
S1Pの使用
医薬化合物を製造するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、S1P(スフィンゴシン−1−リン酸)の使用に関する。本発明の他の形態は、医薬のスクリーニング方法、および、痛みを治療する方法に関する。
【背景技術】
【0002】
痛みは、実際の、または潜在的な組織のダメージに反応して起こる複雑で主観的な感覚であり、それに対する感情的な応答である。急性の痛みは、潜在的な、または実際の外傷を示す生理学的なシグナルである。慢性の痛みは、体性性(器質性)または心因性のいずれかであり得る。慢性の痛みはしばしば、自律神経性の徴候を伴うか、または、その後に自律神経性の徴候が続くため、場合によっては、うつ状態に陥る。
【0003】
体性性の痛みは、侵害受容性、炎症性、または、神経障害性によるものと考えられる。侵害受容性疼痛は、体性または内臓の痛みを感じる神経線維の継続的な活性化と同じと判断されている。神経障害性の痛みは、神経系の機能障害から生じる;末梢神経系、CNS、または、その両方における異常な体性感覚のプロセスによって持続すると考えられている。(痛みのメカニズムの総論については、例えば、ScholzおよびWoolf,2002年;JuliusおよびBasbaum,2001年,WoolfおよびMannion,1999年;Wood,J.D.,2000年;WoolfおよびSalter,2000年を参照)。
【0004】
慢性の痛みは、個人が被る、加えて社会的、経済的な途方もない費用を発生させる。現存する薬理学的な痛みの治療は、有効性と安全性の両方に関してはなはだ満足のいくものではない。
【0005】
これまで、痛みの治療には、2つのクラスの鎮痛薬が主として用いられている:非オピオイド鎮痛薬、大部分がアセトアミノフェンとNSAID(非ステロイド系抗炎症薬)、および、オピオイド(麻酔性)アゴニスト(ここにおいて、「オピオイド」は、CNS中の特異的なオピオイド受容体に結合し、アゴニスト作用を生じさせる天然または合成物質の一般名称である)。遺憾ながら、両方の鎮痛薬クラスであるオピオイドおよび非オピオイドは、いくつかの不要な副作用がある。オピオイドの最も重篤な副作用は、呼吸器系を阻害する可能性と、長期の治療の後に依存症になる可能性である(Schaible H.G.,Vanegas H.,2000年)。一方で、非オピオイドの主要なクラスであるNSAIDは、多種多様な潰瘍や出血のような胃腸の合併症、加えて腎臓障害を誘導する可能性がある(Schaible H.G.,Vanegas H.;2000年)。米国では毎年、従来のNSAIDによって引き起こされる重度の胃腸の合併症のために約16,000人の患者が死亡していると推測されている。
【発明の開示】
【発明が解決しようとする課題】
【0006】
最先端の痛みの治療に関連する深刻な欠点を考慮すると、痛みを調節する薬物の新規のクラスの多大な必要性がある。特別には、急速に進む痛みの神経生物学の理解と、最先端の治療の欠点がない有効な治療を提供するという未だ満たされていない臨床上の必要性との間にある大きなギャップを考慮すると、新規の鎮痛薬のクラスに関する新しい標的の発見に努力を向ける必要がある。従って、本発明の目的は、痛みを調節する薬物の新しいクラスを開発し、提供する新しい手段を提供することである。
【0007】
この目的は、痛みを調節する医薬化合物を製造するための、S1Pまたはそれらの機能
的フラグメントもしくは誘導体の使用によって解決される。
【課題を解決するための手段】
【0008】
本発明は、侵害受容過程におけるS1Pの関連と、その痛みを減少させる能力を初めて実証した発明者等の発見に基づく。S1Pに関して機能的な、という用語、または、S1Pの機能という用語は、S1Pが、その受容体の少なくとも1種と相互作用する能力、好ましくはその受容体を活性化する能力、または、細胞内のcAMPレベルを低める能力、または、PAM(Myc関連タンパク質)の小胞体から細胞膜へのトランスロケーションを媒介する能力を意味し;より好ましくは、S1Pの機能という用語は、そのPAM活性を増強する能力(すなわち、PAMが、ACと相互作用する能力、および/または、AC活性を低める能力、および/または、痛みを減少させる能力)、および/または、AC活性を阻害する能力、さらにより好ましくはその痛みを減少させる能力を意味する。S1P受容体に関して機能的という用語、または、S1P受容体の機能という用語は、S1P受容体が、S1Pと相互作用する能力、より特定には、S1Pの相互作用によって発生する前記受容体に典型的なシグナルを媒介する能力、さらに特定には、S1Pの相互作用によって発生する痛みのプロセシングに影響を与える能力を意味する。
【0009】
S1Pのフラグメントは、図15に記載の野生型分子より小さいあらゆるフラグメントであり得る。PAMのフラグメントは、対応する野生型より短いあらゆるポリペプチド、または、ポリヌクレオチド断片であり得る。S1P受容体のフラグメントは、対応する野生型より短いあらゆるポリペプチド、または、ポリヌクレオチド断片であり得る。
【0010】
S1PまたはS1Pフラグメントの誘導体は、S1Pの機能を有する分子のあらゆる改変、または、化学的もしくは生物学的な改変のようなその他のあらゆる種類の改変を含んでもよく、このような改変としては、例えば、分子を安定化すること、または、それらの例えば所定の細胞への特異的なターゲティングを調節すること、または、その細胞への進入もしくは細胞による摂取を容易にすることが挙げられ;既知の改変の一つとしては、S1Pの水酸化またはメチル化が挙げられる。それらの必要な部位へのターゲティングと細胞への進入を確実にする、または、容易にするのに適した改変または添加剤が有用である。一方で、それらの脊髄へのターゲティングを確実にするために、適切なカテーテルなどを用いた脊髄内への適用のような局所的な適用なども考えられる。その他の有用な添加剤としては、それらの安定化等のための、塩(生理学的に許容できる有機または無機塩のための塩、例えば、Remington's Pharmaceutical Sciences,1418頁,1985年を参照)、緩衝液などが挙げられる。
【0011】
S1Pは、特異的な受容体を介して細胞に吸収されるため、外部からの適用が可能であり、特異的に吸収されると予想される。S1Pターゲティングの調節は、例えば、S1P受容体をクローニングし、所望の細胞で発現させることにより実現することができる。細胞型に特異的な発現は、適切な遺伝子のプロモーター/エンハンサーを用いて確実にすることができ、これらは、当業界で既知である。
【0012】
本発明は、発明者等の脊髄および後根神経節(DRG)内での感作メカニズムにおけるS1Pの驚くべき関連を初めて実証した研究に基づく。
【0013】
S1P(スフィンゴシン−1−リン酸)は、スフィンゴシンのリン酸化された誘導体であり、全てのスフィンゴ脂質の構造上の主鎖である。この細胞外(血清介在)スフィンゴ脂質は、異なる組織で差異的に発現され、それぞれ特異的な細胞の作用を調節するS1P1〜S1P5という5種の特異的なGタンパク質共役受容体(GPCR)の一つに結合することによって多種多様な細胞性プロセスを調節することがわかっている(総論のために、例えばPayne等,2002年;および、SpiegelおよびMilstien,2
000年を参照)。細胞外S1Pの既知の機能としては、例えば、細胞の移動、細胞の生存または血管新生の調節が挙げられる。その細胞外の作用のほかに、細胞内メッセンジャーとして作用することも知られている(総論のために、例えばPayne等,2002年;および、SpiegelおよびMilstien,2000年を参照)。しかしながら、その侵害受容プロセスにおける関連は未だわかっていない。
【0014】
PAM(Myc関連タンパク質)は、510kDaの巨大タンパク質である。PAMのタンパク質、ゲノムおよびコードポリヌクレオチド配列は当業界で既知であり、例えば、NCBI(National Centre for Biotechnology Information;National Library of Medicine,Building 38A,ベセスダ,メリーランド州20894,米国;www.ncbi.nhm.nih.gov)データベースより、登録番号AAC39928(コード配列;配列番号1)、AF075587(タンパク質配列;配列番号2)で公けに利用可能である。ヒトPAMは、染色体13q22上に位置する;そのゲノム配列は、NT_024524.11(始点:24679861位;終点:24962245位;配列番号3)で、公けに利用可能である。あるいは、これらのタンパク質およびコード配列は、KIAA0916、タンパク質登録番号NP_055872(タンパク質配列)、および、NM_015057(コード配列)で、公けに利用可能である。
【0015】
ラットPAMについて、以下のEST−クローンをコードする配列が公開された形で利用可能である:
AW921303(hscDNAのbp960〜1394に対応する;配列番号4)、
AW918711(hscDNAのbp8188〜8632に対応する;配列番号5)、
BQ201485(hscDNAのbp8966〜9633に対応する;配列番号6)、
BE112881(hscDNAのbp10311〜10830に対応する;配列番号7)、
AW441131(hscDNAのbp13569〜14152に対応する;配列番号8)、
BF409872(hscDNAのbp13569〜14807に対応する;配列番号9)。
【0016】
PAMはもともと、MycのN末端における転写活性化ドメインと特異的に相互作用する能力によって同定された(Guo Q.等,1998年)。PAMは近年、強力なAC活性阻害剤と説明されているが(Scholich K.,Pierre S.,Patel T.B.:Protein associated with myc(PAM)is a potent inhibitor of adenylyl cyclase. J.Biol.Chem.2001年12月14日;276(50):47583〜9)、侵害受容過程および感作におけるそれらの機能に関する証拠は未だ示されていない。
【0017】
むしろ、PAMは、シナプス前部の成長の調節において役割を果たすと考えられている:PAMのmRNAは、海馬、歯状回および小脳などの特定の解剖学的な領域で高度に発現されることがわかっている。成体ラットおよびマウスの脳におけるPAMおよびMyc発現の両方は、小脳の成熟したプルキンエ細胞、ならびに、海馬の顆粒および錐体細胞に限定される(Ruppert C.等,1986年;Yang H.等,2002年)。しかしながら、これら細胞型のいずれも、痛みのプロセシングおよび感作に関与することはわかっていない。
【0018】
ショウジョウバエにおけるPAM相同体(highwire)、および、C.エレガンスにおけるPAM相同体(rpm−1)は、シナプス前終末の組織化(Zhen等,20
00年)、シナプスの成長の調節(Wan等,2000年)、シナプス形成、ならびに、軸索成長およびターゲティング(Schaefer等,2000年)において重要な役割を果たすことが示されている。これらの発見により、highwire、rpm−1およびそれらの哺乳動物相同体PAMは、シナプスの成長の負の調節因子として作用する可能性があるという仮説に至った(Chang等,2000年;Jin Y.2002年)。それに沿って、小脳、海馬および歯状回におけるPAM発現の劇的な増加が、これら構造体における主要なシナプス形成期の際に観察された(Yang等,2002年)。
【0019】
齧歯類における脳の発達の際に、PAM発現は、誕生直後に開始し、最初の2週間の間中アップレギュレートされ、その後、PAM発現は、成人期の間中高いレベルを保つ(Yang等,2002年)。これまで、脊髄およびDRGにおけるPAMの発現および調節、ならびに、痛みの感作メカニズムおよび調節におけるその機能についてはわかっていない。
【0020】
これまで、ヒトPAMは、サイクリックAMP(cAMP)シグナル伝達の強力な調節因子であり、数種のアデニリルシクラーゼ(AC;E.C.4.6.1.1)アイソフォームの酵素活性をナノモル濃度で阻害する(Scholich等,2001年)ことが実証されている。
【0021】
遍在的なサイクリックAMP(cAMP)のセカンドメッセンジャーシステムは、細胞外の刺激を細胞内のシグナルおよび応答に翻訳する様々なシグナル変換メカニズムの一つである。細胞外の刺激を受けると、Gタンパク質共役受容体(GPCR)は、三量体GTP結合調節タンパク質(Gタンパク質)を介して細胞質膜に結合した酵素またはイオンチャンネルを調節する。GPCRによって活性が調節される酵素の一つは、アデニリルシクラーゼ(AC)、cAMP生成酵素である。このようにして、入ってくる細胞外の刺激は、細胞内のメディエイターであるサイクリックAMPの細胞内の濃度に影響を与える。cAMPレベルの上昇は、タンパク質キナーゼA(PKA)を刺激し、特異的な細胞内の標的タンパク質をリン酸化し、それによってそれらの活性を改変することによって細胞に影響を与える。
【0022】
それぞれのタイプの細胞は、特徴的な一連のGPCR、これらGPCRによって調節される酵素、アデニリルシクラーゼ(AC)の特異的なサブセットおよび標的タンパク質を有し、これらは、比較的非特異的な、または一般的に生じるプレイヤー(例えば遍在的なcAMP)と共に作用して、各細胞が、入ってくる細胞外シグナルに対してそれらに特有の応答を生じさせるようにできる。例えば、サイクリックAMP(cAMP)セカンドメッセンジャーは、シナプス形成性の調節において主要な役割を果たすことがわかっている(Bailey等,1996年;Xia等,1997年;Brandon等,1997年);一方で、cAMPは、代謝プロセスおよび細胞の増殖に関与する。このようにして、遍在的なcAMPメッセンジャーシステムの役割と、その様々な成分は、様々な組織および細胞型の様々な特殊化に従って多様である。
【0023】
これまで、S1P受容体として作用する、S1P1〜S1P5という5種の異なるGPCRは当業界で既知である(総論のために、例えば、Spiegel,S.およびMilstien,S.,2000年を参照)。様々なS1P受容体のタンパク質およびコードポリヌクレオチド配列が当業界で既知であり、例えばNCBI(National Centre for Biotechnology Information;National Library of Medicine,Building 38A,ベセスダ,メリーランド州20894,米国;www.ncbi.nhm.nih.gov)データベースより、登録番号:NM_001400(配列番号32;ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体、1mRNA(EDG1/S1P1)のヌクレオチド配列;NP_001391(配列番号31,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体(EDG1/S1P1)のタンパク質配列;NM:_004230(配列番号34,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体、5(EDG5/S1P2)mRNAのヌクレオチド配列;NP_004221(配列番号33,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体、5(EDG5/S1P2)のタンパク質配列;NM_005226(配列番号36,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体3(EDG3/S1P3)mRNAのヌクレオチド配列;NP:005217(配列番号35,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体3(EDG3/S1P3)のタンパク質配列;NM:003775(配列番号38,ホモサピエンスの内皮分化、Gタンパク質共役受容体6(EDG6/S1P4)mRNAのヌクレオチド配列;CAA04118(配列番号37,ホモサピエンスの内皮分化、13−タンパク質共役受容体6(EDG6/S1P4)のタンパク質配列;NM_030760(配列番号40,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体8(EDG8/S1P5)mRNのAヌクレオチド配列;NP_110387(配列番号39,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体8(EDG8/S1P5)のタンパク質配列で公けに利用可能である。
【0024】
本発明のその他の形態は、医薬品、好ましくは痛みを予防または治療するための医薬品として使用するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体に関する。
【0025】
本発明のさらなる形態は、痛みの調節のための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用に関する。この調節は、好ましくは軽減または予防または全体的な抑制である。
【0026】
その上、本発明は、痛みを調節する化合物を同定するためのS1Pまたはそれらの誘導体の機能的フラグメントの使用を包含する。上記調節する化合物は、好ましくは、S1P活性を模擬または増強する化合物である。最も好ましくは、それらは、痛みを予防する、減らす、または、止める能力を有する。
【0027】
上記化合物は、例えば、それらの以下の能力によって同定することが可能である;
a)S1Pの機能(すなわち、その受容体またはそれらのフラグメントの少なくとも1種と相互作用する能力、好ましくは受容体を活性化する能力、または、細胞内のcAMPレベルを低める能力、または、PAMの小胞体から細胞膜へのトランスロケーションを媒介する能力;より好ましくは、そのPAM活性を増強する能力、および/または、AC活性を阻害する能力、さらにより好ましくは、その痛みを減少させる能力)、または、PAMの機能(すなわち、その、細胞内cAMPレベルを低める能力、ACのようなその他のファクター、特にACと相互作用する能力、ACを阻害する能力、または、その痛みの感覚を低める能力)を模擬する、回復させる、活性化する、または、増強することを意味しており、または、
b)S1Pの血清レベルを増加させること(例えば、S1Pの生産を活性化または増強することによって、または、その細胞外の分解を減少させることによって)、または、
c)少なくとも1つのS1P受容体の発現を増強すること(すなわち、その転写、転写物の安定化、翻訳またはその翻訳後プロセシングの活性化によって;その翻訳後修飾の調節によって、または、その安定化の活性化、または、その分解の阻害などによって)、または、
d)S1Pの生産または分解に関与する酵素と相互作用すること。
【0028】
本発明のその他の形態は、十分な量のS1Pまたはそれらの機能的フラグメントもしくは誘導体を個体に投与することを含む、痛みを予防する、または、減らす方法に関する。
【0029】
投与は、S1Pの作用部位(DRGまたは脊髄)へのターゲティングを可能にする方法で適切に予備的に行われるべきであり、例えば、S1P誘導体または製剤の血流への全身投与(例えば、静脈内または経口での適用)によって、または、S1Pまたはそれらのフラグメントもしくは誘導体の局所的な(例えば脊髄内の)適用によってなされる。
【0030】
本発明のその他の形態は、痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法に関し、以下の工程を含む:
a.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、S1Pを含まないサンプルを提供する工程、
b.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、同様に、S1Pを含む第二のサンプルを提供する工程、
c.少なくとも第一のサンプルと、化合物とを接触させる工程、
d.サンプル中のPAM活性を測定する工程、
e.前記化合物のS1Pの機能を模擬する能力を測定する工程。
【0031】
本方法は、前記細胞を、前記化合物の代わりにS1Pと接触させ、上記のc)およびd)に係るPAM活性を、S1Pの存在下でのPAM活性と比較する工程をさらに含んでもよい。
【0032】
その他の例としては、以下の工程を含む方法が示される:
a)S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現する細胞を含む2つのサンプルを提供する工程、および、
b)一方のサンプルと、前記化合物とを接触させる工程、および、
c)両方のサンプル中の受容体活性を測定する工程。
【0033】
本発明のその他の形態に係る方法は、以下の工程を含む:
a)S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現する細胞を含む2つのサンプルを提供する工程、および、
b)一方のサンプルと、S1Pとを接触させる工程、および、
c)他方のサンプルと、化合物とを接触させる工程、および、
d)両方のサンプル中の受容体活性またはS1Pおよび受容体の相互作用を測定する工程。
【0034】
PAMまたはS1P受容体は、本発明の様々な形態に係る特定の目的が実現されるようなあらゆる配列から得られたものが利用可能である。好ましくは、PAMまたはS1P受容体はヒト由来である。
【0035】
本発明の様々な形態に関して、PAMまたはS1P受容体が、単離されたポリペプチド、または、オリゴもしくはポリヌクレオチドである場合も好ましい。本発明の様々な形態の環境で「単離された」とは、自然源から少なくとも部分的に精製されているか、または、組換え分子(これらは当然ながら、精製されていてもよいし、または部分的に精製されていてもよい)を意味する。
【0036】
アッセイとは、生物学的プロセスをモニターすることができるあらゆるタイプの分析方法のことである。薬物のスクリーニングで使用するためには、このようなアッセイは、再現可能でなければならず、好ましくは拡張可能で、頑強でもある。このようなアッセイは、好ましくは、化学物質の、痛みを調節する(好ましくは減少させる)および/または痛みを予防する能力に関するハイスループットスクリーニングに適している。ハイスループットスクリーニングの大部分は、特定の能力に関して異なる約500,000種の化合物をスクリーニングすることを含む。このアッセイのタイプは、例えば、用いられる分子のタイプ(ポリペプチドまたはポリヌクレオチドのいずれか)と「読み出し」、すなわちS1P、PAMまたはS1P受容体活性が測定される方法によって様々である(以下を参照)。
【0037】
このようなアッセイの様々なタイプが一般的に当業界で既知であり、商業的な供給元から市販されている。適切なアッセイは、標識された構成要素と標識されていない構成要素との相互作用を測定するための(例えば、PAMまたはそれらのフラグメントは、標識することができ、それらのACとの相互作用は、モニターすることができる)、放射性同位体分析、または、蛍光分析、例えば蛍光偏光分析を包含する(例えば、パンベラ(Panvera)、パーキン・エルマー・ライフサイエンス(Perkin−Elmer life sciences)(例えばLANCE)、または、パッカード・バイオサイエンス(Packard BioScience)(例えば、HTRFまたはアルファスクリーン(ALPHAscreen)TM)によって商業的に供給されているもの)。
【0038】
例えば医薬候補化合物と、受容体または機能的受容体フラグメントもしくは誘導体との相互作用を測定するためには、簡単な生化学的分析が適切である。また、より手の込んだ分析によれば、化合物が、特定の受容体を活性化し、それによりS1P活性を模倣することができるかどうかを決定することができる。
【0039】
さらなる例としては、細胞に基づくアッセイが挙げられ、この場合、細胞系は、対象の組換えタンパク質を、安定して(誘導的に、または非誘導的に;染色体の、またはエピソームの)、または、一時的に発現する。これらのアッセイは、例えばレポーター遺伝子アッセイを含み、この場合、特定のプロモーター、または、シグナル変換カスケードの構成要素のシグナル伝達経路の調節が、レポーター酵素の活性に従って測定され、このレポーター酵素の発現は、前記特定のプロモーターの制御下にある。このタイプのアッセイにとって、組換え細胞系は、それ自体が調査されるか、または、調査中のシグナル伝達カスケードによって調節される特定のプロモーターの制御下に、レポーター遺伝子が含まれるように構築されていなければならない。適切なレポーター酵素は、当業界で一般的に既知であり、ホタルルシフェラーゼ、ウミシイタケルシフェラーゼ(例えば、パッカード・リージェント(Packard Reagents)より市販されている)、β−ガラクトシダーゼが挙げられる。適切な細胞系は、アッセイ目的に応じて様々であるが、たいていは、トランスフェクトが簡単であり、培養が簡単な細胞系が挙げられ、例えばHeLa、COS、CHO、NIH−3T3などである。
【0040】
細胞内イオン濃度を測定するアッセイとしては、例えばFLIPR(蛍光測定イメージングプレートリーダー、モレキュラーデバイス(Molecular Devices)から市販されている)アッセイが挙げられ、この場合、冷却CCDカメラと連結されたアルゴンレーザー光源は、384ウェルプレートで、細胞(例えば、ニューロンの細胞、または、例えば、組換えもしくは自然に特定のイオンチャンネルを発現する細胞などの細胞)内で、一過性のイオンシグナル(例えばCa2+など)を同時測定することを可能にする。FLIPRアッセイは、例えば、Fluo−3、Fluo−4のような所定の蛍光色素を用いた細胞内のカルシウムのモニタリング、または、BCECFもしくはBCPCFもしくは特異的なFLIPRアッセイキットを用いた細胞内のpHのモニタリング、または、例えばDiBACもしくは特異的なFLIPRアッセイキットを用いた膜電位の変化の検出、または、膜の分極のモニタリングを可能にする。その他の細胞内のイオン、例えば亜鉛またはナトリウムをモニタリングするために、当業界で既知のその他の色素を用いることができる。その他のタイプのアッセイ、および、その他のタイプの読み出しは、一般的に当業者既知である。
【0041】
cAMPレベルの測定のためには、例えばアルファスクリーン、蛍光偏光法またはHTRF技術が適切である。
【0042】
イオンチャンネル活性(これは、例えば細胞内のイオン濃度を制御するため、細胞内のイオン濃度の測定に用いることができる)の測定のためには、例えば、膜電位感応性アッセイおよび色素を用いることができ、例えば、DiBAC、または、FLIPR技術に基づくモレキュラーデバイスの膜電位分析キット;FLIPR技術を用いたミトコンドリア膜の分極を測定するJC−1色素;細胞内のカルシウム濃度測定のための、Fluo−3、Fluo−4のようなイオン感応性色素、または、モレキュラーデバイスのカルシウム分析キット;細胞内のナトリウムを測定するための、ナトリウム感受性色素(例えばモレキュラープローブス製);細胞内のカリウム濃度を決定するための、パッチ−クランピングに基づくアッセイ、または、原子吸光分光分析法に基づくルビジウムイオン流出測定などを用いることができる。さらに、細胞内の所定の変化および状態を検出するための自動装置および分析方法は当業者既知であり、例えばアキュメン・バイオサイエンス(ACUMEN bioscience)のアキュメン(Acumen)検出器(適切に標識された目的物の分布の3次元再構築を可能にする蛍光ベースのレーザースキャニングリーダー)が挙げられる。
【0043】
GPCR活性の測定については、例えばcAMP測定、例えばパッカード・バイオサイエンス製のアルファスクリーンTMcAMP検出システムによる、Ca2+動員アッセイまたはレポーター遺伝子アッセイが適切である。
【0044】
PAMポリペプチドは、好ましくは、配列番号2に記載の配列を含む、または、それからなるポリペプチド、または、配列番号1または3に記載の配列を含む、または、それからなるポリヌクレオチドによってコードされるポリペプチドである。S1P受容体ポリペプチドは、好ましくは配列番号31、33、35、37または39に記載のアミノ酸配列のいずれか一つを含む、または、それからなるポリペプチド、または、配列番号32、34、36、38または40に記載のヌクレオチド配列のいずれか1つを含む、または、それからなるポリヌクレオチドによってコードされるポリペプチド、または、これらmRNA配列に含まれるコード配列によってコードされるポリペプチドである。
【0045】
PAMポリヌクレオチドは、好ましくは配列番号1または3に記載の配列を含む、または、それからなるポリヌクレオチド、または、上記のポリヌクレオチドとストリンジェントな条件下でハイブリダイズできる配列を含む、または、それからなるポリヌクレオチドである。S1P受容体ポリヌクレオチドは、好ましくは配列番号32、34、36、38または40に記載の配列を含む、または、それからなるポリヌクレオチド、配列番号32の244〜1392位、配列番号34の1〜1137位、配列番号36の1〜1062位、配列番号38の23〜1177位、または、配列番号40の10〜1206位を含む、または、それに対応するポリヌクレオチド、または、これらポリヌクレオチドのいずれか一つとストリンジェントな条件下でハイブリダイズできる配列を含む、または、それからなるポリヌクレオチドである。
【0046】
ストリンジェンシーとは、2本の一本鎖の核酸分子のハイブリダイゼーションまたはアニールの特異性に影響を与える反応条件を説明する。ストリンジェンシー、すなわち反応の特異性は、特に、反応に用いられる温度と緩衝液条件に依存する:ストリンジェンシー、すなわち特異性は、例えば、反応温度を高める、および/または、反応緩衝液のイオン強度を低めることによって増加させることができる。低いストリンジェント条件(すなわち低い反応およびハイブリダイゼーション特異性)とは、例えば、室温で、2×SSC溶液中で、ハイブリダイゼーションが行われる場合に生じる。高いストリンジェンシー条件は、例えば、68℃、0.1×SSC、および、0.1%SDS溶液中でのハイブリダイゼーション反応を含む。
【0047】
本発明の様々な形態の範囲内のストリンジェンシー条件下におけるハイブリダイゼーションは、好ましくは、以下のように理解される:
1)標識されたプローブと解析しようとする核酸サンプルとを、65℃で、または、オリゴヌクレオチドプローブの場合、オリゴヌクレオチドとサンプルとからなる二本鎖のアニーリング温度または融解温度より5℃低い温度で(アニーリング温度および融解温度は、以下では同義語とする)、一晩、50mMトリス(pH7.5)、1MのNaCl、1%SDS、10%硫酸デキストラン、0.5mg/mlの変性サケまたはニシン精子DNA中で、ハイブリダイズする。
2)2×SSC中で、室温で10分間洗浄する。
3)1×SSC/0.1%SDS中で、65℃(または、オリゴヌクレオチドの場合:アニーリング温度より5℃低い温度)で、30分間洗浄する。
4)0.1×SSC/0.1%SDS中で、65℃(または、オリゴヌクレオチドの場合:アニーリング温度より5℃低い温度)で、30分間洗浄する。
【0048】
ハイブリダイゼーションプローブとしての使用するためのオリゴヌクレオチドは、ポリヌクレオチドであり、好ましくはDNAフラグメントであって、長さが、15〜30個、好ましくは20個のヌクレオチドを有する。アニーリング温度は、式Tm=2×(A+Tの数)+4×(G+Cの数)℃に従って決定される。
【0049】
2×SSC、または、0.1×SSC(または、その他のあらゆる種類のSSC希釈液)を製造するためには、例えば、20×SSC溶液を適宜希釈する。20×SSCは、3MのNaCl/0.3Mクエン酸Na×2H2Oからなる。
【0050】
ハイブリダイゼーション反応を行う前に、上記ポリヌクレオチドは、必要に応じて、電気泳動による分離を行った後に(次に:サザンブロット(DNA)、または、ノーザンブロット(RNA))、または、電気泳動による分離を行わないで(次に:スロットまたはドットブロット)、適切なメンブレン(例えばナイロンまたはニトロセルロースメンブレン)にトランスファーする。ハイブリダイゼーションは、適切に標識されたプローブを用いて行われる。適切な標識技術は、例えば、放射性標識、または、蛍光色素を用いた標識である。上記プローブは、一本鎖ポリリボまたはポリデスオキシリボヌクレオチドであり、自然状態では一本鎖であるか、または、通常は二本鎖で変性によって一本鎖にされる。このプローブは、DNAまたはRNAサンプル(これらも一本鎖状態である)に、塩基対形成によって結合する。
【0051】
PAMフラグメントは、好ましくは上記の配列番号1、2または3に含まれるフラグメントであり、その誘導体は、好ましくは、上記の配列番号1、2もしくは3、または、それらのフラグメントから得られる。S1P受容体フラグメントは、好ましくは上記の配列番号31〜40に含まれるフラグメント、より好ましくは、配列番号32の244〜1392位、配列番号34の1〜1137位、配列番号36の1〜1062位、配列番号38の23〜1177位、または、配列番号40の10〜1206位を含む、または、それからなるフラグメントであり、その誘導体は、好ましくは、これらの配列から得られる。
【0052】
それらの機能的フラグメントまたは誘導体は、好ましくは、アデニリルシクラーゼ(AC)活性、より好ましくはACのI、VまたはVI型の活性を阻害することができる(S1P受容体に関して、最も好ましくは、S1Pが結合することによって、または、S1Pを模倣する分子が結合することによって活性化される場合に、AC活性を阻害することができる)。
【0053】
本発明の様々な形態の好ましい実施形態によれば、PAMの機能的フラグメントまたは誘導体は、ヒトPAM配列の、好ましくは配列番号2に記載のヒトPAM配列の、アミノ酸400〜1400、好ましくは446〜1062、499〜1065または1028〜1231、より好ましくは1000〜1300、さらにより好ましくは1000〜1100、さらにより好ましくは1028〜1065を含む、もしくは、それからなり、または、それらが上記それぞれのポリヌクレオチドフラグメントによってコードされる場合、特別には、配列番号2もしくは3に記載の配列に含まれる場合である。
【0054】
PAMの機能的フラグメントまたは誘導体が、ポリヌクレオチドである場合、それらは、上記のポリペプチドフラグメントをコードするポリヌクレオチドを含む、または、それからなる場合が好ましい。より特定には、それらが、ヒトPAMcdsの1482〜3332位(アミノ酸446〜1062をコードする)、または、1641〜3341(アミノ酸498〜1066をコードする)、または、3228〜3839(アミノ酸1038〜1231をコードする)を含む、または、それからなる場合が好ましい。上記フラグメントが得られるヒトPAMcdsが、配列番号2に記載の配列を有する場合がさらにより好ましい。
【0055】
痛みを調節する化合物を同定するための本発明の方法の好ましい一実施形態によれば、S1P受容体および/またはPAMを発現する細胞、好ましくは組換えS1P受容体および/またはPAMを発現する細胞が用いられる。
【0056】
上記細胞は、あらゆるタイプの細胞が可能であり、例えば、真核性または原核性の単細胞生物(例えば細菌、例えばE.coli、または、酵母、例えばS.ポンベ(S.pombe)またはS.セレビジエ(S.cerevisiae))、または、多細胞生物から得られた細胞系(例えばHeLa、COS、NIH−3T3、CHOなど)が挙げられ、なかでも哺乳動物細胞系が好ましい。
【0057】
その他の好ましい実施形態によれば、改変されていない状態と比較して低いS1P受容体活性を有する改変された細胞が用いられる。この方法において、このような細胞は、それらの痛みを調節する(好ましくは減少させる)および/または痛みを予防する能力に関して試験しようとする化学物質が、低められた、または完全に止められたS1P受容体活性を増強する、または回復させることができる場合、試験することができる。
【0058】
上記改変は、あらゆるタイプの改変(安定な、または、一過性の、好ましくは安定な)が可能であり、それにより、S1P受容体活性および/またはPAM活性(すなわち、それらの細胞内cAMPレベルを低める能力、PAMのトランスロケーション、ACを阻害する能力、または、それらの痛みの感覚を低める能力)の減少、S1P受容体またはPAM転写物の定常状態レベルの減少(すなわち、S1P受容体またはPAM転写の阻害、または、転写物の安定化によって)、または、S1P受容体またはPAMタンパク質の定常状態レベルの減少(すなわち、S1P受容体、または、PAM翻訳もしくはその翻訳後プロセシングの不活性化によって;その翻訳後修飾の調節によって、または、その安定化の不活性化によって、または、その分解を増加させることによって)が起こる。これは、例えば、S1P受容体またはPAMの優性の負変異体、アンチセンスオリゴヌクレオチド、RNAiコンストラクトを用いることによって、機能的またはゲノムS1P受容体またはPAMノックアウト(これらは、例えば誘導型が可能である)を生成することによって、または、当業界既知のその他の適切な技術によって達成することができる。上記の技術の総論としては、例えば、以下を参照:Current protocols in Molecular biology(2000年)J.G.Seidman,第23章,Supplement52,ジョン・ワイリー&サンズ社(John Wiley and
Sons,Inc.);Gene Targeting:a practical a
pproach(1995年),編集者:A.L.Joyner,IRLプレス(IRL Press);Genetic Manipulation of Receptor Expression and Function,2000年;Antisense Therapeutics,1996年;Scherr等,2003年。好ましい実施形態によれば、PAMノックアウト細胞が用いられる。ノックアウトを作製するのに適した細胞系は当業界周知であり、例えば、Current protocols in Molecular Biology(2000年)J.G.Seidman,第23章,Supplement 52,ジョン・ワイリー&サンズ社;または、GeneTargeting a practical approach.(1995年)編集者A.L.Joyner,IRLプレスが挙げられる。
【0059】
S1P活性は、例えば、その(または、そのフラグメントおよび誘導体の)受容体またはそれらの機能的フラグメントの少なくとも1種と相互作用する能力、または、PAMの細胞膜へのトランスロケーションを開始させる能力によって、直接決定することができ、または、S1P活性は、例えば、その(または、その機能的フラグメントおよび誘導体の)細胞内cAMPレベルを低める能力、ニューロン内のイオン濃度を調節する能力、ACの機能を阻害する能力、または、その痛みの感覚を調節する能力、特別には減少させる能力によって、間接的に決定することもできる。上記のパラメーターを測定するのに適した技術は当業界周知である(上記も参照):cAMPレベルは、例えば、HTRFまたはアルファスクリーンTMによって測定することができ、イオン濃度は、例えば、パッチ−クランピングまたは適切な色素によって推定することができ、痛みの感覚は、例えば、ホルマリン試験、または、機械刺激もしくは温熱性痛覚過敏の試験、または、ホットプレート試験などによって測定することができる。その受容体との相互作用は、例えば、cAMP測定、Ca2+動員またはレポーター遺伝子アッセイによって測定することができる。
【0060】
本発明のその他の形態は、痛みを調節する化合物を同定する方法に関し、以下を含む:
a)試験化合物として、S1Pの活性を調節または模倣する化合物を選択すること、および、
b)前記試験化合物を被検体に投与して、痛みが調節されているかどうかを測定すること。
【0061】
被検体は、痛みを知覚する能力を有するあらゆる被検体が可能であり、好ましくは哺乳動物であり、すなわちヒト以外の哺乳動物またはヒト(すなわち、患者の研究の範囲内で)のいずれかである。
【0062】
上記調節は、好ましくは、痛みを予防、軽減または止めることである。本発明の好ましい一実施形態によれば、上記化合物は、S1P受容体アゴニスト(総論のために、例えば、Mandala等,Science 2002年を参照)、より好ましくはFTY720(2−アミノ−2−(4−オクチルフェニル)エチル)プロパン−1,3−ジオール、図17を参照)、または、それらの機能的な誘導体または類似体(類似体は当業界で既知であり、例えば、Brinkmann等,JBC,2002年を参照)(すなわち、上述の痛みを調節する能力を有する誘導体または類似体)、好ましくはリン酸化された誘導体(Mandala等,Science 2002年を参照)である。また、上記化合物またはその誘導体もしくは類似体の生理学的に許容できる塩も適切である。
