
Pseudomonas細菌感染の処置のためのオリゴ糖およびその結合体
Pseudomonas細菌に関連して、組成物および方法が提供される。その組成物および方法は、Pseudomonas細菌による感染に関連する医学的状態の診断および治療のために使用され得る。そのような伝染病としては、嚢胞性線維症の患者の肺中のPseudomonas aeruginosaが挙げられる。本方法において有用な化合物は、治療薬剤に結合され得る。Pseudomonas細菌は、コロニー形成をブロックすることにより、この細菌の増殖を阻止することにより、またはその細菌を殺すことにより阻害され得る。

【発明の詳細な説明】
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、一般的に、嚢胞性線維症を有する患者の肺の中でのPseudomonas aeruginosaを含む、Pseudomonas細菌による感染およびコロニー形成に関与する、温血動物(例えば、ヒト)における疾患の診断および治療のための化合物、組成物、および方法に関する。本発明は、より具体的には、Pseudomonas細菌に選択的に結合するための1つ以上の化合物の使用に関する。これらの化合物は、Pseudomonas細菌のコロニー形成の診断および/または治療的介入に対して有用であるか、またはPseudomonas細菌を標的とし、Pseudomonas細菌を有効に阻止もしくは殺傷するために薬剤へ結合され得る。
【背景技術】
【0002】
(関連技術の説明)
Pseudomonas感染は、種々の医学的状態において発生し、そして生命を脅かし得る。Pseudomonasは、日和見性細菌である。危険状態にある個体の例としては、嚢胞性線維症患者および熱傷患者が挙げられる。嚢胞性線維症は、Pseudomonas細菌の感染を含み得る医学的状態の代表的な例として以下に記載される。
【0003】
嚢胞性線維症(CF)は、白人集団の間で最も一般的な致死の遺伝疾患である。CFは、嚢胞性線維症膜貫通調節タンパク質(cystic fibrosis transmembrane conductance regulator;CFTR)をコードする遺伝子中の変異によって引き起こされ、塩化物チャネルとして作用する。イオン移動を変化させるCFTRの遺伝的変異もまた、CFTRのN−グルコシル化、および他の細胞表面分子に影響を及ぼす。患者の全ての外分泌腺は、影響を受ける;しかし、肺は、病的状態および死亡の主要な部分である。グルコシル化における一般的な変化は、ルイスのフコシル化における増加およびシアル化における減少を生じる。CF患者由来の唾液のおよび呼吸器のムチン(musin)はまた、シアル化および硫酸化されたルイスx/a構造を含む、より高レベルのルイス型オリゴ糖を含む。
【発明の開示】
【発明が解決しようとする課題】
【0004】
CF患者における病的状態および死亡の主な原因は、この細菌、Pseudomonas aeruginosaによる慢性の肺コロニー形成であり、この細菌は、肺の破壊および死につながる強い好中球性炎症応答の著しい肺感染を生じる。P.aeruginosaによるコロニー形成は、この細菌上の線毛レクチンおよび鞭毛レクチンの、肺細胞表面上のルイス型糖鎖構造への結合によって開始される。PA−ILおよびPA−IILとして知られるこれらのレクチンは、これらのオリゴ糖構造と高い親和性で結合し、そして細菌のコロニー形成の最初の工程を妨げる潜在的分子標的を表す。この細菌によって十分にはコロニー形成されない患者は、良好な長期間の予後を維持する。Pseudomonas細菌による個体中でのコロニー形成の予防についての、当該分野での現在のアプローチにおける難しさのために、化合物、組成物および方法を改良する必要がある。
【課題を解決するための手段】
【0005】
(発明の簡単な要旨)
簡単に述べると、本発明は、Pseudomonas細菌の検出のためにPseudomonas細菌上に発現するレクチンを利用するための化合物、組成物および方法、ならびにPseudomonas細菌に関与する疾患(ヒトの疾患を含む)の診断および治療を提供する。例えば、P.aeruginosa上のレクチンに対して高い親和性の結合性を有するルイス構造の糖模倣物は、CF患者に対して有益な治療効果を有する。さらに、これらの糖模倣物は、例えば、効力を増大させ、そして投与量を低下させるために強い抗生物質と結合体化され得、それによってこれらの強力な抗生物質の周知の有害な副作用が回避され得る。これらの結合部分が、この細菌のコロニー形成および病原性に対して重大であることを考慮すると、この結合体治療に対して抵抗性となる、この標的における変異は、この細菌の非病原性形態を生じるに違いない。
【0006】
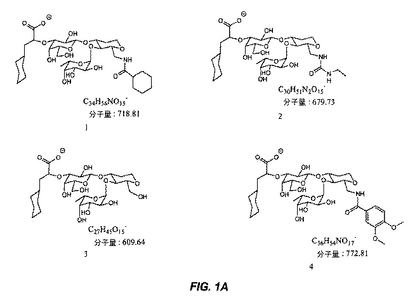
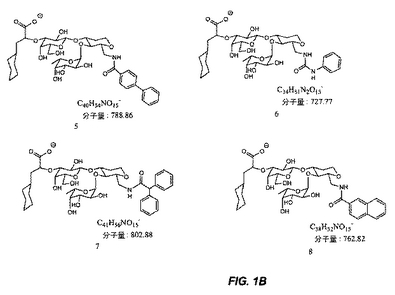
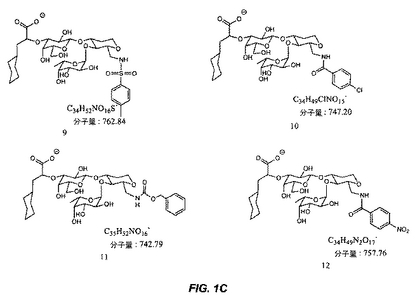
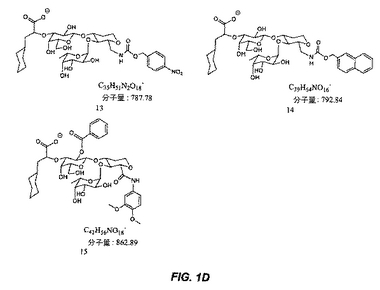
本発明の1つの実施形態は、温血動物におけるPseudomonas細菌を阻害する方法を提供し、この方法は、図1または図2に記載の化合物を含む化合物を、この細菌を阻害するための有効な量で、この動物へ投与する工程を包含する。
【0007】
別の実施形態では、本発明は、図1または図2に記載の化合物と結合した治療薬剤を含む結合体を提供する。
【0008】
別の実施形態では、本発明は、Pseudomonas細菌を検出する方法を提供し、この方法は、図1または図2に記載の化合物を含む化合物に結合された診断薬剤とサンプルを、このサンプル中に存在する場合、この化合物がこの細菌と結合するための十分な条件下で、接触させる工程;およびこのサンプル中に存在するこの薬剤を検出する工程であって、ここで、このサンプル中のこの薬剤の存在は、Pseudomonas細菌の存在を示す、工程を包含する。
【0009】
別の実施形態では、本発明は、固体支持体上にPseudomonas細菌を固定する方法を提供し、この方法は、結合するための十分な条件下で、Pseudomonas細菌含有サンプルと、固体支持体上に固定された図1または図2に記載の化合物を含む化合物とを接触させる工程;およびこの固体支持体からこのサンプルを分離する工程を包含する。
【0010】
他の実施形態では、本明細書に記載の化合物および結合体は、Pseudomonas細菌の阻害のための薬の調製において使用され得る。
【0011】
本発明のこれらの局面および他の局面は、以下の詳細な説明および添付された図面を参照すれば明白になる。本明細書で開示された全ての参考文献は、あたかもそれぞれが個々に援用されたかのように、その全体が参考として援用される。
【発明を実施するための最良の形態】
【0012】
(発明の詳細な説明)
上述で記載のように、本発明は、P.aeruginosaと結合して、疾患の診断および治療において使用され得る、化合物および組成物を提供する。
【0013】
(糖模倣化合物)
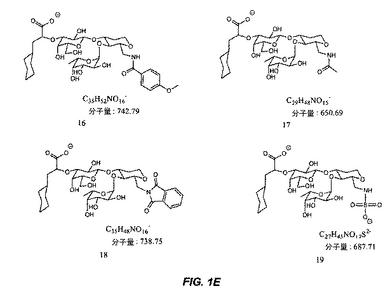
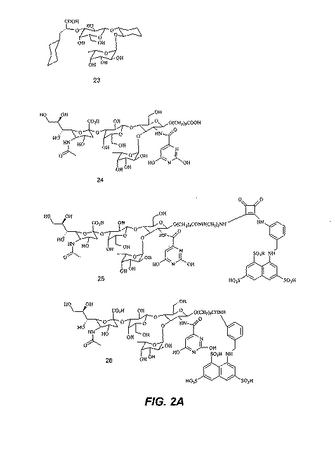
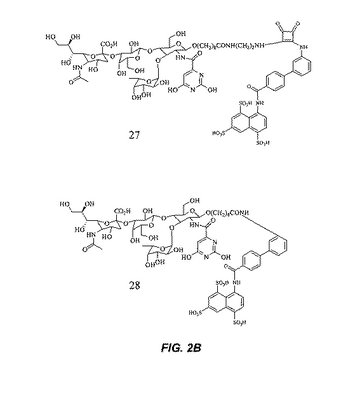
本明細書で使用される場合、用語「糖模倣化合物」とは、P.aeruginosaと特異的に結合する分子をいう。本発明によって包含される糖模倣化合物の構造は、図1および図2において示され、そしてまた、この化合物が、末端のシクロヘキシル乳酸部分として示されるシアル酸についての模倣物を含まないことを除いては、本明細書で開示された化合物を包含する。これは、例えば、ある反応スキームにおいて中間体Eの添加を含む工程を除去することによって達成される。本発明において有用な全ての化合物(またはその結合体)は、生理学的に受容可能な、それらの塩を含む。
【0014】
ある実施形態については、薬物のような診断薬剤もしくは治療薬剤を糖模倣化合物と結合させて結合体(その結合は共有結合である)を形成させることもまた、あるいは、有益であり得る。本明細書で使用される場合、用語「治療薬剤」とは、疾患または他の非所望状態を予防または処置するか、あるいは治療の成功を促進させるための温血動物(例えば、ヒトのような哺乳動物)への投与が意図される、任意の生物活性薬剤をいう。治療薬剤としては、抗生物質、ホルモン、成長因子、タンパク質、ペプチド、遺伝子、非ウイルスベクターおよび他の化合物が挙げられる。
【0015】
(糖模倣化合物の処方物)
本明細書で記載の糖模倣化合物は、薬学的組成物内に存在し得る。薬学的組成物は、1つ以上の薬学的または生理学的に受容可能なキャリア、希釈剤または賦形剤と組み合わされた1つ以上の糖模倣化合物を含む。そのような組成物は、緩衝液(例えば、中性緩衝生理食塩水またはリン酸緩衝生理食塩水)、糖質(例えば、グルコース、マンノース、スクロース、またはデキストラン)、マンニトール、タンパク質、ポリペプチド、またはグリシンのようなアミノ酸、抗酸化剤、キレート剤(例えば、EDTAまたはグルタチオン)、アジュバント(例えば、水酸化アルミニウム)および/または保存剤を含み得る。なおさらなる他の実施形態では、本発明の組成物は、凍結乾燥物(lyophilizate)として処方され得る。本発明の組成物は、例えば、エアロゾル投与、局所投与、経口投与、鼻腔投与、静脈内投与、頭蓋内投与、腹腔内投与、皮下投与、または筋肉投与を含む、任意の適切な投与様式について処方され得る。
【0016】
薬学的組成物はまた、あるいは、糖模倣化合物と結合され得るか、またはこの組成物内で遊離であり得る薬物のような、1つ以上の活性薬剤を含み得る。糖模倣化合物への薬剤の結合は、共有結合または非共有結合であり得る。
【0017】
本明細書で記載の組成物は、持続放出処方物(すなわち、投与後の調節薬剤の、ゆっくりとした放出をもたらすカプセルまたはスポンジのような処方物)の一部として投与され得る。そのような処方物は、一般的に、周知の技術を用いて調製され得、そして例えば、経口移植、直腸移植、または皮下移植、または所望の標的部位での移植によって投与され得る。そのような処方物内での使用のためのキャリアは、生体適合性であり、そしてまた生物分解性であり得;好ましくは、この処方物は、比較的一定のレベルでの調節薬剤の放出を提供する。維持放出処方物内に含まれる糖模倣化合物の量は、移植部位、放出速度、および放出の期待された持続時間、ならびに治療または予防される状態の性質に依存する。
【0018】
糖模倣化合物は、一般的に、治療的有効量で、薬学的組成物内に存在する。治療的有効量は、Pseudomonas感染に関連した状態の測定または観察された応答のような、患者の認識可能な利益をもたらす量である。
【0019】
(糖模倣化合物の使用方法)
一般的に、本明細書に記載の糖模倣化合物は、Pseudomonas(例えば、P.aeruginosa)細菌による感染に関与する疾患(例えばヒトの疾患)をもたらす診断結果および/または治療結果を達成するために使用され得る。そのような診断結果および/または治療結果は、動物、好ましくは、ヒトのような哺乳動物のインビトロおよび/またはインビボにおいて達成され得る。ただし、Pseudomonas(例えば、P.aeruginosa)は、最終的に、認識可能な診断結果または治療結果を達成するために十分な量および時間において、糖模倣化合物と接触される。本発明の背景では、治療結果は、例えば、肺感染の予防と関連する。いくつかの状態では、治療結果は、Pseudomonasの阻害(例えば、P.aeruginosa)(例えば、このような細菌の増殖を阻止するかもしくはこの細菌を殺傷する、またはこの細菌によるコロニー形成を防ぐことを包含する)と関連する。本明細書で使用される場合、治療または治療学的結果は、処置または予防を含む。
【0020】
本発明の糖模倣化合物は、処置または予防されるべき疾患に対する適切な様式において投与され得る。適切な投与量ならびに投与の適切な持続時間および頻度は、患者の状態、患者の疾患の型および重症度ならびに投与方法のような要因によって決定され得る。一般的に、適切な投与量および処置レジメンは、処置および/または予防の利益を提供するために十分な量で、この調節薬剤を提供する。本発明の特に好ましい実施形態では、糖模倣化合物は、0.001mg/kg体重〜1000mg/kg体重(より代表的には0.01mg/kg〜1000mg/kg)の範囲の投与量で、毎日1回または複数回の投与レジメンで投与され得る。適切な投与量は、一般的に、実験モデルおよび/または臨床試験を用いて決定され得る。一般的に、効果的な治療を提供するために十分である最小の投与量の使用が好ましい。患者は、一般的に、当業者が精通している、治療または予防される状態に対して適切なアッセイを用いて、治療的有効性についてモニターされ得る。
【0021】
糖模倣化合物はまた、Pseudomonas細菌(例えば、P.aeruginosa)へ、物質を標的化するために使用され得る。そのような物質としては、治療薬剤および診断薬剤が挙げられる。治療薬剤は、分子、ウイルス、ウイルス成分、細胞、細胞成分、あるいは、標的細胞の性質を変更することが立証され得、その結果障害を治療もしくは予防するため、または患者の生理機能を制御するための利益を提供し得る、任意の他の物質であり得る。治療薬剤はまた、インビボで生物学的活性を有する薬剤を生じるプロドラッグであり得る。治療薬剤であり得る分子は、例えば、ポリペプチド、アミノ酸、核酸、ポリヌクレオチド、ステロイド、多糖または無機化合物であり得る。そのような分子は、任意の種々の方法において機能し得、それらとしては、例えば、酵素、酵素インヒビター、ホルモン、レセプター、アンチセンスオリゴヌクレオチド、触媒性ポリヌクレオチド、抗ウイルス剤、抗腫瘍剤、抗細菌剤、免疫調節剤、および細胞障害性薬剤(例えば、ヨウ素、臭素、鉛、レニウム、ホミウム(homium)、パラジウムまたは銅のような放射性核種)が挙げられる。診断薬剤としては、金属および放射性薬剤のような画像化剤(例えば、ガリウム、テクネチウム、インジウム、ストロンチウム、ヨウ素、バリウム、臭素およびリン含有化合物)、造影剤、色素(例えば、蛍光色素または発蛍光団)ならびに比色定量的反応または蛍光定量的反応を触媒する酵素が挙げられる。一般的に、治療薬剤および診断薬剤は、上述で記載の技術のような種々の技術を用いて、糖模倣化合物に結合され得る。標的化の目的のためには、糖模倣化合物は、本明細書で記載の通りに患者に投与され得る。
【0022】
