
S−アデノシル−L−ホモシステイン加水分解酵素の可逆的阻害剤およびその使用
【課題】Sアデノシル−L−ホモシステイン(SAH)加水分解酵素を可逆的に阻害するための組成物および方法の提供。
【解決手段】本発明の化合物は、抗出血性ウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、または抗新生物薬と併用され得る。本発明の組成物および方法は、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、高ホモシステイン不全症(hyperhomocysteineuria)、循環器病、脳卒中、アルツハイマー病、または糖尿病の予防および処置に用いられ得る。
【解決手段】本発明の化合物は、抗出血性ウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、または抗新生物薬と併用され得る。本発明の組成物および方法は、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、高ホモシステイン不全症(hyperhomocysteineuria)、循環器病、脳卒中、アルツハイマー病、または糖尿病の予防および処置に用いられ得る。
【発明の詳細な説明】
【技術分野】
【0001】
(技術分野)
本願は、米国特許出願番号10/410,879(2003年4月9日出願、現在係属中)および米国特許出願番号PCT/US2004/011229(2004年4月9日出願、現在係属中)(これらの内容は、その全体が本明細書中に参考として援用される)の一部継続である、米国特許出願番号10/964,236(2004年10月13日出願、現在係属中)に関する。
【背景技術】
【0002】
(発明の背景)
SAH加水分解酵素は、多くのウイルスがウイルスタンパク質の効率的な翻訳のためにそのmRNA上に5’−キャップメチル化構造を必要とするという観察に基づく、抗ウイルス薬設計のための魅力的な標的であった。非特許文献1;非特許文献2。SAH加水分解酵素の阻害は、ウイルスmRNAのメチル化を含む、S−アデノシル−L−メチオニン(SAM)依存性メチル化反応の阻害を生じ、したがって、ウイルス複製を阻害する(スキーム1)。
【0003】
【化10】

SAH加水分解酵素の多数の阻害剤が、天然に存在する化合物および合成化合物から同定されている。最も強力な阻害剤は、時間依存的な様式でSAH加水分解酵素を不可逆的に不活化する不可逆的阻害剤である。不可逆的阻害剤は、その激しい細胞傷害作用により、狭い治療ウインドウを生じるのみであることが、研究により示されている(非特許文献3)。SAH加水分解酵素は、非常に緩徐な代謝回転速度(マウス肝臓においてt1/2=24時間)を有する普遍的な細胞性酵素であるので、不可逆的阻害剤は、酵素活性の長期阻害を引き起こし得る。例えば、酵素活性の完全な回復のために最大7日間かかり得、このことが、望ましくない副作用を引き起こし得る。不可逆的阻害剤に関連する激しい細胞傷害性が、これらの阻害剤の臨床的に有用な薬物への開発を損なう主な要因であった。不可逆的阻害剤に関連する細胞傷害性のために、可逆的阻害剤が好ましい。
【0004】
しかしながら、現時点では、インビボで試験した場合に、SAH加水分解酵素に対して実質的な阻害活性を生じるのに十分に強力な、公知の可逆的なSAH加水分解酵素阻害剤は存在しない。例えば、SAH加水分解酵素に対するKi値が3.5μMである可逆的阻害剤(S)−9−(2,3−ジヒドロキシプロピル)アデニン((S)−DHPA)は、阻害能力を欠く。(非特許文献4)。(s)−DHPAは、単離されたAdoHcy加水分解酵素の可逆的阻害剤であることが報告されたが(非特許文献4)、(s)−DHPAはまた、細胞内AdoHcy加水分解酵素の不可逆的阻害剤であることも報告された(非特許文献5)。したがって、望ましくない細胞傷害作用を有することなく効力を発揮するSAH加水分解酵素阻害剤の必要性が残っている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Yuanら、Exp.Opin.Ther.Patents(1999)9:1197−1206
【非特許文献2】Yuanら、Adv.Antiviral Drug Des.,De Clercq(編),,JAI Press,Inc.London,UK(1996)第2巻,pp.41−88
【非特許文献3】WolfeおよびBorchardt,Journal of Medicinal Chemistry(1991)34:1521−1530
【非特許文献4】VotrubaおよびHoly,Coll.Czech.Chem.Commun.(1980)45:3039
【非特許文献5】Schancheら、Molecular Pharmacology(1984)26:553−558
【発明の概要】
【課題を解決するための手段】
【0006】
(発明の簡単な概要)
本発明は、SAH加水分解酵素の新規な可逆的阻害剤を提供する。本発明の化合物は、SAH加水分解酵素を阻害するこれらの化合物の能力に関連する生物学的活性を示す薬剤として有用である。
【0007】
1つの実施形態において、本発明は、式(I):
【0008】
【化11】
[式中、Zは炭素または窒素であり、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、またはハロゲンであり;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、またはヘテロアリールであり;Xは、酸素、窒素、または硫黄であり;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、またはヘテロアリールである]で示される化合物、およびその薬学的に受容可能な塩を提供する。特定の実施形態において、化合物は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0009】
1つの局面において、本発明は、式IA:
【0010】
【化12】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、あるいは式(IB):
【0011】
【化13】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]で示される化合物を提供する。
【0012】
式Iまたは式IAの化合物は、R1、R2、R3およびR4が水素である置換基を有し得る。本発明の1つの局面において、Xは、酸素である。本発明の別の局面において、Yは、水素またはC1−10アルキル基である。本発明のさらに別の局面において、R1、R2、R3およびR4は、水素であり、Xは酸素であり、そしてYは水素またはC1−10アルキル基である。
【0013】
本発明はまた、式I、式IAまたは式IBで示されるSAH加水分解酵素の可逆的阻害剤、およびそれらの薬学的に受容可能な塩も提供する:
いくつかの実施形態において、該SAH加水分解酵素の可逆的阻害剤は、式(II):
【0014】
【化14】
で示される。
【0015】
いくつかの実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(III):
【0016】
【化15】
[式中、R1およびR2は、それぞれ独立して、水素またはヒドロキシである;ただし、R1およびR2がともにヒドロキシであることはない]で示される。いくつかの実施形態において、R1は水素であり、そしてR2はヒドロキシである。いくつかの実施形態において、R1はヒドロキシであり、そしてR2は水素である。いくつかの実施形態において、R1およびR2は、ともに水素である。
【0017】
いくつかの実施形態において、該SAH加水分解酵素の可逆的阻害剤は、式(IV):
【0018】
【化16】
[式中、R1は、NH2、SCH3、またはCH2NH2である]
で示される。
【0019】
いくつかの実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(V):
【0020】
【化17】
[式中、R1は、NH2またはCONH2である]
で示される。
【0021】
なお別の実施形態において、SAH加水分解酵素の可逆的阻害は、式(VI):
【0022】
【化18】
[式中、Wは、Hまたはメトキシである]
で示される。
【0023】
式I、IA、IBおよびII〜VIの化合物は、β炭素でS配置を有し得るか、β炭素でR配置を有し得るか、またはラセミ混合物を含み得る。1つの実施形態において、これらの化合物は、生体媒体(例えば、血清)中の哺乳動物SAH加水分解酵素に対するKi値が100nM未満である。他の実施形態において、これらの化合物は、生体媒体中の哺乳動物SAH加水分解酵素に対するKi値が約1nMと約100nMの間である。これらの化合物は、好ましくは、生体媒体中のヒトSAH加水分解酵素に対して100nM未満のKi値か、または約1nMと約100nMの間のKi値を示す。
【0024】
本発明はまた、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物またはその薬学的に受容可能な塩、ならびに薬学的に受容可能なキャリアまたは希釈剤を含む、薬学的組成物に関する。薬学的組成物は、経口、非経口(例えば、筋肉内、腹腔内、静脈内、嚢内注射もしくは注入、皮下注射、または移植)、吸入スプレー、経鼻、膣内、直腸、舌下、あるいは局所投与経路によって投与され得る。これらの薬学的組成物は、各投与経路に適した適切な投薬単位処方物に処方され得る。
【0025】
本発明は、特定の処方物または特定の投与様式に限定されることを意図するものではない。1つの実施形態において、組成物は、経口投与、非経口投与、鼻腔内投与、局所投与、または注射投与用に処方される。注射用投与の非限定的な例は、空洞内注射、皮下注射、静脈内注射、筋肉内注射および皮内注射である。本薬学的組成物は、1日当たり約0.1〜約20mg/kgの範囲の投薬量で、経口投与用に処方され得る。本薬学的組成物はまた、1日当たり約0.1〜約20mg/kgの範囲の投薬量で、注射投与用に処方され得る。
【0026】
本発明の薬学的組成物は、固体または液体の剤形で処方され得る。例えば、本薬学的組成物は、錠剤、カプセル剤、顆粒剤、散剤、および類似の化合物の形態で、固体として処方され得る。本薬学的組成物はまた、シロップ剤、注射混合物などの形態で、液体として処方され得る。
【0027】
本発明はまた、有効量の本発明の組成物、および該組成物を投与するための指示手段を含む、キットを提供する。
【0028】
さらに、本発明は、S−アデニル−L−ホモシステイン(SAH)加水分解酵素の活性を可逆的に阻害するための方法を提供する。1つの実施形態において、本発明は、哺乳動物におけるS−アデノシル−L−ホモシステイン(SAH)加水分解酵素の活性を可逆的に阻害するための方法を提供し、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、式(I):
【0029】
【化19】
[式中、Zは、炭素および窒素からなる群より選択され、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;Xは、酸素、窒素、および硫黄からなる群より選択され;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
で示される有効量の化合物またはその薬学的に受容可能な塩を投与し、それによって該哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害する工程を包含する。特定の実施形態において、投与される化合物またはその薬学的に受容可能な誘導体は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0030】
1つの局面において、本発明は、式IA
【0031】
【化20】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【0032】
【化21】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される化合物またはその薬学的に受容可能な塩を用いて、SAH加水分解酵素の活性を可逆的に阻害するための方法を提供する。
【0033】
さらに別の局面において、本発明は、上記に規定されるような、式II〜VIのいずれか1つの式で示される化合物またはその薬学的に受容可能な塩を用いて、SAH加水分解酵素の活性を可逆的に阻害するための方法を提供する。
【0034】
好ましい実施形態において、哺乳動物は、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、高ホモシステイン不全症、循環器病、脳卒中、アルツハイマー病、糖尿病、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある。しかしながら、本発明が特定の疾患の予防および処置に限定されることを意図するものではない。
【0035】
出血性ウイルス感染を予防および処置するための方法を提供することは、本発明の目的である。1つの局面において、この方法は、哺乳動物における出血性ウイルス感染の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。特定の実施形態において、出血性ウイルス感染は、ブンヤウイルス科、フィロウイルス科、フラビウイルス科、およびアレナウイルス科のウイルスからなる群より選択されるウイルスによって引き起こされる。他の特定の実施形態において、フィロウイルス科のウイルスは、エボラウイルスである。
【0036】
自己免疫疾患を予防および処置するための方法を提供することもまた、本発明の目的である。1つの局面において、この方法は、哺乳動物における自己免疫疾患の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。
【0037】
同種移植拒絶を予防および処置するための方法を提供することもまた、本発明の目的である。1つの局面において、この方法は、哺乳動物における同種移植拒絶の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。
【0038】
さらに、哺乳動物における血漿ホモシステインを低下させるための方法、あるいは高ホモシステイン不全症を予防または処置するための方法を提供することも、本発明の目的である。1つの局面において、この方法は、哺乳動物における血漿ホモシステインを低下させるために、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。
【0039】
また、新生物を予防または処置するための方法を提供することも、本発明の目的である。1つの局面において、この方法は、哺乳動物における新生物の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。新生物の非限定的な例は、副腎、肛門、聴神経、胆管、膀胱、骨、脳、胸、頬、中枢神経系、頚部、結腸、耳、子宮内膜、食道、眼、眼瞼、ファロピウス管、胃腸管、頭頸部、心臓、腎臓、喉頭、肝臓、肺、下顎骨、下顎関節頭、上顎骨、口、鼻咽頭、鼻、口腔、卵巣、膵臓、耳下腺、陰茎、耳介、脳下垂体、前立腺、直腸、網膜、唾液腺、皮膚、小腸、脊髄、胃、精巣、甲状腺、扁桃腺、尿道、子宮、膣、内耳神経、および外陰の新生物である。
【0040】
本発明はまた、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物と、有効量の抗出血性のウイルス感染薬、免疫抑制剤、血漿ホモシステイン低下薬、および抗新生物薬とを含む組み合わせを提供する。この組み合わせは、薬学的に受容可能なキャリアまたは賦形剤をさらに含み得る。特定の実施形態において、この組み合わせは、(4−アデニン−9−イル)−2−ヒドロキシブタン酸を含まない。
【0041】
特定の実施形態において、抗出血性のウイルス感染薬は、インターロイキン−1(IL−1)、腫瘍壊死因子(TNF)、またはその組み合わせを阻害する。抗出血性のウイルス感染薬は、抗ウイルスワクチン、抗ウイルス抗体、ウイルスによって活性化された免疫細胞、またはウイルスによって活性化された免疫血清であり得る。
【0042】
別の実施形態において、免疫抑制剤は、シクロスポリン、タクロリムス、副腎皮質ステロイド、アザチオプリン、ミコフェノレート、シクロホスファミド、メトトレキサート、クロラムブチル、ビンクリスチン、ビンブラスチン、ダクチノマイシン、抗胸腺細胞グロブリン、ムロモナブ−CD3モノクローナル抗体、RhO(D)免疫グロブリン、メトキサレン、またはサリドマイドである。
【0043】
他の特定の実施形態において、ホモシステイン低下薬は、ビタミンB6、ビタミンB12、または葉酸塩である。
【0044】
なお別の実施形態において、抗新生物薬は、抗血管形成剤、アルキル化剤、代謝拮抗剤、天然物、プラチナ配位錯体、アントラセンジオン、置換尿素、メチルヒドラジン誘導体、副腎皮質の反応抑制薬、ホルモン、アンタゴニスト、癌遺伝子阻害剤、癌抑制遺伝子またはタンパク質、癌抑制遺伝子抗体、あるいは癌抑制遺伝子のアンチセンスオリゴヌクレオチドである。
【0045】
本発明はまた、本発明の有効量の組み合わせ、およびこの組み合わせを投与するための指示手段を含むキットを提供する。
【0046】
さらに、本発明は、哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害するための方法を提供し、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、以下:a)式(I):
【0047】
【化22】
[式中、Zは、炭素および窒素からなる群より選択され、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;Xは、酸素、窒素、および硫黄からなる群より選択され;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]で示される有効量の化合物またはその薬学的に受容可能な塩;ならびにb)抗出血性のウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、および抗新生物薬からなる群より選択される有効量の化合物を含む、有効量の組み合わせを投与し、それによって該哺乳動物における該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する。特定の実施形態において、この投与される組み合わせは、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0048】
本発明はまた、哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害するための方法を提供し、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、以下:a)上記に予め規定されたような、式IA、IB、およびII〜VIのいずれか1つの式で示される有効量の化合物、またはその薬学的に受容可能な塩を含む、有効量の組み合わせを投与する工程を包含する。
【0049】
この組み合わせは、任意の他の薬学的組成物と併用されて、哺乳動物におけるSAH加水分解酵素活性を調節し得る。この組み合わせはまた、上記のように、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、および高ホモシステイン不全症、循環器病、脳卒中、アルツハイマー病、糖尿病、炎症性腸疾患、多発性硬化症または自己免疫性神経炎などの疾患の予防および処置にも用いられ得る。しかしながら、この組み合わせが、特定の疾患の予防および用途に限定されることを意図するものではない。
【0050】
本発明はまた、IL−12の産生および/または放出を可逆的に阻害するための方法を提供し、該方法は、IL−12産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−12産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する。いくつかの実施形態において、IL−12は、IL−12P40、IL−12P35、またはIL−12P70である。いくつかの実施形態において、IL−12産生細胞は、哺乳動物に含まれる。いくつかの実施形態において、哺乳動物はヒトである。いくつかの実施形態において、哺乳動物は、炎症性腸疾患、多発性硬化症および他の自己免疫疾患からなる群より選択される疾患に罹患している疑いがある。
【0051】
本発明は、哺乳動物における遅延型過敏症(DTH)反応を軽減するための方法を提供し、該方法は、このような軽減が必要であるかまたは望ましい哺乳動物に、SAH加水分解酵素の有効量の可逆的阻害剤を投与することによって、該哺乳動物におけるDTH反応を軽減する工程を包含する。1つの実施形態において、哺乳動物はヒトである。別の実施形態において、SAH加水分解酵素の可逆的阻害剤は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0052】
本発明はまた、IL−10の産生および/または放出を維持するかあるいは増加させるための方法を提供し、該方法は、IL−10産生細胞を、SAH加水分解酵素の可逆的阻害剤と接触させることによって、該IL−10産生細胞中の該SAH加水分解酵素を可逆的に阻害する工程を包含する。1つの実施形態において、IL−10産生細胞は、哺乳動物に含まれる。別の実施形態において、SAH加水分解酵素の可逆的阻害剤は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0053】
本発明は、IL−2またはIFN−γの産生および/あるいは放出を可逆的に阻害するための方法をさらに提供し、該方法は、IL−2またはIFN−γ産生細胞を、SAH加水分解酵素の可逆的阻害剤と接触させることによって、該IL−2またはIFN−γ産生細胞中の該SAH加水分解酵素を可逆的に阻害する工程をさらに包含する。1つの実施形態において、IL−2またはIFN−γ産生細胞は、哺乳動物に含まれる。別の実施形態において、SAH加水分解酵素の可逆的阻害剤は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
例えば、本願発明は以下の項目を提供する。
(項目1)
式(IA):
【化1】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【化2】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
を有する化合物、またはその薬学的に受容可能な塩。
(項目2)
項目1に記載の化合物および薬学的に受容可能な賦形剤を含む、薬学的組成物。
(項目3)
有効量の項目2に記載の組成物、および該組成物を投与するための指示手段を含む、キット。
(項目4)
哺乳動物のS−アデノシル−L−ホモシステイン(SAH)加水分解酵素の活性を可逆的に阻害するための方法であって、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、式(I):
【化3】
[式中、Zは、炭素および窒素からなる群より選択され、
R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
を有する化合物またはその薬学的に受容可能な塩の有効量を投与し、それによって該哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目5)
組み合わせであって、以下のa)およびb):
a)式(I):
【化4】
[式中、Zは、炭素および窒素からなる群より選択され、
R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
を有する化合物またはその薬学的に受容可能な塩の有効量;ならびに
b)抗出血性のウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、および抗新生物薬からなる群より選択される化合物の有効量
を含む、組み合わせ。
(項目6)
哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害するための方法であって、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、以下のa)およびb):
a)式(I):
【化5】
[式中、Zは、炭素および窒素からなる群より選択され、
R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
を有する化合物またはその薬学的に受容可能な塩の有効量;ならびに
b)抗出血性のウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、および抗新生物薬からなる群より選択される化合物の有効量
を含む、組み合わせの有効量を投与し、
それによって、該哺乳動物における該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目7)
項目5に記載の組み合わせの有効量、および該組み合わせを投与するための指示手段を含む、キット。
(項目8)
S−アデノシル−L−ホモシステイン加水分解酵素(SAH)活性を阻害し得る候補阻害剤化合物を同定するための方法であって、以下の工程:
a)SAH結合ポケットのコンピュータモデルを構築する工程;
b)以下の構造
【化6】
[式中、Zは、炭素および窒素からなる群より選択される]
を有する複数の化合物をスクリーニングする工程;ならびに
c)該結合ポケットに計算上結合する化合物を同定する工程
を包含する、方法。
(項目9)
SAH加水分解酵素の活性を可逆的に阻害するための方法であって、SAH加水分解酵素を、有効量の項目1に記載の化合物と接触させて、該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目10)
SAH加水分解酵素の活性を可逆的に阻害するための方法であって、SAH加水分解酵素を、有効量の項目5に記載の組み合わせと接触させて、該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目11)
IL−12の産生および/または放出を可逆的に阻害するための方法であって、該方法は、IL−12産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−12産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する、方法。
(項目12)
哺乳動物における遅延型過敏症(DTH)反応を軽減するための方法であって、該方法は、このような軽減が必要であるかまたは望ましい哺乳動物に、SAH加水分解酵素の可逆的阻害剤の有効量を投与し、それによって該哺乳動物におけるDTH反応を軽減する工程を包含する、方法。
(項目13)
前記哺乳動物がヒトである、項目12に記載の方法。
(項目14)
前記SAH加水分解酵素の可逆的阻害剤が(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない、項目12に記載の方法。
(項目15)
IL−10の産生および/または放出を維持または増大させるための方法であって、該方法は、IL−10産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−10産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する、方法。
(項目16)
前記IL−10産生細胞が哺乳動物に含まれる、項目15に記載の方法。
(項目17)
前記SAH加水分解酵素の可逆的阻害剤が(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない、項目15に記載の方法。
(項目18)
IL−2またはIFN−γの産生および/あるいは放出を可逆的に阻害するための方法であって、該方法は、IL−2またはIFN−γ産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−2またはIFN−γ産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する、方法。
(項目19)
前記IL−2またはIFN−γ産生細胞が哺乳動物に含まれる、項目18に記載の方法。
(項目20)
前記SAH加水分解酵素の可逆的阻害剤が(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない、項目18に記載の方法。
(項目21)
前記化合物またはその薬学的に受容可能な塩が、式(IA)
【化7】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]で示される、項目4に記載の方法。
(項目22)
前記化合物またはその薬学的に受容可能な塩が、式(IB)
【化8】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される、項目4に記載の方法。
(項目23)
前記化合物またはその薬学的に受容可能な塩が、
【化9】
[式中、Wは、Hまたはメトキシである]
である、項目4に記載の方法。
(項目24)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目4に記載の方法。
(項目25)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目21に記載の方法。
(項目26)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目22に記載の方法。
(項目27)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目23に記載の方法。
【図面の簡単な説明】
【0054】
【図1】図1は、ヒツジ赤血球の定量的溶血(QHS)アッセイに対するDZ2002の影響を示す。データは、平均値±SDとして表した。**:コントロールと比較してP<0.01。
【図2】図2は、DZ2002が、混合リンパ球反応においてT細胞増殖を抑制することを示す。データは、平均値±SDとして表した。***:コントロールと比較してP<0.001。
【図3】図3は、DZ2002が、脾臓細胞において細胞傷害性を有さないことを示す。
【図4】図4は、Balb/cマウスにおけるDTH耳介腫脹に対するDZ2002の影響を示す。
【図5】図5は、TG誘導性腹膜細胞からのTNF−α産生に対するDZ2002の影響を示す。
【図6A】図6Aは、THP−1細胞におけるMHC−IIの発現に対するDZ2002の影響を示す。
