製した。
【0220】
3本のシンチレーションバイアルに15μlのサンプルを取って、cAMPの正確な濃度を決定した。次いで、4mlのStarscint(登録商標)(シンチレーションカクテル、Perkin Elmerから入手可能)を加え、管をdpmプログラムによるβカウンターでのカウントにかけた。
【0221】
放射性リガンドの濃度は、以下の式によって求める。
【0222】
【数2】
【0223】
次いで、濃度を2で割って、アッセイプレートで2倍希釈が行われていることを計算に入れる。
【0224】
6.6mg/mlのケイ酸イットリウムPDE SPAビーズの調製
ホスホジエステラーゼSPAビーズ(ケイ酸イットリウム)は、Amershamから入手可能である。
【0225】
製造者の推奨に従い、28mlの蒸留水または脱イオン水(約20mg/ml)を使用して、ビーズのバイアルを再構成した。再構成されたビーズは、2〜8℃で保存したとき、1ヶ月間安定である。アッセイ用のビーズを調製するために、再構成されたビーズを無菌二重蒸留水(約6.6mg/ml)中に3倍希釈した。ビーズを安定させることができるので、分注しながら一定にかき混ぜた/攪拌した(stirred/agitated)。
【0226】
約6.6mg/mlのビーズ30μlを30μlのアッセイに加えて、最終ビーズ濃度を約0.2mg/ウェルとする。
【0227】
化合物希釈物および「バックグラウンド」ウェルは、アッセイプレートにおいて必要となるものの30倍にしてあるので、1μlの化合物を29μlの他のアッセイ構成要素(14μlの酵素および15μlの放射性リガンド)によって希釈した。すなわち、最終アッセイ濃度10μMでは、化合物は、化合物添加プレートにおいて300μMでなければならない。化合物の4mM保存物は、100%DMSO中に供給される(または粉末提出物から4mMで作られる)。これには、DMSO中で1/13.33希釈を行う必要がある。
【0228】
アッセイプロトコル
1μlの試験化合物を、試薬のアッセイ付加の直前に好適なマルチウェルアッセイプレートに移し、次いで14μlの酵素溶液をアッセイプレートに加えた後、15μlの基質溶液を加えた(すなわち、最終アッセイ体積30μl、最終スクリーニング化合物濃度1μM)。次いで、プレートシーラーを使用してプレートをシールし、プレート振盪機に載せて室温で45分間インキュベートした。
【0229】
次いで、30μlのケイ酸イットリウムPDE4 SPAビーズを加え、ビーズを一定に攪拌してアッセイプレート中に確実に一様な分布が得られるようにした。次いで、プレートシーラーを使用してプレートをシールし、プレート振盪機に載せて室温で30分間インキュベートした。次いで、ビーズを30分間安定させた後、プレートを200gで1分間回転させた。
【0230】
次いで、好適な放射能カウンター、たとえばNXT−TopCount(商標)(Perkin Elmerから入手可能)によって、関連するプロトコル(ウェルあたり30秒の読み時間)を使用してプレートを読み取った。
【0231】
最小二乗アルゴリズムを使用して、データをシグモイド曲線に適合させた。
【0232】
次のCheng−Prussofの式を使用して、IC50値をKi値に変換した。
【0233】
【数3】
【0234】
実施例1〜7の化合物のPDE7阻害活性を、上記プロトコルに従って試験した。得られたKi値を以下の表4に示す。
【0235】
【表4】
【0236】
ヒト肝細胞データの概要
本出願の実施例1〜7の化合物のヒト肝代謝安定性を、以下で述べるモデルで評価した。最も近い現況技術を表すと考えられるWO02/074754の実施例75の化合物5’−カルボキシプロポキシ−8’−クロロ−スピロ[シクロヘキサン−1−4’−(3’,4’−ジヒドロ)キナゾリン]−2’(1’H)−オン(以下では「化合物A」)を比較として使用した。
【0237】
方法
肝細胞をin vitro系として使用して、肝代謝をモニターする。というのは、これらの無処置細胞は、シトクロムP450オキシダーゼ(CYP)、アルデヒドオキシダーゼ、モノアミンオキシダーゼ(MAO)などの第一相酵素、およびUDP−グルクロニルトランスフェラーゼやスルホトランスフェラーゼなどの第二相酵素を含む、in vivoで見出されるすべての肝酵素を含有している。凍結保存したヒト肝細胞を5人のドナーから調製し、WilliamsのE培地に懸濁させた。4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)を加えて最終濃度を50mMとし、pHを7.4に調整する。試験基質をDMSOに溶解し、肝細胞に加えて、基質濃度を1μMとし、インキュベートにおける最終DMSO濃度は0.1%より低くする。実験は、96または384ウェルプレートにおいて、肝細胞密度を50万生存細胞/mLとして37℃で実施する。10、20、30、60、90、および120分のサンプル採取時間を使用し、分析の定量化はLC−MS/MSによるものとする。次式を使用して、固有クリアランス(見かけ上)を算出する。
CLint,app=[−勾配/0.5M細胞/mL]・1000μL/mL=μL/分/M細胞
【0238】
本発明の化合物のヒト肝代謝安定性を、上記プロトコルに従って試験した。得られた固有クリアランス値を以下の表5に示す。
【0239】
【表5】
【0240】
上の表5に示すデータは、固有の肝代謝安定性に関して、本出願の実施例1〜7の化合物と、最も近い従来技術の化合物Aとの明らかな差異を示す。したがって、上記データに基づき、本出願の実施例1〜7の化合物は、肝クリアランスが低下しているので、化合物Aと比べてヒトにおける半減期の改善された化合物をもたらすと思われる。
【0241】
ラットIV薬物動態の概要
実施例1および2の化合物の薬物動態学的性質を、以下で述べるラットモデルで試験した。最も近い現況技術であると考えられるWO02/074754の実施例75の化合物5’−カルボキシプロポキシ−8’−クロロ−スピロ[シクロヘキサン−1−4’−(3’,4’−ジヒドロ)キナゾリン]−2’(1’H)−オン(以下では「化合物A」)を比較として使用した。
【0242】
方法
試験化合物を雄のラットに1mg/kg(実施例1の化合物については0.08mg/kg)の投与量で尾静脈から投与した(各ラットに1種類の化合物を与える)。投与後の所定の時点で、外科的に移植した頸静脈カニューレを介してラットから血液サンプルを抜き取り、遠心分離して血漿を生成した。血漿中の薬物を定量化するために、血漿サンプルを特定のLC−MS/MSアッセイによって分析した。各化合物の性質を理解するために、得られる血漿濃度−時間曲線を、非コンパートメントの薬物動態分析を使用して調べた。その後、得られる出力を使用して、確からしいヒト薬物動態プロフィールを以下で述べるとおりに推定した。
【0243】
実施例1および2の化合物ならびに化合物Aを1mg/kgで静脈内投与した後の、投与量で正規化した平均薬物動態プロフィールを以下の表6および図1に示す。
【0244】
【表6】
【0245】
表6では次の略語を使用する。
Clはラットクリアランスであり、
T1/2は半減期であり、
Vdはラットにおける分布体積であり、
fupはラット血漿中の非結合画分であり、
Cluは非結合ラット血漿クリアランスであり、Clu=CI/fupである。
【0246】
ヒト薬物動態の推定
ラットにおける上記薬物動態学的データに基づき、実施例1および2の化合物ならびに化合物Aの確からしいヒト薬物動態は、以下のとおりに推定できる。
【0247】
・以下の関係式を使用して、静脈内投与後に観察された非結合ラット血漿クリアランス(Cluラット)を基準化して、非結合ヒト血漿クリアランス(Cluヒト)を推定する。
Cluヒト=Cluラット*(BWヒト/BWラット)0.75
[式中、BWヒトおよびBWラットは、それぞれヒト(70kg)およびラット(0.25kg)の平均体重であり、クリアランスの単位はml/分である。]
【0248】
・Cluヒトをヒトにおける推定合計血液クリアランス(Clヒト)に変換する。
Clヒト=[(Cluヒト)*fup]/B:P
[式中、fupは、血漿中の非結合薬物の遊離画分であり、B:Pは、ヒト血液中の血液対血漿比である。]
【0249】
・次の関係式を使用して導かれるヒト半減期の推定値を各化合物について示す。
T1/2=[ln(2)*Vd]/Cl
[式中、T1/2は時間での推定ヒト半減期であり、Vdはヒトにおける分布体積であり(この系列の物理化学により0.2L/kgであると仮定される)、Clはヒトクリアランスである。]
【0250】
推定ヒト薬物動態の概要を以下の表7に示す。
【0251】
【表7】
【0252】
上記表6および7に示すデータでは、ラットにおける観察された薬物動態および推定ヒト薬物動態の両方に関して、従来技術の化合物Aと比べて、本出願の実施例2の化合物間に明らかな差異が示されている。このことは、約1時間のヒトでの半減期を示すと思われる化合物Aと比べて、実施例2の化合物では10時間であると推定された突出したヒト半減期に現れている。
【0253】
したがって、上記データに基づき、本出願の実施例2の化合物の薬物動態は、臨床現場では1日1回または2回の投薬に相応しいであろうと思われる。本出願の実施例1の化合物の薬物動態は、1日2回または3回の投薬に相応しいといえる。これは、その短い半減期のために同じようにして投与するのには適さないと思われる、最も近い従来技術の化合物Aに優る著しい進歩である。
【0254】
神経因性疼痛の治療における本発明による式(I)の化合物の活性は、以下の試験プロトコルに従って測定することができる。
【0255】
動物:雄のSprague Dawleyラット(平均体重500g)を12のグループにして収容する。動物はすべて、12時間の明/暗サイクル(7時間00分点灯)で、食物および水を自由に与えながら飼育する。実験はすべて、治療について知らない観察者によって、またHome Office Animals(Scientific Procedures)Act 1986に従って実施する。
【0256】
神経因性疼痛の慢性絞縮傷(CCI)ラットモデル
坐骨神経のCCIは、以前に記載されているとおりに実施する(G.J.BennettおよびY.K.Xie、Pain(1988年)第33巻、87〜107ページ)。動物を2%イソフルオラン(isofluorane)/O2混合物で麻酔した。右後部大腿を剪毛し、1%ヨウ素で拭く。次いで、手順を続ける間動物を恒温性のブランケットに移し、手術中はノーズコーンから麻酔を維持する。皮膚を大腿骨の線に沿って切開する。大腿二頭筋の鈍的切開によって総坐骨神経を大腿の中央で露出させる。神経の下に鉗子を挿入して、坐骨の三分岐の近位にある約7mmの神経をほどき、その神経を大腿から穏やかに引き上げる。鉗子を使用して、縫合糸を、神経をくぐらせて引っ張り、わずかな抵抗が感じられるまで一重結びで結び、次いで二重結びする。4本の結紮糸(4−0絹糸)が神経の周りに約1mm間隔で緩く結ばれるまで、この手順を繰り返す。切開を重ねて閉じ、創傷を局所用の抗生物質で処理する。
【0257】
ラットにおけるストレプトゾシン(STZ)誘発糖尿病ニューロパチー
0.9%の無菌食塩水に新たに溶解させたストレプトゾトシン(50mg/kg)の単回腹腔内投与によって、糖尿病を誘発する。ストレプトゾトシン注射は、少なくとも7週間持続する再現可能な機械的異痛症を3週間以内に誘発する(S.R.ChenおよびH.L.Pan.J.Neurophysiol.(2002年)、第87巻、2726〜2733ページ)。
【0258】
静的および動的な異痛症の評価
静的な異痛症
異痛症評価の前に、動物を底面がワイヤーの試験ケージに慣らす。静的な異痛症は、von Frey毛(Stoelting、米国イリノイ州ウッドデール)を弱い力から順に(0.6、1、1.4、2、4、6、8、10、15、および26グラム)後足の足底面に適用して評価する。各von Frey毛を、引っ込め応答が生じるまで最長で6秒間足に適用する。一度von Frey毛に対する引っ込め応答が確立したならば、引っ込め動作を生じたものより細いフィラメントから出発して、その後残りのフィラメントで強い力から順に引っ込め動作が生じなくなるまで足を試験し直す。最高の力である26gは、応答を誘発するだけでなく足を持ち上げ、すなわちカットオフポイントとなる。各動物の両方の後足をこのようにして試験する。応答を誘発するのに必要な最低量の力を足引っ込め動作閾値(PWT)としてグラムで記録する。動物が無処置のラットでは非侵害性である4g以下の刺激に応答したならば、静的な異痛症が存在すると定められている(M.J.Fieldら、Pain(1999年)、第83巻、303〜11ページ)。
【0259】
動的な異痛症
動的な異痛症は、後足の足底表面を綿棒で軽くなでて評価する。一般の運動能を記録しないようにするために、活発でなかった完全に慣らされたラットでこの手順を実施するように注意する。各時点で少なくとも2回の測定を行い、その平均値が足引っ込め動作待ち時間(PWL)となる。15秒以内に反応を示さない場合、手順を終了し、動物をその引っ込め動作時間に割り振る。痛み引っ込め応答はしばしば、繰り返される尻込み動作または足舐め動作を伴う。動物がなで始めてから8秒以内に綿による刺激に応答するならば、動的な異痛症が存在するとみなされる(Fieldら、1999年、前掲書)。
【実施例】
【0260】
1H核磁気共鳴(NMR)スペクトルは、すべての場合において、提案した構造と一致した。特徴的な化学シフト(δ)を、主要なピークの呼称については従来の略語、たとえばs:一重線、d:二重線、t:三重線、q:四重線、m:多重線、br:ブロードを使用して、テトラメチルシランから低磁場方向への百万分率(ppm)で示す。質量スペクトル(m/z)は、エレクトロスプレーイオン化(ESもしくはESI)または大気圧化学イオン化(APCI)のいずれかを使用して記録した。一般的な溶媒については次の略語、すなわち、CDCl3:重水素化クロロホルム、D6−DMSO:6重水素化ジメチルスルホキシドを使用した。
【0261】
単結晶X線回折実験
実施例2の化合物の酢酸溶媒和物の結晶構造を、Bruker SMART APEX単結晶X線回折計およびMo Kα放射線照射を使用する室温での単結晶X線回折によって決定した。SMART v5.622(制御)およびSAINT v6.02(積分)ソフトウェア(Bruker AXS Inc.、米国ウィスコンシン州マディソン、1994年)を使用して、ωにおいて各照射が0.3°をカバーする数本の照射から、照射時間を60秒とし、合計データセットを半球より多くして強度を積分した。マルチスキャン法(SADABS、エリアディテクターデータの基準化および補正のためのプログラム、G.M.Sheldrick、ゲッティゲン大学、1997年;R.H.Blessing、Acta Cryst.1995年、A51、33〜38ページの方法に基づくもの)を使用して、吸収に関してデータを補正した。
【0262】
結晶構造は、空間群C2/cにおいてSHELXS−97(結晶構造解明のためのプログラム、G.M.Sheldrick、ドイツ国ゲッティゲン大学、1997年、リリース97−2)を使用する直接的な方法によって首尾よく解明し、SHELXL−97(結晶構造高精度化のためのプログラム、G.M.Sheldrick、ドイツ国ゲッティゲン大学、1997年、リリース97−2)を使用する最小二乗法によって精度を高めた。この結晶構造高精度化手順によって、非対称ユニット内に実施例2の化合物一分子と酢酸一分子が存在することが明らかになった。したがって、この構造は、実施例2の化合物の1:1の酢酸溶媒和物と呼ぶことができる。
【0263】
実施例2ステップ(b)(酢酸溶媒和物)の結晶構造からの粉末X線回折パターンの算出
実施例2の化合物の酢酸溶媒和物の単結晶構造から、Accelrys MS Modelling(商標)[バージョン3.0]の「反射粉末回折」モジュールを使用して、2θ角度および相対強度(以下の表8を参照されたい)を算出した。関連するシミュレーションパラメータは以下のとおりである。
波長=1.5406Å(Cu Kα)
偏光因子=0.5
Pseudo−Voigtプロフィール(U=0.01、V=−0.001、W=0.002)
【0264】
算出したパターンは、単結晶構造から得られるので、実施例2化合物の酢酸溶媒和物の純粋な相のパターンを表す。算出したパターンと予測したパターンの比較を図2に示すが、大部分に単結晶構造が表われていることを示している。ピーク強度のわずかな不一致は、測定したパターンにおける好ましい配向効果のためであるとすることができる。
【0265】
粉末X線回折
自動サンプル変更装置、θ−θゴニオメーター、自動ビームダイバージェンススリット、およびPSD Vantec−1検出器を装着したBruker−AXS Ltd.D4粉末X線回折計を使用して、実施例2の化合物の溶媒和していない結晶形態(形態A)の粉末X線回折パターンを決定した。正確なピーク位置を測定するために、最初にケイ素でドープ処理することによって、分析用のサンプルを調製し、その後低バックグラウンドのシリコンウェーハ検体取付台に載せた。検体を回転させながら、X線管を40kV/30mAで作動させて銅K−α1X線(波長=1.5406オングストローム)を照射した。分析は、ゴニオメーターを、2°〜55°の2θ範囲にわたり、0.018°の1ステップあたり0.2秒のカウントにセットした連続的な方式で作動させて実施した。真空乾燥した溶媒和物サンプルのPXRDパターンも、この回折計で同じパラメータを使用して収集したが、これらのサンプルはケイ素でドープ処理しなかった。
【0266】
他の粉末X線回折パターンはすべて、平らなシリコンウェーハに載せたサンプルについて、Gobel光学鏡、単一のサンプルステージ、および高感度位置検出器(PSD)を装着したBruker AXS Ltd.D8 Advance粉末X線回折計を使用して記録した。X線管を40kV/40mAで作動させて、各検体に銅K−α1X線(波長=1.5406Å)を照射した。分析は、ゴニオメーターを、3°〜35°の2θ範囲にわたり、0.014°の1ステップあたり0.2秒のカウントにセットした連続的な走査方式で作動させて実施した。
【0267】
示差走査熱量測定(DSC)
DSC測定は、Perkin Elmer Diamond示差走査熱量計を使用して行った。サンプルを50μl容脱気孔付きアルミニウム受皿において周囲温度から300℃に20℃/分で加熱した。流動ガスは40ml/分の窒素とした。
【0268】
熱重量分析(TGA)
溶媒和物のTGA測定は、TA Instruments TGA2950 Hi−Res熱重量分析装置を使用して、窒素パージガスを75cm3/分の速度で使用しながら、20℃/分の加熱速度で周囲温度から脱溶媒和温度(150℃〜180℃)で行った。次いで、サンプルをその後のPXRD分析に向けて室温に冷却した。
【0269】
(実施例1)
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0270】
【化10】
調製例8のアルコール(50mg、0.14mmol)を99.25:0.75のアセトニトリル:水(2ml)に溶かした溶液に、過ヨウ素酸(82mg、0.359mmol)および酸化クロム(VI)(1.6mg、0.016mmol)を99.25:0.75のアセトニトリル:水(2ml)に溶かした溶液を加え、反応温度を5℃より低く保った。反応混合物を室温で18時間攪拌した。反応混合物を濾過し、残渣を99.25:0.75のアセトニトリル:水、2N塩酸:メタノール(5:1)、水、およびメタノールで洗浄した。残渣を真空乾燥して、表題化合物を白色固体(28mg、0.077mmol、55%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.17(m,1H)、1.40〜1.65(m,5H)、1.79(m,2H)、2.16(m,2H)、2.48(m,2H)、2.72(m,3H)、4.64(m,1H)、6.43(d,1H)、7.0(s,1H)、7.21(d,1H)、7.90(s,1H)、12.26(bs,1H)。
LRMS m/z(APCI):365[M+H]+、406[M+CH3CN+H]+
【0271】
(実施例2)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0272】
【化11】
方法A
調製例11のアルコール(2.05g、5.84mmol)を0.75%の水を含有するアセトニトリル(50ml)に溶かした溶液に、酸化クロム(VI)(12mg、0.11mmol)および過ヨウ素酸(3.33g、14.6mmol)の溶液を加え、反応混合物を40℃で96時間攪拌した。水(100ml)を加え、懸濁液を2時間攪拌した。得られる沈殿を濾過によって収集し、水で洗浄し、真空乾燥して、表題化合物(1.90g、5.2mmol、89%)を得た。
1H−NMR(400MHz、D6−DMSO):δ1.2(m,1H)、1.2(m,2H)、1.6(m,2H)、1.8(m,2H)、2.3(m,2H)、2.6(m,2H)、3.1(m,1H)、3.2(s,1H)、4.0(bs,1H)、4.8(m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.9(s,1H)。
LRMS m/z(APCI)365[MH]+
【0273】
方法B
ステップ(a)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸t−ブチルエステル
【0274】
【化12】
調製例27ステップ(b)の化合物(34.1g、130mmol)をDMF(300ml)に懸濁させ、スラリーを35℃に温めた。炭酸セシウム(63g、190mmol)を一度に加えた。調製例22の化合物をDMF(90ml)に溶解させ、反応液に加えた。反応液を1時間かけて90℃に加熱し、8時間維持した。反応液を73℃に冷却し、水(160ml)を加え、その間温度は65℃より高く保った。得られるスラリーを35℃に冷却し、その後酢酸エチル(260ml)を一度に加えた。室温に冷却した後、スラリーを濾過し、生成物を酢酸エチル(2×100ml)で洗浄した。得られる白色固体を60℃で16時間真空乾燥して、表題化合物を白色固体(44.4g、105mmol、82%)として得た。
1H−NMR(300MHz、CDCl3):δ1.3(m,1H)、1.4(m,1H)、1.5(s,9H)、1.6(m,1H)、1.7(bm,1H)、1.8(bm,2H)、1.8(bm,1H)、2.4(m,2H)、2.6(t,2H)、2.7(m,2H)3.1(m,1H)、3.2(m,1H)、4.8(m,1H)、5.5(s,1H)、6.2(d,1H)、6.9(s,1H)、7.1(d,1H)。
LC−MS(ESI):22.6分 11.8(%){シス異性体}m/z 422[MH+]、23.1分 88.2(%){トランス異性体}m/z 422[MH+]。
【0275】
ステップ(b)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸酢酸溶媒和物
【0276】
【化13】
ステップ(a)の生成物(207g、0.492 mol)を酢酸(3100ml)中にスラリー化し、60℃に加熱した。温度を60℃に保ちながら、48%の臭化水素酸(4.93mol)を滴下した。溶液を60℃で30分間攪拌した。温度を55℃より高く保ちながら、水(700ml)を滴下した。スラリーを20℃に冷却し、さらに30分間攪拌し、その後これを濾過し、酢酸:水(2000ml)および水(1000ml)で洗浄した後、真空オーブンに入れて60℃で一晩乾燥させて、表題化合物の白色固体を酢酸溶媒和物(153.6g、73.5%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.2(m,1H)、1.2(m,2H)、1.6(m,2H)、1.8(m,2H)、1.9(s,3H,CH3COOH)、2.3(m,2H)、2.6(m,2H)、3.1(m,1H)、3.2(s,1H)、4.0(bs,1H)、4.8 (m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.9(s,1H)、9.9 (s,1H,CH3COOH)。
LC−MS(ESI):18.0分 1.65(%){シス異性体}m/z 365[MH+]、
18.3分 98.4(%){トランス異性体}m/z 365[MH+]。
【0277】
上記酢酸溶媒和物は、単結晶X線回折の方法によって決定されたその結晶構造からわかるように、1:1の化学量論比で結晶化することがわかった。図2は、上記酢酸溶媒和物バッチについて測定した粉末X線回折(PXRD)パターン(A)、ならびに単結晶構造から予測したシミュレーションによるPXRDパターン(B)を示す。2つのパターンのピーク位置が非常によく一致することがわかる。回折ピークの相対強度および幅に差異があれば、それぞれ好ましい配向効果および粒径効果のためであるとすることができる。上記酢酸溶媒和物についての特徴的な2θX線回折ピークおよびその相対強度を以下の表8で一覧にする。
【0278】
【表8】
【0279】
酢酸溶媒和物は、熱重量分析(TGA)によって示されるように、115℃付近で熱によって脱溶媒することがわかったが、TGAでは、この温度で1モル当量の酢酸に等しい約15%のはっきりした重量減少が認められる。TGAプロットを図3に示す。上記溶媒和物は、脱溶媒すると、図4に示すPXRDプロットによって示されるように、再結晶化して(以下のステップ(c)に記載の)脱溶媒和形態Aになる。
【0280】
ステップ(c)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
ステップ(b)の酢酸溶媒和物(157g、369mmol)を室温で一晩かけて水(5200ml)中にスラリー化した。次いで、スラリーを濾過し、水(4×500ml)で洗浄した後、真空オーブンに入れて60℃で一晩乾燥させて、溶媒和していない表題化合物を白色固体(130g、357mmol、96%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.2(m,1H)、1.2(m,2H)、1.6(m,2H)、1.8(m,2H)、2.3(m,2H)、2.6(m,2H)、3.1(m,1H)、3.2(s,1H)、4.0(bs,1H)、4.8(m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.9(s,1H)。
LRMS m/z(APCI)365[MH]+
【0281】
この化合物は、図5に示す特徴的な粉末X線回折(PXRD)パターンを有する溶媒和していない形態(形態A)で結晶化することもわかった。特徴的な2θX線回折ピークおよびその相対強度を以下の表9で一覧にする。この結晶形態は、図6で例示する示差走査熱量測定(DSC)によって求められるとおり、融点が250℃である。
【0282】
【表9】
【0283】
実施例2の化合物は、ジメチルアセトアミド(DMAC)、ピリジン、テトラヒドロフラン(THF)、およびジメチルスルホキシド(DMSO)との溶媒和物として結晶化することもわかった。これらの溶媒和物はそれぞれ、図7に示すような特徴的なPXRDパターンを有する。
【0284】
これらの溶媒和物のTGA測定では、ピリジンおよびTHF溶媒和物が1:1の化学量論比を有することが明らかになったが(図8および9)、DMAC溶媒和物は、2:1の溶媒対化合物化学量論比を有することが示された(図10)。DMSO溶媒和物は脆い性質であるので、その化学量論比は求められないということになった。
【0285】
ピリジン、THF、DMAC、およびDMSO溶媒和物はそれぞれ、脱溶媒すると、それぞれ図11、12、13、および14に示されるPXRD分析に示すように、無水形態Aに再結晶化する。
【0286】
(実施例3)
3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
【0287】
【化14】
(a)3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸ベンジルエステル
【0288】
【化15】
8’−フルオロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(140mg、0.56mmol)(WO2004/026818に記載のとおりに調製したもの、中間体c)および炭酸セシウム(301mg、0.925mmol)をDMF(2ml)中で合わせ、調製例15の化合物(220mg、0.588mmol)のDMF(2ml)溶液を加え、混合物を80℃で18時間攪拌した。次いで、水(35ml)を加え、生成物を酢酸エチル(2×25ml)で抽出した。有機抽出物を合わせて飽和ブラインで洗浄し、硫酸マグネシウムで乾燥させた。溶媒を蒸発させると、褐色のゴム質である表題化合物がシス異性体とトランス異性体の約5:4の混合物(204mg、80%)として得られた。
1H−NMR(400MHz、CDCl3):δ1.29(m,1H)、1.66(m,7H)、2.21(m,2H)、2.41(m,2H)、2.59(m,2H)、2.8&2.90(2×m,1H)、3.2(m,1H)、3.96&4.00(2×d,2H)、5.13(2×s,2H)、6.6(m,1H)、6.95(m,1H)、7.33(m,5H)。
LRMS m/z(ES)453[MH]+
【0289】
(b)3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
ステップ(a)の生成物(200mg、0.442mmol)をメタノール(2ml)に溶解させ、2M NaOH(2ml、4.0mmol)を加え、褐色の乳濁液を60℃で1.5時間攪拌した後冷却した。2N HCl(2ml、4.0mmol)を加え、得られる懸濁液を1.5時間攪拌した。クリーム色の固体を濾過によって収集し、水でよく洗浄して、真空乾燥後に表題化合物(136mg、85%)を得た。
【0290】
Chiralpak AD−H、15%イソプロパノール:85%ヘキサン+0.1%トリフルオロ酢酸でのキラルHPLCによって、43:57の異性体比率が示される(保持時間13.69分および15.27分)。
1H−NMR(400MHz、CDCl3):δ1.16(m,1H)、1.43(m,2H)、1.57(m,3H)、1.76(m,2H)、2.05(m,2H)、2.28(m,2H)、2.43(m,2H)、2.68(2×m,1H)、3.03(2×m,1H)、3.88&3.97(2×d,2H)、6.48(m,1H)、6.76(m,1H)、6.98(m,1H)、8.79(s,1H)、12.06(br,1H)。
LRMS m/z(ES)363[MH]+
【0291】
(実施例4)
トランス−3−[(8’−シアノ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0292】
【化16】
調製例16の化合物(101mg、0.296mmol)から出発し、酸化クロム(VI)(0.5mg、0.005mmol)および過ヨウ素酸(167mg、0.733mmol)を使用する実施例1の方法によって、表題化合物(76mg、72%)が得られた。
1H−NMR(400MHz、D6−DMSO)δ:1.1〜1.85(m,8H)、2.3〜2.7(m,6H)、3.10(m,1H)、4.92(m,1H)、6.47(d,1H)、7.14(s,1H)、7.51(d,1H)、8.51(s,1H)。
LC−MS:保持時間=2.49分(100%)、LRMS(ESI)m/z 356[MH+]
【0293】
(実施例5)
1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
【0294】
【化17】
(a)1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸メチルエステル
【0295】
【化18】
8’−フルオロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(120mg、0.48mmol)(WO2004/026818に記載のもの)の室温のDMF(1ml)溶液に、炭酸セシウム(234mg、0.72mmol)を加え、混合物を10分間攪拌した後、調製例18の化合物(172mg、0.58mmol)のDMF(1ml)溶液を加えた。反応混合物を18時間かけて80℃に加熱し、次いで室温に冷却した。混合物を酢酸エチル(20ml)および水(20ml)で希釈し、有機層を分離し、水層を酢酸エチル(20ml)で抽出した。有機層を合わせて水(2×20ml)およびブライン(2×20ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。淡褐色の油状物をジエチルエーテル(10ml)から摩砕し、表題化合物を淡褐色の固体として得た(0.3molのDMFを含有する140mgの溶媒和物、0.35mmol、73%)。
1H−NMR(400MHz、D6−DMSO):δ1.16(m,1H)、1.35(m,2H)、1.49(m,3H)、1.72(m,2H)、1.90(m,1H)、2.03(m,3H)、2.26(m,2H)、2.42(m,2H)、3.60(s,3H)、4.22(s,2H)、6.52(dd,1H)、6.79(s,1H)、7.01(t,1H)、8.85(s,1H)。
【0296】
(b)1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
ステップ(a)の生成物(140mg、0.35mmol)をメタノール/水(1:1、2ml)に溶かした部分溶液に、水酸化ナトリウム(28mg、0.70mmol)を加え、反応液を50℃で24時間攪拌した。混合物を室温に冷却し、さらに6日間攪拌した後、2N塩酸水溶液(2ml)で処理した。得られるクリーム色の固体を濾過によって収集し、水で洗浄し、真空乾燥して、表題化合物(83mg、0.23mmol、65%)を得た。
