
TcおよびRe標識放射活性グリコシル化オクトレオチド誘導体
【課題】グリコシル化され、かつソマトスタチンレセプターに結合する新規放射活性オクトレオチド誘導体を提供すること。
【解決手段】核医学における診断および治療応用のための改良されたsst−レセプター結合ペプチド性リガンドを提供する。改良されたリガンドは、N−末端もしくはC−末端もしくはその両端に天然または非天然アミノ酸またはペプチド類似構造、炭水化物単位およびキレート化剤もしくは接合団を含有し、放射性同位体を結合または保持する放射性同位体の錯体形成物を提供する。該sst−またはSSTR−レセプター結合ペプチド性リガンドは、任意にペプチドにカップリング結合した1個以上の多機能リンカー単位、および/または糖部分および/またはキレート化剤および/または接合団を含有していてもよい。
【解決手段】核医学における診断および治療応用のための改良されたsst−レセプター結合ペプチド性リガンドを提供する。改良されたリガンドは、N−末端もしくはC−末端もしくはその両端に天然または非天然アミノ酸またはペプチド類似構造、炭水化物単位およびキレート化剤もしくは接合団を含有し、放射性同位体を結合または保持する放射性同位体の錯体形成物を提供する。該sst−またはSSTR−レセプター結合ペプチド性リガンドは、任意にペプチドにカップリング結合した1個以上の多機能リンカー単位、および/または糖部分および/またはキレート化剤および/または接合団を含有していてもよい。
【発明の詳細な説明】
【技術分野】
【0001】
本発明はグリコシル化され、かつソマトスタチンレセプターに結合する新規放射活性オクトレオチド誘導体に関する。
【背景技術】
【0002】
これらのいわゆるSSTRリガンドは、ソマトスタチンレセプターのインビボ標的化に適しており、また核医学での広い応用が見出されている。
これら分子の必須の部分は、当該分子の生物活性部分に直接またはリンカーを介して接合する糖部分である。対応する非糖質化誘導体に比べて、これらの誘導体は、レニウムおよびテクネチウムなどの放射性同位体で標識し、直接の還元的方法またはトリカルボニル錯体を経て、改善された腫瘍/非腫瘍蓄積比、改善された薬物動態および改善されたインターナリゼーション現象を示す強力なソマトスタチンレセプターリガンドに誘導し得る。
【発明の概要】
【課題を解決するための手段】
【0003】
本発明のソマトスタチンレセプター結合ペプチド性リガンドは天然の、または非天然(合成)リガンドから調製する。これらのリガンドはN−末端またはC−末端またはその両方に構造上の修飾をもつ。当該ペプチドリガンドはsst−レセプターに対し親和性を有し、図示すると以下の構造により表される:
【化1】

ただし、式中のXはリガンドのC末端を示す。本発明の組成物において多重リガンドが存在し得る場合、ペプチド鎖のC末端とN末端が交じり合って結合することにより、二量体および多量体の形成が可能となる。従って、本発明の範囲は、ホモ二量体、ホモ多量体または異なる結合構造のヘテロ二量体およびヘテロ多量体を包含する。二量体および多量体はペプチド鎖のC末端とN末端が交じり合うカップリングにより形成し得る。
【0004】
本発明のリガンド組成は、多機能となり得る少なくとも1個のリンカー単位を任意に含む。かかるリンカー単位は、縮合反応、アシル化反応、置換反応または付加反応を介して、ペプチド、糖部分およびキレート化剤の結合を可能とする。代表的なリンカー単位はペプチド性または他の有機構造、例えば、L−またはD−アミノ酸(リジン、オルニチン、セリン、グルタミン酸、アスパラギン酸、O−アミノセリンなど)、メルカプトプロピオン酸、ヒドロキシ炭酸類、アミノ炭酸類、ハロゲン炭酸類またはポリアミノ酸類などから得られるリガンドを含んでなる。
【0005】
本発明の改良されたソマトスタチンレセプター結合ペプチド性リガンドは、炭水化物、特に、単糖類、二糖類および三糖類などの糖を含んでなる。代表的な適切な糖は、共有結合を介して結合するグルコース、ガラクトース、マルトース、マンノース、マルトロトリオースおよびラクトースなどである。すなわち、該糖はメイラード(Maillard)反応およびアマドリ(Amadori)転移、グリコシド結合、アルキル化、アリル化反応を介して結合させるか、または修飾後の錯体形成、すなわち、炭水化物イソニトリルまたは炭水化物リン酸エステルの形成を経て結合させることが可能である。
【0006】
本発明の改良されたソマトスタチンレセプター結合ペプチド性リガンドのもう一つの成分はキレート化剤である。代表的なキレート化剤は、TcおよびRe放射性同位体の単座もしくは多座配位錯体形成に適したペプチド性または非ペプチド性構造である。本発明組成物において有用なキレート化剤は、その酸化状態と錯体幾何構造に依存的に、放射性金属の錯体形成に必要な数の官能基を有する1個以上の(リガンドおよびコリガンド)有機分子を含んでなる。模範となる適切なキレート化剤は、例えば、ヒスチジン、ピコリルアミン二酢酸、ヒドロキシニコチンアミド(HYNIC)、メルカプトアセチル−グリシル−グリシル−グリシン(MAG3)およびテトラペプチドである。
【0007】
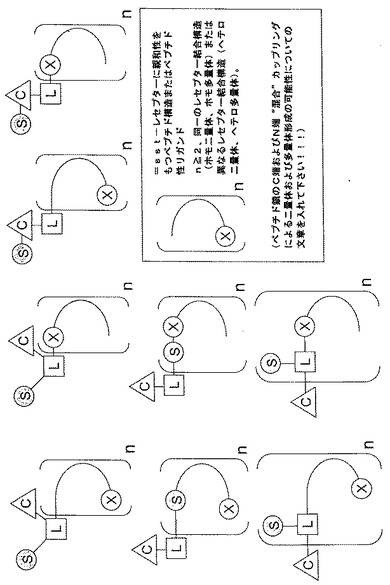
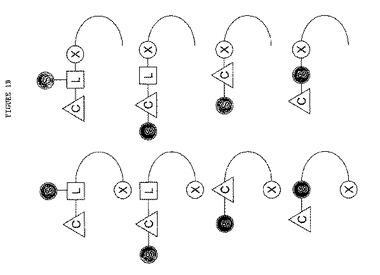
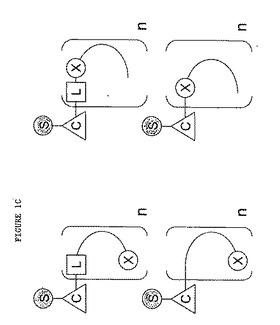
本発明の改良されたソマトスタチンレセプター結合ペプチド性リガンドについてより詳しく記載するために、図1A〜1Cを参照する。図1A〜1Cには、本発明のペプチドリガンドをその中で調製し得る様々な立体配置を図解形式で示す。他の実際の立体配置については本明細書の教示に基づき、当業者が思考し得る。
【0008】
図1A、1Bおよび1Cには、リンカー部分L、糖部分S、およびTcとReの放射性同位体を保持し得るキレート化剤Cを含む単量体、二量体および多量体を示す(ただし、nは2またはそれ以上の整数である)。
【0009】
図1A、1Bおよび1Cにおいては、ペプチドの末端はXによってC末端を表し、その反対側の末端がN末端であるように規定する。従って、薬理作用ペプチドはリンカー、キレート化剤などのC−またはN−端と結合している。上述のように、ペプチドリガンドおよびリンカー(多機能性)はともに天然または非天然のアミノ酸から構成される。勿論、リガンド(すなわち、オクトレオチド)は本発明の組成物においてリンカーとして採用されないであろう。
例えば、本発明は以下の項目を提供する。
(項目1)
天然もしくは非天然アミノ酸またはペプチド類似構造、少なくとも1個の炭水化物、およびTcおよびReから選択される放射性同位体の単座または多座配位錯体形成を可能とする少なくとも1個のキレート化基を含んでなるソマトスタチンレセプター(sst)結合ペプチド性リガンド。
(項目2)
放射性同位体がTcの同位体である項目1記載のリガンド。
(項目3)
該キレート化基がペプチドのN末端に結合し、該炭水化物がキレート化基に結合している項目1記載のリガンド。
(項目4)
該炭水化物がペプチドのN末端に結合し、該キレート化基が該炭水化物に結合している項目1記載のリガンド。
(項目5)
該炭水化物が糖である項目1記載のリガンド。
(項目6)
該糖が単糖類または多糖類である項目5記載のリガンド。
(項目7)
さらに少なくとも1個の多機能リンカー単位を含んでなる項目1記載のリガンド。
(項目8)
該糖がレセプター結合構造、多機能リンカー単位、またはグリコシド結合を介してキレート化剤にカップリング結合する項目7記載のリガンド。
(項目9)
該糖がレセプター結合構造、多機能リンカー単位、またはキレート化剤のアミノ基と、アルドースとの間の、アマドリ反応によるカップリング結合産物である項目7記載のリガンド。
(項目10)
該糖がグルコース、マルトースおよびマルトトリオースからなる群より選択されるものである項目7記載のリガンド。
(項目11)
該リガンドがN末端でグリコシル化されている項目7記載のリガンド。
(項目12)
該放射性同位体が同位体TcおよびReからなる群より選択されるものである項目7記載のリガンド。
(項目13)
該多機能リンカー単位がペプチドのN末端と併合し、該炭水化物と該キレート化基が多機能リンカー単位に結合している項目7記載のリガンド。
(項目14)
該多機能リンカー単位がリガンドのN末端に結合し、該キレート化基が多機能リンカー単位に結合し、該炭水化物がキレート化基に結合している項目7記載のリガンド。
(項目15)
さらに少なくとも1個の多機能リンカー単位および少なくとも2個のsst−レセプター結合構造ペプチド基を含んでなる項目1記載のリガンド。
(項目16)
該2個のペプチド基が多機能リンカー単位に結合し、該キレート化基と該炭水化物双方が多機能リンカーに結合している項目14記載のリガンド。
(項目17)
それぞれがペプチド単位の1個のN末端に結合する2個の炭水化物単位が存在し、当該炭水化物単位それぞれが多機能リンカー単位に結合し、かつ該キレート化基が多機能リンカー単位に結合している項目14記載のリガンド。
(項目18)
それぞれがペプチドのN末端に結合する2個の多機能リンカー単位、多機能リンカー単位に連結する1個のキレート化基、およびそれぞれが多機能リンカー単位の一方に結合する2個の炭水化物とが存在する項目14記載のリガンド。
(項目19)
該放射性同位体がTcおよびReの同位体から選択されるものである項目14記載のリガンド。
(項目20)
該炭水化物が糖であり、糖がレセプター結合構造、多機能リンカー単位、またはキレート化剤のアミノ基と、アルドースとの間の、アマドリ反応によるカップリング結合産物である項目14記載のリガンド。
(項目21)
該糖がレセプター結合構造、多機能リンカー単位、またはグリコシド結合を介してキレート化剤にカップリング結合する項目14記載のリガンド。
(項目22)
項目1記載の組成物と医薬的に許容し得る担体とを含有してなる医薬組成物。
(項目23)
項目7記載の組成物と医薬的に許容し得る担体とを含有してなる医薬組成物。
(項目24)
項目14記載の組成物と医薬的に許容し得る担体とを含有してなる医薬組成物。
(項目25)
項目22記載の組成物を患者に投与することを特徴とするsst−画像化からなる群より選択される画像化方法。
(項目26)
項目23記載の組成物を患者に投与することを特徴とするsst−画像化からなる群より選択される画像化方法。
(項目27)
項目24記載の組成物を患者に投与することを特徴とするsst−画像化からなる群より選択される画像化方法。
(項目28)
化合物[99mTc]Gluc−K0(H)TOC。
(項目29)
化合物[99mTc]Mtr−K0(H)TOC。
【図面の簡単な説明】
【0010】
【図1A】図1Aには、2個以上のペプチドリガンドの多量体を提供する本発明の組成物の様々な立体配置を示す。該多量体は同一のレセプターペプチド結合構造(ホモ二量体、ホモ多量体)または異なるレセプター結合構造(ヘテロ二量体、ヘテロ多量体)を含んでいてもよい。
【図1B】図1Bには、種々量体の立体配置を示すが、その幾つかはリンカー単位(多機能)を含み、その内の4個はリンカー単位を含まない。従って、多機能リンカー単位は本発明の組成物において任意であることが分かる。
【図1C】図1Cには二量体と多量体を示すが、この場合には、多重リンカー単位をペプチドと、勿論、多重ペプチドリガンドの様々な配向で採用する。
【発明を実施するための形態】
【0011】
図1Aおよび図1Cに示す二量体と多量体は、リンカーまたはペプチドリガンドのキレート化剤への共有結合を介して形成されるか、またはリンカーとキレート化剤間の錯体の形成を介して形成される。
本発明を以下の実施例で説明するが、これら実施例は本発明を制限するものではない。(実施例1)
【0012】
ペプチド放射線医薬の合理的な設計:
