
T細胞調節方法
【課題】患者における進行性の免疫抑制の発症により、多様な疾患が特徴づけられ、悪性腫瘍を有する患者における免疫応答障害の治療薬および薬物をスクリーニングする方法を提供する。
【解決手段】調節性T細胞(Treg)は、自己免疫を制限するが、抗病原体および抗腫瘍免疫の程度をも減弱させ得る。Treg機能の機構の理解およびインビボでのTregの治療処置には、Treg選択的な受容体を同定することが必要である。エフェクター/メモリーまたは調節性の表現型のいずれかに分化する抗原特異的なCD4+T細胞からの遺伝子発現アレイの比較解析により、MHCクラスII組織に結合するCD4関連分子のLAG-3(CD223)のTreg選択的な発現。
【解決手段】調節性T細胞(Treg)は、自己免疫を制限するが、抗病原体および抗腫瘍免疫の程度をも減弱させ得る。Treg機能の機構の理解およびインビボでのTregの治療処置には、Treg選択的な受容体を同定することが必要である。エフェクター/メモリーまたは調節性の表現型のいずれかに分化する抗原特異的なCD4+T細胞からの遺伝子発現アレイの比較解析により、MHCクラスII組織に結合するCD4関連分子のLAG-3(CD223)のTreg選択的な発現。
【発明の詳細な説明】
【技術分野】
【0001】
発明の技術分野
本発明は、治療薬および薬物をスクリーニングする方法に関する。
【0002】
本出願は、2003年2月28日に出願された米国特許出願第60/451,039号、2003年6月24日に出願された米国特許出願第60/482,143号、および2003年12月22日に出願された米国特許出願第60/531,704号に対する優先権を主張する。
【0003】
米国政府、国立衛生研究所は、本発明の基礎をなす研究のための資金を提供した(AI39480)。その資金提供協定の期間の下で、米国政府は、本発明に一定の権利を保持する。
【背景技術】
【0004】
発明の背景
患者における進行性の免疫抑制の発症により、多様な疾患が特徴づけられる。悪性腫瘍を有する患者における免疫応答障害の存在は、特によく考証されている。癌患者および腫瘍を有するマウスは、遅延型の過敏症の減少、溶解機能の減少、およびリンパ球の増殖反応などの種々の免疫機能の変化を有することが示された。S. Broder et al., N. Engl. J.Ned. 299: 1281 (1978); E.M. Hersh et al., N. Engl. J. Med., 273: 1006 (1965); North and Burnauker, (1984)。その他の疾患または介入の多くが、免疫応答障害の発症によって特徴づけられる。たとえば、進行性の免疫抑制は、後天性免疫不全症候群(AIDS)、敗血症、ライ病、サイトメガロウイルス感染、マラリアなどを有する患者、並びに化学療法および放射線療法を使用する患者で観察された。しかし、免疫応答のダウンレギュレーションの原因となる機構は、完全に解明されないままである。
【0005】
免疫応答は、複雑な現象である。Tリンパ球(T細胞)は、全ての細胞性免疫反応の発症に重要である。ヘルパーT細胞は、免疫応答の発現を制御し、調整する。細胞障害性T細胞(キラーT細胞)は、感染した標的細胞を溶解することにより、細胞内寄生体およびウイルスに対する免疫反応において重要な役割を果たすエフェクター細胞である。また、細胞障害性T細胞は、免疫監視機構を介して体を癌の発生から保護することに関係している。調節性T細胞は、Tヘルパー細胞の誘導および/または活性をブロックする。T細胞は、通常遊離の抗原を認識しないが、その他の細胞の表面上でそれを認識する。これらのその他の細胞は、T細胞分裂を刺激することができる分化した抗原提供細胞であってもよく、または体内でウイルスに感染した、細胞障害性T細胞の標的になる細胞であってもよい。
【0006】
細胞障害性T細胞は、通常、全ての有核細胞上に発現されているクラスI主要組織適合性抗原(MHC)産物と会合して抗原を認識する。ヘルパーT細胞、およびインビトロで抗原に応答して増殖する大部分のT細胞は、クラスIIMHC産物に会合して抗原を認識する。クラスII産物は、大部分が抗原提供細胞上に、およびいくつかがリンパ球上に発現される。また、T細胞は、モノクローナル抗体によって定義されるこれらの細胞膜糖タンパク質を基礎として2つの主要亜集団に分けることができる。通常62kD糖タンパク質を発現するCD4+サブセットは、クラスII抗原の状況で抗原を認識し、一方でCD8+サブセットは、76Kd糖タンパク質を発現し、クラスI MHCの状況で抗原を認識することに制限される。
【0007】
リンホカインの注入を経た免疫易感染性動物の免疫応答を増強する、養子免疫治療は、不定に適用され、成功が限られていた。この種の治療を改善するための方法が必要である。たとえば、リンパ球、血液、およびその他の細胞の注入は、一定の設定で免疫不全の患者に提供される。しかし、健康なT細胞集団の再構成を促進し、増強することにより、このような患者に対して有意に増大された利益および有効性を提供することができる。
【0008】
いくつかの症状では、有害なT細胞活性を生じる可能性がある。たとえば、T細胞を媒介した自己免疫疾患および炎症性疾患は、自己抗原を認識するT細胞が増殖し、このような抗原を発現する細胞を襲う有害なT細胞の活性によって特徴づけられる。その他の例には、宿主T細胞および移植片対宿主疾患によって媒介される、移植片拒絶の発生を含む。
【0009】
これらの症状を治療するために利用できる既存の免疫抑制療法は、シクロスポリンA、FK506、およびラパマイシンなどの免疫抑制の化合物の投与を含む。しかし、これらの療法は、完全に有効ではなく、腎毒性、肝毒性、高血圧、多毛、および神経毒性などの有意に有害な副作用と関係がある。したがって、これらの条件を治療するために、より副作用を有さずにT細胞活性をより効率的に抑制することができるさらなる療法が必要である。
【0010】
リンパ球恒常性は、厳密に調節された中心的な生物学的プロセスである。Tanchot, C. et al Semin. Immunol. 9: 331-337 (1997); Marrack, P. et al., Nat. Immunol. 1: 107-111 (2000); C. D. Surh, C.D. and Sprent, J., Microbes. Infect. 4: 51-56 (2002); Jameson, S.C., Nat.Rev. Immunol. 2: 547-556 (2002)。このプロセスの分子制御は、十分に理解されていないが、2つのシグナリング経路の媒介に関与する分子が必須であると考えられる。第1に、自己主要組織適合性(MHC)分子の認識は、ナイーブT細胞の恒常性およびメモリーT細胞機能の維持に重要である。Takeda, S. et al., Immunity 5:217-228 (1996); Tanchot, C. et al., Science 276:2057-2062 (1997)。
【0011】
さらに、最近の研究では、ナイーブT細胞が生存し続けるためには、T細胞受溶体(TCR)の発現が必要とされることが証明された。Polic, B. et al., Proc. Natl. Acad. Sci. 98: 8744-8749(2001); Labrecque, N. et al., Immunity 15: 71-82(2001)。第2に、共通のガンマ(γc)鎖を介してシグナルを送るサイトカイン、特にインターロイキン7(IL-7)は、ナイーブT細胞の生存および恒常性に重要である。Schluns, K.S. et al., Nat. Immunol. 1:426-432(2000); Tan, J.T. et al., Proc. Natl. Acad. Sci. 98:8732-8737(2001)。これらの分子は全て、T細胞恒常性を正に調節する。対照的に、CTLA-4およびTGF-βだけが、T細胞の負の調節に関係していたが、これは、リンパ球減少症の宿主にT細胞を導入すること、または新生児の発現の解析によって噴出して確認された。Waterhouse, P. et al., Science 270: 985-988(1995); Tivol, E.A. et al., Immunity 3: 541-547(1995); Lucas, P.J. et al., J. Exp. MEd. 191:1187-1196(2000); Gorelik, L. and Flavell, R.A., Immunity 12: 171-181(2000)。
【0012】
LAG-3は、CD4とのその緊密な関係のために特に興味を生じさせる。LAG-3は、CD4と同様のゲノム機構を有し、同じ染色体位置に存在するBruniquel, D. et al., Immunogenetics 47:96-98 (1997)。LAG-3は、活性化されたCD4+およびCD8+ αβ Tリンパ球およびγδT細胞およびNK細胞のサブセットで発現される。

CD4の様に、LAG-3もMHCクラスII分子に対して、しかも非常に高い親和性で結合する。Huard, B. et al., Immunogenetics 39: 213-217 (1994); Huard, B. et al., Eur.J. Immunol. 25: 2718-2721 (1995)。
【発明の概要】
【0013】
本発明の簡単な概要
本発明の第1の態様において、自己免疫疾患に罹患する患者を治療するための方法が提供される。患者から単離された自己免疫性T細胞には、インビトロでCD223のコード配列を含む発現構築物がトランスフェクトされる。次いで、トランスフェクトされた自己免疫性T細胞は、患者に再注入される。
【0014】
本発明の第2の態様において、組成物が提供される。組成物は、CD223と特異的に結合する抗体および抗癌ワクチンを含む。
【0015】
本発明のもう一つの態様において、キットが提供される。キットは、CD223と特異的に結合する抗体および抗癌ワクチンを含む。
【0016】
本発明の第4の態様において、抗癌ワクチンで癌患者を治療するための改善された方法が提供される。CD223と特異的に結合する抗体が癌患者に投与される。また、抗癌ワクチンが投与される。抗体は、抗癌ワクチンに対する癌患者の抗癌応答の大きさを増加させる。
【0017】
本発明の第5の態様において、抗癌ワクチンに対する免疫応答の抑制を克服するための方法が提供される。CD223と特異的に結合する抗体が、抗癌ワクチンに対する免疫応答を抑制する調節性T細胞を有する癌患者に投与される。また、抗癌ワクチンが患者に投与される。抗体は、抗癌ワクチンに対する癌患者の応答を増大させる。
【0018】
本発明のもう一つの態様において、哺乳類のT細胞の数を増大させるための方法が提供される。CD223タンパク質またはCD223 mRNAに結合する阻害剤が哺乳類に投与される。阻害剤は、CD223の活性または発現を阻害する。
【0019】
本発明のさらにもう一つの態様において、哺乳類のT細胞の数を減少させるための方法が提供される。CD223をコードする発現構築物が哺乳類に投与される。CD223は、発現構築物から発現され、哺乳類中のCD223の濃度が増大される。哺乳類中のT細胞の数は、減少される。
【0020】
本発明のさらにもう一つの態様において、哺乳類中のT細胞の数を減少させるための方法が提供される。CD223+ T細胞集団が哺乳類に投与される。哺乳類中のCD223の濃度が増大され、これにより哺乳類中のT細胞の数が減少される。
【0021】
本発明のもう一つの側面に従って、CD223の50以下の連続したアミノ酸残基からなるポリペプチドが提供される。ポリペプチドは、SEQ ID NO:5で示されるアミノ酸配列KIEELEを含む。
【0022】
本発明のもう一つの態様は、少なくとも2つのセグメントを含む融合ポリペプチドである。第1のセグメントは、CD223の50以下の連続したアミノ酸残基を含む。第1のセグメントは、SEQ ID NO:5で示されるアミノ酸配列KIELLEを含む。第2のセグメントは、SEQ ID NO:2または4で示されるCD223に見いだされないアミノ酸配列を含む。
【0023】
さらなる態様において、癌、自己免疫疾患、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法が提供される。試験物質をCD223タンパク質またはSEQ ID NO:5に示されるアミノ酸配列KIELLEを含むCD223タンパク質断片と接触させる。次いで、試験物質がCD223タンパク質またはCD223タンパク質断片に結合したかどうかを決定する。試験物質がCD223タンパク質またはCD223タンパク質断片に結合した場合には、試験物質を、癌、自己免疫疾患、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定される。
【0024】
本発明のもう一つの態様では、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法が提供される。試験物質は、CD223タンパク質と接触される。試験物質の存在下および非存在下において、CD223活性が決定される。試験物質がCD223活性を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定される。
【0025】
本発明のもう一つの側面に従って、自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験する方法が提供される。試験物質をCD223タンパク質と接触させる。試験物質の存在下および非存在下においてCD223活性を決定する。試験物質がCD223活性を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定される。
【0026】
本発明のもう一つの態様は、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法である。CD223+ T細胞を試験物質と接触させる。試験物質の存在下および非存在下において細胞におけるCD223の発現を決定する。試験物質がT細胞におけるCD223発現を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定される。
【0027】
本発明のさらにもう一つの側面は、自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験するもう一つの方法である。試験物質をCD223+ T細胞と接触させる。試験物質の存在下および非存在下において細胞におけるCD223発現を決定する。試験物質がT細胞におけるCD223発現を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定される。
【0028】
本発明のさらにもう一つの側面は、CD223+ T細胞またはCD223-T細胞を単離する方法である。T細胞の混合集団を、SEQ ID NO:2または4に記載のCD223と特異的に結合する抗体と接触させる。抗体に結合していないT細胞から抗体に結合するT細胞を分離する。これによりCD223+ T細胞の集団およびCD223-T細胞の集団を形成する。
【0029】
本発明のもう一つの態様は、残基1〜431を含み、残基467〜521を欠いている、単離された可溶性マウスCD223タンパク質である。
【0030】
本発明のもう一つの側面は、残基1〜440を含み、残基475〜525を欠いている、単離された可溶性ヒトCD223タンパク質である。
【0031】
本発明のさらにもう一つの側面は、哺乳類においてT細胞数を減少するための方法である。可溶性CD223タンパク質を哺乳類に投与する。これにより、哺乳類におけるMHCクラスII拘束性/CD4+ T細胞応答が調整される。
【図面の簡単な説明】
【0032】
【図1】HA特異的なCD4+ T細胞は、寛容になり、C3-HAhighトランスジェニックマウスに養子性の導入をすることにより、調節性T細胞活性を発生する。(図1A)C3-HAhighトランスジェニックマウスは、種々の上皮区画に高レベルのHAを発現し、肺上皮において最高レベルで発現される。C3-HAhighのレシピエントは、2.5×106のHA特異的TCRトランスジェニック(6.5)CD4+ T細胞の養子性の導入の4〜7日後に、アネルギー性の表現型の発症前に生じる一過性エフェクター期の活性化と関係する間質性肺炎により死亡した。少数の6.5 CD4+ T細胞の導入により、重篤でない肺病態を生じ、C3-HAhighレシピエントは生き残る。残りの6.5 T細胞は、アネルギー性になる(これらは、γ-インターフェロンを産生できず、またはインビトロでHA抗原に対して増殖することができないことによって定義される)。6.5 T細胞の亜致死量を受けているマウスは、その後の2.5×106のナイーブ6.5T細胞の注入から保護される。従って、最初に寛容化されたT細胞は、Treg活性を発生し、第2の高用量の6.5T細胞によって誘導される致死性の間質性肺炎を抑制する。(図1B〜1E)C3-HAhighマウスにおけるエフェクター/メモリーT細胞、対、抑制T細胞の局在。ナイーブ6.5T細胞(Thy1.1+/1.2-)には、6.5アネルギー性/Treg細胞(Thy 1.1-/1.2+)の非存在下または存在下のいずれかにおいて、C3-HAhighレシピエント(Thy 1.1-/1.2+)に養子性に導入した。養子性の導入の3日後に脾臓および肺を回収し、Thy 1.1+細胞を免疫組織化学によって染色した。Treg細胞の非存在下では、Tエフェクター細胞は、脾臓の小胞に点在し(図1B)、肺血管を浸潤する(図1C)。Treg細胞の存在下では、抑制されたHA-特異的な6.5T細胞は、脾臓の動脈周囲のリンパ性外筒に隔絶されて(図1D)、肺血管を浸潤することができない(図1E)。
【図2】LAG-3は、アネルギー性/Treg細胞とエフェクター/メモリーCD4+ T細胞との間で差次的に発現され、アネルギー性/Treg CD4+ T細胞のLAG-3発現は、IL-10発現と相関される。遺伝子チップ解析によって明らかになった差次的発現を定量的リアルタイムRT-PCRによって確認した(図2A)。早期の日(2日〜4日)のLAG-3の差次的発現は、養子性の導入の30日後まで及ぶ。(図2B)細胞表面LAG-3タンパク質レベルは、抗体染色によって評価した。脾細胞をC3-HAhigh、Vac-HAで免疫した野生型B10.D2マウス、または6.5+/-Thy1.1+/-脾細胞をi.v.注射の5日後のwt B10.D2マウスから収集し、単個細胞懸濁液に調製する。全ての試料を最初にFc受容体をブロックするために全ラットIgGと共にインキュベートした。細胞をTCR特異的な項-6.5-ビオチン+SA-APC、LAG-3-PE、または対応するアイソタイプ対照で染色した。細胞を総リンパ球集団および6.5陽性リンパ球で二重ゲートした。アイソタイプ対照-破線、ナイーブ細胞-明灰色線、エフェクター/メモリー細胞-暗灰色線、アネルギー性/Treg細胞-黒線。(図2C)多くの実験にわたってアネルギー性/Treg集団の複数の試料を解析することにより、LAG-3レベルとIL-10 mRNAレベルとの間の直接的な相互関係を確認する。
【図3】LAG-3は、CD25とは独立して誘導されたTreg細胞上に発現され、Treg機能のマーカーである。(図3A)導入の5日後のC3-HAhighレシピエント脾臓からのアネルギー性/Treg 6.5 CD4+ T細胞をLAG-3およびCD25発現について染色し、アイソタイプ対照と比較した。(図3B)これらの表面LAG-3およびCD25染色に基づいて、細胞を4集団に選別した:LAG-3highCD25high、LAG-3highCD25low、LAG-3lowCD25high、およびLAG-3 lowCD25low。インビトロでの抑制アッセイ法において、サプレッサーとして1×105の細胞の各々の異なる選別されたサブセットを、応答者として1×104ナイーブ6.5 CD4+を添加した。LAG-3lowCD25low細胞は、最も抑制性が少ない。LAG-3highCD25high、LAG-3highCD25low、およびLAG-3lowCD25lowは、最も抑制活性を示すLAG-3highCD25high二重陽性細胞と抑制活性が匹敵する。これは、3回の再現実験の代表的結果である。
【図4】抗LAG-3抗体は、インビトロでTreg活性をブロックする。モノクローナル抗-LAG-3抗体を2μg/mlの濃度でインビトロ抑制アッセイに添加し、0.04:1のサプレッサー:応答者の比率で6.5CD4+サプレッサーによるインビトロでのナイーブ6.5 CD4+ T細胞増殖の抑制を完全に逆転させる。
【図5】抗LAG-3抗体は、直接Treg細胞を阻害することによって、6.5CD4+ Treg細胞によるインビボ抑制を排除する。(図5A)8,000の6.5 CD4+ T細胞によって前処理されたC3-HAhighマウスは、Treg集団の最初の導入確立の4日後に与えられる2.5×106の6.5 CD4+ T細胞によるその後のチャレンジでも生存した(w/保護)。致死下での前処理を伴わずに、C3-HAhighレシピエントは、致死的チャレンジの4〜6日後に死んだ(保護無し)。C3-HAhighマウスに対して、これらを8,000の6.5 CD4+ T細胞で前処理し、200μgの別の用量を2日後に与えた4日後に6.5のT細胞の致死量と共に、モノクローナル抗LAG-3抗体(200μg)をC3-HAhighマウスにi.v.で与えた。抗LAG-3抗体処理されたマウスは、もはやその後の致死チャレンジに耐えることができない(保護+aLAG-3)。対照的に、アイソタイプ対照抗体ラットIgGIでの処理では、インビボ抑制を排除することができない(保護+ラットIgGI)。抗-LAG-3mAbは、Tregの非存在下でナイーブ6.5 CD4+ T細胞を超活性化(hyper-activate)しない(図5Bおよび5C)。C3-HAhighマウスには、2.5×105(亜致死量;図5B)または8×105(導入後7〜14日の間に部分的な死亡率;図5C)のナイーブ6.5 CD4+ T細胞を、抗LAG-3抗体、対照ラットIgGIと組み合わせて、または抗体無しで受けさせた。8×105用量での死亡率は、抗LAG-3抗体による影響を受けなかったのに対して、2.5×105用量での死亡率は、抗LAG-3抗体注入で観察されなかった。
【図6】天然のCD4+CD25+ T細胞におけるLAG-3の役割。(図6A)天然のCD4+CD25+ T細胞は、これらのCD4+CD25-対応物と比較して高レベルのLAG-3 mRNA発現を有する。CD4+CD25+およびCD4+CD25-T細胞は、野生型BALB/cリンパ節から精製した。CD4+CD25+ T細胞(天然の調節性T細胞を含むことが実証されている集団)は、CD25およびLAG-3について、並びにCTLA-4、GITR、およびFoxp3について、これらのCD4+CD25-対応物(CD4+CD25-サブセットのそれぞれのmRNAの発現は、1の値に標準化した)と比較して、有意に高いmRNAレベルを有する。(図6B)LAG-3の表面染色では、これらのCD4+CD25-対応物と同様に、CD4+CD25+の天然調節性T細胞で陰性である。しかし、LAG-3の細胞内染色では、CD4+CD25+において陽性集団が示されるが、CD4+CD25-T細胞においては示されない。(図6C)インビトロ抑制アッセイ法では、T細胞刺激としての抗CD3抗体(0.5μg/ml)と共に、BALB/cマウスリンパ節に由来する選別されたCD4+CD25+ T細胞を応答者として使用し、CD4+CD25-T細胞をサプレッサーとして使用した(0.04:1のサプレッサー:エフェクター比率)。50μg/mlの濃度の抗LAG-3抗体では、天然のCD4+CD25+調節性T細胞のインビトロでの抑制を逆転するが、アイソタイプ対照抗体では、逆転しない。(図6D)Cでの抑制アッセイの後、CD4+CD25+細胞(Thy1.2マーキングによってエフェクター細胞から区別される)は、抗LAG-3またはアイソタイプ対照抗体で染色した。
【図7】CD25枯渇6.5 CD4+ T細胞における、変異型でなく野生型のLAG-3の異所性の発現は、強力なインビトロ調節活性を与える。6.5 CD4+ T細胞は、最初にあらゆるCD25+「天然の」Tregを枯渇させ、次いでGFP単独、GFP +野生型LAG-3、またはGFP+変異型LAG-3.Y73FΔCY(MHCクラスII組織に対する結合が減弱され、かつ下流のシグナリングを媒介することができない)のいずれかをコードするMSCVに基づいたレトロウイルスベクターを形質導入した。活動休止期の10日の後、MSCV-GFPベクターを形質導入したGFP+ 6.5CD4+ T細胞では、本質的に内因性LAG-3の染色は観察されなかったが、一方、MSCV-LAG-3/GFPおよびMSCV-LAG-3.Y73FΔCY/GFPベクターを形質導入したGFP+6.5の細胞では、高レベルのLAG-3染色が観察された。MSCV-LAG-3/GFPおよびMSCV-LAG-3.Y73FΔCY/GFPの形質導入に由来するGFP+細胞は、抗LAG-3抗体で明るく染色されたが、MSCV-GFP形質導入された細胞は、実質的にLAG-3染色を示さなかった。それぞれの群に由来するGFP+細胞を選別し、増殖アッセイ法において、異なる比率でAPC、5μg/ml HA110-120ペプチド、およびナイーブ6.5 CD4+CD25-細胞と混合した。
【図8】フォギン(Phogin)特異的なT細胞クローンでのLAG-3の異所性の発現が、NODマウス由来の脾細胞(splenyocytes)を同時導入後に、糖尿病からの保護を与えることを示す。107のプレ糖尿病性NOD脾細胞を単独で、またはベクター(MIG)LAG-3、もしくはシグナリング欠損がある変異体のLAG-3□Kを形質導入したPhogrin T細胞クローン4(John Huttonから得られた)細胞と組み合わせて、NOD/SCIDマウスに導入した。NOD/SCIDマウス(5/群)を糖尿病についてモニターした。
【発明を実施するための形態】
【0033】
発明の詳細な説明
LAG-3は、CD4関連の活性化で誘導される細胞表面分子であり、MHCクラスII組織と高親和性で結合する。本発明者らは、老いたLAG-3欠損マウスが、野生型対照よりも2倍のCD4+ およびCD8+ T細胞を有することを見いだした。LAG-3欠損T細胞は、MHCクラスII分子のLAG-3ライゲーションに依存的な、リンパ球減少宿主の恒常性の拡張の増強を示す。これは、野生型LAG-3の異所性の発現によって消失したが、シグナリング欠損変異体によっては消失されなかった。このT細胞恒常性の調節解除により、複数の細胞型の拡張を生じる。本発明者らのデータは、LAG-3がCD4+およびCD8+ T細胞の恒常性をネガティブに調整し、骨髄移植に続くT細胞移植を促進するための治療標的としてLAG-3が存在することを示唆する。
【0034】
CD223は、リンパ球抗原遺伝子-3またはLAG-3としても知られ、MHCクラスII分子と高親和性で結合する、CD4関連の活性化で誘導される細胞表面タンパク質である。Baixeras, E. et al., J. Exp. Med. 176:327 (1992)。URLアドレス: http file type, www host server, domain name ncbi.nlm.gov, directory PROW, subdirectory guide, document name 165481751_g.htm.のTriebel, F., "Lag-3 (CD223)", Protein Reviews on the Web (PROW) 3: 15-18 (2002); Triebel, F. et al.,"LAG-3, a novel lymphocyte activation gene closely related to CD4", J. Exp. Med. 171: 1393-1405 (1990)を参照されたい。CD223の代表的なマウスDNAおよびアミノ酸配列は、それぞれSEQ ID NOS:1および2に記載されている。また、GenBankアクセッションコードX9113も参照されたい。CD223の代表的なヒトのDNAおよびアミノ酸配列は、それぞれSEQ ID NOS:3および4に記載されている。また、GenBankアクセッション番号X51985も参照されたい。これらの配列は、一個体に由来する。位置の約5%未満が異なる対立遺伝子変異体が、集団に存在することが予想される。このような対立遺伝子変異体は、マウスまたはヒト起源のCD223の意味の範囲内に含まれる。
【0035】
調節性T細胞は、エフェクターT細胞を阻害することによって機能するT細胞のサブグループである。調節性T細胞は、CD223+であり、典型的には、CD4+CD25+でもある。調節性T細胞は、自己免疫の寛容および免疫応答のバランスをとる際に中心的役割を果たす。このような細胞は、抗体および当該技術分野において既知の分離技術を使用してCD223細胞から単離することができる。これらは、免疫アフィニティー・クロマトグラフィー、FACS、免疫沈降、その他を含むが、これらに限定されない。CD223+細胞は、自己免疫疾患、アレルギー、または喘息の患者に対して投与することができる。自己免疫疾患患者の場合、細胞を自己抗原でプレ活性化することができる。CD223-細胞は、同様に癌患者、細菌もしくはウイルス感染患者、またはAIDS患者に導入することができる。
【0036】
エフェクター/メモリーの表現型または抑制性の表現型のいずれかに分化する抗原特異的なCD4+ T細胞からの遺伝子発現アレイの比較解析により、LAG-3(MHCクラスII組織を結合するCD4相同体)のTreg特異的な発現が明らかにされた。LAG-3high CD4+ T細胞は、インビトロでサプレッサー活性を示し、LAG-3に対する抗体は、インビトロおよびインビボの両者における抑制を阻害する。これらの知見は、Treg特異的な受容体または補助受容体がサプレッサー活性を調整するものとしてLAG-3を同定する。従って、LAG-3を経たTreg細胞の操作は、自己免疫疾患、癌、および感染症の免疫療法を増強するために、並びにドナーリンパ球の注入、骨髄移植、および養子性(adoptive)のT細胞導入の環境においてリンパ球移植を増強するために使用することができる。
【0037】
CD223は、調節性T細胞の機能を調節する調節性T細胞特異的な細胞表面分子である。調節性T細胞の機能は、CD223活性を増強もしくは増大することによって、またはT細胞集団中のCD223+細胞の数を増大することによって増強されるであろう。被検者が自己免疫疾患に罹患しているなどの、このような反応が望ましくない状況において免疫T細胞反応を制限するために、生物体の調節性T細胞の機能の増強を使用してもよい。逆に、CD223活性を阻害することによって、またはT細胞集団中のCD223+細胞の数を減少させることによって、調節性T細胞の機能を阻害してもよい。癌、慢性感染症に罹患している患者、または骨髄移植レシピエントにおいてなど、このような反応が望ましい状況の免疫T細胞反応を増強するために、生物体の調節性T細胞の機能の阻害を使用してもよい。
【0038】
CD223タンパク質またはmRNAと結合する阻害剤で癌患者を治療するときに、抗腫瘍ワクチンを選択的に同時投与してもよい。このようなワクチンは、単離された抗原もしくは抗原群に、または全腫瘍細胞に対して作製されたものであってよい。化学療法薬と共に阻害剤を投与することが望ましいであろう。薬剤の混合物を使用して、複数の薬剤での治療を行うことは必要ではなく、別々の製剤を使用して行ってもよい。製剤は、同じ正確な時間で送達することは必要ではなく、同じ治療期間、すなわち1週もしくは1ヶ月、または相互に、患者に送達されるように調整してもよい。したがって、2つの活性成分を含む組成物が、患者の体内で構成されてもよい。いずれの適切な抗腫瘍治療も、CD223に対してターゲットされる本発明の治療と共に調整することができる。同様に、感染症を有する患者を治療する場合、その他の抗感染症薬を、CD223にターゲットされる本発明の治療と共に調整することができる。このような薬剤は、小分子薬物、ワクチン、抗体、その他であってもよい。
【0039】
T細胞集団におけるCD223+細胞の数は、CD223に対して選択的に結合する抗体またはその他の薬剤を使用して修飾することができる。CD223+細胞は、調節性T細胞の濃縮された集団を示し、調節性T細胞機能を増強するためにこれをT細胞の本来の供与源へ、または別の適合性の宿主に導入することができる。または、CD223-細胞は、調節性T細胞活性を欠損したT細胞の集団を表し、一般的なT細胞活性を保持したままで調節性T細胞機能を阻害または減少させるために、T細胞の本来の供与源に、または別の適合性の宿主に再び導入することができる。
【0040】
CD223活性を増大または減少する(調整する)ためのいずれの所望の手段も本発明の方法に使用することができる。これは、直接CD223タンパク質の機能を調整すること、CD223のシグナル伝達を調整すること、および転写もしくは翻訳または両方ともを調整することによってT細胞におけるCD223の発現を調整することを含む。これらのCD223活性を選択的に調整する手段は、非選択的なモジュレーターよりも好ましい。また、一時的なT細胞欠損を治療するためには、T細胞の集団に一過性CD223欠損を生じさせ、次いで正常レベルのCD223活性に戻る阻害手段が好ましいであろう。一過性に欠損したT細胞を使用して、CD223に関して遺伝的に正常であると考えられるT細胞と共に、減弱されたT細胞集団を再構成してもよい。このような一過性のT細胞欠損は、たとえば筋切除後に幹細胞導入を受けている患者において生じる。CD223活性の調整は、細胞に対してインビトロで、または動物全体に対して(インビボで)行うことができる。インビトロで処理した細胞を、細胞の本来の供与源または無関係な個体の患者のいずれかに投与することもできる。
