
RrhJ1IIヌクレアーゼおよびその遺伝子
【課題】遺伝子工学の分野で有用な新規なヌクレアーゼとその遺伝子、及びこれを利用した前記ヌクレアーゼの製造方法の提供。
【解決手段】ロドコッカスロドクロウス(Rhodococcusrhodochrous)J−1株からのニッキング酵素活性を有する新規ヌクレアーゼの単離、該酵素のアミノ酸配列の決定、およびこれをコードする遺伝子の塩基配列の決定。前記新規ヌクレアーゼ遺伝子を発現する組換え菌によるRrhJ1IIヌクレアーゼの製造方法。
【解決手段】ロドコッカスロドクロウス(Rhodococcusrhodochrous)J−1株からのニッキング酵素活性を有する新規ヌクレアーゼの単離、該酵素のアミノ酸配列の決定、およびこれをコードする遺伝子の塩基配列の決定。前記新規ヌクレアーゼ遺伝子を発現する組換え菌によるRrhJ1IIヌクレアーゼの製造方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、遺伝子工学試薬として有用な新規ヌクレアーゼ、該酵素タンパク質をコードする遺伝子、及び該酵素の製造方法に関する。
【背景技術】
【0002】
ヌクレアーゼ(Nuclease)は核酸の糖とリン酸の間のホスホジエステル結合を加水分解してヌクレオチドとする酵素である。分解の型式により、エンドヌクレアーゼとエキソヌクレアーゼに分類できる。
【0003】
エキソヌクレアーゼ(Exonuclease)は核酸配列の外側(exo−)から、すなわち核酸の5’端または3’端から削るように分解する。一方エンドヌクレアーゼ(Endonuclease)は核酸配列の内部(endo−)で核酸を切断する酵素であり、制限酵素は代表的なエンドヌクレアーゼである。
【0004】
分子生物学、または生化学等の発展により、DNAが遺伝子を司る本体であることが明らかになって以来、ヌクレアーゼは遺伝子操作等の遺伝子工学技術において現在幅広く用いられている有用な酵素である。
【0005】
ニッキング酵素(Nicking enzymeあるいはNicking endonuclease)は、二本鎖DNAのうち一方の鎖だけホスホジエステル結合が切断されたニックを生じさせるエンドヌクレアーゼであるが、その特性を利用して核酸増幅に用いられている。たとえば、ニッキング酵素を用いて二本鎖DNAの片方にニックを形成させ、生じたニックをプライミングサイトとして、鎖置換型DNAポリメラーゼにより、15塩基程度の短い核酸を恒温増幅する方法が知られている(非特許文献1)。また、この改良技術として、鎖置換型DNAポリメラーゼとニッキング酵素を用いて、21塩基以上の核酸を恒温増幅する方法も開発されている(特許文献1)。
【0006】
これまでにニッキング酵素は400種以上が報告されているが、ロドコッカス(Rhodococcus)属に属する細菌において、ニッキング酵素は4例が報告されているに過ぎない(非特許文献2)。
【0007】
発明者らは、これまでロドコッカス属ロドクロウスJ−1株から制限酵素と、これに対応する修飾酵素を単離しているが、この制限酵素にはニッキング酵素活性はない(特許文献2)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2008−136451号公報
【特許文献2】特開2007−259853号公報
【非特許文献】
【0009】
【非特許文献1】Jeffrey Van Ness,et al.,(2003)“Isothermal reactions for the amplification of oligonucleotides”Proc.Natl.Acad.Sci.USA, Vol.100, No.8, 4504−4509
【非特許文献2】Letek M,et al.,(2010)”The genome of a pathogenic rhodococcus: cooptive virulence underpinned by key gene acquisitions” PLoS Genet.2010 Sep 30;6(9).
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の目的は、遺伝子工学の分野で有用な新規なヌクレアーゼとその遺伝子を提供するとともに、これを利用した前記ヌクレアーゼの製造方法を提供することにある。
【課題を解決するための手段】
【0011】
発明者らは上記課題を解決するために鋭意研究を行った結果、ロドコッカス属に属する細菌:ロドコッカス ロドクロウス(Rhodococcus rhodochrous)J−1株より、新規なヌクレアーゼRrhJ1IIを単離した。
【0012】
すなわち、本発明は、ロドコッカス属微生物由来の新規ヌクレアーゼに関する。
本発明の新規ヌクレアーゼタンパク質は、以下の(A)、(B)または(C)のタンパク質である。
(A)配列番号2記載のアミノ酸配列を含むタンパク質
(B)配列番号2記載のアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質
(C)配列番号2記載のアミノ酸配列と相同性が80%以上のアミノ酸配列からなり、かつニッキング酵素活性を有するタンパク質
【0013】
また、上記新規ヌクレアーゼタンパク質は、上記ニッキング酵素活性が、二重鎖デオキシリボ核酸中の下記式1:
【化1】

(式中、Tはチミン、Gはグアニン、Cはシトシン、Sはグアニンまたはシトシン、Yはシトシンまたはチミン、Rはアデニンまたはグアニンを表す)で表される塩基配列に特異的であり、かつ、式1中のT/Gミスマッチに対するニッキング酵素活性であることを特徴とするタンパク質であってもよい。
【0014】
本発明は、上記新規ヌクレアーゼタンパク質をコードする遺伝子も提供する。
具体的には、本発明の新規ヌクレアーゼ遺伝子は、以下の(a)、(b)または(c)のDNAを含む。
(a)配列番号1記載の塩基配列からなるDNA
(b)配列番号1記載の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつニッキング酵素活性を有するタンパク質をコードするDNA
(c)配列番号1記載の塩基配列からなるDNAと相同性が80%以上の塩基配列からなり、かつニッキング酵素活性を有するタンパク質をコードするDNA
【0015】
また本発明は、本発明の新規ヌクレアーゼ遺伝子を含む組換ベクター、および前記組換ベクターを含む形質転換体も提供する。
【0016】
さらに本発明は、本発明の新規ヌクレアーゼタンパク質の製造方法も提供する。前記製造方法は、たとえば、上記した形質転換体を培養し、得られる培養物からニッキング酵素活性を有するタンパク質を採取することにより実施できる。好ましくは、前記製造方法は、細胞を用いない無細胞タンパク質合成法を用いて実施する。
【発明の効果】
【0017】
本発明により、遺伝子工学の分野で有用なニッキング酵素活性を有するエンドヌクレアーゼおよびその遺伝子が提供される。本発明の新規ヌクレアーゼは、前記遺伝子を用いた組換え製造により、簡便かつ大量に生産することができる。
【0018】
本発明の新規ヌクレアーゼは、ニッキング酵素活性を有するため、鎖置換型恒温核酸増幅や、染色体DNAの蛍光標識による可視化、生体内遺伝子ターゲティングのための相同組換えを誘発するための一本鎖DNA切断反応などに利用できる。
【図面の簡単な説明】
【0019】
【図1】ロドコッカス属細菌の産生するエンドヌクレアーゼのホモロジー解析結果を示す図である。
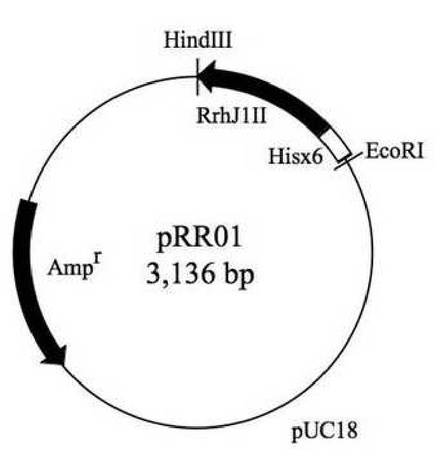
【図2】RrhJ1II発現プラスミドpRR01の構造を示す模式図である。
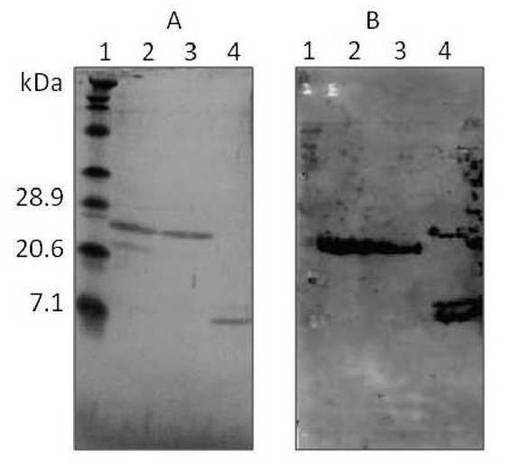
【図3】RrhJ1II発現プラスミドpRR01を含む組換大腸菌から精製したRrhJ1IIタンパク質に対するHis−tag特異的抗体を用いたウェスタンブロッティングの結果を示す図である。
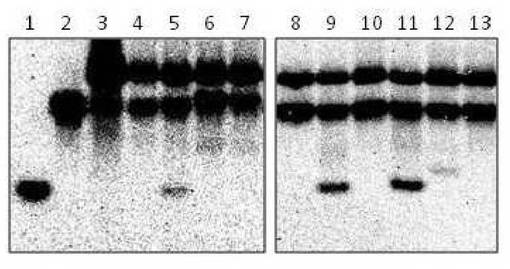
【図4】RrhJ1II発現プラスミドpRR01を含む組換大腸菌から精製したRrhJ1IIタンパク質のヌクレアーゼ活性を示す図である。
【発明を実施するための形態】
【0020】
以下、本発明の実施の形態について詳細に説明するが、本実施の形態は、本発明を説明するための例示であり、本発明をこの実施の形態のみに限定させるものではない。
【0021】
1.本発明の新規ヌクレアーゼ(RrhJ1IIヌクレアーゼ)
本発明の新規ヌクレアーゼは、ロドコッカス ロドクロウス(Rhodococcus rhodochrous)J−1株から、発明者らにより初めて単離された。ロドコッカス ロドクロウスJ−1株(以下、「J1菌」ともいう)はFERM BP−1478として独立行政法人産業技術総合研究所 特許生物寄託センター(茨城県つくば市東1丁目1番地1 中央第6)に寄託されている菌株である。
【0022】
発明者らは、このJ1菌由来の新規ヌクレアーゼを「RrhJ1II」と命名した。本明細書中では、このRrhJ1IIヌクレアーゼが有するニッキング酵素活性を「RrhJ1IIヌクレアーゼ活性」と記載する。
【0023】
本明細書において、「エンドヌクレアーゼ活性」とは、核酸配列の内部(endo−)で核酸を切断する酵素活性を意味し、「ニッキング酵素活性」とは、特定の配列を有する二本鎖DNAの片方の鎖のみを切断するエンドヌクレアーゼ活性を意味する。
【0024】
本明細書において、ニッキング酵素活性は、RrhJ1IIヌクレアーゼをDNAと接触させ、接触後のDNAの分子量またはDNA断片数を測定することにより評価することができる。当業者であれば、基質DNA、接触時の酵素量、温度、溶液組成または接触時間などの条件は適宜設定することができる。DNAの分子量は、例えばアガロース電気泳動によって測定することができる。接触前のDNAの分子量と接触後のDNAの分子量、または接触前のDNAの断片数と接触後のDNA断片数とを比較することで、ニッキング酵素活性を評価することができる。
【0025】
発明者らは、J1菌由来のRrhJ1IIヌクレアーゼが配列番号2に示されるアミノ酸配列を有することを同定した。
【0026】
しかしながら、本発明のRrhJ1IIヌクレアーゼタンパク質は、上記配列を有するものに限定されるものではなく、配列番号2記載のアミノ酸配列と約50%以上、好ましくは約60%以上、より好ましくは約70%以上、さらに好ましくは約80%以上、特に好ましくは約90%以上、さらに特に好ましくは約95%以上、最も好ましくは約98%以上の相同性あるいは同一性を有するアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質も本発明のRrhJ1IIヌクレアーゼに含まれる。
【0027】
また、本発明のRrhJ1IIヌクレアーゼタンパク質には、配列番号2記載のアミノ酸配列において、1個または数個のアミノ酸が欠失、置換または付加されたアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質も含まれる。
【0028】
具体的には、(i)配列番号2記載のアミノ酸配列において、1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が欠失したアミノ酸配列、(ii)配列番号2記載のアミノ酸配列の1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が他のアミノ酸に置換されたアミノ酸配列、(iii)配列番号2記載のアミノ酸配列に1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が付加したアミノ酸配列、(iv)配列番号2記載のアミノ酸配列に1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が挿入されたアミノ酸配列、(v)上記(i)〜(iv)を組み合わせたアミノ酸配列が挙げられる。
【0029】
アミノ酸置換は、類似するアミノ酸残基間の保存的置換が好ましい。例えばアミノ酸は、その側鎖の性質に基づいて、疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、水酸基含有側鎖を有するアミノ酸(S、T、Y)、硫黄原子含有側鎖を有するアミノ酸(C、M)、カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、塩基含有側鎖を有するアミノ酸(R、K、H)、芳香族含有側鎖を有するアミノ酸(H、F、Y、W)に分類される。各群に分類されたアミノ酸は、相互に置換したときに、当該ポリペプチドの活性が維持される可能性が高いことが知られており、そのようなアミノ酸相互の置換が好ましい。例えば、グリシンとプロリン、グリシンとアラニンまたはバリン、ロイシンとイソロイシン、グルタミン酸とグルタミン、アスパラギン酸とアスパラギン、システインとスレオニン、スレオニンとセリン又はアラニン、リジンとアルギニン間での置換を挙げることができる。
