
TAF4bプロモーターおよびその利用
【課題】細胞増殖因子かつ転写因子であるMycの直接の標的およびそれを用いた真核生物の組織特異的事象の調節手段の提供。
【解決手段】Mycによる組織特異的発現を調節するためのヌクレオチドであって、TATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフおよび/またはCACGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチド、前記ヌクレオチドを含有してなるベクター、前記ヌクレオチドおよびレポーター遺伝子を含有してなるレポータープラスミド、前記ベクターもしくはレポータープラスミドを導入してなる形質転換体またはトランスジェニック非ヒト動物、ならびに前記レポータープラスミドまたは形質転換体を用いることを特徴とする、細胞増殖の調節剤または妊娠調節剤をスクリーニングする方法。
【解決手段】Mycによる組織特異的発現を調節するためのヌクレオチドであって、TATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフおよび/またはCACGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチド、前記ヌクレオチドを含有してなるベクター、前記ヌクレオチドおよびレポーター遺伝子を含有してなるレポータープラスミド、前記ベクターもしくはレポータープラスミドを導入してなる形質転換体またはトランスジェニック非ヒト動物、ならびに前記レポータープラスミドまたは形質転換体を用いることを特徴とする、細胞増殖の調節剤または妊娠調節剤をスクリーニングする方法。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、TAF4bプロモーターおよびその利用に関する。詳しくは、組織特異的な転写調節または転写増強が可能なヌクレオチドおよび真核生物の様々な事象の解明に寄与する当該ヌクレオチドの利用などに関する。
【背景技術】
【0002】
真核細胞(真核生物)において、タンパク質をコードする遺伝子はRNAポリメラーゼII(RNA Pol II)によって転写される。特定の遺伝子の転写を開始するためにRNA Pol IIを導く分子機構は、調節タンパク質の複数のクラスから構成される(非特許文献1)。TFIIDは、RNA Pol II制御機構の中核となる要素であり、酵母(Saccharomyces cerevisiae)からヒトに至るまで保存されており、TATAボックス結合タンパク質(TBP)と複数のTBP関連因子(TAFs)からなる大きな多量体タンパク質複合体である。TFIIDは、初期の頃には発現および機能において遍在性(ユビキタス)であると考えられていたが、組織特異的TAFsの同定は、特殊化したTFIID複合体が遺伝子発現の組織特異的プログラムの調節において直接的な役割を果たしうるということを示唆した。同定された最初の細胞型特異的TFIIDサブユニットは、TAF4b(以前はTAFII105と呼ばれていた)であった(非特許文献2)。TAF4bは、広範囲に発現される因子であるTAF4(以前はTAFII130と呼ばれていた)のホモロガスタンパク質である。TAF4bの細胞型特異的機能は、TAF4bがマウスの精巣および卵巣で高度に発現しており、TAF4bを欠損したマウスは、生存可能であるがオス・メスともに不妊である(非特許文献3、4)という発見によって支持された。
【0003】
一次Bリンパ球が細菌のリポ多糖によって活性化された場合、TAF4ではなくTAF4bの発現が増加した(非特許文献5)。最近では、TAF4およびTAF4bを含むTFIID複合体が別個の標的遺伝子特異性を有しているということが報告されている。TAF4を不活性化すると、多くの一連の遺伝子の発現に影響を及ぼした。これらの中で、TAF4bの不活性化は、TGFβ1、TGFβ3、CTGF、ITGα6およびHB−EGFを含む多くの成長因子を誘導した。これらの成長因子は、TAF4bの過剰発現によっても誘導された。これらの結果は、TAF4とTAF4bとの平衡が細胞増殖の制御に関連する遺伝子の調節に作用しているということ(非特許文献6)を示唆しているとともに、TAF4bの発現は厳密に制御されるべきであることを示している。しかしながら、TAF4b発現の制御メカニズムは、未だ解明されていない。
【0004】
プロトオンコジーンのmycファミリーは、3つの主要な遺伝子:c−myc、N−mycおよびL−mycから構成されている(非特許文献7、8、9)。この3つの遺伝子は、異なる発現パターンを示すが、基本的には同じ生物学的活性を有しているように見える。c−mycまたはN−mycを欠損したマウスの胎児は、多臓器発育不全になり、胚形成の中間の段階で死ぬ(非特許文献10、11)。c−mycの発現が一定量減少する対立形質を有する一連のマウスを作製した場合、in vivoでのc−mycレベルの減少は、多臓器発育不全が原因となって、c−mycのレベルに依存してマウスの体重を減少させる(非特許文献12)。これらの結果は、Mycの量が細胞増殖に直接的に関与していることを示すものである。これらの蓄積されたデータは、mycファミリー遺伝子が細胞増殖の調節因子の中心となっていることを示している。
【0005】
mycがコードするタンパク質は、塩基性へリックス・ループ・へリックス−ロイシンジッパー転写因子のメンバーである(非特許文献8、13)。Mycタンパク質とその必須パートナーMaxタンパク質との二量体化が起こると、E−ボックス部位に結合するヘテロ二量体が形成される。このE−ボックス部位は、通常は基準的なもの(CACGTG)であるが、まれにはCGCGTGを含む非基準的なものである。Myc−Maxヘテロ二量体は、オルニチンデカルボキシラーゼ(非特許文献14)、RCC1(非特許文献15)、ヌクレオリン(非特許文献16)、サイクリンD2(非特許文献17)、mina53(非特許文献18)、およびmimitin(非特許文献19)を含む、種々の遺伝子の転写を活性化する。mycに関しては、その研究に多大な努力がなされているが、いまだなぞのままであり、Mycによって制御されるさらなる遺伝子に関する情報は、mycの機能の解明に役立てることができる。
【0006】
mycプロトオンコジーンファミリーのタンパク質以外にも、USF(非特許文献20)およびTFE3(非特許文献21)を含むE−ボックス結合転写因子の他のクラスが存在する。これらの因子も、E−ボックスを介して遺伝子発現をトランスに活性化するが、これらの生物学的な機能およびこれらに対応する遺伝子は、Mycとは異なっている。しかしながら、それぞれのE−ボックス結合転写因子におけるこれらの相違を創り出すメカニズムは、未だ明らかになっていない。
【非特許文献1】Lemon, B. et al., Genes Dev. 14:2551-2569, 2000
【非特許文献2】Dikstein, R. et al., Cell 87:137-146, 1996
【非特許文献3】Falender, A. E. et al., Genes Dev. 19:794-803, 2005
【非特許文献4】Freiman, R. N. et al., Science 293:2084-2087, 2001
【非特許文献5】Freiman, R. N.et al., Mol. Cell. Biol. 22:6564-6572, 2002
【非特許文献6】Mengus, G. et al., EMBO J. 24:2753-2767, 2005
【非特許文献7】DePinho, R. A. et al., Adv. Cancer Res. 57:1-46, 1991
【非特許文献8】Grandori, C. et al., Annu. Rev. Cell Dev. Biol. 16:653-699, 2000
【非特許文献9】Henriksson, M. et al., Adv. Cancer Res. 68:109-182, 1996
【非特許文献10】Davis, A. C. et al., Genes Dev. 7:671-682, 1993
【非特許文献11】Stanton, B. R. et al., Genes Dev. 6:2235-2247, 1992
【非特許文献12】Trumpp, A. et al., Nature 414:768-773, 2001
【非特許文献13】Luscher, B. et al., Gene 277:1-14, 2001
【非特許文献14】Bello-Fernandez, C. et al., Proc. Natl. Acad. Sci. U. S. A. 90:7804-7808, 1993
【非特許文献15】Tsuneoka, M. et al., Oncogene 14:2301-2311, 1997
【非特許文献16】Greasley, P. J. et al., Nucleic Acids Res. 28:446-453, 2000
【非特許文献17】Bouchard, C. et al., Genes Dev. 15:2042-2047, 2001
【非特許文献18】Tsuneoka, M. et al., J. Biol. Chem. 277:35450-35459, 2002
【非特許文献19】Tsuneoka, M. et al., J. Biol. Chem. 280:19977-19985, 2005
【非特許文献20】Gregor, P. D. et al., Genes Dev. 4:1730-1740, 1990
【非特許文献21】Beckmann, H. et al., Genes Dev. 4:167-179, 1990
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的は、細胞増殖因子かつ転写因子であるMycの直接の標的およびそれを用いた真核生物の組織特異的事象の調節手段の提供である。
【課題を解決するための手段】
【0008】
本発明者は、上記観点から鋭意検討した結果、Mycの直接の標的となる核酸配列および当該配列中の必須配列を見出し、本発明を完成するに至った。即ち、本願発明は、以下に示す通りである。
〔1〕 Mycによる組織特異的発現を調節するためのヌクレオチドであって、TATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフおよび/またはCACGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチド。
〔2〕 前記ヌクレオチドが
(a)配列番号3に記載の塩基配列、
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列、
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列、
(g)配列番号3の1081〜1126位の領域の塩基配列を必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列および/または当該必須配列の下流に1127〜1157位の領域の塩基配列が配置されてなる塩基配列を含む塩基配列、または
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列、
からなるものである、前記〔1〕記載のヌクレオチド。
〔3〕 (a)配列番号4に記載の塩基配列、または
(b)配列番号4に記載の塩基配列において、51位〜57位のTATAボックスおよび43〜48位のCACGTGモチーフ以外の塩基配列と70%以上の同一性を有し、かつTATAボックスとその1〜50塩基上流に位置するCACGTGモチーフとを含む塩基配列
を含有するものである、前記〔1〕に記載のヌクレオチド。
〔4〕 前記CGCGTGモチーフおよび/またはCACGTGモチーフが、下記:
(i)塩基配列GTGにおける少なくとも1塩基の置換、
(ii)塩基配列CACにおける少なくとも1塩基の置換もしくは挿入
のいずれかまたは両方の変異を有するものである、前記〔1〕〜〔3〕いずれか1項に記載のヌクレオチド。
〔5〕 前記〔1〕〜〔4〕いずれか1項に記載のヌクレオチドを含有してなるベクター。
〔6〕 前記〔1〕〜〔4〕いずれか1項に記載のヌクレオチドおよびレポーター遺伝子を含有してなる、レポータープラスミド。
〔7〕 前記〔5〕に記載のベクターまたは前記〔6〕に記載のレポータープラスミドを導入してなる形質転換体。
〔8〕 前記〔5〕に記載のベクターまたは前記〔6〕に記載のレポータープラスミドを導入してなるトランスジェニック非ヒト動物。
〔9〕 前記〔6〕に記載のレポータープラスミドまたは前記〔7〕に記載の形質転換体を用いることを特徴とする、細胞増殖の調節剤をスクリーニングする方法。
〔10〕 前記〔6〕に記載のレポータープラスミド、前記〔7〕に記載の形質転換体または前記〔8〕に記載のトランスジェニック非ヒト動物を用いることを特徴とする、妊娠調節剤をスクリーニングする方法。
〔11〕 Mycによる組織特異的発現の調節剤であって、下記いずれかのペプチドを有効成分として含有する調節剤:
(1)配列番号1に記載の塩基配列からコードされるペプチド、
(2)配列番号2に記載のアミノ酸配列からなるペプチド、
(3)前記(1)または(2)に記載のペプチドと実質的に同一のアミノ酸配列からなるペプチド。
〔12〕 妊娠の調節に用いられる前記〔11〕に記載の調節剤。
〔13〕 (a)配列番号5に記載の塩基配列、または
(b)配列番号5に記載の塩基配列において210位〜216位のTATAボックスおよび202〜207位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号5に記載の塩基配列と実質的に同一な塩基配列
を含有するサルTAF4bプロモーター。
〔14〕 (a)配列番号6に記載の塩基配列、または
(b)配列番号6に記載の塩基配列において778位〜784位のTATAボックスおよび770〜775位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号6に記載の塩基配列と実質的に同一な塩基配列
を含有するラットTAF4bプロモーター。
【発明の効果】
【0009】
本発明のヌクレオチドによると、Mycによる組織特異的発現を調節するために必要十分な塩基配列を提供することが可能となり、真核生物におけるMycを介した転写のさらなる解明のみならず、細胞増殖や組織特異的な転写制御の調節に寄与することができる。本発明のベクターによると、本発明のヌクレオチドを様々な用途に供する際に便利なツールを提供することができる。本発明のレポータープラスミドによると、Mycの活性を測るための新しいツールとして利用することができる。
【0010】
本発明のベクター、レポータープラスミド、形質転換体およびトランスジェニック非ヒト動物によると、真核生物におけるMycを介した転写のさらなる解明ならびに細胞増殖や組織特異的な転写制御の調節をインビトロまたはインビボで調べるためのツールを提供することができる。本発明のスクリーニング方法によると、細胞増殖のシグナル伝達の中核に位置するMycの活性を調節可能な細胞増殖調節剤を簡便および高感度に選別することができる。また、本発明のスクリーニング方法によると、今までに知られていない作用機序を有する妊娠調節剤を選別することができる。
【0011】
本発明のMycによる組織特異的発現の調節剤によると、Mycが介在する真核生物の発生および維持に関する様々な事象の調節に用いることができ、好ましくは妊娠の調節に用いられうる。本発明のサルまたはラットTAF4bプロモーターによると、サルまたはラット等の実験動物を用いた転写調節の研究のみならず、哺乳動物を始めとする多細胞生物全体の転写調節の解明にも寄与することができる。
【発明を実施するための最良の形態】
【0012】
本発明は、Mycによる組織特異的発現を調節するためのヌクレオチドを提供する。前記ヌクレオチドは、TATAボックスとその1〜50塩基上流に位置するCACGTGモチーフおよび/またはCGCGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチドである。
【0013】
本発明において「Mycによる組織特異的発現」とは、転写因子であるMycが特定の塩基配列を標的とすることにより、真核生物の特定の組織で当該特定の塩基配列の下流に位置する遺伝子が転写されることをいう。
【0014】
本発明におけるMycとは、c−Myc、N−MycおよびL−Mycを包含するものであり、特に限定されるものではないが、種々の癌において遺伝子増幅、過剰発現や転座が報告されているc−Mycを中心に説明する。
【0015】
本発明における組織とは、真核生物を構成するものである限り特に限定されるものではないが、例えば、脳、神経、骨髄、骨、筋肉、脂肪、心臓、肺、食道、胃、腸、腎臓、副腎、肝臓、膵臓、リンパ節、リンパ管、胸腺、脾臓、血管、血球、臍帯、皮膚、感覚器官、およびその前駆体、生殖組織、幹細胞などが好ましい。また、前記組織中に含まれる細胞、組織中から単離された細胞、これら細胞から樹立した細胞株も組織に含まれる。
【0016】
本発明において「調節」とは、本発明のヌクレオチドの下流に位置する遺伝子の転写の開始、転写増強、転写抑制などをいう。
【0017】
本発明のヌクレオチドとは、その下流に位置する遺伝子の転写開始部位を決定し、その頻度を直接的に調節する核酸をいい、通常プロモーターと称される用語と同義である。
【0018】
本発明のヌクレオチドは、具体的には、TATAボックスとその1〜50塩基上流に位置するCACGTGモチーフおよび/またはCGCGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するものであり、本発明によって初めて解明された、Mycの直接の標的である。
【0019】
「TAF4bプロモーターに由来する」とは、TAF4bとして公知の、例えばGenBank Accession No.XM_290809.4のヒトTAF4b中のプロモーター領域の塩基配列そのものであってもよく、本発明の目的を達成しうるプロモーター活性を有する限りTAF4bプロモーター領域内の塩基配列が改変されていてもよい。プロモーター活性は、後述するレポーターアッセイによりレポーターの発現強度を指標にして調べることができる。
【0020】
本発明のヌクレオチドの必須配列である「TATAボックス」とは、真核細胞のmRNAをコードする遺伝子の転写開始位置より約20〜80塩基上流にあって、通常「TATAT(A)AA(T)」と一般化されているDNA配列をいう。
【0021】
本発明のヌクレオチドの必須配列である「CACGTG」モチーフまたは「CGCGTG」モチーフとは、Mycが直接結合する部位のコンセンサス配列であり、本発明によりMycによる組織特異的発現がこれらのモチーフの使い分けにより調節されていることを初めて解明したものである。
【0022】
本発明のヌクレオチド中に含まれる前記CACGTGモチーフおよび/またはCGCGTGモチーフは、いずれかのモチーフ1つのみでもよく、同じ配列のモチーフが2つでもよく、異なる配列のモチーフが1つずつでもよい。異なる配列のモチーフを含む場合、モチーフ間の上流、下流の関係は特に限定されるものではない。これらのモチーフの位置関係は、前記TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではない。
【0023】
別の側面によると、本発明のヌクレオチドは、
(a)配列番号3に記載の塩基配列、
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列、
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列、
(g)配列番号3の1081〜1126位の領域の塩基配列を必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列および/または当該必須配列の下流に1127〜1157位の領域の塩基配列が配置されてなる塩基配列を含む塩基配列、または
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列、
からなるものであることが好ましい。
【0024】
同一性(%)は、当該分野で慣用のホモロジー検索プログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。本発明において、塩基配列の「同一性」とは、比較する2種の塩基配列を整列(アラインメント)させ、整列により一致した塩基配列の数を基準となる塩基配列の総数で除して算出した割合を%で示した数字である。なお、整列により生じたギャップは、不一致と見なして算出する。
【0025】
(a)配列番号3に記載の塩基配列
本発明によりMycの直接の標的となる配列としてクローニングされた塩基配列である。
【0026】
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列
ここで「実質的に同一」とは、1090〜1096位のTATAボックスと1082〜1087位のCGCGTGモチーフとがプロモーターの構成要素として機能する限り、それ以外の塩基配列は特に限定されないことを意味する。「実質的に同一」か否かの判断は、後述するレポーターアッセイによる前記(a)の塩基配列と(b)の塩基配列との間のレポーターの発現量を比較し、同程度の発現量であることを指標にすればよい。
【0027】
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列
前記TATAボックスおよびCGCGTGモチーフを必須の塩基配列として含み、配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。前記必須配列は、配列番号3の1040〜1126位の領域と対応する塩基配列中に含まれていることが好ましく、対応する塩基配列中に含まれていない場合は、対応配列外に機能可能に連結される。CGCGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0028】
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列
前記TATAボックスおよびCGCGTGモチーフを必須の塩基配列として含み、配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。前記必須配列は、配列番号3の1082〜1156位の領域と対応する塩基配列中に含まれていることが好ましく、対応する塩基配列中に含まれていない場合は、対応配列外に機能可能に連結される。CGCGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0029】
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列
前記TATAボックスおよびCGCGTGモチーフを必須の塩基配列として含み、配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。前記必須配列は、配列番号3の1040〜1120位の領域と対応する塩基配列中に含まれていることが好ましく、対応する塩基配列中に含まれていない場合は、対応配列外に機能可能に連結される。CGCGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0030】
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列
配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置した塩基配列である。
【0031】
(g)配列番号3の1081〜1126位の領域の塩基配列Bを必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列Aおよび/または当該必須配列の下流に1127〜1157位の領域の塩基配列Cが配置されてなる塩基配列を含む塩基配列
ここでは、前記塩基配列Bを必須とするものであり、塩基配列AB(左から右に5’→3’と連結される、以下同様)、塩基配列BC、塩基配列ABCの3通りの組合せがあげられる。すなわち、塩基配列Bは必須であるがこの塩基配列のみではMycによる組織特異的発現が十分ではなく、発現を上昇させる目的には、塩基配列ABまたはBCが好ましく、塩基配列ABCがより好ましい。
【0032】
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列
当該断片は、前記(g)の項の塩基配列AB、塩基配列BC、塩基配列ABCに対応するものであり、これらの塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列を含む塩基配列である。
【0033】
Mycによる組織特異的発現を増強する目的に使用する場合、本発明のヌクレオチドは、
(a)配列番号4に記載の塩基配列、または
(b)配列番号4に記載の塩基配列において、51位〜57位のTATAボックスおよび43〜48位のCACGTGモチーフ以外の塩基配列と70%以上の同一性を有し、かつTATAボックスとその1〜50塩基上流に位置するCACGTGモチーフとを含む塩基配列
を含有することが好ましい。
【0034】
前記(a)は、本発明により、Mycの直接の標的となる配列の中でもMycによって発現がさらに増強されうる配列として見出された塩基配列である。したがって、当該配列は、Mycの存在量を高感度に検出するプロモーターとなりうる。
【0035】
前記(b)は、TATAボックスおよびCACGTGモチーフを必須の塩基配列として含み、必須配列以外の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。CACGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0036】
本発明のヌクレオチド中に含まれる前記CGCGTGモチーフおよび/またはCACGTGモチーフは、本発明のヌクレオチドをプロモーターとして用いた場合に、当該モチーフ内の塩基を変異させることによりMycによる発現の程度を維持または抑制することができる。具体的には前記モチーフは、下記:
(i)塩基配列GTGにおける少なくとも1塩基の置換、
(ii)塩基配列CACにおける少なくとも1塩基の置換もしくは挿入
のいずれかまたは両方の変異を有するものである場合、変異前のMycによる発現を同等またはそれ以下に調節することができる。変異モチーフの具体例としては、CACCTG、CATGCG、CACGCG、CAACGTG、CACGAGモチーフなどがあげられる。
【0037】
本発明のヌクレオチドを細胞に導入する場合、ベクターの形で導入されることが好ましい。本発明は、前記ヌクレオチドを含有するベクターを提供する。
【0038】
基本骨格となるベクターは特に限定されず、形質転換を行う細胞(例えば、大腸菌)中で自己複製可能なものであればよい。例えば、市販のpBR322、pUC、pBluescript、pGL2、pGL3、pGL4 (プロメガ社)等が使用可能である。
【0039】
本発明のベクターは、本発明のヌクレオチドを自体公知の方法により前記基本骨格となるベクターにクローニングすることにより得られる。このようにして得られたベクターは、シークエンス等により所望の位置に所望の方向で本発明のヌクレオチドがクローニングされていることを確認することができる。
【0040】
本発明のヌクレオチドのプロモーター活性を調べるためには、レポーター遺伝子を用いることが好ましい。本発明は、本発明のヌクレオチドおよびレポーター遺伝子を含有するレポータープラスミドを提供する。
【0041】
