
WT−1とGATA−1のエピトープを用いた免疫治療方法
【課題】従来のロールプリンティング法は、ロールプリンティングによるパターン転写時にブランケットから基板に円滑にパターンを転写するために表面張力の低いPDMS(polydimethylsilosane)などの材料をブランケットとして用いるため、インクの表面張力がブランケット材料より低くならないと良好なコーティング結果を得ることができない。よって、ロールプリンティング用インク組成物がブランケット表面より低い表面張力を有するようにするためには、インク組成物内に表面張力を下げる効果に優れた界面活性剤を添加して使わなければならない。
【解決手段】完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列RMFPNAPYLを含むペプチドまたはその部分または異型、完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列CMTWNQMNLを含むペプチドまたはその部分または異型、または、完全なヒトgata−1ポリペプチドではないという条件で、アミノ酸配列HLMPFPGPLLを含むペプチドまたはその部分または異型、並びに、これらのペプチドをコードするポリヌクレオチド。これらのペプチドおよびポリヌクレオチドは、ガンのワクチンとして使用できる。
【解決手段】完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列RMFPNAPYLを含むペプチドまたはその部分または異型、完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列CMTWNQMNLを含むペプチドまたはその部分または異型、または、完全なヒトgata−1ポリペプチドではないという条件で、アミノ酸配列HLMPFPGPLLを含むペプチドまたはその部分または異型、並びに、これらのペプチドをコードするポリヌクレオチド。これらのペプチドおよびポリヌクレオチドは、ガンのワクチンとして使用できる。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、免疫治療法、および、免疫治療法に使用される分子および細胞に関する。特に、本発明は、白血病を含むガンの治療に関する。
【背景技術】
【0002】
抗腫瘍細胞毒性Tリンパ球(CTL)がin vivoにおいて重要な役割を演じるという証拠がある。腫瘍反応性CTLが腫瘍後退を媒介することが、動物モデル(Kastら(1989) Cell 59, 603-614)およびヒト(カワカミら(1994) Proc.Natl.Acad.Sci.USA91,6458-6462)において示された。抗腫瘍治療の全てのタイプに関して、克服される必要のある問題は、この治療が有益な程度まで標的腫瘍細胞を破壊または不活性化しなければならないが、この治療が非腫瘍細胞を有害な程度まで破壊または不活性化してはならないことである。言い換えれば、この治療が腫瘍細胞に対して有益な程度にまで選択的であれば好ましい。
【0003】
ガンの免疫治療に対する現在の研究のほとんどは、一部の腫瘍が、等価の非腫瘍組織に発現されないポリペプチドを発現するという事実を利用するか、腫瘍が、非腫瘍組織に発現されないポリペプチドの変異形態を発現するという事実を利用する。しかしながら、このカテゴリーに入る腫瘍のポリペプチドを常に同定できるわけではなく、それゆえ、免疫治療アプローチのベースを形成する別の標的ポリペプチドが同定されてきた。
【0004】
メラノーマ患者における研究は、メラノーマ反応性CTLによって認識されたペプチドエピトープが組織特異的分化抗原からしばしば誘導されることを示した。正常組織に発現される分化抗原の認識は、免疫学的寛容のルールを壊すようにも見える;しかしながら、メラノーマ関連分化抗原のCTL認識は、それらが通常は、免疫学的に特別に許可された部位において比較的少数で存在するメラノサイトにおいて発現されるのみであり、かくして寛容を確立しないという事実によって説明される。また、前立腺特異的分化抗原が、前立腺の腫瘍に対するCTLの標的として作用しうることも示されている。発生的に制御された転写因子が、どの程度まで、これらの因子を異常に発現する腫瘍に対してCTLの標的として機能するのかは、現在のところ不明である。
【0005】
Gata−1は、赤血球、巨核球、エオシン好性白血球およびマスト細胞直系、並びに、多型潜在性先祖において通常発現される転写因子である。この厳しくコントロールされた転写因子の異常発現は、CMLおよびAMLを含む白血球において観察される(Shimamotoら(1995)Blood 86,3173-3180)。
【0006】
成人では、WT1、胚の転写因子の発現が、腎の有足突起、精巣、卵巣、胸部筋上皮細胞、そして骨髄の一部のCD34+幹細胞に観察されている。異常発現は、乳ガン、卵巣ガン、メラノーマおよび、CMLおよびAMLを含む白血病に観察された(例えば、Menssenら(1995) Leukaemia 9,1060-1067;Inoueら(1997) Blood 89,1405-1412; Inoueら(1996) Blood 88, 2267-2278; Inoueら (1998) Blood 91, 2969-2976; Menssenら(1997) Int.J.Cancer 70, 518-523; Menssenら(1995) Leukemia 9, 1060-1067; Ogawaら(1998) Transplant 21,527-527; Rodeckら(1994) Int.J.Cancer 59, 78-82; Silbersteinら(1997) Proc.Natl.Acad.Sci.USA 94, 8132-8137; Tamakiら (1996) Blood 88, 4396-4398; およびVielら(1994) Int.J.Cancer 57,515-521)。
【0007】
米国特許第5726288号は、Wilmsの腫瘍遺伝子の位置および特徴に関する。4つのアミノ酸配列が開示されており(配列番号2、4、5および6)、配列RMFPNAPYLまたはCMTWMNQMNLのいずれかを含むが、これらの配列に対応するペプチドもしくは免疫治療におけるそれらの用途については全く開示されていない。
【0008】
アロ-MHC-制限CTLを用いる通常でないアプローチを用いて、タンパクWT−1およびgata−1のペプチドエピトープを驚くべきことに同定した。これらは、HLA−A0201クラスI分子によって提示され、これらのタンパクを内因的に発現する腫瘍細胞表面に提示される。HLA−A0201陰性は、HLA−A0201クラスI分子によって提示されたペプチドに特異的なCTLの源として用いられ、このアプローチは、自己のCTLの可能な寛容から独立したHLA−A0201提示ペプチドの同定を可能にする。
【0009】
HLA−A0201は、最も一般的なHLA−Aハプロタイプである。
【0010】
疑いを除くために、用語HLAおよびMHCは本明細書で交換可能に用いられる。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】米国特許第5726288号
【非特許文献】
【0012】
【非特許文献1】Kastら(1989) Cell 59, 603-614
【非特許文献2】カワカミら(1994) Proc.Natl.Acad.Sci.USA91,6458-6462
【非特許文献3】Shimamotoら(1995)Blood 86,3173-3180
【非特許文献4】Menssenら(1995) Leukaemia 9,1060-1067
【非特許文献5】Inoueら(1997) Blood 89,1405-1412
【非特許文献6】Inoueら(1996) Blood 88, 2267-2278
【非特許文献7】Inoueら (1998) Blood 91, 2969-2976
【非特許文献8】Menssenら(1997) Int.J.Cancer 70, 518-523
【非特許文献9】Menssenら(1995) Leukemia 9, 1060-1067
【非特許文献10】Ogawaら(1998) Transplant 21,527-527
【非特許文献11】Rodeckら(1994) Int.J.Cancer 59, 78-82
【非特許文献12】Silbersteinら(1997) Proc.Natl.Acad.Sci.USA 94, 8132-8137
【非特許文献13】Tamakiら (1996) Blood 88, 4396-4398
【非特許文献14】Vielら(1994) Int.J.Cancer 57,515-521
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の第一の態様は、完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列RMFPNAPYLを含むペプチドまたはその部分または異型を提供する。
【課題を解決するための手段】
【0014】
本発明の第二の態様は、完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列CMTWNQMNLを含むペプチドまたはその部分または異型を提供する。
【0015】
本発明の第三の態様は、完全なヒトgata−1ポリペプチドではないという条件で、アミノ酸配列HLMPFPGPLLを含むペプチドまたはその部分または異型を提供する。
【0016】
“ペプチド”により、アミノ酸残基がペプチド結合(-CO-NH-)によって結合した分子のみでなく、ペプチド結合が転換された分子をも含める。かかるレトロ-インバーソ(retro-inverso)ペプチド模倣(peptidomimetics)は、当該技術分野において周知の方法、例えば、参照として含めるMeziereら(1997) J.Immunol.159,3230-3237に記載された方法を用いて調製することができる。かかるアプローチは、側鎖の配向ではなく、バックボーンを含む変化を含む偽ペプチドを調製することを含む。Meziereら(1997)は、少なくともMHCクラスIIおよびTヘルパー細胞応答に関して、これらの偽ペプチドが使用できることを示している。CO−NHペプチド結合の代わりにNH−CO結合を含むレトロ-インバースペプチドは、タンパク分解に対してはるかに耐性である。
【0017】
同様に、アミノ酸残基のCα原子間の空間を維持する適切なリンカー部が用いられるという条件で、ペプチド結合が省かれてもよく、リンカー部がペプチド結合の実質的に同一の平面と実質的に同一のチャージ分布とを有することが特に好ましい。
【0018】
ペプチドがそのN-またはC-末端において都合よくブロックされて、外タンパク分解(exoproteolytic)切断に対する感度を低減することが好ましい。
【0019】
所定のアミノ酸配列の“部分(portion)”により、ペプチドが所定のアミノ酸配列からなるペプチドと実質的に同様にHLA分子に結合できるような、所定の配列の少なくとも6つの連続するアミノ酸を意味する。
【0020】
所定のアミノ酸配列の“異型(variant)”により、例えば1または2つのアミノ酸残基の側鎖が、ペプチドが所定のアミノ酸配列からなるペプチドと実質的に同様にHLA分子に結合できるように、変更(例えば、別の天然に生じるアミノ酸残基の側鎖または他の側鎖で置換することによる)されていることを意味する。例えば、ペプチドは、改善しないにしても、HLA−A0201のような適切なMHC分子と相互作用して結合する能力を少なくとも維持するように、そして、改善しないにしても、本発明の第一、第二または第三の態様に定義したようなアミノ酸配列を含むポリペプチド(例えば、WT1またはgata−1)を異常に発現する細胞を認識して殺すことができる活性化CTLを産生する能力を少なくとも維持するように、修飾されてもよい。HLA−A2結合ノナマーの2位および9位は、通常、アンカー残基である。これらの残基およびHLA−A2を結合することに関わる他の残基の修飾は、CTL認識を変更することなく結合を増強させることができる(例えば、Tourdotら(1997) J.Immunol.159,2391-2398参照)。
【0021】
T細胞レセプターと相互作用するのに必須でないアミノ酸残基は、取り込みがT細胞反応性に実質的に影響を与えず、関連MHCへの結合を排除しない別のアミノ酸で置換することによって修飾することができる。
【0022】
かくして、所定の条件から離れて、本発明のペプチドは、上記のアミノ酸配列または部分または異型を含むあらゆるペプチド(この用語はオリゴペプチドまたはポリペプチドも含む)とすることができる。通常、本発明のペプチドは、抗原提示細胞において発現されると、適切なMHC分子に結合できるフラグメントが形成されるように処理され、適切な細胞によって提示されて、適切なT細胞応答を導き出すペプチドである。このペプチドから産生されたフラグメントも本発明のペプチドであると認められる。都合よく、本発明のペプチドは、所定のアミノ酸配列を含む部分またはその部分または異型、そしてさらに、一部の所望の特性を与えるさらなる部分を含む。例えば、前記さらなる部分とは、さらなるT細胞エピトープを含むか(最初のT細胞エピトープ含有部分と同じポリペプチドから誘導されてもそうでなくてもよい)、キャリアータンパクまたはペプチドを含むことができる。かくして、一つの実施態様において、本発明のペプチドは、ヒトWT−1部分がアミノ酸配列RMFPNAPYLまたはCMTWNQMNLまたはこれらの両方を含むという条件において、切断されたヒトWT−1タンパクまたはWT−1タンパクフラグメントと他のポリペプチド部分との融合タンパクである。別の実施態様では、本発明のペプチドは、ヒトgata−1部分がアミノ酸配列HLMPFPGPLLを含むという条件において、切断されたヒトgata−1タンパクまたはヒトgata−1タンパクフラグメントと他のポリペプチド部分との融合タンパクである。
【0023】
特に好ましい実施態様では、本発明のペプチドは、本発明の第一、第二または第三の態様に与えられたアミノ酸配列、並びに少なくとも一つのさらなるT細胞エピトープを含み、ここでさらなるT細胞エピトープは、WT−1またはgata−1を異常に発現する腫瘍のタイプに向けられたT細胞応答の産生を容易にすることができるものである。かくして、本発明のペプチドは、ワクチンとして用いられる、いわゆる“ビーズ・オン・ストリング(beads on a string)”ポリペプチドを含む。
【0024】
本発明のペプチドは、あらゆる大きさとすることができるが、通常は、分子量が100000未満、好ましくは50000未満、さらに好ましくは10000未満、そして通常は約5000とすることができる。アミノ酸残基の数としては、本発明のペプチドは1000残基未満、好ましくは500残基未満、さらに好ましくは100残基未満とすることができる。
【図面の簡単な説明】
【0025】
【図1】図1は、抗WT126−34CTLの殺傷活性を示す。T2はATCCからCatalogue No CRL 1992のもとに入手可能なペプチド結合欠陥を有するヒト細胞系統である。これらは、WT126−34ペプチド(RMFPNAPYL)(T2+WT12)、または、HLA−A2−結合ペプチド(T2+E7)であるE7コントロールペプチド(TLGIVCPI)を用いて結合される。K562は、白血病細胞系統であり、K562−A2はHLA−A0201でトランスフェクションされたK562白血病細胞系統である。E:T比は、エフェクター:ターゲット細胞比である。比溶解(%)は、参照としてここに含める、Sadovnikova & Stauss(1996) Proc.Natl.Acad.Sci.USA 93, 13114-13118に記載されているような、標準的なCTLアッセイで測定した。
【図2】図2は、抗huWT126−34CTLの殺傷活性を示す。Leuk-697、MV441およびBV173は白血病細胞系統である。P12はWT126−34ペプチド(RMFPNAPYL)を示す。
【図3】図3は、抗hug378−87CTLの殺傷活性を示す。hug378−87は、ペプチドHLMPFPGPLLであり、hug14はHLA−A2−結合コントロールペプチド(RLSPDLLTL)である。C1R−A2はヒトBリンパ球細胞系統である。K562−A2ヒトは、白血病細胞系統である。
【図4A】図4は、WT−1誘導ペプチドP126に対して生じたアロ制限CTLの特異性を示す。CTLを、P126ペプチドで被覆されたHLA−A0201+刺激細胞で刺激されたHLA−A0201−ドナーからTリンパ球バルク培養物の制限希釈クローニングによって単離した。(A)単離されたCTL系統は、免疫化P126ペプチドで被覆されたTAP欠陥T2ターゲット細胞を殺傷したが、HLA−A0201結合E7コントロールペプチドで被覆されたT2細胞を殺さなかった。
【図4B】(B)ペプチド滴定実験は、3つの抗P126CTL系統が低ピコモーラーのP126を認識する高い親和力であり、3つのCTL系統が低い親和力であったことを示す。これは、ナノモーラーP126濃度が、ターゲット細胞認識に必要であったからである。示された濃度のP126で被覆されたT2細胞は、CTLターゲットとして使用できた。低親和性CTLがWT1を内因的に発現するターゲット細胞を認識しなかったことから、高親和性CTLを、次の全ての実験に用いた。
【図4C】(C)抗親和性CTLは、HLA−A0201+白血病細胞系統BV173,697を殺傷するが、HLA−A0201+、EBV−トランスフォームBリンパ球細胞C1R−A2を殺傷しなかった。C1R−A2のP126でのコーティングは、効率的なCTL殺傷をもたらした。HLA−A0201-白血病細胞系統K562は、HLA−A0201遺伝子でトランスフェクションされない限りCTLによって殺傷されない。
