
PSMAのペプチド模倣体阻害剤、それらを含む化合物、およびそれらの使用方法
前立腺特異的膜抗原(PMSA)の強力阻害剤である、式:A−L−Bの化合物であって、式中、Aがグルタメートまたはグルタメート類似体であり;Lがホスホルアミデートまたはホスホルアミデート類似体であり;Bがセリンまたはセリン類似体である、化合物が記載される。このような化合物は、前立腺癌の治療に有用であり、蛍光色素に化学的に結合する場合、蛍光イメージングのために前立腺癌細胞を効率的かつ選択的に標識することができる。
【発明の詳細な説明】
【技術分野】
【0001】
(関連出願への相互参照)
本願は、2006年3月14日に出願された米国仮特許出願第60/782,211号の優先権の利益を請求し、その全体は、本明細書中に参考として援用される。
【0002】
(発明の分野)
本発明は、前立腺特異的膜抗原(PSMA)に対する高い親和性および特異性を有する小分子、ならびに診断目的および治療目的のためにそれらを使用する方法に関する。
【背景技術】
【0003】
(関連技術の要約)
前立腺特異的膜抗原(PSMA)は、前立腺癌細胞の表面ならびに種々の固形腫瘍の新生血管系に特異的に過剰発現する。そのため、PSMAは、前立腺癌の検出および管理のための臨床的バイオマーカーとして注目を集めてきた。一般的に、これらの手法は、イメージング剤または治療剤を方向づけるためにPSMAを特異的に標的とする抗体を利用する。例えば、ProstaScint(Cytogen、米国ペンシルベニア州フィラデルフィア)は、前立腺癌の検出およびイメージングのためにFDAの承認を得たものであるが、キレート化された放射性同位元素(インジウム(III))を送達するために抗体を利用する。しかし、ProstaScint技術は死細胞の検出に限定されているため、その臨床的な重要性が疑わしいことが現在認められている。
【0004】
抗体を使用する癌の診断および治療は、これらの分子を血液から排除するのが遅い点や、血管透過性に劣っている点などの問題により、十分な成果をあげていない。さらに、細胞表面の標的に結合する大きな抗体は、その後隣接した細胞表面部位に他の抗体が結合する際の障害となるため、細胞表面の標識化が低減してしまう。
【0005】
診断剤または治療剤を送達する抗体の細胞表面標的として機能することに加えて、PSMAのほとんど見過ごされている固有の特性に、その酵素活性がある。すなわち、PSMAは、ジペプチドと同程度の小さな分子を認識し、プロセッシングすることができる。この特性を有するにもかかわらず、PSMAは、新規の診断戦略および治療戦略の開においてほとんど考察されてこなかった。PSMAの標識小分子阻害剤を使用して前立腺癌細胞を検出した結果について記載した参考文献の例は、最近では数例しかない。
【発明の開示】
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、高親和性および特異性を有する前立腺特異的膜抗原(PSMA)と結合する化合物を含む。これらの特性により、本発明の化合物は、PSMAを提示する細胞に診断剤または治療剤を送達するのに有用であるか、あるいは本発明の化合物を固体担体に直接的または間接的に固定する時などにこのような細胞を捕捉および検出するのに有用である。それためこれに付随して、本発明はまた、PSMAを提示する細胞を検出および/または同定する診断方法であって、PSMAを提示することが疑われる細胞を検出可能なマーカーまたは検出素子に結合する本発明の化合物と接触させる(またはこれらの接触を引き起こす)工程と、前記化合物と細胞とが結合しているかどうかを判定する工程を含む、方法も含む。本発明はまた、本発明の化合物を、医薬的に許容される担体、賦形剤および/または希釈剤とともに含む組成物も含む。本発明はさらに、前立腺癌を抑制または治療する方法であって、前立腺癌治療剤(またはその組成物)に結合する治療有効量の本発明の化合物を前立腺癌の羅患体に投与する工程を含む、方法も含む。
【0007】
小分子を調製および精製することは抗体を調製することよりも効率性がよく、費用対効果も高いことから、本発明の小分子は、PMSA抗体を使用する手法よりも利点がある。
【発明を実施するための最良の形態】
【0008】
(発明の詳細な説明)
小分子が腫瘍細胞に対して十分な親和性を有する場合は、小分子によって腫瘍の標的化を達成できることが認識されている。抗体に基づく送達剤と拮抗し、実質的に同等となるには、小分子の親和性の範囲は、標的に対する抗体の親和性と同じでなければならない。
【0009】
第1の態様において、本発明は、この基準を満たす、PSMA(好ましくはPSMAの酵素認識部位)を標的とする小分子を含む。本発明の化合物の固有の特徴は、それらの化合物が中心的なホスホルアミデートコアに主に由来することである。具体的には、このような化学化合物には、図1(CCS、JM139および2−PMPAを除く)、図2および図7〜12に示すすべての化合物が含まれる。本発明の化合物は、実施例(下記参照)に記載の検定に従って測定した場合に、一般的には5μM未満、好ましくは1μM未満、より好ましくは100nM未満のIC50を有する。
【0010】
本発明の化合物は、PSMA発現細胞に診断剤または治療剤を送達するために官能基化することもできるPMSAの強力なペプチド模倣体阻害剤を生じる以下の3つの成分を有する:1)P1’位のグルタメートまたはグルタメート類似体、2)亜鉛結合基でしての中心的なホスホルアミデートまたはホスホルアミデート類似体、および3)側鎖を介して結合し、好ましくはそのN末端またはN末端等価物において疎水基を有するP1位のセリンまたはセリン類似体。
【0011】
従って、第1の態様の実施形態において、本発明は、以下の式Iの化合物であって:
A−L−B (I)
(式中、Aがグルタメートまたはグルタメート類似体であり;Lがホスホルアミデートまたはホスホルアミデート類似体であり;Bがセリンまたはセリン類似体である、化合物を提供する。好ましくは、セリンまたはセリン類似体は、N末端またはN末端等価物において疎水基を有する。本明細書に開示される本発明のすべての化合物の医薬的に許容される塩もまた、本発明の態様である。
【0012】
第1の態様の好ましい実施形態において、本発明は、Aが以下の式(Ia)に記載のものである式Iの化合物であって:
【0013】
【化9】

式中、
nが1、2、3、4、5または6であり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−であって、式中、QがLに結合し;
R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
それぞれが独立して−HまたはC1−C6アルキルである、
化合物;ならびにその医薬的に許容される塩を提供する。
【0014】
第1の態様の別の実施形態において、本発明は、Bが以下の式(Ib)である式Iの化合物であって:
【0015】
【化10】
式中、
nが1、2、3、4、5または6であり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であって、式中、TがLに結合し;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が、−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−HまたはC1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
各R3が独立して−HまたはC1−C6アルキルである、
化合物;ならびにその医薬的に許容される塩を提供する。
【0016】
第1の態様の別の実施形態において、本発明は、式Iの化合物であって、式中、Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であって、式中、各R3が独立して−HまたはC1−C6アルキルである、化合物(ならびにその医薬的に許容される塩)を提供する。
【0017】
第1の態様の好ましい実施形態において、本発明は、以下の式(II)の化合物であって:
【0018】
【化11】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−HまたはC1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
化合物;ならびにその医薬的に許容される塩を提供する。
【0019】
第1の態様の好ましい実施形態において、本発明は、以下の式(III)の化合物であって:
【0020】
【化12】
式中、各変数が式(II)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0021】
第1の態様の好ましい実施形態において、本発明は、以下の式(IV)の化合物であって:
【0022】
【化13】
式中、各変数が式(II)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0023】
式(II)〜(IV)の好ましい実施形態において、R1およびR2はそれぞれ−C(O)OHである。
【0024】
式(II)〜(IV)の好ましい実施形態において、R4は−C(O)OHである。
【0025】
式(II)〜(IV)のより好ましい実施形態において、R1、R2およびR4はそれぞれ−C(O)OHである。
【0026】
式(II)〜(IV)の好ましい実施形態において、R10は−C(O)−フェニルである。
【0027】
式(II)〜(IV)のより好ましい実施形態において、R10はR7である。
【0028】
式(II)〜(IV)のより好ましい実施形態において、R10はR7であって、式中、R7が−L1−R8であって、式中、
L1が、−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHである。
【0029】
式(II)〜(IV)のより好ましい実施形態において、R10はR7であって、式中、R7が、−L1−R8であって、式中、
L1が、−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHである。
【0030】
式(II)〜(IV)のより好ましい実施形態において、R10は、ペプチド、デンドリマーまたはペプチドデンドリマーである。
【0031】
第1の態様のより好ましい実施形態において、本発明は、式(I)の化合物であって、以下の通りである、化合物を提供する。
【0032】
【化14】
【0033】
【化15】
第2の態様において、本発明は、検出可能な標識と共有結合する本発明の第1の態様の化合物、治療剤、または固体担体に結合する生体分子アンカーを含むキメラ化合物を含む。固体担体の例には、ポリリシンがコーティングされた、無水マレイン酸がコーティングされた、またはストレプトアビジンがコーティングされた、市販の96ウェルプレートが含まれる。他の固体担体には、金がコーティングされた、または官能基化した金がコーティングされた市販のセンサーチップが含まれる。
【0034】
一実施形態において、検出可能な標識は蛍光標識である。標準的な蛍光標識には、Alexa Fluor染料、BODIPY染料、フルオレセイン系染料、ローダミン系染料、クマリン系染料、およびピレン系染料が含まれる。
【0035】
別の実施形態において、検出可能な標識は、特異的結合対の片側、例えば、ビオチンとストレプトアビジンの結合対の内のビオチンである。代表的な結合対の薬剤および生体分子アンカーには、ビオチン、DNAもしくはRNAのオリゴヌクレオチド、または脂質が含まれる。
【0036】
別の実施形態において、検出可能な標識は、99Tcなどの放射性同位元素またはGdなどのMRI造影剤に結合することが可能なキレート構造である。標準的なキレート剤には、DOTA、DTPA、CHX−A”、PCTA、およびDO3Aが含まれる。
【0037】
本明細書に記載されるような標準的な蛍光標識、結合対、キレート剤および生体分子アンカーが使用される場合がある。当業者に既知の標準的な方法は、このような薬剤およびアンカーならびに治療剤に本発明の化合物を結合するために使用される場合がある。
【0038】
治療剤は、好ましくは、PSMAを提示する細胞の1つ以上の生物学的プロセスに干渉し、それによってPMSA提示細胞に関連する疾患を治療または抑制する、化合物である。治療剤は、90Yまたは188Reなどのキレート化または共有結合した細胞障害性放射性同位元素を場合により含む場合があり、このようなキレート化または共有結合は、当業者に既知の手段により行うことができる。
【0039】
治療剤には、癌細胞の細胞表面に直接結合するか、癌細胞における抗原ペプチドの発現を調節するかのいずれかによって、腫瘍細胞の免疫原性を増大させるものが含まれる。共有結合のために選択される薬剤は、既知の抗癌性、抗増殖性、または細胞障害性を有する。あるいは、このような剤は、T細胞免疫監視機構の標的として細胞の免疫原性を促進するか、または増大させる既知の特性を有する。
【0040】
治療剤にはまた、2−メトキシエストラジオールなどのステロイド系剤や、ミフェプリストーン、タモキシフェン、レチノイン酸などのアポトーシス誘導物質、酪酸などのヒストンデアセチラーゼ阻害剤、Plk1 siRNAなどのアポトーシス誘導または細胞障害性siRNA、ドキソルビシンなどの有糸分裂阻害剤、メトトレキサートなどの代謝拮抗剤、および細胞毒をカプセル化するように設計されたナノ粒子またはリポソームが含まれるが、これらに限定されない。
【0041】
第2の態様の実施形態において、本発明は、式:A−L−Bの化合物であって、式中、
Aがグルタメートまたはグルタメート類似体であり;
Lがホスホルアミデートまたはホスホルアミデート類似体であり;
Bがセリンまたはセリン類似体であり;
前記化合物が、前記化合物の任意の置換可能な位置で、検出可能な標識、治療剤、または固体担体に結合する生体分子アンカーに、2価のリンカーを介して共有結合する、
化合物;ならびにその医薬的に許容される塩を提供する。
【0042】
第2の態様の実施形態において、前記2価のリンカーは、アミノ酸、オリゴペプチド、ポリ(エチレン)グリコール、オリゴエチレングリコールなどに由来する。
【0043】
第2の態様の実施形態において、前記化合物は、検出可能な標識に共有結合する。
【0044】
第2の態様の別の実施形態において、前記化合物は、治療剤に結合共有結合する。
【0045】
第2の態様の別の実施形態において、前記化合物は、固体担体に結合する生体分子アンカーに共有結合する。
【0046】
第2の態様の好ましい実施形態において、前記化合物は、検出可能な標識に共有結合し、前記検出可能な標識は蛍光標識である。
【0047】
第2の態様の別の好ましい実施形態において、前記化合物は、検出可能な標識に共有結合し、前記検出可能な標識は、放射性同位元素または磁気共鳴映像法用造影剤に結合するキレート構造である。
【0048】
第2の態様の別の好ましい実施形態において、本発明は、以下の式(VI)の化合物であって:
【0049】
【化16】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R7が、−X−R8または−L1−R8であって、式中、
Xが、−C(O)−、−S(O)2、−O−、−S−または−N(R3)−であり;
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)、−C(O)−L2−、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、治療剤、検出可能な標識、または固体担体に結合する生体分子アンカーであり;
Qが、−O−、−S−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
化合物;ならびにその医薬的に許容される塩を提供する。
【0050】
第2の態様の好ましい実施形態において、本発明は、以下の式(VII)の化合物であって:
【0051】
【化17】
式中、各変数が式(VI)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0052】
第2の態様の好ましい実施形態において、本発明は、以下の式(VIII)の化合物であって:
【0053】
【化18】
式中、各変数が式(VI)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0054】
式(VI)〜(VIII)の好ましい実施形態において、R1およびR2はそれぞれ−C(O)OHである。
【0055】
式(VI)〜(VIII)の好ましい実施形態において、R4は−C(O)OHである。
【0056】
式(VI)〜(VIII)のより好ましい実施形態において、R1、R2およびR4はそれぞれ−C(O)OHである。
【0057】
式(VI)〜(VIII)のより好ましい実施形態において、L1は−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とする。
【0058】
式(VI)〜(VIII)のより好ましい実施形態において、L1は−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−または−N(R3)−で置換されないことを条件とする。
【0059】
式(VI)〜(VIII)のより好ましい実施形態において、L1は、ペプチド、デンドリマーまたはペプチドデンドリマーである。
【0060】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識である。
【0061】
式(VI)〜(VIII)の好ましい実施形態において、R8は固体担体に結合する生体分子アンカーである。
【0062】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は蛍光色素である。
【0063】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は蛍光標識であって、前記蛍光標識は、Alexa Fluor、BODIPY、フルオレセイン、ローダミン、クマリンまたはピレン系染料である。
【0064】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は蛍光色素であって、前記蛍光色素は、フルオレセインまたはフルオレセイン誘導体である。
【0065】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識はキレート剤である。
【0066】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識はキレート剤であって、前記キレート剤は、DOTA、DTPA、CHX−A”、PCTAおよびDO3Aである。
【0067】
式(VI)〜(VIII)のより好ましい実施形態において、R8はR9であって、式中、
R9が、C1−C6アルキル、アリールまたはC1−C6アルキル−アリールであって、式中、R9が、独立して−COOHまたはN(R91)2である1〜3個の基で置換され、式中、
各R91が独立して、独立して−COOHまたは−N(R92)2である1〜3個の基で置換される−HまたはC1−C6アルキルであって、式中、
