
PPR−γ受容体の活性化による自己免疫疾患およびアルツハイマー病の治療に有用なAndrographispaniculataから抽出されたラブダンジテルペン類の組成物
Andrographis paniculataからの貴重な植物種の乾燥薬草から特殊な抽出法により得られるジテルペンラブダン類と同定された分子から構成される、ヒトおよび動物の自己免疫その他の免疫学的疾患の治療のための、低毒性および重大な副作用のないことを特徴とする組成物。同様に、この新規な組成物はアルツハイマー病の予防および治療のために有用な医薬組成物を調製するためにも有用である。本組成物はPPARγ受容体を活性化してNFκBを減らす結果、炎症誘発性サイトカインの合成を阻止する。それ自身の治療特性に加えて、この新規な組成物は、重要な毒性レベルを有しかつ/または望ましくない副作用を引き起こす現在の製品、薬および処置の不便を解決する。本発明は、免疫薬理学、臨床および前臨床のヒトおよび獣医科の薬理学、製薬技術、リウマチ学および臨床免疫学、免疫療法、臓器および組織移植、ならびにエイズのような免疫不全疾患に用途を有する。
【発明の詳細な説明】
【発明の詳細な説明】
【0001】
説明
免疫薬理学および生物薬剤学の主な目的は、免疫疾患の症状を治療しその経過を修飾する新規な治療解決策を継続的に探索することである。
【0002】
発明の背景
自己免疫疾患は、それ自身の生体に対する免疫系の自発的反応を特徴とする。これらの反応は、液性(自己抗体生産)および細胞性(リンパ球およびマクロファージの細胞毒性活性の増加)の免疫反応を起動させる役割を担うTリンパ球による自己抗原の認識により生じる。自己免疫疾患には、リウマチ様疾患、乾癬、全身の皮膚筋炎、多発性硬化症、紅斑性狼瘡(lupus erythematous)、または抗原により悪化される免疫応答、即ち喘息、薬剤および食物に対するアレルギー、その他が含まれる。全てのこれらの疾患は制限的、慢性的であり、場合によっては致死性であるが、それらを治療する有効な治療法は現在存在しない。したがって、疾患の経過において寛解または減少をもたらすことができるあらゆる薬剤、医薬または媒体は、患者の健康のための重要な解決策となる。
【0003】
自己免疫疾患のための治療法の探索は、適切な薬剤および方法を見つける重要な努力をもたらした。
現在、これらの疾患の治療は、主に糖質コルチコイド、カルシニューリン阻害剤および抗増殖剤−代謝拮抗物質などの免疫抑制剤の使用に基づいている。しかし、これらの薬理学的療法は多くの異なる標的に作用するので、それらは免疫機能を全体として低下させるか、または、長期使用では異なる細胞傷害作用という不利を有する可能性があり、したがって、非特異的に免疫系を抑制して患者を感染症および癌のリスクにさらす可能性がある。カルシニューリンおよび糖質コルチコイドは、それらの腎毒性および糖尿病誘発作用のために、いくつかの臨床状態(例えば腎不全、糖尿病)でのそれらの有用性を制限するさらなる不利を示す。
【0004】
免疫抑制における最新の治療上の進歩は抗CD3モノクローナル抗体、即ち抗IL−2受容体モノクローナル抗体および抗TNFαモノクローナル抗体である。これらの治療法は著しい免疫抑制作用、アナフィラキシー反応、日和見感染(結核)および新生物を示すにもかかわらず、熱、じんま疹、低血圧症、呼吸困難がこれらの薬と関連し、前記組成物および医薬品の適用において深刻な問題となっている。注射用途の場合、患者3人中1人は痒み、腫脹および疼痛を示しうる。
【0005】
発明の簡単な要約
本発明で請求する組成物は、自己免疫疾患、アレルギーの特徴である免疫反応を減少させて疾患の症状および経過を緩和し、「免疫寛容」を維持することができる。
【0006】
言い換えると、本発明で開示される組成物は、それがもたらす免疫寛容によって本質的に特徴づけられ、これは、抗原に対する特異的な反応が欠如した活性状態に対応し、現行の免疫抑制薬剤の副作用を引き起こすことがない。
【0007】
具体的には、この組成物は、PPARγ受容体を刺激してNFκB因子を減らすことにより、インターフェロンγ、IL−2の合成および発現を阻害する。
したがってこのジテルペン性ラブダン類の新規な組成物は、自己免疫疾患の病因に関係するサイトカイン過剰発現を選択的に減少させることを特徴とする。
【0008】
急性および慢性炎症性疾患および癌に関係するメディエーターの科学的理解における最近の進歩は、有効な治療法の探索における新規な戦略をもたらした。伝統的な手法としては、特異的抗体、受容体アンタゴニストまたは酵素阻害剤の使用など、直接的標的介入があり、これらは全て重大レベルの副作用(例えばアレルギー、胃腸の潰瘍、出血、その他)を伴う。様々なメディエーターの転写および翻訳に関係する調節機構の解明が最近進展したことにより、遺伝子転写レベル(例えば、COX2、iNOS、IL1β、TNFα、ICAM、その他)に向けられた治療手段に対する関心が増加した。
【0009】
最も重要なメディエーターの1つは、Rel/NF−κBファミリーのポリペプチドの様々な組合せから構成される緊密に関連したダイマー転写因子複合体ファミリーに属している、NFκBである。このファミリーは、哺乳類の5つの個々の遺伝子産物である、RelA(p65)、NF−κB1(p50/p105)、NF−κB2(p49/p100)、c−RelおよびRelBからなり、その全てはヘテロダイマーまたはホモダイマーを形成することができる。これらのタンパク質は、DNA結合および二量体化ドメインを含有する相同の高い300アミノ酸の「Rel相同性ドメイン」を共有する。Rel相同性ドメインの最C末端には、細胞質から核へのNF−κBの輸送において重要な核転座配列がある。さらに、p65およびcRelは、それらのC末端に強力なトランス活性化ドメインを有する。
【0010】
NF−κBの活性は、阻害剤IκBファミリーのタンパク質のメンバーとの相互作用によって調節される。この相互作用は効果的にNF−κBタンパク質上の核移行配列を遮断することにより、核への二量体の移動を防止する。多種多様な刺激が、おそらくは複数のシグナル伝達経路を介して、NF−κBを活性化する。これには細菌生成物(LPS)、一部のウイルス(HIV−1、HTLV−1)、炎症性サイトカイン(TNFα、IL−1)および環境ストレスが含まれる。しかし、表面上全ての刺激に共通するのは、リン酸化および以降のIκBの分解である。IκBは、最近特定されたIκBキナーゼ(IKK−αおよびIKK−β)によって、2つのN末端セリン上でリン酸化される。部位特異的突然変異誘発研究によると、一度リン酸化されるとタンパク質はユビキチン−プロテアソーム経路による分解のために目印をつけられることから、これらのリン酸化はNF−κBの後の活性化のために重要であることが示されている。IκBから遊離すると、活性NF−κB複合体は核に移行して、そこで好ましい遺伝子特異的エンハンサー配列に選択的に結合することができる。NF−κBによって調節される遺伝子には、いくつかのサイトカイン、細胞接着分子および急性期タンパク質が含まれる。
【0011】
NF−κBが、IL−6およびIL−8などのサイトカイン、ICAMおよびVCAMなどの細胞接着分子、ならびに誘導性酸化窒素シンターゼ(iNOS)を含む多数の炎症誘発性メディエーターの調節された発現で重要な役割を果たすことは、周知である。そのようなメディエーターは炎症部位で白血球を補充する役をつとめることが知られており、iNOSの場合は、一部の炎症性疾患および自己免疫疾患では臓器破壊をもたらす可能性がある。
【0012】
炎症性障害におけるNF−κBの重要性は、NF−κBが活性化されることが明らかにされている喘息を含む気道炎症の研究においてさらに強調されている。この活性化は、これらの障害の特徴であるサイトカイン生産の増加および白血球浸潤の根底にあるかもしれない。さらに、吸入されたステロイドは喘息の気道で気道過敏症を減少させ、炎症性反応を抑えることが知られている。糖質コルチコイドのNF−κB阻害に関する最近の知見を考慮すると、これらの効果はNF−κBの阻害により媒介されると推測することができる。
【0013】
炎症性障害におけるNF−κBの役割のさらなる証拠は、リウマチの滑膜に関する研究からもたらされる。NF−κBは通常不活性の細胞質複合体として存在するが、近年の免疫組織化学的研究は、リウマチ性滑膜を含む細胞ではNF−κBが核に存在し、したがって活性であることを示している。さらに、NF−κBはTNFαによる刺激に応じてヒト滑膜細胞で活性化されることが示された。そのような分布は、この組織の特徴であるサイトカインおよびエイコサノイド産生の増加の根底にあるメカニズムであるかもしれない。Roshak、A.K.ら、J.Biol.Chem.、271、31496〜31501(1996)を参照。
【0014】
炎症性障害におけるNF−κBの役割のさらなる証拠は、リウマチの滑膜に関する研究からもたらされる。NF−κBは通常不活性の細胞質複合体として存在するが、近年の免疫組織化学的研究は、リウマチ性滑膜を含む細胞ではNF−κBが核に存在し、したがって活性であることを示している。さらに、NF−κBはTNFαによる刺激に応じてヒト滑膜細胞で活性化されることが示された。そのような分布は、この組織の特徴であるサイトカインおよびエイコサノイド産生の増加の根底にあるメカニズムであるかもしれない。Roshak、A.K.ら、J.Biol.Chem.、271、31496〜31501(1996)を参照。
【0015】
NF−κB/RelおよびIκBタンパク質も、悪性形質転換で重要な役割を果たしているようである。ファミリーメンバーは、過剰発現、遺伝子増幅、遺伝子再構成または転座の結果としてin vitroおよびin vivoでの細胞形質転換と関連している。さらに、これらのタンパク質をエンコードしている遺伝子の再構成および/または増幅が、ある種のヒトリンパ性腫瘍の20〜25%で見られる。さらに、アポトーシスの調節におけるNF−κBの役割が報告され、細胞増殖の制御におけるこの転写因子の役割を強化している。
【0016】
NF−κBの最初の植物由来のモジュレーターは、サリチル酸ナトリウムおよびその半合成誘導体のアスピリンを特定したKoppとGhosh(1994)によって、ほぼ10年前に報告された。この発見の後、異なる化学クラスのいくつかの新規な天然物が、NF−κB阻害活性を示した。
【0017】
いくつかのNF−κB阻害剤が文献:C.Wahlら、J.Clin.Invest.101(5)、1163〜1174(1998);R.W.Sullivanら、J.Med.Chem.41、413〜419(1998);J.W.Pierceら、J.Biol.Chem.272、21096〜21103(1997)に記載されている。海洋性天然物ヒメニアルジシン(hymenialdisine)は、NF−κBを阻害することが知られている:Roshak、Aら、JPET、283、955〜961(1997)。Breton、J.JおよびChabot−Fletcher、M.C.、JPET、282、459〜466(1997)。サリチルアニリドは公知の化合物であり、M.T.Clark、R.A.Coburn、R.T.Evans、R.J.Genco、J.Med.Chem.、1986、29、25〜29によって記載されている。
【0018】
最近、NF−κB阻害の重要なメカニズムにより、ペルオキシソーム受容体の活性化の可能性が示唆されている。
「ペルオキシソーム増殖因子活性化受容体」(PPAR)[この用語の通常の使用に注目するときには、科学の領域で公知のその略語で表し、当該略語はこの受容体を特定するために使用されるものとする]として知られるペルオキシソームの受容体は、自己免疫疾患および他の疾患、即ち糖尿病、心血管および胃腸の疾患ならびにアルツハイマー病との関係が示唆されている。PPARγアゴニストを含む現在の医薬剤はまだ実験段階であり、その作用機構のために人には深刻な副作用を起こす。したがって、上述の疾患または病態を予防、治療および/または軽減するために、より正確にこれらの受容体をモジュレートすることができるより毒性の低い新規な薬剤を開発する必要がある。
【0019】
この新規な組成物はこれらの受容体をより正確にモジュレートするので、患者に望ましくない副作用を引き起こすことなく、自己免疫疾患をより有効に予防、治療および/または軽減することを可能にする。
【0020】
ペルオキシソーム増殖因子活性化受容体(PPAR)は、遺伝子発現を調節するリガンド活性化転写因子である核ホルモン受容体スーパーファミリーのメンバーである。PPARの様々な亜型が発見されている。これらには、PPARα、PPARβまたはNUC1、PPARγおよびPPARδが含まれる。
【0021】
PPARγは、当初脂肪細胞分化および脂質代謝の重要な調節因子と特徴づけされた。PPARγの発現は異なる複数のプロモーターによって指示される、3つのPPARγアイソフォームが生じる。PPARγは繊維芽細胞、筋細胞、乳房細胞、ラット脾臓の白色および赤色脾髄、ヒト骨髄前駆体ならびにマクロファージ/単球を含む他の細胞型でも見られることが、現在明らかとなっている。さらに、PPARγはアテローム硬化型プラーク内のマクロファージ泡沫細胞で見られた。糖代謝におけるPPARγの重要な役割は、あるクラスの抗糖尿病薬であるチアゾリジンジオンが高親和性のPPARγリガンドであることが証明されたときに特定された。チアゾリジンジオンは、当初、インスリン抵抗性の齧歯類モデルでグルコースレベル(および循環脂肪酸のレベル)を下げるそれらの能力に基づき、2型糖尿病の治療のために開発された。チアゾリジンジオンはPPARγとの直接相互作用を通してそれらの治療効果を媒介するとの知見により、PPARγがグルコースおよび脂質のホメオスタシスの重要な調節因子であると立証された。当初は脂質およびグルコースの代謝の調節因子として記載されたにもかかわらず、近年、PPARγは細胞増殖および悪性腫瘍における役割を有することも立証された。PPARγのリガンドは、細胞増殖および悪性腫瘍に対する正および負の作用を媒介することが示された。
【0022】
チアゾリジンジオンクラスの抗糖尿病薬に加えて、様々な非ステロイド性抗炎症薬もPPARγリガンドとして機能することができるが、後者の親和性は比較的低い。
プロスタグランジンD2(PGD2)脱水物PGJ2は、最初に発見されたPPARγの内因性リガンドであった。別のPGD2脱水物15−デオキシ−Δ12,14−PGJ2(15d−PGJ2)も直接PPARγと結合する天然物質であり、PPARγ活性化のための強力なリガンドである。
【0023】
PPARおよびマクロファージを関連づける最も初期の知見の1つは、PPARγがヒトおよびマウスのアテローム硬化性病変のマクロファージ由来泡沫細胞で高度に発現されたことであった。その後、PPARγがヒトおよびマウスの単球/マクロファージで発現されることが証明された。機能的に、PPARγは、単球の分化および活性化で、また炎症性作用の調節において役割を果たすことが示された。
【0024】
多くの研究は、PPARγリガンドがマクロファージ炎症性応答を阻害することを証明している。PPARγ活性化の抗炎症効果は、ヒトおよびマウスの単球/マクロファージおよび単球/マクロファージ系で証明された。マクロファージの活性化は、通常いくつかの異なる炎症誘発性メディエーターの分泌をもたらす。15d−PGJ2またはチアゾリジンジオンによる治療は、これらのメディエーター(ゲラチナーゼB、IL−6、TNF−αおよびIL−1βを含む)の多くの分泌を阻止し、また誘導可能なNOS(iNOS)の誘導性発現およびスカベンジャー受容体A遺伝子の転写を減少させることが見出された。
【0025】
PPARγの関連性は、いくつかのヒト自己免疫疾患および自己免疫疾患の動物モデルで研究された。Kawahitoらは、慢性関節リウマチ(RA)患者で滑膜組織がPPARγを発現することを証明した。PPARγはマクロファージで高度に発現されることが発見され、中程度の発現が滑膜内層(synovial-lining)れた。15d−PGJ2およびトログリタゾンによるPPARγの活性化は、in vitroでRA滑膜細胞アポトーシスを誘導した。
【0026】
PPARγは新たに単離されたT細胞で機能的に関連しているか、活性化の初期に機能的に関連するようになることが示唆されている。これらの研究で、PPARγの2つのリガンドがT細胞クローンによるIL−2分泌の阻止を媒介し、そのようなクローンのIL−2誘導性増殖を阻止しないことが証明された。
【0027】