糖模倣化合物はまた、(例えば、種々の周知の細胞培養方法および細胞分離方法で)インビトロで使用され得る。例えば、糖模倣化合物は、培養中でのスクリーニング、アッセイまたは増殖のためにPseudomonas細菌を固定する際に使用するために固体支持体上に固定され得る。(例えば、組織培養プレートまたは他の細胞培養支持体の内部表面と連結される)。そのような連結は、上述に記載の方法のような任意の適切な技術、および他の標準的技術によって行われ得る。また、糖模倣化合物の使用は、細胞の同定およびインビトロでの選別を容易にし、そのような細菌細胞の選択を許容し得る。好ましくは、そのような方法における使用のための糖模倣化合物は、検出可能なマーカーである診断薬剤に連結される。適切なマーカーは、当該分野で周知であり、それらとして、放射性核種、発光基、蛍光基、酵素、色素、免疫グロブリンの定常ドメイン、およびビオチンが挙げられる。1つの好ましい実施形態では、フルオレセインのような蛍光マーカーに連結された糖模倣化合物は、細胞と接触され、次いで、蛍光活性化細胞選別器(FACS)によって分析される。
【0023】
そのようなインビトロでの方法は、一般的に、サンプル(例えば、生物学的調製物)と、糖模倣化合物の任意の1個とを接触させる工程、およびこのサンプル中のこの化合物を検出する工程を包含する。所望される場合、1回以上の洗浄工程が方法に加えられ得る。例えば、この化合物の検出より先にサンプルと糖模倣化合物とを接触させ、その後、このサンプルは、洗浄され得る(すなわち、流体と接触させ、次いでこの流体を除去し、結合しない糖模倣化合物を除去する)。あるいは、または加えて、洗浄工程は、この検出プロセスの間にさらに加えられ得る。例えば、糖模倣化合物が、検出可能である物質と結合し得るマーカー(診断薬剤)を所有する場合、この検出の前に、このサンプルと検出可能な物質とを接触させた後に、このサンプルを洗浄することが所望され得る。本明細書で使用される場合、語句「このサンプル中の化合物(または薬剤)の検出」は、この化合物(もしくは薬剤)がこのサンプルと結合したままであるこの化合物(もしくは薬剤)を検出すること、またはこの化合物(もしくは薬剤)がこのサンプルから分離された後に、このサンプルと結合した化合物(もしくは薬剤)を検出することを包含する。
【0024】
以下の実施例は、例証の目的で提示されるのであり、限定の目的で提示されるのではない。
【実施例】
【0025】
(実施例1)
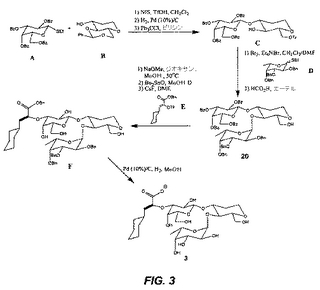
(3の合成(図3))
(中間体Cの形成:)
化合物A(5.00g、12.74mmol)およびB(4.50g、19.11mmol)ならびにNIS(3.58g、15.93mmol)をCH2Cl2(50ml)中に溶解し、0℃に冷却する。トリフルオロメタンスルホン酸の溶液(CH2Cl2中の0.15M)を、攪拌しながら滴下する。その溶液が橙色から暗褐色に色を変えたら、TMS−OHの添加を止める。この溶液を次いで飽和NaHCO3(30ml)で洗浄し、そしてその有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。得られるシロップをシリカゲルクロマトグラフィー(ヘキサン/エーテル、1:1)により精製し、次の工程に使用する。
【0026】
上記で得られる化合物をTHF(40ml)中に溶解し、炭素担持Pd(10%)(質量で1/10)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、4位および6位がOHの化合物を与える。この化合物を、次いでピリジン(25ml)中に溶解し、0℃に冷却する。Ph3CCl(1.2当量)を滴下し、この反応を室温で6時間進行させる。酢酸エチル(50ml)を次いで添加し、この溶液を0.1N HCl(50mlで2回)、飽和NaHCO3(50mlで1回)および飽和NaCl(50mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、乾燥状態までエバポレートする。中間体Cを、シリカゲルクロマトグラフィーにより得る。
【0027】
(20の形成:)
化合物C(800mg、1.41mmol)およびEt4NBr(353mg、1.69mmol)をDMF/CH2Cl2(10ml、1:1、モレキュラーシーブスを含む)中に溶解し、0℃に冷却する。Br2(298mg、1.86mmol、CH2Cl2中)を、0℃で、CH2Cl2中の化合物D(808mg、1.69mmol)の別の溶液に滴下する。30分後、そのBr2/D溶液をシクロヘキセン(0.2ml)でクエンチし、そして直ちに(10分以内に)上記Cの溶液に添加する。この混合物を、室温で65時間反応させる。酢酸エチル(100ml)を添加し、この溶液を濾過し、そしてこの濾液を、飽和NaS2O3(50mlで2回)および飽和NaCl(50mlで2回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。得られるシロップを、次いでエーテル(50ml)に溶解し、そして攪拌しながらギ酸(10ml)を添加する。その反応の完了後(TLCにより確認されるように)、この溶液を、飽和NaHCO3(50mlで2回)および飽和NaCl(50mlで1回)で洗浄する。次いで、この有機層をNa2SO4で乾燥し、次いで乾燥状態までエバポレートする。化合物20を、次いでシリカゲルクロマトグラフィーにより精製する。
【0028】
(中間体Fの形成:)
化合物20(1g、1.02mmol)をMeOH/ジオキサン(10ml、20:1)中に溶解し、NaOMe(0.10mmol)を攪拌しながら添加する。この反応を、50℃で20時間進行させ、次いで2滴の酢酸を添加する。この溶液を乾燥状態までエバポレートし、エチルエーテル(25ml)に溶解し、飽和NaCl(50mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、乾燥状態までエバポレートする。最終生成物を、シリカゲルクロマトグラフィーにより精製する。この生成物(0.980mmol)およびBu2Sn(1.08mmol)をMeOH(15ml)中に懸濁し、加熱して2時間還流させる。その結果得られる清澄な溶液を、次いで乾燥状態までエバポレートし、ペンタン(10ml)中に溶解し、エバポレートし無色の泡状物を与える。その泡状物を1,2−ジメトキシエタン(DME、15ml)に溶解し、化合物E(1.96mmol)およびCsF(1.18mmol)を添加し、その反応物を室温で2時間攪拌する。2時間後、1M KH2PO4(50ml)およびKF(1g)を攪拌しながら添加し、続いて酢酸エチルで抽出する(25mlで2回)。その有機層を10%KF(50mlで2回)および飽和NaCl(50mlで2回)で洗浄し、Na2SO4で乾燥し、減圧下で乾燥状態までエバポレートする。化合物Fをシリカゲルクロマトグラフィーを経由して得る。
【0029】
(3の形成:)
化合物FをCH3OH(50ml)に溶解し、炭素担持Pd(10%)(質量で1/10)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、化合物3を与える。
【0030】
(実施例2)
(21の合成(図4))
(中間体Hの形成:)
G(15.0g、66.9mmol)およびBu2SnO(20.0g、80.3mmol)をMeOH(450ml)中に懸濁し、加熱して2時間還流させる。その結果得られる清澄な溶液を、次いで乾燥状態までエバポレートし、ペンタン中に溶解し、再度エバポレートし無色の泡状物を与える。その泡状物を1,2−ジメトキシエタン(DME、120ml)に溶解し、E(39.6g、100.3mmol)およびCsF(12.2g、80.3mmol)を添加し、その反応物を室温で2時間攪拌する。2時間後、1M KH2PO4(700ml)およびKF(25g)を攪拌しながら添加し、続いて酢酸エチルで抽出する(250mlで3回)。その有機層を10%KF(250mlで2回)および飽和NaCl(250mlで1回)で洗浄し、Na2SO4で乾燥し、減圧下で乾燥状態までエバポレートする。その化合物(19.3g、41.2mmol)をシリカゲルクロマトグラフィーにより精製し、直ちに結晶のDMAPとともにピリジン(210ml)に溶解する。この溶液を0℃に冷却し、塩化ベンゾイル(52.1g、370.7mmol)を、攪拌しながら滴下する。この溶液を、室温までゆっくり温め、この反応を室温で20分間進行させる。この溶液を乾燥状態までエバポレートし、酢酸エチル(500ml)に溶解し、0.1M HCl溶液(250mlで2回)、飽和NaHCO3溶液(250mlで2回)および飽和NaCl溶液(250mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。Hを、シリカゲルクロマトグラフィーによって得る。
【0031】
(中間体Iの形成:)
H(10.0g、12.82mmol)およびB(6.05g、25.64mmol)をCH2Cl2(75ml)に溶解し、−10℃で、攪拌しながら、0.15M CF3SO3H(CH2Cl2中)を滴下する。その橙色溶液が褐色に変わると、添加を停止する。酢酸エチル(500ml)を添加し、この溶液を飽和NaHCO3(250mlで4回)および飽和NaCl(250ml)で洗浄する。次いで、この有機層をNa2SO4で乾燥し、減圧下でエバポレートする。その化合物(7.96g、9.19mmol)を、次いでシリカゲルクロマトグラフィーにより精製し、そして次いでDMF(55ml)中に溶解する。TBDMS−Cl(1.52g、10.1mmol)およびイミダゾール(0.94g、13.8mmol)を次いで添加し、そしてその反応を室温で1時間進行させる。酢酸エチル(250ml)を添加し、この溶液を飽和NaHCO3(250mlで5回)および飽和NaCl(250mlで1回)で洗浄する。この有機層を次いでNa2SO4で乾燥し、シリカゲルクロマトグラフィーにより精製し、化合物Iを与える。
【0032】
(中間体Jの形成:)
化合物I(7.71g、7.87mmol)およびEt4NBr(2.00g、9.45mmol)をDMF/CH2Cl2(60ml、1:1、12gのモレキュラーシーブスを含む)中に溶解し、0℃に冷却する。CH2Cl2(11ml)中のBr2(1.90g、11.8mmol)を、0℃で、CH2Cl2中の化合物D(4.5g、9.45mmol)の別の溶液に滴下する。30分後、そのBr2/D溶液をシクロヘキセン(2.5ml)でクエンチし、そして直ちに(10分以内に)上記Iの溶液に添加する。この混合物を、室温で65時間反応させる。CH2Cl2(250ml)を添加し、この溶液を濾過し、そしてこの濾液を、飽和NaHCO3(50mlで2回)、0.5M HCl(250mlで2回)および飽和NaCl(250mlで2回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。この混合物を、室温でMeCN(85ml)に溶解し、そしてMeCN(17ml)中のEt3N(0.21ml)およびH2SiF6(1.3ml、35%)を添加し、2時間攪拌する。CH2Cl2(250ml)を添加し、この溶液を、飽和NaHCO3(250mlで3回)および飽和NaCl(250mlで1回)で洗浄する。次いで、この有機層をNa2SO4で乾燥し、次いで乾燥状態までエバポレートし、Jをシリカゲルクロマトグラフィーにより精製する。
【0033】
(中間体Kの形成:)
J(12.5g、9.75mmol)をピリジン(80ml)中に溶解し、塩化メタンスルホニル(3.35g、29.2mmol)を攪拌しながら5分にわたって滴下する。その反応を30分間進行させ、次いで酢酸エチル(500ml)を添加する。この溶液を1N HCl(250ml)で洗浄する。この有機層をNa2SO4で乾燥し、エバポレートする。その得られるシロップ(12.95g、9.52mmol)をDMF(40ml)中に溶解しNaN3(4.64g、74.4mmol)を添加する。この反応を、アルゴン雰囲気下、65℃で35時間進行させる。この溶液を酢酸エチル(500ml)で希釈し、H2O(300ml)および飽和NaCl(150ml)で洗浄する。この有機層をNa2SO3で乾燥し、そして乾燥状態までエバポレートする。その化合物をシリカゲルクロマトグラフィーにより精製する。その精製した生成物(12.2g、9.33mmol)を次いでMeOH/H2O(200ml/20ml)溶液中に懸濁し、そしてLiOH−H2O(5.1g、121.3mmol)を添加した。この反応を、65℃で20時間進行させる。酢酸エチル(500ml)を添加した。そして、この溶液を飽和NaCl(200ml)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。化合物Kを、シリカゲルクロマトグラフィーによって精製する。
【0034】
(21の形成:)
化合物K(8.45g、9.33mmol)をジオキサン/H2O(250ml/50ml)に溶解し、炭素担持Pd(10%)(3.4g)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、化合物21を与える。
【0035】
(実施例3)
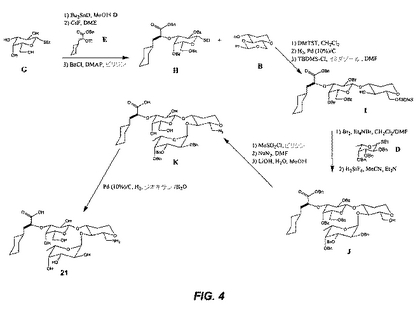
(15の合成(図5))
(中間体Lの形成:)
化合物20(10mmol)をCH2Cl2(30ml)中に溶解し、DMSO(20mmol)を添加し、そしてこの溶液を−60℃に冷却する。塩化オキサリル(11mmol)を20の攪拌された溶液にゆっくり添加する。その反応をN2雰囲気下で30分間進行させる。その溶液を0.1M HCl、飽和NaHCO3および飽和NaClで洗浄する。その有機層をNa2SO4で乾燥し、減圧下で乾燥状態までエバポレートする。その得られるシロップをtBuOH(20ml)中に入れ、2−メチル−2−ブテン(10ml)およびNaH2PO4(20mmol)を、攪拌しながら添加する。この反応を3時間進行させ、次いでエバポレートし、CH2Cl2中に溶解し、そして0.1M HCl、飽和NaHCO3および飽和NaClで洗浄する。その得られる化合物を、シリカゲルクロマトグラフィーにより精製し、化合物Lを与える。
【0036】
(中間体Nの形成:)
化合物L(10mmol)を、DMF(15ml)に溶解し、化合物M(10mmol)、HBTU(12mmol)およびEt3N(20mmol)を攪拌しながら添加する。この反応を、室温で24時間進行させる。酢酸エチル(100ml)を添加し、そしてこの溶液を0.1M HCl(100mlで1回)、飽和NaHCO3(100mlで1回)および飽和NaCl(100mlで1回)で洗浄する。その有機層をNa2SO4で乾燥し、乾燥状態までエバポレートする。化合物Nを、シリカゲルクロマトグラフィーによって単離する。
【0037】
(中間体Oの形成:)
