
T細胞活性化
【課題】T細胞をサイトカインとの組み合わせとを接触させることを包含する、抗原非依存的なT細胞の活性化の方法が提供。
【解決手段】T細胞をサイトカインの組み合わせと接触させることを包含する、T細胞の抗原非依存的活性化の方法であって、一実施形態において、T細胞が少なくともi)インターロイキン-2;ii)インターロイキン-6;およびiii) 腫瘍壊死因子αの2つと接触されるか、あるいは、それらの機能的に等価なフラグメントと接触される、方法。
【解決手段】T細胞をサイトカインの組み合わせと接触させることを包含する、T細胞の抗原非依存的活性化の方法であって、一実施形態において、T細胞が少なくともi)インターロイキン-2;ii)インターロイキン-6;およびiii) 腫瘍壊死因子αの2つと接触されるか、あるいは、それらの機能的に等価なフラグメントと接触される、方法。
【発明の詳細な説明】
【技術分野】
【0001】
発明の分野
本発明はT細胞の活性化のための抗原非依存的方法に関する。本発明はまたT細胞培養におけるリンホカイン生産を増大させるための方法、および疾病の処置における治療用途を有するインビボの特定部位における免疫応答を増大させる方法に関する。
【背景技術】
【0002】
発明の背景
T細胞は、このような応答として免疫応答に関わり、そして、主に細胞性免疫に関わっているウイルス感染した細胞、菌類、寄生虫、および外来組織に対する防御が挙げられる。
【0003】
簡潔には、T細胞は抗原提示マクロファージに結合することによって活性化される。しかし、T細胞レセプターはマクロファージの表面に提示された抗原および主要組織適合性抗原複合体(MHC)タンパク質と特異的に複合体を形成するに違いない。
【0004】
この結合は、マクロファージにインターロイキン-1、ポリペプチド成長因子の放出を
誘導し、これは結合T細胞を刺激して増殖および分化を起こさせる。この増殖および分化は、T細胞自己刺激性分泌インターロイキン-2によって増強される。このT細胞は多数
の異なった表現型に分化し得る。この表現型としては、例えば、抗原提示宿主細胞を特異的に標的化しそしてその細胞を溶解し得る細胞傷害性T細胞、細胞傷害性T細胞を活性化することおよびB細胞と協同して抗体を産生させることに関わるヘルパーT細胞、およびそれらと同種の抗原に再度遭遇したとき非記憶T細胞よりも速い速度で増殖する記憶T細胞が挙げられる。
【0005】
T細胞の活性化が免疫応答において重要な工程であることは当業者には明らかである。T細胞の活性化を操作することにより、有用な免疫産物を得ることおよびより効率的な治療技術を開発することが可能である。
【発明の開示】
【発明が解決しようとする課題】
【0006】
以前は、T細胞活性化を達成するために、抗原およびMHCタンパク質を提示するマクロ
ファージが必要とされた。多数の問題および欠点は、このことと関連し、主な欠点は抗原に対して特異的なT細胞のみが活性化されることである。抗原に特異的ではない他のT細胞は不活性なままである。もし所望の抗原を得ることが困難または作用が運任せ(hazardous)である場合、他の問題が生じ得る。さらに、もし抗原が活性化を達成するために細胞
培養物に使用され、そしてそれは容易に除去されないならば、混入の問題が生じ得る。
【0007】
同じ問題がインビボで生じ、そして個体に抗原性物質が感染することは明らかに望ましくない。
【0008】
抗原非依存的なT細胞の活性化を達成することにより、抗原を単離する必要も、そしてマクロファージの表面に抗原を提示する必要もなく、T細胞集団を活性化することが可能である。
【0009】
インターロイキン-2は強力なTリンパ球増殖エンハンサーであることが知られ、そし
てインターロイキン-2のアジュバントとしての使用が記載されている。この役割におい
て、インターロイキン-2は既に活性化されたTリンパ球の集団の増殖賦活剤として機能
すると考えられた。しかし、インターロイキン-2が(他のサイトカインとの組み合わせにおいて)、抗原非依存的な様式でTリンパ球を特異的に活性化し得ることは知られていな
かった。
【課題を解決するための手段】
【0010】
1.T細胞をサイトカインの組み合わせと接触させることを包含する、T細胞の抗原非依存的活性化の方法。
2.前記T細胞が少なくとも以下の2つと接触される、項目1に記載の方法:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいは、それらの機能的に等価なフラグメント。
3.前記T細胞がナイーブT細胞および/または記憶休止T細胞である、項目1または2
に記載の方法。
4.前記T細胞がナイーブCD45RA+細胞および/または記憶休止CD45RO+細胞である、項目
1から3のいずれか1項に記載の方法。
5.前記インターロイキン-2の濃度が100から400U/mlであり、前記インターロイキン-6の濃度が400から600U/mlであり、そして腫瘍壊死因子αの濃度が15から35ng/mlである、
前述の項目のいずれか1項に記載の方法。
6.前記インターロイキン-2の濃度が200から300U/mlであり、前記インターロイキン-6の濃度が約500U/mlであり、そして腫瘍壊死因子αの濃度が約25ng/mlである、前述の項目のいずれか1項に記載の方法。
7.前記T細胞がインビトロで活性化される、前述の項目のいずれか1項に記載の方法。8.T細胞培養物から増加したリンホカイン産生を得る方法であって、項目7に記載の方法を用いるT細胞の活性化を包含する方法。
9.前記T細胞がインビボで活性化される、項目1から6のいずれか1項に記載の方法。10.前記T細胞のインビボでの活性化が、免疫応答の増強に導く、項目9に記載の方法。
11.項目9または10に記載の方法を用いる、ヒトまたは動物被験体T細胞の活性化を包含する治療法。
発明の要旨
本発明に従って、T細胞をサイトカインとの組み合わせとを接触させることを包含する、抗原非依存的なT細胞の活性化の方法が提供される。
【0011】
好ましくは、T細胞は以下の少なくとも2つと接触される:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいはそれらの機能的に等価なフラグメント。
【0012】
T細胞は、ナイーブ(naive)T細胞および/または記憶休止T細胞、最も適切にはナイーブCD45RA+細胞および/または記憶休止CD45RO+細胞であり得る。
【0013】
適切には、インターロイキン-2の濃度は100U/mlから400U/mlであり、インターロイキ
ン-6の濃度は400U/mlから600U/mlであり、そして腫瘍壊死因子αの濃度は15ng/mlから35ng/mlである。より好ましくは、インターロイキン-2の濃度は200U/mlから300U/ml、インターロイキン-6の濃度は約500U/mlそして腫瘍壊死因子αの濃度は約25ng/mlである。
【0014】
T細胞は、インビトロで、例えば、T細胞培養物から増加したリンホカイン産生を得る
ための方法において活性化され得、そしてそれは本発明に従うT細胞の活性化を包含する。
【0015】
T細胞は、インビボで活性化されて、免疫応答の増強に導き得、そしてそれは本発明に従う方法を用いるヒトまたは動物被験体T細胞の活性化を包含する治療法に使用され得る。
【0016】
本発明のこの局面において、サイトカインの組合わせは、T細胞応答を増強しそしてそれにより免疫応答を増強するアジュバントとして作用する。
【0017】
T細胞は、活性化され、インターロイキン、インターフェロン、およびコロニー刺激因子のような、細胞媒介性免疫応答に有用な所望のリンホカインを、抗原依存的活性化と関連した問題なしに、産生し得る。
【0018】
さらに、抗原を使用することなく、特定の免疫学的に興味のある領域において、単離されたT細胞の活性化およびエフェクターT細胞の補充(recruitment)を達成することが可
能である。それゆえ、これはHIVおよび肝炎のような多数の疾病および感染のインビボで
の処置に非常に有用である。
【0019】
本発明は、単に1つの特定の刺激抗原に特異的ではなく、「バイスタンダー(bystander)」T細胞を活性化する利点を有し、それゆえ、より大きな免疫応答が生じ、より多いリ
ンホカインの産生、および引き続くB細胞によるより多い免疫グロブリンの産生に導く。
【0020】
