
T細胞媒介性疾患の処置
【課題】T細胞媒介性疾患を処置する方法、およびT細胞の活性化を抑制する方法を提供する。
【解決手段】動物において通常に見出されるペプチドまたはタンパク質の使用であって、上記ペプチドまたはタンパク質は、ペプチドまたはタンパク質から誘導される少なくとも1つのジケトピペラジンを含むように処理される方法。及びジケトピペラジンを合成する方法、および特定のジケトピペラジンを含む薬学的組成物を合成する方法を提供する。さらに、薬学的組成物中のジケトピペラジンの量を増加または減少させることにより、タンパク質およびペプチドの改善された薬学的組成物を製造する方法、および得られた薬学的組成物。
【解決手段】動物において通常に見出されるペプチドまたはタンパク質の使用であって、上記ペプチドまたはタンパク質は、ペプチドまたはタンパク質から誘導される少なくとも1つのジケトピペラジンを含むように処理される方法。及びジケトピペラジンを合成する方法、および特定のジケトピペラジンを含む薬学的組成物を合成する方法を提供する。さらに、薬学的組成物中のジケトピペラジンの量を増加または減少させることにより、タンパク質およびペプチドの改善された薬学的組成物を製造する方法、および得られた薬学的組成物。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、特定のジケトピペラジンを使用する、T細胞媒介性疾患の処置、およびT細胞の活性化の抑制に関する。本発明はまた、特定のジケトピペラジンを含む薬学的組成物、およびジケトピペラジンを合成する方法に関する。本発明はさらに、薬学的組成物中のジケトピペラジンの量を増加または減少させることにより、タンパク質およびペプチドの改善された薬学的組成物を製造する方法、および、得られた、改善された薬学的組成物に関する。
【背景技術】
【0002】
T細胞媒介性疾患は、多くの免疫系障害を示す。特に、T細胞は、自己免疫性疾患を開始させかつ永続させる細胞であると考えられている。自己免疫性疾患は、合衆国だけでも何百万もの人々を苦しめている80の深刻な慢性疾患の一群である。自己免疫疾患は、内因性(自己)抗原に対する免疫系の反応によって特徴付けられる。自己抗原に対するこれらの免疫応答は、自己反応性T細胞の持続的または反回性の活性化によって維持され、そして、直接的にまたは間接的に、この自己反応性T細胞は、自己免疫性疾患に見られる特徴的な組織の損傷および破壊の原因である。自己免疫性疾患および他のT細胞媒介性疾患に対する多くの処置が提案されているが、さらなる処置の必要性がなお存在する。
【発明の概要】
【0003】
本発明は、T細胞媒介性疾患を処置する方法を提供する。本方法は、以下の式:
【0004】
【化1】

【0005】
を有するジケトピペラジンまたはその生理学的に受容可能な塩の有効量を、該ジケトピペラジンまたはその塩を必要とする動物に投与する工程を含む、T細胞媒介性疾患を処置する方法であって、
ここで:
R1およびR2は同一であっても異なってもよく、R1およびR2の各々は、
(a)アミノ酸の側鎖であって、該アミノ酸がグリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、2,4−ジアミノ酪酸、2,3−ジアミノ酪酸、ロイシン、イソロイシン、ノルロイシン、セリン、ホモセリン、スレオニン、アスパラギン酸、アスパラギン、グルタミン酸、グルタミン、リジン、ヒドロキシリジン、ヒスチジン、アルギニン、ホモアルギニン、シトルリン、フェニルアラニン、p−アミノフェニルアラニン、チロシン、トリプトファン、チロキシン、システイン、ホモシステイン、メチオニン、ペニシラミン、またはオルニチンであるが、R1がアスパラギンまたはグルタミンの側鎖である場合は、R2はリジンまたはオルニチンの側鎖ではあり得ず、R1がリジンまたはオルニチンの側鎖の場合は、R2はアスパラギンまたはグルタミンの側鎖ではあり得ない、アミノ酸の側鎖であるか;
(b)R1は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、R2は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、または、R1およびR2の両方がそれぞれ独立して−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか;あるいは
(c)アミノ酸側鎖の誘導体であって、ここで、該アミノ酸は上記(a)に記載のアミノ酸の1つであり、該誘導体化された側鎖は、以下:
(i)−NHR3基または−N(R3)2基に置き換えられた−NH2基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−NHR3基または−N(R3)2基に置き換えられた−NH2基、
(ii)−OPO3H2基または−OR3基に置き換えられた−OH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−OPO3H2基または−OR3基に置き換えられた−OH基
(iii)−COOR3基に置き換えられた−COOH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−COOR3基に置き換えられた−COOH基、
(iv)−CON(R4)2基に置き換えられた−COOH基であって、R4の各々は独立して、Hあるいは置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−CON(R4)2基に置き換えられた−COOH基、
(v)−S−S−CH2−CH(NH2)−COOHまたは−S−S−CH2−CH2−CH(NH2)−COOHに置き換えられた−SH基、
(vi)−CH(NH2)−または−CH(OH)−基に置き換えられた−CH2−基、
(vii)−CH2−NH2または−CH2−OH基に置き換えられた−CH3基、
および/または
(viii)ハロゲンに置き換えられた炭素原子に結合しているH
を有する。
【0006】
本発明はまた、T細胞の活性化を抑制する方法を提供する。本方法は、式Iのジケトピペラジンまたはその生理学的に受容可能な塩の有効量を、当該ジケトピペラジンまたはその塩を必要とする動物に投与する工程を含む。
【0007】
本発明はさらに、薬学的に受容可能な担体および以下の式:
【0008】
【化2】
【0009】
を有するジケトピペラジンまたはその生理学的に受容可能な塩を含む薬学的組成物を提供し、
ここで:
R5およびR6は同一であっても異なってもよく、R5およびR6の々は、
(a)アミノ酸の側鎖であって、該アミノ酸がグリシン、アラニン、バリン、ノルバリン、αアミノイソ酪酸、2,4−ジアミノ酪酸、2,3−ジアミノ酪酸、ロイシン、イソロイシン、ノルロイシン、セリン、ホモセリン、スレオニン、リジン、ヒドロキシリジン、ヒスチジン、アルギニン、ホモアルギニン、シトルリン、フェニルアラニン、pアミノフェニルアラニン、チロシン、トリプトファン、チロキシン、またはオルニチンであるが、R5がアスパラギンまたはグルタミンの側鎖である場合は、R6はリジンまたはオルニチンの側鎖ではあり得ず、R5がリジンまたはオルニチンの側鎖の場合は、R6はアスパラギンまたはグルタミンの側鎖ではあり得ない、アミノ酸の側鎖であるか;
(b)R5は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、R6は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、または、R5およびR6の両方がそれぞれ独立して−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか;あるいは
(c)アミノ酸側鎖の誘導体であって、ここで、該アミノ酸は上記(a)に記載のアミノ酸の1つであり、該誘導体化された側鎖は、以下:
(i)−NHR3基または−N(R3)2基に置き換えられた−NH2基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−NHR3基または−N(R3)2基に置き換えられた−NH2基、
(ii)−OPO3H2基または−OR3基に置き換えられた−OH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−OPO3H2基または−OR3基に置き換えられた−OH基、
(iii)−CH(NH2)−または−CH(OH)−基に置き換えられた−CH2−基、
(iv)−CH2−NH2または−CH2−OH基に置き換えられた−CH3基、
および/または
(v)ハロゲンに置き換えられた炭素原子に結合しているH
を有する。
【0010】
本発明は、T細胞媒介性疾患を処置する別の方法を提供する。本方法は、動物において正常に見出されるタンパク質またはペプチドを含む薬学的組成物の有効量を、当該薬学的組成物を必要とする動物に投与する工程を含み、また薬学的組成物が該タンパク質またはペプチドから誘導される少なくとも1つのジケトピペラジンをまた含むように該タンパク質またはペプチドが処置される。
【0011】
本発明はさらに、T細胞の活性化を抑制する方法を提供する。本方法は、動物において正常に見出されるタンパク質またはペプチドを含む薬学的組成物の有効量を、当該薬学的組成物を必要とする動物に投与する工程を含み、また該薬学的組成物が該タンパク質またはペプチドから誘導される少なくとも1つのジケトピペラジンをまた含むように該タンパク質またはペプチドが処置される。
【0012】
さらに、本発明は、ジケトピペラジンを合成する方法を提供する。1つの実施形態においては、本方法は、ジケトピペラジンの形成を効率的に生じさせる条件下でタンパク質またはペプチドの溶液を加熱する工程を含む。第二の実施形態においては、本方法は、タンパク質またはペプチドの溶液を酵素に接触させる工程を含み、該酵素は、ジケトピペラジンを効率的に生成させる条件下で該タンパク質またはペプチドのN末端の2つのアミノ酸またはC末端の2つのアミノ酸を切断する。
【0013】
本発明はまた、タンパク質およびペプチドの改善された薬学的組成物を提供する。この改善は、該組成物におけるジケトピペラジンの減量を含む。
【0014】
さらに、本発明は、タンパク質およびペプチドの改善された薬学的組成物を生成する方法を提供する。本方法は、該組成物中に存在する少なくともいくらかのジケトピペラジンを該化合物から取り除く工程を含む。
【0015】
本発明はさらに、タンパク質およびペプチドの改善された薬学的組成物を製造する方法を提供する。本方法は、当該組成物中のジケトピペラジンの量が増加するように該タンパク質またはペプチドの溶液を処置する工程を含む。
【0016】
本発明はまた、タンパク質およびペプチドの改善された薬学的組成物を提供する。この改善は、該組成物におけるジケトピペラジンの増量を含む。
【図面の簡単な説明】
【0017】
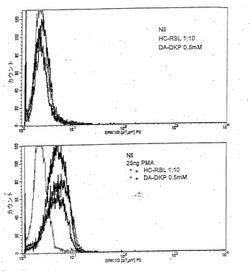
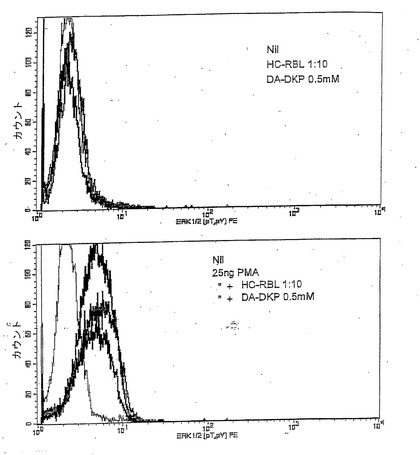
【図1】抗CD3 OKT3抗体を用いた刺激後20日目に単離し、25ngのホルボールミリスチン酸(PMA)、HC−RBL(3kD未満の分子量の、加熱されたヒト初乳の画分(MR−DKPを含む))を1:10希釈、および0,5mMDA−DKPとともに、37℃で15分間インキュベートしたTriPS細胞(インフルエンザの予防接種を受けたドナーから単離された、ヘマグルチニンに特有のCD4+T細胞株)についての、ERK1/2のカウント対濃度をトレースしたものである。
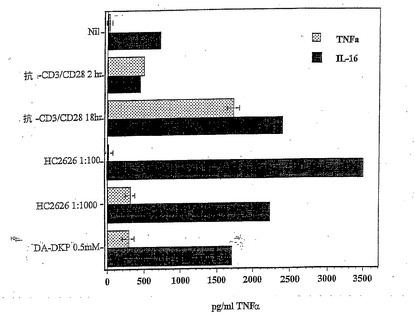
【図2】抗CD3 OKT3抗体を用いた刺激12日後の、腫瘍壊死因子α(TNFα)およびIL−16の、TriPS細胞からの分泌の抑制を示す棒グラフであり、ヒト初乳(HC)2626(MR−DKPを含む)バンドDA−DKPからの、TNFαおよびIL−16の両方の分泌の抑制を示しており、HC2626を1:100希釈および1:1000希釈で用いて観察された最大放出は、高濃度のヒト初乳による溶解効果に起因している。0.5mM DA−DKPを使用しても、溶解は観察されず、TNFαおよびIL−16の分泌は低減されている。
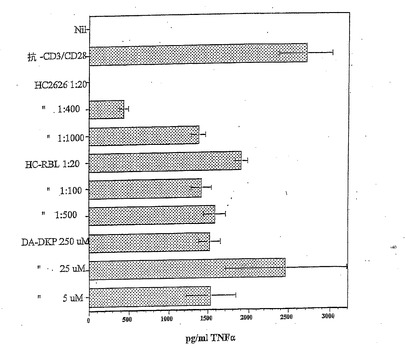
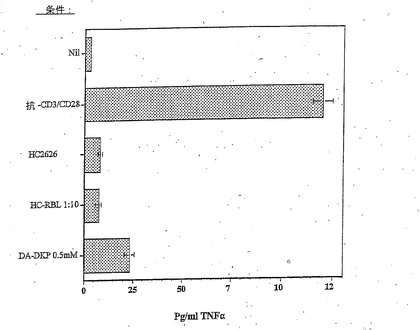
【図3】抗CD3 OKT3抗体を用いた刺激10日後の、TriPS細胞からのTNFαの分泌の抑制を示す棒グラフであり、HC RBLおよびDA−DKPは、HC2626を用いて観察されるような滴定可能な応答に関してさらに調査する必要があることを示しており、強力な活性が示され得る。
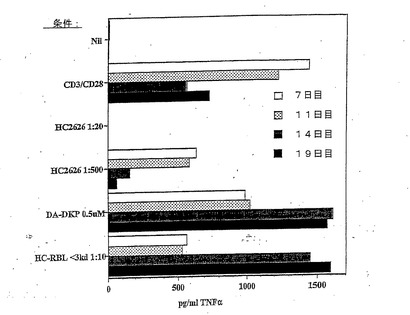
【図4】抗CD3 OKT3抗体を用いた刺激後の種々の時点での、TriPS細胞からのTNFαの分泌の抑制を示す棒グラフであり、刺激周期の初期においてはDA−DKPおよびHC RBLの効果は抑制性であるが、周期の後期(14日目)においては刺激性であることを示しており、HC2626は常に抑制しており、おそらくは、他の構成因子に起因する。
【図5】抗CD3 OKT3抗体を用いた刺激後の7〜10日目における、H4#9,25細胞(ミエリン塩基性タンパク質に特有の、多発性硬化症患者の検死用脳組織から単離したCD4+T細胞株)からのTNFαの分泌の抑制を示す棒グラフであり、このT細胞株からのTNFαの分泌はまた、HC2626、HCRBLおよびDA−DKPによって抑制されることを示している。
【発明を実施するための形態】
【0018】
本発明は、T細胞媒介性疾患を処置する方法を提供する。「処置」は、疾患の症状、継続期間および重篤度を(完全にまたは部分的に)低減させること(疾患が治癒することを含む)、および疾患を予防することを意味するように本明細書中で使用される。
【0019】
T細胞媒介性疾患としては、組織不適合性、移殖片対宿主疾患、望ましくない遅延型超過敏反応(たとえば遅延型アレルギー反応)、T細胞媒介性肺疾患または自己免疫疾患が挙げられる。T細胞媒介性肺疾患としては、サルコイドーシス、過敏性肺炎、急性間質性肺炎、歯槽骨炎、肺線維症、特発性肺線維症、および炎症性肺損傷によって特徴付けられる他の疾患が挙げられる。自己免疫疾患としては、多発性硬化症、神経炎、多発性筋炎、乾癬、白斑、シェーグレン症候群、慢性間接リウマチ、1型糖尿病、自己免疫性膵炎、炎症性腸疾患(たとえばクローン病および潰瘍性大腸炎)、セリアック病、糸球体腎炎、強皮症、サルコイドーシス、自己免疫性甲状腺疾患(たとえば橋本甲状腺炎および甲状腺機能亢進症)、重症筋無力症、アジソン病、自己免疫性ぶどう膜網膜炎、尋常性天疱瘡、原発性胆汁性肝硬変、悪性貧血および結合線維組織増殖症候群が挙げられる。
【0020】
このT細胞媒介性疾患は、以下の式:
【0021】
【化3】
【0022】
を有するジケトピペラジンまたはその生理学的に受容可能な塩の有効量を、該ジケトピペラジンまたはその塩を必要とする動物に投与する工程を含む、T細胞媒介性疾患を処置する方法であって、
ここで:
R1およびR2は同一であっても異なってもよく、R1およびR2の各々は、
(a)アミノ酸の側鎖であって、該アミノ酸がグリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、2,4−ジアミノ酪酸、2,3−ジアミノ酪酸、ロイシン、イソロイシン、ノルロイシン、セリン、ホモセリン、スレオニン、アスパラギン酸、アスパラギン、グルタミン酸、グルタミン、リジン、ヒドロキシリジン、ヒスチジン、アルギニン、ホモアルギニン、シトルリン、フェニルアラニン、p−アミノフェニルアラニン、チロシン、トリプトファン、チロキシン、システイン、ホモシステイン、メチオニン、ペニシラミン、またはオルニチンであるが、R1がアスパラギンまたはグルタミンの側鎖である場合は、R2はリジンまたはオルニチンの側鎖ではあり得ず、R1がリジンまたはオルニチンの側鎖の場合は、R2はアスパラギンまたはグルタミンの側鎖ではあり得ない、アミノ酸の側鎖であるか;
(b)R1は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、R2は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、または、R1およびR2の両方がそれぞれ独立して−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか;あるいは
(c)アミノ酸側鎖の誘導体であって、ここで、該アミノ酸は上記(a)に記載のアミノ酸の1つであり、該誘導体化された側鎖は、以下:
(i)−NHR3基または−N(R3)2基に置き換えられた−NH2基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−NHR3基または−N(R3)2基に置き換えられた−NH2基、
(ii)−OPO3H2基または−OR3基に置き換えられた−OH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−OPO3H2基または−OR3基に置き換えられた−OH基
(iii)−COOR3基に置き換えられた−COOH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−COOR3基に置き換えられた−COOH基、
(iv)−CON(R4)2基に置き換えられた−COOH基であって、R4の各々は独立して、Hあるいは置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−CON(R4)2基に置き換えられた−COOH基、
(v)−S−S−CH2−CH(NH2)−COOHまたは−S−S−CH2−CH2−CH(NH2)−COOHに置き換えられた−SH基、
(vi)−CH(NH2)−または−CH(OH)−基に置き換えられた−CH2−基、
(vii)−CH2−NH2または−CH2−OH基に置き換えられた−CH3基、
および/または
(viii)ハロゲンに置き換えられた炭素原子に結合しているH
を有する。
【0023】