【図5】白血病患者と正常ドナーから単離されたCD34+およびCD34-細胞集団および白血病細胞系統におけるタンパク発現とWT1 RNAを示す。(A)白血病細胞系統およびBリンパ球細胞系統C1R−A2におけるWT1 RNAを測定するRT−PCR。図4CのCTLターゲットと同じ細胞系統を用いた。増幅されたWT1産物は482bpであり、ハウスキーピングABL遺伝子のRNAを増幅して、各サンプルのRNAの量を示した。ABL産物は385bpの長さである。(B)4人のCML患者および3人の正常ドナー由来の、精製されたCD34+とCD34−細胞集団におけるWT1 RNA発現を測定するRT−PCR。白血病細胞系統BV173は、WT1発現の陽性コントロールとして提供された。さらに6人のCML患者からのサンプルを用いて類似の結果が得られた。(C)2人のCML患者および2人の正常ドナー由来の精製されたCD34+およびCD34−細胞集団および白血病細胞系統におけるWT1タンパク発現を測定するウェスタンブロッティング。ハウスキーピングアクチンタンパクの発現は、各サンプル中に存在するタンパクの量を制御するためのインジケーターとして用いられた。WT1タンパクは約54kDaの大きさであり、アクチンタンパクは約42kDaであった。
【図6A】図6は、白血病患者および正常ドナーから精製されたCD34+細胞集団のコロニー形成の阻害およびCTL媒介殺傷の分析である。(A)HLA−A0201+またはA0201-であるCML患者から単離された精製CD34+およびCD34-細胞集団に対する抗P126CTLによる殺傷レベルを示す代表的な実験である。CD34-/A0201+細胞は、P126ペプチドでコートされない限りCTLに認識されなかった。P126またはコントロールE7ペプチドで被覆されたTAP欠陥T2細胞および白血病細胞系統BV173は、全ての実験で陽性および陰性のコントロールとして用いられた。
【図6B】(B)11人の異なるHLA−A0201+CML患者と6人の正常ドナーに由来する精製CD34+細胞の特異的CTL殺傷レベルの平均。陽性コントロール細胞BV173に対する、CML患者から精製されたCD34-細胞の殺傷レベルも示されている。この図は、特異的CTL殺傷の平均値と標準偏差を示す。
【図6C】(C)HLA−A0201+正常ドナーから単離された精製CD34+とCD34−細胞集団の抗P126CTLによる殺傷レベルを示す代表例である。CTLは、ターゲット細胞がP126ペプチドで被覆されない限り検出されなかった。
【図6D】(D)エフェクター/ターゲット細胞比10:1においてCTLで4時間共培養された精製CD34+細胞によるコロニー形成のCTL媒介阻害。未処理コントロールCD34+細胞を、CTLを含まずに同じ条件下で培養した。CTL処理および未処理コントロール細胞をメチルセルロース中におき、14日後にCFU−GMの数をカウントした。未処理コントロール中に観察されたCFU−GMを100%基準として用いてCTL処理後のCFU−GMのパーセントを示す。この図は、9人の異なるHLA−A0201+CML患者、7人の異なる正常ドナーに由来するCD34+、および5人の異なるHLA−A0201-CML患者に由来するCD34+細胞を用いた、独立した実験の平均と標準偏差を示す。
【図6E】(E)高親和性P126特異的CTL(ライン81)または低親和性CTL(ライン85)で、プレーティングの前に、4時間処理されたまたは未処理のA0201+/CD34+CML細胞によるコロニー形成。未処理コントロールに観察されたコロニーの数を100%の基準として用いてCFU−GMおよびBFU−Eを示す。
【図6F】(F)コールドターゲット競合実験。30倍過剰のコールドBV173およびC1R−A2ターゲットの存在下または不在下で、A0201+CML患者に由来するクロムラベルCD34+ターゲットに対する抗P126CTLによる殺傷を示す。クロムラベルBV173およびC1R−A2の殺傷は、比較のために示されている。
【発明を実施するための形態】
【0026】
以下から、一部の適用において、本発明のペプチドが直接的に用いられることがわかる(すなわち、患者の細胞または患者に与えられた細胞においてポリヌクレオチドの発現により産生されるものではない);そのような適用においては、ペプチドが100残基未満を有することが好ましい。
【0027】
本発明のポリペプチドが、HLA−A0201に結合できることが好ましい;しなしながら、このペプチドは他のHLAタイプおよびHLA−A0201に結合してもよい。特に、ペプチドがHLA−A0201に選択的に結合できることが好ましい。
【0028】
本発明のペプチドは、特に、WT1ポリペプチド(Wilms腫瘍遺伝子産物)またはgata−1ポリペプチドを異常に発現する細胞を標的とし殺す免疫治療方法において特に使用できる。gata−1ポリペプチドは、一部の遺伝子のプロモーター領域に存在するgata−ボックスに結合する転写因子であることから、そのように命名された。それはまた、NF−e1、GF−1およびEryf−1とも呼ばれる。アミノ酸配列RMFPNAPYLおよびCMTWNQMNLは、それぞれWT1の126−134および235−243残基に見られ、アミノ酸配列HLMPFPGPLLは、gata−1の378−387残基に見られる。WT−1アミノ酸配列は、Gesslerら(1990) Nature 343, 774-778に与えられており、gata−1アミノ酸配列は、Zonら(1990) Proc.Natl.Acad.Sci.USA 87, 668-672に与えられており、これらの刊行物の両方を参照としてここに含める。
【0029】
所定のアミノ酸配列からなるこれらの特異的ペプチドは、HLA−A0201に結合するので、本発明のペプチドはHLA−A0201に結合し、結合した際に、HLA−A0201−ペプチド複合体が、適切な抗原提示細胞の表面に提示された際に、本発明の第一および第二の態様のペプチドに関してWT1ポリペプチドのような、そして、本発明の第三の態様のペプチドに関してgata−1ポリペプチドのような、所定のアミノ酸配列を含むポリペプチドを異常に発現する細胞を認識するCTLの産生を引き出すことができるペプチドであることが好ましい。
【0030】
WT1ポリペプチドは、白血病、乳ガン、メラノーマおよび卵巣ガンにおいて異常に発現され、gata−1ポリペプチドは白血病において異常に発現される。
【0031】
“異常に発現”により、ポリペプチドが発現の通常レベルと比較して過剰に発現されること、もしくは、腫瘍においては発現されるが、腫瘍が誘導される組織において遺伝子がサイレントであることを含む。“過剰発現”により、ポリペプチドが、正常細胞に存在するレベルの少なくとも1.2倍、好ましくは少なくとも2倍、さらに好ましくは少なくとも5倍または10倍のレベルで存在することを意味する。
【0032】
HLA分子に結合するペプチドの最適な長さが約8ないし12アミノ酸、好ましくは9アミノ酸であることは周知である。
【0033】
本発明の特に好ましいペプチドは、アミノ酸配列RMFPNAPYLまたはCMTWNQMNLまたはHLMPFPGPLLからなるものである。
【0034】
12アミノ酸残基より大きいペプチドがMHC分子との結合に直接用いられる場合には、コアHLA結合領域に隣接する残基が、ペプチドがMHC分子に結合する能力またはCTLにそのペプチドを提示する能力に実質的に影響を与えないものであることが好ましい。しかしながら、特にポリヌクレオチドによってコードされる場合に、より大きなペプチドが用いられることがわかる。なぜなら、これらの大きなペプチドは、適切な抗原提示細胞によって断片化されうるからである。
【0035】
ペプチド(少なくとも、アミノ酸残基間にペプチド結合を含むもの)は、Luら(1981) J.Org.Chem.46,3433とそこに記載された参考文献に記載されている固相ペプチド合成のFmoc-ポリアミドモード(Fmoc-polyamide mode)によって合成されてもよい。一時的Nアミノ基保護は、9−フルオレニルメチルオキシカルボニル(Fmoc)基によって供給される。この高度にベース-置換活性保護基の反復切断は、N,N-ジメチルホルムアミド中の20%ピペリジンを用いて実行される。側鎖官能性は、そのブチルエーテル(セリン、スレオニンおよびチロシンの場合)、ブチルエステル(グルタミン酸およびアスパラギン酸の場合)、ブチルオキシカルボニル誘導体(リシンおよびヒスチジンの場合)、トリチル誘導体(システインの場合)および4-メトキシ-2,3,6-トリメチルベンゼンスルホニル誘導体(アルギニンの場合)として保護することができる。グルタミンまたはアスパラギンがC末端残基である場合には、側鎖アミド官能性の保護には4,4'-ジメトキシベンズヒドリル基を使用する。固相支持体は、三つのモノマーであるジメチルアクリルアミド(骨格モノマー)、ビスアクリロイルエチレンジアミン(架橋剤)およびアクリロイルサルコシンメチルエステル(官能化剤)から構成されたポリジメチル−アクリルアミドポリマーをベースとする。使用されたペプチド−樹脂切断可能連結剤は、酸不安定性4-ヒドロキシメチル-フェノキシ酢酸誘導体である。全てのアミノ酸誘導体は、アスパラギンとグルタミンを除いて、予め形成された対称的な無水物誘導体として添加され、これらは、リバースN,N-ジシクロヘキシル-カルボジイミド/1-ヒドロキシベンゾトリアゾル媒介カップリング処理を用いて加えられる。全てのカップリングおよび脱保護化反応は、ニンヒドリン、トリニトロベンゼンスルホン酸、またはイソチン(isotin)テスト処理を用いて観察される。合成の完了時に、50%のスカベンジャーミックスを含む95%のトリフルオロ酢酸で処理することにより、側鎖保護基の付随した除去とともに、ペプチドを樹脂支持体から切断する。一般的に用いられるスカベンジャーは、エタンジチオール、フェノール、アニソールおよび水であり、その的確な選択は合成されるペプチドの構成アミノ酸に基づく。ト
リフルオロ酢酸は、真空で蒸発させることによって除去され、その後のジエチルエーテルを用いた摩砕が粗製ペプチドを与える。存在する全てのスカベンジャーを単純な抽出方法で除去する。水相の凍結乾燥により、スカベンジャーを含まない粗製ペプチドを与える。ペプチド合成のための試薬は、一般的にCalbiochem-Novabiochem(UK)Ltd,Nottingham NG7 2QJ, UKから入手できる。精製は、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィーおよび、(主に)逆相高速液体クロマトグラフィーのような技術のいずれか一つまたは組み合わせにより行うことができる。ペプチドの分析は、薄相クロマトグラフィー、逆相高速液体クロマトグラフィー、酸加水分解後のアミノ酸分析および高速原子衝撃(FAB)質量分析を用いて行うことができる。
【0036】
本発明のさらなる態様は、本発明の第一、第二または第三の態様に定義されたペプチドをコードするポリヌクレオチドを提供する。このポリヌクレオチドはDNAまたはRNAとすることができ、前記ペプチドをコードする限りイントロンを含んでも含まなくてもよい。もちろん、それは、ポリヌクレオチドにコードされうる、天然に生じるペプチド結合によって結合された天然に生じるアミノ酸残基を含むペプチドのみである。
【0037】
本発明のさらなる態様は、本発明の第一または第二または第三の態様にかかるポリペプチドを発現することができる発現ベクターを提供する。
【0038】
例えば相補付着末端を介して、ベクターに、DNAのようなポリヌクレオチドを結合するために、種々の方法が開発されている。例えば、相補的ホモポリマー領域を、ベクターDNAに挿入されるDNAセグメントに付加することができる。次いで、ベクターおよびDNAセグメントが、相補ホモポリメリックテイル間の水素結合によって結合され、組み換えDNA分子を形成する。
【0039】
一以上の制限部位を有する合成リンカーは、DNAセグメントをベクターに結合させる別の方法を提供する。上記のエンドヌクレアーゼ制限切断によって生じたDNAセグメントを、バクテリオファージT4 DNAポリメラーゼまたはE.coli DNAポリメラーゼIで処理する。これらの酵素は、突出した3’一本鎖末端を3’-5’エキソヌクレアーゼ活性で除去し、欠けた3’端をポリメラーゼ活性で満たす。
【0040】
これらの活性の組み合わせにより、平滑末端のDNAセグメントを形成することができる。この平滑末端セグメントを、バクテリオファージT4DNAリガーゼのような、平滑末端DNA分子のライゲーションを触媒することができる酵素の存在下で大過剰のリンカー分子と共にインキュベートする。かくして、この反応産物は、端部にポリメリックリンカー配列を備えたDNAセグメントである。これらのDNAセグメントは、適切な制限酵素で切断され、DNAセグメントの端部と適合する端部を生じる酵素で切断された発現ベクターにライゲートされる。
【0041】
種々の制限エンドヌクレアーゼ部位を含む合成リンカーは、International Biotechnologies Inc,New Haven, CN,USAを含む多数の源から商業的に入手可能である。
【0042】
本発明のポリペプチドをコードするDNAを修飾するのに望ましい方法は、Saikiら(1988) Science 239,487-491に記載されたポリメラーゼ連鎖反応を用いることである。この方法は、例えば、適切な制限部位において工作することにより、適切なベクターにDNAを導入するために用いることができ、また、当該技術分野において公知であるような他の使用できる方法でDNAを修飾するために用いることができる。
【0043】
この方法では、酵素により増幅されるDNAは、増幅されたDNAにそれ自身が取り込まれる二つの特異的プライマーによって隣接される。この特異的プライマーは、当該技術分野において公知の方法を用いて発現ベクターにクローニングするために用いられる制限エンドヌクレアーゼ認識部位を含む。
【0044】
DNA(または、レトロウイルスベクターの場合にはRNA)を、本発明の化合物を含むポリペプチドを産生する適切な宿主において発現させる。かくして、本発明の化合物を構成するポリペプチドをコードするDNAが、ここに含まれる教示から適切に変更された公知の技術に従って、発現ベクターを構成するために用いられてもよい。この発現ベクターは、本発明のポリペプチドの発現および産生に適した宿主細胞をトランスフォームするために用いられる。かかる技術は、Rutterらに1984年4月3日に発せられた米国特許第4440859号、Weissmanに1985年7月23日に発せられた第4530901号、Crowlに1986年4月15日に発せられた第4582800号、Markらに1987年6月30日に発せられた第4677063号、Goeddelに1987年7月7日に発せられた第4678751号、Itakuraらに1987年11月3日に発せられた第4704362号、Murrayに1987年12月1日に発せられた第4710463号、Toole,Jr.らに1988年7月12日に発せられた第4757006号、Goeddelらに1988年8月23日に発せられた第4766075号、およびStalkerに1989年3月7日に発せられた第4810648号に開示された技術を含み、これらの全てを参照としてここに含める。
【0045】
本発明の化合物を構成するポリペプチドをコードするDNA(レトロウイルスベクターの場合はRNA)が、適切な宿主に導入するために広範囲の他のDNA配列に結合されてもよい。コンパニオンDNAは、宿主の性質、宿主へのDNAの導入方法、およびエピソームメンテナンスまたはインテグレーションが望ましいかどうかに依存する。
【0046】
一般に、DNAは、プラスミドのような発現ベクターに、発現に適切な配向および正しい読み枠で挿入される。必要であれば、DNAは、所望の宿主によって認識される適切な転写および翻訳制御コントロールヌクレオチド配列に結合されてもよいが、かかるコントロールは一般的に発現ベクターにおいて利用可能である。次いで、このベクターを、通常の技術を介して宿主に導入する。一般的に、全ての宿主がベクターによりトランスフォームされるわけではない。それゆえ、トランスフォームされた宿主細胞を選別する必要がある。選別技術の一つは、必要なコントロール成分と共に、抗生物質耐性のような、トランスフォームされた細胞において選別可能な特徴をコードするDNA配列を発現ベクターに取り込むことを含む。あるいは、かかる選別可能な性質の遺伝子が、所望の宿主細胞をコトランスフォームするために用いられる別のベクターに存在してもよい。
【0047】
本発明の組み換えDNAによりトランスフォームされた宿主細胞を、ここに開示した技術の観点から当業者に公知の適切な条件下および十分な時間にわたって培養して、ポリペプチドを発現させ、次いで回収する。
【0048】
細菌(例えば、E.coliおよびBacillus subtilis)酵母(例えば、Saccharomyces cerevisiae)、糸状菌類(例えば、Aspergillus)、植物細胞、動物細胞および昆虫細胞を含む、多くの発現システムが知られている。
【0049】
通常、ベクターは、原核生物における増殖のために、そして、他の非原核細胞タイプにおける発現のために用いられる場合でさえも、ColE1 oriのような原核生物のレプリコンを含む。ベクターは、形質転換された、E.coliのような細菌性宿主細胞において遺伝子を発現(転写および翻訳)させることができる原核プロモーターのような適切なプロモーターを含むこともできる。
【0050】
プロモーターとは、RNAポリメラーゼを結合させ、転写を開始させるDNA配列によって形成された発現コントロール成分である。典型的な細菌性宿主と適合するプロモーター配列は、本発明のDNAセグメントの挿入に都合のよい制限部位を含むプラスミドベクターに設けられる。
【0051】
通常の原核生物のベクタープラスミドは、Biorad Laboratories(Richmond,CA,USA)から入手可能なpUC18、pUC19、pBR322およびpBR329と、Pharmacia, Piscataway,NJ,USAから入手可能なpTrc99AおよびpKK223−3である。
【0052】
通常の哺乳動物細胞ベクタープラスミドは、Pharmacia,Piscataway,NJ,USAから入手可能なpSVLである。このベクターは、クローン化遺伝子の発現を誘導するためにSV40後期プロモーターを用い、最高レベルの発現が、COS-1細胞のようなT抗原産生細胞に見られる。
【0053】
誘導できる哺乳動物発現ベクターの例はpMSGであり、これもPharmaciaから入手できる。このベクターは、クローン化遺伝子の発現を引き起こすために、マウス乳房腫瘍ウイルス長期リピートのグルココルチコイド誘導プロモーターを用いる。
【0054】
