
RNA干渉を媒介する短鎖RNA分子
【課題】標的特異的RNA干渉またはDNAメチル化のような他の標的特異的な核酸改変を媒介することができる新規の物質を提供すること。
【解決手段】単離された二本鎖RNA分子であって、各鎖が39〜52塩基長を有し、該RNA分子は標的特異的なRNA干渉が可能な1以上の二本鎖断片にプロセシングされることができ、該RNA分子は、第1の鎖が予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が該第1の鎖に相補的な配列からなる、上記RNA分子、ならびに該RNA分子の使用。
【解決手段】単離された二本鎖RNA分子であって、各鎖が39〜52塩基長を有し、該RNA分子は標的特異的なRNA干渉が可能な1以上の二本鎖断片にプロセシングされることができ、該RNA分子は、第1の鎖が予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が該第1の鎖に相補的な配列からなる、上記RNA分子、ならびに該RNA分子の使用。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、RNA干渉および/またはDNAメチル化などの標的特異的な核酸改変を媒介するのに必要な二本鎖(ds)RNAの配列および構造的特徴に関する。
【背景技術】
【0002】
「RNA干渉(RNAインターフェアランス)」(RNAi)は、線虫(C.elegans)にdsRNAを注入すると、送達したdsRNAと配列が高度に相同的な遺伝子の特異的サイレンシングが起こるという発見後、造り出された用語である(Fireら、1998)。その後、RNAiは、昆虫、カエル(Oelgeschlagerら、2000)、ならびに、マウスを含むその他の動物(Svobodaら、2000;WiannyおよびZernicka-Goetz, 2000)でも観察されており、ヒトにも存在すると考えられる。RNAiは、植物における共抑制および真菌における抑制の転写後遺伝子サイレンシング(PTGS)機構と密接に関連しており(Catalanottoら、2000;CogoniおよびMacino, 1999;Dalmayら、2000;KettingおよびPlasterk, 2000;Mourrainら、2000;Smardonら、2000)、また、RNAi機構の構成要素には、共抑制による転写後サイレンシングに必要なものもある(Catalanottoら、2000;Dernburgら、2000;KettingおよびPlasterk, 2000)。これについては、近年再考されている(Bass, 2000;BosherおよびLabouesse, 2000;Fire, 1999;PlasterkおよびKetting, 2000;Sharp, 1999;SijenおよびKooter, 2000)。また、Plant Molecular Biology、第43巻、2/3号(2000)全体も参照されたい。
【0003】
植物では、PTGSに加えて、導入されたトランスジーンも、シトシンのRNA指令DNAメチル化を介した転写遺伝子サイレンシングを誘導することができる(Wassenegger, 2000の文献を参照)。植物では、30bpという短いゲノム標的がRNA指令方式でメチル化される(Pelissier, 2000)。DNAメチル化は哺乳動物にも存在する。
【0004】
RNAiおよび共抑制の天然の機能は、活性となったとき、宿主細胞に異常RNAまたはdsRNAを産生するレトロトランスポゾンやウイルスなどの可動性遺伝的エレメントによる侵入に対してゲノムを保護することであると考えられる(Jensenら、1999;Kettingら、1999;Ratcliffら、1999;Tabaraら、1999)。特異的mRNA分解により、トランスポゾンおよびウイルスの複製が阻止されるが、PTGSを抑制するタンパク質を発現させることにより、この過程を克服または阻止することができる(Lucyら、2000;Voinnetら、2000)。
【0005】
dsRNAは、dsRNAとの同一性の領域内でしか相同性RNAの特異的分解を誘発しない(Zamoreら、2000)。dsRNAは21〜23塩基のRNA断片にプロセシングされ、標的RNA切断部位は、通常、21〜23塩基の間隔である。従って、21〜23塩基の断片が、標的認識のためのガイド(guide)RNAであると考えられてきた(Zamoreら、2000)。これらの短いRNAは、細胞溶解の前にdsRNAでトランスフェクトしたキイロショウジョウバエシュナイダー2細胞から調製した抽出物でも検出されている(Hammondら、2000)が、配列特異的ヌクレアーゼ活性を呈示する画分も、残留dsRNAの大きな画分を含んでいた。mRNA切断の指示における21〜23塩基断片の役割は、プロセシングされたdsRNAから単離された21〜23塩基断片が、特異的mRNA分解をある程度まで媒介することができるという研究結果によってさらに支持される(Zamoreら、2000)。類似サイズのRNA分子は、PTGSを示す植物組織にも蓄積する(HamiltonおよびBaulcombe, 1999)。
【発明の概要】
【発明が解決しようとする課題】
【0006】
ここで、本発明者は、確立されたショウジョウバエin vitro系(Tuschlら、1999;Zamoreら、2000)を用いて、RNAiの機構をさらに探究した。本発明者は、21および22塩基の短いRNAが、3’突出末端と塩基対合すると、配列特異的mRNA分解のガイドRNAとして作用することを証明する。30bpの短いdsRNAは、もはや21および22塩基のRNAにプロセシングされることはないため、この系でRNAiを媒介することはできない。さらに、本発明者は、21および22塩基という短鎖の干渉作用を有するRNA(siRNA)に対する標的RNA切断部位を特定し、dsRNAプロセシングの方向が、生成されるsiRNPエンドヌクレアーゼ複合体により、センスまたはアンチセンス標的RNAが切断されうるか否かを決定する証拠を提供する。siRNAはまた、転写調節、例えば、DNAメチル化を指示することによって哺乳動物遺伝子のサイレンシングのための重要なツールにもなりうる。
【0007】
ヒトin vivo細胞培養系(HeLa細胞)でさらに実験したところ、好ましくは19〜25塩基長の二本鎖RNA分子がRNAi活性を有することがわかった。従って、ショウジョウバエからの結果とは対照的に、24および25塩基長の二本鎖RNA分子もRNAiに有効である。
【0008】
本発明の目的は、標的特異的RNA干渉またはDNAメチル化のような他の標的特異的な核酸改変を媒介することができる新規の物質を提供することであり、この物質は、従来の技術による物質と比べて、効力および安全性が改善されている。
【課題を解決するための手段】
【0009】
上記課題は、各RNA鎖が19〜25、特に19〜23塩基長を有し、かつ、該RNA分子が、標的特異的な核酸改変、特にRNA干渉および/またはDNAメチル化を媒介することが可能な、単離された二本鎖RNA分子により解決される。少なくとも1本の鎖が、好ましくは1〜5塩基、さらに好ましくは1〜3塩基、最も好ましくは2塩基からなる3’突出部を有する。他方の鎖は、平滑末端であるか、あるいは、6塩基以下の3’突出部を有する。また、dsRNAの両鎖が厳密に21または22塩基の場合には、両末端が平滑である(0塩基の突出部)とき、ある程度のRNA干渉を観察することができる。RNA分子は、細胞抽出物中に存在する不純物、例えば、ショウジョウバエ胚を実質的に含まない合成RNA分子であることが好ましい。さらに、RNA分子は、好ましくは、非標的特異的不純物、特に非標的特異的RNA分子、例えば、細胞抽出物中に存在する不純物を実質的に含まない。
【0010】
さらには、本発明は、哺乳動物細胞、特にヒト細胞における標的特異的な核酸改変、特にRNAiを媒介することを目的とした、各RNA鎖が19〜25塩基長を有する単離された二本鎖RNA分子の使用に関する。
【0011】
具体的には、本発明は、以下の特徴を有する。
〔1〕単離された二本鎖RNA分子であって、各鎖が39〜52塩基長を有し、該RNA分子は標的特異的なRNA干渉が可能な1以上の二本鎖断片にプロセシングされることができ、該RNA分子は、第1の鎖が予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が該第1の鎖に相補的な配列からなる、上記RNA分子。
〔2〕鎖が39塩基長を有する、上記〔1〕に記載のRNA分子。
〔3〕単離された二本鎖RNA分子であって、該RNA分子は標的特異的なRNA干渉が可能なものであり、第1の鎖が19〜25塩基長を有し、予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が25塩基長を有する、上記RNA分子。
〔4〕なくとも1つの修飾されたリボヌクレオチドを含む、上記〔1〕〜〔3〕のいずれか1つに記載のRNA分子。
〔5〕修飾リボヌクレオチドが、糖、骨格鎖または核酸塩基修飾リボヌクレオチドから選択される、上記〔4〕に記載のRNA分子。
〔6〕修飾リボヌクレオチドが、糖修飾リボヌクレオチドであり、2’−OH基が、H、OR、R、ハロ、SH、SR1、NH2、NHR、NR2またはCNから選択される基で置換され、Rは、C1−C6アルキル、アルケニルまたはアルキニルであり、ハロは、F、Cl、BrまたはIである、上記〔5〕に記載のRNA分子。
〔7〕飾リボヌクレオチドが、ホスホロチオエート基を含む骨格鎖修飾リボヌクレオチドである、上記〔5〕に記載のRNA分子。
〔8〕前記RNA分子の第1の鎖が、予め決定したmRNA標的分子に対して、少なくとも85%の同一性を有する配列からなる、上記〔1〕〜〔7〕のいずれか1つに記載のRNA分子。
〔9〕下記のステップを含む、請求項1〜8のいずれか1項に記載の二本鎖RNA分子の作製方法:
(a)各々が39〜52または19〜25塩基長を有する2本のRNA鎖を合成するステップであって、該RNA鎖は二本鎖RNA分子を形成することができるものである、上記ステップ、
(b)二本鎖RNA分子が形成される条件下で合成RNA鎖を結合させるステップであって、得られる二本鎖RNA分子は標的特異的なRNA干渉が可能なものである、上記ステップ。
〔10〕RNA鎖が化学的に合成される、上記〔9〕に記載の方法。
〔11〕RNA鎖が酵素により合成される、上記〔9〕に記載の方法。
〔12〕下記のステップを含む、動物細胞において標的特異的なRNA干渉を媒介する方法:
(a)標的特異的なRNA干渉が起こりうる条件下で、上記細胞を請求項1〜8のいずれか1項に記載の二本鎖RNA分子と接触させるステップ、および
(b)上記二本鎖RNAと同一の配列部分を有する標的核酸に対する、上記二本鎖RNAにより引き起こされる標的特異的なRNA干渉を媒介するステップ。
〔13〕接触ステップが、二本鎖RNA分子を標的細胞に導入し、そこで標的特異的なRNA干渉を起こさせうるステップを含む、上記〔12〕に記載の方法。
〔14〕導入ステップが、キャリア媒介による送達または注射を含む、上記〔13〕に記載の方法。
〔15〕動物細胞における遺伝子の機能を決定するための、上記〔12〕〜〔14〕のいずれか1つに記載の方法の使用。
〔16〕動物細胞における遺伝子の機能を抑制するための、上記〔12〕〜〔14〕のいずれか1つに記載の方法の使用。
〔17〕遺伝子が病理的状態と関連する、上記〔15〕または〔16〕に記載の使用。
〔18〕遺伝子が病原体関連遺伝子である、上記〔17〕に記載の使用。
〔19〕遺伝子がウイルス遺伝子である、上記〔18〕に記載の使用。
〔20〕遺伝子が腫瘍関連遺伝子である、上記〔17〕に記載の使用。
〔21〕遺伝子が自己免疫疾患関連遺伝子である、上記〔17〕に記載の使用。
〔22〕標的遺伝子特異的ノックアウト表現型を示す動物細胞であって、該細胞が、内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子、または少なくとも1つの内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子をコードするDNAでトランスフェクトされており、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定されたmRNA標的分子に対して100%の同一性を有する配列からなる、上記細胞。
〔23〕哺乳動物細胞である、上記〔22〕に記載の細胞。
〔24〕ヒト細胞である、上記〔23〕に記載の細胞。
〔25〕標的タンパク質、または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸でさらにトランスフェクトされた、上記〔22〕〜〔24〕のいずれか1つに記載の細胞であって、該外因性標的核酸は、二本鎖RNA分子による該外因性標的核酸の発現の阻害が、内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記細胞。
〔26〕外因性標的核酸が、検出可能なペプチドまたはポリペプチドをコードするさらなる核酸配列と融合されている、上記〔25〕に記載の細胞。
〔27〕分析手法のための、上記〔22〕〜〔26〕のいずれか1つに記載の細胞の使用。
〔28〕遺伝子発現プロフィールを分析するための、上記〔27〕に記載の使用。
〔29〕プロテオーム分析のための、上記〔27〕に記載の使用。
〔30〕外因性標的核酸によってコードされる標識タンパク質の変異体または突然変異形態の分析が実施される、上記〔27〕〜〔29〕のいずれか1つに記載の使用。
〔31〕標的タンパク質の機能ドメインを同定するための、上記〔30〕に記載の使用。
〔32〕下記のものから選択される少なくとも2つの動物細胞の比較を実施する、上記〔27〕〜〔31〕のいずれか1つに記載の使用:
(i)標的遺伝子阻害を含まない対照細胞、
(ii)標的遺伝子阻害を含む細胞、および
(iii)標的遺伝子阻害と、外因性標的核酸による標的遺伝子の相補を含む細胞。
〔33〕分析が、機能および/または表現型の分析を含む、上記〔27〕〜〔32〕のいずれか1つに記載の使用。
〔34〕調製手法のための、上記〔22〕〜〔26〕のいずれか1つに記載の細胞の使用。
〔35〕真核細胞からタンパク質またはタンパク質複合体を単離するための、上記〔34〕に記載の使用。
〔36〕高分子量タンパク質複合体を単離するための、上記〔35〕に記載の使用。
〔37〕高分子量タンパク質複合体が核酸を含む、上記〔36〕に記載の使用。
〔38〕薬理学的物質を同定および/または特性決定する手法における、〔27〕〜〔37〕のいずれか1つに記載の使用。
〔39〕下記の(a)〜(c)を含む、少なくとも1つの標的タンパク質に作用する薬理学的物質の同定および/または特性決定システム:
(a)少なくとも1つの標的タンパク質をコードする少なくとも1つの標的遺伝子を発現することができる、動物細胞、
(b)上記少なくとも1つの内因性標的遺伝子の発現を阻害することができる、少なくとも1つの二本鎖RNA分子であって、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定したmRNA標的分子に対して100%の同一性を有する配列からなる、上記RNA分子、および
(c)薬理学的特性を同定および/または特性決定しようとする、試験物質または試験物質のコレクション。
〔40〕さらに下記の(d)を含む、上記〔39〕に記載のシステム:
(d)上記標的タンパク質または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸であって、該外因性標的核酸は、二本鎖RNA分子による発現の阻害が、上記内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記外因性標的核酸。
【図面の簡単な説明】
【0012】
【図1A】Pp−luc mRNAをターゲッティングするのに用いたdsRNAのグラフである。
【図1B】RNA干渉アッセイを示す図である。
【図2】ショウジョウバエ溶解物における内部32P標識dsRNA(5nM)のプロセシングからの21〜23mer形成の時間経過と、dsRNAの長さおよび供給源を示す図。
【図3A】5’切断産物の変性ゲル泳動を示す図である。
【図3B】センスおよびアンチセンス標的RNA上の切断部位の位置を示す図である。
【図4A】dsRNAプロセシング後の約21塩基RNAの配列を示す図である。
【図4B】約21塩基RNAのヌクレオチド組成の二次元TLCの分析を示す図である。
【図5A】対称52bpのdsRNAならびに合成21および22塩基のdsRNAをグラフで示す図である。
【図5B】センスおよびアンチセンス標的RNA上の切断部位の位置を示す図である。
【図6A】52bpのdsRNA構築物をグラフで示す図である。
【図6B】センスおよびアンチセンス標的RNA上の切断部位の位置を示す図である。
【図7】RNAiのモデル案を示す図である。
【図8a】プラスミドpGL2−Control、pGL−3−ControlおよびpRL−TK(Promega)からのホタル(Pp−luc)およびウミシイタケ(Rr−luc)ルシフェラーゼリポーター遺伝子領域を示す図である。
【図8b】GL2、GL3およびRLルシフェラーゼをターゲッティングするsiRNA二本鎖のセンス(上)およびアンチセンス(下)配列を示す図である。
【図9】siRNA二本鎖によるRNA干渉を示す図である。
【図10】pGL2−ControlとpRL−TKリポータープラスミドを用いて実施した実験を示す。
【図11】(A)実験戦略の概要を示す図である。(B)対照ルシフェラーゼに対する、標的ルシフェラーゼの正規化相対ルミネセンスを示す図である。(C〜J)8系統の21塩基のsiRNA二本鎖について正規化した干渉比を示す図である。
【図12】siRNA二本鎖のセンス鎖の長さ変動を示す図である。
【図13】保存された2塩基3’突出部を有するsiRNA二本鎖の長さ変動を示す図である。
【図14】siRNAリボース残基の2’−ヒドロキシ基の置換を示す図である。
【図15A】32P(星印)キャップ標識センスおよびアンチセンス標的RNAと、siRNA二本鎖をグラフで示す図である。
【図15B】標的RNA切断部位のマッピングを示す図である。
【図16】(AおよびB)実験戦略をグラフで示す図である。(CおよびD)キャップ標識センスまたはアンチセンス標的RNAを用いた標識RNA切断の分析を示す図である。
【図17】siRNA二本鎖の3’突出部の配列変動を示す図である。
【図18】標的認識の配列特異性を示す図である。
【図19】保存された2塩基3’突出部を有するsiRNA二本鎖の長さ変動を示す図である。
【発明を実施するための形態】
【0013】
驚くべきことに、特に、3’突出末端を有する短い合成二本鎖RNA分子が、RNAiの配列特異的媒介物質であり、有効な標的RNA切断を媒介し、その際、この切断部位は、短鎖RNAが指示(ガイド;guide)する範囲の領域の中心近くに位置することがわかった。
【0014】
RNA分子の各鎖は、20〜22塩基長(または哺乳動物細胞では20〜25塩基長)を有するのが好ましく、各鎖の長さは同じでも異なっていてもよい。好ましくは、3’突出部は、1〜3塩基の範囲にあり、この突出部の長さは、鎖によって同じでも異なっていてもよい。RNA鎖は、3’ヒドロキシル基を有するのが好ましい。5’末端は、好ましくは、リン酸、二リン酸、三リン酸またはヒドロキシル基を含む。最も有効なdsRNAは、1〜3塩基、特に2塩基の3’突出部がdsRNAの両端に存在するように、対合した2本の21塩基の鎖から構成される。
【0015】
siRNAにより指示される標的RNA切断反応は、高度に配列特異的である。しかし、siRNAの全ての位置が等しく標的認識に寄与するわけではない。siRNA二本鎖の中心におけるミスマッチが最も重要で、標的RNA切断をほとんど破壊する。これに対し、一本鎖標的RNAと相補的なsiRNA鎖の3’ヌクレオチド(例えば、位置21)は、標的認識の特異性に寄与しない。さらに、アンチセンスsiRNA鎖だけが標的認識を指示するため、標的RNAと同じ極性を有するsiRNAの非対合2塩基3’突出部の配列は、標的RNA切断に重要ではない。従って、一本鎖突出塩基(ヌクレオチド)から、アンチセンスsiRNAの最後から2番目の位置(例えば、位置20)だけが標的センスmRNAと一致する必要がある。
【0016】
驚くべきことに、本発明の二本鎖RNA分子は、血清または細胞培養用の増殖培地において高いin vivo安定性を呈示する。安定性をさらに高めるため、3’突出部を分解に対して安定化させる、例えば、これらが、プリンヌクレオチド、特にアデノシンまたはグアノシンヌクレオチドからなるように、選択することができる。あるいは、修飾類似体によるピリミジンヌクレオチドの置換、例えば、2’−デオキシチミジンによるウリジン2塩基の3’突出部の置換は許容されるものであり、RNA緩衝の効率に影響しない。また、2’ヒドロキシルの欠如により、組織培地中の突出部のヌクレアーゼ耐性が有意に増強される。
【0017】
本発明の特に好ましい実施形態では、RNA分子は、少なくとも1つの修飾ヌクレオチド類似体を含むことができる。ヌクレオチド類似体は、標的特異的活性、例えば、RNAi媒介活性が実質的に影響を受けない位置、例えば、二本鎖RNA分子の5’末端および/または3’末端の領域内に配置することができる。特に、修飾ヌクレオチド類似体を組み込むことにより、突出部を安定化させることができる。
【0018】
好ましいヌクレオチド類似体は、糖または骨格鎖修飾リボヌクレオチドから選択する。しかし、核酸塩基が修飾されたリボヌクレオチド、すなわち、天然に存在する核酸塩基ではなく、下記のように天然に存在しない核酸塩基を含むリボヌクレオチドも好適であることに留意すべきである:すなわち、天然に存在しない核酸塩基としては、5位置で修飾されたウリジンまたはシチジン、例えば、5−(2−アミノ)プロピルウリジン、5−ブロモウリジン;8位で修飾されたアデノシンおよびグアノシン、例えば、8−ブロモグアノシン;デアザヌクレオチド、例えば、7−デアザ−アデノシン;O−およびN−アルキル化ヌクレオチド、例えば、N6−メチルアデノシンなどである。好ましい糖修飾リボヌクレオチドでは、H、OR、ハロ、SH、SR、NH2、NHR、NR2またはCNからなる群より選択される基で2’OH基を置換するが、ここで、Rは、C1〜C6アルキル、アルケニルまたはアルキニルであり、ハロは、F、Cl、BrまたはIである。好ましい骨格鎖修飾リボヌクレオチドでは、隣接するリボヌクレオチドを結合するホスホエステル基を、例えば、ホスホチオエート基の修飾基で置換する。前述した修飾を組み合わせてもよいことに留意されたい。
【0019】
標的特異的RNAiおよび/またはDNAメチル化を媒介するためには、本発明の二本鎖RNA分子の配列は、核酸標的分子に対し十分な同一性を有していなければならない。好ましくは、この配列は、RNA分子の二本鎖部分における所望の標的分子に対して、少なくとも50%、特に少なくとも70%の同一性を有する。さらに好ましくは、同一性は、RNA分子の二本鎖部分において少なくとも85%、最も好ましくは100%である。予め決定した核酸標的分子、例えば、mRNA標的分子に対する二本鎖RNA分子の同一性は、下記のように決定することができる:
【数1】

【0020】
(式中、Iは、同一性のパーセンテージであり、nは、dsRNA分子の二本鎖部分における同一塩基数であり、Lは、dsRNAの二本鎖部分と標的との配列重複部分の長さである)。
【0021】
これ以外に、3’突出部、特に、1〜3塩基長を有する突出部を含む二本鎖RNA分子の標的配列に対する同一性も決定することができる。この場合、配列同一性は、標的配列に対して、好ましくは少なくとも50%、さらに好ましくは少なくとも70%、最も好ましくは少なくとも85%である。例えば、二本鎖の3’突出部からの塩基、5’および/または3’末端からの2以下の塩基は、活性を有意に失うことなく修飾することができる。
【0022】
本発明の二本鎖RNA分子は、下記のステップを含む方法によって作製することができる:
(a)各々が19〜25塩基長、例えば19〜23塩基長を有する2本のRNA鎖を合成するステップであって、このRNA鎖は二本鎖RNA分子を形成することができるものであり、好ましくは少なくとも1本が1〜5塩基の3’突出部を有する、上記ステップ、
(b)二本鎖RNA分子が形成される条件下で合成RNA鎖を結合させるステップであって、得られる二本鎖RNA分子は標的特異的な核酸改変、特にRNA干渉および/またはDNAメチル化を媒介することが可能なものである、上記ステップ。
【0023】
RNA分子を合成する方法は、当業者には公知である。本発明では、特に、VermaおよびEckstein(1998)に記載されている化学的合成方法を例示する。
【0024】
一本鎖RNAは、合成DNA鋳型、または組換え細菌から単離したDNAプラスミドからの酵素の転写により作製することができる。典型的には、T7、T3またはSP6RNAポリメラーゼのようなファージRNAポリメラーゼを用いる(MilliganおよびUhlenbeck(1989))。
【0025】
本発明の別の態様は、以下のステップを含む、細胞または生物における標的特異的な核酸改変、特にRNA干渉および/またはDNAメチル化を媒介する方法に関する:
(a)標的特異的な核酸改変が起こりうる条件下で、上記細胞または生物を本発明の二本鎖RNA分子と接触させるステップ、
(b)上記二本鎖RNAと実質的に対応する配列部分を有する標的核酸に対する、上記二本鎖RNAにより引き起こされる標的特異的な核酸改変を媒介するステップ。
【0026】
好ましくは、接触ステップ(a)は、標的細胞(例えば細胞培養物中の、例えば単離した標的細胞)、単細胞微生物、または多細胞生物内の標的細胞もしくは複数の標的細胞に、二本鎖RNA分子を導入することを含む。さらに好ましくは、導入ステップは、例えば、リポソームキャリアまたは注射によるキャリア媒介の送達を含む。
【0027】
本発明の方法を用いて、RNA干渉を媒介することが可能な細胞または生物における遺伝子の機能を決定する、あるいは、細胞または生物における遺伝子の機能を調節することさえできる。上記細胞は、好ましくは真核細胞または細胞系、例えば植物細胞または動物細胞(哺乳動物細胞など)、例えば胚細胞、多能性幹細胞、腫瘍細胞、例えば奇形癌細胞またはウイルス感染細胞である。上記生物は、好ましくは真核生物、例えば、植物または動物、例えば哺乳動物、特にヒトなどである。
【0028】
本発明のRNA分子が指令する対象となる標的遺伝子は、病理的状態と関連するものでもよい。例えば、遺伝子は、病原体関連遺伝子、例えば、ウイルス遺伝子、腫瘍関連遺伝子または自己免疫疾患関連遺伝子としうる。標的遺伝子はまた、組換え細胞または遺伝的に改変された生物において発現された異種遺伝子であってもよい。このような遺伝子の機能を決定または調節する、特に阻害することにより、農業または医学もしくは獣医学の分野で有用な情報および治療利益が得られると考えられる。
【0029】
dsRNAは、通常、医薬組成物として投与される。この投与は、核酸を所望の標的細胞にin vitroまたはin vivoで導入する公知の方法により実施される。通常用いられる遺伝子伝達法として、リン酸カルシウム法、DEAE−デキストラン法、エレクトロポレーションおよびマイクロインジェクション、ならびに、ウイルス法が挙げられる(Graham, F.L.およびvan der Eb, A.J.(1973)Virol. 52, 456;McCutchan, J.H.およびPagano, J.S.(1968)J. Natl. Cancer Inst. 41, 351;Chu, G.ら(1987)Nucl. Acids Res. 15, 1311;Fraley, Rら(1980)J. Biol. Chem. 255, 10431;Capechi, M.R.(1980)Cell 22, 479)。細胞へのDNAの導入方法の技術群に、近年、カチオンリポソームの使用が加わった(Felgner, P.L.ら(1987), Proc. Natl. Acad. Sci. USA 84, 7413)。市販されているカチオン脂質製剤として、例えば、Tfx50(Promega)またはリポフェクトアミン2000(Life Technologies)がある。
【0030】
従って、本発明はまた、有効物質として前述した少なくとも1つの二本鎖RNA分子と、薬学的キャリアを含む医薬組成物に関する。この組成物は、ヒト医学または獣医学における診断および治療用途に用いることができる。
【0031】
診断または治療用途の場合、組成物は、溶剤(例えば注射液)、クリーム剤、軟膏、錠剤、懸濁剤などの形態とすることができる。組成物は、好適な方法、例えば注射、経口、局所、鼻内、直腸投与などにより、投与することができる。キャリアは、好適な薬学的キャリアであればいずれでもよい。好ましくは、RNA分子が標的細胞に侵入する効率を高めることができるキャリアを用いる。このようなキャリアの好適な例として、リポソーム、特にカチオンリポソームが挙げられる。さらに好ましい投与方法は注射である。
【0032】
RNAi法のさらに好ましい用途は、真核細胞、またはヒト以外の真核生物、好ましくは哺乳動物細胞もしくは生物、最も好ましくはヒト細胞、例えばHeLaもしくは293などの細胞系、またはげっ歯類、例えばラットおよびマウスの機能分析である。予め決定した標的遺伝子と相同的である好適な二本鎖RNA分子、または好適な二本鎖RNA分子をコードするDNA分子を用いたトランスフェクションにより、標的細胞(例えば、細胞培養物)または標的生物において、特異的ノックアウト表現型を獲得することができる。驚くべきことに、短い二本鎖RNA分子が存在しても、宿主細胞または宿主生物からのインターフェロン応答は起こらない。
【0033】
従って、本発明のさらに別の目的は、少なくとも1つの内因性標的遺伝子の少なくとも部分的に欠失した発現を含む標的遺伝子特異的ノックアウト表現型を示す真核細胞またはヒト以外の真核生物であり、この細胞または生物は、少なくとも1つの内因性標的遺伝子の発現を阻害することが可能な少なくとも1つの二本鎖RNA分子、あるいは、少なくとも1つの内因性標的遺伝子の発現を阻害することが可能な少なくとも1つの二本鎖RNA分子をコードするDNAを用いてトランスフェクトされている。本発明により、RNAiの特異性のために、複数の異なる内因性遺伝子の標的特異的ノックアウトが可能となることに留意すべきである。
【0034】
細胞またはヒト以外の生物、特にヒト細胞または非ヒト哺乳動物の遺伝子特異的ノックアウト表現型は、分析方法、例えば遺伝子発現プロフィールおよび/またはプロテオームの分析などの、複雑な生理学的過程の機能および/または表現型分析に用いることができる。例えば、培養細胞中で、選択的スプライシング過程の調節因子と推定されるヒト遺伝子のノックアウト表現型を作製することができる。このような遺伝子として、特に、SRスプライシング因子ファミリーのメンバー、例えば、ASF/SF2,SC35、SRp20、SRp40またはSRp55が挙げられる。さらに、CD44のように、予め決定した、選択的にスプライシングされる遺伝子のmRNAプロフィールに対するSRタンパク質の作用を分析することができる。好ましくは、オリゴヌクレオチドを含むチップを用いたハイスループット方法により、分析を実施する。
【0035】
RNAiによるノックアウト技法を用いて、標的細胞または標的生物における内因性標的遺伝子の発現を阻害することができる。内因性標的遺伝子は、標的タンパク質、または標的タンパク質の変異体もしくは突然変異形態をコードする内因性標的核酸、例えば、遺伝子またはcDNAによって相補することができ、このような核酸は、場合によっては、検出可能なペプチドまたはポリペプチドをコードする別の核酸配列、例えばアフィニティータグ、特に多重アフィニティータグと融合させてもよい。標的遺伝子の変異体または突然変異形態は、これらが、1または複数のアミノ酸の置換、挿入および/または欠失により、アミノ酸レベルで内因性遺伝子産物とは異なる遺伝子産物をコードする点で、内因性標的遺伝子とは異なる。変異体または突然変異形態は、内因性標的遺伝子と同じ生物活性を有しうる。これに対し、変異体または突然変異形態は、内因性標的遺伝子の生物活性とは異なる生物活性、例えば、部分的に欠失した活性、完全に欠失した活性、増強された活性などを有していてもよい。