いくつかの研究は、自己免疫疾患の動物モデルを修飾する際のPPARγリガンドの役割を調査した。Suらは、炎症性腸疾患のマウスモデルにおいて、チアゾリジンジオンは結腸炎症を著しく減少させたことを示した。この効果は、PPARγを高度に発現して炎症性サイトカインを生産することができる結腸上皮細胞に及ぼす直接的効果の結果であるかもしれないと、提唱された。Kawahitoらは、PPARリガンド、15d−PGJ2およびトログリタゾンの腹腔内投与はアジュバント誘導性の関節炎を寛解させることを示した。Ninoらは実験的アレルギー性脳脊髄炎に及ぼすチアゾリジンジオンの効果を調べ、この療法が多発性硬化症のこのマウスモデルで炎症を弱め、臨床症状を減少させることを発見した。最後に、Reillyらは、腎臓糸球体メサンギウム細胞がループス腎炎の炎症性応答の重要なモジュレーターであり、活性化されるとNOおよびシクロオキシゲナーゼ生成物を含む炎症伝達物質を分泌することにより局所炎症性応答を永続させることを証明した。上記研究から、炎症性または自己免疫性病因を有する疾患におけるPPARの関連性およびPPARアゴニストによる治療の有用性は、おそらく研究の焦点であり続けるであろう。
【0028】
近年では、PPARγに対する15d−PGJ2の特異性の問題は、少なくとも部分的に解明された。NF−κBは、炎症および免疫に関係する遺伝子の重要な活性化物質である。この活性化では、IκBキナーゼ複合体(IKK)はNF−κB阻害剤(IκBタンパク質)をリン酸化し、ユビキチンとのコンジュゲーションおよびその後のプロテオソームによる分解へと導く。これは次に、遊離NF−κB二量体が核に移行して標的遺伝子を誘導することを可能にする。Rossiらは、15d−PGJ2を含むシクロペンテノンPGがIKKのIKK2サブユニットを直接阻害して修飾することを証明した。これは、次に、抑制性IκBタンパク質のリン酸化を防止するこれらのタンパク質をユビキチンコンジュゲーションおよび分解の標的にする。これは次にNF−κBの活性化を防止する。同様に、CastrilloらはLPSおよびIFN−αで処理したRAW264.7マクロファージ細胞において、15d−PGJ2とのインキュベーションがIKK2活性のかなりの阻害および抑制性IκBタンパク質の分解の阻害をもたらすことを示した。これは、次に、NF−κB活性の部分阻害、および2型NOSおよびシクロオキシゲナーゼ2などのNF−κB活性化を必要とする遺伝子の損なわれたた発現を引き起こした。
【0029】
したがって、PPARγおよびNF−κBは自己免疫疾患に関係する重要なメディエーターであり、医薬産業がこれらのメディエーターに影響を及ぼす新規な選択的薬剤および薬を探索する刺激をもたらすと結論することができる。
【0030】
一方、アルツハイマー病(AD)は、脳内のβアミロイド原線維の細胞外沈着およびアミロイドプラークと関連しているミクログリア細胞の活性化を特徴とする。活性化されたミクログリアは、その後広範な炎症性生成物を分泌する。Kitamuraらは特異抗体を使って正常およびADの脳内のPPARγおよびCOX−1、COX−2の発生を評価し、AD脳でのこれらの分子の発現増加を発見した。非ステロイド系抗炎症薬(NSAID)は、ADの発生率およびリスクを低下させ、疾患の進行を遅延させる効果を有することが示された。Combsらは、その全てはPPARγアゴニストであるNSAID、チアゾリジンジオンおよびPGJ2は、ミクログリアおよび単球によるβ−アミロイド刺激による炎症性生成物の分泌を阻害することを証明した。PPARγアゴニストは、β−アミロイド刺激によるIL−6およびTNF−αの遺伝子の発現およびCOX−2の発現を阻害することが示された。Henekaらは、ラット小脳へのLPSおよびIFN−αのマイクロインジェクションは、小脳顆粒細胞でのiNOS発現およびその後の細胞死を誘導することを証明した。PPARγアゴニスト(トログリタゾンおよび15d−PGJ2を含む)の同時注入はiNOSの発現および細胞死を減少させたが、選択的なCOX阻害剤の同時注入は効果がなかった。全体として、ADの研究はPPARγアゴニストが脳内の炎症性応答をモジュレートすることができること、およびNSAIDはPPARγに対するそれらの効果の結果としてADで役に立ちうることを示唆しているようである。
【0031】
これまで本明細書で開示したことから、以下の結論が得られる。
現在まで、薬草から単離されたPPARγアゴニスト化合物の前例がない。今日、自己免疫疾患の治療のためのこれらの特性を有する薬剤、組成物又は薬は存在しない。
【0032】
一方、この新規な組成物は、自己免疫疾患および神経変性疾患で増加する炎症誘発性サイトカインの生産を減らすことができる。
さらに、本発明の組成物は低毒性であり、いかなる有害な副作用も示さない。
【0033】
現在の科学の「技術の現状」を考慮すると、前記組成物の使用は当業者が推測できるものではなく、前記組成物は前記特性を有して上述の疾患を対象にし、これらの疾患のために現在使用されている他の物質で起こるような悪影響を引き起こすことなく免疫寛容を維持する。
【0034】
Andrographis paniculata(Nees)はアジア、インド、マレーシア、中国、韓国およびその他で自生するキツネノマゴ科に属する薬草である。これらの国では、異なる疾患、例えば風邪、肝臓状態、糖尿病、その他における生植物および乾燥植物またはその成分の有益効果のために、この薬草は広く使用されてきた。
【0035】
発明の詳しい説明
本出願では、本明細書で開示されている手法を適用することによってAndrographis paniculataから抽出されたアンドログラホリド混合物を用いてPPAR−γアゴニスト効果を誘導し、転写因子NF−κBの活性化を阻害する新規な組成物が記載されている。
【0036】
アンドログラホリド組成物の説明
本出願で請求されている組成物は、Andrographis paniculata乾燥抽出物の抽出から得られ、以下の一般式を有するジテルペン系ラブダン類の混合物を含む:
C20H30O5 アンドログラホリド(Andrographolide)
C20H30O4 14−デオキシアンドログラホリド(14−Deoxyandrographolide)
C26H41O8 ネオアンドログラホリド(Neoandrographolide)
これらアンドログラホリド化合物の化学構造および特徴を以下に示す。
・アンドログラホリド
i.一般式:C20H30O5
ii.分子量:350.46
iii.分子名:3−[2−[デカヒドロ−6−ヒドロキシ−5−(ヒドロキシメチル)−5,ha−ジメチル−2−メチレン−1−ナフタレニル]エチリデン]ジヒドロ−4−ヒドロキシ−2(3h)−フラノン。
【0037】
iv.分子構造:
【0038】
【化1】

【0039】
・14−デオキシアンドログラホリド
i.一般式:C20H30O4
ii.分子量:336.46
iii.分子構造:
【0040】
【化2】
【0041】
・ネオアンドログラホリド
i.一般式:C20H41O8
ii.分子量:326.46
iii.分子構造:
【0042】
【化3】
【0043】
本組成物の代表的抽出物は、次のアンドログラホリド類:アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドの混合物で構成される。前記個々の成分は、乾燥抽出物中、アンドログラホリドが約20〜40w/w%、14−デオキシアンドログラホリドが約3〜6w/w%およびネオアンドログラホリドが約0.2〜0.8w/w%含まれる。好ましくは、これらの化合物は、最終的な抽出物中にアンドログラホリドが約25から35w/w%、14−デオキシアンドログラホリドが約4.5から5.5w/w%およびネオアンドログラホリドが約0.4から0.8w/w%含まれる。
【0044】
より好ましい態様では、本発明の新規な抽出物は以下を含む:
アンドログラホリド 24.6%
14−デオキシアンドログラホリド 4.8%
ネオアンドログラホリド 0.6%
前記製剤は、薬学的に許容される担体と共に投与され得る、即ち錠剤の形態で投与され得る、アンドログラホリド混合物の約1〜6.5mg/kg体重/日を含む用量で投与されうる医薬品を製造することが許容される。
【0045】
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【0046】
本明細書で開示されている実施例によると、他の製剤および投与の態様に影響することなく、本明細書で開示されている態様は、既に言及されている自己免疫疾患の治療のために、加えてアルツハイマー病の治療のために効率的にかつ効果的に寄与する。
【0047】
したがって、本組成物およびその医薬製剤は、特に錠剤の形態で上記の用量で投与される場合に、炎症性障害特に糖尿病、炎症性腸疾患、自己免疫疾患(紅斑性狼瘡、多発性硬化症および慢性関節リウマチ)などの様々な自己免疫疾患を治療するための薬剤を提供する。
【0048】
その作用機構のために、本明細書で開示されている化合物および医薬組成物は、AIDSおよび組織及び臓器の拒絶反応を治療するためにも有用でありうる。
本発明の組成で、特に開示されている処方により製造することができる医薬組成物は、適切な経腸、非経口、皮膚、目、鼻、耳、直腸、膣、尿道、舌下、咽頭−気管−気管支の医薬形態に対応することができる。
【0049】
Andrographis paniculata原材料の取得法および分析法
有効成分:Andrographis paniculata Nees(Burm.f.)
科:キツネノマゴ科
使用する部分:地上部(herba)
発明者の管理下で有機栽培された緑葉、茎および地上部を、種を含めて天日乾燥する。全ての異物を手で除去し、原材料は1〜1.5cmの断片に切断して、換気のよい場所で保存する。同一性を調査するために慣用の分析を実行する。肉眼および顕微鏡による分析、官能性パラメータおよびTLC分析(薄層クロマトグラフィー)を、ヨーロッパ薬局方に従い実施する。
【0050】
Andrographis paniculata乾燥抽出物の取得方法
A.paniculataの抽出は、パート1の極性溶媒を使用した粉砕乾燥植物(地上部)の連続パーコレーションによって実施される。
【0051】
適切に分析された薬剤材料を、ナイフハンマーミル(0.8cm2)で適切な粒径に粉砕する。粉砕された材料はステンレス鋼パーコレーターに入れ、抽出溶液を50℃の温度で加える。パーコレーション時間は、2サイクルの抽出で約6日(6×24時間)である。パーコレーションが終了するまで、パーコレートをステンレス鋼タンクで収集する。パーコレートは、溶媒および大部分の水を除去するために、蒸発ユニットへ直接移す。蒸発は、140〜158°F(60〜70℃)および0.65〜0.85bar(65kPa〜85kPa)の真空で、LUWA薄膜エバポレーター内で実施する。蒸発工程は3〜4サイクルで実施され、その間、抽出物は1日につき30分間に4回、混合され続ける。spissum抽出物の水分含量が正常なとき、以下の分析をする:灰分、HCl−灰、乾燥減量、pH、TLC同定およびHPLC(高速液体クロマトグラフィー);アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドの分析。その後、spissum抽出物を乾燥ユニットへ移す。乾燥の前に、最終乾燥抽出物を以降の分析のためにファイバードラム内のプラスチック袋に詰める。
【0052】
Andrographis paniculata30%抽出物の調製法
Andrographis paniculataの切断および篩い分けされた葉/茎を、発明者の直接監督下で農場から収集する。地上部は前述のとおり同定のために分析し、次に抽出のために採取する。
【0053】
地上部は、真空下のステンレス鋼抽出装置内でn−ヘキサンまたはクロロホルムなどの低極性溶媒(A)で抽出する。連続抽出の後に溶媒を除去し、絞り滓はPetエーテル40:60または酢酸エチルなどの第2のより高い極性を有するの溶媒(B)で処理し、1回抽出の後溶媒を除去して、絞り滓はエタノールまたは水などのより大きな極性の溶媒(C)で処理する。
【0054】
第3の溶媒を回収して蒸発させると水分が30〜40%の塊が残り、その絞り滓は前に記載した低極性の溶媒で処理し、塊は次に濾過して水分が5%未満になるまで真空下で乾燥する。その粒状体をアンドログラホリド類として計算されるラブダンジテルペン類を30%以上有する微粉に粉砕する。
【0055】
抽出の詳細:
ステップ1:Andrographis paniculataの細かく切断された葉茎を重量比で3〜5倍の溶媒AとともにS S反応器に詰める。
【0056】
ステップ2:その草は4〜6時間繰り返し抽出して、溶媒を除去する。
ステップ3:次に絞り滓は、溶媒Aより高い極性を有する溶媒Bで処理して、3〜5時間で一度抽出する。
【0057】
ステップ4:溶媒Bを除去し、次に絞り滓を溶媒AまたはBより高い極性を有する溶媒Cで抽出する。
ステップ5:溶媒Cが3〜5時間絞り滓内に循環されて真空下でS S蒸発装置に取り出され、絞り滓は溶媒Cで再び抽出し、この過程を3〜4回繰り返す。
【0058】
ステップ6:溶媒Cを全ての洗液から回収して、生じる塊は一緒にプールする
ステップ7:ステップ4〜7から得られる塊は、次に溶媒AまたはBで処理し、残留物は60℃以下の真空下で乾燥させる
ステップ8:ステップ7から得られる乾燥塊は、100〜200ASTMのステンレス鋼メッシュを有するGMPグラインダーを使って粉末にする。
【0059】
ステップ9:ステップ8から得られる粉体を自動篩い機で篩いにかけ、直ちに密封できる殺菌PPバッグに直接詰める。
上述したように得た粉体を本明細書で記載のプロトコルに従って分析すると、最終抽出物中にアンドログラホリドは25から35w/w%、14−デオキシアンドログラホリドは約4.5から5.5w/w%およびネオアンドログラホリドは約0.4から0.8w/w%含まれる。
【0060】
Andrographis paniculataの同一性−TLC
試液:1gの薬草抽出物に20mlのメタノールを加えて約1時間振とうさせ、メタノールはフィルターを通してデカントする。残留物を20mlのメタノールで振とうし、濾過してから最初の抽出物と混合する(40mlの試液となる)。
【0061】
標準液:1.アンドログラホリド(A)、14−デオキシアンドログラホリド(DA)およびネオアンドログラホリド(NA)のメタノール溶液。2.試験抽出物と同様に処理された標準抽出物。20〜30μlの試液をTLCプレート(コーティング物質としてシリカゲルGF254)に付し、77容量部の酢酸エチル、15容量部のメタノールおよび8容量部の水(77:15:8)の混合液を使って15cm展開する。その後、プレートを風乾させてUV(254nM)下で検査する。クロマトグラムの少数の黒点は、Rf:0.65〜07がアンドログラホリドに、Rf:0.75〜0.8が14−デオキシアンドログラホリドに、およびRf:0.60〜0.65がネオアンドログラホリドに対応する。
【0062】
ジテルペンラブダン類の定量化のためのHPLC法
それら3化合物をアセトン(4:1)で抽出して、次に逆相RP−C18 licrospherカラム(4×125mm)を使用してHPLCによって分析する。移動相は26%アセトニトリルおよび0.5%リン酸からなり1.1ml/分の速度であり、Burgosら、1999、Acta Hort.(ISHS)501:83〜86頁に従って228nmで検出される。
【0063】
Andrographis paniculata乾燥抽出物は最低30%の総アンドログラホリド類に標準化されるが、これは、アンドログラホリドを約20〜40w/w%、14−デオキシアンドログラホリドを3〜6w/w%およびネオアンドログラホリドを0.2〜0.8w/w%含む。
【0064】
本発明による組成物はこれまで現在の科学における「技術の現状」では開示されておらず、自己免疫疾患およびADに関して記載されている方法論上の問題を解決するためのその利用に関しては前例がない。
【0065】
本発明の医薬組成物は経口的にまたは非経口的に投与することができ、非経口投与としては静脈内注射、皮下注射、筋肉内注射および関節内注射がある。
本発明の医薬組成物の正しい投薬量は、特定の製剤、投与様式、患者の年齢、体重および性別、食事、患者の疾患状態、補完的薬剤ならびに有害反応によって変動する。通常の熟練医師ならば、この医薬組成物の正しい投薬量を容易に決定して処方することができるものと理解される。好ましくは、この医薬組成物の日用量は、体重1kgにつきアンドログラホリド混合物が1から6.5mgの範囲である。