本発明の別の利点は、記憶T細胞の末梢血プールの維持である。なぜなら、記憶T細胞は、クローンサイズを維持するために特異的抗原刺激を必要とせずに、拡大(増殖)され得るためである。またナイーブT細胞レパートリーが維持され得る。なぜなら、本発明は、抗原刺激の場合とは異なり、それらを記憶表現型に切り換えずにナイーブT細胞の増殖を可能にするためである。
【0021】
本発明のさらなる局面に従って、以下の2つ以上を含み、必要に応じて1つ以上の薬学的に受容可能な賦形剤と組み合わされる、薬学的組成物が提供される:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいはそれらの機能的に等価なフラグメント。
【0022】
薬学的組成物は、それ自身T細胞の治療学的活性化に有用であり得、またはワクチンのようなさらなる治療剤と共に投与され得る。投与は同時または断続的であり得る。
【0023】
本発明に従って、以下の2つ以上をコードする遺伝子を有するベクターを投与する工程を包含する遺伝子治療の方法を提供する:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいは、それらの機能的に等価なフラグメント。
【0024】
そのような適切なベクターは当該分野1で周知である。
【0025】
本発明のさらなる局面に従って、以下
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいは、それらの機能的に等価なフラグメントの1つ以上をコードする遺伝子を有するベクター、および1つ以上の
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
のタンパク質あるいはそれらの機能的に等価なフラグメントの同時投与(coadministration)を包含する、組合わせ治療方法を提供する。
【0026】
特異的なT細胞タイプのそのような維持は、T細胞培養物と共に作用させる場合、非常に有利である。
【0027】
その他多くの使用および利点が本発明から理解され得、そしてそのような使用および利点は当業者に明らかである。
【発明の効果】
【0028】
本発明に従って、T細胞をサイトカインとの組み合わせとを接触させることを包含する、抗原非依存的なT細胞の活性化の方法が提供された。
【発明を実施するための最良の形態】
【0029】
実施態様の詳細な説明
材料および方法
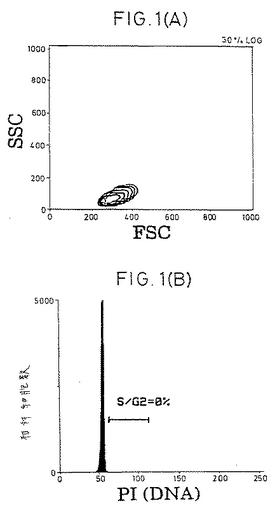
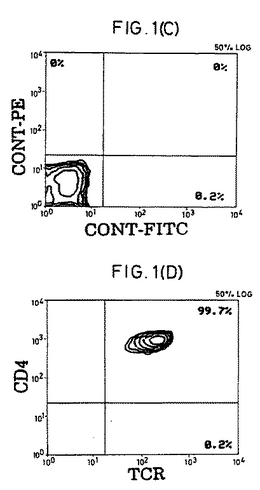
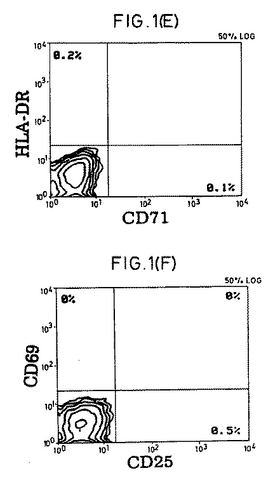
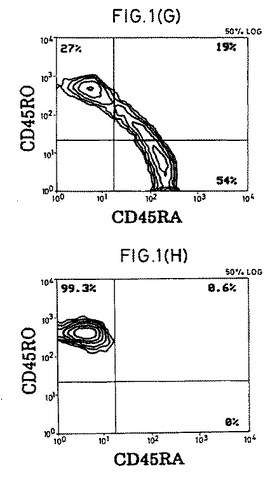
休止T細胞の単離.健常な提供者のバフィーコート由来PBMCをフィコール-ハイパック(Pharmacia製)分離後、大部分のマクロファージをプラスチック接着によって除去した。純粋な休止CD4+T細胞集団を得るために、細胞を、以下のものに対するmAbのカクテルと共にインキュベートした。以下とは、HLA-DR(L-243;American Type Culture Collection [ATCC],Rockville,MD)、CD19(4GT)、CD16(B73.1)、CD56(MY31)、CD57(HNK-1,ATCC)、CD8(OKT8,ATCC)、CD11b(OKM-1,ATCC)、CD14(Mφ-P9)、TCR-c/δ(B1,G.De Libero,ZLF Basel,Switzerlandの贈与)、CD25(2A3)、CD69(L78)、およびCD71(LO1.1)である。氷上での30分間のインキュベーションの後、細胞を2回洗浄し、そして1:4の標的/ビーズ比で、ヤギ抗マウスIgGおよびラット抗マウスIgMを結合させた磁気ビーズ(Dynabeads;Dynal,Oslo,Norway)と共にインキュベートした。インキュベーション30分後に、ビーズ結合細胞を希土類磁石(Advanced Magnetics,Inc.,Cambridge,MA)を用いて除去した。残っている細胞は、漸増標的/ビーズ比(1:10から1:100)でビーズと共にさらに4回インキュベーションすることで、さらに精製された。最終的な集団は、その集団の>99.3%がTCRα/β+(WT/31)およびCD4+(Leu3a)であり(FACScan(R)フローサイトメーター(Becton Dickinson & Co.,Mountain View,CA)を用いる免疫蛍光分析によって決定された)、そして以下の特徴を満たした場合、休止CD4+T細胞源として使用した。以下の特徴とは、(a)FACS(R)散乱が小サイズであること;(b)活性化マーカー(CD69、CD71、MHC-DR、およびIL-2レセプターp55鎖(CD25))がFACS(R)検出可能レベルで存在しないこと;(c)細胞周期のSおよびG2/M期の細胞が存在しないこと;および(d)IL-2に曝露した場合に有意な[3H]チミジンの取り込みがないことである。いくつかの実験においては、休止細胞を、CD45RO-(mABUCH1-1を添加する)またはCD45RA-(mAB L48を添加する)としてさらにネガティブに選別した。他に示さない限り、全てのmAbはBecton Dickinson & Co.製である。
【0030】
上清の調製.破傷風トキソイド(TT)特異的クローン由来T細胞(5×103/ml)を、TT(3μg/ml)(Biocine Sclavo,Siena,Italy)存在下または非存在下でプレパルスした自己マクロファージ(2.5×105/ml)と共に培養した。16時間後、上清を採集し、そして0.2μmフィルターで濾過した。培養培地は5%ヒト血清または血漿を用い、以前に(3)に記載された。効果的な上清を、5%ヒト血清(フィレンツェ血液バンク製)または無血清培地(HL-1:Ventrex,Portland,OR)のいずれかを有する培地を用いて調製した。同様の結果が、6つの異な
った健常個体のPBMC由来の休止T細胞、および4人の異なったヒト由来の異なった特異性(精製タンパク質誘導体[PPD]または百日咳毒素)を有する活性化CD4+T細胞由来の上清で
得られた(図2を参照のこと、およびデータは示されていない)。
【0031】
細胞周期分析.これは抗CD4 mAb(FITC標識)染色と組み合わせて、ヨウ化プロピジウム
を用いて、(4)に記載のように実施した。分析はFACScan(R)LysisIIソフトウエアおよび
二重識別プログラム(Becton Dickinson & Co)を用いて実施した。
【0032】
B細胞の精製.PBMC由来B細胞をFITC標識抗CD19mAbで染色し、そしてFACStar(R)(Becton Dickinson & Co)を用いたポジティブ選別によって精製した。精製度は、抗CD20およ
び抗Igを用いた染色によって決定したように>98%であった。
【0033】
ヘルパーアッセイ.非同族ヘルパーアッセイを以前に記載(5)のように実施した。簡潔には、精製自己PBMC由来B細胞(2×103/ウエル)を、記載したような(図3を参照のこと)
サイトカイン組合わせの存在下あるいは抗CD3でコートしたプレート上で、CD4+CD45RO+休止T細胞(3×104/ウエル)と共に12日間共培養した。B細胞分化におけるサイトカインの
影響を回避するため、プレートは4日培養後に洗浄し、そしてサイトカイン組合わせをIL-2単独と置換した。上清中のIgはELISAによって測定した(5)。
【0034】
上清による休止T細胞の活性化.休止T細胞を、96ウエルの平底プレート(5×104/ウエル)中で、抗原、培地、またはT細胞上清中に見出される濃度に対応する濃度(すなわち200〜300U/ml)のrIL-2(Cetus Corp.,Emeryville,CA)で、プレパルスした自己マクロファージと共に培養したT細胞クローン由来の上清(50%vol/vol)と、共に培養した。活性化は種々の時点で、[3H]チミジン取り込みのCD69およびCD25の発現として測定した。いくつかの実験において、休止CD45RO+またはCD45RA+T細胞の[3H]チミジン取り込みは、異なった濃度のIL-2と1μg/ml LPS(Difco,Detroit,MI)またはLPS活性化マクロファージ由来の上清(50%vol/vol)のいずれかとの存在下で測定した。活性化マクロファージ上清の調製には、5×105マクロファージを1μg/mlLPSで刺激した(6〜8時間) 。[3H]チミジン取り込み実験は、(5)に記載のように実施した。結果は3組ウエルの平均を表し、そしてSDは常に15%であった。
