
PPAR調節因子
【課題】糖尿病、癌、炎症、神経変性障害および感染等のPPAR調節に依存する疾患または状態の治療における、2,4−ジフェニル−1,3−ジオキサンの使用を提供すること。
【解決手段】1,3−ジオキサン誘導体、ならびに糖尿病、癌、炎症、神経変性障害および感染等のPPAR調節に依存する疾患または状態の治療におけるそれらの使用が記載される。本発明はまた、PPAR−γ活性の調節に反応性のある疾患または状態、具体的には本明細書中で後述されるPPAR−γ反応性疾患または状態のいずれかの治療のための薬物の調製のための、上記で定義されるような化合物の使用に関する。
【解決手段】1,3−ジオキサン誘導体、ならびに糖尿病、癌、炎症、神経変性障害および感染等のPPAR調節に依存する疾患または状態の治療におけるそれらの使用が記載される。本発明はまた、PPAR−γ活性の調節に反応性のある疾患または状態、具体的には本明細書中で後述されるPPAR−γ反応性疾患または状態のいずれかの治療のための薬物の調製のための、上記で定義されるような化合物の使用に関する。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明は、糖尿病、癌、炎症、神経変性障害および感染等のPPAR調節に依存する疾患または状態の治療における、2,4−ジフェニル−1,3−ジオキサンの使用を対象とする。
【背景技術】
【0002】
発明の背景
ペルオキシソーム増殖因子活性化受容体(PPAR)は、核内ホルモン受容体である。PPAR受容体は、レチノイドX受容体(RXRとして公知)とのヘテロ二量体の形態でペルオキシソーム増殖因子応答エレメント(PPRE)として公知のDNA配列の要素に結合することによって、転写を活性化する。3つのサブタイプのヒトPPARが同定および記載されている:PPAR−α、PPAR−γおよびPPAR−δ(またはNUCI)。PPAR−αは主に肝臓中で発現されるが、PPAR−δは遍在する。PPAR−γは、脂肪細胞の分化の制御に関与しており、そこで高度に発現される。これはまた、全身性の脂質恒常性において重要な役割を有する。糖尿病の治療において使用されているチアゾリジンジオンを含んで、PPARの活性を調節する多数の化合物もまた、同定されている。
【0003】
PPAR−γサブタイプのDNA配列は、Elbrechtら、BBRC 224、431−437(1996)に記載されている。フィブラートおよび脂肪酸を含むペルオキシソーム増殖因子は、PPARの転写活性を活性化する。
【0004】
PPARが、これらの核内受容体を発現している細胞に関連した多様な疾患または病的状態に密接に関与していることを説明する文献中で、多数の例が提供されている。より具体的には、PPARは、血中のグルコース、コレステロールおよびトリグリセリドレベルを減少させるための方法における薬物標的として有用であり、したがって、インスリン抵抗性、脂質異常症、ならびに肥満およびアテローム性動脈硬化症(Duezら、2001年、J.Cardiovasc.Risk、8、185−186)、冠動脈疾患およびある種の他の心血管障害を含むシンドロームX(「メタボリックシンドロームとも命名されている)(国際公開第97/25042号、国際公開第97/10813号、国際公開第97/28149号、Kaplanら、2001年、J.Cardiovasc Risk、8、211−7も参照のこと)に関連した他の障害の、治療および/または予防のために使用されている。さらに、PPARは、皮膚病(Smithら、2001年、J.Cutan.Med.Surg.、5、231−43参照)、胃腸疾患(国際公開第98/43081号)、または糸球体腎炎、糸球体硬化症、ネフローゼ症候群および高血圧性腎硬化症を含む腎疾患等の、ある種の炎症性疾患の治療のための潜在的な標的であると示されている。同様に、PPARは、神経系疾患(LandrethおよびHeneka、2001年、Neurobiol Aging、22、937−44)もしくは認知症において認知機能を向上させるため、乾癬、多嚢胞性卵巣症候群(PCOS)を治療するため、または骨量減少、例えば骨粗鬆症を予防および治療するために、有用である(例えば、米国特許第5981586号または米国特許第6291496号参照)。
【0005】
したがって、PPARは、治療用化合物の開発のための、興味深い標的である。疾患または病的状態を治療および/または予防するための種々の方法の状況において観察される反応が奨励されているが(例えば、チアゾリジンジオン(TZD)クラスの薬物療法、例えば、トログリタゾン、ロシグリタゾンまたはピオグリタゾンは、明らかに、2型糖尿病の患者において、インスリン感受性の向上において重要な役割を果たし;Cheng laiおよびLevine、2000年、Heart Dis.、2、326−333参照)、それらは、多数の深刻な望ましくない副作用の発生のために(例えば、体重増加、高血圧、心肥大、血液希釈、肝毒性および浮腫;Haskinsら、2001年、Arch Toxicol.、75、425−438、Yamamotoら、2001年、Life Sci.、70、471−482、Scheen、2001年、Diabetes Metab.、27、305−313、Gale、2001年、Lancet、357、1870−1875、Formanら、2000年、Ann.Intern.Med.、132、118−121ならびにAl Salmanら、2000年、Ann.Intern.Med.、132、121−124参照)、完全に満足できる治療ではない。結果として、PPAR核内受容体を発現する細胞型と関連した疾患または病的状態の治療および/または予防を可能にする、新規な改良された生成物および/または新規な方法を同定することが望ましい。より具体的には、TZD誘導体で見られる副作用のほとんどは前記化合物の完全アゴニスト特性に起因し、したがって、必ずしも完全アゴニストでない新しい化合物を同定することが望ましい。
ある種の4−フェニル−1,3−ジオキサン−5−イルアルケノン酸誘導体は、トロンボキサン受容体アンタゴニストまたはトロンボキサンA2合成の阻害剤として記載されている。トロンボキサン受容体は、効力のある血小板の凝集因子であり、血管収縮、ならびに気管支および気管平滑筋収縮に関わってきた(例えば、特許文献1、特許文献2および特許文献3参照)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許出願公開第94239号明細書
【特許文献2】欧州特許出願公開第0266980号明細書
【特許文献3】米国特許第4895962号明細書
【発明の概要】
【課題を解決するための手段】
【0007】
発明の要旨
本発明のある態様は、1,3−ジオキサン誘導体、またはその薬学的に許容され得る塩の治療有効量を投与することを含む、個体においてPPARの活性を調節するための方法に関し、誘導体は式:
【0008】
【化4】

【0009】
によって表され、
式中:
Aは2つまでの二重結合を有する、3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されている、フェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaおよびRbは、独立して、水素、2〜6Cアルケニル、場合により3つまでのハロゲノ置換基を有する1〜8Cアルキル、ペンタフルオロフェニル、アリールもしくはアリール(1〜4C)アルキルであり、前記アリールもしくはアリール(1〜4C)アルキル置換基は、場合によりハロゲノ、(1〜6C)アルキル、分枝もしくは直鎖状(1〜6C)アルコキシ、(1〜4C)アルキレンジオキシ、トリフルオロメチル、シアノ、ニトロ、ヒドロキシル、(2〜6C)アルカノイルオキシ、(1〜6C)アルキルチオ、(1〜6C)アルカンスルホニル、(1〜6C)アルカノイルアミノおよび2〜4個の炭素原子のオキサポリメチレンで置換されているか、または、RaおよびRbは、場合により1または2個の(1〜4C)アルキル置換基を有する2〜7個の炭素原子のポリメチレンをともに形成する。
【0010】
2,4−ジフェニル−1,3−ジオキサン誘導体またはその薬学的に許容され得る塩を投与することによって、PPAR反応性疾患または状態を治療するための方法もまた、提供され、誘導体は、式II:
【0011】
【化5】
【0012】
によって表され、
式中Xは、フルオロ、クロロ、ブロモ、トリフルオロメチル、場合により置換されたフェニル、シアノ、メトキシおよびニトロから選択されるか、またはフェニル−X基は、場合により置換されたクロメン誘導体でもよく;YおよびZは、それぞれ水素またはハロゲノである。
【0013】
1,3−ジオキサン誘導体、それらの薬学的に許容され得る塩、および式:
【0014】
【化6】
【0015】
によって表される誘導体を含む薬学的組成物もまた、本発明によって提供され、
式中:
Aは2つまでの二重結合を有する3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されているフェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaはHであり、Rbは場合によりハロゲン、OH、O−アルキル、O−アリール、アミノまたはN−モノアルキルもしくはN−ジアルキルまたはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキルまたはオキソからなる群より選択される3つの異なる置換基で置換されている、アリール基または複素環である。具体的な目的の化合物は、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸および4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸、またはRbがビフェニルである化合物である。
【0016】
本発明はまた、PPAR−γ活性の調節に反応性のある疾患または状態、具体的には本明細書中で後述されるPPAR−γ反応性疾患または状態のいずれかの治療のための薬物の調製のための、上記で定義されるような化合物の使用に関する。
【0017】
化合物は、メタボリックシンドローム、肥満、インスリン抵抗性、糖尿病前症、糖尿病、脂質異常症、多発性硬化症、乾癬、アトピー性皮膚炎、喘息および潰瘍性大腸炎等の自己免疫疾患、脂肪肉腫、神経芽細胞腫、膀胱癌、乳癌、結腸癌、肺癌、膵癌および前立腺癌等の癌、炎症、感染、AIDSおよび創傷治癒を含む多数の臨床状態の治療または予防のための薬物の調製において有用である。
例えば、本発明は、以下の項目を提供する。
(項目1)
1,3−ジオキサン誘導体、またはその薬学的に許容され得る塩の治療有効量を個体に投与することを含む、個体においてペルオキシソーム増殖因子活性化受容体(PPAR)の活性を調節するための方法であって、
前記誘導体は式:
【化1】
によって表され、
式中:
Aは2つまでの二重結合を有する、3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されている、フェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaおよびRbは、独立して、水素、2〜6Cアルケニル、場合により3つまでのハロゲノ置換基を有する1〜8Cアルキル、ペンタフルオロフェニル、アリールもしくはアリール(1〜4C)アルキルであり、前記アリールもしくはアリール(1〜4C)アルキル置換基は、場合によりハロゲノ、(1〜6C)アルキル、分枝もしくは直鎖状(1〜6C)アルコキシ、(1〜4C)アルキレンジオキシ、トリフルオロメチル、シアノ、ニトロ、ヒドロキシル、(2〜6C)アルカノイルオキシ、(1〜6C)アルキルチオ、(1〜6C)アルカンスルホニル、(1〜6C)アルカノイルアミノおよび2〜4個の炭素原子のオキサポリメチレンで置換されているか、または、RaおよびRbは、場合により1または2個の(1〜4C)アルキル置換基を有する2〜7個の炭素原子のポリメチレンをともに形成する、方法。
(項目2)
Aが3〜7個の炭素の直鎖状炭素鎖であり、WがCOOHである、項目1に記載の方法。
(項目3)
前記炭素鎖がC2−C3またはC3−C4の間の二重結合を含む、項目2に記載の方法。
(項目4)
Arが2−OHまたは2−OMe置換フェニルまたはナフチル基である、項目1〜3のいずれか一項に記載の方法。
(項目5)
RaがHであり、Rbが、それぞれ場合によりハロゲン、OH、O−アルキル、アミノ、N−モノアルキル、N−ジアルキル、ニトロアルキルまたはチオアルキルで置換されているフェニル、ベンジル、2−または3−または4−ピリジン、フラン、ビフェニル1−ナフチルおよび2−ナフチルからなる群より選択されるアリール基である、項目1〜4のいずれか一項に記載の方法。
(項目6)
前記化合物が、2,4−ジフェニル−1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であり、前記誘導体は式II:
【化2】
によって表され、
式中Xは、フルオロ、クロロ、ブロモ、トリフルオロメチル、場合により置換されたフェニル、シアノ、メトキシおよびニトロから選択されるか、またはフェニル−X基は、場合により置換されたクロメン誘導体でもよく;YおよびZは、それぞれ水素またはハロゲノである、項目1に記載の方法。
(項目7)
式IIによって表される前記誘導体中のジオキサン環の2、4および5位の基がシス−相対立体化学を有する、項目5に記載の方法。
(項目8)
Xが2−フルオロ、2−クロロ、2−ブロモ、2−シアノ、2−トリフルオロメチル、3−フルオロ、3−クロロ、3−シアノ、3−ニトロ、3−メトキシ、4−クロロ、4−シアノ、4−ニトロおよび4−メトキシから選択され;Yが水素またはフルオロであり;Zが水素である、項目5または6に記載の方法。
(項目9)
Xが2−クロロ、3−クロロ、2−シアノ、4−シアノ、3−ニトロおよび4−ニトロから選択され;YおよびZが水素である、項目7に記載の方法。
(項目10)
前記誘導体が4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸またはその薬学的に許容され得る塩である、項目8に記載の方法。
(項目11)
Aが2つまでの二重結合を有する3〜7個の炭素の分枝または直鎖状炭素鎖であり、
WがCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されているフェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arがフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaがHであり、Rbがアリール基、または場合によりハロゲン、OH、O−アルキル、O−アリール、アミノもしくはN−モノアルキルもしくはN−ジアルキルもしくはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキルもしくはオキソからなる群より選択される3つの異なる置換基で置換されている複素環である、
項目1に記載の方法。
(項目12)
Aが1つの二重結合を有する5炭素直鎖状鎖であり、WがCOOHであり、Arがo−位でOHまたはOMeによって置換されているフェニルであり、Rbが複素環、またはO−アリールで置換されているフェニル基である、項目11に記載の方法。
(項目13)
前記誘導体が、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸またはその薬学的に許容され得る塩である、項目11に記載の方法。
(項目14)
前記誘導体が、4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸またはその薬学的に許容され得る塩である、項目11に記載の誘導体。
(項目15)
前記誘導体が、アルカリ金属およびアルカリ土類金属塩、アルミニウムおよびアンモニウム塩、ならびに生理学的に許容され得る陽イオンを形成する有機アミンおよび第四級塩基の塩から選択される薬学的に許容され得る塩として存在する、前記項目のいずれか一項に記載の方法。
(項目16)
前記方法が、PPAR−γ反応性疾患または状態の予防または治療のためである、項目1〜15のいずれか一項に記載の方法。
(項目17)
前記疾患または状態がインスリン抵抗性である、項目1〜16のいずれか一項に記載の方法。
(項目18)
前記疾患または状態が糖尿病である、項目1〜17のいずれか一項に記載の方法。
(項目19)
前記疾患または状態が、肥満の個体における糖尿病である、項目1〜18のいずれか一項に記載の方法。
(項目20)
前記疾患または状態が、PPAR−γによって媒介される慢性炎症性障害である、項目1〜19のいずれか一項に記載の方法。
(項目21)
前記疾患または状態が炎症性腸疾患、潰瘍性大腸炎またはクローン病である、項目1〜20のいずれか一項に記載の方法。
(項目22)
前記疾患または状態が関節炎、特に関節リウマチ、多発性関節炎および喘息である、項目1〜21のいずれか一項に記載の方法。
(項目23)
前記疾患が眼の炎症または眼乾燥疾患である、項目1〜22のいずれか一項に記載の方法。
(項目24)
前記疾患が皮膚障害、特に乾癬である、項目1〜23のいずれか一項に記載の方法。
(項目25)
前記疾患が高脂血症である、項目1〜24のいずれか一項に記載の方法。
(項目26)
前記疾患または状態が癌である、項目1〜25のいずれか一項に記載の方法。
(項目27)
前記癌が脂肪肉腫、前立腺癌、子宮頸癌、乳癌、多発性骨髄腫、膵癌、神経芽細胞腫または膀胱癌である、項目1〜26のいずれか一項に記載の方法。
(項目28)
PPAR媒介性疾患または状態である対象における臨床状態を治療または予防するために有用な薬物組成物を調製するための、項目1〜14のいずれか一項において定義される化合物の使用。
(項目29)
糖尿病、癌、炎症、AIDS、メタボリックシンドローム、肥満、糖尿病前症、高血圧および脂質異常症からなる群より選択される臨床状態の治療または予防のための薬物の調製のための、項目28に記載の化合物の使用。
(項目30)
1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であって、前記誘導体が式:
【化3】
によって表され、
式中:
Aは2つまでの二重結合を有する3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されているフェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaはHであり、Rbは、アリール基、または場合によりハロゲン、OH、O−アルキル、O−アリール、アミノまたはN−モノアルキルもしくはN−ジアルキルもしくはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキルもしくはオキソからなる群より選択される3つの異なる置換基で置換されている複素環である、誘導体。
(項目31)
Aが1つの二重結合を有する5炭素直鎖状鎖であり、WがCOOHであり、Arがo−位でOHまたはOMeによって置換されているフェニルであり、Rbが、O−アリールで置換されている複素環またはフェニルである、項目30に記載の誘導体。
(項目32)
Rbが(4−メトキシフェノキシ)−フェニルである、項目31に記載の誘導体。
(項目33)
前記誘導体が、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸である、項目32に記載の誘導体。
(項目34)
Rbが6−クロロ−4H−クロメン−4−オンである、項目31に記載の誘導体。
(項目35)
前記誘導体が、4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸である、項目34に記載の誘導体。
(項目36)
Rbがビフェニルである、項目31に記載の誘導体。
(項目37)
項目30〜項目36の誘導体の1つ以上を含む、医薬組成物。
(項目38)
さらなる医薬成分を含む、項目37に記載の医薬組成物。
【図面の簡単な説明】
【0018】
【図1】図1は、それぞれ完全アゴニストおよび部分アゴニストのPPAR活性化のモデルを説明する。
【図2】図2は、PPAR−δ、PPARαおよびRxRと比較した、ロシグリタゾンおよび4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸によるPPAR−γの選択的活性化を説明する。
【図3】図3は、ロシグリタゾンおよび4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸による、全長PPAR−γの活性化を説明する。
【図4】図4は、4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸が全長PPARδトランス活性化に対して効果を有さないことを説明する。
【図5】図5は、部分PPARγ競合アッセイの結果を説明する。
【図6】図6は、PPARγリガンド置換アッセイの結果を説明する。
【図7】図7は、グルコース取り込みアッセイの結果を説明する。
【図8】図8は、化学式II〜Vを説明する。
【図9】図9は、化学式VI〜VIIIを説明する。
【図10】図10は、化学反応スキームIを説明する。
【発明を実施するための形態】
【0019】
定義
一般に、本明細書中で使用される用語は、本発明の理解において薬学、生物学および化学分野の当業者が採用する、それらの標準的な定義を有する。以下の用語は、示される意味を有し、他の用語が本明細書中で提供される場合がある。
アルキル:用語「アルキル」は、一般に1〜22個の、示された数の炭素原子を有する、一価の飽和脂肪族炭化水素ラジカルをいう。例えば、「1〜8Cアルキル」または「1〜8個の炭素のアルキル」または「Alk1〜8」は、構造中に1〜8個の炭素を含む任意のアルキル基をいう。アルキルは、直鎖(すなわち直鎖状)または分枝鎖であってもよい。低級アルキルは、1〜6個の炭素のアルキルをいう。低級アルキルラジカルのそれぞれの例としては、メチル、エチル、n−プロピル、n−ブチル、n−ペンチル、n−ヘキシル、イソプロピル、イソブチル、イソペンチル、アミル、sec−ブチル、tert−ブチル、sec−アミル、tert−ペンチル、2−エチルブチル、2,3−ジメチルブチル等が挙げられる。高級アルキルは、7つ以上の炭素のアルキルをいう。これらのものとしては、n−ヘプチル、n−オクチル、n−ノニル、n−デシル、n−ドデシル、n−テトラデシル、n−ヘキサデシル、n−オクタデシル、n−エイコシル等とともに、それらの分枝のバリエーションが挙げられる。3〜7個の炭素の直鎖状炭素鎖は鎖長をいい、枝上にあるいかなる炭素も含まない。ラジカルは、場合により置換されていてもよい。
【0020】
アルケニル:用語「アルケニル」は、少なくとも1つの炭素−炭素二重結合を有し、示された数の炭素原子を有する、一価の脂肪族炭化水素ラジカルをいう。例えば、「C2〜6アルケニル」または「1〜6個の炭素のアルケニル」、または「アルケニル1〜6」は、構造中に1〜6個の炭素原子を含むアルケニル基をいう。アルケニルは、直鎖(すなわち、直鎖状)または分枝鎖であってもよい。低級アルケニルは、1〜6個の炭素のアルケニルをいう。低級アルケニルラジカルのそれぞれの例としては、エテニル、1−プロペニル、1−ブテニル、1−ペンテニル、1−ヘキセニル、イソプロペニル、イソブテニル等が挙げられる。高級アルケニルは、7つ以上の炭素のアルケニルをいう。これらのものとしては、1−ヘプテニル、1−オクテニル、1−ノネニル、1−デセニル、1−ドデセニル、1−テトラデセニル、1−ヘキサデセニル、1−オクタデセニル、1−エイコセニル等とともに、それらの分枝のバリエーションが挙げられる。ラジカルは、場合により置換されていてもよい。
【0021】
アルコキシ:用語「アルコキシ」は、Rが本明細書中で定義されるようなアルキルである、式RO−の一価のラジカルをいう。低級アルコキシは、1〜6個の炭素原子のアルコキシまたは(1〜6)アルコキシをいう。それぞれの低級アルコキシラジカルとしては、メトキシ、エトキシ、n−プロポキシ、n−ブトキシ、n−ペンチルオキシ、n−ヘキシルオキシ、イソプロポキシ、イソブトキシ、イソペンチルオキシ、アミルオキシ、sec−ブトキシ、tert−ブトキシ、tert−ペンチルオキシ等が挙げられる。ラジカルは、場合により置換されていてもよい。
【0022】
アリール:用語「アリール」は、本明細書中で使用される場合、場合により、他に示されない限りは、ヒドロキシ、チオ、シアノ、アルキル、アルコキシ、低級ハロアルコキシ、アルキルチオ、オキソ、ハロゲン、ハロアルキル、ヒドロキシアルキル、ニトロ、アルコキシカルボニル、アミノ、アルキルアミノ、ジアルキルアミノ、アミノアルキル、アルキルアミノアルキル、およびジアルキルアミノアルキル、チオアルキル、アルキルスルホニル、アリールスルフィニル、アルキルアミノスルホニル、アリールアミノスルホニル、アルキルスルホニルアミノ、アリールスルホニルアミノ、カルバモイル、アルキルカルバモイルおよびジアルキルカルバモイル、アリールカルバモイル、アルキルカルボニルアミノ、アリールカルボニルアミノから選択される独立して選択される1つ以上の、好ましくは1または3個の置換基で置換されていてもよい、1つの個々の環、または少なくとも1つの環が天然に芳香族である1つ以上の縮合環からなる、5〜15個の炭素原子を含む一価の芳香族炭素環ラジカルを示す。あるいは、アリール環の2つの隣接する原子は、メチレンジオキシまたはエチレンジオキシ基で置換されていてもよい。したがって、二環式アリール置換基は、複素環式またはヘテロアリール環に縮合していてもよい。しかしながら、二環式アリール置換基の付着点は、炭素環芳香環上にある。アリールラジカルの例としては、フェニル、ナフチル、ベンジル、ビフェニル、フラニル、ピリジニル、インダニル、アントラキノリル、テトラヒドロナフチル、3,4−メチレンジオキシフェニル、1,2,3,4−テトラヒドロキノリン−7−イル、1,2,3,4−テトラヒドロイソキノリン−7−イル、1,3ジオキソランラジカル、安息香酸ラジカル、、フラン−2−カルボン酸ラジカル、2−(イソキサゾール−5−イル)酢酸ラジカル、3−ヒドロキシ−2−メチルピリジン−4−カルボン酸ラジカル等が挙げられる。
【0023】
ハロ:「ハロ」置換基は、クロロ、ブロモ、ヨード、およびフルオロから選択される、一価のハロゲンラジカルである。「ハロゲン化」化合物は、1つ以上のハロ置換基で置換されたものである。
【0024】
フェニル:「フェニル」は、ベンゼン環からの水素の除去によって形成されたラジカルである。フェニルは、場合により置換されていてもよい。
【0025】
フェノキシ:「フェノキシ」基は、式RO−のラジカルであり、式中Rはフェニルラジカルである。
【0026】
ベンジル:「ベンジル」基は、式R−CH2−のラジカルであり、式中Rはフェニルラジカルである。
【0027】
ベンジルオキシ:「ベンジルオキシ」基は、式RO−のラジカルであり、式中Rはベンジルラジカルである。
【0028】
複素環:「複素環」または「複素環式実体」は、炭素および少なくとも1つの他の元素、一般に窒素、酸素、または硫黄を含む、5または6員閉環の一価のラジカルであり、完全に飽和されているか、部分的に飽和されているか、または不飽和(すなわち、天然に芳香族)でもよい。一般に、複素環は、わずか2つのヘテロ原子を含む。1つのヘテロ原子しか有さない不飽和5員複素環のそれぞれの代表的な例としては、2−または3−ピロリル、2−または3−フラニル、および2−または3−チオフェニルが挙げられる。対応する、部分的に飽和された、または完全に飽和されたラジカルとしては、3−ピロリン−2−イル、2−または3−ピロリンジニル、2−または3−テトラヒドロフラニル、および2−または3−テトラヒドロチオフェニルが挙げられる。2つのヘテロ原子を有するそれぞれの不飽和5員複素環ラジカルの代表としては、イミダゾリル、オキサゾリル、チアゾリル、ピラゾリル等が挙げられる。対応する、完全に飽和された、および部分的に飽和されたラジカルもまた、含まれる。1つのヘテロ原子しか有さない不飽和6員複素環のそれぞれの代表的な例としては、2−、3−、または4−ピリジニル、2H−ピラニル、および4H−ピラニルが挙げられる。対応する、部分的に飽和された、または完全に飽和されたラジカルとしては、2−、3−、または4−ピペリジニル、2−、3−、または4−テトラヒドロピラニル等が挙げられる。2つのヘテロ原子を有するそれぞれの不飽和6員複素環ラジカルの代表としては、3−または4−ピリダジニル、2−、4−、または5−ピリミジニル、2−ピラジニル、モルホリノ等が挙げられる。対応する、完全に飽和された、および部分的に飽和されたラジカルもまた含まれ、例えば2−ピペラジンである。複素環式ラジカルは、複素環中の利用可能な炭素原子もしくはヘテロ原子を通じて実体に直接結合しているか、または、メチレンもしくはエチレン等のアルキレン等のリンカーを通じて結合している。複素環は、場合により、アリール基と同じ様式で置換されていてもよい。
【0029】
場合により置換され:ラジカルが、「場合により置換され」という場合、これは、ラジカルが非置換であるか、またはラジカルの少なくとも1つの−Hが除去されており、別の置換基がその場所に挿入されていることを意味する。ラジカルは、場合により、本発明の範囲内に含まれる化合物の調製を有意に妨げず、化合物の生物学的活性に有意に悪影響を与えない位置で、置換基で置換されていてもよい。ラジカルは、場合により、ハロ、低級アルコキシ、ヒドロキシル、シアノ、ニトロ、アミノ、ハロ低級アルキル、ハロ低級アルコキシ、ヒドロキシカルボニル、低級アルコキシカルボニル、低級アルキルカルボニルオキシ、および低級アルキルカルボニルアミノからなる群より独立して選択されるか、または本明細書中で上述された、1、2、3、4または5個の置換基で置換されていてもよい。
【0030】
用語「ヒドロキシカルボニル」は、式−C(O)OHを有する一価のラジカルである。
【0031】
用語「低級アルコキシカルボニル」は、式−C(O)OAlkを有する一価のラジカルであり、ここでAlkは低級アルキルである。
【0032】
用語「低級アルキルカルボキシルオキシ」は、式−OC(O)Alkの一価のラジカルであり、ここでAlkは低級アルキルである。
【0033】
本明細書中で使用される場合、「糖」は、単糖、二糖または多糖を意味する。適した単糖としては、ペントース、ヘキソース、またはヘプトース残基が挙げられる。ペントースの非限定的な例としては、アラビノース、リボース、リブロース、キシロース、リキソース、およびキシルロースが挙げられる。ヘキソースの非限定的な例としては、グルコース、ガラクトース、フルクトース、フコース、マンノース、アロース、アルトロース、タロース、イドース、プシコース、ソルボース、およびタガトースが挙げられる。ヘプトースの非限定的な例としては、マンノヘプツロースおよびセドヘプツロースが挙げられる。糖部分は、アミドまたはエステル結合を形成することができる糖環の任意の位置で化合物に連結されていてもよい。好ましい糖類は、β−グリコシル糖類である。
【0034】
PPAR調節は、天然の状態、すなわち、リガンドなしでの標的遺伝子のPPAR依存的転写の基礎レベルを参考にして定義され、ここで、PPAR活性の調節は、PPAR活性を調節することができる化合物の存在下での前記転写の基礎レベルの減少または増大に反映される。一般に、前記転写の増大は、PPAR活性の増強と関連があり、活性化因子またはアゴニストという名前の化合物に関する。反対に、前記転写の減少は、PPAR活性の阻害と関連があり、阻害剤またはアンタゴニストという名前の化合物に関する。部分アゴニストは、標的遺伝子のサブセットのPPAR依存的転写を生じるが、他のPPAR標的遺伝子に影響を及ぼさない化合物である。部分アゴニストは、生化学的または生理学的観点から見ることができる。部分アゴニストの生化学的見識は、完全アゴニストと競合することができ、完全アゴニストと比べて低いレベルのトランス活性化を有する化合物である。部分アゴニストの生理学的見識は、異なるサブセットの遺伝子の活性化に関する(いくつかの標的遺伝子の完全な活性化があっても、他のPPAR標的遺伝子の活性化はなし)。これは、完全アゴニストの生理学的効果のいくつかのみを生じ、これは大いに望ましい。
【0035】
化合物の「治療有効量」は、それを必要とする対象に投与したときに、治療される状態に対して、経時的に所望の結果を生じる量を意味する。本願において、所望の効果はPPAR調節、およびそれと関連する生物学的活性である。
【0036】
本発明において有用な化合物は、本願中で種々の式によって示される。式を見ることによって、化合物が、しばしばキラル中心、すなわち4つの異なる基が付着している炭素を有し、これらが鏡像異性体として存在する場合があることが明らかであろう。また、いくつかの場合における二重結合の存在によって、化合物は、回転障害を有する。したがって、化合物は幾何異性を示し、すなわち、空間中で原子が方向付けされる様式で、2つの形態は互いに異なる場合がある。二重結合に関して、互いに鏡像でない立体異性体が存在し、ジアステレオマーと呼ばれる。本願中の化合物の命名において、R,Sシステムにおける鏡像異性体の命名において行われるように、二重結合の各末端に付着した2つの基が優先順位番号を提供される化学情報検索サービス機関システムが用いられる。より高い優先順位番号の2つの基が同じ側にある場合、分子はZ異性体である。(ドイツ語−zusammen、ともに)。反対の側にある場合、分子はEである。(ドイツ語−entgegen、反対)。式は、単独であれ混合物中であれ、可能性のある鏡像異性体およびジアステレオマーの全てを包含するよう意図されることが、理解されるべきである。
【0037】
本発明において有用な化合物
本発明は、部分的に、ある種の化合物が少なくとも1つのPPARサブタイプ、例えばPPARγまたはPPARβ/δの活性を調節することができるという発見に基づく。この発見は、かかるPPARの機能によって媒介されるヒト等の哺乳動物における状態または疾患を治療するための、これらの化合物の使用につながる。
【0038】
本発明において有用な化合物は、1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であり、ここで誘導体は、式I:
【0039】
【化7】
【0040】
によって表され、
式中:
Aは、場合により1または2個の二重結合を含む(それぞれはシスでもトランスでもよい)、3〜7個の炭素の分枝または直鎖状炭素鎖であり、
Wは、COOH、OH、NH2、SO3H、OSO3H、場合により例えばCOOH、OHまたはNH2で置換されている、フェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジン等の、しかしこれらに限定されない芳香族基;または2位を通して連結された1,3ジオキソラン基であり、
Arは、フェニル、または2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニル等の、しかしこれらに限定されない5または6員の複素環式芳香族基であり、フェニルおよびナフチル部分は場合により、オルト、メタおよび/もしくはパラ位でOHまたはOMeで置換されているが、好ましくは、置換されている場合、オルト位においてOHまたはOMeで一置換されており、
RaおよびRbは、独立して、水素、2〜6Cアルケニル、場合により3つまでのハロゲノ置換基を有する1〜8Cアルキル、ペンタフルオロフェニル、アリールもしくはアリール(1〜4C)アルキルであり、その最後の2つは場合によりハロゲノ、(1〜6C)アルキル、分枝もしくは直鎖状(1〜6C)アルコキシ、(1〜4C)アルキレンジオキシ、トリフルオロメチル、シアノ、ニトロ、ヒドロキシル、(2〜6C)アルカノイルオキシ、(1〜6C)アルキルチオ、(1〜6C)アルカンスルホニル、(1〜6C)アルカノイルアミノおよび2〜4個の炭素原子のオキサポリメチレンから選択される5つまでの置換基を有するか、またはRaおよびRbは、場合により1または2個の(1〜4C)アルキル置換基を有する、2〜7個の炭素原子のポリメチレンをともに形成する。
【0041】
いくつかの実施形態において、Aは、3〜7個の炭素の直鎖状炭素鎖であり、WはCOOHである。かかる炭素鎖は、例えば、C2−C3またはC3−C4の間に二重結合を含んでもよい。
【0042】
ある実施形態において、RaはHであり、Rbは、ハロゲン、OH、O−アルキル、アミノ、N−モノアルキル、N−ジアルキル(分枝または直鎖状)、ニトロおよびチオアルキル(分枝または直鎖状)からなる群より選択される5つまでの異なる置換基を有する、フェニル、ベンジル、2−または3−または4−ピリジン、フラン、ビフェニル、1−または2−ナフチル等の芳香族部分である。
【0043】
いくつかの実施形態において、誘導体は、2,4−ジフェニル−1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であり、ここで誘導体は、式II:
【0044】
【化8】
【0045】
によって表され、
式中Xは、フルオロ、クロロ、ブロモ、トリフルオロメチル、場合により置換されたフェニル、シアノ、メトキシおよびニトロから選択されるか、またはフェニル−X基は、場合により置換されたクロメン誘導体であってもよく;YおよびZは、個々に、水素またはハロゲノである。
【0046】
典型的には、式Iによって表される誘導体中のジオキサン環の2、4および5位の基は、シス−相対立体化学を有する。
【0047】
特に興味深いXを有するフェニル部分の他の特定の置換基としては、例えば、2−フルオロ−、2−クロロ−、2−ブロモ−、2−シアノ−、2−トリフルオロメチル−、3−フルオロ−、3−クロロ−、3−シアノ−、3−ニトロ−、3−メトキシ−、4−クロロ−、4−シアノ−、4−ニトロ−および4−メトキシ−フェニルが挙げられる。
【0048】
Yについての好ましい置換は、水素またはフルオロであり、Zは水素である。
【0049】
本発明において有用な化合物の好ましい基は、X1が2−クロロ、3−クロロ、2−シアノ、4−シアノ、3−ニトロおよび4−ニトロから選択され;ジオキサン環の2、4および5位の基がシス−相対立体化学を有する、式III(本明細書中に下記で示す)の化合物;ならびにその薬学的に許容され得る塩を含む。
【0050】