【0063】
本発明のさらなるその他の観点によれば、痛みを減らす、または予防する方法は、少なくとも1種のS1P受容体に結合し、活性化する能力、および/または、PAMの機能を活性化する活性を有する医薬化合物の十分な量を個体に投与することを含み、これも、本願の範囲内に含まれる。このような化合物の適切な例の一つは、S1P受容体アゴニスト(総論のために、例えば、Mandala等,Science 2002年を参照)、よ
り好ましくはFTY720、または、上述したような機能的な誘導体または類似体、好ましくはそれらのリン酸化された誘導体、または、上記化合物またはその機能的な誘導体もしくは類似体の生理学的に許容できる塩である。
【0064】
以下で、本発明を実施例と図によってより詳細に説明する。しかしながら、これら実施例は、本発明の範囲を限定するものではない。
【実施例】
【0065】
実施例:PAM発現パターン、ならびに、PAMおよびS1Pの機能の調査
1.材料
S1Pは、トクリス(Tocris,エリスヴィル,ミズーリ州)から購入し、抗Hsp70抗体と抗カルネキシン抗体は、BDトランスダクション・ラボ(Transduction Labs)(ベッドフォード,マサチューセッツ州)から購入した。抗活性ERK1/2抗体を、プロメガ(Promega)(マディソン,ウィスコンシン州)から得て、百日咳毒素、U0126、U73122、および、ワートマニンは、トクリス(エリスヴィル,ミズーリ州)から得て、RO31−223、BAPTA−AM、および、GF109203Xは、シグマ(Sigma)(セントルイス,ミズーリ州)から得た。
【0066】
2.動物切片標本の製造:
野生型スプラギー・ダーレー(Sprague Dawley)ラットを、チャールス・リバー・ウィガ社(Charles River Wiga GmbH,ズルツフェルト,ドイツ)から購入した。実験の前は、動物は、えさと水を自由に摂取できるようにした。それらを環境と光を制御した部屋(24+0.5℃)で維持した。各動物は、1回の実験のみで用いられた。全ての実験において、意識のある動物を研究するための倫理ガイドラインに従い、その手法は、地域倫理委員会で認証された。殺した後に、成体ラットを、0.1Mリン酸緩衝食塩水(PBS,pH7.2)中の4%パラホルムアルデヒドで、1時間潅流することによって固定した。組織を、クライオスタットを用いて、水平面で厚さ14〜16μmの切片標本にした。切片標本を、スーパーフロスト・プラス・スライド(Superfrost Plus Slides)(フィッシャー・サイエンティフィック社(Fisher Scientific Co.),ピッツバーグ,ペンシルベニア州)にマウントし、使用するまで−80℃で保存した。
【0067】
3.リボプローブの製造:
リボプローブを、これまで説明した通りにして作成した(Yang等,2002年)。ラットPAMのアンチセンス、および、センスリボプローブを、プラスミドをHindIII(アンチセンス)およびBamHI(センス)で線状化した後に、それぞれT7およびT3ポリメラーゼを用いて得た(Yang等,2002年を参照)。インビトロでの転写を、製造元(プロメガ、マディソン,ウィスコンシン州)の推奨に従って、[35S]UTP−αS(ICN,アーバイン,カリフォルニア州)、線状化したPAMcDNA、NTPの存在下で、37℃で1時間行った。RNAの転写物を、RNAプローブ精製キット(Pequlab,エルランゲン,ドイツ)を用いて精製した。
【0068】
4.インサイチュハイブリダイゼーション:
インサイチュハイブリダイゼーションを、前述した通りに行った(Yang等,2002年):切片標本を、0.1Mリン酸緩衝食塩水(pH7.2)中の4%パラホルムアルデヒドで固定し、0.25%無水酢酸と0.1Mトリエタノールアミンで前処理し、0.2×SSCでリンスし、アルコール濃度を連続的に高めて脱水させた。切片標本を、プレハイブリダイゼーション溶液(50%脱イオン化ホルムアミド、0.6M塩化ナトリウム、10mMトリスHCl(pH7.6)、50mMのEDTA、0.025%ピロリン酸ナトリウム、0.02%フィコール、0.02%BSA、0.02%ポリビニルピロリドン、10mMのDTT、および、熱変性させた異種核酸(0.005%の酵母tRNA、タイプX、0.05%の酵母全RNA、タイプI、0.05%サケ精巣DNA、タイプIII))で、室温で2時間プレハイブリダイズさせ;ハイブリダイゼーション溶液(2.5×106cpm/切片標本)、50%脱イオン化ホルムアミド、および、50%ハイブリダイゼーション緩衝液[0.6M塩化ナトリウム、10mMトリスHCl(pH7.6)、50mMのEDTA、0.025%ピロリン酸ナトリウム、0.02%フィコール、0.02%BSA、0.02%ポリビニルピロリドン、熱変性させた異種核酸(0.005%の酵母tRNA、タイプX、0.005%の酵母全RNA、タイプI、0.05%サケ精巣DNA、タイプIII)、100mMのDTT、0.0005%ポリアデニル酸、10%硫酸デキストランを含む]中で、50℃で一晩、リボプローブとハイブリダイズさせた。切片標本を、RT(室温)で、2×、1×、0.5×SSCでリンスした。20μg/mlのRNアーゼA(シグマ,セントルイス,ミズーリ州)中で消化させた後、切片標本を、室温で、1×RNアーゼ緩衝液、2×、1×、0.5×SSC中で、および、45℃で、0.1×SSC中で一晩洗浄した。切片標本をアルコール濃度を連続的に高めて脱水させ、コダック(Kodak)のバイオマックスMRフィルム(Biomax MR film)(コダック,ロチェスター,ニューヨーク州)に−80℃で3〜7日間露光した。
【0069】
5.抗体の生成、および、免疫蛍光法による染色:
ヒトPAMのアミノ酸残基135〜153および4601〜4614(それぞれ配列番号1に対応する)からなるペプチド(バイオトレンド(BioTrend),ケルン,ドイツ)を用いて、抗血清をウサギで商業的に生成させた。その抗血清を、標準的な手法に従って、バイオトレンド(ケルン,ドイツ)で商業的に生産させた。脊髄とDRGの切片におけるPAMの分布をモニターするために、切片を、0.1%トリトンX−100中で5分間浸透させた。切片を、PBS中の3%BSA中で1時間ブロッキングし、次に、抗PAM抗血清(1:50希釈)と共に1時間インキュベートした。続いて、これを、3%BSAを含むPBS中のFITC標識したヤギ抗ウサギ抗体と共にインキュベートした。次に、切片をPBSで洗浄し、フルオマウント(fluoromount)TMを用いてマウントした。
【0070】
6.RT−PCR:
ラットの脊髄およびDRGからの全RNAを、グアニジンイソチオシアネート/フェノール/クロロホルム抽出によって単離した(ChomczynskiおよびSacchi,1987年)。全RNA2μgを、0.6μMのそれぞれのオリゴ(dT)プライマーとアニールし、逆転写酵素(プロメガ,マディソン,ウィスコンシン州)を用いて37℃で30分間逆転写した。次に、即座にcDNAを増幅に用いた。ラットGADPHの増幅に用いられるオリゴヌクレオチドプライマーは、5’−GAAGGGTGGGGCCAAAAG−3’(センス;配列番号10)、および、5’−GGATGCAGGGATGATGTTCT−3’(アンチセンス;配列番号11;Trajkovic等,2000年)であった。ACアイソフォームを増幅するためのオリゴヌクレオチドプライマーを、Xu等によって公開されているようにして選択した(Xu等,2001年)。ラットPAMのためのプライマーは、5’−GGTGGTGAAGCTCGCTGTGATGCT−3’(センス;配列番号12)、および、5’−CGTGTGAGCATTTCTGCACACTCC−3’(アンチセンス;配列番号13)であった。そのPCR産物は、ヒトPAMcDNAヌクレオチド13692〜14064に対応する。それに対応するラット配列は、ESTクローンAW441131(配列番号8)から得た。半定量PCRのために、SAWDAY DNAポリメラーゼ(Peqlab,エルランゲン,ドイツ)を用いた。最初の95℃で5分間での変性工程の後に、95℃で1分間、55℃で30秒間、および、72℃で10秒間の30サイクルを行い、その後、最終に72℃で10分の伸長工程を行った。定量PCRを、TaqManTMシステムおよび試薬(アプライド・バイオシステムズ,ヴァイターシュタット,ドイツ)を製造元の説明書に従って用いて行った。
【0071】
7.全長PAMの精製:
以前に公開されたように、ただし数箇所を改変してPAM精製を行った(Scholich等。2001年)。簡単に言えば、HeLa細胞を、10%ウシ胎仔血清と1%ペニシリン/ストレプトマイシンを含むDMEM培地中で増殖させた。40個の150mm培養皿の密集した細胞を、1×PBS、1mMのEDTAを用いて回収し、400×gで5分間沈殿させた。細胞を、125mMのNaCl、20μg/mlのアプロチニン、20μg/mlのロイペプチン、1mMのベンズアミジン、5μg/mlのダイズトリプシンインヒビターを含むTED緩衝液(50mMトリスHCl(pH8.0)、1mMのEDTA、1mMのDTT)に再懸濁させ、超音波破砕を2×5秒間行い溶解させた。ホモジネートを、27000×gで、4℃で30分間で遠心分離し、上清を、Q−セファロースXK16カラム(アマシャム・ファルマシア(Amersham Pharmacia),ピスカタウェイ,ニュージャージー州)にローディングし、製造元の説明書に従って、TED中で、150〜350mMのNaClの濃度勾配で溶出させた。標準的な手法に従ってウェスタンブロッティングで分画を解析した;陽性の分画をプールし、そのNaCl濃度を1Mに調節した。次に、タンパク質を、フェニル−セファロースXK16カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングし、製造元の説明書に従って、TED中の300mMのNaClで洗浄した。流出液分画および洗浄分画にPAMが含まれていた。それらをプールし、製造元の説明書に従って、セントリコン(Centricon)50(アミコン(Amicon),ビバリー,マサチューセッツ州)を用いて緩衝液を上述の100mMのNaClを含むTED緩衝液で交換した。次に、タンパク質を、モノS(Mono S)5/5FPLCカラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングし、製造元の説明書に従って、ローディング緩衝液(TED中、100mMのNaCl)で洗浄した。流出液を回収し、モノQ(Mono Q)5/5FPLCカラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にアプライした。タンパク質を、TED中の150〜400mMのNaClの濃度勾配で溶出させた。陽性の分画をプールし、セントリコン(Centricon)50(アミコン(Amicon),ビバリー,マサチューセッツ州)を用いて緩衝液を50mMトリスHCl(pH8.0)、1mMのDTTで交換し、−80℃で保存した。保存したPAMは、3週間以内に用いた。
【0072】
8.組換えGsαの発現および精製:
ヘキサヒスチジルタグを有する構成的に活性なGsαのQ213L突然変異体(Gsα*)を、発現させ、Graziano等,1991年で説明されている通りに精製した。Gsα*の最大活性化を確認するために、AC活性分析で使用する前に、MgCl2(25mM)の存在下で、30分間、Gタンパク質を、1μMのGTPγSとインキュベートした。
【0073】
9.アデニリルシクラーゼ活性アッセイ(AC活性アッセイ):
脊髄を、25mMのHepes(pH7.4)、1mMのEGTA中で溶解させ、KassisおよびFishmanによって説明された通りに細胞膜を調製した。アリコートを使用するまで−80℃で保存した。AC活性分析を、以前に説明された通りに、容積100μlで、室温で15分間、100μMのMgCl2の存在下で行った(Patel等,2002年)。Gsα*(80nM)、または、フォルスコリン(100μM)を用いて、膜中のAC酵素活性(タンパク質10μg)を刺激した。
【0074】
10.PAMのアンチセンス、および、センスオリゴヌクレオチドの脊髄への搬送:
ラットを、ケタミン(60mg/kg,腹腔内)と、ミダゾラム(0.5〜1mg/kg,腹腔内)で麻酔した。椎骨Th13からL3までの脊柱の上で皮膚を切開した。L2
〜3周辺の筋肉組織を取り除いた。L3の棘突起を除去し、L2で椎弓切除術を施した。次に、ポリエチレン製カテーテル(ID0.28mm、OD0.61mm)を、カテーテルの先端がTh9〜10に達するように硬膜外腔に挿入した。カテーテルをシアノアクリレート系接着剤で固定し、頚部の領域で外部に露出させ、皮膚を縫合した。
【0075】
11.PAMオリゴヌクレオチドの注入:
オリゴデオキシヌクレオチド(ODN)の配列を、以下のようにラットPAM配列から選択した。センス:5’−GACTGGTTTAGCAATGGC−3’(配列番号14)、アンチセンス:5’−GCCATTGCTAAACCAGTC−3’(配列番号15)、および、3つの突然変異(3M−as;突然変異は下線で示した)を含むアンチセンスODN:5’−GCAATTGCTAAATCAGTA−3’(配列番号16)。外科手術の3日後に、ラットを「自由行動系」(CMA,ストックホルム,スウェーデン)の状態に置き、アンチセンス(n=5)、または、センス(n=5)オリゴヌクレオチド(人工髄液中、2.5mg/ml)をカテーテルを通じて、微量注入ポンプ(CMA,ストックホルム,スウェーデン)を用いて、0.05〜0.1μl/分の低速で、100時間注入した。
【0076】
12.ホルマリン試験:
注入を止めた後15分以内に、ホルマリン試験を行った。5%ホルムアルデヒド溶液50μlを、1本の後肢の背面の皮下に(s.c.)注射した。ホルマリン注射の直後に開始して、1分間のインターバルで60分間まで、フリンチ行動をカウントした。5分間のインターバルのフリンチ行動を、1分あたりの平均フリンチ行動として要約した。グループ間の侵害受容の挙動を比較するために、フリンチ行動の、1時間の観察期間の第二相の合計を、スチューデントのt検定で処理した。aを0.05に設定した。
【0077】
ホルマリン試験の最後にラットを殺し、腰髄および後根神経節(DRG)を切り出し、液体窒素中で急速冷凍し、さらなる解析まで−80℃で保存した。PAM発現を決定するために、脊髄切片を、上記の抗PAM抗体を用いて免疫組織化学的に解析した。
【0078】
13.ザイモサン誘発性の炎症:
炎症を誘発させるために、0.03Mのリン酸緩衝食塩水(PBS,pH7.5)30μlに懸濁したザイモサンA2.5mg(シグマ,セントルイス,ミズーリ州)を、右の後肢の足底の中央部の領域に皮下注射した。このような足底内部へのザイモサン注射は、信頼できる温熱性および機械刺激痛覚過敏ラットのモデルを誘導することがわかっている(MellerおよびGebhart,1997年)。ラットを、ザイモサン注射の24〜96時間後に、深いイソフルラン麻酔下で心臓穿刺を行うことにより殺した。腰髄および後根神経節(DRG)を切り出し、液体窒素中で急速冷凍し、さらなる解析まで−80℃で保存した。
【0079】
14.免疫蛍光法による染色:
HeLa細胞におけるPAMの分布をモニターするために、カバーガラス上で、10%FBSおよび1%ペニシリン/ストレプトマイシン(ギブコ(Gibco),カールスルーエ,ドイツ)を含むDMEM中で細胞を増殖させた。HeLaにおけるPAMのトランスロケーションを可視化するために、細胞を、一晩、血清欠乏状態にし、その後、10%血清で2時間処理した。指示されている場合は、細胞を、様々な阻害剤濃度の存在下で30分間プレインキュベートした。PBS中の4%パラホルムアルデヒド(シグマ,タウフキルヒェン,ドイツ)で、細胞を10分間固定し、次に、さらに5分間、0.1%トリトンX−100中に浸透させた。カバーガラスを、PBS中の3%BSAで1時間ブロッキングし、次に、抗PAM抗体(1:50希釈)と共に1時間インキュベートした。続いて、これを、3%BSAを含むPBS中で、FITC標識したヤギ抗ウサギ抗体と共にインキュベートした。次に、細胞をPBSで洗浄し、マウントした。解析のために、共焦点(バイオ・ラッド,ハーキュリーズ,カリフォルニア州)、および、標準蛍光顕微鏡(ニコン(Nikon),デュッセルドルフ,ドイツ)を用いた。
【0080】
15.cAMPの蓄積
脊髄サンプルを超音波破砕し、18.000×gで、4℃で20分間遠心分離した。上清をcAMP測定に用いた。細胞中のcAMPの蓄積を、cAMP検出キット(Assay Design Inc,アナーバー,ミシガン州)によって、製造元の説明書に従って決定した。
【0081】
16.アンチセンスオリゴデオキシヌクレオチド
オリゴデオキシヌクレオチド(ODN)の配列を、以前に公開されたようにして選択した(Scholich等,2001年)。センス:5’−CTGTTCATGCCGGTT−3’、アンチセンス:5’−AACCGGCATGAACAG−3’、および、3つの突然変異(3M−as;突然変異は下線で示した)を含むアンチセンスODN:5’−AATCCGTATGAACAC−3’。HeLa−細胞を35mm培養皿で平板培養し(300,000細胞)、10%FBSおよび1%ペニシリン/ストレプトマイシンを含むDMEM培地(ギブコ,カールスルーエ,ドイツ)中で24時間増殖させた。ODN(それぞれ3μM)を、無血清培地1ml中で、Tf×20(プロメガ,マディソン,ウィスコンシン州)を製造元の説明書に従って用いてトランスフェクトすることによって細胞に導入した。2時間後、10%FBSを含むDMEM(ギブコ,カールスルーエ,ドイツ)1mlを添加した。次に、細胞を6時間インキュベートした。次に、培地を無血清DMEM(ギブコ,カールスルーエ,ドイツ)で交換し、続いて、無血清DMEM(ギブコ,カールスルーエ,ドイツ)中で16時間インキュベートし、その後、10%血清または500MのS1Pで処理した。次に、細胞を、沸騰した1×レムリー緩衝液を添加することによってウェスタンブロットに用い、または、上述のようなAC活性分析のためにそれらを回収した。
【0082】
17.精製またはPAMの血清因子の活性化:
ウシ胎仔血清(ギブコ,カールスルーエ,ドイツ)188mlを、NaClの最終濃度が0.3Mになるように調節した。次に、血清を、フェニル−セファロース15/10カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングした。このカラムから溶出させた後に、同様に、それに続く全てのカラムの後に、流出液と全ての溶出した分画を回収し、そのHeLa細胞でPAMのトランスロケーションを誘導する能力について解析した。次に、流出液を、Q−セファロース15/10カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングした。カラムを、TE(50mMトリス/Cl(pH7.4)、0.5mMのEDTA)中の400mMのNaClで洗浄し、TE中の1MのNaClで溶出させた。溶出液を、スーパーデックス(Superdex)200pgゲルろ過カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングした。製造元の説明書に従ってタンパク質をTEで溶出させ、分画を、HeLa細胞中でPAMのトランスロケーションを誘導する能力に関して解析した。陽性の分画をプールし、モノQ5/5FPLC.カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングし、TE中の400mMのNaClで洗浄した。タンパク質をTED中、NaClが400〜1000mMの濃度勾配で溶出させた。陽性の分画をプールし、スーパーデックス50pgゲルろ過カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)に製造元の説明書に従ってローディングした。タンパク質を、製造元の説明書に従ってTEで溶出させ、分画を、HeLa細胞中でPAMのトランスロケーションを誘導する能力に関して解析した。陽性の分画をプールし、−80℃で保存し、マススペクトロメトリーまたは生化学的アッセイのためには2週間以内に用いた。フタルアルデヒド標識を用いてS1Pを検出し、続いて、Caligan等によって説明されているように、HPLCで分離した。
【0083】
18.侵害受容過程におけるPAMの関連を示す結果:
上記の発明者等によるRT−PCR、免疫組織化学およびインサイチュハイブリダイゼーションを用いた実験で、PAMは、成体ラットの脊髄の感覚ニューロン、同様に、後根神経節(DRG)で発現されることがが初めて実証された。RT−PCRによれば、PAMのmRNAは、脊髄と後根神経節とで発達を通して(E14〜成体)同様のレベルで検出された。PAM発現は、ウェスタンブロットおよびRT−PCRで示されるように、ラットのザイモサン処理の24〜48時間後にアップレギュレートされる。
【0084】
脊髄とDRGで発現される主要なアデニリルシクラーゼアイソフォームは、それぞれAC5および6型、ならびに、AC4および6型である。脊髄をザイモサン処理した後は、ACアイソフォーム発現における主要な変化は観察されなかった。従って、脊髄およびDRGからの膜調製物におけるGαsによって刺激されたAC活性は、PAMによって阻害された。その結果として、成体ラットにおいて、PAMに対するセンスオリゴヌクレオチドではなくアンチセンスオリゴヌクレオチドでの処理により、ホルマリンで誘発される足のフリンチ行動が増加したことを発見した。従って、PAMに対するアンチセンスオリゴヌクレオチドで処理したラットの脊髄におけるcAMPの蓄積は、コントロールラットと比較して上昇した。
【0085】
精製されたPAMを脊髄溶解産物へ添加することにより、コントロールとザイモサン処理動物からの脊髄溶解産物のGαsで刺激されたAC活性が阻害された(図5b)。30nMのGαsで刺激されたAC活性は、コントロール動物の脊髄溶解産物において50%、ザイモサンで96時間処理されたラットから得られた溶解産物で70%減少した。
【0086】
脊髄でPAMが発現されるかどうかを測定するために、第一のインサイチュハイブリダイゼーションを行った。これにより、成体ラットの脊髄の白質ではなく灰白質の至る所に、PAMのmRNAに関する明らかなシグナルが検出された(図1)。脊髄およびDRGでPAMを発現する細胞集団をより正確に明らかにするために、ヒトPAMのアミノ酸残基135〜153および4601〜4614に対応するペプチドを用いて、PAMに対する抗体を作成した。免疫組織化学的な解析により、PAMは、抗GFAP免疫反応性ではなく、抗NeuN免疫反応性と共存することが明らかになった(図2a)。より特定には、PAM発現は、後角ニューロンで優勢に検出されたが(図2a)、ニューロンではない細胞集団では極めてわずかなPAM発現しか示さなかった。特に、DRGニューロンでは、高いPAM発現を検出することができた(図2b)。ここで、PAMの免疫反応性は、軸索に、同様に、直径が大きいニューロンと小さいニューロンの両方の細胞体に存在していた(図2b)。興味深いことに、抗ヒストン抗体で共染色することによって実証されたように、細胞の核では、PAMは検出されなかった(図2b)。脊髄でも観察されるように、GFAP発現細胞では、PAM発現は、検出されなかった。
【0087】
ラットおよびマウスの発達中に、脳におけるPAM発現は差異的に調節されるために(Yang等,2002年)、発明者等は、ラットの脊髄およびDRGの発達中にPAMのmRNA発現も変化するかどうかを調査した。この目的を達成するために、PAMのmRNA発現を、定量RT−PCRを用いて決定した。PAMのmRNAは、後期胚形成期(E16)から、誕生直後まで(P0.5;図3)、脊髄とDRGで高度に発現されることを発見した。興味深いことに、発現は、誕生直後に胚発生の30〜40%に減少し、続いて、成人期中は一定を保った(図3)。
【0088】
次に、脊髄におけるPAM発現は、侵害受容の刺激によって調節されるかどうかを試験した。そこで、成体ラットの後肢へのザイモサンおよびホルマリン注射の後に、脊髄のPAM発現をモニターした。ザイモサン処理の24および48時間後に、PAMのmRNAが約2倍アップレギュレートされた(図4a)。それにより、ザイモサン注射の24時間後に、PAM発現は、タンパク質レベルでアップレギュレートされ、96時間上昇し続けた(図4b)。ザイモサン注射の96時間後に、PAMのmRNA発現は減少した。このPAMのmRNA発現における減少は、タンパク質レベルには反映されていなかった(図4aおよびbを比較)。特に、ラットの脊髄においても、ホルマリン注射の1時間後にPAMのmRNAがアップレギュレートされた(図4c)。
【0089】
PAMは、アデニリルシクラーゼ1、5および6型の強力な阻害剤であることがわかっているため(Scholich等。2001年)、PAMが、脊髄およびDRG溶解産物中でAC活性を阻害できるかどうかを調べた。脊髄において、2種の主要なACアイソフォームが半定量RT−PCRにより検出された。これらのACアイソフォームは、5および6型である(図5a)。特に、アイソフォームはいずれも、ナノモル濃度のPAMで阻害される(Scholich等,2001年)。ザイモサン注射の24〜96時間後に、脊髄におけるACアイソフォーム発現パターンは、有意に改変されなかった。興味深いことに、ホルマリン注射の1時間後に、ACアイソフォーム発現におけるシフトが検出された(図5a)。AC5型のmRNAは、ダウンレギュレートされ、AC3および9型のmRNAはアップレギュレートされる。
【0090】
上記の実験により示されたように、精製されたPAMを脊髄溶解産物へ添加することにより、脊髄調製物において、Gαsで刺激されたAC活性が阻害された。30nMのPAMの添加により、Gαsで刺激されたAC活性が、コントロール動物の脊髄溶解産物において49%減少した(図5b)。ザイモサン注射の96時間後の、ラットから得られた脊髄溶解産物におけるGαsで刺激されたAC活性は、PAMの阻害に対して、未処理動物と比較して高い感受性を示した(30nMで、70%阻害;図5b)。それに対して、ホルマリンで1時間処理された動物からの脊髄溶解産物におけるPAMによるAC活性の阻害の程度は、より少なかった(30nMで25%阻害;図5b)。
【0091】
DRGにおいて、優勢に発現されたACアイソフォームは、AC4および6型である(図5a)。DRG溶解産物におけるGαsで刺激されたAC活性は、脊髄溶解産物と比較して30nMのPAMの存在下で32%減少した(図5b)。
【0092】
脊髄の侵害受容伝達におけるPAMの可能性のある役割を試験するために、ホルマリン分析を行う前に、動物に、PAMのセンスおよびアンチセンスオリゴヌクレオチドを、腰髄内へのカテーテルにより注入した。免疫組織化学で観察されたように、脊髄ニューロンにおいて、PAM発現は減少した(図6a)。PAMのアンチセンスオリゴヌクレオチドの注入により、ホルマリン注射後の侵害刺激反応が、PAMのセンス処理と比較して顕著に増加した(p=0.007;図6bおよびc)。PAMのアンチセンス処理ラットにおける痛覚過敏には、足をなめる挙動、および噛みつき挙動の増加が伴った。PAMは、AC活性阻害剤であるため(Scholich等,2001年)、センスおよびアンチセンスODNで処理されたラットの脊髄溶解産物における、基礎的なGαsおよびフォルスコリン刺激によるAC活性を測定した。この実験により、アンチセンスで処理されたラットにおいて、基礎的なAC活性の顕著な増加(20.7%)が示された(表1)。それに対して、Gαsおよびフォルスコリン刺激によるAC活性において顕著な変化は検出されず、これは、ODN処理によってACの総量が変化しなかったことを示す(表1)。
【0093】
上記の実験より、PAMは、脊髄とDRGニューロンの両方の細胞体と軸索に局在化していることが示された(図2a,b)。細胞核では、ほんのわずかな免疫活性しか検出で
きず、これは、ニューロンおよびガン細胞系において、PAMに関する機能が異なることを示している。
【0094】
持続的な侵害受容の刺激の後の中枢性感作は、脊髄におけるニューロンおよびシナプスの変化に基づく(WoolfおよびCostigan 1999年;WoolfおよびSalter2000年;JiおよびWoolf 2001年)。発明者等による、PAMは、脊髄およびDRGの感覚ニューロンで発現されるという発見により、PAMは、脊髄の侵害受容過程の際のシナプスの変化に関与する可能性があるという新たな仮説が構築された。上記の、PAMは、侵害受容の刺激の後にアップレギュレートされるという発見は(図4a)、この、脊髄の侵害受容過程の際のシナプスの変化においてPAMは役割を果たす可能性があるという仮説を裏付けている。
【0095】
さらに、発明者等の、PAMは、脊髄およびDRGの知覚神経で発現されるという驚くべき発見により、同様に、脊髄およびDRGにおいて、PAMは、AC活性を阻害することができるかどうかという疑問に至った。
【0096】
上記の実験より、PAMは、脊髄調製物におけるGαsで刺激されたAC活性の有力な阻害剤であることが初めて示された(図5b)。AC活性は、30nMのPAMを添加した後に、50%減少した。抑制性Gタンパク質のαサブユニット、Gαiを用いて匹敵する阻害を達成するために、200〜800nMのGαiを用いなければならない(Wittpoth等,1999年)。ザイモサンで96時間処理された動物の脊髄調製物において、PAMの阻害作用はいっそう強く(図5b)、ザイモサン注射の後に、脊髄における内因性PAMの量が上昇したことで説明することができる(図4a,b)。ホルマリンで処理した動物からの脊髄調製物におけるGαsで刺激されたAC活性の阻害(25%阻害)は、コントロール動物またはザイモサンで処理した動物と比較してそれほど顕著ではなかった(それぞれ50%および75%)。
【0097】
特に、ホルマリンで1時間処理した動物において、ACアイソフォーム発現におけるシフトを観察した(図5a)。AC3および9型はアップレギュレートされるが、AC型5は、ダウンレギュレートされた。これまで、PAMが、AC3および9型の阻害剤であるかどうかはわかっていない。それゆえに、これらアイソフォームは、PAMによって阻害されないか、または、試験されたPAM濃度が、阻害作用を達成するには低すぎる可能性がある。PAMは、510kDaの巨大タンパク質であるため、30nMを超えるPAM濃度を試験することは技術的に不可能である。それにもかかわらず、図5bに示される用量反応曲線によれば、試験された脊髄調製物において、より高いPAM濃度により、Gαsで刺激されたAC活性のより強い阻害が引き起こされる可能性がある。
【0098】
興味深いことに、PAMは、DRGにおいて、脊髄調製物においてより有効性が弱いAC酵素活性の阻害剤であった。脊髄およびDRG調製物においてPAMの阻害効率が異なるのは、観察されたACアイソフォーム発現の差による可能性が最も高い。脊髄で発現される主要なACアイソフォームは、5および6型であり、いずれもPAMによって強く阻害される(図5a;(Scholich等,2001年))。DRGにおいて、AC4および6型は、優勢なACアイソフォームである(図5a)。PAMがAC4型を阻害するかどうかはわかっていないため、このアイソフォームは、PAMによって阻害されないか、または、それに加えて、試験されたPAM濃度が、阻害作用を達成するには低すぎる。しかしながら、図5bに示される用量反応曲線によれば、DRG調製物において、より高いPAM濃度により、Gαsで刺激されたAC活性のより強い阻害が引き起こされる可能性が高いようである。
【0099】
しかしながら、最も驚くべきことは、PAM活性は、試験動物の侵害受容の挙動に影響
を与えるという発見であった:これは、基礎的なAC活性における顕著な増加(表1)、および、より重要なことは、ホルマリン注射後、侵害刺激反応がPAMのセンス処理と比較して顕著に増加したこと(図6bおよびc)、その際、脊髄におけるPAMの内因性の発現は、動物にPAMのアンチセンスオリゴヌクレオチドを注入することによって減少したこと(図6a)を示す発明者等の実験によって初めて実証された。
【0100】
19.PAMの鎮痛作用の測定
上述したPAMの鎮痛作用に関する証拠は、例えば、以下の仮想的実験によって裏付けることができるだろう:例えば急性の痛みのホルマリンモデルにおけるPAMの鎮痛作用は、例えば、アミノ酸残基1028〜1065に対応するペプチドの髄腔内への適用によって直接決定することができるだろう。このペプチドは、酵母ツーハイブリッドシステムとAC活性分析で測定されるように、PAM−アデニリルシクラーゼの相互作用に介在できることが発見された最小の領域を示す。このペプチドは、バイオポーター(bioporter)リポフェクション試薬(Peqlab,ドイツで市販されている)との複合体として適用され得る。このアプローチにより、ペプチドを組織に進入させ、ACに対する生理学的なPAMの作用を模擬することが可能になる。
【0101】
20.PAMシグナル伝達に対するS1Pの影響を示す結果
HeLa細胞におけるPAM発現と局在化を調査するために、ヒトPAMのアミノ酸残基135〜153および4601〜4614に対応するペプチドに対して向けられた2種のPAMに対する抗体が用いられた。ラット脳の免疫組織学的染色の比較により、両方の抗体が、同じ脳の領域(同様にPAMのmRNA発現を示す)を認識することが示された(Yang等,2002年)。血清欠乏状態にしたHeLa細胞において、両方の抗体は、PAMとカルネキシン、小胞体マーカーとの共存を示した(図18a)。血清を細胞に添加した後に、部分的なPAMの細胞質膜へのトランスロケーションが観察された(図18b)。PAMは、血清処理の20〜30分後に膜に出現し、血清とインキュベートして1時間後に膜から消失し始めた。用いられた抗体で観察されたHeLaにおけるPAMの細胞分布は、Guo等によって説明されている細胞分布とは異なっている。Guo等は、PAMの一部を用いて、核タンパク質に共通のモチーフを含む抗体を生産させているため、この抗体による核タンパク質との交差反応が起こる可能性がある。
【0102】
PAMは、AC酵素活性の強力な阻害剤なので、次に、ERから細胞質膜へのPAMのトランスロケーションにより、AC活性が阻害されるかどうかを調べた。HeLa細胞の血清処理により、細胞内cAMPの蓄積が減少した(図19a)。加えて、血清処理により、Gαsおよびフォルスコリン刺激によるAC活性が、未処理細胞と比較してそれぞれ56.7%および64.7%減少した(図19b)。観察されたAC活性の減少は、ACアイソフォームのmRNA発現における変化が検出されなかったことから、ACアイソフォーム発現における変化、または、AC発現の増加によるものではなかった(図19c)。これまでにScholich等,2001年で説明された通りに、刺激したAC活性の減少に、PAMが介在するかどうかを決定するために、PAMに対するアンチセンスオリゴヌクレオチドを用いて内因性PAMの量を減少させた。図19dに示すように、アンチセンスODNで処理されたHeLa細胞において、PAMの量(ウェスタンブロット解析によって決定された)は、センスまたは突然変異アンチセンスODNで処理した細胞と比較して減少した。抗Hsp70抗体を用いて同じブロットを再度検査したところ、タンパク質のローディングは同じであることが示された(図19d)。しかしながら、アンチセンスODNでHeLa細胞を処理すると、Gαsおよびフォルスコリン刺激によるAC活性の血清誘発性の阻害が、著しく減少した(図19e)。重要なのは、HeLa細胞のセンスまたは突然変異ODNでのトランスフェクトは、Gαsおよびフォルスコリン刺激によるAC活性の血清誘発性の阻害に影響しないことである(図19e)。これらのデータにより、血清でHeLa細胞を刺激した後、内因性PAMは、AC活性に阻害作用を与えることが示される。
【0103】
PAMの細胞質膜へのトランスロケーションを誘発する血清因子を同定するために、血清因子を、逆相カラム、陰イオン交換カラム、およびゲルろ過カラムを用いて精製した。各精製工程の後に、分画のPAMのトランスロケーションを誘導しACを阻害する能力に関して試した。精製の特性によれば、血清因子は、わずかに疎水性であり(フェニル−セファロースカラムから0.3MのNaClで溶出)、強い負電荷を有し(モノQ、および、Q−セファロースカラムから0.7MのNaClで溶出した)、および、スーパーデックス30ゲルろ過カラムでの保持時間によれば推定分子量が500未満と同定された。物理的な特性に従って、数種の候補物質が試験されたが、そのうちスフィンゴシン−1−リン酸のみがPAMのトランスロケーションを誘発した。
【0104】
S1Pは、5種のGタンパク質共役受容体のファミリーに結合できる。それゆえに、半定量的RT−PCRで、HeLa細胞がS1P受容体を発現するかどうかを調べた。HeLa細胞で、5種のS1P受容体アイソフォームのうち4種のmRNA(S1P1〜S1P4)が検出された(図20a)。次に、精製されたS1Pが、PAMの活性化/トランスロケーションに対して血清と同じ特性を示すかどうかを試した。まず、精製されたS1Pの濃度を徐々に高めてHeLa細胞を処理した。0.1〜5μMのS1Pで処理した細胞で処理した細胞の70〜90%で、PAMの細胞質膜へのトランスロケーションが起こった。PAMは、500nMのS1Pと10分間インキュベートした後に細胞質膜で出現し、インキュベートして1時間後に消失し始めた(図3b,c)。最も重要なことには、HeLa細胞を0.5μMのS1Pで処理することによって、細胞内のcAMP含量が減少し(図21a)、同様に、Gαsで刺激されたAC活性が減少した(図21b)。Gαsで刺激したAC活性は、S1Pとインキュベートした後3分以内、かつ部分的に回復する前に減少した。S1P処理を開始して5〜10分後に、Gαsで刺激したAC活性は、再び減少した(図4b)。PAMに対するアンチセンスODNは、S1Pとインキュベートして60分後に、Gαsで刺激されたAC活性の阻害を消滅させた(図21c)。総合すると、これらデータにより、HeLa細胞におけるS1PによるAC活性の阻害は、2つの異なるメカニズム:迅速なPAM非依存性のAC阻害(3〜10分のS1P処理)、および、遅延性のPAM依存性のAC阻害(10〜60分のS1P処理)によって達成されることが初めて実証された。
【0105】
それぞれの受容体に結合することによって、S1Pは、4種の異なるGタンパク質、Gi、Gq、G12およびG13を活性化する可能性を有することが実証された(Hla等,Science,2001年;Kluk等,BBA,2002年;SiehlerおよびManning,BBA,2002年;SpiegelおよびMilstein,JBC,2002年)。これらから、Giのみが百日咳毒素感応性である。百日咳毒素処理は、Gαsで刺激されたAC活性に対するS1Pの阻害作用(図22a)、および、PAMの細胞質膜へのトランスロケーション(図22b)を消滅させた。このようにして、抑制性Gタンパク質Giは、S1Pによる迅速なPAM非依存性阻害に関与する可能性が高いようである。
【0106】
次に、PAMのトランスロケーションと活性化を引き起こすシグナル伝達経路のさらなる構成要素を研究した。S1P1〜S1P4受容体は、Gi、GqおよびG12/13に共役しており(Hla等,2001年;Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)、PAMのトランスロケーション、同様に、AC阻害は百日咳毒素依存性であると説明されているため(図22a)、上記のデータにより、HeLa細胞におけるPAMの活性化は、Gi活性化に依存していることが示される。これまで、S1Pは、ホスホリパーゼC(PLC)、同様に、Giの活性化を介したERK1/2シグナル伝達を活性化できることが説明されている(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)。このようにして、PLC活性化が、PAMのトランスロケーションに関与しているかどうかを試したところ、PLC阻害剤U73122の存在下で、PAMのトランスロケーションと後期のAC阻害が止められることを発見できた(図22a)。PLCは、ホスファチジルイノシトール4,5−二リン酸を、イノシトール1,4,5−三リン酸(IP3)、カルシウム動員セカンドメッセンジャーと、1,2−ジアシルグリセロール(DAG)、タンパク質キナーゼC(PKC)の活性化因子に変換する(Rebecchi等,2000年;Wilde等,2001年)。カルシウムのイメージングにより、S1Pは、HeLa細胞におけるPLC依存性のカルシウムの増加を誘発することが示された。しかしながら、BAPTA−AMでの前処理は、PAMのトランスロケーションを妨害しないため、PAMのトランスロケーションにこのようなカルシウムの減少は必要ない(図22a)。その上、PKC阻害剤GF109203Xと、RO31−8220は、それぞれPAMのトランスロケーションとAC阻害を消滅させた(図22a)。これらのデータにより、S1Pは、抑制性Gタンパク質Giを介してPLCを活性化することが示される。その後、PLC作用により、PAMのトランスロケーションを媒介するのに必要なカルシウム非依存性のPKC活性化と、遅延性のS1P誘発性AC阻害が起こる。