【0066】
当業者に公知の慣用技術によると、本発明の医薬組成物は先に述べたような薬学的に許容される担体および/またはビヒクルと共に、例えば単位用量剤形として製剤化することができる。製剤のそれには非限定的な例としては、無菌液、溶液、懸濁液またはエマルション、抽出物、エリキシル、散剤、顆粒剤、錠剤、カプセル、塗り薬、ローションおよび軟膏があるが、これらに限定されない。
【0067】
本発明は、アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドのラブダン化合物を、そのような組成物を調製する際に通常使用される薬学的に許容される担体と組み合わせて含有する医薬組成物も包含する。
【0068】
本発明の医薬組成物において、薬学的に許容される担体は医薬製剤のために記載されている慣用的ないかなるものでもよく、例えばラクトース、デキストロース、ショ糖、ソルビトール、マンニトール、澱粉、アラビアゴム、リン酸カルシウム、アルギン酸塩、ゼラチン、ケイ酸カルシウム、微結晶性セルロース、ポリビニルピロリドン、セルロース、水、シロップ、メチルセルロース、ヒドロキシプロピルメチルセルロース(HPMC)、メチルヒドロキシベンゾエート、プロピルヒドロキシベンゾエート、タルク、ステアリン酸、マグネシウムおよび鉱油などがあるが、それには限定されない。さらに、本発明の医薬組成物は、湿潤剤、甘味剤、乳化剤、懸濁化剤、保存剤、着香剤、香料、滑沢剤またはこれらの物質の混合物のいずれも含有することができる。
【0069】
典型的には、本医薬組成物は混合物のアンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドのラブダン類を20〜40%、好ましくは25〜35%、最も好ましくは30w/w%、ならびに薬学的に許容される担体を含有する。
【0070】
本発明の医薬組成物は、それを必要とする哺乳類に対して単回で、または分割して経口ルートで投与することができる。したがって、経口投与の場合、化合物を適切な固体担体と組み合わせてカプセル、錠剤、散剤を形成することができる。さらに、本医薬組成物は、着香剤、甘味料、賦形剤等のような他の成分を含有することができる。
【0071】
さらに、本発明はアンドログラホリド混合物を含む組成物で患者を治療するための、それを必要とする患者に対して本発明の組成物を含む溶液の静脈内投与および錠剤の経口投与することを含む方法を提供する。注射液製剤の好ましい投薬量は、当該組成物が約60〜210mg/日、最も好ましくは60〜80mg/日であり、1回、2回または3回の注射で投与される。
【0072】
注射液形態の本製剤は、1mlにつき当該組成物を約8〜16mg含む。患者に投与するときは、当該組成物は好ましくは0.9%生理食塩水で約1:5から1:10の容量比に希釈する。
【0073】
以下の実施例は例示が目的であり、本発明の範囲を限定するものではない。分別のある職人ならば思いつくような理にかなった変更は、本発明の範囲から逸脱することなく本明細書に加えることができる。
【0074】
本発明の医薬組成物は、医薬産業で典型的である規模ならびにより小さな手段で調製するのに適切である所望の経口用または非経口用製品を提供する。錠剤の形態のためには、適宜必要に応じて湿式造粒法、乾式造粒法、直接圧縮法、流動床造粒法を含む医薬産業で慣用の技術に従う。
【0075】
以下の実施例で示されるパーセンテージは全て重量に基づく。
【実施例】
【0076】
活性剤を含む錠剤を調製するための典型的方法の例は、先ず活性剤をゼラチン、エチルセルロースその他の結合剤と混合することである。その場合、混合は標準的なVブレンダー内で、通常無水条件下で適切に行われる。次に、調製された直後の混合物は従来の錠剤機で強打し、スラッグを錠剤に成形することができる。調製された直後の錠剤は、シェラック、メチルセルロース、カルナバワックス、スチレン−マレイン酸共重合体などの適切なコーティングで、コーティングされる。
【0077】
経口投与のために、アンドログラホリド混合物を30mgから40mgまで含む圧縮錠剤が、上で開示されかつRemington’s Pharmaceutical Science、第39章、Mack Publishing Co.、1965年、で示されている当技術分野で公知の製造方法に従って製造される。
【0078】
本発明製剤の好ましい医薬組成物は、以下の実施例のいくつかで示されている。
実施例1
薬草Andrographis paniculata Neesから得られた乾燥抽出物に含有されるアンドログラホリド混合物を用いた、本発明の錠剤を調製するための医薬組成物
【0079】
【表1】
【0080】
一様に錠剤を製剤化するために、乾燥抽出物(アンドログラホリド混合物)の活性化合物、ジャガイモ澱粉、タルク、ゼラチン、ヒドロキシプロピルメチルセルロース、無水二酸化ケイ素、ポリエチレングリコールおよび炭酸カルシウムを、全ての成分が一様に混合されるまで乾燥条件下で慣用のVブレンダーで混合する。混合物を次に標準的な軽メッシュスクリーンに通し、無水環境下で乾燥させ、次にステアリン酸マグネシウムとブレンドして錠剤に圧縮成形し、シェラックでコーティングする。116から162mgを含有する他の錠剤は、同様に調製される。
【0081】
実施例2
薬草Andrographis paniculata Neesから得られた乾燥抽出物を用いた、本発明のカプセルを調製するための医薬組成物
【0082】
【表2】
【0083】
30mgから40mgのアンドログラホリド混合物を含む経口用カプセルの製造は、乾燥抽出物(アンドログラホリド混合物)を担体と混合して、この混合物を通常ゼラチンその他のポリマーシースに封入することから本質的になる。カプセルは食用の適合する担体中の細かく分散した化合物を封入することによって作られる当技術分野で公知のソフト形態のカプセルでよく、または、カプセルはタルク、ステアリン酸カルシウム、炭酸カルシウム、その他の無毒性固体と混合した新規な組成物から本質的になる硬カプセル剤でもよい。治療使用のために30mgまたは40mgを含むカプセルを用いる典型的な使用例が指示されている。
【0084】
単回投与であれ複数回投与であれ、または日ごとの用量であれ、投与される用量は、化合物の効力が変動するため本発明のどの化合物が使用されるかにより、選択される投与経路により、患者のサイズおよび病状の性質により当然ながら変動する。投与される用量は、1日80から160mgの一般経口投与量に対応し、経口投与量は通常1日120mgを3回;通常の静脈内用量は60から80mgであり、指示があれば後に70から100mgを投与し;通常の筋肉内用量は24時間ごとの70から100mgであり1日に1〜2回注射される。
【0085】
本発明の乾燥抽出物(アンドログラホリド混合物を含有する)を含む新規の有用な医薬組成物は、皮膚送達系、胃腸薬剤送達用デバイスなどの薬剤送達系からの予想される生理効果のための投与に適応可能であり、この送達用デバイスは天然および合成のポリマー材から製造される。制御された薬剤投与のために化合物を含有する薬剤送達系の製造で許容される材料の代表例としては、ポリ塩化ビニル、ポリイソプレン、ポリブタジエン、ポリエチレン、エチレン−酢酸ビニルコポリマー、ポリジメチルシロキサン、アクリルおよびメタクリル酸のエステルの親水性ヒドロゲル、ポリ酢酸ビニル、プロピレン酢酸ビニル共重合体などの材料がある。
【0086】
実施例3
上記組成物を含有するシェラックで被覆された錠剤は、錠剤の剤形のための混合、顆粒化、および必要に応じて圧縮する工程を含む、医薬産業で慣用される技術に従って調製する。
【0087】
具体的には、実施例1の組成物を十分な量のAndrographis paniculata乾燥抽出物と完全に混合する。アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドを含む錠剤の製造のためには、その混合物を実施例1で言及した不活性成分と共に直接的に圧縮し、その後しかるべくシェラックで被覆する。
【0088】
実施例4
本組成物によって誘導されるHL−60細胞の分化
化学物質:May Grunwald−Giemsa、NBT、レチノイン酸、サイトカラシンB、ペニシリン、ストレプトマイシン、グルタミン、ウシ胎仔血清(Sigma)。RPMI1640(GIBCO)、Boehringer Mannheimからのウシ胎仔血清、全トランスレチノイン酸およびアンドログラホリドは、Aldrichから入手。他の分離系統は、Amsar Pvt.Ltd.、インド、から入手した。FURA2−AMはMolecular Probes(アメリカ合衆国)から購入した。ニトロブルーテトラゾリウムはSigmaから入手。
【0089】
細胞培養:HL−60細胞は、20%熱不活化ウシ胎仔血清、2mMグルタミン、100IU/mlペニシリンおよび100pg/mlストレプトマイシンを補ったRPMI1640培地で、5%のCO2を含有する37℃の加湿環境で増殖させた。細胞は3×105細胞/mlで、週2回播種した。100nMの全トランスレチノイン酸を単独でまたは組成物(17.5μgml)と組み合わせて加えることによって分化を誘導し、May Grunwald−Giemsa染色の後の形態変化およびNBT’(11)を還元する能力により評価した。未分化型培養物は、3%未満のNBT陽性細胞を含んでいた。分化した培養物は、レチノイン酸処理の5日後に調査した。
【0090】
カルシウム測定[Ca2+]c
HL−60顆粒球(2×107ml)にCa2+培地+0.1%ウシ血清アルブミン中で2μMのfura−2/AMを37℃で45分間ロードし、次に107細胞/mlに希釈して氷上で保存した。使用直前に、この細胞懸濁液の0.5mlを遠心分離にかけ、5μg/mlサイトカラシンBを含む示された培地の2.4mlで再懸濁した。Fura−2蛍光(F)は、温度自動調節されたキュベット内(37℃)(LS55蛍光計、パーキン−エルマー社)で340および380nmの励起および505nmの発光波長で測定した。
【0091】
実施例5
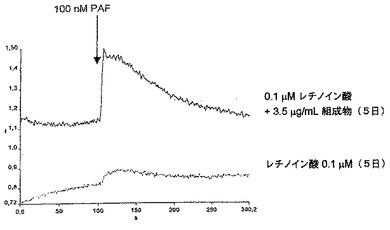
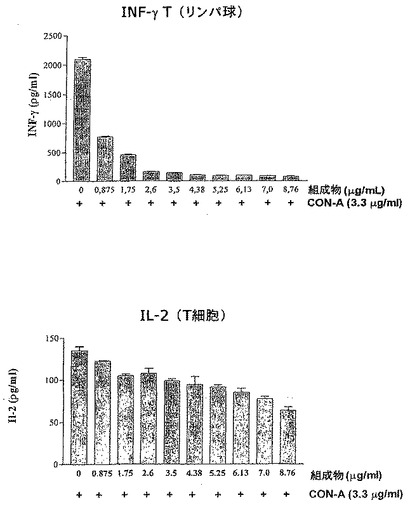
本組成物によるT細胞内のIL−2およびIFN−γ生産の阻害
化学物質:SigmaからのコンカナバリンAおよびRPMI1640培地
細胞培養:Rockefellerマウスをエーテルによって屠殺し、膝窩の神経節および脾臓を5mlの培地RPMI1640を含むペトリプラークに置いた。無菌のRPMI1640溶液中でこれらの臓器を破壊することによってリンパ球を得、これらのリンパ球は1mlのRPMI1640培地で再懸濁し、ヌーバウアーチャンバーで定量化した。最後に、リンパ球の懸濁液を1mlのRPMIにつき4×106細胞数の濃度に調節した。得られたならば、リンパ球は組成物の存在下または非存在下で培養した。この目的のために、1mlの細胞および異なる濃度の組成物および1mlのマイトジェンコンカナバリンA(CONA、0μg/mlおよび10μg/ml)を含む、24穴(それぞれ2ml)のポリスチレンの培養プラークを使用した。
【0092】
プラークは湿度5%およびCO2の環境で37℃のオーブンで24時間インキュベートし、次にそれぞれ2mlの試料を加えて3200rpmで1分間遠心分離した。その後、細胞を0.6mlに小分けして冷凍し、サイトカインをELISA(酵素結合免疫吸着検定法)で検出した。
【0093】
IL−2およびIFN−γのためのELISA
化学物質:IL−2とIFNγはPharmingenから入手。TMBはPierceから、H2O2およびH2SO4はMerckから入手。
【0094】
サイトカイン(IL−2およびIFN)の測定のために、抗抗原を捕捉する第1の抗体、ペルオキシダーゼ酵素とコンジュゲートする第2の抗体、および検量線のための標準溶液を使用した。ELISA「高結合」96穴ポリスチレンプレートを使用した。ウェルへの付着を助長するために1ウェルにつき100μlの第1の抗体を炭酸緩衝液pH9.5で希釈し、固体部分への結合を完全にするために4℃で一晩インキュベートした。その後、ウェルの内容物を除去して1ウェルにつき300μlのTween20、0.05%p/vおよびPBS pH7.0で3回洗浄した。その後、ウェルを200μlの無脂肪ミルク5%およびPBSでブロックし、室温で1時間インキュベートした。完了後、既に説明したようにウェル内容を除去して洗浄した。その後、1ウェルにつき100μlの抗原を含む検査試料を2反復で、およびサイトカインに特異的な校正曲線を100μl加え、室温で2時間インキュベートした。その後、後者のプロトコルに従ってウェル内容を除去して5回洗浄した。その後、第2のコンジュゲート抗体にペルオキシダーゼ酵素を加え、PBSおよびSBF溶液10%に希釈し、1ウェルにつき100μlをプレーティングして室温で1時間放置した。その後、7回洗浄して、1ウェルにつき100μlのTMB溶液およびH2O2に暴露させ、30分後に暗所で展開し、反応は1ウェルにつき50μlの2MのH2SO4で停止させた。反応の結果は、450nmフィルター(Elx800ユニバーサルマイクロプレートリーダー、Biotek)を使用してELISAで測定した。
【0095】
実施例6
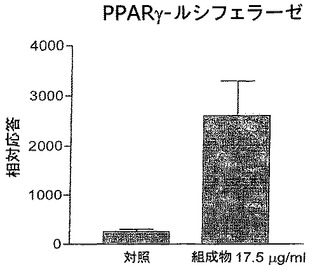
本組成物によるPPAR−γ受容体の刺激
化学物質:ジメチルスルホキシドはMerckから購入した。他の全ての試薬は、Promegaから購入した。
【0096】
転写アッセイ:HL−60細胞を、サイトメガロウイルスプロモーターの制御下のヒトPPARγ1発現ベクターであるpCMX−hPPARγ1でトランスフェクションした。ルシフェラーゼ活性およびβ−ガラクトシダーゼ活性を測定し、ルシフェラーゼ活性はHL−60中のβ−ガラクトシダーゼ標準にノーマライズした。
【0097】
プラスミド:GAL4−DNA結合ドメイン(DBD)およびmPPARγリガンド結合ドメイン(pGAL4DBD−mPPARγ)を発現しているプラスミドは、pGBTmPPARγ1からのScaI/BamHI断片として単離されたマウスPPARγ1リガンド結合ドメイン(アミノ酸162〜475から)をpCMXGal4 DBDへインフレーム挿入することによって構築した。
【0098】
細胞はDMSOまたは17.5μg/mlの本発明の組成物で処理し、ルシフェラーゼ活性は化学発光により測定した。
実施例7
本組成物による好中球中のNF−κB阻害
化学物質:ジメチルスルホキシド(DMSO)はMerckから入手。ウシ胎児血清およびRPMI−1640培地はGibco、米国、から入手。ニトロテトラゾリウムブルーはSigmaから、pRL−TK、pGL3および二重ルシフェラーゼリポーターアッセイ系はPromegaから。Fugene6はRocheから入手。
【0099】
細胞培養
急性骨髄性白血病からの細胞骨髄性のHL−60系を使用した。この細胞は、ジメチルスルホキシド1.3%(DMSO)の存在下で分化することができる(Santos−Beneitら、2000)。細胞は、2mMのL−グルタミン、10%の熱不活化ウシ胎児血清および抗生物質を補ったRPMI−1640培地中で、5%CO2と共に37℃に保たれる。細胞は、好中球を1.3%DMSOで4日間インキュベートすることによって好中球に分化する。分化細胞は、ニトロテトラゾリウムブルー(NBT)で分析する。
【0100】
HL−60細胞へのNFκB−pGL3ベクターのトランスフェクションおよびルシフェラーゼ測定
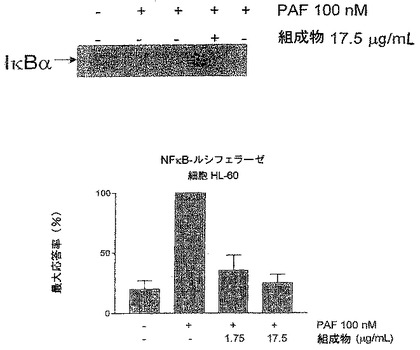
他で記載されているように、HL−60細胞を培養して4日間で好中球に分化させた。4日後、細胞をpGL3−NFκBベクターでトランスフェクションし、トランスフェクションの内部標準としてpRL−TK(Promega)ベクターを使用したが、これはRenillaルシフェラーゼの中レベルの発現を可能にする単純ヘルペスウイルスのチミジンキナーゼプロモーターを含む発現ベクターである。これらのベクターは、リポソームに基づく系(Fugene6、Roche)によって細胞にトランスフェクションされる。トランスフェクションの後、細胞を24時間保ち、次に本発明の組成物の存在下または非存在下で異なる時間にPAFまたはfMLPで刺激する。その後、細胞抽出物は、ルシフェラーゼ活性の測定まで−70℃に保つ。ルシフェラーゼの活性は、蛍(pGL3)およびRenilla(pRL)ルシフェラーゼ酵素の基質を有する市販の系デュアルルシフェラーゼリポーターアッセイ系(Promega)により、化学発光で測定する。