「置き換えられた(置き換えられる)」によって、上記アミノ酸側鎖の式を参照して、特定の基が他の特定の基に置き換えられることが意図される。例えば、イソロイシンの側鎖の式は−CH(CH3)−CH2−CH3である。末端の−CH3基が、−CH2−OH基に置き換えられた場合、得られる誘導体化されたイソロイシンの側鎖の式は、−CH(CH3)−CH2−CH2−OHである。別の例として、アラニンの側鎖の式は−CH3である。水素原子の1つが塩素原子に置き換えられた場合、得られる誘導体化されたアラニンの側鎖の式は、−CH2−Clである。グリシンの側鎖は−Hであり、このHが塩素原子(または他のハロゲン原子)に置き換えられた場合、得られる側鎖は、−Clであり、この塩素原子は、炭素環に結合される(例えば、R1=−Cl)。
【0024】
R1、R2、またはその両方が、アスパラギン酸またはグルタミン酸の側鎖か、または、このような側鎖の誘導体であるジケトピペラジンが好ましく、ここで、−COOH基は−COOR3基または−CON(R4)2基に置き換えられており、ここで、R3およびR4は上記で規定されている。この化合物の基について最も好ましくは、アスパラギン酸およびアラニンの側鎖(AsP−AlaDKPまたはDA−DKP)、グルタミン酸およびアラニンの側鎖(Glu−AlaDKPまたはEA−DKP)、チロシンおよびアスパラギン酸の側鎖(Tyr−AsPDKPまたはYD−DKP)、チロシンおよびグルタミン酸の側鎖(Tyr−GluDKPまたはYE−DKP)、およびこれら4つのジケトピペラジンのアスパラギン酸もしくはグルタミン酸の誘導体を含むジケトピペラジンであり、ここで、該−COOH基は−COOR3基または−CON(R4)2基に置き換えられており、ここで、R3およびR4は上記で規定されている。
【0025】
また、R1およびR2の両方が疎水性の側鎖(例えば、フェニルアラニンの側鎖)または疎水性の側鎖の誘導体であるジケトピペラジンが好ましい。「疎水性の側鎖の誘導体」によって、疎水性である誘導体化された側鎖が意図される。特に、以下のようなジケトピペラジンが好ましい:ここで、R1およびR2は同一であっても異なってもよく、R1およびR2の々は、グリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、ロイシン、イソロイシン、ノルロイシンまたはフェニルアラニンの側鎖であるか;R1は−CH2−CH2−CH2−であり、かつ隣接する環窒素と一緒になってプロリンを形成し、R2は−CH2−CH2−CH2−であり、かつ隣接する環窒素と一緒になってプロリンを形成するか;あるいは、R1は、グリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、ロイシン、イソロイシン、ノルロイシンまたはフェニルアラニンの側鎖であり、R2は−CH2−CH2−CH2−であり、かつ隣接する環窒素と一緒になってプロリンを形成する、ジケトピペラジンが好ましい。この化合物の基の中でもっとも好ましいのは、グリシンおよびロイシンの側鎖(Gly−LeuDKPまたはGL−DKP)、プロリンおよびフェニルアラニンの側鎖(Pro−PheDKPまたはPF−DKP)およびアラニンおよびプロリンの側鎖(Ala−ProDKPまたはAP−DKP)を含む。
【0026】
R1、R2、またはその両方が、メチオニンの側鎖、アルギニンの側鎖またはメチオニンの側鎖もしくはアルギニンの側鎖の誘導体であるジケトピペラジンが、さらに好ましい。この基について、R1がメチオニンの側鎖であり、R2がアルギニンの側鎖(Met−ArgDKPまたはMR−DKP)であるジケトピペラジンが最も好ましい。
【0027】
アミノ酸の「側鎖」によって、上記に列挙されたアミノ酸全てに共通するNH2−CH−COOH骨格に結合しているアミノ酸の一部が意図される。例えば、グリシンの側鎖は−Hであり、アラニンの側鎖は−CH3であり、セリンの側鎖は−CH2−OHである。
【0028】
「疎水性」によって、生理学的pHにおいて荷電されず、水溶液により忌避される(repel)側鎖または側鎖の誘導体が意図される。
【0029】
「アルキル」によって、飽和した直鎖炭化水素または分枝した炭化水素が意図され、これらは、1〜10個の炭素原子、好ましくは1〜6個の炭素原子を含む。「低級アルキル」によって、飽和した直鎖炭化水素または分枝した炭化水素が意図され、これらは、1〜6個の炭素原子を含む。
【0030】
「シクロアルキル」によって、少なくとも1つの環を含む、飽和した環状炭化水素が意図され、各環は、少なくとも3つの炭素原子を含む。好ましくは、このシクロアルキルは、4〜8の炭素原子の環を1つ含む。
【0031】
「ヘテロシクロアルキル」によって、1つ以上の炭素環を有するシクロアルキルが意図され、これらの環の少なくとも1つは、O、S、またはNによって置き換えられている。
【0032】
「アリール」によって、少なくとも1つの芳香環(例えば、フェニル基)を有する芳香族基が意図される。
【0033】
「アルキルアリール」によって、アリール(例えば、−CH2C6H5または−CH3CH(C6H5)CH3)によって置き換えられたHを有する低級アルキルが意図される。
【0034】
「アリールアルキル」によって、低級アルキル基(例えば、−C6H4−CH3)によって置き換えられたHを有するアリールが意図される。
【0035】
「ヘテロアリール」によって、1つ以上の炭素環を有するアリールが意図され、これらの環の少なくとも1つは、O、S、またはNによって置き換えられている。
【0036】
「置換された」によって、構成部分が以下の基の中より選択される1つ以上の置換基によって置き換えられていることが意図される:−OH、NH2、−SH、−COOHおよび/またはハロゲン原子。
【0037】
「ハロゲン」によって、塩素、フッ素、臭素、またはヨウ素が意図される。好ましくは、塩素または臭素である。
【0038】
式Iのジケトピペラジンは、T細胞媒介性疾患の処置に有効である。なぜなら、これらがT細胞の活性化を抑制するからである。従って、式Iのジケトピペラジンはまた、活性化されたT細胞によって引き起こされるかまたは悪化される炎症または炎症性疾患を処置するためにも使用され得る。本明細書において、「抑制」は、(完全にまたは部分的に)低減させること、または予防することを意図して使用される。
【0039】
ジケトピペラジンを生成する方法は、当該分野において周知であり、これらの方法は、本発明のジケトピペラジンを合成する際に使用され得る。例えば、米国特許第4,694,081号、同第5,817,751号、同第5,990,112号、同第5,932,579号、および同第6,555,543号、米国特許出願公開番号2004/0024180、PCT出願WO96/00391およびWO97/48685、ならびにSmithら、Bioorg.Med.Chem,Letters,8,2369−2374(1998)を参照のこと。これらの開示は、その全体が参考として本明細書において援用される。
【0040】
例えば、ジケトピペラジンは、初めにジペプチドを合成することによって調製され得る。このジペプチドは、L−アミノ酸、D−アミノ酸、またはL−アミノ酸とD−アミノ酸とを組合せたものを使用する、当該分野において周知の方法によって合成され得る。固相ペプチド合成法が好ましい。もちろん、ジペプチドはまた、多数の供給源から市販されており、この供給源としては、DMI Synthesis Ltd.、Cardiff、UK(custom synthesis)、Sigma−Aldrich、St.Louis、MO(primarily custom synthesis)、Phoenix Pharmaceuticals, Inc.、Belmont、CA(custom synthesis)、Fisher Scientific(custom synthesis)、およびAdvanced ChemTech, Louisville, KYが挙げられる。
【0041】
一旦、ジペプチドが合成されるかまたは購入されると、ジペプチドは環化してジケトピペラジンを形成する。このことは、種々の技術により達成され得る。
【0042】
例えば、米国特許出願公開番号2004/0024180は、ジペプチドを環化させる方法について記載している。つまり、ジペプチドは、蒸留によって水分を除去する間、有機溶媒中で加熱される。好ましくは、この有機溶媒は、アセトニトリル、アリルアルコール、ベンゼン、ベンジルアルコール、n−ブタノール、2−ブタノール、第三ブタノール、酢酸ブチル、四塩化炭素、クロロベンゼンクロロホルム、シクロヘキサン、1,2ジクロロエタン、ジエチルアセタール、ジメチルアセタール、酢酸エチルエステル、ヘプタン、メチルイソブチルケトン、3ペンタノール、トルエンおよびキシレンのような、低沸点の、水との共沸混合物である。温度は、環化が行われる反応速度、および使用される共沸剤(azeotroping agent)の型に依存する。反応は、好ましくは50〜200℃で、より好ましくは80〜150℃で行なわれる。環化が行われるpH範囲は、当業者により容易に決定され、有利には2〜9であり、好ましくは3〜7である。
【0043】
ジペプチドのアミノ酸の一方または両方が、その側鎖上にカルボキシル基を有するか、または有するように誘導体化されている場合(例えば、アスパラギン酸またはグルタミン酸)、このジペプチドは、米国特許第6,555,543号に記載されているように環化されることが好ましい。つまり、ジペプチドは、側鎖カルボキシルがなお保護されており、中性条件下で加熱される。典型的には、ジペプチドは、約80℃〜約180℃で加熱され、好ましくは約120℃で加熱される。溶媒は、中性溶媒である。例えば、溶媒は、アルコール(例えば、ブタノール、メタノール、エタノール、および高級アルコール(フェノールではない))、および共沸性の共溶媒(例えば、トルエン、ベンゼン、またはキシレン)を含み得る。好ましくは、アルコールはブタン−2−olであり、共沸性の共溶媒はトルエンである。加熱は、反応が完了するまで継続され、そのような時間は経験的に決定される。典型的には、ジペプチドは、それを8〜24時間還流することにより環化され、好ましくは18時間還流される。最終的に、保護基がジケトピペラジンから除去される。このように行なう際に、最終化合物のキラリティーを維持するために、強酸(硫酸または塩化水素酸のような鉱酸)、強塩基(水酸化カリウムまたは水酸化ナトリウムのようなアルカリ性塩基)および強力な還元剤(例えば、水素化アルミニウムリチウム)の使用は避けられるべきである。
【0044】
固相樹脂上で生成されたジペプチドは、1工程で環化されかつ該樹脂から放出され得る。例えば、米国特許第5,817,751号を参照のこと。例えば、結合したN−アルキル化されたジペプチドは、酢酸(例えば1%)またはトリエチルアミン(例えば4%)の存在下で、トルエンまたはトルエン/エタノール中に懸濁される。典型的には、より速い環化時間のためには、塩基性環化条件が好ましい。
【0045】
アミノ酸側鎖が誘導体化された、式IおよびIIのジケトピペラジンを調製するために、ジペプチドの合成にアミノ酸誘導体が使用され得、当該分野において公知のように、このジペプチドは誘導体化され得、および/または、ジケトピペラジンは誘導体化され得る。例えば、上記に列挙された参考文献を参照のこと。
【0046】
ジペプチドを環化する他の方法およびジケトピペラジンを生成する他の方法は、当該分野において公知であり、本発明の実施において有用なジケトピペラジンを調製する際に使用され得る。例えば、上記に列挙された参考文献を参照のこと。さらに、本発明における使用に適切な多くのジケトピペラジンは、以下に記載されるように、タンパク質およびペプチドから生成され得る。さらに、本発明の実施に使用するためのジケトピペラジンは、例えば、DMI Synthesis Ltd.、Cardiff、UK(カスタム合成)から商業的に取得され得る。
【0047】
式IおよびIIのジケトピペラジンは、個々のキラル中心、軸または表面の構成を変更することにより得られる、予想される全ての立体異性体を含む。すなわち、式IおよびIIのジケトピペラジンは、予想される全てのジアスタレオマー、および全ての光学異性体(鏡像異性体)を含む。
【0048】
本発明のジケトピペラジンの生理学的に受容可能な塩は、本発明の実施においてまた使用され得る。生理学的に受容可能な塩としては、慣用的な非毒性の塩(例えば、無機酸(塩化水素酸、臭化水素酸、硫酸、リン酸、硝酸など)、有機酸(酢酸、プロピオン酸、琥珀酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、グルタミン酸、アスパラギン酸、安息香酸、サリチル酸、シュウ酸、アスコルビン酸など)、または塩基(Nジベンジルエチレンジアミン、Dグルコサミン、またはエチレンジアミン由来の、薬学的に受容可能な金属カチオンまたは有機カチオンの、水酸化物、炭酸、または重炭酸)から誘導される塩)が挙げられる。これらの塩は、慣用的な様式で(例えば、化合物の遊離塩基形態を酸で中和することによって)調製される。
【0049】
上述したように、本発明のジケトピペラジンまたはその生理学的に受容可能な塩は、T細胞媒介性疾患を処置するか、または、T細胞の活性化を抑制するために使用され得る。このように使用するために、ジケトピペラジンまたはその生理学的に受容可能な塩は、処置を必要とする動物に投与される。この動物は、ウサギ、ヤギ、イヌ、ネコ、ウマ、またはヒトのような哺乳動物が好ましい。本発明の化合物についての効果的な投薬形態、投与様式、および投薬量は、経験的に決定され得、このような決定を下すことは当業者の範囲内である。投薬量は、使用される特定の化合物、処置されるべき疾患または状態、疾患または状態の重篤度、投与経路、化合物の排泄速度、処置の継続期間、この動物に投与されている他の薬物の同定、この動物の年齢、サイズおよび種類、ならびに医学分野および獣医学分野で知られている他の要因とともに変動することは、当業者によって理解される。一般に、本発明の化合物の好適な1日投与量は、治療効果を生成するために効果的な最少の投与量の、化合物の量である。しかしながら、この1日投与量は、主治医である医師または獣医師により適切な医学的判断の範囲内で決定される。所望される場合、有効な1日投与量は、2個、3個、4個、5個、6個またはそれ以上の副投与量(sub−dose)として投与され得、1日を通して適切な間隔を空けて別々に投与され得る。化合物の投与は、受容可能な応答が達成されるまで継続されるべきである。
【0050】
本発明の化合物(すなわち、ジケトピペラジンまたはその生理学的に受容可能な塩)は、任意の適切な投与経路によって治療のために患者(患畜)に投与され得、その投与経路としては、経口、経鼻、経直腸、経膣、非経口(たとえば、静脈内注入、髄腔内注入、腹腔内注入、皮下または筋肉注射)、大槽内、経皮、頭蓋内、脳内、および局所(頬側および舌下を含む)が挙げられる。好ましい投与経路は、経口および静脈内である。
【0051】
本発明の化合物は単独で投与されることも可能であるが、この化合物を薬学的処方物として投与することが好ましい。本発明の薬学的組成物は、1つ以上の薬学的に受容可能な担体、および必要に応じて、1つ以上の他の化合物、薬物または他の物質との混合物中の活性成分として本発明の化合物(単数または複数)を含む。各担体は、この処方物の他の成分と適合性であり、動物に対して有害ではないという観点において「受容可能」でなければならない。薬学的に受容可能な担体は当該分野において周知である。選択された投与経路にかかわらず、本発明の化合物は、当業者に公知の慣用的な方法によって、薬学的に受容可能な投薬形態に処方される。例えば、Remington’s Pharmaceutical Scienceを参照のこと。
【0052】
経口投与に適切な本発明の処方物は、カプセル、カシュ剤、丸剤、錠剤、散剤、顆粒、または水性もしくは非水性の液体中での溶剤もしくは懸濁液として、または水中油乳剤もしくは油中水乳剤、またはエリキシル剤もしくはシロップ剤、またはトローチ剤(不活性なベース(例えば、ゼラチンおよびグリセリン)、またはスクロースおよびアカシアを使用する)などの形態であり得、これらの各々は、所定量の本発明の化合物(単数または複数)を活性成分として含む。本発明の化合物(単数または複数)はまた、ボーラス、舐剤またはペーストであり得る。
【0053】
経口投与のための本発明の固形の投薬形態(カプセル、錠剤、丸剤、糖衣剤、散剤、顆粒など)において、活性成分(すなわち、1つ以上の本発明のジケトピペラジンおよび/またはその生理学的に受容可能な塩)は、1つ以上の薬学的に受容可能な担体(例えば、クエン酸ナトリウム、リン酸二カルシウム)、および/または以下のいずれかと混合される:(1)充填剤または増量剤(例えば、デンプン、ラクトース、スクロース、グルコース、マンニトールおよび/またはケイ酸);(2)結合剤(例えば、カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロースおよび/またはアカシア);(3)湿潤剤(例えば、グリセロール);(4)崩壊剤(例えば、寒天、炭酸カルシウム、ジャガイモデンプンまたはタピオカデンプン、アルギニン酸、特定のケイ酸塩、および炭酸ナトリウム);(5)溶液遅延剤(例えば、パラフィン);(6)吸収促進剤(例えば、第四アンモニウム化合物);(7)湿潤剤(例えば、セチルアルコールおよびグリセロールモノステアレート);(8)吸収剤(例えば、カオリンおよびベントナイト土);(9)滑剤(例えば、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、充実ポリエチレングリコール、ラウリル硫酸ナトリウムおよびこれらの混合物);(10)着色剤。カプセル、錠剤および丸剤の場合、薬学的組成物は、緩衝剤を含み得る。同様の型の固体組成物は、ラククトースまたは乳糖および高分子量のポリエチレングリコールなどのような賦形剤を使用して、柔和に充填されたゼラチンカプセルおよび堅固に充填されたゼラチンカプセルにおける充填剤として使用され得る。
【0054】
錠剤は、必要に応じて1つ以上の副成分を圧縮または成形することにより生成され得る。圧縮された錠剤は、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース)、滑剤、不活性な希釈剤、防腐剤、崩壊剤(例えば、ナトリウムデンプングリコレートまたは架橋したカルボキシルメチルセルロースナトリウム)、界面活性剤または分散剤を使用して調製され得る。成形された錠剤は、不活性な液体希釈剤で湿らされた粉末状の化合物の混合物を適切な機械中で成形することによって生成され得る。