【0041】
CD223の機能を阻害するために、CD223抗体または小分子阻害剤を使用することができる。このために有用な抗体または抗体断片は、CD223と結合して、これが機能する能力をブロックすることができるものである。このような抗体は、ポリクローナル抗体、モノクローナル抗体(たとえば、Workman, C. J. et al., "Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3)", Eur. J. Immunol. 32:2255-2263 (2002)を参照されたい)、キメラ抗体、ヒト化抗体、単鎖抗体、可溶性MHCクラスII分子、抗体断片、その他であってもよい。
【0042】
CD223ポリペプチドに対して作製される抗体は、CD223ポリペプチドを直接動物に注射することによって、またはCD223ポリペプチドを動物に、好ましくはヒト以外に投与することによって得ることができる。次いで、このようにして得られた抗体は、CD223ポリペプチド自体に結合すると考えられる。このように、CD223ポリペプチドの断片のみをコードする配列さえ、完全な天然のCD223ポリペプチドに結合する抗体を作製するために使用することができる。
【0043】
モノクローナル抗体の調製のためには、連続的な株化細胞培養によって産生される抗体を提供するいずれの技術を使用することもできる。例には、ハイブリドーマ技術(Kohler and Milstein, 1975, Nature, 256:495-497)、トリオーマ技法、ヒトB細胞ハイブリドーマ技術(Kozbor et al., 1983, Immunology Today 4:72)、およびヒト・モノクローナル抗体を産生するためのEBV-ハイブリドーマ技術(Cole, et al., 1985, in Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., pp. 77-96)を含む。
【0044】
一本鎖抗体を産生するために記載された技術(米国特許第4,946,778号)をCD223ポリペプチドに対する一本鎖抗体を作製するために容易に使用することができる。また、免疫原性のCD223ポリペプチドに対するヒト化抗体を発現するために、トランスジェニックマウスを使用してもよい。
【0045】
CD223の機能を増強または活性化するために、CD223のレベルまたはT細胞中の既存のCD223の活性を増大させる何らかの薬剤を使用してもよい。このような薬剤は、以下に記載されているスクリーニングアッセイ法を使用して同定してもよい。また、遺伝子量を増大させるために、CD223をコードする発現ベクターを投与することもできる。発現ベクターは、当該技術分野において既知のプラスミドベクターまたはウイルスベクターであることができる。特に望ましい性質について、実務者によりいずれのベクターを選択することもできる。
【0046】
本発明に従った治療を受け入れられる自己免疫疾患は、自己免疫溶血性貧血、自己免疫性血小板減少症紫斑病、グッドパスチャー症候群、尋常性天疱瘡、急性リウマチ熱、混合型本態性クリオグロブリン血症、全身性エリテマトーデス、インスリン依存型糖尿病、慢性関節リウマチ、グレーブス病、橋本甲状腺炎、重症筋無力症、および多発性硬化症を含む。当該技術分野において既知のように、自己免疫性T細胞は、自己免疫疾患患者から単離することができる。これらには、CD223のコード配列をトランスフェクトすることができる。いずれの好ましい発現ベクターも、CD223を発現するために使用することができる。これらには、プラスミドおよびウイルスベクターを含むが、限定されない。発現調節シグナルは、CD223自体に、またはその他の遺伝子に由来することができる。CD223発現構築物のトランスフェクション後、T細胞を患者に再び導入することができる。患者に血液細胞を注入するための方法は当該技術分野において周知である。
【0047】
特異的にCD223と結合する抗体、および抗癌ワクチンの混合物を含む組成物は、インビトロで作製することができる。好ましくは、組成物は、薬学的組成物として使用するために適した条件下で作製される。薬学的組成物は、無菌でも、発熱性物質なしでもよい。また、組成物の成分は、同時にこれら両方が患者の体内にあるような期間内に別々に患者に投与することもできる。このような時間を離した投与では、患者内で抗体およびワクチンの混合物の形成が引き起こされる。抗体およびワクチンが、時間を離した様式で投与される場合、これらは、キット内に共に供給してもよい。キット内において、成分は、別々にパックされても、または別々に含まれていてもよい。賦形剤、担体、その他の免疫変調成分またはアジュバント、抗体およびワクチンの投与のための説明書、並びに注射装置などのその他の成分も同様にキットに供給することができる。説明書は、書面、ビデオ、または音声形態であることができ、紙、電子媒体、またはウェブサイトもしくは参照マニュアルなどのさらに別の供与源に対する参照として含まれることができる。
【0048】
本発明の抗CD223抗体は、抗癌ワクチンに対する癌患者の抗癌応答の大きさを増大させるために使用することができる。また、癌患者の集団の応答者の数を増大させるために使用することができる。したがって、抗体は、抗癌ワクチンに耐性の患者で見いだされる免疫抑制を克服するために使用することができる。抗癌ワクチンは、完全な腫瘍細胞ワクチン、腫瘍抗原の1つもしくは複数のエピトープを含む単離された腫瘍抗原またはポリペプチドを含む(しかし、限定されない)、当該技術分野において既知のいずれのものであることもできる。
【0049】
T細胞におけるCD223の発現は、転写または翻訳のレベルで調整することができる。このような調整が可能な薬剤は、以下に記載されているスクリーニングアッセイ法を使用して同定することができる。
【0050】
CD223 mRNAの翻訳は、リボザイム、アンチセンス分子、小さな干渉RNA(siRNA; Elbashir, S. M. et al., "Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells", Nature 411:494-498 (2001)を参照されたい)、またはCD223 mRNAをターゲットするこのプロセスの小分子阻害剤を使用して阻害することができる。アンチセンス技術を使用して、トリプル-ヘリックス形成またはアンチセンスDNAもしくはRNAを介して遺伝子発現を制御することができ、これらの両方法ともDNAまたはRNAに対するポリヌクレオチドの結合に基づいている。たとえば、本発明の成熟ポリペプチドをコードするポリヌクレオチド配列の5'をコードする部分を、長さが約10〜40塩基対のアンチセンスRNAオリゴヌクレオチドをデザインするために使用する。DNAオリゴヌクレオチドを、転写に関与する遺伝子の領域に対して相補的となるようにデザインし(トリプル-ヘリックス−Lee et al., Nucl. Acids Res. , 6: 3073 (1979); Cooney et al, Science, 241: 456 (1988);および Dervan et al., Science, 251: 1360 (1991)を参照されたい)、これによりCD223の転写および産生を防げる。アンチセンスRNAオリゴヌクレオチドは、インビボでmRNAにハイブリダイズして、mRNA分子のCD223ポリペプチドへの翻訳をブロックする(アンチセンス−Okano, J. Neurochem., 56:560 (1991); Oligodeoxynucleotides as Antisense Inhibitors of Gene Expression, CRC Press, Boca Raton, Fla. (1988))。また、上記したオリゴヌクレオチドは、アンチセンスRNAまたはDNAがインビボで発現してCD223の産生を阻害するようなアンチセンス発現構築物によって細胞に送達することができる。このような構築物は、当該技術分野において周知である。
【0051】
アンチセンス構築物、アンチセンスオリゴヌクレオチド、RNA干渉構築物、またはsiRNA二重鎖RNA分子は、CD223の発現を妨害するために使用することができる。典型的には、アンチセンス分子には、少なくとも15、17、19、または21のCD223 mRNA配列の相補ヌクレオチドで十分である。典型的には、RNA干渉分子には、少なくとも19、21、22、または23のCD223のヌクレオチドで十分である。好ましくは、RNA干渉分子は、2つのヌクレオチドの3'オーバーハングを有するであろう。RNA干渉分子が細胞において構築物から、たとえばヘアピン分子から、または所望のCD223配列の逆方向反復から発現される場合、内因性の細胞機構がオーバーハングを作製する。siRNA分子は、化学合成、インビトロでの転写、またはRnase IIIもしくはDicerによる長いdsRNAの消化によって調整することができる。これらは、トランスフェクション、電気穿孔法、または当該技術分野において既知のその他の方法によって細胞に導入することができる。
を参照されたい。
【0052】
既知のモジュレーターに加えて、本発明の方法に有用であるCD223活性のさらなるモジュレーターは、CD223に結合する能力について候補分子をスクリーニングすること、または細胞培養中においてCD223活性を阻害する化合物をスクリーニングすることなどの、ツーハイブリッド・スクリーン、従来の生化学的アプローチ、および細胞に基づいたスクリーニング技術を使用して同定することができる。一例として、本発明者らは、活性化後でさえCD223を発現しない、ニワトリ卵白リゾチーム(HEL)の48-62特異的、H-2Ak拘束性マウスCD4+ T細胞ハイブリドーマ3A9を同定した。野生型CD223の異所性の発現(しかし、シグナリング欠損ではない)は、その特異的なペプチドに対するこのT細胞ハイブリドーマのIL-2反応を有意に減少させた。これは、CD223活性モジュレーターをスクリーニングするための簡単なインビトロ・アッセイ法の系を提供する。この後者の方法では、直接CD223と相互作用し、かつ調整する薬剤、並びにCD223シグナル伝達経路の工程に影響を及ぼすことによって間接的にCD223活性を調整する薬剤が同定されるであろう。
【0053】
CD223を発現する細胞を使用する細胞に基づいたアッセイ法では、哺乳類から単離されかつ天然にCD223を発現する細胞を使用することができる。または、CD223を発現するように遺伝的に操作された細胞を使用することもできる。好ましくは、遺伝的に操作された細胞は、T細胞である。
【0054】
CD223遺伝子発現を調整することによってCD223活性を調整する薬剤は、候補薬剤の存在下および非存在下で細胞のCD223タンパク質の量を測定することによって、細胞に基づいたスクリーニングアッセイ法で同定することができる。CD223タンパク質は、たとえば、抗CD223特異的なモノクローナル抗体を使用するフローサイトメトリーによって検出し、測定することができる。また、CD223 mRNAは、ノーザンブロット、RT-PCR、およびアレイ・ハイブリダイゼーションを含む(しかし、限定されない)当該技術分野において既知の技術を使用して検出し、測定することができる。
【0055】
CD223モジュレーターを同定するための1つの特に有用な標的配列はインビトロおよびインビボでのCD223機能に必須である、CD223の細胞質ドメインのアミノ酸のモチーフKIEELEである(SEQ ID NO:5)。このモチーフに結合する薬剤のためのスクリーニングアッセイ法により、候補CD223モジュレーターを同定し、CD223の阻害剤または活性化因子としてのその活性を、細胞に基づいたアッセイ法においてなど、さらなる試験によってさらに特徴づけることができる。このモチーフは、CD223の50以下の連続したアミノ酸残基からなるポリペプチドに含まれることができる。または、モチーフは、CD223の一部および第2の(非CD223)タンパク質の全てまたは一部を含む融合タンパク質に含まれることができる。第2のタンパク質は、天然のタンパク質であってもよく、またはたとえばヒスチジンタグ、もしくはその他の有用なポリペプチドの特徴を含む合成ポリペプチドであることができる。タンパク質-タンパク質結合アッセイ法は、当該技術分野において周知の、および任意の種々の技術および形式を使用することができる。
【0056】
CD223は、タンパク質の可溶性形態を得るために翻訳後にプロセシングすることができる。可溶性形態は、少なくともマウスのCD223のアミノ酸残基1〜431、および少なくともヒトのCD223のアミノ酸残基1〜440を含む。いずれの場合においても細胞質内末端部が失われている。同様に、膜貫通領域の全部または一部も失われている。この可溶性形態は、MHC クラスII-拘束性/CD4+ T細胞の反応を調整する。したがって、可溶性形態は、たとえば、自己免疫疾患患者、アレルギー患者、喘息患者、または癌患者に投与するために有用であろう。可溶性形態の投与は、注入、局所的、または静脈内の投与を含むいずれの便利な手段によるものであってもよい。
【0057】
本発明の教示によれば、CD223阻害剤は、生物体中のT細胞の数を増大させるために、生物体に投与されてもよい。この方法は、低いT細胞集団を生じる症状に罹患する生物体を治療するために有用であろう。このような症状は、AIDSなどの免疫不全によって生じる疾患、並びに外来微生物(細菌もしくはウイルス)の侵入などの不必要な細胞の侵入または増殖を含む障害、または腫瘍成長もしくは癌を含む。
【0058】
また、このようなT細胞欠損は、筋切除後に幹細胞転移を受けた患者にも予想される危険である。このような患者のT細胞は欠陥があり、これらが健康なドナーT細胞によって置換することができるように、破壊の対象となる。幹細胞から健康なT細胞集団を再構成するプロセスは、数ヶ月かかり、その期間の間に、患者は、生命の脅威となり得る日和見感染に非常に感受性である。ドナーT細胞のCD223を阻害するか、または、CD223欠損について選択もしくは操作されたドナーT細胞を使用することによって、T細胞分裂が増強され、T細胞再構成プロセスを促進することができ、T細胞欠損の期間を減少させることができる。
【0059】
また、CD223阻害剤は、T細胞増殖感受性の障害を治療するために従来の治療と組み合わせて投与されるときにも有用であろう。たとえば腫瘍は、T細胞増殖感受性の障害であり、従来は、急速に分裂している細胞を死滅させることによって機能する化学療法薬によって治療されている。本発明のCD223阻害剤は、化学療法薬と組み合わせて投与されるときに、T細胞増殖を刺激して腫瘍細胞の免疫拒絶を増強することによって化学療法薬の殺腫瘍効果を増強する。
【0060】
本発明の教示によれば、CD223活性化因子または発現エンハンサーは、生物体中のT細胞の数を減少させ、これにより有害なT細胞活性を減少させるために生物体に投与してもよい。この方法は、異常に多いT細胞集団または有害なT細胞活性を生じている症状たとえば、宿主T細胞によって媒介される移植片拒絶、移植片対宿主疾患、並びに慢性関節リウマチ、1型糖尿病、筋肉硬化症、その他などのT細胞を媒介した自己免疫疾患および炎症性疾患、に罹患した生物体を治療するために有用であろう。本発明の方法は、CD223を発現するT細胞を含むいずれの生物体に適用してもよい。これは、いずれの哺乳類も含むが、限定されず、特にヒトおよびマウスを含む。
【0061】
本発明の方法がインビボで行われるときは、使用されるCD223モジュレーターの有効な量は、使用する特定のモジュレーター、治療される特定の症状、治療されている被検者の年齢および身体症状、症状の重篤さ、治療の期間、併用療法の性質(ある場合)、投与の特異的な経路、並びに健康な開業医の知識および専門的意見の範囲内の同様の因子によって変更される。たとえば、有効な量は、個体が異常に抑制されたレベルのT細胞を有する程度に依存することができる。
【0062】
投与されるときに、本発明の薬学的製剤は、薬学的に許容される量で、および薬学的に許容される組成物内で適用される。このような製剤は、通常、塩、緩衝剤、防腐剤、適合性の担体、および任意にその他の治療薬を含んでいてもよい。医薬に使用するときに、塩類は、薬学的に許容されるべきであるが、薬学的に許容されない塩類を、これらの薬学的に許容される塩類を調製するために都合よく使用してもよく、本発明の範囲から除外されない。このような薬理学的に、および薬学的に許容される塩類は、以下の酸から調製されるものを含むが、限定されない:塩酸、臭化水素酸、硫酸、硝酸、リン酸、マレイン酸、酢酸、サリチル酸、クエン酸、ギ酸、マロン酸、コハク酸など。また、ナトリウム、カリウム、もしくはカルシウム塩などのアルカリ金属またはアルカリ土類塩類として薬学的に許容される塩類を調製することができる。
【0063】
CD223モジュレーターは、任意に薬学的に許容される担体と組み合わせてもよい。「薬学的に許容される担体」の用語は、本明細書に使用されるものとして、ヒトに投与するために適した1つまたは複数の適合性の固体もしくは液体の充填材、希釈液、またはカプセル化物質を意味する。「担体」の用語は、適用を容易にするために活性成分が組み合わせられる有機または無機の成分で、天然または合成のものを意味する。また、薬学的組成物の成分は、本発明の分子と、互いに所望の薬学的有効性を実質的に損なう相互作用がない様な方法で、共に混合することができる。
【0064】
薬学的組成物は、以下を含む適切な緩衝剤を含んでいてもよい:酢酸の塩溶液;クエン酸の塩溶液;ホウ酸の塩溶液;およびリン酸の塩溶液。また薬学的組成物は、任意に塩化ベンザルコニウム;クロロブタノール;パラベン、およびチメロサールなどの適切な防腐剤を含んでいてもよい。
【0065】
非経口投与のために適した組成物は、都合よくは抗炎症薬の無菌の水性製剤を含み、これは、レシピエントの血液と好ましくは等張性である。この水性製剤は、適切な分散剤または湿潤剤および懸濁剤を使用して、既知の方法に従って処方してもよい。また、無菌注射用製剤も、たとえば1,3-ブタンジオール溶液として、無毒の非経口的に許容される希釈液または溶媒中の無菌の注射用の溶液または懸濁液であってもよい。許容されるもののうち、使用してもよい媒体および溶媒は、水、リンゲル溶液、および等張食塩液である。加えて、従来には、無菌の不揮発性油が溶媒または懸濁媒として使用されている。このために、合成のモノ-またはジ-グリセリドを含むいずれの無刺激性の不揮発性油を使用してもよい。加えて、オレイン酸などの脂肪酸は、注射可能薬物の調製に使用してもよい。経口、皮下、静脈内、筋肉内、その他の投与のために適した担体製剤は、Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa.において見いだされる。
【0066】
種々の投与ルートを利用することができる。選択される特定の様式は、もちろん、選択される特定の薬物、治療される症状の重篤さ、および治療有効性のために必要とされる用量に依存する。本発明の方法は、一般的に言って、医学的に許容される(臨床的に容認できない副作用を生じさせることなく活性化合物の有効レベルを生じるいずれの方法をも意味する)いずれの投与方法を使用して実施してもよい。このような投与方法は、経口、直腸、局所的、経鼻、真皮内、または非経口的な経路を含む。「非経口的」の用語は、皮下、静脈内、筋肉内、または注入を含む。静脈内または筋肉内の経路は、特に長期の療法および予防には適していない。しかし、これらは、緊急事態には好ましいであろう。経口投与は、患者並びに投薬スケジュールに対して都合が良いため好ましいであろう。
【0067】
薬学的組成物は、単位用量形態で提供されるのが都合が良く、薬学の技術分野において周知のいずれの方法によって調製してもよい。全ての方法で、活性薬剤を1つまたは複数の補助成分を構成する担体と会合させる工程を含む。一般に、組成物は、均一に、かつ均質に、活性薬剤を液体担体、微粉固体担体、またはその両方と会合させて、次いで、必要に応じて産物を成形することによって調製される。
【0068】
経口投与に適した組成物は、それぞれが活性薬剤の予め定められた量を含む、カプセル、タブレット、ロゼンジなどの分離した単位として提供されてもよい。その他の組成物は、シロップ、エリキシル、またはエマルジョンなどの水性液体または非水性液体の懸濁液を含む。
【0069】
その他の送達システムは、徐放性、遅放性、または持効性の送達システムを含むことができる。このようなシステムにより、活性薬剤の反復投与を回避することができ、被検者および医師の便宜を増大させる。多くのタイプの放出送達システムが利用でき、当業者に既知である。これらは、ポリ(ラクチド-グリコリド)、コポリオキサラート、ポリカプロラクトン、ポリエステルアミド、ポリオルトエステル、ポリヒドロキシ酪酸、およびポリ無水物などの重合体塩基システムを含む。たとえば、薬物を含む前述の重合体のマイクロカプセルは、米国特許第5,075,109号に記載されている。また、送達システムには、非重合体系:コレステロール、コレステロールエステル、および脂肪酸などのステロールを含む脂質、またはモノ-、ジ-、およびトリ-グリセリドなどの中性脂肪;ヒドロゲル放出システム;シラスティック(sylastic)システム;ペプチドに基づいたシステム;ワックス被覆;従来の結合剤および賦形剤を使用する圧縮錠剤;部分的に融合されたインプラント;などを含む。具体例は、以下を含むが、これらに限定されない:(a)米国特許第4,452,775号、第4,667,014号、第4,748,034号および第5,239,660号に記載されているものなどの、抗炎症薬がマトリックス内の形態に含まれる浸食システム、並びに(b)米国特許第3,832,253号、および第3,854,480号に記載されているものなどの、活性成分が重合体から制御された割合で浸透する拡散システム。加えて、ポンプに基づいたハードウェア・送達システムを使用することもでき、これらのうちのいくつかは、移植に適応される。
【0070】
長期の持効性インプラントの使用は、慢性症状の治療のために特に適しているであろう。長期の放出とは、本明細書に使用されるものとして、インプラントが、少なくとも30日、および好ましくは60日間、治療レベルの活性成分を送達するように構築され、並べられることを意味する。長期の持効性インプラントは、当業者に周知であり、上記で記載されているいくつかの放出システムを含む。
【0071】
本発明では、目下のところ本発明を実施する好ましい様式を含む具体例に関して記載したが、当業者であれば、上記したシステムおよび技術の多数の変形および並び替えが、添付の請求の範囲に記載した本発明の趣旨および範囲の範囲内であることを理解すると考えられる。
【実施例】
【0072】
実施例1-LAG-3(CD223)によるT細胞恒常性のネガティブ制御
以下の実施例は、LAG-3(CD223)がCD4+およびCD8+ T細胞恒常性を負に調節することを示し、骨髄移植後のT細胞移植を促進するための新規の治療的な標的としてこれが同定されたことを支持する。
【0073】
野生型C57BL/6マウスは、4〜52週齢のαβ+ T細胞を一定数有する。前に報告された通り、若い4週齢のLAG-3-/-マウスは、正常なT細胞数を有する。Miyazaki, T. et al., Science 272: 405-408 (1996)。対照的に、LAG-3-/-マウスのαβ+ T細胞の数は、3ヶ月齢から、野生型マウスよりも2倍多くの数まで着実に増大する。この違いは、αβ+ T細胞数の厳密な恒常性の制御が非常に低い標準偏差によって明示されたことを考えると、非常に有意である。CD4+およびCD8+細胞はいずれもLAG-3-/-マウスにおいて増大されたが、CD4:CD8の比率は、不変であった。同様に、OT-II TCRをトランスジェニックしたLAG-3-/-マウス(卵白アルブミン326〜339特異的、H-2Ab拘束性)では、これらの違いが5週間目に明らかであったことを除き、野生型対照OT-IIトランスジェニックマウスと比較して、CD4+ Vα2+ T細胞の数が増大された。野生型マウスでは、ほぼ20%のαβ+ T細胞およびCD49b+ NK細胞が、LAG-3を構成的に発現し(Workman, C.J. et al., Eur. J. Immunol. 32: 2255-2263 (2002))、これらの数も、LAG-3-/-マウスにおいて有意に増大した。驚くべきことに、LAG-3を発現しないB220+B細胞、Gr-1+顆粒球、およびMac-1+マクロファージのようないくつかのその他の細胞タイプでも、対照マウスと比較してLAG-3-/-が増大された。LAG-3-/-マウスで観察される細胞数の増大は、インビボの分裂しているBrdU+細胞数の〜50%の増大に匹敵した。LAG-3-/-と野生型マウスの間で観察される細胞数の相違は、非常に一貫しており、再現性があった。LAG-3がないと、LAG-3-/-マウスに由来するT細胞の細胞表面表現系に対して有意な効果を有さないようにみえる。これらのデータは、LAG-3がマウスのT細胞数を調節し、一般に白血球数に間接的に影響を及ぼしているという考えを支持する。
【0074】
LAG-3がリンパ球減少環境においてT細胞の恒常的発現に影響を及ぼしているかどうかを決定するために、精製したT細胞を、T細胞およびB細胞を欠いたRAG-/-マウスに養子性に(adoptively)導入し、脾臓のT細胞数を導入の15日後に決定した。野生型対照と比較してLAG-3-/-T細胞数の2.8倍の増大があった。注目すべきことに、LAG-3の非存在がT細胞増殖に対して明らかに有効であるにもかかわらず、野生型T細胞のわずかな割合のみがLAG-3を発現した。これにより、LAG-3の短期の一過性の発現が、これが分裂細胞に対してその効果を及ぼすには十分であろうことが推論される。CD4+およびCD8+T細胞の増殖の増大が観察されたことから、両細胞タイプがLAG-3の不存在によって同等の影響を受けたことが証明される。興味深いことに、また、LAG-3-/-対LAG-3+/+ T細胞のレシピエントにおいてαβ-宿主由来細胞数の2倍の増加があった。これは、操作されていないLAG-3-/-マウスで観察されたマクロファージおよび顆粒球の数の増大と一致した。RAG-/-で観察されるLAG-3-/- T細胞の増殖の増大は、抗原特異性から独立していることを確認するために、本発明者らは、OVA [Ovalbumin 257-264-特異的、H-2Kb-拘束性;Hogquist, K.A. et al., Cell 76: 17-27 (1994)]およびOT-II [Ovalbumin 326-339-特異的、H-2Ab-拘束性; Barnden, M.J. et al., Immunol. Cell Biol. 76: 34-40 (1998)]トランスジェニック・マウスから精製したT細胞を使用した。野生型CD4+ Vα2+ OT-II T細胞は、以前の報告と一致してRAG-/-マウスにおいて十分に増殖せずに、これらの細胞がリンパ球減少性宿主においてほとんど恒常的な増殖を示さないことを示している。Ernst, B. et al., Immunity 11: 173-181 (1999)。対照的に、この制限は、LAG-3-/- OT-IIトランスジェニックマウスからのT細胞には適用されず、これは、リンパ球減少性の宿主において活発に、導入後15日までに野生型T細胞の3.2倍以上の数に増殖した。同様に、RAG-1-/-マウスから回収したLAG-3-/- CD8+ Vα2+ OVAトランスジェニックT細胞の数は、野生型の対照OVA T細胞よりも4倍高かった。特筆すべきは、この相違は、導入後少なくとも1ヶ月間持続されたことである。これらのデータは、また、CD4+およびCD8+ T細胞がLAG-3の減少により同様に影響を受けることを証明した。MHCクラスII分子によるLAG-3のライゲーションの重要性を評価するために、LAG-3-/-および野生型OVAトランスジェニックT細胞を、MHCクラスIおよびクラスII分子(β2m-/- x H-2Aβb-/-)を両方とも欠いているマウスに導入した。データは、LAG-3-/- T細胞の増殖の増大がMHCクラスII分子の非存在下で消滅されることを明らかに示しており、この相互作用の重要性が証明された。
【0075】
LAG3-/-マウスまたはLAG-3-/- T細胞の養子性のレシピエントは、B細胞およびマクロファージなどの通常LAG-3陰性である細胞の数を増大した。これは、LAG-3の非存在によるT細胞の恒常性の制御の変化は、その他の白血球細胞型の制御を直接変化させるという考えを支持する。直接これを試験するために、B細胞を、LAG-3-/-または野生型T細胞のいずれかと共にRAG-/-マウスに同時導入した。また、本発明者らは、T細胞恒常性を調節する際のMHCクラスII分子の対照的な役割を評価するためにも、このアプローチを利用した。以前の研究では、CD4+ T細胞の恒常的な増殖および長期の生存には、MHCクラスII分子との周期的な相互作用が必要であることを明らかに証明した。Takeda, S. et al., Immunity 5: 217-228 (1996); Rooke, R., et al., Immunity 7:123-134 (1997)。対照的に、LAG-3とMHCクラスII分子との間の相互作用は、逆の効果を有する可能性もある。以前に示したように、MHCクラスII-/- B細胞を導入したときに、野生型対照と比較してLAG-3-/- T細胞の数に3.0倍の増大がある。しかし、野生型B細胞の存在下では、LAG-3-/-とLAG-3+/+ Tとの間の細胞数の違いは、4.9倍に増大した。LAG-3-/- T細胞数の増大は、MHC:TCRの相互作用が増大され、したがって増殖が増強されるためである可能性が高い。対照的に、LAG-3+/+ T細胞は、正(MHCを経て:TCR相互作用)および負(MHCを経て:LAG-3相互作用)の恒常性調節の両方に供されて、野生型T細胞に匹敵する増殖を生じるであろう。
【0076】
野生型T細胞の存在下において、B細胞の数は、導入7日後に脾臓から回収したB細胞の数は、B細胞単独を受けているマウスと同じであった。対照的に、LAG-3-/- T細胞レシピエントから回収したB細胞の数には、2.7倍の増加があり、B細胞数の増大は、LAG-3-/- T細胞の「調節解除」によるものであることの直接の実証を提供する。興味深いことに、野生型T細胞の存在下において、B細胞単独を受けているマウスと比較して、MHCクラスII-/- B細胞の数が増大する。これは、LAG-3:MHCクラスIIの相互作用が「局部的」になくなると、レシピエントRAG-/-マウスは、脾臓内にMHCクラスII+マクロファージおよび樹状細胞を有するものの、野生型T細胞の一過性の調節解除により、B細胞増殖の増大を生じ得るという考えを支持する。別の可能性としては、LAG-3によるMHCクラスII分子のライゲーションが負の調節シグナルをB細胞に送達することにより、増殖を妨げることである。これは、B細胞に対してはもっともらしいが、これによっては、LAG-3-/-マウスにおける顆粒球などのMHCクラスII細胞の数の増大が説明されない。現在調査している1つの可能性としては、LAG-3-/- T細胞の調節解除された増殖により、多くの細胞タイプの広範な増殖を誘導するサイトカインの生産をこれらが生じることである。
【0077】
リンパ球減少性マウスにおける恒常的な増殖に対するLAG-3発現の影響は、ナイーブT細胞に限定されない。また、抗原を受けた「メモリー」OT-II T細胞の導入により、LAG-3T3-/-T細胞は、野生型の対照細胞と比較して、実質的に増殖の促進を生じた[7.2倍]。LAG-3が、この「調節解除された」T細胞増殖の直接の原因であり、本来のターゲティング戦略によって破壊される密接に連関した遺伝子ではないことを確かめることが、非常に重要であった。従ってLAG-3-/- OT-II T細胞には、野生型LAG-3またはシグナリング欠損がある変異体LAG-3.ΔKMのいずれかを含むマウス幹細胞ウイルス(MSCV)に基づいたレトロウイルスを形質導入した。Workman, C. J. et al., Eur. J. Immunol. 32: 2255-2263 (2002)。また、ベクターは、形質導入された細胞の解析を容易にするために、内部リボソーム導入部位(IRES)および緑色蛍光タンパク質(GFP)カセットを含んだ。Persons, D. A. et al., Blood 90: 1777-1786 (1997)。また、LAG-3-/-およびLAG-3+/+ OT-11のT細胞には、「空」ベクター/GFP単独の対照を形質導入した。形質導入された細胞をRAG-1-/-レシピエントに導入して、導入の15日に回収したOT-II T細胞の数を決定した。予想されるとおり、LAG-3-/- GFP単独対照T細胞では、野生型GFP細胞よりも増殖した[2.8倍]。LAG-3の異所性の発現は、OT-11 T細胞の数を野生型対照と同等のレベルに減少させたが、一方のLAG-3シグナリング欠損がある変異体の発現は、恒常的な増殖に対して効果を有さない。これらのデータは、LAG-3が観察される効果の直接の原因となることを証明する。
【0078】