インビトロでの研究はリガンド誘発SST−2インターナリゼーションに対するオクトレオチドの糖質化およびC末端酸化の相乗作用を示す。
【0013】
目的:その薬物動態による外に、画像化用および治療目的のレセプターに基づくトレーサーの適性は、主としてその薬物動態プロフィルにより決定する。本研究の目的はSSTR細胞内取り込みと排出(インターナリゼーションとエクスターナリゼーション)および再細胞取り込み(再循環)に対するオクトレオチド(octreotide)とオクトレオテート(octreotate)の糖質化の影響を研究することであった。
【0014】
方法:[125I]Tyr3−オクトレオチド(TOC)、[125I]Tyr3−オクトレオテート(TOCA)およびそのグルコース(Gluc)−とマルトトリオース(Mtr)−誘導体のインターナリゼーション、エクスターナリゼーションおよび再循環は、AR42J細胞(SSTR2)の集密単層を用いて検討した。細胞は放射性リガンドと120分までインキュベートした(n=3)。各時点で、上清の活性、表面結合およびインターナリゼーション活性を測定し、TOC値に標準化した。エクスターナリゼーションおよび再循環は120分のインキュベーション後に2時間にわたり検討した。結合リガンドの特異性を10Mサンドスタチン(sandostatin)との競合実験により検討した。
【0015】
結果;インキュベーション2時間後の取り込みリガンド量[取り込まれたTOC%]は以下のとおりであった:Mtr−TOC(35±4%)<Gluc−TOC(121%)<TOCA(154%)<Mtr−TOCA(549%)<Gluc−TOCA(637%)。競合実験において、すべての化合物のインターナリゼーションが、加えた放射活性の<0.1±0.02%(30分)に低下した。リガンドの再循環を可能とするエクスターナリゼーション実験において、TOCAおよびグリケーションしたTOCAは、再循環しない実験と比較して、細胞外に局在する放射活性の2/3を示したが、一方、約80%はTOC、Gluc−TOCおよびMtr−TOCに見出された。TOCの糖質化は細胞表面上のリガンドの利用能に対して有意な作用を示さなかったが、TOCAおよびMtr−TOCAの表面濃度は、TOCに比較してそれぞれ2.1および2.3のファクター増加する。Gluc−TOCAはTOCに比較して、細胞表面上のトレーサー利用能の5倍の増加を示す。TOCのインターナリゼーション率(取り込み/表面結合活性)はグリコシル化により有意な影響を受けないが、TOCAは1.4倍の増加を示す。Gluc−TOCAおよびMtr−TOCAについて、我々はTOCに比べて186%および171%のインターナリゼーション率を観察した。
【0016】
結論:AR42J−細胞を用いて、TOCAを糖質化することにより、放射活性リガンドの細胞表面濃度とインターナリゼーション率を有意に増大させた。
(実施例2)
【0017】
SPECT用糖質化Tc−99M−オクトレオチド誘導体
合成、放射活性標識およびインビボデータ
過去10年にわたり、SPECTおよびPETによるSSTR−シンチグラフィー用の99mTC−および18F−標識オクトレオチド誘導体についての様々な方法が研究されている。Decristoforoらは、EDDAをもつ99mTc−標識HYNIC−Tyr3−オクトレオチドがコリガンドとして好適な生物動力学とマウスにおける高い腫瘍取り込みを示すことを示した。これまでに知られた唯一の18F−標識オクトレオチドである2−[18F]フルオロプロピオニル−D−Phe1−オクトレオチドは優勢な肝胆汁分泌を示すが、これはインビトロSSTR画像化に適用する上での欠点の一つである。
【0018】
本発明者らは、放射活性ヨウ素化Tyr3−オクトレオチド(TOC)とそのThr8−誘導体Tyr3−オクトレオテート(TOCA)をN末端グリコシル化が、生体分布の有意な改善、すなわち、腫瘍への蓄積増大をもたらすことを見出した。この原理の一般的応用を検討するために、我々は99mTc−標識化用のグリコシル化オクトレオチド−およびオクトレテート−誘導体を合成し、評価した(図2)。Lys0(N−His)−TOC(K0(H)TOC)のN−グリコシル化誘導体は、有機金属水和イオン[99mTc(H2O)3(CO)3]+−法1[EgliA. et al. J. Nucl. Med. 40: 1913-1917 (1999)]を用いる99mTc−標識化用前駆体として使用した。該ペプチドは標準Fmoc−SPPSプロトコールに従って合成した。グルコース(Gluc)およびマルトトリオース(Mtr)との接合はアマドリ反応経由で実施した。
Lys5−脱保護ペプチドを99mTc−標識化すると、水和イオンに基づき>97%の放射化学収率で[99mTc]Gluc−K0(H)TOCおよび[99mTc]Mtr−K0(H)TOCを得た。
【0019】
99mTc−標識化誘導体(注射30および120分後)の生体分布の検討はAR42J−腫瘍担持ヌードマウス(n=3〜4)にて実施した。インターナリゼーションおよびエクスターナリゼーションの実験は同じ細胞株を用いて実施した。
【0020】
インターナリゼーションの増加は、対照の[125I]TOCに比較して、[99mTc]Gluc−K0(H)TOCについては2.3±0.8のファクター、また[99mTc]Mtr−K0(H)TOCについては3.6±0.4のファクターで見出された。[125I]TOCはインキュベーションにより急速に細胞から排出されるが、[99mTc]Gluc−K0(H)TOCは120分まで細胞内に取り込まれたままである。
【0021】
[99mTc]Gluc−K0(H)TOCおよび[99mTc]Mtr−K0(H)TOCについて、注射2時間後の生体分布を表1に示す。
【0022】
表1:AR42J腫瘍担持ヌードマウスにおける[99mTc]S1−Lys0(His)TOCおよび[99mTc]S3−Lys0(His)TOC注射2時間後の組織蓄積[%iD/g](n=3〜4)
【表1】
【0023】
グリコシル化化合物は共に注射120分後にsst2陽性組織に高い蓄積性を示す。排泄器官での比較的高い非特異的取り込みならびに血液クリアランスの遅延は、二座ヒスチジンリガンドによる99mTc−コアの不十分な錯体形成によるものと我々は思考する。残りの金属配位部位はチオール含有の本来のタンパク質によりインビボで飽和され、それが血液、肝臓およびその他の排泄器官でのこれら錯体の急速な捕捉に至らしめる。[99mTc(H2O)3(CO)3]+−水和イオンとN−Ac−Hisなどの三座リガンドとの飽和した錯体が形成されると、非腫瘍組織での活性蓄積は有意に低下する。
【0024】
結論として、オクトレオチド(−テート)の炭水化物接合および[99mTc(H2O)3(CO)3]+−水和イオンの三座錯体形成の組合わせに基づく導入トレーサーの設計は新しい一連の非常に有望なSSTR−トレーサーの基礎となると言える。
(実施例3)
【0025】
SST−レセプター−アゴニストのグリケーション:放射活性標識ソマトスタチンアゴニストの動的リガンド移動の改善
糖質化は放射活性標識化オクトレオチドおよび∀v∃3類似体の薬物動態改善のための強力な手段であることが今回判明した。オクトレオチドおよびオクトレオテートのグリケーションは肝胆汁排泄および腎吸収を有意に低下させ、腫瘍取り込みと腫瘍/組織比を上昇させる。放射活性sst2−アゴニストの腫瘍蓄積は、レセプター介在インターナリゼーション、崩壊および引き続く細胞内蓄積に、またはリガンドおよび/または代謝産物の両方の再循環に左右される。各段階で定量分析すると、決定的に重要なことは、sst2発現腫瘍細胞におけるトレーサーの蓄積を如何に制御するかであることが理解される。従って、本研究の目的は、放射活性標識オクトレオチドとオクトレオテートのインターナリゼーションおよび再循環動力学に対するグリケーションの影響を試験することであった。
【0026】
Tyr3−オクトレオチド(TOC)、Tyr3−オクトレオテート(TOCA)およびその誘導体であるグルコース(Gluc)、マルトース(Malt)およびマルトトリオース(Mtr)誘導体は、Fmoc−SPPSおよび引き続く炭水化物接合を経て合成した。放射活性ヨウ素化はヨードゲン法により実施した。放射化学収率はRP−HPLC精製後、50〜84%の範囲であった。99mTc−標識化(Gluc−Lys0(N−His)TOC(Gluc−K(H)−TOC)およびMtr−Lys0(N−His)TOC(Mtr−K(H)−TOC))のための前駆体は、上記のペプチド同様に調製した。99mTcでの標識化はすでに記載した[99mTc(H2O)3(CO)3]+−水和イオンプロトコールに従って実施した[EgliA. et al. J. Nucl. Med. 40: 1913-1917 (1999)]。比較のために、我々は[111In]オクトレオスキャン(Octreoscan)および[111In]ドタトック(DOTATOC)をも我々の検討に含めた。
【0027】
インターナリゼーションとエクスターナリゼーションの実験は、sst2発現ラット膵臓腫瘍細胞株AR42Jを用いて実施した。遊離の表面結合内部に取込まれた放射活性は、放射活性リガンドと37℃でインキュベートし、その10、30、60、90および120分後に定量した。リガンド再循環を可能とするエクスターナリゼーション実験では、10、30、60、90および120分の間に上清に放出された放射活性フラクションならびに細胞中に残存する放射活性を定量した。我々はまた引き続く5回のインキュベーション(10、20および3×30分)に際して、培地の中間変化(制限再循環)を伴って放出された活性を測定した。
【0028】
インターナリゼーションに対するTOC糖質化の影響は、使用した糖の大きさに平行する。[125I]TOCに比較して、グルコース接合体のみが細胞への取込みを上昇させた。対照的に、[99mTc]Gluc−TOCAおよび[99mTc]Mtr−TOCA両方の細胞内蓄積(図3および表2)は有意に増大した([125I]TOCに比較して、7.36±0.50および5.68±0.38倍)。TOCとTOCAのインターナリゼーション特性およびそれぞれのグリコシル化誘導体のインターナリゼーション特性を比較して、我々は、表面利用能およびインターナリゼーション率に対する両方の構造的修飾(Thr8によるThr(ol)8の置換および糖質化)の相乗作用を観察した。[111In]オクトレオスキャン(Octreoscan)および[111In]ドタトック(DOTATOC)に比較して、両方の99mTc−標識化糖質化誘導体のインターナリゼーションは意外に高かった。
【0029】
10μMサンドスタチン(sanndostatin)による事前処理実験は、リガンドの取込みを対照の最大5%まで低下させ、i)sst2特異取込みであって、ii)炭水化物関連取込みメカニズムは関与しないことを示した(表2)。検討したトレーサーすべてについて、内部取込み活性はリガンドの表面利用能と強力に相関する。
【0030】
表2:AR42J細胞と60分間インキュベーション後に得られたインターナリゼーション・データ(インターナリゼーション値ならびに比は対照の[125I]TOCに標準化する)
a:表面利用能=%表面結合リガンド/%遊離放射活性リガンド
b:インターナリゼーション率=%内部取込みリガンド/%表面結合リガンド
*:10Mサンドスタチンでの事前処理実験は細胞リガンド取込みを最小95%低下させた。
【表2】
【0031】
