
UDP−グルコース:N−アシルスフィンゴシングルコシルトランスフェラーゼインヒビターの合成
【課題】より経済的かつ効率的であり、公知の合成よりも少ない工程を含む、アミノセラミド様化合物の鏡像選択的合成方法、該方法により得られるアミノセラミド様化合物を提供すること。
【解決手段】以下の構造式:
【化1】

で表される化合物またはその生理学的に許容され得る塩。
【解決手段】以下の構造式:
【化1】
で表される化合物またはその生理学的に許容され得る塩。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、UDP-グルコース:N-アシルスフィンゴシングルコシルトランスフェラーゼインヒビター、およびその合成方法に関する。
【背景技術】
【0002】
発明の背景
グリコスフィンゴリピド(GSL)は、細胞増殖、細胞分化、細胞間接着または細胞とマトリックスタンパク質との間の接着、微生物およびウイルスの細胞への結合ならびに腫瘍細胞の転移を促進する能力を含む、多くの生物学的機能を有する天然化合物の一群である。GSLは、セラミドおよびUDP-グルコースから酵素UDP-グルコース:N-アシルスフィンゴシングルコシルトランスフェラーゼ(GlcCerシンターゼ)により生成されるグルコシルセラミド(GlcCer)に由来する。セラミドの構造を以下に示す。
【0003】
【化1】
【0004】
GSLの蓄積は、テイ−サックス病、ゴーシェ病、およびファブリー病を含む、いくつかの疾患と関連している(例えば、特許文献1参照)。GSLはまた、特定の癌と関連している。例えば、特定のGSLが腫瘍でのみ発生したり、腫瘍において異常に高い濃度で発生すること;培養培地内の腫瘍細胞に添加すると、腫瘍増殖に対して顕著な刺激性または阻害性作用を及ぼすこと;および腫瘍のそばを通り周囲の細胞外液内に散らばると身体の正常な免疫防御システムを阻害することがわかっている。腫瘍が次第に悪性になるにつれて腫瘍のGSLの組成が変化し、特定のGSLに対する抗体が腫瘍の成長を阻害する。
【0005】
GlcCerシンターゼを阻害する化合物は、GSL濃度を低下させ得、前述の疾患の1つを有する被験体を処置するのに有用であることが報告されている。いくつかのGlcCerの強力なインヒビター(本明細書では「アミノセラミド様化合物」という)が特許文献1、特許文献2、特許文献3、特許文献4および特許文献5に開示されている。用語「セラミド様化合物」は、1)第一級アルコールが置換アミノ基で置換され、2)アルケニル基がアリール基、好ましくはフェニルまたは置換フェニルで置換された、セラミドのアナログをいう。対応するN-脱アシル化化合物を「スフィンゴシン様化合物」とよぶ。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許第6,051,598号明細書
【特許文献2】米国特許第5,952,370号明細書
【特許文献3】米国特許第5,945,442号明細書
【特許文献4】米国特許第5,916,911号明細書
【特許文献5】米国特許第6,030,995号明細書
【発明の概要】
【発明が解決しようとする課題】
【0007】
残念ながら、アミノセラミド様化合物を調製する公知の方法は、工業的規模での製造にあまり適していない。2つのキラル中心により、公知の合成のほとんどは、4つのジアステレオマーを生じ、これは、クロマトグラフィーによりジアステレオマーを分離し、光学活性剤、例えばジベンゾイル酒石酸異性体で誘導体化した後、結晶化によって所望のエナンチオマーを単離する必要性を生じる(例えば、Inokuchi および Radin, Journal of Lipid Research 28:565 (1987)参照)。いずれのプロセスも大規模調製には不向きである。偏左右選択的還元を用いるアミノセラミド様化合物の鏡像選択的合成が報告されている(Mitchell ら, J. Org. Chem. 63:8837 (1998)およびNishidaら, SYNLETT 1998:389 (1998))が、10を超える工程を必要とし、そのうちいくつかの工程では、水素化ジイソブチルアルミニウム(DIABAL)およびガーナーアルデヒド(tert-ブチル(R)-(+)-4ホルミル-2,2-ジメチル-3-オキサゾリジンカルボキシレート)などの高価な試薬を使用する。したがって、より経済的かつ効率的であり、公知の合成よりも少ない工程を含む、アミノセラミド様化合物の鏡像選択的合成の大いなる必要性が存在する。
【課題を解決するための手段】
【0008】
即ち、本発明の要旨は、
〔1〕以下の構造式:
【化2】
で表される化合物またはその生理学的に許容され得る塩
に関する。
【発明の効果】
【0009】
本発明により、より経済的かつ効率的であり、公知の合成よりも少ない工程を含む、アミノセラミド様化合物の鏡像選択的合成方法、該方法により得られるアミノセラミド様化合物が提供される。
【図面の簡単な説明】
【0010】
【図1】図1は、本明細書に開示された方法および中間体を用いた、構造式(I)により表されるセラミド様化合物の合成を示す概略図である。
【図2】図2は、本明細書に開示された方法を用いた、セラミド様化合物(5)の合成を示す概略図である。
【図3】図3は、本明細書に開示された方法を用いた、セラミド様化合物(13)の合成を示す概略図である。
【図4】図4は、化合物(5)〜(8)の構造を示す。
【発明を実施するための形態】
【0011】
発明の概要
本明細書において、アミノセラミド様化合物の効率的かつ高度に鏡像選択的な合成が提供される。このアミノセラミド様化合物の合成は、公知の化合物からわずか5つの工程しか含まない。例えば、図2において「化合物5」として示したセラミド様化合物は、少なくとも99.6%のエナンチオマー過剰で、全収率9%で作製された(実施例1および2参照)。この合成過程中に調製された新規な中間体もまた開示する。
【0012】
本発明は、構造式(I):
【0013】
【化3】
【0014】
により表されるセラミド様化合物の調製方法に関する。
【0015】
R1は、置換または非置換の芳香族基であり; 好ましくは、R1は、置換または非置換のフェニル基, より好ましくは、メタ/パラ位が-OCH2O-、-OCH2CH2O-で置換されているか、またはパラ位がハロ、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノ、低級ジアルキルアミノもしくは-O(低級アルキル)で置換されたフェニルである;
【0016】
R2およびR3は、独立して-H、置換もしくは非置換の脂肪族基または、これらが結合した窒素原子と一緒になって置換もしくは非置換の非芳香族複素環式環である。
【0017】
R7は、置換または非置換の脂肪族基、好ましくは、C1〜C30直鎖非置換脂肪族基、または1つ以上のC1〜C2アルキル基、より好ましくは非置換C1〜C30直鎖アルキルもしくはアルケニル基で置換されたC1〜C30直鎖脂肪族基、さらにより好ましくは非置換C7〜C10もしくはC10〜C16直鎖アルキルもしくはアルケニル基である。
【0018】
構造式(I)により表されるセラミド様化合物の調製方法は、
アミン化合物HNR2R3を、構造式(II):
【0019】
【化4】
【0020】
により表される環式出発物質と反応させる第1工程を含む。
【0021】
アミン化合物HNR2R3と構造式(II)により表される環式出発物質との間の反応は、構造式(III):
【0022】
【化5】
【0023】
により表されるアミド中間体を形成する。構造式(II)および(III)においてR1〜R3は、構造式(I)について記載したとおりであり;R5は、置換または非置換の芳香族基、好ましくは置換または非置換のフェニル基である。
【0024】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(III)により表される中間体のアミノアセタール基を加水分解して、構造式(IV)
【0025】
【化6】
【0026】
により表される非環式化合物を形成する第2工程を含む。構造式(IV)中のR1、R2、R3およびR5は、構造式(I)〜(III)について規定したとおりである。
【0027】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(IV)により表される非環式前駆体化合物をアミド還元剤と反応させて、構造式(V):
【0028】
【化7】
【0029】
により表される化合物を形成する第3工程を含む。構造式(V)中のR1、R2、R3およびR5は、構造式(I)〜(IV)について規定したとおりである。
【0030】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(V)により表されるアミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化して、構造式(VI):
【0031】
【化8】
【0032】
により表されるスフィンゴシン様化合物を形成する第4工程を含む。好ましくは、脱ベンジル化は水素化により達成される。R1、R2およびR3は、構造式(I)〜(V)について記載したとおりである。
【0033】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(VI)により表されるスフィンゴシン様化合物をアシル化して、構造式(I)により表されるセラミド様化合物を形成する第5工程を含む。
【0034】
本発明の他の態様としては、単独で採用、および他の反応と組み合わせた、上述の個々の反応のそれぞれが挙げられる。
【0035】
本発明の他の態様は、本明細書に開示された方法による、構造式(I)により表されるセラミド様化合物の調製における中間体である。一例において、本発明は、構造式(VII):
【0036】
【化9】
【0037】
R1〜R3およびR5は、構造式(I)〜(VI)について上述したとおりであり、
およびR4は、-H2または=Oである
により表される中間体に関する。
【0038】
別の態様において、本発明は、構造式(VIII):
【0039】
【化10】
【0040】
R4は、-H2または=Oであり、
R6は、構造式(IX):
【0041】
【化11】
【0042】
により表される、により表される中間体に関する。構造式(IX)中のフェニル環Aは、置換または非置換である。しかしながら、好ましくは、フェニル環Aは、非置換である。あるいはまた、構造式(VIII)中のR4はH2であり、R6は-Hである。
【0043】
別の態様において、本発明は、構造式(X):
【0044】
【化12】
【0045】
により表される中間体に関する。構造式(X)中のR5は、構造式(I)について規定したとおりである。
【0046】
本発明の方法を利用して、酵素GlcCerシンターゼを阻害するセラミド様化合物を、公知の出発物質から5つの工程で調製することができる。この合成は、高効率であり、通常8%より高い全収率をもたらし、典型的には99%より高いエナンチオマー過剰をもたらす。この合成は、安価な試薬を用い、したがってGlcCerシンターゼの強力なインヒビターへの経済的なルートを提供する。
【0047】
発明の詳細な説明
本明細書において、公知の出発物質からのアミノセラミド様化合物の5工程合成を記載する。この合成は、構造式(II)で表される環式出発物質の調製から始める。該環式出発物質を適当なアミンと反応させ、それによりラクトン環を開裂し、構造式(III)で表されるアミド中間体を形成する。アミド中間体内のアミノアセタールを加水分解し、構造式(IV)で表される非環式化合物を形成する。この非環式化合物のアミドをアミド還元剤で還元し、構造式(V)で表されるアミン化合物を形成し、これを今度は、脱ベンジル化し、構造式(VI)で表されるスフィンゴシン様化合物を形成する。構造式(VI)で表されるスフィンゴシン様化合物の第一級アミンを、次いでアシル化し、アミノセラミド様化合物を形成し得る。この合成を図1に概略的に示す。合成における各反応の詳細を以下に記載する。
【0048】
構造式(II)で表される環式出発物質を、Alkerら, Tetrahedron 54:6089 (1998)ならびにHarwoodおよびRobertson, Chem. Commun. 1998:2641 (1998)に記載の方法にしたがって調製する。具体的には、(5S)-5-フェニルモルホリン-2-オンを少なくとも2当量、好ましくは約2.5〜約5.0当量のアリールアルデヒドR1CHO と脱水条件下で反応させる。R1は、構造式(I)において規定した通りである。「脱水条件」とは、反応混合物から水が除去される条件をいう。水の除去は、例えば、水と反応する(例えば、分子ふるい)が、反応混合物中に存在する他の試薬に対しては実質的に不活性である試薬(「脱水試薬」)の存在下で反応を行なうことにより達成され得、または、水の除去は、トルエンなどの溶剤との共沸によっても達成され得る。充分な脱水試薬を用い、反応中に放出される水2当量(環式出発物質に対して)が除去される。試薬の濃度は、典型的には約0.01M〜5.0Mの間、より典型的には約0.1M〜1.0Mの間であり(if)、好適な反応温度は約50℃〜約150℃の間、好ましくは約100℃〜約120℃の間の範囲である。
【0049】
環式出発物質は、エステルをアミンでアミド化するのに適した条件下で、環式出発物質をアミンNHR2R3と反応させることにより、構造式(II)により表されるアミド中間体に変換される。かかる条件は、当該技術分野において周知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 375-76頁およびそこに引用された文献に記載されている。いずれかの試薬を過剰に使用することができるが、環式出発物質はより一般的には限定試薬である。通常、環式出発物質に対して約15当量まで、典型的には約8当量までのアミンを使用する。反応は純系で(neat)行なわれ得るが、より一般的には、0.01Mという薄いアミン濃度で、非プロトン性非求核性溶媒中にて行なわれる。しかしながら、アミン濃度は、より典型的には約0.4M〜約4.0Mの間である。好適な溶媒としては、クロロホルム、ジクロロメタンおよび1,2-ジクロロエタンなどのハロゲン化溶媒、アセトニトリル、ジメチルホルムアミド(DMF)、ジエチルエーテル、テトラヒドロフラン(THF)および1,4-ジオキサンなどのエーテル系溶媒ならびにベンゼンおよびトルエンなどの芳香族系溶媒が挙げられる。好適な反応温度は通常約0℃〜約100℃の範囲であり、典型的には約25℃〜約35℃の間である。
【0050】
アミノアセタールを加水分解するための条件は当該技術分野において公知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 第 329-32 頁およびそこに引用された文献に記載されている。例えば、構造式(III)により表されるアミド中間体のアミノアセタール基を希無機酸水溶液で加水分解することができる。好適な酸としては、塩酸、硫酸またはリン酸が挙げられるが、塩酸が最も一般的な選択肢である。酢酸およびスルホン酸(例えば、メタスルホン酸、トルエンスルホン酸、トリフルオロ(trifluor)メチルスルホン酸など)などの有機酸も使用することができる。典型的には中間体に対して少なくとも1当量の酸を使用するが、完全な加水分解のためには過剰、例えば、少なくとも10倍過剰、好ましくは約2〜約3倍過剰、より好ましくは約10〜50%の間の酸が好ましい。反応混合物における酸の濃度は、通常、約0.05M〜約1.0Mの間、典型的には約0.1M〜約0.5Mの間である。しばしば水と混和性の有機共存溶媒を用いて中間体を可溶化する。例としては、メタノールまたはエタノールなどのアルコールおよびDMFが挙げられる。水に対する有機溶媒の一般的な溶媒比は、約1:1〜約8:1の範囲である。好適な反応温度は周囲温度から約100℃の範囲、好ましくは約60℃〜約80℃の間である。あるいはまた、March(上記)に記載のようにアミノアセタールをヨウ化トリメチルシリル、湿性シリカゲルまたは湿性アセトニトリル中のLiBF4などのルイス酸を用いて加水分解することができる。
【0051】
「アミド還元剤」は、アミドをアミンに還元し得る試薬である。かかる試薬は当該技術分野において公知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 第 1099-1100 頁、Brown および Krishnamurthy, Aldrichimica Acta 12:3 (1979) ならびにそこに引用された文献に開示されている。例としては、水素化アルミニウムリチウム、水素化ホウ素トリエチルリチウム、ボラン試薬(例えば、ボラン・テトラヒドロフラン、ボラン・メチルスルフィド、ジシアミルボランなど)、水素化アルミニウム、水素化アルミニウムトリメトキシリチウムおよびトリエチルオキソニウムフルオロボレート/水素化ホウ素ナトリウムが挙げられる。本発明の方法において、水素化アルミニウムリチウムが最もよく使用されるアミド還元剤である。アミド出発物質に対して0.5当量ほどの水素化アルミニウムリチウムを使用することができるが、過剰の、しばしば約5当量までを使用するのがより一般的である。好ましくは、アミン出発物質に対して約1.5当量〜約2.5当量の水素化アルミニウムリチウムを使用する。典型的にはエーテル系溶媒が還元に使用される;例としては、ジエチルエーテル、THF、グリム(glyme)、ジグリムおよび1,4−ジオキサンが挙げられる。好適な濃度の還元剤は通常、約0.1M〜約5.0Mの間、より典型的には約0.8M〜約1.5Mの間である。還元は、最も一般的には周囲温度で行なわれるが、約0℃〜約80℃または100℃の間の温度も使用できる。
【0052】
構造式(VI)により表されるスフィンゴシン様化合物を形成するため、構造式(V)により表されるアミン化合物を脱ベンジル化する。本明細書で使用される「脱ベンジル化」という用語は、基-NH-CH2Z(式中、Zはアリール基、好ましくはフェニルである)の炭素−窒素結合の切断をいう。任意に、メチレン基はメチン基と置換され得る。構造式(VI)により表されるスフィンゴシン様化合物に関しては、「脱ベンジル化」は、-NHCH(-CH2OH)R5基の-NH2への変換をいう。脱ベンジル化条件は当該技術分野において周知であり、例えば、GreeneおよびWuts, 「Protective Groups in Organic Synthesis」, John Wiley & Sons (1991), 第384-86頁ならびにそこに引用された文献に開示されている。
【0053】
好ましくは、脱ベンジル化は、水素雰囲気下および水素化触媒の存在下での水素化により行われる。好適な水素圧は通常、ほぼ大気圧から約1000ポンド/平方インチの間である。他の水素源(例えば、ギ酸、ギ酸アンモニウム、シクロヘキサンなど)も使用し得る。好適な水素化触媒としては、カーボン上20%水酸化パラジウム(Parlman触媒)、塩化パラジウム、パラジウム、酸化白金およびカーボン上パラジウムが挙げられる。典型的には、アミン化合物に対して約10%〜約100%重量/重量(w/w)が使用される。ほとんどの場合、ギ酸、酢酸もしくはトリフルオロ酢酸などの有機酸または塩酸もしくは硫酸などの無機酸がアミン化合物に対して、例えば約1〜約5当量、好ましくは約1.6〜約2.4当量で存在する。反応は、最も一般的には、共存溶媒としての水およびメタノールまたはエタノールなどのアルコール系溶媒(例えば、0%〜約50%容積/容積(v/v)の間、約5%〜約15% v/vの間)中で行なわれる。反応温度は、約0℃〜約50℃の間が適当であり、好ましくは約25℃〜約40℃の間である。
【0054】
水素化以外の多くの脱ベンジル化条件は当該技術分野において公知であり、本発明に包含される。例としては、金属ナトリウムおよびNH3(例えば、du VigneaudおよびBehrens, J. Biol. Chem. 117:27 (1937)を参照のこと)、CCl3CH2OCOCl、CH3CN(例えば、Rawalら, J. Org. Chem., 52:19 (1987)を参照のこと)、Me3SiCH2CH2OCOCl、THF、-50℃、次いで一晩25℃(例えば、Campbellら, Tetrahedron Lett., 28:2331 (1987)を参照のこと)、α-クロロエチルクロロホルメートおよび水酸化ナトリウム(例えば、Olofsonら, J. Org. Chem. 49:2081 (1984)ならびにDeShongおよびKell, Tetrahedron Lett., 27:3979 (1986)を参照のこと)、ビニルクロロホルメート(例えば、Olofsonら, Tetrahedron Lett., 1977:1567 (1977)ならびにCooleyおよびEvain, Synthesis, 1989:1 (1989)を参照のこと)、RuO4、NH3、H2O(例えば、GaoおよびJones, J. Am. Chem. Soc., 109:1275 (1987)を参照のこと)ならびにm-クロロ酸化過安息香酸後、FeCl2,−10℃(例えば、Monkovic ら, Synthesis, 1985:770 (1985)を参照のこと)が挙げられる。
【0055】
構造式(VI)により表されるスフィンゴシン様化合物は、遊離アミンをアシル化することによりセラミド様化合物に変換される。アミン基のアシル化は当該技術分野において周知であり、例えば、アミンをアシル化剤R7C(O)-Xと反応させることにより行なわれ得る。R7は、構造式(I)について上記した通りであり、Xは、第一級アミンにより容易に置換される脱離基である。この反応の条件は、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985およびそこに引用された文献に記載されている。好適なアシル化剤の例としては、酸ハロゲン化物、無水物またはエステルが挙げられる。好ましくは、アミンは酸ハロゲン化物によりアシル化される。通常、当モル量のスフィンゴシン様化合物および酸塩化物を、酸塩化物に対して少過剰のトリエチルアミン、ジイソプロピルエチルアミン、ジメチルアミノピリジンまたはピリジンなどの第三級アミンの存在下で使用する。しかしながら、スフィンゴシン様化合物が限られる場合は過剰(典型的には約10〜50%)の酸塩化物を使用することができ、逆の場合も同様である。反応混合物中における試薬の濃度は、通常、約0.005M〜約5.0Mの間で変化し、好ましくは約0.05M〜約0.5Mの間である。過剰のアミン塩基は、約100%超であり得るが、典型的には約5%〜約25%の間である。ハロゲン化溶媒などの非プロトン性溶媒が好ましい(例えば、クロロホルム、塩化メチレンおよび1,2−ジクロロメタン)が、エーテル系溶媒および炭化水素溶媒などの他の非プロトン性溶媒は好適な代用物であり得る。反応には、通常、周囲温度が好ましいが、約0℃〜約50℃の温度も使用され得る。