化合物N(10mmol)をMeOH(35ml)中に溶解し、攪拌しながらNaOMe(1mmol)を添加する。その反応を室温で20時間進行させる。その溶液を乾燥状態までエバポレートし、エチルエーテル(50ml)に溶解し、飽和NaCl(50mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。その最終生成物をシリカゲルクロマトグラフィーにより精製する。その生成物(0.980mmol)およびBu2Sn(1.08mmol)をMeOH(15ml)中に懸濁し、加熱して2時間還流させる。その結果得られる清澄な溶液を、次いで乾燥状態までエバポレートし、ペンタン(10ml)中に溶解し、エバポレートし無色の泡状物を与える。その泡状物を1,2−ジメトキシエタン(DME、15ml)に溶解し、化合物E(1.96mmol)およびCsF(1.18mmol)を添加し、その反応を室温で2時間攪拌する。2時間後、1M KH2PO4(50ml)およびKF(1g)を攪拌しながら添加し、続いて酢酸エチルで抽出する(25mlで2回)。その有機層を10%KF(50mlで2回)および飽和NaCl(50mlで2回)で洗浄し、Na2SO4で乾燥し、そして減圧下で乾燥状態までエバポレートする。化合物Oをシリカゲルクロマトグラフィーによって得る。
【0038】
(15の形成:)
化合物O(9mmol)をMeOH(200ml)に溶解し、炭素担持Pd(10%)(3g)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、化合物15を与える。
【0039】
(実施例4)
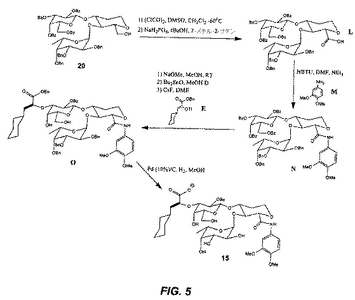
(21のアシル化(図6))
(21の酸塩化物との反応:)
化合物21(20mg、0.033mmol)を、1N NaOHを含むTHF/H2O(2ml、1:1)溶液中(pHは8〜10の間に調整される)に溶解し、0℃に冷却する。塩化シクロヘキシルカルボニル(0.049mmol)を、次いで攪拌しながら滴下する。この反応を0℃で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物1を逆相クロマトグラフィーにより精製する。
【0040】
(21のイソシアネートとの反応:)
化合物21(30mg、0.049mmol)を、0.5N NaOH水溶液(1ml)中に溶解し、0℃に冷却する。エチルイソシアネート(1.2当量)を次いで攪拌しながら滴下する。この反応を室温で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物2を逆相クロマトグラフィーにより精製する。
【0041】
(21のクロロオルトホルメートとの反応:)
化合物21(20mg、0.033mmol)を、NaOHを含むTHF/H2O(2ml、1:1)溶液中(pHは8〜10の間に調整される)に溶解し、0℃に冷却する。ベンジルクロロオルトホルメート(0.049mmol)を次いで攪拌しながら滴下する。この反応を0℃で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物11を逆相クロマトグラフィーにより精製する。
【0042】
(21の塩化スルホニルとの反応:)
化合物21(20mg、0.033mmol)を、飽和NaHCO3/トルエン(2ml、1:1)水溶液中に溶解し、0℃に冷却する。塩化p−トルエンスルホニル(0.049mmol)を次いで攪拌しながら滴下する。この反応を0℃で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物9を逆相クロマトグラフィーにより精製する。
【0043】
(実施例5)
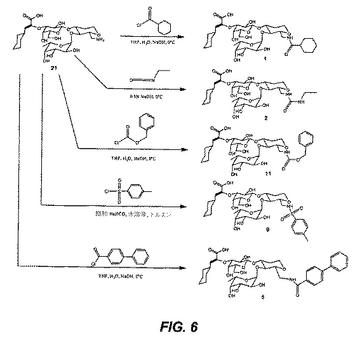
(化合物22の合成(図7))
化合物1(2.77mmol)を、化合物2(5.55mmol)を含むジエチルエーテル/ジクロロメタン(1ml、1:1混合物)の溶液に溶解する。0.1M TMSOTf(2.8ml)溶液を、次いで攪拌しながら添加する。30分後、この溶液を、炭酸水素ナトリウム(0.5g)の添加により中和し、濾過し、そして乾燥状態まで濃縮する。化合物3を、カラムクロマトグラフィー(トルエン/アセトン、4:1混合物)によって精製する。
【0044】
化合物3(1.96mmol)を、4A モレキュラーシーブス、TBABr(968mmol)およびCuBr2(529mmol)とともに、DMF/ジクロロメタン(50ml、1:5混合物)中で1時間攪拌する。化合物4(2.94mmol)をジクロロメタン(5ml)中に溶解し、上記溶液に滴下する。この溶液を24時間反応させ、濾過し、そして飽和炭酸水素ナトリウムおよび水(各々50ml)で洗浄する。その有機層を単離し、硫酸ナトリウムで乾燥し、乾燥状態までエバポレートする。化合物5を、カラムクロマトグラフィー(ヘキサン/酢酸エチル、4:1混合物)によって精製する。
【0045】
化合物22を、化合物5のメタノール中への溶解および続く1M ナトリウムメトキシド溶液の添加により得る。その溶液を3〜5時間反応させ、そして次いでAmberlite IR−120樹脂で中和し、濾過し、そして真空中で濃縮する。化合物22を、カラムクロマトグラフィー(ジクロロメタン/メタノール、20:1混合物)により精製する。
【0046】
(実施例6)
(化合物4、6、7、8、10、12、13、14、16、17、18、19および20の合成)
化合物21(実施例2;図4)から出発して、これらの化合物を文献(Helvetica Chimica Acta Vol.83,pp.2893〜2907,2000;Angew Chem.Int.Ed.Vol.40,No.19,pp.3644〜3647、2001)に記載される手順に従って合成する。
【0047】
(実施例7)
(化合物23、24、25、26、27および28の合成)
23という中間体の合成:化合物i(2.77mmol)を、化合物2(5.55mmol)を含むジエチルエーテル/ジクロロメタン(1ml、1:1混合物)の溶液に溶解する。0.1M TMSOTf(2.8ml)溶液を、次いで攪拌しながら滴下する。30分後、この溶液を、炭酸水素ナトリウム(0.5g)の添加により中和し、濾過し、そして乾燥状態まで濃縮する。3を、シリカゲルクロマトグラフィーにより精製し、化合物iiiを与える。
【0048】
化合物iii(1.96mmol)を、4A モレキュラーシーブス、TBABr(968mmol)およびCuBr2(529mmol)とともに、DMF/ジクロロメタン(50ml、1:5混合物)中で1時間攪拌する。化合物xv(2.94mmol)をジクロロメタン(5ml)中に溶解し、上記溶液に滴下する。この溶液を24時間反応させ、濾過し、そして飽和炭酸水素ナトリウムおよび水(各々50ml)で洗浄する。その有機層を単離し、硫酸ナトリウムで乾燥し、乾燥状態までエバポレートする。ivを、次いでシリカゲルクロマトグラフィーにより精製する。
【0049】
化合物ivを、MeOH中の0.1N NaOMeを用いて、脱O−アセチル化しvを与え、そして次いで還流条件下でMeOH中のジブチルスズオキシドで処理し、(上記溶媒のエバポレート後)viを与え、そのviをさらなる精製なしで次の工程に使用する。
【0050】
化合物vi(1mmol)を、DME(20ml)中に溶解し、次いで化合物vii(2.5mmol)およびCsF(1.4mmol)を添加する。その得られる反応混合物を室温で8時間攪拌し、酢酸エチルで希釈し、その有機層を水で洗浄する。この有機層を乾燥状態まで濃縮し、シリカゲルクロマトグラフィーにより精製し、化合物viiiを与える。
【0051】
化合物23の合成:化合物viiiを炭素中のパラジウムの存在下で水素化し、化合物23を与える。
【0052】
中間体xxの合成:N−アセチルグルコサミン(5,50g)から出発して、化合物xx(50%の全工程収率)を、刊行された手順(Bioorg.Med.Chem.Lett.11、pp.923〜925,2001;Carbohydr.Res.197,75、1990)に従って合成する。
【0053】
中間体xvの合成:化合物xv(15g)を、文献(Carbohydr.Res.201,15〜30、1990)に記載される手順に従って、L−フコースから合成する。
【0054】
中間体xixの合成:化合物xixを、記載されるように(WO9701569;Chem.Astr.,126 186312,1997)、商業的に入手可能なβ−D−ガラクトースペンタアセテートから調製する。
【0055】
中間体xxxivの合成:商業的に入手可能なN−アセチルノイラミン酸(xxx、10g)を、MeOH−H2O(60ml;9:1)中に懸濁し、そして、炭酸セシウムの水溶液を添加することにより、そのpHを8.1に調整する。その溶媒を除去し、そしてその残渣をエタノールを用い、次いでヘキサンを用いて繰返しエバポレートする。その物質をDMF(65ml)中に溶解させ、20分以内に臭化ベンジル(3.5ml)を添加する。その混合物を16時間攪拌し、その後ジクロロメタン(100ml)を添加し、水(50ml)で洗浄する。その溶媒を留去し、シリカゲルクロマトグラフィーにより精製し、xxxiを68%の収率で与える。
【0056】
ピリジン(50ml)中の化合物xxxi(7g)の溶液に、無水酢酸(48ml)を添加し、その反応混合物を室温で16時間攪拌する。溶媒を留去し、そしてその残渣(xxxii)を乾燥DMF(25ml)中に溶解する。その混合物に粉砕された炭酸アンモニウム(2g)を添加し、その混合物をセ氏28度で12時間攪拌する。この混合物を水(50ml)中の1N HClの氷冷溶液に添加し、そしてジクロロメタン(100ml)を添加する。溶媒抽出の後で、有機層を留去し、次いで真空下で24時間乾燥状態する。その残渣をシリカゲルクロマトグラフィーにより精製し、xxxiiiを71%の収率で与える。
【0057】
化合物xxxiiiを乾燥ジクロロメタン中に溶解し、2,6−ジ−tert−ブチルピリジン(5g)を添加する。その溶液をセ氏−20度に冷却し、トリフルオロメタンスルホン酸無水物(7g)を、10分間のうちに少しずつ添加する。その混合物を4時間攪拌し、ジクロロメタン(100ml)で希釈し、そしてリン酸水素カリウムの溶液(500ml)に添加する。その層を分離し、その有機層を乾燥し(硫酸ナトリウム)、溶媒を留去しxxxivを与え、このxxxivをさらなる精製なしで次の工程に使用する。
【0058】
中間体xxxvの合成:ジクロロメタン(100ml)中の化合物xx(10g)および化合物xv(15g)の混合物へ、モレキュラーシーブス(4A、8g)を添加する。室温で1時間攪拌し、その後臭化テトラエチルアンモニウム(5g)を添加する。ジクロロメタン(25ml)中の臭素(1g)の溶液を、1時間の間、滴下する。その反応混合物を3時間攪拌し続け、セライト床を通して濾過し、そして冷水、炭酸水素ナトリウムの飽和水溶液および水で逐次洗浄する。溶媒を留去し、シリカゲルクロマトグラフィーに供する。
【0059】
その生成物を、THF中のシアノ水素化ホウ素ナトリウムおよびエーテル中のHClで処理し、シリカゲルクロマトグラフィーの後、70%の全工程収率で化合物xxxvを与えた。
【0060】
中間体xxxviの合成:ジクロロメタン(80ml)中のxxxv(10g)およびxix(7g)の混合物へ、N−ヨードスクシンイミド(15g)およびモレキュラーシーブス(4A、8g)を添加する。その反応混合物を氷浴中に置く。その溶液を0〜5度で30分間攪拌し、そしてジクロロメタン(25ml)中のトリフルオロメタンスルホン酸(0.2ml)の溶液を、1時間の間、攪拌しながら滴下する。攪拌を2時間継続し、セライト床を通して濾過し、そして冷水、炭酸水素ナトリウムの冷たい飽和溶液および水で逐次洗浄する。溶媒を留去し、シリカゲルクロマトグラフィーにより精製し、68%の収率で化合物xxxviを与える。
【0061】
中間体xxxviiの合成:化合物xxxvi(8g)をMeOH(100ml)中の0.05N NaOEtで4時間処理し、IR120(水素型)樹脂での中和後、その反応混合物を濾別する。溶媒を留去し、96%収率で化合物xxxviiを与える。
【0062】
中間体xxxviiiの合成:化合物xxxvii(5g)をMeOH中のジブチルスズオキシド(1g)を用いて、還流下で4時間処理する。その溶媒を留去し、そしてトルエンとともに、数回、共エバポレートし(co−evaporated)、そして最終的にその残渣を高真空下で24時間乾燥する。
【0063】
その粗反応混合物をジメトキシエタン(DME、100ml)中に溶解し、そしてCsF(1.7g)および化合物xxxiv(2.5g)を添加する。その反応混合物を室温で8時間攪拌し、酢酸エチル(100ml)を添加する。有機層を水で洗浄し、有機溶媒を留去する。その生成物をシリカゲルクロマトグラフィーにより精製し、64%の収率でxxxviiiを与える。
【0064】
中間体xxxixの合成:化合物xxxviii(2g)を、MeOH中の0.01N NaOMe(100ml、1時間)を用いて、脱O−アセチル化し、粗反応混合物をIR120(水素型)樹脂で中和し、溶媒を留去する。
【0065】
上記反応からの生成物をジオキサン−水(1:1、50ml)中に溶解し、そして10%PD−Cを添加する。その反応混合物を、水素雰囲気下で22時間激しく攪拌し、セライト床を通して濾過し、その溶媒を留去する。得られるシロップのシリカゲルクロマトグラフィーは、77%の収率でxxxixを与える。
【0066】
化合物24の合成:MeOH−H2O中の化合物xxxixの溶液に、MeOH中のNaOMeの溶液を添加し、その反応混合物を室温で2時間攪拌する。IR120樹脂による中和および乾燥状態までのエバポレーションは化合物24を与える。
【0067】
xxxxの合成:化合物xxxixを、セ氏70度で4時間、エチレンジアミンで処理する(500mg)。溶媒を留去し、そのシロップ状残渣をシリカゲルクロマトグラフィーにより精製し、化合物xxxxを77%の収率で与える。
【0068】
化合物xxxxvの合成:ヨウ化3−ニトロベンジルを商業的に入手可能な8−アミノナフタレン−1,3,5−トリスルホン酸(xxxxxi)の水溶液(pH11)に、室温で攪拌しながら添加する。その溶液のpHを1に調整し、その溶媒のエバポレーション後に、生成物xxxxiiiをエタノールから沈殿させる。
【0069】
化合物xxxxiiiの白金触媒水素化は、化合物xxxxivを96%の収率で与える。
【0070】
リン酸塩緩衝液(pH7.1)中の化合物xxxxivの溶液に、商業的に入手可能なスクエア酸を添加し、その反応混合物を室温で4時間攪拌する。次いでそれを逆相HPLCにより精製しxxxxvを与える。
【0071】
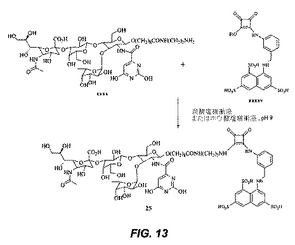