エクスターナリゼーションに関しては、グリコシル化が影響していないように思われる(図4)。細胞からの放射リガンドのエクスターナリゼーションの度合いは、主としてペプチド(TOCまたはTOCA)の性質により、また標識の細胞内運命により判定する。リガンドの再循環を可能とするエクスターナリゼーション実験において、TOCAとそのグリケーション誘導体は再循環なしの実験に比較して、細胞外に局在する放射活性の約2/3を示した。一方、約80%がTOC、Gluc−TOCおよびMtr−TOCに見出された。[99mTc]Gluc−K0(H)TOCならびに両方の111In−標識化合物は、再循環条件下、120分以内に細胞中略定量的な保持を示した。対照的に、[111In]オクトレオスキャン(Octreoscan)および[111In]ドタトック(DOTATOC)の細胞内活性は、再循環を制限した場合、120分間に33%および49%消尽した。
(実施例4)
【0032】
[Tc−99m](CO)3−標識糖質化SSTR−リガンド:
合成、インターナリゼーション動力学およびラット膵臓腫瘍モデルでの生体分布
目的:本研究はSSTR発現腫瘍の特異的高レベル標的化用トレーサーの設計を示すためのものである。これを目的として、我々は薬物動態を最適化し、SSTR特異インターナリゼーションと腫瘍保持を改善するために、糖質化オクトレオチド誘導体を導入した。本明細書にて我々が記載する化合物は一連の新規糖質化99mTc−SSTRリガンドである。
【0033】
方法:グルコース(Gluc)およびマルトトリオース(Mtr)をN−(Boc−His(Boc))Lys0−Tyr3−Lys5(Dde)−オクトレオチド(保護K(H)TOC)のN−Lys0に結合させ、最終的に脱保護し、Gluc−およびMtr−K(H)TOCを得た。99mTc−標識化は[99mTc(H2O)3(CO)3]+を用いて実施した。インターナリゼーションとエクスターナリゼーションの検討(<2時間)はSSTR2発現ラット膵臓細胞株AR42Jおよび対照としての[125I]Tyr3−オクトレオチド([125I]TOC)を用いて実施した。生体分布はAR42J腫瘍担持ヌードマウスにて、0.5時間および2時間のインキュベーション(n=4)により評価した。
【0034】
結果:放射活性標識は効率的に(0.5時間、RCY>97%)均一の産物を生じた。[125I]TOCに比較して、両方の炭水化物導入ペプチドは有意により高い細胞間活性レベルを示した;例えば、[99mTc]Gluc−K(H)TOCについては2時間で2.3±0.2倍高いレベル、また[99mTc]Mtr−K(H)TOCについては3.6±0.4倍高いレベルを示した。エクスターナリゼーションの検討では、2時間で[125I]TOCに対し約50%の細胞内活性の低下を明らかにした。一方、[99mTc]Gluc−K(H)TOCの細胞間レベルは2時間まで殆ど安定であり、このトレーサーが保持されていることを示した。インビボでは、[99mTc]Gluc−K(H)TOC(T/肝臓0.77、T/腎層0.63)および[99mTc]Mtr−K(H)TOC(T/肝臓0.97、T/腎臓0.84)に対して、腫瘍の蓄積は注射2時間後にそれぞれ12.2±1.0および14.0±6.3%ID/gに達し、血中レベルは1.5±0.2%ID/gおよび4.4±1.0%ID/gであった。
結論:これらの結果はSSTR標的化改善の手段として糖質化が有用であることを証明している。
(実施例5)
【0035】
[Tc−99m](CO)3−標識糖質化SSTR−リガンド:
合成、インターナリゼーション動力学およびラット膵臓腫瘍モデルでの生体分布
目的:本研究はSSTR発現腫瘍の特異的高レベル標的化用トレーサーの設計を示すための我々の努力の一部である。この目的のために、我々は薬物動態を最適化し、SSTR特異インターナリゼーションと腫瘍保持を改善するのに適当な糖質化部分を使用した。本明細書に我々は初めて[99mTc(H2O)3(CO)3]+水和イオン法により標識化した糖質化SSTR結合ペプチドを記載する。
【0036】
方法:標準的なFmoc化学により、N−(Boc−His(Boc))Lys0−Tyr3−Lys5(Dde)−オクトレオチド(保護K(H)TOC)を合成した。グルコース(Gluc)およびマルトトリオース(Mtr)をN−Lys0に結合させ、最終的に脱保護し、K(H)TOCのGluc−およびMtr−誘導体を得た。Tc−99m−標識化は有機金属水和イオン[99mTc(H2O)3(CO)3]+(1)を用いて実施した。インターナリゼーションとエクスターナリゼーションの検討(<120分)はSSTR2発現細胞株AR42Jを用い、両化合物について実施した。インビトロの検討のために、対照として[125I]Tyr3−オクトレオチド([125I]TOC)を用いた。静脈内投与した[99mTc]Gluc−K(H)TOCおよび[99mTc]Mtr−K(H)TOCの生体分布はAR42Jラット膵臓腫瘍担持ヌードマウスにて、注射30分および120分(n=4)後に評価した。
【0037】
結果:本標識手法は効率的に(30分間、RCY>97%)均一の産物を生じた(HPLC)。[125I]TOCに比較して、両方の糖質化ペプチドは検討したすべての時点で有意により高いインターナリゼーションを示した;例えば、[99mTc]Gluc−K(H)TOCについては2時間で2.3±0.2倍高いレベル、また[99mTc]Gluc−K(H)TOCについては3.6±0.4倍高いレベル、また[99mTc]Mtr−K(H)TOCについては3.6±0.4倍高いレベルを示した。さらに、エクスターナリゼーションの検討では、120分で対照化合物の約50%の細胞内活性の低下を明らかにした。一方、細胞内[99mTc]Gluc−K(H)TOCの高いレベルは2時間まで殆ど安定であり、このトレーサーが検討した細胞株に保持されていることを示した。インビボでは、[99mTc]Gluc−K(H)TOC(T/肝臓0.77、T/腎層0.63)および[99mTc]Mtr−K(H)TOC(T/肝臓0.97、T/腎臓0.84)に対して、腫瘍の蓄積が注射120分後にそれぞれ12.2±1.0および14.0±6.3%ID/gに達し、対応する血中レベルは1.5±0.2%ID/gおよび4.4±1.0%ID/gであった。
結論:これらの結果はSSTR標的化改善の手段として炭水化物導入が有用であることを示している。
(実施例6)
【0038】
固相ペプチド合成
1. Fmoc−Lys0−Tyr3−Lys(Dde)5−オクトレオチド(Fmoc−Lys0−TOC(Dde))Fmoc−Thr(tBu)−オール Fmoc−Thr(tBu)−OH(1.391g、3.5mmol)をテトラヒドロフラン(THF)30mlに溶かした溶液を−10℃に冷却した。N−メチルモルホリン(386μl、3.5mmol)およびクロロギ酸エチル(333μl、3.5mmol)を引き続き加えた。反応混合物を−10℃で30分間撹拌した。次いで、ホウ素化水素ナトリウム(396mg、10.5mmol)を加えた。30分を要してメタノール60mlをゆっくりと反応混合物に加え、次いで、0℃で3時間撹拌した。
【0039】
1N−HClを50〜70ml添加した後(濁りのある反応混合物が透明にならなければならない)、有機溶媒を蒸発させ、残りの水相はジクロロメタン(DCM)で2回抽出した。抽出効率は両相の薄層クロマトグラフィー(酢酸エチル(AcOEt)による産物のRf=0.83)によりモニターした。併合した有機層を硫酸マグネシウム上乾燥し、濾過、減圧濃縮し、黄色油状物を得た。粗生成物をAcOEtによるフラッシュクロマトグラフィーにより精製した。産物は油状物(1.19g、89%)として得た。
単一同位体質量;C23H29NO4として計算値=383.21;測定値:m/z406.1[M+Na]+
【0040】
Fmoc−Lys0−Dphe1−Cys2−Tyr3−DTrp4−Lys(Dde)5−Thr6−Cys7−Thr−オール8 DHP−HM(3,4−ジヒドロ−2H−ピラン−2−イルメトキシメチルポリスチロール)樹脂(1.000g、0.94mmol/g)を10mlの乾燥DCE(1,2−ジクロロエタン)中、1時間、予め膨潤させた。次いで、Fmoc−Thr(tBu)−オール(1.266g、3.3mmol)とp−トルエンスルホン酸ピリジニウム(414mg、1.75mmol)を7mlの乾燥DCEに溶かした溶液を加え、その反応混合物をアルゴン下、80℃で一夜撹拌した。遊離の官能基を樹脂に取り付けるために、ピリジン5mlを加え、懸濁液を室温で15分間撹拌した。負荷した樹脂を次いで濾過し、DMF(N,N−ジメチルホルムアミド)で2回、およびDCMで洗浄し、乾燥器中で乾燥した。樹脂の最終負荷物は0.834mmol/gであった。ペプチド配列は標準Fmoc−プロトコールを用いて樹脂結合アミノアルコール上で推定した。簡単に説明すると、N−末端FmocをDMF中の20%ピペリジンで除去し、樹脂支持ペプチドの樹脂をNMP(N−メチルピロリドン)で洗浄した。カップリング反応はNMP中、1.5当量のアミノ酸誘導体と、1.5当量のHOBt(1−ヒドロキシベンゾトリアゾール)、1.5当量のTBTU(O−(1H−ベンゾールトリアゾール−1−イル)−N,N,N',N'−テトラメチルウロニウム−テトラフルオロボレート)および3.5当量のDIPEA(N−エチル−ジイソプロピルアミン)と反応させることにより実施した。活性化したアミノ酸を反応容器に加えた後、適当な時間(通常40〜60分)、樹脂を撹拌した。次いで、樹脂を追加のNMPで洗浄した。TOC合成のための添加の順序は、Fmoc−Cys(Trt)、Fmoc−Thr(tBu)、Fmoc−Lys(Dde)、Fmoc−DTrp、Fmoc−Tyr、Fmoc−Cys(Trt)、Fmoc−DpheおよびFmoc−Lys(Boc)の順であった。
【0041】
最終アミノ酸を配列にカップリング結合させた後、樹脂を数回NMPおよびDCMで洗浄した。樹脂からの脱離およびThr−、Cys−およびLys0−側鎖の脱保護は、TFA(トリフルオロ酢酸)/TIBS(トリイソブチルシラン)/水(95:2.5:2.5)およびDCMの混合物(1:1)を用いて実施した。90分後に樹脂を濾取し、DCMで2回洗浄した。併合した濾液をロータリーエバポレーターにて濃縮し、粗製の産物をエーテルで沈殿させた。我々は828mgの粗生成物を得た。収率83%。
【0042】
粗製のペプチドは(ぺプチド300mgあたり)50mlのTHFに再懸濁し、澄明な溶液が得られるまで、5mM酢酸アンモニウムを添加した。飽和NaHCO3溶液を滴下することでpH7に調整した。次いで、過酸化水素水(30%)100μl(ペプチド300mgあたり)を加えた。室温で30分間撹拌した後、環化を終了した(勾配HPLC−コントロール:30〜80%B、15分)。溶媒を蒸発させ、環化した生成物を凍結乾燥した。収率は定量的であった。
単一同位体質量;C80H100N12O16S2として計算値=1548.68;測定値:m/z1549.5[M+H]+、m/z=1571.5[M+Na]+
【0043】
2. Fmoc−Lys0−Tyr3−Lys(Dde)5−オクトレオチド(Fmoc−Lys0−TATE(Dde))Fmoc−Lys0−Dphe1−Cys2−Tyr3−DTrp4−Lys(Dde)5−Thr6−Cys7−Thr8 乾燥TCP−樹脂1.502gに、Fmoc−Thr(tBu)894mg(2.25mmol)、DIPEA321μl(1.87mmol)および乾燥DCM15mlからなる溶液を加えた。5分間撹拌した後、追加のDIPEA642μl(3.76mmol)を加えた。室温で90分間、激しく撹拌した後、メタノール1.5mlを加えて、未反応の塩化トリチル基を遮断した。15分後、樹脂を濾取し、DCM、DMFおよびメタノールでそれぞれ2回洗浄し、減圧乾燥した。樹脂の最終的負荷は0.77mmol/gであった。樹脂結合アミノ酸上のペプチド配列、Cys(Trt)−Thr(tBu)−Lys(Dde)−DTrp−Tyr−Cys(Trt)−DPhe−Lys(Boc)−Fmocの合成、脱保護、および樹脂からの切断、ならびに環化反応は、Fmoc−Lys0−TOC(Dde)について記載したのと同様に実施した。