ここで、レポーター遺伝子とは、本発明のヌクレオチドのプロモーター活性を調べるために組み込まれる目印用の遺伝子をいい、公知のあらゆるレポーター遺伝子を制限なく用いることができる。検出が簡単で定量化も可能であるという観点から、レポーター遺伝子は発光タンパク質遺伝子または蛍光タンパク質遺伝子が好ましい。
【0042】
前記発光タンパク質としては、ホタル由来のルシフェラーゼ、Renilla(ウミシイタケ)由来のルシフェラーゼなどがあげられる。これら発光タンパク質および当該タンパク質をコードする核酸は、公知である。
【0043】
前記蛍光タンパク質としては、GFP、YFP、CFP、BFP、Venusなどのオワンクラゲ由来蛍光タンパク質およびそれらの類似体、ウミシイタケ由来蛍光タンパク質およびその類似体、DsRed、HcRed、AsRed、ZsGreen、ZsYellow、AmCyan、AcGFP、Kaedeなどのサンゴ由来蛍光タンパク質およびそれらの類似体などがあげられる。これら蛍光タンパク質およびそれらの類似体ならびに当該タンパク質をコードする核酸は、公知である。
【0044】
前記レポーター遺伝子は、公知の塩基配列に基づいて、常法により調製することができる。
【0045】
本発明のレポータープラスミドにおいて、構成要素である本発明のヌクレオチドおよびレポーター遺伝子は、直接結合していてもよく、本発明の目的を達成しうる限りにおいて、任意の塩基配列が挿入されていてもよい。かかる挿入配列としては、例えば、クローニングの過程で付加される制限酵素切断部位から生じる塩基配列などがあげられ、通常、1〜1000塩基程度の長さを有するものである。
【0046】
本発明のレポータープラスミドの基本骨格となるプラスミドは特に限定されず、形質転換を行う細胞(例えば、大腸菌)中で自己複製可能なものであればよい。例えば、市販のpBR322、pUC、pBluescript、pGL2、pGL3、pGL4(プロメガ社)等が使用可能である。
【0047】
本発明のレポータープラスミドは、前記ヌクレオチドおよび前記レポーター遺伝子を自体公知の方法により前記基本骨格となるプラスミドにクローニングすることにより得られる。このようにして得られたレポータープラスミドは、シークエンス等によりヌクレオチドおよびレポーター遺伝子が所望の位置に所望の方向でクローニングされていることを確認することができる。
【0048】
このようにして構築された本発明のベクターまたはレポータープラスミドを用いて、形質転換体を製造することができる。本発明は、かかる形質転換体を提供する。
【0049】
宿主としては、例えば、エシェリヒア属菌、バチルス属菌、酵母、動物細胞などが用いられる。エシェリヒア属菌は、本発明のヌクレオチド、ベクターまたはレポータープラスミドを調製するためにもっぱら用いられ、その他の宿主は本発明のヌクレオチドの活性等を調べるために用いられる。
エシェリヒア属菌としては、エシェリヒア・コリ(Escherichia coli)K12・DH1、JM103、JA221、HB101、C600などが用いられる。
バチルス属菌としては、例えば、バチルス・ズブチルス(Bacillus subtilis)MI114などが用いられる。
酵母としては、例えば、サッカロマイセス セレビシエ(Saccharomyces cerevisiae)AH22,AH22R−,NA87−11A,DKD−5D,20B−12、シゾサッカロマイセス ポンベ(Schizosaccharomyces pombe)NCYC1913,NCYC2036、ピキア パストリス(Pichia pastoris)などが用いられる。
動物細胞としては、例えば、サル細胞COS−7、Vero、チャイニーズハムスター細胞CHO、dhfr遺伝子欠損チャイニーズハムスター細胞CHO(CHO(dhfr−))、マウスL細胞、マウスAtT−20、マウスミエローマ細胞、ラットGH3、ヒトFL細胞などが用いられる。
【0050】
エシェリヒア属菌を形質転換するには、例えば、Proc. Natl. Acad. Sci. USA),69巻,2110(1972)やGene,17巻,107(1982)などに記載の方法に従って行なうことができる。
バチルス属菌を形質転換するには、例えば、Molecular & General Genetics,168巻,111(1979)などに記載の方法に従って行なうことができる。
酵母を形質転換するには、例えば、Methods in Enzymology,194巻,182−187(1991)、Proc. Natl. Acad. Sci. USA,75巻,1929(1978)などに記載の方法に従って行なうことができる。
動物細胞を形質転換するには、例えば、細胞工学別冊8新細胞工学実験プロトコール.263−267(1995)(秀潤社発行)、Virology,52巻,456(1973)に記載の方法に従って行なうことができる。
【0051】
前記ベクターまたはレポータープラスミド(以下、「ベクター等」と略す場合がある)を導入することにより、トランスジェニック非ヒト動物を作出することができる。本発明は、かかるトランスジェニック非ヒト動物を提供する。
【0052】
本発明のトランスジェニック非ヒト動物には、前記ヌクレオチドおよび場合によってはレポーター遺伝子が対立遺伝子の両方に導入されたホモ接合動物、対立遺伝子の片方に導入されたヘテロ接合動物およびそれらの出生前の胎仔も含まれる。前記ホモ接合動物は、前記ヘテロ接合動物を交配することにより得られるものである。
【0053】
用いられる非ヒト動物(本発明においては、単に動物と略す場合がある。)としては、ヒト以外の動物であれば特に限定されるものではない。好適な動物としては、マウス、ラット、ハムスター、モルモット、ウサギ、イヌ、ネコ、サル等の実験動物ならびにウシ、ヒツジ、ウマ、ブタ等の家畜があげられるが、遺伝子工学的に利用が容易であるところから、マウスがより好ましい。
【0054】
本発明のトランスジェニック非ヒト動物は、自体公知の方法により製造することができる。次に、本発明のトランスジェニック非ヒト動物の作出方法について説明する。
【0055】
本発明のトランスジェニック非ヒト動物は、例えば下記の工程(a)〜(c)を含む方法により製造することができる。
(a)前記ベクター等を調製する工程;
(b)前記ベクター等を受精卵に導入し、遺伝子導入受精卵を仮親動物に移植する工程;
(c)前記動物から出生した子孫からトランスジェニック動物を選別する工程;および
(d)前記選別した動物(ファウンダー)から系統を樹立する工程。
【0056】
前記工程(a)は、本発明のベクターまたはレポータープラスミドにおいて記載した通りである。得られたベクター等は、制限酵素等により線状化して工程(b)に供することが好ましい。
【0057】
前記工程(b)において、前記ベクター等を受精卵に導入する方法は、例えば、交配後の雌の卵管を洗浄して受精卵を採取し、精子または卵子由来の前核にマイクロインジェクション法により前記ベクター等を直接注入する方法があげられる。この受精卵を偽妊娠させた仮親の輸卵管に移植し、子宮内で発生を続けさせる。
【0058】
前記工程(c)において、工程(b)で移植した動物が出生した子孫からトランスジェニック動物を選別する方法としては、例えば、注入したベクター等が染色体DNAに組み込まれているか否かについて、産仔の尾部等より分離抽出した染色体DNAをサザンブロット法またはPCR法によりレポーター遺伝子の組み込みを確認する方法があげられる。レポーター遺伝子が蛍光タンパク質遺伝子の場合、レポーター遺伝子が発現している生体の部位に紫外線等を照射して蛍光を調べることにより確認することができる。
【0059】
前記工程(d)において、工程(c)で選別した動物(ファウンダー)から遺伝的背景の均一な系統を樹立する方法としては、ベクター等が組み込まれた動物とC57BL/6、FVB、BALB/cなどの近交系の野生型動物とを戻し交配をする方法があげられる。
【0060】
このようにして得られた本発明のトランスジェニック非ヒト動物をさらに交配して得られる子孫動物、これら動物に由来する組織または細胞も本発明に含まれる。前記組織としては、すべての組織があげられ、例えば、脳、神経、骨髄、骨、筋肉、脂肪、心臓、肺、食道、胃、腸、腎臓、副腎、肝臓、膵臓、リンパ節、リンパ管、胸腺、脾臓、血管、血球、臍帯、皮膚、感覚器官およびその前駆体、生殖組織、幹細胞などが好ましい。また、前記細胞としては、前記組織中に含まれる細胞、組織中から単離された細胞、これら細胞から樹立した細胞株があげられる。
【0061】
本発明のトランスジェニック非ヒト動物は、本発明のヌクレオチド(TAF4bプロモーターに由来する塩基配列)によってレポータータンパク質の発現が調節されているものが好ましい。本発明のトランスジェニック非ヒト動物は、Mycに感受性が高い状態で、様々な用途に利用することができる。好ましくは、本発明のトランスジェニック非ヒト動物は、生体内で代謝された後の物質の生体中の各組織への影響を調べるものとして使用される。
【0062】
本発明は、前記レポータープラスミドまたは前記形質転換体を用いることを特徴とする、細胞増殖の調節剤をスクリーニングする方法(スクリーニング方法I)を提供する。
【0063】
[スクリーニング方法I]
本発明のスクリーニング方法Iは、具体的には、下記工程を含む:
(a)レポータープラスミド導入細胞または形質転換体と被験物質とを接触させる工程、
(b)前記被験物質を接触させた細胞または形質転換体におけるレポーター遺伝子の発現量を調べ、被験物質を接触させない細胞または形質転換体における発現量と比較する工程、および
(c)前記比較結果に基づいて、細胞増殖を調節しうる被験物質を選択する工程。
【0064】
前記(a)において、被験物質とは、いかなる公知物質および新規物質であってもよく、例えば、核酸、糖質、脂質、蛋白質、ペプチド、有機低分子化合物、コンビナトリアルケミストリー技術を用いて作製された化合物ライブラリー、固相合成やファージディスプレイ法により作製されたランダムペプチドライブラリー、あるいは微生物、動植物、海洋生物等由来の天然成分などがあげられる。また、これらの化合物の2種以上の混合物を試料として供することもできる。
【0065】
被験物質と接触させる前に、レポータープラスミドの場合はレポーター遺伝子が発現できるように所望の細胞に一過性に導入し、形質転換体の場合は宿主に適した培地および培養条件で培養しておく。用いる細胞または宿主の種類は、好ましくは、ヒト子宮頚部癌細胞HeLa、ヒトクリオブラストーマ細胞T98G、大腸癌細胞SW620、HCT116などである。被験物質との接触方法としては、例えば、前記細胞または形質転換体を適当な培地中に入れ、約25〜40℃のインキュベーター中で生存または培養させ、次に、前記培地中に被験物質を添加し、インキュベートを続けることで接触がなされうる。
【0066】
前記被験物質の添加量は、有効成分の種類、培地に対する溶解性、細胞または形質転換体の感受性等によって適宜設定することができる。
【0067】
工程(b)において、前記被験物質を接触させた細胞または形質転換体におけるレポーターの発現量は、各レポーター遺伝子に応じて自体公知の方法により調べることができる。例えば、蛍光タンパク質の場合、各蛍光タンパク質に応じた励起波長を照射して検出される蛍光を測定することにより行う方法などがあげられる。
【0068】
前記工程(b)において、被験物質を接触させない細胞または形質転換体におけるレポーター遺伝子の発現量も同時にまたは別途調べ、接触した場合の結果と接触しない場合の結果とを比較する。
【0069】
前記工程(c)において、工程(b)で得られた比較結果に基づき、細胞増殖を調節しうる被験物質を選択する。選択する基準は、レポーターの発現が上昇または低下していることを指標にすればよい。レポーターの発現が上昇している場合、被験物質は、細胞増殖を促進させる作用を有する。レポーターの発現が低下している場合、被験物質は、細胞増殖を抑制させる作用を有する。
【0070】
このようにして選択された被験物質は、Mycと本発明のヌクレオチドとの結合に特異的に作用して、あるいはMyc発現量に影響して、あるいはMycによる転写制御機構に作用して機能する作用機序の明確な細胞増殖調節剤の候補となりうる。
【0071】
[スクリーニング方法II]
また、本発明は、本発明のレポータープラスミド、形質転換体またはトランスジェニック非ヒト動物を用いることを特徴とする、妊娠調節剤をスクリーニングする方法(スクリーニング方法II)を提供する。スクリーニング方法IIは、下記工程:
(a)レポータープラスミド導入細胞、形質転換体またはトランスジェニック非ヒト動物と被験物質とを接触させる工程、
(b)前記被験物質を接触させた細胞、形質転換体または動物におけるレポーターの発現量を調べ、被験物質を接触させない細胞、形質転換体または動物における発現量と比較する工程、および
(c)前記比較結果に基づいて、妊娠調節剤の候補となる被験物質を選択する工程を含む。
スクリーニング方法IIにおいて、レポータープラスミド導入細胞または形質転換体を用いる場合は、工程(a)および(b)はスクリーニング方法Iと同様である。ここでは、本発明のトランスジェニック非ヒト動物を用いる場合について説明する。
【0072】
工程(a)において、被験物質は、スクリーニング方法Iと同様のものがあげられる。
【0073】
前記被験物質を本発明のトランスジェニック動物と接触させる方法は特に限定されるものではないが、経口的または非経口的に投与され得る。非経口的投与経路としては、例えば、静脈内、動脈内、筋肉内、腹腔内等の全身投与、または標的細胞付近への局所投与等があげられる。
【0074】
前記被験物質の投与量は、有効成分の種類、分子の大きさ、投与経路、投与対象となる動物種、投与対象の薬物受容性、体重、年齢等によって適宜設定することができる。
【0075】
前記工程(b)において、被験物質を投与した動物におけるレポーター遺伝子の発現量を調べる方法としては、各レポーター遺伝子に応じて自体公知の方法により行うことができる、例えば、蛍光タンパク質の場合、各蛍光タンパク質に応じた励起波長を照射して検出される蛍光を測定することにより行う方法などがあげられる。
【0076】
前記工程(b)において、被験物質を投与しない動物におけるレポーター遺伝子の発現量も同時にまたは別途調べ、投与動物の結果と非投与動物の結果とを比較する。
【0077】
前記工程(c)において、工程(b)で得られた比較結果に基づき、妊娠調節剤の候補となる被験物質を選択する。選択する基準は、レポーターの発現が上昇または低下していることを指標にすればよい。
【0078】
このようにして選択された被験物質は、Mycと本発明のヌクレオチドとの結合に特異的に作用して、、あるいはMyc発現量に影響して、あるいはMycによる転写制御機構に作用して機能する作用機序の明確な妊娠調節剤の候補となりうる。
【0079】
また、本発明は、下記いずれかのペプチド:
(1)配列番号1に記載の塩基配列からコードされるペプチド、
(2)配列番号2に記載のアミノ酸配列からなるペプチド、
(3)前記(1)または(2)に記載のペプチドと実質的に同一のアミノ酸配列を有するペプチド
を有効成分として含有する、Mycによる組織特異的発現の調節剤を提供する。前記調節剤は、妊娠の調節に用いられることが好ましい。
【0080】
「実質的に同一のアミノ酸配列」とは、配列番号2に示されるアミノ酸配列において1もしくは2以上(好ましくは1〜50個、より好ましくは1〜30個、いっそう好ましくは1〜10個、最も好ましくは1〜5個)のアミノ酸が置換、欠失、挿入、付加または修飾されたアミノ酸配列であって、同質の活性を有するものである。
【0081】
また、「実質的に同一のアミノ酸配列」として、配列番号2に示されるアミノ酸配列に対して約70%以上、好ましくは約80%以上、より好ましくは約90%以上、さらにより好ましくは約95%以上、いっそうより好ましくは約97%以上、最も好ましくは99%以上のアミノ酸同一性を有するアミノ酸配列を用いることもできる。同一性(%)は、前記塩基配列の同一性と同様に決定することができる。
【0082】
「同質の活性」とは活性が定性的に同等であることを意味し、定量的にも同等であることが好ましいが、許容し得る範囲(例えば、約0.5〜約2倍)で異なっていてもよい。ここでTAF4bの活性としては、TBPまたはTAFs(TBP associated factors)との結合があげられる。
【0083】
また、本発明は、下記:
(a)配列番号5に記載の塩基配列、または
(b)配列番号5に記載の塩基配列において210位〜216位のTATAボックスおよび202〜207位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号5に記載の塩基配列と実質的に同一な塩基配列
を含有するサルTAF4bプロモーターを提供する。
【0084】
ここで「実質的に同一」とは、210〜216位のTATAボックスと202〜207位のCGCGTGモチーフとがプロモーターの構成要素として機能する限り、それ以外の塩基配列は特に限定されないことを意味する。「実質的に同一」か否かの判断は、後述するレポーターアッセイによる前記(a)の塩基配列と(b)の塩基配列との間のレポーターの発現量を比較し、同程度の発現量であることを指標にすればよい。
【0085】
また、本発明は、下記:
(a)配列番号6に記載の塩基配列、または
(b)配列番号6に記載の塩基配列において778位〜784位のTATAボックスおよび770〜775位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号6に記載の塩基配列と実質的に同一な塩基配列
を含有するラットTAF4bプロモーターを提供する。
【0086】
ここで「実質的に同一」とは、778〜784位のTATAボックスと770〜775位のCGCGTGモチーフとがプロモーターの構成要素として機能する限り、それ以外の塩基配列は特に限定されないことを意味する。「実質的に同一」か否かの判断は、後述するレポーターアッセイによる前記(a)の塩基配列と(b)の塩基配列との間のレポーターの発現量を比較し、同程度の発現量であることを指標にすればよい。
【実施例】
【0087】
以下に実施例を挙げて、本発明をさらに詳しく説明するが、本発明はこれらに限定されるものではない。
【0088】
細胞培養
ヒト膠芽腫細胞株T98G細胞、およびその派生体であり、c−MycERキメラタンパク質を発現するT98Gmycer−2細胞(Tsuneoka M. et al., J. Biol. Chem. 277:35450-35459 (2002))を、非必須アミノ酸および10%ウシ胎児血清(FCS)を含むイーグル培地中で培養した。c−MycERキメラタンパク質を発現するラット線維芽細胞株3Y1の派生体、3Y1MycER(Tsuneoka M. et al., Oncogene 14:2301-2311 (1997))およびアフリカミドリサルの腎線維芽細胞株Cos−7を、10%FCSを含むダルベッコ変法イーグル培地中で培養した。ヒト前骨髄性白血病HL60細胞を、20%FCSを含むRPMI 1640培地中で培養した。ラット大腸がん細胞株RCN−9(理研細胞バンク)およびヒト赤白血病細胞株HEL(ヘルスサイエンスリサーチ資源バンク製)を、10%FCSを含むRPMI 1640培地中で培養した。
【0089】
ポリメラーゼ連鎖反応(PCR)
PCR増幅は、各プライマーを10pmoles、EX Taq DNAポリメラーゼを1.2U、およびdNTPを200μM含む50μl EX Taqバッファー(タカラバイオ製)中で行った。
【0090】
5’−RACE法による解析
HEL細胞のpoly(A)+RNA(1μg)からの逆転写反応、二本鎖cDNA合成、およびアダプターライゲーションは、マラソン(Marathon)cDNA増幅キット(クロンテック社製)を用いて行った(Tsuneoka M. et. al., J. Biol. Chem. 277:35450-35459 (2002),Tsuneoka M. et. al., J. Biol. Chem. 280:19977-19985 (2005))。第1PCRは、プライマーとして、AA287145-1st(5’−TTCTTATGCCTCTTCCTTTCTCCTG−3’(配列番号7)、ESTクローンAA287145の5’末端領域の配列)、および供給者によって提供されたAP1プライマーを使用して行った。温度プロファイルは、94℃で1分間の最初の熱変性後、96℃で15秒間の熱変性工程ならびに68℃で3分間のアニーリング工程および伸張工程を1サイクルとする25サイクルであった。上記第1RACE−PCR産物を1000倍希釈した液1μlをネストRACE−PCRの鋳型として使用した。ネストRACE−PCRは、AA287145ネストプライマー(5’−TTGGCAAATGAGGCAGGTAGTAACA−3’(配列番号8)、ESTクローンAA287145のAA287145-1stの上流領域配列に対応するもの)と、供給者によって提供されたAP2プライマーとを使用して行った。温度プロファイルは、94℃で1分間の最初の熱変性後、96℃で15秒間の熱変性工程ならびに68℃で3分間のアニーリング工程および伸張工程を1サイクルとする25サイクルであった。増幅したDNA断片をpGEM-Tベクター(Promega社製)にクローニング(pT/fragmentTAF4b)し、シークエンスした。シークエンスの結果、AA287145はTAF4b遺伝子の3’−UTRの一部であることが確認された。
【0091】
TAF4bのmRNAの5’末端に関する情報を得るために、5’−RACE増幅が上述のように実施された。ここでは、プライマーとして、TAF4bRACE−1(5’−GGTCTCGGCTCTTGTTACAGTTTGCTG−3’(配列番号9)、文献(Dikstein R. et. al., Cell 87:137-146 (1996))において以前に報告したTAF4b cDNAの5’末端領域の配列)と第1PCR用に供給者によって提供されたAP1プライマー、TAF4bRACE−2プライマー(5’−GGATTGTCGTGGTGTTGGGGGCTTTCA−3’(配列番号10)、TAF4bRACE−1の上流領域の配列に対応するもの)およびネストPCR用に供給者によって提供されたAP2プライマーを使用した。増幅したDNA断片をpGEM-Tベクター(Promega社製)にクローニングし、シークエンスした。
【0092】
逆転写PCR法(RT−PCR)
HEL細胞の一本鎖cDNAの合成は、スーパースクリプト(Superscript)First-strand Synthesis system(インビトロジェン製)を使用して、全RNA(1μg)で行った。得られた一本鎖cDNA1μl(全量20μl)をPCRの鋳型として使用した。増幅用のRT−PCRプライマーは、
TAF4bコードU(5’−CGGAATTCGAAGCTGCGAGAGGTCGGGCGGGTGTCG−3’(配列番号11)、5’非翻訳領域にEcoRI部位を付加した配列)、および、
TAF4bコードL(5’−CGGAATTCGGCAGTAAATAGCAAGGATGTGGATGGA−3’(配列番号12)、 3’非翻訳領域にEcoRI部位を付加した配列)であった。温度プロファイルは、98℃での15秒間の熱変性工程と、65℃での1分間のアニーリング工程および72℃での2分30秒間の伸張工程を1サイクルとして35サイクルであった。増幅したDNA断片をpGEM-Tベクター(プロメガ社製)にクローニングしてpT/TAF4bを作製し、その配列を決定した。当該配列は、増幅された断片を使用してダイレクトシークエンスによって確認された。
【0093】
TAF4bゲノムDNA断片を有するレポータープラスミド
ヒトTAF4b遺伝子のゲノムDNA断片は、プロモーター領域からエキソン1(exon 1)へと伸長しており、プライマー
5’−TTTTACCATAACCTCACTTGCTGGAAGGGG−3’(配列番号13)、および、
5’−TGTTGGGGGCTTTCACGGCGACTATCTG−3’(配列番号14)
を使用したPCRによって増幅した。2.2kbの増幅された断片を、SacIおよびNcoIで切断し、SacIとNcoIで切断したホタルルシフェラーゼを含むpGL3(Promega社製)の4.8kb断片に挿入し、pTAF4b(W)luciを作製した。SmaIとNcoIで切断したpTAF4b(W)luciの0.6kbの断片を、SmaIとNcoIで切断したpGL3の4.8kpの断片に挿入し、pTAF4b(dSma)luciを作製した。XhoIとNcoIで切断したpTAF4b(W)luciの0.6kpの断片を、XhoIとNcoIで切断したpGL3の4.8kpの断片に挿入し、pTAF4b(dXho)luciを作製した。pTAF4b(dSma)luciを、XhoIとNcoIで切断した後、Klenow酵素で平滑末端化し、セルフライゲートして、pTAF4b(Sma−Xho)luciを作製した。また、pTAF4b(dSma)luciを、PvuIIとNcoIで切断した後、Klenow酵素で平滑末端化し、セルフライゲートして、pTAF4b(Sma−Pvu)luciを作製した。
【0094】
さらに、欠失変異体pTAF4b(117Pvu)luci、pTAF4b(114Pvu)luci、pTAF4b(102Pvu)luci、pTAF4b(73Pvu)luciおよびpTAF4b(43Pvu)luciを、鋳型としてpTAF4b(Sma−Pvu)luciを使用し、適当なプライマーを用いたPCR、および、その後に行われるpGL3ベクターへの増幅断片の導入によって作製した。
【0095】
突然変異は、Gene EditorTMインビトロ部位指向突然変異システム(プロメガ社製)を用いて、pTAF4b(W)luciのE−ボックス部位に導入し、2つのE−ボックス(CACGTGエレメント)がCACCTGに変異したpTAF4b(mE1/2)luciを作製した。また、pTAF4b(Sma−Pvu)luciの非基準型E(NCE)−ボックス部位にも突然変異を導入し、pTAF4b(Sma−Pvu mNCE1/2)luci、pTAF4b(Sma−Pvu mNCE1)luciおよびpTAF4b(Sma−Pvu mNCE2)luciを作製した。これにより、1または2個のNCE−ボックス(CGCGTGエレメント)がCGCCTGに変異した。pTAF4b(Sma−Pvu)luciにおける非基準型E(NCE)−ボックス部位2(NCE−box2)を、CACGTGに変異させ、pTAF4b(Sma−Pvu CanoE2)luciを作製した。作製されたプラスミドのDNA配列は、シークエンスによって確認した。
【0096】
ラットおよびサルのTAF4bプロモーターの配列
ラットTAF4bプロモーターの1.2kp断片を、以下のようにして単離した。
まず、ヒトTAF4b遺伝子について、マウスのDNA配列のホモログを選択した(ジーンバンクアクセッション番号:AK012135)。マウスのDNA配列においては、ヒトTAF4bのゲノム配列に正確に適合するヌクレオチド配列として2つの領域を選択し、以下のオリゴヌクレオチド、
ラットTAFII−F(5’−GCGCACGTGTGAGCGCCGCTGAGG−3’(配列番号15))、および、ラットTAFIIPF−1(5’−CTGACAGGAGCCTTAGTCAC−3’(配列番号16))を合成した。これらのオリゴヌクレオチドをプライマーとして使用し、ラット結腸癌細胞RCN−9ゲノムDNAを鋳型として用いて、ラットTAF4bゲノムDNAを増幅した。増幅されたDNA断片をシークエンスし、この配列の上流領域を、promoter find kit(クロンテック社製)によってさらにクローニングした。なお、このクローニングには、鋳型としてDraIおよびSspIライブラリーが用いられ、プライマーとして