本発明者らのデータは、LAG-3がT細胞の恒常的な増殖を負に調節することを明らかに示す。また、これらは、T細胞が多くの細胞型の恒常性に関与しているであろういう考えを支持する。LAG-3の非存在下で、ノックアウトマウスにおけるT細胞数、およびリンパ球減少性マウスにおけるT細胞の増殖に対する明白な効果を有したにもかかわらず、T細胞の非常に少ない割合だけがLAG-3を発現したことが目立った。興味深いことに、全てのT細胞でのLAG-3の異所性の発現が、恒常的な増殖に対して、野生型細胞で見られるLAG-3の低レベルの一過性発現よりも優れた効果を有するというわけではなかった。これは、LAG-3シグナリングの閾値が非常に低いであろうこと、およびLAG-3シグナリングの効果を制限するその他の因子があるかもしれないことを示唆する。LAG-3と相互作用する下流のシグナリング分子(群)を同定し、LAG-3が恒常的な増殖を調節する機構を決定することは、将来の研究の明らかに重要な焦点である。
【0079】
B細胞およびマクロファージなどのLAG-3を発現しない細胞数の増大は、驚くべき観察であった。同時導入実験では、T細胞上にLAG-3が存在しないことが観察したその他の細胞タイプの増加の原因であったことを明らかに証明した。これは、その他の細胞タイプの数および/または増殖を制限するLAG-3シグナリングによって誘導されるか、またはその他の細胞タイプの数および/または増殖を制限するLAG-3によって負の調節が存在しないために産生される、可溶性または細胞表面タンパク質によるものであろう。この傍観者(bystander)の増殖およびその生理的役割の正確な性質は、決定されないままである。
【0080】
骨髄移植またはメガ用量幹細胞移植を受けている患者は、リンパ球再構成の速度が緩徐であるために、最初の4〜6ヶ月において特に感染症に感受性である。本発明者らの研究は、LAG-3が生存可能な治療の標的であること、およびLAG-3の発現またはその機能のブロッキングがT細胞移植を促進し、この感受性のウインドウを有意に減少させるという考えを支持する。
【0081】
実施例2−材料および方法
本実施例は、実施例1のための実験法および材料を提供する。
【0082】
マウス:以下のマウスを使用した:LAG-3-/- [Christophe Benoist and Diane Mathis, Joslin Diabetes Center, Boston, MA; Miyazaki, T. et al., Science 272: 405-408 (1996)からの許可によって、Yueh-Hsiu Chen, Stanford University, Palo Alto, CA,から得られる]; C57BL/6J [Jackson Labs, Bar Harbor, ME]; B6.PL-Thylα/Cy (Thy1.1類遺伝子性) [Jackson Labs]; RAG-1-/- [Jackson Labs, Bar Harbor, ME; Mombaerts, P. et al., Cell 68: 869-877 (1992)]; MHCクラスII-/- [Peter Doherty, St. Jude Children's Research Hospital, Memphis, TN; Grusby, M.J. et al., Science 253:1417-1420 (1991)によって提供された]; MHCクラスI-/-/II-/-[Taconic, Germantown, NY; Grusby, M.J. et al., Proc. Natl. Acad. Sci. U.S.A 90: 3913-3917 (1993)];OT-II TCRトランスジェニック・マウス[William Heath, Walter and Eliza Hall Institute, Parkville, Victoria Australia; Barnden, M.J. et al., Immunol Cell Biol. 76: 34-40 (1998)からの許可とともに、Stephen Schoenberger, La Jolla Institute for Allergy and Immunology, La Jolla, CA, によって提供された]、およびOT-I(OVA)TCRトランスジェニックマウス[Jackson Labs; Hogquist, K.A. et al., Cell 76: 17-27 (1994)]。ゲノム全般のマイクロサテライト解析では、LAG-3-/-マウスに関して試験した88の遺伝マーカーの97%がB6マウスに由来することを証明した(Charles River Laboratories, Troy, NY)。LAG-3-/-、MHCクラスII-/-、OT-I. LAG-3-/-およびOT-II. LAG-3-/-コロニーは、St. Jude Animal Resource Centerで維持した。全ての動物実験は、国家、州、および施設の指針に従って、AAALAC公認のSPF施設で行った。動物プロトコルは、St. Jude IACUCによって承認された。
【0083】
LAG-3構築物およびレトロウイルスの形質導入:LAG-3構築物は、記載されているとおりに組換えPCRを使用して作製した(Vignaali, D.A.A. and K. M. Vignali, J.Immunol. 162: 1431-1439 (1999))。LAG-3.WTおよびLAG-3.ΔKM(細胞質尾部の保存されたKIEELEモチーフを欠失するLAG-3)は、記載されている(Workman, C.J. et al., J. Immunol. 169: 5392-5395 (2002))。LAG-3構築物を、内部リボソーム導入部位(IRES)および緑色蛍光タンパク質(GFP)を含むマウス幹細胞ウイルス(MSCV)に基づいたレトロウイルスベクターにクローニングし、記載されているようにレトロウイルスを作製した(Persons, D.A. et al., Blood 90: 1777-1786 (1997); Persons, D.A. et al., Blood Cells Mol Dis. 24: 167-182 (1998))。次いで、24時間後に105/ml以上のウイルス力価が得られるまで、GPE+86細胞を繰り返して(7〜10回)形質導入することによってレトロウイルス産生株の株化細胞を作製した(Markowitz, D. et al., J Virol. 62: 1120-1124 (1988))。
【0084】
フローサイトメトリー:単個細胞懸濁液を脾臓から作製し、RBCをゲイ液で溶解した。脾細胞は、まずFc Block、抗CD16/CD32(2.4G2)(BD PharMingen, San Diego, CA)と共に氷上で10分間染色した。次いで、細胞をBD PharMingenからの種々の抱合型抗体を使用して以下の細胞表面マーカーを染色した:
。LAG-3発現は、ビオチン化されたラット抗LAG-3 mAb(C9B7W, IgG1κ; Workman, C.J. et al., Eur. J. Immunol. 32: 2255-2263(2002))またはPE抱合体として得た同じ抗体(BD PharMingen)によって評価した。次いで、細胞をフローサイトメトリー(Becton Dickinson, San Jose, CA)によって解析した。
【0085】
ブロモデオキシウリジン取り込み:5、16、28、および52週間目に、LAG-3+/+、LAG-3-/-、OTII.LAG-3+/+、およびOTll.LAG-3-/-マウスには、(0.8mg/ml)それらの飲料水中にBrdU(Sigma, St. Louis, MO)を8日間与えた。次いで、マウスをCO2吸入によって屠殺して、脾臓を取り除いた。BrdU取り込みの染色は記載されているように行った(Flynn, K. J. et al., Proc. Natl. Acad. Sci. U.S.A 96: 8597-8602 (1999))。簡潔には、LAG-3-/-およびLAG-3+/+脾細胞をTCRαβ、CD4、CD8、およびB220発現のために染色した。OTII.LAG-3+/+およびOTII.LAG-3-/-脾細胞は、Vα2およびCD4発現のために染色した(PharMingen)。次いで、細胞を1.2mlの氷冷95%エタノールで氷上で30分間固定した。次いで、細胞を洗浄し、PBS +1%パラホルムアルデヒド +0.01% Tween 20で室温において1時間透過化処理した。次いで、細胞を洗浄し、50KUのデオキシリボヌクレアーゼ(Sigma)の0.15M NaCl+4.2mM MgCl2(pH 5.0)溶液で37℃において10分間インキュベートした。BrdUを抗BrdU-FITC(Becton Dickinson)をRTで30分添加することによって検出し、次いでフローサイトメトリーによって解析した。
【0086】
養子性の導入実験:脾細胞からのT細胞および/またはB細胞をFACSによって陽性選別するか、または磁気ビーズ細胞選別(MACS)によってネガティブに選別した。FACS精製のために、脾細胞をTCR□□、CD4、およびCD8発現について染色し、およびMoFlow(Cytomation, Ft. Collins, CO)で陽性選択によって選別した。ネガティブMACS精製のためには、脾細胞をPE結合抗B220、抗Gr1、抗Mac1、抗TER119(赤血球)、抗CD244.2(NK細胞)、および抗CD8(OTII トランスジェニックT細胞のネガティブ精製のために)で染色した。次いで、細胞を抗PE抗体と結合された磁気ビーズ(Miltenyi Biotech, Auburn, CA)と共にインキュベートし、自動MACS(Miltenyi Biotech, Auburn, CA)で90〜95%の純度に選別した。いくつかの実験では、T細胞をカルボキシフルオレセインジアセテートスクシンイミジルエステル(CFSE)でラベルした。細胞をPBSで2回洗浄し、1×107細胞/mlでPBS+0.1% BSAに再懸濁し、5μM CFSEと共に37℃で10分間インキュベートした。細胞をPBS+0.1% BSAで2回洗浄した。CFSE標識または無標識の精製T細胞(5x106または1×107)、および場合によってはB細胞(5×106)をRAG-1-/-または Thyl.1+(B6.PL)マウスにi.v.注射した。
【0087】
正常T細胞のレトロウイルスによる形質導入:OTII.LAG-3+/+およびOTII.LAG3-/-マウスからの脾臓を取り出し、単細胞懸濁液を2.5×106細胞/mlで作製した。脾細胞をOVA 326-339ペプチド[10μM]で2日間培養液中で活性化した。次いで、活性化された脾細胞をGFP単独、LAG-3.WT/GFP、またはLAG-3.ΔKM/GFPレトロウイルス産生株細胞の単層上で、ポリブレンの存在下において2日間インキュベートした。細胞を10日間静止させ、次いでFACSによってVα2+/CD4+/GFP+発現を選別した。細胞をさらに2日間静止させ、次いで5×106細胞を、尾静脈を経てRAG-/-マウスに注射した。導入の15日後、マウスをCO2吸入によって屠殺し、脾臓を取り除いた。脾細胞を染色し、フローサイトメトリーによって解析した。
【0088】
実施例3−強力な調節活性で誘導されるTreg細胞
Treg特異的な分子を同定するために、本発明者らは、ウイルス感染に応答してエフェクター/メモリー細胞か、または自己抗原としての同族抗原に遭遇することによってTreg細胞のいずれかに分化する抗原特異的なT細胞の差次的遺伝子発現解析を行った。この解析により、LAG-3遺伝子がTreg細胞において選択的にアップレギュレートされることが明らかになった。LAG-3(MHCクラスIIに結合するCD4相同体)の生理的な役割は、はっきり解明されなかった。いくつかのインビトロでの実験では、LAG-3が負の調節機能を有しているかもしれないことを示唆した(Hannier et al., 1998; Huard et al., 1994; Workman et al., 2002a; Workman et al., 2002b)。ここで、本発明者らは、LAG-3の膜発現がCD25とは独立して選択的にTreg細胞をマークすること、およびLAG-3がインビトロおよびインビボの両方でTreg細胞の抑制性の活性を調整することを示した。
【0089】
T細胞エフェクター/メモリーと寛容誘導との間の相違を検討するために、本発明者らは、モデル抗原の赤血球凝集素(HA)に特異的なT細胞受容体(TCR)トランスジェニックCD4+ T細胞(クローン6.5)の養子性の導入を利用した。組換えHA発現ワクシニアウイルス(Vac-HA)を感染させた野生型マウスでは、養子性に導入したHA特異的な6.5 CD4+ T細胞が、HAと遭遇することによってエフェクター/メモリー細胞に分化した。エフェクター/メモリー反応は、典型的な増殖/収縮期およびメモリーマーカーの発生によって特徴づけられる。養子性に導入された動物から取り出したときに、これらのエフェクター/メモリー細胞は、抗原特異的な増殖反応およびγ-インターフェロン生産によってアッセイすると、ナイーブ6.5 CD4+ T細胞と比較して、インビトロでHAに対して応答性亢進である。このメモリー反応は、養子性の導入後に何ヶ月も持続する。対照的に、複数の上皮組織にHAを発現するC3-HAトランスジェニックマウスへの6.5 CD4+ T細胞の養子性導入では、寛容を生じる(Adler et al., 2000; Adler et al., 1998)。エフェクター/メモリー反応と同様に、γ-インターフェロンなどのエフェクターサイトカインの増殖および発現によって特徴づけられる迅速な増殖/活性化期がある。しかし、活性化期の後に、全HA特異的T細胞プールが縮小し、残りの6.5細胞は、養子性導入4〜7日後に抗原刺激によってγ-インターフェロンを産生したり、またはインビトロで増殖することができない(Adler et al., 2000; Huang et al., 2003)。IL-2およびγ-インターフェロンなどのリンホカインを産生して抗原に対する応答を急増する能力の消滅は、アネルギー性の表現型の標準的な実効の定義を表す。
【0090】
寛容の誘導に先だつC3-HAマウスにおける最初のインビボエフェクター期の強度は、養子性に導入した6.5 CD4+ T細胞の数、並びにレシピエントマウスでのHA抗原の発現レベルに比例する。従って、C3-HAlowマウスは、2.5×106 6.5 CD4+ T細胞の導入をかなりよく許容するが、C3-HAlowマウスよりも1000倍多くのHA発現を有するC3-HAhighマウスでは、2.5×106の6.5 CD4+ T細胞の導入後4〜7日以内に死亡する(図1A)。死因は、HA発現が最も高い肺におけるトランスジェニック6.5 CD4+ T細胞の浸潤による致死肺血管炎である。2.5×105未満の6.5 CD4+ T細胞をC3-HAhighマウスに養子性導入すると、肺血管炎の重篤さの軽減が引き起こされて、レシピエントが生存する(図1A)(Huang et al., 2003)。興味深いことに、亜致死量で導入された6.5 CD4+ T細胞では、その後の保護されていないC3-HAhighマウスにおいて致死量の6.5 CD4+ T細胞であると考えられる注入による死からマウスを保護することができるので、これらは節性の表現型を獲得する。わずか8,000細胞の導入(致死量の0.3%)ほどでその後の2.5×106のナイーブ6.5 CD4+ T細胞の注入による死から動物を完全に保護するので、このインビボでの調節機能は極めて強力である。保護は、最初の導入の4日後という早い時期に観察され、6ヶ月まで活性なままである(図1A)。養子性導入前のCD4+ T細胞(しかし、CD8+ T細胞ではない)の枯渇により、保護作用が全くなくなり、これによりアネルギーとなったクローン型(clonotypic)6.5 CD4+ T細胞のTreg表現型を定義する。
【0091】
致死間質性肺炎の抑制は、肺における最初の投入(Treg)の6.5T細胞の蓄積、および第2の注入からの浸潤するTエフェクター細胞の数の急激な減少を伴う。肺に蓄積する代わりに、Treg細胞がない場合に生じるように、Tエフェクター細胞は、脾臓の動脈周囲のリンパ性外筒に蓄積する(図1B)。アネルギー細胞がTreg機能を証明するというさらなる証拠は、これらがインビボで細胞障害性のHA特異的CD8+ T細胞の活性化を阻害するという知見から生じる(データ示さず)。第1の(保護)養子性導入より前にCD25+ T細胞を除去しても、その後の6.5 T細胞の致死的なチャレンジから動物を保護することができるTreg細胞の発生には影響を及ぼさなかった。従って、最初の投入のT細胞のTreg表現型は、養子性に導入された集団の中の天然に存在するTreg細胞の結果とは反対に、養子性の導入の後で獲得された可能性が高い。これらの知見は、発行されたVon Boehmerおよび同僚の知見(彼らは、6.5 CD4+ T細胞が、実際にTreg機能を示すB細胞区画にHAを発現するトランスジェニックマウスに導入された後、寛容になることを証明した)と非常に一致している。(Jooss et al., 2001)。
【0092】
実施例4−LAG-3は、誘導されたTreg細胞上に差次的に発現される
本発明者らのインビボの系でアネルギー性/Treg表現型と関係する遺伝子を同定するために、本発明者らは、非トランスジェニックレシピエント内に養子性に導入し、続いてVac-HA免疫化を行ってエフェクター/メモリーT細胞を作製した後の、または、C3-HAhighマウス内に導入してアネルギー性の/Treg細胞を作製した後の、いずれかの精製した6.5 CD4+ T細胞に対してアフィメトリックス・チップ解析を行った。Thy1.1(+)Thy1.2(-)類遺伝子性の6.5 T細胞は、CD8+ T細胞、B細胞、およびThy1.2(+) T細胞のMACS Column枯渇、続いてフローサイトメーターによって>95%の純度に選別することを含む経時的な単離手順を使用して、野生型(エフェクタ/メモリー)またはC3HAhigh(アネルギー性/Treg)レシピエントを感染させたThy1.1(-)Thy1.2(+) Vac-HAから精製した。このプロトコルでは、TCRまたはCD4依存的な遺伝子発現パターンを潜在的に変更し得るTCR特異的またはCD4補助受容体特異的な抗体の使用を避けている。
【0093】
RNAは、チップ解析のために、0日の試料としてナイーブ6.5 CD4+ T細胞から単離し、また養子性導入後2、3、および4日での6.5 CD4+ T細胞から単離した。アネルギー性/Treg集団とエフェクター/メモリー集団との間で差次的に発現された遺伝子を0〜4日からのこれらの差次的発現を合計したアルゴリズムに従って順序づけられた順位にした。驚くほど多数の遺伝子が、アネルギー性/Treg集団において、これらの養子性導入後の初期の時点でさえも、選択的に活性化された。これらの遺伝子の多くは、既知の機能のないESTを示した。以前に同定された遺伝子の中で、LAG-3は、エフェクター/メモリー集団と比較してアネルギー性/Treg集団において最も差次的に発現するものの一つであった。その後、この結果を、養子性導入の1ヶ月後まで及ぶ種々の時点について、LAG-3プライマー-プローブ対での定量的RT-PCR解析によって確認した。エフェクター/メモリー細胞における最小の最初の増大後、LAG-3発現は、養子性導入後20日までにベースラインに戻った。著しく対照的に、LAG-3の発現は、アネルギー性/Treg細胞群の中で最初の5日にわたって20〜50倍増大し、その後の4週の解析にわたって高いままである(図2A)。対照的に、FoxP3、GITR、およびCTLA-4のレベルは、最初の4〜5日にわたって適度な増大を示し(1.5〜4倍)、エフェクター/メモリー細胞および誘導されたアネルギー性/Treg細胞の両方においても同様であった(データ示さず)。
【0094】
次いで、エフェクター/メモリー6.5 CD4+ T細胞と比較した、アネルギー性/Treg 6.5 CD4+ T細胞の集団でのLAG-3の細胞表面発現を、抗LAG-3-モノクローナル抗体を使用して解析した(Workman et al., 2002b)(図2B)。エフェクター/メモリー細胞に対するLAG-3染色は非常に低レベルであるが、C3-HAhighトランスジェニックマウスからのアネルギー性/Treg細胞のうちの有意な比率が、遺伝子発現の結果と一致して、適度から高度のレベルのLAG-3染色を示した。IL-10は、一般にTregの分化および機能と関連しているので(Moore et al., 2001)、本発明者らは、IL-10 mRNAの内因性レベルおよびこれらのC3-HAhighトランスジェニックマウスからのCD4+ T細胞サブセット(アネルギー性/Treg 6.5 CD4+ T細胞)のLAG-3 mRNAのレベルとの相関を解析した。多くの実験にわたる複数の試料のアネルギー/Treg集団の解析により、LAG-3 mRNAレベルとIL-10 mRNAレベルの間の相関が明らかとなり、0.87の相関係数(R2)を有した(図2C)。
【0095】
実施例5−LAG-3は、最大のTreg機能のために必要とされる
アネルギー性/Treg 6.5 CD4+ T細胞の集団でのLAG-3およびCD25の細胞表面発現を抗LAG-3および抗CD25モノクローナル抗体を使用して協調的に解析した。同比率のエフェクター/メモリーおよびアネルギー性/Treg細胞がCD25を発現するが(データ示さず)、アネルギー性/Treg細胞でのLAG-3およびCD25発現は、完全には調和性でなかった(図3A)。従って、本発明者らは、細胞をLAG-3high CD25high、LAG-3high CD25low、LAG-3low CD25high、およびLAG-3low CD25low集団に選別し、標準的なインビトロ抑制アッセイ法でこれらの調節活性を解析した。ナイーブ6.5 CD4+細胞の中での増殖反応のインビトロ抑制では、LAG-3high CD25high集団が最高の抑制活性を示すこと、およびLAG-3low CD25low集団が最も低いが、LAG-3high CD25lowおよびLAG-3low CD25low細胞の抑制活性は、同等であったことを示した(図3B)。これらの結果は、誘導されたTreg細胞の中で、LAG-3およびCD25の組み合わせが、最も抑制活性を有するTreg細胞を特徴付けるであろうことを示唆する。
【0096】
誘導されたTreg細胞によって抑制を調節する際のLAG-3の直接的な役割をさらに評価するために、本発明者らは、まずナイーブHA特異的T細胞のインビトロでの増殖反応をLAG-3発現細胞が抑制する能力を、抗LAG-3抗体がブロックすることができるかどうかを決定した。2μg/mlの濃度の抗LAG-3抗体は、インビトロアッセイ系でTreg 6.5 CD4+ T細胞による抑制を阻害する(図4)。2日のアッセイ期間にわたって、抗LAG-3抗体は、Tregの非存在下で刺激される6.5T細胞の増殖反応に影響を及ぼさず、抗LAG-3抗体の効果が、実際にTreg細胞に対するものであり、エフェクター細胞ではなかったことを確認した(データ示さず)。抗LAG-3抗体がTreg細胞によるインビトロでの抑制をブロックする能力は、LAG-3が単にTreg選択マーカーでなく、Treg活性を調整する分子であることを証明する。
【0097】
実施例6−LAG-3は、インビボで誘導されるTreg活性に必要とされる
本発明者らは、次に、抗LAG-3抗体の投与がC3-HAhighマウスにおいてTregによる致死的間質性肺炎の抑制をブロックすることができるかどうかを決定することによって、インビボでのTreg機能を調整する際のLAG-3の役割を評価した。C3-HAhighマウスを8,000(亜致死量)の6.5 CD4+ T細胞で前処理し、続いて最初の導入の4日後に、その後の2.5×106の用量のナイーブ6.5 CD4+ T細胞を前処理した。上述のように、Tregは、この時点ですでに発生していた。抗LAG-3抗体(200□g)をその後の2.5×106の6.5細胞のチャレンジと共にi.v.投与し、2日後に別に200□gを与えた。この抗体治療では、完全にTreg細胞のインビボ抑制活性を除去し、マウスは、保護的な亜致死量の6.5の前処理を伴わずに致死的にチャレンジしたC3-HAhighマウスと同等の時間枠で死んだ。これに反して、アイソタイプ対照抗体(ラットIgG1)または抗体なしで処理した確立されたTregを有するマウスは、その後の2.5×106ナイーブ6.5 T細胞でのチャレンジでも生存した(図5A)。これらの結果は、抗LAG-3抗体がインビボでTreg活性をブロックしたことを示唆するが、他の形式では、直接Treg細胞を阻害するのではなく、抗LAG-3抗体は、チャレンジ集団のT細胞を超活性化(hyper-activate)し、その結果これらがTregの阻害作用を克服した可能性がある。この可能性を除外するために、本発明者らは、わずかに死亡率閾値以下の6.5 T細胞の用量と共に、抗LAG-3抗体をインビボ投与すると、予め確立されたTreg集団の非存在下での致死性を引き起こすかどうかを問うた。従って、本発明者らは、2.5×105の6.5 T細胞(死亡率を生じさせないであろう最大量)または8.0×105の6.5 T細胞(導入の7〜14日後におよそ50%の死亡率)を、抗LAG-3抗体またはアイソタイプ対照と共にC3-HAhighマウスに投与した。図5Bは、抗LAG-3処理では、2.5×105の6.5T細胞の用量でも致死性にならず、また8.0×105の6.5T細胞の用量の部分的な死亡率も増強しなかったことを証明する。従って、図5Aの実験の抗LAG-3抗体の効果は、Treg細胞の直接阻害であった。
【0098】
実施例7−LAG-3は、天然のTreg細胞によって発現され、抑制活性に必要とされる
合わせて考えると、これらのデータは、誘導されたTregの抑制機能を媒介する際のLAG-3の重要な役割を確信させる。誘導されたTregと天然のTregの間の関係は、不明なままであることを考えると、LAG-3は、野生型マウスからのCD4+CD25+ T細胞に選択的に発現したかどうかを調べることに興味が持たれる。LAG-3 mRNA(CTLA-4、FoxP3、およびGITR mRNAとともに)は、実際にCD4+CD25-細胞と比較してCD4+CD25+細胞上に選択的に発現されている(図6A)。この再現性の知見にもかかわらず、本発明者らは、CD4+CD25+またはCD4+CD25-細胞上のいずれにおいても、抗体染色によって表面LAG-3を検出することができなかった。しかし、透過化処理された細胞の抗体染色では、10〜20%のCD4+CD25+細胞がLAG-3の細胞内貯臓を表したことを明らかに示した。対照的に、透過化処理されたCD4+CD25-細胞の染色では、絶対にLAG-3+集団がないことを証明した(図6B)。これらの知見は、少なくともいくつかの天然のTregがLAG-3の細胞内貯臓を有し、これが、同族の抗原と遭遇することによって細胞表面に迅速に動員され、その後に抑制を媒介するのであろうことを示唆する。天然のTregは、CD4およびCD25によって定義されるT細胞群に含まれるが、実際のTreg細胞は、細胞内LAG-3を発現するものである可能性も実際にはある。天然のTregの調節機能におけるLAG-3の役割を直接評価するために、本発明者らは、抗LAG-3抗体が、精製されたCD4+CD25+細胞によって媒介されるインビトロでの抑制を阻害することができるかどうかを調べた。図6Cは、抗LAG-3抗体が、精製されたCD4+CD25+細胞によって媒介される抑制を実際にブロックすることを証明しており、LAG-3は、天然並びに誘導されたTregによって媒介される抑制において役割を果たしていることを示唆する。インビトロでの抑制アッセイ法の最後にCD4+CD25+細胞を染色すると、およぼ20%が現在もそれらの表面上に高レベルのLAG-3を発現することが明らかになり、細胞内LAG-3がTCRの結合環境下で表面に動員され、調節活性を媒介するという考えを支持する(図6D)。
【0099】
実施例8−LAG-3の異所性の発現は、調節活性を与える
図5および6のブロッキング実験は、LAG-3が最大のTreg機能のために必要とされることを示唆する。この結論をさらに確証するために、本発明者らは、T細胞上のLAG-3の異所性の発現が調節活性を与えるかどうかを決定するための一連の形質導入実験を行った。これらの実験のために、6.5 CD4+ T細胞から最初にCD25+「天然」のTregを枯渇させ、次いでGFP単独、GFP+野生型LAG-3、またはGFP+変異体LAG-3.Y73FΔCY(実質的にMHCクラスIIに対する親和性が減少され、かつ下流のシグナリングを媒介することができない(Workman et al., 2002a))のいずれかをコードするMSCVに基づいたレトロウイルスベクターを形質導入した。10日の休止期の後、MSCV-GFPベクターを形質導入したGFP+6.5 CD4+ T細胞では、本質的に内因性LAG-3の染色は観察されなかったが、MSCV-LAG-3/GFPおよびMSCV-LAG-3. Y73FΔCY/GFPを形質導入したGFP+ 6.5細胞では、高レベルのLAG-3染色が観察された。増殖アッセイ法では、それぞれの群に由来するGFP+細胞を選別して、APC、HA110-120ペプチド、およびナイーブ6.5CD4+CD25-細胞と種々の比率で混合した。図7に示したとおり、野生型LAG-3を発現する6.5細胞は、ナイーブ6.5細胞の増殖を強力に抑制したが、対照GFP導入6.5細胞または非機能的LAG-3.Y73FΔCY変異体を発現する6.5細胞では観察されなかった。本アッセイ法において、ナイーブ6.5細胞に加えてGFPおよびLAG-3.Y73FΔCYを導入した6.5細胞自体も増殖したので、総増殖は、実際にこれらの2つの後者の群でやや増大した。実際に、野生型LAG-3を導入したT細胞自体は、非導入6.5細胞の増殖阻害とは別に、増殖応答の有意な減少を示した。これらの結果は、抑制の際のLAG-3の機能的な役割を確証させる直接の証拠を提供する。興味深いことに、LAG-3導入によっても、Foxp3、CD25、CD103、およびGITRを含むその他のTreg関連遺伝子は誘導しなかった(データ示さず)。この結果は、エフェクター/メモリー対アネルギー/Treg表現形に分化する6.5T細胞の間にこれらの遺伝子の有意な差次的発現がないことに加え、LAG-3は、Foxp3経路とは独立に調節性T細胞機能の異なった経路を媒介するであろうことを示唆する。
【0100】
実施例9−考察
これらの知見から、LAG-3は、Treg細胞で選択的にアップレギュレートされ、Treg機能を媒介する際に直接関与しているであろう細胞表面分子であると同定される。天然および誘導性のTreg活性が定義されている多くの系を考えると、LAG-3は、「普遍的な」Tregマーカーであるか、または選択的に特定のTregサブセットのみをマークするかは決定されていないままである。本発明者らの結果は、CD4+ Treg細胞を誘導することに加えて、LAG-3は、天然のCD4+CD25+Treg細胞による抑制を媒介する際に少なくともいくらかの役割を果たしていることを示唆する。さらに、その他の実験データからも、天然のTregによる恒常性リンパ球増殖の調節におけるLAG-3の役割も証明される(Workman and Vignali、添付論文)。LAG-3発現が野生型マウス由来のCD4+CD25+ T細胞の中で有意に多いという知見は、これが天然のTreg、並びに誘導されたTregの機能において役割を果たしているであろうこと示唆する。図3の実験によって示唆されるように、LAG-3とCD25の組み合わせは、最も強力なサプレッサー活性を有するTregサブセットを定義するであろう。本発明者らは、LAG-3は、インビトロでのアッセイ法での調節活性の大きさと相関する可変レベルで発現されるので、Tregの「系統マーカー」であることを提案しない。実際に、Tregが非細胞自律的様式で寛容を促進することができる安定な系統または分化状態を表すかは明白でない(von Boehmer, 2003)。異なる系のTreg機能について、異なる機構が同定された(Shevach, 2002に概説)。LAG-3high細胞は、IL-10の量の増大を生じ、インビトロでの抑制活性の増強を示すが、本発明者らの系において抑制機能を媒介する際のIL-10の役割は、決定されていないままである。LAG-3に対する抗体は、インビトロおよびインビボの両方でTreg細胞のサプレッサー活性を阻害する。従って、本発明者らは、LAG-3がこのT細胞サブセットのサプレッサー活性を調整するTreg特異的な受容体または補助受容体であることを提案する。
【0101】
LAG-3について細胞自立的な抑制的な役割が多くの研究で示唆されているが(Huard et al., 1994; Workman et al., 2002b)、LAG-3 KOマウスによる最初の研究では、明白な自己免疫または高免疫についての何らかの証拠を発見することができなかった(Miyazaki et al., 1996)。Treg機能におけるLAG-3についての本発明者らが提案した役割を考えると、LAG-3ノックアウトマウスは、これらのマウスにおいて報告されなかった多システムの自己免疫を示すであろうことが予測されるだろう(すなわち、Foxp3ノックアウトまたはふけマウス(scurfy mice)と同様)。しかし、細胞恒常性の調節の欠陥などの、LAG-3ノックアウトマウスによって示される調節性T細胞の欠損が明らかに存在した(Workman and Vignali、添付論文)。本発明者らは、PD-1ノックアウトマウスで観察したように(Nishimura et al., 1999; Nishimura et al., 2001)、遅発性の自己免疫のより微妙な証拠について以前のLAG-3ノックアウトマウスを再検査している途中である。また、その他の調節機構がLAG-3発現の減少を補うようにこれらのマウスにおいて増強されるかもしれないことが考えられる。
