
RNAおよびDNAの並行単離および/または並行精製のためのプロセス
本発明は、同じ固定生物学的サンプルからの、RNAおよびDNAの並行単離および/または精製のためのプロセス、本発明によるプロセスによって単離された核酸の定量化および分析、固定サンプルからのRNAおよびDNAの並行単離および/または精製のためのキット、ならびに疾患の診断、予後、治療に関する決定、および/または治療のモニタリングのためのこのキットの使用に関する。溶解した画分を未溶解画分から分離した後、その画分を望ましいように別々に処理し得る。例えば、RNAを未溶解画分から単離し得、そしてDNAを未溶解画分から単離し得る。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、同じ固定生物学的サンプルからの、RNAおよびDNAの並行単離および/または並行精製のためのプロセス、本発明によるプロセスによって単離された核酸の定量化および分析、固定サンプルからのRNAおよびDNAの並行単離および/または精製のためのキット、ならびに疾患の診断、予後、治療に関する決定、および/または治療のモニタリングのための、このキットの使用に関する。
【背景技術】
【0002】
もし、例えば組織断片または単離細胞のような生物学的材料を、生きた生物から除去した場合、その細胞は、短期間で死ぬ。非常に迅速に、死んだ細胞は、まず自己融解/発酵によって、そして次いで細菌によって分解され、もとの細胞および組織構造は破壊される。従って、もし細胞または組織断片を、組織学的調査のために生物から除去するなら、分解を防止するために、取った生物学的サンプルを固定することが推奨される。理想的には、固定は、サンプルの構造を、実質的に変化させないで、その組織学的評価を可能にする。さらに固定は、サンプルの長期保存および記録保管を可能にする。これらの理由のために、多くの形態学的調査は、固定した材料に基づいてのみ可能である。
【0003】
通常、例えば酸、アルコール、ケトン、またはアルデヒド、特にグルタルアルデヒドまたはホルムアルデヒドのような、タンパク質沈殿またはタンパク質架橋化合物を用いて、固定を達成する。ここで、ホルムアルデヒド(「ホルマリン」と呼ばれる水溶液の形態で使用される)による固定に続く、固定サンプルのパラフィンへの包埋は、細胞および組織構造が特に良好に保存されるので、特に病理学において非常に重要である。本明細書の以下で、この方式で固定した材料を、「ホルマリン固定、パラフィン包埋材料」または「FFPE材料」と呼ぶ。
【0004】
しかし、特にホルマリンによるサンプルの固定は、ホルムアルデヒドの架橋効果のために、タンパク質だけでなく、サンプル中に存在する核酸を含む様々な他の生体分子も、お互いに共有結合し、そして結果として、そのようなサンプルからの核酸(DNAまたはRNA)の単離は、非常に困難であるという欠点を有する。しかし、分子レベルにおける多くの研究に関して、核酸の単離は非常に重要である。
【0005】
そのような固定サンプルから核酸を単離する1つの方法が、特許文献1において記載されている。そこに記載された方法は、生物学的サンプルにおいて固定によって形成された架橋を切断し、そして1つの型の核酸、すなわちDNAまたはRNAのいずれかを単離することを可能にし、続いて例えばPCRまたはRT−PCR分析を行い得る。
【0006】
例えば腫瘍障害の診断または予後に関する分子病理学の分野において、DNAに基づく分析、およびRNAに基づく分析の両方が使用される。同じ固定サンプル、例えば腫瘍サンプルについてDNAおよびRNAに基づく分析の両方を可能にするために、例えば生検からの組織切片のような1つのサンプルからのDNAおよびRNAの並行単離を可能にするプロセスが必要である。まず、通常非常に少量のサンプル材料しか入手可能でなく、複数の別個の精製のためには不十分であるので、そのような単一のサンプルからのDNAおよびRNAの並行単離は、非常に望ましい。2番目に、サンプル材料の組成は、一般的に不均一である;例えば健康な細胞のマトリックス中には非常に少数の腫瘍細胞しか存在しない。この場合、異なる細胞のお互いに対する比が、それぞれの部分的なサンプルにおいて同じであることを保証することは不可能であるので、サンプルを分けることは望ましくない。単一の、分割していないサンプルからのDNAおよびRNAの並行単離のみが、研究する全ての被検体が同じ比で存在し、そして同じ組成のサンプル由来であることを保証する。
【0007】
特許文献1において記載されたプロセスは、固定の間に導入された架橋を切断することによって、両方の型の核酸、DNAおよびRNAを等しく放出する。従ってこのプロセスは、DNAまたはRNAのいずれか、または両方の核酸の混合物の単離のみを可能にする;しかし、それは別の画分におけるDNAおよびRNAの並行単離を可能にしない。2つの型の核酸(DNAおよびRNA)を分離するために、特許文献1は、単離後に、精製の間に同時に放出された2つの型の核酸の1つの、選択的沈殿または選択的吸着を示唆する。あるいは、それぞれの不必要な型の核酸を酵素的に分解することが可能である。
【0008】
選択的吸着による、サンプルからのDNAおよびRNAの並行精製の1つの方法が公知であり、そして例えば市販で入手可能なAllprep DNA/RNAキット(Qiagen、Hilden、Germany)を用いて行い得る。ここで、そのサンプルを最初に、いかなるアルコールも含まないカオトロープを含む溶解バッファーで溶解し、そして溶解産物中に存在するDNAはシリカマトリックスに結合し、一方これも溶解産物に存在するRNAは結合しないで溶液中に残る。残った溶解産物にアルコールを加えた後、RNAはさらなるシリカマトリックスに結合し得る。このプロセスは、非固定サンプルに関してよく作用する。しかし、ここでDNAは最初のシリカマトリックスに定量的に結合せず、多くの量が残った溶解産物に残り、そしてRNAと共に精製されるので、それは依然としてホルマリン固定サンプルに最適に適用できないことが見出された。従って、このプロセスは、RNAおよびDNAの別々の精製を可能にしない。
【0009】
特許文献2は、FFPEサンプルを含む生物学的サンプルから、DNAおよびRNAを同時抽出するための方法を記載する。そのFFPEサンプルを脱パラフィン化し、そしてカオトロピック剤、イオン性界面活性剤、およびタンパク質分解酵素を含む溶解バッファーを用いて消化する。RNAおよびDNAを放出するために、少なくとも5時間、好ましくは10時間サンプルを消化し、フェノール−クロロホルムを加え、そして相を分離する。水相は主にRNAを含み、有機相は主にDNAを含む。次いでアルコール沈殿を用いて、RNAを水相から回収し得る。DNAを有機相から回収する。この方法は、特に、上記核酸を有機相および水相から別々に単離する前に、DNAからRNAを分離し得るために、フェノールを用いて放出された核酸を抽出する必要があるという欠点を有する。
【0010】
同様のプロトコールが、O’Sheaら、「Analysis of Tcell receptor beta chain CDR3 size using RNA extracted from formalin fixed paraffin wax embedded tissue」J Clin Pathol 1997;50:811−814において記載されている。
【0011】
DNAおよびRNAの混合物、またはDNAまたはRNAのいずれかを、FFPEサンプルから単離するための、市販で入手可能なキットが同様に公知である。FFPE RNA/DNA Purification Kit(Norgen、Biotek Corp.、Thorold、Canada)は、1つの溶出液中で、RNAおよびDNAの混合物の単離を可能にする。ここで、DNAのみ、またはRNAのみのいずれかを得るために、特に長いプロテアーゼK消化およびRNAse処理を行ってDNAを単離し得る、または短いプロテアーゼK消化およびDNAse処理を行ってRNAを単離し得る。しかし、このキットは、同じサンプルから、両方の型の核酸の、同時であるが別々の精製を可能にしない。
【0012】
Agencourt FormaPure Kit(Beckman Coulter Genomics GmbH、Danvers、Massachusetts、USA)も、1つの溶出液におけるRNAおよびDNAの混合物の単離、またはDNAse消化後のRNAの単離を可能にするが、DNAおよびDNAの同時であるが別々の単離を可能にしない。
【0013】
対応するヌクレアーゼ消化によって、または適当な加熱インキュベーションによって、DNAまたはRNAのいずれかを得るためのオプションも、Ambion Recover AII FFPE Kit(Applied Biosystems,Inc.、Foster City、California、USA)およびQuickExtract FFPE RNA Extraction Kit(Epicentre Biotechnologies、Madison、Wisconsin、USA)によって提供される。しかし、これらの市販で入手可能なキットはいずれも、同じ固定サンプルからDNAおよびRNA両方を別々に精製することを可能にしない。
【0014】
さらに、特許文献2は、同じサンプルから、両方の型の核酸(RNAおよびDNA)を同時に抽出するためのプロセスを記載し、それはまたとりわけ固定サンプルであり得る。このプロセスは、サンプルの溶解および芳香族アルコールを用いた酵素不活性化の後、適当な抽出プロセスによる2つの型の核酸の分離を含む。しかし、このプロセスで得られた2つの相(RNAを含む水相、およびDNAを含む有機相)から、次いでさらなる精製および/または単離の前に、適当な沈殿剤の添加によって、核酸を沈殿させなければならない。まず、これはそのプロセスを時間のかかるものにし、そして2番目に、沈殿のために、物質の損失のリスクおよび/または核酸に沈殿剤が混入するリスクが存在する。
【0015】
よって、架橋によって固定された同じサンプルから、DNAおよびRNAの両方を別々に精製することを可能にするプロセスを提供することが本発明の目的であり、DNAのRNAからの分離は、有機溶媒も、核酸を結合するための固体マトリックスも必要としない。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】国際公開第2007/068764号
【特許文献2】国際公開第2005/075642号
【発明の概要】
【課題を解決するための手段】
【0017】
本発明は、とりわけ少なくとも1つのタンパク質分解活性化合物を用いた、架橋によって固定された生物学的サンプルが有するタンパク質を含む成分の部分的なタンパク質分解は、RNAをサンプルの溶解画分へ選択的に放出することを可能にし、一方DNAは主に上記サンプルの未溶解残渣に残るという発見に基づく。サンプルの上記部分的消化は、別々の画分を得ることを可能にし、ここで溶解画分は主にRNAを含み、そして未溶解残渣は主にDNAを含む。主にRNAを含む溶解画分を、主にDNAを含む未溶解残渣から、例えば遠心分離プロセスを用いて容易に分離し得る。
【0018】
溶解した画分を未溶解画分から分離した後、その画分を望ましいように別々に処理し得る。例えば、RNAを未溶解画分から単離し得、そしてDNAを未溶解画分から単離し得る。個々の画分から核酸を単離する前に、主にRNAを含む溶解画分を、主にDNAを含む未溶解画分から分離することは、同じ架橋サンプルから良好な収率でRNAおよびDNAを効率的に単離することを可能にする。1つの画分からのみ核酸を単離し、そして他の画分を廃棄することも、本発明の範囲内である。例えば、RNAの単離が関心の的であるなら、DNAを含む未溶解画分を、分離後に廃棄し得る。
【0019】
本出願の他の目的、特徴、利点および局面は、以下の記載および添付の特許請求の範囲から当業者に明らかになる。しかし、以下の記載、添付の特許請求の範囲、および特定の実施例は、本出願の好ましい実施態様を示すが、例示としてのみ提供されることが理解されるべきである。開示された本発明の意図および範囲内の様々な変化および改変は、以下を読むことから当業者に容易に明らかになる。
【図面の簡単な説明】
【0020】
【図1】図1は、それぞれTAE−アガロースゲル(DNA)およびホルムアルデヒド−アガロースゲル(RNA)における、エチジウムブロミドによる染色後の、本発明によるプロセスによって上清およびペレットからそれぞれ単離されたRNAおよびDNAの分離を示す(実施例1)。サイズ比較のために、サイズマーカーLambda/HindIII(Invitrogen、Carlsbad、California、USA)を、レーンMにアプライした。
【図2】図2は、タンパク質分解活性化合物の反応時間の関数として、画分A(上清)および画分B(ペレット)から単離された核酸の全収量を示す(実施例3)。
【図3】図3は、タンパク質分解活性化合物の反応時間の関数としての、TAE−アガロースゲルにおける、画分B中のRNA含有量の分析の結果を示す(実施例3)。
【図4】図4は、ct値の変化としての、定量的リアルタイムPCRによる、画分Bから得られたDNAの増幅に対する、タンパク質分解活性化合物の反応時間の影響を示す(実施例3)。
【図5】図5は、市販で入手可能なキットを用いて行ったプロセスにおける収量と比較して、UV分光法によって決定された、本発明によるプロセスを用いて、各場合において示される期間保存された様々な型の組織から単離し得るDNAの収量を示す。(実施例5)。
【図6】図6は、TAE−アガロースゲルにおける、実施例5によって得られたDNAの分析を示す。サイズ比較のために、サイズマーカーLambda/HindIII(Invitrogen、Carlsbad、California、USA)を、レーンMにアプライした。
【図7】図7は、リアルタイムPCR分析による、実施例5によって得られたDNAの分析を示す。
【図8】図8は、単離したRNAのAgilent Bioanalyzer分析を示す。1−5:実施例1において記載された前処理バッファー1−5を用いて、DNAseにより前処理したサンプル。oc:DNAse前処理なしであるが、先行技術において通常であるように、オンカラムDNAse処理したサンプル。
【発明を実施するための形態】
【0021】
上記で議論したように、架橋によって固定された同じサンプルからDNAおよびRNAの両方を別々に精製することを可能にするプロセスを提供することが、本発明の1つの目的であり、DNAのRNAからの分離は、有機溶媒も、核酸を結合するための固体マトリックスも必要としない。
【0022】
この目的を、架橋によって固定された同じ生物学的サンプルからの、リボ核酸(RNA)およびデオキシリボ核酸(DNA)の並行単離および/または精製のためのプロセスによって達成し、それは以下の工程:
a)少なくとも1つのタンパク質分解活性化合物を用いた、サンプルが有するタンパク質を含む成分の部分的なタンパク質分解と同時に、水性緩衝溶液中においてサンプルを部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット;画分B)を得る工程;
b)溶解画分を、未溶解残渣から分離する工程、
を含み、
ここで、溶解画分における核酸の全量に基づいて、その溶解画分は主にRNAを含み、そして未溶解残渣における核酸の全量に基づいて、その未溶解残渣は主にDNAを含み、
そしてここで、主にRNAを含む画分の、主にDNAを含む画分からの分離は、有機溶媒による1つまたは両方の型の核酸の沈殿も抽出も、1つまたは両方の型の核酸の固体マトリックスに対する選択的結合も必要としない。
【0023】
本発明によるプロセスの目的のために、並行単離および/または精製は、2つの型の核酸、RNAおよびDNAの単離および/または精製が、お互いに空間的に離れて起こる、1つは主にRNAを含み、そして他方は主にDNAを含む、その2つの画分AおよびBの処理が、同時に、または異なる時点で起こり得る、単離および/または精製を意味すると理解される。
【0024】
本発明の目的のために、架橋によって固定された同じ生物学的サンプルは、工程(a)において部分的溶解を受けるサンプル全体を意味すると理解される。
【0025】
本発明の目的のために、「部分的溶解」および「部分的タンパク質分解」または「部分的消化」という用語はそれぞれ、下記でより詳細に説明するように、サンプルまたはサンプルの個々の成分の部分的溶解、およびサンプルが有するタンパク質を含む成分の部分的分解を意味すると理解される。
【0026】
本発明の目的のために、もし画分が主に1つの型の核酸を含むと呼ばれる場合、それは、この画分における核酸の全量(すなわち、2つの型の核酸の合計)に基づいて、50重量%超のこの型の核酸を含む。1つの実施態様によって、主に1つの型の核酸を含む上記画分は、この画分における核酸の全量に基づいて、少なくとも60重量%、好ましくは少なくとも70重量%、より好ましくは少なくとも80重量%のこの型の核酸を含む。
【0027】
本発明によるプロセスは、別々の画分における、同じ固定サンプルからのDNAおよびRNAの並行単離、および高感度で、定性的、および/または定量的方法による、それらの引き続く分析を可能にし、ここで例えば直径数mmの中空の針による臨床生検の顕微鏡的に分析可能な切片から得られるような、少量のサンプルでさえも、サンプル材料として適当である。本発明によるプロセスにおいて、市販で入手可能なAllprep DNA/RNA Kit(Qiagen、Hilden、Germany)と対照的に、DNAを含む、およびRNAを含む画分への分離は、核酸の実際の精製の前に実施する。固定した生物学的サンプルから、本発明によるプロセスは、主にRNAを含む溶解画分および主にDNAを含む未溶解画分という2つの画分を生じる。さらなる工程において、これらの画分を、それぞれの核酸のさらなる抽出および/または精製のために利用し得る。部分的消化およびRNAのDNAからの、主にRNAを含む溶解画分および主にDNAを含む未溶解画分への引き続く分離はまた、本発明による方法を、DNAをRNAから分離するためにフェノール/クロロホルム抽出に基づく先行技術の方法から差別化する。それぞれのフェノール/クロロホルムに基づく方法において、DNAおよびRNAはどちらも溶解産物に放出され、そして従って、両方とも溶解画分に存在する。フェノール−クロロホルム抽出および相分離の後、RNAは水相に溶解し、そしてDNAは有機相に溶解した形態で存在する。従って、先行技術の分離原理は、本発明によるプロセスと根本的に異なる。本発明によるプロセスはRNAをDNAから分離するためのフェノール/クロロホルム抽出を必要とせず、とりわけ架橋したサンプルの部分的消化に依拠してDNAを主に未溶解画分に維持する一方、RNAを溶解画分に放出する。
【0028】
最初の工程において、FFPEサンプルを、好ましくはプロテアーゼ処理にかける。驚くべきことに、この最初のプロテアーゼ処理において、プロテアーゼを用いることによるタンパク質のこのタンパク質分解(酵素的「消化」)の条件を調整する最適化によって、サンプルからDNAではなく、RNAのみを選択的に放出することが可能であることが見出された。適当な分離プロセス、例えば遠心分離を用いて、サンプルの本発明による不完全な「消化」後に、DNAを含む依然として未溶解の画分を、RNAを含む上清から分離することが可能である。
【0029】
ここで、2つの画分の溶解画分(A)および未溶解画分(B)への分離を、例えばろ過、沈降、デカンテーション、遠心分離等のような、液体および固体成分を分離するために適当であるとして当業者に公知のあらゆる方法を用いて行い得る。本明細書の以下で、この工程で得られる未溶解残渣はまた、ペレットと呼ばれ、ここで本発明の目的のために、この用語は、明確に遠心分離によってサンプルの液体成分から分離した未溶解残渣に限定されず、他の手段によって分離した未溶解残渣、例えばろ過の後にフィルターに残った固体物質も含む。
【0030】
未溶解画分をペレットにすることは、2つの画分の容易なおよび効率的な分離を可能にするので、有用である。
【0031】
RNAを単離するために、RNAを含む上清を、現在の技術水準から公知である習慣的なプロセスによって、例えば特許出願WO2007/068764において記載されたプロセスによって処理し得、そのプロセスは残った架橋を除去するための求核試薬を含む溶液中における加熱インキュベーションを含み、ここでRNAを次いで、例えばRNeasy FFPE Kit(QIAGEN、Hilden、Germany)を用いて、例えばシリカマトリックスに結合させることによって単離し得る。
【0032】
DNAおよび不完全に消化されたサンプルが有する他の未溶解成分を含む未溶解画分を、DNAを単離するために使用する。ここで、そのペレットは依然として本質的に固定サンプルの性質を有するので、DNAを固定サンプルから単離するための適当な、またはそのために習慣的な現在の技術水準によるあらゆる方法を用いることが可能である。特に、先行する不完全なプロテアーゼ消化は、サンプルから相当量のDNAを除去しなかった、および/または相当量のDNA架橋物を除去しなかった。このために、別のまたはさらなる酵素プロテアーゼ消化を有利に行ってサンプルを完全に溶解し、続いて例えばWO2007/068764において記載されたような求核試薬を含む溶液中で加熱インキュベーションする。この方式において放出されたDNAを、次いであらゆる適当な方法の助けによって、例えばQIAamp FFPE Kit(QIAGEN)を用いて、例えばシリカマトリックスへの結合によって、さらに精製し得る。
【0033】
この方式において、両方の型の核酸を、1つの工程で単一のサンプルから前もって分画し、そして次いでお互いに別々に単離し、そしてさらなる分析方法のために使用可能にする。
【0034】
本発明の目的のために、核酸という用語は、当業者に公知の全ての核酸、例えば天然または合成核酸、およびサンプルに人工的に導入された核酸、1本鎖および2本鎖核酸、直鎖、分枝、または環状核酸、RNA、特にmRNA、siRNA、miRNA、snRNA、tRNA、hnRNA、またはリボザイム、DNA、特にゲノムまたは色素体DNA、または細胞小器官のDNA、および感染由来の核酸も含む。
【0035】
適当な生物学的サンプルは、例えば、血液、精子、脳脊髄液、唾液、痰または尿などの細胞含有体液、白血球画分、バフィーコート、糞便、表面生検材料、吸引物、皮膚断片、生物全体、例えば剖検材料、生検材料、細針吸引物、または組織切片の形態の後生動物、好ましくは昆虫および哺乳類の、特にヒトの臓器および組織、例えば接着または懸濁した細胞培養物の形態の単離細胞、植物、植物の一部、植物組織または植物細胞、細菌、ウイルス、酵母および真菌のような、固定に適当な全ての生物学的サンプルである。
【0036】
本発明によるプロセスの最初の工程a)において、固定サンプルを、好ましくはタンパク質分解活性化合物の活性を可能にする水溶液と接触させ、そしてまた1つまたはそれより多いタンパク質分解活性化合物とも接触させる。
【0037】
本発明の目的のために、タンパク質分解活性化合物は、全てのタンパク質切断化合物、好ましくはプロテアーゼおよび熱安定性プロテアーゼ、特に好ましくはプロテイナーゼK、トリプシン、キモトリプシン、パパイン、ペプシン、プロナーゼ、およびエンドプロテアーゼLys−C、特にプロテイナーゼKのようなタンパク質分解活性酵素、およびまた臭化シアンのような、タンパク質を切断するために適当な非酵素的物質、またはこれらの物質の混合物である。
【0038】
水溶液中のタンパク質分解活性化合物の濃度は、一般的にそのタンパク質分解活性化合物の性質、およびその生物学的サンプルの性質および量に依存し、そしてその濃度は簡単な日常的な実験を用いて当業者が決定し得る。水溶液中のプロテアーゼ酵素の濃度は、それぞれの場合において水溶液の全重量に基づいて、好ましくは0.001重量%から5重量、特に好ましくは0.01重量%〜2.5重量%、そして特に0.05重量%〜0.2重量%の範囲である。ここで、ある特定のサンプルのために使用するタンパク質分解活性化合物の量または濃度は、そのタンパク質分解活性化合物の性質、および当業者が精通している幾つかのもの、pH、補助因子、インキュベーション温度、およびインキュベーション時間などの、選択した反応条件に依存する。そのタンパク質分解活性化合物の適当な量または濃度を、日常的な実験によって、簡単な方式で決定し得る。さらに、本発明によるプロセスにおいて、いかなる場合でも、タンパク質分解活性化合物の量または濃度を特に調整することは決定的ではなく、それは核酸の収量またはその完全性に悪い影響を与えることなく、ある特定のバンド幅で変動し得ることが見出された(実施例2)。
【0039】
その水溶液は、好ましくは、例えばカオトロピック剤、および/または好ましくは界面活性剤のような、生物学的組織の分解および/または細胞の溶解を促進するさらなる物質を含む。
【0040】
本発明によるプロセスにおいて使用するために適当な界面活性剤は、当業者に公知であり、そして細胞を溶解するために適当な全ての界面活性剤である;ここで陰イオン性または非イオン性界面活性剤が好ましい。
【0041】
好ましい界面活性剤は、ドデシル硫酸ナトリウム(SDS)、デオキシコール酸ナトリウム、3−(3−コールアミドプロピル)ジメチルアンモニウム−1−プロパンスルホネート(CHAPS)、例えばTritonX−100、TweenまたはNP−40の商品名で入手可能な界面活性剤などのポリエチレングリコールフェニルエーテル、またはこれらの混合物を含む群から選択される化合物であり、好ましい界面活性剤は、SDS、NP−40、およびTritonX−100(9から10のエトキシ化度を有するポリエチレングリコール(1,1,3,3−テトラメチルブチル)フェニルエーテル)である。生物学的サンプルに存在する細胞の溶解を補助するために使用される界面活性剤の量は、その生物学的サンプルの性質および量に依存し、そして簡単な日常的な実験を用いて当業者が決定し得る。
【0042】
その水溶液はさらに、好ましくは緩衝溶液であり、そのpHは、溶液中に存在する少なくとも1つの緩衝物質によって、6から9、好ましくは6.5から8.5、および特に好ましくは6.8から7.5の範囲に安定化される。よって、その水性緩衝溶液は好ましくは、好ましくはTris、Hepes、Pipes、Mops、アルカリ金属酢酸塩/酢酸等を含む群から選択される、少なくとも1つの緩衝物質、および/または好ましくは、好ましくはドデシル硫酸ナトリウム(SDS)、デオキシコール酸ナトリウム、3−(3−コールアミドプロピル)ジメチルアンモニウム−1−プロパンスルホネート(CHAPS)、ポリエチレングリコールフェニルエーテル、またはこれらの混合物、特に好ましくは、ドデシル硫酸ナトリウム、Tergitol−type NP−40の商品名で入手可能な、40のエトキシ化度を有するポリエチレングリコールノニルフェニルエーテル、および/または9〜10のエトキシ化度を有するポリエチレングリコール(1,1,3,3−テトラメチルブチル)フェニルエーテルを含む群から選択される、少なくとも1つの界面活性剤を含む。
【0043】
その水溶液はさらに、溶解を補助する、核酸を分解要素(constituent)に対して保護する、および/または水溶液を安定化する、さらなる成分、例えば錯体形成剤(complex former)、還元剤、または他の緩衝物質を含み得、ここで当業者は溶解バッファーのための可能性のある添加物の性質および量に精通している、またはそれらを簡単な日常的な実験によって決定し得る。好ましい実施態様において、その水性緩衝溶液はさらに、以下のものを含む群から選択される、少なくとも1つの物質を含む:
・錯体形成剤、好ましくはエチレンジアミン−N,N,N’,N’−4酢酸(EDTA)、エチレングリコールビス(2−アミノエチルエーテル)−N,N,N’,N’−4酢酸(EGTA)、クエン酸ナトリウム、またはこれらの混合物、
・好ましくは0.1から10Mの濃度の、好ましくは塩酸グアニジン、チオシアン酸グアニジン、イソチオシアン酸グアニジン、過塩素酸塩、NaI、KIおよび尿素を含む群から選択される、カオトロピック剤、
・好ましくはジチオスレイトール(DTT)、ジチオエリスリトール(DTE)、チオ硫酸ナトリウム、β−メルカプトエタノール、またはこれらの混合物を含む群から選択される、還元剤、および
・無機塩、好ましくは例えばNaCl、KCl、またはLiClのようなアルカリ金属ハロゲン化物、例えばCaCl2、またはMgCl2のようなアルカリ土類金属ハロゲン化物、例えば塩化アンモニウムまたは硫酸アンモニウムのようなアンモニウム塩、硫酸リチウム、またはこれらの混合物。
【0044】
1つの実施態様によって、その水性緩衝溶液は、界面活性剤、好ましくはSDSのような非イオン性界面活性剤、および好ましくは緩衝剤、好ましくはTRISを含む。その水性緩衝溶液はまた、EDTAのようなキレート剤を含み得る。
【0045】
本発明によるプロセスのプロセス工程a)において、架橋によって固定された生物学的サンプルを、少なくとも1つのタンパク質分解活性化合物を含む水溶液と接触させ、そして適当な温度でインキュベートする。ここで、その温度は一般的に、使用されるタンパク質分解活性化合物の性質に依存する。酵素の場合、その酵素が活性であることを可能にする温度を選択しなければならない。一般的に、低すぎる温度は酵素活性を不活性な段階まで抑制し、一方高過ぎる温度は、変性によって酵素を不活性化し得る。問題の酵素によって許容される温度範囲または最適な反応温度は、それぞれの酵素に依存して変動し、そして当業者に公知である、または簡単な日常的な実験によって決定し得る。例えばプロテイナーゼKを用いる場合、その反応を約95℃まで、好ましくは18℃および80℃の間、特に好ましくは50および65℃の間の温度で行い得る。
【0046】