【0030】
2.本発明の新規ヌクレアーゼ遺伝子(RrhJ1IIヌクレアーゼ遺伝子)
本発明のRrhJ1IIヌクレアーゼ遺伝子は、上記したRrhJ1IIヌクレアーゼタンパク質をコードする遺伝子である。本発明のRrhJ1IIヌクレアーゼ遺伝子は、例えば配列番号1に記載の塩基配列からなるDNAを含む。
【0031】
本発明のRrhJ1IIヌクレアーゼ遺伝子は上記配列に限定されるものではなく、配列番号1記載の塩基配列と約50%以上、好ましくは約60%以上、より好ましくは約70%以上、さらに好ましくは約80%以上、特に好ましくは約90%以上、さらに特に好ましくは約95%以上、最も好ましくは約98%以上の相同性(同一性)を有する塩基配列を有するDNAも、それがRrhJ1IIヌクレアーゼとしての機能(ニッキング酵素活性)を有するタンパク質をコードする限り、本発明の遺伝子に含まれる。
【0032】
また、前述したアミノ酸配列の欠失、置換または付加に対応して、配列番号1記載の塩基配列において、数個の塩基に欠失、置換または付加等の変異が生じた塩基配列も、それが本発明のRrhJ1IIヌクレアーゼとしての機能(ニッキング酵素活性)を有するタンパク質をコードする限り、本発明のRrhJ1IIヌクレアーゼ遺伝子に含まれる。なお、欠失、置換または付加される塩基の個数は、30個以下、好ましくは15個以下、特に好ましくは6個以下である。
【0033】
さらに、配列番号1に記載の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAも、これがニッキング酵素活性を有するタンパク質をコードする限り、本発明の遺伝子に含まれる。
【0034】
ここで、ストリンジェントな条件としては、例えばDNAを固定したナイロン膜を、6×SSC(1×SSCは塩化ナトリウム8.76g、クエン酸ナトリウム4.41gを1リットルの水に溶かしたもの)、1%SDS、100μg/mlサケ精子DNA、0.1%ウシ血清アルブミン、0.1%ポリビニルピロリドン、0.1%フィコールを含む溶液中で65℃にて20時間プローブとともに保温してハイブリダイゼーションを行う条件を挙げることができるが、これに限定されるわけではない。当業者であれば、このような緩衝液の塩濃度、温度等の条件に加えて、その他のプローブ濃度、プローブの長さ、反応時間等の諸条件を加味し、ハイブリダイゼーションの条件を設定することができる。
【0035】
ハイブリダイゼーション法の詳細な手順については、Molecular Cloning, A Laboratory Manual 2nd ed.(Cold Spring Harbor Laboratory Press (1989))等を参照することができる。
【0036】
以下に、ハイブリダイゼーションによりRrhJ1IIヌクレアーゼ遺伝子を得る方法の一例を示すが、これに限定されるわけではない。
【0037】
まず、適当な遺伝子源から得たDNAを定法に従ってプラスミドやファージベクターに接続してDNAライブラリを作製する。このライブラリを適当な宿主に導入して得られる形質転換体をプレート上で培養し、生育したコロニーまたはプラークをニトロセルロースやナイロンの膜にうつしとり、変性処理の後にDNAを膜に固定する。この膜をあらかじめ32P等で標識したプローブを含む上記の組成の溶液中、上記のストリンジェントな条件で保温し、ハイブリダイゼーションを行う。プローブとしては、配列番号2に記載したアミノ酸配列の全部または一部をコードするポリヌクレオチドを使用することができる。
【0038】
ハイブリダイゼーションの終了後、非特異的に吸着したプローブを洗い流し、オートラジオグラフィ等によりプローブとハイブリッドを形成したクローンを同定する。この操作をハイブリッド形成クローンが単離できるまで繰り返す。最後に、得られたクローンの中から、目的の酵素活性を有するタンパク質をコードする遺伝子を選択する。遺伝子の単離は、アルカリ法等の公知のポリヌクレオチド抽出法により実施できる。
【0039】
本発明のRrhJ1IIヌクレアーゼ遺伝子は、当該酵素を発現する微生物から単離することもできる。例えば、ロドコッカス ロドクロウスJ−1株由来のゲノムDNAを鋳型として、既知のアミノ酸配列情報から遺伝子の縮重を考慮して設計したプライマーもしくはプローブまたは既知の塩基配列情報に基づいて設計したプライマーまたはプローブを用いたPCRまたはハイブリダイゼーション法により、前記微生物のゲノムから目的の遺伝子を単離することができる。
【0040】
このように種々のDNAが本発明のRrhJ1IIヌクレアーゼ遺伝子の範囲内に含まれるのは、コドンの縮重に由来する。すなわち、遺伝子上でアミノ酸を指定するコドン(3つの塩基の組み合わせ)は、アミノ酸の種類ごとに1〜6種類存在することが知られている。従って、あるアミノ酸配列をコードする遺伝子は多数存在しうる。
【0041】
遺伝子は自然界において安定に存在しているものではなく、その塩基配列に変異が起こることは稀ではない。遺伝子上に起こった変異によっては、コードされるアミノ酸配列に変化を与えない変異(サイレント変異と呼ばれる)もあり、この場合には同じアミノ酸配列をコードする、異なる遺伝子が生じたと言える。従って、ある特定のアミノ酸配列をコードする遺伝子が単離されても、それを含有する生物が継代されていくうちに同じアミノ酸配列をコードする多種類の遺伝子ができて行く可能性は否定できない。
【0042】
さらに、同じアミノ酸配列をコードする多種類の遺伝子を人為的に作製することは、種々の遺伝子工学的手法を用いれば困難なことではない。例えば、遺伝子工学的なタンパク質の生産において、目的のタンパク質をコードする本来の遺伝子上で使用されているコドンが宿主中では使用頻度の低いものであった場合には、タンパク質の発現量が低いことがある。このような場合にはコードされているアミノ酸配列に変化を与えることなく、コドンを宿主で繁用されているものに人為的に変換することにより、目的タンパク質の高発現を図ることが行われている。
【0043】
このように、特定のアミノ酸配列をコードする多種類の遺伝子は人為的に作製可能なことは言うまでもなく、自然界においても生成されうるものである。従って、本発明中に開示された塩基配列と同一の遺伝子ではなくても、RrhJ1IIと同等の活性を示すタンパク質をコードするDNAである限り、本発明のRrhJ1IIヌクレアーゼ遺伝子に含まれる。
【0044】
遺伝子に変異を導入し、人為的にアミノ酸配列を改変する方法としては、Kunkel法やGapped duplex法等の公知手法や、部位特異的突然変異誘発法を利用した変異導入キット、例えばQuikChangeTM Site−Directed Mutagenesis Kit(ストラタジーン社)、GeneTailorTM Site−Directed Mutagenesis System(インビトロジェン社)、TaKaRa Site−Directed Mutagenesis System(Mutan−K,Mutan−Super Express Km等:タカラバイオ社)等を用いることができる。
【0045】
DNAの塩基配列の確認は、慣用の方法により配列決定することにより行うことができる。例えば、ジデオキシヌクレオチドチェーンターミネーション法(Sanger et al.(1977)Proc.Natl.Acad.Sci.USA 74:5463)等により行うことができる。また、適当なDNAシークエンサーを利用して配列を解析することも可能である。
【0046】
塩基配列の決定は、プラスミドベクターを用いて作製された形質転換体の場合、宿主がエシェリヒア コリ(Escherichia coli)であれば試験管等で培養を行い、定法に従ってプラスミドを調製する。得られたプラスミドをそのまま鋳型とするか、あるいは挿入断片を取り出してM13ファージベクター等にサブクローニングした後に、ジデオキシ法により塩基配列を決定する。ファージベクターで作製された形質転換体の場合も基本的に同様な操作により塩基配列を決定することができる。これら培養から塩基配列決定までの基本的な実験法については、例えば前述のT.ManiatisらのMolecular Cloning,A Laboratory Manual等に記載されている。
【0047】
得られた遺伝子が目的のRrhJ1IIヌクレアーゼをコードする遺伝子であるかどうかの確認は、決定された塩基配列を配列番号1に記載の塩基配列と比較して行うことができる。あるいは決定された塩基配列より推定されるアミノ酸配列を配列番号2に記載のアミノ酸配列と比較して行うことができる。
【0048】
3.本発明の新規ヌクレアーゼ遺伝子を含む組換えベクター
本発明は、RrhJ1IIヌクレアーゼ遺伝子を含む組換えベクターも提供する。本発明の組換えベクターは、上記酵素をコードする遺伝子の上流に転写プロモーター、場合によっては下流にターミネーターを挿入して発現カセットを構築し、このカセットを発現ベクターに挿入することにより作製することができる。あるいは、発現ベクターに転写プロモーターおよび/またはターミネーターがすでに存在する場合には、発現カセットを構築することなく、ベクター中のプロモーターおよび/またはターミネーターを利用して、その間に当該酵素をコードする遺伝子を挿入すればよい。
【0049】
本明細書において、プロモーターは、例えば、trcプロモーター、lacプロモーターなどをあげることができるが、これに限定されるわけではない。
本明細書において、ターミネーターは、例えば、trpオペロンターミネータをあげることができるが、これに限定されるわけではない。
【0050】
ベクターに本発明の遺伝子を挿入するには、制限酵素を用いる方法、トポイソメラーゼを用いる方法等を利用することができる。また、挿入の際に必要であれば、適当なリンカーを付加してもよい。また、アミノ酸への翻訳にとって重要な塩基配列として、SD配列やKozak配列などのリボソーム結合配列が知られており、これらの配列を遺伝子の上流に挿入することもできる。挿入にともない、遺伝子がコードするアミノ酸配列の一部を置換してもよい。
【0051】
本発明において使用されるベクターは、本発明の遺伝子を保持するものであれば特に限定されず、それぞれの宿主に適したベクターを用いることができる。ベクターとしては、例えば、プラスミドDNA、バクテリオファージDNA、レトロトランスポゾンDNA、人工染色体DNAなどが挙げられる。例えば、大腸菌を宿主とする場合には、pTrc99A(GEヘルスケア バイオサイエンス)、pACYC184(ニッポンジーン)、pMW118(ニッポンジーン)などを挙げることができる。また、必要に応じて、これらのベクターを改変したものを用いることもできる。
【0052】
4.本発明のベクターを導入した形質転換体
本発明の組換えベクターを宿主に導入することで、本発明の形質転換体を作製することができる。
【0053】
形質転換体に使用する宿主は、上記組換えベクターが導入された後、目的の制限酵素または修飾酵素を発現することができる限り、特に限定されるものではない。宿主としては、例えば、大腸菌(エシェリヒア・コリ)、枯草菌(バチルス・ズブチリス(Bacillus subtilis))、ロドコッカス菌(Rhodococcus)、放線菌などの細菌、酵母(サッカロミセス・セレビシエ(Saccharomyces cerevisiae))、カビ、動物細胞、植物細胞、昆虫細胞などが挙げられる。本発明において、宿主は好ましくは大腸菌である。
【0054】
本発明において、大腸菌は、例えば、大腸菌K12株やB株、あるいはそれらの野生株由来の派生株であるJM109株、XL1−Blue株(例えば、XL1−Blue MRF’)、K802株、C600株などを挙げることができる。
【0055】
宿主への組換えベクターの導入方法としては、宿主に適した方法であれば特に限定されるものではなく、当業者であれば公知技術から適宜選択することができる。このような方法としては、例えば、エレクトロポレーション法、カルシウムイオンを用いる方法、スフェロプラスト法、酢酸リチウム法、リン酸カルシウム法、リポフェクション法等が挙げられる。
【0056】
5.本発明のRrhJ1IIヌクレアーゼの製造方法
5−1 細胞系による製造
本発明のRrhJ1IIヌクレアーゼは、形質転換体を培養して、培養物中よりRrhJ1IIヌクレアーゼの特徴(例えば、ニッキング酵素活性)を有する活性を有するタンパク質を採取することによりRrhJ1IIヌクレアーゼを製造することができる。
【0057】