【0036】
相補は、外因性核酸によりコードされるポリペプチド、例えば、標的タンパク質とアフィニティータグを含む融合タンパク質と、標的細胞において内因性遺伝子をノックアウトする二本鎖RNA分子を共発現させることにより、達成することができる。この共発現は、外因性核酸によりコードされるポリペプチド、例えば、タグで修飾した標的タンパク質と、二本鎖RNA分子の両方を発現する好適な発現ベクターを用いて、あるいは、発現ベクターの組合せを用いて、達成することができる。標的細胞において新たに合成されたタンパク質およびタンパク質複合体は、外因性遺伝子産物、例えば、修飾された融合タンパク質を含むことになる。RNAi二本鎖分子による外因性遺伝子産物発現の抑制を防止するため、外因性核酸をコードする塩基配列において、DNAレベルで(アミノ酸レベルでの突然変異誘発を含むまたは含まない)、二本鎖RNA分子と相同的な配列の部分を改変することができる。あるいは、内因性標的遺伝子を、他の種、例えばマウス由来の対応する塩基配列により相補してもよい。
【0037】
本発明の細胞または生物の好ましい用途は、遺伝子発現プロフィールおよび/またはプロテオームの分析である。特に好ましい実施形態では、1または複数の標的タンパク質の変異体または突然変異体形態の分析を実施するが、その際、前述したような外因性標的核酸により、この変異体または突然変異体形態を上記細胞または生物に再導入する。内因性遺伝子のノックアウトと、突然変異した、例えば部分的に欠失した外因性標的を用いることによる救済の組合せには、ノックアウト細胞の使用と比べて利点がある。さらに、この方法は、標的タンパク質の機能ドメインを同定するのに特に適している。さらに好ましい実施形態では、少なくとも2つの細胞または生物の、例えば遺伝子発現プロフィールおよび/またはプロテオームおよび/または表現型の特徴を比較する。これらの生物は、下記のものから選択する:
(i)標的遺伝子阻害を含まない対照細胞または対照生物、
(ii)標的遺伝子阻害を含む細胞または生物、および
(iii)標的遺伝子阻害と、外因性標的核酸による標的遺伝子の相補を含む細胞または生物。
【0038】
本発明の方法および細胞は、薬理学的物質の同定および/または特定決定、例えば、試験物質のコレクションからの新規な薬理学的物質の同定ならびに/または既知薬理学的物質の作用および/もしくは副作用の機構の特性決定を行なう手法にも適している。
【0039】
従って、本発明はまた、下記の(a)〜(c)を含む、少なくとも1つの標的タンパク質に作用する薬理学的物質の同定および/または特性決定システムに関する:
(a)上記標的タンパク質をコードする少なくとも1つの内因性標的遺伝子を発現することができる、真核細胞またはヒト以外の真核生物、
(b)上記少なくとも1つの内因性標的遺伝子の発現を阻害することができる、少なくとも1つの二本鎖RNA分子、および
(c)薬理学的特性を同定および/または特性決定しようとする、試験物質または試験物質のコレクション。
【0040】
さらに、前記システムは、好ましくは、下記の(d)を含む:
(d)上記標的タンパク質または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸であって、この外因性標的核酸は、二本鎖RNA分子による発現の阻害が、上記内因性標的遺伝子の発現より実質的に低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記外因性標的核酸。
【0041】
さらに、RNAノックアウト相補方法は、調製目的、例えば真核細胞、特に哺乳動物細胞、さらに具体的にはヒト細胞由来のタンパク質またはタンパク質複合体のアフィニティー精製に用いることもできる。本発明のこの実施形態では、外因性標的核酸は、アフィニティータグと融合した標的タンパク質をコードするのが好ましい。
【0042】
上記調製方法を用いて、高分子量のタンパク質複合体を精製することができ、これらの複合体は、質量が、好ましくは150kD以上、さらに好ましくは500kD以上であり、これらは、場合に応じて、RNAのような核酸を含んでいてもよい。具体的例として、U4/U6 snRNP粒子の20kD、60kDおよび90kDタンパク質からなるヘテロ三量体タンパク質複合体、分子量が14、49、120、145および155kDの5つのタンパク質からなる17S U2 snRNP由来のスプライシング因子SF3b、ならびに、U4、U5およびU6 snRNA分子と約30のタンパク質を含む25S U4/U6/U5 tri−snRNP粒子(分子量は約1.7MD)が挙げられる。
【0043】
この方法は、哺乳動物細胞、特にヒト細胞における機能的プロテオーム分析に適している。
【0044】
以下の図面および実施例を参照にしながら、本発明をさらに詳しく説明する。
【0045】
図面の説明
図1:38bpという短い二本鎖RNAは、RNAiを媒介することができる。
【0046】
(A)Pp−luc mRNAをターゲッティングするのに用いたdsRNAのグラフ。29〜504bpの範囲にある3系統の平滑末端dsRNAを調製した。dsRNAのセンス鎖の第1ヌクレオチドの位置は、Pp−luc mRNA(p1)の開始コドンを基準として示した。
【0047】
(B)RNA干渉アッセイ(Tuschlら、1999)。標的Pp−lucと対照Rr−luc活性の比は、バッファー対照(黒棒)に対して正規化した。dsRNA(5nM)をショウジョウバエ溶解物において25℃で15分プレインキュベートした後、7−メチル−グアノシン−キャップを有するPp−lucおよびRr−luc mRNA(約50pM)を添加した。インキュベーションをさらに1時間継続した後、二重ルシフェラーゼアッセイ(Promega)により分析した。データは、少なくとも4回の独立した実験からの平均値±標準偏差である。
【0048】
図2:29bpのdsRNAは、もはや21〜23塩基の断片にプロセシングされない。ショウジョウバエ溶解物における内部32P標識dsRNA(5nM)のプロセシングからの21〜23mer形成の時間経過。dsRNAの長さおよび供給源を示す。RNAサイズマーカー(M)が左レーンにロードされており、断片のサイズを示す。時間0で認められる2つのバンドは、不完全に変性したdsRNAによるものである。
【0049】
図3:短いdsRNAは、mRNA標的を1回しか切断しない。
【0050】
(A)ショウジョウバエ溶解物におけるp133系統の10nM dsRNAと一緒に、キャップを32Pで標識した10nMセンスまたはアンチセンスRNAを1時間インキュベートすることにより生成された安定な5’切断産物の変性ゲル電気泳動。長さマーカーは、キャップ標識した標的RNAの部分的ヌクレアーゼT1消化および部分的アルカリ加水分解(OH)により作製した。dsRNAによりターゲッティングされる領域は、両側が黒棒として示される。長さが111bpのdsRNAでは、優勢切断部位同士の間隔は、20〜23塩基であることがわかる。水平方向の矢印は、RNAiによるものではない非特異的切断を示す。
【0051】
(B)センスおよびアンチセンス標的RNA上の切断部位の位置。キャップを有する177塩基のセンスおよび180塩基のアンチセンス標的RNAの配列は、逆平行配向をしており、相補配列が互いに向き合っている。様々なdsRNAによりターゲッティングされる領域は、センスおよびアンチセンス標的配列の間に位置する様々な色の棒によって示す。切断部位は丸で示す:大きな丸は強い切断を、小さな丸は弱い切断をそれぞれ示す。32P放射性標識リン酸基は、星印で示す。
【0052】
図4:RNaseIII様機構により21および22塩基のRNA断片を作製する。
【0053】
(A)dsRNAプロセシング後の約21塩基RNAの配列。dsRNAプロセシングにより作製した約21塩基のRNA断片を定方向にクローニングした後、配列決定した。dsRNAのセンス鎖に由来するオリゴリボヌクレオチドを青色の線で示し、アンチセンス鎖に由来するものを赤色の線で示す。同じ配列が複数のクローンに存在する場合には太い棒を用い、右側の数字は頻度を示す。dsRNAが媒介する標的RNA切断部位はオレンジ色の丸で示し、大きな丸は強い切断を、また小さな丸は弱い切断をそれぞれ表す(図3B参照)。センス鎖の上部にある丸はセンス標的内の切断部位を、dsRNAの下にある丸はアンチセンス標的内の切断部位をそれぞれ示す。5以下の更なるヌクレオチドが、dsRNAの3’末端由来の約21塩基断片に確認された。これらのヌクレオチドは、主としてC、GまたはA残基のランダムな組合せであり、dsRNA構成鎖のT7転写中に、非鋳型様式で付加されたと考えられる。
【0054】
(B)約21塩基のRNAのヌクレオチド組成の二次元TLC分析。ショウジョウバエ溶解物における内部放射性標識504bp Pp−luc dsRNAのインキュベーションにより、約21塩基のRNAを作製し、ゲル精製した後、ヌクレアーゼP1(上列)またはリボヌクレアーゼT2(下列)でモノヌクレオチドに消化した。表示したα32Pヌクレオシド三リン酸の1つの存在下での転写により、dsRNAを内部標識した。リン光イメージング(phosphorimaging)により放射活性を検出した。ヌクレオシド5’一リン酸、ヌクレオシド3’一リン酸、ヌクレオシド5’,3’二リン酸、および無機リン酸をそれぞれpN、Np、pNp、およびpiで表す。黒丸は、非放射能キャリアヌクレオチドからのUV吸収斑を示す。3’,5’ビス−リン酸(赤丸)は、T4ポリヌクレオチドキナーゼとγ32P−ATPによるヌクレオシド3’一リン酸の5’リン酸化により調製された放射性標識標準との同時泳動により確認した。
【0055】
図5:合成21および22塩基のRNAは、標的RNA切断を媒介する。
【0056】
(A)対照52bp dsRNA並びに合成21および22塩基dsRNAをグラフで表示した図。21および22塩基の短鎖干渉RNA(siRNA)のセンス鎖を青色で、アンチセンス鎖を赤色で示す。siRNAの配列は、二本鎖5の22塩基アンチセンス鎖を除いて、52および111bp dsRNAのクローン化断片(図4A)に由来する。二本鎖6および7のsiRNAは、111bp dsRNAプロセシング反応に特有のものであった。緑色で示す2つの3’突出塩基は、二本鎖1および3の合成アンチセンス鎖の配列に存在する。対照52bp dsRNAの両鎖は、in vitro転写により作製したため、転写物の画分には、非鋳型3’ヌクレオチド付加が含まれることもある。siRNA二本鎖により指令される標的RNA切断部位は、オレンジ色の丸で示し(図4Aの説明を参照)、図5Bに示したように決定した。
【0057】
(B)センスおよびアンチセンス標的RNA上の切断部位の位置。標的RNA配列は、図3Bに説明した通りである。ショウジョウバエ溶解物において、対照52bp dsRNA(10nM)または21および22塩基のRNA二本鎖1〜7(100nM)を標的RNAと一緒に25℃で2.5時間インキュベートした。安定した5’切断産物がゲル上で分離された。切断部位を図5Aに示す。52bp dsRNAまたはセンス(s)もしくはアンチセンス(as)鎖は、ゲルの横に黒棒で示す。切断部位はすべてdsRNAの同一性領域内に位置する。アンチセンス鎖の切断部位の正確な決定のために、低いパーセンテージのゲルを用いた。
【0058】
図6:短いdsRNA上の長い3’突出部がRNAiを阻害する。
【0059】
(A)52bp dsRNA構築物をグラフで示した図。センスおよびアンチセンス鎖の3’延長部分をそれぞれ青色および赤色で示す。標的RNA上で観察された切断部位を図4Aと同様のオレンジ色の丸で表し、これらを図6Bで示したように決定した。
【0060】
(B)センスおよびアンチセンス標的RNA上の切断部位の位置。標的RNA配列は図3Bに説明した通りである。ショウジョウバエ溶解物において、dsRNA(10nM)を標的RNAと一緒に25℃で2.5時間インキュベートした。安定した5’切断産物をゲル上で分離させた。主要な切断部位は水平方向の矢印で示し、図6Aでも表示する。52bp dsRNAによりターゲッティングされる領域は、ゲル両側の黒棒で表示する。
【0061】
図7:RNAiのモデル案
RNAiは、主として21および22塩基の短鎖干渉RNA(siRNA)を生成するdsRNA(センス鎖は黒、アンチセンス鎖は赤)のプロセシングで開始すると推定される。短い3’突出塩基が、dsRNA上に存在すれば、これは、短鎖dsRNAのプロセシングに有益であると考えられる。これから特性決定しようとするdsRNAプロセシングタンパク質は、緑色および青色の長円形として表し、非対称にdsRNA上に集合させる。本発明のモデルでは、これは、推定上の青色タンパク質またはタンパク質ドメインと、3’から5’方向のsiRNA鎖との結合により説明されるのに対し、推定上の緑色タンパク質またはタンパク質ドメインは、向き合うsiRNA鎖と必ず結合する。これらのタンパク質またはサブセットは、siRNA二本鎖と会合したままであり、dsRNAプロセシング反応の方向により決定される配向を保存している。青色タンパク質と会合したsiRNA配列だけが標的RNA切断を指示することができる。エンドヌクレアーゼ複合体は、短い干渉リボ核タンパク質複合体またはsiRNPと呼ばれる。本発明では、dsRNAを切断するエンドヌクレアーゼは、恐らく、標的認識に使用されない受動siRNAを一時的に置換することにより、標的RNAも切断することができると推定する。次に、この標的RNAは、配列相補的ガイドsiRNAにより認識される領域の中心部で切断される。
【0062】
図8:リポーター構築物とsiRNA二本鎖。
【0063】
(a)プラスミドpGL2−Control、pGL−3−ControlおよびpRL−TK(Promega)からのホタル(Pp−luc)およびウミシイタケ(Rr−luc)ルシフェラーゼリポーター遺伝子領域を示す。SV40調節エレメント、HSVチミジンキナーゼプロモーターと、2つのイントロン(斜線)を示す。GL3ルシフェラーゼの配列はGL2と95%同一であるが、RLは、両者とまったく関連性がない。pGL2からのルシフェラーゼ発現は、トランスフェクトした哺乳動物細胞におけるpGL3からの発現に対して約10分の1低い。siRNA二本鎖によりターゲッティングされる領域は、ルシフェラーゼ遺伝子のコード領域下方の黒棒で表す。
【0064】
(b)GL2、GL3およびRLルシフェラーゼをターゲッティングするsiRNA二本鎖のセンス(上)およびアンチセンス(下)配列を示す。GL2およびGL3siRNA二本鎖は、3つの一塩基置換が異なるにすぎない(灰色で囲んだ箇所)。非特異的対照として、逆転GL2配列、すなわち、invGL2を有する二本鎖を合成した。2’−デオキシチミジンの2塩基3’突出部をTTとして表す。uGL2は、GL2siRNAと類似しているが、リボ−ウリジン3’突出部を含んでいる。
【0065】
図9:siRNA二本鎖によるRNA干渉。
【0066】
標的対照ルシフェラーゼの比をバッファー対照(bu、黒棒)に対して正規化した;灰色の棒は、フォティナス・ピラリス(Photinus pyralis)(Pp−luc)GL2またはGL3ルシフェラーゼと、レニラ・レニフォルミス(Renilla reniformis)(Rr−luc)RLルシフェラーゼの比を示し(左軸)、白色の棒は、RLとGL2またはGL3比を示す(右軸)。a、c、e、gおよびiの各表は、pGL2−ControlとpRL−TKリポータープラスミドの組合せを用いて実施した実験を示し、表b、d、f、hおよびjは、pGL3−ControlとpRL−TKリポータープラスミドを用いたものを示す。干渉実験に用いた細胞系は、各表の上部に示す。バッファー対照(bu)に対するPp−luc/Rr−lucの比は、正規化の前に各種被検細胞系間で、pGL2/pRLでは0.5〜10、また、pGL3/pRLについては0.03〜1の範囲をそれぞれ変動した。プロットしたデータは、3つの独立した実験の平均値±S.D.である。
【0067】
図10:HeLa細胞におけるルシフェラーゼ発現に対する21塩基のsiRNA、50bpおよび500bpのdsRNAの影響。
【0068】
長いdsRNAの正確な長さを棒の下に示す。表a、cおよびeは、pGL2−ControlとpRL−TKリポータープラスミドを用いて実施した実験を示し、表b、dおよびfは、pGL3−ControlとpRL−TKリポータープラスミドを用いたものを示す。データは、2つの独立した実験の平均値±S.D.である。
【0069】
(a)および(b)任意のルミネセンス単位でプロットした絶対Pp−luc発現。
【0070】
(c)および(d)任意のルミネセンス単位でプロットしたRr−luc発現。
【0071】
(e)および(f)正規化した標的と対照ルシフェラーゼの比。siRNA二本鎖についてのルシフェラーゼ活性の比は、バッファー対照(bu、黒棒)に対して正規化した;50または500bp dsRNAについてのルミネセンス比は、ヒト化GFP(hG、黒棒)由来の50および500bp dsRNAについて認められたそれぞれの比に対して正規化した。GL2およびGL3をターゲッティングする49および484bpのdsRNA間の配列の全相違は、GL2およびGL3標的間に特異性を賦与するのに十分ではなかった(49bp断片では43塩基の連続した同一性、484bp断片では239塩基の最長連続同一性)。
【0072】
図11:21塩基siRNA二本鎖の3’突出部の変動
(A)実験戦略の概要。キャップを有し、かつポリアデニル化されたセンス標的mRNAを描くと共に、センスおよびアンチセンスsiRNAの相対位置を示す。8つの異なるアンチセンス鎖に従って8系統の二本鎖を用意した。siRNA配列と、突出ヌクレオチドの数を1塩基ずつ変化させた。
【0073】
(B)5nM平滑末端dsRNAの存在下で、キイロショウジョウバエ胚溶解物において、対照ルシフェラーゼ(Renilla reniformis、Rr−luc)に対する、標的ルシフェラーゼ(Photinus pyralis、Pp−luc)の正規化相対ルミネセンス。dsRNAの存在下で決定したルミネセンス比は、バッファー対照で得られた比(bu、黒棒)に対して正規化した。1より小さい正規化比は、特異的干渉を意味する。
【0074】
(C〜J)8系統の21塩基siRNA二本鎖について正規化した干渉比。siRNA二本鎖の配列を棒グラフの上方に表示する。各表は、所与のアンチセンスガイド(guide)siRNAと、5つの異なるセンスsiRNAにより形成された二本鎖のセットについての干渉比を示す。突出塩基の数(3’突出部は正の数;5’突出部は負の数)をX軸に示す。データ点は、少なくとも3回の独立した実験から平均したものであり、エラーバー(error bar)は標準偏差を表す。
【0075】
図12:siRNA二本鎖のセンス鎖の長さ変動。
【0076】
(A)実験をグラフで示す図。3つの21塩基アンチセンス鎖を8つのセンスsiRNAと対合させた。siRNAは、その3’末端で長さを変化させた。アンチセンスsiRNAの3’突出部は、1塩基(B)、2塩基(C)、または3塩基(D)であるのに対し、センスsiRNAの突出部は、各系統で変動させた。siRNA二本鎖の配列および対応する干渉比を表示する。
【0077】
図13:保存された2塩基3’突出部を有するsiRNA二本鎖の長さの変動。
【0078】
(A)実験をグラフで示す図。21塩基のsiRNA二本鎖は、図11Hまたは12Cに示したものと配列が同じである。siRNA二本鎖は、センスsiRNAの3’側(B)、またはセンスsiRNAの5’側(C)まで延長させた。siRNA二本鎖の配列およびそれぞれの干渉比を表示する。
【0079】
図14:siRNAリボース残基の2’−ヒドロキシル基の置換。
【0080】
siRNA二本鎖の鎖における2’−ヒドロキシル基(OH)は、2’−デオキシ(d)または2’−O−メチル(Me)によって置換した。3’末端での2塩基および4塩基2’−デオキシ置換をそれぞれ2塩基dおよび4塩基dとして表示する。ウリジン残基は2’−デオキシチミジンで置換した。
【0081】
図15:2塩基3’突出部を有する21塩基のsiRNA二本鎖によるセンスおよびアンチセンス標的RNA切断のマッピング
(A)32P(星印)キャップ標識センスおよびアンチセンス標的RNAと、siRNA二本鎖をグラフで示した図。センスおよびアンチセンス標的RNA切断の位置は、siRNA二本鎖の上方および下方に、それぞれ三角形で示す。
【0082】
(B)標的RNA切断部位のマッピング。キイロショウジョウバエ胚溶解物において、100nM siRNA二本鎖と一緒に10nM標的を2時間インキュベートした後、5’キャップ標識基質と5’切断産物を配列決定用ゲル上で分離させた。標的RNAの部分的RNaseT1消化(T1)と部分的アルカリ加水分解(OH−)により、長さマーカーを作製した。画像左側の太線は、標的と同じ配向のsiRNA鎖1および5が占める領域を示す。
【0083】
図16:ガイドsiRNAの5’末端が、標的RNA切断の位置を画定する。
【0084】
(A、B)実験戦略をグラフで示した図。アンチセンスsiRNAは、すべてのsiRNA二本鎖で同じであるが、センス鎖は、3’末端を改変することにより18〜25塩基までの間を(A)、または5’末端を改変することにより18〜23塩基までの間(B)を変動させた。センスおよびアンチセンス標的RNA切断の位置は、siRNA二本鎖の上方および下方に、それぞれ三角形で示す。
【0085】
(C、D)キャップ標識センス(上の表)またはアンチセンス(下の表)標的RNAを用いた標的RNA切断の分析。キャップ標識の5’切断産物だけを示す。siRNA二本鎖の配列を表示すると共に、センスsiRNA鎖の長さを表の上部に示す。パネル(C)においてダッシュで示した対照レーンは、siRNAの不在下でインキュベートした標的RNAを示す。マーカーは、図15に記載した通りである。(D)の下部パネルの矢印は、1塩基だけ異なる標的RNA切断部位を示す。
【0086】
図17:siRNA二本鎖の3’突出部の配列変動
2塩基3’突出部(灰色のNN)は、表示したように配列および組成を改変した(T、2’−デオキシチミジン、dG、2’−デオキシグアノシン;星印、野生型siRNA二本鎖)。正規化した干渉比は、図11で記載したように決定した。野生型配列は図14に示したものと同じである。
【0087】
図18:標的認識の配列特異性。
【0088】
ミスマッチsiRNA二本鎖の配列を示し、修飾した配列部分または単一塩基を灰色で示す。基準二本鎖(ref)とsiRNA二本鎖1〜7は、2’−デオキシチミジン2塩基突出部を含む。チミジンを修飾した基準二本鎖のサイレンシング効率は、野生型配列と同等であった(図17)。正規化した干渉比は、図11に記載したのと同様に決定した。
【0089】
図19:保存された2塩基3’突出部を有するsiRNA二本鎖の長さ変動。
【0090】
siRNA二本鎖は、センスsiRNAの3’側(A)、またはセンスsiRNAの5’側(B)まで延長した。siRNA二本鎖配列と、それぞれの干渉比を示す。HeLa SS6細胞については、GL2ルシフェラーゼをターゲッティングするsiRNA二本鎖(0.84μg)を、pGL2−ControlおよびpRL−TKプラスミドで一緒にトランスフェクトした。比較のため、キイロショウジョウバエ胚溶解物で試験したsiRNA二本鎖のin vitro RNAi活性を示す。
【実施例1】
【0091】
短い合成RNAにより媒介されるRNA干渉
1.1.実験手順
1.1.1 in vitro RNAi
以前記載されている(Tuschlら、1999;Zamoreら、2000)ように、in vitro RNAiおよび溶解物調製を実施した。最適なATP再生のためには、新しく溶解したクレアチンキナーゼ(Roche)を用いる必要がある。RNAi翻訳アッセイ(図1)は、5nMのdsRNA濃度と、25℃で15分の長いプレインキュベーション時間で実施した後、in vitro転写、キャッピングおよびポリアデニル化したPp−lucおよびRr−lucリポーターmRNAを添加した。インキュベーションを1時間継続した後、二重ルシフェラーゼアッセイ(Promega)およびモノライト3010Cルミノメーター(PharMingen)を用いて、Pp−lucおよびRr−lucタンパク質の相対量を分析した。
【0092】
1.1.2 RNA合成
標準的手順を用いて、T7またはSP6プロモーター配列を有するPCR鋳型からのRNAのin vitro転写を実施した(例えば、Tuschlら、1998を参照)。Expedite RNAホスホルアミダイト(Proligo)を用いて、合成RNAを調製した。ジメトキシトリチル−1,4−ベンゼンジメタノール−スクシニル−アミノプロピル−CPGを用いて、3’アダプターオリゴヌクレオチドを合成した。オリゴリボヌクレオチドを、3mlの32%アンモニア/エタノール(3/1)において、55℃で4時間(Expedite RNA)または55℃で16時間かけて脱保護した(3’および5’アダプターDNA/RNAキメラオリゴヌクレオチド)後、以前記載されている(Tuschlら、1993)ように、脱シリル化してからゲル精製した。長い3’突出部を含むdsRNAを調製するためのRNA転写物は、センス方向にT7プロモーターを、アンチセンス方向にSP6プロモーターを含むPCR鋳型から作製した。5’プライマーとしてGCGTAATACGACTCACTATAGAACAATTGCTTTTACAG(下線部、T7プロモーター)と、3’プライマーとしてATTTAGGTGACACTATAGGCATAAAGAATTGAAGA(下線部、SP6プロモーター)、ならびに、鋳型として線状化Pp−lucプラスミド(pGEM−luc配列)(Tuschlら、1999)を用いて、センスおよびアンチセンス標的RNA用の転写鋳型をPCR増幅した。T7転写センスRNAは、177塩基長であり、開始コドンに対して位置113〜273のPp−luc配列と、これに続いて、3’末端にSP6プロモーター配列の17塩基の相補配列(complement)を含む。平滑末端dsRNA形成用の転写物は、単一のプロモーター配列だけを含む2つの異なるPCR産物からの転写によって調製した。
【0093】
フェノール/クロロホルム抽出により、dsDNAのアニーリングを実施した。0.3M NaOAc(pH6)における等モル濃度のセンスおよびアンチセンスRNA(利用可能な長さおよび量に応じて、50nM〜10μM)を90℃で30秒インキュベートしてから、等量のフェノール/クロロホルムを用いて、室温で抽出した後、クロロホルム抽出により、残留フェノールを除去した。得られたdsRNAは、2.5〜3容量のエタノールの添加により沈降させた。このペレットを溶解バッファー(100mM KCl、30mM HEPES−KOH、pH7.4、2mM Mg(OAc)2)中で溶解させ、1×TAEバッファー中の標準アガロースゲル電気泳動によりdsRNAの品質を確認した。17塩基および20塩基の3’突出部を有する52bpのdsRNA(図6)を95℃で1分インキュベートすることによりアニールしてから、70℃まで急冷した後、3時間かけてゆっくりと室温まで冷却した(50μlアニーリング反応、1μM鎖濃度、300mM NaCl、10mM Tris−HCl、pH7.5)。次に、このdsRNAをフェノール/クロロホルム抽出し、エタノール沈降させた後、溶解バッファー中に溶解させた。
【0094】
dsRNA調製に用いる内部32P放射性標識RNAの転写(図2および4)は、1mM ATP、CTP、GTP、0.1または0.2mM UTP、および0.2〜0.3μM−32P−UTP(3000Ci/mmol)、あるいは、UTP以外の放射性標識ヌクレオシド三リン酸についてのそれぞれの比を用いて実施した。標的RNAのキャップの標識は、既述されている通りに実施した。キャップ標識後、標的RNAをゲル精製した。
【0095】
1.1.3 切断部位マッピング
10nM dsRNAを15分プレインキュベートした後、10nMキャップ標識標的RNAをを添加することにより、標準RNAi反応を実施した。プロテイナーゼK処理(Tuschlら、1999)により、さらに2時間(図2A)または2.5時間(図5Bおよび6B)インキュベートした後、反応を停止した。次に、8または10%配列決定用ゲル上でサンプルを分析した。21および22塩基の合成RNA二本鎖を100nM最終濃度で用いた(図5B)。
【0096】
1.1.4 約21塩基のRNAのクローニング
標的RNAの不在下で、ショウジョウバエ溶解物における放射性標識dsRNAのインキュベーションにより(200μl反応物、1時間インキュベーション、50nM dsP111、または100nM dsP52もしくはdsP39)、21塩基のRNAを生成した。次に、反応混合物をプロテイナーゼKで処理した(Tuschlら、1999)後、dsRNAプロセシング産物を変性15%ポリアクリルアミドゲル上で分離した。少なくとも18〜24塩基のサイズ範囲を含むバンドを除去し、0.3M NaClに4℃で一晩かけて溶離した後、シリコン処理管に導入した。エタノール沈降によりRNAを回収した後、脱リン酸化した(30μl反応物、30分、50℃、10Uアルカリホスファターゼ、Roche)。フェノール/クロロホルム抽出により反応を停止し、RNAをエタノール沈降させた。次に、3’アダプターオリゴヌクレオチド(pUUUaaccgcatccttctcx:大文字はRNA;小文字はDNA;pはリン酸;xは4−ヒドロキシメチルベンジル)を脱リン酸化した約21塩基のRNAと連結させた(20μl反応物、30分、37℃、5μM 3’アダプター、50mM Tris−HCl、pH7.6、10mM MgCl2、0.2mM ATP、0.1mg/mlアセチル化BSA、15%DMSO、25U T4 RNAリガーゼ、Amersham-Pharmacia)(PanおよびUhlenbeck, 1992)。等量の8M尿素/50mM EDTA停止混合物(stopmix)の添加により連結反応を停止した後、15%ゲルに直接ロードした。連結収率は50%を超えた。連結産物をゲルから回収した後、5’−リン酸化した(20μl反応物、30分、37℃、2mM ATP、5U T4ポリヌクレオチドキナーゼ、NEB)。フェノール/クロロホルム抽出によりリン酸化反応を停止した後、エタノール沈降によりRNAを回収した。次に、前記と同様に、5’アダプター(tactaatacgactcactAAA:大文字はRNA;小文字はDNA)をリン酸化連結産物と連結させた。新しい連結産物をゲル精製し、キャリアとして用いる逆転写プライマー(GACTAGCTGGAATTCAAGGATGCGGTTAAA:太字はEcoRI部位)の存在下で、ゲル切片から溶離させた。逆転写(15μl反応物、30分、42℃、150U SuperscriptII逆転写酵素、Life Technologies)の後、5’プライマーCAGCCAACGGAATTCATACGACTCACTAAA(太字はEcoRI部位)と、3’RTプライマーを用いたPCRを実施した。PCR産物をフェノール/クロロホルム抽出により精製した後、エタノール沈降させた。次に、PCR産物をEcoRI(NEB)で消化してから、T4 DNAリガーゼ(高濃度、NEB)を用いてコンカテマー化する。サイズが200〜800bpの範囲にあるコンカテマーを低融点アガロースゲル上で分離し、標準的な融解およびフェノール抽出手順によりゲルから回収した後、エタノール沈降させた。非対合末端を標準的条件下でTaqポリメラーゼと一緒に72℃で15分インキュベートすることにより充填した後、TOPO TAクローニングキット(Invitrogen)を用いて、DNA産物をpCR2.1−TOPOベクターに直接連結した。PCRと、M13−20およびM13逆方向配列決定用プライマーを用いて、コロニーをスクリーニングした。カスタム(custom)配列決定(Sequence Laboratories Gottingen GmbH, ドイツ)のためにPCR産物を直接提出した。平均して、1クローン当たり4〜5個の21mer配列が得られた。
