
TrkAを阻害するペプチド化合物及びその用途
【課題】優れた腫瘍増殖抑制作用及び鎮痛作用を有する、化合物を提供する。
【解決手段】式(I):X−L−Y(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ特定のアミノ酸配列の部分配列)を含む15アミノ酸長以下のペプチド残基で表されるペプチド化合物または薬理学的に許容されるその塩。前記ペプチド化合物または薬理学的に許容されるその塩を含む腫瘍治療剤。前記ペプチド化合物または薬理学的に許容されるその塩を含む疼痛治療剤。
【解決手段】式(I):X−L−Y(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ特定のアミノ酸配列の部分配列)を含む15アミノ酸長以下のペプチド残基で表されるペプチド化合物または薬理学的に許容されるその塩。前記ペプチド化合物または薬理学的に許容されるその塩を含む腫瘍治療剤。前記ペプチド化合物または薬理学的に許容されるその塩を含む疼痛治療剤。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、緩和医療学におけるがん疼痛の治療に用いられるペプチドに関する。
【背景技術】
【0002】
がん疼痛は、慢性腰痛、頸肩腕症候群などと共に、神経障害性疼痛と侵害受容性疼痛を併せ持つ痛みである。侵害受容性疼痛とは、例えば、けがや火傷などによって損傷部位に炎症が起こり、その場所で発痛物質が生成され、それが末梢神経の侵害受容器を刺激することで発生する痛みなどをいい、侵害受容器が活性化することによって引き起こされる疼痛である。一方、神経障害性疼痛は、神経が損傷を受けた結果として起こる痛みなどをいい、痛みが長期間続いたり、通常では何ともない程度の刺激に対して強い痛みを感じるなどの特徴があり、体性感覚系に対する損傷や疾患の直接的結果として生じている疼痛とされている。
【0003】
現在、疼痛抑制のために、モルヒネに代表されるオピオイドや非ステロイド性消炎鎮痛剤(NSAIDs)などが用いられているが、これらの鎮痛剤は、有用性、安全性、常習性等の面で改善の余地があり、また、神経障害性疼痛に関しては、こうした薬剤の効果が十分に得られないのが現状である。従って、安全性・常習性等の問題がなく、かつ神経障害性疼痛にも優れた効果を発揮するような医薬組成物、治療方法が望まれている。
【0004】
侵害受容性疼痛および神経障害性疼痛に関与するメカニズムの一つとして、神経成長因子(NGF)、およびその高親和性受容体であるTrkAが知られている。TrkAは、インスリン受容体ファミリーと呼ばれる細胞膜受容体型チロシンキナーゼの一つであり、その酵素活性の中心部に活性化ループを有する。この活性化ループ中のアミノ酸配列を含む合成ポリペプチドは、インスリン受容体やTrkAのチロシンキナーゼ活性を抑制することが示されている(非特許文献1)。
【0005】
また、TrkAは、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍、ホジキンリンパ腫などで発現していること、NGFがTrkAを介して腫瘍細胞の増殖に関与することが知られており、NGFとTrkAを介した細胞情報伝達系が、腫瘍細胞の増殖に関与する可能性が指摘されてきた。これらのことから、NGFおよびTrkAは癌治療薬および疼痛治療薬のターゲットの一つと考えられている。
【0006】
これまで、腫瘍増殖抑制作用や疼痛治療の目的で、NGFの働きやその高親和性受容体TrkAの活性を阻害するための手段として、TrkAのチロシンキナーゼ活性を抑制する薬剤、抗NGF抗体、抗TrkA抗体などが提案されてきた(特許文献1、特許文献2)。TrkAのチロシンキナーゼ活性を抑制する薬剤としては、K252aが知られているが、K252aは、TrkAのチロシンキナーゼ活性以外のプロテインキナーゼC活性も抑制する作用があるため、腫瘍および疼痛治療薬として臨床応用するには毒性が高く、また、抗体医薬については、膨大な製造コストがかかってしまうのが現状である。これに対し、ペプチドは、タンパク質と比較して分子量が少なく構造も単純であるため、生産コストを比較的抑えることが可能であり、また、不必要な分は体内のプロテイナーゼにより分解されるため、遺伝子治療において予想されるような危険性は少ない。従って、TrkA活性を特異的に抑制するようなペプチドを得ることができれば、臨床上非常に大きなメリットがあると考えられる。
【0007】
本発明者らは、TrkA活性化ループのアミノ酸配列と腫瘍細胞の増殖および痛みとの関係について研究を行った。その結果、TrkA活性化ループのアミノ酸配列(664−GMSRDIYSTDYYRVGGR−680)(配列番号1)の部分配列SRDIYSTDYYR(配列番号2)を有するペプチドを、細胞膜透過性を促進する機能を有するペプチドと連結して腫瘍細胞や神経細胞内へ導入すると、TrkAのチロシンキナーゼ活性が抑制され、腫瘍細胞の増殖が抑制されること(特許文献3、非特許文献1)、さらに鎮痛作用が得られる(侵害受容性疼痛および神経障害性疼痛ともに抑制できる)こと(非特許文献2、非特許文献3)を既に報告している。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2010−162028号公報
【特許文献2】国際公開第2006/137106号パンフレット
【特許文献3】特開2010−6731号公報
【非特許文献】
【0009】
【非特許文献1】Munetaka Hirose et al., J Pharmacol Sci 106,107-113,2008
【非特許文献2】Koyo Ueda et al., J Pharmacol Sci 112,438-443,2010
【非特許文献3】Wei-Ying Ma et al., J Pharmacol Sci 114,79-84,2010
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、TrkA活性を抑制するペプチドを用いた、優れた腫瘍治療薬および疼痛治療薬を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、上記の知見を基に、さらに検討を重ね、TrkA活性を強力に抑制することが報告されているTrkA活性化ループ内の部分配列SRDIYSTDYYR(配列番号2)よりも、N末端側のMSRDIYS(配列番号3)を少なくとも含む領域を用いれば、より強力に、且つ短い長さのペプチドでTrkA活性を抑制し、腫瘍や疼痛を治療し得ることを見出し、更に検討を進めた結果、本発明を完成した。
【0012】
すなわち、本発明は以下のとおりである。
(1)式(I):
X−L−Y (I)
(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ配列番号1で表されるアミノ酸配列の部分配列(ただし、該部分配列は配列番号3で表されるアミノ酸配列を含む)を含む15アミノ酸長以下のペプチド残基である)で表されるペプチド化合物または薬理学的に許容されるその塩。
(2)Yが配列番号4または配列番号5で表されるアミノ酸配列を含むポリペプチド残基である、(1)に記載の、ペプチド化合物または薬理学的に許容されるその塩。
(3)Xが配列番号6〜11で表されるアミノ酸配列のいずれかを有するポリペプチド残基である、(1)または(2)に記載の、ペプチド化合物または薬理学的に許容されるその塩。
(4)Lがε−アミノカプロン酸リンカーである、(1)〜(3)のいずれか1つに記載の、ペプチド化合物または薬理学的に許容されるその塩。
(5)ペプチド化合物が、式(II):
YGRKKRRQRRR−acp−GMSRDIYS−NH2 (II)
または式(III):
YGRKKRRQRRR−acp−MSRDIYST−NH2 (III)
(式中、acpはε−アミノカプロン酸リンカーを示す)で表される化合物である、(1)〜(4)のいずれか1つに記載の、ペプチド化合物または薬理学的に許容されるその塩。
(6)(1)〜(5)のいずれか1つに記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する医薬組成物。
(7)(1)〜(5)のいずれか1つに記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、腫瘍治療剤。
(8)腫瘍が、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫である、(7)に記載の腫瘍治療剤。