本発明において「培養物」とは、菌体、培養液、無細胞抽出液、細胞膜などの培養により得られるものを意味する。無細胞抽出液は、培養後の菌体を、例えばリン酸ナトリウム緩衝液を加えてホモジナイザーなどで物理的に破砕した後、遠心(15,000rpm, 10min, 4℃)し、破砕できない菌体(細胞)が存在しないように上清を回収して得ることができる。細胞膜は、上記遠心で得られたペレットとして得ることができ、また、ペレットを溶解バッファーで懸濁することにより得ることもできる。
【0058】
本発明のRrhJ1IIヌクレアーゼは、培養物をそのまま用いてもよいし、透析や硫安沈殿などの公知の方法、あるいは公知の方法を単独または適宜組み合わせることによって、培養物から濃縮、精製したものを用いてもよい。この場合、酵素活性、分子量、等電点などを指標に濃縮、精製することができる。
【0059】
たとえば、形質転換体の培養物からRrhJ1IIヌクレアーゼを採取するにあたっては、培養物より菌体を集菌後、超音波破砕、超遠心分離等により酵素を抽出し、ついで除核酸法、塩析法、アフィニティクロマトグラフィー法、ゲル濾過法、イオン交換クロマトグラフィ法等を組み合わせて精製すればよい。この方法により、RrhJ1IIヌクレアーゼを大量に得ることができる。
【0060】
用いる発現系によっては、形質転換体中で発現された酵素タンパク質が不溶物[封入体(inclusionbody)]として蓄積される場合がある。この場合にはこの不溶物を回収し、穏和な変性条件、たとえば尿素等の変性剤存在下で可溶化した後に変性剤を除くことによって活性型のタンパク質を得ることができる。さらに上記のようなクロマトグラフィー操作を行って目的とする酵素タンパク質を精製することができる。
【0061】
5−2 無細胞タンパク質合成系による製造
本発明においては、無細胞タンパク質合成系を用いて、本発明のRrhJ1IIヌクレアーゼ遺伝子または本発明のベクターから、RrhJ1IIヌクレアーゼを製造することも可能である。
【0062】
無細胞タンパク質合成系とは、細胞抽出液を用いて試験管等の人工容器内でタンパク質を合成する系である。すなわち、無細胞タンパク質合成系は、生物を用いた組換えタンパク質発現とは異なり、細胞を使用せずに試験管内で転写・翻訳という一連のタンパク質合成の流れを行う合成系であるため、細胞にとって毒性となるタンパク質を生産できるという利点がある。なお、本発明において使用される無細胞タンパク質合成系にはDNAを鋳型としてRNAを合成する無細胞転写系も含まれる。
【0063】
無細胞タンパク質合成系の細胞抽出液としては、真核細胞由来または原核細胞由来の抽出液、例えば小麦胚芽、大腸菌等の抽出液を使用することができる。これらの細胞抽出液は濃縮されたものであっても濃縮されていないものであってもよい。
【0064】
細胞抽出液は、例えば限外濾過、透析、ポリエチレングリコール(PEG)沈殿等によって得ることができる。さらに本発明において、無細胞タンパク質合成は、市販のキットを用いて行うこともできる。そのようなキットとしては、例えば試薬キットPROTEIOSTM(東洋紡)、TNTTM System(プロメガ)、合成装置のPG−MateTM(東洋紡)、RTS(ロシュ ダイアグノスティクス)等が挙げられる。
【0065】
こうした細胞抽出液に代えて、タンパク質合成に関与するすべての因子を精製し、それらをリボソーム、ATP、tRNA、アミノ酸などとともに再構成して作成された「再構成された無細胞タンパク質合成系」を用いることもできる。
【0066】
通常の無細胞タンパク質合成系に用いられる細胞抽出液には、ATPを加水分解する酵素などの混在によりタンパク質合成以外に無駄に消費されるエネルギー量が多く、エネルギー効率が低く、また細胞抽出液中に存在するプロテアーゼやヌクレアーゼなどの阻害因子によりンパク質合成の効率が低下する場合がある。
【0067】
「再構成された無細胞タンパク質合成系」は、こうした阻害的要因の問題がなく、また構成成分を自由に操作することができるため、目的や標的タンパク質に応じた自由なシステム設計が可能である。また系の自由度が高く、反応系を微小化したり、自動化装置と組み合わせてハイスループットにすることも可能である。
【0068】
「再構成された無細胞タンパク質合成系」の一例としてPURESYSTEM(登録商標)を挙げることができる。PURESYSTEM(登録商標)は大腸菌から再構成されているが、無細胞タンパク質合成系で用いられる他の公知の細胞、昆虫細胞、小麦胚芽、ウサギ網状赤血球のいずれを用いても、同様の系は構成可能である。
【0069】
上記のように無細胞タンパク質合成系によって得られる本発明のヌクレアーゼは前述のように適宜クロマトグラフィを選択して、濃縮、精製することができる。
【0070】
6.RrhJ1IIヌクレアーゼ活性の解析
6.1 RrhJ1IIヌクレアーゼの認識配列
RrhJ1IIヌクレアーゼの認識配列を調べるために、たとえば前述のような方法で配列番号1に示される遺伝子を発現可能に組み込んだベクターで宿主細胞を形質転換し、その培養物からRrhJ1IIヌクレアーゼ活性を有するタンパク質を得る。得られたタンパク質を、片方の鎖の5'末端に蛍光標識を持ち、配列内に種々のT/Gミスマッチを持つ二本鎖オリゴDNAを合成して活性測定用の試料と接触させた後、変性剤と加熱によりDNAを一本鎖に変性させてSDS−ポリアクリルアミドゲル電気泳動に供し、化学発光による検出を行って、切断パターンを解析すると、J1菌ヌクレアーゼの認識配列を決定することができる。本発明のRrhJ1IIヌクレアーゼは、以下の二重鎖デオキシリボ核酸中の下記式1
【化2】
(式中、Tはチミン、Gはグアニン、Cはシトシン、Sはグアニンまたはシトシン、Yはシトシンまたはチミン、Rはアデニンまたはグアニンを表す)で表される塩基配列を特異的に認識し、かつ、囲み文字で示した位置のT/Gミスマッチに対するニッキング酵素活性を示すことを特徴としている。ここで、「T/Gミスマッチ」とは、チミジン(T)とグアニン(G)との間で水素結合が形成されたミスマッチ塩基対をいう。
【0071】
式1中、S,Y,Rで表記される部分の配列は、上記説明文中に示される塩基である限り特に限定されないが、囲み文字で示した位置のT/Gミスマッチが解消し、リード鎖のTがCとなった場合に回文配列となることが好ましい。たとえば、下記式2〜4を挙げることができる。
【化3】
【実施例】
【0072】
以下、実施例を用いて本発明をより詳細に説明する。以下は本発明の例示であって本発明を限定する趣旨ではない。
【0073】
[実施例1]
J1菌染色体DNAの調製
ロドコッカス ロドクロウス(Rhodococcus rhodochrous)J−1株を100mlのMYKG培地中、30℃にて72時間振盪培養した。
【0074】
培養後、集菌し、集菌された菌体をSaline−EDTA溶液(0.1M EDTA,0.15M NaCl(pH8.0))4mlに懸濁した。懸濁液にリゾチーム40mgを加えて37℃で1〜2時間振盪した後、−20℃で凍結した。
【0075】
次に、10mlのTris−SDS液(1%SDS,0.1M NaCl,0.1M Tris−HCl(pH9.0))を穏やかに振盪しながら加え、さらにプロテイナーゼK(メルク社)(10mg/ml)を10μl加えて37℃で1時間振盪した。
【0076】
次に、等量のTE(10mM Tris−HCl,1mM EDTA(pH8.0))飽和フェノールを加え、攪拌後、遠心した。上層を採取し、2倍量のエタノールを加えた後、ガラス棒でDNAを巻き取り、90%,80%,70%のエタノールで順次フェノールを取り除いた。
【0077】
次に、DNAを3mlのTE緩衝液に溶解させ、リボヌクレアーゼA溶液(100℃,15分間の加熱処理済)を10μg/mlになるように加え、37℃で30分間振盪した。さらに、プロテイナーゼKを加え37℃で30分間振盪した後、等量のTE飽和フェノールを加えて遠心し、上層と下層に分離させた。
【0078】
上層についてこの操作を2回繰り返した後、同量のクロロホルム(4%イソアミルアルコール含有)を加え、同様の抽出操作を繰り返した。その後、上層に2倍量のエタノールを加え、ガラス棒でDNAを巻き取り回収し、染色体DNA標品を得た。
【0079】
[実施例2]
RrhJ1IIヌクレアーゼ遺伝子のPCR
RrhJ1IIヌクレアーゼ遺伝子をPCRで増幅するためのプライマーを以下の方法で設計した。
【0080】
図1はロドコッカス属細菌の産生するヌクレアーゼのアミノ酸ホモロジー解析結果を示す。図1中でホモロジー解析を行ったヌクレアーゼとその由来は以下のとおりである。
A:DNA mismatch endonuclease Vsr(Rhodococcus equi ATCC33707)
B:NaeI very short patch repair endonuclease(Rhodococcus erythropolis SK121)
C:Very short patch repair protein(Rhodococcus jostii RHA1)
D:DNA mismatch endonuclease (Rhodococcus erythropolis PR4)
E:DNA mismatch endonuclease(Rhodococcus
opacus B4)
【0081】
図1中、特に保存されている2つの領域を選んで配列番号3および配列番号4のデジェネレイトプライマーを設計し、実施例1で調製したJ1菌ゲノムDNAを鋳型としてデジェネレイトPCRを以下の条件で実施した。その結果、約350bのバンドの増幅が確認された。
【0082】
反応液組成
鋳型DNA(J1菌染色体DNA,実施例1) 1μl
10×Ex Buffer(タカラバイオ社) 10μl
150μMプライマーDG−01(配列番号3) 1μl
150μMプライマーDG−02(配列番号4) 1μl
2.5mM dNTP 8μl
DMSO 10μl
滅菌水 18μl
ExTaq DNAポリメラーゼ(タカラバイオ社)1μl
総量 50μl
【0083】
温度サイクル:94℃:30秒、65℃:30秒および72℃:1分の反応を30サイクル
【0084】
プライマー
DG−01:5’−GA(T/C)CCIGCIACITCIGCICGIATG−3’(配列番号3)
DG−02:5’−CCAIACICGIACIACIGTCCA−3’(配列番号4)
【0085】
次に、増幅された配列のダイレクトシークエンシングをPCRで使用したプライマーを用いて実施した。その結果、配列番号5に示す配列が得られ、ホモロジー検索の結果、前述のエンドヌクレアーゼとの相同性が認められた。
【0086】
[実施例3]
RrhJ1IIヌクレアーゼ遺伝子のクローニング
(1)ゲノミックサザンハイブリダイゼーション
ApaLI,BamHI,ClaI,Eco52I,EcoT14I,KpnI,MluI,NcoI,NotI,PvuI,SacI,XbaI,XhoIそれぞれで消化したJ1菌ゲノムDNAに対し、後述の方法で調製したRrhJ1IIのプローブを用いてサザンハイブリダイゼーションを行ったところ、Eco52Iで消化した断片から、約1.2kbの単一シグナルが得られた。
【0087】
なお、RrhJ1IIのプローブは以下のようにして調製した。
実施例2で調製したPCR産物をGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を用いて精製した。精製したPCR産物に対してAlkPhos Direct Labeling kit(GEヘルスケア バイオサイエンス社)を用い、添付のマニュアルにしたがってラベリングを行い、RrhJ1IIのプローブとした。
【0088】
(2)コロニーハイブリダイゼーション
J1菌ゲノムDNAを制限酵素Eco52Iで分解して0.7%アガロースゲル電気泳動で分離し、ゲルからGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を使用して約1.2kbの断片を回収した。得られた断片は、pBluescriptII SK(+)ベクター(Stratagene社製)にDNA ligation kit<Mighty mix>(タカラバイオ社製)を用いて連結した。反応条件は以下の通りである。
【0089】
反応液組成
ligation mighty mix(タカラバイオ社製) 5μl
J1菌ゲノムDNA/Eco52I切断断片 4μl
pBluescriptII SK(+)/Eco52I切断断片 1μl
総量 10μl
【0090】
反応:
16℃,1時間
【0091】
