
RNAの直接検出法
【課題】新規な簡便迅速で実用的な、試料中のRNAを検出する方法を提供すること。
【解決手段】本発明は、生物由来の夾雑物を含む試料中のRNAの検出方法であって、(1)検出対象のRNAに相補的な配列を有する一本鎖DNAを環状化し、環状化一本鎖DNAプローブを作製する工程、(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる工程、(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしてローリングサークル増幅法(RCA)により一本鎖DNAを合成する工程、(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する工程、さらに任意に前記(2)の後に(5)RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程を含む、前記方法に関する。
【解決手段】本発明は、生物由来の夾雑物を含む試料中のRNAの検出方法であって、(1)検出対象のRNAに相補的な配列を有する一本鎖DNAを環状化し、環状化一本鎖DNAプローブを作製する工程、(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる工程、(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしてローリングサークル増幅法(RCA)により一本鎖DNAを合成する工程、(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する工程、さらに任意に前記(2)の後に(5)RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程を含む、前記方法に関する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、試料中のRNAを直接検出する方法、より具体的には、検出対象のRNAをプライマーとして、鎖置換型DNA合成酵素によってDNAを増幅するローリングサークル増幅法(RNA-Primed Rolling Circle Amplification;RPRCA)を用いたRNAの直接検出法およびその応用に関する。
【背景技術】
【0002】
食品のような複雑なサンプルから僅かな微生物を検出し、サンプルに存在する生きた微生物の有無を検査することを目的とする場合、一般的には培養法が用いられる。しかし培養法による微生物の検出には、選択培地によるところが大きく、しかも数日の培養期間が必要であるため、迅速性が求められる食中毒検査や臨床診断等には向いていない。一方、微生物の迅速な検出には、微生物等が有する固有のDNA配列をポリメラーゼ連鎖反応(polymerarse chain reaction;PCR)を利用して検出する方法が知られている。しかし、DNAは化学的に安定な化合物であるため、PCRによって微生物等に固有なDNA配列が検出されたとしても、当該微生物等の生死を判別するには至らない。
【0003】
細胞内のRNAは細胞が死滅すると速やかに分解されるため、RNAを検出することによって微生物などの生きた細胞の検出および生死判別を行う方法が開発されている。特に、RNAを逆転写反応(RT(reverse transcription)反応)によってDNAに変換した後にPCRを行うRT−PCR法はよく知られている(例えば、非特許文献1)。しかし、PCR法はその反応のシステムのため、反応温度を変化させることが可能なサーマルサイクラーなどの高価な装置を必要としており、一般的なPCRにかかる時間は少なくとも2〜3時間程度を必要としている。また、増幅と検出を同時に行うことのできるリアルタイム検出法が可能であるが、かかる検出法には、サーマルサイクラーに検出装置が組み込まれている必要があり、サーマルサイクラーよりもさらに費用が嵩むことになる。
【0004】
一方で、RNAを逆転写した後に、鎖置換型DNA合成酵素を利用したLAMP法(Loop-Mediated Isothermal Amplification)によってRNAを迅速に検出するRT−LAMP法も開発されている(非特許文献2)。RT−LAMP法は、およそ1〜2時間程度での検出が可能であるが、その反応システムは複雑で汎用性に乏しく、また反応システム上、比較的高温(65℃)を維持できる機械を必要としている。PCR法と比較して、低価格の装置でリアルタイム検出も可能であるが、その反応システムは、特殊なプライマー設計の複雑さなどのために現時点では汎用されていない。
【0005】
さらに、RT−PCR法やRT−LAMP法によるRNA検出では、最終的な増幅反応に至るまでに生成するDNAが、次の反応ステップに混入して非特異的な逐次反応を誘発する、いわゆるキャリーオーバーによるクロスコンタミネーションが常に問題となる。そのため、試料中の夾雑物、特にゲノム由来のDNAが微量でも混入した場合には偽陽性の結果を生じやすく、目的となるRNAの検出を困難にするため、DNA分解処理などのRNAを精製する工程を必要とする。したがって、追加の工程や専用の設備等が必要となり、さらには高度に訓練された人員が必要となるため、初期導入コストのみならずランニングコストも高額になる傾向がある。その結果、畜産産業や食品産業のみならず、臨床検査の現場においても、核酸増幅による微生物検出法の採用が忌避される傾向にある。
【0006】
一方、反応装置が簡素なRNA検出方法も開発されており、RT反応を経たあと、RT反応に用いたプライマー領域に設定したバクテリオファージプロモーター領域から、再度バクテリオファージRNA合成酵素によりRNAを増幅する転写媒介増幅法(Transcription-mediated amplification;TMA)(非特許文献3)や、TMA法にリボヌクレアーゼH(RNase H)を組み合わせたNASBA法(Nucleic acid sequence-based amplification;NASBA)がRNA増幅・検出方法として利用されている(非特許文献4)。これらは低温(37℃)における反応であるため、サーマルサイクラー等の特別高価な装置は不要である。しかし、反応生成物がRNAであるため検出には特殊なゲル電気泳動装置等を用いるか、あるいは反応後にRT−PCR法を行う必要があり、操作が煩雑である。
【0007】
上述のように、既存のRNAの増幅・検出法はすべて逆転写反応を経由して行われるため、実験操作の簡素化や反応条件の最適化を行ったとしても、逆転写反応そのものにかかる時間は短縮できないため、検出までに概ね2時間以上の時間を必要とし、検出速度の向上は困難である。かかる問題点は、一層の高速化が求められているRNA検出法の開発、例えば、病原性大腸菌O157や高病原性インフルエンザウイルスのような病原性RNAウイルスの検出法の開発などにおいても大きな障害となっていた。
【0008】
近年、これらの問題を解決する核酸増幅方法として、ローリングサークル増幅(rolling circle amplification;RCA)法が検討されている。RCA法は、一本鎖環状DNAプローブを該DNAプローブに相補的な配列を持つDNAプライマーでハイブリダイズさせた後、該DNAプローブを鋳型とした鎖置換型DNA合成反応によって一本鎖DNAを連続的に生成する反応である。したがって、RCA法ではDNA合成反応後に生成する一本鎖DNAを検出することによって、DNAプライマーもしくはDNAプローブの存在を判定することができる。
【0009】
そしてRNA検出方法として、RNA−プライムドローリングサークル型増幅(RNA-Primed Rolling Circle Amplification;RPRCA)法が、新たなRNA検出法として注目を浴びている。RPRCAは標的RNA分子の3’側配列と相補的配列を有する環状DNAプローブとをハイブリダイズさせた後、当該RNA分子の3’末端をDNA合成の開始点として利用し、一本鎖DNAをRCA法により連続的に合成する。これにより合成された一本鎖または二本鎖DNAを検出することにより、逆転写反応を行わずに目的のRNA、特に原核微生物由来のメッセンジャーRNA(mRNA)や、真核生物のマイクロRNA(miRNA)などのsmall RNA(sRNA)を短時間で検出することが可能となる。
【0010】
これまでに、RCA法を利用したRNAの検出方法として、真核生物のmiRNAとハイブリッド形成させた一本鎖環状DNAプローブを用いて、当該RNAをRCA法によって検出する方法が検討されている(特許文献1)。しかしながら、このRCA法には、非特異的に合成されるDNA副生成物によって検出時のシグナルノイズ比(S/N比)が低下するという問題がある。非特異的なDNA副生成物に起因するシグナルノイズ比の低下を回避するために、特許文献1では、複数のDNAオリゴヌクレオチドを介して予め環状DNAを作成し、未反応の一本鎖DNAの除去を試みているが、複数であるが故に部分的な二本差を形成しやすくなり、二本鎖を形成したオリゴヌクレオチドはエクソヌクレアーゼのみでは除けないため、この方法では困難である。
【0011】
また、特許文献2では環状化反応およびDNA合成において、耐熱性酵素を利用して反応温度を上昇させることにより、非特異的なDNA合成の原因となる非特異的な二本鎖形成の抑制を検討している。しかし、耐熱性酵素のために、非耐熱性酵素に比べてシグナル検出速度は低下する。
【0012】
特許文献3では、直鎖状パドロックDNAプローブ(非特許文献5)を検出対象のRNAとハイブリッド形成させた上で、該パドロックDNAプローブを環状化させ、一本鎖環状DNAプローブを形成させる方法を開示している。同様に、特許文献4および非特許文献6においても、パドロックDNAプローブと検出対象のRNAとのハイブリッドを形成させた後、パドロックDNAプローブの環状化反応を行い、さらに検出用DNAプライマーを用いて、検出対象のRNAの有無が検証可能な方法を報告している。
【0013】
しかしながら、直鎖状パドロックDNAプローブを環状化させる際、反応溶液中の直鎖状パドロックDNAプローブが全て環状化されることはなく、未反応の直鎖状パドロックDNAプローブ自身が非特異的な二本鎖核酸を形成する。また、検出用DNAプライマーの使用は、さらに非特異的なDNAの副生成物を生じさせるため、検出時のS/N比が大きく低下するという問題がある。
【0014】
非特異的な副生成物に由来するS/N比の低下は、検出感度を下げるのみならず、目的RNAの検出においては、偽陽性の結果を生じることにより、不正確な判断を引き起こしかねない。この不正確な判断材料は、特にウイルスを含む感染性微生物の臨床検出において被験者の「生活の質」(Quality Of Life;QOL)の低下を引き起こす可能性がある。
【0015】
さらに、パドロックDNAプローブを用いたRPRCA法においては、目的のRNA上におけるパドロックDNAプローブのT4 DNAライゲースによる環状化は、僅か2.5%程度であり、DNA上で起こるそれと比べ著しく効率が悪化していると報告されている(非特許文献7)。使用するライゲースをT4 RNAライゲースに変更しても、環状化反応は全体の20%程度しか起きないため、RNAのパドロックDNAプローブによる検出効率は、非常に悪いことが明らかにされた。
【0016】
また、従来のRPRCA法の多くは検出対象となるRNAの3’末端側配列をプライマーとして利用するため、検出可能なRNA分子種は、トランスファーRNA(tRNA)やリボソームRNA(rRNA)、および原核生物のmRNAや、真核生物のsRNA、あるいは一部のRNAウイルスのゲノムRNA(gRNA)に限定されていた。特に、RPRCA法による真核生物のmRNAの特異的な検出は、全てのmRNAの3’末端にポリA配列が存在するため、原理的に不可能であった。
【0017】
特許文献4では、多様なRNA種の3’末端側配列、特にmRNAの3’末端側配列をプライマーとして利用するため、ポリA配列をリボザイムによって除去する方法や、RNA−DNAハイブリッドにRNase H処理をしてニックを導入することによって、mRNAの3’末端をプライマーとして伸長可能にする方法が記載されている。しかしながら、同文献中、単に手法が記述されているに過ぎず、実際に反応産物を得たものでも、例えば、ゲル中でバンドとして検出したものでもなかった。したがって、同文献に記載の方法では、上述のS/N比の問題を認識していないため、目的物と非特異的な産物との区別ができず、偽陽性の結果を生じる危険性を包含するものであった。さらにまた、同文献における環状化DNAプローブの作製工程は、極めて煩雑であり、実用に耐えるものではなかった。
【0018】
一方で、検出対象のRNAとパドロックDNAプローブをハイブリダイズさせた後、当該パドロックDNAプローブをT4 DNAライゲースにより環状化し、DNA合成酵素の、特にphi29DNAポリメラーゼの3’−5’エクソリボヌクレアーゼ活性により、ハイブリダイズしていない検出対象のRNAの3’末端配列をハイブリダイズしている位置まで除去した後、ハイブリダイズしたRNA部分をプライマーとしてDNA合成を開始する方法が開発された(非特許文献8)。さらに、二本鎖RNA特異的分解酵素であるRNase IIIを反応液に加えることにより、二次構造(二本鎖領域)を取っているであろう非ハイブリダイズ領域のRNA部分を分解・除去し、RCA反応の向上が認められたことを報告している(非特許文献9)。
【0019】
しかしながら、phi29DNAポリメラーゼの3’−5’エクソリボヌクレアーゼ活性は非常に弱いため、除去できるRNAの長さが制限される。さらに、phi29DNAポリメラーゼの3’−5’エクソリボヌクレアーゼ活性は、RNAの二次構造を破壊できず、二次構造を取りやすいRNA分子ではほとんど分解されないため、ハイブリダイズさせるRNA領域は3’末端側に限定されてしまい、例えば、5’EST配列は現実的に利用できなかった(非特許文献10)。
【0020】
また、非特許文献10においては、RNAの直接検出を断念し、代替方法として逆転写反応時に利用する標的RNA特異的なDNAプライマーの構成残基の一部にLocked nucleic acid (LNA)を使用することにより、ハイブリダイゼーション効率を高めて逆転写効率を向上させ、検出対象のRNA特異的な逆転写産物によるRCA反応を行うことにより、検出感度上昇が可能であることを報告している。
【0021】
しかしながら、この方法は、複数のエクソンで構成されている真核生物のmRNAでは有効であるが、1エクソンで構成されている酵母や原核微生物においては、残存していたゲノムDNAとの区別が厳密にはできない。さらに、非特許文献10では最終的にcDNAではあるが、DNAをプライマーとして用いるために、非特異的なDNA副生成物が生じる。したがって,パドロックDNAプローブと相補性のある検出用DNAプローブのハイブリダイゼーションによってのみ、増幅・検出の特異性が保証されるため、煩雑な操作手順を捨て切れていない。
【0022】
さらに、従来技術においては、上記の低いS/N比の問題からも、RNA試料中に混入するDNA等の夾雑物によるノイズを防ぐ必要があると考えられており、目的のRNAを検出するために、RNA試料をDNaseで処理したり、精製されたRNA試料を用意する等、利用できる試料が制限されていた(特許文献4、非特許文献8および9)。
【0023】
以上のように、これまで検討されてきた方法は、PCR反応の温度サイクルを管理する装置が必要であったり、検出対象のRNAを逆転写によってDNA化したり、設備面に止まらず、検出精度においてもコンタミネーションなどによる増幅反応における副生成物の問題などがあった。
【0024】
一方で、RPRCA法においては、検出対象のRNAの種類および由来によっては検出用プローブの構築ができず、簡便な手順のRPRCA法を実行できないRNA分子種のほうが多く存在していた。さらに、非特異的な副生成物に由来するS/N比の低下によって、目的のRNA検出において不正確な判断を引き起こしかねないため、実用段階には至っていない。
【0025】
したがって、RNAを検出する方法として、高額な装置を必要とせず、ゲノム配列等の配列の一部でも既知であれば、特異的な検出が可能となり、さらに簡便でありながら迅速で、かつ高感度の検出方法の構築が求められていた。
【先行技術文献】
【特許文献】
【0026】
【特許文献1】米国特許出願公開第2009/0181390号明細書
【特許文献2】米国特許出願公開第2006/0188893号明細書
【特許文献3】特表2003−534782号公報
【特許文献4】米国特許出願公開第2005/0112639号明細書
【非特許文献】
【0027】
【非特許文献1】Selvaratnam S, Schoedel BA, McFarland BL, Kulpa CF. Application of reverse transcriptase PCR for monitoring expression of the catabolic dmpN gene in a phenol-degrading sequencing batch reactor. Appl. Environ. Microbiol. 61:3981-3985. (1995).
【非特許文献2】Fukuta S, Iida T, Mizukami Y, Ishida A, Ueda J, Kanbe M, Ishimoto Y. Detection of Japanese yam mosaic virus by RT-LAMP. Arch. Virol. 148:1713-20 (2003).
【非特許文献3】Kwoh DY, Davis GR, Whitfield KM, Chappelle HL, DiMichele LJ, Gingeras TR. Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead-based sandwich hybridization format. Proc. Natl. Acad. Sci. USA. 86:1173-7 (1989).
【非特許文献4】Guatelli JC, Whitfield KM, Kwoh DY, Barringer KJ, Richman DD, Gingeras TR. Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc. Natl. Acad. Sci. USA. 87:1874-1878 (1990).
【非特許文献5】Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary B.P., and Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science 265 2085-8 (1994).
【0028】
【非特許文献6】Wang B, Potter SJ, Lin Y, Cunningham AL, Dwyer DE, Su Y, Ma X, Hou Y, Saksena NK. Rapid and sensitive detection of severe acute respiratory syndrome coronavirus by rolling circle amplification. J. Clin. Microbiol. 43(5):2339-44 (2005)
【非特許文献7】Li N, Jablonowski C, Jin H, Zhong W. Stand-Alone Rolling Circle Amplification Combined with Capillary Electrophoresis for Specific Detection of Small RNA. Anal. Chem. 81, 4906-4913 (2009)
【非特許文献8】Lagunavicius A, Merkiene E, Kiveryte Z, Savaneviciute A, Zimbaite-Ruskuliene V, Radzvilavicius T, Janulaitis A Novel application of Phi29 DNA polymerase: RNA detection and analysis in vitro and in situ by target RNA-primed RCA. RNA 15:765-771 (2009).