【0101】
IκBaイムノブロット
化学物質:fMLP、PMSFおよびPAFは、Calbiochemから購入した。トリス、NaCl、NP−40、デオキシコール酸塩、ドデシル硫酸ナトリウム、イプロテアーゼ(iprotease)阻害剤、メルカプトエタノールはMerckから入手。
【0102】
好中球は10分間プレインキュベートし、その後60分間fMLP(0.1μM)およびPAF(0.1μM)で刺激した。タンパク質の分析のために、細胞は溶解タンポン(50mMトリス、pH8.0、150mM NaCl、1%NP−40、0.5%デオキシコール酸塩、0.1%ドデシル硫酸ナトリウム、1mM Na2VO4、1mM PMSFおよび10μg/mlイプロテアーゼ阻害剤)で溶解した。タンパク質はブラッドフォード法で定量し、変性条件のポリアクリルアミドゲル12%(SDS/PAGE)で電気泳動により分解し、ニトロセルロース膜へ電気的に移動させた。膜を抗IκBα抗体と共にインキュベートし、次に第2のペルオキシダーゼコンジュゲート抗体とインキュベートし、最後に化学発光(ECL)で視覚化した。ゲル内のタンパク質の量の対照として、抗体をストリッピング溶液(100mM 2−メルカプトエタノール、2% SDS、62.5mMトリス−HCl、pH6.7)で処理して抗アクチン抗体とインキュベートした。最後に、各抗体について得られたシグナルの濃度測定分析を実施した。
【0103】
実施例8
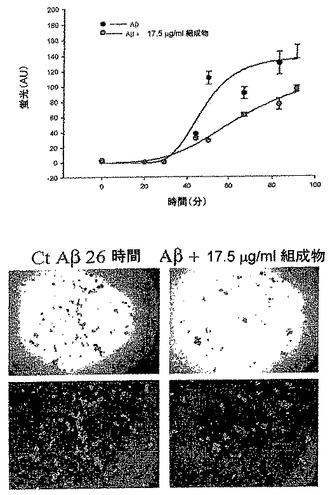
野生型ラットにおける本組成物によるβ−アミロイド形成の阻害
アミロイド形成
アミロイド形成を検査するために、2つの相補性技術、チオフラビン−T蛍光法(Levine、1993;Sotoら、1995;Reyesら、1997;Inestrosaら、2000)およびコンゴーレッド結合法(Alvarezら、1998)を使用した。簡単に説明すると、チオフラビン−Tアッセイは、アミロイド線維と結合するときのチオフラビンの蛍光発光に基づき、450nmで励起されたときの482nmの発光の増加を示す。コンゴ−レッドアッセイは、形成されたアミロイドの量を測定する非常に特異的な定量アッセイである。これらの技術は、特定のアミロイド形成を確認するために、現在使用されている。
【0104】
実施例9
アンドログラホリド組成物からの化合物の説明
本発明の代表組成物は錠剤形態の医薬製剤であり、それは以下の化合物
アンドログラホリド 24.6%
14−デオキシアンドログラホリド 4.8%
ネオアンドログラホリド 0.6%
の混合物をその後の異なる医薬形態の製造のために供給し、以下の用量で適用される:
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【0105】
実施例10
紅斑性狼瘡の治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド類の混合物を使用すると、狼瘡による症状の正常化は3ヵ月間の投与を経て起こる。さらに、本組成物は他の伝統的な非ステロイド系抗炎症剤の通常の再構築(rebuilding)効果に干渉しない。
【0106】
実施例11
多発性硬化症の治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド混合物を使用すると、当該疾患による症状の正常化は本発明の組成物による3ヵ月間の治療を経て起こる。さらに、本組成物は他の治療法に干渉しない。
【0107】
実施例12
関節症および慢性関節リウマチの治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド混合物を使用すると、変形性関節症による関節硬直の正常化は、グルコサミンまたはコンドロイチン硫酸または他の抗炎症薬の有無にかかわらず、3ヵ月を経て起こる。さらに、本組成物は、伝統的な非ステロイド系抗炎症剤と異なり、これら2つのプロテオグリカン構成要素の通常の関節再構築効果に干渉しない。
【0108】
実施例13
糖尿病の治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド混合物を使用すると、糖レベルの正常化は5週を経て起こる。さらに、本組成物は他の糖低下剤の通常の再構築効果に干渉しない。
【0109】
実施例14
AIDSの治療のための経口製剤の臨床的有効性
実施例7で記載されている経口製剤を、HIV陽性の患者に投与する。正常なCD4計数値は、3ヵ月以内の治療で回復する。
【0110】
実施例15
アルツハイマー病の治療のための経口製剤の臨床的有効性
実施例7で記載されている経口製剤を、掛かりつけの開業医に診断され、独立した認定神経科医に確認されたアルツハイマー病(AD)の初期段階を示している患者に投与する。臨床試験の2週間前に、患者はMini Mental Status Exam(MMSE)、アルツハイマー病評価スケール(ADAS)、Boston Naming Test(BNT)およびToken Test(TT)などの適切な精神神経病学的検査を受ける。
【0111】
神経心理学的検査は、臨床試験当日、6週後および3ヵ月後に繰り返し行われる。これらの検査は、患者の治療法を知らない神経心理学者によって実施される。
試験の開始時に、患者は試験製剤またはプラセボに無作為に割付けされる。試験製剤およびプラセボは、1日に1、2回、経口投与される。試験中は、糖尿病、高血圧、その他の病態の治療は許容される。スコアは、各3つの観察期間中に試験製剤とプラセボの間で統計学的に比較される。治療しない場合、ADの自然経過は、臨床試験期間中に検査スコアがかなり悪化する。当該処方で記載されている組成物で治療した患者は、臨床試験期間中、患者のスコアが同じであるか向上した場合に、改善されたとみなされる。
【0112】
患者は、試験の始めに無作為に本組成物またはプラセボを投与される。本組成物およびプラセボは、1日2回投与される。試験の間、患者は糖尿病、高血圧、その他の病態の治療が許可される。本組成物およびプラセボの結果は、全ての試験期間について統計学的に比較される。プラセボを使用する患者は、かなりの認知悪化を示す。本組成物で治療する患者は、検査スコアが相当改善される。
【0113】
前述の説明から、当業者が本発明の本質的特徴を容易に確かめることはできず、また、その精神および範囲から逸脱することなく、様々な用途および条件にそれを適応させるために本発明に様々な変更および/または修正を加えることができることは明らかである。このように、これらの変更および修正は、請求項の均等物の全範囲内に適切に、正当に含まれるものとする。
【図面の簡単な説明】
【0114】
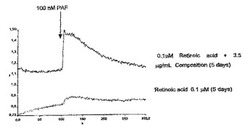
【図1】FURA2−AMインディケーターで標識されたHL−60細胞を用いて340/380nmの比により測定されたPAF誘導性カルシウム置換を示している代表的なグラフである。実施例1〜2で記載されているように、細胞はレチノイン酸だけで、または本発明の組成物(3.5μg/ml)の存在下で分化した。
【図2】PPARγのプロモーターを含むベクターでトランスフェクションしたHL−60細胞内の相対ルシフェラーゼ活性および本発明の組成物の効果を表している棒グラフである。
【図3】本発明の組成物によるコンカナバリンCONAで活性化されたT細胞内のIL−2およびINF−γ濃度の阻害を表している棒グラフである。
【図4】本発明の組成物によるlκBα分解の阻害を示す図であり、棒グラフはNFκBプロモーターを含むベクターでトランスフェクションしたHL−60細胞における相対ルシフェラーゼ活性に対する本発明の組成物の阻害率を表す。
【図5】チオフラビン染色を用いた本発明の組成物によるβアミロイド形成のin vitro阻害を示している顕微鏡写真である。
【発明の詳細な説明】
【0001】
説明
免疫薬理学および生物薬剤学の主な目的は、免疫疾患の症状を治療しその経過を修飾する新規な治療解決策を継続的に探索することである。
【0002】
発明の背景
自己免疫疾患は、それ自身の生体に対する免疫系の自発的反応を特徴とする。これらの反応は、液性(自己抗体生産)および細胞性(リンパ球およびマクロファージの細胞毒性活性の増加)の免疫反応を起動させる役割を担うTリンパ球による自己抗原の認識により生じる。自己免疫疾患には、リウマチ様疾患、乾癬、全身の皮膚筋炎、多発性硬化症、紅斑性狼瘡(lupus erythematous)、または抗原により悪化される免疫応答、即ち喘息、薬剤および食物に対するアレルギー、その他が含まれる。全てのこれらの疾患は制限的、慢性的であり、場合によっては致死性であるが、それらを治療する有効な治療法は現在存在しない。したがって、疾患の経過において寛解または減少をもたらすことができるあらゆる薬剤、医薬または媒体は、患者の健康のための重要な解決策となる。
【0003】
自己免疫疾患のための治療法の探索は、適切な薬剤および方法を見つける重要な努力をもたらした。
現在、これらの疾患の治療は、主に糖質コルチコイド、カルシニューリン阻害剤および抗増殖剤−代謝拮抗物質などの免疫抑制剤の使用に基づいている。しかし、これらの薬理学的療法は多くの異なる標的に作用するので、それらは免疫機能を全体として低下させるか、または、長期使用では異なる細胞傷害作用という不利を有する可能性があり、したがって、非特異的に免疫系を抑制して患者を感染症および癌のリスクにさらす可能性がある。カルシニューリンおよび糖質コルチコイドは、それらの腎毒性および糖尿病誘発作用のために、いくつかの臨床状態(例えば腎不全、糖尿病)でのそれらの有用性を制限するさらなる不利を示す。
【0004】
免疫抑制における最新の治療上の進歩は抗CD3モノクローナル抗体、即ち抗IL−2受容体モノクローナル抗体および抗TNFαモノクローナル抗体である。これらの治療法は著しい免疫抑制作用、アナフィラキシー反応、日和見感染(結核)および新生物を示すにもかかわらず、熱、じんま疹、低血圧症、呼吸困難がこれらの薬と関連し、前記組成物および医薬品の適用において深刻な問題となっている。注射用途の場合、患者3人中1人は痒み、腫脹および疼痛を示しうる。
【0005】
発明の簡単な要約
本発明で請求する組成物は、自己免疫疾患、アレルギーの特徴である免疫反応を減少させて疾患の症状および経過を緩和し、「免疫寛容」を維持することができる。
【0006】
言い換えると、本発明で開示される組成物は、それがもたらす免疫寛容によって本質的に特徴づけられ、これは、抗原に対する特異的な反応が欠如した活性状態に対応し、現行の免疫抑制薬剤の副作用を引き起こすことがない。
【0007】
具体的には、この組成物は、PPARγ受容体を刺激してNFκB因子を減らすことにより、インターフェロンγ、IL−2の合成および発現を阻害する。
したがってこのジテルペン性ラブダン類の新規な組成物は、自己免疫疾患の病因に関係するサイトカイン過剰発現を選択的に減少させることを特徴とする。
【0008】
急性および慢性炎症性疾患および癌に関係するメディエーターの科学的理解における最近の進歩は、有効な治療法の探索における新規な戦略をもたらした。伝統的な手法としては、特異的抗体、受容体アンタゴニストまたは酵素阻害剤の使用など、直接的標的介入があり、これらは全て重大レベルの副作用(例えばアレルギー、胃腸の潰瘍、出血、その他)を伴う。様々なメディエーターの転写および翻訳に関係する調節機構の解明が最近進展したことにより、遺伝子転写レベル(例えば、COX2、iNOS、IL1β、TNFα、ICAM、その他)に向けられた治療手段に対する関心が増加した。
【0009】
最も重要なメディエーターの1つは、Rel/NF−κBファミリーのポリペプチドの様々な組合せから構成される緊密に関連したダイマー転写因子複合体ファミリーに属している、NFκBである。このファミリーは、哺乳類の5つの個々の遺伝子産物である、RelA(p65)、NF−κB1(p50/p105)、NF−κB2(p49/p100)、c−RelおよびRelBからなり、その全てはヘテロダイマーまたはホモダイマーを形成することができる。これらのタンパク質は、DNA結合および二量体化ドメインを含有する相同の高い300アミノ酸の「Rel相同性ドメイン」を共有する。Rel相同性ドメインの最C末端には、細胞質から核へのNF−κBの輸送において重要な核転座配列がある。さらに、p65およびcRelは、それらのC末端に強力なトランス活性化ドメインを有する。
【0010】
NF−κBの活性は、阻害剤IκBファミリーのタンパク質のメンバーとの相互作用によって調節される。この相互作用は効果的にNF−κBタンパク質上の核移行配列を遮断することにより、核への二量体の移動を防止する。多種多様な刺激が、おそらくは複数のシグナル伝達経路を介して、NF−κBを活性化する。これには細菌生成物(LPS)、一部のウイルス(HIV−1、HTLV−1)、炎症性サイトカイン(TNFα、IL−1)および環境ストレスが含まれる。しかし、表面上全ての刺激に共通するのは、リン酸化および以降のIκBの分解である。IκBは、最近特定されたIκBキナーゼ(IKK−αおよびIKK−β)によって、2つのN末端セリン上でリン酸化される。部位特異的突然変異誘発研究によると、一度リン酸化されるとタンパク質はユビキチン−プロテアソーム経路による分解のために目印をつけられることから、これらのリン酸化はNF−κBの後の活性化のために重要であることが示されている。IκBから遊離すると、活性NF−κB複合体は核に移行して、そこで好ましい遺伝子特異的エンハンサー配列に選択的に結合することができる。NF−κBによって調節される遺伝子には、いくつかのサイトカイン、細胞接着分子および急性期タンパク質が含まれる。
【0011】
NF−κBが、IL−6およびIL−8などのサイトカイン、ICAMおよびVCAMなどの細胞接着分子、ならびに誘導性酸化窒素シンターゼ(iNOS)を含む多数の炎症誘発性メディエーターの調節された発現で重要な役割を果たすことは、周知である。そのようなメディエーターは炎症部位で白血球を補充する役をつとめることが知られており、iNOSの場合は、一部の炎症性疾患および自己免疫疾患では臓器破壊をもたらす可能性がある。
【0012】
炎症性障害におけるNF−κBの重要性は、NF−κBが活性化されることが明らかにされている喘息を含む気道炎症の研究においてさらに強調されている。この活性化は、これらの障害の特徴であるサイトカイン生産の増加および白血球浸潤の根底にあるかもしれない。さらに、吸入されたステロイドは喘息の気道で気道過敏症を減少させ、炎症性反応を抑えることが知られている。糖質コルチコイドのNF−κB阻害に関する最近の知見を考慮すると、これらの効果はNF−κBの阻害により媒介されると推測することができる。
【0013】
炎症性障害におけるNF−κBの役割のさらなる証拠は、リウマチの滑膜に関する研究からもたらされる。NF−κBは通常不活性の細胞質複合体として存在するが、近年の免疫組織化学的研究は、リウマチ性滑膜を含む細胞ではNF−κBが核に存在し、したがって活性であることを示している。さらに、NF−κBはTNFαによる刺激に応じてヒト滑膜細胞で活性化されることが示された。そのような分布は、この組織の特徴であるサイトカインおよびエイコサノイド産生の増加の根底にあるメカニズムであるかもしれない。Roshak、A.K.ら、J.Biol.Chem.、271、31496〜31501(1996)を参照。
【0014】
炎症性障害におけるNF−κBの役割のさらなる証拠は、リウマチの滑膜に関する研究からもたらされる。NF−κBは通常不活性の細胞質複合体として存在するが、近年の免疫組織化学的研究は、リウマチ性滑膜を含む細胞ではNF−κBが核に存在し、したがって活性であることを示している。さらに、NF−κBはTNFαによる刺激に応じてヒト滑膜細胞で活性化されることが示された。そのような分布は、この組織の特徴であるサイトカインおよびエイコサノイド産生の増加の根底にあるメカニズムであるかもしれない。Roshak、A.K.ら、J.Biol.Chem.、271、31496〜31501(1996)を参照。
【0015】