上記ライゲーション産物の全量を、後述の方法で調製した大腸菌JM109株コンピテントセル200μlに加え、0℃で30分放置した。続いて、前述コンピテントセルに42℃で45秒間ヒートショックを与え、0℃で2分間冷却した。その後、SOC培地(20mMグルコース、2%バクトトリプトン、0.5%バクトイーストエキス、10mM NaCl,2.5mM KCl,1mM MgSO4,1mM MgCl2)を1ml添加し、37℃にて1時間振盪培養した。培養後の培養液を200μlずつ、LB AIXプレート(100μg/lアンピシリン、100μM IPTG,50μg/l X−galを含むLB寒天培地)に塗布し、37℃で一晩放置した。プレート上に生育した白色の組換コロニーを新しいLB AIXプレートに、プレート1枚に付き94個、プレート10枚分単離した。各プレートにはインサートを含まないpBluescriptII SK(+)で形質転換したJM109株を2コロニー/プレート植菌した。コロニー単離したプレートを37℃で一晩放置した後、Hybond−N+(GEヘルスケア バイオサイエンス社)膜にコロニーを写し取り、実施例3(1)で調製したRrhJ1IIのプローブを用いてコロニーハイブリダイゼーションを行った。
【0092】
検出されたコロニーを培養して得られた培養液を集菌後、QIAprep miniprep kit(QIAGEN社製)を用いて組換えプラスミドを回収した。キャピラリーDNAシーケンサーCEQ2000(ベックマン・コールター社製)を用いて、添付のマニュアルに従って、プラスミド中にクローニングされているゲノムDNA断片の塩基配列を解析した。その結果、配列番号6に示される塩基配列が得られた。配列番号6に示される塩基配列中に、配列番号2に示す450bpのオープンリーディングフレーム(ORF1)を見出した。このORF1のコードするアミノ酸配列は、図1中に示したA〜Eのロドコッカス属細菌由来エンドヌクレアーゼに対して87%〜89%の相同性を持っていることから、ORF1はエンドヌクレアーゼをコードしていることが推定されたため、ORF1のコードするエンドヌクレアーゼをRrhJ1IIヌクレアーゼと命名した。また、本実施例3(2)で得られたORF1を含むプラスミドをpBRrhJ1IIと命名した。
【0093】
なお、大腸菌JM109株のコンピテントセルは以下の方法で調製した。
大腸菌JM109株をLB培地1mlに接種し、37℃で5時間好気的に前培養した。
次に、前培養液0.4mlをSOB培地40ml(2%バクトトリプトン、0.5%バクトイーストエキス、10mM NaCl,2.5mM KCl,1mM MgSO4,1mM MgCl2)に加え、18℃で20時間培養した。得られた培養物を遠心分離(3,700×g,10分間、4℃)により集菌した後、冷TF溶液(20mM PIPES−KOH(pH6.0),200mM KCl,10mM CaCl2,40mM MnCl2)を13ml加え、0℃で10分間放置し、再度遠心分離(3,700×g,10分間、4℃)して上清を除いた。得られた大腸菌菌体を冷TF溶液3.2mlに懸濁し、0.22mlのジメチルスルホキシドを加え、0℃で10分間放置した後、液体窒素を用いて凍結したものをコンピテントセルとした。
【0094】
[実施例4]
大腸菌組換体によるRrhJ1IIヌクレアーゼの生産
(1)RrhJ1IIヌクレアーゼ発現プラスミドの構築
RrhJ1IIヌクレアーゼを得るために、実施例1で得られたJ1菌染色体DNAを鋳型として使用し、以下に示す反応液組成およびプライマーを用いてPCRを行った。この際、RrhJ1IIヌクレアーゼをHis−tag融合タンパクとして発現させるため、RrhJ1IIヌクレアーゼ遺伝子上流にSD配列とHis−tag配列を付加した。
【0095】
反応液組成
鋳型DNA(J1菌染色体DNA) 1μl
2×PCR Buffer KOD FX(東洋紡) 25μl
10μMプライマーN(配列番号7) 1.5μl
10μMプライマーC(配列番号8) 1.5μl
2mM dNTP 10μl
KOD FX DNAポリメラーゼ(東洋紡) 1μl
総量 50μl
【0096】
温度サイクル:94℃:120秒、98℃:10秒および68℃:1分の反応を30サイクル
【0097】
プライマー
N:5'−AGTGAATTCCTTTAAGAAGGAGATATACCATGCATCATCATCATCATCACATGGCGTCGTCGGAT−3'(配列番号7)
C:5'−GCCAAGCTTTCACCCCCGCGCCGGTTT−3'(配列番号8)
【0098】
反応終了後、反応液5μlを1%アガロースゲルにおける電気泳動に供し、約0.5kbのPCR産物の検出を行った。PCR産物を確認した後、反応液からPCR産物をDNA/RNA extaction Kit(VIOGENE社)で精製した。得られたPCR産物を、制限酵素EcoRIとHindIIIで切断した。制限酵素処理を行ったPCR産物を1%アガロースゲルにおける電気泳動に供し、約0.5kb付近のバンドを回収した。回収したPCR産物をベクターpUC18のEcoRI−HindIII部位に連結し、プラスミドを作製した。得られたプラスミドをpRR01と名づけた。図2はプラスミドpRR01の構造を示す模式図である。
【0099】
このプラスミドで大腸菌JM109を形質転換し、100μg/mlアンピシリン、1mM IPTG,50μg/ml X−galを含むLB寒天培地上でコロニーを形成させた。得られた白色のコロニーを100μg/mlアンピシリンを含むLB液体培地に植菌し、37℃で一晩培養した後Mini Plus Plasmid DNA Extraction kit(VIOGENE社)を使用してプラスミドを回収し、EcoRI−HindIIIによる切断とシークエンス解析を行って目的のDNA断片が挿入されていることを確認した。
【0100】
(2)精製酵素の調製
(1)で作製したRrhJ1IIヌクレアーゼ発現ベクターを含む大腸菌組換体(JM109/pRR01)を、100μg/mlアンピシリンを含むLB液体培地10mlで37℃,5時間培養した後、1mlを100μg/mlアンピシリン、1mM IPTGを含むLB液体培地100ml×1本に植菌し、37℃,18時間培養した。培養液を遠心分離によって回収し、回収した菌体を破砕用緩衝液(組成 20mM Sodium Phosphate,0.5M NaCl,20mM イミダゾール、10% グリセロール、pH7.4)に懸濁した後4℃で10分間超音波による破砕を行った。破砕液を遠心分離し、得られた上清を無細胞抽出液とした。この無細胞抽出液をHis−tag精製カラム(His Trap HP:GE Healthcare)を用いて精製した。精製タンパク質はElution Buffer(0.5M NaCl,0.5M イミダゾール、20mM Sodium Phosphate,10%グリセロール、pH7.4)にて溶出し、2つのフラクションに分画してフラクション1,フラクション2とした。
【0101】
得られた精製サンプルをSDS−PAGEおよびHis−tag特異的抗体(GE Healthcare)を用いたウェスタンブロッティングに供し、目的タンパク質が精製されていることを確認した。結果を図3に示す。図3中、A,Bの「1」は分子量マーカー(BioRad Prestained Standard Broad)、「4」はポジティヴコントロール(His×6融合タンパク質、12kDa)である。SDS−PAGEの結果、フラクション1(図3A「2」)とフラクション2(図3A「3」)で約25kDa付近にバンドが出現し、His−tag特異的抗体を用いたウェスタンブロッティングにより、目的タンパク質であることを確認した(図3B「2」「3」)。
【0102】
(3)酵素活性の測定
(2)で調製した精製酵素を用いて、RrhJ1IIヌクレアーゼ活性を調べた。活性は片方の鎖の5'末端にビオチン標識を持ち、配列内にT/Gミスマッチを1箇所持つ二本鎖オリゴDNAを用いてそのニッキング活性を測定した。
【0103】
反応液組成
1.5μM基質DNA(配列番号9〜18) 2μl
100mM Tris−HCl(pH7.5) 2μl
100mM MgCl2 2μl
1mg/ml BSA 2μl
総量 20μl
【0104】
【化4】
【0105】
反応温度:20℃,60分
【0106】
反応終了後、反応液と等量のフェノールでDNAを抽出し、ホルムアミド(終濃度1M)の存在下、100℃で2分間加熱後氷上にて急冷処理し、DNAを一本鎖に変性させた。変性後の一本鎖DNAサンプルを、8M尿素を含む20%ポリアクリルアミドゲル電気泳動に供し、Imaging high chemilumi kit(東洋紡)を使用して化学発光による検出を行って泳動パターンを比較した。
【0107】
結果を図4に示す。図4中、「1」は20merの一本鎖DNA,「2」は40merの一本鎖DNA,「3」はネガティヴコントロール(酵素を含まない反応液でインキュベートと変性処理を行った基質DNA01)である。オリゴヌクレオチド02、06、08は切断され約20merの1本鎖が生成した(図4 「5」,「9」,「11」)。一方、01、03、04、05、07、09、10は切断されなかった(図4 「4」、「6」、「7」、「8」、「10」、「12」、「13」)ことから、RrhJ1IIヌクレアーゼはミスマッチを認識してDNAを切断するエンドヌクレアーゼ活性を持つことを確認した。
【産業上の利用可能性】
【0108】
本発明の新規ヌクレアーゼおよびその遺伝子は、遺伝子工学の分野で有用であり、鎖置換型恒温核酸増幅や、染色体DNAの蛍光標識による可視化、生体内遺伝子ターゲティングのための相同組換えを誘発するための一本鎖DNA切断反応などに利用できる。
【配列表フリーテキスト】
【0109】
配列番号3:プライマーDG−01
配列番号4:プライマーDG−02
配列番号7:プライマーN
配列番号8:プライマーC
配列番号9:二本鎖オリゴDNA01
配列番号10:二本鎖オリゴDNA02
配列番号11:二本鎖オリゴDNA03
配列番号12:二本鎖オリゴDNA04
配列番号13:二本鎖オリゴDNA05
配列番号14:二本鎖オリゴDNA06
配列番号15:二本鎖オリゴDNA07
配列番号16:二本鎖オリゴDNA08
配列番号17:二本鎖オリゴDNA09
配列番号18:二本鎖オリゴDNA10
【技術分野】
【0001】
本発明は、遺伝子工学試薬として有用な新規ヌクレアーゼ、該酵素タンパク質をコードする遺伝子、及び該酵素の製造方法に関する。
【背景技術】
【0002】
ヌクレアーゼ(Nuclease)は核酸の糖とリン酸の間のホスホジエステル結合を加水分解してヌクレオチドとする酵素である。分解の型式により、エンドヌクレアーゼとエキソヌクレアーゼに分類できる。
【0003】
エキソヌクレアーゼ(Exonuclease)は核酸配列の外側(exo−)から、すなわち核酸の5’端または3’端から削るように分解する。一方エンドヌクレアーゼ(Endonuclease)は核酸配列の内部(endo−)で核酸を切断する酵素であり、制限酵素は代表的なエンドヌクレアーゼである。
【0004】
分子生物学、または生化学等の発展により、DNAが遺伝子を司る本体であることが明らかになって以来、ヌクレアーゼは遺伝子操作等の遺伝子工学技術において現在幅広く用いられている有用な酵素である。
【0005】
ニッキング酵素(Nicking enzymeあるいはNicking endonuclease)は、二本鎖DNAのうち一方の鎖だけホスホジエステル結合が切断されたニックを生じさせるエンドヌクレアーゼであるが、その特性を利用して核酸増幅に用いられている。たとえば、ニッキング酵素を用いて二本鎖DNAの片方にニックを形成させ、生じたニックをプライミングサイトとして、鎖置換型DNAポリメラーゼにより、15塩基程度の短い核酸を恒温増幅する方法が知られている(非特許文献1)。また、この改良技術として、鎖置換型DNAポリメラーゼとニッキング酵素を用いて、21塩基以上の核酸を恒温増幅する方法も開発されている(特許文献1)。
【0006】
これまでにニッキング酵素は400種以上が報告されているが、ロドコッカス(Rhodococcus)属に属する細菌において、ニッキング酵素は4例が報告されているに過ぎない(非特許文献2)。
【0007】
発明者らは、これまでロドコッカス属ロドクロウスJ−1株から制限酵素と、これに対応する修飾酵素を単離しているが、この制限酵素にはニッキング酵素活性はない(特許文献2)。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2008−136451号公報
【特許文献2】特開2007−259853号公報
【非特許文献】
【0009】
【非特許文献1】Jeffrey Van Ness,et al.,(2003)“Isothermal reactions for the amplification of oligonucleotides”Proc.Natl.Acad.Sci.USA, Vol.100, No.8, 4504−4509
【非特許文献2】Letek M,et al.,(2010)”The genome of a pathogenic rhodococcus: cooptive virulence underpinned by key gene acquisitions” PLoS Genet.2010 Sep 30;6(9).