【0107】
S1Pは、ERK1/2シグナル伝達経路を活性化できることも示されているため(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)、HeLaにおいて、EK1/2は、S1Pによって活性化されるかどうかを試した。抗活性ERKと抗ホスホTyr183ERK抗体を用いて、HeLaをS1Pとインキュベートした後、ERK1/2のリン酸化が検出できた(図22b)。驚くべきことに、ERK1/2のリン酸化は、PLC活性化(図22b)、GiおよびPKC活性依存性であったが(データ示さず)、細胞内のカルシウムの増加には非依存性であった。しかしながら、ERK1/2の活性化は、PAMのトランスロケーションまたはAC活性の阻害に必要なかった(図22a)。総じて、これら発見により、S1Pは、PAMのトランスロケーション、それに続く、Gi、PLCおよびPKCを含むシグナル伝達カスケードを介したAC酵素活性の阻害を誘発することが初めて示された。興味深いことに、S1Pはさらに、ERK1/2の活性化と、細胞内のカルシウム濃度の増加を誘発し、これらは両方、PAMのトランスロケーションまたはGαiで刺激したAC活性の阻害に必要なかった。
【0108】
S1Pで調節されたシグナル伝達経路は、集中的な調査の焦点である(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)。S1P受容体は、Gi依存性メカニズムによってAC活性を阻害できることは周知である(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)。しかしながら、ここで、遅延性のAC阻害は、PLCおよびPKC活性化に依存しているため、S1Pで刺激した後の長期にわたるAC活性の阻害は、抑制性Gタンパク質によるACの直接的な阻害によるものではないことが初めて示された。その上、発明者等のデータにより、S1P処理後のHeLa細胞における持続性のAC活性の阻害は、PAMの細胞質膜へのトランスロケーション/活性化に依存することが示される。このトランスロケーションは、Gi活性化と、PLC/PKCシグナル伝達によって調節される。PAMは、AC酵素活性の有力な阻害剤であるため、S1P誘導性のPAM依存性のAC阻害は、PAMとACとの直接的な相互作用の結果と考えられるが、このことはなお証明の必要がある。上記の発明者等の実験により、HeLa細胞において、PAMは、小胞体に局在しており、血清処理の後に細胞質膜へトランスロケーションすることが初めて発見された。PAMのトランスロケーションには、未処理のHeLa細胞と比較して、Gαsおよびフォルスコリン刺激によるAC活性の減少が伴った。細胞をPAMに対するアンチセンスオリゴヌクレオチドで前処理すると、AC阻害を妨害したことから、AC阻害にはPAMが介在されていた。以下で、我々は、PAMのトランスロケーションに関与する血清因子としてスフィンゴシン−1−リン酸(S1P)を同定した。HeLa細胞を0.1〜5μMのS1Pで処理することにより、10〜30分以内に細胞の80%においてPAMの細胞質膜へのトランスロケーションが誘発された。2つの別個のメカニズムによって、S1Pは、AC活性を減少させた。最初のAC阻害にPAMは介在していないが、百日咳毒素感応性であった。長期のS1P処理の後、AC阻害は、PAMのトランスロケーションに依存性であった。PAMのトランスロケーションに対するS1Pの作用と、PAMが介在するAC阻害は、百日咳毒素感応性であり、PLCおよびPKCの活性化を必要とした。総合すると、これらデータにより、PAM活性の調節因子が初めて同定された。その上、S1Pによる長期のAC活性の阻害は、ERから細胞質膜へのAC阻害タンパク質PAMのトランスロケーションが介在することを示すことができた。
【0109】
21.S1Pの鎮痛作用の測定
S1Pの鎮痛の作用を測定するために、S1Pを髄腔内への投与によって脊髄へ搬送した。
【0110】
21a)腰髄内へのカテーテル埋め込み:
ラットを、ケタミン(60mg/kg,腹腔内)と、ミダゾラム(0.5〜1mg/kg,腹腔内)で麻酔した。脊椎骨Th13からL3までの脊柱の上で皮膚を切開した。L2〜3周辺の筋肉組織を取り除いた。L3の棘突起を除去し、L2で椎弓切除術を施した。次に、ポリエチレン製カテーテル(ID0.28mm、OD0.61mm)を、カテーテルの先端がTh9〜10に達するように硬膜外腔に挿入した。カテーテルをシアノアクリレート系接着剤で固定し、頚部の領域で外部に置き、皮膚を縫合した。
【0111】
21b)PAMオリゴヌクレオチドの注入。
外科手術の3日後に、ラットを「自由行動系」(CMA,ストックホルム,スウェーデン)の状態に置き、カテーテルを通じて、10μMのS1P20μlを注入した。
【0112】
21c)ホルマリン試験:
注入を止めた後15分以内に、ホルマリン試験を行った。5%ホルムアルデヒド溶液50μlを、1本の後肢の背面の皮下に(s.c.)注射した。ホルマリン注射の直後に開始して、1分間のインターバルで60分間まで、フリンチ行動をカウントした。5分間のインターバルのフリンチ行動を、1分あたりの平均フリンチ行動として要約した。グループ間の侵害受容の挙動を比較するためにフリンチ行動の1時間の観察期間の合計を、スチューデントのt検定で処理した。ホルマリン試験の最後に、腰髄および後根神経節(DRG)を切り出し、液体窒素中で急速冷凍し、さらなる解析まで−80℃で保存した。
【0113】
21d)結果:
成体ラットに、ホルマリン注射の15分前に、10μMのS1P20μlまたはPBS/DMSO20μlを髄腔内へ投与した。次に、5分間のインターバルのフリンチ行動を、60分間にわたりカウントした。第2A相(ホルマリン注射の20〜35分後)における侵害受容性の反応の数が、PBS/DMSO処理動物と比較して、顕著に減少したことが検出された(図24を参照)。これら実験により、外因性のS1Pが、鎮痛薬として作用することが証明された。
【0114】
22.S1P受容体アゴニストの鎮痛/抗侵害受容作用の測定
S1P受容体アゴニスト(例えばFTY720)の鎮痛/抗侵害受容作用、は、例えば、以下の仮想的実験により裏付けることができるだろう:例えばFTY720の、例えば急性の痛みのホルマリンモデルにおける鎮痛/抗侵害受容作用は、例えばFTY720を髄腔内または静脈内投与し、例えばフリンチ試験によってその鎮痛/抗侵害受容作用を連続的に試験することによって直接決定することができるだろう。このアプローチにより、分子を、組織に進入させ、ACに対する生理学的なS1Pの作用を模倣させることが可能になると思われる。
【0115】
文献
Bailey C.H., Bartsch D., Kandel E.R.: Toward a molecular definition of long-term memory storage. Proc. Natl. Acad. Sci. U.S.A. 1996 Nov 26;93(24):13445-52;
Brandon E.P., Idzerda R.L., McKnight G.S.: PKA isoforms, neural pathways, and behaviour: making the connection. Curr. Opin. Neurobiol. 1997 Jun;7(3):397-403;
Bek M.J., Zheng S., Xu J., Yamaguchi I., Asico L.D., Sun X.G. and Jose P.A. (2001) Differential expression of adenylyl cyclases in the rat nephron. Kidney Int 60, 890-899;
Brinkmann, V., Davis, M.D., Heise, C.E., Albert, R., Cottens, S., Hof,R., Bruns, C., Prieschl, E., Baumruker, T., Hiestand, P., Foster, C.A., Zollinger, M. and Lynch, K.R., (2002), The Immune Modulator FTY720 Targets Sphingosine-1-Phosphate Receptors; the Journal of Biological Chemistry, Vol.277, No.24, June 14, p.21453 to 21457;
Caligan, T.B., Peters, K., Ou, J., Wang, E., Saba, J., and Merrill, A.H., Jr.(2000) Anal Biochem 281, 36-44
Chang Q. and Balice-Gordon R.J.(2000) Highwire, rpm-1, and futsch: balancing synaptic growth and stability. Neuron 26, 287-290;
Chen, Z., Nield, H.S., Sun, H., Barbier, A., and Patel, T.B.(1995) J Biol Chem 270, 27525-27530
Chomczynski, P., Sacchi, N.: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, (1987) 156-159;
DiAntonio, A., Haghighi, A.P., Portman, S.L., Lee, J.D., Amaranto, A.M., and Goodman, C.S.(2001) Nature 412, 449-452
Graziano, M.P. FreissmuthM. Gilman A.G.: Purification of recombinant Gs alpha.
Meth. Enz. (1991) 195: 192-215;
Graziano M.P., Freissmuth M. and Gilman A.G. (1991) Purification of recombinant Gs alpha. Methods Enzymol 195, 192-202;
Guo Q., Xie J., Dang C.V., Liu E.T., Bishop J.M.: Identification of a large Myc-binding protein that contains RCC1-like repeats. Proc. Natl. Acad. Sci. U.S.A. 1998 Aug 4;95(16):9172-7);
Grossberger, R., Gieffers, C., Zachariae, W., Podtelejnikov, A.V., Schleiffer,
A., Nasmyth, K., Mann, M., and Peters, J.M.(1999) J Biol Chem 274, 14500-14507;
Hla, T., Lee, M.J., Ancellin, N., Paik, J.H., and Kluk, M.J. (2001) Science 294, 1875-1878;
Jin Y. (2002) Synaptogenesis: insights from worm and fly. Curr Opin Neurobiol 12, 71-79.);
【0116】
Julius and Basbaum“Molecular mechanisms of nociception”, Nature, volume 413,
13. September 2001, pp.203-209;
Kassis, S., and Fishman, P.H.(1982) J Biol Chem 257(9), 5312-5318;
Kind, P.C., and Neumann, P.E. (2001) Trends Neurosci 24, 553-555;
Kluk, M.J., and Hla, T. (2002) Biochim Biophys Acta 1582, 72-80
Mandala, S., Hajdu, R., Bergstrom, J., Quackenbush, E., Xie, J., Milligan, J.,
Thornton, R., Shei, G., Card, D., Keohand, C., Rosenbach, M., Hale, J., Lynch, C.L., Rupprecht, K., Parsons, W. and Rosen, H., (2002), Alteration of Lymphocyte Trafficking by Spingosine-1-Phospate Receptor Agonists, Science, Vol.296, April 2002;
Meller S.T., Gebhart G.F.(1997), intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat; Eur.J. Pain 1, 43-52;
Nair, B.G., Parikh, B., Milligan, G., and Patel, T.B.(1990) J Biol Chem 265(34), 21317-21322;
Nestler, E.J.(2001) Nat Rev Neurosci 2, 119-128
Patel T.B., Wittpoth C., Barbier A.J., Yigzaw Y. and Scholich K. (2002) Functional analyses of type V adenylyl cyclase. Methods Enzymol 345, 160-187. Snyder S.H.(1985) Adenosine as a neuromodulator. Annu Rev Neurosci 8, 103-124;
Payne, S.G., Milstien, S., and Spiegel, S.(2002), Sphingosine-1-phosphate: dual messenger functions; FEBS Letters 531 (2002), p.54 to 57;
Postma, F.R., Jalink, K., Hengeveld, T., and Moolenaar, W.H.(1996) Embo J 15, 2388-2392
Rebecchi, M.J., and Pentyala, S.N.(2000) Physiol Rev 80, 1291-1335
Ruppert C., Goldowitz D., and Wille W.(1986), Proto-oncogene-c-myc is expressed in cerebellar neurons at different developmental stages, Embo J5, 1897-1901;
Sato, K., Tomura, H., Igarashi, Y., Ui, M., and Okajima, F.(1997) Biochem Biophys Res Commun 240, 329-334
Schaefer A.M., Hadwiger G.D. and Nonet M.L.(2000) rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26, 345-356);
【0117】
Schaible H.G., Vanegas H.: How do we manage chronic pain? Baillieres Best. Pract. Res. Clin. Rheumatol. 2000 Dec;14(4):797-811;
Scherr M., Morgan M.A., Eder M., Gene Silencing Mediated by Small Interfering RNAs in Mammalian Cells, Curr Med Chem. 2003, Feb; 10 (3): 245-256;
Scholich, K., Mullenix, J.B., Wittpoth, C., Poppleton, H.M., Pierre, S.C., Lindorfer, M.A., Garrison, J.C., and Patel, T.B. (1999) Science 283, 1328-1331
Scholich K., Pierre S., Patel T.B.: Protein associated with myc (PAM) is a potent inhibitor of adenylyl cyclase. J. Biol. Chem. 2001, Dec 14;276(50):47583-9;
Scholz and Woolf“Can we conquer pain”, Nature neuroscience supplement, volume 5, November 2002, pp.1062-1067;
Siehler, S., and Manning, D.R. (2002) Biochim Biophys Acta 1582, 94-99
Snyder S.H. (1985), Adenosine as a neuromodulator. Annual Reviews of Neuroscience 8, 103-104;
Spiegel, S. and Milstien, S.(2000), Functions of a new family of sphingosine-1-phosphate receptors, Biochimica et Biophysica Acta 1484, p.107 to 116;
Spiegel, S., and Milstien, S.(2002) J. Biol. Chem 277; 25851-25854;
Trajkovic V, Samardzic T, Stosic-Grujicic S, Ramic Z, Mostarica Stojkovic M. Muramyl dipeptide potentiates cytokine-induced activation of inducible nitric oxide synthase in rat astrocytes. Brain Res. 2000 Nov 10;883(1):157-63;
Wan H.I., DiAntonio A., Fetter R.D., Bergstrom K., Strauss R. and Goodman C.S. (2000) Highwire regulates synaptic growth in Drosophila. Neuron 26, 313-329.);
West, A.E., Chen, W.G., Dalva, M.B., Dolmetsch, R.E., Kornhauser, J.M., Shaywitz, A.J., Takasu, M.A., Tao, X., and Greenberg, M.E.(2001) Proc Natl Acad Sci USA 98, 11024-11031;
【0118】
Wilde, J.I., and Watson, S.P.(2001) Cell Signal 13, 691-701
Wittpoth C., Scholich K., Yigzaw Y., Stringfield T.M. and Patel T.B.(1999), R
egions on adenylyl cyclase that are necessary for inhibition of activity by beta
gamma G(iα) subunits of heterotrimeric G proteins. Proc. Natl. Acad. Sci. USA 96, 7723-7730;
Wood, J.D.“Pathobiology of Visceral Pain: Molecular Mechanisms and Therapeutic Implications II. genetic approaches to pain therapy”, American Journal pf Physiological Gastrointestinal Liver Physiology, 2000, volume 278, G507-G512;
Woolf and Mannion“Neuropathic pain: aetiology, symptoms mechanisms, and management”, The LANCET, volume 353, June 5, 1999, pp.1959-1964;
Woolf J. and Salter M.W.“Neuronal Plasticity: Increasing the Gain in Pain”, Science, volume 288, June 9, 2000, pp.1765-1768;
Xia Z., Storm D.R.: Calmodulin-regulated adenylyl cyclases and neuromodulation. Curr. Opin. Neurobiol. 1997 Jun;7(3):391-6;
Xu, D., Isaacs, C., Hall, I.P., and Emala, C.W.(2001) Am J Physiol Lung Cell Mol Physiol 281, L832-843
Yang H., Scholich K., Poser S., Storm D., Patel T.B., Goldowitz D.: Developmental expression of protein associated with myc (PAM) in the rodent brain. Brain Res Dev Brain Res 136, 2002, 35-42;
Zhen M., Huang X., Bamber B. and Jin Y.(2000) Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26, 331-343);
【0119】
実験方法に関する標準的な文献:
特に他の規定がない限り、実験方法は、以下の標準的な文献で列挙された標準的な方法に従って行われた、または、行うことができる:
Sambrook et al. (1989) Molecular Cloning: A Laboratory Manual. Second edition.
Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp or Current Protocols in Molecular Biology;
Current Protocols in Molecular Biology; regularly updated, e.g. Volume 2000; John Wiley & Sons, Inc; Editors: Fred M. Ausubel, Roger Brent, Robert Eg. Kingston, David D. Moore, J.G.Seidman, John A. Smith, Kevin Struhl.
Current Protocols in Human Genetics; regularly uptdated, e.g. Volume 2003; John Wiley & Sons, Inc; Editors: Nicholas C. Dracopoli, Honathan L. Haines, Bruce R. Korf, Cynthia C. Morton, Christine E. Seidman, J.G. Seigman, Douglas R. Smith.
Current Protocols in Protein Science; regularly updated, e.g. Volume 2003; John Wiley & Sons, Inc; Editors: John E. Coligan, Ben M. Dunn, Hidde L. Ploegh, David W. Speicher, Paul T. Wingfield.
Molecular Biology of the Cell; third edition; Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K., Watson, J.D.; Garland Publishing, Inc. New York & London, 1994;
Gene Targeting: a practical approach (1995), Editor: A.L. Joyner, IRL Press
Genetic Manipulation of Receptor Expression and Function; D. Accili, Wiley-Liss., USA, 2000; ISBN: 0-471-35057-5.
Antisense Therapeutics, S. Agrawal, Humana Press, USA, 1996, ISBN: 0-89603-305-8.
Remington's Pharmaceutical Sciences, Edition 17, 1985
【0120】
用いられた略語:
AC、アデニリルシクラーゼ;Gαs、アデニリルシクラーゼの促進性Gタンパク質のαサブユニット、Gαs*、Gαsの構成的に活性な(Q213L)突然変異体;Gαi、抑制性Gタンパク質Giのαサブユニット;Gβγ、ヘテロ三量体Gタンパク質のβγサブユニット;ODN、オリゴデオキシヌクレオチド;PAM、Myc関連タンパク質;RCC1、染色体凝縮の調節因子;S1P、スフィンゴシン−1−リン酸;TED、50mMトリスHCl(pH8.0)、1mMのEDTA、1mMのDTT;RT、室温。
【0121】
図面
図1:PAMは、脊髄ニューロンで高度に発現される。脊髄の水平切片標本を用いたインサイチュハイブリダイゼーションを、上述したように、ラットPAMに対するセンスまたはアンチセンスプローブとハイブリダイズさせた。
図2:PAMは、DRGニューロンで、同様に、ラット脊髄中のニューロンの細胞で発現される。
パネルA:ラットの脊髄切片標本の免疫組織化学的な解析。切片標本を、抗PAM抗体(緑)、および、抗NeuNまたは抗GFAP(赤)で染色し、ニューロンまたはグリア細胞をそれぞれ可視化した。両方のシグナルの重ね合わせは、右のパネルに示される。対物を20×に拡大した。
パネルB:ラットDRG切片標本の免疫組織化学的な解析。切片標本を、抗PAM抗体(緑)、および、抗Neu68、抗ヒストンまたは抗GFAP(赤)で染色し、それぞれニューロン、核またはグリア細胞を可視化した。両方のシグナルの重ね合わせは、右のパネルに示される。対物を40×に拡大した(ただし、ヒストン染色は、63×に拡大した)。
【0122】
図3:PAMは、DRGおよび脊髄において、異なる発生段階で差異的に発現される。定量RT−PCR(TaqmanTM)を用いて、16日齢のラット胎児(E16)、出生後の日数が0.5(P0.5)、3(P3)、5(P5)、9(P9)のラット、および、成体ラットの脊髄およびDRGのRNA(40ng)中のPAMを検出した。少なくとも3回の測定の平均±SEMを示す。
図4:PAMは、ラットの脊髄において、ザイモサンおよびホルマリン処理の後にアップレギュレートされた。
パネルA:コントロール動物、または、ザイモサンで処理して24時間、48時間および96時間後の動物からの脊髄のRNA(40ng)を用いたRT−PCR解析。下部のパネルは、7回の実験の平均±SEMを示す。スチューデントのt検定:*p<0.001。
パネルB:コントロール動物、または、ザイモサンで処理して24時間、48時間および96時間後の動物のラットの脊髄溶解産物(40μg)を用いた、抗PAM抗体および抗ERK1/2を用いた、7%SDS−PAGEゲルを用いたウェスタンブロット解析。
パネルC:コントロール動物、または、ホルマリンで1時間処理した動物からの脊髄のRNA(40ng)を用いた定量RT−PCR解析。
【0123】
図5:PAMは、脊髄溶解産物中でGαsで刺激されたAC活性を阻害する。
パネルA:RT−PCRを用いて、脊髄およびDRGのRNA(40ng)におけるACアイソフォーム発現を決定した。
パネルB:材料および方法で説明したように、脊髄またはDRGの溶解産物(10μg)を、80nMのGαsの存在下で、AC活性に関して分析した。三連で行われた少なくとも3回の測定の平均±SEMを示す。
図6:PAMに対するアンチセンスODNの髄腔内への適用により、侵害受容の挙動が増加する。
パネルA:説明されているように、成体ラットに、センスおよびアンチセンスODNを髄腔内投与した。ホルマリン処理の後に、脊髄を除去し、抗PAM抗体(緑)、または、
抗NeuN(赤)を用いて免疫組織学的な解析で処理した。
パネルB:材料の章において説明されているように、センスまたはアンチセンスODNで処理した動物のホルマリンアッセイ。
1時間にわたるフリンチ行動の合計を示す。少なくとも4回の測定の平均±SEを示す。
パネルC:材料の章において説明されているように、センスまたはアンチセンスODNで処理した動物のホルマリンアッセイ。
1時間にわたるフリンチ行動の回数を示す。
少なくとも4回の測定の平均±SEを示す。
【0124】
図7:NCBI登録番号AAC39928(タンパク質配列;配列番号1)、AF075587(コード配列;配列番号2)による、ヒトPAMのタンパク質、ゲノムおよびコードヌクレオチド配列。ヒトPAMは、染色体13q22上に位置する;そのゲノム配列は、NT_024524.11(始点:24679861位;終点:24962245位;配列番号3)で公開された形で利用可能である;
図7Cは、24679861〜24962245位の連続した配列を示す。
図8:ラットPAMに関するEST−クローンをコードする配列:
図8A:AW921303(hscDNAのbp960〜1394に対応する;配列番号4)
図8B:AW918711(hscDNAのbp8188〜8632に対応する;配列番号5)
図8C:BQ201485(hscDNAのbp8966〜9633に対応する;配列番号6)
図8D:BE112881(hscDNAのbp10311〜10830に対応する;配列番号7)
図8E:AW441131(hscDNAのbp13569〜14152に対応する;配列番号8)
図8F:BF409872(hscDNAのbp13569〜14807に対応する)。(配列番号9)
【0125】
図9:Guo等,1988年/Grossberger等,JBC 1999年、および、Scholich等,JBC 2001年)によるヒトPAMのドメイン構造の概観。
図10:ラットPAM RTPCRのためのPCRプライマー。
図11:ラットPAM発現を阻害するためのアンチセンスオリゴデスオキシヌクレオチド、および、コントロールオリゴヌクレオチド。
図12:本発明の環境で使用するための、様々なPAMhsポリペプチド。これらのポリペプチドは、配列番号2に記載のポリペプチドから得られたフラグメントである。
図13:本発明の環境で使用するための、異なるPAMhsポリヌクレオチド。
図13A:hsPamcDNAのヌクレオチドの1317〜4366位を含む、配列番号17に記載のタンパク質フラグメントをコードするcDNA配列(配列番号24);
図13B:hsPamcDNAのヌクレオチドの1482〜3332位を含む、配列番号18に記載のタンパク質フラグメントをコードするcDNA配列(配列番号25);
図13C:hsPamcDNAのヌクレオチドの1641〜3341位を含む、配列番号19に記載のタンパク質フラグメントをコードするcDNA配列(配列番号26);
図13D:hsPamcDNAのヌクレオチドの3142〜4046位を含む、配列番号20に記載のタンパク質フラグメントをコードするcDNA配列(配列番号27);
図13E:hsPamcDNAのヌクレオチドの3142〜3446位を含む、配列番号21に記載のタンパク質フラグメントをコードするcDNA配列(配列番号28);
図13F:hsPamcDNAのヌクレオチドの3228〜3839位を含む、配列番
号22に記載のタンパク質フラグメントをコードするcDNA配列(配列番号29);
図13G:hsPamcDNAのヌクレオチドの3228〜3341位を含む、配列番号23に記載のタンパク質フラグメントをコードするcDNA配列(配列番号30);
【0126】
図14:上記の発見によるPAMシグナル伝達。
図15:スフィンゴシン−1−リン酸の構造;
図16:S1P受容体のmRNAおよびアミノ酸配列、
16a)S1P1のアミノ酸配列(配列番号31);
16b)S1P1のmRNA配列(配列番号32);コード配列は、244位で始まり、1392位で終わる、
16c)S1P2のアミノ酸配列(配列番号33);
16d)S1P2のmRNA配列(配列番号34);コード配列は、1位で始まで始まり、1062位で終わる;
16e)S1P3のアミノ酸配列(配列番号35)
16f)S1P3のmRNA配列(配列番号36)、コード配列は、1位で始まり、1137位で終わる;
16g)S1P4のアミノ酸配列(配列番号37)
16h)S1P4のmRNA配列(配列番号38)、コード配列は、23位で始まり、1177位で終わる;
16i)S1P5のアミノ酸配列(配列番号39)
16j)S1P5のmRNA配列(配列番号40)、コード配列は、10位で始まり、1206位で終わる:
図17:FT720の構造
図18:PAMは、HeLa細胞において、血清での刺激の後に、ERから細胞質膜へトランスロケーションする。
パネルA:上述したように、血清欠乏状態にしたHeLa細胞(24時間)を固定し、抗PAM抗体(緑)と、抗カルネキシン抗体(赤)で染色した。
パネルB:HeLa細胞を、様々な時間で10%ウシ胎仔血清で処理し、抗PAM抗体で染色し、細胞内局在をモニターした。
【0127】
図19:HeLa細胞の血清処理により、PAM依存性プロセスを介してAC活性が減少する。
パネルA:上述したように、血清の非存在および存在で、HeLa細胞におけるcAMPの蓄積を測定した。3回の測定の平均±SEM(標準誤差)を示す。
パネルB:材料および方法の章で説明したように、血清の非存在および存在で、HeLa細胞溶解産物のアデニリルシクラーゼ活性を測定した。Gsα*(80nM;灰色のバー)で、および、フォルスコリン(100μM;黒いバー)で刺激した、ACVの特異的活性は、それぞれ124±10pmol/mg/分、および、464±89pmol/分/mgであった。基礎的な活性は、11.75±2pmol/mg/分であった。少なくとも2回の実験(それぞれ三連で行われた)の平均±SEを示す。スチューデントのt検定:*p<0.001。
パネルC:血清欠乏状態にしたHeLa細胞において、10%FBSで1時間インキュベートした後の、ACアイソフォーム発現のRT−PCR解析。
パネルD:上述したように、血清欠乏状態にしたHeLa細胞を、アンチセンス、センスおよび3つの点突然変異(3M−as)を含むアンチセンスODNをそれぞれ3□Mで用いてトランスフェクトした。細胞を回収し、7%SDS−PAGE(タンパク質30μg)を用いて、抗PAM抗体および抗Hsp70抗体を用いてウェスタン解析に付した。
パネルE:上述したように、血清欠乏状態にしたHeLa細胞を、センス、アンチセンス、および、3M−as ODNで処理した。
24時間後、細胞を、10%FBSと30分間インキュベートした。上述したように、
細胞を回収し、Gαs*(80nM;灰色のバー)、および、フォルスコリン(100μM;黒いバー)の存在下でAC活性を決定した。少なくとも3回の実験(三連で測定された)からのデータを、平均±SEMとして示す。スチューデントのt検定:*p<0.01、**p<0.05。
【0128】
図20:S1Pは、PAMの細胞質膜へのトランスロケーションを誘発させ、PAM依存性のAC酵素活性を阻害する。
パネルA:血清欠乏状態にしたHeLa細胞におけるS1P受容体アイソフォーム発現のRT−PCR解析。
パネルB:HeLa細胞を、0.5μMのS1Pで30分間処理し、抗PAM抗体で染色し、細胞内局在をモニターした。
パネルC:HeLa細胞におけるPAMのトランスロケーションの時間依存性。血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで様々な時間で処理し、抗PAM抗体で染色し、細胞内局在をモニターした。PAMのトランスロケーションを示す細胞のパーセンテージを示す。
図21:HeLa細胞のS1P処理により、PAM依存性プロセスを介してAC活性が減少する。
パネルA:材料および方法の章で説明したように、0.5μMのS1Pが非存在下(黒いバー)、および、0.5μMのS1Pが存在下(灰色のバー)での、様々な時間での、HeLa細胞におけるcAMPの蓄積を測定した。3回の測定の平均±SEMを示す。
パネルB:血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで様々な時間で処理した。材料および方法で説明したように、細胞を回収し、80nMのGαs*の存在下でAC活性を決定した。少なくとも4回の実験(三連で測定された)からのデータを、平均±SEとして示す。
パネルC:材料および方法の章で説明されているように、血清欠乏状態にしたHeLa細胞を、センス、アンチセンス、および、3M−as ODNで処理した。24時間後、細胞を、0.5μMのS1Pと指定された時間インキュベートした。
細胞を回収し、上述したように、Gαs*(80nM)の存在下でAC活性を決定した。データを、3回の測定の平均±SEMとして示す。
【0129】
図22:PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に、PLC/PKCシグナル伝達経路が介在する。
パネルA:血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで、3分間(灰色のバー)、または、60分間(黒いバー)処理した。
S1Pとのインキュベートの前に、細胞を、1μg/μlの百日咳毒素(PTX)と24時間インキュベートした。80nMのGαs*の存在で、AC活性を決定した。少なくとも2回の実験(三連で測定された)からのデータを、平均±SEとして示す。
パネルB:血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで処理した。S1Pとのインキュベートの前に、細胞を、10μMのU73122、RO31−8220、BAPTA−AMまたはU0126と20分間インキュベートした。80nMのGαs*の存在下で、AC活性を決定した。少なくとも2回の実験(三連で測定された)からのデータを、平均±SEとして示す。
パネルC:0.5μMのS1Pで10分間処理したHeLa細胞のウェスタンブロット解析。S1Pとのインキュベートの前に、細胞を、10μMのU73122、BAPTA−AMまたはU0126と20分間インキュベートした。沸騰したレムリー緩衝液中で細胞を回収し、抗活性ERK1/2抗体を用いてウェスタン解析で処理した。ローディングコントロールとして抗Hsp70抗体を用いた。
パネルD:HeLa細胞を、0.5μMのS1Pで20分間処理し、抗PAM抗体で染色し、細胞内局在をモニターした。S1P処理の前に、細胞を、1μg/μlの百日咳毒素(PTX)と24時間、1μMのGF109203X、10μMのU0126、BAP
TA−AM、U73122またはRo31−223と20分間、インキュベートした。
図23:PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に至る提唱されたシグナル伝達経路の模式図。
図24:ホルマリン分析における、60分間にわたるS1P適用を行った場合と、行わない場合の成体ラットの痛み行動。
【0130】
ホルマリン注射の前に、成体ラットに、10μMのS1P20μl、または、PBS/DMSO20μlを髄腔内へ15分間適用することによって投与した。ホルマリン注射の後に、フリンチ行動を60分間にわたり5分間のインターバルでカウントした。6回の動物実験の平均+SEMを示す。
【0131】
【表1】