1H−NMR(400MHz、D6−DMSO):δ1.25(m,1H)、1.37(m,2H)、1.49(m,3H)、1.71(m,2H)、1.90(m,1H)、2.01(m,3H)、2.38(m,4H)、4.18(s,2H)、6.52(dd,1H)、6.71(s,1H)、7.00(dd,1H)、8.78(s,1H)、12.43(s,1H)。
LRMS m/z(ESI)377[M+H]+
【0297】
(実施例6)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0298】
【化19】
(a)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸エチルエステル
【0299】
【化20】
調製例20の化合物(100mg、0.36mmol)、炭酸カリウム(57mg、0.41mmol)、および18−クラウン−6(110mg、0.41mmol)の80℃のジメチルホルムアミド(3ml)懸濁液に、3−(トルエン−4−スルホニルオキシ)−シス−シクロブタンカルボン酸エチルエステル(調製例22の化合物と類似の方法によって調製したもの)(123mg、0.41mmol)のジメチルホルムアミド(1ml)溶液を加え、反応混合物を80℃で18時間攪拌した。混合物を室温に冷却し、水(30ml)から酢酸エチル(2×20ml)に2回抽出した。有機層を合わせてブライン(2×20ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。油性の残渣をメタノールから再蒸発させ、ジエチルエーテルで摩砕して、表題化合物をクリーム色の固体(75mg、0.18mmol、51%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.19(t,3H)、1.43〜1.77(m,10H)、2.24(m,2H)、2.38(m,2H)、2.63(m,2H)、3.14(m,1H)、4.09(q,2H)、4.82(m,1H)、6.36(d,1H)、7.17(d,1H)、7.29(s,1H)、8.05(s,1H)。
LRMS m/z(ESI)407[MH]+
【0300】
(b)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
ステップ(a)の化合物(70mg、0.17mmol)のメタノール(1ml)懸濁液に、水酸化ナトリウム(14mg、0.35mmol)の水(1ml)溶液を加え、得られる懸濁液を40℃で2時間攪拌した。メタノールを真空中で除去し、得られる溶液のpHを、2N HCl水溶液(5ml)を滴下して約1に調整した。得られる固体を濾別し、イソプロパノール(1.5ml)で洗浄して、表題化合物を白色固体(25mg、0.066mmol、40%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.43〜1.77(m,10H)、2.23(m,2H)、2.34(m,2H)、2.61(m,2H)、3.06(m,1H)、4.81(q,1H)、6.36(d,1H)、7.17(d,1H)、7.29(s,1H)、8.05(s,1H)、12.35(s,1H)。
LRMS m/z(ESI)755[2M−H]−
【0301】
(実施例7)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0302】
【化21】
(a)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸t−ブチルエステル
【0303】
【化22】
(a)調製例24の化合物(300mg、1.14mmol)をDMF(3ml)に溶かした部分溶液に、炭酸セシウム(559mg、1.72mmol)を加え、反応混合物を10分間かけて40℃に加熱した後、調製例22の粗製の化合物(523mg、1.60mmol)のDMF(3mL)溶液を一度に加えた。反応混合物をさらに9時間かけて80℃に加熱し、室温に冷ました。次いで、反応混合物に水(3ml)を加えた後、酢酸エチル(5ml)を加え、得られる沈殿を収集しようとしたが成功しなかった。その後直ちに完全に溶解させ、反応混合物を真空中で2mlに濃縮し、水(5mL)を加えて、結晶化を誘発し、得られる生成物を濾別し、真空乾燥して、表題化合物(310mg、0.76mmol、67%)を得た。LC−MSによって、10%の出発材料フェノールが残存することが示された。この材料をそれ以上精製せずにステップ(b)で使用した。
LRMS m/z(ESI)407[M+H]+
【0304】
(b)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
ステップ(a)の生成物(310mg、0.76mmol)の60℃の酢酸(3ml)溶液に、48%の臭化水素酸水溶液(0.5ml)を加え、反応液を室温で30分間攪拌した。わずかな濁りが認められるまで水(0.1ml)を滴下して、混合物を失活させた。反応液を室温に冷ました後、得られる沈殿を濾別して、淡褐色の固体(130mg)を得た。酢酸(1.5ml)/水(0.1ml)で再結晶化することにより精製を実現して、表題化合物をオフホワイトの固体(35mg、0.09mmol、9%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.70(m,4H)、1.82(m,2H)、2.27(m,2H)、2.60(m,2H)、3.04(m,2H)、4.81(m,1H)、6.35(d,1H)、7.18(d,1H)、7.30(s,1H)、7.98(s,1H)、12.14(s,1H)。
LRMS(ESI)m/z 351[M+H]+
【0305】
調製例
調製例1
3−[(ベンジルオキシ)メチル]−2,2−ジクロロシクロブタノン
【0306】
【化23】
亜鉛末(6.54g、0.1mol)を水(30ml)に懸濁させ、アルゴンを懸濁液に15分間バブルした後、硫酸銅(II)(780mg、3.1mmol)を加えた。反応混合物をアルゴン中にて室温で30分間攪拌した。アルゴン流中で混合物を濾別し、固体を水(100ml)、アセトン(100ml)で洗浄し、4時間真空乾燥した。得られる亜鉛/銅の対をアルゴン中でエチルエーテル:1,2−ジメトキシエタン(70ml:10ml)に懸濁させ、アリルベンジルエーテル(4.6ml、30mmol)を加えた。塩化トリクロロアセチル(9ml、81mmol)のジエチルエーテル:1,2−ジメトキシエタン(58ml:7ml)溶液を45分間かけて滴下し、反応混合物を48時間加熱還流した。反応混合物をCelite(登録商標)で濾過し、塩をジエチルエーテル(3×70ml)で洗浄した。濾液を真空中で蒸発させ、残渣をヘキサン(150ml)に溶解し直した。残りの固体を濾過によって除去し、濾液を炭酸水素ナトリウムの飽和水溶液(2×100ml)、ブライン(80ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、10〜25%のヘキサン:ジエチルエーテルを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物が黄色の油状物(7.03g、27.3mmol、91%)として得られた。
1H−NMR(CDCl3、400MHz):δ3.11〜3.21(m,2H)、3.48(m,1H)、3.70(m,1H)、3.85(m,1H)、7.35(m,5H)、4.58(s,2H)。
【0307】
調製例2
3−[(ベンジルオキシ)メチル]シクロブタノン
【0308】
【化24】
調製例1のジクロロシクロブタノン(5.98g、23.08mmol)を塩化アンモニウムで飽和させたメタノール(90ml)に溶かした溶液に、亜鉛粉末(9.25g、142mmol)を加え、反応混合物を室温で2時間攪拌した。塩化アンモニウムを加え、反応混合物を室温でさらに6時間攪拌した。混合物をCelite(登録商標)で濾過し、塩をジエチルエーテル(50ml)で洗浄した。濾液を真空中で濃縮し、残渣をジエチルエーテル(200ml)と水(100ml)とに分配した。混合物を濾過し、有機相を水で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。表題化合物が黄色の油状物(3.7g、19.5mmol、84%)として得られた。
1H−NMR(CDCl3、400MHz):δ2.69(m,1H)、2.90(m,2H)、3.11(m,2H)、3.60(d,2H)、4.56(s,2H)、7.34(m,5H)。
【0309】
調製例3
シス−3−[(ベンジルオキシ)メチル]シクロブタノール
【0310】
【化25】
調製例2のシクロブタノン(1.166g、6.13mmol)をテトラヒドロフランに溶かした、−70℃で攪拌中の溶液に、トリ−s−ブチル水素化ホウ素リチウムの1Mテトラヒドロフラン(40ml)溶液を滴下し、反応温度を−65℃より低く保った。反応液を18時間かけて室温に温めた。反応混合物を炭酸水素ナトリウムの飽和水溶液(25ml)で失活させ、次いで5℃に冷却した。30%過酸化水素水溶液(4ml)を滴下し、反応温度を10℃より低く保った。混合物を水から酢酸エチル(50ml)中に抽出し、有機相を合わせてブライン(30ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、25〜50%の酢酸エチル:ペンタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、無色の油状物(1.05g、5.5mmol、89%)を得た。1H−NMRでは、15:1の比のシス:トランス異性体が得られたことが示された。
1H−NMR(CDCl3、400MHz):δ1.70(m,2H)、2.10(m,1H)、2.46(m,2H)、3.45(d,2H)、4.15(q,1H)、4.52(s,2H)、7.33(m,5H)。
【0311】
調製例4
4−ニトロ安息香酸トランス−3−[(ベンジルオキシ)メチル]シクロブチル
【0312】
【化26】
調製例3のシクロブチルアルコール(1.05g、5.47mmol)、4−ニトロ安息香酸(1.82g、10.9mmol)、およびトリフェニルホスフィン(3.016g、11.5mmol)をテトラヒドロフラン(20ml)に溶かした攪拌した溶液に、0℃でアゾジカルボン酸ジエチル(2g、11.5mmol)のテトラヒドロフラン(5ml)溶液を滴下した。反応混合物を室温で18時間攪拌した。溶媒を真空中で蒸発にさせ、残渣をジエチルエーテル(30ml)に溶解し直した。残りの固体を濾過によって除去し、濾液を真空中で蒸発させた。粗製材料を、1:10〜1:3の酢酸エチル:ペンタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、無色の油状物(1.64g、4.8mmol、88%)を得た。1H−NMRでは、15:1の比のトランス:シス異性体が得られたことが示された。
1H−NMR(CDCl3、400MHz):δ2.40(m,4H)、2.67(m,1H)、3.53(d,2H)、4.57(s,2H)、5.36(q,1H)、7.37(m,5H)、8.20(d,2H)、8.29(d,2H)。
【0313】
調製例5
トランス−3−[(ベンジルオキシ)メチル]シクロブタノール
【0314】
【化27】
調製例4のp−ニトロエステル(1.64g、4.8mmol)の1,4−ジオキサン(35ml)溶液に、水酸化ナトリウム(385mg、9.6mmol)の水(25ml)溶液を加え、反応混合物を室温で30分間攪拌した。酢酸(0.4ml、7mmol)を加え、混合物を真空中で濃縮した。残渣を、炭酸水素ナトリウムの飽和水溶液から酢酸エチル(20ml)中に抽出し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。表題化合物が黄色の油状物(850mg、4.4mmol、92%)として得られた。
1H−NMR(CDCl3、400MHz):δ2.08(m,2H)、2.20(m,2H)、2.47(m,1H)、3.47(d,2H)、4.39(q,1H)、4.52(s,2H)、7.34(m,5H)。
【0315】
調製例6
p−トルエンスルホン酸トランス−3−[(ベンジルオキシ)メチル]シクロブチル
【0316】
【化28】
調製例5のシクロブタノール(850mg、4.42mmol)をピリジン(5ml)に溶かした攪拌した溶液に、0℃で塩化p−トルエンスルホニル(1.18g、6.2mmol)を少量ずつ加え、反応混合物を室温で18時間攪拌した。溶媒を真空中で濃縮し、残渣酢酸エチル(30ml)に溶解し直し、2N塩酸(30ml)、炭酸水素ナトリウムの飽和水溶液(30ml)、ブライン(30ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、ジクロロメタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物が無色の油状物(1.53g、4.4mmol)として得られた。
1H−NMR(CDCl3、400MHz):δ2.15(m,2H)、2.31(m,2H)、2.44(s,3H)、2.49(m,1H)、3.4(d,2H)、4.49(s,2H)、4.93(q,1H)、7.32(m,7H)、7.75(d,2H)。
【0317】
調製例7
5’−({シス−3−[(ベンジルオキシ)メチル]シクロブチル}オキシ)−8’−クロロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0318】
【化29】
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(Bioorg.Med.Chem.Lett(2004年)、第14巻(18)、4627〜4632ページに記載のとおりに調製したもの)(640mg、2.4mmol)、炭酸カリウム(400mg、2.9mmol)、および18−クラウン−6(767mg、2.9mmol)をジメチルホルムアミド(8ml)中で合わせ、反応混合物を80℃に加熱した。調製例6のトシラート(1g、2.9mmol)のジメチルホルムアミド溶液を3回で加え、混合物をさらに18時間80℃で加熱した。反応混合物を酢酸エチル(100ml)と水(150ml)とに分配し、固体を濾過によって収集した。相を分離し、水相を酢酸エチルで抽出し直し、ブラインで希釈し、再び酢酸エチル中に抽出した。有機相を合わせて真空中で濃縮し、残渣を水およびメタノールで摩砕した。粗生成物を合わせて、ジクロロメタンからジクロロメタン:酢酸エチル(1:1)を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、表題化合物をオフホワイトの固体(685mg、1.156mmol、64%)として得た。
1H−NMR(D6−DMSO、400MHz):δ1.1(m,1H)、1.4(m,2H)、1.6(m,3H)、1.7(m,2H)、1.8(m,2H)、2.3(m,1H)、2.5(m,4H)、3.4(s,2H)、4.4(s,2H)、4.6(m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.3(m,5H)、7.8(s,1H)。
【0319】
調製例8
8’−クロロ−5’−{[シス−3−(ヒドロキシメチル)シクロブチル]オキシ}−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0320】
【化30】
調製例7のベンジルアルコール(400mg、0.9mmol)のジクロロメタン(10ml)懸濁液に、三塩化ホウ素−ジメチルスルフィド錯体の2Mジクロロメタン溶液(1.8ml、3.6mmol)を加え、反応混合物を室温で終夜攪拌した。炭酸水素ナトリウムの飽和水溶液(10ml)を加え、混合物を5分間攪拌した。ジクロロメタンおよび水を加え、得られる固体を濾過によって収集した。表題化合物が白色固体(230mg、0.657mmol、73%)として得られた。
1H−NMR(D6−DMSO、400MHz):δ1.17(m,1H)、1.42(m,2H)、1.57(m,3H)、1.82(m,4H)、2.05(m,1H)、2.45(m,4H)、3.38(t,2H)、4.58(m,2H)、6.41(d,1H)、6.99(s,1H)、7.20(d,1H)、7.86(s,1H)。LRMS m/z(APCI)351[MH]+
【0321】
調製例9
p−トルエンスルホン酸シス−3−[(ベンジルオキシ)メチル]シクロブチル
【0322】
【化31】
調製例3のアルコール(17g、88.4mmol)をジクロロメタン(90ml)に溶かした、5℃で攪拌中の溶液に、ピリジン(14.3ml、176mmol)および塩化p−トルエンスルホニル(20.2g、105.9mmol)を加え、反応混合物を室温で18時間攪拌した。反応混合物をジクロロメタン(50ml)で希釈し、2N塩酸(50ml)、炭酸水素ナトリウムの飽和水溶液(50ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、ペンタン:酢酸エチル(19:1、9:1、4:1)を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物が無色の油状物(24.8g、71.6mmol、81%)として得られた。
1H−NMR(CDCl3、400MHz):δ1.95(m,2H)、2.1(m,1H)、2.35(m,2H)、2.45(s,3H)、3.4(m,2H)、4.5(s,2H)、4.7(m,1H)、7.3(m,7H)、7.8(m,2H)。
LRMS m/z(ESI)347[MH]+
【0323】
調製例10
5’−({トランス−3−[(ベンジルオキシ)メチル]シクロブチル}オキシ)−8’−クロロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0324】
【化32】
方法A
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(500mg、1.87mmol)をジメチルホルムアミド(2ml)に懸濁させた攪拌した懸濁液に、炭酸セシウム(730mg、2.24mmol)を加え、反応混合物を80℃に加熱した。5分後、調製例9のトシラート(710mg、2.05mmol)のジメチルホルムアミド(1ml)溶液を加え、反応混合物を80℃で18時間加熱した。混合物をブライン(60ml)から酢酸エチル(1×80ml、2×30ml)中に抽出し、ブライン(3×100ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。表題化合物が、わずかに不純なクリーム色の固体(800mg、0.96mmol、96%)として得られた。
【0325】
方法B
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(950mg、3.56mmol)をジメチルホルムアミド(12ml)に溶かした、80℃で攪拌中の溶液に、炭酸カリウム(590mg、4.27mmol)および18−クラウン−6(1.1g、4.27mmol)を加えた。反応混合物を10分間攪拌した後、調製例9のトシラート(1.48g、4.27mmol)のジメチルホルムアミド(3ml)溶液を加えた。反応混合物を80℃で24時間加熱した。混合物を水:メタノール(75ml:25ml)上に注ぎ、10分間攪拌し、得られる沈殿を濾過によって収集し、メタノールで洗浄した。固体をジクロロメタンに溶解させ、Celite(登録商標)で濾過し、得られる濾液を真空中で蒸発にさせて、表題化合物をトランス:シス異性体の9:1混合物(887mg、2.0mmol、56%)として得た。
1H−NMR(CDCl3、400MHz):δ1.3(m,1H)、1.5〜1.9(m,9H)、2.4(m,3H)、2.6(m,2H)、3.5(d,2H)、4.6(s,2H)、4.75(m,1H)、5.85(bs,1H)、6.25(d,1H)、7.05(bs,1H)、7.1(d,1H)、7.3〜7.4(m,5H)。
LRMS m/z(ESI)441[MH]+
【0326】
調製例11
8’−クロロ−5’−{[トランス−3−(ヒドロキシメチル)シクロブチル]オキシ}−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0327】
【化33】
三塩化ホウ素−ジメチルスルフィド錯体をジクロロメタン(15ml)に溶かした2Mの溶液を、調製例10のベンジルエーテル(3.5g、7.9mmol)のジクロロメタン(80ml)溶液に滴下し、反応混合物を室温で18時間攪拌した。この混合物を、炭酸水素ナトリウムの飽和水溶液(200ml)中に注ぎ、泡立ちが止むまで攪拌した。混合物をジクロロメタン(1×200ml、2×100ml)中に抽出し、ブライン(50ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料をアセトニトリルから再結晶化して、表題化合物を91:9の比のトランス:シス生成物(2.33g、6.65mmol、84%)として得た。
1H−NMR(CDCl3、400MHz):δ1.3(m,1H)、1.5(m,2H)、1.8(m,5H)、2.4(m,4H)、2.6(m,3H)、3.8(d,2H)、4.8(m,1H)、5.7(bs,1H)、6.25(d,1H)、7.0(bs,1H)、7.1(d,1H)。
LRMS m/z(ESI)351[MH]+
【0328】
調製例12
3−メチレンシクロブタンカルボン酸
【0329】
【化34】
水酸化カリウム(17.37g、214.7mmol)を水(20ml)に溶解させ、エタノール(20ml)を加えた。この溶液を冷たいときに3−メチレン−シクロブタンカルボニトリル(5.0g、53.7mmol)に加え、得られる溶液を2.5時間加熱還流し、冷まし、真空中で蒸発にさせて、クリーム色の固体とした。固体を水(15ml)に溶解させ、氷浴で冷却し、濃HClを加えてpH1とし、ジエチルエーテル(3×20ml)で抽出した。エーテル抽出物を硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて、表題化合物を淡黄色の液体(5.6g、93%)として得た。
1H−NMR(CDCl3、400MHz):δ2.95(m,2H)、3.02(m,2H)、3.17(m,1H)、4.82(m,2H)(交換可能な1プロトンが見られなかった)。
【0330】
調製例13
3−メチレンシクロブタンカルボン酸ベンジルエステル
【0331】
【化35】
調製例12の生成物(1g、8.9mmol)の酢酸エチル(5ml)溶液に、1,1’−カルボニルジイミダゾール(1.59g、9.81mmol)の酢酸エチル(5ml)懸濁液を数回に分けて加えた。穏やかな泡立ちが認められた。混合物を約1.5時間室温で攪拌し、ベンジルアルコール(1.11ml、10.7mmol)を加え、終夜攪拌を続けた。溶液をジエチルエーテル(20ml)で希釈し、水(2×10ml)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて無色の液体とし、これを、ジクロロメタンを溶離液としながら10gのSiO2で濾過して精製して、表題化合物を無色の油状物(1.246g、69%)として得た。
1H−NMR(CDCl3、400MHz):δ2.92(m,2H)、3.02(m,2H)、3.16(m,2H)、4.80(m,2H)、5.15(s,2H)、7.36(m,5H)。
LRMS m/z(ESI)203[MH]+
【0332】
調製例14
3−(ヒドロキシメチル)シクロブタンカルボン酸ベンジルエステル
【0333】
【化36】
ボラン−ジメチルスルフィド(0.07ml、0.72mmol)をTHF(1ml)で希釈し、調製例13の化合物(300mg、1.48mmol)をTHF(1ml)に溶かした攪拌した溶液に室温で滴下した。無色の溶液を室温で1時間攪拌し、次いで過ホウ酸ナトリウム(145mg、1.78mmol)の水(1ml)溶液を、泡立ちを制御する速度で滴下した。加え終えたなら、混合物を1,4−ジオキサン(1ml)で希釈し、得られる溶液を60℃で1時間温め、水(5ml)を加えて失活させ、酢酸エチル(10ml)で抽出した。酢酸エチル抽出物を硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて、無色の油状物(226mg、69%)を得、これを次のステップでそれ自体として使用した。
1H−NMR(CDCl3、400MHz):δ2.05(m,2H)、2.33(m,2H)、2.45(m,1H)、3.11(m,1H)、3.62(dd,2H)、5.13(d,2H)、7.35(m,5H)。
【0334】
調製例15
3−(p−トルエンスルホニルオキシメチル)シクロブタン−1−カルボン酸ベンジルエステル
【0335】
【化37】
調製例14の化合物(275mg、1.25mmol)およびピリジン(0.26ml、3.25mmol)の攪拌した溶液に、室温で塩化p−トルエンスルホニル(309mg、1.62mmol)のジクロロメタン(2ml)溶液を滴下し、3日間攪拌を続けた。混合物をジクロロメタン(20ml)と水(2×20ml)とに分配し、ジクロロメタン抽出物を硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて無色の油状物とし、これを、ジクロロメタンから1:1のジエチルエーテル:ジクロロメタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、表題化合物(226mg、48%)を無色の油状物としてを得た。
1H−NMR(CDCl3、400MHz):δ2.02(m,2H)、2.25〜2.41(m,2H)、2.44(s,3H)、2.55〜2.75(m,1H)、3.07(m,1H)、4.00(2×d,2H)、5.10(d,2H)、7.34(m,7H)、7.78(m,2H)。
【0336】
調製例16
5’−{[トランス−3−(ヒドロキシメチル)シクロブチル]オキシ}−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−8’−カルボニトリル
【0337】
【化38】
調製例11の化合物(100mg、0.285mmol)のN−メチルピロリジノン(1.5mL)懸濁液に、シアン化ナトリウム(27.9mg、0.57mmol)を加えた後、臭化ニッケル(62.3mg、0.285mmol)を加え、反応混合物をマイクロ波反応器に入れて200℃で10分間加熱した。混合物をジエチルエーテル(2×20ml)と水(10mL)とに分配し、有機抽出物を合わせて硫酸マグネシウムで乾燥させ、真空中で濃縮して、表題化合物を淡い橙色/赤色の固体(27.8mg)として得た。
1H−NMR(400MHz、CDCl3):δ1.4〜1.85(m,9H)、2.3〜2.7(m,6H)、3.73(d,2H)、4.83(m,1H)、5.60(s,1H)、6.33(d,1H)、6.98(s,1H)、7.36(d,1H)。
LC−MS:保持時間=2.54分(100%)、LRMS m/z 342[MH+]
【0338】
調製例17
1−(ヒドロキシメチル)−シクロブタンカルボン酸メチルエステル
【0339】
【化39】
1,1−シクロブタンジカルボン酸ジメチルエステル(英国Lancaster Synthesis Ltdから入手可能)(2.0g、11.6mmol)のテトラヒドロフラン(20ml)溶液に、室温で水素化トリ−t−ブトキシアルミニウムリチウム(1Mのテトラヒドロフラン溶液25.5ml、25.5mmol)を10分間かけて滴下した。反応混合物を加熱して、3時間穏やかに還流させ、室温に冷却し、18時間攪拌した。得られる懸濁液を塩化アンモニウム飽和水溶液(30ml)で希釈し、15分間激しく攪拌してから濾過した。固体をジエチルエーテル(50ml)で洗浄し、有機層を分離し、水層をジエチルエーテル(50ml)で抽出した。有機抽出物を合わせて硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発にさせて、表題化合物を無色の油状物(1.8g)として得た。
【0340】
調製例18
1−(p−トルエンスルホニルオキシメチル)−シクロブタンカルボン酸メチルエステル
【0341】
【化40】
調製例17の粗製の化合物(1.6g、11.0mmol)のジクロロメタン(5ml)溶液に、塩化p−トルエンスルホニル(4.2g、22.0mmol)を加えた後、ピリジン(2.7ml、33.0mmol)を加え、溶液を室温で18時間攪拌した。次いで、反応混合物をジクロロメタン(30ml)で希釈し、2N HCl水溶液(2×25ml)、重炭酸ナトリウム飽和水溶液(50ml)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で蒸発させた。粗製の橙色油状物(5g)を、酢酸エチル:ペンタン(1:5)を溶離液とするシリカゲルでのフラッシュクロマトグラフィーによって精製して、表題化合物を無色の油状物(1.7g、5.7mmol、49%)として得た。
1H−NMR(CDCl3、400MHz):δ1.96(m,4H)、2.40(m,2H)、2.45(s,3H)、3.62(s,3H)、4.24(s,2H)、7.35(d,2H)、7.79(d,2H)。
【0342】
調製例19
8’クロロ−5’−メトキシ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−2’(3’H)−オン
【0343】
【化41】
2−クロロ−5−メトキシフェニル尿素(WO02/074754、中間体5)(17.9g、89.5mmol)のシクロヘプタノン(60ml、0.51mol)溶液を、100℃で20分間かけてポリリン酸(213g)に滴下し(127℃までの発熱が認められた)、1時間加熱した。混合物を攪拌しながら水(3リットル)および酢酸エチル(1リットル)中に注いだ。得られる固体を濾過によって収集し、酢酸エチルでよく洗浄し、乾燥させた。乾燥させた固体をクロロホルムに溶解させ、重炭酸ナトリウム水溶液で洗浄し、硫酸ナトリウムで乾燥させ、真空中で濃縮して、表題化合物を白色固体(10.2g、34mmol、39%)として得た。
【0344】
二相性の濾液を分離し、酢酸エチル相を水およびブラインで洗浄し、硫酸ナトリウムで乾燥させ、濃縮した。残渣をt−ブチルメチルエーテルで摩砕し、得られる白色固体を濾過によって収集し、別のt−ブチルメチルエーテルで洗浄し、乾燥させて、次の分の表題化合物(9.0g、30.6mmol、34%)を得た。
1H−NMR(400MHz、CDCl3):δ1.53〜1.84(複合,10H)、2.46(m,2H)、3.81(s,3H)、5.33(br,1H)、6.46(d,1H)、7.03(br,1H)、7.18(d,1H)。
LRMS m/z 295[M+H]+
【0345】
調製例20
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘプタン−1,4’−キナゾリン]−2’(3’H)−オン
【0346】
【化42】
調製例19の化合物(19.0g、64.6mmol)のジクロロメタン(500ml)溶液に、三臭化ホウ素の1Mジクロロメタン溶液(129ml、129mmol)を加え、混合物を室温で3日間攪拌した。反応混合物を水(1.5リットル)および酢酸エチル(1リットル)中に注いだ。白色固体を濾過によって収集し、相を分離し、有機相を硫酸ナトリウムで乾燥させ、真空中で濃縮して、褐色の固体を得た。濾過した白色固体をエタノールから再結晶化して、表題化合物(7.4g、26.4mmol、41%)を得た。再結晶からの母液を褐色の固体と合わせ、混合物をエタノール中で攪拌し、濾過し、乾燥させて、2番目のバッチの生成物(7.0g、25mmol、39%)を得た。
1H−NMR(400MHz、D6−DMSO):δ1.38〜1.78(複合,10H)、2.26(m,2H)、6.40(d,1H)、7.02(d,1H)、7.20(br,1H)、7.80(br,1H)、9.84(br,1H)。
【0347】
調製例21
シス−3−ヒドロキシシクロブチルカルボン酸t−ブチルエステル
【0348】
【化43】
方法A
3−オキソシクロブタンカルボン酸t−ブチル(J.Org.Chem.(1993年)第58巻、110ページ)(3.10g、18.2mmol)を窒素中でテトラヒドロフラン:メタノール(20:1、30ml)に溶解させ、5℃に冷却した。水素化ホウ素ナトリウム(345mg)を数回に分けて加え、得られる透明な溶液を5℃で15分間攪拌した後、水(135ml)、次いで酢酸エチル(135ml)を滴下して希釈した。水相を分離し、酢酸エチル(2×25ml)で洗浄した。有機相を合わせてブライン(20ml)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、表題化合物をかすかに黄色の油状物(3.05g、17.7mmol、97%)として得た。
1H−NMR(CDCl3、400MHz):δ1.46(s,9H)、2.12(m,2H)、2.55(m,3H)、4.17(m,1H)。
GC分析(アセトニトリル中サンプル):保持時間4.57分(91.5%面積)。
【0349】
方法B