(9)(1)〜(5)のいずれか1つに記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、疼痛治療剤。
(10)癌疼痛治療剤である、(9)に記載の疼痛治療剤。
【発明の効果】
【0013】
本発明によれば、優れた腫瘍増殖抑制作用を有し、製造コストの低い、腫瘍治療剤を提供することが出来る。また、本発明によれば、優れた鎮痛作用を有し、製造コストの低い、疼痛治療剤を提供することができる。
【図面の簡単な説明】
【0014】
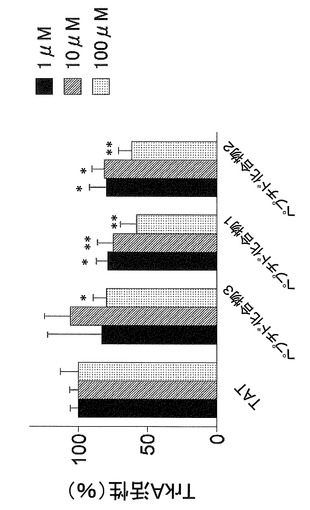
【図1】In vitroにおける、ペプチド化合物によるTrkAチロシンキナーゼ活性抑制作用を示す図である。
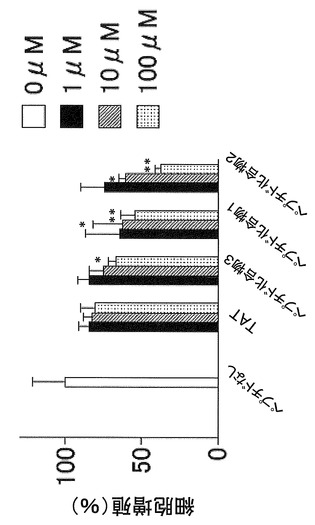
【図2】マウスメラノーマ細胞株(B16−F1)を、血清およびペプチド化合物を加えて培養した場合の、ペプチド化合物による細胞増殖抑制作用を示す図である。
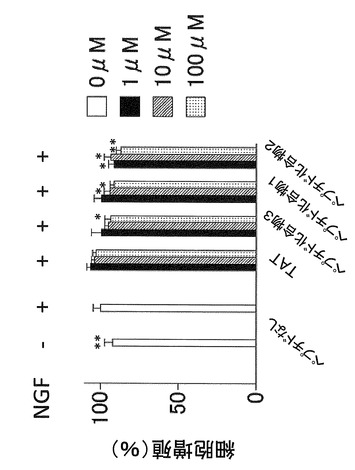
【図3】マウスメラノーマ細胞株(B16−F1)を、血清無しでNGFおよびペプチド化合物を加えて培養した場合の、ペプチド化合物による細胞増殖抑制作用を示す図である。
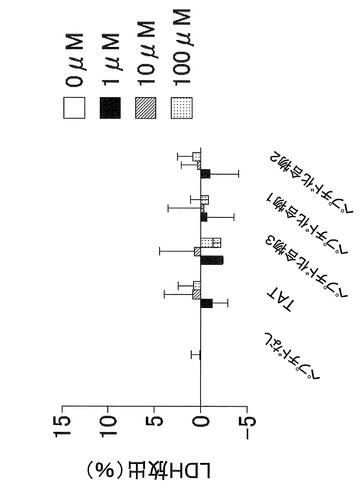
【図4】マウスメラノーマ細胞株(B16−F1)を、血清およびペプチド化合物を加えて培養した場合の、ペプチド化合物による細胞毒性を示す図である。
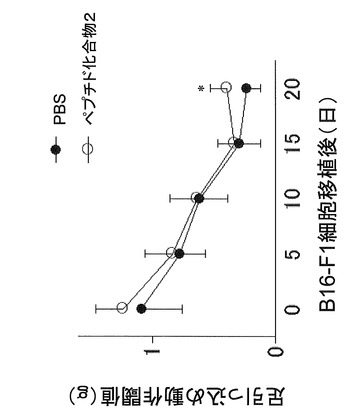
【図5】マウスメラノーマモデルにおける、ペプチド化合物による鎮痛効果を示す図である。
【発明を実施するための形態】
【0015】
本発明は、式(I):
X−L−Y (I)
(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ配列番号1で表されるアミノ酸配列の部分配列を含むペプチド残基である)で表されるペプチド化合物または薬理学的に許容されるその塩を提供するものである。
【0016】
Yで表されるペプチド残基は、TrkAのチロシンキナーゼ活性を抑制する機能を有する。
【0017】
TrkAは、神経成長因子(NGF)の高親和性受容体として知られる公知のタンパク質である。TrkAは、その細胞内ドメインにチロシンキナーゼ活性を有しており、NGFがTrkAに結合すると、該チロシンキナーゼ活性によりTrkAの細胞内ドメインに含まれる5つのチロシン残基が自己リン酸化され、下流の細胞内情報伝達系へ情報伝達を行う。「TrkAのチロシンキナーゼ活性の抑制」とは、このTrkAの自己リン酸化の抑制および下流の細胞内情報伝達系への情報伝達の抑制を意味する。
【0018】
Yで表されるペプチド残基は、配列番号1で表されるアミノ酸配列の部分配列を含む。配列番号1で表されるアミノ酸配列は、ヒトTrkAの活性化ループ部位に相当する。また、該部分配列は、配列番号3で表されるアミノ酸配列を含む。該部分配列の長さは、Yで表されるペプチド残基がTrkAのチロシンキナーゼ活性の抑制作用を有し、且つ式(I)で表されるペプチド化合物が腫瘍細胞の増殖や、疼痛を抑制する機能を発揮し得る限り特に限定されないが、十分な強さのTrkAのチロシンキナーゼ活性抑制機能を確保する観点から、該部分配列の長さは7アミノ酸長以上、好ましくは8アミノ酸長以上である。また、該部分配列の長さは、通常15アミノ酸長以下、好ましくは13アミノ酸長以下、より好ましくは11アミノ酸長以下、更に好ましくは9アミノ酸長以下である。
【0019】
一態様において、該部分配列の長さは、好ましくは7〜9アミノ酸であり、最も好ましくは8アミノ酸である。
【0020】
一方、該ペプチド残基が長すぎると、式(I)で表されるペプチド化合物の細胞内への導入効率が低下する恐れがある。従って、細胞内への高い導入効率を達成する観点から、Yで表されるペプチド残基の全長は、通常15アミノ酸長以下、好ましくは13アミノ酸長以下、より好ましくは11アミノ酸長以下、更に好ましくは9アミノ酸長以下である。
【0021】
Yで表されるペプチド残基は、上述の配列番号1で表されるアミノ酸配列の部分配列以外の領域を含んでいても含んでいなくてもよい。Yで表されるペプチド残基が、上述の配列番号1で表されるアミノ酸配列の部分配列以外の領域を含む場合、当該領域を構成するアミノ酸配列は、Yで表されるペプチド残基がTrkAのチロシンキナーゼ活性を抑制する限り、特に限定されない。好ましくは、Yで表されるペプチド残基は、上述の配列番号1で表されるアミノ酸配列の部分配列からなる。
【0022】
最も好ましくは、Yで表されるペプチド残基は、配列番号4または配列番号5で表されるアミノ酸配列からなる。
【0023】
Xで表されるペプチド残基は、細胞膜透過性を促進する機能を有するアミノ酸配列を含む。「細胞膜透過性を促進する機能」とは、細胞外から細胞内への化合物の透過を促進する機能を意味する。式(I)で表されるペプチド化合物がこのXで表されるペプチド残基を有することにより、生体へ該ペプチド化合物を投与すると、該ペプチド化合物が細胞内へ取り込まれ、該細胞においてTrkAのチロシンキナーゼ活性を抑制し、腫瘍細胞の増殖や、さらに、疼痛を抑制することができる。
【0024】
細胞膜透過性を促進する機能を有するアミノ酸配列としては、公知のものを用いることができる。例えば、配列番号6〜11で表されるアミノ酸配列を好適なものとして挙げることができる。このうち、配列番号6で表されるアミノ酸配列は、ヒト免疫不全ウィルスタイプ1のTAT(転写のトランス活性化因子)に由来するペプチド(47−YGRKKRRQRRR−57)に相当する。
【0025】
Xで表されるペプチド残基の長さは、細胞膜透過性を促進する機能を発揮し得る限り特に限定されないが、式(I)の化合物の細胞内への高い導入効率を達成する観点から通常4〜15アミノ酸長、好ましくは5〜11アミノ酸長である。
【0026】
最も好ましくは、Xで表されるペプチド残基は、配列番号6で表されるアミノ酸配列からなる。
【0027】
Lはリンカーまたはペプチド結合を示す。リンカーとは、共有結合を介して2つの分子を連結する分子をいう。リンカーとしては、XおよびYが、それぞれ細胞膜透過性促進機能およびTrkAのチロシンキナーゼ活性抑制機能を発揮し得る限り特にその種類は限定されない。リンカーとしては、細胞外においては切断され難く、高い可動性を有するものが好適に用いられる。好適なリンカーとしては、ε−アミノカプロン酸リンカーを挙げることができる。
【0028】
Lがペプチド結合である場合、Xで表されるペプチド残基のカルボン酸末端にYで表されるペプチド残基がペプチド結合を介して直接結合することにより、式(I)の化合物はひとつながりのペプチドとなる。
【0029】
最も好ましくは、Lはε−アミノカプロン酸リンカーである。
【0030】
また、式(I)のペプチド化合物は修飾されていてもよい。該修飾としては、アミド化、脂質鎖の付加(脂肪族アシル化(パルミトイル化、ミリストイル化等)、プレニル化(ファルネシル化、ゲラニルゲラニル化等)等)、リン酸化(セリン残基、スレオニン残基、チロシン残基等におけるリン酸化)、アセチル化、糖鎖の付加(N−グリコシル化、O−グリコシル化)等を挙げることができる。好ましくは、式(I)のペプチド化合物はアミド化されている。
【0031】
また、式(I)のペプチド化合物の最も好適な例としては、例えば式(II):
YGRKKRRQRRR−acp−GMSRDIYS−NH2 (II)
または式(III):
YGRKKRRQRRR−acp−MSRDIYST−NH2 (III)
(式中、acpはε−アミノカプロン酸リンカーを示す)で表される化合物を挙げることができる。式(II)および(III)に含まれる「NH2」は、該ペプチド化合物のカルボン酸末端がアミド化されていることを意味する。
【0032】
また、式(I)のペプチド化合物の薬理学的に許容される塩としては、薬理学的に許容される酸(例:無機酸、有機酸)や塩基(例:アルカリ金属塩)などとの塩が用いられ、とりわけ薬理学的に許容される酸付加塩が好ましい。このような塩としては、例えば、無機酸(例えば、塩酸、リン酸、臭化水素酸、硫酸)との塩、あるいは有機酸(例えば、酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、蓚酸、安息香酸、メタンスルホン酸、ベンゼンスルホン酸)との塩などが挙げられる。
【0033】