NF−κB/RelおよびIκBタンパク質も、悪性形質転換で重要な役割を果たしているようである。ファミリーメンバーは、過剰発現、遺伝子増幅、遺伝子再構成または転座の結果としてin vitroおよびin vivoでの細胞形質転換と関連している。さらに、これらのタンパク質をエンコードしている遺伝子の再構成および/または増幅が、ある種のヒトリンパ性腫瘍の20〜25%で見られる。さらに、アポトーシスの調節におけるNF−κBの役割が報告され、細胞増殖の制御におけるこの転写因子の役割を強化している。
【0016】
NF−κBの最初の植物由来のモジュレーターは、サリチル酸ナトリウムおよびその半合成誘導体のアスピリンを特定したKoppとGhosh(1994)によって、ほぼ10年前に報告された。この発見の後、異なる化学クラスのいくつかの新規な天然物が、NF−κB阻害活性を示した。
【0017】
いくつかのNF−κB阻害剤が文献:C.Wahlら、J.Clin.Invest.101(5)、1163〜1174(1998);R.W.Sullivanら、J.Med.Chem.41、413〜419(1998);J.W.Pierceら、J.Biol.Chem.272、21096〜21103(1997)に記載されている。海洋性天然物ヒメニアルジシン(hymenialdisine)は、NF−κBを阻害することが知られている:Roshak、Aら、JPET、283、955〜961(1997)。Breton、J.JおよびChabot−Fletcher、M.C.、JPET、282、459〜466(1997)。サリチルアニリドは公知の化合物であり、M.T.Clark、R.A.Coburn、R.T.Evans、R.J.Genco、J.Med.Chem.、1986、29、25〜29によって記載されている。
【0018】
最近、NF−κB阻害の重要なメカニズムにより、ペルオキシソーム受容体の活性化の可能性が示唆されている。
「ペルオキシソーム増殖因子活性化受容体」(PPAR)[この用語の通常の使用に注目するときには、科学の領域で公知のその略語で表し、当該略語はこの受容体を特定するために使用されるものとする]として知られるペルオキシソームの受容体は、自己免疫疾患および他の疾患、即ち糖尿病、心血管および胃腸の疾患ならびにアルツハイマー病との関係が示唆されている。PPARγアゴニストを含む現在の医薬剤はまだ実験段階であり、その作用機構のために人には深刻な副作用を起こす。したがって、上述の疾患または病態を予防、治療および/または軽減するために、より正確にこれらの受容体をモジュレートすることができるより毒性の低い新規な薬剤を開発する必要がある。
【0019】
この新規な組成物はこれらの受容体をより正確にモジュレートするので、患者に望ましくない副作用を引き起こすことなく、自己免疫疾患をより有効に予防、治療および/または軽減することを可能にする。
【0020】
ペルオキシソーム増殖因子活性化受容体(PPAR)は、遺伝子発現を調節するリガンド活性化転写因子である核ホルモン受容体スーパーファミリーのメンバーである。PPARの様々な亜型が発見されている。これらには、PPARα、PPARβまたはNUC1、PPARγおよびPPARδが含まれる。
【0021】
PPARγは、当初脂肪細胞分化および脂質代謝の重要な調節因子と特徴づけされた。PPARγの発現は異なる複数のプロモーターによって指示される、3つのPPARγアイソフォームが生じる。PPARγは繊維芽細胞、筋細胞、乳房細胞、ラット脾臓の白色および赤色脾髄、ヒト骨髄前駆体ならびにマクロファージ/単球を含む他の細胞型でも見られることが、現在明らかとなっている。さらに、PPARγはアテローム硬化型プラーク内のマクロファージ泡沫細胞で見られた。糖代謝におけるPPARγの重要な役割は、あるクラスの抗糖尿病薬であるチアゾリジンジオンが高親和性のPPARγリガンドであることが証明されたときに特定された。チアゾリジンジオンは、当初、インスリン抵抗性の齧歯類モデルでグルコースレベル(および循環脂肪酸のレベル)を下げるそれらの能力に基づき、2型糖尿病の治療のために開発された。チアゾリジンジオンはPPARγとの直接相互作用を通してそれらの治療効果を媒介するとの知見により、PPARγがグルコースおよび脂質のホメオスタシスの重要な調節因子であると立証された。当初は脂質およびグルコースの代謝の調節因子として記載されたにもかかわらず、近年、PPARγは細胞増殖および悪性腫瘍における役割を有することも立証された。PPARγのリガンドは、細胞増殖および悪性腫瘍に対する正および負の作用を媒介することが示された。
【0022】
チアゾリジンジオンクラスの抗糖尿病薬に加えて、様々な非ステロイド性抗炎症薬もPPARγリガンドとして機能することができるが、後者の親和性は比較的低い。
プロスタグランジンD2(PGD2)脱水物PGJ2は、最初に発見されたPPARγの内因性リガンドであった。別のPGD2脱水物15−デオキシ−Δ12,14−PGJ2(15d−PGJ2)も直接PPARγと結合する天然物質であり、PPARγ活性化のための強力なリガンドである。
【0023】
PPARおよびマクロファージを関連づける最も初期の知見の1つは、PPARγがヒトおよびマウスのアテローム硬化性病変のマクロファージ由来泡沫細胞で高度に発現されたことであった。その後、PPARγがヒトおよびマウスの単球/マクロファージで発現されることが証明された。機能的に、PPARγは、単球の分化および活性化で、また炎症性作用の調節において役割を果たすことが示された。
【0024】
多くの研究は、PPARγリガンドがマクロファージ炎症性応答を阻害することを証明している。PPARγ活性化の抗炎症効果は、ヒトおよびマウスの単球/マクロファージおよび単球/マクロファージ系で証明された。マクロファージの活性化は、通常いくつかの異なる炎症誘発性メディエーターの分泌をもたらす。15d−PGJ2またはチアゾリジンジオンによる治療は、これらのメディエーター(ゲラチナーゼB、IL−6、TNF−αおよびIL−1βを含む)の多くの分泌を阻止し、また誘導可能なNOS(iNOS)の誘導性発現およびスカベンジャー受容体A遺伝子の転写を減少させることが見出された。
【0025】
PPARγの関連性は、いくつかのヒト自己免疫疾患および自己免疫疾患の動物モデルで研究された。Kawahitoらは、慢性関節リウマチ(RA)患者で滑膜組織がPPARγを発現することを証明した。PPARγはマクロファージで高度に発現されることが発見され、中程度の発現が滑膜内層(synovial-lining)れた。15d−PGJ2およびトログリタゾンによるPPARγの活性化は、in vitroでRA滑膜細胞アポトーシスを誘導した。
【0026】
PPARγは新たに単離されたT細胞で機能的に関連しているか、活性化の初期に機能的に関連するようになることが示唆されている。これらの研究で、PPARγの2つのリガンドがT細胞クローンによるIL−2分泌の阻止を媒介し、そのようなクローンのIL−2誘導性増殖を阻止しないことが証明された。
【0027】
いくつかの研究は、自己免疫疾患の動物モデルを修飾する際のPPARγリガンドの役割を調査した。Suらは、炎症性腸疾患のマウスモデルにおいて、チアゾリジンジオンは結腸炎症を著しく減少させたことを示した。この効果は、PPARγを高度に発現して炎症性サイトカインを生産することができる結腸上皮細胞に及ぼす直接的効果の結果であるかもしれないと、提唱された。Kawahitoらは、PPARリガンド、15d−PGJ2およびトログリタゾンの腹腔内投与はアジュバント誘導性の関節炎を寛解させることを示した。Ninoらは実験的アレルギー性脳脊髄炎に及ぼすチアゾリジンジオンの効果を調べ、この療法が多発性硬化症のこのマウスモデルで炎症を弱め、臨床症状を減少させることを発見した。最後に、Reillyらは、腎臓糸球体メサンギウム細胞がループス腎炎の炎症性応答の重要なモジュレーターであり、活性化されるとNOおよびシクロオキシゲナーゼ生成物を含む炎症伝達物質を分泌することにより局所炎症性応答を永続させることを証明した。上記研究から、炎症性または自己免疫性病因を有する疾患におけるPPARの関連性およびPPARアゴニストによる治療の有用性は、おそらく研究の焦点であり続けるであろう。
【0028】
近年では、PPARγに対する15d−PGJ2の特異性の問題は、少なくとも部分的に解明された。NF−κBは、炎症および免疫に関係する遺伝子の重要な活性化物質である。この活性化では、IκBキナーゼ複合体(IKK)はNF−κB阻害剤(IκBタンパク質)をリン酸化し、ユビキチンとのコンジュゲーションおよびその後のプロテオソームによる分解へと導く。これは次に、遊離NF−κB二量体が核に移行して標的遺伝子を誘導することを可能にする。Rossiらは、15d−PGJ2を含むシクロペンテノンPGがIKKのIKK2サブユニットを直接阻害して修飾することを証明した。これは、次に、抑制性IκBタンパク質のリン酸化を防止するこれらのタンパク質をユビキチンコンジュゲーションおよび分解の標的にする。これは次にNF−κBの活性化を防止する。同様に、CastrilloらはLPSおよびIFN−αで処理したRAW264.7マクロファージ細胞において、15d−PGJ2とのインキュベーションがIKK2活性のかなりの阻害および抑制性IκBタンパク質の分解の阻害をもたらすことを示した。これは、次に、NF−κB活性の部分阻害、および2型NOSおよびシクロオキシゲナーゼ2などのNF−κB活性化を必要とする遺伝子の損なわれたた発現を引き起こした。
【0029】
したがって、PPARγおよびNF−κBは自己免疫疾患に関係する重要なメディエーターであり、医薬産業がこれらのメディエーターに影響を及ぼす新規な選択的薬剤および薬を探索する刺激をもたらすと結論することができる。
【0030】
一方、アルツハイマー病(AD)は、脳内のβアミロイド原線維の細胞外沈着およびアミロイドプラークと関連しているミクログリア細胞の活性化を特徴とする。活性化されたミクログリアは、その後広範な炎症性生成物を分泌する。Kitamuraらは特異抗体を使って正常およびADの脳内のPPARγおよびCOX−1、COX−2の発生を評価し、AD脳でのこれらの分子の発現増加を発見した。非ステロイド系抗炎症薬(NSAID)は、ADの発生率およびリスクを低下させ、疾患の進行を遅延させる効果を有することが示された。Combsらは、その全てはPPARγアゴニストであるNSAID、チアゾリジンジオンおよびPGJ2は、ミクログリアおよび単球によるβ−アミロイド刺激による炎症性生成物の分泌を阻害することを証明した。PPARγアゴニストは、β−アミロイド刺激によるIL−6およびTNF−αの遺伝子の発現およびCOX−2の発現を阻害することが示された。Henekaらは、ラット小脳へのLPSおよびIFN−αのマイクロインジェクションは、小脳顆粒細胞でのiNOS発現およびその後の細胞死を誘導することを証明した。PPARγアゴニスト(トログリタゾンおよび15d−PGJ2を含む)の同時注入はiNOSの発現および細胞死を減少させたが、選択的なCOX阻害剤の同時注入は効果がなかった。全体として、ADの研究はPPARγアゴニストが脳内の炎症性応答をモジュレートすることができること、およびNSAIDはPPARγに対するそれらの効果の結果としてADで役に立ちうることを示唆しているようである。
【0031】
これまで本明細書で開示したことから、以下の結論が得られる。
現在まで、薬草から単離されたPPARγアゴニスト化合物の前例がない。今日、自己免疫疾患の治療のためのこれらの特性を有する薬剤、組成物又は薬は存在しない。
【0032】
一方、この新規な組成物は、自己免疫疾患および神経変性疾患で増加する炎症誘発性サイトカインの生産を減らすことができる。
さらに、本発明の組成物は低毒性であり、いかなる有害な副作用も示さない。
【0033】
現在の科学の「技術の現状」を考慮すると、前記組成物の使用は当業者が推測できるものではなく、前記組成物は前記特性を有して上述の疾患を対象にし、これらの疾患のために現在使用されている他の物質で起こるような悪影響を引き起こすことなく免疫寛容を維持する。
【0034】
Andrographis paniculata(Nees)はアジア、インド、マレーシア、中国、韓国およびその他で自生するキツネノマゴ科に属する薬草である。これらの国では、異なる疾患、例えば風邪、肝臓状態、糖尿病、その他における生植物および乾燥植物またはその成分の有益効果のために、この薬草は広く使用されてきた。
【0035】
発明の詳しい説明
本出願では、本明細書で開示されている手法を適用することによってAndrographis paniculataから抽出されたアンドログラホリド混合物を用いてPPAR−γアゴニスト効果を誘導し、転写因子NF−κBの活性化を阻害する新規な組成物が記載されている。
【0036】
アンドログラホリド組成物の説明
本出願で請求されている組成物は、Andrographis paniculata乾燥抽出物の抽出から得られ、以下の一般式を有するジテルペン系ラブダン類の混合物を含む:
C20H30O5 アンドログラホリド(Andrographolide)
C20H30O4 14−デオキシアンドログラホリド(14−Deoxyandrographolide)
C26H41O8 ネオアンドログラホリド(Neoandrographolide)
これらアンドログラホリド化合物の化学構造および特徴を以下に示す。
・アンドログラホリド
i.一般式:C20H30O5
ii.分子量:350.46
iii.分子名:3−[2−[デカヒドロ−6−ヒドロキシ−5−(ヒドロキシメチル)−5,ha−ジメチル−2−メチレン−1−ナフタレニル]エチリデン]ジヒドロ−4−ヒドロキシ−2(3h)−フラノン。
【0037】
iv.分子構造:
【0038】
【化1】
【0039】
・14−デオキシアンドログラホリド
i.一般式:C20H30O4
ii.分子量:336.46
iii.分子構造:
【0040】
【化2】
【0041】
・ネオアンドログラホリド
i.一般式:C20H41O8
ii.分子量:326.46
iii.分子構造:
【0042】
【化3】
【0043】
本組成物の代表的抽出物は、次のアンドログラホリド類:アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドの混合物で構成される。前記個々の成分は、乾燥抽出物中、アンドログラホリドが約20〜40w/w%、14−デオキシアンドログラホリドが約3〜6w/w%およびネオアンドログラホリドが約0.2〜0.8w/w%含まれる。好ましくは、これらの化合物は、最終的な抽出物中にアンドログラホリドが約25から35w/w%、14−デオキシアンドログラホリドが約4.5から5.5w/w%およびネオアンドログラホリドが約0.4から0.8w/w%含まれる。
【0044】
より好ましい態様では、本発明の新規な抽出物は以下を含む:
アンドログラホリド 24.6%
14−デオキシアンドログラホリド 4.8%
ネオアンドログラホリド 0.6%
前記製剤は、薬学的に許容される担体と共に投与され得る、即ち錠剤の形態で投与され得る、アンドログラホリド混合物の約1〜6.5mg/kg体重/日を含む用量で投与されうる医薬品を製造することが許容される。
【0045】
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【0046】
本明細書で開示されている実施例によると、他の製剤および投与の態様に影響することなく、本明細書で開示されている態様は、既に言及されている自己免疫疾患の治療のために、加えてアルツハイマー病の治療のために効率的にかつ効果的に寄与する。
【0047】
したがって、本組成物およびその医薬製剤は、特に錠剤の形態で上記の用量で投与される場合に、炎症性障害特に糖尿病、炎症性腸疾患、自己免疫疾患(紅斑性狼瘡、多発性硬化症および慢性関節リウマチ)などの様々な自己免疫疾患を治療するための薬剤を提供する。
【0048】
その作用機構のために、本明細書で開示されている化合物および医薬組成物は、AIDSおよび組織及び臓器の拒絶反応を治療するためにも有用でありうる。
本発明の組成で、特に開示されている処方により製造することができる医薬組成物は、適切な経腸、非経口、皮膚、目、鼻、耳、直腸、膣、尿道、舌下、咽頭−気管−気管支の医薬形態に対応することができる。
【0049】
Andrographis paniculata原材料の取得法および分析法
有効成分:Andrographis paniculata Nees(Burm.f.)