化合物25の合成:化合物xxxx(0.2g)を炭酸塩緩衝液(2ml、pH8.8)に溶解し、化合物xxxxv(0.4g)を添加する。その反応混合物を室温で24時間攪拌する。別のバッチ(0.2g)の化合物xxxxvを添加し、攪拌を20時間室温で継続する。溶媒を留去し、その混合物を逆相HPLCにより精製し、25を与える。
【0072】
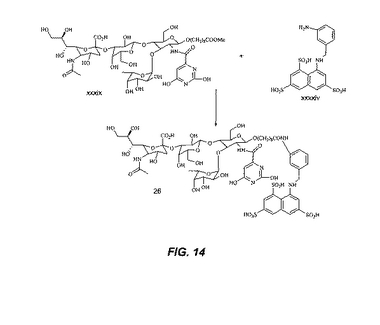
化合物26の合成:化合物xxxix(0.1g)を化合物xxxxivと反応させ、HPLCによる精製後、化合物26を与える。
【0073】
中間体xxxxxiiの合成:商業的に入手可能な化合物xxxxvi(4−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)安息香酸、1当量)およびKOAc(3当量)をTHF(25ml)中に入れる。その生成するスラリーにPdCl2および商業的に入手可能なp−ブロモニトロベンゼン(xxxxvii、1.2当量)を攪拌しながら添加し、そしてその混合物をセ氏80度までゆるやかに加熱する。6時間後、その反応混合物を乾燥状態までエバポレートし、ジクロロメタン(30ml)中に溶解し、蒸留水および炭酸水素ナトリウムの飽和溶液で洗浄する。その得られるビフェニル化合物xxxxviiiを直接次の工程に供する。
【0074】
化合物xxxxviii(1当量)、ジメチルアミノピリジン(触媒量、1個の結晶)およびEDCl(1.05当量)をDMFまたはTHF(20ml)中に溶解し、室温で10分間反応させる。商業的に入手可能な化合物xxxxix(8−アミノナフタレン−1,3,5−トリスルホン酸)をその反応混合物に攪拌しながら添加し、そしてその反応を、48時間、窒素下、室温で進行させる。その反応混合物を、次いで乾燥状態までエバポレートし、そして逆相クロマトグラフィーにより精製し、化合物xxxxxを与える。
【0075】
EtOAc中の化合物xxxxxの溶液に、PD−Cを添加し、そしてその反応混合物を水素雰囲気下で2時間攪拌する。その反応混合物をセライト床を通して濾過し、そして乾燥状態までエバポレートし、化合物xxxxxiを与える。
【0076】
リン酸塩緩衝液(pH7.1)中の化合物xxxxxiの溶液に、商業的に入手可能なスクエア酸を添加し、その反応混合物を室温で4時間攪拌する。次いで、それを逆相HPLCにより精製しxxxxxiiを与える。
【0077】
化合物27の合成:化合物xxxx(0.2g)を炭酸塩緩衝液(2ml、pH8.8)中に溶解し、化合物xxxxxii(0.4g)を添加する。その反応混合物を室温で24時間攪拌する。別のバッチ(0.2g)の化合物xxxxxiiを添加し、そして攪拌を20時間室温で継続する。溶媒を留去し、そしてその混合物を逆相HPLCにより精製し27を与える。
【0078】
化合物28の合成:化合物xxxixを化合物xxxxxiと反応させ、化合物28を与え、その化合物28をHPLCにより精製する。
【0079】
(合成に関する参考文献)
本出願内に含まれる特定の化合物の調製についての合成プロトコルは、以下の参考文献に例示される:Helvetica Chemica Acta Vol.83,pp.2893〜2907(2000)およびAngew.Chem.Int.Ed.Vol.40,No.19,pp.3644〜3647(2001)。
【0080】
(実施例8)
(アッセイ)
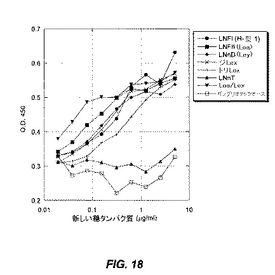
アルブミンに化学的に結合させた精製オリゴ糖(新しい糖タンパク質)を、プラスチック製マイクロタイターウェル中にコーティングする。BSAでブロックした後、そのウェルを次いで精製PA−IILレクチンとともにインキュベートし、精製PA−IILレクチンに結合させる。結合されたPA−IILレクチンを、1M H3PO4による発色を用いて、抗PA−IILウサギ抗血清、続いてHRP標識化抗ウサギIgおよびTMB試薬で検出する(図18)。構造および活性を表1に提示する。
【0081】
(表1.PA−IILレクチンへの結合についてスクリーニングされた固定化中性オリゴ糖の構造)
固定化中性オリゴ糖のスクリーニングの結果を図18に記載し、以下にまとめる。1型ラクト系列鎖(例えば、Lea構造体)は、ラクト系列2型鎖(例えば、Lex構造体)よりもより良好にPA−IILレクチンに結合する。Lea/Lexにおけるように、その1型構造体の伸長は、この系列において最も活性な化合物をもたらす。
【0082】
【表1】
(表2.PA−IILレクチンへの結合についてスクリーニングされた固定化酸性オリゴ糖の構造)
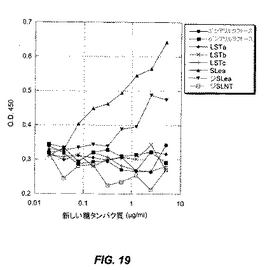
固定化酸性オリゴ糖のスクリーニングの結果を、図19に記載し、以下にまとめる。フコースを含むシアリル化オリゴ糖のみが、PA−IILレクチンに結合する。ジシアリルLea上の第2のシアリル基は、結合を阻害するように見える。
【0083】
【表2】
アルブミンに化学的に結合させた精製オリゴ糖(新しい糖タンパク質)を、プラスチック製マイクロタイターウェル中にコーティングする。BSAでブロックした後、そのウェルを次いで精製PA−IILレクチンとともにインキュベートし、精製PA−IILレクチンに結合させる。結合されたPA−IILレクチンを、1M H3PO4による発色を用いて、抗PA−IILウサギ抗血清、続いてHRP標識化抗ウサギIgおよびTMB試薬で検出する(図19)。構造および活性を表2に提示する。
【0084】
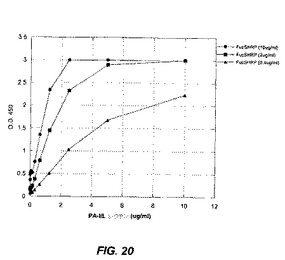
フコースのビオチン化ポリマー(Fuc−PAA−ビオチン、GlycoTech Corp.)を、HRP標識化ストレプトアビジン(KPL labs)と、4℃で終夜結合させる。PA−IILレクチンを、プラスチック製マイクロタイターウェル中に固定化し、HRP標識化フコシル化ポリマーと反応させる。結合をTMB基質で検出し、続いて1M H3PO4を用いる発色により検出する(図20)。そのアッセイのために選ばれた条件は、1mlあたり3μgのPA−IILのコーティングおよび1mlあたり2μgのフコシル化ポリマーを用いるインヒビターのインキュベーションである。
【0085】
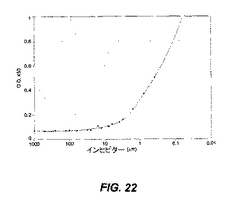
化合物23は、420nMのIC50でPA−IILを阻害したが(図22)、一方で天然の糖類インヒビターであるマンノースは、95μMのIC50でPA−IILレクチンを阻害し、負の対照糖類であるガラクトースは、阻害を示さなかった(データは示されていない)。
【0086】
本発明の特定の実施形態が、例示の目的のために本明細書中で記載されてきたが、本発明の精神および範囲から逸脱することなく、様々な改変がなされ得ることは、上記のことから理解される。
【図面の簡単な説明】
【0087】
【図1A】図1Aは、糖模倣化合物の構造を示す。
【図1B】図1Bは、糖模倣化合物の構造を示す。
【図1C】図1Cは、糖模倣化合物の構造を示す。
【図1D】図1Dは、糖模倣化合物の構造を示す。
【図1E】図1Eは、糖模倣化合物の構造を示す。
【図2A】図2Aは、さらなる糖模倣化合物の構造を示す。
【図2B】図2Bは、さらなる糖模倣化合物の構造を示す。
【図3】図3は、代表的な糖模倣化合物3の合成を図示する。
【図4】図4は、糖模倣化合物21の合成を図示する。
【図5】図5は、代表的な糖模倣化合物15の合成を図示する。
【図6】図6は、種々の代表的な糖模倣化合物を与える中間体21のアシル化を図示する。
【図7】図7は、化合物22の合成を図示する。
【図8A】図8Aは、化合物4、6、7、8、10および12の合成を図示する。
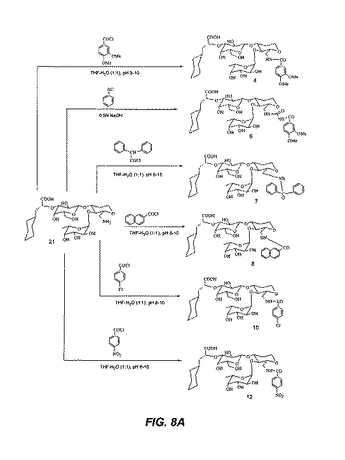
【図8B】図8Bは、化合物13、14、16、17、18および19の合成を図示する。
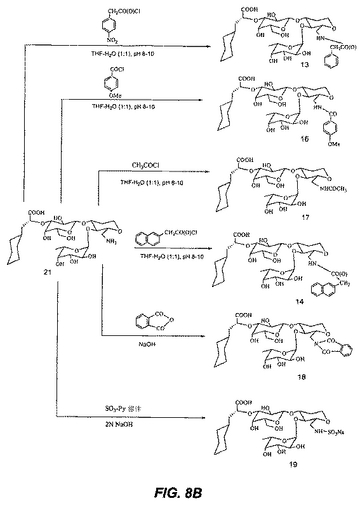
【図9】図9は、化合物22および23の合成を図示する。
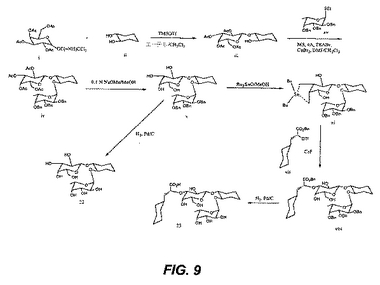
【図10】図10は、中間体の合成を図示する。
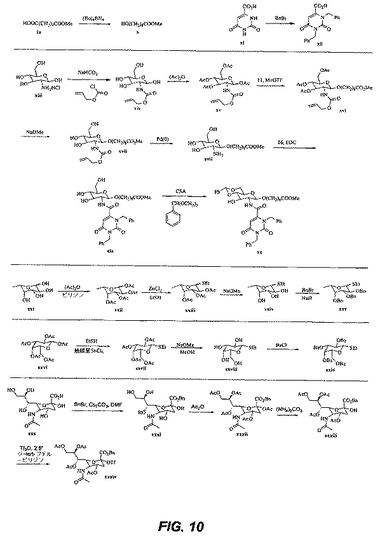
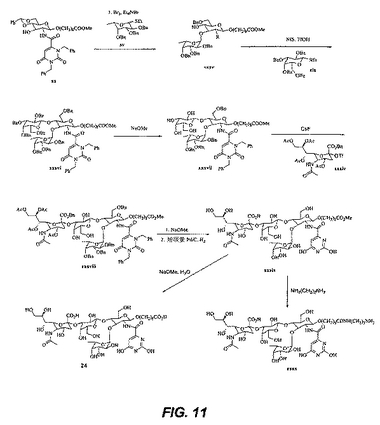
【図11】図11は、中間体および化合物24の合成を図示する。
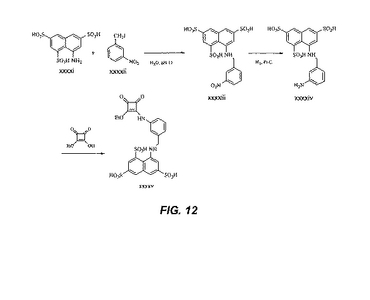
【図12】図12は、中間体xxxxivおよびxxxxvの合成を図示する。
【図13】図13は、化合物25の合成を図示する。
【図14】図14は、化合物26の合成を図示する。
【図15】図15は、中間体xxxxxiおよびxxxxxiiの合成を図示する。
【図16】図16は、化合物27の合成を図示する。
【図17】図17は、化合物28の合成を図示する。
【図18】図18は、PA−IILレクチンの、固定された中性糖鎖構造体への結合を図示する。
【図19】図19は、PA−IILレクチンの、固定された酸性糖鎖構造体への結合を図示する。
【図20】図20は、PA−IILレクチン阻害のIC50値についてのアッセイ条件の決定を図示する。
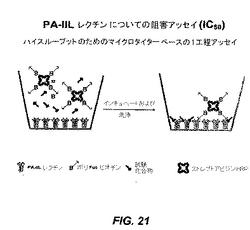
【図21】図21は、PA−IILレクチンの糖模倣インヒビターのIC50値を決定するために開発したアッセイの図式的表示を示す。
【図22】図22は、糖模倣化合物23による、PA−IILレクチンの阻害を図示する。
【技術分野】
【0001】
(発明の背景)
(発明の分野)
本発明は、一般的に、嚢胞性線維症を有する患者の肺の中でのPseudomonas aeruginosaを含む、Pseudomonas細菌による感染およびコロニー形成に関与する、温血動物(例えば、ヒト)における疾患の診断および治療のための化合物、組成物、および方法に関する。本発明は、より具体的には、Pseudomonas細菌に選択的に結合するための1つ以上の化合物の使用に関する。これらの化合物は、Pseudomonas細菌のコロニー形成の診断および/または治療的介入に対して有用であるか、またはPseudomonas細菌を標的とし、Pseudomonas細菌を有効に阻止もしくは殺傷するために薬剤へ結合され得る。
【背景技術】
【0002】
(関連技術の説明)
Pseudomonas感染は、種々の医学的状態において発生し、そして生命を脅かし得る。Pseudomonasは、日和見性細菌である。危険状態にある個体の例としては、嚢胞性線維症患者および熱傷患者が挙げられる。嚢胞性線維症は、Pseudomonas細菌の感染を含み得る医学的状態の代表的な例として以下に記載される。
【0003】
嚢胞性線維症(CF)は、白人集団の間で最も一般的な致死の遺伝疾患である。CFは、嚢胞性線維症膜貫通調節タンパク質(cystic fibrosis transmembrane conductance regulator;CFTR)をコードする遺伝子中の変異によって引き起こされ、塩化物チャネルとして作用する。イオン移動を変化させるCFTRの遺伝的変異もまた、CFTRのN−グルコシル化、および他の細胞表面分子に影響を及ぼす。患者の全ての外分泌腺は、影響を受ける;しかし、肺は、病的状態および死亡の主要な部分である。グルコシル化における一般的な変化は、ルイスのフコシル化における増加およびシアル化における減少を生じる。CF患者由来の唾液のおよび呼吸器のムチン(musin)はまた、シアル化および硫酸化されたルイスx/a構造を含む、より高レベルのルイス型オリゴ糖を含む。
【発明の開示】
【発明が解決しようとする課題】
【0004】
CF患者における病的状態および死亡の主な原因は、この細菌、Pseudomonas aeruginosaによる慢性の肺コロニー形成であり、この細菌は、肺の破壊および死につながる強い好中球性炎症応答の著しい肺感染を生じる。P.aeruginosaによるコロニー形成は、この細菌上の線毛レクチンおよび鞭毛レクチンの、肺細胞表面上のルイス型糖鎖構造への結合によって開始される。PA−ILおよびPA−IILとして知られるこれらのレクチンは、これらのオリゴ糖構造と高い親和性で結合し、そして細菌のコロニー形成の最初の工程を妨げる潜在的分子標的を表す。この細菌によって十分にはコロニー形成されない患者は、良好な長期間の予後を維持する。Pseudomonas細菌による個体中でのコロニー形成の予防についての、当該分野での現在のアプローチにおける難しさのために、化合物、組成物および方法を改良する必要がある。
【課題を解決するための手段】
【0005】
(発明の簡単な要旨)
簡単に述べると、本発明は、Pseudomonas細菌の検出のためにPseudomonas細菌上に発現するレクチンを利用するための化合物、組成物および方法、ならびにPseudomonas細菌に関与する疾患(ヒトの疾患を含む)の診断および治療を提供する。例えば、P.aeruginosa上のレクチンに対して高い親和性の結合性を有するルイス構造の糖模倣物は、CF患者に対して有益な治療効果を有する。さらに、これらの糖模倣物は、例えば、効力を増大させ、そして投与量を低下させるために強い抗生物質と結合体化され得、それによってこれらの強力な抗生物質の周知の有害な副作用が回避され得る。これらの結合部分が、この細菌のコロニー形成および病原性に対して重大であることを考慮すると、この結合体治療に対して抵抗性となる、この標的における変異は、この細菌の非病原性形態を生じるに違いない。
【0006】
本発明の1つの実施形態は、温血動物におけるPseudomonas細菌を阻害する方法を提供し、この方法は、図1または図2に記載の化合物を含む化合物を、この細菌を阻害するための有効な量で、この動物へ投与する工程を包含する。
【0007】
別の実施形態では、本発明は、図1または図2に記載の化合物と結合した治療薬剤を含む結合体を提供する。
【0008】
別の実施形態では、本発明は、Pseudomonas細菌を検出する方法を提供し、この方法は、図1または図2に記載の化合物を含む化合物に結合された診断薬剤とサンプルを、このサンプル中に存在する場合、この化合物がこの細菌と結合するための十分な条件下で、接触させる工程;およびこのサンプル中に存在するこの薬剤を検出する工程であって、ここで、このサンプル中のこの薬剤の存在は、Pseudomonas細菌の存在を示す、工程を包含する。
【0009】
別の実施形態では、本発明は、固体支持体上にPseudomonas細菌を固定する方法を提供し、この方法は、結合するための十分な条件下で、Pseudomonas細菌含有サンプルと、固体支持体上に固定された図1または図2に記載の化合物を含む化合物とを接触させる工程;およびこの固体支持体からこのサンプルを分離する工程を包含する。