本発明はまた、A、WおよびArが上記で定義され、RaがHであり、Rbがアリール部分の1つの炭素原子が1つの酸素もしくは窒素原子で置換されているもの等のアリール基もしくは複素環である、式Iの化合物を提供する。かかるアリールおよび複素環基は、場合により、ハロゲン、OH、O−アルキル(分枝または直鎖状)、O−アリール、アミノまたはN−モノアルキルもしくはN−ジアルキル(分枝または直鎖状)またはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキル(分枝または直鎖状)またはオキソ(例えば、表II中のSN13)からなる群より選択される3つの異なる置換基で置換されていてもよい。RaがHでありRbが複素環、または0−アリールで置換されているフェニル基である式Iの化合物が、特にArがo−位でOHまたはOMeによって置換されたフェニルであり、WがCOOHであり、Aが1つの二重結合を有する5つの炭素の直鎖である場合、PPARγ調節因子として特に興味深い。かかる化合物の例示的な例としては、RaがHであり、Rbが、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸等の、フェニルの2もしくはオルト位を通じてジオキサン環に連結された(4−メトキシフェノキシ)−フェニルであるか;またはRbが、4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸等の6−クロロ−4H−クロメン−4−オン(例えば、クロメン部分の3位を通じてジオキサン環に連結されている)であるか;またはRbが、ビフェニルの2もしくはオルト位を通じてジオキサン環に連結されたビフェニルである、式Iの化合物が挙げられる。したがって、本発明は、単独で、ならびに薬学的担体、および場合によりさらなる治療上活性のある成分と組み合わせて、本実施形態において言及される化合物およびそれらの薬学的に適切な塩を提供する。
【0051】
式Iおよび式IIの化合物が不斉炭素原子を有し、ラセミ形態および光学活性形態で存在および単離することができることが、認識されるであろう。本発明は、PPAR活性を調節することができる、ラセミ形態および任意の光学活性形態の両方(またはそれらの混合物)、ならびにそれらの使用を含み、個々の光学異性体をどのように調製するか(例えば、光学活性な出発物質からの合成、またはラセミ形態のクロマトグラフィー分解によって)および下記で言及される1つ以上のアッセイを用いてどのようにPPAR調節特性を判定するかは当該分野で周知である。
【0052】
文脈から他に明らかでない限りは、本明細書中で言及される化学式は、特定の配置で示されるが、これは、必ずしも、絶対配置に対応しない。
【0053】
式IまたはIIの酸の、特定の薬学的に許容され得る塩は、例えば、リチウム、ナトリウムカリウム、マグネシウムおよびカルシウム塩等のアルカリ金属およびアルカリ土類金属塩、アルミニウムおよびアンモニウム塩、ならびにメチルアミン、ジメチルアミン、トリメチルアミン、エチレンジアミン、ピペリジン、モルホリン、ピロリジン、ピペラジン、エタノールアミン、トリエタノールアミン、N−メチルグルカミン、水酸化テトラメチルアンモニウムおよび水酸化ベンジルトリメチルアンモニウムとの塩等の、生理学的に許容され得る陽イオンを形成する有機アミンおよび第四級塩基の塩である。
【0054】
式Iの化合物は、構造的に類似の化合物の製造のための、当該分野で周知の従来の有機化学の手順によって製造することができる。かかる手順は、例えば、本明細書中に参照によって特に援用される、欧州特許第0094239号、4〜10頁に提供されており、下記に実施例の項で、X、YおよびZが本明細書中に上記で定義された意味を有する以下のプロセスによって、説明される。
【0055】
(A)式IVのアルデヒドを、式R13P=CH(CH2)2CO2−M+(式中R1が(1〜6C)アルキルまたはアリール(特にフェニル)であり、M+が陽イオン、例えばリチウム、ナトリウムまたはカリウム陽イオン等のアルカリ金属陽イオンである)のウィティッヒ試薬と反応させる。いくつかの実施形態において、アルデヒドを、P=CH(CH2)nCOO−M+(式中n=0〜4)に変更されている式のウィティッヒ試薬と反応させる。
【0056】
一般に、プロセスは、二重結合に隣接する置換基が主にシス−相対立体化学を有する、式IIの必要な化合物、すなわち「Z」異性体を生じる。しかしながら、プロセスはまた、所望される場合、クロマトグラフィーまたは結晶化等の従来の手順によって除去することができるトランス−相対立体化学を有する類似化合物を生じる。
【0057】
合成プロセスは、従来、適した溶媒または希釈剤、例えばベンゼン、トルエンもしくはクロロベンゼン等の芳香族溶媒、1,2−ジメトキシエタン、t−ブチルメチルエーテル、ジブチルエーテルもしくはテトラヒドロフラン等のエーテル、ジメチルスルホキシドもしくはテトラメチレンスルホン、またはかかる溶媒もしくは希釈剤の1つ以上の混合物中で行われる。プロセスは、一般に、例えば−80℃〜40℃の範囲の温度で行われるが、従来、室温で、または室温近く、例えば0〜35℃の範囲で行われている。
【0058】
(B)R1が保護基、例えば(1〜6C)アルキル(メチルまたはエチル等)、アシル(アセチル、ベンゾイル、メタンスルホニルまたはp−トルエンスルホニル等)、アリル、テトラヒドロピラン−2−イル、トリメチルシリルであり、脱保護される、式Vのフェノール誘導体。
【0059】
用いられる脱保護条件は、保護基R1の性質に依存する。したがって、例えば、それがメチルまたはエチルである場合、脱保護は、適した溶媒(N,N−ジメチルホルムアミドまたはN,N−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン等)中、例えば50〜160℃の範囲の温度で、ナトリウムチオエトキシド(水素化物およびエタンチオール)とともに加熱することによって行うことができる。あるいは、エチルまたはメチル保護基は、例えば0〜60℃の範囲の温度で、適した溶媒(テトラヒドロフランまたはメチルt−ブチルエーテル等)中でのリチウムジフェニルホスフィドとの反応によって除去することができる。保護基がアシルである場合、これは、例えば、0〜60℃の温度の範囲で、適した水性溶媒[水性(1〜4C)アルカノール等)中で、塩基(水酸化ナトリウムまたは水酸化カリウム等)の存在下での加水分解によって除去することができる。保護基がアリルまたはテトラヒドロピラン−2−イルである場合、これは、例えば、トリフルオロ酢酸等の強酸での処理によって除去することができ、これがトリメチルシリルである場合、これは、例えば、従来の手順を用いて、水性テトラブチルフッ化アンモニウムまたはフッ化ナトリウムとの反応によって除去することができる。
【0060】
(C)式中Q1およびQ2の一方が水素であり、他方が水素または式−CRaRb.OH(式中RaおよびRbは同じまたは異なる(1〜4C)アルキルである)の基である式Vのエリトロ−ジオール誘導体を、式VIIのベンズアルデヒド誘導体、またはアセタール、ヘミアセタールまたはその水和物と反応させる。
【0061】
ベンズアルデヒドVII[またはその水和物、または(1〜4C)アルカノール(メタノールまたはエタノール等)とのそのアセタールもしくはヘミアセタール]は、好都合に、過剰に存在してもよい。
【0062】
反応は、一般に、塩化水素、臭化水素、硫酸、リン酸、メタンスルホン酸またはp−トルエンスルホン酸等の酸触媒の存在下、好都合にトルエン、キシレンまたはエーテル等の適した溶媒または希釈剤、例えばテトラヒドロフラン、ジブチルエーテル、メチルt−ブチルエーテルまたは1,2−ジメトキシエタンの存在下、および例えば0〜80℃の範囲の温度で、行われる。
【0063】
式中Q1およびQ2がともに水素である式VIの出発物質は、例えば、本明細書中のプロセス(A)と類似した手順によって得られる、RaおよびRbがともにメチルまたはエチル等のアルキルである式VIIIの化合物のジオキサン環の、温和な、酸触媒された、加水分解またはアルコール分解によって、得ることができる。加水分解またはアルコール分解は、通常、溶媒としてアルカノイル(エタノールまたは2−プロパノール等)またはエーテル(テトラヒドロフラン等)中、塩酸等の水性鉱酸を用いて、10〜80℃の範囲の温度で行われる。
【0064】
式中Q1およびQ2の一方が水素であり、他方が式−−CRaRb.OHの基である式VIの出発物質は、上述の、式中Q1およびQ2がともに水素である式VIの出発物質の形成における中間体である。しかしながら、前記中間体は、通常、単離または特徴付けされない。したがって、プロセス(C)の有用な変更は、酸触媒の存在下で(上記のもののいずれか等)、好都合に例えば10〜80℃の範囲の温度で、および場合により適した溶媒または希釈剤(上記のもののいずれか等)の存在下で、式中RaおよびRbの一方が水素、メチルまたはエチルであり、他方がメチルまたはエチルである式VIIIの化合物を、過剰な式VIIの化合物(またはその水和物、アセタールもしくはヘミアセタール)と反応させることを含む。
【0065】
上記のプロセスにおける使用のための出発物質は、構造的に関係がある化合物の調製で知られている一般的な有機化学の手順によって生じることができる。したがって、式IVのアルデヒドは、例えば、スキームIに示される方法によって得ることができる。式Vの、保護されたフェノール誘導体は、例えば、式IVのものと類似したアルデヒドを用いるがフェノール基が、例えば脱保護工程(ii)を欠いたスキームIの手順を行うことによって生じたアルデヒド等のR1基で保護されている、上記のプロセス(A)に類似した手順を用いることによって、生じることができる。新規である、式VIIIの出発物質のものは、欧州特許出願、公開第94239号に記載されているものと類似した手順を用いて得ることができる。
【0066】
必要なウィティッヒ試薬は、従来の手順によって、例えば対応するハロゲン化ホスホニウムを水素化ナトリウム、リチウムジイソプロピルアミド、カリウムt−ブトキシド、LiHMDSまたはブチルリチウム等の強塩基で処理することによって、得ることができる。それらは、一般に、上記の縮合プロセス(A)を行う直前に、原位置(in situ)で形成される。
【0067】
式IおよびIIの化合物はまた、当該分野で周知の他の従来の手順によって、例えば対応するエステル、アミドまたはニトリルの塩基触媒加水分解によって得ることができることが、理解されるであろう。
【0068】
式IまたはIIの化合物の塩が必要とされる場合、これは、生理学的に許容され得る陽イオンを提供する適切な塩基との反応によって、または任意の他の従来の手順によって、得られる。
【0069】
さらに、式IまたはIIの化合物の光学活性形態が必要とされる場合、光学活性な出発物質を用いて、前述のプロセスの1つを行ってもよい。あるいは、式IIの化合物のラセミ形態を、光学活性形態の適した有機塩基、例えばエフェドリン、N,N,N−トリメチル(1−フェニルエチル)アンモニウム水酸化物または1−フェニルエチルアミンと反応させ、続いて例えば適した溶媒、例えば(1〜4C)アルカノールからの分別結晶化によって、このようにして得られた塩のジアステレオ異性体混合物の従来の分離を行い、その後従来の手順を用いて、例えば希塩酸等の水性鉱酸を用いて、酸での処理によって前記式IまたはIIの化合物の光学活性形態を遊離させてもよい。
【0070】
また、実施例29は、どのようにしてキラルクロマトグラフィーによって式IおよびIIの化合物のラセミ混合物を単離することができるかを説明する。
【0071】
前述したように、本明細書中に記載されている化合物は、PPAR活性の調節因子である。したがって、上記で概説された構造的特性に加えて、本発明の方法とともに使用されるのに好ましい化合物はまた、PPARアゴニスト、PPARアンタゴニストまたはPPAR部分アゴニスト、好ましくはPPAR部分アゴニストである。PPARアゴニスト/アンタゴニスト/部分アゴニストとしての機能性を判定するための方法は、「化合物の機能性」の項で、本明細書中で後述される。
【0072】
化合物の機能性
PPARの活性を調節することができる化合物(本明細書中でPPARリガンドとも呼ばれる)は、3つの別個のクラス:完全アゴニスト、部分アゴニスト/部分アンタゴニストに、および完全アンタゴニストに分類することができる。アゴニストおよびアンタゴニストは、それらの結合親和性、指示能力/EC50/IC50値、および飽和レベルの化合物の存在下で得られる活性のレベル、すなわち有効性によって、特徴付けられる。部分アゴニスト/部分アンタゴニストはまた、その結合親和性、および有効性によって特徴付けられる。部分アゴニスト/部分アンタゴニストは、同族のPPARを完全に活性化することができず、競合的様式で受容体由来の完全アゴニストを置換することができ、それによって、トランス活性化のレベルを減少させる。完全および部分アゴニストは、さらに、補助因子の異なる補体を補充することができ、所定のPPARサブタイプに対する補充された補助因子の性質は、所定のアゴニストによって活性化された遺伝子のパターンに大いに影響を及ぼす。
【0073】
PPARのリガンド結合ポケットは、他の核内受容体と比較して大きく、これは、部分的に、PPARに結合することができ、活性化することができる、多種多様な化合物を説明することができる。3つのPPARサブタイプの間で、リガンド認識に相当な重複があり、厳密にいうと、サブタイプ特異的リガンドは未だ同定されていない。しかしながら、いくつかの天然および合成リガンドが、高い程度の選択性を示し、今日最も選択的なリガンドは、個々のPPARサブタイプを活性化するのに必要な濃度に関して、3桁より大きく異なる。ステロイド核内受容体についてのアゴニストと同様に、用語、選択的PPAR調節因子(SPPARM)が導入されている(本明細書中で、「部分アゴニストまたはアンタゴニスト」とも呼ばれる)。このクラスのリガンドは、PPAR(複数可)への結合の際に異なる組の活性化補助因子の補充につながる異なる構造を導入する、部分アゴニスト/アンタゴニストを含む。原則として、SPPARMは、PPAR標的遺伝子のサブセットのみを活性化することができ、それによって、おそらく所望の組の遺伝子の特異的発現を促進する。完全アゴニストおよび部分アゴニストによるPPAR活性化のモデルを図1に示す。本発明の好ましい化合物は、部分PPARアゴニストである。
【0074】
PPAR調節活性は、当該分野で公知の任意の数の方法、またはその適応によって、容易に判定することができる。例えば、PPAR調節活性は、トランス活性化アッセイによって判定することができる。PPARγ調節活性を判定するための、有用なトランス活性化アッセイの非限定的な例は、実施例21に記載されており、PPARδ調節活性を判定するための、有用なトランス活性化アッセイの非限定的な例は、実施例22に記載されている。下記の実施例21は、化合物が、PPAR−GAL4(DNA結合ドメイン)融合タンパク質をコードする構築物およびGAL4依存性受容体構築物をコードする構築物でトランスフェクトされた細胞に添加される方法を説明している。異なるDNA結合ドメインを用いて転写または異なるレポーター遺伝子(例えば、蛍光タンパク質、β−ガラクトシダーゼ、ペルオキシダーゼ、ルシフェラーゼ等)を活性化すること等の、任意の数の可能性のある構築物を用いることができることは、当業者に明らかであろう。どのPPAR活性を判定するのが望ましいかに依存して、構築物は好ましくは前記PPARまたはそのリガンド結合ドメインをコードすることは、当業者に明らかであろう。PPARの活性化の際に(すなわち、PPARアゴニストまたは部分アゴニストの存在下で)、PPARは、レポーター構築物を、場合により定量的な様式で、トランス活性化する。
【0075】
PPAR調節因子はまた、少なくとも1つのPPREを含む第二の核酸の制御下で操作可能に、第一の核酸を含むレポーター遺伝子を用いて同定することができる。第一の核酸は、好ましくは、蛍光タンパク質、β−ガラクトシダーゼ、ペルオキシダーゼ、ルシフェラーゼ等のレポータータンパク質をコードする。前記レポーター構築物は、PPARγおよび/またはδ等の1つ以上のPPARを発現している細胞に挿入されるべきである。したがって、PPARアゴニストは、第一の核酸の転写を活性化することができる化合物として同定することができる。
【0076】
本発明の特定の実施形態によると、好ましい化合物は、PPARおよび/またはPPAR LBDアゴニストまたは部分アゴニストである。用語「PPAR LBD」は、PPARのリガンド結合ドメインをいう。好ましい実施形態によると、本発明の化合物および組成物が、PPARγおよび/またはPPARγLBDアゴニストである。「アゴニスト」によって、細胞内受容体と結合したときに受容体に典型的な反応、例えば転写活性化活性を刺激または増大させる、化合物または組成物が意味される。ある実施形態において、前記アゴニストは、PPARγアゴニスト、すなわち、例えば受容体に対する天然の生理学的リガンドを模倣すること等によって、PPARγ受容体の転写活性を促進、刺激、誘導またはそうでなければ増強する、PPARリガンドである。
【0077】
前記PPAR調節活性は、本明細書中で上述されたトランス活性化アッセイを用いて判定することができる。適した標準的なアゴニストとしては、PPARγのためのロシグリタゾンおよびPPARδのための(4−[3−(2−プロピル−3−ヒドロキシ−4−アセチル)フェノキシ]プロピルオキシフェノキシ−酢酸(L165041、市販されている)が挙げられる。標準的なアゴニストより50%未満のトランス活性化を示す潜在的なアゴニストもまた、特に新しい化合物もしくは活性のある誘導体の開発のために、または部分アゴニストの存在の指標として、有用である場合がある。
【0078】
好ましい実施形態によると、本明細書中の化合物または組成物は、PPARおよび/またはPPAR LBD部分アゴニストであり、より具体的には、本発明の化合物および組成物は、PPARγおよび/またはPPARγLBD部分アゴニストである。可能性のある最大効果(すなわち、完全アゴニスト、または参照分子によって生じる最大効果)未満を生じる薬物は、部分アゴニストと呼ばれる。
【0079】
例えば、本発明の化合物および組成物の部分アゴニスト特性は、完全アゴニストであるロシグリタゾン(アバンディア(商標)、Glaxo−SmithKline)を参照して定義することができる。これは具体的には、PPARγ部分アゴニストに当てはまる。部分PPARアゴニスト。具体的には、本発明のPPARγアゴニストは、所望の治療を受けている患者に対して同様またはよりよい有効性を提供するが、より少ない、患者に有害な望ましくない副作用を有する。例えば、SN1(DPD)は、10mMで1mMのアバンディアと同じレベルのグルコース取り込みを誘導するが、より低い脂肪細胞分化の結果として、より少ない副作用が予期される。
【0080】
本発明の化合物および組成物の部分アゴニスト特性はまた、L165041(市販されている)を参照して定義することができる。これは具体的には、PPARδ部分アゴニストに当てはまる。例えば、かかるトランス活性化アッセイの1つは、実施例22に記載されるトランス活性化アッセイである。
【0081】
ある実施形態において、本発明の化合物は、PPARの活性化に対して選択的であることが好ましい。かかる実施形態において、化合物は、RxRおよび/またはRxR LBDトランス活性化を有意に活性化しないことが好ましく、好ましくは、RxR転写は、バックグラウンドレベルの1.5倍未満等、バックグラウンドレベルの2倍より少なく、例えば、バックグラウンドレベルとおよそ等しいか、またはバックグラウンドレベル未満である。RxRトランス活性化は、例えば実施例29に記載されているようなRxRトランス活性化アッセイによって判定することができる。バックグラウンドレベルは、リガンドの添加なしでのトランス活性化である。
【0082】
本発明のある実施形態において、化合物は、PPARアンタゴニストである。「アンタゴニスト」によって、PPARと結合したときに前記PPARに典型的な反応、例えば転写活性を、干渉または減少させる化合物が意味される。一般的な定義として、「PPARアンタゴニスト」は、対応するPPARアゴニストの活性を阻害することができるPPARリガンドを示す。より一般には、これらのアゴニスト/アンタゴニスト/部分アゴニスト活性は、例えば国際公開第99/50664号または国際公開第96/41013号に開示されているもの等の、当業者に広く公知のアッセイによって測定することができる。PPARアンタゴニストの例は、実施例21に提供される。
【0083】
本発明の化合物および組成物は、PPAR活性の調節によって影響を及ぼされる状態または疾患を有する患者に投与されたときの、それらの生物学的活性によって、さらに特徴付けられる。本発明の好ましい化合物は、それを必要とする患者において、以下の生物学的実体:グルコース、トリグリセリド、脂肪酸、コレステロール、胆汁酸等の1つ以上を、当該分野で公知の分子(例えば、チアゾリジンジオン)と比較してよりよい、または同等な有効性および能力で、しかしより低い毒性および/またはより少ない望ましくない副作用の発生で、低下させることができる化合物である。具体的には、前記化合物は、好ましくは、脂肪細胞分化および体重増加の、より少ない誘導につながる。かかる化合物は、具体的には、本明細書中の上記のC.項に記載された化合物のいずれかであってもよい。脂肪細胞分化を判定するための有用な方法は、本明細書中で下記の実施例25に記載されている。より具体的には、それらは、インスリン抵抗性(減少した、インスリンが血糖値を低下させる有効性)および/または脂肪生成に対する有益な活性を提供する。PPARγを活性化する多くの化合物(例えばチアゾリジンジオン)が、脂肪細胞分化をさらに誘導し(すなわち、脂肪生成または脂質生成の効果を示す)、したがって、治療される患者において体重増加を生じることが、示されている。したがって、次世代のかかる化合物は、減少した活性を示し、好ましくはかかる活性を欠いていることが、高度に望ましい。これらの活性は、当該分野で広く用いられている方法(実施例5に記載されているもの等)を用いて評価することができる。より具体的には、これらの活性は、ロシグリタゾン等の、当該分野ですでに同定されている分子を参照して評価される。本発明の好ましい実施形態によると、好ましい化合物は、例えばインスリン抵抗性を患っている患者においてグルコースレベルを判定することによって判定することができるインスリン抵抗性に関するロシグリタゾン特性の、少なくとも約50%、好ましくは少なくとも約60%、より好ましくは少なくとも約70%、さらにより好ましくは少なくとも80%を示す。理想的には、これは100%以上である。本発明の別の好ましい実施形態によると、好ましい化合物は、脂肪細胞分化に対するロシグリタゾン特性の、約80%未満、好ましくは約50%未満、より好ましくは約40%未満、さらにより好ましくは約30%未満を示す。理想的には、これは脂肪細胞分化に対するロシグリタゾン特性の20%未満である。脂肪細胞分化は、例えば、本明細書中に下記で実施例30に記載されているように判定することができる。非常に好ましい実施形態において、好ましい化合物は、上述の特性の両方を有する。あるいは、化合物は、トランス活性化アッセイで判定すると、少なくともロシグリタゾンのものの50%の程度まで、PPAR活性を誘導することができ、脂肪細胞分化に対する上述の特性を示す。
【0084】
血液希釈、浮腫、脂肪細胞分化、または肥満等の望ましくない副作用のインビボでの発生は、例えば欧州特許第1267171号に記載されている方法を用いて、前記化合物の補助因子補充プロフィールによって影響を受ける場合がある。したがって、本発明のある実施形態において、好ましい化合物は、望ましくない副作用の、低いインビボでの発生を有すると予想される、化合物である。
【0085】
PPARγ等の核内受容体が、標的遺伝子のプロモーター領域中の同族配列に結合することならびに活性がクロマチンリモデリング、ヒストンおよび補助因子修飾から基本的な転写機構補充の範囲にわたる多数の補助因子複合体を補充することによって、転写活性化または抑制を達成することは、広く認められている(Glass,およびRosenfeld、2000年、Genes Dev.、14、121−141)。これらの補助因子は、かなりの部分まで、核内受容体の作用の特異性を判定することができ、適した変性が特定の細胞反応につながる刺激のネットワーク中でそれらの作用を統合することができる。したがって、時間および細胞型の関数としての、各核内受容体が携わる複数の連携の判定は、転写調節における核内受容体の活性のよりよい理解につながるであろう。例えば、エストロゲン等のある種のホルモンについて、ホルモンに対する反応は、それぞれの核内ホルモン受容体の存在によって、受容体と相互作用する補助因子の存在によるのとほとんど同じ程度まで判定されることが、公知である。種々のPPAR補助因子が同定されている。p300/CBP(Dowellら、1997年、J.Biol.Chem.272、33435−33433)、SRC−1(Onateら、1995年、Science
270、1354−1357)、TIF2(GRIP−2、Chakravartiら、1996年、Nature、383、99−103)、SRA(Lanzら、1999年、Cell、97、17−27)、AIB−1(Anzickら、1997年、Science、277、965−968)、TRAP220/DRIP205(すなわち、PBP、Zhuら、1997年、J.Biol.Chem.272、25500−25506、Rachezら、1999年、Nature、398、824−828)、PGC−1(Puigserverら、1998年、Cell 92、829−839)、PRIP(Zhuら、2000年、J Biol Chem 275、13510−13516)、PGC−2(Castilloら、1999年、Embo J、18、3676−3687)、ARA70(Heinleinら、1999年、J Biol Chem 274、16147−16152)、RIP140(Treuterら、1998年、Mol Endocrinol 12、864−881)等のいくつかの補助因子は、それらの転写活性を増強するが、SMRT(Lavinskyら、1998年、Proc.Natl.Acad.Sci.USA 95、2920−2925)およびN−CoR(Dowellら、J Biol Chem 274、15901−15907)は、それを抑制する。また、PPARγ補助因子ファミリーのメンバー(例えば、160−kDaタンパク質(SRC−1/TIF2/AIB−1)、CBP/p300またはTRAP220/DRIP205)は、PPARγと直接相互作用し、リガンド依存的様式で核内受容体トランス活性化機能を促進して、用いられるリガンドによって異なり得る生物学的作用または副作用につながることが、示されている(Adamsら、1997年、J.Clin.Invest.、100、3149−3153)。Koderaら、(2000年、J
Biol Chem.、275、33201−33204)は、PPARγと公知の補助因子との間の相互作用が、異なるクラスのPPARγリガンド(天然および合成)によって同じ程度に誘導されるかどうかを調べ、PPARγおよび補助因子複合体の全体的な構造が、関与するリガンドによって異なり、異なる生物学的作用を示す特定の組の標的遺伝子プロモーターの活性化を生じる場合があると結論付けた。
【0086】
PPAR部分アゴニストは、一般に、特定の活性化補助因子補充プロフィールを有し、したがって、特定の活性化補助因子補充プロフィールを有する化合物が好ましい。したがって、特定の実施形態によると、本発明の化合物および組成物は、限定された補助因子(複数可)補充パターンによって、さらに特徴付けられる。好ましい実施形態において、前記パターンは、前記核内受容体の転写活性の調節に対して別個の効果を生じ、特定の代謝プロセスの活性化ならびに望まれない副作用の排除を生じる非常に精密に調整された調節を可能にする。より具体的な実施形態において、本発明の化合物および組成物はさらに、PPAR受容体、より好ましくはPPAR受容体LBDの、補助因子TIF2との相互作用を阻害することができる。別の実施形態によると、本発明の化合物および組成物はまた、PPAR受容体、より好ましくはPPAR受容体LBDの、補助因子SRC−1との相互作用を増強することができる。好ましくは、前記PPAR受容体はPPARγ受容体である。
【0087】
リガンドによる補助因子補充の阻害および/または増強を測定するための方法は、欧州特許1267171に詳述されている。
【0088】
好ましい実施形態において、本発明の化合物は、PPARγに結合したときに、TIF2のものより少なくとも1ログ大きく、少なくとも2ログが好ましい、EC50で、LBDへのSRC1の補充を可能にする。
【0089】
本発明のある実施形態において、PPAR受容体の天然の生理学的リガンド、特にPPARγ受容体のものに対するそれらのアゴニスト、特に部分アゴニスト、またはアンタゴニスト特性のために、好ましい化合物は、PPAR媒介性転写制御およびそれによって生じる付随する生理学的効果の生物学的効果を制御するための医薬品として機能することができる。より具体的には、それらは、細胞生理学を調節して、関連する病理を減少させるか、または予防を提供もしくは増強することができる。
【0090】
本発明のさらに別の実施形態において、好ましい化合物は、1つより多いPPAR、例えばPPARγおよびPPARδの両方の、アゴニスト(または、好ましくは部分アゴニスト)である化合物である。かかるアゴニストは、例えば本明細書中に記載されているように、それぞれPPARγおよびPPARδについてのトランス活性化アッセイによって同定することができる。PPARγおよびPPARδ活性を判定するための非限定的な有用な方法は、本明細書中で、下記でそれぞれ実施例26および27に記載されている。
【0091】
臨床状態
本発明は、上述の化合物を投与することを含む、臨床状態の治療の方法、ならびに臨床状態の治療のための薬物の調製のための前記化合物の使用に関する。
【0092】
PPAR活性の調節因子は、体重管理において使用することができる。したがって、ある実施形態における臨床状態は、神経性食欲不振症(本明細書中で「食欲不振」とも省略される)または過食症等の摂食障害である場合がある。本明細書中に上記で開示される化合物はまた、体重を増大または減少させるための、特に体重を減少させるための方法において、使用することができる。臨床状態は、したがって、肥満である場合がある。脂肪症は、脂肪組織の過剰な累積である。最近の調査から、PPAR、具体的にはPPARγが、脂肪細胞の遺伝子発現および分化において中心的な役割を果たすことが示されている。PPARγサブタイプは、脂肪細胞分化の活性化に関与するが、肝臓におけるペルオキシソーム増殖の刺激において、それほど重要な役割を果たさない。PPARγの活性化は、典型的には、脂肪細胞特異的遺伝子発現を活性化することによって、脂肪細胞分化に寄与する(Lehmann、Moore、Smith−Oliver、Wilkison、Willson、Kliewer、J.Biol.Chem.、270:12953−12956、1995)。したがって、PPARアゴニストを用いて、脂肪組織を獲得することができる。PPAR部分アゴニストは、脂肪組織の過剰な累積の治療において有用な特性について選択することができる。
【0093】
ある好ましい実施形態において、本発明は、本明細書中に上記で記載されている化合物のいずれかを、それを必要とする個体に投与することによってインスリン抵抗性を治療するための方法に関する。本発明はまた、インスリン抵抗性の治療のための薬物の調製のための、前記化合物のいずれかの使用に関する。また、本発明は、前記化合物を投与することによってインスリン感受性を増大させるための方法、ならびにインスリン感受性を増大させるための薬物の調製のための前記化合物の使用に関する。外傷、手術、または心筋梗塞の後に発生し得るもの等のインスリン感受性における急性および一過性の障害は、本明細書中で教示されるように、治療することができる。
【0094】
インスリン抵抗性は、多数の臨床状態に関与している。インスリン抵抗性は、減少した、インスリンが広範囲の濃度にわたって生物学的作用を示す能力によって、顕在化される。インスリン抵抗性の初期の間、体は、異常に高い量のインスリンを分泌して、その欠損を補う。血中インスリンレベルは慢性的に高いが、インスリンに対する、活性のある筋細胞の代謝反応障害によって、それらはグルコースを効果的に取り込むことができなくなる。現在、インスリン抵抗性および結果として生じる高インスリン血症はいくつかの臨床状態、例えばメタボリックシンドローム(シンドロームXとも名付けられている)に寄与する場合があるということが、ますます認識されてきている。メタボリックシンドロームは、高インスリン血症を引き起こす最初のインスリン抵抗性段階、脂質異常症、および減少した糖耐性によって特徴付けられる。メタボリックシンドロームの患者は、心血管疾患および/またはII型糖尿病を発症する危険性が高いことが示されている。
【0095】
患者は、以下の基準の少なくとも3つが当てはまるとき、メタボリックシンドロームを患っているといわれる:
−胴の外周によって測定される中心性/腹部肥満(男性で102cmより大きく、女性で94cmより大きい)
−150mg/dL(1.69mmol/L)以上の空腹時トリグリセリド
−HDLコレステロール[男性−40mg/dL未満(1.04mmol/L)、女性−50mg/dL(1.29mmol/L)未満]
−130/85mmHg以上の血圧
−110mg/dL(6.1mmol/L)以上の空腹時血糖
インスリン抵抗性はまた、脂質生成に対して負の効果を有し、血流中のVLDL(超低密度リポタンパク質)、LDL(低密度リポタンパク質)、およびトリグリセリドレベルの増大ならびにHDL(高密度リポタンパク質)の減少に寄与する。これは、動脈における脂肪の斑の沈着につながる場合があり、これは、経時的に、アテローム性動脈硬化症につながる場合がある。したがって、本発明による臨床状態は、家族性高脂血症等の高脂血症である場合がある。好ましくは、高脂血症は、高コレステロール血症および/または高グリセリド血症によって特徴付けられる。臨床状態はまた、脂質異常症および糖尿病性脂質異常症を含む。本明細書中に含まれる化合物はまた、血清トリグリセリドレベルを低下させるため、またはHDLの血漿レベルを上昇させるために利用することができる。
【0096】
インスリン抵抗性は、血液中の過剰なインスリンおよびグルコースレベルにつながる場合がある。過剰なインスリンは、腎臓によるナトリウム貯留を増大させる場合があり、したがって、本発明の方法は、腎臓によるナトリウム貯留を減少させるために使用することができる。上昇したグルコースレベルによって、血管および腎臓が損傷を受ける場合がある。したがって、本発明の方法を使用して、血管および腎臓への障害を予防することができる。
【0097】
本発明の別の実施形態において、臨床状態は、PPARγによって媒介される炎症性障害である。用語「PPARγによって媒介される」によって、PPARγが状態の顕在化において役割を果たすことが理解されるべきである。例えば、PPARγは、急性炎症等の、好中球活性化と関連した炎症において役割を果たさないと考えられる。理論に拘束されることを望むことなく、PPARγのアゴニストは、NFκB媒介性転写と直接関連して阻害し、したがって例えば誘導型一酸化窒素合成酵素(NOS)およびサイクロオキシゲナーゼ−2(COX−2)の酵素経路等の種々の炎症反応を調節することによって、効果的な抗炎症薬である場合がある(Pineda−Torra,I.ら、1999年、Curr.Opinion in Lipidology、10、151−9)。
【0098】
炎症性障害は、例えば眼の炎症(J Biol Chem.2000年1月28日、275(4):2837−44)または眼乾燥疾患(J Ocul Pharmacol Ther.2003年12月、19(6):579−87)等、急性でも慢性でもよい。慢性的炎症性障害の例示的な例としては、炎症性腸疾患、潰瘍性大腸炎、またはクローン病が挙げられる。慢性炎症性障害はまた、関節炎、特に関節リウマチおよび多発性関節炎である場合がある。慢性炎症性障害はまた、炎症性皮膚疾患、特に尋常性ざ瘡、アトピー性皮膚炎、バリアー機能障害を有する皮膚障害、加齢または乾癬、特に乾癬の皮膚の効果である場合がある。慢性炎症性障害はまた、多発性硬化症またはアルツハイマー病等の炎症性神経変性疾患である場合がある。臨床状態はまた、糸球体腎炎、糸球体硬化症、腎炎症候群、および高血圧性腎硬化症を含む、胃腸疾患および腎疾患である場合がある。
【0099】
本発明の別の実施形態において、臨床状態は、PPARγの活性化に反応性のある癌である。したがって、臨床状態は、例えば、脂肪細胞腫瘍、例えば脂肪腫、線維脂肪腫、脂肪芽細胞腫、脂肪腫症、褐色脂肪腫、血管腫、および/または脂肪肉腫等の、脂肪組織において生じる過形成性または腫瘍性障害等の、PPAR応答性細胞の異常な細胞増殖によって特徴付けられる障害である場合がある。さらに、前立腺、胃、肺および膵臓の、ある種の癌は、PPARγアゴニストでの治療に反応性があることが実証されている。具体的には、ある種の脂肪肉腫、前立腺癌、多発性骨髄腫、および膵癌は、PPARγの活性化に反応性があることが示されているが、少なくともいくつかの結腸直腸癌および乳癌は反応性がない(Rumiら、2004年、Curr.Med.Chem.Anti−Canc Agents、4:465−77)。他の研究から、他の乳癌および結腸癌、ならびに神経芽細胞腫および傍抗癌が、PPARアゴニストに反応性があることが実証されている。癌の治療のためのPPARリガンドの使用は、Levy Kopelovich、2002年、Molecular Cancer Therapeutics、357によって概説されている。
【0100】
しかしながら、ある型の癌はPPARγでの活性化に反応性がある場合があっても、所定の型の全ての癌は反応性がない場合もある。具体的には、PPARγの機能喪失型の変異が癌において頻繁に生じ、かかる癌は、一般に反応性でない。したがって、癌が機能的PPARγを発現することが好ましい。
【0101】
臨床状態はまた、ウイルス感染、特にAIDSまたはHIVによる感染またはC型肝炎ウイルスによる感染等の感染である場合がある。また、本発明のPPARリガンドは、神経系疾患もしくは認知症における認知機能を向上させるために、または多嚢胞性卵巣症候群を治療するために、または骨量減少、例えば骨粗鬆症を予防および治療するために、有用である場合がある。
【0102】
臨床状態はまた、肝疾患、特にC型肝炎ウイルスによる感染、またはC型肝炎ウイルス感染と関連があるかどうかを問わないが、好ましくはPPAR調節に反応性がある、脂肪肝、肝炎、肝臓病変、肝硬変、非アルコール性脂肪性肝炎、もしくは肝癌後である場合がある。
【0103】
記載の多くはPPARγに関するが、本発明の化合物および方法は、PPARγの調節に限定されない。実際、他のPPARサブタイプが疾患において重要な役割を果たすことは、当業者に明らかであろう。例えば、PPARδは、脂肪代謝障害および創傷治癒、具体的には表皮創傷治癒と関連付けられている(Soon Tanら、2004年、Expert Opinion in Molecular Targets、39)。したがって、臨床状態はまた、表皮創傷治癒を含む創傷治癒である場合がある。