各R92が独立して、1〜3個のCOOHで置換される−HまたはC1−C6アルキルである。
【0068】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は特異的結合対の片側である。
【0069】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は特異的結合対の片側であって、前記特異的結合対の前記片側は、ビオチン、DNAもしくはRNAのオリゴヌクレオチド、または脂質である。
【0070】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は特異的結合対の片側であって、前記特異的結合対の前記片側はビオチンである。
【0071】
式(VI)〜(VIII)の好ましい実施形態において、R8は治療剤である。
【0072】
式(VI)〜(VIII)のより好ましい実施形態において、R8は、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基である。
【0073】
第1の態様の別の好ましい実施形態において、本発明は、以下の式(IX)の化合物であって:
【0074】
【化19】
式中、R7が−O−R8であって、式中、R8が、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基であり;各残りの変数が式(VI)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0075】
第1の態様のより好ましい実施形態において、本発明は、式(VI)の化合物であって、以下の通りである:
【0076】
【化20】
【0077】
【化21】
【0078】
【化22】
【0079】
【化23】
を提供する。
【0080】
第3の態様において、本発明は、本発明の第2の態様のキメラ化合物、またはその医薬的に許容される塩、および医薬的に許容される担体、希釈剤または賦形剤を含む組成物を含む。
【0081】
第4の態様において、本発明は、本発明の第2の態様のキメラ化合物を含む診断キットを含む。本発明の本態様の一実施形態において、検出可能な標識または生体分子アンカーは、特異的結合対の一方のメンバー(例えば、ビオチン)であり、前記キットは前記特異的結合対のもう一方のメンバーを含む。
【0082】
第5の態様において、本発明は、PMSA提示細胞を検出する方法であって、PMSAを提示することが疑われる細胞を本発明の第2の態様のキメラ化合物と接触させる工程と、前記検出可能な標識が、PMSA提示細胞に結合する場合にのみ検出される条件下で、前記検出可能な標識の存在を測定する工程とを含む、方法を含む。検出可能な標識を検出する方法は、当然ながら、使用する標識により異なり、当業者に明らかになるであろう。例えば、検出可能な標識が近赤外蛍光標識である場合、標識の検出はin vivo蛍光イメージングにより達成することができる。
【0083】
第6の態様において、本発明は、PMSAを提示する細胞を伴う疾患を阻害または治療する方法であって、前記細胞を本発明の第2の態様の化合物または本発明の第3の態様の組成物と接触させるか、または前記細胞への接触を引き起こす工程を含み、前記キメラ化合物が、治療剤に共有結合する本発明の第1の態様の化合物を含む、方法を含む。本態様の一実施形態において、本発明は、PMSA提示細胞を伴う疾患を有する哺乳類の被験体(好ましくはヒト)に、前記疾患の抑制または治療に有効な量の前記組成物を投与する工程を含む。適切な製剤および投与方法は、標準的な方法を使用して慣例的に決定することができる。
【0084】
第7の態様において、本発明は、PMSA提示細胞を捕捉、検出および定量化する方法であって、PMSAを提示することが疑われる細胞を本発明の第2の態様のキメラ化合物と接触させる工程と、捕捉または固定されたPSMA提示細胞を検出する工程とを含む、方法を含む。第7の態様の好ましい実施形態において、第2の態様のキメラ化合物は、固体担体上の生体分子アンカーに結合する。細胞を検出する方法は、当然ながら、使用する検出素子により異なり、当業者に明らかになるであろう。例えば、本発明の第2の態様のキメラ化合物が固体担体に結合する場合、PSMA提示細胞の検出は、プラズモン共鳴を使用して直接達成することもできれば、あるいは細胞が放出され、フローサイトメトリーを使用して蛍光標識で標識されることで達成することもできる。
【0085】
本発明で使用される場合がある代表的なデンドリマーについては、J.M.J.Frechet,D.A.Tomalia,Dendrimers and Other Dendritic Polymers,John Wiley & Sons,Ltd.NY,NY(2002)に記載されている。
【0086】
本発明の併用に有用な代表的な化合物には、上記の化合物、それらの医薬的に許容される酸、ならびにそれらの塩基付加塩および溶媒和物が含まれる。本発明の化合物が酸付加塩として得られる場合、遊離塩基は、酸性塩の溶液を塩基性化することによって得ることができる。逆に、産物が遊離塩基である場合、付加塩、特に医薬的に許容される付加塩は、塩基化合物から酸付加塩を調製する従来の方法に従って、適切な有機溶媒中に遊離塩基を溶解し、その溶液を酸で処理することによって産生される場合がある。
【0087】
無毒性医薬塩には、例えば、塩酸、リン酸、臭化水素酸、硫酸、スルフィン酸、蟻酸、トルエンスルホン酸、メタンスルホン酸、硝酸、安息香酸、クエン酸、酒石酸、マレイン酸、ヨウ化水素酸、アルカン酸(例えば酢酸)、HOOC−(CH2)n−COOH(式中、nは0〜4である)などの酸の塩が含まれる。無毒性医薬塩基付加塩には、例えば、ナトリウム、カリウム、カルシウム、アンモニウムなどの塩基の塩が含まれる。当業者であれば、多種多様な無毒性の医薬的に許容される付加塩を認識するであろう。
【0088】
本発明の化合物および/またはそれらの組成物は、動物およびヒトのPMSA関連疾患の抑制および/または治療における特定の用途が見出されている。従って、本発明の別の態様には、治療有効量の本発明の1種以上の化合物または本発明の1種以上の化合物を含有する組成物を、このようなPMSA関連疾患の治療を必要とする羅患体に投与することがある。好ましくは、羅患体は哺乳類であり、最も好ましくはヒトである。この状況で使用される場合、前記化合物は、それ自体が投与される場合があるが、通常は医薬組成物の形態に製剤化されて投与される。正確な組成物は、とりわけ投与方法により異なり、当業者に明らかになるであろう。例えば、Remington’s Pharmaceutical Sciences,20th ed.,2001には、多種多様な適切な医薬組成物が記載されている。
【0089】
経口投与に適切な製剤は、(a)希釈液(例えば、水、生理食塩水、またはPEG 400)に懸濁した有効量の活性化合物などの液体溶液;(b)それぞれ液体、固体、顆粒またはゼラチンなどの活性成分を所定量含有する、カプセル剤、小包、または錠剤;(c)適切な液体中の懸濁剤;および(d)適切な乳剤、から構成されてよい。錠剤の形態には、ラクトース、スクロース、マンニトール、ソルビトール、リン酸カルシウム、トウモロコシ澱粉、ジャガイモ澱粉、微結晶性セルロース、ゼラチン、コロイド状二酸化ケイ素、タルク、ステアリン酸マグネシウム、ステアリン酸、ならびに他の賦形剤、顔料、充填剤、結合剤、希釈剤、緩衝剤、湿潤剤、保存剤、着香剤、染料、崩壊剤および医薬的に適合する担体が含まれてよい。口内錠の形態は、香料中の活性成分(例えばスクロース)、ならびに不活性塩基中の活性成分を含む香錠(例えば、活性成分に加えて当該技術分野で既知の担体を含有する、ゼラチンおよびグリセリンまたはスクロースおよびアカシア乳剤、ゲル剤など)を含んでよい。
【0090】
選択化合物は、単独でまたは他の適切な成分と組み合わせて、吸入により投与されるようにエアロゾル製剤に作製することができる(すなわち、「霧状」にすることができる)。エアロゾル製剤は、許容される加圧噴射剤(例えば、ジクロロジフルオロメタン、プロパン、窒素など)に入れることができる。
【0091】
直腸投与に適切な製剤には、例えば、坐剤基剤で包装された核酸から構成される坐剤が含まれる。適切な坐剤基剤には、天然または合成のトリグリセリドまたはパラフィン族炭化水素が含まれる。さらに、基剤と選択化合物との組み合わせから構成されるゼラチン直腸投与カプセル(例えば、液体トリグリセリド、ポリエチレングリコール、およびパラフィン族炭化水素を含む)も使用することができる。
【0092】
例えば、関節内経路(関節中)、静脈内経路、筋肉内経路、皮内経路、腹腔内経路、皮下経路などによる非経口投与に適切な製剤には、製剤を所期のレシピエントの血液と浸透圧が等しくなるようにする抗酸化剤、緩衝液、静菌剤および溶質を含有し得る水溶性および非水溶性の等張性無菌注射溶液、ならびに懸濁化剤、溶解剤、増粘剤、安定剤および保存剤を含み得る水溶性および非水溶性の無菌懸濁液が含まれる。本発明の実施において、組成物は、例えば、静脈内、経口的、局所的、腹腔内、膀胱内、髄腔内に投与することができる。非経口投与、経口投与、皮下投与および静脈内投与が、好ましい投与方法である。適切な溶液製剤の具体例は、水中に約0.5〜100mg/mLの化合物と約1000mg/mLのプロピレングリコールとを含む場合がある。適切な溶液製剤の別の具体例は、水中に約0.5〜100mg/mLの化合物と約800〜1000mg/mLのポリエチレングリコール400(PEG 400)とを含む場合がある。
【0093】
適切な懸濁製剤の具体例は、水中に約0.5〜30mg/mLの化合物と、約200mg/mLのエタノール、約1000mg/mLの植物油(例えばトウモロコシ油)、約600〜1000mg/mLの果汁(例えばグレープフルーツジュース)、約400〜800mg/mLの乳汁、約0.1mg/mLのカルボキシメチルセルロース(または微結晶性セルロース)、約0.5mg/mLのベンジルアルコール(またはベンジルアルコールと塩化ベンザルコニウムとの組み合わせ)および約40〜50mMの緩衝液(pH7)(例えば、リン酸緩衝液、酢酸緩衝液、クエン酸緩衝液、あるいは緩衝液の代わりに5%のデキストロースが使用される場合がある)からなる群から選択される1種以上の賦形剤とを含む場合がある。
【0094】
適切なリポソーム懸濁製剤の具体例は、水中に約0.5〜30mg/mLの化合物と、約100〜200mg/mLのレシチン(または他のリン脂質もしくはリン脂質の混合物)と、場合により5mg/mLのコレステロールとを含む場合がある。化合物9の皮下投与の場合には、100mg/mLのレシチンを有する水中の5mg/mLの化合物と、100mg/mLのレシチンおよび5mg/mLのコレステロールを有する水中の5mg/mLの化合物とを含むリポソーム懸濁製剤が、良好な結果をもたらす。本製剤は、本発明の他の化合物に使用される場合もある。
【0095】
化合物の製剤は、アンプルやバイアルなど、単回用量または複数回用量の密封容器で提供することができる。注射溶液および注射懸濁液は、既述の種類の無菌の粉剤、顆粒および錠剤から調製することができる。
【0096】
前記医薬製剤は、単位投与形態が好ましい。このような形態では、前記製剤は、適切な量の有効成分を含有する単位用量に細分される。単位投与形態は、包装製剤、すなわち、小包にされた錠剤、カプセル剤、およびバイアルまたはアンプルに入った粉剤など、個別の量の製剤を含有する包装物であってよい。また、単位投与形態は、カプセル剤、錠剤、カシェ剤または口内錠自体であってもよければ、あるいは包装形態における適切な数のこれらのいずれかであってもよい。組成物はまた、所望により、以下に詳述する他の適合した治療剤を含有してもよい。
【0097】
別の態様において、本発明は、PMSA関連疾患の治療および/または阻害のための組成物を作製する方法であって、本発明の化合物を、医薬的に許容される担体、希釈剤および/または賦形剤と混合する工程を含む、方法を含む。
【0098】
PMSA関連疾患の治療のための治療的使用において、本発明の医薬的方法で利用される化合物は、治療的有益性を達成するのに適切な投与レベルで、PMSA関連疾患であると診断された罹患体に投与される。治療的有益性とは、化合物の投与が時間とともに罹患体に有益な効果をもたらすことを意味する。治療的有益性はまた、化合物の投与によりPMSA関連疾患に通常伴う有害な症候が緩和するかもしくは完全に消失する場合に達成される。
【0099】
本発明の化合物および/またはそれらの組成物はまた、PMSA関連疾患を発症するリスクがある罹患体に予防的に投与される場合がある。
【0100】
ヒトへの投与に適切な初回投与量は、in vitro検定または動物モデルから決定される場合がある。例えば、初回投与量は、in vitro検定で測定される、投与する特定の化合物のIC50を含む血清中濃度を達成するように製剤化される場合がある。あるいは、ヒトの初回投与量は、PMSA関連疾患の動物モデルにおいて有効であることが見出されている投与量を基準にする場合もある。適切なモデル系の例については、例えば、Muchmore,2001,Immunol.Rev.183:86−93;およびLanford & Bigger,2002,Virology,293:1−9;ならびにそれらで引用された参考文献に記載されている。一例として、初回投与量は、約0.01mg/kg/日〜約200mg/kg/日である場合もあれば、あるいは約0.1mg/kg/日〜約100mg/kg/日、または約1mg/kg/日〜約50mg/kg/日、または約10mg/kg/日〜約50mg/kg/日を使用してもよい。しかし、投与量は、罹患体の必要量、治療する病態の重症度、および使用する化合物により異なる場合がある。投与量は、特定の罹患体における特定の化合物の投与に伴ういずれかの有害副作用の存在、性質および程度によっても決定される。特定の状況に適切な投与量を決定することは、実務家の技術の範囲内に含まれる。一般的に、投与は、化合物の最適な量よりも少ない投与量で開始される。その後、状況下における最適な作用に達するまで、投与量が少しずつ増加される。便宜上、1日の全投与量は、所望により、その日の中で複数回に分けて投与される場合がある。
【0101】
定義
本明細書におけるすべての化合物は、ACD/Name 8.00(Product Release 8.17、Build:04 May 2005;http://www.acdlabs.com;カナダ オンタリオ州トロント)を使用して命名した。
【0102】
本発明の化合物は、不斉中心またはキラル中心が存在する立体異性体として存在することができる。本発明は、種々の立体異性体およびそれらの混合物を企図し、本発明の適用範囲内に具体的に含まれる。立体異性体には、エナンチオマー、ジアステレオマー、エナンチオマーまたはジアステレオマーの混合物が含まれる。
【0103】
本明細書で使用される「アルキル」という用語は、特に定義がない限り、1〜24個の炭素原子を含有する直鎖または分枝鎖の炭化水素を意味する。アルキルの代表的な例には、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチル、n−ペンチル、イソペンチル、ネオペンチル、n−ヘキシル、3−メチルヘキシル、2,2−ジメチルペンチル、2,3−ジメチルペンチル、n−ヘプチル、n−オクチル、n−ノニル、およびn−デシルが含まれるが、これらに限定されない。
【0104】
本明細書で定義されるように、本明細書で使用される「アルキル−アリール」という用語は、本明細書で定義されるようにアルキル基を介して母体部分に結合するアリール基を意味する。アルキル−アリール基の例には、ベンジルおよびフェネチルが含まれるが、これらに限定されない。
【0105】
本明細書で定義されるように、本明細書で使用される「アルキル−ヘテロアリール」という用語は、本明細書で定義されるようにアルキル基を介して母体部分に結合するヘテロアリール基を意味する。アルキル−ヘテロアリール基の例には、ピリジルメチルおよび2−ピリジルエチルが含まれるが、これらに限定されない。
【0106】
本明細書で使用される「アリール」という用語は、フェニルまたは2環式アリールまたは3環系アリールを意味する。2環式アリールはナフチル、またはシクロアルキルに融合するフェニル、またはシクロアルケニルに融合するフェニルである。2環式アリールは、前記2環式アリールの中に含まれる任意の炭素原子を介して母体分子部分に結合する。2環式アリールの代表的な例には、ジヒドロインデニル、インデニル、ナフチル、ジヒドロナフタレニル、およびテトラヒドロナフタレニルが含まれるが、これらに限定されない。3環式アリールは、アンスラセンもしくはフェナントレン、またはシクロアルキルに融合する2環式アリール、またはシクロアルケニルに融合する2環式アリール、またはフェニルに融合する2環式アリールである。3環系アリールは、3環系アリールの中に含まれる任意の炭素原子を介して母体分子部分に結合する。3環式アリール環の代表的な例には、アズレニル、ジヒドロアントラセニル、フルオレニル、およびテトラヒドロフェナントレニルが含まれるが、これらに限定されない。
【0107】
本明細書で使用される「ヘテロアリール」という用語は、単環式ヘテロアリールまたは2環式ヘテロアリールを意味する。単環式ヘテロアリールは、5員環または6員環である。5員環は、2個の二重結合と、1個、2個、3個または4個の窒素原子と、場合により1個の酸素原子または硫黄原子とから構成される。6員環は、3個の二重結合と、1個、2個、3個または4個の窒素原子とから構成される。5員環または6員環のヘテロアリールは、ヘテロアリールの中に含まれる任意の炭素原子または任意の窒素原子を介して母体分子部分に結合する。単環式ヘテロアリールの代表的な例には、フリル、イミダゾリル、イソオキサゾリル、イソチアゾリル、オキサジアゾリル、オキサゾリル、ピリジニル、ピリダジニル、ピリミジニル、ピラジニル、ピラゾリル、ピロリル、テトラゾリル、チアジアゾリル、チアゾリル、チエニル、トリアゾリル、およびトリアジニルが含まれるが、これらに限定されない。2環式ヘテロアリールは、フェニルに融合する単環式ヘテロアリール、またはシクロアルキルに融合する単環式ヘテロアリール、またはシクロアルケニルに融合する単環式ヘテロアリール、または単環式ヘテロアリールに融合する単環式ヘテロアリールである。2環式ヘテロアリールは、2環式ヘテロアリールの中に含まれる任意の炭素原子または任意の窒素原子を介して母体分子部分に結合する。2環式のヘテロアリールの代表的な例には、ベンズイミダゾリル、ベンゾフラニル、ベンゾチエニル、ベンゾオキサジアゾリル、シンノリニル、ジヒドロキノリニル、ジヒドロイソキノリニル、フロピリジニル、インダゾリル、インドリル、イソキノリニル、ナフチリジニル、キノリニル、テトラヒドロキノリニル、およびチエノピリジニルが含まれるが、これらに限定されない。
【0108】
本明細書で使用される「ハロ」または「ハロゲン」という用語は、−Cl、−Br、−Iまたは−Fを意味する。
【0109】
本明細書で使用される「ヒドロキシ」という用語は、−OH基を意味する。
【0110】
本明細書で使用される「メチレン」という用語は、−CH2−基を意味する。
【0111】
本明細書で使用される「オキソ」という用語は、=O部分を意味する。