【非特許文献9】Merkiene E, Gaidamaviciute E, Riauba L, Lagunavicius A. Direct detection of RNA in vitro and in situ by target-primed RCA: the impact of E. coli RNase III on the detection efficiency of RNA sequences distanced far from the 39-end. RNA Published online June 28 (2010)
【非特許文献10】Larsson C, Grundberg I, Soderberg O, Nilsson M. In situ detection and genotyping of individual mRNA molecules. Nat. Methods 7: 395-397 (2010)
【発明の概要】
【発明が解決しようとする課題】
【0029】
本発明の課題は、高度な設備を必要とせず、簡便な操作で、実用的な感度を有しながら迅速に行うことができる、RNAの検出方法であって、生物由来の夾雑物を含む試料中、特にDNAが混入した試料においても、特異的な検出を可能とする方法を提供することである。
【課題を解決するための手段】
【0030】
本発明者らは、上記課題を解決するため、鋭意研究を重ねた結果、検出対象とする目的RNAの3’末端部分配列と相補的なDNAを合成し、一本鎖DNA連結酵素により自己環状化させて、得られた環状化一本鎖DNAプローブを目的RNAとハイブリッド形成させ、さらに目的RNAをプライマーとして一本鎖DNAを合成させると、予め反応系に一本鎖DNA特異的な蛍光色素を添加しておくことにより、目的RNAのリアルタイム検出が可能となることを見出した。
【0031】
また、RNase H処理の工程を加えることにより、3’末端にDNAプローブとの相補配列を有さないRNAであっても検出可能となり、ウイルスや原核生物のみならず真核生物由来の多様なRNAの検出が可能となった。
【0032】
さらに、本発明者らは、本発明のRPRCA法によって、細胞から実際に得られた試料中から目的RNAを検出した知見に基づき、生物由来の夾雑物を含む試料中、例えば、DNAが混入した試料であっても、特異的な目的RNAの検出が可能となることを見出し、本発明を完成させるに至った。
【0033】
すなわち本発明は、以下の方法およびキットに関する。
[1] 生物由来の夾雑物を含む試料中のRNAの検出方法であって、
(1)検出対象のRNAに相補的な配列を有する一本鎖DNAを環状化し、環状化一本鎖DNAプローブを作製する工程、
(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる工程、
(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしてローリングサークル増幅法(RCA;Rolling Circle Amplification)により一本鎖DNAを合成する工程、
(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する工程
を含む、前記方法。
[2] 生物由来の夾雑物が、DNAを含む、[1]に記載の方法。
[3] 試料が、核酸試料である、[1]または[2]に記載の方法。
[4] 試料中のDNAが、分解処理されていない、[2]または[3]に記載の方法。
[5] 前記(1)において、環状化していない未反応の一本鎖DNAを分解することを含む、[1]〜[4]のいずれか一項に記載の方法。
[6] 環状化一本鎖DNAプローブが、検出対象のRNAに相補的な配列のみからなる、[1]〜[5]のいずれか一項に記載の方法。
[7] 環状化一本鎖DNAプローブが、30塩基長〜4500塩基長である、[1]〜[6]のいずれか一項に記載の方法。
[8] 前記(2)の後に、
(5)RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程
をさらに含む、[1]〜[7]のいずれか一項に記載の方法。
[9] 前記(3)および/または(5)が、25℃〜65℃の範囲の一定の温度で行われる、[8]に記載の方法。
[10] 前記(5)において、RNase Hを終濃度1×10−4〜10ユニット/反応で用いる、[8]または[9]に記載の方法。
[11] 前記(5)において、RNase Hを終濃度1×10−7〜0.1ユニット/μlで用いる、[8]または[9]に記載の方法。
[12] RNAが、真核生物、原核生物、ウイルスまたはウイロイド由来である、[1]〜[11]のいずれか一項に記載の方法。
[13] RNAが、ポリA配列を有する、[12]に記載の方法。
[14] RNAが、ノンコーディングRNAである、[12]に記載の方法。
[15] [1]〜[14]のいずれか一項に記載のRNAの検出方法を含む、真核生物、原核生物、ウイルスまたはウイロイドの検出方法。
[16] 微生物を検出する、[15]に記載の方法。
[17] [1]〜[14]のいずれか一項に記載のRNAの検出方法、および/または[15]に記載の真核生物、原核生物、ウイルスまたはウイロイドの検出方法、および/または[16]に記載の微生物の検出方法のために用いるキットであって、検出対象のRNAに相補的な配列を含む環状化一本鎖DNAプローブを含むキット。
[18] アデニン、グアニン、シトシンおよびチミンからなるデオキシヌクレオチド、反応用緩衝液および鎖置換型DNA合成酵素からなる群から選択される1種または2種以上をさらに含む、[17]に記載のキット。
【発明の効果】
【0034】
本発明によれば、RNAの検出の際に検出対象である目的RNAに対して逆転写反応を行う必要がなく、直接RNAをプライマーとして用いてDNAを合成させ、かつ合成された一本鎖DNAに一本鎖特異的蛍光色素を反応させることにより、極めて簡便に、リアルタイム検出が可能である。
また、未反応の一本鎖DNAに由来する非特異的なDNA合成を完全に抑制することが可能となり、高いシグナルノイズ比(S/N比)を実現し、さらに、過去の増幅産物のキャリーオーバーによるクロスコンタミネーションリスクが、他の核酸増幅法に比べ著しく低いため、RNAを簡便な操作で、実用的な感度を有しながら、また高度な設備を必要せず、安価な装置で、短時間で迅速に検出することが可能となる。
【0035】
また、RNase H処理の工程を加えることにより、検出対象となるRNAの3’末端の配列に依存せずに、部分的にでも既知であれば、様々な種類のRNAの検出が可能となり、ウイルスや原核生物由来のRNAだけでなく、真核生物由来のRNAおよびそれら生物自身の検出も可能となった。
【0036】
さらに、本発明によれば、検出感度がよいため、試料中に生物由来の夾雑物を含んでいてもRNAの検出が可能である。また、試料中にDNAを含む場合であっても、DNAの分解処理を要しない。このため本発明の方法は、従来のRNA検出法と比較して、迅速かつ簡便に試料中のRNAを検出することができる。これは、夾雑DNAが、環状化一本鎖DNAプローブとハイブリダイズしても、その3’末端がプライマーとして利用されることが確率的にほとんどありえないことによる。
【0037】
このことは、RNAを特異的に検出するためには試料中の夾雑DNAを除去するという当該分野の技術常識に反する驚くべき知見であり、これによって、生きている細胞由来のRNAの検出が容易になり、実用に十分な感度を有しながらも、簡便でありながら迅速で、かつ特異的なRNAの検出が可能となった。したがって、医学、薬学、農学、食品学などの研究に止まらず、臨床検査などの医療分野や、食品産業における簡便な生菌検査や食中毒における迅速検査など、簡便さ、省力化、迅速さが求められる様々な基礎および応用分野において、とくに有用である。
【図面の簡単な説明】
【0038】
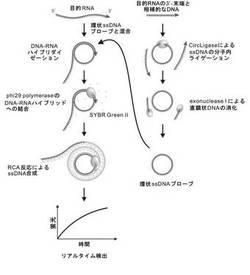
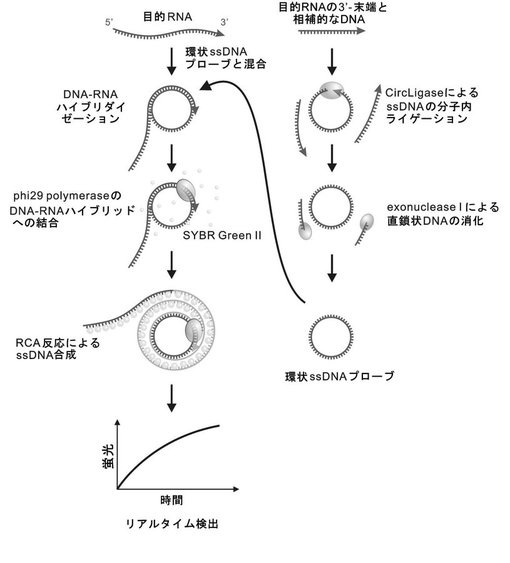
【図1】図1は、本発明のRNA−primed RCAによるRNAの検出を示す概略図である。
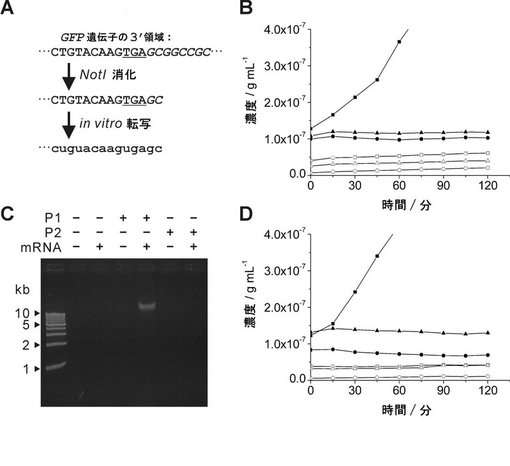
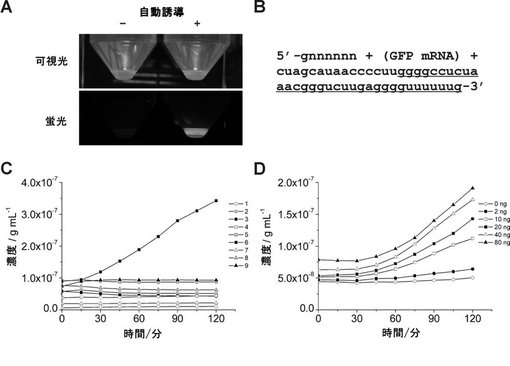
【図2】図2は、RNA−primed RCAによるin vitro転写mRNAの特異的検出を示す:(A)in vitroで転写されたGFPmRNAの3’末端配列を示す。GFPmRNAは、NotI消化後のGFP遺伝子のin vitro転写により調製した。下線およびイタリック文字は、夫々、GFP遺伝子の終止コドンおよびNotI制限酵素サイトを示す。配列中の大文字および小文字は、夫々、DNA残基およびRNA残基を示す。(B)RNA−primed RCAによるin vitro転写mRNAのリアルタイム検出を示す。RNA−primed RCAによる蛍光は、夫々、以下の記号で示す:白丸はmRNAもDNAプローブもなし、黒丸はmRNA単独、白四角はP1(配列番号1で表されるプローブ、表1)単独、黒四角はP1とmRNA、白三角はP2(配列番号2で表されるプローブ、表1)単独、黒三角はP2とmRNA。(C)RNA−primed RCAによるin vitro転写mRNAの電気泳動による検出を示す。15時間のRNA−primed RCAにより増幅されたDNAを分子量マーカー(キロベースラダーマーカー)とともにアガロースゲルで泳動した。(D)RNA−primed RCAにおけるプローブの長さの効果を示す。RNA−primed RCAによる蛍光は、夫々、以下の記号で示す:白丸はmRNAもDNAプローブもなし、黒丸はmRNA単独、白四角はP1単独、黒四角はP1とmRNA、白三角はP3(配列番号3で表されるプローブ、表1)単独、黒三角はP3とmRNA。
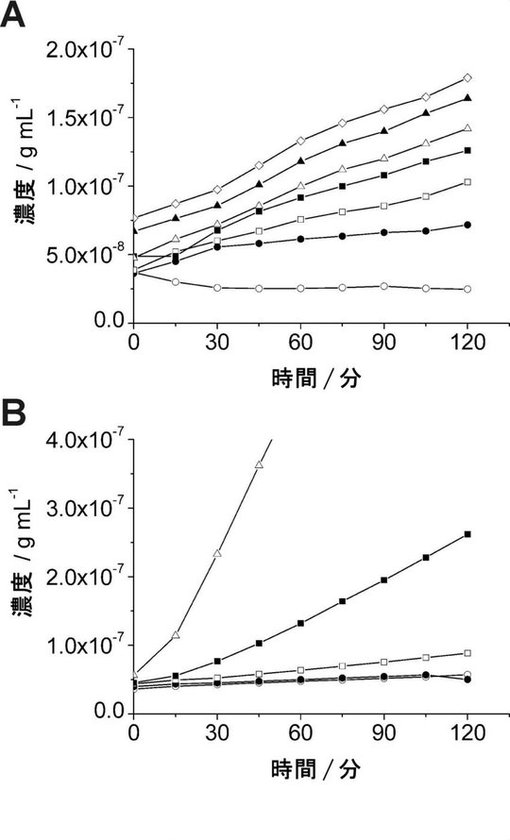
【図3】図3は、RNA−primed RCAの感度を示す:(A)プライマーとしてのin vitro転写GFPとのRNA−primed RCAにおけるphi29DNAポリメラーゼの濃度の効果。酵素濃度は、夫々、以下の記号で示す:白丸は0ユニット、黒丸は10ユニット、白四角は20ユニット、黒四角は40ユニット、白三角は60ユニット、黒三角は80ユニット、白菱形は100ユニット。(B)段階希釈したin vitro転写GFPによるRNA−primed RCAの滴定分析。用いたGFPmRNAの量は、0pg(白丸)、32pg(黒丸)、320pg(白四角)、3.2ng(黒四角)、32ng(白三角)。
【図4】図4は、大腸菌から単離された全RNA中に存在するmRNAの特異的検出を示す:(A)pET-AcGFPを保有する大腸菌におけるGFPの発現。自己誘導ありまたはなしのGFP発現をブルーライト下で可視化した。(B)大腸菌において転写されたGFPmRNAの3’末端配列。GFPmRNAはT7プロモーターの制御下で転写され、かかる転写は、T7ターミネーターで終了した。下線は、環状化プローブP4(配列番号4で表されるプローブ、表1)に対して相補的な配列を示す。(C)RNA−primed RCAによる自己誘導ありまたはなしの大腸菌から単離された全RNAに存在するGFPmRNAのリアルタイム検出。RNA−primed RCAによる蛍光は、以下の記号で示す:(1)白丸はmRNAもDNAプローブもなし、(2)灰丸は誘導なしの大腸菌由来の全RNA、(3)黒丸は誘導ありの大腸菌由来の全RNA、(4)白四角はP4単独、(5)灰四角はP4と誘導なしの大腸菌由来の全RNA、(6)黒四角はP4と誘導ありの大腸菌由来の全RNA、(7)白三角はP3単独、(8)灰三角はP3と誘導なしの大腸菌由来の全RNA、(9)黒三角はP4と誘導ありの大腸菌由来の全RNA。(D)誘導した大腸菌から得られた全RNAの段階希釈したものによるRNA−primed RCAの滴定分析。用いた全RNAの量は、0ng(白丸)、2ng(黒丸)、10ng(白四角)、20ng(黒四角)、40ng(白三角)および80ng(黒三角)。
【0039】
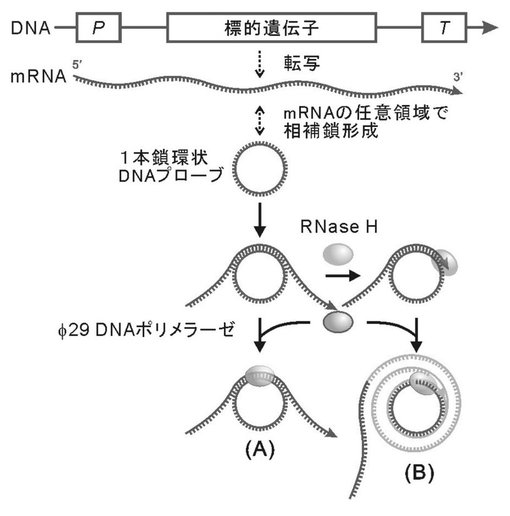
【図5】図5は、RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程をさらに含む、本発明のRNA−primed RCAによるRNAの検出を示す概略図である。(A)標的遺伝子のmRNAの下流(3’末端)は環状化DNAプローブと相補鎖形成しないため、DNAの合成反応が開始しない。(B)標的遺伝子のmRNAと環状化DNAプローブが相補鎖形成した後、RNase Hで処理することによりニックが導入され、RCA反応が開始する。
【図6】図6は、pET-21dにクローン化したGFP遺伝子の全配列を示す。GFPmRNAは、NotI消化後のpET-AcGFPおよびT7RNAポリメラーゼを使用してin vitro転写により調製した。下線(太字)は、GFPmRNA内部領域にて相補鎖形成するように設計された環状化DNAプローブ(P5)の位置を示す。下線のATGおよびTGAは、夫々開始コドンおよび終止コドンを示す。
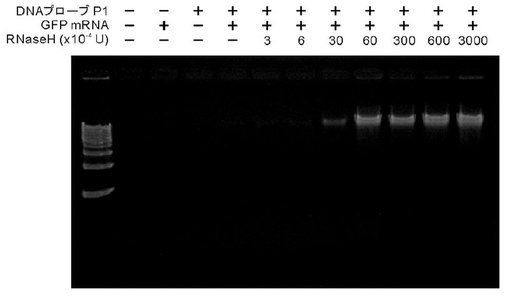
【図7】図7は、RNA−primed RCAによるin vitro転写mRNAの検出における、RNase H処理の効果を示す。15時間のRNA−primed RCAにより増幅されたDNAを分子量マーカー(キロベースラダーマーカー)とともにアガロースゲルで泳動した。また、RNA−primed RCA反応におけるRNase Hの濃度依存性について検討した結果を示す。
【0040】
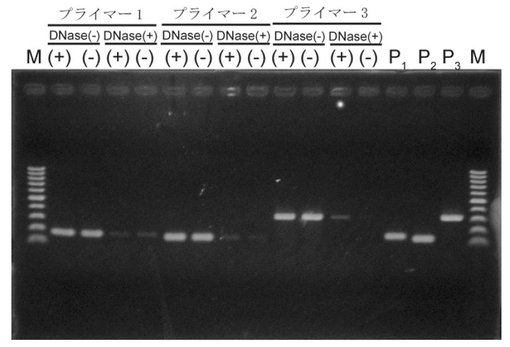
【図8】図8は、PCRによるRNA試料中の夾雑DNA(pET-AcGFP)の検出結果を示す。中段のDNase(+)およびDNase(−)は、DNase処理有りおよび無しをそれぞれ示し、下段の(+)および(−)は発現誘導した菌体および発現誘導していない菌体由来の全RNAをそれぞれ示す。PCR産物は、分子量マーカー(100bpラダー)とともにアガロースゲルで泳動した。
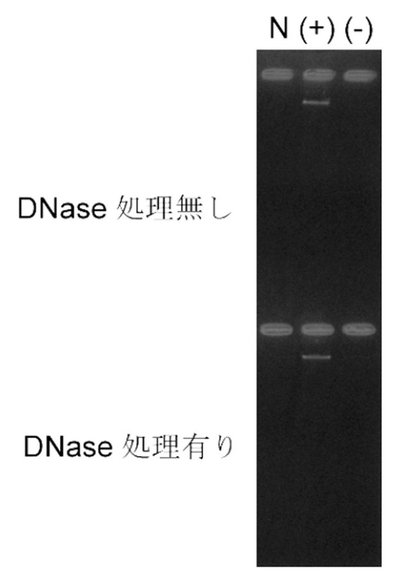
【図9】図9は、夾雑DNAが混入した試料中における、本発明のRNA−primed RCAによる標的RNAの特異的検出を示す。4時間のRNA−primed RCAにより増幅されたDNAを、アガロースゲルで泳動した。DNase処理の有無は、全RNA試料中のGFPmRNAの検出結果に影響を与えない。Nはネガティブコントロールを示し、(+)および(−)はそれぞれ発現誘導した菌体および発現誘導していない菌体由来の全RNAを示す。
【発明を実施するための形態】
【0041】
本発明のRNAの検出方法は、鎖置換型DNA合成酵素を利用したローリングサークル型DNA複製法(Rolling circle amplification;RCA)を利用し、応用した技術を用いる。
したがって、先ず、(1)検出対象のRNAに相補的な配列を有する一本鎖DNA(ssDNA)を環状化し、環状化一本鎖DNAプローブを作製する。
検出対象のRNA(目的RNA)に相補的な配列としては、目的RNA特異的検出の観点から、目的RNAの3’末端側の部分の、例えば、20〜100塩基、とくに、25〜40塩基に相補的な配列が好ましい。RNase H処理によって、DNA−RNAハイブリッド上のRNA鎖にニックを導入する工程を含む場合、検出対象の目的RNAに相補的な配列は、RNAのいずれの位置からも任意に設定することが可能である。
【0042】
相補鎖を形成するssDNAは、100塩基以下の場合は核酸合成機によるDNA合成が好ましいが、100塩基以上の場合は、PCR等により目的DNAを増幅後、公知の方法、例えば熱変性により一本鎖化した後、環状化反応に使用することもできる。例えば、30〜4,500塩基、とくに、50〜700塩基に相当する相補的な配列が好ましい。検出感度はプローブの長さが長い程上昇し、短ければ短いほど減少する。また、非特異的な反応をさらに抑えるために、プローブは検出対象のRNAとハイブリダイズ可能な相補配列のみから構成されてもよい。
【0043】
一本鎖DNAの環状化は、任意の手段によって行うことができるが、例えば、CircLigase(登録商標)、CircLigase II(登録商標)、ssDNA Ligase(Epicentre社)、ThermoPhage ligase(登録商標) single-stranded DNA(Prokzyme社)を用いて行うことができる。環状化は、分子内ライゲーションにより環状化してもよいし、また複数の一本鎖DNAが分子間ライゲーションにより、タンデムに配した状態で環状化してもよい。好ましくは、部分的な二本鎖形成による不完全なエクソヌクレアーゼ処理を防ぐために、環状化反応においてDNAプローブにガイド配列を必要としない種類の一本鎖DNAリガーゼが用いられる。
高いS/N比を得るためにも、環状化していない未反応の一本鎖DNAを分解することが好ましく、とくに好ましくは、エクソヌクレアーゼI(各社)、T5エクソヌクレアーゼ(Epicentre社)、エクソヌクレアーゼVI(Epicentre社)、RecJエクソヌクレアーゼ(Epicentre社)等の一本鎖オリゴヌクレオチドをモノヌクレオチドまで分解可能なエクソヌクレアーゼによる酵素分解が好ましい。ただし、エクソヌクレアーゼIIIは、作成された一本鎖環状DNAを基質とするため使用できない。