【0035】
組換えサイトカインによる休止T細胞の活性化.96ウエル平底マイクロプレート中で、休止T細胞(5×104/ウエル)を、以下のrIL-2(200〜300U/ml)、rIL-6(500U/ml;Ciba-Geigy,Basel,Switzerland;IL-6ユニットはB9アッセイで決定された)、TNF-α(25ng/ml;Genzyme Corp.,Cambridge,MA)、およびLPS刺激マクロファージの上清(50%vol/vol)と共に培養した。チミジンの取り込みおよびCD69発現を、図2に記載のように測定した。IL-2およびTNF-αと組み合わせたIL-1b(100ng/mlまで、Biocine Sclavo Siena,Italy)は全く活性を有さなかった(データは示していない)。2つの異なったソース由来の組換え型サイトカインを用い同様の結果を得た。サイトカインの最適濃度は、予備的な用量-応答実験で確立した。
【0036】
PCR補助mRNA増幅(PCR-assisted mRNA Amplification).精製休止CD4+CD45RO+T細胞をTNF-α+IL-6+IL-2、またはIL-2単独と共に培養した。全RNAは、培養60〜100時間後に、RNAzol*B(Biotecx Laboratories,Houston,TX)によって、5×105個の細胞から単離した。cDNAは、(5)に記載のようにマウス逆転写酵素を用いて合成された。β−アクチン、IL-4、およびIFN-c特異的プライマーペアは、Clontech(Palo Alto,CA)から購入した。PCRは(5)に記載のように実施した。
【0037】
限界希釈分析.CD45RO+休止T細胞を、IL-2単独(300U/ml)あるいはTNF-α(25ng/ml)お
よびIL-6(500U/ml)の組合わせと共に、精製自己照射(2,500ラド)マクロファージ(3×103/ウエル)、抗DRmAb(L243,20μg/ml)の存在下、20μl容でTerasakiプレート(64ウエル/1条
件)に異なる数で播種した。14日目に、培養物の増殖を肉眼で調べた。ランダムに選択し
た増殖ウエルを抗CD4抗体および抗TCR-α/β抗体でポジティブに染色した。頻度分析は最小自乗法(6)によって行った。
結果および考察
本研究の重要な点は、IL-2にのみ応答する活性化T細胞が存在しない休止集団を使用することである。発明者らは休止CD4+T細胞と作用することを選択した。なぜなら、休止表現型を有するいくらかのCD8+またはc/δT細胞を有する変形で、それらの細胞は、p55鎖
欠如下でIL-2レセプターp75鎖を発現しないためである(7)。これはIL-2に対する不要な
増殖応答を担い得(8)、かつ発明者らが選別するための良好な抗体を有していない。それゆえ発明者らは、PBMCから高度に精製された休止CD4+T細胞を得るために、多工程の徹底的な精製を実施した(図1)。予備実験において、休止CD4+T細胞を、抗原でパルスした(Ag-pulsed)マクロファージで活性化されたCD4+T細胞クローン由来の上清と共に培養した
。図2の代表的実験は、休止CD4+T細胞がIL-2によってではなく、上清によって活性化され、CD69(9)(図2A)およびIL-2レセプターp55鎖(図2B)を発現すること、そして[3H]チミ
ジンを取り込むこと(図2C)を示す。
【0038】
活性化上清は、2つの細胞タイプの共培養(coculture)によって生成されるので、発明
者らは、T細胞およびAPCによって産生された可溶性因子の相対的寄与度の決定を試みた
。この実験のために、休止CD4+T細胞を、CD45RO+(記憶)およびCD45RA+(ナイーブ)亜集団(10)としてさらに精製した。なぜならそれらはTCR媒介性活性化について既に報告されて
いるように異なった活性化要求性を有し得るためである(11、12)。図2Dは、LPS活性化マ
クロファージ由来の上清単独ではIL-2単独と同様に活性を有さなかったこと、ところがIL-2と組合わせたマクロファージ上清はCD45RA+およびCD45RO+休止T細胞の両方においてチミジン取り込みを誘導したことを示す。これらの結果は、IL-2とAPCによって産生された
可溶性因子とが休止T細胞の活性化に必要とされることを示す。
【0039】
APC由来因子を同定するために、発明者らはマクロファージによって産生され、T細胞
に対し共刺激活性を有することが公知の組換え型サイトカイン、すなわち、IL-1β、IL-6、およびTNF-α(13〜15)の効果を試験した。IL-2の非存在下では、これらのサイトカインの可能な組合わせ全てが、広範な濃度範囲にわたり、活性を示さなかった(データは示し
ていない)。図3Aは、TNF-αがIL-2との組合わせにおいて休止CD45RO+T細胞にCD69の発現およびチミジンの取り込みを誘導したこと、一方IL-6はIL-2と組合わせても大して効果のないことを示す。注目すべきことに、TNF-αおよびIL-6は、IL-2との組合わせにおいて、より強い活性化に導く相乗効果を有した。IL-2、IL-6、およびTNFαの同様の効果がまた
、CD45RA+休止T細胞において観察されたが(図3B)、この場合、3つのサイトカイン全て
が活性化の誘導に要求された。さらに、図3Cの細胞周期分析は、培養7日目に、CD45RO+
およびCD45RA+T細胞両方の8%が、細胞周期のSまたはG2/M期にあることを示す。活性
化マーカーの発現、チミジン取り込み、あるいは細胞周期へのエントリーとして測定されたサイトカインの活性化は、DR、CD4、またはCD3に特異的なmAbによって決して阻害され
なかった(データは示していない)。それゆえ、TCRシグナリングはこのタイプの活性化に
関与しないことが確認された。
【0040】
サイトカインによって活性化されたCD45RA+T細胞は、TCR連結(engagement)後数日内に起こると報告された(16)ような、表現型をCD45ROに切り換えないことを、本発明者らが観察したことは、注目すべき興味ある点である。IL-2、TNF-α、およびIL-6の組合わせに
よって活性化されたCD45RA+T細胞を、培養23日まで3日おきに抗CD45RA抗体および抗CD45RO抗体を用いて二重染色した。本発明者らは、どの時点においても、シングルポジティ
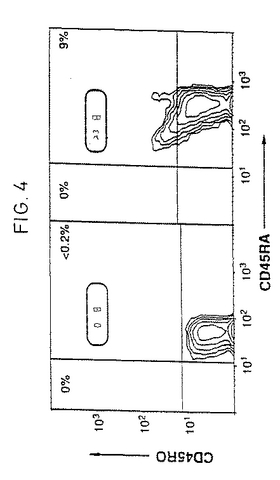
ブなCD45RA+細胞を見出すことはなく、数%のダブルポジティブCD45RA+high/CD45RO+dullを見出すだけであった。実際に、図4は、ナイーブT細胞が、ほとんどの細胞が芽様(blastic)であり、CD69を発現している(データは示していない)とき、サイトカイン活性化23
日後でさえ、主にCD45RA+であることを示す。抗CD3で活性化された同細胞は、数日でCD45RO+CD45RA-表現型に切り換わった(データは示していない)。
【0041】
本発明者らは、次に休止Tリンパ球がサイトカインによって活性化され得、エフェクター機能を示すか否かを問うた。本発明者らはリンホカインについてPCR補助mRNA増幅を実
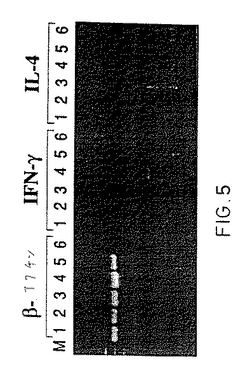
施した。図5は、IFN-cおよびIL-4のmRNA両方がIL-2単独ではなく、IL-2、TNF-α、およ
びIL-6と共に培養したCD45RO+T細胞によって発現されることを示す。さらに、サイトカ
イン組合わせによって活性化されたCD45RO+T細胞は、B細胞をIgを産生することを補助
することにおいて、抗CD3刺激T細胞と同程度効果的である(表1)。
表1.サイトカインによって活性化された休止CD45RO+T細胞はB細胞への補助を提供し得る
【0042】
【表1】

【0043】
T細胞のB細胞への補助が自己反応性細胞の活性化のためである可能性を除外するために、ヘルパーアッセイの終わりに、B細胞を選別によって除去し、そしてCD4+T細胞を自己精製B細胞またはマクロファージに対する増殖において試験した。本発明者らは任意の自己反応性増殖を見出さなかった(データは示していない)。
【0044】
サイトカインも抗CD3もCD45RA+T細胞にIFN-cの産生(<1IU/ml)およびB細胞の補助を誘導しなかった(データは示していない)。それゆえ、本発明者らは、TCR媒介活性化に類
似して(17)、サイトカインは、休止CD45RO+T細胞を活性化し増殖およびエフェクター機
能を表示させるのに対し、CD45RA+T細胞を補充しIg産生を補助しないと結論する。