使用できる酵母プラスミドベクターは、pRS403−406およびpRS413−416であり、一般的に、Stratagene Cloning Systems, La Jolla, CA 92037, USAから入手できる。プラスミドpRS403、pRS404、pRS405、およびpRS406は、酵母統合プラスミド(YIp)であり、酵母選択マーカーHIS3、TRP1、LEU2およびURA3を取り込む。プラスミドpRS413−416は酵母セントロメアプラスミド(Ycp)である。
【0055】
他のベクターおよび発現システムは、種々の宿主細胞を用いた使用について当該技術分野において周知である。
【0056】
また、本発明は、本発明のポリヌクレオチドベクター構成物でトランスフォームされた宿主細胞に関する。宿主細胞は原核生物または真核生物とすることができる。細菌細胞は、一部の状況では原核生物宿主細胞が好ましく、通常は、E.coliの株、例えば、Bethesda Research Laboratories Inc.,Bethesda,MD,USAから入手可能なE.coli株DH5およびRockville,MD,USAのAmerican Type Culture Collection(ATCC)から入手できるRR1(No ATCC 31343)である。好ましい真核生物宿主細胞は、酵母、昆虫および哺乳動物細胞、好ましくは、マウス、ラット、サルまたはヒトの繊維芽細胞および腎臓細胞系統のような脊椎動物細胞を含む。酵母宿主細胞は、Stratagene Cloning Systems, La Jolla, CA 92037,USAから入手できるYPH499、YPH500およびYPH501を含む。好ましい哺乳動物宿主細胞は、CCL61としてATCCから入手可能なチャイニーズハムスター卵巣(CHO)細胞、CRL1658としてATCCから入手可能なNIH Swissマウス胚細胞NIH/3T3、CRL1650としてATCCから入手可能なサル腎臓誘導COS−1細胞、および、ヒト胚腎臓細胞である293細胞を含む。好ましい昆虫細胞は、バキュロウイルス発現ベクターでトランスフェクションされうるSf9細胞である。
【0057】
本発明のDNA構成物を用いた適切な細胞宿主のトランスフォーメーションは、用いられるベクターのタイプに依存する周知の方法によって行われる。原核生物宿主細胞のトランスフォーメーションに関しては、例えば、Cohenら(1972)Proc.Natl.Acad.Sci.USA69,2110およびSambrookら(1989)Molecular Cloning,A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor,NYを参照。酵母細胞のトランスフォーメーションは、Shermanら(1986)Methods In Yeast Genetics, A Laboratory Manual, Cold Spring Harbor, NYに記載されている。Beggs(1978)Nature 275,104-109の方法も使用できる。脊椎動物細胞に関して、かかる細胞をトランスフェクションするのに使用できる試薬、例えば、リン酸カルシウムおよびDEAE-デキストランまたはリポソームフォーミュレーションは、Stratagene Cloning SystemsまたはLife Technologies Inc.,Gaithersburg,MD 20877, USAから入手できる。
【0058】
エレクトロポレーションも、細胞をトランスフォームおよび/またはトランスフェクトするのに使用でき、酵母細胞、細菌細胞、昆虫細胞および脊椎動物細胞をトランスフォームするのに、当該技術分野において周知である。
【0059】
例えば、多くの細菌種が、参照としてここに含めるLuchanskyら(1988)Mol.Microbiol.2,637-646に記載された方法によりトランスフォームされる。最大数のトランスフォーマントは、一貫して、25μFDにおいてcm当たり6250Vを用いて2.5X PEBに懸濁されたDNA-細胞混合物のエレクトロポレーション後に回収される。
【0060】
エレクトロポレーションによる酵母のトランスフォーメーション方法は、Becker&Guarente (1990) Metholds Enzymol.194,182に記載されている。
【0061】
首尾よくトランスフォームした細胞、すなわち本発明のDNA構成物を含む細胞は、周知の技術により同定することができる。例えば、本発明の発現構成物の導入から得られた細胞は、増殖して本発明のポリペプチドを生じる。細胞を回収して溶解し、そのDNA内容物を、Southern(1975)J.Mol.Biol.98,503またはBerentら(1985)Biotech.3,208に記載されている方法を用いてDNAの存在について試験することができる。あるいは、上清におけるタンパクの存在を、以下の抗体を用いて検出することができる。
【0062】
組み換えDNAの存在を直接アッセイすることに加えて、組み換えDNAがタンパクの発現に向けられる場合には、トランスフォーメーションの成功を、周知の免疫学的方法によって確認できる。例えば、発現ベクターで首尾よくトランスフォームした細胞は、適切な抗原性を示すタンパクを産生する。トランスフォームされたと思われる細胞のサンプルを、回収し、適切な抗体を用いてタンパクをアッセイする。
【0063】
かくして、トランスフォームされた宿主細胞自身の他に、本発明は、これらの細胞の培養物、好ましくはモノクローナル(クローンとして均質)培養物、または栄養培地におけるモノクローナル培養から誘導された培養物をも含む。
【0064】
本発明の一部の宿主細胞、例えば、細菌、酵母および昆虫の細胞が、本発明のペプチドの調製に使用できることも考えられる。しかしながら、他の宿主細胞は、一部の治療方法に使用できる。例えば、抗原提示細胞、例えば樹状細胞は、本発明のペプチドを発現し、適切なMHC分子に結合するために使用できる。
【0065】
本発明のさらなる態様は、本発明の第一、第二または第三の態様のペプチドを産生する方法を提供することであり、この方法は、ペプチドをコードするポリヌクレオチドまたは発現ベクターを含む宿主細胞を培養し、宿主細胞または培養培地からペプチドを得ることを含む。
【0066】
本発明のさらなる態様は、薬学的に許容できるキャリアーと、本発明の第一、第二または第三の態様にかかるペプチドまたはかかるペプチドをコードするポリヌクレオチドまたは発現ベクターを含む薬学的組成物を提供する。この薬学的製剤は、患者への投与に適した形態に調製され、無菌でありかつ発熱物質を含まない。本発明のさらなる態様は、本発明の第一、第二または第三の態様にかかるペプチド、またはかかるペプチドをコードするポリヌクレオチドまたは発現ベクターを、薬剤における使用のために提供する。このペプチドまたはポリヌクレオチドまたは発現ベクターは、収容されて、薬剤における使用のために提供される。
【0067】
薬学的組成物、または収容および提示は、適切な形態とすることができる。適切な形態が静脈(i.v.)注射、皮下(s.c.)注射、皮内(i.d.)注射、腹腔内(i.p.)注射、筋肉内(i.m.)注射用であることが考えられる。
【0068】
ペプチド注射の好ましい方法は、s.c.、i.d.、i.p.、i.m.そしてi.v.である。
【0069】
DNA注射の好ましい方法は、i.d.、i.m.、s.c.、i.p.そしてi.v.である。
【0070】
1ないし500mgのペプチドまたはDNAの投与量を与えることができる。
【0071】
本発明のさらなる態様は、本発明の第一、第二または第三態様のいずれかのアミノ酸配列を含むポリペプチドを異常に発現する、患者の標的細胞を殺す方法を提供するものであって、この方法は、患者に、有効量の、本発明の第一、第二または第三態様のいずれかにかかるペプチドを投与すること、あるいは、前記ペプチドをコードする発現ベクターまたはポリヌクレオチドの有効量を投与することを含み、前記ペプチドの量またはポリヌクレオチドまたは発現ベクターの量は、前記患者の抗ターゲット細胞免疫応答を誘発するのに有効な量である。
【0072】
ターゲット細胞は、通常、腫瘍またはガン細胞である。通常、腫瘍またはガン細胞は、WT1またはgata−1を異常に発現する細胞である。
【0073】
ペプチドまたはペプチドをコードする核酸は、腫瘍またはガンワクチンを構成する。患者に直接、影響を受けた器官または全身的に投与されてもよく、患者またはヒト細胞系統から誘導された細胞にex vivoで適用され、その後患者に投与されてもよく、また、患者から誘導された免疫細胞から部分集団を選別するためにin vitroで用いられ、次いで患者に再投与されてもよい。核酸がin vitroで細胞に投与される場合には、細胞が、インターロイキン-2のような免疫刺激サイトカインを共発現するようにトランスフェクションされることが有益である。ペプチドは実質的に純粋であっても、Detoxのような免疫刺激アジュバントと組み合わされてもよく、免疫刺激サイトカインと組み合わせて用いられてもよく、また、リポソームのような適切なデリバリーシステムと共に投与されてもよい。また、ペプチドは、キーホールリンペットヘモシアニン(KLH)またはマンナンのような適切なキャリアーと組み合わされてもよい(WO95/18145およびLongeneckerら(1993)Ann.NY Acad.Sci.690,276-291参照)。また、このペプチドは、ダグが付されるか、融合タンパクとされるか、ハイブリッド分子とされてもよい。本発明の第一、第二または第三態様にその配列が与えられたペプチドは、CD8 CTLを刺激することが期待される。しかしながら、刺激はCD4T細胞によって提供される助けがあるとさらに効率的である。かくして、ハイブリッド分子の融合パートナーまたはセクションは、CD4T細胞を刺激するエピトープを適切に提供する。CD4刺激エピトープは、当該技術分野において周知であり、破傷風トキソイドに同定されるものを含む。ポリヌクレオチドは、実質的に純粋であっても、適切なベクターまたはデリバリーシステムに含まれてもよい。適切なベクターおよびデリバリーシステムは、アデノウイルス、ワクシニアウイルス、レトロウイルス、ヘルペスウイルス、アデノ関連ウイルス、または一以上のウイルスの成分を含むハイブリッドに基づくシステムのようなウイルス性のものを含む。非ウイルス性デリバリーシステムは、DNAデリバリーの分野で周知であるようなカチオン脂質およびカチオンポリマーを含む。物理的デリバリー、例えば、“遺伝子銃”を介するものも使用できる。ペプチドま
たは核酸によってコードされるペプチドは、例えば、CD4T細胞を刺激する破傷風トキソイドのエピトープとの融合タンパクであってもよい。
【0074】
ガンワクチンに使用されるペプチドは、適切なペプチドであってもよい。特に、適切な9マーのペプチド、または適切な7マーまたは8マーまたは10マーのペプチドとすることができる。また、より長いペプチドも適しているが、9マーまたは10マーのペプチドが好ましい。ガンワクチンが、WT1を発現するガンに関して用いられる場合には、本発明の第一および第二の態様の両方のペプチドを用いる、あるいは、本発明の第一および第二の態様に記載された両方の配列を含む一つのペプチドを用いることが有利である。
【0075】
患者に投与される核酸が無菌かつ発熱性物質を含まないことが適切である。裸のDNAは、筋肉内、皮内または皮下に投与できる。ペプチドは、筋肉内、皮内または皮下に投与できる。
【0076】
ワクチン接種は、プロフェッショナル抗原提示細胞により刺激されたCTL応答をもたらし、CTLが刺激されると、腫瘍細胞におけるMHC発現を増強する利点が存在する。
【0077】
注入部位、ターゲッティングベクターおよびデリバリーシステムの使用、または、患者からかかる細胞集団を選択的に精製してペプチドまたは核酸をex vivo投与することのいずれかによって、ワクチンを、例えば抗原提示細胞のような特異的細胞集団に向けることも有益である(例えば、樹状細胞をZhouら(1995) Blood86,3295-3301;Rothら(1996)Scand.J.Immunology 43,646-651に記載されているように選別してもよい)。例えば、ダーゲッティングベクターは、適切な場所において抗原を発現させる組織-または腫瘍-特異的プロモーターを含んでもよい。
【0078】
治療が施される患者は、WT1またはgata−1の過剰発現または異常発現(両方とも異常な発現)のタイプの腫瘍を有しうる。
【0079】
それゆえ、本発明のさらなる態様は、ガン、またはガンまたは腫瘍細胞に有効なワクチンを提供することであり、これは、本発明の第一、第二または第三の態様のいずれか一つにかかるペプチドの有効量を含むか、かかるペプチドをコードする核酸を含む。
【0080】
ワクチンが核酸ワクチンであることが特に好ましい。あるポリペプチドをコードするDNAワクチンのような核酸ワクチンを用いた接種が、T細胞反応を導くことが知られている。
【0081】
都合よく、核酸ワクチンは、適切な核酸デリバリー手段を含んでもよい。核酸、好ましくはDNAは、裸であってもよく(すなわち、投与される他の成分を実質的に含まない)、また、リポソーム中またはウイルスベクターデリバリーシステムの一部としてデリバリーされてもよい。
【0082】
樹状細胞による核酸の取り込みおよびコードされたポリペプチドの発現が、免疫応答の開示のメカニズムであってもよい;しかしながら、樹状細胞はトランスフェクションされなくてもよいが、組織のトランスフェクションされた細胞から発現されたペプチドをピックアップするかもしれないので、重要である。
【0083】
DNAワクチンのようなワクチンを筋肉内に投与することが好ましい。また、皮膚にワクチンが投与されることも好ましい。
【0084】
核酸ワクチンはアジュバントを含まずに投与されてもよい。また、核酸ワクチンは、BCGまたはミョウバンのようなアジュバントと共に投与されてもよい。他の適切なアジュバントは、サポニン、マイコバクテリウム抽出物、および合成細菌性細胞壁模造体から誘導されるAquila QS21スティミュロン(stimulon)(Aquila Biotech,Worcester,MA,USA)およびRibi's Detoxのようなプロプリエトリー(proprietory)アジュバントを含む。別のサポニン誘導アジュバントであるQuil Aも使用できる(Superfos,Denmark)。核酸ワクチンがアジュバントなしで投与されることが好ましい。
【0085】
Freund'sのような別のアジュバントも使用できる。また、好ましくはアジュバントと共に、キーホールリンペットヘモシアニンと複合体形成したペプチドを得ることも有益である。
【0086】
ガンのポリヌクレオチド媒介免疫治療は、Conryら(1996)Seminars in Oncology 23,135-147; Condonら (1996) Nature Medicine 2,1122-1127; Gongら(1997)Nature Medicine 3,558-561; Zhaiら(1996) J.Immunol.156,700-710; Grahamら(1996)Int J. Cancer 65,664-670;およびBurchellら(1996)pp 309-313 In:Breast Cancer, Advances in biology and therapeutics, Calvoら(eds),Jhon Libbey Eurotextに記載されており、これら全てが参照としてここに含められる。
【0087】
本発明のさらなる態様は、本発明の第一、第二または第三の態様にかかるペプチド、もしくはかかるペプチドをコードするポリヌクレオチドまたは発現ベクターの、本発明の第一、第二または第三の態様に定義されたアミノ酸配列を含むポリペプチドを異常に発現する患者のターゲット細胞を殺すための薬剤の製造における使用を提供するものである。
【0088】
かくして、薬剤は、WT1またはgata−1を異常に発現するガンを治療するために使用できる。
【0089】
本発明のさらなる態様は、in vitroにおいて活性化細胞毒性Tリンパ球(CTL)を産生する方法であり、この方法は、in vitroにおいて、前記CTLを抗原特異的に活性化するのに十分な時間、適切な抗原提示細胞の表面に発現された抗原-結合ヒトクラスI MHC分子とCTLとを接触させることを含み、ここで、前記抗原は、本発明の第一、第二または第三の態様にかかるペプチドである。
【0090】
CTLはCD8+細胞であるが、CD4+細胞であってもよい。MHCクラスI分子は、適切な細胞の表面に発現され、その細胞がMHCクラスI分子を自然に発現しないもの(この場合、かかる分子を発現するように細胞がトランスフェクションされる)であるか、抗原処理または抗原提示経路に欠陥を有するものが好ましい。このように、MHCクラスI分子を発現する細胞は、CTLを活性化する前に選択されたペプチド抗原で実質的に完全に開始されうる。
【0091】
抗原提示細胞(または刺激細胞)は、通常、その表面にMHCクラスI分子を有し、好ましくは、自然には、選択された抗原をMHCクラスI分子に結合することができない。以下に詳細に記載するように、MHCクラスI分子は、in vitroにおいて選択された抗原と容易に結合されてもよい。
【0092】
前記抗原提示細胞は、少なくとも一部の前記選択された分子がペプチドである場合、前記MHCクラスI分子に結合されないような、ペプチドトランスポーターの発現に欠陥のある哺乳動物細胞であると都合がよい。
【0093】
好ましくは、哺乳動物細胞が、TAPペプチドトランスポーターを欠失しているか、低下したレベル、または低下した機能で有する。TAPペプチドトランスポーターを欠失した適切な細胞は、T2、RMA-Sおよびショウジョウバエの細胞を含む。TAPとは、抗原処理に関連したトランスポーター(Transporter Associated with antigen Processing)である。
【0094】
かくして、細胞がショウジョウバエのような昆虫細胞であると都合がよい。
【0095】
ヒトペプチド結合欠陥細胞系統T2は、American Type Culture Collection, 12301 Parklawn Drive, Rockville, Maryland 20852, USAからCatalogue No CRL 1992のもとに入手でき、ショウジョウバエ細胞系統Schneider系統2は、ATCCからCatalogue No CRL 19863のもとに入手でき、マウスRMA−S細胞系統は、KarreとLjunggren (1985)J.Exp.Med.162,1745に記載されており、参照としてここに含める。