【0044】
次に、(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる。かかるハイブリダイゼーションの条件は、当業者であれば、環状化一本鎖DNAプローブと目的RNAとの組み合わせを検討し、適宜設定できる。
本発明に用いる試料は、標的となるRNAを含有していれば、特に限定されないが、かかるRNAが精製されていても、精製されていなくても用いることができる。目的RNAの分解を避けるため、リボヌクレアーゼ活性の低減した試料であってもよい。例えば、リボヌクレアーゼ阻害剤で処理し、リボヌクレアーゼ活性を抑制させた試料であってもよい。
【0045】
典型的には、生物由来の夾雑物を含んでいてもよい。生物由来の夾雑物としては、例えば、ゲノムDNA、葉緑体DNA、ミトコンドリアDNAおよびプラスミドDNAなどの各種のDNA、タンパク質、脂質、多糖類などが挙げられる。
また、試料中にDNAが含まれている場合、DNaseなどのDNA分解酵素や、他のDNA分解処理がされていないものであってもよい。特に、本発明は、DNA分解処理を必要としないため、DNAを含む試料に対してむしろ好適に実施することができる。
【0046】
任意に、前記(2)において得られた目的RNA−DNAハイブリッド上で、RNase Hによって目的RNA鎖のみにニックを形成させる工程(5)を含むことができる。RNase Hは一般に用いられている中温性のRNase HI(各社)やRNase HII(NEB社)の他に、Hybridase(登録商標)、Thermostable RNase H(Epicentre社)、Tth RNase H(東洋紡社)等の耐熱をRNase Hを用いることでも行うことができる。RNase Hは反応の初期段階において必要であり、反応中期から後期には必要ないため必要な量はごく少量でもよい。DNAの伸長反応において使用されるRNase Hの量(濃度)は、好ましくは終濃度約1×10−4〜10ユニット/反応、より好ましくは終濃度約1×10−4〜1ユニット/反応で使用する。
または、本発明の方法において、RNase Hは、好ましくは1×10−7〜0.1ユニット/μl、より好ましくは1×10−7〜1×10−2ユニット/μlの範囲において好適に用いられる。
【0047】
さらに、(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしたローリングサークル増幅法(RCA;Rolling Circle Amplification)を行う。RCAは、例えば、phi29 polymerase(各社)、Klenow DNA Polymerase (5’-3’, 3’-5’ exo minus) (各社)、Sequenase(登録商標)Version 2.0 T7 DNA Polymerase (USB社)、Bsu DNA Polymerase, Large Fragment (NEB社)などの中温性の鎖置換型DNA合成酵素や、Bst DNA Polymerase (Large Fragment) (各社)、Bsm DNA Polymerase, Large Fragment (Fermentas社) 、BcaBEST DNA polymerase (TakaraBio社)、Vent DNA polymerase (NEB社)、Deep Vent DNA polymerase (NEB社)、DisplaceAce (登録商標) DNA Polymerase (Epicentre社)等の耐熱性の鎖置換型DNA合成酵素を用いることでも行うことができる。
【0048】
本発明の方法によるDNAの伸長反応は、好ましくは、サーマルサイクラーを用いる必要はなく、例えば、25℃〜65℃の範囲の一定の温度において実施される。反応温度は、酵素の至適温度とプライマー鎖長に基づく変性温度(プライマーが鋳型DNAに結合(アニール)/解離する温度帯)に基づいて通常の手順により適宜設定される。さらに、一定の比較的低温においても実施される。例えば、鎖置換型DNA合成酵素としてphi29DNAポリメラーゼを使用する場合は、好ましくは25℃〜42℃、より好ましくは約30℃で反応する。
【0049】
そして、(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する。 このような標識としては、例えば、放射線(32Pなど)標識、蛍光標識など、検知可能な種々の標識を用いることができるが、とくに、検知の容易さ、安全性およびLED使用等による検出機作成時の簡便さなどの観点から、蛍光標識であることが好ましい。また、一本鎖DNAが部分的に二本鎖を形成するため、一般的な二本鎖DNA検出に利用されるエチジウムブロマイドも、増幅産物の検出に使用できることは、当業者であれば理解できるだろう。
【0050】
標識は、合成された一本鎖DNAに結合すれば、検出対象配列に特異的であっても、特異的でなくてもよい。配列に特異的に結合する標識としては、例えば、放射線標識されたDNA断片やメチル化等の修飾を含むRNA断片、ペプチド核酸(PNA)断片、DNA配列特異的に結合する抗体や金属ナノ粒子などが挙げられる。標識の標的となる特異的なDNA、RNAおよびPNA配列は、検出対象のRNAの配列に相補的な配列であってもよいが、例えば、環状化一本鎖DNAプローブに標識の標的となる配列の相補的な配列を組み込んでおくことも可能である。
また配列に特異的でなくても、DNAに特異的に結合する標識を用いることも可能であり、例えば、SYBR GreenII(Invitrogen社)などの蛍光色素をもちいることができる。
【0051】
標識の検知方法は、標識の種類に応じて適宜選択し得る。例えば、抗体を標識に用いた場合は、二次抗体やカップリング反応を用いて検知すること、また金属ナノ粒子を用いた場合には、粒子間相互作用の変化に伴う物性変化を計測することによる検知が可能である。さらに、クロマトグラフィーや電気泳動などを用いて検知することも可能である。
本発明のRNAの検出方法において、検出対象となるRNAはとくに限定されず、例えば、各種細胞におけるメッセンジャーRNA(mRNA)の発現をリアルタイム検出することや、微生物やウイルス由来のRNAを検出対象とすることも可能である。
【0052】
本発明の一態様においては、本明細書に記載するRNA検出方法のための反応試薬および組成物を含む、RNA検出のためのキットとすることができる。本発明のキットには、本明細書に記載するRNA検出反応のための鋳型となる環状化一本鎖DNAプローブ、鎖置換型DNA合成酵素、RNase H、dNTP混合物、反応用緩衝液、および検出用蛍光色素などを含むことができる。
【0053】
本発明の検出するRNAは、とくに限定されることなく、mRNA、リボソームRNA(rRNA)、トランスファーRNA(tRNA)を含むあらゆる種類のRNAを検出することが可能である。検出対象のmRNAは、ポリA配列を有していても、有していなくてもよい。RNAは、siRNA、miRNA、piRNA、rasiRNA、rRNA、tRNAを含むノンコーディングRNAであってもよく、さらに、ウイルスなどのゲノムRNAであってもよい。また、検出対象のRNAの形状は、直鎖状または環状のどちらでも可能である。
【0054】
本発明のRNA検出の一態様においては、検出対象となるRNAはあらゆる生物種由来の試料から調製または単離されたものでもよい。そのようなRNAを含む試料には、ウイルス、原核生物または真核生物の個体そのもの、あるいはその一部が使用できる。例えば、脊椎動物(ヒトを含む)では、糞便、尿、または汗のような排泄物、血液、精液、唾液、胃液、または胆汁のような体液等が挙げられる。また、外科的に生体から取り出した組織、または体毛のように生体から脱落した組織であってもよい。さらに、食品等の加工物から調整したRNA含有調整物であってもよい。また、前記試料をさらに分画して、その一部を取り出したものから調製したRNA含有調製物であってもよい。
【0055】
本発明はまた、生物の検出方法に関する。
本発明において、生物は、原核生物、真核生物、ウイルス、およびウイロイドを含み、それに由来するRNAを検出することによって検出することができる。検出可能な生物種は、RNAを有するものであればとくに限定されず、RNAの種類も問わない。例えば、ウイルス、細菌、カビ、酵母、昆虫、植物および動物のような生物を対象とすることができる。
【0056】
さらに、本発明の一態様は、微生物の検出方法に関する。
本発明において、微生物は、それに由来するRNAを検出することによって検出することができる。微生物は、構成単位が肉眼では観察できない微小な生物をいい、ウイルスや多細胞の生物も含む。検出可能な微生物としては、例えば、ポリA配列を持たないmRNAを産生する細菌、およびゲノムがRNAであるウイルスまたはウイロイドなどが挙げられる。また、原生動物、およびカビや酵母などの真菌を含む真核微生物を対象とすることもできる。
【0057】
以下に実施例によって本発明を詳述するが、本発明は、各実施例に限定されるものではない。
【実施例】
【0058】
〔材料および方法〕
1.鋳型オリゴヌクレオチドの環状化
表1に示すオリゴヌクレオチドを、1mM ATPを含む1×T4ライゲースバッファー中、10ユニットのT4ポリヌクレオチドキナーゼを用いてリン酸化した。リン酸化したオリゴヌクレオチドを、10ユニットのCircLigase(登録商標)ssDNA Ligase(Epicentre社)を用い、2.5mM MnCl2の1×CircLigaseバッファー中、65℃、1時間で環状化した。環状化されていないオリゴヌクレオチドは、10ユニットのエクソヌクレアーゼIで37℃1時間消化し、続いて80℃20分間処理し、酵素を不活性化した。環状化したオリゴヌクレオチドは、BioSpin(BioRad社)で精製し、濃度をQuant-iT ssDNA BR Assay Kit(Invitrogen社)およびQubit fluorometer(Invitrogen社)を用いて測定した。環状化オリゴヌクレオチドのエクソヌクレアーゼによる消化率と直鎖状のオリゴヌクレオチドのものとの比較をアガロースゲル電気泳動(2.0%、Tris-Borate-EDTAバッファー)により分析し、CircLigaseの反応による環状化が行われたことを確認した。
【0059】
【表1】

【0060】
2.T7発現ベクターpET-21dへのGFP遺伝子のクローニング
2μgのpAcGFP1(Clontech社)からそれぞれ50ユニットのNcoIおよびNotI(タカラバイオ社)で同時に消化し、緑色蛍光タンパク質(green fluorescent protein;GFP)遺伝子を単離した。GFP遺伝子はアガロースゲル電気泳動により精製し、Wizard(登録商標)SV GelおよびPCR Clean-Up Systems(Promega社)を用いて精製した。精製したDNAの濃度は、Quant-iT dsDNA HS Assay Kit(Invitrogen社)およびQubit fluorometer(Invitrogen社)を用いて測定した。精製したGFP遺伝子をNcoIおよびNotIで切断したT7発現ベクターpET-21d(Novagen社)に連結し、組み換えプラスミドpET-AcGFPを作製した。大腸菌(Escherichia coli)DH5α株に形質転換した後、プラスミドDNAを選択したコロニーから精製し、制限酵素処理の後、アガロースゲル電気泳動で確認した。精製したpET-AcGFPの濃度をQuant-iT dsDNA HS Assay KitおよびQubit fluorometerを用いて測定した。
【0061】
3.GFPmRNAのin vitroでの転写
GFPmRNAのin vitroでの転写は、MEGAscript(登録商標)T7 Kit(Ambion社)を用いて行った。1μgのpET-AcGFPをNotIで消化して得られた直鎖状の鋳型DNAを転写反応液に添加し、37℃で2時間インキュベートした。DNaseI処理により鋳型DNAを除去した後、転写したmRNAをMEGAclear(登録商標)Kit(Ambion社)を用いて精製し、濃度を分光分析法により測定した。転写したmRNAを用時まで−80℃で保存した。
【0062】
4.GFPmRNAのin vivoでの発現およびRNAの抽出
大腸菌BL21(DE3)株(Novagen社)をpET-AcGFPで形質転換し、pET-AcGFPの選択のため50μg/mlアンピシリンを含むLB寒天プレート(1%トリプトン、0.5%酵母エキス、1.0%NaClおよび1.5%寒天)で培養した。大腸菌はLB培地で30℃8時間培養した。培養物をOvernight Express(登録商標)Autoinduction System(Novagen社)を含むフレッシュなLB培地で1:1000に希釈し、30℃で16時間振とう培養し、DE3にコードされるT7RNAポリメラーゼによりGFPmRNAの転写を誘導した。大腸菌は、3,500rpmで4℃20分間の遠心分離により50mlのコニカルチューブに回収した。全RNAをTrizol(登録商標)Max Bacterial RNA Isolation kit(Invitrogen社)を用いて単離した。調製した全RNAは、混入したゲノムDNAやプラスミドDNAなどの夾雑DNAを除去するためのDNase処理(例えば、DNaseIによる処理)を行うことなく試料として供した。当該試料中、微量の夾雑DNAが含まれていることをアガロースゲル電気泳動(1.5%、Tris-borate-EDTA緩衝液)によって確認した。なお、全RNAの濃度は分光分析法により測定した。
【0063】
5.RNA−primed RCA検出
in vitroで転写したGFPmRNAまたはpET-AcGFPを保有する大腸菌BL21(DE3)株から単離した全RNAを、10pmolの環状化オリゴヌクレオチド(表1のP1(配列番号1)、P2(配列番号2)、P3(配列番号3)またはP4(配列番号4))とともに、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4および10mM KClを含むバッファー中に混合し、100μlの反応混合物を得た。mRNAと環状化オリゴヌクレオチドとのハイブリダイゼーションを95℃5分間のインキュベーションにより促進し、その後30℃に急冷した。RCA反応は、100μlのハイブリダイゼーションした混合物を、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4、10mM KCl、0.4mMデオキシヌクレオシド三リン酸(dNTPs)、8mMジチオスレイトール(DTT)、SYBR GreenII(Invitrogen社)および適切なユニットのRepliPHI phi29ポリメラーゼ(Epicentre社)を含む100μlの反応液とを混合することで開始した。phi29ポリメラーゼにより合成されたssDNA(一本鎖DNA)に結合した際のSYBR GreenIIの蛍光を15分毎にQubit fluorometerで検出した。30℃、15時間のインキュベーションの後の合成DNA15μlを、20ユニットのS1ヌクレアーゼ(タカラバイオ社)で37℃、30分間消化し、アガロースゲル電気泳動(1.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した。
【0064】
6.RNase Hを利用したRNA−primed RCA検出
in vitroで転写したGFPmRNAを、10pmolの環状化オリゴヌクレオチド(表1のP5、配列番号5)とともに、50mM Tris−HCl(pH 7.5)、10mM MgCl2、20mM(NH4)2SO4および10mM KClを含むバッファー中に混合し、100μLの反応混合物を得た。mRNAと環状化オリゴヌクレオチドとのハイブリダイゼーションを95℃、5分間のインキュベーションにより促進し、その後30℃に急冷した。RCA反応は、100μLのハイブリダイゼーションした混合物を、50mM Tris−HCl(pH 7.5)、10mM MgCl2、20mM(NH4)2SO4および10mM KCl、0.4mM dNTPs、8mM ジチオスレイトール(DTT)、さらに適切なユニット数のRNase HおよびRepliPHI phi29ポリメラーゼ(Epicentre社)を含む100μLの反応液を混合することで開始した。30℃、15時間のインキュベーションの後の合成DNA15μLを、20ユニットのS1ヌクレアーゼ(タカラバイオ社)で37℃、30分間消化し、アガロースゲル電気泳動(1.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した
【0065】
7.PCRによるRNA試料中の夾雑DNAの検出
pET-AcGFPを保有する大腸菌BL21(DE3)株から、全RNAの抽出を行った。この際、GFPの発現を自動誘導により制御し、発現誘導した菌体および発現誘導してない菌体から、Trizol(登録商標)Max Bacterial RNA Isolation kit(Invitrogen社)を用いて、2種類の全RNAをそれぞれ調整した。また、得られた全RNA試料の一部(10μg)について、DNase処理を以下の手順で行った。10μgの全RNA試料を1×DNase緩衝液(50mM Tris−HCl(pH 8.0)、10mM MgSO4、1mM CaCl2)中で、10ユニットのRNase−free DNase I(タカラバイオ社)で37℃、30分間消化後、フェノールクロロホルム抽出によってDNase画分と核酸画分に分離し、エタノール沈殿によってRNAを沈澱物として回収した。回収したRNAはジエチルピロカーボネート処理された滅菌蒸留水で溶解後、RNA濃度をQuant-iT RNA BR Assay Kitで決定した。