また、式(I)のペプチド化合物の製造方法については特に制限なく、公知のペプチド合成法に従って製造することができる。ペプチド合成法は、例えば、固相合成法、液相合成法のいずれであってもよい。ペプチド化合物を構成し得る部分ペプチドもしくはアミノ酸と残余部分とを縮合し、生成物が保護基を有する場合は保護基を脱離することにより目的とするペプチド化合物を製造することができる。
【0034】
また、リンカーによるペプチド同士の連結も、自体公知の方法により行うことができる。例えば、ε−アミノカプロン酸リンカーを用いる場合には、ケミカル・ファーマシューティカル・ブルテン(Chem.Pharm.Bull.),2005年,第53巻,1131−1135頁に記載された方法を参照することにより、Xで表されるペプチドのカルボン酸末端にε−アミノカプロン酸リンカーを介してYで表されるペプチドのアミノ酸末端を連結することができる。
【0035】
式(I)のペプチド化合物またはその薬理学的に許容される塩は、良性または悪性の腫瘍細胞内に取り込まれ、NGFによるTrkAのチロシンキナーゼ活性を抑制することにより、該腫瘍細胞の増殖を強力に抑制し得る。また、式(I)のペプチド化合物またはその薬理学的に許容される塩は、一次知覚神経のニューロンに取り込まれ、NGFによるTrkAのチロシンキナーゼ活性を抑制することにより、疼痛を強力に抑制し得る。式(I)のペプチド化合物またはその薬理学的に許容される塩の有効量を哺乳動物(好ましくはヒト)に投与することにより、該哺乳動物における腫瘍を治療することができる。また、式(I)のペプチド化合物またはその薬理学的に許容される塩の有効量を哺乳動物(好ましくはヒト)に投与することにより、該哺乳動物における疼痛を治療することができる。従って、本発明は、式(I)のペプチド化合物またはその薬理学的に許容される塩を含む医薬(腫瘍治療剤又は疼痛治療剤)を提供するものである。
【0036】
式(I)のペプチド化合物またはその薬理学的に許容される塩は、NGFによるTrkAのチロシンキナーゼ活性を抑制することにより、該腫瘍細胞の増殖を強力に抑制するので、式(I)のペプチド化合物またはその薬理学的に許容される塩を腫瘍の治療に用いる場合、該腫瘍は通常TrkAを発現する腫瘍である。好ましくは、該腫瘍はTrkAを介したNGF刺激により増殖が促進している腫瘍である。褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫においてはNGFがTrkAを介してその増殖を促進することから、本発明の腫瘍治療剤は、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫、とりわけ褐色細胞腫の治療に効果的である。
【0037】
また、式(I)のペプチド化合物またはその薬理学的に許容される塩は、腫瘍細胞の増殖抑制効果と、疼痛抑制効果の双方を併せ持つので、がん疼痛の治療に好適に用いられる。
【0038】
遺伝子治療によりTrkAのチロシンキナーゼ活性を抑制し得るアミノ酸配列を有するペプチドを細胞内で発現させる場合は、正常細胞におけるTrkAのチロシンキナーゼ活性のコントロールができなくなる可能性がある。またTrkAのチロシンキナーゼ活性の、長期間の持続的な抑制は、免疫反応の低下やアルツハイマー病などの発症を促進する危険性がある。しかし、本発明の医薬の有効成分は、ペプチド化合物であるため、不必要な分は体内のプロテイナーゼにより分解され、このような合併症を来たす危険性は少ない。
【0039】
本発明の医薬は、活性成分として式(I)のペプチド化合物または薬理学的に許容されるその塩単独で、あるいは任意の他の治療のための有効成分との混合物として含有することができる。また、本発明の医薬は、製剤学の技術分野において周知の方法により、活性成分を薬理学的に許容される一種もしくはそれ以上の担体と一緒に混合し、医薬組成物として製造することができる。
【0040】
薬理学的に許容される担体としては、製剤素材として慣用の各種有機あるいは無機担体物質が用いられ、その具体例としては、固形製剤における賦形剤、滑沢剤、結合剤、崩壊剤、液状製剤における溶剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、無痛化剤などが挙げられる。製剤化の際には、必要に応じて、防腐剤、抗酸化剤、着色剤、甘味剤などの製剤添加剤を用いてもよい。
【0041】
賦形剤の好適な例としては、乳糖、白糖、D−マンニトール、D−ソルビトール、デンプン、α化デンプン、デキストリン、結晶セルロース、低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロースナトリウム、アラビアゴム、プルラン、軟質無水ケイ酸、合成ケイ酸アルミニウム、メタケイ酸アルミン酸マグネシウム、キシリトール、ソルビトール、エリスリトールなどが挙げられる。
滑沢剤の好適な例としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルク、コロイドシリカ、ポリエチレングリコール6000などが挙げられる。
結合剤の好適な例としては、α化デンプン、ショ糖、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースナトリウム、結晶セルロース、白糖、D−マンニトール、トレハロース、デキストリン、プルラン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドンなどが挙げられる。
崩壊剤の好適な例としては、乳糖、白糖、デンプン、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、軟質無水ケイ酸、炭酸カルシウムなどが挙げられる。
【0042】
溶剤の好適な例としては、注射用水、生理食塩水、リンゲル液、アルコール、プロピレングリコール、ポリエチレングリコール、ゴマ油、トウモロコシ油、オリーブ油、綿実油などが挙げられる。
溶解補助剤の好適な例としては、ポリエチレングリコール、プロピレングリコール、D−マンニトール、トレハロース、安息香酸ベンジル、エタノール、トリスアミノメタン、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、サリチル酸ナトリウム、酢酸ナトリウムなどが挙げられる。
懸濁化剤の好適な例としては、例えば、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウム、モノステアリン酸グリセリンなどの界面活性剤;例えば、ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロースなどの親水性高分子;ポリソルベート類、ポリオキシエチレン硬化ヒマシ油などが挙げられる。
等張化剤の好適な例としては、塩化ナトリウム、グリセリン、D−マンニトール、D−ソルビトール、ブドウ糖、キシリトール、果糖などが挙げられる。
緩衝剤の好適な例としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩などの緩衝液などが挙げられる。
無痛化剤の好適な例としては、プロピレングリコール、塩酸リドカイン、ベンジルアルコールなどが挙げられる。
防腐剤の好適な例としては、パラオキシ安息香酸エステル類、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸などが挙げられる。
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩などが挙げられる。
着色剤の好適な例としては、水溶性着色タール色素(例、食用赤色2号および3号、食用黄色4号および5号、食用青色1号および2号などの食用色素)、不溶性レーキ色素(例、前記水溶性食用タール色素のアルミニウム塩)、天然色素(例、β−カロチン、クロロフィル、ベンガラ)などが挙げられる。
甘味剤の好適な例としては、サッカリンナトリウム、グリチルリチン酸ニカリウム、アスパルテーム、ステビアなどが挙げられる。
【0043】
また、投与経路は、治療に際し最も効果的なものを使用するのが望ましく、経口製剤、注射剤または経皮製剤などで投与可能である。経口製剤としては、錠剤(舌下錠、口腔内崩壊剤を含む)、カプセル剤(ソフトカプセル、マイクロカプセルを含む)、散剤、顆粒剤、トローチ剤、シロップ剤、乳剤、懸濁剤などが挙げられる。また、注射剤としては、皮内注射、皮下注射、静脈内注射、筋肉内注射、脊髄腔内注射、硬膜外注射、局所注射などが挙げられる。また、経皮製剤としては、貼付剤、軟膏剤、散布剤などが挙げられる。これらの製剤は、速放性製剤または徐放性製剤などの放出制御製剤(例、徐放性マイクロカプセル)であってもよい。