【0056】
あるいはまた、アシル化剤は、活性化エステルR7C(O)-OX’(式中、-OX’は容易に第一級アミンで置換される)である。活性化エステルでのアミンのアシル化方法は、当該技術分野において公知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 371-375 頁およびそこに引用された文献に記載されている。多くの活性化エステルは、単離するのに充分安定である。N−ヒドロキシスクシンイミジルエステル(いくつかはAldrich Chemical Co., Milwaukee, WIから市販されている)は、このタイプの活性化エステルの一例である。先の段落で記載した酸塩化物アシル化剤でアミドを形成するのに好適な条件が、安定な活性化エステルと共に典型的に使用され得る。第三級アミンでの活性化を必要とする酸塩化物とは対照的に、活性化エステルは第一級アミンの存在下で直接アミドを形成するように充分反応性である。したがって、活性化エステルを使用する場合は、第三級アミンはアシル化反応から省略され得る。
【0057】
あるいはまた、活性化エステルをインサイチューで形成させる。インサイチューでの活性化エステルの形成は、カルボン酸のヒドロキシル基を、求核性置換を受けやすい基で置換する試薬である「カップリング剤」を必要とする。カップリング剤の例としては、1,1'−カルボニルジイミダゾール(CDI)、イソブチルクロロホルメート、ジメチルアミノプロピルエチル−カルボジイミド(EDC)、ジシクロヘキシルカルボジイミド(DCC, が挙げられる。活性化エステルのインサイチュー生成によりアミド化する場合、カルボン酸またはアミンのいずれかを過剰に使用し得る(典型的には50%過剰、より典型的には約10〜15%過剰)。しかしながら、本発明を実施する際には、限定試薬としてアミン化合物を使用することがより一般的である。通常、カルボン酸1モルあたり約1.0モル〜約10モルのカップリング剤、好ましくはカルボン酸1モルあたり約1.0モル〜約1.5モルのカップリング剤を使用する。反応は、通常、非プロトン性溶媒、例えば、塩化メチレン、ジクロロエタンおよびクロロホルムなどのハロゲン化溶媒、エーテル系溶媒テトラヒドロフラン、1,4−ジオキサンおよびジエチルエーテルならびにジメチルホルムアミド中で行なわれる。好適な反応温度は、通常、約0℃〜約100℃の範囲であるが、反応は好ましくは周囲温度で行なわれる。
【0058】
本明細書に記載の反応を行なうための具体的な条件の例を実施例1および2に提供する。
【0059】
構造式(II)により表される化合物のエナンチオマーを環式出発物質として用いることにより、本明細書に記載の方法を用い、構造式(III)〜(VI)および(I)により表される化合物のエナンチオマーを調製し得る。構造式(III)により表される環式出発物質のエナンチオマーは、上記のような脱水条件下で、(5R)-5-フェニルモルホリン-2-オンを2当量のアルデヒドR1CHOと反応させることにより調製され得る。構造式(III)、(VII)、(VIII)および(X)により表される化合物のエナンチオマーならびに本明細書に開示される手順を用いた構造式(II)〜(VI)および(I)により表される化合物のエナンチオマーの調製方法は本発明に包含される。
【0060】
本明細書で使用される用語「エナンチオマー」およびエナンチオマーを示す構造式は、その光学異性体を含まない「純粋な」エナンチオマーならびにエナンチオマーとその光学異性体との混合物(エナンチオマーが、エナンチオマー過剰、例えば、少なくとも10%、25%、50%、75%、90%、95%、98%または99%エナンチオマー過剰で存在する)
を包含することが意図される。
【0061】
構造式(I)〜(IX)における変数R1〜R5に関して、「脂肪族基」は、非芳香族系であり、炭素と水素のみから構成され、任意に1つ以上の不飽和単位、例えば、二重結合および/または三重結合を含み得る。脂肪族基は、直鎖、分枝鎖または環式であり得る。直鎖または分枝鎖である場合、脂肪族基は、典型的には約1〜約30個の炭素原子、より典型的には約1〜約24個の炭素原子を含む。環式である場合、脂肪族基は、典型的には約3〜約10個の炭素原子、より典型的には約3〜約7個の炭素原子を含む。脂肪族基は、C1〜C30直鎖または分枝鎖の飽和炭化水素、好ましくは、C1〜C24直鎖または分枝鎖の飽和炭化水素を含む低級アルキル基である。例としては、メチル、エチル、n-プロピル、iso-プロピル、n-ブチル、sec-ブチルおよびtert-ブチルが挙げられる。
【0062】
芳香族基としては、フェニル、1-ナフチル、2-ナフチル、1-アントラシルおよび2-アントラシルなどの炭素環式芳香族基、ならびにN-イミダゾリル、2-イミダゾール、2-チエニル、3-チエニル、2-フラニル、3-フラニル、2-ピリジル、3-ピリジル、4-ピリジル、2-ピリミジル、4-ピリミジル、2-ピラニル、3-ピラニル、2-ピラゾリル、3-ピラゾリル、4-ピラゾリル、5-ピラゾリル、2-ピラジニル、2-チアゾール、4-チアゾール、5-チアゾール、2-オキサゾリル、4-オキサゾリルおよび5-オキサゾリルなどの複素環式芳香族基が挙げられる。
【0063】
芳香族基はまた、炭素環式芳香族環またはヘテロアリール環が1つ以上の他のヘテロアリール環に融合された融合多環式芳香族環系も含む。例としては、2-ベンゾチエニル、3-ベンゾチエニル、2-ベンゾフラニル、3-ベンゾフラニル、2-インドリル、3-インドリル、2-キノリニル、3-キノリニル、2-ベンゾチアゾール、2-ベンゾオキサゾール、2-ベンズイミダゾール、2-キノリニル、3-キノリニル、1-イソキノリニル、3-キノリニル、1-イソインドリルおよび3-イソインドリルが挙げられる。
【0064】
非芳香族系複素環式環は、環内に1つ以上窒素、酸素または硫黄などのヘテロ原子を含む非芳香族系炭素環式環である。環は、五、六、七または八員環であり得る。例としては、モルホリニル、チオモルホリニル、ピロリジニル、ピペラジニル、ピペリジニル、アゼチジニル、アザシクロヘプチルまたはN-フェニルピペラジニルが挙げられる。
【0065】
低級アルキル、脂肪族、芳香族、非芳香族、複素環式またはベンジル基上の好適な置換基は、本明細書に記載の反応を実質的に妨げないものである。「反応を妨げる」とは、実質的に収率を低下させる(例えば、50%を超える低下)またはかなりの量の副生成物形成を引き起こすこと(例えば、副生成物が理論収量の少なくとも50%を占める)ことをいう。干渉置換基が最初に保護形態に変換されるならば、干渉置換基を使用し得る。好適な保護基は当該技術分野において公知であり、例えば、GreeneおよびWuts, 「Protective Groups in Organic Synthesis」, John Wiley & Sons (1991) に開示されている。
【0066】
アルキル、脂肪族、芳香族、非芳香族複素環式環またはベンジル基上の好適な置換基としては、例えば、ハロゲン(-Br、-Cl、-Iおよび-F)、-OR、-CN、-NO2、-NR2、-COOR、-CONR2、-SOkR (kは0、1または2)ならびに-NH-C(=NH)-NH2が挙げられる。各Rは独立して−H、脂肪族基、置換脂肪族基、ベンジル基、置換ベンジル基、芳香族基または置換芳香族基、好ましくは-H、低級アルキル基、ベンジル基またはフェニル基である。また、置換非芳香族複素環式環、ベンジル基または芳香族基は、置換基として脂肪族基または置換脂肪族基を有し得る。また、置換アルキルまたは脂肪族基も、置換基として非芳香族複素環式環、ベンジル、置換ベンジル、芳香族または置換芳香族基を有し得る。置換アルキル、置換脂肪族、置換非芳香族複素環式、置換芳香族または置換ベンジル基は1つを超える置換基を有し得る。
【0067】
R1が置換フェニル基である場合、好ましい置換基の例としては、-OCH2O-、-OCH2CH2O-、ハロ、(低級アルキル)O-、低級アルキルチオール、低級ジアルキルアミン、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミンおよび低級アルキルアミノが挙げられる。
【0068】
R5が置換フェニル基である場合、好ましい置換基の例としては、ハロ、(低級アルキル)O-、-O(フェニル)および低級アルキルが挙げられる。
【0069】
本明細書に記載する構造式において、化学基または部分が連結された分子または化合物の残部は、以下の記号:
【0070】
【化13】
【0071】
で表される。例えば、構造式(IX)において対応する変数は、図に示した基(構造式(VIII)においてR6により表される)がベンジル炭素を介して単共有結合により構造式(VIII)におけるアミンに連結されていることを示す。
【0072】
本発明の好ましい態様において、本明細書において使用される記号は、以下のように定義される:R1は、置換または非置換のフェニル基であり;R2およびR3は、独立して、−H、非置換C1〜C5アルキル基、またはこれらが結合している窒素原子と一緒になって非置換C3〜C10非芳香族複素環式環であり;R5は、置換または非置換のフェニル基、好ましくはフェニルであり;R7は、C1〜C30直鎖非置換脂肪族基または1つ以上のC1〜C2アルキル基で置換されたC1〜C30直鎖脂肪族基、より好ましくは、非置換C1〜C30直鎖アルキルまたはアルケニル基である。
【0073】
別の好ましい態様において、-NR2R3は、一緒になってピロリジニルである。より好ましくは、-NR2R3は、一緒になってピロリジニルであり、R5は、R2、R3およびR5をむ化合物において、R1はフェニルである。さらにより好ましくは、R1は、R1、R2、R3およびR5を含む化合物において、置換または非置換フェニル基(好ましくは、メタ/パラ位が-OCH2O-、-OCH2CH2O-で置換されているか、あるいはパラ位がハロ、低級アルキルチオール、-OH、-O(フェニル)、-O-CH2(フェニル)、低級アルキル、アミノ、低級アルキルアミン、低級ジアルキルアミノもしくは-O(低級アルキル)で置換されているフェニルであり、-NR2R3は一緒になってピロリンジニル(pyrrolindinyl)であり、R5はフェニルである。
【0074】
別の好ましい態様において、-NR2R3は、一緒になってピペリジルである。より好ましくは、-NR2R3は、一緒になってピペリジルであり、R5は、R2、R3およびR5を含む化合物において、フェニルである。さらにより好ましくは、R1は、R1、R2、R3およびR5を含む化合物において、R1は置換または非置換フェニル基(好ましくは、メタ/パラ位が-OCH2O-、-OCH2CH2O-で置換されているか、あるいはパラ位がハロ、低級アルキルチオール、-OH、-O(フェニル)、-O-CH2(フェニル)、-O-CH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノ、低級ジアルキルアミノもしくは-O(低級アルキル)で置換されているフェニルであり、-NR2R3は一緒になってピペリジルであり、R5はフェニルである。
【0075】
本発明の方法により調製され得るセラミド様化合物の例は、構造式(XI):
【0076】
【化14】
【0077】
により表される
【0078】
R1は、メタ/パラ位が-OCH2O-もしくは-OCH2CH2O-で置換、またはパラ位がハロ、CH3O-、CH3CH2O-、CH3CH2CH2O-、CH3(CH3)CHO-、CH3-、CH3CH2-、CH3CH2CH2-、CH3(CH3)CH-、-OHもしくは-OCH2(フェニル)で置換されたフェニルであり;R7は、CH3(CH2)n-またはCH3(CH2)n-2CH=CH-(式中、nは、0から約30の整数である) である。好ましくは、nは7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23または24である。より好ましくは、R1は、メタ/パラが-OCH2CH2O-で置換されたフェニルである。
【0079】
充分に酸性な、充分に塩基性な、または両方の官能基を有する本発明の化合物は、したがって、いくつかの無機塩基ならびに無機酸および有機酸と反応して、塩を形成し得る。したがって、本発明はまた、構造式(VII)、(VIII)および(X)により表される中間体の塩も包含する。生理学的に許容され得る塩が好ましい。酸付加塩を形成するために一般的に使用される酸は、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、リン酸などの無機酸、およびp-トルエンスルホン酸、メタンスルホン酸、シュウ酸、p-ブロモフェニル−スルホン酸、カルボン酸、コハク酸、クエン酸、安息香酸、酢酸などの有機酸である。かかる塩の例としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプロン酸塩、ヘプタン酸塩、プロピオール酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオエート、ヘキシン-1,6-ジオエート、安息香酸塩、クロロベンゾエート、メチルベンゾエート、ジニトロベンゾエート、ヒドロキシベンゾエート、メトキシベンゾエート、フタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、γ−ヒドロキシ酪酸塩、グリコール酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン-1-スルホン酸塩、ナフタレン-2-スルホン酸塩、マンデル酸などが挙げられる。
【0080】
塩基付加塩としては、アンモニウムまたはアルカリもしくはアルカリ土類金属水酸化物、炭酸塩、重炭酸塩などの無機塩基由来のものが挙げられる。したがって、本発明の塩の調製に有用なかかる塩基としては、水酸化ナトリウム、水酸化カリウム、水酸化アンモニウム、炭酸カリウムなどが挙げられる。
【0081】
本出願において引用した刊行物の全教示は参照により本明細書に取り込まれる。
【実施例】
【0082】
実施例
実施例1−セラミド様化合物の小規模調製
中間体1
(1R,3S,5S,8aS)-1,3-ビス-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
【0083】
【化15】
【0084】
(5S)-5-フェニルホルホリン-2-オン(2.00 g, 11.3 mmol)(Dellaria, J.F.: Santarsiero, B.D. J. Org. Chem., 1989, 54, 3916 に記載のようにして調製)および1,4-ベンゾジオキサン-6-カルボキサルデヒド(5.56 g, 33.9 mmol)を含むトルエン(125 mL)の攪拌溶液に、4Å分子ふるい(約20 mL)を添加した。混合物を72時間加熱還流し、濾過して分子ふるいを除き、濃縮した。得られたコハク色ゴムをシリカでフラッシュクロマトフ(ジエチルエーテル/ヘキサン)し、淡黄色固体を得た。この物質を、ジエチルエーテルで摩砕してさらに精製し、1.89 g(34%)の生成物を綿毛状白色固体として得た: 1H NMR (CDCl3) δ 7.31-7.17 (m, 5H), 6.95-6.79 (m, 5H), 5.32-5.27 (m, 2H), 4.43-4.28 (m, 2H), 4.24 (s, 4H), 4.18 (m, 4H), 4.16-4.08 (m, 2H) ppm.
【0085】
中間体2
(2S,3R,1''S)-3-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
【0086】
【化16】
【0087】
中間体1(1.80g, 3.69mmol)を含有するクロロホルム(20mL)の撹拌溶液に、ピロリジン(2.0mL, 24mmol)を添加した。溶液を一晩撹拌し、次に濃縮した。得られた無色の粘着性泡沫物をメタノール(16mL)および1N塩酸(4mL)中に取り出した。混合物を1時間還流し、追加の1N塩酸(2mL)と処理し、さらに2時間還流した。反応溶液を濃縮し、得られた残渣を酢酸エチルと炭酸水素ナトリウム水溶液との間で分配した。有機層を乾燥し(硫酸ナトリウム)、濃縮した。得られた淡黄色の粘性ゴムをシリカゲルに通すフラッシュクロマトグラフィー(塩化メチレン/2Nメタノール性アンモニア(methanolic ammonia))により精製し、無色の泡沫状固体として1.40g(92%)の中間体2を得た:1H NMR (CDCl3) δ 7.31-7.13 (m, 5H), 6.93-6.70 (m, 3H), 4.47 (d, J = 8.5, 1H), 4.18 (s, 4H), 3.82 (t, J = 5.9, 1H), 3.74 (d, J = 6.0, 2H), 3.06 (d, J = 8.5, 1H), 3.06-2.97 (m, 1H), 2.92-2.83 (m, 1H), 1.97-1.87 (m, 1H), 1.45-1.15 (m, 4H) ppm.
【0088】
中間体3
(1R,2R,1''S)-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
【0089】
【化17】
【0090】
中間体2(1.38g, 3.35mmol)を含有するテトラヒドロフラン(30mL)の撹拌溶液に、水素化アルミニウムリチウム(0.26g, 6.9mmol)を添加した。泡沫状懸濁液を一晩撹拌し、次に1N水酸化ナトリウム水溶液(13mL)の添加(泡立つのがやむまで滴下)によりクエンチした。混合物を水で希釈して、酢酸エチルで抽出した。有機層を乾燥し(硫酸ナトリウム)、濃縮して無色の粘性ゴムを得た。シリカゲルに通すフラッシュクロマトグラフィー(塩化メチレン/2Nメタノール性アンモニア)により、0.94g(70%)の無色の粘着性泡沫物として生成物を得た: 1H NMR (CDCl3) δ 7.36-7.17 (m, 5H), 6.88-6.74 (m, 3H), 4.42 (d, J = 5.4, 1H), 4.26 (s, 4H), 3.79-3.69 (m, 1H), 3.64-3.56 (m, 1H), 3.55-3.45 (m, 1H), 3.00-2.90 (m, 1H), 2.67-2.57 (m, 1H), 2.43-2.32 (m, 4H), 2.25-2.15 (m, 1H), 1.75-1.65 (m, 4H) ppm.
【0091】
中間体4
(1R,2R)-2-アミノ-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ピロリジン-1-イル-プロパン-1-オール
【0092】
【化18】
【0093】
メカニカルスターラーを備え付けた高圧反応ボンベ中に、中間体3(0.91g, 2.28mmol)を含有する10:1メタノール/水(22mL)溶液、トリフルオロ酢酸(0.18mL, 2.3mmol)および炭素上20%水酸化パラジウム(Perlman触媒;0.91g)を負荷した。反応器を排気し、3倍のアルゴンで埋め戻し、次いで排気し、水素で再び満たした(100 psi)。反応物を2日間撹拌し、次に排気し、窒素でフラッシュした。反応溶液をセライトを通して濾過し、濃縮した。得られた灰緑色のゴムをシリカゲルに通すフラッシュクロマトグラフィーにかけ(塩化メチレン/2Nメタノール性アンモニア)、ほとんど無色のゴムとして0.165g(26%)の生成物を得た: 1HNMR (CDCl3) δ 6.89-6.76 (m, 3H), 4.54 (d, J = 3.7, 1H), 4.25 (s, 4H), 3.43 (s, 1H), 3.14-3.07 (m, 1H), 2.68-2.41 (m, 6H), 1.82-1.71 (m, 4H) ppm.
【0094】
化合物5
(1R,2R)-ヘキサデカン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0095】
【化19】
【0096】
中間体4(0.165g, 0.593mmol)を含有する塩化メチレン(8mL)の撹拌溶液に、塩化パルミトイル(0.18g, 0.59mmol)、次いでN,N-ジイソプロピルエチルアミン(0.11mL, 0.65mmol)を添加した。溶液を2時間攪拌し、次いで濃縮した。残渣を酢酸エチルと炭酸水素ナトリウム水溶液との間で分配した。有機層を乾燥し(硫酸ナトリウム)、濃縮した。得られたオフホワイトの固体を、シリカゲルに通すフラッシュクロマトグラフィーにかけ(塩化メチレン/2Nメタノール性アンモニア)、白色固体として0.174g(57%)の精製物を得た。1H NMR分光学および分析用キラルHPLC(カラム:Chirex(S)-VALおよび(R)-NE, 4.6×250mm;溶離剤:0.5%トリフルオロ酢酸含有67:31:2ヘキサン/塩化メチレン/エタノール;流量:1mL/分;検出280nM)による比較は、この物質が、Poltらの方法(J. Org. Chem., 1998, 63, 8837)により調製されたものと同一の化合物のサンプルと一致したことを説明する。エナンチオマー過剰は99.6%であると決定した。2つの起こりうるジアステレオマーに由来する全汚染は、0.2%であると決定する。1H NMR (CDCl3) δ 6.88-6.73 (m, 3H), 5.84 (d, J = 7.3, 1H), 4.90 (d, J = 3.8, 1H), 4.24 (s, 4H), 4.22-4.15 (m, 1H), 2.86-2.72 (m, 2H), 2.72-2.55 (m, 4H), 2.10 (t, J = 7.5, 2H), 1.82-1.74 (m, 4H), 1.58-1.46 (m, 2H), 1.32-1.16 (m, 24H), 0.88 (t, J = 6.7, 3H) ppm.