【図6B】図6Bは、THP−1細胞におけるCD80の発現に対するDZ2002の影響を示す。
【図6C】図6Cは、THP−1細胞におけるCD86の発現に対するDZ2002の影響を示す。
【図7】図7Aおよび7Bは、THP−1細胞からのIL−12P40およびIL−12P70産生に対するDZ2002の影響を示す。
【図8】図8は、遅延型過敏症(DTH)反応に対するDZ2002の影響を示す。
【図9】図9は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−10産生に対するDZ2002の影響を示す。
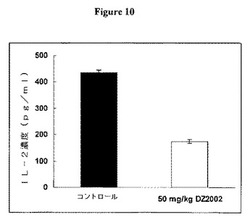
【図10】図10は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−2産生に対するDZ2002の影響を示す。
【図11】図11は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIFN−γ産生に対するDZ2002の影響を示す。
【発明を実施するための形態】
【0055】
(発明の詳細な説明)
開示内容を明確にするためであって、限定のためではなく、本発明の詳細な説明は以下の小節に分けられる。
【0056】
A.定義
他に規定しない限り、本明細書中で使用されるすべての技術用語および科学用語は、本発明が属する技術分野の当業者によって通常理解されるのと同じ意味を有する。本明細書中に引用される全ての特許、出願、公開された出願および他の刊行物は、その全体が、参考として援用される。この節に示される定義が、本明細書中に参考として援用される特許、出願、公開された出願および他の刊行物に示される定義に反するかまたは矛盾する場合、この節に示される定義が、本明細書中に参考として援用される定義より優先する。
【0057】
本明細書中で使用される場合、「a」または「an」は、「少なくとも1つ」または「1つ以上」を意味する。
【0058】
本明細書中で使用される場合、「組成物」とは、2つ以上の生成物または化合物の任意の混合物をいう。組成物は、溶液、懸濁液、液体、粉末、ペースト、水性、非水性、またはそれらの任意の組み合わせであり得る。
【0059】
本明細書中で使用される場合、「組み合わせ」とは、2つのアイテム間かまたはより多くのアイテム間の任意の関連をいう。
【0060】
本明細書中で使用される場合、「ホモシステイン」(Hcy)とは、以下の分子式の化合物をいう:HSCH2CH2CH(NH2)COOH。生物学的には、Hcyは、メチオニンの脱メチル化によって生成され、メチオニンからのシステインの生合成における中間体である。用語「Hcy」は、遊離Hcy(還元型の)および結合Hcy(酸化型の)を包含する。Hcyは、タンパク質、ペプチド、Hcy自体または他のチオールと、ジスルフィド結合で結合体化し得る。
【0061】
本明細書中で使用される場合、「SAH加水分解酵素」とは、SAHのアデノシン(Ado)およびHcyへの加水分解を触媒する酵素をいう。この酵素は、遍在性の真核生物酵素であり、一部の原核生物にも見出される。SAH加水分解酵素はまた、AdoおよびHcyからのSAHの生成も触媒する。SAH加水分解酵素の補酵素は、NAD+/NADHである。SAH加水分解酵素は、いくつかの触媒活性を有し得る。加水分解の方向において、第1の工程は、酵素結合NAD+(E−NAD+)によるSAHの3’−ヒドロキシル基の酸化(3’−酸化活性)、その後のL−Hcyのβ−脱離を包含し、3’−ケト−4’,5’−ジデヒドロ−5’−デオキシ−Adoを生じる。この強固に結合した中間体の5’側の位置への水のMichael付加(5’−加水分解活性)により、3’−ケト−Adoを生じ、これは次いで、酵素結合NADH(E−NADH)によってAdoに還元される(3’−還元活性)。この用語は、活性を実質的に変化させない保存的アミノ酸置換を行ったSAH加水分解酵素を包含することが意図される。
【0062】
本明細書中で使用される場合、本発明の化合物の「薬学的に受容可能な塩」または「薬学的に受容可能な誘導体」との用語は、当業者に容易に調製され得る任意の塩、エステルまたは誘導体を包含する。本発明の化合物の薬学的に受容可能な塩としては、例えば、薬学的に受容可能な無機および有機の酸および塩基から誘導されるものが挙げられる。適切な塩基から誘導される塩としては、アルカリ金属(例えば、ナトリウム)塩、アルカリ土類金属(例えば、マグネシウム)塩、アンモニウム塩およびN(C1−4アルキル)4+塩が挙げられるが、これらに限定されない。適切な酸の例としては、塩酸、臭化水素酸、硫酸、硝酸、過塩素酸、フマル酸、マレイン酸、リン酸、グリコール酸、乳酸、サリチル酸、コハク酸、トルエン−p−スルホン酸、酒石酸、酢酸、クエン酸、メタンスルホン酸、ギ酸、安息香酸、マロン酸、ナフタレン−2−スルホン酸、およびベンゼンスルホン酸が挙げられるが、これらに限定されない。それ自体では薬学的に受容可能でない他の酸(シュウ酸など)は、本発明の化合物およびその薬学的に受容可能な酸性塩を得る際に中間体として有用な塩の調製に用いられ得る。
【0063】
本明細書中で使用される場合、「生物活性」とは、化合物のインビボ活性、または化合物、組成物、もしくは他の混合物のインビボ投与の結果として生じる生理反応をいう。従って、生物活性は、このような化合物、組成物および混合物の治療効果ならびに薬学的活性を包含する。生物学的活性は、このような活性を試験するかまたは用いるように設計されたインビトロ系で認められ得る。
【0064】
本明細書中で使用される場合、「血漿」とは、流体(血液の非細胞部分)をいい、凝血後に得られる血清とは区別される。
【0065】
本明細書中で使用される場合、「血清」とは、フィブリン凝塊および血球の除去後に得られる血液の流体部分をいい、循環血液中の血漿とは区別される。
【0066】
本明細書中で使用される場合、「流体」とは、流動し得る任意の組成物をいう。従って、流体は、半固体、ペースト、溶液、水性混合物、ゲル、ローション、クリームの形態の組成物、および他のこのような組成物を包含する。
【0067】
本明細書中で使用される場合、任意の保護基、アミノ酸および他の化合物の略語は、他に指示がない限り、その一般的用法、認められた略語、またはIUPAC−IUB Commission on Biochemical Nomenclatureによるものである(Biochemistry 11:1726(1972)を参照のこと)。
【0068】
本明細書中で使用される場合、「疾患または障害」とは、特定可能な症状によって特徴付け可能な、生物における病理学的状態をいう。
【0069】
本明細書中で使用される場合、用語「治療剤」とは、当業者に公知の任意の従来の薬物または薬物療法をいい、ワクチンが挙げられるが、これに限定されない。
【0070】
本明細書中で使用される場合、「ワクチン」とは、能動的免疫予防を意図される任意の組成物をいう。ワクチンは、疾患を処置するか、疾患の発症を予防するか、または疾患の重症度を軽減するために、事前にかまたは感染後のいずれかに治療的に用いられ得る。例示的なワクチンとしては、病原性株の死滅させた微生物の調製物、弱毒化(改変または変異)株の生きた微生物の調製物、あるいは微生物、真菌、植物、原生動物、または後生動物の派生物もしくは生成物が挙げられるが、これらに限定されない。この用語はまた、タンパク質/ペプチドおよびヌクレオチドを主成分とするワクチンを包含する。
【0071】
本明細書中で使用される場合、用語「治療有効量」とは、疾患に関連する症状を寛解させるか、または何らかの様式で軽減するのに十分な量をいう。このような量は、単回投薬として、またはレジメンに従って投与され得る。反復投与は、症状の所望の寛解を達成するのに必要な場合がある。
【0072】
本明細書中で使用される場合、用語「投与」または化合物を「投与すること」とは、本発明の化合物または本発明の化合物のプロドラッグを被験体に与える任意の適切な方法をいう。
【0073】
本明細書中で使用される場合、用語「処置」とは、状態、障害または疾患の症状が寛解されるか、あるいは有利に変更される、任意の様式をいう。処置はまた、本明細書における組成物の任意の医薬用途も包含する。特定の障害の症状の寛解とは、組成物の投与に起因し得るかまたは関連し得る、症状の任意の軽減(永久であるか一時的であるかにかかわらず)をいう。
【0074】
本明細書中で使用される場合、用語「置換」とは、化合物中の水素原子を置換基で置換することをいう。
【0075】
本明細書中で使用される場合、用語「アルキル」は、1つ以上の置換基で置換されていてもよいアルキル基を含む、直鎖または分岐鎖アルキル基を包含する。例えば、アルキル基は、ヒドロキシ、ハロゲン、アリール、アルコキシ、アシル、または当該分野で公知の他の置換基で置換されていてもよい。アルキル基のより多くの炭素原子の1つもまた、1つ以上のヘテロ原子で置換されていてもよい。
【0076】
本明細書中で使用される場合、用語「Ki」とは、ICEなどの標的酵素の活性の阻害における化合物の有効性の数値尺度をいう。Kiの値が低いほど、高い有効性を示している。Ki値は、実験的に決定された速度データを、標準酵素反応速度式に当てはめることによって求められる(Segel,Enzyme Kinetics,Wiley−Interscience,1975)。
【0077】
本明細書中で使用される場合、「抗悪性腫瘍薬処置」とは、新生物、腫瘍または癌を、その症状を軽減または寛解することによって処置するように設計された、任意の処置をいう。新生物、腫瘍または癌の発生を予防するか、あるいはこれらの重症度を軽減する処置も、企図される。
【0078】
本明細書中で使用される場合、「新生物(新生物形成)」とは、異常な新生物をいい、従って腫瘍と同じものを意味し、これは良性であっても悪性であってもよい。過形成とは異なり、新生物の増殖は、本来の刺激がない状態においても持続する。
【0079】
本明細書中で使用される場合、「抗新生物薬(抗悪性腫瘍薬、抗腫瘍剤または抗癌剤と交換可能に用いられる)」とは、抗新生物処置に用いられる任意の薬剤をいう。これらとしては、単独かまたは他の化合物と組み合わせて用いられる場合に、新生物、腫瘍または癌と関連する臨床症状もしくは診断マーカーを、緩和するか、軽減するか、寛解するか、予防するか、緩解状態にするかまたは緩解状態を維持し得る、任意の薬剤が挙げられる。本発明と組み合わせて用いられ得る抗新生物薬としては、抗血管形成剤、アルキル化剤、代謝拮抗剤、特定の天然物、プラチナ配位錯体、アントラセンジオン、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、特定のホルモンおよびアンタゴニスト、抗癌性多糖類、ならびに漢方薬草抽出物のような特定の薬草抽出物が挙げられるが、これらに限定されない。
【0080】
本明細書中で使用される場合、「癌抑制遺伝子」(癌抑制遺伝子または癌感受性遺伝子とも呼ばれる)とは、細胞周期を通常負に制御し、かつ、細胞が急速な分裂へと進み得る前に変異させられるかまたは不活化されなければならない産物をコードする遺伝子をいう。例示的な癌抑制遺伝子としては、p16、p21、p53、RB(網膜芽細胞腫)、WT−1(Wilmの腫瘍)、DCC(結腸癌で欠失している)、NF−1(神経線維肉腫)およびAPC(adenomatous polypospis coli)が挙げられるが、これらに限定されない。
【0081】
本明細書中で使用される場合、「癌遺伝子」とは、動物細胞の正常な遺伝子(原癌遺伝子)の変異型および/または過剰発現型をいい、優性で細胞を正常な成長抑制から解放し得る。従って、癌遺伝子は、単独で、または他の変更と連携して、細胞を腫瘍細胞に変える。例示的な癌遺伝子としては、abl、erbA、erbB、ets、fes(fps)、fgr、fms、fos、hst、int1、int2、jun、hit、B−lym、mas、met、mil(raf)、mos、myb、myc、N−myc、neu(ErbB2)、ral(mil)、Ha−ras、Ki−ras、N−ras、rel、ros、sis、src、ski、trkおよびyesが挙げられるが、これらに限定されない。
【0082】
本明細書中で使用される場合、「アンチセンスポリヌクレオチド」とは、mRNAに相補的なヌクレオチド塩基の合成配列または二本鎖DNAのセンス鎖をいう。適切な条件下でのセンスポリヌクレオチドおよびアンチセンスポリヌクレオチドの混合により、これらの2つの分子の結合(すなわち、ハイブリダイゼーション)が生じる。これらのポリヌクレオチドがmRNAに結合(ハイブリダイズ)する場合、タンパク質合成(翻訳)の阻害が起こる。これらのポリヌクレオチドが二本鎖DNAに結合する場合、RNA合成(転写)の阻害が起こる。結果として生じた翻訳および/または転写の阻害により、センス鎖によってコードされるタンパク質の合成の阻害が生じる。
【0083】
本明細書中で使用される場合、「抗体」は、軽鎖および重鎖の可変領域で構成される抗体フラグメント(例えば、Fabフラグメント)を包含する。
【0084】
本明細書中で使用される場合、用語「ヒト化抗体」とは、ヒトに投与されても免疫応答を引き起こさないように、アミノ酸の「ヒト」配列を含むように改変されている抗体をいう。このような抗体を調製するための方法は公知である。例えば、モノクローナル抗体を発現するハイブリドーマは、非可変領域のアミノ酸組成がヒト抗体に基づいている抗体を発現するように、組換えDNA技術によって改変される。コンピュータプログラムは、このような領域を同定するように設計されている。
【0085】
本明細書中で使用される場合、「抗出血性ウイルス剤」または「抗ウイルス性出血剤」とは、出血性ウイルス感染の処置に用いられる任意の薬剤をいう。これらの薬剤としては、単独かまたは他の化合物と組み合わせて、ウイルス性出血疾患もしくは障害に関連する臨床症状または診断マーカーを緩和し得るか、軽減し得るか、寛解し得るか、予防し得るか、あるいは緩解状態を維持し得る、任意の薬剤を包含する。抗ウイルス性出血剤の非限定的な例としては、インターロイキン−1(IL−1)阻害剤、腫瘍壊死因子(TNF)阻害剤、抗ウイルスワクチン、抗ウイルス抗体、ウイルスによって活性化された免疫細胞、およびウイルスによって活性化された免疫血清が挙げられる。
【0086】
本明細書中で使用される場合、「抗出血性ウイルス処置」とは、症状を軽減するかまたは寛解させることによって、出血性ウイルス感染を処置するように設計される任意の処置をいう。感染を予防するかまたはその重症度を軽減する処置もまた企図される。
【0087】
本明細書中で使用される場合、「IL−1阻害剤」は、IL−1の産生、翻訳後修飾、成熟、または放出を阻止するかもしくは減少させる任意の物質、あるいはIL−1とIL−1レセプターの間の相互作用の効力に干渉するかもしくはこれを減少させる任意の物質を包含する。好ましくは、IL−1阻害剤は、抗IL−1抗体、抗IL−1レセプター抗体、IL−1レセプターアンタゴニスト、IL−1産生阻害剤、IL−1レセプター産生阻害剤、またはIL−1放出阻害剤である。
【0088】
本明細書中で使用される場合、「腫瘍壊死因子」(「TNF」)とは、主要組織適合性複合体内にコードされる炎症誘発性サイトカインの群をいう。TNFファミリーメンバーとしては、TNFαおよびTNFR(それぞれ、カケクチンおよびリンホトキシンとしても公知である)が挙げられる。TNAαおよびTNFRをコードする相補的なcDNAクローンが単離されている。従って、「TNF」という用語は、TNFαおよびTNFを含む、TNF遺伝子ファミリーによってコードされるすべてのタンパク質、あるいは任意の他の供給源から得られるかまたは合成的に調製された相当する分子を包含する。活性を実質的に変化させない保存的アミノ酸置換を行ったTNFを包含することが意図される。
【0089】
本明細書中で使用される場合、「TNF阻害剤」は、TNFの産生、翻訳後修飾、成熟、または放出を阻止するかもしくは減少させる任意の物質、あるいはTNFとTNFレセプターの間の相互作用の効力に干渉するかもしくはこれを減少させる任意の物質を包含する。好ましくは、TNF阻害剤は、抗TNF抗体、抗TNFレセプター抗体、TNFレセプターアンタゴニスト、TNF産生阻害剤、TNFレセプター産生阻害剤、またはTNF放出阻害剤である。
【0090】
B.S−アデノシル−L−ホモシステイン加水分解酵素の可逆的阻害剤
作用機序に基づく細胞傷害性を最小にするための1つのアプローチは、SAH加水分解酵素阻害剤が可逆的な阻害活性を示すように、該阻害剤の薬物動態プロファイルを最適化することである。薬物動態プロファイルは、KOff値を最適化することによって最適化され得る。例えば、KOff値は、これらの値が所望の治療効果を生じるのに十分に小さいが、次の投与前に酵素活性の適切な回復を可能にするのに十分に大きいように、最適化される。
【0091】
一般に、SAH加水分解酵素の可逆的阻害剤は、酵素に非共有結合的に結合し、かつ、酵素から容易に遊離される。例えば、酵素に結合した可逆的阻害剤は、簡単な透析または緩衝液の交換もしくはpHの変更によって取り除かれ得る。可逆的阻害剤としては、拮抗阻害剤、非拮抗阻害剤、不拮抗阻害剤、および混合型の阻害剤が挙げられる。
【0092】
拮抗阻害剤は、単に遊離の酵素に結合して、酵素が基質に結合するのを阻止する阻害剤である。通常、拮抗阻害剤は、基質と構造が類似しており、活性部位への基質の接近を妨害するように、活性部位に結合する(すなわち、酵素は、インヒビターを遊離してからでなければ基質に結合できない)。基質および阻害剤の両方が、同じ部位に結合するために競合する。反応速度論に対する拮抗阻害剤の影響は、酵素の最大速度(Vmax)に影響を及ぼすことなく、ミカエリス定数(Km)を増加させることである。拮抗阻害剤の存在下での見かけのKm(Km,app)は、Km(1+[I]/Ki)であり、式中、[I]は阻害剤の濃度であり、そしてKiは阻害剤の解離定数である。
【0093】
非拮抗阻害剤は、酵素が反応を触媒するのを阻止するが、基質が結合するのを妨害しない阻害剤である。基質および阻害剤の両方とも、同時に酵素に結合し得る(すなわち、阻害剤および基質は、同じ部位に結合するために拮抗しない)が、触媒反応は、阻害剤が結合していない場合にのみ生じる。非拮抗阻害剤は、遊離の酵素および酵素−基質複合体に、同じ親和性で結合する。非拮抗阻害剤の反応速度論に対する影響は、Kmに影響を及ぼすことなくVmaxを減少させることである。非拮抗阻害剤の存在下での見かけのVmax(Vmax,app)は、Vmax/(1+[I]/Ki)であり、式中、[I]は阻害剤の濃度であり、そしてKiは阻害剤の解離定数である。
【0094】
不拮抗阻害剤は、単に酵素−基質複合体に結合して、該複合体を不活化する阻害剤である。反応速度論に対する不拮抗阻害剤の影響は、VmaxおよびKmの両方を減少させることである。不拮抗阻害剤の存在下での見かけのKm(Km,app)は、Km(1+[I]/Kib)であり、式中、[I]は阻害剤の濃度であり、そしてKibは酵素−基質複合体に対する阻害剤の解離定数である。不拮抗阻害剤の存在下での見かけのVmax(Vmax,app)は、Vmax/(1+[I]/Kib)であり、式中、[I]は阻害剤の濃度であり、そしてKibは酵素−基質複合体に対する阻害剤の解離定数である。
【0095】
混合型の阻害剤は、遊離の酵素および酵素−基質複合体の両方に結合する阻害剤であり、触媒反応を阻害する。混合型の阻害剤には、活性を完全には無効にせず、単に反応速度を顕著に減少させるだけの阻害剤も含まれる。混合阻害剤の存在下での見かけのKm(Km,app)は、Km(1+[I]/Kia)であり、式中、[I]は阻害剤の濃度であり、そしてKiaは遊離の酵素に対する阻害剤の解離定数である。混合阻害剤の存在下での見かけのVmax(Vmax,app)は、Vmax/(1+[I]/Kib)であり、式中、[I]は阻害剤の濃度であり、そしてKibは酵素−基質複合体に対する阻害剤の解離定数である。
【0096】
酵素(例えば、SAH加水分解酵素)に対する阻害剤のタイプを決定するための方法は、当該分野で公知である。例えば、酵素反応速度論実験を用いて阻害剤が結合する機序および部位を試験し、阻害剤のタイプを決定し得る。KmおよびVmaxは、阻害剤の非存在下で、および2種以上の濃度の阻害剤の存在下で決定され得る。次いで、収集したデータは、阻害剤のタイプを決定するために、見かけのKmまたはVmaxの変化(すなわち、阻害剤濃度の関数としてのパラメータの変化)について分析され得る。
【0097】
強固に結合した阻害剤および作用機序に基づく阻害剤はまた、酵素と阻害剤の間に共有結合が形成されないにもかかわらず、時間依存的阻害パターンを示し得る。これらのタイプの阻害剤によって不活化される酵素は、通常は永久に無効にされ、かつその活性は、例えば、0.5〜5時間のような一定時間のゲル濾過または透析によって、容易に回復することができない。これらのタイプの時間依存的阻害剤は、不可逆的阻害剤とみなされ、本発明において記載される可逆的阻害剤から除外される。
【0098】
SAH加水分解酵素の可逆的阻害剤としてのエリタデニン誘導体
本発明は、可逆的かつ強力なS−アデノシル−L−ホモシステイン組成物の新規阻害剤に関する。例えば、本発明は、Ki値が100nM未満の化合物を提供する。1つの実施形態において、本発明は、4(アデニン−9−イル)−2−ヒドロキシブタン酸、その誘導体、およびその薬学的に受容可能な塩、ならびにこのような化合物を用いてSAH加水分解酵素を可逆的に阻害するための方法を提供する。
【0099】
可逆的阻害剤である、4(アデニン−9−イル)−2−ヒドロキシブタン酸は、エリタデニンのβ炭素でのデオキシル修飾(deoxyl modification)により合成される。エリタデニンは、天然に存在する化合物であり、SAH加水分解酵素の強力な不可逆的阻害剤である。エリタデニンのβ炭素でのデオキシル修飾により、阻害能力を保持しつつ、可逆的阻害剤である化合物を生じる。4(アデニン−9−イル)−2−ヒドロキシブタン酸の誘導体は、当業者に公知の従来の合成方法を用いて合成され得る。(例えば、Yuanら、Adv.Antiviral Drug Des.2:41−88(1996);Holyら、Coll.Czechoslovak Chem.Commun.50:245−279(1985)を参照のこと)。
【0100】
4(アデニン−9−イル)−2−ヒドロキシブタン酸誘導体の例としては、塩基修飾誘導体、および側鎖置換誘導体が挙げられるが、これらに限定されない。塩基修飾誘導体は、アデニル環塩基(adenyl ring base)にて修飾された4(アデニン−9−イル)−2−ヒドロキシブタン酸の誘導体である。アデニル環は、アミノ基にて、種々の修飾基で修飾され得る。アデニル環はまた、アデニル環のC2位およびC8位にて、種々の置換基で修飾され得る。
【0101】
1つの実施形態において、SAH加水分解酵素の可逆的阻害剤は、下記式(I)で示されるもの、およびその薬学的に受容可能な塩である:
【0102】
【化23】
[式中、Zは炭素または窒素であり、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、またはハロゲンであり;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、またはヘテロアリールであり;Xは、酸素、窒素、または硫黄であり;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、またはヘテロアリールである]。
【0103】
1つの局面において、SAH加水分解酵素の可逆的阻害剤は、式IA
【0104】
【化24】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【0105】
【化25】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される。
【0106】
種々のR基が、他の置換基で置換されていてもよい。これらの置換基は、ハロゲン、ヒドロキシ、アルコキシ、ニトロ、シアノ、カルボン酸、アルキル、アルケニル、シクロアルキル、チオール、アミノ、アシル、カルボキシレート、アリール、カルバメート、カルボキサミド、スルホンアミド、複素環基、または当該分野で公知の任意の適切な置換基であり得る。特定の実施形態において、各R基は、水素、またはメチルのような低級直鎖アルキルである。別の実施形態において、アルキルまたはアルコキシ基の中の1つ以上の炭素原子は、1つ以上のヘテロ原子で置換され得る。
【0107】
アミノ基はまた、2回または3回置換されて、2級または3級アミンを形成し得る。置換基の非限定的な例としては、アルキルまたは置換されていてもよいアルキル基;アルケンまたは置換されていてもよいアルケニル基;シクロアルキルまたは置換されていてもよいシクロアルキル基;アリール、複素環;アラルキル(例えば、フェニルC1−4アルキル);フェニル、ピリジン、フェニルメチル、フェネチル、ピリジニルメチル、ピリジニルエチルのようなヘテロアルキル;および他の置換基が挙げられる。複素環基は、1〜4個のヘテロ原子を含む5または6員環であってもよい。
【0108】
アミノ基は、置換されていてもよいC2−4アルカノイル(例えば、アセチル、プロピオニル、ブチリル、イソブチリルなど);C1−4アルキルスルホニル(例えば、メタンスルホニル、エタンスルホニルなど);カルボニルまたはスルホニル置換された芳香族環または複素環(例えば、ベンゼンスルホニル、ベンゾイル、ピリジンスルホニル、ピリジンカルボニルなど)で置換され得る。
【0109】
CO−X−Y基は、置換されていてもよいカルボキシレート基であり得る。置換されていてもよいカルボキシレート基の例としては、置換されていてもよいアルキル(例えば、C1−10アルキル);置換されていてもよいシクロアルキル(例えば、C3−7シクロアルキル);置換されていてもよいアルケニル(例えば、C2−10アルケニル);置換されていてもよいシクロアルケニル(例えば、C3−7シクロアルケニル);置換されていてもよいアリール(例えば、フェニル、ナフチル、ベンジルなどのC1−4アリール);および他の適切な置換基が挙げられるが、これらに限定されない。メトキシメチル、メトキシエチル、および関連する基のような基もまた、包含される。
【0110】
新規のSAH加水分解酵素阻害剤の構造に基づく薬物設計
本発明の化合物を最初の鋳型分子として用いた、構造に基づく薬物設計を提供することもまた、本発明の目的である。最近、SAH加水分解酵素のX線構造は、酵素の「開」および「閉」状態の両方について入手可能になっている。当業者は、構造に基づく設計を用いて、SAH加水分解酵素阻害剤をスクリーニングするための新規の化合物を設計し得る。候補化合物の設計または選択は、SAH加水分解酵素の結合ポケットを満たす種々の部分の選択から開始し得る。(例えば、米国特許第5,756,466号;Klebe,J.Mol.Med.78:69−281(2000);およびMaignanら、Curr.Top.Med.Chem.1:161−174(2001)を参照のこと)。
【0111】
個々の結合ポケットを満たす部分を選択するための方法は、多数存在する。これらとしては、活性部位の物理的モデルまたはコンピュータモデルの外観検査、および選択された部分のモデルを種々の結合ポケットに手動でドッキングすることが挙げられる。当該分野で周知かつ入手可能なモデリングソフトウェアが用いられ得る。これらとしては、QUANTA(Molecular Simulations,Inc.,Burlington,Mass.,1992);SYBYL(Molecular Modeling Software,Tripos Associates,Inc.,St.Louis,Mo.,1992);AMBER(Weinerら、J.Am.Chem.Soc.6:765−784(1984));CHARMM(Brooksら、J.Comp.Chem.4:187−217(1983))が挙げられるが、これらに限定されない。モデリング工程に続いて、CHARMMおよびAMBERのような標準分子力学力場を用いたエネルギー最小化が行なわれ得る。さらに、本発明の結合部分を選択するプロセスを補助する、より専門化したコンピュータプログラムが多数存在する。これらとしては、以下が挙げられるが、これらに限定されない:
1.GRID(Goodford、「A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules」、J.Med.Chem.28:849−857(1985))。GRIDは、Oxford University,Oxford,UKから入手可能である。
【0112】
2.MCSS(Mirankerら、「Functionality Maps of
Binding Sites:A Multiple Copy Simultaneous Search Method」,Proteins:Structure,Function and Genetics」11:29−34(1991))。MCSSは、Molecular Simulations,Burlington,Mass.から入手可能である。
【0113】
3.AUTODOCK(Goodsellら、「Automated Docking
of Substrates to Proteins by Simulated Annealing」,PROTEINS:Structure,Function and Genetics 8:195−202(1990))。AUTODOCKは、Scripps Research Institute,La Jolla,Calif.から入手可能である。
【0114】
4.DOCK(Kuntzら、「A Geometric Approach to Macromolecule−Ligand Interactions」,J.Mol.Biol.161:269−288(1982))。DOCKは、University of California,San Francisco,Calif.から入手可能である。
【0115】
一旦、適切な結合部分が選択されると、これらの結合部分は、単一の阻害剤に構築され得る。この構築は、種々の部分を中心骨格に結合することによって達成され得る。この構築プロセスは、例えば、外観検査に続いて、QUANTAまたはSYBYLのようなソフトウェアを同じく用いた手動モデル構成によってなされ得る。多数の他のプログラムも、種々の部分を結合するための選択方法を補助するために用いられ得る。これらとしては、以下が挙げられるが、これらに限定されない:
1.CAVEAT(Bartlettら、「CAVEAT:A Program to
Facilitate the Structure−Derived Design
of Biologically Active Molecules」,Molecular Recognition in Chemical and Biological Problems,Special Pub.,Royal Chem.Soc.78:182−196(1989))。CAVEATは、University of California,Berkeley,Calif.から入手可能である。
【0116】