ラットTAFIIPF3(5’−GCCGGGGCGGGCGGCAGGAGACTCG−3’(配列番号17))およびTAFIIPF4(5’−CGGGCACTCCCTCAGCGGCGCTCAC−3’(配列番号18))(これらの配列は、先に確認されたラットのTAF4bDNA配列内のもの)、ならびにAP1およびAP2が使用された。増幅されたラットのTAF4bゲノム断片を、シークエンスした。
サルTAF4bプロモーターを、ヒトTAF4b遺伝子と適合するTAF−プライマー1(5’−GACACAAGGAGAGGAACACGGATGC−3’(配列番号19))、およびTAF−プライマー2(5’−GCCTGGGCTGCCCCCGGAGCGACAC−3’(配列番号20))をプライマーとし、Cos−7細胞のゲノムDNAを鋳型として使用して増幅した後、シークエンスした。
【0097】
その他のプラスミド
CMVプロモーターの制御条件下のウミシイタケルシフェラーゼ(Renilla reniformis luciferase)遺伝子を含むpRL−CMVを、Promega社から購入した。CMV−c−Myc、pCMV−USF、およびpCMV−TFE−3は、Martin Eilers博士から供与された(Desbarats L. et. al., Genes Dev. 10:447-460 (1996))。組換え型のGFP−USFおよびGFP−TFE3を作製するために、pCMV−USFおよびpCMV−TFE−3を鋳型としてDNA断片を増幅した。なお、プライマーとしては、
5’−GGCGGATCCAGATGAAGGGGCAGCAGAAAAC−3’(配列番号21) (USF-BamHI-5’)、および、
5’−CGGGAATTCCCCATAGTTAGTTGCTGTC−3’(配列番号22) (USF-EcoRI-3’)、
あるいは、5’−GCCGGATCCATGGCGCTGCTCACCATCGG−3’(配列番号23) (TFE3-BamHI-5’)、および、5’−CGGCTCGAGGGTTCATGGGGAAGGGGCAG−3’(配列番号24) (TFE3-XhoI-3’)を使用した。次いで、得られた断片を、BamHIおよびEcoRIまたはBamHIおよびXhoIで切断し、BamHIおよびEcoRIで切断した細菌発現ベクターpGEX−3X、またはBamHIおよびXhoIで切断した細菌発現ベクターpGEX−4T3にそれぞれ導入し、pGEX−USFまたはpGEX−TFE−3を作製した。
【0098】
RNA調製
全RNAを、DEPC処理されたRNA調製液セット(ナカライテスク社製)を用いて、酸グアニジウム−チオシアネート−フェノール−クロロホルム抽出法によって細胞から単離した。
【0099】
DNAチップを使用した示差ディスプレイ解析(cDNAマイクロアレイ)
cDNAマイクロアレイは、文献(Tsuneoka M. et. al., J. Biol. Chem. 277:35450-35459 (2002))で説明した方法によって実施した。全RNAを、未処理のT98Gmycer−2細胞および4−ヒドロキシタモキシフェン(OHT)で20時間処理した細胞から単離した。poly(A+)RNAを回収し、DNAチップ(インサイト社製)を利用した示差ディスプレイ解析に供した。発現遺伝子配列タグ(EST)クローンを含む約9000種のcDNAがチップ上に搭載された(UniGEM Human V Ver. 2)。
【0100】
ノーザンブロット解析およびプローブDNAの調製
RNAは、ホルムアルデヒド含有アガロースゲルで電気泳動し、ハイボンドN(アマシャム社製)に転写し、32P標識した各cDNAプローブで検出した。各プローブは、市販のMultiprimeラベリングキット(アマシャム社製)を用いて[α−32P]dCTPで標識した。結果はBAS2000イメージアナライザー(富士フィルム社製)を用いて定量した。c−myc用のDNAプローブは、文献(Tsuneoka M. et. al., Oncogene 14:2301-2311 (1997))に示すものであった。TAF4b用のDNAプローブは、pT/fragmentTAF4b由来の0.6kpのSacI断片であった。
【0101】
一過性発現解析
T98Gmycer−2細胞における一過性発現レポーター解析を、文献(Tsuneoka M. et. al., J. Biol. Chem. 277:35450-35459 (2002))に説明する方法で行った。
TAF4bプロモーターにおけるMycおよびその他の転写因子の影響を、当該転写因子をコードしているプラスミドの転写によって調べた。T98G細胞を、10%FCSを含む培地で生育させた。細胞(3×104個)を培養皿(12ウエルのプレート、ウエルの径は22mm)に入れ、20〜24時間培養した。1μgのレポータープラスミド、インターナルマーカーとして20ngのpRL−CMV、および転写因子を発現する様々な量のプラスミドをFuGENE6試薬(ロシュ・ダイアグノステイックス社製)を用いて細胞に導入し、トランスフェクションを行った。各トランスフェクションにおける全DNA量は、空の発現ベクターを加えることによって一定に維持した。一日経過後、細胞を集め、ルシフェラーゼ活性を文献(Tsuneoka M. et. al., Oncogene 14:2301-2311 (1997))に示す方法によって測定した。
【0102】
クロマチン免疫沈降実験
クロマチン免疫沈降実験は、基本的に文献(Tsuneoka, M., et. al., J. Biol. Chem. 277:35450-35459(2002))に示す方法と同様に実施した。免疫沈降したDNA断片を、PCRによって検出した。使用したPCRプライマーは、
5’−GACACAAGGAGAGGAACACGGATGC−3’(配列番号25) (TAF4b-primer 1)、
および、5’−GCCTGGGCTGCCCCCGGAGCGACAC−3’ (配列番号26)(TAF4b-primer 2)である。これらは、ヒトTAF4b遺伝子のプライマー(TAF4b primers)の転写開始部位を含む521bp断片を増幅するプライマーである。
また、他のPCRプライマーとして、
5’−TTACAGGTAAGCCCTCCAATGACC−3’(配列番号27)、
および、5’−GCAAAGCTACCATTTAGGAACCC−3’(配列番号28)を使用した。これらは、第22番染色体中のE−ボックスを含む領域のゲノム配列を増幅するプライマーである(control primers)。このE−ボックスは検出可能な遺伝子を有しない染色体中に位置している(Bouchard, C., et. al., Genes Dev. 15:2042-2047.(2001))。
【0103】
抗体
c−Mycに対する抗体−1は、文献(Tsuneoka, M., et. al., Oncogene 14:2301-2311.(1997))に記載されたものである。抗c−Myc抗体−2(N262)(Santa Cruz Biotechnology, Santa Cruz, CA)、抗Max抗体(Oncogene Science)、抗c−Mycモノクローナル抗体(C−8)(Santa Cruz Biotechnology)、抗TFE3ヤギ抗体(sc−5958)(Santa Cruz Biotechnology)、および、抗USF−1ウサギ抗体(sc−229)(Santa Cruz Biotechnology)は、購入した。
【0104】
ゲルシフトアッセイ
ゲルシフトアッセイは、本質的には文献(Tsuneoka, M., et. al., Oncogene 14:2301-2311(1997))に示す方法で行った。組換え型GST−MycおよびGST−Maxを混合し、室温で30分間インキュベートした。混合物を、ヒトTAF4bプロモーターの合成二本鎖オリゴヌクレオチドへの結合について解析した。二本鎖オリゴヌクレオチドCanoEは、TAF4bプロモーターCGCGTG(NCE−box2)の代わりにCACGTGエレメントを含んでいるが、合成オリゴヌクレオチド
5’−GCGTGCGCGAGGTCTCCACGTGGCTATATAAACATGG−3’(配列番号29)、
および、5’−GCCAGCCATGTTTATATAGCCACGTGGAGACCTCG−3’(配列番号30)
のアニーリングによって作製された。CanoEオリゴヌクレオチドは、Klenow酵素を使用して[32P]dCTPで標識した。
20μlの結合反応溶液内の最終状態は、32P標識されたCanoEオリゴヌクレオチド:1ng、ポリ(dI−dC)ポリ(dI−dC)(Pharmacia Biotech社製):1μg、非特異的一本鎖オリゴヌクレオチド(5’−GAGTCGACGAACACACCAGGTCTTGGAGCG−3’(配列番号31)):1μg、塩化ナトリウム(NaCl):98.5mM、塩化カリウム(KCl):2.1mM、リン酸ナトリウム:4.8mM、Tris(pH7.4):2mM、EDTA:0.1mM、グリセロール:1%であった。
反応を進行させるために、反応混合液を室温で20分間放置した。抗Max(Oncogene Science)、非特異的抗体または抗c−Mycモノクローナル抗体(C−8)(Santa Cruz Biotechnology)をDNA結合反応中に加え、抗体解析を行った。標識されていない二本鎖オリゴヌクレオチドCanoE、標識されていない二本鎖オリゴヌクレオチドnon−CanoE(野生型:5’−GCGTGCGCGAGGTCTCCGCGTGGCTATATAAACATGG−3’(配列番号32)と、
5’−GCCAGCCATGTTTATATAGCCACGCGGAGACCTCG−3’(配列番号33)とのアニーリングによって作製されたもの)、および、
mE(5’−GCGTGCGCGAGGTCTCCGCCTGGCTATATAAACATGG−3’(配列番号34)と5’−GCCAGCCATGTTTATATAGCCAGGCGGAGACCTCG−3’(配列番号35)とのアニーリングによって作製されたもの)は、competitorとして使用された。
グルタチオンS−トランスフェラーゼフュージョンUSFおよびTFE3タンパク質を、E. coli Rosetta(Novagen Inc. Cat# 70953-4)中のpGEX−USFあるいはpGEX−TFE3を用いて発現させ、グルタチオンセファロースカラム(Amersham Biosciences)で単離した。ゲルシフトアッセイを、c−Mycの場合と同様の方法で、USFおよびTFE3について実施した。
【0105】
(結果)
c−Mycは、TAF4bの発現を誘導する。
条件付でc−Myc活性を誘導するために、エストロゲン誘発性Mycシステム(Eilers M. et. al., Nature 340:66-68(1989),Tsuneoka M. et.al., J. Biol. Chem. 277:35450-35459 (2002)参照)を使用した。キメラタンパク質c−MycERは、ヒトc−Mycおよびヒトエストロゲン受容体のエストロゲン結合ドメインから構成されており、エストロゲンの非存在下では細胞骨格成分に結合している。このキメラタンパク質にエストロゲンまたはその類似体4−ヒドロキシタモキシフェン(OHT)が結合すると、細胞骨格成分から遊離し、c−Mycとして機能する。ヒト膠芽腫細胞株T98Gを、異所性c−Myc活性が導入される親細胞として使用した。c−MycERタンパク質を発現するT98G細胞株(T98Gmycer−2細胞)が確立された(Tsuneoka M. et.al., J. Biol. Chem. 277:35450-35459 (2002))。OHTの存在下あるいは非存在下における指数増殖期の20時間のT98Gmycer−2細胞由来の全RNAを、cDNAマイクロアレイ解析に供した。Myc標的遺伝子であるオルニチンデカルボキシラーゼ(Bello-Fernandez C. et. al., Proc. Natl. Acad. Sci. U. S. A. 90:7804-7808 (1993))、ヌクレオリン(Greasley P. J. et. al., Nucleic Acids Res. 28:446-453 (2000))、およびmina53(Tsuneoka M. et.al., J. Biol. Chem. 277:35450-35459 (2002))に対する特異的シグナルは、c−MycERの活性化によって、それぞれ2.6倍、1.6倍、1.9倍に上昇した。これらの結果は、Myc標的遺伝子がこの実験システムにおいて検出されうるということを示唆している。
【0106】
本発明者は、ESTクローンAA287145に対するシグナルが、c−Mycの活性化によって2.3倍に刺激されるということを見出した。ESTクローンAA287145の5’上流領域をコードするcDNAは、ヒト赤白血病HEL細胞ライブラリーから5’−RACEプロトコールを用いて単離した。そして、AA287145は、TAF4b遺伝子(Dikstein, R., et. al., Cell 87:137-146 (1996))においてはTAFII105と呼ばれるもの)の3’非コード領域の一部であることが確認された。ヒトTAF4b遺伝子の翻訳および転写開始部位は以前に報告されていなかったため、本発明者らは、5’−RACE実験によって転写物の5’末端をコードするcDNAを単離した。転写開始部位をTAF4bゲノム配列上にマップした。この部位は、図1Aにおいて矢印を付して示している。TAF4bのmRNAの5’および3’配列と一致するプライマーを使用して、RT−PCRプロトコールに基づいてHEL細胞の全RNAから3.2kb長のcDNAを増幅し、シークエンスした。全体のヌクレオチド配列は文献(Dikstein, R., et. al., Cell 87:137-146.(1996))において報告したものとほぼ同じであったが、既に報告しているTAF4bのN末端のアミノ酸Glyの先に66個のアミノ酸配列が付加されていること(図1A参照)、および、いくつかのアミノ酸が異なっていることが確認された(当該配列は、ジーンバンク(Gen/Bank)にアクセッション番号AB234096として登録されている。)。当該cDNAクローンは、862アミノ酸からなり、推定分子量91090.52Da、等電点9.59のタンパク質をコードしている。
【0107】
TAF4b mRNAの発現
ヒト前骨髄性白血病HL60細胞を、TPA(phorbol 12-myristate 13-acetate)によって末期に分化させると、一時的に刺激された後(Vass, J. K., et al., Differentiation 45:49-54 (1990)参照)にc−mycの発現量が減少する(Hickstein, D. D., et. al., J. Biol. Chem. 264:21812-21817 (1989)、Hozumi, M., Adv. Cancer Res. 38:121-169 (1983)参照)。この実験システムを用いて、Myc標的遺伝子がMycの遮断中に影響されるか否かを調べた(Coller, H. A., et. al., Proc. Natl. Acad. Sci. U. S. A. 97:3260-3265.(2000)、Grandori, C., et. al., EMBO J 15:4344-4357.(1996)参照)。図2Aに示すように、c−mycのmRNAのレベルは、TPAを添加した後1時間で2倍に増加した後、3時間後に減少し始め、7時間後には1/5に減少した。TAF4bのmRNAのレベルは、TPAを添加した後3時間で1.5倍に上昇した後、6時間後に減少し始め、9時間後に1/10に減少した。これらの結果は、TAF4b発現がc−myc発現の後に起こることを示している。
【0108】
次に、MycERシステムを用いて、TAF4bのmRNAに対するc−Myc活性化の影響を調べた(図2B参照)。OHT処理されたT98Gmycer−2細胞において、TAF4bのmRNAのレベルは、3時間で2倍、7時間で3倍にそれぞれ上昇した。T98G親細胞のOHT処理は、TAF4bのmRNAのレベルを刺激しなかった。図2Cに示すように、T98Gmycer−2細胞におけるOHTによるTAF4bのmRNAの誘導は、タンパク質合成阻害剤シクロへキシミドの存在下で維持された。シキロヘキシミドによる処理は、T98G親細胞において、TAF4bのmRNAレベルにほとんど影響を及ぼさなかった(データ示さず)。これらの結果は、TAF4b遺伝子がMycの直接の標的となっていることを示すものである。
【0109】
c−MycはインビボでTAF4bプロモーターの領域に結合する
HL60細胞の増殖中において、生体内での内因性TAF4b遺伝子に結合するc−Mycタンパク質を調べるために、クロマチン免疫沈降を前述した方法(Boyd, K. E., et. al., Mol. Cell. Biol. 17:2529-2537.(1997),Boyd, K. E., et. al., Proc. Natl. Acad. Sci. U. S. A. 95:13887-13892.(1998)参照)により行った。免疫沈降後、各サンプルにおける内因性TAF4b遺伝子断片の強化を、TAF4b遺伝子における転写開始部位周辺のDNAを特異的に増幅させるプライマーを用いたPCRによってモニターした。図1Bに示すように、2つの異なる抗c−Myc抗体が、増殖期のHL60細胞由来のTAF4bDNA断片を免疫沈降した。一方、TPA処理された細胞からは、同じ抗体は検出可能なレベルのDNA断片を免疫沈降しなかった。なお、TPA処理された場合、c−mycの発現は非常に低くなった。非特異的抗体はTAF4b遺伝子断片を免疫沈降しないため、TAF4bゲノムDNA断片の強化は、TAF4b遺伝子に結合するc−Mycに依存する。さらに、c−Mycに対する抗体は、第22番染色体において検出可能な遺伝子を有さない染色体領域に位置するE−ボックスを含むゲノムDNA断片を強化しないため、TAF4b遺伝子において検出されるc−Mycの結合は特異的であるといえる。これらの結果から、転写開始部位周辺のTAF4b遺伝子が、HL60細胞の増殖期で、特異的にc−Mycに結合していることがわかる。
【0110】
c−Mycは、非基準型E−ボックス(CGCGTG)エレメントを介してTAF4bプロモーターを活性化する。
c−MycがどのDNAエレメントを介してTAF4bの発現を制御するかを確認するために、ヒトTAF4b遺伝子のプロモーター領域を単離した。図1Aでは、最初のATGにおける“A”を「+1」とした(星印で示す)。転写開始部位は−460bpであった。この位置は、5’−RACEによって決定し、図1Aにおいては矢じりで示した。TATAボックスの配列は、転写開始部位の約30bp上流付近で見つかった。
TAF4bプロモーターの活性を調べるために、エキソン1の上流領域からエキソン1の一部にかけての1.6kp領域を含むゲノムDNA断片を、ホタルルシフェラーゼcDNAに結合して、レポータープラスミドpTAF4b(W)luciを構築した(図3A参照)。T98Gmycer−2細胞における一過性発現解析により、前記DNA断片がプロモーター活性を有することが示された。OHTによってc−MycERを活性化した後、ルシフェラーゼ活性が2.5倍にまで上昇した(図3A)。この刺激は、T98Gmycer−2細胞においてOHTにより増加したTAF4bのmRNAレベルの増加で観察されたものに匹敵する(図2B参照)。
【0111】
転写開始部位付近には、2つの基準型E−ボックス部位(CACGTGエレメント)が存在する。この第1および第2のCACGTGエレメントを、E−ボックス1、E−ボックス2とそれぞれ命名した。CACGTGエレメントは、多くのMyc標的遺伝子においてMyc応答エレメントとして機能することが報告されていたため、当該E−ボックス部位がc−Mycによってプロモーターの活性化に関与するか否かを調べた。しかしながら、E−ボックス1およびE−ボックス2が変異した場合でも、ルシフェラーゼ活性は野生型レポータープラスミドと同様にMycによって依然として増加した(図3AのpTAF4b(mE1/2)luci)。
【0112】
次に、Myc刺激に応答可能なプロモーター領域を、欠失変異株を用いて調べた(図3A参照)。−1575番目から−535番目までの領域の欠失(pTAF4b(dSma)luci)は、Mycによるプロモーターの刺激を減少させなかったが、−1575番目から−367番目までのDNA断片の欠失(pTAF4b(dXho)luci)は、Myc刺激を消滅させた。この結果から、−534番目から−367番目までの領域が重要であることが示唆される。続いて、2つの追加レポーターを構成した。このレポーターは、−534番目から−360番目までの配列を含むもの(pTAF4b(Sma−Xho)luci)と、−534番目から−377番目までの配列を含むもの(pTAF4b(Sma−Pvu)luci)であった。これら2つのレポーターの発現は、Mycによって十分刺激された。
【0113】
応答部位を見つけるために、レポーター(pTAF4b(Sma−Pvu)luci)におけるDNA断片をさらに短くした(図3B参照)。−534番目と−492番目との間の領域の欠失(pTAF4b(117Pvu)luci)は、Mycによる上昇にほとんど影響を及ぼさないが、−534番目と−490番目との間の欠失(pTAF4b(114Pvu)luci)は、Mycによる上昇を消滅させた。さらに多くの部位を欠失させたレポーター(pTAF4b(102Pvu)luci、pTAF4b(73Pvu)luci、pTAF4b(43Pvu)luci)による発現も、Mycによって上昇しなかった。これらの結果は、−492番目から−490番目までの領域の重要性を示すものである。
【0114】
−492番目から−490番目までの領域は、非基準型E−ボックスエレメントCGCGTG(図1Aの−492番目から−487番目)内に存在した。CGCGTGエレメントが、Myc応答エレメントであるということはほとんど実証されていないが、c−Mycがこのエレメントに結合しうるということは文献(Grandori, C., et. al., Annu. Rev. Cell Dev. Biol. 16:653-699 (2000))に報告されている。そこで我々は、CGCGTGエレメントがMycの活性化に関与するか否かを実験した。TATAボックス配列のすぐ近くには2つのCGCGTGエレメントが存在する(図1Aおよび3C)。我々は、第1および第2のCGCGTGエレメントを、それぞれ非基準型E(NCE)−ボックス1(−509番目から−504番目)、NCE−ボックス2(−492番目から−487番目)と命名した。我々は、レポータープラスミドpTAF4b(Sma−Pvu、mNCE1)luci、pTAF4b(Sma−Pvu、mNCE2)luciおよびpTAF4b(Sma−Pvu、mNCE1/2)luciを作製した。これらのプラスミドにおいて、1個または2個のCGCGTGをCGCCTGに変異させた(図3C)。NCE−ボックス1の変異は、c−Mycによるプロモーター活性の刺激にほとんど影響を及ぼさないが、NCE−ボックス2を変異させた場合、あるいは、NCE−ボックス1およびNCE−ボックス2をともに変異させた場合には、Myc刺激をかなり減少させた(図3C参照)。これらの結果は、c−MycがNCE−ボックス2を介してTAF4bプロモーターをトランス活性化させることを示唆する。
【0115】
TAF4bプロモーターにおけるCGCGTGエレメントは、哺乳類の進化の過程で保存されている。
c−Mycが他の種においてTAF4bの発現を刺激するか否かを調べるために、ラットの細胞でTAF4bの発現に対するc−Mycの影響を調べた。図4Aに示すように、MycERキメラタンパク質を発現するラット3Y1線維芽細胞(Tsuneoka M. et. al., Oncogene 14:2301-2311(1997))において、TAF4bのmRNAレベルは、OHTによって3倍まで上昇した。この誘導は、タンパク質合成阻害剤シクロへキシミドの存在下で維持された。TAF4bはラットにおいてもMyc標的遺伝子であるということが、この結果から示唆される。
【0116】
我々は、プロモーター領域を含むラットTAF4bゲノムDNAの1.2kp断片を単離し、この断片についてシークエンスした。図4bは、ヒトおよびラットTAF4bプロモーター領域間のヌクレオチド配列の相違を示している。10bp間隔においた100bpのウインドウをスライドさせたり、重ねたりした際のヌクレオチドの相違数の平均を用いて、ヌクレオチド多様性を算出した。この解析において、挿入/欠失(indel)を一重変異として扱った。ヌクレオチド多様性の最小値、すなわち最大の相同性は、NCE−ボックス2(位置792)の周囲で観察された。低レベルのヌクレオチド多様性は、位置1000の周辺でも観察された。これは、上記の位置では、実際より小さく見積もられるヌクレオチド多様性を引き起こす大きな挿入/欠失(indel)がいくつか存在するという事実に起因するものである。ヒトTAF4bプロモーター配列を、そのDNA配列がアクセッション番号AC133172.4として最近ジーンバンク(GenBank)に登録されたマウスのTAF4bプロモーター配列と比較すると、似たような結果が得られた(データ示さず)。
【0117】
我々は、アフリカミドリサルの腎臓の線維芽細胞Cos−7からサルTAF4bプロモーターのDNA断片を単離し、それをシークエンスした。ヒト、サル、ラット、およびマウスのNCE−ボックス2周辺のヌクレオチド配列を、図4Cにおいて比較する。NCE−ボックス2およびTATAボックスエレメントを含む28bpの配列が4つの種において完全に保存されていた。
【0118】
CGCGTGおよびCACGTGエレメントは、異なる優先度でE−ボックス結合転写因子に応答する。
非基準型E−ボックス配列の保存は、生理学的な意義を有している可能性がある。NCE−ボックス2(CGCGTG)が基準型E−ボックスに変異した場合、プロモーター活性はc−Mycによって刺激され、野生型プロモーターの活性よりも上昇する(図5A参照)。
次に、Mycの基準型E−ボックスおよび非基準型E−ボックスに対する結合活性を調べた。TAF4bプロモーターの場合においてNCE−ボックス2の位置に基準型E−ボックスを含んでいる、放射標識されたCanoEヌクレオチドを使用したゲルシフトアッセイにおいて、MycとMaxとの結合はシフトしたバンドを生成した(図6A、レーン2参照)。このバンドは抗c−Myc抗体または抗Max抗体の存在下において減少し、スーパーシフトした(図6A、レーン1およびレーン3参照)。標識されていないCanoEオリゴヌクレオチドを過剰に添加した場合、シフトしたバンドの強度は減少した。一方、CACGTGがCGCCTGに変異した非標識のmEオリゴヌクレオチドを過剰に添加することによっては、バンドの強度はほとんど影響されなかった。この結果は、c−MycおよびMaxが、オリゴヌクレオチド内のCACGTGエレメントを特異的に認識することを示すものである。スキャッチャードプロット解析(図6C)によって解離定数を計算すると、24nMであった。
【0119】
基準型E−ボックス部位に対するMyc−Maxのアフィニティを、非基準型E−ボックス部位に対するMyc−Maxのアフィニティと比較するために、標識されていないCanoEオリゴヌクレオチドまたはnon−CanoEオリゴヌクレオチド(野生型E−ボックスCGCGTGを有する)が、反応混合物に対して過剰に加えられた。図5Dは、標識されていないCanoEオリゴヌクレオチドと比較して、約30倍以上の標識されていないnon−CanoEオリゴヌクレオチドが、標識されたCanoEオリゴヌクレオチドおよびMyc−Maxによって生成される特異的シグナルを50%に減少させるために必要であったことを示す。これらの結果から、TAF4bプロモーターにおいて、Myc−Maxは、CACGTGエレメントよりもCGCGTGエレメントに弱く結合することが示唆される。この弱い結合活性によって、Mycによる野生型TAF4bプロモーターの活性は緩やかに上昇すると思われる。
【0120】