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明の目的は、遺伝子工学の分野で有用な新規なヌクレアーゼとその遺伝子を提供するとともに、これを利用した前記ヌクレアーゼの製造方法を提供することにある。
【課題を解決するための手段】
【0011】
発明者らは上記課題を解決するために鋭意研究を行った結果、ロドコッカス属に属する細菌:ロドコッカス ロドクロウス(Rhodococcus rhodochrous)J−1株より、新規なヌクレアーゼRrhJ1IIを単離した。
【0012】
すなわち、本発明は、ロドコッカス属微生物由来の新規ヌクレアーゼに関する。
本発明の新規ヌクレアーゼタンパク質は、以下の(A)、(B)または(C)のタンパク質である。
(A)配列番号2記載のアミノ酸配列を含むタンパク質
(B)配列番号2記載のアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質
(C)配列番号2記載のアミノ酸配列と相同性が80%以上のアミノ酸配列からなり、かつニッキング酵素活性を有するタンパク質
【0013】
また、上記新規ヌクレアーゼタンパク質は、上記ニッキング酵素活性が、二重鎖デオキシリボ核酸中の下記式1:
【化1】
(式中、Tはチミン、Gはグアニン、Cはシトシン、Sはグアニンまたはシトシン、Yはシトシンまたはチミン、Rはアデニンまたはグアニンを表す)で表される塩基配列に特異的であり、かつ、式1中のT/Gミスマッチに対するニッキング酵素活性であることを特徴とするタンパク質であってもよい。
【0014】
本発明は、上記新規ヌクレアーゼタンパク質をコードする遺伝子も提供する。
具体的には、本発明の新規ヌクレアーゼ遺伝子は、以下の(a)、(b)または(c)のDNAを含む。
(a)配列番号1記載の塩基配列からなるDNA
(b)配列番号1記載の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつニッキング酵素活性を有するタンパク質をコードするDNA
(c)配列番号1記載の塩基配列からなるDNAと相同性が80%以上の塩基配列からなり、かつニッキング酵素活性を有するタンパク質をコードするDNA
【0015】
また本発明は、本発明の新規ヌクレアーゼ遺伝子を含む組換ベクター、および前記組換ベクターを含む形質転換体も提供する。
【0016】
さらに本発明は、本発明の新規ヌクレアーゼタンパク質の製造方法も提供する。前記製造方法は、たとえば、上記した形質転換体を培養し、得られる培養物からニッキング酵素活性を有するタンパク質を採取することにより実施できる。好ましくは、前記製造方法は、細胞を用いない無細胞タンパク質合成法を用いて実施する。
【発明の効果】
【0017】
本発明により、遺伝子工学の分野で有用なニッキング酵素活性を有するエンドヌクレアーゼおよびその遺伝子が提供される。本発明の新規ヌクレアーゼは、前記遺伝子を用いた組換え製造により、簡便かつ大量に生産することができる。
【0018】
本発明の新規ヌクレアーゼは、ニッキング酵素活性を有するため、鎖置換型恒温核酸増幅や、染色体DNAの蛍光標識による可視化、生体内遺伝子ターゲティングのための相同組換えを誘発するための一本鎖DNA切断反応などに利用できる。
【図面の簡単な説明】
【0019】
【図1】ロドコッカス属細菌の産生するエンドヌクレアーゼのホモロジー解析結果を示す図である。
【図2】RrhJ1II発現プラスミドpRR01の構造を示す模式図である。
【図3】RrhJ1II発現プラスミドpRR01を含む組換大腸菌から精製したRrhJ1IIタンパク質に対するHis−tag特異的抗体を用いたウェスタンブロッティングの結果を示す図である。
【図4】RrhJ1II発現プラスミドpRR01を含む組換大腸菌から精製したRrhJ1IIタンパク質のヌクレアーゼ活性を示す図である。
【発明を実施するための形態】
【0020】
以下、本発明の実施の形態について詳細に説明するが、本実施の形態は、本発明を説明するための例示であり、本発明をこの実施の形態のみに限定させるものではない。
【0021】
1.本発明の新規ヌクレアーゼ(RrhJ1IIヌクレアーゼ)
本発明の新規ヌクレアーゼは、ロドコッカス ロドクロウス(Rhodococcus rhodochrous)J−1株から、発明者らにより初めて単離された。ロドコッカス ロドクロウスJ−1株(以下、「J1菌」ともいう)はFERM BP−1478として独立行政法人産業技術総合研究所 特許生物寄託センター(茨城県つくば市東1丁目1番地1 中央第6)に寄託されている菌株である。
【0022】
発明者らは、このJ1菌由来の新規ヌクレアーゼを「RrhJ1II」と命名した。本明細書中では、このRrhJ1IIヌクレアーゼが有するニッキング酵素活性を「RrhJ1IIヌクレアーゼ活性」と記載する。
【0023】
本明細書において、「エンドヌクレアーゼ活性」とは、核酸配列の内部(endo−)で核酸を切断する酵素活性を意味し、「ニッキング酵素活性」とは、特定の配列を有する二本鎖DNAの片方の鎖のみを切断するエンドヌクレアーゼ活性を意味する。
【0024】
本明細書において、ニッキング酵素活性は、RrhJ1IIヌクレアーゼをDNAと接触させ、接触後のDNAの分子量またはDNA断片数を測定することにより評価することができる。当業者であれば、基質DNA、接触時の酵素量、温度、溶液組成または接触時間などの条件は適宜設定することができる。DNAの分子量は、例えばアガロース電気泳動によって測定することができる。接触前のDNAの分子量と接触後のDNAの分子量、または接触前のDNAの断片数と接触後のDNA断片数とを比較することで、ニッキング酵素活性を評価することができる。
【0025】
発明者らは、J1菌由来のRrhJ1IIヌクレアーゼが配列番号2に示されるアミノ酸配列を有することを同定した。
【0026】
しかしながら、本発明のRrhJ1IIヌクレアーゼタンパク質は、上記配列を有するものに限定されるものではなく、配列番号2記載のアミノ酸配列と約50%以上、好ましくは約60%以上、より好ましくは約70%以上、さらに好ましくは約80%以上、特に好ましくは約90%以上、さらに特に好ましくは約95%以上、最も好ましくは約98%以上の相同性あるいは同一性を有するアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質も本発明のRrhJ1IIヌクレアーゼに含まれる。
【0027】
また、本発明のRrhJ1IIヌクレアーゼタンパク質には、配列番号2記載のアミノ酸配列において、1個または数個のアミノ酸が欠失、置換または付加されたアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質も含まれる。
【0028】
具体的には、(i)配列番号2記載のアミノ酸配列において、1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が欠失したアミノ酸配列、(ii)配列番号2記載のアミノ酸配列の1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が他のアミノ酸に置換されたアミノ酸配列、(iii)配列番号2記載のアミノ酸配列に1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が付加したアミノ酸配列、(iv)配列番号2記載のアミノ酸配列に1〜20個(例えば1〜10個、好ましくは1〜5個、さらに好ましくは1〜2個)のアミノ酸が挿入されたアミノ酸配列、(v)上記(i)〜(iv)を組み合わせたアミノ酸配列が挙げられる。
【0029】
アミノ酸置換は、類似するアミノ酸残基間の保存的置換が好ましい。例えばアミノ酸は、その側鎖の性質に基づいて、疎水性アミノ酸(A、I、L、M、F、P、W、Y、V)、親水性アミノ酸(R、D、N、C、E、Q、G、H、K、S、T)、脂肪族側鎖を有するアミノ酸(G、A、V、L、I、P)、水酸基含有側鎖を有するアミノ酸(S、T、Y)、硫黄原子含有側鎖を有するアミノ酸(C、M)、カルボン酸及びアミド含有側鎖を有するアミノ酸(D、N、E、Q)、塩基含有側鎖を有するアミノ酸(R、K、H)、芳香族含有側鎖を有するアミノ酸(H、F、Y、W)に分類される。各群に分類されたアミノ酸は、相互に置換したときに、当該ポリペプチドの活性が維持される可能性が高いことが知られており、そのようなアミノ酸相互の置換が好ましい。例えば、グリシンとプロリン、グリシンとアラニンまたはバリン、ロイシンとイソロイシン、グルタミン酸とグルタミン、アスパラギン酸とアスパラギン、システインとスレオニン、スレオニンとセリン又はアラニン、リジンとアルギニン間での置換を挙げることができる。
【0030】
2.本発明の新規ヌクレアーゼ遺伝子(RrhJ1IIヌクレアーゼ遺伝子)
本発明のRrhJ1IIヌクレアーゼ遺伝子は、上記したRrhJ1IIヌクレアーゼタンパク質をコードする遺伝子である。本発明のRrhJ1IIヌクレアーゼ遺伝子は、例えば配列番号1に記載の塩基配列からなるDNAを含む。
【0031】
本発明のRrhJ1IIヌクレアーゼ遺伝子は上記配列に限定されるものではなく、配列番号1記載の塩基配列と約50%以上、好ましくは約60%以上、より好ましくは約70%以上、さらに好ましくは約80%以上、特に好ましくは約90%以上、さらに特に好ましくは約95%以上、最も好ましくは約98%以上の相同性(同一性)を有する塩基配列を有するDNAも、それがRrhJ1IIヌクレアーゼとしての機能(ニッキング酵素活性)を有するタンパク質をコードする限り、本発明の遺伝子に含まれる。
【0032】
また、前述したアミノ酸配列の欠失、置換または付加に対応して、配列番号1記載の塩基配列において、数個の塩基に欠失、置換または付加等の変異が生じた塩基配列も、それが本発明のRrhJ1IIヌクレアーゼとしての機能(ニッキング酵素活性)を有するタンパク質をコードする限り、本発明のRrhJ1IIヌクレアーゼ遺伝子に含まれる。なお、欠失、置換または付加される塩基の個数は、30個以下、好ましくは15個以下、特に好ましくは6個以下である。
【0033】
さらに、配列番号1に記載の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズするDNAも、これがニッキング酵素活性を有するタンパク質をコードする限り、本発明の遺伝子に含まれる。
【0034】
ここで、ストリンジェントな条件としては、例えばDNAを固定したナイロン膜を、6×SSC(1×SSCは塩化ナトリウム8.76g、クエン酸ナトリウム4.41gを1リットルの水に溶かしたもの)、1%SDS、100μg/mlサケ精子DNA、0.1%ウシ血清アルブミン、0.1%ポリビニルピロリドン、0.1%フィコールを含む溶液中で65℃にて20時間プローブとともに保温してハイブリダイゼーションを行う条件を挙げることができるが、これに限定されるわけではない。当業者であれば、このような緩衝液の塩濃度、温度等の条件に加えて、その他のプローブ濃度、プローブの長さ、反応時間等の諸条件を加味し、ハイブリダイゼーションの条件を設定することができる。
【0035】
ハイブリダイゼーション法の詳細な手順については、Molecular Cloning, A Laboratory Manual 2nd ed.(Cold Spring Harbor Laboratory Press (1989))等を参照することができる。
【0036】
以下に、ハイブリダイゼーションによりRrhJ1IIヌクレアーゼ遺伝子を得る方法の一例を示すが、これに限定されるわけではない。
【0037】
まず、適当な遺伝子源から得たDNAを定法に従ってプラスミドやファージベクターに接続してDNAライブラリを作製する。このライブラリを適当な宿主に導入して得られる形質転換体をプレート上で培養し、生育したコロニーまたはプラークをニトロセルロースやナイロンの膜にうつしとり、変性処理の後にDNAを膜に固定する。この膜をあらかじめ32P等で標識したプローブを含む上記の組成の溶液中、上記のストリンジェントな条件で保温し、ハイブリダイゼーションを行う。プローブとしては、配列番号2に記載したアミノ酸配列の全部または一部をコードするポリヌクレオチドを使用することができる。