2.MACCS−3Dなどの3Dデータベースシステム(MDL Information Systems,San Leandro,Calif.)。この領域は、最近、Martinによって概説された(Martin,「3D Database Searching in Drug Design」,J.Med.Chem.35:2145−2154(1992))。
【0117】
3.HOOK(Molecular Simulations,Burlington,Mass.から入手可能)。
【0118】
上記の阻害剤化合物のコンピュータ補助によるモデリングに加えて、本発明の阻害剤は、活性のない部位または公知の阻害剤の一部を必要に応じて含むもののいずれかを用いて、デノボで構築され得る。このような方法は、当該分野で周知である。これらとしては、例えば、以下が挙げられる:
1.LUDI(Bohm,「The Computer Program LUDI:A New Method for the De Novo Design of Enzyme Inhibitors」,J.Comp.Aid.Molec.Design 6:61−78(1992))。LUDIは、Biosym Technologies,San Diego,Calif.から入手可能である。
【0119】
2.LEGEND(Nishibataら、Tetrahedron,47:8985(1991))。LEGENDは、Molecular Simulations,Burlington,Mass.から入手可能である。
【0120】
3.LeapFrog(Tripos associates,St.Louis,Mo.から入手可能)。
【0121】
薬物のモデリングに一般に使用される多数の技術が用いられ得る(例えば、Cohenら、J.Med.Chem.33:883−894(1990)を参照のこと)。同様に、化学技術文献中の多数の例が、特定の薬物設計プロジェクトに適用され得る。(概説について、Naviaら、Curr.Opin.Struc.Biol.2:202−210(1991)を参照のこと)。本発明の工程の新規の組み合わせを用いると、当業者は、時間のかかる高価な実験を有利に回避して、特定の化合物の酵素阻害活性を決定し得る。この方法はまた、SAH加水分解酵素阻害剤、ならびにSAH加水分解酵素媒介性の疾患に対する治療および予防剤の合理的設計を容易にするのにも有用である。従って、本発明は、このような阻害剤、およびこのような阻害剤を同定または選択するための方法に関する。
【0122】
種々の従来の技術を用いて、各々の上記の評価、およびSAH加水分解酵素阻害活性について候補化合物をスクリーニングするのに必要な評価を行い得る。一般に、これらの技術は、所定の部分の位置および結合近似、結合した阻害剤の占有空間、所定の化合物の結合の変形エネルギーおよび静電的相互作用エネルギーを決定する工程を包含する。上記の評価に有用な従来の技術の例としては、量子力学、分子力学、分子動力学、モンテカルロサンプリング、系統的探索およびディスタンスジオメトリー法(Marshall,Ann.Ref.Pharmacol.Toxicol.27:193(1987))が挙げられるが、これらに限定されない。これらの方法を実行するのに用いられる特殊なコンピュータソフトウェアが開発されている。このような用途のために設計されたプログラムの例としては、以下が挙げられる:Gaussian 92(Gaussian,Inc.,Pittsburgh,Pa.);AMBER;QUANTA/CHARMM;およびInsight II/Discover(Biosysm Technologies
Inc.,San Diego,Calif.)。これらのプログラムは、例えば、Silicon Graphics Indigo2ワークステーションまたはIBM RISC/6000ワークステーションモデル550を用いて実行され得る。他のハードウェアシステムおよびソフトウェアパッケージが公知であり、かつ、当業者に明らかな適用性を有する。
【0123】
本発明に従って、様々なクラスの活性なSAH加水分解酵素阻害剤が、SAH加水分解酵素活性部位の種々の結合ポケットと、類似の方法で相互作用し得る。これらの重要な基の空間的配置は、しばしばファルマコフォアと呼ばれる。ファルマコフォアの概念は、文献に十分に記載されている(Mayerら、J.Comp.Aided Molec.Design 1:3−16(1987);Hopfingerら,Concepts and Applications of Molecular Similarity,JohnsonおよびMaggiora(編),Wiley(1990)を参照のこと)。
【0124】
本発明の様々なクラスのSAH加水分解酵素阻害剤はまた、結合に必要な特異的相互作用が得られ得るように、必要な部分が活性部位に配置されることを可能にする、様々な骨格またはコア構造を用い得る。これらの化合物は、ファルマコフォア(すなわち、SAH加水分解酵素の活性部位の形状および性質に対する構造的同一性)にマッチする能力に関して最善に定義される。種々の骨格が、例えば、Klebe,G.,J.Mol.Med.78:269−281(2000);Maignanら、Curr.Top.Med.Chem.1:161−174(2001);およびBemisらに対する米国特許第5,756,466号)に記載されている。
【0125】
阻害されるS−アデノシル−L−ホモシステイン加水分解酵素
本発明の化合物は、任意のSAH加水分解酵素を可逆的に阻害するのに用いられ得る。本発明は、任意の特定のSAH加水分解酵素を可逆的に阻害することに限定されることを意図するものではない。
【0126】
1つの実施形態において、本発明の化合物は、以下のGenBank登録番号のヌクレオチド配列を含む核酸によってコードされるSAH加水分解酵素を、可逆的に阻害するのに用いられ得る:AF129871(Gossypium hirsutum);AQ003753(Cryptosporidium parvum);AF105295(Alexandrium fundyense);AA955402(Rattus norvegicus);AA900229(Rattus norvegicus);AA874914(Rattus norvegicus);AA695679(Drosophila melanogaster卵巣);AA803942(Drosophila melanogaster卵巣;AI187655(Manduca sexta雄触角);U40872(Trichomonas vaginalis);AJ007835(Xenopus laevis);AF080546(Anopheles gambiae);AI069796(T.cruzi epimastigote);Z97059(Arabidopsis thaliana);AF059581(Arabidopsis thaliana);U82761(Homo sapiens);AA754430(Oryza sativa);D49804(Nicotiana tabacum);D45204(Nicotiana tabacum);X95636(D.melanogaster);T18277(Zea mays胚乳);R75259(マウス脳);Z26881(C.roseus);X12523(D.discoideum);X64391(Streptomyces fradiae);W21772(トウモロコシの葉);AH003443(Rattus norvegicus);U14963(Rattus norvegicus);U14962(Rattus
norvegicus);U14961(Rattus norvegicus);U14960(Rattus norvegicus);U14959(Rattus norvegicus);U14937(Rattus norvegicus);U14988(Rattus norvegicus);U14987(Rattus norvegicus);U14986(Rattus norvegicus);U14985(Rattus norvegicus);U14984(Rattus norvegicus);U14983(Rattus norvegicus);U14982(Rattus norvegicus);U14981(Rattus norvegicus);U14980(Rattus norvegicus);U14979(Rattus norvegicus);U14978(Rattus norvegicus);U14977(Rattus norvegicus);U14976(Rattus norvegicus);U14975(Rattus norvegicus);L32836(Mus musculus);L35559(Xenopus laevis);Z19779(ヒト胎児副腎組織);L23836(Rhodobacter
capsulatus);M15185(ラット);L11872(Triticum
aestivum);M19937(粘菌(D.discoideum);M80630(Rhodobacter capsulatus)。
【0127】
別の実施形態において、本発明の化合物は、GenBank登録番号M61831−61832のヌクレオチド配列を含む核酸によってコードされるSAH加水分解酵素を可逆的に阻害するのに用いられ得る(Coulter−KarisおよびHershfield,Ann.Hum.Genet.,53(2):169−175(1989)も参照のこと)。本発明の化合物はまた、米国特許第5,854,023号に示されるヌクレオチド配列またはアミノ酸配列を含む核酸によってコードされるSAH加水分解酵素を可逆的に阻害するのにも用いられ得る。
【0128】
C.治療剤としての用途
4(アデニン−9−イル)−2−ヒドロキシブタン酸、その誘導体、および薬学的に受容可能な塩を用いたSAH加水分解酵素の可逆的な阻害により、その治療効果を保持しつつ、細胞傷害性を顕著に低下させる。本発明の化合物は、その効力および可逆性のため、他の不可逆的阻害剤に付随する強い毒性のない治療剤として用いられ得る。本発明の化合物は、SAH加水分解酵素を阻害する能力に関連する生物学的活性を示す薬剤として有用である。SAH加水分解酵素に対する阻害効果は、阻害剤の存在下または非存在下におけるSAH加水分解の初速度の比を用いるか、あるいは当業者に公知の任意の方法を用いて評価され得る。本発明は、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、高ホモシステイン不全症、循環器病、脳卒中、アルツハイマー病、および糖尿病などの疾患の予防および処置のための組成物ならびに方法を提供する。しかしながら、本発明は、特定の疾患の予防および処置に限定されることを意図するものではない。
【0129】
1.出血熱ウイルス
本発明は、ウイルス性出血熱の処置のための組成物および方法を提供する。本発明の可逆的阻害剤は、出血熱を引き起こす全てのタイプのウイルス(トガウイルス、アレナウイルス、ナイロウイルス、およびハンタウイルスが挙げられるが、これらに限定されない)に対する広域スペクトラムの抗ウイルス剤として機能し得る。広域スペクトラムの抗ウイルス薬は、狭域スペクトラムの薬剤より優れた多くの利点を与える。ウイルスの病原体の臨床診断に伴う困難のために、診断結果から特定の抗ウイルス剤の選択に到達するのが遅すぎる場合が多い。速効作用は、患者の状態が悪化するのを防ぐために、特に、患者が臨床症状を示すや否やウイルス化学療法を開始しなければならない急性感染症において、しばしば必要である。
【0130】
S−アデノシル−L−ホモシステイン(SAH)加水分解酵素の阻害剤は、エボラウイルス感染の処置に有効であると報告されている。本発明の化合物はまた、他の出血性疾患(例えば、WO00/64479に記載される出血性疾患)に対して用いられ得る。阻害の機構は、本発明の方法を実施するのに必要ではないが、本発明の化合物がウイルス複製を阻害する作用機構は、ウイルスのメチル化の阻害に基づき得る。
【0131】
2.自己免疫疾患および免疫抑制に関連する疾患
本発明は、自己免疫疾患を予防および処置するための組成物ならびに方法を企図する。ある人が自己免疫疾患を有する場合、免疫系が、その人自身の体の細胞、組織、および器官を誤って攻撃する。集団として、自己免疫疾患には、何百万人ものアメリカ人が罹患している。ほとんどの自己免疫疾患は、男性よりも女性にしばしば好発する。自己免疫疾患の例は、国立保健研究所(National Institute of Health)、「Understanding Autoimmune Disease」(http://www.niaid.nih.gov/publications/autoimmune/autoimmune.htm.)から見出され得る。
【0132】
SAH加水分解酵素活性を調節する化合物はまた、免疫抑制に関連する疾患の処置に用いられ得る。免疫抑制は、化学療法、放射線治療、創傷治癒の促進、熱傷処置の強化、またはコルチコステロイド治療などの他の薬物治療、あるいは自己免疫疾患および移植片/移植拒絶の処置に用いられる薬物の組み合わせに起因し得る。免疫抑制はまた、レセプター機能の先天性欠損、感染症、寄生虫症、または他の原因にも起因し得る。
【0133】
3.新生物および癌
本発明はまた、副腎、肛門、聴神経、胆管、膀胱、骨、脳、胸、頬(bruccal)、中枢神経系、頚部、結腸、耳、子宮内膜、食道、眼、眼瞼、ファロピウス管、胃腸管、頭部、首、心臓、腎臓、喉頭、肝臓、肺、下顎骨、下顎関節頭、上顎骨、口、鼻咽腔、鼻、口腔、卵巣、膵臓、耳下腺、陰茎、耳介、脳下垂体、前立腺、直腸、網膜、唾液腺、皮膚、小腸、脊髄、胃、精巣、甲状腺、扁桃腺、尿道、子宮、膣、内耳神経、外陰に関連する新生物、および他の器官に関連する新生物が挙げられるが、これらに限定されない新生物を予防および処置するための組成物ならびに方法も企図する。特定の実施形態において、本発明の薬学的組成物は、非小細胞肺癌、肺癌、乳癌、および前立腺癌の処置に有用である。本発明は、癌(固形腫瘍、リンパ腫、転移性腫瘍、膠芽細胞腫瘍、および他の癌腫瘍に関連する癌が挙げられるが、これらに限定されない)を予防および処置するための組成物ならびに方法をさらに企図する。
【0134】
4.高いホモシステインレベルに関連する疾患
さらに、本発明の化合物は、高レベルのホモシステインに関連する疾患の予防および処置のための血漿ホモシステイン低下薬として用いられ得ることが企図される。高いホモシステインレベル(すなわち、高ホモシステイン血症)に関連していることが見出されている疾患としては、循環器病、脳卒中、アルツハイマー病および糖尿病が挙げられるが、これらに限定されない。例えば、高ホモシステイン血症と冠状動脈性心臓病(CHD)、末梢血管疾患、脳卒中、および静脈血栓症との間の関係が、種々の研究で示されている。
【0135】
高ホモシステインレベルによる脳卒中のリスクの増大はまた、アルツハイマー病を発症する可能性を増加させる。アルツハイマー型の痴呆を有する人々が、血中に高レベルのホモシステインを有することも、最近の研究で示されている。(Selhubら、「Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease」、N.Eng.J.Med.46:476−483(2002))。高いホモシステインはまた、糖尿病、狼瘡、および他の慢性疾患における合併症とも関連している。
【0136】
5.IL−12産生および/または放出の可逆的阻害
本発明は、IL−12(IL−12P70、IL−12P35およびIL−12P40が挙げられる)の産生および/または放出を可逆的に阻害するための組成物ならびに方法を提供し、この方法は、IL−12産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−12産生細胞中の該SAH加水分解酵素を可逆的に阻害する工程を包含する。IL−12P70(時には「IL−12P75」と呼ばれる、Abdi,Scand J.Immunol.56:1−11(2002)を参照のこと)は、p35およびp40サブユニットで構成されているヘテロ二量体のサイトカインである。いくつかの実施形態において、IL−12産生細胞は、哺乳動物(例えば、ヒト)に含まれる。これらの方法は、このような可逆的阻害が必要であるかまたは望ましい疾患の処置に用いられ得る。このような疾患としては、炎症性腸疾患、多発性硬化症および他の自己免疫疾患が挙げられるが、これらに限定されない。
【0137】
SAH加水分解酵素の任意の可逆的阻害剤は、例えば、本明細書中に記載される可逆的阻害剤(例えば、B項に記載される阻害剤)が用いられ得る。SAH加水分解酵素の可逆的阻害剤はまた、Yuanら、Exp.Opin.Ther.Patents 9:1197−1206(1999)、ならびに米国特許第5,137,876号、および同第6,541,482号にも記載される。これらの可逆的阻害剤は、例えば、薬学的に受容可能なキャリアまたは希釈剤とともに処方されて、薬学的組成物中に含まれ得る。
【0138】
いくつかの実施形態において、SAH加水分解酵素の可逆的阻害剤は、下記式(I)で示されるもの、および/またはその薬学的に受容可能な塩である:
【0139】
【化26】
[式中、Zは炭素または窒素であり、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、またはハロゲンであり;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、またはヘテロアリールであり;Xは、酸素、窒素、または硫黄であり;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、またはヘテロアリールである]。特定の実施形態において、投与される組み合わせは、(4−アデニン−9−イル)−2−ヒドロキシブタン酸を含まない。
【0140】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式IA
【0141】
【化27】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【0142】
【化28】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される。
【0143】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(II):
【0144】
【化29】
で示される。
【0145】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(III):
【0146】
【化30】
[式中、R1およびR2は、それぞれ独立して、水素またはヒドロキシである;ただし、R1およびR2がともにヒドロキシであることはない]で示される。いくつかの実施形態において、R1は水素であり、そしてR2はヒドロキシである。いくつかの実施形態において、R1はヒドロキシであり、そしてR2は水素である。いくつかの実施形態において、R1およびR2は、ともに水素である。
【0147】
他の実施形態において、該SAH加水分解酵素の可逆的阻害剤は、式(IV):
【0148】
【化31】
[式中、R1は、NH2、SCH3、またはCH2NH2である]で示される。
【0149】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(V):
【0150】
【化32】
[式中、R1は、NH2またはCONH2である]
で示される。
【0151】
なお別の実施形態において、SAH加水分解酵素の可逆的阻害は、式(VI):
【0152】
【化33】
[式中、Wは、Hまたはメトキシである]
で示される。
【0153】
D.薬学的組成物
本発明の薬学的組成物は、本発明の化合物(例えば、本明細書中に記載されるような、式I、IA、IBまたはII〜VIのいずれか1つの式で示される化合物)、およびその薬学的に受容可能な塩のいずれかを、単独かあるいは任意の薬学的に受容可能なキャリア、アジュバントまたはビヒクルと組み合わせて含む。本発明での使用に用いられ得る受容可能な組成物およびそれらの投与方法としては、米国特許第5,736,154号;同第6,197,801号;同第5,741,511号;同第5,886,039号;同第5,941,868号;同第6,258,374号および同第5,686,102号に記載されるものが挙げられるが、これらに限定されない。本発明の薬学的組成物において用いられ得る薬学的に受容可能なキャリア、アジュバントおよびビヒクルの例としては、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、ヒト血清アルブミンのような血清タンパク質、リン酸塩のような緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、塩または電解質、例えば硫酸プロタミン、リン酸水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩、コロイドシリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロース系物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリレート、ワックス、ポリエチレン−ポリオキシプロピレン−ブロックポリマー、ポリエチレングリコールおよび羊毛脂が挙げられるが、これらに限定されない。
【0154】
処方、投薬量および投与経路は、当該分野で公知の方法に従って決定され得る(例えば、Remington:The Science and Practice of Pharmacy,Alfonso R.Gennaro(編者)Mack Publishing Company,1997年4月;Therapeutic Peptides and Proteins:Formulation,Processing,and Delivery Systems,Banga,1999;ならびにPharmaceutical Formulation Development of Peptides and Proteins,HovgaardおよびFrkjr(編),Taylor & Francis,Inc.,2000;Biopharmaceutical Drug Design and Development,Wu−PongおよびRojanasakul(編),Humana Press,1999を参照のこと)。SAH加水分解酵素の調節を必要とする状態の処置または予防において、適切な投薬レベルは、一般に、1日に体重1kg当たり約0.01〜500mgである。好ましくは、投薬レベルは、1日に約0.1〜約250mg/kgである。より好ましい実施形態において、投薬レベルは、1日に約0.1〜約20mg/kgの範囲である。適切な投薬量は、単回または複数回投与で投与され得る。任意の特定の被験体に対する特定の用量レベルおよび投薬の頻度は変動し得、そして種々の因子(例えば、用いられる特定の化合物の活性、その化合物の代謝安定性および作用の長さ、年齢、体重、全身的な健康状態、性別、食餌、投与の様式および時間、排泄速度、薬物の組み合わせ、特定の状態の重症度、ならびに治療を受ける患者)に依存することが理解される。
【0155】
本発明の薬学的組成物は、経口的に、非経口的に、吸入スプレーにより、局所的に、直腸内に、経鼻的に、口腔内に、経膣的に、埋め込みリザーバーを介して、または任意の適切な投与形態で投与され得る。本明細書中で使用される場合、用語「非経口」は、皮下、皮内、静脈内、筋肉内、関節内、滑膜内、胸骨内、くも膜下腔内、病巣内および頭蓋内の注射または注入技術を包含する。任意の所定の場合における最も適切な経路は、処置される状態の性質および重症度、ならびに用いられるSAH加水分解酵素阻害剤の性質に依存する。
【0156】
薬学的組成物は、滅菌注射用調製物の形態(例えば、滅菌注射用水性または油性懸濁液として)であり得る。この懸濁液は、適切な分散剤または湿潤剤(例えば、Tween 80)、および懸濁剤を用いて、当該分野で公知の技術に従って処方され得る。滅菌注射用調製物はまた、無毒の非経口的に受容可能な希釈剤または溶媒中の滅菌注射用溶液または懸濁液であってもよい。例えば、薬学的組成物は、1,3−ブタンジオール中の溶液であってもよい。本発明の組成物中に用いられ得る受容可能なビヒクルおよび溶媒の他の例としては、マンニトール、水、リンガー溶液および生理食塩液が挙げられるが、これらに限定されない。さらに、滅菌不揮発性油は、溶媒または懸濁媒体として慣習的に用いられている。この目的のために、合成モノグリセリドまたはジグリセリドを含めた、任意の無菌性の不揮発性油が用いられ得る。脂肪酸(例えば、オレイン酸)およびそのグリセリド誘導体は、特にポリオキシエチル化型の、天然の薬学的に受容可能な油(例えば、オリーブ油またはヒマシ油)と同様に、注射剤の調製に有用である。これらの油剤または懸濁液はまた、長鎖アルコール希釈剤または分散剤を含み得る。
【0157】
本発明の薬学的組成物は、任意の経口的に受容可能な剤形(カプセル剤、錠剤、ならびに水性懸濁液および溶液が挙げられるが、これらに限定されない)で、経口的に投与され得る。経口用途のための錠剤の場合、一般に使用されるキャリアとしては、ラクトースおよびコーンスターチが挙げられるが、これらに限定されない。潤滑剤(例えば、ステアリン酸マグネシウム)も添加され得る。カプセル形態での経口投与のために、有用な希釈剤としては、ラクトースおよび乾燥コーンスターチが挙げられる。水性懸濁液が経口的に投与される場合、活性成分は、乳化剤および懸濁剤と組み合わせられる。所望であれば、ある種の甘味剤、香料添加剤、および着色剤が添加され得る。
【0158】
本発明の薬学的組成物はまた、直腸投与のための坐薬の形態で投与されてもよい。これらの組成物は、本発明の化合物を適切な非刺激性の賦形剤と混合することによって、調製され得る。特定の実施形態において、賦形剤は、室温で固体であるが、直腸温では液体である。従って、この賦形剤は、直腸内で融解して、活性成分を放出する。このような材料としては、カカオバター、蜜蝋およびポリエチレングリコールが挙げられるが、これらに限定されない。
【0159】
本発明の薬学的組成物は、鼻エアロゾルまたは吸入によって投与されてもよい。このような組成物は、製剤処方の技術分野で周知の技術に従って調製される。例えば、このような組成物は、ベンジルアルコールまたは他の適切な防腐剤、バイオアベイラビリティーを向上させるための吸収促進剤、フルオロカーボン、および/あるいは当該分野で公知の他の可溶化剤または分散剤を用いて、生理食塩水中の溶液として調製され得る。
【0160】
本発明の薬学的組成物はまた、局所的に投与されてもよい。皮膚への局所適用のために、薬学的組成物は、キャリア中に懸濁または溶解された活性成分を含む、適切な軟膏と共に処方され得る。本発明の化合物の局所投与のためのキャリアとしては、ミネラルオイル、鉱油、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化蝋および水が挙げられるが、これらに限定されない。あるいは、薬学的組成物は、キャリア中に懸濁または溶解された活性化合物を含む、適切なローションまたはクリームと共に処方され得る。適切なキャリアとしては、ミネラルオイル、ソルビタンモノステアレート、ポリソルベート60、セチルエステルワックス、セタリルアルコール(cetaryl alcohol)、2−オクチルドデカノール、ベンジルアルコールおよび水が挙げられるが、これらに限定されない。本発明の薬学的組成物はまた、直腸坐薬処方によるかまたは適切な浣腸処方で、下部腸管に局所的に適用されてもよい。局所的経皮パッチもまた、本発明に包含される。
【0161】
他の実施形態において、本発明は、医薬としての使用および/または医薬の製造のための使用のいずれかに関連して、本明細書中に記載される方法のいずれかで用いるための本明細書中に記載されるSAHの可逆的阻害剤を含む組成物を提供する。
【0162】
本発明はまた、本発明の治療レジメンを実行するためのキットを提供する。このようなキットは、治療有効量のSAH加水分解酵素可逆的阻害剤(例えば、式I、IA、IBおよびII〜VIのいずれか1つの式で示される可逆的阻害剤)を、単独かまたは他の薬剤と組み合わせて、薬学的に受容可能な形態で含む。好ましい製薬形態は、滅菌生理食塩水、デキストロース溶液、緩衝溶液、または他の薬学的に受容可能な滅菌流体と組み合わせて阻害剤を含む。あるいは、組成物は、凍結乾燥または乾燥されていてもよい。この場合、キットは、注射用の溶液を形成するために、薬学的に受容可能な溶液(好ましくは滅菌したもの)をさらに含み得る。別の実施形態において、キットは、組成物を注射するための針またはシリンジ(好ましくは、滅菌形態でパッケージ化されたもの)をさらに含み得る。他の実施形態において、キットは、被験体に組成物を投与するための指示手段をさらに含み得る。この指示手段は、書面挿入物、録音テープ、視聴覚テープ、または被験体への組成物の投与を指示する任意の他の手段であり得る。
【0163】
関連する局面において、本発明は、上記のキットの内容を含む製品を提供する。例えば、本発明は、有効量のSAH加水分解酵素可逆的阻害剤(単独かまたは他の薬剤と組み合わせて)、および本明細書中に記載される疾患を処置するための使用を示す指示書を含む製品を提供する。
【0164】
E.SAH加水分解酵素活性を可逆的に阻害するための組み合わせ
本発明はまた、SAH加水分解酵素活性を可逆的に阻害するための組み合わせおよびキットを提供する。1つの実施形態において、本発明は、式I、IA、IBおよびH〜VIのいずれか1つの式で示される有効量の化合物、ならびに有効量の抗出血性のウイルス感染薬、免疫抑制剤、血漿ホモシステイン低下薬、または抗新生物薬を含む、組み合わせを提供する。この組み合わせは、薬学的に受容可能なキャリアまたは賦形剤をさらに含み得る。さらに別の局面において、本発明は、有効量の記載されるような組み合わせ、および該組み合わせを被験体に投与するための指示手段を含む、キットを提供する。
【0165】
ウイルス性の出血性疾患に関連する臨床症状または診断マーカーを軽減するかまたは寛解させ得る任意の薬剤は、本発明の組み合わせに用いられ得る。抗ウイルス治療剤としては、抗ウイルスワクチン、抗ウイルス抗生物質、ウイルスによって活性化された免疫細胞およびウイルスによって活性化された免疫血清が挙げられるが、これらに限定されない。WO00/64479は、本発明の組み合わせに用いられ得る、抗ウイルス治療剤の例を記載する。好ましい実施形態は、ブンヤウイルス科、フィロウイルス科、フラビウイルス科、またはアレナウイルス科ウイルスの感染によって引き起こされるウイルス性の出血性疾患に対する生物活性を示す、抗ウイルス治療剤である。
【0166】