単一同位体質量;C80H98N12O17S2として計算値=1562.66;測定値:m/z= ([M+H]+)
【0044】
溶液相誘導化
1. Lys0(N∈−Boc−His(Boc))−TOC(Dde)
Fmoc−Lys0−TOC(Dde)500mg(0.32mmol)、Boc−His(Boc)170mg(0.48mmol)、HOBt65mg(0.048mmol)、TBTU154mg(0.48mmol)およびDIPEA300μl(1.75mmol)をDMF3mlに溶かした溶液を室温で撹拌した。RP−HPLC分析(勾配:30〜80%B、15分)により、反応は30分以内に終了することが判明した。生成物を水で沈殿させ、減圧下に乾燥した。N末端Fmoc−保護基を除去するために、ペプチドを1mlのピペリジン/DMF(1:4)に溶解し、15分間撹拌した。エーテルで沈殿させ、脱保護産物を86%の収率で得た。
単一同位体質量;C81H113N15O19S2として計算値=1663.78;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+
【0045】
2. Lys0(Boc−His(Boc))−TOC(Dde)のLys0−N−アマドリ誘導体
以前に報告された方法1に基づき、1当量のLys0(Boc−His(Boc))−TOC(Dde)および10当量のそれぞれのアルドース(グルコース=S1、マルトース=S2、マルトトリオース=S3)をメタノール/酢酸(95:5)に溶かし、その反応混合物を60℃で24〜48時間撹拌した。次いで、溶媒を蒸発させ、粗生成物をエーテル添加により沈殿させた。
【0046】
Dde−保護基を除去するために、グリコシル化ペプチドを少量のDMFに再溶解し、2容量%のヒドラジン水和物を添加し、その溶液を室温で15分間撹拌した。脱保護ペプチドをエーテル添加により沈殿させ、エーテルで洗浄して減圧乾燥した。His−残基上のBoc−保護基は、粗生成物を少量のTFA/TIBS/水(95:2.5:2.5)に再溶解し、室温にて15分間撹拌することにより除去した。生成物をエーテルで沈殿させ、調製用RP−HPLC(勾配:10〜60%B,30分)にて精製した。収率は17〜32%の範囲であった。
【0047】
S1−Lys0(His)TOC(Na−(1−デオキシ−D−フルクトシル)−Lys0N−His)−Tyr3−オクトレオチド:
単一同位体質量;C67H95N15O18S2として計算値=1461.60;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+S2 Lys0(His)TOC(N−(O− −D−グルコピラノシル−(1−4)−1−デオキシ−D−フルクトシル)−Lys0(N∈−His)−Tyr3−オクトレオチド):
単一同位体質量;C73H105N15O28S2として計算値=1623.70;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+
S3 Lys0(His)TOC(Na−(O−a−D−グルコピラノシル−(1−4)−O−a−D−グルコピラノシル−(1−4)−1−デオキシ−D−フルクトシル)−Lys0(N−His)−Tyr3−オクトレオチド:
単一同位体質量;C73H115N15O23S2として計算値=1785.75;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+
【0048】
3. Lys0(N−2−ピコリルアミノ−....)TATE(Dde)
2−ピコリルアミンN,N−二酢酸2
イミノ二酢酸(3.25g、24.4mmol)を無水エタノール30mlおよびNaOH(1.95g;49mmol)水溶液(10ml)の混合物に溶解した。水9mlに溶かした塩化ピコリル4.00g(24.4mmol)および水4mlに溶かしたNaOH1.95g(49mmol)を添加した後、その溶液を70℃で2時間撹拌した。追加のNaOHを1.95g(49mmol)添加した後、その溶液をさらに70℃で1.5時間撹拌した。TLC(EtOAc)により反応を追跡し、その時点で反応物が完全に消失していることを確認した。溶媒を減圧下に蒸発させ、残渣を水25mlに再溶解した。溶液のpHを濃塩酸により1.5に調整した後、溶液を再度蒸発乾固した。残渣をメタノールで再懸濁して還流する工程を数回連続して繰返し、懸濁液を熱時濾過し、濾液を減圧下に濃縮して過剰のNaClを成功裏に除去した。ピコリルアミノ二酢酸を残りのメタノール溶液から一夜−20℃で結晶化した。
単一同位体質量;C10H12N2O4として計算値=224.08;測定値:m/z= [M+H]+(二ナトリウム塩)
【0049】
2−ピコリルアミンN,N−二酢酸のFmoc−Lys0−TATE(Dde)へのカップリング反応
2−ピコリルアミンN,N−二酢酸52mg(0.19mmol)、HOBt26mg(0.19mmol)、TBTU62mg(0.19mmol)、およびDIPEA175μl(1.75mmol)を2mlのDMFに溶かした溶液を室温で10分間撹拌した。この溶液をFmoc−Lys0−TATE(Dde)200mg(0.13mmol)のDMF(1ml)溶液に激しく撹拌しながら滴下した。30分後、生成物をジエチルエーテルで沈殿させ、減圧下に乾燥した。N末端Fmoc−保護基を除去するために、ペプチドをピペリジン/DMF(1:4)2mlに溶解し、10分間撹拌した。エーテルで沈殿させることにより、脱保護した産物を84%の収率で得た。
単一同位体質量;C75H98N14O18S2として計算値=1546.66;測定値:m/z=[M+H]+、m/z=[M+Na]+
【0050】
4. Lys0(Pic)TATE(Dde)のLys0−Na−アマドリ誘導体
Lys0(Pic)TATE(Dde)とグルコースの接合、および引き続くDde−脱保護は、Lys0(Boc−His(Boc))−TOC(Dde)のLys0−Na−アマドリ誘導体について記載したと同様に実施した。
単一同位体質量;C71H96N14O21S2として計算値=1544.63;測定値:m/z=[M+H]+、m/z=[M+Na]+
【0051】
放射活性標識化
最近報告された有機金属水和イオン[99mTc(H2O)3(CO)3]+−法3に基づき、10mgのNaBH4、8mgのNa2CO3および41.5mgのNa/K酒石酸塩を、ガラスフラスコ中600μlの[99mTc]ペルテクネテート溶離液に溶解した。次いで、フラスコをCOを満たした風船につなぎ、密封した。75℃で45分間撹拌した後、溶液を氷浴上で冷却し、150μlのリン酸緩衝食塩液(PBS、pH7.4)を加えた。還元剤を分解するために600μlの1N−HClを加えた。次いで、溶液を500μlの飽和NaHCO3溶液で中和した。[99mTc(H2O)3(CO)3]+−水和イオンの存在はTLC(5%conc. HCl/メタノール)にて確認した。
【0052】
S1−Lys0(His)TOC700μgに1mlの[99mTc(H2O)3(CO)3]+−溶液(51MBq)を加えた後、混合物を75℃に30分間加熱した。勾配HPLC精製により、放射化学的に純粋なペプチドNa−(1−デオキシ−D−フルクトシル)−Lys0(N−His[99mTc(H2O)3(CO)3]+)−Tyr3−オクトレオチドを34%の収率で得た。
S3−Lys0(His)TOCは同様の方法で標識した。
【0053】
親油性の定量
放射活性標識ペプチド約300000cpmをPBS(pH7.4)500mlに溶かした溶液に、オクタノール500mlを添加した(n=6)。バイアルを3分間ボルテックス処理し、15000rpmで6分間遠心分離した。PBSとオクタノールの100mlを取り、γ−カウンターにて測定した。
S1−Lys0(His)TOC:logpow=−0.673±0.064
S3−Lys0(His)TOC:logpow=−1.304±0.040
インビトロ実験
AR42J細胞を、10%FCSおよび2mM−L−グルタミン添加RPMI1640中に維持した。
【0054】
インターナリゼーションの検討
1ウエルあたり8×105〜5×106個のAR42J細胞の細胞集団を容れた6穴プレートを、実験に先立ち、新鮮な培地(RPMI1640、1%BSA、2mM−L−グルタミン)とともに1時間インキュベートした。培地10μl中、約300000cpmの放射活性金属ペプチドを各ウエルに加えた(対照化合物[125I]TOC(内部基準)および各対象の放射活性金属化合物について、3組の実験とする)。37℃で10、30、60、90または120分間インキュベートした後、上清を採取し、細胞は冷培地1mlにて1回洗浄した。上清を前工程の上清と併合した。レセプター結合(酸解離可能)放射活性を除くために、細胞を0.02M−NaOAc(酸洗浄)1mlと37℃でインキュベートした。10分後に上清を採取し、細胞を0.02M−NaOAc1mlにて1回洗浄し、その上清を前工程の上清と再度併合した。次いで、細胞を1mlの1N−NaOHで抽出し、ウエルはPBSにて1回洗浄した。このプールしたフラクションはインターナリゼーションの放射活性リガンドを示している。NaOHフラクションならびに上清と酸洗浄フラクションの活性をγ−カウンターにて計測した。
【0055】
エクスターナリゼーションの検討
インターナリゼーションの検討にて記載したように、新鮮な培地(RPMI1640、1%BSA、2mM−L−グルタミン)を容れた6穴プレートを、実験に先立ち、37℃で1時間インキュベートした。培地10μl中、約300000cpmの放射活性金属ペプチドを各ウエルに加えた(対照化合物[125I]TOC(内部基準)および各対象の放射活性金属化合物について、3組の実験とする)。37℃にて120分間インキュベートした後、上清を採取し、細胞は冷培地1mlにて1回洗浄した。上清を前工程の上清と併合した。エクスターナリゼーションの放射活性を採取するために、細胞を1mlの培地とともに37℃でインキュベートした。10、30、60、90および120分後に、それぞれ上清を集め、細胞は1mlの培地で1回洗浄し、その上清を前工程の上清と併合した。次いで、細胞を1mlの1N−NaOHで抽出し、ウエルはPBSにて1回洗浄した。このプールしたフラクションは細胞中になお局在する放射活性リガンドを示している。NaOHフラクションならびに上清とエクスターナリゼーションフラクションの活性をγ−カウンターにて計測した。
【0056】
トレーサー再循環なしのエクスターナリゼーションの検討
前記実験について記載したのと同様に、6穴プレートを放射活性リガンドとともに37℃にて120分間インキュベートした。上清を採取し、細胞は冷培地1mlにて1回洗浄した。上清を前工程の上清と併合した。エクスターナリゼーションの放射活性を採取するために、細胞を1mlの培地とともに37℃でインキュベートした。10分後に上清を採取し、細胞は再度さらに20分間37℃にて培地1mlとインキュベートした。上清を採取し、細胞は1mlの培地とともに37℃にて30分間さらに3回インキュベートした。次いで、細胞を1mlの1N−NaOHで抽出し、ウエルはPBSにて1回洗浄した。このプールしたフラクションは細胞中になお局在する放射活性リガンドを示している。NaOHフラクションならびにすべての時点でのエクスターナリゼーションフラクションの活性をγ−カウンターにて計測した。
【0057】
生体分布の検討
腫瘍増殖を確立するために、サブコンフルエント単層細胞を1mM−EDTAで処理し、懸濁し、遠心分離し、RPMI1640に再懸濁した。
ヌードマウス(オスおよびメス、6〜8週令)の脇腹に、PBS(pH7.4)100μlに懸濁した細胞2.5〜3×107個を皮下接種し、AR42J腫瘍担持ヌードマウスの尾部静脈に注射した。動物(n=3〜4)を注射後30分および120分後に屠殺し、対象臓器を摘出した。秤量した組織サンプルにつきガンマカウンターにて放射活性を測定した。データは%iD/g組織で表す(平均±DS、n=3〜4)。