【0112】
本明細書で使用される「ペプチド」という用語は、2〜10個のアミノ酸残基を有するペプチドを意味する。
【0113】
本明細書で使用される「ステロイド」という用語は、一般的な4環構造を有する基のモノラジカル(monoradical)であって、
【0114】
【化24】
前記4環構造は、完全に飽和する場合もあれば、1個以上の不飽和結合を含有する場合があり、かつ任意の有効な置換可能な位置で母体部分に結合する場合がある。例えば、環式のサブユニットの1個以上は芳香族である場合がある。
【0115】
本明細書で使用される「グルタメート類似体」という用語は、グルタミン酸の構造を模倣するための化学構造を意味する。このような化学構造は、グルタミン酸の構造におけるように相互に関連して同様に配置されるアミノ基またはアミノ基のバイオアイソスター(bioisostere)、ならびに2個のカルボン酸またはカルボン酸のバイオアイソスターから構成されるグルタミン酸の分子骨格の周囲に設計される。例えば、アミノ基は、チオール基、ヒドロキシル基、ヒドロキシルアミノ基、オキシム基またはメチレン基で置換できると考えられる。カルボン酸の一方または両方は、カルボキサミド基、ホスホン酸塩基、リン酸塩基、スルホン酸塩基またはテトラゾール基で置換できると考えられる。グルタメート類似体はまた、環式化学構造および非環式化学構造、ならびに相同構造(短いまたは長い構造)から構成されると考えられる。生物活性を有し、本発明で使用できるこのようなグルタメート類似体の例が最近検討されている(全体が参考として本明細書で援用される、Aspartate and glutamate mimetic structures in biologically active compounds.(Stefanic P,Dolenc MS.Curr Med Chem.2004 Apr;11(8):945−68))。
【0116】
本明細書で使用される「セリン類似体」という用語は、セリンの構造を模倣するための化学構造を意味する。このような化学構造は、セリン自体の構造におけるように相互に関連して同様に位置するアミノ基またはアミノ基のバイオアイソスター、カルボン酸またはカルボン酸のバイオアイソスター、ならびに水酸基または水酸基のバイオアイソスターから構成されるセリンの分子骨格の周囲に設計される。例えば、アミノ基は、チオール基、ヒドロキシル基またはメチレン基で置換できると考えられる。カルボン酸は、カルボキサミド基、ホスホン酸塩基、リン酸塩基、スルホン酸塩基またはテトラゾール基で置換できると考えられる。カルボン酸は完全に除去できると考えられる。水酸基は、アミノ基、チオール基またはメチレン基で置換できると考えられる。セリン類似体はまた、環式化学構造および非環式化学構造、ならびに相同構造(短いまたは長い構造)から構成されると考えられる。
【0117】
本明細書で使用される「ホスホルアミデート類似体」という用語は、亜鉛結合能を有するホスホルアミデートの分子構造を模倣する化学構造を意味する。例えば、ホスホルアミデート類似体は、ホスホルアミデート基、ホスホン酸塩基、リン酸塩基、ホスフィン酸塩基、スルホンアミド基、尿素基、N−ヒドロキシ尿素基、チオ尿素基、カルバミン酸塩基、ヒドロキサメート基、逆のヒドロキサメート基、N−ヒドロキシアミド基またはN−ヒドロキシカルバミン酸塩基であってよい。
【0118】
「フルオレセイン」という用語は、以下の一般式の化合物を指す:
【0119】
【化25】
「フルオレセイン誘導体」という用語は、それぞれが本明細書で定義される、独立してハロゲン基またはアルキル基である1〜3個の基で場合により置換される、本明細書で定義されるフルオレセインを指す。
【実施例】
【0120】
スキーム1は、ホスホルアミデート阻害剤T33の合成についての代表的な合成スキームを示す。この一般的な方法を使用して、複雑性の異なるホスホルアミデートT33の類似体を15個超合成した。このスキームを、標準的な合成方法とともに使用して、本発明の第1の態様の化合物を調製することができる。本発明の第2の態様の化合物は、標準的な合成方法を使用して、これらから慣例的に調製することができる。
【0121】
スキーム1
【0122】
【化26】
(実施例1)
PMSA阻害検定
Maung,J.;Mallari,J.P.;Girtsman,T.A.;Wu,L.Y.;Rowley,J.A.;Santiago,N.M.;Brunelle,A.;Berkman,C.E.Probing for a Hydrophobic a Binding Register in Prostate−Specific Membrane Antigen with Phenylalkylphosphonamidates.Bioorg.Med.Chem.2004,12,4969より引用。
【0123】
基質(N−[4−(フェニルアゾ)ベンゾイル]−グルタミル−g−グルタミン酸、PAB−Glu−γ−Glu)の使用液およびすべての阻害剤を、TRIS緩衝液(50mM、pH7.4)中で作製した。精製したPSMAの使用液を、TRIS緩衝液(50mM、pH7.4)中で適切に希釈し、阻害剤の非存在下において基質から産物へ15%〜20%変換させた。25μLの阻害剤溶液か25μLのTRIS緩衝液(50mM、pH7.4)かのいずれかを試験管内の175μLのTRIS緩衝液(50mM、pH7.4)に添加することによって、一般的なインキュベーション混合物(終量250μL)を調製した。上記の溶液に25μLのPAB−Glu−γ−Glu(100μM)を添加した。25μLのPSMA使用液を添加することより、酵素反応を開始した。いずれの例でも、PAB−Glu−γ−Gluの終濃度は10μMであったが、一方で酵素を5種類の連続希釈した阻害剤濃縮物でインキュベートして、10%〜90%の阻害率を得た。37℃での一定の振盪により反応を15分間継続させた後、25μLのメタノールTFA(メタノール中における2容量%のトリフルオロ酢酸)を添加してボルテックスすることにより反応を終了させた。急冷したインキュベーション混合物を、25μLのK2HPO4(0.1M)を添加して直ちに緩衝し、ボルテックスして、遠心分離させた(7000g、10分間)。その後、得られた上澄液の85μLのアリコートをHPLCにより定量化し、KaleidaGraph 3.6(Synergy Software)を使用してIC50値を算出した。
【0124】
PABGγGおよびその加水分解産物(PABG)を、それぞれ40:60の容積比で、ACN/リン酸カリウムからなる移動相[25mM、pH2.0(H3PO4で調整)]で解析用逆相HPLCカラム(Lichrosphere C18 5μm、150×4.6mm;Phenomenex、米国カリフォルニア州トーランス)を使用して分離し、定量化した。1.0mL/分の流速で、PABGγGおよびその加水分解産物(PABG)を、それぞれ4.8分および6.9分の滞留時間により325nmで検出した。
【0125】
(実施例2)
γ−ジグルタメートのホスホルアミデート類似体の分子剪定によるPSMAの強力阻害剤の同定
本発明者等は、PSMAの阻害のための鉛化合物として、ホスホルアミデートT33を設計し、合成した。その設計は、葉酸類似体のγ−グルタミン酸誘導体などの既知のPSMA基質の構造に基づくものであった。T33の初期スクリーニングにより、T33がPSMAに対して相当な効力を発揮することが示された(IC50<50nM)。PSMAに対する阻害能についてのT33の種々の構造要素の重要性をよりよく理解するために、本発明者等は分子剪定試験を実施し、図1に示すT33類似体のライブラリーを作製した。このライブラリーからの3種の化合物の合成については、既に文献の中で報告されている:CCS、JM139(Lu,H.;Ng,R.;Shieh,C.C.;Martinez,A.R.;Berkman,C.E.Inhibition of Glutamate Carboxypeptidase by Phosphoryl and Thiophosphoryl Derivatives of Glutamic and 2−Hydroxyglutaric Acid.Phosphorus,Sulfur and Silicon,2003,178,17)、および2−PMPA(Jackson,P.F.;Cole,D.C.;Slusher,B.S.;Stetz,S.L.;Ross,L.E.;Donzanti,B.A.;Trainor,D.A.Design,synthesis,and biological activity of a patent inhibitor of the neuropeptidase N−acetylated−linked acidic dipeptidase.J.Med.Chem.1996,39,619−622)。図1、図2および図7〜12の残りの化合物は、本発明の新規の化合物である。
【0126】
図1、図2および図7〜12に記載のライブラリー内の全化合物に対して、実施例1に記載の検定を使用してPSMAの阻害能についてスクリーニングを行った。
【0127】
本発明者等が公表した方法(Purification of Prostate−Specific Membrane Antigen with Conformational Epitope−Specific Antibody−Affinity Chromatography.Liu,T.;Toriyabe,Y.;Berkman,C.E.Prot.Exp.Purif.2006,49,251.)によるLNCaP細胞由来の精製PSMAに対する図1のライブラリーのスクリーニングにより、以下の2つの一般的構造が優れた阻害能を発揮することが示された:無損傷のホスホルアミデートペプチド模倣体(例えばT33およびL36)、ならびに単純なP1’類似体(例えばCCS、JM140、および2−PMPA)。
【0128】
他の親和性エレメントが欠如しているものの、CCS、JM140および2−PMPAの2塩基性ホスホリルモチーフは、PSMAの活性部位の亜鉛原子との強力な相互作用によりPSMAに対する親和性の増大に関与しているとの仮説を立てている。興味深いことに、単純な疎水性類似体MP1Dにかなりの親和性が維持されている。本発明者等がステロイド含有ホスホルアミデート阻害剤のライブラリーを調製してスクリーニングを行ったところ、疎水性を有しかつ空間的に厳しいリガンドを収容するPSMAの能力がさらに確認された(図2)。図2のすべての化合物は、実施例1に記載の検定による測定で1μM未満のIC50を示した。MP1Dおよびステロイド含有ホスホルアミデート阻害剤による阻害検定の結果は、PSMAの中心的触媒機構から離れた疎水性結合ドメインの存在を同定した本発明者等の以前の研究と一致する(Maung,J.;Mallari,J.P.;Girtsman,T.A.;Wu,L.Y.;Rowley,J.A.;Santiago,N.M.;Brunelle,A.;Berkman,C.E.Probing for a Hydrophobic a Binding Register in Prostate−Specific Membrane Antigen with Phenylalkylphosphonamidates.Bioorg.Med.Chem.2004,12,4969)。
【0129】
(実施例3)
T33類似体によるPSMAの時間依存的かつ不可逆的な阻害
驚くべきことに、本発明者等は、T33類似体ライブラリー(図1)のPSMA阻害化合物が、PSMAの強力な阻害剤であるだけでなく、図3に示す通り酵素活性の時間依存的な減損も示したことを見出した。この固有の結果により、本発明者等は、T33およびその類似体が固有の緩慢な結合阻害剤であることを仮定するに至った。場合により、その阻害は不可逆的であると思われ、T33、L36およびMP1Eの場合がこれに当てはまる。というのも、これらの化合物により阻害される酵素の100倍の希釈によってPSMAの活性を回復できないためである(図4)。これらの結果により、T33、L36およびMP1Eは、PSMAのいずれかの機構に基づいた不可逆的な阻害剤であるか、または場合により劇的な配座変化を生じる可能性あるか、もしくは場合により酵素に共有結合による損傷を引き起こす可能性がある、PSMAの機能的に不可逆的な阻害剤であることが強く示唆されている。本発明者等は最近になって、T33、L36およびMP1Eのほかにも、LW−54およびLW−39もまたPSMAの不可逆的な阻害剤であることを明らかにした。文献を検討によれば、「これはPSMAの時間依存的かつ不可逆的な阻害剤の最初の発見」とのことである。リン酸誘導体による亜鉛ペプチダーゼおよび亜鉛プロテアーゼの緩徐な結合阻害は既知の現象であるものの、このような酵素の機序に基づいた不可逆的な阻害剤はまれである。図1の阻害剤は、図5の不可逆的および緩徐に可逆的な阻害剤に分類される。図5に示すすべての化合物は、1μM未満のIC50を示した。
【0130】
(実施例4)
阻害剤により誘導されるホモダイマーの形成
ホスホルアミデートT33およびL36の作用に関して、本発明者等は、これらの阻害剤におるPSMAの治療により、ウェスタンブロット法により示される、共有結合性PSMAホモダイマーの形成が生じることを明らかにした。図6の代表例に示される通り、T33により媒介される共有結合性PSMAダイマーの形成は、濃度依存的でかつ時間依存的である。これらの結果は、上述のようなホスホルアミデートT33およびその類似体によるPSMAの時間依存的な酵素的阻害と一致する。さらに重要なことに、これらのデータは、化合物T33、L36およびMP1Dの不可逆的阻害プロファイルと一致する。これらのデータにより、T33およびL36は、タンパク質間架橋や潜在的なタンパク質間架橋を促進するPSMAの共有結合による修飾に関与することが示されている。本発明者等はT33、L36およびMP1Dの阻害機序を測定しなかったが、これらの薬剤が、PSMA発現癌細胞に特異的な共有結合送達ビヒクルとして使用される場合がある、固有の種類のPSMA阻害剤であることは明らかである。おおまかに文献を考察したところ、これは、PSMAの不可逆的な共有結合調節剤における初めての設計と調製である。
【0131】
(実施例5)
PSMAに対するホスホルアミデート阻害剤の特異性
本発明のホスホルアミデート阻害剤の設計がPSMAに特異的であることを判定するため、マトリックスメタロプロテアーゼ−9(MMP−9)を有するT33類似体ライブラリー(図1)に由来する代表的な化合物により予備試験を実施した。MMP酵素は、細胞外マトリックスの分解に関与する亜鉛依存性エンドペプチダーゼである。NC−2−29などの検討された化合物の系列の中で、1つの化合物(NC−2−29)だけが、MMP−9に対する阻害能を示し、Ki値が5mMであった。T33は最近になり、MMP−2の阻害について検定が行われており、最高10μMまでの濃度では阻害を示さなかった。これらの大部分の陰性の結果により、本発明のT33系阻害剤の一般的な構造骨格は、標的金属ペプチダーゼであるPSMAに対して特異性をもたらすことが示唆される。
【0132】
(実施例6)
PSMA発現前立腺癌細胞の阻害剤に誘導される標識
最近の実験により、イメージングペイロード(例えば有機蛍光団)は、PSMA発現前立腺癌細胞に特異的に送達され得ることが明らかにされている。本発明者等は、それぞれがイメージングペイロードまたは治療的ペイロードの結合点としてN末端アミノ基を有する4種のPSMA阻害剤を設計し、合成した(図8)。
【0133】
その後、これらの化合物を、PSMA発現前立腺癌細胞を特異的に標識するためにアミン反応性蛍光色素で誘導体化した(図9)。蛍光顕微鏡データにより、これらの化合物はそれぞれ、PSMA発現前立腺癌細胞(LNCaP細胞)を特異的に標識し、PSMA(PC3細胞)を発現しない細胞は標識しないことが明らかにされた。チオ尿素結合蛍光阻害剤L6−V1−21は、PSMAに対する85nMのIC50を示した。この結果により、阻害剤のコアがかなりの大きさの構造モチーフで誘導体化される場合、阻害能が消滅しなかったことが明らかとなっている。L6−VI−21で治療したPSMA発現前立腺癌細胞(LNCaP)は、蛍光顕微鏡による観察で特異的な標識を生じた。L6−V1−21およびヨウ化プロピジウムによる治療後、LNCaP前立腺癌細胞およびPC3前立腺癌細胞の両方に共焦点顕微鏡法を行った。
【0134】
LNCaP細胞をL6−V1−21で治療すると、蛍光顕微鏡検査により、濃い緑色の細胞蛍光標識で示されるようにL6−V1−21による細胞表面の広範な標識化が確認される。LNCaP細胞をL6−V1−2およびヨウ化プロピジウムで治療すると、共焦点蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識で示されるようにL6−V1−21による細胞表面の広範な標識化が確認されるとともに、ヨウ化プロピジウム染色から細胞核の赤い蛍光が観察される。PC3細胞をL6−V1−2およびヨウ化プロピジウムで治療すると、細胞表面の緑色の蛍光標識化がPC3細胞において観察されないが、ヨウ化プロピジウム染色から細胞核の赤い蛍光が観察され、共焦点蛍光顕微鏡検査により、PSMA発現細胞(LNCaP)に対するL6−V1−2の特異性が確認される。PC3細胞は、PSMAを発現しないことから、陰性対照として機能した。ヨウ化プロピジウムにより両細胞株の核が染色されたものの、PSMA発現LNCaP 6細胞の表面のみがL6−V1−21で蛍光標識された。これらのLNCaP細胞上の強い標識部位は、PSMAが凝集した位置であると仮定される。
【0135】
蛍光阻害剤LW−54−F5EXにより治療されたLNCaP細胞の蛍光顕微鏡検査により、PSMAの小分子阻害剤は、PSMA発現細胞を送達して蛍光色素で標識する際に、実質的に抗体(3C6)と等価であることが明らかになった。この実験で、フルオレセイン−5−EXでそれぞれが標識されたLW−54および抗体3C6はいずれも、LNCaP細胞の膜を特異的に標識した。LNCaP細胞をLW−54−F5EXまたは蛍光標識抗体3C6−F5EXのいずれかで治療すると、蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識により明らかになるように、いずれの場合も細胞表面の広範な標識化が確認される。LW−54−F5EXがPSMA発現細胞に特異的であることを確認するため、LNCaPおよびPC3細胞をこの化合物で治療した。DAPI(4’,6−ジアミジノ−2−フェニルインドール)は両細胞株内の核DNAを染色したものの、LW−54−F5EXで蛍光標識されたのはLNCaP細胞だけであった。LNCaP細胞をLW−54−F5EXおよびDAPIで治療すると、蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識で示されるようにLW−54−F5EXによる広範わたる細胞表面の標識化が確認されるとともに、DAPI染色から細胞核の青色蛍光が観察される。PC3細胞をLW−54−F5EXおよびDAPIで治療すると、細胞表面の緑色の蛍光標識化がPC3細胞において観察されないものの、DAPI染色から細胞核の青色蛍光が観察され、蛍光顕微鏡検査により、PSMA発現細胞(LNCaP)に対するLW−54−F5EXの特異性が確認される。PC3細胞はPSMAを発現しないことから、これらの結果により、54−F5EXなどの小分子剤はPSMA発現癌細胞に特異的であることが確認される。