【0097】
実施例2−セラミド様化合物の大規模調製
(5S)-5-フェニルモルホリン-2-オン
【0098】
【化20】
【0099】
S-(+)-フェニルグリシノール(Aldrich, 10.17g, 78.12mmol)およびジイソプロピルエチルアミン(Aldrich, 34mL, 195mmol, 2.5当量)の溶液をCH3CN(200mL)中に調製した。この溶液を、CH3CN(50mL)に溶解したフェニル-α-ブロモ酢酸(18.48g, 85.9mnol, 1.1当量)に窒素下で2時間かけて滴下した。得られた溶液を、窒素下で16〜20時間攪拌した。溶媒を、25℃未満の浴温を保ちながら回転蒸発(rotoevaporation)により除去した。油に酢酸エチル(120mL)を添加し、混合物を15分間攪拌した。得られた白色沈殿を濾過して取り出し、固体を酢酸エチル(25mL)で洗浄した。濾液を、25℃以下の浴温を保ちながら油まで回転蒸発した。減圧下で0.5時間乾燥した後、油をCH2Cl2(17mL)に溶解し、シリカゲルカラム(60g充填し、10%酢酸エチル/ヘキサンを用いる)上に負荷した。上の方の副生成物のスポットを10%酢酸エチル/ヘキサンで溶出し、生成物を50%酢酸エチル/ヘキサン〜100%酢酸エチルで溶出した。生成物を含む画分を、25℃以下の浴温を保ちながら油まで回転蒸発した。この油を酢酸エチル(12mL)に溶解し、氷浴中でヘキサン(60mL)をゆっくり添加して、生成物を沈殿させた。得られた沈殿を濾過した。白〜黄色固体を減圧乾燥した。得られた(5S)-5-フェニルモルホリン-2-オン(7.4g, 41.8mmol, 53%)を、次の工程に直接使用した。
【0100】
中間体1
(1R,3S,5S,8aS)-1,3-ビス-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
【0101】
【化21】
【0102】
(5S)-5-フェニルモルホリン-2-オン(7.4g, 41.8mmol)およびベンゾジオキソラン-6-カルボキシアルデヒド(AldrichまたはAlfa Aesar, 20.56g, 125.2mmol, 3.0当量)をトルエン(180mL)に溶解した。溶液を、4Åモレキュラーシーブ(約30g)を充填したソックスレー抽出器に配置した。溶液を窒素下で2〜3日間還流した。室温に冷却した後、溶媒を回転蒸発により除去し、油を酢酸エチル(200mL)に溶解した。重亜硫酸ナトリウム(Aldrich, 50g)の水溶液(100mL)を添加し、2相の混合物を1時間室温で攪拌した。得られた白色固体を濾過して取り出し、酢酸エチルで洗浄した。濾液を分液漏斗に配置し、層を分離した。有機層を水(100mL)および飽和塩化ナトリウム溶液(100mL)で洗浄した。乾燥(Na2SO4)溶液を濾過および回転蒸発し、黄赤色泡沫状油(23.11g)を得た。減圧下で1時間乾燥した後、ジエチルエーテル(350ml)を添加し、混合物を室温で16〜20時間攪拌した。得られた白〜黄色固体を濾過した。固体を減圧下で乾燥した。環状付加物(cycloadduct)を46%収率(9.34g)で得た。
【0103】
中間体2
(2S,3R,1''S)-3-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
【0104】
【化22】
【0105】
塩化メチレン(40mL)に溶解した環状付加物(中間体1, 6.7g, 13.74mmol)に、ピロリジン(Aldrich, 5.7mL, 68.7mmol, 5当量)を添加した。溶液を室温にて16〜18時間窒素下で撹拌した。溶媒を回転蒸発し、黄色泡沫状油を得、0.5時間減圧乾燥した。粗製物をメタノール(115mL)に溶解し、1M HCl水溶液(115mL)を添加した。溶液を4時間還流した。室温に冷却した後、メタノールを回転蒸発により除去した。酢酸エチル(60mL)を添加し、2相系を室温で5〜15分間撹拌した。2層を分離し、有機層を1M HCL(30mL)で抽出した。あわせた水層を2回酢酸エチル(60, 30mL)で洗浄した。飽和重炭酸ナトリウム溶液(150mL)を水層にゆっくり添加した。生成物を塩基性(pH=8〜9)水層から酢酸エチル(60mL)で3回抽出した。生成物を含むあわせた有機層を飽和塩化ナトリウム溶液(30mL)で洗浄した。Na2SO4で乾燥した後、溶液を濾過し、回転蒸発して、黄色固体を得た。中間体2が93%収率(5.26g)で得られた。
【0106】
中間体3
(1R,2R,1''S)-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
【0107】
【化23】
【0108】
滴下漏斗および冷却器を備え付けた三つ口フラスコに、LiAlH4(Aldrich, 1.2g, 31.7mmol, 2.5当量)および無水THF(20mL)を窒素下で添加した。中間体2(5.23g, 12.68mmol)を含有する無水THF(75mL)の溶液を、反応に15〜30分かけて滴下した。反応を窒素下で9時間還流した。反応を氷浴中で冷却し、1M NaOH溶液を注意深く滴下した。室温で15分間撹拌した後、水(50mL)および酢酸エチル(75mL)を添加した。層を分離し、水層を2回酢酸エチル(75mL)で抽出した。あわせた有機層を飽和塩化ナトリウム溶液(25mL)で洗浄した。Na2SO4で乾燥した後、溶液を濾過し、回転蒸発して無色〜黄色の泡沫状油を得た。中間体3を99%収率(5.3g)で得た。
【0109】
中間体4
(1R,2R)-2-アミノ-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ピロリジン-1-イル-プロパン-1-オール
【0110】
【化24】
【0111】
中間体3(5.3g, 13.3mmol)をメタノール(60mL)に溶解した。水(6mL)およびトリフルオロ酢酸(2.05mL, 26.6mmol, 2当量)を添加した。窒素下に配置した後、炭素上20%水酸化パラジウム(Pearlman触媒, LancasterまたはAldrich, 5.3g) を添加した。混合物をガラスインサートを有するParr Pressure Reactor Apparatusに配置した。装置を窒素下、次に水素圧110〜120psi下に配置した。混合物を、室温にて水素圧100〜120psi下で2〜3日間撹拌した。反応を 窒素下に配置し、セライトのパッドを介して濾過した。セライトパッドをメタノール(100mL)および水(100mL)で洗浄した。メタノールを回転蒸発により除去した。水層を酢酸エチルで3回(100, 50, 50mL) 洗浄した。10M NaOH溶液(10mL)を水層(pH=12〜14)に添加した。生成物を、水層から塩化メチレンで3回(100, 100, 50mL)抽出した。あわせた有機層をNa2SO4で乾燥し、濾過し、無色の油に回転蒸発した。泡沫状油を2時間減圧乾燥した。中間体4を90%収率(3.34g)で得た。
【0112】
化合物5
(1R,2R)-ヘキサデカン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]アミド
【0113】
【化25】
【0114】
中間体4(3.34g, 12.0mmol)を含有する塩化メチレン(50mL)の溶液に、パルミチン酸N-ヒドロキシスクシンイミドエステル(Sigma, 4.24g, 12.0mmol)の溶液を窒素下で室温にて15〜30分かけて添加した。溶液を室温にて18〜20時間撹拌した。反応に塩化メチレン(50mL)および1M NaOH溶液(25mL)を添加した。2相系を室温で15〜30分間撹拌した。水(25mL)を添加し、層を分離した。水層を塩化メチレン(25mL)で逆抽出した。あわせた有機層を水(25mL)で2回、飽和塩化ナトリウム溶液(25mL)で1回洗浄した。有機層をNa2SO4で乾燥し、濾過し、淡黄色油に回転蒸発した。粗製物をヘキサン(50mL)から再結晶させた。得られた白色固体(5.46g)を、シリカゲル(300g)上で2%メタノール:塩化メチレン〜4%メタノール:塩化メチレン〜4% 2Mアンモニウム含有メタノール:塩化メチレンを用いて分離した。得られた白色固体をヘキサン(70mL)から再結晶させた。化合物5を66%収率(4.18g)で得た。分析用キラルHPLC(カラム:Chirex (S)-VALおよび(R)-NE, 4.6×250mm;溶離剤:0.5%トリフルオロ酢酸含有67:31:2ヘキサン/塩化メチレン/エタノール;流量:1mL/分;検出:280nM)は、この物質が純度98.98%であり、0.89%のジアステレオマーおよび0.14%のエナンチオマーを有することを示した。
【0115】
実施例3−セラミド様化合物の代替的な大規模調製
(5S)-5-フェニルモルホリン-2-オンHCl塩
フェニルブロモアセテート(Aldrich, 862.17g, 4.0モル, 1.1当量)を含有するアセトニトリル(試薬等級, 1500ml)の溶液を、氷浴(内部温度5℃以下)中で冷却した。これにS-(+)-2-フェニルグリシノール(Aldrich, 500g, 3.65モル, 1当量)およびジイソプロピルエチルアミン(DIPEA)(Aldrich, 1587ml, 9.11モル, 2.5当量)を含有するアセトニトリル(2900ml)の冷スラリー(内部温度5℃以下)を、内部温度を10℃以下に保ちながら、分割して添加した。氷浴を取り除く前に混合物をこの温度で30分間撹拌し、混合物を室温でさらに4時間撹拌した。浴温を25℃で保ちながら、溶媒を減圧除去した。混合物を、酢酸エチル(2×500ml)と同時エバポレートし、淡黄色粘性油を生成した。反応混合物に、酢酸エチル(4500ml)を添加し、フラスコを撹拌しながら氷浴に浸した。混合物を8℃以下に冷却した。固体を濾過し、酢酸エチル(3×250ml)で洗浄した。溶液を5℃以下に冷却した。pHが2以下(湿潤pH紙)になるまで、内部温度を15℃以下に維持しながら乾燥HClガスを溶液にゆっくり通過させた。さらに20分間この温度およびpHで混合物を撹拌した後、固体を吸引濾過した。固体を酢酸エチル(3×200ml)で洗浄し、高減圧下で約20時間乾燥した。収率は、412g(53%)であった。1H NMRは、(5S)-5-フェニルモルホリン-2-オンHCl塩と一致した。
【0116】
中間体1
(1R,3S,5S,8aS)-1,3-ビス-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
(5S)-5-フェニルモルホリン-2-オンHCl塩(381g, 1当量)を含有する15%酢酸エチル含有トルエン(2270ml)の撹拌懸濁液に、重炭酸ナトリウム(1.1当量)を含有する水溶液(2000ml)を添加した。得られた二相溶液を室温で約1時間撹拌した。有機層を、1,4-ベンゾジオキサン-6-カルボキシアルデヒドを含むフラスコに移した。次に、フラスコにDean-Starkユニット、冷却器および窒素入口を備え付けた。約650mlの溶媒(酢酸エチルおよびトルエンの混合物)をDean-Starkユニットを介して回収しながら、混合物を撹拌しつつ加熱して還流した。得られた黄赤色溶液を、反応の間に形成された水をDean-Starkユニット中に回収しながら、約64時間窒素下で還流に供した。次に溶媒のほとんどをDean-Starkユニットを介する大気圧での蒸留を介して除去した。次に残りの溶媒を、ヘプタン(500ml)およびtert-ブチルメチルエーテル(2×725ml)の同時エバポレーションにより除去し、黄色半固体生成物を生成した。半固体生成物を酢酸エチル(3400ml)に溶解した。重亜硫酸ナトリウム(920g)の水溶液(1500ml)を添加し、混合物を室温で約1時間撹拌した。形成された固体を濾過により除去し、酢酸エチル(3×400ml)で洗浄した。濾液を、水(1450ml)、5%鹹水溶液(1450ml)で洗浄し、MgSO4(100g)で乾燥した。溶媒を減圧下で除去し、黄色固体を得た。これにtert-ブチルメチルエーテル(2900ml)を添加し、懸濁液を室温で20〜22時間撹拌した。黄色固体を吸引濾過し、tert-ブチルメチルエーテル(2×600ml)で洗浄して、約22時間室温にて高減圧下で乾燥した。収率は400.5g(58%)であった。1H NMRおよびTLCは、中間体1と一致した。
【0117】
中間体2
(2S,3R,1''S)-3-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
中間体1(312g, 0.64モル)、ピロリジン(267ml, 3.2モル, 5当量)およびテトラヒドロフラン(1350ml)の溶液を4.5時間窒素雰囲気下で加熱して還流した。 溶媒および過剰のピロリジンを減圧下で除去し、粗製中間体を橙色粘性油として生成した。油をメタノール(3000ml)および1M塩酸溶液(3000ml)に溶解した。得られた溶液を約7時間加熱して還流した。次に、溶媒を減圧下で除去し、油および水の混合物を得た。これに酢酸エチル(2000ml)を添加し、水層を分離した。有機層を1M HCl水溶液(1000ml)で抽出した。水層をあわせ、酢酸エチル(2000ml)で洗浄した。水層を氷浴中で冷却した。水層のpHを、10M NaOH水溶液(525ml)で約9(pH紙)に調整した。水層を酢酸エチル(3000ml)で抽出した。有機層を5%鹹水溶液(1000ml)で洗浄し、乾燥した(Na2SO4)。溶媒を減圧下で除去し、黄色粘性油を生成した。収率は、213.4g, 81%であった。1H NMRは中間体2に一致した。
【0118】
中間体3
(1R,2R,1''S)-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
LiAlH4(50.7g, 1.34モル, 2.6当量)を含有するテトラヒドロフラン(700ml)のスラリーに、中間体2(213.34g, 0.517モル)を含有するテトラヒドロフラン(2000ml)の溶液を室温でゆっくり撹拌しながら添加した。混合物を約4時間還流した。TLC解析(10%メタノール含有塩化メチレン, v/v)は、出発物質の消費を示した。反応混合物を氷浴(5℃未満)中で冷却し、内部温度を10℃以下に保ちながら水(135ml)を非常にゆっくり添加した。次に、これに15%NaOH水溶液(70ml)、続いて水(200ml)を添加した。撹拌を続けながら、反応混合物を室温に加温した。次に、塩化メチレン(1000ml)を混合物に添加し、塩をセライトのパッドを介して濾過した。塩を塩化メチレン(2×500ml)で洗浄した。濾液をあわせ、溶媒 を減圧下で除去して黄色油を生成した。油を1M HCl水溶液(1500ml)に溶解し、酢酸エチル(3×500ml)で洗浄した。水層を氷浴中で5℃以下に冷却し、内部温度を10℃以下に保ちながら水層のpHを10M NaOH水溶液(220ml)で12〜13に調整した。混合物を室温に加温した。水層を塩化メチレン(2×500ml)で抽出した。有機層をあわせ、鹹水溶液(500ml)で洗浄し、乾燥し(Na2SO4)、溶媒を減圧下で除去して黄色粘性油を得た。収率は、186.4g(88.5%)であった。1H NMR は中間体3に一致した。
【0119】
中間体4 ジオキサレート塩
(1R,2R)-2-アミノ-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ピロリジン-1-イル-プロパン-1-オールジオキサレート塩
中間体3(358g, 0.90モル)、エタノール(1500ml)、1M HCl溶液(1500ml)および10%Pd(OH)2(32g, 20重量%)の懸濁液を、室温にて約50psiで約36時間水素添加した。混合物をCuonoフィルターを介して濾過した。Cuonoフィルターを10%エタノール含有水(500ml)で洗浄した。濾液をあわせ、エタノールを減圧下で除去した。水層を酢酸エチル(3×600ml)で抽出した。有機層を1M HCl水溶液(700ml)で抽出した。水層をあわせ、氷浴(0〜約5℃)中で冷却した。内部温度10℃以下を保ちながら、水層のpHを10M NaOH水溶液(490ml)で約12(pH紙)に調整した。水層を室温に加温した。水層を塩化メチレン(2×1500ml, 1×750ml)で抽出した。あわせた有機層をMgSO4で乾燥し、溶媒を減圧下で除去し、黄色粘性油を得た。粗製物の重量は、214.3g(86%)であった。1H NMRは中間体4と一致した。
【0120】
シュウ酸(152.4g, 1.693モル, 2.2当量)を含有するメチルイソブチルケトン(2300ml)の溶液を、中間体4(214.3g, 0.77モル, 1当量)を含有するメチルイソブチルケトン(800ml)の溶液に室温で撹拌しながらゆっくり添加した。得られた混合物を室温で約2.5時間撹拌した。固体を濾過し、アセトン(2000ml)で室温にて約16時間粉砕した。固体を濾過し、アセトン(3×100ml)で洗浄し、高減圧下で乾燥し、オフホワイトの固体を生成した。収量は312.5g(89%)であった。1H NMRは、中間体4ジオキサレート塩と一致した。
【0121】
化合物5
(1R,2R)-ヘキサデカン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
中間体4ジオキサレート塩(507g, 1.11モル)を含有する水(10L)の冷却溶液(約5℃)に、内部温度を10℃以下に保ちながら10M NaOH水溶液(500ml)を撹拌しつつ添加した。溶液のpHを約14(pH紙)に維持しながら、溶液を室温に加温した。水層を塩化メチレン(3×6000ml)で抽出した。有機層をあわせ、水(2000ml)で洗浄し、乾燥し(MgSO4)、溶媒を減圧下で除去し、黄色粘性油である中間体4を得た。収量は302g(98%)であった。1H NMRは中間体4に一致した。
【0122】
パルミチン酸NHS-エステル(Sigma , 382.5g, 1.01当量)を含有する塩化メチレン(2500ml)の溶液を、中間体4(302g)を含有する塩化メチレン(1500ml)の溶液に、室温にて1.25時間かけて窒素雰囲気下で添加した。混合物を室温で約18時間撹拌した。1M NaOH水溶液(2425ml)を添加し、混合物を室温で約3時間撹拌した。有機層を分離し、水層を塩化メチレン(800ml)で抽出した。有機層をあわせ、1M NaOH溶液(3×1500ml)および水(1500ml)で洗浄した。有機層をMgSO4で乾燥し、溶媒を減圧下で除去し、半固体を得た。半固体を、ヘプタン(3×100ml)と同時エバポレートした。粗生成物を、12L容の三つ口RBフラスコに移し、ヘプタン(7500ml)を添加した。混合物を、窒素雰囲気下で撹拌しながら還流しつつ加熱した。溶液を約55℃(内部温度)にゆっくり冷却し、別のフラスコに注いだ。溶液を室温で24時間窒素雰囲気下で撹拌した。オフホワイトの固体を濾過し、ヘプタン(2×500ml)で洗浄し、高減圧下で24時間乾燥した。固体(397g)を12L容のRBフラスコに移し、30%酢酸エチル含有ヘプタン(8000ml)を添加した。得られた混合物を撹拌しながら30分間還流しつつ加熱した。溶液を約55℃(内部温度)に冷却し、別のフラスコに注いだ。室温にて窒素雰囲気下で約24時間撹拌を続けた。固体を濾過し、ヘプタン(2×100ml)で洗浄し、高減圧下で乾燥し、オフホワイトの固体を得た。収率は、324g(58%)であった。1H NMRおよびTLCは化合物5と一致した。mp 96.1℃ HPLC解析:キラル純度99.7%, 化学純度99.7%.C31H52N2O4についての解析計算値: C, 72.05; H, 10.14; N, 5.42.実測値C, 72.03; H, 10.19; N, 5.42.
【0123】
実施例4−化合物6〜8の調製
Lapidot, Y. Rappoport, S.およびWolman, Y. Journal of Lipid Research 8, 1967の方法によるかまたは以下に記載されるようにN-ヒドロキシスクシンイミド脂肪酸エステルを調製した:
【0124】
オクタン酸N-ヒドロキシスクシンイミドエステル
N-ヒドロキシスクシンイミド(Aldrich, 20.0g, 173 mmol)およびトリエチルアミン(29mL, 208 mmol)を、氷浴中窒素下で塩化メチレンに溶解した。塩化オクタノイル(Aldrich, 35mL, 205 mmol)を0.5時間かけて滴下した。氷浴を取り除き、白色固体を有する溶液を室温で1時間撹拌した。濾過により白色固体を取り除き、濾液を水(100mL)および飽和重炭酸ナトリウム水溶液(100mL)で洗浄した。有機層を硫酸ナトリウムで乾燥し、濾過し、ヘプタン(100mL)を添加した。溶液を回転蒸発して塩化メチレンのほとんどを除去し、ヘプタン中に無色〜白色の薄片状沈殿が残った。沈殿を濾過し、ヘプタンで洗浄した。乾燥後、オクタン酸N-ヒドロキシスクシンイミドエステルを84%収率(35.4g)で得た:1H NMR (CDCl3) 2.84 (br s, 4H), 2.60 (t, J=7.48 Hz, 2H), 1.78-1.71 (m, 2H), 1.42-1.26 (m, 8H), 0.88 (t, J = 6.7 Hz, 3H) ppm.
【0125】
化合物6
(1R,2R)-オクタン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0126】
【化26】
【0127】
無水塩化メチレン(300mL)に溶解した中間体5(22.36g, 80.33mmol)に、無水塩化メチレン(150mL)に溶解したオクタン酸N-ヒドロキシスクシンイミドエステル(19.4g, 80.39 mmol)の溶液を15〜30分間かけて窒素下で室温にて添加した。溶液を室温で18〜20時間撹拌した。反応に、1M NaOH水溶液(200mL)を添加した。2相系を室温で45分間撹拌した。層を分離し、あわせた有機層を1M NaOHで2回(2×200mL)および水で2回(2×100ml)洗浄した。有機層を硫酸ナトリウムで乾燥し、濾過し、黄色油まで回転蒸発した。粗製物質のほとんどが、還流で5%酢酸エチル含有ヘプタン(1L)に溶解した。40℃に冷却後、濁った溶液を新しいフラスコにデカントすることにより、黄色油から分離した。最初のフラスコを5%酢酸エチル含有ヘプタンで2回(2×250ml)同じプロセス(還流および40℃に冷却および油から溶液のデカント)により濯いだ。あわせた溶液を加熱して還流し、4時間かけて室温まで冷却した。得られた白色固体を濾過し、5%酢酸エチル含有ヘプタン(100mL)およびヘプタン(100mL)で洗浄した。白色固体(13.9g)を減圧下で16〜24時間乾燥した。この固体は、還流で5%酢酸エチル含有ヘプタン(800mL)にほとんど溶解した。50℃に冷却した後、濁った溶液を新しいフラスコにデカントすることにより、黄色油から分離した。最初のフラスコを5%酢酸エチル含有ヘプタン(100mL)で同じプロセス(還流および50℃に冷却および油から溶液のデカント)により濯いだ。あわせた溶液を加熱して還流し、4時間かけて室温に冷却した。得られた白色固体を濾過し、5%酢酸エチル/ヘプタン(50mL)およびヘプタン(50mL)で洗浄した。室温にて減圧下2〜3日間乾燥した後、化合物6を39%収率(12.57g)で得た。分析用キラルHPLC(カラム:Chirex (S)-VALおよび(R)-NE, 4.6×250mm)は、この物質が99.9%所望のR,R異性体であることを示した。分析用HPLCは、この物質が99.6%純粋であることを示した。 mp 87-88℃. 1H NMR (CDCl3) δ 6.86-6.73 (m, 3H), 5.84 (d, J = 7.3 Hz, 1H), 4.91 (d, J = 3.4Hz, 1H), 4.25 (s, 4H), 4.24-4.18 (m, 1H), 2.85-2.75 (m, 2H), 2.69-2.62 (m, 4H), 2.10 (t, J = 7.3 Hz, 2H), 1.55-1.45 (m,2H), 1.70-1.85 (m, 4H), 1.30-1.15 (m, 8H), 0.87 (t, J = 6.9 Hz, 3H) ppm.
【0128】
化合物7
(1R,2R)-ノナン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0129】
【化27】
【0130】
この化合物を、ノナン酸 N-ヒドロキシスクシンイミドエステルを使用し、化合物6について記載した方法により調製した。分析用HPLCは、この物質が98.4 %純粋であることを示した。 mp 74-75℃. 1H NMR (CDCl3) δ6.86-6.76 (m, 3H), 5.83 (d, J = 7.3 Hz, 1H), 4.90 (d, J = 3.3 Hz, 1H), 4.24 (s, 4H), 4.24-4.18 (m, 1H), 2.85-2.75 (m, 2H), 2.69-2.62 (m, 4H), 2.10 (t, J = 7.3 Hz, 2H), 1.55-1.45 (m, 2H), 1.70-1.85 (m, 4H), 1.30-1.15 (m, 10H), 0.87 (t, J = 6.9 Hz, 3H) ppm.