3−オキソシクロブタンカルボン酸(J.Org.Chem.(1993年)第58巻、110ページ)(10.0g、88mmol)をジクロロメタン(20ml)に溶解させ、5℃に冷却した。4−ジメチルアミノピリジン(8.6g、70mmol)を少量ずつ加えた後、t−ブタノール(13.0g、176mmol)を一度に加えた。N,N’−ジシクロヘキシルカルボジイミドの1Mジクロロメタン(96ml、96mmol)溶液を、温度を0℃〜5℃の間に保ちながら滴下した。得られるスラリーを室温に温め、終夜攪拌した。濾過した後、濾液を5℃で2Mの塩酸(50ml)に滴下した。得られる相を分離し、下方の有機層を室温に温め、水(50ml)および重炭酸ナトリウム飽和溶液(50ml)で洗浄した。下方の有機相を蒸留によって濃縮し、溶媒をテトラヒドロフランと交換した。最終反応体積を30mlとした。メタノール(6ml)を加えた。その間、水素化ホウ素ナトリウム(1.65g、44mmol)をテトラヒドロフラン(39ml)に懸濁させ、5℃に冷却した。この中間体のテトラヒドロフラン溶液を、温度を0℃〜5℃の間に保ちながら水素化ホウ素ナトリウムのスラリーに滴下した。次いで、反応液を0〜5℃で2時間攪拌した。温度を0℃〜5℃の間に保ちながら水を滴下した。酢酸エチル(75ml)を加え、相を分離した。下方の水層を室温に温め、酢酸エチル(37ml)で洗浄した。有機層を合わせて濃縮し、ブライン(37ml)、次いで水(37ml)でさらに洗浄した。上方の有機層を真空中で抜き取って、表題化合物を黄色の油状物(10.1g、58.6mmol、67%)として得た。
1H−NMR(CDCl3、400MHz):δ1.46(s,9H)、2.12(m,2H)、2.55(m,3H)、4.17(m,1H)。
GC分析:保持時間9.02分(シス異性体)(87.5%面積)、保持時間9.07分(トランス異性体)(10.0%面積)。
【0350】
調製例22
3−(p−トルエンスルホニルオキシ)−シクロブタンカルボン酸t−ブチルエステル
【0351】
【化44】
方法A
調製例21の化合物(3.02g、17.35mmol)を窒素中でピリジン(15ml)に溶解させ、0℃に冷却した。塩化p−トルエンスルホニル(3.5g、18.4mmol)を一度に加え、溶液を室温で72時間攪拌した。得られる、白色の懸濁した材料を含有するピンク色の溶液を真空中で濃縮し、2N HCl水溶液(30ml)と酢酸エチル(30ml)とに分配し、水層を酢酸エチル(15ml)で再び洗浄した。有機層を合わせて2N HCl水溶液(15ml)、重炭酸ナトリウム飽和水溶液(15ml)、およびブライン(30ml)で洗浄し、真空中で濃縮して、表題化合物を橙色の油状物(5.15g、15.7mmol、90%)として得たが、この油状物は静置すると凝固した。
1H−NMR(400MHz、CDCl3):δ1.43(s,9H)、2.32〜2.57(複合,5H)、2.46(s,3H)、4.73(m,1H)、7.35(d,2H)、7.79(d,2H)。
LC−MS:シス異性体 保持時間21.78分(81%)、LRMS(ESI)m/z 327[MH+]、トランス異性体 保持時間22.08分(7.3%)、LRMS(ESI)m/z 327[MH+]。
【0352】
方法B
調製例21の化合物(10.0、58mmol)を窒素中でピリジン(25ml)に溶解させ、0℃に冷却した。塩化p−トルエンスルホニル(16.6g、87mmol)をピリジン(25ml)に溶解させ、温度を0℃〜5℃の間に保ちながら滴下した。次いで、反応液を室温に温め、終夜攪拌した。
【0353】
反応液を濃縮して固体にし、酢酸エチル(50ml)中にスラリー化した。スラリーを0〜5℃に冷却し、2Mの塩酸(75ml)で洗浄し、相を分離した。下方の水相を再び酢酸エチル(50ml)で抽出した。有機相を合わせて水(50ml)および炭酸水素ナトリウム溶液(50ml)で洗浄した。有機相を濃縮し、0〜5℃に冷却した。N,N−ジメチルエチレンジアミン(3.5g、40mmol)を滴下した。反応混合物を2Mの塩酸(75ml)で洗浄した。上方の有機相を水(75ml)およびブライン(75ml)で洗浄した。有機相を濃縮して淡黄色の油状物としたが、これを静置すると結晶化した(15.5g、47mmol、82%)。
1H−NMR(400MHz、CDCl3):δ1.43(s,9H)、2.32〜2.57(複合,5H)、2.46(s,3H)、4.73(m,1H)、7.35(d,2H)、7.79(d,2H)。
GC分析:保持時間15.98分(シス異性体)(94.1%面積)、保持時間15.82分(トランス異性体)(5.9%面積)。
【0354】
調製例23
8’クロロ−5’−メトキシ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−2’(3’H)−オン
【0355】
【化45】
2−クロロ−5−メトキシフェニル尿素(WO02/074754、中間体5)(22.04g、0.11mol)にEaton試薬(7.7重量%の酸化リン(V)のメタンスルホン酸溶液)(440.8ml)を加えた後、シクロペンタノン(19.5ml、0.22mol)を加え、得られる溶液を85℃で4時間加熱した。反応液を約5℃に冷却し、温度を20〜30℃の間に保ちながら慎重に水を加えた。次いで、ジクロロメタン(全体で400ml)およびブライン(200ml)を加え、相を分離した。水相をジクロロメタン(2×100ml)で洗浄し、有機抽出物を合わせ、真空中で蒸発にさせて、暗色の油状物を得、これを、ジクロロメタン:メタノール(95:5→90:10)を溶離液としながらシリカのクロマトグラフィーカラムで精製して、生成物を暗褐色の固体として得た。固体をジエチルエーテルおよびペンタンで摩砕し、濾過によって収集し、乾燥させて、表題化合物を褐色の固体(27.17g、0.1mol、92%)として得た。
1H−NMR(400MHz、CDCl3):δ1.7〜1.8(m,6H)、2.4〜2.5(m,2H)、3.7(s,3H)、5.75(br s,1H)、6.4(d,1H)、7.05(s,1H)、7.15(d,1H)。
LRMS m/z(APCI)267[M+H]+
【0356】
調製例24
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロペンタン−1,4’−キナゾリン]−2’(3’H)−オン
【0357】
【化46】
調製例23の化合物(25g、0.093mol)に、酢酸(250ml)を加えた後、48%の臭化水素酸水溶液(207ml、1.86mol)を一度に加え、得られる溶液を115℃で7日間攪拌した。反応混合物を100℃に冷却し、水(207ml)を滴下した。混合物を真空中で濃縮して、褐色の固体を沈殿させ、これを濾過によって収集し、水(2×100ml)で洗浄した。静置すると、濾液から次の分の生成物が得られた。合わせた分の生成物を、トルエン(150ml)とのスラリーによって、また真空中で3回溶媒を除去することにより乾燥させて、灰色の固体を得、これをシリカに予め吸収させ、ジクロロメタン:メタノール(98:2→95:5→80:20)を溶離液とするカラムクロマトグラフィーによって精製した。生成物の画分を真空中で濃縮し、得られる固体をペンタンで摩砕し、濾過して、表題化合物を褐色の固体(10g、0.0395mol、42%)として得た。
1H−NMR(400MHz、D6−DMSO)δ1.6〜1.8(m,6H)、2.3〜2.4(m,2H)、6.4(d,1H)、7.11(d,1H)、7.2(s,1H)、7.8(s,1H)、9.9(s,1H)。
LRMS m/z(ESI)253[M+H]+
【0358】
調製例25
(2−クロロ−5−メトキシフェニル)尿素
【0359】
【化47】
塩酸2−クロロ−5−メトキシアニリン(26g、134mmol)を酢酸(117ml)および水(13ml)に加えた。スラリーを30℃に温めた。シアン酸カリウム(13g、161mmol)の水(104ml)溶液を滴下した。40℃で1時間経過後、反応液を20℃に冷却し、濾過し、水(78ml)で洗浄した。生成物を60℃の真空中で一晩乾燥させて、表題化合物を白色固体(21.8g、81%)として得た。
1H−NMR(300MHz、D6−DMSO):δ3.71(s,3H)、6.41(s,2H)、6.53(d,1H)、7.26(d,1H)、7.85(s,1H)、7.98(s,1H)。
LC−MS(ESI):10.9分 99.4(%)、m/z 201[MH+]
【0360】
調製例26
8’−クロロ−5’−メトキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン
【0361】
【化48】
調製例25の化合物(5.0g、25mmol)をEaton試薬(7%w/wのP2O5メタンスルホン酸溶液)(150g)に加えて溶液を生成し、次いでこれを60℃に加熱した。シクロヘキサノン(4.9g、50mmol)を10分間かけて加え、次いで、反応液を80℃に温め、1時間その温度に保った。次いで、反応混合物を5℃に冷却し、水を加えた(150ml)。次いで、反応混合物をジクロロメタン(100ml)で抽出し、上方の水相をジクロロメタン(2×10ml)で洗浄した。有機相を合わせて濃縮した後、2−プロパノール(110ml)を加えた。反応混合物をさらに大気圧下で濃縮して、残りのジクロロメタンを除去した。反応液を冷却して生成物を結晶化し、5℃で1時間攪拌した。生成物を濾過によって収集し、2−プロパノール(15ml)で洗浄した後、50℃で18時間かけて乾燥させて、表題化合物を白色固体(5.2g、19mmol、76%)として得た。
1H−NMR(300MHz、D6−DMSO):δ1.2(m,1H)、1.4(m,2H)、1.5(m,1H)、1.6(m,2H)、1.7(m,1H)、1.8(m,1H)、2.4(t,2H)、3.79(s,3H)、6.64(d,1H)、6.97(s,1H)、7.26(d,1H)、7.91(s,1H)。
LC−MS(ESI):18.5分 97.4(%)、m/z 281[MH+]
【0362】
調製例27
8’−クロロ−5’−ヒドロキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン
【0363】
【化49】
ステップ(a)
8’−クロロ−5’−ヒドロキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン酢酸溶媒和物
調製例26の化合物(350g、1.24mol)を酢酸(3500ml)中にスラリー化した。スラリーに室温で臭化水素酸(水中48%w/w)(2800ml、24.8mol)を加えた。次いで、このスラリーを還流温度に温め、4日間攪拌した。反応液を100℃に冷却し、1時間かけて水(2800ml)を滴下した。スラリーを10℃に冷却し、1時間攪拌した後、濾過し、水(1100ml)で洗浄し、真空オーブンに入れて終夜乾燥させて、表題化合物を白色固体(344g、1.28mol、103%)として得た。
1H−NMR(D6−DMSO、300MHz):δ1.2(m,1H)、1.4(m,2H)、1.5(m,1H)、1.6(m,2H)、1.7(m,1H)、1.8(m,1H)、1.9(s,3H,CH3COOH)、2.4(t,2H)、6.45(d,1H)、6.95(s,1H)、7.1(d,1H)、7.75(s,1H)、9.9(s,1H,CH3COOH)。
LC−MS(ESI):14.2分 99.1(%)、m/z 267[MH+]。
【0364】
ステップ(b)
8’−クロロ−5’−ヒドロキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン
ステップ(a)の酢酸溶媒和物(330g、1.24mol)を、室温で6時間かけてアセトン(730ml)中にスラリー化した。次いで、生成物を濾過によって収集し、アセトン(330ml)で洗浄した後、60℃で18時間乾燥させて、溶媒和していない表題化合物を白色固体(238g、0.89mol、72%)として得た。
1H−NMR(D6−DMSO、300MHz):δ1.2(m,1H)、1.4(m,2H)、1.5(m,1H)、1.6(m,2H)、1.7(m,1H)、1.8(m,1H)、2.4(t,2H)、6.45(d,1H)、6.95(s,1H)、7.1(d,1H)、7.75(s,1H)。
【図面の簡単な説明】
【0365】
【図1】本発明の実施例1および2の化合物、ならびにWO02/074754の実施例75の化合物(化合物A)の、1mg/kgで静脈内投与した後の、投与量で正規化した平均の薬物動態プロフィールを示す濃度/時間のグラフである。
【図2】実施例2の化合物の酢酸溶媒和物についての測定したパターン(A)およびシミュレートしたパターン(B)の粉末X線回折(PXRD)プロットを示す図である。
【図3】実施例2の化合物の酢酸溶媒和物について、熱重量分析(TGA)を使用して加熱した後の質量減少のプロットを示すグラフである(示される15.05%の質量減少は、1モル当量の酢酸に等しい)。
【図4】ケイ素でドープ処理した実施例2の化合物の脱溶媒和形態(形態A)(A)、TGA分析前(B)およびTGA分析後(C)のこの化合物の酢酸溶媒和物のPXRDパターンを示す図である。
【図5】ケイ素標準物質を基準として補正した、実施例2の化合物の溶媒和していない結晶形態(形態A)のPXRDパターンを示す図である。
【図6】実施例2の化合物の溶媒和していない結晶形態(形態A)についての示差走査熱量測定(DSC)トレースである。
【図7】実施例2の化合物の結晶性の溶媒和していない形態(形態A)、ならびにジメチルアセトアミド(DMAC)(B)、ピリジン(C)、テトラヒドロフラン(THF)(D)、ジメチルスルホキシド(DMSO)(E)、および酢酸(F)溶媒和物についてのPXRDプロットを示す図である。
【図8】実施例2の化合物のピリジン溶媒和物のTGAトレースを示すグラフであり、示される17.3%の質量減少は、溶媒対化合物の比が1:1であることに等しい。
【図9】実施例2の化合物のテトラヒドロフラン溶媒和物のTGAトレースを示すグラフであり、14.7%の合計質量減少は、化合物対THF溶媒の比が1:1であることに相当する(加熱後の溶媒減少の段階的な性質は、恐らく、中間体の半THF溶媒和物形態の存在を示唆する)。
【図10】実施例2の化合物のジメチルアセトアミド溶媒和物のTGAトレースを示すグラフであり、33.0%の合計質量減少は、溶媒対化合物の比が2:1であることに等しい。
【図11】実施例2の化合物のピリジン溶媒和物(A)、TGA後のピリジン溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【図12】実施例2の化合物のテトラヒドロフラン溶媒和物(A)、TGA後のTHF溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【図13】実施例2の化合物のジメチルアセトアミド溶媒和物(A)、TGA後のジメチルアセトアミド溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【図14】実施例2の化合物のジメチルスルホキシド溶媒和物(A)、真空乾燥後のDMSO溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【技術分野】
【0001】
本発明は、スピロ環式誘導体、ならびにそのような誘導体の調製方法、その調製で使用する中間体、それを含有する組成物、およびその使用に関する。
【0002】
本発明のスピロ環式誘導体は、PDE7阻害剤であり、特に疼痛、特に神経因性疼痛の治療においていくつかの治療用途を有する。
【背景技術】
【0003】
ホスホジエステラーゼ(PDE)は、セカンドメッセンジャー分子のcAMPおよびcGMPを対応する不活性な5’−一リン酸ヌクレオチドに加水分解し、それによりその生理的なレベルを調節する過程によって、様々な細胞内シグナル伝達過程に影響を及ぼす酵素のファミリーである。二次的なメッセンジャーであるcAMPおよびcGMPは、数多くの細胞内過程の調節を司る。PDEファミリーは、少なくとも11種あり、cAMPに特異的なもの(PDE3、4、7、8)もあれば、cGMPに特異的なもの(PDE5、6、および9)もある。
【0004】
PDE7は、PDEファミリーの一員であり、2種のサブクラスメンバーPDE7AおよびBを含む。PDE7のmRNAは、T細胞関連障害などのいくつかの疾患の病因において重要であることがわかっている様々な組織および細胞型で発現される。特にPDE7Aおよびそのスプライスバリアントは、活性化型T細胞中(L.Li、C.Yee、およびJ.A.Beavo、Science(1999年)、第283巻、848〜851ページ)およびBリンパ球中(R.Lee、S.Wolda、E.Moon、J.Esselstyn、C.Hertel、およびA.Lerner、Cell.Signal(2002年)、第14巻、277〜284ページ)、自己免疫疾患(L.Liら、上記)、ならびに気道疾患(S.J.Smithら、Am.J.Physiol.Lung.Cell.Mol.Physiol.(2003年)、第284巻、L279〜L289ページ)で上方制御される。したがって、PDE7の選択的阻害剤は、免疫抑制剤として、また呼吸器の状態、たとえば慢性閉塞性肺疾患および喘息の治療薬として広範な用途を有することが予想される(N.A.Glavas、C.Ostenson、J.B.Schaefer、V.Vasta、およびJ.A.Beavo.PNAS(2001年)、第98巻、6319〜6324ページ)。
【0005】
ラットでの研究は、PDE7A mRNAが、ラット脳のニューロンおよび非ニューロンの両方の細胞集団中に広く分布しているという知見を示している。最高濃度は嗅球、嗅結節、海馬、小脳、内側手綱核、松果体、最後野、および脈絡叢で認められる。PDE7A mRNAは、他の非脳組織でも広く検出される。これらの結果は、PDE7Aが多くの脳機能においてcAMPシグナル伝達の調節に関与していることと一致し、PDE7Aが、記憶、うつ病、および嘔吐に対して効果を有するかもしれないことを示唆している(X.Miro、S.Perez−Torres、J.M.Palacios、P.Puigdomenech、G.Mengod、Synapse(2001年)、第40巻、201〜214ページ)。アルツハイマー病との関連も示唆されている(S.Perez Torresら、Experimental Neurology(2003年)第182巻、322〜334ページ)。PDE7はさらに、受精障害(WO01/83772)および白血病(R.Leeら、Cell Signalling(2002年)第14巻、277〜284ページ)とも関連付けられている。
【0006】
PDE7Aは、酵母(T.Michaeliら、J.Biol.Chem.(1993年)第268巻、12925〜12932ページ)、ヒト(P.Han、Z.Xiaoyan、およびM.Tamar、J.Biol.Chem.(1997年)第272巻、16152〜16157ページ)、マウス(T.BloomおよびJ.A.Beavo、Proc.Natl.Acad.Sci.USA(1996年)、第93巻、14188〜14192ページ)から単離されており、PDE7Aレベルの上方制御は、ヒトTリンパ球中で見られる(M.IchimuraおよびH.Kase.Biochem.Biophys.Res.Commun.(1993年)、第193巻、985〜990ページ)。
【0007】
PDE7ファミリーの第2のメンバーであるPDE7Bは、C末端の触媒ドメインにおいてPDE7Aと70%のアミノ酸相同性を共有している(N末端ドメインは、PDEファミリー全体に保存されている、リン酸化部位を含有する調節性ドメインである)。PDE7Bは、cAMPに特異的であり、マウス(受託番号−AJ251858)およびヒト(受託番号−AJ251860)供給源からクローン化されている(C.Gardner、N.Robas、D.Cawkill、およびM.Fidock、Biochem.Biophys.Res.Commun.(2000年)、第272巻、186〜192ページ)。PDE7Bは、広範な種類の組織、すなわち脳の尾状核、被殻、および後頭葉、末梢では心臓、卵巣、下垂体、腎臓、肝臓、小腸、および胸腺、さらには骨格筋、大腸、膀胱、子宮、前立腺、胃、副腎、および甲状腺で発現されることが示されている。PDE7Bは、いくつかの一般のPDE阻害剤を識別することも示されている(J.M.Hetman、S.H.Soderling、N.A.Glavas、およびJ.A.Beavo、PNAS(2000年)、第97巻、472〜476ページ)。しかし、ザプリナスト、ロリプラム、ミルリノンなどの多くの標準的PDE阻害剤は、PDE7Bを特異的には阻害しない。
【0008】
PDE7の阻害剤は、様々なPDE7関連疾患の治療で使用されることが知られている。たとえば、WO02/074754は、次式の化合物
【0009】
【化1】
ならびにT細胞関連疾患、自己免疫疾患、骨関節炎、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群、アレルギー、または炎症性腸疾患などのPDE7関連障害の治療におけるその使用を記載している。
【0010】
WO2004/026818は、次式の化合物
【0011】
【化2】
【0012】
およびPDE7関連障害の治療におけるその使用を記載している。
【0013】
WO2006/092691は、神経因性疼痛の治療におけるPDE7阻害剤の使用を記載している。
【発明の開示】
【発明が解決しようとする課題】
【0014】
発明者らは、驚いたことに、WO02/074754の一般の開示内に含まれるが、その中で詳細には開示または例示されていない1クラスの化合物が、WO02/074754で例示されている最も近い化合物と比べて予想外に優れた薬物動態特性を示すことを見出した。これらの化合物は、低い身体からのクリアランスを示すことが予想され、1日1回投与したときに治療効果を実現する可能性を有している。
【課題を解決するための手段】
【0015】
本発明は、式(I)の化合物
【0016】
【化3】
[式中、
mは、0、1、または2であり、
Xは、O、S、またはN−CNであり、
Rは、F、Cl、またはCNであり、
Aは、C1〜4アルキル基で置換されていてもよいC3〜6シクロアルキレン基であり、
Bは、単結合またはC1〜2アルキレン基である]
または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグを提供する。
【発明を実施するための最良の形態】
【0017】
本発明の文脈では、用語「アルキレン」とは、1または2個の炭素原子を有する二価の飽和炭化水素鎖を示す。アルキレン基の例には、メチレン、エチレン、およびメチルメチレンが含まれ、メチレンが好ましい。
【0018】
用語「シクロアルキレン」とは、3〜6個の炭素原子を有する二価の飽和炭素環を示す。シクロアルキレン基の例には、シクロプロピレン(たとえば、1,1−シクロプロピレン、シス−およびトランス−1,2−シクロプロピレン)、シクロブチレン(たとえば、1,1−シクロブチレン、シス−およびトランス−1,2−シクロブチレン、シス−およびトランス−1,3−シクロブチレン)、シクロペンチレン(たとえば、1,1−シクロペンチレン、シス−およびトランス−1,2−シクロペンチレン、シス−およびトランス−1,3−シクロペンチレン)、ならびにシクロヘキシレン(たとえば、1,1−シクロヘキシレン、シス−およびトランス−1,2−シクロヘキシレン、シス−およびトランス−1,3−シクロヘキシレン、シス−およびトランス−1,4−シクロヘキシレン)が含まれる。好ましい例には、シクロブチレンおよびシクロヘキシレン、より好ましくはシクロブチレン、さらにより好ましくは1,3−シクロブチレン、最も好ましくはトランス−1,3−シクロブチレンが含まれる。
【0019】
用語「アルキル」とは、1〜4個の炭素原子を含有している直線状または分枝状の一価の飽和炭化水素鎖を示す。アルキル基の例には、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、s−ブチル、およびt−ブチルが含まれる。好ましい例には、メチルおよびエチル、特にメチルが含まれる。
【0020】
シクロアルキレン基は、C1〜4アルキル基で置換されていてもよい。存在するならば、アルキル置換基は、メチルまたはエチル基、より好ましくはメチル基であることが好ましい。存在するならば、アルキル置換基は、環上のどの位置に存在してもよいが、1位(すなわちカルボン酸基と同位置)に存在することが好ましい。
【0021】
mは、好ましくは1または2、より好ましくは1である。
【0022】
Xは、好ましくはOまたはN−CN、より好ましくはOである。
【0023】
Rは、好ましくはFまたはCl、より好ましくはClである。
【0024】
Aは、好ましくは、メチル基で置換されていてもよいシクロブチレンまたはシクロヘキシレン基である。Aは、より好ましくはシクロブチレン基である。Aは、さらにより好ましくは1,3−シクロブチレン基、特にトランス−1,3−シクロブチレン基である。
【0025】
Bは、好ましくは単結合またはメチレン基である。Bは、より好ましくは単結合である。
【0026】
本発明の特に好ましい化合物には、式(I)中の各可変基が、各可変基について好適かつ/または好ましい基から選択されるものが含まれる。本発明のより一層好ましい化合物には、式(I)中の各可変基が、各可変基についてより好ましいまたは最も好ましい基から選択されるものが含まれる。
【0027】
以下の化合物が特に好ましい。
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−シアノ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
ならびにこれらの薬学的に許容できる塩、溶媒和物、およびプロドラッグ。
【0028】
以下の化合物が特に好ましい。
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
ならびにこれらの薬学的に許容できる塩、溶媒和物、多形体、およびプロドラッグ。
【0029】
特に以下で記載する溶媒和していない結晶形態(A型)として、および以下で記載する酢酸溶媒和物としてのトランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸化合物、ならびに薬学的に許容できるその塩、溶媒和物、多形体、およびプロドラッグが最も好ましい。
【0030】
一実施形態では、本発明は、Cu Kα放射線照射(波長=1.5406Å)を使用して測定したとき以下の粉末X線回折ピーク、すなわち6.3、17.8、21.5、22.1、22.4、26.3(2θ、±0.1°の度数)を特徴とする、溶媒和していない結晶形態(A型)のトランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]−シクロブタンカルボン酸化合物を含む。
【0031】
別の実施形態では、本発明は、Cu Kα放射線照射(波長=1.5406Å)を使用して測定したとき以下の粉末X線回折ピーク、すなわち8.3、10.8、16.6、17.1、19.5、20.5、23.7(2θ、±0.1°の度数)を特徴とする、酢酸溶媒和物としてのトランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]−シクロブタンカルボン酸化合物を含む。
【0032】
本発明はさらに、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグと、薬学的に許容できる担体または希釈剤とを含む医薬組成物を含む。
【0033】
本発明はさらに、医薬として使用するための、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグを含む。
【0034】
本発明はさらに、PDE7阻害剤による治療が適切である疾患または状態を治療する医薬の製造における、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグの使用を含む。
【0035】
本発明はさらに、PDE7阻害剤による治療が適切である疾患または状態の治療方法であって、その最も広い態様もしくは好ましい態様の有効量の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグの投与を含む方法を含む。
【0036】
PDE7阻害剤である式(I)の化合物は、ある範囲の障害の治療において潜在的に有用である。疼痛、詳細には神経因性疼痛の治療が好ましい使用である。
【0037】
生理的な痛みは、外部環境からの有害な可能性のある刺激の危険を警告するように設計された重要な防御機構である。このシステムは、特定のセットの一次感覚ニューロンを通して作動し、侵害刺激によって、末梢の変換機構を介して活性化される(総説については、Millan、Prog.Neurobiol.、(1999年)、第57巻、1〜164ページを参照されたい)。これらの感覚線維は、侵害受容器として知られており、伝導速度の緩徐な直径の小さい軸索を特徴とする。侵害受容器は、侵害刺激の強度、持続時間、および質、さらにはその組織分布的に系統立てられた脊髄への投射によって刺激の位置をコードする。侵害受容器は、Aδ線維(有髄)およびC線維(無随)を主要な2タイプとする侵害受容性神経線維上に見られる。侵害受容器による入力によって生成された活性は、後角での複雑な処理の後、直接にまたは脳幹中継核を介して、視床基底複側、次いで皮質へと伝達され、皮質で痛みの感覚が生じる。
【0038】
疼痛は、一般に急性または慢性として分類することができる。急性痛は、突如始まり、短命(通常は12週間以下)である。急性痛は、通常は特定の損傷などの特定の原因に関連し、しばしば鋭く激しい。急性痛は、手術、歯科作業、挫傷、または捻挫の結果として生じる特定の損傷の後に起こり得る種類の疼痛である。急性痛は一般に、どんな持続的な心理的応答ももたらさない。対照的に、慢性痛は、通常は3ヶ月間より長い間持続し、著しい心理的および情緒的問題をもたらす長期間の疼痛である。慢性痛の一般的な例は、神経因性疼痛(たとえば、有痛性の糖尿病性ニューロパチー、ヘルペス後神経痛)、手根管症候群、背痛、頭痛、癌性疼痛、関節炎疼痛、および慢性の術後疼痛である。
【0039】
疾患または外傷によって体組織に相当な損傷が起こると、侵害受容器活性化の特性は変更され、感作は、末梢で、損傷周囲で局所的に、また侵害受容器が終わる中枢で起こる。これらの影響は、疼痛の感覚を強める。急性痛では、これらの機序は、修復過程が起こるのを一層可能にし得る保護的な挙動を促進する点で有用な場合がある。通常の予想では、損傷が治癒してしまえば、その敏感性が正常に戻るということになる。しかし、多くの慢性疼痛状態では、過敏性は、治癒過程よりはるかに長く残り、しばしば神経系損傷を原因とする。この損傷はしばしば、適応不良および異常な活動に関連する感覚神経繊維の異常をもたらす(Woolf&Salter、Science、(2000年)、第288巻、1765〜1768ページ)。
【0040】
患者の症状の中で不快感および異常な敏感性が特徴となっているとき、臨床上の痛みが存在する。患者は、全く均一でない傾向があり、様々な痛み症状で診察を受けにくる場合がある。そのような症状には、1)鈍い、焼けるような、または刺すようであるといえる自発痛、2)侵害刺激に対する疼痛反応の悪化(痛覚過敏)、および3)通常は非侵害性の刺激によって生じる痛み(異痛症−Meyerら、1994、Textbook of Pain、13〜44ページ)が含まれる。様々な形態の急性痛および慢性痛に罹患している患者が同様の症状を有することがあるとしても、根底にある機序は異なる場合があり、したがって異なる治療戦略が必要となり得る。したがって、痛みは、異なる病態生理に従って、侵害受容性、炎症性、および神経因性の疼痛を含むいくつかの異なるサブタイプに分けることができる。
【0041】
侵害受容性疼痛は、組織損傷、または損傷を引き起こす潜在性のある激しい刺激によって誘発される。痛みの求心性は、損傷部位での侵害受容器による刺激の伝達によって活性化され、それが終結するレベルにある脊髄のニューロンを活性化する。次いで、これが脊髄路を上って脳へと中継され、そこで痛みが知覚される(Meyerら、1994年、Textbook of Pain、13〜44ページ)。侵害受容器の活性化は、2タイプの求心性神経線維を活性化する。有髄のAδ線維は、急速な伝達を行い、鋭く刺すような痛覚を司るのに対し、無髄のC線維は、より緩慢な速度で伝達を行い、鈍いまたはうずくような痛みを伝える。中程度から重度の急性侵害受容性疼痛は、中枢神経系外傷、挫傷/捻挫、火傷、心筋梗塞、および急性膵炎の疼痛、術後疼痛(任意のタイプの手術手順後の疼痛)、外傷後疼痛、腎疝痛、癌性疼痛、および背痛からくる疼痛の顕著な特色である。癌性疼痛は、腫瘍関連疼痛(たとえば、骨痛、頭痛、顔面痛、または内臓痛)や、癌治療に関連する疼痛(たとえば、化学療法後症候群、慢性術後疼痛症候群、または放射線照射後症候群)などの慢性痛であるといえる。癌性疼痛は、化学療法、免疫療法、ホルモン療法、または放射線療法に応答して起こる場合もある。背痛は、椎間板ヘルニアもしくは椎間板破裂、または腰椎椎間関節、仙腸関節、傍脊椎筋、もしくは後縦方向靱帯の異常によるものであるといえる。背痛は、自然に消散することもあるが、これが12週間を超えて持続する一部の患者では、特に消耗性となり得る慢性状態になる。
【0042】
神経因性疼痛は現在、神経系の一次病巣または機能不全によって始まる、または引き起こされる疼痛であると定義されている。神経損傷は、外傷および疾患によって引き起こされる場合があり、したがって用語「神経因性疼痛」は、病因の多様な多くの障害を包含する。これらには、末梢性神経障害、糖尿病性ニューロパチー、ヘルペス後神経痛、三叉神経痛、背痛、癌性ニューロパチー、HIVニューロパチー、幻肢痛、手根管症候群、中心性卒中後痛、ならびに慢性アルコール中毒、甲状腺機能低下、尿毒症、多発性硬化症、脊椎損傷、パーキンソン病、てんかん、およびビタミン欠乏に関連する疼痛が含まれるがこの限りでない。保護的な役割をもたない神経因性疼痛は病的である。神経因性疼痛はしばしば、もとの原因が消失した後も多分に存在し、患者の生活の質を著しく低下させながら、一般的に何年間も持続する(Woolf and Mannion、Lancet、(1999年)第353巻、1959〜1964ページ)。神経因性疼痛の症状は、同じ疾患の患者間でさえもしばしば均一でないので、治療が難しい(Woolf&Decosterd、Pain Supp.(1999年)、第6巻、S141〜S147ページ;WoolfおよびMannion、上記)。神経因性疼痛の症状には、持続的になることのある自発痛、ならびに痛覚過敏(侵害刺激に対する敏感性の増大)や異痛症(通常は非侵害性の刺激に対する敏感性)などの発作性または異常性の誘発痛が含まれる。
