
PI−3キナーゼインヒビタープロドラッグ
【課題】PI−3キナーゼのインヒビターの新規なプロドラッグを提供すること。
【解決手段】この新規な化合物は、LY294002および可逆的に四級化されたアミンを含むそのアナログである。改善された薬物動態および薬力学的特性を有するクラスIのPI−3キナーゼインヒビターを提供する。本発明は、特定の実施形態において、四級窒素を含むプロ化合物、およびその四級窒素の1つの結合が、加水分解された化合物を提供する。これらの化合物を生成する方法、患者を処置する方法も提供される。
【解決手段】この新規な化合物は、LY294002および可逆的に四級化されたアミンを含むそのアナログである。改善された薬物動態および薬力学的特性を有するクラスIのPI−3キナーゼインヒビターを提供する。本発明は、特定の実施形態において、四級窒素を含むプロ化合物、およびその四級窒素の1つの結合が、加水分解された化合物を提供する。これらの化合物を生成する方法、患者を処置する方法も提供される。
【発明の詳細な説明】
【技術分野】
【0001】
発明の背景
1.発明の分野
本発明は、PI−キナーゼインヒビターのプロドラッグおよびこれらのインヒビターを用いる方法に関する。
【背景技術】
【0002】
2.関連技術の説明
PI−3キナーゼは、D3位のホスファチジルイノシトールをリン酸化して重要な二次メッセンジャーであるホスファチジルイノシトール3’−リン酸を生成する、大きいファミリーの脂質キナーゼである。PI−3キナーゼファミリーのメンバーは、配列相同性および酵素触媒によって形成される生成物に基づいて3つのクラスに分類される。クラスIのPI−3キナーゼは2つのサブユニットからなる:110kdの触媒性サブユニットおよび85kdの調節性サブユニット。クラスIのPI−3キナーゼは、サイトカイン、インテグリン、増殖因子および免疫レセプターの下流の重要なシグナル伝達事象に関与しており、これによって、この経路の制御が重要な治療効果をもたらし得ることが示唆される。
【0003】
クラスIのPI−3キナーゼの阻害がアポトーシスを誘導し、インビボにおいて腫瘍誘
導性血管形成をブロックし、そして特定の腫瘍の放射線感受性を増大する。LY294002(2−(4−モルホリニル)−8−フェニル−4H−1−ベンソピラン−4−オン)(化合物1)は、クラスI PI−3キナーゼの周知の特異的なインヒビターであり、そして抗癌特性を有することが実証されている。
【0004】
【化17】

【発明の概要】
【発明が解決しようとする課題】
【0005】
しかし、LY294002の抗癌適用は、それが水溶性を欠くこと、および薬物動態が良くないことによって厳しく制限されている。さらに、LY294002は、組織特異的な特性を有さず、そして動物で急速に代謝されることが実証されている。これらの要因のせいで、LY294002は頻繁な間隔で投与する必要があり、従って正常な細胞でのPI−3キナーゼを阻害する能力も有し、それによって望ましくない副作用をもたらす。
【0006】
改善された薬物動態および薬力学的特性を有するクラスIのPI−3キナーゼインヒビターが依然として必要である。本発明は、これらの必要性をみたし、他の関連の利点を提供する。
【課題を解決するための手段】
【0007】
発明の要旨
本発明は、例えば、以下を提供する。
(項目1)
四級窒素の1つの結合が加水分解可能であり、該結合の加水分解後に、式:
【化1】
の化合物が得られる、四級窒素を含むプロ化合物であって、
ここで、
R1およびR2が独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3がH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5が独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成する、化合物。
(項目2)
R1−環A−R2が:
【化2】
からなる群より選択される、項目1に記載の化合物。
(項目3)
R4−N−R5が:
【化3】
からなる群より選択される、項目1に記載の化合物。
(項目4)
式:
【化4】
の化合物であって、
ここで、
環Aは、ベンゾであり;
Z1およびZ2が独立してSまたはOであり;
R1およびR2が独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3がH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5が独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6が、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
Lがリンカー基である;
化合物。
(項目5)
R1−環A−R2が:
【化5】
からなる群より選択される、項目4に記載の化合物。
(項目6)
R4−N−R5が:
【化6】
からなる群より選択される、項目4に記載の化合物。
(項目7)
R6が:
【化7】
からなる群より選択される、項目4に記載の化合物。
(項目8)
式:
【化8】
の化合物であって、
ここで、
環Aがベンゾであり;
Z1、Z2、Z3およびZ4が独立してSまたはOであり;
R1およびR2が独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3がH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5が独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6が、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
R7が−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−である;
化合物。
(項目9)
R1−環A−R2が:
【化9】
からなる群より選択される、項目8に記載の化合物。
(項目10)
R4−N−R5が:
【化10】
からなる群より選択される、項目8に記載の化合物。
(項目11)
R6が:
【化11】
からなる群より選択される、項目8に記載の化合物。
(項目12)
R7と四級アミンとの間の結合が加水分解可能である、項目8に記載の化合物。
(項目13)
式:
【化12】
の化合物であって、
ここで、
環Aがベンゾであり;
Z1およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
R7が−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;そして
Tが標的化因子を表す;
化合物。
(項目14)
R1−環A−R2が
【化13】
からなる群より選択される、項目13に記載の化合物。
(項目15)
R4−N−R5が:
【化14】
からなる群より選択される、項目13に記載の化合物。
(項目16)
R6が:
【化15】
からなる群より選択される、項目13に記載の化合物。
(項目17)
前記標的化因子が、ビタミン、ペプチド、タンパク質、リポソーム、骨親和性剤および軟骨親和性剤からなる群より選択される、項目13に記載の化合物。
(項目18)
前記ビタミンがフォレートまたはビタミンCである、項目17に記載の化合物。
(項目19)
前記ペプチドが、RGD、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択されるRGD含有ペプチドである、項目17に記載の化合物。
(項目20)
前記タンパク質が腫瘍特異的モノクローナル抗体またはそのフラグメントである、項目17に記載の化合物。
(項目21)
前記骨親和性剤が、ホスホネート、ホスホン酸、アミノメチルホスホン酸、ホスフェート、ポリホスフェートおよびハイドロキシアパタイト結合ポリペプチドからなる群より選択される、項目17に記載の化合物。
(項目22)
前記骨親和性剤が、EDTMP、DOTMP、ABDTMP、BAD、MTX−BP、CF−BP、(Asp)6、(Glu)6、アレンドロネート、パミドロネート、4−アミノブチルホスホン酸、1−ヒドロキシエタン−1、1−二リン酸、アミノメチレンビスホスホン酸、フィチン酸およびN,N−ビス(メチルホスホノ)−4−アミノ安息香酸である、項目21に記載の化合物。
(項目23)
式:
【化16】
の化合物であって、
ここで、
Xがハロ基であり;
Yが−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−を表し;
Z1およびZ2が独立してSまたはOであり;そして
n=0〜1である;
化合物。
(項目24)
XがClまたはIであり;
Yが−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり
Z1およびZ2が各々Oであり;そして
n=0である;
項目23に記載の化合物。
(項目25)
XがClまたはIであり;
Z1およびZ2が各々Oであり;そして
n=1である;
項目23に記載の化合物。
(項目26)
項目1〜21のいずれか1項に記載の化合物を精製する方法であって:
(a)少なくとも0.1(v/v)%の酸を含む溶液に該化合物を含む組成物を添加する工程と;
(b)該化合物を含む(a)の溶液をクロマトグラフィーシステムに加える工程と;
(c)該化合物を単離する工程と;
を包含する、方法。
(項目27)
前記クロマトグラフィーシステムがHPLCである、項目26に記載の方法。
(項目28)
PI−3キナーゼ活性に関連する条件に罹患している患者を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目29)
炎症性疾患を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目30)
前記組成物が1つ以上のさらなる治療剤をさらに含む、項目28に記載の方法。
(項目31)
前記1つ以上のさらなる治療剤が、アルキル化剤、代謝拮抗物質、アスパラギナーゼ、ビンクリスチン、ビンブラスチン、アントラサイクリン、微小管阻害剤、タキソール、ハーセプチン、ゲムシタビンおよびエトポシドからなる群より選択される、項目30に記載の方法。
(項目32)
患者においてアポトーシスを誘導するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目33)
腫瘍細胞の化学感受性を増強するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目34)
腫瘍細胞の放射線感受性を増強するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目35)
腫瘍誘導性の血管形成を阻害するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目36)
癌以外の疾患に関連する血管形成のプロセスを阻害するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目37)
膵炎を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目38)
潰瘍を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目39)
胃癌を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目40)
肝臓癌を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目41)
ステントの能力を改善するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目42)
加齢性黄斑変性症を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目43)
変異PTENに伴う状態を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目44)
高血圧を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目45)
白血球の機能を破壊するための方法であって、白血球と、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量とを接触させる工程を包含する、方法。
(項目46)
前駆細胞の分化を抑制するための方法であって、前駆細胞と、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量とを接触させる工程を包含する、方法。
(項目47)
哺乳動物の細胞全体のホスファチジルイノシトール3キナーゼを阻害するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を投与する工程を包含する、方法。
(項目48)
前記投与が、緩徐なI.V.注入による、項目47に記載の方法。
【0008】
本発明は、四級窒素の1つの結合が加水分解され、この結合の加水分解後に、式:
【0009】
【化18】
の化合物が得られる、四級窒素を含むプロ化合物に関しており、
ここで、
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成する。
【0010】
このプロ化合物は、式:
【0011】
【化19】
の化合物であってもよく、
環Aはベンゾであり;
Z1、およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
Lがリンカー基である。
この標的化因子は、ビタミン、ペプチド、タンパク質、リポソーム、骨親和性剤または軟骨親和性剤であってもよい。
【0012】
本発明はまた、式:
【0013】
【化20】
の中間化合物に関し、
ここで、
Xは、ハロ基、好ましくはClまたはIであり;
Yは、−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;
Z1およびZ2は独立してSまたはOであり;そして
n=0〜1である。
【0014】
本発明はまた、PI−3キナーゼ活性に関連する状態、炎症性疾患、加齢性黄斑変性症、変異PTENに伴う状態、高血圧、膵炎、潰瘍、癌の処置;白血球機能破壊;アポトーシス誘導;腫瘍細胞の化学感受性の増強;腫瘍細胞の放射線感受性の増強;腫瘍誘導性血管形成の阻害;癌以外の疾患に関連する血管形成プロセスの阻害;ステントの能力の改善;患者の細胞全体におけるホスファチジルイノシトール3キナーゼの阻害のためのプロ化合物の使用に関し、これには本発明のプロ化合物を含む組成物の有効量をその必要な患者に投与する工程を包含する。このプロ化合物は、緩徐なI.V.注入によって患者に投与され得る。
【0015】
本発明は、前駆細胞と、本発明のプロ化合物を含む組成物の有効量とを接触させる工程を包含する、前駆細胞の分化を抑制するための方法にも関する。
【0016】
本発明はまた、本発明のプロ化合物の精製に関し、これには少なくとも0.1(v/v)%の酸を含む溶液にプロ化合物を添加する工程を包含する。このプロ化合物を含む溶液を、好ましくはHPLCによってクロマトグラフにかけて、プロ化合物を単離する。
【図面の簡単な説明】
【0017】
【図1】図1は、EDTMP、DOTMP、ABDTMP、BAD、MTX−BPおよびCF−BPの化学構造を示す。
【図2】図2は、潜在的な骨標的化因子の化学構造を示す。
【図3】図3は、骨標的化因子のホスホン酸塩を修飾するための化学反応を示す。
【図4】図4は、骨標的化因子のホスホン酸塩を修飾するためのアルキル化反応を示す。
【図5】図5は、EDTMPおよびDOTMPを化学的に修飾するための概念を示す。
【図6】図6は、J774細胞におけるLY294002による食作用の阻害を示す。カラムは、食作用性係数、または食細胞応答の陽性な細胞の割合を示す。食作用性係数は、100J774細胞1個あたりに見出されるsRBC(ヒツジ赤血球)の数であり、そして食作用性細胞の%は、少なくとも1つのsRBCを食菌したJ774細胞の%である。誤差指示線は、平均の標準偏差に相当する。
【図7】図7は、化合物1126(A036−33)のUVおよびELSクロマトグラムを示す。
【図8】図8は、化合物1126(A036−33)の正の質量スペクトルを示す。
【図9】図9は、Avβ3標的化PI 3キナーゼインヒビターが、Matrigel上でのEDC−CBF1内皮細胞の管腔の形成を抑止することを示す。
【発明を実施するための形態】
【0018】
発明の詳細な説明
本発明の化合物、生成物および組成物、ならびに方法が開示され記載される以前には、本明細書において用いられる専門用語は、特定の実施形態を記載する目的のためだけであって、限定を意図するものではないということが理解されるべきである。本明細書および添付の特許請求の範囲において用いられる場合、単数形「1つの(a)、(an)」および「この、その(the)」とは、文脈が明白に他を示さない限り複数の言及を包含することに注意のこと。
【0019】
本出願を通じて、刊行物が言及される場合、これらの刊行物の開示はその全体が本出願に参考として援用され、本発明が属する当該分野の状況をさらに詳細に記載する。
【0020】
1.定義
本明細書において用いる場合、「分枝した(branched)」という用語は、1〜24個の骨格原子を含む基であって、この基の骨格鎖が、主な鎖から1つ以上の従属する分枝を含む基をいう。本明細書における好ましい分枝した基は1〜12個の骨格原子を含む。分枝した基の例としては、限定はしないが、イソブチル、t−ブチル、イソプロピル、−CH2CH2CH(CH3)CH2CH3、−CH2CH(CH2CH3)CH2CH3、−CH2CH2C(CH3)2CH3、−CH2CH2C(CH3)3などが挙げられる。
【0021】
本明細書において用いる場合、「分枝していない(unbranched)」という用語は、1〜24個の骨格原子を含む基であって、この基の骨格鎖が、直接の線状に伸びる基をいう。本明細書における好ましい分枝していない基は、1〜12個の骨格原子を含む。
【0022】
本明細書において用いる場合、「環状(cyclic)」または「シクロ(cyclo)」という用語は、単独で、または組み合わせて、1つ以上の閉じた環を有する基であって、不飽和であっても飽和であっても、3〜12個の骨格原子、好ましくは3〜7個の骨格原子の環を有する基をいう。
【0023】
本明細書において用いる場合、「低級(lower)」という用語は、1〜6個の骨格原子を有する基をいう。
【0024】
本明細書において用いる場合、「飽和された(saturated)」という用語は、その骨格原子の全ての利用可能な原子価結合が、他の原子に結合されている基をいう。飽和された基の代表的な例としては、限定はしないが、ブチル、シクロヘキシル、ピペリジンなどが挙げられる。
【0025】
本明細書において用いる場合、「不飽和の(unsaturated)」という用語は、2つの隣接する骨格原子の少なくとも1つの利用可能な原子価結合が、他の原子に結合されていない基をいう。不飽和基の代表的な例としては、限定はしないが、−CH2CH2CH=CH2、フェニル、ピロールなどが挙げられる。
【0026】
本明細書において用いる場合、「脂肪族(aliphatic)」という用語は、分枝していない、分枝した、または環状の基を指し、これは置換されてもされなくてもよく、そして飽和されても飽和されなくてもよいが、ただし芳香族ではない。脂肪族という用語はさらに、脂肪族基を包含し、これは、炭化水素骨格の1つ以上の原子を置き換える、酸素、窒素、イオウまたはリン原子を含む。
【0027】
本明細書において用いる場合、「芳香族(aromatic)」という用語は、置換されても置換されなくてもよいが、4n+2の非局在化π(パイ)電子を有する、不飽和環状炭化水素基を指す。芳香族という用語はさらに、炭化水素骨格の1つ以上の炭素を置換する窒素原子を含む、芳香族基を包含する。芳香族基の例としては、限定はしないが、フェニル、ナフチル、チエニル、フラニル、ピリジニル、(イソ)オキサゾイルなどが挙げられる。
【0028】
本明細書において用いる場合、「置換された(substituted)」という用語は、炭素または適切なヘテロ原子を取り外して、さらなる基で置換した、1つ以上の水素または他の原子を有する基をいう。本明細書において、好ましい置換基は、1〜5個、最も好ましくは1〜3個の置換基で置換される。2つの置換基を有する原子は、「ジ(di)」で示されるが、3つ以上の置換基を有する原子は、「ポリ(poly)」と示される。このような置換基の代表的な例としては、限定はしないが、脂肪族基、芳香族基、アルキル、アルケニル、アルキニル、アリール、アルコキシ、ハロ、アリールオキシ、カルボニル、アクリル、シアノ、アミノ、ニトロ、リン酸含有基、イオウ含有基、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルアミノカルボニル、ジアリールアミノカルボニル、アルキルチオカルボニル、アシルアミノ、アミジノ、イミノ、アルキルチオ、アリールチオ、チオカルボキシレート、アルキルスルフィニル、トリフルオロメチル、アジド、ヘテロシクリル、アルキルアリール、ヘテロアリール、セミカルバジド、チオセミカルバジド、マレイミド、オキシミノ、イミダート、シクロアルキル、シクロアルキルカルボニル、ジアルキルアミノ、アリールシクロアルキル、アリールカルボニル、アリールアルキルカルボニル、アリールシクロアルキルカルボニル、アリールホスフィニル、アリールアルキルホスフィニル、アリールシクロアルキルホスフィニル、アリールホスホニル、アリールアルキルホスホニル、アリールシクロアルキルホスホニル、アリールスルホニル、アリールシクロアルキルスルホニル、アクリルシクロアルキルスルホニル、それらの組み合わせ、およびそれらの置換が挙げられる。
【0029】
本明細書において用いる場合、「置換されていない、非置換(unsubstituted)」という用語は、そこに結合された、またはそれに代わって置換されたいかなるさらなる基も有さない基をいう。
【0030】
単独でまたは組み合わせて本明細書において用いる場合、「アルキル」という用語は、分枝した、または分枝していない、飽和脂肪族基をいう。アルキル基の代表的な例としては、限定はしないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、オクチル、デシル、テトラデシル、ヘキサデシル、エイコシル、テトラコシルなどが挙げられる。
【0031】
単独でまたは組み合わせて、本明細書において用いる場合、「アルケニル(alkenyl)」という用語は、鎖にそった任意の適切なポイントで生じ得る、少なくとも1つの炭素間二重結合を含む、分枝した、または未分枝の不飽和の脂肪族基を指す。アルケニル基の代表的な例としては、限定はしないが、エテニル、E−およびZ−ペンテニル、デセニルなどが挙げられる。
【0032】
単独でまたは組み合わせて本明細書において用いる場合、「アルキニル(alkynyl)」という用語は、鎖にそった任意の適切なポイントで生じ得る、少なくとも1つの炭素間三重結合を含む、分枝した、または未分枝の、不飽和の脂肪族基を指す。アルキニル基の代表的な例としては、限定はしないが、エチニル、プロピニル、プロパルジル、ブチニル、ヘキシニル、デシニルなどが挙げられる。
【0033】
単独でまたは組み合わせて本明細書において用いる場合、「アリール(aryl)」という用語は、他の芳香族または非芳香族の環状基に必要に応じて縮合され得る、置換した、または置換されていない芳香族基を指す。アリール基の代表的な例としては、限定はしないが、フェニル、ベンジル、ナフチル、ベンジリジン、キシリル、スチレン、スチリル、フェネチル、フェニレン、ベンゼントリイルなどが挙げられる。
【0034】
単独でまたは組み合わせて本明細書において用いる場合、「アルコキシ(alkoxy)」という用語は、単独の末端エーテル結合を通じて結合されたアルキル、アルケニルまたはアルキニル基をいう。アルコキシ基の例としては、限定はしないが、メトキシ、エトキシ、n−プロポキシ、イソ−プロポキシ、n−ブトキシ、2−ブトキシ、tert−ブトキシ、n−ペントキシ、2−ペントキシ、3−ペントキシ、イソペントキシ、ネオペントキシ、n−ヘキソキシ、2−ヘキソキシ、3−ヘキソキシ、3−メチルペントキシ、フルオロメトキシ、ジフルオロメトキシ、トリフルオロメトキシ、クロロメトキシ、ジクロロメトキシおよびトリクロロメトキシが挙げられる。
【0035】
単独でまたは組み合わせて本明細書において用いる場合、「アリールオキシ(aryloxy)」という用語は、単独の末端エーテル結合を通じて結合されたアリール基をいう。
【0036】
単独でまたは組み合わせて本明細書において用いる場合、「ハロゲン(halogen)」、「ハライド(halide)」または「ハロ(halo)」という用語は、フッ素「F」、塩素「Cl」、臭素「Br」、ヨウ素「I」およびアスタチン「At」を指す。ハロ基の代表的な例としては、限定はしないが、クロロアセトアミド、ブロモアセトアミド、ヨードアセトアミドなどが挙げられる。
【0037】
組み合わせて本明細書において用いる場合、「ヘテロ(hetero)」という用語は、炭素または水素以外の任意の元素の1つ以上の原子を含む基をいう。ヘテロ基の代表的な例としては、限定はしないが、窒素、酸素、イオウおよびリンを含むヘテロ原子を含む基が挙げられる。
【0038】
本明細書において用いる場合、「複素環(heterocycle)」という用語は、ヘテロ原子を含む環状基をいう。複素環の代表的な例としては、限定はしないが、ピリジン、ピペラジン、ピリミジン、ピリダジン、ピペラジン、ピロール、ピロリジノン、ピロリジン、モルホリン、チオモルホリン、インドール、イソインドール、イミダゾール、トリアゾール、テトラゾール、フラン、ベンゾフラン、ジベンゾフラン、チオフェン、チアゾール、ベンゾチアゾール、ベンゾキサゾール、ベンゾチオフェン、キノリン、イソキノリン、アザピン、ナフトピラン、フラノベンゾピラノンなどが挙げられる。
【0039】
単独でまたは組み合わせて、本明細書において用いる場合、「カルボニル(carbonyl)」または「カルボキシ(carboxy)」という用語は、炭素−酸素間二重結合を含む基をいう。カルボニルを含む基の代表的な例としては、限定はしないが、アルデヒド(すなわち、ホルミル)、ケトン(すなわち、アシル)、カルボン酸(すなわち、カルボキシル)、アミド(すなわち、アミド)、イミド(imide)(すなわち、イミド(imido))、エステル、無水物などが挙げられる。
【0040】
単独でまたは組み合わせて本明細書において用いる場合、「アクリル(acryl)」という用語は、Qが脂肪族基または芳香族基である、CH2=C(Q)C(O)O−によって示される基をいう。
【0041】
単独でまたは組み合わせて本明細書において用いる場合、「シアノ(cyano)」、「シアン酸塩(cyanate)」または「シアン化物(cyanide)」という用語は、炭素−窒素間二重結合をいう。シアノ基の代表的な例としては限定はしないが、イソシアン酸塩、イソチオシアン酸塩などが挙げられる。
【0042】
単独でまたは組み合わせて本明細書において用いる場合、「アミノ(amino)」という用語は、骨格の窒素原子を含む基をいう。アミノ基の代表的な例としては、限定はしないがアルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、アルキルアリールアミノ、アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、ウレイドなどが挙げられる。
【0043】
本明細書において用いる場合、「リン酸塩含有基(phosphate−containing group)」という用語は、酸化状態で少なくとも1つのリン原子を含む基をいう。代表的な例としては、限定はしないが、ホスホン酸、ホスフィン酸、リン酸エステル、ホスフィニデン、ホスフィノ、ホスフィニル、ホスフィニリデン、リン酸、ホスホノ、ホスホラニル、ホスホラニリデン、ホスホロソなどが挙げられる。
【0044】
本明細書において用いる場合、「イオウ含有基(sulfur−containing
group)」という用語は、イオウ原子を含む基をいう。代表的な例としては限定はしないが、スルフヒドリル、スルフェノ、スルフィノ、スルフェニル、スルホ、スルホニル、チオ、チオキソなどが挙げられる。
【0045】
本明細書において用いる場合、「必要に応じた(optional)」または「必要に応じて(optionally)」という用語は、引き続いて記載される事象または状況が生じても生じなくてもよいことを意味し、そしてこの説明は、この事象または状況が生じる場合、および起こらない場合を包含する。例えば、「必要に応じて置換されたアルキル(optionally substituted alkyl)」という句は、アルキル基が置換されてもされなくてもよいことを意味し、そしてこの説明は置換されていないアルキルおよび置換のあるアルキルの両方を包含する。
【0046】
本明細書において提供されるような化合物、生成物または組成物に関して用いる場合、「有効量(effective amount)」という用語は、所望の結果を得るために十分な化合物、生成物または組成物の量を意味する。必要な正確な量は、用いられる特定の化合物、生成物または組成物、投与の様式などに依存して変化する。従って、正確な「有効量」を特定することは常に可能ではない。しかし、適切な有効量は、慣用的な実験のみを用いて、本開示から知識を得た当業者によって決定できる。
【0047】
本明細書において用いる場合、「適切な(suitable)」という用語は、言及した目的について本明細書において提供されたような化合物、生成物または組成物と適合性である基をいう。言及した目的についての適切性は、慣用的な条件だけを用いて当業者によって決定できる。
【0048】
本明細書において用いる場合、「加水分解性(hydrolyzable)」という用語は、この基が加水分解できるか、または加水分解しやすい(すなわち、この分子または基の2つ以上の新しい分子または基への分離)。ことをいう。
【0049】
2.化合物
本発明は、四級アミンの1つの結合の切断の際に、式:
【0050】
【化21】
の化合物が生じる化合物を提供するが
ここで、
環Aがベンゾであり;
Z1、およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成する。
好ましくは、四級アミンの1つの結合の切断は、LY294002(化合物1)を生じる。
【0051】
本発明はまた、式:
【0052】
【化22】
の化合物を提供し、
ここで、
環Aがベンゾであり;
Z1、およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;そして
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
Lがリンカー基である。
【0053】
好ましい実施形態では、本発明の化合物2〜3は、R1−環A−R2が以下:
【0054】
【化23】
からなる群より選択される化合物である。
【0055】
好ましい実施形態では、本発明の化合物2〜3は、R4−N−R5が以下:
【0056】
【化24】
からなる群より選択される化合物である。
【0057】
別の実施形態では本発明の化合物2〜3は、R6が以下:
【0058】
【化25】
からなる群より選択される化合物である。
【0059】
a.リンカー
別の実施形態では、本発明の化合物3は、リンカー基が加水分解可能である化合物である。プロドラッグのリンカー基は、酵素切断によって、または好ましくは、生存している動物での水性条件を含むがこれに限定されない生理学的条件下での加水分解によって切断されて、化合物2が生じ得る。生理学的条件下でのリンカー基の加水分解の速度は好ましくは、約1分の半減期から約48時間の半減期である。
【0060】
b.加水分解
本明細書において用いる場合、「加水分解可能、加水分解性(hydrolyzable)」という用語は、約1分の半減期から約48時間の半減期という速度で、加水分解(すなわち、水分子の正味の挿入に起因する、その分子または基の2つ以上の新規な分子または基への分離)され得るかまたは加水分解し易いことをいう。
【0061】
リンカー基とは、化合物2を生じるように加水分解または酵素的に切断されてもよい任意の基であり得る。好ましい実施形態では、リンカー基は、以下の式:
【0062】
【化26】
であり、ここでZ3およびZ4は独立してSまたはOであり;そして
R7が−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−である。
【0063】
化合物2を得るためのプロドラッグのリンカー基の加水分解の代表的な例は、(スキーム1)に示され、ここでZ3およびZ4は独立して各々Oである。R6エステルの加水分解または酵素切断は、R7アルデヒドの遊離で崩壊するヘミアミナル(hemiaminal)を生じ、これによって遊離の三級アミンを含む化合物2が生じる。R6およびR7の両方とも、遊離の三級アミンに戻る種々の率が得られるように選択され得る。例えば、R6もしくはR7での置換の増大、またはその組み合わせが加水分解に向かう安定性を増大し得る。さらに、R6部分の電子求引性基は安定性を低下させる。本明細書に開示されるR6またはR7の変化に加えて、四級アミンの安定性を変化し得るさらなる要因は、その内容が参考として本明細書に援用される、N.Bodor,Journal of Medical Chemistry 1980,vol 23 #5、pp469〜480「Soft Drugs.1.Labile Quaternary Ammonium Salts as Soft Antimicrobials」;およびG.Brouillette et al.;Journal of Pharmaceutical Sciences,1996,vol 85 #6,pp620−623にみることができる。
【0064】
スキーム1
【0065】
【化27】
c.標的化因子
別の実施形態では、本発明の化合物は、R6がさらにそれに共有結合した1つ以上の標的化因子(T)を含む化合物である。標的化因子によって本発明のプロドラッグは特定のタイプの細胞、組織、器官または細胞外構造に選択的に送達されることが可能になる。上記で考察されるとおり、化合物1(LY294002)での処理は、バイオアベイラビリティー、急速な代謝および副作用という点で劣る。なぜなら、この化合物は組織特異的ではないからである。従って、副作用が生じ得る場合、薬物の位置を処置の領域の位置に対して限定すること、または薬物が組織に達することを少なくとも妨げること、そして任意の特定の時間で有効であるが過剰ではない量の薬物が用いられることを確実にすることが極めて所望される。標的化剤の使用によって、本発明のプロドラッグは、身体全体を通じて均一に分布するのではなく、処置の部位で濃縮されること、または早期に代謝されるかもしくはさらに急速に排出されることが可能になる。処置の部位に一旦送達されれば、リンカーを、酵素的に切断してもよいし、または上記のように加水分解して、化合物2を得てもよい。さらに、標的化因子の使用によって、処置の部位で薬物の有効濃度を達成するために投与することが必要な投薬量が制限され得る。標的化因子の使用はまた、処置の部位で薬物の有効濃度を得るために、さらに頻繁な投薬またはさらに別の投与方法を可能にする。
【0066】
標的化因子は、共有結合を介して、本発明の化合物に優先的に結合するが、この共有結合は、リンカー上の(それぞれ)求電子性または求核性の基と共有結合的に反応される、この標的化因子の求核性または求電子性基を含むがこれに限定されない方法によって形成され得る。
【0067】
本発明の1実施形態では、本発明の化合物2〜3は、R6〜Tが以下:
【0068】
【化28】
からなる群より選択される化合物である。
【0069】
本発明のプロドラッグと反応し得る標的化因子としては、限定はしないが、炭水化物、ビタミン、ペプチド、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤および軟骨親和性剤が挙げられる。標的化因子はまた、所望の組織におけるレセプターによって結合されて、レセプター媒介プロセスによって細胞に必要に応じて輸送される分子であってもよい。このような標的化因子の代表的な例としては、脳のグリア細胞に存在する末梢ベンゾジアゼピンレセプター(PBR)に結合するジアゼピンが挙げられるがこれらに限定されない。このようなジアゼピンの代表的な例は、その内容が参考として本明細書に援用される、G.Trapani et al.Bioconjugate Chem.2003.vol 14,pp830−839「Peripheral Benzodiazepine Receptor Ligand−Melphalan Cojugates for Potential Selective Drug Delivery to Brain Tumors」に考察される。
【0070】
標的化因子として用いられ得る代表的なビタミンとしては、限定はしないが、フォレート、ビタミンB12、またはビタミンCが挙げられる。「フォレート(folate)」という用語は、葉酸受容体と結合する能力を有する葉酸誘導体を包含する。標的化因子として用いられ得るフォレートの代表的な例としては、限定はしないが、葉酸、フォリン酸、プテロポリグルタミン酸、および葉酸受容体結合プテリジン、例えば、テトラヒドロプテリン、ジヒドロフォレート、テトラヒドロフォレートおよびそれらのデアザアナログおよびジアザアナログが挙げられる。他の適切なフォレートは、フォレートアナログであり、これには限定はしないが、アミノプテリン、アメトプテリン(メトトレキセート)、N10−メチルフォレート、2−デアミノ−ヒドロキシフォレート、デアザアナログ、例えば、1−デアザメトプテリン、または3−デアザメトプテリンおよび3’5’−ジクロロ4−アミノ−4−デオキシ−N10−メチルプテロイル−グルタミン酸(ジクロロメトトレキセート)が挙げられる。本発明の化合物に対して共有結合するために適切なフォレートに分子を結合させる方法は、その内容が参考として本明細書に援用される、米国特許第6,576,239号、同第5,820,847号、同第5,688,488号、同第5,108,921号、同第5,635,382号および同第5,416,016号に開示される。本発明の化合物に対して共有結合するために適切なビタミンCに分子を結合させる方法は、その内容が参考として本明細書に援用されるS.Manfrdini J.Med.Chem.Vol 45、pp559〜562,2002に開示される。
【0071】
標的化因子として用いられ得る代表的なペプチドおよびペプチド模倣物としては、限定はしないが、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン−レセプターアゴニストおよびソマトスタチンレセプターアンタゴニストからなる群より選択されるRGD含有ペプチドが挙げられる。avb3インテグリンレセプターに結合して、アンタゴニストとして作用する分子は、その内容が本明細書において参考として援用される米国特許第6,552,079号、同第6,426,353B、WO2002/40505A2および米国特許公開2002/0055499、同第2002/0061885、同第2002/0065291、同第2002/0072500、U.S.2002/0072518;W.Arap et al.、Science、vol 279、number 16、1998、pp377〜380;RJ Kok et al.、Biojonjugate Chem.2002,vol 13、pp128〜135;DA Sipkins et al.、Nature Medicine vol
4、number 5、1998、pp623〜626;PM Winter et al.、Cancer Research 2003、vol 63、pp5838〜5843;ならびにJD Hood et al.、Science vol 296、pp2404〜2407に記載されるように標的化因子として用いられ得る。標的化因子として用いられ得る代表的なタンパク質としては、限定はしないが、抗体またはそのフラグメント、例えば、腫瘍特異的モノクローナル抗体またはそのフラグメントが挙げられる。標的化因子として用いられ得る代表的な骨親和性因子としては、限定はしないが、ホスホン酸塩、ホスホン酸、アミノメチルホスホン酸、リン酸塩、ポリリン酸塩およびハイドロキシアパタイト結合ポリペプチドが挙げられる。他のペプチドとしては、クロロトキシン(SU6,429,187B1)および組織因子(G.M.Lanza et al.、「Targeted Antiproliferative Drug Delivery to Vascular Smooth Muscle Cells with a
Magnetic Resonance Imaging Nanoparticle
Contrast Agent」;Circulation,2002、volume
106、pp2842〜2847)が挙げられる。
【0072】
他の適切な標的化因子としては、抗体が挙げられる。抗体は、IgG、IgM、IgA、IgDもしくはIgEのクラスの抗体、またはそのフラグメントもしくは誘導体であってもよく、これには、Fab、F(ab’)2、Fdおよび単鎖抗体、二重特異性抗体(diabody)、二重特異性抗体(bispecific antibody)、二機能性抗体、およびその誘導体が挙げられる。抗体は、所望のエピトープまたはそれに由来する配列に対して十分な結合特異性を示す、モノクローナル抗体、ポリクローナル抗体、アフィニティー精製抗体またはそれらの混合物であってもよい。この抗体はまた、キメラ抗体であってもよい。この抗体は、腫瘍、組織適合性および他の細胞表面抗原、細菌、真菌、ウイルス、酵素、毒素、薬物および他の生理学的に活性な分子と関連するものを含む、種々の抗原性決定基に関するものであり得る。抗体が特異的に反応し得る腫瘍と会合する抗原としては、限定はしないが、このような抗原が挙げられ、そして限定はしないが、癌胎児性抗原(CEA)、ムチン、例えばTAG−72、ヒト乳脂肪小球抗原、前立腺血清抗原(PSA)、前立腺特異的膜抗原(PSMA)、PS(ホスファチジルセリン)、ならびにIL−2、EGF、VEGFおよびトランスフェリンレセプターを含むがこれに限定されないレセプターが挙げられる。腫瘍に会合する他の代表的な抗原としては、限定はしないが、それらの内容が参考として本明細書に援用される、ZalcbergおよびMcKenzie,J.Clin.Oncology,Vol 3;pp876〜82(1985)、WO01/68709A1、および米国特許公開US2004/0009122A1に記載される腫瘍関連抗原が挙げられる。
【0073】
他の適切な標的化因子としては、グルコース、ガラクトース、マンノース、マンノース6−リン酸、ホルモン(例えば、インスリン、成長ホルモンなど)、増殖因子またはサイトカイン(例えば、TGFβ、EGF、インスリン様成長因子、など)、YEE(GalNAcAH).sub.3または誘導体、コバラミン、α−2マクログロブリン、アシアロ糖タンパク質、アルブミン、テキサフィリン、メタロテキサフィリン、抗体、抗体フラグメント(例えば、Fab)、単鎖抗体可変領域(scFv)、トランスフェリン、任意のビタミンおよび任意の補酵素が挙げられる。
【0074】
標的化因子はまた、骨にプロドラッグを送達する因子であってもよい。骨標的化因子としては、限定はしないが、EDTMP DOTMPおよびABEDTMPが挙げられ、これは、その内容が本明細書に参考として援用される、米国特許第4,937,333号、同第4,882,142号、同第5,064,633号およびWO−94/00143に開示される。DOTMPおよびEDTMPは、図3に示されるカップリング化学、および図4に示されるアルキル化化学を含むがこれに限定されない任意の方法によってリンカー部分に結合され得、ここではR基が、リンカー部分の(それぞれ)求核性基または求電子性基と反応する、適切な求電子性基または求核性基を有してもよい。カップリング化学のさらなる詳細は、その内容が参考として本明細書に援用される、Tetrahedron
1999,55,pp12997〜13010に提供される。アルキル化化学のさらなる詳細は、その内容が参考として本明細書に援用される、Proc.SPIE−Int.Soc.Opt.Eng.1999,3600(Biomedical Imagn.Receptors Dyes&Instrumental,pp99〜106;米国特許第5,177,054号;J.Med.Chem.1994,37,498〜511;Tetrahedron Letters,1989,30#51pp7141〜7144;および米国特許第5,955,453号に提供される。
【0075】
標的化因子は、本発明の化合物の徐放性リザーバ部位として骨に対してプロドラッグを送達するように用いられ得る。標的化因子は、四級アミンに結合する酸切断性リンカーを介して本発明の化合物に結合する骨親和性(骨向性(osteotropic))部分であってもよい。酸切断可能リンカーの例としては、限定はしないが、オルト酸−アミド結合が挙げられる。酸性の条件下では、タンパク質−ACL−3アミド結合は容易に切断されて、その内容が参考として援用されるWO−94/00143に記載されるように、アミド官能性の天然のアミノ基を遊離する。酸性媒介性機構に関与する骨破壊性の骨再吸収の間、骨に対してプロドラッグを拘束する結合が切断されて、本発明の化合物が遊離され得る。
【0076】