既に記載したように、驚くべきことに、タンパク質分解活性化合物、好ましくはプロテアーゼを用いた、固定サンプルにおけるタンパク質のタンパク質分解「消化」の条件の調整を最適化することによって、サンプルから、DNAではなく、RNAのみを選択的に除去することが可能であることが見出された。サンプルの本発明によるこの不完全な(部分的な)消化の後、DNAを含む、依然として未溶解の画分を、RNAを含む溶解した上清から分離し得る。所定のタンパク質分解活性化合物に関して、RNAおよびDNAへの分離の質は、その濃度およびインキュベーション温度、および特にインキュベーション時間に依存する。もしそのタンパク質分解活性化合物を、非常に短時間のみ反応させたら、DNAだけでなく、RNAもサンプルから不十分にしか離れず、そして従って可溶性画分において全体で低い収量の核酸しか得られない。対照的に、もしそのタンパク質分解活性化合物を長く反応させすぎると、その結果は、RNAの(ほとんど)完全な溶解であるが、より多くのDNAもペレットから遊離する。しかし、そのタンパク質分解活性化合物の反応時間を適切に調整することによって、可溶性画分中のRNAおよび未溶解画分中のDNAの間を実質的に分離する、サンプルの部分的な(不完全な)溶解を達成することが可能であり、それは実際の精製の前の、2つの型の核酸の「予備分画」である。
【0047】
ここで、最適な反応時間は、まずそのタンパク質分解活性化合物、その水溶液中の濃度、およびインキュベーション温度に依存する。2番目に、その生物学的サンプルの量および厚さ、および他のサンプル特異的なパラメーター、例えば固定の型および期間が、そのタンパク質分解活性化合物の最適な反応時間に影響を有する。
【0048】
ホルマリンで固定したサンプルを、特にそのサンプルをパラフィンに包埋した後に使用することが好ましい。比較的大きい組織塊に関して、サンプルの量の多さおよび厚さのために、より小さいサンプルと比較して、タンパク質分解活性化合物を含むより多い容量の溶液、およびまた有利に、より高い濃度のタンパク質分解活性化合物、およびより長い反応時間を使用する。適当な切断装置、例えばミクロトームを使用して、固定サンプルから組織切片を調製し、ここで、光学顕微鏡で調査するための厚さは、一般的に約5μmから20μmである。さらに、パラフィン包埋サンプルをまた、他の方法を用いて、例えば中空針で穴をあけることによって、またはレーザーキャプチャー法によって、より小さいサンプル断片に分割し得る。より小さい組織断片は、より少量のタンパク質分解活性化合物およびより短い反応時間を必要とする。ここで特に切片の厚さが、組織とタンパク質分解活性化合物の完全な接触に関する制限因子であるので、決定的である。従って、好ましくは5μmから50μmの厚さを有する組織切片、またはもし適当なら、より大きなサンプルを分割またはホモジナイズすることによって得られる、より小さい組織断片を用いることが好ましい。
【0049】
固定時間、すなわち固定剤が生物学的サンプルに対して作用する時間は、生物学的サンプルにおいて生体分子の共有結合的架橋の程度に影響を与え、架橋の程度は、固定時間が長くなるほど増加する。短時間しか固定されなかったサンプルにおいては、従って少ない程度の生体分子の架橋しか存在せず、それは、個々の生体分子のより容易なおよびより迅速な溶解を可能にする。対照的に、長時間固定されたサンプルは、高い程度の架橋を有し、それは、特により大きな生体分子の溶解を遅延させ得る。よって、強く固定された(過固定)サンプルに関して、タンパク質分解活性化合物のより長い反応時間を有することが有用であり得る。ここで、最適な固定時間は、組織片のサイズに依存するので、「短時間固定された」および「長時間固定された」という用語は、相対的に理解される。組織におけるホルマリンの拡散速度は、最初約1mm/時間(h)であり、その速度は、組織深度が増加すると減少する。従って、約5mmの厚さの組織片に関して、ホルマリンがサンプルに完全に浸透するために約8時間が必要である(固定時間)。実際、約12〜24時間の固定時間が通例である;非常に小さなサンプルは、はるかにより短い固定時間を必要とし、そして12時間の固定時間で既に過剰固定である。
【0050】
従って、最適な反応時間は、固定および固定時間、生物学的サンプルの性質、量および厚さのような、サンプル特異的なパラメーターに依存し、そして個々のサンプルそれぞれに関して最適に調整し得る。驚くべきことに、それにも関わらず、多くの可能性のあるサンプルパラメーター、すなわち多くの異なる個々のサンプルに関して、DNAおよびRNAの未溶解および溶解画分への分離を可能にするように、その条件を調整することも可能である。ここで、その反応時間は、30秒および数日の間、好ましくは1分および5時間の間、そして特に好ましくは5分および90分の間、そしてより好ましくは10分および30分の間であり得る。600mAU/mlより大きい活性の、10μlから40μlのプロテイナーゼK溶液を、10μmから20μmの厚さのFFPE組織切片に関して、56℃のインキュベーション温度、および約15分から90分の反応時間で使用する場合、本発明の目的のために、サンプルの部分的溶解に関して非常に良い結果が得られる、すなわち、RNAは(ほとんど)完全に未溶解画分から溶解し、そして溶解画分に移動し、一方DNAは依然として(ほとんど)完全に未溶解画分に存在する(実施例2および3)。
【0051】
1つの実施態様によって、工程a)は、界面活性剤、好ましくは非イオン性界面活性剤を含む水性緩衝溶液中でのサンプルの部分的溶解と同時に、タンパク質分解活性酵素、好ましくはプロテイナーゼKのようなプロテアーゼを用いた、サンプルが有するタンパク質を含む成分の部分的なタンパク質分解を含み、ここでその反応を、18℃および80℃の間、好ましくは50℃から65℃の間の温度で、10分から5時間の間、好ましくは10から90分の間、より好ましくは10分から30分の間の反応時間で行う。この実施態様は、RNAの溶解画分への放出において迅速および有効であるという利点を有する。
【0052】
ここで、生物学的サンプルの固定を、当業者に公知のあらゆる固定剤によって、特に酸、アルコール、ケトン、または特にグルタルアルデヒドまたはホルムアルデヒドのような、他の有機物質によって行い得、ここで、ホルムアルデヒドで固定した生物学的サンプルが特に好ましい。本発明によるプロセスの特に好ましい実施態様によって、ホルムアルデヒド固定、パラフィン包埋生物学的サンプル(FFPEサンプル)を使用する。
【0053】
もしパラフィンに包埋した生物学的サンプルを使用するなら、そのパラフィンを好ましくは最初に、少なくとも部分的に、好ましくは完全に、サンプルから除去する。脱パラフィン化(deparaffinization)は、水性媒体中でサンプルを効率的に溶解するために、生物学的サンプルを包埋するために使用したパラフィンを選択的に除去するように働く。一般的に、パラフィンは核酸の溶解および分画の間、および核酸のさらなる精製および分析の間の両方に干渉し得る。好ましくは前もって行う脱パラフィン化は、プロテアーゼ処理後に本発明によるプロセスにおいて得られるペレットの質、特に固体性に、そして従って核酸の分離および得られる収量に顕著な影響を有し得る。
【0054】
生物学的サンプルからのパラフィンの除去を、原則として当業者に公知の、生物学的サンプルの脱パラフィン化のためのあらゆるプロセスによって行い得る。好ましくは、その脱パラフィン化を、最初にサンプルを疎水性有機溶媒に接触させることによって行う。ここで、生物学的サンプルおよび有機溶媒の混合物を、パラフィンのサンプルからの効率的な溶解を保証するために、例えば実験室振とう機で振とうすること、マグネティックスターラー等を使用することによって、撹拌しながら混合することが有利であり得る。有利に、そのサンプルを続いて遠心分離して、有機溶媒に溶解したパラフィンを、ペレット、すなわち生物学的サンプルから分離する。もし必要なら、生物学的サンプルからパラフィンを溶解する工程を、1回、2回、3回、または10回まで繰り返し得る。その脱パラフィン化を、例えば特許出願WO2007/068764において記載されたように、好ましくは疎水性有機溶媒、好ましくは芳香族炭化水素、特にキシレン中でインキュベーションし、続いてエタノール中でサンプルを再水和することによって行い得る。例えばアルカン類、好ましくは6<n<17である、一般式CnH2n+2の、室温で液体であるアルカン類またはこれらの混合物、特に好ましくはヘプタンのような、他の有機溶媒も、もし適当ならC1〜C5アルコール類、すなわちメタノール、エタノール、プロパノール、イソプロパノール、n−ブタノール、イソブタノール、n−ペンタノール、好ましくはメタノールを添加して、脱パラフィン化のために使用し得る。もし直鎖、すなわち未分枝のアルカンを使用するなら、6個またはそれより少ない炭素原子の鎖長のアルカンのいくつかは、毒性であると分類されており、室温でガス状であって、かつ/またはあまりにも揮発性であり、17個およびそれより多い炭素原子の鎖長のアルカンは、室温で固体であるので、nは好ましくは6より多く17未満である。もし適当なら、アルケン類、芳香族化合物等のような他の化合物が室温で液体であり、そして例えば鉱物油のようなパラフィンを溶解する限り、それらとのアルカンの混合物を使用することもさらに可能である。6個より多く17個未満の炭素原子の鎖長を有するアルカン類中で、特に好ましくはヘプタン中でインキュベートすることによる脱パラフィン化は、本発明に従うプロセスによる可溶性および不溶性画分AおよびBの引き続く分離のために特に有利であることが見出された。C1〜C5アルコール、好ましくはメタノールの、容量で1〜25%、好ましくは容量で5〜10%の量での、疎水性有機溶媒への添加は、不溶性残渣の沈殿を促進し、従って可溶性および不溶性画分の分離をより容易に、そしてより効率的になし得る。好ましい実施態様においては、それゆえそのプロセスは、工程(a)による部分的な溶解の前に、もし適当ならC1〜C5アルコール、好ましくはメタノールの、容量で1〜25%、好ましくは容量で5〜10%の量での添加を伴って、好ましくはサンプルを疎水性有機溶媒に接触させることによる、特に好ましくは6個より多く17個未満の炭素原子の鎖長の非極性脂肪族または芳香族炭化水素;特にキシレン、ヘプタン、および鉱物油を含む群から選択される炭化水素を用いた、パラフィンの選択的除去のための工程(i)を含む。1つの実施態様によって、脱パラフィン化を、アルカン、好ましくはヘプタン、およびアルコール、好ましくはメタノールとのインキュベーションによって達成する。適当な有機溶媒によるパラフィンの溶解に加えて、例えば、Biotechniques、18(1995)768−773頁において、Banerjeeらによって記載されたような、パラフィンの融解のような他のプロセスも、脱パラフィン化のプロセスとして適当である。
【0055】
パラフィンの除去後、生物学的サンプルを再水和することが好ましい場合があり、この再水和を、好ましくはアルコール濃度を次第に減少させた水性アルコール溶液(下降アルコール系列(descending alcohol series))で段階的に洗浄することによって行い、C1からC5アルコールが好ましく、そしてメタノール、エタノールおよびイソプロパノールが特に好ましい。もし使用する脱パラフィン化試薬がキシレンであるなら、そのサンプルを、さらなる処理の前に、通常この方式で再水和する。引き続く核酸単離のためのこの再水和の必要性は、関連する文献において議論されている。もし使用する脱パラフィン化試薬が直鎖脂肪族アルカンであるなら、サンプルの再水和は必要ない。しかし、単一の適当な試薬、例えば市販で入手可能な製品、BioGEnex、California、USAからのEZ−DEWAX(登録商標)で脱パラフィン化および再水和を行うことも可能である。
【0056】
好ましくは、そのサンプルは、脱パラフィン化および再水和の後、最初に、例えば空気への曝露または乾燥オーブンでのインキュベーションによって乾燥する。さらに、必要に応じて脱パラフィン化および再水和した生物学的サンプルを、好ましくは部分的な溶解の前にホモジナイズすることができ、それは特に比較的大きな組織サンプルの場合に有用である。対照的に、20μmの厚さまでの組織切片は、一般的にサンプルのホモジナイゼーションを必要としない。このホモジナイゼーションを、生物学的サンプルを細かく砕くための、当業者に公知のあらゆる装置、特に機械的破砕装置、例えばミル、ローター−ステーターホモジナイザー、Ultra−Turraxホモジナイザーまたは細いカニューレの補助を有する高圧細胞消化を用いて、または超音波ホモジナイザーによって行い得る。
【0057】
従って、好ましい実施態様において、そのプロセスは、工程(i)によるパラフィンの除去の後、および工程(a)による水性緩衝溶液におけるサンプルの部分的溶解の前に、好ましくは少なくとも以下の工程の1つを含む:
(ii)好ましくは、サンプルを、連続的に水含有量を増加させた水性C1〜C5アルコール溶液で繰り返し洗浄することによる、サンプルを再水和する工程、
(iii)サンプルを乾燥する工程および/または
(iv)サンプルをホモジナイズする工程。
【0058】
脱パラフィン化する、そして脱パラフィン化サンプルを作り上げるそれぞれの方法の工程も、先行技術において周知であり、そして従って、ここでさらなる説明を必要としない。
【0059】
1つの実施態様によって、架橋によって固定したサンプルを、脱パラフィン化の後にペレットの形態で得る。好ましくは、部分的溶解工程a)を行うために、水性緩衝溶液を、上記ペレットに加える。さらなる実施態様によって、脱パラフィン化化学物質を含む脱パラフィンされたサンプル(それぞれ脱パラフィン化溶液(deparaffinisation solution))を、工程a)において使用するための水性緩衝溶液と混合し、それによって水相を形成し、その水相に対し、工程a)において少なくとも1つのタンパク質分解活性化合物を用いて、上記サンプルが有するタンパク質を含む成分の部分的タンパク質分解を行なうことで、含まれるRNAを選択的に溶解画分に放出させ、一方含まれるDNAは主に未溶解画分に留まる。ここで、そのタンパク質分解活性化合物、好ましくはタンパク質分解酵素を、その水相に加えることができ、一方脱パラフィン化のために用いた溶液は依然として、水性緩衝溶液を添加したことにより形成された水相の上に存在する。もし、これの代替の分離工程b)を、例えば部分的に消化した架橋サンプルを遠心分離することによって(例えば上記および下記を参照のこと)、行うなら、主にDNAを含む未溶解画分は水相中でペレットを形成する。溶解画分を未溶解画分から分離するために、水相を、例えばピペットを使用することによって、例えば脱パラフィン化溶液によって回収し、他方では、未溶解の、主にDNAを含むペレットを残す。あるいは、脱パラフィン化溶液を、水性緩衝溶液の添加後に得られた水相から分離し、その後、タンパク質分解活性化合物を加え、そして未溶解画分を溶解画分から分離し得る。
【0060】
本発明によるプロセスの工程b)において、出発材料に存在する異なる型の核酸、すなわちRNAおよびDNAを、次いで、主にRNAを含む溶解画分(A)、および主にDNAを含む未溶解画分(B)に分離する。溶解および未溶解成分を含むサンプル全体を、少なくとも2つの画分に分離することも可能であり、次いで、それから様々な生体分子を単離または精製する、またはその中の様々な生体分子を次いで検出または分析し得る;しかし、そのサンプルを、本発明によるプロセスの工程a)の後に、好ましくは少なくとも1つの溶解画分(A)、および少なくとも1つの未溶解画分(B)に分離する。2つの画分を分離する利点は、それぞれの収率を減少させる、またはサンプルが有する様々な細胞型の不均一な分布をもたらす、もとの生物学的サンプルを分離する如何なる必要もなく、これらの2つの画分から、それぞれの場合別々に実質的に1つの型の核酸を単離し得ることである。
【0061】
この方式で得られた画分に対し、次いで別々に核酸の精製を行い得る。1つの画分からのみ核酸を単離し、そして他の画分を廃棄することも、本発明の範囲内である(例えば実施例6および7を参照のこと)。核酸の単離のための、サンプル(複数可)のさらなる処理の間に、そのサンプル(複数可)を、好ましくはタンパク質分解活性化合物の存在下で、50℃〜100℃、好ましくは55℃から95℃、特に好ましくは60℃から90℃、および特に65℃から85℃の範囲の温度に加熱する。
【0062】
未溶解成分の、水溶液からの分離は、特にもしそのタンパク質分解活性化合物が高い温度、すなわち室温より上の温度で活性であるなら、好ましくはタンパク質分解活性化合物の反応時間後に混合物を冷却することによって補助される。冷却を、好ましくはそのサンプルをプロテアーゼ消化の温度より下の温度で、好ましくは室温で、特に4℃で、または例えば−20℃または−80℃のようなさらにより低い温度でインキュベートすることによって行い、ここで、水溶液全体が凍結するのを避けるために、これらの温度で冷却するのは短時間である。従って、冷却を好ましくは15℃またはそれより低い、10℃またはそれより低い、4℃またはそれより低い温度で、または例えば−20℃または−80℃のような、さらにより低い温度で行う。冷却を、分離工程の前および/または分離工程の間に行い得る。冷却は、未溶解画分の分離、特にペレット化がより効率的であるという利点を有する。これは、FFPEサンプルは通常未溶解成分を含み、特にDNAは大量の固体成分ではなくタンパク質に架橋しているので、特に有利である。上記未溶解成分は、通常ペレット化するのが困難である。冷却は、未溶解成分のペレット化を補助し、そして従って分離をより効率的にする。従って、冷却は、個々の画分の分離が改善されるため、主にDNAを含む未溶解画分が、より多くのDNAを含むこと、そしてそれゆえ、RNAを含む溶解画分では、DNAの混入がより少なくなることがもたらされる。少しの細胞材料しか含まない架橋サンプルを処理する場合に、これは特に有用である。
【0063】
1つの実施態様によって、分離により、主にDNAを含む未溶解画分を、小型のペレットの形態で得ることになる。これにより、主にDNAを含むペレットを、主にRNAを含む溶解画分から容易に分離することが可能となる。
【0064】
3番目の工程において、この方式で得られたその溶解画分(A)および未溶解画分(B)を、お互い別々に使用して、存在する生体分子、好ましくは核酸(複数可)を精製し得る。ここで、溶解画分(A)を、好ましくはRNAを単離するために使用し、そして未溶解画分(B)を、好ましくはDNAを単離するために使用する。1つの画分からのみ核酸を単離し、そして他の画分を廃棄することも、本発明の範囲内である(例えば実施例6および7を参照のこと)。
【0065】
ここで、溶解画分(A)を、核酸単離のための適当なプロセスにおいて、さらなる溶解無しに、直接使用し得る。しかし、例えばタンパク質分解活性化合物、好ましくはプロテアーゼによるさらなる溶解を、必要に応じて水溶液中で行い得る。適当なプロセスは、当業者に公知である、核酸、特にRNAを単離するための全てのプロセスおよび方法である。特許出願WO2007/068764、WO2008/021419、WO2005/012523、またはWO2005/054466において記載されたような、固定サンプル材料から核酸を単離するためのプロセス、または別に、市販のキットRNeasyFFPEおよびmiRNeasyFFPE(どちらもQIAGENから)の補助によって行われるプロセスが適当である。後者の場合、シリカ膜へのカオトロープが媒介する結合によって核酸を精製する前に、画分Aを少なくとも1つの加熱工程にかける。RNAのさらなる抽出および精製を、好ましくは特許出願WO2007/068764において記載されたプロセスの補助によって行い得る。もし特許出願WO2007/068764において記載されたようなプロセスを、溶解画分からの核酸の単離のために使用するなら、そのサンプルを、求核試薬の存在下で加熱する。これを、少なくとも1つのタンパク質分解活性化合物および現在溶解した核酸を含む水溶液中で行い得、ここで特許出願WO2007/068764において記載されたプロセスのために必要な求核試薬を、本発明によるプロセスの工程a)(タンパク質分解活性化合物の作用)の後に、その水溶液に加え得る、またはさらには工程a)に従ってサンプルに加える前に、求核試薬がその水溶液に存在し得る。画分の分離を、特許出願WO2007/068764において記載されたようなサンプルの加熱の後に、または好ましくは、さらなる加熱の前の、タンパク質分解活性化合物を作用させた後に直接行い得る。
【0066】
適当な求核試薬は、電子をルイス酸の空の軌道に、または複数の空の軌道に移し得る、全てのルイス塩基である。これらのルイス塩基の中で、陰性の電荷を有する、陰性に分極している、または少なくとも1つの自由電子対を有する、少なくとも1つの官能基を有する試薬が特に好ましい。
【0067】
陰性の電荷を有する官能基を含む化合物は、例えばアルカリ金属アルコキシドまたはアルカリ土類金属アルコキシド、アルカリ金属水酸化物またはアルカリ土類金属水酸化物、アルカリ金属ハロゲン化物またはアルカリ土類金属ハロゲン化物、アルカリ金属シアン化物またはアルカリ土類金属シアン化物等であり、それらに制限されない。
【0068】
陰性に分極した少なくとも1つの官能基を有する試薬は、特にお互いに共有結合した2つの原子を含み、そしてそのAlfredおよびRochowによる電気陰性度が、少なくとも0.25、好ましくは少なくとも0.5、そしてより好ましくは少なくとも1.0異なる、少なくとも1つの官能基を有する試薬である。
【0069】
しかし、本発明によって、1つまたは2つ、特に好ましくは1つの自由電子対(複数可)を有する、少なくとも1つの官能基を有する求核試薬が、特に好ましく、そして今度はこれらの化合物の中で、少なくとも1つの1級アミノ基、2級アミノ基、または構造Iの3級アミノ基を有するものが最も好ましい
【0070】
【化1】

ここで、R1はC1からC20炭化水素基、特に好ましくはC2からC15炭化水素基、そしてより好ましくはC2からC10炭化水素基、少なくとも1つのヘテロ原子を有するC1からC20炭化水素基、少なくとも1つのヘテロ原子を有するC2からC15炭化水素基、そしてより好ましくは少なくとも1つのヘテロ原子を有するC2からC10炭化水素基、または必要に応じてヘテロ原子で置換された芳香環系である、
R2は、C1からC20アルキル基、特に好ましくはC1からC10アルキル基、そしてより好ましくはC1からC2アルキル基、特にメチル基またはエチル基、C1からC20ヒドロキシアルキル基、特に好ましくはC1からC10ヒドロキシアルキル基、そしてより好ましくはC1からC2ヒドロキシアルキル基、または水素原子であり、水素原子が最も好ましい、および
R3は、C1からC20アルキル基、特に好ましくはC1からC10アルキル基、そしてより好ましくはC1からC2アルキル基、特にメチル基またはエチル基、C1からC20ヒドロキシアルキル基、特に好ましくはC1からC10ヒドロキシアルキル基、そしてより好ましくはC1からC2ヒドロキシアルキル基、または水素原子であり、水素原子が最も好ましい。
【0071】
本発明によって、上記で示した構造Iの官能基を有する求核試薬が特に好ましく、それは特に少なくとも1つの構造Iの官能基を有し、構造IのうちででラジカルR2およびR3のうち少なくとも1つ、最も好ましくは両方のラジカルR2およびR3が、水素原子である。それに加えて、少なくとも1つの構造Iの官能基を有する求核試薬が特に好ましく、ここで構造Iの窒素原子は、ラジカルR1、R2およびR3のsp3混成原子に共有結合しているのみである。特に、ラジカルR1、R2およびR3はどれも、それぞれラジカルR1、R2およびR3を超えて窒素原子の自由電子対を非局在化する(delocalising)ことはできない。従って、特に好ましくは、ラジカルR1、R2およびR3はどれも、例えば構造IIを有さない。
【0072】
【化2】
本発明によって、メチルアミン、エチルアミン、エタノールアミン、n−プロピルアミン、n−ブチルアミン、イソブチルアミン、tert−ブチルアミン、ジメチルアミン、ジエチルアミン、ジエタノールアミン、ジ−n−プロピルアミン、ジイソプロピルアミン、ジブチルアミン、トリメチルアミン、トリエチルアミン、トリエタノールアミン、ヘキサメチレンテトラミン、2−エチルヘキシルアミン、2−アミノ−1,3−プロパンジオール、ヘキシルアミン、シクロヘキシルアミン、1,2−ジメトキシプロパンアミン、1−アミノペンタン、2−メチルオキシプロピルアミン、トリ(ヒドロキシメチル)アミノメタン、アミノカルボン酸、特にグリシンまたはヒスチジン、またはアミノグアニジンを含む群から選択される、少なくとも1つの構造Iの官能基を有する求核試薬が特に好ましく、ここで最後に述べたものは、可能であるが、好ましくはない。これらの中で、エタノールアミン、ジエタノールアミン、トリエタノールアミン、アミノ−1,3−プロパンジオールおよびトリ(ヒドロキシメチル)アミノメタンが最も好ましい。少なくとも1つの構造Iの官能基を有する、好ましい求核試薬は、さらにアニリン、トルイジン、ナフチルアミン、ベンジルアミン、キシリジン、キシレンジアミン、ナフタレンジアミン、トルエンジアミン、3,3’−ジメチル−4,4’−ジフェニルジアミン、フェニレンジアミン、2,4’−メチレンジアニリン、4,4’−メチレンジアニリン、スルホニルジアニリン、およびジメチルベンジルアミンを含む群から選択される、芳香族アミンである。
【0073】
その求核試薬が少なくとも1つの構造Iの1級アミノ基を有する、本発明によるプロセスの特定の実施態様によって、その求核試薬は、C1からC6アルキルアミン、C1からC6アルキルジアミン、C1からC6アルキルトリアミン、C1からC15アミノアルコール(aminoalkohol)、C1からC15アミノジオール、またはC1からC15アミノカルボン酸である。
【0074】
本発明によるプロセスの別の特定の実施態様によって、その求核試薬は、窒素原子を含むヘテロ環状化合物であり、そしてピロール、ピリジン、キノリン、インドール、アザシクロペンタン、アザシクロヘキサン、モルホリン、ピペリジン、イミダゾ−ルまたはこれらの化合物の誘導体を含む群から選択され、ここでこれらの化合物の誘導体は、好ましくは水素原子の代わりに、上記で述べた化合物の、1つまたはそれより多い炭素原子、または窒素原子に結合した、C1からC3アルキル基、特に好ましくはメチルまたはエチル基を有する化合物を意味すると理解される。
【0075】
上記で述べた求核試薬の中で、水溶性であるもの、特に25℃の温度、およびpH7において、水中で少なくとも1g/l、特に好ましくは少なくとも10g/l、そしてより好ましくは少なくとも100g/lの溶解度を有するものが好ましい。
【0076】
使用する水溶液中での求核試薬の濃度は、好ましくは0.1から10000mmol/l、より好ましくは1から5000mmol/l、さらにより好ましくは5から2500mmol/l、および最も好ましくは20から1000mmol/lの範囲である。本発明によるプロセスの特に有用な実施態様によって、水溶液中の求核試薬の濃度は、20mmol/l超、特に好ましくは50mmol/l超、そして最も好ましくは100mmol/l超である。
【0077】
1つの実施態様によって、好ましくはタンパク質分解酵素である、タンパク質分解活性化合物による消化を行った後、サンプル中の架橋は、好ましくは少なくとも70℃、より好ましくは少なくとも75℃、最も好ましくは少なくとも80℃、または少なくとも90℃の温度まで、少なくとも5分、好ましくは少なくとも10分、最も好ましくは少なくとも15分の期間加熱することによって、少なくとも部分的に逆行する(reversed)。少なくとも15分間80℃に加熱することが、分解したサンプルのRNAを含む溶解画分において架橋を逆行させるために特に好ましい。少なくとも30分から数時間まで、好ましくは少なくとも1.5、または少なくとも2時間、少なくとも85℃、好ましくは少なくとも90℃に加熱することが、DNAにおいて架橋を逆行させるために好ましい。上記で述べたように、求核試薬の存在下で加熱を行う。適当なインキュベーション時間も、引用された先行技術において記載される。架橋を逆行させるためのこのさらなる加熱工程を、主にRNAを含む溶解画分を、主にDNAを含む未溶解画分から分離する前または後に行い得る。この加熱工程は、さらなるDNAが未溶解画分から放出されることをもたらし得るので、特に続いて未溶解画分からDNAを単離することも意図するなら、画分を分離した後に上記加熱工程を行うことが好ましい。さらに放出されたDNAは、例えばDNase消化を行うことによって分解され得るので、RNAを単離することのみを意図するなら、上記加熱工程を、画分を分離する前にも行い得る。小さいRNA分子も保存する、DNase消化を行うための好ましい実施態様を、下記で詳細に説明する。
【0078】
1つの実施態様によって、DNase消化を、分離した、主にRNAを含む溶解画分に対して行う。架橋サンプルに含まれる主な量のDNAを含む未溶解画分を分離することにより、既に上記サンプルに含まれるDNAの主な部分が除去される。従って、部分的な消化および画分の分離の後に得られる、主にRNAを含む溶解画分は、既にDNAが枯渇している。工程a)において部分的消化の間に放出されたかもしれない、残りの量のDNAを、RNAを含む溶解画分に対して、DNase消化を行うことによって効率的に分解し得る。RNAをDNase消化サンプルから単離することにより、ほとんどまたは全くDNAの混入のない、純粋なRNAが提供される。
【0079】