【0097】
1.1.5 2D−TLC分析
放射性標識し、ゲル精製したsiRNAおよび2D−TLCのヌクレアーゼP1消化を記載されている通りに実施した(Zamoreら、2000)。2μg/μlキャリアtRNAと30UリボヌクレアーゼT2(Life Technologies)を用いて、10mM酢酸アンモニウム(pH4.5)中、10μl反応物において、ヌクレアーゼT2消化を50℃で3時間実施した。非放射性標準の泳動をUVシャドウイング(shadowing)により測定した。ヌクレオシド−3’,5’−二リン酸の同一性は、γ−32P−ATPおよびT4ポリヌクレオチドキナーゼを用いた市販のヌクレオシド3’−一リン酸の5’−32Pリン酸化により調製した標準によるT2消化産物の同時泳動によって確認した(データは示していない)。
【0098】
1.2 結果および考察
1.2.1 21および22塩基のRNA断片のプロセシングに必要な長さ
キイロショウジョウバエ合胞体胚から調製した溶解物は、in vitroにおいてRNAiを再現することから、RNAi機構の生化学的分析の新たなツールを提供するものである(Tuschlら、1999;Zamoreら、2000)。RNAiのためのdsRNAに必要な長さについてのin vitroおよびin vivo分析から、標的mRNAを分解する上で、短いdsRNA(<150bp)は、長いdsRNAより効果が低いことが明らかにされている(Caplenら、2000;Hammondら、2000;Ngoら、1998);Tuschlら、1999)。このmRNA分解効率が低い理由はわかっていない。従って、本発明者は、ショウジョウバエ溶解物における最適化条件下で(Zomoreら、2000)、標的RNA分解のためのdsRNAに必要な正確な長さを調べた。数系統のdsRNAを合成し、ホタルルシフェラーゼ(Pp−luc)リポーターRNAに対して指令させた。標的RNA発現の特異的抑制を二重ルシフェラーゼアッセイ(Tuschlら、1999)によりモニタリングした(図1Aおよび1B)。本発明者は、38bpという短いdsRNAの標的RNA発現の特異的阻害を検出したが、29〜36bpのdsRNAはこの過程において有効ではなかった。効果は、Pp−luc mRNAの標的位置および阻害度とは無関係で、dsRNAの長さと相関していた。すなわち、長鎖dsRNAの方が、短鎖dsRNAより有効であった。
【0099】
dsRNAのプロセシングにより作製された21〜23塩基のRNAは、RNA干渉および共抑制の媒介物質であることが示唆されている(HamiltonおよびBaulcombe, 1999;Hammondら、2000;Zamoreら、2000)。従って、本発明者は、サイズが501〜29bpの範囲にあるdsRNAのサブセットについて、21〜23塩基断片の形成の速度を分析した。ショウジョウバエ溶解物における21〜23塩基断片の形成(図2)は、長さが39〜501bpのdsRNAでは容易に検出することができたが、29bpのdsRNAでは有意な遅延が認められた。この観察結果は、mRNA切断を指示する上での21〜23塩基断片の役割と一致し、30bpのdsRNAによる不十分なRNAiを解明する助けとなった。21〜23mer形成が長さに依存するのは、通常の細胞RNAの短い分子内塩基対合構造によるRNAiの不要な活性化を防止するための、生物学的に関連する制御機構を表すものと考えられる。
【0100】
1.2.2 39bpのdsRNAは、単一部位で標的RNA切断を媒介する
ショウジョウバエ溶解物にdsRNAおよび5’−キャップ標識RNAを添加すると、標的RNAの配列特異的分解が起こる(Tuschlら、1999)。標的mRNAは、dsRNAと同一性のある領域内でしか切断されず、標的切断部位の多くは、21〜23塩基ずつ分離された(Zamoreら、2000)。従って、所与のdsRNAの切断部位の数は、dsRNAの長さを21で割った数とおおまかに対応することが予想された。本発明者は、キャップにおいて5’放射性標識されたセンスおよびアンチセンス標的RNA上の標的切断部位をマッピングした(Zamoreら、2000)(図3Aおよび3B)。配列決定用ゲル上で安定した5’切断産物を分離し、標的RNAからの部分的RNaseT1およびアルカリ性加水分解標準(ladder)との比較により、切断位置を決定した。
【0101】
以前の観察結果(Zamoreら、2000)と一致して、すべての標的RNA切断部位は、dsRNAとの同一性領域内に位置した。センスまたはアンチセンス標的は、39bpのdsRNAによって一回しか切断されなかった。各切断部位は、dsRNAが占める領域の5’末端から10塩基の位置にあった(図3B)。39bpのRNAと同じ5’末端を有する52bpのdsRNAは、第1部位から23および24塩基下流の2つの弱い切断部位に加えて、dsRNAと同一性のある領域の5’末端から10塩基の位置に、センス標的上の同じ切断部位を生成する。アンチセンス標的も、やはり各dsRNAが占める領域の5’末端から10塩基の位置で、1回だけ切断された。図1に示した38〜49bpのdsRNAについての切断部位のマッピングから、第1および優勢切断部位は、常に、dsRNAが占める領域から7〜10塩基下流の位置にあることが明らかにされた(データは示していない)。これは、標的RNA切断点が、dsRNAの末端により決定されることを示唆しており、このことは、21〜23merへのプロセシングが二本鎖の末端から開始することを意味すると考えられる。
【0102】
より長い111bpのdsRNAのセンスおよびアンチセンス標的上の切断部位は、予想よりはるかに頻度が高く、それらのほとんどは、20〜23塩基ずつ分離したクラスターとして現れる(図Aおよび3B)。より短いdsRNAに関しては、センス標的上の第1切断部位は、dsRNAが占める領域の5’末端から10塩基の位置であり、アンチセンス標的上の第1切断位置は、dsRNAが占める領域の5’末端から9塩基の位置である。この不規則な切断の原因は不明であるが、1つには、より長いdsRNAが末端からだけではなく、内部でもプロセシングされるためか、あるいは、まだ理解されていないdsRNAプロセシングの何らかの特異性決定因子があると考えられる。21〜23塩基間隔についてのある程度の不規則性も以前指摘されていた(Zamoreら、2000)。dsRNAプロセシングおよび標的RNA認識の分子レベルでの根本原理をより明瞭に理解するために、本発明者は、ショウジョウバエ溶解物における39、52、および111bpのdsRNAのプロセシングにより生成される21〜23塩基断片の配列を分析することにした。
【0103】
1.2.3 dsRNAをRNaseIII様機構による21および23塩基RNAにプロセシングする
21〜23塩基のRNA断片を特性決定するために、RNA断片の5’および3’末端を調べた。ゲル精製した21〜23塩基のRNAを過ヨウ素酸化した後、β脱離すると、末端2’および3’ヒドロキシル基の存在が示された。21〜23merはまた、アルカリ性ホスファターゼ処理に応答性であり、これは5’末端リン酸基の存在を意味する。5’リン酸および3’ヒドロキシル末端の存在は、dsRNAが大腸菌RNaseIIIと類似した酵素活性によりプロセシングされ得ることを示唆している(確認のため、(Dunn, 1982;Nicholson, 1999;Robertson, 1990:Robertson, 1982)を参照)。
【0104】
T4RNAリガーゼを用いて、3’および5’アダプターオリゴヌクレオチドと精製21〜23merとを連結することにより、21〜23塩基のRNA断片の定方向クローニングを実施した。連結産物を逆転写、PCR増幅、コンカテマー化、クローニングした後、配列決定した。39、52および111bp dsRNA(図4A)のdsRNAプロセシング反応から、220個を超える短いRNAを配列決定した。本発明者は、次のような長さ分布をみいだした:1%18塩基、5%19塩基、12%20塩基、45%21塩基、28%22塩基、6%23塩基、および2%24塩基。プロセシングされた断片の5’末端ヌクレオチドの配列分析から、5’グアノシンを有するオリゴヌクレオチドは過小提示されることがわかった。この偏りは、5’リン酸化グアノシンを供与オリゴヌクレオチドとして区別するT4 RNAリガーゼにより引き起こされると考えられる。尚、3’末端の配列には有意な偏りは認められなかった。二本鎖のセンスまたはアンチセンス鎖の3’末端から得られた約21塩基断片の多くは、T7 RNAポリメラーゼを用いたRNA合成中にヌクレオチドの非鋳型付加により得られる3’ヌクレオチドを含んでいる。興味深いことに、有意な数の内因性ショウジョウバエの約21塩基のRNAもクローニングされ、その中には、LTRおよび非LTRレトロトランスポゾン由来のものもあった(データは示していない)。これは、トランスポゾンサイレンシングにおいてRNAiが果たす可能性のある役割と一致している。
【0105】
約21塩基のRNAは、dsRNA配列全体を含むクラスター形成群として現われる(図4A)。明らかに、プロセシング反応は、付着3’末端を残してdsRNAを切断しており、これはRNaseIII切断のもう一つの特徴である。39bpのdsRNAでは、約21塩基のRNAの2つのクラスターが突出3’末端を含む各dsRNA構成鎖から見出されたが、センスおよびアンチセンス標的には1つの切断部位しか検出されなかった(図3Aおよび3B)。約21塩基の断片が、mRNA分解を媒介する複合体において一本鎖ガイドRNAとして存在するのであれば、少なくとも2つの標的切断部位が存在すると想定できたが、そうではなかった。これは、約21塩基のRNAがエンドヌクレアーゼ複合体において二本鎖形態で存在する可能性があるが、標的RNAの認識および切断には、そのうち一方の鎖しか使用できないことを示唆している。標的切断に約21塩基鎖の一方だけを使用することは、約21塩基の二本鎖が、ヌクレアーゼ複合体と結合している配向によって簡単に決定することができる。この配向は、本来のdsRNAがプロセシングされた方向によって決定される。
【0106】
52bpおよび111bpのdsRNAの場合の約21merクラスターは、39bpのdsRNAと比較して、あまりよく明確にされていない。これらのクラスターは、約21塩基二本鎖の複数の個別小集団を提示する、従って、複数の近傍部位における標的切断を指示すると考えられられる25〜30塩基の領域に分布している。これらの切断領域は、やはり20〜23塩基の間隔で主に分離されている。標準的dsRNAがいかにして約21塩基の断片にプロセッシングされ得るかを決定する法則はいまだに解明されていないが、切断部位同士の約21〜23塩基の間隔は、一連のウリジンによって改変できることがすでに認められている(Zamoreら、2000)。大腸菌RNaseIIIによるdsRNA切断の特異性は、主に、非決定因子(antideterminant)、すなわち、切断部位に対する所与の位置で特異的塩基対を排除することにより、制御されているようである。
【0107】
糖−、塩基−またはキャップ修飾が、プロセシングされた約21塩基のRNA断片に存在するか否かを試験するため、本発明者は、溶解物において放射性標識した505bpのPp−luc dsRNAをインキュベートし、約21塩基産物を単離した後、それをP1またはT2ヌクレアーゼでモノヌクレオチドに消化した。このヌクレオチド混合物を2D薄層クロマトグラフィーにより分析した(図4B)。P1またはT2消化により示されるように、4つの天然リボヌクレオチドのうち、修飾されたものは1つもなかった。本発明者は既に、約21塩基断片におけるアデノシンからイノシンへの変換を分析し(2時間のインキュベーション後)、わずかな程度(<0.7%)の脱アミノ化を検出している(Zamoreら、2000)。溶解物中での短いインキュベーション(1時間)により、このイノシン画分をかろうじて検出可能なレベルまで縮小した。ホスホジエステル結合の3’を切断するRNaseT2は、ヌクレオシド3’−リン酸とヌクレオシド3’,5’−二リン酸を生成したが、これは、5’−末端一リン酸の存在を意味する。4つのヌクレオシド3’,5’−二リン酸の全てが検出されたが、これは、配列特異性がほとんどまたは全くなしに、ヌクレオチド間の結合が切断されたことを示唆している。要約すると、約21塩基断片は、非修飾であり、dsRNAから生成され、その際、5’−末端に5’−一リン酸および3’−ヒドロキシルが存在する。
【0108】
1.2.4 合成21および22塩基RNAは、標的RNA切断を媒介する
dsRNAプロセシングの産物の分析から、RNaseIII切断反応のあらゆる特徴を有する反応によって約21断片が生成されることがわかった(Dunn, 1982;Nicholson, 1999;Robertson, 1990;Robertson, 1982)。RNaseIIIは、dsRNAの両鎖に2つの付着切断を形成し、約2塩基の3’突出部を残す。本発明者は、クローン化した約21塩基断片のいくつかと配列が同じである21および22塩基のRNAを化学的に合成し、標的RNA分解を媒介する能力についてそれらを試験した(図5Aおよび5B)。21および22塩基のRNA二本鎖を、溶解物中100nMの濃度(52bpの対照dsRNAの10倍の濃度)でインキュベートした。これらの条件下で、標的RNA切断は容易に検出可能である。21および22塩基二本鎖の濃度を100から10nMまで下げても、やはり標的RNA切断が起こる。しかし、二本鎖の濃度を100から1,000nMに高めても、標的切断は増加しない。これは恐らく、溶解物内の制限タンパク質因子によるものであろう。
【0109】
RNAiを媒介しなかった29または30bpのdsRNAとは対照的に、2〜4塩基の突出3’末端を有する21および22塩基dsRNAは、標的RNAの有効な分解を媒介した(二本鎖1、3、4、6:図5Aおよび5B)。平滑末端21または22塩基dsRNA(二本鎖2、5および7:図5Aおよび5B)は、標的を分解する能力が低かったが、これは、突出3’末端がRNA−タンパク質ヌクレアーゼ複合体の再構成に重要であることを意味している。約21塩基の二本鎖とタンパク質成分の高親和結合には、一本鎖突出部が必要であると考えられる。5’末端リン酸は、dsRNAプロセシング後も存在するが、標的RNA切断を媒介するのに必要ではなく、短い合成RNAからは欠如していた。
【0110】
合成21および22塩基二本鎖は、短い二本鎖が占める領域内で、センスおよびアンチセンス標的の切断を指示した。これは、約21塩基断片の2対のクラスターを形成する39bpのdsRNA(図2)が、センスまたはアンチセンス標的を2回ではなく1回しか切断しなかったことを考慮すると、重要な結果である。本発明者は、この結果から、約21塩基二本鎖に存在する二本鎖の一方だけが標的RNA切断を指示できること、また、ヌクレアーゼ複合体における約21塩基二本鎖の配向がdsRNAプロセシングの最初の方向によって決定されることを示唆していると解釈した。しかし、すでに完全にプロセシングされた約21塩基の二本鎖をin vitro系に提示すれば、対称的なRNA二本鎖の可能性ある2つの配向で活性配列−特異的ヌクレアーゼ複合体を形成することできる。この結果、21塩基のRNA二本鎖と同一性領域内でセンスおよびアンチセンス標的の切断が達成される。
【0111】
標的切断部位は、21または22塩基のガイド(指示)配列と相補的な第1ヌクレオチドから11または12塩基下流に位置する。すなわち、切断部位は、21または22塩基のRNAが占める領域の中心付近にある(図4Aおよび4B)。22塩基二本鎖のセンス鎖を2塩基ずつ移動すると(図5Aの二本鎖1および3を比較)、アンチセンス標的のみの切断部位が2塩基ずつ移動した。センスおよびアンチセンス鎖を2塩基ずつ移動すると、両方の切断部が2塩基ずつシフトした(二本鎖1および4を比較)。本発明者は、ほぼすべての位置で標的RNAを切断する1対の21または22塩基のRNAを設計することが可能であると推定した。
【0112】
異常な切断部位が検出されないことから、21および22塩基のRNAにより指示される標的RNA切断の特異性は、正確であると考えられる(図5B)。しかし、注意すべきは、21および22塩基のRNA二本鎖の3’突出部に存在するヌクレオチドは、切断部位付近のヌクレオチドと比べて、基質認識に寄与しない可能性があることである。これは、活性二本鎖1または3(図5A)の3’突出部における3’末端(most)ヌクレオチドが標的と相補的ではないという観察結果に基づいている。RNAiの特異性についての詳しい分析は、合成21および22塩基RNAを用いて、容易に実施することができる。
【0113】
突出3’末端を有する合成21および22塩基RNAがRNA干渉を媒介するという証拠に基づいて、本発明者は、約21塩基のRNAを「短鎖の干渉RNA」またはsiRNAと、また、それぞれのRNA−タンパク質複合体を「短い干渉リボ核タンパク質粒子」またはsiRNPと呼称することを提案する。
【0114】
1.2.5 短鎖dsRNA上の20塩基の3’突出部がRNAiを阻害する
本発明者は、短い平滑末端dsRNAが、dsRNAの末端からプロセシングされることを明らかにした。RNAiにおけるdsRNAの長さ依存の研究中に、本発明者が、17〜20塩基の突出3’末端を有するdsRNAも分析したところ、驚くべきことに、これらが、平滑末端dsRNAより効力が弱いことがわかった。長い3’末端の阻害効果は、100bpまでのdsRNAに特に顕著であったが、これより長いdsRNAについてはそれほど強くなかった。未改変ゲル分析(データは示していない)によれば、この効果は、不完全なdsRNA形成によるものではなかった。本発明者は、長い突出3’末端の阻害効果を、短いRNA二本鎖の2つの末端の一方だけに対するdsRNAプロセシングを指令するツールとして使用できるかどうかを試験した。
【0115】
52bpのモデルdsRNAを、平滑末端のもの、センス鎖だけに3’延長部分を有するもの、アンチセンス鎖だけに3’延長部分を有するもの、ならびに、両鎖に二重3’延長部分を有するもの、の4つの組合せで合成し、溶解物中でのインキュベーション後、標的RNA切断部位をマッピングした(図6Aおよび6B)。二本鎖のアンチセンス鎖の3’末端が延長されている場合には、センス標的の第1および優勢切断部位が失われ、その逆もまた同じで、二本鎖のセンス鎖の3’末端が延長された場合には、アンチセンス標的の強力な切断部位が失われた。両鎖の3’延長部分によって、52bpのdsRNAが実質的に不活性になった。約20塩基の3’延長部分によるdsRNA不活性化を説明するものとして、この末端におけるdsRNAプロセシング因子の1つの会合を妨害する一本鎖RNA結合タンパク質の会合が考えられる。この結果は、集合siRNPにおけるsiRNA二本鎖の鎖の一方だけが、標的RNA切断を指示することができる、本発明者のモデルとも一致する。RNA切断を指示する鎖の配向は、dsRNAプロセシング反応の方向によって定められる。3’付着末端の存在が、プロセシング複合体の集合を促進していると考えられる。センス鎖の3’末端のブロッキングによってのみ、アンチセンス鎖の向き合う3’末端からのdsRNAプロセシングが可能となる。これが、今度は、siRNA二本鎖のアンチセンス鎖だけが、センス標的RNA切断を指示することができるsiRNP複合体を生成する。同じことが相互の状況で言える。
【0116】
長鎖のdsRNA(500bp以上、データは示していない)の場合に、長い3’延長部分の阻害効果がそれほど高くないのは、長鎖dsRNAも内部dsRNAプロセシングシグナルを含む、あるいは、複数の切断因子の会合により、協働してプロセシングされる可能性があることを示唆している。
【0117】
1.2.6 dsRNA指令によるmRNA切断のモデル
新しい生化学データを用いて、どのようにしてdsRNAがmRNAをターゲッティングし、破壊するかについてのモデルを更新する(図7)。二本鎖RNAはまず、主に21および22塩基長で、かつ、RNaseIII様反応と類似した付着3’末端を有する短いRNA二本鎖にプロセシングされる(Dunn, 1982;Nicholson, 1999;Robertson, 1982)。プロセシングされたRNA断片の21〜23塩基長に基づき、RNaseIII様活性がRNAiに関与し得ることがすでに想定されていた(Bass, 2000)。この仮定は、RNaseIII反応産物に観察されるように、siRNAの末端に5’リン酸および3’ヒドロキシルが存在することにより支持される(Dunn, 1982;Nicholson, 1999)。細菌RNaseIII、S.セレビシエにおける真核生物相同体Rnt1p、およびS.ポンベにおけるPac1pは、リボソームRNA、ならびに、snRNAおよびsnoRNAのプロセシングにおいて機能することがわかっている(例えば、Chanfreauら、2000参照)。
【0118】
植物、動物またはヒト由来のRNaseIII相同体の生化学についてはほとんどわかっていない。2つのファミリーのRNaseIII酵素が、主にデータベースを利用した配列分析またはcDNAのクローニングにより同定されている。第1のRNaseIIIファミリーの代表は、1327アミノ酸長のキイロショウジョウバエ(D.melanogaster)タンパク質droshaである(アクセッション番号AF116572)。C末端は、2つのRNaseIIIと1つのdsRNA結合ドメインから構成され、N末端の機能は未知である。酷似した相同体が、線虫(C.elegans)(アクセッション番号AF160248)およびヒト(アクセッション番号AF189011)でもみいだされている(Filippovら、2000;Wuら、2000)。drosha様ヒトRNaseIIIは、近年クローン化され、特性決定された(Wuら、2000)。遺伝子はヒト組織および細胞系において偏在的に発現し、タンパク質は細胞の核および核小体に局在化する。アンチセンス阻害に関する研究から推定される結果により、rRNAプロセシングにおけるこのタンパク質の役割が示唆された。第2のクラスの代表は、1822アミノ酸長のタンパク質をコードする線虫遺伝子K12H4.8(アクセッション番号S44849)である。このタンパク質は、N末端RNAヘリカーゼモチーフ、続いて、drosha RNaseIIIファミリーと類似した2つのRNaseIII触媒ドメインおよびdsRNA結合モチーフを有する。S.ポンベ(アクセッション番号Q09884)、シロイヌナズナ(アクセッション番号AF187317)、キイロショウジョウバエ(アクセッション番号AE003740)、およびヒト(アクセッション番号AB028449)にも酷似した相同体がある(Filippovら、2000;Jacobsenら、1999;Matsudaら、2000)。恐らく、K12H4.8 RNaseIII/ヘリカーゼは、RNAiに関与すると考えられる候補であろう。
【0119】
線虫における遺伝子スクリーニングにより、トランスポソン可動化または共抑制に影響を及ぼすことなくRNAiを活性化するのにrde−1およびrde−4が不可欠であると確認された(Dernburgら、2000;Grishokら、2000;KettingおよびPlasterk, 2000;Tabaraら、1999)。このことから、これらの遺伝子が、dsRNAプロセシングに重要であるが、mRNA標的分解には関与しないという仮定が導かれる。両遺伝子の機能はいまだに未知であり、rde−1遺伝子産物は、ウサギタンパク質elF2Cと類似したタンパク質ファミリーメンバーであり(Tabaraら、1999)、rde−4の配列はまだ記載されていない。これらタンパク質の将来の生化学的特性決定によって、これらの分子的機能が明らかにされるに相違ない。
【0120】
siRNA二本鎖へのプロセシングは、平滑末端のdsRNAまたは短い(1〜5塩基)3’突出部を有するdsRNAの末端から開始するようであり、約21〜23塩基ずつ進行する。短鎖dsRNA上の長い(約20塩基)3’付着末端は、恐らく一本鎖RNA結合タンパク質との相互作用によって、RNAiを抑制する。短鎖dsRNAと隣接する一本鎖領域によるRNAiの抑制と、30bpの短いdsRNAからのsiRNA形成の欠如が、mRNAで頻繁に出会う構築領域(structured regions)がRNAiの活性化を起こさない理由を説明しうる。
【0121】
特定の理論に拘束されるわけではないが、本発明者は、dsRNAプロセシングタンパク質またはこれらのサブセットが、プロセシング反応後もsiRNA二本鎖と会合したままであると推定する。これらタンパク質に対するsiRNA二本鎖の配向が、2つの相補鎖のどちらが、標的RNA分解を指示する上で機能するかを決定する。化学的に合成されたsiRNA二本鎖は、考えられる2つの配向のいずれかでタンパク質成分と会合することができるため、センスおよびアンチセンス標的RNAの切断を指示する。
【0122】
21および22塩基の合成siRNA二本鎖が効率的なmRNA分解に使用可能であるという注目すべき知見は、機能遺伝学および生物医学的研究における遺伝子発現の配列特異的調節に新しいツールを提供するものである。siRNAは、PKR応答の活性化のために、長いdsRNAを使用することができない哺乳動物系で有効となり得る(Clemens, 1997)。このように、siRNA二本鎖は、アンチセンスまたはリボザイム治療法に代わる新しい治療法を提供する。
【実施例2】
【0123】
ヒト組織培養物におけるRNA干渉
2.1 方法
2.1.1 RNA調製
Expedite RNAホスホルアミダイトとチミジンホスホルアミダイト(Proligo, Germany)を用いて、21塩基のRNAを化学的に合成した。合成オリゴヌクレオチドを脱保護してからゲル精製した(実施例1)後、Sep−Pak C18カートリッジ(Waters, Milford, MA, USA)により精製を実施した(Tuschl, 1993)。siRNA配列がターゲッティングするGL2(アクセッション番号X65324)とGL3ルシフェラーゼ(アクセッション番号U47296)は、開始コドンの第1ヌクレオチドに対してコード領域153〜173と対応し、siRNAがターゲッティングするRL(アクセッション番号AF025846)は、開始コドン後の領域119〜129と対応した。より長いRNAは、PCR産物からT7RNAポリメラーゼで転写した後、ゲルおよびSep−Pak精製に付した。49および484bpのGL2またはGL3dsRNAは、翻訳の開始に対して、位置113〜161および113〜596にそれぞれ対応した。50および501bpのRL dsRNAは、位置118〜167および118〜618にそれぞれ対応した。ヒト化GFP(hG)をターゲッティングするdsRNA合成のためのPCR鋳型をpAD3から増幅した(Kehlenbach, 1998)ところ、50および501bpのhG dsRNAは、翻訳の開始に対して、位置118〜167および118〜618にそれぞれ対応した。
【0124】
siRNAのアニーリングのために、20μMの一本鎖をアニーリングバッファー(100mM酢酸カリウム、30mM HEPES−KOH(pH7.4)、2mM酢酸マグネシウム)において90℃で1分インキュベートした後、37℃で1時間続けた。37℃のインキュベーションステップを50および500bpのdsRNAについて一晩延長し、これらのアニーリング反応を8.4μMおよび0.84μM鎖濃度でそれぞれ実施した。
【0125】
2.1.2 細胞培養
10%FBS、100単位/mlペニシリンおよび100μg/mlストレプトマイシンを補充したシュナイダーのショウジョウバエ培地(Life Technologies)において25℃で、S2細胞を増殖させた。10%FBS、100単位/mlペニシリンおよび100μg/mlストレプトマイシンを補充したダルベッコの改変イーグル培地において37℃で、293、NIH/3T3、HeLaS3、COS−7細胞を増殖させた。細胞を定期的に継代させて、対数増殖を維持した。約80%密集度でのトランスフェクションの24時間前に、哺乳動物細胞をトリプシン処理してから、抗生物質を含まない新鮮な培地で5倍に希釈した(1〜3×105細胞/ml)後、24ウェルプレート(500μl/ウェル)に移した。S2細胞はトリプシン処理せずに開裂反応に付した。付着細胞系について製造者が記載しているように、リポフェクトアミン2000試薬(Life Technologies)を用いて、トランスフェクションを実施した。1ウェルにつき、リポソームに組み込まれた1.0μgのpGL2−Control(Promega)またはpGL3−Control(Promega)、0.1μgのpRL−TK(Promega)および0.28μgのsiRNA二本鎖またはdsRNAを導入した;最終量は、1ウェル当たり600μlであった。トランスフェクションの後、細胞を20時間インキュベートしたが、その後の細胞は健常のようであった。次に、二重ルシフェラーゼアッセイ(Promega)で、ルシフェラーゼ発現をモニタリングした。1.1μgのhGFPコード化pAD3と0.28μgのinvGL2 inGL2 siRNAの共トランスフェクション後、哺乳動物細胞系の蛍光顕微鏡検査により、トランスフェクション効率を測定したところ、70〜90%であった。リポータープラスミドをXL−1 Blue(Stratagene)において増幅した後、Qiagenエンドフリー・マキシ・プラスミドキット(Qiagen EndoFree Maxi Plasmid Kit)を用いて精製した。
【0126】
2.2 結果と考察
siRNAが、組織培養物においてもRNAiを媒介することができるか否かを試験するために、本発明者は、ウミシイタケ(Renilla reniformis)および、ホタルルシフェラーゼ(Photinus pyralis、GL2およびGL3)の2つの配列種をコードするリポーター遺伝子に対して指令される対称2塩基3’突出部を有する21塩基siRNA二本鎖を合成した(図8a、b)。カチオンリポソームを用いて、siRNA二本鎖をリポータープラスミドの組合せpGL2/pRLまたはpGL3/pRLと一緒に、キイロショウジョウバエ・シュナイダーS2細胞または哺乳動物細胞に共トランスフェクトした。トランスフェクションから20時間後にルシフェラーゼ活性を測定した。試験したすべての細胞系において、本発明者は、相同siRNA二本鎖(cognate siRNA duplex)の存在下でリポーター遺伝子の発現の特異的減弱を観察した(図9a〜j)。驚くことに、絶対ルシフェラーゼ発現レベルは、非相同siRNAによる影響を受けなかった。これは、21塩基のRNA二本鎖(例えば、HeLa細胞については図10a〜d)による有害な副作用がないことを意味している。キイロショウジョウバエS2細胞(図9a、b)において、ルシフェラーゼの特異的阻害は完全であった。リポーター遺伝子が50〜100倍強く発現した哺乳動物細胞では、特異的抑制は完全ではなかった(図9c〜j)。相同siRNAに応答して、GL2発現は、3〜12倍、GL3発現は9〜25倍、またRL発現は1〜3倍減弱した。293細胞については、RL siRNAによるRLルシフェラーゼのターゲッティングは無効であったが、GL2およびGL3標的は、特異的に応答した(図9i、j)。293細胞でRL発現の減弱がないのは、この発現が、試験した他の哺乳動物細胞系と比べて5〜20倍高いこと、および/またはRNA二次構造もしくは会合タンパク質のために標的配列の受容能力が制限されたことによるものであろう。それでも、相同siRNA二本鎖によるGL2およびGL3ルシフェラーゼの特異的ターゲッティングは、RNAiが293細胞においても機能していることを示している。
【0127】