【0096】
好ましい実施態様では、刺激細胞は、MHCクラスI分子を発現することができる核酸分子でトランスフェクションされた宿主細胞(例えば、T2、RMA−Sまたはショウジョウバエ細胞)である。T2およびRMA−S細胞は、HLAクラスI分子をトランスフェクション前に発現するが、ペプチドと結合しない。
【0097】
哺乳動物細胞は、当該技術分野において周知の方法によってトランスフェクションされうる。ショウジョウバエ細胞は、Jacksonら(1992) proc.Natl.Acad.Sci.USA 89, 12117に記載されているようにトランスフェクションすることができ、参照としてここに含める。
【0098】
好ましくは、トランスフェクション前に前記宿主細胞は、実質的にMHCクラスI分子を全く発現しない。
【0099】
刺激細胞が、B7.1、B7.2、ICAM−1およびLFA3のいずれかのようなT細胞共刺激に重要な分子を発現することも好ましい。
【0100】
多数のMHCクラスI分子、および共刺激分子の核酸配列は、GenBankおよびEMBLデータベースから公的に入手可能である。
【0101】
前記刺激細胞の表面に発現された実質的に全てのMHCクラスI分子が、同じタイプであることが特に好ましい。
【0102】
ヒトのHLAクラスIおよび他の動物における等価物は、遺伝的に非常に複雑である。例えば、HLA−B座の少なくとも110のアレルと、HLA−A座の少なくとも90のアレルが存在する。HLAクラスI(または等価)分子は本発明の態様において使用できるが、ヒトの集団において適度に高頻度で生じるHLAクラスI分子において少なくとも一部の選択された分子を示すことが好ましい。HLAクラスIアレルの頻度が、カフカス人、アフリカ人、中国人等の異なる民族のグループ間で異なることが周知である。少なくともカフカス人に関する限り、HLAクラスI分子がHLA−A2アレル、HLA−A1アレル、HLA−A3アレル、またはHLA−B27アレルにコードされる。HLA−A2が特に好ましい。
【0103】
さらなる態様において、HLA分子の組み合わせも用いることができる。例えば、HLA−A2とHLA−A3との組み合わせは、カフカス人の74%をカバーする。
【0104】
多重CD8 CTLエピトープのデリバリーのための組み換えポリエピトープワクチンの使用は、Thomsonら(1996) J.Immunol.157, 822-826およびWO96/03144に記載されており、これらの両方を参照としてここに含める。本発明に関して、一つのワクチンにペプチド(またはペプチドをコードする核酸)を含むことが望ましく、ここでペプチドは、任意の順で、アミノ酸配列RMFPNAPYL、CMTWNQMNL、HLMPFPGPLLおよびCD4 T細胞刺激エピトープ(例えば破傷風トキソイド由来)を含む。かかるワクチンは、WT−1およびgata−1を発現するガンを治療するために特に有益である。かかる“ビーズ・オン・ストリング(bead on a string)”ワクチンは、通常、DNAワクチンである。
【0105】
CTL(CD8+細胞)を活性化する簡単な方法は、WO93/17095に記載されており、参照としてここに含める。WO93/17095の方法は、同系の(すなわち、自己の)HLAクラスI分子によって提示されるペプチドに対してCTLを生じる。
【0106】
別の多数の方法が、in vitroでCTLを生じるために用いられてもよい。例えば、Peoplesら(1995)Proc.Natl.Acad.Sci.USA 92,432-436およびKawakamiら(1992) J.Immunol.148,638-643に記載されている方法は、CTL産生において自己の腫瘍浸潤リンパ球を用いる。Plebanskiら(1995)Eur.J.Immunol.25,1783-1787は、CTLの調製において自己の末梢血液リンパ球(PLB)を利用する。Jochmusら(1997) J.Gen.Virol.78,1689-1695は、樹状細胞をペプチドまたはポリペプチドを用いてパルスすること、あるいは、組み換えウイルスで感染させることによる、自己のCTLの産生を記載する。
【0107】
Hillら(1995) J.Exp.Med.181, 2221-2228およびJeromeら(1993) J.Immunol.151,1654-1662は、自己CTLの産生にB細胞を利用する。さらに、ペプチドまたはポリペプチドでパルスした、あるいは、組み換えウイルスで感染したマクロファージが、自己CTLの調製に使用されてもよい。
【0108】
同種細胞もCTLの調製に使用でき、この方法はWO97/26328に詳細に記載されており、参照としてここに取り込む。例えば、ショウジョウバエ細胞およびT2細胞に加えて、CHO細胞、バキュロウイルス感染昆虫細胞、細菌、酵母、ワクシニア感染ターゲット細胞のような他の細胞が、抗原を提示するために用いられてもよい。さらに、植物ウイルスを用いてもよい(例えば、Portaら(1994) Virology 202,449-955参照、外因ペプチドの提示に関する高い収量のシステムとしてカウピーモザイクウイルスの発達について記載)。
【0109】
CTLが本発明のペプチドに関してアロMHC制限されるように、同種細胞がCTLの調製に用いられることが好ましい。CTLがHLA−A0201陰性患者に由来し、かつ、ペプチドが抗原提示細胞によりHLA−A0201クラスI分子によって提示されることが特に好ましい。
【0110】
外因的に適用されたペプチドは、CTLによる提示のためのMHCクラスI経路に向けるために、HIV tatペプチドに連結されてもよい(例えば、Kimら(1997) J.Immunol. 159, 1666-1668参照)。
【0111】
本発明のペプチドに向けられた活性化CTLを、治療に使用することができる。かくして、本発明のさらなる態様は、本発明の上記方法により得られた活性化CTLを提供する。
【0112】
本発明のさらなる態様は、本発明の第一、第二または第三の態様に記載されたアミノ酸配列を含むポリペプチドを異常に発現する細胞を選択的に認識する活性化CTLを提供する。好ましくは、CTLは、本発明の第一、第二または第三の態様のいずれかに定義されたペプチドに結合することにより前記細胞を認識する。
【0113】
CTLは、患者においてターゲット細胞を殺す方法に使用できる。この方法において、ターゲット細胞は、本発明の第一、第二または第三の態様のいずれか一つに記載されたアミノ酸配列を含むポリペプチドを異常に発現するものであり、患者には、有効数の活性化CTLが投与される。
【0114】
患者に投与されるCTLは、患者から誘導され、上記のように活性化される(すなわち、これらは自己のCTLである)。あるいは、CTLは患者由来ではないが、別の個人から由来するものである。もちろん、その個人が健康な個人であることが好ましい。“健康な個人”により、その個人が一般的に健康であり、好ましくは、反応性の免疫系を備え、さらに好ましくは、容易に試験または検出できるあらゆる疾病を患っていないことを意味する。この実施態様において、CTLは、HLAクラスI分子が患者のそれとミスマッチである個人から誘導されたものである。かくして、CTLがアロ制限(allo-restricted)されることが好ましい。アロ制限されたCTLを用いた治療は、本願出願人の先の出願WO97/26328に記載されており、参照としてここに取り込む。
【0115】
かくして、本発明の方法は、養子免疫療法を含む。
【0116】
活性化CTLは、T細胞レセプター(TCR)を含み、これは、異常なポリペプチドを発現する細胞を認識することに関係する。TCRをコードするcDNAが活性化CTLからクローン化され、発現のためのさらなるCTLに導入されることが有益である。
【0117】
本発明の第一、第二または第三の態様のペプチドに特異的な本発明のCTLクローンのTCR(アロ制限または自己制限)がクローン化される。CTLクローンにおけるTCR用途は、(i)TCR可変領域特異的モノクローナル抗体および(ii)VαおよびVβ遺伝子ファミリーに特異的なプライマーを用いたRT−PCRを用いて調べられる。cDNAライブラリーは、CTLクローンから抽出されたポリA mRNAから調製される。TCRαおよびβ鎖のC末端部位および同定されたVαおよびβセグメントのN末端部位に特異的なプライマーが用いられる。TCRαおよびβ鎖の完全なcDNAを、高い忠実度のDNAポリメラーゼを用いて増幅し、その増幅産物を適切なクローニングベクターにクローン化する。クローン化αおよびβ鎖遺伝子は、Chungら(1994)Proc.Natl.Acad.Sci.USA 91, 12654-12658に記載された方法によって一本鎖TCRにアセンブルされてもよい。この一本鎖構成物において、VαJセグメントの後にVβDJセグメントが存在し、さらにその後にCβセグメント、CD3ζ鎖のトランスメンブレンおよび細胞質セグメントが続く。次いで、この一本鎖TCRを、レトロウイルス発現ベクターに挿入する(ベクターのパネルが、成熟ヒトCD8+Tリンパ球を感染および遺伝子発現を媒介する能力に基づいて用いられてもよい:レトロウイルスベクターシステムKatが好ましい(Finerら(1994) Blood 83,43参照))。高力価両栄養性レトロウイルスが、参照としてここに含めるRobertsら(1994) Blood 84, 2878-2889に記載されているプロトコールに従って、腫瘍患者の末梢血液から単離された精製されたCD8+Tリンパ球を感染するために用いられる。抗CD3抗体は、精製されたCD8+T細胞の増殖を開始するために用いられ、レトロウイルスの統合と一本鎖TCRの安定な発現を容易にする。レトロウイルス形質導入の効率は、一本鎖TCRに特異的な抗体で感染CD8+T細胞を染色することにより調べられる。形質導入されたCD8+T細胞のin vitro分析は、それらが、TCR鎖が最初にクローン化されたアロ制限CTLクローンで見られるものと同じ腫瘍特異的殺傷を示すことを確証する。予期される特異性を備えた形質導入された
CD8+T細胞の集団は、腫瘍患者の養子免疫療法に使用できる。患者は、108〜1011(好ましくは109−1010)の間の自己の、形質導入されたCTLで治療されうる。
【0118】
CTLに遺伝子を導入するのに適した他のシステムは、Moritzら(1994) Proc.Natl.Acad.Sci.USA 91, 4318-4322に記載されており、参照としてここに含める。Eshharら(1993) Proc.Natl.Acad.Sci.USA 90, 720-724とHwuら(1993) J.Exp.Med.178,361-366もCTLのトランスフェクションについて記載している。
【0119】
かくして本発明のさらなる態様は、本発明の第一、第二または第三の態様のいずれか一つに与えられたアミノ酸配列を含むポリペプチドを異常に発現する細胞を認識するTCRを提供するものであり、TCRは活性化されたCTLから得られたものである。
【0120】
TCRに加えて、TCRと機能的に等価な分子が、本発明に含まれる。これらは、TCRと同じ機能を行うことができるTCRに機能的に等価な分子を含む。特に、かかる分子は、参照としてここに含めるChungら(1994)Proc.Natl.Acad.Sci.USA 91,12654-12658に記載された方法により製造された遺伝的に操作された3ドメイン一本鎖TCRを含む。
【0121】
また、本発明は、TCRまたは機能的に等価な分子をコードするポリヌクレオチド、TCRまたは機能的に等価な分子をコードする発現ベクターを含む。本発明のTCRを発現するのに適した発現ベクターは、本発明のペプチドの発現に関して上述したものを含む。しかしながら、発現ベクターが、トランスフェクション後にCTLにおいてTCRを発現できるものであることが好ましい。
【0122】
本発明のさらなる態様は、本発明の第一、第二または第三の態様のいずれかに記載されたアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺す方法を提供するものであり、この方法は、(1)患者からCTLを得る工程、(2)上記の、TCRをコードするポリヌクレオチド、または機能的に等価な分子を、前記細胞に導入する工程、および(3)患者において工程(2)で産生された細胞を導入する工程を含む。
【0123】
本発明のさらなる態様は、本発明の第一、第二または第三の態様に定義されたアミノ酸配列を含むポリペプチドを異常に発現する標的細胞を患者において殺す方法を提供するものであり、この方法は、(1)樹状細胞のような抗原提示細胞を患者から得る工程、(2)前記抗原提示細胞を本発明の第一、第二または第三の態様に記載されたペプチド、またはかかるペプチドをコードするポリヌクレオチドと、ex vivoで接触させる工程、および(3)このように処理された抗原提示細胞を患者に再導入する工程を含む。
【0124】
好ましくは、抗原提示細胞は、樹状細胞である。
【0125】
樹状細胞は、抗原ペプチドでパルスされた自己樹状細胞であることが適切である。抗原ペプチドは、適切なT細胞応答を生じる適切な抗原ペプチドとすることができる。腫瘍関連抗原からのペプチドでパルスされた自己樹状細胞を用いたT細胞治療は、Murphyら(1996)The Prostate 29, 371-380とTjuaら(1997) The Prostate 32, 272-278に記載されている。
【0126】
さらなる態様では、樹状細胞のような抗原提示細胞は、本発明のペプチドをコードするポリヌクレオチドと接触される。ポリヌクレオチドは、適切なポリヌクレオチドとすることができ、樹状細胞を形質導入し、かくしてペプチドの提示および免疫誘導をもたらすことができることが好ましい。
【0127】
都合よく、ポリヌクレオチドは、ウイルスポリヌクレオチドまたはウイルスに含まれてもよい。例えば、アデノウイルス形質導入樹状細胞は、MUC1に関して抗原特異的抗腫瘍免疫を誘導することが示された(Gongら(1997)Gene Ther.4,1023-1028参照)。同様に、アデノウイルスをベースとするシステムが用いられてもよい(例えば、Wanら(1997)Hum.Gene Ther.8, 1355-1363参照)。レトロウイルスシステムが用いられてもよい(Spechtら(1997) J.Exp.Med.186,1213-1221およびSzabolcsら(1997)Blood 90, 2160-2167)。樹状細胞への粒子媒介輸送も使用できる(Tutingら(1997) Eur.J.Immunol.27,2702-2707)。また、RNAも使用できる(Ashleyら(1997) J.Exp.Med.186, 1177-1182)。
【0128】
患者におけるターゲット細胞を殺す方法に関して、ターゲット細胞がガン細胞であることが特に好ましい。
【0129】
WT1ポリペプチドは、アミノ酸配列RMFPNAPYLおよびCMTWNQMNLを含み、白血病、乳ガン、メラノーマおよび卵巣ガンにおいて異常に発現される。
【0130】
gata−1ポリペプチドは、アミノ酸配列HLMPFPGPLLを含み、白血病において異常に発現される。
【0131】
特に、本発明の方法によって治療される患者が、HLA−A0201ハプロタイプを有することが好ましい。かくして、好ましい実施態様では、患者のHLAハプロタイプが、治療前に調べられる。HLAハプロタイプは、適切な方法を用いて行われ、かかる方法は当該技術分野において周知である。
【0132】
本発明は、特に、活性なin vivoでのワクチン接種;in vitroでの自己樹状細胞の操作、その後かかる操作がされた樹状細胞のin vivoでの導入して、CTL応答を活性化;in vitroで自己CTLを活性化して、養子治療(すなわち、このように操作されたCTLを患者に導入する);かつ、健康なドナー(MHC適合またはミスマッチ)のCTLをin vitroで活性化して、養子治療するために、本発明のペプチド(またはそれらをコードするポリヌクレオチド)の使用を含む。
【0133】
本発明を、図面および実施例を参照してより詳細に記載する。
【実施例】
【0134】
実施例1:WT1およびgata−1におけるHLA−A0201提示CTLエピトープの同定 HLA−A2結合モチーフを備えた8つのWT−1ペプチドを分析し、二つが天然のCTLエピトープであることが見出された。HLA−A2結合モチーフを備えた12のgata−1ペプチドを分析し、一つが天然のCTLエピトープであることが見出された。
【0135】
以下のアプローチを用いた:i)HLA−A0201結合アッセイにおける合成ペプチドの分析;iii)HLA−A0201陰性のヒト由来のCTL応答を刺激するためにHLA−A0201結合ぺぷちどを使用;iv)WT−1またはgata−1を内因的に発現する腫瘍細胞に対するペプチド特異的CTLの試験。
【0136】
ペプチド結合アッセイ:5x105 T2細胞を、96ウェルプレートにおいて、5%の煮沸したFCS(プロテアーゼを破壊するため)と種々の濃度の合成ペプチドを含む100mlのRPMI1640培地中で、一晩(o/n)インキュベートした。ペプチドまたは周知のA2−結合ペプチドを含まないT2細胞を含むウェルを、それぞれ、陰性および陽性コントロールとして用いた。一晩インキュベーションした後、細胞を洗浄し、A-2特異的モノクローナル抗体HB54およびHB117(American Type Culture Collection, ATCC)を用いて表面HLA−A2について間接免疫蛍光により染色した。FACS分析を、Coulter Corporationフローサイトメーターで行った(Haiteah, Florida)。
【0137】