【0131】
化合物8
(1R,2R)デカノン[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0132】
【化28】
【0133】
この化合物を、デカン酸N-ヒドロキシスクシンイミドエステルを使用し、化合物6について記載した方法により調製した。分析用HPLCは、この物質が99.3%純粋であることを示した。 mp 97.5〜98.5℃. 1H NMR (CDCl3) δ 6.86-6.76 (m, 3H), 5.83 (d, J = 7.5 Hz, 1H), 4.90 (d, J = 3.4 Hz, 1H), 4.24 (s, 4H), 4.24-4.18 (m, 1H), 2.85-2.75 (m, 2H), 2.69-2.62 (m, 4H), 2.10 (t, J = 7.5 Hz, 2H), 1.55-1.45 (m, 2H), 1.70-1.85 (m, 4H), 1.30-1.15 (m, 12H), 0.87 (t, J = 6.8 Hz, 3H) ppm.
【0134】
実施例5−化合物13の調製
中間体9
(1R,3S,5S,8aS)-1,3-ビス-(4-ベンジルオキシ-フェニル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
【0135】
【化29】
【0136】
(5S)-5-フェニルモルホリン-2-オンHCl塩(57.45, 268.9mmol)を、二相溶液が澄むまで酢酸エチル(500mL)および飽和重炭酸ナトリウム水溶液(250mL)と30分間撹拌した。相を分離し、水層を酢酸エチル(2×250mL)で抽出した。あわせた有機相を飽和塩化ナトリウム溶液(250mL)で洗浄した。有機層を硫酸ナトリウムで乾燥し、濾過し、油まで濃縮し、60分間減圧下で乾燥した。5-(S)-フェニルモルホリン-2-オンを86%収率(40.98g, 231.3 mmol)で得た。
【0137】
5-(S)-フェニルモルホリン-2-オン(40.98g, 231.3 mmol)および4-ベンジルオキシベンズアルデヒド(Aldrich, 147.3g, 694 mmol, 3.0当量)をトルエン(750mL)に溶解した。反応は、Dean Stark Trapおよび還流冷却器を備え付けた。溶液を窒素下で2日間還流した。室温に冷却した後、溶媒を回転蒸発により除去し、油を酢酸エチル(500mL)に溶解した。水(250mL)に溶解した重亜硫酸ナトリウム(Aldrich, 125g)の溶液を添加し、2相混合物を室温で3時間撹拌した。得られた白色固体を濾過して取り出し、酢酸エチルで洗浄した。濾液を分液漏斗に配置し、層を分離した。有機層を水(250mL)、飽和塩化ナトリウム水溶液(250mL)で洗浄し、次いで乾燥し(硫酸ナトリウム)、濾過し、泡沫状油(144g)まで回転蒸発した。減圧下で1時間乾燥した後、tert-ブチルメチルエーテル(1450mL)を添加し、混合物を室温で5時間撹拌した。得られた白〜黄色固体を濾過した。固体を減圧下で乾燥した。中間体9を 27%収率(41.64g, 71.46 mmol)で得た。 1H NMR (CDCl3) δ 7.5-6.8 (m, 23H), 5.0および5.1 (2s, 4H), 4.5-4.3 (m, 2H), 4.2-4.1 (m, 2H) ppm.
【0138】
中間体10
(2S,3R,1''S)-3-(4-ベンジルオキシ-フェニル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
【0139】
【化30】
【0140】
テトラヒドロフラン(250mL)に溶解した中間体9(45.1g, 77.4mmol)に、ピロリジン(Aldrich 33mL, 395mmol, 5.1当量)を添加した。窒素下で室温にて16〜18時間、溶液を撹拌し、キャップした。溶媒を回転蒸発して黄色泡沫状油を得、0.5時間減圧乾燥した。粗製物をメタノール(220mL)に溶解し、1M HCl水溶液(220mL)を添加した。溶液を4時間還流した。室温まで冷却した後、メタノールを回転蒸発により除去した。得られた油に、ゆっくり10M NaOH水溶液(22mLでpHを14に調整)を添加した。生成物を、塩基性水層から塩化メチレンで3回(300,100,100mL)抽出した。硫酸ナトリウムで乾燥した後、あわせた有機層を濾過し、回転蒸発し、黄〜橙色の泡沫状固体を得た。tert-ブチルメチルエーテル(300mL)を添加し、混合物を室温で7時間撹拌した。得られた白〜黄色固体を濾過し、tert-ブチルメチルエーテル(50mL)で洗浄し、減圧乾燥した。中間体10を83%収率(29.77g)で得た。 1H NMR (CDCl3) δ 7.4-7.2 (m, 12H), 6.9-6.8 (m, 2H), 5.05 (AB四重項, 2H), 4.47 (d, J = 8.5, 1H), 3.9-3.3 (m, 3H), 3.05 (d, J = 8.5, 1H), 3.0-2.8 (m, 2H), 2.3-2.2 (m, 1H), 1.85-1.7 (m, 1H), 1.45-1.15 (m, 4H) ppm.
【0141】
中間体11
(1R,2R,1''S)-1-(4-ベンジルオキシ-フェニル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
【0142】
【化31】
【0143】
滴下漏斗および冷却器を備える三ツ口フラスコにおいて、窒素下でLiAlH4(Aldrich、6.3g、166mmol、2.57当量)および無水テトラヒドロフラン(75mL)を添加した。中間体10(29.7g、64.48mmol)を含有する無水テトラヒドロフラン(300mL)の溶液を15〜30分にわたって反応液に滴下した。反応液を窒素下で9時間還流した。反応液を氷浴で冷却し、水(7.0mL)を一滴ずつ非常に注意深く添加した(水素を放出する活発な発熱反応)。15%NaOH水溶液(7.0mL)、次いで水(21mL)を滴下した。最終の水添加の途中で大量の白色固体が形成された。これを塩化メチレン(250mL)の添加により破壊した。室温にて15分間の撹拌後、混合物をセライトプラグ(直径17cm×高さ1cm)を介して濾過した。沈殿を塩化メチレンで洗浄した(2×250mL)。濾液を油まで回転蒸発(rotoevaporated)した。油を1M HCl水溶液(300mL)に溶解した。水層をtert-ブチルメチルエーテルで洗浄した(2×200mL)。氷浴で冷却後、10M NaOH水溶液(35mL)を水層に注意深く添加した(最終pH=14)。生成物を塩化メチレンで3回抽出した(300mL、200mLおよび100mL)。硫酸ナトリウムで乾燥後、溶液を濾過し、回転蒸発して白色固体を得た。乾燥後、中間体11を94%収率で得た(26.9g)。1H NMR(CDCl3) δ 7.46-7.115(m, 12H), 6.98-6.96(m, 2H), 5.08(s, 2H), 4.49(d, J = 4.7, 1H), 3.70-3.65(m, 1H), 3.60-3.55(m, 1H), 3.54-3.45(m, 1H), 3.00-2.90(m, 1H), 2.7-2.6(m, 1H), 2.36(br s, 4H), 2.15-2.05(m, 1H), 1.70(br s, 4H) ppm.
【0144】
中間体12
(1R,2R)-2-アミノ-1-(4-ベンジルオキシ-フェニル)-3-ピロリジン-1-イル-プロパン-1-オール塩化水素塩
【0145】
【化32】
【0146】
中間体11(26.9g、60.24mmol)をメタノール(400mL)に溶解し、1M HCl水溶液(130mL)を添加した。窒素下に置いた後、20%水酸化パラジウム炭素(Pearlman触媒、Aldrich、10.8g)を添加した。反応液をバルーンへの排気および充填により窒素下、次いで水素下に置いた。混合物を水素バルーン下で室温にて48時間撹拌した。反応液を窒素下に置き、セライトパッドを介して濾過した。セライトパッドを10%の水含有メタノール(250mL)および水(50mL)で洗浄した。溶媒を回転蒸発およびトルエンとの同時蒸発(3×100mL)により除去した。泡沫固体を還流でイソプロパノール(300mL)に溶解した。溶液を室温に冷却し、tert-ブチルメチルエーテル(550mL)を添加した。室温にて2時間撹拌後、白色固体を濾過して、tert-ブチルメチルエーテルで洗浄した。乾燥後、中間体12を約99%収率で得た(18g)。 1H NMR(DMSO-d6) δ 9.68(br s, 1H), 8.53(br s, 2H) 7.24(d, J = 8.55 Hz, 2H), 6.80(d , J = 8.55 Hz, 2H), 4.72(d, J = 7.0 Hz, 1H), 3.8-3.6(m, 2H), 3.4-3.6(m, 3H), 3.0-3.2(m,2H), 2.7-2.5(br s, 1H), 2.0-1.7(br s , 4H) ppm.
【0147】
化合物13
(1R, 2R)-ヘキサデカン酸[2-(4-ベンジルオキシ-フェニル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エーテル]-アミド
【0148】
【化33】
【0149】
テトラヒドロフラン(500mL)に懸濁した中間体12(16.17g 49.36mmol)にトリエチルアミン(28mL、4当量)を添加した。テトラヒドロフラン(125mL)に溶解したパルミチン酸N-ヒドロキシスクシンイミドエステル(Sigma、19.2g、54.29mmol)の溶液を室温にて窒素下で30分にわたって添加した。溶液を室温にて18〜20時間撹拌した。白色沈殿を濾過により除去し、濾液を泡沫状のオフホワイト固体まで回転蒸発した(35.5g)。粗物質を塩化メチレン(500mL)に溶解し、水(100mL)および飽和炭酸ナトリウム水溶液(100mL)で洗浄した。硫酸ナトリウムで乾燥後、溶液を濾過、回転蒸発し、オフホワイトの泡沫状固体(24.75g)を得た。この物質を40%酢酸エチル含有ヘプタン(500mL、高温濾過)から再結晶させた。化合物13を61%収率で得た(14.45g)。分析キラルHPLCによりこの物質は99.7%純度、所望のR,Rイソマーであることを示した。分析HPLCにより、この物質は99.6%純度、mp95〜97℃であることを示した。 1H NMR(CDCl3) δ 7.15(d, J= 8.5Hz, 2H), 6.70(d, J= 8.5 Hz , 2H), 6.0(d, J = 7.3, 1H), 4.96(d, J = 3.8, 1H), 4.3-4.2(m, 1H), 2.9-2.7 (m, 2H), 2.65-2.55(m, 4H), 2.10(t, J = 7.5, 2H), 1.75(br s, 4H), 1.58-1.46(m, 2H), 1.32-1.16(m, 24H), 0.9(t, J = 6.7, 3H) ppm.
【0150】
本発明は、その好ましい態様を参照して詳細に示され、記載されているが、形式および詳細における種々の変更は、添付の特許請求の範囲により包含される本発明の範囲から逸脱せずにその中でなされ得ることが当業者により理解される。
【0151】
本発明の態様として、以下のものが挙げられる。
[1]以下の構造式:
【化34】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R4は=OまたはH2であり;
R5は置換または非置換芳香族基である)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[2]R1およびR5が独立して置換または非置換フェニル基であり;
R2およびR3が独立して-H、非置換C1〜C5アルキル基またはそれらが結合される窒素原子と共に非置換C3〜C10非芳香族複素環式環である、[1]記載の化合物。
[3]R5がフェニル基である[2]記載の化合物。
[4]R2およびR3が、それらが結合される窒素原子と共にピロリジニル、ピペラジニル、アゼチジニル、モルホリニル、チオモルホリニル、アザシクロヘプチル、ピペリジニルまたはN-フェニルピペラジニルである[3]記載の化合物。
[5]以下の構造式:
【化35】
(式中、R1は置換または非置換フェニル基であり、R4が-H2または=Oである)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[6]R1で表されるフェニル基が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、-OH、低級アルキルチオール、-O(フェニル)、-OCH2(フェニル)、-O-CH2-(フェニル)、低級アルキル、アミノ、低級アルキルアミノまたは低級ジアルキルアミノで置換される[5]記載の化合物。
[7]以下の構造式:
【化36】
(式中、R4は-H2または=Oであり;かつ
R6は以下の構造式:
【化37】
(式中、フェニル環Aは置換または非置換である)
で表される;または
R4はH2であり、かつR6は-Hである)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[8]フェニル環Aが非置換である[7]記載の化合物。
[9]以下の構造式:
【化38】
(式中、R5は置換または非置換芳香族基である)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[10]R5がフェニルである[9]記載の化合物。
[11]以下の構造式:
【化39】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R5は置換または非置換芳香族基である)
で表される非環式化合物を調製する方法であって、アミン化合物HNR2R3と以下の構造式:
【化40】
で表される環式開始物質とを反応させ、それにより以下の構造式:
【化41】
で表される中間体を形成する工程、
および該中間体のアミノアセタール基を加水分解し、それにより該非環式化合物を形成する工程を含む、方法。
[12]R1およびR5が独立して置換または非置換フェニル基であり;
R2およびR3が独立して-H、非置換C1〜C5アルキル基またはそれらが結合される窒素原子と共に非置換C3〜C10非芳香族複素環式環である、[11]記載の方法。
[13]R5がフェニル基である[12]記載の方法。
[14]R2およびR3が、それらが結合される窒素原子と共にピロリジニル、ピペラジニル、アゼチジニル、モルホリニル、チオモルホリニル、アザシクロヘプチル、ピペリジニルまたはN-フェニルピペラジニルである[13]記載の方法。
[15]R1が置換または非置換フェニル基であり、R2およびR3が、それらが結合される窒素原子と共にピロリジニル基である[14]記載の方法。
[16]R1で表されるフェニル基が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノまたは低級ジアルキルアミノで置換される[15]記載の方法。
[17]アミド還元剤と非環式化合物とを反応させ、それにより以下の構造式:
【化42】
で表されるアミン化合物を形成する工程をさらに含む、[11]記載の方法。
[18]アミド還元剤が水素化アルミニウムリチウムである[17]記載の方法。
[19]アミド還元剤がボラン・テトラヒドロフランである[17]記載の方法。
[20]アミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化し、それにより以下の構造式:
【化43】
で表されるセラミド化合物を形成する工程をさらに含む、[17]記載の方法。
[21]アミン化合物が水素添加により脱ベンジル化される[18]記載の方法。
[22]アミン化合物が水素添加触媒の触媒作用量を用いて水素雰囲気下で水素添加される[20]記載の方法。
[23]アミン化合物の第一級アミン基をアシル化し、それにより以下の構造式:
【化44】
で表されるアシル化セラミド化合物を形成する工程をさらに含む、[19]記載の方法。
[24]セラミド化合物がR7COX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジルであり、RはC1〜C30直鎖アルキルまたはアルケニル基である)でアシル化される[22]記載の方法。
[25]非環式化合物が、以下の構造式:
【化45】
で表される環式ラクトンと少なくとも2当量のアルデヒド化合物R1CHOとを反応させることにより調製される[11]記載の方法。
[26]以下の構造式:
【化46】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R5は置換または非置換芳香族もしくは脂肪族基である)
で表されるアミン化合物を調製する方法であって、アミド還元剤と以下の構造式:
【化47】
で表される非環式前駆体化合物とを反応させる工程を含む、方法。
[27]R1およびR5が独立して置換または非置換フェニル基であり;
R2およびR3が独立して-H、非置換C1〜C5アルキル基またはそれらが結合される窒素原子と共に非置換C3〜C10非芳香族複素環式環である、[25]記載の方法。
[28]R5がフェニル基である[26]記載の方法。
[29]R2およびR3が、それらが結合される窒素原子と共にピロリジニル、ピペラジニル、アゼチジニル、モルホリニル、チオモルホリニル、アザシクロヘプチル、ピペリジニルまたはN-フェニルピペラジニルである[27]記載の方法。
[30]R1が置換または非置換フェニル基であり、R2およびR3が、それらが結合される窒素原子と共にピロリジニル基である[28]記載の方法。
[31]R1で表されるフェニル基が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノまたは低級ジアルキルアミノで置換される[29]記載の方法。
[32]アミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化し、以下の構造式:
【化48】
で表されるスフィンゴシン様化合物を形成する工程をさらに含む、[25]記載の方法。
[33]アミン化合物が水素添加により脱ベンジル化される[31]記載の方法。
[34]アミン化合物が触媒作用量のPd(OH)2を用いて水素雰囲気下で水素添加される[32]記載の方法。
[35]スフィンゴシン様化合物の第一級アミン基をアシル化し、それにより以下の構造式:
【化49】
で表されるセラミド様化合物を形成する工程をさらに含む、[32]記載の方法。
[36]セラミド化合物がR7COX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジル基であり、R7はC1〜C30直鎖アルキルまたはアルケニル基である)でアシル化される[34]記載の方法。
[37]以下の構造式:
【化50】
で表されるスフィンゴシン様化合物を調製する方法であって、以下の構造式:
【化51】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R5は置換または非置換芳香族基である)
で表されるアミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化する工程を含む、方法。
[38]アミン化合物が水素添加により脱ベンジル化される[36]記載の方法。
[39]アミン化合物が触媒作用量のPd(OH)2を用いて水素雰囲気下で水素添加される[37]記載の方法。
[40]スフィンゴシン様化合物の第一級アミン基をアシル化し、それにより以下の構造式:
【化52】
で表されるセラミド様化合物を形成する工程をさらに含む、[37]記載の方法。
[41]セラミド化合物がR7COX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジル基であり、R7はC1〜C30直鎖アルキルまたはアルケニル基である)でアシル化される[39]記載の方法。
[42]以下の構造式:
【化53】
(式中、R1は置換または非置換フェニル基であり:
R7はC7〜C10またはC10〜C16アルキル基である)
で表されるセラミド様化合物を調製する方法であって、
a)環式ラクトンと少なくとも2当量のアルデヒドR1CHOとを反応させ、以下の構造式:
【化54】
で表される第1の中間体を形成する工程であって、該環式ラクトンが以下の構造式:
【化55】
で表される、工程;
b)ピロリジンと該第1の中間体とを反応させ、以下の構造式:
【化56】
で表される第2の中間体を形成する工程;
c)該第2の中間体のアミノアセタール基を加水分解し、以下の構造式:
【化57】
で表される第3の中間体を形成する工程;
d)水素化アルミニウムリチウムまたはボラン・テトラヒドロフランで該第3の中間体を還元し、以下の構造式:
【化58】
で表される第4の中間体を形成する工程;
e)触媒作用量のPd(OH)2を用いて水素雰囲気下で該第4の中間体を水素添加し、以下の構造式:
【化59】
で表されるスフィンゴシン様化合物を形成する工程;ならびに
f)RCOX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジルである)で該スフィンゴシン様化合物をアシル化し、セラミド様化合物を形成する工程
を含む、方法。
[43]R7がC10〜C16アルキル基である[42]記載の方法。
[46]R1が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノおよび低級ジアルキルアミノで置換されたフェニル基である[43]記載の方法。
[45]R1が
【化60】
であり、R7が-(CH2)14CH3である[43]記載の方法。
[46]R1がパラ-ヒドロキシフェニルまたは
【化61】
であり、R7がC7〜C10アルキル基である[42a]記載の方法。
【技術分野】
【0001】
本発明は、UDP-グルコース:N-アシルスフィンゴシングルコシルトランスフェラーゼインヒビター、およびその合成方法に関する。