uGL2を除くすべてのsiRNA二本鎖における2塩基3’突出部は、(2’−デオキシ)チミジンから構成されていた。3’突出部における、チミジンによるウリジンの置換は、キイロショウジョウバエin vitro系で十分に許容されたため、突出部の配列は標的認識に重要ではなかった。組織培地中およびトランスフェクトした細胞内でsiRNAのヌクレアーゼ耐性を増強することが推定されるため、チミジン突出部を選択した。実際に、試験したすべての細胞系で、チミジン修飾GL2 siRNAは、非修飾uGL2 siRNAよりやや強力であった(図9a、c、e、g、i)。3’突出塩基をさらに修飾することにより、siRNA二本鎖の送達および安定性が改善されると考えられる。
【0128】
共トランスフェクション実験では、組織培地の最終量に対して25nMのsiRNA二本鎖を用いた(図9、10)。siRNAの濃度を100nMまで増加しても、特異的サイレンシング効果は増強されなかったが、プラスミドDNAとsiRNA間のリポソームのカプセル化(被包)競合により、トランスフェクション効率に影響を与え始めた(データは示していない)。siRNAの濃度を1.5nMまで低下しても、特異的サイレンシング効果は低下せず(データは示していない)、siRNA濃度がDNAプラスミドより2〜20倍高いだけでもそうであった。これは、siRNAが、遺伝子サイレンシングを媒介する極めて強力な試薬であること、また、siRNAが、通常のアンチセンスまたはリボザイム遺伝子ターゲッティング実験で使用される濃度より数桁低い濃度で有効であることを意味している。
【0129】
より長いdsRNAの哺乳動物細胞に対する効果をモニタリングするため、リポーター遺伝子に対して相同な50〜500bpのdsRNAを調製した。非特異的対照として、ヒト化GFP(hG)由来のdsRNA(Kehlenbach, 1998)を用いた。siRNA二本鎖と同じ量(濃度ではない)のdsRNAを共トランスフェクトすると、リポーター遺伝子発現は強力かつ非特異的に減弱した。この効果は、代表的例としてHeLa細胞について示する(図10a〜d)。絶対ルシフェラーゼ活性は、50bp dsRNAにより、10〜20倍、500bp dsRNA共トランスフェクションにより20〜200倍非特異的にそれぞれ低下した。同様の非特異的効果が、COS−7およびNIH/3T3細胞について観察された。293細胞では、10〜20倍の非特異的減弱は、500bp dsRNAによってしか観察されなかった。dsRNA>30bpによるリポーター遺伝子発現の非特異的減弱は、インターフェロン応答の一部として予想されたものだった。
【0130】
驚くことに、リポーター遺伝子発現の強い非特異的減弱にもかかわらず、本発明者は、別の配列特異的dsRNA媒介サイレンシングを再現的に検出した。しかし、特異的サイレンシング効果は、相対リポーター遺伝子活性をhG dsRNA対照に対して正規化したときしか明らかではなかった(図10e、f)。他の3つの被検哺乳動物細胞系においても、相同dsRNAに応答する2〜10倍の特異的減弱が観察された(データは示していない)。dsRNA(356〜1662bp)による特異的サイレンシング作用はCHO−K1細胞においてすでに報告されているが、2〜4倍の特異的減弱を検出するのに必要なdsRNAの量は、本発明者の実験(Ui-Tei、2000)より約20倍高かった。また、CHO−K1細胞は、インターフェロン応答が欠失しているようである。別の報告では、ルシフェラーゼ/lacZリポーターの組合せ、および829bp特異的lacZまたは717bp非特異的GFPdsRNAを用いて、293、NIH/3T3およびBHK−21細胞をRNAiについて試験した(Caplen、2000)。この例でRNAiを検出できなかったのは、ルシフェラーゼ/lacZリポーターアッセイの感受性が低かったことと、標的および対照dsRNAの長さが異なるためであろう。以上のことを考え合わせると、本発明者の結果から、RNAiが哺乳動物細胞において活性であるが、インターフェロン系がdsRNA>30bpにより活性化されると、サイレンシング効果の検出が難しくなることがわかる。
【0131】
要約すると、本発明者は、哺乳動物細胞におけるsiRNA媒介の遺伝子サイレンシングを最初に証明した。短鎖siRNAの使用により、ヒト組織培養物における遺伝子機能の不活性化、ならびに、遺伝子特異的治療法の開発の可能性が高まる。
【実施例3】
【0132】
RNA干渉による遺伝子発現の特異的阻害
3.1 材料と方法
3.1.1 RNA調製とRNAiアッセイ
実施例1または2、あるいは、これまでの文献(Tuschlら、1999;Zamoreら、2000)に記載されているように、化学的RNA合成、アニーリング、およびルシフェラーゼに基づくRNAiアッセイを実施した。すべてのsiRNA二本鎖はホタルルシフェラーゼに対して指令され、ルシフェラーゼmRNA配列は、記載されている(Tuschlら、1999)ように、pGEM−luc(GenBankアクセッション番号X65316)由来のものであった。キイロショウジョウバエRNAi/翻訳反応において、siRNA二本鎖を15分インキュベートしてから、mRNAを添加した。翻訳に基づくRNAiアッセイを少なくとも3回繰り返して実施した。
【0133】
センス標的RNA切断のマッピングのため、開始コドンに対して位置113から273までのホタルルシフェラーゼ配列と対応する177塩基転写物を作製した後、SP6プロモーター配列の17塩基相補体を作製した。アンチセンス標的RNA切断のマッピングのため、鋳型から166塩基転写物を作製し、5’プライマーTAATACGACTCACTATAGAGCCCATATCGTTTCATA(下線部はT7プロモーター)および3’プライマーAGAGGATGGAACCGCTGGを用いて、上記転写物をPCRによりプラスミド配列から増幅した。標的配列は、開始コドンに対して位置50から215までのホタルルシフェラーゼ配列の相補体と対応する。グアニリルトランスフェラーゼ標識を、すでに記載されている(Zamoreら、2000)ように実施した。標的RNA切断のマッピングのために、キイロショウジョウバエ胚溶解物において、100nMのsiRNA二本鎖を5〜10nM標的RNAと一緒に標準的条件(Zamoreら、2000)下で、25℃で2時間インキュベートした。8容量のプロテイナーゼKバッファー(200mM Tris−HCl(pH7.5)、25mM EDTA、300mM NaCl、2%w/vドデシル硫酸ナトリウム)を添加することにより反応を停止させた。プロテイナーゼK(E.M. Merck、水に溶解)を0.6mg/mlの最終濃度まで添加した。次に、反応を65℃で15分インキュベートしてから、フェノルール/クロロホルム/イソアミルアルコール(25:24:1)で抽出した後、3容量のエタノールで沈降させた。サンプルを6%配列決定用ゲルに載せた。長さ基準は、キャップ標識センスまたはアンチセンス標的RNAの部分的RNaseT1消化および部分的塩基加水分解反応によって作製した。
【0134】
3.2 結果
3.2.1 21塩基のsiRNAの二本鎖における3’突出部の変動
前述のように、siRNA二本鎖の3’末端で2または3非対合ヌクレオチドは、それぞれの平滑末端二本鎖と比べて、標的RNA分解が効率的である。末端ヌクレオチドの機能のさらに総合的分析を実施するために、本発明者は、5つの21塩基センスsiRNA(各々、標的RNAに対する1塩基で表される)と、8つの21塩基アンチセンスsiRNA(各々、標的に対する1塩基で表される)を合成した(図11A)。センスおよびアンチセンスsiRNAを組み合わせることにより、7塩基3’突出部〜4塩基5’突出部の範囲にある合成突出末端を有する8系統のsiRNA二本鎖を作製した。二重ルシフェラーゼアッセイ系(Tuschlら、1999;Zamoreら、2000)を用いて、siRNA二本鎖の干渉を測定した。siRNA二本鎖は、ホタルルシフェラーゼmRNAに対して指令され、ウミシイタケルシフェラーゼmRNAを内部対照として用いた。siRNA二本鎖の存在下で、標的と対照ルシフェラーゼ活性のルミネセンス比を測定した後、dsRNAの不在下で得られた比に対して正規化した。比較のため、長いdsRNA(39〜504bp)の干渉比を図11Bに示す。長鎖dsRNA(図11A)については5nM、また、siRNA二本鎖(図11C〜J)については100nMの濃度で、干渉比を測定した。5nMの504bp dsRNAの完全なプロセシングにより、120nMの全siRNA二本鎖が得られたことから、siRNAについては100nM濃度を選択した。
【0135】
21塩基のsiRNA二本鎖がRNAiを媒介する能力は、形成された突出ヌクレオチドまたは塩基対の数に応じて異なる。4〜6個の3’突出塩基を有する二本鎖では、RNAiを媒介することができなかった(図11C〜F)が、2個以上の5’突出塩基を有する二本鎖は媒介することができた(図11G〜J)。2塩基の3’突出部を有する二本鎖が、RNA干渉を媒介するのに最も効率的であったが、サイレンシングの効率はやはり配列依存的であり、2塩基の3’突出部を有する様々なsiRNA二本鎖について12倍の差が認められた(図11D〜Hを比較)。平滑末端、1塩基5’突出部または1〜3塩基3’突出部を有する二本鎖は、場合により機能的であった。7塩基の3’突出部を有するsiRNA二本鎖について観察されたわずかなサイレンシング効果(図11C)は、RNAiよりもむしろ長い3’突出部のアンチセンス効果によるものであろう。長鎖dsRNA(図11B)と、最も有効な21塩基のsiRNA二本鎖(図11E、G、H)をRNAiの効率について比較すると、100nM濃度の一本鎖siRNA二本鎖が、5nMの504bp dsRNAと同じくらい有効となり得ることがわかる。
【0136】
3.2.2 21塩基のアンチセンスsiRNAと対合するセンスsiRNAの長さ変動
RNAiに対するsiRNAの長さの影響を調べるために、本発明者は、3つの21塩基アンチセンス鎖と8つの18〜25塩基センス鎖とを組み合わせて、3系統のsiRNA二本鎖を作製した。アンチセンスsiRNAの3’突出部は、各siRNA二本鎖系統における1、2または3塩基に固定したのに対し、センスsiRNAは、その3’末端で変動させた(図12A)。センスsiRNAの長さとは無関係に、アンチセンスsiRNAの2塩基の3’突出部を有する二本鎖(図12C)は、1または3塩基の3’突出部を有するもの(図12B、D)より活性が高いことがわかった。アンチセンスsiRNAの1塩基の3’突出部を有する第1の系統では、センスsiRNAの1および2塩基の3’突出部をそれぞれ有する、21および22塩基センスsiRNAの二本鎖の活性が最も高かった。19〜25塩基のセンスsiRNAを有する二本鎖もRNAを媒介することができたが、程度は低かった。同様に、アンチセンスsiRNAの2塩基突出部を有する第2系統では、2塩基の3’突出部を有する21塩基のsiRNA二本鎖の活性が最も高く、18〜25塩基のセンスsiRNAとのその他すべての組合せは、有意な程度まで活性であった。3塩基のアンチセンスsiRNA3’突出部を有する最後の系統では、20塩基のセンスsiRNAおよび2塩基のセンス3’突出部を有する二本鎖だけが標的RNA発現を減弱することができた。以上、これらの結果から、siRNAの長さと共に、3’突出部の長さが重要であることと、2塩基3’突出部を有する21塩基のsiRNAの二本鎖がRNAiには最適であることがわかる。
【0137】
3.2.3 一定の2塩基3’突出部を有するsiRNA二本鎖の長さ変動
次に、本発明者は、対称の2塩基3’突出部を維持しながら、両siRNA鎖の長さを同時に変化させて生じる影響を調べた(図13A)。図11Hの21塩基のsiRNA二本鎖を基準として含む2系統のsiRNA二本鎖を作製した。センスsiRNA(図13B)の3’末端またはアンチセンスsiRNA(図13C)の3’末端で塩基対合部分を延長することにより、二本鎖の長さを20〜25bpで変動させた。20〜23bpの二本鎖は、標的ルシフェラーゼ活性の特異的抑制を起こしたが、21塩基のsiRNA二本鎖は、他の二本鎖のどちらと比較しても少なくとも8倍有効であった。24および25塩基のsiRNA二本鎖は、検出可能な干渉を全く起こさなかった。配列特異的効果は、二本鎖の両末端における変動が同様の効果を生じたため、わずかであった。
【0138】
3.2.4 2’−デオキシおよび2’−O−メチル修飾siRNA二本鎖
RNAiにおけるsiRNAリボース残基の重要性を評価するため、2’−デオキシまたは2’−O−メチル修飾鎖を含む21塩基のsiRNAおよび2塩基3’突出部を有する二本鎖を試験した(図14)。2’−デオキシヌクレオチドによる2塩基3’突出部の置換では影響がなく、対合領域における突出部と隣接した2つの別のリボヌクレオチドの置換でも、有意に活性のsiRNAが生じた。従って、siRNA二本鎖の42塩基のうち8つをDNA残基で置換しても、活性は失われなかった。2’−デオキシ残基による一方または両方のsiRNA鎖の完全な置換では、2’−O−メチル残基による置換と同様に、RNAiを破壊した。
【0139】
3.2.5 標的RNA切断部位の画定
22塩基のsiRNA二本鎖および21塩基/22塩基の二本鎖について、標的RNA切断位置を事前に決定した。標的RNA切断の位置は、siRNA二本鎖が占める領域の中心部、すなわち、21または22塩基のsiRNAガイド配列と相補的な第1塩基から11または12塩基下流に位置することがわかった。2塩基3’突出部を有する5つの別個の21塩基siRNA二本鎖(図15A)を5’キャップ標識センスまたはアンチセンス標的RNAと一緒にキイロショウジョウバエ溶解物中でインキュベートした(Tuschlら、1999;Zamoreら、2000)。5’切断産物を配列決定用ゲル上で分離した(図15B)。切断されたセンス標的RNAの量は、翻訳に基づくアッセイで決定されるsiRNA二本鎖の効率と相関しており、siRNA二本鎖1、2および4(図15Bおよび11H、G、E)は、二本鎖3および5(図15Bおよび11F、D)より速く標的RNAを切断する。注目すべきことに、5’切断産物および投入標的RNAの放射活性の合計は、時間に関して一定ではなく、5’切断産物は蓄積しなかった。恐らく、5’−キャップのポリ(A)テイルのいずれかが欠如しているために、siRNA−エンドヌクレアーゼ複合体から放出した切断産物は急速に分解されるのであろう。
【0140】
センスおよびアンチセンス標的RNAの両方で、切断部位は、siRNA二本鎖が占める領域の中央部に位置した。5つの異なる二本鎖により生成された各標的の切断部位は、標的配列に沿って、二本鎖の1塩基置換により1塩基ずつ変動した。標的は、配列相補的ガイドsiRNAの3’末端(most)ヌクレオチドと相補的な標的位置から正確に11塩基下流で切断された。
【0141】
ガイドsiRNAの5’または3’末端が、標的RNA切断のルーラー(ruler)を設定するか否かを決定するために、本発明者は、図16AおよびBに概要を示す実験戦略を考案した。21塩基アンチセンスsiRNAは、この実験では不変に維持し、5’または3’末端のいずれかにおいて修飾されたセンスsiRNAと対合させた。センスおよびアンチセンス標的RNA切断の位置は前記のように決定した。センスsiRNAの3’末端の変化を、1塩基5’突出部〜6塩基3’突出部でモニタリングしたが、これらはいずれも、センスまたはアンチセンス標的RNA切断の位置に影響を及ぼさなかった(図16C)。センスsiRNAの5’末端における変化は、センス標的RNA切断に影響を及ぼさなかった(図16D、上パネル)が、これは、アンチセンスsiRNAが変化しなかったため、予想されたことであった。しかし、アンチセンス標的RNA切断は影響を受け、センスsiRNAの5’末端に強く依存した(図16D、下パネル)。センスsiRNAサイズが20または21塩基であるとき、アンチセンス標的だけが切断され、切断の位置は1塩基ずつ異なった。このことは、標的を認識する5’末端が標的RNA切断のルーラー(ruler)を設定することを示唆している。この位置は、ガイドsiRNAの5’末端(most)塩基と対合した標的の塩基から上流方向に数えると、塩基10と11の間に位置する(図15Aも参照)。
【0142】
3.2.6 3’突出部における配列の影響および2’−デオキシ置換
2塩基の3’突出部が、siRNA機能には好ましい。本発明者は、突出塩基の配列が、標的認識に寄与するのか、またはこの配列が、エンドヌクレアーゼ複合体(RISCまたはsiRNP)の認識に必要な特徴であるだけなのかを知ろうとした。本発明者は、AA、CC、GG、UUおよびUG3’突出部を有するセンスおよびアンチセンスsiRNAを合成し、2’−デオキシ修飾TdGおよびTTを含有させた。野生型siRNAは、センス3’突出部にAAを、また、アンチセンス3’突出部にUGを含んでいた(AA/UG)。siRNA二本鎖はすべて、干渉アッセイにおいて機能的であり、標的発現を少なくとも5倍減弱した(図17)。10倍以上標的発現を減弱した最も効率的なsiRNA二本鎖は、配列型:NN/UG、NN/UU、NN/TdG、およびNN/TT(Nは任意のヌクレオチドである)のものであった。AA、CCまたはGGのアンチセンスsiRNA3’突出部を有するsiRNA二本鎖は、野生型配列UGまたは突然変異配列UUと比較して、2〜4倍活性が低かった。RNAi効率が低いのは、恐らく、末端から2番目の3’塩基が配列特異的標識認識に寄与するためと考えられる。というのは、3’末端塩基は、GからUに改変しても、影響がないからである。
【0143】
センスsiRNAの3’突出部の配列を改変しても、配列に依存する影響は一切明らかにされなかったが、これは、センスsiRNAはセンス標的mRNA認識に寄与してはならないため、予想されたことだった。
【0144】
3.2.7 標的認識の配列特異性
標的認識の配列特異性を調べるため、本発明者は、siRNA二本鎖の対合部分に配列改変を導入し、サイレンシングの効率を調べた。配列改変は、3または4塩基長の短い部分を逆転させるか、あるいは、点突然変異として導入した(図18)。一方のsiRNA鎖の配列改変は、相補的siRNA鎖において補償され、これによって、塩基対合したsiRNA二本鎖構造の不安定化(pertubing)を回避した。合成にかかるコストを下げるため、2塩基の3’突出部の配列は、すべてTT(T、2’−デオキシチミジン)とした。TT/TT基準siRNA二本鎖は、RNAiについて、野生型siRNA二本鎖AA/UGと同等であった(図17)。リポーターmRNA破壊を媒介する能力は、翻訳に基づくルミネセンスアッセイを用いて定量化した。逆転配列部分有するsiRNAの二本鎖は、ホタルルシフェラーゼリポーターをターゲッティングする能力が著しく低いことがわかった(図18)。アンチセンスsiRNAの3’末端と中央部との間に位置する配列改変は、標的RNA認識を完全に破壊したが、アンチセンスsiRNAの5’末端付近の突然変異は、わずかな程度のサイレンシングを呈示する。推定標的RNA切断部位の反対側に直接、または推定部位から1塩基離れて位置するA/U塩基対のトランスバージョンによって、標的RNA切断が阻止されるが、これは、siRNA二本鎖の中心部内の単一突然変異が、ミスマッチ標的を識別することを示している。
【0145】
3.3 論考
siRNAは、昆虫細胞だけではなく、哺乳動物細胞においても、遺伝子発現を不活性化するための有用な試薬であり、治療用途に極めて有望である。本発明者は、キイロショウジョウバエ胚溶解物において効率的な標的RNA分解を促進するのに必要なsiRNA二本鎖の構造上の決定因子を系統的に分析することにより、最も効力のあるsiRNA二本鎖の設計についての法則を提供した。完全なsiRNA二本鎖は、全RNAの同等量を用いれば、500bpのdsRNAと同等の効率で、遺伝子発現をサイレンシングすることができる。
【0146】
3.4 siRNAユーザーガイド
効率的なサイレンシングsiRNA二本鎖は、21塩基のアンチセンスsiRNAから構成されるため、2塩基3’突出末端を有する19bp二重らせんを形成するように選択しなければならない。2塩基3’突出リボヌクレオチドの2’−デオキシ置換は、RNAiに影響を及ぼさないが、RNA合成にかかるコスト削減に役立ち、siRNA二本鎖のRNase抵抗を増強することができる。ところが、これより広範囲な2’−デオキシまたは2’−O−メチル修飾は、恐らく、siRNAP集合のためのタンパク質会合を妨害するために、siRNAがRNAiを媒介する能力を低下させる。
【0147】
標的認識は、標的に相補的なsiRNAにより媒介される、高度に配列特異的過程である。ガイドsiRNAの3’末端(most)塩基は、標的認識の特異性に寄与しないのに対し、3’突出部から2番目の塩基は、標的RNA切断に影響を与え、また、ミスマッチはRNAiを1/2〜1/4倍に低下させる。ガイドsiRNAの5’末端もまた、3’末端と比べて、ミスマッチ標的RNA認識を広く許容するようである。標的RNA切断部位と向き合って位置する、siRNAの中心のヌクレオチドは、重要な特異性決定因子であり、ただ1つの塩基改変でも、RNAiは検出不可能なレベルまで低下する。これは、siRNA二本鎖が、遺伝子ターゲッティング実験において突然変異体または多型対立遺伝子を区別することができることを示唆しており、このことは、将来の治療法開発にとって重要な特徴となるであろう。
【0148】
センスおよびアンチセンスsiRNAは、エンドヌクレアーゼ複合体またはその拘束(commitment)複合体のタンパク質成分と会合すると、個別の役割を果たすことが示唆されている。この複合体におけるsiRNA二本鎖の相対配向が、どの鎖を標的認識に用いることができるかを決定する。合成siRNA二本鎖は、二重らせん構造に対して二回転対称を有するが、配列に対してはそうではない。キイロショウジョウバエ溶解物におけるRNAiタンパク質とsiRNA二本鎖との会合により、2つの非対称複合体が形成される。このような推定上の複合体では、センスおよびアンチセンスsiRNA、従って、それらの機能について、キラル環境がそれぞれ異なる。この推定は、明らかにパリンドロームのsiRNA配列、またはホモ二量体として会合できるRNAiタンパク質には適用されない。センスおよびアンチセンスターゲッティングsiRNPの比に及ぼす可能性のある配列の影響を最小限にするため、本発明者は、同一の3’突出部配列を有するsiRNA配列を用いることを提案する。本発明者は、センスsiRNAの突出部の配列を、アンチセンス3’突出部の配列に対して調節するよう勧める。なぜなら、センスsiRNAは、典型的ノックダウン実験には標的をもたないからである。センスおよびアンチセンス切断siRNPの再構成における非対称性は、この実験(図14)で用いた2塩基3’突出部を有する様々な21塩基のsiRNA二本鎖について観察したRNAi効率の変動に(部分的に)起因すると考えられる。あるいは、標的部位の塩基配列および/または標的RNA構造の受容能力は、これらsiRNA二本鎖の効率の変動によるものと考えられる。
【0149】
参照文献
【技術分野】
【0001】
本発明は、RNA干渉および/またはDNAメチル化などの標的特異的な核酸改変を媒介するのに必要な二本鎖(ds)RNAの配列および構造的特徴に関する。
【背景技術】
【0002】
「RNA干渉(RNAインターフェアランス)」(RNAi)は、線虫(C.elegans)にdsRNAを注入すると、送達したdsRNAと配列が高度に相同的な遺伝子の特異的サイレンシングが起こるという発見後、造り出された用語である(Fireら、1998)。その後、RNAiは、昆虫、カエル(Oelgeschlagerら、2000)、ならびに、マウスを含むその他の動物(Svobodaら、2000;WiannyおよびZernicka-Goetz, 2000)でも観察されており、ヒトにも存在すると考えられる。RNAiは、植物における共抑制および真菌における抑制の転写後遺伝子サイレンシング(PTGS)機構と密接に関連しており(Catalanottoら、2000;CogoniおよびMacino, 1999;Dalmayら、2000;KettingおよびPlasterk, 2000;Mourrainら、2000;Smardonら、2000)、また、RNAi機構の構成要素には、共抑制による転写後サイレンシングに必要なものもある(Catalanottoら、2000;Dernburgら、2000;KettingおよびPlasterk, 2000)。これについては、近年再考されている(Bass, 2000;BosherおよびLabouesse, 2000;Fire, 1999;PlasterkおよびKetting, 2000;Sharp, 1999;SijenおよびKooter, 2000)。また、Plant Molecular Biology、第43巻、2/3号(2000)全体も参照されたい。
【0003】
植物では、PTGSに加えて、導入されたトランスジーンも、シトシンのRNA指令DNAメチル化を介した転写遺伝子サイレンシングを誘導することができる(Wassenegger, 2000の文献を参照)。植物では、30bpという短いゲノム標的がRNA指令方式でメチル化される(Pelissier, 2000)。DNAメチル化は哺乳動物にも存在する。
【0004】
RNAiおよび共抑制の天然の機能は、活性となったとき、宿主細胞に異常RNAまたはdsRNAを産生するレトロトランスポゾンやウイルスなどの可動性遺伝的エレメントによる侵入に対してゲノムを保護することであると考えられる(Jensenら、1999;Kettingら、1999;Ratcliffら、1999;Tabaraら、1999)。特異的mRNA分解により、トランスポゾンおよびウイルスの複製が阻止されるが、PTGSを抑制するタンパク質を発現させることにより、この過程を克服または阻止することができる(Lucyら、2000;Voinnetら、2000)。
【0005】
dsRNAは、dsRNAとの同一性の領域内でしか相同性RNAの特異的分解を誘発しない(Zamoreら、2000)。dsRNAは21〜23塩基のRNA断片にプロセシングされ、標的RNA切断部位は、通常、21〜23塩基の間隔である。従って、21〜23塩基の断片が、標的認識のためのガイド(guide)RNAであると考えられてきた(Zamoreら、2000)。これらの短いRNAは、細胞溶解の前にdsRNAでトランスフェクトしたキイロショウジョウバエシュナイダー2細胞から調製した抽出物でも検出されている(Hammondら、2000)が、配列特異的ヌクレアーゼ活性を呈示する画分も、残留dsRNAの大きな画分を含んでいた。mRNA切断の指示における21〜23塩基断片の役割は、プロセシングされたdsRNAから単離された21〜23塩基断片が、特異的mRNA分解をある程度まで媒介することができるという研究結果によってさらに支持される(Zamoreら、2000)。類似サイズのRNA分子は、PTGSを示す植物組織にも蓄積する(HamiltonおよびBaulcombe, 1999)。
【発明の概要】
【発明が解決しようとする課題】
【0006】
ここで、本発明者は、確立されたショウジョウバエin vitro系(Tuschlら、1999;Zamoreら、2000)を用いて、RNAiの機構をさらに探究した。本発明者は、21および22塩基の短いRNAが、3’突出末端と塩基対合すると、配列特異的mRNA分解のガイドRNAとして作用することを証明する。30bpの短いdsRNAは、もはや21および22塩基のRNAにプロセシングされることはないため、この系でRNAiを媒介することはできない。さらに、本発明者は、21および22塩基という短鎖の干渉作用を有するRNA(siRNA)に対する標的RNA切断部位を特定し、dsRNAプロセシングの方向が、生成されるsiRNPエンドヌクレアーゼ複合体により、センスまたはアンチセンス標的RNAが切断されうるか否かを決定する証拠を提供する。siRNAはまた、転写調節、例えば、DNAメチル化を指示することによって哺乳動物遺伝子のサイレンシングのための重要なツールにもなりうる。
【0007】
ヒトin vivo細胞培養系(HeLa細胞)でさらに実験したところ、好ましくは19〜25塩基長の二本鎖RNA分子がRNAi活性を有することがわかった。従って、ショウジョウバエからの結果とは対照的に、24および25塩基長の二本鎖RNA分子もRNAiに有効である。
【0008】
本発明の目的は、標的特異的RNA干渉またはDNAメチル化のような他の標的特異的な核酸改変を媒介することができる新規の物質を提供することであり、この物質は、従来の技術による物質と比べて、効力および安全性が改善されている。
【課題を解決するための手段】
【0009】
上記課題は、各RNA鎖が19〜25、特に19〜23塩基長を有し、かつ、該RNA分子が、標的特異的な核酸改変、特にRNA干渉および/またはDNAメチル化を媒介することが可能な、単離された二本鎖RNA分子により解決される。少なくとも1本の鎖が、好ましくは1〜5塩基、さらに好ましくは1〜3塩基、最も好ましくは2塩基からなる3’突出部を有する。他方の鎖は、平滑末端であるか、あるいは、6塩基以下の3’突出部を有する。また、dsRNAの両鎖が厳密に21または22塩基の場合には、両末端が平滑である(0塩基の突出部)とき、ある程度のRNA干渉を観察することができる。RNA分子は、細胞抽出物中に存在する不純物、例えば、ショウジョウバエ胚を実質的に含まない合成RNA分子であることが好ましい。さらに、RNA分子は、好ましくは、非標的特異的不純物、特に非標的特異的RNA分子、例えば、細胞抽出物中に存在する不純物を実質的に含まない。
【0010】
さらには、本発明は、哺乳動物細胞、特にヒト細胞における標的特異的な核酸改変、特にRNAiを媒介することを目的とした、各RNA鎖が19〜25塩基長を有する単離された二本鎖RNA分子の使用に関する。
【0011】
具体的には、本発明は、以下の特徴を有する。
〔1〕単離された二本鎖RNA分子であって、各鎖が39〜52塩基長を有し、該RNA分子は標的特異的なRNA干渉が可能な1以上の二本鎖断片にプロセシングされることができ、該RNA分子は、第1の鎖が予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が該第1の鎖に相補的な配列からなる、上記RNA分子。
〔2〕鎖が39塩基長を有する、上記〔1〕に記載のRNA分子。
〔3〕単離された二本鎖RNA分子であって、該RNA分子は標的特異的なRNA干渉が可能なものであり、第1の鎖が19〜25塩基長を有し、予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が25塩基長を有する、上記RNA分子。
〔4〕なくとも1つの修飾されたリボヌクレオチドを含む、上記〔1〕〜〔3〕のいずれか1つに記載のRNA分子。
〔5〕修飾リボヌクレオチドが、糖、骨格鎖または核酸塩基修飾リボヌクレオチドから選択される、上記〔4〕に記載のRNA分子。
〔6〕修飾リボヌクレオチドが、糖修飾リボヌクレオチドであり、2’−OH基が、H、OR、R、ハロ、SH、SR1、NH2、NHR、NR2またはCNから選択される基で置換され、Rは、C1−C6アルキル、アルケニルまたはアルキニルであり、ハロは、F、Cl、BrまたはIである、上記〔5〕に記載のRNA分子。
〔7〕飾リボヌクレオチドが、ホスホロチオエート基を含む骨格鎖修飾リボヌクレオチドである、上記〔5〕に記載のRNA分子。
〔8〕前記RNA分子の第1の鎖が、予め決定したmRNA標的分子に対して、少なくとも85%の同一性を有する配列からなる、上記〔1〕〜〔7〕のいずれか1つに記載のRNA分子。
〔9〕下記のステップを含む、請求項1〜8のいずれか1項に記載の二本鎖RNA分子の作製方法:
(a)各々が39〜52または19〜25塩基長を有する2本のRNA鎖を合成するステップであって、該RNA鎖は二本鎖RNA分子を形成することができるものである、上記ステップ、
(b)二本鎖RNA分子が形成される条件下で合成RNA鎖を結合させるステップであって、得られる二本鎖RNA分子は標的特異的なRNA干渉が可能なものである、上記ステップ。
〔10〕RNA鎖が化学的に合成される、上記〔9〕に記載の方法。