従って、1つの実施態様によって、DNaseを、分離した主にRNAを含む溶解画分に加える。DNase消化を、RNAを単離する前に効率的に行い得ることは、非常に驚くべきことであった。これは、通常の先行技術の方法ではRNAを精製するときRNAを単離してからDNase消化を行っているように、DNaseが、効率的には機能しないことが予測されたからである。さらに、例えば通常のオンカラムのDNase処理と比較して、特に小さいRNAの量が、本発明によるプロセスを用いる場合に増加するので、RNAを単離する前にDNase消化を行うことはまた、かなりの利点を有する。それぞれのDNase消化を、実施例6および7で行う。好ましくは、そのDNase消化を、上記で記載したように加熱によって架橋を逆行させた後に行う。
【0080】
「DNase」という用語は、DNAにおけるホスホジエステル結合の加水分解切断を触媒するあらゆる酵素を指す。多種多様なデオキシリボヌクレアーゼが公知であり、それらはその基質特異性、化学的メカニズム、および生物学的機能が異なる。「DNase」という用語は、エキソデオキシリボヌクレアーゼおよびエンドデオキシリボヌクレアーゼを指す。特に、DNaseIおよびDNaseIIを使用し得る。DNaseIが好ましい。
【0081】
DNase消化を、DNAの効率的な分解を可能にするために、DNaseが活性である条件下で行う。DNase消化の効率を、例えば分解サンプルに加えるDNaseの量によって、およびさらに、特にMgイオンおよびCaイオンのような、DNaseの活性を促進する添加物を加えることによって、調整し得る。さらに、工程a)において部分的消化を達成するために使用する条件に依存して、分離した、主にRNAを含む溶解画分に対して、DNase消化が高い効率で作用することを保証するために、中間の処理工程が有用であり得る。例えば、DNase消化に干渉し得る成分を、DNase消化を阻害しない濃度まで除去または希釈し得る。DNase消化を、DNaseが活性である濃度のMgイオンおよびCaイオンの存在下で行う。例えば、DNase消化を行うために、MgイオンおよびCaイオンを、例えばMgCl2およびCaCl2の形態で、分解サンプルに加えて、DNase消化混合物において適当な濃度を確立し得る。MgイオンおよびCaイオンの適当な濃度は、サンプル、および特に分解工程a)において使用する溶解条件に依存する。例えば、もしCaイオンおよびMgイオンが、工程a)における消化の間に既に提供され、そして従って、分解サンプルに存在するなら、より少ない量のMgイオンおよびCaイオンをDNase消化のために加えなければならない、またはMgおよびCaの添加はさらに必要でない。もし、例えばEDTAのようなキレート剤を、工程a)の間に使用するなら、DNase消化の間により高い濃度のMgイオンおよびCaイオンを使用することが賢明である。1つの実施態様によって、そのMgイオンおよびCaイオンは、好ましくはMgCl2およびCaCl2の形態で、少なくともそれぞれ0.2mM、少なくともそれぞれ2mM、少なくともそれぞれ5mM、少なくともそれぞれ7.5mM、および好ましくは少なくともそれぞれ10mMから成る群から選択される濃度で、反応組成物中で提供される。さらに、そのCaイオンおよびMgイオンを、0.2mMから1M、2mMから100mM、10mMから50mM、および10mMから25mMから成る群から選択される、各イオンの濃度範囲で提供し得る。DNase、分解サンプルおよび必要に応じて、DNase消化を促進するさらなる添加物を含むDNase消化反応組成物を、適当な時間インキュベートして、DNAが分解されることを可能にする。好ましくは、そのインキュベーションは、少なくとも5分、少なくとも好ましくは10分、または少なくとも15分間行う。適当な範囲は、1分から6時間、5分から120分、10分から60分、および15分から30分を含む。随意選択のDNase消化を行った後、RNAをサンプルから単離し得る。本明細書中で議論するように、基本的にあらゆるRNA単離方法を使用し得る。
【0082】
1つの実施態様によって、そのRNAを、溶解した、必要に応じてDNase処理した画分から、適当な添加物を加えることによって適当な結合条件を確立すること、および核酸結合固相にRNAを結合することによって、単離する。1つの実施態様によって、RNAの単離は、少なくとも以下の工程を含む:
i)少なくとも1つのアルコールおよび/または少なくとも1つのカオトロピック剤、および必要に応じてさらなる添加物を加えて、結合混合物を形成する工程、およびその結合混合物を、核酸結合固相と接触させて、RNAを上記固相に結合させる工程;
ii)必要に応じてRNAが固相に結合している間にRNAを洗浄する工程;および
iii)必要に応じてその固相からRNAを溶出する工程。
【0083】
核酸結合固相として、核酸を結合し得るあらゆる材料を使用し得、そして従って適当な条件下で核酸を結合し得る、様々な材料を含む。本発明と関連して使用し得る代表的な固相は、シリカ粒子、二酸化ケイ素、珪藻土、ガラス、アルキルシリカ、ケイ酸アルミニウム、およびホウケイ酸塩を含むがこれに限らない、シリカおよびケイ質固相を含む化合物、ニトロセルロース;ジアゾ化紙;ヒドロキシアパタイト(ヒドロキシルアパタイトとも呼ばれる);ナイロン、金属酸化物;ジルコニア;アルミナ;ポリマー支持体、ジエチルアミノエチル−およびトリエチルアミノエチル誘導体化支持体、疎水性クロマトグラフィー樹脂(フェニルまたはオクチルSepharoseのような)等を含むがこれに限らない。固相という用語は、その形態またはデザインに関していかなる制限も意味することを意図しない。従って、固相という用語は、膜、フィルター、シート、粒子、磁気粒子、ビーズ、ゲル、粉末、繊維等を含むがこれに限らない、多孔性または非多孔性、透過性または不透過性の、適当な材料を包含する。1つの実施態様によって、その固相の表面は、改変されていない、そして例えば官能基によって改変されていない。好ましい実施態様によって、その核酸結合固相は、カラムに含まれる。本明細書中で使用される「カラム」という用語は、特に少なくとも2つの開口部を有する容器を説明する。それによって、溶液および/またはサンプルが、上記カラムを通過し得る。「カラム」という用語は、特に容器の形に関していかなる制限も意味せず、それは例えば円形または角であり得、および好ましくは円柱状であり得る。しかし、特に多層カラムを使用する場合、他の形も使用し得る。そのカラムは、核酸結合固相を含む。上記カラムに含まれる上記固相は、カラムにアプライした場合に溶液、それぞれサンプルの通過を可能にする。これは、もし例えば遠心力をカラムにかけたら、溶液および/またはサンプルは、遠心力の方向にカラムを通過し得ることを意味する。上記で議論したように、それぞれのカラムに基づく核酸単離手順を用いる場合、そのサンプルは、例えば遠心分離または減圧によって補助されて、カラムを通過し、そして核酸は、上記通過の間に、含まれる核酸固相に結合する。そのカラムを、単一の形式で、または多層の形式で使用し得る。マルチウェルプレートと同様の形式を有し、そして膜のような核酸結合固相を含む、そのような多層カラムは、先行技術において周知である。好ましくは、そのカラムはスピンカラムである。カラムに含まれる核酸結合固相として、通常カラムに基づく核酸単離手順で利用される、あらゆる固相を使用し得る。好ましくは、核酸結合膜、および従って核酸を結合し得る膜を使用する。適当な膜は、親水性膜、疎水性膜、およびイオン交換によって核酸を結合する膜を含むが、これに限らない。例は、シリカ膜、ガラス繊維膜、ナイロン膜、ニトロセルロース膜、改変セルロース膜(例えばアセチル−またはヒドロキシ−)のようなセルロース膜、紙膜、特に改変紙を含むがこれに限らない。好ましくは、その膜は多孔性である。さらに、シリカを含む、またはシリカから成る膜を使用することが好ましい。カラムに含まれる、さらなる通常の核酸結合固相は、シリカ粒子のような、核酸結合粒子の充填、または核酸結合材料(例えばシリカゲル)の層である。例えば、そのシリカ粒子を、不活性なフィルターまたは膜上に層として配置し、それによって核酸結合固相を形成し得る。
【0084】
サンプルの未溶解成分を消化し、そして生体分子の残った架橋を切断するために、もし適当なら、未溶解成分を含む画分(B)に、好ましくはさらなる処理を行う。実質的に依然として未溶解画分(B)にある、溶解可能な核酸、主にDNAを、このさらなる処理工程によって単離し得る。固定組織から核酸を溶解するために公知のあらゆるプロセス、例えばWO2007/068764、WO2008/021419、WO2005/012523、またはWO2005/054466において記載されたようなプロセスが適当である、または他に市販で入手可能なキット、例えばQIAamp DNA FFPE Tissue Kits(QIAGEN)の補助によって行い得る方法が、核酸、特にDNAを、未溶解画分(B)から単離するために適当である。後者の場合、画分Bの未溶解成分に、タンパク質分解剤、例えばプロテアーゼによる、少なくとも1つのさらなる処理、および加熱工程を行う。そのプロテアーゼ処理は、効率的な溶解、および従って溶解可能な核酸の放出を行う。未溶解画分Bは、完全な固定サンプルと比較して、特により容易に溶解可能でない成分しか含まないので、さらなる最適化、例えばさらなるプロテアーゼ工程および加熱工程の延長が有用であり得、そして著しく改善した収量および引き続く(下流の)分析における結果をもたらす。
【0085】
従って、1つの実施態様によって、DNAを、画分の分離後に、未溶解の主にDNAを含む画分から得る。DNAを未溶解画分から得ることは、1つの実施態様によって以下の工程を含む:
i)上記未溶解画分に、同時酵素プロテアーゼ消化による溶解を行うことによって、DNAを、未溶解の、主にDNAを含む画分から放出する工程であって、ここで好ましくは、溶解の間に少なくとも1つの界面活性剤、および必要に応じてさらなる添加物を使用し、そしてここで、その酵素消化を、好ましくは加熱によって補助する(適当な条件を上記で(abive)述べる)工程;
ii)架橋を少なくとも部分的に逆行させるために、主にDNAを含む画分を、好ましくは工程i)の後に、好ましくはサンプルを加熱することによって、少なくとも70℃、より好ましくは少なくとも80℃、最も好ましくは少なくとも85℃、より好ましくは少なくとも90℃の温度まで、求核試薬の存在下で加熱する工程、および
iii)架橋を逆行させた後に、好ましくは適当な添加物を加えることによって結合条件を確立すること、およびDNAを核酸結合固相に結合させることによって、DNAを単離する工程。好ましくは、カオトロピック剤および界面活性剤、好ましくは非イオン性界面活性剤およびアルコールを、結合条件を確立するために加える。適当なDNA単離手順はまた、先行技術において周知である。
【0086】
従って、好ましくは、本発明によるプロセスは、工程b)に続いて、画分Aから得られるRNAおよび/またはペレットBから得られるDNAを、好ましくは沈殿、核酸の適当な結合材料への結合、電気泳動、および/またはクロマトグラフィーまたはその組み合わせによって、別々に精製するためのさらなる工程を含む。
【0087】
さらに、そのプロセスは、好ましくは、単離および/または精製した核酸の分析/検出のための工程を含む。
【0088】
当業者に公知である全ての分析方法、例えばPCR、qPCR、RT−PCR、qRT−PCRおよびゲノムDNA全体の増幅(全ゲノム増幅)のような増幅技術、ゲル電気泳動、ブロッティング技術、特にサザンブロッティングおよびノーザンブロッティング、マイクロアレイ分析、制限酵素断片長多型解析(RFLP分析)、SAGE(遺伝子発現の連続分析(serial analysis of gene expression))、NextGeneration配列決定およびRNA配列決定を含む配列決定、一塩基多型分析(SNP分析)、突然変異分析、エピジェネティック分析、特にメチル化パターンの分析、またはその組み合わせを、本発明によるプロセスによって単離した核酸を分析するために使用し得る。
【0089】
上記の開示から明らかになるように、本発明はまた、以下の工程を含む、溶解画分中のRNAおよび未溶解画分中のDNAを、架橋によって固定した同じ生物学的サンプルから得るためのプロセスを提供する:
a)少なくとも1つのタンパク質分解活性化合物を使用した、サンプルが有するタンパク質を含む成分の部分的なタンパク質分解と同時に、水性緩衝溶液中でサンプルを部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット、画分B)を得る工程、
b)溶解画分を未溶解画分から分離する工程、
ここで、溶解画分における核酸の全量に基づいて、その溶解画分は主にRNAを含み、そして未溶解残渣における核酸の全量に基づいて、その未溶解残渣は主にDNAを含む。
【0090】
1つの実施態様によって、主にRNAを含む画分を、主にDNAを含む画分から分離することは、沈殿も、1つまたは両方の型の核酸の有機溶媒による抽出も、1つまたは両方の型の核酸の固体マトリックスへ選択的結合も必要としない。1つの実施態様によって、画分の分離後、RNAを、主にRNAを含む溶解画分から単離し、および/またはDNAを、主にDNAを含む未溶解画分から単離する。もし、例えばRNAの単離のみを意図するなら、主にDNAを含む未溶解画分を、分離後に廃棄し得る。
【0091】
適当なおよび好ましい実施態様、ならびに本発明による部分的消化および分離工程に関連する利点、ならびに引き続く核酸単離の適当なおよび好ましい実施態様を、同じ架橋サンプルからのRNAおよびDNAの並行単離および/または精製のプロセスに関して、上記で詳細に議論した。それは上記の開示に参照され、それもここで適用する。
【0092】
本発明はさらに、少なくとも(1)タンパク質分解活性化合物、好ましくは上記で述べたタンパク質分解活性化合物の1つ、(2)少なくとも1つの緩衝物質、好ましくは上記で述べた緩衝物質の1つ、および(3)少なくとも1つの界面活性剤、好ましくは上記で述べた界面活性剤の1つ、およびまた好ましくは(4)工程(a)による不完全なタンパク質分解を行うための指示を含む、本発明によるプロセスを行うためのキットを含む。
【0093】
特に好ましい実施態様において、本発明によるキットは、さらに(5)少なくとも1つの核酸結合材料、およびまた必要に応じて(6)核酸単離のためのバッファー、好ましくは結合および/または溶出バッファーを含む。
【0094】
核酸結合材料(5)として使用するために適当なのは、DNAまたはRNAの吸着に関して当業者に公知である全ての材料であり、セルロースに基づく材料、特にカルボキシ官能基(funktional)セルロース材料、またはジエチルアミノエチルセルロース、アガロース、シリカ、ガラス、石英、ゼオライト、または金属酸化物のようなミネラルキャリア、またはイオン交換体材料でコーティングしたキャリアが特に好ましい。記載した材料は、例えば膜、または磁気粒子または非磁気粒子の形態で存在し得る。その核酸結合材料は、好ましくは組み立て式カラムのカラム材料として、または別に懸濁物としてキットに含まれる。材料の型は、分析する核酸の化学的構造に決定的に依存し、当業者は、それぞれの意図する適用、すなわちRNAまたはDNAの分析に関して、各場合において適当な吸着材料に精通している。
【0095】
溶出バッファー(6)として、本発明によるキットは、当業者に公知であり、そして核酸の核酸結合材料からの溶出に習慣的に使用されるあらゆるバッファーを含み得る。その溶出バッファーは、好ましくは水性塩溶液、特に例えばNaCl、KCl、またはLiClのようなアルカリ金属ハロゲン化物、例えばCaCl2またはMgCl2のようなアルカリ土類金属ハロゲン化物、例えば塩化アンモニウムまたは硫酸アンモニウムのようなアンモニウム塩、またはこれらの塩の少なくとも2つの混合物を含む水溶液であり、ここでその溶出バッファーは、必要に応じてまた、例えばアルカリ金属酢酸塩/酢酸に基づくバッファーシステム、またはトリス(ヒドロキシルメチル)アミノメタンに基づくバッファーシステムを含み得る。もしそのキットを、固定組織からRNAを単離するために使用するなら、そして使用するマトリックスがシリカ膜であるなら、溶出バッファーとして水、特にRNaseを含まない水を使用することが特に好ましい。
【0096】
結合バッファー(6)として、本発明によるキットは、当業者に公知であり、そして核酸を核酸結合材料に結合させるために習慣的に使用されるあらゆるバッファーを含み得、ここでその結合バッファーは、使用するそれぞれの核酸結合材料とマッチしていなければならない。もし使用する核酸結合材料がシリカマトリックスであるなら、その結合バッファーは、好ましくはカオトロピック剤、および必要に応じてC1〜C5アルコールをさらに含む。
【0097】
本発明によるキットを、生物学的サンプルに存在する核酸、すなわちDNAおよびRNAの両方を分析および/または定量化するために使用し得る。
【0098】
この理由のために、本発明によるキットをさらに、ヒトまたは動物の体の外のサンプルの、疾患の診断、予後、治療に関する決定、および/または治療のモニタリングのために使用し得る。
【実施例】
【0099】
実施例1:本発明のプロセスによるRNAおよびDNAの分離
使用するサンプルは、包埋の後約4ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasy FFPEキットおよびQIAamp FFPEキットの構成成分を使用した。
【0100】
脱パラフィン化のために、その組織をまず1mlのキシレン中で10分間インキュベートした。サンプルの遠心分離によるペレット化、および上清の除去後、このキシレンによるこの処理を、後2回繰り返した。続いて各場合においてサンプルを無水エタノールで2回、続いて水性エタノール溶液で処理して(まず96%エタノール、および次いで70%エタノール)、そして37℃で10分間乾燥した。
【0101】
この方式で得られた脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTA、および0.2%のSDS(w/v)(pH7)を含む150μlの水溶液で処理し、そしてタンパク質分解活性化合物として10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。得られた混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルを遠心分離し、そして上清(画分A)を、未溶解成分(画分B)を含むペレットから除去した。
【0102】
2つの画分における、核酸の型、RNAおよびDNAの分布を決定するために、上清およびペレット由来のRNAを、各場合4つのサンプル(サンプル1〜4)から単離し、そしてさらなる4つのサンプル(サンプル5〜8)から、上清およびペレット由来のDNAを単離した。
【0103】
サンプル1〜4の上清(画分A)(サンプル1aから4a)からRNAを単離するために、その上清を80℃で15分間インキュベートした。DNA結合のための条件に調整するために、320μlのカオトロピックバッファー、例えばQIAGENからのRBCバッファーを加えた。その混合物を、例えばQIAGENからのgDNA除去カラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによってその膜を通過させた。混合物の組成は、DNAのシリカ膜への選択的結合を引き起こすので、RNAは、カラムの溶出液にある。RNAのための結合条件に調整するために、この溶出液を、エタノールと混合し、そして次いでもう1度、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによってその膜を通過させた。次いでそのシリカ膜を、各場合において500μlのアルコールを含む洗浄バッファーRW2(QIAGEN)で2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そして1分のインキュベーション後、RNAを30μlの水によって溶出した。
【0104】
RNAをサンプル1〜4のペレット(画分B)(サンプル1bから4b)から単離するために、そのペレットを、界面活性剤および求核試薬を含む、QIAGENからのPKDバッファー150μl、および10μlのQIAGENからのプロテイナーゼKと混合した。56℃で15分間インキュベートし、そして次いで80℃で15分間インキュベートした後、その溶解産物を、カオトロープを含む結合バッファー、例えばQIAGENからのRBCバッファーで処理し、そしてその混合物を、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして10000rpmで1分間遠心分離することによって、膜を通過させた。上記で記載したように、そのシリカ膜を、洗浄バッファーRW2で2回洗浄し、そしてRNAを溶出した。
【0105】
DNAをサンプル5〜8の上清(画分A)(サンプル5aから8a)から単離するために、その上清を、界面活性剤を含む溶解バッファーATL(QIAGEN)で、180μlの全容量にし、そして20μlのQIAGENからのプロテイナーゼKを加えた。そのサンプルを、1400rpmで振とうしながら、56℃で1時間インキュベートし、そして次いで90℃で1時間加熱した。存在するあらゆるRNAを分解するために、インキュベーション後に、4μlのRNAseA溶液(100mg/ml)をサンプルに混合した。DNAをさらに精製するために、そのサンプルを、各場合で200μlのカオトロピックバッファー、例えばQIAGENからのALバッファー、およびエタノールと混合した。その混合物を、例えばQIAGENからのQIAamp MinEluteカラムに存在するシリカ膜にアプライし、そして10000rpmで1分間遠心分離することによって膜を通過させた。次いでそのシリカ膜を、500μlのグアニジン塩を含む洗浄バッファーAW1で、そして次いで500μlのQIAGENからのアルコールを含む洗浄バッファーAW2で洗浄した。その膜を、14000rpmで2分間遠心分離することによって乾燥し、そして1分間のインキュベーション後、DNAを30μlのDNA溶出バッファー、例えばQIAGENからのバッファーATEと遠心分離することによって溶出した。
【0106】
DNAを、サンプル5〜8のペレット(画分B)(サンプル5bから8b)から単離するために、そのペレットを、例えば180μlのQIAGENからの界面活性剤を含むバッファーATLのような、習慣的なDNA溶解バッファーで処理した。ペレットは、前の処理によって溶解されなかった成分のみを含むので、20μlのQIAGENからのプロテイナーゼKによるさらなる溶解を、これらの未溶解成分も溶解するように行った。56℃で1時間、および続いて90℃で1時間インキュベートした後、その溶解産物を、カオトロープを含む結合バッファー、例えばQIAGENからのALバッファーおよびまたエタノールで処理した。その混合物を、例えばQIAGENからのQIAamp MinEluteカラムに存在するシリカ膜にアプライし、そして10000rpmで1分間遠心分離することによって、膜を通過させた。上記で記載したように、そのシリカ膜を、バッファーAW1およびAW2で洗浄し、その膜を乾燥し、そしてDNAを溶出した。
【0107】
2つの画分、上清(A)およびペレット(B)からこの方式で単離した核酸の分布を決定するために、両方の画分の核酸を、適当な方法を用いて定量化した。DNAおよびRNAの収率および純度の決定を、サンプルの吸収を260/280nmで測定することによる吸光度(OD)によって行った。各場合の収率を、全収量のパーセントで示した。ここで全収量は、上清およびペレットにおける1つの型の核酸の収量の合計である。表1は、4回の決定の平均値を示す。
【0108】
【表1】
さらなる分析のために、各場合において、それぞれの画分から単離した核酸10μlを、DNAの場合はTAEアガロースゲル(Tris酢酸塩/EDTA)上で、RNAの場合はホルムアルデヒド/アガロースゲル上で、習慣的な方法によって分離し、そしてエチジウムブロミドで染色した。その結果を図1に示す。
【0109】
その結果は、本発明によるプロセスを適用した場合、引き続く精製の前に核酸の明らかな分離/分画が存在することを示す。RNAは主に上清に存在し、一方DNAは主にペレット画分に存在する。
【0110】
実施例2:使用したタンパク質分解活性化合物の量の影響
この実験のために使用したサンプルは、包埋の後約7ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約10μmの厚さの切片を、これらのサンプルから調製した。各場合において反応あたり2つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0111】
脱パラフィン化のために、その組織をまずそれぞれ1mlのヘプタン中で10分間インキュベートした。50μlのメタノールを加え、そして混合した後、そのサンプルを遠心分離し、上清を除去し、そして残渣を室温で5分間風乾した。
【0112】
この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μl、20μl、または40μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで4℃で遠心分離した。RNAのさらなる単離のために、その上清を除去し、そしてペレットを廃棄した。
【0113】
その上清を、続いて80℃で15分間インキュベートした。結合条件に調整するために、320μlのカオトロピックバッファー、例えばQIAGENからのRBCバッファーを次いで加え、そして得た混合物をエタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。次いで500μlのアルコールを含む洗浄バッファーRW2を通過させることによって、そのシリカ膜を2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥した。次いでRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0114】
この方式で単離したRNAを分析するために、260nmにおける吸収を測定することによって、その収量を決定した。その結果を表2に示す。
【0115】
RNAの完全性を、Agilent Bioanalyzerを用いて決定し、そしてRIN値の形式で示した、ここで10のRIN値は、完全にインタクトなRNAを示し、そして0のRIN値は、完全に分解されたRNAを示す。その結果を同様に表2に示す。
【0116】
本発明によるプロセスにおける核酸の単離に対するだけでなく、増幅による引き続く分析に対しても、異なる量のタンパク質分解活性化合物の影響を調査するために、そのRNAを、定量的リアルタイムRT−PCRによって分析した。この目的のために、その単離されたRNAを、各場合2回の決定で、madH7転写物のアンプリコンを検出するために使用した。その溶出液をそれぞれ、水で1:10の比で希釈した。各場合において、5μlのこれらの希釈溶液を、リアルタイム−PCRのために使用した。製造会社の指示によって、例えばQIAGENからのQuantiTect SYBRGreen RT−PCRキットのような、リアルタイムRT−PCRのために適当なマスターミックスによって、25μlの全容量で、増幅をおこなった。例えば、Applied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標) 7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において、増幅をおこなった。測定したct値を用いて、平均値を決定し、それを表2に示す。
【0117】
【表2】
その結果は、使用したプロテイナーゼKの全ての量が、同等の収量、同等のRNA完全性、および同等のリアルタイムRT−PCRの結果をもたらしたことを示す。本発明によるプロセスにおけるタンパク質分解活性化合物の量は、従って広い範囲で変更することができる。
【0118】
実施例3:タンパク質分解活性化合物の反応時間
この実験のために使用したサンプルは、包埋後約5ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0119】
切片の脱パラフィン化、再水和、および乾燥を、実施例1に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で3時間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルを遠心分離し、そしてその上清(画分A)を、未溶解成分(画分B)を含むペレットから除去した。実施例1に記載したように、RNAを上清(画分A)から単離し、そしてDNAをペレット(画分B)から単離した。
【0120】
この方式で単離した核酸を分析するために、260nmにおける吸収を測定することによって、その収量を決定した。その結果を図2に示す。プロテイナーゼの反応時間を増加させると、より長いプロテイナーゼ作用によって、より多くの核酸がペレットから溶解するので、核酸の収量はペレットにおいて減少し、そしてそれに対応して上清において増加する。
【0121】
しかし、核酸のこの全体分布は、画分におけるDNAおよびRNA含有量についての情報は提供しない。従って、各場合において、DNAを含む画分(B)の溶出液10μlを、TAE−アガロースゲルで分析した。その結果を図3に示す。
【0122】
5分のみのプロテイナーゼK反応時間後には、RNAの大部分は依然として未溶解画分、すなわちペレットに留まっていることが明らかである。プロテイナーゼ反応時間を増加させると、この量が減少し、そして反応時間15分から先は、未溶解画分は少量のRNAしか含まないか、またはRNAは実質的にもはや検出されない。対照的に、DNAはかなりより長く(少なくとも90分間)、未溶解画分Bに留まる。従って、タンパク質分解活性化合物の反応時間を最適化することによって、2つの画分における核酸の型の分布を調整することが可能である。