【0058】
表1:AR42J腫瘍担持ヌードマウスにおける[99mTc]S1−Lys0(His)TOCおよび[99mTc]S3−Lys0(His)TOC注射2時間後の組織蓄積[%iD/g](n=3〜4)
【表3】
【技術分野】
【0001】
本発明はグリコシル化され、かつソマトスタチンレセプターに結合する新規放射活性オクトレオチド誘導体に関する。
【背景技術】
【0002】
これらのいわゆるSSTRリガンドは、ソマトスタチンレセプターのインビボ標的化に適しており、また核医学での広い応用が見出されている。
これら分子の必須の部分は、当該分子の生物活性部分に直接またはリンカーを介して接合する糖部分である。対応する非糖質化誘導体に比べて、これらの誘導体は、レニウムおよびテクネチウムなどの放射性同位体で標識し、直接の還元的方法またはトリカルボニル錯体を経て、改善された腫瘍/非腫瘍蓄積比、改善された薬物動態および改善されたインターナリゼーション現象を示す強力なソマトスタチンレセプターリガンドに誘導し得る。
【発明の概要】
【課題を解決するための手段】
【0003】
本発明のソマトスタチンレセプター結合ペプチド性リガンドは天然の、または非天然(合成)リガンドから調製する。これらのリガンドはN−末端またはC−末端またはその両方に構造上の修飾をもつ。当該ペプチドリガンドはsst−レセプターに対し親和性を有し、図示すると以下の構造により表される:
【化1】
ただし、式中のXはリガンドのC末端を示す。本発明の組成物において多重リガンドが存在し得る場合、ペプチド鎖のC末端とN末端が交じり合って結合することにより、二量体および多量体の形成が可能となる。従って、本発明の範囲は、ホモ二量体、ホモ多量体または異なる結合構造のヘテロ二量体およびヘテロ多量体を包含する。二量体および多量体はペプチド鎖のC末端とN末端が交じり合うカップリングにより形成し得る。
【0004】
本発明のリガンド組成は、多機能となり得る少なくとも1個のリンカー単位を任意に含む。かかるリンカー単位は、縮合反応、アシル化反応、置換反応または付加反応を介して、ペプチド、糖部分およびキレート化剤の結合を可能とする。代表的なリンカー単位はペプチド性または他の有機構造、例えば、L−またはD−アミノ酸(リジン、オルニチン、セリン、グルタミン酸、アスパラギン酸、O−アミノセリンなど)、メルカプトプロピオン酸、ヒドロキシ炭酸類、アミノ炭酸類、ハロゲン炭酸類またはポリアミノ酸類などから得られるリガンドを含んでなる。
【0005】
本発明の改良されたソマトスタチンレセプター結合ペプチド性リガンドは、炭水化物、特に、単糖類、二糖類および三糖類などの糖を含んでなる。代表的な適切な糖は、共有結合を介して結合するグルコース、ガラクトース、マルトース、マンノース、マルトロトリオースおよびラクトースなどである。すなわち、該糖はメイラード(Maillard)反応およびアマドリ(Amadori)転移、グリコシド結合、アルキル化、アリル化反応を介して結合させるか、または修飾後の錯体形成、すなわち、炭水化物イソニトリルまたは炭水化物リン酸エステルの形成を経て結合させることが可能である。
【0006】
本発明の改良されたソマトスタチンレセプター結合ペプチド性リガンドのもう一つの成分はキレート化剤である。代表的なキレート化剤は、TcおよびRe放射性同位体の単座もしくは多座配位錯体形成に適したペプチド性または非ペプチド性構造である。本発明組成物において有用なキレート化剤は、その酸化状態と錯体幾何構造に依存的に、放射性金属の錯体形成に必要な数の官能基を有する1個以上の(リガンドおよびコリガンド)有機分子を含んでなる。模範となる適切なキレート化剤は、例えば、ヒスチジン、ピコリルアミン二酢酸、ヒドロキシニコチンアミド(HYNIC)、メルカプトアセチル−グリシル−グリシル−グリシン(MAG3)およびテトラペプチドである。
【0007】
本発明の改良されたソマトスタチンレセプター結合ペプチド性リガンドについてより詳しく記載するために、図1A〜1Cを参照する。図1A〜1Cには、本発明のペプチドリガンドをその中で調製し得る様々な立体配置を図解形式で示す。他の実際の立体配置については本明細書の教示に基づき、当業者が思考し得る。
【0008】
図1A、1Bおよび1Cには、リンカー部分L、糖部分S、およびTcとReの放射性同位体を保持し得るキレート化剤Cを含む単量体、二量体および多量体を示す(ただし、nは2またはそれ以上の整数である)。
【0009】
図1A、1Bおよび1Cにおいては、ペプチドの末端はXによってC末端を表し、その反対側の末端がN末端であるように規定する。従って、薬理作用ペプチドはリンカー、キレート化剤などのC−またはN−端と結合している。上述のように、ペプチドリガンドおよびリンカー(多機能性)はともに天然または非天然のアミノ酸から構成される。勿論、リガンド(すなわち、オクトレオチド)は本発明の組成物においてリンカーとして採用されないであろう。
例えば、本発明は以下の項目を提供する。
(項目1)
天然もしくは非天然アミノ酸またはペプチド類似構造、少なくとも1個の炭水化物、およびTcおよびReから選択される放射性同位体の単座または多座配位錯体形成を可能とする少なくとも1個のキレート化基を含んでなるソマトスタチンレセプター(sst)結合ペプチド性リガンド。
(項目2)
放射性同位体がTcの同位体である項目1記載のリガンド。
(項目3)
該キレート化基がペプチドのN末端に結合し、該炭水化物がキレート化基に結合している項目1記載のリガンド。
(項目4)
該炭水化物がペプチドのN末端に結合し、該キレート化基が該炭水化物に結合している項目1記載のリガンド。
(項目5)
該炭水化物が糖である項目1記載のリガンド。
(項目6)
該糖が単糖類または多糖類である項目5記載のリガンド。
(項目7)
さらに少なくとも1個の多機能リンカー単位を含んでなる項目1記載のリガンド。
(項目8)
該糖がレセプター結合構造、多機能リンカー単位、またはグリコシド結合を介してキレート化剤にカップリング結合する項目7記載のリガンド。
(項目9)
該糖がレセプター結合構造、多機能リンカー単位、またはキレート化剤のアミノ基と、アルドースとの間の、アマドリ反応によるカップリング結合産物である項目7記載のリガンド。
(項目10)
該糖がグルコース、マルトースおよびマルトトリオースからなる群より選択されるものである項目7記載のリガンド。
(項目11)
該リガンドがN末端でグリコシル化されている項目7記載のリガンド。
(項目12)
該放射性同位体が同位体TcおよびReからなる群より選択されるものである項目7記載のリガンド。
(項目13)
該多機能リンカー単位がペプチドのN末端と併合し、該炭水化物と該キレート化基が多機能リンカー単位に結合している項目7記載のリガンド。
(項目14)
該多機能リンカー単位がリガンドのN末端に結合し、該キレート化基が多機能リンカー単位に結合し、該炭水化物がキレート化基に結合している項目7記載のリガンド。
(項目15)
さらに少なくとも1個の多機能リンカー単位および少なくとも2個のsst−レセプター結合構造ペプチド基を含んでなる項目1記載のリガンド。
(項目16)
該2個のペプチド基が多機能リンカー単位に結合し、該キレート化基と該炭水化物双方が多機能リンカーに結合している項目14記載のリガンド。
(項目17)
それぞれがペプチド単位の1個のN末端に結合する2個の炭水化物単位が存在し、当該炭水化物単位それぞれが多機能リンカー単位に結合し、かつ該キレート化基が多機能リンカー単位に結合している項目14記載のリガンド。
(項目18)
それぞれがペプチドのN末端に結合する2個の多機能リンカー単位、多機能リンカー単位に連結する1個のキレート化基、およびそれぞれが多機能リンカー単位の一方に結合する2個の炭水化物とが存在する項目14記載のリガンド。
(項目19)
該放射性同位体がTcおよびReの同位体から選択されるものである項目14記載のリガンド。
(項目20)
該炭水化物が糖であり、糖がレセプター結合構造、多機能リンカー単位、またはキレート化剤のアミノ基と、アルドースとの間の、アマドリ反応によるカップリング結合産物である項目14記載のリガンド。
(項目21)
該糖がレセプター結合構造、多機能リンカー単位、またはグリコシド結合を介してキレート化剤にカップリング結合する項目14記載のリガンド。
(項目22)
項目1記載の組成物と医薬的に許容し得る担体とを含有してなる医薬組成物。
(項目23)
項目7記載の組成物と医薬的に許容し得る担体とを含有してなる医薬組成物。
(項目24)
項目14記載の組成物と医薬的に許容し得る担体とを含有してなる医薬組成物。
(項目25)
項目22記載の組成物を患者に投与することを特徴とするsst−画像化からなる群より選択される画像化方法。
(項目26)
項目23記載の組成物を患者に投与することを特徴とするsst−画像化からなる群より選択される画像化方法。
(項目27)
項目24記載の組成物を患者に投与することを特徴とするsst−画像化からなる群より選択される画像化方法。
(項目28)
化合物[99mTc]Gluc−K0(H)TOC。
(項目29)
化合物[99mTc]Mtr−K0(H)TOC。
【図面の簡単な説明】
【0010】
【図1A】図1Aには、2個以上のペプチドリガンドの多量体を提供する本発明の組成物の様々な立体配置を示す。該多量体は同一のレセプターペプチド結合構造(ホモ二量体、ホモ多量体)または異なるレセプター結合構造(ヘテロ二量体、ヘテロ多量体)を含んでいてもよい。
【図1B】図1Bには、種々量体の立体配置を示すが、その幾つかはリンカー単位(多機能)を含み、その内の4個はリンカー単位を含まない。従って、多機能リンカー単位は本発明の組成物において任意であることが分かる。
【図1C】図1Cには二量体と多量体を示すが、この場合には、多重リンカー単位をペプチドと、勿論、多重ペプチドリガンドの様々な配向で採用する。
【発明を実施するための形態】
【0011】
図1Aおよび図1Cに示す二量体と多量体は、リンカーまたはペプチドリガンドのキレート化剤への共有結合を介して形成されるか、またはリンカーとキレート化剤間の錯体の形成を介して形成される。
本発明を以下の実施例で説明するが、これら実施例は本発明を制限するものではない。(実施例1)
【0012】
ペプチド放射線医薬の合理的な設計:
インビトロでの研究はリガンド誘発SST−2インターナリゼーションに対するオクトレオチドの糖質化およびC末端酸化の相乗作用を示す。
【0013】
目的:その薬物動態による外に、画像化用および治療目的のレセプターに基づくトレーサーの適性は、主としてその薬物動態プロフィルにより決定する。本研究の目的はSSTR細胞内取り込みと排出(インターナリゼーションとエクスターナリゼーション)および再細胞取り込み(再循環)に対するオクトレオチド(octreotide)とオクトレオテート(octreotate)の糖質化の影響を研究することであった。
【0014】
方法:[125I]Tyr3−オクトレオチド(TOC)、[125I]Tyr3−オクトレオテート(TOCA)およびそのグルコース(Gluc)−とマルトトリオース(Mtr)−誘導体のインターナリゼーション、エクスターナリゼーションおよび再循環は、AR42J細胞(SSTR2)の集密単層を用いて検討した。細胞は放射性リガンドと120分までインキュベートした(n=3)。各時点で、上清の活性、表面結合およびインターナリゼーション活性を測定し、TOC値に標準化した。エクスターナリゼーションおよび再循環は120分のインキュベーション後に2時間にわたり検討した。結合リガンドの特異性を10Mサンドスタチン(sandostatin)との競合実験により検討した。
【0015】