【0043】
炎症の過程は、組織損傷または異物の存在に応答して活性化される、腫れおよび痛みをもたらす一連の複雑な生化学的および細胞性の事象である(LevineおよびTaiwo、1994年、Textbook of Pain、45〜56ページ)。関節炎疼痛は、最も一般的な炎症性疼痛である。リウマチ様疾患は、先進諸国において最も一般的な慢性炎症状態の1つであり、関節リウマチは、身体障害の一般的な原因である。関節リウマチの正確な病因は不明であるが、現在の仮説は、遺伝的要因と微生物学的要因の両方が重要である可能性があることを示唆している(Grennan&Jayson、1994年、Textbook of Pain、397〜407ページ)。ほぼ1600万人のアメリカ人が症候性の骨関節炎(OA)または変形性関節疾患に罹患し、その大部分が60才を超えていることが推定されており、集団の年齢が増すにつれてこれが4000万人に増えることが想定され、そのためこれが重要性の非常に大きい公衆衛生問題になっている(Houge & Mersfelder、Ann Pharmacother.(2002年)、第36巻、679〜686ページ;McCarthyら、1994年、Textbook of Pain、387〜395ページ)。ほとんどの変形性関節炎患者は、それに伴う疼痛のために医学的な対応処置を捜し求める。関節炎は、心理社会的および身体的な機能に著しい影響を及ぼし、後の人生で身体障害の主な原因となることが知られている。強直性脊椎炎も、脊椎および仙腸関節の関節炎を引き起こすリウマチ性疾患である。強直性脊椎炎は、生涯にわたって起こる背痛の断続的なエピソードから、脊椎、末梢の関節、および他の身体臓器を襲う重度の慢性疾患に至るまで様々である。
【0044】
別のタイプの炎症性疼痛は内臓痛であり、これには、炎症性腸疾患(IBD)に関連する疼痛が含まれる。内臓痛は、腹腔の臓器を包含する内臓に関連する疼痛である。これらの臓器には、生殖器、脾臓、および消化器系の一部が含まれる。内臓に関連する疼痛は、消化系の内臓痛および非消化系の内臓痛に分けることができる。一般的に遭遇する、疼痛を引き起こす胃腸(GI)障害には、機能性腸障害(FBD)および炎症性腸疾患(IBD)が含まれる。これらのGI障害には、FBDに関しては胃食道逆流症、消化不良、過敏性大腸症候群(IBS)、および機能性腹痛症候群(FAPS)、IBDに関してはクローン病、回腸炎、および潰瘍性大腸炎を含めて、すべてが定期的に内臓痛を生じる、現在中等度にしかコントロールされない広範囲な疾患状態が含まれる。他のタイプの内臓痛には、月経困難症、膀胱炎、および膵炎に関連する疼痛、ならびに骨盤痛が含まれる。
【0045】
一部のタイプの疼痛は、複数の病因を有し、すなわち複数の領域に分類される場合があり、たとえば背痛および癌性疼痛は、侵害受容性および神経障害性の両方の構成要素を有することに留意されたい。
【0046】
他のタイプの疼痛には、以下のものが含まれる。
・筋肉痛、線維筋痛、椎骨炎、血清反応陰性(非リウマチ様)関節症、非関節性リウマチ、ジストロフィノパチー、グリコーゲン分解、多発性筋炎、および化膿性筋炎を含む筋骨格障害の結果として生じる疼痛、
・狭心症、心筋梗塞、僧帽弁狭窄、心外膜炎、レイノー現象、浮腫性硬化症、および骨格筋虚血によって引き起こされる疼痛を含む心臓および血管の疼痛、
・偏頭痛(前兆を伴う偏頭痛および前兆を伴わない偏頭痛を含む)、群発性頭痛、筋緊張型頭痛、混合型頭痛、および血管性障害に関連する頭痛などの頭痛、ならびに
・歯痛、耳痛、口腔内灼熱症候群、および側頭下顎の筋筋膜痛を含む口腔顔面痛。
【0047】
本発明の式(I)の化合物は、疼痛以外の状態の治療においても有用である。特に、本発明の式(I)の化合物は、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、および炎症性腸疾患の治療において有用である。
【0048】
本発明はさらに、疼痛(特に神経因性疼痛)、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、および炎症性腸疾患から選択される状態または障害を治療するための医薬の製造における、その最も広い態様もしくは好ましい態様の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、もしくはプロドラッグの使用を含む。
【0049】
本発明はさらに、疼痛(特に神経因性疼痛)、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、または炎症性腸疾患から選択される疾患または状態の治療方法であって、その最も広い態様もしくは好ましい態様の有効量の式(I)の化合物、または薬学的に許容できるその塩、溶媒和物、もしくはプロドラッグを投与することを含む方法を含む。
【0050】
式(I)の化合物の薬学的に許容できる塩には、その酸付加塩および塩基の塩が含まれる。
【0051】
好適な酸付加塩は、非毒性の塩を形成する酸から生成されるものである。例には、酢酸塩、アジピン酸塩、アスパラギン酸塩、安息香酸塩、ベシル酸塩、炭酸水素塩/炭酸塩、重硫酸塩/硫酸塩、ホウ酸塩、カンシル酸塩、クエン酸塩、シクラミン酸塩、エジシル酸塩、エシル酸塩、ギ酸塩、フマル酸塩、グルセプト酸塩、グルコン酸塩、グルクロン酸塩、ヘキサフルオロリン酸塩、ヒベンズ酸塩、塩酸塩/塩化物、臭化水素酸塩/臭化物、ヨウ化水素酸塩/ヨウ化物、イセチオン酸塩、乳酸塩、リンゴ酸塩、マレイン酸塩、マロン酸塩、メシル酸塩、メチル硫酸塩、ナフチル酸塩、2−ナプシル酸塩、ニコチン酸塩、硝酸塩、オロト酸塩、シュウ酸塩、パルミチン酸塩、パモ酸塩、リン酸塩/リン酸水素塩/リン酸二水素塩、ピログルタミン酸塩、糖酸塩、ステアリン酸塩、コハク酸塩、タンニン酸塩、酒石酸塩、トシル酸塩、トリフルオロ酢酸塩、およびキシノホ酸塩(xinofoate)が含まれる。
【0052】
好適な塩基の塩は、非毒性の塩を形成する塩基から生成されるものである。例には、アルミニウム、アルギニン、ベンザチン、カルシウム、コリン、ジエチルアミン、ジオールアミン、グリシン、リジン、マグネシウム、メグルミン、オールアミン、カリウム、ナトリウム、トロメタミン、および亜鉛の塩が含まれる。
【0053】
酸および塩基の半塩、たとえば半硫酸塩および半カルシウム塩を生成してもよい。
【0054】
好適な塩に関する総説については、StahlおよびWermuthによる「Handbook of Pharmaceutical Salts:Properties,Selection,and Use」(Wiley−VCH、2002年)を参照されたい。
【0055】
式(I)の化合物の薬学的に許容できる塩は、次の3つの方法の1つまたは複数によって調製することができる。
(i)式(I)の化合物を所望の酸または塩基と反応させることによる方法、
(ii)式(I)の化合物の好適な前駆体から、酸もしくは塩基に不安定な保護基を除去する、または所望の酸もしくは塩基を使用して好適な環状前駆体、たとえばラクトンもしくはラクタムを開環することによる方法、または
(iii)式(I)の化合物の塩を、適切な酸もしくは塩基との反応によって、または好適なイオン交換カラムによって別の塩に変換することによる方法。
【0056】
3つの反応はすべて、通常は溶液中で実施する。生じる塩が析出することもあり、濾過によって収集してもよいし、または溶媒を蒸発させて回収してもよい。得られる塩のイオン化の程度は、完全にイオン化したものからほとんどイオン化していないものまで様々となり得る。
【0057】
本発明の化合物は、完全な非晶質から完全な結晶性に至る範囲の一連の固体状態で存在し得る。用語「非晶質」とは、材料が分子レベルで長いきちんとした序列を欠き、温度に応じて固体または液体の物理的性質を示し得る状態を指す。通常、そのような材料は、特有のX線回折パターンを与えず、固体の性質を示しながらもより正式には液体であると記述される。加熱すると、固体の性質から液体の性質への変化が起こるが、これは通常、二次の状態変化(「ガラス転移」)を特徴とする。用語「結晶性」とは、材料が、分子レベルで規則的な整った内部構造を有し、明確なピークを伴う特有のX線回折パターンを与える固相を指す。十分に加熱したとき、そのような材料も液体の性質を示すが、固体から液体への変化は、通常は一次の相変化(「融点」)を特徴とする。
【0058】
本発明の化合物はまた、溶媒和していない形態および溶媒和した形態で存在する場合がある。用語「溶媒和物」は、本明細書では、本発明の化合物と、薬学的に許容できる1つまたは複数の溶媒分子、たとえばエタノールとを含む分子複合体について述べるのに使用する。用語「水和物」は、前記溶媒が水であるときに使用する。本発明は、溶媒和していない形態とすべての溶媒和形態の両方を含む。
【0059】
現在受け入れられている有機水和物の分類系統は、隔離部位水和物、チャネル水和物、または金属イオン配位水和物を規定するものである。K.R.Morrisによる「Polymorphism in Pharmaceutical Solids」(H.G.Brittain編、Marcel Dekker、1995年)を参照されたい。隔離部位水和物は、水分子が介在する有機分子によって互いの直接の接触から隔離されているものである。チャネル水和物では、水分子は格子チャネルにあり、そこで他の水分子と隣り合っている。金属イオン配位水和物では、水分子は金属イオンに結合している。
【0060】
溶媒または水が堅く結合しているとき、複合体は、湿度に関係なく明確な化学量論性を有する。しかし、チャネル溶媒和物および吸湿性化合物でのように溶媒または水が弱く結合しているとき、水/溶媒含有量は湿度および乾燥条件に左右される。そのような場合では、非化学量論性が標準となる。
【0061】
以下では、式(I)の化合物へのすべての言及は、その塩および溶媒和物への言及ならびにその塩の溶媒和物を含む。
【0062】
本発明の化合物には、そのすべての多形体および晶癖、以下で定義するそのプロドラッグおよび異性体(光学異性体、幾何異性体、および互変異性体を含む)、ならびに同位体標識された式(I)の化合物を含めて、上で規定した式(I)の化合物が含まれる。
【0063】
指摘したように、式(I)の化合物のいわゆる「プロドラッグ」も、本発明の範囲内にある。したがって、それ自体は薬理活性をほとんどまたは全くもたなくてもよい式(I)の化合物の特定の誘導体は、身体中または身体上に投与されたとき、たとえば加水分解による切断によって、所望の活性を有する式(I)の化合物に変換され得る。そのような誘導体を「プロドラッグ」と呼ぶ。プロドラッグの使用についてのそれ以上の情報は、「Pro−drugs as Novel Delivery Systems」、第14巻、ACS Symposium Series(T.HiguchiおよびW.Stella)および「Bioreversible Carriers in Drug Design」、Pergamon Press、1987年(E.B.Roche編、米国薬剤師会)で見ることができる。
【0064】
本発明によるプロドラッグは、たとえば、たとえばH.Bundgaardによる「Design of Prodrugs」(Elsevier、1985年)に記載されているように、式(I)の化合物中に存在する適切な官能基を、当業者に「pro−部分」として知られている特定の部分と交換することによって生成することができる。
【0065】
本発明の式(I)の化合物は、カルボン酸官能基(−COOH)を含有する。したがって、好適なプロドラッグは、式(I)の化合物のカルボン酸官能基の水素がエステル残基によって置換されている、そのエステルを含む。用語「エステル残基」とは、加水分解などの生物学的方法によってin vivoで切断することができ、遊離のカルボン酸基を有する式(I)の化合物またはその塩を生成するエステル基を意味する。
【0066】
化合物がそのようなプロドラッグであるか否かは、たとえば、それをラットやマウスなどの実験動物に静脈内注射によって投与し、次いで動物の体液を調査して、式(I)の化合物または薬学的に許容できるその塩が検出できるか否かを判定することによって決定することができる。
【0067】
エステル残基の好ましい例には、以下のものが含まれる。
メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、s−ブチル、t−ブチル、n−ペンチル、イソペンチル、ネオペンチル、ヘキシル、ヘプチル、オクチル、ノニル、デシル、ウンデシル、ドデシル、トリデシル、テトラデシル、ペンタデシル、ヘキサデシル、ヘプタデシル、オクタデシル、ノナデシル、イコサニルなどの直鎖または分枝鎖のアルキル基でよい、C1〜20アルキル基、特にC1〜12アルキル基、好ましくはC1〜8アルキル基、より好ましくはC1〜6アルキル基、最も好ましくは上で定義し例示したものなどのC1〜4アルキル基、
C1〜10ハロアルキル基(1個または複数のハロゲン原子、好ましくはフッ素または塩素原子、より好ましくはフッ素原子で置換されたアルキル基であると定義される)、好ましくはC1〜8ハロアルキル基、より好ましくはC1〜6ハロアルキル基、最も好ましくはC1〜4ハロアルキル基、たとえば、モノ−、ジ−、またはトリフルオロメチル、モノ−、ジ−、またはトリクロロメチル、ブロモメチル、2−フルオロエチル、2,2−ジフルオロエチル、2,2,2−トリフルオロエチル、2−クロロエチル、2,2−ジクロロエチル、2,2,2−トリクロロエチル、ペルフルオロエチル、ペルフルオロプロピル、ペルフルオロブチルなど、
C1〜10ヒドロキシアルキル基(ヒドロキシ(−OH)基で置換されたアルキル基であると定義される)、好ましくはC1〜8ヒドロキシアルキル基、より好ましくはC1〜6ヒドロキシアルキル基、最も好ましくはC1〜4ヒドロキシアルキル基、たとえば、ヒドロキシメチル、1−または2−ヒドロキシエチル、1−、2−、または3−ヒドロキシプロピル、1−、2−、3−、または4−ヒドロキシブチルなど、
(C1〜10アルコキシ)C1〜10アルキル基(アルコキシ基で置換されたアルキル基であると定義される)、好ましくは(C1〜6アルコキシ)C1〜6アルキル基、より好ましくは(C1〜4アルコキシ)C1〜4アルキル基、最も好ましくは(C1〜4アルコキシ)メチル基、たとえば、メトキシメチル、1,1−ジメチル−1−メトキシメチル、エトキシメチル、プロポキシメチル、イソプロポキシメチル、ブトキシメチル、t−ブトキシメチル基など、
C1〜6アルコキシル化された(C1〜6アルコキシ)メチル基、たとえば、2−メトキシエトキシメチル基など、
ハロ(C1〜6アルコキシ)メチル基、たとえば、2,2,2−トリクロロエトキシメチルやビス(2−クロロエトキシ)メチル基など、
C3〜8シクロアルキル基、たとえば、シクロプロピル、シクロブチル、シクロペンチル、シクロヘキシル、シクロヘプチル、シクロオクチル基など、
アラルキル基、たとえば、1〜3個のC6〜14アリール基で置換されたC1〜6アルキル基(アリール部分は、フェニル、ナフチル、アントリル、およびフェナントリルから選択される)、たとえば、ベンジル、α−ナフチルメチル、β−ナフチルメチル、ジフェニルメチル、トリフェニルメチル、α−ナフチルジフェニルメチル、9−アントリルメチル基など;ならびに1〜3個の置換C6〜14アリール基で置換されたC1〜6アルキル基(アリール基の1個または複数は、1個または複数(好ましくは1〜3個、より好ましくは1個のみ)のC1〜6アルキル、C1〜6アルコキシ、ニトロ、ハロゲン、またはシアノ置換基で置換されている)、たとえば、4−メチルベンジル、2,4,6−トリメチルベンジル、3,4,5−トリメチルベンジル、4−メトキシベンジル、4−メトキシフェニルジフェニルメチル、2−ニトロベンジル、4−ニトロベンジル、4−クロロベンジル、4−ブロモベンジル、4−シアノベンジル基など;特にベンジル基、
ハロおよびC1〜6アルコキシから選択される置換基で置換されていてもよいテトラヒドロピラニルまたはテトラヒドロチオピラニル基、たとえば、テトラヒドロピラン−2−イル、3−ブロモテトラヒドロピラン−2−イル、4−メトキシ−テトラヒドロピラン−4−イル、テトラヒドロチオピラン−2−イル、4−メトキシ−テトラヒドロチオピラン−4−イル基など、
ハロおよびC1〜6アルコキシから選択される置換基で置換されていてもよいテトラヒドロフラニルまたはテトラヒドロチオフラニル基、たとえば、テトラヒドロフラン−2−イル基やテトラヒドロチオフラン−2−イル基など、
C2〜10アルケニル基、たとえば、ビニル、プロペニル、ブテニル、ペンテニル、ヘキセニル、ヘプテニル、オクテニル、ノネニル、デセニル基など、ならびに
C2〜10アルキニル基、たとえば、エチニル、プロピニル、ブチニル、ペンチニル、ヘキシニル、ヘプチニル、オクチニル、ノニニル、デシニル基など。
【0068】
前述の例に従う置換基のこれ以上の例、および他のプロドラッグタイプの例は、上述の参考文献で見ることができる。
【0069】
また、特定の式(I)の化合物は、それ自体が他の式(I)の化合物のプロドラッグとして働く場合もある。
【0070】
1個または複数の不斉炭素原子を含有している式(I)の化合物は、2種以上の立体異性体として存在する場合がある。式(I)の化合物は、シクロアルキレン基を含有しているので、CO2H基およびB基が同じ炭素上にないとき、シス/トランス異性体が考えられる。構造異性体が低いエネルギー障壁で相互変換可能である場合、互変異性体異性(「互変異性」)が起こり得る。互変異性は、環状尿素、チオ尿素、またはシアノグアニジン基を含有している式(I)の化合物ではプロトン互変異性の形態、または芳香族部分を含有している化合物ではいわゆる原子価互変異性の形態を取り得る。これは、単一の化合物が1種類に留まらない異性を示す場合もあるということである。
【0071】
本発明の範囲内には、1種類を超える異性を示す化合物を含めて、式(I)の化合物のすべての立体異性体、ジアステレオ異性体(特にシス/トランス異性体)、および互変異性体形態、ならびにこれらの1種または複数の混合物が含まれる。また、酸付加塩または塩基の塩において対イオンが光学活性を有するもの、たとえばd−乳酸塩もしくはl−リジン、またはラセミ体であるもの、たとえばdl−酒石酸塩もしくはdl−アルギニンも含まれる。
【0072】
シス/トランス異性体は、当業者によく知られている従来の技術、たとえば、クロマトグラフィーおよび分別結晶によって分離することができる。
【0073】
個々の鏡像異性体を調製/単離するための従来の技術には、光学的に純粋な好適な前駆体からのキラル合成、またはたとえば、キラルな高圧液体クロマトグラフィー(HPLC)を使用するラセミ体(または塩もしくは誘導体のラセミ体)の分割が含まれる。
【0074】
別法として、ラセミ体(またはラセミ前駆体)を、光学活性を有する好適な化合物、たとえばアルコールと、または式(I)の化合物が酸性もしくは塩基性の部分を含有している場合には1−フェニルエチルアミンや酒石酸などの塩基もしくは酸と反応させることができる。得られるジアステレオ異性体混合物は、クロマトグラフィーおよび/または分別結晶によって分離し、当業者によく知られる方法によってジアステレオ異性体の一方または両方を対応する純粋な鏡像異性体に変換することができる。
【0075】
本発明のキラルな化合物(およびそのキラルな前駆体)は、0〜50体積%、通常は2%〜20%のイソプロパノール、および0〜5体積%のアルキルアミン、通常は0.1%のジエチルアミンを含有する炭化水素、通常はヘプタンまたはヘキサンからなる移動相を用いる不斉樹脂でのクロマトグラフィー、通常はHPLCを使用して、鏡像異性体を豊富に含む形で得ることができる。溶出液を濃縮すると、濃縮された混合物が得られる。
【0076】
ラセミ体が結晶化するとき、異なる2タイプの結晶が考えられる。第1のタイプは、両方の鏡像異性体を等モル量で含有する1つの均質な形態の結晶が生成する、上で言及したラセミ化合物(真のラセミ体)である。第2のタイプは、それぞれ単一の鏡像異性体を含む2形態の結晶が等モル量で生成する、ラセミ混合物または集晶体である。
【0077】
ラセミ混合物中に存在する結晶形は両方とも同一の物理的性質を有するが、これらは真のラセミ体と比較すると異なる物理的性質を有する場合もある。ラセミ混合物は、当業者に知られている従来の技術によって分離することができる。たとえば、E.L.ElielおよびS.H.Wilenによる「Stereochemistry of Organic Compounds」(Wiley、1994年)を参照されたい。
【0078】
本発明は、1個または複数の原子が、原子番号が同じであるが、原子質量または質量数が自然界で優位を占める原子質量または質量数と異なっている原子によって置換されている、薬学的に許容できるすべての同位体標識された式(I)の化合物を含む。
【0079】
本発明の化合物中に含めるのに好適な同位体の例には、2Hや3Hなどの水素、11C、13C、14Cなどの炭素、36Clなどの塩素、18Fなどのフッ素、123Iや125Iなどのヨウ素、13Nや15Nなどの窒素、15O、17O、18Oなどの酸素、32Pなどのリン、および35Sなどの硫黄の同位体が含まれる。
【0080】
特定の同位体標識された式(I)の化合物、たとえば放射性同位体が組み込まれているものは、薬物および/または基質の組織分布調査において有用である。放射性同位体トリチウム、すなわち3H、およびカーボン14、すなわち14Cは、組込みが容易であり、検出手段が整っていることを考えて、この目的に特に有用である。
【0081】
ジュウテリウム、すなわち2Hなどのより重い同位体で置換すると、より高い代謝安定性の結果として生じる特定の治療上の利点、たとえば、in vivo半減期の延長または投与必要量の減少がもたらされる場合もあり、したがってある状況で好ましいといえる。
【0082】
11C、18F、15O、13Nなどの陽電子放射同位体での置換は、基質受容体占有率を調べる陽電子放射断層撮影(PET)研究において有用となり得る。
【0083】
同位体標識された式(I)の化合物は一般に、以前から用いられる標識されていない試薬の代わりに同位体標識された適切な試薬を使用して、当業者に知られている従来の技術によって、または添付の実施例および調製例に記載のものと類似の方法によって調製することができる。
【0084】
本発明による薬学的に許容できる溶媒和物には、結晶化の溶媒が同位体によって置換されているもの、たとえばD2O、d6−アセトン、d6−DMSOでよいものが含まれる。
【0085】
上で規定した式(I)の中間体化合物、上で式(I)の化合物について定義したようなそのすべての塩、溶媒和物、および複合体、ならびにその塩のすべての溶媒和物および複合体も、本発明の範囲内にある。本発明は、上述の化学種およびその晶癖のすべての多形体を含む。
【0086】
本発明に従って式(I)の化合物を調製するとき、当業者は、この目的のために最高の特徴の組合せをもたらす式(I)の化合物の形態を常法どおりに選択することができる。そのような特徴には、融点、溶解性、加工性、および中間体形態の収率、ならびにその結果としての、生成物を単離時に容易に精製できることが含まれる。
【0087】
式(I)の化合物は、提案された適応症を治療するのに最も適切な剤形および投与経路を選択するために、(全pHでの)溶解性および溶液安定性、透過性などのその生物薬剤学的な性質について評価すべきである。
【0088】
医薬としての使用を目的とする本発明の化合物は、結晶性または非晶質の生成物として投与することができる。そのような本発明の化合物は、沈殿、結晶化、凍結乾燥、噴霧乾燥、または蒸発乾燥などの方法によって、たとえば、固体充填物、粉末、またはフィルムとして得ることができる。マイクロ波または高周波乾燥をこの目的のために使用してもよい。
【0089】
化合物は、単独で、または1種または複数の他の本発明化合物と組み合わせて、または1種または複数の他の薬物と組み合わせて(またはこれらの任意の組合せとして)投与することができる。一般に、化合物は、1種または複数の薬学的に許容できる賦形剤と合同で製剤として投与される。用語「賦形剤」は、本明細書では、本発明の化合物以外の任意の成分について述べるのに使用する。賦形剤の選択は、大部分は、特定の投与方式、賦形剤が溶解性および安定性に及ぼす影響、剤形の性質などの要素に応じて決まる。
【0090】
本発明の化合物の送達に好適な医薬組成物およびその調製方法は、当業者には言うまでもない。そのような組成物およびその調製方法は、たとえば、「Remington’s Pharmaceutical Sciences」第19版(Mack Publishing Company、1995年)で見ることができる。
【0091】
経口投与
本発明の化合物は、経口投与することができる。経口投与は、化合物がGIに入るように飲み込むものでもよいし、および/または化合物がそれによって口から直接血流に入る頬側、舌側、もしくは舌下投与を含むものでもよい。
【0092】
経口投与に好適な製剤には、錠剤;多粒子もしくはナノ粒子、液体、または粉末を含有する軟または硬カプセル剤;ロゼンジ(液体充填型を含む);咀嚼剤;ゲル;急速分散型剤形;フィルム;膣坐剤;スプレー;ならびに頬側/粘膜付着性パッチなどの、固体、半固体、および液体の系が含まれる。
【0093】
液体製剤には、懸濁液、溶液、シロップ、およびエリキシルが含まれる。このような製剤は、(たとえばゼラチンまたはヒドロキシプロピルメチルセルロース製の)軟もしくは硬カプセル中に充填剤として用いることもでき、通常は、担体、たとえば、水、エタノール、ポリエチレングリコール、プロピレングリコール、メチルセルロース、または好適な油と、1種または複数の乳化剤および/または懸濁化剤とを含む。液体製剤は、たとえば小袋から出した固体を再構成して、調製することもできる。
【0094】
本発明の化合物は、LiangおよびChenによるExpert Opinion in Therapeutic Patents、第11巻(6)、981〜986ページ(2001年)に記載のものなどの急速溶解急速崩壊型の剤形にして使用してもよい。
【0095】
錠剤剤形では、薬物は、用量に応じて、剤形の1重量%〜80重量%、より典型的な例では剤形の5重量%〜60重量%を占めてよい。錠剤は一般に、薬物に加えて崩壊剤を含有する。崩壊剤の例には、ナトリウムデンプングリコラート、カルボキシメチルセルロースナトリウム、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、クロスポビドン、ポリビニルピロリドン、メチルセルロース、微結晶セルロース、低級アルキル置換されたヒドロキシプロピルセルロース、デンプン、α化デンプン、およびアルギン酸ナトリウムが含まれる。一般に、崩壊剤は、剤形の1重量%〜25重量%、好ましくは5重量%〜20重量%を占める。
【0096】
結合剤は一般に、錠剤製剤に粘着性の性質を付与するために使用する。好適な結合剤には、微結晶セルロース、ゼラチン、糖、ポリエチレングリコール、天然および合成のゴム、ポリビニルピロリドン、α化デンプン、ヒドロキシプロピルセルロース、およびヒドロキシプロピルメチルセルロースが含まれる。錠剤は、ラクトース(一水和物、噴霧乾燥一水和物、無水物など)、マンニトール、キシリトール、デキストロース、スクロース、ソルビトール、微結晶セルロース、デンプン、第二リン酸カルシウム二水和物などの希釈剤も含有する場合がある。
【0097】
錠剤は、ラウリル硫酸ナトリウムやポリソルベート80などの界面活性剤、および二酸化ケイ素やタルクなどの滑剤を場合により含むこともある。存在するとき、界面活性剤は錠剤の0.2重量%〜5重量%を占めてよく、滑剤は錠剤の0.2重量%〜1重量%を占めてよい。
【0098】
錠剤は一般に、ステアリン酸マグネシウム、ステアリン酸カルシウム、ステアリン酸亜鉛、フマル酸ステアリルナトリウム、ステアリン酸マグネシウムとラウリル硫酸ナトリウムの混合物などの滑沢剤も含有する。滑沢剤は一般に、錠剤の0.25重量%〜10重量%、好ましくは0.5重量%〜3重量%を占める。
【0099】
他の考えられる成分には、抗酸化剤、着色剤、着香剤、保存剤、および矯味剤が含まれる。
【0100】
好例となる錠剤は、約80%までの薬物、約10重量%〜約90重量%の結合剤、約0重量%〜約85重量%の希釈剤、約2重量%〜約10重量%の崩壊剤、および約0.25重量%〜約10重量%の滑沢剤を含有する。
【0101】
錠剤ブレンドを直接にまたはローラーによって圧縮すると、錠剤を生成することができる。別法として、錠剤ブレンドまたはブレンドの一部分を、湿式、乾式、もしくは溶融造粒、溶融凝固、または押出し成形処理した後に打錠してもよい。最終製剤は、1重または複数の層を含むこともあり、コーティングされていてもされていなくてもよく、カプセル封入されていてもよい。
【0102】
錠剤の製剤については、H.LiebermanおよびL.Lachmanによる「Pharmaceutical Dosage Forms:Tablets」、第1巻(Marcel Dekker、ニューヨーク、1980年)で論述されている。
【0103】
ヒトまたは獣医学で使用する摂取可能な経口フィルムは通常、急速溶解型または粘膜付着性でよい可撓性の水溶性または水膨張性薄膜剤形であり、通常は式(I)の化合物、フィルム形成ポリマー、結合剤、溶媒、湿潤剤、可塑剤、安定剤または乳化剤、粘度調整剤、ならびに溶媒を含む。製剤の一部の構成要素が複数の機能を果たす場合もある。
【0104】
式(I)の化合物は、水溶性である場合もあれば不溶性である場合もある。水溶性の化合物は通常、溶質の1重量%〜80重量%、より典型的な例では20重量%〜50重量%を占める。それほど可溶性でない化合物は、組成物のより高い割合、通常は溶質の88重量%までを占めてよい。別法として、式(I)の化合物を多粒子ビーズの形にしてもよい。
【0105】
フィルム形成ポリマーは、天然の多糖類、タンパク質、または合成の親水コロイドから選択されるものでよく、通常は0.01〜99重量%の範囲、より典型的な例では30〜80重量%の範囲で存在する。
【0106】
他の考えられる成分には、抗酸化剤、着色剤、着香剤、および香味剤、保存剤、唾液腺刺激剤、冷却剤、共溶媒(油を含む)、緩和剤、増量剤、消泡剤、界面活性剤、および矯味剤が含まれる。
【0107】
本発明によるフィルムは通常、可剥性の支持担体または紙上にコーティングした薄い水性フィルムを蒸発乾燥して調製する。これは、乾燥オーブンもしくは乾燥トンネル、通常は複合塗工乾燥機で、または凍結乾燥もしくは減圧によって行うことができる。
【0108】
経口投与用の固体製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0109】
本発明の目的に好適な変更型放出製剤は、米国特許第6106864号に記載されている。高エネルギー分散や浸透性粒子および被覆粒子などの他の好適な放出技術の詳細は、VermaらによるPharmaceutical Technology On−line、第25巻(2)、1〜14ページ(2001年)で見られる。制御放出を実現するためのチューインガムの使用は、WO00/35298に記載されている。
【0110】
非経口投与
本発明の化合物は、血流中、筋肉中、または内臓中に直接投与することもできる。非経口投与に好適な手段には、静脈内、動脈内、腹腔内、くも膜下腔内、側脳室内、尿道内、胸骨内、脳内、筋肉内、滑液包内、および皮下が含まれる。非経口投与に好適な装置には、(微細針を含む)針注射器、無針注射器、および注入技術が含まれる。
【0111】
非経口製剤は通常、塩、炭水化物、(好ましくはpH3〜9にする)緩衝剤などの賦形剤を含有することもある水溶液であるが、一部の適用例では、無菌の非水性溶液として、または発熱物質を含まない無菌水などの好適な媒体と共に使用する乾燥形態としてより適切に製剤してもよい。
【0112】
たとえば凍結乾燥による無菌条件下での非経口製剤の調製は、当業者によく知られている標準の製薬技術を使用して容易に実現することができる。
【0113】
非経口溶液の調製で使用する式(I)の化合物の溶解性は、溶解性改善剤を混ぜるなどの適切な製剤技術を使用して増大させることができる。
【0114】
非経口投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。したがって、本発明の化合物は、懸濁液として、または活性化合物の変更型放出をもたらす移植デポー剤として投与するための固体、半固体、もしくは揺変性液体として製剤することができる。そのような製剤の例には、薬物でコーティングされたステント、ならびに薬物を載せたdl−乳酸−グリコール酸共重合体(PGLA)ミクロスフェアを含む半固体および懸濁液が含まれる。
【0115】
局所投与
本発明の化合物は、皮膚または粘膜に局所的に、(皮内)皮膚上に、または経皮的に投与することもできる。この目的のための典型的な製剤には、ゲル、ヒドロゲル、ローション、溶液、クリーム、軟膏、散粉剤、包帯剤、フォーム、フィルム、皮膚パッチ、ウェーハ、植込錠、スポンジ、線維、絆創膏、およびマイクロエマルジョンが含まれる。リポソームを使用してもよい。典型的な担体には、アルコール、水、鉱油、流動パラフィン、白色ワセリン、グリセリン、ポリエチレングリコール、およびプロピレングリコールが含まれる。浸透性改善剤を混ぜてもよい。たとえば、FinninおよびMorganによるJ.Pharm.Sci.、第88巻(10)、955〜958ページ(1999年10月)を参照されたい。
【0116】