mycプロトオンコジーンファミリーのタンパク質以外にも、USF(Gregor, P. D., et. al., Genes Dev. 4:1730-1740.(1990)参照)、TFE3(Beckmann, H., et. al., Genes Dev. 4:167-179.(1990)参照)を始めとする他のE−ボックス結合転写因子が存在する。次に、当該E−ボックス結合転写因子が、CACGTGエレメントおよびCGCGTGエレメントにどのように応答するのかについて調べた。図5Bおよび図5Cに示すように、野生型TAF4bプロモーターは、TFE3およびUSFによってほとんど刺激されなかった。これらの結果は、TAF4bプロモーターがMycに特異的に応答することを示唆する。CGCGTGエレメントが基準型(canonical)CACGTGに変異した場合、TAF4bプロモーターからの発現は、USFによって刺激されたが、TFE3は依然として当該レポーターからの発現をほとんど刺激しなかった。
【0121】
USFおよびTFE3のCanoEオリゴヌクレオチドに対する結合活性は、Myc−Maxの場合と同様の方法で決定された(図7参照)。CanoEオリゴヌクレオチドに結合するTFE3およびUSFのKd値は、それぞれ5.6nMおよび4.8nMであった。標識されていないCanoEオリゴヌクレオチドと比較して、約30倍の標識されていないnon−CanoEオリゴヌクレオチドが、標識されたCanoEオリゴヌクレオチドおよびUSFによって生成される特異的シグナルを50%に減少させるために必要とされることが確認された。これらの結果から、TAF4bプロモーターにおいて、USFとCGCGTGエレメントとの結合は、USFとCACGTGエレメントとの結合よりも弱いことが示唆される(図5F参照)。このことから、USFによって野生型TAF4bプロモーター活性は上昇しないことがわかった。
【0122】
TFE3の場合には、標識されたCanoEオリゴヌクレオチドおよびTFE3によって生成される特異的シグナルを50%に減少させるためには標識されていないnon−CanoEオリゴヌクレオチドは、標識されていないCanoEオリゴヌクレオチドの場合と比較して、3倍だけ必要であった。
【0123】
(考察)
TAF4b遺伝子は、Mycの直接の標的である。
我々は、c−MycがTAF4bの発現を直接誘導するということを実証した。HL60細胞において、TPA処理後のTAF4bのmRNAの発現は、c−mycの発現と密接に続いて起こった(図2A参照)。c−Mycがc−MycERキメラタンパク質で活性化されると、TAF4bのmRNAの発現の刺激が観察された(図2B参照)。この誘導は、タンパク質合成阻害剤の存在下でも維持され(図2C参照)、c−MycはTAF4bの発現を直接制御することが示された。c−Mycタンパク質は、増殖中のHL60細胞で、インビボでTAF4bプロモーターに結合した(図1B参照)。TAF4bプロモーター活性は、c−Mycによって上昇した(図3参照)。この活性の上昇は、進化の過程で保存された非基準型E−ボックス(NCE−ボックス2;CGCGTGエレメント)を介することによって起こる(図3および図4参照)。ゲルシフトアッセイでは、TAF4bプロモーターの場合、c−Mycタンパク質はNCE−ボックス2に結合することが示された(図5D参照)。これらの結果をまとめると、c−Mycは進化の過程で保存されたE−ボックス部位を介してTAF4bの発現を直接誘導するということがいえる。
【0124】
TAF4bを欠損したオスおよびメスのマウスは、生殖能力に顕著な異常を示す(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005), Freiman, R. N., et. al., Science 293:2084-2087.(2001)参照)。TAF4bを欠損するメスのマウスは、卵巣での卵胞形成に欠陥を有するために不妊である一方、野生型マウスの卵胞の顆粒膜細胞においては、TAF4bおよびc−mycの発現が見られた(Freiman, R. N., et. al., Science 293:2084-2087.(2001),Li, S., et. al., Endocr J. 41:83-92.(1994),Sato, A., et. al., Eur. J. Endocrinol. 131:319-322.(1994)参照)。成体の精巣において、TAF4bは生殖細胞の増殖に必要とされ(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005)参照)、TAF4bおよびc−mycの発現は減数分裂前の精原細胞において検出される(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005),Koji, T., et. al., Histochem. J. 20:551-557.(1998)参照)。TAF4bは細胞増殖の制御に関与する多くの遺伝子を誘導し得るため(Mengus, G., et. al., EMBO J. 24:2753-2767.(2005)参照)、上記の結果は、c−mycがTAF4b発現の刺激を介して、卵胞形成および精子形成中の細胞増殖に寄与する可能性を示唆する。
【0125】
E−ボックス内の配列:基準型(CACGTG)および非基準型(CGCGTG)エレメント
TAF4bプロモーターにおけるCGCGTGエレメント(NCE−ボックス2)は、c−MycによるTAF4b発現の誘導に必要である。この非基準型エレメントは進化の過程で保存されているが、これは、この配列が生理学的に重要であることを示すものである。CGCGTGエレメントが基準型CACGTGに変異した場合、c−MycのTAF4bプロモーターへの結合は強化され、c−Mycはプロモーター活性をより強くアップレギュレートした(図5参照)。これらの結果は、c−MycのE−ボックスへの結合活性がその転写活性化に直接影響を及ぼすということを示すものである。USFの場合も、同様の現象が観察された(以下参照)。
【0126】
本発明者は、別のE−ボックス結合転写因子であるUSFに関しても、E−ボックス部位における配列変化の影響を調査した。USFは野生型TAF4bプロモーターをほとんど刺激しない。しかしながら、NCE−ボックス2がCACGTGに変異すると、USFとTAF4bプロモーターとの結合はより強固なものとなり、USFはプロモーター活性をアップレギュレートし始めた(図5Cおよび図5F参照)。これらの結果は、非基準型CGCGTG E−ボックスがUSFによる制御を避けるために選択されている可能性を示唆する。
【0127】
マウスの精巣では、幹細胞系列でTAF4bは発現するが、体細胞であるセルトリ細胞ではTAF4bは発現しない(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005)参照)。セルトリ細胞では、USFは高度に発現しており、基準型E−ボックスエレメント(CACGTG)を介してステロイド産生因子1(Daggett, M. A., et. al., Biol. Reprod. 62:670-679.(2000)参照)および卵胞刺激ホルモンレセプター(Heckert, L. L., et. al., Mol. Endocrinol. 14:1836-1848.(2000):参照)の発現を誘導する。TAF4bプロモーターにおいてNCE−ボックス2が基準型であったとすれば、セルトリ細胞において、TAF4bの発現はUSFによって誘導されるであろう。それゆえ、TAF4bプロモーターにおけるCGCGTGエレメントの保存は、哺乳類の精巣においてTAF4bの適度な発現を支持するために必要であるかもしれない。
【0128】
TAF4bプロモーターの場合において、CACGTGエレメントに結合するc−Myc−Max、USF、およびTFE3のKd値は、それぞれ24nM、4.8nM、5.8nMであり、c−Mycは上記のタンパク質の中で最も効率的にTAF4bプロモーターを活性化する。それゆえ、応答するエレメントに対する転写因子の結合活性は、応答する遺伝子を単純に決定するものではない。TFE3のCACGTGエレメントに対する結合活性は、CGCGTGエレメントの結合活性と近似している。そして、TFE3は、CACGTGエレメントを有するTAF4bプロモーターならびにCGCGTGエレメントを有するプロモーターを活性化しなかった。しかしながら、上述したように、USFのE−ボックスエレメントに対する結合活性が上昇すると、結果としてUSFによるTAF4bプロモーターの活性化が起こる。これらの結果は、転写因子のその応答エレメントに対する結合活性の上昇が、応答遺伝子の範囲を拡大しうることを示唆する。
【0129】
TFE3のCGCGTGエレメントに対する結合活性は、CACGTGエレメントに対する結合活性と同等であった。一方、c−Myc−MaxおよびUSFのCGCGTGエレメントに対する結合活性は、CACGTGエレメントに対する結合活性に比べて非常に弱かった(図5参照)。これらの結果から、E−ボックス内の各配列は、E−ボックスに対するE−ボックス結合転写因子の結合活性にそれぞれ異なる影響を与えるということが示唆される。現在までのところ、Myc−Maxに対する非基準型(non-canonical)結合配列がいくつか報告されている(CACCTG、CATGCG、CACGCG、CAACGTG、およびCACGAG)(Grandori, C., et. al., Annu. Rev. Cell Dev. Biol. 16:653-699.(2000):参照)。これらのエレメントは、異なる結合活性でE−ボックス結合転写因子と異なって結合する。これらの違いは、E−ボックス結合転写因子に応答する遺伝子の領域(spectrum)に影響を与える可能性を有している。以上の結果は、E−ボックス転写因子が異なる標的遺伝子特異性を有するというメカニズムに、E−ボックス内の配列が寄与しているという証拠を初めて提供するものである。
【図面の簡単な説明】
【0130】
【図1A】図1Aは、ヒトTAF4bプロモーターおよびエキソン1の一部のゲノムDNA配列である。また、本図では、以前には報告していなかったN末端のアミノ酸配列を、DNA配列の下に一文字表記で示す。転写開始部位は矢印で示され、翻訳開始部位は星印で示されている。図中には、E−ボックス部位、E−ボックス1およびE−ボックス2(破線で囲まれた部分)、非基準型E(NCE)−ボックス部位、NCE−ボックス1およびNCE−ボックス2(下線を引いた部分)、TATAボックス配列(影付きの部分)、SmaI部位(CCCGGG)、PvuII部位(CAGCTG)、およびXhoI部位(CTCGAG)が示されている。
【図1B】図1Bは、抗c−Myc抗体を使用したクロマチン免疫沈降実験の結果を示す図である。増殖期のHL60細胞(−TPA)、および、10nM TPAで24時間処理されたHL60細胞(+TPA)を、1%ホルムアルデヒドで固定した後、核抽出物を集め、c−Mycに対する抗体(c−MycのN末端側と反応する抗c−Myc抗体1、c−MycのC末端側と反応する抗c−Myc抗体2)、又はコントロール抗体を使用して、あるいは抗体を使用せずに(no antibody)、クロマチンを免疫沈降した。TPAの存在下でc−myc発現は大きく低下した(図2A)。核抽出物を添加しないmock免疫沈降実験も実施された(nuclear extract -)。DNAを精製した後、試料を、TAF4bプロモーター領域のDNA断片を増幅するために設計されたプライマー(TAF4bプライマー)、または、第22番染色体のE−ボックスを含むDNA断片を特異的に増幅するためのプライマー(コントロールプライマー)を用いたPCRに付した(Bouchard, C., et. al., Genes Dev. 15:2042-2047.(2001):参照)。
【図2A】図2Aは、TPA処理後のヒト前骨髄性白血病HL60細胞におけるTAF4bおよびc−mycmRNAの変化を示す図である。RNAを10nM TPAによる処理が行われた後、指示された時点で単離し、ノーザンブロッティングで解析してTAF4bおよびc−mycのmRNAを検出した(左側の図参照)。28Sおよび18SのrRNAも検出した。この結果を定量し、プロットした(右側の図参照)。
【図2B】図2Bは、MycERタンパク質の活性化によって、T98G細胞中で増加したTAF4bのmRNAレベルを示す。T98Gmycer−2細胞(T98GMycER)および親細胞(T98G)を、0.25%血清を含む培地で40時間培養した後、200nM OHTの存在下(+)、あるいは非存在下(−)で培養した。RNAを指示された時点で単離し、TAF4bのmRNAを検出するためにノーザンブロッティングによる解析が行われた(左側の図参照)。28Sおよび18SのrRNAも示す。結果を定量し、棒グラフとして表す(右側の図参照)。
【図2C】図2Cは、タンパク質合成阻害剤の存在下で、MycERタンパク質の活性化によってTAF4bのmRNAレベルの増加が維持されたことを示す図である。T98Gmycer−2細胞を、0.25%血清を含む培地で40時間培養した後、200nM OHTで8時間処理した。RNAを単離し、TAF4bのmRNAを検出するためにノーザンブロッティングにより解析した(左側の図参照)。図中、CHXの列で「+」と示されたものは、OHT添加の20分前に20μg/mlのシクロヘキシミドが添加されたものである。28Sおよび18SのrRNAも示す。結果を定量し、棒グラフとして表す(右側の図参照)。
【図3A】図3Aは、レポーターアッセイの結果を示すグラフである。レポータープラスミド(pTAF4b(W)luci)を、TAF4bプロモーター活性を検出するために構築した。pTAF4b(mE1/2)luciにおいて、pTAF4b(W)luciの2つのE−ボックス(CACGTGエレメント)をCACCTGに変異させた(図1A参照)。また、pTAF4b(W)luci、pTAF4b(dSma)luci、pTAF4b(dXho)luci、pTAF4b(Sma−Xho)luci、およびpTAF4b(Sma−Pvu)luciの欠失変異体を作製した。T98Gmycer−2細胞をレポータープラスミドでトランスフェクトした。一日経過後、MycERキメラタンパク質を活性化させるために、最終濃度が200nMになるようにOHTを加え、細胞をさらに17時間培養した。細胞抽出物を、ホタルルシフェラーゼ活性について解析した。プロモーター活性は、MycER活性化なしの活性に対する割合として決定した。MycER活性化を介さないOHTの効果を規準化するために、T98Gmycer−2細胞の細胞数を、T98G親細胞の細胞数で除した。なお、T98G親細胞は、上述したT98Gmycer−2細胞と全く同じ手順で処理した。基本のプロモーターとして、何も挿入されていないpGL3プラスミドを用いた。数値は、4つの別個の実験の結果を平均したものである。バーは、標準誤差を示している。
【図3B】図3Bは、レポーターアッセイの結果を示すグラフである。pTAF4b(Sma−Pvu)luciの配列を欠失させ、pTAF4b(117Pvu)luci、pTAF4b(114Pvu)luci、pTAF4b(102Pvu)luci、pTAF4b(73Pvu)luci、およびpTAF4b(43Pvu)luciを作製した。Mycによるプロモーター活性化は、図3Aで説明したのと同様にして測定した。
【図3C】図3Cは、レポーターアッセイの結果を示すグラフである。pTAF4b(Sma−Pvu)luciのNCE−ボックス1およびNCE−ボックス2(図1A参照)について変異を施し、pTAF4b(Sma−Pvu mNCE1)luci、pTAF4b(Sma−Pvu mNCE2)luci、pTAF4b(Sma−Pvu mNCE1/2)luciを作製した。ここでは、1つまたは両方の非基準型E−ボックス(CGCGTGエレメント)を、CGCCTGに変異した。Mycによるプロモーター活性化は、図3Aで説明したのと同様にして測定した。
【図4A】図4Aは、MycERタンパク質の活性化によって、TAF4bのmRNA量がラットの線維芽細胞3Y1においてどの程度増加するかを示す図である。200nMのOHTで細胞を8時間処理した後、RNAを単離した。図中、CHXの列で「+」と示されたものは、OHT添加の20分前に20μg/mlのシクロヘキシミドが添加されたものである。得られたRNAをノーザンブロッティングで解析し、TAF4bのmRNAを検出した。ここでも、28Sおよび18SのrRNAも示す。
【図4B】図4Bは、ヒトとラットとの間でTAF4bプロモーター配列を比較したものである。この解析において、相同性が高い領域については、数値が低くなった(本文参照)。本図では、NCE−ボックス2(CGCGTG)およびTATAボックス配列(TATA)の位置を示している(図1A参照)。
【図4C】図4Cは、ヒト(HUM)、サル(MON)、ラット(RAT)、およびマウス(MOU)のTAF4bプロモーターにおいて、CGCGTGエレメント周辺の配列を比較したものである。NCE−ボックス2およびTATAボックス配列は、明るい影付きで示している。ヒトTAF4bプロモーターとは異なるヌクレオチドは、暗い影付きで示している。本図では、ヒトプロモーターのSmaI部位も示している。
【図5】図5は、TAF4bプロモーターの特性を示すグラフである。 図5A〜図5Cでは、TAF4bプロモーター活性に対するc−Myc、TFE3、およびUSFの影響を調べた。本図では、pTAF4b(Sma−Pvu)luciを(○)で、NCEW−ボックス2(CGCGTGエレメント)が基準型(CACGTG)に変異されたpTAF4b(Sma−Pvu CACGTG)luciを(●)で示している。T98G細胞を、トランスフェクション効率の基準化用のpRL/CMVとともにレポータープラスミド、および、pCMV/c−myc(A)、pCMV/TFE3(B)、又はpCMV/USF(C)でトランスフェクトした。ルシフェラーゼ活性は、エフェクタープラスミドが無い場合の発現量に対する割合で表す。図中、X軸はエフェクタープラスミドの量を示す。数値は、4つの別個の実験の結果を平均したものである。バー(the bars)は、基準誤差を示している。 図5D〜図5Fは、TAF4bプロモーターにおいて、非基準型(野生型)E−ボックスに対するMyc−Max(D)、TFE3(E)、またはUSF(F)の結合活性を、基準型E−ボックスに対するそれぞれの結合活性と比較した図である。組換えタンパク質を、32P標識されたCanoEオリゴヌクレオチドに、様々な量の非標識のCanoEオリゴヌクレオチド(●で示す)あるいはnon−CanoEオリゴヌクレオチド(野生型;○で示す)を加えたものとともにインキュベートし、その後、実施例ならびに図6で説明したゲルシフトアッセイにより解析した。non−CanoEオリゴヌクレオチドは、野生型TAF4bプロモーターから合成されたオリゴヌクレオチドであり、Myc応答性NCE−ボックス2を有するものであった。CanoEオリゴヌクレオチドにおいて、NCE−ボックス2を基準型CACGTGエレメントに変異させた。反応混合物中、標識されていないcompetitorの量は、標識されたオリゴヌクレオチドとの倍数として表す。
【図6】c−Myc/MaxとTAF4bプロモーターオリゴヌクレオチドとの結合を調べた図である。 図6Aは、c−Myc/Max複合体とのゲルシフトアッセイの結果を示す。組換えタンパク質を32P標識されたCanoEオリゴヌクレオチドとともにインキュベートし、ゲルシフトアッセイにより解析した。シフトしたバンドの特異性をテストするために、ゲルシフトアッセイの前に、ウサギ抗Max抗体(文献1:Beckmann, H., et. al., Genes Dev. 4:167-179.(1990):参照)、非特異的抗体(Bello-Fernandez, C., et. al., Proc. Natl. Acad. Sci. U. S. A. 90:7804-7808.(1993)参照)、あるいは抗c−Mycモノクローナル抗体(Bouchard, C., et. al., Genes Dev. 15:2042-2047.(2001)参照)を反応混合物に加えた。図には、シフトしたバンド(*)および抗体によってスーパーシフトしたバンド(<)が示されている。 図6Bでは、配列特異的結合をテストするために、32P標識されたCanoEオリゴヌクレオチドに、その30倍量(レーン3および5)、100倍量(レーン4および6)、300倍量(レーン7)過剰の非標識CanoEオリゴヌクレオチド(レーン3および4)あるいはCACGTGエレメントがCGCCTGに置き換わったmEオリゴヌクレオチド(レーン5、6、および7)が加えられたものとともに、タンパク質をインキュベートした。その後、上述の方法でゲルシフトによる解析が行われた。 図6Cは、Myc/Maxと32P標識されたCanoEオリゴヌクレオチドとの結合を示す。c−Myc/Max複合体を、反応混合物中で様々な濃度の32P標識されたCanoEオリゴヌクレオチドとともに室温でインキュベートした。次いで、前記混合物を電気泳動し(左側の図参照)、シフトした放射能の強度を定量した。この結果を、スキャッチャードプロットで解析した(右側の図参照)。
【図7】USFおよびTFE3とTAF4bプロモーターオリゴヌクレオチドとの結合を調べた図である。 図7Aでは、CanoEオリゴヌクレオチドに結合するUSFのゲルシフトアッセイを図6と同様にして実施した。シフトしたバンドの特異性をテストするために、抗USF抗体を反応混合物に加えた(レーン3)。配列特異的結合をテストするために、32P標識されたCanoEオリゴヌクレオチドに、その30倍量(レーン4および7)、100倍量(レーン5および8)、300倍量(レーン6および9)過剰の非標識CanoEオリゴヌクレオチド(レーン4、5、および6)あるいはmEオリゴヌクレオチド(レーン7、8、および9)が加えられたものとともに、タンパク質をインキュベートした。 図7Bでは、CanoEオリゴヌクレオチドに対するUSFの結合を解析するため、スキャッチャードプロット解析を図6と同様にして実施した。 図7Cは、CanoEオリゴヌクレオチドに結合するTFE3のゲルシフトアッセイを示す。シフトしたバンドの特異性をテストするために、抗TFE3抗体(レーン3)またはコントロールの抗体をインキュベーション混合物に加えた。配列特異的結合をテストするために、32P標識されたCanoEオリゴヌクレオチドに、その30倍量(レーン5および7)、100倍量(レーン6および8)、300倍量(レーン9)過剰の非標識CanoEオリゴヌクレオチド(レーン5および6)またはmEオリゴヌクレオチド(レーン7、8、および9)が加えられたものとともに、タンパク質をインキュベートした。 図7Dでは、CanoEオリゴヌクレオチドに対するTFE3の結合を解析するために、スキャッチャードプロット解析が図6と同様にして実施された。
【図8】図8は、TAF4bプロモーター領域とその活性および推定される発現上昇因子の結合部位を示す図である。
【技術分野】
【0001】
本発明は、TAF4bプロモーターおよびその利用に関する。詳しくは、組織特異的な転写調節または転写増強が可能なヌクレオチドおよび真核生物の様々な事象の解明に寄与する当該ヌクレオチドの利用などに関する。
【背景技術】
【0002】
真核細胞(真核生物)において、タンパク質をコードする遺伝子はRNAポリメラーゼII(RNA Pol II)によって転写される。特定の遺伝子の転写を開始するためにRNA Pol IIを導く分子機構は、調節タンパク質の複数のクラスから構成される(非特許文献1)。TFIIDは、RNA Pol II制御機構の中核となる要素であり、酵母(Saccharomyces cerevisiae)からヒトに至るまで保存されており、TATAボックス結合タンパク質(TBP)と複数のTBP関連因子(TAFs)からなる大きな多量体タンパク質複合体である。TFIIDは、初期の頃には発現および機能において遍在性(ユビキタス)であると考えられていたが、組織特異的TAFsの同定は、特殊化したTFIID複合体が遺伝子発現の組織特異的プログラムの調節において直接的な役割を果たしうるということを示唆した。同定された最初の細胞型特異的TFIIDサブユニットは、TAF4b(以前はTAFII105と呼ばれていた)であった(非特許文献2)。TAF4bは、広範囲に発現される因子であるTAF4(以前はTAFII130と呼ばれていた)のホモロガスタンパク質である。TAF4bの細胞型特異的機能は、TAF4bがマウスの精巣および卵巣で高度に発現しており、TAF4bを欠損したマウスは、生存可能であるがオス・メスともに不妊である(非特許文献3、4)という発見によって支持された。
【0003】
一次Bリンパ球が細菌のリポ多糖によって活性化された場合、TAF4ではなくTAF4bの発現が増加した(非特許文献5)。最近では、TAF4およびTAF4bを含むTFIID複合体が別個の標的遺伝子特異性を有しているということが報告されている。TAF4を不活性化すると、多くの一連の遺伝子の発現に影響を及ぼした。これらの中で、TAF4bの不活性化は、TGFβ1、TGFβ3、CTGF、ITGα6およびHB−EGFを含む多くの成長因子を誘導した。これらの成長因子は、TAF4bの過剰発現によっても誘導された。これらの結果は、TAF4とTAF4bとの平衡が細胞増殖の制御に関連する遺伝子の調節に作用しているということ(非特許文献6)を示唆しているとともに、TAF4bの発現は厳密に制御されるべきであることを示している。しかしながら、TAF4b発現の制御メカニズムは、未だ解明されていない。
【0004】
プロトオンコジーンのmycファミリーは、3つの主要な遺伝子:c−myc、N−mycおよびL−mycから構成されている(非特許文献7、8、9)。この3つの遺伝子は、異なる発現パターンを示すが、基本的には同じ生物学的活性を有しているように見える。c−mycまたはN−mycを欠損したマウスの胎児は、多臓器発育不全になり、胚形成の中間の段階で死ぬ(非特許文献10、11)。c−mycの発現が一定量減少する対立形質を有する一連のマウスを作製した場合、in vivoでのc−mycレベルの減少は、多臓器発育不全が原因となって、c−mycのレベルに依存してマウスの体重を減少させる(非特許文献12)。これらの結果は、Mycの量が細胞増殖に直接的に関与していることを示すものである。これらの蓄積されたデータは、mycファミリー遺伝子が細胞増殖の調節因子の中心となっていることを示している。
【0005】
mycがコードするタンパク質は、塩基性へリックス・ループ・へリックス−ロイシンジッパー転写因子のメンバーである(非特許文献8、13)。Mycタンパク質とその必須パートナーMaxタンパク質との二量体化が起こると、E−ボックス部位に結合するヘテロ二量体が形成される。このE−ボックス部位は、通常は基準的なもの(CACGTG)であるが、まれにはCGCGTGを含む非基準的なものである。Myc−Maxヘテロ二量体は、オルニチンデカルボキシラーゼ(非特許文献14)、RCC1(非特許文献15)、ヌクレオリン(非特許文献16)、サイクリンD2(非特許文献17)、mina53(非特許文献18)、およびmimitin(非特許文献19)を含む、種々の遺伝子の転写を活性化する。mycに関しては、その研究に多大な努力がなされているが、いまだなぞのままであり、Mycによって制御されるさらなる遺伝子に関する情報は、mycの機能の解明に役立てることができる。
【0006】
mycプロトオンコジーンファミリーのタンパク質以外にも、USF(非特許文献20)およびTFE3(非特許文献21)を含むE−ボックス結合転写因子の他のクラスが存在する。これらの因子も、E−ボックスを介して遺伝子発現をトランスに活性化するが、これらの生物学的な機能およびこれらに対応する遺伝子は、Mycとは異なっている。しかしながら、それぞれのE−ボックス結合転写因子におけるこれらの相違を創り出すメカニズムは、未だ明らかになっていない。