〔11〕RNA鎖が酵素により合成される、上記〔9〕に記載の方法。
〔12〕下記のステップを含む、動物細胞において標的特異的なRNA干渉を媒介する方法:
(a)標的特異的なRNA干渉が起こりうる条件下で、上記細胞を請求項1〜8のいずれか1項に記載の二本鎖RNA分子と接触させるステップ、および
(b)上記二本鎖RNAと同一の配列部分を有する標的核酸に対する、上記二本鎖RNAにより引き起こされる標的特異的なRNA干渉を媒介するステップ。
〔13〕接触ステップが、二本鎖RNA分子を標的細胞に導入し、そこで標的特異的なRNA干渉を起こさせうるステップを含む、上記〔12〕に記載の方法。
〔14〕導入ステップが、キャリア媒介による送達または注射を含む、上記〔13〕に記載の方法。
〔15〕動物細胞における遺伝子の機能を決定するための、上記〔12〕〜〔14〕のいずれか1つに記載の方法の使用。
〔16〕動物細胞における遺伝子の機能を抑制するための、上記〔12〕〜〔14〕のいずれか1つに記載の方法の使用。
〔17〕遺伝子が病理的状態と関連する、上記〔15〕または〔16〕に記載の使用。
〔18〕遺伝子が病原体関連遺伝子である、上記〔17〕に記載の使用。
〔19〕遺伝子がウイルス遺伝子である、上記〔18〕に記載の使用。
〔20〕遺伝子が腫瘍関連遺伝子である、上記〔17〕に記載の使用。
〔21〕遺伝子が自己免疫疾患関連遺伝子である、上記〔17〕に記載の使用。
〔22〕標的遺伝子特異的ノックアウト表現型を示す動物細胞であって、該細胞が、内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子、または少なくとも1つの内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子をコードするDNAでトランスフェクトされており、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定されたmRNA標的分子に対して100%の同一性を有する配列からなる、上記細胞。
〔23〕哺乳動物細胞である、上記〔22〕に記載の細胞。
〔24〕ヒト細胞である、上記〔23〕に記載の細胞。
〔25〕標的タンパク質、または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸でさらにトランスフェクトされた、上記〔22〕〜〔24〕のいずれか1つに記載の細胞であって、該外因性標的核酸は、二本鎖RNA分子による該外因性標的核酸の発現の阻害が、内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記細胞。
〔26〕外因性標的核酸が、検出可能なペプチドまたはポリペプチドをコードするさらなる核酸配列と融合されている、上記〔25〕に記載の細胞。
〔27〕分析手法のための、上記〔22〕〜〔26〕のいずれか1つに記載の細胞の使用。
〔28〕遺伝子発現プロフィールを分析するための、上記〔27〕に記載の使用。
〔29〕プロテオーム分析のための、上記〔27〕に記載の使用。
〔30〕外因性標的核酸によってコードされる標識タンパク質の変異体または突然変異形態の分析が実施される、上記〔27〕〜〔29〕のいずれか1つに記載の使用。
〔31〕標的タンパク質の機能ドメインを同定するための、上記〔30〕に記載の使用。
〔32〕下記のものから選択される少なくとも2つの動物細胞の比較を実施する、上記〔27〕〜〔31〕のいずれか1つに記載の使用:
(i)標的遺伝子阻害を含まない対照細胞、
(ii)標的遺伝子阻害を含む細胞、および
(iii)標的遺伝子阻害と、外因性標的核酸による標的遺伝子の相補を含む細胞。
〔33〕分析が、機能および/または表現型の分析を含む、上記〔27〕〜〔32〕のいずれか1つに記載の使用。
〔34〕調製手法のための、上記〔22〕〜〔26〕のいずれか1つに記載の細胞の使用。
〔35〕真核細胞からタンパク質またはタンパク質複合体を単離するための、上記〔34〕に記載の使用。
〔36〕高分子量タンパク質複合体を単離するための、上記〔35〕に記載の使用。
〔37〕高分子量タンパク質複合体が核酸を含む、上記〔36〕に記載の使用。
〔38〕薬理学的物質を同定および/または特性決定する手法における、〔27〕〜〔37〕のいずれか1つに記載の使用。
〔39〕下記の(a)〜(c)を含む、少なくとも1つの標的タンパク質に作用する薬理学的物質の同定および/または特性決定システム:
(a)少なくとも1つの標的タンパク質をコードする少なくとも1つの標的遺伝子を発現することができる、動物細胞、
(b)上記少なくとも1つの内因性標的遺伝子の発現を阻害することができる、少なくとも1つの二本鎖RNA分子であって、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定したmRNA標的分子に対して100%の同一性を有する配列からなる、上記RNA分子、および
(c)薬理学的特性を同定および/または特性決定しようとする、試験物質または試験物質のコレクション。
〔40〕さらに下記の(d)を含む、上記〔39〕に記載のシステム:
(d)上記標的タンパク質または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸であって、該外因性標的核酸は、二本鎖RNA分子による発現の阻害が、上記内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記外因性標的核酸。
【図面の簡単な説明】
【0012】
【図1A】Pp−luc mRNAをターゲッティングするのに用いたdsRNAのグラフである。
【図1B】RNA干渉アッセイを示す図である。
【図2】ショウジョウバエ溶解物における内部32P標識dsRNA(5nM)のプロセシングからの21〜23mer形成の時間経過と、dsRNAの長さおよび供給源を示す図。
【図3A】5’切断産物の変性ゲル泳動を示す図である。
【図3B】センスおよびアンチセンス標的RNA上の切断部位の位置を示す図である。
【図4A】dsRNAプロセシング後の約21塩基RNAの配列を示す図である。
【図4B】約21塩基RNAのヌクレオチド組成の二次元TLCの分析を示す図である。
【図5A】対称52bpのdsRNAならびに合成21および22塩基のdsRNAをグラフで示す図である。
【図5B】センスおよびアンチセンス標的RNA上の切断部位の位置を示す図である。
【図6A】52bpのdsRNA構築物をグラフで示す図である。
【図6B】センスおよびアンチセンス標的RNA上の切断部位の位置を示す図である。
【図7】RNAiのモデル案を示す図である。
【図8a】プラスミドpGL2−Control、pGL−3−ControlおよびpRL−TK(Promega)からのホタル(Pp−luc)およびウミシイタケ(Rr−luc)ルシフェラーゼリポーター遺伝子領域を示す図である。
【図8b】GL2、GL3およびRLルシフェラーゼをターゲッティングするsiRNA二本鎖のセンス(上)およびアンチセンス(下)配列を示す図である。
【図9】siRNA二本鎖によるRNA干渉を示す図である。
【図10】pGL2−ControlとpRL−TKリポータープラスミドを用いて実施した実験を示す。
【図11】(A)実験戦略の概要を示す図である。(B)対照ルシフェラーゼに対する、標的ルシフェラーゼの正規化相対ルミネセンスを示す図である。(C〜J)8系統の21塩基のsiRNA二本鎖について正規化した干渉比を示す図である。
【図12】siRNA二本鎖のセンス鎖の長さ変動を示す図である。
【図13】保存された2塩基3’突出部を有するsiRNA二本鎖の長さ変動を示す図である。
【図14】siRNAリボース残基の2’−ヒドロキシ基の置換を示す図である。
【図15A】32P(星印)キャップ標識センスおよびアンチセンス標的RNAと、siRNA二本鎖をグラフで示す図である。
【図15B】標的RNA切断部位のマッピングを示す図である。
【図16】(AおよびB)実験戦略をグラフで示す図である。(CおよびD)キャップ標識センスまたはアンチセンス標的RNAを用いた標識RNA切断の分析を示す図である。
【図17】siRNA二本鎖の3’突出部の配列変動を示す図である。
【図18】標的認識の配列特異性を示す図である。
【図19】保存された2塩基3’突出部を有するsiRNA二本鎖の長さ変動を示す図である。
【発明を実施するための形態】
【0013】
驚くべきことに、特に、3’突出末端を有する短い合成二本鎖RNA分子が、RNAiの配列特異的媒介物質であり、有効な標的RNA切断を媒介し、その際、この切断部位は、短鎖RNAが指示(ガイド;guide)する範囲の領域の中心近くに位置することがわかった。
【0014】
RNA分子の各鎖は、20〜22塩基長(または哺乳動物細胞では20〜25塩基長)を有するのが好ましく、各鎖の長さは同じでも異なっていてもよい。好ましくは、3’突出部は、1〜3塩基の範囲にあり、この突出部の長さは、鎖によって同じでも異なっていてもよい。RNA鎖は、3’ヒドロキシル基を有するのが好ましい。5’末端は、好ましくは、リン酸、二リン酸、三リン酸またはヒドロキシル基を含む。最も有効なdsRNAは、1〜3塩基、特に2塩基の3’突出部がdsRNAの両端に存在するように、対合した2本の21塩基の鎖から構成される。
【0015】
siRNAにより指示される標的RNA切断反応は、高度に配列特異的である。しかし、siRNAの全ての位置が等しく標的認識に寄与するわけではない。siRNA二本鎖の中心におけるミスマッチが最も重要で、標的RNA切断をほとんど破壊する。これに対し、一本鎖標的RNAと相補的なsiRNA鎖の3’ヌクレオチド(例えば、位置21)は、標的認識の特異性に寄与しない。さらに、アンチセンスsiRNA鎖だけが標的認識を指示するため、標的RNAと同じ極性を有するsiRNAの非対合2塩基3’突出部の配列は、標的RNA切断に重要ではない。従って、一本鎖突出塩基(ヌクレオチド)から、アンチセンスsiRNAの最後から2番目の位置(例えば、位置20)だけが標的センスmRNAと一致する必要がある。
【0016】
驚くべきことに、本発明の二本鎖RNA分子は、血清または細胞培養用の増殖培地において高いin vivo安定性を呈示する。安定性をさらに高めるため、3’突出部を分解に対して安定化させる、例えば、これらが、プリンヌクレオチド、特にアデノシンまたはグアノシンヌクレオチドからなるように、選択することができる。あるいは、修飾類似体によるピリミジンヌクレオチドの置換、例えば、2’−デオキシチミジンによるウリジン2塩基の3’突出部の置換は許容されるものであり、RNA緩衝の効率に影響しない。また、2’ヒドロキシルの欠如により、組織培地中の突出部のヌクレアーゼ耐性が有意に増強される。
【0017】
本発明の特に好ましい実施形態では、RNA分子は、少なくとも1つの修飾ヌクレオチド類似体を含むことができる。ヌクレオチド類似体は、標的特異的活性、例えば、RNAi媒介活性が実質的に影響を受けない位置、例えば、二本鎖RNA分子の5’末端および/または3’末端の領域内に配置することができる。特に、修飾ヌクレオチド類似体を組み込むことにより、突出部を安定化させることができる。
【0018】
好ましいヌクレオチド類似体は、糖または骨格鎖修飾リボヌクレオチドから選択する。しかし、核酸塩基が修飾されたリボヌクレオチド、すなわち、天然に存在する核酸塩基ではなく、下記のように天然に存在しない核酸塩基を含むリボヌクレオチドも好適であることに留意すべきである:すなわち、天然に存在しない核酸塩基としては、5位置で修飾されたウリジンまたはシチジン、例えば、5−(2−アミノ)プロピルウリジン、5−ブロモウリジン;8位で修飾されたアデノシンおよびグアノシン、例えば、8−ブロモグアノシン;デアザヌクレオチド、例えば、7−デアザ−アデノシン;O−およびN−アルキル化ヌクレオチド、例えば、N6−メチルアデノシンなどである。好ましい糖修飾リボヌクレオチドでは、H、OR、ハロ、SH、SR、NH2、NHR、NR2またはCNからなる群より選択される基で2’OH基を置換するが、ここで、Rは、C1〜C6アルキル、アルケニルまたはアルキニルであり、ハロは、F、Cl、BrまたはIである。好ましい骨格鎖修飾リボヌクレオチドでは、隣接するリボヌクレオチドを結合するホスホエステル基を、例えば、ホスホチオエート基の修飾基で置換する。前述した修飾を組み合わせてもよいことに留意されたい。
【0019】
標的特異的RNAiおよび/またはDNAメチル化を媒介するためには、本発明の二本鎖RNA分子の配列は、核酸標的分子に対し十分な同一性を有していなければならない。好ましくは、この配列は、RNA分子の二本鎖部分における所望の標的分子に対して、少なくとも50%、特に少なくとも70%の同一性を有する。さらに好ましくは、同一性は、RNA分子の二本鎖部分において少なくとも85%、最も好ましくは100%である。予め決定した核酸標的分子、例えば、mRNA標的分子に対する二本鎖RNA分子の同一性は、下記のように決定することができる:
【数1】
【0020】
(式中、Iは、同一性のパーセンテージであり、nは、dsRNA分子の二本鎖部分における同一塩基数であり、Lは、dsRNAの二本鎖部分と標的との配列重複部分の長さである)。
【0021】
これ以外に、3’突出部、特に、1〜3塩基長を有する突出部を含む二本鎖RNA分子の標的配列に対する同一性も決定することができる。この場合、配列同一性は、標的配列に対して、好ましくは少なくとも50%、さらに好ましくは少なくとも70%、最も好ましくは少なくとも85%である。例えば、二本鎖の3’突出部からの塩基、5’および/または3’末端からの2以下の塩基は、活性を有意に失うことなく修飾することができる。
【0022】
本発明の二本鎖RNA分子は、下記のステップを含む方法によって作製することができる:
(a)各々が19〜25塩基長、例えば19〜23塩基長を有する2本のRNA鎖を合成するステップであって、このRNA鎖は二本鎖RNA分子を形成することができるものであり、好ましくは少なくとも1本が1〜5塩基の3’突出部を有する、上記ステップ、
(b)二本鎖RNA分子が形成される条件下で合成RNA鎖を結合させるステップであって、得られる二本鎖RNA分子は標的特異的な核酸改変、特にRNA干渉および/またはDNAメチル化を媒介することが可能なものである、上記ステップ。
【0023】
RNA分子を合成する方法は、当業者には公知である。本発明では、特に、VermaおよびEckstein(1998)に記載されている化学的合成方法を例示する。
【0024】
一本鎖RNAは、合成DNA鋳型、または組換え細菌から単離したDNAプラスミドからの酵素の転写により作製することができる。典型的には、T7、T3またはSP6RNAポリメラーゼのようなファージRNAポリメラーゼを用いる(MilliganおよびUhlenbeck(1989))。
【0025】
本発明の別の態様は、以下のステップを含む、細胞または生物における標的特異的な核酸改変、特にRNA干渉および/またはDNAメチル化を媒介する方法に関する:
(a)標的特異的な核酸改変が起こりうる条件下で、上記細胞または生物を本発明の二本鎖RNA分子と接触させるステップ、
(b)上記二本鎖RNAと実質的に対応する配列部分を有する標的核酸に対する、上記二本鎖RNAにより引き起こされる標的特異的な核酸改変を媒介するステップ。
【0026】
好ましくは、接触ステップ(a)は、標的細胞(例えば細胞培養物中の、例えば単離した標的細胞)、単細胞微生物、または多細胞生物内の標的細胞もしくは複数の標的細胞に、二本鎖RNA分子を導入することを含む。さらに好ましくは、導入ステップは、例えば、リポソームキャリアまたは注射によるキャリア媒介の送達を含む。
【0027】
本発明の方法を用いて、RNA干渉を媒介することが可能な細胞または生物における遺伝子の機能を決定する、あるいは、細胞または生物における遺伝子の機能を調節することさえできる。上記細胞は、好ましくは真核細胞または細胞系、例えば植物細胞または動物細胞(哺乳動物細胞など)、例えば胚細胞、多能性幹細胞、腫瘍細胞、例えば奇形癌細胞またはウイルス感染細胞である。上記生物は、好ましくは真核生物、例えば、植物または動物、例えば哺乳動物、特にヒトなどである。
【0028】
本発明のRNA分子が指令する対象となる標的遺伝子は、病理的状態と関連するものでもよい。例えば、遺伝子は、病原体関連遺伝子、例えば、ウイルス遺伝子、腫瘍関連遺伝子または自己免疫疾患関連遺伝子としうる。標的遺伝子はまた、組換え細胞または遺伝的に改変された生物において発現された異種遺伝子であってもよい。このような遺伝子の機能を決定または調節する、特に阻害することにより、農業または医学もしくは獣医学の分野で有用な情報および治療利益が得られると考えられる。
【0029】
dsRNAは、通常、医薬組成物として投与される。この投与は、核酸を所望の標的細胞にin vitroまたはin vivoで導入する公知の方法により実施される。通常用いられる遺伝子伝達法として、リン酸カルシウム法、DEAE−デキストラン法、エレクトロポレーションおよびマイクロインジェクション、ならびに、ウイルス法が挙げられる(Graham, F.L.およびvan der Eb, A.J.(1973)Virol. 52, 456;McCutchan, J.H.およびPagano, J.S.(1968)J. Natl. Cancer Inst. 41, 351;Chu, G.ら(1987)Nucl. Acids Res. 15, 1311;Fraley, Rら(1980)J. Biol. Chem. 255, 10431;Capechi, M.R.(1980)Cell 22, 479)。細胞へのDNAの導入方法の技術群に、近年、カチオンリポソームの使用が加わった(Felgner, P.L.ら(1987), Proc. Natl. Acad. Sci. USA 84, 7413)。市販されているカチオン脂質製剤として、例えば、Tfx50(Promega)またはリポフェクトアミン2000(Life Technologies)がある。
【0030】
従って、本発明はまた、有効物質として前述した少なくとも1つの二本鎖RNA分子と、薬学的キャリアを含む医薬組成物に関する。この組成物は、ヒト医学または獣医学における診断および治療用途に用いることができる。
【0031】
診断または治療用途の場合、組成物は、溶剤(例えば注射液)、クリーム剤、軟膏、錠剤、懸濁剤などの形態とすることができる。組成物は、好適な方法、例えば注射、経口、局所、鼻内、直腸投与などにより、投与することができる。キャリアは、好適な薬学的キャリアであればいずれでもよい。好ましくは、RNA分子が標的細胞に侵入する効率を高めることができるキャリアを用いる。このようなキャリアの好適な例として、リポソーム、特にカチオンリポソームが挙げられる。さらに好ましい投与方法は注射である。
【0032】
RNAi法のさらに好ましい用途は、真核細胞、またはヒト以外の真核生物、好ましくは哺乳動物細胞もしくは生物、最も好ましくはヒト細胞、例えばHeLaもしくは293などの細胞系、またはげっ歯類、例えばラットおよびマウスの機能分析である。予め決定した標的遺伝子と相同的である好適な二本鎖RNA分子、または好適な二本鎖RNA分子をコードするDNA分子を用いたトランスフェクションにより、標的細胞(例えば、細胞培養物)または標的生物において、特異的ノックアウト表現型を獲得することができる。驚くべきことに、短い二本鎖RNA分子が存在しても、宿主細胞または宿主生物からのインターフェロン応答は起こらない。
【0033】
従って、本発明のさらに別の目的は、少なくとも1つの内因性標的遺伝子の少なくとも部分的に欠失した発現を含む標的遺伝子特異的ノックアウト表現型を示す真核細胞またはヒト以外の真核生物であり、この細胞または生物は、少なくとも1つの内因性標的遺伝子の発現を阻害することが可能な少なくとも1つの二本鎖RNA分子、あるいは、少なくとも1つの内因性標的遺伝子の発現を阻害することが可能な少なくとも1つの二本鎖RNA分子をコードするDNAを用いてトランスフェクトされている。本発明により、RNAiの特異性のために、複数の異なる内因性遺伝子の標的特異的ノックアウトが可能となることに留意すべきである。
【0034】
細胞またはヒト以外の生物、特にヒト細胞または非ヒト哺乳動物の遺伝子特異的ノックアウト表現型は、分析方法、例えば遺伝子発現プロフィールおよび/またはプロテオームの分析などの、複雑な生理学的過程の機能および/または表現型分析に用いることができる。例えば、培養細胞中で、選択的スプライシング過程の調節因子と推定されるヒト遺伝子のノックアウト表現型を作製することができる。このような遺伝子として、特に、SRスプライシング因子ファミリーのメンバー、例えば、ASF/SF2,SC35、SRp20、SRp40またはSRp55が挙げられる。さらに、CD44のように、予め決定した、選択的にスプライシングされる遺伝子のmRNAプロフィールに対するSRタンパク質の作用を分析することができる。好ましくは、オリゴヌクレオチドを含むチップを用いたハイスループット方法により、分析を実施する。
【0035】
RNAiによるノックアウト技法を用いて、標的細胞または標的生物における内因性標的遺伝子の発現を阻害することができる。内因性標的遺伝子は、標的タンパク質、または標的タンパク質の変異体もしくは突然変異形態をコードする内因性標的核酸、例えば、遺伝子またはcDNAによって相補することができ、このような核酸は、場合によっては、検出可能なペプチドまたはポリペプチドをコードする別の核酸配列、例えばアフィニティータグ、特に多重アフィニティータグと融合させてもよい。標的遺伝子の変異体または突然変異形態は、これらが、1または複数のアミノ酸の置換、挿入および/または欠失により、アミノ酸レベルで内因性遺伝子産物とは異なる遺伝子産物をコードする点で、内因性標的遺伝子とは異なる。変異体または突然変異形態は、内因性標的遺伝子と同じ生物活性を有しうる。これに対し、変異体または突然変異形態は、内因性標的遺伝子の生物活性とは異なる生物活性、例えば、部分的に欠失した活性、完全に欠失した活性、増強された活性などを有していてもよい。
【0036】
相補は、外因性核酸によりコードされるポリペプチド、例えば、標的タンパク質とアフィニティータグを含む融合タンパク質と、標的細胞において内因性遺伝子をノックアウトする二本鎖RNA分子を共発現させることにより、達成することができる。この共発現は、外因性核酸によりコードされるポリペプチド、例えば、タグで修飾した標的タンパク質と、二本鎖RNA分子の両方を発現する好適な発現ベクターを用いて、あるいは、発現ベクターの組合せを用いて、達成することができる。標的細胞において新たに合成されたタンパク質およびタンパク質複合体は、外因性遺伝子産物、例えば、修飾された融合タンパク質を含むことになる。RNAi二本鎖分子による外因性遺伝子産物発現の抑制を防止するため、外因性核酸をコードする塩基配列において、DNAレベルで(アミノ酸レベルでの突然変異誘発を含むまたは含まない)、二本鎖RNA分子と相同的な配列の部分を改変することができる。あるいは、内因性標的遺伝子を、他の種、例えばマウス由来の対応する塩基配列により相補してもよい。
【0037】
本発明の細胞または生物の好ましい用途は、遺伝子発現プロフィールおよび/またはプロテオームの分析である。特に好ましい実施形態では、1または複数の標的タンパク質の変異体または突然変異体形態の分析を実施するが、その際、前述したような外因性標的核酸により、この変異体または突然変異体形態を上記細胞または生物に再導入する。内因性遺伝子のノックアウトと、突然変異した、例えば部分的に欠失した外因性標的を用いることによる救済の組合せには、ノックアウト細胞の使用と比べて利点がある。さらに、この方法は、標的タンパク質の機能ドメインを同定するのに特に適している。さらに好ましい実施形態では、少なくとも2つの細胞または生物の、例えば遺伝子発現プロフィールおよび/またはプロテオームおよび/または表現型の特徴を比較する。これらの生物は、下記のものから選択する:
(i)標的遺伝子阻害を含まない対照細胞または対照生物、
(ii)標的遺伝子阻害を含む細胞または生物、および
(iii)標的遺伝子阻害と、外因性標的核酸による標的遺伝子の相補を含む細胞または生物。
【0038】
本発明の方法および細胞は、薬理学的物質の同定および/または特定決定、例えば、試験物質のコレクションからの新規な薬理学的物質の同定ならびに/または既知薬理学的物質の作用および/もしくは副作用の機構の特性決定を行なう手法にも適している。
【0039】
従って、本発明はまた、下記の(a)〜(c)を含む、少なくとも1つの標的タンパク質に作用する薬理学的物質の同定および/または特性決定システムに関する:
(a)上記標的タンパク質をコードする少なくとも1つの内因性標的遺伝子を発現することができる、真核細胞またはヒト以外の真核生物、
(b)上記少なくとも1つの内因性標的遺伝子の発現を阻害することができる、少なくとも1つの二本鎖RNA分子、および
(c)薬理学的特性を同定および/または特性決定しようとする、試験物質または試験物質のコレクション。
【0040】
さらに、前記システムは、好ましくは、下記の(d)を含む:
(d)上記標的タンパク質または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸であって、この外因性標的核酸は、二本鎖RNA分子による発現の阻害が、上記内因性標的遺伝子の発現より実質的に低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記外因性標的核酸。
【0041】
さらに、RNAノックアウト相補方法は、調製目的、例えば真核細胞、特に哺乳動物細胞、さらに具体的にはヒト細胞由来のタンパク質またはタンパク質複合体のアフィニティー精製に用いることもできる。本発明のこの実施形態では、外因性標的核酸は、アフィニティータグと融合した標的タンパク質をコードするのが好ましい。
【0042】
上記調製方法を用いて、高分子量のタンパク質複合体を精製することができ、これらの複合体は、質量が、好ましくは150kD以上、さらに好ましくは500kD以上であり、これらは、場合に応じて、RNAのような核酸を含んでいてもよい。具体的例として、U4/U6 snRNP粒子の20kD、60kDおよび90kDタンパク質からなるヘテロ三量体タンパク質複合体、分子量が14、49、120、145および155kDの5つのタンパク質からなる17S U2 snRNP由来のスプライシング因子SF3b、ならびに、U4、U5およびU6 snRNA分子と約30のタンパク質を含む25S U4/U6/U5 tri−snRNP粒子(分子量は約1.7MD)が挙げられる。
【0043】
この方法は、哺乳動物細胞、特にヒト細胞における機能的プロテオーム分析に適している。
【0044】
以下の図面および実施例を参照にしながら、本発明をさらに詳しく説明する。
【0045】
図面の説明
図1:38bpという短い二本鎖RNAは、RNAiを媒介することができる。
【0046】
(A)Pp−luc mRNAをターゲッティングするのに用いたdsRNAのグラフ。29〜504bpの範囲にある3系統の平滑末端dsRNAを調製した。dsRNAのセンス鎖の第1ヌクレオチドの位置は、Pp−luc mRNA(p1)の開始コドンを基準として示した。
【0047】
(B)RNA干渉アッセイ(Tuschlら、1999)。標的Pp−lucと対照Rr−luc活性の比は、バッファー対照(黒棒)に対して正規化した。dsRNA(5nM)をショウジョウバエ溶解物において25℃で15分プレインキュベートした後、7−メチル−グアノシン−キャップを有するPp−lucおよびRr−luc mRNA(約50pM)を添加した。インキュベーションをさらに1時間継続した後、二重ルシフェラーゼアッセイ(Promega)により分析した。データは、少なくとも4回の独立した実験からの平均値±標準偏差である。
【0048】
図2:29bpのdsRNAは、もはや21〜23塩基の断片にプロセシングされない。ショウジョウバエ溶解物における内部32P標識dsRNA(5nM)のプロセシングからの21〜23mer形成の時間経過。dsRNAの長さおよび供給源を示す。RNAサイズマーカー(M)が左レーンにロードされており、断片のサイズを示す。時間0で認められる2つのバンドは、不完全に変性したdsRNAによるものである。
【0049】
図3:短いdsRNAは、mRNA標的を1回しか切断しない。
【0050】
(A)ショウジョウバエ溶解物におけるp133系統の10nM dsRNAと一緒に、キャップを32Pで標識した10nMセンスまたはアンチセンスRNAを1時間インキュベートすることにより生成された安定な5’切断産物の変性ゲル電気泳動。長さマーカーは、キャップ標識した標的RNAの部分的ヌクレアーゼT1消化および部分的アルカリ加水分解(OH)により作製した。dsRNAによりターゲッティングされる領域は、両側が黒棒として示される。長さが111bpのdsRNAでは、優勢切断部位同士の間隔は、20〜23塩基であることがわかる。水平方向の矢印は、RNAiによるものではない非特異的切断を示す。
【0051】
(B)センスおよびアンチセンス標的RNA上の切断部位の位置。キャップを有する177塩基のセンスおよび180塩基のアンチセンス標的RNAの配列は、逆平行配向をしており、相補配列が互いに向き合っている。様々なdsRNAによりターゲッティングされる領域は、センスおよびアンチセンス標的配列の間に位置する様々な色の棒によって示す。切断部位は丸で示す:大きな丸は強い切断を、小さな丸は弱い切断をそれぞれ示す。32P放射性標識リン酸基は、星印で示す。
【0052】
図4:RNaseIII様機構により21および22塩基のRNA断片を作製する。
【0053】
(A)dsRNAプロセシング後の約21塩基RNAの配列。dsRNAプロセシングにより作製した約21塩基のRNA断片を定方向にクローニングした後、配列決定した。dsRNAのセンス鎖に由来するオリゴリボヌクレオチドを青色の線で示し、アンチセンス鎖に由来するものを赤色の線で示す。同じ配列が複数のクローンに存在する場合には太い棒を用い、右側の数字は頻度を示す。dsRNAが媒介する標的RNA切断部位はオレンジ色の丸で示し、大きな丸は強い切断を、また小さな丸は弱い切断をそれぞれ表す(図3B参照)。センス鎖の上部にある丸はセンス標的内の切断部位を、dsRNAの下にある丸はアンチセンス標的内の切断部位をそれぞれ示す。5以下の更なるヌクレオチドが、dsRNAの3’末端由来の約21塩基断片に確認された。これらのヌクレオチドは、主としてC、GまたはA残基のランダムな組合せであり、dsRNA構成鎖のT7転写中に、非鋳型様式で付加されたと考えられる。
【0054】
(B)約21塩基のRNAのヌクレオチド組成の二次元TLC分析。ショウジョウバエ溶解物における内部放射性標識504bp Pp−luc dsRNAのインキュベーションにより、約21塩基のRNAを作製し、ゲル精製した後、ヌクレアーゼP1(上列)またはリボヌクレアーゼT2(下列)でモノヌクレオチドに消化した。表示したα32Pヌクレオシド三リン酸の1つの存在下での転写により、dsRNAを内部標識した。リン光イメージング(phosphorimaging)により放射活性を検出した。ヌクレオシド5’一リン酸、ヌクレオシド3’一リン酸、ヌクレオシド5’,3’二リン酸、および無機リン酸をそれぞれpN、Np、pNp、およびpiで表す。黒丸は、非放射能キャリアヌクレオチドからのUV吸収斑を示す。3’,5’ビス−リン酸(赤丸)は、T4ポリヌクレオチドキナーゼとγ32P−ATPによるヌクレオシド3’一リン酸の5’リン酸化により調製された放射性標識標準との同時泳動により確認した。
【0055】
図5:合成21および22塩基のRNAは、標的RNA切断を媒介する。
【0056】