【0038】
ハイブリダイゼーションの終了後、非特異的に吸着したプローブを洗い流し、オートラジオグラフィ等によりプローブとハイブリッドを形成したクローンを同定する。この操作をハイブリッド形成クローンが単離できるまで繰り返す。最後に、得られたクローンの中から、目的の酵素活性を有するタンパク質をコードする遺伝子を選択する。遺伝子の単離は、アルカリ法等の公知のポリヌクレオチド抽出法により実施できる。
【0039】
本発明のRrhJ1IIヌクレアーゼ遺伝子は、当該酵素を発現する微生物から単離することもできる。例えば、ロドコッカス ロドクロウスJ−1株由来のゲノムDNAを鋳型として、既知のアミノ酸配列情報から遺伝子の縮重を考慮して設計したプライマーもしくはプローブまたは既知の塩基配列情報に基づいて設計したプライマーまたはプローブを用いたPCRまたはハイブリダイゼーション法により、前記微生物のゲノムから目的の遺伝子を単離することができる。
【0040】
このように種々のDNAが本発明のRrhJ1IIヌクレアーゼ遺伝子の範囲内に含まれるのは、コドンの縮重に由来する。すなわち、遺伝子上でアミノ酸を指定するコドン(3つの塩基の組み合わせ)は、アミノ酸の種類ごとに1〜6種類存在することが知られている。従って、あるアミノ酸配列をコードする遺伝子は多数存在しうる。
【0041】
遺伝子は自然界において安定に存在しているものではなく、その塩基配列に変異が起こることは稀ではない。遺伝子上に起こった変異によっては、コードされるアミノ酸配列に変化を与えない変異(サイレント変異と呼ばれる)もあり、この場合には同じアミノ酸配列をコードする、異なる遺伝子が生じたと言える。従って、ある特定のアミノ酸配列をコードする遺伝子が単離されても、それを含有する生物が継代されていくうちに同じアミノ酸配列をコードする多種類の遺伝子ができて行く可能性は否定できない。
【0042】
さらに、同じアミノ酸配列をコードする多種類の遺伝子を人為的に作製することは、種々の遺伝子工学的手法を用いれば困難なことではない。例えば、遺伝子工学的なタンパク質の生産において、目的のタンパク質をコードする本来の遺伝子上で使用されているコドンが宿主中では使用頻度の低いものであった場合には、タンパク質の発現量が低いことがある。このような場合にはコードされているアミノ酸配列に変化を与えることなく、コドンを宿主で繁用されているものに人為的に変換することにより、目的タンパク質の高発現を図ることが行われている。
【0043】
このように、特定のアミノ酸配列をコードする多種類の遺伝子は人為的に作製可能なことは言うまでもなく、自然界においても生成されうるものである。従って、本発明中に開示された塩基配列と同一の遺伝子ではなくても、RrhJ1IIと同等の活性を示すタンパク質をコードするDNAである限り、本発明のRrhJ1IIヌクレアーゼ遺伝子に含まれる。
【0044】
遺伝子に変異を導入し、人為的にアミノ酸配列を改変する方法としては、Kunkel法やGapped duplex法等の公知手法や、部位特異的突然変異誘発法を利用した変異導入キット、例えばQuikChangeTM Site−Directed Mutagenesis Kit(ストラタジーン社)、GeneTailorTM Site−Directed Mutagenesis System(インビトロジェン社)、TaKaRa Site−Directed Mutagenesis System(Mutan−K,Mutan−Super Express Km等:タカラバイオ社)等を用いることができる。
【0045】
DNAの塩基配列の確認は、慣用の方法により配列決定することにより行うことができる。例えば、ジデオキシヌクレオチドチェーンターミネーション法(Sanger et al.(1977)Proc.Natl.Acad.Sci.USA 74:5463)等により行うことができる。また、適当なDNAシークエンサーを利用して配列を解析することも可能である。
【0046】
塩基配列の決定は、プラスミドベクターを用いて作製された形質転換体の場合、宿主がエシェリヒア コリ(Escherichia coli)であれば試験管等で培養を行い、定法に従ってプラスミドを調製する。得られたプラスミドをそのまま鋳型とするか、あるいは挿入断片を取り出してM13ファージベクター等にサブクローニングした後に、ジデオキシ法により塩基配列を決定する。ファージベクターで作製された形質転換体の場合も基本的に同様な操作により塩基配列を決定することができる。これら培養から塩基配列決定までの基本的な実験法については、例えば前述のT.ManiatisらのMolecular Cloning,A Laboratory Manual等に記載されている。
【0047】
得られた遺伝子が目的のRrhJ1IIヌクレアーゼをコードする遺伝子であるかどうかの確認は、決定された塩基配列を配列番号1に記載の塩基配列と比較して行うことができる。あるいは決定された塩基配列より推定されるアミノ酸配列を配列番号2に記載のアミノ酸配列と比較して行うことができる。
【0048】
3.本発明の新規ヌクレアーゼ遺伝子を含む組換えベクター
本発明は、RrhJ1IIヌクレアーゼ遺伝子を含む組換えベクターも提供する。本発明の組換えベクターは、上記酵素をコードする遺伝子の上流に転写プロモーター、場合によっては下流にターミネーターを挿入して発現カセットを構築し、このカセットを発現ベクターに挿入することにより作製することができる。あるいは、発現ベクターに転写プロモーターおよび/またはターミネーターがすでに存在する場合には、発現カセットを構築することなく、ベクター中のプロモーターおよび/またはターミネーターを利用して、その間に当該酵素をコードする遺伝子を挿入すればよい。
【0049】
本明細書において、プロモーターは、例えば、trcプロモーター、lacプロモーターなどをあげることができるが、これに限定されるわけではない。
本明細書において、ターミネーターは、例えば、trpオペロンターミネータをあげることができるが、これに限定されるわけではない。
【0050】
ベクターに本発明の遺伝子を挿入するには、制限酵素を用いる方法、トポイソメラーゼを用いる方法等を利用することができる。また、挿入の際に必要であれば、適当なリンカーを付加してもよい。また、アミノ酸への翻訳にとって重要な塩基配列として、SD配列やKozak配列などのリボソーム結合配列が知られており、これらの配列を遺伝子の上流に挿入することもできる。挿入にともない、遺伝子がコードするアミノ酸配列の一部を置換してもよい。
【0051】
本発明において使用されるベクターは、本発明の遺伝子を保持するものであれば特に限定されず、それぞれの宿主に適したベクターを用いることができる。ベクターとしては、例えば、プラスミドDNA、バクテリオファージDNA、レトロトランスポゾンDNA、人工染色体DNAなどが挙げられる。例えば、大腸菌を宿主とする場合には、pTrc99A(GEヘルスケア バイオサイエンス)、pACYC184(ニッポンジーン)、pMW118(ニッポンジーン)などを挙げることができる。また、必要に応じて、これらのベクターを改変したものを用いることもできる。
【0052】
4.本発明のベクターを導入した形質転換体
本発明の組換えベクターを宿主に導入することで、本発明の形質転換体を作製することができる。
【0053】
形質転換体に使用する宿主は、上記組換えベクターが導入された後、目的の制限酵素または修飾酵素を発現することができる限り、特に限定されるものではない。宿主としては、例えば、大腸菌(エシェリヒア・コリ)、枯草菌(バチルス・ズブチリス(Bacillus subtilis))、ロドコッカス菌(Rhodococcus)、放線菌などの細菌、酵母(サッカロミセス・セレビシエ(Saccharomyces cerevisiae))、カビ、動物細胞、植物細胞、昆虫細胞などが挙げられる。本発明において、宿主は好ましくは大腸菌である。
【0054】
本発明において、大腸菌は、例えば、大腸菌K12株やB株、あるいはそれらの野生株由来の派生株であるJM109株、XL1−Blue株(例えば、XL1−Blue MRF’)、K802株、C600株などを挙げることができる。
【0055】
宿主への組換えベクターの導入方法としては、宿主に適した方法であれば特に限定されるものではなく、当業者であれば公知技術から適宜選択することができる。このような方法としては、例えば、エレクトロポレーション法、カルシウムイオンを用いる方法、スフェロプラスト法、酢酸リチウム法、リン酸カルシウム法、リポフェクション法等が挙げられる。
【0056】
5.本発明のRrhJ1IIヌクレアーゼの製造方法
5−1 細胞系による製造
本発明のRrhJ1IIヌクレアーゼは、形質転換体を培養して、培養物中よりRrhJ1IIヌクレアーゼの特徴(例えば、ニッキング酵素活性)を有する活性を有するタンパク質を採取することによりRrhJ1IIヌクレアーゼを製造することができる。
【0057】
本発明において「培養物」とは、菌体、培養液、無細胞抽出液、細胞膜などの培養により得られるものを意味する。無細胞抽出液は、培養後の菌体を、例えばリン酸ナトリウム緩衝液を加えてホモジナイザーなどで物理的に破砕した後、遠心(15,000rpm, 10min, 4℃)し、破砕できない菌体(細胞)が存在しないように上清を回収して得ることができる。細胞膜は、上記遠心で得られたペレットとして得ることができ、また、ペレットを溶解バッファーで懸濁することにより得ることもできる。
【0058】
本発明のRrhJ1IIヌクレアーゼは、培養物をそのまま用いてもよいし、透析や硫安沈殿などの公知の方法、あるいは公知の方法を単独または適宜組み合わせることによって、培養物から濃縮、精製したものを用いてもよい。この場合、酵素活性、分子量、等電点などを指標に濃縮、精製することができる。
【0059】
たとえば、形質転換体の培養物からRrhJ1IIヌクレアーゼを採取するにあたっては、培養物より菌体を集菌後、超音波破砕、超遠心分離等により酵素を抽出し、ついで除核酸法、塩析法、アフィニティクロマトグラフィー法、ゲル濾過法、イオン交換クロマトグラフィ法等を組み合わせて精製すればよい。この方法により、RrhJ1IIヌクレアーゼを大量に得ることができる。
【0060】
用いる発現系によっては、形質転換体中で発現された酵素タンパク質が不溶物[封入体(inclusionbody)]として蓄積される場合がある。この場合にはこの不溶物を回収し、穏和な変性条件、たとえば尿素等の変性剤存在下で可溶化した後に変性剤を除くことによって活性型のタンパク質を得ることができる。さらに上記のようなクロマトグラフィー操作を行って目的とする酵素タンパク質を精製することができる。
【0061】
5−2 無細胞タンパク質合成系による製造
本発明においては、無細胞タンパク質合成系を用いて、本発明のRrhJ1IIヌクレアーゼ遺伝子または本発明のベクターから、RrhJ1IIヌクレアーゼを製造することも可能である。
【0062】
無細胞タンパク質合成系とは、細胞抽出液を用いて試験管等の人工容器内でタンパク質を合成する系である。すなわち、無細胞タンパク質合成系は、生物を用いた組換えタンパク質発現とは異なり、細胞を使用せずに試験管内で転写・翻訳という一連のタンパク質合成の流れを行う合成系であるため、細胞にとって毒性となるタンパク質を生産できるという利点がある。なお、本発明において使用される無細胞タンパク質合成系にはDNAを鋳型としてRNAを合成する無細胞転写系も含まれる。
【0063】
無細胞タンパク質合成系の細胞抽出液としては、真核細胞由来または原核細胞由来の抽出液、例えば小麦胚芽、大腸菌等の抽出液を使用することができる。これらの細胞抽出液は濃縮されたものであっても濃縮されていないものであってもよい。
【0064】
細胞抽出液は、例えば限外濾過、透析、ポリエチレングリコール(PEG)沈殿等によって得ることができる。さらに本発明において、無細胞タンパク質合成は、市販のキットを用いて行うこともできる。そのようなキットとしては、例えば試薬キットPROTEIOSTM(東洋紡)、TNTTM System(プロメガ)、合成装置のPG−MateTM(東洋紡)、RTS(ロシュ ダイアグノスティクス)等が挙げられる。