【0066】
PCRによる夾雑DNA(pET-AcGFP)の検出は、以下の手順で行った。各RNA試料20ng、1×AmpliTaq Goldバッファー、0.2mM dNTPs、1μMの各プライマーの組み合わせ(表2)、2mM MgCl2、および0.625ユニットのAmpliTaq Goldポリメラーゼを含む反応液を調製し、95℃で10分間の予備加熱後、95℃で15秒、60℃で30秒、72℃で40秒のサイクルを35回繰り返した後、72℃で7分間の予備伸長を経たのち、4℃で保持した。各プライマーの組み合わせにおけるポジティブコントロール(P1、P2、P3)として、RNA試料の代わりに、1ngのpAcGFP-1(Clontech社)を鋳型として用いた。各PCR産物2μLは、アガロースゲル電気泳動(2.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した。
【表2】
【0067】
8.夾雑DNA含有試料におけるRNA−primed RCAによるRNAの検出
上記(7)のPCRによって夾雑DNAの存在が確認されたRNA試料を用いて、本発明のRPRCAによるGFPmRNAの検出を以下のように行った。各RNA試料20ngを10pmolの環状化オリゴヌクレオチド(表1のP4(配列番号4))とともに、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4および10mM KClを含むバッファー中に混合し、10μlの反応混合物を得た。ネガティブコントロール(N)として、RNA試料を含まない反応混合物を用意した。mRNAと環状化オリゴヌクレオチドとのハイブリダイゼーションを95℃で5分間のインキュベーションにより促進し、その後30℃に急冷した。RCA反応は、ハイブリダイゼーションした混合物10μlを、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4、10mM KCl、0.4mMデオキシヌクレオシド三リン酸(dNTPs)、8mMジチオスレイトール(DTT)、および10ユニットのRepliPHI phi29ポリメラーゼ(Epicentre社)を含む10μlの反応液とを混合することで開始した。反応は30℃、4時間のインキュベーションで行い、その後の合成DNA20μlを、24ユニットのS1ヌクレアーゼ(タカラバイオ社)を用いて37℃、30分間消化し、消化液のうち10μlをアガロースゲル電気泳動(1.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した。
【0068】
〔実験結果のまとめ〕
1.環状化プローブでのRCAを利用したmRNAのリアルタイム検出のためのストラテジー
図1は、RCAによるmRNA検出の基本的なスキームを示す。かかるスキームは、以下の5つの工程を含む:
(1)目的mRNAに相補的な配列を有するssDNAプローブを、CricLigaseなどを用いて、分子内ライゲーションにより環状化する工程、
(2)連続的なRCA反応における非特異的な副生成のDNA合成を避けるため、ライゲーションしていないssDNAプローブをエクソヌクレアーゼIを用いた分解により除去する工程、
(3)環状ssDNAプローブを目的mRNAの3’末端とハイブリダイズさせる工程、
(4)phi29DNAポリメラーゼによって、RNA−DNAハイブリッドからRNA−primed RCA(RNAをプライマーとしたRCA;RPRCA)を行う工程、
(5)SYBR GreenIIなどのssDNAに対する蛍光色素によって、RCAによって合成された結果物であるssDNAの結合による蛍光を得る工程。
これにより、RPRCAを介したmRNAのリアルタイム検出が達成される。
【0069】
2.in vitroで転写されたmRNAのリアルタイム検出
まず最初に、phi29DNAポリメラーゼが、長い配列を有するmRNAをRCA反応のプライマーとして適合させ得るのかについて検討した。プラスミドpET-AcGFPをNotIで、T7ターミネーターとGFP遺伝子の終止コドンの間の領域を消化し、対応するmRNAをT7RNAポリメラーゼでin vitroでの転写によって調製した(図2A)。環状ssDNAプローブ(P1またはP2)もRPRCA用のin vitro転写mRNAと併せて用いた。P1のDNA配列は、GFPの3’末端配列と相補的である(表1)。他方、P2の配列は、P1と長さとGC含量とが同じであるが、配列上、GFPのmRNAと異なるものである(表1)。
【0070】
phi29DNAポリメラーゼによるRCA反応は、P1またはP2を有しないmRNAからは開始せず、反応バッファーにおけるP2のインキュベーションによっても、mRNAが存在してもしなくても、RCA反応を起こすことはなかった(図2BおよびC)。しかしながら、P1単独ではRCA反応に影響しないのに拘らず、mRNAとP1との組み合わせによってRCA反応が促進された。このことから、RPRCAによって、in vitro転写mRNAの3’末端に特異的なssDNAプローブを用いる目的mRNAのリアルタイム検出が行い得ること、および、非特異的環状プローブがRPRCAに影響しないこと判明した(図2BおよびC)。これらのことは、phi29ポリメラーゼが、適切な環状ssDNAプローブの存在下、mRNAをプライマーとして用いるRCA反応を特異的に作用し得ることを示唆している。さらに、エクソヌクレアーゼIで処理した環状ssDNAプローブは、RPRCAにおける長時間の反応期間の後に非特異的な副生成物を産生しなかった。これは、環状プローブの調製におけるエクソヌクレアーゼI処理が特異的で信頼できるRPRCAによる増幅に寄与していることを示す。
【0071】
RPRCAにおける環状ssDNAプローブの長さの効果を、GFPmRNAの3’末端に相補的なDNA領域をカバーするssDNAプローブP3(表1)を用いて行った。図2Dに示すとおり、興味深いことに、同じ長さのGFPmRNAに対して特異的なDNA配列を有するにも拘らず、短いプローブ(P3)ではなく、長いプローブ(P1)で十分なRCA反応が観察された。このことは、プローブの長さが、mRNAと環状プローブとの最初のハイブリダイゼーションに関係し、RPRCAの効率に影響を与えることを示している。
【0072】
まとめると、上記の証拠は、本発明のRPRCAが、適切な環状プローブを用いることで、不規則なDNA増幅副生成物を生成することなく、目的mRNAのリアルタイム検出を可能とすることを示している。
【0073】
3.RPRCAの感度
感度とphi29DNAポリメラーゼの濃度との間の相関関係を明確にするため、RPRCA反応における濃度の影響を調べた。RCA反応は、phi29DNAポリメラーゼの濃度を変化させた条件下で、各サンプルについて、3.2ngのin vitro転写mRNAをプライマーとして添加した場合を観測した。RCA反応は、濃度依存的に促進されることが確認された(図3A)。次いで、図3Aにおいて試験した最も高い酵素濃度(1反応当たり100ユニット)でのRPRCAによるmRNA検出の感度を測定した。その結果、RPRCAは、2時間で320pgのin vitro転写mRNAを検出することができた。
【0074】
4.生きた微生物由来の目的mRNAの特異的検出
RPRCAによって、生きた微生物のmRNAを目的として検出できるかについて検討した。誘導したGFP発現が容易に観察できるため(図4A)、pET-AcGFPで形質転換した大腸菌BL21(DE3)株をRPRCAのモデルとして用いた。生きた細胞において、T7RNAポリメラーゼによって転写されたGFPmRNAの一次構造は、7塩基の5’非翻訳領域(UTR)、GFPmRNA、およびそれに続く3’UTRの48塩基の隣接残基から構成される(図4B)。この実験においては、ssDNAプローブP4を合成した。前記P4は、図4Bに下線で示した3’末端に対応するDNA配列が含まれるように合成されている(表1)。栄養増殖している大腸菌において全RNAの大部分はrRNAおよびtRNAが占めているので、両RNAともRPRCAに影響を及ぼし得る。どの程度の量のrRNAおよびtRNAがRPRCAに影響したかを調べるため、誘導した大腸菌および誘導していない大腸菌の両方から抽出した後、全RNAをRPRCAに用いた。誘導ありまたは誘導なしの大腸菌由来の全RNA200ngをP2またはP4とともにインキュベートした場合、誘導した大腸菌由来の全RNAと環状プローブP4とを組み合わせたものだけ、十分なRPRCAが観察された(図4C)。全RNA単独または環状プローブ単独ではRCA反応は起きなかった。さらに、段階希釈した全RNAを用いた2時間のRPRCAによって、GFPmRNAが10ngの全RNAから検出可能であることが明らかになった(図4D)。RNAの全量(400μg)が1.5×1010細胞の密度の大腸菌から抽出されたことを考慮すると、10ngの全RNAは、およそ4×105細胞に相当する。これらの結果は、細菌性全RNAと適切なssDNAプローブとの組合せを用いたRPRCAは、in vitro転写mRNAの場合(図2)と同様に、GFPmRNAを特異的に検出できるばかりでなく、発現したGFPmRNAと全RNAの大部分を構成するrRNAやtRNAのような他のRNA種とを明確に区別することができることが示された。
【0075】
また、試料中に夾雑DNAが含まれていても(試料中のDNAの分解処理をしていなくても)、良好にRNAの検出ができたことを示すものであった。すなわち、試料中のDNAの分解処理を行わなくても、目的RNAが発現していない試料(図4の「−」(ネガティブコントロール))においては夾雑DNAによる不規則なDNA増幅生成物を生成することがなかったことからも明らかなとおり、本方法においては高いシグナルノイズ比(S/N比)を実現することができた。
【0076】
5.RPRCAのRNase Hを利用したRNA検出のためのストラテジー
図5は、RNase H処理工程を含むRCAによるRNA検出の基本的なスキームを示す。かかるスキームは、以下の6つの工程を含む:
(1)目的mRNAに相補的な配列を有するssDNAプローブを、CricLigaseなどを用いて、分子内ライゲーションにより環状化する工程、
(2)連続的なRCA反応における非特異的な副生成のDNA合成を避けるため、ライゲーションしていないssDNAプローブをエクソヌクレアーゼIによる分解・除去する工程、
(3)環状ssDNAプローブに目的RNAをハイブリダイズさせる工程、
(4)環状ssDNAプローブにハイブリダイズした領域の目的RNAに、RNase Hにより、ニックサイトを形成させる工程、
(5)phi29DNAポリメラーゼによって、ニッキングサイトのRNAの3‘末端からRPRCAを行う工程、
(6)RCAによって合成された結果物であるssDNAを検出する工程。
【0077】
6.RNAase Hを利用したRPRCAによるin vitroで転写されたmRNAの検出
GFPmRNAの内部領域と相補性のあるDNAプローブを利用した場合、RPRCA反応が起こりえるかどうか検討した。プラスミドpET-AcGFPをNotIで、T7ターミネーターとGFP遺伝子の終止コドンの間の領域を消化し、対応するmRNAをT7RNAポリメラーゼによるin vitro転写によって調製した(図6)。転写したGFPmRNAは環状ssDNAプローブ(P5)と併せて用いた。プローブP1のDNA配列は、GFPの内部領域と相補的である(図6)。
【0078】
環状化一本鎖プローブが存在しない場合、phi29DNAポリメラーゼによるRCA反応は、促進されず一切のDNA産物が確認できなかった(図7)。プローブP5を利用した場合、RNase Hを添加しない条件のRPRCA反応では非特許文献8の報告とは異なり、一切のDNA産物は確認できなかった(図7)。このことから、本願のRNase H処理工程を含むRPRCA法では、目的RNAの内部配列をDNAプローブとハイブリダイズさせても、目的RNAを検出できることを示している。さらに、特許文献4とは異なり、ゲル中で単一のバンドとして検出される本願発明は、非特異的な副産物が生じていないことを確認し、その検出精度の高さが認められる。
また、添加するRNase Hの濃度依存性についても検討した結果、RNase Hの添加によってRPRCA反応が促進されることが示された。
【0079】
上記の証拠は、本発明のRNA検出方法は、RPRCAの工程において、適切な濃度のRNase Hで処理することによって、目的RNAの3’末端配列の情報に依存せずに、非特異的なDNA増幅副生成物を生成しないで特異的な検出が可能であることを示している。
【0080】
7.PCRによるRNA試料中の夾雑DNAの検出
GFP遺伝子に特異的なプライマー3セット(表2)を使用して、RNA試料中の夾雑DNAをPCRにより検出した。その際、GFPの発現を自動誘導により制御し、発現誘導した菌体から調整した全RNA(+)と、発現誘導していない菌体から調整した全RNA(−)の2種類を得た。さらに、その一部をそれぞれDNase Iを用いてDNA分解処理をし、発現誘導有りまたは無しの全RNAであって、DNase処理有りまたは無しの合計4種類の全RNAを得た。そして、各全RNAを用いて夾雑DNAの検出を試みた。
【0081】
DNase処理を行っていないRNA試料からは、いずれのプライマーセットにおいても、GFPの誘導・非誘導にかかわらず目的DNA増幅産物が、各プライマーセットのポジティブコントロール(P1、P2、P3)と同様に確認された(図8)。一方で、DNase処理を行ったRNA試料からは、誘導・非誘導で共に目的DNA増幅産物が明らかに減少した(図8)。以上のことから、DNase処理によって夾雑DNAの大部分を除去することが可能であることが示された。しかし、DNase処理を行っても、わずかにバンドが検出されたことから、一般的なDNase処理だけでは、RNA試料から完全に夾雑DNAを取り除くことが難しいことも同時に示された。この夾雑DNAがRT−PCRにおいても同様に、ノイズとして検出される可能性が高いことは、当業者であれば理解できる。
【0082】
8.夾雑DNA含有試料におけるRNA−primed RCAによるRNAの検出
上記(7)のDNase処理有りまたは処理無しのRNA試料を用いて、RPRCAによるGFPmRNAの検出を試みた。その結果、DNase処理の有無、すなわち夾雑DNAの量に拘わらず、発現誘導した菌体から調整したRNA試料から、RPRCAにより増幅したDNAが十分な量で確認された(図9)。また、DNase処理の有無に拘わらず、発現誘導していない菌体から調整したRNA試料では、RPRCAによるDNAの増幅は認められなかった(図9)。以上のことから、本発明のRPRCAにおいては、RNA試料から夾雑DNAを除去することなく、また、RNA試料中の夾雑DNA量の影響を受けることなく、目的RNAのみを高いS/N比で検出できることが示された。
【産業上の利用可能性】
【0083】
本発明は、鎖置換型DNA合成酵素を利用したRNAプライムドローリングサークル型DNA複製法(RNA-Primed Rolling circle amplification;RPRCA)を応用し、RNAの種類、3’末端配列情報等に影響を受けずに、簡便な操作で、逆転写反応を経由せずに、生物由来の夾雑物を含む試料中から直接RNAをin situやリアルタイムで検出可能な方法に関するものであり、研究、医療・臨床検査、食品産業など幅広い応用範囲において貢献するものである。
【技術分野】
【0001】
本発明は、試料中のRNAを直接検出する方法、より具体的には、検出対象のRNAをプライマーとして、鎖置換型DNA合成酵素によってDNAを増幅するローリングサークル増幅法(RNA-Primed Rolling Circle Amplification;RPRCA)を用いたRNAの直接検出法およびその応用に関する。
【背景技術】
【0002】
食品のような複雑なサンプルから僅かな微生物を検出し、サンプルに存在する生きた微生物の有無を検査することを目的とする場合、一般的には培養法が用いられる。しかし培養法による微生物の検出には、選択培地によるところが大きく、しかも数日の培養期間が必要であるため、迅速性が求められる食中毒検査や臨床診断等には向いていない。一方、微生物の迅速な検出には、微生物等が有する固有のDNA配列をポリメラーゼ連鎖反応(polymerarse chain reaction;PCR)を利用して検出する方法が知られている。しかし、DNAは化学的に安定な化合物であるため、PCRによって微生物等に固有なDNA配列が検出されたとしても、当該微生物等の生死を判別するには至らない。
【0003】