【0104】
インスリン感受性改善薬、例えばグリタゾンは、インスリン抵抗性を治療するために使用されているが、インスリン感受性を増強するが、それらはまた、体重を減少させるよりは増加させる。肥満および肉体的不活発は、インスリン抵抗性を悪化させる。グリタゾン(チアゾリジンジオンまたはTZD)は、脂肪組織、肝臓、および筋肉においてインスリン感受性を向上させることによって作用する。前記薬剤での治療は、いくつかの糖尿病の動物モデルにおいて試験されており、いかなる低血糖反応の発生もなしに、上昇したグルコース、トリグリセリド、および非エステル型遊離脂肪酸の血漿レベルの完全な矯正を生じる(Cheng LaiおよびLevine、2000年、Heart Dis.、2、326−333)。これらのチアゾリジンジオンの例は、ロシグリタゾン、ピオグリタゾンおよびトログリタゾンである。しかしながら、魅力的な治療効果を提供するが、これらの化合物は、血液希釈(浮腫を含む)、肝毒性、体重増加(増大した脂肪細胞分化からの体脂肪増大、血漿量増大、および心肥大を含む)、中程度だが有意なLDL−コレステロール増大および貧血を含む多数の深刻な望ましくない副作用を被っている(概説については、Lebovits、2002年、Diabetes Metab.Res.Rev、18、補足2、S23−9参照)。実際、糖尿病の多くの利用可能な治療は、体重増加、疾患の長期の管理の高い有意性の問題と関連がある。したがって、肥満、糖尿病ならびに心血管および肝疾患等の一般に関連のある障害の管理において使用することができる、代替的な、より効果的な治療剤の、大きな必要性がある。
【0105】
したがって、ある実施形態において、本発明は、本発明の化合物を、
i.肥満および糖尿病を患っている、または
ii.糖尿病を得る危険性があり、肥満になりつつある、または
iii.肥満を患っており、糖尿病を得る危険性がある、または
iv.糖尿病を患っており、肥満になる危険性がある
個体に投与することによる、肥満および糖尿病の同時の治療および/または予防に関する。
【0106】
本発明はまた、肥満および糖尿病の同時の治療および/または予防のための薬物の調製のための、本発明の化合物の使用に関する。化合物は、本明細書中で上述される化合物のいずれであってもよい。この実施形態の範囲内で、糖尿病は、好ましくは、II型糖尿病である。糖尿病を得る危険性がある前記個体は、例えば、本明細書中で上述されているメタボリックシンドロームを患っている個体である場合がある。肥満になる危険性がある前記個体は、例えば、体重増加の副作用を有する抗糖尿病薬での医学的治療下にある個体である場合がある。
【0107】
本発明はまた、臨床状態の治療または予防のための薬物の調製のための、上述の特定の化合物のいずれかの使用に関する。臨床状態は、メタボリックシンドローム、脂質異常症、肥満、真性糖尿病、インスリン抵抗性、または上述のインスリン抵抗性と関連する状態のいずれか、高血圧、心血管疾患、冠状動脈再狭窄、自己免疫疾患(喘息、多発性硬化症、乾癬、局所的皮膚炎、および潰瘍性大腸炎等)、癌、炎症、創傷治癒、脂肪代謝障害、肝疾患(C型肝炎ウイルスによる感染、またはC型肝炎ウイルス感染と関連があるかないかを問わず、脂肪肝、肝炎、肝臓病変、肝硬変もしくは肝癌後)、胃腸もしくは腎疾患(糸球体腎炎、糸球体硬化症、腎炎症候群、または高血圧性腎硬化症等)、感染(具体的にはウイルス感染)、認知機能障害(神経系の障害または認知症等)、多嚢胞性卵巣症候群、骨量減少(骨粗鬆症等)ならびにAIDSからなる群より選択してもよい。
【0108】
癌は、任意の癌、例えば以下のもののいずれかである場合がある:癌腫、肉腫、白血病、およびリンパ腫、腫瘍血管新生および転移、骨胃形成症、肝障害、ならびに造血疾患および/または骨髄増殖性疾患。例示的な障害としては、線維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊索腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、結腸癌、膵癌、乳癌、卵巣癌、前立腺癌、扁平上皮癌、基底細胞癌、腺癌、汗腺癌、皮脂腺癌、乳頭癌、乳頭状腺癌、嚢胞腺癌、髄様癌、気管支原性肺癌、腎細胞癌、肝細胞腫、胆管癌、絨毛癌、セミノーマ、胚性癌腫、ウィルムス腺癌、子宮頸癌、精巣腫瘍、肺癌、小細胞肺癌、膀胱癌、上皮癌、グリオーマ、星細胞腫、髄芽腫、頭蓋咽頭癌、上皮腫、松果体腫、血管芽腫、聴神経腫瘍、乏突起グリオーマ、髄膜腫、メラノーマ、神経芽細胞腫、または網膜芽細胞腫が挙げられるが、これらに限定されない。好ましくは、癌は、PPARγの活性化に反応性のある、上述の癌の1つである。
【0109】
心血管疾患は、例えば、アテローム発生、アテローム性動脈硬化症もしくはアテローム硬化性障害、血管再狭窄、心筋症、または心筋線維症、または上述の心血管疾患のいずれかである場合がある。
【0110】
炎症は、例えば、慢性炎症、好ましくは本明細書中で上述された慢性炎症のいずれかである場合がある。
【0111】
真性糖尿病は、複数の病因因子由来の疾患プロセスをいい、上昇した血液中のグルコースのレベル、または高血糖によって特徴付けられる。制御されていない高血糖は、増大した、および早発性の罹患率および死亡率によって特徴付けられる。少なくとも2つの型の真性糖尿病が同定されている:(i)正常な生理学的条件下でグルコース利用を制御するホルモンであるインスリンの完全な欠如の結果である、I型糖尿病、またはインスリン依存性糖尿病(IDDM)、および(ii)II型糖尿病、またはインスリン非依存性糖尿病(NIDDM)。NIDDMは、いくつかの場合において循環インスリンレベルを増大させることによって対処することができる、複数の病因因子由来の複雑な疾患である。
【0112】
医薬製剤および投与の方法
本発明の化合物および組成物に従って、当業者に理解されるように、本明細書中に記載されている化合物の1つ以上を、選択された投与の経路に依存して、種々の形態で哺乳動物に投与してもよい。本発明の組成物は、経口または非経口で投与してもよく、後者の経路は、静脈内および皮下投与を含む。非経口投与は、選択された期間にわたる持続点滴によるものであってもよい。
【0113】
注射可能な使用のための形態としては、無菌水溶液または分散および無菌の注射可能溶液または分散の即席の調製のための無菌の粉末が挙げられる。全ての場合において、形態は、無菌でなければならず、容易な注射可能性が存在する程度まで流動性でなければならない。
【0114】
患者による投与の容易さのために、経口または他の非侵襲性の投与の様式、例えばパッチ、坐剤等の様式が好ましい。化合物は、不活性な希釈剤もしくは吸収できる食用担体とともに経口投与してもよく、またはハードもしくはソフトシェルゼラチンカプセル中に封入するか、圧縮して錠剤にするか、もしくは食事の食物と直接組み合わせてもよい。経口の治療的投与のために、化合物は、賦形剤と組み合わされ、摂取可能な錠剤、バッカル錠、トローチ、カプセル剤、エリキシル剤、懸濁液、シロップ、ウェーハ等の形態で使用してもよい。
【0115】
1つ以上の本発明の化合物を含む組成物はまた、リポソームと呼ばれるリン脂質ベシクル内に含まれた溶液または乳濁液中で投与してもよい。リポソームは、単層でも多層でもよく、ホスファチジルコリン、ジパルミトイルホスファチジルコリン、コレステロール、ホスファチジルエタノールアミン、ホスファチジルセリン、ジミリストイルホスファチジルコリンおよびそれらの組み合わせから選択される成分で形成される。多層リポソームは、単層ベシクルと同様の組成の多層ベシクルを含むが、溶液または乳濁液中の化合物が封入される複数の区画を生じるように調製される。また、他の補助剤および修飾物質が、ポリエチレングリコール、または他の物質等のリポソーム製剤中に含まれる場合がある。
【0116】
組成物を含むリポソームはまた、それらの送達を標的化するために、リポソームの表面に固定された抗体を有すること等の変更を有してもよい。
【0117】
「臨床状態」の項で上述された臨床状態の1つを治療するための、インビボの投与に適した生物学的に適合性のある形態の、対象への投与のための医薬組成物が、本発明のある実施形態にあり、前記方法は、安全で効果的な量の化合物を単独で、または他の薬剤および/もしくは薬学的担体との組み合わせを含む。例えば、フィブラートクラスの薬物、例えばベザフィブラート等の脂質異常症に対して効果的な薬剤を組み合わせて本発明の化合物を用いて、インスリン抵抗性および/または糖尿病を治療することができる。いくつかの他の薬剤の例は、インスリン感受性改善薬、PPARγアゴニスト、グリタゾン、トログリタゾン、ピオグリタゾン、エングリタゾン、MCC−555、BRL 49653、ビグアナイド、メトホルミン、フェンホルミン、インスリン、インスリン模倣剤、スホニル尿素、トルブタミド、グリピジド、α−グルコシダーゼ阻害剤、アカルボース、コレステロール降下薬、HMG−CoA還元酵素阻害剤、ロバスタチン、シンバスタチン、プラバスタチン、フルバスタチン、アトルバスタチン、リバスタチン、他のスタチン、シクエストレート、コレスチラミン、コレスチポール、架橋デキストランのジアルキルアミノアルキル誘導体、ニコチニルアルコール、ニコチン酸:ニコチン酸塩、PPARαアゴニスト、フェノフィブリン酸誘導体、ゲムフィブロジル、クロフィブラート、フェノフィブラート、コレステロール吸収の阻害剤、β−シトステロール、アクリルCoA:コレステロールアシルトランスフェラーゼ阻害剤、メリナミド、プロブコール、PPARδアゴニスト、抗肥満化合物、フェンフルラミン、デクスフェンフルラミン、フェンチラミン、スルビトラミン、オーリスタット、神経ペプチドY5阻害剤、β3アドレナリン受容体アゴニスト、および回腸胆汁酸トランスポーター阻害剤である。
【0118】
組成物は、組成物がインビボで有効性を有するので、ヒトおよび動物を含む、かかる治療を必要とする任意の生物に投与することができる。安全で有効、によって、本明細書中で使用される場合、深刻な副作用を避けながら、対象に影響を及ぼす疾患を減少、予防、改善、または治療するために十分な能力を提供することが意味される。安全で有効な量は、対象の年齢、治療される対象の身体的条件、障害の重症度、治療の期間および任意の併用療法の性質に依存して変化し、その決定は、通常の医師の能力の範囲内である。組成物は、本明細書中に記載されているのと同じ一般的な様式で、製剤化および投与される。本発明の化合物は、単独で、または1つ以上のさらなる活性薬剤と組み合わせて、効果的に用いることができる。併用療法は、本発明の化合物および1つ以上のさらなる活性薬剤を含む単一の薬剤の投薬組成物の投与、ならびに本発明の化合物の投与および各活性薬剤をそれぞれ別々に投薬することを含む。例えば、本発明の化合物およびスルホニル尿素、チアゾリジンジオン、ビグアナイド、メグリチニド、インスリンまたはα−グルコシダーゼ阻害剤等のインスリン分泌促進物質を、カプセル剤もしくは錠剤等の単回経口投薬組成物でともに患者に投与するか、または各薬剤を別々の経口投薬で投与することができる。別々の投薬が使用される場合、本発明の化合物および1つ以上のさらなる活性薬剤を、本質的に同時に、すなわち同時または別々に交互の回で、すなわち連続して投与することができ、併用療法は、これら全ての投与計画を含むと理解される。
【0119】
本発明の医薬組成物の治療有効量はまた、対象の疾患状態、年齢、性別、および体重、ならびに化合物が対象において所望の反応を誘発する能力等の因子によって変化してもよい。投与計画は、最適な治療反応を提供するよう調節してもよい。例えば、いくつかの分割された用量を毎日投与してもよく、または、治療状況の要求によって示されるように、用量が比例して減少してもよい。
【0120】
約4mg/kgの用量は、哺乳動物に適した最初の投薬量であるようであり、この投薬量は、安全で有効な量を提供するように、必要に応じて調節してもよい。したがって、投薬量は、最初は、典型的には0.1〜20mg/kg、好ましくは0.5〜10mg/kg、より好ましくは1〜5mg/kgである。
【0121】
薬学的に許容され得る担体は、本明細書中で使用される場合、1つ以上の適合性のある固体または液体送達系が、対象に投与されたときに非侵害性の生理学的反応を有することを意味する。いくつかの例としては、デンプン、糖、セルロースおよびその誘導体、粉末化トラガカント、麦芽、ゼラチン、コラーゲン、滑石、ステアリン酸、ステアリン酸マグネシウム、硫酸カルシウム、植物油、ポリオール、寒天、アルギン酸、発熱物質不含水、等張生理食塩水、リン酸バッファー、および医薬製剤において用いられる他の適した非毒性物質が挙げられるが、これらに限定されない。湿潤剤および潤滑剤、錠剤化剤、安定剤、抗酸化剤、ならびに保存料等の等の他の賦形剤もまた、企図される。
【0122】
本明細書中に記載されている組成物は、化合物または類似体の有効量が薬学的に許容され得る担体と混合物中で組み合わされるように、対象に投与することができる薬学的に許容され得る組成物の調製のための公知の方法によって調製することができる。適した担体は、例えば、Remington’s Pharmaceutical Sciences(Mack Publishing Company、米国ペンシルバニア州イーストン、1985年)に記載されている。これに基づいて、組成物は、排他的ではないが、1つ以上の薬学的に許容され得るビヒクルまたは希釈剤と関連した化合物の溶液を含み、適したpHを有し、生理学的液体と等浸透圧の緩衝液中に含まれる。
【0123】
以下の実施例は、本明細書中の特定の態様を記載して、本発明を説明し、実施例は単に本発明の理解および実施において有用な特定の方法論を提供するだけなので、本発明を限定すると解釈されるべきではない。
【実施例】
【0124】
実施例1
4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸の合成
この実施例は、スキーム2に従って、本明細書中でDPDとも呼ばれる、4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸の合成を説明する(表II中のSN1)。
【0125】
スキーム2
【0126】
【化9】
【0127】
2−メトキシ−パラコン酸の合成(2−3):20Lの二重ジャケットガラスリアクターに260gのo−メトキシベンズアルデヒド、286gの無水コハク酸、572gの無水塩化亜鉛、および2600mLの無水DCMを充填した。混合物を撹拌し、2℃に冷却した。533mLの量のトリエチルアミンを30分間にわたって添加した。次いで、混合物を周囲温度で24時間撹拌させた。1690mLの量の2M HClを添加し、次いで2600mLの酢酸エチルを添加した。混合物を5分間活発に撹拌した。2000mLの酢酸エチルで水相を抽出した。合わせた有機性抽出物を650mLの飽和塩水で洗浄し、次いで3×2600mLの飽和重炭酸ナトリウムで洗浄した。次いで、合わせた水性抽出物を酢酸エチルで洗浄した。濃HClを用いて、水性抽出物をpH2まで酸性化した。黄色の油を分離した。混合物を、2000mLの酢酸エチルを用いて2回抽出した。有機相を1000mLの塩水で4回洗浄し、45℃の加熱浴温度を使用したBuchi R220ロータバップで蒸発させた。残りの残渣に4000mLのトルエンを添加した。混合物を110℃に加熱した。1Lのトルエンを蒸発させて除いた。残りを室温まで冷却させ、48時間放置し、その間純粋な2−メトキシ−パラコン酸が結晶化した。結晶物質を集め、ろ過し、一定の重量まで、真空オーブン中で、真空中、45℃で乾燥させた。
【0128】
収量:220g(49%)。シス/トランス比:46/54
シス−およびトランスラセミメトキシ−パラコン酸から全トランス−2−メトキシパラコン酸への転換(2−4):1020gのメトキシパラコン酸を、1729mLの濃硫酸および2570mLの水の混合物に添加した。混合物を室温で18時間撹拌させた。33:64のシス/トランス比が得られた。次いで、混合物を2.5時間、60℃に加熱した。11:89のシス/トランス比が得られた(HPLCによる分析)。次いで、混合物を室温に冷却させ、続いてろ過した。固体物質を酢酸エチル中に再び溶解させ、水および塩水で洗浄した。有機相をMgSO4で乾燥させ、蒸発させた。6:94のシス/トランス比が得られた。固体物質を熱いトルエンから再結晶化させた。得られた結晶物質を、40℃で48時間、真空中で乾燥させた。
【0129】
収量:855g(84%)。シス/トランス比:8/92。融点:132〜133℃
【0130】
【化10】
【0131】
メトキシ−パラコン酸、ラセミ化合物のエステル化(2−5):193gのメトキシ−パラコン酸を、600mLのTHF中に溶解させた。混合物に、145gのCDI(899mmol、1.1当量)を添加し、混合物を10分間撹拌した。65mLの量の無水エタノール(またはメチルエステルを生じるためのメタノール)を添加し、完了するまで(約120分間)混合物を撹拌した。酢酸エチルおよび飽和重炭酸ナトリウムを用いて、粗反応混合物を抽出した。有機相を0.5N HClおよび塩水で洗浄した。蒸発の後、188gの量の所望のエチルエステルが得られた。
【0132】
ラセミメトキシ−パラコン酸、エチルエステルの還元(2−6):ラセミラクトールの調製:5℃で、700mLのトルエン中の105gのエチルエステル(397mmol)。3当量のDIBAL−H(1.19mol、1.19L 1M溶液)を添加した。室温で60分間撹拌した。メタノールで反応停止させた。2.5Lの酢酸エチルを加えた。700mLの水。水相をEtOAcで抽出した。有機層を塩水で洗浄した。クロロホルム/ヘキサンから油状の残渣を蒸発、再結晶化させた。固体をろ過し、真空中で乾燥させた。
【0133】
収量:53g(237mmol、59%)。
【0134】
ラセミジオールのラセミラクトール合成に使用されるウィティッヒ反応(2−8):191gの量のカルボキシプロピルトリフェニルホスホニウムブロマイド、1000mLの無水トルエンおよび100gのカリウムt−ブトキシドを、80℃で30分間混合した。混合物を室温に冷却した。180mLの無水THFに予め溶解させた25gの量の精製ラセミラクトール(114.5mmol)を、ゆっくり添加した。反応を60分間継続させた。粗反応混合物を1500mLの冷水に注ぎ、300mLの酢酸エチルを添加した。水相を300mLの酢酸エチルで再抽出した。次いで、水相を2N HClで酸性化し、300mLの酢酸エチルを用いて3回抽出した。形成された固体をろ過して除いた。有機相を蒸発させた。蒸発した残渣に、500mLのジエチルエーテルを添加した。フラスコを10分間回転させ、固体をろ過して除いた。ろ液を、飽和重炭酸ナトリウム溶液で3回抽出した。次いで、2M HClを用いて水相をpH4まで酸性化した。次いで、水相を、200mLの酢酸エチルを使用して3回抽出した。有機相を合わせて、MgSO4で乾燥させ、蒸発させて、45gの物質を生じた。
【0135】
カラムクロマトグラフィー:ラセミジオールをシリカゲル(35cmカラム長、4cm直径)で精製した。ラセミジオールを最少の酢酸エチルに溶解させ、カラムに適用した。1Lの酢酸エチル(60%)/ヘキサン(40%)を、容積測定シリンダーに加えた。300mLのEtOAc/ヘキサンをシリンダーから採取し、カラムに加えた。酢酸エチルを用いて、シリンダー中の残りの700mLのEtOAc/ヘキサンを1Lまで希釈した。次いで、300mLの新しいEtOAc/ヘキサン溶液をカラムに加え、カラムを通過させた。酢酸エチルを用いて、シリンダー中の残りの700mLのEtOAc/ヘキサンを、再び1Lまで希釈した。次いで、300mLの新しいEtOAc/ヘキサン溶液をカラムに加え、カラムを通過させた。酢酸エチルを用いて、シリンダー中の残りの700mLのEtOAc/ヘキサンを、もう1回1Lまで希釈した。次いで、300mLの新しいEtOAc/ヘキサン溶液をカラムに加え、カラムを通過させた。ラセミジオールの純粋な画分を集め、蒸発させて、26gの純粋なラセミジオールを生じた。
【0136】
収量:26g(88.3mmol、79%)
ラセミジオールの、ラセミアセトニドへの転換(2−10):26g(88mmol)の精製ジオールを、260mLのジメトキシプロパンおよび26mgのp−TsOHと混合した。混合物を、周囲温度で一晩撹拌させた。3滴のトリエチルアミンを添加し、混合物を蒸発させた。残りの残渣に150mLのヘキサンを添加し、混合物を一晩撹拌した。固体をろ過して除き、乾燥させて25g(75mmol)のラセミアセトニドを生じた。
【0137】
収率:85%
ラセミアセトニドの脱メチル化(2−12):16.7gのエタンチオールを、375mLのDMPU中の21.5gのNaHの混合物に添加することによって、水素化ナトリウムおよびエタンチオールの懸濁液を調製した。懸濁液を80℃に加熱し、周囲温度まで冷却させた。
【0138】
15gのラセミアセトニドを75mLのDMPU中に溶解させ、EtSH/NaHの懸濁液に添加した。混合物を130℃で2時間加熱した。次いで、反応混合物を氷水に注ぎ、DCMで抽出した。2N HClを用いて水層を酸性化し、酢酸エチルで抽出した。有機層を塩水で洗浄し、蒸発乾固させた。
【0139】
収量:16.5g(粗)。
【0140】
ラセミの調製(2−14):28mmolの量の脱メチル化されたラセミアセトニドを、15mLの2−クロロベンズアルデヒド、0.5gのp−TsOH、および60mLのトルエンと混合した。混合物を24時間撹拌し、蒸発させた。Biotage Horizon(登録商標)クロマトグラフィー装置を使用したシリカゲルクロマトグラフィーを用いて、粗反応混合物を精製した。DCM(19)/メタノール(1)を用いて混合物を精製して、蒸発の後に6.5gの固体を生じた。
【0141】
収量:6.5g(16.7mmol、59%)
スキーム3:表I(および表II)に記載されているPPAR調節因子のための合成経路
【0142】
【化11】
【0143】
【化12】
【0144】
実施例2
PPAR調節因子2(SN2、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子2(SN2、表II)の合成を説明する。実施例1に記載されているような100mg(0.31mmol)の量のラセミアセトニド(6−[4−(2−ヒドロキシフェニル)−2,2−ジメチル−[1,3]ジオキサン−5−イル]−ヘキス−4−エノン酸)2−12を、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、108mg)の2,3−ジクロロベンズアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析よって分析した。
【0145】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=435
実施例3
PPAR調節因子6(SN6、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子6(SN6、表II)の合成を説明する。100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、70mg)のシクロヘキサノンを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0146】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=359
実施例4
PPAR調節因子8(SN8、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子8(SN8、表II)の合成を説明する。
【0147】
N−Boc 4−オキソピペリジンの調製:2gの量の4−オキソピペリジンを、20mLのジオキサン/水に溶解させた。混合物に、2当量の重炭酸ナトリウム、次いで1.0当量のBoc2Oを添加した。混合物を4時間撹拌した。酢酸エチル(100mL)を添加した。0.2N HClおよび塩水を用いて、有機相を2回洗浄した。次いで、有機相を硫酸マグネシウムで乾燥させ、蒸発させて、結晶化した油を生じた。100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol)のN−Boc−4−オキソピペリジンを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物を微小のシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0148】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=460
本明細書中で、下記で言及される生物学的アッセイは、スピロ−ピペリジン環上に依然として存在するBoc−保護基を含む反応生成物で行った。
【0149】
実施例5
PPAR調節因子9(SN9、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子9(SN9、表II)の合成を説明する。
【0150】
1−ナフタレンカルボキシアルデヒドの調製 3.87mLの量の塩化オキサリル(44.24mmol)を、100mLのDCMに溶解させた。混合物を−60℃に冷却した。注射器を用いて、5.6mLの量のDMSOを滴下した。混合物を15分間撹拌した。75mLのDCM中の5gのナフタレン−1−メタノールの溶液を滴下した。反応を、−60℃で1時間継続させた。20mLの量のトリエチルアミンを添加した。混合物を15℃に達させた。混合物を抽出漏斗に移し、水、1M HCl、水および塩水で洗浄した。次いで、有機層を硫酸マグネシウムで乾燥させ、蒸発させた。粗生成物は、次の工程においてさらなる精製なしで使用するのに十分純粋であった。
【0151】
実施例1に従って調製された100mg(0.31mmol)の量のラセミアセトニド(上述のような2−12)を、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、97mg)の1−ナフタレンカルボキシアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0152】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=417
実施例6
PPAR調節因子11(SN11、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子11(SN11、表II)の合成を説明する。
【0153】
100mg(0.31mmol)の量のラセミアセトニド(実施例1の2−12)を、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、62mg)のヘキサナールを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0154】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=361
実施例7
PPAR調節因子13(SN13、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子13(SN13、表II)の合成を説明する。
【0155】
100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、130mg)のo−2−オキソ−4a,8a−ジヒドロ−2H−クロメン−4−カルバルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物を微小なシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0156】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=469
実施例8
PPAR調節因子19(SN19、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子19(SN19、表II)の合成を説明する。
【0157】
100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、103mg)の2,3−ジメトキシベンズアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0158】
HPLC:保持時間(グラジエントA):15.23、15.39分、95%(異性体の1:1混合物の合計)
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=427.2
実施例9
PPAR調節因子14(SN14、表II)の合成 この実施例は、スキーム3に従う、スキーム3に従う、PPAR調節因子14(SN14、表II)の合成を説明する。
【0159】
室温で、撹拌された、CH2Cl2(10mL)中のジオール(0.08g、0.27mmol)の溶液に、2,6−ジクロロベンズアルデヒド(0.07g、0.40mmol)およびpTSA(3mg)を添加した。反応混合物を8時間撹拌し、トリエチルアミン(2〜3滴)で反応停止させた後、真空中で濃縮した。残渣を、ヘキサン/EtOAc(8:2)で希釈したシリカゲル上でのカラムクロマトグラフィーによって精製して、無色の油として、アセタール(100mg、45%収率)を得た。
【0160】
【化13】
【0161】
実施例10
PPAR調節因子15(SN15、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子15(SN15、表II)の合成を説明する。スキーム−2、実施例1の化合物2−12(100mg、0.31mmol)を、THF(5mL)中に溶解させ、触媒量のpTsOH(5mg)を室温で添加した。反応混合物を室温で8時間撹拌し続けた。2,3,4,5,6−ペンタフルオロベンズアルデヒド(158mg、0.62mmol)を反応塊に添加し、もう1回触媒量のpTsOHを添加した。反応条件をさらに24時間維持した。反応の完了後に、乾燥したEt3Nを添加して、pH=7に調節した。真空下で溶媒を除去し、得られた粗混合物をカラムクロマトグラフィーによって精製して、最終生成物15を得た。
【0162】
スキーム4:表III(および表II)に記載されている化合物の合成経路
【0163】
【化14】
【0164】
【化15】
【0165】
実施例10
PPAR調節因子23(SN23、表II)の合成
この実施例は、スキーム4に従う、PPAR調節因子23(SN23、表II)の合成を説明する。
【0166】
ウィティッヒ反応 10g(22.56mmol)の量のBrPPh3(CH2)4CO2Hを、60mLの乾燥トルエン中の5.06g(45mmol)のKOtBuと混合した。混合物を30分間80℃に加熱し、周囲温度まで冷却させた。10mLの無水THF中の1.26gの量のラセミラクトールを添加し、反応を2時間継続させた。スキーム−2の2−8の調製についての後処理。
【0167】
収量:3.3g(粗、次の工程で使用する)。
【0168】
アセトニドの調製 前の工程で得られたジオールを、40mLのジメトキシプロパン中に溶解させた。25mgの量のp−TsOHを添加し、反応を24時間撹拌した。1滴のトリエチルアミンを添加し、混合物を蒸発させた。粗混合物を、小さいシリカゲルカラムでろ過した。
【0169】
収量:1.6g
アセトニドの脱メチル化 1.91mLの量のエタンチオール(25.83mmol)を、50mLのDMPUと混合した。51.7mmolの量のNaH(2.06gの、油中の60%分散)を添加し、混合物を30分間80℃に加熱した。混合物を周囲温度まで冷却した。7.5mLのDMPU中の1.5gのアセトニド(4.3mmol)の溶液を添加し、混合物を125℃で2時間撹拌した。粗混合物を氷水に注ぎ、2×50mLのDCMで抽出した。水相を2N HClで酸性化し、次いで、EtOAcで抽出し、最後に塩水で洗浄した。有機相を蒸発させて、1.6gのヒドロキシ−アセトニドを生じた。
【0170】
2−クロロベンズアルデヒドとの反応 ヒドロキシ−アセトニド(1.6g、粗)を、2mLの2−クロロベンズアルデヒド、8mLの無水トルエン、および50mgのpTsOHと混合した。混合物を24時間撹拌し、蒸発させ、シリカゲルカラムクロマトグラフィーによって精製した。
【0171】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=415
実施例11
PPAR調節因子17(SN17、表II)の合成
この実施例は、スキーム4に従う、PPAR調節因子17(SN17、表II)の合成を説明する。
【0172】
100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、122mg)の4−フェニルアミノベンズアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物を微小なシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0173】
o−メトキシパラコン酸のメチルエステルの合成:a)撹拌された、乾燥DMF(150mL)中の酸(5g、21.28mmol)の溶液に、K2CO3(29.2g、211.8mmol)を添加し、次いでヨウ化メチル(6.01g、42.3mmol)を0℃で添加し、反応混合物を室温で5時間撹拌した。反応混合物を水(10mL)、CH2Cl2(30mL)で希釈し、有機層を分離した。CH2Cl2(2×15mL)で水層を抽出し、合わせた有機相を塩水(2×20mL)で洗浄し、Na2SO4で乾燥させ、真空中で蒸発させて、エステルを得たが、これはさらなる反応において使用するのに十分に純粋であった。(収量:4.34g、81.9%)。
【0174】
b)撹拌された、乾燥エーテル(60mL)中の酸5(10g、42.3mmol)の溶液に、新しく調製したジアゾメタン溶液(200mL溶液、10gのニトロソメチル尿素から生じた)を0℃で添加し、室温で30分間撹拌した。出発物質が完全に消えた後で、エーテルを蒸発させて、エステル6を得て、これを、精製なしで、さらなる反応のために使用した。(収量:9.8g、98%)。黄色の、粘性のある油
【0175】
【化16】
【0176】
実施例12
PPAR調節因子34(SN34、表II)の合成
この実施例は、PPAR調節因子34(SN34、表II)の合成を説明する。
【0177】
窒素雰囲気下、触媒のp−トルエンスルホン酸(約5mg)の存在下で、乾燥トルエン(2mL)中のジオール7(0.4g、1.36mmol)およびクロロベンズアルデヒドジメチルアセタール(0.28g、1.63mmol)の混合物を一晩撹拌した。出発物質が完全に消えた後で、反応混合物を固体NaHCO3で中和した。溶液を反応混合物から傾しゃし、ロータリーエバポレーターで濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、ベンジリデンアセタールを生じた。
【0178】
収量:0.31g(59%)
【0179】
【化17】
【0180】
THF:H2O(4mL、3:1)中のこの化合物(0.2g、0.48mmol)の溶液に、LiOH.H2O(0.3g、7.15mmol)、メタノール(0.5mL)を添加し、室温で一晩撹拌した。反応混合物をNaHSO4の飽和水溶液で中和し、CH2Cl2(2×10mL)で抽出した。合わせた有機層を塩水(10mL)で洗浄し、ロータリーエバポレーターで蒸発させ、粗生成物をカラムクロマトグラフィーによって精製した。
【0181】
収量:95mg(51%)
【0182】
【化18】
【0183】
スキーム−4a
【0184】
【化19】
【0185】
実施例13
PPAR調節因子25(SN25、表II)の合成
この実施例は、表IIのPPAR調節因子25の合成を説明する。
【0186】
エタンチオール(0.7mL、9.5mmol)を、撹拌された、乾燥DMF(8mL)中のNaH(0.38g、9.5mmol)の懸濁液(油中60%)に、0〜5℃で添加し、20〜30分間撹拌した。温度を維持しながら、乾燥DMF(2mL)中の化合物4a−1(0.25g、0.95mmol)を、上述の混合物にゆっくり滴下した。反応塊温度を120〜130℃に上昇させ、6〜8時間維持した。反応の完了後に、塊を0〜5℃に冷却し、pH4〜5に調節することによって、1N HCl(1mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、純粋な4a−2を得た(酢酸エチル:ヘキサン、2.5:7.5)。
【0187】
収量:155mg(65.6%収率)。
【0188】
4a−2の化合物(0.15g、0.6mmol)を、8:2の比のアセトンおよび水の混合物(10mL)中に溶解させた。上述の混合物に、OsO4(触媒量、0.05M)およびNMO(0.14g、1.2mmol)を添加し、室温で8〜10時間撹拌し続けた。反応の進行をTLCによってモニタリングした。反応の完了後に、飽和Na2S2O5溶液(3mL)によって反応停止させた。溶媒アセトンを真空下で除去し、酢酸エチル(3×10mL)で化合物を抽出した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。
【0189】
THF:H2O(8:2、10mL)混合物中の粗生成物(スキーム中に示されていないジオール)の溶液に、NaI4(0.27g、1.8mmol)を室温で添加し、1時間撹拌し続けた。反応の完了後に、飽和Na2SO4溶液(3mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物4a−3を、さらなる反応のために処理した。
【0190】
乾燥ベンゼン(10mL)中の化合物4a−3の溶液に、C2−ウィティッヒイリド(0.15g、0.42mmol)を添加した。反応塊を、不活性な条件下、室温で2〜3時間撹拌し続けた。ロータリーエバポレーター中で過剰なベンゼンを除去し、このようにして得られた粗化合物4a−5をカラムクロマトグラフィーによる精製に供した。(酢酸エチル:ヘキサン、0.5:9.5)。
【0191】
収量:0.11g g(95%)
【0192】
【化20】
【0193】
THF:H2O(8:2、5mL)の混合物中の化合物4a−5(0.11g、0.34mmol)の溶液に、LiOH.H2O(0.1g、2.4mmol)を室温で添加し、8〜10時間撹拌し続けた。反応の完了後に、pHを4〜5に調節することによって、飽和NaHSO4溶液(1mL)によって反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で蒸発させ、真空で濃縮した。粗生成物4a−6を、さらなる反応のために処理した。