【0065】
こうした細胞抽出液に代えて、タンパク質合成に関与するすべての因子を精製し、それらをリボソーム、ATP、tRNA、アミノ酸などとともに再構成して作成された「再構成された無細胞タンパク質合成系」を用いることもできる。
【0066】
通常の無細胞タンパク質合成系に用いられる細胞抽出液には、ATPを加水分解する酵素などの混在によりタンパク質合成以外に無駄に消費されるエネルギー量が多く、エネルギー効率が低く、また細胞抽出液中に存在するプロテアーゼやヌクレアーゼなどの阻害因子によりンパク質合成の効率が低下する場合がある。
【0067】
「再構成された無細胞タンパク質合成系」は、こうした阻害的要因の問題がなく、また構成成分を自由に操作することができるため、目的や標的タンパク質に応じた自由なシステム設計が可能である。また系の自由度が高く、反応系を微小化したり、自動化装置と組み合わせてハイスループットにすることも可能である。
【0068】
「再構成された無細胞タンパク質合成系」の一例としてPURESYSTEM(登録商標)を挙げることができる。PURESYSTEM(登録商標)は大腸菌から再構成されているが、無細胞タンパク質合成系で用いられる他の公知の細胞、昆虫細胞、小麦胚芽、ウサギ網状赤血球のいずれを用いても、同様の系は構成可能である。
【0069】
上記のように無細胞タンパク質合成系によって得られる本発明のヌクレアーゼは前述のように適宜クロマトグラフィを選択して、濃縮、精製することができる。
【0070】
6.RrhJ1IIヌクレアーゼ活性の解析
6.1 RrhJ1IIヌクレアーゼの認識配列
RrhJ1IIヌクレアーゼの認識配列を調べるために、たとえば前述のような方法で配列番号1に示される遺伝子を発現可能に組み込んだベクターで宿主細胞を形質転換し、その培養物からRrhJ1IIヌクレアーゼ活性を有するタンパク質を得る。得られたタンパク質を、片方の鎖の5'末端に蛍光標識を持ち、配列内に種々のT/Gミスマッチを持つ二本鎖オリゴDNAを合成して活性測定用の試料と接触させた後、変性剤と加熱によりDNAを一本鎖に変性させてSDS−ポリアクリルアミドゲル電気泳動に供し、化学発光による検出を行って、切断パターンを解析すると、J1菌ヌクレアーゼの認識配列を決定することができる。本発明のRrhJ1IIヌクレアーゼは、以下の二重鎖デオキシリボ核酸中の下記式1
【化2】
(式中、Tはチミン、Gはグアニン、Cはシトシン、Sはグアニンまたはシトシン、Yはシトシンまたはチミン、Rはアデニンまたはグアニンを表す)で表される塩基配列を特異的に認識し、かつ、囲み文字で示した位置のT/Gミスマッチに対するニッキング酵素活性を示すことを特徴としている。ここで、「T/Gミスマッチ」とは、チミジン(T)とグアニン(G)との間で水素結合が形成されたミスマッチ塩基対をいう。
【0071】
式1中、S,Y,Rで表記される部分の配列は、上記説明文中に示される塩基である限り特に限定されないが、囲み文字で示した位置のT/Gミスマッチが解消し、リード鎖のTがCとなった場合に回文配列となることが好ましい。たとえば、下記式2〜4を挙げることができる。
【化3】
【実施例】
【0072】
以下、実施例を用いて本発明をより詳細に説明する。以下は本発明の例示であって本発明を限定する趣旨ではない。
【0073】
[実施例1]
J1菌染色体DNAの調製
ロドコッカス ロドクロウス(Rhodococcus rhodochrous)J−1株を100mlのMYKG培地中、30℃にて72時間振盪培養した。
【0074】
培養後、集菌し、集菌された菌体をSaline−EDTA溶液(0.1M EDTA,0.15M NaCl(pH8.0))4mlに懸濁した。懸濁液にリゾチーム40mgを加えて37℃で1〜2時間振盪した後、−20℃で凍結した。
【0075】
次に、10mlのTris−SDS液(1%SDS,0.1M NaCl,0.1M Tris−HCl(pH9.0))を穏やかに振盪しながら加え、さらにプロテイナーゼK(メルク社)(10mg/ml)を10μl加えて37℃で1時間振盪した。
【0076】
次に、等量のTE(10mM Tris−HCl,1mM EDTA(pH8.0))飽和フェノールを加え、攪拌後、遠心した。上層を採取し、2倍量のエタノールを加えた後、ガラス棒でDNAを巻き取り、90%,80%,70%のエタノールで順次フェノールを取り除いた。
【0077】
次に、DNAを3mlのTE緩衝液に溶解させ、リボヌクレアーゼA溶液(100℃,15分間の加熱処理済)を10μg/mlになるように加え、37℃で30分間振盪した。さらに、プロテイナーゼKを加え37℃で30分間振盪した後、等量のTE飽和フェノールを加えて遠心し、上層と下層に分離させた。
【0078】
上層についてこの操作を2回繰り返した後、同量のクロロホルム(4%イソアミルアルコール含有)を加え、同様の抽出操作を繰り返した。その後、上層に2倍量のエタノールを加え、ガラス棒でDNAを巻き取り回収し、染色体DNA標品を得た。
【0079】
[実施例2]
RrhJ1IIヌクレアーゼ遺伝子のPCR
RrhJ1IIヌクレアーゼ遺伝子をPCRで増幅するためのプライマーを以下の方法で設計した。
【0080】
図1はロドコッカス属細菌の産生するヌクレアーゼのアミノ酸ホモロジー解析結果を示す。図1中でホモロジー解析を行ったヌクレアーゼとその由来は以下のとおりである。
A:DNA mismatch endonuclease Vsr(Rhodococcus equi ATCC33707)
B:NaeI very short patch repair endonuclease(Rhodococcus erythropolis SK121)
C:Very short patch repair protein(Rhodococcus jostii RHA1)
D:DNA mismatch endonuclease (Rhodococcus erythropolis PR4)
E:DNA mismatch endonuclease(Rhodococcus
opacus B4)
【0081】
図1中、特に保存されている2つの領域を選んで配列番号3および配列番号4のデジェネレイトプライマーを設計し、実施例1で調製したJ1菌ゲノムDNAを鋳型としてデジェネレイトPCRを以下の条件で実施した。その結果、約350bのバンドの増幅が確認された。
【0082】
反応液組成
鋳型DNA(J1菌染色体DNA,実施例1) 1μl
10×Ex Buffer(タカラバイオ社) 10μl
150μMプライマーDG−01(配列番号3) 1μl
150μMプライマーDG−02(配列番号4) 1μl
2.5mM dNTP 8μl
DMSO 10μl
滅菌水 18μl
ExTaq DNAポリメラーゼ(タカラバイオ社)1μl
総量 50μl
【0083】
温度サイクル:94℃:30秒、65℃:30秒および72℃:1分の反応を30サイクル
【0084】
プライマー
DG−01:5’−GA(T/C)CCIGCIACITCIGCICGIATG−3’(配列番号3)
DG−02:5’−CCAIACICGIACIACIGTCCA−3’(配列番号4)
【0085】
次に、増幅された配列のダイレクトシークエンシングをPCRで使用したプライマーを用いて実施した。その結果、配列番号5に示す配列が得られ、ホモロジー検索の結果、前述のエンドヌクレアーゼとの相同性が認められた。
【0086】
[実施例3]
RrhJ1IIヌクレアーゼ遺伝子のクローニング
(1)ゲノミックサザンハイブリダイゼーション
ApaLI,BamHI,ClaI,Eco52I,EcoT14I,KpnI,MluI,NcoI,NotI,PvuI,SacI,XbaI,XhoIそれぞれで消化したJ1菌ゲノムDNAに対し、後述の方法で調製したRrhJ1IIのプローブを用いてサザンハイブリダイゼーションを行ったところ、Eco52Iで消化した断片から、約1.2kbの単一シグナルが得られた。
【0087】
なお、RrhJ1IIのプローブは以下のようにして調製した。
実施例2で調製したPCR産物をGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を用いて精製した。精製したPCR産物に対してAlkPhos Direct Labeling kit(GEヘルスケア バイオサイエンス社)を用い、添付のマニュアルにしたがってラベリングを行い、RrhJ1IIのプローブとした。
【0088】
(2)コロニーハイブリダイゼーション
J1菌ゲノムDNAを制限酵素Eco52Iで分解して0.7%アガロースゲル電気泳動で分離し、ゲルからGFX PCR DNA band and Gel Band Purification kit(GEヘルスケア バイオサイエンス社)を使用して約1.2kbの断片を回収した。得られた断片は、pBluescriptII SK(+)ベクター(Stratagene社製)にDNA ligation kit<Mighty mix>(タカラバイオ社製)を用いて連結した。反応条件は以下の通りである。
【0089】
反応液組成
ligation mighty mix(タカラバイオ社製) 5μl
J1菌ゲノムDNA/Eco52I切断断片 4μl
pBluescriptII SK(+)/Eco52I切断断片 1μl
総量 10μl
【0090】
反応:
16℃,1時間
【0091】
上記ライゲーション産物の全量を、後述の方法で調製した大腸菌JM109株コンピテントセル200μlに加え、0℃で30分放置した。続いて、前述コンピテントセルに42℃で45秒間ヒートショックを与え、0℃で2分間冷却した。その後、SOC培地(20mMグルコース、2%バクトトリプトン、0.5%バクトイーストエキス、10mM NaCl,2.5mM KCl,1mM MgSO4,1mM MgCl2)を1ml添加し、37℃にて1時間振盪培養した。培養後の培養液を200μlずつ、LB AIXプレート(100μg/lアンピシリン、100μM IPTG,50μg/l X−galを含むLB寒天培地)に塗布し、37℃で一晩放置した。プレート上に生育した白色の組換コロニーを新しいLB AIXプレートに、プレート1枚に付き94個、プレート10枚分単離した。各プレートにはインサートを含まないpBluescriptII SK(+)で形質転換したJM109株を2コロニー/プレート植菌した。コロニー単離したプレートを37℃で一晩放置した後、Hybond−N+(GEヘルスケア バイオサイエンス社)膜にコロニーを写し取り、実施例3(1)で調製したRrhJ1IIのプローブを用いてコロニーハイブリダイゼーションを行った。
【0092】
検出されたコロニーを培養して得られた培養液を集菌後、QIAprep miniprep kit(QIAGEN社製)を用いて組換えプラスミドを回収した。キャピラリーDNAシーケンサーCEQ2000(ベックマン・コールター社製)を用いて、添付のマニュアルに従って、プラスミド中にクローニングされているゲノムDNA断片の塩基配列を解析した。その結果、配列番号6に示される塩基配列が得られた。配列番号6に示される塩基配列中に、配列番号2に示す450bpのオープンリーディングフレーム(ORF1)を見出した。このORF1のコードするアミノ酸配列は、図1中に示したA〜Eのロドコッカス属細菌由来エンドヌクレアーゼに対して87%〜89%の相同性を持っていることから、ORF1はエンドヌクレアーゼをコードしていることが推定されたため、ORF1のコードするエンドヌクレアーゼをRrhJ1IIヌクレアーゼと命名した。また、本実施例3(2)で得られたORF1を含むプラスミドをpBRrhJ1IIと命名した。
【0093】
なお、大腸菌JM109株のコンピテントセルは以下の方法で調製した。
大腸菌JM109株をLB培地1mlに接種し、37℃で5時間好気的に前培養した。
次に、前培養液0.4mlをSOB培地40ml(2%バクトトリプトン、0.5%バクトイーストエキス、10mM NaCl,2.5mM KCl,1mM MgSO4,1mM MgCl2)に加え、18℃で20時間培養した。得られた培養物を遠心分離(3,700×g,10分間、4℃)により集菌した後、冷TF溶液(20mM PIPES−KOH(pH6.0),200mM KCl,10mM CaCl2,40mM MnCl2)を13ml加え、0℃で10分間放置し、再度遠心分離(3,700×g,10分間、4℃)して上清を除いた。得られた大腸菌菌体を冷TF溶液3.2mlに懸濁し、0.22mlのジメチルスルホキシドを加え、0℃で10分間放置した後、液体窒素を用いて凍結したものをコンピテントセルとした。