【背景技術】
【0002】
発明の背景
グリコスフィンゴリピド(GSL)は、細胞増殖、細胞分化、細胞間接着または細胞とマトリックスタンパク質との間の接着、微生物およびウイルスの細胞への結合ならびに腫瘍細胞の転移を促進する能力を含む、多くの生物学的機能を有する天然化合物の一群である。GSLは、セラミドおよびUDP-グルコースから酵素UDP-グルコース:N-アシルスフィンゴシングルコシルトランスフェラーゼ(GlcCerシンターゼ)により生成されるグルコシルセラミド(GlcCer)に由来する。セラミドの構造を以下に示す。
【0003】
【化1】
【0004】
GSLの蓄積は、テイ−サックス病、ゴーシェ病、およびファブリー病を含む、いくつかの疾患と関連している(例えば、特許文献1参照)。GSLはまた、特定の癌と関連している。例えば、特定のGSLが腫瘍でのみ発生したり、腫瘍において異常に高い濃度で発生すること;培養培地内の腫瘍細胞に添加すると、腫瘍増殖に対して顕著な刺激性または阻害性作用を及ぼすこと;および腫瘍のそばを通り周囲の細胞外液内に散らばると身体の正常な免疫防御システムを阻害することがわかっている。腫瘍が次第に悪性になるにつれて腫瘍のGSLの組成が変化し、特定のGSLに対する抗体が腫瘍の成長を阻害する。
【0005】
GlcCerシンターゼを阻害する化合物は、GSL濃度を低下させ得、前述の疾患の1つを有する被験体を処置するのに有用であることが報告されている。いくつかのGlcCerの強力なインヒビター(本明細書では「アミノセラミド様化合物」という)が特許文献1、特許文献2、特許文献3、特許文献4および特許文献5に開示されている。用語「セラミド様化合物」は、1)第一級アルコールが置換アミノ基で置換され、2)アルケニル基がアリール基、好ましくはフェニルまたは置換フェニルで置換された、セラミドのアナログをいう。対応するN-脱アシル化化合物を「スフィンゴシン様化合物」とよぶ。
【先行技術文献】
【特許文献】
【0006】
【特許文献1】米国特許第6,051,598号明細書
【特許文献2】米国特許第5,952,370号明細書
【特許文献3】米国特許第5,945,442号明細書
【特許文献4】米国特許第5,916,911号明細書
【特許文献5】米国特許第6,030,995号明細書
【発明の概要】
【発明が解決しようとする課題】
【0007】
残念ながら、アミノセラミド様化合物を調製する公知の方法は、工業的規模での製造にあまり適していない。2つのキラル中心により、公知の合成のほとんどは、4つのジアステレオマーを生じ、これは、クロマトグラフィーによりジアステレオマーを分離し、光学活性剤、例えばジベンゾイル酒石酸異性体で誘導体化した後、結晶化によって所望のエナンチオマーを単離する必要性を生じる(例えば、Inokuchi および Radin, Journal of Lipid Research 28:565 (1987)参照)。いずれのプロセスも大規模調製には不向きである。偏左右選択的還元を用いるアミノセラミド様化合物の鏡像選択的合成が報告されている(Mitchell ら, J. Org. Chem. 63:8837 (1998)およびNishidaら, SYNLETT 1998:389 (1998))が、10を超える工程を必要とし、そのうちいくつかの工程では、水素化ジイソブチルアルミニウム(DIABAL)およびガーナーアルデヒド(tert-ブチル(R)-(+)-4ホルミル-2,2-ジメチル-3-オキサゾリジンカルボキシレート)などの高価な試薬を使用する。したがって、より経済的かつ効率的であり、公知の合成よりも少ない工程を含む、アミノセラミド様化合物の鏡像選択的合成の大いなる必要性が存在する。
【課題を解決するための手段】
【0008】
即ち、本発明の要旨は、
〔1〕以下の構造式:
【化2】
で表される化合物またはその生理学的に許容され得る塩
に関する。
【発明の効果】
【0009】
本発明により、より経済的かつ効率的であり、公知の合成よりも少ない工程を含む、アミノセラミド様化合物の鏡像選択的合成方法、該方法により得られるアミノセラミド様化合物が提供される。
【図面の簡単な説明】
【0010】
【図1】図1は、本明細書に開示された方法および中間体を用いた、構造式(I)により表されるセラミド様化合物の合成を示す概略図である。
【図2】図2は、本明細書に開示された方法を用いた、セラミド様化合物(5)の合成を示す概略図である。
【図3】図3は、本明細書に開示された方法を用いた、セラミド様化合物(13)の合成を示す概略図である。
【図4】図4は、化合物(5)〜(8)の構造を示す。
【発明を実施するための形態】
【0011】
発明の概要
本明細書において、アミノセラミド様化合物の効率的かつ高度に鏡像選択的な合成が提供される。このアミノセラミド様化合物の合成は、公知の化合物からわずか5つの工程しか含まない。例えば、図2において「化合物5」として示したセラミド様化合物は、少なくとも99.6%のエナンチオマー過剰で、全収率9%で作製された(実施例1および2参照)。この合成過程中に調製された新規な中間体もまた開示する。
【0012】
本発明は、構造式(I):
【0013】
【化3】
【0014】
により表されるセラミド様化合物の調製方法に関する。
【0015】
R1は、置換または非置換の芳香族基であり; 好ましくは、R1は、置換または非置換のフェニル基, より好ましくは、メタ/パラ位が-OCH2O-、-OCH2CH2O-で置換されているか、またはパラ位がハロ、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノ、低級ジアルキルアミノもしくは-O(低級アルキル)で置換されたフェニルである;
【0016】
R2およびR3は、独立して-H、置換もしくは非置換の脂肪族基または、これらが結合した窒素原子と一緒になって置換もしくは非置換の非芳香族複素環式環である。
【0017】
R7は、置換または非置換の脂肪族基、好ましくは、C1〜C30直鎖非置換脂肪族基、または1つ以上のC1〜C2アルキル基、より好ましくは非置換C1〜C30直鎖アルキルもしくはアルケニル基で置換されたC1〜C30直鎖脂肪族基、さらにより好ましくは非置換C7〜C10もしくはC10〜C16直鎖アルキルもしくはアルケニル基である。
【0018】
構造式(I)により表されるセラミド様化合物の調製方法は、
アミン化合物HNR2R3を、構造式(II):
【0019】
【化4】
【0020】
により表される環式出発物質と反応させる第1工程を含む。
【0021】
アミン化合物HNR2R3と構造式(II)により表される環式出発物質との間の反応は、構造式(III):
【0022】
【化5】
【0023】
により表されるアミド中間体を形成する。構造式(II)および(III)においてR1〜R3は、構造式(I)について記載したとおりであり;R5は、置換または非置換の芳香族基、好ましくは置換または非置換のフェニル基である。
【0024】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(III)により表される中間体のアミノアセタール基を加水分解して、構造式(IV)
【0025】
【化6】
【0026】
により表される非環式化合物を形成する第2工程を含む。構造式(IV)中のR1、R2、R3およびR5は、構造式(I)〜(III)について規定したとおりである。
【0027】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(IV)により表される非環式前駆体化合物をアミド還元剤と反応させて、構造式(V):
【0028】
【化7】
【0029】
により表される化合物を形成する第3工程を含む。構造式(V)中のR1、R2、R3およびR5は、構造式(I)〜(IV)について規定したとおりである。
【0030】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(V)により表されるアミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化して、構造式(VI):
【0031】
【化8】
【0032】
により表されるスフィンゴシン様化合物を形成する第4工程を含む。好ましくは、脱ベンジル化は水素化により達成される。R1、R2およびR3は、構造式(I)〜(V)について記載したとおりである。
【0033】
構造式(I)により表されるセラミド様化合物の調製方法は、構造式(VI)により表されるスフィンゴシン様化合物をアシル化して、構造式(I)により表されるセラミド様化合物を形成する第5工程を含む。
【0034】
本発明の他の態様としては、単独で採用、および他の反応と組み合わせた、上述の個々の反応のそれぞれが挙げられる。
【0035】
本発明の他の態様は、本明細書に開示された方法による、構造式(I)により表されるセラミド様化合物の調製における中間体である。一例において、本発明は、構造式(VII):
【0036】
【化9】
【0037】
R1〜R3およびR5は、構造式(I)〜(VI)について上述したとおりであり、
およびR4は、-H2または=Oである
により表される中間体に関する。
【0038】
別の態様において、本発明は、構造式(VIII):
【0039】
【化10】
【0040】
R4は、-H2または=Oであり、
R6は、構造式(IX):
【0041】
【化11】
【0042】
により表される、により表される中間体に関する。構造式(IX)中のフェニル環Aは、置換または非置換である。しかしながら、好ましくは、フェニル環Aは、非置換である。あるいはまた、構造式(VIII)中のR4はH2であり、R6は-Hである。
【0043】
別の態様において、本発明は、構造式(X):
【0044】
【化12】
【0045】
により表される中間体に関する。構造式(X)中のR5は、構造式(I)について規定したとおりである。
【0046】
本発明の方法を利用して、酵素GlcCerシンターゼを阻害するセラミド様化合物を、公知の出発物質から5つの工程で調製することができる。この合成は、高効率であり、通常8%より高い全収率をもたらし、典型的には99%より高いエナンチオマー過剰をもたらす。この合成は、安価な試薬を用い、したがってGlcCerシンターゼの強力なインヒビターへの経済的なルートを提供する。
【0047】
発明の詳細な説明
本明細書において、公知の出発物質からのアミノセラミド様化合物の5工程合成を記載する。この合成は、構造式(II)で表される環式出発物質の調製から始める。該環式出発物質を適当なアミンと反応させ、それによりラクトン環を開裂し、構造式(III)で表されるアミド中間体を形成する。アミド中間体内のアミノアセタールを加水分解し、構造式(IV)で表される非環式化合物を形成する。この非環式化合物のアミドをアミド還元剤で還元し、構造式(V)で表されるアミン化合物を形成し、これを今度は、脱ベンジル化し、構造式(VI)で表されるスフィンゴシン様化合物を形成する。構造式(VI)で表されるスフィンゴシン様化合物の第一級アミンを、次いでアシル化し、アミノセラミド様化合物を形成し得る。この合成を図1に概略的に示す。合成における各反応の詳細を以下に記載する。
【0048】
構造式(II)で表される環式出発物質を、Alkerら, Tetrahedron 54:6089 (1998)ならびにHarwoodおよびRobertson, Chem. Commun. 1998:2641 (1998)に記載の方法にしたがって調製する。具体的には、(5S)-5-フェニルモルホリン-2-オンを少なくとも2当量、好ましくは約2.5〜約5.0当量のアリールアルデヒドR1CHO と脱水条件下で反応させる。R1は、構造式(I)において規定した通りである。「脱水条件」とは、反応混合物から水が除去される条件をいう。水の除去は、例えば、水と反応する(例えば、分子ふるい)が、反応混合物中に存在する他の試薬に対しては実質的に不活性である試薬(「脱水試薬」)の存在下で反応を行なうことにより達成され得、または、水の除去は、トルエンなどの溶剤との共沸によっても達成され得る。充分な脱水試薬を用い、反応中に放出される水2当量(環式出発物質に対して)が除去される。試薬の濃度は、典型的には約0.01M〜5.0Mの間、より典型的には約0.1M〜1.0Mの間であり(if)、好適な反応温度は約50℃〜約150℃の間、好ましくは約100℃〜約120℃の間の範囲である。
【0049】
環式出発物質は、エステルをアミンでアミド化するのに適した条件下で、環式出発物質をアミンNHR2R3と反応させることにより、構造式(II)により表されるアミド中間体に変換される。かかる条件は、当該技術分野において周知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 375-76頁およびそこに引用された文献に記載されている。いずれかの試薬を過剰に使用することができるが、環式出発物質はより一般的には限定試薬である。通常、環式出発物質に対して約15当量まで、典型的には約8当量までのアミンを使用する。反応は純系で(neat)行なわれ得るが、より一般的には、0.01Mという薄いアミン濃度で、非プロトン性非求核性溶媒中にて行なわれる。しかしながら、アミン濃度は、より典型的には約0.4M〜約4.0Mの間である。好適な溶媒としては、クロロホルム、ジクロロメタンおよび1,2-ジクロロエタンなどのハロゲン化溶媒、アセトニトリル、ジメチルホルムアミド(DMF)、ジエチルエーテル、テトラヒドロフラン(THF)および1,4-ジオキサンなどのエーテル系溶媒ならびにベンゼンおよびトルエンなどの芳香族系溶媒が挙げられる。好適な反応温度は通常約0℃〜約100℃の範囲であり、典型的には約25℃〜約35℃の間である。
【0050】
アミノアセタールを加水分解するための条件は当該技術分野において公知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 第 329-32 頁およびそこに引用された文献に記載されている。例えば、構造式(III)により表されるアミド中間体のアミノアセタール基を希無機酸水溶液で加水分解することができる。好適な酸としては、塩酸、硫酸またはリン酸が挙げられるが、塩酸が最も一般的な選択肢である。酢酸およびスルホン酸(例えば、メタスルホン酸、トルエンスルホン酸、トリフルオロ(trifluor)メチルスルホン酸など)などの有機酸も使用することができる。典型的には中間体に対して少なくとも1当量の酸を使用するが、完全な加水分解のためには過剰、例えば、少なくとも10倍過剰、好ましくは約2〜約3倍過剰、より好ましくは約10〜50%の間の酸が好ましい。反応混合物における酸の濃度は、通常、約0.05M〜約1.0Mの間、典型的には約0.1M〜約0.5Mの間である。しばしば水と混和性の有機共存溶媒を用いて中間体を可溶化する。例としては、メタノールまたはエタノールなどのアルコールおよびDMFが挙げられる。水に対する有機溶媒の一般的な溶媒比は、約1:1〜約8:1の範囲である。好適な反応温度は周囲温度から約100℃の範囲、好ましくは約60℃〜約80℃の間である。あるいはまた、March(上記)に記載のようにアミノアセタールをヨウ化トリメチルシリル、湿性シリカゲルまたは湿性アセトニトリル中のLiBF4などのルイス酸を用いて加水分解することができる。
【0051】
「アミド還元剤」は、アミドをアミンに還元し得る試薬である。かかる試薬は当該技術分野において公知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 第 1099-1100 頁、Brown および Krishnamurthy, Aldrichimica Acta 12:3 (1979) ならびにそこに引用された文献に開示されている。例としては、水素化アルミニウムリチウム、水素化ホウ素トリエチルリチウム、ボラン試薬(例えば、ボラン・テトラヒドロフラン、ボラン・メチルスルフィド、ジシアミルボランなど)、水素化アルミニウム、水素化アルミニウムトリメトキシリチウムおよびトリエチルオキソニウムフルオロボレート/水素化ホウ素ナトリウムが挙げられる。本発明の方法において、水素化アルミニウムリチウムが最もよく使用されるアミド還元剤である。アミド出発物質に対して0.5当量ほどの水素化アルミニウムリチウムを使用することができるが、過剰の、しばしば約5当量までを使用するのがより一般的である。好ましくは、アミン出発物質に対して約1.5当量〜約2.5当量の水素化アルミニウムリチウムを使用する。典型的にはエーテル系溶媒が還元に使用される;例としては、ジエチルエーテル、THF、グリム(glyme)、ジグリムおよび1,4−ジオキサンが挙げられる。好適な濃度の還元剤は通常、約0.1M〜約5.0Mの間、より典型的には約0.8M〜約1.5Mの間である。還元は、最も一般的には周囲温度で行なわれるが、約0℃〜約80℃または100℃の間の温度も使用できる。
【0052】
構造式(VI)により表されるスフィンゴシン様化合物を形成するため、構造式(V)により表されるアミン化合物を脱ベンジル化する。本明細書で使用される「脱ベンジル化」という用語は、基-NH-CH2Z(式中、Zはアリール基、好ましくはフェニルである)の炭素−窒素結合の切断をいう。任意に、メチレン基はメチン基と置換され得る。構造式(VI)により表されるスフィンゴシン様化合物に関しては、「脱ベンジル化」は、-NHCH(-CH2OH)R5基の-NH2への変換をいう。脱ベンジル化条件は当該技術分野において周知であり、例えば、GreeneおよびWuts, 「Protective Groups in Organic Synthesis」, John Wiley & Sons (1991), 第384-86頁ならびにそこに引用された文献に開示されている。
【0053】
好ましくは、脱ベンジル化は、水素雰囲気下および水素化触媒の存在下での水素化により行われる。好適な水素圧は通常、ほぼ大気圧から約1000ポンド/平方インチの間である。他の水素源(例えば、ギ酸、ギ酸アンモニウム、シクロヘキサンなど)も使用し得る。好適な水素化触媒としては、カーボン上20%水酸化パラジウム(Parlman触媒)、塩化パラジウム、パラジウム、酸化白金およびカーボン上パラジウムが挙げられる。典型的には、アミン化合物に対して約10%〜約100%重量/重量(w/w)が使用される。ほとんどの場合、ギ酸、酢酸もしくはトリフルオロ酢酸などの有機酸または塩酸もしくは硫酸などの無機酸がアミン化合物に対して、例えば約1〜約5当量、好ましくは約1.6〜約2.4当量で存在する。反応は、最も一般的には、共存溶媒としての水およびメタノールまたはエタノールなどのアルコール系溶媒(例えば、0%〜約50%容積/容積(v/v)の間、約5%〜約15% v/vの間)中で行なわれる。反応温度は、約0℃〜約50℃の間が適当であり、好ましくは約25℃〜約40℃の間である。
【0054】
水素化以外の多くの脱ベンジル化条件は当該技術分野において公知であり、本発明に包含される。例としては、金属ナトリウムおよびNH3(例えば、du VigneaudおよびBehrens, J. Biol. Chem. 117:27 (1937)を参照のこと)、CCl3CH2OCOCl、CH3CN(例えば、Rawalら, J. Org. Chem., 52:19 (1987)を参照のこと)、Me3SiCH2CH2OCOCl、THF、-50℃、次いで一晩25℃(例えば、Campbellら, Tetrahedron Lett., 28:2331 (1987)を参照のこと)、α-クロロエチルクロロホルメートおよび水酸化ナトリウム(例えば、Olofsonら, J. Org. Chem. 49:2081 (1984)ならびにDeShongおよびKell, Tetrahedron Lett., 27:3979 (1986)を参照のこと)、ビニルクロロホルメート(例えば、Olofsonら, Tetrahedron Lett., 1977:1567 (1977)ならびにCooleyおよびEvain, Synthesis, 1989:1 (1989)を参照のこと)、RuO4、NH3、H2O(例えば、GaoおよびJones, J. Am. Chem. Soc., 109:1275 (1987)を参照のこと)ならびにm-クロロ酸化過安息香酸後、FeCl2,−10℃(例えば、Monkovic ら, Synthesis, 1985:770 (1985)を参照のこと)が挙げられる。
【0055】
構造式(VI)により表されるスフィンゴシン様化合物は、遊離アミンをアシル化することによりセラミド様化合物に変換される。アミン基のアシル化は当該技術分野において周知であり、例えば、アミンをアシル化剤R7C(O)-Xと反応させることにより行なわれ得る。R7は、構造式(I)について上記した通りであり、Xは、第一級アミンにより容易に置換される脱離基である。この反応の条件は、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985およびそこに引用された文献に記載されている。好適なアシル化剤の例としては、酸ハロゲン化物、無水物またはエステルが挙げられる。好ましくは、アミンは酸ハロゲン化物によりアシル化される。通常、当モル量のスフィンゴシン様化合物および酸塩化物を、酸塩化物に対して少過剰のトリエチルアミン、ジイソプロピルエチルアミン、ジメチルアミノピリジンまたはピリジンなどの第三級アミンの存在下で使用する。しかしながら、スフィンゴシン様化合物が限られる場合は過剰(典型的には約10〜50%)の酸塩化物を使用することができ、逆の場合も同様である。反応混合物中における試薬の濃度は、通常、約0.005M〜約5.0Mの間で変化し、好ましくは約0.05M〜約0.5Mの間である。過剰のアミン塩基は、約100%超であり得るが、典型的には約5%〜約25%の間である。ハロゲン化溶媒などの非プロトン性溶媒が好ましい(例えば、クロロホルム、塩化メチレンおよび1,2−ジクロロメタン)が、エーテル系溶媒および炭化水素溶媒などの他の非プロトン性溶媒は好適な代用物であり得る。反応には、通常、周囲温度が好ましいが、約0℃〜約50℃の温度も使用され得る。