【0132】
アンチセンス処理ラットの脊髄溶解産物において、基礎的なAC活性は上昇する。上述したように、脊髄溶解産物(20μg)を、80nMのGαs、または、100μMのフォルスコリンの非存在下、または存在下でAC活性に関して分析した。グループあたり少なくとも3匹のラットからの脊髄溶解産物の平均AC活性±SEM(それぞれ三連で2回測定された)を示す(ns=有意差なし)。
【図面の簡単な説明】
【0133】
【図1】PAMは、脊髄ニューロンで高度に発現される。
【図2A】PAMは、DRGニューロンで、同様に、ラット脊髄中のニューロンの細胞で発現される。
【図2B】PAMは、DRGニューロンで、同様に、ラット脊髄中のニューロンの細胞で発現される。
【図3】PAMは、DRGおよび脊髄において、異なる発生段階で差異的に発現される。
【図4】PAMは、ラットの脊髄において、ザイモサンおよびホルマリン処理の後にアップレギュレートされた。
【図5】PAMは、脊髄溶解産物中でGαsで刺激されたAC活性を阻害する。
【図6】PAMに対するアンチセンスODNの髄腔内への適用により、侵害受容の挙動が増加する。
【図6−1】図6の続きである。
【図7】NCBI登録番号AAC39928(タンパク質配列;配列番号1)、AF075587(コード配列;配列番号2)による、ヒトPAMのタンパク質、ゲノムおよびコードヌクレオチド配列である。
【図7−1】図7の続きである。
【図7−2】図7−1の続きである。
【図7−3】図7−2の続きである。
【図7−4】図7−3の続きである。
【図7−5】図7−4の続きである。
【図7−6】図7−5の続きである。
【図7−7】図7−6の続きである。
【図7−8】図7−7の続きである。
【図7−9】図7−8の続きである。
【図7−10】図7−9の続きである。
【図7−11】図7−10の続きである。
【図7−12】図7−11の続きである。
【図7−13】図7−12の続きである。
【図7−14】図7−13の続きである。
【図7−15】図7−14の続きである。
【図7−16】図7−15の続きである。
【図7−17】図7−16の続きである。
【図7−18】図7−17の続きである。
【図7−19】図7−18の続きである。
【図7−20】図7−19の続きである。
【図7−21】図7−20の続きである。
【図7−22】図7−21の続きである。
【図7−23】図7−22の続きである。
【図7−24】図7−23の続きである。
【図7−25】図7−24の続きである。
【図7−26】図7−25の続きである。
【図7−27】図7−26の続きである。
【図7−28】図7−27の続きである。
【図7−29】図7−28の続きである。
【図7−30】図7−29の続きである。
【図7−31】図7−30の続きである。
【図7−32】図7−31の続きである。
【図7−33】図7−32の続きである。
【図7−34】図7−33の続きである。
【図7−35】図7−34の続きである。
【図7−36】図7−35の続きである。
【図7−37】図7−36の続きである。
【図7−38】図7−37の続きである。
【図7−39】図7−38の続きである。
【図7−40】図7−39の続きである。
【図7−41】図7−40の続きである。
【図7−42】図7−41の続きである。
【図7−43】図7−42の続きである。
【図7−44】図7−43の続きである。
【図7−45】図7−44の続きである。
【図7−46】図7−45の続きである。
【図7−47】図7−46の続きである。
【図7−48】図7−47の続きである。
【図7−49】図7−48の続きである。
【図7−50】図7−49の続きである。
【図7−51】図7−50の続きである。
【図7−52】図7−51の続きである。
【図7−53】図7−52の続きである。
【図7−54】図7−53の続きである。
【図7−55】図7−54の続きである。
【図7−56】図7−55の続きである。
【図7−57】図7−56の続きである。
【図7−58】図7−57の続きである。
【図7−59】図7−58の続きである。
【図7−60】図7−59の続きである。
【図7−61】図7−60の続きである。
【図7−62】図7−61の続きである。
【図7−63】図7−62の続きである。
【図7−64】図7−63の続きである。
【図7−65】図7−64の続きである。
【図7−66】図7−65の続きである。
【図7−67】図7−66の続きである。
【図7−68】図7−67の続きである。
【図7−69】図7−68の続きである。
【図7−70】図7−69の続きである。
【図7−71】図7−70の続きである。
【図7−72】図7−71の続きである。
【図7−73】図7−72の続きである。
【図7−74】図7−73の続きである。
【図7−75】図7−74の続きである。
【図7−76】図7−75の続きである。
【図7−77】図7−76の続きである。
【図7−78】図7−77の続きである。
【図7−79】図7−78の続きである。
【図7−80】図7−79の続きである。
【図7−81】図7−80の続きである。
【図7−82】図7−81の続きである。
【図7−83】図7−82の続きである。
【図7−84】図7−83の続きである。
【図7−85】図7−84の続きである。
【図7−86】図7−85の続きである。
【図7−87】図7−86の続きである。
【図7−88】図7−87の続きである。
【図7−89】図7−88の続きである。
【図7−90】図7−89の続きである。
【図7−91】図7−90の続きである。
【図7−92】図7−91の続きである。
【図7−93】図7−92の続きである。
【図7−94】図7−93の続きである。
【図7−95】図7−94の続きである。
【図7−96】図7−95の続きである。
【図7−97】図7−96の続きである。
【図7−98】図7−97の続きである。
【図7−99】図7−98の続きである。
【図7−100】図7−99の続きである。
【図7−101】図7−100の続きである。
【図7−102】図7−101の続きである。
【図7−103】図7−102の続きである。
【図7−104】図7−103の続きである。
【図7−105】図7−104の続きである。
【図7−106】図7−105の続きである。
【図7−107】図7−106の続きである。
【図7−108】図7−107の続きである。
【図7−109】図7−108の続きである。
【図7−110】図7−109の続きである。
【図7−111】図7−110の続きである。
【図7−112】図7−111の続きである。
【図7−113】図7−112の続きである。
【図7−114】図7−113の続きである。
【図7−115】図7−114の続きである。
【図7−116】図7−115の続きである。
【図7−117】図7−116の続きである。
【図7−118】図7−117の続きである。
【図7−119】図7−118の続きである。
【図7−120】図7−119の続きである。
【図7−121】図7−120の続きである。
【図7−122】図7−121の続きである。
【図7−123】図7−122の続きである。
【図7−124】図7−123の続きである。
【図7−125】図7−124の続きである。
【図7−126】図7−125の続きである。
【図8】ラットPAMに関するEST−クローンをコードする配列である。
【図8−1】図8の続きである。
【図8−2】図8−1の続きである。
【図9】Guo等,1988年/Grossberger等,JBC 1999年、および、Scholich等,JBC 2001年)によるヒトPAMのドメイン構造の概観である。
【図10】ラットPAMRT−PCRのためのPCRプライマーである。
【図11】ラットPAM発現を阻害するためのアンチセンスオリゴデゾキシヌクレオチド、および、コントロールオリゴヌクレオチドである。
【図12】本発明の環境で使用するための、様々なPAMhsポリペプチド。これらのポリペプチドは、配列番号2に記載のポリペプチドから得られたフラグメントであるである。
【図12−1】図12の続きである。
【図12−2】図12−1の続きである。
【図13】本発明の環境で使用するための、異なるPAMhsポリヌクレオチドである。
【図13−1】図13の続きである。
【図13−2】図13−1の続きである。
【図13−3】図13−2の続きである。
【図13−4】図13−3の続きである。
【図14】上記の発見によるPAMシグナル伝達である。
【図15】スフィンゴシン−1−リン酸の構造である。
【図16】S1P受容体のmDNAおよびアミノ酸配列である。
【図16−1】図16の続きである。
【図16−2】図16−1の続きである。
【図16−3】図16−2の続きである。
【図16−4】図16−3の続きである。
【図16−5】図16−4の続きである。
【図16−6】図16−5の続きである。
【図17】FT720の構造である。
【図18】PAMは、HeLa細胞において、血清での刺激の後に、ERから細胞質膜へトランスロケーションする。
【図19】HeLa細胞の血清処理により、PAM依存性プロセスを介してAC活性が減少する。
【図19−1】HeLa細胞の血清処理により、PAM依存性プロセスを介してAC活性が減少する。
【図20】S1Pは、PAMの細胞質膜へのトランスロケーションを誘発させ、PAM依存性のAC酵素活性を阻害する。
【図21】HeLa細胞のS1P処理により、PAM依存性プロセスを介してAC活性が減少する。
【図22】PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に、PLC/PKCシグナル伝達経路が介在する。
【図22−1】図22の続きである。
【図23】PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に至る提唱されたシグナル伝達経路の模式図である。
【図24】ホルマリン分析における、60分間にわたるS1P適用を行った場合と、行わない場合の成体ラットの痛み行動である。
【技術分野】
【0001】
本発明は、S1P(スフィンゴシン−1−リン酸)の使用に関する。本発明の他の形態は、医薬のスクリーニング方法、および、痛みを治療する方法に関する。
【背景技術】
【0002】
痛みは、実際の、または潜在的な組織のダメージに反応して起こる複雑で主観的な感覚であり、それに対する感情的な応答である。急性の痛みは、潜在的な、または実際の外傷を示す生理学的なシグナルである。慢性の痛みは、体性性(器質性)または心因性のいずれかであり得る。慢性の痛みはしばしば、自律神経性の徴候を伴うか、または、その後に自律神経性の徴候が続くため、場合によっては、うつ状態に陥る。
【0003】
体性性の痛みは、侵害受容性、炎症性、または、神経障害性によるものと考えられる。侵害受容性疼痛は、体性または内臓の痛みを感じる神経線維の継続的な活性化と同じと判断されている。神経障害性の痛みは、神経系の機能障害から生じる;末梢神経系、CNS、または、その両方における異常な体性感覚のプロセスによって持続すると考えられている。(痛みのメカニズムの総論については、例えば、ScholzおよびWoolf,2002年;JuliusおよびBasbaum,2001年,WoolfおよびMannion,1999年;Wood,J.D.,2000年;WoolfおよびSalter,2000年を参照)。
【0004】
慢性の痛みは、個人が被る、加えて社会的、経済的な途方もない費用を発生させる。現存する薬理学的な痛みの治療は、有効性と安全性の両方に関してはなはだ満足のいくものではない。
【0005】
これまで、痛みの治療には、2つのクラスの鎮痛薬が主として用いられている:非オピオイド鎮痛薬、大部分がアセトアミノフェンとNSAID(非ステロイド系抗炎症薬)、および、オピオイド(麻酔性)アゴニスト(ここにおいて、「オピオイド」は、CNS中の特異的なオピオイド受容体に結合し、アゴニスト作用を生じさせる天然または合成物質の一般名称である)。遺憾ながら、両方の鎮痛薬クラスであるオピオイドおよび非オピオイドは、いくつかの不要な副作用がある。オピオイドの最も重篤な副作用は、呼吸器系を阻害する可能性と、長期の治療の後に依存症になる可能性である(Schaible H.G.,Vanegas H.,2000年)。一方で、非オピオイドの主要なクラスであるNSAIDは、多種多様な潰瘍や出血のような胃腸の合併症、加えて腎臓障害を誘導する可能性がある(Schaible H.G.,Vanegas H.;2000年)。米国では毎年、従来のNSAIDによって引き起こされる重度の胃腸の合併症のために約16,000人の患者が死亡していると推測されている。
【発明の開示】
【発明が解決しようとする課題】
【0006】
最先端の痛みの治療に関連する深刻な欠点を考慮すると、痛みを調節する薬物の新規のクラスの多大な必要性がある。特別には、急速に進む痛みの神経生物学の理解と、最先端の治療の欠点がない有効な治療を提供するという未だ満たされていない臨床上の必要性との間にある大きなギャップを考慮すると、新規の鎮痛薬のクラスに関する新しい標的の発見に努力を向ける必要がある。従って、本発明の目的は、痛みを調節する薬物の新しいクラスを開発し、提供する新しい手段を提供することである。
【0007】
この目的は、痛みを調節する医薬化合物を製造するための、S1Pまたはそれらの機能
的フラグメントもしくは誘導体の使用によって解決される。
【課題を解決するための手段】
【0008】
本発明は、侵害受容過程におけるS1Pの関連と、その痛みを減少させる能力を初めて実証した発明者等の発見に基づく。S1Pに関して機能的な、という用語、または、S1Pの機能という用語は、S1Pが、その受容体の少なくとも1種と相互作用する能力、好ましくはその受容体を活性化する能力、または、細胞内のcAMPレベルを低める能力、または、PAM(Myc関連タンパク質)の小胞体から細胞膜へのトランスロケーションを媒介する能力を意味し;より好ましくは、S1Pの機能という用語は、そのPAM活性を増強する能力(すなわち、PAMが、ACと相互作用する能力、および/または、AC活性を低める能力、および/または、痛みを減少させる能力)、および/または、AC活性を阻害する能力、さらにより好ましくはその痛みを減少させる能力を意味する。S1P受容体に関して機能的という用語、または、S1P受容体の機能という用語は、S1P受容体が、S1Pと相互作用する能力、より特定には、S1Pの相互作用によって発生する前記受容体に典型的なシグナルを媒介する能力、さらに特定には、S1Pの相互作用によって発生する痛みのプロセシングに影響を与える能力を意味する。
【0009】
S1Pのフラグメントは、図15に記載の野生型分子より小さいあらゆるフラグメントであり得る。PAMのフラグメントは、対応する野生型より短いあらゆるポリペプチド、または、ポリヌクレオチド断片であり得る。S1P受容体のフラグメントは、対応する野生型より短いあらゆるポリペプチド、または、ポリヌクレオチド断片であり得る。
【0010】
S1PまたはS1Pフラグメントの誘導体は、S1Pの機能を有する分子のあらゆる改変、または、化学的もしくは生物学的な改変のようなその他のあらゆる種類の改変を含んでもよく、このような改変としては、例えば、分子を安定化すること、または、それらの例えば所定の細胞への特異的なターゲティングを調節すること、または、その細胞への進入もしくは細胞による摂取を容易にすることが挙げられ;既知の改変の一つとしては、S1Pの水酸化またはメチル化が挙げられる。それらの必要な部位へのターゲティングと細胞への進入を確実にする、または、容易にするのに適した改変または添加剤が有用である。一方で、それらの脊髄へのターゲティングを確実にするために、適切なカテーテルなどを用いた脊髄内への適用のような局所的な適用なども考えられる。その他の有用な添加剤としては、それらの安定化等のための、塩(生理学的に許容できる有機または無機塩のための塩、例えば、Remington's Pharmaceutical Sciences,1418頁,1985年を参照)、緩衝液などが挙げられる。
【0011】
S1Pは、特異的な受容体を介して細胞に吸収されるため、外部からの適用が可能であり、特異的に吸収されると予想される。S1Pターゲティングの調節は、例えば、S1P受容体をクローニングし、所望の細胞で発現させることにより実現することができる。細胞型に特異的な発現は、適切な遺伝子のプロモーター/エンハンサーを用いて確実にすることができ、これらは、当業界で既知である。
【0012】
本発明は、発明者等の脊髄および後根神経節(DRG)内での感作メカニズムにおけるS1Pの驚くべき関連を初めて実証した研究に基づく。
【0013】
S1P(スフィンゴシン−1−リン酸)は、スフィンゴシンのリン酸化された誘導体であり、全てのスフィンゴ脂質の構造上の主鎖である。この細胞外(血清介在)スフィンゴ脂質は、異なる組織で差異的に発現され、それぞれ特異的な細胞の作用を調節するS1P1〜S1P5という5種の特異的なGタンパク質共役受容体(GPCR)の一つに結合することによって多種多様な細胞性プロセスを調節することがわかっている(総論のために、例えばPayne等,2002年;および、SpiegelおよびMilstien,2
000年を参照)。細胞外S1Pの既知の機能としては、例えば、細胞の移動、細胞の生存または血管新生の調節が挙げられる。その細胞外の作用のほかに、細胞内メッセンジャーとして作用することも知られている(総論のために、例えばPayne等,2002年;および、SpiegelおよびMilstien,2000年を参照)。しかしながら、その侵害受容プロセスにおける関連は未だわかっていない。
【0014】
PAM(Myc関連タンパク質)は、510kDaの巨大タンパク質である。PAMのタンパク質、ゲノムおよびコードポリヌクレオチド配列は当業界で既知であり、例えば、NCBI(National Centre for Biotechnology Information;National Library of Medicine,Building 38A,ベセスダ,メリーランド州20894,米国;www.ncbi.nhm.nih.gov)データベースより、登録番号AAC39928(コード配列;配列番号1)、AF075587(タンパク質配列;配列番号2)で公けに利用可能である。ヒトPAMは、染色体13q22上に位置する;そのゲノム配列は、NT_024524.11(始点:24679861位;終点:24962245位;配列番号3)で、公けに利用可能である。あるいは、これらのタンパク質およびコード配列は、KIAA0916、タンパク質登録番号NP_055872(タンパク質配列)、および、NM_015057(コード配列)で、公けに利用可能である。
【0015】
ラットPAMについて、以下のEST−クローンをコードする配列が公開された形で利用可能である:
AW921303(hscDNAのbp960〜1394に対応する;配列番号4)、
AW918711(hscDNAのbp8188〜8632に対応する;配列番号5)、
BQ201485(hscDNAのbp8966〜9633に対応する;配列番号6)、
BE112881(hscDNAのbp10311〜10830に対応する;配列番号7)、
AW441131(hscDNAのbp13569〜14152に対応する;配列番号8)、
BF409872(hscDNAのbp13569〜14807に対応する;配列番号9)。
【0016】
PAMはもともと、MycのN末端における転写活性化ドメインと特異的に相互作用する能力によって同定された(Guo Q.等,1998年)。PAMは近年、強力なAC活性阻害剤と説明されているが(Scholich K.,Pierre S.,Patel T.B.:Protein associated with myc(PAM)is a potent inhibitor of adenylyl cyclase. J.Biol.Chem.2001年12月14日;276(50):47583〜9)、侵害受容過程および感作におけるそれらの機能に関する証拠は未だ示されていない。
【0017】
むしろ、PAMは、シナプス前部の成長の調節において役割を果たすと考えられている:PAMのmRNAは、海馬、歯状回および小脳などの特定の解剖学的な領域で高度に発現されることがわかっている。成体ラットおよびマウスの脳におけるPAMおよびMyc発現の両方は、小脳の成熟したプルキンエ細胞、ならびに、海馬の顆粒および錐体細胞に限定される(Ruppert C.等,1986年;Yang H.等,2002年)。しかしながら、これら細胞型のいずれも、痛みのプロセシングおよび感作に関与することはわかっていない。
【0018】
ショウジョウバエにおけるPAM相同体(highwire)、および、C.エレガンスにおけるPAM相同体(rpm−1)は、シナプス前終末の組織化(Zhen等,20
00年)、シナプスの成長の調節(Wan等,2000年)、シナプス形成、ならびに、軸索成長およびターゲティング(Schaefer等,2000年)において重要な役割を果たすことが示されている。これらの発見により、highwire、rpm−1およびそれらの哺乳動物相同体PAMは、シナプスの成長の負の調節因子として作用する可能性があるという仮説に至った(Chang等,2000年;Jin Y.2002年)。それに沿って、小脳、海馬および歯状回におけるPAM発現の劇的な増加が、これら構造体における主要なシナプス形成期の際に観察された(Yang等,2002年)。
【0019】
齧歯類における脳の発達の際に、PAM発現は、誕生直後に開始し、最初の2週間の間中アップレギュレートされ、その後、PAM発現は、成人期の間中高いレベルを保つ(Yang等,2002年)。これまで、脊髄およびDRGにおけるPAMの発現および調節、ならびに、痛みの感作メカニズムおよび調節におけるその機能についてはわかっていない。
【0020】
これまで、ヒトPAMは、サイクリックAMP(cAMP)シグナル伝達の強力な調節因子であり、数種のアデニリルシクラーゼ(AC;E.C.4.6.1.1)アイソフォームの酵素活性をナノモル濃度で阻害する(Scholich等,2001年)ことが実証されている。
【0021】
遍在的なサイクリックAMP(cAMP)のセカンドメッセンジャーシステムは、細胞外の刺激を細胞内のシグナルおよび応答に翻訳する様々なシグナル変換メカニズムの一つである。細胞外の刺激を受けると、Gタンパク質共役受容体(GPCR)は、三量体GTP結合調節タンパク質(Gタンパク質)を介して細胞質膜に結合した酵素またはイオンチャンネルを調節する。GPCRによって活性が調節される酵素の一つは、アデニリルシクラーゼ(AC)、cAMP生成酵素である。このようにして、入ってくる細胞外の刺激は、細胞内のメディエイターであるサイクリックAMPの細胞内の濃度に影響を与える。cAMPレベルの上昇は、タンパク質キナーゼA(PKA)を刺激し、特異的な細胞内の標的タンパク質をリン酸化し、それによってそれらの活性を改変することによって細胞に影響を与える。
【0022】
それぞれのタイプの細胞は、特徴的な一連のGPCR、これらGPCRによって調節される酵素、アデニリルシクラーゼ(AC)の特異的なサブセットおよび標的タンパク質を有し、これらは、比較的非特異的な、または一般的に生じるプレイヤー(例えば遍在的なcAMP)と共に作用して、各細胞が、入ってくる細胞外シグナルに対してそれらに特有の応答を生じさせるようにできる。例えば、サイクリックAMP(cAMP)セカンドメッセンジャーは、シナプス形成性の調節において主要な役割を果たすことがわかっている(Bailey等,1996年;Xia等,1997年;Brandon等,1997年);一方で、cAMPは、代謝プロセスおよび細胞の増殖に関与する。このようにして、遍在的なcAMPメッセンジャーシステムの役割と、その様々な成分は、様々な組織および細胞型の様々な特殊化に従って多様である。
【0023】
これまで、S1P受容体として作用する、S1P1〜S1P5という5種の異なるGPCRは当業界で既知である(総論のために、例えば、Spiegel,S.およびMilstien,S.,2000年を参照)。様々なS1P受容体のタンパク質およびコードポリヌクレオチド配列が当業界で既知であり、例えばNCBI(National Centre for Biotechnology Information;National Library of Medicine,Building 38A,ベセスダ,メリーランド州20894,米国;www.ncbi.nhm.nih.gov)データベースより、登録番号:NM_001400(配列番号32;ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体、1mRNA(EDG1/S1P1)のヌクレオチド配列;NP_001391(配列番号31,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体(EDG1/S1P1)のタンパク質配列;NM:_004230(配列番号34,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体、5(EDG5/S1P2)mRNAのヌクレオチド配列;NP_004221(配列番号33,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体、5(EDG5/S1P2)のタンパク質配列;NM_005226(配列番号36,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体3(EDG3/S1P3)mRNAのヌクレオチド配列;NP:005217(配列番号35,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体3(EDG3/S1P3)のタンパク質配列;NM:003775(配列番号38,ホモサピエンスの内皮分化、Gタンパク質共役受容体6(EDG6/S1P4)mRNAのヌクレオチド配列;CAA04118(配列番号37,ホモサピエンスの内皮分化、13−タンパク質共役受容体6(EDG6/S1P4)のタンパク質配列;NM_030760(配列番号40,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体8(EDG8/S1P5)mRNのAヌクレオチド配列;NP_110387(配列番号39,ホモサピエンスの内皮分化、スフィンゴ脂質Gタンパク質共役受容体8(EDG8/S1P5)のタンパク質配列で公けに利用可能である。
【0024】
本発明のその他の形態は、医薬品、好ましくは痛みを予防または治療するための医薬品として使用するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体に関する。
【0025】
本発明のさらなる形態は、痛みの調節のための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用に関する。この調節は、好ましくは軽減または予防または全体的な抑制である。
【0026】
その上、本発明は、痛みを調節する化合物を同定するためのS1Pまたはそれらの誘導体の機能的フラグメントの使用を包含する。上記調節する化合物は、好ましくは、S1P活性を模擬または増強する化合物である。最も好ましくは、それらは、痛みを予防する、減らす、または、止める能力を有する。
【0027】
上記化合物は、例えば、それらの以下の能力によって同定することが可能である;
a)S1Pの機能(すなわち、その受容体またはそれらのフラグメントの少なくとも1種と相互作用する能力、好ましくは受容体を活性化する能力、または、細胞内のcAMPレベルを低める能力、または、PAMの小胞体から細胞膜へのトランスロケーションを媒介する能力;より好ましくは、そのPAM活性を増強する能力、および/または、AC活性を阻害する能力、さらにより好ましくは、その痛みを減少させる能力)、または、PAMの機能(すなわち、その、細胞内cAMPレベルを低める能力、ACのようなその他のファクター、特にACと相互作用する能力、ACを阻害する能力、または、その痛みの感覚を低める能力)を模擬する、回復させる、活性化する、または、増強することを意味しており、または、
b)S1Pの血清レベルを増加させること(例えば、S1Pの生産を活性化または増強することによって、または、その細胞外の分解を減少させることによって)、または、
c)少なくとも1つのS1P受容体の発現を増強すること(すなわち、その転写、転写物の安定化、翻訳またはその翻訳後プロセシングの活性化によって;その翻訳後修飾の調節によって、または、その安定化の活性化、または、その分解の阻害などによって)、または、
d)S1Pの生産または分解に関与する酵素と相互作用すること。
【0028】
本発明のその他の形態は、十分な量のS1Pまたはそれらの機能的フラグメントもしくは誘導体を個体に投与することを含む、痛みを予防する、または、減らす方法に関する。
【0029】
投与は、S1Pの作用部位(DRGまたは脊髄)へのターゲティングを可能にする方法で適切に予備的に行われるべきであり、例えば、S1P誘導体または製剤の血流への全身投与(例えば、静脈内または経口での適用)によって、または、S1Pまたはそれらのフラグメントもしくは誘導体の局所的な(例えば脊髄内の)適用によってなされる。
【0030】
本発明のその他の形態は、痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法に関し、以下の工程を含む:
a.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、S1Pを含まないサンプルを提供する工程、
b.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、同様に、S1Pを含む第二のサンプルを提供する工程、
c.少なくとも第一のサンプルと、化合物とを接触させる工程、
d.サンプル中のPAM活性を測定する工程、
e.前記化合物のS1Pの機能を模擬する能力を測定する工程。
【0031】
本方法は、前記細胞を、前記化合物の代わりにS1Pと接触させ、上記のc)およびd)に係るPAM活性を、S1Pの存在下でのPAM活性と比較する工程をさらに含んでもよい。
【0032】
その他の例としては、以下の工程を含む方法が示される:
a)S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現する細胞を含む2つのサンプルを提供する工程、および、
b)一方のサンプルと、前記化合物とを接触させる工程、および、
c)両方のサンプル中の受容体活性を測定する工程。
【0033】
本発明のその他の形態に係る方法は、以下の工程を含む:
a)S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現する細胞を含む2つのサンプルを提供する工程、および、
b)一方のサンプルと、S1Pとを接触させる工程、および、
c)他方のサンプルと、化合物とを接触させる工程、および、
d)両方のサンプル中の受容体活性またはS1Pおよび受容体の相互作用を測定する工程。
【0034】
PAMまたはS1P受容体は、本発明の様々な形態に係る特定の目的が実現されるようなあらゆる配列から得られたものが利用可能である。好ましくは、PAMまたはS1P受容体はヒト由来である。
【0035】
本発明の様々な形態に関して、PAMまたはS1P受容体が、単離されたポリペプチド、または、オリゴもしくはポリヌクレオチドである場合も好ましい。本発明の様々な形態の環境で「単離された」とは、自然源から少なくとも部分的に精製されているか、または、組換え分子(これらは当然ながら、精製されていてもよいし、または部分的に精製されていてもよい)を意味する。
【0036】
アッセイとは、生物学的プロセスをモニターすることができるあらゆるタイプの分析方法のことである。薬物のスクリーニングで使用するためには、このようなアッセイは、再現可能でなければならず、好ましくは拡張可能で、頑強でもある。このようなアッセイは、好ましくは、化学物質の、痛みを調節する(好ましくは減少させる)および/または痛みを予防する能力に関するハイスループットスクリーニングに適している。ハイスループットスクリーニングの大部分は、特定の能力に関して異なる約500,000種の化合物をスクリーニングすることを含む。このアッセイのタイプは、例えば、用いられる分子のタイプ(ポリペプチドまたはポリヌクレオチドのいずれか)と「読み出し」、すなわちS1P、PAMまたはS1P受容体活性が測定される方法によって様々である(以下を参照)。
【0037】
このようなアッセイの様々なタイプが一般的に当業界で既知であり、商業的な供給元から市販されている。適切なアッセイは、標識された構成要素と標識されていない構成要素との相互作用を測定するための(例えば、PAMまたはそれらのフラグメントは、標識することができ、それらのACとの相互作用は、モニターすることができる)、放射性同位体分析、または、蛍光分析、例えば蛍光偏光分析を包含する(例えば、パンベラ(Panvera)、パーキン・エルマー・ライフサイエンス(Perkin−Elmer life sciences)(例えばLANCE)、または、パッカード・バイオサイエンス(Packard BioScience)(例えば、HTRFまたはアルファスクリーン(ALPHAscreen)TM)によって商業的に供給されているもの)。
【0038】
例えば医薬候補化合物と、受容体または機能的受容体フラグメントもしくは誘導体との相互作用を測定するためには、簡単な生化学的分析が適切である。また、より手の込んだ分析によれば、化合物が、特定の受容体を活性化し、それによりS1P活性を模倣することができるかどうかを決定することができる。
【0039】
さらなる例としては、細胞に基づくアッセイが挙げられ、この場合、細胞系は、対象の組換えタンパク質を、安定して(誘導的に、または非誘導的に;染色体の、またはエピソームの)、または、一時的に発現する。これらのアッセイは、例えばレポーター遺伝子アッセイを含み、この場合、特定のプロモーター、または、シグナル変換カスケードの構成要素のシグナル伝達経路の調節が、レポーター酵素の活性に従って測定され、このレポーター酵素の発現は、前記特定のプロモーターの制御下にある。このタイプのアッセイにとって、組換え細胞系は、それ自体が調査されるか、または、調査中のシグナル伝達カスケードによって調節される特定のプロモーターの制御下に、レポーター遺伝子が含まれるように構築されていなければならない。適切なレポーター酵素は、当業界で一般的に既知であり、ホタルルシフェラーゼ、ウミシイタケルシフェラーゼ(例えば、パッカード・リージェント(Packard Reagents)より市販されている)、β−ガラクトシダーゼが挙げられる。適切な細胞系は、アッセイ目的に応じて様々であるが、たいていは、トランスフェクトが簡単であり、培養が簡単な細胞系が挙げられ、例えばHeLa、COS、CHO、NIH−3T3などである。
【0040】
細胞内イオン濃度を測定するアッセイとしては、例えばFLIPR(蛍光測定イメージングプレートリーダー、モレキュラーデバイス(Molecular Devices)から市販されている)アッセイが挙げられ、この場合、冷却CCDカメラと連結されたアルゴンレーザー光源は、384ウェルプレートで、細胞(例えば、ニューロンの細胞、または、例えば、組換えもしくは自然に特定のイオンチャンネルを発現する細胞などの細胞)内で、一過性のイオンシグナル(例えばCa2+など)を同時測定することを可能にする。FLIPRアッセイは、例えば、Fluo−3、Fluo−4のような所定の蛍光色素を用いた細胞内のカルシウムのモニタリング、または、BCECFもしくはBCPCFもしくは特異的なFLIPRアッセイキットを用いた細胞内のpHのモニタリング、または、例えばDiBACもしくは特異的なFLIPRアッセイキットを用いた膜電位の変化の検出、または、膜の分極のモニタリングを可能にする。その他の細胞内のイオン、例えば亜鉛またはナトリウムをモニタリングするために、当業界で既知のその他の色素を用いることができる。その他のタイプのアッセイ、および、その他のタイプの読み出しは、一般的に当業者既知である。
【0041】
cAMPレベルの測定のためには、例えばアルファスクリーン、蛍光偏光法またはHTRF技術が適切である。
【0042】
イオンチャンネル活性(これは、例えば細胞内のイオン濃度を制御するため、細胞内のイオン濃度の測定に用いることができる)の測定のためには、例えば、膜電位感応性アッセイおよび色素を用いることができ、例えば、DiBAC、または、FLIPR技術に基づくモレキュラーデバイスの膜電位分析キット;FLIPR技術を用いたミトコンドリア膜の分極を測定するJC−1色素;細胞内のカルシウム濃度測定のための、Fluo−3、Fluo−4のようなイオン感応性色素、または、モレキュラーデバイスのカルシウム分析キット;細胞内のナトリウムを測定するための、ナトリウム感受性色素(例えばモレキュラープローブス製);細胞内のカリウム濃度を決定するための、パッチ−クランピングに基づくアッセイ、または、原子吸光分光分析法に基づくルビジウムイオン流出測定などを用いることができる。さらに、細胞内の所定の変化および状態を検出するための自動装置および分析方法は当業者既知であり、例えばアキュメン・バイオサイエンス(ACUMEN bioscience)のアキュメン(Acumen)検出器(適切に標識された目的物の分布の3次元再構築を可能にする蛍光ベースのレーザースキャニングリーダー)が挙げられる。
【0043】
GPCR活性の測定については、例えばcAMP測定、例えばパッカード・バイオサイエンス製のアルファスクリーンTMcAMP検出システムによる、Ca2+動員アッセイまたはレポーター遺伝子アッセイが適切である。
【0044】
PAMポリペプチドは、好ましくは、配列番号2に記載の配列を含む、または、それからなるポリペプチド、または、配列番号1または3に記載の配列を含む、または、それからなるポリヌクレオチドによってコードされるポリペプチドである。S1P受容体ポリペプチドは、好ましくは配列番号31、33、35、37または39に記載のアミノ酸配列のいずれか一つを含む、または、それからなるポリペプチド、または、配列番号32、34、36、38または40に記載のヌクレオチド配列のいずれか1つを含む、または、それからなるポリヌクレオチドによってコードされるポリペプチド、または、これらmRNA配列に含まれるコード配列によってコードされるポリペプチドである。
【0045】
PAMポリヌクレオチドは、好ましくは配列番号1または3に記載の配列を含む、または、それからなるポリヌクレオチド、または、上記のポリヌクレオチドとストリンジェントな条件下でハイブリダイズできる配列を含む、または、それからなるポリヌクレオチドである。S1P受容体ポリヌクレオチドは、好ましくは配列番号32、34、36、38または40に記載の配列を含む、または、それからなるポリヌクレオチド、配列番号32の244〜1392位、配列番号34の1〜1137位、配列番号36の1〜1062位、配列番号38の23〜1177位、または、配列番号40の10〜1206位を含む、または、それに対応するポリヌクレオチド、または、これらポリヌクレオチドのいずれか一つとストリンジェントな条件下でハイブリダイズできる配列を含む、または、それからなるポリヌクレオチドである。
【0046】
ストリンジェンシーとは、2本の一本鎖の核酸分子のハイブリダイゼーションまたはアニールの特異性に影響を与える反応条件を説明する。ストリンジェンシー、すなわち反応の特異性は、特に、反応に用いられる温度と緩衝液条件に依存する:ストリンジェンシー、すなわち特異性は、例えば、反応温度を高める、および/または、反応緩衝液のイオン強度を低めることによって増加させることができる。低いストリンジェント条件(すなわち低い反応およびハイブリダイゼーション特異性)とは、例えば、室温で、2×SSC溶液中で、ハイブリダイゼーションが行われる場合に生じる。高いストリンジェンシー条件は、例えば、68℃、0.1×SSC、および、0.1%SDS溶液中でのハイブリダイゼーション反応を含む。
【0047】
本発明の様々な形態の範囲内のストリンジェンシー条件下におけるハイブリダイゼーションは、好ましくは、以下のように理解される:
1)標識されたプローブと解析しようとする核酸サンプルとを、65℃で、または、オリゴヌクレオチドプローブの場合、オリゴヌクレオチドとサンプルとからなる二本鎖のアニーリング温度または融解温度より5℃低い温度で(アニーリング温度および融解温度は、以下では同義語とする)、一晩、50mMトリス(pH7.5)、1MのNaCl、1%SDS、10%硫酸デキストラン、0.5mg/mlの変性サケまたはニシン精子DNA中で、ハイブリダイズする。
2)2×SSC中で、室温で10分間洗浄する。
3)1×SSC/0.1%SDS中で、65℃(または、オリゴヌクレオチドの場合:アニーリング温度より5℃低い温度)で、30分間洗浄する。
4)0.1×SSC/0.1%SDS中で、65℃(または、オリゴヌクレオチドの場合:アニーリング温度より5℃低い温度)で、30分間洗浄する。
【0048】
ハイブリダイゼーションプローブとしての使用するためのオリゴヌクレオチドは、ポリヌクレオチドであり、好ましくはDNAフラグメントであって、長さが、15〜30個、好ましくは20個のヌクレオチドを有する。アニーリング温度は、式Tm=2×(A+Tの数)+4×(G+Cの数)℃に従って決定される。
【0049】
2×SSC、または、0.1×SSC(または、その他のあらゆる種類のSSC希釈液)を製造するためには、例えば、20×SSC溶液を適宜希釈する。20×SSCは、3MのNaCl/0.3Mクエン酸Na×2H2Oからなる。
【0050】
ハイブリダイゼーション反応を行う前に、上記ポリヌクレオチドは、必要に応じて、電気泳動による分離を行った後に(次に:サザンブロット(DNA)、または、ノーザンブロット(RNA))、または、電気泳動による分離を行わないで(次に:スロットまたはドットブロット)、適切なメンブレン(例えばナイロンまたはニトロセルロースメンブレン)にトランスファーする。ハイブリダイゼーションは、適切に標識されたプローブを用いて行われる。適切な標識技術は、例えば、放射性標識、または、蛍光色素を用いた標識である。上記プローブは、一本鎖ポリリボまたはポリデスオキシリボヌクレオチドであり、自然状態では一本鎖であるか、または、通常は二本鎖で変性によって一本鎖にされる。このプローブは、DNAまたはRNAサンプル(これらも一本鎖状態である)に、塩基対形成によって結合する。
【0051】
PAMフラグメントは、好ましくは上記の配列番号1、2または3に含まれるフラグメントであり、その誘導体は、好ましくは、上記の配列番号1、2もしくは3、または、それらのフラグメントから得られる。S1P受容体フラグメントは、好ましくは上記の配列番号31〜40に含まれるフラグメント、より好ましくは、配列番号32の244〜1392位、配列番号34の1〜1137位、配列番号36の1〜1062位、配列番号38の23〜1177位、または、配列番号40の10〜1206位を含む、または、それからなるフラグメントであり、その誘導体は、好ましくは、これらの配列から得られる。
【0052】
それらの機能的フラグメントまたは誘導体は、好ましくは、アデニリルシクラーゼ(AC)活性、より好ましくはACのI、VまたはVI型の活性を阻害することができる(S1P受容体に関して、最も好ましくは、S1Pが結合することによって、または、S1Pを模倣する分子が結合することによって活性化される場合に、AC活性を阻害することができる)。
【0053】
本発明の様々な形態の好ましい実施形態によれば、PAMの機能的フラグメントまたは誘導体は、ヒトPAM配列の、好ましくは配列番号2に記載のヒトPAM配列の、アミノ酸400〜1400、好ましくは446〜1062、499〜1065または1028〜1231、より好ましくは1000〜1300、さらにより好ましくは1000〜1100、さらにより好ましくは1028〜1065を含む、もしくは、それからなり、または、それらが上記それぞれのポリヌクレオチドフラグメントによってコードされる場合、特別には、配列番号2もしくは3に記載の配列に含まれる場合である。
【0054】
PAMの機能的フラグメントまたは誘導体が、ポリヌクレオチドである場合、それらは、上記のポリペプチドフラグメントをコードするポリヌクレオチドを含む、または、それからなる場合が好ましい。より特定には、それらが、ヒトPAMcdsの1482〜3332位(アミノ酸446〜1062をコードする)、または、1641〜3341(アミノ酸498〜1066をコードする)、または、3228〜3839(アミノ酸1038〜1231をコードする)を含む、または、それからなる場合が好ましい。上記フラグメントが得られるヒトPAMcdsが、配列番号2に記載の配列を有する場合がさらにより好ましい。
【0055】
痛みを調節する化合物を同定するための本発明の方法の好ましい一実施形態によれば、S1P受容体および/またはPAMを発現する細胞、好ましくは組換えS1P受容体および/またはPAMを発現する細胞が用いられる。
【0056】
上記細胞は、あらゆるタイプの細胞が可能であり、例えば、真核性または原核性の単細胞生物(例えば細菌、例えばE.coli、または、酵母、例えばS.ポンベ(S.pombe)またはS.セレビジエ(S.cerevisiae))、または、多細胞生物から得られた細胞系(例えばHeLa、COS、NIH−3T3、CHOなど)が挙げられ、なかでも哺乳動物細胞系が好ましい。
【0057】
その他の好ましい実施形態によれば、改変されていない状態と比較して低いS1P受容体活性を有する改変された細胞が用いられる。この方法において、このような細胞は、それらの痛みを調節する(好ましくは減少させる)および/または痛みを予防する能力に関して試験しようとする化学物質が、低められた、または完全に止められたS1P受容体活性を増強する、または回復させることができる場合、試験することができる。
【0058】
上記改変は、あらゆるタイプの改変(安定な、または、一過性の、好ましくは安定な)が可能であり、それにより、S1P受容体活性および/またはPAM活性(すなわち、それらの細胞内cAMPレベルを低める能力、PAMのトランスロケーション、ACを阻害する能力、または、それらの痛みの感覚を低める能力)の減少、S1P受容体またはPAM転写物の定常状態レベルの減少(すなわち、S1P受容体またはPAM転写の阻害、または、転写物の安定化によって)、または、S1P受容体またはPAMタンパク質の定常状態レベルの減少(すなわち、S1P受容体、または、PAM翻訳もしくはその翻訳後プロセシングの不活性化によって;その翻訳後修飾の調節によって、または、その安定化の不活性化によって、または、その分解を増加させることによって)が起こる。これは、例えば、S1P受容体またはPAMの優性の負変異体、アンチセンスオリゴヌクレオチド、RNAiコンストラクトを用いることによって、機能的またはゲノムS1P受容体またはPAMノックアウト(これらは、例えば誘導型が可能である)を生成することによって、または、当業界既知のその他の適切な技術によって達成することができる。上記の技術の総論としては、例えば、以下を参照:Current protocols in Molecular biology(2000年)J.G.Seidman,第23章,Supplement52,ジョン・ワイリー&サンズ社(John Wiley and
Sons,Inc.);Gene Targeting:a practical a
pproach(1995年),編集者:A.L.Joyner,IRLプレス(IRL Press);Genetic Manipulation of Receptor Expression and Function,2000年;Antisense Therapeutics,1996年;Scherr等,2003年。好ましい実施形態によれば、PAMノックアウト細胞が用いられる。ノックアウトを作製するのに適した細胞系は当業界周知であり、例えば、Current protocols in Molecular Biology(2000年)J.G.Seidman,第23章,Supplement 52,ジョン・ワイリー&サンズ社;または、GeneTargeting a practical approach.(1995年)編集者A.L.Joyner,IRLプレスが挙げられる。
【0059】
S1P活性は、例えば、その(または、そのフラグメントおよび誘導体の)受容体またはそれらの機能的フラグメントの少なくとも1種と相互作用する能力、または、PAMの細胞膜へのトランスロケーションを開始させる能力によって、直接決定することができ、または、S1P活性は、例えば、その(または、その機能的フラグメントおよび誘導体の)細胞内cAMPレベルを低める能力、ニューロン内のイオン濃度を調節する能力、ACの機能を阻害する能力、または、その痛みの感覚を調節する能力、特別には減少させる能力によって、間接的に決定することもできる。上記のパラメーターを測定するのに適した技術は当業界周知である(上記も参照):cAMPレベルは、例えば、HTRFまたはアルファスクリーンTMによって測定することができ、イオン濃度は、例えば、パッチ−クランピングまたは適切な色素によって推定することができ、痛みの感覚は、例えば、ホルマリン試験、または、機械刺激もしくは温熱性痛覚過敏の試験、または、ホットプレート試験などによって測定することができる。その受容体との相互作用は、例えば、cAMP測定、Ca2+動員またはレポーター遺伝子アッセイによって測定することができる。
【0060】
本発明のその他の形態は、痛みを調節する化合物を同定する方法に関し、以下を含む:
a)試験化合物として、S1Pの活性を調節または模倣する化合物を選択すること、および、
b)前記試験化合物を被検体に投与して、痛みが調節されているかどうかを測定すること。
【0061】
被検体は、痛みを知覚する能力を有するあらゆる被検体が可能であり、好ましくは哺乳動物であり、すなわちヒト以外の哺乳動物またはヒト(すなわち、患者の研究の範囲内で)のいずれかである。
【0062】
上記調節は、好ましくは、痛みを予防、軽減または止めることである。本発明の好ましい一実施形態によれば、上記化合物は、S1P受容体アゴニスト(総論のために、例えば、Mandala等,Science 2002年を参照)、より好ましくはFTY720(2−アミノ−2−(4−オクチルフェニル)エチル)プロパン−1,3−ジオール、図17を参照)、または、それらの機能的な誘導体または類似体(類似体は当業界で既知であり、例えば、Brinkmann等,JBC,2002年を参照)(すなわち、上述の痛みを調節する能力を有する誘導体または類似体)、好ましくはリン酸化された誘導体(Mandala等,Science 2002年を参照)である。また、上記化合物またはその誘導体もしくは類似体の生理学的に許容できる塩も適切である。
【0063】
本発明のさらなるその他の観点によれば、痛みを減らす、または予防する方法は、少なくとも1種のS1P受容体に結合し、活性化する能力、および/または、PAMの機能を活性化する活性を有する医薬化合物の十分な量を個体に投与することを含み、これも、本願の範囲内に含まれる。このような化合物の適切な例の一つは、S1P受容体アゴニスト(総論のために、例えば、Mandala等,Science 2002年を参照)、よ
り好ましくはFTY720、または、上述したような機能的な誘導体または類似体、好ましくはそれらのリン酸化された誘導体、または、上記化合物またはその機能的な誘導体もしくは類似体の生理学的に許容できる塩である。
【0064】