【0123】
核酸の単離に対するだけでなく、増幅による分析にも対する、本発明によるプロセスにおけるタンパク質分解活性化合物の反応時間の長さの影響を調査するために、そのDNAを定量的リアルタイムPCRによって分析した。この目的のために、各場合において、等容量の単離DNA溶出液を、各場合2回の決定で、prnp遺伝子のアンプリコンを検出するために使用した。例えばQIAGENからのQuantiTect SYBRGreen PCRキットのような、リアルタイムRT−PCRに適当なマスターミックスを用いて、製造会社の指示に従って、25μlの全量で増幅を行った。例えば、Applied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標) 7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において、増幅をおこなった。ct値から決定した平均値を、図4に示す。
【0124】
その結果は、反応時間が増加すると、ct値が増加することを示し、それは溶出液中のDNAの量が減少したためである。反応時間を増加させると、RNAだけでなくDNAも徐々に上清へ移るが、ここでRNAは、DNAよりも著しくより迅速に、上清に溶解および移動する(encounter)。ゲルは、15分の反応時間後でも、RNAは既に実質的に完全にペレットから溶解し、そして上清に移動していることを示すが、未溶解画分BのDNAの量は、ゲルおよびリアルタイムPCRによると、90分超後にのみ有意に減少する。
【0125】
実施例4:本発明によるプロセスにおける、異なる水溶液の使用
この実験のために使用したサンプルは、包埋後約7ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0126】
切片の脱パラフィン化、再水和、および乾燥を、実施例2に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液1(pH7)、50mMのTris、25mMのEDTA、1%のSDS、0.1%のNonidet NP40、および500mMのNaClを含む水溶液2(pH7.4)、または50mMのTris、100mMのEDTA、3%のSDSおよび10mMのNaClを含む水溶液3(pH8.2)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。その混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで4℃で遠心分離した。DNAを、実施例1で記載したようにペレット(画分B)から単離し、そのペレットを50mMのTris、25mMのEDTA、1%のSDS、0.1%のNonidet P−40、および500mMのNaClを含むバッファー(pH7.4)中に溶解した。その収量を、260nmにおける吸収を測定することによって決定した。その結果を表3に示す。
【0127】
【表3】
使用した3つの水溶液は全て、高い収量のDNAを与え、一定の変動がサンプルの不均一性によって生じた。本実施例は、多数の異なる水溶液を、本発明によるプロセスのために使用し得、その成分および濃度の両方を変動させることが可能であることを示す。従って、使用する水溶液を、使用するタンパク質分解活性化合物に適合させることが可能である。
【0128】
実施例5:本発明によるプロセスの補助による、異なる型の組織からのDNAの単離
使用したサンプルは、室温で異なる期間保存されていた、ラット由来のFFPEサンプルであった:腎臓(保存期間約13ヶ月)、肝臓(保存期間約6ヶ月)、脾臓(保存期間約19ヶ月)、心臓(保存期間約13ヶ月)、および肺(保存期間約6ヶ月)。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルそれぞれから調製した。各場合において、反応あたり1つの切片を使用した。本発明による方法の補助による、処理したサンプルからの引き続く核酸の単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0129】
本発明によるプロセスの補助によるDNAの単離を、FFPEサンプルからのDNAの精製のために特に確立されたプロセスと比較するために、各場合において同じサンプルの切片を、製造会社(QIAGEN)の指示による、QIAampFFPEキットによるDNA単離のために、およびコントロールサンプルとして使用した。
【0130】
切片の脱パラフィン化、再水和、および乾燥を、実施例2に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。その混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで遠心分離した。DNAを、90℃で2時間インキュベートして、実施例1で記載したようにペレット(画分B)から単離した。
【0131】
得られたDNAの分析のために、その収量を、260nmにおける吸収を測定することによって決定した。2組の決定の平均値および標準偏差を図5に示す。本発明によるプロセスの補助によって、DNAを全てのサンプルから単離し得、全ての場合の収量は、コントロールプロセスで得られた収量を超えた。
【0132】
さらに、各場合において、10μlのDNA溶出液を、TAE−アガロースゲルで分離し、そしてエチジウムブロミドで染色した。その結果を図6に示す。全ての場合において、本発明による方法の補助によって単離されたDNAは、コントロール方法で単離されたDNAと、およそ同じ分子サイズ分布を示した。
【0133】
本発明によるプロセスによって単離されたDNAの増幅分析に対する適合性を調査するために、この方式で得られたDNAを、定量的リアルタイムPCRアッセイにおいて使用した。等容量の単離DNA溶出液を、pmp遺伝子の465塩基対のアンプリコンを検出するために、2組の決定において使用した。
【0134】
FFPEサンプルにおいて、そのDNAは、原則として断片化形態で存在し、単離し得るDNA断片の断片化の程度および従ってスペクトルは、特に固定および包埋の性質に依存するが、またサンプルの種類およびサンプルの保存にも依存する。さらに、核酸の単離後に残るDNAの架橋の程度が、増幅、および特にアンプリコンの可能性のあるサイズを制限する。これにも関わらず効率的な増幅を保証するためには、原則として小さいアンプリコンが好ましい。ここで使用した465bpというアンプリコンサイズは、FFPEサンプルに関しては非常に大きく、そしてそれを本発明によるプロセスによって単離されたDNAの質および適合性を試験するために選択した。
【0135】
増幅を、例えば、QIAGENからのQuantiTect SYBRGreen PCRキットのような、リアルタイムRT−PCRに適当なマスターミックスを用いて、製造会社の指示に従って、25μlの全容量で行った。増幅を、例えばApplied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標)7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において、増幅をおこなった。測定したct値を用いて、本発明によって単離されたDNAの平均値および標準偏差を決定し、それを図7に示す。
【0136】
全ての場合において、ct値は、コントロールDNAのものと同等であるか、またはさらにより低く、そのことはより良い増幅能力(amplifiability)および/またはより高い収量を確実にする。
【0137】
全体として、その結果は、本発明によるプロセスにより、FFPEサンプルからのDNAの単離が可能となること、収量、質、断片サイズ、および増幅分析への適合性に関し、FFPEサンプルからのDNAの特異的単離について、先行技術から公知のプロセスによって単離したDNAと少なくとも同じ、またはより良好であるということを示す。
【0138】
実施例6:本発明によるプロセスによる、異なる型の組織からのRNAの単離
この実験のために使用したサンプルは、異なる期間室温で保存されていた、ラット由来のFFPEサンプルであった:腎臓(保存期間約5ヶ月)、肝臓(保存期間約24ヶ月)、心臓(保存期間約24ヶ月)、および肺(保存期間約24ヶ月)。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。本発明のプロセスの補助による、FFPE切片からの引き続く核酸の単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0139】
本発明によるプロセスの補助によるRNAの単離を、FFPEサンプルからのRNAの精製のために特に確立されたプロセスと比較するために、同じサンプルの切片を、製造会社(QIAGEN)の指示による、RNeasy FFPEキットによるRNAの単離のために、およびコントロールサンプルとして使用した。
【0140】
切片の脱パラフィン化、再水和、および乾燥を、実施例2に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで遠心分離した。RNAのさらなる単離のために、その上清(画分A)を除去し、そしてペレットを廃棄した。
【0141】
その上清を続いて80℃で15分間インキュベートした。そのサンプルを室温で5分間冷却し、その後20μlの従来のDNAseバッファー(例えば0.46MのTris−HCl(pH7.5)、114mMのNaCl、114mMのMgCl2、114mMのCaCl2を含む)、15μlの脱イオン水、および5μlのQIAGENからのDNAseI溶液を加え、そしてその混合物を室温で15分間インキュベートした。400μlのカオトロピックバッファー、例えばQIAGENからのRLTバッファーを次いで加え、その混合物をエタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。500μlのアルコールを含む洗浄バッファーRW2(QIAGEN)で、そのシリカ膜を2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そしてRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0142】
この方式で単離したRNAを分析するために、260nmにおける吸収を測定することによってその収量を決定した。2組の決定の平均値を、表4に示す。
【0143】
【表4】
本発明によるプロセスの補助によって、RNAを全てのサンプルから単離することが可能であり、ここで、全ての場合において、本発明によるプロセスによって得られた収量は、コントロールのものと同等、またはそれより高かった。
【0144】
本発明によるプロセスによって単離されたRNAの、増幅分析のための適合性を調査するために、そのRNAを、定量的リアルタイムRT−PCRアッセイにおいて使用した。等容量の単離RNA溶出液を、各場合においてmadH7転写産物およびc−jun転写産物のアンプリコンを検出するために、2組の決定において使用した。増幅を、例えば、QIAGENからのQuantiTect SYBRGreen RT−PCRキットのような、リアルタイムRT−PCRに適当なマスターミックスを用いて、製造会社の指示に従って、25μlの全容量で行った。増幅を、例えばApplied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標)7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において行った。それに加えて、microRNA16(miR16)を、リアルタイムRT−PCRによって、製造会社(QIAGEN)の指示に従って、miScript PCRシステムを用いて、RNA溶出液において検出した。測定したct値から得た平均値を、表5に示す。
【0145】
【表5】
全ての場合において、本発明によって処理したサンプルの測定したct値は、コントロールサンプルのものと同等であるか、またはさらにより低く、そのことはよりよい増幅能力またはより多いRNA量によるものである。
【0146】
実施例7:効率的なmiRNA精製のためのDNAse処理
この実験のために、異なる期間室温で保存していたラット由来のFFPEサンプルを使用した:脳(保存期間約5ヶ月)および心臓(保存期間約18ヶ月)。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。本発明のプロセスの補助による、FFPE切片からの引き続く核酸の単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0147】
本発明によるプロセスの補助によるmiRNAの単離を、FFPEサンプルからのmiRNAの精製のために特に確立されたプロセスと比較するために、同じサンプルの切片を、製造会社(QIAGEN)の指示による、miRNeasy FFPEキットによるmiRNAの単離のために、およびコントロールサンプルとして使用した。
【0148】
この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。主にRNAを含む溶解画分(A)を、主にDNAを含む未溶解画分(B)から分離するために、そのサンプルをまず氷上で3分間冷却し、そして次いで遠心分離した。miRNAを含むRNAのさらなる単離のために、上清(画分A)を除去し、そしてDNAを含むペレットを廃棄した。
【0149】
続いて上清を80℃で15分間インキュベートして、架橋を逆行させた。そのサンプルを室温で5分間冷却し、その後DNase活性を促進するための異なるバッファー(前処理バッファー1〜5、下記を参照のこと)を20μl、15μlの水、および5μlのQIAGENからのDNAseI溶液を加えた。この実験のために以下のバッファーを使用した:
前処理バッファー1:0.46MのTris−HCl(pH7.5)、114mMのNaCl、114mMのMgCl2、114mMのCaCl2
前処理バッファー2:0.46MのTris−HCl(pH7.5)、114mMのMgCl2、114mMのCaCl2
前処理バッファー3:46mMのTris−HCl(pH7.5)、11.4mMのNaCl、11.4mMのMgCl2、11.4mMのCaCl2
前処理バッファー4:20mMのTris−HCl(pH7.5)、100mMのMgCl2、10mMのCaCl2
前処理バッファー5:20mMのTris−HCl(pH7.5)、100mMのMgCl2、2.5mMのCaCl2
その混合物を、室温で15分間インキュベートした。次いでDNase消化サンプルから、マイクロRNAのような小さいRNAを含むRNAを単離するために、400μlのカオトロピックバッファー、例えばQIAGENからのRLTバッファーを加え、その混合物を1400μlの96〜100%エタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。500μlのアルコールを含む洗浄バッファーRPE(QIAGEN)で、そのシリカ膜を2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そしてRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0150】
比較のために、DNAse前処理をしないが、RNAの膜への結合の後に通常のオンカラムDNAse処理を行う、小さいRNAを含むRNAの精製のために、同じサンプルを使用した。脱パラフィン化およびプロテイナーゼK消化を、上記で記載したように行った。その後、320μlのカオトロピックバッファー、例えばQIAGENからのRLTバッファーを次いで加え、その混合物を1120μlの96〜100%エタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。バッファーRWT(QIAGEN)のような、カオトロピック試薬およびエタノールを含む350μlの洗浄バッファーで、そのシリカ膜を洗浄した。10μlのDNase1および適当なDNAseバッファー(例えばバッファーRDD(QIAGEN))を含む80μlの混合物を、次いでその膜にアプライし、そして室温で15分間インキュベートした。その後、その膜を再びバッファーRWTで洗浄し、そして500μlのアルコールを含む洗浄バッファーRPE(QIAGEN)で2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そしてRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0151】
この方式で単離したRNAを分析するために、代表的な脳のRNAを、RNA分子をサイズに依存して分離する、Agilent Bioanalyzerを用いて分析した。図8は、Bioanalyzer測定の結果を示す。FFPEサンプル由来のRNAは常に、部分的に分解されており、そして分解の程度は、サンプルの固定、包埋および保存、およびRNAの抽出方法のような複数の因子に依存する。従って、RNAのゲル様の視覚化は、全ての場合において、部分的に分解したRNAを示す(図8を参照のこと)。28SrRNAは明らかでなく、そして18SrRNAは弱く現れるのみである。それに加えて、多数のRNA断片が、28srRNAのバンドのサイズから低分子量まで生じる。通常のオンカラムDNAse処理は、miRNAを含む最も小さいRNA集団の、非常に低い収量をもたらす(矢印を参照のこと)。対照的に、本発明によるカラムに添加する前のDNAse前処理は、非常に低分子量のRNAの多量の単離を可能にする。
【0152】
特にmiRNAの精製の効率を決定するために、miScript PCR Systemを用いて、製造会社(QIAGEN)の指示に従って、リアルタイムRT−PCRによって、精製RNAを、miRNAの検出および定量に関して分析した。測定したct値から得られる平均値を、表1に示す。
【0153】
【表6】
全ての場合において、測定したct値は、DNAse前処理をしたサンプルにおいてより低く、一方オンカラムDNAse処理は、有意により高いct値を与える。より低いct値は、より多量のmiRNAを示し、ct値の1の差異は、検出されたmiRNAの約2倍の量を示す。従って、RNAを単離する前のDNAse前処理は、従来技術によるオンカラムDNase消化に対して、miRNA精製効率を有意に増強する。
【0154】
全体として、その結果は、本発明によるプロセスにより、FFPEサンプルからのRNAの単離が可能となること、収量、質、断片サイズおよび増幅分析への適合性に関し、先行技術から公知の単離プロセスによって単離したRNAと、少なくとも同程度に良好であり、かつFFPEサンプルからのRNAの単離に特異的であることを示す。
【技術分野】
【0001】
本発明は、同じ固定生物学的サンプルからの、RNAおよびDNAの並行単離および/または並行精製のためのプロセス、本発明によるプロセスによって単離された核酸の定量化および分析、固定サンプルからのRNAおよびDNAの並行単離および/または精製のためのキット、ならびに疾患の診断、予後、治療に関する決定、および/または治療のモニタリングのための、このキットの使用に関する。
【背景技術】
【0002】
もし、例えば組織断片または単離細胞のような生物学的材料を、生きた生物から除去した場合、その細胞は、短期間で死ぬ。非常に迅速に、死んだ細胞は、まず自己融解/発酵によって、そして次いで細菌によって分解され、もとの細胞および組織構造は破壊される。従って、もし細胞または組織断片を、組織学的調査のために生物から除去するなら、分解を防止するために、取った生物学的サンプルを固定することが推奨される。理想的には、固定は、サンプルの構造を、実質的に変化させないで、その組織学的評価を可能にする。さらに固定は、サンプルの長期保存および記録保管を可能にする。これらの理由のために、多くの形態学的調査は、固定した材料に基づいてのみ可能である。
【0003】
通常、例えば酸、アルコール、ケトン、またはアルデヒド、特にグルタルアルデヒドまたはホルムアルデヒドのような、タンパク質沈殿またはタンパク質架橋化合物を用いて、固定を達成する。ここで、ホルムアルデヒド(「ホルマリン」と呼ばれる水溶液の形態で使用される)による固定に続く、固定サンプルのパラフィンへの包埋は、細胞および組織構造が特に良好に保存されるので、特に病理学において非常に重要である。本明細書の以下で、この方式で固定した材料を、「ホルマリン固定、パラフィン包埋材料」または「FFPE材料」と呼ぶ。
【0004】
しかし、特にホルマリンによるサンプルの固定は、ホルムアルデヒドの架橋効果のために、タンパク質だけでなく、サンプル中に存在する核酸を含む様々な他の生体分子も、お互いに共有結合し、そして結果として、そのようなサンプルからの核酸(DNAまたはRNA)の単離は、非常に困難であるという欠点を有する。しかし、分子レベルにおける多くの研究に関して、核酸の単離は非常に重要である。
【0005】
そのような固定サンプルから核酸を単離する1つの方法が、特許文献1において記載されている。そこに記載された方法は、生物学的サンプルにおいて固定によって形成された架橋を切断し、そして1つの型の核酸、すなわちDNAまたはRNAのいずれかを単離することを可能にし、続いて例えばPCRまたはRT−PCR分析を行い得る。
【0006】
例えば腫瘍障害の診断または予後に関する分子病理学の分野において、DNAに基づく分析、およびRNAに基づく分析の両方が使用される。同じ固定サンプル、例えば腫瘍サンプルについてDNAおよびRNAに基づく分析の両方を可能にするために、例えば生検からの組織切片のような1つのサンプルからのDNAおよびRNAの並行単離を可能にするプロセスが必要である。まず、通常非常に少量のサンプル材料しか入手可能でなく、複数の別個の精製のためには不十分であるので、そのような単一のサンプルからのDNAおよびRNAの並行単離は、非常に望ましい。2番目に、サンプル材料の組成は、一般的に不均一である;例えば健康な細胞のマトリックス中には非常に少数の腫瘍細胞しか存在しない。この場合、異なる細胞のお互いに対する比が、それぞれの部分的なサンプルにおいて同じであることを保証することは不可能であるので、サンプルを分けることは望ましくない。単一の、分割していないサンプルからのDNAおよびRNAの並行単離のみが、研究する全ての被検体が同じ比で存在し、そして同じ組成のサンプル由来であることを保証する。
【0007】
特許文献1において記載されたプロセスは、固定の間に導入された架橋を切断することによって、両方の型の核酸、DNAおよびRNAを等しく放出する。従ってこのプロセスは、DNAまたはRNAのいずれか、または両方の核酸の混合物の単離のみを可能にする;しかし、それは別の画分におけるDNAおよびRNAの並行単離を可能にしない。2つの型の核酸(DNAおよびRNA)を分離するために、特許文献1は、単離後に、精製の間に同時に放出された2つの型の核酸の1つの、選択的沈殿または選択的吸着を示唆する。あるいは、それぞれの不必要な型の核酸を酵素的に分解することが可能である。
【0008】
選択的吸着による、サンプルからのDNAおよびRNAの並行精製の1つの方法が公知であり、そして例えば市販で入手可能なAllprep DNA/RNAキット(Qiagen、Hilden、Germany)を用いて行い得る。ここで、そのサンプルを最初に、いかなるアルコールも含まないカオトロープを含む溶解バッファーで溶解し、そして溶解産物中に存在するDNAはシリカマトリックスに結合し、一方これも溶解産物に存在するRNAは結合しないで溶液中に残る。残った溶解産物にアルコールを加えた後、RNAはさらなるシリカマトリックスに結合し得る。このプロセスは、非固定サンプルに関してよく作用する。しかし、ここでDNAは最初のシリカマトリックスに定量的に結合せず、多くの量が残った溶解産物に残り、そしてRNAと共に精製されるので、それは依然としてホルマリン固定サンプルに最適に適用できないことが見出された。従って、このプロセスは、RNAおよびDNAの別々の精製を可能にしない。
【0009】
特許文献2は、FFPEサンプルを含む生物学的サンプルから、DNAおよびRNAを同時抽出するための方法を記載する。そのFFPEサンプルを脱パラフィン化し、そしてカオトロピック剤、イオン性界面活性剤、およびタンパク質分解酵素を含む溶解バッファーを用いて消化する。RNAおよびDNAを放出するために、少なくとも5時間、好ましくは10時間サンプルを消化し、フェノール−クロロホルムを加え、そして相を分離する。水相は主にRNAを含み、有機相は主にDNAを含む。次いでアルコール沈殿を用いて、RNAを水相から回収し得る。DNAを有機相から回収する。この方法は、特に、上記核酸を有機相および水相から別々に単離する前に、DNAからRNAを分離し得るために、フェノールを用いて放出された核酸を抽出する必要があるという欠点を有する。
【0010】
同様のプロトコールが、O’Sheaら、「Analysis of Tcell receptor beta chain CDR3 size using RNA extracted from formalin fixed paraffin wax embedded tissue」J Clin Pathol 1997;50:811−814において記載されている。
【0011】
DNAおよびRNAの混合物、またはDNAまたはRNAのいずれかを、FFPEサンプルから単離するための、市販で入手可能なキットが同様に公知である。FFPE RNA/DNA Purification Kit(Norgen、Biotek Corp.、Thorold、Canada)は、1つの溶出液中で、RNAおよびDNAの混合物の単離を可能にする。ここで、DNAのみ、またはRNAのみのいずれかを得るために、特に長いプロテアーゼK消化およびRNAse処理を行ってDNAを単離し得る、または短いプロテアーゼK消化およびDNAse処理を行ってRNAを単離し得る。しかし、このキットは、同じサンプルから、両方の型の核酸の、同時であるが別々の精製を可能にしない。
【0012】
Agencourt FormaPure Kit(Beckman Coulter Genomics GmbH、Danvers、Massachusetts、USA)も、1つの溶出液におけるRNAおよびDNAの混合物の単離、またはDNAse消化後のRNAの単離を可能にするが、DNAおよびDNAの同時であるが別々の単離を可能にしない。
【0013】
対応するヌクレアーゼ消化によって、または適当な加熱インキュベーションによって、DNAまたはRNAのいずれかを得るためのオプションも、Ambion Recover AII FFPE Kit(Applied Biosystems,Inc.、Foster City、California、USA)およびQuickExtract FFPE RNA Extraction Kit(Epicentre Biotechnologies、Madison、Wisconsin、USA)によって提供される。しかし、これらの市販で入手可能なキットはいずれも、同じ固定サンプルからDNAおよびRNA両方を別々に精製することを可能にしない。
【0014】
さらに、特許文献2は、同じサンプルから、両方の型の核酸(RNAおよびDNA)を同時に抽出するためのプロセスを記載し、それはまたとりわけ固定サンプルであり得る。このプロセスは、サンプルの溶解および芳香族アルコールを用いた酵素不活性化の後、適当な抽出プロセスによる2つの型の核酸の分離を含む。しかし、このプロセスで得られた2つの相(RNAを含む水相、およびDNAを含む有機相)から、次いでさらなる精製および/または単離の前に、適当な沈殿剤の添加によって、核酸を沈殿させなければならない。まず、これはそのプロセスを時間のかかるものにし、そして2番目に、沈殿のために、物質の損失のリスクおよび/または核酸に沈殿剤が混入するリスクが存在する。
【0015】
よって、架橋によって固定された同じサンプルから、DNAおよびRNAの両方を別々に精製することを可能にするプロセスを提供することが本発明の目的であり、DNAのRNAからの分離は、有機溶媒も、核酸を結合するための固体マトリックスも必要としない。
【先行技術文献】
【特許文献】
【0016】
【特許文献1】国際公開第2007/068764号
【特許文献2】国際公開第2005/075642号
【発明の概要】
【課題を解決するための手段】
【0017】
本発明は、とりわけ少なくとも1つのタンパク質分解活性化合物を用いた、架橋によって固定された生物学的サンプルが有するタンパク質を含む成分の部分的なタンパク質分解は、RNAをサンプルの溶解画分へ選択的に放出することを可能にし、一方DNAは主に上記サンプルの未溶解残渣に残るという発見に基づく。サンプルの上記部分的消化は、別々の画分を得ることを可能にし、ここで溶解画分は主にRNAを含み、そして未溶解残渣は主にDNAを含む。主にRNAを含む溶解画分を、主にDNAを含む未溶解残渣から、例えば遠心分離プロセスを用いて容易に分離し得る。
【0018】
溶解した画分を未溶解画分から分離した後、その画分を望ましいように別々に処理し得る。例えば、RNAを未溶解画分から単離し得、そしてDNAを未溶解画分から単離し得る。個々の画分から核酸を単離する前に、主にRNAを含む溶解画分を、主にDNAを含む未溶解画分から分離することは、同じ架橋サンプルから良好な収率でRNAおよびDNAを効率的に単離することを可能にする。1つの画分からのみ核酸を単離し、そして他の画分を廃棄することも、本発明の範囲内である。例えば、RNAの単離が関心の的であるなら、DNAを含む未溶解画分を、分離後に廃棄し得る。