【0044】
本発明の医薬中の式(I)のペプチド化合物の含有量は、例えば、0.1〜100重量%である。
【0045】
次に経口製剤の製造法について説明する。経口製剤は、活性成分に、例えば、賦形剤、崩壊剤、結合剤または滑沢剤などを添加して圧縮形成することにより製造される。
さらに、味のマスキング、腸溶化あるいは徐放化を目的として、自体公知の方法により、経口製剤にコーティングを行ってもよい。コーティング剤としては、例えば、腸溶性ポリマー(例、酢酸フタル酸セルロース、メタアクリル酸コポリマーL、メタアクリル酸コポリマーLD、メタアクリル酸コポリマーS、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、カルボキシメチルエチルセルロース)、胃溶性ポリマー(例、ポリビニルアセタールジエチルアミノアセテート、アミノアルキルメタアクリレートコポリマーE)、水溶性ポリマー(例、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース)、水不溶性ポリマー(例、エチルセルロース、アミノアルキルメタアクリレートコポリマーRS、アクリル酸エチル・メタアクリル酸メチル共重合体)、ワックスなどが用いられる。コーティングを行う場合、前記コーティング剤とともに、ポリエチレングリコール等の可塑剤;酸化チタン、三二酸化鉄等の遮光剤を用いてもよい。
【0046】
また、注射剤は、活性成分を分散剤(例、ツィーン(Tween)80(アトラスパウダー社製、米国)、HCO60(日光ケミカルズ社製)、ポリエチレングリコール、カルボキシメチルセルロース、アルギン酸ナトリウム)、保存剤(例、メチルパラベン、プロピルパラベン、ベンジルアルコール、クロロブタノール、フェノール)、等張化剤などと共に、水性溶剤(例、蒸留水、生理的食塩水、リンゲル液)あるいは油性溶剤(例、オリーブ油、ゴマ油、綿実油、コーン油などの植物油;プロピレングリコール、マクロゴール、トリカプリリン)などに溶解、懸濁あるいは乳化することにより製造される。この際、所望により溶解補助剤、懸濁化剤、緩衝剤、安定剤(例、ヒト血清アルブミン)、無痛化剤、防腐剤等の添加剤を用いてもよい。
【0047】
また、経皮製剤は、活性成分を固状、半固状または液状の組成物とすることにより製造される。例えば固状の組成物は、活性成分をそのまま、あるいは賦形剤、増粘剤などを添加、混合して粉状とすることにより製造される。液状の組成物は、注射剤の場合とほとんど同様にして製造される。半固状の組成物は、水性または油性のゲル剤あるいは軟膏状のものがよい。また、これらの組成物は、いずれもpH調整剤、防腐剤などを含んでいてもよい。
【0048】
本発明の医薬の投与量および投与回数は、投与形態、患者の年齢、体重、治療すべき症状の性質もしくは重篤度により異なるが、通常、静脈内投与の場合、成人一人当たり約10mg〜1gを一日一回ないし数回投与する。より好ましくは、注射剤による静脈内投与で、成人一人当たり30mg〜0.3gを一日1〜3回で1〜2週間の継続投与を1クールとし、これを3〜4クール繰り返す。
なお、これら投与量および投与回数に関しては、前述の種々の条件により変動する。
【0049】
以下、本発明について、実施例を挙げてさらに具体的に説明する。本発明はこれらにより何ら限定されるものではない。
【実施例1】
【0050】
ATPによるリコンビナントTrkAのチロシンリン酸化を、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)、ペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)がどの程度抑制するか、濃度を振って(1μM、10μM、100μM)検討した。抗リン酸化チロシン抗体を用いて、細胞膜透過性促進部位のペプチドTAT(YGRKKRRQRRR−acp)を用いた場合のTrkA活性を100%としてin vitroで明らかにした。
図1より、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)およびペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)はペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)よりもTrkAのチロシンキナーゼ活性を抑制する作用が強いことがわかる。
【実施例2】
【0051】
マウスのメラノーマ細胞株(B16−F1)を、血清を加えて培養した場合と、血清なしでNGFのみ加えて培養した場合において、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)、ペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)を濃度を振って(0μM、1μM、10μM、100μM)、それぞれ2日間暴露したのち細胞増殖率を測定した。
またマウスのメラノーマ細胞株(B16−F1)を、血清を加えて、それぞれ濃度を振った(0μM、1μM、10μM、100μM)ペプチド化合物と2日間培養し、培養液中のLDH濃度を測定することにより、細胞毒性を評価した。
図2および図3より、マウスのメラノーマ細胞株(B16−F1)において、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)およびペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)はペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)よりも細胞増殖抑制作用が強いことがわかる。また、図4より、ペプチド化合物100μM以下では細胞毒性が生じなかったこともわかる。
【実施例3】
【0052】
B16−F1細胞をC57BL/6Jマウスの左足底皮下に移植し、移植後5−9日目まで、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)10mg/kgを1日1回腹腔内投与し、移植後0−20日までの痛み反応(機械刺激過敏反応)を調査した。その結果、移植後20日目の痛み反応を、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)は抑制することが示された(図5)。この結果より、本発明のペプチド化合物が優れた疼痛抑制効果を有することが示唆された。
【産業上の利用可能性】
【0053】
本発明によれば、優れた腫瘍増殖抑制作用を有し、製造コストの低い、腫瘍治療剤を提供することが出来る。また、本発明によれば、優れた鎮痛作用を有し、製造コストの低い、疼痛治療剤を提供することができる。
【技術分野】
【0001】
本発明は、緩和医療学におけるがん疼痛の治療に用いられるペプチドに関する。
【背景技術】
【0002】
がん疼痛は、慢性腰痛、頸肩腕症候群などと共に、神経障害性疼痛と侵害受容性疼痛を併せ持つ痛みである。侵害受容性疼痛とは、例えば、けがや火傷などによって損傷部位に炎症が起こり、その場所で発痛物質が生成され、それが末梢神経の侵害受容器を刺激することで発生する痛みなどをいい、侵害受容器が活性化することによって引き起こされる疼痛である。一方、神経障害性疼痛は、神経が損傷を受けた結果として起こる痛みなどをいい、痛みが長期間続いたり、通常では何ともない程度の刺激に対して強い痛みを感じるなどの特徴があり、体性感覚系に対する損傷や疾患の直接的結果として生じている疼痛とされている。
【0003】
現在、疼痛抑制のために、モルヒネに代表されるオピオイドや非ステロイド性消炎鎮痛剤(NSAIDs)などが用いられているが、これらの鎮痛剤は、有用性、安全性、常習性等の面で改善の余地があり、また、神経障害性疼痛に関しては、こうした薬剤の効果が十分に得られないのが現状である。従って、安全性・常習性等の問題がなく、かつ神経障害性疼痛にも優れた効果を発揮するような医薬組成物、治療方法が望まれている。
【0004】
侵害受容性疼痛および神経障害性疼痛に関与するメカニズムの一つとして、神経成長因子(NGF)、およびその高親和性受容体であるTrkAが知られている。TrkAは、インスリン受容体ファミリーと呼ばれる細胞膜受容体型チロシンキナーゼの一つであり、その酵素活性の中心部に活性化ループを有する。この活性化ループ中のアミノ酸配列を含む合成ポリペプチドは、インスリン受容体やTrkAのチロシンキナーゼ活性を抑制することが示されている(非特許文献1)。