【0045】
サイトカインによって増殖するように刺激され得る記憶表現型を有する休止T細胞の頻度を評価するため、本発明者らは限界希釈実験を実施した。CD45RO+CD4+休止T細胞を、
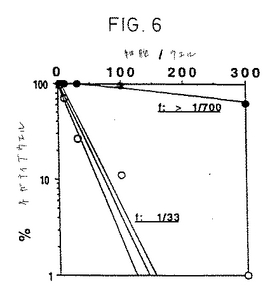
自己反応性応答を防ぐため自己照射マクロファージおよび抗DR抗体の存在下で、IL-2単独あるいはTNF-αおよびIL-6との組合わせと共に培養した。図6は33の休止CD45RO+CD4+T
細胞のうち1つがIL-2、TNF-α、およびIL-6に応答して目に見えるクローンに増殖したことを示す。現在のところ、どうして細胞の3%のみがサイトカインに応答して増殖したの
かは不明である。増殖した細胞は、休止T細胞のサブセットであったかあるいは成熟化/
活性化の異なる時期にあった可能性があった。多くの細胞(約20%)がサイトカインに応答し、そして活性化マーカーを発現する可能性がある。これらの細胞のいくらかはエフェクター機能を表し、そして少数(3%)のみがインビトロで増殖し得、目に見えるサイズのクローンになる。
【0046】
TNF-αおよびIL-6は共にT細胞上のIL-2Rをアップレギュレートすることが示されてき
た(15、18)。これは、本サイトカイン組合わせによる休止T細胞の活性化のための1つの可能な機構であり得た。しかし、TNF-αおよびIL-6と共に1〜3日間培養し、そして洗浄しIL-2と共にさらに4〜5日培養した休止T細胞は、FACS(R)で検出可能なレベルのIL-2R(p55)を示さず(データは示していない)、一方IL-2Rは、培養の開始から、TNF-α、IL-6、およびIL-2と共に培養した約20%の同細胞上に発現された。しかし、この実験は、FACS(R)感
度以下の低いレベルのIL-2Rが発現し機能的に関連した可能性を除外してはいない。実際
、IL-2がTNF-αまたはIL-6によるIL-2Rの誘導に必要とされることが報告された(19)。さ
らに、IL-2はそれ自身のレセプターの発現を増強する(20)のみならず、TNF-αレセプターのアップレギュレート(21)もまた行っている。サイトカインによる休止T細胞の活性化の機構の解明にはさらなる生化学的分析および分子的分析が必要である。
【0047】
この新規な抗原非依存的T細胞活性化経路はインビボにおいて、2つの重要な役割、すなわち免疫応答部位におけるエフェクターT細胞の補充および記憶T細胞の末梢プールの維持を果たし得る。休止T細胞は、抗原特異的応答の部位で、特定のT細胞およびマクロファージによって産生されたサイトカインにより活性化されて、増殖および他のリンホカインを分泌し、そしてそのリンホカインが応答をさらに増幅し得るという1つのシナリオが描かれ得る。実際、サイトカイン組合わせに応答する休止CD45RO+T細胞の頻度は、い
ずれの公知の抗原により感作されたT細胞の通常の頻度よりも明確に高い。
【0048】
記憶は、抗原の持続、再発的な感染、あるいは環境抗原の交差反応性により、繰り返しの断続的な刺激を必要とする短命の細胞からなる長命のクローンによって保有され得ることが仮定されている(22〜24)。本発明者らの結果に照らしてみれば、記憶表現型(CD45RO+)を有する休止T細胞が無関係の抗原に対する応答の間に分泌されたサイトカインによっ
て拡大され得るため、記憶T細胞はそれらのクローンサイズを維持するのに抗原刺激を必要としなくてもよいと推測されがちである。他方、サイトカインは記憶表現型への切り換えなしにナイーブ細胞の増殖を誘導し得、従ってナイーブ(CD45RA+)T細胞レパートリー
の維持を補助し得る。
【0049】
本発明は例示のみによって上述され、発明の範囲および意図内で改変がなされ得ることが理解される。
【0050】
【数1−1】
【0051】
【数1−2】
【0052】
【数1−3】
【0053】
【数1−4】
【図面の簡単な説明】
【0054】
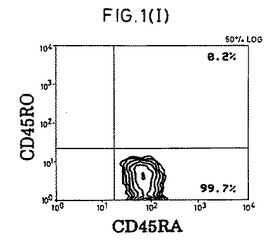
【図1−1】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−2】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−3】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−4】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−5】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
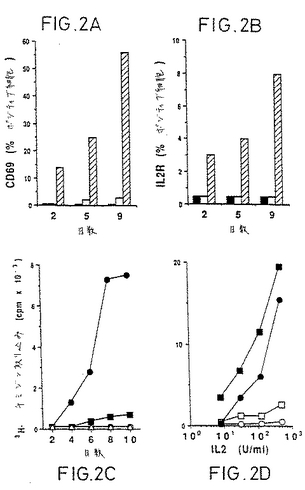
【図2】可溶性因子による休止CD4+T細胞の活性化。(AおよびB)抗原(Ag)(斜線棒)または培地(黒棒)、またはrIL-2(白棒)でプレパルスした自己マクロファージと共に培養したT細胞クローン由来の上清で培養した休止T細胞上の活性化マーカーの発現。CD69またはCD25の発現を抗CD4を用いる二重染色で分析した。(C)培地のみ(三角)、rIL-2(四角)、あるいは抗原(黒丸)または培地(白丸)でプレパルスしたマクロファージと共に培養したT細胞クローン由来の上清と共に培養した、AおよびBの同細胞の[3H]チミジンの取り込み。(D)種々の濃度のIL-2+1μg/mlLPS(白記号)、または種々の濃度のIL-2とLPS活性化マクロファージ由来上清(黒記号)存在下での、休止CD45RO+T細胞(四角)、休止CD45RA+T細胞(丸)の[3H]チミジンの取り込み。
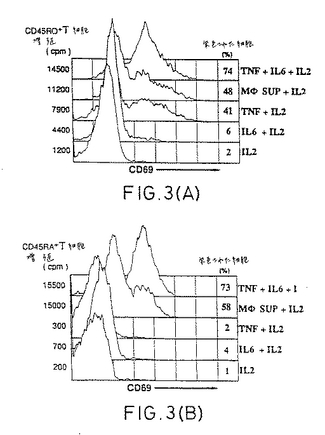
【図3−1】IL-2、TNF-α、およびIL-6の組合わせは、休止T細胞を活性化する。CD45RO+(A)あるいはCD45RA+(B)休止T細胞を、以下の種々の組合わせと共に8日間培養した:rIL-2、rIL-6、TNF-αおよびLPS刺激マクロファージ由来の上清。チミジンの取り込みおよびCD69発現を図1に記載のように測定した。(C)IL-2単独(白記号)あるいはTNF-αおよびIL-6と組合わせた(黒記号)存在下での、休止CD45RO+T細胞(四角)あるいは休止CD45RA+T細胞(丸)の細胞周期分析。
【図3−2】IL-2、TNF-α、およびIL-6の組合わせは、休止T細胞を活性化する。CD45RO+(A)あるいはCD45RA+(B)休止T細胞を、以下の種々の組合わせと共に8日間培養した:rIL-2、rIL-6、TNF-αおよびLPS刺激マクロファージ由来の上清。チミジンの取り込みおよびCD69発現を図1に記載のように測定した。(C)IL-2単独(白記号)あるいはTNF-αおよびIL-6と組合わせた(黒記号)存在下での、休止CD45RO+T細胞(四角)あるいは休止CD45RA+T細胞(丸)の細胞周期分析。
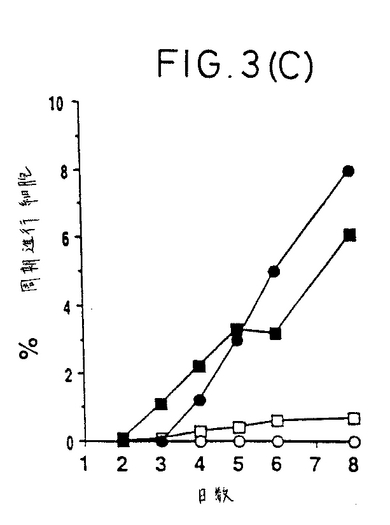
【図4】サイトカインにより活性化されたCD45RA+T細胞はそれらの表現型をCD45ROに切り換えない。CD45RA+T細胞をIL-2、TNF-α、およびIL-6の組合わせで活性化し、そして23日後、抗CD45RA-FITC抗体および抗CD45RO-PE抗体で二重染色した。
【図5】サイトカイン活性化T細胞によるIFNcおよびIL-4mRNAの発現。精製CD4+ CD45RO+休止T細胞を、材料および方法に記載のように、IL-2単独と共に60時間(レーン1)および100時間(レーン3)、あるいはIL-2、TNF-α、およびIL-6と共に60時間(レーン2)および100時間(レーン4)培養する。ポジティブテンプレート(レーン5);ネガティブコントロール(レーン6)。
【図6】サイトカイン組合わせに応答して増殖する休止T細胞の頻度。CD45RO+休止T細胞を、IL-2単独(黒丸)あるいはTNF-αおよびIL-6の組合わせ(白丸)と共に精製自己マクロファージ、抗DRmAb存在下で播種した。95%信頼限界(破線)。
【技術分野】
【0001】
発明の分野
本発明はT細胞の活性化のための抗原非依存的方法に関する。本発明はまたT細胞培養におけるリンホカイン生産を増大させるための方法、および疾病の処置における治療用途を有するインビボの特定部位における免疫応答を増大させる方法に関する。
【背景技術】
【0002】
発明の背景
T細胞は、このような応答として免疫応答に関わり、そして、主に細胞性免疫に関わっているウイルス感染した細胞、菌類、寄生虫、および外来組織に対する防御が挙げられる。