【0055】
本発明の薬学的組成物の錠剤および他の固形の投薬形態(例えば、糖衣錠、カプセル、丸剤、および顆粒)は、必要に応じて、コーティングおよびシェル(shell)(例えば、腸溶性コーティングおよび薬学的処方物の分野において周知の他のコーティング)を用いて獲得(score)または調製され得る。本発明の薬学的組成物の錠剤および他の固形の投薬形態はまた、例えば、所望の放出プロファイルを提供するための種々の割合のヒドロキシプロピルメチルセルロース、他のポリマーマトリクス、リポソームおよび/またはミクロスフェアを用いて、組成物中の活性成分を徐放(slow or controlled release)し得るように処方され得る。本発明の薬学的組成物の錠剤および他の固形の投薬形態は、たとえば細菌保持フィルタ(bacteria−retaining filter)を通してろ過することによって滅菌され得る。これらの組成物はまた、必要に応じて、乳白剤を含み得、または必要に応じて遅延された形態で、好ましくは胃腸管の特定の部分に、活性成分のみを放出する組成物であり得る。使用され得る包埋型の組成物としては、ポリマー性物質およびワックスが挙げられる。この活性成分はまた、マイクロカプセル化された形態中にあり得る。
【0056】
本発明の化合物の経口投与のための液体投薬形態は、薬学的に受容可能な乳剤、マイクロエマルジョン、溶液、懸濁液、シロップ剤、およびエリキシル剤を含む。活性成分に加えて、液体投薬形態は、当該分野においては一般に使用される不活性な希釈剤(例えば、水または他の溶媒、可溶化剤および乳化剤(例えば、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(具体的には、綿実油、ラッカセイ油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油、およびゴマ油)、グリセロール、テトラヒドロフルフリルアルコール、ポリエチレングリコール、ソルビタン脂肪酸エステル、およびこれらの混合物))を含み得る。
【0057】
不活性な希釈剤の他に、経口組成物はまた、アジュバント(例えば、湿潤剤、乳化剤および懸濁化剤)、甘味剤、着香料、着色料、香料および保存料を含み得る。
【0058】
活性成分に加えて、懸濁液は、懸濁剤(例えば、エトキシル化イソステリアルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム(aluminum metahydroxide)、ベントナイト、寒天、トラガカント、およびこれらの混合物)を含み得る。
【0059】
経直腸投与または経膣投与のための本発明の薬学的組成物の処方物は、坐剤として提供され得、1つ以上の本発明の化合物を、例えば、カカオ脂、ポリエチレングリコール、坐材のワックスまたはサリチル酸塩のような、室温では固体であるが体温では液体になり、よって、直腸または膣腔では溶解して活性成分を放出する、1つ以上の適切な無刺激の賦形剤または担体と混合することによって調製され得る。経膣投与に適切な本発明の処方物としてはまた、ペッサリー、タンポン、クリーム、ゲル、ペースト、発泡体またはスプレー処方物(当該分野において適当であることが知られている担体を含む)が挙げられる。
【0060】
本発明の化合物の局所投与または経皮投与のための投薬形態としては、散剤、スプレー、軟膏、ペースト、クリーム、ローション、ゲル、溶液、膏薬、点滴薬および吸入剤(inhalant)が挙げられる。活性成分は、滅菌条件下で、薬学的に受容可能な担体、および任意の緩衝液または必要とされ得る推進剤と混合され得る。
【0061】
軟膏、ペースト、クリーム、およびゲルは、活性成分に加えて、賦形剤(例えば、動物性および植物性脂肪、油、ワックス、パラフィン、デンプン、トラガカント、セルロース誘導体、ポリエチレングリコール、シリコン、ベントナイト、ケイ酸、タルクおよび酸化亜鉛、またはこれらの混合物)を含み得る。
【0062】
散剤およびスプレーは、活性成分に加えて、例えば、ラクトース、タルク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウム、ポリアミド散剤、およびこれらの物質の混合物を含み得る。スプレーはさらに、推進剤(例えば、フロン(chlorofluorohydrocarbon))および揮発性の非置換の炭化水素(例えば、ブタンおよびプロパン)を慣用的に含み得る。
【0063】
経皮的膏薬は、本発明の化合物の身体への制御された送達を提供するというさらなる利点を有する。このような投薬形態は、本発明の1つ以上の化合物を、好ましい媒体(例えば、エラストマーマトリクス材(elastomeric matrix material))に、溶解させるか、分散させるか、または組み込むかによって製造され得る。また、吸収促進剤を使用して、皮膚を超えるこの化合物の流量を増加させ得る。この流量の速度は、速度制御された膜を設けるか、またはポリマーマトリクスまたはゲル中に化合物を分散することによって制御され得る。
【0064】
薬学的処方物としては、吸入(inhalation)投与もしくは通気(insufflation)投与、または経鼻投与もしくは眼内投与に好適な処方物が挙げられる。吸入による上気道(経鼻)または下気道への投与のために、本発明の上記化合物は、吸入器、噴霧器、もしくは加圧パック、またはエアロゾルスプレー送達の他の簡便な手段エアロゾルから簡便に送達される。可圧パックは、適切な推進剤(例えば、ジクロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、炭酸ガスまたは他の適切なガス)を含み得る。加圧型エアロゾルの場合は、測定された量を送達するために弁を設けることにより、処方単位が決定され得る。
【0065】
あるいは、吸入または通気によって投与するために、組成物は、乾燥粉末の形態(例えば、1つ以上の本発明の化合物および適切な粉末ベース(例えば、ラクトースまたはデンプン)の粉体混合物)をとり得る。この粉末組成物は、例えば、カプセルもしくは薬包、または例えば、ゲラチンもしくは発泡剤パック中に、単位投与量形態で提供され得、吸入器(inhalator)、通気器、または測定された投与量の吸入器(inhaler)を用いてそこから粉末が投与され得る。
【0066】
経鼻投与のために、本発明の化合物は、点鼻剤または液体スプレーの手段(例えば、プラスチックボトル噴霧器または測定された投与量の吸入器)によって投与され得る。典型的な噴霧器はMistometer(Wintrop)およびMedihaler(Riker)である。
【0067】
点眼薬または点鼻薬のようなドロップは、水性または非水性のベースを用いて処方され得、1つ以上の分散剤、可溶化剤、または懸濁剤もまた含む。液体スプレーは、加圧されたパックから簡便に送達され得る。ドロップは、単純な目薬キャップボトルによって、または特別な形状の梱包により滴下される液体内容物を送達するために適合されたプラスチックボトルによって、送達され得る。
【0068】
非経口投与に好適な本発明の薬学的組成物は、1つ以上の本発明の化合物を、1つ以上の薬学的に受容可能な滅菌した等張性の水溶液または非水溶性の水溶液、分散液、懸濁液または乳剤、あるいは滅菌した注射可能な溶液もしくは分散液に使用直前に再構成され得る滅菌粉末とともに、含む。この組成物は、抗酸化剤、緩衝液、溶質を含み得、これらは、目的のレシピエントの血液、懸濁剤または濃化剤を用いてこの組成物を等張性にする。
【0069】
本発明の薬学的組成物に使用され得る適切な水性担体または非水性担体としては、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリセロール、ポリエチレングリコールなど)、およびこれらの適切な混合物、植物油(例えば、オリーブ油)、注入可能な有機エステル(例えば、オレイン酸エチル)が挙げられる。好ましい流動性は、コーティング物質(例えば、レシチン)の使用によって、分散の場合は必要とされる粒子サイズの維持によって、および界面活性剤の使用によって維持され得る。
【0070】
これらの組成物はまた、アジュバント(例えば、湿潤剤、乳化剤、および分散剤)を含み得る。これらの組成物中に、等張化剤(例えば、糖質、塩化ナトリウムなど)を含むことも望ましい。さらに、注入可能な薬学的な形態の延長された吸収は、吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよびゼラチン)の内包により、もたらされ得る。
【0071】
いくつかの場合、薬物の効果を延長させるために、皮下注射または筋肉注射からこの薬物の吸収を遅らせることが望ましい。このことは、水溶性の低い結晶性または非晶質物質の液体懸濁液の使用によって達成され得る。次いで、薬物の吸収速度は、その溶解速度に依存し、これは、結晶サイズおよび結晶形態に依存し得る。あるいは、非経口的に投与された薬物の遅延された吸収は、油ビヒクル中に薬物を溶解または懸濁することによって達成され得る。
【0072】
注入可能な貯蔵形態は、生分解性ポリマー(例えば、ポリラクチド−ポリグリコリド)中で薬物のマイクロカプセル化マトリクスを形成することによって製造される。薬物対ポリマーの比、および使用した特定のポリマーの特性に依存して、薬物放出の速度は、制御され得る。生分解性ポリマーの他の例としては、ポリ(オルソエステル)およびポリ(無水物)が挙げられる。注入可能な貯蔵処方物はまた、生体組織と適合性のリポソームまたはマイクロエマルジョンに薬物を閉じ込めることによって調製される。この注射可能な物質は、例えば、細菌保持フィルタを通してろ過することによって、滅菌され得る。
【0073】
この処方物は、単回用量または複数回用量の封着された容器(例えば、アンプルおよびバイアル)にて提供され得、そして滅菌された液体担体(例えば、注射水)を使用直前に添加することのみが必要とされる凍結乾燥された状態で保存され得る。即時調整の注射用の溶液および懸濁液は、上記した型の滅菌された散剤、顆粒および錠剤から調製され得る。
【0074】
本発明での使用に好適なジケトピペラジンは、アルブミン、免疫グロブリンおよびエリスロポエチンを含む市販されている静脈注射用の薬学的組成物のいくつかにおいて提供されていることは知られている。これらの薬学的調製物にて提供されるジケトピペラジンは、これらの薬学的組成物の製造においてしばしば使用される加熱工程によって形成される。この加熱工程により、タンパク質のN末端の2つのアミノ酸および/またはC末端の2つのアミノ酸の切断および環化が生じて、ジケトピペラジンが形成される。
【0075】
従って、本発明における使用のためのジケトピペラジンは、アルブミン、免疫グロブリン、エリスロポエチン、ならびに他のタンパク質およびペプチドの溶液を加熱する工程によって調製され得る。例えば、中性のpHでのリン酸緩衝液中の、アルブミン、免疫グロブリン、エリスロポエチン、または別のタンパク質もしくはペプチドの溶液は、調製される。好ましくは、この溶液は、N末端アミノ酸および/またはC末端アミノ酸のプロトン化を達成するための濃縮溶液(例えば、約100〜500mM)である。この溶液は、60℃で約2時間〜数日間、好ましくは4日間加熱されて、ジケトピペラジンの形成を生じる。好ましくは、タンパク質の変性は避けられるべきである。このことは、時間を短くすることおよび/またはカプリル酸もしくはN−アセチルトリプトファンを各々に対して約0,02M添加することによって達成される。
【0076】
本発明における使用のためのジケトピペラジンはまた、アルブミン、免疫グロブリン、エリスロポエチン、または別のタンパク質もしくはペプチドの溶液を、このタンパク質もしくはペプチド由来のN末端の2つのアミノ酸を切断し得る酵素(例えば、ジペプチジルペプチダーゼ)またはこのタンパク質もしくはペプチド由来のC末端の2つのアミノ酸を切断し得る酵素(例えば、カルボキシペプチダーゼ)と接触させる工程によって調製され得る。好適なジペプチジルペプチダーゼおよびカルボキシペプチダーゼは、例えば、Sigmaから市販されている。この反応は、pH6〜8、好ましくはリン酸緩衝液のような緩衝液中で、反応を速めるのに充分高いがこのタンパク質が変性されるほど高くはない温度(例えば、37℃)で、行われるべきである。
【0077】
多数のタンパク質およびペプチドのアミノ酸配列が公知であり、所望のN末端配列および/またはC末端配列を有するタンパク質またはペプチドが選択されて、いずれかの方法を用いて、所望のジケトピペラジンを得ることができる。また、所望の配列を有するペプチドは、周知の方法によって合成され得、使用され得る。
【0078】
ジケトピペラジンは、これらを含有する溶液(このような溶液としては、アルブミン、免疫グロブリンおよびエリスロポエチンを含む市販の薬学的組成物が挙げられる。)から、周知の方法(サイズ排除クロマトグラフィー(例えば、Centriconろ過)、アフィニティークロマトグラフィー(例えば、所望のジケトピペラジンに対する抗体(単数または複数)または短縮型のタンパク質もしくはペプチドに対する抗体(単数または複数)がビーズ上に結合されているそのビーズのカラムを使用する)、陰イオン交換または陽イオン交換)によって精製され得る。精製されたジケトピペラジンは使用され得、上記のような薬学的組成物に組み込まれ得る。
【0079】
ジケトピペラジンを精製する代わりに、動物レシピエント中に正常に見出されるアルブミン、免疫グロブリン、エリスロポエチン、ならびに/あるいは他のタンパク質および/またはペプチドを含む薬学的組成物は、T細胞媒介性疾患を処置するために投与され得、T細胞の活性化を抑制するために使用され得る。現在市販されている、これらのタンパク質および/またはペプチドを含む組成物は、これらがジケトピペラジンを含んでいる場合には使用され得るが、、このように改善された組成物を投与する前に、所望のジケトピペラジンの量を増加させるように、アルブミン、免疫グロブリン、エリスロポエチン、ならびに/あるいは他のタンパク質および/またはペプチドを上記のように処置することは、かなり好ましい。この動物は、好ましくはヒトであり、このタンパク質および/またはペプチドは、好ましくはヒトのタンパク質および/またはペプチドである。この組成物(単数または複数)の経口投与が好ましい。
【0080】
上記タンパク質および/またはペプチドの効果的な投薬量は、経験的に決定され得、そのような決定は当業者の範囲内でなされる。特に、上記タンパク質および/またはペプチドの組成物の効果的な投薬量を決定するために、この組成物中に存在する1つ以上のジケトピペラジンの量が測定され得、有効な量のジケトピペラジンが送達されるに十分な量のこの組成物が、上記動物に投与され得る。当業者は、投薬量が、使用される特定の組成物、処置されるべき疾患または状態、疾患または状態の重篤度、投与経路、排出速度、処置の継続期間、この動物に投与されている任意の他の薬物の同定、動物の年齢、サイズおよび種類、ならびに医学分野または獣医学分野において公知の他の要因とともに変動するということは、当業者によって理解される。一般に、タンパク質および/またはペプチドの組成物の適切な1日投与量は、治療効果を生成するに効果的な最少の用量の量である。しかしながら、1日投与量は、主治医である医師または獣医師により適切な医学的判断の範囲内で決定される。所望される場合、効果的な1日投与量は、2個、3個、4個、5個、6個またはそれ以上の副投与量(sub−dose)として投与され得、1日を通して適切な間隔をあけて別々に投与され得る。投与は、受容可能な応答が達成されるまで継続されるべきである。
【0081】
上記のように、ジケトピペラジンは、アルブミン、免疫グロブリンおよびエリスロポエチンの、市販の静脈注射用の薬学的組成物中に見出されることが知られている。ここで、これらの組成物の製造は1回以上の加熱工程(例えば、滅菌)を含む。ジケトピペラジンはまた、おそらくは、タンパク質およびペプチドの他の薬学的組成物中に存在する。ここで、この組成物の製造は加熱工程を含む。上記のように、多くのジケトピペラジンは、T細胞の活性化を抑制するための能力を有する。従って、多くの状況において、ジケトピペラジンを含む、アルブミン、免疫グロブリン、エリスロポエチン、または他のタンパク質もしくはペプチドの組成物を、患者(患畜)に投与することが望ましくないかもしれない。例えば、アルブミンは、しばしば外傷を罹患する患者に投与され、免疫グロブリンは、しばしば感染症または免疫不全症を罹患する患者に投与され、そしてエリスロポエチンは、貧血性の癌を罹患する患者または慢性的体調不良の患者(この患者の免疫系はしばしば障害を生じている。)に投与される。したがって、本発明は、このような組成物から少なくともいくらかの、好ましくは実質的に全てのジケトピペラジンを除去する方法を提供する。ジケトピペラジンを、上記のように(例えば、サイズ排除クロマトグラフィー(例えば、Centriconろ過)、アフィニティークロマトグラフィー(例えば、所望のジケトピペラジンに対する抗体(単数または複数)またはアルブミン、免疫グロブリン、エリスロポエチン、または他のタンパク質もしくはペプチドに対する抗体(単数または複数)がビーズ上に結合されているビーズのカラムを使用する)、陰イオン交換または陽イオン交換)によって除去して、アルブミン、免疫グロブリン、エリスロポエチン、ならびに他のタンパク質およびペプチドの改善された組成物を生成し得る。
【実施例】
【0082】
〔実施例1:ラットの腸からのAsp Ala DKP(DA−DKP)およびGlu Ala DKP(EA−DKP)の吸収〕
肛門括約筋から直腸までのラット腸をわずかに単離し、ウシ血清アルブミンを含む赤血球ベースの灌流液を用いて、張間膜動脈を介して潅流した。消化管から流出した赤血球を、門脈の挿管によって回収し、(再酸化後)再循環した。平衡化の期間後、約1mgのAsp−Alaジケトピペラジン(DA−DKP)または約1,4mgのGlu−Alaジケトピペラジン(EA−DKP)を含む溶液(約1ml)を、十二指腸の内腔に注射によって投与した。
【0083】
投薬後、灌流液の一連のサンプルを、間隔をおいて投与2時間後まで回収した。これらのサンプルを遠心分離機し、両方の環状ペプチドについてタンデム液体クロマトグラフフィー質量分法(tandem liquid chromatography mass spectrometry)によって、血漿をアッセイした。