アロ制限CTL系統およびクローンの産生:HLA−A2陰性バフィーコート血液パックからのPBMCを、レスポンダーとして用いた。24ウェルプレートの各ウェルに、2x106Ficoll分離PBMCと2x105刺激細胞を与えた。刺激細胞は、5%の煮沸FCSを含むRPMI中の100μMペプチドでT2細胞を一晩インキュベートすることによって調製された。5日目に、T細胞を回収し、フィーダーとして2x106の自己の照射したPBMC、2x105ペプチドコート照射T2またはC1R−A2細胞、500nMペプチドおよび10%QS4120培養上清を加えた24ウェルプレートにおいてウェル当たり5x105の密度で新鮮なT細胞培地中に移し、CD4T細胞の成長を抑制する。この培養物を、刺激因子として免疫ペプチドで被覆されたHLA−A2陽性細胞系統を用いて、2週間毎にフィードした。このバルク培養を、T細胞培地に、ウェル当たり1、10および30細胞の密度でクローン化した。104ペプチド被覆T2細胞、2x105HLA−A2陰性フィーダー、および2UのIL−2を各ウェルに添加した。ウェル当たり1細胞でまかれたミクロ培養から得られたCTLを、ウェル増殖細胞のパーセントが30%を越えない場合には、さらにクローン化される。
【0138】
CTLアッセイ:106T2細胞を37℃で100μMの合成ペプチドを含む200μlのアッセイ培地(5% 熱不活性化FCSを含むRPMI 1640)において1時間インキュベートした。ペプチド被覆および未被覆の細胞を、さらに1時間51Crラベルし、洗浄し、丸底96ウェルプレートにおいて連続する二倍希釈のエフェクター細胞に添加して、200μl/ウェルの全体量とした。CTLバルク系統を、51Crラベル化ターゲット細胞当たり30のコールドK562細胞の存在下で分析し、NK細胞により引き起こされたバックグラウンド殺傷を低減を低減した。T細胞クローンの感度を試験するために、アッセイ培地における一連の希釈のペプチドを、96ウェルプレートに調製した。5x103の51Crラベル化T2細胞を各ウェルに添加し、100μlの全体量として、1時間インキュベートした。エフェクター細胞を、最大CTL殺傷に十分なE:T比で添加した。アッセイプレートを37℃、5%CO2でインキュベートし、4時間後、100μlの上清を各ウェルから回収し、Wallac Gamma Counterを用いてカウントした。比溶解(specific lysis)を、式(実験放出自然放出)/(最大放出自然放出)x100%によって計算した。
【0139】
結果
WT−1タンパクにおいて、WT−1発現腫瘍細胞において発現された二つのCTL認識ペプチドエピトープが同定された:WT126−34:RMFPNAPYLWT235−43:CMTWNQMNL
【0140】
これらは9アミノ酸の長さのペプチドであり、CTLによる有効な認識に必要な最小エピトープを示すと考えられる。
【0141】
WT126−34に対するCTLの殺傷活性を、以下の表に記載する。
【0142】
【表1】

【0143】
表のデータは、WT126−34ペプチドに対するCTLが、WT−1およびHLA−A0201を発現する腫瘍細胞を殺傷することを示す。WT−1陰性Bリンパ芽球細胞は殺傷されない。図1および2は、抗WT126−34CTLの殺傷活性を示す代表例を示す。
【0144】
抗WT235−43CTLを用いて得られたデータは、WT126−34CTLを用いて得られたデータほど大規模ではない。しかしながら、WT235−43CTLがWT−1を内因的に発現するHLA−A0201陽性腫瘍細胞を殺傷することが明らかである。
【0145】
gata−1タンパクにおいて、gata−1発現腫瘍細胞において発現されたCTL認識ペプチドエピトープが同定された:Hug378−87 HLMPFPGPLL。
【0146】
これは10アミノ酸の長さのペプチドであり、CTLによる有効な認識に必要とされる最小エピトープを示すと考えられる。
【0147】
このペプチドに対するCTLは、内因的にgata−1を発現するHLA−A0201形質転換K562白血病細胞系統を認識できるが、形質転換されていないK562細胞は認識されない。
【0148】
結論
WT−1およびgata−1のCTL認識ペプチドエピトープが同定された。これらのエピトープは、これらのタンパクを異常に発現する腫瘍細胞に提示される。WT−1およびgata−1の生理学的発現は、比較的少数の正常細胞に制限されている。かくして、自己のCTLがこれらのタンパクに対して制限された寛容を示すことができ、かつ、同定されたCTLエピトープが、異常なWT−1およびgata−1発現を有する腫瘍に対してワクチン接種、および別の免疫治療法に使用することができる。
【0149】
実施例2:クラスI分子とWT1ペプチド抗原RMFPNAPYLを用いた活性化された細胞傷害性リンパ球(CTL)の産生およびその投与 活性化された細胞傷害性Tリンパ球(CTL)を、HLA−A2クラスI分子とWT1由来のノナマーペプチド:RMFPNAPYLを用いて産生した。
【0150】
PCT特許出願WO93/17095に記載された方法を、CTLを作製するために使用した。ショウジョウバエ細胞をCTLにペプチド抗原を提示するために使用した。HLA−A2分子は、ショウジョウバエ細胞において発現された。
【0151】
ペプチドは、Applied Biosystems synthesiser, ABI 431A(Foster City, CA, USA)で合成し、HPLCで精製した。
【0152】
WO93/17095に詳細に記載されているように、特異的な細胞傷害性T細胞の産生のためのin vitro条件を最適化するために、刺激細胞の培養が適切な培地で維持された。刺激細胞は、WO93/17095に記載されているようなショウジョウバエ細胞であり、好ましくは血清を含まない培地に維持される(例えば、Excell 400)。
【0153】
前駆体CD8細胞のような活性化される細胞と刺激細胞のインキュベーションの前に、ヒトクラスI分子に結合して、刺激細胞の表面に提示されるのに十分な量の所定量の抗原性ペプチドが、刺激細胞培養物に添加される。十分な量のペプチドとは、ペプチドを結合したヒトクラスI MHC分子が約200,好ましくは200以上、各刺激細胞の表面に提示される量である。刺激細胞は通常、>20μg/mlのペプチドとインキュベートされる。
【0154】
休止または前駆体CD8細胞を、CD8細胞を活性化するのに十分な時間、適切な刺激細胞と培養においてインキュベートした。CD8細胞は、抗原特異的方法で活性化される。休止または前駆体CD8(エフェクター)細胞の刺激細胞に対する比率は、個々に異なってよく、培養条件に対する個人のリンパ球の感度のような変異性に依存してもよい。リンパ球:刺激細胞(ショウジョウバエ細胞)比は、通常、約30:1から300:1の範囲である。例えば、3x107ヒトPBLと1x106ショウジョウバエ細胞が混合され、20mlのRPMI 1640培養培地に維持される。
【0155】
エフェクター/刺激培養物を、治療に使用できるまたは有効な数のCD8細胞を刺激するのに必要な時間にわたって維持する。最適な時間は、通常、“プラトー(plateau)”、すなわち“最大”特異的CD8活性化レベルであり、通常培養の5日後に見られる、で約1〜5日の間である。CD8細胞のin vitro活性化は、通常、細胞系統のトランスフェクション後、短い期間内に検出される。CD8細胞を活性化することができるトランスフェクションされた細胞系統における一過性の発現は、トランスフェクションから48時間以内に検出される。これは、ヒトクラスI MHC分子を発現する形質転換された細胞の安定または一過性の培養が、CD8細胞を活性化するのに有効であることを、はっきりと示す。
【0156】
活性化されたCD8細胞は、刺激細胞、刺激細胞上に結合されたペプチド、またはCD8細胞(またはそのセグメント)に特異的なモノクローナル抗体を用いて、その適切な相補的リガンドに結合させることにより、刺激(ショウジョウバエ)細胞から有効に分離することができる。次いで、抗体タグ化した分子は、免疫沈降またはイムノアッセイ方法を解して刺激−エフェクター細胞混合物から抽出される。
【0157】
有効な、細胞傷害的な量の活性化CD8細胞は、in vitroおよびin vivoでの使用の間で、これらの傷害細胞の究極のターゲットである細胞の量およびタイプによって変化することができ、約1x106および1x1012の活性化されたCTLが、成人に対して用いられる。
【0158】
活性化されたCD8細胞は、処置される個人にCD8細胞を投与する前に、ショウジョウバエ細胞培養から回収される。しかしながら、他の存在しかつ提案された治療モダリティーとは異なって、この実施例に記載した方法が、腫瘍形成性でない細胞培養システム(すなわちショウジョウバエ細胞)を用いないことに注意することが重要である。それゆえ、ショウジョウバエ細胞と活性化されたCD8細胞との完全な分離が達成されない場合でも、少量のショウジョウバエ細胞の投与に関連することが知られた固有の危険性は存在しない。一方、哺乳動物の腫瘍促進細胞の投与は危険である。
【0159】
例えば、Honsikらの米国特許第4844893号およびRosenbergの米国特許第4690915号に例示されているような、細胞性成分を再導入する方法が用いられる。例えば、静脈注入による活性化CD8細胞の投与が適切である。
【0160】
実施例3:乳ガンを処置するための、WT1ペプチドCMTWNQMNLを用いてパルスした樹状細胞 乳ガンは、真に局在かしている場合にのみ治療可能性がある。最も一般的な問題は、明確な転移に関する遅い発表か、多くは、局所治癒後の全身的転移の発達である。転移性乳ガンは、高度に化学的感受性であり、有効な化学療法は、きまって疾患の軽減を誘導し、二次疾患の開始を遅延し、広範囲の疾患の症状を緩和する。
【0161】
この種の免疫治療は、腫瘍増殖および転移が、新生細胞による抗原提示の低下または患者の免疫における一般的な衰えのいずれかによる、免疫学的監視の欠陥を反映するという考えに基づく。両方のメカニズムが乳ガンにおいて生じ、特に樹状細胞(DC)機能に重要な欠陥が存在するという証拠が存在する(Gabrilovichら(1997) Clin.Cancer Res.3, 483-490)。細胞傷害性T細胞応答は、WT1ペプチド:CMTWNQMNLのような免疫原性ペプチドに対してin vitroで証明されている。DCは、プロフェッショナル抗原-処理および-提示細胞である。これは、腫瘍MHC制限T細胞免疫の発達に重要である。これらは、骨髄のCD34+前駆体に由来するが、末梢血液中のポストコロニー形成ユニットCD14+中間体からも誘導される。DCは、皮膚、粘膜、脾臓および胸腺において末梢部位に移動する。これらは、同種移植片拒絶、アトピー性疾患、自己免疫および抗腫瘍免疫を含む、種々の臨床的に重要な処理に関係している。
【0162】
患者は、HLA−A2に分類される。
【0163】
DCは、主にGM-CSF、IL−4およびTNFαのようなサイトカインを用いて、CD34+ステム細胞またはCD14+末梢血液単球からex vivoで培養される。これら両方の源に由来するDCは免疫応答性であり、外因的に提示された抗原を取り込み、処理して、細胞傷害性T細胞にそれを提示することができる(Grabbeら (1995) Immunology Today 16, 117-121; Girolomoni & Ricciardi-Castagnoli (1997) Immunology Today 18, 102-104)。最近の研究は、DCがin vivoで生成された抗原特異的腫瘍免疫を運搬しうること(Kwakら(1995) Lancet 345, 1016-1020)、および、ex vivoで腫瘍抗原を用いてパルスされた自己のDCが、無視できない抗腫瘍効果を誘導しうること(Hsuら (1996) Nature Medicine 2, 52-58)を証明した。DCは、粗製腫瘍膜溶解物、精製されたペプチドまたはペプチドフラグメントを用いて効率的にパルスされうる。
【0164】
WT1は、乳ガンによって発現されるポリペプチドである。
【0165】
キーホールリンペットヘモシアニン(KLH)は、これと類似した研究において患者の免疫応答性に対する無害の陽性コントロールとして用いられる免疫原性タンパクである(Hsuら (1996) Nature Medicine 2, 52-58)。
【0166】
WT1ペプチドCMTWNQMNLの精製された調製物と結合し、かつ、養子免疫療法として再注入された、再発性乳ガン患者由来のex vivo拡大自己樹状細胞を用いる実現性は、以下のように確立される。
【0167】
記載された研究は、再発性乳ガン患者の末梢血液からのex vivo拡大(expansion)による自己DCの最適な生成方法論を確立し;外因性WT1ペプチドをDCに載せる(結合させる:load)可能性を評価し;自己再注入の急性寛容および毒性を評価し;WT1ペプチドまたはKLHディベロップに対して免疫が反応するか否かを調べ;かつ、無視できない腫瘍バルクへの効果を調べる。
【0168】
養子免疫療法は、バルク疾患の低減よりも、最小の残余疾患のコントロールまたは消失において最も効果的であろう。免疫療法は、細胞傷害性Tリンパ球の流入によりバルク疾患の大きさを一時的に増大させうると思われる。疾患の程度およびバルクは、治療後に監視されるが、正式な終点として用いられない。患者は、決まった方法で長期にわたり追跡調査され、長期にわたって、不利な症状が現れないことを確認する。
【0169】
正常な志願者由来の樹状細胞培養 CD14+末梢血液単球を組織培養フラスコに付着させ、1%AB血清、GM−CSF(400ng/ml)およびIL−4(400IU/ml)の存在下で7日間培養した。これは、DCの多型性を備えた細胞を与え、抗原を取り込んで提示するDCの未熟形態を示すCD1a+マーカーで49%の平均を与えた。次いで、これらの細胞を、DCを細胞傷害性T細胞に対して抗原提示可能にする、TNFα(15ng/ml)の添加によりCD83+細胞に成熟化される。7%の細胞が、1日以内にCD83+になるが、最大効果のためには少なくとも3日必要である。単球調整培地が、1%AB血清と置換できる。
【0170】
再発した乳ガン患者由来の樹状細胞培養 救助療法前後の、再発した転移性疾患の6人の患者からDCを生成した(末梢血液の12サンプルの合計、それぞれ50ml)。
【0171】
臨床研究 標準的なプロトコールに従って、単一ユニットの自己の血液を患者が献血した。血液輸血サービス医師によって献血前に患者を評価した。自己の献血を、決まったウイルスマーカー(HIV、HBV、HCVおよび梅毒)について同種献血と同様に選別した。患者は、参加を望めば、適切なカウンセリングの後に、これに同意した。この警戒は、臨床および実験室スタッフを可能性のある感染から保護し、交差汚染の可能性から決まった血液供給を保護する。この血液を、決まったクワドパックに入れる。これは、赤血球、バフィコートおよびプラスマの自動化された分離を可能にする。このバフィコートは、約670x106の単核白血球を与え、これは現在の技術を用いて約47x106のDCを与える。患者当たり8−128x106のDCの投与範囲を用いる。末梢血液単球を二つの分注量に分け、1から10日間、WT1ペプチドおよびKLHを用いてパルスした。血清を含まない培養条件または自己プラスマを、同種AB血清に優先して用いた。培養されたDCを集め、洗浄し、100mlの生理食塩水に再懸濁した後に、1時間かけて注入した。自己赤血球濃縮物は、標準臨床示度以外、患者に戻さない。ex vivo DC培養方法を、以下の良好な製造処理に従って実施した。
【0172】
最初の血液サンプルを献血する患者は、このときまでに、救助療法を受けており、臨床的寛解にあってもそうでなくてもよい。再発転移性疾患患者は、化学療法を受ける前に処置を受ける。二つの処置プログラムがある。
(1)転移性再発、標準治療後、養子免疫療法;
(2)転移性再発、養子免疫療法後、標準治療。
【0173】
治療のための患者を含める基準:限局性の再発または転移性乳ガン患者。
細胞毒性化学療法またはホルモン治療を用いて以前に処置。
評価可能な疾患(UICC基準)。
>12週であると予想される生存。
自己血液献血の基準を満たす(HgB>120g/Iを含む)。
告知に基づく同意。
年齢が18歳〜70歳。
【0174】
処置から患者を除く基準:妊婦。
CNS転移。
以前または共同性転移。
告知に基づく同意が得られない。
同意拒否。
年齢が<18歳または>70歳。
【0175】
産物の注入は、必要であれば蘇生および支持的な便宜を利用できる寝椅子の上で、監視のもとで、経験のある医師の直接的監督のもとに行われた。
【0176】
実施例4:WT1に特異的な細胞傷害性Tリンパ球による白血病CD34+始原細胞の選択的除去 例えば、急性および慢性骨髄性白血病のような血液学悪性腫瘍は、未熟なCD34+始原細胞の悪性トランスフォーメーションによって特徴付けされる。トランスフォーメーションは、WT1転写因子の増加した発現と関係する。ここで、WT1が、白血病性始原細胞に対する鋭敏な特異性を備えたCTLのターゲットとして作用しうることを証明した。WT1に特異的なHLA−A0201制限CTLは白血病性細胞系統を殺し、CML患者から単離された形質転換されたCD34+始原細胞によるコロニー形成を阻害したが、正常なCD34+始原細胞によるコロニー形成は影響されなかった。
【0177】
かくして、組織特異的転写因子WT1は、in vitroでの白血病性始原細胞のCTL媒介パージング、および、in vivoにおける白血病およびWT1発現悪性腫瘍の抗原特異的治療の、理想のターゲットである。
【0178】
造血系の細胞を、自己再生および分化可能な幹細胞(HSC)から誘導した。ヒトとマウスにおける移植実験は、CD34+細胞集団が、骨髄切除受容者において赤血球、脊髄およびリンパ球の直系を再構成可能であるHSCを含むことを示した1。さらに、マウスの宿主を再構成することができるHSCが、CD34-/lin-骨髄細胞の稀な集団において最近証明された2。
【0179】
CMLとAMLにおける重要なトランスフォーメーションが、未熟なCD34+始原細胞に影響するという強い証拠がある。大多数の白血病性芽細胞が、制限された増殖能を有するので、悪性疾患は、強い増殖力と自己新生力を備えた白血病性始原細胞の部分集団によって維持されなければならない3、4。AML患者由来の精製された細胞を用いた移植研究は、未熟なCD34+細胞のみが、免疫無防備状態のマウス受容者において白血病を開始することができることを示した5。同様に、CML患者由来の精製されたCD34+細胞は、マウス受容者において効率的に白血病を開始した6、7。
【0180】