【0102】
Treg細胞に高レベルで発現されているので、LAG-3は、癌および自己免疫疾患の両方を治療するためにTreg活性を選択的に操作するための優れた潜在的な標的を提供する。CD25「ゴールドスタンダード(gold standard)」Tregマーカーは、IL-2受容体複合体の重要な成分であり、活性化された細胞において高レベルで誘導されている。CD4+CD25+細胞がTreg活性の際に濃縮されていることの明らかな理由は、CD25がTreg機能に特異的であるためではなく、むしろTreg細胞が、末梢において自己抗原と持続的に遭遇することによって慢性的に刺激されるためである。より最近では、TNF受容体スーパーファミリーメンバー18分子(GITRとも呼ばれる)が、Treg細胞上でアップレギュレートされることが証明された。さらに、GITRに対する抗体は、インビボおよびインビトロの両方でTreg活性を阻害することが報告された。しかし、GITRは、活性化されたT細胞上で同等にアップレギュレートされており、したがって、明らかにTreg細胞のマーカーとしてCD25よりも選択的ではない(McHugh et al., 2002; Shimizu et al., 2002)。そのうえ、CD4+CD25-細胞群は、一定の免疫機能を抑制することができるという多数の報告がある(Annacker et al., 2001; Apostolou et al., 2002; Curotto de Lafaille et al., 2001; Graca et al., 2002; Shimizu and Moriizumi, 2003; Stephens and Mason, 2000)。それにもかかわらず、CD25highLAG-3high細胞が最も優れた抑制活性を示すという知見は、これらの細胞表面分子の両方に対する抗体を、Treg活性を操作するために協調的に使用してもよいことを示唆する。
【0103】
本発明者らのデータは、LAG-3が天然および誘導されたTreg細胞の最大の抑制活性に必要とされることを示す。しかし、それは十分であるか?これまでに、活性化されたT細胞上の調節活性を与えることが示された唯一の分子は、Foxp3である(Fontenot et al., 2003; Hori et al., 2003)。重要なことに、本発明者らは、ここでLAG-3(しかし、機能的に欠損がある変異体でない)のCD4+ T細胞におけるその異所性の発現が調節活性を与えることができることも示した。
【0104】
もう一つの重要な疑問は、Treg細胞が直接的なT-T相互作用を介して、またはDC中間体を介してCD4+およびCD8+エフェクター細胞の反応性を抑制するかどうかということである。MHCクラスII結合分子のLAG-3のTreg選択的および機能的な発現の同定により、これらの細胞が重要な役割を果たしている疾患のために、機構を分析し、およびTreg機能を操作するに際して新たな足掛かりを提供するはずである。
【0105】
実施例10−実験法
トランスジェニックマウス
C3-HAトランスジェニックマウスは、以前に記載されている(Adler et al., 2000; Adler et al., 1998)。手短には、インフルエンザウイルスA/PR/8/34(Mount Sinai strain)に由来する赤血球凝集素(HA)遺伝子をラットC3(1)プロモーターの制御下に配置した。2つの初代系統をB10.D2遺伝的バックグラウンドで確立した。これらの2つの初代系統、C3-HAhighおよびC3-HAlowは、それぞれ30〜50および3つの導入遺伝子コピーを含み、肺および前立腺を含む非リンパ系の組織の同一セットにおいてC3-HAハイブリッドmRNAを発現する。C3-HAhighとC3-HAlowとの間の総HAタンパク質発現の相違は、発現レベルが最も高い肺および前立腺では直接測定しなかったが、相違は、ハイブリドーマのサイトカイン放出が誘導された組織抽出物の生物検定によって示すと、およそ1000倍である。
【0106】
I-Ed-制限されたHAエピトープ(110SFERFEIEPKE120; SEQ ID NO:7)を認識するTCRを発現するTCRトランスジェニックマウス系統6.5株(Dr. Harald von Boehmer, Harvard University, Boston, MAによって寛大に提供された)をB10.D2遺伝的バックグラウンドに対して9世代戻し交配した。Kd拘束性HAエピトープを認識するTCRを発現するその他のTCRトランスジェニックマウス系統クローン-4(518IYSTVASSL526;SEQ ID NO:8)(Dr. Linda Sherman, Scripps Research Institute, La Jolla, CAによって寛大に提供された)も、Thy 1.1/1.1 B10.D2遺伝的バックグラウンドに対して9世代を超えて戻し交配した。クローン型(clonotypic)抗体は、クローン-4 TCRに利用できないので、Thy 1.1を代わりのマーカーとして使用した。クローン-4マウスの成熟CD8+ T細胞のほとんど全てがKd制限されたHAエピトープを直接認識するので、Thy 1.2/1.2レシピエントへの養子性導入に続き、本発明者らは、全てのThy 1.1+CD8+ T細胞がHA-特異的なクローン型TCRを発現するとみなすことができる(Morgan et al., 1996)。
【0107】
実験に使用されるトランスジェニックマウスは、8〜24週の間の年齢であった。マウスの使用を含む全ての実験は、Johns Hopkins University School of Medicineの動物実験委員会(Animal Care and Use Committee)によって承認されたプロトコルに従って行われた。
【0108】
養子性の導入
クローン型CD4+またはCD8+ T細胞は、6.5またはクローン-4トランスジェニックマウスのプールされた脾臓およびリンパ節から調製した。クローン型の割合は、フローサイトメトリー解析によって決定した。活性化マーカーCD44を解析して、これらのクローン型細胞がドナーマウスにおいて活性化されず、表現型がナイーブであったことを確認した。HBSSで3回洗浄した後に、尾静脈を介してi.v.注射するために、適切な数の細胞を0.2mlのHBSSに再懸濁した。
【0109】
免疫組織化学
組織を養子性の導入の3日後にマウスから集めた。組織をイムノヒストフィックス(Immuno Histo Fix)(A Phase sprl, Belgium)に4℃で3日間固定し、次いでイムノヒストワックス(Immuno Histo Wax)(A Phase sprl, Belgium)に埋め込んだ。ビオチンラベルした抗Thy1.1 mAb(PharMingen, San Diego, CA)を使用して連続切片を染色した。ベクタステインABCキット(Vectastain ABC kit:Vector, Burlingame, CA)およびNovaRed(Vector)は、発色のために使用した。切片をヘマトキシリンQS(Vector)で対比染色した。ニコンEclipse E800を使用して切片を解析した。最終的な画像処理は、Adobe PhotoShop (Mountain View, CA)を使用して行った。
【0110】
インビボで初回抗原刺激した6.5 CD4+ T細胞の濃縮および精製
インビボで養子性導入の後に、エフェクター/メモリーまたは寛容のいずれかを誘導すると、レシピエントマウスの脾臓の6.5 CD4+ T細胞のクローン型の割合は、0.2%〜5%だけである。アフィメトリックス遺伝子チップ解析のためなど、さらなる研究のために十分なクローン型CD4+ T細胞を得るためには、慎重な濃縮および精製が必須である。ドナー6.5 T細胞をThy1.1(+)Thy1.2(-)遺伝的バックグラウンドに対して交配した(これは、Thy1.1(-)/Thy1.2(+)レシピエントに養子性の導入後に2工程の濃縮および精製手順でできる)。CD8+ T細胞、B細胞、およびレシピエントT細胞(Thy1.2+)を枯渇させるために、最初にビオチン化された抗CD8(Ly-2、53-6.7)、抗B220(RA3-6B2)、および抗Thy1.2(30-H12)抗体(全てBD Biosciences PharMingen, San Diego, CAから購入)、並びにMACSストレプトアビジン・ミクロビーズおよびMACS LS分離カラム(Miltenyi Biotech, Auburn, CA)を使用して6.5 CD4+ T細胞を濃縮した。CD4+ T細胞およびCD8+ T細胞は、Thy1.1を有する唯一の集団であるので、またCD8+ T細胞が濃縮の間に減少したために、 FACSVantage SE細胞ソーター(BD Biosciences)を使用してThy1.1(+)細胞をソーティングすることにより、非常に精製された6.5 CD4+ T細胞(95%)にした。この技術では、TCRまたはCD4依存的な遺伝子発現パターンを変更する可能性のあるTCR特異的またはCD4補助受容体特異的な抗体の使用を回避する。
【0111】
遺伝子チップ解析
選別した細胞を、Qiashredderカラム(Qiagen, Valencia CA)で剪断し、続いてRNeasyキット(Qiagen)を使用して総RNAを単離した。cDNAは、Superscript Choiceキット(Gibco/BRL)およびHPLCで精製したT7-DTプライマー(Proligo, Boulder, CO)を使用して合成した。ビオチン化したcRNAプローブは、ENZO BioArray RNA転写物キット(Affymetrix, Santa Clara, CA)を使用して調製した。マウスの遺伝子チップU174A、B、およびCには、標準的なアフィメトリックス・プロトコルに従ってハイブリダイズさせて解析した。
【0112】
アネルギー/Treg誘導とエフェクター/メモリー誘導との間のCD4+ T細胞の遺伝子の差次的発現のランク付け
養子性の導入後の種々の日に、精製したナイーブ6.5クローン型CD4+ T細胞、並びにアネルギー性/Tregおよびエフェクター/メモリー6.5クローン型のCD4+ T細胞から調製したmRNAをアフィメトリックス遺伝子チップによって解析した。アネルギー/Treg誘導とエフェクター/メモリー誘導との間の遺伝子の差次的発現は、「距離」によってランクを付けた。距離は、養子性の導入後の2日(|d1|)、3日(|d2|)、および4日(|d3|)目の、アネルギー性T細胞とエフェクター/メモリーT細胞との間の発現の絶対的な相違の合計として定義し、標準化のためのナイーブCD4+ T細胞(n)の値で割った。
【0113】
抗体および染色
以下の抗体を使用した。精製したか、またはPE結合したかのいずれかの抗LAG-3(C9B7W、PharMingenから)(Workman et al., 2002b);精製したか、またはFITC結合したかのいずれかの抗CD25(7D4、PharMingenから);および抗GITR(R&D Systemsから購入したポリクローナル抗体)。LAG-3およびCD25の細胞表面染色のために、6.5+/-Thy1.1+/-トランスジェニックマウスからの脾細胞を単離し、CD4+ネガティブ選択単離キット(Miltenyi Biotec)を使用してCD4+について濃縮した。フローサイトメトリーによって決定した約2.5×106のクローン型6.5細胞(総CD4+細胞の16%)をHBSSに再懸濁し、137(C3-HA high)または野生型B10.D2内に尾静脈を介して注射した。B10.D2マウスの1群には、5×106のVac-HAを処理し、その一方で、その他の群には、ナイーブ対照のために無処置のままにした。脾細胞および鼠径部および腋窩リンパ節を5日後に集め、単個細胞懸濁液に調製した。RBCをACK溶解緩衝液で溶解した。直ちに細胞を5μgの全ラットIgG(Sigma)で15分間ブロックした後に、抗6.5 TCR-ビオチン+SA-APC、LAG-3-PE、およびCD25-FITC、または対応するアイソタイプ対照で染色した。抗6.5ビオチン以外は、全ての染色試薬をPharmingenから購入した(San Diego, CA)。短いインキュベーション後に、試料をPBS+1% FBS溶液中で一度洗浄し、FACScalibur機に読み込んだ(BD, San Jose, CA)。
【0114】
誘導された6.5調節性T細胞のインビトロでの抑制アッセイ法
1×104の精製したナイーブ6.5 CD4+ T細胞(応答者)および1×105の3000-radを照射した同系のB10.D2脾細胞(抗原提示細胞)を異なる数のサプレッサー6.5 CD4+ T細胞と共に混合し、10μg/mlのHAクラスII(110SFERFEIFPKE120;SEQ No:7)ペプチドと共に丸底96穴組織培養プレートにおいて、200μlのCTL培地中でインキュベートした。48〜72時間後、培養液を1μCiの3H-チミジンでパルスし、さらに16時間インキュベート後、パッカード・マイクロメート細胞収穫器(Packard Micromate cell harvester)で収集した。取り込まれた放射性カウントの量の決定は、パッカード・マトリックス96ダイレクトベータカウンター(Packard Matrix 96 direct beta counter:Packard Biosciences, Meriden, CT)で行った。
【0115】
天然の調節性T細胞のインビトロでの抑制アッセイ法
野生型BALB/cマウスを天然のTregアッセイ法に使用した。5×104のフローサイトメトリーで選別したCD4+CD25-T細胞(応答者)および5×104の3000-radの照射を受けたBALB/c脾細胞(抗原提示細胞)を、フローサイトメトリーで選別した異なる数のCD4+CD25+サプレッサーT細胞と共に混合し、0.5μg/mlの抗CD3抗体を有する丸底96穴組織培養プレートにおいて、200μlのCTL培地中でインキュベートした。48〜72時間後、培養液を1μCiの3H-チミジンでパルスし、さらに16時間インキュベート後、パッカード・マイクロメート細胞収穫器(Packard Micromate cell harvester)で収集した。取り込まれた放射性カウントの量の決定は、パッカード・マトリックス96ダイレクトベータカウンター(Packard Matrix 96 direct beta counter:Packard Biosciences, Meriden, CT)で行った。
【0116】
定量的リアルタイムPCR解析
Trizol試薬を使用して、RNA抽出のために選別した6.5 CD4+ T細胞を直ちに使用した(Invitrogen, Carlsbad, CA)。逆転写は、スーパースクリプト・第1鎖標準合成系(Superscript First Strand Synthesis System:Invitrogen, Carlsbad, CA)によって行った。cDNAレベルは、Taqman系でのリアルタイム定量的PCRによって解析された(Applied Biosystems, Foster City, CA)。それぞれの試料は、タックマン・ユニバーサル・PCRマスターミックス(Taqman Universal PCR Master Mix)およびABIプリズム7700配列検出システム(ABI Prism 7700 Sequence Detection system)を使用して、25μlの最終反応体積で、内部基準として18S rRNAと共に標的遺伝子について2回または3回アッセイした。IL-10およびIL-2の検出のためには、予め作製した反応試薬(PDARs)をApplied Biosystemsから購入した。LAG-3、CD25、GITR、およびIFN-γについては、プライマー対およびプローブセットは、プライマー・エキスプレス(Primer Express)ソフトウェアを使用してデザインし、次いでApplied Biosystemsによって合成した。Foxp3に使用するプライマーおよびプローブセットは、文献(S4)からのものを引用した。相対的なmRNA頻度は、内部対照18S RNAに対し基準化することによって決定した。簡単には、本発明者らは、標的遺伝子と18S RNAとの間の閾値サイクル(Ct)の相違:ΔCtsample=(Ctsample-Ct18s)を使用してそれぞれの試料セットを基準化した。較正試料は、アッセイ法(ΔCtcalibration)のそれぞれのセットにおいて最も高いΔCtを有する試料として割り当てた。相対的mRNA頻度は、2ΔΔCtとして算出し、式中ΔΔCt=(ΔCtcalibration-ΔCtsample)である。使用したプライマーおよびプローブセットは以下の通りである:
。
【0117】
LAG-3構築物およびレトロウイルス産生株の株化細胞
LAG-3構築物は、記載されたとおりに組換えPCRを使用して作製した(Vignali and Vignali, 1999)。Lag-3.WTおよび機能的ヌル変異体LAG-3.Y73F.ΔCY(LAG-3のMHCクラスIIを結合する能力を非常に減少させる点変異を有する細胞質末端のLAG-3)は記載されている(Workman et al., 2002a)。LAG-3構築物を、内部リボソーム導入部位(IRES)および緑色蛍光タンパク質(GFP)を含むマウス幹細胞ウイルス(MSCV)に基づいたレトロウイルスベクターにクローニングし、記載されているようにレトロウイルスを作製した(Persons et al., 1997; Persons et al., 1998)。次いで、24時間後に105/ml以上のウイルス力価が得られるまで、GPE+86細胞を繰り返して(7〜10)回形質導入することによってレトロウイルス産生株の株化細胞を作製した(Markowitz et al., 1988)。
【0118】
CD4+/CD25- T細胞のレトロウイルスによる形質導入およびインビトロでの抑制アッセイ法
6.5マウスからの脾細胞をビオチンラベルした抗B220、抗Gr1、抗Mac1、抗TER119、抗CD49b、抗CD8、および抗CD25抗体(PharMingen, San Diego,CA)で染色した。次いで、細胞をストレプトアビジンに結合させた磁気ビーズと共にインキュベートして、90〜95%の純度のCD4+/CD25- T細胞になるよう自動MACS(autoMACS: Miltenyi Biotech, Auburn CA)でネガティブに選別した。精製した6.5 CD4+/CD25- T細胞を、プレートに結合した抗CD3(2C11)および抗CD28(35.71)によって活性化した。刺激の2日および3日後に、活性化されたT細胞(4×105細胞/ml)には、上記したベクター単独、LAG-3.WT/GFP、またはLAG-3.Y73F.ΔCY/GFP レトロウイルスGPE+86産生株株化細胞(6μg/ml)プラスIL-2およびポリブレンからのウイルス上清をスピン形質導入(spin transduce)(90min、3000rpm)した。細胞を10日間静止することができ、次いで上位30〜35% GFP+/Thy1.2+ T細胞に対して選別した。
【0119】
インビトロ抑制アッセイ法については、精製したGFP+ T細胞(2.5×104で開始して2倍希釈)を、2.5×104のCD4+/CD25- 6.5T細胞(ネガティブに自動MACSによって精製した)、5×104の照射(3000rads)を受けた脾細胞、および5μg/mlのHA110-120と共に96穴丸底プレートにおいて培養した。細胞を72時間培養し、培養の最後の7〜8時間1μCi/ウェルの[3H]チミジンでパルスした(Du Pont, Wilmington, DE)。
【0120】
実施例11
CD223は、細胞表面から切断されて、可溶性形態(sLAG-3)で放出される。これは、インビトロで活性化されたT細胞によって有意な量で産生され(5μg/ml)、またマウスの血清中にも見いだされる(80ng/ml)。これは、細胞表面プロテアーゼによって産生される可能性が高い。本発明者らは、ウエスタンブロットによってsLAG-3を検出した。切断は、膜貫通領域(たとえば、SEQ ID NO:2のアミノ酸442〜466)に、またはアミノ末期の直前のコネクタ領域(たとえば、SEQ ID NO:2のアミノ酸432〜441)に生じる。
【0121】
実施例12
上記のように、LAG-3は、最大の調節性T細胞(Treg)機能に必要かつ十分である。言い換えると、LAG-3単独の発現でも、細胞を活性化されたエフェクターT細胞から調節性T細胞に変換するために十分である。
【0122】
本発明者らは、次に、LAG-3を異所性に発現する細胞もインビボで調節性の機能を示し、疾患状況において保護されるかどうかを決定したいと考えた。本発明者らは、自己抗原特異的なT細胞上のLAG-3の異所性の発現が、マウスを1型糖尿病から保護することができるかどうかについて問うことを選択した。この実験系では、糖尿病を起こしやすいNODマウスからの脾細胞を、リンパ球を欠如するNOD-Scidマウスに養子性に導入した。全てのマウスが3ヶ月以内に糖尿病を発症する。本発明者らの予備解析では、NOD脾細胞によって誘導される糖尿病の発症が、LAG-3を形質導入したホグリン(phogrin)特異的なT細胞によって防止されるが、シグナリング欠損がある変異体またはGFP対照では防止されないことを示唆する。これらのデータは、LAG-3を有する自己抗原特異的なT細胞の異所性の発現を、多くの自己免疫疾患、喘息、およびアレルギーの治療のための新規治療に使用するという考えを支持する。
【0123】
参考文献
【技術分野】
【0001】
発明の技術分野
本発明は、治療薬および薬物をスクリーニングする方法に関する。
【0002】
本出願は、2003年2月28日に出願された米国特許出願第60/451,039号、2003年6月24日に出願された米国特許出願第60/482,143号、および2003年12月22日に出願された米国特許出願第60/531,704号に対する優先権を主張する。
【0003】
米国政府、国立衛生研究所は、本発明の基礎をなす研究のための資金を提供した(AI39480)。その資金提供協定の期間の下で、米国政府は、本発明に一定の権利を保持する。
【背景技術】
【0004】
発明の背景
患者における進行性の免疫抑制の発症により、多様な疾患が特徴づけられる。悪性腫瘍を有する患者における免疫応答障害の存在は、特によく考証されている。癌患者および腫瘍を有するマウスは、遅延型の過敏症の減少、溶解機能の減少、およびリンパ球の増殖反応などの種々の免疫機能の変化を有することが示された。S. Broder et al., N. Engl. J.Ned. 299: 1281 (1978); E.M. Hersh et al., N. Engl. J. Med., 273: 1006 (1965); North and Burnauker, (1984)。その他の疾患または介入の多くが、免疫応答障害の発症によって特徴づけられる。たとえば、進行性の免疫抑制は、後天性免疫不全症候群(AIDS)、敗血症、ライ病、サイトメガロウイルス感染、マラリアなどを有する患者、並びに化学療法および放射線療法を使用する患者で観察された。しかし、免疫応答のダウンレギュレーションの原因となる機構は、完全に解明されないままである。
【0005】
免疫応答は、複雑な現象である。Tリンパ球(T細胞)は、全ての細胞性免疫反応の発症に重要である。ヘルパーT細胞は、免疫応答の発現を制御し、調整する。細胞障害性T細胞(キラーT細胞)は、感染した標的細胞を溶解することにより、細胞内寄生体およびウイルスに対する免疫反応において重要な役割を果たすエフェクター細胞である。また、細胞障害性T細胞は、免疫監視機構を介して体を癌の発生から保護することに関係している。調節性T細胞は、Tヘルパー細胞の誘導および/または活性をブロックする。T細胞は、通常遊離の抗原を認識しないが、その他の細胞の表面上でそれを認識する。これらのその他の細胞は、T細胞分裂を刺激することができる分化した抗原提供細胞であってもよく、または体内でウイルスに感染した、細胞障害性T細胞の標的になる細胞であってもよい。
【0006】
細胞障害性T細胞は、通常、全ての有核細胞上に発現されているクラスI主要組織適合性抗原(MHC)産物と会合して抗原を認識する。ヘルパーT細胞、およびインビトロで抗原に応答して増殖する大部分のT細胞は、クラスIIMHC産物に会合して抗原を認識する。クラスII産物は、大部分が抗原提供細胞上に、およびいくつかがリンパ球上に発現される。また、T細胞は、モノクローナル抗体によって定義されるこれらの細胞膜糖タンパク質を基礎として2つの主要亜集団に分けることができる。通常62kD糖タンパク質を発現するCD4+サブセットは、クラスII抗原の状況で抗原を認識し、一方でCD8+サブセットは、76Kd糖タンパク質を発現し、クラスI MHCの状況で抗原を認識することに制限される。
【0007】
リンホカインの注入を経た免疫易感染性動物の免疫応答を増強する、養子免疫治療は、不定に適用され、成功が限られていた。この種の治療を改善するための方法が必要である。たとえば、リンパ球、血液、およびその他の細胞の注入は、一定の設定で免疫不全の患者に提供される。しかし、健康なT細胞集団の再構成を促進し、増強することにより、このような患者に対して有意に増大された利益および有効性を提供することができる。
【0008】
いくつかの症状では、有害なT細胞活性を生じる可能性がある。たとえば、T細胞を媒介した自己免疫疾患および炎症性疾患は、自己抗原を認識するT細胞が増殖し、このような抗原を発現する細胞を襲う有害なT細胞の活性によって特徴づけられる。その他の例には、宿主T細胞および移植片対宿主疾患によって媒介される、移植片拒絶の発生を含む。
【0009】
これらの症状を治療するために利用できる既存の免疫抑制療法は、シクロスポリンA、FK506、およびラパマイシンなどの免疫抑制の化合物の投与を含む。しかし、これらの療法は、完全に有効ではなく、腎毒性、肝毒性、高血圧、多毛、および神経毒性などの有意に有害な副作用と関係がある。したがって、これらの条件を治療するために、より副作用を有さずにT細胞活性をより効率的に抑制することができるさらなる療法が必要である。
【0010】
リンパ球恒常性は、厳密に調節された中心的な生物学的プロセスである。Tanchot, C. et al Semin. Immunol. 9: 331-337 (1997); Marrack, P. et al., Nat. Immunol. 1: 107-111 (2000); C. D. Surh, C.D. and Sprent, J., Microbes. Infect. 4: 51-56 (2002); Jameson, S.C., Nat.Rev. Immunol. 2: 547-556 (2002)。このプロセスの分子制御は、十分に理解されていないが、2つのシグナリング経路の媒介に関与する分子が必須であると考えられる。第1に、自己主要組織適合性(MHC)分子の認識は、ナイーブT細胞の恒常性およびメモリーT細胞機能の維持に重要である。Takeda, S. et al., Immunity 5:217-228 (1996); Tanchot, C. et al., Science 276:2057-2062 (1997)。
【0011】
さらに、最近の研究では、ナイーブT細胞が生存し続けるためには、T細胞受溶体(TCR)の発現が必要とされることが証明された。Polic, B. et al., Proc. Natl. Acad. Sci. 98: 8744-8749(2001); Labrecque, N. et al., Immunity 15: 71-82(2001)。第2に、共通のガンマ(γc)鎖を介してシグナルを送るサイトカイン、特にインターロイキン7(IL-7)は、ナイーブT細胞の生存および恒常性に重要である。Schluns, K.S. et al., Nat. Immunol. 1:426-432(2000); Tan, J.T. et al., Proc. Natl. Acad. Sci. 98:8732-8737(2001)。これらの分子は全て、T細胞恒常性を正に調節する。対照的に、CTLA-4およびTGF-βだけが、T細胞の負の調節に関係していたが、これは、リンパ球減少症の宿主にT細胞を導入すること、または新生児の発現の解析によって噴出して確認された。Waterhouse, P. et al., Science 270: 985-988(1995); Tivol, E.A. et al., Immunity 3: 541-547(1995); Lucas, P.J. et al., J. Exp. MEd. 191:1187-1196(2000); Gorelik, L. and Flavell, R.A., Immunity 12: 171-181(2000)。
【0012】
LAG-3は、CD4とのその緊密な関係のために特に興味を生じさせる。LAG-3は、CD4と同様のゲノム機構を有し、同じ染色体位置に存在するBruniquel, D. et al., Immunogenetics 47:96-98 (1997)。LAG-3は、活性化されたCD4+およびCD8+ αβ Tリンパ球およびγδT細胞およびNK細胞のサブセットで発現される。
CD4の様に、LAG-3もMHCクラスII分子に対して、しかも非常に高い親和性で結合する。Huard, B. et al., Immunogenetics 39: 213-217 (1994); Huard, B. et al., Eur.J. Immunol. 25: 2718-2721 (1995)。
【発明の概要】
【0013】
本発明の簡単な概要
本発明の第1の態様において、自己免疫疾患に罹患する患者を治療するための方法が提供される。患者から単離された自己免疫性T細胞には、インビトロでCD223のコード配列を含む発現構築物がトランスフェクトされる。次いで、トランスフェクトされた自己免疫性T細胞は、患者に再注入される。
【0014】
本発明の第2の態様において、組成物が提供される。組成物は、CD223と特異的に結合する抗体および抗癌ワクチンを含む。
【0015】
本発明のもう一つの態様において、キットが提供される。キットは、CD223と特異的に結合する抗体および抗癌ワクチンを含む。
【0016】
本発明の第4の態様において、抗癌ワクチンで癌患者を治療するための改善された方法が提供される。CD223と特異的に結合する抗体が癌患者に投与される。また、抗癌ワクチンが投与される。抗体は、抗癌ワクチンに対する癌患者の抗癌応答の大きさを増加させる。
【0017】
本発明の第5の態様において、抗癌ワクチンに対する免疫応答の抑制を克服するための方法が提供される。CD223と特異的に結合する抗体が、抗癌ワクチンに対する免疫応答を抑制する調節性T細胞を有する癌患者に投与される。また、抗癌ワクチンが患者に投与される。抗体は、抗癌ワクチンに対する癌患者の応答を増大させる。
【0018】
本発明のもう一つの態様において、哺乳類のT細胞の数を増大させるための方法が提供される。CD223タンパク質またはCD223 mRNAに結合する阻害剤が哺乳類に投与される。阻害剤は、CD223の活性または発現を阻害する。
【0019】
本発明のさらにもう一つの態様において、哺乳類のT細胞の数を減少させるための方法が提供される。CD223をコードする発現構築物が哺乳類に投与される。CD223は、発現構築物から発現され、哺乳類中のCD223の濃度が増大される。哺乳類中のT細胞の数は、減少される。
【0020】
本発明のさらにもう一つの態様において、哺乳類中のT細胞の数を減少させるための方法が提供される。CD223+ T細胞集団が哺乳類に投与される。哺乳類中のCD223の濃度が増大され、これにより哺乳類中のT細胞の数が減少される。
【0021】
本発明のもう一つの側面に従って、CD223の50以下の連続したアミノ酸残基からなるポリペプチドが提供される。ポリペプチドは、SEQ ID NO:5で示されるアミノ酸配列KIEELEを含む。
【0022】
本発明のもう一つの態様は、少なくとも2つのセグメントを含む融合ポリペプチドである。第1のセグメントは、CD223の50以下の連続したアミノ酸残基を含む。第1のセグメントは、SEQ ID NO:5で示されるアミノ酸配列KIELLEを含む。第2のセグメントは、SEQ ID NO:2または4で示されるCD223に見いだされないアミノ酸配列を含む。
【0023】
さらなる態様において、癌、自己免疫疾患、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法が提供される。試験物質をCD223タンパク質またはSEQ ID NO:5に示されるアミノ酸配列KIELLEを含むCD223タンパク質断片と接触させる。次いで、試験物質がCD223タンパク質またはCD223タンパク質断片に結合したかどうかを決定する。試験物質がCD223タンパク質またはCD223タンパク質断片に結合した場合には、試験物質を、癌、自己免疫疾患、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定される。
【0024】
本発明のもう一つの態様では、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法が提供される。試験物質は、CD223タンパク質と接触される。試験物質の存在下および非存在下において、CD223活性が決定される。試験物質がCD223活性を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定される。
【0025】