【非特許文献1】Lemon, B. et al., Genes Dev. 14:2551-2569, 2000
【非特許文献2】Dikstein, R. et al., Cell 87:137-146, 1996
【非特許文献3】Falender, A. E. et al., Genes Dev. 19:794-803, 2005
【非特許文献4】Freiman, R. N. et al., Science 293:2084-2087, 2001
【非特許文献5】Freiman, R. N.et al., Mol. Cell. Biol. 22:6564-6572, 2002
【非特許文献6】Mengus, G. et al., EMBO J. 24:2753-2767, 2005
【非特許文献7】DePinho, R. A. et al., Adv. Cancer Res. 57:1-46, 1991
【非特許文献8】Grandori, C. et al., Annu. Rev. Cell Dev. Biol. 16:653-699, 2000
【非特許文献9】Henriksson, M. et al., Adv. Cancer Res. 68:109-182, 1996
【非特許文献10】Davis, A. C. et al., Genes Dev. 7:671-682, 1993
【非特許文献11】Stanton, B. R. et al., Genes Dev. 6:2235-2247, 1992
【非特許文献12】Trumpp, A. et al., Nature 414:768-773, 2001
【非特許文献13】Luscher, B. et al., Gene 277:1-14, 2001
【非特許文献14】Bello-Fernandez, C. et al., Proc. Natl. Acad. Sci. U. S. A. 90:7804-7808, 1993
【非特許文献15】Tsuneoka, M. et al., Oncogene 14:2301-2311, 1997
【非特許文献16】Greasley, P. J. et al., Nucleic Acids Res. 28:446-453, 2000
【非特許文献17】Bouchard, C. et al., Genes Dev. 15:2042-2047, 2001
【非特許文献18】Tsuneoka, M. et al., J. Biol. Chem. 277:35450-35459, 2002
【非特許文献19】Tsuneoka, M. et al., J. Biol. Chem. 280:19977-19985, 2005
【非特許文献20】Gregor, P. D. et al., Genes Dev. 4:1730-1740, 1990
【非特許文献21】Beckmann, H. et al., Genes Dev. 4:167-179, 1990
【発明の開示】
【発明が解決しようとする課題】
【0007】
本発明の目的は、細胞増殖因子かつ転写因子であるMycの直接の標的およびそれを用いた真核生物の組織特異的事象の調節手段の提供である。
【課題を解決するための手段】
【0008】
本発明者は、上記観点から鋭意検討した結果、Mycの直接の標的となる核酸配列および当該配列中の必須配列を見出し、本発明を完成するに至った。即ち、本願発明は、以下に示す通りである。
〔1〕 Mycによる組織特異的発現を調節するためのヌクレオチドであって、TATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフおよび/またはCACGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチド。
〔2〕 前記ヌクレオチドが
(a)配列番号3に記載の塩基配列、
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列、
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列、
(g)配列番号3の1081〜1126位の領域の塩基配列を必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列および/または当該必須配列の下流に1127〜1157位の領域の塩基配列が配置されてなる塩基配列を含む塩基配列、または
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列、
からなるものである、前記〔1〕記載のヌクレオチド。
〔3〕 (a)配列番号4に記載の塩基配列、または
(b)配列番号4に記載の塩基配列において、51位〜57位のTATAボックスおよび43〜48位のCACGTGモチーフ以外の塩基配列と70%以上の同一性を有し、かつTATAボックスとその1〜50塩基上流に位置するCACGTGモチーフとを含む塩基配列
を含有するものである、前記〔1〕に記載のヌクレオチド。
〔4〕 前記CGCGTGモチーフおよび/またはCACGTGモチーフが、下記:
(i)塩基配列GTGにおける少なくとも1塩基の置換、
(ii)塩基配列CACにおける少なくとも1塩基の置換もしくは挿入
のいずれかまたは両方の変異を有するものである、前記〔1〕〜〔3〕いずれか1項に記載のヌクレオチド。
〔5〕 前記〔1〕〜〔4〕いずれか1項に記載のヌクレオチドを含有してなるベクター。
〔6〕 前記〔1〕〜〔4〕いずれか1項に記載のヌクレオチドおよびレポーター遺伝子を含有してなる、レポータープラスミド。
〔7〕 前記〔5〕に記載のベクターまたは前記〔6〕に記載のレポータープラスミドを導入してなる形質転換体。
〔8〕 前記〔5〕に記載のベクターまたは前記〔6〕に記載のレポータープラスミドを導入してなるトランスジェニック非ヒト動物。
〔9〕 前記〔6〕に記載のレポータープラスミドまたは前記〔7〕に記載の形質転換体を用いることを特徴とする、細胞増殖の調節剤をスクリーニングする方法。
〔10〕 前記〔6〕に記載のレポータープラスミド、前記〔7〕に記載の形質転換体または前記〔8〕に記載のトランスジェニック非ヒト動物を用いることを特徴とする、妊娠調節剤をスクリーニングする方法。
〔11〕 Mycによる組織特異的発現の調節剤であって、下記いずれかのペプチドを有効成分として含有する調節剤:
(1)配列番号1に記載の塩基配列からコードされるペプチド、
(2)配列番号2に記載のアミノ酸配列からなるペプチド、
(3)前記(1)または(2)に記載のペプチドと実質的に同一のアミノ酸配列からなるペプチド。
〔12〕 妊娠の調節に用いられる前記〔11〕に記載の調節剤。
〔13〕 (a)配列番号5に記載の塩基配列、または
(b)配列番号5に記載の塩基配列において210位〜216位のTATAボックスおよび202〜207位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号5に記載の塩基配列と実質的に同一な塩基配列
を含有するサルTAF4bプロモーター。
〔14〕 (a)配列番号6に記載の塩基配列、または
(b)配列番号6に記載の塩基配列において778位〜784位のTATAボックスおよび770〜775位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号6に記載の塩基配列と実質的に同一な塩基配列
を含有するラットTAF4bプロモーター。
【発明の効果】
【0009】
本発明のヌクレオチドによると、Mycによる組織特異的発現を調節するために必要十分な塩基配列を提供することが可能となり、真核生物におけるMycを介した転写のさらなる解明のみならず、細胞増殖や組織特異的な転写制御の調節に寄与することができる。本発明のベクターによると、本発明のヌクレオチドを様々な用途に供する際に便利なツールを提供することができる。本発明のレポータープラスミドによると、Mycの活性を測るための新しいツールとして利用することができる。
【0010】
本発明のベクター、レポータープラスミド、形質転換体およびトランスジェニック非ヒト動物によると、真核生物におけるMycを介した転写のさらなる解明ならびに細胞増殖や組織特異的な転写制御の調節をインビトロまたはインビボで調べるためのツールを提供することができる。本発明のスクリーニング方法によると、細胞増殖のシグナル伝達の中核に位置するMycの活性を調節可能な細胞増殖調節剤を簡便および高感度に選別することができる。また、本発明のスクリーニング方法によると、今までに知られていない作用機序を有する妊娠調節剤を選別することができる。
【0011】
本発明のMycによる組織特異的発現の調節剤によると、Mycが介在する真核生物の発生および維持に関する様々な事象の調節に用いることができ、好ましくは妊娠の調節に用いられうる。本発明のサルまたはラットTAF4bプロモーターによると、サルまたはラット等の実験動物を用いた転写調節の研究のみならず、哺乳動物を始めとする多細胞生物全体の転写調節の解明にも寄与することができる。
【発明を実施するための最良の形態】
【0012】
本発明は、Mycによる組織特異的発現を調節するためのヌクレオチドを提供する。前記ヌクレオチドは、TATAボックスとその1〜50塩基上流に位置するCACGTGモチーフおよび/またはCGCGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチドである。
【0013】
本発明において「Mycによる組織特異的発現」とは、転写因子であるMycが特定の塩基配列を標的とすることにより、真核生物の特定の組織で当該特定の塩基配列の下流に位置する遺伝子が転写されることをいう。
【0014】
本発明におけるMycとは、c−Myc、N−MycおよびL−Mycを包含するものであり、特に限定されるものではないが、種々の癌において遺伝子増幅、過剰発現や転座が報告されているc−Mycを中心に説明する。
【0015】
本発明における組織とは、真核生物を構成するものである限り特に限定されるものではないが、例えば、脳、神経、骨髄、骨、筋肉、脂肪、心臓、肺、食道、胃、腸、腎臓、副腎、肝臓、膵臓、リンパ節、リンパ管、胸腺、脾臓、血管、血球、臍帯、皮膚、感覚器官、およびその前駆体、生殖組織、幹細胞などが好ましい。また、前記組織中に含まれる細胞、組織中から単離された細胞、これら細胞から樹立した細胞株も組織に含まれる。
【0016】
本発明において「調節」とは、本発明のヌクレオチドの下流に位置する遺伝子の転写の開始、転写増強、転写抑制などをいう。
【0017】
本発明のヌクレオチドとは、その下流に位置する遺伝子の転写開始部位を決定し、その頻度を直接的に調節する核酸をいい、通常プロモーターと称される用語と同義である。
【0018】
本発明のヌクレオチドは、具体的には、TATAボックスとその1〜50塩基上流に位置するCACGTGモチーフおよび/またはCGCGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するものであり、本発明によって初めて解明された、Mycの直接の標的である。
【0019】
「TAF4bプロモーターに由来する」とは、TAF4bとして公知の、例えばGenBank Accession No.XM_290809.4のヒトTAF4b中のプロモーター領域の塩基配列そのものであってもよく、本発明の目的を達成しうるプロモーター活性を有する限りTAF4bプロモーター領域内の塩基配列が改変されていてもよい。プロモーター活性は、後述するレポーターアッセイによりレポーターの発現強度を指標にして調べることができる。
【0020】
本発明のヌクレオチドの必須配列である「TATAボックス」とは、真核細胞のmRNAをコードする遺伝子の転写開始位置より約20〜80塩基上流にあって、通常「TATAT(A)AA(T)」と一般化されているDNA配列をいう。
【0021】
本発明のヌクレオチドの必須配列である「CACGTG」モチーフまたは「CGCGTG」モチーフとは、Mycが直接結合する部位のコンセンサス配列であり、本発明によりMycによる組織特異的発現がこれらのモチーフの使い分けにより調節されていることを初めて解明したものである。
【0022】
本発明のヌクレオチド中に含まれる前記CACGTGモチーフおよび/またはCGCGTGモチーフは、いずれかのモチーフ1つのみでもよく、同じ配列のモチーフが2つでもよく、異なる配列のモチーフが1つずつでもよい。異なる配列のモチーフを含む場合、モチーフ間の上流、下流の関係は特に限定されるものではない。これらのモチーフの位置関係は、前記TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではない。
【0023】
別の側面によると、本発明のヌクレオチドは、
(a)配列番号3に記載の塩基配列、
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列、
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列、
(g)配列番号3の1081〜1126位の領域の塩基配列を必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列および/または当該必須配列の下流に1127〜1157位の領域の塩基配列が配置されてなる塩基配列を含む塩基配列、または
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列、
からなるものであることが好ましい。
【0024】
同一性(%)は、当該分野で慣用のホモロジー検索プログラム(例えば、BLAST、FASTA等)を初期設定で用いて決定することができる。本発明において、塩基配列の「同一性」とは、比較する2種の塩基配列を整列(アラインメント)させ、整列により一致した塩基配列の数を基準となる塩基配列の総数で除して算出した割合を%で示した数字である。なお、整列により生じたギャップは、不一致と見なして算出する。
【0025】
(a)配列番号3に記載の塩基配列
本発明によりMycの直接の標的となる配列としてクローニングされた塩基配列である。
【0026】
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列
ここで「実質的に同一」とは、1090〜1096位のTATAボックスと1082〜1087位のCGCGTGモチーフとがプロモーターの構成要素として機能する限り、それ以外の塩基配列は特に限定されないことを意味する。「実質的に同一」か否かの判断は、後述するレポーターアッセイによる前記(a)の塩基配列と(b)の塩基配列との間のレポーターの発現量を比較し、同程度の発現量であることを指標にすればよい。
【0027】
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列
前記TATAボックスおよびCGCGTGモチーフを必須の塩基配列として含み、配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。前記必須配列は、配列番号3の1040〜1126位の領域と対応する塩基配列中に含まれていることが好ましく、対応する塩基配列中に含まれていない場合は、対応配列外に機能可能に連結される。CGCGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0028】
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列
前記TATAボックスおよびCGCGTGモチーフを必須の塩基配列として含み、配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。前記必須配列は、配列番号3の1082〜1156位の領域と対応する塩基配列中に含まれていることが好ましく、対応する塩基配列中に含まれていない場合は、対応配列外に機能可能に連結される。CGCGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0029】
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列
前記TATAボックスおよびCGCGTGモチーフを必須の塩基配列として含み、配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。前記必須配列は、配列番号3の1040〜1120位の領域と対応する塩基配列中に含まれていることが好ましく、対応する塩基配列中に含まれていない場合は、対応配列外に機能可能に連結される。CGCGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0030】
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列
配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置した塩基配列である。
【0031】
(g)配列番号3の1081〜1126位の領域の塩基配列Bを必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列Aおよび/または当該必須配列の下流に1127〜1157位の領域の塩基配列Cが配置されてなる塩基配列を含む塩基配列
ここでは、前記塩基配列Bを必須とするものであり、塩基配列AB(左から右に5’→3’と連結される、以下同様)、塩基配列BC、塩基配列ABCの3通りの組合せがあげられる。すなわち、塩基配列Bは必須であるがこの塩基配列のみではMycによる組織特異的発現が十分ではなく、発現を上昇させる目的には、塩基配列ABまたはBCが好ましく、塩基配列ABCがより好ましい。
【0032】
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列
当該断片は、前記(g)の項の塩基配列AB、塩基配列BC、塩基配列ABCに対応するものであり、これらの塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列を含む塩基配列である。
【0033】
Mycによる組織特異的発現を増強する目的に使用する場合、本発明のヌクレオチドは、
(a)配列番号4に記載の塩基配列、または
(b)配列番号4に記載の塩基配列において、51位〜57位のTATAボックスおよび43〜48位のCACGTGモチーフ以外の塩基配列と70%以上の同一性を有し、かつTATAボックスとその1〜50塩基上流に位置するCACGTGモチーフとを含む塩基配列
を含有することが好ましい。
【0034】
前記(a)は、本発明により、Mycの直接の標的となる配列の中でもMycによって発現がさらに増強されうる配列として見出された塩基配列である。したがって、当該配列は、Mycの存在量を高感度に検出するプロモーターとなりうる。
【0035】
前記(b)は、TATAボックスおよびCACGTGモチーフを必須の塩基配列として含み、必須配列以外の塩基配列と70%以上の同一性、好ましくは75%以上、より好ましくは80%以上、さらにより好ましくは85%以上、さらにいっそうより好ましくは90%以上、最も好ましくは95%以上の同一性を有する塩基配列からなるものである。CACGTGモチーフは、TATAボックスの1〜50塩基上流に位置する限り特に限定されるものではなく、モチーフの数は1つまたは2つでもよい。
【0036】
本発明のヌクレオチド中に含まれる前記CGCGTGモチーフおよび/またはCACGTGモチーフは、本発明のヌクレオチドをプロモーターとして用いた場合に、当該モチーフ内の塩基を変異させることによりMycによる発現の程度を維持または抑制することができる。具体的には前記モチーフは、下記:
(i)塩基配列GTGにおける少なくとも1塩基の置換、
(ii)塩基配列CACにおける少なくとも1塩基の置換もしくは挿入
のいずれかまたは両方の変異を有するものである場合、変異前のMycによる発現を同等またはそれ以下に調節することができる。変異モチーフの具体例としては、CACCTG、CATGCG、CACGCG、CAACGTG、CACGAGモチーフなどがあげられる。
【0037】
本発明のヌクレオチドを細胞に導入する場合、ベクターの形で導入されることが好ましい。本発明は、前記ヌクレオチドを含有するベクターを提供する。
【0038】
基本骨格となるベクターは特に限定されず、形質転換を行う細胞(例えば、大腸菌)中で自己複製可能なものであればよい。例えば、市販のpBR322、pUC、pBluescript、pGL2、pGL3、pGL4 (プロメガ社)等が使用可能である。
【0039】
本発明のベクターは、本発明のヌクレオチドを自体公知の方法により前記基本骨格となるベクターにクローニングすることにより得られる。このようにして得られたベクターは、シークエンス等により所望の位置に所望の方向で本発明のヌクレオチドがクローニングされていることを確認することができる。
【0040】
本発明のヌクレオチドのプロモーター活性を調べるためには、レポーター遺伝子を用いることが好ましい。本発明は、本発明のヌクレオチドおよびレポーター遺伝子を含有するレポータープラスミドを提供する。
【0041】
ここで、レポーター遺伝子とは、本発明のヌクレオチドのプロモーター活性を調べるために組み込まれる目印用の遺伝子をいい、公知のあらゆるレポーター遺伝子を制限なく用いることができる。検出が簡単で定量化も可能であるという観点から、レポーター遺伝子は発光タンパク質遺伝子または蛍光タンパク質遺伝子が好ましい。
【0042】
前記発光タンパク質としては、ホタル由来のルシフェラーゼ、Renilla(ウミシイタケ)由来のルシフェラーゼなどがあげられる。これら発光タンパク質および当該タンパク質をコードする核酸は、公知である。
【0043】
前記蛍光タンパク質としては、GFP、YFP、CFP、BFP、Venusなどのオワンクラゲ由来蛍光タンパク質およびそれらの類似体、ウミシイタケ由来蛍光タンパク質およびその類似体、DsRed、HcRed、AsRed、ZsGreen、ZsYellow、AmCyan、AcGFP、Kaedeなどのサンゴ由来蛍光タンパク質およびそれらの類似体などがあげられる。これら蛍光タンパク質およびそれらの類似体ならびに当該タンパク質をコードする核酸は、公知である。
【0044】
前記レポーター遺伝子は、公知の塩基配列に基づいて、常法により調製することができる。
【0045】
本発明のレポータープラスミドにおいて、構成要素である本発明のヌクレオチドおよびレポーター遺伝子は、直接結合していてもよく、本発明の目的を達成しうる限りにおいて、任意の塩基配列が挿入されていてもよい。かかる挿入配列としては、例えば、クローニングの過程で付加される制限酵素切断部位から生じる塩基配列などがあげられ、通常、1〜1000塩基程度の長さを有するものである。
【0046】
本発明のレポータープラスミドの基本骨格となるプラスミドは特に限定されず、形質転換を行う細胞(例えば、大腸菌)中で自己複製可能なものであればよい。例えば、市販のpBR322、pUC、pBluescript、pGL2、pGL3、pGL4(プロメガ社)等が使用可能である。
【0047】
本発明のレポータープラスミドは、前記ヌクレオチドおよび前記レポーター遺伝子を自体公知の方法により前記基本骨格となるプラスミドにクローニングすることにより得られる。このようにして得られたレポータープラスミドは、シークエンス等によりヌクレオチドおよびレポーター遺伝子が所望の位置に所望の方向でクローニングされていることを確認することができる。
【0048】
このようにして構築された本発明のベクターまたはレポータープラスミドを用いて、形質転換体を製造することができる。本発明は、かかる形質転換体を提供する。
【0049】
宿主としては、例えば、エシェリヒア属菌、バチルス属菌、酵母、動物細胞などが用いられる。エシェリヒア属菌は、本発明のヌクレオチド、ベクターまたはレポータープラスミドを調製するためにもっぱら用いられ、その他の宿主は本発明のヌクレオチドの活性等を調べるために用いられる。
エシェリヒア属菌としては、エシェリヒア・コリ(Escherichia coli)K12・DH1、JM103、JA221、HB101、C600などが用いられる。
バチルス属菌としては、例えば、バチルス・ズブチルス(Bacillus subtilis)MI114などが用いられる。
酵母としては、例えば、サッカロマイセス セレビシエ(Saccharomyces cerevisiae)AH22,AH22R−,NA87−11A,DKD−5D,20B−12、シゾサッカロマイセス ポンベ(Schizosaccharomyces pombe)NCYC1913,NCYC2036、ピキア パストリス(Pichia pastoris)などが用いられる。
動物細胞としては、例えば、サル細胞COS−7、Vero、チャイニーズハムスター細胞CHO、dhfr遺伝子欠損チャイニーズハムスター細胞CHO(CHO(dhfr−))、マウスL細胞、マウスAtT−20、マウスミエローマ細胞、ラットGH3、ヒトFL細胞などが用いられる。
【0050】
エシェリヒア属菌を形質転換するには、例えば、Proc. Natl. Acad. Sci. USA),69巻,2110(1972)やGene,17巻,107(1982)などに記載の方法に従って行なうことができる。
バチルス属菌を形質転換するには、例えば、Molecular & General Genetics,168巻,111(1979)などに記載の方法に従って行なうことができる。
酵母を形質転換するには、例えば、Methods in Enzymology,194巻,182−187(1991)、Proc. Natl. Acad. Sci. USA,75巻,1929(1978)などに記載の方法に従って行なうことができる。
動物細胞を形質転換するには、例えば、細胞工学別冊8新細胞工学実験プロトコール.263−267(1995)(秀潤社発行)、Virology,52巻,456(1973)に記載の方法に従って行なうことができる。
【0051】
前記ベクターまたはレポータープラスミド(以下、「ベクター等」と略す場合がある)を導入することにより、トランスジェニック非ヒト動物を作出することができる。本発明は、かかるトランスジェニック非ヒト動物を提供する。
【0052】
本発明のトランスジェニック非ヒト動物には、前記ヌクレオチドおよび場合によってはレポーター遺伝子が対立遺伝子の両方に導入されたホモ接合動物、対立遺伝子の片方に導入されたヘテロ接合動物およびそれらの出生前の胎仔も含まれる。前記ホモ接合動物は、前記ヘテロ接合動物を交配することにより得られるものである。
【0053】
用いられる非ヒト動物(本発明においては、単に動物と略す場合がある。)としては、ヒト以外の動物であれば特に限定されるものではない。好適な動物としては、マウス、ラット、ハムスター、モルモット、ウサギ、イヌ、ネコ、サル等の実験動物ならびにウシ、ヒツジ、ウマ、ブタ等の家畜があげられるが、遺伝子工学的に利用が容易であるところから、マウスがより好ましい。
【0054】
本発明のトランスジェニック非ヒト動物は、自体公知の方法により製造することができる。次に、本発明のトランスジェニック非ヒト動物の作出方法について説明する。
【0055】
本発明のトランスジェニック非ヒト動物は、例えば下記の工程(a)〜(c)を含む方法により製造することができる。
(a)前記ベクター等を調製する工程;
(b)前記ベクター等を受精卵に導入し、遺伝子導入受精卵を仮親動物に移植する工程;
(c)前記動物から出生した子孫からトランスジェニック動物を選別する工程;および
(d)前記選別した動物(ファウンダー)から系統を樹立する工程。
【0056】
前記工程(a)は、本発明のベクターまたはレポータープラスミドにおいて記載した通りである。得られたベクター等は、制限酵素等により線状化して工程(b)に供することが好ましい。
【0057】