【0136】
また、蛍光阻害剤LW−54−5FAMXで治療したLNCaP細胞の蛍光顕微鏡検査により、PSMAの小分子阻害剤は、PSMA発現細胞を送達して蛍光色素で標識する際に、実質的に抗体と等価であることが明らかになった。この実験では、LNCaP細胞およびPC3細胞の両方をLW−54−5FAMXで治療した。DAPI(4’,6−ジアミジノ−2−フェニルインドール)は両細胞株内の核DNAを染色したものの、LW−54−F5EXで蛍光標識されたのはLNCaP細胞だけであった。LNCaP細胞をLW−54−5FAMXおよびDAPIで治療すると、蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識で示されるようにLW−54−5FAMXによる細胞表面の広範な標識化が確認されるとともに、DAPI染色から細胞核の青色蛍光が観察される。PC3細胞をLW−54−5FAMXおよびDAPIで治療すると、細胞表面の緑色の蛍光標識化がPC3細胞において観察されないものの、DAPI染色から細胞核の青色蛍光が観察され、蛍光顕微鏡検査により、PSMA発現細胞(LNCaP)に対するLW−54−5FAMXの特異性が確認される。これらの結果により、LW−54−5FAMXなどの小分子剤はPSMA発現癌細胞に特異的であることが確認される。
【0137】
細胞標識化試験の上記の結果により、LNCaPに特異的な標識化は、PSMAの小分子非生物学的阻害剤によって達成できることが確認される。これらの結果は、PSMAの小分子阻害剤がPSMA発現癌細胞にイメージングペイロードまたは治療的ペイロードを特異的に送達できるという証明となる。
【0138】
以上の詳細な説明および添付の実施例は、単に例示的なものであって、添付の特許請求の範囲で定義される本発明の適用範囲を限定するものとは見なされないことが理解される。開示した実施形態の種々の変更および改変は当業者に明らかになるであろう。本発明の化学構造、置換基、誘導体、中間体、合成および/または使用方法を含むがこれらに限定されないこのような変更および改変は、本発明の趣旨および適用範囲から逸脱しない範囲で行われる場合がある。
【0139】
本明細書で言及する各雑誌論文、書籍、特許および特許出願は、全体が参考として本明細書で援用される。
【図面の簡単な説明】
【0140】
【図1】T33の分子剪定類似体を示す。
【図2】PSMAのステロイド含有ホスホルアミデート阻害剤を示す。
【図3】T33による時間依存的なPSMAの阻害を示す。
【図4】T33および代表的分子剪定類似体の不可逆的および緩徐な可逆的阻害を示す。
【図5】PSMAの不可逆的、緩徐な可逆的、および急速な可逆的阻害剤としてのT33およびその分子剪定類似体の分類を示す。
【図6】(A)時間の増加および(B)濃度の増加に伴うT33治療PSMAのウェスタンブロットを示す。
【図7】MP1Dのビフェニル類似体を示す。
【図8】N末端アミノ基における誘導体化と、前立腺癌細胞へのイメージングペイロードおよび治療的ペイロードの送達との両方のために設計される合成PSMA阻害剤を示す。
【図9】PSMA発現前立腺癌細胞を特異的に標識することが知られている蛍光標識PSMA阻害剤を示す。
【図10】蛍光標識PSMA阻害剤の代表的な調製物を示す。
【図11】キレート剤含有PSMA阻害剤TL−LW−54−BnDTPAの調製物を示す。
【図12】代表的なビオチン標識PSMA阻害剤を示す。
【技術分野】
【0001】
(関連出願への相互参照)
本願は、2006年3月14日に出願された米国仮特許出願第60/782,211号の優先権の利益を請求し、その全体は、本明細書中に参考として援用される。
【0002】
(発明の分野)
本発明は、前立腺特異的膜抗原(PSMA)に対する高い親和性および特異性を有する小分子、ならびに診断目的および治療目的のためにそれらを使用する方法に関する。
【背景技術】
【0003】
(関連技術の要約)
前立腺特異的膜抗原(PSMA)は、前立腺癌細胞の表面ならびに種々の固形腫瘍の新生血管系に特異的に過剰発現する。そのため、PSMAは、前立腺癌の検出および管理のための臨床的バイオマーカーとして注目を集めてきた。一般的に、これらの手法は、イメージング剤または治療剤を方向づけるためにPSMAを特異的に標的とする抗体を利用する。例えば、ProstaScint(Cytogen、米国ペンシルベニア州フィラデルフィア)は、前立腺癌の検出およびイメージングのためにFDAの承認を得たものであるが、キレート化された放射性同位元素(インジウム(III))を送達するために抗体を利用する。しかし、ProstaScint技術は死細胞の検出に限定されているため、その臨床的な重要性が疑わしいことが現在認められている。
【0004】
抗体を使用する癌の診断および治療は、これらの分子を血液から排除するのが遅い点や、血管透過性に劣っている点などの問題により、十分な成果をあげていない。さらに、細胞表面の標的に結合する大きな抗体は、その後隣接した細胞表面部位に他の抗体が結合する際の障害となるため、細胞表面の標識化が低減してしまう。
【0005】
診断剤または治療剤を送達する抗体の細胞表面標的として機能することに加えて、PSMAのほとんど見過ごされている固有の特性に、その酵素活性がある。すなわち、PSMAは、ジペプチドと同程度の小さな分子を認識し、プロセッシングすることができる。この特性を有するにもかかわらず、PSMAは、新規の診断戦略および治療戦略の開においてほとんど考察されてこなかった。PSMAの標識小分子阻害剤を使用して前立腺癌細胞を検出した結果について記載した参考文献の例は、最近では数例しかない。
【発明の開示】
【課題を解決するための手段】
【0006】
(発明の要旨)
本発明は、高親和性および特異性を有する前立腺特異的膜抗原(PSMA)と結合する化合物を含む。これらの特性により、本発明の化合物は、PSMAを提示する細胞に診断剤または治療剤を送達するのに有用であるか、あるいは本発明の化合物を固体担体に直接的または間接的に固定する時などにこのような細胞を捕捉および検出するのに有用である。それためこれに付随して、本発明はまた、PSMAを提示する細胞を検出および/または同定する診断方法であって、PSMAを提示することが疑われる細胞を検出可能なマーカーまたは検出素子に結合する本発明の化合物と接触させる(またはこれらの接触を引き起こす)工程と、前記化合物と細胞とが結合しているかどうかを判定する工程を含む、方法も含む。本発明はまた、本発明の化合物を、医薬的に許容される担体、賦形剤および/または希釈剤とともに含む組成物も含む。本発明はさらに、前立腺癌を抑制または治療する方法であって、前立腺癌治療剤(またはその組成物)に結合する治療有効量の本発明の化合物を前立腺癌の羅患体に投与する工程を含む、方法も含む。
【0007】
小分子を調製および精製することは抗体を調製することよりも効率性がよく、費用対効果も高いことから、本発明の小分子は、PMSA抗体を使用する手法よりも利点がある。
【発明を実施するための最良の形態】
【0008】
(発明の詳細な説明)
小分子が腫瘍細胞に対して十分な親和性を有する場合は、小分子によって腫瘍の標的化を達成できることが認識されている。抗体に基づく送達剤と拮抗し、実質的に同等となるには、小分子の親和性の範囲は、標的に対する抗体の親和性と同じでなければならない。
【0009】
第1の態様において、本発明は、この基準を満たす、PSMA(好ましくはPSMAの酵素認識部位)を標的とする小分子を含む。本発明の化合物の固有の特徴は、それらの化合物が中心的なホスホルアミデートコアに主に由来することである。具体的には、このような化学化合物には、図1(CCS、JM139および2−PMPAを除く)、図2および図7〜12に示すすべての化合物が含まれる。本発明の化合物は、実施例(下記参照)に記載の検定に従って測定した場合に、一般的には5μM未満、好ましくは1μM未満、より好ましくは100nM未満のIC50を有する。
【0010】
本発明の化合物は、PSMA発現細胞に診断剤または治療剤を送達するために官能基化することもできるPMSAの強力なペプチド模倣体阻害剤を生じる以下の3つの成分を有する:1)P1’位のグルタメートまたはグルタメート類似体、2)亜鉛結合基でしての中心的なホスホルアミデートまたはホスホルアミデート類似体、および3)側鎖を介して結合し、好ましくはそのN末端またはN末端等価物において疎水基を有するP1位のセリンまたはセリン類似体。
【0011】
従って、第1の態様の実施形態において、本発明は、以下の式Iの化合物であって:
A−L−B (I)
(式中、Aがグルタメートまたはグルタメート類似体であり;Lがホスホルアミデートまたはホスホルアミデート類似体であり;Bがセリンまたはセリン類似体である、化合物を提供する。好ましくは、セリンまたはセリン類似体は、N末端またはN末端等価物において疎水基を有する。本明細書に開示される本発明のすべての化合物の医薬的に許容される塩もまた、本発明の態様である。
【0012】
第1の態様の好ましい実施形態において、本発明は、Aが以下の式(Ia)に記載のものである式Iの化合物であって:
【0013】
【化9】
式中、
nが1、2、3、4、5または6であり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−であって、式中、QがLに結合し;
R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
それぞれが独立して−HまたはC1−C6アルキルである、
化合物;ならびにその医薬的に許容される塩を提供する。
【0014】
第1の態様の別の実施形態において、本発明は、Bが以下の式(Ib)である式Iの化合物であって:
【0015】
【化10】
式中、
nが1、2、3、4、5または6であり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であって、式中、TがLに結合し;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が、−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−HまたはC1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
各R3が独立して−HまたはC1−C6アルキルである、
化合物;ならびにその医薬的に許容される塩を提供する。
【0016】
第1の態様の別の実施形態において、本発明は、式Iの化合物であって、式中、Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であって、式中、各R3が独立して−HまたはC1−C6アルキルである、化合物(ならびにその医薬的に許容される塩)を提供する。
【0017】
第1の態様の好ましい実施形態において、本発明は、以下の式(II)の化合物であって:
【0018】
【化11】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−HまたはC1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
化合物;ならびにその医薬的に許容される塩を提供する。
【0019】
第1の態様の好ましい実施形態において、本発明は、以下の式(III)の化合物であって:
【0020】
【化12】
式中、各変数が式(II)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0021】
第1の態様の好ましい実施形態において、本発明は、以下の式(IV)の化合物であって:
【0022】
【化13】
式中、各変数が式(II)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0023】
式(II)〜(IV)の好ましい実施形態において、R1およびR2はそれぞれ−C(O)OHである。
【0024】
式(II)〜(IV)の好ましい実施形態において、R4は−C(O)OHである。
【0025】
式(II)〜(IV)のより好ましい実施形態において、R1、R2およびR4はそれぞれ−C(O)OHである。
【0026】
式(II)〜(IV)の好ましい実施形態において、R10は−C(O)−フェニルである。
【0027】
式(II)〜(IV)のより好ましい実施形態において、R10はR7である。
【0028】
式(II)〜(IV)のより好ましい実施形態において、R10はR7であって、式中、R7が−L1−R8であって、式中、
L1が、−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHである。
【0029】
式(II)〜(IV)のより好ましい実施形態において、R10はR7であって、式中、R7が、−L1−R8であって、式中、
L1が、−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHである。
【0030】
式(II)〜(IV)のより好ましい実施形態において、R10は、ペプチド、デンドリマーまたはペプチドデンドリマーである。
【0031】
第1の態様のより好ましい実施形態において、本発明は、式(I)の化合物であって、以下の通りである、化合物を提供する。
【0032】
【化14】
【0033】
【化15】
第2の態様において、本発明は、検出可能な標識と共有結合する本発明の第1の態様の化合物、治療剤、または固体担体に結合する生体分子アンカーを含むキメラ化合物を含む。固体担体の例には、ポリリシンがコーティングされた、無水マレイン酸がコーティングされた、またはストレプトアビジンがコーティングされた、市販の96ウェルプレートが含まれる。他の固体担体には、金がコーティングされた、または官能基化した金がコーティングされた市販のセンサーチップが含まれる。
【0034】
一実施形態において、検出可能な標識は蛍光標識である。標準的な蛍光標識には、Alexa Fluor染料、BODIPY染料、フルオレセイン系染料、ローダミン系染料、クマリン系染料、およびピレン系染料が含まれる。
【0035】
別の実施形態において、検出可能な標識は、特異的結合対の片側、例えば、ビオチンとストレプトアビジンの結合対の内のビオチンである。代表的な結合対の薬剤および生体分子アンカーには、ビオチン、DNAもしくはRNAのオリゴヌクレオチド、または脂質が含まれる。
【0036】
別の実施形態において、検出可能な標識は、99Tcなどの放射性同位元素またはGdなどのMRI造影剤に結合することが可能なキレート構造である。標準的なキレート剤には、DOTA、DTPA、CHX−A”、PCTA、およびDO3Aが含まれる。
【0037】
本明細書に記載されるような標準的な蛍光標識、結合対、キレート剤および生体分子アンカーが使用される場合がある。当業者に既知の標準的な方法は、このような薬剤およびアンカーならびに治療剤に本発明の化合物を結合するために使用される場合がある。
【0038】
治療剤は、好ましくは、PSMAを提示する細胞の1つ以上の生物学的プロセスに干渉し、それによってPMSA提示細胞に関連する疾患を治療または抑制する、化合物である。治療剤は、90Yまたは188Reなどのキレート化または共有結合した細胞障害性放射性同位元素を場合により含む場合があり、このようなキレート化または共有結合は、当業者に既知の手段により行うことができる。
【0039】
治療剤には、癌細胞の細胞表面に直接結合するか、癌細胞における抗原ペプチドの発現を調節するかのいずれかによって、腫瘍細胞の免疫原性を増大させるものが含まれる。共有結合のために選択される薬剤は、既知の抗癌性、抗増殖性、または細胞障害性を有する。あるいは、このような剤は、T細胞免疫監視機構の標的として細胞の免疫原性を促進するか、または増大させる既知の特性を有する。
【0040】
治療剤にはまた、2−メトキシエストラジオールなどのステロイド系剤や、ミフェプリストーン、タモキシフェン、レチノイン酸などのアポトーシス誘導物質、酪酸などのヒストンデアセチラーゼ阻害剤、Plk1 siRNAなどのアポトーシス誘導または細胞障害性siRNA、ドキソルビシンなどの有糸分裂阻害剤、メトトレキサートなどの代謝拮抗剤、および細胞毒をカプセル化するように設計されたナノ粒子またはリポソームが含まれるが、これらに限定されない。
【0041】
第2の態様の実施形態において、本発明は、式:A−L−Bの化合物であって、式中、
Aがグルタメートまたはグルタメート類似体であり;
Lがホスホルアミデートまたはホスホルアミデート類似体であり;
Bがセリンまたはセリン類似体であり;
前記化合物が、前記化合物の任意の置換可能な位置で、検出可能な標識、治療剤、または固体担体に結合する生体分子アンカーに、2価のリンカーを介して共有結合する、
化合物;ならびにその医薬的に許容される塩を提供する。
【0042】
第2の態様の実施形態において、前記2価のリンカーは、アミノ酸、オリゴペプチド、ポリ(エチレン)グリコール、オリゴエチレングリコールなどに由来する。
【0043】
第2の態様の実施形態において、前記化合物は、検出可能な標識に共有結合する。
【0044】
第2の態様の別の実施形態において、前記化合物は、治療剤に結合共有結合する。
【0045】
第2の態様の別の実施形態において、前記化合物は、固体担体に結合する生体分子アンカーに共有結合する。
【0046】
第2の態様の好ましい実施形態において、前記化合物は、検出可能な標識に共有結合し、前記検出可能な標識は蛍光標識である。
【0047】
第2の態様の別の好ましい実施形態において、前記化合物は、検出可能な標識に共有結合し、前記検出可能な標識は、放射性同位元素または磁気共鳴映像法用造影剤に結合するキレート構造である。
【0048】
第2の態様の別の好ましい実施形態において、本発明は、以下の式(VI)の化合物であって:
【0049】
【化16】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R7が、−X−R8または−L1−R8であって、式中、
Xが、−C(O)−、−S(O)2、−O−、−S−または−N(R3)−であり;
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)、−C(O)−L2−、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、治療剤、検出可能な標識、または固体担体に結合する生体分子アンカーであり;
Qが、−O−、−S−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
化合物;ならびにその医薬的に許容される塩を提供する。
【0050】
第2の態様の好ましい実施形態において、本発明は、以下の式(VII)の化合物であって:
【0051】
【化17】
式中、各変数が式(VI)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0052】
第2の態様の好ましい実施形態において、本発明は、以下の式(VIII)の化合物であって:
【0053】
【化18】