未調整始原細胞増殖を導く分子事象は完全には理解されていない。t(9;22)染色体転座に関連するBCR/ABL融合タンパクはCMLの証明であるが、BCR/ABL転写物は、健康な個人にも観察され、さらなるファクターが白血病の発達に必要であることを示唆する8。WT1転写因子は、白血病誘発に寄与する候補タンパクである。この転写因子は、通常、未熟なCD34+始原細胞に発現され、分化はWT1ダウンレギュレーションに関与する9、10。WT1発現の高められたレベルは、AMLおよびCML患者由来の精製されたCD34+細胞および未分離の単核細胞において観察された11、12。増大したWT1発現が正常な分化をブロックし、造血始原細胞の増殖を増強することを示すin vitro研究は、白血病誘発に寄与するWT1の能力の説明を与える13、14。
【0181】
最近の研究結果は、CD34+始原細胞に特異的なTリンパ球が、CML患者における抗白血病性効果を媒介することに極めて重要であるということを示唆した15。この研究において、白血病性始原細胞に対して細胞傷害性Tリンパ球を向けるために、ターゲット分子としてWT1を開発する可能性を調査した。正常なCD34+始原細胞ではなくCMLが、CTL攻撃を開始するのに十分なWT1タンパクを発現するという仮説を調べた。
【0182】
方法:細胞系:急性転化状態の女性のCML患者の胸水からK562細胞を確立した16。急性転化状態の男性のCML患者の末梢血からBV173細胞を確立した17。急性リンパ性白血病を有する12歳の少年の骨髄から細胞系697を確立した18。C1R細胞系は、HLA−A0201陰性EBVトランスフォームリンパ芽球細胞系である19。T2細胞系は、TAP(transporter associated with antigen processing)をコードする遺伝子の欠失について選択され、内因性ペプチドを有するHLAクラスI分子を不十分にしか運搬しないことを引き起こす20。従って、T2細胞のHLA−A0201分子は、外因性ペプチドを効率的に運搬する。HLA−A0201、ヒトβ−2ミクログロブリン、B7.1及びICAM−1でトランスフェクトされたDrosophila細胞は、Dr. M. Jacksonからの好意で得た。
合成ペプチド:ヒトウィルムス主要抗原1P126から由来するペプチド(RMFPNAPYL)及びヒトパピローマウイルスタイプ16のE7タンパク質から由来するコントロールHLA−A0201結合ペプチドを、フルオレニルメトキシカルボニル化学を使用して、Imperial College Medical Schoolの中央ペプチド合成研究室によって合成した。ペプチドの質をHPLC分析によって評価し、予想される分子量をマトリックスアシストレーザー放出マススペクトロメトリーを使用して観察した。ペプチドをPBS(pH7.4)に溶解し、2mMの濃度を得て−20℃で貯蔵した。
【0183】
アロ−HLA−制限CTL系の世代:PBMCをFicoll勾配遠心分離を使用してバッフィーコートパックから分離し、モノクローナル抗体HB54(抗HLA−A2,B17)及びHB117(抗HLA−A2,A28)で染色した。A2陰性PBMCを応答因子として使用した。HLA−A0201、ヒトβ2−ミクログロブリン、B7.1及びICAM−1でトランスフェクトされたペプチドコートDrosophila細胞を、初期刺激因子として使用した。Drosophila細胞を48時間100mMのCuSO4中で誘導し、培地で3回洗浄し、4時間100μMの濃度でペプチドを乗せた。24穴プレートの各ウェルは、2mlのT細胞培地中に2×106の応答因子PBMCと2×105の刺激因子細胞を受け取った。5日目でT細胞を回収し、餌として2×106の自己発光PBMC、2×105の発光ペプチドコートT2細胞、10%QS4120培養上清(抗CD4抗体を含む)、及び10U/mlhu−rIL−2(Boehringer)を添加して、ウェル当たり5×105の密度で新鮮なT細胞培地中に蒔いた。刺激因子として免疫化ペプチドでコートされたT2細部を使用して、培養物を弱く再刺激化した。2−3サイクルの刺激の後、大きな培養物がウェル当たり1,10及び30細胞の密度で96穴プレート中にクローン化した。104ペプチドコートT2細胞、2×105HLA−A2陰性PBMCフィーダー、及び2U/mlIL−2を、各ウェルに加えた。各ウェルの細胞毒性を、免疫化ペプチドまたはコントロールHLA−A0201結合ペプチドでコートされたT2標的細胞に対して試験した。ペプチド特異的ミクロ培養物を増殖させ、2×106のフィーダー、2×105の刺激因子細胞、及び10U/mlIL−2を加えることによって、24穴プレート中で弱く再刺激化した。T細胞系77(図4B参照)を、培養物中で1年間維持し、この研究でのほとんどの実験についてCTLのソースとして機能した。この系はCD4+及びCD8+細胞より成るため、特異的な殺傷活性がCD8+CTLによって介在されることを示すため、CD8+サブクローンを使用した。親の77系とは異なり、CD8+サブクローンのin vitroでのライフスパンは数カ月に制限された。
【0184】
CTLアッセイ:CTLアッセイを記載されているように実施した。略記すると、106T2細胞を、37℃で100μMの合成ペプチドを有する200μlのアッセイ培地(5%熱不活性化FCSを有するRPMI1640)中で1時間培養した。ペプチドコートT2細胞または腫瘍細胞を1時間51Crラベルし、洗浄し、丸底96穴プレート中に連続的な2倍希釈のエフェクター細胞に加え、200μl/ウェルの全容量を得た。アッセイプレートを37℃で5%CO2で4時間インキュベートした。100μlの上清を回収し、Wallac Gamma Counterを使用してカウントした。特異的な溶解を、以下の式:(実験的放出−自発的放出)/(最大放出−自発的放出)×100%によって計算した。
【0185】
造血CD34+細胞の精製:正常CD34+細胞のソースとして、成人で健康なドナー由来のヒト骨髄(n=5)、疾患回復期の幹細胞不朽固化腫瘍患者からの白血球搬出産物(n=2)及び臍帯血(n=1)を使用した。臍帯血のサンプルは、滅菌回収チューブ内への血液の排出によって廃棄された胎盤及びへそのお組織から得られた。これらの細胞の使用についてのインフォームドコンセントを、ドナー及び患者から適切なように取得した。白血病CD34+細胞のソースとして、少なくとも3ヶ月間インターフェロンで処理されていない慢性状態のCML患者から末梢血を得た。
【0186】
サンプルをHanks緩衝塩溶液(HBSS)中で1:2に希釈し、密度勾配遠心分離(Lymphoprep 1.077g/ml, Nycomed, UK)によって単核細胞で富化し、回収した単核分画を、Minimacsシステムを使用して製造者の説明書に従って(Miltenyi Biotec, UK)、CD34+分画の単離のための磁性ミクロビーズ選択にかけた。抗ヒトCD34フィコエリスリン(PE)マウスモノクローナル抗体(Becton Dickinson)を使用したFACS分析によって見積もると、細胞集団の純度は80−95%の範囲であった。
【0187】
RNA抽出及びRT−PCR:106細胞の全RNAを、RNAzolTMBプロトコール(AMS Bio, UK)に従って単離した。全RNAペレットのcDNA合成を、40μl反応物で実施した。溶解RNAペレットを初めに65℃で10分2μgのオリゴdT12−18プライマー(Life Technologies, Scotland)でインキュベートし、次いで50Uの齧歯類白血病ウイルス(MuLV)逆転写酵素、10mMのジチオスレイトール、1mMのdNTP(Boehringer Mannheim, UK)、40UのRAアーゼインヒビター(Promega, UK)の混合物で、42℃で1時間インキュベーションした。5μlのcDNA調製物を、2.5UのTaqDNAポリメラーゼ(Qiagen, UK)、1mMdNTP、20OD/mlのプライマーを含む最終反応混合物の50μl容量中でPCR増幅のために使用した。ヒトWT1コード領域の増幅は、エクソン7に位置するセンスプライマー(21マー、5'-ggc atc tga gac cag tga gaa-3')及びエクソン10のアンチセンスプライマー(22マー、5'-gag agt cag act tga aag cag t-3')を使用して達成した。WT1PCR産物の予想サイズは482bpである。RNAの完全性を、22マーセンス5'-ccc aac ctt ttc gtt gca ctg t-3';22マーアンチセンス5'-cgg ctc tcg gag gag acg atg a-3'といったイントロンにまたがるプライマーを使用して全てのサンプル中のヒトc−abl遺伝子を増幅することによって確認した。c−ABLPCR産物の予想サイズは385bpである。ホットスタートPCRを、以下の条件(ABL及びWT1増幅について同じ):95℃で1分の変性、56℃で1分のプライマーアニリング、及び72℃で2分の鎖伸張の下で、熱サイクラー(Techne Genius, Cambridge)で35サイクル実施した。サイクリングは、95℃で5分の変性工程で開始し、逆転写酵素を熱不活性化し、72℃で10分の最終伸張によって終結した。全てのRT−PCRを少なくとも2回実施し、陰性コントロール(cDNA含まず)及び陽性コントロール(WT1発現白血病細胞系BV173から得たcDNA)を全ての実験で含ませた。PCR産物を1.5%アガロースゲルで電気泳動した。
【0188】
ウエスタンブロット分析:分離したCD34+細胞(2×105細胞)をPBSで洗浄し、Laemmli Buffer中で溶解した。細胞溶解物を12%SDSポリアクリルアミドゲル電気泳動(PAGE)によって分画し、湿潤トランスファーによりニトロセルロース膜(Amersham, UK)にトランスファーした。次いで膜を、0.01%Tween20及び5%ノンファットドライミルクを含むPBS中で室温で1時間ブロックし、ウサギ抗ヒトWT−1C19ポリクローナル抗体(Santa Cruz CA, ブロッカー中で1:200)で最初に4℃で一晩インキュベーションし、次いでウサギ抗アクチンポリクローナル血清(Sigma UK, ブロッカー中で1:500)で室温で30分インキュベーションした。セイヨウワサビペルオキシダーゼ接合ブタ抗ウサギ抗体(DAKO UK, 1:1000)での膜のインキュベーションと製造者の説明書に従ったECL反応(Amersham UK)により、シグナルを検出した。
【0189】
祖先アッセイ(CFUアッセイ):以下の組換えヒト成長因子(Stem Cell Technologies):幹細胞因子(SCF、50ng/ml)、インターロイキン−3(IL−3,20ng/ml)、インターロイキン−6(IL−6,20ng/ml)、顆粒球マクロファージコロニー刺激因子(GM−CSF、20ng/ml)、顆粒球コロニー刺激因子(G−CSF)を補ったメチルセルロース培地中に、1000−3000CD34+細胞を蒔くことによってCFUアッセイを実施した。培養物を5%CO2の湿潤環境で37℃で14日間インキュベートし、コロニー形成単位顆粒球マクロファージ(CFU−GM)の発達を可能にした。
【0190】
結果:WT−1特異的CTLの世代:成人におけるWT1転写因子の発現は、腎有足突起、精巣セルトーリ細胞、卵巣顆粒層細胞、及びCD34+骨髄細胞で検出可能である21。WT1に対する可能性のある免疫寛容を避けるために、MHCミスマッチドナーから得られたペプチド特異的CTLを生産する以前に記載されたアプローチを使用した。このアプローチは、免疫寛容と独立した腫瘍細胞で過剰発現されるいずれかのタンパク質に対してCTLを生産するのに適している22,23。9のアミノ酸長のWT−1由来ペプチドエピトープP126(RMFPNAPYL)を、CVTL標的として選択した。このペプチドは、コーカサス人で見出される最も顕著なクラスI対立遺伝子であるHLA−A0201クラスI分子(実施例1参照)に結合する。HLA−A0201-ドナーから得た応答因子白血球を、P126ペプチドを提示するHLA−A0201+刺激因子細胞と共にin vitroで培養し、希釈培養物の制限を、ペプチド特異的CTL細胞を単離するために使用した。ペプチドコートT2標的細胞での実験により、CTLがP126ペプチドに対して非常に特異的であることが示された(図4A)。ペプチド滴定により、CTLが低いピコモーラーのペプチド濃度を認識可能な非常に渇望した系と、低いピコモーラーのペプチド濃度を認識するあまり渇望していない系に分割できることが示された(図4B)。非常に渇望したCTL系を、さらなる実験のために選択した。
【0191】
WT−1特異的CTL殺傷白血病細胞系:白血病細胞系のパネルの分析により、P126特異的CTLが、HLA−A0201+細胞BV173及び697を殺傷することが明らかとなった(図4C)。HLA−A0201-白血病細胞系K562は、HLA−A0201でのトランスフェクションの後のみで殺傷される。対照的に、細胞がP126ペプチドでコートされていないと、HLA−A0201+EBVトランスフォームB細胞系C1R−A2は殺傷されなかった(図4C;同じ結果は他のEBVトランスフォーム細胞でも見られた)。CTL標的細胞中でのWT1の発現を、RNA及びタンパク質レベルで分析した。RT−PCRにより、白血病細胞系はWT1RNAを発現するが、EBVトランスフォームC1R−A2細胞は発現しないことが示された(図5A)。同じ結果は、WT1タンパク質が白血病細胞でのみ発現され、C1R−A2では発現されないことを示すウエスタンブロットによっても得られた(図5C)。このデータを総合して、CTLはA0201+白血病細胞系を認識し、CTL殺傷はWT1発現と相関することが示された。
【0192】
WT1特異的CTL殺傷新鮮白血病CD34+細胞:慢性状態のCML患者の末梢血から得た単核細胞を、未成熟CD34+と成熟CD34-集団に分類した。予想されたように、HLA−A0201-患者から単離された細胞は、P126特異的CTLによって認識されなかった(図6A)。HLA−A0201+CML患者から得られた細胞を分析した場合、CTLはCD34+細胞集団を選択的に認識する一方で、より成熟したCD34-集団の殺傷は観察されなかった(図6A)。CD34-細胞の殺傷の欠如は、外因性P126ペプチドを有するこれらの細胞のコーティングがCTL殺傷を引き起こすため、WT1由来標的ペプチドの不十分な発現のためである可能性が高い(図6A)。RT−PCR分析により、CD34+細胞における強力なWT1RNA発現と、CD34-細胞における低い発現が明らかとなった(図5B)。ウエスタンブロッティングにより検出可能なWT1タンパク質の発現はCD34+に制限され、CD34-細胞ではタンパク質は検出不可能であった(図5C)。RT−PCRよウエスタン分析の両者により、白血病CD34+細胞集団におけるWT1発現のレベルの変異が示された。かくして、WT1発現のレベルにおける観察された変異が、CTL殺傷のレベルの変異を生ずるかどうかを調査した。しかしながら11の異なるCML患者の分析により、CTLはCD34+集団の20%(SD5%)を一貫して溶解することが示された(図5B)。この結果は、WT1発現がCD34+細胞の約20%のサブ集団に制限され、全ての11のCML患者における発現レベルが、これらの細胞のほとんどをCTL殺傷に感受性にするのには不十分である可能性を示唆した。それ故、CTLによって認識されるサブ集団が、顆粒球/マクロファージ直系のコロニーに高めることができるクローン原性の始原細胞を含むかどうかを調査した。実際、9のCML患者から単離されたCD34+集団をP126特異的CTLで処理した場合、これはコロニー形成の80−100%の阻害を生ずる(図5D)。このことは、コロニー形成始原細胞の大多数がP126特異的CTLによって除去されることを示した。CTL処理サンプルで見られる「エスケープ」コロニーは、非処理サンプル中のコロニーと比較した場合小さかった。これは、「エスケープ」コロニーが、WT1発現の下流調節と関連する顆粒球/マクロファージ直系に向けた分化をすでに開始している始原細胞から由来する可能性と一致する。上記部分的に分化した始原細胞は、WT1特異的CTLによる認識を避け、CFUアッセイで観察される小さいサイズのコロニーは、これらの始原細胞の減少したクローン増大サイズを反映しているであろう。
【0193】
重要なことに、HLA−A0201-CML患者から得たCD34+細胞によるコロニー形成は影響されず、コロニー形成活性を有する始原細胞の消失は、CTLによるHLA制限抗原認識に依存することを示した。
【0194】
最後に、CTLが白血病細胞と正常なCD34+始原細胞の間を識別するかどうかを調査した。正常CD34+細胞を、HLA−A0201+ドナーの骨髄、末梢血または臍帯血から単離し、CTL標的として使用した。CD34+細胞のソースとは独立して、抗P126CTLはこれらの細胞によってコロニー形成を阻害しなかった(図5D)。さらに正常CD34+細胞は、細胞毒性アッセイにおいて標的として使用された場合殺傷されなかった(図6B及びC)。白血病対正常CD34+細胞の選択的なCTL殺傷は、WT1発現における差異によって説明できる。WT1RNA発現は、正常CD34+細胞と比較して白血病細胞で高く(図5B)、ウエスタンブロット分析は白血病細胞でのみWT1タンパク質を検出し、正常CD34+細胞集団においては検出しなかった(図5C)。
【0195】
以前の自己幹細胞移植の後に再発した患者に対するドナーリンパ球(DLI)の注射は、強力なグラフト対白血病効果(GVL)を引き起こすことができる24,25。最近の研究により、DLIを経験したCML患者における完全な回復は、白血病CD34+始原細胞を認識するT細胞の増大した割合と関連することが示された15。対照的に、CML患者におけるより成熟したCD34-細胞のT細胞認識は、好ましい臨床上の応答と関連しなかった。このことはCD34+CML始原細胞に対して特異性を有するTリンパ球が、in vivoでの抗白血病効果を介在する点で必須に重要であることを示唆した。しかしながら、白血病CD34+始原細胞に対して選択的にT細胞応答を向けることができる標的抗原の性質に関する情報は現在存在しない。
【0196】
上記抗原を同定するために、免疫寛容と独立して、トランスフォーム細胞において上昇したレベルで発現されるいずれかのタンパク質に対してCTLを与えるのに適した、自己制限CTLアプローチを使用した22,23,26。細胞毒性エフェクター機能の引き金を引くことは初めの現象であるために、トランスフォーム細胞における上昇した標的タンパク質レベルによってのみ引き金を引かれ、正常細胞におけるタンパク質の生理学的レベルによっては引かれないCTLを選択することが可能である23。