骨に対してプロドラッグを送達するために用いられる標的化因子は、ノッチレセプターと結合する分子であってもよい。ノッチシグナル伝達は、種々の造血系列の発達および分化に重要な役割を果たす。Jundt et al.、Blood、102(11):928a(2003)に考察されるように、リガンド誘導性ノッチシグナル伝達は、多発性骨髄腫細胞の新規な増殖因子であり、そしてインビボにおいて多発性骨髄腫のリンパ種発生にこれらの相互作用が寄与することが示唆される。
【0077】
骨標的化因子は、骨の主な構成要素である、ヒドロキシアパタイト中のカルシウムイオンに高い親和性を有し得る。本発明の化合物は、骨以外の体の領域におけるカルシウム沈着、例えば、動脈、心臓、腎臓または膀胱のカルシウム沈着に標的化され得る。しかし、骨標的化因子は理想的には骨組織に選択的に結合する。本発明の骨標的化因子は、被験体の骨組織に結合され、好ましくは骨以外の組織よりも高い親和性で骨に結合し、そして特定の時間にわたって結合を残し、これによって骨環境に対してこの組成物を送達する。言い換えれば、骨標的化因子は好ましくは、骨以外の組織に対してこの骨標的化因子が結合するよりも少なくとも2倍大きい(例えば、少なくとも3倍、少なくとも5倍、少なくとも10倍、または少なくとも25倍大きい親和性)親和性で骨組織に結合する。骨標的化因子は骨組織に可逆的に結合するが、このことはこの骨標的化因子が最終的には骨から遊離されて、身体から排出されることを意味する。
【0078】
骨標的化因子は、四級プロドラッグを加水分解することが可能になるのに十分な時間、骨組織に対する結合を維持し得、それによって標的細胞(例えば、骨髄細胞)に対して活性薬物を送達する。骨標的化因子は、約1日(例えば、約2日、約3日、または約7日)〜約1年(例えば、約330日、約365日、または約400日)間にわたって骨に対する結合を維持し得、その後、骨標的化因子が身体から排出される。骨標的化因子は、約7日(例えば、約7日、約14日または約21日)〜約6ヶ月(例えば、約90日、約120日、または約150日)間にわたって骨に対する結合を維持してもよい。例えば、骨標的化プロドラッグは、30日間骨に対して結合を維持し得、その間この薬物が放出される。約45日後、骨標的化因子は骨から遊離されて、最終的に排泄される。従って、本発明における使用のための骨標的化因子は、骨組織に対する結合動態に基づいて選択され得る。候補の骨標的化因子は、例えばマルチウェル形式で、骨組織(例えば、ハイドロキシアパタイト)に対する親和性を決定することによって、インビトロでスクリーニングできる。候補の骨標的化因子はまた、身体からの候補の骨標的化因子の排泄の速度およびタイミングを評価することによってインビボでスクリーニングされ得る。これに関して、骨標的化因子は好ましくは、腎臓を介して身体から排出される。
【0079】
骨標的化因子は望ましくは、リン酸塩、ホスホン酸塩、ビスホスホン酸塩、ヒドロキシビスホスホン酸塩、アミノメチレンホスホン酸、および酸性ペプチドからなる群より選択される。本発明の骨標的化因子は、これらの群のうち1つ、2つ以上またはその混合物を担持してもよい。例えば、骨標的化因子は、ホスホン酸塩であってもよく、このことは、この骨標的化因子は、ホスホン酸塩であってもよく、このことはこの骨標的化因子が1つのホスホン酸塩、2つのホスホン酸塩、または3つ以上のホスホン酸塩を含んでもよいことを意味する。本発明における使用のための適切な骨標的化因子の1つは、疼痛緩和のために骨転移に選択的な放射線用量を送達するための放射性の153Sm複合体として、EDTMP(その化学構造は、図1に示され、現在FDAが承認している、エチレンジアミン−N,N,N’,N’−テトラキス(メチレンホスホン酸)(Quadramet(商標)))である。EDTMPは、4つのホスホン酸基を含むリン酸塩であり、従って、四リン酸塩である。153Sm−EDTMPのような化合物は、腫瘍が存在する骨、対正常な骨に選択的に、10:1より大きい比で局在する。なぜなら好ましくはカルシウムの代謝回転は転移性領域において極めて高いからである。報告によれば、153Sm−EDTMPは、造骨細胞の骨転移において骨格によって急速に取り込まれて血漿から排出される。骨格において蓄積しない化合物の一部は、急速に排出され、排泄は投与後6時間内でほぼ完全である(Jimonet et al.、Heterocycles,36,2745(1993))。疼痛緩和は、隣接する転移性の腫瘍細胞にある程度の効果を有する、造骨細胞の骨転移に対して結合した同位体に由来する放射線に起因していると考えられる。別の臨床上有用な骨標的化システムは、多発性骨髄腫の処置のために骨髄に対して高用量の放射線量を選択的に送達するように設計された放射性の166Ho複合体として、DOTMP(その化学構造は、図1に示され、現在第III相臨床試験中の(STR、骨格標的化放射線(skeletal targeted radiation)と名付けられた)である。放射性の166Ho−DOTMP複合体は、骨格系に局在して、悪性の骨髄腫細胞を有する近接する骨髄を照射することに注目すべきである。153Sm−EDTMP系と同様に、骨に局在しないホスホン酸塩は、尿によって身体の外に排出される。一般には、骨格の取り込みは、注射された用量の約20〜約50%であり、そして腫瘍浸潤を有する骨の領域への局在は、Bayouth et al.,J.Nucl.Med.36,730(1995)の図7に図示される。
【0080】
好ましくは、骨標的化因子は、ポリホスホン酸である。ポリホスホン酸は、骨組織に対して生物学的に活性な分子を首尾よく標的することが実証されている。例えば、成長因子(骨形成を刺激する)に対する、ABDTMP(その化学構造は、図1に示される)のようなポリアミノホスホン酸の結合(イソチオシアネート化学による)によって、ラットの骨に対する増殖因子の標的化が首尾よく生じる(例えば、国際特許出願WO94/00145を参照のこと)。同様に、骨標的化因子は、タンパク質に結合している。例えば、ヒト血清アルブミンに対して結合するビスホスホン酸塩は、インビトロ(Biotechnol.Prog.16,258(2000))およびインビボ(Biotechnol.Prog.,16,1116(2000))において骨にタンパク質を首尾よく送達した。骨親和性因子の有用性は、骨へのタンパク質の送達を上回り、そして例えば、低分子の治療用分子を含む。骨親和性ビスホスホン酸塩およびアルキル化剤、例えば、BAD(その化学構造は図1に示す、を含む結合体が生成されている(例えば、Wingen et
al.,J.Cancer Res.Clin.Oncol.,111,209(1986)を参照のこと)。この分子では、アルキル化剤は、その標的(DNA)との相互作用において特異的ではなく、従って、ビスホスホン酸塩(すなわち、骨親和性剤)とアルキル化部分との間で切断の必要はない。ビスホスホン酸−アルキル化剤は、BADを用いるラット骨肉腫モデルにおいて有効性を実証した。MTX−BPと命名され、図1に示される、ビスホスホン酸塩に共有結合されている葉酸代謝拮抗、抗腫瘍性剤であるメトトレキセートを用いて、別の一連の研究を行なった(例えば、Sturtz et al.,Eur.J.Med.Chem.,27.825(1992);Sturtz et al.,Eur.J.Med.Chem.,28,899(1993);およびHosain et al.,J.Nucl.Med.37,105(1996)を参照のこと)。Tc−99m標識したMTX−BPを用いて、約15%の注射用量が約4時間後に骨格に局在し、このときこの用量の約61%が排出されていることが確認された(Hosain、前出)。MTX−BPはさらに、移植された骨肉腫の動物モデルにおいてメトトレキセート単独と比較して5倍大きい抗癌効果を実証した(Sturtz 1992、前出)。化学構造が図1に記載されている、付加されたビスホスホン酸塩とのカルボキシフルオレセイン基の結合体CF−BPを用いて、同様の研究が記載されている(Fujisaki
et al.,Journal of Drug Targeting,4,117(1994))。この分子では、CF基は、薬物動態および生体分布を定量するための蛍光性マーカーであり、インビボで加水分解しやすいエステル結合を通じて骨標的化因子に結合される。ラットでの静脈内注射の研究によって、CF−BPが骨に局在し、そしてエステル結合の一般的加水分解を介して、生成されたCFの徐放性機構として機能することが示された(Fujisaki、前出)。
【0081】
別の実施形態では、骨親和性因子は、(Asp)6および(Glu)6のようなペプチドであってもよい。骨および象牙質で富んでいることが見出されている糖タンパク質オステオネクチンの酸に富むペプチド配列は、ヒドロキシアパタイトに強い親和性を有する(Fujisawa et al.,Biochimica et Biophysica
Acta,53,1292(1996))。従って、酸性アミノ酸を含むペプチドリガンドは、骨標的化因子の理想的な候補物である。実際、(Glu)10は、ビオチンに結合された場合、標識されたストレプトアビジンを首尾よく補強した(Chu and Orgel、Bioconjugate Chem,8,103(1997)および国際特許出願WO98/35703にさらに記載される)。さらに、(Asp)6に結合したフルオレセインイソチオシアネートの生物学的半減期は、大腿骨では14日であり(Kasugai et al.,Journal of Bone and Mineral Research,15(5),936(2000))、これは、本発明の骨標的化因子にとって受容可能な半減期である。同様に、エストラジオール−(Asp)6結合体の骨への送達は、卵巣切除された動物で実証されており、ここでは骨粗鬆的なタイプの骨の損失の阻害を伴う(Kasugai et al.,Journal of Bone and Mineral Research(Suppl 1),14,S534(1999))。骨に対する(Asp)6結合は、破骨細胞によって媒介される骨吸収プロセスの間に代謝されると考えられる。従って、酸性のペプチドリガンドによって、骨に対する化合物の補充の手段が得られるだけでなく、骨細胞および周囲の組織に対して化合物を緩徐に放出する機構も得られる。
【0082】
骨標的化因子の他の例としては、限定はしないが、アミノおよびヒドロキシル−アルキルホスホン酸およびジホスホン酸;アレンドロン酸、パミドロン酸、4−アミノブチルホスホン酸、1−ヒドロキシエタン−1,1−ジホスホン酸およびアミノメチレンビスホスホン酸を含む、ヒドロキシルビスホスホン酸;フィチン酸のようなホスホン酸;ならびにアミノメチレンホスホン酸、例えば、N,N−ビス(メチルホスホノ)−4−アミノ安息香酸およびニトリロトリ(メチルホスホン酸);が挙げられる。このような骨標的化因子の非限定的な例は、図2に示される。
【0083】
好ましくは、骨標的化因子は、アミノメチレンホスホン酸である。「アミノメチレンホスホン酸(aminomethylenephosphonic acid)」とは、−NCH2PO3H部分を含む化合物であって、アミノ基に1、2または3つのメチレンホスホン酸基が結合されており、そして他の化学部分でさらに置換されてもよい化合物を意味する。アミノメチレンホスホン酸は、1つ以上のホスホン酸基および1つ以上のアミノ基を含んでもよい。これらのアミノメチレンホスホン酸の例としては、限定はしないが、図2に示される化合物F〜Nが挙げられる。
【0084】
これらの骨標的化因子および他の骨標的化因子は、ヘテロ原子の1つを通じて、またはさらなる結合ポイントを導入する化学的修飾によって結合されてもよいと考えられる。例えば、EDTMPを、図3に例示されるように、リン酸素のうちの1つによってリンカーに結合してホスホン酸結合を作製してもよい(例えば、Viera de Almediaら.,Tetrahedron,55,12997〜13010(1999)を参照のこと)。リン酸素はまた、図4に示されるようにアルキル化され得、ここでは例えば、活性化されたPEGに対する結合のための二次結合ポイントを得るために、R基が、例えば、側鎖のアミノ基を有してもよい。本発明において利用できる他のタイプのアルキル化としては、限定はしないが、Chavez et al.,Biomedical Imaging:Reporters,Dyes,& Instrumentation,Contag & Sevick−Muracia編、Proc.SPIE,第3600,99〜106(1999年,July)にさらに記載されているとおり、また、例えば、米国特許第5,177,064号、米国特許第5,955,453号、de Lombaert et al.,J Med.Chem.,37,498〜511(1994)およびIyer et al.,Tetrahedron Letters,30(51),7141〜7144(1989)にさらに記載される他のホスホン酸について示されるように、DOTMPに関与するのと同様の例が挙げられる。あるいは、化学的修飾のために、EDTMPを、例えば、修飾して、アニリン基の導入によってABDTMPを生成してもよい(例えば、国際特許出願WO94/00145の図1にさらに記載されるように)。次いでアニリンアミンを利用して、例えばアミド結合を形成する。DOMTPは図5に概要をまとめたように、同様に修飾されてもよい。
【0085】
「ホスホン酸塩、リン酸塩およびアミノメチレンホスホン酸塩(phosphonate、phosphate、and aminomethylenephosphonate)」という用語は、それぞれ、ホスホン酸、リン酸およびアミノメチレンホスホン酸、ならびにその任意の塩、加水分解性エステル、およびリン酸ベースの酸のプロドラッグを包含することを意味する。血中での7.4という生物学的pHで、または骨の周囲でのそれより酸性のpHでは、骨標的化因子のリン酸塩またはホスホン酸塩の特定の一部が脱プロトン化されて、対イオンで置換され得る。さらに、カルシウムのプロトンの交換は、本発明におけるヒドロキシアパタイトに対する骨標的化因子の結合の内在する事象である。しかし、骨標的化因子を含む組成物の調製および投与は、その中でリン酸の完全なプロトン化は要しても要さなくてもよい。従って、ホスホン酸、リン酸およびアミノメチレンホスホン酸は、ホスホン酸塩、リン酸塩およびアミノメチレンホスホン酸塩と交換可能に記載され利用される。特に好ましくはないが、リン酸ベースの酸の生物学的に加水分解可能なエステルは、骨標的化プロドラッグのインビボ使用においても利用できる。同様に、組成物の酸性度をマスクするために、例えば処方および投与の間、リン酸ベースの酸のプロドラッグをインビボで利用してもよい。
【0086】
標的化因子はまた、特定の組織の特性に基づいて標的する因子であってもよい。このような標的化因子の代表的な例としては、限定はしないが、その内容が参考として本明細書に援用される、H.Maeda et al.「Tumor vascular permeability and the EPR effect in macromolecular therapeutics:A Review」;Journal of
Controlled Release,2000 vol 63、pp271−284に記載されているように、EPR効果(浸透性および保持の増強)に起因して腫瘍組織に選択的に局在するポリマーが挙げられる。他の代表的なポリマーは、N−(2−ヒドロキシプロピル)メタクリルアミド(HPMA)および(ポリ)L−グルタミン酸である。
【0087】
標的化因子はまた、RGD部分を含んでもよい。Curnis et al.,Cancer Research,64(2):565〜571(2004)に考察されるように、RGD部分は、αvβ3インテグリンを含む細胞接着レセプターとの相互作用と相互作用することによって脈管構造に対してRGD融合タンパク質を標的する。
【0088】
3.合成
a.主な環系
本発明の化合物は、出発生成物としてLY294002(化合物1)を用いて合成され得る。LY294002(化合物1)は、商業的に入手されても、または実施例1に記載のように、もしくはその内容が参考として本明細書に援用される米国特許第5,703,075号に記載のように合成してもよい。当業者はまた、出発生成物として化合物2を用いて本発明の化合物を合成し得る。
【0089】
b.主な環系の誘導体の調製
化合物2および3の主な環系は、LY294002(化合物1)の主な環系の誘導体であってもよい。化合物3の主要環系の誘導体は、LY294002(化合物1)の主な環誘導体の調製のために、その内容が本明細書において参考として援用される、米国特許第5,703,075号に開示されるように、調製され得る。化合物3の主要環系の誘導体はまた、置換2−ヒドロキシ−アセトフェノンを含むがこれに限定されない市販の化合物を用いることによって調製され得る。
【0090】
c.モルホリン環の誘導体の調製
化合物3のアミン誘導体は、室温から強制条件(過剰な求核試薬および110℃への加熱)への範囲におよぶ条件下での実施例1のチオアルキル基の置換によって調製できる。任意の一級または二級の窒素含有求核試薬は、モルホリン環構造(異なるモルホリンアナログを含む)に別のアミン置換基を与えるように反応し得る。化合物3のこのようなアミン誘導体の合成の代表的な例は、本明細書の実施例に記載される。
【0091】
d.エステルの調製
上記のとおり、エステルは、本発明の四級化された化合物を形成するために使用され得る。本発明の四級化化合物は好ましくは、ハロエステルを用いて形成される。1つの好ましい実施形態では、本発明の四級化化合物は、クロロメチルエステルを用いて形成される。本発明の化合物の調製において有用な多数のクロロメチルエステルは市販の供給源から入手可能である。さらに、クロロメチルエステルは、その内容が本明細書において援用される、WO02/42265、WO94/23724および米国特許第4,444,686号、同第4,264,765号および同第4,342,768号に記載されるように合成され得る。
【0092】
e.四級化
本発明のプロドラッグは、例えば、実施例4および実施例6に記載のように、ハロメチルエステルでの化合物1または化合物2の三級アミンの四級化によって調製され得る。四級化されたアミン化合物は一般には、温和な条件では可逆性ではない。しかし、本発明の四級化合物は上記で考察されたように容易に加水分解される。化合物1または化合物2の三級アミンを四級化するために用いられ得るハロメチルエステルは市販されており、または以下の実施例に記載されるように調製されてもよい。
【0093】
f.リンカー
本発明のプロドラッグはまた、少なくとも2つの官能基を含むリンカーで化合物1または化合物2の三級アミンを四級化することによって調製されてもよい。このリンカーは三級アミンを四級化可能であり、または標的分子に共有結合可能であるか、または標的分子に容易に結合され得る、任意の天然または合成のリンカーであってもよい。
【0094】
リンカーは好ましくは、酸素もしくはイオウのような原子、−NH−、−CH2−、−C(O)−、−C(O)NH−のような単位、または原子の鎖からなった。リンカーの分子量は代表的には、約14〜200の範囲、好ましくは、14〜96の長さから約6原子までの範囲である。リンカーの代表的な例としては、限定はしないが、必要に応じて置換されており、鎖のうち1または2つの飽和炭素が必要に応じて−C(O)−、−C(O)C(O)−、−CONH−、−CONHNH−、−C(O)O−、−OC(O)−、−NHCO2−、−O−、−NHCONH−、−OC(O)NH−、−NHNH−、−NHCO−、−S−、−SO−、−SO2−、−NH−、−SO2NH−、または−NHSO2−で置換されている、飽和または不飽和の脂肪族基が挙げられる。
【0095】
リンカーの第一の官能基を用いて、上記で考察される三級アミンを四級化する。好ましい第一の官能基は、限定はしないがクロロメチルエステルおよびヨードメチルエステルを含むハロメチルエステルである。リンカーの第二の官能基は、標的化因子に共有結合され得る。
【0096】
第二の官能基は、求電子基または求核性基であってもよい。標的基に共有結合するために好ましい第二の官能基はイソチオシアネート、ハロアセトアミドマレイミド、イミドエステル、チオフタルイミド、N−ヒドロキシスクシンイミルエステル、ピリジルジスルフィド、フェニルアジド、カルボキシル(およびその酸塩化物)、アミノ、アシルヒドロジド、セミカルバジド、チオセミカルバジド、ジアゾニウム、ヒドラジン、アジド、アミノアルキル尿素、アミノアルキルチオ尿素、ハロトリアジン、およびメタ(ジヒドロキシボリル)フェニルチオ尿素である。標的化因子に対して本発明のプロドラッグを共有結合するために適切であり得る他の適切な反応性部分としては、ジスルフィド、ニトレン、スルホンアミド、カルボジイミド、塩化スルホニル、ベンズイミデート、−COCH3および−SO3Hが挙げられる。
【0097】
適切な第二の官能基は、それとの共有結合が形成される標的化因子の官能基に依存し、そして結果として所定のタイプの結合を形成する生物学的活性の損失に対するその感受性による。標的化因子がタンパク質である場合、第二の官能基は、ポリペプチド骨格を構成するアミノ酸の側鎖基と反応し得る。このような側鎖基としては、アスパラギン酸およびグルタミン酸残基のカルボキシル基、リジン残基のアミノ基、チロシンおよびヒスチジンの芳香族基、およびシステイン残基のスルフヒドリル基が挙げられる。
【0098】
ポリペプチド骨格のような標的化因子によって提示されるカルボキシル側鎖基は、可溶性のカルボジイミド反応によってアミン第二官能基と反応され得る。標的化因子によって提示されるアミン側鎖基は、イソチオシアネート、イソシアネートまたはハロトリアジン第二官能基と反応されて、本発明のプロドラッグに対する結合を発揮し得る。あるいは、標的化因子上のアミノ側鎖基は、ジアルデヒドおよびイミドエステルのような二機能性の因子によって、アミン反応性基を有する本発明のプロドラッグ化合物に連結され得る。標的化因子によって提示される芳香族基は、ジアゾニウム誘導体を介して本発明のプロドラッグに結合され得る。標的化因子の分子上のスルフヒドリル基は、マレイミドと、またはハロアルキル標的化因子反応性基、例えばヨードアセトアミドと反応し得る。このような反応に適切な遊離のスルフヒドリル基は、タンパク質である免疫グロブリンのジスルフィド結合から生成されてもよく、または化学的誘導体によって導入されてもよい。免疫グロブリンの重鎖領域内で生成された遊離のスルフヒドリル基に対する結合は、免疫グロブリンの抗原結合部位を妨害しないが、抗体が補体を活性化できなくし得る。
【0099】
標的因子がグリコシル化タンパク質である場合、ポリペプチド骨格を介して本発明の化合物に対する結合を形成する代わりは、McKearn et al.,EPO 88,695のような方法に従って糖タンパク質の炭水化物側鎖との共有結合を形成することである。従って、抗体の炭水化物側鎖は、選択的に酸化されてアルデヒドを生成してもよく、これが次にアミン反応性基と反応してSchiff塩基を形成するか、またはヒドラジン、セミカルバジドもしくはチオカルバジド反応性基と反応して、対応するヒドラゾン、セミカルバゾンもしくはチオセミカルバゾン結合を得ることができる。またこれらの同じ方法を使用して、本発明のプロドラッグを、炭水化物および多糖類のような非タンパク質性の標的化因子に結合してもよい。
【0100】
事前の酸化の必要性なしに炭水化物および多糖類に結合するのに有用な別の標的化因子反応性部分は、ジヒドロキシボリル基であり、例えば、これはメタ(ジヒドロキシボリル)フェニルチオ尿素誘導体に存在する。この基は、1,2−シス−ジオールを含む標的化因子と反応して、5−員環の環状ホウ酸塩エステルを形成し、従ってこの基を含む炭水化物、多糖類および糖タンパク質と使用される。Rosenberg et al.,Biochemistry,11,3623〜28(1972)に開示されるように、リボースは1,2−シス−ジオール基を2’,3’位置で含むので、ジヒドロキシボリル誘導体はまた、本発明のプロドラッグをリボヌクレオシド、リボヌクレオチドおよびリボ核酸に連結するためにも用いられ得る。デオキシリボヌクレオチドおよびDNA標的化因子は、3’ヒドロキシル基が存在しない場合、この方式で本発明のプロドラッグには連結され得ない。しかし、後者の標的化因子は、Engelhardt et al.,EPO97,373に開示されるように、デオキシリボヌクレオチドのアリルアミン誘導体を最初に形成することによって、プロドラッグのイソチオシアネート誘導体に結合し得る。
【0101】
本発明のプロドラッグと結合される標的化因子がインタクトな細胞である場合、ポリペプチド反応性部分または炭水化物反応性部分のいずれが使用されてもよい。HwangおよびWase,Biochim.Biophys.Acta,512,54〜71(1978)は、インジウム−111を用いて赤血球および血小板を標識する、Sundberg et al.,J.Med.Chem.,17,1304(1974)の二機能性EDTAキレーターのジアゾニウム誘導体の使用を開示する。ジヒドロキシボリル基は、種々の細菌、ウイルスおよび微生物と反応する。Zittle,Advan.Enzym.,12 493(1951)およびBurnett et al.,Biochem.Biophys.Res.Comm.,96,157〜62(1980)を参照のこと。
【0102】
化合物1または化合物2の三級アミンを共有結合的に四級化するために用いられ得る好ましいリンカーは、式:
【0103】
【化29】
(化合物4)
のリンカーであり、
ここで、
Xはハロ基であり;
Yは−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり
Z1およびZ2は独立してSまたはOであり;そして
n=0〜4である。
【0104】
1実施形態では、本発明の化合物4は、それらの化合物であり、ここで、
XはClまたはIであり;
Yは−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;
Z1およびZ2は独立してOであり;そして
n=0である。
【0105】
別の実施形態では、本発明の化合物4は、それらの化合物であり、ここで、
XはClまたはIであり;
Yは−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;
Z1およびZ2は独立してOであり;そして
n=1である。
【0106】
化合物4は、最終の四級窒素の切断率に変動を与える、アルキルおよびアリールカルボン酸骨格の両方とのリンカーを提供する。化合物4のリンカーは、実施例5に記載されるような市販の出発製品を用いて調製され得る。
【0107】
g.精製
本発明の化合物は、標準的な精製方法を用いて単離され得る。本発明の化合物の加水分解性の結合は、この化合物の精製の間に加水分解しやすくてもよい。
【0108】
本発明はまた、本発明の化合物を精製する方法に関しており、この方法は、化合物を可溶化するために少なくとも0.1%の酸(v/v)を含む溶液にこの化合物を添加する工程を包含する。次いでこの化合物を、クロマトグラフィー、好ましくはHPLCを行うことによって精製する。
【0109】
h.試験
本発明のプロドラッグは、時間の関数として、切断条件に曝露されたプロドラッグのHPLC分析を行なうことによって、加水分解性結合の加水分解の速度および加水分解の生成物を決定するように試験され得る。本発明の化合物の生物学的活性は、実施例17に記載されるようにマクロファージ細胞株J774細胞において食作用をブロックする工程を含むがこれに限定されない方法によって測定され得る。本発明の化合物の生物学的活性はまた、その内容が参考として援用される、米国特許第5,480,906号;K.Fuchikami et al.,J.Biomol Screen,2002 Oct.pp441〜450;VI Silveria et al.,J.Biomol.Screen,2002,Dec.7(6),507〜514;BE Drees Combinatorial Chemistry and Highthroughput Screening 2003,vol 6、321〜330に記載されるように、PI−3キナーゼ酵素アッセイによって測定され得る。
【0110】
i.塩
本発明の化合物は、種々の薬学的に受容可能な塩型で有用である。「薬学的に受容可能な塩(pharmaceutically acceptable salt)」という用語は、薬剤師によって理解される塩型、すなわち、実質的に毒性でなく、所望の薬物動態学的特性、嗜好性、吸収、分布、代謝または排出を提供する塩型をいう。選択にも重要であり、天然にはさらに重要な他の要因は、原料の費用、結晶化の容易さ、収率、安定性、吸湿性および得られたバルク薬物の流動性である。好都合には、薬学的組成物は、活性成分またはその薬学的に受容可能な塩から、薬学的に受容可能なキャリアと組み合わせて調製されてもよい。
【0111】
本発明の方法および組成物における使用に適切である、本発明の化合物の薬学的に受容可能な塩としては、限定はしないが、種々の有機酸および無機酸、例えば、塩化水素、スルホン酸ヒドロキシメタン、臭化水素、メタンスルホン酸、硫酸、酢酸、トリフルオロ酢酸、マレイン酸、ベンゼンスルホン酸、トルエンスルホン酸、フルファミン酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、パモン酸、スルファニル酸、2−アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、イセチオン酸と形成される塩が挙げられ、そして種々の他の薬学的に受容可能な塩、例えば、硝酸塩、リン酸塩、ホウ酸塩、酒石酸塩、クエン酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、サリチル酸塩などが挙げられる。四級アンモニウムイオンのような陽イオンは、陰イオン性部分についての薬学的に受容可能な対イオンと考えられる。
【0112】
本発明の化合物の好ましい塩としては、塩酸塩、メタンスルホン酸塩およびトリフルオロ酢酸塩が挙げられるが、メタンスルホン酸塩がより好ましい。さらに、本発明の化合物の薬学的に受容可能な塩は、ナトリウム、カリウムおよびリチウムのようなアルカリ金属;アルカリ土類金属、例えば、カルシウムおよびマグネシウム;有機塩基、例えば、ジシクロヘキシルアミン、トリブチルアミンおよびピリジン;ならびにアルギニン、リジンなどのようなアミノ酸と形成されてもよい。
【0113】
本発明の薬学的に受容可能な塩は、従来の化学的方法によって合成され得る。一般には、塩は、適切な溶媒または溶媒の組み合わせ中での、遊離の塩基または酸と、化学量論的な量または過剰な、所望の塩を形成する無機もしくは有機の酸もしくは塩との反応によって調製される。
【0114】
一般には、本発明の化合物の塩の対イオンは、この合成された化合物に対して用いた反応物によって決定される。反応物に依存して、塩の対イオンの混合物が存在し得る。例えば、反応を促進するためにNaIが添加される場合、対イオンはClおよびIの対の陰イオンの混合物であってもよい。さらに分取HPLCでは、酢酸が溶出物に存在する場合、酢酸塩によって交換されるもとの対イオンを生じ得る。塩の対イオンは、異なる対イオンに交換されてもよい。対イオンは好ましくは、薬学的に受容可能な対イオンと交換されて、上記の塩を形成する。対イオンを交換するための手順は、その内容が参考として本明細書に援用される、WO 2002/042265、WO2002/042276およびS.D.Clas,「Quaternized Colestipol,an improved bile salt adsorbent:In Vitro studies」Journal of Pharmaceutical Sciences,80(2):128〜131(1991)に記載される。明快な理由によって、対イオンは、本明細書の化学構造に明確に示されず、そしてこの化合物の特徴づけは、同定された四級陽イオンに基づく。
【0115】
4.組成物
本明細書はまた、本発明の1つ以上の化合物を含む組成物も包含する。本発明の組成物はさらに、1つ以上の薬学的に受容可能なさらなる成分(単数または複数)、例えば、ミョウバン、安定化剤、抗菌剤、緩衝液、着色剤、香料、アジュバントなどを含んでもよい。
【0116】
a.処方物
本発明の組成物は、従来の方式で処方される錠剤またはトローチ剤の形態であってもよい。例えば、経口投与のための錠剤およびカプセルは、従来の賦形剤を含んでもよく、これには、限定はしないが結合剤、充填剤、潤滑剤、崩壊剤および湿潤剤が挙げられる。結合剤としては、限定はしないが、シロップ、アカシア、ゼラチン、ソルビトール、トラガカント、デンプンの粘液およびポリビニルピロリドンが挙げられる。充填剤としては、限定はしないが、ラクトース、糖、微結晶性セルロース、トウモロコシデンプン、リン酸カルシウムおよびソルビトールが挙げられる。潤滑剤としては、限定はしないが、ステアリン酸マグネシウム、ステアリン酸、滑石、ポリエチレングリコールおよびシリカが挙げられる。崩壊剤としては限定はしないが、ジャガイモデンプンおよびデンプングリコール酸ナトリウムが挙げられる。湿潤剤としては、限定はしないが、ラウリル硫酸ナトリウムが挙げられる。錠剤は、当該分野で周知の方法に従ってコーティングされ得る。
【0117】
本発明の組成物はまた、水性または油性の懸濁液、溶液、エマルジョン、シロップおよびエリキシルを含むがこれに限定されない液体処方物であってもよい。この組成物はまた、使用前の水または他の適切なビヒクルとの構成のために乾燥生成物として処方され得る。このような液体調製物は、懸濁剤、乳化剤、非水性のビヒクルおよび防腐剤を含むがこれらに限定されない添加物を含んでもよい。懸濁剤としては、限定はしないが、ソルビトールシロップ、メチルセルロース、グルコース/糖シロップ、ゼラチン、ヒドロキシエチルセルロース、カルボキシメチルセルロース、ステアリン酸アルミニウムゲルおよび硬化食用油脂が挙げられる。乳化剤としては、限定はしないが、レシチン、ソルビタンモノオレエート、およびアカシアが挙げられる。非水性のビヒクルとしては、限定はしないが、食用油脂、アーモンドオイル、分留ココナッツオイル、油状エステル、プロピレングリコールおよびエチルアルコールが挙げられる。防腐剤としては限定はしないが、メチルまたはプロピルp−ヒドロキシ安息香酸塩およびソルビン酸が挙げられる。
【0118】
本発明の組成物はまた、座剤として処方され得るが、これは座剤基剤を含んでもよく、この基剤としては、カカオバターまたはグリセリドが挙げられるがこれらに限定されない。本発明の組成物はまた、吸引のために処方され得、これは乾燥粉末として投与され得る溶液、懸濁液もしくはエマルジョンを含むがこれに限定されない形態、または噴霧剤、例えば、ジクロロジフルオロメタンもしくはトリクロロフルオロメタンを用いるエアロゾルの形態であってもよい。本発明の組成物はまた、クリーム、軟膏、ローション、ペースト、薬用絆創膏、パッチまたはメンブレンを含むがこれに限定されない、水性または非水性のビヒクルを含む、処方された経皮処方物であってもよい。
【0119】
本発明の組成物はまた、注射または連続注入による投与を含むがこれに限定されない、非経口投与のために処方されてもよい。注射の処方物は、油状または水性のビヒクル中の懸濁液、溶液またはエマルジョンの形態であってもよく、そして懸濁剤、安定化剤および分散剤が挙げられるがこれらに限定されない処方剤を含んでもよい。この組成物はまた、滅菌の、発熱物質のない水を含むがこれに限定されない安定なビヒクルでの再構成のために粉末形態で提供され得る。
【0120】
本発明の組成物はまた、移植によってまたは筋肉内注射によって投与され得るデポ調製物として処方されてもよい。この組成物は、適切なポリマーまたは疎水性物質(例えば、受容可能なオイル中のエマルジョンとして)、イオン交換樹脂とともに、または溶解度の劣る誘導体として(例えば、溶解度の劣る塩として)処方され得る。
【0121】
本発明の組成物はまた、リポソーム調製物として処方され得る。リポソーム調製物は、リポソームを含んでもよく、これが目的の細胞または角質層に浸透して、細胞膜と融合し、このリポソームの内容物の細胞への送達が得られる。例えば、Yaroshの米国特許第5,077,211号、Redziniakらの米国特許第4,621,023号、またはRedziniakらの米国特許第4,508,703号に記載されるようなリポソームを用いてもよい。本発明の組成物は、皮膚状態を標的することを意図しており、UVまたは酸化障害を生じる因子に対する哺乳動物の皮膚の曝露の前、間または後に投与され得る。他の適切な処方物はニオゾームを使用してもよい。ニオゾームはリポソームと同様の脂質小胞であり、その膜は主に非イオン性の脂質から構成され、その形態のいくつかは角質層を横切って化合物を輸送するために有効である。
【0122】
5.処置
本発明はまた、PI−3キナーゼ活性に関連する条件に罹患している患者を処置する方法を包含する。PI−3キナーゼ活性は、異常、過剰または構成的に活性であってもよい。本発明はまた、炎症性疾患を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。不適切なPI−3キナーゼシグナル伝達活性に寄与し得る、このような疾患および有害な健康上の影響は、当該分野において、例えば、その内容が参考として援用される、U.S.2002/0150954A1;US5,504,103;US6,518,277B1;U.S.6,403,588;U.S.6,482,623;U.S.6,518,277;U.S.6,667,300;U.S.20030216389;U.S.20030195211;U.S.20020037276およびU.S.5,703,075で開示されている。
【0123】
本発明はまた、p53媒介性のプログラムされた細胞死を増強するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0124】
本発明はまた、腫瘍細胞の化学感受性を増強するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0125】
本発明はまた、腫瘍細胞の放射線感受性を増強するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0126】
本発明はまた、腫瘍誘導性血管形成を阻害するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0127】
本発明はまた、癌以外の疾患に関連する血管形成プロセスを阻害するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0128】
本発明はまた、癌の処置のための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0129】
この化合物は、化学療法および放射線療法のような他の抗癌処置と同時にまたは周期的に投与されてもよい。本明細書において用いる場合、「同時(simultaneous)」または「同時に(simultaneously)」という用語は、他の抗癌処置および本発明の化合物が、お互いの48時間、好ましくは24時間、さらに好ましくは12時間、それよりさらに好ましくは6時間、そして最も好ましくは3時間以下内に投与されたことを意味する。本明細書において用いる場合、「周期的に(metronomically)」という用語は、化学療法から種々の時点での、そして反復投与および/または化学療法レジメンに対して特定の頻度でのこの化合物の投与を意味する。
【0130】
化学療法処置は、細胞毒性剤もしくは細胞増殖抑制剤、またはそれらの組み合わせの投与を包含し得る。細胞毒性因子は:(1)細胞がDNAを複製する能力を妨害すること、および(2)癌細胞における細胞死および/またはアポトーシスを誘導することによって癌細胞が増殖することを妨げる。細胞増殖抑制剤は、細胞増殖を時には低い連続レベルに調節する、細胞シグナル伝達のプロセスを調節、妨害または阻害することを介して作用する。
【0131】
細胞毒性因子として用いられ得る化合物のクラスは、以下を含む:アルキル化剤(限定はしないが、ナイトロジェンマスタード、エチレンイミン誘導体、スルフォン酸アルキル、ニトロソ尿素およびトリアゼンを含む);ウラシルマスタード、クロルメチン、シクロホスファミド(Cytoxan(登録商標))、イホスファミド、メルファラン、クロランブシル、ピポブロマン、トリエチレン−メラミン、トリエチレンチオホスホラミン、ブスルファン、カルムスチン、ロムスチン、ストレプトゾシン、デカルバジンおよびテモゾラミド;代謝拮抗物質(限定はしないが、葉酸拮抗薬、ピリミジンアナログ、プリンアナログおよびアデノシンデアミナーゼインヒビターを含む):メトトレキセート、5−フルオロウラシル、フロクスウリジン、シタラビン、6−メルカプトプリン、6−チオグアニン、リン酸フルダラビン、ペントスタチンおよびゲムシタビン;天然産物およびそれらの誘導体(例えば、ビンカアルカロイド、抗腫瘍抗生物質、酵素、リンホカインおよびエピポドフィロトキシン);ビンブラスチン、ビンクリスチン、ビンデシン、ブレオマイシン、ダクチノマイシン、ダウノルビシン、ドキソルビシン、エピルビシン、イダルビシン、ara−c、パクリタキセル(パクリタキセルはTaxol(登録商標)として市販されている)、ミトラマイシン、デオキシコ−ホルマイシン、マイトマイシン−c、l−アスパラギナーゼ、インターフェロン(好ましくはIFN−α)、エトポシド、およびテニポシド。
【0132】
他の増殖性の細胞毒性因子は、ナベルベン、CPT−11、アナストラゾール、レトラゾール、カペシタビン、レロキサフィン、シクロホスファミド、イホスファミドおよびドロロキサフィンである。
【0133】
微小管に影響する因子は、細胞の有糸分裂を妨害し、そしてその細胞毒性活性については当該分野で周知である。本発明において有用な微小管影響因子としては、限定はしないがアロコルヒチン(NSC406042)、ハリコンドリンB(NSC609395)、コルヒチン(NSC757)、コルヒチン誘導体(例えば、NSC33410)、ドラスタチン10(NSC376128)、マイタンシン(NSC153858)、リゾキシン(NSC332598)、パクリタキセル(Taxol(登録商標)、NSC125973)、タキソール(登録商標)誘導体(例えば、誘導体(例えば、NSC608832)、チオコルヒチンNSC361792)、トリチルシステイン(NSC83265)、硫酸ビンブラスチン(NSC49842)、硫酸ビンクリスチン(NSC67574)、エポチロンA、エポチロンBおよびを含むがこれに限定されない天然および合成のエポチロン、ならびにジスコデルモライド(Service,(1996)Science,274:2009)エストラムスチン、ノコダゾール、MAP4などが挙げられる。このような因子の例はまた、Bulinski(1997)J.Cell Sci.110:3055 3064;Panda(1997)Proc.Natl.Acad.Sci.USA 94:10560〜10564;Muhlradt(1997)Cancer Res.57:3344〜3346;Nicolaou(1997)Nature 387:268〜272;Vasquez(1997)Mol.Biol.Cell.8:973〜985;およびPanda(1996)J.Biol.Chem.271:29807〜29812にも記載される。
【0134】
また適切なのは、細胞毒性因子、例えば、エピドフィロトキシン;抗悪性腫瘍酵素;トポイソメラーゼインヒビター;プロカルバジン;ミトキサントロン;白金配位錯体、例えば、シス−プラチンおよびカルボプラチン;生物学的応答修飾因子;増殖インヒビター;抗ホルモン治療剤;ロイコボリン;テガフール;および造血成長因子である。
【0135】
用いられ得る細胞増殖抑制剤としては、限定はしないが、ホルモンおよびステロイド(合成アナログを含む):17α−エチニルエストラジオール、ジエチルスチルベストロール、テストステロン、プレドニゾン、フルオキシメステロン、ドロモスタノロンプロピオネート、テストラクトン、メゲストロラセテート、メチルプレドニゾロン、メチル−テストステロン、プレドニゾロン、トリアムシノロン、ハロトリアニセン(hlorotrianisene)、ヒドロキシプロゲステロン、アミノグルテチミド、エストラムスチン、メドロキシプロゲステロンアセテート、リュープロリド、フルタミド、トレミフェン、ゾラデックスが挙げられる。
【0136】
他の細胞増殖抑制剤は、血管新生阻害剤、例えば、マトリックスメタロプロテイナーゼインヒビターであり、そして他のVEGFインヒビター、例えば、抗VEGF抗体および低分子、例えばZD6474およびSU6668も含まれる。Genetechの抗Her2抗体も利用できる。適切なEGFRインヒビターはEKB−569(不可逆性インヒビター)である。また、EGFRに免疫特異性のImclone抗体C225およびsrcインヒビターも含まれる。
【0137】
細胞増殖抑制剤として用いるためにも適切なのはCasodex(登録商標)(bicalutamide,Astra Zeneca)であり、これはアンドロゲン依存性の癌腫を非増殖性にさせる。細胞増殖抑制剤のさらに別の例は、抗エストロゲンであるTamoxifen(登録商標)であり、これはエストロゲン依存性乳癌の増殖または成長を阻害する。細胞増殖シグナルの伝達のインヒビターは、細胞増殖抑制剤である。代表的な例としては、上皮細胞成長因子インヒビター、Her−2インヒビター、MEK−1キナーゼインヒビター、MAPKキナーゼインヒビター、PI3インヒビター、SrcキナーゼインヒビターおよびPDGFインヒビターが挙げられる。
【0138】