式中、各変数が式(VI)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0054】
式(VI)〜(VIII)の好ましい実施形態において、R1およびR2はそれぞれ−C(O)OHである。
【0055】
式(VI)〜(VIII)の好ましい実施形態において、R4は−C(O)OHである。
【0056】
式(VI)〜(VIII)のより好ましい実施形態において、R1、R2およびR4はそれぞれ−C(O)OHである。
【0057】
式(VI)〜(VIII)のより好ましい実施形態において、L1は−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とする。
【0058】
式(VI)〜(VIII)のより好ましい実施形態において、L1は−C(O)−L2−であって、式中、
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−または−N(R3)−で置換されないことを条件とする。
【0059】
式(VI)〜(VIII)のより好ましい実施形態において、L1は、ペプチド、デンドリマーまたはペプチドデンドリマーである。
【0060】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識である。
【0061】
式(VI)〜(VIII)の好ましい実施形態において、R8は固体担体に結合する生体分子アンカーである。
【0062】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は蛍光色素である。
【0063】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は蛍光標識であって、前記蛍光標識は、Alexa Fluor、BODIPY、フルオレセイン、ローダミン、クマリンまたはピレン系染料である。
【0064】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は蛍光色素であって、前記蛍光色素は、フルオレセインまたはフルオレセイン誘導体である。
【0065】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識はキレート剤である。
【0066】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識はキレート剤であって、前記キレート剤は、DOTA、DTPA、CHX−A”、PCTAおよびDO3Aである。
【0067】
式(VI)〜(VIII)のより好ましい実施形態において、R8はR9であって、式中、
R9が、C1−C6アルキル、アリールまたはC1−C6アルキル−アリールであって、式中、R9が、独立して−COOHまたはN(R91)2である1〜3個の基で置換され、式中、
各R91が独立して、独立して−COOHまたは−N(R92)2である1〜3個の基で置換される−HまたはC1−C6アルキルであって、式中、
各R92が独立して、1〜3個のCOOHで置換される−HまたはC1−C6アルキルである。
【0068】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は特異的結合対の片側である。
【0069】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は特異的結合対の片側であって、前記特異的結合対の前記片側は、ビオチン、DNAもしくはRNAのオリゴヌクレオチド、または脂質である。
【0070】
式(VI)〜(VIII)の好ましい実施形態において、R8は検出可能な標識であって、前記標識は特異的結合対の片側であって、前記特異的結合対の前記片側はビオチンである。
【0071】
式(VI)〜(VIII)の好ましい実施形態において、R8は治療剤である。
【0072】
式(VI)〜(VIII)のより好ましい実施形態において、R8は、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基である。
【0073】
第1の態様の別の好ましい実施形態において、本発明は、以下の式(IX)の化合物であって:
【0074】
【化19】
式中、R7が−O−R8であって、式中、R8が、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基であり;各残りの変数が式(VI)に定義される通りである、化合物;ならびにその医薬的に許容される塩を提供する。
【0075】
第1の態様のより好ましい実施形態において、本発明は、式(VI)の化合物であって、以下の通りである:
【0076】
【化20】
【0077】
【化21】
【0078】
【化22】
【0079】
【化23】
を提供する。
【0080】
第3の態様において、本発明は、本発明の第2の態様のキメラ化合物、またはその医薬的に許容される塩、および医薬的に許容される担体、希釈剤または賦形剤を含む組成物を含む。
【0081】
第4の態様において、本発明は、本発明の第2の態様のキメラ化合物を含む診断キットを含む。本発明の本態様の一実施形態において、検出可能な標識または生体分子アンカーは、特異的結合対の一方のメンバー(例えば、ビオチン)であり、前記キットは前記特異的結合対のもう一方のメンバーを含む。
【0082】
第5の態様において、本発明は、PMSA提示細胞を検出する方法であって、PMSAを提示することが疑われる細胞を本発明の第2の態様のキメラ化合物と接触させる工程と、前記検出可能な標識が、PMSA提示細胞に結合する場合にのみ検出される条件下で、前記検出可能な標識の存在を測定する工程とを含む、方法を含む。検出可能な標識を検出する方法は、当然ながら、使用する標識により異なり、当業者に明らかになるであろう。例えば、検出可能な標識が近赤外蛍光標識である場合、標識の検出はin vivo蛍光イメージングにより達成することができる。
【0083】
第6の態様において、本発明は、PMSAを提示する細胞を伴う疾患を阻害または治療する方法であって、前記細胞を本発明の第2の態様の化合物または本発明の第3の態様の組成物と接触させるか、または前記細胞への接触を引き起こす工程を含み、前記キメラ化合物が、治療剤に共有結合する本発明の第1の態様の化合物を含む、方法を含む。本態様の一実施形態において、本発明は、PMSA提示細胞を伴う疾患を有する哺乳類の被験体(好ましくはヒト)に、前記疾患の抑制または治療に有効な量の前記組成物を投与する工程を含む。適切な製剤および投与方法は、標準的な方法を使用して慣例的に決定することができる。
【0084】
第7の態様において、本発明は、PMSA提示細胞を捕捉、検出および定量化する方法であって、PMSAを提示することが疑われる細胞を本発明の第2の態様のキメラ化合物と接触させる工程と、捕捉または固定されたPSMA提示細胞を検出する工程とを含む、方法を含む。第7の態様の好ましい実施形態において、第2の態様のキメラ化合物は、固体担体上の生体分子アンカーに結合する。細胞を検出する方法は、当然ながら、使用する検出素子により異なり、当業者に明らかになるであろう。例えば、本発明の第2の態様のキメラ化合物が固体担体に結合する場合、PSMA提示細胞の検出は、プラズモン共鳴を使用して直接達成することもできれば、あるいは細胞が放出され、フローサイトメトリーを使用して蛍光標識で標識されることで達成することもできる。
【0085】
本発明で使用される場合がある代表的なデンドリマーについては、J.M.J.Frechet,D.A.Tomalia,Dendrimers and Other Dendritic Polymers,John Wiley & Sons,Ltd.NY,NY(2002)に記載されている。
【0086】
本発明の併用に有用な代表的な化合物には、上記の化合物、それらの医薬的に許容される酸、ならびにそれらの塩基付加塩および溶媒和物が含まれる。本発明の化合物が酸付加塩として得られる場合、遊離塩基は、酸性塩の溶液を塩基性化することによって得ることができる。逆に、産物が遊離塩基である場合、付加塩、特に医薬的に許容される付加塩は、塩基化合物から酸付加塩を調製する従来の方法に従って、適切な有機溶媒中に遊離塩基を溶解し、その溶液を酸で処理することによって産生される場合がある。
【0087】
無毒性医薬塩には、例えば、塩酸、リン酸、臭化水素酸、硫酸、スルフィン酸、蟻酸、トルエンスルホン酸、メタンスルホン酸、硝酸、安息香酸、クエン酸、酒石酸、マレイン酸、ヨウ化水素酸、アルカン酸(例えば酢酸)、HOOC−(CH2)n−COOH(式中、nは0〜4である)などの酸の塩が含まれる。無毒性医薬塩基付加塩には、例えば、ナトリウム、カリウム、カルシウム、アンモニウムなどの塩基の塩が含まれる。当業者であれば、多種多様な無毒性の医薬的に許容される付加塩を認識するであろう。
【0088】
本発明の化合物および/またはそれらの組成物は、動物およびヒトのPMSA関連疾患の抑制および/または治療における特定の用途が見出されている。従って、本発明の別の態様には、治療有効量の本発明の1種以上の化合物または本発明の1種以上の化合物を含有する組成物を、このようなPMSA関連疾患の治療を必要とする羅患体に投与することがある。好ましくは、羅患体は哺乳類であり、最も好ましくはヒトである。この状況で使用される場合、前記化合物は、それ自体が投与される場合があるが、通常は医薬組成物の形態に製剤化されて投与される。正確な組成物は、とりわけ投与方法により異なり、当業者に明らかになるであろう。例えば、Remington’s Pharmaceutical Sciences,20th ed.,2001には、多種多様な適切な医薬組成物が記載されている。
【0089】
経口投与に適切な製剤は、(a)希釈液(例えば、水、生理食塩水、またはPEG 400)に懸濁した有効量の活性化合物などの液体溶液;(b)それぞれ液体、固体、顆粒またはゼラチンなどの活性成分を所定量含有する、カプセル剤、小包、または錠剤;(c)適切な液体中の懸濁剤;および(d)適切な乳剤、から構成されてよい。錠剤の形態には、ラクトース、スクロース、マンニトール、ソルビトール、リン酸カルシウム、トウモロコシ澱粉、ジャガイモ澱粉、微結晶性セルロース、ゼラチン、コロイド状二酸化ケイ素、タルク、ステアリン酸マグネシウム、ステアリン酸、ならびに他の賦形剤、顔料、充填剤、結合剤、希釈剤、緩衝剤、湿潤剤、保存剤、着香剤、染料、崩壊剤および医薬的に適合する担体が含まれてよい。口内錠の形態は、香料中の活性成分(例えばスクロース)、ならびに不活性塩基中の活性成分を含む香錠(例えば、活性成分に加えて当該技術分野で既知の担体を含有する、ゼラチンおよびグリセリンまたはスクロースおよびアカシア乳剤、ゲル剤など)を含んでよい。
【0090】
選択化合物は、単独でまたは他の適切な成分と組み合わせて、吸入により投与されるようにエアロゾル製剤に作製することができる(すなわち、「霧状」にすることができる)。エアロゾル製剤は、許容される加圧噴射剤(例えば、ジクロロジフルオロメタン、プロパン、窒素など)に入れることができる。
【0091】
直腸投与に適切な製剤には、例えば、坐剤基剤で包装された核酸から構成される坐剤が含まれる。適切な坐剤基剤には、天然または合成のトリグリセリドまたはパラフィン族炭化水素が含まれる。さらに、基剤と選択化合物との組み合わせから構成されるゼラチン直腸投与カプセル(例えば、液体トリグリセリド、ポリエチレングリコール、およびパラフィン族炭化水素を含む)も使用することができる。
【0092】
例えば、関節内経路(関節中)、静脈内経路、筋肉内経路、皮内経路、腹腔内経路、皮下経路などによる非経口投与に適切な製剤には、製剤を所期のレシピエントの血液と浸透圧が等しくなるようにする抗酸化剤、緩衝液、静菌剤および溶質を含有し得る水溶性および非水溶性の等張性無菌注射溶液、ならびに懸濁化剤、溶解剤、増粘剤、安定剤および保存剤を含み得る水溶性および非水溶性の無菌懸濁液が含まれる。本発明の実施において、組成物は、例えば、静脈内、経口的、局所的、腹腔内、膀胱内、髄腔内に投与することができる。非経口投与、経口投与、皮下投与および静脈内投与が、好ましい投与方法である。適切な溶液製剤の具体例は、水中に約0.5〜100mg/mLの化合物と約1000mg/mLのプロピレングリコールとを含む場合がある。適切な溶液製剤の別の具体例は、水中に約0.5〜100mg/mLの化合物と約800〜1000mg/mLのポリエチレングリコール400(PEG 400)とを含む場合がある。
【0093】
適切な懸濁製剤の具体例は、水中に約0.5〜30mg/mLの化合物と、約200mg/mLのエタノール、約1000mg/mLの植物油(例えばトウモロコシ油)、約600〜1000mg/mLの果汁(例えばグレープフルーツジュース)、約400〜800mg/mLの乳汁、約0.1mg/mLのカルボキシメチルセルロース(または微結晶性セルロース)、約0.5mg/mLのベンジルアルコール(またはベンジルアルコールと塩化ベンザルコニウムとの組み合わせ)および約40〜50mMの緩衝液(pH7)(例えば、リン酸緩衝液、酢酸緩衝液、クエン酸緩衝液、あるいは緩衝液の代わりに5%のデキストロースが使用される場合がある)からなる群から選択される1種以上の賦形剤とを含む場合がある。
【0094】
適切なリポソーム懸濁製剤の具体例は、水中に約0.5〜30mg/mLの化合物と、約100〜200mg/mLのレシチン(または他のリン脂質もしくはリン脂質の混合物)と、場合により5mg/mLのコレステロールとを含む場合がある。化合物9の皮下投与の場合には、100mg/mLのレシチンを有する水中の5mg/mLの化合物と、100mg/mLのレシチンおよび5mg/mLのコレステロールを有する水中の5mg/mLの化合物とを含むリポソーム懸濁製剤が、良好な結果をもたらす。本製剤は、本発明の他の化合物に使用される場合もある。
【0095】
化合物の製剤は、アンプルやバイアルなど、単回用量または複数回用量の密封容器で提供することができる。注射溶液および注射懸濁液は、既述の種類の無菌の粉剤、顆粒および錠剤から調製することができる。
【0096】
前記医薬製剤は、単位投与形態が好ましい。このような形態では、前記製剤は、適切な量の有効成分を含有する単位用量に細分される。単位投与形態は、包装製剤、すなわち、小包にされた錠剤、カプセル剤、およびバイアルまたはアンプルに入った粉剤など、個別の量の製剤を含有する包装物であってよい。また、単位投与形態は、カプセル剤、錠剤、カシェ剤または口内錠自体であってもよければ、あるいは包装形態における適切な数のこれらのいずれかであってもよい。組成物はまた、所望により、以下に詳述する他の適合した治療剤を含有してもよい。
【0097】
別の態様において、本発明は、PMSA関連疾患の治療および/または阻害のための組成物を作製する方法であって、本発明の化合物を、医薬的に許容される担体、希釈剤および/または賦形剤と混合する工程を含む、方法を含む。
【0098】
PMSA関連疾患の治療のための治療的使用において、本発明の医薬的方法で利用される化合物は、治療的有益性を達成するのに適切な投与レベルで、PMSA関連疾患であると診断された罹患体に投与される。治療的有益性とは、化合物の投与が時間とともに罹患体に有益な効果をもたらすことを意味する。治療的有益性はまた、化合物の投与によりPMSA関連疾患に通常伴う有害な症候が緩和するかもしくは完全に消失する場合に達成される。
【0099】
本発明の化合物および/またはそれらの組成物はまた、PMSA関連疾患を発症するリスクがある罹患体に予防的に投与される場合がある。
【0100】
ヒトへの投与に適切な初回投与量は、in vitro検定または動物モデルから決定される場合がある。例えば、初回投与量は、in vitro検定で測定される、投与する特定の化合物のIC50を含む血清中濃度を達成するように製剤化される場合がある。あるいは、ヒトの初回投与量は、PMSA関連疾患の動物モデルにおいて有効であることが見出されている投与量を基準にする場合もある。適切なモデル系の例については、例えば、Muchmore,2001,Immunol.Rev.183:86−93;およびLanford & Bigger,2002,Virology,293:1−9;ならびにそれらで引用された参考文献に記載されている。一例として、初回投与量は、約0.01mg/kg/日〜約200mg/kg/日である場合もあれば、あるいは約0.1mg/kg/日〜約100mg/kg/日、または約1mg/kg/日〜約50mg/kg/日、または約10mg/kg/日〜約50mg/kg/日を使用してもよい。しかし、投与量は、罹患体の必要量、治療する病態の重症度、および使用する化合物により異なる場合がある。投与量は、特定の罹患体における特定の化合物の投与に伴ういずれかの有害副作用の存在、性質および程度によっても決定される。特定の状況に適切な投与量を決定することは、実務家の技術の範囲内に含まれる。一般的に、投与は、化合物の最適な量よりも少ない投与量で開始される。その後、状況下における最適な作用に達するまで、投与量が少しずつ増加される。便宜上、1日の全投与量は、所望により、その日の中で複数回に分けて投与される場合がある。
【0101】
定義
本明細書におけるすべての化合物は、ACD/Name 8.00(Product Release 8.17、Build:04 May 2005;http://www.acdlabs.com;カナダ オンタリオ州トロント)を使用して命名した。
【0102】
本発明の化合物は、不斉中心またはキラル中心が存在する立体異性体として存在することができる。本発明は、種々の立体異性体およびそれらの混合物を企図し、本発明の適用範囲内に具体的に含まれる。立体異性体には、エナンチオマー、ジアステレオマー、エナンチオマーまたはジアステレオマーの混合物が含まれる。
【0103】
本明細書で使用される「アルキル」という用語は、特に定義がない限り、1〜24個の炭素原子を含有する直鎖または分枝鎖の炭化水素を意味する。アルキルの代表的な例には、メチル、エチル、n−プロピル、イソプロピル、n−ブチル、sec−ブチル、イソブチル、tert−ブチル、n−ペンチル、イソペンチル、ネオペンチル、n−ヘキシル、3−メチルヘキシル、2,2−ジメチルペンチル、2,3−ジメチルペンチル、n−ヘプチル、n−オクチル、n−ノニル、およびn−デシルが含まれるが、これらに限定されない。
【0104】
本明細書で定義されるように、本明細書で使用される「アルキル−アリール」という用語は、本明細書で定義されるようにアルキル基を介して母体部分に結合するアリール基を意味する。アルキル−アリール基の例には、ベンジルおよびフェネチルが含まれるが、これらに限定されない。
【0105】
本明細書で定義されるように、本明細書で使用される「アルキル−ヘテロアリール」という用語は、本明細書で定義されるようにアルキル基を介して母体部分に結合するヘテロアリール基を意味する。アルキル−ヘテロアリール基の例には、ピリジルメチルおよび2−ピリジルエチルが含まれるが、これらに限定されない。
【0106】