【0197】
白血病CD34+細胞におけるWT1の過剰発現の証拠が存在する12。上昇したWT1発現は、トランスフォーメーションに寄与できる13,14。正常なWT1発現は、出生後の人生において少数の細胞に制限される21。白血病に加えて、上昇したWT1発現はまた、腎細胞カルシノーマ、卵巣ガン、進行性乳ガン、及びメラノーマにおいて観察されている27-30。それ故、WT1過剰発現悪性細胞を選択的に認識するCTLは、白血病、並びに他のより一般的な悪性疾患の抗原特異的な治療に対して貴重な試薬である。さらに、WT1発現の下流調節による腫瘍エスケープは、もし過剰発現がトランスフォームされた表現型を維持するために必要である場合生じないようである31-33。
【0198】
ここに記載される自己制限CTLは、HLA−A0201-ドナーから単離され、それらはHLA−A0201クラスI分子に関してWT1由来P126ペプチドを提示する白血病始原細胞に対して特異的であった。異なるHLA−A0201-ドナーから由来するリンパ球のin vitroの刺激は、ペプチド特異的HLA−A0201制限CTLを一貫して誘導するため、P126ペプチドは非常に免疫原性である。それ故P126特異的CTLは、HLA−A0201対立遺伝子に関して一つのローカスHLAミスマッチを提示するドナーから幹細胞移植を受けているHLA−A0201+白血病患者の抗原特異的治療のための新規な試薬である。一つのローカスHLAミスマッチは、一つのローカスミスマッチを有しHLAマッチを有する非関連ドナーから移植を受けた白血病患者における比較可能な予後を示す最近の研究に示されているように、臨床上許容可能である34。かくして一つのローカスミスマッチ移植は、ドナーから由来する自己制限CTLでの抗原特異的治療のための理想的なセッティングを提供する。P126ペプチドを含むHLA−A0201テトラマーでの染色による、関連CTLの選択と組み合わせたここに記載されるin vitro刺激プロトコールは、採用可能な治療のためのP126特異的CTLの迅速な単離を可能にするであろう。
【0199】
さらに、WT1は自己セッティングにおける抗原特異的治療について調査可能である。これは、P126特異的CTLがHLA−A0201+ドナーから単離できるという観察によって支持される。成人におけるWT1発現は、比較的小さい数の細胞(例えばCD34+骨髄細胞、腎有足突起、精巣セリトール細胞及び卵巣顆粒層細胞)に制限されるため、WT1に対する自己Tリンパ球の寛容はおそらく不完全である。それ故、白血病、並びに腎細胞カルシノーマ、卵巣ガン、メラノーマ、及び乳ガンのような、上昇したWT1発現を有する他の悪性疾患に対してCTL応答を刺激するために、抗WT1ワクチン調製物のデザインのために同定されたP126エピトープを利用することが可能であろう。
【0200】
in vivoでの治療に加えて、WT1特異的CTLは、白血病患者から回収された自己骨髄細胞のin vitroでの浄化のためのツールを提供する。CTLは、顆粒球/マクロファージ直系の白血病始原細胞(図6D)及び赤血球直系の始原細胞(示さず)を除去する。対照的にCTLは、これらの直系の正常の始原細胞は認識しない。トランスフォームCD34+始原細胞の選択的な除去は、白血病始原細胞を再注射する危険を減少するはずであり、かくして自己幹細胞移植の主要な制限を解消する35。
【0201】
今日まで、組織特異的マイナー組織適合性抗原及びプロテイナーゼ3のような直系特異的抗原は、白血病反応性CTLに対する潜在的案標的として研究されている36-39。WT−1転写因子は、白血病始原細胞に対してCTL応答を選択的に向けることが可能な最初の標的抗原である。
【0202】
実施例4の参考文献
[参考文献]
【技術分野】
【0001】
本発明は、免疫治療法、および、免疫治療法に使用される分子および細胞に関する。特に、本発明は、白血病を含むガンの治療に関する。
【背景技術】
【0002】
抗腫瘍細胞毒性Tリンパ球(CTL)がin vivoにおいて重要な役割を演じるという証拠がある。腫瘍反応性CTLが腫瘍後退を媒介することが、動物モデル(Kastら(1989) Cell 59, 603-614)およびヒト(カワカミら(1994) Proc.Natl.Acad.Sci.USA91,6458-6462)において示された。抗腫瘍治療の全てのタイプに関して、克服される必要のある問題は、この治療が有益な程度まで標的腫瘍細胞を破壊または不活性化しなければならないが、この治療が非腫瘍細胞を有害な程度まで破壊または不活性化してはならないことである。言い換えれば、この治療が腫瘍細胞に対して有益な程度にまで選択的であれば好ましい。
【0003】
ガンの免疫治療に対する現在の研究のほとんどは、一部の腫瘍が、等価の非腫瘍組織に発現されないポリペプチドを発現するという事実を利用するか、腫瘍が、非腫瘍組織に発現されないポリペプチドの変異形態を発現するという事実を利用する。しかしながら、このカテゴリーに入る腫瘍のポリペプチドを常に同定できるわけではなく、それゆえ、免疫治療アプローチのベースを形成する別の標的ポリペプチドが同定されてきた。
【0004】
メラノーマ患者における研究は、メラノーマ反応性CTLによって認識されたペプチドエピトープが組織特異的分化抗原からしばしば誘導されることを示した。正常組織に発現される分化抗原の認識は、免疫学的寛容のルールを壊すようにも見える;しかしながら、メラノーマ関連分化抗原のCTL認識は、それらが通常は、免疫学的に特別に許可された部位において比較的少数で存在するメラノサイトにおいて発現されるのみであり、かくして寛容を確立しないという事実によって説明される。また、前立腺特異的分化抗原が、前立腺の腫瘍に対するCTLの標的として作用しうることも示されている。発生的に制御された転写因子が、どの程度まで、これらの因子を異常に発現する腫瘍に対してCTLの標的として機能するのかは、現在のところ不明である。
【0005】
Gata−1は、赤血球、巨核球、エオシン好性白血球およびマスト細胞直系、並びに、多型潜在性先祖において通常発現される転写因子である。この厳しくコントロールされた転写因子の異常発現は、CMLおよびAMLを含む白血球において観察される(Shimamotoら(1995)Blood 86,3173-3180)。
【0006】
成人では、WT1、胚の転写因子の発現が、腎の有足突起、精巣、卵巣、胸部筋上皮細胞、そして骨髄の一部のCD34+幹細胞に観察されている。異常発現は、乳ガン、卵巣ガン、メラノーマおよび、CMLおよびAMLを含む白血病に観察された(例えば、Menssenら(1995) Leukaemia 9,1060-1067;Inoueら(1997) Blood 89,1405-1412; Inoueら(1996) Blood 88, 2267-2278; Inoueら (1998) Blood 91, 2969-2976; Menssenら(1997) Int.J.Cancer 70, 518-523; Menssenら(1995) Leukemia 9, 1060-1067; Ogawaら(1998) Transplant 21,527-527; Rodeckら(1994) Int.J.Cancer 59, 78-82; Silbersteinら(1997) Proc.Natl.Acad.Sci.USA 94, 8132-8137; Tamakiら (1996) Blood 88, 4396-4398; およびVielら(1994) Int.J.Cancer 57,515-521)。
【0007】
米国特許第5726288号は、Wilmsの腫瘍遺伝子の位置および特徴に関する。4つのアミノ酸配列が開示されており(配列番号2、4、5および6)、配列RMFPNAPYLまたはCMTWMNQMNLのいずれかを含むが、これらの配列に対応するペプチドもしくは免疫治療におけるそれらの用途については全く開示されていない。
【0008】
アロ-MHC-制限CTLを用いる通常でないアプローチを用いて、タンパクWT−1およびgata−1のペプチドエピトープを驚くべきことに同定した。これらは、HLA−A0201クラスI分子によって提示され、これらのタンパクを内因的に発現する腫瘍細胞表面に提示される。HLA−A0201陰性は、HLA−A0201クラスI分子によって提示されたペプチドに特異的なCTLの源として用いられ、このアプローチは、自己のCTLの可能な寛容から独立したHLA−A0201提示ペプチドの同定を可能にする。
【0009】
HLA−A0201は、最も一般的なHLA−Aハプロタイプである。
【0010】
疑いを除くために、用語HLAおよびMHCは本明細書で交換可能に用いられる。
【先行技術文献】
【特許文献】
【0011】
【特許文献1】米国特許第5726288号
【非特許文献】
【0012】
【非特許文献1】Kastら(1989) Cell 59, 603-614
【非特許文献2】カワカミら(1994) Proc.Natl.Acad.Sci.USA91,6458-6462
【非特許文献3】Shimamotoら(1995)Blood 86,3173-3180
【非特許文献4】Menssenら(1995) Leukaemia 9,1060-1067
【非特許文献5】Inoueら(1997) Blood 89,1405-1412
【非特許文献6】Inoueら(1996) Blood 88, 2267-2278
【非特許文献7】Inoueら (1998) Blood 91, 2969-2976
【非特許文献8】Menssenら(1997) Int.J.Cancer 70, 518-523
【非特許文献9】Menssenら(1995) Leukemia 9, 1060-1067
【非特許文献10】Ogawaら(1998) Transplant 21,527-527
【非特許文献11】Rodeckら(1994) Int.J.Cancer 59, 78-82
【非特許文献12】Silbersteinら(1997) Proc.Natl.Acad.Sci.USA 94, 8132-8137
【非特許文献13】Tamakiら (1996) Blood 88, 4396-4398
【非特許文献14】Vielら(1994) Int.J.Cancer 57,515-521
【発明の概要】
【発明が解決しようとする課題】
【0013】
本発明の第一の態様は、完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列RMFPNAPYLを含むペプチドまたはその部分または異型を提供する。
【課題を解決するための手段】
【0014】
本発明の第二の態様は、完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列CMTWNQMNLを含むペプチドまたはその部分または異型を提供する。
【0015】
本発明の第三の態様は、完全なヒトgata−1ポリペプチドではないという条件で、アミノ酸配列HLMPFPGPLLを含むペプチドまたはその部分または異型を提供する。
【0016】
“ペプチド”により、アミノ酸残基がペプチド結合(-CO-NH-)によって結合した分子のみでなく、ペプチド結合が転換された分子をも含める。かかるレトロ-インバーソ(retro-inverso)ペプチド模倣(peptidomimetics)は、当該技術分野において周知の方法、例えば、参照として含めるMeziereら(1997) J.Immunol.159,3230-3237に記載された方法を用いて調製することができる。かかるアプローチは、側鎖の配向ではなく、バックボーンを含む変化を含む偽ペプチドを調製することを含む。Meziereら(1997)は、少なくともMHCクラスIIおよびTヘルパー細胞応答に関して、これらの偽ペプチドが使用できることを示している。CO−NHペプチド結合の代わりにNH−CO結合を含むレトロ-インバースペプチドは、タンパク分解に対してはるかに耐性である。
【0017】
同様に、アミノ酸残基のCα原子間の空間を維持する適切なリンカー部が用いられるという条件で、ペプチド結合が省かれてもよく、リンカー部がペプチド結合の実質的に同一の平面と実質的に同一のチャージ分布とを有することが特に好ましい。
【0018】
ペプチドがそのN-またはC-末端において都合よくブロックされて、外タンパク分解(exoproteolytic)切断に対する感度を低減することが好ましい。
【0019】
所定のアミノ酸配列の“部分(portion)”により、ペプチドが所定のアミノ酸配列からなるペプチドと実質的に同様にHLA分子に結合できるような、所定の配列の少なくとも6つの連続するアミノ酸を意味する。
【0020】
所定のアミノ酸配列の“異型(variant)”により、例えば1または2つのアミノ酸残基の側鎖が、ペプチドが所定のアミノ酸配列からなるペプチドと実質的に同様にHLA分子に結合できるように、変更(例えば、別の天然に生じるアミノ酸残基の側鎖または他の側鎖で置換することによる)されていることを意味する。例えば、ペプチドは、改善しないにしても、HLA−A0201のような適切なMHC分子と相互作用して結合する能力を少なくとも維持するように、そして、改善しないにしても、本発明の第一、第二または第三の態様に定義したようなアミノ酸配列を含むポリペプチド(例えば、WT1またはgata−1)を異常に発現する細胞を認識して殺すことができる活性化CTLを産生する能力を少なくとも維持するように、修飾されてもよい。HLA−A2結合ノナマーの2位および9位は、通常、アンカー残基である。これらの残基およびHLA−A2を結合することに関わる他の残基の修飾は、CTL認識を変更することなく結合を増強させることができる(例えば、Tourdotら(1997) J.Immunol.159,2391-2398参照)。
【0021】
T細胞レセプターと相互作用するのに必須でないアミノ酸残基は、取り込みがT細胞反応性に実質的に影響を与えず、関連MHCへの結合を排除しない別のアミノ酸で置換することによって修飾することができる。
【0022】
かくして、所定の条件から離れて、本発明のペプチドは、上記のアミノ酸配列または部分または異型を含むあらゆるペプチド(この用語はオリゴペプチドまたはポリペプチドも含む)とすることができる。通常、本発明のペプチドは、抗原提示細胞において発現されると、適切なMHC分子に結合できるフラグメントが形成されるように処理され、適切な細胞によって提示されて、適切なT細胞応答を導き出すペプチドである。このペプチドから産生されたフラグメントも本発明のペプチドであると認められる。都合よく、本発明のペプチドは、所定のアミノ酸配列を含む部分またはその部分または異型、そしてさらに、一部の所望の特性を与えるさらなる部分を含む。例えば、前記さらなる部分とは、さらなるT細胞エピトープを含むか(最初のT細胞エピトープ含有部分と同じポリペプチドから誘導されてもそうでなくてもよい)、キャリアータンパクまたはペプチドを含むことができる。かくして、一つの実施態様において、本発明のペプチドは、ヒトWT−1部分がアミノ酸配列RMFPNAPYLまたはCMTWNQMNLまたはこれらの両方を含むという条件において、切断されたヒトWT−1タンパクまたはWT−1タンパクフラグメントと他のポリペプチド部分との融合タンパクである。別の実施態様では、本発明のペプチドは、ヒトgata−1部分がアミノ酸配列HLMPFPGPLLを含むという条件において、切断されたヒトgata−1タンパクまたはヒトgata−1タンパクフラグメントと他のポリペプチド部分との融合タンパクである。
【0023】
特に好ましい実施態様では、本発明のペプチドは、本発明の第一、第二または第三の態様に与えられたアミノ酸配列、並びに少なくとも一つのさらなるT細胞エピトープを含み、ここでさらなるT細胞エピトープは、WT−1またはgata−1を異常に発現する腫瘍のタイプに向けられたT細胞応答の産生を容易にすることができるものである。かくして、本発明のペプチドは、ワクチンとして用いられる、いわゆる“ビーズ・オン・ストリング(beads on a string)”ポリペプチドを含む。
【0024】
本発明のペプチドは、あらゆる大きさとすることができるが、通常は、分子量が100000未満、好ましくは50000未満、さらに好ましくは10000未満、そして通常は約5000とすることができる。アミノ酸残基の数としては、本発明のペプチドは1000残基未満、好ましくは500残基未満、さらに好ましくは100残基未満とすることができる。
【図面の簡単な説明】
【0025】
【図1】図1は、抗WT126−34CTLの殺傷活性を示す。T2はATCCからCatalogue No CRL 1992のもとに入手可能なペプチド結合欠陥を有するヒト細胞系統である。これらは、WT126−34ペプチド(RMFPNAPYL)(T2+WT12)、または、HLA−A2−結合ペプチド(T2+E7)であるE7コントロールペプチド(TLGIVCPI)を用いて結合される。K562は、白血病細胞系統であり、K562−A2はHLA−A0201でトランスフェクションされたK562白血病細胞系統である。E:T比は、エフェクター:ターゲット細胞比である。比溶解(%)は、参照としてここに含める、Sadovnikova & Stauss(1996) Proc.Natl.Acad.Sci.USA 93, 13114-13118に記載されているような、標準的なCTLアッセイで測定した。
【図2】図2は、抗huWT126−34CTLの殺傷活性を示す。Leuk-697、MV441およびBV173は白血病細胞系統である。P12はWT126−34ペプチド(RMFPNAPYL)を示す。
【図3】図3は、抗hug378−87CTLの殺傷活性を示す。hug378−87は、ペプチドHLMPFPGPLLであり、hug14はHLA−A2−結合コントロールペプチド(RLSPDLLTL)である。C1R−A2はヒトBリンパ球細胞系統である。K562−A2ヒトは、白血病細胞系統である。
【図4A】図4は、WT−1誘導ペプチドP126に対して生じたアロ制限CTLの特異性を示す。CTLを、P126ペプチドで被覆されたHLA−A0201+刺激細胞で刺激されたHLA−A0201−ドナーからTリンパ球バルク培養物の制限希釈クローニングによって単離した。(A)単離されたCTL系統は、免疫化P126ペプチドで被覆されたTAP欠陥T2ターゲット細胞を殺傷したが、HLA−A0201結合E7コントロールペプチドで被覆されたT2細胞を殺さなかった。