前記工程(b)において、前記ベクター等を受精卵に導入する方法は、例えば、交配後の雌の卵管を洗浄して受精卵を採取し、精子または卵子由来の前核にマイクロインジェクション法により前記ベクター等を直接注入する方法があげられる。この受精卵を偽妊娠させた仮親の輸卵管に移植し、子宮内で発生を続けさせる。
【0058】
前記工程(c)において、工程(b)で移植した動物が出生した子孫からトランスジェニック動物を選別する方法としては、例えば、注入したベクター等が染色体DNAに組み込まれているか否かについて、産仔の尾部等より分離抽出した染色体DNAをサザンブロット法またはPCR法によりレポーター遺伝子の組み込みを確認する方法があげられる。レポーター遺伝子が蛍光タンパク質遺伝子の場合、レポーター遺伝子が発現している生体の部位に紫外線等を照射して蛍光を調べることにより確認することができる。
【0059】
前記工程(d)において、工程(c)で選別した動物(ファウンダー)から遺伝的背景の均一な系統を樹立する方法としては、ベクター等が組み込まれた動物とC57BL/6、FVB、BALB/cなどの近交系の野生型動物とを戻し交配をする方法があげられる。
【0060】
このようにして得られた本発明のトランスジェニック非ヒト動物をさらに交配して得られる子孫動物、これら動物に由来する組織または細胞も本発明に含まれる。前記組織としては、すべての組織があげられ、例えば、脳、神経、骨髄、骨、筋肉、脂肪、心臓、肺、食道、胃、腸、腎臓、副腎、肝臓、膵臓、リンパ節、リンパ管、胸腺、脾臓、血管、血球、臍帯、皮膚、感覚器官およびその前駆体、生殖組織、幹細胞などが好ましい。また、前記細胞としては、前記組織中に含まれる細胞、組織中から単離された細胞、これら細胞から樹立した細胞株があげられる。
【0061】
本発明のトランスジェニック非ヒト動物は、本発明のヌクレオチド(TAF4bプロモーターに由来する塩基配列)によってレポータータンパク質の発現が調節されているものが好ましい。本発明のトランスジェニック非ヒト動物は、Mycに感受性が高い状態で、様々な用途に利用することができる。好ましくは、本発明のトランスジェニック非ヒト動物は、生体内で代謝された後の物質の生体中の各組織への影響を調べるものとして使用される。
【0062】
本発明は、前記レポータープラスミドまたは前記形質転換体を用いることを特徴とする、細胞増殖の調節剤をスクリーニングする方法(スクリーニング方法I)を提供する。
【0063】
[スクリーニング方法I]
本発明のスクリーニング方法Iは、具体的には、下記工程を含む:
(a)レポータープラスミド導入細胞または形質転換体と被験物質とを接触させる工程、
(b)前記被験物質を接触させた細胞または形質転換体におけるレポーター遺伝子の発現量を調べ、被験物質を接触させない細胞または形質転換体における発現量と比較する工程、および
(c)前記比較結果に基づいて、細胞増殖を調節しうる被験物質を選択する工程。
【0064】
前記(a)において、被験物質とは、いかなる公知物質および新規物質であってもよく、例えば、核酸、糖質、脂質、蛋白質、ペプチド、有機低分子化合物、コンビナトリアルケミストリー技術を用いて作製された化合物ライブラリー、固相合成やファージディスプレイ法により作製されたランダムペプチドライブラリー、あるいは微生物、動植物、海洋生物等由来の天然成分などがあげられる。また、これらの化合物の2種以上の混合物を試料として供することもできる。
【0065】
被験物質と接触させる前に、レポータープラスミドの場合はレポーター遺伝子が発現できるように所望の細胞に一過性に導入し、形質転換体の場合は宿主に適した培地および培養条件で培養しておく。用いる細胞または宿主の種類は、好ましくは、ヒト子宮頚部癌細胞HeLa、ヒトクリオブラストーマ細胞T98G、大腸癌細胞SW620、HCT116などである。被験物質との接触方法としては、例えば、前記細胞または形質転換体を適当な培地中に入れ、約25〜40℃のインキュベーター中で生存または培養させ、次に、前記培地中に被験物質を添加し、インキュベートを続けることで接触がなされうる。
【0066】
前記被験物質の添加量は、有効成分の種類、培地に対する溶解性、細胞または形質転換体の感受性等によって適宜設定することができる。
【0067】
工程(b)において、前記被験物質を接触させた細胞または形質転換体におけるレポーターの発現量は、各レポーター遺伝子に応じて自体公知の方法により調べることができる。例えば、蛍光タンパク質の場合、各蛍光タンパク質に応じた励起波長を照射して検出される蛍光を測定することにより行う方法などがあげられる。
【0068】
前記工程(b)において、被験物質を接触させない細胞または形質転換体におけるレポーター遺伝子の発現量も同時にまたは別途調べ、接触した場合の結果と接触しない場合の結果とを比較する。
【0069】
前記工程(c)において、工程(b)で得られた比較結果に基づき、細胞増殖を調節しうる被験物質を選択する。選択する基準は、レポーターの発現が上昇または低下していることを指標にすればよい。レポーターの発現が上昇している場合、被験物質は、細胞増殖を促進させる作用を有する。レポーターの発現が低下している場合、被験物質は、細胞増殖を抑制させる作用を有する。
【0070】
このようにして選択された被験物質は、Mycと本発明のヌクレオチドとの結合に特異的に作用して、あるいはMyc発現量に影響して、あるいはMycによる転写制御機構に作用して機能する作用機序の明確な細胞増殖調節剤の候補となりうる。
【0071】
[スクリーニング方法II]
また、本発明は、本発明のレポータープラスミド、形質転換体またはトランスジェニック非ヒト動物を用いることを特徴とする、妊娠調節剤をスクリーニングする方法(スクリーニング方法II)を提供する。スクリーニング方法IIは、下記工程:
(a)レポータープラスミド導入細胞、形質転換体またはトランスジェニック非ヒト動物と被験物質とを接触させる工程、
(b)前記被験物質を接触させた細胞、形質転換体または動物におけるレポーターの発現量を調べ、被験物質を接触させない細胞、形質転換体または動物における発現量と比較する工程、および
(c)前記比較結果に基づいて、妊娠調節剤の候補となる被験物質を選択する工程を含む。
スクリーニング方法IIにおいて、レポータープラスミド導入細胞または形質転換体を用いる場合は、工程(a)および(b)はスクリーニング方法Iと同様である。ここでは、本発明のトランスジェニック非ヒト動物を用いる場合について説明する。
【0072】
工程(a)において、被験物質は、スクリーニング方法Iと同様のものがあげられる。
【0073】
前記被験物質を本発明のトランスジェニック動物と接触させる方法は特に限定されるものではないが、経口的または非経口的に投与され得る。非経口的投与経路としては、例えば、静脈内、動脈内、筋肉内、腹腔内等の全身投与、または標的細胞付近への局所投与等があげられる。
【0074】
前記被験物質の投与量は、有効成分の種類、分子の大きさ、投与経路、投与対象となる動物種、投与対象の薬物受容性、体重、年齢等によって適宜設定することができる。
【0075】
前記工程(b)において、被験物質を投与した動物におけるレポーター遺伝子の発現量を調べる方法としては、各レポーター遺伝子に応じて自体公知の方法により行うことができる、例えば、蛍光タンパク質の場合、各蛍光タンパク質に応じた励起波長を照射して検出される蛍光を測定することにより行う方法などがあげられる。
【0076】
前記工程(b)において、被験物質を投与しない動物におけるレポーター遺伝子の発現量も同時にまたは別途調べ、投与動物の結果と非投与動物の結果とを比較する。
【0077】
前記工程(c)において、工程(b)で得られた比較結果に基づき、妊娠調節剤の候補となる被験物質を選択する。選択する基準は、レポーターの発現が上昇または低下していることを指標にすればよい。
【0078】
このようにして選択された被験物質は、Mycと本発明のヌクレオチドとの結合に特異的に作用して、、あるいはMyc発現量に影響して、あるいはMycによる転写制御機構に作用して機能する作用機序の明確な妊娠調節剤の候補となりうる。
【0079】
また、本発明は、下記いずれかのペプチド:
(1)配列番号1に記載の塩基配列からコードされるペプチド、
(2)配列番号2に記載のアミノ酸配列からなるペプチド、
(3)前記(1)または(2)に記載のペプチドと実質的に同一のアミノ酸配列を有するペプチド
を有効成分として含有する、Mycによる組織特異的発現の調節剤を提供する。前記調節剤は、妊娠の調節に用いられることが好ましい。
【0080】
「実質的に同一のアミノ酸配列」とは、配列番号2に示されるアミノ酸配列において1もしくは2以上(好ましくは1〜50個、より好ましくは1〜30個、いっそう好ましくは1〜10個、最も好ましくは1〜5個)のアミノ酸が置換、欠失、挿入、付加または修飾されたアミノ酸配列であって、同質の活性を有するものである。
【0081】
また、「実質的に同一のアミノ酸配列」として、配列番号2に示されるアミノ酸配列に対して約70%以上、好ましくは約80%以上、より好ましくは約90%以上、さらにより好ましくは約95%以上、いっそうより好ましくは約97%以上、最も好ましくは99%以上のアミノ酸同一性を有するアミノ酸配列を用いることもできる。同一性(%)は、前記塩基配列の同一性と同様に決定することができる。
【0082】
「同質の活性」とは活性が定性的に同等であることを意味し、定量的にも同等であることが好ましいが、許容し得る範囲(例えば、約0.5〜約2倍)で異なっていてもよい。ここでTAF4bの活性としては、TBPまたはTAFs(TBP associated factors)との結合があげられる。
【0083】
また、本発明は、下記:
(a)配列番号5に記載の塩基配列、または
(b)配列番号5に記載の塩基配列において210位〜216位のTATAボックスおよび202〜207位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号5に記載の塩基配列と実質的に同一な塩基配列
を含有するサルTAF4bプロモーターを提供する。
【0084】
ここで「実質的に同一」とは、210〜216位のTATAボックスと202〜207位のCGCGTGモチーフとがプロモーターの構成要素として機能する限り、それ以外の塩基配列は特に限定されないことを意味する。「実質的に同一」か否かの判断は、後述するレポーターアッセイによる前記(a)の塩基配列と(b)の塩基配列との間のレポーターの発現量を比較し、同程度の発現量であることを指標にすればよい。
【0085】
また、本発明は、下記:
(a)配列番号6に記載の塩基配列、または
(b)配列番号6に記載の塩基配列において778位〜784位のTATAボックスおよび770〜775位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号6に記載の塩基配列と実質的に同一な塩基配列
を含有するラットTAF4bプロモーターを提供する。
【0086】
ここで「実質的に同一」とは、778〜784位のTATAボックスと770〜775位のCGCGTGモチーフとがプロモーターの構成要素として機能する限り、それ以外の塩基配列は特に限定されないことを意味する。「実質的に同一」か否かの判断は、後述するレポーターアッセイによる前記(a)の塩基配列と(b)の塩基配列との間のレポーターの発現量を比較し、同程度の発現量であることを指標にすればよい。
【実施例】
【0087】
以下に実施例を挙げて、本発明をさらに詳しく説明するが、本発明はこれらに限定されるものではない。
【0088】
細胞培養
ヒト膠芽腫細胞株T98G細胞、およびその派生体であり、c−MycERキメラタンパク質を発現するT98Gmycer−2細胞(Tsuneoka M. et al., J. Biol. Chem. 277:35450-35459 (2002))を、非必須アミノ酸および10%ウシ胎児血清(FCS)を含むイーグル培地中で培養した。c−MycERキメラタンパク質を発現するラット線維芽細胞株3Y1の派生体、3Y1MycER(Tsuneoka M. et al., Oncogene 14:2301-2311 (1997))およびアフリカミドリサルの腎線維芽細胞株Cos−7を、10%FCSを含むダルベッコ変法イーグル培地中で培養した。ヒト前骨髄性白血病HL60細胞を、20%FCSを含むRPMI 1640培地中で培養した。ラット大腸がん細胞株RCN−9(理研細胞バンク)およびヒト赤白血病細胞株HEL(ヘルスサイエンスリサーチ資源バンク製)を、10%FCSを含むRPMI 1640培地中で培養した。
【0089】
ポリメラーゼ連鎖反応(PCR)
PCR増幅は、各プライマーを10pmoles、EX Taq DNAポリメラーゼを1.2U、およびdNTPを200μM含む50μl EX Taqバッファー(タカラバイオ製)中で行った。
【0090】
5’−RACE法による解析
HEL細胞のpoly(A)+RNA(1μg)からの逆転写反応、二本鎖cDNA合成、およびアダプターライゲーションは、マラソン(Marathon)cDNA増幅キット(クロンテック社製)を用いて行った(Tsuneoka M. et. al., J. Biol. Chem. 277:35450-35459 (2002),Tsuneoka M. et. al., J. Biol. Chem. 280:19977-19985 (2005))。第1PCRは、プライマーとして、AA287145-1st(5’−TTCTTATGCCTCTTCCTTTCTCCTG−3’(配列番号7)、ESTクローンAA287145の5’末端領域の配列)、および供給者によって提供されたAP1プライマーを使用して行った。温度プロファイルは、94℃で1分間の最初の熱変性後、96℃で15秒間の熱変性工程ならびに68℃で3分間のアニーリング工程および伸張工程を1サイクルとする25サイクルであった。上記第1RACE−PCR産物を1000倍希釈した液1μlをネストRACE−PCRの鋳型として使用した。ネストRACE−PCRは、AA287145ネストプライマー(5’−TTGGCAAATGAGGCAGGTAGTAACA−3’(配列番号8)、ESTクローンAA287145のAA287145-1stの上流領域配列に対応するもの)と、供給者によって提供されたAP2プライマーとを使用して行った。温度プロファイルは、94℃で1分間の最初の熱変性後、96℃で15秒間の熱変性工程ならびに68℃で3分間のアニーリング工程および伸張工程を1サイクルとする25サイクルであった。増幅したDNA断片をpGEM-Tベクター(Promega社製)にクローニング(pT/fragmentTAF4b)し、シークエンスした。シークエンスの結果、AA287145はTAF4b遺伝子の3’−UTRの一部であることが確認された。
【0091】
TAF4bのmRNAの5’末端に関する情報を得るために、5’−RACE増幅が上述のように実施された。ここでは、プライマーとして、TAF4bRACE−1(5’−GGTCTCGGCTCTTGTTACAGTTTGCTG−3’(配列番号9)、文献(Dikstein R. et. al., Cell 87:137-146 (1996))において以前に報告したTAF4b cDNAの5’末端領域の配列)と第1PCR用に供給者によって提供されたAP1プライマー、TAF4bRACE−2プライマー(5’−GGATTGTCGTGGTGTTGGGGGCTTTCA−3’(配列番号10)、TAF4bRACE−1の上流領域の配列に対応するもの)およびネストPCR用に供給者によって提供されたAP2プライマーを使用した。増幅したDNA断片をpGEM-Tベクター(Promega社製)にクローニングし、シークエンスした。
【0092】
逆転写PCR法(RT−PCR)
HEL細胞の一本鎖cDNAの合成は、スーパースクリプト(Superscript)First-strand Synthesis system(インビトロジェン製)を使用して、全RNA(1μg)で行った。得られた一本鎖cDNA1μl(全量20μl)をPCRの鋳型として使用した。増幅用のRT−PCRプライマーは、
TAF4bコードU(5’−CGGAATTCGAAGCTGCGAGAGGTCGGGCGGGTGTCG−3’(配列番号11)、5’非翻訳領域にEcoRI部位を付加した配列)、および、
TAF4bコードL(5’−CGGAATTCGGCAGTAAATAGCAAGGATGTGGATGGA−3’(配列番号12)、 3’非翻訳領域にEcoRI部位を付加した配列)であった。温度プロファイルは、98℃での15秒間の熱変性工程と、65℃での1分間のアニーリング工程および72℃での2分30秒間の伸張工程を1サイクルとして35サイクルであった。増幅したDNA断片をpGEM-Tベクター(プロメガ社製)にクローニングしてpT/TAF4bを作製し、その配列を決定した。当該配列は、増幅された断片を使用してダイレクトシークエンスによって確認された。
【0093】
TAF4bゲノムDNA断片を有するレポータープラスミド
ヒトTAF4b遺伝子のゲノムDNA断片は、プロモーター領域からエキソン1(exon 1)へと伸長しており、プライマー
5’−TTTTACCATAACCTCACTTGCTGGAAGGGG−3’(配列番号13)、および、
5’−TGTTGGGGGCTTTCACGGCGACTATCTG−3’(配列番号14)
を使用したPCRによって増幅した。2.2kbの増幅された断片を、SacIおよびNcoIで切断し、SacIとNcoIで切断したホタルルシフェラーゼを含むpGL3(Promega社製)の4.8kb断片に挿入し、pTAF4b(W)luciを作製した。SmaIとNcoIで切断したpTAF4b(W)luciの0.6kbの断片を、SmaIとNcoIで切断したpGL3の4.8kpの断片に挿入し、pTAF4b(dSma)luciを作製した。XhoIとNcoIで切断したpTAF4b(W)luciの0.6kpの断片を、XhoIとNcoIで切断したpGL3の4.8kpの断片に挿入し、pTAF4b(dXho)luciを作製した。pTAF4b(dSma)luciを、XhoIとNcoIで切断した後、Klenow酵素で平滑末端化し、セルフライゲートして、pTAF4b(Sma−Xho)luciを作製した。また、pTAF4b(dSma)luciを、PvuIIとNcoIで切断した後、Klenow酵素で平滑末端化し、セルフライゲートして、pTAF4b(Sma−Pvu)luciを作製した。
【0094】
さらに、欠失変異体pTAF4b(117Pvu)luci、pTAF4b(114Pvu)luci、pTAF4b(102Pvu)luci、pTAF4b(73Pvu)luciおよびpTAF4b(43Pvu)luciを、鋳型としてpTAF4b(Sma−Pvu)luciを使用し、適当なプライマーを用いたPCR、および、その後に行われるpGL3ベクターへの増幅断片の導入によって作製した。
【0095】
突然変異は、Gene EditorTMインビトロ部位指向突然変異システム(プロメガ社製)を用いて、pTAF4b(W)luciのE−ボックス部位に導入し、2つのE−ボックス(CACGTGエレメント)がCACCTGに変異したpTAF4b(mE1/2)luciを作製した。また、pTAF4b(Sma−Pvu)luciの非基準型E(NCE)−ボックス部位にも突然変異を導入し、pTAF4b(Sma−Pvu mNCE1/2)luci、pTAF4b(Sma−Pvu mNCE1)luciおよびpTAF4b(Sma−Pvu mNCE2)luciを作製した。これにより、1または2個のNCE−ボックス(CGCGTGエレメント)がCGCCTGに変異した。pTAF4b(Sma−Pvu)luciにおける非基準型E(NCE)−ボックス部位2(NCE−box2)を、CACGTGに変異させ、pTAF4b(Sma−Pvu CanoE2)luciを作製した。作製されたプラスミドのDNA配列は、シークエンスによって確認した。
【0096】
ラットおよびサルのTAF4bプロモーターの配列
ラットTAF4bプロモーターの1.2kp断片を、以下のようにして単離した。
まず、ヒトTAF4b遺伝子について、マウスのDNA配列のホモログを選択した(ジーンバンクアクセッション番号:AK012135)。マウスのDNA配列においては、ヒトTAF4bのゲノム配列に正確に適合するヌクレオチド配列として2つの領域を選択し、以下のオリゴヌクレオチド、
ラットTAFII−F(5’−GCGCACGTGTGAGCGCCGCTGAGG−3’(配列番号15))、および、ラットTAFIIPF−1(5’−CTGACAGGAGCCTTAGTCAC−3’(配列番号16))を合成した。これらのオリゴヌクレオチドをプライマーとして使用し、ラット結腸癌細胞RCN−9ゲノムDNAを鋳型として用いて、ラットTAF4bゲノムDNAを増幅した。増幅されたDNA断片をシークエンスし、この配列の上流領域を、promoter find kit(クロンテック社製)によってさらにクローニングした。なお、このクローニングには、鋳型としてDraIおよびSspIライブラリーが用いられ、プライマーとして
ラットTAFIIPF3(5’−GCCGGGGCGGGCGGCAGGAGACTCG−3’(配列番号17))およびTAFIIPF4(5’−CGGGCACTCCCTCAGCGGCGCTCAC−3’(配列番号18))(これらの配列は、先に確認されたラットのTAF4bDNA配列内のもの)、ならびにAP1およびAP2が使用された。増幅されたラットのTAF4bゲノム断片を、シークエンスした。
サルTAF4bプロモーターを、ヒトTAF4b遺伝子と適合するTAF−プライマー1(5’−GACACAAGGAGAGGAACACGGATGC−3’(配列番号19))、およびTAF−プライマー2(5’−GCCTGGGCTGCCCCCGGAGCGACAC−3’(配列番号20))をプライマーとし、Cos−7細胞のゲノムDNAを鋳型として使用して増幅した後、シークエンスした。
【0097】
その他のプラスミド
CMVプロモーターの制御条件下のウミシイタケルシフェラーゼ(Renilla reniformis luciferase)遺伝子を含むpRL−CMVを、Promega社から購入した。CMV−c−Myc、pCMV−USF、およびpCMV−TFE−3は、Martin Eilers博士から供与された(Desbarats L. et. al., Genes Dev. 10:447-460 (1996))。組換え型のGFP−USFおよびGFP−TFE3を作製するために、pCMV−USFおよびpCMV−TFE−3を鋳型としてDNA断片を増幅した。なお、プライマーとしては、
5’−GGCGGATCCAGATGAAGGGGCAGCAGAAAAC−3’(配列番号21) (USF-BamHI-5’)、および、
5’−CGGGAATTCCCCATAGTTAGTTGCTGTC−3’(配列番号22) (USF-EcoRI-3’)、
あるいは、5’−GCCGGATCCATGGCGCTGCTCACCATCGG−3’(配列番号23) (TFE3-BamHI-5’)、および、5’−CGGCTCGAGGGTTCATGGGGAAGGGGCAG−3’(配列番号24) (TFE3-XhoI-3’)を使用した。次いで、得られた断片を、BamHIおよびEcoRIまたはBamHIおよびXhoIで切断し、BamHIおよびEcoRIで切断した細菌発現ベクターpGEX−3X、またはBamHIおよびXhoIで切断した細菌発現ベクターpGEX−4T3にそれぞれ導入し、pGEX−USFまたはpGEX−TFE−3を作製した。
【0098】
RNA調製
全RNAを、DEPC処理されたRNA調製液セット(ナカライテスク社製)を用いて、酸グアニジウム−チオシアネート−フェノール−クロロホルム抽出法によって細胞から単離した。
【0099】
DNAチップを使用した示差ディスプレイ解析(cDNAマイクロアレイ)
cDNAマイクロアレイは、文献(Tsuneoka M. et. al., J. Biol. Chem. 277:35450-35459 (2002))で説明した方法によって実施した。全RNAを、未処理のT98Gmycer−2細胞および4−ヒドロキシタモキシフェン(OHT)で20時間処理した細胞から単離した。poly(A+)RNAを回収し、DNAチップ(インサイト社製)を利用した示差ディスプレイ解析に供した。発現遺伝子配列タグ(EST)クローンを含む約9000種のcDNAがチップ上に搭載された(UniGEM Human V Ver. 2)。
【0100】
ノーザンブロット解析およびプローブDNAの調製
RNAは、ホルムアルデヒド含有アガロースゲルで電気泳動し、ハイボンドN(アマシャム社製)に転写し、32P標識した各cDNAプローブで検出した。各プローブは、市販のMultiprimeラベリングキット(アマシャム社製)を用いて[α−32P]dCTPで標識した。結果はBAS2000イメージアナライザー(富士フィルム社製)を用いて定量した。c−myc用のDNAプローブは、文献(Tsuneoka M. et. al., Oncogene 14:2301-2311 (1997))に示すものであった。TAF4b用のDNAプローブは、pT/fragmentTAF4b由来の0.6kpのSacI断片であった。
【0101】
一過性発現解析
T98Gmycer−2細胞における一過性発現レポーター解析を、文献(Tsuneoka M. et. al., J. Biol. Chem. 277:35450-35459 (2002))に説明する方法で行った。
TAF4bプロモーターにおけるMycおよびその他の転写因子の影響を、当該転写因子をコードしているプラスミドの転写によって調べた。T98G細胞を、10%FCSを含む培地で生育させた。細胞(3×104個)を培養皿(12ウエルのプレート、ウエルの径は22mm)に入れ、20〜24時間培養した。1μgのレポータープラスミド、インターナルマーカーとして20ngのpRL−CMV、および転写因子を発現する様々な量のプラスミドをFuGENE6試薬(ロシュ・ダイアグノステイックス社製)を用いて細胞に導入し、トランスフェクションを行った。各トランスフェクションにおける全DNA量は、空の発現ベクターを加えることによって一定に維持した。一日経過後、細胞を集め、ルシフェラーゼ活性を文献(Tsuneoka M. et. al., Oncogene 14:2301-2311 (1997))に示す方法によって測定した。
【0102】
クロマチン免疫沈降実験
クロマチン免疫沈降実験は、基本的に文献(Tsuneoka, M., et. al., J. Biol. Chem. 277:35450-35459(2002))に示す方法と同様に実施した。免疫沈降したDNA断片を、PCRによって検出した。使用したPCRプライマーは、
5’−GACACAAGGAGAGGAACACGGATGC−3’(配列番号25) (TAF4b-primer 1)、
および、5’−GCCTGGGCTGCCCCCGGAGCGACAC−3’ (配列番号26)(TAF4b-primer 2)である。これらは、ヒトTAF4b遺伝子のプライマー(TAF4b primers)の転写開始部位を含む521bp断片を増幅するプライマーである。
また、他のPCRプライマーとして、
5’−TTACAGGTAAGCCCTCCAATGACC−3’(配列番号27)、
および、5’−GCAAAGCTACCATTTAGGAACCC−3’(配列番号28)を使用した。これらは、第22番染色体中のE−ボックスを含む領域のゲノム配列を増幅するプライマーである(control primers)。このE−ボックスは検出可能な遺伝子を有しない染色体中に位置している(Bouchard, C., et. al., Genes Dev. 15:2042-2047.(2001))。
【0103】
抗体