(A)対照52bp dsRNA並びに合成21および22塩基dsRNAをグラフで表示した図。21および22塩基の短鎖干渉RNA(siRNA)のセンス鎖を青色で、アンチセンス鎖を赤色で示す。siRNAの配列は、二本鎖5の22塩基アンチセンス鎖を除いて、52および111bp dsRNAのクローン化断片(図4A)に由来する。二本鎖6および7のsiRNAは、111bp dsRNAプロセシング反応に特有のものであった。緑色で示す2つの3’突出塩基は、二本鎖1および3の合成アンチセンス鎖の配列に存在する。対照52bp dsRNAの両鎖は、in vitro転写により作製したため、転写物の画分には、非鋳型3’ヌクレオチド付加が含まれることもある。siRNA二本鎖により指令される標的RNA切断部位は、オレンジ色の丸で示し(図4Aの説明を参照)、図5Bに示したように決定した。
【0057】
(B)センスおよびアンチセンス標的RNA上の切断部位の位置。標的RNA配列は、図3Bに説明した通りである。ショウジョウバエ溶解物において、対照52bp dsRNA(10nM)または21および22塩基のRNA二本鎖1〜7(100nM)を標的RNAと一緒に25℃で2.5時間インキュベートした。安定した5’切断産物がゲル上で分離された。切断部位を図5Aに示す。52bp dsRNAまたはセンス(s)もしくはアンチセンス(as)鎖は、ゲルの横に黒棒で示す。切断部位はすべてdsRNAの同一性領域内に位置する。アンチセンス鎖の切断部位の正確な決定のために、低いパーセンテージのゲルを用いた。
【0058】
図6:短いdsRNA上の長い3’突出部がRNAiを阻害する。
【0059】
(A)52bp dsRNA構築物をグラフで示した図。センスおよびアンチセンス鎖の3’延長部分をそれぞれ青色および赤色で示す。標的RNA上で観察された切断部位を図4Aと同様のオレンジ色の丸で表し、これらを図6Bで示したように決定した。
【0060】
(B)センスおよびアンチセンス標的RNA上の切断部位の位置。標的RNA配列は図3Bに説明した通りである。ショウジョウバエ溶解物において、dsRNA(10nM)を標的RNAと一緒に25℃で2.5時間インキュベートした。安定した5’切断産物をゲル上で分離させた。主要な切断部位は水平方向の矢印で示し、図6Aでも表示する。52bp dsRNAによりターゲッティングされる領域は、ゲル両側の黒棒で表示する。
【0061】
図7:RNAiのモデル案
RNAiは、主として21および22塩基の短鎖干渉RNA(siRNA)を生成するdsRNA(センス鎖は黒、アンチセンス鎖は赤)のプロセシングで開始すると推定される。短い3’突出塩基が、dsRNA上に存在すれば、これは、短鎖dsRNAのプロセシングに有益であると考えられる。これから特性決定しようとするdsRNAプロセシングタンパク質は、緑色および青色の長円形として表し、非対称にdsRNA上に集合させる。本発明のモデルでは、これは、推定上の青色タンパク質またはタンパク質ドメインと、3’から5’方向のsiRNA鎖との結合により説明されるのに対し、推定上の緑色タンパク質またはタンパク質ドメインは、向き合うsiRNA鎖と必ず結合する。これらのタンパク質またはサブセットは、siRNA二本鎖と会合したままであり、dsRNAプロセシング反応の方向により決定される配向を保存している。青色タンパク質と会合したsiRNA配列だけが標的RNA切断を指示することができる。エンドヌクレアーゼ複合体は、短い干渉リボ核タンパク質複合体またはsiRNPと呼ばれる。本発明では、dsRNAを切断するエンドヌクレアーゼは、恐らく、標的認識に使用されない受動siRNAを一時的に置換することにより、標的RNAも切断することができると推定する。次に、この標的RNAは、配列相補的ガイドsiRNAにより認識される領域の中心部で切断される。
【0062】
図8:リポーター構築物とsiRNA二本鎖。
【0063】
(a)プラスミドpGL2−Control、pGL−3−ControlおよびpRL−TK(Promega)からのホタル(Pp−luc)およびウミシイタケ(Rr−luc)ルシフェラーゼリポーター遺伝子領域を示す。SV40調節エレメント、HSVチミジンキナーゼプロモーターと、2つのイントロン(斜線)を示す。GL3ルシフェラーゼの配列はGL2と95%同一であるが、RLは、両者とまったく関連性がない。pGL2からのルシフェラーゼ発現は、トランスフェクトした哺乳動物細胞におけるpGL3からの発現に対して約10分の1低い。siRNA二本鎖によりターゲッティングされる領域は、ルシフェラーゼ遺伝子のコード領域下方の黒棒で表す。
【0064】
(b)GL2、GL3およびRLルシフェラーゼをターゲッティングするsiRNA二本鎖のセンス(上)およびアンチセンス(下)配列を示す。GL2およびGL3siRNA二本鎖は、3つの一塩基置換が異なるにすぎない(灰色で囲んだ箇所)。非特異的対照として、逆転GL2配列、すなわち、invGL2を有する二本鎖を合成した。2’−デオキシチミジンの2塩基3’突出部をTTとして表す。uGL2は、GL2siRNAと類似しているが、リボ−ウリジン3’突出部を含んでいる。
【0065】
図9:siRNA二本鎖によるRNA干渉。
【0066】
標的対照ルシフェラーゼの比をバッファー対照(bu、黒棒)に対して正規化した;灰色の棒は、フォティナス・ピラリス(Photinus pyralis)(Pp−luc)GL2またはGL3ルシフェラーゼと、レニラ・レニフォルミス(Renilla reniformis)(Rr−luc)RLルシフェラーゼの比を示し(左軸)、白色の棒は、RLとGL2またはGL3比を示す(右軸)。a、c、e、gおよびiの各表は、pGL2−ControlとpRL−TKリポータープラスミドの組合せを用いて実施した実験を示し、表b、d、f、hおよびjは、pGL3−ControlとpRL−TKリポータープラスミドを用いたものを示す。干渉実験に用いた細胞系は、各表の上部に示す。バッファー対照(bu)に対するPp−luc/Rr−lucの比は、正規化の前に各種被検細胞系間で、pGL2/pRLでは0.5〜10、また、pGL3/pRLについては0.03〜1の範囲をそれぞれ変動した。プロットしたデータは、3つの独立した実験の平均値±S.D.である。
【0067】
図10:HeLa細胞におけるルシフェラーゼ発現に対する21塩基のsiRNA、50bpおよび500bpのdsRNAの影響。
【0068】
長いdsRNAの正確な長さを棒の下に示す。表a、cおよびeは、pGL2−ControlとpRL−TKリポータープラスミドを用いて実施した実験を示し、表b、dおよびfは、pGL3−ControlとpRL−TKリポータープラスミドを用いたものを示す。データは、2つの独立した実験の平均値±S.D.である。
【0069】
(a)および(b)任意のルミネセンス単位でプロットした絶対Pp−luc発現。
【0070】
(c)および(d)任意のルミネセンス単位でプロットしたRr−luc発現。
【0071】
(e)および(f)正規化した標的と対照ルシフェラーゼの比。siRNA二本鎖についてのルシフェラーゼ活性の比は、バッファー対照(bu、黒棒)に対して正規化した;50または500bp dsRNAについてのルミネセンス比は、ヒト化GFP(hG、黒棒)由来の50および500bp dsRNAについて認められたそれぞれの比に対して正規化した。GL2およびGL3をターゲッティングする49および484bpのdsRNA間の配列の全相違は、GL2およびGL3標的間に特異性を賦与するのに十分ではなかった(49bp断片では43塩基の連続した同一性、484bp断片では239塩基の最長連続同一性)。
【0072】
図11:21塩基siRNA二本鎖の3’突出部の変動
(A)実験戦略の概要。キャップを有し、かつポリアデニル化されたセンス標的mRNAを描くと共に、センスおよびアンチセンスsiRNAの相対位置を示す。8つの異なるアンチセンス鎖に従って8系統の二本鎖を用意した。siRNA配列と、突出ヌクレオチドの数を1塩基ずつ変化させた。
【0073】
(B)5nM平滑末端dsRNAの存在下で、キイロショウジョウバエ胚溶解物において、対照ルシフェラーゼ(Renilla reniformis、Rr−luc)に対する、標的ルシフェラーゼ(Photinus pyralis、Pp−luc)の正規化相対ルミネセンス。dsRNAの存在下で決定したルミネセンス比は、バッファー対照で得られた比(bu、黒棒)に対して正規化した。1より小さい正規化比は、特異的干渉を意味する。
【0074】
(C〜J)8系統の21塩基siRNA二本鎖について正規化した干渉比。siRNA二本鎖の配列を棒グラフの上方に表示する。各表は、所与のアンチセンスガイド(guide)siRNAと、5つの異なるセンスsiRNAにより形成された二本鎖のセットについての干渉比を示す。突出塩基の数(3’突出部は正の数;5’突出部は負の数)をX軸に示す。データ点は、少なくとも3回の独立した実験から平均したものであり、エラーバー(error bar)は標準偏差を表す。
【0075】
図12:siRNA二本鎖のセンス鎖の長さ変動。
【0076】
(A)実験をグラフで示す図。3つの21塩基アンチセンス鎖を8つのセンスsiRNAと対合させた。siRNAは、その3’末端で長さを変化させた。アンチセンスsiRNAの3’突出部は、1塩基(B)、2塩基(C)、または3塩基(D)であるのに対し、センスsiRNAの突出部は、各系統で変動させた。siRNA二本鎖の配列および対応する干渉比を表示する。
【0077】
図13:保存された2塩基3’突出部を有するsiRNA二本鎖の長さの変動。
【0078】
(A)実験をグラフで示す図。21塩基のsiRNA二本鎖は、図11Hまたは12Cに示したものと配列が同じである。siRNA二本鎖は、センスsiRNAの3’側(B)、またはセンスsiRNAの5’側(C)まで延長させた。siRNA二本鎖の配列およびそれぞれの干渉比を表示する。
【0079】
図14:siRNAリボース残基の2’−ヒドロキシル基の置換。
【0080】
siRNA二本鎖の鎖における2’−ヒドロキシル基(OH)は、2’−デオキシ(d)または2’−O−メチル(Me)によって置換した。3’末端での2塩基および4塩基2’−デオキシ置換をそれぞれ2塩基dおよび4塩基dとして表示する。ウリジン残基は2’−デオキシチミジンで置換した。
【0081】
図15:2塩基3’突出部を有する21塩基のsiRNA二本鎖によるセンスおよびアンチセンス標的RNA切断のマッピング
(A)32P(星印)キャップ標識センスおよびアンチセンス標的RNAと、siRNA二本鎖をグラフで示した図。センスおよびアンチセンス標的RNA切断の位置は、siRNA二本鎖の上方および下方に、それぞれ三角形で示す。
【0082】
(B)標的RNA切断部位のマッピング。キイロショウジョウバエ胚溶解物において、100nM siRNA二本鎖と一緒に10nM標的を2時間インキュベートした後、5’キャップ標識基質と5’切断産物を配列決定用ゲル上で分離させた。標的RNAの部分的RNaseT1消化(T1)と部分的アルカリ加水分解(OH−)により、長さマーカーを作製した。画像左側の太線は、標的と同じ配向のsiRNA鎖1および5が占める領域を示す。
【0083】
図16:ガイドsiRNAの5’末端が、標的RNA切断の位置を画定する。
【0084】
(A、B)実験戦略をグラフで示した図。アンチセンスsiRNAは、すべてのsiRNA二本鎖で同じであるが、センス鎖は、3’末端を改変することにより18〜25塩基までの間を(A)、または5’末端を改変することにより18〜23塩基までの間(B)を変動させた。センスおよびアンチセンス標的RNA切断の位置は、siRNA二本鎖の上方および下方に、それぞれ三角形で示す。
【0085】
(C、D)キャップ標識センス(上の表)またはアンチセンス(下の表)標的RNAを用いた標的RNA切断の分析。キャップ標識の5’切断産物だけを示す。siRNA二本鎖の配列を表示すると共に、センスsiRNA鎖の長さを表の上部に示す。パネル(C)においてダッシュで示した対照レーンは、siRNAの不在下でインキュベートした標的RNAを示す。マーカーは、図15に記載した通りである。(D)の下部パネルの矢印は、1塩基だけ異なる標的RNA切断部位を示す。
【0086】
図17:siRNA二本鎖の3’突出部の配列変動
2塩基3’突出部(灰色のNN)は、表示したように配列および組成を改変した(T、2’−デオキシチミジン、dG、2’−デオキシグアノシン;星印、野生型siRNA二本鎖)。正規化した干渉比は、図11で記載したように決定した。野生型配列は図14に示したものと同じである。
【0087】
図18:標的認識の配列特異性。
【0088】
ミスマッチsiRNA二本鎖の配列を示し、修飾した配列部分または単一塩基を灰色で示す。基準二本鎖(ref)とsiRNA二本鎖1〜7は、2’−デオキシチミジン2塩基突出部を含む。チミジンを修飾した基準二本鎖のサイレンシング効率は、野生型配列と同等であった(図17)。正規化した干渉比は、図11に記載したのと同様に決定した。
【0089】
図19:保存された2塩基3’突出部を有するsiRNA二本鎖の長さ変動。
【0090】
siRNA二本鎖は、センスsiRNAの3’側(A)、またはセンスsiRNAの5’側(B)まで延長した。siRNA二本鎖配列と、それぞれの干渉比を示す。HeLa SS6細胞については、GL2ルシフェラーゼをターゲッティングするsiRNA二本鎖(0.84μg)を、pGL2−ControlおよびpRL−TKプラスミドで一緒にトランスフェクトした。比較のため、キイロショウジョウバエ胚溶解物で試験したsiRNA二本鎖のin vitro RNAi活性を示す。
【実施例1】
【0091】
短い合成RNAにより媒介されるRNA干渉
1.1.実験手順
1.1.1 in vitro RNAi
以前記載されている(Tuschlら、1999;Zamoreら、2000)ように、in vitro RNAiおよび溶解物調製を実施した。最適なATP再生のためには、新しく溶解したクレアチンキナーゼ(Roche)を用いる必要がある。RNAi翻訳アッセイ(図1)は、5nMのdsRNA濃度と、25℃で15分の長いプレインキュベーション時間で実施した後、in vitro転写、キャッピングおよびポリアデニル化したPp−lucおよびRr−lucリポーターmRNAを添加した。インキュベーションを1時間継続した後、二重ルシフェラーゼアッセイ(Promega)およびモノライト3010Cルミノメーター(PharMingen)を用いて、Pp−lucおよびRr−lucタンパク質の相対量を分析した。
【0092】
1.1.2 RNA合成
標準的手順を用いて、T7またはSP6プロモーター配列を有するPCR鋳型からのRNAのin vitro転写を実施した(例えば、Tuschlら、1998を参照)。Expedite RNAホスホルアミダイト(Proligo)を用いて、合成RNAを調製した。ジメトキシトリチル−1,4−ベンゼンジメタノール−スクシニル−アミノプロピル−CPGを用いて、3’アダプターオリゴヌクレオチドを合成した。オリゴリボヌクレオチドを、3mlの32%アンモニア/エタノール(3/1)において、55℃で4時間(Expedite RNA)または55℃で16時間かけて脱保護した(3’および5’アダプターDNA/RNAキメラオリゴヌクレオチド)後、以前記載されている(Tuschlら、1993)ように、脱シリル化してからゲル精製した。長い3’突出部を含むdsRNAを調製するためのRNA転写物は、センス方向にT7プロモーターを、アンチセンス方向にSP6プロモーターを含むPCR鋳型から作製した。5’プライマーとしてGCGTAATACGACTCACTATAGAACAATTGCTTTTACAG(下線部、T7プロモーター)と、3’プライマーとしてATTTAGGTGACACTATAGGCATAAAGAATTGAAGA(下線部、SP6プロモーター)、ならびに、鋳型として線状化Pp−lucプラスミド(pGEM−luc配列)(Tuschlら、1999)を用いて、センスおよびアンチセンス標的RNA用の転写鋳型をPCR増幅した。T7転写センスRNAは、177塩基長であり、開始コドンに対して位置113〜273のPp−luc配列と、これに続いて、3’末端にSP6プロモーター配列の17塩基の相補配列(complement)を含む。平滑末端dsRNA形成用の転写物は、単一のプロモーター配列だけを含む2つの異なるPCR産物からの転写によって調製した。
【0093】
フェノール/クロロホルム抽出により、dsDNAのアニーリングを実施した。0.3M NaOAc(pH6)における等モル濃度のセンスおよびアンチセンスRNA(利用可能な長さおよび量に応じて、50nM〜10μM)を90℃で30秒インキュベートしてから、等量のフェノール/クロロホルムを用いて、室温で抽出した後、クロロホルム抽出により、残留フェノールを除去した。得られたdsRNAは、2.5〜3容量のエタノールの添加により沈降させた。このペレットを溶解バッファー(100mM KCl、30mM HEPES−KOH、pH7.4、2mM Mg(OAc)2)中で溶解させ、1×TAEバッファー中の標準アガロースゲル電気泳動によりdsRNAの品質を確認した。17塩基および20塩基の3’突出部を有する52bpのdsRNA(図6)を95℃で1分インキュベートすることによりアニールしてから、70℃まで急冷した後、3時間かけてゆっくりと室温まで冷却した(50μlアニーリング反応、1μM鎖濃度、300mM NaCl、10mM Tris−HCl、pH7.5)。次に、このdsRNAをフェノール/クロロホルム抽出し、エタノール沈降させた後、溶解バッファー中に溶解させた。
【0094】
dsRNA調製に用いる内部32P放射性標識RNAの転写(図2および4)は、1mM ATP、CTP、GTP、0.1または0.2mM UTP、および0.2〜0.3μM−32P−UTP(3000Ci/mmol)、あるいは、UTP以外の放射性標識ヌクレオシド三リン酸についてのそれぞれの比を用いて実施した。標的RNAのキャップの標識は、既述されている通りに実施した。キャップ標識後、標的RNAをゲル精製した。
【0095】
1.1.3 切断部位マッピング
10nM dsRNAを15分プレインキュベートした後、10nMキャップ標識標的RNAをを添加することにより、標準RNAi反応を実施した。プロテイナーゼK処理(Tuschlら、1999)により、さらに2時間(図2A)または2.5時間(図5Bおよび6B)インキュベートした後、反応を停止した。次に、8または10%配列決定用ゲル上でサンプルを分析した。21および22塩基の合成RNA二本鎖を100nM最終濃度で用いた(図5B)。
【0096】
1.1.4 約21塩基のRNAのクローニング
標的RNAの不在下で、ショウジョウバエ溶解物における放射性標識dsRNAのインキュベーションにより(200μl反応物、1時間インキュベーション、50nM dsP111、または100nM dsP52もしくはdsP39)、21塩基のRNAを生成した。次に、反応混合物をプロテイナーゼKで処理した(Tuschlら、1999)後、dsRNAプロセシング産物を変性15%ポリアクリルアミドゲル上で分離した。少なくとも18〜24塩基のサイズ範囲を含むバンドを除去し、0.3M NaClに4℃で一晩かけて溶離した後、シリコン処理管に導入した。エタノール沈降によりRNAを回収した後、脱リン酸化した(30μl反応物、30分、50℃、10Uアルカリホスファターゼ、Roche)。フェノール/クロロホルム抽出により反応を停止し、RNAをエタノール沈降させた。次に、3’アダプターオリゴヌクレオチド(pUUUaaccgcatccttctcx:大文字はRNA;小文字はDNA;pはリン酸;xは4−ヒドロキシメチルベンジル)を脱リン酸化した約21塩基のRNAと連結させた(20μl反応物、30分、37℃、5μM 3’アダプター、50mM Tris−HCl、pH7.6、10mM MgCl2、0.2mM ATP、0.1mg/mlアセチル化BSA、15%DMSO、25U T4 RNAリガーゼ、Amersham-Pharmacia)(PanおよびUhlenbeck, 1992)。等量の8M尿素/50mM EDTA停止混合物(stopmix)の添加により連結反応を停止した後、15%ゲルに直接ロードした。連結収率は50%を超えた。連結産物をゲルから回収した後、5’−リン酸化した(20μl反応物、30分、37℃、2mM ATP、5U T4ポリヌクレオチドキナーゼ、NEB)。フェノール/クロロホルム抽出によりリン酸化反応を停止した後、エタノール沈降によりRNAを回収した。次に、前記と同様に、5’アダプター(tactaatacgactcactAAA:大文字はRNA;小文字はDNA)をリン酸化連結産物と連結させた。新しい連結産物をゲル精製し、キャリアとして用いる逆転写プライマー(GACTAGCTGGAATTCAAGGATGCGGTTAAA:太字はEcoRI部位)の存在下で、ゲル切片から溶離させた。逆転写(15μl反応物、30分、42℃、150U SuperscriptII逆転写酵素、Life Technologies)の後、5’プライマーCAGCCAACGGAATTCATACGACTCACTAAA(太字はEcoRI部位)と、3’RTプライマーを用いたPCRを実施した。PCR産物をフェノール/クロロホルム抽出により精製した後、エタノール沈降させた。次に、PCR産物をEcoRI(NEB)で消化してから、T4 DNAリガーゼ(高濃度、NEB)を用いてコンカテマー化する。サイズが200〜800bpの範囲にあるコンカテマーを低融点アガロースゲル上で分離し、標準的な融解およびフェノール抽出手順によりゲルから回収した後、エタノール沈降させた。非対合末端を標準的条件下でTaqポリメラーゼと一緒に72℃で15分インキュベートすることにより充填した後、TOPO TAクローニングキット(Invitrogen)を用いて、DNA産物をpCR2.1−TOPOベクターに直接連結した。PCRと、M13−20およびM13逆方向配列決定用プライマーを用いて、コロニーをスクリーニングした。カスタム(custom)配列決定(Sequence Laboratories Gottingen GmbH, ドイツ)のためにPCR産物を直接提出した。平均して、1クローン当たり4〜5個の21mer配列が得られた。
【0097】
1.1.5 2D−TLC分析
放射性標識し、ゲル精製したsiRNAおよび2D−TLCのヌクレアーゼP1消化を記載されている通りに実施した(Zamoreら、2000)。2μg/μlキャリアtRNAと30UリボヌクレアーゼT2(Life Technologies)を用いて、10mM酢酸アンモニウム(pH4.5)中、10μl反応物において、ヌクレアーゼT2消化を50℃で3時間実施した。非放射性標準の泳動をUVシャドウイング(shadowing)により測定した。ヌクレオシド−3’,5’−二リン酸の同一性は、γ−32P−ATPおよびT4ポリヌクレオチドキナーゼを用いた市販のヌクレオシド3’−一リン酸の5’−32Pリン酸化により調製した標準によるT2消化産物の同時泳動によって確認した(データは示していない)。
【0098】
1.2 結果および考察
1.2.1 21および22塩基のRNA断片のプロセシングに必要な長さ
キイロショウジョウバエ合胞体胚から調製した溶解物は、in vitroにおいてRNAiを再現することから、RNAi機構の生化学的分析の新たなツールを提供するものである(Tuschlら、1999;Zamoreら、2000)。RNAiのためのdsRNAに必要な長さについてのin vitroおよびin vivo分析から、標的mRNAを分解する上で、短いdsRNA(<150bp)は、長いdsRNAより効果が低いことが明らかにされている(Caplenら、2000;Hammondら、2000;Ngoら、1998);Tuschlら、1999)。このmRNA分解効率が低い理由はわかっていない。従って、本発明者は、ショウジョウバエ溶解物における最適化条件下で(Zomoreら、2000)、標的RNA分解のためのdsRNAに必要な正確な長さを調べた。数系統のdsRNAを合成し、ホタルルシフェラーゼ(Pp−luc)リポーターRNAに対して指令させた。標的RNA発現の特異的抑制を二重ルシフェラーゼアッセイ(Tuschlら、1999)によりモニタリングした(図1Aおよび1B)。本発明者は、38bpという短いdsRNAの標的RNA発現の特異的阻害を検出したが、29〜36bpのdsRNAはこの過程において有効ではなかった。効果は、Pp−luc mRNAの標的位置および阻害度とは無関係で、dsRNAの長さと相関していた。すなわち、長鎖dsRNAの方が、短鎖dsRNAより有効であった。
【0099】
dsRNAのプロセシングにより作製された21〜23塩基のRNAは、RNA干渉および共抑制の媒介物質であることが示唆されている(HamiltonおよびBaulcombe, 1999;Hammondら、2000;Zamoreら、2000)。従って、本発明者は、サイズが501〜29bpの範囲にあるdsRNAのサブセットについて、21〜23塩基断片の形成の速度を分析した。ショウジョウバエ溶解物における21〜23塩基断片の形成(図2)は、長さが39〜501bpのdsRNAでは容易に検出することができたが、29bpのdsRNAでは有意な遅延が認められた。この観察結果は、mRNA切断を指示する上での21〜23塩基断片の役割と一致し、30bpのdsRNAによる不十分なRNAiを解明する助けとなった。21〜23mer形成が長さに依存するのは、通常の細胞RNAの短い分子内塩基対合構造によるRNAiの不要な活性化を防止するための、生物学的に関連する制御機構を表すものと考えられる。
【0100】
1.2.2 39bpのdsRNAは、単一部位で標的RNA切断を媒介する
ショウジョウバエ溶解物にdsRNAおよび5’−キャップ標識RNAを添加すると、標的RNAの配列特異的分解が起こる(Tuschlら、1999)。標的mRNAは、dsRNAと同一性のある領域内でしか切断されず、標的切断部位の多くは、21〜23塩基ずつ分離された(Zamoreら、2000)。従って、所与のdsRNAの切断部位の数は、dsRNAの長さを21で割った数とおおまかに対応することが予想された。本発明者は、キャップにおいて5’放射性標識されたセンスおよびアンチセンス標的RNA上の標的切断部位をマッピングした(Zamoreら、2000)(図3Aおよび3B)。配列決定用ゲル上で安定した5’切断産物を分離し、標的RNAからの部分的RNaseT1およびアルカリ性加水分解標準(ladder)との比較により、切断位置を決定した。
【0101】
以前の観察結果(Zamoreら、2000)と一致して、すべての標的RNA切断部位は、dsRNAとの同一性領域内に位置した。センスまたはアンチセンス標的は、39bpのdsRNAによって一回しか切断されなかった。各切断部位は、dsRNAが占める領域の5’末端から10塩基の位置にあった(図3B)。39bpのRNAと同じ5’末端を有する52bpのdsRNAは、第1部位から23および24塩基下流の2つの弱い切断部位に加えて、dsRNAと同一性のある領域の5’末端から10塩基の位置に、センス標的上の同じ切断部位を生成する。アンチセンス標的も、やはり各dsRNAが占める領域の5’末端から10塩基の位置で、1回だけ切断された。図1に示した38〜49bpのdsRNAについての切断部位のマッピングから、第1および優勢切断部位は、常に、dsRNAが占める領域から7〜10塩基下流の位置にあることが明らかにされた(データは示していない)。これは、標的RNA切断点が、dsRNAの末端により決定されることを示唆しており、このことは、21〜23merへのプロセシングが二本鎖の末端から開始することを意味すると考えられる。
【0102】
より長い111bpのdsRNAのセンスおよびアンチセンス標的上の切断部位は、予想よりはるかに頻度が高く、それらのほとんどは、20〜23塩基ずつ分離したクラスターとして現れる(図Aおよび3B)。より短いdsRNAに関しては、センス標的上の第1切断部位は、dsRNAが占める領域の5’末端から10塩基の位置であり、アンチセンス標的上の第1切断位置は、dsRNAが占める領域の5’末端から9塩基の位置である。この不規則な切断の原因は不明であるが、1つには、より長いdsRNAが末端からだけではなく、内部でもプロセシングされるためか、あるいは、まだ理解されていないdsRNAプロセシングの何らかの特異性決定因子があると考えられる。21〜23塩基間隔についてのある程度の不規則性も以前指摘されていた(Zamoreら、2000)。dsRNAプロセシングおよび標的RNA認識の分子レベルでの根本原理をより明瞭に理解するために、本発明者は、ショウジョウバエ溶解物における39、52、および111bpのdsRNAのプロセシングにより生成される21〜23塩基断片の配列を分析することにした。
【0103】
1.2.3 dsRNAをRNaseIII様機構による21および23塩基RNAにプロセシングする
21〜23塩基のRNA断片を特性決定するために、RNA断片の5’および3’末端を調べた。ゲル精製した21〜23塩基のRNAを過ヨウ素酸化した後、β脱離すると、末端2’および3’ヒドロキシル基の存在が示された。21〜23merはまた、アルカリ性ホスファターゼ処理に応答性であり、これは5’末端リン酸基の存在を意味する。5’リン酸および3’ヒドロキシル末端の存在は、dsRNAが大腸菌RNaseIIIと類似した酵素活性によりプロセシングされ得ることを示唆している(確認のため、(Dunn, 1982;Nicholson, 1999;Robertson, 1990:Robertson, 1982)を参照)。
【0104】
T4RNAリガーゼを用いて、3’および5’アダプターオリゴヌクレオチドと精製21〜23merとを連結することにより、21〜23塩基のRNA断片の定方向クローニングを実施した。連結産物を逆転写、PCR増幅、コンカテマー化、クローニングした後、配列決定した。39、52および111bp dsRNA(図4A)のdsRNAプロセシング反応から、220個を超える短いRNAを配列決定した。本発明者は、次のような長さ分布をみいだした:1%18塩基、5%19塩基、12%20塩基、45%21塩基、28%22塩基、6%23塩基、および2%24塩基。プロセシングされた断片の5’末端ヌクレオチドの配列分析から、5’グアノシンを有するオリゴヌクレオチドは過小提示されることがわかった。この偏りは、5’リン酸化グアノシンを供与オリゴヌクレオチドとして区別するT4 RNAリガーゼにより引き起こされると考えられる。尚、3’末端の配列には有意な偏りは認められなかった。二本鎖のセンスまたはアンチセンス鎖の3’末端から得られた約21塩基断片の多くは、T7 RNAポリメラーゼを用いたRNA合成中にヌクレオチドの非鋳型付加により得られる3’ヌクレオチドを含んでいる。興味深いことに、有意な数の内因性ショウジョウバエの約21塩基のRNAもクローニングされ、その中には、LTRおよび非LTRレトロトランスポゾン由来のものもあった(データは示していない)。これは、トランスポゾンサイレンシングにおいてRNAiが果たす可能性のある役割と一致している。
【0105】
約21塩基のRNAは、dsRNA配列全体を含むクラスター形成群として現われる(図4A)。明らかに、プロセシング反応は、付着3’末端を残してdsRNAを切断しており、これはRNaseIII切断のもう一つの特徴である。39bpのdsRNAでは、約21塩基のRNAの2つのクラスターが突出3’末端を含む各dsRNA構成鎖から見出されたが、センスおよびアンチセンス標的には1つの切断部位しか検出されなかった(図3Aおよび3B)。約21塩基の断片が、mRNA分解を媒介する複合体において一本鎖ガイドRNAとして存在するのであれば、少なくとも2つの標的切断部位が存在すると想定できたが、そうではなかった。これは、約21塩基のRNAがエンドヌクレアーゼ複合体において二本鎖形態で存在する可能性があるが、標的RNAの認識および切断には、そのうち一方の鎖しか使用できないことを示唆している。標的切断に約21塩基鎖の一方だけを使用することは、約21塩基の二本鎖が、ヌクレアーゼ複合体と結合している配向によって簡単に決定することができる。この配向は、本来のdsRNAがプロセシングされた方向によって決定される。
【0106】