以下で、本発明を実施例と図によってより詳細に説明する。しかしながら、これら実施例は、本発明の範囲を限定するものではない。
【実施例】
【0065】
実施例:PAM発現パターン、ならびに、PAMおよびS1Pの機能の調査
1.材料
S1Pは、トクリス(Tocris,エリスヴィル,ミズーリ州)から購入し、抗Hsp70抗体と抗カルネキシン抗体は、BDトランスダクション・ラボ(Transduction Labs)(ベッドフォード,マサチューセッツ州)から購入した。抗活性ERK1/2抗体を、プロメガ(Promega)(マディソン,ウィスコンシン州)から得て、百日咳毒素、U0126、U73122、および、ワートマニンは、トクリス(エリスヴィル,ミズーリ州)から得て、RO31−223、BAPTA−AM、および、GF109203Xは、シグマ(Sigma)(セントルイス,ミズーリ州)から得た。
【0066】
2.動物切片標本の製造:
野生型スプラギー・ダーレー(Sprague Dawley)ラットを、チャールス・リバー・ウィガ社(Charles River Wiga GmbH,ズルツフェルト,ドイツ)から購入した。実験の前は、動物は、えさと水を自由に摂取できるようにした。それらを環境と光を制御した部屋(24+0.5℃)で維持した。各動物は、1回の実験のみで用いられた。全ての実験において、意識のある動物を研究するための倫理ガイドラインに従い、その手法は、地域倫理委員会で認証された。殺した後に、成体ラットを、0.1Mリン酸緩衝食塩水(PBS,pH7.2)中の4%パラホルムアルデヒドで、1時間潅流することによって固定した。組織を、クライオスタットを用いて、水平面で厚さ14〜16μmの切片標本にした。切片標本を、スーパーフロスト・プラス・スライド(Superfrost Plus Slides)(フィッシャー・サイエンティフィック社(Fisher Scientific Co.),ピッツバーグ,ペンシルベニア州)にマウントし、使用するまで−80℃で保存した。
【0067】
3.リボプローブの製造:
リボプローブを、これまで説明した通りにして作成した(Yang等,2002年)。ラットPAMのアンチセンス、および、センスリボプローブを、プラスミドをHindIII(アンチセンス)およびBamHI(センス)で線状化した後に、それぞれT7およびT3ポリメラーゼを用いて得た(Yang等,2002年を参照)。インビトロでの転写を、製造元(プロメガ、マディソン,ウィスコンシン州)の推奨に従って、[35S]UTP−αS(ICN,アーバイン,カリフォルニア州)、線状化したPAMcDNA、NTPの存在下で、37℃で1時間行った。RNAの転写物を、RNAプローブ精製キット(Pequlab,エルランゲン,ドイツ)を用いて精製した。
【0068】
4.インサイチュハイブリダイゼーション:
インサイチュハイブリダイゼーションを、前述した通りに行った(Yang等,2002年):切片標本を、0.1Mリン酸緩衝食塩水(pH7.2)中の4%パラホルムアルデヒドで固定し、0.25%無水酢酸と0.1Mトリエタノールアミンで前処理し、0.2×SSCでリンスし、アルコール濃度を連続的に高めて脱水させた。切片標本を、プレハイブリダイゼーション溶液(50%脱イオン化ホルムアミド、0.6M塩化ナトリウム、10mMトリスHCl(pH7.6)、50mMのEDTA、0.025%ピロリン酸ナトリウム、0.02%フィコール、0.02%BSA、0.02%ポリビニルピロリドン、10mMのDTT、および、熱変性させた異種核酸(0.005%の酵母tRNA、タイプX、0.05%の酵母全RNA、タイプI、0.05%サケ精巣DNA、タイプIII))で、室温で2時間プレハイブリダイズさせ;ハイブリダイゼーション溶液(2.5×106cpm/切片標本)、50%脱イオン化ホルムアミド、および、50%ハイブリダイゼーション緩衝液[0.6M塩化ナトリウム、10mMトリスHCl(pH7.6)、50mMのEDTA、0.025%ピロリン酸ナトリウム、0.02%フィコール、0.02%BSA、0.02%ポリビニルピロリドン、熱変性させた異種核酸(0.005%の酵母tRNA、タイプX、0.005%の酵母全RNA、タイプI、0.05%サケ精巣DNA、タイプIII)、100mMのDTT、0.0005%ポリアデニル酸、10%硫酸デキストランを含む]中で、50℃で一晩、リボプローブとハイブリダイズさせた。切片標本を、RT(室温)で、2×、1×、0.5×SSCでリンスした。20μg/mlのRNアーゼA(シグマ,セントルイス,ミズーリ州)中で消化させた後、切片標本を、室温で、1×RNアーゼ緩衝液、2×、1×、0.5×SSC中で、および、45℃で、0.1×SSC中で一晩洗浄した。切片標本をアルコール濃度を連続的に高めて脱水させ、コダック(Kodak)のバイオマックスMRフィルム(Biomax MR film)(コダック,ロチェスター,ニューヨーク州)に−80℃で3〜7日間露光した。
【0069】
5.抗体の生成、および、免疫蛍光法による染色:
ヒトPAMのアミノ酸残基135〜153および4601〜4614(それぞれ配列番号1に対応する)からなるペプチド(バイオトレンド(BioTrend),ケルン,ドイツ)を用いて、抗血清をウサギで商業的に生成させた。その抗血清を、標準的な手法に従って、バイオトレンド(ケルン,ドイツ)で商業的に生産させた。脊髄とDRGの切片におけるPAMの分布をモニターするために、切片を、0.1%トリトンX−100中で5分間浸透させた。切片を、PBS中の3%BSA中で1時間ブロッキングし、次に、抗PAM抗血清(1:50希釈)と共に1時間インキュベートした。続いて、これを、3%BSAを含むPBS中のFITC標識したヤギ抗ウサギ抗体と共にインキュベートした。次に、切片をPBSで洗浄し、フルオマウント(fluoromount)TMを用いてマウントした。
【0070】
6.RT−PCR:
ラットの脊髄およびDRGからの全RNAを、グアニジンイソチオシアネート/フェノール/クロロホルム抽出によって単離した(ChomczynskiおよびSacchi,1987年)。全RNA2μgを、0.6μMのそれぞれのオリゴ(dT)プライマーとアニールし、逆転写酵素(プロメガ,マディソン,ウィスコンシン州)を用いて37℃で30分間逆転写した。次に、即座にcDNAを増幅に用いた。ラットGADPHの増幅に用いられるオリゴヌクレオチドプライマーは、5’−GAAGGGTGGGGCCAAAAG−3’(センス;配列番号10)、および、5’−GGATGCAGGGATGATGTTCT−3’(アンチセンス;配列番号11;Trajkovic等,2000年)であった。ACアイソフォームを増幅するためのオリゴヌクレオチドプライマーを、Xu等によって公開されているようにして選択した(Xu等,2001年)。ラットPAMのためのプライマーは、5’−GGTGGTGAAGCTCGCTGTGATGCT−3’(センス;配列番号12)、および、5’−CGTGTGAGCATTTCTGCACACTCC−3’(アンチセンス;配列番号13)であった。そのPCR産物は、ヒトPAMcDNAヌクレオチド13692〜14064に対応する。それに対応するラット配列は、ESTクローンAW441131(配列番号8)から得た。半定量PCRのために、SAWDAY DNAポリメラーゼ(Peqlab,エルランゲン,ドイツ)を用いた。最初の95℃で5分間での変性工程の後に、95℃で1分間、55℃で30秒間、および、72℃で10秒間の30サイクルを行い、その後、最終に72℃で10分の伸長工程を行った。定量PCRを、TaqManTMシステムおよび試薬(アプライド・バイオシステムズ,ヴァイターシュタット,ドイツ)を製造元の説明書に従って用いて行った。
【0071】
7.全長PAMの精製:
以前に公開されたように、ただし数箇所を改変してPAM精製を行った(Scholich等。2001年)。簡単に言えば、HeLa細胞を、10%ウシ胎仔血清と1%ペニシリン/ストレプトマイシンを含むDMEM培地中で増殖させた。40個の150mm培養皿の密集した細胞を、1×PBS、1mMのEDTAを用いて回収し、400×gで5分間沈殿させた。細胞を、125mMのNaCl、20μg/mlのアプロチニン、20μg/mlのロイペプチン、1mMのベンズアミジン、5μg/mlのダイズトリプシンインヒビターを含むTED緩衝液(50mMトリスHCl(pH8.0)、1mMのEDTA、1mMのDTT)に再懸濁させ、超音波破砕を2×5秒間行い溶解させた。ホモジネートを、27000×gで、4℃で30分間で遠心分離し、上清を、Q−セファロースXK16カラム(アマシャム・ファルマシア(Amersham Pharmacia),ピスカタウェイ,ニュージャージー州)にローディングし、製造元の説明書に従って、TED中で、150〜350mMのNaClの濃度勾配で溶出させた。標準的な手法に従ってウェスタンブロッティングで分画を解析した;陽性の分画をプールし、そのNaCl濃度を1Mに調節した。次に、タンパク質を、フェニル−セファロースXK16カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングし、製造元の説明書に従って、TED中の300mMのNaClで洗浄した。流出液分画および洗浄分画にPAMが含まれていた。それらをプールし、製造元の説明書に従って、セントリコン(Centricon)50(アミコン(Amicon),ビバリー,マサチューセッツ州)を用いて緩衝液を上述の100mMのNaClを含むTED緩衝液で交換した。次に、タンパク質を、モノS(Mono S)5/5FPLCカラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングし、製造元の説明書に従って、ローディング緩衝液(TED中、100mMのNaCl)で洗浄した。流出液を回収し、モノQ(Mono Q)5/5FPLCカラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にアプライした。タンパク質を、TED中の150〜400mMのNaClの濃度勾配で溶出させた。陽性の分画をプールし、セントリコン(Centricon)50(アミコン(Amicon),ビバリー,マサチューセッツ州)を用いて緩衝液を50mMトリスHCl(pH8.0)、1mMのDTTで交換し、−80℃で保存した。保存したPAMは、3週間以内に用いた。
【0072】
8.組換えGsαの発現および精製:
ヘキサヒスチジルタグを有する構成的に活性なGsαのQ213L突然変異体(Gsα*)を、発現させ、Graziano等,1991年で説明されている通りに精製した。Gsα*の最大活性化を確認するために、AC活性分析で使用する前に、MgCl2(25mM)の存在下で、30分間、Gタンパク質を、1μMのGTPγSとインキュベートした。
【0073】
9.アデニリルシクラーゼ活性アッセイ(AC活性アッセイ):
脊髄を、25mMのHepes(pH7.4)、1mMのEGTA中で溶解させ、KassisおよびFishmanによって説明された通りに細胞膜を調製した。アリコートを使用するまで−80℃で保存した。AC活性分析を、以前に説明された通りに、容積100μlで、室温で15分間、100μMのMgCl2の存在下で行った(Patel等,2002年)。Gsα*(80nM)、または、フォルスコリン(100μM)を用いて、膜中のAC酵素活性(タンパク質10μg)を刺激した。
【0074】
10.PAMのアンチセンス、および、センスオリゴヌクレオチドの脊髄への搬送:
ラットを、ケタミン(60mg/kg,腹腔内)と、ミダゾラム(0.5〜1mg/kg,腹腔内)で麻酔した。椎骨Th13からL3までの脊柱の上で皮膚を切開した。L2
〜3周辺の筋肉組織を取り除いた。L3の棘突起を除去し、L2で椎弓切除術を施した。次に、ポリエチレン製カテーテル(ID0.28mm、OD0.61mm)を、カテーテルの先端がTh9〜10に達するように硬膜外腔に挿入した。カテーテルをシアノアクリレート系接着剤で固定し、頚部の領域で外部に露出させ、皮膚を縫合した。
【0075】
11.PAMオリゴヌクレオチドの注入:
オリゴデオキシヌクレオチド(ODN)の配列を、以下のようにラットPAM配列から選択した。センス:5’−GACTGGTTTAGCAATGGC−3’(配列番号14)、アンチセンス:5’−GCCATTGCTAAACCAGTC−3’(配列番号15)、および、3つの突然変異(3M−as;突然変異は下線で示した)を含むアンチセンスODN:5’−GCAATTGCTAAATCAGTA−3’(配列番号16)。外科手術の3日後に、ラットを「自由行動系」(CMA,ストックホルム,スウェーデン)の状態に置き、アンチセンス(n=5)、または、センス(n=5)オリゴヌクレオチド(人工髄液中、2.5mg/ml)をカテーテルを通じて、微量注入ポンプ(CMA,ストックホルム,スウェーデン)を用いて、0.05〜0.1μl/分の低速で、100時間注入した。
【0076】
12.ホルマリン試験:
注入を止めた後15分以内に、ホルマリン試験を行った。5%ホルムアルデヒド溶液50μlを、1本の後肢の背面の皮下に(s.c.)注射した。ホルマリン注射の直後に開始して、1分間のインターバルで60分間まで、フリンチ行動をカウントした。5分間のインターバルのフリンチ行動を、1分あたりの平均フリンチ行動として要約した。グループ間の侵害受容の挙動を比較するために、フリンチ行動の、1時間の観察期間の第二相の合計を、スチューデントのt検定で処理した。aを0.05に設定した。
【0077】
ホルマリン試験の最後にラットを殺し、腰髄および後根神経節(DRG)を切り出し、液体窒素中で急速冷凍し、さらなる解析まで−80℃で保存した。PAM発現を決定するために、脊髄切片を、上記の抗PAM抗体を用いて免疫組織化学的に解析した。
【0078】
13.ザイモサン誘発性の炎症:
炎症を誘発させるために、0.03Mのリン酸緩衝食塩水(PBS,pH7.5)30μlに懸濁したザイモサンA2.5mg(シグマ,セントルイス,ミズーリ州)を、右の後肢の足底の中央部の領域に皮下注射した。このような足底内部へのザイモサン注射は、信頼できる温熱性および機械刺激痛覚過敏ラットのモデルを誘導することがわかっている(MellerおよびGebhart,1997年)。ラットを、ザイモサン注射の24〜96時間後に、深いイソフルラン麻酔下で心臓穿刺を行うことにより殺した。腰髄および後根神経節(DRG)を切り出し、液体窒素中で急速冷凍し、さらなる解析まで−80℃で保存した。
【0079】
14.免疫蛍光法による染色:
HeLa細胞におけるPAMの分布をモニターするために、カバーガラス上で、10%FBSおよび1%ペニシリン/ストレプトマイシン(ギブコ(Gibco),カールスルーエ,ドイツ)を含むDMEM中で細胞を増殖させた。HeLaにおけるPAMのトランスロケーションを可視化するために、細胞を、一晩、血清欠乏状態にし、その後、10%血清で2時間処理した。指示されている場合は、細胞を、様々な阻害剤濃度の存在下で30分間プレインキュベートした。PBS中の4%パラホルムアルデヒド(シグマ,タウフキルヒェン,ドイツ)で、細胞を10分間固定し、次に、さらに5分間、0.1%トリトンX−100中に浸透させた。カバーガラスを、PBS中の3%BSAで1時間ブロッキングし、次に、抗PAM抗体(1:50希釈)と共に1時間インキュベートした。続いて、これを、3%BSAを含むPBS中で、FITC標識したヤギ抗ウサギ抗体と共にインキュベートした。次に、細胞をPBSで洗浄し、マウントした。解析のために、共焦点(バイオ・ラッド,ハーキュリーズ,カリフォルニア州)、および、標準蛍光顕微鏡(ニコン(Nikon),デュッセルドルフ,ドイツ)を用いた。
【0080】
15.cAMPの蓄積
脊髄サンプルを超音波破砕し、18.000×gで、4℃で20分間遠心分離した。上清をcAMP測定に用いた。細胞中のcAMPの蓄積を、cAMP検出キット(Assay Design Inc,アナーバー,ミシガン州)によって、製造元の説明書に従って決定した。
【0081】
16.アンチセンスオリゴデオキシヌクレオチド
オリゴデオキシヌクレオチド(ODN)の配列を、以前に公開されたようにして選択した(Scholich等,2001年)。センス:5’−CTGTTCATGCCGGTT−3’、アンチセンス:5’−AACCGGCATGAACAG−3’、および、3つの突然変異(3M−as;突然変異は下線で示した)を含むアンチセンスODN:5’−AATCCGTATGAACAC−3’。HeLa−細胞を35mm培養皿で平板培養し(300,000細胞)、10%FBSおよび1%ペニシリン/ストレプトマイシンを含むDMEM培地(ギブコ,カールスルーエ,ドイツ)中で24時間増殖させた。ODN(それぞれ3μM)を、無血清培地1ml中で、Tf×20(プロメガ,マディソン,ウィスコンシン州)を製造元の説明書に従って用いてトランスフェクトすることによって細胞に導入した。2時間後、10%FBSを含むDMEM(ギブコ,カールスルーエ,ドイツ)1mlを添加した。次に、細胞を6時間インキュベートした。次に、培地を無血清DMEM(ギブコ,カールスルーエ,ドイツ)で交換し、続いて、無血清DMEM(ギブコ,カールスルーエ,ドイツ)中で16時間インキュベートし、その後、10%血清または500MのS1Pで処理した。次に、細胞を、沸騰した1×レムリー緩衝液を添加することによってウェスタンブロットに用い、または、上述のようなAC活性分析のためにそれらを回収した。
【0082】
17.精製またはPAMの血清因子の活性化:
ウシ胎仔血清(ギブコ,カールスルーエ,ドイツ)188mlを、NaClの最終濃度が0.3Mになるように調節した。次に、血清を、フェニル−セファロース15/10カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングした。このカラムから溶出させた後に、同様に、それに続く全てのカラムの後に、流出液と全ての溶出した分画を回収し、そのHeLa細胞でPAMのトランスロケーションを誘導する能力について解析した。次に、流出液を、Q−セファロース15/10カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングした。カラムを、TE(50mMトリス/Cl(pH7.4)、0.5mMのEDTA)中の400mMのNaClで洗浄し、TE中の1MのNaClで溶出させた。溶出液を、スーパーデックス(Superdex)200pgゲルろ過カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングした。製造元の説明書に従ってタンパク質をTEで溶出させ、分画を、HeLa細胞中でPAMのトランスロケーションを誘導する能力に関して解析した。陽性の分画をプールし、モノQ5/5FPLC.カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)にローディングし、TE中の400mMのNaClで洗浄した。タンパク質をTED中、NaClが400〜1000mMの濃度勾配で溶出させた。陽性の分画をプールし、スーパーデックス50pgゲルろ過カラム(アマシャム・ファルマシア,ピスカタウェイ,ニュージャージー州)に製造元の説明書に従ってローディングした。タンパク質を、製造元の説明書に従ってTEで溶出させ、分画を、HeLa細胞中でPAMのトランスロケーションを誘導する能力に関して解析した。陽性の分画をプールし、−80℃で保存し、マススペクトロメトリーまたは生化学的アッセイのためには2週間以内に用いた。フタルアルデヒド標識を用いてS1Pを検出し、続いて、Caligan等によって説明されているように、HPLCで分離した。
【0083】
18.侵害受容過程におけるPAMの関連を示す結果:
上記の発明者等によるRT−PCR、免疫組織化学およびインサイチュハイブリダイゼーションを用いた実験で、PAMは、成体ラットの脊髄の感覚ニューロン、同様に、後根神経節(DRG)で発現されることがが初めて実証された。RT−PCRによれば、PAMのmRNAは、脊髄と後根神経節とで発達を通して(E14〜成体)同様のレベルで検出された。PAM発現は、ウェスタンブロットおよびRT−PCRで示されるように、ラットのザイモサン処理の24〜48時間後にアップレギュレートされる。
【0084】
脊髄とDRGで発現される主要なアデニリルシクラーゼアイソフォームは、それぞれAC5および6型、ならびに、AC4および6型である。脊髄をザイモサン処理した後は、ACアイソフォーム発現における主要な変化は観察されなかった。従って、脊髄およびDRGからの膜調製物におけるGαsによって刺激されたAC活性は、PAMによって阻害された。その結果として、成体ラットにおいて、PAMに対するセンスオリゴヌクレオチドではなくアンチセンスオリゴヌクレオチドでの処理により、ホルマリンで誘発される足のフリンチ行動が増加したことを発見した。従って、PAMに対するアンチセンスオリゴヌクレオチドで処理したラットの脊髄におけるcAMPの蓄積は、コントロールラットと比較して上昇した。
【0085】
精製されたPAMを脊髄溶解産物へ添加することにより、コントロールとザイモサン処理動物からの脊髄溶解産物のGαsで刺激されたAC活性が阻害された(図5b)。30nMのGαsで刺激されたAC活性は、コントロール動物の脊髄溶解産物において50%、ザイモサンで96時間処理されたラットから得られた溶解産物で70%減少した。
【0086】
脊髄でPAMが発現されるかどうかを測定するために、第一のインサイチュハイブリダイゼーションを行った。これにより、成体ラットの脊髄の白質ではなく灰白質の至る所に、PAMのmRNAに関する明らかなシグナルが検出された(図1)。脊髄およびDRGでPAMを発現する細胞集団をより正確に明らかにするために、ヒトPAMのアミノ酸残基135〜153および4601〜4614に対応するペプチドを用いて、PAMに対する抗体を作成した。免疫組織化学的な解析により、PAMは、抗GFAP免疫反応性ではなく、抗NeuN免疫反応性と共存することが明らかになった(図2a)。より特定には、PAM発現は、後角ニューロンで優勢に検出されたが(図2a)、ニューロンではない細胞集団では極めてわずかなPAM発現しか示さなかった。特に、DRGニューロンでは、高いPAM発現を検出することができた(図2b)。ここで、PAMの免疫反応性は、軸索に、同様に、直径が大きいニューロンと小さいニューロンの両方の細胞体に存在していた(図2b)。興味深いことに、抗ヒストン抗体で共染色することによって実証されたように、細胞の核では、PAMは検出されなかった(図2b)。脊髄でも観察されるように、GFAP発現細胞では、PAM発現は、検出されなかった。
【0087】
ラットおよびマウスの発達中に、脳におけるPAM発現は差異的に調節されるために(Yang等,2002年)、発明者等は、ラットの脊髄およびDRGの発達中にPAMのmRNA発現も変化するかどうかを調査した。この目的を達成するために、PAMのmRNA発現を、定量RT−PCRを用いて決定した。PAMのmRNAは、後期胚形成期(E16)から、誕生直後まで(P0.5;図3)、脊髄とDRGで高度に発現されることを発見した。興味深いことに、発現は、誕生直後に胚発生の30〜40%に減少し、続いて、成人期中は一定を保った(図3)。
【0088】
次に、脊髄におけるPAM発現は、侵害受容の刺激によって調節されるかどうかを試験した。そこで、成体ラットの後肢へのザイモサンおよびホルマリン注射の後に、脊髄のPAM発現をモニターした。ザイモサン処理の24および48時間後に、PAMのmRNAが約2倍アップレギュレートされた(図4a)。それにより、ザイモサン注射の24時間後に、PAM発現は、タンパク質レベルでアップレギュレートされ、96時間上昇し続けた(図4b)。ザイモサン注射の96時間後に、PAMのmRNA発現は減少した。このPAMのmRNA発現における減少は、タンパク質レベルには反映されていなかった(図4aおよびbを比較)。特に、ラットの脊髄においても、ホルマリン注射の1時間後にPAMのmRNAがアップレギュレートされた(図4c)。
【0089】
PAMは、アデニリルシクラーゼ1、5および6型の強力な阻害剤であることがわかっているため(Scholich等。2001年)、PAMが、脊髄およびDRG溶解産物中でAC活性を阻害できるかどうかを調べた。脊髄において、2種の主要なACアイソフォームが半定量RT−PCRにより検出された。これらのACアイソフォームは、5および6型である(図5a)。特に、アイソフォームはいずれも、ナノモル濃度のPAMで阻害される(Scholich等,2001年)。ザイモサン注射の24〜96時間後に、脊髄におけるACアイソフォーム発現パターンは、有意に改変されなかった。興味深いことに、ホルマリン注射の1時間後に、ACアイソフォーム発現におけるシフトが検出された(図5a)。AC5型のmRNAは、ダウンレギュレートされ、AC3および9型のmRNAはアップレギュレートされる。
【0090】
上記の実験により示されたように、精製されたPAMを脊髄溶解産物へ添加することにより、脊髄調製物において、Gαsで刺激されたAC活性が阻害された。30nMのPAMの添加により、Gαsで刺激されたAC活性が、コントロール動物の脊髄溶解産物において49%減少した(図5b)。ザイモサン注射の96時間後の、ラットから得られた脊髄溶解産物におけるGαsで刺激されたAC活性は、PAMの阻害に対して、未処理動物と比較して高い感受性を示した(30nMで、70%阻害;図5b)。それに対して、ホルマリンで1時間処理された動物からの脊髄溶解産物におけるPAMによるAC活性の阻害の程度は、より少なかった(30nMで25%阻害;図5b)。
【0091】
DRGにおいて、優勢に発現されたACアイソフォームは、AC4および6型である(図5a)。DRG溶解産物におけるGαsで刺激されたAC活性は、脊髄溶解産物と比較して30nMのPAMの存在下で32%減少した(図5b)。
【0092】
脊髄の侵害受容伝達におけるPAMの可能性のある役割を試験するために、ホルマリン分析を行う前に、動物に、PAMのセンスおよびアンチセンスオリゴヌクレオチドを、腰髄内へのカテーテルにより注入した。免疫組織化学で観察されたように、脊髄ニューロンにおいて、PAM発現は減少した(図6a)。PAMのアンチセンスオリゴヌクレオチドの注入により、ホルマリン注射後の侵害刺激反応が、PAMのセンス処理と比較して顕著に増加した(p=0.007;図6bおよびc)。PAMのアンチセンス処理ラットにおける痛覚過敏には、足をなめる挙動、および噛みつき挙動の増加が伴った。PAMは、AC活性阻害剤であるため(Scholich等,2001年)、センスおよびアンチセンスODNで処理されたラットの脊髄溶解産物における、基礎的なGαsおよびフォルスコリン刺激によるAC活性を測定した。この実験により、アンチセンスで処理されたラットにおいて、基礎的なAC活性の顕著な増加(20.7%)が示された(表1)。それに対して、Gαsおよびフォルスコリン刺激によるAC活性において顕著な変化は検出されず、これは、ODN処理によってACの総量が変化しなかったことを示す(表1)。
【0093】
上記の実験より、PAMは、脊髄とDRGニューロンの両方の細胞体と軸索に局在化していることが示された(図2a,b)。細胞核では、ほんのわずかな免疫活性しか検出で
きず、これは、ニューロンおよびガン細胞系において、PAMに関する機能が異なることを示している。
【0094】
持続的な侵害受容の刺激の後の中枢性感作は、脊髄におけるニューロンおよびシナプスの変化に基づく(WoolfおよびCostigan 1999年;WoolfおよびSalter2000年;JiおよびWoolf 2001年)。発明者等による、PAMは、脊髄およびDRGの感覚ニューロンで発現されるという発見により、PAMは、脊髄の侵害受容過程の際のシナプスの変化に関与する可能性があるという新たな仮説が構築された。上記の、PAMは、侵害受容の刺激の後にアップレギュレートされるという発見は(図4a)、この、脊髄の侵害受容過程の際のシナプスの変化においてPAMは役割を果たす可能性があるという仮説を裏付けている。
【0095】
さらに、発明者等の、PAMは、脊髄およびDRGの知覚神経で発現されるという驚くべき発見により、同様に、脊髄およびDRGにおいて、PAMは、AC活性を阻害することができるかどうかという疑問に至った。
【0096】
上記の実験より、PAMは、脊髄調製物におけるGαsで刺激されたAC活性の有力な阻害剤であることが初めて示された(図5b)。AC活性は、30nMのPAMを添加した後に、50%減少した。抑制性Gタンパク質のαサブユニット、Gαiを用いて匹敵する阻害を達成するために、200〜800nMのGαiを用いなければならない(Wittpoth等,1999年)。ザイモサンで96時間処理された動物の脊髄調製物において、PAMの阻害作用はいっそう強く(図5b)、ザイモサン注射の後に、脊髄における内因性PAMの量が上昇したことで説明することができる(図4a,b)。ホルマリンで処理した動物からの脊髄調製物におけるGαsで刺激されたAC活性の阻害(25%阻害)は、コントロール動物またはザイモサンで処理した動物と比較してそれほど顕著ではなかった(それぞれ50%および75%)。
【0097】
特に、ホルマリンで1時間処理した動物において、ACアイソフォーム発現におけるシフトを観察した(図5a)。AC3および9型はアップレギュレートされるが、AC型5は、ダウンレギュレートされた。これまで、PAMが、AC3および9型の阻害剤であるかどうかはわかっていない。それゆえに、これらアイソフォームは、PAMによって阻害されないか、または、試験されたPAM濃度が、阻害作用を達成するには低すぎる可能性がある。PAMは、510kDaの巨大タンパク質であるため、30nMを超えるPAM濃度を試験することは技術的に不可能である。それにもかかわらず、図5bに示される用量反応曲線によれば、試験された脊髄調製物において、より高いPAM濃度により、Gαsで刺激されたAC活性のより強い阻害が引き起こされる可能性がある。
【0098】
興味深いことに、PAMは、DRGにおいて、脊髄調製物においてより有効性が弱いAC酵素活性の阻害剤であった。脊髄およびDRG調製物においてPAMの阻害効率が異なるのは、観察されたACアイソフォーム発現の差による可能性が最も高い。脊髄で発現される主要なACアイソフォームは、5および6型であり、いずれもPAMによって強く阻害される(図5a;(Scholich等,2001年))。DRGにおいて、AC4および6型は、優勢なACアイソフォームである(図5a)。PAMがAC4型を阻害するかどうかはわかっていないため、このアイソフォームは、PAMによって阻害されないか、または、それに加えて、試験されたPAM濃度が、阻害作用を達成するには低すぎる。しかしながら、図5bに示される用量反応曲線によれば、DRG調製物において、より高いPAM濃度により、Gαsで刺激されたAC活性のより強い阻害が引き起こされる可能性が高いようである。
【0099】
しかしながら、最も驚くべきことは、PAM活性は、試験動物の侵害受容の挙動に影響
を与えるという発見であった:これは、基礎的なAC活性における顕著な増加(表1)、および、より重要なことは、ホルマリン注射後、侵害刺激反応がPAMのセンス処理と比較して顕著に増加したこと(図6bおよびc)、その際、脊髄におけるPAMの内因性の発現は、動物にPAMのアンチセンスオリゴヌクレオチドを注入することによって減少したこと(図6a)を示す発明者等の実験によって初めて実証された。
【0100】
19.PAMの鎮痛作用の測定
上述したPAMの鎮痛作用に関する証拠は、例えば、以下の仮想的実験によって裏付けることができるだろう:例えば急性の痛みのホルマリンモデルにおけるPAMの鎮痛作用は、例えば、アミノ酸残基1028〜1065に対応するペプチドの髄腔内への適用によって直接決定することができるだろう。このペプチドは、酵母ツーハイブリッドシステムとAC活性分析で測定されるように、PAM−アデニリルシクラーゼの相互作用に介在できることが発見された最小の領域を示す。このペプチドは、バイオポーター(bioporter)リポフェクション試薬(Peqlab,ドイツで市販されている)との複合体として適用され得る。このアプローチにより、ペプチドを組織に進入させ、ACに対する生理学的なPAMの作用を模擬することが可能になる。
【0101】
20.PAMシグナル伝達に対するS1Pの影響を示す結果
HeLa細胞におけるPAM発現と局在化を調査するために、ヒトPAMのアミノ酸残基135〜153および4601〜4614に対応するペプチドに対して向けられた2種のPAMに対する抗体が用いられた。ラット脳の免疫組織学的染色の比較により、両方の抗体が、同じ脳の領域(同様にPAMのmRNA発現を示す)を認識することが示された(Yang等,2002年)。血清欠乏状態にしたHeLa細胞において、両方の抗体は、PAMとカルネキシン、小胞体マーカーとの共存を示した(図18a)。血清を細胞に添加した後に、部分的なPAMの細胞質膜へのトランスロケーションが観察された(図18b)。PAMは、血清処理の20〜30分後に膜に出現し、血清とインキュベートして1時間後に膜から消失し始めた。用いられた抗体で観察されたHeLaにおけるPAMの細胞分布は、Guo等によって説明されている細胞分布とは異なっている。Guo等は、PAMの一部を用いて、核タンパク質に共通のモチーフを含む抗体を生産させているため、この抗体による核タンパク質との交差反応が起こる可能性がある。
【0102】
PAMは、AC酵素活性の強力な阻害剤なので、次に、ERから細胞質膜へのPAMのトランスロケーションにより、AC活性が阻害されるかどうかを調べた。HeLa細胞の血清処理により、細胞内cAMPの蓄積が減少した(図19a)。加えて、血清処理により、Gαsおよびフォルスコリン刺激によるAC活性が、未処理細胞と比較してそれぞれ56.7%および64.7%減少した(図19b)。観察されたAC活性の減少は、ACアイソフォームのmRNA発現における変化が検出されなかったことから、ACアイソフォーム発現における変化、または、AC発現の増加によるものではなかった(図19c)。これまでにScholich等,2001年で説明された通りに、刺激したAC活性の減少に、PAMが介在するかどうかを決定するために、PAMに対するアンチセンスオリゴヌクレオチドを用いて内因性PAMの量を減少させた。図19dに示すように、アンチセンスODNで処理されたHeLa細胞において、PAMの量(ウェスタンブロット解析によって決定された)は、センスまたは突然変異アンチセンスODNで処理した細胞と比較して減少した。抗Hsp70抗体を用いて同じブロットを再度検査したところ、タンパク質のローディングは同じであることが示された(図19d)。しかしながら、アンチセンスODNでHeLa細胞を処理すると、Gαsおよびフォルスコリン刺激によるAC活性の血清誘発性の阻害が、著しく減少した(図19e)。重要なのは、HeLa細胞のセンスまたは突然変異ODNでのトランスフェクトは、Gαsおよびフォルスコリン刺激によるAC活性の血清誘発性の阻害に影響しないことである(図19e)。これらのデータにより、血清でHeLa細胞を刺激した後、内因性PAMは、AC活性に阻害作用を与えることが示される。
【0103】
PAMの細胞質膜へのトランスロケーションを誘発する血清因子を同定するために、血清因子を、逆相カラム、陰イオン交換カラム、およびゲルろ過カラムを用いて精製した。各精製工程の後に、分画のPAMのトランスロケーションを誘導しACを阻害する能力に関して試した。精製の特性によれば、血清因子は、わずかに疎水性であり(フェニル−セファロースカラムから0.3MのNaClで溶出)、強い負電荷を有し(モノQ、および、Q−セファロースカラムから0.7MのNaClで溶出した)、および、スーパーデックス30ゲルろ過カラムでの保持時間によれば推定分子量が500未満と同定された。物理的な特性に従って、数種の候補物質が試験されたが、そのうちスフィンゴシン−1−リン酸のみがPAMのトランスロケーションを誘発した。
【0104】
S1Pは、5種のGタンパク質共役受容体のファミリーに結合できる。それゆえに、半定量的RT−PCRで、HeLa細胞がS1P受容体を発現するかどうかを調べた。HeLa細胞で、5種のS1P受容体アイソフォームのうち4種のmRNA(S1P1〜S1P4)が検出された(図20a)。次に、精製されたS1Pが、PAMの活性化/トランスロケーションに対して血清と同じ特性を示すかどうかを試した。まず、精製されたS1Pの濃度を徐々に高めてHeLa細胞を処理した。0.1〜5μMのS1Pで処理した細胞で処理した細胞の70〜90%で、PAMの細胞質膜へのトランスロケーションが起こった。PAMは、500nMのS1Pと10分間インキュベートした後に細胞質膜で出現し、インキュベートして1時間後に消失し始めた(図3b,c)。最も重要なことには、HeLa細胞を0.5μMのS1Pで処理することによって、細胞内のcAMP含量が減少し(図21a)、同様に、Gαsで刺激されたAC活性が減少した(図21b)。Gαsで刺激したAC活性は、S1Pとインキュベートした後3分以内、かつ部分的に回復する前に減少した。S1P処理を開始して5〜10分後に、Gαsで刺激したAC活性は、再び減少した(図4b)。PAMに対するアンチセンスODNは、S1Pとインキュベートして60分後に、Gαsで刺激されたAC活性の阻害を消滅させた(図21c)。総合すると、これらデータにより、HeLa細胞におけるS1PによるAC活性の阻害は、2つの異なるメカニズム:迅速なPAM非依存性のAC阻害(3〜10分のS1P処理)、および、遅延性のPAM依存性のAC阻害(10〜60分のS1P処理)によって達成されることが初めて実証された。
【0105】
それぞれの受容体に結合することによって、S1Pは、4種の異なるGタンパク質、Gi、Gq、G12およびG13を活性化する可能性を有することが実証された(Hla等,Science,2001年;Kluk等,BBA,2002年;SiehlerおよびManning,BBA,2002年;SpiegelおよびMilstein,JBC,2002年)。これらから、Giのみが百日咳毒素感応性である。百日咳毒素処理は、Gαsで刺激されたAC活性に対するS1Pの阻害作用(図22a)、および、PAMの細胞質膜へのトランスロケーション(図22b)を消滅させた。このようにして、抑制性Gタンパク質Giは、S1Pによる迅速なPAM非依存性阻害に関与する可能性が高いようである。
【0106】
次に、PAMのトランスロケーションと活性化を引き起こすシグナル伝達経路のさらなる構成要素を研究した。S1P1〜S1P4受容体は、Gi、GqおよびG12/13に共役しており(Hla等,2001年;Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)、PAMのトランスロケーション、同様に、AC阻害は百日咳毒素依存性であると説明されているため(図22a)、上記のデータにより、HeLa細胞におけるPAMの活性化は、Gi活性化に依存していることが示される。これまで、S1Pは、ホスホリパーゼC(PLC)、同様に、Giの活性化を介したERK1/2シグナル伝達を活性化できることが説明されている(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)。このようにして、PLC活性化が、PAMのトランスロケーションに関与しているかどうかを試したところ、PLC阻害剤U73122の存在下で、PAMのトランスロケーションと後期のAC阻害が止められることを発見できた(図22a)。PLCは、ホスファチジルイノシトール4,5−二リン酸を、イノシトール1,4,5−三リン酸(IP3)、カルシウム動員セカンドメッセンジャーと、1,2−ジアシルグリセロール(DAG)、タンパク質キナーゼC(PKC)の活性化因子に変換する(Rebecchi等,2000年;Wilde等,2001年)。カルシウムのイメージングにより、S1Pは、HeLa細胞におけるPLC依存性のカルシウムの増加を誘発することが示された。しかしながら、BAPTA−AMでの前処理は、PAMのトランスロケーションを妨害しないため、PAMのトランスロケーションにこのようなカルシウムの減少は必要ない(図22a)。その上、PKC阻害剤GF109203Xと、RO31−8220は、それぞれPAMのトランスロケーションとAC阻害を消滅させた(図22a)。これらのデータにより、S1Pは、抑制性Gタンパク質Giを介してPLCを活性化することが示される。その後、PLC作用により、PAMのトランスロケーションを媒介するのに必要なカルシウム非依存性のPKC活性化と、遅延性のS1P誘発性AC阻害が起こる。
【0107】
S1Pは、ERK1/2シグナル伝達経路を活性化できることも示されているため(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)、HeLaにおいて、EK1/2は、S1Pによって活性化されるかどうかを試した。抗活性ERKと抗ホスホTyr183ERK抗体を用いて、HeLaをS1Pとインキュベートした後、ERK1/2のリン酸化が検出できた(図22b)。驚くべきことに、ERK1/2のリン酸化は、PLC活性化(図22b)、GiおよびPKC活性依存性であったが(データ示さず)、細胞内のカルシウムの増加には非依存性であった。しかしながら、ERK1/2の活性化は、PAMのトランスロケーションまたはAC活性の阻害に必要なかった(図22a)。総じて、これら発見により、S1Pは、PAMのトランスロケーション、それに続く、Gi、PLCおよびPKCを含むシグナル伝達カスケードを介したAC酵素活性の阻害を誘発することが初めて示された。興味深いことに、S1Pはさらに、ERK1/2の活性化と、細胞内のカルシウム濃度の増加を誘発し、これらは両方、PAMのトランスロケーションまたはGαiで刺激したAC活性の阻害に必要なかった。
【0108】
S1Pで調節されたシグナル伝達経路は、集中的な調査の焦点である(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)。S1P受容体は、Gi依存性メカニズムによってAC活性を阻害できることは周知である(Kluk等,2002年;Siehler等,2002年;Spiegel等,2002年)。しかしながら、ここで、遅延性のAC阻害は、PLCおよびPKC活性化に依存しているため、S1Pで刺激した後の長期にわたるAC活性の阻害は、抑制性Gタンパク質によるACの直接的な阻害によるものではないことが初めて示された。その上、発明者等のデータにより、S1P処理後のHeLa細胞における持続性のAC活性の阻害は、PAMの細胞質膜へのトランスロケーション/活性化に依存することが示される。このトランスロケーションは、Gi活性化と、PLC/PKCシグナル伝達によって調節される。PAMは、AC酵素活性の有力な阻害剤であるため、S1P誘導性のPAM依存性のAC阻害は、PAMとACとの直接的な相互作用の結果と考えられるが、このことはなお証明の必要がある。上記の発明者等の実験により、HeLa細胞において、PAMは、小胞体に局在しており、血清処理の後に細胞質膜へトランスロケーションすることが初めて発見された。PAMのトランスロケーションには、未処理のHeLa細胞と比較して、Gαsおよびフォルスコリン刺激によるAC活性の減少が伴った。細胞をPAMに対するアンチセンスオリゴヌクレオチドで前処理すると、AC阻害を妨害したことから、AC阻害にはPAMが介在されていた。以下で、我々は、PAMのトランスロケーションに関与する血清因子としてスフィンゴシン−1−リン酸(S1P)を同定した。HeLa細胞を0.1〜5μMのS1Pで処理することにより、10〜30分以内に細胞の80%においてPAMの細胞質膜へのトランスロケーションが誘発された。2つの別個のメカニズムによって、S1Pは、AC活性を減少させた。最初のAC阻害にPAMは介在していないが、百日咳毒素感応性であった。長期のS1P処理の後、AC阻害は、PAMのトランスロケーションに依存性であった。PAMのトランスロケーションに対するS1Pの作用と、PAMが介在するAC阻害は、百日咳毒素感応性であり、PLCおよびPKCの活性化を必要とした。総合すると、これらデータにより、PAM活性の調節因子が初めて同定された。その上、S1Pによる長期のAC活性の阻害は、ERから細胞質膜へのAC阻害タンパク質PAMのトランスロケーションが介在することを示すことができた。
【0109】
21.S1Pの鎮痛作用の測定
S1Pの鎮痛の作用を測定するために、S1Pを髄腔内への投与によって脊髄へ搬送した。
【0110】
21a)腰髄内へのカテーテル埋め込み:
ラットを、ケタミン(60mg/kg,腹腔内)と、ミダゾラム(0.5〜1mg/kg,腹腔内)で麻酔した。脊椎骨Th13からL3までの脊柱の上で皮膚を切開した。L2〜3周辺の筋肉組織を取り除いた。L3の棘突起を除去し、L2で椎弓切除術を施した。次に、ポリエチレン製カテーテル(ID0.28mm、OD0.61mm)を、カテーテルの先端がTh9〜10に達するように硬膜外腔に挿入した。カテーテルをシアノアクリレート系接着剤で固定し、頚部の領域で外部に置き、皮膚を縫合した。
【0111】
21b)PAMオリゴヌクレオチドの注入。
外科手術の3日後に、ラットを「自由行動系」(CMA,ストックホルム,スウェーデン)の状態に置き、カテーテルを通じて、10μMのS1P20μlを注入した。
【0112】
21c)ホルマリン試験:
注入を止めた後15分以内に、ホルマリン試験を行った。5%ホルムアルデヒド溶液50μlを、1本の後肢の背面の皮下に(s.c.)注射した。ホルマリン注射の直後に開始して、1分間のインターバルで60分間まで、フリンチ行動をカウントした。5分間のインターバルのフリンチ行動を、1分あたりの平均フリンチ行動として要約した。グループ間の侵害受容の挙動を比較するためにフリンチ行動の1時間の観察期間の合計を、スチューデントのt検定で処理した。ホルマリン試験の最後に、腰髄および後根神経節(DRG)を切り出し、液体窒素中で急速冷凍し、さらなる解析まで−80℃で保存した。
【0113】
21d)結果:
成体ラットに、ホルマリン注射の15分前に、10μMのS1P20μlまたはPBS/DMSO20μlを髄腔内へ投与した。次に、5分間のインターバルのフリンチ行動を、60分間にわたりカウントした。第2A相(ホルマリン注射の20〜35分後)における侵害受容性の反応の数が、PBS/DMSO処理動物と比較して、顕著に減少したことが検出された(図24を参照)。これら実験により、外因性のS1Pが、鎮痛薬として作用することが証明された。
【0114】
22.S1P受容体アゴニストの鎮痛/抗侵害受容作用の測定
S1P受容体アゴニスト(例えばFTY720)の鎮痛/抗侵害受容作用、は、例えば、以下の仮想的実験により裏付けることができるだろう:例えばFTY720の、例えば急性の痛みのホルマリンモデルにおける鎮痛/抗侵害受容作用は、例えばFTY720を髄腔内または静脈内投与し、例えばフリンチ試験によってその鎮痛/抗侵害受容作用を連続的に試験することによって直接決定することができるだろう。このアプローチにより、分子を、組織に進入させ、ACに対する生理学的なS1Pの作用を模倣させることが可能になると思われる。
【0115】
文献
Bailey C.H., Bartsch D., Kandel E.R.: Toward a molecular definition of long-term memory storage. Proc. Natl. Acad. Sci. U.S.A. 1996 Nov 26;93(24):13445-52;
Brandon E.P., Idzerda R.L., McKnight G.S.: PKA isoforms, neural pathways, and behaviour: making the connection. Curr. Opin. Neurobiol. 1997 Jun;7(3):397-403;
Bek M.J., Zheng S., Xu J., Yamaguchi I., Asico L.D., Sun X.G. and Jose P.A. (2001) Differential expression of adenylyl cyclases in the rat nephron. Kidney Int 60, 890-899;
Brinkmann, V., Davis, M.D., Heise, C.E., Albert, R., Cottens, S., Hof,R., Bruns, C., Prieschl, E., Baumruker, T., Hiestand, P., Foster, C.A., Zollinger, M. and Lynch, K.R., (2002), The Immune Modulator FTY720 Targets Sphingosine-1-Phosphate Receptors; the Journal of Biological Chemistry, Vol.277, No.24, June 14, p.21453 to 21457;
Caligan, T.B., Peters, K., Ou, J., Wang, E., Saba, J., and Merrill, A.H., Jr.(2000) Anal Biochem 281, 36-44
Chang Q. and Balice-Gordon R.J.(2000) Highwire, rpm-1, and futsch: balancing synaptic growth and stability. Neuron 26, 287-290;
Chen, Z., Nield, H.S., Sun, H., Barbier, A., and Patel, T.B.(1995) J Biol Chem 270, 27525-27530
Chomczynski, P., Sacchi, N.: Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal. Biochem. 162, (1987) 156-159;
DiAntonio, A., Haghighi, A.P., Portman, S.L., Lee, J.D., Amaranto, A.M., and Goodman, C.S.(2001) Nature 412, 449-452
Graziano, M.P. FreissmuthM. Gilman A.G.: Purification of recombinant Gs alpha.
Meth. Enz. (1991) 195: 192-215;
Graziano M.P., Freissmuth M. and Gilman A.G. (1991) Purification of recombinant Gs alpha. Methods Enzymol 195, 192-202;
Guo Q., Xie J., Dang C.V., Liu E.T., Bishop J.M.: Identification of a large Myc-binding protein that contains RCC1-like repeats. Proc. Natl. Acad. Sci. U.S.A. 1998 Aug 4;95(16):9172-7);
Grossberger, R., Gieffers, C., Zachariae, W., Podtelejnikov, A.V., Schleiffer,
A., Nasmyth, K., Mann, M., and Peters, J.M.(1999) J Biol Chem 274, 14500-14507;