細胞内のRNAは細胞が死滅すると速やかに分解されるため、RNAを検出することによって微生物などの生きた細胞の検出および生死判別を行う方法が開発されている。特に、RNAを逆転写反応(RT(reverse transcription)反応)によってDNAに変換した後にPCRを行うRT−PCR法はよく知られている(例えば、非特許文献1)。しかし、PCR法はその反応のシステムのため、反応温度を変化させることが可能なサーマルサイクラーなどの高価な装置を必要としており、一般的なPCRにかかる時間は少なくとも2〜3時間程度を必要としている。また、増幅と検出を同時に行うことのできるリアルタイム検出法が可能であるが、かかる検出法には、サーマルサイクラーに検出装置が組み込まれている必要があり、サーマルサイクラーよりもさらに費用が嵩むことになる。
【0004】
一方で、RNAを逆転写した後に、鎖置換型DNA合成酵素を利用したLAMP法(Loop-Mediated Isothermal Amplification)によってRNAを迅速に検出するRT−LAMP法も開発されている(非特許文献2)。RT−LAMP法は、およそ1〜2時間程度での検出が可能であるが、その反応システムは複雑で汎用性に乏しく、また反応システム上、比較的高温(65℃)を維持できる機械を必要としている。PCR法と比較して、低価格の装置でリアルタイム検出も可能であるが、その反応システムは、特殊なプライマー設計の複雑さなどのために現時点では汎用されていない。
【0005】
さらに、RT−PCR法やRT−LAMP法によるRNA検出では、最終的な増幅反応に至るまでに生成するDNAが、次の反応ステップに混入して非特異的な逐次反応を誘発する、いわゆるキャリーオーバーによるクロスコンタミネーションが常に問題となる。そのため、試料中の夾雑物、特にゲノム由来のDNAが微量でも混入した場合には偽陽性の結果を生じやすく、目的となるRNAの検出を困難にするため、DNA分解処理などのRNAを精製する工程を必要とする。したがって、追加の工程や専用の設備等が必要となり、さらには高度に訓練された人員が必要となるため、初期導入コストのみならずランニングコストも高額になる傾向がある。その結果、畜産産業や食品産業のみならず、臨床検査の現場においても、核酸増幅による微生物検出法の採用が忌避される傾向にある。
【0006】
一方、反応装置が簡素なRNA検出方法も開発されており、RT反応を経たあと、RT反応に用いたプライマー領域に設定したバクテリオファージプロモーター領域から、再度バクテリオファージRNA合成酵素によりRNAを増幅する転写媒介増幅法(Transcription-mediated amplification;TMA)(非特許文献3)や、TMA法にリボヌクレアーゼH(RNase H)を組み合わせたNASBA法(Nucleic acid sequence-based amplification;NASBA)がRNA増幅・検出方法として利用されている(非特許文献4)。これらは低温(37℃)における反応であるため、サーマルサイクラー等の特別高価な装置は不要である。しかし、反応生成物がRNAであるため検出には特殊なゲル電気泳動装置等を用いるか、あるいは反応後にRT−PCR法を行う必要があり、操作が煩雑である。
【0007】
上述のように、既存のRNAの増幅・検出法はすべて逆転写反応を経由して行われるため、実験操作の簡素化や反応条件の最適化を行ったとしても、逆転写反応そのものにかかる時間は短縮できないため、検出までに概ね2時間以上の時間を必要とし、検出速度の向上は困難である。かかる問題点は、一層の高速化が求められているRNA検出法の開発、例えば、病原性大腸菌O157や高病原性インフルエンザウイルスのような病原性RNAウイルスの検出法の開発などにおいても大きな障害となっていた。
【0008】
近年、これらの問題を解決する核酸増幅方法として、ローリングサークル増幅(rolling circle amplification;RCA)法が検討されている。RCA法は、一本鎖環状DNAプローブを該DNAプローブに相補的な配列を持つDNAプライマーでハイブリダイズさせた後、該DNAプローブを鋳型とした鎖置換型DNA合成反応によって一本鎖DNAを連続的に生成する反応である。したがって、RCA法ではDNA合成反応後に生成する一本鎖DNAを検出することによって、DNAプライマーもしくはDNAプローブの存在を判定することができる。
【0009】
そしてRNA検出方法として、RNA−プライムドローリングサークル型増幅(RNA-Primed Rolling Circle Amplification;RPRCA)法が、新たなRNA検出法として注目を浴びている。RPRCAは標的RNA分子の3’側配列と相補的配列を有する環状DNAプローブとをハイブリダイズさせた後、当該RNA分子の3’末端をDNA合成の開始点として利用し、一本鎖DNAをRCA法により連続的に合成する。これにより合成された一本鎖または二本鎖DNAを検出することにより、逆転写反応を行わずに目的のRNA、特に原核微生物由来のメッセンジャーRNA(mRNA)や、真核生物のマイクロRNA(miRNA)などのsmall RNA(sRNA)を短時間で検出することが可能となる。
【0010】
これまでに、RCA法を利用したRNAの検出方法として、真核生物のmiRNAとハイブリッド形成させた一本鎖環状DNAプローブを用いて、当該RNAをRCA法によって検出する方法が検討されている(特許文献1)。しかしながら、このRCA法には、非特異的に合成されるDNA副生成物によって検出時のシグナルノイズ比(S/N比)が低下するという問題がある。非特異的なDNA副生成物に起因するシグナルノイズ比の低下を回避するために、特許文献1では、複数のDNAオリゴヌクレオチドを介して予め環状DNAを作成し、未反応の一本鎖DNAの除去を試みているが、複数であるが故に部分的な二本差を形成しやすくなり、二本鎖を形成したオリゴヌクレオチドはエクソヌクレアーゼのみでは除けないため、この方法では困難である。
【0011】
また、特許文献2では環状化反応およびDNA合成において、耐熱性酵素を利用して反応温度を上昇させることにより、非特異的なDNA合成の原因となる非特異的な二本鎖形成の抑制を検討している。しかし、耐熱性酵素のために、非耐熱性酵素に比べてシグナル検出速度は低下する。
【0012】
特許文献3では、直鎖状パドロックDNAプローブ(非特許文献5)を検出対象のRNAとハイブリッド形成させた上で、該パドロックDNAプローブを環状化させ、一本鎖環状DNAプローブを形成させる方法を開示している。同様に、特許文献4および非特許文献6においても、パドロックDNAプローブと検出対象のRNAとのハイブリッドを形成させた後、パドロックDNAプローブの環状化反応を行い、さらに検出用DNAプライマーを用いて、検出対象のRNAの有無が検証可能な方法を報告している。
【0013】
しかしながら、直鎖状パドロックDNAプローブを環状化させる際、反応溶液中の直鎖状パドロックDNAプローブが全て環状化されることはなく、未反応の直鎖状パドロックDNAプローブ自身が非特異的な二本鎖核酸を形成する。また、検出用DNAプライマーの使用は、さらに非特異的なDNAの副生成物を生じさせるため、検出時のS/N比が大きく低下するという問題がある。
【0014】
非特異的な副生成物に由来するS/N比の低下は、検出感度を下げるのみならず、目的RNAの検出においては、偽陽性の結果を生じることにより、不正確な判断を引き起こしかねない。この不正確な判断材料は、特にウイルスを含む感染性微生物の臨床検出において被験者の「生活の質」(Quality Of Life;QOL)の低下を引き起こす可能性がある。
【0015】
さらに、パドロックDNAプローブを用いたRPRCA法においては、目的のRNA上におけるパドロックDNAプローブのT4 DNAライゲースによる環状化は、僅か2.5%程度であり、DNA上で起こるそれと比べ著しく効率が悪化していると報告されている(非特許文献7)。使用するライゲースをT4 RNAライゲースに変更しても、環状化反応は全体の20%程度しか起きないため、RNAのパドロックDNAプローブによる検出効率は、非常に悪いことが明らかにされた。
【0016】
また、従来のRPRCA法の多くは検出対象となるRNAの3’末端側配列をプライマーとして利用するため、検出可能なRNA分子種は、トランスファーRNA(tRNA)やリボソームRNA(rRNA)、および原核生物のmRNAや、真核生物のsRNA、あるいは一部のRNAウイルスのゲノムRNA(gRNA)に限定されていた。特に、RPRCA法による真核生物のmRNAの特異的な検出は、全てのmRNAの3’末端にポリA配列が存在するため、原理的に不可能であった。
【0017】
特許文献4では、多様なRNA種の3’末端側配列、特にmRNAの3’末端側配列をプライマーとして利用するため、ポリA配列をリボザイムによって除去する方法や、RNA−DNAハイブリッドにRNase H処理をしてニックを導入することによって、mRNAの3’末端をプライマーとして伸長可能にする方法が記載されている。しかしながら、同文献中、単に手法が記述されているに過ぎず、実際に反応産物を得たものでも、例えば、ゲル中でバンドとして検出したものでもなかった。したがって、同文献に記載の方法では、上述のS/N比の問題を認識していないため、目的物と非特異的な産物との区別ができず、偽陽性の結果を生じる危険性を包含するものであった。さらにまた、同文献における環状化DNAプローブの作製工程は、極めて煩雑であり、実用に耐えるものではなかった。
【0018】
一方で、検出対象のRNAとパドロックDNAプローブをハイブリダイズさせた後、当該パドロックDNAプローブをT4 DNAライゲースにより環状化し、DNA合成酵素の、特にphi29DNAポリメラーゼの3’−5’エクソリボヌクレアーゼ活性により、ハイブリダイズしていない検出対象のRNAの3’末端配列をハイブリダイズしている位置まで除去した後、ハイブリダイズしたRNA部分をプライマーとしてDNA合成を開始する方法が開発された(非特許文献8)。さらに、二本鎖RNA特異的分解酵素であるRNase IIIを反応液に加えることにより、二次構造(二本鎖領域)を取っているであろう非ハイブリダイズ領域のRNA部分を分解・除去し、RCA反応の向上が認められたことを報告している(非特許文献9)。
【0019】
しかしながら、phi29DNAポリメラーゼの3’−5’エクソリボヌクレアーゼ活性は非常に弱いため、除去できるRNAの長さが制限される。さらに、phi29DNAポリメラーゼの3’−5’エクソリボヌクレアーゼ活性は、RNAの二次構造を破壊できず、二次構造を取りやすいRNA分子ではほとんど分解されないため、ハイブリダイズさせるRNA領域は3’末端側に限定されてしまい、例えば、5’EST配列は現実的に利用できなかった(非特許文献10)。
【0020】
また、非特許文献10においては、RNAの直接検出を断念し、代替方法として逆転写反応時に利用する標的RNA特異的なDNAプライマーの構成残基の一部にLocked nucleic acid (LNA)を使用することにより、ハイブリダイゼーション効率を高めて逆転写効率を向上させ、検出対象のRNA特異的な逆転写産物によるRCA反応を行うことにより、検出感度上昇が可能であることを報告している。
【0021】
しかしながら、この方法は、複数のエクソンで構成されている真核生物のmRNAでは有効であるが、1エクソンで構成されている酵母や原核微生物においては、残存していたゲノムDNAとの区別が厳密にはできない。さらに、非特許文献10では最終的にcDNAではあるが、DNAをプライマーとして用いるために、非特異的なDNA副生成物が生じる。したがって,パドロックDNAプローブと相補性のある検出用DNAプローブのハイブリダイゼーションによってのみ、増幅・検出の特異性が保証されるため、煩雑な操作手順を捨て切れていない。
【0022】
さらに、従来技術においては、上記の低いS/N比の問題からも、RNA試料中に混入するDNA等の夾雑物によるノイズを防ぐ必要があると考えられており、目的のRNAを検出するために、RNA試料をDNaseで処理したり、精製されたRNA試料を用意する等、利用できる試料が制限されていた(特許文献4、非特許文献8および9)。
【0023】
以上のように、これまで検討されてきた方法は、PCR反応の温度サイクルを管理する装置が必要であったり、検出対象のRNAを逆転写によってDNA化したり、設備面に止まらず、検出精度においてもコンタミネーションなどによる増幅反応における副生成物の問題などがあった。
【0024】
一方で、RPRCA法においては、検出対象のRNAの種類および由来によっては検出用プローブの構築ができず、簡便な手順のRPRCA法を実行できないRNA分子種のほうが多く存在していた。さらに、非特異的な副生成物に由来するS/N比の低下によって、目的のRNA検出において不正確な判断を引き起こしかねないため、実用段階には至っていない。
【0025】
したがって、RNAを検出する方法として、高額な装置を必要とせず、ゲノム配列等の配列の一部でも既知であれば、特異的な検出が可能となり、さらに簡便でありながら迅速で、かつ高感度の検出方法の構築が求められていた。
【先行技術文献】
【特許文献】
【0026】
【特許文献1】米国特許出願公開第2009/0181390号明細書
【特許文献2】米国特許出願公開第2006/0188893号明細書
【特許文献3】特表2003−534782号公報
【特許文献4】米国特許出願公開第2005/0112639号明細書
【非特許文献】
【0027】
【非特許文献1】Selvaratnam S, Schoedel BA, McFarland BL, Kulpa CF. Application of reverse transcriptase PCR for monitoring expression of the catabolic dmpN gene in a phenol-degrading sequencing batch reactor. Appl. Environ. Microbiol. 61:3981-3985. (1995).
【非特許文献2】Fukuta S, Iida T, Mizukami Y, Ishida A, Ueda J, Kanbe M, Ishimoto Y. Detection of Japanese yam mosaic virus by RT-LAMP. Arch. Virol. 148:1713-20 (2003).
【非特許文献3】Kwoh DY, Davis GR, Whitfield KM, Chappelle HL, DiMichele LJ, Gingeras TR. Transcription-based amplification system and detection of amplified human immunodeficiency virus type 1 with a bead-based sandwich hybridization format. Proc. Natl. Acad. Sci. USA. 86:1173-7 (1989).
【非特許文献4】Guatelli JC, Whitfield KM, Kwoh DY, Barringer KJ, Richman DD, Gingeras TR. Isothermal, in vitro amplification of nucleic acids by a multienzyme reaction modeled after retroviral replication. Proc. Natl. Acad. Sci. USA. 87:1874-1878 (1990).
【非特許文献5】Nilsson M, Malmgren H, Samiotaki M, Kwiatkowski M, Chowdhary B.P., and Landegren U. Padlock probes: circularizing oligonucleotides for localized DNA detection. Science 265 2085-8 (1994).
【0028】
【非特許文献6】Wang B, Potter SJ, Lin Y, Cunningham AL, Dwyer DE, Su Y, Ma X, Hou Y, Saksena NK. Rapid and sensitive detection of severe acute respiratory syndrome coronavirus by rolling circle amplification. J. Clin. Microbiol. 43(5):2339-44 (2005)
【非特許文献7】Li N, Jablonowski C, Jin H, Zhong W. Stand-Alone Rolling Circle Amplification Combined with Capillary Electrophoresis for Specific Detection of Small RNA. Anal. Chem. 81, 4906-4913 (2009)
【非特許文献8】Lagunavicius A, Merkiene E, Kiveryte Z, Savaneviciute A, Zimbaite-Ruskuliene V, Radzvilavicius T, Janulaitis A Novel application of Phi29 DNA polymerase: RNA detection and analysis in vitro and in situ by target RNA-primed RCA. RNA 15:765-771 (2009).