【0019】
本出願の他の目的、特徴、利点および局面は、以下の記載および添付の特許請求の範囲から当業者に明らかになる。しかし、以下の記載、添付の特許請求の範囲、および特定の実施例は、本出願の好ましい実施態様を示すが、例示としてのみ提供されることが理解されるべきである。開示された本発明の意図および範囲内の様々な変化および改変は、以下を読むことから当業者に容易に明らかになる。
【図面の簡単な説明】
【0020】
【図1】図1は、それぞれTAE−アガロースゲル(DNA)およびホルムアルデヒド−アガロースゲル(RNA)における、エチジウムブロミドによる染色後の、本発明によるプロセスによって上清およびペレットからそれぞれ単離されたRNAおよびDNAの分離を示す(実施例1)。サイズ比較のために、サイズマーカーLambda/HindIII(Invitrogen、Carlsbad、California、USA)を、レーンMにアプライした。
【図2】図2は、タンパク質分解活性化合物の反応時間の関数として、画分A(上清)および画分B(ペレット)から単離された核酸の全収量を示す(実施例3)。
【図3】図3は、タンパク質分解活性化合物の反応時間の関数としての、TAE−アガロースゲルにおける、画分B中のRNA含有量の分析の結果を示す(実施例3)。
【図4】図4は、ct値の変化としての、定量的リアルタイムPCRによる、画分Bから得られたDNAの増幅に対する、タンパク質分解活性化合物の反応時間の影響を示す(実施例3)。
【図5】図5は、市販で入手可能なキットを用いて行ったプロセスにおける収量と比較して、UV分光法によって決定された、本発明によるプロセスを用いて、各場合において示される期間保存された様々な型の組織から単離し得るDNAの収量を示す。(実施例5)。
【図6】図6は、TAE−アガロースゲルにおける、実施例5によって得られたDNAの分析を示す。サイズ比較のために、サイズマーカーLambda/HindIII(Invitrogen、Carlsbad、California、USA)を、レーンMにアプライした。
【図7】図7は、リアルタイムPCR分析による、実施例5によって得られたDNAの分析を示す。
【図8】図8は、単離したRNAのAgilent Bioanalyzer分析を示す。1−5:実施例1において記載された前処理バッファー1−5を用いて、DNAseにより前処理したサンプル。oc:DNAse前処理なしであるが、先行技術において通常であるように、オンカラムDNAse処理したサンプル。
【発明を実施するための形態】
【0021】
上記で議論したように、架橋によって固定された同じサンプルからDNAおよびRNAの両方を別々に精製することを可能にするプロセスを提供することが、本発明の1つの目的であり、DNAのRNAからの分離は、有機溶媒も、核酸を結合するための固体マトリックスも必要としない。
【0022】
この目的を、架橋によって固定された同じ生物学的サンプルからの、リボ核酸(RNA)およびデオキシリボ核酸(DNA)の並行単離および/または精製のためのプロセスによって達成し、それは以下の工程:
a)少なくとも1つのタンパク質分解活性化合物を用いた、サンプルが有するタンパク質を含む成分の部分的なタンパク質分解と同時に、水性緩衝溶液中においてサンプルを部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット;画分B)を得る工程;
b)溶解画分を、未溶解残渣から分離する工程、
を含み、
ここで、溶解画分における核酸の全量に基づいて、その溶解画分は主にRNAを含み、そして未溶解残渣における核酸の全量に基づいて、その未溶解残渣は主にDNAを含み、
そしてここで、主にRNAを含む画分の、主にDNAを含む画分からの分離は、有機溶媒による1つまたは両方の型の核酸の沈殿も抽出も、1つまたは両方の型の核酸の固体マトリックスに対する選択的結合も必要としない。
【0023】
本発明によるプロセスの目的のために、並行単離および/または精製は、2つの型の核酸、RNAおよびDNAの単離および/または精製が、お互いに空間的に離れて起こる、1つは主にRNAを含み、そして他方は主にDNAを含む、その2つの画分AおよびBの処理が、同時に、または異なる時点で起こり得る、単離および/または精製を意味すると理解される。
【0024】
本発明の目的のために、架橋によって固定された同じ生物学的サンプルは、工程(a)において部分的溶解を受けるサンプル全体を意味すると理解される。
【0025】
本発明の目的のために、「部分的溶解」および「部分的タンパク質分解」または「部分的消化」という用語はそれぞれ、下記でより詳細に説明するように、サンプルまたはサンプルの個々の成分の部分的溶解、およびサンプルが有するタンパク質を含む成分の部分的分解を意味すると理解される。
【0026】
本発明の目的のために、もし画分が主に1つの型の核酸を含むと呼ばれる場合、それは、この画分における核酸の全量(すなわち、2つの型の核酸の合計)に基づいて、50重量%超のこの型の核酸を含む。1つの実施態様によって、主に1つの型の核酸を含む上記画分は、この画分における核酸の全量に基づいて、少なくとも60重量%、好ましくは少なくとも70重量%、より好ましくは少なくとも80重量%のこの型の核酸を含む。
【0027】
本発明によるプロセスは、別々の画分における、同じ固定サンプルからのDNAおよびRNAの並行単離、および高感度で、定性的、および/または定量的方法による、それらの引き続く分析を可能にし、ここで例えば直径数mmの中空の針による臨床生検の顕微鏡的に分析可能な切片から得られるような、少量のサンプルでさえも、サンプル材料として適当である。本発明によるプロセスにおいて、市販で入手可能なAllprep DNA/RNA Kit(Qiagen、Hilden、Germany)と対照的に、DNAを含む、およびRNAを含む画分への分離は、核酸の実際の精製の前に実施する。固定した生物学的サンプルから、本発明によるプロセスは、主にRNAを含む溶解画分および主にDNAを含む未溶解画分という2つの画分を生じる。さらなる工程において、これらの画分を、それぞれの核酸のさらなる抽出および/または精製のために利用し得る。部分的消化およびRNAのDNAからの、主にRNAを含む溶解画分および主にDNAを含む未溶解画分への引き続く分離はまた、本発明による方法を、DNAをRNAから分離するためにフェノール/クロロホルム抽出に基づく先行技術の方法から差別化する。それぞれのフェノール/クロロホルムに基づく方法において、DNAおよびRNAはどちらも溶解産物に放出され、そして従って、両方とも溶解画分に存在する。フェノール−クロロホルム抽出および相分離の後、RNAは水相に溶解し、そしてDNAは有機相に溶解した形態で存在する。従って、先行技術の分離原理は、本発明によるプロセスと根本的に異なる。本発明によるプロセスはRNAをDNAから分離するためのフェノール/クロロホルム抽出を必要とせず、とりわけ架橋したサンプルの部分的消化に依拠してDNAを主に未溶解画分に維持する一方、RNAを溶解画分に放出する。
【0028】
最初の工程において、FFPEサンプルを、好ましくはプロテアーゼ処理にかける。驚くべきことに、この最初のプロテアーゼ処理において、プロテアーゼを用いることによるタンパク質のこのタンパク質分解(酵素的「消化」)の条件を調整する最適化によって、サンプルからDNAではなく、RNAのみを選択的に放出することが可能であることが見出された。適当な分離プロセス、例えば遠心分離を用いて、サンプルの本発明による不完全な「消化」後に、DNAを含む依然として未溶解の画分を、RNAを含む上清から分離することが可能である。
【0029】
ここで、2つの画分の溶解画分(A)および未溶解画分(B)への分離を、例えばろ過、沈降、デカンテーション、遠心分離等のような、液体および固体成分を分離するために適当であるとして当業者に公知のあらゆる方法を用いて行い得る。本明細書の以下で、この工程で得られる未溶解残渣はまた、ペレットと呼ばれ、ここで本発明の目的のために、この用語は、明確に遠心分離によってサンプルの液体成分から分離した未溶解残渣に限定されず、他の手段によって分離した未溶解残渣、例えばろ過の後にフィルターに残った固体物質も含む。
【0030】
未溶解画分をペレットにすることは、2つの画分の容易なおよび効率的な分離を可能にするので、有用である。
【0031】
RNAを単離するために、RNAを含む上清を、現在の技術水準から公知である習慣的なプロセスによって、例えば特許出願WO2007/068764において記載されたプロセスによって処理し得、そのプロセスは残った架橋を除去するための求核試薬を含む溶液中における加熱インキュベーションを含み、ここでRNAを次いで、例えばRNeasy FFPE Kit(QIAGEN、Hilden、Germany)を用いて、例えばシリカマトリックスに結合させることによって単離し得る。
【0032】
DNAおよび不完全に消化されたサンプルが有する他の未溶解成分を含む未溶解画分を、DNAを単離するために使用する。ここで、そのペレットは依然として本質的に固定サンプルの性質を有するので、DNAを固定サンプルから単離するための適当な、またはそのために習慣的な現在の技術水準によるあらゆる方法を用いることが可能である。特に、先行する不完全なプロテアーゼ消化は、サンプルから相当量のDNAを除去しなかった、および/または相当量のDNA架橋物を除去しなかった。このために、別のまたはさらなる酵素プロテアーゼ消化を有利に行ってサンプルを完全に溶解し、続いて例えばWO2007/068764において記載されたような求核試薬を含む溶液中で加熱インキュベーションする。この方式において放出されたDNAを、次いであらゆる適当な方法の助けによって、例えばQIAamp FFPE Kit(QIAGEN)を用いて、例えばシリカマトリックスへの結合によって、さらに精製し得る。
【0033】
この方式において、両方の型の核酸を、1つの工程で単一のサンプルから前もって分画し、そして次いでお互いに別々に単離し、そしてさらなる分析方法のために使用可能にする。
【0034】
本発明の目的のために、核酸という用語は、当業者に公知の全ての核酸、例えば天然または合成核酸、およびサンプルに人工的に導入された核酸、1本鎖および2本鎖核酸、直鎖、分枝、または環状核酸、RNA、特にmRNA、siRNA、miRNA、snRNA、tRNA、hnRNA、またはリボザイム、DNA、特にゲノムまたは色素体DNA、または細胞小器官のDNA、および感染由来の核酸も含む。
【0035】
適当な生物学的サンプルは、例えば、血液、精子、脳脊髄液、唾液、痰または尿などの細胞含有体液、白血球画分、バフィーコート、糞便、表面生検材料、吸引物、皮膚断片、生物全体、例えば剖検材料、生検材料、細針吸引物、または組織切片の形態の後生動物、好ましくは昆虫および哺乳類の、特にヒトの臓器および組織、例えば接着または懸濁した細胞培養物の形態の単離細胞、植物、植物の一部、植物組織または植物細胞、細菌、ウイルス、酵母および真菌のような、固定に適当な全ての生物学的サンプルである。
【0036】
本発明によるプロセスの最初の工程a)において、固定サンプルを、好ましくはタンパク質分解活性化合物の活性を可能にする水溶液と接触させ、そしてまた1つまたはそれより多いタンパク質分解活性化合物とも接触させる。
【0037】
本発明の目的のために、タンパク質分解活性化合物は、全てのタンパク質切断化合物、好ましくはプロテアーゼおよび熱安定性プロテアーゼ、特に好ましくはプロテイナーゼK、トリプシン、キモトリプシン、パパイン、ペプシン、プロナーゼ、およびエンドプロテアーゼLys−C、特にプロテイナーゼKのようなタンパク質分解活性酵素、およびまた臭化シアンのような、タンパク質を切断するために適当な非酵素的物質、またはこれらの物質の混合物である。
【0038】
水溶液中のタンパク質分解活性化合物の濃度は、一般的にそのタンパク質分解活性化合物の性質、およびその生物学的サンプルの性質および量に依存し、そしてその濃度は簡単な日常的な実験を用いて当業者が決定し得る。水溶液中のプロテアーゼ酵素の濃度は、それぞれの場合において水溶液の全重量に基づいて、好ましくは0.001重量%から5重量、特に好ましくは0.01重量%〜2.5重量%、そして特に0.05重量%〜0.2重量%の範囲である。ここで、ある特定のサンプルのために使用するタンパク質分解活性化合物の量または濃度は、そのタンパク質分解活性化合物の性質、および当業者が精通している幾つかのもの、pH、補助因子、インキュベーション温度、およびインキュベーション時間などの、選択した反応条件に依存する。そのタンパク質分解活性化合物の適当な量または濃度を、日常的な実験によって、簡単な方式で決定し得る。さらに、本発明によるプロセスにおいて、いかなる場合でも、タンパク質分解活性化合物の量または濃度を特に調整することは決定的ではなく、それは核酸の収量またはその完全性に悪い影響を与えることなく、ある特定のバンド幅で変動し得ることが見出された(実施例2)。
【0039】
その水溶液は、好ましくは、例えばカオトロピック剤、および/または好ましくは界面活性剤のような、生物学的組織の分解および/または細胞の溶解を促進するさらなる物質を含む。
【0040】
本発明によるプロセスにおいて使用するために適当な界面活性剤は、当業者に公知であり、そして細胞を溶解するために適当な全ての界面活性剤である;ここで陰イオン性または非イオン性界面活性剤が好ましい。
【0041】
好ましい界面活性剤は、ドデシル硫酸ナトリウム(SDS)、デオキシコール酸ナトリウム、3−(3−コールアミドプロピル)ジメチルアンモニウム−1−プロパンスルホネート(CHAPS)、例えばTritonX−100、TweenまたはNP−40の商品名で入手可能な界面活性剤などのポリエチレングリコールフェニルエーテル、またはこれらの混合物を含む群から選択される化合物であり、好ましい界面活性剤は、SDS、NP−40、およびTritonX−100(9から10のエトキシ化度を有するポリエチレングリコール(1,1,3,3−テトラメチルブチル)フェニルエーテル)である。生物学的サンプルに存在する細胞の溶解を補助するために使用される界面活性剤の量は、その生物学的サンプルの性質および量に依存し、そして簡単な日常的な実験を用いて当業者が決定し得る。
【0042】
その水溶液はさらに、好ましくは緩衝溶液であり、そのpHは、溶液中に存在する少なくとも1つの緩衝物質によって、6から9、好ましくは6.5から8.5、および特に好ましくは6.8から7.5の範囲に安定化される。よって、その水性緩衝溶液は好ましくは、好ましくはTris、Hepes、Pipes、Mops、アルカリ金属酢酸塩/酢酸等を含む群から選択される、少なくとも1つの緩衝物質、および/または好ましくは、好ましくはドデシル硫酸ナトリウム(SDS)、デオキシコール酸ナトリウム、3−(3−コールアミドプロピル)ジメチルアンモニウム−1−プロパンスルホネート(CHAPS)、ポリエチレングリコールフェニルエーテル、またはこれらの混合物、特に好ましくは、ドデシル硫酸ナトリウム、Tergitol−type NP−40の商品名で入手可能な、40のエトキシ化度を有するポリエチレングリコールノニルフェニルエーテル、および/または9〜10のエトキシ化度を有するポリエチレングリコール(1,1,3,3−テトラメチルブチル)フェニルエーテルを含む群から選択される、少なくとも1つの界面活性剤を含む。
【0043】
その水溶液はさらに、溶解を補助する、核酸を分解要素(constituent)に対して保護する、および/または水溶液を安定化する、さらなる成分、例えば錯体形成剤(complex former)、還元剤、または他の緩衝物質を含み得、ここで当業者は溶解バッファーのための可能性のある添加物の性質および量に精通している、またはそれらを簡単な日常的な実験によって決定し得る。好ましい実施態様において、その水性緩衝溶液はさらに、以下のものを含む群から選択される、少なくとも1つの物質を含む:
・錯体形成剤、好ましくはエチレンジアミン−N,N,N’,N’−4酢酸(EDTA)、エチレングリコールビス(2−アミノエチルエーテル)−N,N,N’,N’−4酢酸(EGTA)、クエン酸ナトリウム、またはこれらの混合物、
・好ましくは0.1から10Mの濃度の、好ましくは塩酸グアニジン、チオシアン酸グアニジン、イソチオシアン酸グアニジン、過塩素酸塩、NaI、KIおよび尿素を含む群から選択される、カオトロピック剤、
・好ましくはジチオスレイトール(DTT)、ジチオエリスリトール(DTE)、チオ硫酸ナトリウム、β−メルカプトエタノール、またはこれらの混合物を含む群から選択される、還元剤、および
・無機塩、好ましくは例えばNaCl、KCl、またはLiClのようなアルカリ金属ハロゲン化物、例えばCaCl2、またはMgCl2のようなアルカリ土類金属ハロゲン化物、例えば塩化アンモニウムまたは硫酸アンモニウムのようなアンモニウム塩、硫酸リチウム、またはこれらの混合物。
【0044】
1つの実施態様によって、その水性緩衝溶液は、界面活性剤、好ましくはSDSのような非イオン性界面活性剤、および好ましくは緩衝剤、好ましくはTRISを含む。その水性緩衝溶液はまた、EDTAのようなキレート剤を含み得る。
【0045】
本発明によるプロセスのプロセス工程a)において、架橋によって固定された生物学的サンプルを、少なくとも1つのタンパク質分解活性化合物を含む水溶液と接触させ、そして適当な温度でインキュベートする。ここで、その温度は一般的に、使用されるタンパク質分解活性化合物の性質に依存する。酵素の場合、その酵素が活性であることを可能にする温度を選択しなければならない。一般的に、低すぎる温度は酵素活性を不活性な段階まで抑制し、一方高過ぎる温度は、変性によって酵素を不活性化し得る。問題の酵素によって許容される温度範囲または最適な反応温度は、それぞれの酵素に依存して変動し、そして当業者に公知である、または簡単な日常的な実験によって決定し得る。例えばプロテイナーゼKを用いる場合、その反応を約95℃まで、好ましくは18℃および80℃の間、特に好ましくは50および65℃の間の温度で行い得る。
【0046】
既に記載したように、驚くべきことに、タンパク質分解活性化合物、好ましくはプロテアーゼを用いた、固定サンプルにおけるタンパク質のタンパク質分解「消化」の条件の調整を最適化することによって、サンプルから、DNAではなく、RNAのみを選択的に除去することが可能であることが見出された。サンプルの本発明によるこの不完全な(部分的な)消化の後、DNAを含む、依然として未溶解の画分を、RNAを含む溶解した上清から分離し得る。所定のタンパク質分解活性化合物に関して、RNAおよびDNAへの分離の質は、その濃度およびインキュベーション温度、および特にインキュベーション時間に依存する。もしそのタンパク質分解活性化合物を、非常に短時間のみ反応させたら、DNAだけでなく、RNAもサンプルから不十分にしか離れず、そして従って可溶性画分において全体で低い収量の核酸しか得られない。対照的に、もしそのタンパク質分解活性化合物を長く反応させすぎると、その結果は、RNAの(ほとんど)完全な溶解であるが、より多くのDNAもペレットから遊離する。しかし、そのタンパク質分解活性化合物の反応時間を適切に調整することによって、可溶性画分中のRNAおよび未溶解画分中のDNAの間を実質的に分離する、サンプルの部分的な(不完全な)溶解を達成することが可能であり、それは実際の精製の前の、2つの型の核酸の「予備分画」である。
【0047】
ここで、最適な反応時間は、まずそのタンパク質分解活性化合物、その水溶液中の濃度、およびインキュベーション温度に依存する。2番目に、その生物学的サンプルの量および厚さ、および他のサンプル特異的なパラメーター、例えば固定の型および期間が、そのタンパク質分解活性化合物の最適な反応時間に影響を有する。
【0048】
ホルマリンで固定したサンプルを、特にそのサンプルをパラフィンに包埋した後に使用することが好ましい。比較的大きい組織塊に関して、サンプルの量の多さおよび厚さのために、より小さいサンプルと比較して、タンパク質分解活性化合物を含むより多い容量の溶液、およびまた有利に、より高い濃度のタンパク質分解活性化合物、およびより長い反応時間を使用する。適当な切断装置、例えばミクロトームを使用して、固定サンプルから組織切片を調製し、ここで、光学顕微鏡で調査するための厚さは、一般的に約5μmから20μmである。さらに、パラフィン包埋サンプルをまた、他の方法を用いて、例えば中空針で穴をあけることによって、またはレーザーキャプチャー法によって、より小さいサンプル断片に分割し得る。より小さい組織断片は、より少量のタンパク質分解活性化合物およびより短い反応時間を必要とする。ここで特に切片の厚さが、組織とタンパク質分解活性化合物の完全な接触に関する制限因子であるので、決定的である。従って、好ましくは5μmから50μmの厚さを有する組織切片、またはもし適当なら、より大きなサンプルを分割またはホモジナイズすることによって得られる、より小さい組織断片を用いることが好ましい。
【0049】
固定時間、すなわち固定剤が生物学的サンプルに対して作用する時間は、生物学的サンプルにおいて生体分子の共有結合的架橋の程度に影響を与え、架橋の程度は、固定時間が長くなるほど増加する。短時間しか固定されなかったサンプルにおいては、従って少ない程度の生体分子の架橋しか存在せず、それは、個々の生体分子のより容易なおよびより迅速な溶解を可能にする。対照的に、長時間固定されたサンプルは、高い程度の架橋を有し、それは、特により大きな生体分子の溶解を遅延させ得る。よって、強く固定された(過固定)サンプルに関して、タンパク質分解活性化合物のより長い反応時間を有することが有用であり得る。ここで、最適な固定時間は、組織片のサイズに依存するので、「短時間固定された」および「長時間固定された」という用語は、相対的に理解される。組織におけるホルマリンの拡散速度は、最初約1mm/時間(h)であり、その速度は、組織深度が増加すると減少する。従って、約5mmの厚さの組織片に関して、ホルマリンがサンプルに完全に浸透するために約8時間が必要である(固定時間)。実際、約12〜24時間の固定時間が通例である;非常に小さなサンプルは、はるかにより短い固定時間を必要とし、そして12時間の固定時間で既に過剰固定である。
【0050】
従って、最適な反応時間は、固定および固定時間、生物学的サンプルの性質、量および厚さのような、サンプル特異的なパラメーターに依存し、そして個々のサンプルそれぞれに関して最適に調整し得る。驚くべきことに、それにも関わらず、多くの可能性のあるサンプルパラメーター、すなわち多くの異なる個々のサンプルに関して、DNAおよびRNAの未溶解および溶解画分への分離を可能にするように、その条件を調整することも可能である。ここで、その反応時間は、30秒および数日の間、好ましくは1分および5時間の間、そして特に好ましくは5分および90分の間、そしてより好ましくは10分および30分の間であり得る。600mAU/mlより大きい活性の、10μlから40μlのプロテイナーゼK溶液を、10μmから20μmの厚さのFFPE組織切片に関して、56℃のインキュベーション温度、および約15分から90分の反応時間で使用する場合、本発明の目的のために、サンプルの部分的溶解に関して非常に良い結果が得られる、すなわち、RNAは(ほとんど)完全に未溶解画分から溶解し、そして溶解画分に移動し、一方DNAは依然として(ほとんど)完全に未溶解画分に存在する(実施例2および3)。
【0051】
1つの実施態様によって、工程a)は、界面活性剤、好ましくは非イオン性界面活性剤を含む水性緩衝溶液中でのサンプルの部分的溶解と同時に、タンパク質分解活性酵素、好ましくはプロテイナーゼKのようなプロテアーゼを用いた、サンプルが有するタンパク質を含む成分の部分的なタンパク質分解を含み、ここでその反応を、18℃および80℃の間、好ましくは50℃から65℃の間の温度で、10分から5時間の間、好ましくは10から90分の間、より好ましくは10分から30分の間の反応時間で行う。この実施態様は、RNAの溶解画分への放出において迅速および有効であるという利点を有する。
【0052】
ここで、生物学的サンプルの固定を、当業者に公知のあらゆる固定剤によって、特に酸、アルコール、ケトン、または特にグルタルアルデヒドまたはホルムアルデヒドのような、他の有機物質によって行い得、ここで、ホルムアルデヒドで固定した生物学的サンプルが特に好ましい。本発明によるプロセスの特に好ましい実施態様によって、ホルムアルデヒド固定、パラフィン包埋生物学的サンプル(FFPEサンプル)を使用する。
【0053】
もしパラフィンに包埋した生物学的サンプルを使用するなら、そのパラフィンを好ましくは最初に、少なくとも部分的に、好ましくは完全に、サンプルから除去する。脱パラフィン化(deparaffinization)は、水性媒体中でサンプルを効率的に溶解するために、生物学的サンプルを包埋するために使用したパラフィンを選択的に除去するように働く。一般的に、パラフィンは核酸の溶解および分画の間、および核酸のさらなる精製および分析の間の両方に干渉し得る。好ましくは前もって行う脱パラフィン化は、プロテアーゼ処理後に本発明によるプロセスにおいて得られるペレットの質、特に固体性に、そして従って核酸の分離および得られる収量に顕著な影響を有し得る。
【0054】
生物学的サンプルからのパラフィンの除去を、原則として当業者に公知の、生物学的サンプルの脱パラフィン化のためのあらゆるプロセスによって行い得る。好ましくは、その脱パラフィン化を、最初にサンプルを疎水性有機溶媒に接触させることによって行う。ここで、生物学的サンプルおよび有機溶媒の混合物を、パラフィンのサンプルからの効率的な溶解を保証するために、例えば実験室振とう機で振とうすること、マグネティックスターラー等を使用することによって、撹拌しながら混合することが有利であり得る。有利に、そのサンプルを続いて遠心分離して、有機溶媒に溶解したパラフィンを、ペレット、すなわち生物学的サンプルから分離する。もし必要なら、生物学的サンプルからパラフィンを溶解する工程を、1回、2回、3回、または10回まで繰り返し得る。その脱パラフィン化を、例えば特許出願WO2007/068764において記載されたように、好ましくは疎水性有機溶媒、好ましくは芳香族炭化水素、特にキシレン中でインキュベーションし、続いてエタノール中でサンプルを再水和することによって行い得る。例えばアルカン類、好ましくは6<n<17である、一般式CnH2n+2の、室温で液体であるアルカン類またはこれらの混合物、特に好ましくはヘプタンのような、他の有機溶媒も、もし適当ならC1〜C5アルコール類、すなわちメタノール、エタノール、プロパノール、イソプロパノール、n−ブタノール、イソブタノール、n−ペンタノール、好ましくはメタノールを添加して、脱パラフィン化のために使用し得る。もし直鎖、すなわち未分枝のアルカンを使用するなら、6個またはそれより少ない炭素原子の鎖長のアルカンのいくつかは、毒性であると分類されており、室温でガス状であって、かつ/またはあまりにも揮発性であり、17個およびそれより多い炭素原子の鎖長のアルカンは、室温で固体であるので、nは好ましくは6より多く17未満である。もし適当なら、アルケン類、芳香族化合物等のような他の化合物が室温で液体であり、そして例えば鉱物油のようなパラフィンを溶解する限り、それらとのアルカンの混合物を使用することもさらに可能である。6個より多く17個未満の炭素原子の鎖長を有するアルカン類中で、特に好ましくはヘプタン中でインキュベートすることによる脱パラフィン化は、本発明に従うプロセスによる可溶性および不溶性画分AおよびBの引き続く分離のために特に有利であることが見出された。C1〜C5アルコール、好ましくはメタノールの、容量で1〜25%、好ましくは容量で5〜10%の量での、疎水性有機溶媒への添加は、不溶性残渣の沈殿を促進し、従って可溶性および不溶性画分の分離をより容易に、そしてより効率的になし得る。好ましい実施態様においては、それゆえそのプロセスは、工程(a)による部分的な溶解の前に、もし適当ならC1〜C5アルコール、好ましくはメタノールの、容量で1〜25%、好ましくは容量で5〜10%の量での添加を伴って、好ましくはサンプルを疎水性有機溶媒に接触させることによる、特に好ましくは6個より多く17個未満の炭素原子の鎖長の非極性脂肪族または芳香族炭化水素;特にキシレン、ヘプタン、および鉱物油を含む群から選択される炭化水素を用いた、パラフィンの選択的除去のための工程(i)を含む。1つの実施態様によって、脱パラフィン化を、アルカン、好ましくはヘプタン、およびアルコール、好ましくはメタノールとのインキュベーションによって達成する。適当な有機溶媒によるパラフィンの溶解に加えて、例えば、Biotechniques、18(1995)768−773頁において、Banerjeeらによって記載されたような、パラフィンの融解のような他のプロセスも、脱パラフィン化のプロセスとして適当である。
【0055】
パラフィンの除去後、生物学的サンプルを再水和することが好ましい場合があり、この再水和を、好ましくはアルコール濃度を次第に減少させた水性アルコール溶液(下降アルコール系列(descending alcohol series))で段階的に洗浄することによって行い、C1からC5アルコールが好ましく、そしてメタノール、エタノールおよびイソプロパノールが特に好ましい。もし使用する脱パラフィン化試薬がキシレンであるなら、そのサンプルを、さらなる処理の前に、通常この方式で再水和する。引き続く核酸単離のためのこの再水和の必要性は、関連する文献において議論されている。もし使用する脱パラフィン化試薬が直鎖脂肪族アルカンであるなら、サンプルの再水和は必要ない。しかし、単一の適当な試薬、例えば市販で入手可能な製品、BioGEnex、California、USAからのEZ−DEWAX(登録商標)で脱パラフィン化および再水和を行うことも可能である。