科:キツネノマゴ科
使用する部分:地上部(herba)
発明者の管理下で有機栽培された緑葉、茎および地上部を、種を含めて天日乾燥する。全ての異物を手で除去し、原材料は1〜1.5cmの断片に切断して、換気のよい場所で保存する。同一性を調査するために慣用の分析を実行する。肉眼および顕微鏡による分析、官能性パラメータおよびTLC分析(薄層クロマトグラフィー)を、ヨーロッパ薬局方に従い実施する。
【0050】
Andrographis paniculata乾燥抽出物の取得方法
A.paniculataの抽出は、パート1の極性溶媒を使用した粉砕乾燥植物(地上部)の連続パーコレーションによって実施される。
【0051】
適切に分析された薬剤材料を、ナイフハンマーミル(0.8cm2)で適切な粒径に粉砕する。粉砕された材料はステンレス鋼パーコレーターに入れ、抽出溶液を50℃の温度で加える。パーコレーション時間は、2サイクルの抽出で約6日(6×24時間)である。パーコレーションが終了するまで、パーコレートをステンレス鋼タンクで収集する。パーコレートは、溶媒および大部分の水を除去するために、蒸発ユニットへ直接移す。蒸発は、140〜158°F(60〜70℃)および0.65〜0.85bar(65kPa〜85kPa)の真空で、LUWA薄膜エバポレーター内で実施する。蒸発工程は3〜4サイクルで実施され、その間、抽出物は1日につき30分間に4回、混合され続ける。spissum抽出物の水分含量が正常なとき、以下の分析をする:灰分、HCl−灰、乾燥減量、pH、TLC同定およびHPLC(高速液体クロマトグラフィー);アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドの分析。その後、spissum抽出物を乾燥ユニットへ移す。乾燥の前に、最終乾燥抽出物を以降の分析のためにファイバードラム内のプラスチック袋に詰める。
【0052】
Andrographis paniculata30%抽出物の調製法
Andrographis paniculataの切断および篩い分けされた葉/茎を、発明者の直接監督下で農場から収集する。地上部は前述のとおり同定のために分析し、次に抽出のために採取する。
【0053】
地上部は、真空下のステンレス鋼抽出装置内でn−ヘキサンまたはクロロホルムなどの低極性溶媒(A)で抽出する。連続抽出の後に溶媒を除去し、絞り滓はPetエーテル40:60または酢酸エチルなどの第2のより高い極性を有するの溶媒(B)で処理し、1回抽出の後溶媒を除去して、絞り滓はエタノールまたは水などのより大きな極性の溶媒(C)で処理する。
【0054】
第3の溶媒を回収して蒸発させると水分が30〜40%の塊が残り、その絞り滓は前に記載した低極性の溶媒で処理し、塊は次に濾過して水分が5%未満になるまで真空下で乾燥する。その粒状体をアンドログラホリド類として計算されるラブダンジテルペン類を30%以上有する微粉に粉砕する。
【0055】
抽出の詳細:
ステップ1:Andrographis paniculataの細かく切断された葉茎を重量比で3〜5倍の溶媒AとともにS S反応器に詰める。
【0056】
ステップ2:その草は4〜6時間繰り返し抽出して、溶媒を除去する。
ステップ3:次に絞り滓は、溶媒Aより高い極性を有する溶媒Bで処理して、3〜5時間で一度抽出する。
【0057】
ステップ4:溶媒Bを除去し、次に絞り滓を溶媒AまたはBより高い極性を有する溶媒Cで抽出する。
ステップ5:溶媒Cが3〜5時間絞り滓内に循環されて真空下でS S蒸発装置に取り出され、絞り滓は溶媒Cで再び抽出し、この過程を3〜4回繰り返す。
【0058】
ステップ6:溶媒Cを全ての洗液から回収して、生じる塊は一緒にプールする
ステップ7:ステップ4〜7から得られる塊は、次に溶媒AまたはBで処理し、残留物は60℃以下の真空下で乾燥させる
ステップ8:ステップ7から得られる乾燥塊は、100〜200ASTMのステンレス鋼メッシュを有するGMPグラインダーを使って粉末にする。
【0059】
ステップ9:ステップ8から得られる粉体を自動篩い機で篩いにかけ、直ちに密封できる殺菌PPバッグに直接詰める。
上述したように得た粉体を本明細書で記載のプロトコルに従って分析すると、最終抽出物中にアンドログラホリドは25から35w/w%、14−デオキシアンドログラホリドは約4.5から5.5w/w%およびネオアンドログラホリドは約0.4から0.8w/w%含まれる。
【0060】
Andrographis paniculataの同一性−TLC
試液:1gの薬草抽出物に20mlのメタノールを加えて約1時間振とうさせ、メタノールはフィルターを通してデカントする。残留物を20mlのメタノールで振とうし、濾過してから最初の抽出物と混合する(40mlの試液となる)。
【0061】
標準液:1.アンドログラホリド(A)、14−デオキシアンドログラホリド(DA)およびネオアンドログラホリド(NA)のメタノール溶液。2.試験抽出物と同様に処理された標準抽出物。20〜30μlの試液をTLCプレート(コーティング物質としてシリカゲルGF254)に付し、77容量部の酢酸エチル、15容量部のメタノールおよび8容量部の水(77:15:8)の混合液を使って15cm展開する。その後、プレートを風乾させてUV(254nM)下で検査する。クロマトグラムの少数の黒点は、Rf:0.65〜07がアンドログラホリドに、Rf:0.75〜0.8が14−デオキシアンドログラホリドに、およびRf:0.60〜0.65がネオアンドログラホリドに対応する。
【0062】
ジテルペンラブダン類の定量化のためのHPLC法
それら3化合物をアセトン(4:1)で抽出して、次に逆相RP−C18 licrospherカラム(4×125mm)を使用してHPLCによって分析する。移動相は26%アセトニトリルおよび0.5%リン酸からなり1.1ml/分の速度であり、Burgosら、1999、Acta Hort.(ISHS)501:83〜86頁に従って228nmで検出される。
【0063】
Andrographis paniculata乾燥抽出物は最低30%の総アンドログラホリド類に標準化されるが、これは、アンドログラホリドを約20〜40w/w%、14−デオキシアンドログラホリドを3〜6w/w%およびネオアンドログラホリドを0.2〜0.8w/w%含む。
【0064】
本発明による組成物はこれまで現在の科学における「技術の現状」では開示されておらず、自己免疫疾患およびADに関して記載されている方法論上の問題を解決するためのその利用に関しては前例がない。
【0065】
本発明の医薬組成物は経口的にまたは非経口的に投与することができ、非経口投与としては静脈内注射、皮下注射、筋肉内注射および関節内注射がある。
本発明の医薬組成物の正しい投薬量は、特定の製剤、投与様式、患者の年齢、体重および性別、食事、患者の疾患状態、補完的薬剤ならびに有害反応によって変動する。通常の熟練医師ならば、この医薬組成物の正しい投薬量を容易に決定して処方することができるものと理解される。好ましくは、この医薬組成物の日用量は、体重1kgにつきアンドログラホリド混合物が1から6.5mgの範囲である。
【0066】
当業者に公知の慣用技術によると、本発明の医薬組成物は先に述べたような薬学的に許容される担体および/またはビヒクルと共に、例えば単位用量剤形として製剤化することができる。製剤のそれには非限定的な例としては、無菌液、溶液、懸濁液またはエマルション、抽出物、エリキシル、散剤、顆粒剤、錠剤、カプセル、塗り薬、ローションおよび軟膏があるが、これらに限定されない。
【0067】
本発明は、アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドのラブダン化合物を、そのような組成物を調製する際に通常使用される薬学的に許容される担体と組み合わせて含有する医薬組成物も包含する。
【0068】
本発明の医薬組成物において、薬学的に許容される担体は医薬製剤のために記載されている慣用的ないかなるものでもよく、例えばラクトース、デキストロース、ショ糖、ソルビトール、マンニトール、澱粉、アラビアゴム、リン酸カルシウム、アルギン酸塩、ゼラチン、ケイ酸カルシウム、微結晶性セルロース、ポリビニルピロリドン、セルロース、水、シロップ、メチルセルロース、ヒドロキシプロピルメチルセルロース(HPMC)、メチルヒドロキシベンゾエート、プロピルヒドロキシベンゾエート、タルク、ステアリン酸、マグネシウムおよび鉱油などがあるが、それには限定されない。さらに、本発明の医薬組成物は、湿潤剤、甘味剤、乳化剤、懸濁化剤、保存剤、着香剤、香料、滑沢剤またはこれらの物質の混合物のいずれも含有することができる。
【0069】
典型的には、本医薬組成物は混合物のアンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドのラブダン類を20〜40%、好ましくは25〜35%、最も好ましくは30w/w%、ならびに薬学的に許容される担体を含有する。
【0070】
本発明の医薬組成物は、それを必要とする哺乳類に対して単回で、または分割して経口ルートで投与することができる。したがって、経口投与の場合、化合物を適切な固体担体と組み合わせてカプセル、錠剤、散剤を形成することができる。さらに、本医薬組成物は、着香剤、甘味料、賦形剤等のような他の成分を含有することができる。
【0071】
さらに、本発明はアンドログラホリド混合物を含む組成物で患者を治療するための、それを必要とする患者に対して本発明の組成物を含む溶液の静脈内投与および錠剤の経口投与することを含む方法を提供する。注射液製剤の好ましい投薬量は、当該組成物が約60〜210mg/日、最も好ましくは60〜80mg/日であり、1回、2回または3回の注射で投与される。
【0072】
注射液形態の本製剤は、1mlにつき当該組成物を約8〜16mg含む。患者に投与するときは、当該組成物は好ましくは0.9%生理食塩水で約1:5から1:10の容量比に希釈する。
【0073】
以下の実施例は例示が目的であり、本発明の範囲を限定するものではない。分別のある職人ならば思いつくような理にかなった変更は、本発明の範囲から逸脱することなく本明細書に加えることができる。
【0074】
本発明の医薬組成物は、医薬産業で典型的である規模ならびにより小さな手段で調製するのに適切である所望の経口用または非経口用製品を提供する。錠剤の形態のためには、適宜必要に応じて湿式造粒法、乾式造粒法、直接圧縮法、流動床造粒法を含む医薬産業で慣用の技術に従う。
【0075】
以下の実施例で示されるパーセンテージは全て重量に基づく。
【実施例】
【0076】
活性剤を含む錠剤を調製するための典型的方法の例は、先ず活性剤をゼラチン、エチルセルロースその他の結合剤と混合することである。その場合、混合は標準的なVブレンダー内で、通常無水条件下で適切に行われる。次に、調製された直後の混合物は従来の錠剤機で強打し、スラッグを錠剤に成形することができる。調製された直後の錠剤は、シェラック、メチルセルロース、カルナバワックス、スチレン−マレイン酸共重合体などの適切なコーティングで、コーティングされる。
【0077】
経口投与のために、アンドログラホリド混合物を30mgから40mgまで含む圧縮錠剤が、上で開示されかつRemington’s Pharmaceutical Science、第39章、Mack Publishing Co.、1965年、で示されている当技術分野で公知の製造方法に従って製造される。
【0078】
本発明製剤の好ましい医薬組成物は、以下の実施例のいくつかで示されている。
実施例1
薬草Andrographis paniculata Neesから得られた乾燥抽出物に含有されるアンドログラホリド混合物を用いた、本発明の錠剤を調製するための医薬組成物
【0079】
【表1】
【0080】
一様に錠剤を製剤化するために、乾燥抽出物(アンドログラホリド混合物)の活性化合物、ジャガイモ澱粉、タルク、ゼラチン、ヒドロキシプロピルメチルセルロース、無水二酸化ケイ素、ポリエチレングリコールおよび炭酸カルシウムを、全ての成分が一様に混合されるまで乾燥条件下で慣用のVブレンダーで混合する。混合物を次に標準的な軽メッシュスクリーンに通し、無水環境下で乾燥させ、次にステアリン酸マグネシウムとブレンドして錠剤に圧縮成形し、シェラックでコーティングする。116から162mgを含有する他の錠剤は、同様に調製される。
【0081】
実施例2
薬草Andrographis paniculata Neesから得られた乾燥抽出物を用いた、本発明のカプセルを調製するための医薬組成物
【0082】
【表2】
【0083】
30mgから40mgのアンドログラホリド混合物を含む経口用カプセルの製造は、乾燥抽出物(アンドログラホリド混合物)を担体と混合して、この混合物を通常ゼラチンその他のポリマーシースに封入することから本質的になる。カプセルは食用の適合する担体中の細かく分散した化合物を封入することによって作られる当技術分野で公知のソフト形態のカプセルでよく、または、カプセルはタルク、ステアリン酸カルシウム、炭酸カルシウム、その他の無毒性固体と混合した新規な組成物から本質的になる硬カプセル剤でもよい。治療使用のために30mgまたは40mgを含むカプセルを用いる典型的な使用例が指示されている。
【0084】
単回投与であれ複数回投与であれ、または日ごとの用量であれ、投与される用量は、化合物の効力が変動するため本発明のどの化合物が使用されるかにより、選択される投与経路により、患者のサイズおよび病状の性質により当然ながら変動する。投与される用量は、1日80から160mgの一般経口投与量に対応し、経口投与量は通常1日120mgを3回;通常の静脈内用量は60から80mgであり、指示があれば後に70から100mgを投与し;通常の筋肉内用量は24時間ごとの70から100mgであり1日に1〜2回注射される。
【0085】
本発明の乾燥抽出物(アンドログラホリド混合物を含有する)を含む新規の有用な医薬組成物は、皮膚送達系、胃腸薬剤送達用デバイスなどの薬剤送達系からの予想される生理効果のための投与に適応可能であり、この送達用デバイスは天然および合成のポリマー材から製造される。制御された薬剤投与のために化合物を含有する薬剤送達系の製造で許容される材料の代表例としては、ポリ塩化ビニル、ポリイソプレン、ポリブタジエン、ポリエチレン、エチレン−酢酸ビニルコポリマー、ポリジメチルシロキサン、アクリルおよびメタクリル酸のエステルの親水性ヒドロゲル、ポリ酢酸ビニル、プロピレン酢酸ビニル共重合体などの材料がある。
【0086】
実施例3
上記組成物を含有するシェラックで被覆された錠剤は、錠剤の剤形のための混合、顆粒化、および必要に応じて圧縮する工程を含む、医薬産業で慣用される技術に従って調製する。
【0087】
具体的には、実施例1の組成物を十分な量のAndrographis paniculata乾燥抽出物と完全に混合する。アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドを含む錠剤の製造のためには、その混合物を実施例1で言及した不活性成分と共に直接的に圧縮し、その後しかるべくシェラックで被覆する。
【0088】
実施例4
本組成物によって誘導されるHL−60細胞の分化
化学物質:May Grunwald−Giemsa、NBT、レチノイン酸、サイトカラシンB、ペニシリン、ストレプトマイシン、グルタミン、ウシ胎仔血清(Sigma)。RPMI1640(GIBCO)、Boehringer Mannheimからのウシ胎仔血清、全トランスレチノイン酸およびアンドログラホリドは、Aldrichから入手。他の分離系統は、Amsar Pvt.Ltd.、インド、から入手した。FURA2−AMはMolecular Probes(アメリカ合衆国)から購入した。ニトロブルーテトラゾリウムはSigmaから入手。
【0089】
細胞培養:HL−60細胞は、20%熱不活化ウシ胎仔血清、2mMグルタミン、100IU/mlペニシリンおよび100pg/mlストレプトマイシンを補ったRPMI1640培地で、5%のCO2を含有する37℃の加湿環境で増殖させた。細胞は3×105細胞/mlで、週2回播種した。100nMの全トランスレチノイン酸を単独でまたは組成物(17.5μgml)と組み合わせて加えることによって分化を誘導し、May Grunwald−Giemsa染色の後の形態変化およびNBT’(11)を還元する能力により評価した。未分化型培養物は、3%未満のNBT陽性細胞を含んでいた。分化した培養物は、レチノイン酸処理の5日後に調査した。
【0090】
カルシウム測定[Ca2+]c
HL−60顆粒球(2×107ml)にCa2+培地+0.1%ウシ血清アルブミン中で2μMのfura−2/AMを37℃で45分間ロードし、次に107細胞/mlに希釈して氷上で保存した。使用直前に、この細胞懸濁液の0.5mlを遠心分離にかけ、5μg/mlサイトカラシンBを含む示された培地の2.4mlで再懸濁した。Fura−2蛍光(F)は、温度自動調節されたキュベット内(37℃)(LS55蛍光計、パーキン−エルマー社)で340および380nmの励起および505nmの発光波長で測定した。
【0091】
実施例5
本組成物によるT細胞内のIL−2およびIFN−γ生産の阻害
化学物質:SigmaからのコンカナバリンAおよびRPMI1640培地