【非特許文献9】Merkiene E, Gaidamaviciute E, Riauba L, Lagunavicius A. Direct detection of RNA in vitro and in situ by target-primed RCA: the impact of E. coli RNase III on the detection efficiency of RNA sequences distanced far from the 39-end. RNA Published online June 28 (2010)
【非特許文献10】Larsson C, Grundberg I, Soderberg O, Nilsson M. In situ detection and genotyping of individual mRNA molecules. Nat. Methods 7: 395-397 (2010)
【発明の概要】
【発明が解決しようとする課題】
【0029】
本発明の課題は、高度な設備を必要とせず、簡便な操作で、実用的な感度を有しながら迅速に行うことができる、RNAの検出方法であって、生物由来の夾雑物を含む試料中、特にDNAが混入した試料においても、特異的な検出を可能とする方法を提供することである。
【課題を解決するための手段】
【0030】
本発明者らは、上記課題を解決するため、鋭意研究を重ねた結果、検出対象とする目的RNAの3’末端部分配列と相補的なDNAを合成し、一本鎖DNA連結酵素により自己環状化させて、得られた環状化一本鎖DNAプローブを目的RNAとハイブリッド形成させ、さらに目的RNAをプライマーとして一本鎖DNAを合成させると、予め反応系に一本鎖DNA特異的な蛍光色素を添加しておくことにより、目的RNAのリアルタイム検出が可能となることを見出した。
【0031】
また、RNase H処理の工程を加えることにより、3’末端にDNAプローブとの相補配列を有さないRNAであっても検出可能となり、ウイルスや原核生物のみならず真核生物由来の多様なRNAの検出が可能となった。
【0032】
さらに、本発明者らは、本発明のRPRCA法によって、細胞から実際に得られた試料中から目的RNAを検出した知見に基づき、生物由来の夾雑物を含む試料中、例えば、DNAが混入した試料であっても、特異的な目的RNAの検出が可能となることを見出し、本発明を完成させるに至った。
【0033】
すなわち本発明は、以下の方法およびキットに関する。
[1] 生物由来の夾雑物を含む試料中のRNAの検出方法であって、
(1)検出対象のRNAに相補的な配列を有する一本鎖DNAを環状化し、環状化一本鎖DNAプローブを作製する工程、
(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる工程、
(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしてローリングサークル増幅法(RCA;Rolling Circle Amplification)により一本鎖DNAを合成する工程、
(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する工程
を含む、前記方法。
[2] 生物由来の夾雑物が、DNAを含む、[1]に記載の方法。
[3] 試料が、核酸試料である、[1]または[2]に記載の方法。
[4] 試料中のDNAが、分解処理されていない、[2]または[3]に記載の方法。
[5] 前記(1)において、環状化していない未反応の一本鎖DNAを分解することを含む、[1]〜[4]のいずれか一項に記載の方法。
[6] 環状化一本鎖DNAプローブが、検出対象のRNAに相補的な配列のみからなる、[1]〜[5]のいずれか一項に記載の方法。
[7] 環状化一本鎖DNAプローブが、30塩基長〜4500塩基長である、[1]〜[6]のいずれか一項に記載の方法。
[8] 前記(2)の後に、
(5)RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程
をさらに含む、[1]〜[7]のいずれか一項に記載の方法。
[9] 前記(3)および/または(5)が、25℃〜65℃の範囲の一定の温度で行われる、[8]に記載の方法。
[10] 前記(5)において、RNase Hを終濃度1×10−4〜10ユニット/反応で用いる、[8]または[9]に記載の方法。
[11] 前記(5)において、RNase Hを終濃度1×10−7〜0.1ユニット/μlで用いる、[8]または[9]に記載の方法。
[12] RNAが、真核生物、原核生物、ウイルスまたはウイロイド由来である、[1]〜[11]のいずれか一項に記載の方法。
[13] RNAが、ポリA配列を有する、[12]に記載の方法。
[14] RNAが、ノンコーディングRNAである、[12]に記載の方法。
[15] [1]〜[14]のいずれか一項に記載のRNAの検出方法を含む、真核生物、原核生物、ウイルスまたはウイロイドの検出方法。
[16] 微生物を検出する、[15]に記載の方法。
[17] [1]〜[14]のいずれか一項に記載のRNAの検出方法、および/または[15]に記載の真核生物、原核生物、ウイルスまたはウイロイドの検出方法、および/または[16]に記載の微生物の検出方法のために用いるキットであって、検出対象のRNAに相補的な配列を含む環状化一本鎖DNAプローブを含むキット。
[18] アデニン、グアニン、シトシンおよびチミンからなるデオキシヌクレオチド、反応用緩衝液および鎖置換型DNA合成酵素からなる群から選択される1種または2種以上をさらに含む、[17]に記載のキット。
【発明の効果】
【0034】
本発明によれば、RNAの検出の際に検出対象である目的RNAに対して逆転写反応を行う必要がなく、直接RNAをプライマーとして用いてDNAを合成させ、かつ合成された一本鎖DNAに一本鎖特異的蛍光色素を反応させることにより、極めて簡便に、リアルタイム検出が可能である。
また、未反応の一本鎖DNAに由来する非特異的なDNA合成を完全に抑制することが可能となり、高いシグナルノイズ比(S/N比)を実現し、さらに、過去の増幅産物のキャリーオーバーによるクロスコンタミネーションリスクが、他の核酸増幅法に比べ著しく低いため、RNAを簡便な操作で、実用的な感度を有しながら、また高度な設備を必要せず、安価な装置で、短時間で迅速に検出することが可能となる。
【0035】
また、RNase H処理の工程を加えることにより、検出対象となるRNAの3’末端の配列に依存せずに、部分的にでも既知であれば、様々な種類のRNAの検出が可能となり、ウイルスや原核生物由来のRNAだけでなく、真核生物由来のRNAおよびそれら生物自身の検出も可能となった。
【0036】
さらに、本発明によれば、検出感度がよいため、試料中に生物由来の夾雑物を含んでいてもRNAの検出が可能である。また、試料中にDNAを含む場合であっても、DNAの分解処理を要しない。このため本発明の方法は、従来のRNA検出法と比較して、迅速かつ簡便に試料中のRNAを検出することができる。これは、夾雑DNAが、環状化一本鎖DNAプローブとハイブリダイズしても、その3’末端がプライマーとして利用されることが確率的にほとんどありえないことによる。
【0037】
このことは、RNAを特異的に検出するためには試料中の夾雑DNAを除去するという当該分野の技術常識に反する驚くべき知見であり、これによって、生きている細胞由来のRNAの検出が容易になり、実用に十分な感度を有しながらも、簡便でありながら迅速で、かつ特異的なRNAの検出が可能となった。したがって、医学、薬学、農学、食品学などの研究に止まらず、臨床検査などの医療分野や、食品産業における簡便な生菌検査や食中毒における迅速検査など、簡便さ、省力化、迅速さが求められる様々な基礎および応用分野において、とくに有用である。
【図面の簡単な説明】
【0038】
【図1】図1は、本発明のRNA−primed RCAによるRNAの検出を示す概略図である。
【図2】図2は、RNA−primed RCAによるin vitro転写mRNAの特異的検出を示す:(A)in vitroで転写されたGFPmRNAの3’末端配列を示す。GFPmRNAは、NotI消化後のGFP遺伝子のin vitro転写により調製した。下線およびイタリック文字は、夫々、GFP遺伝子の終止コドンおよびNotI制限酵素サイトを示す。配列中の大文字および小文字は、夫々、DNA残基およびRNA残基を示す。(B)RNA−primed RCAによるin vitro転写mRNAのリアルタイム検出を示す。RNA−primed RCAによる蛍光は、夫々、以下の記号で示す:白丸はmRNAもDNAプローブもなし、黒丸はmRNA単独、白四角はP1(配列番号1で表されるプローブ、表1)単独、黒四角はP1とmRNA、白三角はP2(配列番号2で表されるプローブ、表1)単独、黒三角はP2とmRNA。(C)RNA−primed RCAによるin vitro転写mRNAの電気泳動による検出を示す。15時間のRNA−primed RCAにより増幅されたDNAを分子量マーカー(キロベースラダーマーカー)とともにアガロースゲルで泳動した。(D)RNA−primed RCAにおけるプローブの長さの効果を示す。RNA−primed RCAによる蛍光は、夫々、以下の記号で示す:白丸はmRNAもDNAプローブもなし、黒丸はmRNA単独、白四角はP1単独、黒四角はP1とmRNA、白三角はP3(配列番号3で表されるプローブ、表1)単独、黒三角はP3とmRNA。
【図3】図3は、RNA−primed RCAの感度を示す:(A)プライマーとしてのin vitro転写GFPとのRNA−primed RCAにおけるphi29DNAポリメラーゼの濃度の効果。酵素濃度は、夫々、以下の記号で示す:白丸は0ユニット、黒丸は10ユニット、白四角は20ユニット、黒四角は40ユニット、白三角は60ユニット、黒三角は80ユニット、白菱形は100ユニット。(B)段階希釈したin vitro転写GFPによるRNA−primed RCAの滴定分析。用いたGFPmRNAの量は、0pg(白丸)、32pg(黒丸)、320pg(白四角)、3.2ng(黒四角)、32ng(白三角)。
【図4】図4は、大腸菌から単離された全RNA中に存在するmRNAの特異的検出を示す:(A)pET-AcGFPを保有する大腸菌におけるGFPの発現。自己誘導ありまたはなしのGFP発現をブルーライト下で可視化した。(B)大腸菌において転写されたGFPmRNAの3’末端配列。GFPmRNAはT7プロモーターの制御下で転写され、かかる転写は、T7ターミネーターで終了した。下線は、環状化プローブP4(配列番号4で表されるプローブ、表1)に対して相補的な配列を示す。(C)RNA−primed RCAによる自己誘導ありまたはなしの大腸菌から単離された全RNAに存在するGFPmRNAのリアルタイム検出。RNA−primed RCAによる蛍光は、以下の記号で示す:(1)白丸はmRNAもDNAプローブもなし、(2)灰丸は誘導なしの大腸菌由来の全RNA、(3)黒丸は誘導ありの大腸菌由来の全RNA、(4)白四角はP4単独、(5)灰四角はP4と誘導なしの大腸菌由来の全RNA、(6)黒四角はP4と誘導ありの大腸菌由来の全RNA、(7)白三角はP3単独、(8)灰三角はP3と誘導なしの大腸菌由来の全RNA、(9)黒三角はP4と誘導ありの大腸菌由来の全RNA。(D)誘導した大腸菌から得られた全RNAの段階希釈したものによるRNA−primed RCAの滴定分析。用いた全RNAの量は、0ng(白丸)、2ng(黒丸)、10ng(白四角)、20ng(黒四角)、40ng(白三角)および80ng(黒三角)。
【0039】
【図5】図5は、RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程をさらに含む、本発明のRNA−primed RCAによるRNAの検出を示す概略図である。(A)標的遺伝子のmRNAの下流(3’末端)は環状化DNAプローブと相補鎖形成しないため、DNAの合成反応が開始しない。(B)標的遺伝子のmRNAと環状化DNAプローブが相補鎖形成した後、RNase Hで処理することによりニックが導入され、RCA反応が開始する。
【図6】図6は、pET-21dにクローン化したGFP遺伝子の全配列を示す。GFPmRNAは、NotI消化後のpET-AcGFPおよびT7RNAポリメラーゼを使用してin vitro転写により調製した。下線(太字)は、GFPmRNA内部領域にて相補鎖形成するように設計された環状化DNAプローブ(P5)の位置を示す。下線のATGおよびTGAは、夫々開始コドンおよび終止コドンを示す。
【図7】図7は、RNA−primed RCAによるin vitro転写mRNAの検出における、RNase H処理の効果を示す。15時間のRNA−primed RCAにより増幅されたDNAを分子量マーカー(キロベースラダーマーカー)とともにアガロースゲルで泳動した。また、RNA−primed RCA反応におけるRNase Hの濃度依存性について検討した結果を示す。
【0040】
【図8】図8は、PCRによるRNA試料中の夾雑DNA(pET-AcGFP)の検出結果を示す。中段のDNase(+)およびDNase(−)は、DNase処理有りおよび無しをそれぞれ示し、下段の(+)および(−)は発現誘導した菌体および発現誘導していない菌体由来の全RNAをそれぞれ示す。PCR産物は、分子量マーカー(100bpラダー)とともにアガロースゲルで泳動した。
【図9】図9は、夾雑DNAが混入した試料中における、本発明のRNA−primed RCAによる標的RNAの特異的検出を示す。4時間のRNA−primed RCAにより増幅されたDNAを、アガロースゲルで泳動した。DNase処理の有無は、全RNA試料中のGFPmRNAの検出結果に影響を与えない。Nはネガティブコントロールを示し、(+)および(−)はそれぞれ発現誘導した菌体および発現誘導していない菌体由来の全RNAを示す。
【発明を実施するための形態】
【0041】
本発明のRNAの検出方法は、鎖置換型DNA合成酵素を利用したローリングサークル型DNA複製法(Rolling circle amplification;RCA)を利用し、応用した技術を用いる。
したがって、先ず、(1)検出対象のRNAに相補的な配列を有する一本鎖DNA(ssDNA)を環状化し、環状化一本鎖DNAプローブを作製する。
検出対象のRNA(目的RNA)に相補的な配列としては、目的RNA特異的検出の観点から、目的RNAの3’末端側の部分の、例えば、20〜100塩基、とくに、25〜40塩基に相補的な配列が好ましい。RNase H処理によって、DNA−RNAハイブリッド上のRNA鎖にニックを導入する工程を含む場合、検出対象の目的RNAに相補的な配列は、RNAのいずれの位置からも任意に設定することが可能である。
【0042】
相補鎖を形成するssDNAは、100塩基以下の場合は核酸合成機によるDNA合成が好ましいが、100塩基以上の場合は、PCR等により目的DNAを増幅後、公知の方法、例えば熱変性により一本鎖化した後、環状化反応に使用することもできる。例えば、30〜4,500塩基、とくに、50〜700塩基に相当する相補的な配列が好ましい。検出感度はプローブの長さが長い程上昇し、短ければ短いほど減少する。また、非特異的な反応をさらに抑えるために、プローブは検出対象のRNAとハイブリダイズ可能な相補配列のみから構成されてもよい。
【0043】
一本鎖DNAの環状化は、任意の手段によって行うことができるが、例えば、CircLigase(登録商標)、CircLigase II(登録商標)、ssDNA Ligase(Epicentre社)、ThermoPhage ligase(登録商標) single-stranded DNA(Prokzyme社)を用いて行うことができる。環状化は、分子内ライゲーションにより環状化してもよいし、また複数の一本鎖DNAが分子間ライゲーションにより、タンデムに配した状態で環状化してもよい。好ましくは、部分的な二本鎖形成による不完全なエクソヌクレアーゼ処理を防ぐために、環状化反応においてDNAプローブにガイド配列を必要としない種類の一本鎖DNAリガーゼが用いられる。
高いS/N比を得るためにも、環状化していない未反応の一本鎖DNAを分解することが好ましく、とくに好ましくは、エクソヌクレアーゼI(各社)、T5エクソヌクレアーゼ(Epicentre社)、エクソヌクレアーゼVI(Epicentre社)、RecJエクソヌクレアーゼ(Epicentre社)等の一本鎖オリゴヌクレオチドをモノヌクレオチドまで分解可能なエクソヌクレアーゼによる酵素分解が好ましい。ただし、エクソヌクレアーゼIIIは、作成された一本鎖環状DNAを基質とするため使用できない。
【0044】
次に、(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる。かかるハイブリダイゼーションの条件は、当業者であれば、環状化一本鎖DNAプローブと目的RNAとの組み合わせを検討し、適宜設定できる。
本発明に用いる試料は、標的となるRNAを含有していれば、特に限定されないが、かかるRNAが精製されていても、精製されていなくても用いることができる。目的RNAの分解を避けるため、リボヌクレアーゼ活性の低減した試料であってもよい。例えば、リボヌクレアーゼ阻害剤で処理し、リボヌクレアーゼ活性を抑制させた試料であってもよい。
【0045】
典型的には、生物由来の夾雑物を含んでいてもよい。生物由来の夾雑物としては、例えば、ゲノムDNA、葉緑体DNA、ミトコンドリアDNAおよびプラスミドDNAなどの各種のDNA、タンパク質、脂質、多糖類などが挙げられる。
また、試料中にDNAが含まれている場合、DNaseなどのDNA分解酵素や、他のDNA分解処理がされていないものであってもよい。特に、本発明は、DNA分解処理を必要としないため、DNAを含む試料に対してむしろ好適に実施することができる。
【0046】
任意に、前記(2)において得られた目的RNA−DNAハイブリッド上で、RNase Hによって目的RNA鎖のみにニックを形成させる工程(5)を含むことができる。RNase Hは一般に用いられている中温性のRNase HI(各社)やRNase HII(NEB社)の他に、Hybridase(登録商標)、Thermostable RNase H(Epicentre社)、Tth RNase H(東洋紡社)等の耐熱をRNase Hを用いることでも行うことができる。RNase Hは反応の初期段階において必要であり、反応中期から後期には必要ないため必要な量はごく少量でもよい。DNAの伸長反応において使用されるRNase Hの量(濃度)は、好ましくは終濃度約1×10−4〜10ユニット/反応、より好ましくは終濃度約1×10−4〜1ユニット/反応で使用する。
または、本発明の方法において、RNase Hは、好ましくは1×10−7〜0.1ユニット/μl、より好ましくは1×10−7〜1×10−2ユニット/μlの範囲において好適に用いられる。
【0047】