【0056】
好ましくは、そのサンプルは、脱パラフィン化および再水和の後、最初に、例えば空気への曝露または乾燥オーブンでのインキュベーションによって乾燥する。さらに、必要に応じて脱パラフィン化および再水和した生物学的サンプルを、好ましくは部分的な溶解の前にホモジナイズすることができ、それは特に比較的大きな組織サンプルの場合に有用である。対照的に、20μmの厚さまでの組織切片は、一般的にサンプルのホモジナイゼーションを必要としない。このホモジナイゼーションを、生物学的サンプルを細かく砕くための、当業者に公知のあらゆる装置、特に機械的破砕装置、例えばミル、ローター−ステーターホモジナイザー、Ultra−Turraxホモジナイザーまたは細いカニューレの補助を有する高圧細胞消化を用いて、または超音波ホモジナイザーによって行い得る。
【0057】
従って、好ましい実施態様において、そのプロセスは、工程(i)によるパラフィンの除去の後、および工程(a)による水性緩衝溶液におけるサンプルの部分的溶解の前に、好ましくは少なくとも以下の工程の1つを含む:
(ii)好ましくは、サンプルを、連続的に水含有量を増加させた水性C1〜C5アルコール溶液で繰り返し洗浄することによる、サンプルを再水和する工程、
(iii)サンプルを乾燥する工程および/または
(iv)サンプルをホモジナイズする工程。
【0058】
脱パラフィン化する、そして脱パラフィン化サンプルを作り上げるそれぞれの方法の工程も、先行技術において周知であり、そして従って、ここでさらなる説明を必要としない。
【0059】
1つの実施態様によって、架橋によって固定したサンプルを、脱パラフィン化の後にペレットの形態で得る。好ましくは、部分的溶解工程a)を行うために、水性緩衝溶液を、上記ペレットに加える。さらなる実施態様によって、脱パラフィン化化学物質を含む脱パラフィンされたサンプル(それぞれ脱パラフィン化溶液(deparaffinisation solution))を、工程a)において使用するための水性緩衝溶液と混合し、それによって水相を形成し、その水相に対し、工程a)において少なくとも1つのタンパク質分解活性化合物を用いて、上記サンプルが有するタンパク質を含む成分の部分的タンパク質分解を行なうことで、含まれるRNAを選択的に溶解画分に放出させ、一方含まれるDNAは主に未溶解画分に留まる。ここで、そのタンパク質分解活性化合物、好ましくはタンパク質分解酵素を、その水相に加えることができ、一方脱パラフィン化のために用いた溶液は依然として、水性緩衝溶液を添加したことにより形成された水相の上に存在する。もし、これの代替の分離工程b)を、例えば部分的に消化した架橋サンプルを遠心分離することによって(例えば上記および下記を参照のこと)、行うなら、主にDNAを含む未溶解画分は水相中でペレットを形成する。溶解画分を未溶解画分から分離するために、水相を、例えばピペットを使用することによって、例えば脱パラフィン化溶液によって回収し、他方では、未溶解の、主にDNAを含むペレットを残す。あるいは、脱パラフィン化溶液を、水性緩衝溶液の添加後に得られた水相から分離し、その後、タンパク質分解活性化合物を加え、そして未溶解画分を溶解画分から分離し得る。
【0060】
本発明によるプロセスの工程b)において、出発材料に存在する異なる型の核酸、すなわちRNAおよびDNAを、次いで、主にRNAを含む溶解画分(A)、および主にDNAを含む未溶解画分(B)に分離する。溶解および未溶解成分を含むサンプル全体を、少なくとも2つの画分に分離することも可能であり、次いで、それから様々な生体分子を単離または精製する、またはその中の様々な生体分子を次いで検出または分析し得る;しかし、そのサンプルを、本発明によるプロセスの工程a)の後に、好ましくは少なくとも1つの溶解画分(A)、および少なくとも1つの未溶解画分(B)に分離する。2つの画分を分離する利点は、それぞれの収率を減少させる、またはサンプルが有する様々な細胞型の不均一な分布をもたらす、もとの生物学的サンプルを分離する如何なる必要もなく、これらの2つの画分から、それぞれの場合別々に実質的に1つの型の核酸を単離し得ることである。
【0061】
この方式で得られた画分に対し、次いで別々に核酸の精製を行い得る。1つの画分からのみ核酸を単離し、そして他の画分を廃棄することも、本発明の範囲内である(例えば実施例6および7を参照のこと)。核酸の単離のための、サンプル(複数可)のさらなる処理の間に、そのサンプル(複数可)を、好ましくはタンパク質分解活性化合物の存在下で、50℃〜100℃、好ましくは55℃から95℃、特に好ましくは60℃から90℃、および特に65℃から85℃の範囲の温度に加熱する。
【0062】
未溶解成分の、水溶液からの分離は、特にもしそのタンパク質分解活性化合物が高い温度、すなわち室温より上の温度で活性であるなら、好ましくはタンパク質分解活性化合物の反応時間後に混合物を冷却することによって補助される。冷却を、好ましくはそのサンプルをプロテアーゼ消化の温度より下の温度で、好ましくは室温で、特に4℃で、または例えば−20℃または−80℃のようなさらにより低い温度でインキュベートすることによって行い、ここで、水溶液全体が凍結するのを避けるために、これらの温度で冷却するのは短時間である。従って、冷却を好ましくは15℃またはそれより低い、10℃またはそれより低い、4℃またはそれより低い温度で、または例えば−20℃または−80℃のような、さらにより低い温度で行う。冷却を、分離工程の前および/または分離工程の間に行い得る。冷却は、未溶解画分の分離、特にペレット化がより効率的であるという利点を有する。これは、FFPEサンプルは通常未溶解成分を含み、特にDNAは大量の固体成分ではなくタンパク質に架橋しているので、特に有利である。上記未溶解成分は、通常ペレット化するのが困難である。冷却は、未溶解成分のペレット化を補助し、そして従って分離をより効率的にする。従って、冷却は、個々の画分の分離が改善されるため、主にDNAを含む未溶解画分が、より多くのDNAを含むこと、そしてそれゆえ、RNAを含む溶解画分では、DNAの混入がより少なくなることがもたらされる。少しの細胞材料しか含まない架橋サンプルを処理する場合に、これは特に有用である。
【0063】
1つの実施態様によって、分離により、主にDNAを含む未溶解画分を、小型のペレットの形態で得ることになる。これにより、主にDNAを含むペレットを、主にRNAを含む溶解画分から容易に分離することが可能となる。
【0064】
3番目の工程において、この方式で得られたその溶解画分(A)および未溶解画分(B)を、お互い別々に使用して、存在する生体分子、好ましくは核酸(複数可)を精製し得る。ここで、溶解画分(A)を、好ましくはRNAを単離するために使用し、そして未溶解画分(B)を、好ましくはDNAを単離するために使用する。1つの画分からのみ核酸を単離し、そして他の画分を廃棄することも、本発明の範囲内である(例えば実施例6および7を参照のこと)。
【0065】
ここで、溶解画分(A)を、核酸単離のための適当なプロセスにおいて、さらなる溶解無しに、直接使用し得る。しかし、例えばタンパク質分解活性化合物、好ましくはプロテアーゼによるさらなる溶解を、必要に応じて水溶液中で行い得る。適当なプロセスは、当業者に公知である、核酸、特にRNAを単離するための全てのプロセスおよび方法である。特許出願WO2007/068764、WO2008/021419、WO2005/012523、またはWO2005/054466において記載されたような、固定サンプル材料から核酸を単離するためのプロセス、または別に、市販のキットRNeasyFFPEおよびmiRNeasyFFPE(どちらもQIAGENから)の補助によって行われるプロセスが適当である。後者の場合、シリカ膜へのカオトロープが媒介する結合によって核酸を精製する前に、画分Aを少なくとも1つの加熱工程にかける。RNAのさらなる抽出および精製を、好ましくは特許出願WO2007/068764において記載されたプロセスの補助によって行い得る。もし特許出願WO2007/068764において記載されたようなプロセスを、溶解画分からの核酸の単離のために使用するなら、そのサンプルを、求核試薬の存在下で加熱する。これを、少なくとも1つのタンパク質分解活性化合物および現在溶解した核酸を含む水溶液中で行い得、ここで特許出願WO2007/068764において記載されたプロセスのために必要な求核試薬を、本発明によるプロセスの工程a)(タンパク質分解活性化合物の作用)の後に、その水溶液に加え得る、またはさらには工程a)に従ってサンプルに加える前に、求核試薬がその水溶液に存在し得る。画分の分離を、特許出願WO2007/068764において記載されたようなサンプルの加熱の後に、または好ましくは、さらなる加熱の前の、タンパク質分解活性化合物を作用させた後に直接行い得る。
【0066】
適当な求核試薬は、電子をルイス酸の空の軌道に、または複数の空の軌道に移し得る、全てのルイス塩基である。これらのルイス塩基の中で、陰性の電荷を有する、陰性に分極している、または少なくとも1つの自由電子対を有する、少なくとも1つの官能基を有する試薬が特に好ましい。
【0067】
陰性の電荷を有する官能基を含む化合物は、例えばアルカリ金属アルコキシドまたはアルカリ土類金属アルコキシド、アルカリ金属水酸化物またはアルカリ土類金属水酸化物、アルカリ金属ハロゲン化物またはアルカリ土類金属ハロゲン化物、アルカリ金属シアン化物またはアルカリ土類金属シアン化物等であり、それらに制限されない。
【0068】
陰性に分極した少なくとも1つの官能基を有する試薬は、特にお互いに共有結合した2つの原子を含み、そしてそのAlfredおよびRochowによる電気陰性度が、少なくとも0.25、好ましくは少なくとも0.5、そしてより好ましくは少なくとも1.0異なる、少なくとも1つの官能基を有する試薬である。
【0069】
しかし、本発明によって、1つまたは2つ、特に好ましくは1つの自由電子対(複数可)を有する、少なくとも1つの官能基を有する求核試薬が、特に好ましく、そして今度はこれらの化合物の中で、少なくとも1つの1級アミノ基、2級アミノ基、または構造Iの3級アミノ基を有するものが最も好ましい
【0070】
【化1】
ここで、R1はC1からC20炭化水素基、特に好ましくはC2からC15炭化水素基、そしてより好ましくはC2からC10炭化水素基、少なくとも1つのヘテロ原子を有するC1からC20炭化水素基、少なくとも1つのヘテロ原子を有するC2からC15炭化水素基、そしてより好ましくは少なくとも1つのヘテロ原子を有するC2からC10炭化水素基、または必要に応じてヘテロ原子で置換された芳香環系である、
R2は、C1からC20アルキル基、特に好ましくはC1からC10アルキル基、そしてより好ましくはC1からC2アルキル基、特にメチル基またはエチル基、C1からC20ヒドロキシアルキル基、特に好ましくはC1からC10ヒドロキシアルキル基、そしてより好ましくはC1からC2ヒドロキシアルキル基、または水素原子であり、水素原子が最も好ましい、および
R3は、C1からC20アルキル基、特に好ましくはC1からC10アルキル基、そしてより好ましくはC1からC2アルキル基、特にメチル基またはエチル基、C1からC20ヒドロキシアルキル基、特に好ましくはC1からC10ヒドロキシアルキル基、そしてより好ましくはC1からC2ヒドロキシアルキル基、または水素原子であり、水素原子が最も好ましい。
【0071】
本発明によって、上記で示した構造Iの官能基を有する求核試薬が特に好ましく、それは特に少なくとも1つの構造Iの官能基を有し、構造IのうちででラジカルR2およびR3のうち少なくとも1つ、最も好ましくは両方のラジカルR2およびR3が、水素原子である。それに加えて、少なくとも1つの構造Iの官能基を有する求核試薬が特に好ましく、ここで構造Iの窒素原子は、ラジカルR1、R2およびR3のsp3混成原子に共有結合しているのみである。特に、ラジカルR1、R2およびR3はどれも、それぞれラジカルR1、R2およびR3を超えて窒素原子の自由電子対を非局在化する(delocalising)ことはできない。従って、特に好ましくは、ラジカルR1、R2およびR3はどれも、例えば構造IIを有さない。
【0072】
【化2】
本発明によって、メチルアミン、エチルアミン、エタノールアミン、n−プロピルアミン、n−ブチルアミン、イソブチルアミン、tert−ブチルアミン、ジメチルアミン、ジエチルアミン、ジエタノールアミン、ジ−n−プロピルアミン、ジイソプロピルアミン、ジブチルアミン、トリメチルアミン、トリエチルアミン、トリエタノールアミン、ヘキサメチレンテトラミン、2−エチルヘキシルアミン、2−アミノ−1,3−プロパンジオール、ヘキシルアミン、シクロヘキシルアミン、1,2−ジメトキシプロパンアミン、1−アミノペンタン、2−メチルオキシプロピルアミン、トリ(ヒドロキシメチル)アミノメタン、アミノカルボン酸、特にグリシンまたはヒスチジン、またはアミノグアニジンを含む群から選択される、少なくとも1つの構造Iの官能基を有する求核試薬が特に好ましく、ここで最後に述べたものは、可能であるが、好ましくはない。これらの中で、エタノールアミン、ジエタノールアミン、トリエタノールアミン、アミノ−1,3−プロパンジオールおよびトリ(ヒドロキシメチル)アミノメタンが最も好ましい。少なくとも1つの構造Iの官能基を有する、好ましい求核試薬は、さらにアニリン、トルイジン、ナフチルアミン、ベンジルアミン、キシリジン、キシレンジアミン、ナフタレンジアミン、トルエンジアミン、3,3’−ジメチル−4,4’−ジフェニルジアミン、フェニレンジアミン、2,4’−メチレンジアニリン、4,4’−メチレンジアニリン、スルホニルジアニリン、およびジメチルベンジルアミンを含む群から選択される、芳香族アミンである。
【0073】
その求核試薬が少なくとも1つの構造Iの1級アミノ基を有する、本発明によるプロセスの特定の実施態様によって、その求核試薬は、C1からC6アルキルアミン、C1からC6アルキルジアミン、C1からC6アルキルトリアミン、C1からC15アミノアルコール(aminoalkohol)、C1からC15アミノジオール、またはC1からC15アミノカルボン酸である。
【0074】
本発明によるプロセスの別の特定の実施態様によって、その求核試薬は、窒素原子を含むヘテロ環状化合物であり、そしてピロール、ピリジン、キノリン、インドール、アザシクロペンタン、アザシクロヘキサン、モルホリン、ピペリジン、イミダゾ−ルまたはこれらの化合物の誘導体を含む群から選択され、ここでこれらの化合物の誘導体は、好ましくは水素原子の代わりに、上記で述べた化合物の、1つまたはそれより多い炭素原子、または窒素原子に結合した、C1からC3アルキル基、特に好ましくはメチルまたはエチル基を有する化合物を意味すると理解される。
【0075】
上記で述べた求核試薬の中で、水溶性であるもの、特に25℃の温度、およびpH7において、水中で少なくとも1g/l、特に好ましくは少なくとも10g/l、そしてより好ましくは少なくとも100g/lの溶解度を有するものが好ましい。
【0076】
使用する水溶液中での求核試薬の濃度は、好ましくは0.1から10000mmol/l、より好ましくは1から5000mmol/l、さらにより好ましくは5から2500mmol/l、および最も好ましくは20から1000mmol/lの範囲である。本発明によるプロセスの特に有用な実施態様によって、水溶液中の求核試薬の濃度は、20mmol/l超、特に好ましくは50mmol/l超、そして最も好ましくは100mmol/l超である。
【0077】
1つの実施態様によって、好ましくはタンパク質分解酵素である、タンパク質分解活性化合物による消化を行った後、サンプル中の架橋は、好ましくは少なくとも70℃、より好ましくは少なくとも75℃、最も好ましくは少なくとも80℃、または少なくとも90℃の温度まで、少なくとも5分、好ましくは少なくとも10分、最も好ましくは少なくとも15分の期間加熱することによって、少なくとも部分的に逆行する(reversed)。少なくとも15分間80℃に加熱することが、分解したサンプルのRNAを含む溶解画分において架橋を逆行させるために特に好ましい。少なくとも30分から数時間まで、好ましくは少なくとも1.5、または少なくとも2時間、少なくとも85℃、好ましくは少なくとも90℃に加熱することが、DNAにおいて架橋を逆行させるために好ましい。上記で述べたように、求核試薬の存在下で加熱を行う。適当なインキュベーション時間も、引用された先行技術において記載される。架橋を逆行させるためのこのさらなる加熱工程を、主にRNAを含む溶解画分を、主にDNAを含む未溶解画分から分離する前または後に行い得る。この加熱工程は、さらなるDNAが未溶解画分から放出されることをもたらし得るので、特に続いて未溶解画分からDNAを単離することも意図するなら、画分を分離した後に上記加熱工程を行うことが好ましい。さらに放出されたDNAは、例えばDNase消化を行うことによって分解され得るので、RNAを単離することのみを意図するなら、上記加熱工程を、画分を分離する前にも行い得る。小さいRNA分子も保存する、DNase消化を行うための好ましい実施態様を、下記で詳細に説明する。
【0078】
1つの実施態様によって、DNase消化を、分離した、主にRNAを含む溶解画分に対して行う。架橋サンプルに含まれる主な量のDNAを含む未溶解画分を分離することにより、既に上記サンプルに含まれるDNAの主な部分が除去される。従って、部分的な消化および画分の分離の後に得られる、主にRNAを含む溶解画分は、既にDNAが枯渇している。工程a)において部分的消化の間に放出されたかもしれない、残りの量のDNAを、RNAを含む溶解画分に対して、DNase消化を行うことによって効率的に分解し得る。RNAをDNase消化サンプルから単離することにより、ほとんどまたは全くDNAの混入のない、純粋なRNAが提供される。
【0079】
従って、1つの実施態様によって、DNaseを、分離した主にRNAを含む溶解画分に加える。DNase消化を、RNAを単離する前に効率的に行い得ることは、非常に驚くべきことであった。これは、通常の先行技術の方法ではRNAを精製するときRNAを単離してからDNase消化を行っているように、DNaseが、効率的には機能しないことが予測されたからである。さらに、例えば通常のオンカラムのDNase処理と比較して、特に小さいRNAの量が、本発明によるプロセスを用いる場合に増加するので、RNAを単離する前にDNase消化を行うことはまた、かなりの利点を有する。それぞれのDNase消化を、実施例6および7で行う。好ましくは、そのDNase消化を、上記で記載したように加熱によって架橋を逆行させた後に行う。
【0080】
「DNase」という用語は、DNAにおけるホスホジエステル結合の加水分解切断を触媒するあらゆる酵素を指す。多種多様なデオキシリボヌクレアーゼが公知であり、それらはその基質特異性、化学的メカニズム、および生物学的機能が異なる。「DNase」という用語は、エキソデオキシリボヌクレアーゼおよびエンドデオキシリボヌクレアーゼを指す。特に、DNaseIおよびDNaseIIを使用し得る。DNaseIが好ましい。
【0081】
DNase消化を、DNAの効率的な分解を可能にするために、DNaseが活性である条件下で行う。DNase消化の効率を、例えば分解サンプルに加えるDNaseの量によって、およびさらに、特にMgイオンおよびCaイオンのような、DNaseの活性を促進する添加物を加えることによって、調整し得る。さらに、工程a)において部分的消化を達成するために使用する条件に依存して、分離した、主にRNAを含む溶解画分に対して、DNase消化が高い効率で作用することを保証するために、中間の処理工程が有用であり得る。例えば、DNase消化に干渉し得る成分を、DNase消化を阻害しない濃度まで除去または希釈し得る。DNase消化を、DNaseが活性である濃度のMgイオンおよびCaイオンの存在下で行う。例えば、DNase消化を行うために、MgイオンおよびCaイオンを、例えばMgCl2およびCaCl2の形態で、分解サンプルに加えて、DNase消化混合物において適当な濃度を確立し得る。MgイオンおよびCaイオンの適当な濃度は、サンプル、および特に分解工程a)において使用する溶解条件に依存する。例えば、もしCaイオンおよびMgイオンが、工程a)における消化の間に既に提供され、そして従って、分解サンプルに存在するなら、より少ない量のMgイオンおよびCaイオンをDNase消化のために加えなければならない、またはMgおよびCaの添加はさらに必要でない。もし、例えばEDTAのようなキレート剤を、工程a)の間に使用するなら、DNase消化の間により高い濃度のMgイオンおよびCaイオンを使用することが賢明である。1つの実施態様によって、そのMgイオンおよびCaイオンは、好ましくはMgCl2およびCaCl2の形態で、少なくともそれぞれ0.2mM、少なくともそれぞれ2mM、少なくともそれぞれ5mM、少なくともそれぞれ7.5mM、および好ましくは少なくともそれぞれ10mMから成る群から選択される濃度で、反応組成物中で提供される。さらに、そのCaイオンおよびMgイオンを、0.2mMから1M、2mMから100mM、10mMから50mM、および10mMから25mMから成る群から選択される、各イオンの濃度範囲で提供し得る。DNase、分解サンプルおよび必要に応じて、DNase消化を促進するさらなる添加物を含むDNase消化反応組成物を、適当な時間インキュベートして、DNAが分解されることを可能にする。好ましくは、そのインキュベーションは、少なくとも5分、少なくとも好ましくは10分、または少なくとも15分間行う。適当な範囲は、1分から6時間、5分から120分、10分から60分、および15分から30分を含む。随意選択のDNase消化を行った後、RNAをサンプルから単離し得る。本明細書中で議論するように、基本的にあらゆるRNA単離方法を使用し得る。
【0082】
1つの実施態様によって、そのRNAを、溶解した、必要に応じてDNase処理した画分から、適当な添加物を加えることによって適当な結合条件を確立すること、および核酸結合固相にRNAを結合することによって、単離する。1つの実施態様によって、RNAの単離は、少なくとも以下の工程を含む:
i)少なくとも1つのアルコールおよび/または少なくとも1つのカオトロピック剤、および必要に応じてさらなる添加物を加えて、結合混合物を形成する工程、およびその結合混合物を、核酸結合固相と接触させて、RNAを上記固相に結合させる工程;
ii)必要に応じてRNAが固相に結合している間にRNAを洗浄する工程;および
iii)必要に応じてその固相からRNAを溶出する工程。
【0083】
核酸結合固相として、核酸を結合し得るあらゆる材料を使用し得、そして従って適当な条件下で核酸を結合し得る、様々な材料を含む。本発明と関連して使用し得る代表的な固相は、シリカ粒子、二酸化ケイ素、珪藻土、ガラス、アルキルシリカ、ケイ酸アルミニウム、およびホウケイ酸塩を含むがこれに限らない、シリカおよびケイ質固相を含む化合物、ニトロセルロース;ジアゾ化紙;ヒドロキシアパタイト(ヒドロキシルアパタイトとも呼ばれる);ナイロン、金属酸化物;ジルコニア;アルミナ;ポリマー支持体、ジエチルアミノエチル−およびトリエチルアミノエチル誘導体化支持体、疎水性クロマトグラフィー樹脂(フェニルまたはオクチルSepharoseのような)等を含むがこれに限らない。固相という用語は、その形態またはデザインに関していかなる制限も意味することを意図しない。従って、固相という用語は、膜、フィルター、シート、粒子、磁気粒子、ビーズ、ゲル、粉末、繊維等を含むがこれに限らない、多孔性または非多孔性、透過性または不透過性の、適当な材料を包含する。1つの実施態様によって、その固相の表面は、改変されていない、そして例えば官能基によって改変されていない。好ましい実施態様によって、その核酸結合固相は、カラムに含まれる。本明細書中で使用される「カラム」という用語は、特に少なくとも2つの開口部を有する容器を説明する。それによって、溶液および/またはサンプルが、上記カラムを通過し得る。「カラム」という用語は、特に容器の形に関していかなる制限も意味せず、それは例えば円形または角であり得、および好ましくは円柱状であり得る。しかし、特に多層カラムを使用する場合、他の形も使用し得る。そのカラムは、核酸結合固相を含む。上記カラムに含まれる上記固相は、カラムにアプライした場合に溶液、それぞれサンプルの通過を可能にする。これは、もし例えば遠心力をカラムにかけたら、溶液および/またはサンプルは、遠心力の方向にカラムを通過し得ることを意味する。上記で議論したように、それぞれのカラムに基づく核酸単離手順を用いる場合、そのサンプルは、例えば遠心分離または減圧によって補助されて、カラムを通過し、そして核酸は、上記通過の間に、含まれる核酸固相に結合する。そのカラムを、単一の形式で、または多層の形式で使用し得る。マルチウェルプレートと同様の形式を有し、そして膜のような核酸結合固相を含む、そのような多層カラムは、先行技術において周知である。好ましくは、そのカラムはスピンカラムである。カラムに含まれる核酸結合固相として、通常カラムに基づく核酸単離手順で利用される、あらゆる固相を使用し得る。好ましくは、核酸結合膜、および従って核酸を結合し得る膜を使用する。適当な膜は、親水性膜、疎水性膜、およびイオン交換によって核酸を結合する膜を含むが、これに限らない。例は、シリカ膜、ガラス繊維膜、ナイロン膜、ニトロセルロース膜、改変セルロース膜(例えばアセチル−またはヒドロキシ−)のようなセルロース膜、紙膜、特に改変紙を含むがこれに限らない。好ましくは、その膜は多孔性である。さらに、シリカを含む、またはシリカから成る膜を使用することが好ましい。カラムに含まれる、さらなる通常の核酸結合固相は、シリカ粒子のような、核酸結合粒子の充填、または核酸結合材料(例えばシリカゲル)の層である。例えば、そのシリカ粒子を、不活性なフィルターまたは膜上に層として配置し、それによって核酸結合固相を形成し得る。
【0084】
サンプルの未溶解成分を消化し、そして生体分子の残った架橋を切断するために、もし適当なら、未溶解成分を含む画分(B)に、好ましくはさらなる処理を行う。実質的に依然として未溶解画分(B)にある、溶解可能な核酸、主にDNAを、このさらなる処理工程によって単離し得る。固定組織から核酸を溶解するために公知のあらゆるプロセス、例えばWO2007/068764、WO2008/021419、WO2005/012523、またはWO2005/054466において記載されたようなプロセスが適当である、または他に市販で入手可能なキット、例えばQIAamp DNA FFPE Tissue Kits(QIAGEN)の補助によって行い得る方法が、核酸、特にDNAを、未溶解画分(B)から単離するために適当である。後者の場合、画分Bの未溶解成分に、タンパク質分解剤、例えばプロテアーゼによる、少なくとも1つのさらなる処理、および加熱工程を行う。そのプロテアーゼ処理は、効率的な溶解、および従って溶解可能な核酸の放出を行う。未溶解画分Bは、完全な固定サンプルと比較して、特により容易に溶解可能でない成分しか含まないので、さらなる最適化、例えばさらなるプロテアーゼ工程および加熱工程の延長が有用であり得、そして著しく改善した収量および引き続く(下流の)分析における結果をもたらす。
【0085】
従って、1つの実施態様によって、DNAを、画分の分離後に、未溶解の主にDNAを含む画分から得る。DNAを未溶解画分から得ることは、1つの実施態様によって以下の工程を含む:
i)上記未溶解画分に、同時酵素プロテアーゼ消化による溶解を行うことによって、DNAを、未溶解の、主にDNAを含む画分から放出する工程であって、ここで好ましくは、溶解の間に少なくとも1つの界面活性剤、および必要に応じてさらなる添加物を使用し、そしてここで、その酵素消化を、好ましくは加熱によって補助する(適当な条件を上記で(abive)述べる)工程;
ii)架橋を少なくとも部分的に逆行させるために、主にDNAを含む画分を、好ましくは工程i)の後に、好ましくはサンプルを加熱することによって、少なくとも70℃、より好ましくは少なくとも80℃、最も好ましくは少なくとも85℃、より好ましくは少なくとも90℃の温度まで、求核試薬の存在下で加熱する工程、および
iii)架橋を逆行させた後に、好ましくは適当な添加物を加えることによって結合条件を確立すること、およびDNAを核酸結合固相に結合させることによって、DNAを単離する工程。好ましくは、カオトロピック剤および界面活性剤、好ましくは非イオン性界面活性剤およびアルコールを、結合条件を確立するために加える。適当なDNA単離手順はまた、先行技術において周知である。
【0086】
従って、好ましくは、本発明によるプロセスは、工程b)に続いて、画分Aから得られるRNAおよび/またはペレットBから得られるDNAを、好ましくは沈殿、核酸の適当な結合材料への結合、電気泳動、および/またはクロマトグラフィーまたはその組み合わせによって、別々に精製するためのさらなる工程を含む。
【0087】
さらに、そのプロセスは、好ましくは、単離および/または精製した核酸の分析/検出のための工程を含む。
【0088】
当業者に公知である全ての分析方法、例えばPCR、qPCR、RT−PCR、qRT−PCRおよびゲノムDNA全体の増幅(全ゲノム増幅)のような増幅技術、ゲル電気泳動、ブロッティング技術、特にサザンブロッティングおよびノーザンブロッティング、マイクロアレイ分析、制限酵素断片長多型解析(RFLP分析)、SAGE(遺伝子発現の連続分析(serial analysis of gene expression))、NextGeneration配列決定およびRNA配列決定を含む配列決定、一塩基多型分析(SNP分析)、突然変異分析、エピジェネティック分析、特にメチル化パターンの分析、またはその組み合わせを、本発明によるプロセスによって単離した核酸を分析するために使用し得る。