【0056】
あるいはまた、アシル化剤は、活性化エステルR7C(O)-OX’(式中、-OX’は容易に第一級アミンで置換される)である。活性化エステルでのアミンのアシル化方法は、当該技術分野において公知であり、例えば、March,「Advanced Organic Chemistry - Reactions, Mechanisms and Structure」, 第3版, John Wiley & Sons, 1985, 371-375 頁およびそこに引用された文献に記載されている。多くの活性化エステルは、単離するのに充分安定である。N−ヒドロキシスクシンイミジルエステル(いくつかはAldrich Chemical Co., Milwaukee, WIから市販されている)は、このタイプの活性化エステルの一例である。先の段落で記載した酸塩化物アシル化剤でアミドを形成するのに好適な条件が、安定な活性化エステルと共に典型的に使用され得る。第三級アミンでの活性化を必要とする酸塩化物とは対照的に、活性化エステルは第一級アミンの存在下で直接アミドを形成するように充分反応性である。したがって、活性化エステルを使用する場合は、第三級アミンはアシル化反応から省略され得る。
【0057】
あるいはまた、活性化エステルをインサイチューで形成させる。インサイチューでの活性化エステルの形成は、カルボン酸のヒドロキシル基を、求核性置換を受けやすい基で置換する試薬である「カップリング剤」を必要とする。カップリング剤の例としては、1,1'−カルボニルジイミダゾール(CDI)、イソブチルクロロホルメート、ジメチルアミノプロピルエチル−カルボジイミド(EDC)、ジシクロヘキシルカルボジイミド(DCC, が挙げられる。活性化エステルのインサイチュー生成によりアミド化する場合、カルボン酸またはアミンのいずれかを過剰に使用し得る(典型的には50%過剰、より典型的には約10〜15%過剰)。しかしながら、本発明を実施する際には、限定試薬としてアミン化合物を使用することがより一般的である。通常、カルボン酸1モルあたり約1.0モル〜約10モルのカップリング剤、好ましくはカルボン酸1モルあたり約1.0モル〜約1.5モルのカップリング剤を使用する。反応は、通常、非プロトン性溶媒、例えば、塩化メチレン、ジクロロエタンおよびクロロホルムなどのハロゲン化溶媒、エーテル系溶媒テトラヒドロフラン、1,4−ジオキサンおよびジエチルエーテルならびにジメチルホルムアミド中で行なわれる。好適な反応温度は、通常、約0℃〜約100℃の範囲であるが、反応は好ましくは周囲温度で行なわれる。
【0058】
本明細書に記載の反応を行なうための具体的な条件の例を実施例1および2に提供する。
【0059】
構造式(II)により表される化合物のエナンチオマーを環式出発物質として用いることにより、本明細書に記載の方法を用い、構造式(III)〜(VI)および(I)により表される化合物のエナンチオマーを調製し得る。構造式(III)により表される環式出発物質のエナンチオマーは、上記のような脱水条件下で、(5R)-5-フェニルモルホリン-2-オンを2当量のアルデヒドR1CHOと反応させることにより調製され得る。構造式(III)、(VII)、(VIII)および(X)により表される化合物のエナンチオマーならびに本明細書に開示される手順を用いた構造式(II)〜(VI)および(I)により表される化合物のエナンチオマーの調製方法は本発明に包含される。
【0060】
本明細書で使用される用語「エナンチオマー」およびエナンチオマーを示す構造式は、その光学異性体を含まない「純粋な」エナンチオマーならびにエナンチオマーとその光学異性体との混合物(エナンチオマーが、エナンチオマー過剰、例えば、少なくとも10%、25%、50%、75%、90%、95%、98%または99%エナンチオマー過剰で存在する)
を包含することが意図される。
【0061】
構造式(I)〜(IX)における変数R1〜R5に関して、「脂肪族基」は、非芳香族系であり、炭素と水素のみから構成され、任意に1つ以上の不飽和単位、例えば、二重結合および/または三重結合を含み得る。脂肪族基は、直鎖、分枝鎖または環式であり得る。直鎖または分枝鎖である場合、脂肪族基は、典型的には約1〜約30個の炭素原子、より典型的には約1〜約24個の炭素原子を含む。環式である場合、脂肪族基は、典型的には約3〜約10個の炭素原子、より典型的には約3〜約7個の炭素原子を含む。脂肪族基は、C1〜C30直鎖または分枝鎖の飽和炭化水素、好ましくは、C1〜C24直鎖または分枝鎖の飽和炭化水素を含む低級アルキル基である。例としては、メチル、エチル、n-プロピル、iso-プロピル、n-ブチル、sec-ブチルおよびtert-ブチルが挙げられる。
【0062】
芳香族基としては、フェニル、1-ナフチル、2-ナフチル、1-アントラシルおよび2-アントラシルなどの炭素環式芳香族基、ならびにN-イミダゾリル、2-イミダゾール、2-チエニル、3-チエニル、2-フラニル、3-フラニル、2-ピリジル、3-ピリジル、4-ピリジル、2-ピリミジル、4-ピリミジル、2-ピラニル、3-ピラニル、2-ピラゾリル、3-ピラゾリル、4-ピラゾリル、5-ピラゾリル、2-ピラジニル、2-チアゾール、4-チアゾール、5-チアゾール、2-オキサゾリル、4-オキサゾリルおよび5-オキサゾリルなどの複素環式芳香族基が挙げられる。
【0063】
芳香族基はまた、炭素環式芳香族環またはヘテロアリール環が1つ以上の他のヘテロアリール環に融合された融合多環式芳香族環系も含む。例としては、2-ベンゾチエニル、3-ベンゾチエニル、2-ベンゾフラニル、3-ベンゾフラニル、2-インドリル、3-インドリル、2-キノリニル、3-キノリニル、2-ベンゾチアゾール、2-ベンゾオキサゾール、2-ベンズイミダゾール、2-キノリニル、3-キノリニル、1-イソキノリニル、3-キノリニル、1-イソインドリルおよび3-イソインドリルが挙げられる。
【0064】
非芳香族系複素環式環は、環内に1つ以上窒素、酸素または硫黄などのヘテロ原子を含む非芳香族系炭素環式環である。環は、五、六、七または八員環であり得る。例としては、モルホリニル、チオモルホリニル、ピロリジニル、ピペラジニル、ピペリジニル、アゼチジニル、アザシクロヘプチルまたはN-フェニルピペラジニルが挙げられる。
【0065】
低級アルキル、脂肪族、芳香族、非芳香族、複素環式またはベンジル基上の好適な置換基は、本明細書に記載の反応を実質的に妨げないものである。「反応を妨げる」とは、実質的に収率を低下させる(例えば、50%を超える低下)またはかなりの量の副生成物形成を引き起こすこと(例えば、副生成物が理論収量の少なくとも50%を占める)ことをいう。干渉置換基が最初に保護形態に変換されるならば、干渉置換基を使用し得る。好適な保護基は当該技術分野において公知であり、例えば、GreeneおよびWuts, 「Protective Groups in Organic Synthesis」, John Wiley & Sons (1991) に開示されている。
【0066】
アルキル、脂肪族、芳香族、非芳香族複素環式環またはベンジル基上の好適な置換基としては、例えば、ハロゲン(-Br、-Cl、-Iおよび-F)、-OR、-CN、-NO2、-NR2、-COOR、-CONR2、-SOkR (kは0、1または2)ならびに-NH-C(=NH)-NH2が挙げられる。各Rは独立して−H、脂肪族基、置換脂肪族基、ベンジル基、置換ベンジル基、芳香族基または置換芳香族基、好ましくは-H、低級アルキル基、ベンジル基またはフェニル基である。また、置換非芳香族複素環式環、ベンジル基または芳香族基は、置換基として脂肪族基または置換脂肪族基を有し得る。また、置換アルキルまたは脂肪族基も、置換基として非芳香族複素環式環、ベンジル、置換ベンジル、芳香族または置換芳香族基を有し得る。置換アルキル、置換脂肪族、置換非芳香族複素環式、置換芳香族または置換ベンジル基は1つを超える置換基を有し得る。
【0067】
R1が置換フェニル基である場合、好ましい置換基の例としては、-OCH2O-、-OCH2CH2O-、ハロ、(低級アルキル)O-、低級アルキルチオール、低級ジアルキルアミン、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミンおよび低級アルキルアミノが挙げられる。
【0068】
R5が置換フェニル基である場合、好ましい置換基の例としては、ハロ、(低級アルキル)O-、-O(フェニル)および低級アルキルが挙げられる。
【0069】
本明細書に記載する構造式において、化学基または部分が連結された分子または化合物の残部は、以下の記号:
【0070】
【化13】
【0071】
で表される。例えば、構造式(IX)において対応する変数は、図に示した基(構造式(VIII)においてR6により表される)がベンジル炭素を介して単共有結合により構造式(VIII)におけるアミンに連結されていることを示す。
【0072】
本発明の好ましい態様において、本明細書において使用される記号は、以下のように定義される:R1は、置換または非置換のフェニル基であり;R2およびR3は、独立して、−H、非置換C1〜C5アルキル基、またはこれらが結合している窒素原子と一緒になって非置換C3〜C10非芳香族複素環式環であり;R5は、置換または非置換のフェニル基、好ましくはフェニルであり;R7は、C1〜C30直鎖非置換脂肪族基または1つ以上のC1〜C2アルキル基で置換されたC1〜C30直鎖脂肪族基、より好ましくは、非置換C1〜C30直鎖アルキルまたはアルケニル基である。
【0073】
別の好ましい態様において、-NR2R3は、一緒になってピロリジニルである。より好ましくは、-NR2R3は、一緒になってピロリジニルであり、R5は、R2、R3およびR5をむ化合物において、R1はフェニルである。さらにより好ましくは、R1は、R1、R2、R3およびR5を含む化合物において、置換または非置換フェニル基(好ましくは、メタ/パラ位が-OCH2O-、-OCH2CH2O-で置換されているか、あるいはパラ位がハロ、低級アルキルチオール、-OH、-O(フェニル)、-O-CH2(フェニル)、低級アルキル、アミノ、低級アルキルアミン、低級ジアルキルアミノもしくは-O(低級アルキル)で置換されているフェニルであり、-NR2R3は一緒になってピロリンジニル(pyrrolindinyl)であり、R5はフェニルである。
【0074】
別の好ましい態様において、-NR2R3は、一緒になってピペリジルである。より好ましくは、-NR2R3は、一緒になってピペリジルであり、R5は、R2、R3およびR5を含む化合物において、フェニルである。さらにより好ましくは、R1は、R1、R2、R3およびR5を含む化合物において、R1は置換または非置換フェニル基(好ましくは、メタ/パラ位が-OCH2O-、-OCH2CH2O-で置換されているか、あるいはパラ位がハロ、低級アルキルチオール、-OH、-O(フェニル)、-O-CH2(フェニル)、-O-CH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノ、低級ジアルキルアミノもしくは-O(低級アルキル)で置換されているフェニルであり、-NR2R3は一緒になってピペリジルであり、R5はフェニルである。
【0075】
本発明の方法により調製され得るセラミド様化合物の例は、構造式(XI):
【0076】
【化14】
【0077】
により表される
【0078】
R1は、メタ/パラ位が-OCH2O-もしくは-OCH2CH2O-で置換、またはパラ位がハロ、CH3O-、CH3CH2O-、CH3CH2CH2O-、CH3(CH3)CHO-、CH3-、CH3CH2-、CH3CH2CH2-、CH3(CH3)CH-、-OHもしくは-OCH2(フェニル)で置換されたフェニルであり;R7は、CH3(CH2)n-またはCH3(CH2)n-2CH=CH-(式中、nは、0から約30の整数である) である。好ましくは、nは7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23または24である。より好ましくは、R1は、メタ/パラが-OCH2CH2O-で置換されたフェニルである。
【0079】
充分に酸性な、充分に塩基性な、または両方の官能基を有する本発明の化合物は、したがって、いくつかの無機塩基ならびに無機酸および有機酸と反応して、塩を形成し得る。したがって、本発明はまた、構造式(VII)、(VIII)および(X)により表される中間体の塩も包含する。生理学的に許容され得る塩が好ましい。酸付加塩を形成するために一般的に使用される酸は、塩酸、臭化水素酸、ヨウ化水素酸、硫酸、リン酸などの無機酸、およびp-トルエンスルホン酸、メタンスルホン酸、シュウ酸、p-ブロモフェニル−スルホン酸、カルボン酸、コハク酸、クエン酸、安息香酸、酢酸などの有機酸である。かかる塩の例としては、硫酸塩、ピロ硫酸塩、重硫酸塩、亜硫酸塩、重亜硫酸塩、リン酸塩、一水素リン酸塩、二水素リン酸塩、メタリン酸塩、ピロリン酸塩、塩化物、臭化物、ヨウ化物、酢酸塩、プロピオン酸塩、デカン酸塩、カプリル酸塩、アクリル酸塩、ギ酸塩、イソ酪酸塩、カプロン酸塩、ヘプタン酸塩、プロピオール酸塩、シュウ酸塩、マロン酸塩、コハク酸塩、スベリン酸塩、セバシン酸塩、フマル酸塩、マレイン酸塩、ブチン−1,4−ジオエート、ヘキシン-1,6-ジオエート、安息香酸塩、クロロベンゾエート、メチルベンゾエート、ジニトロベンゾエート、ヒドロキシベンゾエート、メトキシベンゾエート、フタル酸塩、スルホン酸塩、キシレンスルホン酸塩、フェニル酢酸塩、フェニルプロピオン酸塩、フェニル酪酸塩、クエン酸塩、乳酸塩、γ−ヒドロキシ酪酸塩、グリコール酸塩、酒石酸塩、メタンスルホン酸塩、プロパンスルホン酸塩、ナフタレン-1-スルホン酸塩、ナフタレン-2-スルホン酸塩、マンデル酸などが挙げられる。
【0080】
塩基付加塩としては、アンモニウムまたはアルカリもしくはアルカリ土類金属水酸化物、炭酸塩、重炭酸塩などの無機塩基由来のものが挙げられる。したがって、本発明の塩の調製に有用なかかる塩基としては、水酸化ナトリウム、水酸化カリウム、水酸化アンモニウム、炭酸カリウムなどが挙げられる。
【0081】
本出願において引用した刊行物の全教示は参照により本明細書に取り込まれる。
【実施例】
【0082】
実施例
実施例1−セラミド様化合物の小規模調製
中間体1
(1R,3S,5S,8aS)-1,3-ビス-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
【0083】
【化15】
【0084】
(5S)-5-フェニルホルホリン-2-オン(2.00 g, 11.3 mmol)(Dellaria, J.F.: Santarsiero, B.D. J. Org. Chem., 1989, 54, 3916 に記載のようにして調製)および1,4-ベンゾジオキサン-6-カルボキサルデヒド(5.56 g, 33.9 mmol)を含むトルエン(125 mL)の攪拌溶液に、4Å分子ふるい(約20 mL)を添加した。混合物を72時間加熱還流し、濾過して分子ふるいを除き、濃縮した。得られたコハク色ゴムをシリカでフラッシュクロマトフ(ジエチルエーテル/ヘキサン)し、淡黄色固体を得た。この物質を、ジエチルエーテルで摩砕してさらに精製し、1.89 g(34%)の生成物を綿毛状白色固体として得た: 1H NMR (CDCl3) δ 7.31-7.17 (m, 5H), 6.95-6.79 (m, 5H), 5.32-5.27 (m, 2H), 4.43-4.28 (m, 2H), 4.24 (s, 4H), 4.18 (m, 4H), 4.16-4.08 (m, 2H) ppm.
【0085】
中間体2
(2S,3R,1''S)-3-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
【0086】
【化16】
【0087】
中間体1(1.80g, 3.69mmol)を含有するクロロホルム(20mL)の撹拌溶液に、ピロリジン(2.0mL, 24mmol)を添加した。溶液を一晩撹拌し、次に濃縮した。得られた無色の粘着性泡沫物をメタノール(16mL)および1N塩酸(4mL)中に取り出した。混合物を1時間還流し、追加の1N塩酸(2mL)と処理し、さらに2時間還流した。反応溶液を濃縮し、得られた残渣を酢酸エチルと炭酸水素ナトリウム水溶液との間で分配した。有機層を乾燥し(硫酸ナトリウム)、濃縮した。得られた淡黄色の粘性ゴムをシリカゲルに通すフラッシュクロマトグラフィー(塩化メチレン/2Nメタノール性アンモニア(methanolic ammonia))により精製し、無色の泡沫状固体として1.40g(92%)の中間体2を得た:1H NMR (CDCl3) δ 7.31-7.13 (m, 5H), 6.93-6.70 (m, 3H), 4.47 (d, J = 8.5, 1H), 4.18 (s, 4H), 3.82 (t, J = 5.9, 1H), 3.74 (d, J = 6.0, 2H), 3.06 (d, J = 8.5, 1H), 3.06-2.97 (m, 1H), 2.92-2.83 (m, 1H), 1.97-1.87 (m, 1H), 1.45-1.15 (m, 4H) ppm.
【0088】
中間体3
(1R,2R,1''S)-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
【0089】
【化17】
【0090】
中間体2(1.38g, 3.35mmol)を含有するテトラヒドロフラン(30mL)の撹拌溶液に、水素化アルミニウムリチウム(0.26g, 6.9mmol)を添加した。泡沫状懸濁液を一晩撹拌し、次に1N水酸化ナトリウム水溶液(13mL)の添加(泡立つのがやむまで滴下)によりクエンチした。混合物を水で希釈して、酢酸エチルで抽出した。有機層を乾燥し(硫酸ナトリウム)、濃縮して無色の粘性ゴムを得た。シリカゲルに通すフラッシュクロマトグラフィー(塩化メチレン/2Nメタノール性アンモニア)により、0.94g(70%)の無色の粘着性泡沫物として生成物を得た: 1H NMR (CDCl3) δ 7.36-7.17 (m, 5H), 6.88-6.74 (m, 3H), 4.42 (d, J = 5.4, 1H), 4.26 (s, 4H), 3.79-3.69 (m, 1H), 3.64-3.56 (m, 1H), 3.55-3.45 (m, 1H), 3.00-2.90 (m, 1H), 2.67-2.57 (m, 1H), 2.43-2.32 (m, 4H), 2.25-2.15 (m, 1H), 1.75-1.65 (m, 4H) ppm.
【0091】
中間体4
(1R,2R)-2-アミノ-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ピロリジン-1-イル-プロパン-1-オール
【0092】
【化18】
【0093】
メカニカルスターラーを備え付けた高圧反応ボンベ中に、中間体3(0.91g, 2.28mmol)を含有する10:1メタノール/水(22mL)溶液、トリフルオロ酢酸(0.18mL, 2.3mmol)および炭素上20%水酸化パラジウム(Perlman触媒;0.91g)を負荷した。反応器を排気し、3倍のアルゴンで埋め戻し、次いで排気し、水素で再び満たした(100 psi)。反応物を2日間撹拌し、次に排気し、窒素でフラッシュした。反応溶液をセライトを通して濾過し、濃縮した。得られた灰緑色のゴムをシリカゲルに通すフラッシュクロマトグラフィーにかけ(塩化メチレン/2Nメタノール性アンモニア)、ほとんど無色のゴムとして0.165g(26%)の生成物を得た: 1HNMR (CDCl3) δ 6.89-6.76 (m, 3H), 4.54 (d, J = 3.7, 1H), 4.25 (s, 4H), 3.43 (s, 1H), 3.14-3.07 (m, 1H), 2.68-2.41 (m, 6H), 1.82-1.71 (m, 4H) ppm.
【0094】
化合物5
(1R,2R)-ヘキサデカン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0095】
【化19】
【0096】
中間体4(0.165g, 0.593mmol)を含有する塩化メチレン(8mL)の撹拌溶液に、塩化パルミトイル(0.18g, 0.59mmol)、次いでN,N-ジイソプロピルエチルアミン(0.11mL, 0.65mmol)を添加した。溶液を2時間攪拌し、次いで濃縮した。残渣を酢酸エチルと炭酸水素ナトリウム水溶液との間で分配した。有機層を乾燥し(硫酸ナトリウム)、濃縮した。得られたオフホワイトの固体を、シリカゲルに通すフラッシュクロマトグラフィーにかけ(塩化メチレン/2Nメタノール性アンモニア)、白色固体として0.174g(57%)の精製物を得た。1H NMR分光学および分析用キラルHPLC(カラム:Chirex(S)-VALおよび(R)-NE, 4.6×250mm;溶離剤:0.5%トリフルオロ酢酸含有67:31:2ヘキサン/塩化メチレン/エタノール;流量:1mL/分;検出280nM)による比較は、この物質が、Poltらの方法(J. Org. Chem., 1998, 63, 8837)により調製されたものと同一の化合物のサンプルと一致したことを説明する。エナンチオマー過剰は99.6%であると決定した。2つの起こりうるジアステレオマーに由来する全汚染は、0.2%であると決定する。1H NMR (CDCl3) δ 6.88-6.73 (m, 3H), 5.84 (d, J = 7.3, 1H), 4.90 (d, J = 3.8, 1H), 4.24 (s, 4H), 4.22-4.15 (m, 1H), 2.86-2.72 (m, 2H), 2.72-2.55 (m, 4H), 2.10 (t, J = 7.5, 2H), 1.82-1.74 (m, 4H), 1.58-1.46 (m, 2H), 1.32-1.16 (m, 24H), 0.88 (t, J = 6.7, 3H) ppm.