他の局所投与手段には、電気穿孔法、イオン導入法、音波泳動法、超音波導入法、ならびに微細針または無針(たとえばPowderject(商標)、Bioject(商標)など)注射による送達が含まれる。
【0117】
局所投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0118】
吸入投与/鼻腔内投与
本発明の化合物は通常、乾燥粉末吸入器から(単独で、たとえばラクトースとの乾燥ブレンドにした混合物として、またはたとえばホスファチジルコリンなどのリン脂質と混合した混合型成分粒子としての)乾燥粉末の形で、加圧容器、ポンプ、スプレー、アトマイザー(好ましくは電気水力学を使用して微細な霧を生成するアトマイザー)、もしくはネブライザーから、1,1,1,2−テトラフルオロエタンや1,1,1,2,3,3,3−ヘプタフルオロプロパンなどの好適な噴射剤を使用しまたは使用せずにエアロゾルスプレーとして、または点鼻液として、鼻腔内にまたは吸入によって投与することもできる。鼻腔内の使用では、粉末は、生体接着剤、たとえばキトサンまたはシクロデキストリンを含んでもよい。
【0119】
加圧容器、ポンプ、スプレー、アトマイザー、またはネブライザーは、たとえばエタノール、エタノール水溶液、または活性物を分散させ、可溶化し、もしくはその放出を延長するのに好適な別の薬剤を含む本発明の化合物の溶液または懸濁液、溶媒としての噴射剤、ならびにトリオレイン酸ソルビタン、オレイン酸、オリゴ乳酸などのオプションの界面活性剤を含有する。
【0120】
薬物製品は、乾燥粉末または懸濁液製剤中に使用する前に、吸入による送達に好適なサイズ(通常は5ミクロン未満)に微粒子化する。これは、スパイラルジェット粉砕、流動層ジェット粉砕、ナノ粒子を生成するための超臨界流体処理、高圧ホモジナイズ、噴霧乾燥などの任意の適切な微粉砕法によって実現することができる。
【0121】
吸入器または注入器に入れて使用する(たとえばゼラチンまたはヒドロキシプロピルメチルセルロース製の)カプセル、ブリスター、およびカートリッジは、本発明の化合物、ラクトースやデンプンなどの好適な粉末基剤、およびl−ロイシン、マンニトール、ステアリン酸マグネシウムなどの性能調節剤の粉末混合物を含有するように製剤することができる。ラクトースは、無水でも一水和物の形でもよいが、後者が好ましい。他の好適な賦形剤には、デキストラン、グルコース、マルトース、ソルビトール、キシリトール、フルクトース、スクロース、およびトレハロースが含まれる。
【0122】
電気水力学を使用して微細な霧を生成するアトマイザーに入れて使用するのに好適な溶液製剤は、1作動あたり1μg〜20mgの本発明の化合物を含有するものでよく、作動体積は1μl〜100μlと様々でよい。典型的な製剤は、式(I)の化合物、プロピレングリコール、滅菌水、エタノール、および塩化ナトリウムを含む。プロピレングリコールの代わりに使用することのできる別の溶媒には、グリセロールおよびポリエチレングリコールが含まれる。
【0123】
メントールやl−メントールなどの好適な着香剤、またはサッカリンやサッカリンナトリウムなどの甘味剤を、吸入投与/鼻腔内投与を目的とする本発明の製剤に加えてもよい。
【0124】
吸入投与/鼻腔内投与用の製剤は、たとえばPGLAを使用して、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0125】
直腸/膣内投与
本発明の化合物は、たとえば坐剤、膣坐剤、または浣腸の形で直腸にまたは経膣的に投与することができる。カカオ脂が伝統的な坐剤基剤であるが、様々な代替品を適宜使用することができる。
【0126】
直腸/経膣投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、およびプログラム放出が含まれる。
【0127】
眼/耳への投与
本発明の化合物は、通常は、等張性のpH調整された無菌食塩水中の微粒子化された懸濁液または溶液の液滴の形で眼または耳に直接投与することもできる。眼および耳への投与に好適な他の製剤には、軟膏、ゲル、生分解性(たとえば吸収性のゲルスポンジ、コラーゲン)および非生分解性(たとえばケイ素樹脂)の植込錠、ウェーハ、レンズ、ならびにニオソームやリポソームなどの微粒子系またはベシクル系が含まれる。架橋ポリアクリル酸、ポリビニルアルコール、ヒアルロン酸などのポリマー;セルロース系ポリマー、たとえば、ヒドロキシプロピルメチルセルロース、ヒドロキシエチルセルロース、もしくはメチルセルロース;またはヘテロ多糖ポリマー、たとえばゲランガムを、塩化ベンザルコニウムなどの保存剤と共に混ぜることができる。このような製剤は、イオン導入法によって送達してもよい。
【0128】
眼/耳への投与用の製剤は、即時型および/または変更型の放出がなされるように製剤することができる。変更型放出製剤には、遅延放出、持続放出、パルス放出、制御放出、標的指向性放出、またはプログラム放出が含まれる。
【0129】
他の技術
本発明の化合物は、上述の投与様式のいずれかで使用するため、その溶解性、溶出速度、矯味、バイオアベイラビリティおよび/または安定性を改善するためにシクロデキストリンおよびその好適な誘導体やポリエチレングリコール含有ポリマーなどの可溶性の高分子実在物と混ぜ合わせてもよい。
【0130】
たとえば、薬物−シクロデキストリン複合体は、一般にほとんどの剤形および投与経路に有用であることがわかっている。包接複合体および非包接複合体の両方を使用することができる。シクロデキストリンは、薬物との直接の複合体形成に代わるものとして、補助添加剤、すなわち担体、希釈剤、または可溶化剤として使用してもよい。これらの目的のために最も一般に使用されるのは、α、β、およびγシクロデキストリンであり、その例は、WO91/11172、WO94/02518、およびWO98/55148で見ることができる。
【0131】
成分キット
たとえばある特定の疾患または状態を治療する目的のために活性化合物の組合せを投与することが望ましい場合もあるので、その少なくとも1種が本発明による化合物を含有する2種以上の医薬組成物を、組成物の共投与に好適なキットの形で好都合に組み合わせてよいことは、本発明の範囲内である。
【0132】
したがって、本発明のキットは、その少なくとも1種が本発明による式(I)の化合物を含有する2種以上の別個の医薬組成物と、容器、分割されたボトル、または分割されたホイル製袋などの、前記組成物を別々に保持するための手段とを含む。そのようなキットの例は、錠剤、カプセルなどの包装に使用される見慣れたブリスターパックである。
【0133】
本発明のキットは、たとえば経口と非経口の異なる剤形を投与する、別個の組成物を異なる投与間隔で投与する、または別個の組成物を互いに対して滴定するのに特に好適である。服薬遵守を援助するために、キットは通常、投与の説明書を含み、いわゆるメモリーエイドを添えて提供することができる。
【0134】
投与量
ヒト患者への投与について、本発明の化合物の合計1日量は通常、当然ながら投与方式に応じて10mg〜1000mgの範囲にある。たとえば、経口投与には10mg〜1000mgの合計1日量が必要である場合があり、静脈内用量には10mg〜1000mgが必要となる場合がある。合計1日量は、1回で、または数回に分けて投与することができ、医師の裁量で、本明細書で示す典型的な範囲の範囲外になることもある。
【0135】
これらの投与量は、体重が約60kg〜70kgである平均的なヒト対象に基づく。医師は、小児や高齢者などの、体重がこの範囲外にある対象のための用量を容易に決定することができよう。
【0136】
疑義を避けるために、本明細書における「治療」への言及は、治療的、姑息的、および予防的な治療への言及を含む。
【0137】
式(I)の化合物はすべて、以下で述べる一般法に記載の手順によって、または実施例の項および調製例の項に記載の特定の方法によって、またはその常法どおりの変更形態によって調製することができる。本発明はまた、式(I)の化合物の調製方法のいずれか1つまたは複数に加えて、その中で使用する任意の新規な中間体も包含する。
【0138】
一般法
以下の略語を使用する。
DMF=ジメチルホルムアミド
DMSO=ジメチルスルホキシド
TEMPO=2,2,6,6−テトラメチルピペリジン−N−オキシド
THF=テトラヒドロフラン
DCM=ジクロロメタン
【0139】
式(I)の化合物は、以下のスキーム1に示すとおりに調製することができる。
【0140】
【化4】
【0141】
スキーム1において、Pはヒドロキシ保護基を表し、その好適な例は、T.W.GreeneおよびP.Wutsによる「Protective Groups in Organic Synthesis」、Wiley and Sons、1991年に記載されており、LGは、ハロゲン、(C1〜6アルキル)スルホニルオキシ(たとえばメタン−スルホニルオキシ)、(C1〜6ハロアルキル)スルホニルオキシ(たとえばトリフルオロメタンスルホニルオキシ)、またはベンゼンもしくはトルエンスルホニルオキシ(たとえばp−トルエンスルホニルオキシ)などの好適な脱離基を表す。Pがベンジルであり、LGがp−トルエンスルホニルオキシであることが好ましい。
【0142】
ステップ(a):式(III)の化合物は、化合物(II)と、ヒドロキシ基を脱離基に変換することのできる適切な薬剤、通常はスルホニル化試薬(たとえば塩化メタンスルホニルまたは塩化p−トルエンスルホニル)とから、塩基(たとえばトリエチルアミンまたはピリジン)の存在下、好適な溶媒(たとえばピリジンまたはジクロロメタン)中にて、0℃から室温で15分間〜24時間かけて調製することができる。
【0143】
好ましい条件は次のとおりである。ジクロロメタン中の1当量の化合物(II)、1.2当量の塩化p−トルエンスルホニル、2当量のピリジン、室温で18時間。
【0144】
ステップ(b):式(IV)の化合物は、化合物(III)と式(VI)のヒドロキシ化合物とから、好適な溶媒(たとえばDMF、DMSO)中にて、好適な塩基(たとえばCs2CO3、K2CO3)の存在下、場合によりクラウンエーテル(たとえば18−クラウン−6)の存在下、50〜120℃で一晩かけて調製することができる。
【0145】
好ましい条件は次のとおりである。1当量の化合物(VI)、1.1当量の化合物(III)、1.2当量のCs2CO3、DMF中にて80℃で24時間。
【0146】
式(VI)の化合物は、WO02/074754に一般に記載されている。XがOであり、mが1であり、RがClである特定の式(VI)の化合物は、Bioorg.Med.Chem.Lett.(2004年)、第14巻(18)、4627〜32ページに記載のとおり、または以下のスキーム5で概略を述べるとおりに調製することができる。
【0147】
ステップ(c):式(IV)の化合物を好適な溶媒中で脱保護剤と反応させて脱保護すると、式(V)の化合物を得ることができる。好適な試薬および方法は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。Pがベンジルであるとき、好適な試薬の例には、三塩化ホウ素または塩化鉄(III)が含まれる。
【0148】
好ましい条件は次のとおりである。ジクロロメタン中の1当量の化合物(IV)、4当量のBCl3、室温で18時間。
【0149】
ステップ(d):好適な溶媒中で酸化剤を使用して式(V)の化合物を酸化することにより、式(I)の化合物を調製することができる。典型的な試薬および条件としては、三酸化クロムおよび過ヨウ素酸(H5IO6)を触媒とし、アセトニトリルなどの溶媒中にて室温から50℃で18〜36時間、または別法としてNaOClおよびNaClO2、触媒のTEMPOの存在下、アセトニトリルなどの溶媒中にて0℃〜室温で18〜36時間が挙げられる。
【0150】
好ましい条件は次のとおりである。1当量の化合物(V)、2.5当量の過ヨウ素酸、0.02当量のCrO3、0.75%アセトニトリル水溶液中にて40℃で24時間。
【0151】
別法として、式(I)の化合物は、スキーム2に示すように、式(V)の化合物を、式(VII)のアルデヒドを介する二段階の手順で酸化して調製することができる。
【0152】
【化5】
【0153】
ステップ(a):アルコール(V)をアルデヒド(VII)にする酸化は通常、触媒としてのTEMPOと共にNaOClを使用し、好適な溶媒、たとえばアセトニトリル、アセトン中にて0℃から室温で2〜18時間、または別法として、DMSOと共に三酸化物硫黄−ピリジン錯体を使用し、THFなどの溶媒中にて0℃から室温で2〜18時間実施する。
【0154】
ステップ(b):アルデヒド(VII)を酸(I)にするさらなる酸化は通常、NaClO2を使用し、リン酸カリウムの存在下、t−ブタノール水溶液などの溶媒中にて0℃から室温で2〜18時間、または別法として、触媒としてのTEMPOと共にトリクロロイソシアニル酸を使用し、好適な溶媒、たとえばアセトンまたはアセトニトリル中にて0℃から室温で2〜18時間実施する。
【0155】
式(II)の化合物は文献で知られている。たとえば、Aがシス−1,3−シクロブチレン基であり、Bが単結合である式(II)の化合物は、J.Chem.Soc.、Perkin Trans.1(1995年)、第18巻、2281〜7ページに記載のとおりに調製することができる。
【0156】
別法として、Aがシス−またはトランス−1,3−シクロブチレン基であり、Bが単結合である式(I)の化合物である式(Ib)の化合物は、化合物(VIII)または化合物(IX)から、スキーム3に示すような標準の方法によって調製することができる。トランス化合物(II)および(X)は、それぞれシス化合物(II)および(X)から、Synthesis(1981年)、第1巻に記載のものと類似の光延化学を使用する反転によって得ることができる。
【0157】
【化6】
【0158】
スキーム3において、Raはエステル残基であり、その好適な例は、上でプロドラッグに関して述べ、また「Protective Groups in Organic Synthesis」(前掲書)に記載されており(たとえば、(C1〜6)アルキル、ベンジル、または(+)もしくは(−)−メンチル)、LGは、ハロゲン、(C1〜6アルキル)スルホニルオキシ(たとえばメタンスルホニルオキシ)、(C1〜6ハロアルキル)スルホニルオキシ(たとえばトリフルオロメタンスルホニルオキシ)、またはベンゼン−もしくはトルエンスルホニルオキシ(たとえばp−トルエンスルホニルオキシ)などの脱離基である。
【0159】
ステップ(a):式(IX)の化合物は、化合物(VIII)と式RaOHの好適なアルコール(たとえばメタノール、t−ブタノール、ベンジルアルコール、または(−)メントール)とを、様々な条件下で反応させて調製することができ、その好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。
【0160】
好ましい条件は次のとおりである。1当量の化合物(VIII)、1.1当量の1,1’−カルボニルジイミダゾール、酢酸エチル中にて還流温度で1時間、その後1当量のRaOH、室温で4時間。
【0161】
ステップ(b):化合物(IX)をアルコール(X)にする還元は、THFなどの好適な溶媒中で、好適な還元剤、たとえば水素化ホウ素ナトリウムまたはL−Selectride(登録商標)を使用して実施することができる。
【0162】
好ましい条件は次のとおりである。1当量の化合物(IX)、0.5当量のNaBH4、20:1のTHF:メタノール中にて0℃で20分間。
【0163】
ステップ(c):式(XI)の化合物は、化合物(X)から、スキーム1ステップ(a)に記載のものと同様の試薬および条件を使用して調製することができる。
【0164】
好ましい条件は次のとおりである。1当量の化合物(X)、1.05当量の塩化p−トルエンスルホニル、ピリジン中、0℃から室温。
【0165】
ステップ(d):式(Ia)の化合物は、化合物(XI)と式(VI)のヒドロキシ化合物とから、スキーム1ステップ(b)に記載のものと同様の試薬および条件を使用して調製することができる。
【0166】
好ましい条件は次のとおりである。1.2当量の化合物(XI)、1.0当量の化合物(VI)、1.5当量のCs2CO3、DMF中にて80℃で18時間。
【0167】
ステップ(e):式(Ia)の化合物を加水分解すると、式(Ib)の化合物を得ることができる。この反応は、様々な条件下で実現することができ、その好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。好ましい条件は次のとおりである。化合物(Ia)、2当量のNaOH、1:1のエタノール:水中にて60℃で2時間。
【0168】
化合物(VIII)は、J.Org.Chem.、(1981年)、第53巻、3841〜43ページに記載されており、Raがメチル基である化合物(IX)は、J.Org.Chem.、(1994年)、第59巻、2132〜34ページに記載されている。
【0169】
【化7】
【0170】
Bがメチレン基である式(I)の化合物である式(Id)の化合物は、スキーム4に示すとおりに調製することができる。
【0171】
【化8】
【0172】
スキーム4において、Raはエステル残基であり、その好適な例は、上でプロドラッグに関して述べており、また「Protective Groups in Organic Synthesis」(前掲書)に記載されており(たとえば(C1〜6)アルキルまたはベンジル)、LGは、ハロゲン、(C1〜6アルキル)スルホニルオキシ(たとえばメタンスルホニルオキシ)、(C1〜6ハロアルキル)スルホニルオキシ(たとえばトリフルオロメタンスルホニルオキシ)、またはベンゼン−もしくはトルエンスルホニルオキシ(たとえばp−トルエンスルホニルオキシ)などの脱離基である。Raがベンジルであり、LGがp−トルエンスルホニルオキシであることが好ましい。式(XII)の化合物は、市販品として得ることができる。
【0173】
ステップ(a):式(XIII)の化合物は、酸性または塩基性条件下、たとえば、メタノール、エタノール、1,4−ジオキサンなどを好適な共溶媒とする水酸化ナトリウム水溶液中で、またはエタノールや1,4−ジオキサンなどを場合により好適な共溶媒とする塩酸もしくは硫酸水溶液中で、式(XII)の化合物を加水分解することにより調製できる。
【0174】
好ましい条件は次のとおりである。1当量の化合物(XII)、4当量のNaOH、1:1のエタノール:水中にて還流温度で2.5時間。
【0175】
ステップ(b):式(XIV)の化合物は、式(XIII)の化合物と式RaOHの好適なアルコール(たとえばメタノール、t−ブタノール、ベンジルアルコール)とを、様々な条件下で反応させて調製することができ、その条件の好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。好ましい条件は次のとおりである。1当量の化合物(XIII)、1.1当量の1,1’−カルボニルジイミダゾール、酢酸エチル中、約1時間、その後1.2当量のベンジルアルコール、室温で18時間。
【0176】
ステップ(c):式(XV)の化合物は、THFなどの好適な溶媒中にて、0℃から室温で、式(XIV)の化合物をボラン−ジメチルスルフィド、カテコールボラン、9−ボラビシクロ[3.3.1]ノナン(9−BBN)などのヒドロホウ素化剤で処理した後、in situで、室温から60℃にて、過酸化水素、過ホウ酸ナトリウム、トリメチルアミン−N−オキシドなどの酸化剤で酸化して調製することができる。
【0177】
好ましい条件は次のとおりである。1当量の化合物(XIV)、0.5当量のボラン−ジメチルスルフィド、THF中にて室温で1時間、その後1.2当量の過ホウ酸ナトリウム、60℃で1時間の加熱。
【0178】
ステップ(d):式(XVI)の化合物は、式(XV)の化合物から、スキーム1ステップ(a)に記載のものと同様の試薬および条件を使用して調製することができる。
【0179】
好ましい条件は次のとおりである。1当量の化合物(XV)、1.3当量の塩化p−トルエンスルホニル、2.6当量のピリジン、DCM中、0℃から室温。
【0180】
ステップ(e):式(Ic)の化合物は、式(XVI)の化合物と式(VI)のヒドロキシ化合物とから、スキーム1ステップ(b)に記載のものと同様の試薬および条件を使用して調製することができる。
【0181】
好ましい条件は次のとおりである。1.2当量の化合物(XVI)、1.0当量の化合物(VI)、1.5当量のCs2CO3、DMF中にて80℃で18時間。
【0182】
ステップ(f):式(Ic)の化合物を加水分解すると、式(Id)の化合物を得ることができる。この反応は、様々な条件下で実現することができ、その好適な例は、「Protective Groups in Organic Synthesis」(前掲書)に記載されている。
【0183】
好ましい条件は次のとおりである。化合物(Ic)、過剰のNaOH、1:1のエタノール:水中にて60℃で2時間。
【0184】
式(VI)の化合物は、WO02/074754に一般に記載されている。XがOまたはSである式(VI)の化合物である特定の式(VIa)の化合物は、Bioorg.Med.Chem.Lett.、(2004年)、第14巻(18)、4627〜32ページに記載のとおりに、または以下のスキーム5で概略を述べるとおりに調製することができる。
【0185】
【化9】
【0186】
スキーム5において、Rbは(C1〜6)アルキルまたはベンジルである。
【0187】
ステップ(a):式(XVIII)の化合物は、アニリン(XVII)とナトリウムまたはカリウムのシアン酸塩またはチオシアン酸塩とを、マレイン酸や酢酸などの酸の存在下、好適な溶媒または溶媒混合物中、たとえばジクロロメタンまたは酢酸:水中で反応させて調製することができる。別法として、式(XVIII)の化合物は、アニリン(XVII)とイソシアン酸またはチオシアン酸塩トリメチルシリルとを、ジクロロメタンなどの溶媒中で反応させた後、in situで水によって加水分解して調製することができる。
【0188】
XがOであるときの好ましい条件は次のとおりである。1当量の化合物(XVII)、酢酸:水(9:1)中、その後1.2当量のシアン酸カリウム、水中、滴下、40℃で1時間保つ。
【0189】
ステップ(b):式(XIX)の化合物は、式(XVIII)の尿素と適切なケトンとを、ポリリン酸やEaton試薬(メタンスルホン酸中7.5%P2O5)などの脱水剤の存在下、50℃〜100℃の間で反応させて調製することができる。
【0190】
好ましい条件は次のとおりである。1当量の化合物(XVIII)、Eaton試薬(30g/g)、60℃、その後2当量のケトン、80℃で1時間の加熱。
【0191】
ステップ(c):式(VIa)の化合物は、式(XIX)の化合物を、ジクロロメタンなどの好適な溶媒中にて室温で三臭化ホウ素などのルイス酸と反応させ、または高温で強酸と、たとえば110℃で臭化水素酸と反応させることにより調製できる。
【0192】
好ましい条件は次のとおりである。1当量の化合物(XIX)、20当量の48%臭化水素水溶液、酢酸中にて110℃で4日間。
【0193】
式(I)のPDE7阻害剤は、特に疼痛の治療において、もう1種の薬理活性のある化合物、または2種以上の他の薬理活性のある化合物と有益に組み合わせることができる。たとえば、上で定義した、式(I)のPDE7阻害剤、または薬学的に許容できるその塩、溶媒和物、もしくはプロドラッグは、以下のものから選択される1種または複数の薬剤と組み合わせて、同時、逐次、または別々に投与することができる。
・オピオイド鎮痛薬、たとえば、モルヒネ、ヘロイン、ヒドロモルフォン、オキシモルフォン、レボルファノール、レバロルファン、メサドン、メペリジン、フェンタニル、コカイン、コデイン、ジヒドロコデイン、オキシコドン、ハイドロコドン、プロポキシフェン、ナルメフェン、ナロルフィン、ナロキソン、ナルトレキソン、ブプレノルフィン、ブトルファノール、ナルブフィン、またはペンタゾシン、
・非ステロイド性抗炎症薬(NSAID)、たとえば、アスピリン、ジクロフェナク、ジフルシナル(diflusinal)、エトドラク、フェンブフェン、フェノプロフェン、フルフェニサール(flufenisal)、フルルビプロフェン、イブプロフェン、インドメタシン、ケトプロフェン、ケトロラック、メクロフェナム酸、メフェナム酸、メロキシカム、ナブメトン、ナプロキセン、ニメスリド、ニトロフルルビプロフェン、オルサラジン、オキサプロジン、フェニルブタゾン、ピロキシカム、スルファサラジン、スリンダク、トルメチン、またはゾメピラク、
・バルビツール酸系鎮静薬、たとえば、アモバルビタール、アプロバルビタール、ブタバルビタール、ブタビタール(butabital)、メフォバルビタール、メタルビタール、メトヘキシタール、ペントバルビタール、フェノバルチタール(phenobartital)、セコバルビタール、タルブタール、テアミラル(theamylal)、またはチオペンタール、
・鎮静作用を有するベンゾジアゼピン、たとえば、クロルジアゼポキシド、クロラゼプ酸、ジアゼパム、フルラゼパム、ロラゼパム、オキサゼパム、テマゼパム、またはトリアゾラム、
・鎮静作用を有するH1拮抗薬、たとえば、ジフェンヒドラミン、ピリラミン、プロメタジン、クロルフェニラミン、またはクロルシクリジン、
・鎮静薬、たとえば、グルテチミド、メプロバメート、メタカロン、またはジクロラールフェナゾンなど、
・骨格筋弛緩薬、たとえば、バクロフェン、カリソプロドール、クロルゾキサゾン、シクロベンザプリン、メトカルバモール、またはオルフレナジン(orphrenadine)、
・NMDA受容体拮抗薬、たとえば、デキストロメトルファン((+)−3−ヒドロキシ−N−メチルモルヒナン)もしくはその代謝産物デキストロルファン((+)−3−ヒドロキシ−N−メチルモルヒナン)、ケタミン、メマンチン、ピロロキノリンキニン、シス−4−(ホスホノメチル)−2−ピペリジンカルボン酸、ブジピン、EN−3231(MorphiDex(登録商標)、モルヒネとデキストロメトルファンの合剤)、トピラメート、ネラメキサン(neramexane)、またはペルジンフォテル(perzinfotel)(NR2B拮抗薬、たとえば、イフェンプロジル、トラキソプロジル(traxoprodil)、または(−)−(R)−6−{2−[4−(3−フルオロフェニル)−4−ヒドロキシ−1−ピペリジニル]−1−ヒドロキシエチル−3,4−ジヒドロ−2(1H)−キノリノンを含む)、
・α−アドレナリン作動薬、たとえば、ドキサゾシン、タムスロシン、クロニジン、グァンファシン、デクスメタトミジン(dexmetatomidine)、モダフィニル、または4−アミノ−6,7−ジメトキシ−2−(5−メタン−スルホンアミド−1,2,3,4−テトラヒドロイソキノール−2−イル)−5−(2−ピリジル)キナゾリン、
・三環系抗うつ薬、たとえば、デシプラミン、イミプラミン、アミトリプチリン、またはノルトリプチリン、
・抗痙攣薬、たとえば、カルバマゼピン、ラモトリジン、トピラトメート(topiratmate)、またはバルプロエート、
・タキキニン(NK)拮抗薬、特にNK−3、NK−2、またはNK−1拮抗薬、たとえば、(αR,9R)−7−[3,5−ビス(トリフルオロメチル)ベンジル]−8,9,10,11−テトラヒドロ−9−メチル−5−(4−メチルフェニル)−7H−[1,4]ジアゾシノ[2,1−g][1,7]−ナフチリジン−6−13−ジオン(TAK−637)、5−[[(2R,3S)−2−[(1R)−1−[3,5−ビス(トリフルオロメチル)フェニル]エトキシ−3−(4−フルオロフェニル)−4−モルホリニル]−メチル]−1,2−ジヒドロ−3H−1,2,4−トリアゾール−3−オン(MK−869)、アプレピタント、ラネピタント、ダピタント、または3−[[2−メトキシ−5−(トリフルオロメトキシ)フェニル]−メチルアミノ]−2−フェニルピペリジン(2S,3S)、
・ムスカリン性拮抗薬、たとえば、オキシブチニン、トルテロジン、プロピベリン、塩化トロプシウム(tropsium chloride)、ダリフェナシン、ソリフェナシン、テミベリン、およびイプラトロピウム、
・COX−2選択的阻害剤、たとえば、セレコキシブ、ロフェコキシブ、パレコキシブ、バルデコキシブ、デラコキシブ(deracoxib)、エトリコキシブ、またはルミラコキシブ、
・コールタール鎮痛薬、特にパラセタモール、
・神経弛緩薬、たとえば、ドロペリドール、クロルプロマジン、ハロペリドール、ペルフェナジン、チオリダジン、メソリダジン、トリフルオペラジン、フルフェナジン、クロザピン、オランザピン、リスペリドン、ジプラシドン、クエチアピン、セルチンドール、アリピプラゾール、ソネピプラゾール、ブロナンセリン、イロペリドン、ペロスピロン、ラクロプライド、ゾテピン、ビフェプルノックス、アセナピン、ルラシドン、アミスルプリド、バラペリドン、パリンドーレ(palindore)、エプリバンセリン、オサネタント、リモナバント、メクリネルタント(meclinertant)、Miraxion(登録商標)、サリゾタン(sarizotan)など、
・バニロイド受容体作動薬(たとえばレシンフェラトキシン(resinferatoxin))または拮抗薬(たとえばカプサゼピン)、
・プロプラノロールなどのβ−アドレナリン作動薬、
・メキシレチンなどの局所麻酔薬、
・デキサメタゾンなどの副腎皮質ステロイド、
・5−HT受容体作動薬または拮抗薬、特に、エレトリプタン、スマトリプタン、ナラトリプタン、ゾルミトリプタン、リザトリプタンなどの5−HT1B/1D作動薬、
・R(+)−α−(2,3−ジメトキシ−フェニル)−1−[2−(4−フルオロフェニルエチル)]−4−ピペリジンメタノール(MDL−100907)などの5−HT2A受容体拮抗薬、
・コリン作用性(ニコチン性)鎮痛薬、たとえば、イスプロニクリン(TC−1734)、(E)−N−メチル−4−(3−ピリジニル)−3−ブテン−1−アミン(RJR−2403)、(R)−5−(2−アゼチジニルメトキシ)−2−クロロピリジン(ABT−594)、ニコチンなど、
・Tramadol(登録商標)、
・PDEV阻害剤、たとえば、5−[2−エトキシ−5−(4−メチル−1−ピペラジニル−スルホニル)フェニル]−1−メチル−3−n−プロピル−1,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン(シルデナフィル)、(6R,12aR)−2,3,6,7,12,12a−ヘキサヒドロ−2−メチル−6−(3,4−メチレンジオキシフェニル)−ピラジノ[2’,1’:6,1]−ピリド[3,4−b]インドール−1,4−ジオン(IC−351またはタダラフィル)、2−[2−エトキシ−5−(4−エチル−ピペラジン−1−イル−1−スルホニル)−フェニル]−5−メチル−7−プロピル−3H−イミダゾ[5,1−f][1,2,4]トリアジン−4−オン(バルデナフィル)、5−(5−アセチル−2−ブトキシ−3−ピリジニル)−3−エチル−2−(1−エチル−3−アゼチジニル)−2,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン、5−(5−アセチル−2−プロポキシ−3−ピリジニル)−3−エチル−2−(1−イソプロピル−3−アゼチジニル)−2,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン、5−[2−エトキシ−5−(4−エチルピペラジン−1−イルスルホニル)ピリジン−3−イル]−3−エチル−2−[2−メトキシエチル]−2,6−ジヒドロ−7H−ピラゾロ[4,3−d]ピリミジン−7−オン、4−[(3−クロロ−4−メトキシベンジル)アミノ]−2−[(2S)−2−(ヒドロキシメチル)ピロリジン−1−イル]−N−(ピリミジン−2−イルメチル)ピリミジン−5−カルボキサミド、3−(1−メチル−7−オキソ−3−プロピル−6,7−ジヒドロ−1H−ピラゾロ[4,3−d]ピリミジン−5−イル)−N−[2−(1−メチルピロリジン−2−イル)エチル]−4−プロポキシベンゼンスルホンアミドなど、
・α−2−δリガンド、たとえば、ギャバペンチン、プレガバリン、3−メチルギャバペンチン、(1α,3α,5α)(3−アミノ−メチル−ビシクロ[3.2.0]ヘプタ−3−イル)−酢酸、(3S,5R)−3−アミノメチル−5−メチル−ヘプタン酸、(3S,5R)−3−アミノ−5−メチル−ヘプタン酸、(3S,5R)−3−アミノ−5−メチル−オクタン酸、(2S,4S)−4−(3−クロロフェノキシ)プロリン、(2S,4S)−4−(3−フルオロベンジル)−プロリン、[(1R,5R,6S)−6−(アミノメチル)ビシクロ[3.2.0]ヘプタ−6−イル]酢酸、3−(1−アミノメチル−シクロヘキシルメチル)−4H−[1,2,4]オキサジアゾール−5−オン、C−[1−(1H−テトラゾール−5−イルメチル)−シクロヘプチル]−メチルアミン、(3S,4S)−(1−アミノメチル−3,4−ジメチル−シクロペンチル)−酢酸、(3S,5R)−3−アミノメチル−5−メチル−オクタン酸、(3S,5R)−3−アミノ−5−メチル−ノナン酸、(3S,5R)−3−アミノ−5−メチル−オクタン酸、(3R,4R,5R)−3−アミノ−4,5−ジメチル−ヘプタン酸、(3R,4R,5R)−3−アミノ−4,5−ジメチル−オクタン酸など、
・カンナビノイド、
・代謝調節型グルタミン酸サブタイプ1受容体(mGluR1)拮抗薬、
・セロトニン再取込み阻害剤、たとえばセルトラリン、セルトラリン代謝産物のデメチルセルトラリン、フルオキセチン、ノルフルオキセチン(フルオキセチンデスメチル代謝産物)、フルボキサミン、パロキセチン、シタロプラム、シタロプラム代謝産物のデスメチルシタロプラム、エスシタロプラム、d,l−フェンフルラミン、フェモキセチン、イフォキセチン(ifoxetine)、シアノドチエピン(cyanodothiepin)、リトキセチン、ダポキセチン、ネファゾドン、セリクラミン、トラゾドンなど、