種々の癌を本発明に従って処置することが可能であり、この癌としては、限定はしないが以下:膀胱(加速性および転移性の膀胱癌を含む)、乳房、結腸(結腸直腸癌を含む)、腎臓、肝臓、肺(小細胞および非小細胞肺癌および肺の腺癌を含む)、卵巣、前立腺、精巣、尿生殖路、リンパ系、直腸、喉頭、膵臓(膵外分泌癌腫を含む)、食道、胃、膀胱、頸部、甲状腺、および皮膚(扁平上皮癌を含む)の癌;白血病、急性リンパ性白血病、急性リンパ芽球性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、ヘアリー細胞リンパ腫、組織球性リンパ腫およびバーキットリンパ腫を含むリンパ系の造血性腫瘍;急性および慢性の骨髄性白血病、骨髄異形成症候群、骨髄性白血病および前骨髄球性白血病を含む骨髄系列の造血性腫瘍;星細胞腫、神経芽細胞腫、神経膠腫およびシュワン細胞腫を含む中枢および末梢神経系の腫瘍;線維肉腫、横紋筋肉腫および骨肉種を含む間葉起源の腫瘍;ならびに黒色腫、色素性乾皮症、角化棘細胞腫、精上皮腫、甲状腺濾胞性癌および奇形癌を含む他の腫瘍;を含む癌腫が挙げられる。
【0139】
最も好ましくは、本発明は、膀胱の加速性または転移性の癌、膵臓癌、前立腺癌、非小細胞肺癌、結腸直腸癌および乳癌を処置するために用いられる。
【0140】
本発明はまた、膵臓を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。Gukovsky et
al.,Gastroenterology、126(2):554〜66(2004)に考察されるように、PI−3キナーゼの阻害は、膵炎を妨げ得る。
【0141】
本発明はまた、潰瘍を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。本発明はまた、胃癌(gastric cancer)、例えば、胃癌(stomach cancer)を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。Bacon et al.,Digestive Disease Week Abstracts and Itinerary Planner、Vol.2003,Abstract No.M921(2003)およびRokutan et al.,Digestive Disease Week Abstracts and Itinerary Planner,Vo.2003,Abstract No.354(2003)に考察されるように、PI−3キナーゼは、胃の細胞へのHelicobactor Pyloriの結合に関与する。さらに、Osaki
et al.Journal of Cancer Research and Clinical Oncology、130(1):8〜14(2004)は、PI−3キナーゼインヒビター、例えばLY294002が、胃癌の抗腫瘍剤として有用であり得ることを示す。
【0142】
本発明はまた、心血管ステントのようなステントを有する患者に対して本発明の化合物の治療上有効な量を投与する工程を包含する、ステントの能力を改善する方法を包含する。Zhouら、Arteriosclerosis Thrombosis and Vascular Biology,23(11):2015〜2020(2003)に考察されるように、PI−3キナーゼの阻害は、血管のステント位置に伴う「伸展(stretch)」障害を妨げ得る。ステントまたはそのポリマーマトリックス中の本発明の化合物は、ステントコーティングマトリックスにおける可溶性を改善し得、水/血清の溶解度を改善し得、またはステント位置に隣接する細胞への灌流を改善し得る。
【0143】
本発明はまた、加齢性黄斑変性症(AMD)を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。Retina,Feburary 18,2004に考察されるように、VEGFの阻害は、AMDに関連する血管の過剰増殖を阻害する。本発明の化合物は、血管形成を阻害することによってAMDを処置し得る。
【0144】
本発明はまた、高血圧を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。Notthcott and Watts,Hypertension,43(1):125〜130(2004)に考察されるように、PI−3キナーゼの阻害は、高血圧に関連するMg2+の低い細胞外濃度を妨げ得る。
【0145】
本発明はまた、前駆細胞、例えば、骨髄前駆細胞の分化を抑制するための方法を包含するが、この方法は、本発明の化合物の有効量を前駆細胞に投与する工程を包含する。LewisらExperimental Hematology、32(1):36〜44(2004)に考察されるように、PI−3キナーゼ経路の阻害は、骨髄前駆細胞を抑制する。
【0146】
本発明はまた、肝臓癌を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。Leng et al.,Hepatology 38(4)Suppl 1:401A(2003)に考察されるように、LY294002は、ヒト肝臓組織の指標であるAkt(セリン/トレオニンプロテインキナーゼB)のリン酸化を阻害する。
【0147】
本発明はまた、変異PTENに関連する状態を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。PTENは、コーデン病を有する患者で同定されている染色体10q23に位置する癌抑制遺伝子である。Vegaら、Journal of Investigative Dermatology,121(6):1356〜1359(2003)に考察されるように、変異PTENは、プロトオンコジーンAktの活性化を阻害する能力が低くなっている。PI−3キナーゼのインヒビターは、Aktのリン酸化を阻害し得、それによって変異PTENの影響を軽減する。
【0148】
a.投与
本発明の組成物は、限定はしないが経口、非経口、舌下、経皮、直腸、経粘膜、局所、吸引、口腔内投与、またはそれらの組み合わせを含む任意の方法で投与されてもよい。非経口投与としては、限定はしないが、静脈内、動脈内、腹腔内、皮下、筋肉内、クモ膜下腔内および関節内が挙げられる。本発明の組成物はまた、この組成物の緩徐な放出および緩徐な制御されたi.v.注入を可能にするインプラントの形態で投与されてもよい。
【0149】
b.投薬量
治療での使用に必要な化合物の治療上有効な量は、処置される条件の性質、活性が所望される時間の長さ、ならびに患者の年齢および条件によって変化し、そして担当医によって最終的に決定される。しかし、一般には、成人の処置に使用される用量は代表的には、1日あたり0.001mg/kg〜約200mg/kgの範囲である。この用量は、1日あたり約1μg/kg〜約100μg/kgであってもよい。所望の用量は、単回用量で、または適切な間隔で投与される複数の用量として、例えば、1日あたり2、3、4またはそれ以上の低用量として都合よく投与されてもよい。複数回の用量もしばしば所望されるか、または必要である。
【0150】
多数の要因によって、本発明の化合物は広範な用量で投与されることになり得る。他の治療剤と組み合わせて用いる場合、本発明の化合物の投薬量は、比較的低い投薬量で与えられてもよい。さらに、標的化因子の使用によって必要な投薬量を比較的低くすることができる。本発明の特定の化合物は、限定はしないが、低い毒性、高いクリアランス、三級アミンの切断率の低さを含む要因に起因して比較的高用量で投与されてもよい。結果として、本発明の化合物の投薬量は、約1ng/kg〜約100mg/kgの範囲であってもよい。本発明の化合物の投薬量は、限定はしないが約1μg/kg、25μg/kg、50μg/kg、75μg/kg、100μg/kg、125μg/kg、150μg/kg、175μg/kg、200μg/kg、225μg/kg、250μg/kg、275μg/kg、300μg/kg、325μg/kg、350μg/kg、375μg/kg、400μg/kg、425μg/kg、450μg/kg、475μg/kg、500μg/kg、525μg/kg、550μl/kg、575μg/kg、600μg/kg、625μg/kg、650μg/kg、675μg/kg、700μg/kg、725μg/kg、750μg/kg、775μg/kg、800μg/kg、825μg/kg、850μg/kg、875μg/kg、900μg/kg、925μg/kg、950μg/kg、975μg/kg、1mg/kg、5mg/kg、10mg/kg、15mg/kg、20mg/kg、25mg/kg、30mg/kg、35mg/kg、40mg/kg、45mg/kg、50mg/kg、60mg/kg、70mg/kg、80mg/kg、90mg/kgまたは100mg/kgを含む任意の投薬量であってもよい。
【0151】
本発明は、以下の非限定的な実施例によって例示される多数の局面を有する。
【実施例】
【0152】
実施例1
LY294002の調製
Ly294002の10gのサンプルは、その内容が参考として援用される、Vlahos et al.、J.Biol.Chem.269(7):5241(1994)に記載される手順に基づくスキーム2に従って調製した。アミンによる12のようなチオクロムのチオメチル基の置換は、前に記載されており(その内容が参考として援用される、Bantickら、J.Heterocyclic Chem,18:679(1981))、チオールアニオンのアルキル化を伴う二硫化炭素での11のようなメチルフェニルケトンの環化を有する(Vlahos et al.and Bantick et al.)。カルボン酸(10)からの1工程反応のメチルケトン(例えば、11)の調製は、その内容が参考として本明細書に援用される、Rubottom et al.,J.Org.Chem.,48:1550(1983)に記載された手順を用いて行なった。
【0153】
スキーム2
【0154】
【化30】
実施例2
LY294002の四級アナログの調製
スキーム3の手順後、LY294002の三級アミンを、強制条件下でヨードメタンまたは塩化ベンジルを用いて四級化して、化合物A052−10および化合物13Bを得た。実施例56は、メチル四級プロドラッグA052−10の合成を記載する。実施例57は、フタルイミド四級プロドラッグA052−08の合成を記載する。実施例58は、パラカルボキシルベンジル四級A044−78の合成を記載する。[0199][0215]は、パラ−scn−ベンジル四級プロドラッグA044−80の合成を記載する。
【0155】
スキーム3
【0156】
【化31】
実施例3
クロロメチルエステルの調製
クロロメチル中間体を、Tsujihara,Synth Commun,24,767,1994に記載の手順に従って調製した。要するに、適切なカルボン酸を、ジクロロメタン/水の50/50混合物中に希釈した。この混合物を氷水槽中で冷却して、炭酸水素ナトリウム(4当量)およびn−テトラブチル硫酸水素アンモニウム(0.05当量)を添加した。5分間の撹拌後、クロロメチルクロロサルフェート(1.1当量)を添加した。この溶液を一晩激しく撹拌した。この混合物をさらなるジクロロメタンとともに分液漏斗に移して、飽和塩化ナトリウム溶液で洗浄した。有機物を硫酸ナトリウム乾燥させて、溶媒を除去して生成物を得た。この物質をLC−MSによって、そしてある場合には1H NMR分光法によって特徴付けた。この一般的な手順によって、以下の代表的なクロロメチルエステルを対応するカルボン酸から調製した:
表1
【0157】
【化32】
【0158】
【化32A】
【0159】
【化32B】
*UV検出を用いるHPLC−MS保持時間;
**UV検出を用いる出発カルボン酸のHPLC−MS保持時間;
UD=UV吸収がないこと、およびMSによるイオン化がないことに起因して検出されない。
【0160】
実施例4
四級プロドラッグへのLY294002の変換
LY294002(化合物1)をアセトニトリルに溶解し、次いで実施例3由来の各々のクロロメチルエステル(1〜1.5当量)を、ヨウ化ナトリウムの1〜2当量とともに添加した。室温ではこの反応はクロロメチルエステルでごくゆっくり進行して、塩化ナトリウムの沈殿とともにごく少量の四級化されたアミン生成物が得られた。65℃では、この反応は通常4時間で完了した。この反応が完全に濾過された場合(LC−MSによる分析で判定した);濃縮し、ついで逆相HPLCで精製した。この画分を収集して、凍結乾燥し、ふわふわした粉末として所望の生成物を得た。この方式で調製および精製した実施例は、下の表に示す(対イオンは示していないが、その塩化物、ヨウ化物、酢酸塩またはそれらの混合物を含む)。
【0161】
表2
【0162】
【化33】
【0163】
【化34】
【0164】
【化35】
実施例5
ハロメチルエステルリンカー
実施例3および実施例4の結果に基づいて、ハロメチルエステルリンカーを調製した(スキーム4およびチャート)。実施例3に記載されるように、化合物Bは、化合物A(市販される)から調製した。この化合物は、アセトンまたは2−ブタノンへの溶解、次いで2〜5当量のヨードナトリウムに溶解することによるFinklestein反応により、さらに反応性のヨードメチルエステル(化合物C)に変換され、ここでは塩化ナトリウムが沈殿して、ヨードメチルエステル(化合物C)が溶液中で生成された。化合物Cは、溶媒の剥ぎ取りおよび水不混和性の溶媒、例えば塩化メチレンへの溶解、および残りのヨードナトリウムを除去するための水での抽出によって単離された。
【0165】
化合物Eは化合物D(市販されている)から調製し、化合物FおよびGは、それぞれ、化合物BおよびCの生成物と同様の方式で調製した。
【0166】
スキーム4
【0167】
【化36】
実施例6
ハロメチルリンカーでのLY294402の四級化
ハロメチルエステルは、実施例5のものを含むが、これを、実施例4の方法論と同様の条件を用いてLY294002を四級化するために用いた。遊離の官能基とのリンカーを含む代表的なプロドラッグとしては以下が挙げられる:
【0168】
【化37】
【0169】
【化38】
化合物1105は、アセトニトリル中に化合物Cとともに化合物1101を混合することによって調製され、ここではこの両方が可溶性であり、そして生成物化合物1105は、3日間にわたって沈殿し、少量のアセトニトリルで洗浄されて実質的に純粋な化合物1105が得られた(LCMSによって確認された)。
【0170】
実施例7
化合物1111を用いるプロドラッグの調製
化合物1111は、スキーム5に示す方法によって生成した。化合物1110は、1〜3時間にわたってニートなトリフルオロ酢酸で処理し、TFAをアルゴンで吹き飛ばして減圧下で乾燥し、化合物1113からなるガラス状の固体を得た。次いで、化合物1113を1〜3mlの塩化チオニルに溶解して、65℃で3〜8時間加熱した。塩化チオニルをアルゴンで吹き飛ばし、ついで減圧下で乾燥して、ガラス状の黄色固体として良好な収率で化合物1111を得た。化合物1111は、例えば、メタノールへの単なる溶解によって、種々の窒素含有およびヒドロキシル含有求核試薬との代表的な酸塩化物として反応されて、相当するメチルエステル化合物1112を得ることができる。
【0171】
スキーム5
【0172】
【化39】
化合物1111のサンプルを、アセトニトリルに溶解して、別々のバイアルに含まれる少なくとも5当量の種々のアルコールで処理した。1時間後、サンプルをHPLC−MSによって分析したところ、表3に示されて特徴付けられるとおり、90%より多い化合物1111の対応するエステルへの良好な変換が示された。
【0173】
表3
【0174】
【化40】
【0175】
【化41】
*UV 214nm
実施例8
化合物1111を用いるタンパク質結合プロドラッグの調製
修飾されるアミノ基またはヒドロキシル基に対して2〜10倍過剰な化合物1111を用いて大量の水溶液(pH7〜9)(炭酸塩緩衝液に対してリン酸塩緩衝液)中でタンパク質を結合体化する。酸塩化物の化合物1111は、混合した有機の水溶液(50/50の水/アセトニトリルまたは50/50の水/THFなど)に導入してもよいし、または1〜24時間、室温で2相の反応系で塩化メチレン中に撹拌してもよい。タンパク質結合体は、透析または限外濾過によって精製して、直接用いることができる。
【0176】
50mMの炭酸水素ナトリウム緩衝液に含まれる5mg/mlのトランスフェリンタンパク質(Sigma)の500μlのアリコートを、実施例12に従ってDMSO中に調製した、100倍の30mM A024−79(100モル当量)と混合した。室温での1時間および20分の反応の後、50倍のサンプルを取り出して、Sephadex G−10(700モル重量のカットオフ)カラムを通過させて、低分子からタンパク質を分離した。次いで、精製された結合されたタンパク質溶出物のアリコートをアセトニトリルで抽出したところ、LC−MSでは化合物1は検出されなかった。精製された結合体化されたタンパク質溶出物を室温で39時間静置させて、その時点でタンパク質混合物をアセトニトリルで再度溶出したところ、この時点で化合物1の最大理論量の15%が検出された。これらの結果によって、プロドラッグの15モルというモル比がトランスフェリンの1モルあたりに結合されたことが示される。これらの結果によって、代表的なタンパク質に対する求電子性リンカー保有プロドラッグの結合が実証され、そして経時的に、PI3キナーゼインヒビター(化合物1)のかなりの量が水性の条件下でタンパク質から遊離されたことが実証された。
【0177】
実施例9
化合物1111を用いる樹脂結合プロドラッグの調製
全て天然のアミノ酸を用いる標準的なFMOC/HOBTカップリングペプチド化学を用いて、wang樹脂上でペプチドarg−gly−asp−ser(RGDS)を調製した。樹脂結合したペプチドを1〜24時間DMF中で化合物1111と反応させて、濾過して、樹脂をDMFで、次いで、塩化メチレンで洗浄し、次いでトリフルオロ酢酸で処理して、樹脂から結合体化合物1126を切断した(スキーム6)。実施例55は、化合物1111の大規模化調製を記載する。
【0178】
スキーム6
【0179】
【化42】
実施例10
化合物1111を用いるフォレート標的化因子でのプロドラッグの調製
化合物1111は、求電子性基を有し、この基が軽度に塩基性の有機または水性の条件(すなわち、20mM〜500mMの炭酸水素ナトリウム)下で求核性アミノ基と反応して非可逆性のチオ尿素結合を形成し得る。適切な求核性アミノ基は、標的化生体高分子フォレートに存在する。フォレート分子AおよびCを、塩基トリメチルアミンまたはジイソプロピルエチルアミンの存在下でほぼ等しい割合で混合することによってDMF中のアミノ基を介して化合物1111に結合して、化合物BおよびDを得た(スキーム7)。
【0180】
スキーム7
【0181】
【化43】
実施例11
化合物1111を用いる抗体標的化因子でのプロドラッグの調製
化合物1111は、pH7〜9の水溶液中でモノクローナル抗体に結合体化し、次いで限外濾過、または低分子からタンパク質結合体を分離する他の標準的な方法によって分離する。行なわれた結合体化は実施例8に従って調製され得る。
【0182】
実施例12
N−ヒドロキシスクシンイミドエステルを用いるプロドラッグの調製
化合物1111よりも反応性の低いエステルは、化合物1113のN−ヒドロキシスクシンイミド活性エステルを生成することによって調製した(スキーム8)。化合物1113(A024−67)の100mgのサンプルを、53mgのN−ヒドロキシスクシンイミド(2当量)とともに1mlの乾燥THFに溶解した。塩化メチレン(2当量)に含まれる1Mジクロヘキシカルボジイミドの45のアリコートを撹拌しながら一挙に添加した。3分内に重い白色沈殿が形成され、このことはカップリング反応が生じたことを示した。反応物を23時間撹拌させた後、この反応混合物を濾過して、濾液から溶媒を除去して、172mgの粗活性エステル生成物を、化合物A024−79と命名された濃い黄色の油状物として得たが、これは2.334分という保持時間を示し、M+=535という予想質量がこのピークについて見出された。
【0183】
スキーム8
【0184】
【化44】
化合物1111について上記したのと同じ化学を用いて、実施例8に記載されたように標的タンパク質に対して結合体化するためにA024−79を用い、そしてポリマーに対して結合体化するために用いたのは実施例74に記載する。
【0185】
実施例13
化合物1105を用いるプロドラッグの調製
化合物1105は、求電子性基を有し、この基は、軽度に塩基性の有機または水性の条件(すなわち、20mM〜500mMの炭酸水素ナトリウム)下で求核性アミノ基と反応して非可逆性のチオ尿素結合を形成し得る。適切な求核性アミノ基は、ビタミン誘導体のようなアミン基を保有するペプチド、タンパク質および低分子のような生体高分子に存在する(スキーム7のAおよびC)。このような生成物の代表的な例としては、化合物BおよびDが挙げられる。
【0186】
スキーム9
【0187】
【化45】
実施例14
モルホリン環の誘導体の調製
スキーム1のチオメチル化合物は、実施例1に記載のように調製した。この化合物を、適切な溶媒中において触媒量の酢酸の有無のもとで、そして過剰な求核性のアミン化合物とともに、ほとんどのチオメチル化合物が消費されるまで加熱した。この混合物を分取逆相LC−MSに供して、所望のモルホリンアナログを単離した。この方式で調製した化合物は、それらの調製条件とともに表4に示し、特徴および単離は表5に示す。化合物のNMRデータは表6に示す。
【0188】
表4
【0189】
【化46】
【0190】
【化47】
表5
【0191】
【化48】
表6
【0192】
【化49】
実施例15
HPLC分析
HPLC分析を島津LCMS−2010で行い、流速は3ml/分を、そして出発B濃度は5%を使用した。B溶媒は5.0分で95%濃度まで直線的に増大させて、6.0分まで95%で保持し、次いで6.5分で5%に直線的に減少させ、これを7.5分で流し終わるまで保持する。他に注記しない限り、これが本実施例で用いた方法である。方法Bは、極性の化合物のための緩い勾配の方法であって、これには3ml/分の流速および0%の出発B濃度を使用して、これを最初の1分間保持する。溶媒Bは3.0分で10%の濃度まで直線的に増大させ、次いで5.0分で95%まで直線的に減少させて、ここで6.0分まで保持し、次いで6.5分で5%まで直線的に減少させて、これを7.5分で流し終わるまで保持する。質量検出に加えて、LC検出は、3つのチャネルからなった;254nmのUV吸収、214nmのUV吸収、そして蒸発光散乱(Alltech ELSD 2000)。蒸発光散乱検出器は、1分あたり1.5リットルの窒素流を用いて50℃で行なった。CDL(化学脱溶媒ライン)および島津LCMS−2010のブロック温度は両方とも300℃であって、窒素ネブライザーガス流は4.5L/分であった。正および負の質量スペクトルが50〜2000m/zで検出された。カラムはYMC CombiScreen ODS−AQ,S−5μ粒子サイズ、50mm長、内径4.6mmであった。移動相Aは、0.1%(v/v)HOAcを添加したHPLC等級のB&Jの水を用いて作製し、移動相Bは0.1%(v/v)HOAcを添加したHPLC等級のB&Jのアセトニトリルを用いて作製した。このシステムによって、参照標準として用いた標準の市販の物質(4−ヒドロキシフェニル酢酸;Aldrich Catalog H5000−4;融点149〜151℃)について1.50〜1.60分(tR=1.50〜1.60)という保持時間が得られる。
【0193】
実施例16
分取HPLC
勾配分取HPLCを、SIL−10A自動サンプリング装置に接続した2つのLC−8Aポンプを備える島津システムで行い、逆相カラム(YMC,カタログCCAQSOSO52OWT;ODS−AQ CombiPrep,20mm×50mm)で溶出し、次いでMRA可変容積分配器を通過させた;次いでより小さいストリームを、LC−10ADVP補給ポンプ(MeOH)を用いて3ml/分まで作製し、溶出液を可変2チャンネル波長UV検出器を通過させ、次いで蒸発光散乱検出器(1分あたり1.5リットルの窒素流を用いて50Cで実施)と島津2010質量検出器とにほぼ6:1に分けた;次いで、MRA分配器からの大きいストリームを、質量、UV吸収またはELSピークサイズによって誘発される分画収集器として機能するGilson 215液体処理装置に流した。
【0194】
種々の勾配を、さらに水性の溶媒Aから出発して流して、種々の濃度のBまで傾斜させた。移動相Aは0.1%(v/v)HOAcを添加したHPLC等級のB&Jの水を用いて作製し、移動相Bは0.1%(v/v)HOAcを添加したHPLC等級のB&Jのアセトニトリルを用いて作製した。
【0195】
実施例17
加水分解されたプロドラッグの生物活性
LY294002(化合物1)の生物活性は、クラスI PI−3キナーゼ依存性経路である、J774マクロファージ中の食作用を評価することによって決定した。要するに、J774細胞を、10%FCSを添加したDMEM中で1時間、適切なDMSOコントロールとともに、10、1および0.1μMの濃度のLY294002で処理して、次いで感作されたsRBC(ヒツジ赤血球)を、100:1の標的対エフェクター比で37℃で30分間添加した。細胞を低張性ショックに曝して赤血球を取り除き、細胞溶出液中のヘモグロビン濃度の測定によって食作用を決定した。図1に示されるように、LY294002は、全ての濃度で用量依存性の様式で食作用を有意にブロックした。これらの結果によって、J774細胞系は、本発明の化合物がPI−3キナーゼ活性を阻害する能力を迅速にかつ容易にアッセイするために用いられ得ることが示される。PI−3キナーゼ活性をアッセイするこの方法を用いて、標的されたプロドラッグ化合物1126を、種々のプレインキュベーション時間を用いて5μMの濃度で試験して、プロドラッグを活性薬物(化合物1)にインサイチュで変換させた。コントロールのサンプル(化合物1126のプレインキュベーションなしの時間ゼロ)は、140という食作用係数(PGI;PI−3キナーゼの阻害がない結果として生じる食作用の程度の指標)を示したが、化合物1126は水性でpH=7であり、2、5および10時間のインキュベーション時間で、それぞれPGIは88、78および37を示した。本実施例によって、最初のプロドラッグ1126は、PI−3キナーゼ阻害活性をほとんどまたは全く有さず、そして有意なPI−3キナーゼ阻害を示す生物活性薬物に経時的に変換されることが実証される。別の実験では、20μM濃度の化合物1126は曝露限界の設定(試験溶液に対する20分の曝露、次いで試験溶液の除去)で、溶媒ブランクについて163、化合物1について190、そしてRGDS(化合物1126の標的部分であるテトラペプチド)についての170に対して、50というPGIを有した。本実施例は、曝露時間限界の設定での標的されたPI−3キナーゼインヒビターの利点を示した。この効果はさらに、実験を繰り返すことによって評価したが、ここでは、化合物1126の10μM、3μM、1μMおよび0μMという漸減する用量を用いて、化合物の除去後20分のインキュベーション時間、続いて食作用の2時間のインキュベーションによって、それぞれ33、143、206および213というPGIが得られた。本実施例によって化合物1126は用量曝露限界の設定で、用量依存性の様式でPI−3キナーゼを阻害したことが実証される。
【0196】
実施例18
骨標的化基A030−84の調製
500mgの4−[(N−BOC)アミノエチル]アニリン(Aldrich)を含む10mlのジオキサンの溶液をパラホルムアルデヒド(400mol%、270mg)および亜リン酸トリメチル(400mol%、1.12g)で処理した。この混合物を95℃で一晩加熱した。次いでさらにパラホルムアルデヒド(270mg)および亜リン酸トリメチル(1.12g)を添加して、これを再度95℃で一晩加熱した。この溶液を冷却して、クロロホルム(20mL)にとり、飽和塩化ナトリウム(20mL)および水(20mL)で洗浄した。有機物を硫酸ナトリウムで乾燥して、溶媒および過剰の亜リン酸トリメチルを80℃のロータリーエバポレーションによって除去して、1.723gの透明な油状物を得た。ロット番号A030−74と命名された表題の化合物の存在を、エレクトロスプレーHPLC−MSによって確認したところ、これはtR=2.9分の保持時間、そして所望の質量[M=C18H32N2O8P2]について実測した、467m/z[M+H]+および489m/z[M+H]+を示した。
【0197】
10mLのジクロロメタン中で上記のように調製した870mgのA030−74の溶液をブロモトリメチルシラン(690モル%、1.97g)で処理した。この溶液を一晩撹拌した。メタノール(10mL)を添加して、溶液を15分撹拌し、次いで濃縮して1.12gのオレンジ色の油状物を得た。表題の化合物の存在をエレクトロスプレーLC−MSによって確認した。この勾配を用いる保持時間は、tR=0.85分であることが見出され、そして所望の生成物[M=C9H16N2O6P2]についての質量スペクトルは、ネガティブモードで操作する予想m/z 309[M−H]−で見出した。この生成物には参照番号A030−84を与えた。
【0198】
【化50】
実施例19
化合物A014−52の合成
【0199】
【化50A】
1.0gの4−カルボキシフェニルイソチオシアナートにジクロロメタン(15mL)および蒸留水(15mL)を添加した。フラスコを氷水槽中で冷却して、炭酸水素ナトリウム(4.0当量)およびn−硫酸水素テトラブチルアンモニウム(0.05当量)を添加した。10分後、クロロメチルクロロサルフェート(1.2当量)を添加した。この溶液を一晩激しく撹拌して、ジクロロメタン(10mL)で補助して分液漏斗に移した。この相を分離して有機物を飽和塩化ナトリウム(20mL)で洗浄した。有機物を硫酸ナトリウムで乾燥させ、溶媒を除去して、1.10gの黄褐色の固体を得た。表題の化合物の存在は、生成物(4.2分)対、出発のカルボン酸(3.2分)の保持時間のシフトによって示された。この化合物をプロトンNMR分光法によって確認した:1H(CDCl3)δ:8.08(d,2H,J8.8Hz),7.30(d,2H,J8.8Hz),5.95(s,2H)。
【0200】
実施例20
化合物A014−48の合成
【0201】
【化51】
250mgのクロロメチルエステル(A014−52に記載の手順を介して調製)を含む2mLのアセトンの溶液を、ヨウ化ナトリウム(1.2当量)で処理して、この溶液を一晩撹拌した。この溶液を濾過して、溶媒を除去し、その残留物をジクロロメタン(10mL)にとった。この溶液を10%(w/v)の亜硫酸ナトリウム(10mL)、5%(w/v)炭酸水素ナトリウム(10mL)および水(10mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去して137mgの薄緑色の固体を得た。表題の化合物の存在は、ヨードメチルエステル生成物(4.4分)対出発クロロメチルエステル(4.2分)の保持時間のシフトで示された。この化合物はまたプロトンNMR分光法によって確認した:1H(CDCl3)δ:8.04(d,2H,J8.8Hz),7.29(d,2H,J8.1Hz),6.15(s,2H)。
【0202】
実施例21
化合物A014−76の合成
【0203】
【化52】
387mgのクロロメチルエステル(A014−52に記載の手順を介して調製)を含む6mLの2−ブタノンの溶液を、ヨウ化ナトリウム(1.2当量)で処理して、この溶液を10時間加熱した。この溶液を濾過して、溶媒を除去し、その残留物をジクロロメタン(10mL)にとった。この溶液を10%(w/v)の亜硫酸ナトリウム(10mL)、5%(w/v)炭酸水素ナトリウム(10mL)および水(5mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去して310mgの黄褐色色の固体を得た。表題の化合物の存在は、ヨードメチルエステル生成物(4.4分)対出発クロロメチルエステル(4.2分)の保持時間のシフトで示された。
【0204】
実施例22
化合物A018−24の合成
【0205】
【化53】
64mgの2−p−ニトロベンジル−1,4,7,10−テトラアザサイクロドデカン(Macrocyclics)を含む500μLのジオキサンの溶液を、パラホルムアルデヒド(50mg)および亜リン酸トリメチル(207mg)で処理した。この混合物を85℃で加熱して、次いで溶媒を75℃のロータリーエバポレーションによって除去した。クロロホルム(10mL)を添加して、溶液を飽和塩化ナトリウム(2×10mL)および水(2×10mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥し、溶媒を除去して褐色の油状物を得た。これをLCを介して精製して、所望の物質を得た。表題の化合物の存在をエレクトロスプレーLC−MS Aによって確認した;tR=1.8分。MS[M=C27H53N5O14P4]m/z796(MH+),818(MNa+)。
【0206】
実施例23
化合物A022−32の合成
【0207】
【化54】
33mgのホスホン酸大環状化合物(A018−24で調製)を含む700μlのジクロロメタンの溶液をブロモトリメチルシラン(72mg)で処理した。この混合物を一晩撹拌し、次いでさらなるブロモトリメチルシラン(36mg)を添加して、これをさらに3日撹拌した。メタノール(500mL)を添加して溶液を1時間撹拌し、次いで揮発性物質を除去して、褐色の油状物を得た。メタノールの添加で褐色の固体を沈殿させ、これを濾過して乾燥させた。これを、LCを介して精製して、2.7mgの所望の物質を得た。表題の化合物の存在をエレクトロスプレーLC−MS Aによって確認した;tR=1.5分。MS[M=C19H37N5O14P4]m/z 682(M−H−),340(M−2H)/2)2−]。
【0208】
ニトロ基は標準的な還元方法を用いて、例えば、純粋な水素の雰囲気下でのメタノール中の炭素触媒上で5%パラジウムとともに撹拌することによって還元する。次いでこの混合物を濾過して(触媒に対する空気の曝露を妨げるように注意する)、溶媒をエバポレートしてアミンを得る。
【0209】
実施例24
化合物A022−56の合成
【0210】
【化55】
リン酸(1.26g)、6M塩酸(19.5mL)およびp−キシレンジアミン(1.0g)の混合物を100℃に加熱した。これに37%(wt/wt)のホルムアルデヒド水溶液(1.15mL)を添加して、この混合物を100℃で一晩撹拌した。この混合物を濾過して、80℃でのロータリーエバポレーションによって水を除去して、2.11gの白色固体を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=1.8分。MS[M=C10H18N2O6P2]m/z 325(MH+)。
【0211】
実施例25
化合物A018−12の合成
【0212】
【化56】
928mg N−BOC−1,4−ジアミノブタンを含む10mLのジオキサンの溶液を、パラホルムアルデヒド(592mg)および亜リン酸トリメチル(2.44g)で処理した。この混合物を108℃で一晩撹拌して、75℃のロータリーエバポレーションによって溶媒を除去した。クロロホルム(10mL)を添加してこの溶液を飽和塩化ナトリウム(2×10mL)および水(2×10mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥し、溶媒を除去して1.55gの油状物を得た。表題の化合物の存在をエレクトロスプレーLC−MSによって確認した;tR=2.4分。MS[M=C15H34N2O8P2]m/z 455(MNa+)。
【0213】
実施例26
化合物A026−92の合成
【0214】
【化57】
783mgのホスホン酸塩(A018−24で調製)を含む18mLのジクロロメタンの溶液をブロモトリメチルシラン(2.2g)で処理した。この溶液を一晩撹拌し、メタノール(10mL)を添加してその混合物を2時間撹拌した。揮発性物質を除去して、1.22gの黄色油状物を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.4分。MS[M=C6H18N2O6P2]m/z 275(M−H−)。
【0215】
実施例27
化合物A030−74の合成
【0216】
【化58】
500mg 4−[(N−BOC)アミノエチル]アニリンを含む10mLのジオキサンの溶液をパラホルムアルデヒド(270mg)および亜リン酸トリメチル(1.12g)で処理した。この混合物を95℃に一晩加熱した。次いでさらなるパラホルムアルデヒド(270mg)および亜リン酸トリメチル(1.12g)を添加して、これを再度、一晩95℃で加熱した。この溶液を冷却して、クロロホルム(20mL)にとり、飽和塩化ナトリウム(20mL)および水(20mL)で洗浄した。有機物を硫酸ナトリウムで乾燥させて、溶媒および過剰の亜リン酸トリメチルを80℃のロータリーエバポレーションによって除去して、1.72gの透明な油状物を得た。表題の化合物の存在を、エレクトロスプレーLC−MSによって確認した;tR=2.9分。MS[M=C18H32N2O8P2]m/z 467(MH+);489(MNa+)。
【0217】
実施例28
化合物A030−84の合成
【0218】
【化59】
870mgのA030−74を含む10mLのジクロロメタンの溶液をブロモトリメチルシラン(1.97g)で処理した。この溶液を一晩撹拌した。メタノール(10mL)を添加してその溶液を15分撹拌し、次いで濃縮して1.12gのオレンジ色の油状物を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.85分。MS[M=C9H16N2O6P2]m/zの実測309[M−H]−。
【0219】
実施例29
化合物A035−66の合成
【0220】
【化60】
2.0g部の4−アミノメチル安息香酸(Aldrich)を、0.64gの固体NaOHを含む20mLの水に溶解した。3.18g部のBoc無水物(Aldrich)を添加して、その混合物を一晩撹拌させた。この混合物に15mLの2N HClを注意深く添加することによってpH=2に調節した。得られた白色固体を濾過して、乾燥させ、2.9997gの生成物を得た。この生成物をLCMS(保持時間2.901分、所望のM−H質量イオン250m/zで観察)によって特徴付けた。
【0221】
実施例30
化合物A035−6の合成
【0222】
【化61】
1.5部のA35−66を17mLの乾燥THF中に、0.69gのN−ヒドロキシスクシンイミド(Aldrich)とともに溶解し、次いで1Mジシクロヘキシカルボジイミド(Aldrich)を含む6mLのジクロロメタンとともに撹拌して一度に処理した。2日後、白色沈殿(ジシクロヘキシル尿素)を濾過して除き、次いで濾液を減圧下でロータリーエバポレートして、2.8146gの白色固体を得て、LCMSによって特徴付けた(保持時間3.299分、そして349m/zで所望のM+Hを観察)。
【0223】
実施例31
化合物A035−14の合成
【0224】
【化62】
A035−6の500mg部を、5mLの乾燥THFに溶解して、1.002mL(10当量)のエチレンジアミン(EDA)で処理して2時間撹拌させた。次いでこの溶液を形成された固体からデカントした。次いで溶媒および過剰のEDAを、減圧下でロータリーエバポレーションによってデカントされた溶液から除去し、0.8728gの白色固体を得た;この生成物をLCMSによって特徴付けた(保持時間1.608分、そして294m/zで所望のM+Hを観察)。
【0225】
実施例32
化合物A032−24の合成
【0226】
【化63】
872mgのアミン(A035−14で調製)を含む10mLのジオキサンの溶液をパラホルムアルデヒド(535mg)および亜リン酸トリメチル(2.21g)で処理した。この混合物を100℃で一晩加熱し、次いで溶媒を80℃のロータリーエバポレーションによって除去して、褐色の固体を得た。クロロホルム(25mL)を添加してその溶液を水(15mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥させて、溶媒を除去して241mgの黄色半固体を得た。これを、LCを介して精製して、58.8mgの所望の物質を得た。表題の化合物の存在を、エレクトロスプレーLC−MSによって確認した;tR=2.6分。MS[M=C21H37N23O9P2]m/z 538(MH+),560(MNa+)。
【0227】
実施例33
化合物A032−40の合成
【0228】
【化64】
54.6mgのホスホン酸塩(A032−24で調製)を含む1mLのジクロロメタンの溶液をブロモトリメチルシラン(156mg)で処理した。この混合物を一晩撹拌させた。エタノール(0.5mL)および水(3滴)を添加して、これを1時間撹拌し、次いで揮発性物質を除去して、その物質を減圧下で乾燥させた。これを水(1mL)にとって、凍結乾燥させ、59mgの黄褐色固体を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.4分。MS[M=C12H21N3O7P2]m/z 380(M−H−),382(MH+),404(MNa+)。
【0229】
実施例34
化合物A026−60の合成
【0230】
【化65】
Kantoci,D.,Kenike,J.K.,Wechter,W.J.Syn.Commun.,1996,26(10),2037の手順を介してA026−60を調製した:N−ベンジル−N−メチルアミン(20.0g)、亜リン酸ジエチル(70.7g)およびオルトギ酸トリエチル(29.3g)の混合物をアルゴン還流下(150℃)で5時間撹拌した。エタノールを70℃のロータリーエバポレートを介して除去して、その混合物を再度、一晩還流して加熱した。この溶液を600mLクロロホルムで希釈して、1Mの水酸化ナトリウム(3×100mL)および飽和塩化ナトリウム(3×150mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去し、74.0gの淡黄色油状物を得た。この物質の10.0gを、溶出液として14:4:1の酢酸エチル:ヘキサン:メタノールを用いるシリカゲルカラムクロマトグラフィーに供した。これによって、6.08gの透明な油状物を得た。表題の化合物の存在を、エレクトロスプレーLC−MSによって確認した;tR=3.4分。MS[M=C17H31NO6P2]m/z 408(MH+),430(MNa+),471(MNa−CH3CN+)。
【0231】
実施例35
化合物A030−54の合成
【0232】
【化66】
Kantoci,D.,Kenike,J.K.,Wechter,W.J.Syn.Commun.,1996,26(10),2037の手順を介してA030−54(CAS#80475−00−9として公知の化合物)を調製した:4.52gのホスホン酸化ベンジルアミン(A026−60で調製)を含むメタノール(45mL)の溶液を10%パラジウムとともに炭素(200mg)上で処理して、水素雰囲気に一晩供した。パラジウム/炭素を濾過して、2.98gの淡黄色の油状物を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=1.9分。MS[M=C10H25NO6P2]m/z 318(MH+)。
【0233】
実施例36
化合物A039−16の合成
【0234】
【化67】
A039−16:500mgのサンプルの4−クロロメチル安息香酸(Aldrich)を、8mLのTHFに溶解し、5当量のエチレンジアミン(Aldrich)(983μL)とともに一度に処理した。24時間後、溶媒を高真空下で除去して、白色固体(93%)をLCMSによって特徴付けた(保持時間0.4分、そして195m/zで所望のM+Hを観察)。
【0235】
実施例37
化合物A038−24の合成
【0236】
【化68】
ジオキサン(10mL)中の679mgのアミノ酸(A039−16で調製)、37%(wt/wt)ホルムアルデヒド水溶液(1.04mL)、亜リン酸(1.15g)および濃(12.1M)塩酸(2.3mL)の混合物を100℃で一晩撹拌した。この溶媒を75℃でロータリーエバポレートを介して除去して、混合物を遠心分離して固体を廃棄した。この液体にさらにホルムアルデヒド溶液(1.04mL)、亜リン酸(1.15g)、濃塩酸(2mL)およびジオキサン(10mL)を添加して、これを再度100℃で一晩撹拌させた。この溶媒を75℃のロータリーエバポレーションによって除去して、濃い油状物を得た。これをLCを介して精製して、276mgの黄褐色の固体を得た。表題の化合物の存在を方法Aを用いてエレクトロスプレーLCMSによって確認した;tR=0.6分。MS[M=C13H23N2O11P3]m/z 475(MH−),477(MH+)。
【0237】
実施例38
化合物A038−50の合成
【0238】
【化69】
6.0gのFmoc−Lys−OH(Advanced ChemTech)を含むメタノール(25mL)および水(25mL)の混合物を37%(wt/wt)ホルムアルデヒド水溶液(6.06mL)および亜リン酸ジメチル(8.96g)で処理した。この混合物を80℃で2時間撹拌して、冷却し、ジクロロメタン(1×100mL、2×50mL)で抽出した。この有機物を飽和塩化ナトリウム(50mL)で洗浄し、硫酸マグネシウムで30分間乾燥し、溶媒を除去して、10.17gの薄緑色の油状物を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.3分。MS[M=C27H38N2O10P2]m/z 613(MH+),636(MNa+)。
【0239】
実施例39
化合物A038−66の合成
【0240】
【化70】