身体の免疫系が疾患と戦う能力を抑制する任意の薬剤は、本発明の組み合わせに用いられ得る。免疫抑制剤の非限定的な例は、シクロスポリン、プレドニシロン(prednisilone)、アザチオプリン、タクロリムス、副腎皮質ステロイド、ミコフェノレート、シクロホスファミド、メトトレキサート、クロラムブチル、ビンクリスチン、ビンブラスチン、ダクチノマイシン、抗胸腺細胞グロブリン、ムロモナブ−CD3モノクローナル抗体、RhO(D)免疫グロブリン、メトキサレン、およびサリドマイドである(Goodman & Gilman’s The Pharmacological Basis of Therapeutics、(第9版)McGraw−Hill 1996、1294−1304頁を参照のこと)。免疫抑制剤は、薬物の組み合わせとして服用され得る。例えば、大抵の人は、薬物の組み合わせ(例えば、シクロスポリン、アザチオプリン、およびプレドニシロンの組み合わせ)を移植後に開始する。長い期間にわたって、各薬物の用量および服用される薬物の数は、拒絶のリスクが低下するにつれて減少され得る。
【0167】
ホモシステインレベルを低下させる任意の薬剤は、本発明の組み合わせに用いられ得る。葉酸は、有効なホモシステイン低下薬であることが知られている。他のホモシステイン低下薬としては、ベタイン、トリメチルグリシン、シアノコバラミン、および他のビタミンB群が挙げられるが、これらに限定されない。この組み合わせはまた、ホモシステインを低下させるのに用いられる任意の総合ビタミンおよびミネラル補充剤も含み得る。本発明の組み合わせに用いられ得る総合ビタミンおよびミネラル補充剤の例としては、米国特許第6,361,800号;同第6,353,003号;同第6,323,188号;同第6,274,170号;同第6,210,686号;同第6,203,818号;および同第5,668,173号に記載されるものが挙げられるが、これらに限定されない。
【0168】
任意の抗新生物薬が、本発明の組み合わせに用いられ得る。本発明の組成物および方法に用いられ得る抗新生物薬の例は、米国特許出願第2002/044919号に記載される。1つの実施形態において、用いられる抗新生物薬は、抗血管形成剤である。抗血管形成剤は、基底膜分解の阻害剤、細胞遊走の阻害剤、内皮細胞増殖の阻害剤、ならびに3次元組織化および効力の確立の阻害剤であり得る。このような抗血管形成剤の例は、AuerbachおよびAuerbach,Pharmacol.Ther.,63:265−311(1994);O’Reilly,Investigational New Drugs,15:5−13(1997);J.Nat’l Cancer Instit.,88:786−788(1996);ならびに米国特許第5,593,990号;同第5,629,327号および同第5,712,291号に示される。別の実施形態において、用いられる抗新生物薬は、アルキル化剤、代謝拮抗剤、天然物、プラチナ配位錯体、アントラセンジオン、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、ホルモン、およびアンタゴニストである。
【0169】
他の抗新生物薬としては、シチジン、アラビノシルアデニン(araC)、ダウノマイシン、ドキソルビシン、メトトレキサート(MTX)、5−フルオロウラシル(5−FU)などのフッ化ピリミジン、ヒドロキシ尿素、6−メルカプトプリン、ビンクリスチン(VCR)、VP−16およびビンブラスチン(VLB)などの植物アルカロイド、アルキル化剤、シスプラチン、ナイトロジェンマスタード、トリスアミン、プロカルバジン、ブレオマイシン、マイトマイシンC、アクチノマイシンD、またはL−アスパラギナーゼなどの酵素が挙げられるが、これらに限定されない。抗新生物薬はまた、癌抑制遺伝子抗体または抗癌遺伝子アンチセンスオリゴヌクレオチドのような癌遺伝子阻害剤であり得る。別の実施形態において、抗悪性腫瘍薬は、抗細胞マトリックス抗体または抗細胞マトリックスアンチセンスオリゴヌクレオチドのような細胞マトリックス阻害剤である。例えば、カベオリン−1、デコリン、カドヘリン、カテニン、インテグリン、および他の細胞マトリックスまたは細胞マトリックス遺伝子に対する抗体ならびにアンチセンスオリゴヌクレオチドが用いられ得る。
【0170】
特定の実施形態において、組み合わせは、腫瘍内治療および遺伝子治療の併用のための癌抑制遺伝子をさらに含む。遺伝子は、遺伝子送達系の成分として、裸のDNA、複合DNA、cDNA、プラスミドDNA、RNAまたはそれらの他の混合物の形態で用いられ得る。別の実施形態において、癌抑制遺伝子は、ウイルスベクター中に含まれる。遺伝子治療に適している任意のウイルスベクターが、組み合わせて用いられ得る。例えば、アデノウイルスベクター(米国特許第5,869,305号)、シミアンウイルスベクター(米国特許第5,962,274号)、条件付き複製可能型ヒト免疫不全ウイルスベクター(米国特許第5,888,767号)、レトロウイルス、SV40、単純ヘルペスウイルスアンプリコンベクターおよびワクシニアウイルスベクターが用いられ得る。さらに、遺伝子は、リポソーム(ここでは、脂質が、凝固時の酸化からDNAまたは他の生体材料を保護する)のような非ウイルスベクター系で送達され得る。
【実施例】
【0171】
F.実施例
実施例1
SAH加水分解酵素の可逆的阻害剤の合成
特に断りのない限り、1H(Me4Si)NMRスペクトルを、400MHzでCDCl3中、100.6MHzで13C(Me4Si)中の溶液を用いて決定した。質量スペクトル(MS)を、大気圧化学イオン化(APCI)技術によって得た。試薬等級の薬品を使用した。THF以外の溶媒は、アルゴン雰囲気下、CaH2で還流し、そしてCaH2から蒸留することによって乾燥させ、THFは、ベンゾフェノンおよびカリウムから蒸留した。TLCは、MeOH/CHCl3(1:9)およびEtOAc/MeOH(95:5)を展開系として用いてMerck kieselgel 60−F254上で行い、生成物を254nm光で検出した。Merck kieselgel 60(230−400メッシュ)を、カラムクロマトグラフィーに用いた。
【0172】
元素分析は、Galbraith Laboratories,Knoxville,TNで決定した。化合物を単離するためのスペクトルデータは、報告されたデータと一致した。(Holy,Coll.Czech.Chem.Commun.43,3444−3464(1978);Holyら、Coll.Czech.Chem.Commun.50:262−279(1985);日本特許第69−50781号;Chem.Abstr.1972:514811)。この合成を、スキーム2に概略的に示す。
【0173】
9−(3,4−O−イソプロピリデン−3,4−ジヒドロキシブチル)アデニン(l)
【0174】
【化34】
9−(3,4−ジヒドロキシブチル)アデニン(2)
CF3COOH/H2O(9:1)(5ml)中の1(110mg、0.18mmol)の溶液を、約0℃で20分間攪拌した。揮発物を蒸発させ、トルエン(3×)およびEtOH(2×)と同時蒸発させて、EtOHからの結晶化後に報告されたスペクトルデータを示す2(73mg、78%)を得た。
【0175】
9−(4−O−t−ブチルジメチルシリル−3,4−ジヒドロキシブチル)アデニン(3)
TBDMS−Cl(186mg、1.23mmol)およびイミダゾール(168mg、2.46mmol)を、無水DMF(8mL)中の2(250mg、1.12mmol)の攪拌溶液に添加した。混合物を周囲温度で5時間攪拌し、次いで、この反応混合物をEtOAc/NH4Cl/H2Oの間で分配した。水層を隣の部分のEtOAcで抽出した。合わせた有機相を洗浄し(ブライン)、乾燥し(Na2SO4)、蒸発させ、そして残渣をカラムクロマトグラフィー(CHCl3/MeOH;97:3)に付して、3(234mg、62%)を得た:
【0176】
【化35】
9−[4−O−t−ブチルジメチルシリル−3−O−(1−エトキシエチル)−3,4−ジヒドロキシブチル]アデニン(4)
エチルビニルエーテル(214mg、0.28mL、2.96mmol)およびp−トルエンスルホン酸ピリジニウム(15mg、mmol)を無水CH2Cl2(30mL)中の3(250mg、0.74mmol)の溶液に添加し、この混合物を周囲温度にてN2下で、出発物質がTLCで検出されなくなるまで(通常5〜6日間)攪拌した。次いで、反応混合物を水で洗浄し、乾燥し(Na2SO4)、そして蒸発させた。カラムクロマトグラフィー(EtOAc/MeOH;97:3)により、4(160mg、53%)をジアステレオ異性体の約1:1混合物として得た:
【0177】
【化36】
9−[3−O−(1−エトキシエチル)−3,4−ジヒドロキシブチル]アデニン(5)−手順A
TBAF/THF(0.88mL、1M)を、無水THF(6mL)中の4(180mg、0.44mmol)の溶液に添加し、この混合物を周囲温度で20分間攪拌した。揮発物を蒸発させ、残渣をカラムクロマトグラフィー(EtOAc/MeOH;78:12)に付して、5(120mg、92%)をジアステレオ異性体の約1:1混合物として得た:
【0178】
【化37】
4−(アデニン−9−イル)−2−ヒドロキシ酪酸メチル(6)−手順B
CH3CN/CCl4/H2O(1:1:1.5;1.5mL)中の5(90mg、0.31mmol)の懸濁液に、NaHCO3(161mg、0.88mmol)、NaIO4(353mg、1.65mmol)およびRuCl3(微量)を添加した。この混合物を、出発物質がTLCで検出されなくなるまで、周囲温度で48時間攪拌した。次いで、水(5mL)およびCHCl3(4mL)を添加し、これらの2つの相を分離し、そして水相をCHCl3(3mL)で洗浄した。水層をpHが約4になるまでHClで酸性化し、Dowex 50W×2(H+)のカラムにアプライした。カラムを200mLの水で洗浄し、次いで、生成物を2.5%NH4OH/H2Oで溶出した。合わせたUV吸収性のアンモニア溶出物を蒸発させ、そしてMeOH(2×)と同時蒸発させた。残渣をMeOH(5mL)中に溶解し、ジエチルエーテル中のCH2N2の溶液を、ジアゾメタンの黄色が数分間維持されるまで添加した。この溶液を濃縮し、カラムクロマトグラフィー(CHCl3/MeOH;95:5)に付して、6(DZ2002)(31mg、41%)を報告されたものと同一のデータを示す白色固体として得た。副産物6’の生成を回避するために、反応混合物中に所望の量のNaHCO3を維持することが重要である。
【0179】
4−(アデニン−9−イル)−2−ヒドロキシブタン酸(7)−手順C
NaOH/H2O(1mL、0.1M)を、MeOH/H2O(2.0mL)中の6(10mg、0.04mmol)の溶液に添加した。この混合物を、出発物質がTLCで検出されなくなるまで、周囲温度で6時間攪拌した。次いで、反応混合物を、pHが約4になるまでHClで酸性化し、Dowex 50W×2(H+)のカラムにアプライした。カラムを水(100mL)で洗浄し、次いで、生成物を2.5%NH4OHで溶出した。合わせたUV吸収性のアンモニア溶出物を蒸発させて、7を、報告されたデータを示すアンモニウム塩(7.6mg、75%)として得た。
【0180】
9−(3,4−O−ジ−t−ブチルジメチルシリル−3,4−ジヒドロキシブチル)アデニン(8)
TBDMS−Cl(593mg、3.92mmol)およびイミダゾール(534mg、7.85mmol)を、無水DMF(8mL)中の2(350mg、1.57mmol)の攪拌溶液に添加し、この混合物を周囲温度で一晩攪拌した。次いで、反応混合物をEtOAc/NH4Cl/H2Oの間で分配した。水層をEtOAcで抽出した。合わせた有機相を洗浄し(ブライン)、乾燥し(Na2SO4)、そして蒸発させた。カラムクロマトグラフィー(CHCl3→3%MeOH/CHCl3)により、8(610mg、86%)を得た:
【0181】
【化38】
9−(3−O−t−ブチルジメチルシリル−3,4−ジヒドロキシブチル)アデニン(9)
化合物8(400mg、0.887mmol)を、CH3CO2H/H2O/THF(13:7:3;8mL)の溶液に添加し、この混合物を、化合物2のより極性の高いスポットがTLC上に出現し始めるまで、周囲温度で攪拌した。次いで、反応混合物を分配し(EtOAc//NaHCO3/H2O)、そして水層を隣の部分のEtOAcで抽出した。合わせた有機相を洗浄し(NaHCO3、ブライン)、乾燥し(Na2SO4)、蒸発させ、そしてカラムクロマトグラフィーに付して(CHCl3→4%MeOH/CHCl3)、回収した8(140mg、35%)および9(155mg、52%)を得た:
【0182】
【化39】
3−(アデニン−9−イル)プロピオン酸メチル(6’)
9(50mg、0.148mmol)を手順B(カラムクロマトグラフィー:CHCl3/MeOH 97:3)によって処理することにより、報告されたデータを示す6’(9mg、27%)を得た。
【0183】
3−(アデニン−9−イル)プロピオン酸(7’)
6’(10mg、0.045mmol)を手順C(カラムクロマトグラフィー:CHCl3/MeOH 97:3)によって処理することにより、報告されたデータを示す6’(7.3mg、78%)を得た。
【0184】
スキーム2
【0185】
【化40】
a(a)CF3COOH/H2O;(b)TBDMSCl/イミダゾール/DMF;(c)エチルビニルエーテル/CH2Cl2/(p)CH3C6H4SO3H−C5H10N;(d)TBAF/THF;(e)(i)NaIO4/NaHCO3/RuCl3/CH3CN/CCl4/H2O、(ii)CH2N2MeOH;(f)CH3CO2H/H2O/THF;(g)NaOH/H2O/MeOH。
【0186】
実施例2
DZ2002は免疫抑制作用を媒介する
材料および方法
試薬
AdoHcy加水分解酵素阻害剤DZ2002(スキーム2中の化合物6)は、Diazyme Laboratoriesで合成された。Con A(Concanavalin A)、LPS(Escherichia coli 055:B5)およびSac(Staphylococcus aureus Cowan strain1)は、Pansorbin(登録商標)cells,Biosciences,inc.(La Jolla.,CA92039,USA)から入手した。RPMI1640およびウシ胎仔血清(FBS)は、GIBCOから入手した。精製ラット抗マウスIL−10、IL−12p70、IL−12p40、IFN−γおよびビオチン標識抗マウスIL−10、IL−12p70、IL−12p40、IFN−γ、FITC−抗マウス−CD11b(Mac−1)、Phycorythrin(PE)−抗マウスI−Ad、PE−抗ヒト−CD14、PE−抗ヒト−ABC、PE−抗ヒト−DR、PE−抗ヒト−CD80およびPE−抗ヒト−CD86は、Pharmingen製品であった。チオグリコレート(TG)は、Sigma−Aldrichから入手可能である。
【0187】
動物
近交系BALB/Cマウス(6〜8週齢)を、認可番号99−003で、中国科学院上海実験動物センター(Shanghai Experimental Animal Center of Chinese Academy of Sciences)により提供された。これらのマウスを、室温24±2℃、12時間の明/暗サイクルで、特定病原体感染防止(SPF)条件に収容し、滅菌飼料および水を自由に与えた。
【0188】
細胞
Balb/cマウスから脾臓を無菌的に切除し、プールし、そしてPBS中の単細胞懸濁液を調製した。赤血球を、トリス緩衝化塩化アンモニウム(0.155M NH4CL、0.0165Mトリス、PH7.2)で処理することによって溶解した。単核細胞をPBSで洗浄し、そしてベンジルペニシリン100000U・L−1およびストレプトマイシン100mg・L−1を補充したRPMI−1640培地中に再懸濁した。細胞の生存度および濃度を、トリパンブルー排除により決定した。
【0189】
腹腔滲出細胞を、0.5mlの3%TGの腹膜腔内注射によってBALB/Cマウスに誘導した。4日後に、腹膜滲出細胞を滅菌洗浄によって収集した。
【0190】
THP−1(American Type Culture Collection,Manassas,VA)は、ヒト単球性白血病である。THP−1細胞を、10%FBSを補充したRPMI1640培地中の懸濁培養で維持した。培養を、空気中5%CO2の加湿雰囲気下、37℃で維持し、5〜6日ごとに1/10希釈で継代培養した。
【0191】
脾臓リンパ球への[3H]−チミジン取込み
マウス脾臓リンパ球を、10%FBSを補充したRPMI1640中で、インビトロで培養した。細胞を、種々の濃度のDZ2002の存在下または非存在下で、5μg/mlのCon Aまたは10μg/mlのLPSとともに、37℃で48時間、加湿CO2インキュベーター中、1×105細胞/200μl/ウェルにて96ウェルプレート中でインキュベートした。40時間のインキュベーションの後、細胞を0.5μCi/ウェルの[3H]−チミジンでパルスし、さらに8時間培養した。次いで、細胞をガラス繊維フィルター上に収集し、取り込まれた放射能をBeta Scintillator(MicroBeta Trilux,PerkinElmer Life Sciences)を用いてカウントした。
【0192】
脾臓リンパ球のMTTアッセイ
細胞傷害性をMTTアッセイで評価した。マウス脾臓リンパ球を、種々の濃度のDZ2002の存在下または非存在下で、37℃で48時間、加湿CO2インキュベーター中、9×104細胞/180μl/ウェルにて96ウェルプレート中でインキュベートした。培養終了前に15μlの5mg/mlのMTTを4時間パルスし(合計190μl)、次いで、80μlの溶媒(10%SDS、50%N、N−ジメチルホルムアミド、PH7.2)を添加した。7時間インキュベートし、マイクロプレートリーダー(Bio−rad
Model 550 Japan)でOD590を読み取る。
【0193】
サイトカイン産生
マウスの脾臓単核球(5×106)を、種々の濃度のDZ2002の存在下または非存在下、Sac(1:10000)、ConA(5ug/ml)またはLPS(10ug/ml)の存在下で、24ウェルプレート中、2ml/ウェルの容量で培養した。24時間後、無細胞の上清を収集し、−20℃で凍結した。IL−12p40、IL−12p70、IL−10およびTNF−αの濃度を、マウスのサイトカインに特異的なELISAで決定した。
【0194】
マウスの腹腔滲出細胞(6.25×105)を、24ウェルプレート中、1ml/ウェルの容量で2時間培養した。接着細胞を氷冷RPMI1640で洗浄し、そして接着細胞を、種々の濃度のDZ2002の存在下または非存在下、IFN−γ(2.5ng/ml)およびLPS(1μg/ml)の存在下で、2ml/ウェルの容量で培養した。24時間後、無細胞の上清を収集し、−20℃で凍結した。IL−12p40、IL−12p70、IL−10およびTNF−αの濃度を、ELISAで決定した。
【0195】
THP−1細胞(6×105)を、1.2%の存在下および種々の濃度のDZ2002の存在下または非存在下で、24ウェルプレート中、2ml/ウェルの容量で培養した。24時間後にIFN−γ(500U/ml)を添加し、さらに16時間後にLPS(1μg/ml)を添加した。無細胞の上清を24時間後に収集し、−20℃で凍結した。IL−12p40、IL−12p70、IL−10およびTNF−αの濃度を、ELISAで決定した。
【0196】
ヒツジ赤血球の定量的溶血(QHS)アッセイ
雌性Babl/cマウスを、4日目に0.2mlの16.7%のSRBCを腹腔内注射することによって免疫した。ビヒクル、デキサメタゾンおよびDZ2002を、1〜7日目の7日間連続して、腹腔内注射によって各群(n=6)に投与した。8日目に、マウスを屠殺して、2×106細胞/mlの脾臓細胞の混合懸濁液を作製した。1mlの細胞懸濁液を、1mlの0.5%SRBCおよび1mlのモルモット補体の1:10希釈物とともに、37℃で1時間インキュベートし、次いで、遠心分離し(3分、3000g)、そして一部改変したSimpsonら、J.Immunol.Methods.,21(1−2):159−65.(1978)に従って、413nmで上清の溶血を決定した。各群を3連で行った。
【0197】
混合リンパ球反応(MLR)増殖アッセイ
Balb/cマウス脾臓細胞を107細胞/mlの懸濁液に調製し、50μg/mlのマイトマイシンとともに2時間培養した。次いで、細胞を洗浄し、新鮮なC57/B6マウス脾細胞とともに、等しく最終濃度1.0×10細胞/mlで、種々の濃度のDZ2002の存在下または非存在下で培養した。48時間インキュベーションの後、細胞を0.5μCi/ウェルの[3H]−チミジンでパルスし、さらに24時間培養した。次いで、細胞をガラス繊維フィルター上に収集し、取り込まれた放射能をBeta Scintillator(MicroBeta Trilux,PerkinElmer Life
Sciences)を用いてカウントした。
【0198】
DNFB誘導性の遅延型過敏症(DTH)応答
雌性Balb/cマウスを、0日目および1日目に各後足に、20μlのアセトン−オリーブ油(4:1)中に溶解された0.6%DNFBで感作した。7日目にマウスを、一部改変したPhanuphak(1974)に従う方法で、左耳の両側に10μlの0.5%DNFBで攻撃した。ビヒクル、CsA、およびDZ2002(1、3、10mg/kg)を、攻撃の1時間前および12時間後、24時間後に、腹腔内注射によって各群(n=10)に投与した。耳介腫脹を、攻撃の30時間後に特定の8mmパンチで作製した左右の耳パッチの重量差として表した。
【0199】
フローサイトメトリー
マウスの腹腔滲出細胞またはTHP−1細胞を、冷PBS(0.1%NaN3、1%FBSを含む染色緩衝液(PH7.2))中で洗浄した。細胞を、冷染色緩衝液中に2.0×107/mlで最懸濁した。最適濃度の各蛍光色素標識抗体を、50μLの細胞に添加した。Fcレセプターを、10μL正常マウス血清を用いてブロックした。細胞を暗所で4℃にて30分間インキュベートし、2.0mLの染色緩衝液で2回洗浄し、そして0.5mLのPBS(PH7.2)中に再懸濁した。細胞を4℃で暗所に保存し、FACScanフローサイトメーター(Becton Dickinson,San Jose,CA)で分析した。データをCellQuest(商標)ソフトウェア(Becton Dickinson,San Jose,CA)を用いて分析した。
【0200】
統計分析
結果をx±sとして表し、独立両側t検定を行って、0.05未満のP値を有意であるとみなした。各実験を少なくとも3回繰り返した。
【0201】
結果
AdoHcy加水分解酵素阻害剤による脾臓リンパ球への[3H]−チミジン取込みの阻害
培養の48時間後、DZ2002(0.1〜10μmol・L−1)は、ConAによって誘導されるリンパ球増殖に対して影響を及ぼさない。DZ2002(10μmol・L−1)は、LPSによって誘導されるリンパ球増殖を阻害した。
【0202】
Sac刺激マウス脾細胞からのIL−10、IL−12P40およびIL−12P70産生に対するDZ2002の影響
Sac刺激は、休止脾細胞と比較して、マウス脾細胞からのIL−10、IL−12P40およびIFN−γ産生の著しい増加を誘導した。DZ2002(μmol・L−1)は、IL−12P40、IL/−12P70およびTNF−αの放出を用量依存的に阻害した(データは示さず)。
【0203】
ヒツジ赤血球の定量的溶血(QHS)アッセイに対するDZ2002の影響
SRBCの定量的溶血は、抗原刺激に対する応答における一次抗体産生のモデルである。図3に示すように、5mg/kgのデキサメタゾンのQHS阻害が38.1%であるのと比較して、DZ2002の連続7日間の腹腔内注射により、0.08mg/kgおよび2mg/kgの用量で、それぞれQHSを24.5%および18.4%阻害した(ビヒクルコントロール群と比較してすべての実験群についてp<0.05)(図1)。
【0204】
DZ2002は混合リンパ球反応におけるT細胞増殖を抑制する
マイトマイシン処理Balb/c(H−2d)脾臓細胞を、C57BL/6(H−2b)脾臓細胞増殖に対する同種刺激因子として適用した。DZ2002は、それぞれ0.1、1および10μmol/Lの用量で3日間培養して、MLRに対する強力な抑制(40.2%、36.9%および42.3%)を示した。(図2)。
【0205】
DZ2002は、脾臓細胞において細胞傷害性を有さない
2日間の培養において、0.1〜10μmol/LのDZ2002は、脾臓細胞に対する細胞傷害性を示さなかった。DZ2002とともにインキュベートした細胞のOD値は、コントロールのOD値と差異がなかった。(図3)。
【0206】
DZ2002は、エタノール消費によって誘導されるマウスDTH応答の抑制を逆転させた
各群につき9匹のマウスを準備した。マウスを、0日目および1日目に各後足に、無水アセトン/オリーブ油(4:1)中の0.5%DNFB溶液(20μl)で感作した。初回感作の5日後に、マウスを、軽いメトファン(Metofane)麻酔下で左耳の両側に0.2%DNFB(10μl)で攻撃した。右耳をビヒクル単独で処理した。DNFB攻撃の1時間前に、DZ2002をマウスに経口投与した。耳介腫脹度を、イヤーパンチャーおよび重量(mg)を測定するために分析天秤を用いて、攻撃の24時間後に測定した。結果を、左右の耳の重量差として表した。耳介腫脹の測定後に、各群の4匹のマウスから脾臓を取り出し、分析まで凍結した。
【0207】
常在腹膜細胞およびTG誘導性腹膜細胞におけるMHC−IIの発現に対するDZ2002の影響
腹腔マクロファージによるMHC−II発現を、培地単独かまたは100U/mlのIFN−γとともに48時間インキュベーションした後に評価した。0.1および1μmol/LのDZ2002の存在下でIFN−γとともにインキュベートした細胞は、常在腹膜細胞のMHC−II発現のレベルを高め、そして10μmol/LのDZ2002は、MHC−II発現のレベルを低下させる。(データは示さず)。TG誘導性の腹膜細胞におけるように、1および10μmol/LのDZ2002は、培地単独でインキュベートする場合、Mac−1+パーセントを低下させる。そして、IFN−γとともにインキュベートした細胞のMHC−II発現のレベルは、0.1〜10μmol/LのDZ2002の存在下で用量依存的に低下した。(データは示さず)。
【0208】
TG誘導性の腹膜細胞からのIL−10、IL−12P40およびTNF−α産生に対するDZ2002の影響
腹腔マクロファージによって産生されたサイトカインを、25U/mlのIFN−γおよび1μg/mlのLPSとともに24時間インキュベーションした後に評価した。常在腹膜細胞は、IFN−γおよびLPSとともにインキュベートすると、一部のIL−10を除く低レベルのサイトカインを産生する(データは示さず)。TG誘導性の腹腔マクロファージについては、DZ2002は、0.1〜10μmol/Lの用量でIL−12P40およびTNF−αの放出を阻害したが、IL−10産生に対して影響を及ぼさない(図5)。
【0209】
DZ2002は、THP−1細胞におけるMHC−II、CD80およびCD86の発現を阻害する
THP−1細胞によるMHC−II、CD80およびCD86発現は、培地単独かまたは100U/mlのIFN−γとともに48時間インキュベーションした後に評価した。10μmol/LのDZ2002の存在下でIFN−γとともにインキュベートした細胞は、THP−1細胞のMHC−II発現のレベルを穏やかに低下させ、そして0.1〜10μmol/LのDZ2002は、用量依存的な方法でCD80およびCD86発現のレベルを低下させる。(図6A−C)。
【0210】
THP−1細胞からのIL−10、IL−12P40およびTNF−α産生に対するDZ2002の影響
THP−1細胞によって産生されたサイトカインを、500U/mlのIFN−γおよび1μg/mlのLPSとともに24時間インキュベーションした後に評価した。図7に示すように、DZ2002は、0.1〜10μmol/Lの用量で、IL−12P40およびTNF−αの放出を阻害した。
【0211】
実施例3
DZ2002は、遅延型過敏症(DTH)反応を軽減した
図8に示すように、DZ2002は、遅延型過敏症(DTH)反応を用量依存的な様式で有意に軽減した。BALB/cマウスを、0日目および1日目にDNFB(ジフルオロニトロベンゼン)に初回感作させ、次いで9日目にDNFBで再度攻撃した。DZ2002を、9日目の攻撃の1時間前または24時間後に、i.p.(腹腔内)投与した。耳介腫脹を、攻撃の40時間後に特定の8mmパンチによって決定した。データを平均±SDとして表す。
【0212】
実施例4
DZ2002は、MBPで刺激した脾細胞からのIL−10産生を維持または増大させた
図9に示すように、DZ2002は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−10産生を維持または増大させた。SJL/Jマウスを0日目にMBPで免疫し、次いで2日目に百日咳毒素を投与した。DZ2002をマウス腹腔内に14日目から開始して毎日投与した。脾細胞を21日目に採取し、そしてMBPとともに培養した。培養上清をIL−10についてELISAで分析した。データを平均±SDとして表す。
【0213】
実施例5
DZ2002は、MBPで刺激した脾細胞からのIL−2産生を阻害した
図10に示すように、DZ2002は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−2産生を阻害した。SJL/Jマウスを0日目にMBPで免疫し、次いで2日目に百日咳毒素を投与した。DZ2002をマウス腹腔内に14日目から開始して毎日投与した。脾細胞を21日目に採取し、そしてMBPとともに培養した。培養上清をIL−2についてELISAで分析した。データを平均±SDとして表す。
【0214】
実施例6
DZ2002は、MBPで刺激した脾細胞からのIFN−γ産生を阻害した
図11に示すように、DZ2002は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIFN−γ産生を阻害した。SJL/Jマウスを0日目にMBPで免疫し、次いで2日目に百日咳毒素を投与した。DZ2002をマウス腹腔内に14日目から開始して毎日投与した。脾細胞を21日目に採取し、そしてMBPとともに培養した。培養上清をIFN−γについてELISAで分析した。データを平均±SDとして表す。
【0215】
上記の実施例は、単に例示の目的で挙げられるものであり、本発明の範囲を限定することを意図するものではない。上記の実施例に対する多様な変形が可能である。上記の実施例に対する改変および変形が当業者に明らかであるので、本発明は、添付の特許請求の範囲によってのみ限定されることが意図される。
【技術分野】
【0001】
(技術分野)
本願は、米国特許出願番号10/410,879(2003年4月9日出願、現在係属中)および米国特許出願番号PCT/US2004/011229(2004年4月9日出願、現在係属中)(これらの内容は、その全体が本明細書中に参考として援用される)の一部継続である、米国特許出願番号10/964,236(2004年10月13日出願、現在係属中)に関する。
【背景技術】
【0002】
(発明の背景)