本発明のもう一つの側面に従って、自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験する方法が提供される。試験物質をCD223タンパク質と接触させる。試験物質の存在下および非存在下においてCD223活性を決定する。試験物質がCD223活性を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定される。
【0026】
本発明のもう一つの態様は、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法である。CD223+ T細胞を試験物質と接触させる。試験物質の存在下および非存在下において細胞におけるCD223の発現を決定する。試験物質がT細胞におけるCD223発現を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定される。
【0027】
本発明のさらにもう一つの側面は、自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験するもう一つの方法である。試験物質をCD223+ T細胞と接触させる。試験物質の存在下および非存在下において細胞におけるCD223発現を決定する。試験物質がT細胞におけるCD223発現を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定される。
【0028】
本発明のさらにもう一つの側面は、CD223+ T細胞またはCD223-T細胞を単離する方法である。T細胞の混合集団を、SEQ ID NO:2または4に記載のCD223と特異的に結合する抗体と接触させる。抗体に結合していないT細胞から抗体に結合するT細胞を分離する。これによりCD223+ T細胞の集団およびCD223-T細胞の集団を形成する。
【0029】
本発明のもう一つの態様は、残基1〜431を含み、残基467〜521を欠いている、単離された可溶性マウスCD223タンパク質である。
【0030】
本発明のもう一つの側面は、残基1〜440を含み、残基475〜525を欠いている、単離された可溶性ヒトCD223タンパク質である。
【0031】
本発明のさらにもう一つの側面は、哺乳類においてT細胞数を減少するための方法である。可溶性CD223タンパク質を哺乳類に投与する。これにより、哺乳類におけるMHCクラスII拘束性/CD4+ T細胞応答が調整される。
【図面の簡単な説明】
【0032】
【図1】HA特異的なCD4+ T細胞は、寛容になり、C3-HAhighトランスジェニックマウスに養子性の導入をすることにより、調節性T細胞活性を発生する。(図1A)C3-HAhighトランスジェニックマウスは、種々の上皮区画に高レベルのHAを発現し、肺上皮において最高レベルで発現される。C3-HAhighのレシピエントは、2.5×106のHA特異的TCRトランスジェニック(6.5)CD4+ T細胞の養子性の導入の4〜7日後に、アネルギー性の表現型の発症前に生じる一過性エフェクター期の活性化と関係する間質性肺炎により死亡した。少数の6.5 CD4+ T細胞の導入により、重篤でない肺病態を生じ、C3-HAhighレシピエントは生き残る。残りの6.5 T細胞は、アネルギー性になる(これらは、γ-インターフェロンを産生できず、またはインビトロでHA抗原に対して増殖することができないことによって定義される)。6.5 T細胞の亜致死量を受けているマウスは、その後の2.5×106のナイーブ6.5T細胞の注入から保護される。従って、最初に寛容化されたT細胞は、Treg活性を発生し、第2の高用量の6.5T細胞によって誘導される致死性の間質性肺炎を抑制する。(図1B〜1E)C3-HAhighマウスにおけるエフェクター/メモリーT細胞、対、抑制T細胞の局在。ナイーブ6.5T細胞(Thy1.1+/1.2-)には、6.5アネルギー性/Treg細胞(Thy 1.1-/1.2+)の非存在下または存在下のいずれかにおいて、C3-HAhighレシピエント(Thy 1.1-/1.2+)に養子性に導入した。養子性の導入の3日後に脾臓および肺を回収し、Thy 1.1+細胞を免疫組織化学によって染色した。Treg細胞の非存在下では、Tエフェクター細胞は、脾臓の小胞に点在し(図1B)、肺血管を浸潤する(図1C)。Treg細胞の存在下では、抑制されたHA-特異的な6.5T細胞は、脾臓の動脈周囲のリンパ性外筒に隔絶されて(図1D)、肺血管を浸潤することができない(図1E)。
【図2】LAG-3は、アネルギー性/Treg細胞とエフェクター/メモリーCD4+ T細胞との間で差次的に発現され、アネルギー性/Treg CD4+ T細胞のLAG-3発現は、IL-10発現と相関される。遺伝子チップ解析によって明らかになった差次的発現を定量的リアルタイムRT-PCRによって確認した(図2A)。早期の日(2日〜4日)のLAG-3の差次的発現は、養子性の導入の30日後まで及ぶ。(図2B)細胞表面LAG-3タンパク質レベルは、抗体染色によって評価した。脾細胞をC3-HAhigh、Vac-HAで免疫した野生型B10.D2マウス、または6.5+/-Thy1.1+/-脾細胞をi.v.注射の5日後のwt B10.D2マウスから収集し、単個細胞懸濁液に調製する。全ての試料を最初にFc受容体をブロックするために全ラットIgGと共にインキュベートした。細胞をTCR特異的な項-6.5-ビオチン+SA-APC、LAG-3-PE、または対応するアイソタイプ対照で染色した。細胞を総リンパ球集団および6.5陽性リンパ球で二重ゲートした。アイソタイプ対照-破線、ナイーブ細胞-明灰色線、エフェクター/メモリー細胞-暗灰色線、アネルギー性/Treg細胞-黒線。(図2C)多くの実験にわたってアネルギー性/Treg集団の複数の試料を解析することにより、LAG-3レベルとIL-10 mRNAレベルとの間の直接的な相互関係を確認する。
【図3】LAG-3は、CD25とは独立して誘導されたTreg細胞上に発現され、Treg機能のマーカーである。(図3A)導入の5日後のC3-HAhighレシピエント脾臓からのアネルギー性/Treg 6.5 CD4+ T細胞をLAG-3およびCD25発現について染色し、アイソタイプ対照と比較した。(図3B)これらの表面LAG-3およびCD25染色に基づいて、細胞を4集団に選別した:LAG-3highCD25high、LAG-3highCD25low、LAG-3lowCD25high、およびLAG-3 lowCD25low。インビトロでの抑制アッセイ法において、サプレッサーとして1×105の細胞の各々の異なる選別されたサブセットを、応答者として1×104ナイーブ6.5 CD4+を添加した。LAG-3lowCD25low細胞は、最も抑制性が少ない。LAG-3highCD25high、LAG-3highCD25low、およびLAG-3lowCD25lowは、最も抑制活性を示すLAG-3highCD25high二重陽性細胞と抑制活性が匹敵する。これは、3回の再現実験の代表的結果である。
【図4】抗LAG-3抗体は、インビトロでTreg活性をブロックする。モノクローナル抗-LAG-3抗体を2μg/mlの濃度でインビトロ抑制アッセイに添加し、0.04:1のサプレッサー:応答者の比率で6.5CD4+サプレッサーによるインビトロでのナイーブ6.5 CD4+ T細胞増殖の抑制を完全に逆転させる。
【図5】抗LAG-3抗体は、直接Treg細胞を阻害することによって、6.5CD4+ Treg細胞によるインビボ抑制を排除する。(図5A)8,000の6.5 CD4+ T細胞によって前処理されたC3-HAhighマウスは、Treg集団の最初の導入確立の4日後に与えられる2.5×106の6.5 CD4+ T細胞によるその後のチャレンジでも生存した(w/保護)。致死下での前処理を伴わずに、C3-HAhighレシピエントは、致死的チャレンジの4〜6日後に死んだ(保護無し)。C3-HAhighマウスに対して、これらを8,000の6.5 CD4+ T細胞で前処理し、200μgの別の用量を2日後に与えた4日後に6.5のT細胞の致死量と共に、モノクローナル抗LAG-3抗体(200μg)をC3-HAhighマウスにi.v.で与えた。抗LAG-3抗体処理されたマウスは、もはやその後の致死チャレンジに耐えることができない(保護+aLAG-3)。対照的に、アイソタイプ対照抗体ラットIgGIでの処理では、インビボ抑制を排除することができない(保護+ラットIgGI)。抗-LAG-3mAbは、Tregの非存在下でナイーブ6.5 CD4+ T細胞を超活性化(hyper-activate)しない(図5Bおよび5C)。C3-HAhighマウスには、2.5×105(亜致死量;図5B)または8×105(導入後7〜14日の間に部分的な死亡率;図5C)のナイーブ6.5 CD4+ T細胞を、抗LAG-3抗体、対照ラットIgGIと組み合わせて、または抗体無しで受けさせた。8×105用量での死亡率は、抗LAG-3抗体による影響を受けなかったのに対して、2.5×105用量での死亡率は、抗LAG-3抗体注入で観察されなかった。
【図6】天然のCD4+CD25+ T細胞におけるLAG-3の役割。(図6A)天然のCD4+CD25+ T細胞は、これらのCD4+CD25-対応物と比較して高レベルのLAG-3 mRNA発現を有する。CD4+CD25+およびCD4+CD25-T細胞は、野生型BALB/cリンパ節から精製した。CD4+CD25+ T細胞(天然の調節性T細胞を含むことが実証されている集団)は、CD25およびLAG-3について、並びにCTLA-4、GITR、およびFoxp3について、これらのCD4+CD25-対応物(CD4+CD25-サブセットのそれぞれのmRNAの発現は、1の値に標準化した)と比較して、有意に高いmRNAレベルを有する。(図6B)LAG-3の表面染色では、これらのCD4+CD25-対応物と同様に、CD4+CD25+の天然調節性T細胞で陰性である。しかし、LAG-3の細胞内染色では、CD4+CD25+において陽性集団が示されるが、CD4+CD25-T細胞においては示されない。(図6C)インビトロ抑制アッセイ法では、T細胞刺激としての抗CD3抗体(0.5μg/ml)と共に、BALB/cマウスリンパ節に由来する選別されたCD4+CD25+ T細胞を応答者として使用し、CD4+CD25-T細胞をサプレッサーとして使用した(0.04:1のサプレッサー:エフェクター比率)。50μg/mlの濃度の抗LAG-3抗体では、天然のCD4+CD25+調節性T細胞のインビトロでの抑制を逆転するが、アイソタイプ対照抗体では、逆転しない。(図6D)Cでの抑制アッセイの後、CD4+CD25+細胞(Thy1.2マーキングによってエフェクター細胞から区別される)は、抗LAG-3またはアイソタイプ対照抗体で染色した。
【図7】CD25枯渇6.5 CD4+ T細胞における、変異型でなく野生型のLAG-3の異所性の発現は、強力なインビトロ調節活性を与える。6.5 CD4+ T細胞は、最初にあらゆるCD25+「天然の」Tregを枯渇させ、次いでGFP単独、GFP +野生型LAG-3、またはGFP+変異型LAG-3.Y73FΔCY(MHCクラスII組織に対する結合が減弱され、かつ下流のシグナリングを媒介することができない)のいずれかをコードするMSCVに基づいたレトロウイルスベクターを形質導入した。活動休止期の10日の後、MSCV-GFPベクターを形質導入したGFP+ 6.5CD4+ T細胞では、本質的に内因性LAG-3の染色は観察されなかったが、一方、MSCV-LAG-3/GFPおよびMSCV-LAG-3.Y73FΔCY/GFPベクターを形質導入したGFP+6.5の細胞では、高レベルのLAG-3染色が観察された。MSCV-LAG-3/GFPおよびMSCV-LAG-3.Y73FΔCY/GFPの形質導入に由来するGFP+細胞は、抗LAG-3抗体で明るく染色されたが、MSCV-GFP形質導入された細胞は、実質的にLAG-3染色を示さなかった。それぞれの群に由来するGFP+細胞を選別し、増殖アッセイ法において、異なる比率でAPC、5μg/ml HA110-120ペプチド、およびナイーブ6.5 CD4+CD25-細胞と混合した。
【図8】フォギン(Phogin)特異的なT細胞クローンでのLAG-3の異所性の発現が、NODマウス由来の脾細胞(splenyocytes)を同時導入後に、糖尿病からの保護を与えることを示す。107のプレ糖尿病性NOD脾細胞を単独で、またはベクター(MIG)LAG-3、もしくはシグナリング欠損がある変異体のLAG-3□Kを形質導入したPhogrin T細胞クローン4(John Huttonから得られた)細胞と組み合わせて、NOD/SCIDマウスに導入した。NOD/SCIDマウス(5/群)を糖尿病についてモニターした。
【発明を実施するための形態】
【0033】
発明の詳細な説明
LAG-3は、CD4関連の活性化で誘導される細胞表面分子であり、MHCクラスII組織と高親和性で結合する。本発明者らは、老いたLAG-3欠損マウスが、野生型対照よりも2倍のCD4+ およびCD8+ T細胞を有することを見いだした。LAG-3欠損T細胞は、MHCクラスII分子のLAG-3ライゲーションに依存的な、リンパ球減少宿主の恒常性の拡張の増強を示す。これは、野生型LAG-3の異所性の発現によって消失したが、シグナリング欠損変異体によっては消失されなかった。このT細胞恒常性の調節解除により、複数の細胞型の拡張を生じる。本発明者らのデータは、LAG-3がCD4+およびCD8+ T細胞の恒常性をネガティブに調整し、骨髄移植に続くT細胞移植を促進するための治療標的としてLAG-3が存在することを示唆する。
【0034】
CD223は、リンパ球抗原遺伝子-3またはLAG-3としても知られ、MHCクラスII分子と高親和性で結合する、CD4関連の活性化で誘導される細胞表面タンパク質である。Baixeras, E. et al., J. Exp. Med. 176:327 (1992)。URLアドレス: http file type, www host server, domain name ncbi.nlm.gov, directory PROW, subdirectory guide, document name 165481751_g.htm.のTriebel, F., "Lag-3 (CD223)", Protein Reviews on the Web (PROW) 3: 15-18 (2002); Triebel, F. et al.,"LAG-3, a novel lymphocyte activation gene closely related to CD4", J. Exp. Med. 171: 1393-1405 (1990)を参照されたい。CD223の代表的なマウスDNAおよびアミノ酸配列は、それぞれSEQ ID NOS:1および2に記載されている。また、GenBankアクセッションコードX9113も参照されたい。CD223の代表的なヒトのDNAおよびアミノ酸配列は、それぞれSEQ ID NOS:3および4に記載されている。また、GenBankアクセッション番号X51985も参照されたい。これらの配列は、一個体に由来する。位置の約5%未満が異なる対立遺伝子変異体が、集団に存在することが予想される。このような対立遺伝子変異体は、マウスまたはヒト起源のCD223の意味の範囲内に含まれる。
【0035】
調節性T細胞は、エフェクターT細胞を阻害することによって機能するT細胞のサブグループである。調節性T細胞は、CD223+であり、典型的には、CD4+CD25+でもある。調節性T細胞は、自己免疫の寛容および免疫応答のバランスをとる際に中心的役割を果たす。このような細胞は、抗体および当該技術分野において既知の分離技術を使用してCD223細胞から単離することができる。これらは、免疫アフィニティー・クロマトグラフィー、FACS、免疫沈降、その他を含むが、これらに限定されない。CD223+細胞は、自己免疫疾患、アレルギー、または喘息の患者に対して投与することができる。自己免疫疾患患者の場合、細胞を自己抗原でプレ活性化することができる。CD223-細胞は、同様に癌患者、細菌もしくはウイルス感染患者、またはAIDS患者に導入することができる。
【0036】
エフェクター/メモリーの表現型または抑制性の表現型のいずれかに分化する抗原特異的なCD4+ T細胞からの遺伝子発現アレイの比較解析により、LAG-3(MHCクラスII組織を結合するCD4相同体)のTreg特異的な発現が明らかにされた。LAG-3high CD4+ T細胞は、インビトロでサプレッサー活性を示し、LAG-3に対する抗体は、インビトロおよびインビボの両者における抑制を阻害する。これらの知見は、Treg特異的な受容体または補助受容体がサプレッサー活性を調整するものとしてLAG-3を同定する。従って、LAG-3を経たTreg細胞の操作は、自己免疫疾患、癌、および感染症の免疫療法を増強するために、並びにドナーリンパ球の注入、骨髄移植、および養子性(adoptive)のT細胞導入の環境においてリンパ球移植を増強するために使用することができる。
【0037】
CD223は、調節性T細胞の機能を調節する調節性T細胞特異的な細胞表面分子である。調節性T細胞の機能は、CD223活性を増強もしくは増大することによって、またはT細胞集団中のCD223+細胞の数を増大することによって増強されるであろう。被検者が自己免疫疾患に罹患しているなどの、このような反応が望ましくない状況において免疫T細胞反応を制限するために、生物体の調節性T細胞の機能の増強を使用してもよい。逆に、CD223活性を阻害することによって、またはT細胞集団中のCD223+細胞の数を減少させることによって、調節性T細胞の機能を阻害してもよい。癌、慢性感染症に罹患している患者、または骨髄移植レシピエントにおいてなど、このような反応が望ましい状況の免疫T細胞反応を増強するために、生物体の調節性T細胞の機能の阻害を使用してもよい。
【0038】
CD223タンパク質またはmRNAと結合する阻害剤で癌患者を治療するときに、抗腫瘍ワクチンを選択的に同時投与してもよい。このようなワクチンは、単離された抗原もしくは抗原群に、または全腫瘍細胞に対して作製されたものであってよい。化学療法薬と共に阻害剤を投与することが望ましいであろう。薬剤の混合物を使用して、複数の薬剤での治療を行うことは必要ではなく、別々の製剤を使用して行ってもよい。製剤は、同じ正確な時間で送達することは必要ではなく、同じ治療期間、すなわち1週もしくは1ヶ月、または相互に、患者に送達されるように調整してもよい。したがって、2つの活性成分を含む組成物が、患者の体内で構成されてもよい。いずれの適切な抗腫瘍治療も、CD223に対してターゲットされる本発明の治療と共に調整することができる。同様に、感染症を有する患者を治療する場合、その他の抗感染症薬を、CD223にターゲットされる本発明の治療と共に調整することができる。このような薬剤は、小分子薬物、ワクチン、抗体、その他であってもよい。
【0039】
T細胞集団におけるCD223+細胞の数は、CD223に対して選択的に結合する抗体またはその他の薬剤を使用して修飾することができる。CD223+細胞は、調節性T細胞の濃縮された集団を示し、調節性T細胞機能を増強するためにこれをT細胞の本来の供与源へ、または別の適合性の宿主に導入することができる。または、CD223-細胞は、調節性T細胞活性を欠損したT細胞の集団を表し、一般的なT細胞活性を保持したままで調節性T細胞機能を阻害または減少させるために、T細胞の本来の供与源に、または別の適合性の宿主に再び導入することができる。
【0040】
CD223活性を増大または減少する(調整する)ためのいずれの所望の手段も本発明の方法に使用することができる。これは、直接CD223タンパク質の機能を調整すること、CD223のシグナル伝達を調整すること、および転写もしくは翻訳または両方ともを調整することによってT細胞におけるCD223の発現を調整することを含む。これらのCD223活性を選択的に調整する手段は、非選択的なモジュレーターよりも好ましい。また、一時的なT細胞欠損を治療するためには、T細胞の集団に一過性CD223欠損を生じさせ、次いで正常レベルのCD223活性に戻る阻害手段が好ましいであろう。一過性に欠損したT細胞を使用して、CD223に関して遺伝的に正常であると考えられるT細胞と共に、減弱されたT細胞集団を再構成してもよい。このような一過性のT細胞欠損は、たとえば筋切除後に幹細胞導入を受けている患者において生じる。CD223活性の調整は、細胞に対してインビトロで、または動物全体に対して(インビボで)行うことができる。インビトロで処理した細胞を、細胞の本来の供与源または無関係な個体の患者のいずれかに投与することもできる。
【0041】
CD223の機能を阻害するために、CD223抗体または小分子阻害剤を使用することができる。このために有用な抗体または抗体断片は、CD223と結合して、これが機能する能力をブロックすることができるものである。このような抗体は、ポリクローナル抗体、モノクローナル抗体(たとえば、Workman, C. J. et al., "Phenotypic analysis of the murine CD4-related glycoprotein, CD223 (LAG-3)", Eur. J. Immunol. 32:2255-2263 (2002)を参照されたい)、キメラ抗体、ヒト化抗体、単鎖抗体、可溶性MHCクラスII分子、抗体断片、その他であってもよい。
【0042】
CD223ポリペプチドに対して作製される抗体は、CD223ポリペプチドを直接動物に注射することによって、またはCD223ポリペプチドを動物に、好ましくはヒト以外に投与することによって得ることができる。次いで、このようにして得られた抗体は、CD223ポリペプチド自体に結合すると考えられる。このように、CD223ポリペプチドの断片のみをコードする配列さえ、完全な天然のCD223ポリペプチドに結合する抗体を作製するために使用することができる。
【0043】
モノクローナル抗体の調製のためには、連続的な株化細胞培養によって産生される抗体を提供するいずれの技術を使用することもできる。例には、ハイブリドーマ技術(Kohler and Milstein, 1975, Nature, 256:495-497)、トリオーマ技法、ヒトB細胞ハイブリドーマ技術(Kozbor et al., 1983, Immunology Today 4:72)、およびヒト・モノクローナル抗体を産生するためのEBV-ハイブリドーマ技術(Cole, et al., 1985, in Monoclonal Antibodies and Cancer Therapy, Alan R. Liss, Inc., pp. 77-96)を含む。
【0044】
一本鎖抗体を産生するために記載された技術(米国特許第4,946,778号)をCD223ポリペプチドに対する一本鎖抗体を作製するために容易に使用することができる。また、免疫原性のCD223ポリペプチドに対するヒト化抗体を発現するために、トランスジェニックマウスを使用してもよい。
【0045】
CD223の機能を増強または活性化するために、CD223のレベルまたはT細胞中の既存のCD223の活性を増大させる何らかの薬剤を使用してもよい。このような薬剤は、以下に記載されているスクリーニングアッセイ法を使用して同定してもよい。また、遺伝子量を増大させるために、CD223をコードする発現ベクターを投与することもできる。発現ベクターは、当該技術分野において既知のプラスミドベクターまたはウイルスベクターであることができる。特に望ましい性質について、実務者によりいずれのベクターを選択することもできる。
【0046】
本発明に従った治療を受け入れられる自己免疫疾患は、自己免疫溶血性貧血、自己免疫性血小板減少症紫斑病、グッドパスチャー症候群、尋常性天疱瘡、急性リウマチ熱、混合型本態性クリオグロブリン血症、全身性エリテマトーデス、インスリン依存型糖尿病、慢性関節リウマチ、グレーブス病、橋本甲状腺炎、重症筋無力症、および多発性硬化症を含む。当該技術分野において既知のように、自己免疫性T細胞は、自己免疫疾患患者から単離することができる。これらには、CD223のコード配列をトランスフェクトすることができる。いずれの好ましい発現ベクターも、CD223を発現するために使用することができる。これらには、プラスミドおよびウイルスベクターを含むが、限定されない。発現調節シグナルは、CD223自体に、またはその他の遺伝子に由来することができる。CD223発現構築物のトランスフェクション後、T細胞を患者に再び導入することができる。患者に血液細胞を注入するための方法は当該技術分野において周知である。
【0047】
特異的にCD223と結合する抗体、および抗癌ワクチンの混合物を含む組成物は、インビトロで作製することができる。好ましくは、組成物は、薬学的組成物として使用するために適した条件下で作製される。薬学的組成物は、無菌でも、発熱性物質なしでもよい。また、組成物の成分は、同時にこれら両方が患者の体内にあるような期間内に別々に患者に投与することもできる。このような時間を離した投与では、患者内で抗体およびワクチンの混合物の形成が引き起こされる。抗体およびワクチンが、時間を離した様式で投与される場合、これらは、キット内に共に供給してもよい。キット内において、成分は、別々にパックされても、または別々に含まれていてもよい。賦形剤、担体、その他の免疫変調成分またはアジュバント、抗体およびワクチンの投与のための説明書、並びに注射装置などのその他の成分も同様にキットに供給することができる。説明書は、書面、ビデオ、または音声形態であることができ、紙、電子媒体、またはウェブサイトもしくは参照マニュアルなどのさらに別の供与源に対する参照として含まれることができる。
【0048】
本発明の抗CD223抗体は、抗癌ワクチンに対する癌患者の抗癌応答の大きさを増大させるために使用することができる。また、癌患者の集団の応答者の数を増大させるために使用することができる。したがって、抗体は、抗癌ワクチンに耐性の患者で見いだされる免疫抑制を克服するために使用することができる。抗癌ワクチンは、完全な腫瘍細胞ワクチン、腫瘍抗原の1つもしくは複数のエピトープを含む単離された腫瘍抗原またはポリペプチドを含む(しかし、限定されない)、当該技術分野において既知のいずれのものであることもできる。
【0049】
T細胞におけるCD223の発現は、転写または翻訳のレベルで調整することができる。このような調整が可能な薬剤は、以下に記載されているスクリーニングアッセイ法を使用して同定することができる。
【0050】
CD223 mRNAの翻訳は、リボザイム、アンチセンス分子、小さな干渉RNA(siRNA; Elbashir, S. M. et al., "Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells", Nature 411:494-498 (2001)を参照されたい)、またはCD223 mRNAをターゲットするこのプロセスの小分子阻害剤を使用して阻害することができる。アンチセンス技術を使用して、トリプル-ヘリックス形成またはアンチセンスDNAもしくはRNAを介して遺伝子発現を制御することができ、これらの両方法ともDNAまたはRNAに対するポリヌクレオチドの結合に基づいている。たとえば、本発明の成熟ポリペプチドをコードするポリヌクレオチド配列の5'をコードする部分を、長さが約10〜40塩基対のアンチセンスRNAオリゴヌクレオチドをデザインするために使用する。DNAオリゴヌクレオチドを、転写に関与する遺伝子の領域に対して相補的となるようにデザインし(トリプル-ヘリックス−Lee et al., Nucl. Acids Res. , 6: 3073 (1979); Cooney et al, Science, 241: 456 (1988);および Dervan et al., Science, 251: 1360 (1991)を参照されたい)、これによりCD223の転写および産生を防げる。アンチセンスRNAオリゴヌクレオチドは、インビボでmRNAにハイブリダイズして、mRNA分子のCD223ポリペプチドへの翻訳をブロックする(アンチセンス−Okano, J. Neurochem., 56:560 (1991); Oligodeoxynucleotides as Antisense Inhibitors of Gene Expression, CRC Press, Boca Raton, Fla. (1988))。また、上記したオリゴヌクレオチドは、アンチセンスRNAまたはDNAがインビボで発現してCD223の産生を阻害するようなアンチセンス発現構築物によって細胞に送達することができる。このような構築物は、当該技術分野において周知である。
【0051】
アンチセンス構築物、アンチセンスオリゴヌクレオチド、RNA干渉構築物、またはsiRNA二重鎖RNA分子は、CD223の発現を妨害するために使用することができる。典型的には、アンチセンス分子には、少なくとも15、17、19、または21のCD223 mRNA配列の相補ヌクレオチドで十分である。典型的には、RNA干渉分子には、少なくとも19、21、22、または23のCD223のヌクレオチドで十分である。好ましくは、RNA干渉分子は、2つのヌクレオチドの3'オーバーハングを有するであろう。RNA干渉分子が細胞において構築物から、たとえばヘアピン分子から、または所望のCD223配列の逆方向反復から発現される場合、内因性の細胞機構がオーバーハングを作製する。siRNA分子は、化学合成、インビトロでの転写、またはRnase IIIもしくはDicerによる長いdsRNAの消化によって調整することができる。これらは、トランスフェクション、電気穿孔法、または当該技術分野において既知のその他の方法によって細胞に導入することができる。
を参照されたい。
【0052】
既知のモジュレーターに加えて、本発明の方法に有用であるCD223活性のさらなるモジュレーターは、CD223に結合する能力について候補分子をスクリーニングすること、または細胞培養中においてCD223活性を阻害する化合物をスクリーニングすることなどの、ツーハイブリッド・スクリーン、従来の生化学的アプローチ、および細胞に基づいたスクリーニング技術を使用して同定することができる。一例として、本発明者らは、活性化後でさえCD223を発現しない、ニワトリ卵白リゾチーム(HEL)の48-62特異的、H-2Ak拘束性マウスCD4+ T細胞ハイブリドーマ3A9を同定した。野生型CD223の異所性の発現(しかし、シグナリング欠損ではない)は、その特異的なペプチドに対するこのT細胞ハイブリドーマのIL-2反応を有意に減少させた。これは、CD223活性モジュレーターをスクリーニングするための簡単なインビトロ・アッセイ法の系を提供する。この後者の方法では、直接CD223と相互作用し、かつ調整する薬剤、並びにCD223シグナル伝達経路の工程に影響を及ぼすことによって間接的にCD223活性を調整する薬剤が同定されるであろう。
【0053】
CD223を発現する細胞を使用する細胞に基づいたアッセイ法では、哺乳類から単離されかつ天然にCD223を発現する細胞を使用することができる。または、CD223を発現するように遺伝的に操作された細胞を使用することもできる。好ましくは、遺伝的に操作された細胞は、T細胞である。
【0054】
CD223遺伝子発現を調整することによってCD223活性を調整する薬剤は、候補薬剤の存在下および非存在下で細胞のCD223タンパク質の量を測定することによって、細胞に基づいたスクリーニングアッセイ法で同定することができる。CD223タンパク質は、たとえば、抗CD223特異的なモノクローナル抗体を使用するフローサイトメトリーによって検出し、測定することができる。また、CD223 mRNAは、ノーザンブロット、RT-PCR、およびアレイ・ハイブリダイゼーションを含む(しかし、限定されない)当該技術分野において既知の技術を使用して検出し、測定することができる。
【0055】
CD223モジュレーターを同定するための1つの特に有用な標的配列はインビトロおよびインビボでのCD223機能に必須である、CD223の細胞質ドメインのアミノ酸のモチーフKIEELEである(SEQ ID NO:5)。このモチーフに結合する薬剤のためのスクリーニングアッセイ法により、候補CD223モジュレーターを同定し、CD223の阻害剤または活性化因子としてのその活性を、細胞に基づいたアッセイ法においてなど、さらなる試験によってさらに特徴づけることができる。このモチーフは、CD223の50以下の連続したアミノ酸残基からなるポリペプチドに含まれることができる。または、モチーフは、CD223の一部および第2の(非CD223)タンパク質の全てまたは一部を含む融合タンパク質に含まれることができる。第2のタンパク質は、天然のタンパク質であってもよく、またはたとえばヒスチジンタグ、もしくはその他の有用なポリペプチドの特徴を含む合成ポリペプチドであることができる。タンパク質-タンパク質結合アッセイ法は、当該技術分野において周知の、および任意の種々の技術および形式を使用することができる。
【0056】
CD223は、タンパク質の可溶性形態を得るために翻訳後にプロセシングすることができる。可溶性形態は、少なくともマウスのCD223のアミノ酸残基1〜431、および少なくともヒトのCD223のアミノ酸残基1〜440を含む。いずれの場合においても細胞質内末端部が失われている。同様に、膜貫通領域の全部または一部も失われている。この可溶性形態は、MHC クラスII-拘束性/CD4+ T細胞の反応を調整する。したがって、可溶性形態は、たとえば、自己免疫疾患患者、アレルギー患者、喘息患者、または癌患者に投与するために有用であろう。可溶性形態の投与は、注入、局所的、または静脈内の投与を含むいずれの便利な手段によるものであってもよい。
【0057】