【0003】
簡潔には、T細胞は抗原提示マクロファージに結合することによって活性化される。しかし、T細胞レセプターはマクロファージの表面に提示された抗原および主要組織適合性抗原複合体(MHC)タンパク質と特異的に複合体を形成するに違いない。
【0004】
この結合は、マクロファージにインターロイキン-1、ポリペプチド成長因子の放出を
誘導し、これは結合T細胞を刺激して増殖および分化を起こさせる。この増殖および分化は、T細胞自己刺激性分泌インターロイキン-2によって増強される。このT細胞は多数
の異なった表現型に分化し得る。この表現型としては、例えば、抗原提示宿主細胞を特異的に標的化しそしてその細胞を溶解し得る細胞傷害性T細胞、細胞傷害性T細胞を活性化することおよびB細胞と協同して抗体を産生させることに関わるヘルパーT細胞、およびそれらと同種の抗原に再度遭遇したとき非記憶T細胞よりも速い速度で増殖する記憶T細胞が挙げられる。
【0005】
T細胞の活性化が免疫応答において重要な工程であることは当業者には明らかである。T細胞の活性化を操作することにより、有用な免疫産物を得ることおよびより効率的な治療技術を開発することが可能である。
【発明の開示】
【発明が解決しようとする課題】
【0006】
以前は、T細胞活性化を達成するために、抗原およびMHCタンパク質を提示するマクロ
ファージが必要とされた。多数の問題および欠点は、このことと関連し、主な欠点は抗原に対して特異的なT細胞のみが活性化されることである。抗原に特異的ではない他のT細胞は不活性なままである。もし所望の抗原を得ることが困難または作用が運任せ(hazardous)である場合、他の問題が生じ得る。さらに、もし抗原が活性化を達成するために細胞
培養物に使用され、そしてそれは容易に除去されないならば、混入の問題が生じ得る。
【0007】
同じ問題がインビボで生じ、そして個体に抗原性物質が感染することは明らかに望ましくない。
【0008】
抗原非依存的なT細胞の活性化を達成することにより、抗原を単離する必要も、そしてマクロファージの表面に抗原を提示する必要もなく、T細胞集団を活性化することが可能である。
【0009】
インターロイキン-2は強力なTリンパ球増殖エンハンサーであることが知られ、そし
てインターロイキン-2のアジュバントとしての使用が記載されている。この役割におい
て、インターロイキン-2は既に活性化されたTリンパ球の集団の増殖賦活剤として機能
すると考えられた。しかし、インターロイキン-2が(他のサイトカインとの組み合わせにおいて)、抗原非依存的な様式でTリンパ球を特異的に活性化し得ることは知られていな
かった。
【課題を解決するための手段】
【0010】
1.T細胞をサイトカインの組み合わせと接触させることを包含する、T細胞の抗原非依存的活性化の方法。
2.前記T細胞が少なくとも以下の2つと接触される、項目1に記載の方法:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいは、それらの機能的に等価なフラグメント。
3.前記T細胞がナイーブT細胞および/または記憶休止T細胞である、項目1または2
に記載の方法。
4.前記T細胞がナイーブCD45RA+細胞および/または記憶休止CD45RO+細胞である、項目
1から3のいずれか1項に記載の方法。
5.前記インターロイキン-2の濃度が100から400U/mlであり、前記インターロイキン-6の濃度が400から600U/mlであり、そして腫瘍壊死因子αの濃度が15から35ng/mlである、
前述の項目のいずれか1項に記載の方法。
6.前記インターロイキン-2の濃度が200から300U/mlであり、前記インターロイキン-6の濃度が約500U/mlであり、そして腫瘍壊死因子αの濃度が約25ng/mlである、前述の項目のいずれか1項に記載の方法。
7.前記T細胞がインビトロで活性化される、前述の項目のいずれか1項に記載の方法。8.T細胞培養物から増加したリンホカイン産生を得る方法であって、項目7に記載の方法を用いるT細胞の活性化を包含する方法。
9.前記T細胞がインビボで活性化される、項目1から6のいずれか1項に記載の方法。10.前記T細胞のインビボでの活性化が、免疫応答の増強に導く、項目9に記載の方法。
11.項目9または10に記載の方法を用いる、ヒトまたは動物被験体T細胞の活性化を包含する治療法。
発明の要旨
本発明に従って、T細胞をサイトカインとの組み合わせとを接触させることを包含する、抗原非依存的なT細胞の活性化の方法が提供される。
【0011】
好ましくは、T細胞は以下の少なくとも2つと接触される:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいはそれらの機能的に等価なフラグメント。
【0012】
T細胞は、ナイーブ(naive)T細胞および/または記憶休止T細胞、最も適切にはナイーブCD45RA+細胞および/または記憶休止CD45RO+細胞であり得る。
【0013】
適切には、インターロイキン-2の濃度は100U/mlから400U/mlであり、インターロイキ
ン-6の濃度は400U/mlから600U/mlであり、そして腫瘍壊死因子αの濃度は15ng/mlから35ng/mlである。より好ましくは、インターロイキン-2の濃度は200U/mlから300U/ml、インターロイキン-6の濃度は約500U/mlそして腫瘍壊死因子αの濃度は約25ng/mlである。
【0014】
T細胞は、インビトロで、例えば、T細胞培養物から増加したリンホカイン産生を得る
ための方法において活性化され得、そしてそれは本発明に従うT細胞の活性化を包含する。
【0015】
T細胞は、インビボで活性化されて、免疫応答の増強に導き得、そしてそれは本発明に従う方法を用いるヒトまたは動物被験体T細胞の活性化を包含する治療法に使用され得る。
【0016】
本発明のこの局面において、サイトカインの組合わせは、T細胞応答を増強しそしてそれにより免疫応答を増強するアジュバントとして作用する。
【0017】
T細胞は、活性化され、インターロイキン、インターフェロン、およびコロニー刺激因子のような、細胞媒介性免疫応答に有用な所望のリンホカインを、抗原依存的活性化と関連した問題なしに、産生し得る。
【0018】
さらに、抗原を使用することなく、特定の免疫学的に興味のある領域において、単離されたT細胞の活性化およびエフェクターT細胞の補充(recruitment)を達成することが可
能である。それゆえ、これはHIVおよび肝炎のような多数の疾病および感染のインビボで
の処置に非常に有用である。
【0019】
本発明は、単に1つの特定の刺激抗原に特異的ではなく、「バイスタンダー(bystander)」T細胞を活性化する利点を有し、それゆえ、より大きな免疫応答が生じ、より多いリ
ンホカインの産生、および引き続くB細胞によるより多い免疫グロブリンの産生に導く。
【0020】
本発明の別の利点は、記憶T細胞の末梢血プールの維持である。なぜなら、記憶T細胞は、クローンサイズを維持するために特異的抗原刺激を必要とせずに、拡大(増殖)され得るためである。またナイーブT細胞レパートリーが維持され得る。なぜなら、本発明は、抗原刺激の場合とは異なり、それらを記憶表現型に切り換えずにナイーブT細胞の増殖を可能にするためである。
【0021】
本発明のさらなる局面に従って、以下の2つ以上を含み、必要に応じて1つ以上の薬学的に受容可能な賦形剤と組み合わされる、薬学的組成物が提供される:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいはそれらの機能的に等価なフラグメント。
【0022】
薬学的組成物は、それ自身T細胞の治療学的活性化に有用であり得、またはワクチンのようなさらなる治療剤と共に投与され得る。投与は同時または断続的であり得る。
【0023】
本発明に従って、以下の2つ以上をコードする遺伝子を有するベクターを投与する工程を包含する遺伝子治療の方法を提供する:
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいは、それらの機能的に等価なフラグメント。
【0024】
そのような適切なベクターは当該分野1で周知である。
【0025】
本発明のさらなる局面に従って、以下
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