23.9mgのホスホン酸化Fmoc−Lys−OH(A038−50で調製)を含むジクロロメタン(1mL)の溶液を、ブロモトリメチルシラン(60mg)で処理した。この混合物を一晩撹拌した。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.4分。MS[M=C23H30N2O10P2]m/z 555(M−H−)。
【0241】
実施例40
化合物A038−76の合成
【0242】
【化71】
112.9mgのホスホン酸化Fmoc−Lys−OH(A038−50で調製)を含む6M塩酸(3mL)の溶液を、80℃で2日間撹拌した。水(9mL)を添加してさらに2日後、この混合物を遠心分離して液体をデカントした。この固体を減圧下で乾燥させて、86.8mgの灰色の固体を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.1分。MS[M=C23H30N2O10P2]m/z 555(M−H−)。
【0243】
実施例41
化合物A038−90の合成
【0244】
【化72】
500mgのFmoc−Lys−OH(Advanced Chem Tech)を含むジオキサン(5mL)の溶液を、37%(wt/wt)ホルムアルデヒド水溶液(303μL)、亜リン酸(333mg)および濃(12.1M)塩酸(674μL)で処理した。この混合物を90℃で一晩撹拌して、この溶媒を75℃のロータリーエバポレーションによって除去した。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=5.4分。MS[M=C23H30N2O10P2]m/z 555(M−H−)。
【0245】
実施例42
化合物A042−18の合成
【0246】
【化73】
1.29gのBoc−保護アミノ酸(上記ロットA035−66と同じく調製)を含む15mLのテトラヒドロフランの溶液をN−ヒドロキシスクシンイミド(623mg)および1.0M 1,3−ジクロロヘキシルカルボジイミドを含むジクロロメタン(5.4mL)で処理した。この混合物を一晩撹拌して、白色沈殿を濾過して、上清を濃縮し、1.89gの白色固体を得た。表題の化合物の存在は、3.3分のUV信号の存在によって示された。
【0247】
実施例43
化合物A042−26の合成
【0248】
【化74】
2.1gのtris−(2−アミノエチル)アミンを含む20mLのテトラヒドロフランの溶液に、1.0gの活性化エステル(A042−18によって調製)を含む20mLのテトラヒドロフランの溶液を40分間にわたって滴下して加えた。この混合物を一晩撹拌して、沈殿物を得て、これを濾過してロータリーエバポレーションによって濃縮し、2.10gの黄色油状物を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=1.4分。MS[M=C19H33N5O3]m/z 380(M−H+)、402(MNa+)。
【0249】
実施例44
化合物A042−32の合成
【0250】
【化75】
2.08gのアミン(A042−26で調製)を含むジオキセン(20mL)の溶液を、パラホルムアルデヒド(1.50g)および亜リン酸ジメチル(6.85g)で処理した。この混合物を90℃で一晩撹拌して、その溶媒を70℃のロータリーエバポレーションによって除去した。ジメチルメタン(50mL)を添加して、これを飽和塩化ナトリウム(25mL)および水(25mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去した。この残滓をLCで精製して、123.8mgの黄色油状物を得た。表題の化合物の存在を、方法Dを用いてエレクトロスプレーLC−MSによって確認した;tR=2.2分。MS[M=C31H61N5O15P4]m/z 868(MH+)。
【0251】
実施例45
化合物A042−70の合成
【0252】
【化76】
111.1mgのホスホン酸化ジアミン(A042−32を介して調製)を含む1mLのジクロロメタンの溶液を、194mgのブロモトリメチルシランで処理した。5時間後、メタノール(1mL)を添加した。この混合物を1時間撹拌して、溶媒を除去して113.9mgの黄褐色の固体を得た。表題の化合物の存在は、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=1.0分。MS[M=C18H37N5O13P4]m/z 328(M+2H/2)2+)]、656(MH+)。この化合物をまたプロトンNMR分光法によって分析した:1H(CDCl3)δ:7.77(d,2H,J8.1Hz),7.43(d,2H,J8.2Hz),4.1〜3.3(m,33H)。
【0253】
実施例46
化合物A026−94の合成
【0254】
【化77】
750mgのホスホン酸塩(A030−54を介して調製)を含む24mLのジクロロメタンの溶液を、ブロモトリメチルシラン(2.89g)で処理した。この溶液を一晩撹拌した。メタノール(10mL)を添加して、その溶液を2時間撹拌し、その溶媒を除去して、黄色の油状物を得て、これを凍結乾燥し、364mgの白色固体を得た。表題の化合物の存在は、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.6分。MS[M=C2H9NO6P2]m/z 204(M−H)−)。
【0255】
実施例47
化合物A026−96の合成
【0256】
【化78】
A042−96(tert−亜リン酸ブチル):4.10gの亜リン酸を含む100mLのテトラヒドロフランの溶液を2−メチル−2−プロパノール(7.41g)で処理した。1,3−ジシクロヘキシルカルボジイミドを含むジクロロメタン(100.0mL)の1.0M溶液を添加して、白色固体の形成を生じた。この混合物を一晩撹拌して、固体を濾過し、溶媒を除去して6.82gの黄色固体を得た。表題の化合物の存在は、GC−MSによって確認した。以下のフラグメントを見出した:57[(CH3)3C+]、83[HP(OH)3+]、123[(HO)2PC(CH3)2+]。
【0257】
実施例48
化合物A042−98の合成
【0258】
【化79】
エタノールアミン(858mg)、パラホルムアルデヒド(1.05g)、tert−亜リン酸ブチル(A042−96,6.82gを介して調製)を含むベンゼン(100mL)の混合物を一晩90℃で加熱して、濃い油状物の上に液体を生じた。この液体をデカントして、ロータリーエバポレーションを介して濃縮し、油状物を得て、これを10%メタノールを含有するジクロロメタンを溶出液として用いるシリカゲルカラムクロマトグラフィーに供した。透明な油状物(436mg)を得た。表題の化合物の存在は、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.6分。MS[M=C20H45NO7P2]m/z 496(MNa+)。
【0259】
実施例49
化合物A029−34の合成
【0260】
【化80】
4−イソチオシアナートフェニル酢酸を調製するために、1.3ml(13.5mmol、2当量)のチオホスゲンを、20mlの乾燥THFに懸濁した1.0gの4−アミノフェニル酢酸(6.61mmol)[Aldrich]および3.73gの無水炭酸カリウム(4当量)に加えた。この懸濁物を室温で15分間撹拌して、続いて85℃のシリコーン油槽中で4時間加熱した。この溶液を冷却して、濾過シリンジ中で1インチのセライト層を通した。この溶液を丸底フラスコ中で収集して、溶媒を減圧下で除去した。
【0261】
このサンプルを真空デシケーター中に2時間保管し、次いでアセトン−水混合物に溶解し、凍結して凍結乾燥し、1.65gの黒っぽい固体を得た。この化合物は、LCMSクロマトグラムの保持時間の3.32分へのシフトによって同定した。
【0262】
実施例50
化合物A040−22の合成
【0263】
【化81】
4−イソチオシアナートフェニル酢酸クロロメチルエステルを調製するために、0.530gの4−イソシアナートフェニル酢酸(2.7mmol)をガラスバイアル中で5.0mlの塩化メチレンに溶解して、これに、5.0mlの水に溶解した0.044gのテトラ−n−ブチル硫酸水素アンモニウム(相間移動触媒−0.05当量)および0.866gの炭酸水素ナトリウム(4当量)を添加した。この溶液を氷浴中で10分間撹拌した。この冷たい混合物に0.520gのクロロメチルクロロサルフェート(ACROS Chemical−1.2当量)を添加して、室温まで徐々に温度上昇させながら4時間撹拌した。この有機層を分液漏斗で分離して、10mlの飽和ブラインで洗浄し、無水硫酸ナトリウムで乾燥させた。この溶媒を減圧下で除去して、0.703gの粗油状物を得た。この生成物は、LCMSで新しいピークの形成によって同定され、この保持時間は4.268分であり、出発材料に相当するピークの消失があった。
【0264】
実施例51
化合物A040−26の合成
【0265】
【化82】
化合物1101の4−イソチオシアナートフェニル酢酸四級塩誘導体を調製するために、化合物1101(0.300g、1.0mmol)および0.582gのヨウ化ナトリウム(4当量)を、1ドラムのバイアル中で4.0ml乾燥アセトニトリルに溶解した。この4−イソチオシアナートフェニル酢酸クロロメチルエステル(化合物A040−22)を含む2.0mlの乾燥アセトニトリルを撹拌しながら添加した。この混合物をシリコーン油浴上で、65℃で5時間加熱して、LCMSによって反応の進行をモニタリングした。化合物1101出発材料のほとんどを消費した場合、この反応混合物を、3.019分、m+=513の分取LCMS保持時間を用いて精製した。94%純粋な生成物の194.1mgの収量を得て、LCMSによって同定した。次いで化合物A040−26を、化合物1105を利用する実施例に記載された方法によって記載されるように求核試薬保有標的化因子と反応させて、有用な標的プロドラッグを調製する。
【0266】
実施例52
化合物A044−52の合成
【0267】
【化83】
N−ベンジル−N−メチルカルボニルクロロメチルエステルを調製するために、0.300mg(324μL,d=0.942)ベンジルメチルアミンおよび640μLジイソプロピルエチルアミン(1.5当量)を2.0mlの乾燥塩化メチレンに溶解して、氷浴中で冷却した。この溶液が冷却された場合、330μLのクロロメチルクロロホルメート(1.5当量)を含む2.0mlの乾燥塩化メチレンを添加して、2時間撹拌し、この溶液を徐々に室温に戻させた。黄色溶液を分液漏斗に入れて、2×10ml 1N HCl、1×10ml水および2×10ml 1N 重炭酸ナトリウムを用いて洗浄した。この有機層を硫酸ナトリウムで乾燥して、減圧下で濃縮した。カルバミン酸クロロメチルを、0.95gの黄色油状物として得て、LCMSにおいてuv活性成分として3.520分の保持時間で同定した。
【0268】
化合物1101のN−ベンジル,N−メチルカルバモイルメチル四級塩を調製するために、0.5gの化合物1101および0.5gのヨウ化ナトリウム(20当量)を、ガラスバイアル中で5.0ml乾燥アセトニトリルに溶解した。このN−ベンジル、N−メチルカルバモイルクロロメチルエステルを溶液に追加し、次いで一晩65℃の油浴に入れた。この生成物を、LCMSによって同定して、分取LCMSによって精製し、保持時間2.731分、M+=485、そして90%の純度で、A044−52と命名された169.5mg(理論収率21.5%)の固体を得た。
【0269】
実施例53
化合物A044−62の合成
【0270】
【化84】
化合物1157のN−ベンジル,N−メチルカルバモイルメチル四級塩を調製するために、22mgの化合物1157、20mgのヨウ化ナトリウム(2.0当量)および30.7mgのN−ベンジル−N−メチルカルボンモイルクロロメチルメステル(2.0当量)を、ガラスバイアル中で500μLの乾燥アセトニトリルに混合し、一晩65℃の油浴中で加熱した。この生成物を、LCMSによって同定して、分取LCMSによって精製した。保持時間2.810分、M+=483および98%純度で5.4mgの化合物(理論収率15.5%)を得た。
【0271】
実施例54
化合物A044−28の合成
【0272】
【化85】
化合物1101のベンジルホルモイル−1−エチル四級塩を調製するために、200μL 1−クロロエチルクロロホルメートを、ガラスバイアル中で100μLのベンジルアルコールを含む2.0mlの乾燥塩化メチレンに添加して、氷浴中で0℃でインキュベートした。冷却された溶液に200μLのピリジンを添加して、2分内に白色沈殿を形成させた。撹拌を室温で一晩継続した。この混合物に10mlの塩化メチレンを添加し、次いで1×10ml 0.5M HCl、1×10ml水および1×10ml 0.5N重炭酸ナトリウム溶液を用いて洗浄した。この有機層を硫酸ナトリウムで乾燥させて、減圧下で溶媒を除去した。ベンジル−1−ギ酸クロロメチルを、3.933分の保持時間のuv活性成分として同定した。
【0273】
1.0mlの乾燥アセトニトリルに溶解した100mgの化合物1101および100mgのNaI(20当量)に107mg(1.5当量)のベンジル−1−ギ酸クロロメチルを添加した。この混合物を65℃で一晩加熱した。LCMSで出発物質の存在が示されたため、さらなる107mg(1.5当量)のベンジル−1−ギ酸クロロエーテルを添加して、さらに24時間加熱を続けた。LCMSによって、緩慢勾配クロマトグラフィーで出発物質から分離され得る生成物を同定した。所望の化合物を分取LCMSを用いて単離して、保持時間4.690分、M+=488、98.6%純度で13.7mg(8.7%理論収率)の化合物を得た。
【0274】
実施例55
化合物1126の合成
クロロメチル−t−ブチルスクシネートを調製するために、モノt−ブチルスクシネート(Aldrich)、2.0gを8.0mlの塩化メチレンに溶解したものを、4.0gの炭酸カリウムと0.24gのテトラn−ブチル硫酸水素アンモニウムを8.0mlの水に含むガラスバイアルに添加して、氷浴で撹拌した。15分後、1.3mLのクロロメチルクロロサルフェート(Acros)を塩化メチレン層に添加して、反応混合物をゆっくり室温になる温度で撹拌した。有機層を分離して1×10mlの水および1×10mlの飽和ブライン溶液で洗浄した。この溶液を無水硫酸ナトリウムで乾燥して、溶媒を減圧下で除去した。3gの淡黄色油状物を得て、ロット番号A047−71とした。
【0275】
上記のA047−71の3gを36mLのアセトニトリルに溶解して、1.8gの化合物1101および1.8gのNaIで処理し、撹拌しながら16時間ヒーター上に置いた。反応混合物(沈殿を含む)を水と塩化メチレンとの間で分画して、塩化メチレン層を分離して、ブラインで洗浄し、硫酸ナトリウムで乾燥しエバポレートして、暗い油状物を得た。この油状物を5mLのアセトニトリルに溶解して、2日間冷蔵庫に保管した。次いで、形成した黄色沈殿を濾過して3mLのアセトニトリルで洗浄し乾燥して、ロット番号A046−67Aとして、2.8435gの95%を超える純度の、保持時間2.719分、494m/zの[M+]という生成物を得た。この生成物の全てを10mLの塩化チオニルに溶解して、65℃に4時間加熱した。過剰の塩化チオニルを減圧下で除去して、黄色油状物を高真空下で乾燥して1.9906gの酸塩化物(化合物1111)を黄色のバリバリした固体として得た。この固体を引き続く反応に直接用いた。
【0276】
化合物1101酸塩化物およびde−FMOCed RGDSペプチドをカップリングするために、化合物1111の1.26gの酸塩化物および5.6g de−FMOC除去RGDSペプチド(両方とも五酸化リン上で、真空デシケーターで乾燥させた)をアルゴンガス下で50mlの丸底フラスコ中に混合した。この固体に270μLの乾燥ピリジン含有28ml塩化メチレンを添加して、この混合物を振盪して酸塩化物を溶解した。この混合物をオービタルシェイカーに1時間おいた。この溶液をガラス質プラスチックシリンジで排液して、2×10mlの塩化メチレンで洗浄して、溶媒を排液した。この樹脂に500μLのアニソール、続いて20mlの50/50 TFA/塩化メチレン溶液を添加した。この樹脂を時折撹拌しながら、TFA溶液中に3時間静置させた。TFA溶液を樹脂から排液した。この樹脂をTFA溶液と組み合わせた10mlの塩化メチレンで洗浄した。TFA溶液を4つのバイアルに入れて、これをアルゴンガスで風乾した。各々のバイアルを複数回のエーテル洗浄で処理し、再度アルゴンガスで風乾した。この生成物をLCMSによって同定し、そして分取逆相LCMSを用いて17回の実施で精製した。この組み合わせた実施によって、163.7mgの96%純度の生成物(ロット番号a036−33と指定)を保持時間1.768分、M+=853そして427m/zで[M+H]/2で得た。この化合物についてのLC−MSクロマトグラムおよび質量スペクトルは、図7および図8に示す。図7では、x軸が分単位の時間であり、そして上のクロマトグラムのy軸は254nmでのUV検出器のミリ吸光度単位であり、そして下のクロマトグラムのy軸は、蒸発光散乱検出器によって検出されるミリボルトである。図8では、x軸は質量対電荷比(m/z)であり、y軸は質量イオンカウントの強度である。
【0277】
スキーム10
【0278】
【化86】
実施例56
化合物A052−10の合成
【0279】
【化87】
化合物1101のフタルイミドメチル四級塩を調製するために、100mgの化合物1101および100mgのヨウ化ナトリウム(2.0当量)の混合物を3.0mlの乾燥アセトニトリルに溶解した。この混合物に128mgのクロロメチルフタルイミド(Aldrich)を添加して、このバイアルを55℃の油浴中で4日間加熱した。この生成物を、保持時間3.984分、M+=467で新しいピークとしてLCMSによって同定した。
【0280】
実施例57
化合物A052−08の合成
【0281】
【化88】
化合物1101のフタルイミドメチル四級塩を調製するために、100mgの化合物1101および100mgのヨウ化ナトリウム(2.0当量)の混合物を3.0mlの乾燥アセトニトリルに溶解した。この混合物に128mgのクロロメチルフタルイミドを添加して、このバイアルを55℃の油浴中で4日間加熱した。この生成物を、LCMSによって、Rf=3.984、M+=467で新しいピークとして同定した。
【0282】
実施例58
化合物A044−78の合成
【0283】
【化89】
化合物1101の4−カルボキシベンジル四級塩を調製するために、300mgの化合物1101、400mgのヨウ化ナトリウムおよび500mgのクロロメチル安息香酸を、ガラスバイアル中で混合して、4.0mlの乾燥アセトニトリル中で懸濁した。この反応物を、65℃で油浴において加熱して、LCMSによって2週間モニターした。この溶液をガラス質プラスチックシリンジで濾過してさらなる2ミクロンのフィルターと適合させた。この所望の化合物を分取LCMCを用いて単離したが、これは3.717分の保持時間、M+=442であった。96%純度の27.7mgの収量を得た。化合物A044−78のカルボン酸基を、a)NHS活性エステルを調製するための実施例31の方法によって記載されるようなN−ヒドロキシスクシンイミドとの反応、またはb)実施例56の方法によって記載されるような酸塩化物への変換によって反応性の基に変換する。次いで、これらの反応性基のいずれかを、前の実施例に記載した方法を用いて標的化因子の求核アミンまたはアルコール基と反応させて標的化プロドラッグ結合体を得る。
【0284】
実施例59
化合物A044−80の合成
【0285】
【化90】
化合物1101の4−イソシアナトベンジル四級塩を調製するため、300mgの化合物1101、450mgのヨウ化ナトリウム(3.0当量)および490mg(3.0当量)の4−クロロメチルベンゼンイソシアナートの混合物を4.0mlの乾燥アセトニトリルに溶解して、油浴中で65℃で2日間加熱した。LCMSによって、この反応は、出発材料(化合物1101)がないために終了したことが示された。この生成物は、4.577分の保持時間、M+=439で新しいピークとして同定された。化合物A044−80は、種々の標的化因子の求核性基と反応して、化合物1105およびA040−26を利用する実施例の方法によって、標的化因子に対するカルバミン酸塩または尿素結合を生じる。
【0286】
実施例60
化合物A044−4の合成
【0287】
【化91】
化合物1101のピボロイルメチル四級塩を調製するために、100mgの塩化ピボロイル(2.0当量)を、100mgの化合物1101および100mgのヨウ化ナトリウム(2.0当量)を含む2.0mlの乾燥アセトニトリルの混合物に滴下して加えた。この混合物を65℃の油浴で2時間加熱した。この固体は2ミクロンフィルターを装着したガラス質のプラスチックシリンジを用いて濾過した。この化合物を同定して、分取LCMSを用いて単離した。保持時間2.735分、M+=422の黄色固体を得たが、これは93.7%の純度で102.3mgの収率であった。
【0288】
実施例61
化合物A040−70の合成
【0289】
【化92】
化合物1101のアセトキシメチル四級塩を調製するために、1.0gの化合物1101を、ガラスバイアル中で10ml乾燥アセトニトリルに溶解して、1.0gの酢酸ブロモメチル(2.0当量)を添加して、室温で一晩撹拌させた。LCMSによって、出発物質の存在が示され、そして反応物を65℃で、油浴中で8時間加熱した。母液を固体からデカントして、固体を少量の冷たいアセトニトリルで洗浄した。この固体を真空デシケーターで一晩乾燥させた。この生成物を、97%の純度である、2.204分の保持時間、M+=380の264mgの白色固体として同定した。この化合物は、極めて高い水溶性を有することが見出された;リン酸緩衝化生理食塩水中のこの化合物の32.5ミリモル溶液を、1.865mLのリン酸緩衝化生理食塩水中で27mgを溶解することによって調製して約4というpHを得た。この溶液の50μLのアリコート(これはプロドラッグとして化合物1101の1.625μモルを含む)を、ヌードマウスの尾静脈に注射したが、有害な観察可能な影響はなく、このプロドラッグ形態の毒性のないことが示されたが、化合物1101の1.04μモルの注射では3匹のマウス中3匹で即時的な死亡が生じた。さらに、化合物A040−70のこの32.5mMの溶液の250μLを、経口胃管によってヌードマウスに投与した。5、15および30分後、40μLのマウス血液を得て、LC−MSによって分析し(アセトニトリルを用いる抽出後)、プロドラッグA040−70および化合物1の組み合わせた血液レベルが、それぞれの時点で1.8、4.97および4.80マイクロモルであることが実証された。この結果によって、インビボにおけるプロドラッグから活性薬物の経口のバイオアベイラビリティーが実証される。少量の水にこの化合物A040−70を溶解すること、およびAldrichから入手可能なDowex22(塩化物型)のような陰イオン交換樹脂層を通過させること、そして次に水および凍結乾燥の混合性溶離剤を用いて洗浄して臭化物イオンが塩化物イオンで交換された固体を得ることによって塩化物を得る。
【0290】
実施例62
非イオン性水溶性のためのポリオキシエチレン保有プロドラッグの調製
【0291】
【化93】
化合物1の78mg部分を76mgのNaIとともに2mLのアセトニトリルに溶解して、144mgのクロロメチルエステルA029−62で一度に処理して、65℃で5時間撹拌した。この反応物を、逆相LC−MSによって精製して、2.30分の保持時間で72mg(51%収率)の黄色固体A027−85を得たが、これはC30H38NO9によって期待されるとおりM+=556を示す質量スペクトルを有することによって特徴づけられた。わずかなサンプルをpH=7.4のリン酸緩衝液、続いてLC−MSに経時的に導入して、ここでこのプロドラッグのかなりの部分が、約3時間という変換の推定半減期で、化合物1に戻された。
【0292】
【化94】
実施例63
化合物1の骨標的化プロドラッグの調製
75mモル A042−70(1.5μモル)の20μL部分を含む水を、500mモルのリン酸緩衝液を含むバイアル(0.5単位で増す3.0〜8.0にわたる11の種々のpHの11のバイアル)に添加した。次いで、各々のバイアルを混合した後、60μモル化合物1111を含むアセトニトリルの50μL(3.0μモル=骨標的化因子のアミノ基に対して2当量)で処理して、振盪することによって混合した。1時間後、各々のバイアルの3μLのアリコートをHPLCに注入して、UVピーク領域を出発物質、ならびに化合物1101の加水分解の所望の生成物および副産物について決定した。6、6.5、7、7.5および8というpHのサンプルのみが、所望の骨標的化プロドラッグA046−89P(保持時間2.50分;[M+]1075m/z C42H59N6O19P4について実測)[M+2]/2=538 m/zも実測)の有意な量を有することが見出された。本実施例によって、これらの条件下の骨標的化プロドラッグの合成のための最適pHはpH=7.0であり、4つのホスホン酸基を有する化合物1の所望の骨標的化プロドラッグの約42%という理論収率が得られることが実証される。24時間後、pH=7.0の時間を示す同じ溶液の分析によって、標的化プロドラッグが化合物1に完全に戻されたことが示され、これによって生理学的に関連する条件下での可逆性が実証される。
【0293】
実施例64
非小細胞肺癌に対する化合物1126のインビボ有用性
約30グラムの体重の4〜6週齢の雄性ヌードマウスに、0日目に5百万の腫瘍細胞(ヒト非小細胞肺癌細胞:H1299)を右脇腹に皮下に接種した。腫瘍細胞を増殖させた14日後、動物を各5匹の3群に分けた。1群にはビヒクルコントロールのみを与えた。1群にはプロドラッグ(すなわち、化合物1)の活性成分の25mg/kg/日用量レベルに対応して、24.4ミリモル溶液の化合物1126を含むリン酸緩衝化生理食塩水の50μLの容積の1日2回尾静脈(i.v)注射を与えた。最終群には、プロドラッグ(化合物1)の活性成分の5mg/kg/日の投薬レベルに対応して、4.9ミリモル溶液の化合物1126を含むリン酸緩衝化生理食塩水の50μLの容積の1日2回尾静脈(i.v)注射を与えた。ノギスを用いて3日ごとに腫瘍を測定して、腫瘍容積を決定して、27日で動物を屠殺したときに動物の重量を記録した。その結果を表7に示すが、これによって処置のわずか3日後に最初のデータポイントで(17日目)、そしてこの研究の終わりまで続く、両方の用量レベルについてのコントロールに対する強力な腫瘍容積の低下が示される:
表7
【0294】
【化95】
*ビヒクルのみのコントロール動物に対して比較
1日2回の用量は、2週間の投与期間にわたって十分耐容された。上記の有効性の結果ではまた、コントロール群とこの処置群との間で動物の体重の統計学的に有意な相違を伴わず、これによって、全体の毒性の一般的な測定として、標的されたプロドラッグが所望のとおり毒性を欠失していたことが示される。
【0295】
実施例65
脳の癌に対する化合物1126のインビボ有効性
ヒト脳癌細胞株(U87MG)を用い、初回の腫瘍容積測定を10日目に行なうように7日目に処置を開始すること以外は上記の実施例に記載のとおり動物実験を行なった。この癌細胞株に対する化合物1126の結果を、表8に示しており、そして有効性および望まれるような毒性の欠失が示された:
表8
【0296】
【化96】
*ビヒクルのみのコントロール動物に対して比較
実施例66
α v標的化PI 3キナーゼインヒビターは、Matrigel上でEDC−CBF1内皮細胞の管腔形成を抑制した
管腔形成は、インビボである程度の血管形成の形成を示す。本実施例では、どの程度PI 3キナーゼインヒビター(標的化PI3キナーゼインヒビタープロドラッグを含む)が管腔形成を阻害できるかを決定した。Matrigelを12ウェルプレートウェルにプレートして、37℃で2時間固めた。次いで1×105 EDC−CBF1内皮細胞をPBS、RADfV(環状陰性コントロールペプチド)、RGDfV(環状陽性コントロールペプチド)、RADS(直線状陰性コントロールペプチド)、化合物1または化合物1126の存在下でMatrigel層の上に20°M濃度で一晩置いた。次いで顕微鏡を用いて写真を撮った。十分形成された管腔はPBSコントロールウェル中で可視化することができる(図の上左側のパネル)。これらはPBSコントロールに比較してRGDfV、RADfVまたはRGDS含有ウェルでそれほど異なることはなかった。管腔形成は、化合物1101および化合物1126を含有するウェルでは有意に少なかった。
【0297】
実施例67
標的化PI3キナーゼインヒビターは、HBECにおいてp53転写活性を誘導した
本実験で、p53ルシフェラーゼ活性の誘導に対するPI 3キナーゼインヒビター(化合物1および化合物1の標的化プロドラッグバージョン;化合物1126)の効果を試験した。このトランスフェクション手順は、p53転写をモニターするために文献に記載された手順と同様であった。化合物1(6時間曝露)は、コントロールよりも2倍高いルシフェラーゼ活性を誘導し、そして化合物1の標的化バージョン(化合物1126)はp53ルシフェラーゼ活性を誘導する能力がさらに優れていた(ほぼ3倍)。p53機能のこの誘導は、p53インヒビターであるピフィスリン(pifithrin)αの20μM濃度によって抑止されることが実証された。触媒活性Aktの同時トランスフェクションはまた、これらの化合物によって誘導されたp53機能を阻害した。この結果によって、PI3キナーゼインヒビターによって誘導されたp53転写が、全シグナル伝達カスケードにおいてAktの下流であり、標的化プロドラッグ化合物1126が未標的薬物である化合物1に対してp53の誘導を増強したことが示される。
【0298】
実施例68
化合物1126の精製
反応混合物A044−84(2.33g)を、別々の0.33gのサンプルに秤量して、1容積部のアセトニトリル、1容積部の水、および1容積%の酢酸を含む800μlの溶液中に分取クロマトグラフィーの直前に溶解した。この溶液の400μlを各々の分取クロマトグラフィー実施について注入した。ポンプA溶離剤は0.1%酢酸を添加したB&J水(365−4)であって、ポンプB溶離剤は、0.1%酢酸を添加したB&Jアセトニトリル(015−4)であった。最初に、溶離剤は10%Bであって、次いで4分の期間にわたって直線的に34%Bに増大し、次いで4.25分で直線的に95%Bまで増大し、5.25分まで保持し、次いで5.50分で10%Bの開始濃度に直線的に低下して戻った。総ポンプ流量は20mL/分であった。自動サンプリング装置が次回のためにサンプリングしている間にシステムの再平衡を達成した。この勾配を用いて、正の質量スペクトルピークを853(m/z=1)および427(m/z=2)を有する生成物が3.37分で溶出した。ELSDで検出されたシグナルが10mvを超えた場合、分取クロマトグラフィー実施の間に画分を収集した。生成物を含む画分は、2倍過剰の水(容積で)を用いて溶出して、収集の直後にドライアイス−アセトン浴を用いて凍結乾燥容器中で凍結させた。24〜48時間にわたる凍結乾燥後、95%の純度を有する白色のフワフワした固体として化合物1126の全部で180mgを得た。本実施例によって、注意深いpH制御で不安定なプロドラッグ1126は、水性ベースの逆相分離方法を用いて高い純度で単離できることが実証される。
【0299】
実施例69
SCN反応性プロドラッグの調製
【0300】
【化97】
実施例20の方法によって調製したA014−48の184mg部を、154mgの化合物1とともに12mLのアセトニトリルに溶解して、室温で撹拌した。16時間後、LC−MSによって、所望の四級アンモニウム化合物への約65%の変換が示されたので、さらに45mgのヨード化合物を添加して、さらに22時間後に反応は80%終了したので、さらに40mgのヨード化合物を添加して、さらに24時間撹拌させた。次いでこの固体を遠心分離して、アセトニトリルで洗浄して、2.89分の保持時間で特徴付けられる、282.6mg(45%収率)の所望の生成物が高純度で得られ、これは、化合物COMPOUND1105としてC28H23N2O5Sについて499という所望のM+を示した。
【0301】
メタノールに入れた際、化合物1105はメタノールときれいに反応して、保持時間2.78分であってC29H27N2O6Sについて531という予想されるM+で化合物A013−94が得られた。これによってアルコールとのイソチオシアネートプロドラッグの反応がリンカーに対するカルバミン酸塩結合を生じることが示される。
【0302】
このイソチオシアネート中間プロドラッグはまた、塩化メチレン(必要に応じてトリメチルアミンの存在)中で二級アミンを含む種々のアミンと反応してA017−55(C35H38N3O7Sについて644m/zという所望のM+を示す、保持時間2.84分);A017−57(C33H34N3O7Sについて616m/zという所望のM+を示す、保持時間2.46分)およびA027−15(C38H48N3O11P2Sについて816m/zという所望のM+を示す、保持時間2.82分)のような尿素生成物が生じる。これらの実施例によって、所望の連結されたプロドラッグを生じるための求核試薬の多様な基を有するイソチオシアネートプロドラッグの良好な収率での反応が実証される。A017−55化合物はさらに、トリフルオロ酢酸と反応して、驚くべき事にt−ブチル基の切断の経過の間に環状化合物A017−59が得られた(C31H28N3O6Sについて570m/zという所望のM+を示す保持時間2.47分)。本実施例によって、さらなる化学がプロドラッグ化合物のリンカー基上で行なわれ得るということが実証された。ホスホン酸塩緩衝液中でpH=7.4で、LC−MSによって追跡したところ、この化合物のかなりの量が17時間にわたって化合物1に戻った。
【0303】
実施例70
化合物1対標的化プロドラッグの急性毒性
静脈内に投与された化合物1は3匹のヌードマウスにおいて5分内の投与で16mg/kgという用量レベルで100%の死亡率を有することが確認された。実施例56の方法によって調製された化合物1126は、3匹のヌードマウスに37%より高い用量レベル(22mg/kg)で静脈内注射された場合、観察の1時間後でさえ0%の死亡率を示した。本実施例は、このプロドラッグの処方物の能力の改善および化合物1のプロドラッグの毒性の減少を示す。
【0304】
実施例71
化合物1のアナログ
【0305】
【化98】
実施例56の化合物1126について記載されたような中間体A046−67SMの調製の間、副産物が観察された。この物質をLC−MSによって精製して、単独の化合物A037−94を得たが、この化合物は提案された構造のプロトンNMRと一致し、3.00分という保持時間を与え、そして質量スペクトルは338m/z(極めて低値)という予想される[M+H]+および379m/zのそれより大きな[M+H+41(ACN)]を示した。本実施例によって、さらなるプロドラッグ修飾に適切な化合物1の新規なアナログの単離が示される。
【0306】
実施例72
マウスにおいて化合物1を送達するためのプロドラッグの使用
マウスに100万個の非小細胞肺癌細胞(H1299)を皮下注射して、腫瘍塊が約10〜15mm×7〜9mmの直径になるまで約7日間増殖させた。動物に標的化プロドラッグ化合物1126を、i.v.(50μL)またはi.p.(50μL)のいずれかで、32.6ミリモル溶液の化合物1126を含むリン酸緩衝化生理食塩水とともに注射した。60分後、マウスを屠殺して、腫瘍を取り出した。腫瘍の3つの小片を取り出して切り刻んだ。プロドラッグの全てを化合物1に変換させるために24時間経過した後、腫瘍サンプルをアセトニトリルで抽出した。LC−MSによる定量によって、抽出可能な化合物1の濃度(遊離の化合物1および化合物1126由来の合計として)は、I.P.注射のための腫瘍片中で157±7ナノモル、そしてI.V.注射のための腫瘍片中で271±17ナノモルであったことが示された。本実施例によって、標的化プロドラッグを用いる腫瘍組織への化合物1の送達が実証される。
【0307】
実施例73
化合物1を形成するためのプロドラッグの可逆性
プロドラッグ化合物を、水またはDMSOに(水に自由には溶解しない場合)溶解し、次いで、50mMリン酸緩衝液pH=7.4またはpH=4.8中に少なくとも10倍に希釈して、室温で静置させた。この化合物の最終濃度は水性環境中で50〜500μモルに及んだ。アリコートを経時的に採取して、LC−MSによって分析し、プロドラッグの消失の確認および薬物(化合物1)の出現の確認の両方を行なった。化合物1126は、pH=7.5で約1時間の半減期、そしてpH=4.8で約64時間の半減期を有することが見出された。化合物1110は、pH=7.4で約10時間、そしてpH=4.8で120時間を超える半減期を有することが見出された。化合物A040−70は、pH=7.4で約10時間の半減期を有することが見出された。これらの実験によって、化学的にプロドラッグが薬物(化合物1)に変換し、このプロドラッグの消失は極めてpH依存性であり、この変換は生理学的なpHで極めて急速に生じ、そして酸性のpHで実質的に遅いことが実証される。
【0308】
実施例74
腫瘍に局在する結合体の合成
求電子性の基を保有する化合物(例えば、化合物A036−48B、1105、1107、1111、A024−79および1113)は、アルコール、アミノおよびチオール基のような求核性基を保有するポリマーと反応し得る。2000〜100,000の分子量を有するN−(2−ヒドロキシプロピル)メチルアクリルアミド(HPMA)は、トリエチルアミンまたはジイソプロピルエチレンアミンの存在下で塩化メチレンまたはテトラヒドロフランのような非プロトン性有機溶媒中で過剰の化合物1111と反応され、次いでサイズ排除クロマトグラフィー、超遠心分離、またはメタノールもしくはエーテルのような別の溶媒中での沈殿によって分離される。このように沈殿または分離されたポリマーは実質的に1111を含まず、そして腫瘍の近位で経時的に活性な化合物1を遊離して、抗腫瘍効果および血管新生阻害効果を生じる、腫瘍に局在する結合体として用いられる。同様にポリグルタミン酸は、カルボジイミドカップリングを用いる、カルボン酸と過剰なジアミンとの反応によってポリ求核性保有基に変換され得、続いてサイズ排除クロマトグラフィーまたは逆相HPLC精製によってポリグルタミン酸のポリ求核性バージョンが得られる。次いで、これらのポリマーは、トリエチルアミンまたはジイソプロピルエチレンアミンの存在下で塩化メチレンまたはテトラヒドロフランのような非プロトン性有機溶媒中で過剰部分の化合物1111または1105またはA036−48BまたはA024−79と直接的に反応され得、次いでサイズ排除クロマトグラフィー、超遠心分離、またはメタノールもしくはエーテルのような別の溶媒中での沈殿によって分離され得る。このように沈殿または分離されたポリ結合体化ポリマーは実質的に低分子量の残留プロドラッグを含まず、そして腫瘍の近位で経時的に活性な化合物1を遊離して、抗腫瘍効果および血管新生阻害効果を生じる、腫瘍に局在する結合体として用いられる。
【技術分野】
【0001】
発明の背景
1.発明の分野
本発明は、PI−キナーゼインヒビターのプロドラッグおよびこれらのインヒビターを用いる方法に関する。
【背景技術】
【0002】
2.関連技術の説明
PI−3キナーゼは、D3位のホスファチジルイノシトールをリン酸化して重要な二次メッセンジャーであるホスファチジルイノシトール3’−リン酸を生成する、大きいファミリーの脂質キナーゼである。PI−3キナーゼファミリーのメンバーは、配列相同性および酵素触媒によって形成される生成物に基づいて3つのクラスに分類される。クラスIのPI−3キナーゼは2つのサブユニットからなる:110kdの触媒性サブユニットおよび85kdの調節性サブユニット。クラスIのPI−3キナーゼは、サイトカイン、インテグリン、増殖因子および免疫レセプターの下流の重要なシグナル伝達事象に関与しており、これによって、この経路の制御が重要な治療効果をもたらし得ることが示唆される。
【0003】
クラスIのPI−3キナーゼの阻害がアポトーシスを誘導し、インビボにおいて腫瘍誘
導性血管形成をブロックし、そして特定の腫瘍の放射線感受性を増大する。LY294002(2−(4−モルホリニル)−8−フェニル−4H−1−ベンソピラン−4−オン)(化合物1)は、クラスI PI−3キナーゼの周知の特異的なインヒビターであり、そして抗癌特性を有することが実証されている。
【0004】
【化17】
【発明の概要】
【発明が解決しようとする課題】
【0005】
しかし、LY294002の抗癌適用は、それが水溶性を欠くこと、および薬物動態が良くないことによって厳しく制限されている。さらに、LY294002は、組織特異的な特性を有さず、そして動物で急速に代謝されることが実証されている。これらの要因のせいで、LY294002は頻繁な間隔で投与する必要があり、従って正常な細胞でのPI−3キナーゼを阻害する能力も有し、それによって望ましくない副作用をもたらす。
【0006】
改善された薬物動態および薬力学的特性を有するクラスIのPI−3キナーゼインヒビターが依然として必要である。本発明は、これらの必要性をみたし、他の関連の利点を提供する。
【課題を解決するための手段】
【0007】
発明の要旨
本発明は、例えば、以下を提供する。
(項目1)
四級窒素の1つの結合が加水分解可能であり、該結合の加水分解後に、式:
【化1】
の化合物が得られる、四級窒素を含むプロ化合物であって、
ここで、
R1およびR2が独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3がH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5が独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成する、化合物。
(項目2)
R1−環A−R2が:
【化2】
からなる群より選択される、項目1に記載の化合物。
(項目3)
R4−N−R5が:
【化3】
からなる群より選択される、項目1に記載の化合物。
(項目4)
式:
【化4】
の化合物であって、
ここで、
環Aは、ベンゾであり;
Z1およびZ2が独立してSまたはOであり;
R1およびR2が独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3がH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5が独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6が、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
Lがリンカー基である;
化合物。
(項目5)
R1−環A−R2が:
【化5】
からなる群より選択される、項目4に記載の化合物。
(項目6)
R4−N−R5が:
【化6】
からなる群より選択される、項目4に記載の化合物。
(項目7)
R6が:
【化7】
からなる群より選択される、項目4に記載の化合物。
(項目8)
式:
【化8】
の化合物であって、
ここで、
環Aがベンゾであり;
Z1、Z2、Z3およびZ4が独立してSまたはOであり;
R1およびR2が独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3がH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5が独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6が、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
R7が−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−である;
化合物。
(項目9)
R1−環A−R2が:
【化9】
からなる群より選択される、項目8に記載の化合物。
(項目10)
R4−N−R5が:
【化10】
からなる群より選択される、項目8に記載の化合物。
(項目11)
R6が:
【化11】
からなる群より選択される、項目8に記載の化合物。
(項目12)
R7と四級アミンとの間の結合が加水分解可能である、項目8に記載の化合物。
(項目13)
式:
【化12】
の化合物であって、
ここで、
環Aがベンゾであり;
Z1およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
R7が−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;そして
Tが標的化因子を表す;
化合物。
(項目14)
R1−環A−R2が
【化13】
からなる群より選択される、項目13に記載の化合物。
(項目15)
R4−N−R5が:
【化14】
からなる群より選択される、項目13に記載の化合物。
(項目16)
R6が:
【化15】
からなる群より選択される、項目13に記載の化合物。
(項目17)
前記標的化因子が、ビタミン、ペプチド、タンパク質、リポソーム、骨親和性剤および軟骨親和性剤からなる群より選択される、項目13に記載の化合物。
(項目18)
前記ビタミンがフォレートまたはビタミンCである、項目17に記載の化合物。
(項目19)
前記ペプチドが、RGD、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択されるRGD含有ペプチドである、項目17に記載の化合物。
(項目20)
前記タンパク質が腫瘍特異的モノクローナル抗体またはそのフラグメントである、項目17に記載の化合物。
(項目21)
前記骨親和性剤が、ホスホネート、ホスホン酸、アミノメチルホスホン酸、ホスフェート、ポリホスフェートおよびハイドロキシアパタイト結合ポリペプチドからなる群より選択される、項目17に記載の化合物。