Hla, T., Lee, M.J., Ancellin, N., Paik, J.H., and Kluk, M.J. (2001) Science 294, 1875-1878;
Jin Y. (2002) Synaptogenesis: insights from worm and fly. Curr Opin Neurobiol 12, 71-79.);
【0116】
Julius and Basbaum“Molecular mechanisms of nociception”, Nature, volume 413,
13. September 2001, pp.203-209;
Kassis, S., and Fishman, P.H.(1982) J Biol Chem 257(9), 5312-5318;
Kind, P.C., and Neumann, P.E. (2001) Trends Neurosci 24, 553-555;
Kluk, M.J., and Hla, T. (2002) Biochim Biophys Acta 1582, 72-80
Mandala, S., Hajdu, R., Bergstrom, J., Quackenbush, E., Xie, J., Milligan, J.,
Thornton, R., Shei, G., Card, D., Keohand, C., Rosenbach, M., Hale, J., Lynch, C.L., Rupprecht, K., Parsons, W. and Rosen, H., (2002), Alteration of Lymphocyte Trafficking by Spingosine-1-Phospate Receptor Agonists, Science, Vol.296, April 2002;
Meller S.T., Gebhart G.F.(1997), intraplantar zymosan as a reliable, quantifiable model of thermal and mechanical hyperalgesia in the rat; Eur.J. Pain 1, 43-52;
Nair, B.G., Parikh, B., Milligan, G., and Patel, T.B.(1990) J Biol Chem 265(34), 21317-21322;
Nestler, E.J.(2001) Nat Rev Neurosci 2, 119-128
Patel T.B., Wittpoth C., Barbier A.J., Yigzaw Y. and Scholich K. (2002) Functional analyses of type V adenylyl cyclase. Methods Enzymol 345, 160-187. Snyder S.H.(1985) Adenosine as a neuromodulator. Annu Rev Neurosci 8, 103-124;
Payne, S.G., Milstien, S., and Spiegel, S.(2002), Sphingosine-1-phosphate: dual messenger functions; FEBS Letters 531 (2002), p.54 to 57;
Postma, F.R., Jalink, K., Hengeveld, T., and Moolenaar, W.H.(1996) Embo J 15, 2388-2392
Rebecchi, M.J., and Pentyala, S.N.(2000) Physiol Rev 80, 1291-1335
Ruppert C., Goldowitz D., and Wille W.(1986), Proto-oncogene-c-myc is expressed in cerebellar neurons at different developmental stages, Embo J5, 1897-1901;
Sato, K., Tomura, H., Igarashi, Y., Ui, M., and Okajima, F.(1997) Biochem Biophys Res Commun 240, 329-334
Schaefer A.M., Hadwiger G.D. and Nonet M.L.(2000) rpm-1, a conserved neuronal gene that regulates targeting and synaptogenesis in C. elegans. Neuron 26, 345-356);
【0117】
Schaible H.G., Vanegas H.: How do we manage chronic pain? Baillieres Best. Pract. Res. Clin. Rheumatol. 2000 Dec;14(4):797-811;
Scherr M., Morgan M.A., Eder M., Gene Silencing Mediated by Small Interfering RNAs in Mammalian Cells, Curr Med Chem. 2003, Feb; 10 (3): 245-256;
Scholich, K., Mullenix, J.B., Wittpoth, C., Poppleton, H.M., Pierre, S.C., Lindorfer, M.A., Garrison, J.C., and Patel, T.B. (1999) Science 283, 1328-1331
Scholich K., Pierre S., Patel T.B.: Protein associated with myc (PAM) is a potent inhibitor of adenylyl cyclase. J. Biol. Chem. 2001, Dec 14;276(50):47583-9;
Scholz and Woolf“Can we conquer pain”, Nature neuroscience supplement, volume 5, November 2002, pp.1062-1067;
Siehler, S., and Manning, D.R. (2002) Biochim Biophys Acta 1582, 94-99
Snyder S.H. (1985), Adenosine as a neuromodulator. Annual Reviews of Neuroscience 8, 103-104;
Spiegel, S. and Milstien, S.(2000), Functions of a new family of sphingosine-1-phosphate receptors, Biochimica et Biophysica Acta 1484, p.107 to 116;
Spiegel, S., and Milstien, S.(2002) J. Biol. Chem 277; 25851-25854;
Trajkovic V, Samardzic T, Stosic-Grujicic S, Ramic Z, Mostarica Stojkovic M. Muramyl dipeptide potentiates cytokine-induced activation of inducible nitric oxide synthase in rat astrocytes. Brain Res. 2000 Nov 10;883(1):157-63;
Wan H.I., DiAntonio A., Fetter R.D., Bergstrom K., Strauss R. and Goodman C.S. (2000) Highwire regulates synaptic growth in Drosophila. Neuron 26, 313-329.);
West, A.E., Chen, W.G., Dalva, M.B., Dolmetsch, R.E., Kornhauser, J.M., Shaywitz, A.J., Takasu, M.A., Tao, X., and Greenberg, M.E.(2001) Proc Natl Acad Sci USA 98, 11024-11031;
【0118】
Wilde, J.I., and Watson, S.P.(2001) Cell Signal 13, 691-701
Wittpoth C., Scholich K., Yigzaw Y., Stringfield T.M. and Patel T.B.(1999), R
egions on adenylyl cyclase that are necessary for inhibition of activity by beta
gamma G(iα) subunits of heterotrimeric G proteins. Proc. Natl. Acad. Sci. USA 96, 7723-7730;
Wood, J.D.“Pathobiology of Visceral Pain: Molecular Mechanisms and Therapeutic Implications II. genetic approaches to pain therapy”, American Journal pf Physiological Gastrointestinal Liver Physiology, 2000, volume 278, G507-G512;
Woolf and Mannion“Neuropathic pain: aetiology, symptoms mechanisms, and management”, The LANCET, volume 353, June 5, 1999, pp.1959-1964;
Woolf J. and Salter M.W.“Neuronal Plasticity: Increasing the Gain in Pain”, Science, volume 288, June 9, 2000, pp.1765-1768;
Xia Z., Storm D.R.: Calmodulin-regulated adenylyl cyclases and neuromodulation. Curr. Opin. Neurobiol. 1997 Jun;7(3):391-6;
Xu, D., Isaacs, C., Hall, I.P., and Emala, C.W.(2001) Am J Physiol Lung Cell Mol Physiol 281, L832-843
Yang H., Scholich K., Poser S., Storm D., Patel T.B., Goldowitz D.: Developmental expression of protein associated with myc (PAM) in the rodent brain. Brain Res Dev Brain Res 136, 2002, 35-42;
Zhen M., Huang X., Bamber B. and Jin Y.(2000) Regulation of presynaptic terminal organization by C. elegans RPM-1, a putative guanine nucleotide exchanger with a RING-H2 finger domain. Neuron 26, 331-343);
【0119】
実験方法に関する標準的な文献:
特に他の規定がない限り、実験方法は、以下の標準的な文献で列挙された標準的な方法に従って行われた、または、行うことができる:
Sambrook et al. (1989) Molecular Cloning: A Laboratory Manual. Second edition.
Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. 545 pp or Current Protocols in Molecular Biology;
Current Protocols in Molecular Biology; regularly updated, e.g. Volume 2000; John Wiley & Sons, Inc; Editors: Fred M. Ausubel, Roger Brent, Robert Eg. Kingston, David D. Moore, J.G.Seidman, John A. Smith, Kevin Struhl.
Current Protocols in Human Genetics; regularly uptdated, e.g. Volume 2003; John Wiley & Sons, Inc; Editors: Nicholas C. Dracopoli, Honathan L. Haines, Bruce R. Korf, Cynthia C. Morton, Christine E. Seidman, J.G. Seigman, Douglas R. Smith.
Current Protocols in Protein Science; regularly updated, e.g. Volume 2003; John Wiley & Sons, Inc; Editors: John E. Coligan, Ben M. Dunn, Hidde L. Ploegh, David W. Speicher, Paul T. Wingfield.
Molecular Biology of the Cell; third edition; Alberts, B., Bray, D., Lewis, J., Raff, M., Roberts, K., Watson, J.D.; Garland Publishing, Inc. New York & London, 1994;
Gene Targeting: a practical approach (1995), Editor: A.L. Joyner, IRL Press
Genetic Manipulation of Receptor Expression and Function; D. Accili, Wiley-Liss., USA, 2000; ISBN: 0-471-35057-5.
Antisense Therapeutics, S. Agrawal, Humana Press, USA, 1996, ISBN: 0-89603-305-8.
Remington's Pharmaceutical Sciences, Edition 17, 1985
【0120】
用いられた略語:
AC、アデニリルシクラーゼ;Gαs、アデニリルシクラーゼの促進性Gタンパク質のαサブユニット、Gαs*、Gαsの構成的に活性な(Q213L)突然変異体;Gαi、抑制性Gタンパク質Giのαサブユニット;Gβγ、ヘテロ三量体Gタンパク質のβγサブユニット;ODN、オリゴデオキシヌクレオチド;PAM、Myc関連タンパク質;RCC1、染色体凝縮の調節因子;S1P、スフィンゴシン−1−リン酸;TED、50mMトリスHCl(pH8.0)、1mMのEDTA、1mMのDTT;RT、室温。
【0121】
図面
図1:PAMは、脊髄ニューロンで高度に発現される。脊髄の水平切片標本を用いたインサイチュハイブリダイゼーションを、上述したように、ラットPAMに対するセンスまたはアンチセンスプローブとハイブリダイズさせた。
図2:PAMは、DRGニューロンで、同様に、ラット脊髄中のニューロンの細胞で発現される。
パネルA:ラットの脊髄切片標本の免疫組織化学的な解析。切片標本を、抗PAM抗体(緑)、および、抗NeuNまたは抗GFAP(赤)で染色し、ニューロンまたはグリア細胞をそれぞれ可視化した。両方のシグナルの重ね合わせは、右のパネルに示される。対物を20×に拡大した。
パネルB:ラットDRG切片標本の免疫組織化学的な解析。切片標本を、抗PAM抗体(緑)、および、抗Neu68、抗ヒストンまたは抗GFAP(赤)で染色し、それぞれニューロン、核またはグリア細胞を可視化した。両方のシグナルの重ね合わせは、右のパネルに示される。対物を40×に拡大した(ただし、ヒストン染色は、63×に拡大した)。
【0122】
図3:PAMは、DRGおよび脊髄において、異なる発生段階で差異的に発現される。定量RT−PCR(TaqmanTM)を用いて、16日齢のラット胎児(E16)、出生後の日数が0.5(P0.5)、3(P3)、5(P5)、9(P9)のラット、および、成体ラットの脊髄およびDRGのRNA(40ng)中のPAMを検出した。少なくとも3回の測定の平均±SEMを示す。
図4:PAMは、ラットの脊髄において、ザイモサンおよびホルマリン処理の後にアップレギュレートされた。
パネルA:コントロール動物、または、ザイモサンで処理して24時間、48時間および96時間後の動物からの脊髄のRNA(40ng)を用いたRT−PCR解析。下部のパネルは、7回の実験の平均±SEMを示す。スチューデントのt検定:*p<0.001。
パネルB:コントロール動物、または、ザイモサンで処理して24時間、48時間および96時間後の動物のラットの脊髄溶解産物(40μg)を用いた、抗PAM抗体および抗ERK1/2を用いた、7%SDS−PAGEゲルを用いたウェスタンブロット解析。
パネルC:コントロール動物、または、ホルマリンで1時間処理した動物からの脊髄のRNA(40ng)を用いた定量RT−PCR解析。
【0123】
図5:PAMは、脊髄溶解産物中でGαsで刺激されたAC活性を阻害する。
パネルA:RT−PCRを用いて、脊髄およびDRGのRNA(40ng)におけるACアイソフォーム発現を決定した。
パネルB:材料および方法で説明したように、脊髄またはDRGの溶解産物(10μg)を、80nMのGαsの存在下で、AC活性に関して分析した。三連で行われた少なくとも3回の測定の平均±SEMを示す。
図6:PAMに対するアンチセンスODNの髄腔内への適用により、侵害受容の挙動が増加する。
パネルA:説明されているように、成体ラットに、センスおよびアンチセンスODNを髄腔内投与した。ホルマリン処理の後に、脊髄を除去し、抗PAM抗体(緑)、または、
抗NeuN(赤)を用いて免疫組織学的な解析で処理した。
パネルB:材料の章において説明されているように、センスまたはアンチセンスODNで処理した動物のホルマリンアッセイ。
1時間にわたるフリンチ行動の合計を示す。少なくとも4回の測定の平均±SEを示す。
パネルC:材料の章において説明されているように、センスまたはアンチセンスODNで処理した動物のホルマリンアッセイ。
1時間にわたるフリンチ行動の回数を示す。
少なくとも4回の測定の平均±SEを示す。
【0124】
図7:NCBI登録番号AAC39928(タンパク質配列;配列番号1)、AF075587(コード配列;配列番号2)による、ヒトPAMのタンパク質、ゲノムおよびコードヌクレオチド配列。ヒトPAMは、染色体13q22上に位置する;そのゲノム配列は、NT_024524.11(始点:24679861位;終点:24962245位;配列番号3)で公開された形で利用可能である;
図7Cは、24679861〜24962245位の連続した配列を示す。
図8:ラットPAMに関するEST−クローンをコードする配列:
図8A:AW921303(hscDNAのbp960〜1394に対応する;配列番号4)
図8B:AW918711(hscDNAのbp8188〜8632に対応する;配列番号5)
図8C:BQ201485(hscDNAのbp8966〜9633に対応する;配列番号6)
図8D:BE112881(hscDNAのbp10311〜10830に対応する;配列番号7)
図8E:AW441131(hscDNAのbp13569〜14152に対応する;配列番号8)
図8F:BF409872(hscDNAのbp13569〜14807に対応する)。(配列番号9)
【0125】
図9:Guo等,1988年/Grossberger等,JBC 1999年、および、Scholich等,JBC 2001年)によるヒトPAMのドメイン構造の概観。
図10:ラットPAM RTPCRのためのPCRプライマー。
図11:ラットPAM発現を阻害するためのアンチセンスオリゴデスオキシヌクレオチド、および、コントロールオリゴヌクレオチド。
図12:本発明の環境で使用するための、様々なPAMhsポリペプチド。これらのポリペプチドは、配列番号2に記載のポリペプチドから得られたフラグメントである。
図13:本発明の環境で使用するための、異なるPAMhsポリヌクレオチド。
図13A:hsPamcDNAのヌクレオチドの1317〜4366位を含む、配列番号17に記載のタンパク質フラグメントをコードするcDNA配列(配列番号24);
図13B:hsPamcDNAのヌクレオチドの1482〜3332位を含む、配列番号18に記載のタンパク質フラグメントをコードするcDNA配列(配列番号25);
図13C:hsPamcDNAのヌクレオチドの1641〜3341位を含む、配列番号19に記載のタンパク質フラグメントをコードするcDNA配列(配列番号26);
図13D:hsPamcDNAのヌクレオチドの3142〜4046位を含む、配列番号20に記載のタンパク質フラグメントをコードするcDNA配列(配列番号27);
図13E:hsPamcDNAのヌクレオチドの3142〜3446位を含む、配列番号21に記載のタンパク質フラグメントをコードするcDNA配列(配列番号28);
図13F:hsPamcDNAのヌクレオチドの3228〜3839位を含む、配列番
号22に記載のタンパク質フラグメントをコードするcDNA配列(配列番号29);
図13G:hsPamcDNAのヌクレオチドの3228〜3341位を含む、配列番号23に記載のタンパク質フラグメントをコードするcDNA配列(配列番号30);
【0126】
図14:上記の発見によるPAMシグナル伝達。
図15:スフィンゴシン−1−リン酸の構造;
図16:S1P受容体のmRNAおよびアミノ酸配列、
16a)S1P1のアミノ酸配列(配列番号31);
16b)S1P1のmRNA配列(配列番号32);コード配列は、244位で始まり、1392位で終わる、
16c)S1P2のアミノ酸配列(配列番号33);
16d)S1P2のmRNA配列(配列番号34);コード配列は、1位で始まで始まり、1062位で終わる;
16e)S1P3のアミノ酸配列(配列番号35)
16f)S1P3のmRNA配列(配列番号36)、コード配列は、1位で始まり、1137位で終わる;
16g)S1P4のアミノ酸配列(配列番号37)
16h)S1P4のmRNA配列(配列番号38)、コード配列は、23位で始まり、1177位で終わる;
16i)S1P5のアミノ酸配列(配列番号39)
16j)S1P5のmRNA配列(配列番号40)、コード配列は、10位で始まり、1206位で終わる:
図17:FT720の構造
図18:PAMは、HeLa細胞において、血清での刺激の後に、ERから細胞質膜へトランスロケーションする。
パネルA:上述したように、血清欠乏状態にしたHeLa細胞(24時間)を固定し、抗PAM抗体(緑)と、抗カルネキシン抗体(赤)で染色した。
パネルB:HeLa細胞を、様々な時間で10%ウシ胎仔血清で処理し、抗PAM抗体で染色し、細胞内局在をモニターした。
【0127】
図19:HeLa細胞の血清処理により、PAM依存性プロセスを介してAC活性が減少する。
パネルA:上述したように、血清の非存在および存在で、HeLa細胞におけるcAMPの蓄積を測定した。3回の測定の平均±SEM(標準誤差)を示す。
パネルB:材料および方法の章で説明したように、血清の非存在および存在で、HeLa細胞溶解産物のアデニリルシクラーゼ活性を測定した。Gsα*(80nM;灰色のバー)で、および、フォルスコリン(100μM;黒いバー)で刺激した、ACVの特異的活性は、それぞれ124±10pmol/mg/分、および、464±89pmol/分/mgであった。基礎的な活性は、11.75±2pmol/mg/分であった。少なくとも2回の実験(それぞれ三連で行われた)の平均±SEを示す。スチューデントのt検定:*p<0.001。
パネルC:血清欠乏状態にしたHeLa細胞において、10%FBSで1時間インキュベートした後の、ACアイソフォーム発現のRT−PCR解析。
パネルD:上述したように、血清欠乏状態にしたHeLa細胞を、アンチセンス、センスおよび3つの点突然変異(3M−as)を含むアンチセンスODNをそれぞれ3□Mで用いてトランスフェクトした。細胞を回収し、7%SDS−PAGE(タンパク質30μg)を用いて、抗PAM抗体および抗Hsp70抗体を用いてウェスタン解析に付した。
パネルE:上述したように、血清欠乏状態にしたHeLa細胞を、センス、アンチセンス、および、3M−as ODNで処理した。
24時間後、細胞を、10%FBSと30分間インキュベートした。上述したように、
細胞を回収し、Gαs*(80nM;灰色のバー)、および、フォルスコリン(100μM;黒いバー)の存在下でAC活性を決定した。少なくとも3回の実験(三連で測定された)からのデータを、平均±SEMとして示す。スチューデントのt検定:*p<0.01、**p<0.05。
【0128】
図20:S1Pは、PAMの細胞質膜へのトランスロケーションを誘発させ、PAM依存性のAC酵素活性を阻害する。
パネルA:血清欠乏状態にしたHeLa細胞におけるS1P受容体アイソフォーム発現のRT−PCR解析。
パネルB:HeLa細胞を、0.5μMのS1Pで30分間処理し、抗PAM抗体で染色し、細胞内局在をモニターした。
パネルC:HeLa細胞におけるPAMのトランスロケーションの時間依存性。血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで様々な時間で処理し、抗PAM抗体で染色し、細胞内局在をモニターした。PAMのトランスロケーションを示す細胞のパーセンテージを示す。
図21:HeLa細胞のS1P処理により、PAM依存性プロセスを介してAC活性が減少する。
パネルA:材料および方法の章で説明したように、0.5μMのS1Pが非存在下(黒いバー)、および、0.5μMのS1Pが存在下(灰色のバー)での、様々な時間での、HeLa細胞におけるcAMPの蓄積を測定した。3回の測定の平均±SEMを示す。
パネルB:血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで様々な時間で処理した。材料および方法で説明したように、細胞を回収し、80nMのGαs*の存在下でAC活性を決定した。少なくとも4回の実験(三連で測定された)からのデータを、平均±SEとして示す。
パネルC:材料および方法の章で説明されているように、血清欠乏状態にしたHeLa細胞を、センス、アンチセンス、および、3M−as ODNで処理した。24時間後、細胞を、0.5μMのS1Pと指定された時間インキュベートした。
細胞を回収し、上述したように、Gαs*(80nM)の存在下でAC活性を決定した。データを、3回の測定の平均±SEMとして示す。
【0129】
図22:PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に、PLC/PKCシグナル伝達経路が介在する。
パネルA:血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで、3分間(灰色のバー)、または、60分間(黒いバー)処理した。
S1Pとのインキュベートの前に、細胞を、1μg/μlの百日咳毒素(PTX)と24時間インキュベートした。80nMのGαs*の存在で、AC活性を決定した。少なくとも2回の実験(三連で測定された)からのデータを、平均±SEとして示す。
パネルB:血清欠乏状態にしたHeLa細胞を、0.5μMのS1Pで処理した。S1Pとのインキュベートの前に、細胞を、10μMのU73122、RO31−8220、BAPTA−AMまたはU0126と20分間インキュベートした。80nMのGαs*の存在下で、AC活性を決定した。少なくとも2回の実験(三連で測定された)からのデータを、平均±SEとして示す。
パネルC:0.5μMのS1Pで10分間処理したHeLa細胞のウェスタンブロット解析。S1Pとのインキュベートの前に、細胞を、10μMのU73122、BAPTA−AMまたはU0126と20分間インキュベートした。沸騰したレムリー緩衝液中で細胞を回収し、抗活性ERK1/2抗体を用いてウェスタン解析で処理した。ローディングコントロールとして抗Hsp70抗体を用いた。
パネルD:HeLa細胞を、0.5μMのS1Pで20分間処理し、抗PAM抗体で染色し、細胞内局在をモニターした。S1P処理の前に、細胞を、1μg/μlの百日咳毒素(PTX)と24時間、1μMのGF109203X、10μMのU0126、BAP
TA−AM、U73122またはRo31−223と20分間、インキュベートした。
図23:PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に至る提唱されたシグナル伝達経路の模式図。
図24:ホルマリン分析における、60分間にわたるS1P適用を行った場合と、行わない場合の成体ラットの痛み行動。
【0130】
ホルマリン注射の前に、成体ラットに、10μMのS1P20μl、または、PBS/DMSO20μlを髄腔内へ15分間適用することによって投与した。ホルマリン注射の後に、フリンチ行動を60分間にわたり5分間のインターバルでカウントした。6回の動物実験の平均+SEMを示す。
【0131】
【表1】
【0132】
アンチセンス処理ラットの脊髄溶解産物において、基礎的なAC活性は上昇する。上述したように、脊髄溶解産物(20μg)を、80nMのGαs、または、100μMのフォルスコリンの非存在下、または存在下でAC活性に関して分析した。グループあたり少なくとも3匹のラットからの脊髄溶解産物の平均AC活性±SEM(それぞれ三連で2回測定された)を示す(ns=有意差なし)。
【図面の簡単な説明】
【0133】
【図1】PAMは、脊髄ニューロンで高度に発現される。
【図2A】PAMは、DRGニューロンで、同様に、ラット脊髄中のニューロンの細胞で発現される。
【図2B】PAMは、DRGニューロンで、同様に、ラット脊髄中のニューロンの細胞で発現される。
【図3】PAMは、DRGおよび脊髄において、異なる発生段階で差異的に発現される。
【図4】PAMは、ラットの脊髄において、ザイモサンおよびホルマリン処理の後にアップレギュレートされた。
【図5】PAMは、脊髄溶解産物中でGαsで刺激されたAC活性を阻害する。
【図6】PAMに対するアンチセンスODNの髄腔内への適用により、侵害受容の挙動が増加する。
【図6−1】図6の続きである。
【図7】NCBI登録番号AAC39928(タンパク質配列;配列番号1)、AF075587(コード配列;配列番号2)による、ヒトPAMのタンパク質、ゲノムおよびコードヌクレオチド配列である。
【図7−1】図7の続きである。
【図7−2】図7−1の続きである。
【図7−3】図7−2の続きである。
【図7−4】図7−3の続きである。
【図7−5】図7−4の続きである。
【図7−6】図7−5の続きである。
【図7−7】図7−6の続きである。
【図7−8】図7−7の続きである。
【図7−9】図7−8の続きである。
【図7−10】図7−9の続きである。
【図7−11】図7−10の続きである。
【図7−12】図7−11の続きである。
【図7−13】図7−12の続きである。
【図7−14】図7−13の続きである。
【図7−15】図7−14の続きである。
【図7−16】図7−15の続きである。
【図7−17】図7−16の続きである。
【図7−18】図7−17の続きである。
【図7−19】図7−18の続きである。
【図7−20】図7−19の続きである。
【図7−21】図7−20の続きである。
【図7−22】図7−21の続きである。
【図7−23】図7−22の続きである。
【図7−24】図7−23の続きである。
【図7−25】図7−24の続きである。
【図7−26】図7−25の続きである。
【図7−27】図7−26の続きである。
【図7−28】図7−27の続きである。
【図7−29】図7−28の続きである。
【図7−30】図7−29の続きである。
【図7−31】図7−30の続きである。
【図7−32】図7−31の続きである。
【図7−33】図7−32の続きである。
【図7−34】図7−33の続きである。
【図7−35】図7−34の続きである。
【図7−36】図7−35の続きである。
【図7−37】図7−36の続きである。
【図7−38】図7−37の続きである。
【図7−39】図7−38の続きである。
【図7−40】図7−39の続きである。
【図7−41】図7−40の続きである。
【図7−42】図7−41の続きである。
【図7−43】図7−42の続きである。
【図7−44】図7−43の続きである。
【図7−45】図7−44の続きである。
【図7−46】図7−45の続きである。
【図7−47】図7−46の続きである。
【図7−48】図7−47の続きである。
【図7−49】図7−48の続きである。
【図7−50】図7−49の続きである。
【図7−51】図7−50の続きである。
【図7−52】図7−51の続きである。
【図7−53】図7−52の続きである。
【図7−54】図7−53の続きである。
【図7−55】図7−54の続きである。
【図7−56】図7−55の続きである。
【図7−57】図7−56の続きである。
【図7−58】図7−57の続きである。
【図7−59】図7−58の続きである。
【図7−60】図7−59の続きである。
【図7−61】図7−60の続きである。
【図7−62】図7−61の続きである。
【図7−63】図7−62の続きである。
【図7−64】図7−63の続きである。
【図7−65】図7−64の続きである。
【図7−66】図7−65の続きである。
【図7−67】図7−66の続きである。
【図7−68】図7−67の続きである。
【図7−69】図7−68の続きである。
【図7−70】図7−69の続きである。
【図7−71】図7−70の続きである。
【図7−72】図7−71の続きである。
【図7−73】図7−72の続きである。
【図7−74】図7−73の続きである。
【図7−75】図7−74の続きである。
【図7−76】図7−75の続きである。
【図7−77】図7−76の続きである。
【図7−78】図7−77の続きである。
【図7−79】図7−78の続きである。
【図7−80】図7−79の続きである。
【図7−81】図7−80の続きである。
【図7−82】図7−81の続きである。
【図7−83】図7−82の続きである。
【図7−84】図7−83の続きである。
【図7−85】図7−84の続きである。
【図7−86】図7−85の続きである。
【図7−87】図7−86の続きである。
【図7−88】図7−87の続きである。
【図7−89】図7−88の続きである。
【図7−90】図7−89の続きである。
【図7−91】図7−90の続きである。
【図7−92】図7−91の続きである。
【図7−93】図7−92の続きである。
【図7−94】図7−93の続きである。
【図7−95】図7−94の続きである。
【図7−96】図7−95の続きである。
【図7−97】図7−96の続きである。
【図7−98】図7−97の続きである。
【図7−99】図7−98の続きである。
【図7−100】図7−99の続きである。
【図7−101】図7−100の続きである。
【図7−102】図7−101の続きである。
【図7−103】図7−102の続きである。
【図7−104】図7−103の続きである。
【図7−105】図7−104の続きである。
【図7−106】図7−105の続きである。
【図7−107】図7−106の続きである。
【図7−108】図7−107の続きである。
【図7−109】図7−108の続きである。
【図7−110】図7−109の続きである。
【図7−111】図7−110の続きである。
【図7−112】図7−111の続きである。
【図7−113】図7−112の続きである。
【図7−114】図7−113の続きである。
【図7−115】図7−114の続きである。
【図7−116】図7−115の続きである。
【図7−117】図7−116の続きである。
【図7−118】図7−117の続きである。
【図7−119】図7−118の続きである。
【図7−120】図7−119の続きである。
【図7−121】図7−120の続きである。
【図7−122】図7−121の続きである。
【図7−123】図7−122の続きである。
【図7−124】図7−123の続きである。
【図7−125】図7−124の続きである。
【図7−126】図7−125の続きである。
【図8】ラットPAMに関するEST−クローンをコードする配列である。
【図8−1】図8の続きである。
【図8−2】図8−1の続きである。
【図9】Guo等,1988年/Grossberger等,JBC 1999年、および、Scholich等,JBC 2001年)によるヒトPAMのドメイン構造の概観である。
【図10】ラットPAMRT−PCRのためのPCRプライマーである。
【図11】ラットPAM発現を阻害するためのアンチセンスオリゴデゾキシヌクレオチド、および、コントロールオリゴヌクレオチドである。
【図12】本発明の環境で使用するための、様々なPAMhsポリペプチド。これらのポリペプチドは、配列番号2に記載のポリペプチドから得られたフラグメントであるである。
【図12−1】図12の続きである。
【図12−2】図12−1の続きである。
【図13】本発明の環境で使用するための、異なるPAMhsポリヌクレオチドである。
【図13−1】図13の続きである。
【図13−2】図13−1の続きである。
【図13−3】図13−2の続きである。
【図13−4】図13−3の続きである。
【図14】上記の発見によるPAMシグナル伝達である。
【図15】スフィンゴシン−1−リン酸の構造である。
【図16】S1P受容体のmDNAおよびアミノ酸配列である。
【図16−1】図16の続きである。
【図16−2】図16−1の続きである。
【図16−3】図16−2の続きである。
【図16−4】図16−3の続きである。
【図16−5】図16−4の続きである。
【図16−6】図16−5の続きである。
【図17】FT720の構造である。
【図18】PAMは、HeLa細胞において、血清での刺激の後に、ERから細胞質膜へトランスロケーションする。
【図19】HeLa細胞の血清処理により、PAM依存性プロセスを介してAC活性が減少する。
【図19−1】HeLa細胞の血清処理により、PAM依存性プロセスを介してAC活性が減少する。
【図20】S1Pは、PAMの細胞質膜へのトランスロケーションを誘発させ、PAM依存性のAC酵素活性を阻害する。
【図21】HeLa細胞のS1P処理により、PAM依存性プロセスを介してAC活性が減少する。
【図22】PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に、PLC/PKCシグナル伝達経路が介在する。
【図22−1】図22の続きである。
【図23】PAMのトランスロケーション、および、AC酵素活性のPAM依存性阻害に至る提唱されたシグナル伝達経路の模式図である。
【図24】ホルマリン分析における、60分間にわたるS1P適用を行った場合と、行わない場合の成体ラットの痛み行動である。
【特許請求の範囲】
【請求項1】
医薬化合物を製造するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用。
【請求項2】
痛みの調節のための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用。
【請求項3】
痛みを調節する化合物を同定するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用。
【請求項4】
十分な量のS1Pまたはそれらの機能的フラグメントもしくは誘導体を、個体に投与することを含む、痛みを軽減する方法。
【請求項5】
医薬品として使用するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体。
【請求項6】
痛み、および/または、炎症を予防または治療するための医薬品として使用するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体。
【請求項7】
医薬化合物は、痛み、および/または、炎症を予防または治療するための化合物である、請求項1に記載の使用。
【請求項8】
化合物は、痛みを軽減するか、または止める、請求項3に記載の使用。
【請求項9】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、S1Pを含まないサンプルを提供する工程、
b.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、同様に、S1Pも含む第二のサンプルを提供する工程、
c.両方のサンプル中のPAM活性を測定する工程、
d.第一のサンプルと化合物とを接触させる工程、
e.上記化合物のS1Pの機能を模擬する能力を測定する工程
を含む、上記方法。
【請求項10】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a)S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現する細胞を含む2つのサンプルを提供すること、
b)一方のサンプルと、上記化合物とを接触させる工程、および、
c)両方のサンプル中の受容体活性を測定する工程
を含む、上記方法。
【請求項11】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a)S1P受容体またはそれらの機能的フラグメントまたは誘導体を発現する細胞を含む2つのサンプルを提供する工程、および、
b)サンプルと、S1Pとを接触させる工程、および、
c)他方のサンプルと、化合物とを接触させる工程、および、
d)両方のサンプル中のS1Pおよび受容体の受容体活性または相互作用を測定する工程
を含む、上記方法。
【請求項12】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a.S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現し、そして、PAMまたはそれらの機能的フラグメントもしくは誘導体を発現する細胞を提供する工程、
b.上記細胞と化合物とを接触させる工程、
c.化合物の存在下でS1P活性を測定する工程、
d.化合物の非存在下でS1P活性を測定する工程、および、
e.c)によるS1P活性と、d)によるS1P活性とを比較する工程
を含む、上記方法。
【請求項13】
細胞を、化合物の代わりにS1Pと接触させ、c)およびd)に記載のPAM活性を、S1Pの存在下でのPAM活性と比較する工程をさらに含む、請求項10に記載の方法。
【請求項14】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a.2つのサンプル(S1Pまたはそれらの機能的フラグメントもしくは誘導体を含むサンプル、および、S1P受容体またはそれらの機能的フラグメントもしくは誘導体を含むサンプル)を提供する工程、
b.上記サンプルの一方と、化合物とを接触させる工程、
c.両方のサンプル中のS1P活性を測定し、比較する工程
を含む、上記方法。
【請求項15】
機能的フラグメントまたは誘導体は、アデニリルシクラーゼ活性を阻害することができる、請求項1〜14のいずれか一項に記載の使用、方法またはS1P。
【請求項16】
アデニリルシクラーゼは、アデニリルシクラーゼI、VまたはVI型である、請求項10に記載の使用、方法またはS1P。
【請求項17】
S1P活性の測定は、S1P受容体と相互作用して、および/または、S1P受容体を活性化して、PAMのトランスロケーションを媒介する、PAMの機能を活性化する、または、ACの機能を不活性化する能力に関する、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
痛みを調節する化合物を同定する方法であって、
a)試験化合物として、S1Pの活性を調節または模擬する化合物を選択すること、および、
b)上記試験化合物を被検体に投与すること、および、
c)被検体において痛みが調節されているかどうかを測定すること
を含む、上記方法。
【請求項19】
痛みを調節または予防する医薬品を製造するための、少なくとも1種のS1P受容体を活性化する能力を有する、および/または、PAMの機能を活性化する能力を有する化合物の使用。
【請求項20】
調節は、痛みを軽減すること、または、止めることである、請求項19に記載の使用。
【請求項21】
化合物は、FTY720、もしくはそれらの機能的な誘導体、好ましくはリン酸化された誘導体、または、該化合物もしくは誘導体の塩である、請求項19または20に記載の使用。
【請求項22】
少なくとも1種のS1P受容体に結合し、活性化する能力を有する、および/または、PAMの機能を活性化する活性を有する医薬化合物の十分な量を、個体に投与することを含む、痛みを軽減する、または予防する方法。
【請求項23】
化合物は、FTY720、もしくは、それらの機能的な誘導体、好ましくはリン酸化された誘導体であるか、または、該化合物または誘導体の生理学的に許容できる塩である、請求項22に記載の方法。
【請求項1】
医薬化合物を製造するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用。
【請求項2】
痛みの調節のための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用。
【請求項3】
痛みを調節する化合物を同定するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体の使用。
【請求項4】
十分な量のS1Pまたはそれらの機能的フラグメントもしくは誘導体を、個体に投与することを含む、痛みを軽減する方法。
【請求項5】
医薬品として使用するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体。
【請求項6】
痛み、および/または、炎症を予防または治療するための医薬品として使用するための、S1Pまたはそれらの機能的フラグメントもしくは誘導体。
【請求項7】
医薬化合物は、痛み、および/または、炎症を予防または治療するための化合物である、請求項1に記載の使用。
【請求項8】
化合物は、痛みを軽減するか、または止める、請求項3に記載の使用。
【請求項9】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、S1Pを含まないサンプルを提供する工程、
b.PAMまたはそれらの機能的フラグメントもしくは誘導体を含み、同様に、S1Pも含む第二のサンプルを提供する工程、
c.両方のサンプル中のPAM活性を測定する工程、
d.第一のサンプルと化合物とを接触させる工程、
e.上記化合物のS1Pの機能を模擬する能力を測定する工程
を含む、上記方法。
【請求項10】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a)S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現する細胞を含む2つのサンプルを提供すること、
b)一方のサンプルと、上記化合物とを接触させる工程、および、
c)両方のサンプル中の受容体活性を測定する工程
を含む、上記方法。
【請求項11】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a)S1P受容体またはそれらの機能的フラグメントまたは誘導体を発現する細胞を含む2つのサンプルを提供する工程、および、
b)サンプルと、S1Pとを接触させる工程、および、
c)他方のサンプルと、化合物とを接触させる工程、および、
d)両方のサンプル中のS1Pおよび受容体の受容体活性または相互作用を測定する工程
を含む、上記方法。
【請求項12】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a.S1P受容体またはそれらの機能的フラグメントもしくは誘導体を発現し、そして、PAMまたはそれらの機能的フラグメントもしくは誘導体を発現する細胞を提供する工程、
b.上記細胞と化合物とを接触させる工程、
c.化合物の存在下でS1P活性を測定する工程、
d.化合物の非存在下でS1P活性を測定する工程、および、
e.c)によるS1P活性と、d)によるS1P活性とを比較する工程
を含む、上記方法。
【請求項13】
細胞を、化合物の代わりにS1Pと接触させ、c)およびd)に記載のPAM活性を、S1Pの存在下でのPAM活性と比較する工程をさらに含む、請求項10に記載の方法。
【請求項14】
痛みを調節、および/または、予防するのに有用な医薬をスクリーニングする方法であって、
a.2つのサンプル(S1Pまたはそれらの機能的フラグメントもしくは誘導体を含むサンプル、および、S1P受容体またはそれらの機能的フラグメントもしくは誘導体を含むサンプル)を提供する工程、
b.上記サンプルの一方と、化合物とを接触させる工程、
c.両方のサンプル中のS1P活性を測定し、比較する工程
を含む、上記方法。
【請求項15】
機能的フラグメントまたは誘導体は、アデニリルシクラーゼ活性を阻害することができる、請求項1〜14のいずれか一項に記載の使用、方法またはS1P。
【請求項16】
アデニリルシクラーゼは、アデニリルシクラーゼI、VまたはVI型である、請求項10に記載の使用、方法またはS1P。
【請求項17】
S1P活性の測定は、S1P受容体と相互作用して、および/または、S1P受容体を活性化して、PAMのトランスロケーションを媒介する、PAMの機能を活性化する、または、ACの機能を不活性化する能力に関する、請求項1〜16のいずれか一項に記載の方法。
【請求項18】
痛みを調節する化合物を同定する方法であって、
a)試験化合物として、S1Pの活性を調節または模擬する化合物を選択すること、および、
b)上記試験化合物を被検体に投与すること、および、
c)被検体において痛みが調節されているかどうかを測定すること
を含む、上記方法。
【請求項19】
痛みを調節または予防する医薬品を製造するための、少なくとも1種のS1P受容体を活性化する能力を有する、および/または、PAMの機能を活性化する能力を有する化合物の使用。
【請求項20】
調節は、痛みを軽減すること、または、止めることである、請求項19に記載の使用。
【請求項21】
化合物は、FTY720、もしくはそれらの機能的な誘導体、好ましくはリン酸化された誘導体、または、該化合物もしくは誘導体の塩である、請求項19または20に記載の使用。
【請求項22】
少なくとも1種のS1P受容体に結合し、活性化する能力を有する、および/または、PAMの機能を活性化する活性を有する医薬化合物の十分な量を、個体に投与することを含む、痛みを軽減する、または予防する方法。
【請求項23】
化合物は、FTY720、もしくは、それらの機能的な誘導体、好ましくはリン酸化された誘導体であるか、または、該化合物または誘導体の生理学的に許容できる塩である、請求項22に記載の方法。
【図1】