【0084】
この結果は、灌流のわずか2時間後には、腸内の内腔から循環へ吸収されたDA−DKPおよびEA−DKPの量は、投与された用量のそれぞれ95%および100%(実際には112%)に相当したことを示した。
【0085】
したがって、両方の環状ペプチドは、腸内の内腔から血液へ迅速にかつ効果的に吸収され、腸壁を超えて輸送される間に代謝される証拠はない。よって、これらの潜在的治療薬は経口で与えられ得る。
【0086】
単離され、灌流されたラット肝臓における両方の化合物の、初回通過肝クリアランス(first pass hepatic clearance)の欠如(図示せず)と組み合わせて、変化していないDA−DKPおよびEA−DKPの胃腸管から血液への迅速な吸収は、前全身性クリアランス(pre−systemic clearance)が低いことを示す。したがって、経口的な投薬は理想的な投薬経路である。
【0087】
さらに、単離され、灌流されたラット腎臓の研究は、腎ペプチダーゼによって広範に代謝される多くの直鎖ペプチドとは異なり、両方の環状ジペプチドの腎クリアランスが比較的遅いことを示している。
【0088】
このデータをひとまとめにすると、少ない1日投与量のジケトピペラジンの投薬レジメンは、治療目的に適しているであろうということが示唆される。
【0089】
経口投与後のラットにおける予備的な薬物動態学的データは、両方の環状ジペプチドについて上述したものと一致していた:1,1〜3,7mg/kg体重(DA−DKP)および1,5〜4,8mg/kg体重(EA−DKP)で経口的に投薬した後の、Tmax値が30分〜60分、Cmax値が4〜6μg/ml(DA−DKP)および0,6〜1,1μg/ml(EA−DKP)(Tmaxは、濃度が最大に達する時間であり、Cmaxは、到達する最大濃度である。これらの両方を、得られたデータについてのカーブフィット方程式(curve fit equation)から計算した。)。
【0090】
予備的データは、DA−DKPおよび他のジケトピペラジンが血液脳関門を超えることを示唆している。従って、本発明のDA−DKPおよび他のジケトピペラジンは、多発性硬化症のような神経性障害を処置するために役に立つはずである。
【0091】
〔実施例2:Met−Arg DKP(MR−DKP)を含むヒト初乳画分およびAsp−Ala DKP(DA−DKP)による、ヒトTリンパ球サイトカイン産生のインビトロでの抑制〕
〔A.材料〕
本実施例は、DA−DKP、MR−DKPを含むヒト初乳(HC2626)、およびまたMR−DKPを含むヒト初乳(HCRBL;脱脂した初乳のCentriconろ過によって調製した、3000未満の分子量の成分を含むヒト初乳画分)の低分子量画分がヒトTリンパ球サイトカイン産生を抑制したことを実証する。DA−DKPおよびMR−DKPを、DMI Synthesis,Ltd.,Cardiff,UKから入手した。これら2つのジケトピペラジンは、炎症に対する生理学的応答の間に生成される、天然に存在する小さな化合物である。これらはまた、時折、ヒト静脈内免疫グロブリン(IVIg)、ヒトアルブミン、および他の生物学的調製物中に見出される。
【0092】
〔B.Tリンパ球サイトカイン産生の抑制〕
異なる2つのCD4−陽性ヒトTリンパ球クローンを試験した。細胞株の1つ(TRiPS)を、インフルエンザの予防接種を受けたドナーから単離した。この細胞は、ヘマグルチニンペプチド307−319について特有のものである。他の細胞株(H4#9,25)を、複数の硬化症ドナーの検死用脳組織から単離した。この細胞は、ミエリン塩基性タンパク質(アミノ酸87−99)について特有のものである。Tリンパ球クローンの両方が、(1)特異的抗原およびHLA−DR2−陽性提示細胞、または(2)抗−CD3抗体および抗−CD28抗体、のいずれかを用いるインビトロ刺激後に、インターロイキン8(IL−8)、IL−16、インターフェロン−ガンマ(IFN−γ)、および腫瘍壊死因子α(TNF−α)を生成する。
【0093】
これらのT細胞株を、刺激後18〜20日目に約4×105個の細胞を使用する継代のために刺激した。細胞を、10%ウシ胎仔血清(FBS;American Type Culture Collection(ATCC))を含む冷Iscove’s Modified Dulbecco Minimal Essential Medeium(IMDM,Sigma)中で洗浄し、抗−CD3モノクローナル抗体OKT3(ラット腹水から調製した)の1:500希釈物を含む、1.0mlの冷IMDM培地中で再懸濁した。細胞を抗体とともに氷上で30分間インキュベートし、次いで、FBSを含まない冷培地で洗浄し、50U/mlのヒトIL−2(Xenometrix)を含む培地中で、フィーダー細胞として、4000R照射した約2×106個の正常ヒトドナーの末梢血白血球(PBL)と結合させた。3日目に、FBSおよびIL−2を加えた新鮮なIMDM培地の添加によって、培養物を播いた。培養の日数を、OKT3を用いる刺激の日から測定した。細胞を、7日目(最大増殖時)、典型的には14日目(再刺激に対し最も感受性)および21日目(残りの細胞が老化に近づく)まで使用し得る。
【0094】
活性化実験を、細胞のアリコートを回収し、加温した(37℃)IMDM培地で2度洗浄することにより、実行した。各特異的アッセイのために、2×105個の生存細胞を、特定量の治療添加物(例えば、HC2626、DA−DKP、PMAなど)を含む、全量0.9mlの加温したIMDM培地で、37℃で15分間予めインキュベートした。次いで、活性化刺激としての0,1mlの加温したIMDM中の2×105個のCD3/CD28 Dynabeads(Dynal)のアリコートを添加し、、この培養物を一晩(18時間)37℃でインキュベートした。細胞培養物の上清を、遠心分離によって細胞をペレット化した後に回収した。サイトカイン量を、特異的ELISA(例えば、TNFα、IFNγ、IL−8、IL−16;Endrogen)によってアッセイした。
【0095】
図1〜5に示すように、ヒト初乳(HC2626)は、用量依存的様式で両方のTリンパ球細胞株によるインビトロでのサイトカイン産生を抑制した。また、図1〜5に示すように、HC RBLおよびDA−DKPは、促進サイクルの初期において、用量依存的様式で両方のTリンパ球細胞株によるインビトロでのサイトカイン産生を抑制した。しかしながら、周期後期(14日目以降)におけるHC RBLおよびDA−DKPの効果は、刺激性であった(図4参照)。HC RBLおよびDA−DKPの両方が、MR−DKPを含んでいる(質量分析法によって決定されたように)が、HC2626は、MR−DKP(細胞周期の後期における抑制性効果の原因であり得る)に加えて他の構成成分(★2003年11月25日出願の係属中の出願10/723,247に記載されるようなカゼイン(これは、相対的な脱リン酸化タンパク質であり、よって抗炎症性であり得る)を含む)を含む。したがって、HCRBLおよびHC2626(いずれもMR−DKPを含んでいる)、MR−DKPおよびDA−DKPは、全て刺激周期の初期においてT細胞によるサイトカイン産生を抑制するため、T細胞媒介性疾患および/または自己免疫性疾患(例えば、多発性硬化症)における炎症性サイトカイン応答を下方調節する際に有用であるはずである。これらの結果はまた、HC RBL、HC2626、MR−DKPおよびDA−DKPが、残りのT細胞に影響することなく、抗原特異的T細胞に選択的に影響を与えることを示唆する。
【0096】
〔C.作用機構〕
DA−DKPおよびHC2626(MR−DKPを含む)の作用機構を試験した。試験するために、何も加えず(「Nil」)、CD3/CD28 Dynabeads(CD3/CD28ビーズ)、CD3/CD28ビーズおよび0.5mMDA−DKP、またはCD3/CD28ビーズおよびHC2626の1:500希釈物のいずれかとともに、1×106個の18日目のTRiPS細胞を、37℃で30分間インキュベートした。インキュベーション後、これらの細胞をCell−Lytic Mammalian Cell Extraction Reagent(Sigma)中で溶解した。
【0097】
次いで、細胞抽出物を、2連のHypromatrix Arraysとともに、室温で2時間別々にインキュベートし、続いて、製造者(Hypromatrix)のプロトコルに従って2回洗浄した。Hypromatrix Arraysは、表1に列挙される転写調節因子に対する抗体(Hypromatrixによりカスタム製造された)を用いてブロットされたナイロン膜である。リン酸化チロシン、リン酸化セリン、およびリン酸化スレオニンに特異的な抗体カクテル(Zymed)を添加して、1時間インキュベートした。次いで、ビオチンで標識された抗免疫グロブリン抗体を添加した。抗免疫グロブリンビオチンを洗浄した後、スプレプトアビジン−ペルオキシダーゼを添加し、このアレイを、ペルオキシダーゼ−反応性発光基材を加える前に最終洗浄に供した。
【0098】
結果をフィルムに曝露することにより視覚化し、表2が示すように、0(陰性)または+から++++(陽性)とスコア付けした。表2に示すように、いくつかのサイトカイニン転写因子活性化(ERK1/2)および予め形成されたサイトカイニンの放出が、HC2626(MR−DKPを含む)およびDA−DKPによって抑制された。
【0099】
【表1】
【0100】
【表2】
【0101】
〔実施例3:Gly−Leu DKP(GL−DKP)およびAla−Pro DKP(AP−DKP)による、インビトロでのヒトTリンパ球サイトカイン産生抑制〕
GL−DKPおよびAP−DKP(DMI Synthesis, Ltd., Cardiff, UKから入手した)を、実施例2に記載したように、TRiPS細胞株およびH4#9.25細胞株を使用して試験した。GL−DKPおよびAP−DKPが、用量依存的様式でこれらのTリンパ球細胞株の両方によるインビトロでのサイトカイン産生を抑制することを見出した。この作用機構は、実施例2に記載したように、現在調査中であり、サイトカイン転写調節因子の活性化および予め形成されたサイトカインの放出の両方が影響を受けたようである。
【0102】
〔実施例4:Asp Ala DKP(DA−DKP)およびTyr Glu DKP(YE−DKP)による、インビトロでのヒトTリンパ球サイトカイン産生の抑制〕
正常なヒトリンパ球を、Histopaque(Sigma)を用いて、正常なヒトドナーの末梢血白血球から単離した。次いで、3〜4×105個のリンパ球を、血清を含まない1mlのIMDM培地中で懸濁した。これらの細胞を、1:2000希釈した抗CD3抗体希釈物(Pharmingen, San Diego, CA )25μlを添加し、37℃で18時間インキュベートすることによって刺激した。
【0103】
次いで、3つのDKP製剤のうちの1つおよびデキサメタゾン(最終濃度10−5M)を3連の培養物に添加した。これらの3つのDKP調製物は、以下の通りである:
1.DA−DKP(DMI Synthesis,Ltd.,Cardiff, UK)より入手;培養液中の最終濃度25μg/ml)
2.DKP−ZLB、60℃で4日間加熱した後に質量分析法による測定にて0,5mM DA−DKPを含むことがわかった25%アルブミン調製物(ZLB Bioplasma,AG 3000 Berne 22 Switzerlandより入手)(培養液中の最終濃度14μg/ml DA−DKP)
3.DKP−γ−glob、リン酸緩衝化生理食塩水(pH7.4)中に12mg/mlのγ−グロブリンを含むγ−グロブリン調製物(Sigmaより入手、番号G−4386)を、Centricon 3000フィルタを使用してろ過し、このろ液(3000未満のMWを有する成分を含む)を使用した。このろ液は質量292(これは、Tyr−Glu DKP(YE−DKP)の質量である)を含んでいた。この質量を、陰性電気スプレイ質量分析法と結合した陰イオン交換HPLCによって測定した。このろ液を、培養液中にて1:4の最終希釈で使用した。
【0104】
DKP調製物またはデキサメタゾンを添加した後、培養物を、37℃で18時間インキュベートした。次いで、各培養液に放出されたIL−2、IFNγ、およびTNFαの量を、ELISA(Pierce Biotechnology, Rockford, IL61105)によって測定した。
【0105】
結果を、以下の表3に示す。表に見られるように、3つ全てのサイトカインの放出の減少は、DKP−γ−グロブリンを用いて得られた。CD69+T細胞(CD69は活性化されたT細胞上に見られるマーカーである。)の数を調べるフローサイトメトリーもまた、デキサメタゾンによる約50%の低減と比較した場合に、DKP−γ−globは、T細胞受容体複合体が内部移行するにもかかわらず、CD69+T細胞の数を約90%まで減少させたことを示した。
【0106】
【表3】
【技術分野】
【0001】
本発明は、特定のジケトピペラジンを使用する、T細胞媒介性疾患の処置、およびT細胞の活性化の抑制に関する。本発明はまた、特定のジケトピペラジンを含む薬学的組成物、およびジケトピペラジンを合成する方法に関する。本発明はさらに、薬学的組成物中のジケトピペラジンの量を増加または減少させることにより、タンパク質およびペプチドの改善された薬学的組成物を製造する方法、および、得られた、改善された薬学的組成物に関する。
【背景技術】
【0002】
T細胞媒介性疾患は、多くの免疫系障害を示す。特に、T細胞は、自己免疫性疾患を開始させかつ永続させる細胞であると考えられている。自己免疫性疾患は、合衆国だけでも何百万もの人々を苦しめている80の深刻な慢性疾患の一群である。自己免疫疾患は、内因性(自己)抗原に対する免疫系の反応によって特徴付けられる。自己抗原に対するこれらの免疫応答は、自己反応性T細胞の持続的または反回性の活性化によって維持され、そして、直接的にまたは間接的に、この自己反応性T細胞は、自己免疫性疾患に見られる特徴的な組織の損傷および破壊の原因である。自己免疫性疾患および他のT細胞媒介性疾患に対する多くの処置が提案されているが、さらなる処置の必要性がなお存在する。
【発明の概要】
【0003】
本発明は、T細胞媒介性疾患を処置する方法を提供する。本方法は、以下の式:
【0004】
【化1】
【0005】
を有するジケトピペラジンまたはその生理学的に受容可能な塩の有効量を、該ジケトピペラジンまたはその塩を必要とする動物に投与する工程を含む、T細胞媒介性疾患を処置する方法であって、
ここで:
R1およびR2は同一であっても異なってもよく、R1およびR2の各々は、
(a)アミノ酸の側鎖であって、該アミノ酸がグリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、2,4−ジアミノ酪酸、2,3−ジアミノ酪酸、ロイシン、イソロイシン、ノルロイシン、セリン、ホモセリン、スレオニン、アスパラギン酸、アスパラギン、グルタミン酸、グルタミン、リジン、ヒドロキシリジン、ヒスチジン、アルギニン、ホモアルギニン、シトルリン、フェニルアラニン、p−アミノフェニルアラニン、チロシン、トリプトファン、チロキシン、システイン、ホモシステイン、メチオニン、ペニシラミン、またはオルニチンであるが、R1がアスパラギンまたはグルタミンの側鎖である場合は、R2はリジンまたはオルニチンの側鎖ではあり得ず、R1がリジンまたはオルニチンの側鎖の場合は、R2はアスパラギンまたはグルタミンの側鎖ではあり得ない、アミノ酸の側鎖であるか;
(b)R1は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、R2は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、または、R1およびR2の両方がそれぞれ独立して−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか;あるいは
(c)アミノ酸側鎖の誘導体であって、ここで、該アミノ酸は上記(a)に記載のアミノ酸の1つであり、該誘導体化された側鎖は、以下:
(i)−NHR3基または−N(R3)2基に置き換えられた−NH2基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−NHR3基または−N(R3)2基に置き換えられた−NH2基、
(ii)−OPO3H2基または−OR3基に置き換えられた−OH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−OPO3H2基または−OR3基に置き換えられた−OH基
(iii)−COOR3基に置き換えられた−COOH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−COOR3基に置き換えられた−COOH基、
(iv)−CON(R4)2基に置き換えられた−COOH基であって、R4の各々は独立して、Hあるいは置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−CON(R4)2基に置き換えられた−COOH基、
(v)−S−S−CH2−CH(NH2)−COOHまたは−S−S−CH2−CH2−CH(NH2)−COOHに置き換えられた−SH基、
(vi)−CH(NH2)−または−CH(OH)−基に置き換えられた−CH2−基、
(vii)−CH2−NH2または−CH2−OH基に置き換えられた−CH3基、
および/または
(viii)ハロゲンに置き換えられた炭素原子に結合しているH
を有する。
【0006】
本発明はまた、T細胞の活性化を抑制する方法を提供する。本方法は、式Iのジケトピペラジンまたはその生理学的に受容可能な塩の有効量を、当該ジケトピペラジンまたはその塩を必要とする動物に投与する工程を含む。
【0007】
本発明はさらに、薬学的に受容可能な担体および以下の式:
【0008】
【化2】
【0009】
を有するジケトピペラジンまたはその生理学的に受容可能な塩を含む薬学的組成物を提供し、
ここで:
R5およびR6は同一であっても異なってもよく、R5およびR6の々は、