52bpおよび111bpのdsRNAの場合の約21merクラスターは、39bpのdsRNAと比較して、あまりよく明確にされていない。これらのクラスターは、約21塩基二本鎖の複数の個別小集団を提示する、従って、複数の近傍部位における標的切断を指示すると考えられられる25〜30塩基の領域に分布している。これらの切断領域は、やはり20〜23塩基の間隔で主に分離されている。標準的dsRNAがいかにして約21塩基の断片にプロセッシングされ得るかを決定する法則はいまだに解明されていないが、切断部位同士の約21〜23塩基の間隔は、一連のウリジンによって改変できることがすでに認められている(Zamoreら、2000)。大腸菌RNaseIIIによるdsRNA切断の特異性は、主に、非決定因子(antideterminant)、すなわち、切断部位に対する所与の位置で特異的塩基対を排除することにより、制御されているようである。
【0107】
糖−、塩基−またはキャップ修飾が、プロセシングされた約21塩基のRNA断片に存在するか否かを試験するため、本発明者は、溶解物において放射性標識した505bpのPp−luc dsRNAをインキュベートし、約21塩基産物を単離した後、それをP1またはT2ヌクレアーゼでモノヌクレオチドに消化した。このヌクレオチド混合物を2D薄層クロマトグラフィーにより分析した(図4B)。P1またはT2消化により示されるように、4つの天然リボヌクレオチドのうち、修飾されたものは1つもなかった。本発明者は既に、約21塩基断片におけるアデノシンからイノシンへの変換を分析し(2時間のインキュベーション後)、わずかな程度(<0.7%)の脱アミノ化を検出している(Zamoreら、2000)。溶解物中での短いインキュベーション(1時間)により、このイノシン画分をかろうじて検出可能なレベルまで縮小した。ホスホジエステル結合の3’を切断するRNaseT2は、ヌクレオシド3’−リン酸とヌクレオシド3’,5’−二リン酸を生成したが、これは、5’−末端一リン酸の存在を意味する。4つのヌクレオシド3’,5’−二リン酸の全てが検出されたが、これは、配列特異性がほとんどまたは全くなしに、ヌクレオチド間の結合が切断されたことを示唆している。要約すると、約21塩基断片は、非修飾であり、dsRNAから生成され、その際、5’−末端に5’−一リン酸および3’−ヒドロキシルが存在する。
【0108】
1.2.4 合成21および22塩基RNAは、標的RNA切断を媒介する
dsRNAプロセシングの産物の分析から、RNaseIII切断反応のあらゆる特徴を有する反応によって約21断片が生成されることがわかった(Dunn, 1982;Nicholson, 1999;Robertson, 1990;Robertson, 1982)。RNaseIIIは、dsRNAの両鎖に2つの付着切断を形成し、約2塩基の3’突出部を残す。本発明者は、クローン化した約21塩基断片のいくつかと配列が同じである21および22塩基のRNAを化学的に合成し、標的RNA分解を媒介する能力についてそれらを試験した(図5Aおよび5B)。21および22塩基のRNA二本鎖を、溶解物中100nMの濃度(52bpの対照dsRNAの10倍の濃度)でインキュベートした。これらの条件下で、標的RNA切断は容易に検出可能である。21および22塩基二本鎖の濃度を100から10nMまで下げても、やはり標的RNA切断が起こる。しかし、二本鎖の濃度を100から1,000nMに高めても、標的切断は増加しない。これは恐らく、溶解物内の制限タンパク質因子によるものであろう。
【0109】
RNAiを媒介しなかった29または30bpのdsRNAとは対照的に、2〜4塩基の突出3’末端を有する21および22塩基dsRNAは、標的RNAの有効な分解を媒介した(二本鎖1、3、4、6:図5Aおよび5B)。平滑末端21または22塩基dsRNA(二本鎖2、5および7:図5Aおよび5B)は、標的を分解する能力が低かったが、これは、突出3’末端がRNA−タンパク質ヌクレアーゼ複合体の再構成に重要であることを意味している。約21塩基の二本鎖とタンパク質成分の高親和結合には、一本鎖突出部が必要であると考えられる。5’末端リン酸は、dsRNAプロセシング後も存在するが、標的RNA切断を媒介するのに必要ではなく、短い合成RNAからは欠如していた。
【0110】
合成21および22塩基二本鎖は、短い二本鎖が占める領域内で、センスおよびアンチセンス標的の切断を指示した。これは、約21塩基断片の2対のクラスターを形成する39bpのdsRNA(図2)が、センスまたはアンチセンス標的を2回ではなく1回しか切断しなかったことを考慮すると、重要な結果である。本発明者は、この結果から、約21塩基二本鎖に存在する二本鎖の一方だけが標的RNA切断を指示できること、また、ヌクレアーゼ複合体における約21塩基二本鎖の配向がdsRNAプロセシングの最初の方向によって決定されることを示唆していると解釈した。しかし、すでに完全にプロセシングされた約21塩基の二本鎖をin vitro系に提示すれば、対称的なRNA二本鎖の可能性ある2つの配向で活性配列−特異的ヌクレアーゼ複合体を形成することできる。この結果、21塩基のRNA二本鎖と同一性領域内でセンスおよびアンチセンス標的の切断が達成される。
【0111】
標的切断部位は、21または22塩基のガイド(指示)配列と相補的な第1ヌクレオチドから11または12塩基下流に位置する。すなわち、切断部位は、21または22塩基のRNAが占める領域の中心付近にある(図4Aおよび4B)。22塩基二本鎖のセンス鎖を2塩基ずつ移動すると(図5Aの二本鎖1および3を比較)、アンチセンス標的のみの切断部位が2塩基ずつ移動した。センスおよびアンチセンス鎖を2塩基ずつ移動すると、両方の切断部が2塩基ずつシフトした(二本鎖1および4を比較)。本発明者は、ほぼすべての位置で標的RNAを切断する1対の21または22塩基のRNAを設計することが可能であると推定した。
【0112】
異常な切断部位が検出されないことから、21および22塩基のRNAにより指示される標的RNA切断の特異性は、正確であると考えられる(図5B)。しかし、注意すべきは、21および22塩基のRNA二本鎖の3’突出部に存在するヌクレオチドは、切断部位付近のヌクレオチドと比べて、基質認識に寄与しない可能性があることである。これは、活性二本鎖1または3(図5A)の3’突出部における3’末端(most)ヌクレオチドが標的と相補的ではないという観察結果に基づいている。RNAiの特異性についての詳しい分析は、合成21および22塩基RNAを用いて、容易に実施することができる。
【0113】
突出3’末端を有する合成21および22塩基RNAがRNA干渉を媒介するという証拠に基づいて、本発明者は、約21塩基のRNAを「短鎖の干渉RNA」またはsiRNAと、また、それぞれのRNA−タンパク質複合体を「短い干渉リボ核タンパク質粒子」またはsiRNPと呼称することを提案する。
【0114】
1.2.5 短鎖dsRNA上の20塩基の3’突出部がRNAiを阻害する
本発明者は、短い平滑末端dsRNAが、dsRNAの末端からプロセシングされることを明らかにした。RNAiにおけるdsRNAの長さ依存の研究中に、本発明者が、17〜20塩基の突出3’末端を有するdsRNAも分析したところ、驚くべきことに、これらが、平滑末端dsRNAより効力が弱いことがわかった。長い3’末端の阻害効果は、100bpまでのdsRNAに特に顕著であったが、これより長いdsRNAについてはそれほど強くなかった。未改変ゲル分析(データは示していない)によれば、この効果は、不完全なdsRNA形成によるものではなかった。本発明者は、長い突出3’末端の阻害効果を、短いRNA二本鎖の2つの末端の一方だけに対するdsRNAプロセシングを指令するツールとして使用できるかどうかを試験した。
【0115】
52bpのモデルdsRNAを、平滑末端のもの、センス鎖だけに3’延長部分を有するもの、アンチセンス鎖だけに3’延長部分を有するもの、ならびに、両鎖に二重3’延長部分を有するもの、の4つの組合せで合成し、溶解物中でのインキュベーション後、標的RNA切断部位をマッピングした(図6Aおよび6B)。二本鎖のアンチセンス鎖の3’末端が延長されている場合には、センス標的の第1および優勢切断部位が失われ、その逆もまた同じで、二本鎖のセンス鎖の3’末端が延長された場合には、アンチセンス標的の強力な切断部位が失われた。両鎖の3’延長部分によって、52bpのdsRNAが実質的に不活性になった。約20塩基の3’延長部分によるdsRNA不活性化を説明するものとして、この末端におけるdsRNAプロセシング因子の1つの会合を妨害する一本鎖RNA結合タンパク質の会合が考えられる。この結果は、集合siRNPにおけるsiRNA二本鎖の鎖の一方だけが、標的RNA切断を指示することができる、本発明者のモデルとも一致する。RNA切断を指示する鎖の配向は、dsRNAプロセシング反応の方向によって定められる。3’付着末端の存在が、プロセシング複合体の集合を促進していると考えられる。センス鎖の3’末端のブロッキングによってのみ、アンチセンス鎖の向き合う3’末端からのdsRNAプロセシングが可能となる。これが、今度は、siRNA二本鎖のアンチセンス鎖だけが、センス標的RNA切断を指示することができるsiRNP複合体を生成する。同じことが相互の状況で言える。
【0116】
長鎖のdsRNA(500bp以上、データは示していない)の場合に、長い3’延長部分の阻害効果がそれほど高くないのは、長鎖dsRNAも内部dsRNAプロセシングシグナルを含む、あるいは、複数の切断因子の会合により、協働してプロセシングされる可能性があることを示唆している。
【0117】
1.2.6 dsRNA指令によるmRNA切断のモデル
新しい生化学データを用いて、どのようにしてdsRNAがmRNAをターゲッティングし、破壊するかについてのモデルを更新する(図7)。二本鎖RNAはまず、主に21および22塩基長で、かつ、RNaseIII様反応と類似した付着3’末端を有する短いRNA二本鎖にプロセシングされる(Dunn, 1982;Nicholson, 1999;Robertson, 1982)。プロセシングされたRNA断片の21〜23塩基長に基づき、RNaseIII様活性がRNAiに関与し得ることがすでに想定されていた(Bass, 2000)。この仮定は、RNaseIII反応産物に観察されるように、siRNAの末端に5’リン酸および3’ヒドロキシルが存在することにより支持される(Dunn, 1982;Nicholson, 1999)。細菌RNaseIII、S.セレビシエにおける真核生物相同体Rnt1p、およびS.ポンベにおけるPac1pは、リボソームRNA、ならびに、snRNAおよびsnoRNAのプロセシングにおいて機能することがわかっている(例えば、Chanfreauら、2000参照)。
【0118】
植物、動物またはヒト由来のRNaseIII相同体の生化学についてはほとんどわかっていない。2つのファミリーのRNaseIII酵素が、主にデータベースを利用した配列分析またはcDNAのクローニングにより同定されている。第1のRNaseIIIファミリーの代表は、1327アミノ酸長のキイロショウジョウバエ(D.melanogaster)タンパク質droshaである(アクセッション番号AF116572)。C末端は、2つのRNaseIIIと1つのdsRNA結合ドメインから構成され、N末端の機能は未知である。酷似した相同体が、線虫(C.elegans)(アクセッション番号AF160248)およびヒト(アクセッション番号AF189011)でもみいだされている(Filippovら、2000;Wuら、2000)。drosha様ヒトRNaseIIIは、近年クローン化され、特性決定された(Wuら、2000)。遺伝子はヒト組織および細胞系において偏在的に発現し、タンパク質は細胞の核および核小体に局在化する。アンチセンス阻害に関する研究から推定される結果により、rRNAプロセシングにおけるこのタンパク質の役割が示唆された。第2のクラスの代表は、1822アミノ酸長のタンパク質をコードする線虫遺伝子K12H4.8(アクセッション番号S44849)である。このタンパク質は、N末端RNAヘリカーゼモチーフ、続いて、drosha RNaseIIIファミリーと類似した2つのRNaseIII触媒ドメインおよびdsRNA結合モチーフを有する。S.ポンベ(アクセッション番号Q09884)、シロイヌナズナ(アクセッション番号AF187317)、キイロショウジョウバエ(アクセッション番号AE003740)、およびヒト(アクセッション番号AB028449)にも酷似した相同体がある(Filippovら、2000;Jacobsenら、1999;Matsudaら、2000)。恐らく、K12H4.8 RNaseIII/ヘリカーゼは、RNAiに関与すると考えられる候補であろう。
【0119】
線虫における遺伝子スクリーニングにより、トランスポソン可動化または共抑制に影響を及ぼすことなくRNAiを活性化するのにrde−1およびrde−4が不可欠であると確認された(Dernburgら、2000;Grishokら、2000;KettingおよびPlasterk, 2000;Tabaraら、1999)。このことから、これらの遺伝子が、dsRNAプロセシングに重要であるが、mRNA標的分解には関与しないという仮定が導かれる。両遺伝子の機能はいまだに未知であり、rde−1遺伝子産物は、ウサギタンパク質elF2Cと類似したタンパク質ファミリーメンバーであり(Tabaraら、1999)、rde−4の配列はまだ記載されていない。これらタンパク質の将来の生化学的特性決定によって、これらの分子的機能が明らかにされるに相違ない。
【0120】
siRNA二本鎖へのプロセシングは、平滑末端のdsRNAまたは短い(1〜5塩基)3’突出部を有するdsRNAの末端から開始するようであり、約21〜23塩基ずつ進行する。短鎖dsRNA上の長い(約20塩基)3’付着末端は、恐らく一本鎖RNA結合タンパク質との相互作用によって、RNAiを抑制する。短鎖dsRNAと隣接する一本鎖領域によるRNAiの抑制と、30bpの短いdsRNAからのsiRNA形成の欠如が、mRNAで頻繁に出会う構築領域(structured regions)がRNAiの活性化を起こさない理由を説明しうる。
【0121】
特定の理論に拘束されるわけではないが、本発明者は、dsRNAプロセシングタンパク質またはこれらのサブセットが、プロセシング反応後もsiRNA二本鎖と会合したままであると推定する。これらタンパク質に対するsiRNA二本鎖の配向が、2つの相補鎖のどちらが、標的RNA分解を指示する上で機能するかを決定する。化学的に合成されたsiRNA二本鎖は、考えられる2つの配向のいずれかでタンパク質成分と会合することができるため、センスおよびアンチセンス標的RNAの切断を指示する。
【0122】
21および22塩基の合成siRNA二本鎖が効率的なmRNA分解に使用可能であるという注目すべき知見は、機能遺伝学および生物医学的研究における遺伝子発現の配列特異的調節に新しいツールを提供するものである。siRNAは、PKR応答の活性化のために、長いdsRNAを使用することができない哺乳動物系で有効となり得る(Clemens, 1997)。このように、siRNA二本鎖は、アンチセンスまたはリボザイム治療法に代わる新しい治療法を提供する。
【実施例2】
【0123】
ヒト組織培養物におけるRNA干渉
2.1 方法
2.1.1 RNA調製
Expedite RNAホスホルアミダイトとチミジンホスホルアミダイト(Proligo, Germany)を用いて、21塩基のRNAを化学的に合成した。合成オリゴヌクレオチドを脱保護してからゲル精製した(実施例1)後、Sep−Pak C18カートリッジ(Waters, Milford, MA, USA)により精製を実施した(Tuschl, 1993)。siRNA配列がターゲッティングするGL2(アクセッション番号X65324)とGL3ルシフェラーゼ(アクセッション番号U47296)は、開始コドンの第1ヌクレオチドに対してコード領域153〜173と対応し、siRNAがターゲッティングするRL(アクセッション番号AF025846)は、開始コドン後の領域119〜129と対応した。より長いRNAは、PCR産物からT7RNAポリメラーゼで転写した後、ゲルおよびSep−Pak精製に付した。49および484bpのGL2またはGL3dsRNAは、翻訳の開始に対して、位置113〜161および113〜596にそれぞれ対応した。50および501bpのRL dsRNAは、位置118〜167および118〜618にそれぞれ対応した。ヒト化GFP(hG)をターゲッティングするdsRNA合成のためのPCR鋳型をpAD3から増幅した(Kehlenbach, 1998)ところ、50および501bpのhG dsRNAは、翻訳の開始に対して、位置118〜167および118〜618にそれぞれ対応した。
【0124】
siRNAのアニーリングのために、20μMの一本鎖をアニーリングバッファー(100mM酢酸カリウム、30mM HEPES−KOH(pH7.4)、2mM酢酸マグネシウム)において90℃で1分インキュベートした後、37℃で1時間続けた。37℃のインキュベーションステップを50および500bpのdsRNAについて一晩延長し、これらのアニーリング反応を8.4μMおよび0.84μM鎖濃度でそれぞれ実施した。
【0125】
2.1.2 細胞培養
10%FBS、100単位/mlペニシリンおよび100μg/mlストレプトマイシンを補充したシュナイダーのショウジョウバエ培地(Life Technologies)において25℃で、S2細胞を増殖させた。10%FBS、100単位/mlペニシリンおよび100μg/mlストレプトマイシンを補充したダルベッコの改変イーグル培地において37℃で、293、NIH/3T3、HeLaS3、COS−7細胞を増殖させた。細胞を定期的に継代させて、対数増殖を維持した。約80%密集度でのトランスフェクションの24時間前に、哺乳動物細胞をトリプシン処理してから、抗生物質を含まない新鮮な培地で5倍に希釈した(1〜3×105細胞/ml)後、24ウェルプレート(500μl/ウェル)に移した。S2細胞はトリプシン処理せずに開裂反応に付した。付着細胞系について製造者が記載しているように、リポフェクトアミン2000試薬(Life Technologies)を用いて、トランスフェクションを実施した。1ウェルにつき、リポソームに組み込まれた1.0μgのpGL2−Control(Promega)またはpGL3−Control(Promega)、0.1μgのpRL−TK(Promega)および0.28μgのsiRNA二本鎖またはdsRNAを導入した;最終量は、1ウェル当たり600μlであった。トランスフェクションの後、細胞を20時間インキュベートしたが、その後の細胞は健常のようであった。次に、二重ルシフェラーゼアッセイ(Promega)で、ルシフェラーゼ発現をモニタリングした。1.1μgのhGFPコード化pAD3と0.28μgのinvGL2 inGL2 siRNAの共トランスフェクション後、哺乳動物細胞系の蛍光顕微鏡検査により、トランスフェクション効率を測定したところ、70〜90%であった。リポータープラスミドをXL−1 Blue(Stratagene)において増幅した後、Qiagenエンドフリー・マキシ・プラスミドキット(Qiagen EndoFree Maxi Plasmid Kit)を用いて精製した。
【0126】
2.2 結果と考察
siRNAが、組織培養物においてもRNAiを媒介することができるか否かを試験するために、本発明者は、ウミシイタケ(Renilla reniformis)および、ホタルルシフェラーゼ(Photinus pyralis、GL2およびGL3)の2つの配列種をコードするリポーター遺伝子に対して指令される対称2塩基3’突出部を有する21塩基siRNA二本鎖を合成した(図8a、b)。カチオンリポソームを用いて、siRNA二本鎖をリポータープラスミドの組合せpGL2/pRLまたはpGL3/pRLと一緒に、キイロショウジョウバエ・シュナイダーS2細胞または哺乳動物細胞に共トランスフェクトした。トランスフェクションから20時間後にルシフェラーゼ活性を測定した。試験したすべての細胞系において、本発明者は、相同siRNA二本鎖(cognate siRNA duplex)の存在下でリポーター遺伝子の発現の特異的減弱を観察した(図9a〜j)。驚くことに、絶対ルシフェラーゼ発現レベルは、非相同siRNAによる影響を受けなかった。これは、21塩基のRNA二本鎖(例えば、HeLa細胞については図10a〜d)による有害な副作用がないことを意味している。キイロショウジョウバエS2細胞(図9a、b)において、ルシフェラーゼの特異的阻害は完全であった。リポーター遺伝子が50〜100倍強く発現した哺乳動物細胞では、特異的抑制は完全ではなかった(図9c〜j)。相同siRNAに応答して、GL2発現は、3〜12倍、GL3発現は9〜25倍、またRL発現は1〜3倍減弱した。293細胞については、RL siRNAによるRLルシフェラーゼのターゲッティングは無効であったが、GL2およびGL3標的は、特異的に応答した(図9i、j)。293細胞でRL発現の減弱がないのは、この発現が、試験した他の哺乳動物細胞系と比べて5〜20倍高いこと、および/またはRNA二次構造もしくは会合タンパク質のために標的配列の受容能力が制限されたことによるものであろう。それでも、相同siRNA二本鎖によるGL2およびGL3ルシフェラーゼの特異的ターゲッティングは、RNAiが293細胞においても機能していることを示している。
【0127】
uGL2を除くすべてのsiRNA二本鎖における2塩基3’突出部は、(2’−デオキシ)チミジンから構成されていた。3’突出部における、チミジンによるウリジンの置換は、キイロショウジョウバエin vitro系で十分に許容されたため、突出部の配列は標的認識に重要ではなかった。組織培地中およびトランスフェクトした細胞内でsiRNAのヌクレアーゼ耐性を増強することが推定されるため、チミジン突出部を選択した。実際に、試験したすべての細胞系で、チミジン修飾GL2 siRNAは、非修飾uGL2 siRNAよりやや強力であった(図9a、c、e、g、i)。3’突出塩基をさらに修飾することにより、siRNA二本鎖の送達および安定性が改善されると考えられる。
【0128】
共トランスフェクション実験では、組織培地の最終量に対して25nMのsiRNA二本鎖を用いた(図9、10)。siRNAの濃度を100nMまで増加しても、特異的サイレンシング効果は増強されなかったが、プラスミドDNAとsiRNA間のリポソームのカプセル化(被包)競合により、トランスフェクション効率に影響を与え始めた(データは示していない)。siRNAの濃度を1.5nMまで低下しても、特異的サイレンシング効果は低下せず(データは示していない)、siRNA濃度がDNAプラスミドより2〜20倍高いだけでもそうであった。これは、siRNAが、遺伝子サイレンシングを媒介する極めて強力な試薬であること、また、siRNAが、通常のアンチセンスまたはリボザイム遺伝子ターゲッティング実験で使用される濃度より数桁低い濃度で有効であることを意味している。
【0129】
より長いdsRNAの哺乳動物細胞に対する効果をモニタリングするため、リポーター遺伝子に対して相同な50〜500bpのdsRNAを調製した。非特異的対照として、ヒト化GFP(hG)由来のdsRNA(Kehlenbach, 1998)を用いた。siRNA二本鎖と同じ量(濃度ではない)のdsRNAを共トランスフェクトすると、リポーター遺伝子発現は強力かつ非特異的に減弱した。この効果は、代表的例としてHeLa細胞について示する(図10a〜d)。絶対ルシフェラーゼ活性は、50bp dsRNAにより、10〜20倍、500bp dsRNA共トランスフェクションにより20〜200倍非特異的にそれぞれ低下した。同様の非特異的効果が、COS−7およびNIH/3T3細胞について観察された。293細胞では、10〜20倍の非特異的減弱は、500bp dsRNAによってしか観察されなかった。dsRNA>30bpによるリポーター遺伝子発現の非特異的減弱は、インターフェロン応答の一部として予想されたものだった。
【0130】
驚くことに、リポーター遺伝子発現の強い非特異的減弱にもかかわらず、本発明者は、別の配列特異的dsRNA媒介サイレンシングを再現的に検出した。しかし、特異的サイレンシング効果は、相対リポーター遺伝子活性をhG dsRNA対照に対して正規化したときしか明らかではなかった(図10e、f)。他の3つの被検哺乳動物細胞系においても、相同dsRNAに応答する2〜10倍の特異的減弱が観察された(データは示していない)。dsRNA(356〜1662bp)による特異的サイレンシング作用はCHO−K1細胞においてすでに報告されているが、2〜4倍の特異的減弱を検出するのに必要なdsRNAの量は、本発明者の実験(Ui-Tei、2000)より約20倍高かった。また、CHO−K1細胞は、インターフェロン応答が欠失しているようである。別の報告では、ルシフェラーゼ/lacZリポーターの組合せ、および829bp特異的lacZまたは717bp非特異的GFPdsRNAを用いて、293、NIH/3T3およびBHK−21細胞をRNAiについて試験した(Caplen、2000)。この例でRNAiを検出できなかったのは、ルシフェラーゼ/lacZリポーターアッセイの感受性が低かったことと、標的および対照dsRNAの長さが異なるためであろう。以上のことを考え合わせると、本発明者の結果から、RNAiが哺乳動物細胞において活性であるが、インターフェロン系がdsRNA>30bpにより活性化されると、サイレンシング効果の検出が難しくなることがわかる。
【0131】
要約すると、本発明者は、哺乳動物細胞におけるsiRNA媒介の遺伝子サイレンシングを最初に証明した。短鎖siRNAの使用により、ヒト組織培養物における遺伝子機能の不活性化、ならびに、遺伝子特異的治療法の開発の可能性が高まる。
【実施例3】
【0132】
RNA干渉による遺伝子発現の特異的阻害
3.1 材料と方法
3.1.1 RNA調製とRNAiアッセイ
実施例1または2、あるいは、これまでの文献(Tuschlら、1999;Zamoreら、2000)に記載されているように、化学的RNA合成、アニーリング、およびルシフェラーゼに基づくRNAiアッセイを実施した。すべてのsiRNA二本鎖はホタルルシフェラーゼに対して指令され、ルシフェラーゼmRNA配列は、記載されている(Tuschlら、1999)ように、pGEM−luc(GenBankアクセッション番号X65316)由来のものであった。キイロショウジョウバエRNAi/翻訳反応において、siRNA二本鎖を15分インキュベートしてから、mRNAを添加した。翻訳に基づくRNAiアッセイを少なくとも3回繰り返して実施した。
【0133】
センス標的RNA切断のマッピングのため、開始コドンに対して位置113から273までのホタルルシフェラーゼ配列と対応する177塩基転写物を作製した後、SP6プロモーター配列の17塩基相補体を作製した。アンチセンス標的RNA切断のマッピングのため、鋳型から166塩基転写物を作製し、5’プライマーTAATACGACTCACTATAGAGCCCATATCGTTTCATA(下線部はT7プロモーター)および3’プライマーAGAGGATGGAACCGCTGGを用いて、上記転写物をPCRによりプラスミド配列から増幅した。標的配列は、開始コドンに対して位置50から215までのホタルルシフェラーゼ配列の相補体と対応する。グアニリルトランスフェラーゼ標識を、すでに記載されている(Zamoreら、2000)ように実施した。標的RNA切断のマッピングのために、キイロショウジョウバエ胚溶解物において、100nMのsiRNA二本鎖を5〜10nM標的RNAと一緒に標準的条件(Zamoreら、2000)下で、25℃で2時間インキュベートした。8容量のプロテイナーゼKバッファー(200mM Tris−HCl(pH7.5)、25mM EDTA、300mM NaCl、2%w/vドデシル硫酸ナトリウム)を添加することにより反応を停止させた。プロテイナーゼK(E.M. Merck、水に溶解)を0.6mg/mlの最終濃度まで添加した。次に、反応を65℃で15分インキュベートしてから、フェノルール/クロロホルム/イソアミルアルコール(25:24:1)で抽出した後、3容量のエタノールで沈降させた。サンプルを6%配列決定用ゲルに載せた。長さ基準は、キャップ標識センスまたはアンチセンス標的RNAの部分的RNaseT1消化および部分的塩基加水分解反応によって作製した。
【0134】
3.2 結果
3.2.1 21塩基のsiRNAの二本鎖における3’突出部の変動
前述のように、siRNA二本鎖の3’末端で2または3非対合ヌクレオチドは、それぞれの平滑末端二本鎖と比べて、標的RNA分解が効率的である。末端ヌクレオチドの機能のさらに総合的分析を実施するために、本発明者は、5つの21塩基センスsiRNA(各々、標的RNAに対する1塩基で表される)と、8つの21塩基アンチセンスsiRNA(各々、標的に対する1塩基で表される)を合成した(図11A)。センスおよびアンチセンスsiRNAを組み合わせることにより、7塩基3’突出部〜4塩基5’突出部の範囲にある合成突出末端を有する8系統のsiRNA二本鎖を作製した。二重ルシフェラーゼアッセイ系(Tuschlら、1999;Zamoreら、2000)を用いて、siRNA二本鎖の干渉を測定した。siRNA二本鎖は、ホタルルシフェラーゼmRNAに対して指令され、ウミシイタケルシフェラーゼmRNAを内部対照として用いた。siRNA二本鎖の存在下で、標的と対照ルシフェラーゼ活性のルミネセンス比を測定した後、dsRNAの不在下で得られた比に対して正規化した。比較のため、長いdsRNA(39〜504bp)の干渉比を図11Bに示す。長鎖dsRNA(図11A)については5nM、また、siRNA二本鎖(図11C〜J)については100nMの濃度で、干渉比を測定した。5nMの504bp dsRNAの完全なプロセシングにより、120nMの全siRNA二本鎖が得られたことから、siRNAについては100nM濃度を選択した。
【0135】
21塩基のsiRNA二本鎖がRNAiを媒介する能力は、形成された突出ヌクレオチドまたは塩基対の数に応じて異なる。4〜6個の3’突出塩基を有する二本鎖では、RNAiを媒介することができなかった(図11C〜F)が、2個以上の5’突出塩基を有する二本鎖は媒介することができた(図11G〜J)。2塩基の3’突出部を有する二本鎖が、RNA干渉を媒介するのに最も効率的であったが、サイレンシングの効率はやはり配列依存的であり、2塩基の3’突出部を有する様々なsiRNA二本鎖について12倍の差が認められた(図11D〜Hを比較)。平滑末端、1塩基5’突出部または1〜3塩基3’突出部を有する二本鎖は、場合により機能的であった。7塩基の3’突出部を有するsiRNA二本鎖について観察されたわずかなサイレンシング効果(図11C)は、RNAiよりもむしろ長い3’突出部のアンチセンス効果によるものであろう。長鎖dsRNA(図11B)と、最も有効な21塩基のsiRNA二本鎖(図11E、G、H)をRNAiの効率について比較すると、100nM濃度の一本鎖siRNA二本鎖が、5nMの504bp dsRNAと同じくらい有効となり得ることがわかる。
【0136】
3.2.2 21塩基のアンチセンスsiRNAと対合するセンスsiRNAの長さ変動