【0194】
化合物4a−6(85mg、0.29mmol)をTHF中に溶解させ、触媒量のpTSOHを室温で添加した。反応混合物を室温で6〜8時間撹拌し続けた。o−クロロベンズアルデヒド(81mg、0.58mmol)を反応塊に添加した。もう1回触媒量のpTSOHを添加した。反応条件をさらに5〜6時間維持した。反応の完了後に、pH=7に調節することによって、乾燥Et3Nを添加した。真空下で溶媒を除去し、得られた粗混合物をカラムクロマトグラフィーによって精製して、最終生成物25(表II)を得た。(酢酸エチル:ヘキサン、3.5:6.5)。
【0195】
収量:35mg(32%)
上述の化合物HPLC純度は79.2%であり、分取HPLCによってこれをさらに精製して、純粋な化合物(98%純度、15mg)を得た。
【0196】
【化21】
【0197】
スキーム5:
【0198】
【化22】
【0199】
実施例14
PPAR調節因子37(SN37、表II)の合成
この実施例は、スキーム5に従う、表IIのPPAR調節因子37の合成を説明する。
【0200】
スキーム5中のマロン酸水素エチル5−2の調製:撹拌された、エタノール(100mL)中のマロン酸ジエチル(20g、0.125モル)の溶液に、室温で、時々冷却しながら、エタノール(30mL)中の85%KOH(7g、0.125モル)の溶液を添加した。20分後に、混合物を濃HClで酸性化し、ろ過した。ろ過ケーキ(KCl)をエタノール(50mL)で洗浄した。合わせたろ液を濃縮し、残りの液体をカラムクロマトグラフィー(ヘキサン−EtOAc:85:15)に供して、1(14.7g、87%)を得た。
【0201】
【化23】
【0202】
ケト−エステル5−4の調製:
乾燥THF(100mL)中の14.34gのマグネシウムエトキシド(0.13モル)の懸濁液に、10.5gのマロン酸水素エチル1 5−2(0.08モル)を添加し、90分間還流した。反応混合物を0℃に冷却し、乾燥THF(25mL)中の2−メトキシベンゾイルクロライド2(14.59g:0.085モル)の溶液を、反応温度が5℃を超えない速度で添加した。反応混合物を室温で一晩撹拌し、2日間放置した。塩化アンモニウムの飽和溶液を添加し、反応混合物をEtOAc(3×50mL)で抽出した。合わせた有機層を水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を蒸発させて、油として生成物を得て、カラムクロマトグラフィー(ヘキサン−EtOAc:95:5)によって精製して、生成物(4.33g、25%収率)を生じた。生成物をスペクトルデータによって確認した。
【0203】
【化24】
【0204】
撹拌された、乾燥THF(40mL)中の水素化ナトリウム(60%、0.9g、0.08モル)の懸濁液に、アセチル化エステル(4.33g、0.019モル)を0〜10℃で滴下し、15分間撹拌した。臭化アリル3(2.30g、0.019モル)を室温で反応混合物に添加し、反応混合物を3〜4時間還流した。反応混合物を冷却し、塩化アンモニウム溶液を添加し、EtOAc(3×30mL)で抽出した。有機層を水で洗浄し、無水硫酸ナトリウムで乾燥させ、蒸発させた。粗生成物をカラムクロマトグラフィー(n−へキサン−EtOAc:95:5)によって精製して、4(2.5g、50%収率)を得た。
【0205】
【化25】
【0206】
ジオールの調製(5−7):
温度が10℃を超えない速度で、冷却した、乾燥THF(15mL)中の水素化ホウ素リチウム(0.83g、0.038モル)の懸濁液に、冷却した、乾燥THF(25mL)中のアルキル化ケトエステル4 5−4(2.5g、0.0095モル)の溶液を添加した。撹拌を室温で4〜5時間継続し、反応の進行をTLCによってモニタリングした。2N HClの添加によって反応混合物をpH2に酸性化し、水を反応混合物に添加し、EtOAc(3×25mL)で抽出した。合わせた有機層を塩水、水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を蒸発させて、ジオール5 5−7(2g、94%収率)のシスおよびトランス反応混合物を生じ、精製なしで次の工程に進んだ。HPLC:シス77.318%、反応時間20.244分、トランス:22.682%、反応時間22.337分(HPLC条件は、クロマトグラムに関して言及される)。
【0207】
アセトニド5−10の調製:
ジオール5−7(2g、0.009モル)、2,2−ジメチルプロパン(15mL、0.12モル)、触媒量のpTSA(10mg)の溶液を、室温で4〜5時間撹拌し、反応の進行をTLCによってモニタリングした。反応混合物をEt3Nで中和した。過剰なDMPを真空下で除去し、油状のシス−トランス混合物をカラムクロマトグラフィーによって分離して、6b 5−10(0.925g、収率40%)を得た。
【0208】
【化26】
【0209】
1,3−ジオキサンアルデヒドの調製(オゾン分解)5−11:
永続的な青色が生じるまで、O3を−78℃で乾燥DCM(10mL)中のアセトニド5−10(0.93g、0.004モル)の溶液に通過させた。青色が消えるまで、反応混合物を0℃で撹拌した。DCM(5mL)中のTPP(1.1g、0.0043モル)の溶液を、無色の溶液に0℃で添加し、反応混合物を室温まで温め、1時間撹拌した。反応混合物の進行をTLCによってモニタリングした。溶媒を除去し、生成物をフラッシュクロマトグラフィーによって精製して、アルデヒド7(0.5g、収率54%)を生じた。
【0210】
【化27】
【0211】
ウィティッヒ塩5−12の調製:
乾燥アセトニトリル(50mL)中の6−ブロモヘキセン酸(3g、0.0512モル)およびトリフェニルホスフィン4.8g、0.018モル)の溶液を20〜24時間還流し、過剰な溶媒を減圧下で除去して、無色の油を得て、これを乾燥ベンゼンで粉末状にし、乾燥ベンゼンおよびエーテルの連続(3回ずつ)で洗浄した。洗浄手順の間に、減圧下で乾燥する結晶化した物質から、白色の微結晶性粉末としてウィティッヒ塩が提供された(3.6g、55%収率)。
【0212】
2,4−置換(1,3−ジオキサン−5−イル)カルボン酸(SN37、表II)](ウィティッヒ生成物)5−13の調製:
乾燥THF(5mL)中の水素化ナトリウムの溶液(60%、0.364g、0.0152モル)を、撹拌された、乾燥THF(10mL)中のウィティッヒ塩8 5−12(0.95g、0.002モル)の懸濁液に、0℃で、N2雰囲気下で添加した。混合物を30分間撹拌した。次いで、アルデヒド7 5−11の溶液(0.5g、0.0019モル)を添加した。反応を、撹拌しながら36時間継続させた。反応の完了後に、水を添加し、溶媒を減圧下で除去した。水溶液を酢酸エチルで洗浄し、5%HCLでpH2まで酸性化し、酢酸エチルで抽出した。合わせた有機性抽出物を飽和塩水で洗浄し、無水Na2SO4で乾燥させ、蒸発させた。得られた油をフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル−75:25)によって精製して、ウィティッヒ生成物9 5−13(0.2g、35%収率)を得た。
【0213】
【化28】
【0214】
実施例15
PPAR調節因子36(SN36、表II)の合成
この実施例は、表IIのPPAR調節因子36(2,2−ジメチル−4−(2−ヒドロキシフェニル)−1,3−ジオキサン−5−イル−カルボン酸)(5−15)の合成(メトキシ基の脱保護)を説明する:
エタンチオール(0.13g、0.00203モル)を、0℃、N2雰囲気下で、撹拌された、DMPU(5mL)中の水素化ナトリウム(60%、1g、0.004167モル)の懸濁液に添加した。30分後に、DMPU中の化合物5−13(0.00055モル)の溶液を添加した。混合物を120℃で2〜3時間加熱し、冷却し、氷水に注ぎ、水性混合物をDCMで洗浄した。5%HClで水層をpH5まで酸性化し、酢酸エチルで抽出した。合わせた有機性抽出物を飽和塩水で洗浄し、乾燥させ、蒸発させた。得られた油をフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル−75:25)によって精製して、ウィティッヒ生成物5−15(0.6g、66%収率)を得た。
【0215】
【化29】
【0216】
実施例16
PPAR調節因子35(SN35、表II)の合成
この実施例は、表IIのPPAR調節因子35([2,4,5−シス]−2−o−クロロフェニル−4−o−メトキシフェニル−1,3−ジオキサン5−イル)オクテン酸−(スキーム5の5−20)の合成を説明する。
【0217】
2−クロロベンズアルデヒド(50.48mg、0.359mmol)、p−TSA(触媒の、5mg)およびアセトニド化合物5−15(130mg、0.359mmol)の混合物を、4mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−20(55mg、34.3%収率)を得た。
【0218】
【化30】
【0219】
実施例17
PPAR調節因子38(SN38、表II)の合成
この実施例は、表IIのPPAR調節因子38([2,4,5,−シス]−2−o−クロロフェニル−4−o−メトキシフェニル−1,3−ジオキサン5−イル)オクタン酸(5−21)の合成を説明する:
5mLの乾燥酢酸エチル中のオレフィン化合物5−13(150mg、0.414mmol)の溶液に、10モル%の10%Pd−C(44mg)を注意深く添加した。反応混合物をH2雰囲気下で1.5〜2時間撹拌させた。反応が完了した後に、混合物をセライトおよびケーキ(Pd−C)を通してろ過し、乾燥酢酸エチル(2×5mL)で洗浄した。合わせたろ液を濃縮し、残りの液体をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−21(SN38)(80mg、53.03%収率)を得た。
【0220】
【化31】
【0221】
2−クロロベンズアルデヒド(34.25mg、0.247mmol)、p−TSA(触媒の、3mg)およびアセトニド化合物12 5−21(80mg、0.34mmol)の混合物を、3mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物13 5−22(50mg、45.09%収率)を得た。
【0222】
【化32】
【0223】
LC−MS:純度87.53%および質量464(M+18)。
(実施例18)
【0224】
PPAR調節因子24(SN24、表II)の合成
この実施例は、表IIfのPPAR調節因子24([2,4,5−シス]−2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン5−イル)オクテン酸(5−17)の合成を説明する。
【0225】
2−クロロベンズアルデヒド(48.47mg、0.344mmol)、p−TSA(触媒の、5mg)およびアセトニド化合物(120mg、0.344mmol)の混合物を、4mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−17(40mg、27%収率)を得た。
【0226】
【化33】
【0227】
LC−MS:純度は94.43%(2つのジアステレオマー、それぞれ80.12+14.31)および質量448(M+18)
実施例19
PPAR調節因子31(SN31、表II)の合成
この実施例は、表IIのPPAR調節因子31([2,4,5−シス]−2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン5−イル)オクタン酸の合成を説明する。(5−19)
3mLの乾燥酢酸エチル中のオレフィン5−15(60mg、0.172mmol)の溶液に、10モル%(19mg、0.0172mmol)の10%Pd−Cを添加した。反応混合物をH2雰囲気下で8時間撹拌させた。反応が完了した後に、セライトおよびケーキ(Pd−C)を通して混合物をろ過し、乾燥酢酸エチル(2×5mL)で洗浄した。合わせたろ液を濃縮し、残りの液体をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−18(40mg、66.29%収率)を得た。
【0228】
【化34】
【0229】
2−クロロベンズアルデヒド(16mg、0.114mmol)、p−TSA(触媒の、3mg)およびアセトニド化合物15 5−18(40mg、0.114mmol)の混合物を、3mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物16 5−19(20mg、40.28%収率)を得た。
【0230】
【化35】
【0231】
LC−MS:純度85%および質量450(M+18)
スキーム6
【0232】
【化36】
【0233】
実施例20
PPAR調節因子32(SN32、表II)の合成
この実施例は、表IIのPPAR調節因子32の合成を説明する。
【0234】
乾燥THF(10mL)中のアルケン6−10(前述の5−10)(1.2g、4.5mmol)の溶液に、BH3.DMS(0.34g、4.5)を0℃で添加し、反応混合物を同じ温度で2時間撹拌した。混合物に、3N NaOH(3mL)および30%H2O2(1mL)を0℃で添加し、室温でさらに1時間撹拌し続けた。反応混合物を水で希釈し、酢酸エチル(2×30mL)で抽出した。合わせた有機層を減圧下で蒸発させ、粗生成物をカラムクロマトグラフィー(酢酸エチル:ヘキサン、2:8)によって精製した。
【0235】
収量:0.9g(70%)
【0236】
【化37】
【0237】
6−12の調製:IBX(0.525g、1.87mmol)を乾燥DMSO(1mL)中に溶解させ、室温で10分間撹拌し、0℃に冷却した。これに、アルコール6(0.35g、1.25mmol)を乾燥THF(9mL)中、窒素雰囲気下で添加し、室温で1.5時間撹拌した。反応混合物をエーテル(10mL)で希釈し、20分間撹拌し、ろ過した。ろ液を水(2×10mL)で洗浄し、エーテル層を乾燥させ(Na2SO4)、ロータリーエバポレーターで濃縮した。生成物をフィルターカラムによって精製して、アルデヒド6−12を得た(酢酸エチル:ヘキサン、2:8)。
【0238】
収量:300mg(86.4%収率)。
【0239】
【化38】
【0240】
6−14の調製:(3−カルボキシプロピル)トリフェニルホスホニウムブロマイドを真空下、100℃で2〜3時間乾燥させた。撹拌された、乾燥トルエン(10mL)中の(3−カルボキシプロピル)トリフェニルホスホニウムブロマイド(1.47g、3.43mmol)の溶液に、カリウムter−ブトキシド(0.774g、6.89mmol)を一部ずつ、室温で、不活性な条件下で添加した。混合物を80℃に加熱し、温度を30〜40分間維持した。反応塊を50℃に冷却し、乾燥THF(2mL)中のアルデヒド(化合物6−12)(0.3g、1.07mmol)を上述の混合物に滴下した。反応の進行をTLCによってモニタリングした。反応の完了後に、塊を0〜5℃に冷却し、pH4〜5に調節することによって、1N HCl(1mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、純粋な化合物6−14を得た(酢酸エチル:ヘキサン、2:8)。
【0241】
収量:325mg(86%)。
【0242】
【化39】
【0243】
質量:371.1(M++Na)。
【0244】
6−16の調製:エタンチオール(0.44g、7.17mmol)を、撹拌された、乾燥DMF(10mL)中の60%NaH(0.287g、7.17mmol)の懸濁液に0〜5℃で添加し、20〜30分間撹拌した。乾燥DMF(2mL)中の化合物8 6−14(0.25g、0.71mmol)を、温度を維持しながら上述の混合物にゆっくり滴下した。反応塊温度を120〜130℃に上昇させ、6〜8時間維持した。反応の完了後に、塊を0〜5℃に冷却し、pH4〜5に調節することによって、1N HCl(1mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、純粋な6−16を得た(酢酸エチル:ヘキサン、2.5:7.5)。
【0245】
収量:155mg(65%)。
【0246】
化合物6−16(130mg、0.38mmol)をTHF(8mL)中に溶解させ、触媒量のpTsOHを室温で添加した。反応混合物を室温で6〜8時間撹拌し続けた。o−クロロベンズアルデヒド(109mg、0.77mmol)を反応塊に添加し、もう1回触媒量のpTsOHを添加した。反応条件をさらに5〜6時間維持した。反応の完了後に、pH=7に調節することによって、乾燥Et3Nを添加した。溶媒を真空下で除去し、得られた粗混合物をカラムクロマトグラフィーによって精製して、最終生成物SN32(表II)6−19を得た。(酢酸エチル:ヘキサン、3.5:6.5)。
【0247】
収量:65mg(40%)。
【0248】
カラムクロマトグラフィーの後のHPLC純度は、より近い不純物を有して、80.3%であった。分取HPLCによってこれをさらに精製して、92.4%HPLC純粋化合物(20mg得られた)を得た。
【0249】
【化40】
【0250】
質量:415.1(M+−H)
HPLC純度:92.48%(カラム:水 ノバパック3.9×300mm、移動相:70%CH3CN+30%酢酸アンモニウムバッファー、RT:2.472)。
【0251】
さらなる類似体を、以下のようにして合成した:化合物6−14(90mg、0.25mmol)を、THF:0.2N HCl(10mL、9:1)の混合物中に溶解させ、室温で2時間撹拌した。反応の完了後に、混合物を酢酸エチル(2×10mL)で抽出し、Na2SO4で乾燥させ、ロータリーエバポレーターで蒸発させて、68mgの粗生成物を得て、これを、精製なしでさらなる反応のために利用した。
【0252】
上述の粗化合物(68mg、0.22mmol)を、乾燥THF中に溶解させ、この混合物に2−クロロベンズアルデヒド(61mg、0.44mmol)および触媒量のpTSA(約4mg)を室温で添加した。反応混合物を、窒素雰囲気下で、同じ温度で5時間撹拌した。出発物質が消えた後に、混合物を乾燥トリエチルアミンで中和し(pH=7に調節することによって)、溶媒を蒸発させた。粗生成物をカラムクロマトグラフィー(ヘキサン中の25%酢酸エチル)によって精製して、化合物11を得た。
【0253】
収量:32mg(34%)。
【0254】
【化41】
【0255】
質量:429.1(M+−H)
HPLC純度:97.84%(カラム:水 ノバパック3.9×300mm、移動相:70%CH3CN+30%酢酸アンモニウムバッファー(0.05%AcOH、RT:7.428)。
【0256】
実施例21
PPARγトランス活性化アッセイ
細胞培養物、プラスミドおよびトランスフェクション
この実施例は、PPARγ調節活性を判定するためのトランス活性化アッセイの例である。
【0257】
これらのアッセイにおいて、Gal4−PPARγLBD(Helledieら2000年)、UASx4−TK luc(ChenおよびEvans、1995年)およびCMV−β−ガラクトシダーゼ(市販されている、例えばClontech)を用いて、PPARγトランス活性化を示した。UASx4−TK−luc受容体構築物(ここでUASは「上流活性化因子配列」をいう)は、4つのGal4−応答エレメントを含む。プラスミドGal4−PPARγLBDは、UASに結合することによってUASx4−TK−lucレポータープラスミドをトランス活性化することができるGal4−DBD−PPARγ−LBD融合タンパク質(すなわち、PPARγのリガンド結合ドメイン、LBDに融合したGal4のDNA結合ドメイン、DBD)をコードする。CMV−β−ガラクトシダーゼプラスミド(ここでCMVはサイトメガロウイルスである)は、実験値の正規化のために使用される。
【0258】
マウス胎仔線維芽細胞(MEF)を、7.5%AmniomaxサプリメントC−100(Gibco)、7.5%ウシ胎仔血清(FBS)、2mMグルタミン、62.5μg/mlペニシリンおよび100μg/mlストレプトマイシンを補充したAmnioMax基本培地(Gibco)(増殖培地)中で増殖させた。あるいは、ME3細胞(Hansenら、1999年)を、10%仔ウシ血清(CS)、62.5μg/mlペニシリンおよび100μg/mlストレプトマイシンを補充したDMEM(増殖培地)中で増殖させた。トランスフェクションの時間に細胞が50〜70%コンフルエントになっているように、細胞を、典型的には24ウェルプレートに、再び播種した。
【0259】
製造業者の使用説明書に従ってLipofectamine Plus(Invitrogen)またはMetaffectane(Biontex)を用いて、細胞をGal4−PPARγLBD(Helledieら2000年)、UASx4−TK luc(ChenおよびEvans、1995年)およびCMV−β−ガラクトシダーゼ(市販されている、例えばClontech)でトランスフェクトした。簡潔にいうと、24ウェルプレートのウェルあたりに、30μLのDMEM(血清および抗生物質を含まない)中のUASx4TKluc(0.2μg)Gal4−PPARγLBD(またはpM−hPPARγ−LBD、0.1μg)およびCMV−β−ガラクトシダーゼ(0.05μg)を、1μLのメタフェクテネインを含む30μLのDMEM(血清および抗生物質を含まない)と混合した。混合物を室温で20分間インキュベートして、核酸−脂質複合体の形成を可能にし、次いで、およそ60mLを、50〜60%コンフルエント細胞を含む各ウェルに添加した。次いで、細胞をCO2インキュベータ内で、37℃で6〜12時間インキュベートし、次いで、培地を、抗生物質および目的の物質(例えば、DMSO中に溶解させた、本明細書中でDPDとも呼ばれる4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸、または陽性対照としてのロシグリタゾン(アバンディア))を補充した培地、または匹敵する体積のDMSO(全細胞培養物体積の<0.5%)で置換した。DPDは市販されているか、または、実施例1に従って合成することができる。12〜24時間後に細胞を採取し、標準的なプロトコルに従って、ルシフェラーゼおよびβ−ガラクトシダーゼ活性を測定した。
【0260】
PPARトランス活性化は、DMSO単独より、ロシグリタゾン(公知のPPARγアゴニスト)で40倍より大きく高く、10μM 4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸(図2中の「DPD」参照)で約10倍高かった。したがって、4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸は、PPARγアゴニストである。
【0261】
キラルHPLCによって精製されたDPD鏡像異性体のいずれかの間に、PPARγトランス活性化活性に違いは見られなかった。
【0262】
表IIは、DPD(SN1)の種々の類似体で得られた結果を要約し、それらの活性を比較している。「P」は、10μMのDPD(表II中のSN1)と比較して、10μMでの類似したPPARγ活性化活性を表し、「P−」は、10μMのDPDと比較して、30μMでの試験物質での、同じレベルの活性(すなわち、より能力が低い)を表し、「P+」は、「P」より大きい活性のレベル、および10μMのDPDに匹敵する3μMでの活性のレベル(すなわち、より能力が高い)を表し、「P++」は、3μMでは10μMのDPDのものよりも高いが1μMでは10μMのDPDのものより低い、活性のレベルを表し、「P+++」は、10μMのDPDに匹敵する、1μMでの活性のレベルを表し、「−」は、試験が行われなかったことを意味する。
【0263】
このアッセイが、本質的に上述の通りだが、PPARγリガンド結合ドメインでなく全長ヒトPPARγで行われる場合、全長hPPARg2の活性化は64%までのアバンディア陽性対照で見られる(図3)。
【0264】
実施例22
PPARδトランス活性化アッセイ
この実施例は、PPARδ調節活性を判定するためのトランス活性化アッセイを説明する。
【0265】
DPDおよび他のリガンドを、それらがPPARδを本質的に実施例1に記載されているようにトランス活性化する能力について試験する。しかしながら、トランス活性化構築物は、mPPARδLBDであり、ここでPPARδリガンド結合ドメインがPPARγのものに置き換わり、L165041(市販されている)がロシグリタゾンの代わりの選択的PPARδアゴニストとして使用される。用いられた条件下で、L165041はPPARδトランス活性化を50倍より大きく増大させることが示されたが、DPDはトランス活性化の増大を生じないか、わずかしか生じなかった(図2参照)。したがって、DPDはPPARγに対する選択性を示す。
【0266】
このアッセイが、本質的に上述の通りだが、PPARδリガンド結合ドメインでなく全長ヒトPPARδで行われる場合、全長hPPARδの活性化、活性化は見られず(図4)、PPARγに対するDPDの選択性が確認された。
【0267】
実施例23
PPARαトランス活性化アッセイ
この実施例は、PPARα調節活性を判定するためのトランス活性化アッセイを説明する。
【0268】
DPDおよび他のリガンドを、それらがPPARαをトランス活性化する能力について、本質的に実施例1に記載されているように試験する。しかしながら、トランス活性化構築物はmPPARαLBDであり、ここでPPARαリガンド結合ドメインがPPARγのものに置き換わり、GW7647(市販されている)をロシグリタゾンの代わりの選択的PPARαアゴニストとして使用する。用いられた条件下で、GW7647は2倍より大きくPPARαトランス活性化を増大させることが示されたが、DPDはトランス活性化を生じなかった(図2参照)。したがって、DPDはPPARγに対する選択性を示す。
【0269】
実施例24
RXRトランス活性化アッセイ
この実施例は、レチノイン酸X受容体トランス活性化アッセイを説明する。
【0270】
化合物を、それらがRXRをトランス活性化する能力について、本質的に実施例1に記載されているように試験する。しかしながら、トランス活性化構築物はhRXRαLBDであり、ここでRXRリガンド結合ドメインがPPARγのものに置き換わり、{4−[1−(3,5,5,8,8−ペンタメチル−5,6,7,8−テトラヒドロ−2−ナフチル)エテニル]安息香酸}(LG1069、市販されている)を、ロシグリタゾンの代わりの選択的RXRアゴニストとして使用する。用いられる条件下で、LG1069は5倍より大きくRXRトランス活性化を増大させることが示されるが、DPDはトランス活性化を生じない(図2参照)。したがって、DPDは、ここでもPPARγに対する選択性を示す。
【0271】
実施例25
脂肪細胞分化アッセイ
この実施例は、脂肪細胞分化を判定するためのアッセイを説明する。化合物を、それらが脂肪細胞分化を誘導するかどうかを調べるため、およびそれらが脂肪細胞分化を阻害するかどうかを調べるために、試験する。
【0272】
細胞培養物および分化
MEFを、7.5%AmniomaxサプリメントC−100(Gibco)、7.5%ウシ胎仔血清(FBS)、2mMグルタミン、62.5μg/mlペニシリンおよび100μg/mlストレプトマイシンを補充したAmnioMax基本培地(Gibco)(増殖培地)中で増殖させた。コンフルエンスで、MEFを、DMSO中に溶解させた1μMのデキサメタゾン(Sigma)0.5mMのイソブチルメチルキサンチン(Sigma)、5μg/mlのインスリン(Sigma)および10μMの試験化合物を添加した増殖培地中、またはDMSO単独の中で、分化に誘導した。続いて、培地に48時間ごとに、5μg/mlのインスリンおよびリガンドを補充した増殖培地またはDMSOを新たに供給した。
【0273】
簡潔にいうと、脂肪細胞分化の誘導因子として化合物を試験するために、典型的には24ウェルディッシュ中の、10%仔ウシ血清(CS)を含むDMEM中で、3T3−L1をコンフルエンスまで増殖させる。コンフルエンス後2日目(0日目)に、10%ウシ胎仔血清(FBS)、1μMのデキサメタゾンおよび試験化合物(01、1および10μM)を補充したDMEMで、細胞を分化するよう誘導する。BRL49653(100%Me2SO中に溶解させた1.0μM)を陽性対照として使用する。48時間後に、細胞に、試験化合物または陽性対照を補充した、10%FBSを含むDMEMを再び供給する。4日目から、細胞を、10%FBSを含むDMEM中で増殖させ、8日目まで、2日目ごとに交換する。8日目に、後述されるように、細胞をOil Red Oで染色する。
【0274】
脂肪細胞分化の阻害剤として化合物を試験するために、コンフルエンス後2日目(0日目と名付けられる)まで、3T3−L1細胞を上述のように増殖させ、その後、10%ウシ胎仔血清(FBS)、1μMデキサメタゾン(Sigma)、0.5mMメチルイソブチルキサンチン(Sigma)、1μg/mlインスリン(Roche Molecular Biochemicals)および試験化合物を含むDMEMで、細胞を分化するよう誘導する。試験化合物の溶媒の存在下で分化するよう誘導された細胞を、陽性対照として使用する。48時間後に、細胞に、試験化合物または陽性対照を補充した、10%FBSを含むDMEMを新たに供給した。4日目から、細胞を、10%FBSを含むDMEM中で増殖させ、8日目まで、2日目ごとに交換する。8日目に、後述されるように、細胞をOil Red Oで染色する。
【0275】
Oil Red O染色
上述のように培養された細胞を、Oil Red染色のために使用する。PBS中でディッシュを洗浄し、細胞を3.7%パラホルムアルデヒド中で1時間固定し、(Hansenら1999年)に記載されているようにoil red Oで染色した。0.5gのOil Red O(Sigma)を100mlのイソプロパノール中に溶解させることによって、Oil Red O溶液ストック溶液を調製する。ストック溶液を水で希釈し(6:4)、続いてろ過することによって、Oil red O希釈標準溶液を調製する。
【0276】
DPDは、誘導アッセイにおいて、非常に少ない赤色染色しか誘導しなかった。赤色染色は脂肪細胞の存在の指標であるので、DPDが脂肪細胞分化を誘導しないことが推測できる。しかしながら、DPDは、脂肪細胞分化の阻害において有意な効果を示さなかったので、脂肪細胞を阻害しない。表IIは、DPD(SN1)の種々の類似体で得られた結果を要約し、「0」はDPDと類似した結果を表し、「−1」はさらに低い脂肪細胞分化を表し、「+1」はより大きい脂肪細胞分化を表し(ともにDPDと比較して)、「−」はアッセイされなかったことを表す。
【0277】
本質的に上述の通りだがヒト前脂肪細胞を用いて行われたアッセイからも、DMSOと同様に、DPDが非常に少ない赤色染色しか誘導しなかったことが実証された。
【0278】
実施例26
部分アゴニスト対完全アゴニストの同定
この実施例は、部分PPARアゴニストを判定するためのアッセイを説明し、これは医薬品として特に望ましい。
【0279】
簡潔にいうと、トランス活性化アッセイを、本質的に実施例1に記載されている通りだが100nMアバンディア(完全アゴニスト)を、増大する濃度の試験化合物(または対照としての試験化合物なし)とともに、各ウェルに添加して行う。アバンディアによってトランス活性化が減少する化合物は、PPAR部分アゴニストである。結果を図5に示す。
【0280】
PPARγ部分アゴニストを、それらがPPARγ、例えばこの場合アバンディアに結合することから、公知のPPARγアゴニストに置き換わる能力によって、この実施例中で同定する。あるいは、他の公知の完全アゴニスト、例えば300nM L165041を用いて、本質的に実施例2に記載されている通りにトランス活性化アッセイを用い、PPARδの部分アゴニストを同定することができる。
【0281】
実施例27
部分PPARγアゴニストリガンド置換アッセイ
以下のように、PPARγリガンド結合ポケットへの結合の分析のために、Invitrogen POLARSCREEN PPAR競合アッセイを用いて、部分PPARγアゴニストを同定するためのさらなるアッセイを行う。
1.20μLの2X試験化合物をキュベットに分注する
2.20μlの2XPPAR−LBD/Fluormone Green Complexを添加して混合する
3.暗黒中で2時間インキュベートする
4.蛍光偏光度を測定する
5.対照として、ロシグリタゾン(アバンディア)を使用する
AVANDIAと比較したDPDでの結果を図6に示す。
【0282】
実施例28
グルコース取り込みアッセイ
この実施例は、PPARγアゴニストとして同定された化合物が、PPARγアゴニストに期待される、細胞アッセイにおける生理学的効果、特にグルコース取り込みに対する効果も生じることを説明する。グルコース取り込みアッセイは、インスリン抵抗性の治療のための化合物の適合性を確立するために重要である。
【0283】
簡潔にいうと、3T3−L1前脂肪細胞を、コンフルエンスまで12ウェルプレート中で増殖させる。細胞を、血清不含DMEMで洗浄し、1mlの同じ培地とともに37℃で1〜2時間インキュベートする。次いで、細胞をクレブス・リンガー−Hepes(KRP)バッファーで洗浄し、0.9mlのKRPバッファーとともに37℃で30分間インキュベートする。0、0.3、1および3nMの終濃度でインスリンを添加し、37℃で15分間インキュベートする。10mM[3H]2−デオキシ−D−グルコース(1mCi/l)を補充した0.1mlのクレブス・リンガーリン酸(KRP)バッファーの添加によって、グルコース取り込みを開始する。37℃で10分間のインキュベーションの後に、培地を吸引し、プレートを氷冷PBSで洗浄して、誘導されたグルコース取り込みと終了させる。0.5mlの1%Triton X−100で細胞を溶解させ、シンチレーションカウンターを用いて放射活性レベルを測定する。結果を図7に示す。
【0284】
要約すると、DPDおよび表IIで言及される多数の類似体は、PPARγアゴニストであると実証されるが、物質10はPPARγアンタゴニストであるようである。物質7および13は、脂肪細胞分化をあまり引き起こさないか、引き起こさずに、より高い可能性を示すので、特に興味深い類似体である。
【0285】
本明細書中で言及される全ての刊行物および特許出願は、個々の刊行物または特許出願が、特に、および個々に参照によって援用されていると示されるのと同じ程度に、参照によって本明細書中に援用される。本発明はここで完全に説明されているが、添付の特許請求の範囲の精神または範囲から逸脱することなく、これに多くの変化および変更を行うことができることが、当業者に明らかであろう。
【0286】
【化42】
【0287】
【化43】
【0288】
【化44】
【0289】
【化45】
【0290】
【化46】
【0291】
実施例29
トロンボキサン受容体活性
この実施例は、物質1の両方の鏡像異性体がPPARアゴニスト活性を有するが、一方のみがトロンボキサン受容体アンタゴニストとして作用することを実証する。
【0292】
以下の条件下で、キラルクロマトグラフィーによって、DPD(表II中のSN1)の各鏡像異性体を単離した:
カラム:250×4.6mm Chiralpak AD−H5 □m
移動相:80/20/0.1 n−ヘプタン/エタノール/トリフルオロ酢酸
流速:1ml/分
検出:230nmでのUV
温度:25C
試料を80/20 n−ヘプタン/エタノール中に溶解させた。
【0293】
鏡像異性体1が最初にキラルカラムで溶出し、鏡像異性体2が2番目にキラルカラム上で溶出した。
【0294】
本質的にHedbergら(1988年)J Pharmacol.Exp.Ther.245:786−792およびSaussyら(1986年)J.Biol.Chem.261:3025−3029によって記載されているように、トロンボキサン受容体結合についての放射性リガンド結合アッセイで、鏡像異性体1および2を試験した。鏡像異性体1は潜在的なトロンボキサン受容体結合剤である(IC50、0.841nM)とわかったが、鏡像異性体2は結合しないようであった(IC50>10nM)。
【0295】
トロンボキサン受容体アンタゴニストとして作用する鏡像異性体で、ある種の患者集団を治療することが望ましい場合があり、この場合、鏡像異性体1が投与される(例えば、トロンボキサン受容体アンタゴニストの心血管の利益が同時に望ましい場合)。トロンボキサン受容体に対する効果を有さないことが望ましい場合(例えば、患者がかかる治療を必要としない場合、または患者がすでに異なる治療を受けている場合等の、さらなる心血管効果が必要でない場合)、鏡像異性体2が有益に投与される。
【0296】
実施例30
癌細胞増殖の阻害
この実施例は、癌細胞の増殖に対するDPDの抗増殖性効果を実証し、癌の治療のための、本明細書中に記載されている類似体の有用性を説明する。
【0297】
組換えバイオセンサーレポーター活性を安定して発現するよう操作されたヒト子宮頸癌細胞系(HeLa)および非癌性細胞系、HaCaT(Boukampら、1988年、J.Cell Biol 106(3):761−771)を、Caspa Tagキット(例えばChemiconから、市販されている)とともに用いた。ハイコンテンツスクリーニング自動フローサイトメーターを用いて、増殖の検出を行った。
【0298】
1%DMSOに調節された、血清のない細胞培地中で、DPD(物質1)を20μMまで希釈した。HTSフローサイトメーター(FACS Calibur HTS、Becton Dickenson)のFL2(増殖マーカーの希釈)チャネル中の処理の48時間後に、96ウェルプレート中、2連で、細胞事象の検出を行った。1日目に、細胞を96ウェルプレートに播種した。2日目に細胞をDPDおよび適切な対照で処理し、4日目に分析を行った。
【0299】
DPDは、20μMで、HeLa細胞の増殖を90%阻害した。DPDは、非癌性細胞系HaCaTに効果を有さず、したがって、DPD(表II中のSN1)による癌細胞に対する選択的抗増殖性効果が確立された。
【技術分野】
【0001】
発明の分野
本発明は、糖尿病、癌、炎症、神経変性障害および感染等のPPAR調節に依存する疾患または状態の治療における、2,4−ジフェニル−1,3−ジオキサンの使用を対象とする。
【背景技術】
【0002】
発明の背景
ペルオキシソーム増殖因子活性化受容体(PPAR)は、核内ホルモン受容体である。PPAR受容体は、レチノイドX受容体(RXRとして公知)とのヘテロ二量体の形態でペルオキシソーム増殖因子応答エレメント(PPRE)として公知のDNA配列の要素に結合することによって、転写を活性化する。3つのサブタイプのヒトPPARが同定および記載されている:PPAR−α、PPAR−γおよびPPAR−δ(またはNUCI)。PPAR−αは主に肝臓中で発現されるが、PPAR−δは遍在する。PPAR−γは、脂肪細胞の分化の制御に関与しており、そこで高度に発現される。これはまた、全身性の脂質恒常性において重要な役割を有する。糖尿病の治療において使用されているチアゾリジンジオンを含んで、PPARの活性を調節する多数の化合物もまた、同定されている。