あるいは、それらの機能的に等価なフラグメントの1つ以上をコードする遺伝子を有するベクター、および1つ以上の
i) インターロイキン-2;
ii) インターロイキン-6;および
iii) 腫瘍壊死因子α
のタンパク質あるいはそれらの機能的に等価なフラグメントの同時投与(coadministration)を包含する、組合わせ治療方法を提供する。
【0026】
特異的なT細胞タイプのそのような維持は、T細胞培養物と共に作用させる場合、非常に有利である。
【0027】
その他多くの使用および利点が本発明から理解され得、そしてそのような使用および利点は当業者に明らかである。
【発明の効果】
【0028】
本発明に従って、T細胞をサイトカインとの組み合わせとを接触させることを包含する、抗原非依存的なT細胞の活性化の方法が提供された。
【発明を実施するための最良の形態】
【0029】
実施態様の詳細な説明
材料および方法
休止T細胞の単離.健常な提供者のバフィーコート由来PBMCをフィコール-ハイパック(Pharmacia製)分離後、大部分のマクロファージをプラスチック接着によって除去した。純粋な休止CD4+T細胞集団を得るために、細胞を、以下のものに対するmAbのカクテルと共にインキュベートした。以下とは、HLA-DR(L-243;American Type Culture Collection [ATCC],Rockville,MD)、CD19(4GT)、CD16(B73.1)、CD56(MY31)、CD57(HNK-1,ATCC)、CD8(OKT8,ATCC)、CD11b(OKM-1,ATCC)、CD14(Mφ-P9)、TCR-c/δ(B1,G.De Libero,ZLF Basel,Switzerlandの贈与)、CD25(2A3)、CD69(L78)、およびCD71(LO1.1)である。氷上での30分間のインキュベーションの後、細胞を2回洗浄し、そして1:4の標的/ビーズ比で、ヤギ抗マウスIgGおよびラット抗マウスIgMを結合させた磁気ビーズ(Dynabeads;Dynal,Oslo,Norway)と共にインキュベートした。インキュベーション30分後に、ビーズ結合細胞を希土類磁石(Advanced Magnetics,Inc.,Cambridge,MA)を用いて除去した。残っている細胞は、漸増標的/ビーズ比(1:10から1:100)でビーズと共にさらに4回インキュベーションすることで、さらに精製された。最終的な集団は、その集団の>99.3%がTCRα/β+(WT/31)およびCD4+(Leu3a)であり(FACScan(R)フローサイトメーター(Becton Dickinson & Co.,Mountain View,CA)を用いる免疫蛍光分析によって決定された)、そして以下の特徴を満たした場合、休止CD4+T細胞源として使用した。以下の特徴とは、(a)FACS(R)散乱が小サイズであること;(b)活性化マーカー(CD69、CD71、MHC-DR、およびIL-2レセプターp55鎖(CD25))がFACS(R)検出可能レベルで存在しないこと;(c)細胞周期のSおよびG2/M期の細胞が存在しないこと;および(d)IL-2に曝露した場合に有意な[3H]チミジンの取り込みがないことである。いくつかの実験においては、休止細胞を、CD45RO-(mABUCH1-1を添加する)またはCD45RA-(mAB L48を添加する)としてさらにネガティブに選別した。他に示さない限り、全てのmAbはBecton Dickinson & Co.製である。
【0030】
上清の調製.破傷風トキソイド(TT)特異的クローン由来T細胞(5×103/ml)を、TT(3μg/ml)(Biocine Sclavo,Siena,Italy)存在下または非存在下でプレパルスした自己マクロファージ(2.5×105/ml)と共に培養した。16時間後、上清を採集し、そして0.2μmフィルターで濾過した。培養培地は5%ヒト血清または血漿を用い、以前に(3)に記載された。効果的な上清を、5%ヒト血清(フィレンツェ血液バンク製)または無血清培地(HL-1:Ventrex,Portland,OR)のいずれかを有する培地を用いて調製した。同様の結果が、6つの異な
った健常個体のPBMC由来の休止T細胞、および4人の異なったヒト由来の異なった特異性(精製タンパク質誘導体[PPD]または百日咳毒素)を有する活性化CD4+T細胞由来の上清で
得られた(図2を参照のこと、およびデータは示されていない)。
【0031】
細胞周期分析.これは抗CD4 mAb(FITC標識)染色と組み合わせて、ヨウ化プロピジウム
を用いて、(4)に記載のように実施した。分析はFACScan(R)LysisIIソフトウエアおよび
二重識別プログラム(Becton Dickinson & Co)を用いて実施した。
【0032】
B細胞の精製.PBMC由来B細胞をFITC標識抗CD19mAbで染色し、そしてFACStar(R)(Becton Dickinson & Co)を用いたポジティブ選別によって精製した。精製度は、抗CD20およ
び抗Igを用いた染色によって決定したように>98%であった。
【0033】
ヘルパーアッセイ.非同族ヘルパーアッセイを以前に記載(5)のように実施した。簡潔には、精製自己PBMC由来B細胞(2×103/ウエル)を、記載したような(図3を参照のこと)
サイトカイン組合わせの存在下あるいは抗CD3でコートしたプレート上で、CD4+CD45RO+休止T細胞(3×104/ウエル)と共に12日間共培養した。B細胞分化におけるサイトカインの
影響を回避するため、プレートは4日培養後に洗浄し、そしてサイトカイン組合わせをIL-2単独と置換した。上清中のIgはELISAによって測定した(5)。
【0034】
上清による休止T細胞の活性化.休止T細胞を、96ウエルの平底プレート(5×104/ウエル)中で、抗原、培地、またはT細胞上清中に見出される濃度に対応する濃度(すなわち200〜300U/ml)のrIL-2(Cetus Corp.,Emeryville,CA)で、プレパルスした自己マクロファージと共に培養したT細胞クローン由来の上清(50%vol/vol)と、共に培養した。活性化は種々の時点で、[3H]チミジン取り込みのCD69およびCD25の発現として測定した。いくつかの実験において、休止CD45RO+またはCD45RA+T細胞の[3H]チミジン取り込みは、異なった濃度のIL-2と1μg/ml LPS(Difco,Detroit,MI)またはLPS活性化マクロファージ由来の上清(50%vol/vol)のいずれかとの存在下で測定した。活性化マクロファージ上清の調製には、5×105マクロファージを1μg/mlLPSで刺激した(6〜8時間) 。[3H]チミジン取り込み実験は、(5)に記載のように実施した。結果は3組ウエルの平均を表し、そしてSDは常に15%であった。
【0035】
組換えサイトカインによる休止T細胞の活性化.96ウエル平底マイクロプレート中で、休止T細胞(5×104/ウエル)を、以下のrIL-2(200〜300U/ml)、rIL-6(500U/ml;Ciba-Geigy,Basel,Switzerland;IL-6ユニットはB9アッセイで決定された)、TNF-α(25ng/ml;Genzyme Corp.,Cambridge,MA)、およびLPS刺激マクロファージの上清(50%vol/vol)と共に培養した。チミジンの取り込みおよびCD69発現を、図2に記載のように測定した。IL-2およびTNF-αと組み合わせたIL-1b(100ng/mlまで、Biocine Sclavo Siena,Italy)は全く活性を有さなかった(データは示していない)。2つの異なったソース由来の組換え型サイトカインを用い同様の結果を得た。サイトカインの最適濃度は、予備的な用量-応答実験で確立した。
【0036】
PCR補助mRNA増幅(PCR-assisted mRNA Amplification).精製休止CD4+CD45RO+T細胞をTNF-α+IL-6+IL-2、またはIL-2単独と共に培養した。全RNAは、培養60〜100時間後に、RNAzol*B(Biotecx Laboratories,Houston,TX)によって、5×105個の細胞から単離した。cDNAは、(5)に記載のようにマウス逆転写酵素を用いて合成された。β−アクチン、IL-4、およびIFN-c特異的プライマーペアは、Clontech(Palo Alto,CA)から購入した。PCRは(5)に記載のように実施した。
【0037】
限界希釈分析.CD45RO+休止T細胞を、IL-2単独(300U/ml)あるいはTNF-α(25ng/ml)お