【0005】
また、TrkAは、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍、ホジキンリンパ腫などで発現していること、NGFがTrkAを介して腫瘍細胞の増殖に関与することが知られており、NGFとTrkAを介した細胞情報伝達系が、腫瘍細胞の増殖に関与する可能性が指摘されてきた。これらのことから、NGFおよびTrkAは癌治療薬および疼痛治療薬のターゲットの一つと考えられている。
【0006】
これまで、腫瘍増殖抑制作用や疼痛治療の目的で、NGFの働きやその高親和性受容体TrkAの活性を阻害するための手段として、TrkAのチロシンキナーゼ活性を抑制する薬剤、抗NGF抗体、抗TrkA抗体などが提案されてきた(特許文献1、特許文献2)。TrkAのチロシンキナーゼ活性を抑制する薬剤としては、K252aが知られているが、K252aは、TrkAのチロシンキナーゼ活性以外のプロテインキナーゼC活性も抑制する作用があるため、腫瘍および疼痛治療薬として臨床応用するには毒性が高く、また、抗体医薬については、膨大な製造コストがかかってしまうのが現状である。これに対し、ペプチドは、タンパク質と比較して分子量が少なく構造も単純であるため、生産コストを比較的抑えることが可能であり、また、不必要な分は体内のプロテイナーゼにより分解されるため、遺伝子治療において予想されるような危険性は少ない。従って、TrkA活性を特異的に抑制するようなペプチドを得ることができれば、臨床上非常に大きなメリットがあると考えられる。
【0007】
本発明者らは、TrkA活性化ループのアミノ酸配列と腫瘍細胞の増殖および痛みとの関係について研究を行った。その結果、TrkA活性化ループのアミノ酸配列(664−GMSRDIYSTDYYRVGGR−680)(配列番号1)の部分配列SRDIYSTDYYR(配列番号2)を有するペプチドを、細胞膜透過性を促進する機能を有するペプチドと連結して腫瘍細胞や神経細胞内へ導入すると、TrkAのチロシンキナーゼ活性が抑制され、腫瘍細胞の増殖が抑制されること(特許文献3、非特許文献1)、さらに鎮痛作用が得られる(侵害受容性疼痛および神経障害性疼痛ともに抑制できる)こと(非特許文献2、非特許文献3)を既に報告している。
【先行技術文献】
【特許文献】
【0008】
【特許文献1】特開2010−162028号公報
【特許文献2】国際公開第2006/137106号パンフレット
【特許文献3】特開2010−6731号公報
【非特許文献】
【0009】
【非特許文献1】Munetaka Hirose et al., J Pharmacol Sci 106,107-113,2008
【非特許文献2】Koyo Ueda et al., J Pharmacol Sci 112,438-443,2010
【非特許文献3】Wei-Ying Ma et al., J Pharmacol Sci 114,79-84,2010
【発明の概要】
【発明が解決しようとする課題】
【0010】
本発明は、TrkA活性を抑制するペプチドを用いた、優れた腫瘍治療薬および疼痛治療薬を提供することを目的とする。
【課題を解決するための手段】
【0011】
本発明者らは、上記の知見を基に、さらに検討を重ね、TrkA活性を強力に抑制することが報告されているTrkA活性化ループ内の部分配列SRDIYSTDYYR(配列番号2)よりも、N末端側のMSRDIYS(配列番号3)を少なくとも含む領域を用いれば、より強力に、且つ短い長さのペプチドでTrkA活性を抑制し、腫瘍や疼痛を治療し得ることを見出し、更に検討を進めた結果、本発明を完成した。
【0012】
すなわち、本発明は以下のとおりである。
(1)式(I):
X−L−Y (I)
(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ配列番号1で表されるアミノ酸配列の部分配列(ただし、該部分配列は配列番号3で表されるアミノ酸配列を含む)を含む15アミノ酸長以下のペプチド残基である)で表されるペプチド化合物または薬理学的に許容されるその塩。
(2)Yが配列番号4または配列番号5で表されるアミノ酸配列を含むポリペプチド残基である、(1)に記載の、ペプチド化合物または薬理学的に許容されるその塩。
(3)Xが配列番号6〜11で表されるアミノ酸配列のいずれかを有するポリペプチド残基である、(1)または(2)に記載の、ペプチド化合物または薬理学的に許容されるその塩。
(4)Lがε−アミノカプロン酸リンカーである、(1)〜(3)のいずれか1つに記載の、ペプチド化合物または薬理学的に許容されるその塩。
(5)ペプチド化合物が、式(II):
YGRKKRRQRRR−acp−GMSRDIYS−NH2 (II)
または式(III):
YGRKKRRQRRR−acp−MSRDIYST−NH2 (III)
(式中、acpはε−アミノカプロン酸リンカーを示す)で表される化合物である、(1)〜(4)のいずれか1つに記載の、ペプチド化合物または薬理学的に許容されるその塩。
(6)(1)〜(5)のいずれか1つに記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する医薬組成物。
(7)(1)〜(5)のいずれか1つに記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、腫瘍治療剤。
(8)腫瘍が、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫である、(7)に記載の腫瘍治療剤。
(9)(1)〜(5)のいずれか1つに記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、疼痛治療剤。
(10)癌疼痛治療剤である、(9)に記載の疼痛治療剤。
【発明の効果】
【0013】
本発明によれば、優れた腫瘍増殖抑制作用を有し、製造コストの低い、腫瘍治療剤を提供することが出来る。また、本発明によれば、優れた鎮痛作用を有し、製造コストの低い、疼痛治療剤を提供することができる。
【図面の簡単な説明】
【0014】
【図1】In vitroにおける、ペプチド化合物によるTrkAチロシンキナーゼ活性抑制作用を示す図である。
【図2】マウスメラノーマ細胞株(B16−F1)を、血清およびペプチド化合物を加えて培養した場合の、ペプチド化合物による細胞増殖抑制作用を示す図である。
【図3】マウスメラノーマ細胞株(B16−F1)を、血清無しでNGFおよびペプチド化合物を加えて培養した場合の、ペプチド化合物による細胞増殖抑制作用を示す図である。
【図4】マウスメラノーマ細胞株(B16−F1)を、血清およびペプチド化合物を加えて培養した場合の、ペプチド化合物による細胞毒性を示す図である。
【図5】マウスメラノーマモデルにおける、ペプチド化合物による鎮痛効果を示す図である。
【発明を実施するための形態】
【0015】
本発明は、式(I):
X−L−Y (I)
(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ配列番号1で表されるアミノ酸配列の部分配列を含むペプチド残基である)で表されるペプチド化合物または薬理学的に許容されるその塩を提供するものである。
【0016】
Yで表されるペプチド残基は、TrkAのチロシンキナーゼ活性を抑制する機能を有する。
【0017】
TrkAは、神経成長因子(NGF)の高親和性受容体として知られる公知のタンパク質である。TrkAは、その細胞内ドメインにチロシンキナーゼ活性を有しており、NGFがTrkAに結合すると、該チロシンキナーゼ活性によりTrkAの細胞内ドメインに含まれる5つのチロシン残基が自己リン酸化され、下流の細胞内情報伝達系へ情報伝達を行う。「TrkAのチロシンキナーゼ活性の抑制」とは、このTrkAの自己リン酸化の抑制および下流の細胞内情報伝達系への情報伝達の抑制を意味する。
【0018】
Yで表されるペプチド残基は、配列番号1で表されるアミノ酸配列の部分配列を含む。配列番号1で表されるアミノ酸配列は、ヒトTrkAの活性化ループ部位に相当する。また、該部分配列は、配列番号3で表されるアミノ酸配列を含む。該部分配列の長さは、Yで表されるペプチド残基がTrkAのチロシンキナーゼ活性の抑制作用を有し、且つ式(I)で表されるペプチド化合物が腫瘍細胞の増殖や、疼痛を抑制する機能を発揮し得る限り特に限定されないが、十分な強さのTrkAのチロシンキナーゼ活性抑制機能を確保する観点から、該部分配列の長さは7アミノ酸長以上、好ましくは8アミノ酸長以上である。また、該部分配列の長さは、通常15アミノ酸長以下、好ましくは13アミノ酸長以下、より好ましくは11アミノ酸長以下、更に好ましくは9アミノ酸長以下である。