本発明の教示によれば、CD223阻害剤は、生物体中のT細胞の数を増大させるために、生物体に投与されてもよい。この方法は、低いT細胞集団を生じる症状に罹患する生物体を治療するために有用であろう。このような症状は、AIDSなどの免疫不全によって生じる疾患、並びに外来微生物(細菌もしくはウイルス)の侵入などの不必要な細胞の侵入または増殖を含む障害、または腫瘍成長もしくは癌を含む。
【0058】
また、このようなT細胞欠損は、筋切除後に幹細胞転移を受けた患者にも予想される危険である。このような患者のT細胞は欠陥があり、これらが健康なドナーT細胞によって置換することができるように、破壊の対象となる。幹細胞から健康なT細胞集団を再構成するプロセスは、数ヶ月かかり、その期間の間に、患者は、生命の脅威となり得る日和見感染に非常に感受性である。ドナーT細胞のCD223を阻害するか、または、CD223欠損について選択もしくは操作されたドナーT細胞を使用することによって、T細胞分裂が増強され、T細胞再構成プロセスを促進することができ、T細胞欠損の期間を減少させることができる。
【0059】
また、CD223阻害剤は、T細胞増殖感受性の障害を治療するために従来の治療と組み合わせて投与されるときにも有用であろう。たとえば腫瘍は、T細胞増殖感受性の障害であり、従来は、急速に分裂している細胞を死滅させることによって機能する化学療法薬によって治療されている。本発明のCD223阻害剤は、化学療法薬と組み合わせて投与されるときに、T細胞増殖を刺激して腫瘍細胞の免疫拒絶を増強することによって化学療法薬の殺腫瘍効果を増強する。
【0060】
本発明の教示によれば、CD223活性化因子または発現エンハンサーは、生物体中のT細胞の数を減少させ、これにより有害なT細胞活性を減少させるために生物体に投与してもよい。この方法は、異常に多いT細胞集団または有害なT細胞活性を生じている症状たとえば、宿主T細胞によって媒介される移植片拒絶、移植片対宿主疾患、並びに慢性関節リウマチ、1型糖尿病、筋肉硬化症、その他などのT細胞を媒介した自己免疫疾患および炎症性疾患、に罹患した生物体を治療するために有用であろう。本発明の方法は、CD223を発現するT細胞を含むいずれの生物体に適用してもよい。これは、いずれの哺乳類も含むが、限定されず、特にヒトおよびマウスを含む。
【0061】
本発明の方法がインビボで行われるときは、使用されるCD223モジュレーターの有効な量は、使用する特定のモジュレーター、治療される特定の症状、治療されている被検者の年齢および身体症状、症状の重篤さ、治療の期間、併用療法の性質(ある場合)、投与の特異的な経路、並びに健康な開業医の知識および専門的意見の範囲内の同様の因子によって変更される。たとえば、有効な量は、個体が異常に抑制されたレベルのT細胞を有する程度に依存することができる。
【0062】
投与されるときに、本発明の薬学的製剤は、薬学的に許容される量で、および薬学的に許容される組成物内で適用される。このような製剤は、通常、塩、緩衝剤、防腐剤、適合性の担体、および任意にその他の治療薬を含んでいてもよい。医薬に使用するときに、塩類は、薬学的に許容されるべきであるが、薬学的に許容されない塩類を、これらの薬学的に許容される塩類を調製するために都合よく使用してもよく、本発明の範囲から除外されない。このような薬理学的に、および薬学的に許容される塩類は、以下の酸から調製されるものを含むが、限定されない:塩酸、臭化水素酸、硫酸、硝酸、リン酸、マレイン酸、酢酸、サリチル酸、クエン酸、ギ酸、マロン酸、コハク酸など。また、ナトリウム、カリウム、もしくはカルシウム塩などのアルカリ金属またはアルカリ土類塩類として薬学的に許容される塩類を調製することができる。
【0063】
CD223モジュレーターは、任意に薬学的に許容される担体と組み合わせてもよい。「薬学的に許容される担体」の用語は、本明細書に使用されるものとして、ヒトに投与するために適した1つまたは複数の適合性の固体もしくは液体の充填材、希釈液、またはカプセル化物質を意味する。「担体」の用語は、適用を容易にするために活性成分が組み合わせられる有機または無機の成分で、天然または合成のものを意味する。また、薬学的組成物の成分は、本発明の分子と、互いに所望の薬学的有効性を実質的に損なう相互作用がない様な方法で、共に混合することができる。
【0064】
薬学的組成物は、以下を含む適切な緩衝剤を含んでいてもよい:酢酸の塩溶液;クエン酸の塩溶液;ホウ酸の塩溶液;およびリン酸の塩溶液。また薬学的組成物は、任意に塩化ベンザルコニウム;クロロブタノール;パラベン、およびチメロサールなどの適切な防腐剤を含んでいてもよい。
【0065】
非経口投与のために適した組成物は、都合よくは抗炎症薬の無菌の水性製剤を含み、これは、レシピエントの血液と好ましくは等張性である。この水性製剤は、適切な分散剤または湿潤剤および懸濁剤を使用して、既知の方法に従って処方してもよい。また、無菌注射用製剤も、たとえば1,3-ブタンジオール溶液として、無毒の非経口的に許容される希釈液または溶媒中の無菌の注射用の溶液または懸濁液であってもよい。許容されるもののうち、使用してもよい媒体および溶媒は、水、リンゲル溶液、および等張食塩液である。加えて、従来には、無菌の不揮発性油が溶媒または懸濁媒として使用されている。このために、合成のモノ-またはジ-グリセリドを含むいずれの無刺激性の不揮発性油を使用してもよい。加えて、オレイン酸などの脂肪酸は、注射可能薬物の調製に使用してもよい。経口、皮下、静脈内、筋肉内、その他の投与のために適した担体製剤は、Remington's Pharmaceutical Sciences, Mack Publishing Co., Easton, Pa.において見いだされる。
【0066】
種々の投与ルートを利用することができる。選択される特定の様式は、もちろん、選択される特定の薬物、治療される症状の重篤さ、および治療有効性のために必要とされる用量に依存する。本発明の方法は、一般的に言って、医学的に許容される(臨床的に容認できない副作用を生じさせることなく活性化合物の有効レベルを生じるいずれの方法をも意味する)いずれの投与方法を使用して実施してもよい。このような投与方法は、経口、直腸、局所的、経鼻、真皮内、または非経口的な経路を含む。「非経口的」の用語は、皮下、静脈内、筋肉内、または注入を含む。静脈内または筋肉内の経路は、特に長期の療法および予防には適していない。しかし、これらは、緊急事態には好ましいであろう。経口投与は、患者並びに投薬スケジュールに対して都合が良いため好ましいであろう。
【0067】
薬学的組成物は、単位用量形態で提供されるのが都合が良く、薬学の技術分野において周知のいずれの方法によって調製してもよい。全ての方法で、活性薬剤を1つまたは複数の補助成分を構成する担体と会合させる工程を含む。一般に、組成物は、均一に、かつ均質に、活性薬剤を液体担体、微粉固体担体、またはその両方と会合させて、次いで、必要に応じて産物を成形することによって調製される。
【0068】
経口投与に適した組成物は、それぞれが活性薬剤の予め定められた量を含む、カプセル、タブレット、ロゼンジなどの分離した単位として提供されてもよい。その他の組成物は、シロップ、エリキシル、またはエマルジョンなどの水性液体または非水性液体の懸濁液を含む。
【0069】
その他の送達システムは、徐放性、遅放性、または持効性の送達システムを含むことができる。このようなシステムにより、活性薬剤の反復投与を回避することができ、被検者および医師の便宜を増大させる。多くのタイプの放出送達システムが利用でき、当業者に既知である。これらは、ポリ(ラクチド-グリコリド)、コポリオキサラート、ポリカプロラクトン、ポリエステルアミド、ポリオルトエステル、ポリヒドロキシ酪酸、およびポリ無水物などの重合体塩基システムを含む。たとえば、薬物を含む前述の重合体のマイクロカプセルは、米国特許第5,075,109号に記載されている。また、送達システムには、非重合体系:コレステロール、コレステロールエステル、および脂肪酸などのステロールを含む脂質、またはモノ-、ジ-、およびトリ-グリセリドなどの中性脂肪;ヒドロゲル放出システム;シラスティック(sylastic)システム;ペプチドに基づいたシステム;ワックス被覆;従来の結合剤および賦形剤を使用する圧縮錠剤;部分的に融合されたインプラント;などを含む。具体例は、以下を含むが、これらに限定されない:(a)米国特許第4,452,775号、第4,667,014号、第4,748,034号および第5,239,660号に記載されているものなどの、抗炎症薬がマトリックス内の形態に含まれる浸食システム、並びに(b)米国特許第3,832,253号、および第3,854,480号に記載されているものなどの、活性成分が重合体から制御された割合で浸透する拡散システム。加えて、ポンプに基づいたハードウェア・送達システムを使用することもでき、これらのうちのいくつかは、移植に適応される。
【0070】
長期の持効性インプラントの使用は、慢性症状の治療のために特に適しているであろう。長期の放出とは、本明細書に使用されるものとして、インプラントが、少なくとも30日、および好ましくは60日間、治療レベルの活性成分を送達するように構築され、並べられることを意味する。長期の持効性インプラントは、当業者に周知であり、上記で記載されているいくつかの放出システムを含む。
【0071】
本発明では、目下のところ本発明を実施する好ましい様式を含む具体例に関して記載したが、当業者であれば、上記したシステムおよび技術の多数の変形および並び替えが、添付の請求の範囲に記載した本発明の趣旨および範囲の範囲内であることを理解すると考えられる。
【実施例】
【0072】
実施例1-LAG-3(CD223)によるT細胞恒常性のネガティブ制御
以下の実施例は、LAG-3(CD223)がCD4+およびCD8+ T細胞恒常性を負に調節することを示し、骨髄移植後のT細胞移植を促進するための新規の治療的な標的としてこれが同定されたことを支持する。
【0073】
野生型C57BL/6マウスは、4〜52週齢のαβ+ T細胞を一定数有する。前に報告された通り、若い4週齢のLAG-3-/-マウスは、正常なT細胞数を有する。Miyazaki, T. et al., Science 272: 405-408 (1996)。対照的に、LAG-3-/-マウスのαβ+ T細胞の数は、3ヶ月齢から、野生型マウスよりも2倍多くの数まで着実に増大する。この違いは、αβ+ T細胞数の厳密な恒常性の制御が非常に低い標準偏差によって明示されたことを考えると、非常に有意である。CD4+およびCD8+細胞はいずれもLAG-3-/-マウスにおいて増大されたが、CD4:CD8の比率は、不変であった。同様に、OT-II TCRをトランスジェニックしたLAG-3-/-マウス(卵白アルブミン326〜339特異的、H-2Ab拘束性)では、これらの違いが5週間目に明らかであったことを除き、野生型対照OT-IIトランスジェニックマウスと比較して、CD4+ Vα2+ T細胞の数が増大された。野生型マウスでは、ほぼ20%のαβ+ T細胞およびCD49b+ NK細胞が、LAG-3を構成的に発現し(Workman, C.J. et al., Eur. J. Immunol. 32: 2255-2263 (2002))、これらの数も、LAG-3-/-マウスにおいて有意に増大した。驚くべきことに、LAG-3を発現しないB220+B細胞、Gr-1+顆粒球、およびMac-1+マクロファージのようないくつかのその他の細胞タイプでも、対照マウスと比較してLAG-3-/-が増大された。LAG-3-/-マウスで観察される細胞数の増大は、インビボの分裂しているBrdU+細胞数の〜50%の増大に匹敵した。LAG-3-/-と野生型マウスの間で観察される細胞数の相違は、非常に一貫しており、再現性があった。LAG-3がないと、LAG-3-/-マウスに由来するT細胞の細胞表面表現系に対して有意な効果を有さないようにみえる。これらのデータは、LAG-3がマウスのT細胞数を調節し、一般に白血球数に間接的に影響を及ぼしているという考えを支持する。
【0074】
LAG-3がリンパ球減少環境においてT細胞の恒常的発現に影響を及ぼしているかどうかを決定するために、精製したT細胞を、T細胞およびB細胞を欠いたRAG-/-マウスに養子性に(adoptively)導入し、脾臓のT細胞数を導入の15日後に決定した。野生型対照と比較してLAG-3-/-T細胞数の2.8倍の増大があった。注目すべきことに、LAG-3の非存在がT細胞増殖に対して明らかに有効であるにもかかわらず、野生型T細胞のわずかな割合のみがLAG-3を発現した。これにより、LAG-3の短期の一過性の発現が、これが分裂細胞に対してその効果を及ぼすには十分であろうことが推論される。CD4+およびCD8+T細胞の増殖の増大が観察されたことから、両細胞タイプがLAG-3の不存在によって同等の影響を受けたことが証明される。興味深いことに、また、LAG-3-/-対LAG-3+/+ T細胞のレシピエントにおいてαβ-宿主由来細胞数の2倍の増加があった。これは、操作されていないLAG-3-/-マウスで観察されたマクロファージおよび顆粒球の数の増大と一致した。RAG-/-で観察されるLAG-3-/- T細胞の増殖の増大は、抗原特異性から独立していることを確認するために、本発明者らは、OVA [Ovalbumin 257-264-特異的、H-2Kb-拘束性;Hogquist, K.A. et al., Cell 76: 17-27 (1994)]およびOT-II [Ovalbumin 326-339-特異的、H-2Ab-拘束性; Barnden, M.J. et al., Immunol. Cell Biol. 76: 34-40 (1998)]トランスジェニック・マウスから精製したT細胞を使用した。野生型CD4+ Vα2+ OT-II T細胞は、以前の報告と一致してRAG-/-マウスにおいて十分に増殖せずに、これらの細胞がリンパ球減少性宿主においてほとんど恒常的な増殖を示さないことを示している。Ernst, B. et al., Immunity 11: 173-181 (1999)。対照的に、この制限は、LAG-3-/- OT-IIトランスジェニックマウスからのT細胞には適用されず、これは、リンパ球減少性の宿主において活発に、導入後15日までに野生型T細胞の3.2倍以上の数に増殖した。同様に、RAG-1-/-マウスから回収したLAG-3-/- CD8+ Vα2+ OVAトランスジェニックT細胞の数は、野生型の対照OVA T細胞よりも4倍高かった。特筆すべきは、この相違は、導入後少なくとも1ヶ月間持続されたことである。これらのデータは、また、CD4+およびCD8+ T細胞がLAG-3の減少により同様に影響を受けることを証明した。MHCクラスII分子によるLAG-3のライゲーションの重要性を評価するために、LAG-3-/-および野生型OVAトランスジェニックT細胞を、MHCクラスIおよびクラスII分子(β2m-/- x H-2Aβb-/-)を両方とも欠いているマウスに導入した。データは、LAG-3-/- T細胞の増殖の増大がMHCクラスII分子の非存在下で消滅されることを明らかに示しており、この相互作用の重要性が証明された。
【0075】
LAG3-/-マウスまたはLAG-3-/- T細胞の養子性のレシピエントは、B細胞およびマクロファージなどの通常LAG-3陰性である細胞の数を増大した。これは、LAG-3の非存在によるT細胞の恒常性の制御の変化は、その他の白血球細胞型の制御を直接変化させるという考えを支持する。直接これを試験するために、B細胞を、LAG-3-/-または野生型T細胞のいずれかと共にRAG-/-マウスに同時導入した。また、本発明者らは、T細胞恒常性を調節する際のMHCクラスII分子の対照的な役割を評価するためにも、このアプローチを利用した。以前の研究では、CD4+ T細胞の恒常的な増殖および長期の生存には、MHCクラスII分子との周期的な相互作用が必要であることを明らかに証明した。Takeda, S. et al., Immunity 5: 217-228 (1996); Rooke, R., et al., Immunity 7:123-134 (1997)。対照的に、LAG-3とMHCクラスII分子との間の相互作用は、逆の効果を有する可能性もある。以前に示したように、MHCクラスII-/- B細胞を導入したときに、野生型対照と比較してLAG-3-/- T細胞の数に3.0倍の増大がある。しかし、野生型B細胞の存在下では、LAG-3-/-とLAG-3+/+ Tとの間の細胞数の違いは、4.9倍に増大した。LAG-3-/- T細胞数の増大は、MHC:TCRの相互作用が増大され、したがって増殖が増強されるためである可能性が高い。対照的に、LAG-3+/+ T細胞は、正(MHCを経て:TCR相互作用)および負(MHCを経て:LAG-3相互作用)の恒常性調節の両方に供されて、野生型T細胞に匹敵する増殖を生じるであろう。
【0076】
野生型T細胞の存在下において、B細胞の数は、導入7日後に脾臓から回収したB細胞の数は、B細胞単独を受けているマウスと同じであった。対照的に、LAG-3-/- T細胞レシピエントから回収したB細胞の数には、2.7倍の増加があり、B細胞数の増大は、LAG-3-/- T細胞の「調節解除」によるものであることの直接の実証を提供する。興味深いことに、野生型T細胞の存在下において、B細胞単独を受けているマウスと比較して、MHCクラスII-/- B細胞の数が増大する。これは、LAG-3:MHCクラスIIの相互作用が「局部的」になくなると、レシピエントRAG-/-マウスは、脾臓内にMHCクラスII+マクロファージおよび樹状細胞を有するものの、野生型T細胞の一過性の調節解除により、B細胞増殖の増大を生じ得るという考えを支持する。別の可能性としては、LAG-3によるMHCクラスII分子のライゲーションが負の調節シグナルをB細胞に送達することにより、増殖を妨げることである。これは、B細胞に対してはもっともらしいが、これによっては、LAG-3-/-マウスにおける顆粒球などのMHCクラスII細胞の数の増大が説明されない。現在調査している1つの可能性としては、LAG-3-/- T細胞の調節解除された増殖により、多くの細胞タイプの広範な増殖を誘導するサイトカインの生産をこれらが生じることである。
【0077】
リンパ球減少性マウスにおける恒常的な増殖に対するLAG-3発現の影響は、ナイーブT細胞に限定されない。また、抗原を受けた「メモリー」OT-II T細胞の導入により、LAG-3T3-/-T細胞は、野生型の対照細胞と比較して、実質的に増殖の促進を生じた[7.2倍]。LAG-3が、この「調節解除された」T細胞増殖の直接の原因であり、本来のターゲティング戦略によって破壊される密接に連関した遺伝子ではないことを確かめることが、非常に重要であった。従ってLAG-3-/- OT-II T細胞には、野生型LAG-3またはシグナリング欠損がある変異体LAG-3.ΔKMのいずれかを含むマウス幹細胞ウイルス(MSCV)に基づいたレトロウイルスを形質導入した。Workman, C. J. et al., Eur. J. Immunol. 32: 2255-2263 (2002)。また、ベクターは、形質導入された細胞の解析を容易にするために、内部リボソーム導入部位(IRES)および緑色蛍光タンパク質(GFP)カセットを含んだ。Persons, D. A. et al., Blood 90: 1777-1786 (1997)。また、LAG-3-/-およびLAG-3+/+ OT-11のT細胞には、「空」ベクター/GFP単独の対照を形質導入した。形質導入された細胞をRAG-1-/-レシピエントに導入して、導入の15日に回収したOT-II T細胞の数を決定した。予想されるとおり、LAG-3-/- GFP単独対照T細胞では、野生型GFP細胞よりも増殖した[2.8倍]。LAG-3の異所性の発現は、OT-11 T細胞の数を野生型対照と同等のレベルに減少させたが、一方のLAG-3シグナリング欠損がある変異体の発現は、恒常的な増殖に対して効果を有さない。これらのデータは、LAG-3が観察される効果の直接の原因となることを証明する。
【0078】
本発明者らのデータは、LAG-3がT細胞の恒常的な増殖を負に調節することを明らかに示す。また、これらは、T細胞が多くの細胞型の恒常性に関与しているであろういう考えを支持する。LAG-3の非存在下で、ノックアウトマウスにおけるT細胞数、およびリンパ球減少性マウスにおけるT細胞の増殖に対する明白な効果を有したにもかかわらず、T細胞の非常に少ない割合だけがLAG-3を発現したことが目立った。興味深いことに、全てのT細胞でのLAG-3の異所性の発現が、恒常的な増殖に対して、野生型細胞で見られるLAG-3の低レベルの一過性発現よりも優れた効果を有するというわけではなかった。これは、LAG-3シグナリングの閾値が非常に低いであろうこと、およびLAG-3シグナリングの効果を制限するその他の因子があるかもしれないことを示唆する。LAG-3と相互作用する下流のシグナリング分子(群)を同定し、LAG-3が恒常的な増殖を調節する機構を決定することは、将来の研究の明らかに重要な焦点である。
【0079】
B細胞およびマクロファージなどのLAG-3を発現しない細胞数の増大は、驚くべき観察であった。同時導入実験では、T細胞上にLAG-3が存在しないことが観察したその他の細胞タイプの増加の原因であったことを明らかに証明した。これは、その他の細胞タイプの数および/または増殖を制限するLAG-3シグナリングによって誘導されるか、またはその他の細胞タイプの数および/または増殖を制限するLAG-3によって負の調節が存在しないために産生される、可溶性または細胞表面タンパク質によるものであろう。この傍観者(bystander)の増殖およびその生理的役割の正確な性質は、決定されないままである。
【0080】
骨髄移植またはメガ用量幹細胞移植を受けている患者は、リンパ球再構成の速度が緩徐であるために、最初の4〜6ヶ月において特に感染症に感受性である。本発明者らの研究は、LAG-3が生存可能な治療の標的であること、およびLAG-3の発現またはその機能のブロッキングがT細胞移植を促進し、この感受性のウインドウを有意に減少させるという考えを支持する。
【0081】
実施例2−材料および方法
本実施例は、実施例1のための実験法および材料を提供する。
【0082】
マウス:以下のマウスを使用した:LAG-3-/- [Christophe Benoist and Diane Mathis, Joslin Diabetes Center, Boston, MA; Miyazaki, T. et al., Science 272: 405-408 (1996)からの許可によって、Yueh-Hsiu Chen, Stanford University, Palo Alto, CA,から得られる]; C57BL/6J [Jackson Labs, Bar Harbor, ME]; B6.PL-Thylα/Cy (Thy1.1類遺伝子性) [Jackson Labs]; RAG-1-/- [Jackson Labs, Bar Harbor, ME; Mombaerts, P. et al., Cell 68: 869-877 (1992)]; MHCクラスII-/- [Peter Doherty, St. Jude Children's Research Hospital, Memphis, TN; Grusby, M.J. et al., Science 253:1417-1420 (1991)によって提供された]; MHCクラスI-/-/II-/-[Taconic, Germantown, NY; Grusby, M.J. et al., Proc. Natl. Acad. Sci. U.S.A 90: 3913-3917 (1993)];OT-II TCRトランスジェニック・マウス[William Heath, Walter and Eliza Hall Institute, Parkville, Victoria Australia; Barnden, M.J. et al., Immunol Cell Biol. 76: 34-40 (1998)からの許可とともに、Stephen Schoenberger, La Jolla Institute for Allergy and Immunology, La Jolla, CA, によって提供された]、およびOT-I(OVA)TCRトランスジェニックマウス[Jackson Labs; Hogquist, K.A. et al., Cell 76: 17-27 (1994)]。ゲノム全般のマイクロサテライト解析では、LAG-3-/-マウスに関して試験した88の遺伝マーカーの97%がB6マウスに由来することを証明した(Charles River Laboratories, Troy, NY)。LAG-3-/-、MHCクラスII-/-、OT-I. LAG-3-/-およびOT-II. LAG-3-/-コロニーは、St. Jude Animal Resource Centerで維持した。全ての動物実験は、国家、州、および施設の指針に従って、AAALAC公認のSPF施設で行った。動物プロトコルは、St. Jude IACUCによって承認された。
【0083】
LAG-3構築物およびレトロウイルスの形質導入:LAG-3構築物は、記載されているとおりに組換えPCRを使用して作製した(Vignaali, D.A.A. and K. M. Vignali, J.Immunol. 162: 1431-1439 (1999))。LAG-3.WTおよびLAG-3.ΔKM(細胞質尾部の保存されたKIEELEモチーフを欠失するLAG-3)は、記載されている(Workman, C.J. et al., J. Immunol. 169: 5392-5395 (2002))。LAG-3構築物を、内部リボソーム導入部位(IRES)および緑色蛍光タンパク質(GFP)を含むマウス幹細胞ウイルス(MSCV)に基づいたレトロウイルスベクターにクローニングし、記載されているようにレトロウイルスを作製した(Persons, D.A. et al., Blood 90: 1777-1786 (1997); Persons, D.A. et al., Blood Cells Mol Dis. 24: 167-182 (1998))。次いで、24時間後に105/ml以上のウイルス力価が得られるまで、GPE+86細胞を繰り返して(7〜10回)形質導入することによってレトロウイルス産生株の株化細胞を作製した(Markowitz, D. et al., J Virol. 62: 1120-1124 (1988))。
【0084】
フローサイトメトリー:単個細胞懸濁液を脾臓から作製し、RBCをゲイ液で溶解した。脾細胞は、まずFc Block、抗CD16/CD32(2.4G2)(BD PharMingen, San Diego, CA)と共に氷上で10分間染色した。次いで、細胞をBD PharMingenからの種々の抱合型抗体を使用して以下の細胞表面マーカーを染色した:
。LAG-3発現は、ビオチン化されたラット抗LAG-3 mAb(C9B7W, IgG1κ; Workman, C.J. et al., Eur. J. Immunol. 32: 2255-2263(2002))またはPE抱合体として得た同じ抗体(BD PharMingen)によって評価した。次いで、細胞をフローサイトメトリー(Becton Dickinson, San Jose, CA)によって解析した。
【0085】
ブロモデオキシウリジン取り込み:5、16、28、および52週間目に、LAG-3+/+、LAG-3-/-、OTII.LAG-3+/+、およびOTll.LAG-3-/-マウスには、(0.8mg/ml)それらの飲料水中にBrdU(Sigma, St. Louis, MO)を8日間与えた。次いで、マウスをCO2吸入によって屠殺して、脾臓を取り除いた。BrdU取り込みの染色は記載されているように行った(Flynn, K. J. et al., Proc. Natl. Acad. Sci. U.S.A 96: 8597-8602 (1999))。簡潔には、LAG-3-/-およびLAG-3+/+脾細胞をTCRαβ、CD4、CD8、およびB220発現のために染色した。OTII.LAG-3+/+およびOTII.LAG-3-/-脾細胞は、Vα2およびCD4発現のために染色した(PharMingen)。次いで、細胞を1.2mlの氷冷95%エタノールで氷上で30分間固定した。次いで、細胞を洗浄し、PBS +1%パラホルムアルデヒド +0.01% Tween 20で室温において1時間透過化処理した。次いで、細胞を洗浄し、50KUのデオキシリボヌクレアーゼ(Sigma)の0.15M NaCl+4.2mM MgCl2(pH 5.0)溶液で37℃において10分間インキュベートした。BrdUを抗BrdU-FITC(Becton Dickinson)をRTで30分添加することによって検出し、次いでフローサイトメトリーによって解析した。
【0086】
養子性の導入実験:脾細胞からのT細胞および/またはB細胞をFACSによって陽性選別するか、または磁気ビーズ細胞選別(MACS)によってネガティブに選別した。FACS精製のために、脾細胞をTCR□□、CD4、およびCD8発現について染色し、およびMoFlow(Cytomation, Ft. Collins, CO)で陽性選択によって選別した。ネガティブMACS精製のためには、脾細胞をPE結合抗B220、抗Gr1、抗Mac1、抗TER119(赤血球)、抗CD244.2(NK細胞)、および抗CD8(OTII トランスジェニックT細胞のネガティブ精製のために)で染色した。次いで、細胞を抗PE抗体と結合された磁気ビーズ(Miltenyi Biotech, Auburn, CA)と共にインキュベートし、自動MACS(Miltenyi Biotech, Auburn, CA)で90〜95%の純度に選別した。いくつかの実験では、T細胞をカルボキシフルオレセインジアセテートスクシンイミジルエステル(CFSE)でラベルした。細胞をPBSで2回洗浄し、1×107細胞/mlでPBS+0.1% BSAに再懸濁し、5μM CFSEと共に37℃で10分間インキュベートした。細胞をPBS+0.1% BSAで2回洗浄した。CFSE標識または無標識の精製T細胞(5x106または1×107)、および場合によってはB細胞(5×106)をRAG-1-/-または Thyl.1+(B6.PL)マウスにi.v.注射した。
【0087】
正常T細胞のレトロウイルスによる形質導入:OTII.LAG-3+/+およびOTII.LAG3-/-マウスからの脾臓を取り出し、単細胞懸濁液を2.5×106細胞/mlで作製した。脾細胞をOVA 326-339ペプチド[10μM]で2日間培養液中で活性化した。次いで、活性化された脾細胞をGFP単独、LAG-3.WT/GFP、またはLAG-3.ΔKM/GFPレトロウイルス産生株細胞の単層上で、ポリブレンの存在下において2日間インキュベートした。細胞を10日間静止させ、次いでFACSによってVα2+/CD4+/GFP+発現を選別した。細胞をさらに2日間静止させ、次いで5×106細胞を、尾静脈を経てRAG-/-マウスに注射した。導入の15日後、マウスをCO2吸入によって屠殺し、脾臓を取り除いた。脾細胞を染色し、フローサイトメトリーによって解析した。
【0088】
実施例3−強力な調節活性で誘導されるTreg細胞
Treg特異的な分子を同定するために、本発明者らは、ウイルス感染に応答してエフェクター/メモリー細胞か、または自己抗原としての同族抗原に遭遇することによってTreg細胞のいずれかに分化する抗原特異的なT細胞の差次的遺伝子発現解析を行った。この解析により、LAG-3遺伝子がTreg細胞において選択的にアップレギュレートされることが明らかになった。LAG-3(MHCクラスIIに結合するCD4相同体)の生理的な役割は、はっきり解明されなかった。いくつかのインビトロでの実験では、LAG-3が負の調節機能を有しているかもしれないことを示唆した(Hannier et al., 1998; Huard et al., 1994; Workman et al., 2002a; Workman et al., 2002b)。ここで、本発明者らは、LAG-3の膜発現がCD25とは独立して選択的にTreg細胞をマークすること、およびLAG-3がインビトロおよびインビボの両方でTreg細胞の抑制性の活性を調整することを示した。
【0089】
T細胞エフェクター/メモリーと寛容誘導との間の相違を検討するために、本発明者らは、モデル抗原の赤血球凝集素(HA)に特異的なT細胞受容体(TCR)トランスジェニックCD4+ T細胞(クローン6.5)の養子性の導入を利用した。組換えHA発現ワクシニアウイルス(Vac-HA)を感染させた野生型マウスでは、養子性に導入したHA特異的な6.5 CD4+ T細胞が、HAと遭遇することによってエフェクター/メモリー細胞に分化した。エフェクター/メモリー反応は、典型的な増殖/収縮期およびメモリーマーカーの発生によって特徴づけられる。養子性に導入された動物から取り出したときに、これらのエフェクター/メモリー細胞は、抗原特異的な増殖反応およびγ-インターフェロン生産によってアッセイすると、ナイーブ6.5 CD4+ T細胞と比較して、インビトロでHAに対して応答性亢進である。このメモリー反応は、養子性の導入後に何ヶ月も持続する。対照的に、複数の上皮組織にHAを発現するC3-HAトランスジェニックマウスへの6.5 CD4+ T細胞の養子性導入では、寛容を生じる(Adler et al., 2000; Adler et al., 1998)。エフェクター/メモリー反応と同様に、γ-インターフェロンなどのエフェクターサイトカインの増殖および発現によって特徴づけられる迅速な増殖/活性化期がある。しかし、活性化期の後に、全HA特異的T細胞プールが縮小し、残りの6.5細胞は、養子性導入4〜7日後に抗原刺激によってγ-インターフェロンを産生したり、またはインビトロで増殖することができない(Adler et al., 2000; Huang et al., 2003)。IL-2およびγ-インターフェロンなどのリンホカインを産生して抗原に対する応答を急増する能力の消滅は、アネルギー性の表現型の標準的な実効の定義を表す。