(項目22)
前記骨親和性剤が、EDTMP、DOTMP、ABDTMP、BAD、MTX−BP、CF−BP、(Asp)6、(Glu)6、アレンドロネート、パミドロネート、4−アミノブチルホスホン酸、1−ヒドロキシエタン−1、1−二リン酸、アミノメチレンビスホスホン酸、フィチン酸およびN,N−ビス(メチルホスホノ)−4−アミノ安息香酸である、項目21に記載の化合物。
(項目23)
式:
【化16】
の化合物であって、
ここで、
Xがハロ基であり;
Yが−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−を表し;
Z1およびZ2が独立してSまたはOであり;そして
n=0〜1である;
化合物。
(項目24)
XがClまたはIであり;
Yが−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり
Z1およびZ2が各々Oであり;そして
n=0である;
項目23に記載の化合物。
(項目25)
XがClまたはIであり;
Z1およびZ2が各々Oであり;そして
n=1である;
項目23に記載の化合物。
(項目26)
項目1〜21のいずれか1項に記載の化合物を精製する方法であって:
(a)少なくとも0.1(v/v)%の酸を含む溶液に該化合物を含む組成物を添加する工程と;
(b)該化合物を含む(a)の溶液をクロマトグラフィーシステムに加える工程と;
(c)該化合物を単離する工程と;
を包含する、方法。
(項目27)
前記クロマトグラフィーシステムがHPLCである、項目26に記載の方法。
(項目28)
PI−3キナーゼ活性に関連する条件に罹患している患者を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目29)
炎症性疾患を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目30)
前記組成物が1つ以上のさらなる治療剤をさらに含む、項目28に記載の方法。
(項目31)
前記1つ以上のさらなる治療剤が、アルキル化剤、代謝拮抗物質、アスパラギナーゼ、ビンクリスチン、ビンブラスチン、アントラサイクリン、微小管阻害剤、タキソール、ハーセプチン、ゲムシタビンおよびエトポシドからなる群より選択される、項目30に記載の方法。
(項目32)
患者においてアポトーシスを誘導するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目33)
腫瘍細胞の化学感受性を増強するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目34)
腫瘍細胞の放射線感受性を増強するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目35)
腫瘍誘導性の血管形成を阻害するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目36)
癌以外の疾患に関連する血管形成のプロセスを阻害するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目37)
膵炎を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目38)
潰瘍を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目39)
胃癌を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目40)
肝臓癌を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目41)
ステントの能力を改善するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目42)
加齢性黄斑変性症を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目43)
変異PTENに伴う状態を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目44)
高血圧を処置するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量を投与する工程を包含する、方法。
(項目45)
白血球の機能を破壊するための方法であって、白血球と、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量とを接触させる工程を包含する、方法。
(項目46)
前駆細胞の分化を抑制するための方法であって、前駆細胞と、項目1〜21のいずれか1項に記載の化合物を含む組成物の有効量とを接触させる工程を包含する、方法。
(項目47)
哺乳動物の細胞全体のホスファチジルイノシトール3キナーゼを阻害するための方法であって、その必要な患者に、項目1〜21のいずれか1項に記載の化合物を投与する工程を包含する、方法。
(項目48)
前記投与が、緩徐なI.V.注入による、項目47に記載の方法。
【0008】
本発明は、四級窒素の1つの結合が加水分解され、この結合の加水分解後に、式:
【0009】
【化18】
の化合物が得られる、四級窒素を含むプロ化合物に関しており、
ここで、
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成する。
【0010】
このプロ化合物は、式:
【0011】
【化19】
の化合物であってもよく、
環Aはベンゾであり;
Z1、およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
Lがリンカー基である。
この標的化因子は、ビタミン、ペプチド、タンパク質、リポソーム、骨親和性剤または軟骨親和性剤であってもよい。
【0012】
本発明はまた、式:
【0013】
【化20】
の中間化合物に関し、
ここで、
Xは、ハロ基、好ましくはClまたはIであり;
Yは、−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;
Z1およびZ2は独立してSまたはOであり;そして
n=0〜1である。
【0014】
本発明はまた、PI−3キナーゼ活性に関連する状態、炎症性疾患、加齢性黄斑変性症、変異PTENに伴う状態、高血圧、膵炎、潰瘍、癌の処置;白血球機能破壊;アポトーシス誘導;腫瘍細胞の化学感受性の増強;腫瘍細胞の放射線感受性の増強;腫瘍誘導性血管形成の阻害;癌以外の疾患に関連する血管形成プロセスの阻害;ステントの能力の改善;患者の細胞全体におけるホスファチジルイノシトール3キナーゼの阻害のためのプロ化合物の使用に関し、これには本発明のプロ化合物を含む組成物の有効量をその必要な患者に投与する工程を包含する。このプロ化合物は、緩徐なI.V.注入によって患者に投与され得る。
【0015】
本発明は、前駆細胞と、本発明のプロ化合物を含む組成物の有効量とを接触させる工程を包含する、前駆細胞の分化を抑制するための方法にも関する。
【0016】
本発明はまた、本発明のプロ化合物の精製に関し、これには少なくとも0.1(v/v)%の酸を含む溶液にプロ化合物を添加する工程を包含する。このプロ化合物を含む溶液を、好ましくはHPLCによってクロマトグラフにかけて、プロ化合物を単離する。
【図面の簡単な説明】
【0017】
【図1】図1は、EDTMP、DOTMP、ABDTMP、BAD、MTX−BPおよびCF−BPの化学構造を示す。
【図2】図2は、潜在的な骨標的化因子の化学構造を示す。
【図3】図3は、骨標的化因子のホスホン酸塩を修飾するための化学反応を示す。
【図4】図4は、骨標的化因子のホスホン酸塩を修飾するためのアルキル化反応を示す。
【図5】図5は、EDTMPおよびDOTMPを化学的に修飾するための概念を示す。
【図6】図6は、J774細胞におけるLY294002による食作用の阻害を示す。カラムは、食作用性係数、または食細胞応答の陽性な細胞の割合を示す。食作用性係数は、100J774細胞1個あたりに見出されるsRBC(ヒツジ赤血球)の数であり、そして食作用性細胞の%は、少なくとも1つのsRBCを食菌したJ774細胞の%である。誤差指示線は、平均の標準偏差に相当する。
【図7】図7は、化合物1126(A036−33)のUVおよびELSクロマトグラムを示す。
【図8】図8は、化合物1126(A036−33)の正の質量スペクトルを示す。
【図9】図9は、Avβ3標的化PI 3キナーゼインヒビターが、Matrigel上でのEDC−CBF1内皮細胞の管腔の形成を抑止することを示す。
【発明を実施するための形態】
【0018】
発明の詳細な説明
本発明の化合物、生成物および組成物、ならびに方法が開示され記載される以前には、本明細書において用いられる専門用語は、特定の実施形態を記載する目的のためだけであって、限定を意図するものではないということが理解されるべきである。本明細書および添付の特許請求の範囲において用いられる場合、単数形「1つの(a)、(an)」および「この、その(the)」とは、文脈が明白に他を示さない限り複数の言及を包含することに注意のこと。
【0019】
本出願を通じて、刊行物が言及される場合、これらの刊行物の開示はその全体が本出願に参考として援用され、本発明が属する当該分野の状況をさらに詳細に記載する。
【0020】
1.定義
本明細書において用いる場合、「分枝した(branched)」という用語は、1〜24個の骨格原子を含む基であって、この基の骨格鎖が、主な鎖から1つ以上の従属する分枝を含む基をいう。本明細書における好ましい分枝した基は1〜12個の骨格原子を含む。分枝した基の例としては、限定はしないが、イソブチル、t−ブチル、イソプロピル、−CH2CH2CH(CH3)CH2CH3、−CH2CH(CH2CH3)CH2CH3、−CH2CH2C(CH3)2CH3、−CH2CH2C(CH3)3などが挙げられる。
【0021】
本明細書において用いる場合、「分枝していない(unbranched)」という用語は、1〜24個の骨格原子を含む基であって、この基の骨格鎖が、直接の線状に伸びる基をいう。本明細書における好ましい分枝していない基は、1〜12個の骨格原子を含む。
【0022】
本明細書において用いる場合、「環状(cyclic)」または「シクロ(cyclo)」という用語は、単独で、または組み合わせて、1つ以上の閉じた環を有する基であって、不飽和であっても飽和であっても、3〜12個の骨格原子、好ましくは3〜7個の骨格原子の環を有する基をいう。
【0023】
本明細書において用いる場合、「低級(lower)」という用語は、1〜6個の骨格原子を有する基をいう。
【0024】
本明細書において用いる場合、「飽和された(saturated)」という用語は、その骨格原子の全ての利用可能な原子価結合が、他の原子に結合されている基をいう。飽和された基の代表的な例としては、限定はしないが、ブチル、シクロヘキシル、ピペリジンなどが挙げられる。
【0025】
本明細書において用いる場合、「不飽和の(unsaturated)」という用語は、2つの隣接する骨格原子の少なくとも1つの利用可能な原子価結合が、他の原子に結合されていない基をいう。不飽和基の代表的な例としては、限定はしないが、−CH2CH2CH=CH2、フェニル、ピロールなどが挙げられる。
【0026】
本明細書において用いる場合、「脂肪族(aliphatic)」という用語は、分枝していない、分枝した、または環状の基を指し、これは置換されてもされなくてもよく、そして飽和されても飽和されなくてもよいが、ただし芳香族ではない。脂肪族という用語はさらに、脂肪族基を包含し、これは、炭化水素骨格の1つ以上の原子を置き換える、酸素、窒素、イオウまたはリン原子を含む。
【0027】
本明細書において用いる場合、「芳香族(aromatic)」という用語は、置換されても置換されなくてもよいが、4n+2の非局在化π(パイ)電子を有する、不飽和環状炭化水素基を指す。芳香族という用語はさらに、炭化水素骨格の1つ以上の炭素を置換する窒素原子を含む、芳香族基を包含する。芳香族基の例としては、限定はしないが、フェニル、ナフチル、チエニル、フラニル、ピリジニル、(イソ)オキサゾイルなどが挙げられる。
【0028】
本明細書において用いる場合、「置換された(substituted)」という用語は、炭素または適切なヘテロ原子を取り外して、さらなる基で置換した、1つ以上の水素または他の原子を有する基をいう。本明細書において、好ましい置換基は、1〜5個、最も好ましくは1〜3個の置換基で置換される。2つの置換基を有する原子は、「ジ(di)」で示されるが、3つ以上の置換基を有する原子は、「ポリ(poly)」と示される。このような置換基の代表的な例としては、限定はしないが、脂肪族基、芳香族基、アルキル、アルケニル、アルキニル、アリール、アルコキシ、ハロ、アリールオキシ、カルボニル、アクリル、シアノ、アミノ、ニトロ、リン酸含有基、イオウ含有基、ヒドロキシル、アルキルカルボニルオキシ、アリールカルボニルオキシ、アルコキシカルボニルオキシ、アリールオキシカルボニルオキシ、アルキルカルボニル、アリールカルボニル、アルコキシカルボニル、アミノカルボニル、アルキルアミノカルボニル、ジアリールアミノカルボニル、アルキルチオカルボニル、アシルアミノ、アミジノ、イミノ、アルキルチオ、アリールチオ、チオカルボキシレート、アルキルスルフィニル、トリフルオロメチル、アジド、ヘテロシクリル、アルキルアリール、ヘテロアリール、セミカルバジド、チオセミカルバジド、マレイミド、オキシミノ、イミダート、シクロアルキル、シクロアルキルカルボニル、ジアルキルアミノ、アリールシクロアルキル、アリールカルボニル、アリールアルキルカルボニル、アリールシクロアルキルカルボニル、アリールホスフィニル、アリールアルキルホスフィニル、アリールシクロアルキルホスフィニル、アリールホスホニル、アリールアルキルホスホニル、アリールシクロアルキルホスホニル、アリールスルホニル、アリールシクロアルキルスルホニル、アクリルシクロアルキルスルホニル、それらの組み合わせ、およびそれらの置換が挙げられる。
【0029】
本明細書において用いる場合、「置換されていない、非置換(unsubstituted)」という用語は、そこに結合された、またはそれに代わって置換されたいかなるさらなる基も有さない基をいう。
【0030】
単独でまたは組み合わせて本明細書において用いる場合、「アルキル」という用語は、分枝した、または分枝していない、飽和脂肪族基をいう。アルキル基の代表的な例としては、限定はしないが、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、イソブチル、sec−ブチル、tert−ブチル、オクチル、デシル、テトラデシル、ヘキサデシル、エイコシル、テトラコシルなどが挙げられる。
【0031】
単独でまたは組み合わせて、本明細書において用いる場合、「アルケニル(alkenyl)」という用語は、鎖にそった任意の適切なポイントで生じ得る、少なくとも1つの炭素間二重結合を含む、分枝した、または未分枝の不飽和の脂肪族基を指す。アルケニル基の代表的な例としては、限定はしないが、エテニル、E−およびZ−ペンテニル、デセニルなどが挙げられる。
【0032】
単独でまたは組み合わせて本明細書において用いる場合、「アルキニル(alkynyl)」という用語は、鎖にそった任意の適切なポイントで生じ得る、少なくとも1つの炭素間三重結合を含む、分枝した、または未分枝の、不飽和の脂肪族基を指す。アルキニル基の代表的な例としては、限定はしないが、エチニル、プロピニル、プロパルジル、ブチニル、ヘキシニル、デシニルなどが挙げられる。
【0033】
単独でまたは組み合わせて本明細書において用いる場合、「アリール(aryl)」という用語は、他の芳香族または非芳香族の環状基に必要に応じて縮合され得る、置換した、または置換されていない芳香族基を指す。アリール基の代表的な例としては、限定はしないが、フェニル、ベンジル、ナフチル、ベンジリジン、キシリル、スチレン、スチリル、フェネチル、フェニレン、ベンゼントリイルなどが挙げられる。
【0034】
単独でまたは組み合わせて本明細書において用いる場合、「アルコキシ(alkoxy)」という用語は、単独の末端エーテル結合を通じて結合されたアルキル、アルケニルまたはアルキニル基をいう。アルコキシ基の例としては、限定はしないが、メトキシ、エトキシ、n−プロポキシ、イソ−プロポキシ、n−ブトキシ、2−ブトキシ、tert−ブトキシ、n−ペントキシ、2−ペントキシ、3−ペントキシ、イソペントキシ、ネオペントキシ、n−ヘキソキシ、2−ヘキソキシ、3−ヘキソキシ、3−メチルペントキシ、フルオロメトキシ、ジフルオロメトキシ、トリフルオロメトキシ、クロロメトキシ、ジクロロメトキシおよびトリクロロメトキシが挙げられる。
【0035】
単独でまたは組み合わせて本明細書において用いる場合、「アリールオキシ(aryloxy)」という用語は、単独の末端エーテル結合を通じて結合されたアリール基をいう。
【0036】
単独でまたは組み合わせて本明細書において用いる場合、「ハロゲン(halogen)」、「ハライド(halide)」または「ハロ(halo)」という用語は、フッ素「F」、塩素「Cl」、臭素「Br」、ヨウ素「I」およびアスタチン「At」を指す。ハロ基の代表的な例としては、限定はしないが、クロロアセトアミド、ブロモアセトアミド、ヨードアセトアミドなどが挙げられる。
【0037】
組み合わせて本明細書において用いる場合、「ヘテロ(hetero)」という用語は、炭素または水素以外の任意の元素の1つ以上の原子を含む基をいう。ヘテロ基の代表的な例としては、限定はしないが、窒素、酸素、イオウおよびリンを含むヘテロ原子を含む基が挙げられる。
【0038】
本明細書において用いる場合、「複素環(heterocycle)」という用語は、ヘテロ原子を含む環状基をいう。複素環の代表的な例としては、限定はしないが、ピリジン、ピペラジン、ピリミジン、ピリダジン、ピペラジン、ピロール、ピロリジノン、ピロリジン、モルホリン、チオモルホリン、インドール、イソインドール、イミダゾール、トリアゾール、テトラゾール、フラン、ベンゾフラン、ジベンゾフラン、チオフェン、チアゾール、ベンゾチアゾール、ベンゾキサゾール、ベンゾチオフェン、キノリン、イソキノリン、アザピン、ナフトピラン、フラノベンゾピラノンなどが挙げられる。
【0039】
単独でまたは組み合わせて、本明細書において用いる場合、「カルボニル(carbonyl)」または「カルボキシ(carboxy)」という用語は、炭素−酸素間二重結合を含む基をいう。カルボニルを含む基の代表的な例としては、限定はしないが、アルデヒド(すなわち、ホルミル)、ケトン(すなわち、アシル)、カルボン酸(すなわち、カルボキシル)、アミド(すなわち、アミド)、イミド(imide)(すなわち、イミド(imido))、エステル、無水物などが挙げられる。
【0040】
単独でまたは組み合わせて本明細書において用いる場合、「アクリル(acryl)」という用語は、Qが脂肪族基または芳香族基である、CH2=C(Q)C(O)O−によって示される基をいう。
【0041】
単独でまたは組み合わせて本明細書において用いる場合、「シアノ(cyano)」、「シアン酸塩(cyanate)」または「シアン化物(cyanide)」という用語は、炭素−窒素間二重結合をいう。シアノ基の代表的な例としては限定はしないが、イソシアン酸塩、イソチオシアン酸塩などが挙げられる。
【0042】
単独でまたは組み合わせて本明細書において用いる場合、「アミノ(amino)」という用語は、骨格の窒素原子を含む基をいう。アミノ基の代表的な例としては、限定はしないがアルキルアミノ、ジアルキルアミノ、アリールアミノ、ジアリールアミノ、アルキルアリールアミノ、アルキルカルボニルアミノ、アリールカルボニルアミノ、カルバモイル、ウレイドなどが挙げられる。
【0043】
本明細書において用いる場合、「リン酸塩含有基(phosphate−containing group)」という用語は、酸化状態で少なくとも1つのリン原子を含む基をいう。代表的な例としては、限定はしないが、ホスホン酸、ホスフィン酸、リン酸エステル、ホスフィニデン、ホスフィノ、ホスフィニル、ホスフィニリデン、リン酸、ホスホノ、ホスホラニル、ホスホラニリデン、ホスホロソなどが挙げられる。
【0044】
本明細書において用いる場合、「イオウ含有基(sulfur−containing
group)」という用語は、イオウ原子を含む基をいう。代表的な例としては限定はしないが、スルフヒドリル、スルフェノ、スルフィノ、スルフェニル、スルホ、スルホニル、チオ、チオキソなどが挙げられる。
【0045】
本明細書において用いる場合、「必要に応じた(optional)」または「必要に応じて(optionally)」という用語は、引き続いて記載される事象または状況が生じても生じなくてもよいことを意味し、そしてこの説明は、この事象または状況が生じる場合、および起こらない場合を包含する。例えば、「必要に応じて置換されたアルキル(optionally substituted alkyl)」という句は、アルキル基が置換されてもされなくてもよいことを意味し、そしてこの説明は置換されていないアルキルおよび置換のあるアルキルの両方を包含する。
【0046】
本明細書において提供されるような化合物、生成物または組成物に関して用いる場合、「有効量(effective amount)」という用語は、所望の結果を得るために十分な化合物、生成物または組成物の量を意味する。必要な正確な量は、用いられる特定の化合物、生成物または組成物、投与の様式などに依存して変化する。従って、正確な「有効量」を特定することは常に可能ではない。しかし、適切な有効量は、慣用的な実験のみを用いて、本開示から知識を得た当業者によって決定できる。
【0047】
本明細書において用いる場合、「適切な(suitable)」という用語は、言及した目的について本明細書において提供されたような化合物、生成物または組成物と適合性である基をいう。言及した目的についての適切性は、慣用的な条件だけを用いて当業者によって決定できる。
【0048】
本明細書において用いる場合、「加水分解性(hydrolyzable)」という用語は、この基が加水分解できるか、または加水分解しやすい(すなわち、この分子または基の2つ以上の新しい分子または基への分離)。ことをいう。
【0049】
2.化合物
本発明は、四級アミンの1つの結合の切断の際に、式:
【0050】
【化21】
の化合物が生じる化合物を提供するが
ここで、
環Aがベンゾであり;
Z1、およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成する。
好ましくは、四級アミンの1つの結合の切断は、LY294002(化合物1)を生じる。
【0051】
本発明はまた、式:
【0052】
【化22】
の化合物を提供し、
ここで、
環Aがベンゾであり;
Z1、およびZ2は独立してSまたはOであり;
R1およびR2は独立してH、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノであるか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3はH、必要に応じて置換された脂肪族および必要に応じて置換されたアリールであり;そして
R4およびR5は独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、カルボキシであるか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;そして
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、アルコキシ、カルボキシ、アミノ、複素環式、アリールオキシ、およびそこで必要に応じて置換された標的化因子であり;そして
Lがリンカー基である。
【0053】
好ましい実施形態では、本発明の化合物2〜3は、R1−環A−R2が以下:
【0054】
【化23】
からなる群より選択される化合物である。
【0055】
好ましい実施形態では、本発明の化合物2〜3は、R4−N−R5が以下:
【0056】
【化24】
からなる群より選択される化合物である。
【0057】
別の実施形態では本発明の化合物2〜3は、R6が以下:
【0058】
【化25】
からなる群より選択される化合物である。
【0059】
a.リンカー
別の実施形態では、本発明の化合物3は、リンカー基が加水分解可能である化合物である。プロドラッグのリンカー基は、酵素切断によって、または好ましくは、生存している動物での水性条件を含むがこれに限定されない生理学的条件下での加水分解によって切断されて、化合物2が生じ得る。生理学的条件下でのリンカー基の加水分解の速度は好ましくは、約1分の半減期から約48時間の半減期である。
【0060】
b.加水分解
本明細書において用いる場合、「加水分解可能、加水分解性(hydrolyzable)」という用語は、約1分の半減期から約48時間の半減期という速度で、加水分解(すなわち、水分子の正味の挿入に起因する、その分子または基の2つ以上の新規な分子または基への分離)され得るかまたは加水分解し易いことをいう。
【0061】
リンカー基とは、化合物2を生じるように加水分解または酵素的に切断されてもよい任意の基であり得る。好ましい実施形態では、リンカー基は、以下の式:
【0062】
【化26】
であり、ここでZ3およびZ4は独立してSまたはOであり;そして
R7が−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−である。
【0063】
化合物2を得るためのプロドラッグのリンカー基の加水分解の代表的な例は、(スキーム1)に示され、ここでZ3およびZ4は独立して各々Oである。R6エステルの加水分解または酵素切断は、R7アルデヒドの遊離で崩壊するヘミアミナル(hemiaminal)を生じ、これによって遊離の三級アミンを含む化合物2が生じる。R6およびR7の両方とも、遊離の三級アミンに戻る種々の率が得られるように選択され得る。例えば、R6もしくはR7での置換の増大、またはその組み合わせが加水分解に向かう安定性を増大し得る。さらに、R6部分の電子求引性基は安定性を低下させる。本明細書に開示されるR6またはR7の変化に加えて、四級アミンの安定性を変化し得るさらなる要因は、その内容が参考として本明細書に援用される、N.Bodor,Journal of Medical Chemistry 1980,vol 23 #5、pp469〜480「Soft Drugs.1.Labile Quaternary Ammonium Salts as Soft Antimicrobials」;およびG.Brouillette et al.;Journal of Pharmaceutical Sciences,1996,vol 85 #6,pp620−623にみることができる。
【0064】
スキーム1
【0065】
【化27】
c.標的化因子
別の実施形態では、本発明の化合物は、R6がさらにそれに共有結合した1つ以上の標的化因子(T)を含む化合物である。標的化因子によって本発明のプロドラッグは特定のタイプの細胞、組織、器官または細胞外構造に選択的に送達されることが可能になる。上記で考察されるとおり、化合物1(LY294002)での処理は、バイオアベイラビリティー、急速な代謝および副作用という点で劣る。なぜなら、この化合物は組織特異的ではないからである。従って、副作用が生じ得る場合、薬物の位置を処置の領域の位置に対して限定すること、または薬物が組織に達することを少なくとも妨げること、そして任意の特定の時間で有効であるが過剰ではない量の薬物が用いられることを確実にすることが極めて所望される。標的化剤の使用によって、本発明のプロドラッグは、身体全体を通じて均一に分布するのではなく、処置の部位で濃縮されること、または早期に代謝されるかもしくはさらに急速に排出されることが可能になる。処置の部位に一旦送達されれば、リンカーを、酵素的に切断してもよいし、または上記のように加水分解して、化合物2を得てもよい。さらに、標的化因子の使用によって、処置の部位で薬物の有効濃度を達成するために投与することが必要な投薬量が制限され得る。標的化因子の使用はまた、処置の部位で薬物の有効濃度を得るために、さらに頻繁な投薬またはさらに別の投与方法を可能にする。
【0066】
標的化因子は、共有結合を介して、本発明の化合物に優先的に結合するが、この共有結合は、リンカー上の(それぞれ)求電子性または求核性の基と共有結合的に反応される、この標的化因子の求核性または求電子性基を含むがこれに限定されない方法によって形成され得る。
【0067】
本発明の1実施形態では、本発明の化合物2〜3は、R6〜Tが以下:
【0068】
【化28】
からなる群より選択される化合物である。
【0069】
本発明のプロドラッグと反応し得る標的化因子としては、限定はしないが、炭水化物、ビタミン、ペプチド、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤および軟骨親和性剤が挙げられる。標的化因子はまた、所望の組織におけるレセプターによって結合されて、レセプター媒介プロセスによって細胞に必要に応じて輸送される分子であってもよい。このような標的化因子の代表的な例としては、脳のグリア細胞に存在する末梢ベンゾジアゼピンレセプター(PBR)に結合するジアゼピンが挙げられるがこれらに限定されない。このようなジアゼピンの代表的な例は、その内容が参考として本明細書に援用される、G.Trapani et al.Bioconjugate Chem.2003.vol 14,pp830−839「Peripheral Benzodiazepine Receptor Ligand−Melphalan Cojugates for Potential Selective Drug Delivery to Brain Tumors」に考察される。
【0070】
標的化因子として用いられ得る代表的なビタミンとしては、限定はしないが、フォレート、ビタミンB12、またはビタミンCが挙げられる。「フォレート(folate)」という用語は、葉酸受容体と結合する能力を有する葉酸誘導体を包含する。標的化因子として用いられ得るフォレートの代表的な例としては、限定はしないが、葉酸、フォリン酸、プテロポリグルタミン酸、および葉酸受容体結合プテリジン、例えば、テトラヒドロプテリン、ジヒドロフォレート、テトラヒドロフォレートおよびそれらのデアザアナログおよびジアザアナログが挙げられる。他の適切なフォレートは、フォレートアナログであり、これには限定はしないが、アミノプテリン、アメトプテリン(メトトレキセート)、N10−メチルフォレート、2−デアミノ−ヒドロキシフォレート、デアザアナログ、例えば、1−デアザメトプテリン、または3−デアザメトプテリンおよび3’5’−ジクロロ4−アミノ−4−デオキシ−N10−メチルプテロイル−グルタミン酸(ジクロロメトトレキセート)が挙げられる。本発明の化合物に対して共有結合するために適切なフォレートに分子を結合させる方法は、その内容が参考として本明細書に援用される、米国特許第6,576,239号、同第5,820,847号、同第5,688,488号、同第5,108,921号、同第5,635,382号および同第5,416,016号に開示される。本発明の化合物に対して共有結合するために適切なビタミンCに分子を結合させる方法は、その内容が参考として本明細書に援用されるS.Manfrdini J.Med.Chem.Vol 45、pp559〜562,2002に開示される。
【0071】
標的化因子として用いられ得る代表的なペプチドおよびペプチド模倣物としては、限定はしないが、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン−レセプターアゴニストおよびソマトスタチンレセプターアンタゴニストからなる群より選択されるRGD含有ペプチドが挙げられる。avb3インテグリンレセプターに結合して、アンタゴニストとして作用する分子は、その内容が本明細書において参考として援用される米国特許第6,552,079号、同第6,426,353B、WO2002/40505A2および米国特許公開2002/0055499、同第2002/0061885、同第2002/0065291、同第2002/0072500、U.S.2002/0072518;W.Arap et al.、Science、vol 279、number 16、1998、pp377〜380;RJ Kok et al.、Biojonjugate Chem.2002,vol 13、pp128〜135;DA Sipkins et al.、Nature Medicine vol
4、number 5、1998、pp623〜626;PM Winter et al.、Cancer Research 2003、vol 63、pp5838〜5843;ならびにJD Hood et al.、Science vol 296、pp2404〜2407に記載されるように標的化因子として用いられ得る。標的化因子として用いられ得る代表的なタンパク質としては、限定はしないが、抗体またはそのフラグメント、例えば、腫瘍特異的モノクローナル抗体またはそのフラグメントが挙げられる。標的化因子として用いられ得る代表的な骨親和性因子としては、限定はしないが、ホスホン酸塩、ホスホン酸、アミノメチルホスホン酸、リン酸塩、ポリリン酸塩およびハイドロキシアパタイト結合ポリペプチドが挙げられる。他のペプチドとしては、クロロトキシン(SU6,429,187B1)および組織因子(G.M.Lanza et al.、「Targeted Antiproliferative Drug Delivery to Vascular Smooth Muscle Cells with a
Magnetic Resonance Imaging Nanoparticle
Contrast Agent」;Circulation,2002、volume
106、pp2842〜2847)が挙げられる。
【0072】
他の適切な標的化因子としては、抗体が挙げられる。抗体は、IgG、IgM、IgA、IgDもしくはIgEのクラスの抗体、またはそのフラグメントもしくは誘導体であってもよく、これには、Fab、F(ab’)2、Fdおよび単鎖抗体、二重特異性抗体(diabody)、二重特異性抗体(bispecific antibody)、二機能性抗体、およびその誘導体が挙げられる。抗体は、所望のエピトープまたはそれに由来する配列に対して十分な結合特異性を示す、モノクローナル抗体、ポリクローナル抗体、アフィニティー精製抗体またはそれらの混合物であってもよい。この抗体はまた、キメラ抗体であってもよい。この抗体は、腫瘍、組織適合性および他の細胞表面抗原、細菌、真菌、ウイルス、酵素、毒素、薬物および他の生理学的に活性な分子と関連するものを含む、種々の抗原性決定基に関するものであり得る。抗体が特異的に反応し得る腫瘍と会合する抗原としては、限定はしないが、このような抗原が挙げられ、そして限定はしないが、癌胎児性抗原(CEA)、ムチン、例えばTAG−72、ヒト乳脂肪小球抗原、前立腺血清抗原(PSA)、前立腺特異的膜抗原(PSMA)、PS(ホスファチジルセリン)、ならびにIL−2、EGF、VEGFおよびトランスフェリンレセプターを含むがこれに限定されないレセプターが挙げられる。腫瘍に会合する他の代表的な抗原としては、限定はしないが、それらの内容が参考として本明細書に援用される、ZalcbergおよびMcKenzie,J.Clin.Oncology,Vol 3;pp876〜82(1985)、WO01/68709A1、および米国特許公開US2004/0009122A1に記載される腫瘍関連抗原が挙げられる。
【0073】
他の適切な標的化因子としては、グルコース、ガラクトース、マンノース、マンノース6−リン酸、ホルモン(例えば、インスリン、成長ホルモンなど)、増殖因子またはサイトカイン(例えば、TGFβ、EGF、インスリン様成長因子、など)、YEE(GalNAcAH).sub.3または誘導体、コバラミン、α−2マクログロブリン、アシアロ糖タンパク質、アルブミン、テキサフィリン、メタロテキサフィリン、抗体、抗体フラグメント(例えば、Fab)、単鎖抗体可変領域(scFv)、トランスフェリン、任意のビタミンおよび任意の補酵素が挙げられる。
【0074】
標的化因子はまた、骨にプロドラッグを送達する因子であってもよい。骨標的化因子としては、限定はしないが、EDTMP DOTMPおよびABEDTMPが挙げられ、これは、その内容が本明細書に参考として援用される、米国特許第4,937,333号、同第4,882,142号、同第5,064,633号およびWO−94/00143に開示される。DOTMPおよびEDTMPは、図3に示されるカップリング化学、および図4に示されるアルキル化化学を含むがこれに限定されない任意の方法によってリンカー部分に結合され得、ここではR基が、リンカー部分の(それぞれ)求核性基または求電子性基と反応する、適切な求電子性基または求核性基を有してもよい。カップリング化学のさらなる詳細は、その内容が参考として本明細書に援用される、Tetrahedron
1999,55,pp12997〜13010に提供される。アルキル化化学のさらなる詳細は、その内容が参考として本明細書に援用される、Proc.SPIE−Int.Soc.Opt.Eng.1999,3600(Biomedical Imagn.Receptors Dyes&Instrumental,pp99〜106;米国特許第5,177,054号;J.Med.Chem.1994,37,498〜511;Tetrahedron Letters,1989,30#51pp7141〜7144;および米国特許第5,955,453号に提供される。
【0075】
標的化因子は、本発明の化合物の徐放性リザーバ部位として骨に対してプロドラッグを送達するように用いられ得る。標的化因子は、四級アミンに結合する酸切断性リンカーを介して本発明の化合物に結合する骨親和性(骨向性(osteotropic))部分であってもよい。酸切断可能リンカーの例としては、限定はしないが、オルト酸−アミド結合が挙げられる。酸性の条件下では、タンパク質−ACL−3アミド結合は容易に切断されて、その内容が参考として援用されるWO−94/00143に記載されるように、アミド官能性の天然のアミノ基を遊離する。酸性媒介性機構に関与する骨破壊性の骨再吸収の間、骨に対してプロドラッグを拘束する結合が切断されて、本発明の化合物が遊離され得る。
【0076】
骨に対してプロドラッグを送達するために用いられる標的化因子は、ノッチレセプターと結合する分子であってもよい。ノッチシグナル伝達は、種々の造血系列の発達および分化に重要な役割を果たす。Jundt et al.、Blood、102(11):928a(2003)に考察されるように、リガンド誘導性ノッチシグナル伝達は、多発性骨髄腫細胞の新規な増殖因子であり、そしてインビボにおいて多発性骨髄腫のリンパ種発生にこれらの相互作用が寄与することが示唆される。
【0077】
骨標的化因子は、骨の主な構成要素である、ヒドロキシアパタイト中のカルシウムイオンに高い親和性を有し得る。本発明の化合物は、骨以外の体の領域におけるカルシウム沈着、例えば、動脈、心臓、腎臓または膀胱のカルシウム沈着に標的化され得る。しかし、骨標的化因子は理想的には骨組織に選択的に結合する。本発明の骨標的化因子は、被験体の骨組織に結合され、好ましくは骨以外の組織よりも高い親和性で骨に結合し、そして特定の時間にわたって結合を残し、これによって骨環境に対してこの組成物を送達する。言い換えれば、骨標的化因子は好ましくは、骨以外の組織に対してこの骨標的化因子が結合するよりも少なくとも2倍大きい(例えば、少なくとも3倍、少なくとも5倍、少なくとも10倍、または少なくとも25倍大きい親和性)親和性で骨組織に結合する。骨標的化因子は骨組織に可逆的に結合するが、このことはこの骨標的化因子が最終的には骨から遊離されて、身体から排出されることを意味する。
【0078】
骨標的化因子は、四級プロドラッグを加水分解することが可能になるのに十分な時間、骨組織に対する結合を維持し得、それによって標的細胞(例えば、骨髄細胞)に対して活性薬物を送達する。骨標的化因子は、約1日(例えば、約2日、約3日、または約7日)〜約1年(例えば、約330日、約365日、または約400日)間にわたって骨に対する結合を維持し得、その後、骨標的化因子が身体から排出される。骨標的化因子は、約7日(例えば、約7日、約14日または約21日)〜約6ヶ月(例えば、約90日、約120日、または約150日)間にわたって骨に対する結合を維持してもよい。例えば、骨標的化プロドラッグは、30日間骨に対して結合を維持し得、その間この薬物が放出される。約45日後、骨標的化因子は骨から遊離されて、最終的に排泄される。従って、本発明における使用のための骨標的化因子は、骨組織に対する結合動態に基づいて選択され得る。候補の骨標的化因子は、例えばマルチウェル形式で、骨組織(例えば、ハイドロキシアパタイト)に対する親和性を決定することによって、インビトロでスクリーニングできる。候補の骨標的化因子はまた、身体からの候補の骨標的化因子の排泄の速度およびタイミングを評価することによってインビボでスクリーニングされ得る。これに関して、骨標的化因子は好ましくは、腎臓を介して身体から排出される。
【0079】
骨標的化因子は望ましくは、リン酸塩、ホスホン酸塩、ビスホスホン酸塩、ヒドロキシビスホスホン酸塩、アミノメチレンホスホン酸、および酸性ペプチドからなる群より選択される。本発明の骨標的化因子は、これらの群のうち1つ、2つ以上またはその混合物を担持してもよい。例えば、骨標的化因子は、ホスホン酸塩であってもよく、このことは、この骨標的化因子は、ホスホン酸塩であってもよく、このことはこの骨標的化因子が1つのホスホン酸塩、2つのホスホン酸塩、または3つ以上のホスホン酸塩を含んでもよいことを意味する。本発明における使用のための適切な骨標的化因子の1つは、疼痛緩和のために骨転移に選択的な放射線用量を送達するための放射性の153Sm複合体として、EDTMP(その化学構造は、図1に示され、現在FDAが承認している、エチレンジアミン−N,N,N’,N’−テトラキス(メチレンホスホン酸)(Quadramet(商標)))である。EDTMPは、4つのホスホン酸基を含むリン酸塩であり、従って、四リン酸塩である。153Sm−EDTMPのような化合物は、腫瘍が存在する骨、対正常な骨に選択的に、10:1より大きい比で局在する。なぜなら好ましくはカルシウムの代謝回転は転移性領域において極めて高いからである。報告によれば、153Sm−EDTMPは、造骨細胞の骨転移において骨格によって急速に取り込まれて血漿から排出される。骨格において蓄積しない化合物の一部は、急速に排出され、排泄は投与後6時間内でほぼ完全である(Jimonet et al.、Heterocycles,36,2745(1993))。疼痛緩和は、隣接する転移性の腫瘍細胞にある程度の効果を有する、造骨細胞の骨転移に対して結合した同位体に由来する放射線に起因していると考えられる。別の臨床上有用な骨標的化システムは、多発性骨髄腫の処置のために骨髄に対して高用量の放射線量を選択的に送達するように設計された放射性の166Ho複合体として、DOTMP(その化学構造は、図1に示され、現在第III相臨床試験中の(STR、骨格標的化放射線(skeletal targeted radiation)と名付けられた)である。放射性の166Ho−DOTMP複合体は、骨格系に局在して、悪性の骨髄腫細胞を有する近接する骨髄を照射することに注目すべきである。153Sm−EDTMP系と同様に、骨に局在しないホスホン酸塩は、尿によって身体の外に排出される。一般には、骨格の取り込みは、注射された用量の約20〜約50%であり、そして腫瘍浸潤を有する骨の領域への局在は、Bayouth et al.,J.Nucl.Med.36,730(1995)の図7に図示される。
【0080】
好ましくは、骨標的化因子は、ポリホスホン酸である。ポリホスホン酸は、骨組織に対して生物学的に活性な分子を首尾よく標的することが実証されている。例えば、成長因子(骨形成を刺激する)に対する、ABDTMP(その化学構造は、図1に示される)のようなポリアミノホスホン酸の結合(イソチオシアネート化学による)によって、ラットの骨に対する増殖因子の標的化が首尾よく生じる(例えば、国際特許出願WO94/00145を参照のこと)。同様に、骨標的化因子は、タンパク質に結合している。例えば、ヒト血清アルブミンに対して結合するビスホスホン酸塩は、インビトロ(Biotechnol.Prog.16,258(2000))およびインビボ(Biotechnol.Prog.,16,1116(2000))において骨にタンパク質を首尾よく送達した。骨親和性因子の有用性は、骨へのタンパク質の送達を上回り、そして例えば、低分子の治療用分子を含む。骨親和性ビスホスホン酸塩およびアルキル化剤、例えば、BAD(その化学構造は図1に示す、を含む結合体が生成されている(例えば、Wingen et