【0010】
他の実施形態では、本明細書に記載の化合物および結合体は、Pseudomonas細菌の阻害のための薬の調製において使用され得る。
【0011】
本発明のこれらの局面および他の局面は、以下の詳細な説明および添付された図面を参照すれば明白になる。本明細書で開示された全ての参考文献は、あたかもそれぞれが個々に援用されたかのように、その全体が参考として援用される。
【発明を実施するための最良の形態】
【0012】
(発明の詳細な説明)
上述で記載のように、本発明は、P.aeruginosaと結合して、疾患の診断および治療において使用され得る、化合物および組成物を提供する。
【0013】
(糖模倣化合物)
本明細書で使用される場合、用語「糖模倣化合物」とは、P.aeruginosaと特異的に結合する分子をいう。本発明によって包含される糖模倣化合物の構造は、図1および図2において示され、そしてまた、この化合物が、末端のシクロヘキシル乳酸部分として示されるシアル酸についての模倣物を含まないことを除いては、本明細書で開示された化合物を包含する。これは、例えば、ある反応スキームにおいて中間体Eの添加を含む工程を除去することによって達成される。本発明において有用な全ての化合物(またはその結合体)は、生理学的に受容可能な、それらの塩を含む。
【0014】
ある実施形態については、薬物のような診断薬剤もしくは治療薬剤を糖模倣化合物と結合させて結合体(その結合は共有結合である)を形成させることもまた、あるいは、有益であり得る。本明細書で使用される場合、用語「治療薬剤」とは、疾患または他の非所望状態を予防または処置するか、あるいは治療の成功を促進させるための温血動物(例えば、ヒトのような哺乳動物)への投与が意図される、任意の生物活性薬剤をいう。治療薬剤としては、抗生物質、ホルモン、成長因子、タンパク質、ペプチド、遺伝子、非ウイルスベクターおよび他の化合物が挙げられる。
【0015】
(糖模倣化合物の処方物)
本明細書で記載の糖模倣化合物は、薬学的組成物内に存在し得る。薬学的組成物は、1つ以上の薬学的または生理学的に受容可能なキャリア、希釈剤または賦形剤と組み合わされた1つ以上の糖模倣化合物を含む。そのような組成物は、緩衝液(例えば、中性緩衝生理食塩水またはリン酸緩衝生理食塩水)、糖質(例えば、グルコース、マンノース、スクロース、またはデキストラン)、マンニトール、タンパク質、ポリペプチド、またはグリシンのようなアミノ酸、抗酸化剤、キレート剤(例えば、EDTAまたはグルタチオン)、アジュバント(例えば、水酸化アルミニウム)および/または保存剤を含み得る。なおさらなる他の実施形態では、本発明の組成物は、凍結乾燥物(lyophilizate)として処方され得る。本発明の組成物は、例えば、エアロゾル投与、局所投与、経口投与、鼻腔投与、静脈内投与、頭蓋内投与、腹腔内投与、皮下投与、または筋肉投与を含む、任意の適切な投与様式について処方され得る。
【0016】
薬学的組成物はまた、あるいは、糖模倣化合物と結合され得るか、またはこの組成物内で遊離であり得る薬物のような、1つ以上の活性薬剤を含み得る。糖模倣化合物への薬剤の結合は、共有結合または非共有結合であり得る。
【0017】
本明細書で記載の組成物は、持続放出処方物(すなわち、投与後の調節薬剤の、ゆっくりとした放出をもたらすカプセルまたはスポンジのような処方物)の一部として投与され得る。そのような処方物は、一般的に、周知の技術を用いて調製され得、そして例えば、経口移植、直腸移植、または皮下移植、または所望の標的部位での移植によって投与され得る。そのような処方物内での使用のためのキャリアは、生体適合性であり、そしてまた生物分解性であり得;好ましくは、この処方物は、比較的一定のレベルでの調節薬剤の放出を提供する。維持放出処方物内に含まれる糖模倣化合物の量は、移植部位、放出速度、および放出の期待された持続時間、ならびに治療または予防される状態の性質に依存する。
【0018】
糖模倣化合物は、一般的に、治療的有効量で、薬学的組成物内に存在する。治療的有効量は、Pseudomonas感染に関連した状態の測定または観察された応答のような、患者の認識可能な利益をもたらす量である。
【0019】
(糖模倣化合物の使用方法)
一般的に、本明細書に記載の糖模倣化合物は、Pseudomonas(例えば、P.aeruginosa)細菌による感染に関与する疾患(例えばヒトの疾患)をもたらす診断結果および/または治療結果を達成するために使用され得る。そのような診断結果および/または治療結果は、動物、好ましくは、ヒトのような哺乳動物のインビトロおよび/またはインビボにおいて達成され得る。ただし、Pseudomonas(例えば、P.aeruginosa)は、最終的に、認識可能な診断結果または治療結果を達成するために十分な量および時間において、糖模倣化合物と接触される。本発明の背景では、治療結果は、例えば、肺感染の予防と関連する。いくつかの状態では、治療結果は、Pseudomonasの阻害(例えば、P.aeruginosa)(例えば、このような細菌の増殖を阻止するかもしくはこの細菌を殺傷する、またはこの細菌によるコロニー形成を防ぐことを包含する)と関連する。本明細書で使用される場合、治療または治療学的結果は、処置または予防を含む。
【0020】
本発明の糖模倣化合物は、処置または予防されるべき疾患に対する適切な様式において投与され得る。適切な投与量ならびに投与の適切な持続時間および頻度は、患者の状態、患者の疾患の型および重症度ならびに投与方法のような要因によって決定され得る。一般的に、適切な投与量および処置レジメンは、処置および/または予防の利益を提供するために十分な量で、この調節薬剤を提供する。本発明の特に好ましい実施形態では、糖模倣化合物は、0.001mg/kg体重〜1000mg/kg体重(より代表的には0.01mg/kg〜1000mg/kg)の範囲の投与量で、毎日1回または複数回の投与レジメンで投与され得る。適切な投与量は、一般的に、実験モデルおよび/または臨床試験を用いて決定され得る。一般的に、効果的な治療を提供するために十分である最小の投与量の使用が好ましい。患者は、一般的に、当業者が精通している、治療または予防される状態に対して適切なアッセイを用いて、治療的有効性についてモニターされ得る。
【0021】
糖模倣化合物はまた、Pseudomonas細菌(例えば、P.aeruginosa)へ、物質を標的化するために使用され得る。そのような物質としては、治療薬剤および診断薬剤が挙げられる。治療薬剤は、分子、ウイルス、ウイルス成分、細胞、細胞成分、あるいは、標的細胞の性質を変更することが立証され得、その結果障害を治療もしくは予防するため、または患者の生理機能を制御するための利益を提供し得る、任意の他の物質であり得る。治療薬剤はまた、インビボで生物学的活性を有する薬剤を生じるプロドラッグであり得る。治療薬剤であり得る分子は、例えば、ポリペプチド、アミノ酸、核酸、ポリヌクレオチド、ステロイド、多糖または無機化合物であり得る。そのような分子は、任意の種々の方法において機能し得、それらとしては、例えば、酵素、酵素インヒビター、ホルモン、レセプター、アンチセンスオリゴヌクレオチド、触媒性ポリヌクレオチド、抗ウイルス剤、抗腫瘍剤、抗細菌剤、免疫調節剤、および細胞障害性薬剤(例えば、ヨウ素、臭素、鉛、レニウム、ホミウム(homium)、パラジウムまたは銅のような放射性核種)が挙げられる。診断薬剤としては、金属および放射性薬剤のような画像化剤(例えば、ガリウム、テクネチウム、インジウム、ストロンチウム、ヨウ素、バリウム、臭素およびリン含有化合物)、造影剤、色素(例えば、蛍光色素または発蛍光団)ならびに比色定量的反応または蛍光定量的反応を触媒する酵素が挙げられる。一般的に、治療薬剤および診断薬剤は、上述で記載の技術のような種々の技術を用いて、糖模倣化合物に結合され得る。標的化の目的のためには、糖模倣化合物は、本明細書で記載の通りに患者に投与され得る。
【0022】
糖模倣化合物はまた、(例えば、種々の周知の細胞培養方法および細胞分離方法で)インビトロで使用され得る。例えば、糖模倣化合物は、培養中でのスクリーニング、アッセイまたは増殖のためにPseudomonas細菌を固定する際に使用するために固体支持体上に固定され得る。(例えば、組織培養プレートまたは他の細胞培養支持体の内部表面と連結される)。そのような連結は、上述に記載の方法のような任意の適切な技術、および他の標準的技術によって行われ得る。また、糖模倣化合物の使用は、細胞の同定およびインビトロでの選別を容易にし、そのような細菌細胞の選択を許容し得る。好ましくは、そのような方法における使用のための糖模倣化合物は、検出可能なマーカーである診断薬剤に連結される。適切なマーカーは、当該分野で周知であり、それらとして、放射性核種、発光基、蛍光基、酵素、色素、免疫グロブリンの定常ドメイン、およびビオチンが挙げられる。1つの好ましい実施形態では、フルオレセインのような蛍光マーカーに連結された糖模倣化合物は、細胞と接触され、次いで、蛍光活性化細胞選別器(FACS)によって分析される。
【0023】
そのようなインビトロでの方法は、一般的に、サンプル(例えば、生物学的調製物)と、糖模倣化合物の任意の1個とを接触させる工程、およびこのサンプル中のこの化合物を検出する工程を包含する。所望される場合、1回以上の洗浄工程が方法に加えられ得る。例えば、この化合物の検出より先にサンプルと糖模倣化合物とを接触させ、その後、このサンプルは、洗浄され得る(すなわち、流体と接触させ、次いでこの流体を除去し、結合しない糖模倣化合物を除去する)。あるいは、または加えて、洗浄工程は、この検出プロセスの間にさらに加えられ得る。例えば、糖模倣化合物が、検出可能である物質と結合し得るマーカー(診断薬剤)を所有する場合、この検出の前に、このサンプルと検出可能な物質とを接触させた後に、このサンプルを洗浄することが所望され得る。本明細書で使用される場合、語句「このサンプル中の化合物(または薬剤)の検出」は、この化合物(もしくは薬剤)がこのサンプルと結合したままであるこの化合物(もしくは薬剤)を検出すること、またはこの化合物(もしくは薬剤)がこのサンプルから分離された後に、このサンプルと結合した化合物(もしくは薬剤)を検出することを包含する。
【0024】
以下の実施例は、例証の目的で提示されるのであり、限定の目的で提示されるのではない。
【実施例】
【0025】
(実施例1)
(3の合成(図3))
(中間体Cの形成:)
化合物A(5.00g、12.74mmol)およびB(4.50g、19.11mmol)ならびにNIS(3.58g、15.93mmol)をCH2Cl2(50ml)中に溶解し、0℃に冷却する。トリフルオロメタンスルホン酸の溶液(CH2Cl2中の0.15M)を、攪拌しながら滴下する。その溶液が橙色から暗褐色に色を変えたら、TMS−OHの添加を止める。この溶液を次いで飽和NaHCO3(30ml)で洗浄し、そしてその有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。得られるシロップをシリカゲルクロマトグラフィー(ヘキサン/エーテル、1:1)により精製し、次の工程に使用する。
【0026】
上記で得られる化合物をTHF(40ml)中に溶解し、炭素担持Pd(10%)(質量で1/10)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、4位および6位がOHの化合物を与える。この化合物を、次いでピリジン(25ml)中に溶解し、0℃に冷却する。Ph3CCl(1.2当量)を滴下し、この反応を室温で6時間進行させる。酢酸エチル(50ml)を次いで添加し、この溶液を0.1N HCl(50mlで2回)、飽和NaHCO3(50mlで1回)および飽和NaCl(50mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、乾燥状態までエバポレートする。中間体Cを、シリカゲルクロマトグラフィーにより得る。
【0027】
(20の形成:)
化合物C(800mg、1.41mmol)およびEt4NBr(353mg、1.69mmol)をDMF/CH2Cl2(10ml、1:1、モレキュラーシーブスを含む)中に溶解し、0℃に冷却する。Br2(298mg、1.86mmol、CH2Cl2中)を、0℃で、CH2Cl2中の化合物D(808mg、1.69mmol)の別の溶液に滴下する。30分後、そのBr2/D溶液をシクロヘキセン(0.2ml)でクエンチし、そして直ちに(10分以内に)上記Cの溶液に添加する。この混合物を、室温で65時間反応させる。酢酸エチル(100ml)を添加し、この溶液を濾過し、そしてこの濾液を、飽和NaS2O3(50mlで2回)および飽和NaCl(50mlで2回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。得られるシロップを、次いでエーテル(50ml)に溶解し、そして攪拌しながらギ酸(10ml)を添加する。その反応の完了後(TLCにより確認されるように)、この溶液を、飽和NaHCO3(50mlで2回)および飽和NaCl(50mlで1回)で洗浄する。次いで、この有機層をNa2SO4で乾燥し、次いで乾燥状態までエバポレートする。化合物20を、次いでシリカゲルクロマトグラフィーにより精製する。
【0028】
(中間体Fの形成:)
化合物20(1g、1.02mmol)をMeOH/ジオキサン(10ml、20:1)中に溶解し、NaOMe(0.10mmol)を攪拌しながら添加する。この反応を、50℃で20時間進行させ、次いで2滴の酢酸を添加する。この溶液を乾燥状態までエバポレートし、エチルエーテル(25ml)に溶解し、飽和NaCl(50mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、乾燥状態までエバポレートする。最終生成物を、シリカゲルクロマトグラフィーにより精製する。この生成物(0.980mmol)およびBu2Sn(1.08mmol)をMeOH(15ml)中に懸濁し、加熱して2時間還流させる。その結果得られる清澄な溶液を、次いで乾燥状態までエバポレートし、ペンタン(10ml)中に溶解し、エバポレートし無色の泡状物を与える。その泡状物を1,2−ジメトキシエタン(DME、15ml)に溶解し、化合物E(1.96mmol)およびCsF(1.18mmol)を添加し、その反応物を室温で2時間攪拌する。2時間後、1M KH2PO4(50ml)およびKF(1g)を攪拌しながら添加し、続いて酢酸エチルで抽出する(25mlで2回)。その有機層を10%KF(50mlで2回)および飽和NaCl(50mlで2回)で洗浄し、Na2SO4で乾燥し、減圧下で乾燥状態までエバポレートする。化合物Fをシリカゲルクロマトグラフィーを経由して得る。