・ノルアドレナリン(ノルエピネフリン)再取込み阻害剤、たとえば、マプロチリン、ロフェプラミン、ミルタゼピン(mirtazepine)、オキサプロチリン、フェゾラミン、トモキセチン、ミアンセリン、ブプロプリオン、ブプロプリオン代謝産物のヒドロキシブプロプリオン、ノミフェンシン、ビロキサジン(Vivalan(登録商標))など、特に選択的ノルアドレナリン再取込み阻害剤、たとえばレボキセチン、特に(S,S)−レボキセチンなど、
・セロトニン−ノルアドレナリン再取込み二重阻害剤、たとえば、ベンラフェキシン、ベンラフェキシン代謝産物のO−デスメチルベンラフェキシン、クロミプラミン、クロミプラミン代謝産物のデスメチルクロミプラミン、デュロキセチン、ミルナシプラン、イミプラミンなど、
・誘導型一酸化窒素合成酵素(iNOS)阻害剤、たとえば、S−[2−[(1−イミノエチル)アミノ]エチル]−L−ホモシステイン、S−[2−[(1−イミノエチル)−アミノ]エチル]−4,4−ジオキソ−L−システイン、S−[2−[(1−イミノエチル)アミノ]エチル]−2−メチル−L−システイン、(2S,5Z)−2−アミノ−2−メチル−7−[(1−イミノエチル)アミノ]−5−ヘプテン酸、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)−ブチル]チオ]−5−クロロ−3−ピリジンカルボニトリル、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)ブチル]チオ]−4−クロロベンゾニトリル、(2S,4R)−2−アミノ−4−[[2−クロロ−5−(トリフルオロメチル)フェニル]チオ]−5−チアゾールブタノール、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)ブチル]チオ]−6−(トリフルオロメチル)−3ピリジンカルボニトリル、2−[[(1R,3S)−3−アミノ−4−ヒドロキシ−1−(5−チアゾリル)ブチル]チオ]−5−クロロベンゾニトリル、N−[4−[2−(3−クロロベンジルアミノ)エチル]フェニル]チオフェン−2−カルボキサミジン、グアニジノエチルジスルフィドなど、
・ドネペジルなどのアセチルコリンエステラーゼ阻害剤、
・プロスタグランジンE2サブタイプ4(EP4)拮抗薬、たとえば、N−[({2−[4−(2−エチル−4,6−ジメチル−1H−イミダゾ[4,5−c]ピリジン−1−イル)フェニル]エチル}アミノ)−カルボニル]−4−メチルベンゼンスルホンアミドや4−[(1S)−1−({[5−クロロ−2−(3−フルオロフェノキシ)ピリジン−3−イル]カルボニル}アミノ)エチル]安息香酸など、
・ロイコトリエンB4拮抗薬、たとえば、1−(3−ビフェニル−4−イルメチル−4−ヒドロキシ−クロマン−7−イル)−シクロペンタンカルボン酸(CP−105696)、5−[2−(2−カルボキシエチル)−3−[6−(4−メトキシフェニル)−5E−ヘキセニル]オキシフェノキシ]−吉草酸(ONO−4057)、DPC−11870など、
・5−リポキシゲナーゼ阻害剤、たとえば、ジロートン、6−[(3−フルオロ−5−[4−メトキシ−3,4,5,6−テトラヒドロ−2H−ピラン−4−イル])フェノキシ−メチル]−1−メチル−2−キノロン(ZD−2138)、2,3,5−トリメチル−6−(3−ピリジルメチル),1,4−ベンゾキノン(CV−6504)など、
・リドカインなどのナトリウムチャネル遮断薬、
・オンダンセトロンなどの5−HT3拮抗薬、
ならびにこれらの薬学的に許容できる塩および溶媒和物。
【0194】
式(I)の化合物のPDE7阻害能は、以下のアッセイプロトコルを使用して測定することができる。
【0195】
PDE7AおよびPDE7B酵素は、3’,5’−環状アデノシン一リン酸(cAMP)を5’アデノシン一リン酸酸、すなわち5’AMPにする加水分解を触媒する。マルチウェルプレートにおいて、PDE酵素、[3H]−cAMP、および試験化合物を室温でインキュベートする。硫酸亜鉛を含有する市販のケイ酸イットリウムシンチレーション近接アッセイ(SPA)ビーズを加えて、インキュベートを終了する。ケイ酸イットリウムビーズは、直鎖状のヌクレオチドを優先的に結合し、したがって酵素反応の産物である[3H]−5’AMPがこのビーズに結合して、光シグナルを生成し、これがシンチレーション計数器によって検出される。生成されたシグナルの量は、生成された産物の量、すなわち酵素の活性と直接に相関する。酵素および基質を単独でインキュベートする場合に最大のシグナルが得られる。酵素を含有しないウェル、または最大上濃度の既知のPDE7A/B阻害剤を含有するウェルからのバックグラウンドシグナルを測定する。精製した各酵素バッチを品質管理下に置き、そのKm、Vmax、および比活性を動力学的研究から求めた後、化合物阻害研究で使用する。試験化合物による酵素の阻害を、最大反応およびバックグラウンド反応と比較して算出する。これらのデータを使用し、得られた最大値および最小値と比較して%阻害値を算出する。
【0196】
作用溶液の調製
以下の表1に示す成分から、1000mlの保存緩衝液を調製した。
【0197】
【表1】
【0198】
保存緩衝液を室温でpH7.4に調整し、次いで0.2μmフィルターで濾過した。保存緩衝液は、4℃で調製日から1ヶ月間安定である。
【0199】
実験日に、ウシ血清アルブミン(BSA、Sigmaから入手可能)を必要な体積の緩衝液に加えて、0.00625%のBSA最終溶液を作った。これは、保存10%BSA溶液を次のように調製することにより実現した。
【0200】
保存10%BSA溶液の調製
1gのBSAを10mlの精製水に溶解させ、反転させて混合し、確実に均質になるようにし、適切にラベルを貼った管に100μl体積で等分した。10%BSA溶液は、−20℃で最長6ヶ月間安定である。
【0201】
一定分量の保存10%BSA保存液を保管所から取り出し、使用する前に室温で解凍して、以下の表2に示すようにBSA作用溶液を作った。
【0202】
10mlの作用BSAアッセイ緩衝液の調製
【0203】
【表2】
【0204】
標準化合物および対照の調製
WO02/074754の実施例75の化合物、すなわち5’−カルボキシプロポキシ−8’−クロロ−スピロ[シクロヘキサン−1−4’−(3’,4’−ジヒドロ)キナゾリン]−2’(1’H)−オン(以下では「化合物A」)を標準物質として使用した。
【0205】
100%DMSO中に調製した4mMの保存液は、4℃で保存することができる。DMSOの体積は、次のように算出することができる。
【0206】
【数1】
【0207】
30倍最大対照は、100%DMSOの溶液である。酵素活性をもたらさない30倍最小対照は、30μMの100%DMSO中化合物Aを使用して実現する。100%DMSO4.962mlを4mMの化合物A37.5μlに加えて、化合物Aの30μM溶液5mlを調製することができる。
【0208】
方法
アッセイ日に、前もって詳述したとおりに1倍の最終アッセイ緩衝液を調製し、必要になるまで氷上で保存した。
【0209】
動力学的研究
それぞれの新たな酵素バッチについて、Kmを求め、反応進行曲線の線形部分に残る間、45分間で約1000cpmのシグナルを得るのに必要な酵素の量を評価した。アッセイの過程の間に<10%の利用可能な[3H]−cAMPが加水分解されることが理想的である。
【0210】
酵素溶液
このアッセイの最適化は、全長PDE7AおよびPDE7B酵素を含有する細胞溶解液を使用して実施している。この細胞溶解物サンプル中の酵素の濃度は不明であるので、細胞溶解物の比活性を尺度として用いて、濃度/活性のバッチごとのどんな差異にもかかわらず、確実にウェルあたり同一の活性が使用されるようにする。
【0211】
PDE7A/B酵素の調製
PDE7保存酵素を調製し、凍結/解凍サイクルの数を減らすために適切にサイズ分けされた一定分量にして−20℃で保存した。以下の表3は、9mlのPDE7A/B酵素溶液を生成するのに必要な体積を示す。PDE7Aは1/8000に、PDE7Bは1/10000に希釈する。
【0212】
【表3】
【0213】
酵素溶液を調製したならば、それを使用前に氷上で保存した。
【0214】
50nMのアデノシン3’,5’環状リン酸(cAMP)基質溶液の調製
基質は、未標識cAMPとトリチウムで放射標識したcAMP([3H]−cAMP)の混合物から構成されるものである。[3H]−cAMP保存物の規格によって、使用する体積が決まる。
【0215】
1mCi/mlおよび24Ci/mmol(したがって41.66μM)である[3H]−cAMP保存物を使用する9mlの基質溶液の調製について以下で述べる。
【0216】
酵素バッチのKmは、これまでのところ以下のとおりである。
PDE7A−20nM PDE7B−100nM
【0217】
アッセイでは、15μlの基質溶液が、分注されて30μlの合計アッセイ体積になる、すなわちアッセイプレートで2倍希釈が行われる必要がある。
【0218】
約25nMの最終アッセイ[cAMP]が必要となるので、約50nMの[3H]−cAMPを調製した。
【0219】
10.8μlの[3H]−cAMP(Amershamから入手可能)と8975μlのアッセイ緩衝液とを混合して、9mlの基質溶液を調製した。
【0220】
3本のシンチレーションバイアルに15μlのサンプルを取って、cAMPの正確な濃度を決定した。次いで、4mlのStarscint(登録商標)(シンチレーションカクテル、Perkin Elmerから入手可能)を加え、管をdpmプログラムによるβカウンターでのカウントにかけた。
【0221】
放射性リガンドの濃度は、以下の式によって求める。
【0222】
【数2】
【0223】
次いで、濃度を2で割って、アッセイプレートで2倍希釈が行われていることを計算に入れる。
【0224】
6.6mg/mlのケイ酸イットリウムPDE SPAビーズの調製
ホスホジエステラーゼSPAビーズ(ケイ酸イットリウム)は、Amershamから入手可能である。
【0225】
製造者の推奨に従い、28mlの蒸留水または脱イオン水(約20mg/ml)を使用して、ビーズのバイアルを再構成した。再構成されたビーズは、2〜8℃で保存したとき、1ヶ月間安定である。アッセイ用のビーズを調製するために、再構成されたビーズを無菌二重蒸留水(約6.6mg/ml)中に3倍希釈した。ビーズを安定させることができるので、分注しながら一定にかき混ぜた/攪拌した(stirred/agitated)。
【0226】
約6.6mg/mlのビーズ30μlを30μlのアッセイに加えて、最終ビーズ濃度を約0.2mg/ウェルとする。
【0227】
化合物希釈物および「バックグラウンド」ウェルは、アッセイプレートにおいて必要となるものの30倍にしてあるので、1μlの化合物を29μlの他のアッセイ構成要素(14μlの酵素および15μlの放射性リガンド)によって希釈した。すなわち、最終アッセイ濃度10μMでは、化合物は、化合物添加プレートにおいて300μMでなければならない。化合物の4mM保存物は、100%DMSO中に供給される(または粉末提出物から4mMで作られる)。これには、DMSO中で1/13.33希釈を行う必要がある。
【0228】
アッセイプロトコル
1μlの試験化合物を、試薬のアッセイ付加の直前に好適なマルチウェルアッセイプレートに移し、次いで14μlの酵素溶液をアッセイプレートに加えた後、15μlの基質溶液を加えた(すなわち、最終アッセイ体積30μl、最終スクリーニング化合物濃度1μM)。次いで、プレートシーラーを使用してプレートをシールし、プレート振盪機に載せて室温で45分間インキュベートした。
【0229】
次いで、30μlのケイ酸イットリウムPDE4 SPAビーズを加え、ビーズを一定に攪拌してアッセイプレート中に確実に一様な分布が得られるようにした。次いで、プレートシーラーを使用してプレートをシールし、プレート振盪機に載せて室温で30分間インキュベートした。次いで、ビーズを30分間安定させた後、プレートを200gで1分間回転させた。
【0230】
次いで、好適な放射能カウンター、たとえばNXT−TopCount(商標)(Perkin Elmerから入手可能)によって、関連するプロトコル(ウェルあたり30秒の読み時間)を使用してプレートを読み取った。
【0231】
最小二乗アルゴリズムを使用して、データをシグモイド曲線に適合させた。
【0232】
次のCheng−Prussofの式を使用して、IC50値をKi値に変換した。
【0233】
【数3】
【0234】
実施例1〜7の化合物のPDE7阻害活性を、上記プロトコルに従って試験した。得られたKi値を以下の表4に示す。
【0235】
【表4】
【0236】
ヒト肝細胞データの概要
本出願の実施例1〜7の化合物のヒト肝代謝安定性を、以下で述べるモデルで評価した。最も近い現況技術を表すと考えられるWO02/074754の実施例75の化合物5’−カルボキシプロポキシ−8’−クロロ−スピロ[シクロヘキサン−1−4’−(3’,4’−ジヒドロ)キナゾリン]−2’(1’H)−オン(以下では「化合物A」)を比較として使用した。
【0237】
方法
肝細胞をin vitro系として使用して、肝代謝をモニターする。というのは、これらの無処置細胞は、シトクロムP450オキシダーゼ(CYP)、アルデヒドオキシダーゼ、モノアミンオキシダーゼ(MAO)などの第一相酵素、およびUDP−グルクロニルトランスフェラーゼやスルホトランスフェラーゼなどの第二相酵素を含む、in vivoで見出されるすべての肝酵素を含有している。凍結保存したヒト肝細胞を5人のドナーから調製し、WilliamsのE培地に懸濁させた。4−(2−ヒドロキシエチル)−1−ピペラジンエタンスルホン酸(HEPES)を加えて最終濃度を50mMとし、pHを7.4に調整する。試験基質をDMSOに溶解し、肝細胞に加えて、基質濃度を1μMとし、インキュベートにおける最終DMSO濃度は0.1%より低くする。実験は、96または384ウェルプレートにおいて、肝細胞密度を50万生存細胞/mLとして37℃で実施する。10、20、30、60、90、および120分のサンプル採取時間を使用し、分析の定量化はLC−MS/MSによるものとする。次式を使用して、固有クリアランス(見かけ上)を算出する。
CLint,app=[−勾配/0.5M細胞/mL]・1000μL/mL=μL/分/M細胞
【0238】
本発明の化合物のヒト肝代謝安定性を、上記プロトコルに従って試験した。得られた固有クリアランス値を以下の表5に示す。
【0239】
【表5】
【0240】
上の表5に示すデータは、固有の肝代謝安定性に関して、本出願の実施例1〜7の化合物と、最も近い従来技術の化合物Aとの明らかな差異を示す。したがって、上記データに基づき、本出願の実施例1〜7の化合物は、肝クリアランスが低下しているので、化合物Aと比べてヒトにおける半減期の改善された化合物をもたらすと思われる。
【0241】
ラットIV薬物動態の概要
実施例1および2の化合物の薬物動態学的性質を、以下で述べるラットモデルで試験した。最も近い現況技術であると考えられるWO02/074754の実施例75の化合物5’−カルボキシプロポキシ−8’−クロロ−スピロ[シクロヘキサン−1−4’−(3’,4’−ジヒドロ)キナゾリン]−2’(1’H)−オン(以下では「化合物A」)を比較として使用した。
【0242】
方法
試験化合物を雄のラットに1mg/kg(実施例1の化合物については0.08mg/kg)の投与量で尾静脈から投与した(各ラットに1種類の化合物を与える)。投与後の所定の時点で、外科的に移植した頸静脈カニューレを介してラットから血液サンプルを抜き取り、遠心分離して血漿を生成した。血漿中の薬物を定量化するために、血漿サンプルを特定のLC−MS/MSアッセイによって分析した。各化合物の性質を理解するために、得られる血漿濃度−時間曲線を、非コンパートメントの薬物動態分析を使用して調べた。その後、得られる出力を使用して、確からしいヒト薬物動態プロフィールを以下で述べるとおりに推定した。
【0243】
実施例1および2の化合物ならびに化合物Aを1mg/kgで静脈内投与した後の、投与量で正規化した平均薬物動態プロフィールを以下の表6および図1に示す。
【0244】
【表6】
【0245】
表6では次の略語を使用する。
Clはラットクリアランスであり、
T1/2は半減期であり、
Vdはラットにおける分布体積であり、
fupはラット血漿中の非結合画分であり、
Cluは非結合ラット血漿クリアランスであり、Clu=CI/fupである。
【0246】
ヒト薬物動態の推定
ラットにおける上記薬物動態学的データに基づき、実施例1および2の化合物ならびに化合物Aの確からしいヒト薬物動態は、以下のとおりに推定できる。
【0247】
・以下の関係式を使用して、静脈内投与後に観察された非結合ラット血漿クリアランス(Cluラット)を基準化して、非結合ヒト血漿クリアランス(Cluヒト)を推定する。
Cluヒト=Cluラット*(BWヒト/BWラット)0.75
[式中、BWヒトおよびBWラットは、それぞれヒト(70kg)およびラット(0.25kg)の平均体重であり、クリアランスの単位はml/分である。]
【0248】
・Cluヒトをヒトにおける推定合計血液クリアランス(Clヒト)に変換する。
Clヒト=[(Cluヒト)*fup]/B:P
[式中、fupは、血漿中の非結合薬物の遊離画分であり、B:Pは、ヒト血液中の血液対血漿比である。]
【0249】
・次の関係式を使用して導かれるヒト半減期の推定値を各化合物について示す。
T1/2=[ln(2)*Vd]/Cl
[式中、T1/2は時間での推定ヒト半減期であり、Vdはヒトにおける分布体積であり(この系列の物理化学により0.2L/kgであると仮定される)、Clはヒトクリアランスである。]
【0250】
推定ヒト薬物動態の概要を以下の表7に示す。
【0251】
【表7】
【0252】
上記表6および7に示すデータでは、ラットにおける観察された薬物動態および推定ヒト薬物動態の両方に関して、従来技術の化合物Aと比べて、本出願の実施例2の化合物間に明らかな差異が示されている。このことは、約1時間のヒトでの半減期を示すと思われる化合物Aと比べて、実施例2の化合物では10時間であると推定された突出したヒト半減期に現れている。
【0253】
したがって、上記データに基づき、本出願の実施例2の化合物の薬物動態は、臨床現場では1日1回または2回の投薬に相応しいであろうと思われる。本出願の実施例1の化合物の薬物動態は、1日2回または3回の投薬に相応しいといえる。これは、その短い半減期のために同じようにして投与するのには適さないと思われる、最も近い従来技術の化合物Aに優る著しい進歩である。
【0254】
神経因性疼痛の治療における本発明による式(I)の化合物の活性は、以下の試験プロトコルに従って測定することができる。
【0255】
動物:雄のSprague Dawleyラット(平均体重500g)を12のグループにして収容する。動物はすべて、12時間の明/暗サイクル(7時間00分点灯)で、食物および水を自由に与えながら飼育する。実験はすべて、治療について知らない観察者によって、またHome Office Animals(Scientific Procedures)Act 1986に従って実施する。
【0256】
神経因性疼痛の慢性絞縮傷(CCI)ラットモデル
坐骨神経のCCIは、以前に記載されているとおりに実施する(G.J.BennettおよびY.K.Xie、Pain(1988年)第33巻、87〜107ページ)。動物を2%イソフルオラン(isofluorane)/O2混合物で麻酔した。右後部大腿を剪毛し、1%ヨウ素で拭く。次いで、手順を続ける間動物を恒温性のブランケットに移し、手術中はノーズコーンから麻酔を維持する。皮膚を大腿骨の線に沿って切開する。大腿二頭筋の鈍的切開によって総坐骨神経を大腿の中央で露出させる。神経の下に鉗子を挿入して、坐骨の三分岐の近位にある約7mmの神経をほどき、その神経を大腿から穏やかに引き上げる。鉗子を使用して、縫合糸を、神経をくぐらせて引っ張り、わずかな抵抗が感じられるまで一重結びで結び、次いで二重結びする。4本の結紮糸(4−0絹糸)が神経の周りに約1mm間隔で緩く結ばれるまで、この手順を繰り返す。切開を重ねて閉じ、創傷を局所用の抗生物質で処理する。
【0257】
ラットにおけるストレプトゾシン(STZ)誘発糖尿病ニューロパチー
0.9%の無菌食塩水に新たに溶解させたストレプトゾトシン(50mg/kg)の単回腹腔内投与によって、糖尿病を誘発する。ストレプトゾトシン注射は、少なくとも7週間持続する再現可能な機械的異痛症を3週間以内に誘発する(S.R.ChenおよびH.L.Pan.J.Neurophysiol.(2002年)、第87巻、2726〜2733ページ)。
【0258】
静的および動的な異痛症の評価
静的な異痛症
異痛症評価の前に、動物を底面がワイヤーの試験ケージに慣らす。静的な異痛症は、von Frey毛(Stoelting、米国イリノイ州ウッドデール)を弱い力から順に(0.6、1、1.4、2、4、6、8、10、15、および26グラム)後足の足底面に適用して評価する。各von Frey毛を、引っ込め応答が生じるまで最長で6秒間足に適用する。一度von Frey毛に対する引っ込め応答が確立したならば、引っ込め動作を生じたものより細いフィラメントから出発して、その後残りのフィラメントで強い力から順に引っ込め動作が生じなくなるまで足を試験し直す。最高の力である26gは、応答を誘発するだけでなく足を持ち上げ、すなわちカットオフポイントとなる。各動物の両方の後足をこのようにして試験する。応答を誘発するのに必要な最低量の力を足引っ込め動作閾値(PWT)としてグラムで記録する。動物が無処置のラットでは非侵害性である4g以下の刺激に応答したならば、静的な異痛症が存在すると定められている(M.J.Fieldら、Pain(1999年)、第83巻、303〜11ページ)。
【0259】
動的な異痛症
動的な異痛症は、後足の足底表面を綿棒で軽くなでて評価する。一般の運動能を記録しないようにするために、活発でなかった完全に慣らされたラットでこの手順を実施するように注意する。各時点で少なくとも2回の測定を行い、その平均値が足引っ込め動作待ち時間(PWL)となる。15秒以内に反応を示さない場合、手順を終了し、動物をその引っ込め動作時間に割り振る。痛み引っ込め応答はしばしば、繰り返される尻込み動作または足舐め動作を伴う。動物がなで始めてから8秒以内に綿による刺激に応答するならば、動的な異痛症が存在するとみなされる(Fieldら、1999年、前掲書)。
【実施例】
【0260】
1H核磁気共鳴(NMR)スペクトルは、すべての場合において、提案した構造と一致した。特徴的な化学シフト(δ)を、主要なピークの呼称については従来の略語、たとえばs:一重線、d:二重線、t:三重線、q:四重線、m:多重線、br:ブロードを使用して、テトラメチルシランから低磁場方向への百万分率(ppm)で示す。質量スペクトル(m/z)は、エレクトロスプレーイオン化(ESもしくはESI)または大気圧化学イオン化(APCI)のいずれかを使用して記録した。一般的な溶媒については次の略語、すなわち、CDCl3:重水素化クロロホルム、D6−DMSO:6重水素化ジメチルスルホキシドを使用した。
【0261】
単結晶X線回折実験
実施例2の化合物の酢酸溶媒和物の結晶構造を、Bruker SMART APEX単結晶X線回折計およびMo Kα放射線照射を使用する室温での単結晶X線回折によって決定した。SMART v5.622(制御)およびSAINT v6.02(積分)ソフトウェア(Bruker AXS Inc.、米国ウィスコンシン州マディソン、1994年)を使用して、ωにおいて各照射が0.3°をカバーする数本の照射から、照射時間を60秒とし、合計データセットを半球より多くして強度を積分した。マルチスキャン法(SADABS、エリアディテクターデータの基準化および補正のためのプログラム、G.M.Sheldrick、ゲッティゲン大学、1997年;R.H.Blessing、Acta Cryst.1995年、A51、33〜38ページの方法に基づくもの)を使用して、吸収に関してデータを補正した。
【0262】
結晶構造は、空間群C2/cにおいてSHELXS−97(結晶構造解明のためのプログラム、G.M.Sheldrick、ドイツ国ゲッティゲン大学、1997年、リリース97−2)を使用する直接的な方法によって首尾よく解明し、SHELXL−97(結晶構造高精度化のためのプログラム、G.M.Sheldrick、ドイツ国ゲッティゲン大学、1997年、リリース97−2)を使用する最小二乗法によって精度を高めた。この結晶構造高精度化手順によって、非対称ユニット内に実施例2の化合物一分子と酢酸一分子が存在することが明らかになった。したがって、この構造は、実施例2の化合物の1:1の酢酸溶媒和物と呼ぶことができる。
【0263】
実施例2ステップ(b)(酢酸溶媒和物)の結晶構造からの粉末X線回折パターンの算出
実施例2の化合物の酢酸溶媒和物の単結晶構造から、Accelrys MS Modelling(商標)[バージョン3.0]の「反射粉末回折」モジュールを使用して、2θ角度および相対強度(以下の表8を参照されたい)を算出した。関連するシミュレーションパラメータは以下のとおりである。
波長=1.5406Å(Cu Kα)
偏光因子=0.5
Pseudo−Voigtプロフィール(U=0.01、V=−0.001、W=0.002)
【0264】
算出したパターンは、単結晶構造から得られるので、実施例2化合物の酢酸溶媒和物の純粋な相のパターンを表す。算出したパターンと予測したパターンの比較を図2に示すが、大部分に単結晶構造が表われていることを示している。ピーク強度のわずかな不一致は、測定したパターンにおける好ましい配向効果のためであるとすることができる。
【0265】
粉末X線回折
自動サンプル変更装置、θ−θゴニオメーター、自動ビームダイバージェンススリット、およびPSD Vantec−1検出器を装着したBruker−AXS Ltd.D4粉末X線回折計を使用して、実施例2の化合物の溶媒和していない結晶形態(形態A)の粉末X線回折パターンを決定した。正確なピーク位置を測定するために、最初にケイ素でドープ処理することによって、分析用のサンプルを調製し、その後低バックグラウンドのシリコンウェーハ検体取付台に載せた。検体を回転させながら、X線管を40kV/30mAで作動させて銅K−α1X線(波長=1.5406オングストローム)を照射した。分析は、ゴニオメーターを、2°〜55°の2θ範囲にわたり、0.018°の1ステップあたり0.2秒のカウントにセットした連続的な方式で作動させて実施した。真空乾燥した溶媒和物サンプルのPXRDパターンも、この回折計で同じパラメータを使用して収集したが、これらのサンプルはケイ素でドープ処理しなかった。
【0266】
他の粉末X線回折パターンはすべて、平らなシリコンウェーハに載せたサンプルについて、Gobel光学鏡、単一のサンプルステージ、および高感度位置検出器(PSD)を装着したBruker AXS Ltd.D8 Advance粉末X線回折計を使用して記録した。X線管を40kV/40mAで作動させて、各検体に銅K−α1X線(波長=1.5406Å)を照射した。分析は、ゴニオメーターを、3°〜35°の2θ範囲にわたり、0.014°の1ステップあたり0.2秒のカウントにセットした連続的な走査方式で作動させて実施した。
【0267】
示差走査熱量測定(DSC)
DSC測定は、Perkin Elmer Diamond示差走査熱量計を使用して行った。サンプルを50μl容脱気孔付きアルミニウム受皿において周囲温度から300℃に20℃/分で加熱した。流動ガスは40ml/分の窒素とした。
【0268】
熱重量分析(TGA)
溶媒和物のTGA測定は、TA Instruments TGA2950 Hi−Res熱重量分析装置を使用して、窒素パージガスを75cm3/分の速度で使用しながら、20℃/分の加熱速度で周囲温度から脱溶媒和温度(150℃〜180℃)で行った。次いで、サンプルをその後のPXRD分析に向けて室温に冷却した。
【0269】
(実施例1)
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0270】
【化10】
調製例8のアルコール(50mg、0.14mmol)を99.25:0.75のアセトニトリル:水(2ml)に溶かした溶液に、過ヨウ素酸(82mg、0.359mmol)および酸化クロム(VI)(1.6mg、0.016mmol)を99.25:0.75のアセトニトリル:水(2ml)に溶かした溶液を加え、反応温度を5℃より低く保った。反応混合物を室温で18時間攪拌した。反応混合物を濾過し、残渣を99.25:0.75のアセトニトリル:水、2N塩酸:メタノール(5:1)、水、およびメタノールで洗浄した。残渣を真空乾燥して、表題化合物を白色固体(28mg、0.077mmol、55%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.17(m,1H)、1.40〜1.65(m,5H)、1.79(m,2H)、2.16(m,2H)、2.48(m,2H)、2.72(m,3H)、4.64(m,1H)、6.43(d,1H)、7.0(s,1H)、7.21(d,1H)、7.90(s,1H)、12.26(bs,1H)。
LRMS m/z(APCI):365[M+H]+、406[M+CH3CN+H]+
【0271】
(実施例2)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0272】
【化11】
方法A
調製例11のアルコール(2.05g、5.84mmol)を0.75%の水を含有するアセトニトリル(50ml)に溶かした溶液に、酸化クロム(VI)(12mg、0.11mmol)および過ヨウ素酸(3.33g、14.6mmol)の溶液を加え、反応混合物を40℃で96時間攪拌した。水(100ml)を加え、懸濁液を2時間攪拌した。得られる沈殿を濾過によって収集し、水で洗浄し、真空乾燥して、表題化合物(1.90g、5.2mmol、89%)を得た。
1H−NMR(400MHz、D6−DMSO):δ1.2(m,1H)、1.2(m,2H)、1.6(m,2H)、1.8(m,2H)、2.3(m,2H)、2.6(m,2H)、3.1(m,1H)、3.2(s,1H)、4.0(bs,1H)、4.8(m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.9(s,1H)。
LRMS m/z(APCI)365[MH]+
【0273】
方法B
ステップ(a)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸t−ブチルエステル
【0274】
【化12】
調製例27ステップ(b)の化合物(34.1g、130mmol)をDMF(300ml)に懸濁させ、スラリーを35℃に温めた。炭酸セシウム(63g、190mmol)を一度に加えた。調製例22の化合物をDMF(90ml)に溶解させ、反応液に加えた。反応液を1時間かけて90℃に加熱し、8時間維持した。反応液を73℃に冷却し、水(160ml)を加え、その間温度は65℃より高く保った。得られるスラリーを35℃に冷却し、その後酢酸エチル(260ml)を一度に加えた。室温に冷却した後、スラリーを濾過し、生成物を酢酸エチル(2×100ml)で洗浄した。得られる白色固体を60℃で16時間真空乾燥して、表題化合物を白色固体(44.4g、105mmol、82%)として得た。
1H−NMR(300MHz、CDCl3):δ1.3(m,1H)、1.4(m,1H)、1.5(s,9H)、1.6(m,1H)、1.7(bm,1H)、1.8(bm,2H)、1.8(bm,1H)、2.4(m,2H)、2.6(t,2H)、2.7(m,2H)3.1(m,1H)、3.2(m,1H)、4.8(m,1H)、5.5(s,1H)、6.2(d,1H)、6.9(s,1H)、7.1(d,1H)。
LC−MS(ESI):22.6分 11.8(%){シス異性体}m/z 422[MH+]、23.1分 88.2(%){トランス異性体}m/z 422[MH+]。
【0275】
ステップ(b)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸酢酸溶媒和物
【0276】
【化13】
ステップ(a)の生成物(207g、0.492 mol)を酢酸(3100ml)中にスラリー化し、60℃に加熱した。温度を60℃に保ちながら、48%の臭化水素酸(4.93mol)を滴下した。溶液を60℃で30分間攪拌した。温度を55℃より高く保ちながら、水(700ml)を滴下した。スラリーを20℃に冷却し、さらに30分間攪拌し、その後これを濾過し、酢酸:水(2000ml)および水(1000ml)で洗浄した後、真空オーブンに入れて60℃で一晩乾燥させて、表題化合物の白色固体を酢酸溶媒和物(153.6g、73.5%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.2(m,1H)、1.2(m,2H)、1.6(m,2H)、1.8(m,2H)、1.9(s,3H,CH3COOH)、2.3(m,2H)、2.6(m,2H)、3.1(m,1H)、3.2(s,1H)、4.0(bs,1H)、4.8 (m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.9(s,1H)、9.9 (s,1H,CH3COOH)。
LC−MS(ESI):18.0分 1.65(%){シス異性体}m/z 365[MH+]、
18.3分 98.4(%){トランス異性体}m/z 365[MH+]。
【0277】
上記酢酸溶媒和物は、単結晶X線回折の方法によって決定されたその結晶構造からわかるように、1:1の化学量論比で結晶化することがわかった。図2は、上記酢酸溶媒和物バッチについて測定した粉末X線回折(PXRD)パターン(A)、ならびに単結晶構造から予測したシミュレーションによるPXRDパターン(B)を示す。2つのパターンのピーク位置が非常によく一致することがわかる。回折ピークの相対強度および幅に差異があれば、それぞれ好ましい配向効果および粒径効果のためであるとすることができる。上記酢酸溶媒和物についての特徴的な2θX線回折ピークおよびその相対強度を以下の表8で一覧にする。