よびIL-6(500U/ml)の組合わせと共に、精製自己照射(2,500ラド)マクロファージ(3×103/ウエル)、抗DRmAb(L243,20μg/ml)の存在下、20μl容でTerasakiプレート(64ウエル/1条
件)に異なる数で播種した。14日目に、培養物の増殖を肉眼で調べた。ランダムに選択し
た増殖ウエルを抗CD4抗体および抗TCR-α/β抗体でポジティブに染色した。頻度分析は最小自乗法(6)によって行った。
結果および考察
本研究の重要な点は、IL-2にのみ応答する活性化T細胞が存在しない休止集団を使用することである。発明者らは休止CD4+T細胞と作用することを選択した。なぜなら、休止表現型を有するいくらかのCD8+またはc/δT細胞を有する変形で、それらの細胞は、p55鎖
欠如下でIL-2レセプターp75鎖を発現しないためである(7)。これはIL-2に対する不要な
増殖応答を担い得(8)、かつ発明者らが選別するための良好な抗体を有していない。それゆえ発明者らは、PBMCから高度に精製された休止CD4+T細胞を得るために、多工程の徹底的な精製を実施した(図1)。予備実験において、休止CD4+T細胞を、抗原でパルスした(Ag-pulsed)マクロファージで活性化されたCD4+T細胞クローン由来の上清と共に培養した
。図2の代表的実験は、休止CD4+T細胞がIL-2によってではなく、上清によって活性化され、CD69(9)(図2A)およびIL-2レセプターp55鎖(図2B)を発現すること、そして[3H]チミ
ジンを取り込むこと(図2C)を示す。
【0038】
活性化上清は、2つの細胞タイプの共培養(coculture)によって生成されるので、発明
者らは、T細胞およびAPCによって産生された可溶性因子の相対的寄与度の決定を試みた
。この実験のために、休止CD4+T細胞を、CD45RO+(記憶)およびCD45RA+(ナイーブ)亜集団(10)としてさらに精製した。なぜならそれらはTCR媒介性活性化について既に報告されて
いるように異なった活性化要求性を有し得るためである(11、12)。図2Dは、LPS活性化マ
クロファージ由来の上清単独ではIL-2単独と同様に活性を有さなかったこと、ところがIL-2と組合わせたマクロファージ上清はCD45RA+およびCD45RO+休止T細胞の両方においてチミジン取り込みを誘導したことを示す。これらの結果は、IL-2とAPCによって産生された
可溶性因子とが休止T細胞の活性化に必要とされることを示す。
【0039】
APC由来因子を同定するために、発明者らはマクロファージによって産生され、T細胞
に対し共刺激活性を有することが公知の組換え型サイトカイン、すなわち、IL-1β、IL-6、およびTNF-α(13〜15)の効果を試験した。IL-2の非存在下では、これらのサイトカインの可能な組合わせ全てが、広範な濃度範囲にわたり、活性を示さなかった(データは示し
ていない)。図3Aは、TNF-αがIL-2との組合わせにおいて休止CD45RO+T細胞にCD69の発現およびチミジンの取り込みを誘導したこと、一方IL-6はIL-2と組合わせても大して効果のないことを示す。注目すべきことに、TNF-αおよびIL-6は、IL-2との組合わせにおいて、より強い活性化に導く相乗効果を有した。IL-2、IL-6、およびTNFαの同様の効果がまた
、CD45RA+休止T細胞において観察されたが(図3B)、この場合、3つのサイトカイン全て
が活性化の誘導に要求された。さらに、図3Cの細胞周期分析は、培養7日目に、CD45RO+
およびCD45RA+T細胞両方の8%が、細胞周期のSまたはG2/M期にあることを示す。活性
化マーカーの発現、チミジン取り込み、あるいは細胞周期へのエントリーとして測定されたサイトカインの活性化は、DR、CD4、またはCD3に特異的なmAbによって決して阻害され
なかった(データは示していない)。それゆえ、TCRシグナリングはこのタイプの活性化に
関与しないことが確認された。
【0040】
サイトカインによって活性化されたCD45RA+T細胞は、TCR連結(engagement)後数日内に起こると報告された(16)ような、表現型をCD45ROに切り換えないことを、本発明者らが観察したことは、注目すべき興味ある点である。IL-2、TNF-α、およびIL-6の組合わせに
よって活性化されたCD45RA+T細胞を、培養23日まで3日おきに抗CD45RA抗体および抗CD45RO抗体を用いて二重染色した。本発明者らは、どの時点においても、シングルポジティ
ブなCD45RA+細胞を見出すことはなく、数%のダブルポジティブCD45RA+high/CD45RO+dullを見出すだけであった。実際に、図4は、ナイーブT細胞が、ほとんどの細胞が芽様(blastic)であり、CD69を発現している(データは示していない)とき、サイトカイン活性化23
日後でさえ、主にCD45RA+であることを示す。抗CD3で活性化された同細胞は、数日でCD45RO+CD45RA-表現型に切り換わった(データは示していない)。
【0041】
本発明者らは、次に休止Tリンパ球がサイトカインによって活性化され得、エフェクター機能を示すか否かを問うた。本発明者らはリンホカインについてPCR補助mRNA増幅を実
施した。図5は、IFN-cおよびIL-4のmRNA両方がIL-2単独ではなく、IL-2、TNF-α、およ
びIL-6と共に培養したCD45RO+T細胞によって発現されることを示す。さらに、サイトカ
イン組合わせによって活性化されたCD45RO+T細胞は、B細胞をIgを産生することを補助
することにおいて、抗CD3刺激T細胞と同程度効果的である(表1)。
表1.サイトカインによって活性化された休止CD45RO+T細胞はB細胞への補助を提供し得る
【0042】
【表1】
【0043】
T細胞のB細胞への補助が自己反応性細胞の活性化のためである可能性を除外するために、ヘルパーアッセイの終わりに、B細胞を選別によって除去し、そしてCD4+T細胞を自己精製B細胞またはマクロファージに対する増殖において試験した。本発明者らは任意の自己反応性増殖を見出さなかった(データは示していない)。
【0044】
サイトカインも抗CD3もCD45RA+T細胞にIFN-cの産生(<1IU/ml)およびB細胞の補助を誘導しなかった(データは示していない)。それゆえ、本発明者らは、TCR媒介活性化に類
似して(17)、サイトカインは、休止CD45RO+T細胞を活性化し増殖およびエフェクター機
能を表示させるのに対し、CD45RA+T細胞を補充しIg産生を補助しないと結論する。
【0045】
サイトカインによって増殖するように刺激され得る記憶表現型を有する休止T細胞の頻度を評価するため、本発明者らは限界希釈実験を実施した。CD45RO+CD4+休止T細胞を、
自己反応性応答を防ぐため自己照射マクロファージおよび抗DR抗体の存在下で、IL-2単独あるいはTNF-αおよびIL-6との組合わせと共に培養した。図6は33の休止CD45RO+CD4+T
細胞のうち1つがIL-2、TNF-α、およびIL-6に応答して目に見えるクローンに増殖したことを示す。現在のところ、どうして細胞の3%のみがサイトカインに応答して増殖したの
かは不明である。増殖した細胞は、休止T細胞のサブセットであったかあるいは成熟化/
活性化の異なる時期にあった可能性があった。多くの細胞(約20%)がサイトカインに応答し、そして活性化マーカーを発現する可能性がある。これらの細胞のいくらかはエフェクター機能を表し、そして少数(3%)のみがインビトロで増殖し得、目に見えるサイズのクローンになる。
【0046】
TNF-αおよびIL-6は共にT細胞上のIL-2Rをアップレギュレートすることが示されてき
た(15、18)。これは、本サイトカイン組合わせによる休止T細胞の活性化のための1つの可能な機構であり得た。しかし、TNF-αおよびIL-6と共に1〜3日間培養し、そして洗浄しIL-2と共にさらに4〜5日培養した休止T細胞は、FACS(R)で検出可能なレベルのIL-2R(p55)を示さず(データは示していない)、一方IL-2Rは、培養の開始から、TNF-α、IL-6、およびIL-2と共に培養した約20%の同細胞上に発現された。しかし、この実験は、FACS(R)感
度以下の低いレベルのIL-2Rが発現し機能的に関連した可能性を除外してはいない。実際
、IL-2がTNF-αまたはIL-6によるIL-2Rの誘導に必要とされることが報告された(19)。さ