SAH加水分解酵素は、多くのウイルスがウイルスタンパク質の効率的な翻訳のためにそのmRNA上に5’−キャップメチル化構造を必要とするという観察に基づく、抗ウイルス薬設計のための魅力的な標的であった。非特許文献1;非特許文献2。SAH加水分解酵素の阻害は、ウイルスmRNAのメチル化を含む、S−アデノシル−L−メチオニン(SAM)依存性メチル化反応の阻害を生じ、したがって、ウイルス複製を阻害する(スキーム1)。
【0003】
【化10】
SAH加水分解酵素の多数の阻害剤が、天然に存在する化合物および合成化合物から同定されている。最も強力な阻害剤は、時間依存的な様式でSAH加水分解酵素を不可逆的に不活化する不可逆的阻害剤である。不可逆的阻害剤は、その激しい細胞傷害作用により、狭い治療ウインドウを生じるのみであることが、研究により示されている(非特許文献3)。SAH加水分解酵素は、非常に緩徐な代謝回転速度(マウス肝臓においてt1/2=24時間)を有する普遍的な細胞性酵素であるので、不可逆的阻害剤は、酵素活性の長期阻害を引き起こし得る。例えば、酵素活性の完全な回復のために最大7日間かかり得、このことが、望ましくない副作用を引き起こし得る。不可逆的阻害剤に関連する激しい細胞傷害性が、これらの阻害剤の臨床的に有用な薬物への開発を損なう主な要因であった。不可逆的阻害剤に関連する細胞傷害性のために、可逆的阻害剤が好ましい。
【0004】
しかしながら、現時点では、インビボで試験した場合に、SAH加水分解酵素に対して実質的な阻害活性を生じるのに十分に強力な、公知の可逆的なSAH加水分解酵素阻害剤は存在しない。例えば、SAH加水分解酵素に対するKi値が3.5μMである可逆的阻害剤(S)−9−(2,3−ジヒドロキシプロピル)アデニン((S)−DHPA)は、阻害能力を欠く。(非特許文献4)。(s)−DHPAは、単離されたAdoHcy加水分解酵素の可逆的阻害剤であることが報告されたが(非特許文献4)、(s)−DHPAはまた、細胞内AdoHcy加水分解酵素の不可逆的阻害剤であることも報告された(非特許文献5)。したがって、望ましくない細胞傷害作用を有することなく効力を発揮するSAH加水分解酵素阻害剤の必要性が残っている。
【先行技術文献】
【非特許文献】
【0005】
【非特許文献1】Yuanら、Exp.Opin.Ther.Patents(1999)9:1197−1206
【非特許文献2】Yuanら、Adv.Antiviral Drug Des.,De Clercq(編),,JAI Press,Inc.London,UK(1996)第2巻,pp.41−88
【非特許文献3】WolfeおよびBorchardt,Journal of Medicinal Chemistry(1991)34:1521−1530
【非特許文献4】VotrubaおよびHoly,Coll.Czech.Chem.Commun.(1980)45:3039
【非特許文献5】Schancheら、Molecular Pharmacology(1984)26:553−558
【発明の概要】
【課題を解決するための手段】
【0006】
(発明の簡単な概要)
本発明は、SAH加水分解酵素の新規な可逆的阻害剤を提供する。本発明の化合物は、SAH加水分解酵素を阻害するこれらの化合物の能力に関連する生物学的活性を示す薬剤として有用である。
【0007】
1つの実施形態において、本発明は、式(I):
【0008】
【化11】
[式中、Zは炭素または窒素であり、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、またはハロゲンであり;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、またはヘテロアリールであり;Xは、酸素、窒素、または硫黄であり;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、またはヘテロアリールである]で示される化合物、およびその薬学的に受容可能な塩を提供する。特定の実施形態において、化合物は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0009】
1つの局面において、本発明は、式IA:
【0010】
【化12】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、あるいは式(IB):
【0011】
【化13】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]で示される化合物を提供する。
【0012】
式Iまたは式IAの化合物は、R1、R2、R3およびR4が水素である置換基を有し得る。本発明の1つの局面において、Xは、酸素である。本発明の別の局面において、Yは、水素またはC1−10アルキル基である。本発明のさらに別の局面において、R1、R2、R3およびR4は、水素であり、Xは酸素であり、そしてYは水素またはC1−10アルキル基である。
【0013】
本発明はまた、式I、式IAまたは式IBで示されるSAH加水分解酵素の可逆的阻害剤、およびそれらの薬学的に受容可能な塩も提供する:
いくつかの実施形態において、該SAH加水分解酵素の可逆的阻害剤は、式(II):
【0014】
【化14】
で示される。
【0015】
いくつかの実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(III):
【0016】
【化15】
[式中、R1およびR2は、それぞれ独立して、水素またはヒドロキシである;ただし、R1およびR2がともにヒドロキシであることはない]で示される。いくつかの実施形態において、R1は水素であり、そしてR2はヒドロキシである。いくつかの実施形態において、R1はヒドロキシであり、そしてR2は水素である。いくつかの実施形態において、R1およびR2は、ともに水素である。
【0017】
いくつかの実施形態において、該SAH加水分解酵素の可逆的阻害剤は、式(IV):
【0018】
【化16】
[式中、R1は、NH2、SCH3、またはCH2NH2である]
で示される。
【0019】
いくつかの実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(V):
【0020】
【化17】
[式中、R1は、NH2またはCONH2である]
で示される。
【0021】
なお別の実施形態において、SAH加水分解酵素の可逆的阻害は、式(VI):
【0022】
【化18】
[式中、Wは、Hまたはメトキシである]
で示される。
【0023】
式I、IA、IBおよびII〜VIの化合物は、β炭素でS配置を有し得るか、β炭素でR配置を有し得るか、またはラセミ混合物を含み得る。1つの実施形態において、これらの化合物は、生体媒体(例えば、血清)中の哺乳動物SAH加水分解酵素に対するKi値が100nM未満である。他の実施形態において、これらの化合物は、生体媒体中の哺乳動物SAH加水分解酵素に対するKi値が約1nMと約100nMの間である。これらの化合物は、好ましくは、生体媒体中のヒトSAH加水分解酵素に対して100nM未満のKi値か、または約1nMと約100nMの間のKi値を示す。
【0024】
本発明はまた、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物またはその薬学的に受容可能な塩、ならびに薬学的に受容可能なキャリアまたは希釈剤を含む、薬学的組成物に関する。薬学的組成物は、経口、非経口(例えば、筋肉内、腹腔内、静脈内、嚢内注射もしくは注入、皮下注射、または移植)、吸入スプレー、経鼻、膣内、直腸、舌下、あるいは局所投与経路によって投与され得る。これらの薬学的組成物は、各投与経路に適した適切な投薬単位処方物に処方され得る。
【0025】
本発明は、特定の処方物または特定の投与様式に限定されることを意図するものではない。1つの実施形態において、組成物は、経口投与、非経口投与、鼻腔内投与、局所投与、または注射投与用に処方される。注射用投与の非限定的な例は、空洞内注射、皮下注射、静脈内注射、筋肉内注射および皮内注射である。本薬学的組成物は、1日当たり約0.1〜約20mg/kgの範囲の投薬量で、経口投与用に処方され得る。本薬学的組成物はまた、1日当たり約0.1〜約20mg/kgの範囲の投薬量で、注射投与用に処方され得る。
【0026】
本発明の薬学的組成物は、固体または液体の剤形で処方され得る。例えば、本薬学的組成物は、錠剤、カプセル剤、顆粒剤、散剤、および類似の化合物の形態で、固体として処方され得る。本薬学的組成物はまた、シロップ剤、注射混合物などの形態で、液体として処方され得る。
【0027】
本発明はまた、有効量の本発明の組成物、および該組成物を投与するための指示手段を含む、キットを提供する。
【0028】
さらに、本発明は、S−アデニル−L−ホモシステイン(SAH)加水分解酵素の活性を可逆的に阻害するための方法を提供する。1つの実施形態において、本発明は、哺乳動物におけるS−アデノシル−L−ホモシステイン(SAH)加水分解酵素の活性を可逆的に阻害するための方法を提供し、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、式(I):
【0029】
【化19】
[式中、Zは、炭素および窒素からなる群より選択され、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;Xは、酸素、窒素、および硫黄からなる群より選択され;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
で示される有効量の化合物またはその薬学的に受容可能な塩を投与し、それによって該哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害する工程を包含する。特定の実施形態において、投与される化合物またはその薬学的に受容可能な誘導体は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0030】
1つの局面において、本発明は、式IA
【0031】
【化20】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【0032】
【化21】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される化合物またはその薬学的に受容可能な塩を用いて、SAH加水分解酵素の活性を可逆的に阻害するための方法を提供する。
【0033】
さらに別の局面において、本発明は、上記に規定されるような、式II〜VIのいずれか1つの式で示される化合物またはその薬学的に受容可能な塩を用いて、SAH加水分解酵素の活性を可逆的に阻害するための方法を提供する。
【0034】
好ましい実施形態において、哺乳動物は、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、高ホモシステイン不全症、循環器病、脳卒中、アルツハイマー病、糖尿病、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある。しかしながら、本発明が特定の疾患の予防および処置に限定されることを意図するものではない。
【0035】
出血性ウイルス感染を予防および処置するための方法を提供することは、本発明の目的である。1つの局面において、この方法は、哺乳動物における出血性ウイルス感染の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。特定の実施形態において、出血性ウイルス感染は、ブンヤウイルス科、フィロウイルス科、フラビウイルス科、およびアレナウイルス科のウイルスからなる群より選択されるウイルスによって引き起こされる。他の特定の実施形態において、フィロウイルス科のウイルスは、エボラウイルスである。
【0036】
自己免疫疾患を予防および処置するための方法を提供することもまた、本発明の目的である。1つの局面において、この方法は、哺乳動物における自己免疫疾患の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。
【0037】
同種移植拒絶を予防および処置するための方法を提供することもまた、本発明の目的である。1つの局面において、この方法は、哺乳動物における同種移植拒絶の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。
【0038】
さらに、哺乳動物における血漿ホモシステインを低下させるための方法、あるいは高ホモシステイン不全症を予防または処置するための方法を提供することも、本発明の目的である。1つの局面において、この方法は、哺乳動物における血漿ホモシステインを低下させるために、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。
【0039】
また、新生物を予防または処置するための方法を提供することも、本発明の目的である。1つの局面において、この方法は、哺乳動物における新生物の処置において、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物を投与する工程を包含する。新生物の非限定的な例は、副腎、肛門、聴神経、胆管、膀胱、骨、脳、胸、頬、中枢神経系、頚部、結腸、耳、子宮内膜、食道、眼、眼瞼、ファロピウス管、胃腸管、頭頸部、心臓、腎臓、喉頭、肝臓、肺、下顎骨、下顎関節頭、上顎骨、口、鼻咽頭、鼻、口腔、卵巣、膵臓、耳下腺、陰茎、耳介、脳下垂体、前立腺、直腸、網膜、唾液腺、皮膚、小腸、脊髄、胃、精巣、甲状腺、扁桃腺、尿道、子宮、膣、内耳神経、および外陰の新生物である。
【0040】
本発明はまた、式I、IA、IBおよびII〜VIのいずれか1つの式で示される有効量の化合物と、有効量の抗出血性のウイルス感染薬、免疫抑制剤、血漿ホモシステイン低下薬、および抗新生物薬とを含む組み合わせを提供する。この組み合わせは、薬学的に受容可能なキャリアまたは賦形剤をさらに含み得る。特定の実施形態において、この組み合わせは、(4−アデニン−9−イル)−2−ヒドロキシブタン酸を含まない。
【0041】
特定の実施形態において、抗出血性のウイルス感染薬は、インターロイキン−1(IL−1)、腫瘍壊死因子(TNF)、またはその組み合わせを阻害する。抗出血性のウイルス感染薬は、抗ウイルスワクチン、抗ウイルス抗体、ウイルスによって活性化された免疫細胞、またはウイルスによって活性化された免疫血清であり得る。
【0042】
別の実施形態において、免疫抑制剤は、シクロスポリン、タクロリムス、副腎皮質ステロイド、アザチオプリン、ミコフェノレート、シクロホスファミド、メトトレキサート、クロラムブチル、ビンクリスチン、ビンブラスチン、ダクチノマイシン、抗胸腺細胞グロブリン、ムロモナブ−CD3モノクローナル抗体、RhO(D)免疫グロブリン、メトキサレン、またはサリドマイドである。
【0043】
他の特定の実施形態において、ホモシステイン低下薬は、ビタミンB6、ビタミンB12、または葉酸塩である。
【0044】
なお別の実施形態において、抗新生物薬は、抗血管形成剤、アルキル化剤、代謝拮抗剤、天然物、プラチナ配位錯体、アントラセンジオン、置換尿素、メチルヒドラジン誘導体、副腎皮質の反応抑制薬、ホルモン、アンタゴニスト、癌遺伝子阻害剤、癌抑制遺伝子またはタンパク質、癌抑制遺伝子抗体、あるいは癌抑制遺伝子のアンチセンスオリゴヌクレオチドである。
【0045】
本発明はまた、本発明の有効量の組み合わせ、およびこの組み合わせを投与するための指示手段を含むキットを提供する。
【0046】
さらに、本発明は、哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害するための方法を提供し、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、以下:a)式(I):
【0047】
【化22】
[式中、Zは、炭素および窒素からなる群より選択され、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;Xは、酸素、窒素、および硫黄からなる群より選択され;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]で示される有効量の化合物またはその薬学的に受容可能な塩;ならびにb)抗出血性のウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、および抗新生物薬からなる群より選択される有効量の化合物を含む、有効量の組み合わせを投与し、それによって該哺乳動物における該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する。特定の実施形態において、この投与される組み合わせは、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0048】
本発明はまた、哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害するための方法を提供し、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、以下:a)上記に予め規定されたような、式IA、IB、およびII〜VIのいずれか1つの式で示される有効量の化合物、またはその薬学的に受容可能な塩を含む、有効量の組み合わせを投与する工程を包含する。
【0049】
この組み合わせは、任意の他の薬学的組成物と併用されて、哺乳動物におけるSAH加水分解酵素活性を調節し得る。この組み合わせはまた、上記のように、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、および高ホモシステイン不全症、循環器病、脳卒中、アルツハイマー病、糖尿病、炎症性腸疾患、多発性硬化症または自己免疫性神経炎などの疾患の予防および処置にも用いられ得る。しかしながら、この組み合わせが、特定の疾患の予防および用途に限定されることを意図するものではない。
【0050】
本発明はまた、IL−12の産生および/または放出を可逆的に阻害するための方法を提供し、該方法は、IL−12産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−12産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する。いくつかの実施形態において、IL−12は、IL−12P40、IL−12P35、またはIL−12P70である。いくつかの実施形態において、IL−12産生細胞は、哺乳動物に含まれる。いくつかの実施形態において、哺乳動物はヒトである。いくつかの実施形態において、哺乳動物は、炎症性腸疾患、多発性硬化症および他の自己免疫疾患からなる群より選択される疾患に罹患している疑いがある。
【0051】
本発明は、哺乳動物における遅延型過敏症(DTH)反応を軽減するための方法を提供し、該方法は、このような軽減が必要であるかまたは望ましい哺乳動物に、SAH加水分解酵素の有効量の可逆的阻害剤を投与することによって、該哺乳動物におけるDTH反応を軽減する工程を包含する。1つの実施形態において、哺乳動物はヒトである。別の実施形態において、SAH加水分解酵素の可逆的阻害剤は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0052】
本発明はまた、IL−10の産生および/または放出を維持するかあるいは増加させるための方法を提供し、該方法は、IL−10産生細胞を、SAH加水分解酵素の可逆的阻害剤と接触させることによって、該IL−10産生細胞中の該SAH加水分解酵素を可逆的に阻害する工程を包含する。1つの実施形態において、IL−10産生細胞は、哺乳動物に含まれる。別の実施形態において、SAH加水分解酵素の可逆的阻害剤は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
【0053】
本発明は、IL−2またはIFN−γの産生および/あるいは放出を可逆的に阻害するための方法をさらに提供し、該方法は、IL−2またはIFN−γ産生細胞を、SAH加水分解酵素の可逆的阻害剤と接触させることによって、該IL−2またはIFN−γ産生細胞中の該SAH加水分解酵素を可逆的に阻害する工程をさらに包含する。1つの実施形態において、IL−2またはIFN−γ産生細胞は、哺乳動物に含まれる。別の実施形態において、SAH加水分解酵素の可逆的阻害剤は、(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない。
例えば、本願発明は以下の項目を提供する。
(項目1)
式(IA):
【化1】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【化2】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
を有する化合物、またはその薬学的に受容可能な塩。
(項目2)
項目1に記載の化合物および薬学的に受容可能な賦形剤を含む、薬学的組成物。
(項目3)
有効量の項目2に記載の組成物、および該組成物を投与するための指示手段を含む、キット。
(項目4)
哺乳動物のS−アデノシル−L−ホモシステイン(SAH)加水分解酵素の活性を可逆的に阻害するための方法であって、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、式(I):
【化3】
[式中、Zは、炭素および窒素からなる群より選択され、
R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
を有する化合物またはその薬学的に受容可能な塩の有効量を投与し、それによって該哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目5)
組み合わせであって、以下のa)およびb):
a)式(I):
【化4】
[式中、Zは、炭素および窒素からなる群より選択され、
R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
を有する化合物またはその薬学的に受容可能な塩の有効量;ならびに
b)抗出血性のウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、および抗新生物薬からなる群より選択される化合物の有効量
を含む、組み合わせ。
(項目6)
哺乳動物におけるSAH加水分解酵素の活性を可逆的に阻害するための方法であって、該方法は、このような可逆的阻害が必要であるかまたは望ましい哺乳動物に、以下のa)およびb):
a)式(I):
【化5】
[式中、Zは、炭素および窒素からなる群より選択され、
R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]
を有する化合物またはその薬学的に受容可能な塩の有効量;ならびに
b)抗出血性のウイルス感染薬、免疫抑制剤、ホモシステイン低下薬、および抗新生物薬からなる群より選択される化合物の有効量
を含む、組み合わせの有効量を投与し、
それによって、該哺乳動物における該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目7)
項目5に記載の組み合わせの有効量、および該組み合わせを投与するための指示手段を含む、キット。
(項目8)
S−アデノシル−L−ホモシステイン加水分解酵素(SAH)活性を阻害し得る候補阻害剤化合物を同定するための方法であって、以下の工程:
a)SAH結合ポケットのコンピュータモデルを構築する工程;
b)以下の構造
【化6】
[式中、Zは、炭素および窒素からなる群より選択される]
を有する複数の化合物をスクリーニングする工程;ならびに
c)該結合ポケットに計算上結合する化合物を同定する工程
を包含する、方法。
(項目9)
SAH加水分解酵素の活性を可逆的に阻害するための方法であって、SAH加水分解酵素を、有効量の項目1に記載の化合物と接触させて、該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目10)
SAH加水分解酵素の活性を可逆的に阻害するための方法であって、SAH加水分解酵素を、有効量の項目5に記載の組み合わせと接触させて、該SAH加水分解酵素の活性を可逆的に阻害する工程を包含する、方法。
(項目11)
IL−12の産生および/または放出を可逆的に阻害するための方法であって、該方法は、IL−12産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−12産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する、方法。
(項目12)
哺乳動物における遅延型過敏症(DTH)反応を軽減するための方法であって、該方法は、このような軽減が必要であるかまたは望ましい哺乳動物に、SAH加水分解酵素の可逆的阻害剤の有効量を投与し、それによって該哺乳動物におけるDTH反応を軽減する工程を包含する、方法。
(項目13)
前記哺乳動物がヒトである、項目12に記載の方法。
(項目14)
前記SAH加水分解酵素の可逆的阻害剤が(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない、項目12に記載の方法。
(項目15)
IL−10の産生および/または放出を維持または増大させるための方法であって、該方法は、IL−10産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−10産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する、方法。
(項目16)
前記IL−10産生細胞が哺乳動物に含まれる、項目15に記載の方法。
(項目17)
前記SAH加水分解酵素の可逆的阻害剤が(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない、項目15に記載の方法。
(項目18)
IL−2またはIFN−γの産生および/あるいは放出を可逆的に阻害するための方法であって、該方法は、IL−2またはIFN−γ産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−2またはIFN−γ産生細胞における該SAH加水分解酵素を可逆的に阻害する工程を包含する、方法。
(項目19)
前記IL−2またはIFN−γ産生細胞が哺乳動物に含まれる、項目18に記載の方法。
(項目20)
前記SAH加水分解酵素の可逆的阻害剤が(4−アデニン−9−イル)−2−ヒドロキシブタン酸ではない、項目18に記載の方法。
(項目21)
前記化合物またはその薬学的に受容可能な塩が、式(IA)
【化7】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]で示される、項目4に記載の方法。
(項目22)
前記化合物またはその薬学的に受容可能な塩が、式(IB)
【化8】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される、項目4に記載の方法。
(項目23)
前記化合物またはその薬学的に受容可能な塩が、
【化9】
[式中、Wは、Hまたはメトキシである]
である、項目4に記載の方法。
(項目24)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目4に記載の方法。
(項目25)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目21に記載の方法。
(項目26)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目22に記載の方法。
(項目27)
前記哺乳動物が、炎症性腸疾患、多発性硬化症および自己免疫性神経炎からなる群より選択される疾患に罹患している疑いがある、項目23に記載の方法。
【図面の簡単な説明】
【0054】
【図1】図1は、ヒツジ赤血球の定量的溶血(QHS)アッセイに対するDZ2002の影響を示す。データは、平均値±SDとして表した。**:コントロールと比較してP<0.01。
【図2】図2は、DZ2002が、混合リンパ球反応においてT細胞増殖を抑制することを示す。データは、平均値±SDとして表した。***:コントロールと比較してP<0.001。
【図3】図3は、DZ2002が、脾臓細胞において細胞傷害性を有さないことを示す。
【図4】図4は、Balb/cマウスにおけるDTH耳介腫脹に対するDZ2002の影響を示す。
【図5】図5は、TG誘導性腹膜細胞からのTNF−α産生に対するDZ2002の影響を示す。
【図6A】図6Aは、THP−1細胞におけるMHC−IIの発現に対するDZ2002の影響を示す。
【図6B】図6Bは、THP−1細胞におけるCD80の発現に対するDZ2002の影響を示す。
【図6C】図6Cは、THP−1細胞におけるCD86の発現に対するDZ2002の影響を示す。
【図7】図7Aおよび7Bは、THP−1細胞からのIL−12P40およびIL−12P70産生に対するDZ2002の影響を示す。
【図8】図8は、遅延型過敏症(DTH)反応に対するDZ2002の影響を示す。
【図9】図9は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−10産生に対するDZ2002の影響を示す。
【図10】図10は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−2産生に対するDZ2002の影響を示す。
【図11】図11は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIFN−γ産生に対するDZ2002の影響を示す。
【発明を実施するための形態】
【0055】
(発明の詳細な説明)
開示内容を明確にするためであって、限定のためではなく、本発明の詳細な説明は以下の小節に分けられる。
【0056】
A.定義
他に規定しない限り、本明細書中で使用されるすべての技術用語および科学用語は、本発明が属する技術分野の当業者によって通常理解されるのと同じ意味を有する。本明細書中に引用される全ての特許、出願、公開された出願および他の刊行物は、その全体が、参考として援用される。この節に示される定義が、本明細書中に参考として援用される特許、出願、公開された出願および他の刊行物に示される定義に反するかまたは矛盾する場合、この節に示される定義が、本明細書中に参考として援用される定義より優先する。
【0057】
本明細書中で使用される場合、「a」または「an」は、「少なくとも1つ」または「1つ以上」を意味する。
【0058】
本明細書中で使用される場合、「組成物」とは、2つ以上の生成物または化合物の任意の混合物をいう。組成物は、溶液、懸濁液、液体、粉末、ペースト、水性、非水性、またはそれらの任意の組み合わせであり得る。
【0059】
本明細書中で使用される場合、「組み合わせ」とは、2つのアイテム間かまたはより多くのアイテム間の任意の関連をいう。
【0060】