【0089】
上記の開示から明らかになるように、本発明はまた、以下の工程を含む、溶解画分中のRNAおよび未溶解画分中のDNAを、架橋によって固定した同じ生物学的サンプルから得るためのプロセスを提供する:
a)少なくとも1つのタンパク質分解活性化合物を使用した、サンプルが有するタンパク質を含む成分の部分的なタンパク質分解と同時に、水性緩衝溶液中でサンプルを部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット、画分B)を得る工程、
b)溶解画分を未溶解画分から分離する工程、
ここで、溶解画分における核酸の全量に基づいて、その溶解画分は主にRNAを含み、そして未溶解残渣における核酸の全量に基づいて、その未溶解残渣は主にDNAを含む。
【0090】
1つの実施態様によって、主にRNAを含む画分を、主にDNAを含む画分から分離することは、沈殿も、1つまたは両方の型の核酸の有機溶媒による抽出も、1つまたは両方の型の核酸の固体マトリックスへ選択的結合も必要としない。1つの実施態様によって、画分の分離後、RNAを、主にRNAを含む溶解画分から単離し、および/またはDNAを、主にDNAを含む未溶解画分から単離する。もし、例えばRNAの単離のみを意図するなら、主にDNAを含む未溶解画分を、分離後に廃棄し得る。
【0091】
適当なおよび好ましい実施態様、ならびに本発明による部分的消化および分離工程に関連する利点、ならびに引き続く核酸単離の適当なおよび好ましい実施態様を、同じ架橋サンプルからのRNAおよびDNAの並行単離および/または精製のプロセスに関して、上記で詳細に議論した。それは上記の開示に参照され、それもここで適用する。
【0092】
本発明はさらに、少なくとも(1)タンパク質分解活性化合物、好ましくは上記で述べたタンパク質分解活性化合物の1つ、(2)少なくとも1つの緩衝物質、好ましくは上記で述べた緩衝物質の1つ、および(3)少なくとも1つの界面活性剤、好ましくは上記で述べた界面活性剤の1つ、およびまた好ましくは(4)工程(a)による不完全なタンパク質分解を行うための指示を含む、本発明によるプロセスを行うためのキットを含む。
【0093】
特に好ましい実施態様において、本発明によるキットは、さらに(5)少なくとも1つの核酸結合材料、およびまた必要に応じて(6)核酸単離のためのバッファー、好ましくは結合および/または溶出バッファーを含む。
【0094】
核酸結合材料(5)として使用するために適当なのは、DNAまたはRNAの吸着に関して当業者に公知である全ての材料であり、セルロースに基づく材料、特にカルボキシ官能基(funktional)セルロース材料、またはジエチルアミノエチルセルロース、アガロース、シリカ、ガラス、石英、ゼオライト、または金属酸化物のようなミネラルキャリア、またはイオン交換体材料でコーティングしたキャリアが特に好ましい。記載した材料は、例えば膜、または磁気粒子または非磁気粒子の形態で存在し得る。その核酸結合材料は、好ましくは組み立て式カラムのカラム材料として、または別に懸濁物としてキットに含まれる。材料の型は、分析する核酸の化学的構造に決定的に依存し、当業者は、それぞれの意図する適用、すなわちRNAまたはDNAの分析に関して、各場合において適当な吸着材料に精通している。
【0095】
溶出バッファー(6)として、本発明によるキットは、当業者に公知であり、そして核酸の核酸結合材料からの溶出に習慣的に使用されるあらゆるバッファーを含み得る。その溶出バッファーは、好ましくは水性塩溶液、特に例えばNaCl、KCl、またはLiClのようなアルカリ金属ハロゲン化物、例えばCaCl2またはMgCl2のようなアルカリ土類金属ハロゲン化物、例えば塩化アンモニウムまたは硫酸アンモニウムのようなアンモニウム塩、またはこれらの塩の少なくとも2つの混合物を含む水溶液であり、ここでその溶出バッファーは、必要に応じてまた、例えばアルカリ金属酢酸塩/酢酸に基づくバッファーシステム、またはトリス(ヒドロキシルメチル)アミノメタンに基づくバッファーシステムを含み得る。もしそのキットを、固定組織からRNAを単離するために使用するなら、そして使用するマトリックスがシリカ膜であるなら、溶出バッファーとして水、特にRNaseを含まない水を使用することが特に好ましい。
【0096】
結合バッファー(6)として、本発明によるキットは、当業者に公知であり、そして核酸を核酸結合材料に結合させるために習慣的に使用されるあらゆるバッファーを含み得、ここでその結合バッファーは、使用するそれぞれの核酸結合材料とマッチしていなければならない。もし使用する核酸結合材料がシリカマトリックスであるなら、その結合バッファーは、好ましくはカオトロピック剤、および必要に応じてC1〜C5アルコールをさらに含む。
【0097】
本発明によるキットを、生物学的サンプルに存在する核酸、すなわちDNAおよびRNAの両方を分析および/または定量化するために使用し得る。
【0098】
この理由のために、本発明によるキットをさらに、ヒトまたは動物の体の外のサンプルの、疾患の診断、予後、治療に関する決定、および/または治療のモニタリングのために使用し得る。
【実施例】
【0099】
実施例1:本発明のプロセスによるRNAおよびDNAの分離
使用するサンプルは、包埋の後約4ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasy FFPEキットおよびQIAamp FFPEキットの構成成分を使用した。
【0100】
脱パラフィン化のために、その組織をまず1mlのキシレン中で10分間インキュベートした。サンプルの遠心分離によるペレット化、および上清の除去後、このキシレンによるこの処理を、後2回繰り返した。続いて各場合においてサンプルを無水エタノールで2回、続いて水性エタノール溶液で処理して(まず96%エタノール、および次いで70%エタノール)、そして37℃で10分間乾燥した。
【0101】
この方式で得られた脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTA、および0.2%のSDS(w/v)(pH7)を含む150μlの水溶液で処理し、そしてタンパク質分解活性化合物として10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。得られた混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルを遠心分離し、そして上清(画分A)を、未溶解成分(画分B)を含むペレットから除去した。
【0102】
2つの画分における、核酸の型、RNAおよびDNAの分布を決定するために、上清およびペレット由来のRNAを、各場合4つのサンプル(サンプル1〜4)から単離し、そしてさらなる4つのサンプル(サンプル5〜8)から、上清およびペレット由来のDNAを単離した。
【0103】
サンプル1〜4の上清(画分A)(サンプル1aから4a)からRNAを単離するために、その上清を80℃で15分間インキュベートした。DNA結合のための条件に調整するために、320μlのカオトロピックバッファー、例えばQIAGENからのRBCバッファーを加えた。その混合物を、例えばQIAGENからのgDNA除去カラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによってその膜を通過させた。混合物の組成は、DNAのシリカ膜への選択的結合を引き起こすので、RNAは、カラムの溶出液にある。RNAのための結合条件に調整するために、この溶出液を、エタノールと混合し、そして次いでもう1度、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによってその膜を通過させた。次いでそのシリカ膜を、各場合において500μlのアルコールを含む洗浄バッファーRW2(QIAGEN)で2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そして1分のインキュベーション後、RNAを30μlの水によって溶出した。
【0104】
RNAをサンプル1〜4のペレット(画分B)(サンプル1bから4b)から単離するために、そのペレットを、界面活性剤および求核試薬を含む、QIAGENからのPKDバッファー150μl、および10μlのQIAGENからのプロテイナーゼKと混合した。56℃で15分間インキュベートし、そして次いで80℃で15分間インキュベートした後、その溶解産物を、カオトロープを含む結合バッファー、例えばQIAGENからのRBCバッファーで処理し、そしてその混合物を、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして10000rpmで1分間遠心分離することによって、膜を通過させた。上記で記載したように、そのシリカ膜を、洗浄バッファーRW2で2回洗浄し、そしてRNAを溶出した。
【0105】
DNAをサンプル5〜8の上清(画分A)(サンプル5aから8a)から単離するために、その上清を、界面活性剤を含む溶解バッファーATL(QIAGEN)で、180μlの全容量にし、そして20μlのQIAGENからのプロテイナーゼKを加えた。そのサンプルを、1400rpmで振とうしながら、56℃で1時間インキュベートし、そして次いで90℃で1時間加熱した。存在するあらゆるRNAを分解するために、インキュベーション後に、4μlのRNAseA溶液(100mg/ml)をサンプルに混合した。DNAをさらに精製するために、そのサンプルを、各場合で200μlのカオトロピックバッファー、例えばQIAGENからのALバッファー、およびエタノールと混合した。その混合物を、例えばQIAGENからのQIAamp MinEluteカラムに存在するシリカ膜にアプライし、そして10000rpmで1分間遠心分離することによって膜を通過させた。次いでそのシリカ膜を、500μlのグアニジン塩を含む洗浄バッファーAW1で、そして次いで500μlのQIAGENからのアルコールを含む洗浄バッファーAW2で洗浄した。その膜を、14000rpmで2分間遠心分離することによって乾燥し、そして1分間のインキュベーション後、DNAを30μlのDNA溶出バッファー、例えばQIAGENからのバッファーATEと遠心分離することによって溶出した。
【0106】
DNAを、サンプル5〜8のペレット(画分B)(サンプル5bから8b)から単離するために、そのペレットを、例えば180μlのQIAGENからの界面活性剤を含むバッファーATLのような、習慣的なDNA溶解バッファーで処理した。ペレットは、前の処理によって溶解されなかった成分のみを含むので、20μlのQIAGENからのプロテイナーゼKによるさらなる溶解を、これらの未溶解成分も溶解するように行った。56℃で1時間、および続いて90℃で1時間インキュベートした後、その溶解産物を、カオトロープを含む結合バッファー、例えばQIAGENからのALバッファーおよびまたエタノールで処理した。その混合物を、例えばQIAGENからのQIAamp MinEluteカラムに存在するシリカ膜にアプライし、そして10000rpmで1分間遠心分離することによって、膜を通過させた。上記で記載したように、そのシリカ膜を、バッファーAW1およびAW2で洗浄し、その膜を乾燥し、そしてDNAを溶出した。
【0107】
2つの画分、上清(A)およびペレット(B)からこの方式で単離した核酸の分布を決定するために、両方の画分の核酸を、適当な方法を用いて定量化した。DNAおよびRNAの収率および純度の決定を、サンプルの吸収を260/280nmで測定することによる吸光度(OD)によって行った。各場合の収率を、全収量のパーセントで示した。ここで全収量は、上清およびペレットにおける1つの型の核酸の収量の合計である。表1は、4回の決定の平均値を示す。
【0108】
【表1】
さらなる分析のために、各場合において、それぞれの画分から単離した核酸10μlを、DNAの場合はTAEアガロースゲル(Tris酢酸塩/EDTA)上で、RNAの場合はホルムアルデヒド/アガロースゲル上で、習慣的な方法によって分離し、そしてエチジウムブロミドで染色した。その結果を図1に示す。
【0109】
その結果は、本発明によるプロセスを適用した場合、引き続く精製の前に核酸の明らかな分離/分画が存在することを示す。RNAは主に上清に存在し、一方DNAは主にペレット画分に存在する。
【0110】
実施例2:使用したタンパク質分解活性化合物の量の影響
この実験のために使用したサンプルは、包埋の後約7ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約10μmの厚さの切片を、これらのサンプルから調製した。各場合において反応あたり2つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0111】
脱パラフィン化のために、その組織をまずそれぞれ1mlのヘプタン中で10分間インキュベートした。50μlのメタノールを加え、そして混合した後、そのサンプルを遠心分離し、上清を除去し、そして残渣を室温で5分間風乾した。
【0112】
この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μl、20μl、または40μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで4℃で遠心分離した。RNAのさらなる単離のために、その上清を除去し、そしてペレットを廃棄した。
【0113】
その上清を、続いて80℃で15分間インキュベートした。結合条件に調整するために、320μlのカオトロピックバッファー、例えばQIAGENからのRBCバッファーを次いで加え、そして得た混合物をエタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。次いで500μlのアルコールを含む洗浄バッファーRW2を通過させることによって、そのシリカ膜を2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥した。次いでRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0114】
この方式で単離したRNAを分析するために、260nmにおける吸収を測定することによって、その収量を決定した。その結果を表2に示す。
【0115】
RNAの完全性を、Agilent Bioanalyzerを用いて決定し、そしてRIN値の形式で示した、ここで10のRIN値は、完全にインタクトなRNAを示し、そして0のRIN値は、完全に分解されたRNAを示す。その結果を同様に表2に示す。
【0116】
本発明によるプロセスにおける核酸の単離に対するだけでなく、増幅による引き続く分析に対しても、異なる量のタンパク質分解活性化合物の影響を調査するために、そのRNAを、定量的リアルタイムRT−PCRによって分析した。この目的のために、その単離されたRNAを、各場合2回の決定で、madH7転写物のアンプリコンを検出するために使用した。その溶出液をそれぞれ、水で1:10の比で希釈した。各場合において、5μlのこれらの希釈溶液を、リアルタイム−PCRのために使用した。製造会社の指示によって、例えばQIAGENからのQuantiTect SYBRGreen RT−PCRキットのような、リアルタイムRT−PCRのために適当なマスターミックスによって、25μlの全容量で、増幅をおこなった。例えば、Applied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標) 7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において、増幅をおこなった。測定したct値を用いて、平均値を決定し、それを表2に示す。
【0117】
【表2】
その結果は、使用したプロテイナーゼKの全ての量が、同等の収量、同等のRNA完全性、および同等のリアルタイムRT−PCRの結果をもたらしたことを示す。本発明によるプロセスにおけるタンパク質分解活性化合物の量は、従って広い範囲で変更することができる。
【0118】
実施例3:タンパク質分解活性化合物の反応時間
この実験のために使用したサンプルは、包埋後約5ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0119】
切片の脱パラフィン化、再水和、および乾燥を、実施例1に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で3時間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルを遠心分離し、そしてその上清(画分A)を、未溶解成分(画分B)を含むペレットから除去した。実施例1に記載したように、RNAを上清(画分A)から単離し、そしてDNAをペレット(画分B)から単離した。
【0120】
この方式で単離した核酸を分析するために、260nmにおける吸収を測定することによって、その収量を決定した。その結果を図2に示す。プロテイナーゼの反応時間を増加させると、より長いプロテイナーゼ作用によって、より多くの核酸がペレットから溶解するので、核酸の収量はペレットにおいて減少し、そしてそれに対応して上清において増加する。
【0121】
しかし、核酸のこの全体分布は、画分におけるDNAおよびRNA含有量についての情報は提供しない。従って、各場合において、DNAを含む画分(B)の溶出液10μlを、TAE−アガロースゲルで分析した。その結果を図3に示す。
【0122】
5分のみのプロテイナーゼK反応時間後には、RNAの大部分は依然として未溶解画分、すなわちペレットに留まっていることが明らかである。プロテイナーゼ反応時間を増加させると、この量が減少し、そして反応時間15分から先は、未溶解画分は少量のRNAしか含まないか、またはRNAは実質的にもはや検出されない。対照的に、DNAはかなりより長く(少なくとも90分間)、未溶解画分Bに留まる。従って、タンパク質分解活性化合物の反応時間を最適化することによって、2つの画分における核酸の型の分布を調整することが可能である。
【0123】
核酸の単離に対するだけでなく、増幅による分析にも対する、本発明によるプロセスにおけるタンパク質分解活性化合物の反応時間の長さの影響を調査するために、そのDNAを定量的リアルタイムPCRによって分析した。この目的のために、各場合において、等容量の単離DNA溶出液を、各場合2回の決定で、prnp遺伝子のアンプリコンを検出するために使用した。例えばQIAGENからのQuantiTect SYBRGreen PCRキットのような、リアルタイムRT−PCRに適当なマスターミックスを用いて、製造会社の指示に従って、25μlの全量で増幅を行った。例えば、Applied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標) 7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において、増幅をおこなった。ct値から決定した平均値を、図4に示す。
【0124】
その結果は、反応時間が増加すると、ct値が増加することを示し、それは溶出液中のDNAの量が減少したためである。反応時間を増加させると、RNAだけでなくDNAも徐々に上清へ移るが、ここでRNAは、DNAよりも著しくより迅速に、上清に溶解および移動する(encounter)。ゲルは、15分の反応時間後でも、RNAは既に実質的に完全にペレットから溶解し、そして上清に移動していることを示すが、未溶解画分BのDNAの量は、ゲルおよびリアルタイムPCRによると、90分超後にのみ有意に減少する。
【0125】
実施例4:本発明によるプロセスにおける、異なる水溶液の使用
この実験のために使用したサンプルは、包埋後約7ヵ月間室温で保存されていたラット肝臓由来の、ホルマリン固定およびパラフィン包埋組織サンプル(FFPEサンプル)であった。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。処理したサンプルからのDNAおよびRNAの引き続く単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0126】
切片の脱パラフィン化、再水和、および乾燥を、実施例2に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液1(pH7)、50mMのTris、25mMのEDTA、1%のSDS、0.1%のNonidet NP40、および500mMのNaClを含む水溶液2(pH7.4)、または50mMのTris、100mMのEDTA、3%のSDSおよび10mMのNaClを含む水溶液3(pH8.2)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。その混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで4℃で遠心分離した。DNAを、実施例1で記載したようにペレット(画分B)から単離し、そのペレットを50mMのTris、25mMのEDTA、1%のSDS、0.1%のNonidet P−40、および500mMのNaClを含むバッファー(pH7.4)中に溶解した。その収量を、260nmにおける吸収を測定することによって決定した。その結果を表3に示す。
【0127】
【表3】
使用した3つの水溶液は全て、高い収量のDNAを与え、一定の変動がサンプルの不均一性によって生じた。本実施例は、多数の異なる水溶液を、本発明によるプロセスのために使用し得、その成分および濃度の両方を変動させることが可能であることを示す。従って、使用する水溶液を、使用するタンパク質分解活性化合物に適合させることが可能である。
【0128】
実施例5:本発明によるプロセスの補助による、異なる型の組織からのDNAの単離
使用したサンプルは、室温で異なる期間保存されていた、ラット由来のFFPEサンプルであった:腎臓(保存期間約13ヶ月)、肝臓(保存期間約6ヶ月)、脾臓(保存期間約19ヶ月)、心臓(保存期間約13ヶ月)、および肺(保存期間約6ヶ月)。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルそれぞれから調製した。各場合において、反応あたり1つの切片を使用した。本発明による方法の補助による、処理したサンプルからの引き続く核酸の単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0129】
本発明によるプロセスの補助によるDNAの単離を、FFPEサンプルからのDNAの精製のために特に確立されたプロセスと比較するために、各場合において同じサンプルの切片を、製造会社(QIAGEN)の指示による、QIAampFFPEキットによるDNA単離のために、およびコントロールサンプルとして使用した。
【0130】
切片の脱パラフィン化、再水和、および乾燥を、実施例2に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。その混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで遠心分離した。DNAを、90℃で2時間インキュベートして、実施例1で記載したようにペレット(画分B)から単離した。
【0131】
得られたDNAの分析のために、その収量を、260nmにおける吸収を測定することによって決定した。2組の決定の平均値および標準偏差を図5に示す。本発明によるプロセスの補助によって、DNAを全てのサンプルから単離し得、全ての場合の収量は、コントロールプロセスで得られた収量を超えた。
【0132】
さらに、各場合において、10μlのDNA溶出液を、TAE−アガロースゲルで分離し、そしてエチジウムブロミドで染色した。その結果を図6に示す。全ての場合において、本発明による方法の補助によって単離されたDNAは、コントロール方法で単離されたDNAと、およそ同じ分子サイズ分布を示した。
【0133】
本発明によるプロセスによって単離されたDNAの増幅分析に対する適合性を調査するために、この方式で得られたDNAを、定量的リアルタイムPCRアッセイにおいて使用した。等容量の単離DNA溶出液を、pmp遺伝子の465塩基対のアンプリコンを検出するために、2組の決定において使用した。
【0134】
FFPEサンプルにおいて、そのDNAは、原則として断片化形態で存在し、単離し得るDNA断片の断片化の程度および従ってスペクトルは、特に固定および包埋の性質に依存するが、またサンプルの種類およびサンプルの保存にも依存する。さらに、核酸の単離後に残るDNAの架橋の程度が、増幅、および特にアンプリコンの可能性のあるサイズを制限する。これにも関わらず効率的な増幅を保証するためには、原則として小さいアンプリコンが好ましい。ここで使用した465bpというアンプリコンサイズは、FFPEサンプルに関しては非常に大きく、そしてそれを本発明によるプロセスによって単離されたDNAの質および適合性を試験するために選択した。
【0135】
増幅を、例えば、QIAGENからのQuantiTect SYBRGreen PCRキットのような、リアルタイムRT−PCRに適当なマスターミックスを用いて、製造会社の指示に従って、25μlの全容量で行った。増幅を、例えばApplied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標)7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において、増幅をおこなった。測定したct値を用いて、本発明によって単離されたDNAの平均値および標準偏差を決定し、それを図7に示す。
【0136】
全ての場合において、ct値は、コントロールDNAのものと同等であるか、またはさらにより低く、そのことはより良い増幅能力(amplifiability)および/またはより高い収量を確実にする。
【0137】
全体として、その結果は、本発明によるプロセスにより、FFPEサンプルからのDNAの単離が可能となること、収量、質、断片サイズ、および増幅分析への適合性に関し、FFPEサンプルからのDNAの特異的単離について、先行技術から公知のプロセスによって単離したDNAと少なくとも同じ、またはより良好であるということを示す。
【0138】
実施例6:本発明によるプロセスによる、異なる型の組織からのRNAの単離
この実験のために使用したサンプルは、異なる期間室温で保存されていた、ラット由来のFFPEサンプルであった:腎臓(保存期間約5ヶ月)、肝臓(保存期間約24ヶ月)、心臓(保存期間約24ヶ月)、および肺(保存期間約24ヶ月)。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。本発明のプロセスの補助による、FFPE切片からの引き続く核酸の単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0139】
本発明によるプロセスの補助によるRNAの単離を、FFPEサンプルからのRNAの精製のために特に確立されたプロセスと比較するために、同じサンプルの切片を、製造会社(QIAGEN)の指示による、RNeasy FFPEキットによるRNAの単離のために、およびコントロールサンプルとして使用した。
【0140】
切片の脱パラフィン化、再水和、および乾燥を、実施例2に記載したように行った。この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。溶解画分(A)を、未溶解画分(B)から分離するために、そのサンプルをまず氷上で5分間冷却し、そして次いで遠心分離した。RNAのさらなる単離のために、その上清(画分A)を除去し、そしてペレットを廃棄した。
【0141】
その上清を続いて80℃で15分間インキュベートした。そのサンプルを室温で5分間冷却し、その後20μlの従来のDNAseバッファー(例えば0.46MのTris−HCl(pH7.5)、114mMのNaCl、114mMのMgCl2、114mMのCaCl2を含む)、15μlの脱イオン水、および5μlのQIAGENからのDNAseI溶液を加え、そしてその混合物を室温で15分間インキュベートした。400μlのカオトロピックバッファー、例えばQIAGENからのRLTバッファーを次いで加え、その混合物をエタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。500μlのアルコールを含む洗浄バッファーRW2(QIAGEN)で、そのシリカ膜を2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そしてRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0142】
この方式で単離したRNAを分析するために、260nmにおける吸収を測定することによってその収量を決定した。2組の決定の平均値を、表4に示す。
【0143】
【表4】
本発明によるプロセスの補助によって、RNAを全てのサンプルから単離することが可能であり、ここで、全ての場合において、本発明によるプロセスによって得られた収量は、コントロールのものと同等、またはそれより高かった。
【0144】