結果;インキュベーション2時間後の取り込みリガンド量[取り込まれたTOC%]は以下のとおりであった:Mtr−TOC(35±4%)<Gluc−TOC(121%)<TOCA(154%)<Mtr−TOCA(549%)<Gluc−TOCA(637%)。競合実験において、すべての化合物のインターナリゼーションが、加えた放射活性の<0.1±0.02%(30分)に低下した。リガンドの再循環を可能とするエクスターナリゼーション実験において、TOCAおよびグリケーションしたTOCAは、再循環しない実験と比較して、細胞外に局在する放射活性の2/3を示したが、一方、約80%はTOC、Gluc−TOCおよびMtr−TOCに見出された。TOCの糖質化は細胞表面上のリガンドの利用能に対して有意な作用を示さなかったが、TOCAおよびMtr−TOCAの表面濃度は、TOCに比較してそれぞれ2.1および2.3のファクター増加する。Gluc−TOCAはTOCに比較して、細胞表面上のトレーサー利用能の5倍の増加を示す。TOCのインターナリゼーション率(取り込み/表面結合活性)はグリコシル化により有意な影響を受けないが、TOCAは1.4倍の増加を示す。Gluc−TOCAおよびMtr−TOCAについて、我々はTOCに比べて186%および171%のインターナリゼーション率を観察した。
【0016】
結論:AR42J−細胞を用いて、TOCAを糖質化することにより、放射活性リガンドの細胞表面濃度とインターナリゼーション率を有意に増大させた。
(実施例2)
【0017】
SPECT用糖質化Tc−99M−オクトレオチド誘導体
合成、放射活性標識およびインビボデータ
過去10年にわたり、SPECTおよびPETによるSSTR−シンチグラフィー用の99mTC−および18F−標識オクトレオチド誘導体についての様々な方法が研究されている。Decristoforoらは、EDDAをもつ99mTc−標識HYNIC−Tyr3−オクトレオチドがコリガンドとして好適な生物動力学とマウスにおける高い腫瘍取り込みを示すことを示した。これまでに知られた唯一の18F−標識オクトレオチドである2−[18F]フルオロプロピオニル−D−Phe1−オクトレオチドは優勢な肝胆汁分泌を示すが、これはインビトロSSTR画像化に適用する上での欠点の一つである。
【0018】
本発明者らは、放射活性ヨウ素化Tyr3−オクトレオチド(TOC)とそのThr8−誘導体Tyr3−オクトレオテート(TOCA)をN末端グリコシル化が、生体分布の有意な改善、すなわち、腫瘍への蓄積増大をもたらすことを見出した。この原理の一般的応用を検討するために、我々は99mTc−標識化用のグリコシル化オクトレオチド−およびオクトレテート−誘導体を合成し、評価した(図2)。Lys0(N−His)−TOC(K0(H)TOC)のN−グリコシル化誘導体は、有機金属水和イオン[99mTc(H2O)3(CO)3]+−法1[EgliA. et al. J. Nucl. Med. 40: 1913-1917 (1999)]を用いる99mTc−標識化用前駆体として使用した。該ペプチドは標準Fmoc−SPPSプロトコールに従って合成した。グルコース(Gluc)およびマルトトリオース(Mtr)との接合はアマドリ反応経由で実施した。
Lys5−脱保護ペプチドを99mTc−標識化すると、水和イオンに基づき>97%の放射化学収率で[99mTc]Gluc−K0(H)TOCおよび[99mTc]Mtr−K0(H)TOCを得た。
【0019】
99mTc−標識化誘導体(注射30および120分後)の生体分布の検討はAR42J−腫瘍担持ヌードマウス(n=3〜4)にて実施した。インターナリゼーションおよびエクスターナリゼーションの実験は同じ細胞株を用いて実施した。
【0020】
インターナリゼーションの増加は、対照の[125I]TOCに比較して、[99mTc]Gluc−K0(H)TOCについては2.3±0.8のファクター、また[99mTc]Mtr−K0(H)TOCについては3.6±0.4のファクターで見出された。[125I]TOCはインキュベーションにより急速に細胞から排出されるが、[99mTc]Gluc−K0(H)TOCは120分まで細胞内に取り込まれたままである。
【0021】
[99mTc]Gluc−K0(H)TOCおよび[99mTc]Mtr−K0(H)TOCについて、注射2時間後の生体分布を表1に示す。
【0022】
表1:AR42J腫瘍担持ヌードマウスにおける[99mTc]S1−Lys0(His)TOCおよび[99mTc]S3−Lys0(His)TOC注射2時間後の組織蓄積[%iD/g](n=3〜4)
【表1】
【0023】
グリコシル化化合物は共に注射120分後にsst2陽性組織に高い蓄積性を示す。排泄器官での比較的高い非特異的取り込みならびに血液クリアランスの遅延は、二座ヒスチジンリガンドによる99mTc−コアの不十分な錯体形成によるものと我々は思考する。残りの金属配位部位はチオール含有の本来のタンパク質によりインビボで飽和され、それが血液、肝臓およびその他の排泄器官でのこれら錯体の急速な捕捉に至らしめる。[99mTc(H2O)3(CO)3]+−水和イオンとN−Ac−Hisなどの三座リガンドとの飽和した錯体が形成されると、非腫瘍組織での活性蓄積は有意に低下する。
【0024】
結論として、オクトレオチド(−テート)の炭水化物接合および[99mTc(H2O)3(CO)3]+−水和イオンの三座錯体形成の組合わせに基づく導入トレーサーの設計は新しい一連の非常に有望なSSTR−トレーサーの基礎となると言える。
(実施例3)
【0025】
SST−レセプター−アゴニストのグリケーション:放射活性標識ソマトスタチンアゴニストの動的リガンド移動の改善
糖質化は放射活性標識化オクトレオチドおよび∀v∃3類似体の薬物動態改善のための強力な手段であることが今回判明した。オクトレオチドおよびオクトレオテートのグリケーションは肝胆汁排泄および腎吸収を有意に低下させ、腫瘍取り込みと腫瘍/組織比を上昇させる。放射活性sst2−アゴニストの腫瘍蓄積は、レセプター介在インターナリゼーション、崩壊および引き続く細胞内蓄積に、またはリガンドおよび/または代謝産物の両方の再循環に左右される。各段階で定量分析すると、決定的に重要なことは、sst2発現腫瘍細胞におけるトレーサーの蓄積を如何に制御するかであることが理解される。従って、本研究の目的は、放射活性標識オクトレオチドとオクトレオテートのインターナリゼーションおよび再循環動力学に対するグリケーションの影響を試験することであった。
【0026】
Tyr3−オクトレオチド(TOC)、Tyr3−オクトレオテート(TOCA)およびその誘導体であるグルコース(Gluc)、マルトース(Malt)およびマルトトリオース(Mtr)誘導体は、Fmoc−SPPSおよび引き続く炭水化物接合を経て合成した。放射活性ヨウ素化はヨードゲン法により実施した。放射化学収率はRP−HPLC精製後、50〜84%の範囲であった。99mTc−標識化(Gluc−Lys0(N−His)TOC(Gluc−K(H)−TOC)およびMtr−Lys0(N−His)TOC(Mtr−K(H)−TOC))のための前駆体は、上記のペプチド同様に調製した。99mTcでの標識化はすでに記載した[99mTc(H2O)3(CO)3]+−水和イオンプロトコールに従って実施した[EgliA. et al. J. Nucl. Med. 40: 1913-1917 (1999)]。比較のために、我々は[111In]オクトレオスキャン(Octreoscan)および[111In]ドタトック(DOTATOC)をも我々の検討に含めた。
【0027】
インターナリゼーションとエクスターナリゼーションの実験は、sst2発現ラット膵臓腫瘍細胞株AR42Jを用いて実施した。遊離の表面結合内部に取込まれた放射活性は、放射活性リガンドと37℃でインキュベートし、その10、30、60、90および120分後に定量した。リガンド再循環を可能とするエクスターナリゼーション実験では、10、30、60、90および120分の間に上清に放出された放射活性フラクションならびに細胞中に残存する放射活性を定量した。我々はまた引き続く5回のインキュベーション(10、20および3×30分)に際して、培地の中間変化(制限再循環)を伴って放出された活性を測定した。
【0028】
インターナリゼーションに対するTOC糖質化の影響は、使用した糖の大きさに平行する。[125I]TOCに比較して、グルコース接合体のみが細胞への取込みを上昇させた。対照的に、[99mTc]Gluc−TOCAおよび[99mTc]Mtr−TOCA両方の細胞内蓄積(図3および表2)は有意に増大した([125I]TOCに比較して、7.36±0.50および5.68±0.38倍)。TOCとTOCAのインターナリゼーション特性およびそれぞれのグリコシル化誘導体のインターナリゼーション特性を比較して、我々は、表面利用能およびインターナリゼーション率に対する両方の構造的修飾(Thr8によるThr(ol)8の置換および糖質化)の相乗作用を観察した。[111In]オクトレオスキャン(Octreoscan)および[111In]ドタトック(DOTATOC)に比較して、両方の99mTc−標識化糖質化誘導体のインターナリゼーションは意外に高かった。
【0029】
10μMサンドスタチン(sanndostatin)による事前処理実験は、リガンドの取込みを対照の最大5%まで低下させ、i)sst2特異取込みであって、ii)炭水化物関連取込みメカニズムは関与しないことを示した(表2)。検討したトレーサーすべてについて、内部取込み活性はリガンドの表面利用能と強力に相関する。
【0030】
表2:AR42J細胞と60分間インキュベーション後に得られたインターナリゼーション・データ(インターナリゼーション値ならびに比は対照の[125I]TOCに標準化する)
a:表面利用能=%表面結合リガンド/%遊離放射活性リガンド
b:インターナリゼーション率=%内部取込みリガンド/%表面結合リガンド
*:10Mサンドスタチンでの事前処理実験は細胞リガンド取込みを最小95%低下させた。
【表2】
【0031】
エクスターナリゼーションに関しては、グリコシル化が影響していないように思われる(図4)。細胞からの放射リガンドのエクスターナリゼーションの度合いは、主としてペプチド(TOCまたはTOCA)の性質により、また標識の細胞内運命により判定する。リガンドの再循環を可能とするエクスターナリゼーション実験において、TOCAとそのグリケーション誘導体は再循環なしの実験に比較して、細胞外に局在する放射活性の約2/3を示した。一方、約80%がTOC、Gluc−TOCおよびMtr−TOCに見出された。[99mTc]Gluc−K0(H)TOCならびに両方の111In−標識化合物は、再循環条件下、120分以内に細胞中略定量的な保持を示した。対照的に、[111In]オクトレオスキャン(Octreoscan)および[111In]ドタトック(DOTATOC)の細胞内活性は、再循環を制限した場合、120分間に33%および49%消尽した。
(実施例4)
【0032】
[Tc−99m](CO)3−標識糖質化SSTR−リガンド:
合成、インターナリゼーション動力学およびラット膵臓腫瘍モデルでの生体分布
目的:本研究はSSTR発現腫瘍の特異的高レベル標的化用トレーサーの設計を示すためのものである。これを目的として、我々は薬物動態を最適化し、SSTR特異インターナリゼーションと腫瘍保持を改善するために、糖質化オクトレオチド誘導体を導入した。本明細書にて我々が記載する化合物は一連の新規糖質化99mTc−SSTRリガンドである。
【0033】
方法:グルコース(Gluc)およびマルトトリオース(Mtr)をN−(Boc−His(Boc))Lys0−Tyr3−Lys5(Dde)−オクトレオチド(保護K(H)TOC)のN−Lys0に結合させ、最終的に脱保護し、Gluc−およびMtr−K(H)TOCを得た。99mTc−標識化は[99mTc(H2O)3(CO)3]+を用いて実施した。インターナリゼーションとエクスターナリゼーションの検討(<2時間)はSSTR2発現ラット膵臓細胞株AR42Jおよび対照としての[125I]Tyr3−オクトレオチド([125I]TOC)を用いて実施した。生体分布はAR42J腫瘍担持ヌードマウスにて、0.5時間および2時間のインキュベーション(n=4)により評価した。