(a)アミノ酸の側鎖であって、該アミノ酸がグリシン、アラニン、バリン、ノルバリン、αアミノイソ酪酸、2,4−ジアミノ酪酸、2,3−ジアミノ酪酸、ロイシン、イソロイシン、ノルロイシン、セリン、ホモセリン、スレオニン、リジン、ヒドロキシリジン、ヒスチジン、アルギニン、ホモアルギニン、シトルリン、フェニルアラニン、pアミノフェニルアラニン、チロシン、トリプトファン、チロキシン、またはオルニチンであるが、R5がアスパラギンまたはグルタミンの側鎖である場合は、R6はリジンまたはオルニチンの側鎖ではあり得ず、R5がリジンまたはオルニチンの側鎖の場合は、R6はアスパラギンまたはグルタミンの側鎖ではあり得ない、アミノ酸の側鎖であるか;
(b)R5は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、R6は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、または、R5およびR6の両方がそれぞれ独立して−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか;あるいは
(c)アミノ酸側鎖の誘導体であって、ここで、該アミノ酸は上記(a)に記載のアミノ酸の1つであり、該誘導体化された側鎖は、以下:
(i)−NHR3基または−N(R3)2基に置き換えられた−NH2基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−NHR3基または−N(R3)2基に置き換えられた−NH2基、
(ii)−OPO3H2基または−OR3基に置き換えられた−OH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−OPO3H2基または−OR3基に置き換えられた−OH基、
(iii)−CH(NH2)−または−CH(OH)−基に置き換えられた−CH2−基、
(iv)−CH2−NH2または−CH2−OH基に置き換えられた−CH3基、
および/または
(v)ハロゲンに置き換えられた炭素原子に結合しているH
を有する。
【0010】
本発明は、T細胞媒介性疾患を処置する別の方法を提供する。本方法は、動物において正常に見出されるタンパク質またはペプチドを含む薬学的組成物の有効量を、当該薬学的組成物を必要とする動物に投与する工程を含み、また薬学的組成物が該タンパク質またはペプチドから誘導される少なくとも1つのジケトピペラジンをまた含むように該タンパク質またはペプチドが処置される。
【0011】
本発明はさらに、T細胞の活性化を抑制する方法を提供する。本方法は、動物において正常に見出されるタンパク質またはペプチドを含む薬学的組成物の有効量を、当該薬学的組成物を必要とする動物に投与する工程を含み、また該薬学的組成物が該タンパク質またはペプチドから誘導される少なくとも1つのジケトピペラジンをまた含むように該タンパク質またはペプチドが処置される。
【0012】
さらに、本発明は、ジケトピペラジンを合成する方法を提供する。1つの実施形態においては、本方法は、ジケトピペラジンの形成を効率的に生じさせる条件下でタンパク質またはペプチドの溶液を加熱する工程を含む。第二の実施形態においては、本方法は、タンパク質またはペプチドの溶液を酵素に接触させる工程を含み、該酵素は、ジケトピペラジンを効率的に生成させる条件下で該タンパク質またはペプチドのN末端の2つのアミノ酸またはC末端の2つのアミノ酸を切断する。
【0013】
本発明はまた、タンパク質およびペプチドの改善された薬学的組成物を提供する。この改善は、該組成物におけるジケトピペラジンの減量を含む。
【0014】
さらに、本発明は、タンパク質およびペプチドの改善された薬学的組成物を生成する方法を提供する。本方法は、該組成物中に存在する少なくともいくらかのジケトピペラジンを該化合物から取り除く工程を含む。
【0015】
本発明はさらに、タンパク質およびペプチドの改善された薬学的組成物を製造する方法を提供する。本方法は、当該組成物中のジケトピペラジンの量が増加するように該タンパク質またはペプチドの溶液を処置する工程を含む。
【0016】
本発明はまた、タンパク質およびペプチドの改善された薬学的組成物を提供する。この改善は、該組成物におけるジケトピペラジンの増量を含む。
【図面の簡単な説明】
【0017】
【図1】抗CD3 OKT3抗体を用いた刺激後20日目に単離し、25ngのホルボールミリスチン酸(PMA)、HC−RBL(3kD未満の分子量の、加熱されたヒト初乳の画分(MR−DKPを含む))を1:10希釈、および0,5mMDA−DKPとともに、37℃で15分間インキュベートしたTriPS細胞(インフルエンザの予防接種を受けたドナーから単離された、ヘマグルチニンに特有のCD4+T細胞株)についての、ERK1/2のカウント対濃度をトレースしたものである。
【図2】抗CD3 OKT3抗体を用いた刺激12日後の、腫瘍壊死因子α(TNFα)およびIL−16の、TriPS細胞からの分泌の抑制を示す棒グラフであり、ヒト初乳(HC)2626(MR−DKPを含む)バンドDA−DKPからの、TNFαおよびIL−16の両方の分泌の抑制を示しており、HC2626を1:100希釈および1:1000希釈で用いて観察された最大放出は、高濃度のヒト初乳による溶解効果に起因している。0.5mM DA−DKPを使用しても、溶解は観察されず、TNFαおよびIL−16の分泌は低減されている。
【図3】抗CD3 OKT3抗体を用いた刺激10日後の、TriPS細胞からのTNFαの分泌の抑制を示す棒グラフであり、HC RBLおよびDA−DKPは、HC2626を用いて観察されるような滴定可能な応答に関してさらに調査する必要があることを示しており、強力な活性が示され得る。
【図4】抗CD3 OKT3抗体を用いた刺激後の種々の時点での、TriPS細胞からのTNFαの分泌の抑制を示す棒グラフであり、刺激周期の初期においてはDA−DKPおよびHC RBLの効果は抑制性であるが、周期の後期(14日目)においては刺激性であることを示しており、HC2626は常に抑制しており、おそらくは、他の構成因子に起因する。
【図5】抗CD3 OKT3抗体を用いた刺激後の7〜10日目における、H4#9,25細胞(ミエリン塩基性タンパク質に特有の、多発性硬化症患者の検死用脳組織から単離したCD4+T細胞株)からのTNFαの分泌の抑制を示す棒グラフであり、このT細胞株からのTNFαの分泌はまた、HC2626、HCRBLおよびDA−DKPによって抑制されることを示している。
【発明を実施するための形態】
【0018】
本発明は、T細胞媒介性疾患を処置する方法を提供する。「処置」は、疾患の症状、継続期間および重篤度を(完全にまたは部分的に)低減させること(疾患が治癒することを含む)、および疾患を予防することを意味するように本明細書中で使用される。
【0019】
T細胞媒介性疾患としては、組織不適合性、移殖片対宿主疾患、望ましくない遅延型超過敏反応(たとえば遅延型アレルギー反応)、T細胞媒介性肺疾患または自己免疫疾患が挙げられる。T細胞媒介性肺疾患としては、サルコイドーシス、過敏性肺炎、急性間質性肺炎、歯槽骨炎、肺線維症、特発性肺線維症、および炎症性肺損傷によって特徴付けられる他の疾患が挙げられる。自己免疫疾患としては、多発性硬化症、神経炎、多発性筋炎、乾癬、白斑、シェーグレン症候群、慢性間接リウマチ、1型糖尿病、自己免疫性膵炎、炎症性腸疾患(たとえばクローン病および潰瘍性大腸炎)、セリアック病、糸球体腎炎、強皮症、サルコイドーシス、自己免疫性甲状腺疾患(たとえば橋本甲状腺炎および甲状腺機能亢進症)、重症筋無力症、アジソン病、自己免疫性ぶどう膜網膜炎、尋常性天疱瘡、原発性胆汁性肝硬変、悪性貧血および結合線維組織増殖症候群が挙げられる。
【0020】
このT細胞媒介性疾患は、以下の式:
【0021】
【化3】
【0022】
を有するジケトピペラジンまたはその生理学的に受容可能な塩の有効量を、該ジケトピペラジンまたはその塩を必要とする動物に投与する工程を含む、T細胞媒介性疾患を処置する方法であって、
ここで:
R1およびR2は同一であっても異なってもよく、R1およびR2の各々は、
(a)アミノ酸の側鎖であって、該アミノ酸がグリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、2,4−ジアミノ酪酸、2,3−ジアミノ酪酸、ロイシン、イソロイシン、ノルロイシン、セリン、ホモセリン、スレオニン、アスパラギン酸、アスパラギン、グルタミン酸、グルタミン、リジン、ヒドロキシリジン、ヒスチジン、アルギニン、ホモアルギニン、シトルリン、フェニルアラニン、p−アミノフェニルアラニン、チロシン、トリプトファン、チロキシン、システイン、ホモシステイン、メチオニン、ペニシラミン、またはオルニチンであるが、R1がアスパラギンまたはグルタミンの側鎖である場合は、R2はリジンまたはオルニチンの側鎖ではあり得ず、R1がリジンまたはオルニチンの側鎖の場合は、R2はアスパラギンまたはグルタミンの側鎖ではあり得ない、アミノ酸の側鎖であるか;
(b)R1は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、R2は−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか、または、R1およびR2の両方がそれぞれ独立して−CH2−CH2−CH2−もしくは−CH2−CH(OH)−CH2−であり、かつ隣接する環窒素と一緒になってプロリンもしくはヒドロキシプロリンを形成するか;あるいは
(c)アミノ酸側鎖の誘導体であって、ここで、該アミノ酸は上記(a)に記載のアミノ酸の1つであり、該誘導体化された側鎖は、以下:
(i)−NHR3基または−N(R3)2基に置き換えられた−NH2基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−NHR3基または−N(R3)2基に置き換えられた−NH2基、
(ii)−OPO3H2基または−OR3基に置き換えられた−OH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−OPO3H2基または−OR3基に置き換えられた−OH基
(iii)−COOR3基に置き換えられた−COOH基であって、ここで、R3の各々は独立して、置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−COOR3基に置き換えられた−COOH基、
(iv)−CON(R4)2基に置き換えられた−COOH基であって、R4の各々は独立して、Hあるいは置換されたまたは非置換のアルキル、シクロアルキル、ヘテロシクロアルキル、アリール、アルキルアリール、アリールアルキル、またはヘテロアリールであってもよい、−CON(R4)2基に置き換えられた−COOH基、
(v)−S−S−CH2−CH(NH2)−COOHまたは−S−S−CH2−CH2−CH(NH2)−COOHに置き換えられた−SH基、
(vi)−CH(NH2)−または−CH(OH)−基に置き換えられた−CH2−基、
(vii)−CH2−NH2または−CH2−OH基に置き換えられた−CH3基、
および/または
(viii)ハロゲンに置き換えられた炭素原子に結合しているH
を有する。
【0023】
「置き換えられた(置き換えられる)」によって、上記アミノ酸側鎖の式を参照して、特定の基が他の特定の基に置き換えられることが意図される。例えば、イソロイシンの側鎖の式は−CH(CH3)−CH2−CH3である。末端の−CH3基が、−CH2−OH基に置き換えられた場合、得られる誘導体化されたイソロイシンの側鎖の式は、−CH(CH3)−CH2−CH2−OHである。別の例として、アラニンの側鎖の式は−CH3である。水素原子の1つが塩素原子に置き換えられた場合、得られる誘導体化されたアラニンの側鎖の式は、−CH2−Clである。グリシンの側鎖は−Hであり、このHが塩素原子(または他のハロゲン原子)に置き換えられた場合、得られる側鎖は、−Clであり、この塩素原子は、炭素環に結合される(例えば、R1=−Cl)。
【0024】
R1、R2、またはその両方が、アスパラギン酸またはグルタミン酸の側鎖か、または、このような側鎖の誘導体であるジケトピペラジンが好ましく、ここで、−COOH基は−COOR3基または−CON(R4)2基に置き換えられており、ここで、R3およびR4は上記で規定されている。この化合物の基について最も好ましくは、アスパラギン酸およびアラニンの側鎖(AsP−AlaDKPまたはDA−DKP)、グルタミン酸およびアラニンの側鎖(Glu−AlaDKPまたはEA−DKP)、チロシンおよびアスパラギン酸の側鎖(Tyr−AsPDKPまたはYD−DKP)、チロシンおよびグルタミン酸の側鎖(Tyr−GluDKPまたはYE−DKP)、およびこれら4つのジケトピペラジンのアスパラギン酸もしくはグルタミン酸の誘導体を含むジケトピペラジンであり、ここで、該−COOH基は−COOR3基または−CON(R4)2基に置き換えられており、ここで、R3およびR4は上記で規定されている。
【0025】
また、R1およびR2の両方が疎水性の側鎖(例えば、フェニルアラニンの側鎖)または疎水性の側鎖の誘導体であるジケトピペラジンが好ましい。「疎水性の側鎖の誘導体」によって、疎水性である誘導体化された側鎖が意図される。特に、以下のようなジケトピペラジンが好ましい:ここで、R1およびR2は同一であっても異なってもよく、R1およびR2の々は、グリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、ロイシン、イソロイシン、ノルロイシンまたはフェニルアラニンの側鎖であるか;R1は−CH2−CH2−CH2−であり、かつ隣接する環窒素と一緒になってプロリンを形成し、R2は−CH2−CH2−CH2−であり、かつ隣接する環窒素と一緒になってプロリンを形成するか;あるいは、R1は、グリシン、アラニン、バリン、ノルバリン、α−アミノイソ酪酸、ロイシン、イソロイシン、ノルロイシンまたはフェニルアラニンの側鎖であり、R2は−CH2−CH2−CH2−であり、かつ隣接する環窒素と一緒になってプロリンを形成する、ジケトピペラジンが好ましい。この化合物の基の中でもっとも好ましいのは、グリシンおよびロイシンの側鎖(Gly−LeuDKPまたはGL−DKP)、プロリンおよびフェニルアラニンの側鎖(Pro−PheDKPまたはPF−DKP)およびアラニンおよびプロリンの側鎖(Ala−ProDKPまたはAP−DKP)を含む。
【0026】
R1、R2、またはその両方が、メチオニンの側鎖、アルギニンの側鎖またはメチオニンの側鎖もしくはアルギニンの側鎖の誘導体であるジケトピペラジンが、さらに好ましい。この基について、R1がメチオニンの側鎖であり、R2がアルギニンの側鎖(Met−ArgDKPまたはMR−DKP)であるジケトピペラジンが最も好ましい。
【0027】
アミノ酸の「側鎖」によって、上記に列挙されたアミノ酸全てに共通するNH2−CH−COOH骨格に結合しているアミノ酸の一部が意図される。例えば、グリシンの側鎖は−Hであり、アラニンの側鎖は−CH3であり、セリンの側鎖は−CH2−OHである。
【0028】
「疎水性」によって、生理学的pHにおいて荷電されず、水溶液により忌避される(repel)側鎖または側鎖の誘導体が意図される。
【0029】
「アルキル」によって、飽和した直鎖炭化水素または分枝した炭化水素が意図され、これらは、1〜10個の炭素原子、好ましくは1〜6個の炭素原子を含む。「低級アルキル」によって、飽和した直鎖炭化水素または分枝した炭化水素が意図され、これらは、1〜6個の炭素原子を含む。
【0030】
「シクロアルキル」によって、少なくとも1つの環を含む、飽和した環状炭化水素が意図され、各環は、少なくとも3つの炭素原子を含む。好ましくは、このシクロアルキルは、4〜8の炭素原子の環を1つ含む。
【0031】
「ヘテロシクロアルキル」によって、1つ以上の炭素環を有するシクロアルキルが意図され、これらの環の少なくとも1つは、O、S、またはNによって置き換えられている。
【0032】
「アリール」によって、少なくとも1つの芳香環(例えば、フェニル基)を有する芳香族基が意図される。
【0033】
「アルキルアリール」によって、アリール(例えば、−CH2C6H5または−CH3CH(C6H5)CH3)によって置き換えられたHを有する低級アルキルが意図される。
【0034】
「アリールアルキル」によって、低級アルキル基(例えば、−C6H4−CH3)によって置き換えられたHを有するアリールが意図される。
【0035】
「ヘテロアリール」によって、1つ以上の炭素環を有するアリールが意図され、これらの環の少なくとも1つは、O、S、またはNによって置き換えられている。
【0036】
「置換された」によって、構成部分が以下の基の中より選択される1つ以上の置換基によって置き換えられていることが意図される:−OH、NH2、−SH、−COOHおよび/またはハロゲン原子。
【0037】
「ハロゲン」によって、塩素、フッ素、臭素、またはヨウ素が意図される。好ましくは、塩素または臭素である。
【0038】
式Iのジケトピペラジンは、T細胞媒介性疾患の処置に有効である。なぜなら、これらがT細胞の活性化を抑制するからである。従って、式Iのジケトピペラジンはまた、活性化されたT細胞によって引き起こされるかまたは悪化される炎症または炎症性疾患を処置するためにも使用され得る。本明細書において、「抑制」は、(完全にまたは部分的に)低減させること、または予防することを意図して使用される。
【0039】
ジケトピペラジンを生成する方法は、当該分野において周知であり、これらの方法は、本発明のジケトピペラジンを合成する際に使用され得る。例えば、米国特許第4,694,081号、同第5,817,751号、同第5,990,112号、同第5,932,579号、および同第6,555,543号、米国特許出願公開番号2004/0024180、PCT出願WO96/00391およびWO97/48685、ならびにSmithら、Bioorg.Med.Chem,Letters,8,2369−2374(1998)を参照のこと。これらの開示は、その全体が参考として本明細書において援用される。
【0040】
例えば、ジケトピペラジンは、初めにジペプチドを合成することによって調製され得る。このジペプチドは、L−アミノ酸、D−アミノ酸、またはL−アミノ酸とD−アミノ酸とを組合せたものを使用する、当該分野において周知の方法によって合成され得る。固相ペプチド合成法が好ましい。もちろん、ジペプチドはまた、多数の供給源から市販されており、この供給源としては、DMI Synthesis Ltd.、Cardiff、UK(custom synthesis)、Sigma−Aldrich、St.Louis、MO(primarily custom synthesis)、Phoenix Pharmaceuticals, Inc.、Belmont、CA(custom synthesis)、Fisher Scientific(custom synthesis)、およびAdvanced ChemTech, Louisville, KYが挙げられる。