本明細書中で使用される場合、「ホモシステイン」(Hcy)とは、以下の分子式の化合物をいう:HSCH2CH2CH(NH2)COOH。生物学的には、Hcyは、メチオニンの脱メチル化によって生成され、メチオニンからのシステインの生合成における中間体である。用語「Hcy」は、遊離Hcy(還元型の)および結合Hcy(酸化型の)を包含する。Hcyは、タンパク質、ペプチド、Hcy自体または他のチオールと、ジスルフィド結合で結合体化し得る。
【0061】
本明細書中で使用される場合、「SAH加水分解酵素」とは、SAHのアデノシン(Ado)およびHcyへの加水分解を触媒する酵素をいう。この酵素は、遍在性の真核生物酵素であり、一部の原核生物にも見出される。SAH加水分解酵素はまた、AdoおよびHcyからのSAHの生成も触媒する。SAH加水分解酵素の補酵素は、NAD+/NADHである。SAH加水分解酵素は、いくつかの触媒活性を有し得る。加水分解の方向において、第1の工程は、酵素結合NAD+(E−NAD+)によるSAHの3’−ヒドロキシル基の酸化(3’−酸化活性)、その後のL−Hcyのβ−脱離を包含し、3’−ケト−4’,5’−ジデヒドロ−5’−デオキシ−Adoを生じる。この強固に結合した中間体の5’側の位置への水のMichael付加(5’−加水分解活性)により、3’−ケト−Adoを生じ、これは次いで、酵素結合NADH(E−NADH)によってAdoに還元される(3’−還元活性)。この用語は、活性を実質的に変化させない保存的アミノ酸置換を行ったSAH加水分解酵素を包含することが意図される。
【0062】
本明細書中で使用される場合、本発明の化合物の「薬学的に受容可能な塩」または「薬学的に受容可能な誘導体」との用語は、当業者に容易に調製され得る任意の塩、エステルまたは誘導体を包含する。本発明の化合物の薬学的に受容可能な塩としては、例えば、薬学的に受容可能な無機および有機の酸および塩基から誘導されるものが挙げられる。適切な塩基から誘導される塩としては、アルカリ金属(例えば、ナトリウム)塩、アルカリ土類金属(例えば、マグネシウム)塩、アンモニウム塩およびN(C1−4アルキル)4+塩が挙げられるが、これらに限定されない。適切な酸の例としては、塩酸、臭化水素酸、硫酸、硝酸、過塩素酸、フマル酸、マレイン酸、リン酸、グリコール酸、乳酸、サリチル酸、コハク酸、トルエン−p−スルホン酸、酒石酸、酢酸、クエン酸、メタンスルホン酸、ギ酸、安息香酸、マロン酸、ナフタレン−2−スルホン酸、およびベンゼンスルホン酸が挙げられるが、これらに限定されない。それ自体では薬学的に受容可能でない他の酸(シュウ酸など)は、本発明の化合物およびその薬学的に受容可能な酸性塩を得る際に中間体として有用な塩の調製に用いられ得る。
【0063】
本明細書中で使用される場合、「生物活性」とは、化合物のインビボ活性、または化合物、組成物、もしくは他の混合物のインビボ投与の結果として生じる生理反応をいう。従って、生物活性は、このような化合物、組成物および混合物の治療効果ならびに薬学的活性を包含する。生物学的活性は、このような活性を試験するかまたは用いるように設計されたインビトロ系で認められ得る。
【0064】
本明細書中で使用される場合、「血漿」とは、流体(血液の非細胞部分)をいい、凝血後に得られる血清とは区別される。
【0065】
本明細書中で使用される場合、「血清」とは、フィブリン凝塊および血球の除去後に得られる血液の流体部分をいい、循環血液中の血漿とは区別される。
【0066】
本明細書中で使用される場合、「流体」とは、流動し得る任意の組成物をいう。従って、流体は、半固体、ペースト、溶液、水性混合物、ゲル、ローション、クリームの形態の組成物、および他のこのような組成物を包含する。
【0067】
本明細書中で使用される場合、任意の保護基、アミノ酸および他の化合物の略語は、他に指示がない限り、その一般的用法、認められた略語、またはIUPAC−IUB Commission on Biochemical Nomenclatureによるものである(Biochemistry 11:1726(1972)を参照のこと)。
【0068】
本明細書中で使用される場合、「疾患または障害」とは、特定可能な症状によって特徴付け可能な、生物における病理学的状態をいう。
【0069】
本明細書中で使用される場合、用語「治療剤」とは、当業者に公知の任意の従来の薬物または薬物療法をいい、ワクチンが挙げられるが、これに限定されない。
【0070】
本明細書中で使用される場合、「ワクチン」とは、能動的免疫予防を意図される任意の組成物をいう。ワクチンは、疾患を処置するか、疾患の発症を予防するか、または疾患の重症度を軽減するために、事前にかまたは感染後のいずれかに治療的に用いられ得る。例示的なワクチンとしては、病原性株の死滅させた微生物の調製物、弱毒化(改変または変異)株の生きた微生物の調製物、あるいは微生物、真菌、植物、原生動物、または後生動物の派生物もしくは生成物が挙げられるが、これらに限定されない。この用語はまた、タンパク質/ペプチドおよびヌクレオチドを主成分とするワクチンを包含する。
【0071】
本明細書中で使用される場合、用語「治療有効量」とは、疾患に関連する症状を寛解させるか、または何らかの様式で軽減するのに十分な量をいう。このような量は、単回投薬として、またはレジメンに従って投与され得る。反復投与は、症状の所望の寛解を達成するのに必要な場合がある。
【0072】
本明細書中で使用される場合、用語「投与」または化合物を「投与すること」とは、本発明の化合物または本発明の化合物のプロドラッグを被験体に与える任意の適切な方法をいう。
【0073】
本明細書中で使用される場合、用語「処置」とは、状態、障害または疾患の症状が寛解されるか、あるいは有利に変更される、任意の様式をいう。処置はまた、本明細書における組成物の任意の医薬用途も包含する。特定の障害の症状の寛解とは、組成物の投与に起因し得るかまたは関連し得る、症状の任意の軽減(永久であるか一時的であるかにかかわらず)をいう。
【0074】
本明細書中で使用される場合、用語「置換」とは、化合物中の水素原子を置換基で置換することをいう。
【0075】
本明細書中で使用される場合、用語「アルキル」は、1つ以上の置換基で置換されていてもよいアルキル基を含む、直鎖または分岐鎖アルキル基を包含する。例えば、アルキル基は、ヒドロキシ、ハロゲン、アリール、アルコキシ、アシル、または当該分野で公知の他の置換基で置換されていてもよい。アルキル基のより多くの炭素原子の1つもまた、1つ以上のヘテロ原子で置換されていてもよい。
【0076】
本明細書中で使用される場合、用語「Ki」とは、ICEなどの標的酵素の活性の阻害における化合物の有効性の数値尺度をいう。Kiの値が低いほど、高い有効性を示している。Ki値は、実験的に決定された速度データを、標準酵素反応速度式に当てはめることによって求められる(Segel,Enzyme Kinetics,Wiley−Interscience,1975)。
【0077】
本明細書中で使用される場合、「抗悪性腫瘍薬処置」とは、新生物、腫瘍または癌を、その症状を軽減または寛解することによって処置するように設計された、任意の処置をいう。新生物、腫瘍または癌の発生を予防するか、あるいはこれらの重症度を軽減する処置も、企図される。
【0078】
本明細書中で使用される場合、「新生物(新生物形成)」とは、異常な新生物をいい、従って腫瘍と同じものを意味し、これは良性であっても悪性であってもよい。過形成とは異なり、新生物の増殖は、本来の刺激がない状態においても持続する。
【0079】
本明細書中で使用される場合、「抗新生物薬(抗悪性腫瘍薬、抗腫瘍剤または抗癌剤と交換可能に用いられる)」とは、抗新生物処置に用いられる任意の薬剤をいう。これらとしては、単独かまたは他の化合物と組み合わせて用いられる場合に、新生物、腫瘍または癌と関連する臨床症状もしくは診断マーカーを、緩和するか、軽減するか、寛解するか、予防するか、緩解状態にするかまたは緩解状態を維持し得る、任意の薬剤が挙げられる。本発明と組み合わせて用いられ得る抗新生物薬としては、抗血管形成剤、アルキル化剤、代謝拮抗剤、特定の天然物、プラチナ配位錯体、アントラセンジオン、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、特定のホルモンおよびアンタゴニスト、抗癌性多糖類、ならびに漢方薬草抽出物のような特定の薬草抽出物が挙げられるが、これらに限定されない。
【0080】
本明細書中で使用される場合、「癌抑制遺伝子」(癌抑制遺伝子または癌感受性遺伝子とも呼ばれる)とは、細胞周期を通常負に制御し、かつ、細胞が急速な分裂へと進み得る前に変異させられるかまたは不活化されなければならない産物をコードする遺伝子をいう。例示的な癌抑制遺伝子としては、p16、p21、p53、RB(網膜芽細胞腫)、WT−1(Wilmの腫瘍)、DCC(結腸癌で欠失している)、NF−1(神経線維肉腫)およびAPC(adenomatous polypospis coli)が挙げられるが、これらに限定されない。
【0081】
本明細書中で使用される場合、「癌遺伝子」とは、動物細胞の正常な遺伝子(原癌遺伝子)の変異型および/または過剰発現型をいい、優性で細胞を正常な成長抑制から解放し得る。従って、癌遺伝子は、単独で、または他の変更と連携して、細胞を腫瘍細胞に変える。例示的な癌遺伝子としては、abl、erbA、erbB、ets、fes(fps)、fgr、fms、fos、hst、int1、int2、jun、hit、B−lym、mas、met、mil(raf)、mos、myb、myc、N−myc、neu(ErbB2)、ral(mil)、Ha−ras、Ki−ras、N−ras、rel、ros、sis、src、ski、trkおよびyesが挙げられるが、これらに限定されない。
【0082】
本明細書中で使用される場合、「アンチセンスポリヌクレオチド」とは、mRNAに相補的なヌクレオチド塩基の合成配列または二本鎖DNAのセンス鎖をいう。適切な条件下でのセンスポリヌクレオチドおよびアンチセンスポリヌクレオチドの混合により、これらの2つの分子の結合(すなわち、ハイブリダイゼーション)が生じる。これらのポリヌクレオチドがmRNAに結合(ハイブリダイズ)する場合、タンパク質合成(翻訳)の阻害が起こる。これらのポリヌクレオチドが二本鎖DNAに結合する場合、RNA合成(転写)の阻害が起こる。結果として生じた翻訳および/または転写の阻害により、センス鎖によってコードされるタンパク質の合成の阻害が生じる。
【0083】
本明細書中で使用される場合、「抗体」は、軽鎖および重鎖の可変領域で構成される抗体フラグメント(例えば、Fabフラグメント)を包含する。
【0084】
本明細書中で使用される場合、用語「ヒト化抗体」とは、ヒトに投与されても免疫応答を引き起こさないように、アミノ酸の「ヒト」配列を含むように改変されている抗体をいう。このような抗体を調製するための方法は公知である。例えば、モノクローナル抗体を発現するハイブリドーマは、非可変領域のアミノ酸組成がヒト抗体に基づいている抗体を発現するように、組換えDNA技術によって改変される。コンピュータプログラムは、このような領域を同定するように設計されている。
【0085】
本明細書中で使用される場合、「抗出血性ウイルス剤」または「抗ウイルス性出血剤」とは、出血性ウイルス感染の処置に用いられる任意の薬剤をいう。これらの薬剤としては、単独かまたは他の化合物と組み合わせて、ウイルス性出血疾患もしくは障害に関連する臨床症状または診断マーカーを緩和し得るか、軽減し得るか、寛解し得るか、予防し得るか、あるいは緩解状態を維持し得る、任意の薬剤を包含する。抗ウイルス性出血剤の非限定的な例としては、インターロイキン−1(IL−1)阻害剤、腫瘍壊死因子(TNF)阻害剤、抗ウイルスワクチン、抗ウイルス抗体、ウイルスによって活性化された免疫細胞、およびウイルスによって活性化された免疫血清が挙げられる。
【0086】
本明細書中で使用される場合、「抗出血性ウイルス処置」とは、症状を軽減するかまたは寛解させることによって、出血性ウイルス感染を処置するように設計される任意の処置をいう。感染を予防するかまたはその重症度を軽減する処置もまた企図される。
【0087】
本明細書中で使用される場合、「IL−1阻害剤」は、IL−1の産生、翻訳後修飾、成熟、または放出を阻止するかもしくは減少させる任意の物質、あるいはIL−1とIL−1レセプターの間の相互作用の効力に干渉するかもしくはこれを減少させる任意の物質を包含する。好ましくは、IL−1阻害剤は、抗IL−1抗体、抗IL−1レセプター抗体、IL−1レセプターアンタゴニスト、IL−1産生阻害剤、IL−1レセプター産生阻害剤、またはIL−1放出阻害剤である。
【0088】
本明細書中で使用される場合、「腫瘍壊死因子」(「TNF」)とは、主要組織適合性複合体内にコードされる炎症誘発性サイトカインの群をいう。TNFファミリーメンバーとしては、TNFαおよびTNFR(それぞれ、カケクチンおよびリンホトキシンとしても公知である)が挙げられる。TNAαおよびTNFRをコードする相補的なcDNAクローンが単離されている。従って、「TNF」という用語は、TNFαおよびTNFを含む、TNF遺伝子ファミリーによってコードされるすべてのタンパク質、あるいは任意の他の供給源から得られるかまたは合成的に調製された相当する分子を包含する。活性を実質的に変化させない保存的アミノ酸置換を行ったTNFを包含することが意図される。
【0089】
本明細書中で使用される場合、「TNF阻害剤」は、TNFの産生、翻訳後修飾、成熟、または放出を阻止するかもしくは減少させる任意の物質、あるいはTNFとTNFレセプターの間の相互作用の効力に干渉するかもしくはこれを減少させる任意の物質を包含する。好ましくは、TNF阻害剤は、抗TNF抗体、抗TNFレセプター抗体、TNFレセプターアンタゴニスト、TNF産生阻害剤、TNFレセプター産生阻害剤、またはTNF放出阻害剤である。
【0090】
B.S−アデノシル−L−ホモシステイン加水分解酵素の可逆的阻害剤
作用機序に基づく細胞傷害性を最小にするための1つのアプローチは、SAH加水分解酵素阻害剤が可逆的な阻害活性を示すように、該阻害剤の薬物動態プロファイルを最適化することである。薬物動態プロファイルは、KOff値を最適化することによって最適化され得る。例えば、KOff値は、これらの値が所望の治療効果を生じるのに十分に小さいが、次の投与前に酵素活性の適切な回復を可能にするのに十分に大きいように、最適化される。
【0091】
一般に、SAH加水分解酵素の可逆的阻害剤は、酵素に非共有結合的に結合し、かつ、酵素から容易に遊離される。例えば、酵素に結合した可逆的阻害剤は、簡単な透析または緩衝液の交換もしくはpHの変更によって取り除かれ得る。可逆的阻害剤としては、拮抗阻害剤、非拮抗阻害剤、不拮抗阻害剤、および混合型の阻害剤が挙げられる。
【0092】
拮抗阻害剤は、単に遊離の酵素に結合して、酵素が基質に結合するのを阻止する阻害剤である。通常、拮抗阻害剤は、基質と構造が類似しており、活性部位への基質の接近を妨害するように、活性部位に結合する(すなわち、酵素は、インヒビターを遊離してからでなければ基質に結合できない)。基質および阻害剤の両方が、同じ部位に結合するために競合する。反応速度論に対する拮抗阻害剤の影響は、酵素の最大速度(Vmax)に影響を及ぼすことなく、ミカエリス定数(Km)を増加させることである。拮抗阻害剤の存在下での見かけのKm(Km,app)は、Km(1+[I]/Ki)であり、式中、[I]は阻害剤の濃度であり、そしてKiは阻害剤の解離定数である。
【0093】
非拮抗阻害剤は、酵素が反応を触媒するのを阻止するが、基質が結合するのを妨害しない阻害剤である。基質および阻害剤の両方とも、同時に酵素に結合し得る(すなわち、阻害剤および基質は、同じ部位に結合するために拮抗しない)が、触媒反応は、阻害剤が結合していない場合にのみ生じる。非拮抗阻害剤は、遊離の酵素および酵素−基質複合体に、同じ親和性で結合する。非拮抗阻害剤の反応速度論に対する影響は、Kmに影響を及ぼすことなくVmaxを減少させることである。非拮抗阻害剤の存在下での見かけのVmax(Vmax,app)は、Vmax/(1+[I]/Ki)であり、式中、[I]は阻害剤の濃度であり、そしてKiは阻害剤の解離定数である。
【0094】
不拮抗阻害剤は、単に酵素−基質複合体に結合して、該複合体を不活化する阻害剤である。反応速度論に対する不拮抗阻害剤の影響は、VmaxおよびKmの両方を減少させることである。不拮抗阻害剤の存在下での見かけのKm(Km,app)は、Km(1+[I]/Kib)であり、式中、[I]は阻害剤の濃度であり、そしてKibは酵素−基質複合体に対する阻害剤の解離定数である。不拮抗阻害剤の存在下での見かけのVmax(Vmax,app)は、Vmax/(1+[I]/Kib)であり、式中、[I]は阻害剤の濃度であり、そしてKibは酵素−基質複合体に対する阻害剤の解離定数である。
【0095】
混合型の阻害剤は、遊離の酵素および酵素−基質複合体の両方に結合する阻害剤であり、触媒反応を阻害する。混合型の阻害剤には、活性を完全には無効にせず、単に反応速度を顕著に減少させるだけの阻害剤も含まれる。混合阻害剤の存在下での見かけのKm(Km,app)は、Km(1+[I]/Kia)であり、式中、[I]は阻害剤の濃度であり、そしてKiaは遊離の酵素に対する阻害剤の解離定数である。混合阻害剤の存在下での見かけのVmax(Vmax,app)は、Vmax/(1+[I]/Kib)であり、式中、[I]は阻害剤の濃度であり、そしてKibは酵素−基質複合体に対する阻害剤の解離定数である。
【0096】
酵素(例えば、SAH加水分解酵素)に対する阻害剤のタイプを決定するための方法は、当該分野で公知である。例えば、酵素反応速度論実験を用いて阻害剤が結合する機序および部位を試験し、阻害剤のタイプを決定し得る。KmおよびVmaxは、阻害剤の非存在下で、および2種以上の濃度の阻害剤の存在下で決定され得る。次いで、収集したデータは、阻害剤のタイプを決定するために、見かけのKmまたはVmaxの変化(すなわち、阻害剤濃度の関数としてのパラメータの変化)について分析され得る。
【0097】
強固に結合した阻害剤および作用機序に基づく阻害剤はまた、酵素と阻害剤の間に共有結合が形成されないにもかかわらず、時間依存的阻害パターンを示し得る。これらのタイプの阻害剤によって不活化される酵素は、通常は永久に無効にされ、かつその活性は、例えば、0.5〜5時間のような一定時間のゲル濾過または透析によって、容易に回復することができない。これらのタイプの時間依存的阻害剤は、不可逆的阻害剤とみなされ、本発明において記載される可逆的阻害剤から除外される。
【0098】
SAH加水分解酵素の可逆的阻害剤としてのエリタデニン誘導体
本発明は、可逆的かつ強力なS−アデノシル−L−ホモシステイン組成物の新規阻害剤に関する。例えば、本発明は、Ki値が100nM未満の化合物を提供する。1つの実施形態において、本発明は、4(アデニン−9−イル)−2−ヒドロキシブタン酸、その誘導体、およびその薬学的に受容可能な塩、ならびにこのような化合物を用いてSAH加水分解酵素を可逆的に阻害するための方法を提供する。
【0099】
可逆的阻害剤である、4(アデニン−9−イル)−2−ヒドロキシブタン酸は、エリタデニンのβ炭素でのデオキシル修飾(deoxyl modification)により合成される。エリタデニンは、天然に存在する化合物であり、SAH加水分解酵素の強力な不可逆的阻害剤である。エリタデニンのβ炭素でのデオキシル修飾により、阻害能力を保持しつつ、可逆的阻害剤である化合物を生じる。4(アデニン−9−イル)−2−ヒドロキシブタン酸の誘導体は、当業者に公知の従来の合成方法を用いて合成され得る。(例えば、Yuanら、Adv.Antiviral Drug Des.2:41−88(1996);Holyら、Coll.Czechoslovak Chem.Commun.50:245−279(1985)を参照のこと)。
【0100】
4(アデニン−9−イル)−2−ヒドロキシブタン酸誘導体の例としては、塩基修飾誘導体、および側鎖置換誘導体が挙げられるが、これらに限定されない。塩基修飾誘導体は、アデニル環塩基(adenyl ring base)にて修飾された4(アデニン−9−イル)−2−ヒドロキシブタン酸の誘導体である。アデニル環は、アミノ基にて、種々の修飾基で修飾され得る。アデニル環はまた、アデニル環のC2位およびC8位にて、種々の置換基で修飾され得る。
【0101】
1つの実施形態において、SAH加水分解酵素の可逆的阻害剤は、下記式(I)で示されるもの、およびその薬学的に受容可能な塩である:
【0102】
【化23】
[式中、Zは炭素または窒素であり、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、またはハロゲンであり;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、またはヘテロアリールであり;Xは、酸素、窒素、または硫黄であり;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、またはヘテロアリールである]。
【0103】
1つの局面において、SAH加水分解酵素の可逆的阻害剤は、式IA
【0104】
【化24】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【0105】
【化25】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される。
【0106】
種々のR基が、他の置換基で置換されていてもよい。これらの置換基は、ハロゲン、ヒドロキシ、アルコキシ、ニトロ、シアノ、カルボン酸、アルキル、アルケニル、シクロアルキル、チオール、アミノ、アシル、カルボキシレート、アリール、カルバメート、カルボキサミド、スルホンアミド、複素環基、または当該分野で公知の任意の適切な置換基であり得る。特定の実施形態において、各R基は、水素、またはメチルのような低級直鎖アルキルである。別の実施形態において、アルキルまたはアルコキシ基の中の1つ以上の炭素原子は、1つ以上のヘテロ原子で置換され得る。
【0107】
アミノ基はまた、2回または3回置換されて、2級または3級アミンを形成し得る。置換基の非限定的な例としては、アルキルまたは置換されていてもよいアルキル基;アルケンまたは置換されていてもよいアルケニル基;シクロアルキルまたは置換されていてもよいシクロアルキル基;アリール、複素環;アラルキル(例えば、フェニルC1−4アルキル);フェニル、ピリジン、フェニルメチル、フェネチル、ピリジニルメチル、ピリジニルエチルのようなヘテロアルキル;および他の置換基が挙げられる。複素環基は、1〜4個のヘテロ原子を含む5または6員環であってもよい。
【0108】
アミノ基は、置換されていてもよいC2−4アルカノイル(例えば、アセチル、プロピオニル、ブチリル、イソブチリルなど);C1−4アルキルスルホニル(例えば、メタンスルホニル、エタンスルホニルなど);カルボニルまたはスルホニル置換された芳香族環または複素環(例えば、ベンゼンスルホニル、ベンゾイル、ピリジンスルホニル、ピリジンカルボニルなど)で置換され得る。
【0109】
CO−X−Y基は、置換されていてもよいカルボキシレート基であり得る。置換されていてもよいカルボキシレート基の例としては、置換されていてもよいアルキル(例えば、C1−10アルキル);置換されていてもよいシクロアルキル(例えば、C3−7シクロアルキル);置換されていてもよいアルケニル(例えば、C2−10アルケニル);置換されていてもよいシクロアルケニル(例えば、C3−7シクロアルケニル);置換されていてもよいアリール(例えば、フェニル、ナフチル、ベンジルなどのC1−4アリール);および他の適切な置換基が挙げられるが、これらに限定されない。メトキシメチル、メトキシエチル、および関連する基のような基もまた、包含される。
【0110】
新規のSAH加水分解酵素阻害剤の構造に基づく薬物設計
本発明の化合物を最初の鋳型分子として用いた、構造に基づく薬物設計を提供することもまた、本発明の目的である。最近、SAH加水分解酵素のX線構造は、酵素の「開」および「閉」状態の両方について入手可能になっている。当業者は、構造に基づく設計を用いて、SAH加水分解酵素阻害剤をスクリーニングするための新規の化合物を設計し得る。候補化合物の設計または選択は、SAH加水分解酵素の結合ポケットを満たす種々の部分の選択から開始し得る。(例えば、米国特許第5,756,466号;Klebe,J.Mol.Med.78:69−281(2000);およびMaignanら、Curr.Top.Med.Chem.1:161−174(2001)を参照のこと)。
【0111】
個々の結合ポケットを満たす部分を選択するための方法は、多数存在する。これらとしては、活性部位の物理的モデルまたはコンピュータモデルの外観検査、および選択された部分のモデルを種々の結合ポケットに手動でドッキングすることが挙げられる。当該分野で周知かつ入手可能なモデリングソフトウェアが用いられ得る。これらとしては、QUANTA(Molecular Simulations,Inc.,Burlington,Mass.,1992);SYBYL(Molecular Modeling Software,Tripos Associates,Inc.,St.Louis,Mo.,1992);AMBER(Weinerら、J.Am.Chem.Soc.6:765−784(1984));CHARMM(Brooksら、J.Comp.Chem.4:187−217(1983))が挙げられるが、これらに限定されない。モデリング工程に続いて、CHARMMおよびAMBERのような標準分子力学力場を用いたエネルギー最小化が行なわれ得る。さらに、本発明の結合部分を選択するプロセスを補助する、より専門化したコンピュータプログラムが多数存在する。これらとしては、以下が挙げられるが、これらに限定されない:
1.GRID(Goodford、「A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules」、J.Med.Chem.28:849−857(1985))。GRIDは、Oxford University,Oxford,UKから入手可能である。
【0112】
2.MCSS(Mirankerら、「Functionality Maps of
Binding Sites:A Multiple Copy Simultaneous Search Method」,Proteins:Structure,Function and Genetics」11:29−34(1991))。MCSSは、Molecular Simulations,Burlington,Mass.から入手可能である。
【0113】
3.AUTODOCK(Goodsellら、「Automated Docking
of Substrates to Proteins by Simulated Annealing」,PROTEINS:Structure,Function and Genetics 8:195−202(1990))。AUTODOCKは、Scripps Research Institute,La Jolla,Calif.から入手可能である。
【0114】
4.DOCK(Kuntzら、「A Geometric Approach to Macromolecule−Ligand Interactions」,J.Mol.Biol.161:269−288(1982))。DOCKは、University of California,San Francisco,Calif.から入手可能である。
【0115】
一旦、適切な結合部分が選択されると、これらの結合部分は、単一の阻害剤に構築され得る。この構築は、種々の部分を中心骨格に結合することによって達成され得る。この構築プロセスは、例えば、外観検査に続いて、QUANTAまたはSYBYLのようなソフトウェアを同じく用いた手動モデル構成によってなされ得る。多数の他のプログラムも、種々の部分を結合するための選択方法を補助するために用いられ得る。これらとしては、以下が挙げられるが、これらに限定されない:
1.CAVEAT(Bartlettら、「CAVEAT:A Program to
Facilitate the Structure−Derived Design
of Biologically Active Molecules」,Molecular Recognition in Chemical and Biological Problems,Special Pub.,Royal Chem.Soc.78:182−196(1989))。CAVEATは、University of California,Berkeley,Calif.から入手可能である。
【0116】
2.MACCS−3Dなどの3Dデータベースシステム(MDL Information Systems,San Leandro,Calif.)。この領域は、最近、Martinによって概説された(Martin,「3D Database Searching in Drug Design」,J.Med.Chem.35:2145−2154(1992))。
【0117】
3.HOOK(Molecular Simulations,Burlington,Mass.から入手可能)。
【0118】
上記の阻害剤化合物のコンピュータ補助によるモデリングに加えて、本発明の阻害剤は、活性のない部位または公知の阻害剤の一部を必要に応じて含むもののいずれかを用いて、デノボで構築され得る。このような方法は、当該分野で周知である。これらとしては、例えば、以下が挙げられる:
1.LUDI(Bohm,「The Computer Program LUDI:A New Method for the De Novo Design of Enzyme Inhibitors」,J.Comp.Aid.Molec.Design 6:61−78(1992))。LUDIは、Biosym Technologies,San Diego,Calif.から入手可能である。
【0119】
2.LEGEND(Nishibataら、Tetrahedron,47:8985(1991))。LEGENDは、Molecular Simulations,Burlington,Mass.から入手可能である。
【0120】
3.LeapFrog(Tripos associates,St.Louis,Mo.から入手可能)。
【0121】
薬物のモデリングに一般に使用される多数の技術が用いられ得る(例えば、Cohenら、J.Med.Chem.33:883−894(1990)を参照のこと)。同様に、化学技術文献中の多数の例が、特定の薬物設計プロジェクトに適用され得る。(概説について、Naviaら、Curr.Opin.Struc.Biol.2:202−210(1991)を参照のこと)。本発明の工程の新規の組み合わせを用いると、当業者は、時間のかかる高価な実験を有利に回避して、特定の化合物の酵素阻害活性を決定し得る。この方法はまた、SAH加水分解酵素阻害剤、ならびにSAH加水分解酵素媒介性の疾患に対する治療および予防剤の合理的設計を容易にするのにも有用である。従って、本発明は、このような阻害剤、およびこのような阻害剤を同定または選択するための方法に関する。