さらに、(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしたローリングサークル増幅法(RCA;Rolling Circle Amplification)を行う。RCAは、例えば、phi29 polymerase(各社)、Klenow DNA Polymerase (5’-3’, 3’-5’ exo minus) (各社)、Sequenase(登録商標)Version 2.0 T7 DNA Polymerase (USB社)、Bsu DNA Polymerase, Large Fragment (NEB社)などの中温性の鎖置換型DNA合成酵素や、Bst DNA Polymerase (Large Fragment) (各社)、Bsm DNA Polymerase, Large Fragment (Fermentas社) 、BcaBEST DNA polymerase (TakaraBio社)、Vent DNA polymerase (NEB社)、Deep Vent DNA polymerase (NEB社)、DisplaceAce (登録商標) DNA Polymerase (Epicentre社)等の耐熱性の鎖置換型DNA合成酵素を用いることでも行うことができる。
【0048】
本発明の方法によるDNAの伸長反応は、好ましくは、サーマルサイクラーを用いる必要はなく、例えば、25℃〜65℃の範囲の一定の温度において実施される。反応温度は、酵素の至適温度とプライマー鎖長に基づく変性温度(プライマーが鋳型DNAに結合(アニール)/解離する温度帯)に基づいて通常の手順により適宜設定される。さらに、一定の比較的低温においても実施される。例えば、鎖置換型DNA合成酵素としてphi29DNAポリメラーゼを使用する場合は、好ましくは25℃〜42℃、より好ましくは約30℃で反応する。
【0049】
そして、(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する。 このような標識としては、例えば、放射線(32Pなど)標識、蛍光標識など、検知可能な種々の標識を用いることができるが、とくに、検知の容易さ、安全性およびLED使用等による検出機作成時の簡便さなどの観点から、蛍光標識であることが好ましい。また、一本鎖DNAが部分的に二本鎖を形成するため、一般的な二本鎖DNA検出に利用されるエチジウムブロマイドも、増幅産物の検出に使用できることは、当業者であれば理解できるだろう。
【0050】
標識は、合成された一本鎖DNAに結合すれば、検出対象配列に特異的であっても、特異的でなくてもよい。配列に特異的に結合する標識としては、例えば、放射線標識されたDNA断片やメチル化等の修飾を含むRNA断片、ペプチド核酸(PNA)断片、DNA配列特異的に結合する抗体や金属ナノ粒子などが挙げられる。標識の標的となる特異的なDNA、RNAおよびPNA配列は、検出対象のRNAの配列に相補的な配列であってもよいが、例えば、環状化一本鎖DNAプローブに標識の標的となる配列の相補的な配列を組み込んでおくことも可能である。
また配列に特異的でなくても、DNAに特異的に結合する標識を用いることも可能であり、例えば、SYBR GreenII(Invitrogen社)などの蛍光色素をもちいることができる。
【0051】
標識の検知方法は、標識の種類に応じて適宜選択し得る。例えば、抗体を標識に用いた場合は、二次抗体やカップリング反応を用いて検知すること、また金属ナノ粒子を用いた場合には、粒子間相互作用の変化に伴う物性変化を計測することによる検知が可能である。さらに、クロマトグラフィーや電気泳動などを用いて検知することも可能である。
本発明のRNAの検出方法において、検出対象となるRNAはとくに限定されず、例えば、各種細胞におけるメッセンジャーRNA(mRNA)の発現をリアルタイム検出することや、微生物やウイルス由来のRNAを検出対象とすることも可能である。
【0052】
本発明の一態様においては、本明細書に記載するRNA検出方法のための反応試薬および組成物を含む、RNA検出のためのキットとすることができる。本発明のキットには、本明細書に記載するRNA検出反応のための鋳型となる環状化一本鎖DNAプローブ、鎖置換型DNA合成酵素、RNase H、dNTP混合物、反応用緩衝液、および検出用蛍光色素などを含むことができる。
【0053】
本発明の検出するRNAは、とくに限定されることなく、mRNA、リボソームRNA(rRNA)、トランスファーRNA(tRNA)を含むあらゆる種類のRNAを検出することが可能である。検出対象のmRNAは、ポリA配列を有していても、有していなくてもよい。RNAは、siRNA、miRNA、piRNA、rasiRNA、rRNA、tRNAを含むノンコーディングRNAであってもよく、さらに、ウイルスなどのゲノムRNAであってもよい。また、検出対象のRNAの形状は、直鎖状または環状のどちらでも可能である。
【0054】
本発明のRNA検出の一態様においては、検出対象となるRNAはあらゆる生物種由来の試料から調製または単離されたものでもよい。そのようなRNAを含む試料には、ウイルス、原核生物または真核生物の個体そのもの、あるいはその一部が使用できる。例えば、脊椎動物(ヒトを含む)では、糞便、尿、または汗のような排泄物、血液、精液、唾液、胃液、または胆汁のような体液等が挙げられる。また、外科的に生体から取り出した組織、または体毛のように生体から脱落した組織であってもよい。さらに、食品等の加工物から調整したRNA含有調整物であってもよい。また、前記試料をさらに分画して、その一部を取り出したものから調製したRNA含有調製物であってもよい。
【0055】
本発明はまた、生物の検出方法に関する。
本発明において、生物は、原核生物、真核生物、ウイルス、およびウイロイドを含み、それに由来するRNAを検出することによって検出することができる。検出可能な生物種は、RNAを有するものであればとくに限定されず、RNAの種類も問わない。例えば、ウイルス、細菌、カビ、酵母、昆虫、植物および動物のような生物を対象とすることができる。
【0056】
さらに、本発明の一態様は、微生物の検出方法に関する。
本発明において、微生物は、それに由来するRNAを検出することによって検出することができる。微生物は、構成単位が肉眼では観察できない微小な生物をいい、ウイルスや多細胞の生物も含む。検出可能な微生物としては、例えば、ポリA配列を持たないmRNAを産生する細菌、およびゲノムがRNAであるウイルスまたはウイロイドなどが挙げられる。また、原生動物、およびカビや酵母などの真菌を含む真核微生物を対象とすることもできる。
【0057】
以下に実施例によって本発明を詳述するが、本発明は、各実施例に限定されるものではない。
【実施例】
【0058】
〔材料および方法〕
1.鋳型オリゴヌクレオチドの環状化
表1に示すオリゴヌクレオチドを、1mM ATPを含む1×T4ライゲースバッファー中、10ユニットのT4ポリヌクレオチドキナーゼを用いてリン酸化した。リン酸化したオリゴヌクレオチドを、10ユニットのCircLigase(登録商標)ssDNA Ligase(Epicentre社)を用い、2.5mM MnCl2の1×CircLigaseバッファー中、65℃、1時間で環状化した。環状化されていないオリゴヌクレオチドは、10ユニットのエクソヌクレアーゼIで37℃1時間消化し、続いて80℃20分間処理し、酵素を不活性化した。環状化したオリゴヌクレオチドは、BioSpin(BioRad社)で精製し、濃度をQuant-iT ssDNA BR Assay Kit(Invitrogen社)およびQubit fluorometer(Invitrogen社)を用いて測定した。環状化オリゴヌクレオチドのエクソヌクレアーゼによる消化率と直鎖状のオリゴヌクレオチドのものとの比較をアガロースゲル電気泳動(2.0%、Tris-Borate-EDTAバッファー)により分析し、CircLigaseの反応による環状化が行われたことを確認した。
【0059】
【表1】
【0060】
2.T7発現ベクターpET-21dへのGFP遺伝子のクローニング
2μgのpAcGFP1(Clontech社)からそれぞれ50ユニットのNcoIおよびNotI(タカラバイオ社)で同時に消化し、緑色蛍光タンパク質(green fluorescent protein;GFP)遺伝子を単離した。GFP遺伝子はアガロースゲル電気泳動により精製し、Wizard(登録商標)SV GelおよびPCR Clean-Up Systems(Promega社)を用いて精製した。精製したDNAの濃度は、Quant-iT dsDNA HS Assay Kit(Invitrogen社)およびQubit fluorometer(Invitrogen社)を用いて測定した。精製したGFP遺伝子をNcoIおよびNotIで切断したT7発現ベクターpET-21d(Novagen社)に連結し、組み換えプラスミドpET-AcGFPを作製した。大腸菌(Escherichia coli)DH5α株に形質転換した後、プラスミドDNAを選択したコロニーから精製し、制限酵素処理の後、アガロースゲル電気泳動で確認した。精製したpET-AcGFPの濃度をQuant-iT dsDNA HS Assay KitおよびQubit fluorometerを用いて測定した。
【0061】
3.GFPmRNAのin vitroでの転写
GFPmRNAのin vitroでの転写は、MEGAscript(登録商標)T7 Kit(Ambion社)を用いて行った。1μgのpET-AcGFPをNotIで消化して得られた直鎖状の鋳型DNAを転写反応液に添加し、37℃で2時間インキュベートした。DNaseI処理により鋳型DNAを除去した後、転写したmRNAをMEGAclear(登録商標)Kit(Ambion社)を用いて精製し、濃度を分光分析法により測定した。転写したmRNAを用時まで−80℃で保存した。
【0062】
4.GFPmRNAのin vivoでの発現およびRNAの抽出
大腸菌BL21(DE3)株(Novagen社)をpET-AcGFPで形質転換し、pET-AcGFPの選択のため50μg/mlアンピシリンを含むLB寒天プレート(1%トリプトン、0.5%酵母エキス、1.0%NaClおよび1.5%寒天)で培養した。大腸菌はLB培地で30℃8時間培養した。培養物をOvernight Express(登録商標)Autoinduction System(Novagen社)を含むフレッシュなLB培地で1:1000に希釈し、30℃で16時間振とう培養し、DE3にコードされるT7RNAポリメラーゼによりGFPmRNAの転写を誘導した。大腸菌は、3,500rpmで4℃20分間の遠心分離により50mlのコニカルチューブに回収した。全RNAをTrizol(登録商標)Max Bacterial RNA Isolation kit(Invitrogen社)を用いて単離した。調製した全RNAは、混入したゲノムDNAやプラスミドDNAなどの夾雑DNAを除去するためのDNase処理(例えば、DNaseIによる処理)を行うことなく試料として供した。当該試料中、微量の夾雑DNAが含まれていることをアガロースゲル電気泳動(1.5%、Tris-borate-EDTA緩衝液)によって確認した。なお、全RNAの濃度は分光分析法により測定した。
【0063】
5.RNA−primed RCA検出
in vitroで転写したGFPmRNAまたはpET-AcGFPを保有する大腸菌BL21(DE3)株から単離した全RNAを、10pmolの環状化オリゴヌクレオチド(表1のP1(配列番号1)、P2(配列番号2)、P3(配列番号3)またはP4(配列番号4))とともに、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4および10mM KClを含むバッファー中に混合し、100μlの反応混合物を得た。mRNAと環状化オリゴヌクレオチドとのハイブリダイゼーションを95℃5分間のインキュベーションにより促進し、その後30℃に急冷した。RCA反応は、100μlのハイブリダイゼーションした混合物を、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4、10mM KCl、0.4mMデオキシヌクレオシド三リン酸(dNTPs)、8mMジチオスレイトール(DTT)、SYBR GreenII(Invitrogen社)および適切なユニットのRepliPHI phi29ポリメラーゼ(Epicentre社)を含む100μlの反応液とを混合することで開始した。phi29ポリメラーゼにより合成されたssDNA(一本鎖DNA)に結合した際のSYBR GreenIIの蛍光を15分毎にQubit fluorometerで検出した。30℃、15時間のインキュベーションの後の合成DNA15μlを、20ユニットのS1ヌクレアーゼ(タカラバイオ社)で37℃、30分間消化し、アガロースゲル電気泳動(1.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した。
【0064】
6.RNase Hを利用したRNA−primed RCA検出
in vitroで転写したGFPmRNAを、10pmolの環状化オリゴヌクレオチド(表1のP5、配列番号5)とともに、50mM Tris−HCl(pH 7.5)、10mM MgCl2、20mM(NH4)2SO4および10mM KClを含むバッファー中に混合し、100μLの反応混合物を得た。mRNAと環状化オリゴヌクレオチドとのハイブリダイゼーションを95℃、5分間のインキュベーションにより促進し、その後30℃に急冷した。RCA反応は、100μLのハイブリダイゼーションした混合物を、50mM Tris−HCl(pH 7.5)、10mM MgCl2、20mM(NH4)2SO4および10mM KCl、0.4mM dNTPs、8mM ジチオスレイトール(DTT)、さらに適切なユニット数のRNase HおよびRepliPHI phi29ポリメラーゼ(Epicentre社)を含む100μLの反応液を混合することで開始した。30℃、15時間のインキュベーションの後の合成DNA15μLを、20ユニットのS1ヌクレアーゼ(タカラバイオ社)で37℃、30分間消化し、アガロースゲル電気泳動(1.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した
【0065】
7.PCRによるRNA試料中の夾雑DNAの検出
pET-AcGFPを保有する大腸菌BL21(DE3)株から、全RNAの抽出を行った。この際、GFPの発現を自動誘導により制御し、発現誘導した菌体および発現誘導してない菌体から、Trizol(登録商標)Max Bacterial RNA Isolation kit(Invitrogen社)を用いて、2種類の全RNAをそれぞれ調整した。また、得られた全RNA試料の一部(10μg)について、DNase処理を以下の手順で行った。10μgの全RNA試料を1×DNase緩衝液(50mM Tris−HCl(pH 8.0)、10mM MgSO4、1mM CaCl2)中で、10ユニットのRNase−free DNase I(タカラバイオ社)で37℃、30分間消化後、フェノールクロロホルム抽出によってDNase画分と核酸画分に分離し、エタノール沈殿によってRNAを沈澱物として回収した。回収したRNAはジエチルピロカーボネート処理された滅菌蒸留水で溶解後、RNA濃度をQuant-iT RNA BR Assay Kitで決定した。
【0066】
PCRによる夾雑DNA(pET-AcGFP)の検出は、以下の手順で行った。各RNA試料20ng、1×AmpliTaq Goldバッファー、0.2mM dNTPs、1μMの各プライマーの組み合わせ(表2)、2mM MgCl2、および0.625ユニットのAmpliTaq Goldポリメラーゼを含む反応液を調製し、95℃で10分間の予備加熱後、95℃で15秒、60℃で30秒、72℃で40秒のサイクルを35回繰り返した後、72℃で7分間の予備伸長を経たのち、4℃で保持した。各プライマーの組み合わせにおけるポジティブコントロール(P1、P2、P3)として、RNA試料の代わりに、1ngのpAcGFP-1(Clontech社)を鋳型として用いた。各PCR産物2μLは、アガロースゲル電気泳動(2.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した。
【表2】
【0067】
8.夾雑DNA含有試料におけるRNA−primed RCAによるRNAの検出
上記(7)のPCRによって夾雑DNAの存在が確認されたRNA試料を用いて、本発明のRPRCAによるGFPmRNAの検出を以下のように行った。各RNA試料20ngを10pmolの環状化オリゴヌクレオチド(表1のP4(配列番号4))とともに、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4および10mM KClを含むバッファー中に混合し、10μlの反応混合物を得た。ネガティブコントロール(N)として、RNA試料を含まない反応混合物を用意した。mRNAと環状化オリゴヌクレオチドとのハイブリダイゼーションを95℃で5分間のインキュベーションにより促進し、その後30℃に急冷した。RCA反応は、ハイブリダイゼーションした混合物10μlを、50mM Tris−HCl(pH7.5)、10mM MgCl2、20mM (NH4)2SO4、10mM KCl、0.4mMデオキシヌクレオシド三リン酸(dNTPs)、8mMジチオスレイトール(DTT)、および10ユニットのRepliPHI phi29ポリメラーゼ(Epicentre社)を含む10μlの反応液とを混合することで開始した。反応は30℃、4時間のインキュベーションで行い、その後の合成DNA20μlを、24ユニットのS1ヌクレアーゼ(タカラバイオ社)を用いて37℃、30分間消化し、消化液のうち10μlをアガロースゲル電気泳動(1.0%、Tris-Borate-EDTAバッファー)を用いて連続的に分析し、エチジウムブロマイドで染色して可視化した。
【0068】
〔実験結果のまとめ〕
1.環状化プローブでのRCAを利用したmRNAのリアルタイム検出のためのストラテジー
図1は、RCAによるmRNA検出の基本的なスキームを示す。かかるスキームは、以下の5つの工程を含む:
(1)目的mRNAに相補的な配列を有するssDNAプローブを、CricLigaseなどを用いて、分子内ライゲーションにより環状化する工程、
(2)連続的なRCA反応における非特異的な副生成のDNA合成を避けるため、ライゲーションしていないssDNAプローブをエクソヌクレアーゼIを用いた分解により除去する工程、
(3)環状ssDNAプローブを目的mRNAの3’末端とハイブリダイズさせる工程、
(4)phi29DNAポリメラーゼによって、RNA−DNAハイブリッドからRNA−primed RCA(RNAをプライマーとしたRCA;RPRCA)を行う工程、
(5)SYBR GreenIIなどのssDNAに対する蛍光色素によって、RCAによって合成された結果物であるssDNAの結合による蛍光を得る工程。
これにより、RPRCAを介したmRNAのリアルタイム検出が達成される。
【0069】
2.in vitroで転写されたmRNAのリアルタイム検出
まず最初に、phi29DNAポリメラーゼが、長い配列を有するmRNAをRCA反応のプライマーとして適合させ得るのかについて検討した。プラスミドpET-AcGFPをNotIで、T7ターミネーターとGFP遺伝子の終止コドンの間の領域を消化し、対応するmRNAをT7RNAポリメラーゼでin vitroでの転写によって調製した(図2A)。環状ssDNAプローブ(P1またはP2)もRPRCA用のin vitro転写mRNAと併せて用いた。P1のDNA配列は、GFPの3’末端配列と相補的である(表1)。他方、P2の配列は、P1と長さとGC含量とが同じであるが、配列上、GFPのmRNAと異なるものである(表1)。