【0090】
寛容の誘導に先だつC3-HAマウスにおける最初のインビボエフェクター期の強度は、養子性に導入した6.5 CD4+ T細胞の数、並びにレシピエントマウスでのHA抗原の発現レベルに比例する。従って、C3-HAlowマウスは、2.5×106 6.5 CD4+ T細胞の導入をかなりよく許容するが、C3-HAlowマウスよりも1000倍多くのHA発現を有するC3-HAhighマウスでは、2.5×106の6.5 CD4+ T細胞の導入後4〜7日以内に死亡する(図1A)。死因は、HA発現が最も高い肺におけるトランスジェニック6.5 CD4+ T細胞の浸潤による致死肺血管炎である。2.5×105未満の6.5 CD4+ T細胞をC3-HAhighマウスに養子性導入すると、肺血管炎の重篤さの軽減が引き起こされて、レシピエントが生存する(図1A)(Huang et al., 2003)。興味深いことに、亜致死量で導入された6.5 CD4+ T細胞では、その後の保護されていないC3-HAhighマウスにおいて致死量の6.5 CD4+ T細胞であると考えられる注入による死からマウスを保護することができるので、これらは節性の表現型を獲得する。わずか8,000細胞の導入(致死量の0.3%)ほどでその後の2.5×106のナイーブ6.5 CD4+ T細胞の注入による死から動物を完全に保護するので、このインビボでの調節機能は極めて強力である。保護は、最初の導入の4日後という早い時期に観察され、6ヶ月まで活性なままである(図1A)。養子性導入前のCD4+ T細胞(しかし、CD8+ T細胞ではない)の枯渇により、保護作用が全くなくなり、これによりアネルギーとなったクローン型(clonotypic)6.5 CD4+ T細胞のTreg表現型を定義する。
【0091】
致死間質性肺炎の抑制は、肺における最初の投入(Treg)の6.5T細胞の蓄積、および第2の注入からの浸潤するTエフェクター細胞の数の急激な減少を伴う。肺に蓄積する代わりに、Treg細胞がない場合に生じるように、Tエフェクター細胞は、脾臓の動脈周囲のリンパ性外筒に蓄積する(図1B)。アネルギー細胞がTreg機能を証明するというさらなる証拠は、これらがインビボで細胞障害性のHA特異的CD8+ T細胞の活性化を阻害するという知見から生じる(データ示さず)。第1の(保護)養子性導入より前にCD25+ T細胞を除去しても、その後の6.5 T細胞の致死的なチャレンジから動物を保護することができるTreg細胞の発生には影響を及ぼさなかった。従って、最初の投入のT細胞のTreg表現型は、養子性に導入された集団の中の天然に存在するTreg細胞の結果とは反対に、養子性の導入の後で獲得された可能性が高い。これらの知見は、発行されたVon Boehmerおよび同僚の知見(彼らは、6.5 CD4+ T細胞が、実際にTreg機能を示すB細胞区画にHAを発現するトランスジェニックマウスに導入された後、寛容になることを証明した)と非常に一致している。(Jooss et al., 2001)。
【0092】
実施例4−LAG-3は、誘導されたTreg細胞上に差次的に発現される
本発明者らのインビボの系でアネルギー性/Treg表現型と関係する遺伝子を同定するために、本発明者らは、非トランスジェニックレシピエント内に養子性に導入し、続いてVac-HA免疫化を行ってエフェクター/メモリーT細胞を作製した後の、または、C3-HAhighマウス内に導入してアネルギー性の/Treg細胞を作製した後の、いずれかの精製した6.5 CD4+ T細胞に対してアフィメトリックス・チップ解析を行った。Thy1.1(+)Thy1.2(-)類遺伝子性の6.5 T細胞は、CD8+ T細胞、B細胞、およびThy1.2(+) T細胞のMACS Column枯渇、続いてフローサイトメーターによって>95%の純度に選別することを含む経時的な単離手順を使用して、野生型(エフェクタ/メモリー)またはC3HAhigh(アネルギー性/Treg)レシピエントを感染させたThy1.1(-)Thy1.2(+) Vac-HAから精製した。このプロトコルでは、TCRまたはCD4依存的な遺伝子発現パターンを潜在的に変更し得るTCR特異的またはCD4補助受容体特異的な抗体の使用を避けている。
【0093】
RNAは、チップ解析のために、0日の試料としてナイーブ6.5 CD4+ T細胞から単離し、また養子性導入後2、3、および4日での6.5 CD4+ T細胞から単離した。アネルギー性/Treg集団とエフェクター/メモリー集団との間で差次的に発現された遺伝子を0〜4日からのこれらの差次的発現を合計したアルゴリズムに従って順序づけられた順位にした。驚くほど多数の遺伝子が、アネルギー性/Treg集団において、これらの養子性導入後の初期の時点でさえも、選択的に活性化された。これらの遺伝子の多くは、既知の機能のないESTを示した。以前に同定された遺伝子の中で、LAG-3は、エフェクター/メモリー集団と比較してアネルギー性/Treg集団において最も差次的に発現するものの一つであった。その後、この結果を、養子性導入の1ヶ月後まで及ぶ種々の時点について、LAG-3プライマー-プローブ対での定量的RT-PCR解析によって確認した。エフェクター/メモリー細胞における最小の最初の増大後、LAG-3発現は、養子性導入後20日までにベースラインに戻った。著しく対照的に、LAG-3の発現は、アネルギー性/Treg細胞群の中で最初の5日にわたって20〜50倍増大し、その後の4週の解析にわたって高いままである(図2A)。対照的に、FoxP3、GITR、およびCTLA-4のレベルは、最初の4〜5日にわたって適度な増大を示し(1.5〜4倍)、エフェクター/メモリー細胞および誘導されたアネルギー性/Treg細胞の両方においても同様であった(データ示さず)。
【0094】
次いで、エフェクター/メモリー6.5 CD4+ T細胞と比較した、アネルギー性/Treg 6.5 CD4+ T細胞の集団でのLAG-3の細胞表面発現を、抗LAG-3-モノクローナル抗体を使用して解析した(Workman et al., 2002b)(図2B)。エフェクター/メモリー細胞に対するLAG-3染色は非常に低レベルであるが、C3-HAhighトランスジェニックマウスからのアネルギー性/Treg細胞のうちの有意な比率が、遺伝子発現の結果と一致して、適度から高度のレベルのLAG-3染色を示した。IL-10は、一般にTregの分化および機能と関連しているので(Moore et al., 2001)、本発明者らは、IL-10 mRNAの内因性レベルおよびこれらのC3-HAhighトランスジェニックマウスからのCD4+ T細胞サブセット(アネルギー性/Treg 6.5 CD4+ T細胞)のLAG-3 mRNAのレベルとの相関を解析した。多くの実験にわたる複数の試料のアネルギー/Treg集団の解析により、LAG-3 mRNAレベルとIL-10 mRNAレベルの間の相関が明らかとなり、0.87の相関係数(R2)を有した(図2C)。
【0095】
実施例5−LAG-3は、最大のTreg機能のために必要とされる
アネルギー性/Treg 6.5 CD4+ T細胞の集団でのLAG-3およびCD25の細胞表面発現を抗LAG-3および抗CD25モノクローナル抗体を使用して協調的に解析した。同比率のエフェクター/メモリーおよびアネルギー性/Treg細胞がCD25を発現するが(データ示さず)、アネルギー性/Treg細胞でのLAG-3およびCD25発現は、完全には調和性でなかった(図3A)。従って、本発明者らは、細胞をLAG-3high CD25high、LAG-3high CD25low、LAG-3low CD25high、およびLAG-3low CD25low集団に選別し、標準的なインビトロ抑制アッセイ法でこれらの調節活性を解析した。ナイーブ6.5 CD4+細胞の中での増殖反応のインビトロ抑制では、LAG-3high CD25high集団が最高の抑制活性を示すこと、およびLAG-3low CD25low集団が最も低いが、LAG-3high CD25lowおよびLAG-3low CD25low細胞の抑制活性は、同等であったことを示した(図3B)。これらの結果は、誘導されたTreg細胞の中で、LAG-3およびCD25の組み合わせが、最も抑制活性を有するTreg細胞を特徴付けるであろうことを示唆する。
【0096】
誘導されたTreg細胞によって抑制を調節する際のLAG-3の直接的な役割をさらに評価するために、本発明者らは、まずナイーブHA特異的T細胞のインビトロでの増殖反応をLAG-3発現細胞が抑制する能力を、抗LAG-3抗体がブロックすることができるかどうかを決定した。2μg/mlの濃度の抗LAG-3抗体は、インビトロアッセイ系でTreg 6.5 CD4+ T細胞による抑制を阻害する(図4)。2日のアッセイ期間にわたって、抗LAG-3抗体は、Tregの非存在下で刺激される6.5T細胞の増殖反応に影響を及ぼさず、抗LAG-3抗体の効果が、実際にTreg細胞に対するものであり、エフェクター細胞ではなかったことを確認した(データ示さず)。抗LAG-3抗体がTreg細胞によるインビトロでの抑制をブロックする能力は、LAG-3が単にTreg選択マーカーでなく、Treg活性を調整する分子であることを証明する。
【0097】
実施例6−LAG-3は、インビボで誘導されるTreg活性に必要とされる
本発明者らは、次に、抗LAG-3抗体の投与がC3-HAhighマウスにおいてTregによる致死的間質性肺炎の抑制をブロックすることができるかどうかを決定することによって、インビボでのTreg機能を調整する際のLAG-3の役割を評価した。C3-HAhighマウスを8,000(亜致死量)の6.5 CD4+ T細胞で前処理し、続いて最初の導入の4日後に、その後の2.5×106の用量のナイーブ6.5 CD4+ T細胞を前処理した。上述のように、Tregは、この時点ですでに発生していた。抗LAG-3抗体(200□g)をその後の2.5×106の6.5細胞のチャレンジと共にi.v.投与し、2日後に別に200□gを与えた。この抗体治療では、完全にTreg細胞のインビボ抑制活性を除去し、マウスは、保護的な亜致死量の6.5の前処理を伴わずに致死的にチャレンジしたC3-HAhighマウスと同等の時間枠で死んだ。これに反して、アイソタイプ対照抗体(ラットIgG1)または抗体なしで処理した確立されたTregを有するマウスは、その後の2.5×106ナイーブ6.5 T細胞でのチャレンジでも生存した(図5A)。これらの結果は、抗LAG-3抗体がインビボでTreg活性をブロックしたことを示唆するが、他の形式では、直接Treg細胞を阻害するのではなく、抗LAG-3抗体は、チャレンジ集団のT細胞を超活性化(hyper-activate)し、その結果これらがTregの阻害作用を克服した可能性がある。この可能性を除外するために、本発明者らは、わずかに死亡率閾値以下の6.5 T細胞の用量と共に、抗LAG-3抗体をインビボ投与すると、予め確立されたTreg集団の非存在下での致死性を引き起こすかどうかを問うた。従って、本発明者らは、2.5×105の6.5 T細胞(死亡率を生じさせないであろう最大量)または8.0×105の6.5 T細胞(導入の7〜14日後におよそ50%の死亡率)を、抗LAG-3抗体またはアイソタイプ対照と共にC3-HAhighマウスに投与した。図5Bは、抗LAG-3処理では、2.5×105の6.5T細胞の用量でも致死性にならず、また8.0×105の6.5T細胞の用量の部分的な死亡率も増強しなかったことを証明する。従って、図5Aの実験の抗LAG-3抗体の効果は、Treg細胞の直接阻害であった。
【0098】
実施例7−LAG-3は、天然のTreg細胞によって発現され、抑制活性に必要とされる
合わせて考えると、これらのデータは、誘導されたTregの抑制機能を媒介する際のLAG-3の重要な役割を確信させる。誘導されたTregと天然のTregの間の関係は、不明なままであることを考えると、LAG-3は、野生型マウスからのCD4+CD25+ T細胞に選択的に発現したかどうかを調べることに興味が持たれる。LAG-3 mRNA(CTLA-4、FoxP3、およびGITR mRNAとともに)は、実際にCD4+CD25-細胞と比較してCD4+CD25+細胞上に選択的に発現されている(図6A)。この再現性の知見にもかかわらず、本発明者らは、CD4+CD25+またはCD4+CD25-細胞上のいずれにおいても、抗体染色によって表面LAG-3を検出することができなかった。しかし、透過化処理された細胞の抗体染色では、10〜20%のCD4+CD25+細胞がLAG-3の細胞内貯臓を表したことを明らかに示した。対照的に、透過化処理されたCD4+CD25-細胞の染色では、絶対にLAG-3+集団がないことを証明した(図6B)。これらの知見は、少なくともいくつかの天然のTregがLAG-3の細胞内貯臓を有し、これが、同族の抗原と遭遇することによって細胞表面に迅速に動員され、その後に抑制を媒介するのであろうことを示唆する。天然のTregは、CD4およびCD25によって定義されるT細胞群に含まれるが、実際のTreg細胞は、細胞内LAG-3を発現するものである可能性も実際にはある。天然のTregの調節機能におけるLAG-3の役割を直接評価するために、本発明者らは、抗LAG-3抗体が、精製されたCD4+CD25+細胞によって媒介されるインビトロでの抑制を阻害することができるかどうかを調べた。図6Cは、抗LAG-3抗体が、精製されたCD4+CD25+細胞によって媒介される抑制を実際にブロックすることを証明しており、LAG-3は、天然並びに誘導されたTregによって媒介される抑制において役割を果たしていることを示唆する。インビトロでの抑制アッセイ法の最後にCD4+CD25+細胞を染色すると、およぼ20%が現在もそれらの表面上に高レベルのLAG-3を発現することが明らかになり、細胞内LAG-3がTCRの結合環境下で表面に動員され、調節活性を媒介するという考えを支持する(図6D)。
【0099】
実施例8−LAG-3の異所性の発現は、調節活性を与える
図5および6のブロッキング実験は、LAG-3が最大のTreg機能のために必要とされることを示唆する。この結論をさらに確証するために、本発明者らは、T細胞上のLAG-3の異所性の発現が調節活性を与えるかどうかを決定するための一連の形質導入実験を行った。これらの実験のために、6.5 CD4+ T細胞から最初にCD25+「天然」のTregを枯渇させ、次いでGFP単独、GFP+野生型LAG-3、またはGFP+変異体LAG-3.Y73FΔCY(実質的にMHCクラスIIに対する親和性が減少され、かつ下流のシグナリングを媒介することができない(Workman et al., 2002a))のいずれかをコードするMSCVに基づいたレトロウイルスベクターを形質導入した。10日の休止期の後、MSCV-GFPベクターを形質導入したGFP+6.5 CD4+ T細胞では、本質的に内因性LAG-3の染色は観察されなかったが、MSCV-LAG-3/GFPおよびMSCV-LAG-3. Y73FΔCY/GFPを形質導入したGFP+ 6.5細胞では、高レベルのLAG-3染色が観察された。増殖アッセイ法では、それぞれの群に由来するGFP+細胞を選別して、APC、HA110-120ペプチド、およびナイーブ6.5CD4+CD25-細胞と種々の比率で混合した。図7に示したとおり、野生型LAG-3を発現する6.5細胞は、ナイーブ6.5細胞の増殖を強力に抑制したが、対照GFP導入6.5細胞または非機能的LAG-3.Y73FΔCY変異体を発現する6.5細胞では観察されなかった。本アッセイ法において、ナイーブ6.5細胞に加えてGFPおよびLAG-3.Y73FΔCYを導入した6.5細胞自体も増殖したので、総増殖は、実際にこれらの2つの後者の群でやや増大した。実際に、野生型LAG-3を導入したT細胞自体は、非導入6.5細胞の増殖阻害とは別に、増殖応答の有意な減少を示した。これらの結果は、抑制の際のLAG-3の機能的な役割を確証させる直接の証拠を提供する。興味深いことに、LAG-3導入によっても、Foxp3、CD25、CD103、およびGITRを含むその他のTreg関連遺伝子は誘導しなかった(データ示さず)。この結果は、エフェクター/メモリー対アネルギー/Treg表現形に分化する6.5T細胞の間にこれらの遺伝子の有意な差次的発現がないことに加え、LAG-3は、Foxp3経路とは独立に調節性T細胞機能の異なった経路を媒介するであろうことを示唆する。
【0100】
実施例9−考察
これらの知見から、LAG-3は、Treg細胞で選択的にアップレギュレートされ、Treg機能を媒介する際に直接関与しているであろう細胞表面分子であると同定される。天然および誘導性のTreg活性が定義されている多くの系を考えると、LAG-3は、「普遍的な」Tregマーカーであるか、または選択的に特定のTregサブセットのみをマークするかは決定されていないままである。本発明者らの結果は、CD4+ Treg細胞を誘導することに加えて、LAG-3は、天然のCD4+CD25+Treg細胞による抑制を媒介する際に少なくともいくらかの役割を果たしていることを示唆する。さらに、その他の実験データからも、天然のTregによる恒常性リンパ球増殖の調節におけるLAG-3の役割も証明される(Workman and Vignali、添付論文)。LAG-3発現が野生型マウス由来のCD4+CD25+ T細胞の中で有意に多いという知見は、これが天然のTreg、並びに誘導されたTregの機能において役割を果たしているであろうこと示唆する。図3の実験によって示唆されるように、LAG-3とCD25の組み合わせは、最も強力なサプレッサー活性を有するTregサブセットを定義するであろう。本発明者らは、LAG-3は、インビトロでのアッセイ法での調節活性の大きさと相関する可変レベルで発現されるので、Tregの「系統マーカー」であることを提案しない。実際に、Tregが非細胞自律的様式で寛容を促進することができる安定な系統または分化状態を表すかは明白でない(von Boehmer, 2003)。異なる系のTreg機能について、異なる機構が同定された(Shevach, 2002に概説)。LAG-3high細胞は、IL-10の量の増大を生じ、インビトロでの抑制活性の増強を示すが、本発明者らの系において抑制機能を媒介する際のIL-10の役割は、決定されていないままである。LAG-3に対する抗体は、インビトロおよびインビボの両方でTreg細胞のサプレッサー活性を阻害する。従って、本発明者らは、LAG-3がこのT細胞サブセットのサプレッサー活性を調整するTreg特異的な受容体または補助受容体であることを提案する。
【0101】
LAG-3について細胞自立的な抑制的な役割が多くの研究で示唆されているが(Huard et al., 1994; Workman et al., 2002b)、LAG-3 KOマウスによる最初の研究では、明白な自己免疫または高免疫についての何らかの証拠を発見することができなかった(Miyazaki et al., 1996)。Treg機能におけるLAG-3についての本発明者らが提案した役割を考えると、LAG-3ノックアウトマウスは、これらのマウスにおいて報告されなかった多システムの自己免疫を示すであろうことが予測されるだろう(すなわち、Foxp3ノックアウトまたはふけマウス(scurfy mice)と同様)。しかし、細胞恒常性の調節の欠陥などの、LAG-3ノックアウトマウスによって示される調節性T細胞の欠損が明らかに存在した(Workman and Vignali、添付論文)。本発明者らは、PD-1ノックアウトマウスで観察したように(Nishimura et al., 1999; Nishimura et al., 2001)、遅発性の自己免疫のより微妙な証拠について以前のLAG-3ノックアウトマウスを再検査している途中である。また、その他の調節機構がLAG-3発現の減少を補うようにこれらのマウスにおいて増強されるかもしれないことが考えられる。
【0102】
Treg細胞に高レベルで発現されているので、LAG-3は、癌および自己免疫疾患の両方を治療するためにTreg活性を選択的に操作するための優れた潜在的な標的を提供する。CD25「ゴールドスタンダード(gold standard)」Tregマーカーは、IL-2受容体複合体の重要な成分であり、活性化された細胞において高レベルで誘導されている。CD4+CD25+細胞がTreg活性の際に濃縮されていることの明らかな理由は、CD25がTreg機能に特異的であるためではなく、むしろTreg細胞が、末梢において自己抗原と持続的に遭遇することによって慢性的に刺激されるためである。より最近では、TNF受容体スーパーファミリーメンバー18分子(GITRとも呼ばれる)が、Treg細胞上でアップレギュレートされることが証明された。さらに、GITRに対する抗体は、インビボおよびインビトロの両方でTreg活性を阻害することが報告された。しかし、GITRは、活性化されたT細胞上で同等にアップレギュレートされており、したがって、明らかにTreg細胞のマーカーとしてCD25よりも選択的ではない(McHugh et al., 2002; Shimizu et al., 2002)。そのうえ、CD4+CD25-細胞群は、一定の免疫機能を抑制することができるという多数の報告がある(Annacker et al., 2001; Apostolou et al., 2002; Curotto de Lafaille et al., 2001; Graca et al., 2002; Shimizu and Moriizumi, 2003; Stephens and Mason, 2000)。それにもかかわらず、CD25highLAG-3high細胞が最も優れた抑制活性を示すという知見は、これらの細胞表面分子の両方に対する抗体を、Treg活性を操作するために協調的に使用してもよいことを示唆する。
【0103】
本発明者らのデータは、LAG-3が天然および誘導されたTreg細胞の最大の抑制活性に必要とされることを示す。しかし、それは十分であるか?これまでに、活性化されたT細胞上の調節活性を与えることが示された唯一の分子は、Foxp3である(Fontenot et al., 2003; Hori et al., 2003)。重要なことに、本発明者らは、ここでLAG-3(しかし、機能的に欠損がある変異体でない)のCD4+ T細胞におけるその異所性の発現が調節活性を与えることができることも示した。
【0104】
もう一つの重要な疑問は、Treg細胞が直接的なT-T相互作用を介して、またはDC中間体を介してCD4+およびCD8+エフェクター細胞の反応性を抑制するかどうかということである。MHCクラスII結合分子のLAG-3のTreg選択的および機能的な発現の同定により、これらの細胞が重要な役割を果たしている疾患のために、機構を分析し、およびTreg機能を操作するに際して新たな足掛かりを提供するはずである。
【0105】
実施例10−実験法
トランスジェニックマウス
C3-HAトランスジェニックマウスは、以前に記載されている(Adler et al., 2000; Adler et al., 1998)。手短には、インフルエンザウイルスA/PR/8/34(Mount Sinai strain)に由来する赤血球凝集素(HA)遺伝子をラットC3(1)プロモーターの制御下に配置した。2つの初代系統をB10.D2遺伝的バックグラウンドで確立した。これらの2つの初代系統、C3-HAhighおよびC3-HAlowは、それぞれ30〜50および3つの導入遺伝子コピーを含み、肺および前立腺を含む非リンパ系の組織の同一セットにおいてC3-HAハイブリッドmRNAを発現する。C3-HAhighとC3-HAlowとの間の総HAタンパク質発現の相違は、発現レベルが最も高い肺および前立腺では直接測定しなかったが、相違は、ハイブリドーマのサイトカイン放出が誘導された組織抽出物の生物検定によって示すと、およそ1000倍である。
【0106】
I-Ed-制限されたHAエピトープ(110SFERFEIEPKE120; SEQ ID NO:7)を認識するTCRを発現するTCRトランスジェニックマウス系統6.5株(Dr. Harald von Boehmer, Harvard University, Boston, MAによって寛大に提供された)をB10.D2遺伝的バックグラウンドに対して9世代戻し交配した。Kd拘束性HAエピトープを認識するTCRを発現するその他のTCRトランスジェニックマウス系統クローン-4(518IYSTVASSL526;SEQ ID NO:8)(Dr. Linda Sherman, Scripps Research Institute, La Jolla, CAによって寛大に提供された)も、Thy 1.1/1.1 B10.D2遺伝的バックグラウンドに対して9世代を超えて戻し交配した。クローン型(clonotypic)抗体は、クローン-4 TCRに利用できないので、Thy 1.1を代わりのマーカーとして使用した。クローン-4マウスの成熟CD8+ T細胞のほとんど全てがKd制限されたHAエピトープを直接認識するので、Thy 1.2/1.2レシピエントへの養子性導入に続き、本発明者らは、全てのThy 1.1+CD8+ T細胞がHA-特異的なクローン型TCRを発現するとみなすことができる(Morgan et al., 1996)。
【0107】
実験に使用されるトランスジェニックマウスは、8〜24週の間の年齢であった。マウスの使用を含む全ての実験は、Johns Hopkins University School of Medicineの動物実験委員会(Animal Care and Use Committee)によって承認されたプロトコルに従って行われた。
【0108】
養子性の導入
クローン型CD4+またはCD8+ T細胞は、6.5またはクローン-4トランスジェニックマウスのプールされた脾臓およびリンパ節から調製した。クローン型の割合は、フローサイトメトリー解析によって決定した。活性化マーカーCD44を解析して、これらのクローン型細胞がドナーマウスにおいて活性化されず、表現型がナイーブであったことを確認した。HBSSで3回洗浄した後に、尾静脈を介してi.v.注射するために、適切な数の細胞を0.2mlのHBSSに再懸濁した。
【0109】
免疫組織化学
組織を養子性の導入の3日後にマウスから集めた。組織をイムノヒストフィックス(Immuno Histo Fix)(A Phase sprl, Belgium)に4℃で3日間固定し、次いでイムノヒストワックス(Immuno Histo Wax)(A Phase sprl, Belgium)に埋め込んだ。ビオチンラベルした抗Thy1.1 mAb(PharMingen, San Diego, CA)を使用して連続切片を染色した。ベクタステインABCキット(Vectastain ABC kit:Vector, Burlingame, CA)およびNovaRed(Vector)は、発色のために使用した。切片をヘマトキシリンQS(Vector)で対比染色した。ニコンEclipse E800を使用して切片を解析した。最終的な画像処理は、Adobe PhotoShop (Mountain View, CA)を使用して行った。
【0110】
インビボで初回抗原刺激した6.5 CD4+ T細胞の濃縮および精製
インビボで養子性導入の後に、エフェクター/メモリーまたは寛容のいずれかを誘導すると、レシピエントマウスの脾臓の6.5 CD4+ T細胞のクローン型の割合は、0.2%〜5%だけである。アフィメトリックス遺伝子チップ解析のためなど、さらなる研究のために十分なクローン型CD4+ T細胞を得るためには、慎重な濃縮および精製が必須である。ドナー6.5 T細胞をThy1.1(+)Thy1.2(-)遺伝的バックグラウンドに対して交配した(これは、Thy1.1(-)/Thy1.2(+)レシピエントに養子性の導入後に2工程の濃縮および精製手順でできる)。CD8+ T細胞、B細胞、およびレシピエントT細胞(Thy1.2+)を枯渇させるために、最初にビオチン化された抗CD8(Ly-2、53-6.7)、抗B220(RA3-6B2)、および抗Thy1.2(30-H12)抗体(全てBD Biosciences PharMingen, San Diego, CAから購入)、並びにMACSストレプトアビジン・ミクロビーズおよびMACS LS分離カラム(Miltenyi Biotech, Auburn, CA)を使用して6.5 CD4+ T細胞を濃縮した。CD4+ T細胞およびCD8+ T細胞は、Thy1.1を有する唯一の集団であるので、またCD8+ T細胞が濃縮の間に減少したために、 FACSVantage SE細胞ソーター(BD Biosciences)を使用してThy1.1(+)細胞をソーティングすることにより、非常に精製された6.5 CD4+ T細胞(95%)にした。この技術では、TCRまたはCD4依存的な遺伝子発現パターンを変更する可能性のあるTCR特異的またはCD4補助受容体特異的な抗体の使用を回避する。
【0111】
遺伝子チップ解析
選別した細胞を、Qiashredderカラム(Qiagen, Valencia CA)で剪断し、続いてRNeasyキット(Qiagen)を使用して総RNAを単離した。cDNAは、Superscript Choiceキット(Gibco/BRL)およびHPLCで精製したT7-DTプライマー(Proligo, Boulder, CO)を使用して合成した。ビオチン化したcRNAプローブは、ENZO BioArray RNA転写物キット(Affymetrix, Santa Clara, CA)を使用して調製した。マウスの遺伝子チップU174A、B、およびCには、標準的なアフィメトリックス・プロトコルに従ってハイブリダイズさせて解析した。
【0112】
アネルギー/Treg誘導とエフェクター/メモリー誘導との間のCD4+ T細胞の遺伝子の差次的発現のランク付け
養子性の導入後の種々の日に、精製したナイーブ6.5クローン型CD4+ T細胞、並びにアネルギー性/Tregおよびエフェクター/メモリー6.5クローン型のCD4+ T細胞から調製したmRNAをアフィメトリックス遺伝子チップによって解析した。アネルギー/Treg誘導とエフェクター/メモリー誘導との間の遺伝子の差次的発現は、「距離」によってランクを付けた。距離は、養子性の導入後の2日(|d1|)、3日(|d2|)、および4日(|d3|)目の、アネルギー性T細胞とエフェクター/メモリーT細胞との間の発現の絶対的な相違の合計として定義し、標準化のためのナイーブCD4+ T細胞(n)の値で割った。
【0113】
抗体および染色
以下の抗体を使用した。精製したか、またはPE結合したかのいずれかの抗LAG-3(C9B7W、PharMingenから)(Workman et al., 2002b);精製したか、またはFITC結合したかのいずれかの抗CD25(7D4、PharMingenから);および抗GITR(R&D Systemsから購入したポリクローナル抗体)。LAG-3およびCD25の細胞表面染色のために、6.5+/-Thy1.1+/-トランスジェニックマウスからの脾細胞を単離し、CD4+ネガティブ選択単離キット(Miltenyi Biotec)を使用してCD4+について濃縮した。フローサイトメトリーによって決定した約2.5×106のクローン型6.5細胞(総CD4+細胞の16%)をHBSSに再懸濁し、137(C3-HA high)または野生型B10.D2内に尾静脈を介して注射した。B10.D2マウスの1群には、5×106のVac-HAを処理し、その一方で、その他の群には、ナイーブ対照のために無処置のままにした。脾細胞および鼠径部および腋窩リンパ節を5日後に集め、単個細胞懸濁液に調製した。RBCをACK溶解緩衝液で溶解した。直ちに細胞を5μgの全ラットIgG(Sigma)で15分間ブロックした後に、抗6.5 TCR-ビオチン+SA-APC、LAG-3-PE、およびCD25-FITC、または対応するアイソタイプ対照で染色した。抗6.5ビオチン以外は、全ての染色試薬をPharmingenから購入した(San Diego, CA)。短いインキュベーション後に、試料をPBS+1% FBS溶液中で一度洗浄し、FACScalibur機に読み込んだ(BD, San Jose, CA)。
【0114】
誘導された6.5調節性T細胞のインビトロでの抑制アッセイ法
1×104の精製したナイーブ6.5 CD4+ T細胞(応答者)および1×105の3000-radを照射した同系のB10.D2脾細胞(抗原提示細胞)を異なる数のサプレッサー6.5 CD4+ T細胞と共に混合し、10μg/mlのHAクラスII(110SFERFEIFPKE120;SEQ No:7)ペプチドと共に丸底96穴組織培養プレートにおいて、200μlのCTL培地中でインキュベートした。48〜72時間後、培養液を1μCiの3H-チミジンでパルスし、さらに16時間インキュベート後、パッカード・マイクロメート細胞収穫器(Packard Micromate cell harvester)で収集した。取り込まれた放射性カウントの量の決定は、パッカード・マトリックス96ダイレクトベータカウンター(Packard Matrix 96 direct beta counter:Packard Biosciences, Meriden, CT)で行った。