【0019】
一態様において、該部分配列の長さは、好ましくは7〜9アミノ酸であり、最も好ましくは8アミノ酸である。
【0020】
一方、該ペプチド残基が長すぎると、式(I)で表されるペプチド化合物の細胞内への導入効率が低下する恐れがある。従って、細胞内への高い導入効率を達成する観点から、Yで表されるペプチド残基の全長は、通常15アミノ酸長以下、好ましくは13アミノ酸長以下、より好ましくは11アミノ酸長以下、更に好ましくは9アミノ酸長以下である。
【0021】
Yで表されるペプチド残基は、上述の配列番号1で表されるアミノ酸配列の部分配列以外の領域を含んでいても含んでいなくてもよい。Yで表されるペプチド残基が、上述の配列番号1で表されるアミノ酸配列の部分配列以外の領域を含む場合、当該領域を構成するアミノ酸配列は、Yで表されるペプチド残基がTrkAのチロシンキナーゼ活性を抑制する限り、特に限定されない。好ましくは、Yで表されるペプチド残基は、上述の配列番号1で表されるアミノ酸配列の部分配列からなる。
【0022】
最も好ましくは、Yで表されるペプチド残基は、配列番号4または配列番号5で表されるアミノ酸配列からなる。
【0023】
Xで表されるペプチド残基は、細胞膜透過性を促進する機能を有するアミノ酸配列を含む。「細胞膜透過性を促進する機能」とは、細胞外から細胞内への化合物の透過を促進する機能を意味する。式(I)で表されるペプチド化合物がこのXで表されるペプチド残基を有することにより、生体へ該ペプチド化合物を投与すると、該ペプチド化合物が細胞内へ取り込まれ、該細胞においてTrkAのチロシンキナーゼ活性を抑制し、腫瘍細胞の増殖や、さらに、疼痛を抑制することができる。
【0024】
細胞膜透過性を促進する機能を有するアミノ酸配列としては、公知のものを用いることができる。例えば、配列番号6〜11で表されるアミノ酸配列を好適なものとして挙げることができる。このうち、配列番号6で表されるアミノ酸配列は、ヒト免疫不全ウィルスタイプ1のTAT(転写のトランス活性化因子)に由来するペプチド(47−YGRKKRRQRRR−57)に相当する。
【0025】
Xで表されるペプチド残基の長さは、細胞膜透過性を促進する機能を発揮し得る限り特に限定されないが、式(I)の化合物の細胞内への高い導入効率を達成する観点から通常4〜15アミノ酸長、好ましくは5〜11アミノ酸長である。
【0026】
最も好ましくは、Xで表されるペプチド残基は、配列番号6で表されるアミノ酸配列からなる。
【0027】
Lはリンカーまたはペプチド結合を示す。リンカーとは、共有結合を介して2つの分子を連結する分子をいう。リンカーとしては、XおよびYが、それぞれ細胞膜透過性促進機能およびTrkAのチロシンキナーゼ活性抑制機能を発揮し得る限り特にその種類は限定されない。リンカーとしては、細胞外においては切断され難く、高い可動性を有するものが好適に用いられる。好適なリンカーとしては、ε−アミノカプロン酸リンカーを挙げることができる。
【0028】
Lがペプチド結合である場合、Xで表されるペプチド残基のカルボン酸末端にYで表されるペプチド残基がペプチド結合を介して直接結合することにより、式(I)の化合物はひとつながりのペプチドとなる。
【0029】
最も好ましくは、Lはε−アミノカプロン酸リンカーである。
【0030】
また、式(I)のペプチド化合物は修飾されていてもよい。該修飾としては、アミド化、脂質鎖の付加(脂肪族アシル化(パルミトイル化、ミリストイル化等)、プレニル化(ファルネシル化、ゲラニルゲラニル化等)等)、リン酸化(セリン残基、スレオニン残基、チロシン残基等におけるリン酸化)、アセチル化、糖鎖の付加(N−グリコシル化、O−グリコシル化)等を挙げることができる。好ましくは、式(I)のペプチド化合物はアミド化されている。
【0031】
また、式(I)のペプチド化合物の最も好適な例としては、例えば式(II):
YGRKKRRQRRR−acp−GMSRDIYS−NH2 (II)
または式(III):
YGRKKRRQRRR−acp−MSRDIYST−NH2 (III)
(式中、acpはε−アミノカプロン酸リンカーを示す)で表される化合物を挙げることができる。式(II)および(III)に含まれる「NH2」は、該ペプチド化合物のカルボン酸末端がアミド化されていることを意味する。
【0032】
また、式(I)のペプチド化合物の薬理学的に許容される塩としては、薬理学的に許容される酸(例:無機酸、有機酸)や塩基(例:アルカリ金属塩)などとの塩が用いられ、とりわけ薬理学的に許容される酸付加塩が好ましい。このような塩としては、例えば、無機酸(例えば、塩酸、リン酸、臭化水素酸、硫酸)との塩、あるいは有機酸(例えば、酢酸、ギ酸、プロピオン酸、フマル酸、マレイン酸、コハク酸、酒石酸、クエン酸、リンゴ酸、蓚酸、安息香酸、メタンスルホン酸、ベンゼンスルホン酸)との塩などが挙げられる。
【0033】
また、式(I)のペプチド化合物の製造方法については特に制限なく、公知のペプチド合成法に従って製造することができる。ペプチド合成法は、例えば、固相合成法、液相合成法のいずれであってもよい。ペプチド化合物を構成し得る部分ペプチドもしくはアミノ酸と残余部分とを縮合し、生成物が保護基を有する場合は保護基を脱離することにより目的とするペプチド化合物を製造することができる。
【0034】
また、リンカーによるペプチド同士の連結も、自体公知の方法により行うことができる。例えば、ε−アミノカプロン酸リンカーを用いる場合には、ケミカル・ファーマシューティカル・ブルテン(Chem.Pharm.Bull.),2005年,第53巻,1131−1135頁に記載された方法を参照することにより、Xで表されるペプチドのカルボン酸末端にε−アミノカプロン酸リンカーを介してYで表されるペプチドのアミノ酸末端を連結することができる。
【0035】
式(I)のペプチド化合物またはその薬理学的に許容される塩は、良性または悪性の腫瘍細胞内に取り込まれ、NGFによるTrkAのチロシンキナーゼ活性を抑制することにより、該腫瘍細胞の増殖を強力に抑制し得る。また、式(I)のペプチド化合物またはその薬理学的に許容される塩は、一次知覚神経のニューロンに取り込まれ、NGFによるTrkAのチロシンキナーゼ活性を抑制することにより、疼痛を強力に抑制し得る。式(I)のペプチド化合物またはその薬理学的に許容される塩の有効量を哺乳動物(好ましくはヒト)に投与することにより、該哺乳動物における腫瘍を治療することができる。また、式(I)のペプチド化合物またはその薬理学的に許容される塩の有効量を哺乳動物(好ましくはヒト)に投与することにより、該哺乳動物における疼痛を治療することができる。従って、本発明は、式(I)のペプチド化合物またはその薬理学的に許容される塩を含む医薬(腫瘍治療剤又は疼痛治療剤)を提供するものである。
【0036】
式(I)のペプチド化合物またはその薬理学的に許容される塩は、NGFによるTrkAのチロシンキナーゼ活性を抑制することにより、該腫瘍細胞の増殖を強力に抑制するので、式(I)のペプチド化合物またはその薬理学的に許容される塩を腫瘍の治療に用いる場合、該腫瘍は通常TrkAを発現する腫瘍である。好ましくは、該腫瘍はTrkAを介したNGF刺激により増殖が促進している腫瘍である。褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫においてはNGFがTrkAを介してその増殖を促進することから、本発明の腫瘍治療剤は、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫、とりわけ褐色細胞腫の治療に効果的である。
【0037】
また、式(I)のペプチド化合物またはその薬理学的に許容される塩は、腫瘍細胞の増殖抑制効果と、疼痛抑制効果の双方を併せ持つので、がん疼痛の治療に好適に用いられる。
【0038】
遺伝子治療によりTrkAのチロシンキナーゼ活性を抑制し得るアミノ酸配列を有するペプチドを細胞内で発現させる場合は、正常細胞におけるTrkAのチロシンキナーゼ活性のコントロールができなくなる可能性がある。またTrkAのチロシンキナーゼ活性の、長期間の持続的な抑制は、免疫反応の低下やアルツハイマー病などの発症を促進する危険性がある。しかし、本発明の医薬の有効成分は、ペプチド化合物であるため、不必要な分は体内のプロテイナーゼにより分解され、このような合併症を来たす危険性は少ない。
【0039】