RNAiに対するsiRNAの長さの影響を調べるために、本発明者は、3つの21塩基アンチセンス鎖と8つの18〜25塩基センス鎖とを組み合わせて、3系統のsiRNA二本鎖を作製した。アンチセンスsiRNAの3’突出部は、各siRNA二本鎖系統における1、2または3塩基に固定したのに対し、センスsiRNAは、その3’末端で変動させた(図12A)。センスsiRNAの長さとは無関係に、アンチセンスsiRNAの2塩基の3’突出部を有する二本鎖(図12C)は、1または3塩基の3’突出部を有するもの(図12B、D)より活性が高いことがわかった。アンチセンスsiRNAの1塩基の3’突出部を有する第1の系統では、センスsiRNAの1および2塩基の3’突出部をそれぞれ有する、21および22塩基センスsiRNAの二本鎖の活性が最も高かった。19〜25塩基のセンスsiRNAを有する二本鎖もRNAを媒介することができたが、程度は低かった。同様に、アンチセンスsiRNAの2塩基突出部を有する第2系統では、2塩基の3’突出部を有する21塩基のsiRNA二本鎖の活性が最も高く、18〜25塩基のセンスsiRNAとのその他すべての組合せは、有意な程度まで活性であった。3塩基のアンチセンスsiRNA3’突出部を有する最後の系統では、20塩基のセンスsiRNAおよび2塩基のセンス3’突出部を有する二本鎖だけが標的RNA発現を減弱することができた。以上、これらの結果から、siRNAの長さと共に、3’突出部の長さが重要であることと、2塩基3’突出部を有する21塩基のsiRNAの二本鎖がRNAiには最適であることがわかる。
【0137】
3.2.3 一定の2塩基3’突出部を有するsiRNA二本鎖の長さ変動
次に、本発明者は、対称の2塩基3’突出部を維持しながら、両siRNA鎖の長さを同時に変化させて生じる影響を調べた(図13A)。図11Hの21塩基のsiRNA二本鎖を基準として含む2系統のsiRNA二本鎖を作製した。センスsiRNA(図13B)の3’末端またはアンチセンスsiRNA(図13C)の3’末端で塩基対合部分を延長することにより、二本鎖の長さを20〜25bpで変動させた。20〜23bpの二本鎖は、標的ルシフェラーゼ活性の特異的抑制を起こしたが、21塩基のsiRNA二本鎖は、他の二本鎖のどちらと比較しても少なくとも8倍有効であった。24および25塩基のsiRNA二本鎖は、検出可能な干渉を全く起こさなかった。配列特異的効果は、二本鎖の両末端における変動が同様の効果を生じたため、わずかであった。
【0138】
3.2.4 2’−デオキシおよび2’−O−メチル修飾siRNA二本鎖
RNAiにおけるsiRNAリボース残基の重要性を評価するため、2’−デオキシまたは2’−O−メチル修飾鎖を含む21塩基のsiRNAおよび2塩基3’突出部を有する二本鎖を試験した(図14)。2’−デオキシヌクレオチドによる2塩基3’突出部の置換では影響がなく、対合領域における突出部と隣接した2つの別のリボヌクレオチドの置換でも、有意に活性のsiRNAが生じた。従って、siRNA二本鎖の42塩基のうち8つをDNA残基で置換しても、活性は失われなかった。2’−デオキシ残基による一方または両方のsiRNA鎖の完全な置換では、2’−O−メチル残基による置換と同様に、RNAiを破壊した。
【0139】
3.2.5 標的RNA切断部位の画定
22塩基のsiRNA二本鎖および21塩基/22塩基の二本鎖について、標的RNA切断位置を事前に決定した。標的RNA切断の位置は、siRNA二本鎖が占める領域の中心部、すなわち、21または22塩基のsiRNAガイド配列と相補的な第1塩基から11または12塩基下流に位置することがわかった。2塩基3’突出部を有する5つの別個の21塩基siRNA二本鎖(図15A)を5’キャップ標識センスまたはアンチセンス標的RNAと一緒にキイロショウジョウバエ溶解物中でインキュベートした(Tuschlら、1999;Zamoreら、2000)。5’切断産物を配列決定用ゲル上で分離した(図15B)。切断されたセンス標的RNAの量は、翻訳に基づくアッセイで決定されるsiRNA二本鎖の効率と相関しており、siRNA二本鎖1、2および4(図15Bおよび11H、G、E)は、二本鎖3および5(図15Bおよび11F、D)より速く標的RNAを切断する。注目すべきことに、5’切断産物および投入標的RNAの放射活性の合計は、時間に関して一定ではなく、5’切断産物は蓄積しなかった。恐らく、5’−キャップのポリ(A)テイルのいずれかが欠如しているために、siRNA−エンドヌクレアーゼ複合体から放出した切断産物は急速に分解されるのであろう。
【0140】
センスおよびアンチセンス標的RNAの両方で、切断部位は、siRNA二本鎖が占める領域の中央部に位置した。5つの異なる二本鎖により生成された各標的の切断部位は、標的配列に沿って、二本鎖の1塩基置換により1塩基ずつ変動した。標的は、配列相補的ガイドsiRNAの3’末端(most)ヌクレオチドと相補的な標的位置から正確に11塩基下流で切断された。
【0141】
ガイドsiRNAの5’または3’末端が、標的RNA切断のルーラー(ruler)を設定するか否かを決定するために、本発明者は、図16AおよびBに概要を示す実験戦略を考案した。21塩基アンチセンスsiRNAは、この実験では不変に維持し、5’または3’末端のいずれかにおいて修飾されたセンスsiRNAと対合させた。センスおよびアンチセンス標的RNA切断の位置は前記のように決定した。センスsiRNAの3’末端の変化を、1塩基5’突出部〜6塩基3’突出部でモニタリングしたが、これらはいずれも、センスまたはアンチセンス標的RNA切断の位置に影響を及ぼさなかった(図16C)。センスsiRNAの5’末端における変化は、センス標的RNA切断に影響を及ぼさなかった(図16D、上パネル)が、これは、アンチセンスsiRNAが変化しなかったため、予想されたことであった。しかし、アンチセンス標的RNA切断は影響を受け、センスsiRNAの5’末端に強く依存した(図16D、下パネル)。センスsiRNAサイズが20または21塩基であるとき、アンチセンス標的だけが切断され、切断の位置は1塩基ずつ異なった。このことは、標的を認識する5’末端が標的RNA切断のルーラー(ruler)を設定することを示唆している。この位置は、ガイドsiRNAの5’末端(most)塩基と対合した標的の塩基から上流方向に数えると、塩基10と11の間に位置する(図15Aも参照)。
【0142】
3.2.6 3’突出部における配列の影響および2’−デオキシ置換
2塩基の3’突出部が、siRNA機能には好ましい。本発明者は、突出塩基の配列が、標的認識に寄与するのか、またはこの配列が、エンドヌクレアーゼ複合体(RISCまたはsiRNP)の認識に必要な特徴であるだけなのかを知ろうとした。本発明者は、AA、CC、GG、UUおよびUG3’突出部を有するセンスおよびアンチセンスsiRNAを合成し、2’−デオキシ修飾TdGおよびTTを含有させた。野生型siRNAは、センス3’突出部にAAを、また、アンチセンス3’突出部にUGを含んでいた(AA/UG)。siRNA二本鎖はすべて、干渉アッセイにおいて機能的であり、標的発現を少なくとも5倍減弱した(図17)。10倍以上標的発現を減弱した最も効率的なsiRNA二本鎖は、配列型:NN/UG、NN/UU、NN/TdG、およびNN/TT(Nは任意のヌクレオチドである)のものであった。AA、CCまたはGGのアンチセンスsiRNA3’突出部を有するsiRNA二本鎖は、野生型配列UGまたは突然変異配列UUと比較して、2〜4倍活性が低かった。RNAi効率が低いのは、恐らく、末端から2番目の3’塩基が配列特異的標識認識に寄与するためと考えられる。というのは、3’末端塩基は、GからUに改変しても、影響がないからである。
【0143】
センスsiRNAの3’突出部の配列を改変しても、配列に依存する影響は一切明らかにされなかったが、これは、センスsiRNAはセンス標的mRNA認識に寄与してはならないため、予想されたことだった。
【0144】
3.2.7 標的認識の配列特異性
標的認識の配列特異性を調べるため、本発明者は、siRNA二本鎖の対合部分に配列改変を導入し、サイレンシングの効率を調べた。配列改変は、3または4塩基長の短い部分を逆転させるか、あるいは、点突然変異として導入した(図18)。一方のsiRNA鎖の配列改変は、相補的siRNA鎖において補償され、これによって、塩基対合したsiRNA二本鎖構造の不安定化(pertubing)を回避した。合成にかかるコストを下げるため、2塩基の3’突出部の配列は、すべてTT(T、2’−デオキシチミジン)とした。TT/TT基準siRNA二本鎖は、RNAiについて、野生型siRNA二本鎖AA/UGと同等であった(図17)。リポーターmRNA破壊を媒介する能力は、翻訳に基づくルミネセンスアッセイを用いて定量化した。逆転配列部分有するsiRNAの二本鎖は、ホタルルシフェラーゼリポーターをターゲッティングする能力が著しく低いことがわかった(図18)。アンチセンスsiRNAの3’末端と中央部との間に位置する配列改変は、標的RNA認識を完全に破壊したが、アンチセンスsiRNAの5’末端付近の突然変異は、わずかな程度のサイレンシングを呈示する。推定標的RNA切断部位の反対側に直接、または推定部位から1塩基離れて位置するA/U塩基対のトランスバージョンによって、標的RNA切断が阻止されるが、これは、siRNA二本鎖の中心部内の単一突然変異が、ミスマッチ標的を識別することを示している。
【0145】
3.3 論考
siRNAは、昆虫細胞だけではなく、哺乳動物細胞においても、遺伝子発現を不活性化するための有用な試薬であり、治療用途に極めて有望である。本発明者は、キイロショウジョウバエ胚溶解物において効率的な標的RNA分解を促進するのに必要なsiRNA二本鎖の構造上の決定因子を系統的に分析することにより、最も効力のあるsiRNA二本鎖の設計についての法則を提供した。完全なsiRNA二本鎖は、全RNAの同等量を用いれば、500bpのdsRNAと同等の効率で、遺伝子発現をサイレンシングすることができる。
【0146】
3.4 siRNAユーザーガイド
効率的なサイレンシングsiRNA二本鎖は、21塩基のアンチセンスsiRNAから構成されるため、2塩基3’突出末端を有する19bp二重らせんを形成するように選択しなければならない。2塩基3’突出リボヌクレオチドの2’−デオキシ置換は、RNAiに影響を及ぼさないが、RNA合成にかかるコスト削減に役立ち、siRNA二本鎖のRNase抵抗を増強することができる。ところが、これより広範囲な2’−デオキシまたは2’−O−メチル修飾は、恐らく、siRNAP集合のためのタンパク質会合を妨害するために、siRNAがRNAiを媒介する能力を低下させる。
【0147】
標的認識は、標的に相補的なsiRNAにより媒介される、高度に配列特異的過程である。ガイドsiRNAの3’末端(most)塩基は、標的認識の特異性に寄与しないのに対し、3’突出部から2番目の塩基は、標的RNA切断に影響を与え、また、ミスマッチはRNAiを1/2〜1/4倍に低下させる。ガイドsiRNAの5’末端もまた、3’末端と比べて、ミスマッチ標的RNA認識を広く許容するようである。標的RNA切断部位と向き合って位置する、siRNAの中心のヌクレオチドは、重要な特異性決定因子であり、ただ1つの塩基改変でも、RNAiは検出不可能なレベルまで低下する。これは、siRNA二本鎖が、遺伝子ターゲッティング実験において突然変異体または多型対立遺伝子を区別することができることを示唆しており、このことは、将来の治療法開発にとって重要な特徴となるであろう。
【0148】
センスおよびアンチセンスsiRNAは、エンドヌクレアーゼ複合体またはその拘束(commitment)複合体のタンパク質成分と会合すると、個別の役割を果たすことが示唆されている。この複合体におけるsiRNA二本鎖の相対配向が、どの鎖を標的認識に用いることができるかを決定する。合成siRNA二本鎖は、二重らせん構造に対して二回転対称を有するが、配列に対してはそうではない。キイロショウジョウバエ溶解物におけるRNAiタンパク質とsiRNA二本鎖との会合により、2つの非対称複合体が形成される。このような推定上の複合体では、センスおよびアンチセンスsiRNA、従って、それらの機能について、キラル環境がそれぞれ異なる。この推定は、明らかにパリンドロームのsiRNA配列、またはホモ二量体として会合できるRNAiタンパク質には適用されない。センスおよびアンチセンスターゲッティングsiRNPの比に及ぼす可能性のある配列の影響を最小限にするため、本発明者は、同一の3’突出部配列を有するsiRNA配列を用いることを提案する。本発明者は、センスsiRNAの突出部の配列を、アンチセンス3’突出部の配列に対して調節するよう勧める。なぜなら、センスsiRNAは、典型的ノックダウン実験には標的をもたないからである。センスおよびアンチセンス切断siRNPの再構成における非対称性は、この実験(図14)で用いた2塩基3’突出部を有する様々な21塩基のsiRNA二本鎖について観察したRNAi効率の変動に(部分的に)起因すると考えられる。あるいは、標的部位の塩基配列および/または標的RNA構造の受容能力は、これらsiRNA二本鎖の効率の変動によるものと考えられる。
【0149】
参照文献
【特許請求の範囲】
【請求項1】
単離された二本鎖RNA分子であって、各鎖が39〜52塩基長を有し、該RNA分子は標的特異的なRNA干渉が可能な1以上の二本鎖断片にプロセシングされることができ、該RNA分子は、第1の鎖が予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が該第1の鎖に相補的な配列からなる、上記RNA分子。
【請求項2】
各鎖が39塩基長を有する、請求項1に記載のRNA分子。
【請求項3】
単離された二本鎖RNA分子であって、該RNA分子は標的特異的なRNA干渉が可能なものであり、第1の鎖が19〜25塩基長を有し、予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が25塩基長を有する、上記RNA分子。
【請求項4】
少なくとも1つの修飾されたリボヌクレオチドを含む、請求項1〜3のいずれか1項に記載のRNA分子。
【請求項5】
修飾リボヌクレオチドが、糖、骨格鎖または核酸塩基修飾リボヌクレオチドから選択される、請求項4に記載のRNA分子。
【請求項6】
修飾リボヌクレオチドが、糖修飾リボヌクレオチドであり、2’−OH基が、H、OR、R、ハロ、SH、SR1、NH2、NHR、NR2またはCNから選択される基で置換され、Rは、C1−C6アルキル、アルケニルまたはアルキニルであり、ハロは、F、Cl、BrまたはIである、請求項5に記載のRNA分子。
【請求項7】
修飾リボヌクレオチドが、ホスホロチオエート基を含む骨格鎖修飾リボヌクレオチドである、請求項5に記載のRNA分子。
【請求項8】
前記RNA分子の第1の鎖が、予め決定したmRNA標的分子に対して、少なくとも85%の同一性を有する配列からなる、請求項1〜7のいずれか1項に記載のRNA分子。
【請求項9】
下記のステップを含む、請求項1〜8のいずれか1項に記載の二本鎖RNA分子の作製方法:
(a)各々が39〜52または19〜25塩基長を有する2本のRNA鎖を合成するステップであって、該RNA鎖は二本鎖RNA分子を形成することができるものである、上記ステップ、
(b)二本鎖RNA分子が形成される条件下で合成RNA鎖を結合させるステップであって、得られる二本鎖RNA分子は標的特異的なRNA干渉が可能なものである、上記ステップ。
【請求項10】
RNA鎖が化学的に合成される、請求項9に記載の方法。
【請求項11】
RNA鎖が酵素により合成される、請求項9に記載の方法。
【請求項12】
下記のステップを含む、動物細胞において標的特異的なRNA干渉を媒介する方法:
(a)標的特異的なRNA干渉が起こりうる条件下で、上記細胞を請求項1〜8のいずれか1項に記載の二本鎖RNA分子と接触させるステップ、および
(b)上記二本鎖RNAと同一の配列部分を有する標的核酸に対する、上記二本鎖RNAにより引き起こされる標的特異的なRNA干渉を媒介するステップ。
【請求項13】
接触ステップが、二本鎖RNA分子を標的細胞に導入し、そこで標的特異的なRNA干渉を起こさせうるステップを含む、請求項12に記載の方法。
【請求項14】
導入ステップが、キャリア媒介による送達または注射を含む、請求項13に記載の方法。
【請求項15】
動物細胞における遺伝子の機能を決定するための、請求項12〜14のいずれか1項に記載の方法の使用。
【請求項16】
動物細胞における遺伝子の機能を抑制するための、請求項12〜14のいずれか1項に記載の方法の使用。
【請求項17】
遺伝子が病理的状態と関連する、請求項15または16に記載の使用。
【請求項18】
遺伝子が病原体関連遺伝子である、請求項17に記載の使用。
【請求項19】
遺伝子がウイルス遺伝子である、請求項18に記載の使用。
【請求項20】
遺伝子が腫瘍関連遺伝子である、請求項17に記載の使用。
【請求項21】
遺伝子が自己免疫疾患関連遺伝子である、請求項17に記載の使用。
【請求項22】
標的遺伝子特異的ノックアウト表現型を示す動物細胞であって、該細胞が、内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子、または少なくとも1つの内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子をコードするDNAでトランスフェクトされており、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定されたmRNA標的分子に対して100%の同一性を有する配列からなる、上記細胞。
【請求項23】
哺乳動物細胞である、請求項22に記載の細胞。
【請求項24】
ヒト細胞である、請求項23に記載の細胞。
【請求項25】
標的タンパク質、または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸でさらにトランスフェクトされた、請求項22〜24のいずれか1項に記載の細胞であって、該外因性標的核酸は、二本鎖RNA分子による該外因性標的核酸の発現の阻害が、内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記細胞。
【請求項26】
外因性標的核酸が、検出可能なペプチドまたはポリペプチドをコードするさらなる核酸配列と融合されている、請求項25に記載の細胞。
【請求項27】
分析手法のための、請求項22〜26のいずれか1項に記載の細胞の使用。
【請求項28】
遺伝子発現プロフィールを分析するための、請求項27に記載の使用。
【請求項29】
プロテオーム分析のための、請求項27に記載の使用。
【請求項30】
外因性標的核酸によってコードされる標識タンパク質の変異体または突然変異形態の分析が実施される、請求項27〜29のいずれか1項に記載の使用。
【請求項31】
標的タンパク質の機能ドメインを同定するための、請求項30に記載の使用。
【請求項32】
下記のものから選択される少なくとも2つの動物細胞の比較を実施する、請求項27〜31のいずれか1項に記載の使用:
(i)標的遺伝子阻害を含まない対照細胞、
(ii)標的遺伝子阻害を含む細胞、および
(iii)標的遺伝子阻害と、外因性標的核酸による標的遺伝子の相補を含む細胞。
【請求項33】
分析が、機能および/または表現型の分析を含む、請求項27〜32のいずれか1項に記載の使用。
【請求項34】
調製手法のための、請求項22〜26のいずれか1項に記載の細胞の使用。
【請求項35】
真核細胞からタンパク質またはタンパク質複合体を単離するための、請求項34に記載の使用。
【請求項36】
高分子量タンパク質複合体を単離するための、請求項35に記載の使用。
【請求項37】
高分子量タンパク質複合体が核酸を含む、請求項36に記載の使用。
【請求項38】
薬理学的物質を同定および/または特性決定する手法における、請求項27〜37のいずれか1項に記載の使用。
【請求項39】
下記の(a)〜(c)を含む、少なくとも1つの標的タンパク質に作用する薬理学的物質の同定および/または特性決定システム:
(a)少なくとも1つの標的タンパク質をコードする少なくとも1つの標的遺伝子を発現することができる、動物細胞、
(b)上記少なくとも1つの内因性標的遺伝子の発現を阻害することができる、少なくとも1つの二本鎖RNA分子であって、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定したmRNA標的分子に対して100%の同一性を有する配列からなる、上記RNA分子、および
(c)薬理学的特性を同定および/または特性決定しようとする、試験物質または試験物質のコレクション。
【請求項40】
さらに下記の(d)を含む、請求項39に記載のシステム:
(d)上記標的タンパク質または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸であって、該外因性標的核酸は、二本鎖RNA分子による発現の阻害が、上記内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記外因性標的核酸。
【請求項1】
単離された二本鎖RNA分子であって、各鎖が39〜52塩基長を有し、該RNA分子は標的特異的なRNA干渉が可能な1以上の二本鎖断片にプロセシングされることができ、該RNA分子は、第1の鎖が予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が該第1の鎖に相補的な配列からなる、上記RNA分子。
【請求項2】
各鎖が39塩基長を有する、請求項1に記載のRNA分子。
【請求項3】
単離された二本鎖RNA分子であって、該RNA分子は標的特異的なRNA干渉が可能なものであり、第1の鎖が19〜25塩基長を有し、予め決定したmRNA標的分子に対して少なくとも70%の同一性を有する配列からなり、第2の鎖が25塩基長を有する、上記RNA分子。
【請求項4】
少なくとも1つの修飾されたリボヌクレオチドを含む、請求項1〜3のいずれか1項に記載のRNA分子。
【請求項5】
修飾リボヌクレオチドが、糖、骨格鎖または核酸塩基修飾リボヌクレオチドから選択される、請求項4に記載のRNA分子。
【請求項6】
修飾リボヌクレオチドが、糖修飾リボヌクレオチドであり、2’−OH基が、H、OR、R、ハロ、SH、SR1、NH2、NHR、NR2またはCNから選択される基で置換され、Rは、C1−C6アルキル、アルケニルまたはアルキニルであり、ハロは、F、Cl、BrまたはIである、請求項5に記載のRNA分子。
【請求項7】
修飾リボヌクレオチドが、ホスホロチオエート基を含む骨格鎖修飾リボヌクレオチドである、請求項5に記載のRNA分子。
【請求項8】
前記RNA分子の第1の鎖が、予め決定したmRNA標的分子に対して、少なくとも85%の同一性を有する配列からなる、請求項1〜7のいずれか1項に記載のRNA分子。
【請求項9】
下記のステップを含む、請求項1〜8のいずれか1項に記載の二本鎖RNA分子の作製方法:
(a)各々が39〜52または19〜25塩基長を有する2本のRNA鎖を合成するステップであって、該RNA鎖は二本鎖RNA分子を形成することができるものである、上記ステップ、
(b)二本鎖RNA分子が形成される条件下で合成RNA鎖を結合させるステップであって、得られる二本鎖RNA分子は標的特異的なRNA干渉が可能なものである、上記ステップ。
【請求項10】
RNA鎖が化学的に合成される、請求項9に記載の方法。
【請求項11】
RNA鎖が酵素により合成される、請求項9に記載の方法。
【請求項12】
下記のステップを含む、動物細胞において標的特異的なRNA干渉を媒介する方法:
(a)標的特異的なRNA干渉が起こりうる条件下で、上記細胞を請求項1〜8のいずれか1項に記載の二本鎖RNA分子と接触させるステップ、および
(b)上記二本鎖RNAと同一の配列部分を有する標的核酸に対する、上記二本鎖RNAにより引き起こされる標的特異的なRNA干渉を媒介するステップ。
【請求項13】
接触ステップが、二本鎖RNA分子を標的細胞に導入し、そこで標的特異的なRNA干渉を起こさせうるステップを含む、請求項12に記載の方法。
【請求項14】
導入ステップが、キャリア媒介による送達または注射を含む、請求項13に記載の方法。
【請求項15】
動物細胞における遺伝子の機能を決定するための、請求項12〜14のいずれか1項に記載の方法の使用。
【請求項16】
動物細胞における遺伝子の機能を抑制するための、請求項12〜14のいずれか1項に記載の方法の使用。
【請求項17】
遺伝子が病理的状態と関連する、請求項15または16に記載の使用。
【請求項18】
遺伝子が病原体関連遺伝子である、請求項17に記載の使用。
【請求項19】
遺伝子がウイルス遺伝子である、請求項18に記載の使用。
【請求項20】
遺伝子が腫瘍関連遺伝子である、請求項17に記載の使用。
【請求項21】
遺伝子が自己免疫疾患関連遺伝子である、請求項17に記載の使用。
【請求項22】
標的遺伝子特異的ノックアウト表現型を示す動物細胞であって、該細胞が、内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子、または少なくとも1つの内因性標的遺伝子の発現を阻害しうる少なくとも1つの二本鎖RNA分子をコードするDNAでトランスフェクトされており、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定されたmRNA標的分子に対して100%の同一性を有する配列からなる、上記細胞。
【請求項23】
哺乳動物細胞である、請求項22に記載の細胞。
【請求項24】
ヒト細胞である、請求項23に記載の細胞。
【請求項25】
標的タンパク質、または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸でさらにトランスフェクトされた、請求項22〜24のいずれか1項に記載の細胞であって、該外因性標的核酸は、二本鎖RNA分子による該外因性標的核酸の発現の阻害が、内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記細胞。
【請求項26】
外因性標的核酸が、検出可能なペプチドまたはポリペプチドをコードするさらなる核酸配列と融合されている、請求項25に記載の細胞。
【請求項27】
分析手法のための、請求項22〜26のいずれか1項に記載の細胞の使用。
【請求項28】
遺伝子発現プロフィールを分析するための、請求項27に記載の使用。
【請求項29】
プロテオーム分析のための、請求項27に記載の使用。
【請求項30】
外因性標的核酸によってコードされる標識タンパク質の変異体または突然変異形態の分析が実施される、請求項27〜29のいずれか1項に記載の使用。
【請求項31】
標的タンパク質の機能ドメインを同定するための、請求項30に記載の使用。
【請求項32】
下記のものから選択される少なくとも2つの動物細胞の比較を実施する、請求項27〜31のいずれか1項に記載の使用:
(i)標的遺伝子阻害を含まない対照細胞、
(ii)標的遺伝子阻害を含む細胞、および
(iii)標的遺伝子阻害と、外因性標的核酸による標的遺伝子の相補を含む細胞。
【請求項33】
分析が、機能および/または表現型の分析を含む、請求項27〜32のいずれか1項に記載の使用。
【請求項34】
調製手法のための、請求項22〜26のいずれか1項に記載の細胞の使用。
【請求項35】
真核細胞からタンパク質またはタンパク質複合体を単離するための、請求項34に記載の使用。
【請求項36】
高分子量タンパク質複合体を単離するための、請求項35に記載の使用。
【請求項37】
高分子量タンパク質複合体が核酸を含む、請求項36に記載の使用。
【請求項38】
薬理学的物質を同定および/または特性決定する手法における、請求項27〜37のいずれか1項に記載の使用。
【請求項39】
下記の(a)〜(c)を含む、少なくとも1つの標的タンパク質に作用する薬理学的物質の同定および/または特性決定システム:
(a)少なくとも1つの標的タンパク質をコードする少なくとも1つの標的遺伝子を発現することができる、動物細胞、
(b)上記少なくとも1つの内因性標的遺伝子の発現を阻害することができる、少なくとも1つの二本鎖RNA分子であって、該二本鎖RNA分子は、各RNA鎖が39〜52または19〜25塩基長を有し、少なくとも一方の鎖が1〜3塩基の3’突出部を有し、3’突出部を除く該RNA分子の一方の鎖が、予め決定したmRNA標的分子に対して100%の同一性を有する配列からなる、上記RNA分子、および
(c)薬理学的特性を同定および/または特性決定しようとする、試験物質または試験物質のコレクション。
【請求項40】
さらに下記の(d)を含む、請求項39に記載のシステム:
(d)上記標的タンパク質または該標的タンパク質の変異体もしくは突然変異形態をコードする少なくとも1つの外因性標的核酸であって、該外因性標的核酸は、二本鎖RNA分子による発現の阻害が、上記内因性標的遺伝子の発現より低いという点で、該内因性標的遺伝子とは核酸レベルで異なるものである、上記外因性標的核酸。
【図1A】


【図1B】
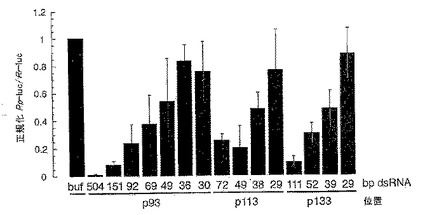
【図2】

【図3A】

【図3B】
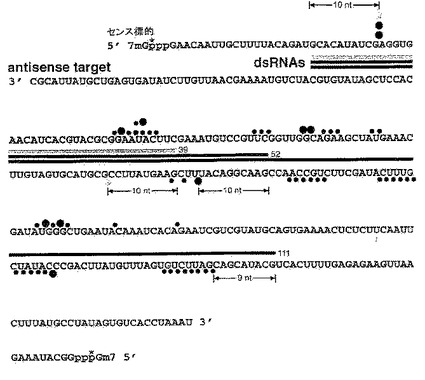
【図4A】

【図4B】

【図5A】
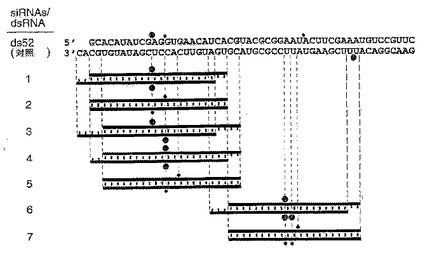
【図5B】
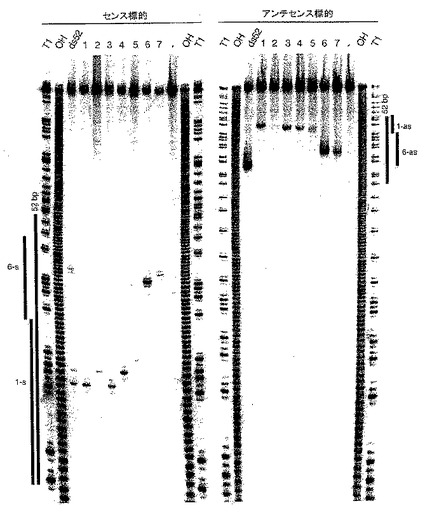
【図6A】
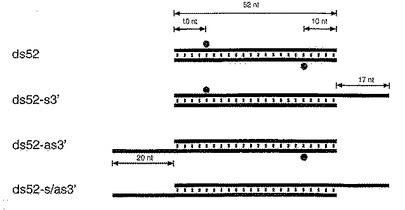
【図6B】
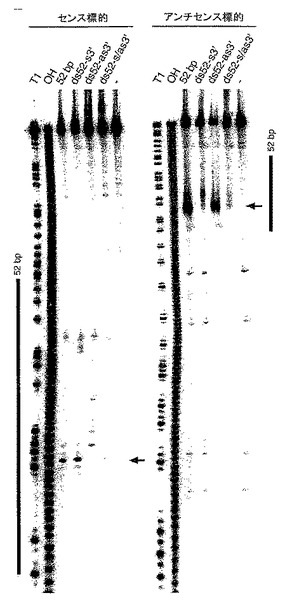
【図7】

【図8a】
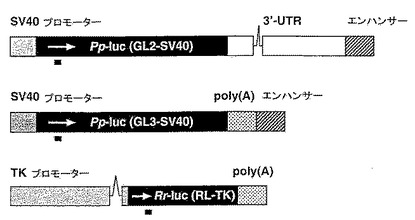
【図8b】
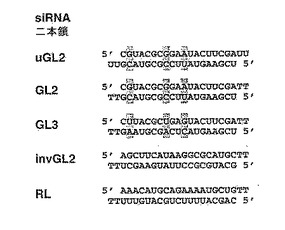
【図9】

【図10】
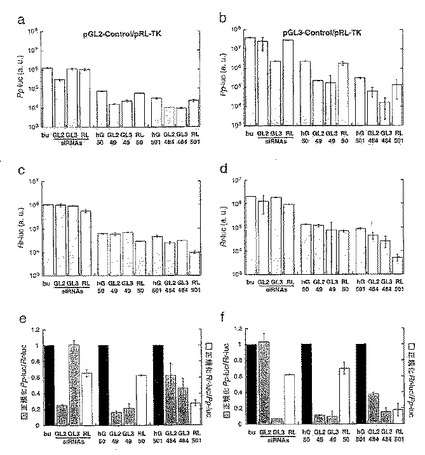
【図11】
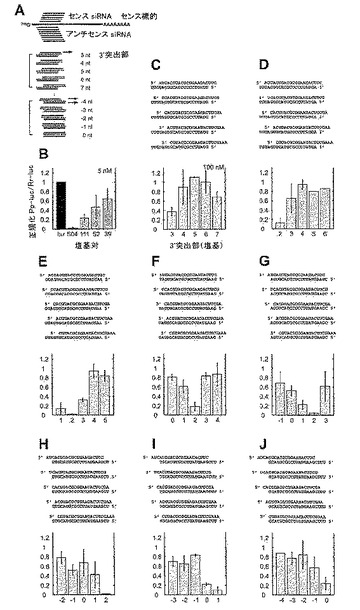
【図12】

【図13】

【図14】
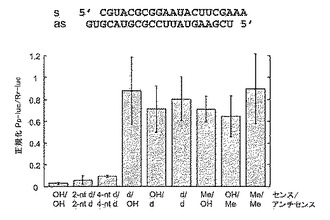
【図15A】

【図15B】
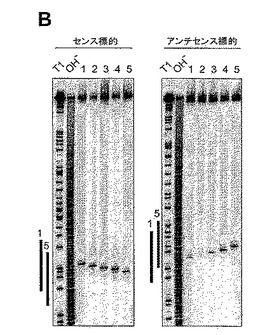
【図16】
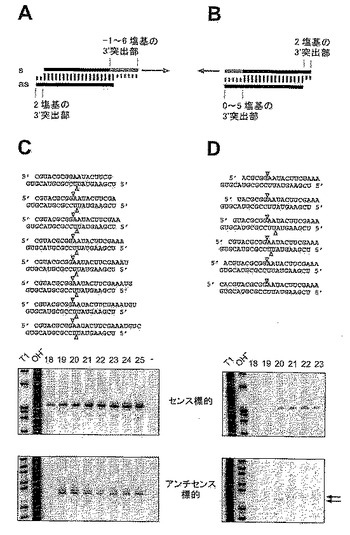
【図17】

【図18】
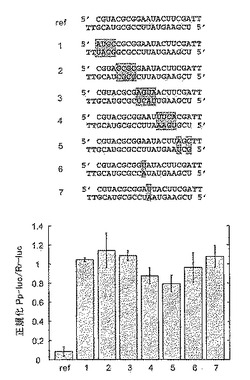
【図19】
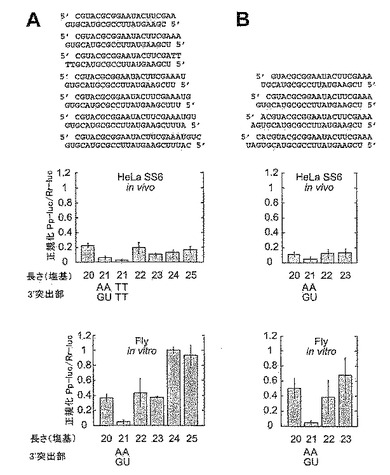
【図1B】
【図2】
【図3A】
【図3B】
【図4A】
【図4B】
【図5A】
【図5B】
【図6A】
【図6B】
【図7】
【図8a】
【図8b】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15A】
【図15B】
【図16】
【図17】
【図18】
【図19】
【公開番号】特開2009−284915(P2009−284915A)
【公開日】平成21年12月10日(2009.12.10)
【国際特許分類】
【出願番号】特願2009−210276(P2009−210276)
【出願日】平成21年9月11日(2009.9.11)
【分割の表示】特願2006−317758(P2006−317758)の分割
【原出願日】平成13年11月29日(2001.11.29)
【出願人】(596070445)マックス−プランク−ゲゼルシャフト ツール フォーデルング デル ヴィッセンシャフテン エー.ヴェー. (13)
【出願人】(500262197)ヨーロピアン モレキュラー バイオロジー ラボラトリー (13)
【Fターム(参考)】
【公開日】平成21年12月10日(2009.12.10)
【国際特許分類】
【出願日】平成21年9月11日(2009.9.11)
【分割の表示】特願2006−317758(P2006−317758)の分割
【原出願日】平成13年11月29日(2001.11.29)
【出願人】(596070445)マックス−プランク−ゲゼルシャフト ツール フォーデルング デル ヴィッセンシャフテン エー.ヴェー. (13)
【出願人】(500262197)ヨーロピアン モレキュラー バイオロジー ラボラトリー (13)
【Fターム(参考)】
[ Back to top ]