【0094】
[実施例4]
大腸菌組換体によるRrhJ1IIヌクレアーゼの生産
(1)RrhJ1IIヌクレアーゼ発現プラスミドの構築
RrhJ1IIヌクレアーゼを得るために、実施例1で得られたJ1菌染色体DNAを鋳型として使用し、以下に示す反応液組成およびプライマーを用いてPCRを行った。この際、RrhJ1IIヌクレアーゼをHis−tag融合タンパクとして発現させるため、RrhJ1IIヌクレアーゼ遺伝子上流にSD配列とHis−tag配列を付加した。
【0095】
反応液組成
鋳型DNA(J1菌染色体DNA) 1μl
2×PCR Buffer KOD FX(東洋紡) 25μl
10μMプライマーN(配列番号7) 1.5μl
10μMプライマーC(配列番号8) 1.5μl
2mM dNTP 10μl
KOD FX DNAポリメラーゼ(東洋紡) 1μl
総量 50μl
【0096】
温度サイクル:94℃:120秒、98℃:10秒および68℃:1分の反応を30サイクル
【0097】
プライマー
N:5'−AGTGAATTCCTTTAAGAAGGAGATATACCATGCATCATCATCATCATCACATGGCGTCGTCGGAT−3'(配列番号7)
C:5'−GCCAAGCTTTCACCCCCGCGCCGGTTT−3'(配列番号8)
【0098】
反応終了後、反応液5μlを1%アガロースゲルにおける電気泳動に供し、約0.5kbのPCR産物の検出を行った。PCR産物を確認した後、反応液からPCR産物をDNA/RNA extaction Kit(VIOGENE社)で精製した。得られたPCR産物を、制限酵素EcoRIとHindIIIで切断した。制限酵素処理を行ったPCR産物を1%アガロースゲルにおける電気泳動に供し、約0.5kb付近のバンドを回収した。回収したPCR産物をベクターpUC18のEcoRI−HindIII部位に連結し、プラスミドを作製した。得られたプラスミドをpRR01と名づけた。図2はプラスミドpRR01の構造を示す模式図である。
【0099】
このプラスミドで大腸菌JM109を形質転換し、100μg/mlアンピシリン、1mM IPTG,50μg/ml X−galを含むLB寒天培地上でコロニーを形成させた。得られた白色のコロニーを100μg/mlアンピシリンを含むLB液体培地に植菌し、37℃で一晩培養した後Mini Plus Plasmid DNA Extraction kit(VIOGENE社)を使用してプラスミドを回収し、EcoRI−HindIIIによる切断とシークエンス解析を行って目的のDNA断片が挿入されていることを確認した。
【0100】
(2)精製酵素の調製
(1)で作製したRrhJ1IIヌクレアーゼ発現ベクターを含む大腸菌組換体(JM109/pRR01)を、100μg/mlアンピシリンを含むLB液体培地10mlで37℃,5時間培養した後、1mlを100μg/mlアンピシリン、1mM IPTGを含むLB液体培地100ml×1本に植菌し、37℃,18時間培養した。培養液を遠心分離によって回収し、回収した菌体を破砕用緩衝液(組成 20mM Sodium Phosphate,0.5M NaCl,20mM イミダゾール、10% グリセロール、pH7.4)に懸濁した後4℃で10分間超音波による破砕を行った。破砕液を遠心分離し、得られた上清を無細胞抽出液とした。この無細胞抽出液をHis−tag精製カラム(His Trap HP:GE Healthcare)を用いて精製した。精製タンパク質はElution Buffer(0.5M NaCl,0.5M イミダゾール、20mM Sodium Phosphate,10%グリセロール、pH7.4)にて溶出し、2つのフラクションに分画してフラクション1,フラクション2とした。
【0101】
得られた精製サンプルをSDS−PAGEおよびHis−tag特異的抗体(GE Healthcare)を用いたウェスタンブロッティングに供し、目的タンパク質が精製されていることを確認した。結果を図3に示す。図3中、A,Bの「1」は分子量マーカー(BioRad Prestained Standard Broad)、「4」はポジティヴコントロール(His×6融合タンパク質、12kDa)である。SDS−PAGEの結果、フラクション1(図3A「2」)とフラクション2(図3A「3」)で約25kDa付近にバンドが出現し、His−tag特異的抗体を用いたウェスタンブロッティングにより、目的タンパク質であることを確認した(図3B「2」「3」)。
【0102】
(3)酵素活性の測定
(2)で調製した精製酵素を用いて、RrhJ1IIヌクレアーゼ活性を調べた。活性は片方の鎖の5'末端にビオチン標識を持ち、配列内にT/Gミスマッチを1箇所持つ二本鎖オリゴDNAを用いてそのニッキング活性を測定した。
【0103】
反応液組成
1.5μM基質DNA(配列番号9〜18) 2μl
100mM Tris−HCl(pH7.5) 2μl
100mM MgCl2 2μl
1mg/ml BSA 2μl
総量 20μl
【0104】
【化4】
【0105】
反応温度:20℃,60分
【0106】
反応終了後、反応液と等量のフェノールでDNAを抽出し、ホルムアミド(終濃度1M)の存在下、100℃で2分間加熱後氷上にて急冷処理し、DNAを一本鎖に変性させた。変性後の一本鎖DNAサンプルを、8M尿素を含む20%ポリアクリルアミドゲル電気泳動に供し、Imaging high chemilumi kit(東洋紡)を使用して化学発光による検出を行って泳動パターンを比較した。
【0107】
結果を図4に示す。図4中、「1」は20merの一本鎖DNA,「2」は40merの一本鎖DNA,「3」はネガティヴコントロール(酵素を含まない反応液でインキュベートと変性処理を行った基質DNA01)である。オリゴヌクレオチド02、06、08は切断され約20merの1本鎖が生成した(図4 「5」,「9」,「11」)。一方、01、03、04、05、07、09、10は切断されなかった(図4 「4」、「6」、「7」、「8」、「10」、「12」、「13」)ことから、RrhJ1IIヌクレアーゼはミスマッチを認識してDNAを切断するエンドヌクレアーゼ活性を持つことを確認した。
【産業上の利用可能性】
【0108】
本発明の新規ヌクレアーゼおよびその遺伝子は、遺伝子工学の分野で有用であり、鎖置換型恒温核酸増幅や、染色体DNAの蛍光標識による可視化、生体内遺伝子ターゲティングのための相同組換えを誘発するための一本鎖DNA切断反応などに利用できる。
【配列表フリーテキスト】
【0109】
配列番号3:プライマーDG−01
配列番号4:プライマーDG−02
配列番号7:プライマーN
配列番号8:プライマーC
配列番号9:二本鎖オリゴDNA01
配列番号10:二本鎖オリゴDNA02
配列番号11:二本鎖オリゴDNA03
配列番号12:二本鎖オリゴDNA04
配列番号13:二本鎖オリゴDNA05
配列番号14:二本鎖オリゴDNA06
配列番号15:二本鎖オリゴDNA07
配列番号16:二本鎖オリゴDNA08
配列番号17:二本鎖オリゴDNA09
配列番号18:二本鎖オリゴDNA10
【特許請求の範囲】
【請求項1】
以下の(A)、(B)または(C)のタンパク質。
(A)配列番号2記載のアミノ酸配列を含むタンパク質
(B)配列番号2記載のアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質
(C)配列番号2記載のアミノ酸配列と相同性が80%以上のアミノ酸配列からなり、かつニッキング酵素活性を有するタンパク質
【請求項2】
請求項1に記載のニッキング酵素活性が、二重鎖デオキシリボ核酸中の下記式1:
【化1】
(式中、Tはチミン、Gはグアニン、Cはシトシン、Sはグアニンまたはシトシン、Yはシトシンまたはチミン、Rはアデニンまたはグアニンを表す)で表される塩基配列に特異的であり、かつ、式1中のT/Gミスマッチに対するニッキング酵素活性であることを特徴とする、請求項1に記載のタンパク質。
【請求項3】
請求項1または2に記載のタンパク質をコードする遺伝子。
【請求項4】
以下の(a)、(b)または(c)のDNAを含む遺伝子。
(a)配列番号1記載の塩基配列からなるDNA
(b)配列番号1記載の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつニッキング酵素活性を有するタンパク質をコードするDNA
(c)配列番号1記載の塩基配列からなるDNAと相同性が80%以上の塩基配列からなり、かつニッキング酵素活性を有するタンパク質をコードするDNA
【請求項5】
請求項3または4に記載の遺伝子を含む組換ベクター。
【請求項6】
請求項5記載の組換ベクターを含む形質転換体。
【請求項7】
請求項6記載の形質転換体を培養し、得られる培養物からニッキング酵素活性を有するタンパク質を採取することを特徴とする、タンパク質の製造方法。
【請求項8】
請求項1または2記載のニッキング酵素活性を有するタンパク質を、無細胞タンパク質合成法を用いて合成し、反応液より該ニッキング酵素活性を有するタンパク質を採取することを特徴とする、タンパク質の製造方法。
【請求項1】
以下の(A)、(B)または(C)のタンパク質。
(A)配列番号2記載のアミノ酸配列を含むタンパク質
(B)配列番号2記載のアミノ酸配列において、1もしくは数個のアミノ酸が欠失、置換もしくは付加されたアミノ酸配列を含み、かつニッキング酵素活性を有するタンパク質
(C)配列番号2記載のアミノ酸配列と相同性が80%以上のアミノ酸配列からなり、かつニッキング酵素活性を有するタンパク質
【請求項2】
請求項1に記載のニッキング酵素活性が、二重鎖デオキシリボ核酸中の下記式1:
【化1】
(式中、Tはチミン、Gはグアニン、Cはシトシン、Sはグアニンまたはシトシン、Yはシトシンまたはチミン、Rはアデニンまたはグアニンを表す)で表される塩基配列に特異的であり、かつ、式1中のT/Gミスマッチに対するニッキング酵素活性であることを特徴とする、請求項1に記載のタンパク質。
【請求項3】
請求項1または2に記載のタンパク質をコードする遺伝子。
【請求項4】
以下の(a)、(b)または(c)のDNAを含む遺伝子。
(a)配列番号1記載の塩基配列からなるDNA
(b)配列番号1記載の塩基配列からなるDNAと相補的な塩基配列からなるDNAとストリンジェントな条件下でハイブリダイズし、かつニッキング酵素活性を有するタンパク質をコードするDNA
(c)配列番号1記載の塩基配列からなるDNAと相同性が80%以上の塩基配列からなり、かつニッキング酵素活性を有するタンパク質をコードするDNA
【請求項5】
請求項3または4に記載の遺伝子を含む組換ベクター。
【請求項6】
請求項5記載の組換ベクターを含む形質転換体。
【請求項7】
請求項6記載の形質転換体を培養し、得られる培養物からニッキング酵素活性を有するタンパク質を採取することを特徴とする、タンパク質の製造方法。
【請求項8】
請求項1または2記載のニッキング酵素活性を有するタンパク質を、無細胞タンパク質合成法を用いて合成し、反応液より該ニッキング酵素活性を有するタンパク質を採取することを特徴とする、タンパク質の製造方法。
【図1】


【図2】
【図3】
【図4】
【図2】
【図3】
【図4】
【公開番号】特開2013−5791(P2013−5791A)
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願番号】特願2012−38067(P2012−38067)
【出願日】平成24年2月24日(2012.2.24)
【出願人】(000006035)三菱レイヨン株式会社 (2,875)
【Fターム(参考)】
【公開日】平成25年1月10日(2013.1.10)
【国際特許分類】
【出願日】平成24年2月24日(2012.2.24)
【出願人】(000006035)三菱レイヨン株式会社 (2,875)
【Fターム(参考)】
[ Back to top ]