本発明の医薬は、活性成分として式(I)のペプチド化合物または薬理学的に許容されるその塩単独で、あるいは任意の他の治療のための有効成分との混合物として含有することができる。また、本発明の医薬は、製剤学の技術分野において周知の方法により、活性成分を薬理学的に許容される一種もしくはそれ以上の担体と一緒に混合し、医薬組成物として製造することができる。
【0040】
薬理学的に許容される担体としては、製剤素材として慣用の各種有機あるいは無機担体物質が用いられ、その具体例としては、固形製剤における賦形剤、滑沢剤、結合剤、崩壊剤、液状製剤における溶剤、溶解補助剤、懸濁化剤、等張化剤、緩衝剤、無痛化剤などが挙げられる。製剤化の際には、必要に応じて、防腐剤、抗酸化剤、着色剤、甘味剤などの製剤添加剤を用いてもよい。
【0041】
賦形剤の好適な例としては、乳糖、白糖、D−マンニトール、D−ソルビトール、デンプン、α化デンプン、デキストリン、結晶セルロース、低置換度ヒドロキシプロピルセルロース、カルボキシメチルセルロースナトリウム、アラビアゴム、プルラン、軟質無水ケイ酸、合成ケイ酸アルミニウム、メタケイ酸アルミン酸マグネシウム、キシリトール、ソルビトール、エリスリトールなどが挙げられる。
滑沢剤の好適な例としては、ステアリン酸マグネシウム、ステアリン酸カルシウム、タルク、コロイドシリカ、ポリエチレングリコール6000などが挙げられる。
結合剤の好適な例としては、α化デンプン、ショ糖、ゼラチン、アラビアゴム、メチルセルロース、カルボキシメチルセルロース、カルボキシメチルセルロースナトリウム、結晶セルロース、白糖、D−マンニトール、トレハロース、デキストリン、プルラン、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース、ポリビニルピロリドンなどが挙げられる。
崩壊剤の好適な例としては、乳糖、白糖、デンプン、カルボキシメチルセルロース、カルボキシメチルセルロースカルシウム、クロスカルメロースナトリウム、カルボキシメチルスターチナトリウム、低置換度ヒドロキシプロピルセルロース、軟質無水ケイ酸、炭酸カルシウムなどが挙げられる。
【0042】
溶剤の好適な例としては、注射用水、生理食塩水、リンゲル液、アルコール、プロピレングリコール、ポリエチレングリコール、ゴマ油、トウモロコシ油、オリーブ油、綿実油などが挙げられる。
溶解補助剤の好適な例としては、ポリエチレングリコール、プロピレングリコール、D−マンニトール、トレハロース、安息香酸ベンジル、エタノール、トリスアミノメタン、コレステロール、トリエタノールアミン、炭酸ナトリウム、クエン酸ナトリウム、サリチル酸ナトリウム、酢酸ナトリウムなどが挙げられる。
懸濁化剤の好適な例としては、例えば、ステアリルトリエタノールアミン、ラウリル硫酸ナトリウム、ラウリルアミノプロピオン酸、レシチン、塩化ベンザルコニウム、塩化ベンゼトニウム、モノステアリン酸グリセリンなどの界面活性剤;例えば、ポリビニルアルコール、ポリビニルピロリドン、カルボキシメチルセルロースナトリウム、メチルセルロース、ヒドロキシメチルセルロース、ヒドロキシエチルセルロース、ヒドロキシプロピルセルロースなどの親水性高分子;ポリソルベート類、ポリオキシエチレン硬化ヒマシ油などが挙げられる。
等張化剤の好適な例としては、塩化ナトリウム、グリセリン、D−マンニトール、D−ソルビトール、ブドウ糖、キシリトール、果糖などが挙げられる。
緩衝剤の好適な例としては、リン酸塩、酢酸塩、炭酸塩、クエン酸塩などの緩衝液などが挙げられる。
無痛化剤の好適な例としては、プロピレングリコール、塩酸リドカイン、ベンジルアルコールなどが挙げられる。
防腐剤の好適な例としては、パラオキシ安息香酸エステル類、クロロブタノール、ベンジルアルコール、フェネチルアルコール、デヒドロ酢酸、ソルビン酸などが挙げられる。
抗酸化剤の好適な例としては、亜硫酸塩、アスコルビン酸塩などが挙げられる。
着色剤の好適な例としては、水溶性着色タール色素(例、食用赤色2号および3号、食用黄色4号および5号、食用青色1号および2号などの食用色素)、不溶性レーキ色素(例、前記水溶性食用タール色素のアルミニウム塩)、天然色素(例、β−カロチン、クロロフィル、ベンガラ)などが挙げられる。
甘味剤の好適な例としては、サッカリンナトリウム、グリチルリチン酸ニカリウム、アスパルテーム、ステビアなどが挙げられる。
【0043】
また、投与経路は、治療に際し最も効果的なものを使用するのが望ましく、経口製剤、注射剤または経皮製剤などで投与可能である。経口製剤としては、錠剤(舌下錠、口腔内崩壊剤を含む)、カプセル剤(ソフトカプセル、マイクロカプセルを含む)、散剤、顆粒剤、トローチ剤、シロップ剤、乳剤、懸濁剤などが挙げられる。また、注射剤としては、皮内注射、皮下注射、静脈内注射、筋肉内注射、脊髄腔内注射、硬膜外注射、局所注射などが挙げられる。また、経皮製剤としては、貼付剤、軟膏剤、散布剤などが挙げられる。これらの製剤は、速放性製剤または徐放性製剤などの放出制御製剤(例、徐放性マイクロカプセル)であってもよい。
【0044】
本発明の医薬中の式(I)のペプチド化合物の含有量は、例えば、0.1〜100重量%である。
【0045】
次に経口製剤の製造法について説明する。経口製剤は、活性成分に、例えば、賦形剤、崩壊剤、結合剤または滑沢剤などを添加して圧縮形成することにより製造される。
さらに、味のマスキング、腸溶化あるいは徐放化を目的として、自体公知の方法により、経口製剤にコーティングを行ってもよい。コーティング剤としては、例えば、腸溶性ポリマー(例、酢酸フタル酸セルロース、メタアクリル酸コポリマーL、メタアクリル酸コポリマーLD、メタアクリル酸コポリマーS、ヒドロキシプロピルメチルセルロースフタレート、ヒドロキシプロピルメチルセルロースアセテートサクシネート、カルボキシメチルエチルセルロース)、胃溶性ポリマー(例、ポリビニルアセタールジエチルアミノアセテート、アミノアルキルメタアクリレートコポリマーE)、水溶性ポリマー(例、ヒドロキシプロピルセルロース、ヒドロキシプロピルメチルセルロース)、水不溶性ポリマー(例、エチルセルロース、アミノアルキルメタアクリレートコポリマーRS、アクリル酸エチル・メタアクリル酸メチル共重合体)、ワックスなどが用いられる。コーティングを行う場合、前記コーティング剤とともに、ポリエチレングリコール等の可塑剤;酸化チタン、三二酸化鉄等の遮光剤を用いてもよい。
【0046】
また、注射剤は、活性成分を分散剤(例、ツィーン(Tween)80(アトラスパウダー社製、米国)、HCO60(日光ケミカルズ社製)、ポリエチレングリコール、カルボキシメチルセルロース、アルギン酸ナトリウム)、保存剤(例、メチルパラベン、プロピルパラベン、ベンジルアルコール、クロロブタノール、フェノール)、等張化剤などと共に、水性溶剤(例、蒸留水、生理的食塩水、リンゲル液)あるいは油性溶剤(例、オリーブ油、ゴマ油、綿実油、コーン油などの植物油;プロピレングリコール、マクロゴール、トリカプリリン)などに溶解、懸濁あるいは乳化することにより製造される。この際、所望により溶解補助剤、懸濁化剤、緩衝剤、安定剤(例、ヒト血清アルブミン)、無痛化剤、防腐剤等の添加剤を用いてもよい。
【0047】
また、経皮製剤は、活性成分を固状、半固状または液状の組成物とすることにより製造される。例えば固状の組成物は、活性成分をそのまま、あるいは賦形剤、増粘剤などを添加、混合して粉状とすることにより製造される。液状の組成物は、注射剤の場合とほとんど同様にして製造される。半固状の組成物は、水性または油性のゲル剤あるいは軟膏状のものがよい。また、これらの組成物は、いずれもpH調整剤、防腐剤などを含んでいてもよい。
【0048】
本発明の医薬の投与量および投与回数は、投与形態、患者の年齢、体重、治療すべき症状の性質もしくは重篤度により異なるが、通常、静脈内投与の場合、成人一人当たり約10mg〜1gを一日一回ないし数回投与する。より好ましくは、注射剤による静脈内投与で、成人一人当たり30mg〜0.3gを一日1〜3回で1〜2週間の継続投与を1クールとし、これを3〜4クール繰り返す。
なお、これら投与量および投与回数に関しては、前述の種々の条件により変動する。
【0049】
以下、本発明について、実施例を挙げてさらに具体的に説明する。本発明はこれらにより何ら限定されるものではない。
【実施例1】
【0050】
ATPによるリコンビナントTrkAのチロシンリン酸化を、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)、ペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)がどの程度抑制するか、濃度を振って(1μM、10μM、100μM)検討した。抗リン酸化チロシン抗体を用いて、細胞膜透過性促進部位のペプチドTAT(YGRKKRRQRRR−acp)を用いた場合のTrkA活性を100%としてin vitroで明らかにした。