本明細書で使用される「アリール」という用語は、フェニルまたは2環式アリールまたは3環系アリールを意味する。2環式アリールはナフチル、またはシクロアルキルに融合するフェニル、またはシクロアルケニルに融合するフェニルである。2環式アリールは、前記2環式アリールの中に含まれる任意の炭素原子を介して母体分子部分に結合する。2環式アリールの代表的な例には、ジヒドロインデニル、インデニル、ナフチル、ジヒドロナフタレニル、およびテトラヒドロナフタレニルが含まれるが、これらに限定されない。3環式アリールは、アンスラセンもしくはフェナントレン、またはシクロアルキルに融合する2環式アリール、またはシクロアルケニルに融合する2環式アリール、またはフェニルに融合する2環式アリールである。3環系アリールは、3環系アリールの中に含まれる任意の炭素原子を介して母体分子部分に結合する。3環式アリール環の代表的な例には、アズレニル、ジヒドロアントラセニル、フルオレニル、およびテトラヒドロフェナントレニルが含まれるが、これらに限定されない。
【0107】
本明細書で使用される「ヘテロアリール」という用語は、単環式ヘテロアリールまたは2環式ヘテロアリールを意味する。単環式ヘテロアリールは、5員環または6員環である。5員環は、2個の二重結合と、1個、2個、3個または4個の窒素原子と、場合により1個の酸素原子または硫黄原子とから構成される。6員環は、3個の二重結合と、1個、2個、3個または4個の窒素原子とから構成される。5員環または6員環のヘテロアリールは、ヘテロアリールの中に含まれる任意の炭素原子または任意の窒素原子を介して母体分子部分に結合する。単環式ヘテロアリールの代表的な例には、フリル、イミダゾリル、イソオキサゾリル、イソチアゾリル、オキサジアゾリル、オキサゾリル、ピリジニル、ピリダジニル、ピリミジニル、ピラジニル、ピラゾリル、ピロリル、テトラゾリル、チアジアゾリル、チアゾリル、チエニル、トリアゾリル、およびトリアジニルが含まれるが、これらに限定されない。2環式ヘテロアリールは、フェニルに融合する単環式ヘテロアリール、またはシクロアルキルに融合する単環式ヘテロアリール、またはシクロアルケニルに融合する単環式ヘテロアリール、または単環式ヘテロアリールに融合する単環式ヘテロアリールである。2環式ヘテロアリールは、2環式ヘテロアリールの中に含まれる任意の炭素原子または任意の窒素原子を介して母体分子部分に結合する。2環式のヘテロアリールの代表的な例には、ベンズイミダゾリル、ベンゾフラニル、ベンゾチエニル、ベンゾオキサジアゾリル、シンノリニル、ジヒドロキノリニル、ジヒドロイソキノリニル、フロピリジニル、インダゾリル、インドリル、イソキノリニル、ナフチリジニル、キノリニル、テトラヒドロキノリニル、およびチエノピリジニルが含まれるが、これらに限定されない。
【0108】
本明細書で使用される「ハロ」または「ハロゲン」という用語は、−Cl、−Br、−Iまたは−Fを意味する。
【0109】
本明細書で使用される「ヒドロキシ」という用語は、−OH基を意味する。
【0110】
本明細書で使用される「メチレン」という用語は、−CH2−基を意味する。
【0111】
本明細書で使用される「オキソ」という用語は、=O部分を意味する。
【0112】
本明細書で使用される「ペプチド」という用語は、2〜10個のアミノ酸残基を有するペプチドを意味する。
【0113】
本明細書で使用される「ステロイド」という用語は、一般的な4環構造を有する基のモノラジカル(monoradical)であって、
【0114】
【化24】
前記4環構造は、完全に飽和する場合もあれば、1個以上の不飽和結合を含有する場合があり、かつ任意の有効な置換可能な位置で母体部分に結合する場合がある。例えば、環式のサブユニットの1個以上は芳香族である場合がある。
【0115】
本明細書で使用される「グルタメート類似体」という用語は、グルタミン酸の構造を模倣するための化学構造を意味する。このような化学構造は、グルタミン酸の構造におけるように相互に関連して同様に配置されるアミノ基またはアミノ基のバイオアイソスター(bioisostere)、ならびに2個のカルボン酸またはカルボン酸のバイオアイソスターから構成されるグルタミン酸の分子骨格の周囲に設計される。例えば、アミノ基は、チオール基、ヒドロキシル基、ヒドロキシルアミノ基、オキシム基またはメチレン基で置換できると考えられる。カルボン酸の一方または両方は、カルボキサミド基、ホスホン酸塩基、リン酸塩基、スルホン酸塩基またはテトラゾール基で置換できると考えられる。グルタメート類似体はまた、環式化学構造および非環式化学構造、ならびに相同構造(短いまたは長い構造)から構成されると考えられる。生物活性を有し、本発明で使用できるこのようなグルタメート類似体の例が最近検討されている(全体が参考として本明細書で援用される、Aspartate and glutamate mimetic structures in biologically active compounds.(Stefanic P,Dolenc MS.Curr Med Chem.2004 Apr;11(8):945−68))。
【0116】
本明細書で使用される「セリン類似体」という用語は、セリンの構造を模倣するための化学構造を意味する。このような化学構造は、セリン自体の構造におけるように相互に関連して同様に位置するアミノ基またはアミノ基のバイオアイソスター、カルボン酸またはカルボン酸のバイオアイソスター、ならびに水酸基または水酸基のバイオアイソスターから構成されるセリンの分子骨格の周囲に設計される。例えば、アミノ基は、チオール基、ヒドロキシル基またはメチレン基で置換できると考えられる。カルボン酸は、カルボキサミド基、ホスホン酸塩基、リン酸塩基、スルホン酸塩基またはテトラゾール基で置換できると考えられる。カルボン酸は完全に除去できると考えられる。水酸基は、アミノ基、チオール基またはメチレン基で置換できると考えられる。セリン類似体はまた、環式化学構造および非環式化学構造、ならびに相同構造(短いまたは長い構造)から構成されると考えられる。
【0117】
本明細書で使用される「ホスホルアミデート類似体」という用語は、亜鉛結合能を有するホスホルアミデートの分子構造を模倣する化学構造を意味する。例えば、ホスホルアミデート類似体は、ホスホルアミデート基、ホスホン酸塩基、リン酸塩基、ホスフィン酸塩基、スルホンアミド基、尿素基、N−ヒドロキシ尿素基、チオ尿素基、カルバミン酸塩基、ヒドロキサメート基、逆のヒドロキサメート基、N−ヒドロキシアミド基またはN−ヒドロキシカルバミン酸塩基であってよい。
【0118】
「フルオレセイン」という用語は、以下の一般式の化合物を指す:
【0119】
【化25】
「フルオレセイン誘導体」という用語は、それぞれが本明細書で定義される、独立してハロゲン基またはアルキル基である1〜3個の基で場合により置換される、本明細書で定義されるフルオレセインを指す。
【実施例】
【0120】
スキーム1は、ホスホルアミデート阻害剤T33の合成についての代表的な合成スキームを示す。この一般的な方法を使用して、複雑性の異なるホスホルアミデートT33の類似体を15個超合成した。このスキームを、標準的な合成方法とともに使用して、本発明の第1の態様の化合物を調製することができる。本発明の第2の態様の化合物は、標準的な合成方法を使用して、これらから慣例的に調製することができる。
【0121】
スキーム1
【0122】
【化26】
(実施例1)
PMSA阻害検定
Maung,J.;Mallari,J.P.;Girtsman,T.A.;Wu,L.Y.;Rowley,J.A.;Santiago,N.M.;Brunelle,A.;Berkman,C.E.Probing for a Hydrophobic a Binding Register in Prostate−Specific Membrane Antigen with Phenylalkylphosphonamidates.Bioorg.Med.Chem.2004,12,4969より引用。
【0123】
基質(N−[4−(フェニルアゾ)ベンゾイル]−グルタミル−g−グルタミン酸、PAB−Glu−γ−Glu)の使用液およびすべての阻害剤を、TRIS緩衝液(50mM、pH7.4)中で作製した。精製したPSMAの使用液を、TRIS緩衝液(50mM、pH7.4)中で適切に希釈し、阻害剤の非存在下において基質から産物へ15%〜20%変換させた。25μLの阻害剤溶液か25μLのTRIS緩衝液(50mM、pH7.4)かのいずれかを試験管内の175μLのTRIS緩衝液(50mM、pH7.4)に添加することによって、一般的なインキュベーション混合物(終量250μL)を調製した。上記の溶液に25μLのPAB−Glu−γ−Glu(100μM)を添加した。25μLのPSMA使用液を添加することより、酵素反応を開始した。いずれの例でも、PAB−Glu−γ−Gluの終濃度は10μMであったが、一方で酵素を5種類の連続希釈した阻害剤濃縮物でインキュベートして、10%〜90%の阻害率を得た。37℃での一定の振盪により反応を15分間継続させた後、25μLのメタノールTFA(メタノール中における2容量%のトリフルオロ酢酸)を添加してボルテックスすることにより反応を終了させた。急冷したインキュベーション混合物を、25μLのK2HPO4(0.1M)を添加して直ちに緩衝し、ボルテックスして、遠心分離させた(7000g、10分間)。その後、得られた上澄液の85μLのアリコートをHPLCにより定量化し、KaleidaGraph 3.6(Synergy Software)を使用してIC50値を算出した。
【0124】
PABGγGおよびその加水分解産物(PABG)を、それぞれ40:60の容積比で、ACN/リン酸カリウムからなる移動相[25mM、pH2.0(H3PO4で調整)]で解析用逆相HPLCカラム(Lichrosphere C18 5μm、150×4.6mm;Phenomenex、米国カリフォルニア州トーランス)を使用して分離し、定量化した。1.0mL/分の流速で、PABGγGおよびその加水分解産物(PABG)を、それぞれ4.8分および6.9分の滞留時間により325nmで検出した。
【0125】
(実施例2)
γ−ジグルタメートのホスホルアミデート類似体の分子剪定によるPSMAの強力阻害剤の同定
本発明者等は、PSMAの阻害のための鉛化合物として、ホスホルアミデートT33を設計し、合成した。その設計は、葉酸類似体のγ−グルタミン酸誘導体などの既知のPSMA基質の構造に基づくものであった。T33の初期スクリーニングにより、T33がPSMAに対して相当な効力を発揮することが示された(IC50<50nM)。PSMAに対する阻害能についてのT33の種々の構造要素の重要性をよりよく理解するために、本発明者等は分子剪定試験を実施し、図1に示すT33類似体のライブラリーを作製した。このライブラリーからの3種の化合物の合成については、既に文献の中で報告されている:CCS、JM139(Lu,H.;Ng,R.;Shieh,C.C.;Martinez,A.R.;Berkman,C.E.Inhibition of Glutamate Carboxypeptidase by Phosphoryl and Thiophosphoryl Derivatives of Glutamic and 2−Hydroxyglutaric Acid.Phosphorus,Sulfur and Silicon,2003,178,17)、および2−PMPA(Jackson,P.F.;Cole,D.C.;Slusher,B.S.;Stetz,S.L.;Ross,L.E.;Donzanti,B.A.;Trainor,D.A.Design,synthesis,and biological activity of a patent inhibitor of the neuropeptidase N−acetylated−linked acidic dipeptidase.J.Med.Chem.1996,39,619−622)。図1、図2および図7〜12の残りの化合物は、本発明の新規の化合物である。
【0126】
図1、図2および図7〜12に記載のライブラリー内の全化合物に対して、実施例1に記載の検定を使用してPSMAの阻害能についてスクリーニングを行った。
【0127】
本発明者等が公表した方法(Purification of Prostate−Specific Membrane Antigen with Conformational Epitope−Specific Antibody−Affinity Chromatography.Liu,T.;Toriyabe,Y.;Berkman,C.E.Prot.Exp.Purif.2006,49,251.)によるLNCaP細胞由来の精製PSMAに対する図1のライブラリーのスクリーニングにより、以下の2つの一般的構造が優れた阻害能を発揮することが示された:無損傷のホスホルアミデートペプチド模倣体(例えばT33およびL36)、ならびに単純なP1’類似体(例えばCCS、JM140、および2−PMPA)。
【0128】
他の親和性エレメントが欠如しているものの、CCS、JM140および2−PMPAの2塩基性ホスホリルモチーフは、PSMAの活性部位の亜鉛原子との強力な相互作用によりPSMAに対する親和性の増大に関与しているとの仮説を立てている。興味深いことに、単純な疎水性類似体MP1Dにかなりの親和性が維持されている。本発明者等がステロイド含有ホスホルアミデート阻害剤のライブラリーを調製してスクリーニングを行ったところ、疎水性を有しかつ空間的に厳しいリガンドを収容するPSMAの能力がさらに確認された(図2)。図2のすべての化合物は、実施例1に記載の検定による測定で1μM未満のIC50を示した。MP1Dおよびステロイド含有ホスホルアミデート阻害剤による阻害検定の結果は、PSMAの中心的触媒機構から離れた疎水性結合ドメインの存在を同定した本発明者等の以前の研究と一致する(Maung,J.;Mallari,J.P.;Girtsman,T.A.;Wu,L.Y.;Rowley,J.A.;Santiago,N.M.;Brunelle,A.;Berkman,C.E.Probing for a Hydrophobic a Binding Register in Prostate−Specific Membrane Antigen with Phenylalkylphosphonamidates.Bioorg.Med.Chem.2004,12,4969)。
【0129】
(実施例3)
T33類似体によるPSMAの時間依存的かつ不可逆的な阻害
驚くべきことに、本発明者等は、T33類似体ライブラリー(図1)のPSMA阻害化合物が、PSMAの強力な阻害剤であるだけでなく、図3に示す通り酵素活性の時間依存的な減損も示したことを見出した。この固有の結果により、本発明者等は、T33およびその類似体が固有の緩慢な結合阻害剤であることを仮定するに至った。場合により、その阻害は不可逆的であると思われ、T33、L36およびMP1Eの場合がこれに当てはまる。というのも、これらの化合物により阻害される酵素の100倍の希釈によってPSMAの活性を回復できないためである(図4)。これらの結果により、T33、L36およびMP1Eは、PSMAのいずれかの機構に基づいた不可逆的な阻害剤であるか、または場合により劇的な配座変化を生じる可能性あるか、もしくは場合により酵素に共有結合による損傷を引き起こす可能性がある、PSMAの機能的に不可逆的な阻害剤であることが強く示唆されている。本発明者等は最近になって、T33、L36およびMP1Eのほかにも、LW−54およびLW−39もまたPSMAの不可逆的な阻害剤であることを明らかにした。文献を検討によれば、「これはPSMAの時間依存的かつ不可逆的な阻害剤の最初の発見」とのことである。リン酸誘導体による亜鉛ペプチダーゼおよび亜鉛プロテアーゼの緩徐な結合阻害は既知の現象であるものの、このような酵素の機序に基づいた不可逆的な阻害剤はまれである。図1の阻害剤は、図5の不可逆的および緩徐に可逆的な阻害剤に分類される。図5に示すすべての化合物は、1μM未満のIC50を示した。
【0130】
(実施例4)
阻害剤により誘導されるホモダイマーの形成
ホスホルアミデートT33およびL36の作用に関して、本発明者等は、これらの阻害剤におるPSMAの治療により、ウェスタンブロット法により示される、共有結合性PSMAホモダイマーの形成が生じることを明らかにした。図6の代表例に示される通り、T33により媒介される共有結合性PSMAダイマーの形成は、濃度依存的でかつ時間依存的である。これらの結果は、上述のようなホスホルアミデートT33およびその類似体によるPSMAの時間依存的な酵素的阻害と一致する。さらに重要なことに、これらのデータは、化合物T33、L36およびMP1Dの不可逆的阻害プロファイルと一致する。これらのデータにより、T33およびL36は、タンパク質間架橋や潜在的なタンパク質間架橋を促進するPSMAの共有結合による修飾に関与することが示されている。本発明者等はT33、L36およびMP1Dの阻害機序を測定しなかったが、これらの薬剤が、PSMA発現癌細胞に特異的な共有結合送達ビヒクルとして使用される場合がある、固有の種類のPSMA阻害剤であることは明らかである。おおまかに文献を考察したところ、これは、PSMAの不可逆的な共有結合調節剤における初めての設計と調製である。
【0131】
(実施例5)
PSMAに対するホスホルアミデート阻害剤の特異性
本発明のホスホルアミデート阻害剤の設計がPSMAに特異的であることを判定するため、マトリックスメタロプロテアーゼ−9(MMP−9)を有するT33類似体ライブラリー(図1)に由来する代表的な化合物により予備試験を実施した。MMP酵素は、細胞外マトリックスの分解に関与する亜鉛依存性エンドペプチダーゼである。NC−2−29などの検討された化合物の系列の中で、1つの化合物(NC−2−29)だけが、MMP−9に対する阻害能を示し、Ki値が5mMであった。T33は最近になり、MMP−2の阻害について検定が行われており、最高10μMまでの濃度では阻害を示さなかった。これらの大部分の陰性の結果により、本発明のT33系阻害剤の一般的な構造骨格は、標的金属ペプチダーゼであるPSMAに対して特異性をもたらすことが示唆される。
【0132】
(実施例6)
PSMA発現前立腺癌細胞の阻害剤に誘導される標識
最近の実験により、イメージングペイロード(例えば有機蛍光団)は、PSMA発現前立腺癌細胞に特異的に送達され得ることが明らかにされている。本発明者等は、それぞれがイメージングペイロードまたは治療的ペイロードの結合点としてN末端アミノ基を有する4種のPSMA阻害剤を設計し、合成した(図8)。
【0133】
その後、これらの化合物を、PSMA発現前立腺癌細胞を特異的に標識するためにアミン反応性蛍光色素で誘導体化した(図9)。蛍光顕微鏡データにより、これらの化合物はそれぞれ、PSMA発現前立腺癌細胞(LNCaP細胞)を特異的に標識し、PSMA(PC3細胞)を発現しない細胞は標識しないことが明らかにされた。チオ尿素結合蛍光阻害剤L6−V1−21は、PSMAに対する85nMのIC50を示した。この結果により、阻害剤のコアがかなりの大きさの構造モチーフで誘導体化される場合、阻害能が消滅しなかったことが明らかとなっている。L6−VI−21で治療したPSMA発現前立腺癌細胞(LNCaP)は、蛍光顕微鏡による観察で特異的な標識を生じた。L6−V1−21およびヨウ化プロピジウムによる治療後、LNCaP前立腺癌細胞およびPC3前立腺癌細胞の両方に共焦点顕微鏡法を行った。
【0134】