【0029】
(3の形成:)
化合物FをCH3OH(50ml)に溶解し、炭素担持Pd(10%)(質量で1/10)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、化合物3を与える。
【0030】
(実施例2)
(21の合成(図4))
(中間体Hの形成:)
G(15.0g、66.9mmol)およびBu2SnO(20.0g、80.3mmol)をMeOH(450ml)中に懸濁し、加熱して2時間還流させる。その結果得られる清澄な溶液を、次いで乾燥状態までエバポレートし、ペンタン中に溶解し、再度エバポレートし無色の泡状物を与える。その泡状物を1,2−ジメトキシエタン(DME、120ml)に溶解し、E(39.6g、100.3mmol)およびCsF(12.2g、80.3mmol)を添加し、その反応物を室温で2時間攪拌する。2時間後、1M KH2PO4(700ml)およびKF(25g)を攪拌しながら添加し、続いて酢酸エチルで抽出する(250mlで3回)。その有機層を10%KF(250mlで2回)および飽和NaCl(250mlで1回)で洗浄し、Na2SO4で乾燥し、減圧下で乾燥状態までエバポレートする。その化合物(19.3g、41.2mmol)をシリカゲルクロマトグラフィーにより精製し、直ちに結晶のDMAPとともにピリジン(210ml)に溶解する。この溶液を0℃に冷却し、塩化ベンゾイル(52.1g、370.7mmol)を、攪拌しながら滴下する。この溶液を、室温までゆっくり温め、この反応を室温で20分間進行させる。この溶液を乾燥状態までエバポレートし、酢酸エチル(500ml)に溶解し、0.1M HCl溶液(250mlで2回)、飽和NaHCO3溶液(250mlで2回)および飽和NaCl溶液(250mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。Hを、シリカゲルクロマトグラフィーによって得る。
【0031】
(中間体Iの形成:)
H(10.0g、12.82mmol)およびB(6.05g、25.64mmol)をCH2Cl2(75ml)に溶解し、−10℃で、攪拌しながら、0.15M CF3SO3H(CH2Cl2中)を滴下する。その橙色溶液が褐色に変わると、添加を停止する。酢酸エチル(500ml)を添加し、この溶液を飽和NaHCO3(250mlで4回)および飽和NaCl(250ml)で洗浄する。次いで、この有機層をNa2SO4で乾燥し、減圧下でエバポレートする。その化合物(7.96g、9.19mmol)を、次いでシリカゲルクロマトグラフィーにより精製し、そして次いでDMF(55ml)中に溶解する。TBDMS−Cl(1.52g、10.1mmol)およびイミダゾール(0.94g、13.8mmol)を次いで添加し、そしてその反応を室温で1時間進行させる。酢酸エチル(250ml)を添加し、この溶液を飽和NaHCO3(250mlで5回)および飽和NaCl(250mlで1回)で洗浄する。この有機層を次いでNa2SO4で乾燥し、シリカゲルクロマトグラフィーにより精製し、化合物Iを与える。
【0032】
(中間体Jの形成:)
化合物I(7.71g、7.87mmol)およびEt4NBr(2.00g、9.45mmol)をDMF/CH2Cl2(60ml、1:1、12gのモレキュラーシーブスを含む)中に溶解し、0℃に冷却する。CH2Cl2(11ml)中のBr2(1.90g、11.8mmol)を、0℃で、CH2Cl2中の化合物D(4.5g、9.45mmol)の別の溶液に滴下する。30分後、そのBr2/D溶液をシクロヘキセン(2.5ml)でクエンチし、そして直ちに(10分以内に)上記Iの溶液に添加する。この混合物を、室温で65時間反応させる。CH2Cl2(250ml)を添加し、この溶液を濾過し、そしてこの濾液を、飽和NaHCO3(50mlで2回)、0.5M HCl(250mlで2回)および飽和NaCl(250mlで2回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。この混合物を、室温でMeCN(85ml)に溶解し、そしてMeCN(17ml)中のEt3N(0.21ml)およびH2SiF6(1.3ml、35%)を添加し、2時間攪拌する。CH2Cl2(250ml)を添加し、この溶液を、飽和NaHCO3(250mlで3回)および飽和NaCl(250mlで1回)で洗浄する。次いで、この有機層をNa2SO4で乾燥し、次いで乾燥状態までエバポレートし、Jをシリカゲルクロマトグラフィーにより精製する。
【0033】
(中間体Kの形成:)
J(12.5g、9.75mmol)をピリジン(80ml)中に溶解し、塩化メタンスルホニル(3.35g、29.2mmol)を攪拌しながら5分にわたって滴下する。その反応を30分間進行させ、次いで酢酸エチル(500ml)を添加する。この溶液を1N HCl(250ml)で洗浄する。この有機層をNa2SO4で乾燥し、エバポレートする。その得られるシロップ(12.95g、9.52mmol)をDMF(40ml)中に溶解しNaN3(4.64g、74.4mmol)を添加する。この反応を、アルゴン雰囲気下、65℃で35時間進行させる。この溶液を酢酸エチル(500ml)で希釈し、H2O(300ml)および飽和NaCl(150ml)で洗浄する。この有機層をNa2SO3で乾燥し、そして乾燥状態までエバポレートする。その化合物をシリカゲルクロマトグラフィーにより精製する。その精製した生成物(12.2g、9.33mmol)を次いでMeOH/H2O(200ml/20ml)溶液中に懸濁し、そしてLiOH−H2O(5.1g、121.3mmol)を添加した。この反応を、65℃で20時間進行させる。酢酸エチル(500ml)を添加した。そして、この溶液を飽和NaCl(200ml)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。化合物Kを、シリカゲルクロマトグラフィーによって精製する。
【0034】
(21の形成:)
化合物K(8.45g、9.33mmol)をジオキサン/H2O(250ml/50ml)に溶解し、炭素担持Pd(10%)(3.4g)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、化合物21を与える。
【0035】
(実施例3)
(15の合成(図5))
(中間体Lの形成:)
化合物20(10mmol)をCH2Cl2(30ml)中に溶解し、DMSO(20mmol)を添加し、そしてこの溶液を−60℃に冷却する。塩化オキサリル(11mmol)を20の攪拌された溶液にゆっくり添加する。その反応をN2雰囲気下で30分間進行させる。その溶液を0.1M HCl、飽和NaHCO3および飽和NaClで洗浄する。その有機層をNa2SO4で乾燥し、減圧下で乾燥状態までエバポレートする。その得られるシロップをtBuOH(20ml)中に入れ、2−メチル−2−ブテン(10ml)およびNaH2PO4(20mmol)を、攪拌しながら添加する。この反応を3時間進行させ、次いでエバポレートし、CH2Cl2中に溶解し、そして0.1M HCl、飽和NaHCO3および飽和NaClで洗浄する。その得られる化合物を、シリカゲルクロマトグラフィーにより精製し、化合物Lを与える。
【0036】
(中間体Nの形成:)
化合物L(10mmol)を、DMF(15ml)に溶解し、化合物M(10mmol)、HBTU(12mmol)およびEt3N(20mmol)を攪拌しながら添加する。この反応を、室温で24時間進行させる。酢酸エチル(100ml)を添加し、そしてこの溶液を0.1M HCl(100mlで1回)、飽和NaHCO3(100mlで1回)および飽和NaCl(100mlで1回)で洗浄する。その有機層をNa2SO4で乾燥し、乾燥状態までエバポレートする。化合物Nを、シリカゲルクロマトグラフィーによって単離する。
【0037】
(中間体Oの形成:)
化合物N(10mmol)をMeOH(35ml)中に溶解し、攪拌しながらNaOMe(1mmol)を添加する。その反応を室温で20時間進行させる。その溶液を乾燥状態までエバポレートし、エチルエーテル(50ml)に溶解し、飽和NaCl(50mlで1回)で洗浄する。この有機層をNa2SO4で乾燥し、そして乾燥状態までエバポレートする。その最終生成物をシリカゲルクロマトグラフィーにより精製する。その生成物(0.980mmol)およびBu2Sn(1.08mmol)をMeOH(15ml)中に懸濁し、加熱して2時間還流させる。その結果得られる清澄な溶液を、次いで乾燥状態までエバポレートし、ペンタン(10ml)中に溶解し、エバポレートし無色の泡状物を与える。その泡状物を1,2−ジメトキシエタン(DME、15ml)に溶解し、化合物E(1.96mmol)およびCsF(1.18mmol)を添加し、その反応を室温で2時間攪拌する。2時間後、1M KH2PO4(50ml)およびKF(1g)を攪拌しながら添加し、続いて酢酸エチルで抽出する(25mlで2回)。その有機層を10%KF(50mlで2回)および飽和NaCl(50mlで2回)で洗浄し、Na2SO4で乾燥し、そして減圧下で乾燥状態までエバポレートする。化合物Oをシリカゲルクロマトグラフィーによって得る。
【0038】
(15の形成:)
化合物O(9mmol)をMeOH(200ml)に溶解し、炭素担持Pd(10%)(3g)を添加する。この溶液を脱気し、H2の雰囲気を発生させる。この反応を、出発物質の消失がTLCで確認されるまで、室温で進行させる。この溶液をセライト床を通して濾過し、そしてその濾液を真空中で濃縮し、化合物15を与える。
【0039】
(実施例4)
(21のアシル化(図6))
(21の酸塩化物との反応:)
化合物21(20mg、0.033mmol)を、1N NaOHを含むTHF/H2O(2ml、1:1)溶液中(pHは8〜10の間に調整される)に溶解し、0℃に冷却する。塩化シクロヘキシルカルボニル(0.049mmol)を、次いで攪拌しながら滴下する。この反応を0℃で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物1を逆相クロマトグラフィーにより精製する。
【0040】
(21のイソシアネートとの反応:)
化合物21(30mg、0.049mmol)を、0.5N NaOH水溶液(1ml)中に溶解し、0℃に冷却する。エチルイソシアネート(1.2当量)を次いで攪拌しながら滴下する。この反応を室温で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物2を逆相クロマトグラフィーにより精製する。
【0041】
(21のクロロオルトホルメートとの反応:)
化合物21(20mg、0.033mmol)を、NaOHを含むTHF/H2O(2ml、1:1)溶液中(pHは8〜10の間に調整される)に溶解し、0℃に冷却する。ベンジルクロロオルトホルメート(0.049mmol)を次いで攪拌しながら滴下する。この反応を0℃で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物11を逆相クロマトグラフィーにより精製する。
【0042】
(21の塩化スルホニルとの反応:)
化合物21(20mg、0.033mmol)を、飽和NaHCO3/トルエン(2ml、1:1)水溶液中に溶解し、0℃に冷却する。塩化p−トルエンスルホニル(0.049mmol)を次いで攪拌しながら滴下する。この反応を0℃で3時間継続させる。この溶液を氷でクエンチし、その溶液を乾燥状態までエバポレートする。化合物9を逆相クロマトグラフィーにより精製する。
【0043】
(実施例5)
(化合物22の合成(図7))
化合物1(2.77mmol)を、化合物2(5.55mmol)を含むジエチルエーテル/ジクロロメタン(1ml、1:1混合物)の溶液に溶解する。0.1M TMSOTf(2.8ml)溶液を、次いで攪拌しながら添加する。30分後、この溶液を、炭酸水素ナトリウム(0.5g)の添加により中和し、濾過し、そして乾燥状態まで濃縮する。化合物3を、カラムクロマトグラフィー(トルエン/アセトン、4:1混合物)によって精製する。
【0044】
化合物3(1.96mmol)を、4A モレキュラーシーブス、TBABr(968mmol)およびCuBr2(529mmol)とともに、DMF/ジクロロメタン(50ml、1:5混合物)中で1時間攪拌する。化合物4(2.94mmol)をジクロロメタン(5ml)中に溶解し、上記溶液に滴下する。この溶液を24時間反応させ、濾過し、そして飽和炭酸水素ナトリウムおよび水(各々50ml)で洗浄する。その有機層を単離し、硫酸ナトリウムで乾燥し、乾燥状態までエバポレートする。化合物5を、カラムクロマトグラフィー(ヘキサン/酢酸エチル、4:1混合物)によって精製する。
【0045】
化合物22を、化合物5のメタノール中への溶解および続く1M ナトリウムメトキシド溶液の添加により得る。その溶液を3〜5時間反応させ、そして次いでAmberlite IR−120樹脂で中和し、濾過し、そして真空中で濃縮する。化合物22を、カラムクロマトグラフィー(ジクロロメタン/メタノール、20:1混合物)により精製する。
【0046】
(実施例6)
(化合物4、6、7、8、10、12、13、14、16、17、18、19および20の合成)
化合物21(実施例2;図4)から出発して、これらの化合物を文献(Helvetica Chimica Acta Vol.83,pp.2893〜2907,2000;Angew Chem.Int.Ed.Vol.40,No.19,pp.3644〜3647、2001)に記載される手順に従って合成する。
【0047】
(実施例7)
(化合物23、24、25、26、27および28の合成)
23という中間体の合成:化合物i(2.77mmol)を、化合物2(5.55mmol)を含むジエチルエーテル/ジクロロメタン(1ml、1:1混合物)の溶液に溶解する。0.1M TMSOTf(2.8ml)溶液を、次いで攪拌しながら滴下する。30分後、この溶液を、炭酸水素ナトリウム(0.5g)の添加により中和し、濾過し、そして乾燥状態まで濃縮する。3を、シリカゲルクロマトグラフィーにより精製し、化合物iiiを与える。
【0048】
化合物iii(1.96mmol)を、4A モレキュラーシーブス、TBABr(968mmol)およびCuBr2(529mmol)とともに、DMF/ジクロロメタン(50ml、1:5混合物)中で1時間攪拌する。化合物xv(2.94mmol)をジクロロメタン(5ml)中に溶解し、上記溶液に滴下する。この溶液を24時間反応させ、濾過し、そして飽和炭酸水素ナトリウムおよび水(各々50ml)で洗浄する。その有機層を単離し、硫酸ナトリウムで乾燥し、乾燥状態までエバポレートする。ivを、次いでシリカゲルクロマトグラフィーにより精製する。
【0049】