c−Mycに対する抗体−1は、文献(Tsuneoka, M., et. al., Oncogene 14:2301-2311.(1997))に記載されたものである。抗c−Myc抗体−2(N262)(Santa Cruz Biotechnology, Santa Cruz, CA)、抗Max抗体(Oncogene Science)、抗c−Mycモノクローナル抗体(C−8)(Santa Cruz Biotechnology)、抗TFE3ヤギ抗体(sc−5958)(Santa Cruz Biotechnology)、および、抗USF−1ウサギ抗体(sc−229)(Santa Cruz Biotechnology)は、購入した。
【0104】
ゲルシフトアッセイ
ゲルシフトアッセイは、本質的には文献(Tsuneoka, M., et. al., Oncogene 14:2301-2311(1997))に示す方法で行った。組換え型GST−MycおよびGST−Maxを混合し、室温で30分間インキュベートした。混合物を、ヒトTAF4bプロモーターの合成二本鎖オリゴヌクレオチドへの結合について解析した。二本鎖オリゴヌクレオチドCanoEは、TAF4bプロモーターCGCGTG(NCE−box2)の代わりにCACGTGエレメントを含んでいるが、合成オリゴヌクレオチド
5’−GCGTGCGCGAGGTCTCCACGTGGCTATATAAACATGG−3’(配列番号29)、
および、5’−GCCAGCCATGTTTATATAGCCACGTGGAGACCTCG−3’(配列番号30)
のアニーリングによって作製された。CanoEオリゴヌクレオチドは、Klenow酵素を使用して[32P]dCTPで標識した。
20μlの結合反応溶液内の最終状態は、32P標識されたCanoEオリゴヌクレオチド:1ng、ポリ(dI−dC)ポリ(dI−dC)(Pharmacia Biotech社製):1μg、非特異的一本鎖オリゴヌクレオチド(5’−GAGTCGACGAACACACCAGGTCTTGGAGCG−3’(配列番号31)):1μg、塩化ナトリウム(NaCl):98.5mM、塩化カリウム(KCl):2.1mM、リン酸ナトリウム:4.8mM、Tris(pH7.4):2mM、EDTA:0.1mM、グリセロール:1%であった。
反応を進行させるために、反応混合液を室温で20分間放置した。抗Max(Oncogene Science)、非特異的抗体または抗c−Mycモノクローナル抗体(C−8)(Santa Cruz Biotechnology)をDNA結合反応中に加え、抗体解析を行った。標識されていない二本鎖オリゴヌクレオチドCanoE、標識されていない二本鎖オリゴヌクレオチドnon−CanoE(野生型:5’−GCGTGCGCGAGGTCTCCGCGTGGCTATATAAACATGG−3’(配列番号32)と、
5’−GCCAGCCATGTTTATATAGCCACGCGGAGACCTCG−3’(配列番号33)とのアニーリングによって作製されたもの)、および、
mE(5’−GCGTGCGCGAGGTCTCCGCCTGGCTATATAAACATGG−3’(配列番号34)と5’−GCCAGCCATGTTTATATAGCCAGGCGGAGACCTCG−3’(配列番号35)とのアニーリングによって作製されたもの)は、competitorとして使用された。
グルタチオンS−トランスフェラーゼフュージョンUSFおよびTFE3タンパク質を、E. coli Rosetta(Novagen Inc. Cat# 70953-4)中のpGEX−USFあるいはpGEX−TFE3を用いて発現させ、グルタチオンセファロースカラム(Amersham Biosciences)で単離した。ゲルシフトアッセイを、c−Mycの場合と同様の方法で、USFおよびTFE3について実施した。
【0105】
(結果)
c−Mycは、TAF4bの発現を誘導する。
条件付でc−Myc活性を誘導するために、エストロゲン誘発性Mycシステム(Eilers M. et. al., Nature 340:66-68(1989),Tsuneoka M. et.al., J. Biol. Chem. 277:35450-35459 (2002)参照)を使用した。キメラタンパク質c−MycERは、ヒトc−Mycおよびヒトエストロゲン受容体のエストロゲン結合ドメインから構成されており、エストロゲンの非存在下では細胞骨格成分に結合している。このキメラタンパク質にエストロゲンまたはその類似体4−ヒドロキシタモキシフェン(OHT)が結合すると、細胞骨格成分から遊離し、c−Mycとして機能する。ヒト膠芽腫細胞株T98Gを、異所性c−Myc活性が導入される親細胞として使用した。c−MycERタンパク質を発現するT98G細胞株(T98Gmycer−2細胞)が確立された(Tsuneoka M. et.al., J. Biol. Chem. 277:35450-35459 (2002))。OHTの存在下あるいは非存在下における指数増殖期の20時間のT98Gmycer−2細胞由来の全RNAを、cDNAマイクロアレイ解析に供した。Myc標的遺伝子であるオルニチンデカルボキシラーゼ(Bello-Fernandez C. et. al., Proc. Natl. Acad. Sci. U. S. A. 90:7804-7808 (1993))、ヌクレオリン(Greasley P. J. et. al., Nucleic Acids Res. 28:446-453 (2000))、およびmina53(Tsuneoka M. et.al., J. Biol. Chem. 277:35450-35459 (2002))に対する特異的シグナルは、c−MycERの活性化によって、それぞれ2.6倍、1.6倍、1.9倍に上昇した。これらの結果は、Myc標的遺伝子がこの実験システムにおいて検出されうるということを示唆している。
【0106】
本発明者は、ESTクローンAA287145に対するシグナルが、c−Mycの活性化によって2.3倍に刺激されるということを見出した。ESTクローンAA287145の5’上流領域をコードするcDNAは、ヒト赤白血病HEL細胞ライブラリーから5’−RACEプロトコールを用いて単離した。そして、AA287145は、TAF4b遺伝子(Dikstein, R., et. al., Cell 87:137-146 (1996))においてはTAFII105と呼ばれるもの)の3’非コード領域の一部であることが確認された。ヒトTAF4b遺伝子の翻訳および転写開始部位は以前に報告されていなかったため、本発明者らは、5’−RACE実験によって転写物の5’末端をコードするcDNAを単離した。転写開始部位をTAF4bゲノム配列上にマップした。この部位は、図1Aにおいて矢印を付して示している。TAF4bのmRNAの5’および3’配列と一致するプライマーを使用して、RT−PCRプロトコールに基づいてHEL細胞の全RNAから3.2kb長のcDNAを増幅し、シークエンスした。全体のヌクレオチド配列は文献(Dikstein, R., et. al., Cell 87:137-146.(1996))において報告したものとほぼ同じであったが、既に報告しているTAF4bのN末端のアミノ酸Glyの先に66個のアミノ酸配列が付加されていること(図1A参照)、および、いくつかのアミノ酸が異なっていることが確認された(当該配列は、ジーンバンク(Gen/Bank)にアクセッション番号AB234096として登録されている。)。当該cDNAクローンは、862アミノ酸からなり、推定分子量91090.52Da、等電点9.59のタンパク質をコードしている。
【0107】
TAF4b mRNAの発現
ヒト前骨髄性白血病HL60細胞を、TPA(phorbol 12-myristate 13-acetate)によって末期に分化させると、一時的に刺激された後(Vass, J. K., et al., Differentiation 45:49-54 (1990)参照)にc−mycの発現量が減少する(Hickstein, D. D., et. al., J. Biol. Chem. 264:21812-21817 (1989)、Hozumi, M., Adv. Cancer Res. 38:121-169 (1983)参照)。この実験システムを用いて、Myc標的遺伝子がMycの遮断中に影響されるか否かを調べた(Coller, H. A., et. al., Proc. Natl. Acad. Sci. U. S. A. 97:3260-3265.(2000)、Grandori, C., et. al., EMBO J 15:4344-4357.(1996)参照)。図2Aに示すように、c−mycのmRNAのレベルは、TPAを添加した後1時間で2倍に増加した後、3時間後に減少し始め、7時間後には1/5に減少した。TAF4bのmRNAのレベルは、TPAを添加した後3時間で1.5倍に上昇した後、6時間後に減少し始め、9時間後に1/10に減少した。これらの結果は、TAF4b発現がc−myc発現の後に起こることを示している。
【0108】
次に、MycERシステムを用いて、TAF4bのmRNAに対するc−Myc活性化の影響を調べた(図2B参照)。OHT処理されたT98Gmycer−2細胞において、TAF4bのmRNAのレベルは、3時間で2倍、7時間で3倍にそれぞれ上昇した。T98G親細胞のOHT処理は、TAF4bのmRNAのレベルを刺激しなかった。図2Cに示すように、T98Gmycer−2細胞におけるOHTによるTAF4bのmRNAの誘導は、タンパク質合成阻害剤シクロへキシミドの存在下で維持された。シキロヘキシミドによる処理は、T98G親細胞において、TAF4bのmRNAレベルにほとんど影響を及ぼさなかった(データ示さず)。これらの結果は、TAF4b遺伝子がMycの直接の標的となっていることを示すものである。
【0109】
c−MycはインビボでTAF4bプロモーターの領域に結合する
HL60細胞の増殖中において、生体内での内因性TAF4b遺伝子に結合するc−Mycタンパク質を調べるために、クロマチン免疫沈降を前述した方法(Boyd, K. E., et. al., Mol. Cell. Biol. 17:2529-2537.(1997),Boyd, K. E., et. al., Proc. Natl. Acad. Sci. U. S. A. 95:13887-13892.(1998)参照)により行った。免疫沈降後、各サンプルにおける内因性TAF4b遺伝子断片の強化を、TAF4b遺伝子における転写開始部位周辺のDNAを特異的に増幅させるプライマーを用いたPCRによってモニターした。図1Bに示すように、2つの異なる抗c−Myc抗体が、増殖期のHL60細胞由来のTAF4bDNA断片を免疫沈降した。一方、TPA処理された細胞からは、同じ抗体は検出可能なレベルのDNA断片を免疫沈降しなかった。なお、TPA処理された場合、c−mycの発現は非常に低くなった。非特異的抗体はTAF4b遺伝子断片を免疫沈降しないため、TAF4bゲノムDNA断片の強化は、TAF4b遺伝子に結合するc−Mycに依存する。さらに、c−Mycに対する抗体は、第22番染色体において検出可能な遺伝子を有さない染色体領域に位置するE−ボックスを含むゲノムDNA断片を強化しないため、TAF4b遺伝子において検出されるc−Mycの結合は特異的であるといえる。これらの結果から、転写開始部位周辺のTAF4b遺伝子が、HL60細胞の増殖期で、特異的にc−Mycに結合していることがわかる。
【0110】
c−Mycは、非基準型E−ボックス(CGCGTG)エレメントを介してTAF4bプロモーターを活性化する。
c−MycがどのDNAエレメントを介してTAF4bの発現を制御するかを確認するために、ヒトTAF4b遺伝子のプロモーター領域を単離した。図1Aでは、最初のATGにおける“A”を「+1」とした(星印で示す)。転写開始部位は−460bpであった。この位置は、5’−RACEによって決定し、図1Aにおいては矢じりで示した。TATAボックスの配列は、転写開始部位の約30bp上流付近で見つかった。
TAF4bプロモーターの活性を調べるために、エキソン1の上流領域からエキソン1の一部にかけての1.6kp領域を含むゲノムDNA断片を、ホタルルシフェラーゼcDNAに結合して、レポータープラスミドpTAF4b(W)luciを構築した(図3A参照)。T98Gmycer−2細胞における一過性発現解析により、前記DNA断片がプロモーター活性を有することが示された。OHTによってc−MycERを活性化した後、ルシフェラーゼ活性が2.5倍にまで上昇した(図3A)。この刺激は、T98Gmycer−2細胞においてOHTにより増加したTAF4bのmRNAレベルの増加で観察されたものに匹敵する(図2B参照)。
【0111】
転写開始部位付近には、2つの基準型E−ボックス部位(CACGTGエレメント)が存在する。この第1および第2のCACGTGエレメントを、E−ボックス1、E−ボックス2とそれぞれ命名した。CACGTGエレメントは、多くのMyc標的遺伝子においてMyc応答エレメントとして機能することが報告されていたため、当該E−ボックス部位がc−Mycによってプロモーターの活性化に関与するか否かを調べた。しかしながら、E−ボックス1およびE−ボックス2が変異した場合でも、ルシフェラーゼ活性は野生型レポータープラスミドと同様にMycによって依然として増加した(図3AのpTAF4b(mE1/2)luci)。
【0112】
次に、Myc刺激に応答可能なプロモーター領域を、欠失変異株を用いて調べた(図3A参照)。−1575番目から−535番目までの領域の欠失(pTAF4b(dSma)luci)は、Mycによるプロモーターの刺激を減少させなかったが、−1575番目から−367番目までのDNA断片の欠失(pTAF4b(dXho)luci)は、Myc刺激を消滅させた。この結果から、−534番目から−367番目までの領域が重要であることが示唆される。続いて、2つの追加レポーターを構成した。このレポーターは、−534番目から−360番目までの配列を含むもの(pTAF4b(Sma−Xho)luci)と、−534番目から−377番目までの配列を含むもの(pTAF4b(Sma−Pvu)luci)であった。これら2つのレポーターの発現は、Mycによって十分刺激された。
【0113】
応答部位を見つけるために、レポーター(pTAF4b(Sma−Pvu)luci)におけるDNA断片をさらに短くした(図3B参照)。−534番目と−492番目との間の領域の欠失(pTAF4b(117Pvu)luci)は、Mycによる上昇にほとんど影響を及ぼさないが、−534番目と−490番目との間の欠失(pTAF4b(114Pvu)luci)は、Mycによる上昇を消滅させた。さらに多くの部位を欠失させたレポーター(pTAF4b(102Pvu)luci、pTAF4b(73Pvu)luci、pTAF4b(43Pvu)luci)による発現も、Mycによって上昇しなかった。これらの結果は、−492番目から−490番目までの領域の重要性を示すものである。
【0114】
−492番目から−490番目までの領域は、非基準型E−ボックスエレメントCGCGTG(図1Aの−492番目から−487番目)内に存在した。CGCGTGエレメントが、Myc応答エレメントであるということはほとんど実証されていないが、c−Mycがこのエレメントに結合しうるということは文献(Grandori, C., et. al., Annu. Rev. Cell Dev. Biol. 16:653-699 (2000))に報告されている。そこで我々は、CGCGTGエレメントがMycの活性化に関与するか否かを実験した。TATAボックス配列のすぐ近くには2つのCGCGTGエレメントが存在する(図1Aおよび3C)。我々は、第1および第2のCGCGTGエレメントを、それぞれ非基準型E(NCE)−ボックス1(−509番目から−504番目)、NCE−ボックス2(−492番目から−487番目)と命名した。我々は、レポータープラスミドpTAF4b(Sma−Pvu、mNCE1)luci、pTAF4b(Sma−Pvu、mNCE2)luciおよびpTAF4b(Sma−Pvu、mNCE1/2)luciを作製した。これらのプラスミドにおいて、1個または2個のCGCGTGをCGCCTGに変異させた(図3C)。NCE−ボックス1の変異は、c−Mycによるプロモーター活性の刺激にほとんど影響を及ぼさないが、NCE−ボックス2を変異させた場合、あるいは、NCE−ボックス1およびNCE−ボックス2をともに変異させた場合には、Myc刺激をかなり減少させた(図3C参照)。これらの結果は、c−MycがNCE−ボックス2を介してTAF4bプロモーターをトランス活性化させることを示唆する。
【0115】
TAF4bプロモーターにおけるCGCGTGエレメントは、哺乳類の進化の過程で保存されている。
c−Mycが他の種においてTAF4bの発現を刺激するか否かを調べるために、ラットの細胞でTAF4bの発現に対するc−Mycの影響を調べた。図4Aに示すように、MycERキメラタンパク質を発現するラット3Y1線維芽細胞(Tsuneoka M. et. al., Oncogene 14:2301-2311(1997))において、TAF4bのmRNAレベルは、OHTによって3倍まで上昇した。この誘導は、タンパク質合成阻害剤シクロへキシミドの存在下で維持された。TAF4bはラットにおいてもMyc標的遺伝子であるということが、この結果から示唆される。
【0116】
我々は、プロモーター領域を含むラットTAF4bゲノムDNAの1.2kp断片を単離し、この断片についてシークエンスした。図4bは、ヒトおよびラットTAF4bプロモーター領域間のヌクレオチド配列の相違を示している。10bp間隔においた100bpのウインドウをスライドさせたり、重ねたりした際のヌクレオチドの相違数の平均を用いて、ヌクレオチド多様性を算出した。この解析において、挿入/欠失(indel)を一重変異として扱った。ヌクレオチド多様性の最小値、すなわち最大の相同性は、NCE−ボックス2(位置792)の周囲で観察された。低レベルのヌクレオチド多様性は、位置1000の周辺でも観察された。これは、上記の位置では、実際より小さく見積もられるヌクレオチド多様性を引き起こす大きな挿入/欠失(indel)がいくつか存在するという事実に起因するものである。ヒトTAF4bプロモーター配列を、そのDNA配列がアクセッション番号AC133172.4として最近ジーンバンク(GenBank)に登録されたマウスのTAF4bプロモーター配列と比較すると、似たような結果が得られた(データ示さず)。
【0117】
我々は、アフリカミドリサルの腎臓の線維芽細胞Cos−7からサルTAF4bプロモーターのDNA断片を単離し、それをシークエンスした。ヒト、サル、ラット、およびマウスのNCE−ボックス2周辺のヌクレオチド配列を、図4Cにおいて比較する。NCE−ボックス2およびTATAボックスエレメントを含む28bpの配列が4つの種において完全に保存されていた。
【0118】
CGCGTGおよびCACGTGエレメントは、異なる優先度でE−ボックス結合転写因子に応答する。
非基準型E−ボックス配列の保存は、生理学的な意義を有している可能性がある。NCE−ボックス2(CGCGTG)が基準型E−ボックスに変異した場合、プロモーター活性はc−Mycによって刺激され、野生型プロモーターの活性よりも上昇する(図5A参照)。
次に、Mycの基準型E−ボックスおよび非基準型E−ボックスに対する結合活性を調べた。TAF4bプロモーターの場合においてNCE−ボックス2の位置に基準型E−ボックスを含んでいる、放射標識されたCanoEヌクレオチドを使用したゲルシフトアッセイにおいて、MycとMaxとの結合はシフトしたバンドを生成した(図6A、レーン2参照)。このバンドは抗c−Myc抗体または抗Max抗体の存在下において減少し、スーパーシフトした(図6A、レーン1およびレーン3参照)。標識されていないCanoEオリゴヌクレオチドを過剰に添加した場合、シフトしたバンドの強度は減少した。一方、CACGTGがCGCCTGに変異した非標識のmEオリゴヌクレオチドを過剰に添加することによっては、バンドの強度はほとんど影響されなかった。この結果は、c−MycおよびMaxが、オリゴヌクレオチド内のCACGTGエレメントを特異的に認識することを示すものである。スキャッチャードプロット解析(図6C)によって解離定数を計算すると、24nMであった。
【0119】
基準型E−ボックス部位に対するMyc−Maxのアフィニティを、非基準型E−ボックス部位に対するMyc−Maxのアフィニティと比較するために、標識されていないCanoEオリゴヌクレオチドまたはnon−CanoEオリゴヌクレオチド(野生型E−ボックスCGCGTGを有する)が、反応混合物に対して過剰に加えられた。図5Dは、標識されていないCanoEオリゴヌクレオチドと比較して、約30倍以上の標識されていないnon−CanoEオリゴヌクレオチドが、標識されたCanoEオリゴヌクレオチドおよびMyc−Maxによって生成される特異的シグナルを50%に減少させるために必要であったことを示す。これらの結果から、TAF4bプロモーターにおいて、Myc−Maxは、CACGTGエレメントよりもCGCGTGエレメントに弱く結合することが示唆される。この弱い結合活性によって、Mycによる野生型TAF4bプロモーターの活性は緩やかに上昇すると思われる。
【0120】
mycプロトオンコジーンファミリーのタンパク質以外にも、USF(Gregor, P. D., et. al., Genes Dev. 4:1730-1740.(1990)参照)、TFE3(Beckmann, H., et. al., Genes Dev. 4:167-179.(1990)参照)を始めとする他のE−ボックス結合転写因子が存在する。次に、当該E−ボックス結合転写因子が、CACGTGエレメントおよびCGCGTGエレメントにどのように応答するのかについて調べた。図5Bおよび図5Cに示すように、野生型TAF4bプロモーターは、TFE3およびUSFによってほとんど刺激されなかった。これらの結果は、TAF4bプロモーターがMycに特異的に応答することを示唆する。CGCGTGエレメントが基準型(canonical)CACGTGに変異した場合、TAF4bプロモーターからの発現は、USFによって刺激されたが、TFE3は依然として当該レポーターからの発現をほとんど刺激しなかった。
【0121】
USFおよびTFE3のCanoEオリゴヌクレオチドに対する結合活性は、Myc−Maxの場合と同様の方法で決定された(図7参照)。CanoEオリゴヌクレオチドに結合するTFE3およびUSFのKd値は、それぞれ5.6nMおよび4.8nMであった。標識されていないCanoEオリゴヌクレオチドと比較して、約30倍の標識されていないnon−CanoEオリゴヌクレオチドが、標識されたCanoEオリゴヌクレオチドおよびUSFによって生成される特異的シグナルを50%に減少させるために必要とされることが確認された。これらの結果から、TAF4bプロモーターにおいて、USFとCGCGTGエレメントとの結合は、USFとCACGTGエレメントとの結合よりも弱いことが示唆される(図5F参照)。このことから、USFによって野生型TAF4bプロモーター活性は上昇しないことがわかった。
【0122】
TFE3の場合には、標識されたCanoEオリゴヌクレオチドおよびTFE3によって生成される特異的シグナルを50%に減少させるためには標識されていないnon−CanoEオリゴヌクレオチドは、標識されていないCanoEオリゴヌクレオチドの場合と比較して、3倍だけ必要であった。
【0123】
(考察)
TAF4b遺伝子は、Mycの直接の標的である。
我々は、c−MycがTAF4bの発現を直接誘導するということを実証した。HL60細胞において、TPA処理後のTAF4bのmRNAの発現は、c−mycの発現と密接に続いて起こった(図2A参照)。c−Mycがc−MycERキメラタンパク質で活性化されると、TAF4bのmRNAの発現の刺激が観察された(図2B参照)。この誘導は、タンパク質合成阻害剤の存在下でも維持され(図2C参照)、c−MycはTAF4bの発現を直接制御することが示された。c−Mycタンパク質は、増殖中のHL60細胞で、インビボでTAF4bプロモーターに結合した(図1B参照)。TAF4bプロモーター活性は、c−Mycによって上昇した(図3参照)。この活性の上昇は、進化の過程で保存された非基準型E−ボックス(NCE−ボックス2;CGCGTGエレメント)を介することによって起こる(図3および図4参照)。ゲルシフトアッセイでは、TAF4bプロモーターの場合、c−Mycタンパク質はNCE−ボックス2に結合することが示された(図5D参照)。これらの結果をまとめると、c−Mycは進化の過程で保存されたE−ボックス部位を介してTAF4bの発現を直接誘導するということがいえる。
【0124】
TAF4bを欠損したオスおよびメスのマウスは、生殖能力に顕著な異常を示す(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005), Freiman, R. N., et. al., Science 293:2084-2087.(2001)参照)。TAF4bを欠損するメスのマウスは、卵巣での卵胞形成に欠陥を有するために不妊である一方、野生型マウスの卵胞の顆粒膜細胞においては、TAF4bおよびc−mycの発現が見られた(Freiman, R. N., et. al., Science 293:2084-2087.(2001),Li, S., et. al., Endocr J. 41:83-92.(1994),Sato, A., et. al., Eur. J. Endocrinol. 131:319-322.(1994)参照)。成体の精巣において、TAF4bは生殖細胞の増殖に必要とされ(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005)参照)、TAF4bおよびc−mycの発現は減数分裂前の精原細胞において検出される(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005),Koji, T., et. al., Histochem. J. 20:551-557.(1998)参照)。TAF4bは細胞増殖の制御に関与する多くの遺伝子を誘導し得るため(Mengus, G., et. al., EMBO J. 24:2753-2767.(2005)参照)、上記の結果は、c−mycがTAF4b発現の刺激を介して、卵胞形成および精子形成中の細胞増殖に寄与する可能性を示唆する。
【0125】
E−ボックス内の配列:基準型(CACGTG)および非基準型(CGCGTG)エレメント
TAF4bプロモーターにおけるCGCGTGエレメント(NCE−ボックス2)は、c−MycによるTAF4b発現の誘導に必要である。この非基準型エレメントは進化の過程で保存されているが、これは、この配列が生理学的に重要であることを示すものである。CGCGTGエレメントが基準型CACGTGに変異した場合、c−MycのTAF4bプロモーターへの結合は強化され、c−Mycはプロモーター活性をより強くアップレギュレートした(図5参照)。これらの結果は、c−MycのE−ボックスへの結合活性がその転写活性化に直接影響を及ぼすということを示すものである。USFの場合も、同様の現象が観察された(以下参照)。
【0126】
本発明者は、別のE−ボックス結合転写因子であるUSFに関しても、E−ボックス部位における配列変化の影響を調査した。USFは野生型TAF4bプロモーターをほとんど刺激しない。しかしながら、NCE−ボックス2がCACGTGに変異すると、USFとTAF4bプロモーターとの結合はより強固なものとなり、USFはプロモーター活性をアップレギュレートし始めた(図5Cおよび図5F参照)。これらの結果は、非基準型CGCGTG E−ボックスがUSFによる制御を避けるために選択されている可能性を示唆する。
【0127】