LNCaP細胞をL6−V1−21で治療すると、蛍光顕微鏡検査により、濃い緑色の細胞蛍光標識で示されるようにL6−V1−21による細胞表面の広範な標識化が確認される。LNCaP細胞をL6−V1−2およびヨウ化プロピジウムで治療すると、共焦点蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識で示されるようにL6−V1−21による細胞表面の広範な標識化が確認されるとともに、ヨウ化プロピジウム染色から細胞核の赤い蛍光が観察される。PC3細胞をL6−V1−2およびヨウ化プロピジウムで治療すると、細胞表面の緑色の蛍光標識化がPC3細胞において観察されないが、ヨウ化プロピジウム染色から細胞核の赤い蛍光が観察され、共焦点蛍光顕微鏡検査により、PSMA発現細胞(LNCaP)に対するL6−V1−2の特異性が確認される。PC3細胞は、PSMAを発現しないことから、陰性対照として機能した。ヨウ化プロピジウムにより両細胞株の核が染色されたものの、PSMA発現LNCaP 6細胞の表面のみがL6−V1−21で蛍光標識された。これらのLNCaP細胞上の強い標識部位は、PSMAが凝集した位置であると仮定される。
【0135】
蛍光阻害剤LW−54−F5EXにより治療されたLNCaP細胞の蛍光顕微鏡検査により、PSMAの小分子阻害剤は、PSMA発現細胞を送達して蛍光色素で標識する際に、実質的に抗体(3C6)と等価であることが明らかになった。この実験で、フルオレセイン−5−EXでそれぞれが標識されたLW−54および抗体3C6はいずれも、LNCaP細胞の膜を特異的に標識した。LNCaP細胞をLW−54−F5EXまたは蛍光標識抗体3C6−F5EXのいずれかで治療すると、蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識により明らかになるように、いずれの場合も細胞表面の広範な標識化が確認される。LW−54−F5EXがPSMA発現細胞に特異的であることを確認するため、LNCaPおよびPC3細胞をこの化合物で治療した。DAPI(4’,6−ジアミジノ−2−フェニルインドール)は両細胞株内の核DNAを染色したものの、LW−54−F5EXで蛍光標識されたのはLNCaP細胞だけであった。LNCaP細胞をLW−54−F5EXおよびDAPIで治療すると、蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識で示されるようにLW−54−F5EXによる広範わたる細胞表面の標識化が確認されるとともに、DAPI染色から細胞核の青色蛍光が観察される。PC3細胞をLW−54−F5EXおよびDAPIで治療すると、細胞表面の緑色の蛍光標識化がPC3細胞において観察されないものの、DAPI染色から細胞核の青色蛍光が観察され、蛍光顕微鏡検査により、PSMA発現細胞(LNCaP)に対するLW−54−F5EXの特異性が確認される。PC3細胞はPSMAを発現しないことから、これらの結果により、54−F5EXなどの小分子剤はPSMA発現癌細胞に特異的であることが確認される。
【0136】
また、蛍光阻害剤LW−54−5FAMXで治療したLNCaP細胞の蛍光顕微鏡検査により、PSMAの小分子阻害剤は、PSMA発現細胞を送達して蛍光色素で標識する際に、実質的に抗体と等価であることが明らかになった。この実験では、LNCaP細胞およびPC3細胞の両方をLW−54−5FAMXで治療した。DAPI(4’,6−ジアミジノ−2−フェニルインドール)は両細胞株内の核DNAを染色したものの、LW−54−F5EXで蛍光標識されたのはLNCaP細胞だけであった。LNCaP細胞をLW−54−5FAMXおよびDAPIで治療すると、蛍光顕微鏡検査により、細胞表面の濃い緑色の蛍光標識で示されるようにLW−54−5FAMXによる細胞表面の広範な標識化が確認されるとともに、DAPI染色から細胞核の青色蛍光が観察される。PC3細胞をLW−54−5FAMXおよびDAPIで治療すると、細胞表面の緑色の蛍光標識化がPC3細胞において観察されないものの、DAPI染色から細胞核の青色蛍光が観察され、蛍光顕微鏡検査により、PSMA発現細胞(LNCaP)に対するLW−54−5FAMXの特異性が確認される。これらの結果により、LW−54−5FAMXなどの小分子剤はPSMA発現癌細胞に特異的であることが確認される。
【0137】
細胞標識化試験の上記の結果により、LNCaPに特異的な標識化は、PSMAの小分子非生物学的阻害剤によって達成できることが確認される。これらの結果は、PSMAの小分子阻害剤がPSMA発現癌細胞にイメージングペイロードまたは治療的ペイロードを特異的に送達できるという証明となる。
【0138】
以上の詳細な説明および添付の実施例は、単に例示的なものであって、添付の特許請求の範囲で定義される本発明の適用範囲を限定するものとは見なされないことが理解される。開示した実施形態の種々の変更および改変は当業者に明らかになるであろう。本発明の化学構造、置換基、誘導体、中間体、合成および/または使用方法を含むがこれらに限定されないこのような変更および改変は、本発明の趣旨および適用範囲から逸脱しない範囲で行われる場合がある。
【0139】
本明細書で言及する各雑誌論文、書籍、特許および特許出願は、全体が参考として本明細書で援用される。
【図面の簡単な説明】
【0140】
【図1】T33の分子剪定類似体を示す。
【図2】PSMAのステロイド含有ホスホルアミデート阻害剤を示す。
【図3】T33による時間依存的なPSMAの阻害を示す。
【図4】T33および代表的分子剪定類似体の不可逆的および緩徐な可逆的阻害を示す。
【図5】PSMAの不可逆的、緩徐な可逆的、および急速な可逆的阻害剤としてのT33およびその分子剪定類似体の分類を示す。
【図6】(A)時間の増加および(B)濃度の増加に伴うT33治療PSMAのウェスタンブロットを示す。
【図7】MP1Dのビフェニル類似体を示す。
【図8】N末端アミノ基における誘導体化と、前立腺癌細胞へのイメージングペイロードおよび治療的ペイロードの送達との両方のために設計される合成PSMA阻害剤を示す。
【図9】PSMA発現前立腺癌細胞を特異的に標識することが知られている蛍光標識PSMA阻害剤を示す。
【図10】蛍光標識PSMA阻害剤の代表的な調製物を示す。
【図11】キレート剤含有PSMA阻害剤TL−LW−54−BnDTPAの調製物を示す。
【図12】代表的なビオチン標識PSMA阻害剤を示す。
【特許請求の範囲】
【請求項1】
以下の式の化合物であって:
A−L−B
式中、
Aがグルタメートまたはグルタメート類似体であり;
Lがホスホルアミデートまたはホスホルアミデート類似体であり;
Bがセリンまたはセリン類似体である、
化合物、あるいはその医薬的に許容される塩。
【請求項2】
請求項1に記載の化合物、あるいはその医薬的に許容される塩であって、
式中、Aが、以下の式に記載のものであり:
【化1】
式中、
nが1、2、3、4、5または6であり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−であって、式中、QがLに結合し;
R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルである、
化合物、あるいはその医薬的に許容される塩。
【請求項3】
請求項1に記載の化合物、あるいはその医薬的に許容される塩であって、
式中、Bが、以下の式に記載のものであり:
【化2】
式中、
nが1、2、3、4、5または6であり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であって、式中、TがLに結合し;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が、−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−Hまたは−C1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
各R3が独立して−HまたはC1−C6アルキルである、
記載の化合物、あるいはその医薬的に許容される塩。
【請求項4】
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であって、式中、各R3が独立して−HまたはC1−C6アルキルである、請求項1に記載の化合物、あるいはその医薬的に許容される塩。
【請求項5】
以下の式に記載の請求項1に記載の化合物、あるいはその医薬的に許容される塩であって、
【化3】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール、C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−HまたはC1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
化合物、あるいはその医薬的に許容される塩。
【請求項6】
以下の式に記載のものである:
【化4】
請求項5に記載の化合物、またはその医薬的に許容される塩。
【請求項7】
以下の式に記載のものである:
【化5】
請求項5に記載の化合物、またはその医薬的に許容される塩。
【請求項8】
R1およびR2が、それぞれ−C(O)OHである、請求項7に記載の化合物、またはその医薬的に許容される塩。
【請求項9】
R10が、−C(O)−フェニルである、請求項7に記載の化合物、またはその医薬的に許容される塩。
【請求項10】
R10がR7である、請求項7に記載の化合物、またはその医薬的に許容される塩。
【請求項11】
N−{[(2S)−2−(ベンゾイルアミノ)−2−カルボキシエトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{(2S)−2−[ベンゾイル(メチル)アミノ]−2−カルボキシエトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−{[2−(ベンゾイルアミノ)エトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{2−[ベンゾイル(メチル)アミノ]エトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[(ビフェニル−4−イルメトキシ)(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[(2−カルボキシ−4−フェニルブトキシ)(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[ヒドロキシ(4−フェニルブトキシ)ホスホリル]−L−グルタミン酸;
N−{[(2S)−2−{[4−(アミノメチル)ベンゾイル]アミノ}−2−カルボキシエトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−{[(2S)−2−アミノ−2−カルボキシエトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{[(2S)−20−アミノ−2−カルボキシ−4,8−ジオキソ−6,12,15,18−テトラオキサ−3,9−ジアザイコス−1−イル]オキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸である、
請求項1に記載の化合物、あるいはその医薬的に許容される塩。
【請求項12】
以下の式の化合物であって:
A−L−B
式中、
Aがグルタメートまたはグルタメート類似体であり;
Lがホスホルアミデートまたはホスホルアミデート類似体であり;
Bがセリンまたはセリン類似体であり;
前記化合物が、前記化合物の任意の置換可能な位置で、検出可能な標識、治療剤、または固体担体に結合する生体分子アンカーに、2価のリンカーを介して共有結合する、
化合物、あるいはその医薬的に許容される塩。
【請求項13】
以下の式に記載の請求項12に記載の化合物、あるいはその医薬的に許容される塩であって、
【化6】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R7が、−X−R8または−L1−R8であって、式中、
Xが、−O−、−S−または−N(R3)−であり;
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)−、−C(O)−L2−、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、治療剤、検出可能な標識、または固体担体に結合する生体分子アンカーであり;
Qが、−O−、−S−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
請求項12に記載の化合物、あるいはその医薬的に許容される塩。
【請求項14】
以下の式に記載のものである:
【化7】
請求項13に記載の化合物、またはその医薬的に許容される塩。
【請求項15】
R8が治療剤である、請求項14に記載の化合物、またはその医薬的に許容される塩。
【請求項16】
R8が、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基である、請求項15に記載の化合物、あるいはその医薬的に許容される塩。
【請求項17】
以下の式に記載のものであり:
【化8】
式中、
R7が、−O−R8であって、式中、
R8が、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基である、
請求項13に記載の化合物、あるいはその医薬的に許容される塩。
【請求項18】
R8が検出可能な標識である、請求項14に記載の化合物、またはその医薬的に許容される塩。
【請求項19】
R8が蛍光標識である、請求項18に記載の化合物、またはその医薬的に許容される塩。
【請求項20】
式中、R8がフルオレセインまたはフルオレセイン誘導体である、請求項19に記載の化合物、あるいはその医薬的に許容される塩。
【請求項21】
R8がキレート剤である、請求項14に記載の化合物、またはその医薬的に許容される塩。
【請求項22】
R8がR9であって、式中、
R9が、C1−C6アルキル、アリールまたはC1−C6アルキル−アリールであって、式中、R9が、独立して−COOHまたはN(R91)2である1〜3個の基で置換され、式中、
各R91が独立して、独立して−COOHまたは−N(R92)2である1〜3個の基で置換される−HまたはC1−C6アルキルであって、式中、
各R92が独立して、−Hまたは1〜3個のCOOHで置換されるC1−C6アルキルである、
請求項21に記載の化合物、あるいはその医薬的に許容される塩。
【請求項23】
N−{[(2S)−2−カルボキシ−2−({4−[({3−[(2−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)フェニル]アミノ}−2−オキソエチル)チオ]プロパノイル}アミノ)メチル]ベンゾイル}アミノ)エトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{(2S)−2−カルボキシ−2−[(4−{[(6−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)ベンゾイル]アミノ}ヘキサノイル)アミノ]メチル}ベンゾイル)アミノ]エトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−{3−[(2−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)フェニル]アミノ}−2−オキソエチル)チオ]プロパノイル}−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−(6−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)ベンゾイル]アミノ}ヘキサノイル)−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−[{(2S)−2−カルボキシ−2−[({[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)フェニル]アミノ}カルボノチオイル)アミノ]エトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−{[(4−{2−[ビス(カルボキシメチル)アミノ]−3−[{2−[ビス(カルボキシメチル)アミノ]エチル}(カルボキシメチル)アミノ]プロピル}フェニル)アミノ]カルボノチオイル}−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−{6−[(6−{[5−(2−オキソヘキサヒドロ−1H−チエノ[3,4−d]イミダゾール−4−イル)ペンタノイル]アミノ}ヘキサノイル)アミノ]ヘキサノイル}−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−{[(2S)−2−カルボキシ−2−({4−[({6−[(6−{[5−(2−オキソヘキサヒドロ−1H−チエノ[3,4−d]イミダゾール−4−イル)ペンタノイル]アミノ}ヘキサノイル)アミノ]ヘキサノイル}アミノ)メチル]ベンゾイル}アミノ)エトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{3−[(3β,8ξ,9ξ,14ξ,17ξ,20ξ)−コレスタン−3−イルオキシ]プロポキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[ヒドロキシ(3−{[(3β,8ξ,9ξ,14ξ)−17−オキソアンドロスタン−3−イル]オキシ}プロポキシ)ホスホリル]−L−グルタミン酸;
N−{[(3β,8ξ,9ξ,14ξ,17ξ,20ξ)−コレスタン−3−イルオキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−(ヒドロキシ{[(3β,8ξ,9ξ,14ξ)−17−オキソアンドロスタン−3−イル]オキシ}ホスホリル)−L−グルタミン酸;
N−[ヒドロキシ(3−{[17−オキソエストラ−1(10),2,4−トリエン−3−イル]オキシ}プロポキシ)ホスホリル]−L−グルタミン酸;
N−[(3−{[3−(ベンゾイルオキシ)エストラ−1(10),2,4−トリエン−17−イル]オキシ}プロポキシ)(ヒドロキシ)ホスホリル]−L−グルタミン酸である、
請求項13に記載の化合物、あるいはその医薬的に許容される塩。
【請求項24】
請求項13に記載の化合物と、医薬的に許容される賦形剤、担体または希釈剤とを含む、組成物。
【請求項25】
請求項13に記載の化合物を含む、診断キット。
【請求項26】
前立腺特異的膜抗原(PSMA)を阻害するための方法であって、PMSAを発現する細胞を、請求項24に記載の有効阻害量の医薬組成物と接触させる工程を含む、方法。
【請求項27】
前立腺特異的膜抗原提示細胞を検出するための方法であって、該細胞を請求項13に記載の化合物と接触させる工程と、PMSA提示細胞のみの検出が可能となる条件下で検出可能な標識を検出する工程とを含む、方法。