al.,J.Cancer Res.Clin.Oncol.,111,209(1986)を参照のこと)。この分子では、アルキル化剤は、その標的(DNA)との相互作用において特異的ではなく、従って、ビスホスホン酸塩(すなわち、骨親和性剤)とアルキル化部分との間で切断の必要はない。ビスホスホン酸−アルキル化剤は、BADを用いるラット骨肉腫モデルにおいて有効性を実証した。MTX−BPと命名され、図1に示される、ビスホスホン酸塩に共有結合されている葉酸代謝拮抗、抗腫瘍性剤であるメトトレキセートを用いて、別の一連の研究を行なった(例えば、Sturtz et al.,Eur.J.Med.Chem.,27.825(1992);Sturtz et al.,Eur.J.Med.Chem.,28,899(1993);およびHosain et al.,J.Nucl.Med.37,105(1996)を参照のこと)。Tc−99m標識したMTX−BPを用いて、約15%の注射用量が約4時間後に骨格に局在し、このときこの用量の約61%が排出されていることが確認された(Hosain、前出)。MTX−BPはさらに、移植された骨肉腫の動物モデルにおいてメトトレキセート単独と比較して5倍大きい抗癌効果を実証した(Sturtz 1992、前出)。化学構造が図1に記載されている、付加されたビスホスホン酸塩とのカルボキシフルオレセイン基の結合体CF−BPを用いて、同様の研究が記載されている(Fujisaki
et al.,Journal of Drug Targeting,4,117(1994))。この分子では、CF基は、薬物動態および生体分布を定量するための蛍光性マーカーであり、インビボで加水分解しやすいエステル結合を通じて骨標的化因子に結合される。ラットでの静脈内注射の研究によって、CF−BPが骨に局在し、そしてエステル結合の一般的加水分解を介して、生成されたCFの徐放性機構として機能することが示された(Fujisaki、前出)。
【0081】
別の実施形態では、骨親和性因子は、(Asp)6および(Glu)6のようなペプチドであってもよい。骨および象牙質で富んでいることが見出されている糖タンパク質オステオネクチンの酸に富むペプチド配列は、ヒドロキシアパタイトに強い親和性を有する(Fujisawa et al.,Biochimica et Biophysica
Acta,53,1292(1996))。従って、酸性アミノ酸を含むペプチドリガンドは、骨標的化因子の理想的な候補物である。実際、(Glu)10は、ビオチンに結合された場合、標識されたストレプトアビジンを首尾よく補強した(Chu and Orgel、Bioconjugate Chem,8,103(1997)および国際特許出願WO98/35703にさらに記載される)。さらに、(Asp)6に結合したフルオレセインイソチオシアネートの生物学的半減期は、大腿骨では14日であり(Kasugai et al.,Journal of Bone and Mineral Research,15(5),936(2000))、これは、本発明の骨標的化因子にとって受容可能な半減期である。同様に、エストラジオール−(Asp)6結合体の骨への送達は、卵巣切除された動物で実証されており、ここでは骨粗鬆的なタイプの骨の損失の阻害を伴う(Kasugai et al.,Journal of Bone and Mineral Research(Suppl 1),14,S534(1999))。骨に対する(Asp)6結合は、破骨細胞によって媒介される骨吸収プロセスの間に代謝されると考えられる。従って、酸性のペプチドリガンドによって、骨に対する化合物の補充の手段が得られるだけでなく、骨細胞および周囲の組織に対して化合物を緩徐に放出する機構も得られる。
【0082】
骨標的化因子の他の例としては、限定はしないが、アミノおよびヒドロキシル−アルキルホスホン酸およびジホスホン酸;アレンドロン酸、パミドロン酸、4−アミノブチルホスホン酸、1−ヒドロキシエタン−1,1−ジホスホン酸およびアミノメチレンビスホスホン酸を含む、ヒドロキシルビスホスホン酸;フィチン酸のようなホスホン酸;ならびにアミノメチレンホスホン酸、例えば、N,N−ビス(メチルホスホノ)−4−アミノ安息香酸およびニトリロトリ(メチルホスホン酸);が挙げられる。このような骨標的化因子の非限定的な例は、図2に示される。
【0083】
好ましくは、骨標的化因子は、アミノメチレンホスホン酸である。「アミノメチレンホスホン酸(aminomethylenephosphonic acid)」とは、−NCH2PO3H部分を含む化合物であって、アミノ基に1、2または3つのメチレンホスホン酸基が結合されており、そして他の化学部分でさらに置換されてもよい化合物を意味する。アミノメチレンホスホン酸は、1つ以上のホスホン酸基および1つ以上のアミノ基を含んでもよい。これらのアミノメチレンホスホン酸の例としては、限定はしないが、図2に示される化合物F〜Nが挙げられる。
【0084】
これらの骨標的化因子および他の骨標的化因子は、ヘテロ原子の1つを通じて、またはさらなる結合ポイントを導入する化学的修飾によって結合されてもよいと考えられる。例えば、EDTMPを、図3に例示されるように、リン酸素のうちの1つによってリンカーに結合してホスホン酸結合を作製してもよい(例えば、Viera de Almediaら.,Tetrahedron,55,12997〜13010(1999)を参照のこと)。リン酸素はまた、図4に示されるようにアルキル化され得、ここでは例えば、活性化されたPEGに対する結合のための二次結合ポイントを得るために、R基が、例えば、側鎖のアミノ基を有してもよい。本発明において利用できる他のタイプのアルキル化としては、限定はしないが、Chavez et al.,Biomedical Imaging:Reporters,Dyes,& Instrumentation,Contag & Sevick−Muracia編、Proc.SPIE,第3600,99〜106(1999年,July)にさらに記載されているとおり、また、例えば、米国特許第5,177,064号、米国特許第5,955,453号、de Lombaert et al.,J Med.Chem.,37,498〜511(1994)およびIyer et al.,Tetrahedron Letters,30(51),7141〜7144(1989)にさらに記載される他のホスホン酸について示されるように、DOTMPに関与するのと同様の例が挙げられる。あるいは、化学的修飾のために、EDTMPを、例えば、修飾して、アニリン基の導入によってABDTMPを生成してもよい(例えば、国際特許出願WO94/00145の図1にさらに記載されるように)。次いでアニリンアミンを利用して、例えばアミド結合を形成する。DOMTPは図5に概要をまとめたように、同様に修飾されてもよい。
【0085】
「ホスホン酸塩、リン酸塩およびアミノメチレンホスホン酸塩(phosphonate、phosphate、and aminomethylenephosphonate)」という用語は、それぞれ、ホスホン酸、リン酸およびアミノメチレンホスホン酸、ならびにその任意の塩、加水分解性エステル、およびリン酸ベースの酸のプロドラッグを包含することを意味する。血中での7.4という生物学的pHで、または骨の周囲でのそれより酸性のpHでは、骨標的化因子のリン酸塩またはホスホン酸塩の特定の一部が脱プロトン化されて、対イオンで置換され得る。さらに、カルシウムのプロトンの交換は、本発明におけるヒドロキシアパタイトに対する骨標的化因子の結合の内在する事象である。しかし、骨標的化因子を含む組成物の調製および投与は、その中でリン酸の完全なプロトン化は要しても要さなくてもよい。従って、ホスホン酸、リン酸およびアミノメチレンホスホン酸は、ホスホン酸塩、リン酸塩およびアミノメチレンホスホン酸塩と交換可能に記載され利用される。特に好ましくはないが、リン酸ベースの酸の生物学的に加水分解可能なエステルは、骨標的化プロドラッグのインビボ使用においても利用できる。同様に、組成物の酸性度をマスクするために、例えば処方および投与の間、リン酸ベースの酸のプロドラッグをインビボで利用してもよい。
【0086】
標的化因子はまた、特定の組織の特性に基づいて標的する因子であってもよい。このような標的化因子の代表的な例としては、限定はしないが、その内容が参考として本明細書に援用される、H.Maeda et al.「Tumor vascular permeability and the EPR effect in macromolecular therapeutics:A Review」;Journal of
Controlled Release,2000 vol 63、pp271−284に記載されているように、EPR効果(浸透性および保持の増強)に起因して腫瘍組織に選択的に局在するポリマーが挙げられる。他の代表的なポリマーは、N−(2−ヒドロキシプロピル)メタクリルアミド(HPMA)および(ポリ)L−グルタミン酸である。
【0087】
標的化因子はまた、RGD部分を含んでもよい。Curnis et al.,Cancer Research,64(2):565〜571(2004)に考察されるように、RGD部分は、αvβ3インテグリンを含む細胞接着レセプターとの相互作用と相互作用することによって脈管構造に対してRGD融合タンパク質を標的する。
【0088】
3.合成
a.主な環系
本発明の化合物は、出発生成物としてLY294002(化合物1)を用いて合成され得る。LY294002(化合物1)は、商業的に入手されても、または実施例1に記載のように、もしくはその内容が参考として本明細書に援用される米国特許第5,703,075号に記載のように合成してもよい。当業者はまた、出発生成物として化合物2を用いて本発明の化合物を合成し得る。
【0089】
b.主な環系の誘導体の調製
化合物2および3の主な環系は、LY294002(化合物1)の主な環系の誘導体であってもよい。化合物3の主要環系の誘導体は、LY294002(化合物1)の主な環誘導体の調製のために、その内容が本明細書において参考として援用される、米国特許第5,703,075号に開示されるように、調製され得る。化合物3の主要環系の誘導体はまた、置換2−ヒドロキシ−アセトフェノンを含むがこれに限定されない市販の化合物を用いることによって調製され得る。
【0090】
c.モルホリン環の誘導体の調製
化合物3のアミン誘導体は、室温から強制条件(過剰な求核試薬および110℃への加熱)への範囲におよぶ条件下での実施例1のチオアルキル基の置換によって調製できる。任意の一級または二級の窒素含有求核試薬は、モルホリン環構造(異なるモルホリンアナログを含む)に別のアミン置換基を与えるように反応し得る。化合物3のこのようなアミン誘導体の合成の代表的な例は、本明細書の実施例に記載される。
【0091】
d.エステルの調製
上記のとおり、エステルは、本発明の四級化された化合物を形成するために使用され得る。本発明の四級化化合物は好ましくは、ハロエステルを用いて形成される。1つの好ましい実施形態では、本発明の四級化化合物は、クロロメチルエステルを用いて形成される。本発明の化合物の調製において有用な多数のクロロメチルエステルは市販の供給源から入手可能である。さらに、クロロメチルエステルは、その内容が本明細書において援用される、WO02/42265、WO94/23724および米国特許第4,444,686号、同第4,264,765号および同第4,342,768号に記載されるように合成され得る。
【0092】
e.四級化
本発明のプロドラッグは、例えば、実施例4および実施例6に記載のように、ハロメチルエステルでの化合物1または化合物2の三級アミンの四級化によって調製され得る。四級化されたアミン化合物は一般には、温和な条件では可逆性ではない。しかし、本発明の四級化合物は上記で考察されたように容易に加水分解される。化合物1または化合物2の三級アミンを四級化するために用いられ得るハロメチルエステルは市販されており、または以下の実施例に記載されるように調製されてもよい。
【0093】
f.リンカー
本発明のプロドラッグはまた、少なくとも2つの官能基を含むリンカーで化合物1または化合物2の三級アミンを四級化することによって調製されてもよい。このリンカーは三級アミンを四級化可能であり、または標的分子に共有結合可能であるか、または標的分子に容易に結合され得る、任意の天然または合成のリンカーであってもよい。
【0094】
リンカーは好ましくは、酸素もしくはイオウのような原子、−NH−、−CH2−、−C(O)−、−C(O)NH−のような単位、または原子の鎖からなった。リンカーの分子量は代表的には、約14〜200の範囲、好ましくは、14〜96の長さから約6原子までの範囲である。リンカーの代表的な例としては、限定はしないが、必要に応じて置換されており、鎖のうち1または2つの飽和炭素が必要に応じて−C(O)−、−C(O)C(O)−、−CONH−、−CONHNH−、−C(O)O−、−OC(O)−、−NHCO2−、−O−、−NHCONH−、−OC(O)NH−、−NHNH−、−NHCO−、−S−、−SO−、−SO2−、−NH−、−SO2NH−、または−NHSO2−で置換されている、飽和または不飽和の脂肪族基が挙げられる。
【0095】
リンカーの第一の官能基を用いて、上記で考察される三級アミンを四級化する。好ましい第一の官能基は、限定はしないがクロロメチルエステルおよびヨードメチルエステルを含むハロメチルエステルである。リンカーの第二の官能基は、標的化因子に共有結合され得る。
【0096】
第二の官能基は、求電子基または求核性基であってもよい。標的基に共有結合するために好ましい第二の官能基はイソチオシアネート、ハロアセトアミドマレイミド、イミドエステル、チオフタルイミド、N−ヒドロキシスクシンイミルエステル、ピリジルジスルフィド、フェニルアジド、カルボキシル(およびその酸塩化物)、アミノ、アシルヒドロジド、セミカルバジド、チオセミカルバジド、ジアゾニウム、ヒドラジン、アジド、アミノアルキル尿素、アミノアルキルチオ尿素、ハロトリアジン、およびメタ(ジヒドロキシボリル)フェニルチオ尿素である。標的化因子に対して本発明のプロドラッグを共有結合するために適切であり得る他の適切な反応性部分としては、ジスルフィド、ニトレン、スルホンアミド、カルボジイミド、塩化スルホニル、ベンズイミデート、−COCH3および−SO3Hが挙げられる。
【0097】
適切な第二の官能基は、それとの共有結合が形成される標的化因子の官能基に依存し、そして結果として所定のタイプの結合を形成する生物学的活性の損失に対するその感受性による。標的化因子がタンパク質である場合、第二の官能基は、ポリペプチド骨格を構成するアミノ酸の側鎖基と反応し得る。このような側鎖基としては、アスパラギン酸およびグルタミン酸残基のカルボキシル基、リジン残基のアミノ基、チロシンおよびヒスチジンの芳香族基、およびシステイン残基のスルフヒドリル基が挙げられる。
【0098】
ポリペプチド骨格のような標的化因子によって提示されるカルボキシル側鎖基は、可溶性のカルボジイミド反応によってアミン第二官能基と反応され得る。標的化因子によって提示されるアミン側鎖基は、イソチオシアネート、イソシアネートまたはハロトリアジン第二官能基と反応されて、本発明のプロドラッグに対する結合を発揮し得る。あるいは、標的化因子上のアミノ側鎖基は、ジアルデヒドおよびイミドエステルのような二機能性の因子によって、アミン反応性基を有する本発明のプロドラッグ化合物に連結され得る。標的化因子によって提示される芳香族基は、ジアゾニウム誘導体を介して本発明のプロドラッグに結合され得る。標的化因子の分子上のスルフヒドリル基は、マレイミドと、またはハロアルキル標的化因子反応性基、例えばヨードアセトアミドと反応し得る。このような反応に適切な遊離のスルフヒドリル基は、タンパク質である免疫グロブリンのジスルフィド結合から生成されてもよく、または化学的誘導体によって導入されてもよい。免疫グロブリンの重鎖領域内で生成された遊離のスルフヒドリル基に対する結合は、免疫グロブリンの抗原結合部位を妨害しないが、抗体が補体を活性化できなくし得る。
【0099】
標的因子がグリコシル化タンパク質である場合、ポリペプチド骨格を介して本発明の化合物に対する結合を形成する代わりは、McKearn et al.,EPO 88,695のような方法に従って糖タンパク質の炭水化物側鎖との共有結合を形成することである。従って、抗体の炭水化物側鎖は、選択的に酸化されてアルデヒドを生成してもよく、これが次にアミン反応性基と反応してSchiff塩基を形成するか、またはヒドラジン、セミカルバジドもしくはチオカルバジド反応性基と反応して、対応するヒドラゾン、セミカルバゾンもしくはチオセミカルバゾン結合を得ることができる。またこれらの同じ方法を使用して、本発明のプロドラッグを、炭水化物および多糖類のような非タンパク質性の標的化因子に結合してもよい。
【0100】
事前の酸化の必要性なしに炭水化物および多糖類に結合するのに有用な別の標的化因子反応性部分は、ジヒドロキシボリル基であり、例えば、これはメタ(ジヒドロキシボリル)フェニルチオ尿素誘導体に存在する。この基は、1,2−シス−ジオールを含む標的化因子と反応して、5−員環の環状ホウ酸塩エステルを形成し、従ってこの基を含む炭水化物、多糖類および糖タンパク質と使用される。Rosenberg et al.,Biochemistry,11,3623〜28(1972)に開示されるように、リボースは1,2−シス−ジオール基を2’,3’位置で含むので、ジヒドロキシボリル誘導体はまた、本発明のプロドラッグをリボヌクレオシド、リボヌクレオチドおよびリボ核酸に連結するためにも用いられ得る。デオキシリボヌクレオチドおよびDNA標的化因子は、3’ヒドロキシル基が存在しない場合、この方式で本発明のプロドラッグには連結され得ない。しかし、後者の標的化因子は、Engelhardt et al.,EPO97,373に開示されるように、デオキシリボヌクレオチドのアリルアミン誘導体を最初に形成することによって、プロドラッグのイソチオシアネート誘導体に結合し得る。
【0101】
本発明のプロドラッグと結合される標的化因子がインタクトな細胞である場合、ポリペプチド反応性部分または炭水化物反応性部分のいずれが使用されてもよい。HwangおよびWase,Biochim.Biophys.Acta,512,54〜71(1978)は、インジウム−111を用いて赤血球および血小板を標識する、Sundberg et al.,J.Med.Chem.,17,1304(1974)の二機能性EDTAキレーターのジアゾニウム誘導体の使用を開示する。ジヒドロキシボリル基は、種々の細菌、ウイルスおよび微生物と反応する。Zittle,Advan.Enzym.,12 493(1951)およびBurnett et al.,Biochem.Biophys.Res.Comm.,96,157〜62(1980)を参照のこと。
【0102】
化合物1または化合物2の三級アミンを共有結合的に四級化するために用いられ得る好ましいリンカーは、式:
【0103】
【化29】
(化合物4)
のリンカーであり、
ここで、
Xはハロ基であり;
Yは−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり
Z1およびZ2は独立してSまたはOであり;そして
n=0〜4である。
【0104】
1実施形態では、本発明の化合物4は、それらの化合物であり、ここで、
XはClまたはIであり;
Yは−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;
Z1およびZ2は独立してOであり;そして
n=0である。
【0105】
別の実施形態では、本発明の化合物4は、それらの化合物であり、ここで、
XはClまたはIであり;
Yは−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−またはCH(CH(CH3)2)−であり;
Z1およびZ2は独立してOであり;そして
n=1である。
【0106】
化合物4は、最終の四級窒素の切断率に変動を与える、アルキルおよびアリールカルボン酸骨格の両方とのリンカーを提供する。化合物4のリンカーは、実施例5に記載されるような市販の出発製品を用いて調製され得る。
【0107】
g.精製
本発明の化合物は、標準的な精製方法を用いて単離され得る。本発明の化合物の加水分解性の結合は、この化合物の精製の間に加水分解しやすくてもよい。
【0108】
本発明はまた、本発明の化合物を精製する方法に関しており、この方法は、化合物を可溶化するために少なくとも0.1%の酸(v/v)を含む溶液にこの化合物を添加する工程を包含する。次いでこの化合物を、クロマトグラフィー、好ましくはHPLCを行うことによって精製する。
【0109】
h.試験
本発明のプロドラッグは、時間の関数として、切断条件に曝露されたプロドラッグのHPLC分析を行なうことによって、加水分解性結合の加水分解の速度および加水分解の生成物を決定するように試験され得る。本発明の化合物の生物学的活性は、実施例17に記載されるようにマクロファージ細胞株J774細胞において食作用をブロックする工程を含むがこれに限定されない方法によって測定され得る。本発明の化合物の生物学的活性はまた、その内容が参考として援用される、米国特許第5,480,906号;K.Fuchikami et al.,J.Biomol Screen,2002 Oct.pp441〜450;VI Silveria et al.,J.Biomol.Screen,2002,Dec.7(6),507〜514;BE Drees Combinatorial Chemistry and Highthroughput Screening 2003,vol 6、321〜330に記載されるように、PI−3キナーゼ酵素アッセイによって測定され得る。
【0110】
i.塩
本発明の化合物は、種々の薬学的に受容可能な塩型で有用である。「薬学的に受容可能な塩(pharmaceutically acceptable salt)」という用語は、薬剤師によって理解される塩型、すなわち、実質的に毒性でなく、所望の薬物動態学的特性、嗜好性、吸収、分布、代謝または排出を提供する塩型をいう。選択にも重要であり、天然にはさらに重要な他の要因は、原料の費用、結晶化の容易さ、収率、安定性、吸湿性および得られたバルク薬物の流動性である。好都合には、薬学的組成物は、活性成分またはその薬学的に受容可能な塩から、薬学的に受容可能なキャリアと組み合わせて調製されてもよい。
【0111】
本発明の方法および組成物における使用に適切である、本発明の化合物の薬学的に受容可能な塩としては、限定はしないが、種々の有機酸および無機酸、例えば、塩化水素、スルホン酸ヒドロキシメタン、臭化水素、メタンスルホン酸、硫酸、酢酸、トリフルオロ酢酸、マレイン酸、ベンゼンスルホン酸、トルエンスルホン酸、フルファミン酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、パモン酸、スルファニル酸、2−アセトキシ安息香酸、フマル酸、トルエンスルホン酸、メタンスルホン酸、エタンジスルホン酸、シュウ酸、イセチオン酸と形成される塩が挙げられ、そして種々の他の薬学的に受容可能な塩、例えば、硝酸塩、リン酸塩、ホウ酸塩、酒石酸塩、クエン酸塩、コハク酸塩、安息香酸塩、アスコルビン酸塩、サリチル酸塩などが挙げられる。四級アンモニウムイオンのような陽イオンは、陰イオン性部分についての薬学的に受容可能な対イオンと考えられる。
【0112】
本発明の化合物の好ましい塩としては、塩酸塩、メタンスルホン酸塩およびトリフルオロ酢酸塩が挙げられるが、メタンスルホン酸塩がより好ましい。さらに、本発明の化合物の薬学的に受容可能な塩は、ナトリウム、カリウムおよびリチウムのようなアルカリ金属;アルカリ土類金属、例えば、カルシウムおよびマグネシウム;有機塩基、例えば、ジシクロヘキシルアミン、トリブチルアミンおよびピリジン;ならびにアルギニン、リジンなどのようなアミノ酸と形成されてもよい。
【0113】
本発明の薬学的に受容可能な塩は、従来の化学的方法によって合成され得る。一般には、塩は、適切な溶媒または溶媒の組み合わせ中での、遊離の塩基または酸と、化学量論的な量または過剰な、所望の塩を形成する無機もしくは有機の酸もしくは塩との反応によって調製される。
【0114】
一般には、本発明の化合物の塩の対イオンは、この合成された化合物に対して用いた反応物によって決定される。反応物に依存して、塩の対イオンの混合物が存在し得る。例えば、反応を促進するためにNaIが添加される場合、対イオンはClおよびIの対の陰イオンの混合物であってもよい。さらに分取HPLCでは、酢酸が溶出物に存在する場合、酢酸塩によって交換されるもとの対イオンを生じ得る。塩の対イオンは、異なる対イオンに交換されてもよい。対イオンは好ましくは、薬学的に受容可能な対イオンと交換されて、上記の塩を形成する。対イオンを交換するための手順は、その内容が参考として本明細書に援用される、WO 2002/042265、WO2002/042276およびS.D.Clas,「Quaternized Colestipol,an improved bile salt adsorbent:In Vitro studies」Journal of Pharmaceutical Sciences,80(2):128〜131(1991)に記載される。明快な理由によって、対イオンは、本明細書の化学構造に明確に示されず、そしてこの化合物の特徴づけは、同定された四級陽イオンに基づく。
【0115】
4.組成物
本明細書はまた、本発明の1つ以上の化合物を含む組成物も包含する。本発明の組成物はさらに、1つ以上の薬学的に受容可能なさらなる成分(単数または複数)、例えば、ミョウバン、安定化剤、抗菌剤、緩衝液、着色剤、香料、アジュバントなどを含んでもよい。
【0116】
a.処方物
本発明の組成物は、従来の方式で処方される錠剤またはトローチ剤の形態であってもよい。例えば、経口投与のための錠剤およびカプセルは、従来の賦形剤を含んでもよく、これには、限定はしないが結合剤、充填剤、潤滑剤、崩壊剤および湿潤剤が挙げられる。結合剤としては、限定はしないが、シロップ、アカシア、ゼラチン、ソルビトール、トラガカント、デンプンの粘液およびポリビニルピロリドンが挙げられる。充填剤としては、限定はしないが、ラクトース、糖、微結晶性セルロース、トウモロコシデンプン、リン酸カルシウムおよびソルビトールが挙げられる。潤滑剤としては、限定はしないが、ステアリン酸マグネシウム、ステアリン酸、滑石、ポリエチレングリコールおよびシリカが挙げられる。崩壊剤としては限定はしないが、ジャガイモデンプンおよびデンプングリコール酸ナトリウムが挙げられる。湿潤剤としては、限定はしないが、ラウリル硫酸ナトリウムが挙げられる。錠剤は、当該分野で周知の方法に従ってコーティングされ得る。
【0117】
本発明の組成物はまた、水性または油性の懸濁液、溶液、エマルジョン、シロップおよびエリキシルを含むがこれに限定されない液体処方物であってもよい。この組成物はまた、使用前の水または他の適切なビヒクルとの構成のために乾燥生成物として処方され得る。このような液体調製物は、懸濁剤、乳化剤、非水性のビヒクルおよび防腐剤を含むがこれらに限定されない添加物を含んでもよい。懸濁剤としては、限定はしないが、ソルビトールシロップ、メチルセルロース、グルコース/糖シロップ、ゼラチン、ヒドロキシエチルセルロース、カルボキシメチルセルロース、ステアリン酸アルミニウムゲルおよび硬化食用油脂が挙げられる。乳化剤としては、限定はしないが、レシチン、ソルビタンモノオレエート、およびアカシアが挙げられる。非水性のビヒクルとしては、限定はしないが、食用油脂、アーモンドオイル、分留ココナッツオイル、油状エステル、プロピレングリコールおよびエチルアルコールが挙げられる。防腐剤としては限定はしないが、メチルまたはプロピルp−ヒドロキシ安息香酸塩およびソルビン酸が挙げられる。
【0118】
本発明の組成物はまた、座剤として処方され得るが、これは座剤基剤を含んでもよく、この基剤としては、カカオバターまたはグリセリドが挙げられるがこれらに限定されない。本発明の組成物はまた、吸引のために処方され得、これは乾燥粉末として投与され得る溶液、懸濁液もしくはエマルジョンを含むがこれに限定されない形態、または噴霧剤、例えば、ジクロロジフルオロメタンもしくはトリクロロフルオロメタンを用いるエアロゾルの形態であってもよい。本発明の組成物はまた、クリーム、軟膏、ローション、ペースト、薬用絆創膏、パッチまたはメンブレンを含むがこれに限定されない、水性または非水性のビヒクルを含む、処方された経皮処方物であってもよい。
【0119】
本発明の組成物はまた、注射または連続注入による投与を含むがこれに限定されない、非経口投与のために処方されてもよい。注射の処方物は、油状または水性のビヒクル中の懸濁液、溶液またはエマルジョンの形態であってもよく、そして懸濁剤、安定化剤および分散剤が挙げられるがこれらに限定されない処方剤を含んでもよい。この組成物はまた、滅菌の、発熱物質のない水を含むがこれに限定されない安定なビヒクルでの再構成のために粉末形態で提供され得る。
【0120】
本発明の組成物はまた、移植によってまたは筋肉内注射によって投与され得るデポ調製物として処方されてもよい。この組成物は、適切なポリマーまたは疎水性物質(例えば、受容可能なオイル中のエマルジョンとして)、イオン交換樹脂とともに、または溶解度の劣る誘導体として(例えば、溶解度の劣る塩として)処方され得る。
【0121】
本発明の組成物はまた、リポソーム調製物として処方され得る。リポソーム調製物は、リポソームを含んでもよく、これが目的の細胞または角質層に浸透して、細胞膜と融合し、このリポソームの内容物の細胞への送達が得られる。例えば、Yaroshの米国特許第5,077,211号、Redziniakらの米国特許第4,621,023号、またはRedziniakらの米国特許第4,508,703号に記載されるようなリポソームを用いてもよい。本発明の組成物は、皮膚状態を標的することを意図しており、UVまたは酸化障害を生じる因子に対する哺乳動物の皮膚の曝露の前、間または後に投与され得る。他の適切な処方物はニオゾームを使用してもよい。ニオゾームはリポソームと同様の脂質小胞であり、その膜は主に非イオン性の脂質から構成され、その形態のいくつかは角質層を横切って化合物を輸送するために有効である。
【0122】
5.処置
本発明はまた、PI−3キナーゼ活性に関連する条件に罹患している患者を処置する方法を包含する。PI−3キナーゼ活性は、異常、過剰または構成的に活性であってもよい。本発明はまた、炎症性疾患を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。不適切なPI−3キナーゼシグナル伝達活性に寄与し得る、このような疾患および有害な健康上の影響は、当該分野において、例えば、その内容が参考として援用される、U.S.2002/0150954A1;US5,504,103;US6,518,277B1;U.S.6,403,588;U.S.6,482,623;U.S.6,518,277;U.S.6,667,300;U.S.20030216389;U.S.20030195211;U.S.20020037276およびU.S.5,703,075で開示されている。
【0123】
本発明はまた、p53媒介性のプログラムされた細胞死を増強するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0124】
本発明はまた、腫瘍細胞の化学感受性を増強するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0125】
本発明はまた、腫瘍細胞の放射線感受性を増強するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0126】
本発明はまた、腫瘍誘導性血管形成を阻害するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0127】
本発明はまた、癌以外の疾患に関連する血管形成プロセスを阻害するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0128】
本発明はまた、癌の処置のための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。
【0129】
この化合物は、化学療法および放射線療法のような他の抗癌処置と同時にまたは周期的に投与されてもよい。本明細書において用いる場合、「同時(simultaneous)」または「同時に(simultaneously)」という用語は、他の抗癌処置および本発明の化合物が、お互いの48時間、好ましくは24時間、さらに好ましくは12時間、それよりさらに好ましくは6時間、そして最も好ましくは3時間以下内に投与されたことを意味する。本明細書において用いる場合、「周期的に(metronomically)」という用語は、化学療法から種々の時点での、そして反復投与および/または化学療法レジメンに対して特定の頻度でのこの化合物の投与を意味する。
【0130】
化学療法処置は、細胞毒性剤もしくは細胞増殖抑制剤、またはそれらの組み合わせの投与を包含し得る。細胞毒性因子は:(1)細胞がDNAを複製する能力を妨害すること、および(2)癌細胞における細胞死および/またはアポトーシスを誘導することによって癌細胞が増殖することを妨げる。細胞増殖抑制剤は、細胞増殖を時には低い連続レベルに調節する、細胞シグナル伝達のプロセスを調節、妨害または阻害することを介して作用する。
【0131】
細胞毒性因子として用いられ得る化合物のクラスは、以下を含む:アルキル化剤(限定はしないが、ナイトロジェンマスタード、エチレンイミン誘導体、スルフォン酸アルキル、ニトロソ尿素およびトリアゼンを含む);ウラシルマスタード、クロルメチン、シクロホスファミド(Cytoxan(登録商標))、イホスファミド、メルファラン、クロランブシル、ピポブロマン、トリエチレン−メラミン、トリエチレンチオホスホラミン、ブスルファン、カルムスチン、ロムスチン、ストレプトゾシン、デカルバジンおよびテモゾラミド;代謝拮抗物質(限定はしないが、葉酸拮抗薬、ピリミジンアナログ、プリンアナログおよびアデノシンデアミナーゼインヒビターを含む):メトトレキセート、5−フルオロウラシル、フロクスウリジン、シタラビン、6−メルカプトプリン、6−チオグアニン、リン酸フルダラビン、ペントスタチンおよびゲムシタビン;天然産物およびそれらの誘導体(例えば、ビンカアルカロイド、抗腫瘍抗生物質、酵素、リンホカインおよびエピポドフィロトキシン);ビンブラスチン、ビンクリスチン、ビンデシン、ブレオマイシン、ダクチノマイシン、ダウノルビシン、ドキソルビシン、エピルビシン、イダルビシン、ara−c、パクリタキセル(パクリタキセルはTaxol(登録商標)として市販されている)、ミトラマイシン、デオキシコ−ホルマイシン、マイトマイシン−c、l−アスパラギナーゼ、インターフェロン(好ましくはIFN−α)、エトポシド、およびテニポシド。
【0132】
他の増殖性の細胞毒性因子は、ナベルベン、CPT−11、アナストラゾール、レトラゾール、カペシタビン、レロキサフィン、シクロホスファミド、イホスファミドおよびドロロキサフィンである。
【0133】
微小管に影響する因子は、細胞の有糸分裂を妨害し、そしてその細胞毒性活性については当該分野で周知である。本発明において有用な微小管影響因子としては、限定はしないがアロコルヒチン(NSC406042)、ハリコンドリンB(NSC609395)、コルヒチン(NSC757)、コルヒチン誘導体(例えば、NSC33410)、ドラスタチン10(NSC376128)、マイタンシン(NSC153858)、リゾキシン(NSC332598)、パクリタキセル(Taxol(登録商標)、NSC125973)、タキソール(登録商標)誘導体(例えば、誘導体(例えば、NSC608832)、チオコルヒチンNSC361792)、トリチルシステイン(NSC83265)、硫酸ビンブラスチン(NSC49842)、硫酸ビンクリスチン(NSC67574)、エポチロンA、エポチロンBおよびを含むがこれに限定されない天然および合成のエポチロン、ならびにジスコデルモライド(Service,(1996)Science,274:2009)エストラムスチン、ノコダゾール、MAP4などが挙げられる。このような因子の例はまた、Bulinski(1997)J.Cell Sci.110:3055 3064;Panda(1997)Proc.Natl.Acad.Sci.USA 94:10560〜10564;Muhlradt(1997)Cancer Res.57:3344〜3346;Nicolaou(1997)Nature 387:268〜272;Vasquez(1997)Mol.Biol.Cell.8:973〜985;およびPanda(1996)J.Biol.Chem.271:29807〜29812にも記載される。
【0134】
また適切なのは、細胞毒性因子、例えば、エピドフィロトキシン;抗悪性腫瘍酵素;トポイソメラーゼインヒビター;プロカルバジン;ミトキサントロン;白金配位錯体、例えば、シス−プラチンおよびカルボプラチン;生物学的応答修飾因子;増殖インヒビター;抗ホルモン治療剤;ロイコボリン;テガフール;および造血成長因子である。
【0135】
用いられ得る細胞増殖抑制剤としては、限定はしないが、ホルモンおよびステロイド(合成アナログを含む):17α−エチニルエストラジオール、ジエチルスチルベストロール、テストステロン、プレドニゾン、フルオキシメステロン、ドロモスタノロンプロピオネート、テストラクトン、メゲストロラセテート、メチルプレドニゾロン、メチル−テストステロン、プレドニゾロン、トリアムシノロン、ハロトリアニセン(hlorotrianisene)、ヒドロキシプロゲステロン、アミノグルテチミド、エストラムスチン、メドロキシプロゲステロンアセテート、リュープロリド、フルタミド、トレミフェン、ゾラデックスが挙げられる。
【0136】
他の細胞増殖抑制剤は、血管新生阻害剤、例えば、マトリックスメタロプロテイナーゼインヒビターであり、そして他のVEGFインヒビター、例えば、抗VEGF抗体および低分子、例えばZD6474およびSU6668も含まれる。Genetechの抗Her2抗体も利用できる。適切なEGFRインヒビターはEKB−569(不可逆性インヒビター)である。また、EGFRに免疫特異性のImclone抗体C225およびsrcインヒビターも含まれる。
【0137】
細胞増殖抑制剤として用いるためにも適切なのはCasodex(登録商標)(bicalutamide,Astra Zeneca)であり、これはアンドロゲン依存性の癌腫を非増殖性にさせる。細胞増殖抑制剤のさらに別の例は、抗エストロゲンであるTamoxifen(登録商標)であり、これはエストロゲン依存性乳癌の増殖または成長を阻害する。細胞増殖シグナルの伝達のインヒビターは、細胞増殖抑制剤である。代表的な例としては、上皮細胞成長因子インヒビター、Her−2インヒビター、MEK−1キナーゼインヒビター、MAPKキナーゼインヒビター、PI3インヒビター、SrcキナーゼインヒビターおよびPDGFインヒビターが挙げられる。
【0138】
種々の癌を本発明に従って処置することが可能であり、この癌としては、限定はしないが以下:膀胱(加速性および転移性の膀胱癌を含む)、乳房、結腸(結腸直腸癌を含む)、腎臓、肝臓、肺(小細胞および非小細胞肺癌および肺の腺癌を含む)、卵巣、前立腺、精巣、尿生殖路、リンパ系、直腸、喉頭、膵臓(膵外分泌癌腫を含む)、食道、胃、膀胱、頸部、甲状腺、および皮膚(扁平上皮癌を含む)の癌;白血病、急性リンパ性白血病、急性リンパ芽球性白血病、B細胞リンパ腫、T細胞リンパ腫、ホジキンリンパ腫、非ホジキンリンパ腫、ヘアリー細胞リンパ腫、組織球性リンパ腫およびバーキットリンパ腫を含むリンパ系の造血性腫瘍;急性および慢性の骨髄性白血病、骨髄異形成症候群、骨髄性白血病および前骨髄球性白血病を含む骨髄系列の造血性腫瘍;星細胞腫、神経芽細胞腫、神経膠腫およびシュワン細胞腫を含む中枢および末梢神経系の腫瘍;線維肉腫、横紋筋肉腫および骨肉種を含む間葉起源の腫瘍;ならびに黒色腫、色素性乾皮症、角化棘細胞腫、精上皮腫、甲状腺濾胞性癌および奇形癌を含む他の腫瘍;を含む癌腫が挙げられる。
【0139】
最も好ましくは、本発明は、膀胱の加速性または転移性の癌、膵臓癌、前立腺癌、非小細胞肺癌、結腸直腸癌および乳癌を処置するために用いられる。
【0140】
本発明はまた、膵臓を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。Gukovsky et
al.,Gastroenterology、126(2):554〜66(2004)に考察されるように、PI−3キナーゼの阻害は、膵炎を妨げ得る。
【0141】
本発明はまた、潰瘍を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。本発明はまた、胃癌(gastric cancer)、例えば、胃癌(stomach cancer)を処置するための方法を包含するが、この方法は、本発明の化合物の治療上有効な量をその必要な患者に投与する工程を包含する。Bacon et al.,Digestive Disease Week Abstracts and Itinerary Planner、Vol.2003,Abstract No.M921(2003)およびRokutan et al.,Digestive Disease Week Abstracts and Itinerary Planner,Vo.2003,Abstract No.354(2003)に考察されるように、PI−3キナーゼは、胃の細胞へのHelicobactor Pyloriの結合に関与する。さらに、Osaki
et al.Journal of Cancer Research and Clinical Oncology、130(1):8〜14(2004)は、PI−3キナーゼインヒビター、例えばLY294002が、胃癌の抗腫瘍剤として有用であり得ることを示す。
【0142】
本発明はまた、心血管ステントのようなステントを有する患者に対して本発明の化合物の治療上有効な量を投与する工程を包含する、ステントの能力を改善する方法を包含する。Zhouら、Arteriosclerosis Thrombosis and Vascular Biology,23(11):2015〜2020(2003)に考察されるように、PI−3キナーゼの阻害は、血管のステント位置に伴う「伸展(stretch)」障害を妨げ得る。ステントまたはそのポリマーマトリックス中の本発明の化合物は、ステントコーティングマトリックスにおける可溶性を改善し得、水/血清の溶解度を改善し得、またはステント位置に隣接する細胞への灌流を改善し得る。
【0143】
本発明はまた、加齢性黄斑変性症(AMD)を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。Retina,Feburary 18,2004に考察されるように、VEGFの阻害は、AMDに関連する血管の過剰増殖を阻害する。本発明の化合物は、血管形成を阻害することによってAMDを処置し得る。
【0144】
本発明はまた、高血圧を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。Notthcott and Watts,Hypertension,43(1):125〜130(2004)に考察されるように、PI−3キナーゼの阻害は、高血圧に関連するMg2+の低い細胞外濃度を妨げ得る。
【0145】
本発明はまた、前駆細胞、例えば、骨髄前駆細胞の分化を抑制するための方法を包含するが、この方法は、本発明の化合物の有効量を前駆細胞に投与する工程を包含する。LewisらExperimental Hematology、32(1):36〜44(2004)に考察されるように、PI−3キナーゼ経路の阻害は、骨髄前駆細胞を抑制する。
【0146】
本発明はまた、肝臓癌を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。Leng et al.,Hepatology 38(4)Suppl 1:401A(2003)に考察されるように、LY294002は、ヒト肝臓組織の指標であるAkt(セリン/トレオニンプロテインキナーゼB)のリン酸化を阻害する。
【0147】
本発明はまた、変異PTENに関連する状態を処置するための方法を包含するが、この方法は、本発明の治療上有効な量の化合物をその必要な患者に投与する工程を包含する。PTENは、コーデン病を有する患者で同定されている染色体10q23に位置する癌抑制遺伝子である。Vegaら、Journal of Investigative Dermatology,121(6):1356〜1359(2003)に考察されるように、変異PTENは、プロトオンコジーンAktの活性化を阻害する能力が低くなっている。PI−3キナーゼのインヒビターは、Aktのリン酸化を阻害し得、それによって変異PTENの影響を軽減する。
【0148】
a.投与
本発明の組成物は、限定はしないが経口、非経口、舌下、経皮、直腸、経粘膜、局所、吸引、口腔内投与、またはそれらの組み合わせを含む任意の方法で投与されてもよい。非経口投与としては、限定はしないが、静脈内、動脈内、腹腔内、皮下、筋肉内、クモ膜下腔内および関節内が挙げられる。本発明の組成物はまた、この組成物の緩徐な放出および緩徐な制御されたi.v.注入を可能にするインプラントの形態で投与されてもよい。
【0149】
b.投薬量
治療での使用に必要な化合物の治療上有効な量は、処置される条件の性質、活性が所望される時間の長さ、ならびに患者の年齢および条件によって変化し、そして担当医によって最終的に決定される。しかし、一般には、成人の処置に使用される用量は代表的には、1日あたり0.001mg/kg〜約200mg/kgの範囲である。この用量は、1日あたり約1μg/kg〜約100μg/kgであってもよい。所望の用量は、単回用量で、または適切な間隔で投与される複数の用量として、例えば、1日あたり2、3、4またはそれ以上の低用量として都合よく投与されてもよい。複数回の用量もしばしば所望されるか、または必要である。
【0150】