本発明によるプロセスによって単離されたRNAの、増幅分析のための適合性を調査するために、そのRNAを、定量的リアルタイムRT−PCRアッセイにおいて使用した。等容量の単離RNA溶出液を、各場合においてmadH7転写産物およびc−jun転写産物のアンプリコンを検出するために、2組の決定において使用した。増幅を、例えば、QIAGENからのQuantiTect SYBRGreen RT−PCRキットのような、リアルタイムRT−PCRに適当なマスターミックスを用いて、製造会社の指示に従って、25μlの全容量で行った。増幅を、例えばApplied Biosystems(Carlsbad、California、USA)からの、ABI PRISM(登録商標)7900HT Sequence Detection Systemのような、適当なリアルタイム増幅装置において行った。それに加えて、microRNA16(miR16)を、リアルタイムRT−PCRによって、製造会社(QIAGEN)の指示に従って、miScript PCRシステムを用いて、RNA溶出液において検出した。測定したct値から得た平均値を、表5に示す。
【0145】
【表5】
全ての場合において、本発明によって処理したサンプルの測定したct値は、コントロールサンプルのものと同等であるか、またはさらにより低く、そのことはよりよい増幅能力またはより多いRNA量によるものである。
【0146】
実施例7:効率的なmiRNA精製のためのDNAse処理
この実験のために、異なる期間室温で保存していたラット由来のFFPEサンプルを使用した:脳(保存期間約5ヶ月)および心臓(保存期間約18ヶ月)。ミクロトームの補助によって、約20μmの厚さの切片を、これらのサンプルから調製した。各場合において、反応あたり1つの切片を使用した。本発明のプロセスの補助による、FFPE切片からの引き続く核酸の単離のために、QIAGENからのRNeasyFFPEキットおよびQIAampFFPEキットの構成成分を使用した。
【0147】
本発明によるプロセスの補助によるmiRNAの単離を、FFPEサンプルからのmiRNAの精製のために特に確立されたプロセスと比較するために、同じサンプルの切片を、製造会社(QIAGEN)の指示による、miRNeasy FFPEキットによるmiRNAの単離のために、およびコントロールサンプルとして使用した。
【0148】
この方式で得た脱パラフィン化サンプルペレットを、20mMのTris、2mMのEDTAおよび0.2%のSDSを含む150μlの水溶液(pH7)で処理し、そしてタンパク質分解活性化合物として、10μlのプロテイナーゼK溶液(>600mAU/ml)と混合した。この混合物を、1400rpmで振とうしながら、56℃で15分間インキュベートした。主にRNAを含む溶解画分(A)を、主にDNAを含む未溶解画分(B)から分離するために、そのサンプルをまず氷上で3分間冷却し、そして次いで遠心分離した。miRNAを含むRNAのさらなる単離のために、上清(画分A)を除去し、そしてDNAを含むペレットを廃棄した。
【0149】
続いて上清を80℃で15分間インキュベートして、架橋を逆行させた。そのサンプルを室温で5分間冷却し、その後DNase活性を促進するための異なるバッファー(前処理バッファー1〜5、下記を参照のこと)を20μl、15μlの水、および5μlのQIAGENからのDNAseI溶液を加えた。この実験のために以下のバッファーを使用した:
前処理バッファー1:0.46MのTris−HCl(pH7.5)、114mMのNaCl、114mMのMgCl2、114mMのCaCl2
前処理バッファー2:0.46MのTris−HCl(pH7.5)、114mMのMgCl2、114mMのCaCl2
前処理バッファー3:46mMのTris−HCl(pH7.5)、11.4mMのNaCl、11.4mMのMgCl2、11.4mMのCaCl2
前処理バッファー4:20mMのTris−HCl(pH7.5)、100mMのMgCl2、10mMのCaCl2
前処理バッファー5:20mMのTris−HCl(pH7.5)、100mMのMgCl2、2.5mMのCaCl2
その混合物を、室温で15分間インキュベートした。次いでDNase消化サンプルから、マイクロRNAのような小さいRNAを含むRNAを単離するために、400μlのカオトロピックバッファー、例えばQIAGENからのRLTバッファーを加え、その混合物を1400μlの96〜100%エタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。500μlのアルコールを含む洗浄バッファーRPE(QIAGEN)で、そのシリカ膜を2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そしてRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0150】
比較のために、DNAse前処理をしないが、RNAの膜への結合の後に通常のオンカラムDNAse処理を行う、小さいRNAを含むRNAの精製のために、同じサンプルを使用した。脱パラフィン化およびプロテイナーゼK消化を、上記で記載したように行った。その後、320μlのカオトロピックバッファー、例えばQIAGENからのRLTバッファーを次いで加え、その混合物を1120μlの96〜100%エタノールと混合し、例えばQIAGENからのRNeasy MinEluteカラムに存在するシリカ膜にアプライし、そして14000rpmで1分間遠心分離することによって膜を通過させた。バッファーRWT(QIAGEN)のような、カオトロピック試薬およびエタノールを含む350μlの洗浄バッファーで、そのシリカ膜を洗浄した。10μlのDNase1および適当なDNAseバッファー(例えばバッファーRDD(QIAGEN))を含む80μlの混合物を、次いでその膜にアプライし、そして室温で15分間インキュベートした。その後、その膜を再びバッファーRWTで洗浄し、そして500μlのアルコールを含む洗浄バッファーRPE(QIAGEN)で2回洗浄した。その膜を、14000rpmで5分間遠心分離することによって乾燥し、そしてRNAを、1分間インキュベートした後、30μlの水を加えることによって遠心分離によって溶出した。
【0151】
この方式で単離したRNAを分析するために、代表的な脳のRNAを、RNA分子をサイズに依存して分離する、Agilent Bioanalyzerを用いて分析した。図8は、Bioanalyzer測定の結果を示す。FFPEサンプル由来のRNAは常に、部分的に分解されており、そして分解の程度は、サンプルの固定、包埋および保存、およびRNAの抽出方法のような複数の因子に依存する。従って、RNAのゲル様の視覚化は、全ての場合において、部分的に分解したRNAを示す(図8を参照のこと)。28SrRNAは明らかでなく、そして18SrRNAは弱く現れるのみである。それに加えて、多数のRNA断片が、28srRNAのバンドのサイズから低分子量まで生じる。通常のオンカラムDNAse処理は、miRNAを含む最も小さいRNA集団の、非常に低い収量をもたらす(矢印を参照のこと)。対照的に、本発明によるカラムに添加する前のDNAse前処理は、非常に低分子量のRNAの多量の単離を可能にする。
【0152】
特にmiRNAの精製の効率を決定するために、miScript PCR Systemを用いて、製造会社(QIAGEN)の指示に従って、リアルタイムRT−PCRによって、精製RNAを、miRNAの検出および定量に関して分析した。測定したct値から得られる平均値を、表1に示す。
【0153】
【表6】
全ての場合において、測定したct値は、DNAse前処理をしたサンプルにおいてより低く、一方オンカラムDNAse処理は、有意により高いct値を与える。より低いct値は、より多量のmiRNAを示し、ct値の1の差異は、検出されたmiRNAの約2倍の量を示す。従って、RNAを単離する前のDNAse前処理は、従来技術によるオンカラムDNase消化に対して、miRNA精製効率を有意に増強する。
【0154】
全体として、その結果は、本発明によるプロセスにより、FFPEサンプルからのRNAの単離が可能となること、収量、質、断片サイズおよび増幅分析への適合性に関し、先行技術から公知の単離プロセスによって単離したRNAと、少なくとも同程度に良好であり、かつFFPEサンプルからのRNAの単離に特異的であることを示す。
【特許請求の範囲】
【請求項1】
架橋によって固定された同じ生物学的サンプルからの、リボ核酸(RNA)およびデオキシリボ核酸(DNA)の並行単離および/または並行精製のためのプロセスであって、該プロセスは、以下の工程:
a)少なくとも1つのタンパク質分解活性化合物を用いた、該サンプルのタンパク質含有成分の部分的なタンパク質分解と同時に、該サンプルを水性緩衝溶液中に部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット;画分B)を得る工程;
b)該溶解画分を、該未溶解残渣から分離する工程、
を含み、
ここで、該溶解画分における核酸の全量に基づいて、該溶解画分は主にRNAを含み、そして該未溶解残渣における核酸の全量に基づいて、該未溶解残渣は主にDNAを含み、
そして、主にRNAを含む該画分の、主にDNAを含む該画分からの分離は、有機溶媒による1つまたは両方の型の核酸の沈殿も抽出も、1つまたは両方の型の核酸の固体マトリックスへの選択的結合のいずれも必要としない、プロセス。
【請求項2】
前記水性緩衝溶液は、少なくとも1つの緩衝物質、および/または、好ましくは少なくとも1つの界面活性剤を含み、
該緩衝物質は、Tris、Hepes、Pipes、Mops、およびアルカリ金属酢酸塩/酢酸を含む群から好ましくは選択され、
該界面活性剤は、ドデシル硫酸ナトリウム(SDS)、デオキシコール酸ナトリウム、3−(3−コールアミドプロピル)ジメチルアンモニウム−1−プロパンスルホネート(CHAPS)、ポリエチレングリコールフェニルエーテル、またはこれらの混合物、特に好ましくは、ドデシル硫酸ナトリウム、40のエトキシ化度を有するポリエチレングリコールノニルフェニルエーテル、および/または9〜10のエトキシ化度を有するポリエチレングリコール(1,1,3,3−テトラメチルブチル)フェニルエーテルを含む群から好ましくは選択される、請求項1に記載のプロセス。
【請求項3】
前記水性緩衝溶液は、以下:
・錯体形成剤、好ましくはエチレンジアミン−N,N,N’,N’−4酢酸(EDTA)、エチレングリコールビス(2−アミノエチルエーテル)−N,N,N’,N’−4酢酸(EGTA)、クエン酸ナトリウム、またはこれらの混合物、
・好ましくは0.1から10Mの濃度の、塩酸グアニジン、チオシアン酸グアニジン、イソチオシアン酸グアニジン、過塩素酸塩、NaI、KIおよび尿素を含む群から好ましくは選択される、カオトロピック剤、
・ジチオスレイトール(DTT)、ジチオエリスリトール(DTE)、チオ硫酸ナトリウム、β−メルカプトエタノール、またはこれらの混合物を含む群から好ましくは選択される、還元剤、および
・無機塩、好ましくはアルカリ金属ハロゲン化物、特に好ましくはNaCl、KCl、またはLiCl、好ましくはアルカリ土類金属ハロゲン化物、特に好ましくはCaCl2、またはMgCl2、好ましくはアンモニウム塩、特に好ましくは塩化アンモニウムまたは硫酸アンモニウム、好ましくは硫酸リチウム、またはこれらの混合物
を含む群から選択される、少なくとも1つの物質をさらに含み、そして
該水性緩衝溶液は、6から9の範囲のpH、好ましくは6.5から8.5の範囲のpH、特に好ましくは6.8から7.5の範囲のpHを好ましくはさらに有する、請求項2に記載のプロセス。
【請求項4】
前記タンパク質分解活性化合物は、プロテアーゼおよび非酵素的タンパク質分解活性化合物、好ましくはプロテイナーゼK、トリプシン、キモトリプシン、パパイン、ペプシン、プロナーゼ、エンドプロテアーゼLys−C、および臭化シアン、またはこれらの混合物、特にプロテイナーゼKを含む群から選択され、前記水溶液中の該タンパク質分解活性化合物の総濃度は、該水溶液の全重量に基づいて、好ましくは0.001重量%から5重量%、特に好ましくは0.01重量%〜2.5重量%、そして特に0.05重量%〜0.2重量%の範囲である、請求項1〜3のいずれかに記載のプロセス。
【請求項5】
前記水性緩衝溶液中の前記サンプルの前記部分的溶解は、好ましくは18℃から80℃の温度、特に50℃から65℃の温度で、そして好ましくは30秒から5日の期間、特に好ましくは1分から5時間の期間、より好ましくは5分から90分の期間、そして特に10分から30分の期間、該水性緩衝溶液中の該サンプルのインキュベーションにより生じる、請求項1〜4のいずれかに記載のプロセス。
【請求項6】
架橋により固定された前記生物学的サンプルは、パラフィン包埋サンプル、好ましくはホルマリン固定パラフィン包埋サンプル(FFPEサンプル)である、請求項1〜5のいずれかに記載のプロセス。
【請求項7】
前記プロセスは、工程(a)による前記部分的溶解の前に、好ましくは前記サンプルを疎水性有機溶媒に接触させることによって、特に好ましくは、C1〜C5アルコールの添加を必要に応じて伴って、6個より多く17個未満の炭素原子の鎖長の非極性の脂肪族もしくは芳香族炭化水素またはこれらの混合物を使用して;特に1〜25容量%のメタノールの添加を必要に応じて伴って、キシレン、ヘプタン、および鉱物油を含む群から選択される炭化水素または炭化水素混合物を使用して、前記パラフィンを選択的に除去するための工程(i)を含む、請求項6に記載のプロセス。
【請求項8】
工程(i)による前記パラフィンの除去の後、および工程(a)による前記水性緩衝溶液における前記サンプルの前記部分的溶解の前に、以下の工程:
(ii)好ましくは、該サンプルを、連続的に水含有量を増加させた水性C1〜C5アルコール溶液で繰り返し洗浄することによって、該サンプルを再水和する工程、
(iii)該サンプルを乾燥する工程、および/または
(iv)該サンプルをホモジナイズする工程
のうちの少なくとも1つを含む、請求項7に記載のプロセス。
【請求項9】
得られた前記未溶解残渣からの前記DNAの続いて起こる放出のために、該未溶解残渣は、同時酵素プロテアーゼ消化による溶解に供される、請求項1〜8のいずれかに記載のプロセス。
【請求項10】
請求項1による工程(b)に続いて、前記溶解画分Aから得られる前記RNAおよび/またはペレットBから得られる前記DNAを、好ましくは沈殿、核酸結合材料への結合、電気泳動、および/またはクロマトグラフィーによって、別々に精製するためのさらなる工程を含む、請求項1〜9のいずれかに記載のプロセス。
【請求項11】
主にDNAを含む前記未溶解画分は、分離後に廃棄される、請求項1〜10のいずれかに記載のプロセス。
【請求項12】
主にRNAを含む前記溶解画分からの該RNAの単離の前に、DNase消化が、該画分に対して行われる、請求項1〜11の一つ以上に記載のプロセス。
【請求項13】
増幅技術、特にPCR、qPCR、RT−PCR、qRT−PCRおよびゲノムDNA全体の増幅(全ゲノム増幅)、ゲル電気泳動、ブロッティング技術、特にサザンブロッティングおよびノーザンブロッティング、マイクロアレイ分析、制限酵素断片長多型解析(RFLP分析)、SAGE(遺伝子発現の連続分析)、NextGeneration配列決定およびRNA配列決定を含む配列決定、一塩基多型分析(SNP分析)、突然変異分析、エピジェネティック分析、特にメチル化パターンの分析、またはそれらの組み合わせを含む群から好ましくは選択される、単離かつ/または精製された核酸の検出のための工程をさらに含む、請求項1〜12のいずれかに記載のプロセス。
【請求項14】
(1)タンパク質分解活性化合物、好ましくは請求項4に記載のタンパク質分解活性化合物、(2)少なくとも1つの緩衝物質、好ましくは請求項2に記載の緩衝物質、および(3)少なくとも1つの界面活性剤、好ましくは請求項2に記載の界面活性剤の1つ、ならびに好ましくは(4)工程(a)による不完全なタンパク質分解を行うための指示、を少なくとも含む、請求項1〜13のいずれかに記載のプロセスを行うためのキット。
【請求項15】
(5)少なくとも1つの核酸結合材料、およびまた必要に応じて(6)核酸精製のためのバッファー、好ましくは結合および/または溶出バッファーをさらに含む、請求項14に記載のキット。
【請求項16】
生物学的サンプル中に存在する核酸の分析および/または定量化のための、請求項14または15に記載のキットの使用。
【請求項17】
ヒトの身体外または動物の身体外のサンプルを用いた、疾患の診断、予後、治療に関する決定、および/または治療のモニタリングのための、請求項14または15に記載のキットの使用。
【請求項18】
架橋によって固定された同じ生物学的サンプルから、溶解画分中のRNA、および未溶解画分中のDNAを得るためのプロセスであって、該プロセスは、以下の工程:
a)少なくとも1つのタンパク質分解活性化合物を用いた、該サンプルのタンパク質含有成分の部分的なタンパク質分解と同時に、該サンプルを水性緩衝溶液中に部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット;画分B)を得る工程;
b)該溶解画分を、該未溶解残渣から分離する工程、
を含み、
ここで、該溶解画分における核酸の全量に基づいて、該溶解画分は主にRNAを含み、そして該未溶解残渣における核酸の全量に基づいて、該未溶解残渣は主にDNAを含む、プロセス。
【請求項19】
主にRNAを含む前記画分の、主にDNAを含む前記画分からの分離は、有機溶媒による1つまたは両方の型の核酸の沈殿も抽出も、1つまたは両方の型の核酸の固体マトリックスへの選択的結合のいずれも必要としない、請求項18に記載のプロセス。
【請求項20】
前記画分の分離後、前記RNAは、主にRNAを含む前記溶解画分から単離され、そして/またはDNAは、主にDNAを含む前記未溶解画分から単離される、請求項18または19に記載のプロセス。
【請求項21】
主にDNAを含む前記未溶解画分は、分離後に廃棄される、請求項18〜20のうちの一つ以上に記載のプロセス。
【請求項22】
請求項1〜13のうちの一つ以上で規定される工程および/または特徴のうちの一つ以上を含む、請求項18〜21のうちの一つ以上に記載のプロセス。
【請求項1】
架橋によって固定された同じ生物学的サンプルからの、リボ核酸(RNA)およびデオキシリボ核酸(DNA)の並行単離および/または並行精製のためのプロセスであって、該プロセスは、以下の工程:
a)少なくとも1つのタンパク質分解活性化合物を用いた、該サンプルのタンパク質含有成分の部分的なタンパク質分解と同時に、該サンプルを水性緩衝溶液中に部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット;画分B)を得る工程;
b)該溶解画分を、該未溶解残渣から分離する工程、
を含み、
ここで、該溶解画分における核酸の全量に基づいて、該溶解画分は主にRNAを含み、そして該未溶解残渣における核酸の全量に基づいて、該未溶解残渣は主にDNAを含み、
そして、主にRNAを含む該画分の、主にDNAを含む該画分からの分離は、有機溶媒による1つまたは両方の型の核酸の沈殿も抽出も、1つまたは両方の型の核酸の固体マトリックスへの選択的結合のいずれも必要としない、プロセス。
【請求項2】
前記水性緩衝溶液は、少なくとも1つの緩衝物質、および/または、好ましくは少なくとも1つの界面活性剤を含み、
該緩衝物質は、Tris、Hepes、Pipes、Mops、およびアルカリ金属酢酸塩/酢酸を含む群から好ましくは選択され、
該界面活性剤は、ドデシル硫酸ナトリウム(SDS)、デオキシコール酸ナトリウム、3−(3−コールアミドプロピル)ジメチルアンモニウム−1−プロパンスルホネート(CHAPS)、ポリエチレングリコールフェニルエーテル、またはこれらの混合物、特に好ましくは、ドデシル硫酸ナトリウム、40のエトキシ化度を有するポリエチレングリコールノニルフェニルエーテル、および/または9〜10のエトキシ化度を有するポリエチレングリコール(1,1,3,3−テトラメチルブチル)フェニルエーテルを含む群から好ましくは選択される、請求項1に記載のプロセス。
【請求項3】
前記水性緩衝溶液は、以下:
・錯体形成剤、好ましくはエチレンジアミン−N,N,N’,N’−4酢酸(EDTA)、エチレングリコールビス(2−アミノエチルエーテル)−N,N,N’,N’−4酢酸(EGTA)、クエン酸ナトリウム、またはこれらの混合物、
・好ましくは0.1から10Mの濃度の、塩酸グアニジン、チオシアン酸グアニジン、イソチオシアン酸グアニジン、過塩素酸塩、NaI、KIおよび尿素を含む群から好ましくは選択される、カオトロピック剤、
・ジチオスレイトール(DTT)、ジチオエリスリトール(DTE)、チオ硫酸ナトリウム、β−メルカプトエタノール、またはこれらの混合物を含む群から好ましくは選択される、還元剤、および
・無機塩、好ましくはアルカリ金属ハロゲン化物、特に好ましくはNaCl、KCl、またはLiCl、好ましくはアルカリ土類金属ハロゲン化物、特に好ましくはCaCl2、またはMgCl2、好ましくはアンモニウム塩、特に好ましくは塩化アンモニウムまたは硫酸アンモニウム、好ましくは硫酸リチウム、またはこれらの混合物
を含む群から選択される、少なくとも1つの物質をさらに含み、そして
該水性緩衝溶液は、6から9の範囲のpH、好ましくは6.5から8.5の範囲のpH、特に好ましくは6.8から7.5の範囲のpHを好ましくはさらに有する、請求項2に記載のプロセス。
【請求項4】
前記タンパク質分解活性化合物は、プロテアーゼおよび非酵素的タンパク質分解活性化合物、好ましくはプロテイナーゼK、トリプシン、キモトリプシン、パパイン、ペプシン、プロナーゼ、エンドプロテアーゼLys−C、および臭化シアン、またはこれらの混合物、特にプロテイナーゼKを含む群から選択され、前記水溶液中の該タンパク質分解活性化合物の総濃度は、該水溶液の全重量に基づいて、好ましくは0.001重量%から5重量%、特に好ましくは0.01重量%〜2.5重量%、そして特に0.05重量%〜0.2重量%の範囲である、請求項1〜3のいずれかに記載のプロセス。
【請求項5】
前記水性緩衝溶液中の前記サンプルの前記部分的溶解は、好ましくは18℃から80℃の温度、特に50℃から65℃の温度で、そして好ましくは30秒から5日の期間、特に好ましくは1分から5時間の期間、より好ましくは5分から90分の期間、そして特に10分から30分の期間、該水性緩衝溶液中の該サンプルのインキュベーションにより生じる、請求項1〜4のいずれかに記載のプロセス。
【請求項6】
架橋により固定された前記生物学的サンプルは、パラフィン包埋サンプル、好ましくはホルマリン固定パラフィン包埋サンプル(FFPEサンプル)である、請求項1〜5のいずれかに記載のプロセス。
【請求項7】
前記プロセスは、工程(a)による前記部分的溶解の前に、好ましくは前記サンプルを疎水性有機溶媒に接触させることによって、特に好ましくは、C1〜C5アルコールの添加を必要に応じて伴って、6個より多く17個未満の炭素原子の鎖長の非極性の脂肪族もしくは芳香族炭化水素またはこれらの混合物を使用して;特に1〜25容量%のメタノールの添加を必要に応じて伴って、キシレン、ヘプタン、および鉱物油を含む群から選択される炭化水素または炭化水素混合物を使用して、前記パラフィンを選択的に除去するための工程(i)を含む、請求項6に記載のプロセス。
【請求項8】
工程(i)による前記パラフィンの除去の後、および工程(a)による前記水性緩衝溶液における前記サンプルの前記部分的溶解の前に、以下の工程:
(ii)好ましくは、該サンプルを、連続的に水含有量を増加させた水性C1〜C5アルコール溶液で繰り返し洗浄することによって、該サンプルを再水和する工程、
(iii)該サンプルを乾燥する工程、および/または
(iv)該サンプルをホモジナイズする工程
のうちの少なくとも1つを含む、請求項7に記載のプロセス。
【請求項9】
得られた前記未溶解残渣からの前記DNAの続いて起こる放出のために、該未溶解残渣は、同時酵素プロテアーゼ消化による溶解に供される、請求項1〜8のいずれかに記載のプロセス。
【請求項10】
請求項1による工程(b)に続いて、前記溶解画分Aから得られる前記RNAおよび/またはペレットBから得られる前記DNAを、好ましくは沈殿、核酸結合材料への結合、電気泳動、および/またはクロマトグラフィーによって、別々に精製するためのさらなる工程を含む、請求項1〜9のいずれかに記載のプロセス。
【請求項11】
主にDNAを含む前記未溶解画分は、分離後に廃棄される、請求項1〜10のいずれかに記載のプロセス。
【請求項12】
主にRNAを含む前記溶解画分からの該RNAの単離の前に、DNase消化が、該画分に対して行われる、請求項1〜11の一つ以上に記載のプロセス。
【請求項13】
増幅技術、特にPCR、qPCR、RT−PCR、qRT−PCRおよびゲノムDNA全体の増幅(全ゲノム増幅)、ゲル電気泳動、ブロッティング技術、特にサザンブロッティングおよびノーザンブロッティング、マイクロアレイ分析、制限酵素断片長多型解析(RFLP分析)、SAGE(遺伝子発現の連続分析)、NextGeneration配列決定およびRNA配列決定を含む配列決定、一塩基多型分析(SNP分析)、突然変異分析、エピジェネティック分析、特にメチル化パターンの分析、またはそれらの組み合わせを含む群から好ましくは選択される、単離かつ/または精製された核酸の検出のための工程をさらに含む、請求項1〜12のいずれかに記載のプロセス。
【請求項14】
(1)タンパク質分解活性化合物、好ましくは請求項4に記載のタンパク質分解活性化合物、(2)少なくとも1つの緩衝物質、好ましくは請求項2に記載の緩衝物質、および(3)少なくとも1つの界面活性剤、好ましくは請求項2に記載の界面活性剤の1つ、ならびに好ましくは(4)工程(a)による不完全なタンパク質分解を行うための指示、を少なくとも含む、請求項1〜13のいずれかに記載のプロセスを行うためのキット。
【請求項15】
(5)少なくとも1つの核酸結合材料、およびまた必要に応じて(6)核酸精製のためのバッファー、好ましくは結合および/または溶出バッファーをさらに含む、請求項14に記載のキット。
【請求項16】
生物学的サンプル中に存在する核酸の分析および/または定量化のための、請求項14または15に記載のキットの使用。
【請求項17】
ヒトの身体外または動物の身体外のサンプルを用いた、疾患の診断、予後、治療に関する決定、および/または治療のモニタリングのための、請求項14または15に記載のキットの使用。
【請求項18】
架橋によって固定された同じ生物学的サンプルから、溶解画分中のRNA、および未溶解画分中のDNAを得るためのプロセスであって、該プロセスは、以下の工程:
a)少なくとも1つのタンパク質分解活性化合物を用いた、該サンプルのタンパク質含有成分の部分的なタンパク質分解と同時に、該サンプルを水性緩衝溶液中に部分的に溶解して、溶解画分(画分A)および未溶解残渣(ペレット;画分B)を得る工程;
b)該溶解画分を、該未溶解残渣から分離する工程、
を含み、
ここで、該溶解画分における核酸の全量に基づいて、該溶解画分は主にRNAを含み、そして該未溶解残渣における核酸の全量に基づいて、該未溶解残渣は主にDNAを含む、プロセス。
【請求項19】
主にRNAを含む前記画分の、主にDNAを含む前記画分からの分離は、有機溶媒による1つまたは両方の型の核酸の沈殿も抽出も、1つまたは両方の型の核酸の固体マトリックスへの選択的結合のいずれも必要としない、請求項18に記載のプロセス。
【請求項20】
前記画分の分離後、前記RNAは、主にRNAを含む前記溶解画分から単離され、そして/またはDNAは、主にDNAを含む前記未溶解画分から単離される、請求項18または19に記載のプロセス。
【請求項21】
主にDNAを含む前記未溶解画分は、分離後に廃棄される、請求項18〜20のうちの一つ以上に記載のプロセス。
【請求項22】
請求項1〜13のうちの一つ以上で規定される工程および/または特徴のうちの一つ以上を含む、請求項18〜21のうちの一つ以上に記載のプロセス。
【図1】

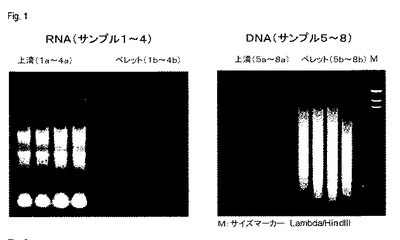
【図2】
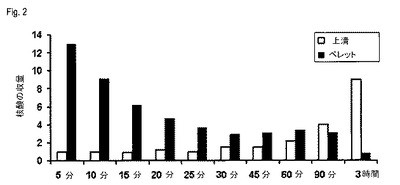
【図3】
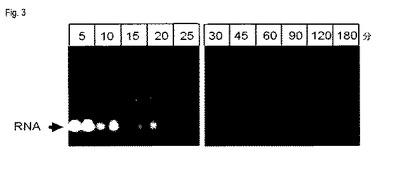
【図4】
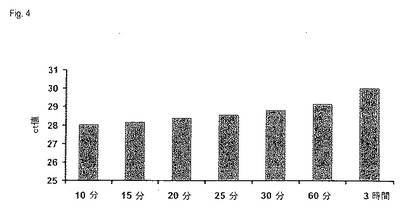
【図5】
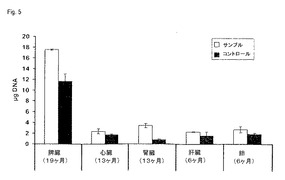
【図7】
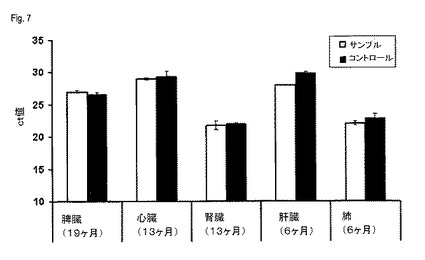
【図8】
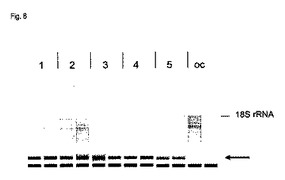
【図6】
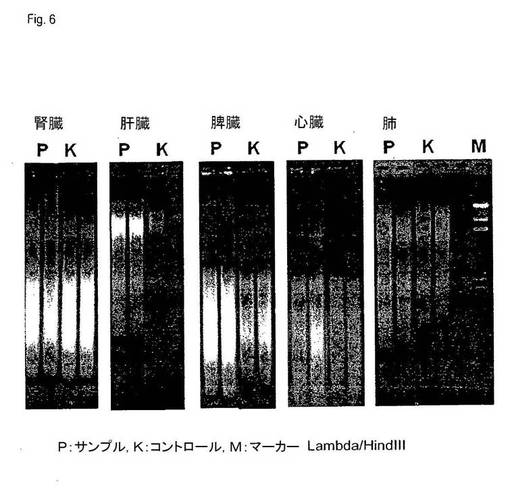
【図2】
【図3】
【図4】
【図5】
【図7】
【図8】
【図6】
【公表番号】特表2013−520187(P2013−520187A)
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願番号】特願2012−554251(P2012−554251)
【出願日】平成23年2月25日(2011.2.25)
【国際出願番号】PCT/EP2011/000920
【国際公開番号】WO2011/104027
【国際公開日】平成23年9月1日(2011.9.1)
【出願人】(507214038)キアゲン ゲーエムベーハー (16)
【Fターム(参考)】
【公表日】平成25年6月6日(2013.6.6)
【国際特許分類】
【出願日】平成23年2月25日(2011.2.25)
【国際出願番号】PCT/EP2011/000920
【国際公開番号】WO2011/104027
【国際公開日】平成23年9月1日(2011.9.1)
【出願人】(507214038)キアゲン ゲーエムベーハー (16)
【Fターム(参考)】
[ Back to top ]