【0034】
結果:放射活性標識は効率的に(0.5時間、RCY>97%)均一の産物を生じた。[125I]TOCに比較して、両方の炭水化物導入ペプチドは有意により高い細胞間活性レベルを示した;例えば、[99mTc]Gluc−K(H)TOCについては2時間で2.3±0.2倍高いレベル、また[99mTc]Mtr−K(H)TOCについては3.6±0.4倍高いレベルを示した。エクスターナリゼーションの検討では、2時間で[125I]TOCに対し約50%の細胞内活性の低下を明らかにした。一方、[99mTc]Gluc−K(H)TOCの細胞間レベルは2時間まで殆ど安定であり、このトレーサーが保持されていることを示した。インビボでは、[99mTc]Gluc−K(H)TOC(T/肝臓0.77、T/腎層0.63)および[99mTc]Mtr−K(H)TOC(T/肝臓0.97、T/腎臓0.84)に対して、腫瘍の蓄積は注射2時間後にそれぞれ12.2±1.0および14.0±6.3%ID/gに達し、血中レベルは1.5±0.2%ID/gおよび4.4±1.0%ID/gであった。
結論:これらの結果はSSTR標的化改善の手段として糖質化が有用であることを証明している。
(実施例5)
【0035】
[Tc−99m](CO)3−標識糖質化SSTR−リガンド:
合成、インターナリゼーション動力学およびラット膵臓腫瘍モデルでの生体分布
目的:本研究はSSTR発現腫瘍の特異的高レベル標的化用トレーサーの設計を示すための我々の努力の一部である。この目的のために、我々は薬物動態を最適化し、SSTR特異インターナリゼーションと腫瘍保持を改善するのに適当な糖質化部分を使用した。本明細書に我々は初めて[99mTc(H2O)3(CO)3]+水和イオン法により標識化した糖質化SSTR結合ペプチドを記載する。
【0036】
方法:標準的なFmoc化学により、N−(Boc−His(Boc))Lys0−Tyr3−Lys5(Dde)−オクトレオチド(保護K(H)TOC)を合成した。グルコース(Gluc)およびマルトトリオース(Mtr)をN−Lys0に結合させ、最終的に脱保護し、K(H)TOCのGluc−およびMtr−誘導体を得た。Tc−99m−標識化は有機金属水和イオン[99mTc(H2O)3(CO)3]+(1)を用いて実施した。インターナリゼーションとエクスターナリゼーションの検討(<120分)はSSTR2発現細胞株AR42Jを用い、両化合物について実施した。インビトロの検討のために、対照として[125I]Tyr3−オクトレオチド([125I]TOC)を用いた。静脈内投与した[99mTc]Gluc−K(H)TOCおよび[99mTc]Mtr−K(H)TOCの生体分布はAR42Jラット膵臓腫瘍担持ヌードマウスにて、注射30分および120分(n=4)後に評価した。
【0037】
結果:本標識手法は効率的に(30分間、RCY>97%)均一の産物を生じた(HPLC)。[125I]TOCに比較して、両方の糖質化ペプチドは検討したすべての時点で有意により高いインターナリゼーションを示した;例えば、[99mTc]Gluc−K(H)TOCについては2時間で2.3±0.2倍高いレベル、また[99mTc]Gluc−K(H)TOCについては3.6±0.4倍高いレベル、また[99mTc]Mtr−K(H)TOCについては3.6±0.4倍高いレベルを示した。さらに、エクスターナリゼーションの検討では、120分で対照化合物の約50%の細胞内活性の低下を明らかにした。一方、細胞内[99mTc]Gluc−K(H)TOCの高いレベルは2時間まで殆ど安定であり、このトレーサーが検討した細胞株に保持されていることを示した。インビボでは、[99mTc]Gluc−K(H)TOC(T/肝臓0.77、T/腎層0.63)および[99mTc]Mtr−K(H)TOC(T/肝臓0.97、T/腎臓0.84)に対して、腫瘍の蓄積が注射120分後にそれぞれ12.2±1.0および14.0±6.3%ID/gに達し、対応する血中レベルは1.5±0.2%ID/gおよび4.4±1.0%ID/gであった。
結論:これらの結果はSSTR標的化改善の手段として炭水化物導入が有用であることを示している。
(実施例6)
【0038】
固相ペプチド合成
1. Fmoc−Lys0−Tyr3−Lys(Dde)5−オクトレオチド(Fmoc−Lys0−TOC(Dde))Fmoc−Thr(tBu)−オール Fmoc−Thr(tBu)−OH(1.391g、3.5mmol)をテトラヒドロフラン(THF)30mlに溶かした溶液を−10℃に冷却した。N−メチルモルホリン(386μl、3.5mmol)およびクロロギ酸エチル(333μl、3.5mmol)を引き続き加えた。反応混合物を−10℃で30分間撹拌した。次いで、ホウ素化水素ナトリウム(396mg、10.5mmol)を加えた。30分を要してメタノール60mlをゆっくりと反応混合物に加え、次いで、0℃で3時間撹拌した。
【0039】
1N−HClを50〜70ml添加した後(濁りのある反応混合物が透明にならなければならない)、有機溶媒を蒸発させ、残りの水相はジクロロメタン(DCM)で2回抽出した。抽出効率は両相の薄層クロマトグラフィー(酢酸エチル(AcOEt)による産物のRf=0.83)によりモニターした。併合した有機層を硫酸マグネシウム上乾燥し、濾過、減圧濃縮し、黄色油状物を得た。粗生成物をAcOEtによるフラッシュクロマトグラフィーにより精製した。産物は油状物(1.19g、89%)として得た。
単一同位体質量;C23H29NO4として計算値=383.21;測定値:m/z406.1[M+Na]+
【0040】
Fmoc−Lys0−Dphe1−Cys2−Tyr3−DTrp4−Lys(Dde)5−Thr6−Cys7−Thr−オール8 DHP−HM(3,4−ジヒドロ−2H−ピラン−2−イルメトキシメチルポリスチロール)樹脂(1.000g、0.94mmol/g)を10mlの乾燥DCE(1,2−ジクロロエタン)中、1時間、予め膨潤させた。次いで、Fmoc−Thr(tBu)−オール(1.266g、3.3mmol)とp−トルエンスルホン酸ピリジニウム(414mg、1.75mmol)を7mlの乾燥DCEに溶かした溶液を加え、その反応混合物をアルゴン下、80℃で一夜撹拌した。遊離の官能基を樹脂に取り付けるために、ピリジン5mlを加え、懸濁液を室温で15分間撹拌した。負荷した樹脂を次いで濾過し、DMF(N,N−ジメチルホルムアミド)で2回、およびDCMで洗浄し、乾燥器中で乾燥した。樹脂の最終負荷物は0.834mmol/gであった。ペプチド配列は標準Fmoc−プロトコールを用いて樹脂結合アミノアルコール上で推定した。簡単に説明すると、N−末端FmocをDMF中の20%ピペリジンで除去し、樹脂支持ペプチドの樹脂をNMP(N−メチルピロリドン)で洗浄した。カップリング反応はNMP中、1.5当量のアミノ酸誘導体と、1.5当量のHOBt(1−ヒドロキシベンゾトリアゾール)、1.5当量のTBTU(O−(1H−ベンゾールトリアゾール−1−イル)−N,N,N',N'−テトラメチルウロニウム−テトラフルオロボレート)および3.5当量のDIPEA(N−エチル−ジイソプロピルアミン)と反応させることにより実施した。活性化したアミノ酸を反応容器に加えた後、適当な時間(通常40〜60分)、樹脂を撹拌した。次いで、樹脂を追加のNMPで洗浄した。TOC合成のための添加の順序は、Fmoc−Cys(Trt)、Fmoc−Thr(tBu)、Fmoc−Lys(Dde)、Fmoc−DTrp、Fmoc−Tyr、Fmoc−Cys(Trt)、Fmoc−DpheおよびFmoc−Lys(Boc)の順であった。
【0041】
最終アミノ酸を配列にカップリング結合させた後、樹脂を数回NMPおよびDCMで洗浄した。樹脂からの脱離およびThr−、Cys−およびLys0−側鎖の脱保護は、TFA(トリフルオロ酢酸)/TIBS(トリイソブチルシラン)/水(95:2.5:2.5)およびDCMの混合物(1:1)を用いて実施した。90分後に樹脂を濾取し、DCMで2回洗浄した。併合した濾液をロータリーエバポレーターにて濃縮し、粗製の産物をエーテルで沈殿させた。我々は828mgの粗生成物を得た。収率83%。
【0042】
粗製のペプチドは(ぺプチド300mgあたり)50mlのTHFに再懸濁し、澄明な溶液が得られるまで、5mM酢酸アンモニウムを添加した。飽和NaHCO3溶液を滴下することでpH7に調整した。次いで、過酸化水素水(30%)100μl(ペプチド300mgあたり)を加えた。室温で30分間撹拌した後、環化を終了した(勾配HPLC−コントロール:30〜80%B、15分)。溶媒を蒸発させ、環化した生成物を凍結乾燥した。収率は定量的であった。
単一同位体質量;C80H100N12O16S2として計算値=1548.68;測定値:m/z1549.5[M+H]+、m/z=1571.5[M+Na]+
【0043】
2. Fmoc−Lys0−Tyr3−Lys(Dde)5−オクトレオチド(Fmoc−Lys0−TATE(Dde))Fmoc−Lys0−Dphe1−Cys2−Tyr3−DTrp4−Lys(Dde)5−Thr6−Cys7−Thr8 乾燥TCP−樹脂1.502gに、Fmoc−Thr(tBu)894mg(2.25mmol)、DIPEA321μl(1.87mmol)および乾燥DCM15mlからなる溶液を加えた。5分間撹拌した後、追加のDIPEA642μl(3.76mmol)を加えた。室温で90分間、激しく撹拌した後、メタノール1.5mlを加えて、未反応の塩化トリチル基を遮断した。15分後、樹脂を濾取し、DCM、DMFおよびメタノールでそれぞれ2回洗浄し、減圧乾燥した。樹脂の最終的負荷は0.77mmol/gであった。樹脂結合アミノ酸上のペプチド配列、Cys(Trt)−Thr(tBu)−Lys(Dde)−DTrp−Tyr−Cys(Trt)−DPhe−Lys(Boc)−Fmocの合成、脱保護、および樹脂からの切断、ならびに環化反応は、Fmoc−Lys0−TOC(Dde)について記載したのと同様に実施した。
単一同位体質量;C80H98N12O17S2として計算値=1562.66;測定値:m/z= ([M+H]+)
【0044】
溶液相誘導化
1. Lys0(N∈−Boc−His(Boc))−TOC(Dde)
Fmoc−Lys0−TOC(Dde)500mg(0.32mmol)、Boc−His(Boc)170mg(0.48mmol)、HOBt65mg(0.048mmol)、TBTU154mg(0.48mmol)およびDIPEA300μl(1.75mmol)をDMF3mlに溶かした溶液を室温で撹拌した。RP−HPLC分析(勾配:30〜80%B、15分)により、反応は30分以内に終了することが判明した。生成物を水で沈殿させ、減圧下に乾燥した。N末端Fmoc−保護基を除去するために、ペプチドを1mlのピペリジン/DMF(1:4)に溶解し、15分間撹拌した。エーテルで沈殿させ、脱保護産物を86%の収率で得た。
単一同位体質量;C81H113N15O19S2として計算値=1663.78;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+
【0045】
2. Lys0(Boc−His(Boc))−TOC(Dde)のLys0−N−アマドリ誘導体
以前に報告された方法1に基づき、1当量のLys0(Boc−His(Boc))−TOC(Dde)および10当量のそれぞれのアルドース(グルコース=S1、マルトース=S2、マルトトリオース=S3)をメタノール/酢酸(95:5)に溶かし、その反応混合物を60℃で24〜48時間撹拌した。次いで、溶媒を蒸発させ、粗生成物をエーテル添加により沈殿させた。