【0003】
PPAR−γサブタイプのDNA配列は、Elbrechtら、BBRC 224、431−437(1996)に記載されている。フィブラートおよび脂肪酸を含むペルオキシソーム増殖因子は、PPARの転写活性を活性化する。
【0004】
PPARが、これらの核内受容体を発現している細胞に関連した多様な疾患または病的状態に密接に関与していることを説明する文献中で、多数の例が提供されている。より具体的には、PPARは、血中のグルコース、コレステロールおよびトリグリセリドレベルを減少させるための方法における薬物標的として有用であり、したがって、インスリン抵抗性、脂質異常症、ならびに肥満およびアテローム性動脈硬化症(Duezら、2001年、J.Cardiovasc.Risk、8、185−186)、冠動脈疾患およびある種の他の心血管障害を含むシンドロームX(「メタボリックシンドロームとも命名されている)(国際公開第97/25042号、国際公開第97/10813号、国際公開第97/28149号、Kaplanら、2001年、J.Cardiovasc Risk、8、211−7も参照のこと)に関連した他の障害の、治療および/または予防のために使用されている。さらに、PPARは、皮膚病(Smithら、2001年、J.Cutan.Med.Surg.、5、231−43参照)、胃腸疾患(国際公開第98/43081号)、または糸球体腎炎、糸球体硬化症、ネフローゼ症候群および高血圧性腎硬化症を含む腎疾患等の、ある種の炎症性疾患の治療のための潜在的な標的であると示されている。同様に、PPARは、神経系疾患(LandrethおよびHeneka、2001年、Neurobiol Aging、22、937−44)もしくは認知症において認知機能を向上させるため、乾癬、多嚢胞性卵巣症候群(PCOS)を治療するため、または骨量減少、例えば骨粗鬆症を予防および治療するために、有用である(例えば、米国特許第5981586号または米国特許第6291496号参照)。
【0005】
したがって、PPARは、治療用化合物の開発のための、興味深い標的である。疾患または病的状態を治療および/または予防するための種々の方法の状況において観察される反応が奨励されているが(例えば、チアゾリジンジオン(TZD)クラスの薬物療法、例えば、トログリタゾン、ロシグリタゾンまたはピオグリタゾンは、明らかに、2型糖尿病の患者において、インスリン感受性の向上において重要な役割を果たし;Cheng laiおよびLevine、2000年、Heart Dis.、2、326−333参照)、それらは、多数の深刻な望ましくない副作用の発生のために(例えば、体重増加、高血圧、心肥大、血液希釈、肝毒性および浮腫;Haskinsら、2001年、Arch Toxicol.、75、425−438、Yamamotoら、2001年、Life Sci.、70、471−482、Scheen、2001年、Diabetes Metab.、27、305−313、Gale、2001年、Lancet、357、1870−1875、Formanら、2000年、Ann.Intern.Med.、132、118−121ならびにAl Salmanら、2000年、Ann.Intern.Med.、132、121−124参照)、完全に満足できる治療ではない。結果として、PPAR核内受容体を発現する細胞型と関連した疾患または病的状態の治療および/または予防を可能にする、新規な改良された生成物および/または新規な方法を同定することが望ましい。より具体的には、TZD誘導体で見られる副作用のほとんどは前記化合物の完全アゴニスト特性に起因し、したがって、必ずしも完全アゴニストでない新しい化合物を同定することが望ましい。
ある種の4−フェニル−1,3−ジオキサン−5−イルアルケノン酸誘導体は、トロンボキサン受容体アンタゴニストまたはトロンボキサンA2合成の阻害剤として記載されている。トロンボキサン受容体は、効力のある血小板の凝集因子であり、血管収縮、ならびに気管支および気管平滑筋収縮に関わってきた(例えば、特許文献1、特許文献2および特許文献3参照)。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】欧州特許出願公開第94239号明細書
【特許文献2】欧州特許出願公開第0266980号明細書
【特許文献3】米国特許第4895962号明細書
【発明の概要】
【課題を解決するための手段】
【0007】
発明の要旨
本発明のある態様は、1,3−ジオキサン誘導体、またはその薬学的に許容され得る塩の治療有効量を投与することを含む、個体においてPPARの活性を調節するための方法に関し、誘導体は式:
【0008】
【化4】
【0009】
によって表され、
式中:
Aは2つまでの二重結合を有する、3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されている、フェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaおよびRbは、独立して、水素、2〜6Cアルケニル、場合により3つまでのハロゲノ置換基を有する1〜8Cアルキル、ペンタフルオロフェニル、アリールもしくはアリール(1〜4C)アルキルであり、前記アリールもしくはアリール(1〜4C)アルキル置換基は、場合によりハロゲノ、(1〜6C)アルキル、分枝もしくは直鎖状(1〜6C)アルコキシ、(1〜4C)アルキレンジオキシ、トリフルオロメチル、シアノ、ニトロ、ヒドロキシル、(2〜6C)アルカノイルオキシ、(1〜6C)アルキルチオ、(1〜6C)アルカンスルホニル、(1〜6C)アルカノイルアミノおよび2〜4個の炭素原子のオキサポリメチレンで置換されているか、または、RaおよびRbは、場合により1または2個の(1〜4C)アルキル置換基を有する2〜7個の炭素原子のポリメチレンをともに形成する。
【0010】
2,4−ジフェニル−1,3−ジオキサン誘導体またはその薬学的に許容され得る塩を投与することによって、PPAR反応性疾患または状態を治療するための方法もまた、提供され、誘導体は、式II:
【0011】
【化5】
【0012】
によって表され、
式中Xは、フルオロ、クロロ、ブロモ、トリフルオロメチル、場合により置換されたフェニル、シアノ、メトキシおよびニトロから選択されるか、またはフェニル−X基は、場合により置換されたクロメン誘導体でもよく;YおよびZは、それぞれ水素またはハロゲノである。
【0013】
1,3−ジオキサン誘導体、それらの薬学的に許容され得る塩、および式:
【0014】
【化6】
【0015】
によって表される誘導体を含む薬学的組成物もまた、本発明によって提供され、
式中:
Aは2つまでの二重結合を有する3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されているフェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaはHであり、Rbは場合によりハロゲン、OH、O−アルキル、O−アリール、アミノまたはN−モノアルキルもしくはN−ジアルキルまたはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキルまたはオキソからなる群より選択される3つの異なる置換基で置換されている、アリール基または複素環である。具体的な目的の化合物は、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸および4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸、またはRbがビフェニルである化合物である。
【0016】
本発明はまた、PPAR−γ活性の調節に反応性のある疾患または状態、具体的には本明細書中で後述されるPPAR−γ反応性疾患または状態のいずれかの治療のための薬物の調製のための、上記で定義されるような化合物の使用に関する。
【0017】
化合物は、メタボリックシンドローム、肥満、インスリン抵抗性、糖尿病前症、糖尿病、脂質異常症、多発性硬化症、乾癬、アトピー性皮膚炎、喘息および潰瘍性大腸炎等の自己免疫疾患、脂肪肉腫、神経芽細胞腫、膀胱癌、乳癌、結腸癌、肺癌、膵癌および前立腺癌等の癌、炎症、感染、AIDSおよび創傷治癒を含む多数の臨床状態の治療または予防のための薬物の調製において有用である。
例えば、本発明は、以下の項目を提供する。
(項目1)
1,3−ジオキサン誘導体、またはその薬学的に許容され得る塩の治療有効量を個体に投与することを含む、個体においてペルオキシソーム増殖因子活性化受容体(PPAR)の活性を調節するための方法であって、
前記誘導体は式:
【化1】
によって表され、
式中:
Aは2つまでの二重結合を有する、3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されている、フェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaおよびRbは、独立して、水素、2〜6Cアルケニル、場合により3つまでのハロゲノ置換基を有する1〜8Cアルキル、ペンタフルオロフェニル、アリールもしくはアリール(1〜4C)アルキルであり、前記アリールもしくはアリール(1〜4C)アルキル置換基は、場合によりハロゲノ、(1〜6C)アルキル、分枝もしくは直鎖状(1〜6C)アルコキシ、(1〜4C)アルキレンジオキシ、トリフルオロメチル、シアノ、ニトロ、ヒドロキシル、(2〜6C)アルカノイルオキシ、(1〜6C)アルキルチオ、(1〜6C)アルカンスルホニル、(1〜6C)アルカノイルアミノおよび2〜4個の炭素原子のオキサポリメチレンで置換されているか、または、RaおよびRbは、場合により1または2個の(1〜4C)アルキル置換基を有する2〜7個の炭素原子のポリメチレンをともに形成する、方法。
(項目2)
Aが3〜7個の炭素の直鎖状炭素鎖であり、WがCOOHである、項目1に記載の方法。
(項目3)
前記炭素鎖がC2−C3またはC3−C4の間の二重結合を含む、項目2に記載の方法。
(項目4)
Arが2−OHまたは2−OMe置換フェニルまたはナフチル基である、項目1〜3のいずれか一項に記載の方法。
(項目5)
RaがHであり、Rbが、それぞれ場合によりハロゲン、OH、O−アルキル、アミノ、N−モノアルキル、N−ジアルキル、ニトロアルキルまたはチオアルキルで置換されているフェニル、ベンジル、2−または3−または4−ピリジン、フラン、ビフェニル1−ナフチルおよび2−ナフチルからなる群より選択されるアリール基である、項目1〜4のいずれか一項に記載の方法。
(項目6)
前記化合物が、2,4−ジフェニル−1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であり、前記誘導体は式II:
【化2】
によって表され、
式中Xは、フルオロ、クロロ、ブロモ、トリフルオロメチル、場合により置換されたフェニル、シアノ、メトキシおよびニトロから選択されるか、またはフェニル−X基は、場合により置換されたクロメン誘導体でもよく;YおよびZは、それぞれ水素またはハロゲノである、項目1に記載の方法。
(項目7)
式IIによって表される前記誘導体中のジオキサン環の2、4および5位の基がシス−相対立体化学を有する、項目5に記載の方法。
(項目8)
Xが2−フルオロ、2−クロロ、2−ブロモ、2−シアノ、2−トリフルオロメチル、3−フルオロ、3−クロロ、3−シアノ、3−ニトロ、3−メトキシ、4−クロロ、4−シアノ、4−ニトロおよび4−メトキシから選択され;Yが水素またはフルオロであり;Zが水素である、項目5または6に記載の方法。
(項目9)
Xが2−クロロ、3−クロロ、2−シアノ、4−シアノ、3−ニトロおよび4−ニトロから選択され;YおよびZが水素である、項目7に記載の方法。
(項目10)
前記誘導体が4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸またはその薬学的に許容され得る塩である、項目8に記載の方法。
(項目11)
Aが2つまでの二重結合を有する3〜7個の炭素の分枝または直鎖状炭素鎖であり、
WがCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されているフェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arがフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaがHであり、Rbがアリール基、または場合によりハロゲン、OH、O−アルキル、O−アリール、アミノもしくはN−モノアルキルもしくはN−ジアルキルもしくはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキルもしくはオキソからなる群より選択される3つの異なる置換基で置換されている複素環である、
項目1に記載の方法。
(項目12)
Aが1つの二重結合を有する5炭素直鎖状鎖であり、WがCOOHであり、Arがo−位でOHまたはOMeによって置換されているフェニルであり、Rbが複素環、またはO−アリールで置換されているフェニル基である、項目11に記載の方法。
(項目13)
前記誘導体が、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸またはその薬学的に許容され得る塩である、項目11に記載の方法。
(項目14)
前記誘導体が、4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸またはその薬学的に許容され得る塩である、項目11に記載の誘導体。
(項目15)
前記誘導体が、アルカリ金属およびアルカリ土類金属塩、アルミニウムおよびアンモニウム塩、ならびに生理学的に許容され得る陽イオンを形成する有機アミンおよび第四級塩基の塩から選択される薬学的に許容され得る塩として存在する、前記項目のいずれか一項に記載の方法。
(項目16)
前記方法が、PPAR−γ反応性疾患または状態の予防または治療のためである、項目1〜15のいずれか一項に記載の方法。
(項目17)
前記疾患または状態がインスリン抵抗性である、項目1〜16のいずれか一項に記載の方法。
(項目18)
前記疾患または状態が糖尿病である、項目1〜17のいずれか一項に記載の方法。
(項目19)
前記疾患または状態が、肥満の個体における糖尿病である、項目1〜18のいずれか一項に記載の方法。
(項目20)
前記疾患または状態が、PPAR−γによって媒介される慢性炎症性障害である、項目1〜19のいずれか一項に記載の方法。
(項目21)
前記疾患または状態が炎症性腸疾患、潰瘍性大腸炎またはクローン病である、項目1〜20のいずれか一項に記載の方法。
(項目22)
前記疾患または状態が関節炎、特に関節リウマチ、多発性関節炎および喘息である、項目1〜21のいずれか一項に記載の方法。
(項目23)
前記疾患が眼の炎症または眼乾燥疾患である、項目1〜22のいずれか一項に記載の方法。
(項目24)
前記疾患が皮膚障害、特に乾癬である、項目1〜23のいずれか一項に記載の方法。
(項目25)
前記疾患が高脂血症である、項目1〜24のいずれか一項に記載の方法。
(項目26)
前記疾患または状態が癌である、項目1〜25のいずれか一項に記載の方法。
(項目27)
前記癌が脂肪肉腫、前立腺癌、子宮頸癌、乳癌、多発性骨髄腫、膵癌、神経芽細胞腫または膀胱癌である、項目1〜26のいずれか一項に記載の方法。
(項目28)
PPAR媒介性疾患または状態である対象における臨床状態を治療または予防するために有用な薬物組成物を調製するための、項目1〜14のいずれか一項において定義される化合物の使用。
(項目29)
糖尿病、癌、炎症、AIDS、メタボリックシンドローム、肥満、糖尿病前症、高血圧および脂質異常症からなる群より選択される臨床状態の治療または予防のための薬物の調製のための、項目28に記載の化合物の使用。
(項目30)
1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であって、前記誘導体が式:
【化3】
によって表され、
式中:
Aは2つまでの二重結合を有する3〜7個の炭素の分枝または直鎖状炭素鎖であり;
WはCOOH、OH、NH2、SO3H、OSO3H、またはそれぞれ場合によりCOOH、OHもしくはNH2で置換されているフェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジンおよびジオキソランからなる群より選択される芳香族基であり;
Arはフェニル、または置換もしくは非置換2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニルから選択される5もしくは6員の複素環式芳香族基であり;
RaはHであり、Rbは、アリール基、または場合によりハロゲン、OH、O−アルキル、O−アリール、アミノまたはN−モノアルキルもしくはN−ジアルキルもしくはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキルもしくはオキソからなる群より選択される3つの異なる置換基で置換されている複素環である、誘導体。
(項目31)
Aが1つの二重結合を有する5炭素直鎖状鎖であり、WがCOOHであり、Arがo−位でOHまたはOMeによって置換されているフェニルであり、Rbが、O−アリールで置換されている複素環またはフェニルである、項目30に記載の誘導体。
(項目32)
Rbが(4−メトキシフェノキシ)−フェニルである、項目31に記載の誘導体。
(項目33)
前記誘導体が、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸である、項目32に記載の誘導体。
(項目34)
Rbが6−クロロ−4H−クロメン−4−オンである、項目31に記載の誘導体。
(項目35)
前記誘導体が、4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸である、項目34に記載の誘導体。
(項目36)
Rbがビフェニルである、項目31に記載の誘導体。
(項目37)
項目30〜項目36の誘導体の1つ以上を含む、医薬組成物。
(項目38)
さらなる医薬成分を含む、項目37に記載の医薬組成物。
【図面の簡単な説明】
【0018】
【図1】図1は、それぞれ完全アゴニストおよび部分アゴニストのPPAR活性化のモデルを説明する。
【図2】図2は、PPAR−δ、PPARαおよびRxRと比較した、ロシグリタゾンおよび4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸によるPPAR−γの選択的活性化を説明する。
【図3】図3は、ロシグリタゾンおよび4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸による、全長PPAR−γの活性化を説明する。
【図4】図4は、4(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸が全長PPARδトランス活性化に対して効果を有さないことを説明する。
【図5】図5は、部分PPARγ競合アッセイの結果を説明する。
【図6】図6は、PPARγリガンド置換アッセイの結果を説明する。
【図7】図7は、グルコース取り込みアッセイの結果を説明する。
【図8】図8は、化学式II〜Vを説明する。
【図9】図9は、化学式VI〜VIIIを説明する。
【図10】図10は、化学反応スキームIを説明する。
【発明を実施するための形態】
【0019】
定義
一般に、本明細書中で使用される用語は、本発明の理解において薬学、生物学および化学分野の当業者が採用する、それらの標準的な定義を有する。以下の用語は、示される意味を有し、他の用語が本明細書中で提供される場合がある。
アルキル:用語「アルキル」は、一般に1〜22個の、示された数の炭素原子を有する、一価の飽和脂肪族炭化水素ラジカルをいう。例えば、「1〜8Cアルキル」または「1〜8個の炭素のアルキル」または「Alk1〜8」は、構造中に1〜8個の炭素を含む任意のアルキル基をいう。アルキルは、直鎖(すなわち直鎖状)または分枝鎖であってもよい。低級アルキルは、1〜6個の炭素のアルキルをいう。低級アルキルラジカルのそれぞれの例としては、メチル、エチル、n−プロピル、n−ブチル、n−ペンチル、n−ヘキシル、イソプロピル、イソブチル、イソペンチル、アミル、sec−ブチル、tert−ブチル、sec−アミル、tert−ペンチル、2−エチルブチル、2,3−ジメチルブチル等が挙げられる。高級アルキルは、7つ以上の炭素のアルキルをいう。これらのものとしては、n−ヘプチル、n−オクチル、n−ノニル、n−デシル、n−ドデシル、n−テトラデシル、n−ヘキサデシル、n−オクタデシル、n−エイコシル等とともに、それらの分枝のバリエーションが挙げられる。3〜7個の炭素の直鎖状炭素鎖は鎖長をいい、枝上にあるいかなる炭素も含まない。ラジカルは、場合により置換されていてもよい。
【0020】
アルケニル:用語「アルケニル」は、少なくとも1つの炭素−炭素二重結合を有し、示された数の炭素原子を有する、一価の脂肪族炭化水素ラジカルをいう。例えば、「C2〜6アルケニル」または「1〜6個の炭素のアルケニル」、または「アルケニル1〜6」は、構造中に1〜6個の炭素原子を含むアルケニル基をいう。アルケニルは、直鎖(すなわち、直鎖状)または分枝鎖であってもよい。低級アルケニルは、1〜6個の炭素のアルケニルをいう。低級アルケニルラジカルのそれぞれの例としては、エテニル、1−プロペニル、1−ブテニル、1−ペンテニル、1−ヘキセニル、イソプロペニル、イソブテニル等が挙げられる。高級アルケニルは、7つ以上の炭素のアルケニルをいう。これらのものとしては、1−ヘプテニル、1−オクテニル、1−ノネニル、1−デセニル、1−ドデセニル、1−テトラデセニル、1−ヘキサデセニル、1−オクタデセニル、1−エイコセニル等とともに、それらの分枝のバリエーションが挙げられる。ラジカルは、場合により置換されていてもよい。
【0021】
アルコキシ:用語「アルコキシ」は、Rが本明細書中で定義されるようなアルキルである、式RO−の一価のラジカルをいう。低級アルコキシは、1〜6個の炭素原子のアルコキシまたは(1〜6)アルコキシをいう。それぞれの低級アルコキシラジカルとしては、メトキシ、エトキシ、n−プロポキシ、n−ブトキシ、n−ペンチルオキシ、n−ヘキシルオキシ、イソプロポキシ、イソブトキシ、イソペンチルオキシ、アミルオキシ、sec−ブトキシ、tert−ブトキシ、tert−ペンチルオキシ等が挙げられる。ラジカルは、場合により置換されていてもよい。
【0022】
アリール:用語「アリール」は、本明細書中で使用される場合、場合により、他に示されない限りは、ヒドロキシ、チオ、シアノ、アルキル、アルコキシ、低級ハロアルコキシ、アルキルチオ、オキソ、ハロゲン、ハロアルキル、ヒドロキシアルキル、ニトロ、アルコキシカルボニル、アミノ、アルキルアミノ、ジアルキルアミノ、アミノアルキル、アルキルアミノアルキル、およびジアルキルアミノアルキル、チオアルキル、アルキルスルホニル、アリールスルフィニル、アルキルアミノスルホニル、アリールアミノスルホニル、アルキルスルホニルアミノ、アリールスルホニルアミノ、カルバモイル、アルキルカルバモイルおよびジアルキルカルバモイル、アリールカルバモイル、アルキルカルボニルアミノ、アリールカルボニルアミノから選択される独立して選択される1つ以上の、好ましくは1または3個の置換基で置換されていてもよい、1つの個々の環、または少なくとも1つの環が天然に芳香族である1つ以上の縮合環からなる、5〜15個の炭素原子を含む一価の芳香族炭素環ラジカルを示す。あるいは、アリール環の2つの隣接する原子は、メチレンジオキシまたはエチレンジオキシ基で置換されていてもよい。したがって、二環式アリール置換基は、複素環式またはヘテロアリール環に縮合していてもよい。しかしながら、二環式アリール置換基の付着点は、炭素環芳香環上にある。アリールラジカルの例としては、フェニル、ナフチル、ベンジル、ビフェニル、フラニル、ピリジニル、インダニル、アントラキノリル、テトラヒドロナフチル、3,4−メチレンジオキシフェニル、1,2,3,4−テトラヒドロキノリン−7−イル、1,2,3,4−テトラヒドロイソキノリン−7−イル、1,3ジオキソランラジカル、安息香酸ラジカル、、フラン−2−カルボン酸ラジカル、2−(イソキサゾール−5−イル)酢酸ラジカル、3−ヒドロキシ−2−メチルピリジン−4−カルボン酸ラジカル等が挙げられる。
【0023】
ハロ:「ハロ」置換基は、クロロ、ブロモ、ヨード、およびフルオロから選択される、一価のハロゲンラジカルである。「ハロゲン化」化合物は、1つ以上のハロ置換基で置換されたものである。
【0024】
フェニル:「フェニル」は、ベンゼン環からの水素の除去によって形成されたラジカルである。フェニルは、場合により置換されていてもよい。
【0025】
フェノキシ:「フェノキシ」基は、式RO−のラジカルであり、式中Rはフェニルラジカルである。
【0026】
ベンジル:「ベンジル」基は、式R−CH2−のラジカルであり、式中Rはフェニルラジカルである。
【0027】
ベンジルオキシ:「ベンジルオキシ」基は、式RO−のラジカルであり、式中Rはベンジルラジカルである。
【0028】
複素環:「複素環」または「複素環式実体」は、炭素および少なくとも1つの他の元素、一般に窒素、酸素、または硫黄を含む、5または6員閉環の一価のラジカルであり、完全に飽和されているか、部分的に飽和されているか、または不飽和(すなわち、天然に芳香族)でもよい。一般に、複素環は、わずか2つのヘテロ原子を含む。1つのヘテロ原子しか有さない不飽和5員複素環のそれぞれの代表的な例としては、2−または3−ピロリル、2−または3−フラニル、および2−または3−チオフェニルが挙げられる。対応する、部分的に飽和された、または完全に飽和されたラジカルとしては、3−ピロリン−2−イル、2−または3−ピロリンジニル、2−または3−テトラヒドロフラニル、および2−または3−テトラヒドロチオフェニルが挙げられる。2つのヘテロ原子を有するそれぞれの不飽和5員複素環ラジカルの代表としては、イミダゾリル、オキサゾリル、チアゾリル、ピラゾリル等が挙げられる。対応する、完全に飽和された、および部分的に飽和されたラジカルもまた、含まれる。1つのヘテロ原子しか有さない不飽和6員複素環のそれぞれの代表的な例としては、2−、3−、または4−ピリジニル、2H−ピラニル、および4H−ピラニルが挙げられる。対応する、部分的に飽和された、または完全に飽和されたラジカルとしては、2−、3−、または4−ピペリジニル、2−、3−、または4−テトラヒドロピラニル等が挙げられる。2つのヘテロ原子を有するそれぞれの不飽和6員複素環ラジカルの代表としては、3−または4−ピリダジニル、2−、4−、または5−ピリミジニル、2−ピラジニル、モルホリノ等が挙げられる。対応する、完全に飽和された、および部分的に飽和されたラジカルもまた含まれ、例えば2−ピペラジンである。複素環式ラジカルは、複素環中の利用可能な炭素原子もしくはヘテロ原子を通じて実体に直接結合しているか、または、メチレンもしくはエチレン等のアルキレン等のリンカーを通じて結合している。複素環は、場合により、アリール基と同じ様式で置換されていてもよい。
【0029】
場合により置換され:ラジカルが、「場合により置換され」という場合、これは、ラジカルが非置換であるか、またはラジカルの少なくとも1つの−Hが除去されており、別の置換基がその場所に挿入されていることを意味する。ラジカルは、場合により、本発明の範囲内に含まれる化合物の調製を有意に妨げず、化合物の生物学的活性に有意に悪影響を与えない位置で、置換基で置換されていてもよい。ラジカルは、場合により、ハロ、低級アルコキシ、ヒドロキシル、シアノ、ニトロ、アミノ、ハロ低級アルキル、ハロ低級アルコキシ、ヒドロキシカルボニル、低級アルコキシカルボニル、低級アルキルカルボニルオキシ、および低級アルキルカルボニルアミノからなる群より独立して選択されるか、または本明細書中で上述された、1、2、3、4または5個の置換基で置換されていてもよい。
【0030】
用語「ヒドロキシカルボニル」は、式−C(O)OHを有する一価のラジカルである。
【0031】
用語「低級アルコキシカルボニル」は、式−C(O)OAlkを有する一価のラジカルであり、ここでAlkは低級アルキルである。
【0032】
用語「低級アルキルカルボキシルオキシ」は、式−OC(O)Alkの一価のラジカルであり、ここでAlkは低級アルキルである。
【0033】
本明細書中で使用される場合、「糖」は、単糖、二糖または多糖を意味する。適した単糖としては、ペントース、ヘキソース、またはヘプトース残基が挙げられる。ペントースの非限定的な例としては、アラビノース、リボース、リブロース、キシロース、リキソース、およびキシルロースが挙げられる。ヘキソースの非限定的な例としては、グルコース、ガラクトース、フルクトース、フコース、マンノース、アロース、アルトロース、タロース、イドース、プシコース、ソルボース、およびタガトースが挙げられる。ヘプトースの非限定的な例としては、マンノヘプツロースおよびセドヘプツロースが挙げられる。糖部分は、アミドまたはエステル結合を形成することができる糖環の任意の位置で化合物に連結されていてもよい。好ましい糖類は、β−グリコシル糖類である。
【0034】
PPAR調節は、天然の状態、すなわち、リガンドなしでの標的遺伝子のPPAR依存的転写の基礎レベルを参考にして定義され、ここで、PPAR活性の調節は、PPAR活性を調節することができる化合物の存在下での前記転写の基礎レベルの減少または増大に反映される。一般に、前記転写の増大は、PPAR活性の増強と関連があり、活性化因子またはアゴニストという名前の化合物に関する。反対に、前記転写の減少は、PPAR活性の阻害と関連があり、阻害剤またはアンタゴニストという名前の化合物に関する。部分アゴニストは、標的遺伝子のサブセットのPPAR依存的転写を生じるが、他のPPAR標的遺伝子に影響を及ぼさない化合物である。部分アゴニストは、生化学的または生理学的観点から見ることができる。部分アゴニストの生化学的見識は、完全アゴニストと競合することができ、完全アゴニストと比べて低いレベルのトランス活性化を有する化合物である。部分アゴニストの生理学的見識は、異なるサブセットの遺伝子の活性化に関する(いくつかの標的遺伝子の完全な活性化があっても、他のPPAR標的遺伝子の活性化はなし)。これは、完全アゴニストの生理学的効果のいくつかのみを生じ、これは大いに望ましい。
【0035】
化合物の「治療有効量」は、それを必要とする対象に投与したときに、治療される状態に対して、経時的に所望の結果を生じる量を意味する。本願において、所望の効果はPPAR調節、およびそれと関連する生物学的活性である。
【0036】
本発明において有用な化合物は、本願中で種々の式によって示される。式を見ることによって、化合物が、しばしばキラル中心、すなわち4つの異なる基が付着している炭素を有し、これらが鏡像異性体として存在する場合があることが明らかであろう。また、いくつかの場合における二重結合の存在によって、化合物は、回転障害を有する。したがって、化合物は幾何異性を示し、すなわち、空間中で原子が方向付けされる様式で、2つの形態は互いに異なる場合がある。二重結合に関して、互いに鏡像でない立体異性体が存在し、ジアステレオマーと呼ばれる。本願中の化合物の命名において、R,Sシステムにおける鏡像異性体の命名において行われるように、二重結合の各末端に付着した2つの基が優先順位番号を提供される化学情報検索サービス機関システムが用いられる。より高い優先順位番号の2つの基が同じ側にある場合、分子はZ異性体である。(ドイツ語−zusammen、ともに)。反対の側にある場合、分子はEである。(ドイツ語−entgegen、反対)。式は、単独であれ混合物中であれ、可能性のある鏡像異性体およびジアステレオマーの全てを包含するよう意図されることが、理解されるべきである。
【0037】
本発明において有用な化合物
本発明は、部分的に、ある種の化合物が少なくとも1つのPPARサブタイプ、例えばPPARγまたはPPARβ/δの活性を調節することができるという発見に基づく。この発見は、かかるPPARの機能によって媒介されるヒト等の哺乳動物における状態または疾患を治療するための、これらの化合物の使用につながる。
【0038】
本発明において有用な化合物は、1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であり、ここで誘導体は、式I:
【0039】
【化7】
【0040】
によって表され、
式中:
Aは、場合により1または2個の二重結合を含む(それぞれはシスでもトランスでもよい)、3〜7個の炭素の分枝または直鎖状炭素鎖であり、
Wは、COOH、OH、NH2、SO3H、OSO3H、場合により例えばCOOH、OHまたはNH2で置換されている、フェニル、1−もしくは2−ナフチル、ピリジン、フラン、2−メチルピリジン等の、しかしこれらに限定されない芳香族基;または2位を通して連結された1,3ジオキソラン基であり、
Arは、フェニル、または2−ピリジン、3−ピリジン、チオフェン、フラン、1−ナフチル、2−ナフチル、ビフェニルおよび(4−メトキシフェノキシ)−フェニル等の、しかしこれらに限定されない5または6員の複素環式芳香族基であり、フェニルおよびナフチル部分は場合により、オルト、メタおよび/もしくはパラ位でOHまたはOMeで置換されているが、好ましくは、置換されている場合、オルト位においてOHまたはOMeで一置換されており、
RaおよびRbは、独立して、水素、2〜6Cアルケニル、場合により3つまでのハロゲノ置換基を有する1〜8Cアルキル、ペンタフルオロフェニル、アリールもしくはアリール(1〜4C)アルキルであり、その最後の2つは場合によりハロゲノ、(1〜6C)アルキル、分枝もしくは直鎖状(1〜6C)アルコキシ、(1〜4C)アルキレンジオキシ、トリフルオロメチル、シアノ、ニトロ、ヒドロキシル、(2〜6C)アルカノイルオキシ、(1〜6C)アルキルチオ、(1〜6C)アルカンスルホニル、(1〜6C)アルカノイルアミノおよび2〜4個の炭素原子のオキサポリメチレンから選択される5つまでの置換基を有するか、またはRaおよびRbは、場合により1または2個の(1〜4C)アルキル置換基を有する、2〜7個の炭素原子のポリメチレンをともに形成する。
【0041】
いくつかの実施形態において、Aは、3〜7個の炭素の直鎖状炭素鎖であり、WはCOOHである。かかる炭素鎖は、例えば、C2−C3またはC3−C4の間に二重結合を含んでもよい。
【0042】
ある実施形態において、RaはHであり、Rbは、ハロゲン、OH、O−アルキル、アミノ、N−モノアルキル、N−ジアルキル(分枝または直鎖状)、ニトロおよびチオアルキル(分枝または直鎖状)からなる群より選択される5つまでの異なる置換基を有する、フェニル、ベンジル、2−または3−または4−ピリジン、フラン、ビフェニル、1−または2−ナフチル等の芳香族部分である。
【0043】
いくつかの実施形態において、誘導体は、2,4−ジフェニル−1,3−ジオキサン誘導体またはその薬学的に許容され得る塩であり、ここで誘導体は、式II:
【0044】
【化8】
【0045】
によって表され、
式中Xは、フルオロ、クロロ、ブロモ、トリフルオロメチル、場合により置換されたフェニル、シアノ、メトキシおよびニトロから選択されるか、またはフェニル−X基は、場合により置換されたクロメン誘導体であってもよく;YおよびZは、個々に、水素またはハロゲノである。
【0046】
典型的には、式Iによって表される誘導体中のジオキサン環の2、4および5位の基は、シス−相対立体化学を有する。
【0047】
特に興味深いXを有するフェニル部分の他の特定の置換基としては、例えば、2−フルオロ−、2−クロロ−、2−ブロモ−、2−シアノ−、2−トリフルオロメチル−、3−フルオロ−、3−クロロ−、3−シアノ−、3−ニトロ−、3−メトキシ−、4−クロロ−、4−シアノ−、4−ニトロ−および4−メトキシ−フェニルが挙げられる。
【0048】
Yについての好ましい置換は、水素またはフルオロであり、Zは水素である。
【0049】
本発明において有用な化合物の好ましい基は、X1が2−クロロ、3−クロロ、2−シアノ、4−シアノ、3−ニトロおよび4−ニトロから選択され;ジオキサン環の2、4および5位の基がシス−相対立体化学を有する、式III(本明細書中に下記で示す)の化合物;ならびにその薬学的に許容され得る塩を含む。
【0050】
本発明はまた、A、WおよびArが上記で定義され、RaがHであり、Rbがアリール部分の1つの炭素原子が1つの酸素もしくは窒素原子で置換されているもの等のアリール基もしくは複素環である、式Iの化合物を提供する。かかるアリールおよび複素環基は、場合により、ハロゲン、OH、O−アルキル(分枝または直鎖状)、O−アリール、アミノまたはN−モノアルキルもしくはN−ジアルキル(分枝または直鎖状)またはN−モノアリールもしくはN−ジアリール、ニトロ、チオアルキル(分枝または直鎖状)またはオキソ(例えば、表II中のSN13)からなる群より選択される3つの異なる置換基で置換されていてもよい。RaがHでありRbが複素環、または0−アリールで置換されているフェニル基である式Iの化合物が、特にArがo−位でOHまたはOMeによって置換されたフェニルであり、WがCOOHであり、Aが1つの二重結合を有する5つの炭素の直鎖である場合、PPARγ調節因子として特に興味深い。かかる化合物の例示的な例としては、RaがHであり、Rbが、4(Z)−6−(2−[4−メトキシフェノキシ−o−フェニル]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸等の、フェニルの2もしくはオルト位を通じてジオキサン環に連結された(4−メトキシフェノキシ)−フェニルであるか;またはRbが、4(Z)−6−(2−3−[6−クロロ−4H−クロメン−4−オン]−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸等の6−クロロ−4H−クロメン−4−オン(例えば、クロメン部分の3位を通じてジオキサン環に連結されている)であるか;またはRbが、ビフェニルの2もしくはオルト位を通じてジオキサン環に連結されたビフェニルである、式Iの化合物が挙げられる。したがって、本発明は、単独で、ならびに薬学的担体、および場合によりさらなる治療上活性のある成分と組み合わせて、本実施形態において言及される化合物およびそれらの薬学的に適切な塩を提供する。
【0051】