【0041】
一旦、ジペプチドが合成されるかまたは購入されると、ジペプチドは環化してジケトピペラジンを形成する。このことは、種々の技術により達成され得る。
【0042】
例えば、米国特許出願公開番号2004/0024180は、ジペプチドを環化させる方法について記載している。つまり、ジペプチドは、蒸留によって水分を除去する間、有機溶媒中で加熱される。好ましくは、この有機溶媒は、アセトニトリル、アリルアルコール、ベンゼン、ベンジルアルコール、n−ブタノール、2−ブタノール、第三ブタノール、酢酸ブチル、四塩化炭素、クロロベンゼンクロロホルム、シクロヘキサン、1,2ジクロロエタン、ジエチルアセタール、ジメチルアセタール、酢酸エチルエステル、ヘプタン、メチルイソブチルケトン、3ペンタノール、トルエンおよびキシレンのような、低沸点の、水との共沸混合物である。温度は、環化が行われる反応速度、および使用される共沸剤(azeotroping agent)の型に依存する。反応は、好ましくは50〜200℃で、より好ましくは80〜150℃で行なわれる。環化が行われるpH範囲は、当業者により容易に決定され、有利には2〜9であり、好ましくは3〜7である。
【0043】
ジペプチドのアミノ酸の一方または両方が、その側鎖上にカルボキシル基を有するか、または有するように誘導体化されている場合(例えば、アスパラギン酸またはグルタミン酸)、このジペプチドは、米国特許第6,555,543号に記載されているように環化されることが好ましい。つまり、ジペプチドは、側鎖カルボキシルがなお保護されており、中性条件下で加熱される。典型的には、ジペプチドは、約80℃〜約180℃で加熱され、好ましくは約120℃で加熱される。溶媒は、中性溶媒である。例えば、溶媒は、アルコール(例えば、ブタノール、メタノール、エタノール、および高級アルコール(フェノールではない))、および共沸性の共溶媒(例えば、トルエン、ベンゼン、またはキシレン)を含み得る。好ましくは、アルコールはブタン−2−olであり、共沸性の共溶媒はトルエンである。加熱は、反応が完了するまで継続され、そのような時間は経験的に決定される。典型的には、ジペプチドは、それを8〜24時間還流することにより環化され、好ましくは18時間還流される。最終的に、保護基がジケトピペラジンから除去される。このように行なう際に、最終化合物のキラリティーを維持するために、強酸(硫酸または塩化水素酸のような鉱酸)、強塩基(水酸化カリウムまたは水酸化ナトリウムのようなアルカリ性塩基)および強力な還元剤(例えば、水素化アルミニウムリチウム)の使用は避けられるべきである。
【0044】
固相樹脂上で生成されたジペプチドは、1工程で環化されかつ該樹脂から放出され得る。例えば、米国特許第5,817,751号を参照のこと。例えば、結合したN−アルキル化されたジペプチドは、酢酸(例えば1%)またはトリエチルアミン(例えば4%)の存在下で、トルエンまたはトルエン/エタノール中に懸濁される。典型的には、より速い環化時間のためには、塩基性環化条件が好ましい。
【0045】
アミノ酸側鎖が誘導体化された、式IおよびIIのジケトピペラジンを調製するために、ジペプチドの合成にアミノ酸誘導体が使用され得、当該分野において公知のように、このジペプチドは誘導体化され得、および/または、ジケトピペラジンは誘導体化され得る。例えば、上記に列挙された参考文献を参照のこと。
【0046】
ジペプチドを環化する他の方法およびジケトピペラジンを生成する他の方法は、当該分野において公知であり、本発明の実施において有用なジケトピペラジンを調製する際に使用され得る。例えば、上記に列挙された参考文献を参照のこと。さらに、本発明における使用に適切な多くのジケトピペラジンは、以下に記載されるように、タンパク質およびペプチドから生成され得る。さらに、本発明の実施に使用するためのジケトピペラジンは、例えば、DMI Synthesis Ltd.、Cardiff、UK(カスタム合成)から商業的に取得され得る。
【0047】
式IおよびIIのジケトピペラジンは、個々のキラル中心、軸または表面の構成を変更することにより得られる、予想される全ての立体異性体を含む。すなわち、式IおよびIIのジケトピペラジンは、予想される全てのジアスタレオマー、および全ての光学異性体(鏡像異性体)を含む。
【0048】
本発明のジケトピペラジンの生理学的に受容可能な塩は、本発明の実施においてまた使用され得る。生理学的に受容可能な塩としては、慣用的な非毒性の塩(例えば、無機酸(塩化水素酸、臭化水素酸、硫酸、リン酸、硝酸など)、有機酸(酢酸、プロピオン酸、琥珀酸、グリコール酸、ステアリン酸、乳酸、リンゴ酸、酒石酸、クエン酸、グルタミン酸、アスパラギン酸、安息香酸、サリチル酸、シュウ酸、アスコルビン酸など)、または塩基(Nジベンジルエチレンジアミン、Dグルコサミン、またはエチレンジアミン由来の、薬学的に受容可能な金属カチオンまたは有機カチオンの、水酸化物、炭酸、または重炭酸)から誘導される塩)が挙げられる。これらの塩は、慣用的な様式で(例えば、化合物の遊離塩基形態を酸で中和することによって)調製される。
【0049】
上述したように、本発明のジケトピペラジンまたはその生理学的に受容可能な塩は、T細胞媒介性疾患を処置するか、または、T細胞の活性化を抑制するために使用され得る。このように使用するために、ジケトピペラジンまたはその生理学的に受容可能な塩は、処置を必要とする動物に投与される。この動物は、ウサギ、ヤギ、イヌ、ネコ、ウマ、またはヒトのような哺乳動物が好ましい。本発明の化合物についての効果的な投薬形態、投与様式、および投薬量は、経験的に決定され得、このような決定を下すことは当業者の範囲内である。投薬量は、使用される特定の化合物、処置されるべき疾患または状態、疾患または状態の重篤度、投与経路、化合物の排泄速度、処置の継続期間、この動物に投与されている他の薬物の同定、この動物の年齢、サイズおよび種類、ならびに医学分野および獣医学分野で知られている他の要因とともに変動することは、当業者によって理解される。一般に、本発明の化合物の好適な1日投与量は、治療効果を生成するために効果的な最少の投与量の、化合物の量である。しかしながら、この1日投与量は、主治医である医師または獣医師により適切な医学的判断の範囲内で決定される。所望される場合、有効な1日投与量は、2個、3個、4個、5個、6個またはそれ以上の副投与量(sub−dose)として投与され得、1日を通して適切な間隔を空けて別々に投与され得る。化合物の投与は、受容可能な応答が達成されるまで継続されるべきである。
【0050】
本発明の化合物(すなわち、ジケトピペラジンまたはその生理学的に受容可能な塩)は、任意の適切な投与経路によって治療のために患者(患畜)に投与され得、その投与経路としては、経口、経鼻、経直腸、経膣、非経口(たとえば、静脈内注入、髄腔内注入、腹腔内注入、皮下または筋肉注射)、大槽内、経皮、頭蓋内、脳内、および局所(頬側および舌下を含む)が挙げられる。好ましい投与経路は、経口および静脈内である。
【0051】
本発明の化合物は単独で投与されることも可能であるが、この化合物を薬学的処方物として投与することが好ましい。本発明の薬学的組成物は、1つ以上の薬学的に受容可能な担体、および必要に応じて、1つ以上の他の化合物、薬物または他の物質との混合物中の活性成分として本発明の化合物(単数または複数)を含む。各担体は、この処方物の他の成分と適合性であり、動物に対して有害ではないという観点において「受容可能」でなければならない。薬学的に受容可能な担体は当該分野において周知である。選択された投与経路にかかわらず、本発明の化合物は、当業者に公知の慣用的な方法によって、薬学的に受容可能な投薬形態に処方される。例えば、Remington’s Pharmaceutical Scienceを参照のこと。
【0052】
経口投与に適切な本発明の処方物は、カプセル、カシュ剤、丸剤、錠剤、散剤、顆粒、または水性もしくは非水性の液体中での溶剤もしくは懸濁液として、または水中油乳剤もしくは油中水乳剤、またはエリキシル剤もしくはシロップ剤、またはトローチ剤(不活性なベース(例えば、ゼラチンおよびグリセリン)、またはスクロースおよびアカシアを使用する)などの形態であり得、これらの各々は、所定量の本発明の化合物(単数または複数)を活性成分として含む。本発明の化合物(単数または複数)はまた、ボーラス、舐剤またはペーストであり得る。
【0053】
経口投与のための本発明の固形の投薬形態(カプセル、錠剤、丸剤、糖衣剤、散剤、顆粒など)において、活性成分(すなわち、1つ以上の本発明のジケトピペラジンおよび/またはその生理学的に受容可能な塩)は、1つ以上の薬学的に受容可能な担体(例えば、クエン酸ナトリウム、リン酸二カルシウム)、および/または以下のいずれかと混合される:(1)充填剤または増量剤(例えば、デンプン、ラクトース、スクロース、グルコース、マンニトールおよび/またはケイ酸);(2)結合剤(例えば、カルボキシメチルセルロース、アルギン酸塩、ゼラチン、ポリビニルピロリドン、スクロースおよび/またはアカシア);(3)湿潤剤(例えば、グリセロール);(4)崩壊剤(例えば、寒天、炭酸カルシウム、ジャガイモデンプンまたはタピオカデンプン、アルギニン酸、特定のケイ酸塩、および炭酸ナトリウム);(5)溶液遅延剤(例えば、パラフィン);(6)吸収促進剤(例えば、第四アンモニウム化合物);(7)湿潤剤(例えば、セチルアルコールおよびグリセロールモノステアレート);(8)吸収剤(例えば、カオリンおよびベントナイト土);(9)滑剤(例えば、タルク、ステアリン酸カルシウム、ステアリン酸マグネシウム、充実ポリエチレングリコール、ラウリル硫酸ナトリウムおよびこれらの混合物);(10)着色剤。カプセル、錠剤および丸剤の場合、薬学的組成物は、緩衝剤を含み得る。同様の型の固体組成物は、ラククトースまたは乳糖および高分子量のポリエチレングリコールなどのような賦形剤を使用して、柔和に充填されたゼラチンカプセルおよび堅固に充填されたゼラチンカプセルにおける充填剤として使用され得る。
【0054】
錠剤は、必要に応じて1つ以上の副成分を圧縮または成形することにより生成され得る。圧縮された錠剤は、結合剤(例えば、ゼラチンまたはヒドロキシプロピルメチルセルロース)、滑剤、不活性な希釈剤、防腐剤、崩壊剤(例えば、ナトリウムデンプングリコレートまたは架橋したカルボキシルメチルセルロースナトリウム)、界面活性剤または分散剤を使用して調製され得る。成形された錠剤は、不活性な液体希釈剤で湿らされた粉末状の化合物の混合物を適切な機械中で成形することによって生成され得る。
【0055】
本発明の薬学的組成物の錠剤および他の固形の投薬形態(例えば、糖衣錠、カプセル、丸剤、および顆粒)は、必要に応じて、コーティングおよびシェル(shell)(例えば、腸溶性コーティングおよび薬学的処方物の分野において周知の他のコーティング)を用いて獲得(score)または調製され得る。本発明の薬学的組成物の錠剤および他の固形の投薬形態はまた、例えば、所望の放出プロファイルを提供するための種々の割合のヒドロキシプロピルメチルセルロース、他のポリマーマトリクス、リポソームおよび/またはミクロスフェアを用いて、組成物中の活性成分を徐放(slow or controlled release)し得るように処方され得る。本発明の薬学的組成物の錠剤および他の固形の投薬形態は、たとえば細菌保持フィルタ(bacteria−retaining filter)を通してろ過することによって滅菌され得る。これらの組成物はまた、必要に応じて、乳白剤を含み得、または必要に応じて遅延された形態で、好ましくは胃腸管の特定の部分に、活性成分のみを放出する組成物であり得る。使用され得る包埋型の組成物としては、ポリマー性物質およびワックスが挙げられる。この活性成分はまた、マイクロカプセル化された形態中にあり得る。
【0056】
本発明の化合物の経口投与のための液体投薬形態は、薬学的に受容可能な乳剤、マイクロエマルジョン、溶液、懸濁液、シロップ剤、およびエリキシル剤を含む。活性成分に加えて、液体投薬形態は、当該分野においては一般に使用される不活性な希釈剤(例えば、水または他の溶媒、可溶化剤および乳化剤(例えば、エチルアルコール、イソプロピルアルコール、炭酸エチル、酢酸エチル、ベンジルアルコール、安息香ベンジル、プロピレングリコール、1,3−ブチレングリコール、油(具体的には、綿実油、ラッカセイ油、トウモロコシ油、胚芽油、オリーブ油、ヒマシ油、およびゴマ油)、グリセロール、テトラヒドロフルフリルアルコール、ポリエチレングリコール、ソルビタン脂肪酸エステル、およびこれらの混合物))を含み得る。
【0057】
不活性な希釈剤の他に、経口組成物はまた、アジュバント(例えば、湿潤剤、乳化剤および懸濁化剤)、甘味剤、着香料、着色料、香料および保存料を含み得る。
【0058】
活性成分に加えて、懸濁液は、懸濁剤(例えば、エトキシル化イソステリアルアルコール、ポリオキシエチレンソルビトールおよびソルビタンエステル、微結晶性セルロース、メタ水酸化アルミニウム(aluminum metahydroxide)、ベントナイト、寒天、トラガカント、およびこれらの混合物)を含み得る。
【0059】
経直腸投与または経膣投与のための本発明の薬学的組成物の処方物は、坐剤として提供され得、1つ以上の本発明の化合物を、例えば、カカオ脂、ポリエチレングリコール、坐材のワックスまたはサリチル酸塩のような、室温では固体であるが体温では液体になり、よって、直腸または膣腔では溶解して活性成分を放出する、1つ以上の適切な無刺激の賦形剤または担体と混合することによって調製され得る。経膣投与に適切な本発明の処方物としてはまた、ペッサリー、タンポン、クリーム、ゲル、ペースト、発泡体またはスプレー処方物(当該分野において適当であることが知られている担体を含む)が挙げられる。
【0060】
本発明の化合物の局所投与または経皮投与のための投薬形態としては、散剤、スプレー、軟膏、ペースト、クリーム、ローション、ゲル、溶液、膏薬、点滴薬および吸入剤(inhalant)が挙げられる。活性成分は、滅菌条件下で、薬学的に受容可能な担体、および任意の緩衝液または必要とされ得る推進剤と混合され得る。
【0061】
軟膏、ペースト、クリーム、およびゲルは、活性成分に加えて、賦形剤(例えば、動物性および植物性脂肪、油、ワックス、パラフィン、デンプン、トラガカント、セルロース誘導体、ポリエチレングリコール、シリコン、ベントナイト、ケイ酸、タルクおよび酸化亜鉛、またはこれらの混合物)を含み得る。
【0062】
散剤およびスプレーは、活性成分に加えて、例えば、ラクトース、タルク、ケイ酸、水酸化アルミニウム、ケイ酸カルシウム、ポリアミド散剤、およびこれらの物質の混合物を含み得る。スプレーはさらに、推進剤(例えば、フロン(chlorofluorohydrocarbon))および揮発性の非置換の炭化水素(例えば、ブタンおよびプロパン)を慣用的に含み得る。
【0063】
経皮的膏薬は、本発明の化合物の身体への制御された送達を提供するというさらなる利点を有する。このような投薬形態は、本発明の1つ以上の化合物を、好ましい媒体(例えば、エラストマーマトリクス材(elastomeric matrix material))に、溶解させるか、分散させるか、または組み込むかによって製造され得る。また、吸収促進剤を使用して、皮膚を超えるこの化合物の流量を増加させ得る。この流量の速度は、速度制御された膜を設けるか、またはポリマーマトリクスまたはゲル中に化合物を分散することによって制御され得る。
【0064】
薬学的処方物としては、吸入(inhalation)投与もしくは通気(insufflation)投与、または経鼻投与もしくは眼内投与に好適な処方物が挙げられる。吸入による上気道(経鼻)または下気道への投与のために、本発明の上記化合物は、吸入器、噴霧器、もしくは加圧パック、またはエアロゾルスプレー送達の他の簡便な手段エアロゾルから簡便に送達される。可圧パックは、適切な推進剤(例えば、ジクロジフルオロメタン、トリクロロフルオロメタン、ジクロロテトラフルオロエタン、炭酸ガスまたは他の適切なガス)を含み得る。加圧型エアロゾルの場合は、測定された量を送達するために弁を設けることにより、処方単位が決定され得る。
【0065】
あるいは、吸入または通気によって投与するために、組成物は、乾燥粉末の形態(例えば、1つ以上の本発明の化合物および適切な粉末ベース(例えば、ラクトースまたはデンプン)の粉体混合物)をとり得る。この粉末組成物は、例えば、カプセルもしくは薬包、または例えば、ゲラチンもしくは発泡剤パック中に、単位投与量形態で提供され得、吸入器(inhalator)、通気器、または測定された投与量の吸入器(inhaler)を用いてそこから粉末が投与され得る。
【0066】
経鼻投与のために、本発明の化合物は、点鼻剤または液体スプレーの手段(例えば、プラスチックボトル噴霧器または測定された投与量の吸入器)によって投与され得る。典型的な噴霧器はMistometer(Wintrop)およびMedihaler(Riker)である。
【0067】
点眼薬または点鼻薬のようなドロップは、水性または非水性のベースを用いて処方され得、1つ以上の分散剤、可溶化剤、または懸濁剤もまた含む。液体スプレーは、加圧されたパックから簡便に送達され得る。ドロップは、単純な目薬キャップボトルによって、または特別な形状の梱包により滴下される液体内容物を送達するために適合されたプラスチックボトルによって、送達され得る。
【0068】
非経口投与に好適な本発明の薬学的組成物は、1つ以上の本発明の化合物を、1つ以上の薬学的に受容可能な滅菌した等張性の水溶液または非水溶性の水溶液、分散液、懸濁液または乳剤、あるいは滅菌した注射可能な溶液もしくは分散液に使用直前に再構成され得る滅菌粉末とともに、含む。この組成物は、抗酸化剤、緩衝液、溶質を含み得、これらは、目的のレシピエントの血液、懸濁剤または濃化剤を用いてこの組成物を等張性にする。
【0069】