【0115】
天然の調節性T細胞のインビトロでの抑制アッセイ法
野生型BALB/cマウスを天然のTregアッセイ法に使用した。5×104のフローサイトメトリーで選別したCD4+CD25-T細胞(応答者)および5×104の3000-radの照射を受けたBALB/c脾細胞(抗原提示細胞)を、フローサイトメトリーで選別した異なる数のCD4+CD25+サプレッサーT細胞と共に混合し、0.5μg/mlの抗CD3抗体を有する丸底96穴組織培養プレートにおいて、200μlのCTL培地中でインキュベートした。48〜72時間後、培養液を1μCiの3H-チミジンでパルスし、さらに16時間インキュベート後、パッカード・マイクロメート細胞収穫器(Packard Micromate cell harvester)で収集した。取り込まれた放射性カウントの量の決定は、パッカード・マトリックス96ダイレクトベータカウンター(Packard Matrix 96 direct beta counter:Packard Biosciences, Meriden, CT)で行った。
【0116】
定量的リアルタイムPCR解析
Trizol試薬を使用して、RNA抽出のために選別した6.5 CD4+ T細胞を直ちに使用した(Invitrogen, Carlsbad, CA)。逆転写は、スーパースクリプト・第1鎖標準合成系(Superscript First Strand Synthesis System:Invitrogen, Carlsbad, CA)によって行った。cDNAレベルは、Taqman系でのリアルタイム定量的PCRによって解析された(Applied Biosystems, Foster City, CA)。それぞれの試料は、タックマン・ユニバーサル・PCRマスターミックス(Taqman Universal PCR Master Mix)およびABIプリズム7700配列検出システム(ABI Prism 7700 Sequence Detection system)を使用して、25μlの最終反応体積で、内部基準として18S rRNAと共に標的遺伝子について2回または3回アッセイした。IL-10およびIL-2の検出のためには、予め作製した反応試薬(PDARs)をApplied Biosystemsから購入した。LAG-3、CD25、GITR、およびIFN-γについては、プライマー対およびプローブセットは、プライマー・エキスプレス(Primer Express)ソフトウェアを使用してデザインし、次いでApplied Biosystemsによって合成した。Foxp3に使用するプライマーおよびプローブセットは、文献(S4)からのものを引用した。相対的なmRNA頻度は、内部対照18S RNAに対し基準化することによって決定した。簡単には、本発明者らは、標的遺伝子と18S RNAとの間の閾値サイクル(Ct)の相違:ΔCtsample=(Ctsample-Ct18s)を使用してそれぞれの試料セットを基準化した。較正試料は、アッセイ法(ΔCtcalibration)のそれぞれのセットにおいて最も高いΔCtを有する試料として割り当てた。相対的mRNA頻度は、2ΔΔCtとして算出し、式中ΔΔCt=(ΔCtcalibration-ΔCtsample)である。使用したプライマーおよびプローブセットは以下の通りである:
。
【0117】
LAG-3構築物およびレトロウイルス産生株の株化細胞
LAG-3構築物は、記載されたとおりに組換えPCRを使用して作製した(Vignali and Vignali, 1999)。Lag-3.WTおよび機能的ヌル変異体LAG-3.Y73F.ΔCY(LAG-3のMHCクラスIIを結合する能力を非常に減少させる点変異を有する細胞質末端のLAG-3)は記載されている(Workman et al., 2002a)。LAG-3構築物を、内部リボソーム導入部位(IRES)および緑色蛍光タンパク質(GFP)を含むマウス幹細胞ウイルス(MSCV)に基づいたレトロウイルスベクターにクローニングし、記載されているようにレトロウイルスを作製した(Persons et al., 1997; Persons et al., 1998)。次いで、24時間後に105/ml以上のウイルス力価が得られるまで、GPE+86細胞を繰り返して(7〜10)回形質導入することによってレトロウイルス産生株の株化細胞を作製した(Markowitz et al., 1988)。
【0118】
CD4+/CD25- T細胞のレトロウイルスによる形質導入およびインビトロでの抑制アッセイ法
6.5マウスからの脾細胞をビオチンラベルした抗B220、抗Gr1、抗Mac1、抗TER119、抗CD49b、抗CD8、および抗CD25抗体(PharMingen, San Diego,CA)で染色した。次いで、細胞をストレプトアビジンに結合させた磁気ビーズと共にインキュベートして、90〜95%の純度のCD4+/CD25- T細胞になるよう自動MACS(autoMACS: Miltenyi Biotech, Auburn CA)でネガティブに選別した。精製した6.5 CD4+/CD25- T細胞を、プレートに結合した抗CD3(2C11)および抗CD28(35.71)によって活性化した。刺激の2日および3日後に、活性化されたT細胞(4×105細胞/ml)には、上記したベクター単独、LAG-3.WT/GFP、またはLAG-3.Y73F.ΔCY/GFP レトロウイルスGPE+86産生株株化細胞(6μg/ml)プラスIL-2およびポリブレンからのウイルス上清をスピン形質導入(spin transduce)(90min、3000rpm)した。細胞を10日間静止することができ、次いで上位30〜35% GFP+/Thy1.2+ T細胞に対して選別した。
【0119】
インビトロ抑制アッセイ法については、精製したGFP+ T細胞(2.5×104で開始して2倍希釈)を、2.5×104のCD4+/CD25- 6.5T細胞(ネガティブに自動MACSによって精製した)、5×104の照射(3000rads)を受けた脾細胞、および5μg/mlのHA110-120と共に96穴丸底プレートにおいて培養した。細胞を72時間培養し、培養の最後の7〜8時間1μCi/ウェルの[3H]チミジンでパルスした(Du Pont, Wilmington, DE)。
【0120】
実施例11
CD223は、細胞表面から切断されて、可溶性形態(sLAG-3)で放出される。これは、インビトロで活性化されたT細胞によって有意な量で産生され(5μg/ml)、またマウスの血清中にも見いだされる(80ng/ml)。これは、細胞表面プロテアーゼによって産生される可能性が高い。本発明者らは、ウエスタンブロットによってsLAG-3を検出した。切断は、膜貫通領域(たとえば、SEQ ID NO:2のアミノ酸442〜466)に、またはアミノ末期の直前のコネクタ領域(たとえば、SEQ ID NO:2のアミノ酸432〜441)に生じる。
【0121】
実施例12
上記のように、LAG-3は、最大の調節性T細胞(Treg)機能に必要かつ十分である。言い換えると、LAG-3単独の発現でも、細胞を活性化されたエフェクターT細胞から調節性T細胞に変換するために十分である。
【0122】
本発明者らは、次に、LAG-3を異所性に発現する細胞もインビボで調節性の機能を示し、疾患状況において保護されるかどうかを決定したいと考えた。本発明者らは、自己抗原特異的なT細胞上のLAG-3の異所性の発現が、マウスを1型糖尿病から保護することができるかどうかについて問うことを選択した。この実験系では、糖尿病を起こしやすいNODマウスからの脾細胞を、リンパ球を欠如するNOD-Scidマウスに養子性に導入した。全てのマウスが3ヶ月以内に糖尿病を発症する。本発明者らの予備解析では、NOD脾細胞によって誘導される糖尿病の発症が、LAG-3を形質導入したホグリン(phogrin)特異的なT細胞によって防止されるが、シグナリング欠損がある変異体またはGFP対照では防止されないことを示唆する。これらのデータは、LAG-3を有する自己抗原特異的なT細胞の異所性の発現を、多くの自己免疫疾患、喘息、およびアレルギーの治療のための新規治療に使用するという考えを支持する。
【0123】
参考文献
【特許請求の範囲】
【請求項1】
自己免疫疾患に罹患している患者を治療する方法であって:
CD223のコード配列を含む発現構築物を有する患者から単離された自己免疫性のT細胞をインビトロでトランスフェクトすること;および
トランスフェクトされた自己免疫性のT細胞を患者に再注入すること
を含む方法。
【請求項2】
患者が多発性硬化症に罹患している、請求項1記載の方法。
【請求項3】
CD223と特異的に結合する抗体;および
抗癌ワクチン
を含む組成物。
【請求項4】
薬学的組成物である、請求項3記載の組成物。
【請求項5】
抗体がモノクローナル抗体である、請求項3記載の組成物。
【請求項6】
組成物が患者への投与後にインビボで形成される、請求項3記載の組成物。
【請求項7】
特異的にCD223と結合する抗体;および
抗癌ワクチン
を含むキット。
【請求項8】
抗体およびワクチンが別々の容器内にある、請求項7記載のキット。
【請求項9】
癌患者に対するキットの成分の投与のための説明書をさらに含む、請求項7記載のキット。
【請求項10】
抗癌ワクチンで癌患者を治療する改善された方法であって:
癌患者に対して、CD223と特異的に結合する抗体を投与すること;および
癌患者に対して、抗癌ワクチンを投与すること
を含み、抗体が、抗癌ワクチンに対する癌患者の抗癌応答の大きさを増大させる、方法。
【請求項11】
抗癌ワクチンに対する免疫応答の抑制を克服する方法であって:
抗癌ワクチンに対する免疫応答を抑制する調節性T細胞を有する癌患者に対して、CD223と特異的に結合する抗体を投与すること;および
癌患者に対して抗癌ワクチンを投与すること
を含み、これにより、抗体が、抗癌ワクチンに対する癌患者の応答を増大させる、方法。
【請求項12】
抗体がモノクローナルである、請求項11記載の方法。
【請求項13】
哺乳類のT細胞の数を増大させるための方法であって:
哺乳類にCD223タンパク質またはCD223 mRNAと結合する阻害剤を投与することを含む方法。
【請求項14】
阻害剤がCD223タンパク質と特異的に結合する抗体である、請求項13記載の方法。
【請求項15】
阻害剤がCD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンスRNA分子を発現するアンチセンス構築物である、請求項13記載の方法。
【請求項16】
阻害剤がCD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンス・オリゴヌクレオチドである、請求項13記載の方法
【請求項17】
阻害剤がCD223 mRNAと特異的に結合するリボザイムである、請求項13記載の方法。
【請求項18】
阻害剤がCD223 mRNAと特異的に結合するRNA干渉分子である、請求項13記載の方法。
【請求項19】
哺乳類が癌患者である、請求項13記載の方法。
【請求項20】
哺乳類が骨髄移植レシピエントである、請求項13記載の方法。
【請求項21】
癌患者を抗腫瘍ワクチンでワクチン接種する工程をさらに含む、請求項19記載の方法。
【請求項22】
腫瘍特異的なT細胞群を癌患者に導入する工程をさらに含む、請求項19記載の方法。
【請求項23】
患者に対して抗癌化学療法剤を投与する工程をさらに含む、請求項19記載の方法。
【請求項24】
患者に対して抗癌抗体を投与する工程をさらに含む、請求項19記載の方法。
【請求項25】
哺乳類が慢性ウイルス感染を有する、請求項13記載の方法。
【請求項26】
哺乳類に対して抗ウイルス・ワクチンを投与する工程をさらに含む、請求項25記載の方法。
【請求項27】
哺乳類が慢性細菌感染を有する、請求項13記載の方法。
【請求項28】
哺乳類に対して抗細菌ワクチンを投与する工程をさらに含む、請求項27記載の方法。
【請求項29】
慢性ウイルス感染がHIVである、請求項25記載の方法。
【請求項30】
T細胞群のT細胞の数を増大させるための方法であって:
T細胞群に対して、CD223タンパク質またはCD223 mRNAと結合する阻害剤をインビトロで投与することを含む方法。
【請求項31】
阻害剤がCD223タンパク質と特異的に結合する抗体である、請求項30記載の方法。
【請求項32】
阻害剤が、CD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンスRNA分子を発現するアンチセンス構築物である、請求項30記載の方法。
【請求項33】
阻害剤が、CD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンス・オリゴヌクレオチドである、請求項30記載の方法。
【請求項34】
阻害剤が、CD223 mRNAと特異的に結合するリボザイムである、請求項30記載の方法。
【請求項35】
阻害剤がCD223 mRNAと特異的に結合するRNA干渉分子である、請求項30記載の方法。
【請求項36】
哺乳類のT細胞の数を減少させるための方法であって:
哺乳類に対して、CD223をコードする発現構築物を投与することを含み、これにより、CD223が発現構築物から発現され、哺乳類のCD223の濃度が増大し、哺乳類のT細胞の数が減少する、方法。
【請求項37】
哺乳類が自己免疫疾患患者である、請求項36記載の方法。
【請求項38】
哺乳類のT細胞の数を減少させるための方法であって:
哺乳類に対して、CD223+ T細胞の集団を投与することを含み、これにより、哺乳類のCD223の濃度が増大し、哺乳類のT細胞の数が減少する、方法。
【請求項39】
哺乳類が自己免疫疾患患者である、請求項38記載の方法。
【請求項40】
CD223+ T細胞が、CD223をコードする発現構築物を形質導入されたCD4+ T細胞である、請求項39記載の方法。
【請求項41】
CD223+ T細胞が、CD223をコードする発現構築物を形質導入された自己抗原特異的なT細胞である、請求項39記載の方法。
【請求項42】
SEQ ID NO:5で示されたアミノ酸配列KIEELEを含む、CD223の50以下の連続したアミノ酸残基からなるポリペプチド。
【請求項43】
少なくとも2つのセグメントを含む融合ポリペプチドであって、第1のセグメントが、CD223の50以下の連続したアミノ酸残基からなり、第1のセグメントが、SEQ ID NO:5で示されたアミノ酸配列KIELLEを含み、第2のセグメントが、SEQ ID NO:2または4に示されたCD223には見いだされないアミノ酸配列を含む、融合ポリペプチド。
【請求項44】
癌、自己免疫疾患、慢性感染症、AIDS、骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法であって:
CD223タンパク質またはSEQ ID NO:5で示されたアミノ酸配列KIELLEを含むCD223タンパク質断片と、試験物質を接触させること;
試験物質がCD223タンパク質またはCD223タンパク質断片に対して結合したかどうかを決定すること;および
試験物質がCD223タンパク質またはCD223タンパク質断片に結合した場合には、試験物質を、癌、自己免疫疾患、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定すること
を含む方法。
【請求項45】
癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法であって:
試験物質をCD223タンパク質と接触させること;
試験物質の存在下および非存在下においてCD223活性を決定すること;および
試験物質がCD223活性を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための潜在的薬物として同定すること
を含む方法。
【請求項46】
自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験する方法であって:
試験物質をCD223タンパク質と接触させること;
試験物質の存在下および非存在下においてCD223活性を決定すること;および
試験物質がCD223活性を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定すること
を含む方法。
【請求項47】
癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法であって:
CD223+ T細胞を試験物質と接触させること;
試験物質の存在下および非存在下において細胞におけるCD223の発現を決定すること;および
試験物質がT細胞におけるCD223発現を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための潜在的薬物として同定すること
を含む方法。
【請求項48】
自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験する方法であって:
試験物質をCD223+ T細胞と接触させること;
試験物質の存在下および非存在下において細胞におけるCD223発現を決定すること;および
試験物質がT細胞におけるCD223発現を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定すること
を含む方法。
【請求項49】
CD223+ T細胞またはCD223- T細胞を単離する方法であって:
T細胞の混合集団を、SEQ ID NO:2または4に記載のCD223と特異的に結合する抗体と接触させること;および
抗体に結合していないT細胞から抗体に結合したT細胞を分離し、これによりCD223+ T細胞の集団およびCD223- T細胞の集団を形成すること
を含む方法。
【請求項50】
CD223+ T細胞の集団を自己免疫疾患患者に導入する工程をさらに含む、請求項49記載の方法。
【請求項51】
集団を自己免疫疾患患者に導入する工程の前に、集団が自己抗原によりインビトロで活性化される、請求項49記載の方法。
【請求項52】
T細胞の混合集団がドナーリンパ球の試料である、請求項49記載の方法。
【請求項53】
CD223- T細胞の集団を癌患者に導入する工程をさらに含む、請求項52記載の方法。
【請求項54】
T細胞の混合集団が腫瘍特異的である、請求項49記載の方法。
【請求項55】
CD223- T細胞の集団を癌患者に導入する工程をさらに含む、請求項54記載の方法。
【請求項56】
残基1〜431を含み、残基467〜521を欠いている、単離された可溶性マウスCD223タンパク質。
【請求項57】
CD223+ T細胞の表面上のプロテアーゼによって産生される、請求項56記載の単離された可溶性CD223タンパク質。
【請求項58】
その他の血清タンパク質がない、請求項56記載の単離された可溶性CD223タンパク質。
【請求項59】
残基1〜440を含み、残基475〜525を欠いている、単離された可溶性ヒトCD223タンパク質。
【請求項60】
CD223+ T細胞の表面上のプロテアーゼによって産生される、請求項59記載の単離された可溶性CD223タンパク質。
【請求項61】
その他の血清タンパク質がない、請求項59記載の単離された可溶性CD223タンパク質。
【請求項62】
哺乳類においてT細胞数を減少させるための方法であって:
哺乳類に対して可溶性CD223タンパク質を投与することを含み、これにより、哺乳類におけるMHCクラスII拘束性/CD4+ T細胞応答が調整される、方法。
【請求項63】
哺乳類が自己免疫疾患患者である、請求項62記載の方法。
【請求項64】
哺乳類がアレルギー患者である、請求項62記載の方法。
【請求項65】
哺乳類が喘息患者である、請求項62記載の方法。
【請求項1】
自己免疫疾患に罹患している患者を治療する方法であって:
CD223のコード配列を含む発現構築物を有する患者から単離された自己免疫性のT細胞をインビトロでトランスフェクトすること;および
トランスフェクトされた自己免疫性のT細胞を患者に再注入すること
を含む方法。
【請求項2】
患者が多発性硬化症に罹患している、請求項1記載の方法。
【請求項3】
CD223と特異的に結合する抗体;および
抗癌ワクチン
を含む組成物。
【請求項4】
薬学的組成物である、請求項3記載の組成物。
【請求項5】
抗体がモノクローナル抗体である、請求項3記載の組成物。
【請求項6】
組成物が患者への投与後にインビボで形成される、請求項3記載の組成物。
【請求項7】
特異的にCD223と結合する抗体;および
抗癌ワクチン
を含むキット。
【請求項8】
抗体およびワクチンが別々の容器内にある、請求項7記載のキット。
【請求項9】
癌患者に対するキットの成分の投与のための説明書をさらに含む、請求項7記載のキット。
【請求項10】
抗癌ワクチンで癌患者を治療する改善された方法であって:
癌患者に対して、CD223と特異的に結合する抗体を投与すること;および
癌患者に対して、抗癌ワクチンを投与すること
を含み、抗体が、抗癌ワクチンに対する癌患者の抗癌応答の大きさを増大させる、方法。
【請求項11】
抗癌ワクチンに対する免疫応答の抑制を克服する方法であって:
抗癌ワクチンに対する免疫応答を抑制する調節性T細胞を有する癌患者に対して、CD223と特異的に結合する抗体を投与すること;および
癌患者に対して抗癌ワクチンを投与すること
を含み、これにより、抗体が、抗癌ワクチンに対する癌患者の応答を増大させる、方法。
【請求項12】
抗体がモノクローナルである、請求項11記載の方法。
【請求項13】
哺乳類のT細胞の数を増大させるための方法であって:
哺乳類にCD223タンパク質またはCD223 mRNAと結合する阻害剤を投与することを含む方法。
【請求項14】
阻害剤がCD223タンパク質と特異的に結合する抗体である、請求項13記載の方法。
【請求項15】
阻害剤がCD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンスRNA分子を発現するアンチセンス構築物である、請求項13記載の方法。
【請求項16】
阻害剤がCD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンス・オリゴヌクレオチドである、請求項13記載の方法
【請求項17】
阻害剤がCD223 mRNAと特異的に結合するリボザイムである、請求項13記載の方法。
【請求項18】
阻害剤がCD223 mRNAと特異的に結合するRNA干渉分子である、請求項13記載の方法。
【請求項19】
哺乳類が癌患者である、請求項13記載の方法。
【請求項20】
哺乳類が骨髄移植レシピエントである、請求項13記載の方法。
【請求項21】
癌患者を抗腫瘍ワクチンでワクチン接種する工程をさらに含む、請求項19記載の方法。
【請求項22】
腫瘍特異的なT細胞群を癌患者に導入する工程をさらに含む、請求項19記載の方法。
【請求項23】
患者に対して抗癌化学療法剤を投与する工程をさらに含む、請求項19記載の方法。
【請求項24】
患者に対して抗癌抗体を投与する工程をさらに含む、請求項19記載の方法。
【請求項25】
哺乳類が慢性ウイルス感染を有する、請求項13記載の方法。
【請求項26】
哺乳類に対して抗ウイルス・ワクチンを投与する工程をさらに含む、請求項25記載の方法。
【請求項27】
哺乳類が慢性細菌感染を有する、請求項13記載の方法。
【請求項28】
哺乳類に対して抗細菌ワクチンを投与する工程をさらに含む、請求項27記載の方法。
【請求項29】
慢性ウイルス感染がHIVである、請求項25記載の方法。
【請求項30】
T細胞群のT細胞の数を増大させるための方法であって:
T細胞群に対して、CD223タンパク質またはCD223 mRNAと結合する阻害剤をインビトロで投与することを含む方法。
【請求項31】
阻害剤がCD223タンパク質と特異的に結合する抗体である、請求項30記載の方法。
【請求項32】
阻害剤が、CD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンスRNA分子を発現するアンチセンス構築物である、請求項30記載の方法。
【請求項33】
阻害剤が、CD223 mRNAの少なくとも7ヌクレオチドに対して相補的であるアンチセンス・オリゴヌクレオチドである、請求項30記載の方法。
【請求項34】
阻害剤が、CD223 mRNAと特異的に結合するリボザイムである、請求項30記載の方法。
【請求項35】
阻害剤がCD223 mRNAと特異的に結合するRNA干渉分子である、請求項30記載の方法。
【請求項36】
哺乳類のT細胞の数を減少させるための方法であって:
哺乳類に対して、CD223をコードする発現構築物を投与することを含み、これにより、CD223が発現構築物から発現され、哺乳類のCD223の濃度が増大し、哺乳類のT細胞の数が減少する、方法。
【請求項37】
哺乳類が自己免疫疾患患者である、請求項36記載の方法。
【請求項38】
哺乳類のT細胞の数を減少させるための方法であって:
哺乳類に対して、CD223+ T細胞の集団を投与することを含み、これにより、哺乳類のCD223の濃度が増大し、哺乳類のT細胞の数が減少する、方法。
【請求項39】
哺乳類が自己免疫疾患患者である、請求項38記載の方法。
【請求項40】
CD223+ T細胞が、CD223をコードする発現構築物を形質導入されたCD4+ T細胞である、請求項39記載の方法。
【請求項41】
CD223+ T細胞が、CD223をコードする発現構築物を形質導入された自己抗原特異的なT細胞である、請求項39記載の方法。
【請求項42】
SEQ ID NO:5で示されたアミノ酸配列KIEELEを含む、CD223の50以下の連続したアミノ酸残基からなるポリペプチド。
【請求項43】
少なくとも2つのセグメントを含む融合ポリペプチドであって、第1のセグメントが、CD223の50以下の連続したアミノ酸残基からなり、第1のセグメントが、SEQ ID NO:5で示されたアミノ酸配列KIELLEを含み、第2のセグメントが、SEQ ID NO:2または4に示されたCD223には見いだされないアミノ酸配列を含む、融合ポリペプチド。
【請求項44】
癌、自己免疫疾患、慢性感染症、AIDS、骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法であって:
CD223タンパク質またはSEQ ID NO:5で示されたアミノ酸配列KIELLEを含むCD223タンパク質断片と、試験物質を接触させること;
試験物質がCD223タンパク質またはCD223タンパク質断片に対して結合したかどうかを決定すること;および
試験物質がCD223タンパク質またはCD223タンパク質断片に結合した場合には、試験物質を、癌、自己免疫疾患、慢性感染症、AIDS、または骨髄移植レシピエントを治療するための潜在的薬物として同定すること
を含む方法。
【請求項45】
癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法であって:
試験物質をCD223タンパク質と接触させること;
試験物質の存在下および非存在下においてCD223活性を決定すること;および
試験物質がCD223活性を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための潜在的薬物として同定すること
を含む方法。
【請求項46】
自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験する方法であって:
試験物質をCD223タンパク質と接触させること;
試験物質の存在下および非存在下においてCD223活性を決定すること;および
試験物質がCD223活性を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定すること
を含む方法。
【請求項47】
癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための薬物としての潜在的活性について物質を試験する方法であって:
CD223+ T細胞を試験物質と接触させること;
試験物質の存在下および非存在下において細胞におけるCD223の発現を決定すること;および
試験物質がT細胞におけるCD223発現を阻害する場合には、試験物質を、癌、慢性感染症、AIDS、および骨髄移植レシピエントを治療するための潜在的薬物として同定すること
を含む方法。
【請求項48】
自己免疫疾患を治療するための薬物としての潜在的活性について物質を試験する方法であって:
試験物質をCD223+ T細胞と接触させること;
試験物質の存在下および非存在下において細胞におけるCD223発現を決定すること;および
試験物質がT細胞におけるCD223発現を増大させる場合には、試験物質を、自己免疫疾患を治療するための潜在的薬物として同定すること
を含む方法。
【請求項49】
CD223+ T細胞またはCD223- T細胞を単離する方法であって:
T細胞の混合集団を、SEQ ID NO:2または4に記載のCD223と特異的に結合する抗体と接触させること;および
抗体に結合していないT細胞から抗体に結合したT細胞を分離し、これによりCD223+ T細胞の集団およびCD223- T細胞の集団を形成すること
を含む方法。
【請求項50】
CD223+ T細胞の集団を自己免疫疾患患者に導入する工程をさらに含む、請求項49記載の方法。
【請求項51】
集団を自己免疫疾患患者に導入する工程の前に、集団が自己抗原によりインビトロで活性化される、請求項49記載の方法。
【請求項52】
T細胞の混合集団がドナーリンパ球の試料である、請求項49記載の方法。
【請求項53】
CD223- T細胞の集団を癌患者に導入する工程をさらに含む、請求項52記載の方法。
【請求項54】
T細胞の混合集団が腫瘍特異的である、請求項49記載の方法。
【請求項55】
CD223- T細胞の集団を癌患者に導入する工程をさらに含む、請求項54記載の方法。
【請求項56】
残基1〜431を含み、残基467〜521を欠いている、単離された可溶性マウスCD223タンパク質。
【請求項57】
CD223+ T細胞の表面上のプロテアーゼによって産生される、請求項56記載の単離された可溶性CD223タンパク質。
【請求項58】
その他の血清タンパク質がない、請求項56記載の単離された可溶性CD223タンパク質。
【請求項59】
残基1〜440を含み、残基475〜525を欠いている、単離された可溶性ヒトCD223タンパク質。
【請求項60】
CD223+ T細胞の表面上のプロテアーゼによって産生される、請求項59記載の単離された可溶性CD223タンパク質。
【請求項61】
その他の血清タンパク質がない、請求項59記載の単離された可溶性CD223タンパク質。
【請求項62】
哺乳類においてT細胞数を減少させるための方法であって:
哺乳類に対して可溶性CD223タンパク質を投与することを含み、これにより、哺乳類におけるMHCクラスII拘束性/CD4+ T細胞応答が調整される、方法。
【請求項63】
哺乳類が自己免疫疾患患者である、請求項62記載の方法。
【請求項64】
哺乳類がアレルギー患者である、請求項62記載の方法。
【請求項65】
哺乳類が喘息患者である、請求項62記載の方法。
【図1】

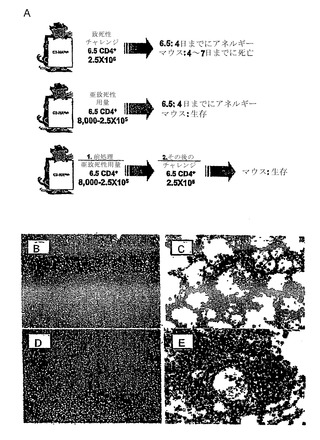
【図2】
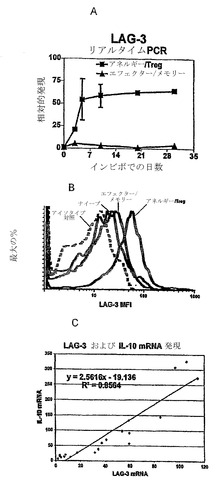
【図3】
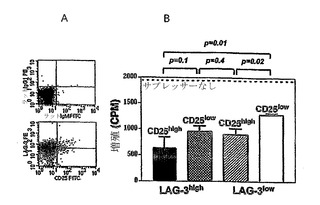
【図4】
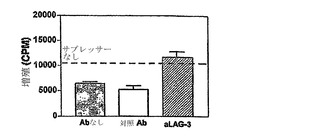
【図5】
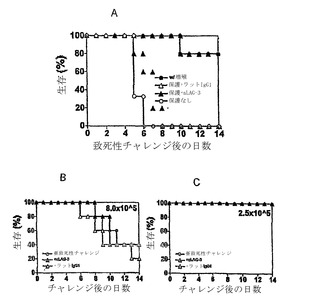
【図6】
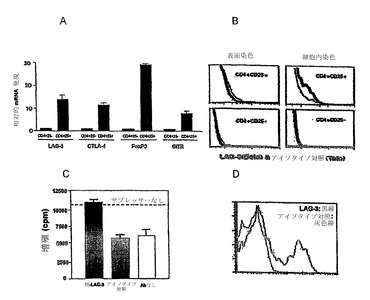
【図7】
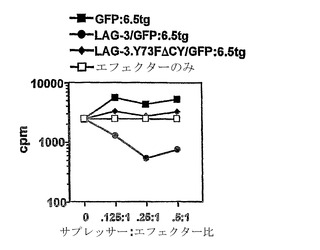
【図8】
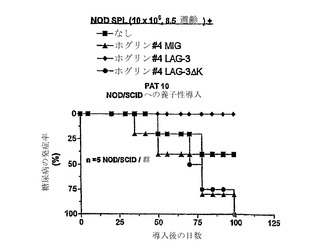
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2011−116755(P2011−116755A)
【公開日】平成23年6月16日(2011.6.16)
【国際特許分類】
【出願番号】特願2010−253326(P2010−253326)
【出願日】平成22年11月12日(2010.11.12)
【分割の表示】特願2006−508934(P2006−508934)の分割
【原出願日】平成16年3月1日(2004.3.1)
【出願人】(504394146)ザ ジョンズ ホプキンス ユニバーシティ (4)
【氏名又は名称原語表記】THE JOHNS HOPKINS UNIVERSITY
【出願人】(505325800)セント ジュード チルドレンズ リサーチ ホスピタル インコーポレイティッド (2)
【Fターム(参考)】
【公開日】平成23年6月16日(2011.6.16)
【国際特許分類】
【出願日】平成22年11月12日(2010.11.12)
【分割の表示】特願2006−508934(P2006−508934)の分割
【原出願日】平成16年3月1日(2004.3.1)
【出願人】(504394146)ザ ジョンズ ホプキンス ユニバーシティ (4)
【氏名又は名称原語表記】THE JOHNS HOPKINS UNIVERSITY
【出願人】(505325800)セント ジュード チルドレンズ リサーチ ホスピタル インコーポレイティッド (2)
【Fターム(参考)】
[ Back to top ]