式Iおよび式IIの化合物が不斉炭素原子を有し、ラセミ形態および光学活性形態で存在および単離することができることが、認識されるであろう。本発明は、PPAR活性を調節することができる、ラセミ形態および任意の光学活性形態の両方(またはそれらの混合物)、ならびにそれらの使用を含み、個々の光学異性体をどのように調製するか(例えば、光学活性な出発物質からの合成、またはラセミ形態のクロマトグラフィー分解によって)および下記で言及される1つ以上のアッセイを用いてどのようにPPAR調節特性を判定するかは当該分野で周知である。
【0052】
文脈から他に明らかでない限りは、本明細書中で言及される化学式は、特定の配置で示されるが、これは、必ずしも、絶対配置に対応しない。
【0053】
式IまたはIIの酸の、特定の薬学的に許容され得る塩は、例えば、リチウム、ナトリウムカリウム、マグネシウムおよびカルシウム塩等のアルカリ金属およびアルカリ土類金属塩、アルミニウムおよびアンモニウム塩、ならびにメチルアミン、ジメチルアミン、トリメチルアミン、エチレンジアミン、ピペリジン、モルホリン、ピロリジン、ピペラジン、エタノールアミン、トリエタノールアミン、N−メチルグルカミン、水酸化テトラメチルアンモニウムおよび水酸化ベンジルトリメチルアンモニウムとの塩等の、生理学的に許容され得る陽イオンを形成する有機アミンおよび第四級塩基の塩である。
【0054】
式Iの化合物は、構造的に類似の化合物の製造のための、当該分野で周知の従来の有機化学の手順によって製造することができる。かかる手順は、例えば、本明細書中に参照によって特に援用される、欧州特許第0094239号、4〜10頁に提供されており、下記に実施例の項で、X、YおよびZが本明細書中に上記で定義された意味を有する以下のプロセスによって、説明される。
【0055】
(A)式IVのアルデヒドを、式R13P=CH(CH2)2CO2−M+(式中R1が(1〜6C)アルキルまたはアリール(特にフェニル)であり、M+が陽イオン、例えばリチウム、ナトリウムまたはカリウム陽イオン等のアルカリ金属陽イオンである)のウィティッヒ試薬と反応させる。いくつかの実施形態において、アルデヒドを、P=CH(CH2)nCOO−M+(式中n=0〜4)に変更されている式のウィティッヒ試薬と反応させる。
【0056】
一般に、プロセスは、二重結合に隣接する置換基が主にシス−相対立体化学を有する、式IIの必要な化合物、すなわち「Z」異性体を生じる。しかしながら、プロセスはまた、所望される場合、クロマトグラフィーまたは結晶化等の従来の手順によって除去することができるトランス−相対立体化学を有する類似化合物を生じる。
【0057】
合成プロセスは、従来、適した溶媒または希釈剤、例えばベンゼン、トルエンもしくはクロロベンゼン等の芳香族溶媒、1,2−ジメトキシエタン、t−ブチルメチルエーテル、ジブチルエーテルもしくはテトラヒドロフラン等のエーテル、ジメチルスルホキシドもしくはテトラメチレンスルホン、またはかかる溶媒もしくは希釈剤の1つ以上の混合物中で行われる。プロセスは、一般に、例えば−80℃〜40℃の範囲の温度で行われるが、従来、室温で、または室温近く、例えば0〜35℃の範囲で行われている。
【0058】
(B)R1が保護基、例えば(1〜6C)アルキル(メチルまたはエチル等)、アシル(アセチル、ベンゾイル、メタンスルホニルまたはp−トルエンスルホニル等)、アリル、テトラヒドロピラン−2−イル、トリメチルシリルであり、脱保護される、式Vのフェノール誘導体。
【0059】
用いられる脱保護条件は、保護基R1の性質に依存する。したがって、例えば、それがメチルまたはエチルである場合、脱保護は、適した溶媒(N,N−ジメチルホルムアミドまたはN,N−ジメチル−3,4,5,6−テトラヒドロ−2(1H)−ピリミジノン等)中、例えば50〜160℃の範囲の温度で、ナトリウムチオエトキシド(水素化物およびエタンチオール)とともに加熱することによって行うことができる。あるいは、エチルまたはメチル保護基は、例えば0〜60℃の範囲の温度で、適した溶媒(テトラヒドロフランまたはメチルt−ブチルエーテル等)中でのリチウムジフェニルホスフィドとの反応によって除去することができる。保護基がアシルである場合、これは、例えば、0〜60℃の温度の範囲で、適した水性溶媒[水性(1〜4C)アルカノール等)中で、塩基(水酸化ナトリウムまたは水酸化カリウム等)の存在下での加水分解によって除去することができる。保護基がアリルまたはテトラヒドロピラン−2−イルである場合、これは、例えば、トリフルオロ酢酸等の強酸での処理によって除去することができ、これがトリメチルシリルである場合、これは、例えば、従来の手順を用いて、水性テトラブチルフッ化アンモニウムまたはフッ化ナトリウムとの反応によって除去することができる。
【0060】
(C)式中Q1およびQ2の一方が水素であり、他方が水素または式−CRaRb.OH(式中RaおよびRbは同じまたは異なる(1〜4C)アルキルである)の基である式Vのエリトロ−ジオール誘導体を、式VIIのベンズアルデヒド誘導体、またはアセタール、ヘミアセタールまたはその水和物と反応させる。
【0061】
ベンズアルデヒドVII[またはその水和物、または(1〜4C)アルカノール(メタノールまたはエタノール等)とのそのアセタールもしくはヘミアセタール]は、好都合に、過剰に存在してもよい。
【0062】
反応は、一般に、塩化水素、臭化水素、硫酸、リン酸、メタンスルホン酸またはp−トルエンスルホン酸等の酸触媒の存在下、好都合にトルエン、キシレンまたはエーテル等の適した溶媒または希釈剤、例えばテトラヒドロフラン、ジブチルエーテル、メチルt−ブチルエーテルまたは1,2−ジメトキシエタンの存在下、および例えば0〜80℃の範囲の温度で、行われる。
【0063】
式中Q1およびQ2がともに水素である式VIの出発物質は、例えば、本明細書中のプロセス(A)と類似した手順によって得られる、RaおよびRbがともにメチルまたはエチル等のアルキルである式VIIIの化合物のジオキサン環の、温和な、酸触媒された、加水分解またはアルコール分解によって、得ることができる。加水分解またはアルコール分解は、通常、溶媒としてアルカノイル(エタノールまたは2−プロパノール等)またはエーテル(テトラヒドロフラン等)中、塩酸等の水性鉱酸を用いて、10〜80℃の範囲の温度で行われる。
【0064】
式中Q1およびQ2の一方が水素であり、他方が式−−CRaRb.OHの基である式VIの出発物質は、上述の、式中Q1およびQ2がともに水素である式VIの出発物質の形成における中間体である。しかしながら、前記中間体は、通常、単離または特徴付けされない。したがって、プロセス(C)の有用な変更は、酸触媒の存在下で(上記のもののいずれか等)、好都合に例えば10〜80℃の範囲の温度で、および場合により適した溶媒または希釈剤(上記のもののいずれか等)の存在下で、式中RaおよびRbの一方が水素、メチルまたはエチルであり、他方がメチルまたはエチルである式VIIIの化合物を、過剰な式VIIの化合物(またはその水和物、アセタールもしくはヘミアセタール)と反応させることを含む。
【0065】
上記のプロセスにおける使用のための出発物質は、構造的に関係がある化合物の調製で知られている一般的な有機化学の手順によって生じることができる。したがって、式IVのアルデヒドは、例えば、スキームIに示される方法によって得ることができる。式Vの、保護されたフェノール誘導体は、例えば、式IVのものと類似したアルデヒドを用いるがフェノール基が、例えば脱保護工程(ii)を欠いたスキームIの手順を行うことによって生じたアルデヒド等のR1基で保護されている、上記のプロセス(A)に類似した手順を用いることによって、生じることができる。新規である、式VIIIの出発物質のものは、欧州特許出願、公開第94239号に記載されているものと類似した手順を用いて得ることができる。
【0066】
必要なウィティッヒ試薬は、従来の手順によって、例えば対応するハロゲン化ホスホニウムを水素化ナトリウム、リチウムジイソプロピルアミド、カリウムt−ブトキシド、LiHMDSまたはブチルリチウム等の強塩基で処理することによって、得ることができる。それらは、一般に、上記の縮合プロセス(A)を行う直前に、原位置(in situ)で形成される。
【0067】
式IおよびIIの化合物はまた、当該分野で周知の他の従来の手順によって、例えば対応するエステル、アミドまたはニトリルの塩基触媒加水分解によって得ることができることが、理解されるであろう。
【0068】
式IまたはIIの化合物の塩が必要とされる場合、これは、生理学的に許容され得る陽イオンを提供する適切な塩基との反応によって、または任意の他の従来の手順によって、得られる。
【0069】
さらに、式IまたはIIの化合物の光学活性形態が必要とされる場合、光学活性な出発物質を用いて、前述のプロセスの1つを行ってもよい。あるいは、式IIの化合物のラセミ形態を、光学活性形態の適した有機塩基、例えばエフェドリン、N,N,N−トリメチル(1−フェニルエチル)アンモニウム水酸化物または1−フェニルエチルアミンと反応させ、続いて例えば適した溶媒、例えば(1〜4C)アルカノールからの分別結晶化によって、このようにして得られた塩のジアステレオ異性体混合物の従来の分離を行い、その後従来の手順を用いて、例えば希塩酸等の水性鉱酸を用いて、酸での処理によって前記式IまたはIIの化合物の光学活性形態を遊離させてもよい。
【0070】
また、実施例29は、どのようにしてキラルクロマトグラフィーによって式IおよびIIの化合物のラセミ混合物を単離することができるかを説明する。
【0071】
前述したように、本明細書中に記載されている化合物は、PPAR活性の調節因子である。したがって、上記で概説された構造的特性に加えて、本発明の方法とともに使用されるのに好ましい化合物はまた、PPARアゴニスト、PPARアンタゴニストまたはPPAR部分アゴニスト、好ましくはPPAR部分アゴニストである。PPARアゴニスト/アンタゴニスト/部分アゴニストとしての機能性を判定するための方法は、「化合物の機能性」の項で、本明細書中で後述される。
【0072】
化合物の機能性
PPARの活性を調節することができる化合物(本明細書中でPPARリガンドとも呼ばれる)は、3つの別個のクラス:完全アゴニスト、部分アゴニスト/部分アンタゴニストに、および完全アンタゴニストに分類することができる。アゴニストおよびアンタゴニストは、それらの結合親和性、指示能力/EC50/IC50値、および飽和レベルの化合物の存在下で得られる活性のレベル、すなわち有効性によって、特徴付けられる。部分アゴニスト/部分アンタゴニストはまた、その結合親和性、および有効性によって特徴付けられる。部分アゴニスト/部分アンタゴニストは、同族のPPARを完全に活性化することができず、競合的様式で受容体由来の完全アゴニストを置換することができ、それによって、トランス活性化のレベルを減少させる。完全および部分アゴニストは、さらに、補助因子の異なる補体を補充することができ、所定のPPARサブタイプに対する補充された補助因子の性質は、所定のアゴニストによって活性化された遺伝子のパターンに大いに影響を及ぼす。
【0073】
PPARのリガンド結合ポケットは、他の核内受容体と比較して大きく、これは、部分的に、PPARに結合することができ、活性化することができる、多種多様な化合物を説明することができる。3つのPPARサブタイプの間で、リガンド認識に相当な重複があり、厳密にいうと、サブタイプ特異的リガンドは未だ同定されていない。しかしながら、いくつかの天然および合成リガンドが、高い程度の選択性を示し、今日最も選択的なリガンドは、個々のPPARサブタイプを活性化するのに必要な濃度に関して、3桁より大きく異なる。ステロイド核内受容体についてのアゴニストと同様に、用語、選択的PPAR調節因子(SPPARM)が導入されている(本明細書中で、「部分アゴニストまたはアンタゴニスト」とも呼ばれる)。このクラスのリガンドは、PPAR(複数可)への結合の際に異なる組の活性化補助因子の補充につながる異なる構造を導入する、部分アゴニスト/アンタゴニストを含む。原則として、SPPARMは、PPAR標的遺伝子のサブセットのみを活性化することができ、それによって、おそらく所望の組の遺伝子の特異的発現を促進する。完全アゴニストおよび部分アゴニストによるPPAR活性化のモデルを図1に示す。本発明の好ましい化合物は、部分PPARアゴニストである。
【0074】
PPAR調節活性は、当該分野で公知の任意の数の方法、またはその適応によって、容易に判定することができる。例えば、PPAR調節活性は、トランス活性化アッセイによって判定することができる。PPARγ調節活性を判定するための、有用なトランス活性化アッセイの非限定的な例は、実施例21に記載されており、PPARδ調節活性を判定するための、有用なトランス活性化アッセイの非限定的な例は、実施例22に記載されている。下記の実施例21は、化合物が、PPAR−GAL4(DNA結合ドメイン)融合タンパク質をコードする構築物およびGAL4依存性受容体構築物をコードする構築物でトランスフェクトされた細胞に添加される方法を説明している。異なるDNA結合ドメインを用いて転写または異なるレポーター遺伝子(例えば、蛍光タンパク質、β−ガラクトシダーゼ、ペルオキシダーゼ、ルシフェラーゼ等)を活性化すること等の、任意の数の可能性のある構築物を用いることができることは、当業者に明らかであろう。どのPPAR活性を判定するのが望ましいかに依存して、構築物は好ましくは前記PPARまたはそのリガンド結合ドメインをコードすることは、当業者に明らかであろう。PPARの活性化の際に(すなわち、PPARアゴニストまたは部分アゴニストの存在下で)、PPARは、レポーター構築物を、場合により定量的な様式で、トランス活性化する。
【0075】
PPAR調節因子はまた、少なくとも1つのPPREを含む第二の核酸の制御下で操作可能に、第一の核酸を含むレポーター遺伝子を用いて同定することができる。第一の核酸は、好ましくは、蛍光タンパク質、β−ガラクトシダーゼ、ペルオキシダーゼ、ルシフェラーゼ等のレポータータンパク質をコードする。前記レポーター構築物は、PPARγおよび/またはδ等の1つ以上のPPARを発現している細胞に挿入されるべきである。したがって、PPARアゴニストは、第一の核酸の転写を活性化することができる化合物として同定することができる。
【0076】
本発明の特定の実施形態によると、好ましい化合物は、PPARおよび/またはPPAR LBDアゴニストまたは部分アゴニストである。用語「PPAR LBD」は、PPARのリガンド結合ドメインをいう。好ましい実施形態によると、本発明の化合物および組成物が、PPARγおよび/またはPPARγLBDアゴニストである。「アゴニスト」によって、細胞内受容体と結合したときに受容体に典型的な反応、例えば転写活性化活性を刺激または増大させる、化合物または組成物が意味される。ある実施形態において、前記アゴニストは、PPARγアゴニスト、すなわち、例えば受容体に対する天然の生理学的リガンドを模倣すること等によって、PPARγ受容体の転写活性を促進、刺激、誘導またはそうでなければ増強する、PPARリガンドである。
【0077】
前記PPAR調節活性は、本明細書中で上述されたトランス活性化アッセイを用いて判定することができる。適した標準的なアゴニストとしては、PPARγのためのロシグリタゾンおよびPPARδのための(4−[3−(2−プロピル−3−ヒドロキシ−4−アセチル)フェノキシ]プロピルオキシフェノキシ−酢酸(L165041、市販されている)が挙げられる。標準的なアゴニストより50%未満のトランス活性化を示す潜在的なアゴニストもまた、特に新しい化合物もしくは活性のある誘導体の開発のために、または部分アゴニストの存在の指標として、有用である場合がある。
【0078】
好ましい実施形態によると、本明細書中の化合物または組成物は、PPARおよび/またはPPAR LBD部分アゴニストであり、より具体的には、本発明の化合物および組成物は、PPARγおよび/またはPPARγLBD部分アゴニストである。可能性のある最大効果(すなわち、完全アゴニスト、または参照分子によって生じる最大効果)未満を生じる薬物は、部分アゴニストと呼ばれる。
【0079】
例えば、本発明の化合物および組成物の部分アゴニスト特性は、完全アゴニストであるロシグリタゾン(アバンディア(商標)、Glaxo−SmithKline)を参照して定義することができる。これは具体的には、PPARγ部分アゴニストに当てはまる。部分PPARアゴニスト。具体的には、本発明のPPARγアゴニストは、所望の治療を受けている患者に対して同様またはよりよい有効性を提供するが、より少ない、患者に有害な望ましくない副作用を有する。例えば、SN1(DPD)は、10mMで1mMのアバンディアと同じレベルのグルコース取り込みを誘導するが、より低い脂肪細胞分化の結果として、より少ない副作用が予期される。
【0080】
本発明の化合物および組成物の部分アゴニスト特性はまた、L165041(市販されている)を参照して定義することができる。これは具体的には、PPARδ部分アゴニストに当てはまる。例えば、かかるトランス活性化アッセイの1つは、実施例22に記載されるトランス活性化アッセイである。
【0081】
ある実施形態において、本発明の化合物は、PPARの活性化に対して選択的であることが好ましい。かかる実施形態において、化合物は、RxRおよび/またはRxR LBDトランス活性化を有意に活性化しないことが好ましく、好ましくは、RxR転写は、バックグラウンドレベルの1.5倍未満等、バックグラウンドレベルの2倍より少なく、例えば、バックグラウンドレベルとおよそ等しいか、またはバックグラウンドレベル未満である。RxRトランス活性化は、例えば実施例29に記載されているようなRxRトランス活性化アッセイによって判定することができる。バックグラウンドレベルは、リガンドの添加なしでのトランス活性化である。
【0082】
本発明のある実施形態において、化合物は、PPARアンタゴニストである。「アンタゴニスト」によって、PPARと結合したときに前記PPARに典型的な反応、例えば転写活性を、干渉または減少させる化合物が意味される。一般的な定義として、「PPARアンタゴニスト」は、対応するPPARアゴニストの活性を阻害することができるPPARリガンドを示す。より一般には、これらのアゴニスト/アンタゴニスト/部分アゴニスト活性は、例えば国際公開第99/50664号または国際公開第96/41013号に開示されているもの等の、当業者に広く公知のアッセイによって測定することができる。PPARアンタゴニストの例は、実施例21に提供される。
【0083】
本発明の化合物および組成物は、PPAR活性の調節によって影響を及ぼされる状態または疾患を有する患者に投与されたときの、それらの生物学的活性によって、さらに特徴付けられる。本発明の好ましい化合物は、それを必要とする患者において、以下の生物学的実体:グルコース、トリグリセリド、脂肪酸、コレステロール、胆汁酸等の1つ以上を、当該分野で公知の分子(例えば、チアゾリジンジオン)と比較してよりよい、または同等な有効性および能力で、しかしより低い毒性および/またはより少ない望ましくない副作用の発生で、低下させることができる化合物である。具体的には、前記化合物は、好ましくは、脂肪細胞分化および体重増加の、より少ない誘導につながる。かかる化合物は、具体的には、本明細書中の上記のC.項に記載された化合物のいずれかであってもよい。脂肪細胞分化を判定するための有用な方法は、本明細書中で下記の実施例25に記載されている。より具体的には、それらは、インスリン抵抗性(減少した、インスリンが血糖値を低下させる有効性)および/または脂肪生成に対する有益な活性を提供する。PPARγを活性化する多くの化合物(例えばチアゾリジンジオン)が、脂肪細胞分化をさらに誘導し(すなわち、脂肪生成または脂質生成の効果を示す)、したがって、治療される患者において体重増加を生じることが、示されている。したがって、次世代のかかる化合物は、減少した活性を示し、好ましくはかかる活性を欠いていることが、高度に望ましい。これらの活性は、当該分野で広く用いられている方法(実施例5に記載されているもの等)を用いて評価することができる。より具体的には、これらの活性は、ロシグリタゾン等の、当該分野ですでに同定されている分子を参照して評価される。本発明の好ましい実施形態によると、好ましい化合物は、例えばインスリン抵抗性を患っている患者においてグルコースレベルを判定することによって判定することができるインスリン抵抗性に関するロシグリタゾン特性の、少なくとも約50%、好ましくは少なくとも約60%、より好ましくは少なくとも約70%、さらにより好ましくは少なくとも80%を示す。理想的には、これは100%以上である。本発明の別の好ましい実施形態によると、好ましい化合物は、脂肪細胞分化に対するロシグリタゾン特性の、約80%未満、好ましくは約50%未満、より好ましくは約40%未満、さらにより好ましくは約30%未満を示す。理想的には、これは脂肪細胞分化に対するロシグリタゾン特性の20%未満である。脂肪細胞分化は、例えば、本明細書中に下記で実施例30に記載されているように判定することができる。非常に好ましい実施形態において、好ましい化合物は、上述の特性の両方を有する。あるいは、化合物は、トランス活性化アッセイで判定すると、少なくともロシグリタゾンのものの50%の程度まで、PPAR活性を誘導することができ、脂肪細胞分化に対する上述の特性を示す。
【0084】
血液希釈、浮腫、脂肪細胞分化、または肥満等の望ましくない副作用のインビボでの発生は、例えば欧州特許第1267171号に記載されている方法を用いて、前記化合物の補助因子補充プロフィールによって影響を受ける場合がある。したがって、本発明のある実施形態において、好ましい化合物は、望ましくない副作用の、低いインビボでの発生を有すると予想される、化合物である。
【0085】
PPARγ等の核内受容体が、標的遺伝子のプロモーター領域中の同族配列に結合することならびに活性がクロマチンリモデリング、ヒストンおよび補助因子修飾から基本的な転写機構補充の範囲にわたる多数の補助因子複合体を補充することによって、転写活性化または抑制を達成することは、広く認められている(Glass,およびRosenfeld、2000年、Genes Dev.、14、121−141)。これらの補助因子は、かなりの部分まで、核内受容体の作用の特異性を判定することができ、適した変性が特定の細胞反応につながる刺激のネットワーク中でそれらの作用を統合することができる。したがって、時間および細胞型の関数としての、各核内受容体が携わる複数の連携の判定は、転写調節における核内受容体の活性のよりよい理解につながるであろう。例えば、エストロゲン等のある種のホルモンについて、ホルモンに対する反応は、それぞれの核内ホルモン受容体の存在によって、受容体と相互作用する補助因子の存在によるのとほとんど同じ程度まで判定されることが、公知である。種々のPPAR補助因子が同定されている。p300/CBP(Dowellら、1997年、J.Biol.Chem.272、33435−33433)、SRC−1(Onateら、1995年、Science
270、1354−1357)、TIF2(GRIP−2、Chakravartiら、1996年、Nature、383、99−103)、SRA(Lanzら、1999年、Cell、97、17−27)、AIB−1(Anzickら、1997年、Science、277、965−968)、TRAP220/DRIP205(すなわち、PBP、Zhuら、1997年、J.Biol.Chem.272、25500−25506、Rachezら、1999年、Nature、398、824−828)、PGC−1(Puigserverら、1998年、Cell 92、829−839)、PRIP(Zhuら、2000年、J Biol Chem 275、13510−13516)、PGC−2(Castilloら、1999年、Embo J、18、3676−3687)、ARA70(Heinleinら、1999年、J Biol Chem 274、16147−16152)、RIP140(Treuterら、1998年、Mol Endocrinol 12、864−881)等のいくつかの補助因子は、それらの転写活性を増強するが、SMRT(Lavinskyら、1998年、Proc.Natl.Acad.Sci.USA 95、2920−2925)およびN−CoR(Dowellら、J Biol Chem 274、15901−15907)は、それを抑制する。また、PPARγ補助因子ファミリーのメンバー(例えば、160−kDaタンパク質(SRC−1/TIF2/AIB−1)、CBP/p300またはTRAP220/DRIP205)は、PPARγと直接相互作用し、リガンド依存的様式で核内受容体トランス活性化機能を促進して、用いられるリガンドによって異なり得る生物学的作用または副作用につながることが、示されている(Adamsら、1997年、J.Clin.Invest.、100、3149−3153)。Koderaら、(2000年、J
Biol Chem.、275、33201−33204)は、PPARγと公知の補助因子との間の相互作用が、異なるクラスのPPARγリガンド(天然および合成)によって同じ程度に誘導されるかどうかを調べ、PPARγおよび補助因子複合体の全体的な構造が、関与するリガンドによって異なり、異なる生物学的作用を示す特定の組の標的遺伝子プロモーターの活性化を生じる場合があると結論付けた。
【0086】
PPAR部分アゴニストは、一般に、特定の活性化補助因子補充プロフィールを有し、したがって、特定の活性化補助因子補充プロフィールを有する化合物が好ましい。したがって、特定の実施形態によると、本発明の化合物および組成物は、限定された補助因子(複数可)補充パターンによって、さらに特徴付けられる。好ましい実施形態において、前記パターンは、前記核内受容体の転写活性の調節に対して別個の効果を生じ、特定の代謝プロセスの活性化ならびに望まれない副作用の排除を生じる非常に精密に調整された調節を可能にする。より具体的な実施形態において、本発明の化合物および組成物はさらに、PPAR受容体、より好ましくはPPAR受容体LBDの、補助因子TIF2との相互作用を阻害することができる。別の実施形態によると、本発明の化合物および組成物はまた、PPAR受容体、より好ましくはPPAR受容体LBDの、補助因子SRC−1との相互作用を増強することができる。好ましくは、前記PPAR受容体はPPARγ受容体である。
【0087】
リガンドによる補助因子補充の阻害および/または増強を測定するための方法は、欧州特許1267171に詳述されている。
【0088】
好ましい実施形態において、本発明の化合物は、PPARγに結合したときに、TIF2のものより少なくとも1ログ大きく、少なくとも2ログが好ましい、EC50で、LBDへのSRC1の補充を可能にする。
【0089】
本発明のある実施形態において、PPAR受容体の天然の生理学的リガンド、特にPPARγ受容体のものに対するそれらのアゴニスト、特に部分アゴニスト、またはアンタゴニスト特性のために、好ましい化合物は、PPAR媒介性転写制御およびそれによって生じる付随する生理学的効果の生物学的効果を制御するための医薬品として機能することができる。より具体的には、それらは、細胞生理学を調節して、関連する病理を減少させるか、または予防を提供もしくは増強することができる。
【0090】
本発明のさらに別の実施形態において、好ましい化合物は、1つより多いPPAR、例えばPPARγおよびPPARδの両方の、アゴニスト(または、好ましくは部分アゴニスト)である化合物である。かかるアゴニストは、例えば本明細書中に記載されているように、それぞれPPARγおよびPPARδについてのトランス活性化アッセイによって同定することができる。PPARγおよびPPARδ活性を判定するための非限定的な有用な方法は、本明細書中で、下記でそれぞれ実施例26および27に記載されている。
【0091】
臨床状態
本発明は、上述の化合物を投与することを含む、臨床状態の治療の方法、ならびに臨床状態の治療のための薬物の調製のための前記化合物の使用に関する。
【0092】
PPAR活性の調節因子は、体重管理において使用することができる。したがって、ある実施形態における臨床状態は、神経性食欲不振症(本明細書中で「食欲不振」とも省略される)または過食症等の摂食障害である場合がある。本明細書中に上記で開示される化合物はまた、体重を増大または減少させるための、特に体重を減少させるための方法において、使用することができる。臨床状態は、したがって、肥満である場合がある。脂肪症は、脂肪組織の過剰な累積である。最近の調査から、PPAR、具体的にはPPARγが、脂肪細胞の遺伝子発現および分化において中心的な役割を果たすことが示されている。PPARγサブタイプは、脂肪細胞分化の活性化に関与するが、肝臓におけるペルオキシソーム増殖の刺激において、それほど重要な役割を果たさない。PPARγの活性化は、典型的には、脂肪細胞特異的遺伝子発現を活性化することによって、脂肪細胞分化に寄与する(Lehmann、Moore、Smith−Oliver、Wilkison、Willson、Kliewer、J.Biol.Chem.、270:12953−12956、1995)。したがって、PPARアゴニストを用いて、脂肪組織を獲得することができる。PPAR部分アゴニストは、脂肪組織の過剰な累積の治療において有用な特性について選択することができる。
【0093】
ある好ましい実施形態において、本発明は、本明細書中に上記で記載されている化合物のいずれかを、それを必要とする個体に投与することによってインスリン抵抗性を治療するための方法に関する。本発明はまた、インスリン抵抗性の治療のための薬物の調製のための、前記化合物のいずれかの使用に関する。また、本発明は、前記化合物を投与することによってインスリン感受性を増大させるための方法、ならびにインスリン感受性を増大させるための薬物の調製のための前記化合物の使用に関する。外傷、手術、または心筋梗塞の後に発生し得るもの等のインスリン感受性における急性および一過性の障害は、本明細書中で教示されるように、治療することができる。
【0094】
インスリン抵抗性は、多数の臨床状態に関与している。インスリン抵抗性は、減少した、インスリンが広範囲の濃度にわたって生物学的作用を示す能力によって、顕在化される。インスリン抵抗性の初期の間、体は、異常に高い量のインスリンを分泌して、その欠損を補う。血中インスリンレベルは慢性的に高いが、インスリンに対する、活性のある筋細胞の代謝反応障害によって、それらはグルコースを効果的に取り込むことができなくなる。現在、インスリン抵抗性および結果として生じる高インスリン血症はいくつかの臨床状態、例えばメタボリックシンドローム(シンドロームXとも名付けられている)に寄与する場合があるということが、ますます認識されてきている。メタボリックシンドロームは、高インスリン血症を引き起こす最初のインスリン抵抗性段階、脂質異常症、および減少した糖耐性によって特徴付けられる。メタボリックシンドロームの患者は、心血管疾患および/またはII型糖尿病を発症する危険性が高いことが示されている。
【0095】
患者は、以下の基準の少なくとも3つが当てはまるとき、メタボリックシンドロームを患っているといわれる:
−胴の外周によって測定される中心性/腹部肥満(男性で102cmより大きく、女性で94cmより大きい)
−150mg/dL(1.69mmol/L)以上の空腹時トリグリセリド
−HDLコレステロール[男性−40mg/dL未満(1.04mmol/L)、女性−50mg/dL(1.29mmol/L)未満]
−130/85mmHg以上の血圧
−110mg/dL(6.1mmol/L)以上の空腹時血糖
インスリン抵抗性はまた、脂質生成に対して負の効果を有し、血流中のVLDL(超低密度リポタンパク質)、LDL(低密度リポタンパク質)、およびトリグリセリドレベルの増大ならびにHDL(高密度リポタンパク質)の減少に寄与する。これは、動脈における脂肪の斑の沈着につながる場合があり、これは、経時的に、アテローム性動脈硬化症につながる場合がある。したがって、本発明による臨床状態は、家族性高脂血症等の高脂血症である場合がある。好ましくは、高脂血症は、高コレステロール血症および/または高グリセリド血症によって特徴付けられる。臨床状態はまた、脂質異常症および糖尿病性脂質異常症を含む。本明細書中に含まれる化合物はまた、血清トリグリセリドレベルを低下させるため、またはHDLの血漿レベルを上昇させるために利用することができる。
【0096】
インスリン抵抗性は、血液中の過剰なインスリンおよびグルコースレベルにつながる場合がある。過剰なインスリンは、腎臓によるナトリウム貯留を増大させる場合があり、したがって、本発明の方法は、腎臓によるナトリウム貯留を減少させるために使用することができる。上昇したグルコースレベルによって、血管および腎臓が損傷を受ける場合がある。したがって、本発明の方法を使用して、血管および腎臓への障害を予防することができる。
【0097】
本発明の別の実施形態において、臨床状態は、PPARγによって媒介される炎症性障害である。用語「PPARγによって媒介される」によって、PPARγが状態の顕在化において役割を果たすことが理解されるべきである。例えば、PPARγは、急性炎症等の、好中球活性化と関連した炎症において役割を果たさないと考えられる。理論に拘束されることを望むことなく、PPARγのアゴニストは、NFκB媒介性転写と直接関連して阻害し、したがって例えば誘導型一酸化窒素合成酵素(NOS)およびサイクロオキシゲナーゼ−2(COX−2)の酵素経路等の種々の炎症反応を調節することによって、効果的な抗炎症薬である場合がある(Pineda−Torra,I.ら、1999年、Curr.Opinion in Lipidology、10、151−9)。
【0098】
炎症性障害は、例えば眼の炎症(J Biol Chem.2000年1月28日、275(4):2837−44)または眼乾燥疾患(J Ocul Pharmacol Ther.2003年12月、19(6):579−87)等、急性でも慢性でもよい。慢性的炎症性障害の例示的な例としては、炎症性腸疾患、潰瘍性大腸炎、またはクローン病が挙げられる。慢性炎症性障害はまた、関節炎、特に関節リウマチおよび多発性関節炎である場合がある。慢性炎症性障害はまた、炎症性皮膚疾患、特に尋常性ざ瘡、アトピー性皮膚炎、バリアー機能障害を有する皮膚障害、加齢または乾癬、特に乾癬の皮膚の効果である場合がある。慢性炎症性障害はまた、多発性硬化症またはアルツハイマー病等の炎症性神経変性疾患である場合がある。臨床状態はまた、糸球体腎炎、糸球体硬化症、腎炎症候群、および高血圧性腎硬化症を含む、胃腸疾患および腎疾患である場合がある。
【0099】
本発明の別の実施形態において、臨床状態は、PPARγの活性化に反応性のある癌である。したがって、臨床状態は、例えば、脂肪細胞腫瘍、例えば脂肪腫、線維脂肪腫、脂肪芽細胞腫、脂肪腫症、褐色脂肪腫、血管腫、および/または脂肪肉腫等の、脂肪組織において生じる過形成性または腫瘍性障害等の、PPAR応答性細胞の異常な細胞増殖によって特徴付けられる障害である場合がある。さらに、前立腺、胃、肺および膵臓の、ある種の癌は、PPARγアゴニストでの治療に反応性があることが実証されている。具体的には、ある種の脂肪肉腫、前立腺癌、多発性骨髄腫、および膵癌は、PPARγの活性化に反応性があることが示されているが、少なくともいくつかの結腸直腸癌および乳癌は反応性がない(Rumiら、2004年、Curr.Med.Chem.Anti−Canc Agents、4:465−77)。他の研究から、他の乳癌および結腸癌、ならびに神経芽細胞腫および傍抗癌が、PPARアゴニストに反応性があることが実証されている。癌の治療のためのPPARリガンドの使用は、Levy Kopelovich、2002年、Molecular Cancer Therapeutics、357によって概説されている。
【0100】
しかしながら、ある型の癌はPPARγでの活性化に反応性がある場合があっても、所定の型の全ての癌は反応性がない場合もある。具体的には、PPARγの機能喪失型の変異が癌において頻繁に生じ、かかる癌は、一般に反応性でない。したがって、癌が機能的PPARγを発現することが好ましい。
【0101】
臨床状態はまた、ウイルス感染、特にAIDSまたはHIVによる感染またはC型肝炎ウイルスによる感染等の感染である場合がある。また、本発明のPPARリガンドは、神経系疾患もしくは認知症における認知機能を向上させるために、または多嚢胞性卵巣症候群を治療するために、または骨量減少、例えば骨粗鬆症を予防および治療するために、有用である場合がある。
【0102】
臨床状態はまた、肝疾患、特にC型肝炎ウイルスによる感染、またはC型肝炎ウイルス感染と関連があるかどうかを問わないが、好ましくはPPAR調節に反応性がある、脂肪肝、肝炎、肝臓病変、肝硬変、非アルコール性脂肪性肝炎、もしくは肝癌後である場合がある。
【0103】
記載の多くはPPARγに関するが、本発明の化合物および方法は、PPARγの調節に限定されない。実際、他のPPARサブタイプが疾患において重要な役割を果たすことは、当業者に明らかであろう。例えば、PPARδは、脂肪代謝障害および創傷治癒、具体的には表皮創傷治癒と関連付けられている(Soon Tanら、2004年、Expert Opinion in Molecular Targets、39)。したがって、臨床状態はまた、表皮創傷治癒を含む創傷治癒である場合がある。
【0104】
インスリン感受性改善薬、例えばグリタゾンは、インスリン抵抗性を治療するために使用されているが、インスリン感受性を増強するが、それらはまた、体重を減少させるよりは増加させる。肥満および肉体的不活発は、インスリン抵抗性を悪化させる。グリタゾン(チアゾリジンジオンまたはTZD)は、脂肪組織、肝臓、および筋肉においてインスリン感受性を向上させることによって作用する。前記薬剤での治療は、いくつかの糖尿病の動物モデルにおいて試験されており、いかなる低血糖反応の発生もなしに、上昇したグルコース、トリグリセリド、および非エステル型遊離脂肪酸の血漿レベルの完全な矯正を生じる(Cheng LaiおよびLevine、2000年、Heart Dis.、2、326−333)。これらのチアゾリジンジオンの例は、ロシグリタゾン、ピオグリタゾンおよびトログリタゾンである。しかしながら、魅力的な治療効果を提供するが、これらの化合物は、血液希釈(浮腫を含む)、肝毒性、体重増加(増大した脂肪細胞分化からの体脂肪増大、血漿量増大、および心肥大を含む)、中程度だが有意なLDL−コレステロール増大および貧血を含む多数の深刻な望ましくない副作用を被っている(概説については、Lebovits、2002年、Diabetes Metab.Res.Rev、18、補足2、S23−9参照)。実際、糖尿病の多くの利用可能な治療は、体重増加、疾患の長期の管理の高い有意性の問題と関連がある。したがって、肥満、糖尿病ならびに心血管および肝疾患等の一般に関連のある障害の管理において使用することができる、代替的な、より効果的な治療剤の、大きな必要性がある。