本発明の薬学的組成物に使用され得る適切な水性担体または非水性担体としては、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリセロール、ポリエチレングリコールなど)、およびこれらの適切な混合物、植物油(例えば、オリーブ油)、注入可能な有機エステル(例えば、オレイン酸エチル)が挙げられる。好ましい流動性は、コーティング物質(例えば、レシチン)の使用によって、分散の場合は必要とされる粒子サイズの維持によって、および界面活性剤の使用によって維持され得る。
【0070】
これらの組成物はまた、アジュバント(例えば、湿潤剤、乳化剤、および分散剤)を含み得る。これらの組成物中に、等張化剤(例えば、糖質、塩化ナトリウムなど)を含むことも望ましい。さらに、注入可能な薬学的な形態の延長された吸収は、吸収を遅延させる薬剤(例えば、モノステアリン酸アルミニウムおよびゼラチン)の内包により、もたらされ得る。
【0071】
いくつかの場合、薬物の効果を延長させるために、皮下注射または筋肉注射からこの薬物の吸収を遅らせることが望ましい。このことは、水溶性の低い結晶性または非晶質物質の液体懸濁液の使用によって達成され得る。次いで、薬物の吸収速度は、その溶解速度に依存し、これは、結晶サイズおよび結晶形態に依存し得る。あるいは、非経口的に投与された薬物の遅延された吸収は、油ビヒクル中に薬物を溶解または懸濁することによって達成され得る。
【0072】
注入可能な貯蔵形態は、生分解性ポリマー(例えば、ポリラクチド−ポリグリコリド)中で薬物のマイクロカプセル化マトリクスを形成することによって製造される。薬物対ポリマーの比、および使用した特定のポリマーの特性に依存して、薬物放出の速度は、制御され得る。生分解性ポリマーの他の例としては、ポリ(オルソエステル)およびポリ(無水物)が挙げられる。注入可能な貯蔵処方物はまた、生体組織と適合性のリポソームまたはマイクロエマルジョンに薬物を閉じ込めることによって調製される。この注射可能な物質は、例えば、細菌保持フィルタを通してろ過することによって、滅菌され得る。
【0073】
この処方物は、単回用量または複数回用量の封着された容器(例えば、アンプルおよびバイアル)にて提供され得、そして滅菌された液体担体(例えば、注射水)を使用直前に添加することのみが必要とされる凍結乾燥された状態で保存され得る。即時調整の注射用の溶液および懸濁液は、上記した型の滅菌された散剤、顆粒および錠剤から調製され得る。
【0074】
本発明での使用に好適なジケトピペラジンは、アルブミン、免疫グロブリンおよびエリスロポエチンを含む市販されている静脈注射用の薬学的組成物のいくつかにおいて提供されていることは知られている。これらの薬学的調製物にて提供されるジケトピペラジンは、これらの薬学的組成物の製造においてしばしば使用される加熱工程によって形成される。この加熱工程により、タンパク質のN末端の2つのアミノ酸および/またはC末端の2つのアミノ酸の切断および環化が生じて、ジケトピペラジンが形成される。
【0075】
従って、本発明における使用のためのジケトピペラジンは、アルブミン、免疫グロブリン、エリスロポエチン、ならびに他のタンパク質およびペプチドの溶液を加熱する工程によって調製され得る。例えば、中性のpHでのリン酸緩衝液中の、アルブミン、免疫グロブリン、エリスロポエチン、または別のタンパク質もしくはペプチドの溶液は、調製される。好ましくは、この溶液は、N末端アミノ酸および/またはC末端アミノ酸のプロトン化を達成するための濃縮溶液(例えば、約100〜500mM)である。この溶液は、60℃で約2時間〜数日間、好ましくは4日間加熱されて、ジケトピペラジンの形成を生じる。好ましくは、タンパク質の変性は避けられるべきである。このことは、時間を短くすることおよび/またはカプリル酸もしくはN−アセチルトリプトファンを各々に対して約0,02M添加することによって達成される。
【0076】
本発明における使用のためのジケトピペラジンはまた、アルブミン、免疫グロブリン、エリスロポエチン、または別のタンパク質もしくはペプチドの溶液を、このタンパク質もしくはペプチド由来のN末端の2つのアミノ酸を切断し得る酵素(例えば、ジペプチジルペプチダーゼ)またはこのタンパク質もしくはペプチド由来のC末端の2つのアミノ酸を切断し得る酵素(例えば、カルボキシペプチダーゼ)と接触させる工程によって調製され得る。好適なジペプチジルペプチダーゼおよびカルボキシペプチダーゼは、例えば、Sigmaから市販されている。この反応は、pH6〜8、好ましくはリン酸緩衝液のような緩衝液中で、反応を速めるのに充分高いがこのタンパク質が変性されるほど高くはない温度(例えば、37℃)で、行われるべきである。
【0077】
多数のタンパク質およびペプチドのアミノ酸配列が公知であり、所望のN末端配列および/またはC末端配列を有するタンパク質またはペプチドが選択されて、いずれかの方法を用いて、所望のジケトピペラジンを得ることができる。また、所望の配列を有するペプチドは、周知の方法によって合成され得、使用され得る。
【0078】
ジケトピペラジンは、これらを含有する溶液(このような溶液としては、アルブミン、免疫グロブリンおよびエリスロポエチンを含む市販の薬学的組成物が挙げられる。)から、周知の方法(サイズ排除クロマトグラフィー(例えば、Centriconろ過)、アフィニティークロマトグラフィー(例えば、所望のジケトピペラジンに対する抗体(単数または複数)または短縮型のタンパク質もしくはペプチドに対する抗体(単数または複数)がビーズ上に結合されているそのビーズのカラムを使用する)、陰イオン交換または陽イオン交換)によって精製され得る。精製されたジケトピペラジンは使用され得、上記のような薬学的組成物に組み込まれ得る。
【0079】
ジケトピペラジンを精製する代わりに、動物レシピエント中に正常に見出されるアルブミン、免疫グロブリン、エリスロポエチン、ならびに/あるいは他のタンパク質および/またはペプチドを含む薬学的組成物は、T細胞媒介性疾患を処置するために投与され得、T細胞の活性化を抑制するために使用され得る。現在市販されている、これらのタンパク質および/またはペプチドを含む組成物は、これらがジケトピペラジンを含んでいる場合には使用され得るが、、このように改善された組成物を投与する前に、所望のジケトピペラジンの量を増加させるように、アルブミン、免疫グロブリン、エリスロポエチン、ならびに/あるいは他のタンパク質および/またはペプチドを上記のように処置することは、かなり好ましい。この動物は、好ましくはヒトであり、このタンパク質および/またはペプチドは、好ましくはヒトのタンパク質および/またはペプチドである。この組成物(単数または複数)の経口投与が好ましい。
【0080】
上記タンパク質および/またはペプチドの効果的な投薬量は、経験的に決定され得、そのような決定は当業者の範囲内でなされる。特に、上記タンパク質および/またはペプチドの組成物の効果的な投薬量を決定するために、この組成物中に存在する1つ以上のジケトピペラジンの量が測定され得、有効な量のジケトピペラジンが送達されるに十分な量のこの組成物が、上記動物に投与され得る。当業者は、投薬量が、使用される特定の組成物、処置されるべき疾患または状態、疾患または状態の重篤度、投与経路、排出速度、処置の継続期間、この動物に投与されている任意の他の薬物の同定、動物の年齢、サイズおよび種類、ならびに医学分野または獣医学分野において公知の他の要因とともに変動するということは、当業者によって理解される。一般に、タンパク質および/またはペプチドの組成物の適切な1日投与量は、治療効果を生成するに効果的な最少の用量の量である。しかしながら、1日投与量は、主治医である医師または獣医師により適切な医学的判断の範囲内で決定される。所望される場合、効果的な1日投与量は、2個、3個、4個、5個、6個またはそれ以上の副投与量(sub−dose)として投与され得、1日を通して適切な間隔をあけて別々に投与され得る。投与は、受容可能な応答が達成されるまで継続されるべきである。
【0081】
上記のように、ジケトピペラジンは、アルブミン、免疫グロブリンおよびエリスロポエチンの、市販の静脈注射用の薬学的組成物中に見出されることが知られている。ここで、これらの組成物の製造は1回以上の加熱工程(例えば、滅菌)を含む。ジケトピペラジンはまた、おそらくは、タンパク質およびペプチドの他の薬学的組成物中に存在する。ここで、この組成物の製造は加熱工程を含む。上記のように、多くのジケトピペラジンは、T細胞の活性化を抑制するための能力を有する。従って、多くの状況において、ジケトピペラジンを含む、アルブミン、免疫グロブリン、エリスロポエチン、または他のタンパク質もしくはペプチドの組成物を、患者(患畜)に投与することが望ましくないかもしれない。例えば、アルブミンは、しばしば外傷を罹患する患者に投与され、免疫グロブリンは、しばしば感染症または免疫不全症を罹患する患者に投与され、そしてエリスロポエチンは、貧血性の癌を罹患する患者または慢性的体調不良の患者(この患者の免疫系はしばしば障害を生じている。)に投与される。したがって、本発明は、このような組成物から少なくともいくらかの、好ましくは実質的に全てのジケトピペラジンを除去する方法を提供する。ジケトピペラジンを、上記のように(例えば、サイズ排除クロマトグラフィー(例えば、Centriconろ過)、アフィニティークロマトグラフィー(例えば、所望のジケトピペラジンに対する抗体(単数または複数)またはアルブミン、免疫グロブリン、エリスロポエチン、または他のタンパク質もしくはペプチドに対する抗体(単数または複数)がビーズ上に結合されているビーズのカラムを使用する)、陰イオン交換または陽イオン交換)によって除去して、アルブミン、免疫グロブリン、エリスロポエチン、ならびに他のタンパク質およびペプチドの改善された組成物を生成し得る。
【実施例】
【0082】
〔実施例1:ラットの腸からのAsp Ala DKP(DA−DKP)およびGlu Ala DKP(EA−DKP)の吸収〕
肛門括約筋から直腸までのラット腸をわずかに単離し、ウシ血清アルブミンを含む赤血球ベースの灌流液を用いて、張間膜動脈を介して潅流した。消化管から流出した赤血球を、門脈の挿管によって回収し、(再酸化後)再循環した。平衡化の期間後、約1mgのAsp−Alaジケトピペラジン(DA−DKP)または約1,4mgのGlu−Alaジケトピペラジン(EA−DKP)を含む溶液(約1ml)を、十二指腸の内腔に注射によって投与した。
【0083】
投薬後、灌流液の一連のサンプルを、間隔をおいて投与2時間後まで回収した。これらのサンプルを遠心分離機し、両方の環状ペプチドについてタンデム液体クロマトグラフフィー質量分法(tandem liquid chromatography mass spectrometry)によって、血漿をアッセイした。
【0084】
この結果は、灌流のわずか2時間後には、腸内の内腔から循環へ吸収されたDA−DKPおよびEA−DKPの量は、投与された用量のそれぞれ95%および100%(実際には112%)に相当したことを示した。
【0085】
したがって、両方の環状ペプチドは、腸内の内腔から血液へ迅速にかつ効果的に吸収され、腸壁を超えて輸送される間に代謝される証拠はない。よって、これらの潜在的治療薬は経口で与えられ得る。
【0086】
単離され、灌流されたラット肝臓における両方の化合物の、初回通過肝クリアランス(first pass hepatic clearance)の欠如(図示せず)と組み合わせて、変化していないDA−DKPおよびEA−DKPの胃腸管から血液への迅速な吸収は、前全身性クリアランス(pre−systemic clearance)が低いことを示す。したがって、経口的な投薬は理想的な投薬経路である。
【0087】
さらに、単離され、灌流されたラット腎臓の研究は、腎ペプチダーゼによって広範に代謝される多くの直鎖ペプチドとは異なり、両方の環状ジペプチドの腎クリアランスが比較的遅いことを示している。
【0088】
このデータをひとまとめにすると、少ない1日投与量のジケトピペラジンの投薬レジメンは、治療目的に適しているであろうということが示唆される。
【0089】
経口投与後のラットにおける予備的な薬物動態学的データは、両方の環状ジペプチドについて上述したものと一致していた:1,1〜3,7mg/kg体重(DA−DKP)および1,5〜4,8mg/kg体重(EA−DKP)で経口的に投薬した後の、Tmax値が30分〜60分、Cmax値が4〜6μg/ml(DA−DKP)および0,6〜1,1μg/ml(EA−DKP)(Tmaxは、濃度が最大に達する時間であり、Cmaxは、到達する最大濃度である。これらの両方を、得られたデータについてのカーブフィット方程式(curve fit equation)から計算した。)。
【0090】
予備的データは、DA−DKPおよび他のジケトピペラジンが血液脳関門を超えることを示唆している。従って、本発明のDA−DKPおよび他のジケトピペラジンは、多発性硬化症のような神経性障害を処置するために役に立つはずである。
【0091】
〔実施例2:Met−Arg DKP(MR−DKP)を含むヒト初乳画分およびAsp−Ala DKP(DA−DKP)による、ヒトTリンパ球サイトカイン産生のインビトロでの抑制〕
〔A.材料〕
本実施例は、DA−DKP、MR−DKPを含むヒト初乳(HC2626)、およびまたMR−DKPを含むヒト初乳(HCRBL;脱脂した初乳のCentriconろ過によって調製した、3000未満の分子量の成分を含むヒト初乳画分)の低分子量画分がヒトTリンパ球サイトカイン産生を抑制したことを実証する。DA−DKPおよびMR−DKPを、DMI Synthesis,Ltd.,Cardiff,UKから入手した。これら2つのジケトピペラジンは、炎症に対する生理学的応答の間に生成される、天然に存在する小さな化合物である。これらはまた、時折、ヒト静脈内免疫グロブリン(IVIg)、ヒトアルブミン、および他の生物学的調製物中に見出される。
【0092】
〔B.Tリンパ球サイトカイン産生の抑制〕
異なる2つのCD4−陽性ヒトTリンパ球クローンを試験した。細胞株の1つ(TRiPS)を、インフルエンザの予防接種を受けたドナーから単離した。この細胞は、ヘマグルチニンペプチド307−319について特有のものである。他の細胞株(H4#9,25)を、複数の硬化症ドナーの検死用脳組織から単離した。この細胞は、ミエリン塩基性タンパク質(アミノ酸87−99)について特有のものである。Tリンパ球クローンの両方が、(1)特異的抗原およびHLA−DR2−陽性提示細胞、または(2)抗−CD3抗体および抗−CD28抗体、のいずれかを用いるインビトロ刺激後に、インターロイキン8(IL−8)、IL−16、インターフェロン−ガンマ(IFN−γ)、および腫瘍壊死因子α(TNF−α)を生成する。
【0093】
これらのT細胞株を、刺激後18〜20日目に約4×105個の細胞を使用する継代のために刺激した。細胞を、10%ウシ胎仔血清(FBS;American Type Culture Collection(ATCC))を含む冷Iscove’s Modified Dulbecco Minimal Essential Medeium(IMDM,Sigma)中で洗浄し、抗−CD3モノクローナル抗体OKT3(ラット腹水から調製した)の1:500希釈物を含む、1.0mlの冷IMDM培地中で再懸濁した。細胞を抗体とともに氷上で30分間インキュベートし、次いで、FBSを含まない冷培地で洗浄し、50U/mlのヒトIL−2(Xenometrix)を含む培地中で、フィーダー細胞として、4000R照射した約2×106個の正常ヒトドナーの末梢血白血球(PBL)と結合させた。3日目に、FBSおよびIL−2を加えた新鮮なIMDM培地の添加によって、培養物を播いた。培養の日数を、OKT3を用いる刺激の日から測定した。細胞を、7日目(最大増殖時)、典型的には14日目(再刺激に対し最も感受性)および21日目(残りの細胞が老化に近づく)まで使用し得る。
【0094】
活性化実験を、細胞のアリコートを回収し、加温した(37℃)IMDM培地で2度洗浄することにより、実行した。各特異的アッセイのために、2×105個の生存細胞を、特定量の治療添加物(例えば、HC2626、DA−DKP、PMAなど)を含む、全量0.9mlの加温したIMDM培地で、37℃で15分間予めインキュベートした。次いで、活性化刺激としての0,1mlの加温したIMDM中の2×105個のCD3/CD28 Dynabeads(Dynal)のアリコートを添加し、、この培養物を一晩(18時間)37℃でインキュベートした。細胞培養物の上清を、遠心分離によって細胞をペレット化した後に回収した。サイトカイン量を、特異的ELISA(例えば、TNFα、IFNγ、IL−8、IL−16;Endrogen)によってアッセイした。
【0095】
図1〜5に示すように、ヒト初乳(HC2626)は、用量依存的様式で両方のTリンパ球細胞株によるインビトロでのサイトカイン産生を抑制した。また、図1〜5に示すように、HC RBLおよびDA−DKPは、促進サイクルの初期において、用量依存的様式で両方のTリンパ球細胞株によるインビトロでのサイトカイン産生を抑制した。しかしながら、周期後期(14日目以降)におけるHC RBLおよびDA−DKPの効果は、刺激性であった(図4参照)。HC RBLおよびDA−DKPの両方が、MR−DKPを含んでいる(質量分析法によって決定されたように)が、HC2626は、MR−DKP(細胞周期の後期における抑制性効果の原因であり得る)に加えて他の構成成分(★2003年11月25日出願の係属中の出願10/723,247に記載されるようなカゼイン(これは、相対的な脱リン酸化タンパク質であり、よって抗炎症性であり得る)を含む)を含む。したがって、HCRBLおよびHC2626(いずれもMR−DKPを含んでいる)、MR−DKPおよびDA−DKPは、全て刺激周期の初期においてT細胞によるサイトカイン産生を抑制するため、T細胞媒介性疾患および/または自己免疫性疾患(例えば、多発性硬化症)における炎症性サイトカイン応答を下方調節する際に有用であるはずである。これらの結果はまた、HC RBL、HC2626、MR−DKPおよびDA−DKPが、残りのT細胞に影響することなく、抗原特異的T細胞に選択的に影響を与えることを示唆する。
【0096】
〔C.作用機構〕