【0278】
【表8】
【0279】
酢酸溶媒和物は、熱重量分析(TGA)によって示されるように、115℃付近で熱によって脱溶媒することがわかったが、TGAでは、この温度で1モル当量の酢酸に等しい約15%のはっきりした重量減少が認められる。TGAプロットを図3に示す。上記溶媒和物は、脱溶媒すると、図4に示すPXRDプロットによって示されるように、再結晶化して(以下のステップ(c)に記載の)脱溶媒和形態Aになる。
【0280】
ステップ(c)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
ステップ(b)の酢酸溶媒和物(157g、369mmol)を室温で一晩かけて水(5200ml)中にスラリー化した。次いで、スラリーを濾過し、水(4×500ml)で洗浄した後、真空オーブンに入れて60℃で一晩乾燥させて、溶媒和していない表題化合物を白色固体(130g、357mmol、96%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.2(m,1H)、1.2(m,2H)、1.6(m,2H)、1.8(m,2H)、2.3(m,2H)、2.6(m,2H)、3.1(m,1H)、3.2(s,1H)、4.0(bs,1H)、4.8(m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.9(s,1H)。
LRMS m/z(APCI)365[MH]+
【0281】
この化合物は、図5に示す特徴的な粉末X線回折(PXRD)パターンを有する溶媒和していない形態(形態A)で結晶化することもわかった。特徴的な2θX線回折ピークおよびその相対強度を以下の表9で一覧にする。この結晶形態は、図6で例示する示差走査熱量測定(DSC)によって求められるとおり、融点が250℃である。
【0282】
【表9】
【0283】
実施例2の化合物は、ジメチルアセトアミド(DMAC)、ピリジン、テトラヒドロフラン(THF)、およびジメチルスルホキシド(DMSO)との溶媒和物として結晶化することもわかった。これらの溶媒和物はそれぞれ、図7に示すような特徴的なPXRDパターンを有する。
【0284】
これらの溶媒和物のTGA測定では、ピリジンおよびTHF溶媒和物が1:1の化学量論比を有することが明らかになったが(図8および9)、DMAC溶媒和物は、2:1の溶媒対化合物化学量論比を有することが示された(図10)。DMSO溶媒和物は脆い性質であるので、その化学量論比は求められないということになった。
【0285】
ピリジン、THF、DMAC、およびDMSO溶媒和物はそれぞれ、脱溶媒すると、それぞれ図11、12、13、および14に示されるPXRD分析に示すように、無水形態Aに再結晶化する。
【0286】
(実施例3)
3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
【0287】
【化14】
(a)3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸ベンジルエステル
【0288】
【化15】
8’−フルオロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(140mg、0.56mmol)(WO2004/026818に記載のとおりに調製したもの、中間体c)および炭酸セシウム(301mg、0.925mmol)をDMF(2ml)中で合わせ、調製例15の化合物(220mg、0.588mmol)のDMF(2ml)溶液を加え、混合物を80℃で18時間攪拌した。次いで、水(35ml)を加え、生成物を酢酸エチル(2×25ml)で抽出した。有機抽出物を合わせて飽和ブラインで洗浄し、硫酸マグネシウムで乾燥させた。溶媒を蒸発させると、褐色のゴム質である表題化合物がシス異性体とトランス異性体の約5:4の混合物(204mg、80%)として得られた。
1H−NMR(400MHz、CDCl3):δ1.29(m,1H)、1.66(m,7H)、2.21(m,2H)、2.41(m,2H)、2.59(m,2H)、2.8&2.90(2×m,1H)、3.2(m,1H)、3.96&4.00(2×d,2H)、5.13(2×s,2H)、6.6(m,1H)、6.95(m,1H)、7.33(m,5H)。
LRMS m/z(ES)453[MH]+
【0289】
(b)3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
ステップ(a)の生成物(200mg、0.442mmol)をメタノール(2ml)に溶解させ、2M NaOH(2ml、4.0mmol)を加え、褐色の乳濁液を60℃で1.5時間攪拌した後冷却した。2N HCl(2ml、4.0mmol)を加え、得られる懸濁液を1.5時間攪拌した。クリーム色の固体を濾過によって収集し、水でよく洗浄して、真空乾燥後に表題化合物(136mg、85%)を得た。
【0290】
Chiralpak AD−H、15%イソプロパノール:85%ヘキサン+0.1%トリフルオロ酢酸でのキラルHPLCによって、43:57の異性体比率が示される(保持時間13.69分および15.27分)。
1H−NMR(400MHz、CDCl3):δ1.16(m,1H)、1.43(m,2H)、1.57(m,3H)、1.76(m,2H)、2.05(m,2H)、2.28(m,2H)、2.43(m,2H)、2.68(2×m,1H)、3.03(2×m,1H)、3.88&3.97(2×d,2H)、6.48(m,1H)、6.76(m,1H)、6.98(m,1H)、8.79(s,1H)、12.06(br,1H)。
LRMS m/z(ES)363[MH]+
【0291】
(実施例4)
トランス−3−[(8’−シアノ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0292】
【化16】
調製例16の化合物(101mg、0.296mmol)から出発し、酸化クロム(VI)(0.5mg、0.005mmol)および過ヨウ素酸(167mg、0.733mmol)を使用する実施例1の方法によって、表題化合物(76mg、72%)が得られた。
1H−NMR(400MHz、D6−DMSO)δ:1.1〜1.85(m,8H)、2.3〜2.7(m,6H)、3.10(m,1H)、4.92(m,1H)、6.47(d,1H)、7.14(s,1H)、7.51(d,1H)、8.51(s,1H)。
LC−MS:保持時間=2.49分(100%)、LRMS(ESI)m/z 356[MH+]
【0293】
(実施例5)
1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
【0294】
【化17】
(a)1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸メチルエステル
【0295】
【化18】
8’−フルオロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(120mg、0.48mmol)(WO2004/026818に記載のもの)の室温のDMF(1ml)溶液に、炭酸セシウム(234mg、0.72mmol)を加え、混合物を10分間攪拌した後、調製例18の化合物(172mg、0.58mmol)のDMF(1ml)溶液を加えた。反応混合物を18時間かけて80℃に加熱し、次いで室温に冷却した。混合物を酢酸エチル(20ml)および水(20ml)で希釈し、有機層を分離し、水層を酢酸エチル(20ml)で抽出した。有機層を合わせて水(2×20ml)およびブライン(2×20ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。淡褐色の油状物をジエチルエーテル(10ml)から摩砕し、表題化合物を淡褐色の固体として得た(0.3molのDMFを含有する140mgの溶媒和物、0.35mmol、73%)。
1H−NMR(400MHz、D6−DMSO):δ1.16(m,1H)、1.35(m,2H)、1.49(m,3H)、1.72(m,2H)、1.90(m,1H)、2.03(m,3H)、2.26(m,2H)、2.42(m,2H)、3.60(s,3H)、4.22(s,2H)、6.52(dd,1H)、6.79(s,1H)、7.01(t,1H)、8.85(s,1H)。
【0296】
(b)1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸
ステップ(a)の生成物(140mg、0.35mmol)をメタノール/水(1:1、2ml)に溶かした部分溶液に、水酸化ナトリウム(28mg、0.70mmol)を加え、反応液を50℃で24時間攪拌した。混合物を室温に冷却し、さらに6日間攪拌した後、2N塩酸水溶液(2ml)で処理した。得られるクリーム色の固体を濾過によって収集し、水で洗浄し、真空乾燥して、表題化合物(83mg、0.23mmol、65%)を得た。
1H−NMR(400MHz、D6−DMSO):δ1.25(m,1H)、1.37(m,2H)、1.49(m,3H)、1.71(m,2H)、1.90(m,1H)、2.01(m,3H)、2.38(m,4H)、4.18(s,2H)、6.52(dd,1H)、6.71(s,1H)、7.00(dd,1H)、8.78(s,1H)、12.43(s,1H)。
LRMS m/z(ESI)377[M+H]+
【0297】
(実施例6)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0298】
【化19】
(a)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸エチルエステル
【0299】
【化20】
調製例20の化合物(100mg、0.36mmol)、炭酸カリウム(57mg、0.41mmol)、および18−クラウン−6(110mg、0.41mmol)の80℃のジメチルホルムアミド(3ml)懸濁液に、3−(トルエン−4−スルホニルオキシ)−シス−シクロブタンカルボン酸エチルエステル(調製例22の化合物と類似の方法によって調製したもの)(123mg、0.41mmol)のジメチルホルムアミド(1ml)溶液を加え、反応混合物を80℃で18時間攪拌した。混合物を室温に冷却し、水(30ml)から酢酸エチル(2×20ml)に2回抽出した。有機層を合わせてブライン(2×20ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。油性の残渣をメタノールから再蒸発させ、ジエチルエーテルで摩砕して、表題化合物をクリーム色の固体(75mg、0.18mmol、51%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.19(t,3H)、1.43〜1.77(m,10H)、2.24(m,2H)、2.38(m,2H)、2.63(m,2H)、3.14(m,1H)、4.09(q,2H)、4.82(m,1H)、6.36(d,1H)、7.17(d,1H)、7.29(s,1H)、8.05(s,1H)。
LRMS m/z(ESI)407[MH]+
【0300】
(b)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
ステップ(a)の化合物(70mg、0.17mmol)のメタノール(1ml)懸濁液に、水酸化ナトリウム(14mg、0.35mmol)の水(1ml)溶液を加え、得られる懸濁液を40℃で2時間攪拌した。メタノールを真空中で除去し、得られる溶液のpHを、2N HCl水溶液(5ml)を滴下して約1に調整した。得られる固体を濾別し、イソプロパノール(1.5ml)で洗浄して、表題化合物を白色固体(25mg、0.066mmol、40%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.43〜1.77(m,10H)、2.23(m,2H)、2.34(m,2H)、2.61(m,2H)、3.06(m,1H)、4.81(q,1H)、6.36(d,1H)、7.17(d,1H)、7.29(s,1H)、8.05(s,1H)、12.35(s,1H)。
LRMS m/z(ESI)755[2M−H]−
【0301】
(実施例7)
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
【0302】
【化21】
(a)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸t−ブチルエステル
【0303】
【化22】
(a)調製例24の化合物(300mg、1.14mmol)をDMF(3ml)に溶かした部分溶液に、炭酸セシウム(559mg、1.72mmol)を加え、反応混合物を10分間かけて40℃に加熱した後、調製例22の粗製の化合物(523mg、1.60mmol)のDMF(3mL)溶液を一度に加えた。反応混合物をさらに9時間かけて80℃に加熱し、室温に冷ました。次いで、反応混合物に水(3ml)を加えた後、酢酸エチル(5ml)を加え、得られる沈殿を収集しようとしたが成功しなかった。その後直ちに完全に溶解させ、反応混合物を真空中で2mlに濃縮し、水(5mL)を加えて、結晶化を誘発し、得られる生成物を濾別し、真空乾燥して、表題化合物(310mg、0.76mmol、67%)を得た。LC−MSによって、10%の出発材料フェノールが残存することが示された。この材料をそれ以上精製せずにステップ(b)で使用した。
LRMS m/z(ESI)407[M+H]+
【0304】
(b)トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
ステップ(a)の生成物(310mg、0.76mmol)の60℃の酢酸(3ml)溶液に、48%の臭化水素酸水溶液(0.5ml)を加え、反応液を室温で30分間攪拌した。わずかな濁りが認められるまで水(0.1ml)を滴下して、混合物を失活させた。反応液を室温に冷ました後、得られる沈殿を濾別して、淡褐色の固体(130mg)を得た。酢酸(1.5ml)/水(0.1ml)で再結晶化することにより精製を実現して、表題化合物をオフホワイトの固体(35mg、0.09mmol、9%)として得た。
1H−NMR(400MHz、D6−DMSO):δ1.70(m,4H)、1.82(m,2H)、2.27(m,2H)、2.60(m,2H)、3.04(m,2H)、4.81(m,1H)、6.35(d,1H)、7.18(d,1H)、7.30(s,1H)、7.98(s,1H)、12.14(s,1H)。
LRMS(ESI)m/z 351[M+H]+
【0305】
調製例
調製例1
3−[(ベンジルオキシ)メチル]−2,2−ジクロロシクロブタノン
【0306】
【化23】
亜鉛末(6.54g、0.1mol)を水(30ml)に懸濁させ、アルゴンを懸濁液に15分間バブルした後、硫酸銅(II)(780mg、3.1mmol)を加えた。反応混合物をアルゴン中にて室温で30分間攪拌した。アルゴン流中で混合物を濾別し、固体を水(100ml)、アセトン(100ml)で洗浄し、4時間真空乾燥した。得られる亜鉛/銅の対をアルゴン中でエチルエーテル:1,2−ジメトキシエタン(70ml:10ml)に懸濁させ、アリルベンジルエーテル(4.6ml、30mmol)を加えた。塩化トリクロロアセチル(9ml、81mmol)のジエチルエーテル:1,2−ジメトキシエタン(58ml:7ml)溶液を45分間かけて滴下し、反応混合物を48時間加熱還流した。反応混合物をCelite(登録商標)で濾過し、塩をジエチルエーテル(3×70ml)で洗浄した。濾液を真空中で蒸発させ、残渣をヘキサン(150ml)に溶解し直した。残りの固体を濾過によって除去し、濾液を炭酸水素ナトリウムの飽和水溶液(2×100ml)、ブライン(80ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、10〜25%のヘキサン:ジエチルエーテルを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物が黄色の油状物(7.03g、27.3mmol、91%)として得られた。
1H−NMR(CDCl3、400MHz):δ3.11〜3.21(m,2H)、3.48(m,1H)、3.70(m,1H)、3.85(m,1H)、7.35(m,5H)、4.58(s,2H)。
【0307】
調製例2
3−[(ベンジルオキシ)メチル]シクロブタノン
【0308】
【化24】
調製例1のジクロロシクロブタノン(5.98g、23.08mmol)を塩化アンモニウムで飽和させたメタノール(90ml)に溶かした溶液に、亜鉛粉末(9.25g、142mmol)を加え、反応混合物を室温で2時間攪拌した。塩化アンモニウムを加え、反応混合物を室温でさらに6時間攪拌した。混合物をCelite(登録商標)で濾過し、塩をジエチルエーテル(50ml)で洗浄した。濾液を真空中で濃縮し、残渣をジエチルエーテル(200ml)と水(100ml)とに分配した。混合物を濾過し、有機相を水で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。表題化合物が黄色の油状物(3.7g、19.5mmol、84%)として得られた。
1H−NMR(CDCl3、400MHz):δ2.69(m,1H)、2.90(m,2H)、3.11(m,2H)、3.60(d,2H)、4.56(s,2H)、7.34(m,5H)。
【0309】
調製例3
シス−3−[(ベンジルオキシ)メチル]シクロブタノール
【0310】
【化25】
調製例2のシクロブタノン(1.166g、6.13mmol)をテトラヒドロフランに溶かした、−70℃で攪拌中の溶液に、トリ−s−ブチル水素化ホウ素リチウムの1Mテトラヒドロフラン(40ml)溶液を滴下し、反応温度を−65℃より低く保った。反応液を18時間かけて室温に温めた。反応混合物を炭酸水素ナトリウムの飽和水溶液(25ml)で失活させ、次いで5℃に冷却した。30%過酸化水素水溶液(4ml)を滴下し、反応温度を10℃より低く保った。混合物を水から酢酸エチル(50ml)中に抽出し、有機相を合わせてブライン(30ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、25〜50%の酢酸エチル:ペンタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、無色の油状物(1.05g、5.5mmol、89%)を得た。1H−NMRでは、15:1の比のシス:トランス異性体が得られたことが示された。
1H−NMR(CDCl3、400MHz):δ1.70(m,2H)、2.10(m,1H)、2.46(m,2H)、3.45(d,2H)、4.15(q,1H)、4.52(s,2H)、7.33(m,5H)。
【0311】
調製例4
4−ニトロ安息香酸トランス−3−[(ベンジルオキシ)メチル]シクロブチル
【0312】
【化26】
調製例3のシクロブチルアルコール(1.05g、5.47mmol)、4−ニトロ安息香酸(1.82g、10.9mmol)、およびトリフェニルホスフィン(3.016g、11.5mmol)をテトラヒドロフラン(20ml)に溶かした攪拌した溶液に、0℃でアゾジカルボン酸ジエチル(2g、11.5mmol)のテトラヒドロフラン(5ml)溶液を滴下した。反応混合物を室温で18時間攪拌した。溶媒を真空中で蒸発にさせ、残渣をジエチルエーテル(30ml)に溶解し直した。残りの固体を濾過によって除去し、濾液を真空中で蒸発させた。粗製材料を、1:10〜1:3の酢酸エチル:ペンタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、無色の油状物(1.64g、4.8mmol、88%)を得た。1H−NMRでは、15:1の比のトランス:シス異性体が得られたことが示された。
1H−NMR(CDCl3、400MHz):δ2.40(m,4H)、2.67(m,1H)、3.53(d,2H)、4.57(s,2H)、5.36(q,1H)、7.37(m,5H)、8.20(d,2H)、8.29(d,2H)。
【0313】
調製例5
トランス−3−[(ベンジルオキシ)メチル]シクロブタノール
【0314】
【化27】
調製例4のp−ニトロエステル(1.64g、4.8mmol)の1,4−ジオキサン(35ml)溶液に、水酸化ナトリウム(385mg、9.6mmol)の水(25ml)溶液を加え、反応混合物を室温で30分間攪拌した。酢酸(0.4ml、7mmol)を加え、混合物を真空中で濃縮した。残渣を、炭酸水素ナトリウムの飽和水溶液から酢酸エチル(20ml)中に抽出し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。表題化合物が黄色の油状物(850mg、4.4mmol、92%)として得られた。
1H−NMR(CDCl3、400MHz):δ2.08(m,2H)、2.20(m,2H)、2.47(m,1H)、3.47(d,2H)、4.39(q,1H)、4.52(s,2H)、7.34(m,5H)。
【0315】
調製例6
p−トルエンスルホン酸トランス−3−[(ベンジルオキシ)メチル]シクロブチル
【0316】
【化28】
調製例5のシクロブタノール(850mg、4.42mmol)をピリジン(5ml)に溶かした攪拌した溶液に、0℃で塩化p−トルエンスルホニル(1.18g、6.2mmol)を少量ずつ加え、反応混合物を室温で18時間攪拌した。溶媒を真空中で濃縮し、残渣酢酸エチル(30ml)に溶解し直し、2N塩酸(30ml)、炭酸水素ナトリウムの飽和水溶液(30ml)、ブライン(30ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、ジクロロメタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物が無色の油状物(1.53g、4.4mmol)として得られた。
1H−NMR(CDCl3、400MHz):δ2.15(m,2H)、2.31(m,2H)、2.44(s,3H)、2.49(m,1H)、3.4(d,2H)、4.49(s,2H)、4.93(q,1H)、7.32(m,7H)、7.75(d,2H)。
【0317】
調製例7
5’−({シス−3−[(ベンジルオキシ)メチル]シクロブチル}オキシ)−8’−クロロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0318】
【化29】
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(Bioorg.Med.Chem.Lett(2004年)、第14巻(18)、4627〜4632ページに記載のとおりに調製したもの)(640mg、2.4mmol)、炭酸カリウム(400mg、2.9mmol)、および18−クラウン−6(767mg、2.9mmol)をジメチルホルムアミド(8ml)中で合わせ、反応混合物を80℃に加熱した。調製例6のトシラート(1g、2.9mmol)のジメチルホルムアミド溶液を3回で加え、混合物をさらに18時間80℃で加熱した。反応混合物を酢酸エチル(100ml)と水(150ml)とに分配し、固体を濾過によって収集した。相を分離し、水相を酢酸エチルで抽出し直し、ブラインで希釈し、再び酢酸エチル中に抽出した。有機相を合わせて真空中で濃縮し、残渣を水およびメタノールで摩砕した。粗生成物を合わせて、ジクロロメタンからジクロロメタン:酢酸エチル(1:1)を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、表題化合物をオフホワイトの固体(685mg、1.156mmol、64%)として得た。
1H−NMR(D6−DMSO、400MHz):δ1.1(m,1H)、1.4(m,2H)、1.6(m,3H)、1.7(m,2H)、1.8(m,2H)、2.3(m,1H)、2.5(m,4H)、3.4(s,2H)、4.4(s,2H)、4.6(m,1H)、6.4(d,1H)、7.0(s,1H)、7.2(d,1H)、7.3(m,5H)、7.8(s,1H)。
【0319】
調製例8
8’−クロロ−5’−{[シス−3−(ヒドロキシメチル)シクロブチル]オキシ}−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0320】
【化30】
調製例7のベンジルアルコール(400mg、0.9mmol)のジクロロメタン(10ml)懸濁液に、三塩化ホウ素−ジメチルスルフィド錯体の2Mジクロロメタン溶液(1.8ml、3.6mmol)を加え、反応混合物を室温で終夜攪拌した。炭酸水素ナトリウムの飽和水溶液(10ml)を加え、混合物を5分間攪拌した。ジクロロメタンおよび水を加え、得られる固体を濾過によって収集した。表題化合物が白色固体(230mg、0.657mmol、73%)として得られた。
1H−NMR(D6−DMSO、400MHz):δ1.17(m,1H)、1.42(m,2H)、1.57(m,3H)、1.82(m,4H)、2.05(m,1H)、2.45(m,4H)、3.38(t,2H)、4.58(m,2H)、6.41(d,1H)、6.99(s,1H)、7.20(d,1H)、7.86(s,1H)。LRMS m/z(APCI)351[MH]+
【0321】
調製例9
p−トルエンスルホン酸シス−3−[(ベンジルオキシ)メチル]シクロブチル
【0322】
【化31】
調製例3のアルコール(17g、88.4mmol)をジクロロメタン(90ml)に溶かした、5℃で攪拌中の溶液に、ピリジン(14.3ml、176mmol)および塩化p−トルエンスルホニル(20.2g、105.9mmol)を加え、反応混合物を室温で18時間攪拌した。反応混合物をジクロロメタン(50ml)で希釈し、2N塩酸(50ml)、炭酸水素ナトリウムの飽和水溶液(50ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料を、ペンタン:酢酸エチル(19:1、9:1、4:1)を溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製した。表題化合物が無色の油状物(24.8g、71.6mmol、81%)として得られた。
1H−NMR(CDCl3、400MHz):δ1.95(m,2H)、2.1(m,1H)、2.35(m,2H)、2.45(s,3H)、3.4(m,2H)、4.5(s,2H)、4.7(m,1H)、7.3(m,7H)、7.8(m,2H)。
LRMS m/z(ESI)347[MH]+
【0323】
調製例10
5’−({トランス−3−[(ベンジルオキシ)メチル]シクロブチル}オキシ)−8’−クロロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0324】
【化32】
方法A
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(500mg、1.87mmol)をジメチルホルムアミド(2ml)に懸濁させた攪拌した懸濁液に、炭酸セシウム(730mg、2.24mmol)を加え、反応混合物を80℃に加熱した。5分後、調製例9のトシラート(710mg、2.05mmol)のジメチルホルムアミド(1ml)溶液を加え、反応混合物を80℃で18時間加熱した。混合物をブライン(60ml)から酢酸エチル(1×80ml、2×30ml)中に抽出し、ブライン(3×100ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。表題化合物が、わずかに不純なクリーム色の固体(800mg、0.96mmol、96%)として得られた。
【0325】
方法B
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン(950mg、3.56mmol)をジメチルホルムアミド(12ml)に溶かした、80℃で攪拌中の溶液に、炭酸カリウム(590mg、4.27mmol)および18−クラウン−6(1.1g、4.27mmol)を加えた。反応混合物を10分間攪拌した後、調製例9のトシラート(1.48g、4.27mmol)のジメチルホルムアミド(3ml)溶液を加えた。反応混合物を80℃で24時間加熱した。混合物を水:メタノール(75ml:25ml)上に注ぎ、10分間攪拌し、得られる沈殿を濾過によって収集し、メタノールで洗浄した。固体をジクロロメタンに溶解させ、Celite(登録商標)で濾過し、得られる濾液を真空中で蒸発にさせて、表題化合物をトランス:シス異性体の9:1混合物(887mg、2.0mmol、56%)として得た。
1H−NMR(CDCl3、400MHz):δ1.3(m,1H)、1.5〜1.9(m,9H)、2.4(m,3H)、2.6(m,2H)、3.5(d,2H)、4.6(s,2H)、4.75(m,1H)、5.85(bs,1H)、6.25(d,1H)、7.05(bs,1H)、7.1(d,1H)、7.3〜7.4(m,5H)。
LRMS m/z(ESI)441[MH]+
【0326】
調製例11
8’−クロロ−5’−{[トランス−3−(ヒドロキシメチル)シクロブチル]オキシ}−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−2’(3’H)−オン
【0327】
【化33】
三塩化ホウ素−ジメチルスルフィド錯体をジクロロメタン(15ml)に溶かした2Mの溶液を、調製例10のベンジルエーテル(3.5g、7.9mmol)のジクロロメタン(80ml)溶液に滴下し、反応混合物を室温で18時間攪拌した。この混合物を、炭酸水素ナトリウムの飽和水溶液(200ml)中に注ぎ、泡立ちが止むまで攪拌した。混合物をジクロロメタン(1×200ml、2×100ml)中に抽出し、ブライン(50ml)で洗浄し、硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発させた。粗製材料をアセトニトリルから再結晶化して、表題化合物を91:9の比のトランス:シス生成物(2.33g、6.65mmol、84%)として得た。
1H−NMR(CDCl3、400MHz):δ1.3(m,1H)、1.5(m,2H)、1.8(m,5H)、2.4(m,4H)、2.6(m,3H)、3.8(d,2H)、4.8(m,1H)、5.7(bs,1H)、6.25(d,1H)、7.0(bs,1H)、7.1(d,1H)。
LRMS m/z(ESI)351[MH]+
【0328】
調製例12
3−メチレンシクロブタンカルボン酸
【0329】
【化34】
水酸化カリウム(17.37g、214.7mmol)を水(20ml)に溶解させ、エタノール(20ml)を加えた。この溶液を冷たいときに3−メチレン−シクロブタンカルボニトリル(5.0g、53.7mmol)に加え、得られる溶液を2.5時間加熱還流し、冷まし、真空中で蒸発にさせて、クリーム色の固体とした。固体を水(15ml)に溶解させ、氷浴で冷却し、濃HClを加えてpH1とし、ジエチルエーテル(3×20ml)で抽出した。エーテル抽出物を硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて、表題化合物を淡黄色の液体(5.6g、93%)として得た。
1H−NMR(CDCl3、400MHz):δ2.95(m,2H)、3.02(m,2H)、3.17(m,1H)、4.82(m,2H)(交換可能な1プロトンが見られなかった)。
【0330】
調製例13
3−メチレンシクロブタンカルボン酸ベンジルエステル
【0331】
【化35】
調製例12の生成物(1g、8.9mmol)の酢酸エチル(5ml)溶液に、1,1’−カルボニルジイミダゾール(1.59g、9.81mmol)の酢酸エチル(5ml)懸濁液を数回に分けて加えた。穏やかな泡立ちが認められた。混合物を約1.5時間室温で攪拌し、ベンジルアルコール(1.11ml、10.7mmol)を加え、終夜攪拌を続けた。溶液をジエチルエーテル(20ml)で希釈し、水(2×10ml)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて無色の液体とし、これを、ジクロロメタンを溶離液としながら10gのSiO2で濾過して精製して、表題化合物を無色の油状物(1.246g、69%)として得た。
1H−NMR(CDCl3、400MHz):δ2.92(m,2H)、3.02(m,2H)、3.16(m,2H)、4.80(m,2H)、5.15(s,2H)、7.36(m,5H)。
LRMS m/z(ESI)203[MH]+
【0332】
調製例14
3−(ヒドロキシメチル)シクロブタンカルボン酸ベンジルエステル
【0333】
【化36】
ボラン−ジメチルスルフィド(0.07ml、0.72mmol)をTHF(1ml)で希釈し、調製例13の化合物(300mg、1.48mmol)をTHF(1ml)に溶かした攪拌した溶液に室温で滴下した。無色の溶液を室温で1時間攪拌し、次いで過ホウ酸ナトリウム(145mg、1.78mmol)の水(1ml)溶液を、泡立ちを制御する速度で滴下した。加え終えたなら、混合物を1,4−ジオキサン(1ml)で希釈し、得られる溶液を60℃で1時間温め、水(5ml)を加えて失活させ、酢酸エチル(10ml)で抽出した。酢酸エチル抽出物を硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて、無色の油状物(226mg、69%)を得、これを次のステップでそれ自体として使用した。
1H−NMR(CDCl3、400MHz):δ2.05(m,2H)、2.33(m,2H)、2.45(m,1H)、3.11(m,1H)、3.62(dd,2H)、5.13(d,2H)、7.35(m,5H)。