【0105】
したがって、ある実施形態において、本発明は、本発明の化合物を、
i.肥満および糖尿病を患っている、または
ii.糖尿病を得る危険性があり、肥満になりつつある、または
iii.肥満を患っており、糖尿病を得る危険性がある、または
iv.糖尿病を患っており、肥満になる危険性がある
個体に投与することによる、肥満および糖尿病の同時の治療および/または予防に関する。
【0106】
本発明はまた、肥満および糖尿病の同時の治療および/または予防のための薬物の調製のための、本発明の化合物の使用に関する。化合物は、本明細書中で上述される化合物のいずれであってもよい。この実施形態の範囲内で、糖尿病は、好ましくは、II型糖尿病である。糖尿病を得る危険性がある前記個体は、例えば、本明細書中で上述されているメタボリックシンドロームを患っている個体である場合がある。肥満になる危険性がある前記個体は、例えば、体重増加の副作用を有する抗糖尿病薬での医学的治療下にある個体である場合がある。
【0107】
本発明はまた、臨床状態の治療または予防のための薬物の調製のための、上述の特定の化合物のいずれかの使用に関する。臨床状態は、メタボリックシンドローム、脂質異常症、肥満、真性糖尿病、インスリン抵抗性、または上述のインスリン抵抗性と関連する状態のいずれか、高血圧、心血管疾患、冠状動脈再狭窄、自己免疫疾患(喘息、多発性硬化症、乾癬、局所的皮膚炎、および潰瘍性大腸炎等)、癌、炎症、創傷治癒、脂肪代謝障害、肝疾患(C型肝炎ウイルスによる感染、またはC型肝炎ウイルス感染と関連があるかないかを問わず、脂肪肝、肝炎、肝臓病変、肝硬変もしくは肝癌後)、胃腸もしくは腎疾患(糸球体腎炎、糸球体硬化症、腎炎症候群、または高血圧性腎硬化症等)、感染(具体的にはウイルス感染)、認知機能障害(神経系の障害または認知症等)、多嚢胞性卵巣症候群、骨量減少(骨粗鬆症等)ならびにAIDSからなる群より選択してもよい。
【0108】
癌は、任意の癌、例えば以下のもののいずれかである場合がある:癌腫、肉腫、白血病、およびリンパ腫、腫瘍血管新生および転移、骨胃形成症、肝障害、ならびに造血疾患および/または骨髄増殖性疾患。例示的な障害としては、線維肉腫、粘液肉腫、脂肪肉腫、軟骨肉腫、骨肉腫、脊索腫、血管肉腫、内皮肉腫、リンパ管肉腫、リンパ管内皮肉腫、滑膜腫、中皮腫、ユーイング腫瘍、平滑筋肉腫、横紋筋肉腫、結腸癌、膵癌、乳癌、卵巣癌、前立腺癌、扁平上皮癌、基底細胞癌、腺癌、汗腺癌、皮脂腺癌、乳頭癌、乳頭状腺癌、嚢胞腺癌、髄様癌、気管支原性肺癌、腎細胞癌、肝細胞腫、胆管癌、絨毛癌、セミノーマ、胚性癌腫、ウィルムス腺癌、子宮頸癌、精巣腫瘍、肺癌、小細胞肺癌、膀胱癌、上皮癌、グリオーマ、星細胞腫、髄芽腫、頭蓋咽頭癌、上皮腫、松果体腫、血管芽腫、聴神経腫瘍、乏突起グリオーマ、髄膜腫、メラノーマ、神経芽細胞腫、または網膜芽細胞腫が挙げられるが、これらに限定されない。好ましくは、癌は、PPARγの活性化に反応性のある、上述の癌の1つである。
【0109】
心血管疾患は、例えば、アテローム発生、アテローム性動脈硬化症もしくはアテローム硬化性障害、血管再狭窄、心筋症、または心筋線維症、または上述の心血管疾患のいずれかである場合がある。
【0110】
炎症は、例えば、慢性炎症、好ましくは本明細書中で上述された慢性炎症のいずれかである場合がある。
【0111】
真性糖尿病は、複数の病因因子由来の疾患プロセスをいい、上昇した血液中のグルコースのレベル、または高血糖によって特徴付けられる。制御されていない高血糖は、増大した、および早発性の罹患率および死亡率によって特徴付けられる。少なくとも2つの型の真性糖尿病が同定されている:(i)正常な生理学的条件下でグルコース利用を制御するホルモンであるインスリンの完全な欠如の結果である、I型糖尿病、またはインスリン依存性糖尿病(IDDM)、および(ii)II型糖尿病、またはインスリン非依存性糖尿病(NIDDM)。NIDDMは、いくつかの場合において循環インスリンレベルを増大させることによって対処することができる、複数の病因因子由来の複雑な疾患である。
【0112】
医薬製剤および投与の方法
本発明の化合物および組成物に従って、当業者に理解されるように、本明細書中に記載されている化合物の1つ以上を、選択された投与の経路に依存して、種々の形態で哺乳動物に投与してもよい。本発明の組成物は、経口または非経口で投与してもよく、後者の経路は、静脈内および皮下投与を含む。非経口投与は、選択された期間にわたる持続点滴によるものであってもよい。
【0113】
注射可能な使用のための形態としては、無菌水溶液または分散および無菌の注射可能溶液または分散の即席の調製のための無菌の粉末が挙げられる。全ての場合において、形態は、無菌でなければならず、容易な注射可能性が存在する程度まで流動性でなければならない。
【0114】
患者による投与の容易さのために、経口または他の非侵襲性の投与の様式、例えばパッチ、坐剤等の様式が好ましい。化合物は、不活性な希釈剤もしくは吸収できる食用担体とともに経口投与してもよく、またはハードもしくはソフトシェルゼラチンカプセル中に封入するか、圧縮して錠剤にするか、もしくは食事の食物と直接組み合わせてもよい。経口の治療的投与のために、化合物は、賦形剤と組み合わされ、摂取可能な錠剤、バッカル錠、トローチ、カプセル剤、エリキシル剤、懸濁液、シロップ、ウェーハ等の形態で使用してもよい。
【0115】
1つ以上の本発明の化合物を含む組成物はまた、リポソームと呼ばれるリン脂質ベシクル内に含まれた溶液または乳濁液中で投与してもよい。リポソームは、単層でも多層でもよく、ホスファチジルコリン、ジパルミトイルホスファチジルコリン、コレステロール、ホスファチジルエタノールアミン、ホスファチジルセリン、ジミリストイルホスファチジルコリンおよびそれらの組み合わせから選択される成分で形成される。多層リポソームは、単層ベシクルと同様の組成の多層ベシクルを含むが、溶液または乳濁液中の化合物が封入される複数の区画を生じるように調製される。また、他の補助剤および修飾物質が、ポリエチレングリコール、または他の物質等のリポソーム製剤中に含まれる場合がある。
【0116】
組成物を含むリポソームはまた、それらの送達を標的化するために、リポソームの表面に固定された抗体を有すること等の変更を有してもよい。
【0117】
「臨床状態」の項で上述された臨床状態の1つを治療するための、インビボの投与に適した生物学的に適合性のある形態の、対象への投与のための医薬組成物が、本発明のある実施形態にあり、前記方法は、安全で効果的な量の化合物を単独で、または他の薬剤および/もしくは薬学的担体との組み合わせを含む。例えば、フィブラートクラスの薬物、例えばベザフィブラート等の脂質異常症に対して効果的な薬剤を組み合わせて本発明の化合物を用いて、インスリン抵抗性および/または糖尿病を治療することができる。いくつかの他の薬剤の例は、インスリン感受性改善薬、PPARγアゴニスト、グリタゾン、トログリタゾン、ピオグリタゾン、エングリタゾン、MCC−555、BRL 49653、ビグアナイド、メトホルミン、フェンホルミン、インスリン、インスリン模倣剤、スホニル尿素、トルブタミド、グリピジド、α−グルコシダーゼ阻害剤、アカルボース、コレステロール降下薬、HMG−CoA還元酵素阻害剤、ロバスタチン、シンバスタチン、プラバスタチン、フルバスタチン、アトルバスタチン、リバスタチン、他のスタチン、シクエストレート、コレスチラミン、コレスチポール、架橋デキストランのジアルキルアミノアルキル誘導体、ニコチニルアルコール、ニコチン酸:ニコチン酸塩、PPARαアゴニスト、フェノフィブリン酸誘導体、ゲムフィブロジル、クロフィブラート、フェノフィブラート、コレステロール吸収の阻害剤、β−シトステロール、アクリルCoA:コレステロールアシルトランスフェラーゼ阻害剤、メリナミド、プロブコール、PPARδアゴニスト、抗肥満化合物、フェンフルラミン、デクスフェンフルラミン、フェンチラミン、スルビトラミン、オーリスタット、神経ペプチドY5阻害剤、β3アドレナリン受容体アゴニスト、および回腸胆汁酸トランスポーター阻害剤である。
【0118】
組成物は、組成物がインビボで有効性を有するので、ヒトおよび動物を含む、かかる治療を必要とする任意の生物に投与することができる。安全で有効、によって、本明細書中で使用される場合、深刻な副作用を避けながら、対象に影響を及ぼす疾患を減少、予防、改善、または治療するために十分な能力を提供することが意味される。安全で有効な量は、対象の年齢、治療される対象の身体的条件、障害の重症度、治療の期間および任意の併用療法の性質に依存して変化し、その決定は、通常の医師の能力の範囲内である。組成物は、本明細書中に記載されているのと同じ一般的な様式で、製剤化および投与される。本発明の化合物は、単独で、または1つ以上のさらなる活性薬剤と組み合わせて、効果的に用いることができる。併用療法は、本発明の化合物および1つ以上のさらなる活性薬剤を含む単一の薬剤の投薬組成物の投与、ならびに本発明の化合物の投与および各活性薬剤をそれぞれ別々に投薬することを含む。例えば、本発明の化合物およびスルホニル尿素、チアゾリジンジオン、ビグアナイド、メグリチニド、インスリンまたはα−グルコシダーゼ阻害剤等のインスリン分泌促進物質を、カプセル剤もしくは錠剤等の単回経口投薬組成物でともに患者に投与するか、または各薬剤を別々の経口投薬で投与することができる。別々の投薬が使用される場合、本発明の化合物および1つ以上のさらなる活性薬剤を、本質的に同時に、すなわち同時または別々に交互の回で、すなわち連続して投与することができ、併用療法は、これら全ての投与計画を含むと理解される。
【0119】
本発明の医薬組成物の治療有効量はまた、対象の疾患状態、年齢、性別、および体重、ならびに化合物が対象において所望の反応を誘発する能力等の因子によって変化してもよい。投与計画は、最適な治療反応を提供するよう調節してもよい。例えば、いくつかの分割された用量を毎日投与してもよく、または、治療状況の要求によって示されるように、用量が比例して減少してもよい。
【0120】
約4mg/kgの用量は、哺乳動物に適した最初の投薬量であるようであり、この投薬量は、安全で有効な量を提供するように、必要に応じて調節してもよい。したがって、投薬量は、最初は、典型的には0.1〜20mg/kg、好ましくは0.5〜10mg/kg、より好ましくは1〜5mg/kgである。
【0121】
薬学的に許容され得る担体は、本明細書中で使用される場合、1つ以上の適合性のある固体または液体送達系が、対象に投与されたときに非侵害性の生理学的反応を有することを意味する。いくつかの例としては、デンプン、糖、セルロースおよびその誘導体、粉末化トラガカント、麦芽、ゼラチン、コラーゲン、滑石、ステアリン酸、ステアリン酸マグネシウム、硫酸カルシウム、植物油、ポリオール、寒天、アルギン酸、発熱物質不含水、等張生理食塩水、リン酸バッファー、および医薬製剤において用いられる他の適した非毒性物質が挙げられるが、これらに限定されない。湿潤剤および潤滑剤、錠剤化剤、安定剤、抗酸化剤、ならびに保存料等の等の他の賦形剤もまた、企図される。
【0122】
本明細書中に記載されている組成物は、化合物または類似体の有効量が薬学的に許容され得る担体と混合物中で組み合わされるように、対象に投与することができる薬学的に許容され得る組成物の調製のための公知の方法によって調製することができる。適した担体は、例えば、Remington’s Pharmaceutical Sciences(Mack Publishing Company、米国ペンシルバニア州イーストン、1985年)に記載されている。これに基づいて、組成物は、排他的ではないが、1つ以上の薬学的に許容され得るビヒクルまたは希釈剤と関連した化合物の溶液を含み、適したpHを有し、生理学的液体と等浸透圧の緩衝液中に含まれる。
【0123】
以下の実施例は、本明細書中の特定の態様を記載して、本発明を説明し、実施例は単に本発明の理解および実施において有用な特定の方法論を提供するだけなので、本発明を限定すると解釈されるべきではない。
【実施例】
【0124】
実施例1
4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸の合成
この実施例は、スキーム2に従って、本明細書中でDPDとも呼ばれる、4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸の合成を説明する(表II中のSN1)。
【0125】
スキーム2
【0126】
【化9】
【0127】
2−メトキシ−パラコン酸の合成(2−3):20Lの二重ジャケットガラスリアクターに260gのo−メトキシベンズアルデヒド、286gの無水コハク酸、572gの無水塩化亜鉛、および2600mLの無水DCMを充填した。混合物を撹拌し、2℃に冷却した。533mLの量のトリエチルアミンを30分間にわたって添加した。次いで、混合物を周囲温度で24時間撹拌させた。1690mLの量の2M HClを添加し、次いで2600mLの酢酸エチルを添加した。混合物を5分間活発に撹拌した。2000mLの酢酸エチルで水相を抽出した。合わせた有機性抽出物を650mLの飽和塩水で洗浄し、次いで3×2600mLの飽和重炭酸ナトリウムで洗浄した。次いで、合わせた水性抽出物を酢酸エチルで洗浄した。濃HClを用いて、水性抽出物をpH2まで酸性化した。黄色の油を分離した。混合物を、2000mLの酢酸エチルを用いて2回抽出した。有機相を1000mLの塩水で4回洗浄し、45℃の加熱浴温度を使用したBuchi R220ロータバップで蒸発させた。残りの残渣に4000mLのトルエンを添加した。混合物を110℃に加熱した。1Lのトルエンを蒸発させて除いた。残りを室温まで冷却させ、48時間放置し、その間純粋な2−メトキシ−パラコン酸が結晶化した。結晶物質を集め、ろ過し、一定の重量まで、真空オーブン中で、真空中、45℃で乾燥させた。
【0128】
収量:220g(49%)。シス/トランス比:46/54
シス−およびトランスラセミメトキシ−パラコン酸から全トランス−2−メトキシパラコン酸への転換(2−4):1020gのメトキシパラコン酸を、1729mLの濃硫酸および2570mLの水の混合物に添加した。混合物を室温で18時間撹拌させた。33:64のシス/トランス比が得られた。次いで、混合物を2.5時間、60℃に加熱した。11:89のシス/トランス比が得られた(HPLCによる分析)。次いで、混合物を室温に冷却させ、続いてろ過した。固体物質を酢酸エチル中に再び溶解させ、水および塩水で洗浄した。有機相をMgSO4で乾燥させ、蒸発させた。6:94のシス/トランス比が得られた。固体物質を熱いトルエンから再結晶化させた。得られた結晶物質を、40℃で48時間、真空中で乾燥させた。
【0129】
収量:855g(84%)。シス/トランス比:8/92。融点:132〜133℃
【0130】
【化10】
【0131】
メトキシ−パラコン酸、ラセミ化合物のエステル化(2−5):193gのメトキシ−パラコン酸を、600mLのTHF中に溶解させた。混合物に、145gのCDI(899mmol、1.1当量)を添加し、混合物を10分間撹拌した。65mLの量の無水エタノール(またはメチルエステルを生じるためのメタノール)を添加し、完了するまで(約120分間)混合物を撹拌した。酢酸エチルおよび飽和重炭酸ナトリウムを用いて、粗反応混合物を抽出した。有機相を0.5N HClおよび塩水で洗浄した。蒸発の後、188gの量の所望のエチルエステルが得られた。
【0132】
ラセミメトキシ−パラコン酸、エチルエステルの還元(2−6):ラセミラクトールの調製:5℃で、700mLのトルエン中の105gのエチルエステル(397mmol)。3当量のDIBAL−H(1.19mol、1.19L 1M溶液)を添加した。室温で60分間撹拌した。メタノールで反応停止させた。2.5Lの酢酸エチルを加えた。700mLの水。水相をEtOAcで抽出した。有機層を塩水で洗浄した。クロロホルム/ヘキサンから油状の残渣を蒸発、再結晶化させた。固体をろ過し、真空中で乾燥させた。
【0133】
収量:53g(237mmol、59%)。
【0134】
ラセミジオールのラセミラクトール合成に使用されるウィティッヒ反応(2−8):191gの量のカルボキシプロピルトリフェニルホスホニウムブロマイド、1000mLの無水トルエンおよび100gのカリウムt−ブトキシドを、80℃で30分間混合した。混合物を室温に冷却した。180mLの無水THFに予め溶解させた25gの量の精製ラセミラクトール(114.5mmol)を、ゆっくり添加した。反応を60分間継続させた。粗反応混合物を1500mLの冷水に注ぎ、300mLの酢酸エチルを添加した。水相を300mLの酢酸エチルで再抽出した。次いで、水相を2N HClで酸性化し、300mLの酢酸エチルを用いて3回抽出した。形成された固体をろ過して除いた。有機相を蒸発させた。蒸発した残渣に、500mLのジエチルエーテルを添加した。フラスコを10分間回転させ、固体をろ過して除いた。ろ液を、飽和重炭酸ナトリウム溶液で3回抽出した。次いで、2M HClを用いて水相をpH4まで酸性化した。次いで、水相を、200mLの酢酸エチルを使用して3回抽出した。有機相を合わせて、MgSO4で乾燥させ、蒸発させて、45gの物質を生じた。
【0135】
カラムクロマトグラフィー:ラセミジオールをシリカゲル(35cmカラム長、4cm直径)で精製した。ラセミジオールを最少の酢酸エチルに溶解させ、カラムに適用した。1Lの酢酸エチル(60%)/ヘキサン(40%)を、容積測定シリンダーに加えた。300mLのEtOAc/ヘキサンをシリンダーから採取し、カラムに加えた。酢酸エチルを用いて、シリンダー中の残りの700mLのEtOAc/ヘキサンを1Lまで希釈した。次いで、300mLの新しいEtOAc/ヘキサン溶液をカラムに加え、カラムを通過させた。酢酸エチルを用いて、シリンダー中の残りの700mLのEtOAc/ヘキサンを、再び1Lまで希釈した。次いで、300mLの新しいEtOAc/ヘキサン溶液をカラムに加え、カラムを通過させた。酢酸エチルを用いて、シリンダー中の残りの700mLのEtOAc/ヘキサンを、もう1回1Lまで希釈した。次いで、300mLの新しいEtOAc/ヘキサン溶液をカラムに加え、カラムを通過させた。ラセミジオールの純粋な画分を集め、蒸発させて、26gの純粋なラセミジオールを生じた。
【0136】
収量:26g(88.3mmol、79%)
ラセミジオールの、ラセミアセトニドへの転換(2−10):26g(88mmol)の精製ジオールを、260mLのジメトキシプロパンおよび26mgのp−TsOHと混合した。混合物を、周囲温度で一晩撹拌させた。3滴のトリエチルアミンを添加し、混合物を蒸発させた。残りの残渣に150mLのヘキサンを添加し、混合物を一晩撹拌した。固体をろ過して除き、乾燥させて25g(75mmol)のラセミアセトニドを生じた。
【0137】
収率:85%
ラセミアセトニドの脱メチル化(2−12):16.7gのエタンチオールを、375mLのDMPU中の21.5gのNaHの混合物に添加することによって、水素化ナトリウムおよびエタンチオールの懸濁液を調製した。懸濁液を80℃に加熱し、周囲温度まで冷却させた。
【0138】
15gのラセミアセトニドを75mLのDMPU中に溶解させ、EtSH/NaHの懸濁液に添加した。混合物を130℃で2時間加熱した。次いで、反応混合物を氷水に注ぎ、DCMで抽出した。2N HClを用いて水層を酸性化し、酢酸エチルで抽出した。有機層を塩水で洗浄し、蒸発乾固させた。
【0139】
収量:16.5g(粗)。
【0140】
ラセミの調製(2−14):28mmolの量の脱メチル化されたラセミアセトニドを、15mLの2−クロロベンズアルデヒド、0.5gのp−TsOH、および60mLのトルエンと混合した。混合物を24時間撹拌し、蒸発させた。Biotage Horizon(登録商標)クロマトグラフィー装置を使用したシリカゲルクロマトグラフィーを用いて、粗反応混合物を精製した。DCM(19)/メタノール(1)を用いて混合物を精製して、蒸発の後に6.5gの固体を生じた。
【0141】
収量:6.5g(16.7mmol、59%)
スキーム3:表I(および表II)に記載されているPPAR調節因子のための合成経路
【0142】
【化11】
【0143】
【化12】
【0144】
実施例2
PPAR調節因子2(SN2、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子2(SN2、表II)の合成を説明する。実施例1に記載されているような100mg(0.31mmol)の量のラセミアセトニド(6−[4−(2−ヒドロキシフェニル)−2,2−ジメチル−[1,3]ジオキサン−5−イル]−ヘキス−4−エノン酸)2−12を、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、108mg)の2,3−ジクロロベンズアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析よって分析した。
【0145】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=435
実施例3
PPAR調節因子6(SN6、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子6(SN6、表II)の合成を説明する。100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、70mg)のシクロヘキサノンを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0146】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=359
実施例4
PPAR調節因子8(SN8、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子8(SN8、表II)の合成を説明する。
【0147】
N−Boc 4−オキソピペリジンの調製:2gの量の4−オキソピペリジンを、20mLのジオキサン/水に溶解させた。混合物に、2当量の重炭酸ナトリウム、次いで1.0当量のBoc2Oを添加した。混合物を4時間撹拌した。酢酸エチル(100mL)を添加した。0.2N HClおよび塩水を用いて、有機相を2回洗浄した。次いで、有機相を硫酸マグネシウムで乾燥させ、蒸発させて、結晶化した油を生じた。100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol)のN−Boc−4−オキソピペリジンを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物を微小のシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0148】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=460
本明細書中で、下記で言及される生物学的アッセイは、スピロ−ピペリジン環上に依然として存在するBoc−保護基を含む反応生成物で行った。
【0149】
実施例5
PPAR調節因子9(SN9、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子9(SN9、表II)の合成を説明する。
【0150】
1−ナフタレンカルボキシアルデヒドの調製 3.87mLの量の塩化オキサリル(44.24mmol)を、100mLのDCMに溶解させた。混合物を−60℃に冷却した。注射器を用いて、5.6mLの量のDMSOを滴下した。混合物を15分間撹拌した。75mLのDCM中の5gのナフタレン−1−メタノールの溶液を滴下した。反応を、−60℃で1時間継続させた。20mLの量のトリエチルアミンを添加した。混合物を15℃に達させた。混合物を抽出漏斗に移し、水、1M HCl、水および塩水で洗浄した。次いで、有機層を硫酸マグネシウムで乾燥させ、蒸発させた。粗生成物は、次の工程においてさらなる精製なしで使用するのに十分純粋であった。
【0151】
実施例1に従って調製された100mg(0.31mmol)の量のラセミアセトニド(上述のような2−12)を、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、97mg)の1−ナフタレンカルボキシアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0152】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=417
実施例6
PPAR調節因子11(SN11、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子11(SN11、表II)の合成を説明する。
【0153】
100mg(0.31mmol)の量のラセミアセトニド(実施例1の2−12)を、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、62mg)のヘキサナールを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0154】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=361
実施例7
PPAR調節因子13(SN13、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子13(SN13、表II)の合成を説明する。
【0155】
100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、130mg)のo−2−オキソ−4a,8a−ジヒドロ−2H−クロメン−4−カルバルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物を微小なシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0156】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=469
実施例8
PPAR調節因子19(SN19、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子19(SN19、表II)の合成を説明する。
【0157】
100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、103mg)の2,3−ジメトキシベンズアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて粗反応混合物をシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0158】
HPLC:保持時間(グラジエントA):15.23、15.39分、95%(異性体の1:1混合物の合計)
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=427.2
実施例9
PPAR調節因子14(SN14、表II)の合成 この実施例は、スキーム3に従う、スキーム3に従う、PPAR調節因子14(SN14、表II)の合成を説明する。
【0159】
室温で、撹拌された、CH2Cl2(10mL)中のジオール(0.08g、0.27mmol)の溶液に、2,6−ジクロロベンズアルデヒド(0.07g、0.40mmol)およびpTSA(3mg)を添加した。反応混合物を8時間撹拌し、トリエチルアミン(2〜3滴)で反応停止させた後、真空中で濃縮した。残渣を、ヘキサン/EtOAc(8:2)で希釈したシリカゲル上でのカラムクロマトグラフィーによって精製して、無色の油として、アセタール(100mg、45%収率)を得た。
【0160】
【化13】
【0161】
実施例10
PPAR調節因子15(SN15、表II)の合成
この実施例は、スキーム3に従う、PPAR調節因子15(SN15、表II)の合成を説明する。スキーム−2、実施例1の化合物2−12(100mg、0.31mmol)を、THF(5mL)中に溶解させ、触媒量のpTsOH(5mg)を室温で添加した。反応混合物を室温で8時間撹拌し続けた。2,3,4,5,6−ペンタフルオロベンズアルデヒド(158mg、0.62mmol)を反応塊に添加し、もう1回触媒量のpTsOHを添加した。反応条件をさらに24時間維持した。反応の完了後に、乾燥したEt3Nを添加して、pH=7に調節した。真空下で溶媒を除去し、得られた粗混合物をカラムクロマトグラフィーによって精製して、最終生成物15を得た。
【0162】
スキーム4:表III(および表II)に記載されている化合物の合成経路
【0163】
【化14】
【0164】
【化15】
【0165】
実施例10
PPAR調節因子23(SN23、表II)の合成
この実施例は、スキーム4に従う、PPAR調節因子23(SN23、表II)の合成を説明する。
【0166】
ウィティッヒ反応 10g(22.56mmol)の量のBrPPh3(CH2)4CO2Hを、60mLの乾燥トルエン中の5.06g(45mmol)のKOtBuと混合した。混合物を30分間80℃に加熱し、周囲温度まで冷却させた。10mLの無水THF中の1.26gの量のラセミラクトールを添加し、反応を2時間継続させた。スキーム−2の2−8の調製についての後処理。
【0167】
収量:3.3g(粗、次の工程で使用する)。
【0168】
アセトニドの調製 前の工程で得られたジオールを、40mLのジメトキシプロパン中に溶解させた。25mgの量のp−TsOHを添加し、反応を24時間撹拌した。1滴のトリエチルアミンを添加し、混合物を蒸発させた。粗混合物を、小さいシリカゲルカラムでろ過した。
【0169】
収量:1.6g
アセトニドの脱メチル化 1.91mLの量のエタンチオール(25.83mmol)を、50mLのDMPUと混合した。51.7mmolの量のNaH(2.06gの、油中の60%分散)を添加し、混合物を30分間80℃に加熱した。混合物を周囲温度まで冷却した。7.5mLのDMPU中の1.5gのアセトニド(4.3mmol)の溶液を添加し、混合物を125℃で2時間撹拌した。粗混合物を氷水に注ぎ、2×50mLのDCMで抽出した。水相を2N HClで酸性化し、次いで、EtOAcで抽出し、最後に塩水で洗浄した。有機相を蒸発させて、1.6gのヒドロキシ−アセトニドを生じた。
【0170】
2−クロロベンズアルデヒドとの反応 ヒドロキシ−アセトニド(1.6g、粗)を、2mLの2−クロロベンズアルデヒド、8mLの無水トルエン、および50mgのpTsOHと混合した。混合物を24時間撹拌し、蒸発させ、シリカゲルカラムクロマトグラフィーによって精製した。
【0171】
質量スペクトル(エレクトロスプレー、ネガティブモード):[M−H]−=415
実施例11
PPAR調節因子17(SN17、表II)の合成
この実施例は、スキーム4に従う、PPAR調節因子17(SN17、表II)の合成を説明する。
【0172】
100mg(0.31mmol)の量のラセミアセトニドを、1mLのトルエンおよび10mgのp−トルエンスルホン酸と混合した。混合物に、2当量(0.62mmol、122mg)の4−フェニルアミノベンズアルデヒドを添加した。混合物を24時間撹拌し、窒素流を用いて蒸発させた。メタノール(1)/DCM(19)を用いて、粗反応混合物を微小なシリカゲルカラムで精製した。純粋な画分をTLCによって同定し、集め、蒸発させ、HPLCおよび質量分析によって分析した。
【0173】
o−メトキシパラコン酸のメチルエステルの合成:a)撹拌された、乾燥DMF(150mL)中の酸(5g、21.28mmol)の溶液に、K2CO3(29.2g、211.8mmol)を添加し、次いでヨウ化メチル(6.01g、42.3mmol)を0℃で添加し、反応混合物を室温で5時間撹拌した。反応混合物を水(10mL)、CH2Cl2(30mL)で希釈し、有機層を分離した。CH2Cl2(2×15mL)で水層を抽出し、合わせた有機相を塩水(2×20mL)で洗浄し、Na2SO4で乾燥させ、真空中で蒸発させて、エステルを得たが、これはさらなる反応において使用するのに十分に純粋であった。(収量:4.34g、81.9%)。
【0174】
b)撹拌された、乾燥エーテル(60mL)中の酸5(10g、42.3mmol)の溶液に、新しく調製したジアゾメタン溶液(200mL溶液、10gのニトロソメチル尿素から生じた)を0℃で添加し、室温で30分間撹拌した。出発物質が完全に消えた後で、エーテルを蒸発させて、エステル6を得て、これを、精製なしで、さらなる反応のために使用した。(収量:9.8g、98%)。黄色の、粘性のある油
【0175】
【化16】
【0176】
実施例12
PPAR調節因子34(SN34、表II)の合成
この実施例は、PPAR調節因子34(SN34、表II)の合成を説明する。
【0177】
窒素雰囲気下、触媒のp−トルエンスルホン酸(約5mg)の存在下で、乾燥トルエン(2mL)中のジオール7(0.4g、1.36mmol)およびクロロベンズアルデヒドジメチルアセタール(0.28g、1.63mmol)の混合物を一晩撹拌した。出発物質が完全に消えた後で、反応混合物を固体NaHCO3で中和した。溶液を反応混合物から傾しゃし、ロータリーエバポレーターで濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、ベンジリデンアセタールを生じた。
【0178】
収量:0.31g(59%)
【0179】
【化17】
【0180】
THF:H2O(4mL、3:1)中のこの化合物(0.2g、0.48mmol)の溶液に、LiOH.H2O(0.3g、7.15mmol)、メタノール(0.5mL)を添加し、室温で一晩撹拌した。反応混合物をNaHSO4の飽和水溶液で中和し、CH2Cl2(2×10mL)で抽出した。合わせた有機層を塩水(10mL)で洗浄し、ロータリーエバポレーターで蒸発させ、粗生成物をカラムクロマトグラフィーによって精製した。
【0181】
収量:95mg(51%)
【0182】
【化18】
【0183】
スキーム−4a
【0184】
【化19】
【0185】
実施例13
PPAR調節因子25(SN25、表II)の合成
この実施例は、表IIのPPAR調節因子25の合成を説明する。
【0186】
エタンチオール(0.7mL、9.5mmol)を、撹拌された、乾燥DMF(8mL)中のNaH(0.38g、9.5mmol)の懸濁液(油中60%)に、0〜5℃で添加し、20〜30分間撹拌した。温度を維持しながら、乾燥DMF(2mL)中の化合物4a−1(0.25g、0.95mmol)を、上述の混合物にゆっくり滴下した。反応塊温度を120〜130℃に上昇させ、6〜8時間維持した。反応の完了後に、塊を0〜5℃に冷却し、pH4〜5に調節することによって、1N HCl(1mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、純粋な4a−2を得た(酢酸エチル:ヘキサン、2.5:7.5)。
【0187】
収量:155mg(65.6%収率)。
【0188】
4a−2の化合物(0.15g、0.6mmol)を、8:2の比のアセトンおよび水の混合物(10mL)中に溶解させた。上述の混合物に、OsO4(触媒量、0.05M)およびNMO(0.14g、1.2mmol)を添加し、室温で8〜10時間撹拌し続けた。反応の進行をTLCによってモニタリングした。反応の完了後に、飽和Na2S2O5溶液(3mL)によって反応停止させた。溶媒アセトンを真空下で除去し、酢酸エチル(3×10mL)で化合物を抽出した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。
【0189】
THF:H2O(8:2、10mL)混合物中の粗生成物(スキーム中に示されていないジオール)の溶液に、NaI4(0.27g、1.8mmol)を室温で添加し、1時間撹拌し続けた。反応の完了後に、飽和Na2SO4溶液(3mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物4a−3を、さらなる反応のために処理した。
【0190】
乾燥ベンゼン(10mL)中の化合物4a−3の溶液に、C2−ウィティッヒイリド(0.15g、0.42mmol)を添加した。反応塊を、不活性な条件下、室温で2〜3時間撹拌し続けた。ロータリーエバポレーター中で過剰なベンゼンを除去し、このようにして得られた粗化合物4a−5をカラムクロマトグラフィーによる精製に供した。(酢酸エチル:ヘキサン、0.5:9.5)。
【0191】
収量:0.11g g(95%)
【0192】
【化20】
【0193】
THF:H2O(8:2、5mL)の混合物中の化合物4a−5(0.11g、0.34mmol)の溶液に、LiOH.H2O(0.1g、2.4mmol)を室温で添加し、8〜10時間撹拌し続けた。反応の完了後に、pHを4〜5に調節することによって、飽和NaHSO4溶液(1mL)によって反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で蒸発させ、真空で濃縮した。粗生成物4a−6を、さらなる反応のために処理した。
【0194】
化合物4a−6(85mg、0.29mmol)をTHF中に溶解させ、触媒量のpTSOHを室温で添加した。反応混合物を室温で6〜8時間撹拌し続けた。o−クロロベンズアルデヒド(81mg、0.58mmol)を反応塊に添加した。もう1回触媒量のpTSOHを添加した。反応条件をさらに5〜6時間維持した。反応の完了後に、pH=7に調節することによって、乾燥Et3Nを添加した。真空下で溶媒を除去し、得られた粗混合物をカラムクロマトグラフィーによって精製して、最終生成物25(表II)を得た。(酢酸エチル:ヘキサン、3.5:6.5)。
【0195】
収量:35mg(32%)
上述の化合物HPLC純度は79.2%であり、分取HPLCによってこれをさらに精製して、純粋な化合物(98%純度、15mg)を得た。
【0196】
【化21】
【0197】
スキーム5:
【0198】
【化22】
【0199】
実施例14
PPAR調節因子37(SN37、表II)の合成
この実施例は、スキーム5に従う、表IIのPPAR調節因子37の合成を説明する。
【0200】
スキーム5中のマロン酸水素エチル5−2の調製:撹拌された、エタノール(100mL)中のマロン酸ジエチル(20g、0.125モル)の溶液に、室温で、時々冷却しながら、エタノール(30mL)中の85%KOH(7g、0.125モル)の溶液を添加した。20分後に、混合物を濃HClで酸性化し、ろ過した。ろ過ケーキ(KCl)をエタノール(50mL)で洗浄した。合わせたろ液を濃縮し、残りの液体をカラムクロマトグラフィー(ヘキサン−EtOAc:85:15)に供して、1(14.7g、87%)を得た。