【0122】
種々の従来の技術を用いて、各々の上記の評価、およびSAH加水分解酵素阻害活性について候補化合物をスクリーニングするのに必要な評価を行い得る。一般に、これらの技術は、所定の部分の位置および結合近似、結合した阻害剤の占有空間、所定の化合物の結合の変形エネルギーおよび静電的相互作用エネルギーを決定する工程を包含する。上記の評価に有用な従来の技術の例としては、量子力学、分子力学、分子動力学、モンテカルロサンプリング、系統的探索およびディスタンスジオメトリー法(Marshall,Ann.Ref.Pharmacol.Toxicol.27:193(1987))が挙げられるが、これらに限定されない。これらの方法を実行するのに用いられる特殊なコンピュータソフトウェアが開発されている。このような用途のために設計されたプログラムの例としては、以下が挙げられる:Gaussian 92(Gaussian,Inc.,Pittsburgh,Pa.);AMBER;QUANTA/CHARMM;およびInsight II/Discover(Biosysm Technologies
Inc.,San Diego,Calif.)。これらのプログラムは、例えば、Silicon Graphics Indigo2ワークステーションまたはIBM RISC/6000ワークステーションモデル550を用いて実行され得る。他のハードウェアシステムおよびソフトウェアパッケージが公知であり、かつ、当業者に明らかな適用性を有する。
【0123】
本発明に従って、様々なクラスの活性なSAH加水分解酵素阻害剤が、SAH加水分解酵素活性部位の種々の結合ポケットと、類似の方法で相互作用し得る。これらの重要な基の空間的配置は、しばしばファルマコフォアと呼ばれる。ファルマコフォアの概念は、文献に十分に記載されている(Mayerら、J.Comp.Aided Molec.Design 1:3−16(1987);Hopfingerら,Concepts and Applications of Molecular Similarity,JohnsonおよびMaggiora(編),Wiley(1990)を参照のこと)。
【0124】
本発明の様々なクラスのSAH加水分解酵素阻害剤はまた、結合に必要な特異的相互作用が得られ得るように、必要な部分が活性部位に配置されることを可能にする、様々な骨格またはコア構造を用い得る。これらの化合物は、ファルマコフォア(すなわち、SAH加水分解酵素の活性部位の形状および性質に対する構造的同一性)にマッチする能力に関して最善に定義される。種々の骨格が、例えば、Klebe,G.,J.Mol.Med.78:269−281(2000);Maignanら、Curr.Top.Med.Chem.1:161−174(2001);およびBemisらに対する米国特許第5,756,466号)に記載されている。
【0125】
阻害されるS−アデノシル−L−ホモシステイン加水分解酵素
本発明の化合物は、任意のSAH加水分解酵素を可逆的に阻害するのに用いられ得る。本発明は、任意の特定のSAH加水分解酵素を可逆的に阻害することに限定されることを意図するものではない。
【0126】
1つの実施形態において、本発明の化合物は、以下のGenBank登録番号のヌクレオチド配列を含む核酸によってコードされるSAH加水分解酵素を、可逆的に阻害するのに用いられ得る:AF129871(Gossypium hirsutum);AQ003753(Cryptosporidium parvum);AF105295(Alexandrium fundyense);AA955402(Rattus norvegicus);AA900229(Rattus norvegicus);AA874914(Rattus norvegicus);AA695679(Drosophila melanogaster卵巣);AA803942(Drosophila melanogaster卵巣;AI187655(Manduca sexta雄触角);U40872(Trichomonas vaginalis);AJ007835(Xenopus laevis);AF080546(Anopheles gambiae);AI069796(T.cruzi epimastigote);Z97059(Arabidopsis thaliana);AF059581(Arabidopsis thaliana);U82761(Homo sapiens);AA754430(Oryza sativa);D49804(Nicotiana tabacum);D45204(Nicotiana tabacum);X95636(D.melanogaster);T18277(Zea mays胚乳);R75259(マウス脳);Z26881(C.roseus);X12523(D.discoideum);X64391(Streptomyces fradiae);W21772(トウモロコシの葉);AH003443(Rattus norvegicus);U14963(Rattus norvegicus);U14962(Rattus
norvegicus);U14961(Rattus norvegicus);U14960(Rattus norvegicus);U14959(Rattus norvegicus);U14937(Rattus norvegicus);U14988(Rattus norvegicus);U14987(Rattus norvegicus);U14986(Rattus norvegicus);U14985(Rattus norvegicus);U14984(Rattus norvegicus);U14983(Rattus norvegicus);U14982(Rattus norvegicus);U14981(Rattus norvegicus);U14980(Rattus norvegicus);U14979(Rattus norvegicus);U14978(Rattus norvegicus);U14977(Rattus norvegicus);U14976(Rattus norvegicus);U14975(Rattus norvegicus);L32836(Mus musculus);L35559(Xenopus laevis);Z19779(ヒト胎児副腎組織);L23836(Rhodobacter
capsulatus);M15185(ラット);L11872(Triticum
aestivum);M19937(粘菌(D.discoideum);M80630(Rhodobacter capsulatus)。
【0127】
別の実施形態において、本発明の化合物は、GenBank登録番号M61831−61832のヌクレオチド配列を含む核酸によってコードされるSAH加水分解酵素を可逆的に阻害するのに用いられ得る(Coulter−KarisおよびHershfield,Ann.Hum.Genet.,53(2):169−175(1989)も参照のこと)。本発明の化合物はまた、米国特許第5,854,023号に示されるヌクレオチド配列またはアミノ酸配列を含む核酸によってコードされるSAH加水分解酵素を可逆的に阻害するのにも用いられ得る。
【0128】
C.治療剤としての用途
4(アデニン−9−イル)−2−ヒドロキシブタン酸、その誘導体、および薬学的に受容可能な塩を用いたSAH加水分解酵素の可逆的な阻害により、その治療効果を保持しつつ、細胞傷害性を顕著に低下させる。本発明の化合物は、その効力および可逆性のため、他の不可逆的阻害剤に付随する強い毒性のない治療剤として用いられ得る。本発明の化合物は、SAH加水分解酵素を阻害する能力に関連する生物学的活性を示す薬剤として有用である。SAH加水分解酵素に対する阻害効果は、阻害剤の存在下または非存在下におけるSAH加水分解の初速度の比を用いるか、あるいは当業者に公知の任意の方法を用いて評価され得る。本発明は、出血性ウイルス感染、自己免疫疾患、自家移植片拒絶、新生物、高ホモシステイン不全症、循環器病、脳卒中、アルツハイマー病、および糖尿病などの疾患の予防および処置のための組成物ならびに方法を提供する。しかしながら、本発明は、特定の疾患の予防および処置に限定されることを意図するものではない。
【0129】
1.出血熱ウイルス
本発明は、ウイルス性出血熱の処置のための組成物および方法を提供する。本発明の可逆的阻害剤は、出血熱を引き起こす全てのタイプのウイルス(トガウイルス、アレナウイルス、ナイロウイルス、およびハンタウイルスが挙げられるが、これらに限定されない)に対する広域スペクトラムの抗ウイルス剤として機能し得る。広域スペクトラムの抗ウイルス薬は、狭域スペクトラムの薬剤より優れた多くの利点を与える。ウイルスの病原体の臨床診断に伴う困難のために、診断結果から特定の抗ウイルス剤の選択に到達するのが遅すぎる場合が多い。速効作用は、患者の状態が悪化するのを防ぐために、特に、患者が臨床症状を示すや否やウイルス化学療法を開始しなければならない急性感染症において、しばしば必要である。
【0130】
S−アデノシル−L−ホモシステイン(SAH)加水分解酵素の阻害剤は、エボラウイルス感染の処置に有効であると報告されている。本発明の化合物はまた、他の出血性疾患(例えば、WO00/64479に記載される出血性疾患)に対して用いられ得る。阻害の機構は、本発明の方法を実施するのに必要ではないが、本発明の化合物がウイルス複製を阻害する作用機構は、ウイルスのメチル化の阻害に基づき得る。
【0131】
2.自己免疫疾患および免疫抑制に関連する疾患
本発明は、自己免疫疾患を予防および処置するための組成物ならびに方法を企図する。ある人が自己免疫疾患を有する場合、免疫系が、その人自身の体の細胞、組織、および器官を誤って攻撃する。集団として、自己免疫疾患には、何百万人ものアメリカ人が罹患している。ほとんどの自己免疫疾患は、男性よりも女性にしばしば好発する。自己免疫疾患の例は、国立保健研究所(National Institute of Health)、「Understanding Autoimmune Disease」(http://www.niaid.nih.gov/publications/autoimmune/autoimmune.htm.)から見出され得る。
【0132】
SAH加水分解酵素活性を調節する化合物はまた、免疫抑制に関連する疾患の処置に用いられ得る。免疫抑制は、化学療法、放射線治療、創傷治癒の促進、熱傷処置の強化、またはコルチコステロイド治療などの他の薬物治療、あるいは自己免疫疾患および移植片/移植拒絶の処置に用いられる薬物の組み合わせに起因し得る。免疫抑制はまた、レセプター機能の先天性欠損、感染症、寄生虫症、または他の原因にも起因し得る。
【0133】
3.新生物および癌
本発明はまた、副腎、肛門、聴神経、胆管、膀胱、骨、脳、胸、頬(bruccal)、中枢神経系、頚部、結腸、耳、子宮内膜、食道、眼、眼瞼、ファロピウス管、胃腸管、頭部、首、心臓、腎臓、喉頭、肝臓、肺、下顎骨、下顎関節頭、上顎骨、口、鼻咽腔、鼻、口腔、卵巣、膵臓、耳下腺、陰茎、耳介、脳下垂体、前立腺、直腸、網膜、唾液腺、皮膚、小腸、脊髄、胃、精巣、甲状腺、扁桃腺、尿道、子宮、膣、内耳神経、外陰に関連する新生物、および他の器官に関連する新生物が挙げられるが、これらに限定されない新生物を予防および処置するための組成物ならびに方法も企図する。特定の実施形態において、本発明の薬学的組成物は、非小細胞肺癌、肺癌、乳癌、および前立腺癌の処置に有用である。本発明は、癌(固形腫瘍、リンパ腫、転移性腫瘍、膠芽細胞腫瘍、および他の癌腫瘍に関連する癌が挙げられるが、これらに限定されない)を予防および処置するための組成物ならびに方法をさらに企図する。
【0134】
4.高いホモシステインレベルに関連する疾患
さらに、本発明の化合物は、高レベルのホモシステインに関連する疾患の予防および処置のための血漿ホモシステイン低下薬として用いられ得ることが企図される。高いホモシステインレベル(すなわち、高ホモシステイン血症)に関連していることが見出されている疾患としては、循環器病、脳卒中、アルツハイマー病および糖尿病が挙げられるが、これらに限定されない。例えば、高ホモシステイン血症と冠状動脈性心臓病(CHD)、末梢血管疾患、脳卒中、および静脈血栓症との間の関係が、種々の研究で示されている。
【0135】
高ホモシステインレベルによる脳卒中のリスクの増大はまた、アルツハイマー病を発症する可能性を増加させる。アルツハイマー型の痴呆を有する人々が、血中に高レベルのホモシステインを有することも、最近の研究で示されている。(Selhubら、「Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease」、N.Eng.J.Med.46:476−483(2002))。高いホモシステインはまた、糖尿病、狼瘡、および他の慢性疾患における合併症とも関連している。
【0136】
5.IL−12産生および/または放出の可逆的阻害
本発明は、IL−12(IL−12P70、IL−12P35およびIL−12P40が挙げられる)の産生および/または放出を可逆的に阻害するための組成物ならびに方法を提供し、この方法は、IL−12産生細胞をSAH加水分解酵素の可逆的阻害剤と接触させて、該IL−12産生細胞中の該SAH加水分解酵素を可逆的に阻害する工程を包含する。IL−12P70(時には「IL−12P75」と呼ばれる、Abdi,Scand J.Immunol.56:1−11(2002)を参照のこと)は、p35およびp40サブユニットで構成されているヘテロ二量体のサイトカインである。いくつかの実施形態において、IL−12産生細胞は、哺乳動物(例えば、ヒト)に含まれる。これらの方法は、このような可逆的阻害が必要であるかまたは望ましい疾患の処置に用いられ得る。このような疾患としては、炎症性腸疾患、多発性硬化症および他の自己免疫疾患が挙げられるが、これらに限定されない。
【0137】
SAH加水分解酵素の任意の可逆的阻害剤は、例えば、本明細書中に記載される可逆的阻害剤(例えば、B項に記載される阻害剤)が用いられ得る。SAH加水分解酵素の可逆的阻害剤はまた、Yuanら、Exp.Opin.Ther.Patents 9:1197−1206(1999)、ならびに米国特許第5,137,876号、および同第6,541,482号にも記載される。これらの可逆的阻害剤は、例えば、薬学的に受容可能なキャリアまたは希釈剤とともに処方されて、薬学的組成物中に含まれ得る。
【0138】
いくつかの実施形態において、SAH加水分解酵素の可逆的阻害剤は、下記式(I)で示されるもの、および/またはその薬学的に受容可能な塩である:
【0139】
【化26】
[式中、Zは炭素または窒素であり、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、またはハロゲンであり;R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、またはヘテロアリールであり;Xは、酸素、窒素、または硫黄であり;そしてYは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、またはヘテロアリールである]。特定の実施形態において、投与される組み合わせは、(4−アデニン−9−イル)−2−ヒドロキシブタン酸を含まない。
【0140】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式IA
【0141】
【化27】
[式中、R1およびR2は、同じかまたは異なって、水素、ヒドロキシ、アルキル、シクロアルキル、アルケニル、アルコキシ、アミノ、アリール、ヘテロアリール、およびハロゲンからなる群より選択され;
R3およびR4は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Xは、酸素、窒素、および硫黄からなる群より選択され;そして
Yは、水素、C1−10アルキル基、アルケニル、ビニル、アリール、およびヘテロアリールからなる群より選択される]、
あるいは式(IB):
【0142】
【化28】
[式中、R5は、シクロアルキル、アルケニルまたはヘテロアリールであり;
R6は、アセチル、アルケニルまたはヘテロアリールであり;
R7およびR8は、同じかまたは異なって、水素、アルキル、アセチル、アルケニル、アリール、およびヘテロアリールからなる群より選択され;
Vは、酸素、窒素または硫黄であり;
そしてWは、H1、C1−10アルキル、アルケニル、ビニルアリール、またはヘテロアリールである]
で示される。
【0143】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(II):
【0144】
【化29】
で示される。
【0145】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(III):
【0146】
【化30】
[式中、R1およびR2は、それぞれ独立して、水素またはヒドロキシである;ただし、R1およびR2がともにヒドロキシであることはない]で示される。いくつかの実施形態において、R1は水素であり、そしてR2はヒドロキシである。いくつかの実施形態において、R1はヒドロキシであり、そしてR2は水素である。いくつかの実施形態において、R1およびR2は、ともに水素である。
【0147】
他の実施形態において、該SAH加水分解酵素の可逆的阻害剤は、式(IV):
【0148】
【化31】
[式中、R1は、NH2、SCH3、またはCH2NH2である]で示される。
【0149】
他の実施形態において、SAH加水分解酵素の可逆的阻害剤は、式(V):
【0150】
【化32】
[式中、R1は、NH2またはCONH2である]
で示される。
【0151】
なお別の実施形態において、SAH加水分解酵素の可逆的阻害は、式(VI):
【0152】
【化33】
[式中、Wは、Hまたはメトキシである]
で示される。
【0153】
D.薬学的組成物
本発明の薬学的組成物は、本発明の化合物(例えば、本明細書中に記載されるような、式I、IA、IBまたはII〜VIのいずれか1つの式で示される化合物)、およびその薬学的に受容可能な塩のいずれかを、単独かあるいは任意の薬学的に受容可能なキャリア、アジュバントまたはビヒクルと組み合わせて含む。本発明での使用に用いられ得る受容可能な組成物およびそれらの投与方法としては、米国特許第5,736,154号;同第6,197,801号;同第5,741,511号;同第5,886,039号;同第5,941,868号;同第6,258,374号および同第5,686,102号に記載されるものが挙げられるが、これらに限定されない。本発明の薬学的組成物において用いられ得る薬学的に受容可能なキャリア、アジュバントおよびビヒクルの例としては、イオン交換体、アルミナ、ステアリン酸アルミニウム、レシチン、ヒト血清アルブミンのような血清タンパク質、リン酸塩のような緩衝物質、グリシン、ソルビン酸、ソルビン酸カリウム、飽和植物性脂肪酸の部分グリセリド混合物、水、塩または電解質、例えば硫酸プロタミン、リン酸水素二ナトリウム、リン酸水素カリウム、塩化ナトリウム、亜鉛塩、コロイドシリカ、三ケイ酸マグネシウム、ポリビニルピロリドン、セルロース系物質、ポリエチレングリコール、カルボキシメチルセルロースナトリウム、ポリアクリレート、ワックス、ポリエチレン−ポリオキシプロピレン−ブロックポリマー、ポリエチレングリコールおよび羊毛脂が挙げられるが、これらに限定されない。
【0154】
処方、投薬量および投与経路は、当該分野で公知の方法に従って決定され得る(例えば、Remington:The Science and Practice of Pharmacy,Alfonso R.Gennaro(編者)Mack Publishing Company,1997年4月;Therapeutic Peptides and Proteins:Formulation,Processing,and Delivery Systems,Banga,1999;ならびにPharmaceutical Formulation Development of Peptides and Proteins,HovgaardおよびFrkjr(編),Taylor & Francis,Inc.,2000;Biopharmaceutical Drug Design and Development,Wu−PongおよびRojanasakul(編),Humana Press,1999を参照のこと)。SAH加水分解酵素の調節を必要とする状態の処置または予防において、適切な投薬レベルは、一般に、1日に体重1kg当たり約0.01〜500mgである。好ましくは、投薬レベルは、1日に約0.1〜約250mg/kgである。より好ましい実施形態において、投薬レベルは、1日に約0.1〜約20mg/kgの範囲である。適切な投薬量は、単回または複数回投与で投与され得る。任意の特定の被験体に対する特定の用量レベルおよび投薬の頻度は変動し得、そして種々の因子(例えば、用いられる特定の化合物の活性、その化合物の代謝安定性および作用の長さ、年齢、体重、全身的な健康状態、性別、食餌、投与の様式および時間、排泄速度、薬物の組み合わせ、特定の状態の重症度、ならびに治療を受ける患者)に依存することが理解される。
【0155】
本発明の薬学的組成物は、経口的に、非経口的に、吸入スプレーにより、局所的に、直腸内に、経鼻的に、口腔内に、経膣的に、埋め込みリザーバーを介して、または任意の適切な投与形態で投与され得る。本明細書中で使用される場合、用語「非経口」は、皮下、皮内、静脈内、筋肉内、関節内、滑膜内、胸骨内、くも膜下腔内、病巣内および頭蓋内の注射または注入技術を包含する。任意の所定の場合における最も適切な経路は、処置される状態の性質および重症度、ならびに用いられるSAH加水分解酵素阻害剤の性質に依存する。
【0156】
薬学的組成物は、滅菌注射用調製物の形態(例えば、滅菌注射用水性または油性懸濁液として)であり得る。この懸濁液は、適切な分散剤または湿潤剤(例えば、Tween 80)、および懸濁剤を用いて、当該分野で公知の技術に従って処方され得る。滅菌注射用調製物はまた、無毒の非経口的に受容可能な希釈剤または溶媒中の滅菌注射用溶液または懸濁液であってもよい。例えば、薬学的組成物は、1,3−ブタンジオール中の溶液であってもよい。本発明の組成物中に用いられ得る受容可能なビヒクルおよび溶媒の他の例としては、マンニトール、水、リンガー溶液および生理食塩液が挙げられるが、これらに限定されない。さらに、滅菌不揮発性油は、溶媒または懸濁媒体として慣習的に用いられている。この目的のために、合成モノグリセリドまたはジグリセリドを含めた、任意の無菌性の不揮発性油が用いられ得る。脂肪酸(例えば、オレイン酸)およびそのグリセリド誘導体は、特にポリオキシエチル化型の、天然の薬学的に受容可能な油(例えば、オリーブ油またはヒマシ油)と同様に、注射剤の調製に有用である。これらの油剤または懸濁液はまた、長鎖アルコール希釈剤または分散剤を含み得る。
【0157】
本発明の薬学的組成物は、任意の経口的に受容可能な剤形(カプセル剤、錠剤、ならびに水性懸濁液および溶液が挙げられるが、これらに限定されない)で、経口的に投与され得る。経口用途のための錠剤の場合、一般に使用されるキャリアとしては、ラクトースおよびコーンスターチが挙げられるが、これらに限定されない。潤滑剤(例えば、ステアリン酸マグネシウム)も添加され得る。カプセル形態での経口投与のために、有用な希釈剤としては、ラクトースおよび乾燥コーンスターチが挙げられる。水性懸濁液が経口的に投与される場合、活性成分は、乳化剤および懸濁剤と組み合わせられる。所望であれば、ある種の甘味剤、香料添加剤、および着色剤が添加され得る。
【0158】
本発明の薬学的組成物はまた、直腸投与のための坐薬の形態で投与されてもよい。これらの組成物は、本発明の化合物を適切な非刺激性の賦形剤と混合することによって、調製され得る。特定の実施形態において、賦形剤は、室温で固体であるが、直腸温では液体である。従って、この賦形剤は、直腸内で融解して、活性成分を放出する。このような材料としては、カカオバター、蜜蝋およびポリエチレングリコールが挙げられるが、これらに限定されない。
【0159】
本発明の薬学的組成物は、鼻エアロゾルまたは吸入によって投与されてもよい。このような組成物は、製剤処方の技術分野で周知の技術に従って調製される。例えば、このような組成物は、ベンジルアルコールまたは他の適切な防腐剤、バイオアベイラビリティーを向上させるための吸収促進剤、フルオロカーボン、および/あるいは当該分野で公知の他の可溶化剤または分散剤を用いて、生理食塩水中の溶液として調製され得る。
【0160】
本発明の薬学的組成物はまた、局所的に投与されてもよい。皮膚への局所適用のために、薬学的組成物は、キャリア中に懸濁または溶解された活性成分を含む、適切な軟膏と共に処方され得る。本発明の化合物の局所投与のためのキャリアとしては、ミネラルオイル、鉱油、白色ワセリン、プロピレングリコール、ポリオキシエチレンポリオキシプロピレン化合物、乳化蝋および水が挙げられるが、これらに限定されない。あるいは、薬学的組成物は、キャリア中に懸濁または溶解された活性化合物を含む、適切なローションまたはクリームと共に処方され得る。適切なキャリアとしては、ミネラルオイル、ソルビタンモノステアレート、ポリソルベート60、セチルエステルワックス、セタリルアルコール(cetaryl alcohol)、2−オクチルドデカノール、ベンジルアルコールおよび水が挙げられるが、これらに限定されない。本発明の薬学的組成物はまた、直腸坐薬処方によるかまたは適切な浣腸処方で、下部腸管に局所的に適用されてもよい。局所的経皮パッチもまた、本発明に包含される。
【0161】
他の実施形態において、本発明は、医薬としての使用および/または医薬の製造のための使用のいずれかに関連して、本明細書中に記載される方法のいずれかで用いるための本明細書中に記載されるSAHの可逆的阻害剤を含む組成物を提供する。
【0162】
本発明はまた、本発明の治療レジメンを実行するためのキットを提供する。このようなキットは、治療有効量のSAH加水分解酵素可逆的阻害剤(例えば、式I、IA、IBおよびII〜VIのいずれか1つの式で示される可逆的阻害剤)を、単独かまたは他の薬剤と組み合わせて、薬学的に受容可能な形態で含む。好ましい製薬形態は、滅菌生理食塩水、デキストロース溶液、緩衝溶液、または他の薬学的に受容可能な滅菌流体と組み合わせて阻害剤を含む。あるいは、組成物は、凍結乾燥または乾燥されていてもよい。この場合、キットは、注射用の溶液を形成するために、薬学的に受容可能な溶液(好ましくは滅菌したもの)をさらに含み得る。別の実施形態において、キットは、組成物を注射するための針またはシリンジ(好ましくは、滅菌形態でパッケージ化されたもの)をさらに含み得る。他の実施形態において、キットは、被験体に組成物を投与するための指示手段をさらに含み得る。この指示手段は、書面挿入物、録音テープ、視聴覚テープ、または被験体への組成物の投与を指示する任意の他の手段であり得る。
【0163】
関連する局面において、本発明は、上記のキットの内容を含む製品を提供する。例えば、本発明は、有効量のSAH加水分解酵素可逆的阻害剤(単独かまたは他の薬剤と組み合わせて)、および本明細書中に記載される疾患を処置するための使用を示す指示書を含む製品を提供する。
【0164】
E.SAH加水分解酵素活性を可逆的に阻害するための組み合わせ
本発明はまた、SAH加水分解酵素活性を可逆的に阻害するための組み合わせおよびキットを提供する。1つの実施形態において、本発明は、式I、IA、IBおよびH〜VIのいずれか1つの式で示される有効量の化合物、ならびに有効量の抗出血性のウイルス感染薬、免疫抑制剤、血漿ホモシステイン低下薬、または抗新生物薬を含む、組み合わせを提供する。この組み合わせは、薬学的に受容可能なキャリアまたは賦形剤をさらに含み得る。さらに別の局面において、本発明は、有効量の記載されるような組み合わせ、および該組み合わせを被験体に投与するための指示手段を含む、キットを提供する。
【0165】
ウイルス性の出血性疾患に関連する臨床症状または診断マーカーを軽減するかまたは寛解させ得る任意の薬剤は、本発明の組み合わせに用いられ得る。抗ウイルス治療剤としては、抗ウイルスワクチン、抗ウイルス抗生物質、ウイルスによって活性化された免疫細胞およびウイルスによって活性化された免疫血清が挙げられるが、これらに限定されない。WO00/64479は、本発明の組み合わせに用いられ得る、抗ウイルス治療剤の例を記載する。好ましい実施形態は、ブンヤウイルス科、フィロウイルス科、フラビウイルス科、またはアレナウイルス科ウイルスの感染によって引き起こされるウイルス性の出血性疾患に対する生物活性を示す、抗ウイルス治療剤である。
【0166】
身体の免疫系が疾患と戦う能力を抑制する任意の薬剤は、本発明の組み合わせに用いられ得る。免疫抑制剤の非限定的な例は、シクロスポリン、プレドニシロン(prednisilone)、アザチオプリン、タクロリムス、副腎皮質ステロイド、ミコフェノレート、シクロホスファミド、メトトレキサート、クロラムブチル、ビンクリスチン、ビンブラスチン、ダクチノマイシン、抗胸腺細胞グロブリン、ムロモナブ−CD3モノクローナル抗体、RhO(D)免疫グロブリン、メトキサレン、およびサリドマイドである(Goodman & Gilman’s The Pharmacological Basis of Therapeutics、(第9版)McGraw−Hill 1996、1294−1304頁を参照のこと)。免疫抑制剤は、薬物の組み合わせとして服用され得る。例えば、大抵の人は、薬物の組み合わせ(例えば、シクロスポリン、アザチオプリン、およびプレドニシロンの組み合わせ)を移植後に開始する。長い期間にわたって、各薬物の用量および服用される薬物の数は、拒絶のリスクが低下するにつれて減少され得る。
【0167】
ホモシステインレベルを低下させる任意の薬剤は、本発明の組み合わせに用いられ得る。葉酸は、有効なホモシステイン低下薬であることが知られている。他のホモシステイン低下薬としては、ベタイン、トリメチルグリシン、シアノコバラミン、および他のビタミンB群が挙げられるが、これらに限定されない。この組み合わせはまた、ホモシステインを低下させるのに用いられる任意の総合ビタミンおよびミネラル補充剤も含み得る。本発明の組み合わせに用いられ得る総合ビタミンおよびミネラル補充剤の例としては、米国特許第6,361,800号;同第6,353,003号;同第6,323,188号;同第6,274,170号;同第6,210,686号;同第6,203,818号;および同第5,668,173号に記載されるものが挙げられるが、これらに限定されない。
【0168】