多数の要因によって、本発明の化合物は広範な用量で投与されることになり得る。他の治療剤と組み合わせて用いる場合、本発明の化合物の投薬量は、比較的低い投薬量で与えられてもよい。さらに、標的化因子の使用によって必要な投薬量を比較的低くすることができる。本発明の特定の化合物は、限定はしないが、低い毒性、高いクリアランス、三級アミンの切断率の低さを含む要因に起因して比較的高用量で投与されてもよい。結果として、本発明の化合物の投薬量は、約1ng/kg〜約100mg/kgの範囲であってもよい。本発明の化合物の投薬量は、限定はしないが約1μg/kg、25μg/kg、50μg/kg、75μg/kg、100μg/kg、125μg/kg、150μg/kg、175μg/kg、200μg/kg、225μg/kg、250μg/kg、275μg/kg、300μg/kg、325μg/kg、350μg/kg、375μg/kg、400μg/kg、425μg/kg、450μg/kg、475μg/kg、500μg/kg、525μg/kg、550μl/kg、575μg/kg、600μg/kg、625μg/kg、650μg/kg、675μg/kg、700μg/kg、725μg/kg、750μg/kg、775μg/kg、800μg/kg、825μg/kg、850μg/kg、875μg/kg、900μg/kg、925μg/kg、950μg/kg、975μg/kg、1mg/kg、5mg/kg、10mg/kg、15mg/kg、20mg/kg、25mg/kg、30mg/kg、35mg/kg、40mg/kg、45mg/kg、50mg/kg、60mg/kg、70mg/kg、80mg/kg、90mg/kgまたは100mg/kgを含む任意の投薬量であってもよい。
【0151】
本発明は、以下の非限定的な実施例によって例示される多数の局面を有する。
【実施例】
【0152】
実施例1
LY294002の調製
Ly294002の10gのサンプルは、その内容が参考として援用される、Vlahos et al.、J.Biol.Chem.269(7):5241(1994)に記載される手順に基づくスキーム2に従って調製した。アミンによる12のようなチオクロムのチオメチル基の置換は、前に記載されており(その内容が参考として援用される、Bantickら、J.Heterocyclic Chem,18:679(1981))、チオールアニオンのアルキル化を伴う二硫化炭素での11のようなメチルフェニルケトンの環化を有する(Vlahos et al.and Bantick et al.)。カルボン酸(10)からの1工程反応のメチルケトン(例えば、11)の調製は、その内容が参考として本明細書に援用される、Rubottom et al.,J.Org.Chem.,48:1550(1983)に記載された手順を用いて行なった。
【0153】
スキーム2
【0154】
【化30】
実施例2
LY294002の四級アナログの調製
スキーム3の手順後、LY294002の三級アミンを、強制条件下でヨードメタンまたは塩化ベンジルを用いて四級化して、化合物A052−10および化合物13Bを得た。実施例56は、メチル四級プロドラッグA052−10の合成を記載する。実施例57は、フタルイミド四級プロドラッグA052−08の合成を記載する。実施例58は、パラカルボキシルベンジル四級A044−78の合成を記載する。[0199][0215]は、パラ−scn−ベンジル四級プロドラッグA044−80の合成を記載する。
【0155】
スキーム3
【0156】
【化31】
実施例3
クロロメチルエステルの調製
クロロメチル中間体を、Tsujihara,Synth Commun,24,767,1994に記載の手順に従って調製した。要するに、適切なカルボン酸を、ジクロロメタン/水の50/50混合物中に希釈した。この混合物を氷水槽中で冷却して、炭酸水素ナトリウム(4当量)およびn−テトラブチル硫酸水素アンモニウム(0.05当量)を添加した。5分間の撹拌後、クロロメチルクロロサルフェート(1.1当量)を添加した。この溶液を一晩激しく撹拌した。この混合物をさらなるジクロロメタンとともに分液漏斗に移して、飽和塩化ナトリウム溶液で洗浄した。有機物を硫酸ナトリウム乾燥させて、溶媒を除去して生成物を得た。この物質をLC−MSによって、そしてある場合には1H NMR分光法によって特徴付けた。この一般的な手順によって、以下の代表的なクロロメチルエステルを対応するカルボン酸から調製した:
表1
【0157】
【化32】
【0158】
【化32A】
【0159】
【化32B】
*UV検出を用いるHPLC−MS保持時間;
**UV検出を用いる出発カルボン酸のHPLC−MS保持時間;
UD=UV吸収がないこと、およびMSによるイオン化がないことに起因して検出されない。
【0160】
実施例4
四級プロドラッグへのLY294002の変換
LY294002(化合物1)をアセトニトリルに溶解し、次いで実施例3由来の各々のクロロメチルエステル(1〜1.5当量)を、ヨウ化ナトリウムの1〜2当量とともに添加した。室温ではこの反応はクロロメチルエステルでごくゆっくり進行して、塩化ナトリウムの沈殿とともにごく少量の四級化されたアミン生成物が得られた。65℃では、この反応は通常4時間で完了した。この反応が完全に濾過された場合(LC−MSによる分析で判定した);濃縮し、ついで逆相HPLCで精製した。この画分を収集して、凍結乾燥し、ふわふわした粉末として所望の生成物を得た。この方式で調製および精製した実施例は、下の表に示す(対イオンは示していないが、その塩化物、ヨウ化物、酢酸塩またはそれらの混合物を含む)。
【0161】
表2
【0162】
【化33】
【0163】
【化34】
【0164】
【化35】
実施例5
ハロメチルエステルリンカー
実施例3および実施例4の結果に基づいて、ハロメチルエステルリンカーを調製した(スキーム4およびチャート)。実施例3に記載されるように、化合物Bは、化合物A(市販される)から調製した。この化合物は、アセトンまたは2−ブタノンへの溶解、次いで2〜5当量のヨードナトリウムに溶解することによるFinklestein反応により、さらに反応性のヨードメチルエステル(化合物C)に変換され、ここでは塩化ナトリウムが沈殿して、ヨードメチルエステル(化合物C)が溶液中で生成された。化合物Cは、溶媒の剥ぎ取りおよび水不混和性の溶媒、例えば塩化メチレンへの溶解、および残りのヨードナトリウムを除去するための水での抽出によって単離された。
【0165】
化合物Eは化合物D(市販されている)から調製し、化合物FおよびGは、それぞれ、化合物BおよびCの生成物と同様の方式で調製した。
【0166】
スキーム4
【0167】
【化36】
実施例6
ハロメチルリンカーでのLY294402の四級化
ハロメチルエステルは、実施例5のものを含むが、これを、実施例4の方法論と同様の条件を用いてLY294002を四級化するために用いた。遊離の官能基とのリンカーを含む代表的なプロドラッグとしては以下が挙げられる:
【0168】
【化37】
【0169】
【化38】
化合物1105は、アセトニトリル中に化合物Cとともに化合物1101を混合することによって調製され、ここではこの両方が可溶性であり、そして生成物化合物1105は、3日間にわたって沈殿し、少量のアセトニトリルで洗浄されて実質的に純粋な化合物1105が得られた(LCMSによって確認された)。
【0170】
実施例7
化合物1111を用いるプロドラッグの調製
化合物1111は、スキーム5に示す方法によって生成した。化合物1110は、1〜3時間にわたってニートなトリフルオロ酢酸で処理し、TFAをアルゴンで吹き飛ばして減圧下で乾燥し、化合物1113からなるガラス状の固体を得た。次いで、化合物1113を1〜3mlの塩化チオニルに溶解して、65℃で3〜8時間加熱した。塩化チオニルをアルゴンで吹き飛ばし、ついで減圧下で乾燥して、ガラス状の黄色固体として良好な収率で化合物1111を得た。化合物1111は、例えば、メタノールへの単なる溶解によって、種々の窒素含有およびヒドロキシル含有求核試薬との代表的な酸塩化物として反応されて、相当するメチルエステル化合物1112を得ることができる。
【0171】
スキーム5
【0172】
【化39】
化合物1111のサンプルを、アセトニトリルに溶解して、別々のバイアルに含まれる少なくとも5当量の種々のアルコールで処理した。1時間後、サンプルをHPLC−MSによって分析したところ、表3に示されて特徴付けられるとおり、90%より多い化合物1111の対応するエステルへの良好な変換が示された。
【0173】
表3
【0174】
【化40】
【0175】
【化41】
*UV 214nm
実施例8
化合物1111を用いるタンパク質結合プロドラッグの調製
修飾されるアミノ基またはヒドロキシル基に対して2〜10倍過剰な化合物1111を用いて大量の水溶液(pH7〜9)(炭酸塩緩衝液に対してリン酸塩緩衝液)中でタンパク質を結合体化する。酸塩化物の化合物1111は、混合した有機の水溶液(50/50の水/アセトニトリルまたは50/50の水/THFなど)に導入してもよいし、または1〜24時間、室温で2相の反応系で塩化メチレン中に撹拌してもよい。タンパク質結合体は、透析または限外濾過によって精製して、直接用いることができる。
【0176】
50mMの炭酸水素ナトリウム緩衝液に含まれる5mg/mlのトランスフェリンタンパク質(Sigma)の500μlのアリコートを、実施例12に従ってDMSO中に調製した、100倍の30mM A024−79(100モル当量)と混合した。室温での1時間および20分の反応の後、50倍のサンプルを取り出して、Sephadex G−10(700モル重量のカットオフ)カラムを通過させて、低分子からタンパク質を分離した。次いで、精製された結合されたタンパク質溶出物のアリコートをアセトニトリルで抽出したところ、LC−MSでは化合物1は検出されなかった。精製された結合体化されたタンパク質溶出物を室温で39時間静置させて、その時点でタンパク質混合物をアセトニトリルで再度溶出したところ、この時点で化合物1の最大理論量の15%が検出された。これらの結果によって、プロドラッグの15モルというモル比がトランスフェリンの1モルあたりに結合されたことが示される。これらの結果によって、代表的なタンパク質に対する求電子性リンカー保有プロドラッグの結合が実証され、そして経時的に、PI3キナーゼインヒビター(化合物1)のかなりの量が水性の条件下でタンパク質から遊離されたことが実証された。
【0177】
実施例9
化合物1111を用いる樹脂結合プロドラッグの調製
全て天然のアミノ酸を用いる標準的なFMOC/HOBTカップリングペプチド化学を用いて、wang樹脂上でペプチドarg−gly−asp−ser(RGDS)を調製した。樹脂結合したペプチドを1〜24時間DMF中で化合物1111と反応させて、濾過して、樹脂をDMFで、次いで、塩化メチレンで洗浄し、次いでトリフルオロ酢酸で処理して、樹脂から結合体化合物1126を切断した(スキーム6)。実施例55は、化合物1111の大規模化調製を記載する。
【0178】
スキーム6
【0179】
【化42】
実施例10
化合物1111を用いるフォレート標的化因子でのプロドラッグの調製
化合物1111は、求電子性基を有し、この基が軽度に塩基性の有機または水性の条件(すなわち、20mM〜500mMの炭酸水素ナトリウム)下で求核性アミノ基と反応して非可逆性のチオ尿素結合を形成し得る。適切な求核性アミノ基は、標的化生体高分子フォレートに存在する。フォレート分子AおよびCを、塩基トリメチルアミンまたはジイソプロピルエチルアミンの存在下でほぼ等しい割合で混合することによってDMF中のアミノ基を介して化合物1111に結合して、化合物BおよびDを得た(スキーム7)。
【0180】
スキーム7
【0181】
【化43】
実施例11
化合物1111を用いる抗体標的化因子でのプロドラッグの調製
化合物1111は、pH7〜9の水溶液中でモノクローナル抗体に結合体化し、次いで限外濾過、または低分子からタンパク質結合体を分離する他の標準的な方法によって分離する。行なわれた結合体化は実施例8に従って調製され得る。
【0182】
実施例12
N−ヒドロキシスクシンイミドエステルを用いるプロドラッグの調製
化合物1111よりも反応性の低いエステルは、化合物1113のN−ヒドロキシスクシンイミド活性エステルを生成することによって調製した(スキーム8)。化合物1113(A024−67)の100mgのサンプルを、53mgのN−ヒドロキシスクシンイミド(2当量)とともに1mlの乾燥THFに溶解した。塩化メチレン(2当量)に含まれる1Mジクロヘキシカルボジイミドの45のアリコートを撹拌しながら一挙に添加した。3分内に重い白色沈殿が形成され、このことはカップリング反応が生じたことを示した。反応物を23時間撹拌させた後、この反応混合物を濾過して、濾液から溶媒を除去して、172mgの粗活性エステル生成物を、化合物A024−79と命名された濃い黄色の油状物として得たが、これは2.334分という保持時間を示し、M+=535という予想質量がこのピークについて見出された。
【0183】
スキーム8
【0184】
【化44】
化合物1111について上記したのと同じ化学を用いて、実施例8に記載されたように標的タンパク質に対して結合体化するためにA024−79を用い、そしてポリマーに対して結合体化するために用いたのは実施例74に記載する。
【0185】
実施例13
化合物1105を用いるプロドラッグの調製
化合物1105は、求電子性基を有し、この基は、軽度に塩基性の有機または水性の条件(すなわち、20mM〜500mMの炭酸水素ナトリウム)下で求核性アミノ基と反応して非可逆性のチオ尿素結合を形成し得る。適切な求核性アミノ基は、ビタミン誘導体のようなアミン基を保有するペプチド、タンパク質および低分子のような生体高分子に存在する(スキーム7のAおよびC)。このような生成物の代表的な例としては、化合物BおよびDが挙げられる。
【0186】
スキーム9
【0187】
【化45】
実施例14
モルホリン環の誘導体の調製
スキーム1のチオメチル化合物は、実施例1に記載のように調製した。この化合物を、適切な溶媒中において触媒量の酢酸の有無のもとで、そして過剰な求核性のアミン化合物とともに、ほとんどのチオメチル化合物が消費されるまで加熱した。この混合物を分取逆相LC−MSに供して、所望のモルホリンアナログを単離した。この方式で調製した化合物は、それらの調製条件とともに表4に示し、特徴および単離は表5に示す。化合物のNMRデータは表6に示す。
【0188】
表4
【0189】
【化46】
【0190】
【化47】
表5
【0191】
【化48】
表6
【0192】
【化49】
実施例15
HPLC分析
HPLC分析を島津LCMS−2010で行い、流速は3ml/分を、そして出発B濃度は5%を使用した。B溶媒は5.0分で95%濃度まで直線的に増大させて、6.0分まで95%で保持し、次いで6.5分で5%に直線的に減少させ、これを7.5分で流し終わるまで保持する。他に注記しない限り、これが本実施例で用いた方法である。方法Bは、極性の化合物のための緩い勾配の方法であって、これには3ml/分の流速および0%の出発B濃度を使用して、これを最初の1分間保持する。溶媒Bは3.0分で10%の濃度まで直線的に増大させ、次いで5.0分で95%まで直線的に減少させて、ここで6.0分まで保持し、次いで6.5分で5%まで直線的に減少させて、これを7.5分で流し終わるまで保持する。質量検出に加えて、LC検出は、3つのチャネルからなった;254nmのUV吸収、214nmのUV吸収、そして蒸発光散乱(Alltech ELSD 2000)。蒸発光散乱検出器は、1分あたり1.5リットルの窒素流を用いて50℃で行なった。CDL(化学脱溶媒ライン)および島津LCMS−2010のブロック温度は両方とも300℃であって、窒素ネブライザーガス流は4.5L/分であった。正および負の質量スペクトルが50〜2000m/zで検出された。カラムはYMC CombiScreen ODS−AQ,S−5μ粒子サイズ、50mm長、内径4.6mmであった。移動相Aは、0.1%(v/v)HOAcを添加したHPLC等級のB&Jの水を用いて作製し、移動相Bは0.1%(v/v)HOAcを添加したHPLC等級のB&Jのアセトニトリルを用いて作製した。このシステムによって、参照標準として用いた標準の市販の物質(4−ヒドロキシフェニル酢酸;Aldrich Catalog H5000−4;融点149〜151℃)について1.50〜1.60分(tR=1.50〜1.60)という保持時間が得られる。
【0193】
実施例16
分取HPLC
勾配分取HPLCを、SIL−10A自動サンプリング装置に接続した2つのLC−8Aポンプを備える島津システムで行い、逆相カラム(YMC,カタログCCAQSOSO52OWT;ODS−AQ CombiPrep,20mm×50mm)で溶出し、次いでMRA可変容積分配器を通過させた;次いでより小さいストリームを、LC−10ADVP補給ポンプ(MeOH)を用いて3ml/分まで作製し、溶出液を可変2チャンネル波長UV検出器を通過させ、次いで蒸発光散乱検出器(1分あたり1.5リットルの窒素流を用いて50Cで実施)と島津2010質量検出器とにほぼ6:1に分けた;次いで、MRA分配器からの大きいストリームを、質量、UV吸収またはELSピークサイズによって誘発される分画収集器として機能するGilson 215液体処理装置に流した。
【0194】
種々の勾配を、さらに水性の溶媒Aから出発して流して、種々の濃度のBまで傾斜させた。移動相Aは0.1%(v/v)HOAcを添加したHPLC等級のB&Jの水を用いて作製し、移動相Bは0.1%(v/v)HOAcを添加したHPLC等級のB&Jのアセトニトリルを用いて作製した。
【0195】
実施例17
加水分解されたプロドラッグの生物活性
LY294002(化合物1)の生物活性は、クラスI PI−3キナーゼ依存性経路である、J774マクロファージ中の食作用を評価することによって決定した。要するに、J774細胞を、10%FCSを添加したDMEM中で1時間、適切なDMSOコントロールとともに、10、1および0.1μMの濃度のLY294002で処理して、次いで感作されたsRBC(ヒツジ赤血球)を、100:1の標的対エフェクター比で37℃で30分間添加した。細胞を低張性ショックに曝して赤血球を取り除き、細胞溶出液中のヘモグロビン濃度の測定によって食作用を決定した。図1に示されるように、LY294002は、全ての濃度で用量依存性の様式で食作用を有意にブロックした。これらの結果によって、J774細胞系は、本発明の化合物がPI−3キナーゼ活性を阻害する能力を迅速にかつ容易にアッセイするために用いられ得ることが示される。PI−3キナーゼ活性をアッセイするこの方法を用いて、標的されたプロドラッグ化合物1126を、種々のプレインキュベーション時間を用いて5μMの濃度で試験して、プロドラッグを活性薬物(化合物1)にインサイチュで変換させた。コントロールのサンプル(化合物1126のプレインキュベーションなしの時間ゼロ)は、140という食作用係数(PGI;PI−3キナーゼの阻害がない結果として生じる食作用の程度の指標)を示したが、化合物1126は水性でpH=7であり、2、5および10時間のインキュベーション時間で、それぞれPGIは88、78および37を示した。本実施例によって、最初のプロドラッグ1126は、PI−3キナーゼ阻害活性をほとんどまたは全く有さず、そして有意なPI−3キナーゼ阻害を示す生物活性薬物に経時的に変換されることが実証される。別の実験では、20μM濃度の化合物1126は曝露限界の設定(試験溶液に対する20分の曝露、次いで試験溶液の除去)で、溶媒ブランクについて163、化合物1について190、そしてRGDS(化合物1126の標的部分であるテトラペプチド)についての170に対して、50というPGIを有した。本実施例は、曝露時間限界の設定での標的されたPI−3キナーゼインヒビターの利点を示した。この効果はさらに、実験を繰り返すことによって評価したが、ここでは、化合物1126の10μM、3μM、1μMおよび0μMという漸減する用量を用いて、化合物の除去後20分のインキュベーション時間、続いて食作用の2時間のインキュベーションによって、それぞれ33、143、206および213というPGIが得られた。本実施例によって化合物1126は用量曝露限界の設定で、用量依存性の様式でPI−3キナーゼを阻害したことが実証される。
【0196】
実施例18
骨標的化基A030−84の調製
500mgの4−[(N−BOC)アミノエチル]アニリン(Aldrich)を含む10mlのジオキサンの溶液をパラホルムアルデヒド(400mol%、270mg)および亜リン酸トリメチル(400mol%、1.12g)で処理した。この混合物を95℃で一晩加熱した。次いでさらにパラホルムアルデヒド(270mg)および亜リン酸トリメチル(1.12g)を添加して、これを再度95℃で一晩加熱した。この溶液を冷却して、クロロホルム(20mL)にとり、飽和塩化ナトリウム(20mL)および水(20mL)で洗浄した。有機物を硫酸ナトリウムで乾燥して、溶媒および過剰の亜リン酸トリメチルを80℃のロータリーエバポレーションによって除去して、1.723gの透明な油状物を得た。ロット番号A030−74と命名された表題の化合物の存在を、エレクトロスプレーHPLC−MSによって確認したところ、これはtR=2.9分の保持時間、そして所望の質量[M=C18H32N2O8P2]について実測した、467m/z[M+H]+および489m/z[M+H]+を示した。
【0197】
10mLのジクロロメタン中で上記のように調製した870mgのA030−74の溶液をブロモトリメチルシラン(690モル%、1.97g)で処理した。この溶液を一晩撹拌した。メタノール(10mL)を添加して、溶液を15分撹拌し、次いで濃縮して1.12gのオレンジ色の油状物を得た。表題の化合物の存在をエレクトロスプレーLC−MSによって確認した。この勾配を用いる保持時間は、tR=0.85分であることが見出され、そして所望の生成物[M=C9H16N2O6P2]についての質量スペクトルは、ネガティブモードで操作する予想m/z 309[M−H]−で見出した。この生成物には参照番号A030−84を与えた。
【0198】
【化50】
実施例19
化合物A014−52の合成
【0199】
【化50A】
1.0gの4−カルボキシフェニルイソチオシアナートにジクロロメタン(15mL)および蒸留水(15mL)を添加した。フラスコを氷水槽中で冷却して、炭酸水素ナトリウム(4.0当量)およびn−硫酸水素テトラブチルアンモニウム(0.05当量)を添加した。10分後、クロロメチルクロロサルフェート(1.2当量)を添加した。この溶液を一晩激しく撹拌して、ジクロロメタン(10mL)で補助して分液漏斗に移した。この相を分離して有機物を飽和塩化ナトリウム(20mL)で洗浄した。有機物を硫酸ナトリウムで乾燥させ、溶媒を除去して、1.10gの黄褐色の固体を得た。表題の化合物の存在は、生成物(4.2分)対、出発のカルボン酸(3.2分)の保持時間のシフトによって示された。この化合物をプロトンNMR分光法によって確認した:1H(CDCl3)δ:8.08(d,2H,J8.8Hz),7.30(d,2H,J8.8Hz),5.95(s,2H)。
【0200】
実施例20
化合物A014−48の合成
【0201】
【化51】
250mgのクロロメチルエステル(A014−52に記載の手順を介して調製)を含む2mLのアセトンの溶液を、ヨウ化ナトリウム(1.2当量)で処理して、この溶液を一晩撹拌した。この溶液を濾過して、溶媒を除去し、その残留物をジクロロメタン(10mL)にとった。この溶液を10%(w/v)の亜硫酸ナトリウム(10mL)、5%(w/v)炭酸水素ナトリウム(10mL)および水(10mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去して137mgの薄緑色の固体を得た。表題の化合物の存在は、ヨードメチルエステル生成物(4.4分)対出発クロロメチルエステル(4.2分)の保持時間のシフトで示された。この化合物はまたプロトンNMR分光法によって確認した:1H(CDCl3)δ:8.04(d,2H,J8.8Hz),7.29(d,2H,J8.1Hz),6.15(s,2H)。
【0202】
実施例21
化合物A014−76の合成
【0203】
【化52】
387mgのクロロメチルエステル(A014−52に記載の手順を介して調製)を含む6mLの2−ブタノンの溶液を、ヨウ化ナトリウム(1.2当量)で処理して、この溶液を10時間加熱した。この溶液を濾過して、溶媒を除去し、その残留物をジクロロメタン(10mL)にとった。この溶液を10%(w/v)の亜硫酸ナトリウム(10mL)、5%(w/v)炭酸水素ナトリウム(10mL)および水(5mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去して310mgの黄褐色色の固体を得た。表題の化合物の存在は、ヨードメチルエステル生成物(4.4分)対出発クロロメチルエステル(4.2分)の保持時間のシフトで示された。
【0204】
実施例22
化合物A018−24の合成
【0205】
【化53】
64mgの2−p−ニトロベンジル−1,4,7,10−テトラアザサイクロドデカン(Macrocyclics)を含む500μLのジオキサンの溶液を、パラホルムアルデヒド(50mg)および亜リン酸トリメチル(207mg)で処理した。この混合物を85℃で加熱して、次いで溶媒を75℃のロータリーエバポレーションによって除去した。クロロホルム(10mL)を添加して、溶液を飽和塩化ナトリウム(2×10mL)および水(2×10mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥し、溶媒を除去して褐色の油状物を得た。これをLCを介して精製して、所望の物質を得た。表題の化合物の存在をエレクトロスプレーLC−MS Aによって確認した;tR=1.8分。MS[M=C27H53N5O14P4]m/z796(MH+),818(MNa+)。
【0206】
実施例23
化合物A022−32の合成
【0207】
【化54】
33mgのホスホン酸大環状化合物(A018−24で調製)を含む700μlのジクロロメタンの溶液をブロモトリメチルシラン(72mg)で処理した。この混合物を一晩撹拌し、次いでさらなるブロモトリメチルシラン(36mg)を添加して、これをさらに3日撹拌した。メタノール(500mL)を添加して溶液を1時間撹拌し、次いで揮発性物質を除去して、褐色の油状物を得た。メタノールの添加で褐色の固体を沈殿させ、これを濾過して乾燥させた。これを、LCを介して精製して、2.7mgの所望の物質を得た。表題の化合物の存在をエレクトロスプレーLC−MS Aによって確認した;tR=1.5分。MS[M=C19H37N5O14P4]m/z 682(M−H−),340(M−2H)/2)2−]。
【0208】
ニトロ基は標準的な還元方法を用いて、例えば、純粋な水素の雰囲気下でのメタノール中の炭素触媒上で5%パラジウムとともに撹拌することによって還元する。次いでこの混合物を濾過して(触媒に対する空気の曝露を妨げるように注意する)、溶媒をエバポレートしてアミンを得る。
【0209】
実施例24
化合物A022−56の合成
【0210】
【化55】
リン酸(1.26g)、6M塩酸(19.5mL)およびp−キシレンジアミン(1.0g)の混合物を100℃に加熱した。これに37%(wt/wt)のホルムアルデヒド水溶液(1.15mL)を添加して、この混合物を100℃で一晩撹拌した。この混合物を濾過して、80℃でのロータリーエバポレーションによって水を除去して、2.11gの白色固体を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=1.8分。MS[M=C10H18N2O6P2]m/z 325(MH+)。
【0211】
実施例25
化合物A018−12の合成
【0212】
【化56】
928mg N−BOC−1,4−ジアミノブタンを含む10mLのジオキサンの溶液を、パラホルムアルデヒド(592mg)および亜リン酸トリメチル(2.44g)で処理した。この混合物を108℃で一晩撹拌して、75℃のロータリーエバポレーションによって溶媒を除去した。クロロホルム(10mL)を添加してこの溶液を飽和塩化ナトリウム(2×10mL)および水(2×10mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥し、溶媒を除去して1.55gの油状物を得た。表題の化合物の存在をエレクトロスプレーLC−MSによって確認した;tR=2.4分。MS[M=C15H34N2O8P2]m/z 455(MNa+)。
【0213】
実施例26
化合物A026−92の合成
【0214】
【化57】
783mgのホスホン酸塩(A018−24で調製)を含む18mLのジクロロメタンの溶液をブロモトリメチルシラン(2.2g)で処理した。この溶液を一晩撹拌し、メタノール(10mL)を添加してその混合物を2時間撹拌した。揮発性物質を除去して、1.22gの黄色油状物を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.4分。MS[M=C6H18N2O6P2]m/z 275(M−H−)。
【0215】
実施例27
化合物A030−74の合成
【0216】
【化58】
500mg 4−[(N−BOC)アミノエチル]アニリンを含む10mLのジオキサンの溶液をパラホルムアルデヒド(270mg)および亜リン酸トリメチル(1.12g)で処理した。この混合物を95℃に一晩加熱した。次いでさらなるパラホルムアルデヒド(270mg)および亜リン酸トリメチル(1.12g)を添加して、これを再度、一晩95℃で加熱した。この溶液を冷却して、クロロホルム(20mL)にとり、飽和塩化ナトリウム(20mL)および水(20mL)で洗浄した。有機物を硫酸ナトリウムで乾燥させて、溶媒および過剰の亜リン酸トリメチルを80℃のロータリーエバポレーションによって除去して、1.72gの透明な油状物を得た。表題の化合物の存在を、エレクトロスプレーLC−MSによって確認した;tR=2.9分。MS[M=C18H32N2O8P2]m/z 467(MH+);489(MNa+)。
【0217】
実施例28
化合物A030−84の合成
【0218】
【化59】
870mgのA030−74を含む10mLのジクロロメタンの溶液をブロモトリメチルシラン(1.97g)で処理した。この溶液を一晩撹拌した。メタノール(10mL)を添加してその溶液を15分撹拌し、次いで濃縮して1.12gのオレンジ色の油状物を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.85分。MS[M=C9H16N2O6P2]m/zの実測309[M−H]−。
【0219】
実施例29
化合物A035−66の合成
【0220】
【化60】
2.0g部の4−アミノメチル安息香酸(Aldrich)を、0.64gの固体NaOHを含む20mLの水に溶解した。3.18g部のBoc無水物(Aldrich)を添加して、その混合物を一晩撹拌させた。この混合物に15mLの2N HClを注意深く添加することによってpH=2に調節した。得られた白色固体を濾過して、乾燥させ、2.9997gの生成物を得た。この生成物をLCMS(保持時間2.901分、所望のM−H質量イオン250m/zで観察)によって特徴付けた。
【0221】
実施例30
化合物A035−6の合成
【0222】
【化61】
1.5部のA35−66を17mLの乾燥THF中に、0.69gのN−ヒドロキシスクシンイミド(Aldrich)とともに溶解し、次いで1Mジシクロヘキシカルボジイミド(Aldrich)を含む6mLのジクロロメタンとともに撹拌して一度に処理した。2日後、白色沈殿(ジシクロヘキシル尿素)を濾過して除き、次いで濾液を減圧下でロータリーエバポレートして、2.8146gの白色固体を得て、LCMSによって特徴付けた(保持時間3.299分、そして349m/zで所望のM+Hを観察)。
【0223】
実施例31
化合物A035−14の合成
【0224】
【化62】
A035−6の500mg部を、5mLの乾燥THFに溶解して、1.002mL(10当量)のエチレンジアミン(EDA)で処理して2時間撹拌させた。次いでこの溶液を形成された固体からデカントした。次いで溶媒および過剰のEDAを、減圧下でロータリーエバポレーションによってデカントされた溶液から除去し、0.8728gの白色固体を得た;この生成物をLCMSによって特徴付けた(保持時間1.608分、そして294m/zで所望のM+Hを観察)。
【0225】
実施例32
化合物A032−24の合成
【0226】
【化63】
872mgのアミン(A035−14で調製)を含む10mLのジオキサンの溶液をパラホルムアルデヒド(535mg)および亜リン酸トリメチル(2.21g)で処理した。この混合物を100℃で一晩加熱し、次いで溶媒を80℃のロータリーエバポレーションによって除去して、褐色の固体を得た。クロロホルム(25mL)を添加してその溶液を水(15mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥させて、溶媒を除去して241mgの黄色半固体を得た。これを、LCを介して精製して、58.8mgの所望の物質を得た。表題の化合物の存在を、エレクトロスプレーLC−MSによって確認した;tR=2.6分。MS[M=C21H37N23O9P2]m/z 538(MH+),560(MNa+)。
【0227】
実施例33
化合物A032−40の合成
【0228】
【化64】
54.6mgのホスホン酸塩(A032−24で調製)を含む1mLのジクロロメタンの溶液をブロモトリメチルシラン(156mg)で処理した。この混合物を一晩撹拌させた。エタノール(0.5mL)および水(3滴)を添加して、これを1時間撹拌し、次いで揮発性物質を除去して、その物質を減圧下で乾燥させた。これを水(1mL)にとって、凍結乾燥させ、59mgの黄褐色固体を得た。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.4分。MS[M=C12H21N3O7P2]m/z 380(M−H−),382(MH+),404(MNa+)。
【0229】
実施例34
化合物A026−60の合成
【0230】
【化65】
Kantoci,D.,Kenike,J.K.,Wechter,W.J.Syn.Commun.,1996,26(10),2037の手順を介してA026−60を調製した:N−ベンジル−N−メチルアミン(20.0g)、亜リン酸ジエチル(70.7g)およびオルトギ酸トリエチル(29.3g)の混合物をアルゴン還流下(150℃)で5時間撹拌した。エタノールを70℃のロータリーエバポレートを介して除去して、その混合物を再度、一晩還流して加熱した。この溶液を600mLクロロホルムで希釈して、1Mの水酸化ナトリウム(3×100mL)および飽和塩化ナトリウム(3×150mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去し、74.0gの淡黄色油状物を得た。この物質の10.0gを、溶出液として14:4:1の酢酸エチル:ヘキサン:メタノールを用いるシリカゲルカラムクロマトグラフィーに供した。これによって、6.08gの透明な油状物を得た。表題の化合物の存在を、エレクトロスプレーLC−MSによって確認した;tR=3.4分。MS[M=C17H31NO6P2]m/z 408(MH+),430(MNa+),471(MNa−CH3CN+)。
【0231】
実施例35
化合物A030−54の合成
【0232】
【化66】
Kantoci,D.,Kenike,J.K.,Wechter,W.J.Syn.Commun.,1996,26(10),2037の手順を介してA030−54(CAS#80475−00−9として公知の化合物)を調製した:4.52gのホスホン酸化ベンジルアミン(A026−60で調製)を含むメタノール(45mL)の溶液を10%パラジウムとともに炭素(200mg)上で処理して、水素雰囲気に一晩供した。パラジウム/炭素を濾過して、2.98gの淡黄色の油状物を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=1.9分。MS[M=C10H25NO6P2]m/z 318(MH+)。
【0233】
実施例36
化合物A039−16の合成
【0234】
【化67】
A039−16:500mgのサンプルの4−クロロメチル安息香酸(Aldrich)を、8mLのTHFに溶解し、5当量のエチレンジアミン(Aldrich)(983μL)とともに一度に処理した。24時間後、溶媒を高真空下で除去して、白色固体(93%)をLCMSによって特徴付けた(保持時間0.4分、そして195m/zで所望のM+Hを観察)。
【0235】
実施例37
化合物A038−24の合成
【0236】
【化68】
ジオキサン(10mL)中の679mgのアミノ酸(A039−16で調製)、37%(wt/wt)ホルムアルデヒド水溶液(1.04mL)、亜リン酸(1.15g)および濃(12.1M)塩酸(2.3mL)の混合物を100℃で一晩撹拌した。この溶媒を75℃でロータリーエバポレートを介して除去して、混合物を遠心分離して固体を廃棄した。この液体にさらにホルムアルデヒド溶液(1.04mL)、亜リン酸(1.15g)、濃塩酸(2mL)およびジオキサン(10mL)を添加して、これを再度100℃で一晩撹拌させた。この溶媒を75℃のロータリーエバポレーションによって除去して、濃い油状物を得た。これをLCを介して精製して、276mgの黄褐色の固体を得た。表題の化合物の存在を方法Aを用いてエレクトロスプレーLCMSによって確認した;tR=0.6分。MS[M=C13H23N2O11P3]m/z 475(MH−),477(MH+)。
【0237】
実施例38
化合物A038−50の合成
【0238】
【化69】
6.0gのFmoc−Lys−OH(Advanced ChemTech)を含むメタノール(25mL)および水(25mL)の混合物を37%(wt/wt)ホルムアルデヒド水溶液(6.06mL)および亜リン酸ジメチル(8.96g)で処理した。この混合物を80℃で2時間撹拌して、冷却し、ジクロロメタン(1×100mL、2×50mL)で抽出した。この有機物を飽和塩化ナトリウム(50mL)で洗浄し、硫酸マグネシウムで30分間乾燥し、溶媒を除去して、10.17gの薄緑色の油状物を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.3分。MS[M=C27H38N2O10P2]m/z 613(MH+),636(MNa+)。
【0239】
実施例39
化合物A038−66の合成
【0240】
【化70】
23.9mgのホスホン酸化Fmoc−Lys−OH(A038−50で調製)を含むジクロロメタン(1mL)の溶液を、ブロモトリメチルシラン(60mg)で処理した。この混合物を一晩撹拌した。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.4分。MS[M=C23H30N2O10P2]m/z 555(M−H−)。
【0241】
実施例40
化合物A038−76の合成
【0242】
【化71】
112.9mgのホスホン酸化Fmoc−Lys−OH(A038−50で調製)を含む6M塩酸(3mL)の溶液を、80℃で2日間撹拌した。水(9mL)を添加してさらに2日後、この混合物を遠心分離して液体をデカントした。この固体を減圧下で乾燥させて、86.8mgの灰色の固体を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.1分。MS[M=C23H30N2O10P2]m/z 555(M−H−)。
【0243】
実施例41
化合物A038−90の合成
【0244】
【化72】
500mgのFmoc−Lys−OH(Advanced Chem Tech)を含むジオキサン(5mL)の溶液を、37%(wt/wt)ホルムアルデヒド水溶液(303μL)、亜リン酸(333mg)および濃(12.1M)塩酸(674μL)で処理した。この混合物を90℃で一晩撹拌して、この溶媒を75℃のロータリーエバポレーションによって除去した。表題の化合物の存在を、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=5.4分。MS[M=C23H30N2O10P2]m/z 555(M−H−)。
【0245】
実施例42
化合物A042−18の合成
【0246】
【化73】
1.29gのBoc−保護アミノ酸(上記ロットA035−66と同じく調製)を含む15mLのテトラヒドロフランの溶液をN−ヒドロキシスクシンイミド(623mg)および1.0M 1,3−ジクロロヘキシルカルボジイミドを含むジクロロメタン(5.4mL)で処理した。この混合物を一晩撹拌して、白色沈殿を濾過して、上清を濃縮し、1.89gの白色固体を得た。表題の化合物の存在は、3.3分のUV信号の存在によって示された。
【0247】
実施例43
化合物A042−26の合成
【0248】
【化74】
2.1gのtris−(2−アミノエチル)アミンを含む20mLのテトラヒドロフランの溶液に、1.0gの活性化エステル(A042−18によって調製)を含む20mLのテトラヒドロフランの溶液を40分間にわたって滴下して加えた。この混合物を一晩撹拌して、沈殿物を得て、これを濾過してロータリーエバポレーションによって濃縮し、2.10gの黄色油状物を得た。表題の化合物の存在を、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=1.4分。MS[M=C19H33N5O3]m/z 380(M−H+)、402(MNa+)。
【0249】
実施例44
化合物A042−32の合成
【0250】
【化75】
2.08gのアミン(A042−26で調製)を含むジオキセン(20mL)の溶液を、パラホルムアルデヒド(1.50g)および亜リン酸ジメチル(6.85g)で処理した。この混合物を90℃で一晩撹拌して、その溶媒を70℃のロータリーエバポレーションによって除去した。ジメチルメタン(50mL)を添加して、これを飽和塩化ナトリウム(25mL)および水(25mL)で洗浄した。この有機物を硫酸ナトリウムで乾燥して、溶媒を除去した。この残滓をLCで精製して、123.8mgの黄色油状物を得た。表題の化合物の存在を、方法Dを用いてエレクトロスプレーLC−MSによって確認した;tR=2.2分。MS[M=C31H61N5O15P4]m/z 868(MH+)。
【0251】
実施例45
化合物A042−70の合成
【0252】
【化76】
111.1mgのホスホン酸化ジアミン(A042−32を介して調製)を含む1mLのジクロロメタンの溶液を、194mgのブロモトリメチルシランで処理した。5時間後、メタノール(1mL)を添加した。この混合物を1時間撹拌して、溶媒を除去して113.9mgの黄褐色の固体を得た。表題の化合物の存在は、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=1.0分。MS[M=C18H37N5O13P4]m/z 328(M+2H/2)2+)]、656(MH+)。この化合物をまたプロトンNMR分光法によって分析した:1H(CDCl3)δ:7.77(d,2H,J8.1Hz),7.43(d,2H,J8.2Hz),4.1〜3.3(m,33H)。
【0253】
実施例46
化合物A026−94の合成
【0254】
【化77】
750mgのホスホン酸塩(A030−54を介して調製)を含む24mLのジクロロメタンの溶液を、ブロモトリメチルシラン(2.89g)で処理した。この溶液を一晩撹拌した。メタノール(10mL)を添加して、その溶液を2時間撹拌し、その溶媒を除去して、黄色の油状物を得て、これを凍結乾燥し、364mgの白色固体を得た。表題の化合物の存在は、方法Bを用いてエレクトロスプレーLC−MSによって確認した;tR=0.6分。MS[M=C2H9NO6P2]m/z 204(M−H)−)。
【0255】
実施例47
化合物A026−96の合成
【0256】
【化78】
A042−96(tert−亜リン酸ブチル):4.10gの亜リン酸を含む100mLのテトラヒドロフランの溶液を2−メチル−2−プロパノール(7.41g)で処理した。1,3−ジシクロヘキシルカルボジイミドを含むジクロロメタン(100.0mL)の1.0M溶液を添加して、白色固体の形成を生じた。この混合物を一晩撹拌して、固体を濾過し、溶媒を除去して6.82gの黄色固体を得た。表題の化合物の存在は、GC−MSによって確認した。以下のフラグメントを見出した:57[(CH3)3C+]、83[HP(OH)3+]、123[(HO)2PC(CH3)2+]。
【0257】
実施例48
化合物A042−98の合成
【0258】
【化79】
エタノールアミン(858mg)、パラホルムアルデヒド(1.05g)、tert−亜リン酸ブチル(A042−96,6.82gを介して調製)を含むベンゼン(100mL)の混合物を一晩90℃で加熱して、濃い油状物の上に液体を生じた。この液体をデカントして、ロータリーエバポレーションを介して濃縮し、油状物を得て、これを10%メタノールを含有するジクロロメタンを溶出液として用いるシリカゲルカラムクロマトグラフィーに供した。透明な油状物(436mg)を得た。表題の化合物の存在は、方法Aを用いてエレクトロスプレーLC−MSによって確認した;tR=3.6分。MS[M=C20H45NO7P2]m/z 496(MNa+)。
【0259】
実施例49
化合物A029−34の合成
【0260】
【化80】
4−イソチオシアナートフェニル酢酸を調製するために、1.3ml(13.5mmol、2当量)のチオホスゲンを、20mlの乾燥THFに懸濁した1.0gの4−アミノフェニル酢酸(6.61mmol)[Aldrich]および3.73gの無水炭酸カリウム(4当量)に加えた。この懸濁物を室温で15分間撹拌して、続いて85℃のシリコーン油槽中で4時間加熱した。この溶液を冷却して、濾過シリンジ中で1インチのセライト層を通した。この溶液を丸底フラスコ中で収集して、溶媒を減圧下で除去した。