化合物ivを、MeOH中の0.1N NaOMeを用いて、脱O−アセチル化しvを与え、そして次いで還流条件下でMeOH中のジブチルスズオキシドで処理し、(上記溶媒のエバポレート後)viを与え、そのviをさらなる精製なしで次の工程に使用する。
【0050】
化合物vi(1mmol)を、DME(20ml)中に溶解し、次いで化合物vii(2.5mmol)およびCsF(1.4mmol)を添加する。その得られる反応混合物を室温で8時間攪拌し、酢酸エチルで希釈し、その有機層を水で洗浄する。この有機層を乾燥状態まで濃縮し、シリカゲルクロマトグラフィーにより精製し、化合物viiiを与える。
【0051】
化合物23の合成:化合物viiiを炭素中のパラジウムの存在下で水素化し、化合物23を与える。
【0052】
中間体xxの合成:N−アセチルグルコサミン(5,50g)から出発して、化合物xx(50%の全工程収率)を、刊行された手順(Bioorg.Med.Chem.Lett.11、pp.923〜925,2001;Carbohydr.Res.197,75、1990)に従って合成する。
【0053】
中間体xvの合成:化合物xv(15g)を、文献(Carbohydr.Res.201,15〜30、1990)に記載される手順に従って、L−フコースから合成する。
【0054】
中間体xixの合成:化合物xixを、記載されるように(WO9701569;Chem.Astr.,126 186312,1997)、商業的に入手可能なβ−D−ガラクトースペンタアセテートから調製する。
【0055】
中間体xxxivの合成:商業的に入手可能なN−アセチルノイラミン酸(xxx、10g)を、MeOH−H2O(60ml;9:1)中に懸濁し、そして、炭酸セシウムの水溶液を添加することにより、そのpHを8.1に調整する。その溶媒を除去し、そしてその残渣をエタノールを用い、次いでヘキサンを用いて繰返しエバポレートする。その物質をDMF(65ml)中に溶解させ、20分以内に臭化ベンジル(3.5ml)を添加する。その混合物を16時間攪拌し、その後ジクロロメタン(100ml)を添加し、水(50ml)で洗浄する。その溶媒を留去し、シリカゲルクロマトグラフィーにより精製し、xxxiを68%の収率で与える。
【0056】
ピリジン(50ml)中の化合物xxxi(7g)の溶液に、無水酢酸(48ml)を添加し、その反応混合物を室温で16時間攪拌する。溶媒を留去し、そしてその残渣(xxxii)を乾燥DMF(25ml)中に溶解する。その混合物に粉砕された炭酸アンモニウム(2g)を添加し、その混合物をセ氏28度で12時間攪拌する。この混合物を水(50ml)中の1N HClの氷冷溶液に添加し、そしてジクロロメタン(100ml)を添加する。溶媒抽出の後で、有機層を留去し、次いで真空下で24時間乾燥状態する。その残渣をシリカゲルクロマトグラフィーにより精製し、xxxiiiを71%の収率で与える。
【0057】
化合物xxxiiiを乾燥ジクロロメタン中に溶解し、2,6−ジ−tert−ブチルピリジン(5g)を添加する。その溶液をセ氏−20度に冷却し、トリフルオロメタンスルホン酸無水物(7g)を、10分間のうちに少しずつ添加する。その混合物を4時間攪拌し、ジクロロメタン(100ml)で希釈し、そしてリン酸水素カリウムの溶液(500ml)に添加する。その層を分離し、その有機層を乾燥し(硫酸ナトリウム)、溶媒を留去しxxxivを与え、このxxxivをさらなる精製なしで次の工程に使用する。
【0058】
中間体xxxvの合成:ジクロロメタン(100ml)中の化合物xx(10g)および化合物xv(15g)の混合物へ、モレキュラーシーブス(4A、8g)を添加する。室温で1時間攪拌し、その後臭化テトラエチルアンモニウム(5g)を添加する。ジクロロメタン(25ml)中の臭素(1g)の溶液を、1時間の間、滴下する。その反応混合物を3時間攪拌し続け、セライト床を通して濾過し、そして冷水、炭酸水素ナトリウムの飽和水溶液および水で逐次洗浄する。溶媒を留去し、シリカゲルクロマトグラフィーに供する。
【0059】
その生成物を、THF中のシアノ水素化ホウ素ナトリウムおよびエーテル中のHClで処理し、シリカゲルクロマトグラフィーの後、70%の全工程収率で化合物xxxvを与えた。
【0060】
中間体xxxviの合成:ジクロロメタン(80ml)中のxxxv(10g)およびxix(7g)の混合物へ、N−ヨードスクシンイミド(15g)およびモレキュラーシーブス(4A、8g)を添加する。その反応混合物を氷浴中に置く。その溶液を0〜5度で30分間攪拌し、そしてジクロロメタン(25ml)中のトリフルオロメタンスルホン酸(0.2ml)の溶液を、1時間の間、攪拌しながら滴下する。攪拌を2時間継続し、セライト床を通して濾過し、そして冷水、炭酸水素ナトリウムの冷たい飽和溶液および水で逐次洗浄する。溶媒を留去し、シリカゲルクロマトグラフィーにより精製し、68%の収率で化合物xxxviを与える。
【0061】
中間体xxxviiの合成:化合物xxxvi(8g)をMeOH(100ml)中の0.05N NaOEtで4時間処理し、IR120(水素型)樹脂での中和後、その反応混合物を濾別する。溶媒を留去し、96%収率で化合物xxxviiを与える。
【0062】
中間体xxxviiiの合成:化合物xxxvii(5g)をMeOH中のジブチルスズオキシド(1g)を用いて、還流下で4時間処理する。その溶媒を留去し、そしてトルエンとともに、数回、共エバポレートし(co−evaporated)、そして最終的にその残渣を高真空下で24時間乾燥する。
【0063】
その粗反応混合物をジメトキシエタン(DME、100ml)中に溶解し、そしてCsF(1.7g)および化合物xxxiv(2.5g)を添加する。その反応混合物を室温で8時間攪拌し、酢酸エチル(100ml)を添加する。有機層を水で洗浄し、有機溶媒を留去する。その生成物をシリカゲルクロマトグラフィーにより精製し、64%の収率でxxxviiiを与える。
【0064】
中間体xxxixの合成:化合物xxxviii(2g)を、MeOH中の0.01N NaOMe(100ml、1時間)を用いて、脱O−アセチル化し、粗反応混合物をIR120(水素型)樹脂で中和し、溶媒を留去する。
【0065】
上記反応からの生成物をジオキサン−水(1:1、50ml)中に溶解し、そして10%PD−Cを添加する。その反応混合物を、水素雰囲気下で22時間激しく攪拌し、セライト床を通して濾過し、その溶媒を留去する。得られるシロップのシリカゲルクロマトグラフィーは、77%の収率でxxxixを与える。
【0066】
化合物24の合成:MeOH−H2O中の化合物xxxixの溶液に、MeOH中のNaOMeの溶液を添加し、その反応混合物を室温で2時間攪拌する。IR120樹脂による中和および乾燥状態までのエバポレーションは化合物24を与える。
【0067】
xxxxの合成:化合物xxxixを、セ氏70度で4時間、エチレンジアミンで処理する(500mg)。溶媒を留去し、そのシロップ状残渣をシリカゲルクロマトグラフィーにより精製し、化合物xxxxを77%の収率で与える。
【0068】
化合物xxxxvの合成:ヨウ化3−ニトロベンジルを商業的に入手可能な8−アミノナフタレン−1,3,5−トリスルホン酸(xxxxxi)の水溶液(pH11)に、室温で攪拌しながら添加する。その溶液のpHを1に調整し、その溶媒のエバポレーション後に、生成物xxxxiiiをエタノールから沈殿させる。
【0069】
化合物xxxxiiiの白金触媒水素化は、化合物xxxxivを96%の収率で与える。
【0070】
リン酸塩緩衝液(pH7.1)中の化合物xxxxivの溶液に、商業的に入手可能なスクエア酸を添加し、その反応混合物を室温で4時間攪拌する。次いでそれを逆相HPLCにより精製しxxxxvを与える。
【0071】
化合物25の合成:化合物xxxx(0.2g)を炭酸塩緩衝液(2ml、pH8.8)に溶解し、化合物xxxxv(0.4g)を添加する。その反応混合物を室温で24時間攪拌する。別のバッチ(0.2g)の化合物xxxxvを添加し、攪拌を20時間室温で継続する。溶媒を留去し、その混合物を逆相HPLCにより精製し、25を与える。
【0072】
化合物26の合成:化合物xxxix(0.1g)を化合物xxxxivと反応させ、HPLCによる精製後、化合物26を与える。
【0073】
中間体xxxxxiiの合成:商業的に入手可能な化合物xxxxvi(4−(4,4,5,5−テトラメチル−[1,3,2]ジオキサボロラン−2−イル)安息香酸、1当量)およびKOAc(3当量)をTHF(25ml)中に入れる。その生成するスラリーにPdCl2および商業的に入手可能なp−ブロモニトロベンゼン(xxxxvii、1.2当量)を攪拌しながら添加し、そしてその混合物をセ氏80度までゆるやかに加熱する。6時間後、その反応混合物を乾燥状態までエバポレートし、ジクロロメタン(30ml)中に溶解し、蒸留水および炭酸水素ナトリウムの飽和溶液で洗浄する。その得られるビフェニル化合物xxxxviiiを直接次の工程に供する。
【0074】
化合物xxxxviii(1当量)、ジメチルアミノピリジン(触媒量、1個の結晶)およびEDCl(1.05当量)をDMFまたはTHF(20ml)中に溶解し、室温で10分間反応させる。商業的に入手可能な化合物xxxxix(8−アミノナフタレン−1,3,5−トリスルホン酸)をその反応混合物に攪拌しながら添加し、そしてその反応を、48時間、窒素下、室温で進行させる。その反応混合物を、次いで乾燥状態までエバポレートし、そして逆相クロマトグラフィーにより精製し、化合物xxxxxを与える。
【0075】
EtOAc中の化合物xxxxxの溶液に、PD−Cを添加し、そしてその反応混合物を水素雰囲気下で2時間攪拌する。その反応混合物をセライト床を通して濾過し、そして乾燥状態までエバポレートし、化合物xxxxxiを与える。
【0076】
リン酸塩緩衝液(pH7.1)中の化合物xxxxxiの溶液に、商業的に入手可能なスクエア酸を添加し、その反応混合物を室温で4時間攪拌する。次いで、それを逆相HPLCにより精製しxxxxxiiを与える。
【0077】
化合物27の合成:化合物xxxx(0.2g)を炭酸塩緩衝液(2ml、pH8.8)中に溶解し、化合物xxxxxii(0.4g)を添加する。その反応混合物を室温で24時間攪拌する。別のバッチ(0.2g)の化合物xxxxxiiを添加し、そして攪拌を20時間室温で継続する。溶媒を留去し、そしてその混合物を逆相HPLCにより精製し27を与える。
【0078】
化合物28の合成:化合物xxxixを化合物xxxxxiと反応させ、化合物28を与え、その化合物28をHPLCにより精製する。
【0079】
(合成に関する参考文献)
本出願内に含まれる特定の化合物の調製についての合成プロトコルは、以下の参考文献に例示される:Helvetica Chemica Acta Vol.83,pp.2893〜2907(2000)およびAngew.Chem.Int.Ed.Vol.40,No.19,pp.3644〜3647(2001)。
【0080】
(実施例8)
(アッセイ)
アルブミンに化学的に結合させた精製オリゴ糖(新しい糖タンパク質)を、プラスチック製マイクロタイターウェル中にコーティングする。BSAでブロックした後、そのウェルを次いで精製PA−IILレクチンとともにインキュベートし、精製PA−IILレクチンに結合させる。結合されたPA−IILレクチンを、1M H3PO4による発色を用いて、抗PA−IILウサギ抗血清、続いてHRP標識化抗ウサギIgおよびTMB試薬で検出する(図18)。構造および活性を表1に提示する。
【0081】
(表1.PA−IILレクチンへの結合についてスクリーニングされた固定化中性オリゴ糖の構造)
固定化中性オリゴ糖のスクリーニングの結果を図18に記載し、以下にまとめる。1型ラクト系列鎖(例えば、Lea構造体)は、ラクト系列2型鎖(例えば、Lex構造体)よりもより良好にPA−IILレクチンに結合する。Lea/Lexにおけるように、その1型構造体の伸長は、この系列において最も活性な化合物をもたらす。
【0082】
【表1】
(表2.PA−IILレクチンへの結合についてスクリーニングされた固定化酸性オリゴ糖の構造)
固定化酸性オリゴ糖のスクリーニングの結果を、図19に記載し、以下にまとめる。フコースを含むシアリル化オリゴ糖のみが、PA−IILレクチンに結合する。ジシアリルLea上の第2のシアリル基は、結合を阻害するように見える。
【0083】
【表2】
アルブミンに化学的に結合させた精製オリゴ糖(新しい糖タンパク質)を、プラスチック製マイクロタイターウェル中にコーティングする。BSAでブロックした後、そのウェルを次いで精製PA−IILレクチンとともにインキュベートし、精製PA−IILレクチンに結合させる。結合されたPA−IILレクチンを、1M H3PO4による発色を用いて、抗PA−IILウサギ抗血清、続いてHRP標識化抗ウサギIgおよびTMB試薬で検出する(図19)。構造および活性を表2に提示する。
【0084】
フコースのビオチン化ポリマー(Fuc−PAA−ビオチン、GlycoTech Corp.)を、HRP標識化ストレプトアビジン(KPL labs)と、4℃で終夜結合させる。PA−IILレクチンを、プラスチック製マイクロタイターウェル中に固定化し、HRP標識化フコシル化ポリマーと反応させる。結合をTMB基質で検出し、続いて1M H3PO4を用いる発色により検出する(図20)。そのアッセイのために選ばれた条件は、1mlあたり3μgのPA−IILのコーティングおよび1mlあたり2μgのフコシル化ポリマーを用いるインヒビターのインキュベーションである。
【0085】
化合物23は、420nMのIC50でPA−IILを阻害したが(図22)、一方で天然の糖類インヒビターであるマンノースは、95μMのIC50でPA−IILレクチンを阻害し、負の対照糖類であるガラクトースは、阻害を示さなかった(データは示されていない)。
【0086】
本発明の特定の実施形態が、例示の目的のために本明細書中で記載されてきたが、本発明の精神および範囲から逸脱することなく、様々な改変がなされ得ることは、上記のことから理解される。
【図面の簡単な説明】
【0087】
【図1A】図1Aは、糖模倣化合物の構造を示す。
【図1B】図1Bは、糖模倣化合物の構造を示す。
【図1C】図1Cは、糖模倣化合物の構造を示す。
【図1D】図1Dは、糖模倣化合物の構造を示す。
【図1E】図1Eは、糖模倣化合物の構造を示す。
【図2A】図2Aは、さらなる糖模倣化合物の構造を示す。
【図2B】図2Bは、さらなる糖模倣化合物の構造を示す。
【図3】図3は、代表的な糖模倣化合物3の合成を図示する。
【図4】図4は、糖模倣化合物21の合成を図示する。
【図5】図5は、代表的な糖模倣化合物15の合成を図示する。