【0097】
実施例2−セラミド様化合物の大規模調製
(5S)-5-フェニルモルホリン-2-オン
【0098】
【化20】
【0099】
S-(+)-フェニルグリシノール(Aldrich, 10.17g, 78.12mmol)およびジイソプロピルエチルアミン(Aldrich, 34mL, 195mmol, 2.5当量)の溶液をCH3CN(200mL)中に調製した。この溶液を、CH3CN(50mL)に溶解したフェニル-α-ブロモ酢酸(18.48g, 85.9mnol, 1.1当量)に窒素下で2時間かけて滴下した。得られた溶液を、窒素下で16〜20時間攪拌した。溶媒を、25℃未満の浴温を保ちながら回転蒸発(rotoevaporation)により除去した。油に酢酸エチル(120mL)を添加し、混合物を15分間攪拌した。得られた白色沈殿を濾過して取り出し、固体を酢酸エチル(25mL)で洗浄した。濾液を、25℃以下の浴温を保ちながら油まで回転蒸発した。減圧下で0.5時間乾燥した後、油をCH2Cl2(17mL)に溶解し、シリカゲルカラム(60g充填し、10%酢酸エチル/ヘキサンを用いる)上に負荷した。上の方の副生成物のスポットを10%酢酸エチル/ヘキサンで溶出し、生成物を50%酢酸エチル/ヘキサン〜100%酢酸エチルで溶出した。生成物を含む画分を、25℃以下の浴温を保ちながら油まで回転蒸発した。この油を酢酸エチル(12mL)に溶解し、氷浴中でヘキサン(60mL)をゆっくり添加して、生成物を沈殿させた。得られた沈殿を濾過した。白〜黄色固体を減圧乾燥した。得られた(5S)-5-フェニルモルホリン-2-オン(7.4g, 41.8mmol, 53%)を、次の工程に直接使用した。
【0100】
中間体1
(1R,3S,5S,8aS)-1,3-ビス-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
【0101】
【化21】
【0102】
(5S)-5-フェニルモルホリン-2-オン(7.4g, 41.8mmol)およびベンゾジオキソラン-6-カルボキシアルデヒド(AldrichまたはAlfa Aesar, 20.56g, 125.2mmol, 3.0当量)をトルエン(180mL)に溶解した。溶液を、4Åモレキュラーシーブ(約30g)を充填したソックスレー抽出器に配置した。溶液を窒素下で2〜3日間還流した。室温に冷却した後、溶媒を回転蒸発により除去し、油を酢酸エチル(200mL)に溶解した。重亜硫酸ナトリウム(Aldrich, 50g)の水溶液(100mL)を添加し、2相の混合物を1時間室温で攪拌した。得られた白色固体を濾過して取り出し、酢酸エチルで洗浄した。濾液を分液漏斗に配置し、層を分離した。有機層を水(100mL)および飽和塩化ナトリウム溶液(100mL)で洗浄した。乾燥(Na2SO4)溶液を濾過および回転蒸発し、黄赤色泡沫状油(23.11g)を得た。減圧下で1時間乾燥した後、ジエチルエーテル(350ml)を添加し、混合物を室温で16〜20時間攪拌した。得られた白〜黄色固体を濾過した。固体を減圧下で乾燥した。環状付加物(cycloadduct)を46%収率(9.34g)で得た。
【0103】
中間体2
(2S,3R,1''S)-3-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
【0104】
【化22】
【0105】
塩化メチレン(40mL)に溶解した環状付加物(中間体1, 6.7g, 13.74mmol)に、ピロリジン(Aldrich, 5.7mL, 68.7mmol, 5当量)を添加した。溶液を室温にて16〜18時間窒素下で撹拌した。溶媒を回転蒸発し、黄色泡沫状油を得、0.5時間減圧乾燥した。粗製物をメタノール(115mL)に溶解し、1M HCl水溶液(115mL)を添加した。溶液を4時間還流した。室温に冷却した後、メタノールを回転蒸発により除去した。酢酸エチル(60mL)を添加し、2相系を室温で5〜15分間撹拌した。2層を分離し、有機層を1M HCL(30mL)で抽出した。あわせた水層を2回酢酸エチル(60, 30mL)で洗浄した。飽和重炭酸ナトリウム溶液(150mL)を水層にゆっくり添加した。生成物を塩基性(pH=8〜9)水層から酢酸エチル(60mL)で3回抽出した。生成物を含むあわせた有機層を飽和塩化ナトリウム溶液(30mL)で洗浄した。Na2SO4で乾燥した後、溶液を濾過し、回転蒸発して、黄色固体を得た。中間体2が93%収率(5.26g)で得られた。
【0106】
中間体3
(1R,2R,1''S)-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
【0107】
【化23】
【0108】
滴下漏斗および冷却器を備え付けた三つ口フラスコに、LiAlH4(Aldrich, 1.2g, 31.7mmol, 2.5当量)および無水THF(20mL)を窒素下で添加した。中間体2(5.23g, 12.68mmol)を含有する無水THF(75mL)の溶液を、反応に15〜30分かけて滴下した。反応を窒素下で9時間還流した。反応を氷浴中で冷却し、1M NaOH溶液を注意深く滴下した。室温で15分間撹拌した後、水(50mL)および酢酸エチル(75mL)を添加した。層を分離し、水層を2回酢酸エチル(75mL)で抽出した。あわせた有機層を飽和塩化ナトリウム溶液(25mL)で洗浄した。Na2SO4で乾燥した後、溶液を濾過し、回転蒸発して無色〜黄色の泡沫状油を得た。中間体3を99%収率(5.3g)で得た。
【0109】
中間体4
(1R,2R)-2-アミノ-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ピロリジン-1-イル-プロパン-1-オール
【0110】
【化24】
【0111】
中間体3(5.3g, 13.3mmol)をメタノール(60mL)に溶解した。水(6mL)およびトリフルオロ酢酸(2.05mL, 26.6mmol, 2当量)を添加した。窒素下に配置した後、炭素上20%水酸化パラジウム(Pearlman触媒, LancasterまたはAldrich, 5.3g) を添加した。混合物をガラスインサートを有するParr Pressure Reactor Apparatusに配置した。装置を窒素下、次に水素圧110〜120psi下に配置した。混合物を、室温にて水素圧100〜120psi下で2〜3日間撹拌した。反応を 窒素下に配置し、セライトのパッドを介して濾過した。セライトパッドをメタノール(100mL)および水(100mL)で洗浄した。メタノールを回転蒸発により除去した。水層を酢酸エチルで3回(100, 50, 50mL) 洗浄した。10M NaOH溶液(10mL)を水層(pH=12〜14)に添加した。生成物を、水層から塩化メチレンで3回(100, 100, 50mL)抽出した。あわせた有機層をNa2SO4で乾燥し、濾過し、無色の油に回転蒸発した。泡沫状油を2時間減圧乾燥した。中間体4を90%収率(3.34g)で得た。
【0112】
化合物5
(1R,2R)-ヘキサデカン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]アミド
【0113】
【化25】
【0114】
中間体4(3.34g, 12.0mmol)を含有する塩化メチレン(50mL)の溶液に、パルミチン酸N-ヒドロキシスクシンイミドエステル(Sigma, 4.24g, 12.0mmol)の溶液を窒素下で室温にて15〜30分かけて添加した。溶液を室温にて18〜20時間撹拌した。反応に塩化メチレン(50mL)および1M NaOH溶液(25mL)を添加した。2相系を室温で15〜30分間撹拌した。水(25mL)を添加し、層を分離した。水層を塩化メチレン(25mL)で逆抽出した。あわせた有機層を水(25mL)で2回、飽和塩化ナトリウム溶液(25mL)で1回洗浄した。有機層をNa2SO4で乾燥し、濾過し、淡黄色油に回転蒸発した。粗製物をヘキサン(50mL)から再結晶させた。得られた白色固体(5.46g)を、シリカゲル(300g)上で2%メタノール:塩化メチレン〜4%メタノール:塩化メチレン〜4% 2Mアンモニウム含有メタノール:塩化メチレンを用いて分離した。得られた白色固体をヘキサン(70mL)から再結晶させた。化合物5を66%収率(4.18g)で得た。分析用キラルHPLC(カラム:Chirex (S)-VALおよび(R)-NE, 4.6×250mm;溶離剤:0.5%トリフルオロ酢酸含有67:31:2ヘキサン/塩化メチレン/エタノール;流量:1mL/分;検出:280nM)は、この物質が純度98.98%であり、0.89%のジアステレオマーおよび0.14%のエナンチオマーを有することを示した。
【0115】
実施例3−セラミド様化合物の代替的な大規模調製
(5S)-5-フェニルモルホリン-2-オンHCl塩
フェニルブロモアセテート(Aldrich, 862.17g, 4.0モル, 1.1当量)を含有するアセトニトリル(試薬等級, 1500ml)の溶液を、氷浴(内部温度5℃以下)中で冷却した。これにS-(+)-2-フェニルグリシノール(Aldrich, 500g, 3.65モル, 1当量)およびジイソプロピルエチルアミン(DIPEA)(Aldrich, 1587ml, 9.11モル, 2.5当量)を含有するアセトニトリル(2900ml)の冷スラリー(内部温度5℃以下)を、内部温度を10℃以下に保ちながら、分割して添加した。氷浴を取り除く前に混合物をこの温度で30分間撹拌し、混合物を室温でさらに4時間撹拌した。浴温を25℃で保ちながら、溶媒を減圧除去した。混合物を、酢酸エチル(2×500ml)と同時エバポレートし、淡黄色粘性油を生成した。反応混合物に、酢酸エチル(4500ml)を添加し、フラスコを撹拌しながら氷浴に浸した。混合物を8℃以下に冷却した。固体を濾過し、酢酸エチル(3×250ml)で洗浄した。溶液を5℃以下に冷却した。pHが2以下(湿潤pH紙)になるまで、内部温度を15℃以下に維持しながら乾燥HClガスを溶液にゆっくり通過させた。さらに20分間この温度およびpHで混合物を撹拌した後、固体を吸引濾過した。固体を酢酸エチル(3×200ml)で洗浄し、高減圧下で約20時間乾燥した。収率は、412g(53%)であった。1H NMRは、(5S)-5-フェニルモルホリン-2-オンHCl塩と一致した。
【0116】
中間体1
(1R,3S,5S,8aS)-1,3-ビス-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
(5S)-5-フェニルモルホリン-2-オンHCl塩(381g, 1当量)を含有する15%酢酸エチル含有トルエン(2270ml)の撹拌懸濁液に、重炭酸ナトリウム(1.1当量)を含有する水溶液(2000ml)を添加した。得られた二相溶液を室温で約1時間撹拌した。有機層を、1,4-ベンゾジオキサン-6-カルボキシアルデヒドを含むフラスコに移した。次に、フラスコにDean-Starkユニット、冷却器および窒素入口を備え付けた。約650mlの溶媒(酢酸エチルおよびトルエンの混合物)をDean-Starkユニットを介して回収しながら、混合物を撹拌しつつ加熱して還流した。得られた黄赤色溶液を、反応の間に形成された水をDean-Starkユニット中に回収しながら、約64時間窒素下で還流に供した。次に溶媒のほとんどをDean-Starkユニットを介する大気圧での蒸留を介して除去した。次に残りの溶媒を、ヘプタン(500ml)およびtert-ブチルメチルエーテル(2×725ml)の同時エバポレーションにより除去し、黄色半固体生成物を生成した。半固体生成物を酢酸エチル(3400ml)に溶解した。重亜硫酸ナトリウム(920g)の水溶液(1500ml)を添加し、混合物を室温で約1時間撹拌した。形成された固体を濾過により除去し、酢酸エチル(3×400ml)で洗浄した。濾液を、水(1450ml)、5%鹹水溶液(1450ml)で洗浄し、MgSO4(100g)で乾燥した。溶媒を減圧下で除去し、黄色固体を得た。これにtert-ブチルメチルエーテル(2900ml)を添加し、懸濁液を室温で20〜22時間撹拌した。黄色固体を吸引濾過し、tert-ブチルメチルエーテル(2×600ml)で洗浄して、約22時間室温にて高減圧下で乾燥した。収率は400.5g(58%)であった。1H NMRおよびTLCは、中間体1と一致した。
【0117】
中間体2
(2S,3R,1''S)-3-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
中間体1(312g, 0.64モル)、ピロリジン(267ml, 3.2モル, 5当量)およびテトラヒドロフラン(1350ml)の溶液を4.5時間窒素雰囲気下で加熱して還流した。 溶媒および過剰のピロリジンを減圧下で除去し、粗製中間体を橙色粘性油として生成した。油をメタノール(3000ml)および1M塩酸溶液(3000ml)に溶解した。得られた溶液を約7時間加熱して還流した。次に、溶媒を減圧下で除去し、油および水の混合物を得た。これに酢酸エチル(2000ml)を添加し、水層を分離した。有機層を1M HCl水溶液(1000ml)で抽出した。水層をあわせ、酢酸エチル(2000ml)で洗浄した。水層を氷浴中で冷却した。水層のpHを、10M NaOH水溶液(525ml)で約9(pH紙)に調整した。水層を酢酸エチル(3000ml)で抽出した。有機層を5%鹹水溶液(1000ml)で洗浄し、乾燥した(Na2SO4)。溶媒を減圧下で除去し、黄色粘性油を生成した。収率は、213.4g, 81%であった。1H NMRは中間体2に一致した。
【0118】
中間体3
(1R,2R,1''S)-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
LiAlH4(50.7g, 1.34モル, 2.6当量)を含有するテトラヒドロフラン(700ml)のスラリーに、中間体2(213.34g, 0.517モル)を含有するテトラヒドロフラン(2000ml)の溶液を室温でゆっくり撹拌しながら添加した。混合物を約4時間還流した。TLC解析(10%メタノール含有塩化メチレン, v/v)は、出発物質の消費を示した。反応混合物を氷浴(5℃未満)中で冷却し、内部温度を10℃以下に保ちながら水(135ml)を非常にゆっくり添加した。次に、これに15%NaOH水溶液(70ml)、続いて水(200ml)を添加した。撹拌を続けながら、反応混合物を室温に加温した。次に、塩化メチレン(1000ml)を混合物に添加し、塩をセライトのパッドを介して濾過した。塩を塩化メチレン(2×500ml)で洗浄した。濾液をあわせ、溶媒 を減圧下で除去して黄色油を生成した。油を1M HCl水溶液(1500ml)に溶解し、酢酸エチル(3×500ml)で洗浄した。水層を氷浴中で5℃以下に冷却し、内部温度を10℃以下に保ちながら水層のpHを10M NaOH水溶液(220ml)で12〜13に調整した。混合物を室温に加温した。水層を塩化メチレン(2×500ml)で抽出した。有機層をあわせ、鹹水溶液(500ml)で洗浄し、乾燥し(Na2SO4)、溶媒を減圧下で除去して黄色粘性油を得た。収率は、186.4g(88.5%)であった。1H NMR は中間体3に一致した。
【0119】
中間体4 ジオキサレート塩
(1R,2R)-2-アミノ-1-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-3-ピロリジン-1-イル-プロパン-1-オールジオキサレート塩
中間体3(358g, 0.90モル)、エタノール(1500ml)、1M HCl溶液(1500ml)および10%Pd(OH)2(32g, 20重量%)の懸濁液を、室温にて約50psiで約36時間水素添加した。混合物をCuonoフィルターを介して濾過した。Cuonoフィルターを10%エタノール含有水(500ml)で洗浄した。濾液をあわせ、エタノールを減圧下で除去した。水層を酢酸エチル(3×600ml)で抽出した。有機層を1M HCl水溶液(700ml)で抽出した。水層をあわせ、氷浴(0〜約5℃)中で冷却した。内部温度10℃以下を保ちながら、水層のpHを10M NaOH水溶液(490ml)で約12(pH紙)に調整した。水層を室温に加温した。水層を塩化メチレン(2×1500ml, 1×750ml)で抽出した。あわせた有機層をMgSO4で乾燥し、溶媒を減圧下で除去し、黄色粘性油を得た。粗製物の重量は、214.3g(86%)であった。1H NMRは中間体4と一致した。
【0120】
シュウ酸(152.4g, 1.693モル, 2.2当量)を含有するメチルイソブチルケトン(2300ml)の溶液を、中間体4(214.3g, 0.77モル, 1当量)を含有するメチルイソブチルケトン(800ml)の溶液に室温で撹拌しながらゆっくり添加した。得られた混合物を室温で約2.5時間撹拌した。固体を濾過し、アセトン(2000ml)で室温にて約16時間粉砕した。固体を濾過し、アセトン(3×100ml)で洗浄し、高減圧下で乾燥し、オフホワイトの固体を生成した。収量は312.5g(89%)であった。1H NMRは、中間体4ジオキサレート塩と一致した。
【0121】
化合物5
(1R,2R)-ヘキサデカン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
中間体4ジオキサレート塩(507g, 1.11モル)を含有する水(10L)の冷却溶液(約5℃)に、内部温度を10℃以下に保ちながら10M NaOH水溶液(500ml)を撹拌しつつ添加した。溶液のpHを約14(pH紙)に維持しながら、溶液を室温に加温した。水層を塩化メチレン(3×6000ml)で抽出した。有機層をあわせ、水(2000ml)で洗浄し、乾燥し(MgSO4)、溶媒を減圧下で除去し、黄色粘性油である中間体4を得た。収量は302g(98%)であった。1H NMRは中間体4に一致した。
【0122】
パルミチン酸NHS-エステル(Sigma , 382.5g, 1.01当量)を含有する塩化メチレン(2500ml)の溶液を、中間体4(302g)を含有する塩化メチレン(1500ml)の溶液に、室温にて1.25時間かけて窒素雰囲気下で添加した。混合物を室温で約18時間撹拌した。1M NaOH水溶液(2425ml)を添加し、混合物を室温で約3時間撹拌した。有機層を分離し、水層を塩化メチレン(800ml)で抽出した。有機層をあわせ、1M NaOH溶液(3×1500ml)および水(1500ml)で洗浄した。有機層をMgSO4で乾燥し、溶媒を減圧下で除去し、半固体を得た。半固体を、ヘプタン(3×100ml)と同時エバポレートした。粗生成物を、12L容の三つ口RBフラスコに移し、ヘプタン(7500ml)を添加した。混合物を、窒素雰囲気下で撹拌しながら還流しつつ加熱した。溶液を約55℃(内部温度)にゆっくり冷却し、別のフラスコに注いだ。溶液を室温で24時間窒素雰囲気下で撹拌した。オフホワイトの固体を濾過し、ヘプタン(2×500ml)で洗浄し、高減圧下で24時間乾燥した。固体(397g)を12L容のRBフラスコに移し、30%酢酸エチル含有ヘプタン(8000ml)を添加した。得られた混合物を撹拌しながら30分間還流しつつ加熱した。溶液を約55℃(内部温度)に冷却し、別のフラスコに注いだ。室温にて窒素雰囲気下で約24時間撹拌を続けた。固体を濾過し、ヘプタン(2×100ml)で洗浄し、高減圧下で乾燥し、オフホワイトの固体を得た。収率は、324g(58%)であった。1H NMRおよびTLCは化合物5と一致した。mp 96.1℃ HPLC解析:キラル純度99.7%, 化学純度99.7%.C31H52N2O4についての解析計算値: C, 72.05; H, 10.14; N, 5.42.実測値C, 72.03; H, 10.19; N, 5.42.
【0123】
実施例4−化合物6〜8の調製
Lapidot, Y. Rappoport, S.およびWolman, Y. Journal of Lipid Research 8, 1967の方法によるかまたは以下に記載されるようにN-ヒドロキシスクシンイミド脂肪酸エステルを調製した:
【0124】
オクタン酸N-ヒドロキシスクシンイミドエステル
N-ヒドロキシスクシンイミド(Aldrich, 20.0g, 173 mmol)およびトリエチルアミン(29mL, 208 mmol)を、氷浴中窒素下で塩化メチレンに溶解した。塩化オクタノイル(Aldrich, 35mL, 205 mmol)を0.5時間かけて滴下した。氷浴を取り除き、白色固体を有する溶液を室温で1時間撹拌した。濾過により白色固体を取り除き、濾液を水(100mL)および飽和重炭酸ナトリウム水溶液(100mL)で洗浄した。有機層を硫酸ナトリウムで乾燥し、濾過し、ヘプタン(100mL)を添加した。溶液を回転蒸発して塩化メチレンのほとんどを除去し、ヘプタン中に無色〜白色の薄片状沈殿が残った。沈殿を濾過し、ヘプタンで洗浄した。乾燥後、オクタン酸N-ヒドロキシスクシンイミドエステルを84%収率(35.4g)で得た:1H NMR (CDCl3) 2.84 (br s, 4H), 2.60 (t, J=7.48 Hz, 2H), 1.78-1.71 (m, 2H), 1.42-1.26 (m, 8H), 0.88 (t, J = 6.7 Hz, 3H) ppm.
【0125】
化合物6
(1R,2R)-オクタン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0126】
【化26】
【0127】
無水塩化メチレン(300mL)に溶解した中間体5(22.36g, 80.33mmol)に、無水塩化メチレン(150mL)に溶解したオクタン酸N-ヒドロキシスクシンイミドエステル(19.4g, 80.39 mmol)の溶液を15〜30分間かけて窒素下で室温にて添加した。溶液を室温で18〜20時間撹拌した。反応に、1M NaOH水溶液(200mL)を添加した。2相系を室温で45分間撹拌した。層を分離し、あわせた有機層を1M NaOHで2回(2×200mL)および水で2回(2×100ml)洗浄した。有機層を硫酸ナトリウムで乾燥し、濾過し、黄色油まで回転蒸発した。粗製物質のほとんどが、還流で5%酢酸エチル含有ヘプタン(1L)に溶解した。40℃に冷却後、濁った溶液を新しいフラスコにデカントすることにより、黄色油から分離した。最初のフラスコを5%酢酸エチル含有ヘプタンで2回(2×250ml)同じプロセス(還流および40℃に冷却および油から溶液のデカント)により濯いだ。あわせた溶液を加熱して還流し、4時間かけて室温まで冷却した。得られた白色固体を濾過し、5%酢酸エチル含有ヘプタン(100mL)およびヘプタン(100mL)で洗浄した。白色固体(13.9g)を減圧下で16〜24時間乾燥した。この固体は、還流で5%酢酸エチル含有ヘプタン(800mL)にほとんど溶解した。50℃に冷却した後、濁った溶液を新しいフラスコにデカントすることにより、黄色油から分離した。最初のフラスコを5%酢酸エチル含有ヘプタン(100mL)で同じプロセス(還流および50℃に冷却および油から溶液のデカント)により濯いだ。あわせた溶液を加熱して還流し、4時間かけて室温に冷却した。得られた白色固体を濾過し、5%酢酸エチル/ヘプタン(50mL)およびヘプタン(50mL)で洗浄した。室温にて減圧下2〜3日間乾燥した後、化合物6を39%収率(12.57g)で得た。分析用キラルHPLC(カラム:Chirex (S)-VALおよび(R)-NE, 4.6×250mm)は、この物質が99.9%所望のR,R異性体であることを示した。分析用HPLCは、この物質が99.6%純粋であることを示した。 mp 87-88℃. 1H NMR (CDCl3) δ 6.86-6.73 (m, 3H), 5.84 (d, J = 7.3 Hz, 1H), 4.91 (d, J = 3.4Hz, 1H), 4.25 (s, 4H), 4.24-4.18 (m, 1H), 2.85-2.75 (m, 2H), 2.69-2.62 (m, 4H), 2.10 (t, J = 7.3 Hz, 2H), 1.55-1.45 (m,2H), 1.70-1.85 (m, 4H), 1.30-1.15 (m, 8H), 0.87 (t, J = 6.9 Hz, 3H) ppm.
【0128】
化合物7
(1R,2R)-ノナン酸[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0129】
【化27】
【0130】
この化合物を、ノナン酸 N-ヒドロキシスクシンイミドエステルを使用し、化合物6について記載した方法により調製した。分析用HPLCは、この物質が98.4 %純粋であることを示した。 mp 74-75℃. 1H NMR (CDCl3) δ6.86-6.76 (m, 3H), 5.83 (d, J = 7.3 Hz, 1H), 4.90 (d, J = 3.3 Hz, 1H), 4.24 (s, 4H), 4.24-4.18 (m, 1H), 2.85-2.75 (m, 2H), 2.69-2.62 (m, 4H), 2.10 (t, J = 7.3 Hz, 2H), 1.55-1.45 (m, 2H), 1.70-1.85 (m, 4H), 1.30-1.15 (m, 10H), 0.87 (t, J = 6.9 Hz, 3H) ppm.
【0131】
化合物8
(1R,2R)デカノン[2-(2',3'-ジヒドロ-ベンゾ[1,4]ジオキシン-6'-イル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エチル]-アミド
【0132】
【化28】
【0133】
この化合物を、デカン酸N-ヒドロキシスクシンイミドエステルを使用し、化合物6について記載した方法により調製した。分析用HPLCは、この物質が99.3%純粋であることを示した。 mp 97.5〜98.5℃. 1H NMR (CDCl3) δ 6.86-6.76 (m, 3H), 5.83 (d, J = 7.5 Hz, 1H), 4.90 (d, J = 3.4 Hz, 1H), 4.24 (s, 4H), 4.24-4.18 (m, 1H), 2.85-2.75 (m, 2H), 2.69-2.62 (m, 4H), 2.10 (t, J = 7.5 Hz, 2H), 1.55-1.45 (m, 2H), 1.70-1.85 (m, 4H), 1.30-1.15 (m, 12H), 0.87 (t, J = 6.8 Hz, 3H) ppm.
【0134】
実施例5−化合物13の調製
中間体9
(1R,3S,5S,8aS)-1,3-ビス-(4-ベンジルオキシ-フェニル)-5-フェニル-テトラヒドロ-オキサゾロ[4,3-c][1,4]オキサジン-8-オン
【0135】
【化29】
【0136】
(5S)-5-フェニルモルホリン-2-オンHCl塩(57.45, 268.9mmol)を、二相溶液が澄むまで酢酸エチル(500mL)および飽和重炭酸ナトリウム水溶液(250mL)と30分間撹拌した。相を分離し、水層を酢酸エチル(2×250mL)で抽出した。あわせた有機相を飽和塩化ナトリウム溶液(250mL)で洗浄した。有機層を硫酸ナトリウムで乾燥し、濾過し、油まで濃縮し、60分間減圧下で乾燥した。5-(S)-フェニルモルホリン-2-オンを86%収率(40.98g, 231.3 mmol)で得た。
【0137】
5-(S)-フェニルモルホリン-2-オン(40.98g, 231.3 mmol)および4-ベンジルオキシベンズアルデヒド(Aldrich, 147.3g, 694 mmol, 3.0当量)をトルエン(750mL)に溶解した。反応は、Dean Stark Trapおよび還流冷却器を備え付けた。溶液を窒素下で2日間還流した。室温に冷却した後、溶媒を回転蒸発により除去し、油を酢酸エチル(500mL)に溶解した。水(250mL)に溶解した重亜硫酸ナトリウム(Aldrich, 125g)の溶液を添加し、2相混合物を室温で3時間撹拌した。得られた白色固体を濾過して取り出し、酢酸エチルで洗浄した。濾液を分液漏斗に配置し、層を分離した。有機層を水(250mL)、飽和塩化ナトリウム水溶液(250mL)で洗浄し、次いで乾燥し(硫酸ナトリウム)、濾過し、泡沫状油(144g)まで回転蒸発した。減圧下で1時間乾燥した後、tert-ブチルメチルエーテル(1450mL)を添加し、混合物を室温で5時間撹拌した。得られた白〜黄色固体を濾過した。固体を減圧下で乾燥した。中間体9を 27%収率(41.64g, 71.46 mmol)で得た。 1H NMR (CDCl3) δ 7.5-6.8 (m, 23H), 5.0および5.1 (2s, 4H), 4.5-4.3 (m, 2H), 4.2-4.1 (m, 2H) ppm.