【0334】
調製例15
3−(p−トルエンスルホニルオキシメチル)シクロブタン−1−カルボン酸ベンジルエステル
【0335】
【化37】
調製例14の化合物(275mg、1.25mmol)およびピリジン(0.26ml、3.25mmol)の攪拌した溶液に、室温で塩化p−トルエンスルホニル(309mg、1.62mmol)のジクロロメタン(2ml)溶液を滴下し、3日間攪拌を続けた。混合物をジクロロメタン(20ml)と水(2×20ml)とに分配し、ジクロロメタン抽出物を硫酸マグネシウムで乾燥させ、真空中で蒸発にさせて無色の油状物とし、これを、ジクロロメタンから1:1のジエチルエーテル:ジクロロメタンを溶離液とするシリカゲルでのカラムクロマトグラフィーによって精製して、表題化合物(226mg、48%)を無色の油状物としてを得た。
1H−NMR(CDCl3、400MHz):δ2.02(m,2H)、2.25〜2.41(m,2H)、2.44(s,3H)、2.55〜2.75(m,1H)、3.07(m,1H)、4.00(2×d,2H)、5.10(d,2H)、7.34(m,7H)、7.78(m,2H)。
【0336】
調製例16
5’−{[トランス−3−(ヒドロキシメチル)シクロブチル]オキシ}−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−8’−カルボニトリル
【0337】
【化38】
調製例11の化合物(100mg、0.285mmol)のN−メチルピロリジノン(1.5mL)懸濁液に、シアン化ナトリウム(27.9mg、0.57mmol)を加えた後、臭化ニッケル(62.3mg、0.285mmol)を加え、反応混合物をマイクロ波反応器に入れて200℃で10分間加熱した。混合物をジエチルエーテル(2×20ml)と水(10mL)とに分配し、有機抽出物を合わせて硫酸マグネシウムで乾燥させ、真空中で濃縮して、表題化合物を淡い橙色/赤色の固体(27.8mg)として得た。
1H−NMR(400MHz、CDCl3):δ1.4〜1.85(m,9H)、2.3〜2.7(m,6H)、3.73(d,2H)、4.83(m,1H)、5.60(s,1H)、6.33(d,1H)、6.98(s,1H)、7.36(d,1H)。
LC−MS:保持時間=2.54分(100%)、LRMS m/z 342[MH+]
【0338】
調製例17
1−(ヒドロキシメチル)−シクロブタンカルボン酸メチルエステル
【0339】
【化39】
1,1−シクロブタンジカルボン酸ジメチルエステル(英国Lancaster Synthesis Ltdから入手可能)(2.0g、11.6mmol)のテトラヒドロフラン(20ml)溶液に、室温で水素化トリ−t−ブトキシアルミニウムリチウム(1Mのテトラヒドロフラン溶液25.5ml、25.5mmol)を10分間かけて滴下した。反応混合物を加熱して、3時間穏やかに還流させ、室温に冷却し、18時間攪拌した。得られる懸濁液を塩化アンモニウム飽和水溶液(30ml)で希釈し、15分間激しく攪拌してから濾過した。固体をジエチルエーテル(50ml)で洗浄し、有機層を分離し、水層をジエチルエーテル(50ml)で抽出した。有機抽出物を合わせて硫酸マグネシウムで乾燥させ、濾過し、真空中で蒸発にさせて、表題化合物を無色の油状物(1.8g)として得た。
【0340】
調製例18
1−(p−トルエンスルホニルオキシメチル)−シクロブタンカルボン酸メチルエステル
【0341】
【化40】
調製例17の粗製の化合物(1.6g、11.0mmol)のジクロロメタン(5ml)溶液に、塩化p−トルエンスルホニル(4.2g、22.0mmol)を加えた後、ピリジン(2.7ml、33.0mmol)を加え、溶液を室温で18時間攪拌した。次いで、反応混合物をジクロロメタン(30ml)で希釈し、2N HCl水溶液(2×25ml)、重炭酸ナトリウム飽和水溶液(50ml)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で蒸発させた。粗製の橙色油状物(5g)を、酢酸エチル:ペンタン(1:5)を溶離液とするシリカゲルでのフラッシュクロマトグラフィーによって精製して、表題化合物を無色の油状物(1.7g、5.7mmol、49%)として得た。
1H−NMR(CDCl3、400MHz):δ1.96(m,4H)、2.40(m,2H)、2.45(s,3H)、3.62(s,3H)、4.24(s,2H)、7.35(d,2H)、7.79(d,2H)。
【0342】
調製例19
8’クロロ−5’−メトキシ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−2’(3’H)−オン
【0343】
【化41】
2−クロロ−5−メトキシフェニル尿素(WO02/074754、中間体5)(17.9g、89.5mmol)のシクロヘプタノン(60ml、0.51mol)溶液を、100℃で20分間かけてポリリン酸(213g)に滴下し(127℃までの発熱が認められた)、1時間加熱した。混合物を攪拌しながら水(3リットル)および酢酸エチル(1リットル)中に注いだ。得られる固体を濾過によって収集し、酢酸エチルでよく洗浄し、乾燥させた。乾燥させた固体をクロロホルムに溶解させ、重炭酸ナトリウム水溶液で洗浄し、硫酸ナトリウムで乾燥させ、真空中で濃縮して、表題化合物を白色固体(10.2g、34mmol、39%)として得た。
【0344】
二相性の濾液を分離し、酢酸エチル相を水およびブラインで洗浄し、硫酸ナトリウムで乾燥させ、濃縮した。残渣をt−ブチルメチルエーテルで摩砕し、得られる白色固体を濾過によって収集し、別のt−ブチルメチルエーテルで洗浄し、乾燥させて、次の分の表題化合物(9.0g、30.6mmol、34%)を得た。
1H−NMR(400MHz、CDCl3):δ1.53〜1.84(複合,10H)、2.46(m,2H)、3.81(s,3H)、5.33(br,1H)、6.46(d,1H)、7.03(br,1H)、7.18(d,1H)。
LRMS m/z 295[M+H]+
【0345】
調製例20
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロヘプタン−1,4’−キナゾリン]−2’(3’H)−オン
【0346】
【化42】
調製例19の化合物(19.0g、64.6mmol)のジクロロメタン(500ml)溶液に、三臭化ホウ素の1Mジクロロメタン溶液(129ml、129mmol)を加え、混合物を室温で3日間攪拌した。反応混合物を水(1.5リットル)および酢酸エチル(1リットル)中に注いだ。白色固体を濾過によって収集し、相を分離し、有機相を硫酸ナトリウムで乾燥させ、真空中で濃縮して、褐色の固体を得た。濾過した白色固体をエタノールから再結晶化して、表題化合物(7.4g、26.4mmol、41%)を得た。再結晶からの母液を褐色の固体と合わせ、混合物をエタノール中で攪拌し、濾過し、乾燥させて、2番目のバッチの生成物(7.0g、25mmol、39%)を得た。
1H−NMR(400MHz、D6−DMSO):δ1.38〜1.78(複合,10H)、2.26(m,2H)、6.40(d,1H)、7.02(d,1H)、7.20(br,1H)、7.80(br,1H)、9.84(br,1H)。
【0347】
調製例21
シス−3−ヒドロキシシクロブチルカルボン酸t−ブチルエステル
【0348】
【化43】
方法A
3−オキソシクロブタンカルボン酸t−ブチル(J.Org.Chem.(1993年)第58巻、110ページ)(3.10g、18.2mmol)を窒素中でテトラヒドロフラン:メタノール(20:1、30ml)に溶解させ、5℃に冷却した。水素化ホウ素ナトリウム(345mg)を数回に分けて加え、得られる透明な溶液を5℃で15分間攪拌した後、水(135ml)、次いで酢酸エチル(135ml)を滴下して希釈した。水相を分離し、酢酸エチル(2×25ml)で洗浄した。有機相を合わせてブライン(20ml)で洗浄し、硫酸マグネシウムで乾燥させ、真空中で濃縮して、表題化合物をかすかに黄色の油状物(3.05g、17.7mmol、97%)として得た。
1H−NMR(CDCl3、400MHz):δ1.46(s,9H)、2.12(m,2H)、2.55(m,3H)、4.17(m,1H)。
GC分析(アセトニトリル中サンプル):保持時間4.57分(91.5%面積)。
【0349】
方法B
3−オキソシクロブタンカルボン酸(J.Org.Chem.(1993年)第58巻、110ページ)(10.0g、88mmol)をジクロロメタン(20ml)に溶解させ、5℃に冷却した。4−ジメチルアミノピリジン(8.6g、70mmol)を少量ずつ加えた後、t−ブタノール(13.0g、176mmol)を一度に加えた。N,N’−ジシクロヘキシルカルボジイミドの1Mジクロロメタン(96ml、96mmol)溶液を、温度を0℃〜5℃の間に保ちながら滴下した。得られるスラリーを室温に温め、終夜攪拌した。濾過した後、濾液を5℃で2Mの塩酸(50ml)に滴下した。得られる相を分離し、下方の有機層を室温に温め、水(50ml)および重炭酸ナトリウム飽和溶液(50ml)で洗浄した。下方の有機相を蒸留によって濃縮し、溶媒をテトラヒドロフランと交換した。最終反応体積を30mlとした。メタノール(6ml)を加えた。その間、水素化ホウ素ナトリウム(1.65g、44mmol)をテトラヒドロフラン(39ml)に懸濁させ、5℃に冷却した。この中間体のテトラヒドロフラン溶液を、温度を0℃〜5℃の間に保ちながら水素化ホウ素ナトリウムのスラリーに滴下した。次いで、反応液を0〜5℃で2時間攪拌した。温度を0℃〜5℃の間に保ちながら水を滴下した。酢酸エチル(75ml)を加え、相を分離した。下方の水層を室温に温め、酢酸エチル(37ml)で洗浄した。有機層を合わせて濃縮し、ブライン(37ml)、次いで水(37ml)でさらに洗浄した。上方の有機層を真空中で抜き取って、表題化合物を黄色の油状物(10.1g、58.6mmol、67%)として得た。
1H−NMR(CDCl3、400MHz):δ1.46(s,9H)、2.12(m,2H)、2.55(m,3H)、4.17(m,1H)。
GC分析:保持時間9.02分(シス異性体)(87.5%面積)、保持時間9.07分(トランス異性体)(10.0%面積)。
【0350】
調製例22
3−(p−トルエンスルホニルオキシ)−シクロブタンカルボン酸t−ブチルエステル
【0351】
【化44】
方法A
調製例21の化合物(3.02g、17.35mmol)を窒素中でピリジン(15ml)に溶解させ、0℃に冷却した。塩化p−トルエンスルホニル(3.5g、18.4mmol)を一度に加え、溶液を室温で72時間攪拌した。得られる、白色の懸濁した材料を含有するピンク色の溶液を真空中で濃縮し、2N HCl水溶液(30ml)と酢酸エチル(30ml)とに分配し、水層を酢酸エチル(15ml)で再び洗浄した。有機層を合わせて2N HCl水溶液(15ml)、重炭酸ナトリウム飽和水溶液(15ml)、およびブライン(30ml)で洗浄し、真空中で濃縮して、表題化合物を橙色の油状物(5.15g、15.7mmol、90%)として得たが、この油状物は静置すると凝固した。
1H−NMR(400MHz、CDCl3):δ1.43(s,9H)、2.32〜2.57(複合,5H)、2.46(s,3H)、4.73(m,1H)、7.35(d,2H)、7.79(d,2H)。
LC−MS:シス異性体 保持時間21.78分(81%)、LRMS(ESI)m/z 327[MH+]、トランス異性体 保持時間22.08分(7.3%)、LRMS(ESI)m/z 327[MH+]。
【0352】
方法B
調製例21の化合物(10.0、58mmol)を窒素中でピリジン(25ml)に溶解させ、0℃に冷却した。塩化p−トルエンスルホニル(16.6g、87mmol)をピリジン(25ml)に溶解させ、温度を0℃〜5℃の間に保ちながら滴下した。次いで、反応液を室温に温め、終夜攪拌した。
【0353】
反応液を濃縮して固体にし、酢酸エチル(50ml)中にスラリー化した。スラリーを0〜5℃に冷却し、2Mの塩酸(75ml)で洗浄し、相を分離した。下方の水相を再び酢酸エチル(50ml)で抽出した。有機相を合わせて水(50ml)および炭酸水素ナトリウム溶液(50ml)で洗浄した。有機相を濃縮し、0〜5℃に冷却した。N,N−ジメチルエチレンジアミン(3.5g、40mmol)を滴下した。反応混合物を2Mの塩酸(75ml)で洗浄した。上方の有機相を水(75ml)およびブライン(75ml)で洗浄した。有機相を濃縮して淡黄色の油状物としたが、これを静置すると結晶化した(15.5g、47mmol、82%)。
1H−NMR(400MHz、CDCl3):δ1.43(s,9H)、2.32〜2.57(複合,5H)、2.46(s,3H)、4.73(m,1H)、7.35(d,2H)、7.79(d,2H)。
GC分析:保持時間15.98分(シス異性体)(94.1%面積)、保持時間15.82分(トランス異性体)(5.9%面積)。
【0354】
調製例23
8’クロロ−5’−メトキシ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−2’(3’H)−オン
【0355】
【化45】
2−クロロ−5−メトキシフェニル尿素(WO02/074754、中間体5)(22.04g、0.11mol)にEaton試薬(7.7重量%の酸化リン(V)のメタンスルホン酸溶液)(440.8ml)を加えた後、シクロペンタノン(19.5ml、0.22mol)を加え、得られる溶液を85℃で4時間加熱した。反応液を約5℃に冷却し、温度を20〜30℃の間に保ちながら慎重に水を加えた。次いで、ジクロロメタン(全体で400ml)およびブライン(200ml)を加え、相を分離した。水相をジクロロメタン(2×100ml)で洗浄し、有機抽出物を合わせ、真空中で蒸発にさせて、暗色の油状物を得、これを、ジクロロメタン:メタノール(95:5→90:10)を溶離液としながらシリカのクロマトグラフィーカラムで精製して、生成物を暗褐色の固体として得た。固体をジエチルエーテルおよびペンタンで摩砕し、濾過によって収集し、乾燥させて、表題化合物を褐色の固体(27.17g、0.1mol、92%)として得た。
1H−NMR(400MHz、CDCl3):δ1.7〜1.8(m,6H)、2.4〜2.5(m,2H)、3.7(s,3H)、5.75(br s,1H)、6.4(d,1H)、7.05(s,1H)、7.15(d,1H)。
LRMS m/z(APCI)267[M+H]+
【0356】
調製例24
8’−クロロ−5’−ヒドロキシ−1’H−スピロ[シクロペンタン−1,4’−キナゾリン]−2’(3’H)−オン
【0357】
【化46】
調製例23の化合物(25g、0.093mol)に、酢酸(250ml)を加えた後、48%の臭化水素酸水溶液(207ml、1.86mol)を一度に加え、得られる溶液を115℃で7日間攪拌した。反応混合物を100℃に冷却し、水(207ml)を滴下した。混合物を真空中で濃縮して、褐色の固体を沈殿させ、これを濾過によって収集し、水(2×100ml)で洗浄した。静置すると、濾液から次の分の生成物が得られた。合わせた分の生成物を、トルエン(150ml)とのスラリーによって、また真空中で3回溶媒を除去することにより乾燥させて、灰色の固体を得、これをシリカに予め吸収させ、ジクロロメタン:メタノール(98:2→95:5→80:20)を溶離液とするカラムクロマトグラフィーによって精製した。生成物の画分を真空中で濃縮し、得られる固体をペンタンで摩砕し、濾過して、表題化合物を褐色の固体(10g、0.0395mol、42%)として得た。
1H−NMR(400MHz、D6−DMSO)δ1.6〜1.8(m,6H)、2.3〜2.4(m,2H)、6.4(d,1H)、7.11(d,1H)、7.2(s,1H)、7.8(s,1H)、9.9(s,1H)。
LRMS m/z(ESI)253[M+H]+
【0358】
調製例25
(2−クロロ−5−メトキシフェニル)尿素
【0359】
【化47】
塩酸2−クロロ−5−メトキシアニリン(26g、134mmol)を酢酸(117ml)および水(13ml)に加えた。スラリーを30℃に温めた。シアン酸カリウム(13g、161mmol)の水(104ml)溶液を滴下した。40℃で1時間経過後、反応液を20℃に冷却し、濾過し、水(78ml)で洗浄した。生成物を60℃の真空中で一晩乾燥させて、表題化合物を白色固体(21.8g、81%)として得た。
1H−NMR(300MHz、D6−DMSO):δ3.71(s,3H)、6.41(s,2H)、6.53(d,1H)、7.26(d,1H)、7.85(s,1H)、7.98(s,1H)。
LC−MS(ESI):10.9分 99.4(%)、m/z 201[MH+]
【0360】
調製例26
8’−クロロ−5’−メトキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン
【0361】
【化48】
調製例25の化合物(5.0g、25mmol)をEaton試薬(7%w/wのP2O5メタンスルホン酸溶液)(150g)に加えて溶液を生成し、次いでこれを60℃に加熱した。シクロヘキサノン(4.9g、50mmol)を10分間かけて加え、次いで、反応液を80℃に温め、1時間その温度に保った。次いで、反応混合物を5℃に冷却し、水を加えた(150ml)。次いで、反応混合物をジクロロメタン(100ml)で抽出し、上方の水相をジクロロメタン(2×10ml)で洗浄した。有機相を合わせて濃縮した後、2−プロパノール(110ml)を加えた。反応混合物をさらに大気圧下で濃縮して、残りのジクロロメタンを除去した。反応液を冷却して生成物を結晶化し、5℃で1時間攪拌した。生成物を濾過によって収集し、2−プロパノール(15ml)で洗浄した後、50℃で18時間かけて乾燥させて、表題化合物を白色固体(5.2g、19mmol、76%)として得た。
1H−NMR(300MHz、D6−DMSO):δ1.2(m,1H)、1.4(m,2H)、1.5(m,1H)、1.6(m,2H)、1.7(m,1H)、1.8(m,1H)、2.4(t,2H)、3.79(s,3H)、6.64(d,1H)、6.97(s,1H)、7.26(d,1H)、7.91(s,1H)。
LC−MS(ESI):18.5分 97.4(%)、m/z 281[MH+]
【0362】
調製例27
8’−クロロ−5’−ヒドロキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン
【0363】
【化49】
ステップ(a)
8’−クロロ−5’−ヒドロキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン酢酸溶媒和物
調製例26の化合物(350g、1.24mol)を酢酸(3500ml)中にスラリー化した。スラリーに室温で臭化水素酸(水中48%w/w)(2800ml、24.8mol)を加えた。次いで、このスラリーを還流温度に温め、4日間攪拌した。反応液を100℃に冷却し、1時間かけて水(2800ml)を滴下した。スラリーを10℃に冷却し、1時間攪拌した後、濾過し、水(1100ml)で洗浄し、真空オーブンに入れて終夜乾燥させて、表題化合物を白色固体(344g、1.28mol、103%)として得た。
1H−NMR(D6−DMSO、300MHz):δ1.2(m,1H)、1.4(m,2H)、1.5(m,1H)、1.6(m,2H)、1.7(m,1H)、1.8(m,1H)、1.9(s,3H,CH3COOH)、2.4(t,2H)、6.45(d,1H)、6.95(s,1H)、7.1(d,1H)、7.75(s,1H)、9.9(s,1H,CH3COOH)。
LC−MS(ESI):14.2分 99.1(%)、m/z 267[MH+]。
【0364】
ステップ(b)
8’−クロロ−5’−ヒドロキシスピロ[シクロヘキサン−1,4’−キナゾリン]−2’(1’H)−オン
ステップ(a)の酢酸溶媒和物(330g、1.24mol)を、室温で6時間かけてアセトン(730ml)中にスラリー化した。次いで、生成物を濾過によって収集し、アセトン(330ml)で洗浄した後、60℃で18時間乾燥させて、溶媒和していない表題化合物を白色固体(238g、0.89mol、72%)として得た。
1H−NMR(D6−DMSO、300MHz):δ1.2(m,1H)、1.4(m,2H)、1.5(m,1H)、1.6(m,2H)、1.7(m,1H)、1.8(m,1H)、2.4(t,2H)、6.45(d,1H)、6.95(s,1H)、7.1(d,1H)、7.75(s,1H)。
【図面の簡単な説明】
【0365】
【図1】本発明の実施例1および2の化合物、ならびにWO02/074754の実施例75の化合物(化合物A)の、1mg/kgで静脈内投与した後の、投与量で正規化した平均の薬物動態プロフィールを示す濃度/時間のグラフである。
【図2】実施例2の化合物の酢酸溶媒和物についての測定したパターン(A)およびシミュレートしたパターン(B)の粉末X線回折(PXRD)プロットを示す図である。
【図3】実施例2の化合物の酢酸溶媒和物について、熱重量分析(TGA)を使用して加熱した後の質量減少のプロットを示すグラフである(示される15.05%の質量減少は、1モル当量の酢酸に等しい)。
【図4】ケイ素でドープ処理した実施例2の化合物の脱溶媒和形態(形態A)(A)、TGA分析前(B)およびTGA分析後(C)のこの化合物の酢酸溶媒和物のPXRDパターンを示す図である。
【図5】ケイ素標準物質を基準として補正した、実施例2の化合物の溶媒和していない結晶形態(形態A)のPXRDパターンを示す図である。
【図6】実施例2の化合物の溶媒和していない結晶形態(形態A)についての示差走査熱量測定(DSC)トレースである。
【図7】実施例2の化合物の結晶性の溶媒和していない形態(形態A)、ならびにジメチルアセトアミド(DMAC)(B)、ピリジン(C)、テトラヒドロフラン(THF)(D)、ジメチルスルホキシド(DMSO)(E)、および酢酸(F)溶媒和物についてのPXRDプロットを示す図である。
【図8】実施例2の化合物のピリジン溶媒和物のTGAトレースを示すグラフであり、示される17.3%の質量減少は、溶媒対化合物の比が1:1であることに等しい。
【図9】実施例2の化合物のテトラヒドロフラン溶媒和物のTGAトレースを示すグラフであり、14.7%の合計質量減少は、化合物対THF溶媒の比が1:1であることに相当する(加熱後の溶媒減少の段階的な性質は、恐らく、中間体の半THF溶媒和物形態の存在を示唆する)。
【図10】実施例2の化合物のジメチルアセトアミド溶媒和物のTGAトレースを示すグラフであり、33.0%の合計質量減少は、溶媒対化合物の比が2:1であることに等しい。
【図11】実施例2の化合物のピリジン溶媒和物(A)、TGA後のピリジン溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【図12】実施例2の化合物のテトラヒドロフラン溶媒和物(A)、TGA後のTHF溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【図13】実施例2の化合物のジメチルアセトアミド溶媒和物(A)、TGA後のジメチルアセトアミド溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【図14】実施例2の化合物のジメチルスルホキシド溶媒和物(A)、真空乾燥後のDMSO溶媒和物(B)、および形態A(C)のPXRDパターンを示す図である。
【特許請求の範囲】
【請求項1】
式(I)の化合物
【化1】
[式中、
mは、0、1、または2であり、
Xは、O、S、またはN−CNであり、
Rは、F、Cl、またはCNであり、
Aは、C1〜4アルキル基で置換されていてもよいC3〜6シクロアルキレン基であり、
Bは、単結合またはC1〜2アルキレン基である]
または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグ。
【請求項2】
mが1である、請求項1に記載の化合物。
【請求項3】
XがOである、請求項1または請求項2に記載の化合物。
【請求項4】
RがClである、請求項1から3のいずれか一項に記載の化合物。
【請求項5】
Aがシクロブチレン基である、請求項1から4のいずれか一項に記載の化合物。
【請求項6】
Aが1,3−シクロブチレン基である、請求項5に記載の化合物。
【請求項7】
Aがトランス−1,3−シクロブチレン基である、請求項6に記載の化合物。
【請求項8】
Bが単結合である、請求項1から7のいずれか一項に記載の化合物。
【請求項9】
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−シアノ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
から選択される化合物または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグ。
【請求項10】
請求項1から9のいずれかに記載の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグと、薬学的に許容できる担体または希釈剤とを含む医薬組成物。
【請求項11】
医薬として使用するための、請求項1から9のいずれか一項に記載の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグ。
【請求項12】
PDE7阻害剤による治療が適切である疾患または状態を治療するための医薬の製造における、請求項1から9のいずれか一項に記載の化合物または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグの使用。
【請求項13】
前記疾患または状態が、疼痛、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、または炎症性腸疾患から選択される、請求項12に記載の使用。
【請求項14】
前記疾患または状態が疼痛である、請求項13に記載の使用。
【請求項15】
前記疼痛が神経因性疼痛である、請求項14に記載の使用。
【請求項16】
有効量の請求項1から9のいずれか一項に記載の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグを投与することを含む、PDE7阻害剤による治療が適切である疾患または状態の治療方法。
【請求項17】
前記疾患または状態が、疼痛、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、または炎症性腸疾患から選択される、請求項16に記載の方法。
【請求項18】
前記疾患または状態が疼痛である、請求項17に記載の方法。
【請求項19】
前記疼痛が神経因性疼痛である、請求項18に記載の方法。
【請求項1】
式(I)の化合物
【化1】
[式中、
mは、0、1、または2であり、
Xは、O、S、またはN−CNであり、
Rは、F、Cl、またはCNであり、
Aは、C1〜4アルキル基で置換されていてもよいC3〜6シクロアルキレン基であり、
Bは、単結合またはC1〜2アルキレン基である]
または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグ。
【請求項2】
mが1である、請求項1に記載の化合物。
【請求項3】
XがOである、請求項1または請求項2に記載の化合物。
【請求項4】
RがClである、請求項1から3のいずれか一項に記載の化合物。
【請求項5】
Aがシクロブチレン基である、請求項1から4のいずれか一項に記載の化合物。
【請求項6】
Aが1,3−シクロブチレン基である、請求項5に記載の化合物。
【請求項7】
Aがトランス−1,3−シクロブチレン基である、請求項6に記載の化合物。
【請求項8】
Bが単結合である、請求項1から7のいずれか一項に記載の化合物。
【請求項9】
シス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
3−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−シアノ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
1−[(8’−フルオロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘキサン−1,4’−キナゾリン]−5’−イル)オキシメチル]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロヘプチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸、
トランス−3−[(8’−クロロ−2’−オキソ−2’,3’−ジヒドロ−1’H−スピロ[シクロペンチル−1,4’−キナゾリン]−5’−イル)オキシ]シクロブタンカルボン酸
から選択される化合物または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグ。
【請求項10】
請求項1から9のいずれかに記載の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグと、薬学的に許容できる担体または希釈剤とを含む医薬組成物。
【請求項11】
医薬として使用するための、請求項1から9のいずれか一項に記載の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグ。
【請求項12】
PDE7阻害剤による治療が適切である疾患または状態を治療するための医薬の製造における、請求項1から9のいずれか一項に記載の化合物または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグの使用。
【請求項13】
前記疾患または状態が、疼痛、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、または炎症性腸疾患から選択される、請求項12に記載の使用。
【請求項14】
前記疾患または状態が疼痛である、請求項13に記載の使用。
【請求項15】
前記疼痛が神経因性疼痛である、請求項14に記載の使用。
【請求項16】
有効量の請求項1から9のいずれか一項に記載の化合物、または薬学的に許容できるその塩、溶媒和物、多形体、もしくはプロドラッグを投与することを含む、PDE7阻害剤による治療が適切である疾患または状態の治療方法。
【請求項17】
前記疾患または状態が、疼痛、T細胞関連疾患、自己免疫疾患、多発性硬化症、骨粗鬆症、慢性閉塞性肺疾患、喘息、癌、後天性免疫不全症候群(AIDS)、アレルギー、または炎症性腸疾患から選択される、請求項16に記載の方法。
【請求項18】
前記疾患または状態が疼痛である、請求項17に記載の方法。
【請求項19】
前記疼痛が神経因性疼痛である、請求項18に記載の方法。
【図1】

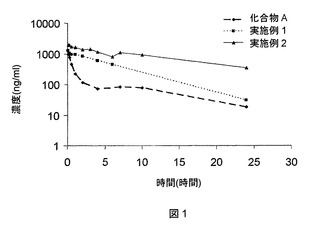
【図2】
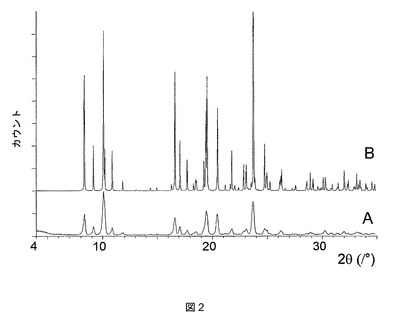
【図3】
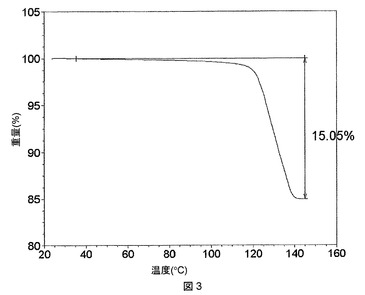
【図4】
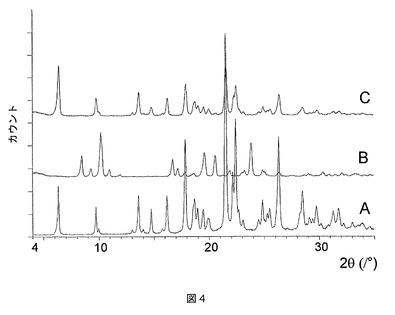
【図5】
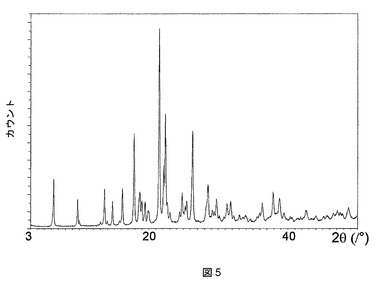
【図6】
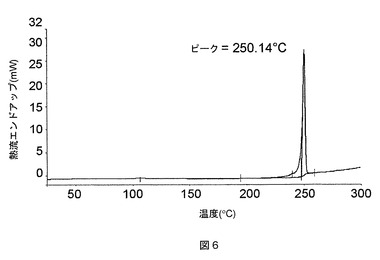
【図7】
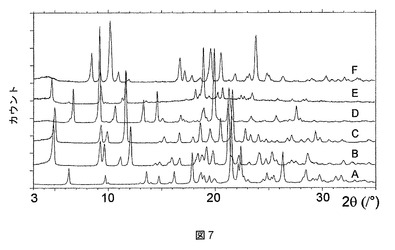
【図8】
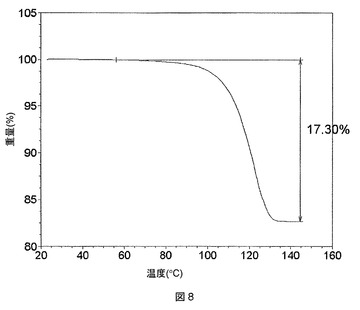
【図9】
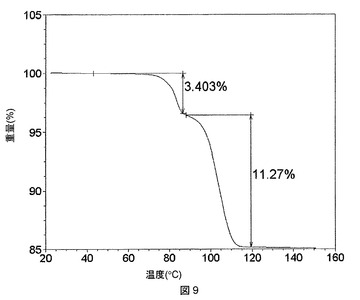
【図10】
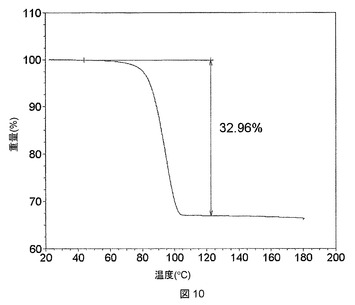
【図11】
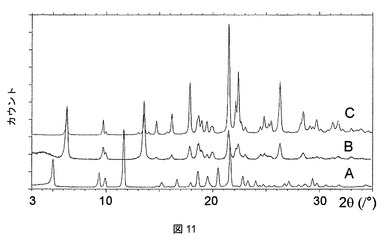
【図12】
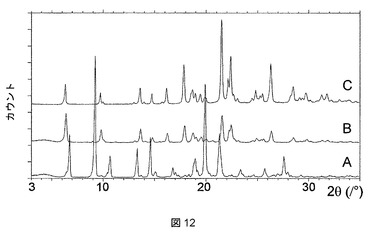
【図13】
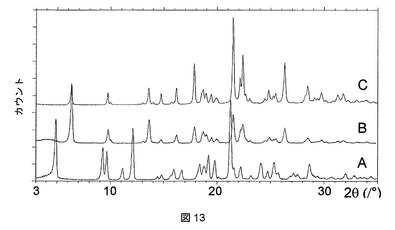
【図14】
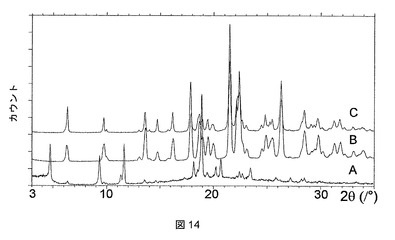
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【公表番号】特表2009−517453(P2009−517453A)
【公表日】平成21年4月30日(2009.4.30)
【国際特許分類】
【出願番号】特願2008−542855(P2008−542855)
【出願日】平成18年11月23日(2006.11.23)
【国際出願番号】PCT/IB2006/003388
【国際公開番号】WO2007/063391
【国際公開日】平成19年6月7日(2007.6.7)
【出願人】(597014501)ファイザー・リミテッド (107)
【氏名又は名称原語表記】Pfizer Limited
【住所又は居所原語表記】Ramsgate Road, Sandwich, Kent, England
【Fターム(参考)】
【公表日】平成21年4月30日(2009.4.30)
【国際特許分類】
【出願日】平成18年11月23日(2006.11.23)
【国際出願番号】PCT/IB2006/003388
【国際公開番号】WO2007/063391
【国際公開日】平成19年6月7日(2007.6.7)
【出願人】(597014501)ファイザー・リミテッド (107)
【氏名又は名称原語表記】Pfizer Limited
【住所又は居所原語表記】Ramsgate Road, Sandwich, Kent, England
【Fターム(参考)】
[ Back to top ]