【図6】図6は、種々の代表的な糖模倣化合物を与える中間体21のアシル化を図示する。
【図7】図7は、化合物22の合成を図示する。
【図8A】図8Aは、化合物4、6、7、8、10および12の合成を図示する。
【図8B】図8Bは、化合物13、14、16、17、18および19の合成を図示する。
【図9】図9は、化合物22および23の合成を図示する。
【図10】図10は、中間体の合成を図示する。
【図11】図11は、中間体および化合物24の合成を図示する。
【図12】図12は、中間体xxxxivおよびxxxxvの合成を図示する。
【図13】図13は、化合物25の合成を図示する。
【図14】図14は、化合物26の合成を図示する。
【図15】図15は、中間体xxxxxiおよびxxxxxiiの合成を図示する。
【図16】図16は、化合物27の合成を図示する。
【図17】図17は、化合物28の合成を図示する。
【図18】図18は、PA−IILレクチンの、固定された中性糖鎖構造体への結合を図示する。
【図19】図19は、PA−IILレクチンの、固定された酸性糖鎖構造体への結合を図示する。
【図20】図20は、PA−IILレクチン阻害のIC50値についてのアッセイ条件の決定を図示する。
【図21】図21は、PA−IILレクチンの糖模倣インヒビターのIC50値を決定するために開発したアッセイの図式的表示を示す。
【図22】図22は、糖模倣化合物23による、PA−IILレクチンの阻害を図示する。
【特許請求の範囲】
【請求項1】
温血動物においてPseudomonas細菌を阻害する方法であって、図1または図2に記載の化合物を含む化合物を、該細菌を阻害するための有効な量で、該動物に投与する工程を包含する、方法。
【請求項2】
前記化合物が、治療薬剤と結合されている、請求項1に記載の方法。
【請求項3】
前記化合物が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項1または2に記載の方法。
【請求項4】
前記細菌が、Pseudomonas aeruginosaである、請求項1に記載の方法。
【請求項5】
図1または図2に記載の化合物と結合された治療薬剤を含む、結合体。
【請求項6】
前記結合体が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項5に記載の結合体。
【請求項7】
Pseudomonas細菌を検出する方法であって、該方法は、図1または図2に記載の化合物を含む化合物に結合された診断薬剤とサンプルを、該サンプル中に存在する場合、該化合物が該細菌と結合するための十分な条件下で、接触させる工程;および該サンプル中に存在する該薬剤を検出する工程であって、ここで、該サンプル中の該薬剤の存在は、Pseudomonas細菌の存在を示す工程を包含する、方法。
【請求項8】
前記細菌は、Pseudomonas aeruginosaである、請求項7に記載の方法。
【請求項9】
固体支持体上にPseudomonas細菌を固定する方法であって、該方法は、結合するための十分な条件下で、Pseudomonas細菌含有サンプルと、固体支持体上に固定された図1または図2に記載の化合物を含む化合物とを接触させる工程;および該固体支持体から該サンプルを分離する工程を包含する、方法。
【請求項10】
Pseudomonas細菌を阻害する方法における使用のための、図1または図2に記載の化合物を含む、化合物。
【請求項11】
薬学的に受容可能なキャリアまたは希釈剤と組み合わされた、請求項10に記載の化合物。
【請求項12】
前記細菌が、Pseudomonas aeruginosaである、請求項10または11に記載の化合物。
【請求項13】
Pseudomonas細菌を阻害する方法における使用のための、請求項5または6に記載の結合体。
【請求項14】
前記細菌が、Pseudomonas aeruginosaである、請求項13に記載の結合体。
【請求項15】
Pseudomonas細菌の阻害のための薬の調製における、図1または図2に記載の化合物を含む化合物の使用。
【請求項16】
Pseudomonas細菌の阻害のための薬の調製における、請求項5による結合体の使用。
【請求項17】
前記細菌が、Pseudomonas aeruginosaである、請求項15または16に記載の使用。
【特許請求の範囲】
【請求項1】
温血動物においてPseudomonas細菌を阻害する方法であって
、図1または図2に記載の化合物を含む化合物を、該細菌を阻害するための有効な量で、該動物に投与する工程を包含する、方法。
【請求項2】
前記化合物が、Pseudomonas細菌を処置するための治療薬剤と結合されている、請求項1に記載の方法。
【請求項3】
前記化合物が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項1または2に記載の方法。
【請求項4】
前記細菌が、Pseudomonas aeruginosaである、請求項1に記載の方法。
【請求項5】
図1または図2に記載の化合物と結合された、Pseudomonas細菌を処置するための治療薬剤を含む、結合体。
【請求項6】
前記結合体が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項5に記載の結合体。
【請求項7】
Pseudomonas細菌を検出する方法であって、該方法は、図1または図2に記載の化合物を含む化合物に結合された検出可能なマーカーとサンプルを、該サンプル中に存在する場合、該化合物が該細菌と結合するための十分な条件下で、接触させる工程;および該サンプル中に存在する該マーカーを検出する工程であって、ここで、該サンプル中の該マーカーの存在は、Pseudomonas細菌の存在を示す工程を包含する、方法。
【請求項8】
前記細菌は、Pseudomonas aeruginosaである、請求項7に記載の方法。
【請求項9】
固体支持体上にPseudomonas細菌を固定する方法であって、該方法は、結合するための十分な条件下で、Pseudomonas細菌含有サンプルと、固体支持体上に固定された図1または図2に記載の化合物を含む化合物とを接触させる工程;および該固体支持体から該サンプルを分離する工程を包含する、方法。
【請求項10】
Pseudomonas細菌を阻害する方法における使用のための、図1または図2に記載の化合物を含む、化合物。
【請求項11】
薬学的に受容可能なキャリアまたは希釈剤と組み合わされた、請求項10に記載の化合物。
【請求項12】
前記細菌が、Pseudomonas aeruginosaである、請求項10または11に記載の化合物。
【請求項13】
Pseudomonas細菌を阻害する方法における使用のための、請求項5または6に記載の結合体。
【請求項14】
前記細菌が、Pseudomonas aeruginosaである、請求項13に記載の結合体。
【請求項15】
Pseudomonas細菌の阻害のための薬の調製における、図1または図2に記載の化合物を含む化合物の使用。
【請求項16】
Pseudomonas細菌の阻害のための薬の調製における、請求項5による結合体の使用。
【請求項17】
前記細菌が、Pseudomonas aeruginosaである、請求項15または16に記載の使用。
【請求項1】
温血動物においてPseudomonas細菌を阻害する方法であって、図1または図2に記載の化合物を含む化合物を、該細菌を阻害するための有効な量で、該動物に投与する工程を包含する、方法。
【請求項2】
前記化合物が、治療薬剤と結合されている、請求項1に記載の方法。
【請求項3】
前記化合物が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項1または2に記載の方法。
【請求項4】
前記細菌が、Pseudomonas aeruginosaである、請求項1に記載の方法。
【請求項5】
図1または図2に記載の化合物と結合された治療薬剤を含む、結合体。
【請求項6】
前記結合体が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項5に記載の結合体。
【請求項7】
Pseudomonas細菌を検出する方法であって、該方法は、図1または図2に記載の化合物を含む化合物に結合された診断薬剤とサンプルを、該サンプル中に存在する場合、該化合物が該細菌と結合するための十分な条件下で、接触させる工程;および該サンプル中に存在する該薬剤を検出する工程であって、ここで、該サンプル中の該薬剤の存在は、Pseudomonas細菌の存在を示す工程を包含する、方法。
【請求項8】
前記細菌は、Pseudomonas aeruginosaである、請求項7に記載の方法。
【請求項9】
固体支持体上にPseudomonas細菌を固定する方法であって、該方法は、結合するための十分な条件下で、Pseudomonas細菌含有サンプルと、固体支持体上に固定された図1または図2に記載の化合物を含む化合物とを接触させる工程;および該固体支持体から該サンプルを分離する工程を包含する、方法。
【請求項10】
Pseudomonas細菌を阻害する方法における使用のための、図1または図2に記載の化合物を含む、化合物。
【請求項11】
薬学的に受容可能なキャリアまたは希釈剤と組み合わされた、請求項10に記載の化合物。
【請求項12】
前記細菌が、Pseudomonas aeruginosaである、請求項10または11に記載の化合物。
【請求項13】
Pseudomonas細菌を阻害する方法における使用のための、請求項5または6に記載の結合体。
【請求項14】
前記細菌が、Pseudomonas aeruginosaである、請求項13に記載の結合体。
【請求項15】
Pseudomonas細菌の阻害のための薬の調製における、図1または図2に記載の化合物を含む化合物の使用。
【請求項16】
Pseudomonas細菌の阻害のための薬の調製における、請求項5による結合体の使用。
【請求項17】
前記細菌が、Pseudomonas aeruginosaである、請求項15または16に記載の使用。
【特許請求の範囲】
【請求項1】
温血動物においてPseudomonas細菌を阻害する方法であって
、図1または図2に記載の化合物を含む化合物を、該細菌を阻害するための有効な量で、該動物に投与する工程を包含する、方法。
【請求項2】
前記化合物が、Pseudomonas細菌を処置するための治療薬剤と結合されている、請求項1に記載の方法。
【請求項3】
前記化合物が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項1または2に記載の方法。
【請求項4】
前記細菌が、Pseudomonas aeruginosaである、請求項1に記載の方法。
【請求項5】
図1または図2に記載の化合物と結合された、Pseudomonas細菌を処置するための治療薬剤を含む、結合体。
【請求項6】
前記結合体が、薬学的に受容可能なキャリアまたは希釈剤と組み合わされている、請求項5に記載の結合体。
【請求項7】
Pseudomonas細菌を検出する方法であって、該方法は、図1または図2に記載の化合物を含む化合物に結合された検出可能なマーカーとサンプルを、該サンプル中に存在する場合、該化合物が該細菌と結合するための十分な条件下で、接触させる工程;および該サンプル中に存在する該マーカーを検出する工程であって、ここで、該サンプル中の該マーカーの存在は、Pseudomonas細菌の存在を示す工程を包含する、方法。
【請求項8】
前記細菌は、Pseudomonas aeruginosaである、請求項7に記載の方法。
【請求項9】
固体支持体上にPseudomonas細菌を固定する方法であって、該方法は、結合するための十分な条件下で、Pseudomonas細菌含有サンプルと、固体支持体上に固定された図1または図2に記載の化合物を含む化合物とを接触させる工程;および該固体支持体から該サンプルを分離する工程を包含する、方法。
【請求項10】
Pseudomonas細菌を阻害する方法における使用のための、図1または図2に記載の化合物を含む、化合物。
【請求項11】
薬学的に受容可能なキャリアまたは希釈剤と組み合わされた、請求項10に記載の化合物。
【請求項12】
前記細菌が、Pseudomonas aeruginosaである、請求項10または11に記載の化合物。
【請求項13】
Pseudomonas細菌を阻害する方法における使用のための、請求項5または6に記載の結合体。
【請求項14】
前記細菌が、Pseudomonas aeruginosaである、請求項13に記載の結合体。
【請求項15】
Pseudomonas細菌の阻害のための薬の調製における、図1または図2に記載の化合物を含む化合物の使用。
【請求項16】
Pseudomonas細菌の阻害のための薬の調製における、請求項5による結合体の使用。
【請求項17】
前記細菌が、Pseudomonas aeruginosaである、請求項15または16に記載の使用。
【図1A】

【図1B】
【図1C】
【図1D】
【図1E】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8B】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【図1B】
【図1C】
【図1D】
【図1E】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8B】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16】
【図17】
【図18】
【図19】
【図20】
【図21】
【図22】
【公表番号】特表2006−515306(P2006−515306A)
【公表日】平成18年5月25日(2006.5.25)
【国際特許分類】
【出願番号】特願2004−563924(P2004−563924)
【出願日】平成15年12月19日(2003.12.19)
【国際出願番号】PCT/US2003/040881
【国際公開番号】WO2004/058304
【国際公開日】平成16年7月15日(2004.7.15)
【出願人】(503443681)グリコミメティクス, インコーポレイテッド (2)
【Fターム(参考)】
【公表日】平成18年5月25日(2006.5.25)
【国際特許分類】
【出願日】平成15年12月19日(2003.12.19)
【国際出願番号】PCT/US2003/040881
【国際公開番号】WO2004/058304
【国際公開日】平成16年7月15日(2004.7.15)
【出願人】(503443681)グリコミメティクス, インコーポレイテッド (2)
【Fターム(参考)】
[ Back to top ]