【0070】
phi29DNAポリメラーゼによるRCA反応は、P1またはP2を有しないmRNAからは開始せず、反応バッファーにおけるP2のインキュベーションによっても、mRNAが存在してもしなくても、RCA反応を起こすことはなかった(図2BおよびC)。しかしながら、P1単独ではRCA反応に影響しないのに拘らず、mRNAとP1との組み合わせによってRCA反応が促進された。このことから、RPRCAによって、in vitro転写mRNAの3’末端に特異的なssDNAプローブを用いる目的mRNAのリアルタイム検出が行い得ること、および、非特異的環状プローブがRPRCAに影響しないこと判明した(図2BおよびC)。これらのことは、phi29ポリメラーゼが、適切な環状ssDNAプローブの存在下、mRNAをプライマーとして用いるRCA反応を特異的に作用し得ることを示唆している。さらに、エクソヌクレアーゼIで処理した環状ssDNAプローブは、RPRCAにおける長時間の反応期間の後に非特異的な副生成物を産生しなかった。これは、環状プローブの調製におけるエクソヌクレアーゼI処理が特異的で信頼できるRPRCAによる増幅に寄与していることを示す。
【0071】
RPRCAにおける環状ssDNAプローブの長さの効果を、GFPmRNAの3’末端に相補的なDNA領域をカバーするssDNAプローブP3(表1)を用いて行った。図2Dに示すとおり、興味深いことに、同じ長さのGFPmRNAに対して特異的なDNA配列を有するにも拘らず、短いプローブ(P3)ではなく、長いプローブ(P1)で十分なRCA反応が観察された。このことは、プローブの長さが、mRNAと環状プローブとの最初のハイブリダイゼーションに関係し、RPRCAの効率に影響を与えることを示している。
【0072】
まとめると、上記の証拠は、本発明のRPRCAが、適切な環状プローブを用いることで、不規則なDNA増幅副生成物を生成することなく、目的mRNAのリアルタイム検出を可能とすることを示している。
【0073】
3.RPRCAの感度
感度とphi29DNAポリメラーゼの濃度との間の相関関係を明確にするため、RPRCA反応における濃度の影響を調べた。RCA反応は、phi29DNAポリメラーゼの濃度を変化させた条件下で、各サンプルについて、3.2ngのin vitro転写mRNAをプライマーとして添加した場合を観測した。RCA反応は、濃度依存的に促進されることが確認された(図3A)。次いで、図3Aにおいて試験した最も高い酵素濃度(1反応当たり100ユニット)でのRPRCAによるmRNA検出の感度を測定した。その結果、RPRCAは、2時間で320pgのin vitro転写mRNAを検出することができた。
【0074】
4.生きた微生物由来の目的mRNAの特異的検出
RPRCAによって、生きた微生物のmRNAを目的として検出できるかについて検討した。誘導したGFP発現が容易に観察できるため(図4A)、pET-AcGFPで形質転換した大腸菌BL21(DE3)株をRPRCAのモデルとして用いた。生きた細胞において、T7RNAポリメラーゼによって転写されたGFPmRNAの一次構造は、7塩基の5’非翻訳領域(UTR)、GFPmRNA、およびそれに続く3’UTRの48塩基の隣接残基から構成される(図4B)。この実験においては、ssDNAプローブP4を合成した。前記P4は、図4Bに下線で示した3’末端に対応するDNA配列が含まれるように合成されている(表1)。栄養増殖している大腸菌において全RNAの大部分はrRNAおよびtRNAが占めているので、両RNAともRPRCAに影響を及ぼし得る。どの程度の量のrRNAおよびtRNAがRPRCAに影響したかを調べるため、誘導した大腸菌および誘導していない大腸菌の両方から抽出した後、全RNAをRPRCAに用いた。誘導ありまたは誘導なしの大腸菌由来の全RNA200ngをP2またはP4とともにインキュベートした場合、誘導した大腸菌由来の全RNAと環状プローブP4とを組み合わせたものだけ、十分なRPRCAが観察された(図4C)。全RNA単独または環状プローブ単独ではRCA反応は起きなかった。さらに、段階希釈した全RNAを用いた2時間のRPRCAによって、GFPmRNAが10ngの全RNAから検出可能であることが明らかになった(図4D)。RNAの全量(400μg)が1.5×1010細胞の密度の大腸菌から抽出されたことを考慮すると、10ngの全RNAは、およそ4×105細胞に相当する。これらの結果は、細菌性全RNAと適切なssDNAプローブとの組合せを用いたRPRCAは、in vitro転写mRNAの場合(図2)と同様に、GFPmRNAを特異的に検出できるばかりでなく、発現したGFPmRNAと全RNAの大部分を構成するrRNAやtRNAのような他のRNA種とを明確に区別することができることが示された。
【0075】
また、試料中に夾雑DNAが含まれていても(試料中のDNAの分解処理をしていなくても)、良好にRNAの検出ができたことを示すものであった。すなわち、試料中のDNAの分解処理を行わなくても、目的RNAが発現していない試料(図4の「−」(ネガティブコントロール))においては夾雑DNAによる不規則なDNA増幅生成物を生成することがなかったことからも明らかなとおり、本方法においては高いシグナルノイズ比(S/N比)を実現することができた。
【0076】
5.RPRCAのRNase Hを利用したRNA検出のためのストラテジー
図5は、RNase H処理工程を含むRCAによるRNA検出の基本的なスキームを示す。かかるスキームは、以下の6つの工程を含む:
(1)目的mRNAに相補的な配列を有するssDNAプローブを、CricLigaseなどを用いて、分子内ライゲーションにより環状化する工程、
(2)連続的なRCA反応における非特異的な副生成のDNA合成を避けるため、ライゲーションしていないssDNAプローブをエクソヌクレアーゼIによる分解・除去する工程、
(3)環状ssDNAプローブに目的RNAをハイブリダイズさせる工程、
(4)環状ssDNAプローブにハイブリダイズした領域の目的RNAに、RNase Hにより、ニックサイトを形成させる工程、
(5)phi29DNAポリメラーゼによって、ニッキングサイトのRNAの3‘末端からRPRCAを行う工程、
(6)RCAによって合成された結果物であるssDNAを検出する工程。
【0077】
6.RNAase Hを利用したRPRCAによるin vitroで転写されたmRNAの検出
GFPmRNAの内部領域と相補性のあるDNAプローブを利用した場合、RPRCA反応が起こりえるかどうか検討した。プラスミドpET-AcGFPをNotIで、T7ターミネーターとGFP遺伝子の終止コドンの間の領域を消化し、対応するmRNAをT7RNAポリメラーゼによるin vitro転写によって調製した(図6)。転写したGFPmRNAは環状ssDNAプローブ(P5)と併せて用いた。プローブP1のDNA配列は、GFPの内部領域と相補的である(図6)。
【0078】
環状化一本鎖プローブが存在しない場合、phi29DNAポリメラーゼによるRCA反応は、促進されず一切のDNA産物が確認できなかった(図7)。プローブP5を利用した場合、RNase Hを添加しない条件のRPRCA反応では非特許文献8の報告とは異なり、一切のDNA産物は確認できなかった(図7)。このことから、本願のRNase H処理工程を含むRPRCA法では、目的RNAの内部配列をDNAプローブとハイブリダイズさせても、目的RNAを検出できることを示している。さらに、特許文献4とは異なり、ゲル中で単一のバンドとして検出される本願発明は、非特異的な副産物が生じていないことを確認し、その検出精度の高さが認められる。
また、添加するRNase Hの濃度依存性についても検討した結果、RNase Hの添加によってRPRCA反応が促進されることが示された。
【0079】
上記の証拠は、本発明のRNA検出方法は、RPRCAの工程において、適切な濃度のRNase Hで処理することによって、目的RNAの3’末端配列の情報に依存せずに、非特異的なDNA増幅副生成物を生成しないで特異的な検出が可能であることを示している。
【0080】
7.PCRによるRNA試料中の夾雑DNAの検出
GFP遺伝子に特異的なプライマー3セット(表2)を使用して、RNA試料中の夾雑DNAをPCRにより検出した。その際、GFPの発現を自動誘導により制御し、発現誘導した菌体から調整した全RNA(+)と、発現誘導していない菌体から調整した全RNA(−)の2種類を得た。さらに、その一部をそれぞれDNase Iを用いてDNA分解処理をし、発現誘導有りまたは無しの全RNAであって、DNase処理有りまたは無しの合計4種類の全RNAを得た。そして、各全RNAを用いて夾雑DNAの検出を試みた。
【0081】
DNase処理を行っていないRNA試料からは、いずれのプライマーセットにおいても、GFPの誘導・非誘導にかかわらず目的DNA増幅産物が、各プライマーセットのポジティブコントロール(P1、P2、P3)と同様に確認された(図8)。一方で、DNase処理を行ったRNA試料からは、誘導・非誘導で共に目的DNA増幅産物が明らかに減少した(図8)。以上のことから、DNase処理によって夾雑DNAの大部分を除去することが可能であることが示された。しかし、DNase処理を行っても、わずかにバンドが検出されたことから、一般的なDNase処理だけでは、RNA試料から完全に夾雑DNAを取り除くことが難しいことも同時に示された。この夾雑DNAがRT−PCRにおいても同様に、ノイズとして検出される可能性が高いことは、当業者であれば理解できる。
【0082】
8.夾雑DNA含有試料におけるRNA−primed RCAによるRNAの検出
上記(7)のDNase処理有りまたは処理無しのRNA試料を用いて、RPRCAによるGFPmRNAの検出を試みた。その結果、DNase処理の有無、すなわち夾雑DNAの量に拘わらず、発現誘導した菌体から調整したRNA試料から、RPRCAにより増幅したDNAが十分な量で確認された(図9)。また、DNase処理の有無に拘わらず、発現誘導していない菌体から調整したRNA試料では、RPRCAによるDNAの増幅は認められなかった(図9)。以上のことから、本発明のRPRCAにおいては、RNA試料から夾雑DNAを除去することなく、また、RNA試料中の夾雑DNA量の影響を受けることなく、目的RNAのみを高いS/N比で検出できることが示された。
【産業上の利用可能性】
【0083】
本発明は、鎖置換型DNA合成酵素を利用したRNAプライムドローリングサークル型DNA複製法(RNA-Primed Rolling circle amplification;RPRCA)を応用し、RNAの種類、3’末端配列情報等に影響を受けずに、簡便な操作で、逆転写反応を経由せずに、生物由来の夾雑物を含む試料中から直接RNAをin situやリアルタイムで検出可能な方法に関するものであり、研究、医療・臨床検査、食品産業など幅広い応用範囲において貢献するものである。
【特許請求の範囲】
【請求項1】
生物由来の夾雑物を含む試料中のRNAの検出方法であって、
(1)検出対象のRNAに相補的な配列を有する一本鎖DNAを環状化し、環状化一本鎖DNAプローブを作製する工程、
(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる工程、
(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしてローリングサークル増幅法(RCA;Rolling Circle Amplification)により一本鎖DNAを合成する工程、
(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する工程
を含む、前記方法。
【請求項2】
生物由来の夾雑物が、DNAを含む、請求項1に記載の方法。
【請求項3】
試料が、核酸試料である、請求項1または2に記載の方法。
【請求項4】
試料中のDNAが、分解処理されていない、請求項2または3に記載の方法。
【請求項5】
前記(1)において、環状化していない未反応の一本鎖DNAを分解することを含む、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
環状化一本鎖DNAプローブが、検出対象のRNAに相補的な配列のみからなる、請求項1〜5のいずれか一項に記載の方法。
【請求項7】
環状化一本鎖DNAプローブが、30塩基長〜4500塩基長である、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
前記(2)の後に、
(5)RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程
をさらに含む、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
前記(3)および/または(5)が、25℃〜65℃の範囲の一定の温度で行われる、請求項8に記載の方法。
【請求項10】
前記(5)において、RNase Hを終濃度1×10−4〜10ユニット/反応で用いる、請求項8または9に記載の方法。
【請求項11】
前記(5)において、RNase Hを終濃度1×10−7〜0.1ユニット/μlで用いる、請求項8または9に記載の方法。
【請求項12】
RNAが、真核生物、原核生物、ウイルスまたはウイロイド由来である、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
RNAが、ポリA配列を有する、請求項12に記載の方法。
【請求項14】
RNAが、ノンコーディングRNAである、請求項12に記載の方法。
【請求項15】
請求項1〜14のいずれか一項に記載のRNAの検出方法を含む、真核生物、原核生物、ウイルスまたはウイロイドの検出方法。
【請求項16】
微生物を検出する、請求項15に記載の方法。
【請求項17】
請求項1〜14のいずれか一項に記載のRNAの検出方法、および/または請求項15に記載の真核生物、原核生物、ウイルスまたはウイロイドの検出方法、および/または請求項16に記載の微生物の検出方法のために用いるキットであって、検出対象のRNAに相補的な配列を含む環状化一本鎖DNAプローブを含むキット。
【請求項18】
アデニン、グアニン、シトシンおよびチミンからなるデオキシヌクレオチド、反応用緩衝液および鎖置換型DNA合成酵素からなる群から選択される1種または2種以上をさらに含む、請求項17に記載のキット。
【請求項1】
生物由来の夾雑物を含む試料中のRNAの検出方法であって、
(1)検出対象のRNAに相補的な配列を有する一本鎖DNAを環状化し、環状化一本鎖DNAプローブを作製する工程、
(2)前記(1)において得られた環状化一本鎖DNAプローブと、試料中のRNAとをハイブリダイズさせる工程、
(3)前記(2)において得られたRNA−DNAハイブリッドから、RNAをプライマーとしてローリングサークル増幅法(RCA;Rolling Circle Amplification)により一本鎖DNAを合成する工程、
(4)前記(3)において合成された一本鎖DNAに結合する標識を添加し、当該標識を検知することによって検出対象のRNAを検出する工程
を含む、前記方法。
【請求項2】
生物由来の夾雑物が、DNAを含む、請求項1に記載の方法。
【請求項3】
試料が、核酸試料である、請求項1または2に記載の方法。
【請求項4】
試料中のDNAが、分解処理されていない、請求項2または3に記載の方法。
【請求項5】
前記(1)において、環状化していない未反応の一本鎖DNAを分解することを含む、請求項1〜4のいずれか一項に記載の方法。
【請求項6】
環状化一本鎖DNAプローブが、検出対象のRNAに相補的な配列のみからなる、請求項1〜5のいずれか一項に記載の方法。
【請求項7】
環状化一本鎖DNAプローブが、30塩基長〜4500塩基長である、請求項1〜6のいずれか一項に記載の方法。
【請求項8】
前記(2)の後に、
(5)RNase HによりRNA/DNAハイブリッドのRNA鎖のみにニックを形成する工程
をさらに含む、請求項1〜7のいずれか一項に記載の方法。
【請求項9】
前記(3)および/または(5)が、25℃〜65℃の範囲の一定の温度で行われる、請求項8に記載の方法。
【請求項10】
前記(5)において、RNase Hを終濃度1×10−4〜10ユニット/反応で用いる、請求項8または9に記載の方法。
【請求項11】
前記(5)において、RNase Hを終濃度1×10−7〜0.1ユニット/μlで用いる、請求項8または9に記載の方法。
【請求項12】
RNAが、真核生物、原核生物、ウイルスまたはウイロイド由来である、請求項1〜11のいずれか一項に記載の方法。
【請求項13】
RNAが、ポリA配列を有する、請求項12に記載の方法。
【請求項14】
RNAが、ノンコーディングRNAである、請求項12に記載の方法。
【請求項15】
請求項1〜14のいずれか一項に記載のRNAの検出方法を含む、真核生物、原核生物、ウイルスまたはウイロイドの検出方法。
【請求項16】
微生物を検出する、請求項15に記載の方法。
【請求項17】
請求項1〜14のいずれか一項に記載のRNAの検出方法、および/または請求項15に記載の真核生物、原核生物、ウイルスまたはウイロイドの検出方法、および/または請求項16に記載の微生物の検出方法のために用いるキットであって、検出対象のRNAに相補的な配列を含む環状化一本鎖DNAプローブを含むキット。
【請求項18】
アデニン、グアニン、シトシンおよびチミンからなるデオキシヌクレオチド、反応用緩衝液および鎖置換型DNA合成酵素からなる群から選択される1種または2種以上をさらに含む、請求項17に記載のキット。
【図1】

【図2】
【図3】
【図4】
【図5】
【図6】

【図7】
【図8】
【図9】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【公開番号】特開2012−80871(P2012−80871A)
【公開日】平成24年4月26日(2012.4.26)
【国際特許分類】
【出願番号】特願2010−271842(P2010−271842)
【出願日】平成22年12月6日(2010.12.6)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り Analytical Biochemistry Volume 401,Issue 2,242−249
【出願人】(501203344)独立行政法人農業・食品産業技術総合研究機構 (827)
【Fターム(参考)】
【公開日】平成24年4月26日(2012.4.26)
【国際特許分類】
【出願日】平成22年12月6日(2010.12.6)
【新規性喪失の例外の表示】特許法第30条第1項適用申請有り Analytical Biochemistry Volume 401,Issue 2,242−249
【出願人】(501203344)独立行政法人農業・食品産業技術総合研究機構 (827)
【Fターム(参考)】
[ Back to top ]