らに、IL-2はそれ自身のレセプターの発現を増強する(20)のみならず、TNF-αレセプターのアップレギュレート(21)もまた行っている。サイトカインによる休止T細胞の活性化の機構の解明にはさらなる生化学的分析および分子的分析が必要である。
【0047】
この新規な抗原非依存的T細胞活性化経路はインビボにおいて、2つの重要な役割、すなわち免疫応答部位におけるエフェクターT細胞の補充および記憶T細胞の末梢プールの維持を果たし得る。休止T細胞は、抗原特異的応答の部位で、特定のT細胞およびマクロファージによって産生されたサイトカインにより活性化されて、増殖および他のリンホカインを分泌し、そしてそのリンホカインが応答をさらに増幅し得るという1つのシナリオが描かれ得る。実際、サイトカイン組合わせに応答する休止CD45RO+T細胞の頻度は、い
ずれの公知の抗原により感作されたT細胞の通常の頻度よりも明確に高い。
【0048】
記憶は、抗原の持続、再発的な感染、あるいは環境抗原の交差反応性により、繰り返しの断続的な刺激を必要とする短命の細胞からなる長命のクローンによって保有され得ることが仮定されている(22〜24)。本発明者らの結果に照らしてみれば、記憶表現型(CD45RO+)を有する休止T細胞が無関係の抗原に対する応答の間に分泌されたサイトカインによっ
て拡大され得るため、記憶T細胞はそれらのクローンサイズを維持するのに抗原刺激を必要としなくてもよいと推測されがちである。他方、サイトカインは記憶表現型への切り換えなしにナイーブ細胞の増殖を誘導し得、従ってナイーブ(CD45RA+)T細胞レパートリー
の維持を補助し得る。
【0049】
本発明は例示のみによって上述され、発明の範囲および意図内で改変がなされ得ることが理解される。
【0050】
【数1−1】
【0051】
【数1−2】
【0052】
【数1−3】
【0053】
【数1−4】
【図面の簡単な説明】
【0054】
【図1−1】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−2】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−3】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−4】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図1−5】精製CD4+休止T細胞の表現型および細胞周期分析。(A)前方散乱および側方散乱プロファイル。(B)細胞周期分析。(C)FITC-またはPE-結合(conjugated)コントロール抗体。(D〜F)CD4+細胞の純度および活性化マーカーの発現。(G)選別されたCD4+細胞上のCD45RA抗原およびCD45RO抗原の発現。(HおよびI)CD45RO+またはCD45RA+亜集団として精製されたCD4+細胞。
【図2】可溶性因子による休止CD4+T細胞の活性化。(AおよびB)抗原(Ag)(斜線棒)または培地(黒棒)、またはrIL-2(白棒)でプレパルスした自己マクロファージと共に培養したT細胞クローン由来の上清で培養した休止T細胞上の活性化マーカーの発現。CD69またはCD25の発現を抗CD4を用いる二重染色で分析した。(C)培地のみ(三角)、rIL-2(四角)、あるいは抗原(黒丸)または培地(白丸)でプレパルスしたマクロファージと共に培養したT細胞クローン由来の上清と共に培養した、AおよびBの同細胞の[3H]チミジンの取り込み。(D)種々の濃度のIL-2+1μg/mlLPS(白記号)、または種々の濃度のIL-2とLPS活性化マクロファージ由来上清(黒記号)存在下での、休止CD45RO+T細胞(四角)、休止CD45RA+T細胞(丸)の[3H]チミジンの取り込み。
【図3−1】IL-2、TNF-α、およびIL-6の組合わせは、休止T細胞を活性化する。CD45RO+(A)あるいはCD45RA+(B)休止T細胞を、以下の種々の組合わせと共に8日間培養した:rIL-2、rIL-6、TNF-αおよびLPS刺激マクロファージ由来の上清。チミジンの取り込みおよびCD69発現を図1に記載のように測定した。(C)IL-2単独(白記号)あるいはTNF-αおよびIL-6と組合わせた(黒記号)存在下での、休止CD45RO+T細胞(四角)あるいは休止CD45RA+T細胞(丸)の細胞周期分析。
【図3−2】IL-2、TNF-α、およびIL-6の組合わせは、休止T細胞を活性化する。CD45RO+(A)あるいはCD45RA+(B)休止T細胞を、以下の種々の組合わせと共に8日間培養した:rIL-2、rIL-6、TNF-αおよびLPS刺激マクロファージ由来の上清。チミジンの取り込みおよびCD69発現を図1に記載のように測定した。(C)IL-2単独(白記号)あるいはTNF-αおよびIL-6と組合わせた(黒記号)存在下での、休止CD45RO+T細胞(四角)あるいは休止CD45RA+T細胞(丸)の細胞周期分析。
【図4】サイトカインにより活性化されたCD45RA+T細胞はそれらの表現型をCD45ROに切り換えない。CD45RA+T細胞をIL-2、TNF-α、およびIL-6の組合わせで活性化し、そして23日後、抗CD45RA-FITC抗体および抗CD45RO-PE抗体で二重染色した。
【図5】サイトカイン活性化T細胞によるIFNcおよびIL-4mRNAの発現。精製CD4+ CD45RO+休止T細胞を、材料および方法に記載のように、IL-2単独と共に60時間(レーン1)および100時間(レーン3)、あるいはIL-2、TNF-α、およびIL-6と共に60時間(レーン2)および100時間(レーン4)培養する。ポジティブテンプレート(レーン5);ネガティブコントロール(レーン6)。
【図6】サイトカイン組合わせに応答して増殖する休止T細胞の頻度。CD45RO+休止T細胞を、IL-2単独(黒丸)あるいはTNF-αおよびIL-6の組合わせ(白丸)と共に精製自己マクロファージ、抗DRmAb存在下で播種した。95%信頼限界(破線)。
【特許請求の範囲】
【請求項1】
(i)インターロイキン2、(ii)インターロイキン6および(iii)腫瘍壊死因子α、あるいは(i)、(ii)および(iii)のいずれかの機能的に等価な断片の、HIV感染または肝炎の処置のための医薬の製造における、使用。
【請求項2】
(i)インターロイキン2、(ii)インターロイキン6および(iii)腫瘍壊死因子α、あるいは(i)、(ii)および(iii)のいずれかの機能的に等価な断片を含む、HIV感染または肝炎の処置のための薬学的組成物。
【請求項3】
本願明細書に開示された薬学的組成物。
【請求項1】
(i)インターロイキン2、(ii)インターロイキン6および(iii)腫瘍壊死因子α、あるいは(i)、(ii)および(iii)のいずれかの機能的に等価な断片の、HIV感染または肝炎の処置のための医薬の製造における、使用。
【請求項2】
(i)インターロイキン2、(ii)インターロイキン6および(iii)腫瘍壊死因子α、あるいは(i)、(ii)および(iii)のいずれかの機能的に等価な断片を含む、HIV感染または肝炎の処置のための薬学的組成物。
【請求項3】
本願明細書に開示された薬学的組成物。
【図1−1】

【図1−2】
【図1−3】
【図1−4】
【図1−5】
【図2】
【図3−1】
【図3−2】
【図4】
【図5】
【図6】
【図1−2】
【図1−3】
【図1−4】
【図1−5】
【図2】
【図3−1】
【図3−2】
【図4】
【図5】
【図6】
【公開番号】特開2006−312640(P2006−312640A)
【公開日】平成18年11月16日(2006.11.16)
【国際特許分類】
【出願番号】特願2006−180501(P2006−180501)
【出願日】平成18年6月29日(2006.6.29)
【分割の表示】特願2004−220905(P2004−220905)の分割
【原出願日】平成7年8月17日(1995.8.17)
【出願人】(592243793)カイロン ソチエタ ア レスポンサビリタ リミタータ (107)
【Fターム(参考)】
【公開日】平成18年11月16日(2006.11.16)
【国際特許分類】
【出願日】平成18年6月29日(2006.6.29)
【分割の表示】特願2004−220905(P2004−220905)の分割
【原出願日】平成7年8月17日(1995.8.17)
【出願人】(592243793)カイロン ソチエタ ア レスポンサビリタ リミタータ (107)
【Fターム(参考)】
[ Back to top ]