【0138】
中間体10
(2S,3R,1''S)-3-(4-ベンジルオキシ-フェニル)-3-ヒドロキシ-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-1-ピロリジン-1-イル-プロパン-1-オン
【0139】
【化30】
【0140】
テトラヒドロフラン(250mL)に溶解した中間体9(45.1g, 77.4mmol)に、ピロリジン(Aldrich 33mL, 395mmol, 5.1当量)を添加した。窒素下で室温にて16〜18時間、溶液を撹拌し、キャップした。溶媒を回転蒸発して黄色泡沫状油を得、0.5時間減圧乾燥した。粗製物をメタノール(220mL)に溶解し、1M HCl水溶液(220mL)を添加した。溶液を4時間還流した。室温まで冷却した後、メタノールを回転蒸発により除去した。得られた油に、ゆっくり10M NaOH水溶液(22mLでpHを14に調整)を添加した。生成物を、塩基性水層から塩化メチレンで3回(300,100,100mL)抽出した。硫酸ナトリウムで乾燥した後、あわせた有機層を濾過し、回転蒸発し、黄〜橙色の泡沫状固体を得た。tert-ブチルメチルエーテル(300mL)を添加し、混合物を室温で7時間撹拌した。得られた白〜黄色固体を濾過し、tert-ブチルメチルエーテル(50mL)で洗浄し、減圧乾燥した。中間体10を83%収率(29.77g)で得た。 1H NMR (CDCl3) δ 7.4-7.2 (m, 12H), 6.9-6.8 (m, 2H), 5.05 (AB四重項, 2H), 4.47 (d, J = 8.5, 1H), 3.9-3.3 (m, 3H), 3.05 (d, J = 8.5, 1H), 3.0-2.8 (m, 2H), 2.3-2.2 (m, 1H), 1.85-1.7 (m, 1H), 1.45-1.15 (m, 4H) ppm.
【0141】
中間体11
(1R,2R,1''S)-1-(4-ベンジルオキシ-フェニル)-2-(2''-ヒドロキシ-1''-フェニル-エチルアミノ)-3-ピロリジン-1-イル-プロパン-1-オール
【0142】
【化31】
【0143】
滴下漏斗および冷却器を備える三ツ口フラスコにおいて、窒素下でLiAlH4(Aldrich、6.3g、166mmol、2.57当量)および無水テトラヒドロフラン(75mL)を添加した。中間体10(29.7g、64.48mmol)を含有する無水テトラヒドロフラン(300mL)の溶液を15〜30分にわたって反応液に滴下した。反応液を窒素下で9時間還流した。反応液を氷浴で冷却し、水(7.0mL)を一滴ずつ非常に注意深く添加した(水素を放出する活発な発熱反応)。15%NaOH水溶液(7.0mL)、次いで水(21mL)を滴下した。最終の水添加の途中で大量の白色固体が形成された。これを塩化メチレン(250mL)の添加により破壊した。室温にて15分間の撹拌後、混合物をセライトプラグ(直径17cm×高さ1cm)を介して濾過した。沈殿を塩化メチレンで洗浄した(2×250mL)。濾液を油まで回転蒸発(rotoevaporated)した。油を1M HCl水溶液(300mL)に溶解した。水層をtert-ブチルメチルエーテルで洗浄した(2×200mL)。氷浴で冷却後、10M NaOH水溶液(35mL)を水層に注意深く添加した(最終pH=14)。生成物を塩化メチレンで3回抽出した(300mL、200mLおよび100mL)。硫酸ナトリウムで乾燥後、溶液を濾過し、回転蒸発して白色固体を得た。乾燥後、中間体11を94%収率で得た(26.9g)。1H NMR(CDCl3) δ 7.46-7.115(m, 12H), 6.98-6.96(m, 2H), 5.08(s, 2H), 4.49(d, J = 4.7, 1H), 3.70-3.65(m, 1H), 3.60-3.55(m, 1H), 3.54-3.45(m, 1H), 3.00-2.90(m, 1H), 2.7-2.6(m, 1H), 2.36(br s, 4H), 2.15-2.05(m, 1H), 1.70(br s, 4H) ppm.
【0144】
中間体12
(1R,2R)-2-アミノ-1-(4-ベンジルオキシ-フェニル)-3-ピロリジン-1-イル-プロパン-1-オール塩化水素塩
【0145】
【化32】
【0146】
中間体11(26.9g、60.24mmol)をメタノール(400mL)に溶解し、1M HCl水溶液(130mL)を添加した。窒素下に置いた後、20%水酸化パラジウム炭素(Pearlman触媒、Aldrich、10.8g)を添加した。反応液をバルーンへの排気および充填により窒素下、次いで水素下に置いた。混合物を水素バルーン下で室温にて48時間撹拌した。反応液を窒素下に置き、セライトパッドを介して濾過した。セライトパッドを10%の水含有メタノール(250mL)および水(50mL)で洗浄した。溶媒を回転蒸発およびトルエンとの同時蒸発(3×100mL)により除去した。泡沫固体を還流でイソプロパノール(300mL)に溶解した。溶液を室温に冷却し、tert-ブチルメチルエーテル(550mL)を添加した。室温にて2時間撹拌後、白色固体を濾過して、tert-ブチルメチルエーテルで洗浄した。乾燥後、中間体12を約99%収率で得た(18g)。 1H NMR(DMSO-d6) δ 9.68(br s, 1H), 8.53(br s, 2H) 7.24(d, J = 8.55 Hz, 2H), 6.80(d , J = 8.55 Hz, 2H), 4.72(d, J = 7.0 Hz, 1H), 3.8-3.6(m, 2H), 3.4-3.6(m, 3H), 3.0-3.2(m,2H), 2.7-2.5(br s, 1H), 2.0-1.7(br s , 4H) ppm.
【0147】
化合物13
(1R, 2R)-ヘキサデカン酸[2-(4-ベンジルオキシ-フェニル)-2-ヒドロキシ-1-ピロリジン-1-イルメチル-エーテル]-アミド
【0148】
【化33】
【0149】
テトラヒドロフラン(500mL)に懸濁した中間体12(16.17g 49.36mmol)にトリエチルアミン(28mL、4当量)を添加した。テトラヒドロフラン(125mL)に溶解したパルミチン酸N-ヒドロキシスクシンイミドエステル(Sigma、19.2g、54.29mmol)の溶液を室温にて窒素下で30分にわたって添加した。溶液を室温にて18〜20時間撹拌した。白色沈殿を濾過により除去し、濾液を泡沫状のオフホワイト固体まで回転蒸発した(35.5g)。粗物質を塩化メチレン(500mL)に溶解し、水(100mL)および飽和炭酸ナトリウム水溶液(100mL)で洗浄した。硫酸ナトリウムで乾燥後、溶液を濾過、回転蒸発し、オフホワイトの泡沫状固体(24.75g)を得た。この物質を40%酢酸エチル含有ヘプタン(500mL、高温濾過)から再結晶させた。化合物13を61%収率で得た(14.45g)。分析キラルHPLCによりこの物質は99.7%純度、所望のR,Rイソマーであることを示した。分析HPLCにより、この物質は99.6%純度、mp95〜97℃であることを示した。 1H NMR(CDCl3) δ 7.15(d, J= 8.5Hz, 2H), 6.70(d, J= 8.5 Hz , 2H), 6.0(d, J = 7.3, 1H), 4.96(d, J = 3.8, 1H), 4.3-4.2(m, 1H), 2.9-2.7 (m, 2H), 2.65-2.55(m, 4H), 2.10(t, J = 7.5, 2H), 1.75(br s, 4H), 1.58-1.46(m, 2H), 1.32-1.16(m, 24H), 0.9(t, J = 6.7, 3H) ppm.
【0150】
本発明は、その好ましい態様を参照して詳細に示され、記載されているが、形式および詳細における種々の変更は、添付の特許請求の範囲により包含される本発明の範囲から逸脱せずにその中でなされ得ることが当業者により理解される。
【0151】
本発明の態様として、以下のものが挙げられる。
[1]以下の構造式:
【化34】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R4は=OまたはH2であり;
R5は置換または非置換芳香族基である)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[2]R1およびR5が独立して置換または非置換フェニル基であり;
R2およびR3が独立して-H、非置換C1〜C5アルキル基またはそれらが結合される窒素原子と共に非置換C3〜C10非芳香族複素環式環である、[1]記載の化合物。
[3]R5がフェニル基である[2]記載の化合物。
[4]R2およびR3が、それらが結合される窒素原子と共にピロリジニル、ピペラジニル、アゼチジニル、モルホリニル、チオモルホリニル、アザシクロヘプチル、ピペリジニルまたはN-フェニルピペラジニルである[3]記載の化合物。
[5]以下の構造式:
【化35】
(式中、R1は置換または非置換フェニル基であり、R4が-H2または=Oである)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[6]R1で表されるフェニル基が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、-OH、低級アルキルチオール、-O(フェニル)、-OCH2(フェニル)、-O-CH2-(フェニル)、低級アルキル、アミノ、低級アルキルアミノまたは低級ジアルキルアミノで置換される[5]記載の化合物。
[7]以下の構造式:
【化36】
(式中、R4は-H2または=Oであり;かつ
R6は以下の構造式:
【化37】
(式中、フェニル環Aは置換または非置換である)
で表される;または
R4はH2であり、かつR6は-Hである)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[8]フェニル環Aが非置換である[7]記載の化合物。
[9]以下の構造式:
【化38】
(式中、R5は置換または非置換芳香族基である)
で表される化合物、該化合物のエナンチオマーおよび該化合物の塩および該エナンチオマーの塩。
[10]R5がフェニルである[9]記載の化合物。
[11]以下の構造式:
【化39】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R5は置換または非置換芳香族基である)
で表される非環式化合物を調製する方法であって、アミン化合物HNR2R3と以下の構造式:
【化40】
で表される環式開始物質とを反応させ、それにより以下の構造式:
【化41】
で表される中間体を形成する工程、
および該中間体のアミノアセタール基を加水分解し、それにより該非環式化合物を形成する工程を含む、方法。
[12]R1およびR5が独立して置換または非置換フェニル基であり;
R2およびR3が独立して-H、非置換C1〜C5アルキル基またはそれらが結合される窒素原子と共に非置換C3〜C10非芳香族複素環式環である、[11]記載の方法。
[13]R5がフェニル基である[12]記載の方法。
[14]R2およびR3が、それらが結合される窒素原子と共にピロリジニル、ピペラジニル、アゼチジニル、モルホリニル、チオモルホリニル、アザシクロヘプチル、ピペリジニルまたはN-フェニルピペラジニルである[13]記載の方法。
[15]R1が置換または非置換フェニル基であり、R2およびR3が、それらが結合される窒素原子と共にピロリジニル基である[14]記載の方法。
[16]R1で表されるフェニル基が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノまたは低級ジアルキルアミノで置換される[15]記載の方法。
[17]アミド還元剤と非環式化合物とを反応させ、それにより以下の構造式:
【化42】
で表されるアミン化合物を形成する工程をさらに含む、[11]記載の方法。
[18]アミド還元剤が水素化アルミニウムリチウムである[17]記載の方法。
[19]アミド還元剤がボラン・テトラヒドロフランである[17]記載の方法。
[20]アミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化し、それにより以下の構造式:
【化43】
で表されるセラミド化合物を形成する工程をさらに含む、[17]記載の方法。
[21]アミン化合物が水素添加により脱ベンジル化される[18]記載の方法。
[22]アミン化合物が水素添加触媒の触媒作用量を用いて水素雰囲気下で水素添加される[20]記載の方法。
[23]アミン化合物の第一級アミン基をアシル化し、それにより以下の構造式:
【化44】
で表されるアシル化セラミド化合物を形成する工程をさらに含む、[19]記載の方法。
[24]セラミド化合物がR7COX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジルであり、RはC1〜C30直鎖アルキルまたはアルケニル基である)でアシル化される[22]記載の方法。
[25]非環式化合物が、以下の構造式:
【化45】
で表される環式ラクトンと少なくとも2当量のアルデヒド化合物R1CHOとを反応させることにより調製される[11]記載の方法。
[26]以下の構造式:
【化46】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R5は置換または非置換芳香族もしくは脂肪族基である)
で表されるアミン化合物を調製する方法であって、アミド還元剤と以下の構造式:
【化47】
で表される非環式前駆体化合物とを反応させる工程を含む、方法。
[27]R1およびR5が独立して置換または非置換フェニル基であり;
R2およびR3が独立して-H、非置換C1〜C5アルキル基またはそれらが結合される窒素原子と共に非置換C3〜C10非芳香族複素環式環である、[25]記載の方法。
[28]R5がフェニル基である[26]記載の方法。
[29]R2およびR3が、それらが結合される窒素原子と共にピロリジニル、ピペラジニル、アゼチジニル、モルホリニル、チオモルホリニル、アザシクロヘプチル、ピペリジニルまたはN-フェニルピペラジニルである[27]記載の方法。
[30]R1が置換または非置換フェニル基であり、R2およびR3が、それらが結合される窒素原子と共にピロリジニル基である[28]記載の方法。
[31]R1で表されるフェニル基が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノまたは低級ジアルキルアミノで置換される[29]記載の方法。
[32]アミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化し、以下の構造式:
【化48】
で表されるスフィンゴシン様化合物を形成する工程をさらに含む、[25]記載の方法。
[33]アミン化合物が水素添加により脱ベンジル化される[31]記載の方法。
[34]アミン化合物が触媒作用量のPd(OH)2を用いて水素雰囲気下で水素添加される[32]記載の方法。
[35]スフィンゴシン様化合物の第一級アミン基をアシル化し、それにより以下の構造式:
【化49】
で表されるセラミド様化合物を形成する工程をさらに含む、[32]記載の方法。
[36]セラミド化合物がR7COX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジル基であり、R7はC1〜C30直鎖アルキルまたはアルケニル基である)でアシル化される[34]記載の方法。
[37]以下の構造式:
【化50】
で表されるスフィンゴシン様化合物を調製する方法であって、以下の構造式:
【化51】
(式中、R1は置換または非置換芳香族基であり;
R2およびR3は独立して-H、置換もしくは非置換脂肪族基、またはそれらが結合される窒素原子と共に置換もしくは非置換非芳香族複素環式環であり;
R5は置換または非置換芳香族基である)
で表されるアミン化合物の-NHCH(-CH2OH)R5基を脱ベンジル化する工程を含む、方法。
[38]アミン化合物が水素添加により脱ベンジル化される[36]記載の方法。
[39]アミン化合物が触媒作用量のPd(OH)2を用いて水素雰囲気下で水素添加される[37]記載の方法。
[40]スフィンゴシン様化合物の第一級アミン基をアシル化し、それにより以下の構造式:
【化52】
で表されるセラミド様化合物を形成する工程をさらに含む、[37]記載の方法。
[41]セラミド化合物がR7COX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジル基であり、R7はC1〜C30直鎖アルキルまたはアルケニル基である)でアシル化される[39]記載の方法。
[42]以下の構造式:
【化53】
(式中、R1は置換または非置換フェニル基であり:
R7はC7〜C10またはC10〜C16アルキル基である)
で表されるセラミド様化合物を調製する方法であって、
a)環式ラクトンと少なくとも2当量のアルデヒドR1CHOとを反応させ、以下の構造式:
【化54】
で表される第1の中間体を形成する工程であって、該環式ラクトンが以下の構造式:
【化55】
で表される、工程;
b)ピロリジンと該第1の中間体とを反応させ、以下の構造式:
【化56】
で表される第2の中間体を形成する工程;
c)該第2の中間体のアミノアセタール基を加水分解し、以下の構造式:
【化57】
で表される第3の中間体を形成する工程;
d)水素化アルミニウムリチウムまたはボラン・テトラヒドロフランで該第3の中間体を還元し、以下の構造式:
【化58】
で表される第4の中間体を形成する工程;
e)触媒作用量のPd(OH)2を用いて水素雰囲気下で該第4の中間体を水素添加し、以下の構造式:
【化59】
で表されるスフィンゴシン様化合物を形成する工程;ならびに
f)RCOX(式中、Xは-ClまたはN-ヒドロキシスクシンイミジルである)で該スフィンゴシン様化合物をアシル化し、セラミド様化合物を形成する工程
を含む、方法。
[43]R7がC10〜C16アルキル基である[42]記載の方法。
[46]R1が-OCH2O-、-OCH2CH2O-、ハロ、-O(低級アルキル)、低級アルキルチオール、-OH、-O(フェニル)、-OCH2(フェニル)、低級アルキル、アミノ、低級アルキルアミノおよび低級ジアルキルアミノで置換されたフェニル基である[43]記載の方法。
[45]R1が
【化60】
であり、R7が-(CH2)14CH3である[43]記載の方法。
[46]R1がパラ-ヒドロキシフェニルまたは
【化61】
であり、R7がC7〜C10アルキル基である[42a]記載の方法。
【特許請求の範囲】
【請求項1】
以下の構造式:
【化1】
で表される化合物またはその生理学的に許容され得る塩。
【請求項1】
以下の構造式:
【化1】
で表される化合物またはその生理学的に許容され得る塩。
【図1】

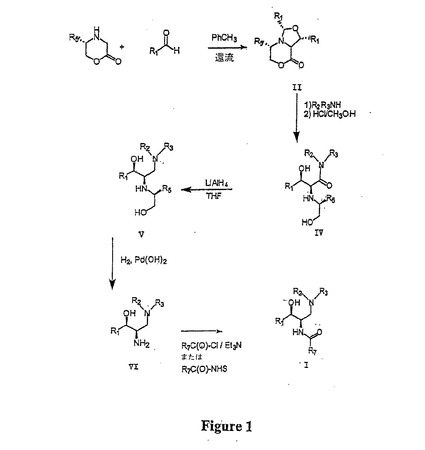
【図2】
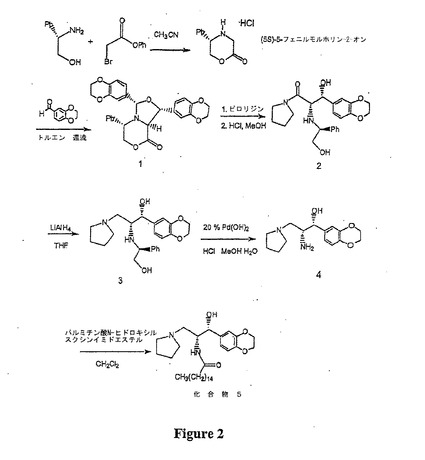
【図3】
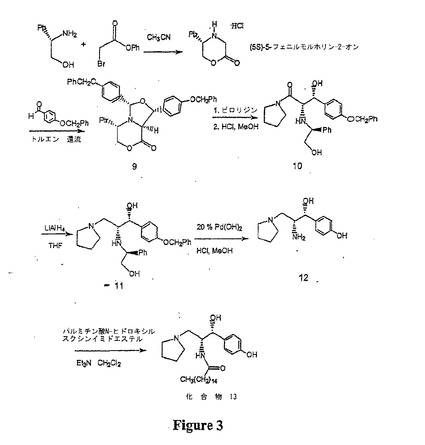
【図4】
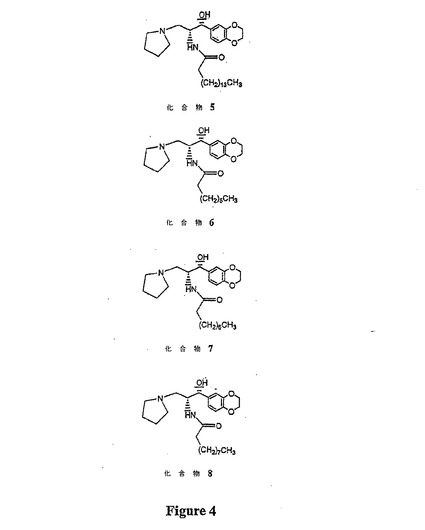
【図2】
【図3】
【図4】
【公開番号】特開2010−95546(P2010−95546A)
【公開日】平成22年4月30日(2010.4.30)
【国際特許分類】
【出願番号】特願2010−20146(P2010−20146)
【出願日】平成22年2月1日(2010.2.1)
【分割の表示】特願2003−513958(P2003−513958)の分割
【原出願日】平成14年7月16日(2002.7.16)
【出願人】(591042816)ジェンザイム コーポレーション (20)
【出願人】(500047572)ザ、リージェンツ、オブ、ザ、ユニバーシティ、オブ、ミシガン (12)
【氏名又は名称原語表記】THE REGENTS OF THE UNIVERSITY OF MICHIGAN
【Fターム(参考)】
【公開日】平成22年4月30日(2010.4.30)
【国際特許分類】
【出願日】平成22年2月1日(2010.2.1)
【分割の表示】特願2003−513958(P2003−513958)の分割
【原出願日】平成14年7月16日(2002.7.16)
【出願人】(591042816)ジェンザイム コーポレーション (20)
【出願人】(500047572)ザ、リージェンツ、オブ、ザ、ユニバーシティ、オブ、ミシガン (12)
【氏名又は名称原語表記】THE REGENTS OF THE UNIVERSITY OF MICHIGAN
【Fターム(参考)】
[ Back to top ]