【請求項28】
治療を必要とする被験体において、前立腺特異的膜抗原(PSMA)を提示する細胞に関与する疾患を阻害または治療するための方法であって、該細胞を、請求項16に記載の有効阻害量の化合物と接触させる工程を含む、方法。
【請求項29】
前記被験体がヒトである、請求項28に記載の方法。
【請求項30】
前立腺特異的膜抗原(PMSA)提示細胞を捕捉、検出および定量化するための方法であって、PMSAを提示することが疑われる細胞を、請求項18に記載の化合物と接触させる工程と、捕捉または固定したPSMA提示細胞の存在を検出する工程とを含む、方法。
【請求項1】
以下の式の化合物であって:
A−L−B
式中、
Aがグルタメートまたはグルタメート類似体であり;
Lがホスホルアミデートまたはホスホルアミデート類似体であり;
Bがセリンまたはセリン類似体である、
化合物、あるいはその医薬的に許容される塩。
【請求項2】
請求項1に記載の化合物、あるいはその医薬的に許容される塩であって、
式中、Aが、以下の式に記載のものであり:
【化1】
式中、
nが1、2、3、4、5または6であり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−であって、式中、QがLに結合し;
R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルである、
化合物、あるいはその医薬的に許容される塩。
【請求項3】
請求項1に記載の化合物、あるいはその医薬的に許容される塩であって、
式中、Bが、以下の式に記載のものであり:
【化2】
式中、
nが1、2、3、4、5または6であり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であって、式中、TがLに結合し;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が、−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−Hまたは−C1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
各R3が独立して−HまたはC1−C6アルキルである、
記載の化合物、あるいはその医薬的に許容される塩。
【請求項4】
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であって、式中、各R3が独立して−HまたはC1−C6アルキルである、請求項1に記載の化合物、あるいはその医薬的に許容される塩。
【請求項5】
以下の式に記載の請求項1に記載の化合物、あるいはその医薬的に許容される塩であって、
【化3】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R10が、−H、−C1−C6アルキル、アリール、−C1−C6アルキル−アリール、−アリール−アリール、−X−R6、−R7、−C(O)R5、−S(O)2R5、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
Xが、−O−、−S−または−N(R3)−であり;
R5が−CH(R51)N(R52)2;
独立して−ハロゲン、COOR53、−N(R52)2である1〜3個の基で場合により置換されるC1−C6アルキル;
アリール;
またはヘテロアリールであって、式中、
R51が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール、C1−C6アルキル−ヘテロアリール、または−OR53、−SR53、−NH2、−N(H)C(=NH)NH2、−COOR53もしくは−C(O)N(R53)2で場合により置換されるC1−C6アルキルであり;
R52が、−H、C1−C6アルキル、−C(O)R53、C(O)OR53、−C(O)NH(C1−C6アルキル)、−C(O)N(R53)2、−C(O)アリールまたは−C(O)ヘテロアリールであり;
R53が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
R6が、−HまたはC1−C6アルキルであり;
R7が、−L1−R8であって、式中、
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)または−C(O)−L2−であって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、−H、−NH2または−OHであり;
Qが、−O−、−S−、−N(R3)−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
化合物、あるいはその医薬的に許容される塩。
【請求項6】
以下の式に記載のものである:
【化4】
請求項5に記載の化合物、またはその医薬的に許容される塩。
【請求項7】
以下の式に記載のものである:
【化5】
請求項5に記載の化合物、またはその医薬的に許容される塩。
【請求項8】
R1およびR2が、それぞれ−C(O)OHである、請求項7に記載の化合物、またはその医薬的に許容される塩。
【請求項9】
R10が、−C(O)−フェニルである、請求項7に記載の化合物、またはその医薬的に許容される塩。
【請求項10】
R10がR7である、請求項7に記載の化合物、またはその医薬的に許容される塩。
【請求項11】
N−{[(2S)−2−(ベンゾイルアミノ)−2−カルボキシエトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{(2S)−2−[ベンゾイル(メチル)アミノ]−2−カルボキシエトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−{[2−(ベンゾイルアミノ)エトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{2−[ベンゾイル(メチル)アミノ]エトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[(ビフェニル−4−イルメトキシ)(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[(2−カルボキシ−4−フェニルブトキシ)(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[ヒドロキシ(4−フェニルブトキシ)ホスホリル]−L−グルタミン酸;
N−{[(2S)−2−{[4−(アミノメチル)ベンゾイル]アミノ}−2−カルボキシエトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−{[(2S)−2−アミノ−2−カルボキシエトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{[(2S)−20−アミノ−2−カルボキシ−4,8−ジオキソ−6,12,15,18−テトラオキサ−3,9−ジアザイコス−1−イル]オキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸である、
請求項1に記載の化合物、あるいはその医薬的に許容される塩。
【請求項12】
以下の式の化合物であって:
A−L−B
式中、
Aがグルタメートまたはグルタメート類似体であり;
Lがホスホルアミデートまたはホスホルアミデート類似体であり;
Bがセリンまたはセリン類似体であり;
前記化合物が、前記化合物の任意の置換可能な位置で、検出可能な標識、治療剤、または固体担体に結合する生体分子アンカーに、2価のリンカーを介して共有結合する、
化合物、あるいはその医薬的に許容される塩。
【請求項13】
以下の式に記載の請求項12に記載の化合物、あるいはその医薬的に許容される塩であって、
【化6】
式中、
各nが独立して1、2、3、4、5または6であり;
各R1およびR2が独立して−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
各R3が独立して−HまたはC1−C6アルキルであり;
R4が、−H、−C(O)OR3、−C(O)N(R3)2、−P(O)(OR3)2、−OP(O)(OR3)2、−S(O)2R3、−S(O)2OR3、−S(O)2N(R3)2またはテトラゾリルであり;
Lが、−P(O)(OR3)−、−P(O)(N(R3)2)−、−S(O)2−、−C(O)−または−C(S)−であり;
MおよびTが独立して−O−、−S−、−N(R3)−または−CH2−であり;
R7が、−X−R8または−L1−R8であって、式中、
Xが、−O−、−S−または−N(R3)−であり;
L1が、−C(O)N(R3)−、−C(S)N(R3)−、−C(O)CH(R21)−、−C(O)(O)−、−C(O)−L2−、ペプチド、デンドリマーまたはペプチドデンドリマーであって、式中、
R21が、−H、アリール、ヘテロアリール;−OHで場合により置換されるC1−C6アルキル−アリール;C1−C6アルキル−ヘテロアリール、または−OR23、−SR23、−NH2、−N(H)C(=NH)NH2、−COOR23もしくは−C(O)N(R23)2で場合により置換されるC1−C6アルキルであり;
R23が、−H、C1−C6アルキルまたはC1−C6アルキル−アリールであり;
L2が、−C1−C24アルキル−または−フェニル−C1−C24アルキル−であって、式中、
各アルキル基が、オキソ、=Sまたは−COOHである1〜4個の基で場合により置換され;
各アルキル基のメチレン基の内の1〜6個が、−O−、−S−または−N(R3)−で場合により置換されるが、但し、2個の隣接するメチレン基がいずれも−O−、−S−または−N(R3)−で置換されないことを条件とし;
R8が、治療剤、検出可能な標識、または固体担体に結合する生体分子アンカーであり;
Qが、−O−、−S−、−N(R3)O−、−ON(R3)−、−CH2−または=NO−である、
請求項12に記載の化合物、あるいはその医薬的に許容される塩。
【請求項14】
以下の式に記載のものである:
【化7】
請求項13に記載の化合物、またはその医薬的に許容される塩。
【請求項15】
R8が治療剤である、請求項14に記載の化合物、またはその医薬的に許容される塩。
【請求項16】
R8が、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基である、請求項15に記載の化合物、あるいはその医薬的に許容される塩。
【請求項17】
以下の式に記載のものであり:
【化8】
式中、
R7が、−O−R8であって、式中、
R8が、C1−C10アルキル、オキソ、ヒドロキシまたはハロゲンからなる群から選択される1〜5個の基で場合により置換されるステロイド基である、
請求項13に記載の化合物、あるいはその医薬的に許容される塩。
【請求項18】
R8が検出可能な標識である、請求項14に記載の化合物、またはその医薬的に許容される塩。
【請求項19】
R8が蛍光標識である、請求項18に記載の化合物、またはその医薬的に許容される塩。
【請求項20】
式中、R8がフルオレセインまたはフルオレセイン誘導体である、請求項19に記載の化合物、あるいはその医薬的に許容される塩。
【請求項21】
R8がキレート剤である、請求項14に記載の化合物、またはその医薬的に許容される塩。
【請求項22】
R8がR9であって、式中、
R9が、C1−C6アルキル、アリールまたはC1−C6アルキル−アリールであって、式中、R9が、独立して−COOHまたはN(R91)2である1〜3個の基で置換され、式中、
各R91が独立して、独立して−COOHまたは−N(R92)2である1〜3個の基で置換される−HまたはC1−C6アルキルであって、式中、
各R92が独立して、−Hまたは1〜3個のCOOHで置換されるC1−C6アルキルである、
請求項21に記載の化合物、あるいはその医薬的に許容される塩。
【請求項23】
N−{[(2S)−2−カルボキシ−2−({4−[({3−[(2−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)フェニル]アミノ}−2−オキソエチル)チオ]プロパノイル}アミノ)メチル]ベンゾイル}アミノ)エトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{(2S)−2−カルボキシ−2−[(4−{[(6−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)ベンゾイル]アミノ}ヘキサノイル)アミノ]メチル}ベンゾイル)アミノ]エトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−{3−[(2−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)フェニル]アミノ}−2−オキソエチル)チオ]プロパノイル}−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−(6−{[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)ベンゾイル]アミノ}ヘキサノイル)−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−[{(2S)−2−カルボキシ−2−[({[3−カルボキシ−4−(6−ヒドロキシ−3−オキソ−9,9a−ジヒドロ−3H−キサンテン−9−イル)フェニル]アミノ}カルボノチオイル)アミノ]エトキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−{[(4−{2−[ビス(カルボキシメチル)アミノ]−3−[{2−[ビス(カルボキシメチル)アミノ]エチル}(カルボキシメチル)アミノ]プロピル}フェニル)アミノ]カルボノチオイル}−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−{6−[(6−{[5−(2−オキソヘキサヒドロ−1H−チエノ[3,4−d]イミダゾール−4−イル)ペンタノイル]アミノ}ヘキサノイル)アミノ]ヘキサノイル}−L−γ−グルタミル−Ο−[{[(1S)−1,3−ジカルボキシプロピル]アミノ}(ヒドロキシ)ホスホリル]−L−セリン;
N−{[(2S)−2−カルボキシ−2−({4−[({6−[(6−{[5−(2−オキソヘキサヒドロ−1H−チエノ[3,4−d]イミダゾール−4−イル)ペンタノイル]アミノ}ヘキサノイル)アミノ]ヘキサノイル}アミノ)メチル]ベンゾイル}アミノ)エトキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−[{3−[(3β,8ξ,9ξ,14ξ,17ξ,20ξ)−コレスタン−3−イルオキシ]プロポキシ}(ヒドロキシ)ホスホリル]−L−グルタミン酸;
N−[ヒドロキシ(3−{[(3β,8ξ,9ξ,14ξ)−17−オキソアンドロスタン−3−イル]オキシ}プロポキシ)ホスホリル]−L−グルタミン酸;
N−{[(3β,8ξ,9ξ,14ξ,17ξ,20ξ)−コレスタン−3−イルオキシ](ヒドロキシ)ホスホリル}−L−グルタミン酸;
N−(ヒドロキシ{[(3β,8ξ,9ξ,14ξ)−17−オキソアンドロスタン−3−イル]オキシ}ホスホリル)−L−グルタミン酸;
N−[ヒドロキシ(3−{[17−オキソエストラ−1(10),2,4−トリエン−3−イル]オキシ}プロポキシ)ホスホリル]−L−グルタミン酸;
N−[(3−{[3−(ベンゾイルオキシ)エストラ−1(10),2,4−トリエン−17−イル]オキシ}プロポキシ)(ヒドロキシ)ホスホリル]−L−グルタミン酸である、
請求項13に記載の化合物、あるいはその医薬的に許容される塩。
【請求項24】
請求項13に記載の化合物と、医薬的に許容される賦形剤、担体または希釈剤とを含む、組成物。
【請求項25】
請求項13に記載の化合物を含む、診断キット。
【請求項26】
前立腺特異的膜抗原(PSMA)を阻害するための方法であって、PMSAを発現する細胞を、請求項24に記載の有効阻害量の医薬組成物と接触させる工程を含む、方法。
【請求項27】
前立腺特異的膜抗原提示細胞を検出するための方法であって、該細胞を請求項13に記載の化合物と接触させる工程と、PMSA提示細胞のみの検出が可能となる条件下で検出可能な標識を検出する工程とを含む、方法。
【請求項28】
治療を必要とする被験体において、前立腺特異的膜抗原(PSMA)を提示する細胞に関与する疾患を阻害または治療するための方法であって、該細胞を、請求項16に記載の有効阻害量の化合物と接触させる工程を含む、方法。
【請求項29】
前記被験体がヒトである、請求項28に記載の方法。
【請求項30】
前立腺特異的膜抗原(PMSA)提示細胞を捕捉、検出および定量化するための方法であって、PMSAを提示することが疑われる細胞を、請求項18に記載の化合物と接触させる工程と、捕捉または固定したPSMA提示細胞の存在を検出する工程とを含む、方法。
【図1】

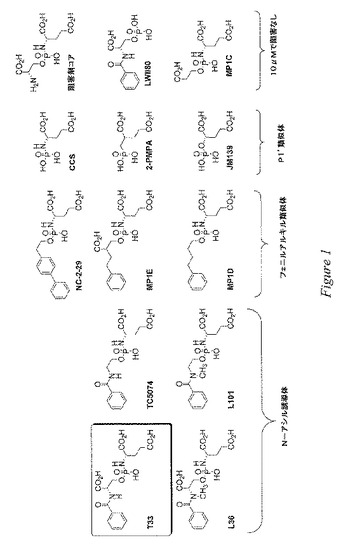
【図2】
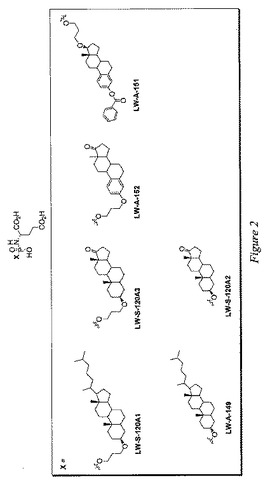
【図3】

【図4】

【図5】
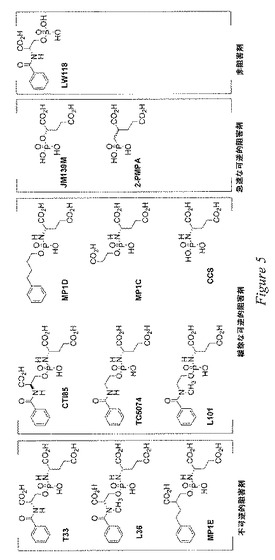
【図6】
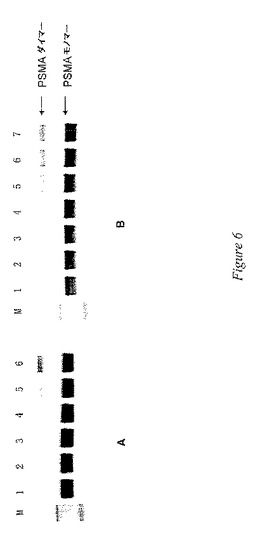
【図7】
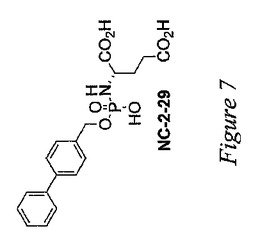
【図8】
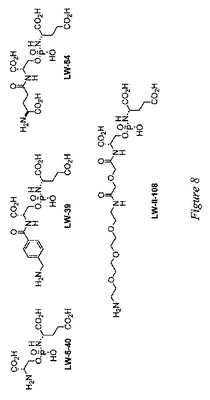
【図9】
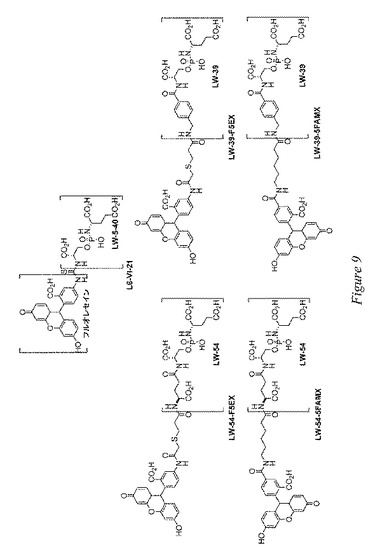
【図10】
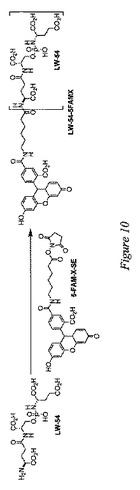
【図11】

【図12】
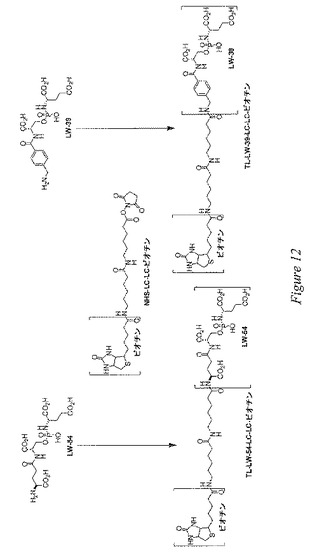
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【公表番号】特表2009−532338(P2009−532338A)
【公表日】平成21年9月10日(2009.9.10)
【国際特許分類】
【出願番号】特願2009−500598(P2009−500598)
【出願日】平成19年3月14日(2007.3.14)
【国際出願番号】PCT/US2007/064002
【国際公開番号】WO2007/106869
【国際公開日】平成19年9月20日(2007.9.20)
【出願人】(508276774)キャンサー ターゲテッド テクノロジー エルエルシー (1)
【Fターム(参考)】
【公表日】平成21年9月10日(2009.9.10)
【国際特許分類】
【出願日】平成19年3月14日(2007.3.14)
【国際出願番号】PCT/US2007/064002
【国際公開番号】WO2007/106869
【国際公開日】平成19年9月20日(2007.9.20)
【出願人】(508276774)キャンサー ターゲテッド テクノロジー エルエルシー (1)
【Fターム(参考)】
[ Back to top ]