【0261】
このサンプルを真空デシケーター中に2時間保管し、次いでアセトン−水混合物に溶解し、凍結して凍結乾燥し、1.65gの黒っぽい固体を得た。この化合物は、LCMSクロマトグラムの保持時間の3.32分へのシフトによって同定した。
【0262】
実施例50
化合物A040−22の合成
【0263】
【化81】
4−イソチオシアナートフェニル酢酸クロロメチルエステルを調製するために、0.530gの4−イソシアナートフェニル酢酸(2.7mmol)をガラスバイアル中で5.0mlの塩化メチレンに溶解して、これに、5.0mlの水に溶解した0.044gのテトラ−n−ブチル硫酸水素アンモニウム(相間移動触媒−0.05当量)および0.866gの炭酸水素ナトリウム(4当量)を添加した。この溶液を氷浴中で10分間撹拌した。この冷たい混合物に0.520gのクロロメチルクロロサルフェート(ACROS Chemical−1.2当量)を添加して、室温まで徐々に温度上昇させながら4時間撹拌した。この有機層を分液漏斗で分離して、10mlの飽和ブラインで洗浄し、無水硫酸ナトリウムで乾燥させた。この溶媒を減圧下で除去して、0.703gの粗油状物を得た。この生成物は、LCMSで新しいピークの形成によって同定され、この保持時間は4.268分であり、出発材料に相当するピークの消失があった。
【0264】
実施例51
化合物A040−26の合成
【0265】
【化82】
化合物1101の4−イソチオシアナートフェニル酢酸四級塩誘導体を調製するために、化合物1101(0.300g、1.0mmol)および0.582gのヨウ化ナトリウム(4当量)を、1ドラムのバイアル中で4.0ml乾燥アセトニトリルに溶解した。この4−イソチオシアナートフェニル酢酸クロロメチルエステル(化合物A040−22)を含む2.0mlの乾燥アセトニトリルを撹拌しながら添加した。この混合物をシリコーン油浴上で、65℃で5時間加熱して、LCMSによって反応の進行をモニタリングした。化合物1101出発材料のほとんどを消費した場合、この反応混合物を、3.019分、m+=513の分取LCMS保持時間を用いて精製した。94%純粋な生成物の194.1mgの収量を得て、LCMSによって同定した。次いで化合物A040−26を、化合物1105を利用する実施例に記載された方法によって記載されるように求核試薬保有標的化因子と反応させて、有用な標的プロドラッグを調製する。
【0266】
実施例52
化合物A044−52の合成
【0267】
【化83】
N−ベンジル−N−メチルカルボニルクロロメチルエステルを調製するために、0.300mg(324μL,d=0.942)ベンジルメチルアミンおよび640μLジイソプロピルエチルアミン(1.5当量)を2.0mlの乾燥塩化メチレンに溶解して、氷浴中で冷却した。この溶液が冷却された場合、330μLのクロロメチルクロロホルメート(1.5当量)を含む2.0mlの乾燥塩化メチレンを添加して、2時間撹拌し、この溶液を徐々に室温に戻させた。黄色溶液を分液漏斗に入れて、2×10ml 1N HCl、1×10ml水および2×10ml 1N 重炭酸ナトリウムを用いて洗浄した。この有機層を硫酸ナトリウムで乾燥して、減圧下で濃縮した。カルバミン酸クロロメチルを、0.95gの黄色油状物として得て、LCMSにおいてuv活性成分として3.520分の保持時間で同定した。
【0268】
化合物1101のN−ベンジル,N−メチルカルバモイルメチル四級塩を調製するために、0.5gの化合物1101および0.5gのヨウ化ナトリウム(20当量)を、ガラスバイアル中で5.0ml乾燥アセトニトリルに溶解した。このN−ベンジル、N−メチルカルバモイルクロロメチルエステルを溶液に追加し、次いで一晩65℃の油浴に入れた。この生成物を、LCMSによって同定して、分取LCMSによって精製し、保持時間2.731分、M+=485、そして90%の純度で、A044−52と命名された169.5mg(理論収率21.5%)の固体を得た。
【0269】
実施例53
化合物A044−62の合成
【0270】
【化84】
化合物1157のN−ベンジル,N−メチルカルバモイルメチル四級塩を調製するために、22mgの化合物1157、20mgのヨウ化ナトリウム(2.0当量)および30.7mgのN−ベンジル−N−メチルカルボンモイルクロロメチルメステル(2.0当量)を、ガラスバイアル中で500μLの乾燥アセトニトリルに混合し、一晩65℃の油浴中で加熱した。この生成物を、LCMSによって同定して、分取LCMSによって精製した。保持時間2.810分、M+=483および98%純度で5.4mgの化合物(理論収率15.5%)を得た。
【0271】
実施例54
化合物A044−28の合成
【0272】
【化85】
化合物1101のベンジルホルモイル−1−エチル四級塩を調製するために、200μL 1−クロロエチルクロロホルメートを、ガラスバイアル中で100μLのベンジルアルコールを含む2.0mlの乾燥塩化メチレンに添加して、氷浴中で0℃でインキュベートした。冷却された溶液に200μLのピリジンを添加して、2分内に白色沈殿を形成させた。撹拌を室温で一晩継続した。この混合物に10mlの塩化メチレンを添加し、次いで1×10ml 0.5M HCl、1×10ml水および1×10ml 0.5N重炭酸ナトリウム溶液を用いて洗浄した。この有機層を硫酸ナトリウムで乾燥させて、減圧下で溶媒を除去した。ベンジル−1−ギ酸クロロメチルを、3.933分の保持時間のuv活性成分として同定した。
【0273】
1.0mlの乾燥アセトニトリルに溶解した100mgの化合物1101および100mgのNaI(20当量)に107mg(1.5当量)のベンジル−1−ギ酸クロロメチルを添加した。この混合物を65℃で一晩加熱した。LCMSで出発物質の存在が示されたため、さらなる107mg(1.5当量)のベンジル−1−ギ酸クロロエーテルを添加して、さらに24時間加熱を続けた。LCMSによって、緩慢勾配クロマトグラフィーで出発物質から分離され得る生成物を同定した。所望の化合物を分取LCMSを用いて単離して、保持時間4.690分、M+=488、98.6%純度で13.7mg(8.7%理論収率)の化合物を得た。
【0274】
実施例55
化合物1126の合成
クロロメチル−t−ブチルスクシネートを調製するために、モノt−ブチルスクシネート(Aldrich)、2.0gを8.0mlの塩化メチレンに溶解したものを、4.0gの炭酸カリウムと0.24gのテトラn−ブチル硫酸水素アンモニウムを8.0mlの水に含むガラスバイアルに添加して、氷浴で撹拌した。15分後、1.3mLのクロロメチルクロロサルフェート(Acros)を塩化メチレン層に添加して、反応混合物をゆっくり室温になる温度で撹拌した。有機層を分離して1×10mlの水および1×10mlの飽和ブライン溶液で洗浄した。この溶液を無水硫酸ナトリウムで乾燥して、溶媒を減圧下で除去した。3gの淡黄色油状物を得て、ロット番号A047−71とした。
【0275】
上記のA047−71の3gを36mLのアセトニトリルに溶解して、1.8gの化合物1101および1.8gのNaIで処理し、撹拌しながら16時間ヒーター上に置いた。反応混合物(沈殿を含む)を水と塩化メチレンとの間で分画して、塩化メチレン層を分離して、ブラインで洗浄し、硫酸ナトリウムで乾燥しエバポレートして、暗い油状物を得た。この油状物を5mLのアセトニトリルに溶解して、2日間冷蔵庫に保管した。次いで、形成した黄色沈殿を濾過して3mLのアセトニトリルで洗浄し乾燥して、ロット番号A046−67Aとして、2.8435gの95%を超える純度の、保持時間2.719分、494m/zの[M+]という生成物を得た。この生成物の全てを10mLの塩化チオニルに溶解して、65℃に4時間加熱した。過剰の塩化チオニルを減圧下で除去して、黄色油状物を高真空下で乾燥して1.9906gの酸塩化物(化合物1111)を黄色のバリバリした固体として得た。この固体を引き続く反応に直接用いた。
【0276】
化合物1101酸塩化物およびde−FMOCed RGDSペプチドをカップリングするために、化合物1111の1.26gの酸塩化物および5.6g de−FMOC除去RGDSペプチド(両方とも五酸化リン上で、真空デシケーターで乾燥させた)をアルゴンガス下で50mlの丸底フラスコ中に混合した。この固体に270μLの乾燥ピリジン含有28ml塩化メチレンを添加して、この混合物を振盪して酸塩化物を溶解した。この混合物をオービタルシェイカーに1時間おいた。この溶液をガラス質プラスチックシリンジで排液して、2×10mlの塩化メチレンで洗浄して、溶媒を排液した。この樹脂に500μLのアニソール、続いて20mlの50/50 TFA/塩化メチレン溶液を添加した。この樹脂を時折撹拌しながら、TFA溶液中に3時間静置させた。TFA溶液を樹脂から排液した。この樹脂をTFA溶液と組み合わせた10mlの塩化メチレンで洗浄した。TFA溶液を4つのバイアルに入れて、これをアルゴンガスで風乾した。各々のバイアルを複数回のエーテル洗浄で処理し、再度アルゴンガスで風乾した。この生成物をLCMSによって同定し、そして分取逆相LCMSを用いて17回の実施で精製した。この組み合わせた実施によって、163.7mgの96%純度の生成物(ロット番号a036−33と指定)を保持時間1.768分、M+=853そして427m/zで[M+H]/2で得た。この化合物についてのLC−MSクロマトグラムおよび質量スペクトルは、図7および図8に示す。図7では、x軸が分単位の時間であり、そして上のクロマトグラムのy軸は254nmでのUV検出器のミリ吸光度単位であり、そして下のクロマトグラムのy軸は、蒸発光散乱検出器によって検出されるミリボルトである。図8では、x軸は質量対電荷比(m/z)であり、y軸は質量イオンカウントの強度である。
【0277】
スキーム10
【0278】
【化86】
実施例56
化合物A052−10の合成
【0279】
【化87】
化合物1101のフタルイミドメチル四級塩を調製するために、100mgの化合物1101および100mgのヨウ化ナトリウム(2.0当量)の混合物を3.0mlの乾燥アセトニトリルに溶解した。この混合物に128mgのクロロメチルフタルイミド(Aldrich)を添加して、このバイアルを55℃の油浴中で4日間加熱した。この生成物を、保持時間3.984分、M+=467で新しいピークとしてLCMSによって同定した。
【0280】
実施例57
化合物A052−08の合成
【0281】
【化88】
化合物1101のフタルイミドメチル四級塩を調製するために、100mgの化合物1101および100mgのヨウ化ナトリウム(2.0当量)の混合物を3.0mlの乾燥アセトニトリルに溶解した。この混合物に128mgのクロロメチルフタルイミドを添加して、このバイアルを55℃の油浴中で4日間加熱した。この生成物を、LCMSによって、Rf=3.984、M+=467で新しいピークとして同定した。
【0282】
実施例58
化合物A044−78の合成
【0283】
【化89】
化合物1101の4−カルボキシベンジル四級塩を調製するために、300mgの化合物1101、400mgのヨウ化ナトリウムおよび500mgのクロロメチル安息香酸を、ガラスバイアル中で混合して、4.0mlの乾燥アセトニトリル中で懸濁した。この反応物を、65℃で油浴において加熱して、LCMSによって2週間モニターした。この溶液をガラス質プラスチックシリンジで濾過してさらなる2ミクロンのフィルターと適合させた。この所望の化合物を分取LCMCを用いて単離したが、これは3.717分の保持時間、M+=442であった。96%純度の27.7mgの収量を得た。化合物A044−78のカルボン酸基を、a)NHS活性エステルを調製するための実施例31の方法によって記載されるようなN−ヒドロキシスクシンイミドとの反応、またはb)実施例56の方法によって記載されるような酸塩化物への変換によって反応性の基に変換する。次いで、これらの反応性基のいずれかを、前の実施例に記載した方法を用いて標的化因子の求核アミンまたはアルコール基と反応させて標的化プロドラッグ結合体を得る。
【0284】
実施例59
化合物A044−80の合成
【0285】
【化90】
化合物1101の4−イソシアナトベンジル四級塩を調製するため、300mgの化合物1101、450mgのヨウ化ナトリウム(3.0当量)および490mg(3.0当量)の4−クロロメチルベンゼンイソシアナートの混合物を4.0mlの乾燥アセトニトリルに溶解して、油浴中で65℃で2日間加熱した。LCMSによって、この反応は、出発材料(化合物1101)がないために終了したことが示された。この生成物は、4.577分の保持時間、M+=439で新しいピークとして同定された。化合物A044−80は、種々の標的化因子の求核性基と反応して、化合物1105およびA040−26を利用する実施例の方法によって、標的化因子に対するカルバミン酸塩または尿素結合を生じる。
【0286】
実施例60
化合物A044−4の合成
【0287】
【化91】
化合物1101のピボロイルメチル四級塩を調製するために、100mgの塩化ピボロイル(2.0当量)を、100mgの化合物1101および100mgのヨウ化ナトリウム(2.0当量)を含む2.0mlの乾燥アセトニトリルの混合物に滴下して加えた。この混合物を65℃の油浴で2時間加熱した。この固体は2ミクロンフィルターを装着したガラス質のプラスチックシリンジを用いて濾過した。この化合物を同定して、分取LCMSを用いて単離した。保持時間2.735分、M+=422の黄色固体を得たが、これは93.7%の純度で102.3mgの収率であった。
【0288】
実施例61
化合物A040−70の合成
【0289】
【化92】
化合物1101のアセトキシメチル四級塩を調製するために、1.0gの化合物1101を、ガラスバイアル中で10ml乾燥アセトニトリルに溶解して、1.0gの酢酸ブロモメチル(2.0当量)を添加して、室温で一晩撹拌させた。LCMSによって、出発物質の存在が示され、そして反応物を65℃で、油浴中で8時間加熱した。母液を固体からデカントして、固体を少量の冷たいアセトニトリルで洗浄した。この固体を真空デシケーターで一晩乾燥させた。この生成物を、97%の純度である、2.204分の保持時間、M+=380の264mgの白色固体として同定した。この化合物は、極めて高い水溶性を有することが見出された;リン酸緩衝化生理食塩水中のこの化合物の32.5ミリモル溶液を、1.865mLのリン酸緩衝化生理食塩水中で27mgを溶解することによって調製して約4というpHを得た。この溶液の50μLのアリコート(これはプロドラッグとして化合物1101の1.625μモルを含む)を、ヌードマウスの尾静脈に注射したが、有害な観察可能な影響はなく、このプロドラッグ形態の毒性のないことが示されたが、化合物1101の1.04μモルの注射では3匹のマウス中3匹で即時的な死亡が生じた。さらに、化合物A040−70のこの32.5mMの溶液の250μLを、経口胃管によってヌードマウスに投与した。5、15および30分後、40μLのマウス血液を得て、LC−MSによって分析し(アセトニトリルを用いる抽出後)、プロドラッグA040−70および化合物1の組み合わせた血液レベルが、それぞれの時点で1.8、4.97および4.80マイクロモルであることが実証された。この結果によって、インビボにおけるプロドラッグから活性薬物の経口のバイオアベイラビリティーが実証される。少量の水にこの化合物A040−70を溶解すること、およびAldrichから入手可能なDowex22(塩化物型)のような陰イオン交換樹脂層を通過させること、そして次に水および凍結乾燥の混合性溶離剤を用いて洗浄して臭化物イオンが塩化物イオンで交換された固体を得ることによって塩化物を得る。
【0290】
実施例62
非イオン性水溶性のためのポリオキシエチレン保有プロドラッグの調製
【0291】
【化93】
化合物1の78mg部分を76mgのNaIとともに2mLのアセトニトリルに溶解して、144mgのクロロメチルエステルA029−62で一度に処理して、65℃で5時間撹拌した。この反応物を、逆相LC−MSによって精製して、2.30分の保持時間で72mg(51%収率)の黄色固体A027−85を得たが、これはC30H38NO9によって期待されるとおりM+=556を示す質量スペクトルを有することによって特徴づけられた。わずかなサンプルをpH=7.4のリン酸緩衝液、続いてLC−MSに経時的に導入して、ここでこのプロドラッグのかなりの部分が、約3時間という変換の推定半減期で、化合物1に戻された。
【0292】
【化94】
実施例63
化合物1の骨標的化プロドラッグの調製
75mモル A042−70(1.5μモル)の20μL部分を含む水を、500mモルのリン酸緩衝液を含むバイアル(0.5単位で増す3.0〜8.0にわたる11の種々のpHの11のバイアル)に添加した。次いで、各々のバイアルを混合した後、60μモル化合物1111を含むアセトニトリルの50μL(3.0μモル=骨標的化因子のアミノ基に対して2当量)で処理して、振盪することによって混合した。1時間後、各々のバイアルの3μLのアリコートをHPLCに注入して、UVピーク領域を出発物質、ならびに化合物1101の加水分解の所望の生成物および副産物について決定した。6、6.5、7、7.5および8というpHのサンプルのみが、所望の骨標的化プロドラッグA046−89P(保持時間2.50分;[M+]1075m/z C42H59N6O19P4について実測)[M+2]/2=538 m/zも実測)の有意な量を有することが見出された。本実施例によって、これらの条件下の骨標的化プロドラッグの合成のための最適pHはpH=7.0であり、4つのホスホン酸基を有する化合物1の所望の骨標的化プロドラッグの約42%という理論収率が得られることが実証される。24時間後、pH=7.0の時間を示す同じ溶液の分析によって、標的化プロドラッグが化合物1に完全に戻されたことが示され、これによって生理学的に関連する条件下での可逆性が実証される。
【0293】
実施例64
非小細胞肺癌に対する化合物1126のインビボ有用性
約30グラムの体重の4〜6週齢の雄性ヌードマウスに、0日目に5百万の腫瘍細胞(ヒト非小細胞肺癌細胞:H1299)を右脇腹に皮下に接種した。腫瘍細胞を増殖させた14日後、動物を各5匹の3群に分けた。1群にはビヒクルコントロールのみを与えた。1群にはプロドラッグ(すなわち、化合物1)の活性成分の25mg/kg/日用量レベルに対応して、24.4ミリモル溶液の化合物1126を含むリン酸緩衝化生理食塩水の50μLの容積の1日2回尾静脈(i.v)注射を与えた。最終群には、プロドラッグ(化合物1)の活性成分の5mg/kg/日の投薬レベルに対応して、4.9ミリモル溶液の化合物1126を含むリン酸緩衝化生理食塩水の50μLの容積の1日2回尾静脈(i.v)注射を与えた。ノギスを用いて3日ごとに腫瘍を測定して、腫瘍容積を決定して、27日で動物を屠殺したときに動物の重量を記録した。その結果を表7に示すが、これによって処置のわずか3日後に最初のデータポイントで(17日目)、そしてこの研究の終わりまで続く、両方の用量レベルについてのコントロールに対する強力な腫瘍容積の低下が示される:
表7
【0294】
【化95】
*ビヒクルのみのコントロール動物に対して比較
1日2回の用量は、2週間の投与期間にわたって十分耐容された。上記の有効性の結果ではまた、コントロール群とこの処置群との間で動物の体重の統計学的に有意な相違を伴わず、これによって、全体の毒性の一般的な測定として、標的されたプロドラッグが所望のとおり毒性を欠失していたことが示される。
【0295】
実施例65
脳の癌に対する化合物1126のインビボ有効性
ヒト脳癌細胞株(U87MG)を用い、初回の腫瘍容積測定を10日目に行なうように7日目に処置を開始すること以外は上記の実施例に記載のとおり動物実験を行なった。この癌細胞株に対する化合物1126の結果を、表8に示しており、そして有効性および望まれるような毒性の欠失が示された:
表8
【0296】
【化96】
*ビヒクルのみのコントロール動物に対して比較
実施例66
α v標的化PI 3キナーゼインヒビターは、Matrigel上でEDC−CBF1内皮細胞の管腔形成を抑制した
管腔形成は、インビボである程度の血管形成の形成を示す。本実施例では、どの程度PI 3キナーゼインヒビター(標的化PI3キナーゼインヒビタープロドラッグを含む)が管腔形成を阻害できるかを決定した。Matrigelを12ウェルプレートウェルにプレートして、37℃で2時間固めた。次いで1×105 EDC−CBF1内皮細胞をPBS、RADfV(環状陰性コントロールペプチド)、RGDfV(環状陽性コントロールペプチド)、RADS(直線状陰性コントロールペプチド)、化合物1または化合物1126の存在下でMatrigel層の上に20°M濃度で一晩置いた。次いで顕微鏡を用いて写真を撮った。十分形成された管腔はPBSコントロールウェル中で可視化することができる(図の上左側のパネル)。これらはPBSコントロールに比較してRGDfV、RADfVまたはRGDS含有ウェルでそれほど異なることはなかった。管腔形成は、化合物1101および化合物1126を含有するウェルでは有意に少なかった。
【0297】
実施例67
標的化PI3キナーゼインヒビターは、HBECにおいてp53転写活性を誘導した
本実験で、p53ルシフェラーゼ活性の誘導に対するPI 3キナーゼインヒビター(化合物1および化合物1の標的化プロドラッグバージョン;化合物1126)の効果を試験した。このトランスフェクション手順は、p53転写をモニターするために文献に記載された手順と同様であった。化合物1(6時間曝露)は、コントロールよりも2倍高いルシフェラーゼ活性を誘導し、そして化合物1の標的化バージョン(化合物1126)はp53ルシフェラーゼ活性を誘導する能力がさらに優れていた(ほぼ3倍)。p53機能のこの誘導は、p53インヒビターであるピフィスリン(pifithrin)αの20μM濃度によって抑止されることが実証された。触媒活性Aktの同時トランスフェクションはまた、これらの化合物によって誘導されたp53機能を阻害した。この結果によって、PI3キナーゼインヒビターによって誘導されたp53転写が、全シグナル伝達カスケードにおいてAktの下流であり、標的化プロドラッグ化合物1126が未標的薬物である化合物1に対してp53の誘導を増強したことが示される。
【0298】
実施例68
化合物1126の精製
反応混合物A044−84(2.33g)を、別々の0.33gのサンプルに秤量して、1容積部のアセトニトリル、1容積部の水、および1容積%の酢酸を含む800μlの溶液中に分取クロマトグラフィーの直前に溶解した。この溶液の400μlを各々の分取クロマトグラフィー実施について注入した。ポンプA溶離剤は0.1%酢酸を添加したB&J水(365−4)であって、ポンプB溶離剤は、0.1%酢酸を添加したB&Jアセトニトリル(015−4)であった。最初に、溶離剤は10%Bであって、次いで4分の期間にわたって直線的に34%Bに増大し、次いで4.25分で直線的に95%Bまで増大し、5.25分まで保持し、次いで5.50分で10%Bの開始濃度に直線的に低下して戻った。総ポンプ流量は20mL/分であった。自動サンプリング装置が次回のためにサンプリングしている間にシステムの再平衡を達成した。この勾配を用いて、正の質量スペクトルピークを853(m/z=1)および427(m/z=2)を有する生成物が3.37分で溶出した。ELSDで検出されたシグナルが10mvを超えた場合、分取クロマトグラフィー実施の間に画分を収集した。生成物を含む画分は、2倍過剰の水(容積で)を用いて溶出して、収集の直後にドライアイス−アセトン浴を用いて凍結乾燥容器中で凍結させた。24〜48時間にわたる凍結乾燥後、95%の純度を有する白色のフワフワした固体として化合物1126の全部で180mgを得た。本実施例によって、注意深いpH制御で不安定なプロドラッグ1126は、水性ベースの逆相分離方法を用いて高い純度で単離できることが実証される。
【0299】
実施例69
SCN反応性プロドラッグの調製
【0300】
【化97】
実施例20の方法によって調製したA014−48の184mg部を、154mgの化合物1とともに12mLのアセトニトリルに溶解して、室温で撹拌した。16時間後、LC−MSによって、所望の四級アンモニウム化合物への約65%の変換が示されたので、さらに45mgのヨード化合物を添加して、さらに22時間後に反応は80%終了したので、さらに40mgのヨード化合物を添加して、さらに24時間撹拌させた。次いでこの固体を遠心分離して、アセトニトリルで洗浄して、2.89分の保持時間で特徴付けられる、282.6mg(45%収率)の所望の生成物が高純度で得られ、これは、化合物COMPOUND1105としてC28H23N2O5Sについて499という所望のM+を示した。
【0301】
メタノールに入れた際、化合物1105はメタノールときれいに反応して、保持時間2.78分であってC29H27N2O6Sについて531という予想されるM+で化合物A013−94が得られた。これによってアルコールとのイソチオシアネートプロドラッグの反応がリンカーに対するカルバミン酸塩結合を生じることが示される。
【0302】
このイソチオシアネート中間プロドラッグはまた、塩化メチレン(必要に応じてトリメチルアミンの存在)中で二級アミンを含む種々のアミンと反応してA017−55(C35H38N3O7Sについて644m/zという所望のM+を示す、保持時間2.84分);A017−57(C33H34N3O7Sについて616m/zという所望のM+を示す、保持時間2.46分)およびA027−15(C38H48N3O11P2Sについて816m/zという所望のM+を示す、保持時間2.82分)のような尿素生成物が生じる。これらの実施例によって、所望の連結されたプロドラッグを生じるための求核試薬の多様な基を有するイソチオシアネートプロドラッグの良好な収率での反応が実証される。A017−55化合物はさらに、トリフルオロ酢酸と反応して、驚くべき事にt−ブチル基の切断の経過の間に環状化合物A017−59が得られた(C31H28N3O6Sについて570m/zという所望のM+を示す保持時間2.47分)。本実施例によって、さらなる化学がプロドラッグ化合物のリンカー基上で行なわれ得るということが実証された。ホスホン酸塩緩衝液中でpH=7.4で、LC−MSによって追跡したところ、この化合物のかなりの量が17時間にわたって化合物1に戻った。
【0303】
実施例70
化合物1対標的化プロドラッグの急性毒性
静脈内に投与された化合物1は3匹のヌードマウスにおいて5分内の投与で16mg/kgという用量レベルで100%の死亡率を有することが確認された。実施例56の方法によって調製された化合物1126は、3匹のヌードマウスに37%より高い用量レベル(22mg/kg)で静脈内注射された場合、観察の1時間後でさえ0%の死亡率を示した。本実施例は、このプロドラッグの処方物の能力の改善および化合物1のプロドラッグの毒性の減少を示す。
【0304】
実施例71
化合物1のアナログ
【0305】
【化98】
実施例56の化合物1126について記載されたような中間体A046−67SMの調製の間、副産物が観察された。この物質をLC−MSによって精製して、単独の化合物A037−94を得たが、この化合物は提案された構造のプロトンNMRと一致し、3.00分という保持時間を与え、そして質量スペクトルは338m/z(極めて低値)という予想される[M+H]+および379m/zのそれより大きな[M+H+41(ACN)]を示した。本実施例によって、さらなるプロドラッグ修飾に適切な化合物1の新規なアナログの単離が示される。
【0306】
実施例72
マウスにおいて化合物1を送達するためのプロドラッグの使用
マウスに100万個の非小細胞肺癌細胞(H1299)を皮下注射して、腫瘍塊が約10〜15mm×7〜9mmの直径になるまで約7日間増殖させた。動物に標的化プロドラッグ化合物1126を、i.v.(50μL)またはi.p.(50μL)のいずれかで、32.6ミリモル溶液の化合物1126を含むリン酸緩衝化生理食塩水とともに注射した。60分後、マウスを屠殺して、腫瘍を取り出した。腫瘍の3つの小片を取り出して切り刻んだ。プロドラッグの全てを化合物1に変換させるために24時間経過した後、腫瘍サンプルをアセトニトリルで抽出した。LC−MSによる定量によって、抽出可能な化合物1の濃度(遊離の化合物1および化合物1126由来の合計として)は、I.P.注射のための腫瘍片中で157±7ナノモル、そしてI.V.注射のための腫瘍片中で271±17ナノモルであったことが示された。本実施例によって、標的化プロドラッグを用いる腫瘍組織への化合物1の送達が実証される。
【0307】
実施例73
化合物1を形成するためのプロドラッグの可逆性
プロドラッグ化合物を、水またはDMSOに(水に自由には溶解しない場合)溶解し、次いで、50mMリン酸緩衝液pH=7.4またはpH=4.8中に少なくとも10倍に希釈して、室温で静置させた。この化合物の最終濃度は水性環境中で50〜500μモルに及んだ。アリコートを経時的に採取して、LC−MSによって分析し、プロドラッグの消失の確認および薬物(化合物1)の出現の確認の両方を行なった。化合物1126は、pH=7.5で約1時間の半減期、そしてpH=4.8で約64時間の半減期を有することが見出された。化合物1110は、pH=7.4で約10時間、そしてpH=4.8で120時間を超える半減期を有することが見出された。化合物A040−70は、pH=7.4で約10時間の半減期を有することが見出された。これらの実験によって、化学的にプロドラッグが薬物(化合物1)に変換し、このプロドラッグの消失は極めてpH依存性であり、この変換は生理学的なpHで極めて急速に生じ、そして酸性のpHで実質的に遅いことが実証される。
【0308】
実施例74
腫瘍に局在する結合体の合成
求電子性の基を保有する化合物(例えば、化合物A036−48B、1105、1107、1111、A024−79および1113)は、アルコール、アミノおよびチオール基のような求核性基を保有するポリマーと反応し得る。2000〜100,000の分子量を有するN−(2−ヒドロキシプロピル)メチルアクリルアミド(HPMA)は、トリエチルアミンまたはジイソプロピルエチレンアミンの存在下で塩化メチレンまたはテトラヒドロフランのような非プロトン性有機溶媒中で過剰の化合物1111と反応され、次いでサイズ排除クロマトグラフィー、超遠心分離、またはメタノールもしくはエーテルのような別の溶媒中での沈殿によって分離される。このように沈殿または分離されたポリマーは実質的に1111を含まず、そして腫瘍の近位で経時的に活性な化合物1を遊離して、抗腫瘍効果および血管新生阻害効果を生じる、腫瘍に局在する結合体として用いられる。同様にポリグルタミン酸は、カルボジイミドカップリングを用いる、カルボン酸と過剰なジアミンとの反応によってポリ求核性保有基に変換され得、続いてサイズ排除クロマトグラフィーまたは逆相HPLC精製によってポリグルタミン酸のポリ求核性バージョンが得られる。次いで、これらのポリマーは、トリエチルアミンまたはジイソプロピルエチレンアミンの存在下で塩化メチレンまたはテトラヒドロフランのような非プロトン性有機溶媒中で過剰部分の化合物1111または1105またはA036−48BまたはA024−79と直接的に反応され得、次いでサイズ排除クロマトグラフィー、超遠心分離、またはメタノールもしくはエーテルのような別の溶媒中での沈殿によって分離され得る。このように沈殿または分離されたポリ結合体化ポリマーは実質的に低分子量の残留プロドラッグを含まず、そして腫瘍の近位で経時的に活性な化合物1を遊離して、抗腫瘍効果および血管新生阻害効果を生じる、腫瘍に局在する結合体として用いられる。
【特許請求の範囲】
【請求項1】
以下の式:
【化100】
の化合物2と、以下の式:
【化101】
のハロメチルエステルとを反応させる工程を包含するプロセスによって作製される化合物であって、
ここで、
halは、ハロゲン原子であり;
Z1およびZ2は、Oを表し;
Z3およびZ4は、Oを表し;
R1およびR2は、独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノを表すか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3は、H、必要に応じて置換された脂肪族および必要に応じて置換されたアリールを表し;
R4およびR5は、独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、アルコキシ、カルボキシを表すか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、アルコキシ、アミノ、またはカルボキシを表し、これらのうちの任意が、必要に応じて、標的化因子(T)で置換されており;そして
R7は、−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−または−CH(CH(CH3)2)−を表す、
化合物。
【請求項2】
請求項1に記載の化合物であって、R1−環A−R2が、以下:
【化102】
からなる群より選択され、ここで、R4−N−R5が、以下:
【化103】
からなる群より選択され、ここで、R6が、以下:
【化104】
からなる群より選択される、化合物。
【請求項3】
請求項1に記載の化合物であって、R6が、標的化因子で置換されて、R6−Tを形成し、以下:
【化105】
からなる群より選択される、化合物。
【請求項4】
請求項1、2、または3に記載の化合物であって、前記標的化因子が、炭水化物、ビタミン、ペプチド、ペプチド模倣物、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤、軟骨親和性剤、ジアゼピン、グルコース、ガラクトース、マンノース、またはマンノース6−リン酸から選択される、化合物。
【請求項5】
請求項4に記載の化合物であって、前記標的化因子がRGD部分であるペプチドである、化合物。
【請求項6】
請求項5に記載の化合物であって、前記RGD部分が、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択される、化合物。
【請求項7】
請求項1に記載の化合物であって、化合物2が、以下の式:
【化106】
である、化合物。
【請求項8】
請求項7に記載の化合物であって、R6が、標的化因子で置換されて、R6−Tを形成し、以下:
【化107】
からなる群より選択される、化合物。
【請求項9】
請求項8に記載の化合物であって、前記標的化因子が、炭水化物、ビタミン、ペプチド、ペプチド模倣物、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤、軟骨親和性剤、ジアゼピン、グルコース、ガラクトース、マンノース、またはマンノース6−リン酸から選択される、化合物。
【請求項10】
請求項9に記載の化合物であって、前記標的化因子がRGD部分であるペプチドである、化合物。
【請求項11】
請求項10に記載の化合物であって、前記RGD部分が、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択される、化合物。
【請求項12】
請求項1に記載の化合物であって、前記ハロメチルエステルが、ハロメチル−t−ブチルスクシネートである、化合物。
【請求項13】
請求項12に記載の化合物であって、前記プロセスが、さらに以下:
(a)化合物2とハロメチル−t−ブチルスクシネートとを反応させることによって作製される生成物からt−ブチル基を除去して、それによって遊離カルボン酸基を生成する工程;
(b)(a)の生成物の該カルボン酸基を酸塩化物または活性エステルに変換する工程;および
(c)(b)の生成物を、遊離アミノ基を含む標的化因子(T)と接触させる工程
を包含する、化合物。
【請求項14】
請求項13に記載の化合物であって、前記ハロメチル−t−ブチルスクシネートがクロロメチル−t−ブチルスクシネートであり、化合物2がLY294002である、化合物。
【請求項15】
請求項13に記載の化合物であって、前記工程(c)の標的化因子(T)が、炭水化物、ビタミン、ペプチド、ペプチド模倣物、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤、軟骨親和性剤、ジアゼピン、グルコース、ガラクトース、マンノース、またはマンノース6−リン酸から選択される、化合物。
【請求項16】
請求項15に記載の化合物であって、前記標的化因子がRGD部分であるペプチドである、化合物。
【請求項17】
請求項16に記載の化合物であって、前記RGD部分が、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択される、化合物。
【請求項18】
請求項16に記載の化合物であって、前記保護されたRGDS部分が遊離アルギニンα−アミノ基を有する、化合物。
【請求項19】
請求項18に記載の化合物であって、前記プロセスが、前記RGDS部分から保護基を除去する工程(d)をさらに包含する、化合物。
【請求項20】
請求項19に記載の化合物であって、前記プロセスが、分取逆相液体クロマトグラフィー質量スペクトル(LCMS)を使用して(d)の生成物を同定および単離する工程をさらに包含し、該単離された生成物が、LCMSの正のモードにおいてM+=853の質量を有する、化合物。
【請求項21】
請求項19に記載の化合物であって、前記プロセスが、任意のアニオン対イオンを塩化物対イオン(塩)に交換する工程をさらに包含する、化合物。
【請求項1】
以下の式:
【化100】
の化合物2と、以下の式:
【化101】
のハロメチルエステルとを反応させる工程を包含するプロセスによって作製される化合物であって、
ここで、
halは、ハロゲン原子であり;
Z1およびZ2は、Oを表し;
Z3およびZ4は、Oを表し;
R1およびR2は、独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、ヒドロキシル、ハロゲン、アルコキシ、複素環式、シアノ、アミノを表すか、または一緒になって、必要に応じて置換された脂環式もしくは必要に応じて置換されたアリールを形成し;
R3は、H、必要に応じて置換された脂肪族および必要に応じて置換されたアリールを表し;
R4およびR5は、独立して、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、アルコキシ、カルボキシを表すか、または一緒になって、必要に応じて置換された複素環式もしくは必要に応じて置換されたヘテロアリールを形成し;
R6は、H、必要に応じて置換された脂肪族、必要に応じて置換されたアリール、複素環式、アリールオキシ、アルコキシ、アミノ、またはカルボキシを表し、これらのうちの任意が、必要に応じて、標的化因子(T)で置換されており;そして
R7は、−CH2−、−CH(CH3)−、−CH(Ph)−、−C(CH3)(COOH)−または−CH(CH(CH3)2)−を表す、
化合物。
【請求項2】
請求項1に記載の化合物であって、R1−環A−R2が、以下:
【化102】
からなる群より選択され、ここで、R4−N−R5が、以下:
【化103】
からなる群より選択され、ここで、R6が、以下:
【化104】
からなる群より選択される、化合物。
【請求項3】
請求項1に記載の化合物であって、R6が、標的化因子で置換されて、R6−Tを形成し、以下:
【化105】
からなる群より選択される、化合物。
【請求項4】
請求項1、2、または3に記載の化合物であって、前記標的化因子が、炭水化物、ビタミン、ペプチド、ペプチド模倣物、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤、軟骨親和性剤、ジアゼピン、グルコース、ガラクトース、マンノース、またはマンノース6−リン酸から選択される、化合物。
【請求項5】
請求項4に記載の化合物であって、前記標的化因子がRGD部分であるペプチドである、化合物。
【請求項6】
請求項5に記載の化合物であって、前記RGD部分が、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択される、化合物。
【請求項7】
請求項1に記載の化合物であって、化合物2が、以下の式:
【化106】
である、化合物。
【請求項8】
請求項7に記載の化合物であって、R6が、標的化因子で置換されて、R6−Tを形成し、以下:
【化107】
からなる群より選択される、化合物。
【請求項9】
請求項8に記載の化合物であって、前記標的化因子が、炭水化物、ビタミン、ペプチド、ペプチド模倣物、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤、軟骨親和性剤、ジアゼピン、グルコース、ガラクトース、マンノース、またはマンノース6−リン酸から選択される、化合物。
【請求項10】
請求項9に記載の化合物であって、前記標的化因子がRGD部分であるペプチドである、化合物。
【請求項11】
請求項10に記載の化合物であって、前記RGD部分が、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択される、化合物。
【請求項12】
請求項1に記載の化合物であって、前記ハロメチルエステルが、ハロメチル−t−ブチルスクシネートである、化合物。
【請求項13】
請求項12に記載の化合物であって、前記プロセスが、さらに以下:
(a)化合物2とハロメチル−t−ブチルスクシネートとを反応させることによって作製される生成物からt−ブチル基を除去して、それによって遊離カルボン酸基を生成する工程;
(b)(a)の生成物の該カルボン酸基を酸塩化物または活性エステルに変換する工程;および
(c)(b)の生成物を、遊離アミノ基を含む標的化因子(T)と接触させる工程
を包含する、化合物。
【請求項14】
請求項13に記載の化合物であって、前記ハロメチル−t−ブチルスクシネートがクロロメチル−t−ブチルスクシネートであり、化合物2がLY294002である、化合物。
【請求項15】
請求項13に記載の化合物であって、前記工程(c)の標的化因子(T)が、炭水化物、ビタミン、ペプチド、ペプチド模倣物、タンパク質、ヌクレオシド、ヌクレオチド、核酸、リポソーム、脂質、骨親和性剤、軟骨親和性剤、ジアゼピン、グルコース、ガラクトース、マンノース、またはマンノース6−リン酸から選択される、化合物。
【請求項16】
請求項15に記載の化合物であって、前記標的化因子がRGD部分であるペプチドである、化合物。
【請求項17】
請求項16に記載の化合物であって、前記RGD部分が、RGDs、c(RGDfK)、ビトロネクチン、フィブロネクチン、ソマトスタチン受容体アゴニストおよびソマトスタチン受容体アンタゴニストからなる群より選択される、化合物。
【請求項18】
請求項16に記載の化合物であって、前記保護されたRGDS部分が遊離アルギニンα−アミノ基を有する、化合物。
【請求項19】
請求項18に記載の化合物であって、前記プロセスが、前記RGDS部分から保護基を除去する工程(d)をさらに包含する、化合物。
【請求項20】
請求項19に記載の化合物であって、前記プロセスが、分取逆相液体クロマトグラフィー質量スペクトル(LCMS)を使用して(d)の生成物を同定および単離する工程をさらに包含し、該単離された生成物が、LCMSの正のモードにおいてM+=853の質量を有する、化合物。
【請求項21】
請求項19に記載の化合物であって、前記プロセスが、任意のアニオン対イオンを塩化物対イオン(塩)に交換する工程をさらに包含する、化合物。
【図1】

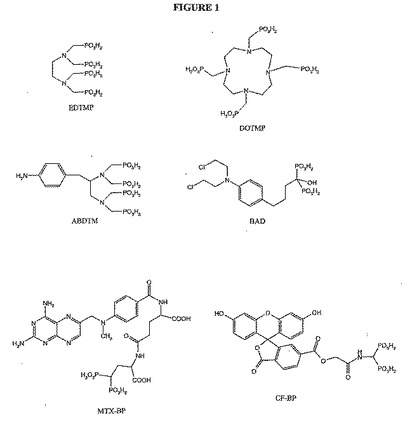
【図2】
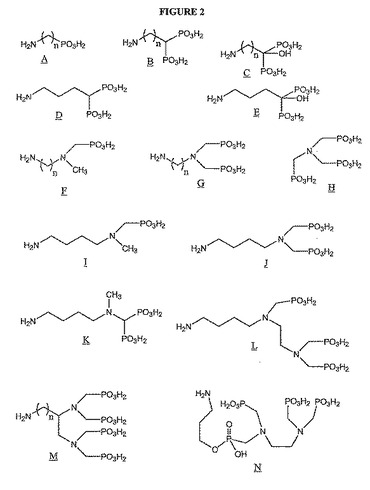
【図3】
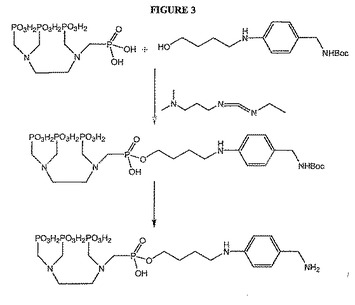
【図4】
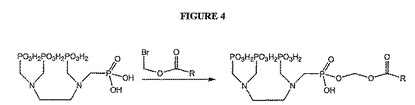
【図5】
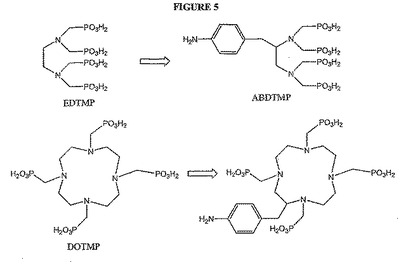
【図6】
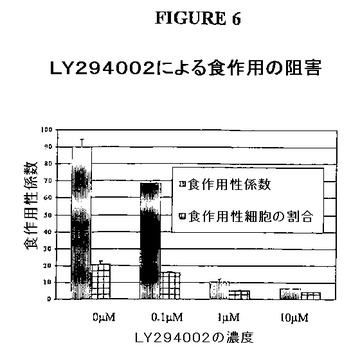
【図7】
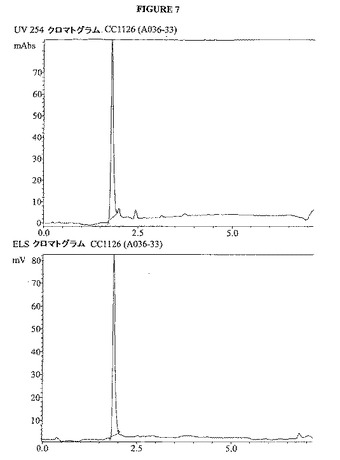
【図8】
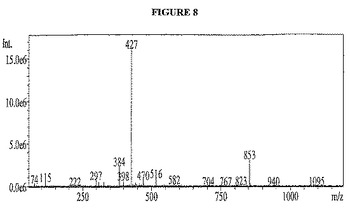
【図9】
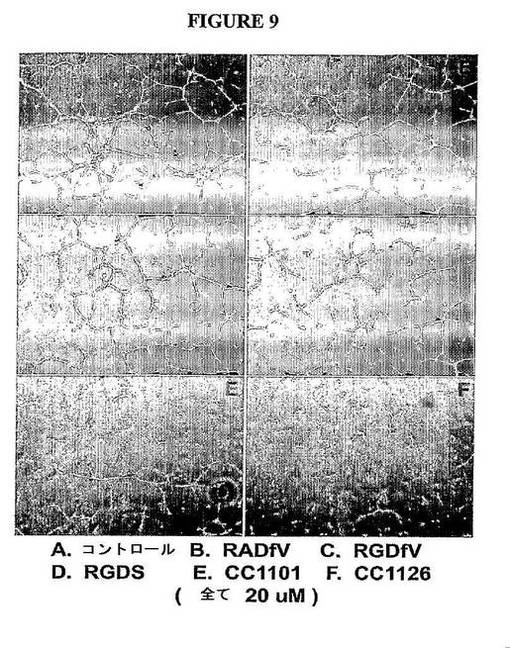
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2011−57686(P2011−57686A)
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【出願番号】特願2010−235985(P2010−235985)
【出願日】平成22年10月20日(2010.10.20)
【分割の表示】特願2006−509693(P2006−509693)の分割
【原出願日】平成16年4月3日(2004.4.3)
【出願人】(505363307)セマフォア ファーマシューティカルズ, インコーポレイテッド (3)
【Fターム(参考)】
【公開日】平成23年3月24日(2011.3.24)
【国際特許分類】
【出願日】平成22年10月20日(2010.10.20)
【分割の表示】特願2006−509693(P2006−509693)の分割
【原出願日】平成16年4月3日(2004.4.3)
【出願人】(505363307)セマフォア ファーマシューティカルズ, インコーポレイテッド (3)
【Fターム(参考)】
[ Back to top ]