図1より、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)およびペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)はペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)よりもTrkAのチロシンキナーゼ活性を抑制する作用が強いことがわかる。
【実施例2】
【0051】
マウスのメラノーマ細胞株(B16−F1)を、血清を加えて培養した場合と、血清なしでNGFのみ加えて培養した場合において、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)、ペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)を濃度を振って(0μM、1μM、10μM、100μM)、それぞれ2日間暴露したのち細胞増殖率を測定した。
またマウスのメラノーマ細胞株(B16−F1)を、血清を加えて、それぞれ濃度を振った(0μM、1μM、10μM、100μM)ペプチド化合物と2日間培養し、培養液中のLDH濃度を測定することにより、細胞毒性を評価した。
図2および図3より、マウスのメラノーマ細胞株(B16−F1)において、ペプチド化合物1(YGRKKRRQRRR−acp−GMSRDIYS−NH2)およびペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)はペプチド化合物3(YGRKKRRQRRR−acp−SRDIYSTDYYR−NH2)よりも細胞増殖抑制作用が強いことがわかる。また、図4より、ペプチド化合物100μM以下では細胞毒性が生じなかったこともわかる。
【実施例3】
【0052】
B16−F1細胞をC57BL/6Jマウスの左足底皮下に移植し、移植後5−9日目まで、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)10mg/kgを1日1回腹腔内投与し、移植後0−20日までの痛み反応(機械刺激過敏反応)を調査した。その結果、移植後20日目の痛み反応を、ペプチド化合物2(YGRKKRRQRRR−acp−MSRDIYST−NH2)は抑制することが示された(図5)。この結果より、本発明のペプチド化合物が優れた疼痛抑制効果を有することが示唆された。
【産業上の利用可能性】
【0053】
本発明によれば、優れた腫瘍増殖抑制作用を有し、製造コストの低い、腫瘍治療剤を提供することが出来る。また、本発明によれば、優れた鎮痛作用を有し、製造コストの低い、疼痛治療剤を提供することができる。
【特許請求の範囲】
【請求項1】
式(I):
X−L−Y (I)
(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ配列番号1で表されるアミノ酸配列の部分配列(ただし、該部分配列は配列番号3で表されるアミノ酸配列を含む)を含む15アミノ酸長以下のペプチド残基である)で表されるペプチド化合物または薬理学的に許容されるその塩。
【請求項2】
Yが配列番号4または配列番号5で表されるアミノ酸配列を含むポリペプチド残基である、請求項1記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項3】
Xが配列番号6〜11で表されるアミノ酸配列のいずれかを有するポリペプチド残基である、請求項1または2に記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項4】
Lがε−アミノカプロン酸リンカーである、請求項1〜3のいずれか1項に記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項5】
ペプチド化合物が、式(II):
YGRKKRRQRRR−acp−GMSRDIYS−NH2 (II)
または式(III):
YGRKKRRQRRR−acp−MSRDIYST−NH2 (III)
(式中、acpはε−アミノカプロン酸リンカーを示す)で表される化合物である、請求項1〜4のいずれか1項に記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項6】
請求項1〜5のいずれか1項に記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する医薬組成物。
【請求項7】
請求項1〜5のいずれか1項に記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、腫瘍治療剤。
【請求項8】
腫瘍が、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫である、請求項7に記載の腫瘍治療剤。
【請求項9】
請求項1〜5のいずれか1項に記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、疼痛治療剤。
【請求項10】
癌疼痛治療剤である、請求項9に記載の疼痛治療剤。
【請求項1】
式(I):
X−L−Y (I)
(式中、Xは細胞膜透過性を促進する機能を有するペプチド残基であり、Lはリンカーまたはペプチド結合であり、YはTrkAのチロシンキナーゼ活性を抑制する機能を有し、且つ配列番号1で表されるアミノ酸配列の部分配列(ただし、該部分配列は配列番号3で表されるアミノ酸配列を含む)を含む15アミノ酸長以下のペプチド残基である)で表されるペプチド化合物または薬理学的に許容されるその塩。
【請求項2】
Yが配列番号4または配列番号5で表されるアミノ酸配列を含むポリペプチド残基である、請求項1記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項3】
Xが配列番号6〜11で表されるアミノ酸配列のいずれかを有するポリペプチド残基である、請求項1または2に記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項4】
Lがε−アミノカプロン酸リンカーである、請求項1〜3のいずれか1項に記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項5】
ペプチド化合物が、式(II):
YGRKKRRQRRR−acp−GMSRDIYS−NH2 (II)
または式(III):
YGRKKRRQRRR−acp−MSRDIYST−NH2 (III)
(式中、acpはε−アミノカプロン酸リンカーを示す)で表される化合物である、請求項1〜4のいずれか1項に記載の、ペプチド化合物または薬理学的に許容されるその塩。
【請求項6】
請求項1〜5のいずれか1項に記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する医薬組成物。
【請求項7】
請求項1〜5のいずれか1項に記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、腫瘍治療剤。
【請求項8】
腫瘍が、褐色細胞腫、神経芽細胞腫、甲状腺癌、肺癌、前立腺癌、尿管癌、膀胱癌、乳癌、膵臓癌、メラノーマ、ウィルムス腫瘍またはホジキンリンパ腫である、請求項7に記載の腫瘍治療剤。
【請求項9】
請求項1〜5のいずれか1項に記載のペプチド化合物、またはそれらの薬理学的に許容される塩を含有する、疼痛治療剤。
【請求項10】
癌疼痛治療剤である、請求項9に記載の疼痛治療剤。
【図1】


【図2】
【図3】
【図4】
【図5】
【図2】
【図3】
【図4】
【図5】
【公開番号】特開2012−201621(P2012−201621A)
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願番号】特願2011−66819(P2011−66819)
【出願日】平成23年3月24日(2011.3.24)
【出願人】(504145320)国立大学法人福井大学 (287)
【Fターム(参考)】
【公開日】平成24年10月22日(2012.10.22)
【国際特許分類】
【出願日】平成23年3月24日(2011.3.24)
【出願人】(504145320)国立大学法人福井大学 (287)
【Fターム(参考)】
[ Back to top ]