【図2A】
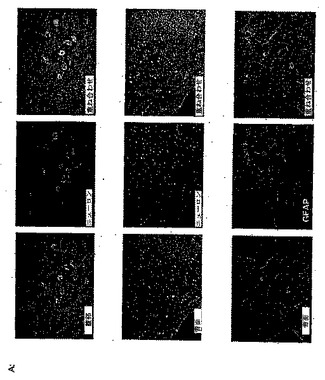
【図2B】
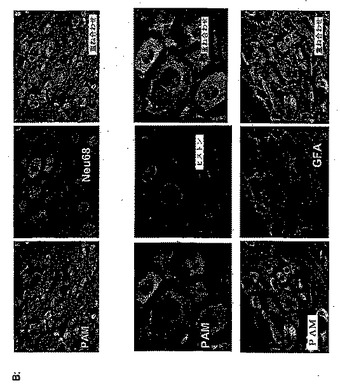
【図3】
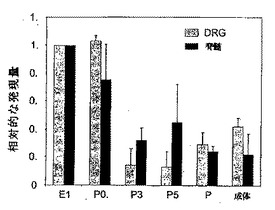
【図4】
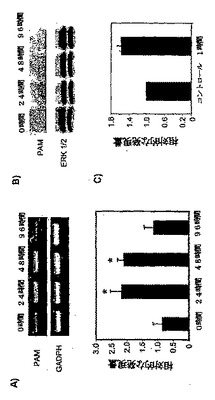
【図5】
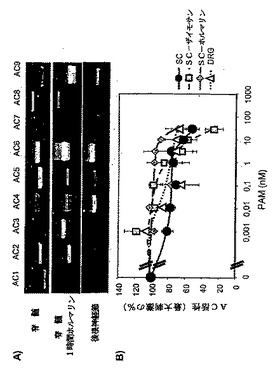
【図6】
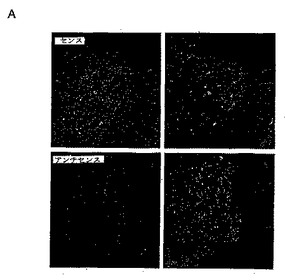
【図6−1】
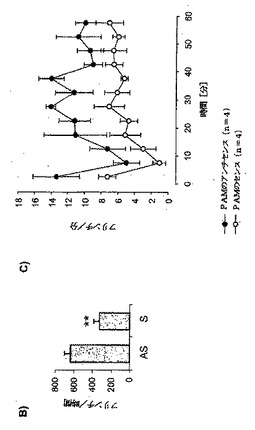
【図7】