マウスの精巣では、幹細胞系列でTAF4bは発現するが、体細胞であるセルトリ細胞ではTAF4bは発現しない(Falender, A. E., et. al., Genes Dev. 19:794-803.(2005)参照)。セルトリ細胞では、USFは高度に発現しており、基準型E−ボックスエレメント(CACGTG)を介してステロイド産生因子1(Daggett, M. A., et. al., Biol. Reprod. 62:670-679.(2000)参照)および卵胞刺激ホルモンレセプター(Heckert, L. L., et. al., Mol. Endocrinol. 14:1836-1848.(2000):参照)の発現を誘導する。TAF4bプロモーターにおいてNCE−ボックス2が基準型であったとすれば、セルトリ細胞において、TAF4bの発現はUSFによって誘導されるであろう。それゆえ、TAF4bプロモーターにおけるCGCGTGエレメントの保存は、哺乳類の精巣においてTAF4bの適度な発現を支持するために必要であるかもしれない。
【0128】
TAF4bプロモーターの場合において、CACGTGエレメントに結合するc−Myc−Max、USF、およびTFE3のKd値は、それぞれ24nM、4.8nM、5.8nMであり、c−Mycは上記のタンパク質の中で最も効率的にTAF4bプロモーターを活性化する。それゆえ、応答するエレメントに対する転写因子の結合活性は、応答する遺伝子を単純に決定するものではない。TFE3のCACGTGエレメントに対する結合活性は、CGCGTGエレメントの結合活性と近似している。そして、TFE3は、CACGTGエレメントを有するTAF4bプロモーターならびにCGCGTGエレメントを有するプロモーターを活性化しなかった。しかしながら、上述したように、USFのE−ボックスエレメントに対する結合活性が上昇すると、結果としてUSFによるTAF4bプロモーターの活性化が起こる。これらの結果は、転写因子のその応答エレメントに対する結合活性の上昇が、応答遺伝子の範囲を拡大しうることを示唆する。
【0129】
TFE3のCGCGTGエレメントに対する結合活性は、CACGTGエレメントに対する結合活性と同等であった。一方、c−Myc−MaxおよびUSFのCGCGTGエレメントに対する結合活性は、CACGTGエレメントに対する結合活性に比べて非常に弱かった(図5参照)。これらの結果から、E−ボックス内の各配列は、E−ボックスに対するE−ボックス結合転写因子の結合活性にそれぞれ異なる影響を与えるということが示唆される。現在までのところ、Myc−Maxに対する非基準型(non-canonical)結合配列がいくつか報告されている(CACCTG、CATGCG、CACGCG、CAACGTG、およびCACGAG)(Grandori, C., et. al., Annu. Rev. Cell Dev. Biol. 16:653-699.(2000):参照)。これらのエレメントは、異なる結合活性でE−ボックス結合転写因子と異なって結合する。これらの違いは、E−ボックス結合転写因子に応答する遺伝子の領域(spectrum)に影響を与える可能性を有している。以上の結果は、E−ボックス転写因子が異なる標的遺伝子特異性を有するというメカニズムに、E−ボックス内の配列が寄与しているという証拠を初めて提供するものである。
【図面の簡単な説明】
【0130】
【図1A】図1Aは、ヒトTAF4bプロモーターおよびエキソン1の一部のゲノムDNA配列である。また、本図では、以前には報告していなかったN末端のアミノ酸配列を、DNA配列の下に一文字表記で示す。転写開始部位は矢印で示され、翻訳開始部位は星印で示されている。図中には、E−ボックス部位、E−ボックス1およびE−ボックス2(破線で囲まれた部分)、非基準型E(NCE)−ボックス部位、NCE−ボックス1およびNCE−ボックス2(下線を引いた部分)、TATAボックス配列(影付きの部分)、SmaI部位(CCCGGG)、PvuII部位(CAGCTG)、およびXhoI部位(CTCGAG)が示されている。
【図1B】図1Bは、抗c−Myc抗体を使用したクロマチン免疫沈降実験の結果を示す図である。増殖期のHL60細胞(−TPA)、および、10nM TPAで24時間処理されたHL60細胞(+TPA)を、1%ホルムアルデヒドで固定した後、核抽出物を集め、c−Mycに対する抗体(c−MycのN末端側と反応する抗c−Myc抗体1、c−MycのC末端側と反応する抗c−Myc抗体2)、又はコントロール抗体を使用して、あるいは抗体を使用せずに(no antibody)、クロマチンを免疫沈降した。TPAの存在下でc−myc発現は大きく低下した(図2A)。核抽出物を添加しないmock免疫沈降実験も実施された(nuclear extract -)。DNAを精製した後、試料を、TAF4bプロモーター領域のDNA断片を増幅するために設計されたプライマー(TAF4bプライマー)、または、第22番染色体のE−ボックスを含むDNA断片を特異的に増幅するためのプライマー(コントロールプライマー)を用いたPCRに付した(Bouchard, C., et. al., Genes Dev. 15:2042-2047.(2001):参照)。
【図2A】図2Aは、TPA処理後のヒト前骨髄性白血病HL60細胞におけるTAF4bおよびc−mycmRNAの変化を示す図である。RNAを10nM TPAによる処理が行われた後、指示された時点で単離し、ノーザンブロッティングで解析してTAF4bおよびc−mycのmRNAを検出した(左側の図参照)。28Sおよび18SのrRNAも検出した。この結果を定量し、プロットした(右側の図参照)。
【図2B】図2Bは、MycERタンパク質の活性化によって、T98G細胞中で増加したTAF4bのmRNAレベルを示す。T98Gmycer−2細胞(T98GMycER)および親細胞(T98G)を、0.25%血清を含む培地で40時間培養した後、200nM OHTの存在下(+)、あるいは非存在下(−)で培養した。RNAを指示された時点で単離し、TAF4bのmRNAを検出するためにノーザンブロッティングによる解析が行われた(左側の図参照)。28Sおよび18SのrRNAも示す。結果を定量し、棒グラフとして表す(右側の図参照)。
【図2C】図2Cは、タンパク質合成阻害剤の存在下で、MycERタンパク質の活性化によってTAF4bのmRNAレベルの増加が維持されたことを示す図である。T98Gmycer−2細胞を、0.25%血清を含む培地で40時間培養した後、200nM OHTで8時間処理した。RNAを単離し、TAF4bのmRNAを検出するためにノーザンブロッティングにより解析した(左側の図参照)。図中、CHXの列で「+」と示されたものは、OHT添加の20分前に20μg/mlのシクロヘキシミドが添加されたものである。28Sおよび18SのrRNAも示す。結果を定量し、棒グラフとして表す(右側の図参照)。
【図3A】図3Aは、レポーターアッセイの結果を示すグラフである。レポータープラスミド(pTAF4b(W)luci)を、TAF4bプロモーター活性を検出するために構築した。pTAF4b(mE1/2)luciにおいて、pTAF4b(W)luciの2つのE−ボックス(CACGTGエレメント)をCACCTGに変異させた(図1A参照)。また、pTAF4b(W)luci、pTAF4b(dSma)luci、pTAF4b(dXho)luci、pTAF4b(Sma−Xho)luci、およびpTAF4b(Sma−Pvu)luciの欠失変異体を作製した。T98Gmycer−2細胞をレポータープラスミドでトランスフェクトした。一日経過後、MycERキメラタンパク質を活性化させるために、最終濃度が200nMになるようにOHTを加え、細胞をさらに17時間培養した。細胞抽出物を、ホタルルシフェラーゼ活性について解析した。プロモーター活性は、MycER活性化なしの活性に対する割合として決定した。MycER活性化を介さないOHTの効果を規準化するために、T98Gmycer−2細胞の細胞数を、T98G親細胞の細胞数で除した。なお、T98G親細胞は、上述したT98Gmycer−2細胞と全く同じ手順で処理した。基本のプロモーターとして、何も挿入されていないpGL3プラスミドを用いた。数値は、4つの別個の実験の結果を平均したものである。バーは、標準誤差を示している。
【図3B】図3Bは、レポーターアッセイの結果を示すグラフである。pTAF4b(Sma−Pvu)luciの配列を欠失させ、pTAF4b(117Pvu)luci、pTAF4b(114Pvu)luci、pTAF4b(102Pvu)luci、pTAF4b(73Pvu)luci、およびpTAF4b(43Pvu)luciを作製した。Mycによるプロモーター活性化は、図3Aで説明したのと同様にして測定した。
【図3C】図3Cは、レポーターアッセイの結果を示すグラフである。pTAF4b(Sma−Pvu)luciのNCE−ボックス1およびNCE−ボックス2(図1A参照)について変異を施し、pTAF4b(Sma−Pvu mNCE1)luci、pTAF4b(Sma−Pvu mNCE2)luci、pTAF4b(Sma−Pvu mNCE1/2)luciを作製した。ここでは、1つまたは両方の非基準型E−ボックス(CGCGTGエレメント)を、CGCCTGに変異した。Mycによるプロモーター活性化は、図3Aで説明したのと同様にして測定した。
【図4A】図4Aは、MycERタンパク質の活性化によって、TAF4bのmRNA量がラットの線維芽細胞3Y1においてどの程度増加するかを示す図である。200nMのOHTで細胞を8時間処理した後、RNAを単離した。図中、CHXの列で「+」と示されたものは、OHT添加の20分前に20μg/mlのシクロヘキシミドが添加されたものである。得られたRNAをノーザンブロッティングで解析し、TAF4bのmRNAを検出した。ここでも、28Sおよび18SのrRNAも示す。
【図4B】図4Bは、ヒトとラットとの間でTAF4bプロモーター配列を比較したものである。この解析において、相同性が高い領域については、数値が低くなった(本文参照)。本図では、NCE−ボックス2(CGCGTG)およびTATAボックス配列(TATA)の位置を示している(図1A参照)。
【図4C】図4Cは、ヒト(HUM)、サル(MON)、ラット(RAT)、およびマウス(MOU)のTAF4bプロモーターにおいて、CGCGTGエレメント周辺の配列を比較したものである。NCE−ボックス2およびTATAボックス配列は、明るい影付きで示している。ヒトTAF4bプロモーターとは異なるヌクレオチドは、暗い影付きで示している。本図では、ヒトプロモーターのSmaI部位も示している。
【図5】図5は、TAF4bプロモーターの特性を示すグラフである。 図5A〜図5Cでは、TAF4bプロモーター活性に対するc−Myc、TFE3、およびUSFの影響を調べた。本図では、pTAF4b(Sma−Pvu)luciを(○)で、NCEW−ボックス2(CGCGTGエレメント)が基準型(CACGTG)に変異されたpTAF4b(Sma−Pvu CACGTG)luciを(●)で示している。T98G細胞を、トランスフェクション効率の基準化用のpRL/CMVとともにレポータープラスミド、および、pCMV/c−myc(A)、pCMV/TFE3(B)、又はpCMV/USF(C)でトランスフェクトした。ルシフェラーゼ活性は、エフェクタープラスミドが無い場合の発現量に対する割合で表す。図中、X軸はエフェクタープラスミドの量を示す。数値は、4つの別個の実験の結果を平均したものである。バー(the bars)は、基準誤差を示している。 図5D〜図5Fは、TAF4bプロモーターにおいて、非基準型(野生型)E−ボックスに対するMyc−Max(D)、TFE3(E)、またはUSF(F)の結合活性を、基準型E−ボックスに対するそれぞれの結合活性と比較した図である。組換えタンパク質を、32P標識されたCanoEオリゴヌクレオチドに、様々な量の非標識のCanoEオリゴヌクレオチド(●で示す)あるいはnon−CanoEオリゴヌクレオチド(野生型;○で示す)を加えたものとともにインキュベートし、その後、実施例ならびに図6で説明したゲルシフトアッセイにより解析した。non−CanoEオリゴヌクレオチドは、野生型TAF4bプロモーターから合成されたオリゴヌクレオチドであり、Myc応答性NCE−ボックス2を有するものであった。CanoEオリゴヌクレオチドにおいて、NCE−ボックス2を基準型CACGTGエレメントに変異させた。反応混合物中、標識されていないcompetitorの量は、標識されたオリゴヌクレオチドとの倍数として表す。
【図6】c−Myc/MaxとTAF4bプロモーターオリゴヌクレオチドとの結合を調べた図である。 図6Aは、c−Myc/Max複合体とのゲルシフトアッセイの結果を示す。組換えタンパク質を32P標識されたCanoEオリゴヌクレオチドとともにインキュベートし、ゲルシフトアッセイにより解析した。シフトしたバンドの特異性をテストするために、ゲルシフトアッセイの前に、ウサギ抗Max抗体(文献1:Beckmann, H., et. al., Genes Dev. 4:167-179.(1990):参照)、非特異的抗体(Bello-Fernandez, C., et. al., Proc. Natl. Acad. Sci. U. S. A. 90:7804-7808.(1993)参照)、あるいは抗c−Mycモノクローナル抗体(Bouchard, C., et. al., Genes Dev. 15:2042-2047.(2001)参照)を反応混合物に加えた。図には、シフトしたバンド(*)および抗体によってスーパーシフトしたバンド(<)が示されている。 図6Bでは、配列特異的結合をテストするために、32P標識されたCanoEオリゴヌクレオチドに、その30倍量(レーン3および5)、100倍量(レーン4および6)、300倍量(レーン7)過剰の非標識CanoEオリゴヌクレオチド(レーン3および4)あるいはCACGTGエレメントがCGCCTGに置き換わったmEオリゴヌクレオチド(レーン5、6、および7)が加えられたものとともに、タンパク質をインキュベートした。その後、上述の方法でゲルシフトによる解析が行われた。 図6Cは、Myc/Maxと32P標識されたCanoEオリゴヌクレオチドとの結合を示す。c−Myc/Max複合体を、反応混合物中で様々な濃度の32P標識されたCanoEオリゴヌクレオチドとともに室温でインキュベートした。次いで、前記混合物を電気泳動し(左側の図参照)、シフトした放射能の強度を定量した。この結果を、スキャッチャードプロットで解析した(右側の図参照)。
【図7】USFおよびTFE3とTAF4bプロモーターオリゴヌクレオチドとの結合を調べた図である。 図7Aでは、CanoEオリゴヌクレオチドに結合するUSFのゲルシフトアッセイを図6と同様にして実施した。シフトしたバンドの特異性をテストするために、抗USF抗体を反応混合物に加えた(レーン3)。配列特異的結合をテストするために、32P標識されたCanoEオリゴヌクレオチドに、その30倍量(レーン4および7)、100倍量(レーン5および8)、300倍量(レーン6および9)過剰の非標識CanoEオリゴヌクレオチド(レーン4、5、および6)あるいはmEオリゴヌクレオチド(レーン7、8、および9)が加えられたものとともに、タンパク質をインキュベートした。 図7Bでは、CanoEオリゴヌクレオチドに対するUSFの結合を解析するため、スキャッチャードプロット解析を図6と同様にして実施した。 図7Cは、CanoEオリゴヌクレオチドに結合するTFE3のゲルシフトアッセイを示す。シフトしたバンドの特異性をテストするために、抗TFE3抗体(レーン3)またはコントロールの抗体をインキュベーション混合物に加えた。配列特異的結合をテストするために、32P標識されたCanoEオリゴヌクレオチドに、その30倍量(レーン5および7)、100倍量(レーン6および8)、300倍量(レーン9)過剰の非標識CanoEオリゴヌクレオチド(レーン5および6)またはmEオリゴヌクレオチド(レーン7、8、および9)が加えられたものとともに、タンパク質をインキュベートした。 図7Dでは、CanoEオリゴヌクレオチドに対するTFE3の結合を解析するために、スキャッチャードプロット解析が図6と同様にして実施された。
【図8】図8は、TAF4bプロモーター領域とその活性および推定される発現上昇因子の結合部位を示す図である。
【特許請求の範囲】
【請求項1】
Mycによる組織特異的発現を調節するためのヌクレオチドであって、TATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフおよび/またはCACGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチド。
【請求項2】
前記ヌクレオチドが
(a)配列番号3に記載の塩基配列、
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列、
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列、
(g)配列番号3の1081〜1126位の領域の塩基配列を必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列および/または当該必須配列の下流に1127〜1157位の領域の塩基配列が配置されてなる塩基配列を含む塩基配列、または
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列、
からなるものである、請求項1記載のヌクレオチド。
【請求項3】
(a)配列番号4に記載の塩基配列、または
(b)配列番号4に記載の塩基配列において、51位〜57位のTATAボックスおよび43〜48位のCACGTGモチーフ以外の塩基配列と70%以上の同一性を有し、かつTATAボックスとその1〜50塩基上流に位置するCACGTGモチーフとを含む塩基配列
を含有するものである、請求項1に記載のヌクレオチド。
【請求項4】
前記CGCGTGモチーフおよび/またはCACGTGモチーフが、下記:
(i)塩基配列GTGにおける少なくとも1塩基の置換、
(ii)塩基配列CACにおける少なくとも1塩基の置換もしくは挿入
のいずれかまたは両方の変異を有するものである、請求項1〜3いずれか1項に記載のヌクレオチド。
【請求項5】
請求項1〜4いずれか1項に記載のヌクレオチドを含有してなるベクター。
【請求項6】
請求項1〜4いずれか1項に記載のヌクレオチドおよびレポーター遺伝子を含有してなる、レポータープラスミド。
【請求項7】
請求項5に記載のベクターまたは請求項6に記載のレポータープラスミドを導入してなる形質転換体。
【請求項8】
請求項5に記載のベクターまたは請求項6に記載のレポータープラスミドを導入してなるトランスジェニック非ヒト動物。
【請求項9】
請求項6に記載のレポータープラスミドまたは請求項7に記載の形質転換体を用いることを特徴とする、細胞増殖の調節剤をスクリーニングする方法。
【請求項10】
請求項6に記載のレポータープラスミド、請求項7に記載の形質転換体または請求項8に記載のトランスジェニック非ヒト動物を用いることを特徴とする、妊娠調節剤をスクリーニングする方法。
【請求項11】
Mycによる組織特異的発現の調節剤であって、下記いずれかのペプチドを有効成分として含有する調節剤:
(1)配列番号1に記載の塩基配列からコードされるペプチド、
(2)配列番号2に記載のアミノ酸配列からなるペプチド、
(3)前記(1)または(2)に記載のペプチドと実質的に同一のアミノ酸配列からなるペプチド。
【請求項12】
妊娠の調節に用いられる請求項11に記載の調節剤。
【請求項13】
(a)配列番号5に記載の塩基配列、または
(b)配列番号5に記載の塩基配列において210位〜216位のTATAボックスおよび202〜207位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号5に記載の塩基配列と実質的に同一な塩基配列
を含有するサルTAF4bプロモーター。
【請求項14】
(a)配列番号6に記載の塩基配列、または
(b)配列番号6に記載の塩基配列において778位〜784位のTATAボックスおよび770〜775位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号6に記載の塩基配列と実質的に同一な塩基配列
を含有するラットTAF4bプロモーター。
【請求項1】
Mycによる組織特異的発現を調節するためのヌクレオチドであって、TATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフおよび/またはCACGTGモチーフとを必須の配列として含むTAF4bプロモーターに由来するヌクレオチド。
【請求項2】
前記ヌクレオチドが
(a)配列番号3に記載の塩基配列、
(b)配列番号3に記載の塩基配列において、1090〜1096位のTATAボックスおよび1082〜1087位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号3に記載の塩基配列と実質的に同一な塩基配列、
(c)配列番号3の1040〜1126位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(d)配列番号3の1082〜1156位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(e)配列番号3の1040〜1120位の領域の塩基配列と70%以上の同一性を有する塩基配列を含み、かつTATAボックスとその1〜50塩基上流に位置するCGCGTGモチーフとを含む塩基配列、
(f)配列番号3の1079〜1107位の領域の塩基配列を必須の配列として含み、かつ1040〜1078位および1108〜1120位の領域の塩基配列とそれぞれ70%以上の同一性を有する塩基配列をそれぞれ当該必須配列の上流および下流に配置してなる塩基配列を含む塩基配列、
(g)配列番号3の1081〜1126位の領域の塩基配列を必須の配列として含み、かつ当該必須配列の上流に1040〜1080位の領域の塩基配列および/または当該必須配列の下流に1127〜1157位の領域の塩基配列が配置されてなる塩基配列を含む塩基配列、または
(h)配列番号3の1040〜1080位の領域と1081〜1126位の領域とからなる断片、1081〜1126位の領域と1127〜1157位の領域とからなる断片、および1040〜1080位の領域と1081〜1126位の領域と1127〜1157位の領域とからなる断片からなる群より選ばれる塩基配列と70%以上の同一性を有する塩基配列を含む塩基配列、
からなるものである、請求項1記載のヌクレオチド。
【請求項3】
(a)配列番号4に記載の塩基配列、または
(b)配列番号4に記載の塩基配列において、51位〜57位のTATAボックスおよび43〜48位のCACGTGモチーフ以外の塩基配列と70%以上の同一性を有し、かつTATAボックスとその1〜50塩基上流に位置するCACGTGモチーフとを含む塩基配列
を含有するものである、請求項1に記載のヌクレオチド。
【請求項4】
前記CGCGTGモチーフおよび/またはCACGTGモチーフが、下記:
(i)塩基配列GTGにおける少なくとも1塩基の置換、
(ii)塩基配列CACにおける少なくとも1塩基の置換もしくは挿入
のいずれかまたは両方の変異を有するものである、請求項1〜3いずれか1項に記載のヌクレオチド。
【請求項5】
請求項1〜4いずれか1項に記載のヌクレオチドを含有してなるベクター。
【請求項6】
請求項1〜4いずれか1項に記載のヌクレオチドおよびレポーター遺伝子を含有してなる、レポータープラスミド。
【請求項7】
請求項5に記載のベクターまたは請求項6に記載のレポータープラスミドを導入してなる形質転換体。
【請求項8】
請求項5に記載のベクターまたは請求項6に記載のレポータープラスミドを導入してなるトランスジェニック非ヒト動物。
【請求項9】
請求項6に記載のレポータープラスミドまたは請求項7に記載の形質転換体を用いることを特徴とする、細胞増殖の調節剤をスクリーニングする方法。
【請求項10】
請求項6に記載のレポータープラスミド、請求項7に記載の形質転換体または請求項8に記載のトランスジェニック非ヒト動物を用いることを特徴とする、妊娠調節剤をスクリーニングする方法。
【請求項11】
Mycによる組織特異的発現の調節剤であって、下記いずれかのペプチドを有効成分として含有する調節剤:
(1)配列番号1に記載の塩基配列からコードされるペプチド、
(2)配列番号2に記載のアミノ酸配列からなるペプチド、
(3)前記(1)または(2)に記載のペプチドと実質的に同一のアミノ酸配列からなるペプチド。
【請求項12】
妊娠の調節に用いられる請求項11に記載の調節剤。
【請求項13】
(a)配列番号5に記載の塩基配列、または
(b)配列番号5に記載の塩基配列において210位〜216位のTATAボックスおよび202〜207位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号5に記載の塩基配列と実質的に同一な塩基配列
を含有するサルTAF4bプロモーター。
【請求項14】
(a)配列番号6に記載の塩基配列、または
(b)配列番号6に記載の塩基配列において778位〜784位のTATAボックスおよび770〜775位のCGCGTGモチーフを必須の塩基配列として含有し、かつ配列番号6に記載の塩基配列と実質的に同一な塩基配列
を含有するラットTAF4bプロモーター。
【図1A】


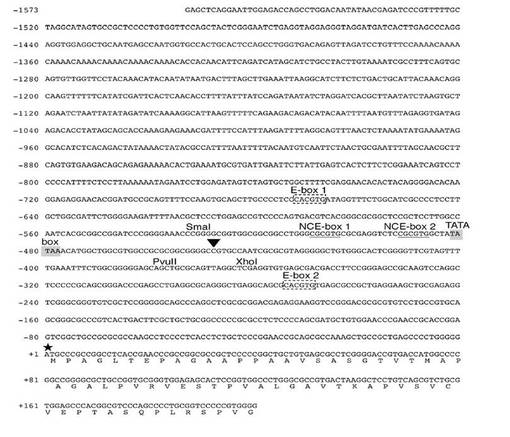
【図1B】
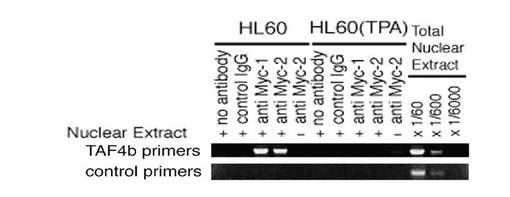
【図2A】
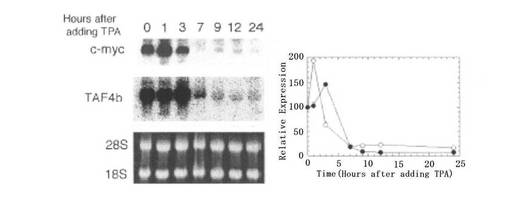
【図2B】
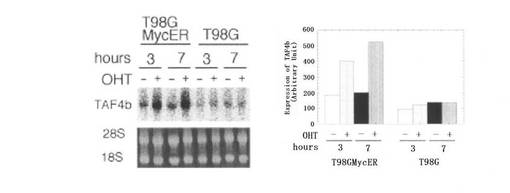
【図2C】
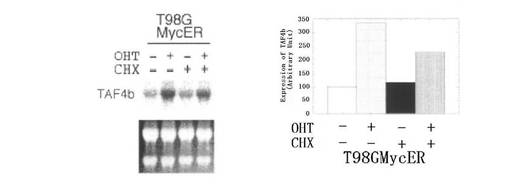
【図3A】
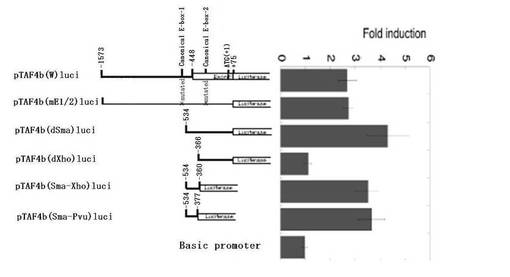
【図3B】
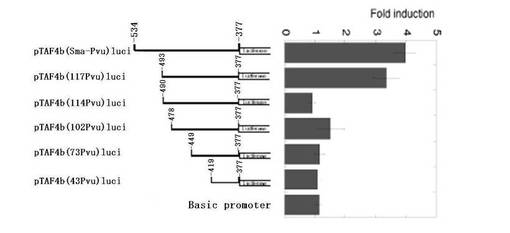
【図3C】
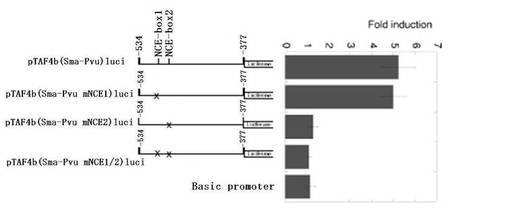
【図4A】
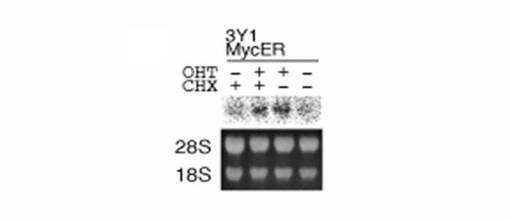
【図4B】
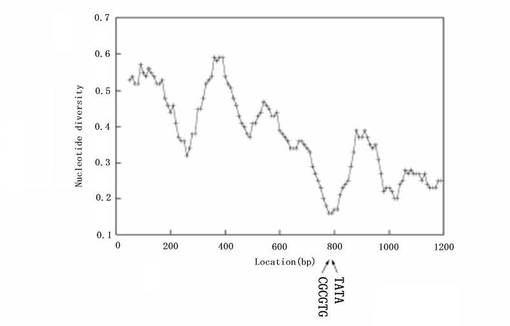
【図4C】
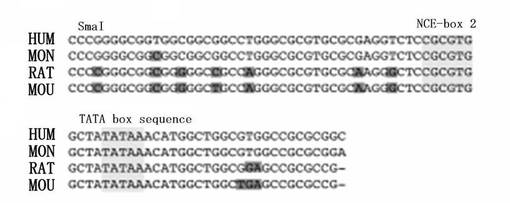
【図5】
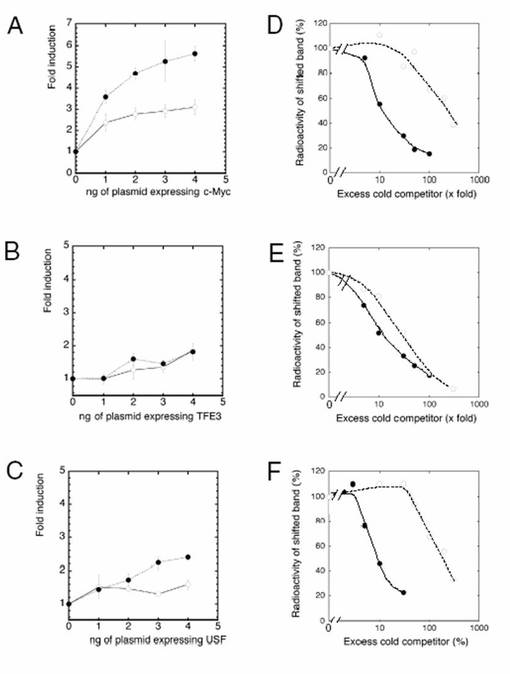
【図6】
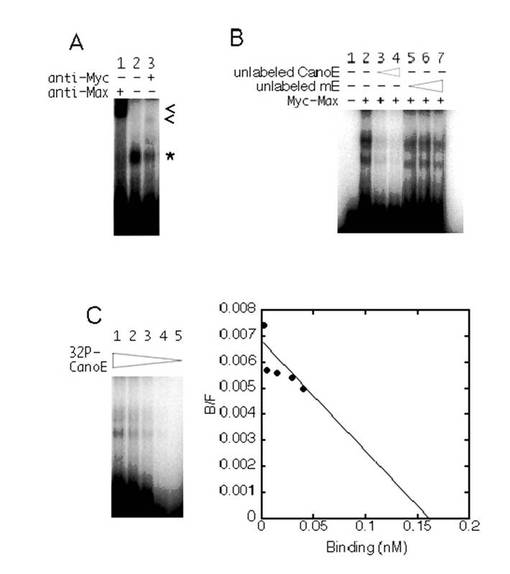
【図7】
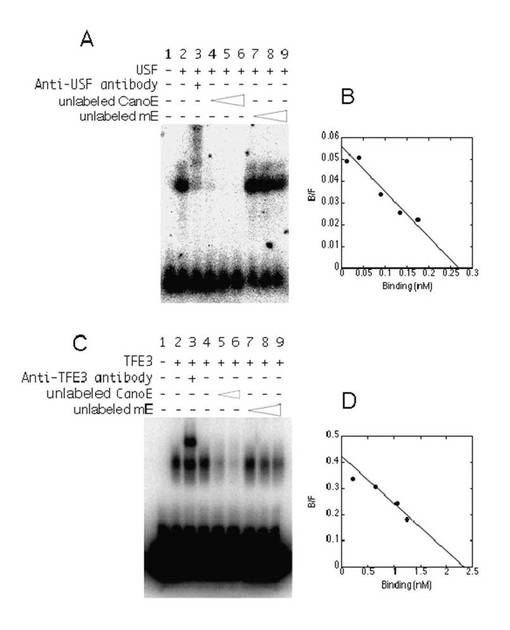
【図8】
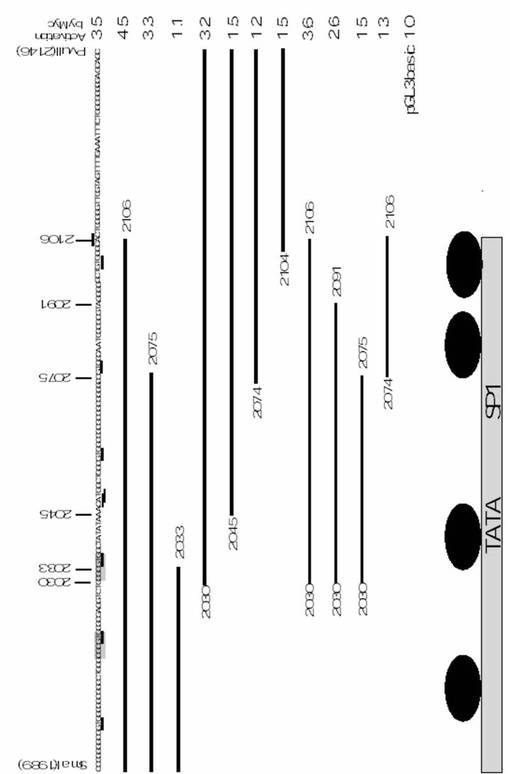
【図1B】
【図2A】
【図2B】
【図2C】
【図3A】
【図3B】
【図3C】
【図4A】
【図4B】
【図4C】
【図5】
【図6】
【図7】
【図8】
【公開番号】特開2007−215486(P2007−215486A)
【公開日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願番号】特願2006−40008(P2006−40008)
【出願日】平成18年2月16日(2006.2.16)
【出願人】(599045903)学校法人 久留米大学 (72)
【Fターム(参考)】
【公開日】平成19年8月30日(2007.8.30)
【国際特許分類】
【出願日】平成18年2月16日(2006.2.16)
【出願人】(599045903)学校法人 久留米大学 (72)
【Fターム(参考)】
[ Back to top ]