DA−DKPおよびHC2626(MR−DKPを含む)の作用機構を試験した。試験するために、何も加えず(「Nil」)、CD3/CD28 Dynabeads(CD3/CD28ビーズ)、CD3/CD28ビーズおよび0.5mMDA−DKP、またはCD3/CD28ビーズおよびHC2626の1:500希釈物のいずれかとともに、1×106個の18日目のTRiPS細胞を、37℃で30分間インキュベートした。インキュベーション後、これらの細胞をCell−Lytic Mammalian Cell Extraction Reagent(Sigma)中で溶解した。
【0097】
次いで、細胞抽出物を、2連のHypromatrix Arraysとともに、室温で2時間別々にインキュベートし、続いて、製造者(Hypromatrix)のプロトコルに従って2回洗浄した。Hypromatrix Arraysは、表1に列挙される転写調節因子に対する抗体(Hypromatrixによりカスタム製造された)を用いてブロットされたナイロン膜である。リン酸化チロシン、リン酸化セリン、およびリン酸化スレオニンに特異的な抗体カクテル(Zymed)を添加して、1時間インキュベートした。次いで、ビオチンで標識された抗免疫グロブリン抗体を添加した。抗免疫グロブリンビオチンを洗浄した後、スプレプトアビジン−ペルオキシダーゼを添加し、このアレイを、ペルオキシダーゼ−反応性発光基材を加える前に最終洗浄に供した。
【0098】
結果をフィルムに曝露することにより視覚化し、表2が示すように、0(陰性)または+から++++(陽性)とスコア付けした。表2に示すように、いくつかのサイトカイニン転写因子活性化(ERK1/2)および予め形成されたサイトカイニンの放出が、HC2626(MR−DKPを含む)およびDA−DKPによって抑制された。
【0099】
【表1】
【0100】
【表2】
【0101】
〔実施例3:Gly−Leu DKP(GL−DKP)およびAla−Pro DKP(AP−DKP)による、インビトロでのヒトTリンパ球サイトカイン産生抑制〕
GL−DKPおよびAP−DKP(DMI Synthesis, Ltd., Cardiff, UKから入手した)を、実施例2に記載したように、TRiPS細胞株およびH4#9.25細胞株を使用して試験した。GL−DKPおよびAP−DKPが、用量依存的様式でこれらのTリンパ球細胞株の両方によるインビトロでのサイトカイン産生を抑制することを見出した。この作用機構は、実施例2に記載したように、現在調査中であり、サイトカイン転写調節因子の活性化および予め形成されたサイトカインの放出の両方が影響を受けたようである。
【0102】
〔実施例4:Asp Ala DKP(DA−DKP)およびTyr Glu DKP(YE−DKP)による、インビトロでのヒトTリンパ球サイトカイン産生の抑制〕
正常なヒトリンパ球を、Histopaque(Sigma)を用いて、正常なヒトドナーの末梢血白血球から単離した。次いで、3〜4×105個のリンパ球を、血清を含まない1mlのIMDM培地中で懸濁した。これらの細胞を、1:2000希釈した抗CD3抗体希釈物(Pharmingen, San Diego, CA )25μlを添加し、37℃で18時間インキュベートすることによって刺激した。
【0103】
次いで、3つのDKP製剤のうちの1つおよびデキサメタゾン(最終濃度10−5M)を3連の培養物に添加した。これらの3つのDKP調製物は、以下の通りである:
1.DA−DKP(DMI Synthesis,Ltd.,Cardiff, UK)より入手;培養液中の最終濃度25μg/ml)
2.DKP−ZLB、60℃で4日間加熱した後に質量分析法による測定にて0,5mM DA−DKPを含むことがわかった25%アルブミン調製物(ZLB Bioplasma,AG 3000 Berne 22 Switzerlandより入手)(培養液中の最終濃度14μg/ml DA−DKP)
3.DKP−γ−glob、リン酸緩衝化生理食塩水(pH7.4)中に12mg/mlのγ−グロブリンを含むγ−グロブリン調製物(Sigmaより入手、番号G−4386)を、Centricon 3000フィルタを使用してろ過し、このろ液(3000未満のMWを有する成分を含む)を使用した。このろ液は質量292(これは、Tyr−Glu DKP(YE−DKP)の質量である)を含んでいた。この質量を、陰性電気スプレイ質量分析法と結合した陰イオン交換HPLCによって測定した。このろ液を、培養液中にて1:4の最終希釈で使用した。
【0104】
DKP調製物またはデキサメタゾンを添加した後、培養物を、37℃で18時間インキュベートした。次いで、各培養液に放出されたIL−2、IFNγ、およびTNFαの量を、ELISA(Pierce Biotechnology, Rockford, IL61105)によって測定した。
【0105】
結果を、以下の表3に示す。表に見られるように、3つ全てのサイトカインの放出の減少は、DKP−γ−グロブリンを用いて得られた。CD69+T細胞(CD69は活性化されたT細胞上に見られるマーカーである。)の数を調べるフローサイトメトリーもまた、デキサメタゾンによる約50%の低減と比較した場合に、DKP−γ−globは、T細胞受容体複合体が内部移行するにもかかわらず、CD69+T細胞の数を約90%まで減少させたことを示した。
【0106】
【表3】
【特許請求の範囲】
【請求項1】
T細胞媒介性疾患を処置するか、またはT細胞の活性化を阻害する薬剤の製造における、動物において通常に見出されるペプチドまたはタンパク質の使用であって、
上記ペプチドまたはタンパク質は、上記薬剤が上記ペプチドまたはタンパク質から誘導される少なくとも1つのジケトピペラジンを含むように処理される、方法。
【請求項2】
上記タンパク質がアルブミンである、請求項1に記載の使用。
【請求項3】
上記タンパク質が免疫グロブリンである、請求項1に記載の使用。
【請求項4】
上記タンパク質がエリスロポエチンである、請求項1に記載の使用。
【請求項5】
上記薬剤が経口投与用に調合されている、請求項1〜4のいずれか1項に記載の使用。
【請求項6】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項1〜5のいずれか1項に記載の使用。
【請求項7】
上記薬剤が炎症または炎症性の疾患もしくは障害の処置に使用される、請求項1〜6のいずれか1項に記載の使用。
【請求項8】
上記薬剤が自己免疫疾患の処置に使用される、請求項1〜6のいずれか1項に記載の使用。
【請求項9】
上記自己免疫疾患が関節炎である、請求項8に記載の使用。
【請求項10】
ジケトピペラジンを合成する方法であって、
a.ジケトピペラジンを形成するのに有効な状況下において、タンパク質もしくはペプチドの溶液を加熱すること;または
b.ジケトピペラジンを生成するために有効な状況下おいて、タンパク質もしくはペプチドの溶液を、N末端もしくはC末端の2つのアミノ酸を切断する酵素と接触させることを包含している、方法。
【請求項11】
上記タンパク質がアルブミンである、請求項10に記載の方法。
【請求項12】
上記タンパク質が免疫グロブリンである、請求項10に記載の方法。
【請求項13】
上記タンパク質がエリスロポエチンである、請求項10に記載の方法。
【請求項14】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項10〜13のいずれか1項に記載の方法。
【請求項15】
上記酵素がジペプチジルペプチダーゼである、請求項10〜14のいずれか1項に記載の方法。
【請求項16】
上記酵素がカルボキシペプチダーゼである、請求項10〜14のいずれか1項に記載の方法。
【請求項17】
上記溶液が60℃において4日間にわたって加熱される、請求項10〜14のいずれか1項に記載の方法。
【請求項18】
上記ジケトピペラジンが上記溶液から精製される、請求項10〜17のいずれか1項に記載の方法。
【請求項19】
上記溶液が、サイズ排除クロマトグラフィーによって分画化されて、3000未満の組成物を含んでいる画分を生成する、請求項10〜18のいずれか1項に記載の方法。
【請求項20】
静脈内投与以外の経路による投与のために調合されるタンパク質またはペプチドの薬学的な組成物であって、上記タンパク質またはペプチドから誘導された少なくとも1つのジケトピペラジンを含んでいる、組成物。
【請求項21】
上記タンパク質がアルブミンである、請求項20に記載の組成物。
【請求項22】
上記タンパク質が免疫グロブリンである、請求項20に記載の組成物。
【請求項23】
上記タンパク質がエリスロポエチンである、請求項20に記載の組成物。
【請求項24】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項20〜23のいずれか1項に記載の組成物。
【請求項25】
経口投与用に調合されている、請求項20〜24のいずれか1項に記載の組成物。
【請求項26】
タンパク質またはペプチドから誘導されるジケトピペラジンを含有しているろ過物を含んでいる薬学的な組成物であって、上記ろ過物が、上記ジケトピペラジンを生成するために処理されている上記タンパク質またはペプチドの溶液のサイズ排除クロマトグラフィーによって生成されている、組成物。
【請求項27】
上記ろ過物が3000未満の分子量を有している、請求項26に記載の組成物。
【請求項28】
タンパク質またはペプチドから誘導されるジケトピペラジンを含んでいる薬学的組成物を製造する方法であって、
タンパク質またはペプチドの上記溶液が、あらかじめ加熱によって処理されているタンパク質またはペプチドの静脈内投与用の溶液であり、当該溶液におけるジケトピペラジンの含有量が処理前の水準を超えて上記処理によって増加させられるという条件の下に、
タンパク質またはペプチドの溶液を準備することを包含しており、その後に
a.ジケトピペラジンを形成するのに有効な状況下において、タンパク質もしくはペプチドの溶液を加熱すること;または
b.ジケトピペラジンを生成するために有効な状況下おいて、タンパク質もしくはペプチドの溶液を、N末端もしくはC末端の2つのアミノ酸を切断する酵素と接触させることを包含している、方法。
【請求項29】
上記タンパク質がアルブミンである、請求項28に記載の方法。
【請求項30】
上記タンパク質が免疫グロブリンである、請求項28に記載の方法。
【請求項31】
上記タンパク質がエリスロポエチンである、請求項28に記載の方法。
【請求項32】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項28〜31のいずれか1項に記載の方法。
【請求項33】
上記酵素がジペプチジルペプチダーゼである、請求項28〜32のいずれか1項に記載の方法。
【請求項34】
上記酵素がカルボキシペプチダーゼである、請求項28〜32のいずれか1項に記載の方法。
【請求項35】
上記溶液が60℃において4日間にわたって加熱される、請求項28〜32に記載の方法。
【請求項36】
上記ジケトピペラジンが上記溶液から精製される、請求項28〜35のいずれか1項に記載の方法。
【請求項37】
上記溶液がサイズ排除クロマトグラフィーによって分画化されて、3000未満の分子量の組成物を含有している画分を生成する、請求項28〜36のいずれか1項に記載の方法。
【請求項1】
T細胞媒介性疾患を処置するか、またはT細胞の活性化を阻害する薬剤の製造における、動物において通常に見出されるペプチドまたはタンパク質の使用であって、
上記ペプチドまたはタンパク質は、上記薬剤が上記ペプチドまたはタンパク質から誘導される少なくとも1つのジケトピペラジンを含むように処理される、方法。
【請求項2】
上記タンパク質がアルブミンである、請求項1に記載の使用。
【請求項3】
上記タンパク質が免疫グロブリンである、請求項1に記載の使用。
【請求項4】
上記タンパク質がエリスロポエチンである、請求項1に記載の使用。
【請求項5】
上記薬剤が経口投与用に調合されている、請求項1〜4のいずれか1項に記載の使用。
【請求項6】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項1〜5のいずれか1項に記載の使用。
【請求項7】
上記薬剤が炎症または炎症性の疾患もしくは障害の処置に使用される、請求項1〜6のいずれか1項に記載の使用。
【請求項8】
上記薬剤が自己免疫疾患の処置に使用される、請求項1〜6のいずれか1項に記載の使用。
【請求項9】
上記自己免疫疾患が関節炎である、請求項8に記載の使用。
【請求項10】
ジケトピペラジンを合成する方法であって、
a.ジケトピペラジンを形成するのに有効な状況下において、タンパク質もしくはペプチドの溶液を加熱すること;または
b.ジケトピペラジンを生成するために有効な状況下おいて、タンパク質もしくはペプチドの溶液を、N末端もしくはC末端の2つのアミノ酸を切断する酵素と接触させることを包含している、方法。
【請求項11】
上記タンパク質がアルブミンである、請求項10に記載の方法。
【請求項12】
上記タンパク質が免疫グロブリンである、請求項10に記載の方法。
【請求項13】
上記タンパク質がエリスロポエチンである、請求項10に記載の方法。
【請求項14】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項10〜13のいずれか1項に記載の方法。
【請求項15】
上記酵素がジペプチジルペプチダーゼである、請求項10〜14のいずれか1項に記載の方法。
【請求項16】
上記酵素がカルボキシペプチダーゼである、請求項10〜14のいずれか1項に記載の方法。
【請求項17】
上記溶液が60℃において4日間にわたって加熱される、請求項10〜14のいずれか1項に記載の方法。
【請求項18】
上記ジケトピペラジンが上記溶液から精製される、請求項10〜17のいずれか1項に記載の方法。
【請求項19】
上記溶液が、サイズ排除クロマトグラフィーによって分画化されて、3000未満の組成物を含んでいる画分を生成する、請求項10〜18のいずれか1項に記載の方法。
【請求項20】
静脈内投与以外の経路による投与のために調合されるタンパク質またはペプチドの薬学的な組成物であって、上記タンパク質またはペプチドから誘導された少なくとも1つのジケトピペラジンを含んでいる、組成物。
【請求項21】
上記タンパク質がアルブミンである、請求項20に記載の組成物。
【請求項22】
上記タンパク質が免疫グロブリンである、請求項20に記載の組成物。
【請求項23】
上記タンパク質がエリスロポエチンである、請求項20に記載の組成物。
【請求項24】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項20〜23のいずれか1項に記載の組成物。
【請求項25】
経口投与用に調合されている、請求項20〜24のいずれか1項に記載の組成物。
【請求項26】
タンパク質またはペプチドから誘導されるジケトピペラジンを含有しているろ過物を含んでいる薬学的な組成物であって、上記ろ過物が、上記ジケトピペラジンを生成するために処理されている上記タンパク質またはペプチドの溶液のサイズ排除クロマトグラフィーによって生成されている、組成物。
【請求項27】
上記ろ過物が3000未満の分子量を有している、請求項26に記載の組成物。
【請求項28】
タンパク質またはペプチドから誘導されるジケトピペラジンを含んでいる薬学的組成物を製造する方法であって、
タンパク質またはペプチドの上記溶液が、あらかじめ加熱によって処理されているタンパク質またはペプチドの静脈内投与用の溶液であり、当該溶液におけるジケトピペラジンの含有量が処理前の水準を超えて上記処理によって増加させられるという条件の下に、
タンパク質またはペプチドの溶液を準備することを包含しており、その後に
a.ジケトピペラジンを形成するのに有効な状況下において、タンパク質もしくはペプチドの溶液を加熱すること;または
b.ジケトピペラジンを生成するために有効な状況下おいて、タンパク質もしくはペプチドの溶液を、N末端もしくはC末端の2つのアミノ酸を切断する酵素と接触させることを包含している、方法。
【請求項29】
上記タンパク質がアルブミンである、請求項28に記載の方法。
【請求項30】
上記タンパク質が免疫グロブリンである、請求項28に記載の方法。
【請求項31】
上記タンパク質がエリスロポエチンである、請求項28に記載の方法。
【請求項32】
上記タンパク質またはペプチドがヒトのタンパク質またはペプチドである、請求項28〜31のいずれか1項に記載の方法。
【請求項33】
上記酵素がジペプチジルペプチダーゼである、請求項28〜32のいずれか1項に記載の方法。
【請求項34】
上記酵素がカルボキシペプチダーゼである、請求項28〜32のいずれか1項に記載の方法。
【請求項35】
上記溶液が60℃において4日間にわたって加熱される、請求項28〜32に記載の方法。
【請求項36】
上記ジケトピペラジンが上記溶液から精製される、請求項28〜35のいずれか1項に記載の方法。
【請求項37】
上記溶液がサイズ排除クロマトグラフィーによって分画化されて、3000未満の分子量の組成物を含有している画分を生成する、請求項28〜36のいずれか1項に記載の方法。
【図1】

【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2011−102309(P2011−102309A)
【公開日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願番号】特願2010−292763(P2010−292763)
【出願日】平成22年12月28日(2010.12.28)
【分割の表示】特願2006−533125(P2006−533125)の分割
【原出願日】平成16年5月14日(2004.5.14)
【出願人】(502113644)ディーエムアイ バイオサイエンシズ インコーポレイテッド (14)
【氏名又は名称原語表記】DMI BIOSCIENCES,INC.
【Fターム(参考)】
【公開日】平成23年5月26日(2011.5.26)
【国際特許分類】
【出願日】平成22年12月28日(2010.12.28)
【分割の表示】特願2006−533125(P2006−533125)の分割
【原出願日】平成16年5月14日(2004.5.14)
【出願人】(502113644)ディーエムアイ バイオサイエンシズ インコーポレイテッド (14)
【氏名又は名称原語表記】DMI BIOSCIENCES,INC.
【Fターム(参考)】
[ Back to top ]