【図7−1】

【図7−2】

【図7−3】

【図7−4】

【図7−5】

【図7−6】

【図7−7】

【図7−8】

【図7−9】

【図7−10】

【図7−11】

【図7−12】

【図7−13】

【図7−14】

【図7−15】

【図7−16】

【図7−17】

【図7−18】

【図7−19】

【図7−20】

【図7−21】

【図7−22】

【図7−23】

【図7−24】

【図7−25】

【図7−26】

【図7−27】

【図7−28】

【図7−29】

【図7−30】

【図7−31】

【図7−32】

【図7−33】

【図7−34】

【図7−35】

【図7−36】

【図7−37】

【図7−38】

【図7−39】

【図7−40】

【図7−41】

【図7−42】

【図7−43】

【図7−44】

【図7−45】

【図7−46】

【図7−47】

【図7−48】

【図7−49】

【図7−50】

【図7−51】

【図7−52】

【図7−53】

【図7−54】

【図7−55】

【図7−56】

【図7−57】

【図7−58】

【図7−59】

【図7−60】

【図7−61】

【図7−62】

【図7−63】

【図7−64】

【図7−65】

【図7−66】

【図7−67】

【図7−68】

【図7−69】

【図7−70】

【図7−71】

【図7−72】

【図7−73】

【図7−74】

【図7−75】

【図7−76】

【図7−77】

【図7−78】

【図7−79】

【図7−80】

【図7−81】

【図7−82】

【図7−83】

【図7−84】

【図7−85】

【図7−86】

【図7−87】

【図7−88】

【図7−89】

【図7−90】

【図7−91】

【図7−92】

【図7−93】

【図7−94】

【図7−95】

【図7−96】

【図7−97】

【図7−98】

【図7−99】

【図7−100】

【図7−101】

【図7−102】

【図7−103】

【図7−104】

【図7−105】

【図7−106】

【図7−107】

【図7−108】

【図7−109】

【図7−110】

【図7−111】

【図7−112】

【図7−113】

【図7−114】

【図7−115】

【図7−116】

【図7−117】

【図7−118】

【図7−119】

【図7−120】

【図7−121】

【図7−122】

【図7−123】

【図7−124】

【図7−125】

【図7−126】

【図8】
【図8−1】

【図8−2】

【図9】
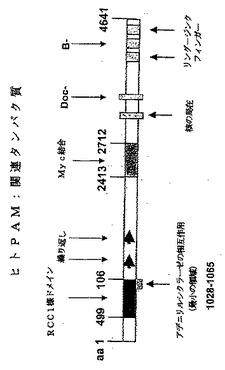
【図10】

【図11】

【図12】

【図12−1】

【図12−2】

【図13】

【図13−1】

【図13−2】

【図13−3】

【図13−4】

【図14】
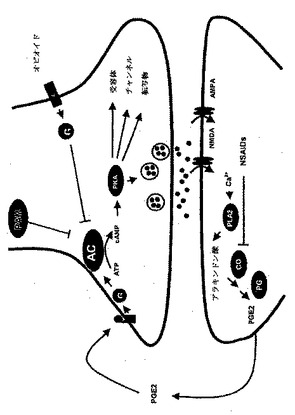
【図15】
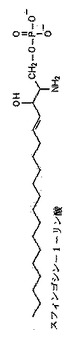
【図16】

【図16−1】

【図16−2】

【図16−3】

【図16−4】

【図16−5】

【図16−6】

【図17】
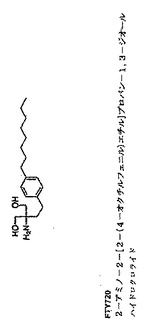
【図18】
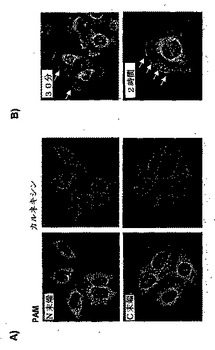
【図19】
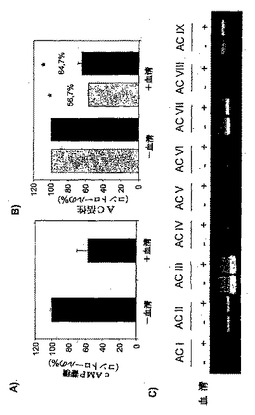
【図19−1】
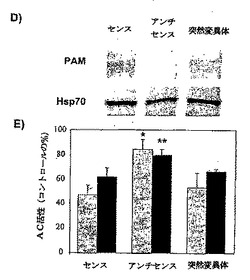
【図20】
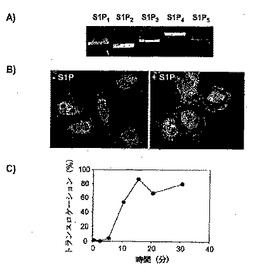
【図21】
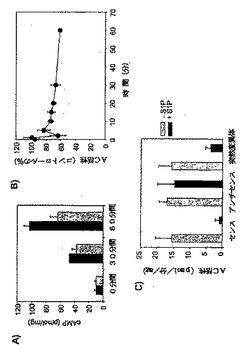
【図22】
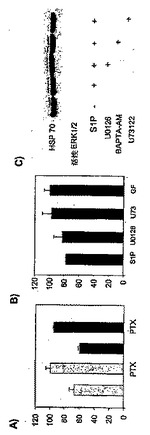
【図22−1】
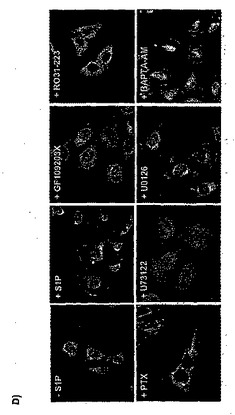
【図23】
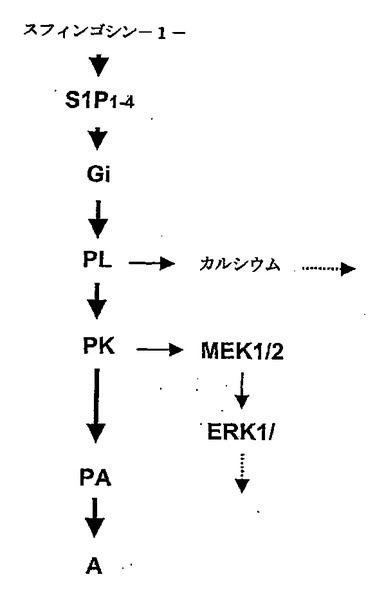
【図24】
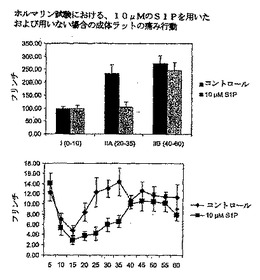
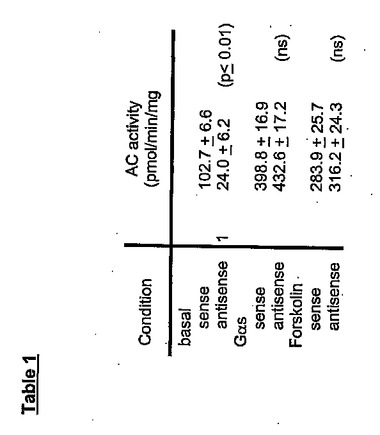
【図2A】
【図2B】
【図3】
【図4】
【図5】
【図6】
【図6−1】
【図7】
【図7−1】
【図7−2】
【図7−3】
【図7−4】
【図7−5】
【図7−6】
【図7−7】
【図7−8】
【図7−9】
【図7−10】
【図7−11】
【図7−12】
【図7−13】
【図7−14】
【図7−15】
【図7−16】
【図7−17】
【図7−18】
【図7−19】
【図7−20】
【図7−21】
【図7−22】
【図7−23】
【図7−24】
【図7−25】
【図7−26】
【図7−27】
【図7−28】
【図7−29】
【図7−30】
【図7−31】
【図7−32】
【図7−33】
【図7−34】
【図7−35】
【図7−36】
【図7−37】
【図7−38】
【図7−39】
【図7−40】
【図7−41】
【図7−42】
【図7−43】
【図7−44】
【図7−45】
【図7−46】
【図7−47】
【図7−48】
【図7−49】
【図7−50】
【図7−51】
【図7−52】
【図7−53】
【図7−54】
【図7−55】
【図7−56】
【図7−57】
【図7−58】
【図7−59】
【図7−60】
【図7−61】
【図7−62】
【図7−63】
【図7−64】
【図7−65】
【図7−66】
【図7−67】
【図7−68】
【図7−69】
【図7−70】
【図7−71】
【図7−72】
【図7−73】
【図7−74】
【図7−75】
【図7−76】
【図7−77】
【図7−78】
【図7−79】
【図7−80】
【図7−81】
【図7−82】
【図7−83】
【図7−84】
【図7−85】
【図7−86】
【図7−87】
【図7−88】
【図7−89】
【図7−90】
【図7−91】
【図7−92】
【図7−93】
【図7−94】
【図7−95】
【図7−96】
【図7−97】
【図7−98】
【図7−99】
【図7−100】
【図7−101】
【図7−102】
【図7−103】
【図7−104】
【図7−105】
【図7−106】
【図7−107】
【図7−108】
【図7−109】
【図7−110】
【図7−111】
【図7−112】
【図7−113】
【図7−114】
【図7−115】
【図7−116】
【図7−117】
【図7−118】
【図7−119】
【図7−120】
【図7−121】
【図7−122】
【図7−123】
【図7−124】
【図7−125】
【図7−126】
【図8】
【図8−1】
【図8−2】
【図9】
【図10】
【図11】
【図12】
【図12−1】
【図12−2】
【図13】
【図13−1】
【図13−2】
【図13−3】
【図13−4】
【図14】
【図15】
【図16】
【図16−1】
【図16−2】
【図16−3】
【図16−4】
【図16−5】
【図16−6】
【図17】
【図18】
【図19】
【図19−1】
【図20】
【図21】
【図22】
【図22−1】
【図23】
【図24】
【公表番号】特表2007−518698(P2007−518698A)
【公表日】平成19年7月12日(2007.7.12)
【国際特許分類】
【出願番号】特願2006−529836(P2006−529836)
【出願日】平成16年5月15日(2004.5.15)
【国際出願番号】PCT/EP2004/005236
【国際公開番号】WO2004/105773
【国際公開日】平成16年12月9日(2004.12.9)
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
【公表日】平成19年7月12日(2007.7.12)
【国際特許分類】
【出願日】平成16年5月15日(2004.5.15)
【国際出願番号】PCT/EP2004/005236
【国際公開番号】WO2004/105773
【国際公開日】平成16年12月9日(2004.12.9)
【出願人】(397056695)サノフィ−アベンティス・ドイチュラント・ゲゼルシャフト・ミット・ベシュレンクテル・ハフツング (456)
【Fターム(参考)】
[ Back to top ]