【0201】
【化23】
【0202】
ケト−エステル5−4の調製:
乾燥THF(100mL)中の14.34gのマグネシウムエトキシド(0.13モル)の懸濁液に、10.5gのマロン酸水素エチル1 5−2(0.08モル)を添加し、90分間還流した。反応混合物を0℃に冷却し、乾燥THF(25mL)中の2−メトキシベンゾイルクロライド2(14.59g:0.085モル)の溶液を、反応温度が5℃を超えない速度で添加した。反応混合物を室温で一晩撹拌し、2日間放置した。塩化アンモニウムの飽和溶液を添加し、反応混合物をEtOAc(3×50mL)で抽出した。合わせた有機層を水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を蒸発させて、油として生成物を得て、カラムクロマトグラフィー(ヘキサン−EtOAc:95:5)によって精製して、生成物(4.33g、25%収率)を生じた。生成物をスペクトルデータによって確認した。
【0203】
【化24】
【0204】
撹拌された、乾燥THF(40mL)中の水素化ナトリウム(60%、0.9g、0.08モル)の懸濁液に、アセチル化エステル(4.33g、0.019モル)を0〜10℃で滴下し、15分間撹拌した。臭化アリル3(2.30g、0.019モル)を室温で反応混合物に添加し、反応混合物を3〜4時間還流した。反応混合物を冷却し、塩化アンモニウム溶液を添加し、EtOAc(3×30mL)で抽出した。有機層を水で洗浄し、無水硫酸ナトリウムで乾燥させ、蒸発させた。粗生成物をカラムクロマトグラフィー(n−へキサン−EtOAc:95:5)によって精製して、4(2.5g、50%収率)を得た。
【0205】
【化25】
【0206】
ジオールの調製(5−7):
温度が10℃を超えない速度で、冷却した、乾燥THF(15mL)中の水素化ホウ素リチウム(0.83g、0.038モル)の懸濁液に、冷却した、乾燥THF(25mL)中のアルキル化ケトエステル4 5−4(2.5g、0.0095モル)の溶液を添加した。撹拌を室温で4〜5時間継続し、反応の進行をTLCによってモニタリングした。2N HClの添加によって反応混合物をpH2に酸性化し、水を反応混合物に添加し、EtOAc(3×25mL)で抽出した。合わせた有機層を塩水、水で洗浄し、無水硫酸ナトリウムで乾燥させた。溶媒を蒸発させて、ジオール5 5−7(2g、94%収率)のシスおよびトランス反応混合物を生じ、精製なしで次の工程に進んだ。HPLC:シス77.318%、反応時間20.244分、トランス:22.682%、反応時間22.337分(HPLC条件は、クロマトグラムに関して言及される)。
【0207】
アセトニド5−10の調製:
ジオール5−7(2g、0.009モル)、2,2−ジメチルプロパン(15mL、0.12モル)、触媒量のpTSA(10mg)の溶液を、室温で4〜5時間撹拌し、反応の進行をTLCによってモニタリングした。反応混合物をEt3Nで中和した。過剰なDMPを真空下で除去し、油状のシス−トランス混合物をカラムクロマトグラフィーによって分離して、6b 5−10(0.925g、収率40%)を得た。
【0208】
【化26】
【0209】
1,3−ジオキサンアルデヒドの調製(オゾン分解)5−11:
永続的な青色が生じるまで、O3を−78℃で乾燥DCM(10mL)中のアセトニド5−10(0.93g、0.004モル)の溶液に通過させた。青色が消えるまで、反応混合物を0℃で撹拌した。DCM(5mL)中のTPP(1.1g、0.0043モル)の溶液を、無色の溶液に0℃で添加し、反応混合物を室温まで温め、1時間撹拌した。反応混合物の進行をTLCによってモニタリングした。溶媒を除去し、生成物をフラッシュクロマトグラフィーによって精製して、アルデヒド7(0.5g、収率54%)を生じた。
【0210】
【化27】
【0211】
ウィティッヒ塩5−12の調製:
乾燥アセトニトリル(50mL)中の6−ブロモヘキセン酸(3g、0.0512モル)およびトリフェニルホスフィン4.8g、0.018モル)の溶液を20〜24時間還流し、過剰な溶媒を減圧下で除去して、無色の油を得て、これを乾燥ベンゼンで粉末状にし、乾燥ベンゼンおよびエーテルの連続(3回ずつ)で洗浄した。洗浄手順の間に、減圧下で乾燥する結晶化した物質から、白色の微結晶性粉末としてウィティッヒ塩が提供された(3.6g、55%収率)。
【0212】
2,4−置換(1,3−ジオキサン−5−イル)カルボン酸(SN37、表II)](ウィティッヒ生成物)5−13の調製:
乾燥THF(5mL)中の水素化ナトリウムの溶液(60%、0.364g、0.0152モル)を、撹拌された、乾燥THF(10mL)中のウィティッヒ塩8 5−12(0.95g、0.002モル)の懸濁液に、0℃で、N2雰囲気下で添加した。混合物を30分間撹拌した。次いで、アルデヒド7 5−11の溶液(0.5g、0.0019モル)を添加した。反応を、撹拌しながら36時間継続させた。反応の完了後に、水を添加し、溶媒を減圧下で除去した。水溶液を酢酸エチルで洗浄し、5%HCLでpH2まで酸性化し、酢酸エチルで抽出した。合わせた有機性抽出物を飽和塩水で洗浄し、無水Na2SO4で乾燥させ、蒸発させた。得られた油をフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル−75:25)によって精製して、ウィティッヒ生成物9 5−13(0.2g、35%収率)を得た。
【0213】
【化28】
【0214】
実施例15
PPAR調節因子36(SN36、表II)の合成
この実施例は、表IIのPPAR調節因子36(2,2−ジメチル−4−(2−ヒドロキシフェニル)−1,3−ジオキサン−5−イル−カルボン酸)(5−15)の合成(メトキシ基の脱保護)を説明する:
エタンチオール(0.13g、0.00203モル)を、0℃、N2雰囲気下で、撹拌された、DMPU(5mL)中の水素化ナトリウム(60%、1g、0.004167モル)の懸濁液に添加した。30分後に、DMPU中の化合物5−13(0.00055モル)の溶液を添加した。混合物を120℃で2〜3時間加熱し、冷却し、氷水に注ぎ、水性混合物をDCMで洗浄した。5%HClで水層をpH5まで酸性化し、酢酸エチルで抽出した。合わせた有機性抽出物を飽和塩水で洗浄し、乾燥させ、蒸発させた。得られた油をフラッシュカラムクロマトグラフィー(ヘキサン:酢酸エチル−75:25)によって精製して、ウィティッヒ生成物5−15(0.6g、66%収率)を得た。
【0215】
【化29】
【0216】
実施例16
PPAR調節因子35(SN35、表II)の合成
この実施例は、表IIのPPAR調節因子35([2,4,5−シス]−2−o−クロロフェニル−4−o−メトキシフェニル−1,3−ジオキサン5−イル)オクテン酸−(スキーム5の5−20)の合成を説明する。
【0217】
2−クロロベンズアルデヒド(50.48mg、0.359mmol)、p−TSA(触媒の、5mg)およびアセトニド化合物5−15(130mg、0.359mmol)の混合物を、4mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−20(55mg、34.3%収率)を得た。
【0218】
【化30】
【0219】
実施例17
PPAR調節因子38(SN38、表II)の合成
この実施例は、表IIのPPAR調節因子38([2,4,5,−シス]−2−o−クロロフェニル−4−o−メトキシフェニル−1,3−ジオキサン5−イル)オクタン酸(5−21)の合成を説明する:
5mLの乾燥酢酸エチル中のオレフィン化合物5−13(150mg、0.414mmol)の溶液に、10モル%の10%Pd−C(44mg)を注意深く添加した。反応混合物をH2雰囲気下で1.5〜2時間撹拌させた。反応が完了した後に、混合物をセライトおよびケーキ(Pd−C)を通してろ過し、乾燥酢酸エチル(2×5mL)で洗浄した。合わせたろ液を濃縮し、残りの液体をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−21(SN38)(80mg、53.03%収率)を得た。
【0220】
【化31】
【0221】
2−クロロベンズアルデヒド(34.25mg、0.247mmol)、p−TSA(触媒の、3mg)およびアセトニド化合物12 5−21(80mg、0.34mmol)の混合物を、3mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物13 5−22(50mg、45.09%収率)を得た。
【0222】
【化32】
【0223】
LC−MS:純度87.53%および質量464(M+18)。
(実施例18)
【0224】
PPAR調節因子24(SN24、表II)の合成
この実施例は、表IIfのPPAR調節因子24([2,4,5−シス]−2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン5−イル)オクテン酸(5−17)の合成を説明する。
【0225】
2−クロロベンズアルデヒド(48.47mg、0.344mmol)、p−TSA(触媒の、5mg)およびアセトニド化合物(120mg、0.344mmol)の混合物を、4mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−17(40mg、27%収率)を得た。
【0226】
【化33】
【0227】
LC−MS:純度は94.43%(2つのジアステレオマー、それぞれ80.12+14.31)および質量448(M+18)
実施例19
PPAR調節因子31(SN31、表II)の合成
この実施例は、表IIのPPAR調節因子31([2,4,5−シス]−2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン5−イル)オクタン酸の合成を説明する。(5−19)
3mLの乾燥酢酸エチル中のオレフィン5−15(60mg、0.172mmol)の溶液に、10モル%(19mg、0.0172mmol)の10%Pd−Cを添加した。反応混合物をH2雰囲気下で8時間撹拌させた。反応が完了した後に、セライトおよびケーキ(Pd−C)を通して混合物をろ過し、乾燥酢酸エチル(2×5mL)で洗浄した。合わせたろ液を濃縮し、残りの液体をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物5−18(40mg、66.29%収率)を得た。
【0228】
【化34】
【0229】
2−クロロベンズアルデヒド(16mg、0.114mmol)、p−TSA(触媒の、3mg)およびアセトニド化合物15 5−18(40mg、0.114mmol)の混合物を、3mLの乾燥トルエン中で24時間撹拌した。溶媒を減圧下で蒸発させ、混合物をカラムクロマトグラフィー(ヘキサン−EtOAc:80:20)に供して、生成物16 5−19(20mg、40.28%収率)を得た。
【0230】
【化35】
【0231】
LC−MS:純度85%および質量450(M+18)
スキーム6
【0232】
【化36】
【0233】
実施例20
PPAR調節因子32(SN32、表II)の合成
この実施例は、表IIのPPAR調節因子32の合成を説明する。
【0234】
乾燥THF(10mL)中のアルケン6−10(前述の5−10)(1.2g、4.5mmol)の溶液に、BH3.DMS(0.34g、4.5)を0℃で添加し、反応混合物を同じ温度で2時間撹拌した。混合物に、3N NaOH(3mL)および30%H2O2(1mL)を0℃で添加し、室温でさらに1時間撹拌し続けた。反応混合物を水で希釈し、酢酸エチル(2×30mL)で抽出した。合わせた有機層を減圧下で蒸発させ、粗生成物をカラムクロマトグラフィー(酢酸エチル:ヘキサン、2:8)によって精製した。
【0235】
収量:0.9g(70%)
【0236】
【化37】
【0237】
6−12の調製:IBX(0.525g、1.87mmol)を乾燥DMSO(1mL)中に溶解させ、室温で10分間撹拌し、0℃に冷却した。これに、アルコール6(0.35g、1.25mmol)を乾燥THF(9mL)中、窒素雰囲気下で添加し、室温で1.5時間撹拌した。反応混合物をエーテル(10mL)で希釈し、20分間撹拌し、ろ過した。ろ液を水(2×10mL)で洗浄し、エーテル層を乾燥させ(Na2SO4)、ロータリーエバポレーターで濃縮した。生成物をフィルターカラムによって精製して、アルデヒド6−12を得た(酢酸エチル:ヘキサン、2:8)。
【0238】
収量:300mg(86.4%収率)。
【0239】
【化38】
【0240】
6−14の調製:(3−カルボキシプロピル)トリフェニルホスホニウムブロマイドを真空下、100℃で2〜3時間乾燥させた。撹拌された、乾燥トルエン(10mL)中の(3−カルボキシプロピル)トリフェニルホスホニウムブロマイド(1.47g、3.43mmol)の溶液に、カリウムter−ブトキシド(0.774g、6.89mmol)を一部ずつ、室温で、不活性な条件下で添加した。混合物を80℃に加熱し、温度を30〜40分間維持した。反応塊を50℃に冷却し、乾燥THF(2mL)中のアルデヒド(化合物6−12)(0.3g、1.07mmol)を上述の混合物に滴下した。反応の進行をTLCによってモニタリングした。反応の完了後に、塊を0〜5℃に冷却し、pH4〜5に調節することによって、1N HCl(1mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、純粋な化合物6−14を得た(酢酸エチル:ヘキサン、2:8)。
【0241】
収量:325mg(86%)。
【0242】
【化39】
【0243】
質量:371.1(M++Na)。
【0244】
6−16の調製:エタンチオール(0.44g、7.17mmol)を、撹拌された、乾燥DMF(10mL)中の60%NaH(0.287g、7.17mmol)の懸濁液に0〜5℃で添加し、20〜30分間撹拌した。乾燥DMF(2mL)中の化合物8 6−14(0.25g、0.71mmol)を、温度を維持しながら上述の混合物にゆっくり滴下した。反応塊温度を120〜130℃に上昇させ、6〜8時間維持した。反応の完了後に、塊を0〜5℃に冷却し、pH4〜5に調節することによって、1N HCl(1mL)で反応停止させた。化合物を酢酸エチル(3×10mL)で抽出し、水層を分離した。合わせた有機性画分を集め、塩水で洗浄し、硫酸ナトリウム(2g)で乾燥させ、真空で濃縮した。粗生成物をカラムクロマトグラフィーによって精製して、純粋な6−16を得た(酢酸エチル:ヘキサン、2.5:7.5)。
【0245】
収量:155mg(65%)。
【0246】
化合物6−16(130mg、0.38mmol)をTHF(8mL)中に溶解させ、触媒量のpTsOHを室温で添加した。反応混合物を室温で6〜8時間撹拌し続けた。o−クロロベンズアルデヒド(109mg、0.77mmol)を反応塊に添加し、もう1回触媒量のpTsOHを添加した。反応条件をさらに5〜6時間維持した。反応の完了後に、pH=7に調節することによって、乾燥Et3Nを添加した。溶媒を真空下で除去し、得られた粗混合物をカラムクロマトグラフィーによって精製して、最終生成物SN32(表II)6−19を得た。(酢酸エチル:ヘキサン、3.5:6.5)。
【0247】
収量:65mg(40%)。
【0248】
カラムクロマトグラフィーの後のHPLC純度は、より近い不純物を有して、80.3%であった。分取HPLCによってこれをさらに精製して、92.4%HPLC純粋化合物(20mg得られた)を得た。
【0249】
【化40】
【0250】
質量:415.1(M+−H)
HPLC純度:92.48%(カラム:水 ノバパック3.9×300mm、移動相:70%CH3CN+30%酢酸アンモニウムバッファー、RT:2.472)。
【0251】
さらなる類似体を、以下のようにして合成した:化合物6−14(90mg、0.25mmol)を、THF:0.2N HCl(10mL、9:1)の混合物中に溶解させ、室温で2時間撹拌した。反応の完了後に、混合物を酢酸エチル(2×10mL)で抽出し、Na2SO4で乾燥させ、ロータリーエバポレーターで蒸発させて、68mgの粗生成物を得て、これを、精製なしでさらなる反応のために利用した。
【0252】
上述の粗化合物(68mg、0.22mmol)を、乾燥THF中に溶解させ、この混合物に2−クロロベンズアルデヒド(61mg、0.44mmol)および触媒量のpTSA(約4mg)を室温で添加した。反応混合物を、窒素雰囲気下で、同じ温度で5時間撹拌した。出発物質が消えた後に、混合物を乾燥トリエチルアミンで中和し(pH=7に調節することによって)、溶媒を蒸発させた。粗生成物をカラムクロマトグラフィー(ヘキサン中の25%酢酸エチル)によって精製して、化合物11を得た。
【0253】
収量:32mg(34%)。
【0254】
【化41】
【0255】
質量:429.1(M+−H)
HPLC純度:97.84%(カラム:水 ノバパック3.9×300mm、移動相:70%CH3CN+30%酢酸アンモニウムバッファー(0.05%AcOH、RT:7.428)。
【0256】
実施例21
PPARγトランス活性化アッセイ
細胞培養物、プラスミドおよびトランスフェクション
この実施例は、PPARγ調節活性を判定するためのトランス活性化アッセイの例である。
【0257】
これらのアッセイにおいて、Gal4−PPARγLBD(Helledieら2000年)、UASx4−TK luc(ChenおよびEvans、1995年)およびCMV−β−ガラクトシダーゼ(市販されている、例えばClontech)を用いて、PPARγトランス活性化を示した。UASx4−TK−luc受容体構築物(ここでUASは「上流活性化因子配列」をいう)は、4つのGal4−応答エレメントを含む。プラスミドGal4−PPARγLBDは、UASに結合することによってUASx4−TK−lucレポータープラスミドをトランス活性化することができるGal4−DBD−PPARγ−LBD融合タンパク質(すなわち、PPARγのリガンド結合ドメイン、LBDに融合したGal4のDNA結合ドメイン、DBD)をコードする。CMV−β−ガラクトシダーゼプラスミド(ここでCMVはサイトメガロウイルスである)は、実験値の正規化のために使用される。
【0258】
マウス胎仔線維芽細胞(MEF)を、7.5%AmniomaxサプリメントC−100(Gibco)、7.5%ウシ胎仔血清(FBS)、2mMグルタミン、62.5μg/mlペニシリンおよび100μg/mlストレプトマイシンを補充したAmnioMax基本培地(Gibco)(増殖培地)中で増殖させた。あるいは、ME3細胞(Hansenら、1999年)を、10%仔ウシ血清(CS)、62.5μg/mlペニシリンおよび100μg/mlストレプトマイシンを補充したDMEM(増殖培地)中で増殖させた。トランスフェクションの時間に細胞が50〜70%コンフルエントになっているように、細胞を、典型的には24ウェルプレートに、再び播種した。
【0259】
製造業者の使用説明書に従ってLipofectamine Plus(Invitrogen)またはMetaffectane(Biontex)を用いて、細胞をGal4−PPARγLBD(Helledieら2000年)、UASx4−TK luc(ChenおよびEvans、1995年)およびCMV−β−ガラクトシダーゼ(市販されている、例えばClontech)でトランスフェクトした。簡潔にいうと、24ウェルプレートのウェルあたりに、30μLのDMEM(血清および抗生物質を含まない)中のUASx4TKluc(0.2μg)Gal4−PPARγLBD(またはpM−hPPARγ−LBD、0.1μg)およびCMV−β−ガラクトシダーゼ(0.05μg)を、1μLのメタフェクテネインを含む30μLのDMEM(血清および抗生物質を含まない)と混合した。混合物を室温で20分間インキュベートして、核酸−脂質複合体の形成を可能にし、次いで、およそ60mLを、50〜60%コンフルエント細胞を含む各ウェルに添加した。次いで、細胞をCO2インキュベータ内で、37℃で6〜12時間インキュベートし、次いで、培地を、抗生物質および目的の物質(例えば、DMSO中に溶解させた、本明細書中でDPDとも呼ばれる4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸、または陽性対照としてのロシグリタゾン(アバンディア))を補充した培地、または匹敵する体積のDMSO(全細胞培養物体積の<0.5%)で置換した。DPDは市販されているか、または、実施例1に従って合成することができる。12〜24時間後に細胞を採取し、標準的なプロトコルに従って、ルシフェラーゼおよびβ−ガラクトシダーゼ活性を測定した。
【0260】
PPARトランス活性化は、DMSO単独より、ロシグリタゾン(公知のPPARγアゴニスト)で40倍より大きく高く、10μM 4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸(図2中の「DPD」参照)で約10倍高かった。したがって、4−(Z)−6−(2−o−クロロフェニル−4−o−ヒドロキシフェニル−1,3−ジオキサン−シス−5−イル)ヘキセン酸は、PPARγアゴニストである。
【0261】
キラルHPLCによって精製されたDPD鏡像異性体のいずれかの間に、PPARγトランス活性化活性に違いは見られなかった。
【0262】
表IIは、DPD(SN1)の種々の類似体で得られた結果を要約し、それらの活性を比較している。「P」は、10μMのDPD(表II中のSN1)と比較して、10μMでの類似したPPARγ活性化活性を表し、「P−」は、10μMのDPDと比較して、30μMでの試験物質での、同じレベルの活性(すなわち、より能力が低い)を表し、「P+」は、「P」より大きい活性のレベル、および10μMのDPDに匹敵する3μMでの活性のレベル(すなわち、より能力が高い)を表し、「P++」は、3μMでは10μMのDPDのものよりも高いが1μMでは10μMのDPDのものより低い、活性のレベルを表し、「P+++」は、10μMのDPDに匹敵する、1μMでの活性のレベルを表し、「−」は、試験が行われなかったことを意味する。
【0263】
このアッセイが、本質的に上述の通りだが、PPARγリガンド結合ドメインでなく全長ヒトPPARγで行われる場合、全長hPPARg2の活性化は64%までのアバンディア陽性対照で見られる(図3)。
【0264】
実施例22
PPARδトランス活性化アッセイ
この実施例は、PPARδ調節活性を判定するためのトランス活性化アッセイを説明する。
【0265】
DPDおよび他のリガンドを、それらがPPARδを本質的に実施例1に記載されているようにトランス活性化する能力について試験する。しかしながら、トランス活性化構築物は、mPPARδLBDであり、ここでPPARδリガンド結合ドメインがPPARγのものに置き換わり、L165041(市販されている)がロシグリタゾンの代わりの選択的PPARδアゴニストとして使用される。用いられた条件下で、L165041はPPARδトランス活性化を50倍より大きく増大させることが示されたが、DPDはトランス活性化の増大を生じないか、わずかしか生じなかった(図2参照)。したがって、DPDはPPARγに対する選択性を示す。
【0266】
このアッセイが、本質的に上述の通りだが、PPARδリガンド結合ドメインでなく全長ヒトPPARδで行われる場合、全長hPPARδの活性化、活性化は見られず(図4)、PPARγに対するDPDの選択性が確認された。
【0267】
実施例23
PPARαトランス活性化アッセイ
この実施例は、PPARα調節活性を判定するためのトランス活性化アッセイを説明する。
【0268】
DPDおよび他のリガンドを、それらがPPARαをトランス活性化する能力について、本質的に実施例1に記載されているように試験する。しかしながら、トランス活性化構築物はmPPARαLBDであり、ここでPPARαリガンド結合ドメインがPPARγのものに置き換わり、GW7647(市販されている)をロシグリタゾンの代わりの選択的PPARαアゴニストとして使用する。用いられた条件下で、GW7647は2倍より大きくPPARαトランス活性化を増大させることが示されたが、DPDはトランス活性化を生じなかった(図2参照)。したがって、DPDはPPARγに対する選択性を示す。
【0269】
実施例24
RXRトランス活性化アッセイ
この実施例は、レチノイン酸X受容体トランス活性化アッセイを説明する。
【0270】
化合物を、それらがRXRをトランス活性化する能力について、本質的に実施例1に記載されているように試験する。しかしながら、トランス活性化構築物はhRXRαLBDであり、ここでRXRリガンド結合ドメインがPPARγのものに置き換わり、{4−[1−(3,5,5,8,8−ペンタメチル−5,6,7,8−テトラヒドロ−2−ナフチル)エテニル]安息香酸}(LG1069、市販されている)を、ロシグリタゾンの代わりの選択的RXRアゴニストとして使用する。用いられる条件下で、LG1069は5倍より大きくRXRトランス活性化を増大させることが示されるが、DPDはトランス活性化を生じない(図2参照)。したがって、DPDは、ここでもPPARγに対する選択性を示す。
【0271】
実施例25
脂肪細胞分化アッセイ
この実施例は、脂肪細胞分化を判定するためのアッセイを説明する。化合物を、それらが脂肪細胞分化を誘導するかどうかを調べるため、およびそれらが脂肪細胞分化を阻害するかどうかを調べるために、試験する。
【0272】
細胞培養物および分化
MEFを、7.5%AmniomaxサプリメントC−100(Gibco)、7.5%ウシ胎仔血清(FBS)、2mMグルタミン、62.5μg/mlペニシリンおよび100μg/mlストレプトマイシンを補充したAmnioMax基本培地(Gibco)(増殖培地)中で増殖させた。コンフルエンスで、MEFを、DMSO中に溶解させた1μMのデキサメタゾン(Sigma)0.5mMのイソブチルメチルキサンチン(Sigma)、5μg/mlのインスリン(Sigma)および10μMの試験化合物を添加した増殖培地中、またはDMSO単独の中で、分化に誘導した。続いて、培地に48時間ごとに、5μg/mlのインスリンおよびリガンドを補充した増殖培地またはDMSOを新たに供給した。
【0273】
簡潔にいうと、脂肪細胞分化の誘導因子として化合物を試験するために、典型的には24ウェルディッシュ中の、10%仔ウシ血清(CS)を含むDMEM中で、3T3−L1をコンフルエンスまで増殖させる。コンフルエンス後2日目(0日目)に、10%ウシ胎仔血清(FBS)、1μMのデキサメタゾンおよび試験化合物(01、1および10μM)を補充したDMEMで、細胞を分化するよう誘導する。BRL49653(100%Me2SO中に溶解させた1.0μM)を陽性対照として使用する。48時間後に、細胞に、試験化合物または陽性対照を補充した、10%FBSを含むDMEMを再び供給する。4日目から、細胞を、10%FBSを含むDMEM中で増殖させ、8日目まで、2日目ごとに交換する。8日目に、後述されるように、細胞をOil Red Oで染色する。
【0274】
脂肪細胞分化の阻害剤として化合物を試験するために、コンフルエンス後2日目(0日目と名付けられる)まで、3T3−L1細胞を上述のように増殖させ、その後、10%ウシ胎仔血清(FBS)、1μMデキサメタゾン(Sigma)、0.5mMメチルイソブチルキサンチン(Sigma)、1μg/mlインスリン(Roche Molecular Biochemicals)および試験化合物を含むDMEMで、細胞を分化するよう誘導する。試験化合物の溶媒の存在下で分化するよう誘導された細胞を、陽性対照として使用する。48時間後に、細胞に、試験化合物または陽性対照を補充した、10%FBSを含むDMEMを新たに供給した。4日目から、細胞を、10%FBSを含むDMEM中で増殖させ、8日目まで、2日目ごとに交換する。8日目に、後述されるように、細胞をOil Red Oで染色する。
【0275】
Oil Red O染色
上述のように培養された細胞を、Oil Red染色のために使用する。PBS中でディッシュを洗浄し、細胞を3.7%パラホルムアルデヒド中で1時間固定し、(Hansenら1999年)に記載されているようにoil red Oで染色した。0.5gのOil Red O(Sigma)を100mlのイソプロパノール中に溶解させることによって、Oil Red O溶液ストック溶液を調製する。ストック溶液を水で希釈し(6:4)、続いてろ過することによって、Oil red O希釈標準溶液を調製する。
【0276】
DPDは、誘導アッセイにおいて、非常に少ない赤色染色しか誘導しなかった。赤色染色は脂肪細胞の存在の指標であるので、DPDが脂肪細胞分化を誘導しないことが推測できる。しかしながら、DPDは、脂肪細胞分化の阻害において有意な効果を示さなかったので、脂肪細胞を阻害しない。表IIは、DPD(SN1)の種々の類似体で得られた結果を要約し、「0」はDPDと類似した結果を表し、「−1」はさらに低い脂肪細胞分化を表し、「+1」はより大きい脂肪細胞分化を表し(ともにDPDと比較して)、「−」はアッセイされなかったことを表す。
【0277】
本質的に上述の通りだがヒト前脂肪細胞を用いて行われたアッセイからも、DMSOと同様に、DPDが非常に少ない赤色染色しか誘導しなかったことが実証された。
【0278】
実施例26
部分アゴニスト対完全アゴニストの同定
この実施例は、部分PPARアゴニストを判定するためのアッセイを説明し、これは医薬品として特に望ましい。
【0279】
簡潔にいうと、トランス活性化アッセイを、本質的に実施例1に記載されている通りだが100nMアバンディア(完全アゴニスト)を、増大する濃度の試験化合物(または対照としての試験化合物なし)とともに、各ウェルに添加して行う。アバンディアによってトランス活性化が減少する化合物は、PPAR部分アゴニストである。結果を図5に示す。
【0280】
PPARγ部分アゴニストを、それらがPPARγ、例えばこの場合アバンディアに結合することから、公知のPPARγアゴニストに置き換わる能力によって、この実施例中で同定する。あるいは、他の公知の完全アゴニスト、例えば300nM L165041を用いて、本質的に実施例2に記載されている通りにトランス活性化アッセイを用い、PPARδの部分アゴニストを同定することができる。
【0281】
実施例27
部分PPARγアゴニストリガンド置換アッセイ
以下のように、PPARγリガンド結合ポケットへの結合の分析のために、Invitrogen POLARSCREEN PPAR競合アッセイを用いて、部分PPARγアゴニストを同定するためのさらなるアッセイを行う。
1.20μLの2X試験化合物をキュベットに分注する
2.20μlの2XPPAR−LBD/Fluormone Green Complexを添加して混合する
3.暗黒中で2時間インキュベートする
4.蛍光偏光度を測定する
5.対照として、ロシグリタゾン(アバンディア)を使用する
AVANDIAと比較したDPDでの結果を図6に示す。
【0282】
実施例28
グルコース取り込みアッセイ
この実施例は、PPARγアゴニストとして同定された化合物が、PPARγアゴニストに期待される、細胞アッセイにおける生理学的効果、特にグルコース取り込みに対する効果も生じることを説明する。グルコース取り込みアッセイは、インスリン抵抗性の治療のための化合物の適合性を確立するために重要である。
【0283】
簡潔にいうと、3T3−L1前脂肪細胞を、コンフルエンスまで12ウェルプレート中で増殖させる。細胞を、血清不含DMEMで洗浄し、1mlの同じ培地とともに37℃で1〜2時間インキュベートする。次いで、細胞をクレブス・リンガー−Hepes(KRP)バッファーで洗浄し、0.9mlのKRPバッファーとともに37℃で30分間インキュベートする。0、0.3、1および3nMの終濃度でインスリンを添加し、37℃で15分間インキュベートする。10mM[3H]2−デオキシ−D−グルコース(1mCi/l)を補充した0.1mlのクレブス・リンガーリン酸(KRP)バッファーの添加によって、グルコース取り込みを開始する。37℃で10分間のインキュベーションの後に、培地を吸引し、プレートを氷冷PBSで洗浄して、誘導されたグルコース取り込みと終了させる。0.5mlの1%Triton X−100で細胞を溶解させ、シンチレーションカウンターを用いて放射活性レベルを測定する。結果を図7に示す。
【0284】
要約すると、DPDおよび表IIで言及される多数の類似体は、PPARγアゴニストであると実証されるが、物質10はPPARγアンタゴニストであるようである。物質7および13は、脂肪細胞分化をあまり引き起こさないか、引き起こさずに、より高い可能性を示すので、特に興味深い類似体である。
【0285】
本明細書中で言及される全ての刊行物および特許出願は、個々の刊行物または特許出願が、特に、および個々に参照によって援用されていると示されるのと同じ程度に、参照によって本明細書中に援用される。本発明はここで完全に説明されているが、添付の特許請求の範囲の精神または範囲から逸脱することなく、これに多くの変化および変更を行うことができることが、当業者に明らかであろう。
【0286】
【化42】
【0287】
【化43】
【0288】
【化44】
【0289】
【化45】
【0290】
【化46】
【0291】
実施例29
トロンボキサン受容体活性
この実施例は、物質1の両方の鏡像異性体がPPARアゴニスト活性を有するが、一方のみがトロンボキサン受容体アンタゴニストとして作用することを実証する。
【0292】
以下の条件下で、キラルクロマトグラフィーによって、DPD(表II中のSN1)の各鏡像異性体を単離した:
カラム:250×4.6mm Chiralpak AD−H5 □m
移動相:80/20/0.1 n−ヘプタン/エタノール/トリフルオロ酢酸
流速:1ml/分
検出:230nmでのUV
温度:25C
試料を80/20 n−ヘプタン/エタノール中に溶解させた。
【0293】
鏡像異性体1が最初にキラルカラムで溶出し、鏡像異性体2が2番目にキラルカラム上で溶出した。
【0294】
本質的にHedbergら(1988年)J Pharmacol.Exp.Ther.245:786−792およびSaussyら(1986年)J.Biol.Chem.261:3025−3029によって記載されているように、トロンボキサン受容体結合についての放射性リガンド結合アッセイで、鏡像異性体1および2を試験した。鏡像異性体1は潜在的なトロンボキサン受容体結合剤である(IC50、0.841nM)とわかったが、鏡像異性体2は結合しないようであった(IC50>10nM)。
【0295】
トロンボキサン受容体アンタゴニストとして作用する鏡像異性体で、ある種の患者集団を治療することが望ましい場合があり、この場合、鏡像異性体1が投与される(例えば、トロンボキサン受容体アンタゴニストの心血管の利益が同時に望ましい場合)。トロンボキサン受容体に対する効果を有さないことが望ましい場合(例えば、患者がかかる治療を必要としない場合、または患者がすでに異なる治療を受けている場合等の、さらなる心血管効果が必要でない場合)、鏡像異性体2が有益に投与される。
【0296】
実施例30
癌細胞増殖の阻害
この実施例は、癌細胞の増殖に対するDPDの抗増殖性効果を実証し、癌の治療のための、本明細書中に記載されている類似体の有用性を説明する。
【0297】
組換えバイオセンサーレポーター活性を安定して発現するよう操作されたヒト子宮頸癌細胞系(HeLa)および非癌性細胞系、HaCaT(Boukampら、1988年、J.Cell Biol 106(3):761−771)を、Caspa Tagキット(例えばChemiconから、市販されている)とともに用いた。ハイコンテンツスクリーニング自動フローサイトメーターを用いて、増殖の検出を行った。
【0298】
1%DMSOに調節された、血清のない細胞培地中で、DPD(物質1)を20μMまで希釈した。HTSフローサイトメーター(FACS Calibur HTS、Becton Dickenson)のFL2(増殖マーカーの希釈)チャネル中の処理の48時間後に、96ウェルプレート中、2連で、細胞事象の検出を行った。1日目に、細胞を96ウェルプレートに播種した。2日目に細胞をDPDおよび適切な対照で処理し、4日目に分析を行った。
【0299】
DPDは、20μMで、HeLa細胞の増殖を90%阻害した。DPDは、非癌性細胞系HaCaTに効果を有さず、したがって、DPD(表II中のSN1)による癌細胞に対する選択的抗増殖性効果が確立された。
【特許請求の範囲】
【請求項1】
本明細書に記載の発明。
【請求項1】
本明細書に記載の発明。
【図1】

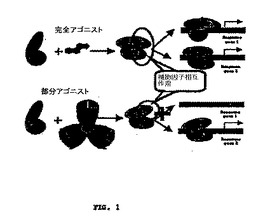
【図2】
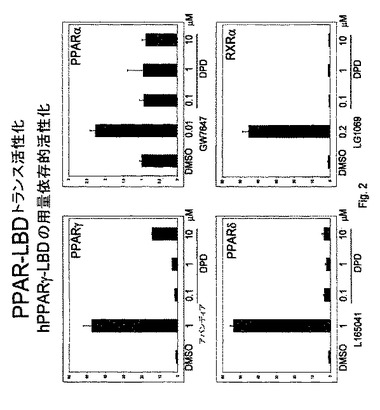
【図3】
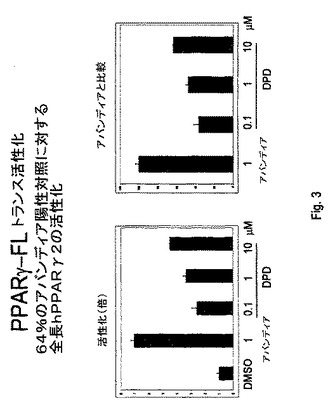
【図4】
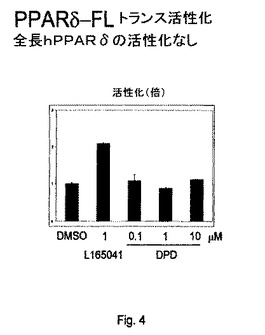
【図5】
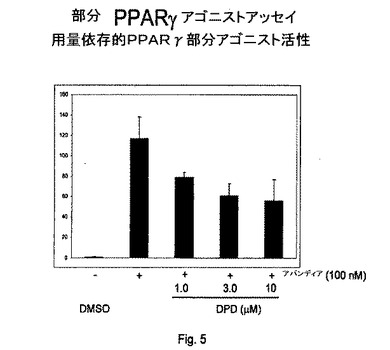
【図6】
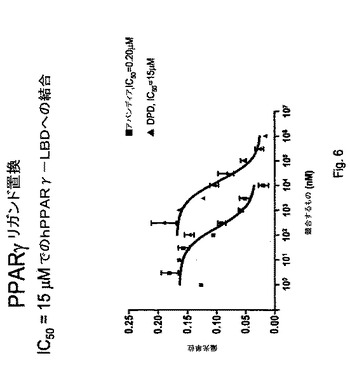
【図7】
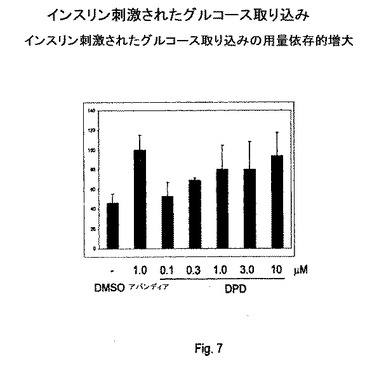
【図8】
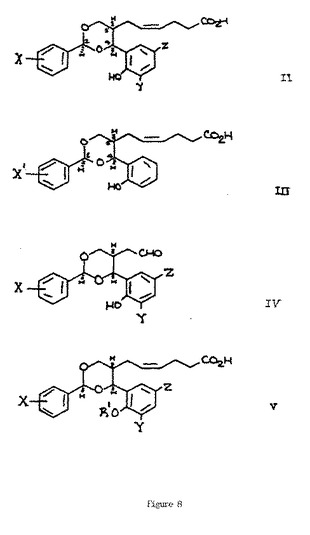
【図9】
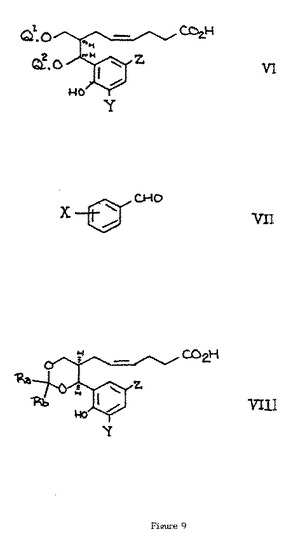
【図10】
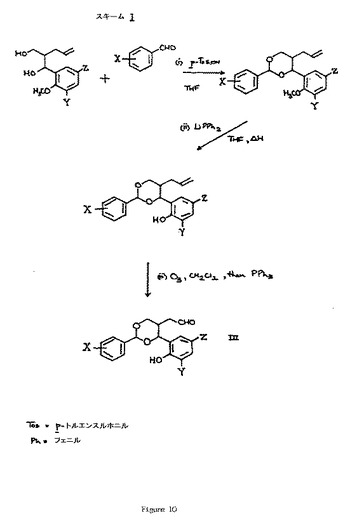
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2013−60465(P2013−60465A)
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−286467(P2012−286467)
【出願日】平成24年12月28日(2012.12.28)
【分割の表示】特願2008−550880(P2008−550880)の分割
【原出願日】平成19年1月18日(2007.1.18)
【出願人】(508217685)
【Fターム(参考)】
【公開日】平成25年4月4日(2013.4.4)
【国際特許分類】
【出願番号】特願2012−286467(P2012−286467)
【出願日】平成24年12月28日(2012.12.28)
【分割の表示】特願2008−550880(P2008−550880)の分割
【原出願日】平成19年1月18日(2007.1.18)
【出願人】(508217685)
【Fターム(参考)】
[ Back to top ]