【0046】
Dde−保護基を除去するために、グリコシル化ペプチドを少量のDMFに再溶解し、2容量%のヒドラジン水和物を添加し、その溶液を室温で15分間撹拌した。脱保護ペプチドをエーテル添加により沈殿させ、エーテルで洗浄して減圧乾燥した。His−残基上のBoc−保護基は、粗生成物を少量のTFA/TIBS/水(95:2.5:2.5)に再溶解し、室温にて15分間撹拌することにより除去した。生成物をエーテルで沈殿させ、調製用RP−HPLC(勾配:10〜60%B,30分)にて精製した。収率は17〜32%の範囲であった。
【0047】
S1−Lys0(His)TOC(Na−(1−デオキシ−D−フルクトシル)−Lys0N−His)−Tyr3−オクトレオチド:
単一同位体質量;C67H95N15O18S2として計算値=1461.60;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+S2 Lys0(His)TOC(N−(O− −D−グルコピラノシル−(1−4)−1−デオキシ−D−フルクトシル)−Lys0(N∈−His)−Tyr3−オクトレオチド):
単一同位体質量;C73H105N15O28S2として計算値=1623.70;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+
S3 Lys0(His)TOC(Na−(O−a−D−グルコピラノシル−(1−4)−O−a−D−グルコピラノシル−(1−4)−1−デオキシ−D−フルクトシル)−Lys0(N−His)−Tyr3−オクトレオチド:
単一同位体質量;C73H115N15O23S2として計算値=1785.75;測定値:m/z=1873.6[M+H]+、m/z=1895.7[M+Na]+
【0048】
3. Lys0(N−2−ピコリルアミノ−....)TATE(Dde)
2−ピコリルアミンN,N−二酢酸2
イミノ二酢酸(3.25g、24.4mmol)を無水エタノール30mlおよびNaOH(1.95g;49mmol)水溶液(10ml)の混合物に溶解した。水9mlに溶かした塩化ピコリル4.00g(24.4mmol)および水4mlに溶かしたNaOH1.95g(49mmol)を添加した後、その溶液を70℃で2時間撹拌した。追加のNaOHを1.95g(49mmol)添加した後、その溶液をさらに70℃で1.5時間撹拌した。TLC(EtOAc)により反応を追跡し、その時点で反応物が完全に消失していることを確認した。溶媒を減圧下に蒸発させ、残渣を水25mlに再溶解した。溶液のpHを濃塩酸により1.5に調整した後、溶液を再度蒸発乾固した。残渣をメタノールで再懸濁して還流する工程を数回連続して繰返し、懸濁液を熱時濾過し、濾液を減圧下に濃縮して過剰のNaClを成功裏に除去した。ピコリルアミノ二酢酸を残りのメタノール溶液から一夜−20℃で結晶化した。
単一同位体質量;C10H12N2O4として計算値=224.08;測定値:m/z= [M+H]+(二ナトリウム塩)
【0049】
2−ピコリルアミンN,N−二酢酸のFmoc−Lys0−TATE(Dde)へのカップリング反応
2−ピコリルアミンN,N−二酢酸52mg(0.19mmol)、HOBt26mg(0.19mmol)、TBTU62mg(0.19mmol)、およびDIPEA175μl(1.75mmol)を2mlのDMFに溶かした溶液を室温で10分間撹拌した。この溶液をFmoc−Lys0−TATE(Dde)200mg(0.13mmol)のDMF(1ml)溶液に激しく撹拌しながら滴下した。30分後、生成物をジエチルエーテルで沈殿させ、減圧下に乾燥した。N末端Fmoc−保護基を除去するために、ペプチドをピペリジン/DMF(1:4)2mlに溶解し、10分間撹拌した。エーテルで沈殿させることにより、脱保護した産物を84%の収率で得た。
単一同位体質量;C75H98N14O18S2として計算値=1546.66;測定値:m/z=[M+H]+、m/z=[M+Na]+
【0050】
4. Lys0(Pic)TATE(Dde)のLys0−Na−アマドリ誘導体
Lys0(Pic)TATE(Dde)とグルコースの接合、および引き続くDde−脱保護は、Lys0(Boc−His(Boc))−TOC(Dde)のLys0−Na−アマドリ誘導体について記載したと同様に実施した。
単一同位体質量;C71H96N14O21S2として計算値=1544.63;測定値:m/z=[M+H]+、m/z=[M+Na]+
【0051】
放射活性標識化
最近報告された有機金属水和イオン[99mTc(H2O)3(CO)3]+−法3に基づき、10mgのNaBH4、8mgのNa2CO3および41.5mgのNa/K酒石酸塩を、ガラスフラスコ中600μlの[99mTc]ペルテクネテート溶離液に溶解した。次いで、フラスコをCOを満たした風船につなぎ、密封した。75℃で45分間撹拌した後、溶液を氷浴上で冷却し、150μlのリン酸緩衝食塩液(PBS、pH7.4)を加えた。還元剤を分解するために600μlの1N−HClを加えた。次いで、溶液を500μlの飽和NaHCO3溶液で中和した。[99mTc(H2O)3(CO)3]+−水和イオンの存在はTLC(5%conc. HCl/メタノール)にて確認した。
【0052】
S1−Lys0(His)TOC700μgに1mlの[99mTc(H2O)3(CO)3]+−溶液(51MBq)を加えた後、混合物を75℃に30分間加熱した。勾配HPLC精製により、放射化学的に純粋なペプチドNa−(1−デオキシ−D−フルクトシル)−Lys0(N−His[99mTc(H2O)3(CO)3]+)−Tyr3−オクトレオチドを34%の収率で得た。
S3−Lys0(His)TOCは同様の方法で標識した。
【0053】
親油性の定量
放射活性標識ペプチド約300000cpmをPBS(pH7.4)500mlに溶かした溶液に、オクタノール500mlを添加した(n=6)。バイアルを3分間ボルテックス処理し、15000rpmで6分間遠心分離した。PBSとオクタノールの100mlを取り、γ−カウンターにて測定した。
S1−Lys0(His)TOC:logpow=−0.673±0.064
S3−Lys0(His)TOC:logpow=−1.304±0.040
インビトロ実験
AR42J細胞を、10%FCSおよび2mM−L−グルタミン添加RPMI1640中に維持した。
【0054】
インターナリゼーションの検討
1ウエルあたり8×105〜5×106個のAR42J細胞の細胞集団を容れた6穴プレートを、実験に先立ち、新鮮な培地(RPMI1640、1%BSA、2mM−L−グルタミン)とともに1時間インキュベートした。培地10μl中、約300000cpmの放射活性金属ペプチドを各ウエルに加えた(対照化合物[125I]TOC(内部基準)および各対象の放射活性金属化合物について、3組の実験とする)。37℃で10、30、60、90または120分間インキュベートした後、上清を採取し、細胞は冷培地1mlにて1回洗浄した。上清を前工程の上清と併合した。レセプター結合(酸解離可能)放射活性を除くために、細胞を0.02M−NaOAc(酸洗浄)1mlと37℃でインキュベートした。10分後に上清を採取し、細胞を0.02M−NaOAc1mlにて1回洗浄し、その上清を前工程の上清と再度併合した。次いで、細胞を1mlの1N−NaOHで抽出し、ウエルはPBSにて1回洗浄した。このプールしたフラクションはインターナリゼーションの放射活性リガンドを示している。NaOHフラクションならびに上清と酸洗浄フラクションの活性をγ−カウンターにて計測した。
【0055】
エクスターナリゼーションの検討
インターナリゼーションの検討にて記載したように、新鮮な培地(RPMI1640、1%BSA、2mM−L−グルタミン)を容れた6穴プレートを、実験に先立ち、37℃で1時間インキュベートした。培地10μl中、約300000cpmの放射活性金属ペプチドを各ウエルに加えた(対照化合物[125I]TOC(内部基準)および各対象の放射活性金属化合物について、3組の実験とする)。37℃にて120分間インキュベートした後、上清を採取し、細胞は冷培地1mlにて1回洗浄した。上清を前工程の上清と併合した。エクスターナリゼーションの放射活性を採取するために、細胞を1mlの培地とともに37℃でインキュベートした。10、30、60、90および120分後に、それぞれ上清を集め、細胞は1mlの培地で1回洗浄し、その上清を前工程の上清と併合した。次いで、細胞を1mlの1N−NaOHで抽出し、ウエルはPBSにて1回洗浄した。このプールしたフラクションは細胞中になお局在する放射活性リガンドを示している。NaOHフラクションならびに上清とエクスターナリゼーションフラクションの活性をγ−カウンターにて計測した。
【0056】
トレーサー再循環なしのエクスターナリゼーションの検討
前記実験について記載したのと同様に、6穴プレートを放射活性リガンドとともに37℃にて120分間インキュベートした。上清を採取し、細胞は冷培地1mlにて1回洗浄した。上清を前工程の上清と併合した。エクスターナリゼーションの放射活性を採取するために、細胞を1mlの培地とともに37℃でインキュベートした。10分後に上清を採取し、細胞は再度さらに20分間37℃にて培地1mlとインキュベートした。上清を採取し、細胞は1mlの培地とともに37℃にて30分間さらに3回インキュベートした。次いで、細胞を1mlの1N−NaOHで抽出し、ウエルはPBSにて1回洗浄した。このプールしたフラクションは細胞中になお局在する放射活性リガンドを示している。NaOHフラクションならびにすべての時点でのエクスターナリゼーションフラクションの活性をγ−カウンターにて計測した。
【0057】
生体分布の検討
腫瘍増殖を確立するために、サブコンフルエント単層細胞を1mM−EDTAで処理し、懸濁し、遠心分離し、RPMI1640に再懸濁した。
ヌードマウス(オスおよびメス、6〜8週令)の脇腹に、PBS(pH7.4)100μlに懸濁した細胞2.5〜3×107個を皮下接種し、AR42J腫瘍担持ヌードマウスの尾部静脈に注射した。動物(n=3〜4)を注射後30分および120分後に屠殺し、対象臓器を摘出した。秤量した組織サンプルにつきガンマカウンターにて放射活性を測定した。データは%iD/g組織で表す(平均±DS、n=3〜4)。
【0058】
表1:AR42J腫瘍担持ヌードマウスにおける[99mTc]S1−Lys0(His)TOCおよび[99mTc]S3−Lys0(His)TOC注射2時間後の組織蓄積[%iD/g](n=3〜4)
【表3】
【特許請求の範囲】
【請求項1】
明細書に記載の発明。
【請求項1】
明細書に記載の発明。
【図1A】

【図1B】
【図1C】
【図1B】
【図1C】
【公開番号】特開2013−100349(P2013−100349A)
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願番号】特願2013−23001(P2013−23001)
【出願日】平成25年2月8日(2013.2.8)
【分割の表示】特願2009−228998(P2009−228998)の分割
【原出願日】平成14年4月23日(2002.4.23)
【出願人】(595181003)マリンクロッド エルエルシー (203)
【Fターム(参考)】
【公開日】平成25年5月23日(2013.5.23)
【国際特許分類】
【出願日】平成25年2月8日(2013.2.8)
【分割の表示】特願2009−228998(P2009−228998)の分割
【原出願日】平成14年4月23日(2002.4.23)
【出願人】(595181003)マリンクロッド エルエルシー (203)
【Fターム(参考)】
[ Back to top ]