細胞培養:Rockefellerマウスをエーテルによって屠殺し、膝窩の神経節および脾臓を5mlの培地RPMI1640を含むペトリプラークに置いた。無菌のRPMI1640溶液中でこれらの臓器を破壊することによってリンパ球を得、これらのリンパ球は1mlのRPMI1640培地で再懸濁し、ヌーバウアーチャンバーで定量化した。最後に、リンパ球の懸濁液を1mlのRPMIにつき4×106細胞数の濃度に調節した。得られたならば、リンパ球は組成物の存在下または非存在下で培養した。この目的のために、1mlの細胞および異なる濃度の組成物および1mlのマイトジェンコンカナバリンA(CONA、0μg/mlおよび10μg/ml)を含む、24穴(それぞれ2ml)のポリスチレンの培養プラークを使用した。
【0092】
プラークは湿度5%およびCO2の環境で37℃のオーブンで24時間インキュベートし、次にそれぞれ2mlの試料を加えて3200rpmで1分間遠心分離した。その後、細胞を0.6mlに小分けして冷凍し、サイトカインをELISA(酵素結合免疫吸着検定法)で検出した。
【0093】
IL−2およびIFN−γのためのELISA
化学物質:IL−2とIFNγはPharmingenから入手。TMBはPierceから、H2O2およびH2SO4はMerckから入手。
【0094】
サイトカイン(IL−2およびIFN)の測定のために、抗抗原を捕捉する第1の抗体、ペルオキシダーゼ酵素とコンジュゲートする第2の抗体、および検量線のための標準溶液を使用した。ELISA「高結合」96穴ポリスチレンプレートを使用した。ウェルへの付着を助長するために1ウェルにつき100μlの第1の抗体を炭酸緩衝液pH9.5で希釈し、固体部分への結合を完全にするために4℃で一晩インキュベートした。その後、ウェルの内容物を除去して1ウェルにつき300μlのTween20、0.05%p/vおよびPBS pH7.0で3回洗浄した。その後、ウェルを200μlの無脂肪ミルク5%およびPBSでブロックし、室温で1時間インキュベートした。完了後、既に説明したようにウェル内容を除去して洗浄した。その後、1ウェルにつき100μlの抗原を含む検査試料を2反復で、およびサイトカインに特異的な校正曲線を100μl加え、室温で2時間インキュベートした。その後、後者のプロトコルに従ってウェル内容を除去して5回洗浄した。その後、第2のコンジュゲート抗体にペルオキシダーゼ酵素を加え、PBSおよびSBF溶液10%に希釈し、1ウェルにつき100μlをプレーティングして室温で1時間放置した。その後、7回洗浄して、1ウェルにつき100μlのTMB溶液およびH2O2に暴露させ、30分後に暗所で展開し、反応は1ウェルにつき50μlの2MのH2SO4で停止させた。反応の結果は、450nmフィルター(Elx800ユニバーサルマイクロプレートリーダー、Biotek)を使用してELISAで測定した。
【0095】
実施例6
本組成物によるPPAR−γ受容体の刺激
化学物質:ジメチルスルホキシドはMerckから購入した。他の全ての試薬は、Promegaから購入した。
【0096】
転写アッセイ:HL−60細胞を、サイトメガロウイルスプロモーターの制御下のヒトPPARγ1発現ベクターであるpCMX−hPPARγ1でトランスフェクションした。ルシフェラーゼ活性およびβ−ガラクトシダーゼ活性を測定し、ルシフェラーゼ活性はHL−60中のβ−ガラクトシダーゼ標準にノーマライズした。
【0097】
プラスミド:GAL4−DNA結合ドメイン(DBD)およびmPPARγリガンド結合ドメイン(pGAL4DBD−mPPARγ)を発現しているプラスミドは、pGBTmPPARγ1からのScaI/BamHI断片として単離されたマウスPPARγ1リガンド結合ドメイン(アミノ酸162〜475から)をpCMXGal4 DBDへインフレーム挿入することによって構築した。
【0098】
細胞はDMSOまたは17.5μg/mlの本発明の組成物で処理し、ルシフェラーゼ活性は化学発光により測定した。
実施例7
本組成物による好中球中のNF−κB阻害
化学物質:ジメチルスルホキシド(DMSO)はMerckから入手。ウシ胎児血清およびRPMI−1640培地はGibco、米国、から入手。ニトロテトラゾリウムブルーはSigmaから、pRL−TK、pGL3および二重ルシフェラーゼリポーターアッセイ系はPromegaから。Fugene6はRocheから入手。
【0099】
細胞培養
急性骨髄性白血病からの細胞骨髄性のHL−60系を使用した。この細胞は、ジメチルスルホキシド1.3%(DMSO)の存在下で分化することができる(Santos−Beneitら、2000)。細胞は、2mMのL−グルタミン、10%の熱不活化ウシ胎児血清および抗生物質を補ったRPMI−1640培地中で、5%CO2と共に37℃に保たれる。細胞は、好中球を1.3%DMSOで4日間インキュベートすることによって好中球に分化する。分化細胞は、ニトロテトラゾリウムブルー(NBT)で分析する。
【0100】
HL−60細胞へのNFκB−pGL3ベクターのトランスフェクションおよびルシフェラーゼ測定
他で記載されているように、HL−60細胞を培養して4日間で好中球に分化させた。4日後、細胞をpGL3−NFκBベクターでトランスフェクションし、トランスフェクションの内部標準としてpRL−TK(Promega)ベクターを使用したが、これはRenillaルシフェラーゼの中レベルの発現を可能にする単純ヘルペスウイルスのチミジンキナーゼプロモーターを含む発現ベクターである。これらのベクターは、リポソームに基づく系(Fugene6、Roche)によって細胞にトランスフェクションされる。トランスフェクションの後、細胞を24時間保ち、次に本発明の組成物の存在下または非存在下で異なる時間にPAFまたはfMLPで刺激する。その後、細胞抽出物は、ルシフェラーゼ活性の測定まで−70℃に保つ。ルシフェラーゼの活性は、蛍(pGL3)およびRenilla(pRL)ルシフェラーゼ酵素の基質を有する市販の系デュアルルシフェラーゼリポーターアッセイ系(Promega)により、化学発光で測定する。
【0101】
IκBaイムノブロット
化学物質:fMLP、PMSFおよびPAFは、Calbiochemから購入した。トリス、NaCl、NP−40、デオキシコール酸塩、ドデシル硫酸ナトリウム、イプロテアーゼ(iprotease)阻害剤、メルカプトエタノールはMerckから入手。
【0102】
好中球は10分間プレインキュベートし、その後60分間fMLP(0.1μM)およびPAF(0.1μM)で刺激した。タンパク質の分析のために、細胞は溶解タンポン(50mMトリス、pH8.0、150mM NaCl、1%NP−40、0.5%デオキシコール酸塩、0.1%ドデシル硫酸ナトリウム、1mM Na2VO4、1mM PMSFおよび10μg/mlイプロテアーゼ阻害剤)で溶解した。タンパク質はブラッドフォード法で定量し、変性条件のポリアクリルアミドゲル12%(SDS/PAGE)で電気泳動により分解し、ニトロセルロース膜へ電気的に移動させた。膜を抗IκBα抗体と共にインキュベートし、次に第2のペルオキシダーゼコンジュゲート抗体とインキュベートし、最後に化学発光(ECL)で視覚化した。ゲル内のタンパク質の量の対照として、抗体をストリッピング溶液(100mM 2−メルカプトエタノール、2% SDS、62.5mMトリス−HCl、pH6.7)で処理して抗アクチン抗体とインキュベートした。最後に、各抗体について得られたシグナルの濃度測定分析を実施した。
【0103】
実施例8
野生型ラットにおける本組成物によるβ−アミロイド形成の阻害
アミロイド形成
アミロイド形成を検査するために、2つの相補性技術、チオフラビン−T蛍光法(Levine、1993;Sotoら、1995;Reyesら、1997;Inestrosaら、2000)およびコンゴーレッド結合法(Alvarezら、1998)を使用した。簡単に説明すると、チオフラビン−Tアッセイは、アミロイド線維と結合するときのチオフラビンの蛍光発光に基づき、450nmで励起されたときの482nmの発光の増加を示す。コンゴ−レッドアッセイは、形成されたアミロイドの量を測定する非常に特異的な定量アッセイである。これらの技術は、特定のアミロイド形成を確認するために、現在使用されている。
【0104】
実施例9
アンドログラホリド組成物からの化合物の説明
本発明の代表組成物は錠剤形態の医薬製剤であり、それは以下の化合物
アンドログラホリド 24.6%
14−デオキシアンドログラホリド 4.8%
ネオアンドログラホリド 0.6%
の混合物をその後の異なる医薬形態の製造のために供給し、以下の用量で適用される:
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【0105】
実施例10
紅斑性狼瘡の治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド類の混合物を使用すると、狼瘡による症状の正常化は3ヵ月間の投与を経て起こる。さらに、本組成物は他の伝統的な非ステロイド系抗炎症剤の通常の再構築(rebuilding)効果に干渉しない。
【0106】
実施例11
多発性硬化症の治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド混合物を使用すると、当該疾患による症状の正常化は本発明の組成物による3ヵ月間の治療を経て起こる。さらに、本組成物は他の治療法に干渉しない。
【0107】
実施例12
関節症および慢性関節リウマチの治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド混合物を使用すると、変形性関節症による関節硬直の正常化は、グルコサミンまたはコンドロイチン硫酸または他の抗炎症薬の有無にかかわらず、3ヵ月を経て起こる。さらに、本組成物は、伝統的な非ステロイド系抗炎症剤と異なり、これら2つのプロテオグリカン構成要素の通常の関節再構築効果に干渉しない。
【0108】
実施例13
糖尿病の治療のための経口製剤の臨床的有効性
実施例7で記載されているアンドログラホリド混合物を使用すると、糖レベルの正常化は5週を経て起こる。さらに、本組成物は他の糖低下剤の通常の再構築効果に干渉しない。
【0109】
実施例14
AIDSの治療のための経口製剤の臨床的有効性
実施例7で記載されている経口製剤を、HIV陽性の患者に投与する。正常なCD4計数値は、3ヵ月以内の治療で回復する。
【0110】
実施例15
アルツハイマー病の治療のための経口製剤の臨床的有効性
実施例7で記載されている経口製剤を、掛かりつけの開業医に診断され、独立した認定神経科医に確認されたアルツハイマー病(AD)の初期段階を示している患者に投与する。臨床試験の2週間前に、患者はMini Mental Status Exam(MMSE)、アルツハイマー病評価スケール(ADAS)、Boston Naming Test(BNT)およびToken Test(TT)などの適切な精神神経病学的検査を受ける。
【0111】
神経心理学的検査は、臨床試験当日、6週後および3ヵ月後に繰り返し行われる。これらの検査は、患者の治療法を知らない神経心理学者によって実施される。
試験の開始時に、患者は試験製剤またはプラセボに無作為に割付けされる。試験製剤およびプラセボは、1日に1、2回、経口投与される。試験中は、糖尿病、高血圧、その他の病態の治療は許容される。スコアは、各3つの観察期間中に試験製剤とプラセボの間で統計学的に比較される。治療しない場合、ADの自然経過は、臨床試験期間中に検査スコアがかなり悪化する。当該処方で記載されている組成物で治療した患者は、臨床試験期間中、患者のスコアが同じであるか向上した場合に、改善されたとみなされる。
【0112】
患者は、試験の始めに無作為に本組成物またはプラセボを投与される。本組成物およびプラセボは、1日2回投与される。試験の間、患者は糖尿病、高血圧、その他の病態の治療が許可される。本組成物およびプラセボの結果は、全ての試験期間について統計学的に比較される。プラセボを使用する患者は、かなりの認知悪化を示す。本組成物で治療する患者は、検査スコアが相当改善される。
【0113】
前述の説明から、当業者が本発明の本質的特徴を容易に確かめることはできず、また、その精神および範囲から逸脱することなく、様々な用途および条件にそれを適応させるために本発明に様々な変更および/または修正を加えることができることは明らかである。このように、これらの変更および修正は、請求項の均等物の全範囲内に適切に、正当に含まれるものとする。
【図面の簡単な説明】
【0114】
【図1】FURA2−AMインディケーターで標識されたHL−60細胞を用いて340/380nmの比により測定されたPAF誘導性カルシウム置換を示している代表的なグラフである。実施例1〜2で記載されているように、細胞はレチノイン酸だけで、または本発明の組成物(3.5μg/ml)の存在下で分化した。
【図2】PPARγのプロモーターを含むベクターでトランスフェクションしたHL−60細胞内の相対ルシフェラーゼ活性および本発明の組成物の効果を表している棒グラフである。
【図3】本発明の組成物によるコンカナバリンCONAで活性化されたT細胞内のIL−2およびINF−γ濃度の阻害を表している棒グラフである。
【図4】本発明の組成物によるlκBα分解の阻害を示す図であり、棒グラフはNFκBプロモーターを含むベクターでトランスフェクションしたHL−60細胞における相対ルシフェラーゼ活性に対する本発明の組成物の阻害率を表す。
【図5】チオフラビン染色を用いた本発明の組成物によるβアミロイド形成のin vitro阻害を示している顕微鏡写真である。
【特許請求の範囲】
【請求項1】
植物アンドログラフィス・パニキュラータ(Andrographis paniculata)の乾燥抽出物から得られ、その一般式が、
C20H30O5 アンドログラホリド
C20H30O4 14−デオキシアンドログラホリド
C26H41O8 ネオアンドログラホリド
であるジテルペンラブダン類の混合物を含む組成物。
【請求項2】
前記アンドログラホリド成分が、
i)一般式:C20H30O5
ii)分子量:350.46
iii)分子名:3−[2−[デカヒドロ−6−ヒドロキシ−5−(ヒドロキシメチル)−5,ha−ジメチル−2−メチレン−1−ナフタレニル]エチリデン]−ジヒドロ−4−ヒドロキシ−2(3h)−フラノン
iv)分子構造:
【化1】
を特徴とする、請求項1に記載の組成物。
【請求項3】
前記14−デオキシアンドログラホリド成分が、
i)一般式:C20H30O4
ii)分子量:336.46
iii)分子名:
iv)分子構造:
【化2】
を特徴とする、請求項1に記載の組成物。
【請求項4】
前記ネオアンドログラホリド成分が、
i)一般式:C26H41O8
ii)分子量:345.89
iii)分子名:
iv)分子構造:
【化3】
を特徴とする、請求項1に記載の組成物。
【請求項5】
薬、薬剤、医薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項6】
自己免疫疾患の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項7】
慢性関節リウマチの治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項8】
紅斑性狼瘡の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項9】
多発性硬化症の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項10】
アルツハイマー病の予防および治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項11】
喘息およびアレルギーの治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項12】
乾癬の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項13】
全身性皮膚筋炎の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項14】
変形性関節症の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項15】
後天性免疫不全症候群(AIDS)の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項16】
糖尿病の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項17】
組織および臓器移植患者における拒絶反応の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項18】
請求項1に記載の組成物および薬学的に許容される担体を含むことを特徴とする医薬組成物。
【請求項19】
前記ジテルペンラブダン混合物が最終乾燥抽出物中にアンドログラホリドを20〜40w/w%、14−デオキシアンドログラホリドを約3〜6w/w%およびネオアンドログラホリドを約0.2〜0.8w/w%含むことを特徴とする、請求項18に記載の医薬組成物。