任意の抗新生物薬が、本発明の組み合わせに用いられ得る。本発明の組成物および方法に用いられ得る抗新生物薬の例は、米国特許出願第2002/044919号に記載される。1つの実施形態において、用いられる抗新生物薬は、抗血管形成剤である。抗血管形成剤は、基底膜分解の阻害剤、細胞遊走の阻害剤、内皮細胞増殖の阻害剤、ならびに3次元組織化および効力の確立の阻害剤であり得る。このような抗血管形成剤の例は、AuerbachおよびAuerbach,Pharmacol.Ther.,63:265−311(1994);O’Reilly,Investigational New Drugs,15:5−13(1997);J.Nat’l Cancer Instit.,88:786−788(1996);ならびに米国特許第5,593,990号;同第5,629,327号および同第5,712,291号に示される。別の実施形態において、用いられる抗新生物薬は、アルキル化剤、代謝拮抗剤、天然物、プラチナ配位錯体、アントラセンジオン、置換尿素、メチルヒドラジン誘導体、副腎皮質抑制剤、ホルモン、およびアンタゴニストである。
【0169】
他の抗新生物薬としては、シチジン、アラビノシルアデニン(araC)、ダウノマイシン、ドキソルビシン、メトトレキサート(MTX)、5−フルオロウラシル(5−FU)などのフッ化ピリミジン、ヒドロキシ尿素、6−メルカプトプリン、ビンクリスチン(VCR)、VP−16およびビンブラスチン(VLB)などの植物アルカロイド、アルキル化剤、シスプラチン、ナイトロジェンマスタード、トリスアミン、プロカルバジン、ブレオマイシン、マイトマイシンC、アクチノマイシンD、またはL−アスパラギナーゼなどの酵素が挙げられるが、これらに限定されない。抗新生物薬はまた、癌抑制遺伝子抗体または抗癌遺伝子アンチセンスオリゴヌクレオチドのような癌遺伝子阻害剤であり得る。別の実施形態において、抗悪性腫瘍薬は、抗細胞マトリックス抗体または抗細胞マトリックスアンチセンスオリゴヌクレオチドのような細胞マトリックス阻害剤である。例えば、カベオリン−1、デコリン、カドヘリン、カテニン、インテグリン、および他の細胞マトリックスまたは細胞マトリックス遺伝子に対する抗体ならびにアンチセンスオリゴヌクレオチドが用いられ得る。
【0170】
特定の実施形態において、組み合わせは、腫瘍内治療および遺伝子治療の併用のための癌抑制遺伝子をさらに含む。遺伝子は、遺伝子送達系の成分として、裸のDNA、複合DNA、cDNA、プラスミドDNA、RNAまたはそれらの他の混合物の形態で用いられ得る。別の実施形態において、癌抑制遺伝子は、ウイルスベクター中に含まれる。遺伝子治療に適している任意のウイルスベクターが、組み合わせて用いられ得る。例えば、アデノウイルスベクター(米国特許第5,869,305号)、シミアンウイルスベクター(米国特許第5,962,274号)、条件付き複製可能型ヒト免疫不全ウイルスベクター(米国特許第5,888,767号)、レトロウイルス、SV40、単純ヘルペスウイルスアンプリコンベクターおよびワクシニアウイルスベクターが用いられ得る。さらに、遺伝子は、リポソーム(ここでは、脂質が、凝固時の酸化からDNAまたは他の生体材料を保護する)のような非ウイルスベクター系で送達され得る。
【実施例】
【0171】
F.実施例
実施例1
SAH加水分解酵素の可逆的阻害剤の合成
特に断りのない限り、1H(Me4Si)NMRスペクトルを、400MHzでCDCl3中、100.6MHzで13C(Me4Si)中の溶液を用いて決定した。質量スペクトル(MS)を、大気圧化学イオン化(APCI)技術によって得た。試薬等級の薬品を使用した。THF以外の溶媒は、アルゴン雰囲気下、CaH2で還流し、そしてCaH2から蒸留することによって乾燥させ、THFは、ベンゾフェノンおよびカリウムから蒸留した。TLCは、MeOH/CHCl3(1:9)およびEtOAc/MeOH(95:5)を展開系として用いてMerck kieselgel 60−F254上で行い、生成物を254nm光で検出した。Merck kieselgel 60(230−400メッシュ)を、カラムクロマトグラフィーに用いた。
【0172】
元素分析は、Galbraith Laboratories,Knoxville,TNで決定した。化合物を単離するためのスペクトルデータは、報告されたデータと一致した。(Holy,Coll.Czech.Chem.Commun.43,3444−3464(1978);Holyら、Coll.Czech.Chem.Commun.50:262−279(1985);日本特許第69−50781号;Chem.Abstr.1972:514811)。この合成を、スキーム2に概略的に示す。
【0173】
9−(3,4−O−イソプロピリデン−3,4−ジヒドロキシブチル)アデニン(l)
【0174】
【化34】
9−(3,4−ジヒドロキシブチル)アデニン(2)
CF3COOH/H2O(9:1)(5ml)中の1(110mg、0.18mmol)の溶液を、約0℃で20分間攪拌した。揮発物を蒸発させ、トルエン(3×)およびEtOH(2×)と同時蒸発させて、EtOHからの結晶化後に報告されたスペクトルデータを示す2(73mg、78%)を得た。
【0175】
9−(4−O−t−ブチルジメチルシリル−3,4−ジヒドロキシブチル)アデニン(3)
TBDMS−Cl(186mg、1.23mmol)およびイミダゾール(168mg、2.46mmol)を、無水DMF(8mL)中の2(250mg、1.12mmol)の攪拌溶液に添加した。混合物を周囲温度で5時間攪拌し、次いで、この反応混合物をEtOAc/NH4Cl/H2Oの間で分配した。水層を隣の部分のEtOAcで抽出した。合わせた有機相を洗浄し(ブライン)、乾燥し(Na2SO4)、蒸発させ、そして残渣をカラムクロマトグラフィー(CHCl3/MeOH;97:3)に付して、3(234mg、62%)を得た:
【0176】
【化35】
9−[4−O−t−ブチルジメチルシリル−3−O−(1−エトキシエチル)−3,4−ジヒドロキシブチル]アデニン(4)
エチルビニルエーテル(214mg、0.28mL、2.96mmol)およびp−トルエンスルホン酸ピリジニウム(15mg、mmol)を無水CH2Cl2(30mL)中の3(250mg、0.74mmol)の溶液に添加し、この混合物を周囲温度にてN2下で、出発物質がTLCで検出されなくなるまで(通常5〜6日間)攪拌した。次いで、反応混合物を水で洗浄し、乾燥し(Na2SO4)、そして蒸発させた。カラムクロマトグラフィー(EtOAc/MeOH;97:3)により、4(160mg、53%)をジアステレオ異性体の約1:1混合物として得た:
【0177】
【化36】
9−[3−O−(1−エトキシエチル)−3,4−ジヒドロキシブチル]アデニン(5)−手順A
TBAF/THF(0.88mL、1M)を、無水THF(6mL)中の4(180mg、0.44mmol)の溶液に添加し、この混合物を周囲温度で20分間攪拌した。揮発物を蒸発させ、残渣をカラムクロマトグラフィー(EtOAc/MeOH;78:12)に付して、5(120mg、92%)をジアステレオ異性体の約1:1混合物として得た:
【0178】
【化37】
4−(アデニン−9−イル)−2−ヒドロキシ酪酸メチル(6)−手順B
CH3CN/CCl4/H2O(1:1:1.5;1.5mL)中の5(90mg、0.31mmol)の懸濁液に、NaHCO3(161mg、0.88mmol)、NaIO4(353mg、1.65mmol)およびRuCl3(微量)を添加した。この混合物を、出発物質がTLCで検出されなくなるまで、周囲温度で48時間攪拌した。次いで、水(5mL)およびCHCl3(4mL)を添加し、これらの2つの相を分離し、そして水相をCHCl3(3mL)で洗浄した。水層をpHが約4になるまでHClで酸性化し、Dowex 50W×2(H+)のカラムにアプライした。カラムを200mLの水で洗浄し、次いで、生成物を2.5%NH4OH/H2Oで溶出した。合わせたUV吸収性のアンモニア溶出物を蒸発させ、そしてMeOH(2×)と同時蒸発させた。残渣をMeOH(5mL)中に溶解し、ジエチルエーテル中のCH2N2の溶液を、ジアゾメタンの黄色が数分間維持されるまで添加した。この溶液を濃縮し、カラムクロマトグラフィー(CHCl3/MeOH;95:5)に付して、6(DZ2002)(31mg、41%)を報告されたものと同一のデータを示す白色固体として得た。副産物6’の生成を回避するために、反応混合物中に所望の量のNaHCO3を維持することが重要である。
【0179】
4−(アデニン−9−イル)−2−ヒドロキシブタン酸(7)−手順C
NaOH/H2O(1mL、0.1M)を、MeOH/H2O(2.0mL)中の6(10mg、0.04mmol)の溶液に添加した。この混合物を、出発物質がTLCで検出されなくなるまで、周囲温度で6時間攪拌した。次いで、反応混合物を、pHが約4になるまでHClで酸性化し、Dowex 50W×2(H+)のカラムにアプライした。カラムを水(100mL)で洗浄し、次いで、生成物を2.5%NH4OHで溶出した。合わせたUV吸収性のアンモニア溶出物を蒸発させて、7を、報告されたデータを示すアンモニウム塩(7.6mg、75%)として得た。
【0180】
9−(3,4−O−ジ−t−ブチルジメチルシリル−3,4−ジヒドロキシブチル)アデニン(8)
TBDMS−Cl(593mg、3.92mmol)およびイミダゾール(534mg、7.85mmol)を、無水DMF(8mL)中の2(350mg、1.57mmol)の攪拌溶液に添加し、この混合物を周囲温度で一晩攪拌した。次いで、反応混合物をEtOAc/NH4Cl/H2Oの間で分配した。水層をEtOAcで抽出した。合わせた有機相を洗浄し(ブライン)、乾燥し(Na2SO4)、そして蒸発させた。カラムクロマトグラフィー(CHCl3→3%MeOH/CHCl3)により、8(610mg、86%)を得た:
【0181】
【化38】
9−(3−O−t−ブチルジメチルシリル−3,4−ジヒドロキシブチル)アデニン(9)
化合物8(400mg、0.887mmol)を、CH3CO2H/H2O/THF(13:7:3;8mL)の溶液に添加し、この混合物を、化合物2のより極性の高いスポットがTLC上に出現し始めるまで、周囲温度で攪拌した。次いで、反応混合物を分配し(EtOAc//NaHCO3/H2O)、そして水層を隣の部分のEtOAcで抽出した。合わせた有機相を洗浄し(NaHCO3、ブライン)、乾燥し(Na2SO4)、蒸発させ、そしてカラムクロマトグラフィーに付して(CHCl3→4%MeOH/CHCl3)、回収した8(140mg、35%)および9(155mg、52%)を得た:
【0182】
【化39】
3−(アデニン−9−イル)プロピオン酸メチル(6’)
9(50mg、0.148mmol)を手順B(カラムクロマトグラフィー:CHCl3/MeOH 97:3)によって処理することにより、報告されたデータを示す6’(9mg、27%)を得た。
【0183】
3−(アデニン−9−イル)プロピオン酸(7’)
6’(10mg、0.045mmol)を手順C(カラムクロマトグラフィー:CHCl3/MeOH 97:3)によって処理することにより、報告されたデータを示す6’(7.3mg、78%)を得た。
【0184】
スキーム2
【0185】
【化40】
a(a)CF3COOH/H2O;(b)TBDMSCl/イミダゾール/DMF;(c)エチルビニルエーテル/CH2Cl2/(p)CH3C6H4SO3H−C5H10N;(d)TBAF/THF;(e)(i)NaIO4/NaHCO3/RuCl3/CH3CN/CCl4/H2O、(ii)CH2N2MeOH;(f)CH3CO2H/H2O/THF;(g)NaOH/H2O/MeOH。
【0186】
実施例2
DZ2002は免疫抑制作用を媒介する
材料および方法
試薬
AdoHcy加水分解酵素阻害剤DZ2002(スキーム2中の化合物6)は、Diazyme Laboratoriesで合成された。Con A(Concanavalin A)、LPS(Escherichia coli 055:B5)およびSac(Staphylococcus aureus Cowan strain1)は、Pansorbin(登録商標)cells,Biosciences,inc.(La Jolla.,CA92039,USA)から入手した。RPMI1640およびウシ胎仔血清(FBS)は、GIBCOから入手した。精製ラット抗マウスIL−10、IL−12p70、IL−12p40、IFN−γおよびビオチン標識抗マウスIL−10、IL−12p70、IL−12p40、IFN−γ、FITC−抗マウス−CD11b(Mac−1)、Phycorythrin(PE)−抗マウスI−Ad、PE−抗ヒト−CD14、PE−抗ヒト−ABC、PE−抗ヒト−DR、PE−抗ヒト−CD80およびPE−抗ヒト−CD86は、Pharmingen製品であった。チオグリコレート(TG)は、Sigma−Aldrichから入手可能である。
【0187】
動物
近交系BALB/Cマウス(6〜8週齢)を、認可番号99−003で、中国科学院上海実験動物センター(Shanghai Experimental Animal Center of Chinese Academy of Sciences)により提供された。これらのマウスを、室温24±2℃、12時間の明/暗サイクルで、特定病原体感染防止(SPF)条件に収容し、滅菌飼料および水を自由に与えた。
【0188】
細胞
Balb/cマウスから脾臓を無菌的に切除し、プールし、そしてPBS中の単細胞懸濁液を調製した。赤血球を、トリス緩衝化塩化アンモニウム(0.155M NH4CL、0.0165Mトリス、PH7.2)で処理することによって溶解した。単核細胞をPBSで洗浄し、そしてベンジルペニシリン100000U・L−1およびストレプトマイシン100mg・L−1を補充したRPMI−1640培地中に再懸濁した。細胞の生存度および濃度を、トリパンブルー排除により決定した。
【0189】
腹腔滲出細胞を、0.5mlの3%TGの腹膜腔内注射によってBALB/Cマウスに誘導した。4日後に、腹膜滲出細胞を滅菌洗浄によって収集した。
【0190】
THP−1(American Type Culture Collection,Manassas,VA)は、ヒト単球性白血病である。THP−1細胞を、10%FBSを補充したRPMI1640培地中の懸濁培養で維持した。培養を、空気中5%CO2の加湿雰囲気下、37℃で維持し、5〜6日ごとに1/10希釈で継代培養した。
【0191】
脾臓リンパ球への[3H]−チミジン取込み
マウス脾臓リンパ球を、10%FBSを補充したRPMI1640中で、インビトロで培養した。細胞を、種々の濃度のDZ2002の存在下または非存在下で、5μg/mlのCon Aまたは10μg/mlのLPSとともに、37℃で48時間、加湿CO2インキュベーター中、1×105細胞/200μl/ウェルにて96ウェルプレート中でインキュベートした。40時間のインキュベーションの後、細胞を0.5μCi/ウェルの[3H]−チミジンでパルスし、さらに8時間培養した。次いで、細胞をガラス繊維フィルター上に収集し、取り込まれた放射能をBeta Scintillator(MicroBeta Trilux,PerkinElmer Life Sciences)を用いてカウントした。
【0192】
脾臓リンパ球のMTTアッセイ
細胞傷害性をMTTアッセイで評価した。マウス脾臓リンパ球を、種々の濃度のDZ2002の存在下または非存在下で、37℃で48時間、加湿CO2インキュベーター中、9×104細胞/180μl/ウェルにて96ウェルプレート中でインキュベートした。培養終了前に15μlの5mg/mlのMTTを4時間パルスし(合計190μl)、次いで、80μlの溶媒(10%SDS、50%N、N−ジメチルホルムアミド、PH7.2)を添加した。7時間インキュベートし、マイクロプレートリーダー(Bio−rad
Model 550 Japan)でOD590を読み取る。
【0193】
サイトカイン産生
マウスの脾臓単核球(5×106)を、種々の濃度のDZ2002の存在下または非存在下、Sac(1:10000)、ConA(5ug/ml)またはLPS(10ug/ml)の存在下で、24ウェルプレート中、2ml/ウェルの容量で培養した。24時間後、無細胞の上清を収集し、−20℃で凍結した。IL−12p40、IL−12p70、IL−10およびTNF−αの濃度を、マウスのサイトカインに特異的なELISAで決定した。
【0194】
マウスの腹腔滲出細胞(6.25×105)を、24ウェルプレート中、1ml/ウェルの容量で2時間培養した。接着細胞を氷冷RPMI1640で洗浄し、そして接着細胞を、種々の濃度のDZ2002の存在下または非存在下、IFN−γ(2.5ng/ml)およびLPS(1μg/ml)の存在下で、2ml/ウェルの容量で培養した。24時間後、無細胞の上清を収集し、−20℃で凍結した。IL−12p40、IL−12p70、IL−10およびTNF−αの濃度を、ELISAで決定した。
【0195】
THP−1細胞(6×105)を、1.2%の存在下および種々の濃度のDZ2002の存在下または非存在下で、24ウェルプレート中、2ml/ウェルの容量で培養した。24時間後にIFN−γ(500U/ml)を添加し、さらに16時間後にLPS(1μg/ml)を添加した。無細胞の上清を24時間後に収集し、−20℃で凍結した。IL−12p40、IL−12p70、IL−10およびTNF−αの濃度を、ELISAで決定した。
【0196】
ヒツジ赤血球の定量的溶血(QHS)アッセイ
雌性Babl/cマウスを、4日目に0.2mlの16.7%のSRBCを腹腔内注射することによって免疫した。ビヒクル、デキサメタゾンおよびDZ2002を、1〜7日目の7日間連続して、腹腔内注射によって各群(n=6)に投与した。8日目に、マウスを屠殺して、2×106細胞/mlの脾臓細胞の混合懸濁液を作製した。1mlの細胞懸濁液を、1mlの0.5%SRBCおよび1mlのモルモット補体の1:10希釈物とともに、37℃で1時間インキュベートし、次いで、遠心分離し(3分、3000g)、そして一部改変したSimpsonら、J.Immunol.Methods.,21(1−2):159−65.(1978)に従って、413nmで上清の溶血を決定した。各群を3連で行った。
【0197】
混合リンパ球反応(MLR)増殖アッセイ
Balb/cマウス脾臓細胞を107細胞/mlの懸濁液に調製し、50μg/mlのマイトマイシンとともに2時間培養した。次いで、細胞を洗浄し、新鮮なC57/B6マウス脾細胞とともに、等しく最終濃度1.0×10細胞/mlで、種々の濃度のDZ2002の存在下または非存在下で培養した。48時間インキュベーションの後、細胞を0.5μCi/ウェルの[3H]−チミジンでパルスし、さらに24時間培養した。次いで、細胞をガラス繊維フィルター上に収集し、取り込まれた放射能をBeta Scintillator(MicroBeta Trilux,PerkinElmer Life
Sciences)を用いてカウントした。
【0198】
DNFB誘導性の遅延型過敏症(DTH)応答
雌性Balb/cマウスを、0日目および1日目に各後足に、20μlのアセトン−オリーブ油(4:1)中に溶解された0.6%DNFBで感作した。7日目にマウスを、一部改変したPhanuphak(1974)に従う方法で、左耳の両側に10μlの0.5%DNFBで攻撃した。ビヒクル、CsA、およびDZ2002(1、3、10mg/kg)を、攻撃の1時間前および12時間後、24時間後に、腹腔内注射によって各群(n=10)に投与した。耳介腫脹を、攻撃の30時間後に特定の8mmパンチで作製した左右の耳パッチの重量差として表した。
【0199】
フローサイトメトリー
マウスの腹腔滲出細胞またはTHP−1細胞を、冷PBS(0.1%NaN3、1%FBSを含む染色緩衝液(PH7.2))中で洗浄した。細胞を、冷染色緩衝液中に2.0×107/mlで最懸濁した。最適濃度の各蛍光色素標識抗体を、50μLの細胞に添加した。Fcレセプターを、10μL正常マウス血清を用いてブロックした。細胞を暗所で4℃にて30分間インキュベートし、2.0mLの染色緩衝液で2回洗浄し、そして0.5mLのPBS(PH7.2)中に再懸濁した。細胞を4℃で暗所に保存し、FACScanフローサイトメーター(Becton Dickinson,San Jose,CA)で分析した。データをCellQuest(商標)ソフトウェア(Becton Dickinson,San Jose,CA)を用いて分析した。
【0200】
統計分析
結果をx±sとして表し、独立両側t検定を行って、0.05未満のP値を有意であるとみなした。各実験を少なくとも3回繰り返した。
【0201】
結果
AdoHcy加水分解酵素阻害剤による脾臓リンパ球への[3H]−チミジン取込みの阻害
培養の48時間後、DZ2002(0.1〜10μmol・L−1)は、ConAによって誘導されるリンパ球増殖に対して影響を及ぼさない。DZ2002(10μmol・L−1)は、LPSによって誘導されるリンパ球増殖を阻害した。
【0202】
Sac刺激マウス脾細胞からのIL−10、IL−12P40およびIL−12P70産生に対するDZ2002の影響
Sac刺激は、休止脾細胞と比較して、マウス脾細胞からのIL−10、IL−12P40およびIFN−γ産生の著しい増加を誘導した。DZ2002(μmol・L−1)は、IL−12P40、IL/−12P70およびTNF−αの放出を用量依存的に阻害した(データは示さず)。
【0203】
ヒツジ赤血球の定量的溶血(QHS)アッセイに対するDZ2002の影響
SRBCの定量的溶血は、抗原刺激に対する応答における一次抗体産生のモデルである。図3に示すように、5mg/kgのデキサメタゾンのQHS阻害が38.1%であるのと比較して、DZ2002の連続7日間の腹腔内注射により、0.08mg/kgおよび2mg/kgの用量で、それぞれQHSを24.5%および18.4%阻害した(ビヒクルコントロール群と比較してすべての実験群についてp<0.05)(図1)。
【0204】
DZ2002は混合リンパ球反応におけるT細胞増殖を抑制する
マイトマイシン処理Balb/c(H−2d)脾臓細胞を、C57BL/6(H−2b)脾臓細胞増殖に対する同種刺激因子として適用した。DZ2002は、それぞれ0.1、1および10μmol/Lの用量で3日間培養して、MLRに対する強力な抑制(40.2%、36.9%および42.3%)を示した。(図2)。
【0205】
DZ2002は、脾臓細胞において細胞傷害性を有さない
2日間の培養において、0.1〜10μmol/LのDZ2002は、脾臓細胞に対する細胞傷害性を示さなかった。DZ2002とともにインキュベートした細胞のOD値は、コントロールのOD値と差異がなかった。(図3)。
【0206】
DZ2002は、エタノール消費によって誘導されるマウスDTH応答の抑制を逆転させた
各群につき9匹のマウスを準備した。マウスを、0日目および1日目に各後足に、無水アセトン/オリーブ油(4:1)中の0.5%DNFB溶液(20μl)で感作した。初回感作の5日後に、マウスを、軽いメトファン(Metofane)麻酔下で左耳の両側に0.2%DNFB(10μl)で攻撃した。右耳をビヒクル単独で処理した。DNFB攻撃の1時間前に、DZ2002をマウスに経口投与した。耳介腫脹度を、イヤーパンチャーおよび重量(mg)を測定するために分析天秤を用いて、攻撃の24時間後に測定した。結果を、左右の耳の重量差として表した。耳介腫脹の測定後に、各群の4匹のマウスから脾臓を取り出し、分析まで凍結した。
【0207】
常在腹膜細胞およびTG誘導性腹膜細胞におけるMHC−IIの発現に対するDZ2002の影響
腹腔マクロファージによるMHC−II発現を、培地単独かまたは100U/mlのIFN−γとともに48時間インキュベーションした後に評価した。0.1および1μmol/LのDZ2002の存在下でIFN−γとともにインキュベートした細胞は、常在腹膜細胞のMHC−II発現のレベルを高め、そして10μmol/LのDZ2002は、MHC−II発現のレベルを低下させる。(データは示さず)。TG誘導性の腹膜細胞におけるように、1および10μmol/LのDZ2002は、培地単独でインキュベートする場合、Mac−1+パーセントを低下させる。そして、IFN−γとともにインキュベートした細胞のMHC−II発現のレベルは、0.1〜10μmol/LのDZ2002の存在下で用量依存的に低下した。(データは示さず)。
【0208】
TG誘導性の腹膜細胞からのIL−10、IL−12P40およびTNF−α産生に対するDZ2002の影響
腹腔マクロファージによって産生されたサイトカインを、25U/mlのIFN−γおよび1μg/mlのLPSとともに24時間インキュベーションした後に評価した。常在腹膜細胞は、IFN−γおよびLPSとともにインキュベートすると、一部のIL−10を除く低レベルのサイトカインを産生する(データは示さず)。TG誘導性の腹腔マクロファージについては、DZ2002は、0.1〜10μmol/Lの用量でIL−12P40およびTNF−αの放出を阻害したが、IL−10産生に対して影響を及ぼさない(図5)。
【0209】
DZ2002は、THP−1細胞におけるMHC−II、CD80およびCD86の発現を阻害する
THP−1細胞によるMHC−II、CD80およびCD86発現は、培地単独かまたは100U/mlのIFN−γとともに48時間インキュベーションした後に評価した。10μmol/LのDZ2002の存在下でIFN−γとともにインキュベートした細胞は、THP−1細胞のMHC−II発現のレベルを穏やかに低下させ、そして0.1〜10μmol/LのDZ2002は、用量依存的な方法でCD80およびCD86発現のレベルを低下させる。(図6A−C)。
【0210】
THP−1細胞からのIL−10、IL−12P40およびTNF−α産生に対するDZ2002の影響
THP−1細胞によって産生されたサイトカインを、500U/mlのIFN−γおよび1μg/mlのLPSとともに24時間インキュベーションした後に評価した。図7に示すように、DZ2002は、0.1〜10μmol/Lの用量で、IL−12P40およびTNF−αの放出を阻害した。
【0211】
実施例3
DZ2002は、遅延型過敏症(DTH)反応を軽減した
図8に示すように、DZ2002は、遅延型過敏症(DTH)反応を用量依存的な様式で有意に軽減した。BALB/cマウスを、0日目および1日目にDNFB(ジフルオロニトロベンゼン)に初回感作させ、次いで9日目にDNFBで再度攻撃した。DZ2002を、9日目の攻撃の1時間前または24時間後に、i.p.(腹腔内)投与した。耳介腫脹を、攻撃の40時間後に特定の8mmパンチによって決定した。データを平均±SDとして表す。
【0212】
実施例4
DZ2002は、MBPで刺激した脾細胞からのIL−10産生を維持または増大させた
図9に示すように、DZ2002は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−10産生を維持または増大させた。SJL/Jマウスを0日目にMBPで免疫し、次いで2日目に百日咳毒素を投与した。DZ2002をマウス腹腔内に14日目から開始して毎日投与した。脾細胞を21日目に採取し、そしてMBPとともに培養した。培養上清をIL−10についてELISAで分析した。データを平均±SDとして表す。
【0213】
実施例5
DZ2002は、MBPで刺激した脾細胞からのIL−2産生を阻害した
図10に示すように、DZ2002は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIL−2産生を阻害した。SJL/Jマウスを0日目にMBPで免疫し、次いで2日目に百日咳毒素を投与した。DZ2002をマウス腹腔内に14日目から開始して毎日投与した。脾細胞を21日目に採取し、そしてMBPとともに培養した。培養上清をIL−2についてELISAで分析した。データを平均±SDとして表す。
【0214】
実施例6
DZ2002は、MBPで刺激した脾細胞からのIFN−γ産生を阻害した
図11に示すように、DZ2002は、ミエリン塩基性タンパク質(MBP)で刺激した脾細胞からのIFN−γ産生を阻害した。SJL/Jマウスを0日目にMBPで免疫し、次いで2日目に百日咳毒素を投与した。DZ2002をマウス腹腔内に14日目から開始して毎日投与した。脾細胞を21日目に採取し、そしてMBPとともに培養した。培養上清をIFN−γについてELISAで分析した。データを平均±SDとして表す。
【0215】
上記の実施例は、単に例示の目的で挙げられるものであり、本発明の範囲を限定することを意図するものではない。上記の実施例に対する多様な変形が可能である。上記の実施例に対する改変および変形が当業者に明らかであるので、本発明は、添付の特許請求の範囲によってのみ限定されることが意図される。
【特許請求の範囲】
【請求項1】
本願明細書に記載された発明。
【請求項1】
本願明細書に記載された発明。
【図1】

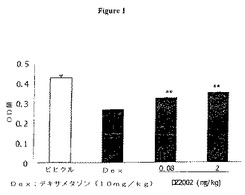
【図2】
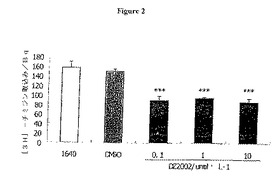
【図3】
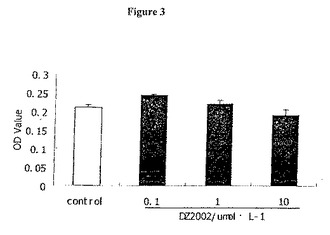
【図4】
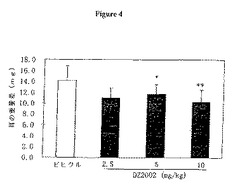
【図5】
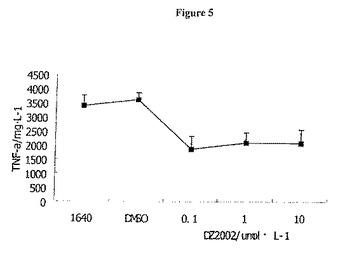
【図6A】
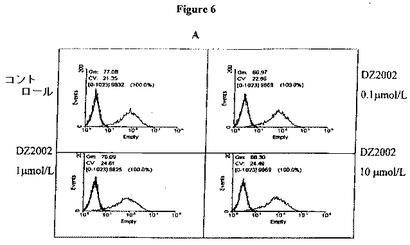
【図6B】
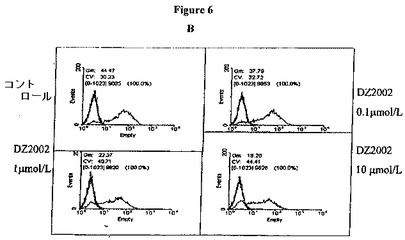
【図6C】
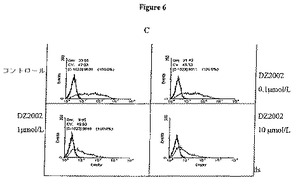
【図7】
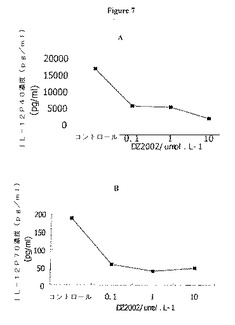
【図8】
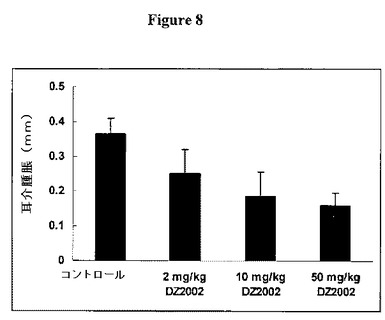
【図9】
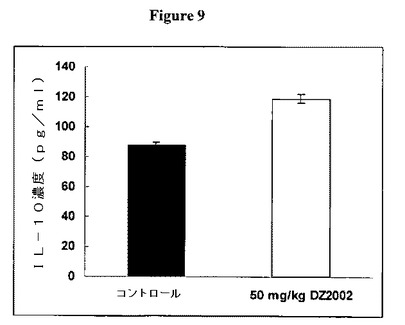
【図10】
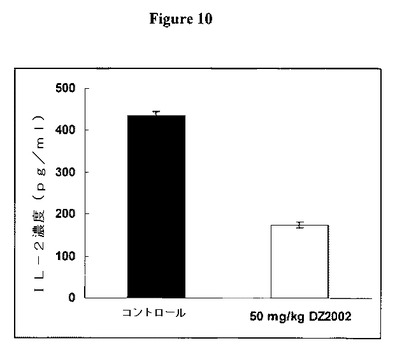
【図11】
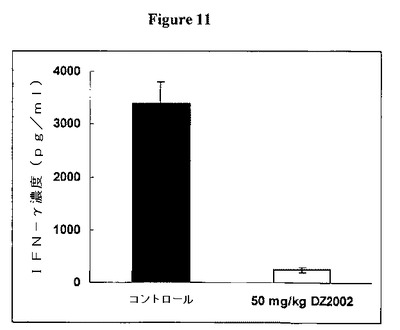
【図2】
【図3】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図7】
【図8】
【図9】
【図10】
【図11】
【公開番号】特開2012−197314(P2012−197314A)
【公開日】平成24年10月18日(2012.10.18)
【国際特許分類】
【外国語出願】
【出願番号】特願2012−153898(P2012−153898)
【出願日】平成24年7月9日(2012.7.9)
【分割の表示】特願2007−536899(P2007−536899)の分割
【原出願日】平成17年10月13日(2005.10.13)
【出願人】(506017089)
【Fターム(参考)】
【公開日】平成24年10月18日(2012.10.18)
【国際特許分類】
【出願番号】特願2012−153898(P2012−153898)
【出願日】平成24年7月9日(2012.7.9)
【分割の表示】特願2007−536899(P2007−536899)の分割
【原出願日】平成17年10月13日(2005.10.13)
【出願人】(506017089)
【Fターム(参考)】
[ Back to top ]