【請求項20】
前記ジテルペンラブダン混合物が最終乾燥抽出物中にアンドログラホリドを約25〜35w/w%、14−デオキシアンドログラホリドを約4.5〜5.5w/w%およびネオアンドログラホリドを約0.4〜0.8w/w%含むことを特徴とする、請求項19に記載の医薬組成物。
【請求項21】
前記ジテルペンラブダン混合物が最終乾燥抽出物に基づいてアンドログラホリドを24.6w/w%、デオキシアンドログラホリドを4.8w/w%およびネオアンドログラホリドを0.6w/w%含むことを特徴とする、請求項20に記載の医薬組成物。
【請求項22】
錠剤の形態の医薬製剤に対応することを特徴とする、請求項18〜21のいずれか1項に記載の医薬組成物。
【請求項23】
経口投与され、以下の分子の以下の用量に寄与することを特徴とする、請求項22に記載の医薬組成物:
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【請求項24】
自己免疫疾患の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項25】
慢性関節リウマチの治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項26】
紅斑性狼瘡の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項27】
多発性硬化症の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項28】
アルツハイマー病の予防および治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項29】
喘息およびアレルギーの治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項30】
乾癬の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項31】
全身性皮膚筋炎の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項32】
変形性関節症の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項33】
後天性免疫不全症候群(AIDS)の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項34】
糖尿病の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項35】
移植患者の臓器および組織の拒絶反応の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項36】
適切な経腸、非経口、皮膚、目、鼻、耳、直腸、膣、尿道、舌下、咽頭−気管−気管支の医薬形態であることができ、アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドのジテルペンラブダン混合物を含有する乾燥抽出物を含むことを特徴とする、請求項18に記載の医薬組成物。
【請求項37】
前記ジテルペンラブダン混合物が最終乾燥抽出物重量に基づいてアンドログラホリドを20〜40w/w%、14−デオキシアンドログラホリドを約3〜6w/w%およびネオアンドログラホリドを約0.2〜0.8w/w%含むことを特徴とする、請求項36に記載の医薬組成物。
【請求項38】
前記ジテルペンラブダン混合物が最終乾燥抽出物重量に基づいてアンドログラホリドを約25〜35w/w%、14−デオキシアンドログラホリドを約4.5〜5.5w/w%およびネオアンドログラホリドを約0.4〜0.8w/w%含むことを特徴とする、請求項37に記載の医薬組成物。
【請求項39】
前記ジテルペンラブダン混合物が最終乾燥抽出物重量に基づいてアンドログラホリドを24.6w/w%、デオキシアンドログラホリドを4.8w/w%およびネオアンドログラホリドを0.6w/w%含むことを特徴とする、請求項38に記載の医薬組成物。
【請求項40】
対応する経路により以下の用量で投与されることを特徴とする、請求項36に記載の医薬組成物:
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【請求項41】
自己免疫疾患の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項42】
慢性関節リウマチの治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項43】
紅斑性狼瘡の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項44】
多発性硬化症の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項45】
アルツハイマー病の予防および治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項46】
喘息およびアレルギーの治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項47】
乾癬の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項48】
全身性皮膚筋炎の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項49】
変形性関節症の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項50】
後天性免疫不全症候群(AIDS)の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項51】
糖尿病の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項52】
移植患者の臓器および組織の拒絶反応の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項1】
植物アンドログラフィス・パニキュラータ(Andrographis paniculata)の乾燥抽出物から得られ、その一般式が、
C20H30O5 アンドログラホリド
C20H30O4 14−デオキシアンドログラホリド
C26H41O8 ネオアンドログラホリド
であるジテルペンラブダン類の混合物を含む組成物。
【請求項2】
前記アンドログラホリド成分が、
i)一般式:C20H30O5
ii)分子量:350.46
iii)分子名:3−[2−[デカヒドロ−6−ヒドロキシ−5−(ヒドロキシメチル)−5,ha−ジメチル−2−メチレン−1−ナフタレニル]エチリデン]−ジヒドロ−4−ヒドロキシ−2(3h)−フラノン
iv)分子構造:
【化1】
を特徴とする、請求項1に記載の組成物。
【請求項3】
前記14−デオキシアンドログラホリド成分が、
i)一般式:C20H30O4
ii)分子量:336.46
iii)分子名:
iv)分子構造:
【化2】
を特徴とする、請求項1に記載の組成物。
【請求項4】
前記ネオアンドログラホリド成分が、
i)一般式:C26H41O8
ii)分子量:345.89
iii)分子名:
iv)分子構造:
【化3】
を特徴とする、請求項1に記載の組成物。
【請求項5】
薬、薬剤、医薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項6】
自己免疫疾患の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項7】
慢性関節リウマチの治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項8】
紅斑性狼瘡の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項9】
多発性硬化症の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項10】
アルツハイマー病の予防および治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項11】
喘息およびアレルギーの治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項12】
乾癬の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項13】
全身性皮膚筋炎の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項14】
変形性関節症の治療に適切な薬を調製するために特に有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項15】
後天性免疫不全症候群(AIDS)の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項16】
糖尿病の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項17】
組織および臓器移植患者における拒絶反応の治療に適切な薬を調製するために有用であることを特徴とする、請求項1に記載の組成物の使用。
【請求項18】
請求項1に記載の組成物および薬学的に許容される担体を含むことを特徴とする医薬組成物。
【請求項19】
前記ジテルペンラブダン混合物が最終乾燥抽出物中にアンドログラホリドを20〜40w/w%、14−デオキシアンドログラホリドを約3〜6w/w%およびネオアンドログラホリドを約0.2〜0.8w/w%含むことを特徴とする、請求項18に記載の医薬組成物。
【請求項20】
前記ジテルペンラブダン混合物が最終乾燥抽出物中にアンドログラホリドを約25〜35w/w%、14−デオキシアンドログラホリドを約4.5〜5.5w/w%およびネオアンドログラホリドを約0.4〜0.8w/w%含むことを特徴とする、請求項19に記載の医薬組成物。
【請求項21】
前記ジテルペンラブダン混合物が最終乾燥抽出物に基づいてアンドログラホリドを24.6w/w%、デオキシアンドログラホリドを4.8w/w%およびネオアンドログラホリドを0.6w/w%含むことを特徴とする、請求項20に記載の医薬組成物。
【請求項22】
錠剤の形態の医薬製剤に対応することを特徴とする、請求項18〜21のいずれか1項に記載の医薬組成物。
【請求項23】
経口投与され、以下の分子の以下の用量に寄与することを特徴とする、請求項22に記載の医薬組成物:
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【請求項24】
自己免疫疾患の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項25】
慢性関節リウマチの治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項26】
紅斑性狼瘡の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項27】
多発性硬化症の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項28】
アルツハイマー病の予防および治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項29】
喘息およびアレルギーの治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項30】
乾癬の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項31】
全身性皮膚筋炎の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項32】
変形性関節症の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項33】
後天性免疫不全症候群(AIDS)の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項34】
糖尿病の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項35】
移植患者の臓器および組織の拒絶反応の治療に有用であることを特徴とする、請求項22または23に記載の医薬組成物の使用。
【請求項36】
適切な経腸、非経口、皮膚、目、鼻、耳、直腸、膣、尿道、舌下、咽頭−気管−気管支の医薬形態であることができ、アンドログラホリド、14−デオキシアンドログラホリドおよびネオアンドログラホリドのジテルペンラブダン混合物を含有する乾燥抽出物を含むことを特徴とする、請求項18に記載の医薬組成物。
【請求項37】
前記ジテルペンラブダン混合物が最終乾燥抽出物重量に基づいてアンドログラホリドを20〜40w/w%、14−デオキシアンドログラホリドを約3〜6w/w%およびネオアンドログラホリドを約0.2〜0.8w/w%含むことを特徴とする、請求項36に記載の医薬組成物。
【請求項38】
前記ジテルペンラブダン混合物が最終乾燥抽出物重量に基づいてアンドログラホリドを約25〜35w/w%、14−デオキシアンドログラホリドを約4.5〜5.5w/w%およびネオアンドログラホリドを約0.4〜0.8w/w%含むことを特徴とする、請求項37に記載の医薬組成物。
【請求項39】
前記ジテルペンラブダン混合物が最終乾燥抽出物重量に基づいてアンドログラホリドを24.6w/w%、デオキシアンドログラホリドを4.8w/w%およびネオアンドログラホリドを0.6w/w%含むことを特徴とする、請求項38に記載の医薬組成物。
【請求項40】
対応する経路により以下の用量で投与されることを特徴とする、請求項36に記載の医薬組成物:
a)1〜5mgのアンドログラホリド/kg/日
b)0.2〜1mgの14−デオキシアンドログラホリド/kg/日
c)0.02〜0.12mgのネオアンドログラホリド/kg/日。
【請求項41】
自己免疫疾患の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項42】
慢性関節リウマチの治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項43】
紅斑性狼瘡の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項44】
多発性硬化症の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項45】
アルツハイマー病の予防および治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項46】
喘息およびアレルギーの治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項47】
乾癬の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項48】
全身性皮膚筋炎の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項49】
変形性関節症の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項50】
後天性免疫不全症候群(AIDS)の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項51】
糖尿病の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【請求項52】
移植患者の臓器および組織の拒絶反応の治療に有用であることを特徴とする、請求項36〜40のいずれか1項に記載の医薬組成物の使用。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公表番号】特表2007−519759(P2007−519759A)
【公表日】平成19年7月19日(2007.7.19)
【国際特許分類】
【出願番号】特願2006−551723(P2006−551723)
【出願日】平成16年5月21日(2004.5.21)
【国際出願番号】PCT/EP2004/005516
【国際公開番号】WO2005/074953
【国際公開日】平成17年8月18日(2005.8.18)
【出願人】(506263985)ウニベルシダドゥ・アウストゥラル・デ・チリ (1)
【Fターム(参考)】
【公表日】平成19年7月19日(2007.7.19)
【国際特許分類】
【出願日】平成16年5月21日(2004.5.21)
【国際出願番号】PCT/EP2004/005516
【国際公開番号】WO2005/074953
【国際公開日】平成17年8月18日(2005.8.18)
【出願人】(506263985)ウニベルシダドゥ・アウストゥラル・デ・チリ (1)
【Fターム(参考)】
[ Back to top ]