【図4B】(B)ペプチド滴定実験は、3つの抗P126CTL系統が低ピコモーラーのP126を認識する高い親和力であり、3つのCTL系統が低い親和力であったことを示す。これは、ナノモーラーP126濃度が、ターゲット細胞認識に必要であったからである。示された濃度のP126で被覆されたT2細胞は、CTLターゲットとして使用できた。低親和性CTLがWT1を内因的に発現するターゲット細胞を認識しなかったことから、高親和性CTLを、次の全ての実験に用いた。
【図4C】(C)抗親和性CTLは、HLA−A0201+白血病細胞系統BV173,697を殺傷するが、HLA−A0201+、EBV−トランスフォームBリンパ球細胞C1R−A2を殺傷しなかった。C1R−A2のP126でのコーティングは、効率的なCTL殺傷をもたらした。HLA−A0201-白血病細胞系統K562は、HLA−A0201遺伝子でトランスフェクションされない限りCTLによって殺傷されない。
【図5】白血病患者と正常ドナーから単離されたCD34+およびCD34-細胞集団および白血病細胞系統におけるタンパク発現とWT1 RNAを示す。(A)白血病細胞系統およびBリンパ球細胞系統C1R−A2におけるWT1 RNAを測定するRT−PCR。図4CのCTLターゲットと同じ細胞系統を用いた。増幅されたWT1産物は482bpであり、ハウスキーピングABL遺伝子のRNAを増幅して、各サンプルのRNAの量を示した。ABL産物は385bpの長さである。(B)4人のCML患者および3人の正常ドナー由来の、精製されたCD34+とCD34−細胞集団におけるWT1 RNA発現を測定するRT−PCR。白血病細胞系統BV173は、WT1発現の陽性コントロールとして提供された。さらに6人のCML患者からのサンプルを用いて類似の結果が得られた。(C)2人のCML患者および2人の正常ドナー由来の精製されたCD34+およびCD34−細胞集団および白血病細胞系統におけるWT1タンパク発現を測定するウェスタンブロッティング。ハウスキーピングアクチンタンパクの発現は、各サンプル中に存在するタンパクの量を制御するためのインジケーターとして用いられた。WT1タンパクは約54kDaの大きさであり、アクチンタンパクは約42kDaであった。
【図6A】図6は、白血病患者および正常ドナーから精製されたCD34+細胞集団のコロニー形成の阻害およびCTL媒介殺傷の分析である。(A)HLA−A0201+またはA0201-であるCML患者から単離された精製CD34+およびCD34-細胞集団に対する抗P126CTLによる殺傷レベルを示す代表的な実験である。CD34-/A0201+細胞は、P126ペプチドでコートされない限りCTLに認識されなかった。P126またはコントロールE7ペプチドで被覆されたTAP欠陥T2細胞および白血病細胞系統BV173は、全ての実験で陽性および陰性のコントロールとして用いられた。
【図6B】(B)11人の異なるHLA−A0201+CML患者と6人の正常ドナーに由来する精製CD34+細胞の特異的CTL殺傷レベルの平均。陽性コントロール細胞BV173に対する、CML患者から精製されたCD34-細胞の殺傷レベルも示されている。この図は、特異的CTL殺傷の平均値と標準偏差を示す。
【図6C】(C)HLA−A0201+正常ドナーから単離された精製CD34+とCD34−細胞集団の抗P126CTLによる殺傷レベルを示す代表例である。CTLは、ターゲット細胞がP126ペプチドで被覆されない限り検出されなかった。
【図6D】(D)エフェクター/ターゲット細胞比10:1においてCTLで4時間共培養された精製CD34+細胞によるコロニー形成のCTL媒介阻害。未処理コントロールCD34+細胞を、CTLを含まずに同じ条件下で培養した。CTL処理および未処理コントロール細胞をメチルセルロース中におき、14日後にCFU−GMの数をカウントした。未処理コントロール中に観察されたCFU−GMを100%基準として用いてCTL処理後のCFU−GMのパーセントを示す。この図は、9人の異なるHLA−A0201+CML患者、7人の異なる正常ドナーに由来するCD34+、および5人の異なるHLA−A0201-CML患者に由来するCD34+細胞を用いた、独立した実験の平均と標準偏差を示す。
【図6E】(E)高親和性P126特異的CTL(ライン81)または低親和性CTL(ライン85)で、プレーティングの前に、4時間処理されたまたは未処理のA0201+/CD34+CML細胞によるコロニー形成。未処理コントロールに観察されたコロニーの数を100%の基準として用いてCFU−GMおよびBFU−Eを示す。
【図6F】(F)コールドターゲット競合実験。30倍過剰のコールドBV173およびC1R−A2ターゲットの存在下または不在下で、A0201+CML患者に由来するクロムラベルCD34+ターゲットに対する抗P126CTLによる殺傷を示す。クロムラベルBV173およびC1R−A2の殺傷は、比較のために示されている。
【発明を実施するための形態】
【0026】
以下から、一部の適用において、本発明のペプチドが直接的に用いられることがわかる(すなわち、患者の細胞または患者に与えられた細胞においてポリヌクレオチドの発現により産生されるものではない);そのような適用においては、ペプチドが100残基未満を有することが好ましい。
【0027】
本発明のポリペプチドが、HLA−A0201に結合できることが好ましい;しなしながら、このペプチドは他のHLAタイプおよびHLA−A0201に結合してもよい。特に、ペプチドがHLA−A0201に選択的に結合できることが好ましい。
【0028】
本発明のペプチドは、特に、WT1ポリペプチド(Wilms腫瘍遺伝子産物)またはgata−1ポリペプチドを異常に発現する細胞を標的とし殺す免疫治療方法において特に使用できる。gata−1ポリペプチドは、一部の遺伝子のプロモーター領域に存在するgata−ボックスに結合する転写因子であることから、そのように命名された。それはまた、NF−e1、GF−1およびEryf−1とも呼ばれる。アミノ酸配列RMFPNAPYLおよびCMTWNQMNLは、それぞれWT1の126−134および235−243残基に見られ、アミノ酸配列HLMPFPGPLLは、gata−1の378−387残基に見られる。WT−1アミノ酸配列は、Gesslerら(1990) Nature 343, 774-778に与えられており、gata−1アミノ酸配列は、Zonら(1990) Proc.Natl.Acad.Sci.USA 87, 668-672に与えられており、これらの刊行物の両方を参照としてここに含める。
【0029】
所定のアミノ酸配列からなるこれらの特異的ペプチドは、HLA−A0201に結合するので、本発明のペプチドはHLA−A0201に結合し、結合した際に、HLA−A0201−ペプチド複合体が、適切な抗原提示細胞の表面に提示された際に、本発明の第一および第二の態様のペプチドに関してWT1ポリペプチドのような、そして、本発明の第三の態様のペプチドに関してgata−1ポリペプチドのような、所定のアミノ酸配列を含むポリペプチドを異常に発現する細胞を認識するCTLの産生を引き出すことができるペプチドであることが好ましい。
【0030】
WT1ポリペプチドは、白血病、乳ガン、メラノーマおよび卵巣ガンにおいて異常に発現され、gata−1ポリペプチドは白血病において異常に発現される。
【0031】
“異常に発現”により、ポリペプチドが発現の通常レベルと比較して過剰に発現されること、もしくは、腫瘍においては発現されるが、腫瘍が誘導される組織において遺伝子がサイレントであることを含む。“過剰発現”により、ポリペプチドが、正常細胞に存在するレベルの少なくとも1.2倍、好ましくは少なくとも2倍、さらに好ましくは少なくとも5倍または10倍のレベルで存在することを意味する。
【0032】
HLA分子に結合するペプチドの最適な長さが約8ないし12アミノ酸、好ましくは9アミノ酸であることは周知である。
【0033】
本発明の特に好ましいペプチドは、アミノ酸配列RMFPNAPYLまたはCMTWNQMNLまたはHLMPFPGPLLからなるものである。
【0034】
12アミノ酸残基より大きいペプチドがMHC分子との結合に直接用いられる場合には、コアHLA結合領域に隣接する残基が、ペプチドがMHC分子に結合する能力またはCTLにそのペプチドを提示する能力に実質的に影響を与えないものであることが好ましい。しかしながら、特にポリヌクレオチドによってコードされる場合に、より大きなペプチドが用いられることがわかる。なぜなら、これらの大きなペプチドは、適切な抗原提示細胞によって断片化されうるからである。
【0035】
ペプチド(少なくとも、アミノ酸残基間にペプチド結合を含むもの)は、Luら(1981) J.Org.Chem.46,3433とそこに記載された参考文献に記載されている固相ペプチド合成のFmoc-ポリアミドモード(Fmoc-polyamide mode)によって合成されてもよい。一時的Nアミノ基保護は、9−フルオレニルメチルオキシカルボニル(Fmoc)基によって供給される。この高度にベース-置換活性保護基の反復切断は、N,N-ジメチルホルムアミド中の20%ピペリジンを用いて実行される。側鎖官能性は、そのブチルエーテル(セリン、スレオニンおよびチロシンの場合)、ブチルエステル(グルタミン酸およびアスパラギン酸の場合)、ブチルオキシカルボニル誘導体(リシンおよびヒスチジンの場合)、トリチル誘導体(システインの場合)および4-メトキシ-2,3,6-トリメチルベンゼンスルホニル誘導体(アルギニンの場合)として保護することができる。グルタミンまたはアスパラギンがC末端残基である場合には、側鎖アミド官能性の保護には4,4'-ジメトキシベンズヒドリル基を使用する。固相支持体は、三つのモノマーであるジメチルアクリルアミド(骨格モノマー)、ビスアクリロイルエチレンジアミン(架橋剤)およびアクリロイルサルコシンメチルエステル(官能化剤)から構成されたポリジメチル−アクリルアミドポリマーをベースとする。使用されたペプチド−樹脂切断可能連結剤は、酸不安定性4-ヒドロキシメチル-フェノキシ酢酸誘導体である。全てのアミノ酸誘導体は、アスパラギンとグルタミンを除いて、予め形成された対称的な無水物誘導体として添加され、これらは、リバースN,N-ジシクロヘキシル-カルボジイミド/1-ヒドロキシベンゾトリアゾル媒介カップリング処理を用いて加えられる。全てのカップリングおよび脱保護化反応は、ニンヒドリン、トリニトロベンゼンスルホン酸、またはイソチン(isotin)テスト処理を用いて観察される。合成の完了時に、50%のスカベンジャーミックスを含む95%のトリフルオロ酢酸で処理することにより、側鎖保護基の付随した除去とともに、ペプチドを樹脂支持体から切断する。一般的に用いられるスカベンジャーは、エタンジチオール、フェノール、アニソールおよび水であり、その的確な選択は合成されるペプチドの構成アミノ酸に基づく。ト
リフルオロ酢酸は、真空で蒸発させることによって除去され、その後のジエチルエーテルを用いた摩砕が粗製ペプチドを与える。存在する全てのスカベンジャーを単純な抽出方法で除去する。水相の凍結乾燥により、スカベンジャーを含まない粗製ペプチドを与える。ペプチド合成のための試薬は、一般的にCalbiochem-Novabiochem(UK)Ltd,Nottingham NG7 2QJ, UKから入手できる。精製は、ゲルろ過クロマトグラフィー、イオン交換クロマトグラフィーおよび、(主に)逆相高速液体クロマトグラフィーのような技術のいずれか一つまたは組み合わせにより行うことができる。ペプチドの分析は、薄相クロマトグラフィー、逆相高速液体クロマトグラフィー、酸加水分解後のアミノ酸分析および高速原子衝撃(FAB)質量分析を用いて行うことができる。
【0036】
本発明のさらなる態様は、本発明の第一、第二または第三の態様に定義されたペプチドをコードするポリヌクレオチドを提供する。このポリヌクレオチドはDNAまたはRNAとすることができ、前記ペプチドをコードする限りイントロンを含んでも含まなくてもよい。もちろん、それは、ポリヌクレオチドにコードされうる、天然に生じるペプチド結合によって結合された天然に生じるアミノ酸残基を含むペプチドのみである。
【0037】
本発明のさらなる態様は、本発明の第一または第二または第三の態様にかかるポリペプチドを発現することができる発現ベクターを提供する。
【0038】
例えば相補付着末端を介して、ベクターに、DNAのようなポリヌクレオチドを結合するために、種々の方法が開発されている。例えば、相補的ホモポリマー領域を、ベクターDNAに挿入されるDNAセグメントに付加することができる。次いで、ベクターおよびDNAセグメントが、相補ホモポリメリックテイル間の水素結合によって結合され、組み換えDNA分子を形成する。
【0039】
一以上の制限部位を有する合成リンカーは、DNAセグメントをベクターに結合させる別の方法を提供する。上記のエンドヌクレアーゼ制限切断によって生じたDNAセグメントを、バクテリオファージT4 DNAポリメラーゼまたはE.coli DNAポリメラーゼIで処理する。これらの酵素は、突出した3’一本鎖末端を3’-5’エキソヌクレアーゼ活性で除去し、欠けた3’端をポリメラーゼ活性で満たす。
【0040】
これらの活性の組み合わせにより、平滑末端のDNAセグメントを形成することができる。この平滑末端セグメントを、バクテリオファージT4DNAリガーゼのような、平滑末端DNA分子のライゲーションを触媒することができる酵素の存在下で大過剰のリンカー分子と共にインキュベートする。かくして、この反応産物は、端部にポリメリックリンカー配列を備えたDNAセグメントである。これらのDNAセグメントは、適切な制限酵素で切断され、DNAセグメントの端部と適合する端部を生じる酵素で切断された発現ベクターにライゲートされる。
【0041】
種々の制限エンドヌクレアーゼ部位を含む合成リンカーは、International Biotechnologies Inc,New Haven, CN,USAを含む多数の源から商業的に入手可能である。
【0042】
本発明のポリペプチドをコードするDNAを修飾するのに望ましい方法は、Saikiら(1988) Science 239,487-491に記載されたポリメラーゼ連鎖反応を用いることである。この方法は、例えば、適切な制限部位において工作することにより、適切なベクターにDNAを導入するために用いることができ、また、当該技術分野において公知であるような他の使用できる方法でDNAを修飾するために用いることができる。
【0043】
この方法では、酵素により増幅されるDNAは、増幅されたDNAにそれ自身が取り込まれる二つの特異的プライマーによって隣接される。この特異的プライマーは、当該技術分野において公知の方法を用いて発現ベクターにクローニングするために用いられる制限エンドヌクレアーゼ認識部位を含む。
【0044】
DNA(または、レトロウイルスベクターの場合にはRNA)を、本発明の化合物を含むポリペプチドを産生する適切な宿主において発現させる。かくして、本発明の化合物を構成するポリペプチドをコードするDNAが、ここに含まれる教示から適切に変更された公知の技術に従って、発現ベクターを構成するために用いられてもよい。この発現ベクターは、本発明のポリペプチドの発現および産生に適した宿主細胞をトランスフォームするために用いられる。かかる技術は、Rutterらに1984年4月3日に発せられた米国特許第4440859号、Weissmanに1985年7月23日に発せられた第4530901号、Crowlに1986年4月15日に発せられた第4582800号、Markらに1987年6月30日に発せられた第4677063号、Goeddelに1987年7月7日に発せられた第4678751号、Itakuraらに1987年11月3日に発せられた第4704362号、Murrayに1987年12月1日に発せられた第4710463号、Toole,Jr.らに1988年7月12日に発せられた第4757006号、Goeddelらに1988年8月23日に発せられた第4766075号、およびStalkerに1989年3月7日に発せられた第4810648号に開示された技術を含み、これらの全てを参照としてここに含める。
【0045】
本発明の化合物を構成するポリペプチドをコードするDNA(レトロウイルスベクターの場合はRNA)が、適切な宿主に導入するために広範囲の他のDNA配列に結合されてもよい。コンパニオンDNAは、宿主の性質、宿主へのDNAの導入方法、およびエピソームメンテナンスまたはインテグレーションが望ましいかどうかに依存する。
【0046】
一般に、DNAは、プラスミドのような発現ベクターに、発現に適切な配向および正しい読み枠で挿入される。必要であれば、DNAは、所望の宿主によって認識される適切な転写および翻訳制御コントロールヌクレオチド配列に結合されてもよいが、かかるコントロールは一般的に発現ベクターにおいて利用可能である。次いで、このベクターを、通常の技術を介して宿主に導入する。一般的に、全ての宿主がベクターによりトランスフォームされるわけではない。それゆえ、トランスフォームされた宿主細胞を選別する必要がある。選別技術の一つは、必要なコントロール成分と共に、抗生物質耐性のような、トランスフォームされた細胞において選別可能な特徴をコードするDNA配列を発現ベクターに取り込むことを含む。あるいは、かかる選別可能な性質の遺伝子が、所望の宿主細胞をコトランスフォームするために用いられる別のベクターに存在してもよい。
【0047】
本発明の組み換えDNAによりトランスフォームされた宿主細胞を、ここに開示した技術の観点から当業者に公知の適切な条件下および十分な時間にわたって培養して、ポリペプチドを発現させ、次いで回収する。
【0048】
細菌(例えば、E.coliおよびBacillus subtilis)酵母(例えば、Saccharomyces cerevisiae)、糸状菌類(例えば、Aspergillus)、植物細胞、動物細胞および昆虫細胞を含む、多くの発現システムが知られている。
【0049】
通常、ベクターは、原核生物における増殖のために、そして、他の非原核細胞タイプにおける発現のために用いられる場合でさえも、ColE1 oriのような原核生物のレプリコンを含む。ベクターは、形質転換された、E.coliのような細菌性宿主細胞において遺伝子を発現(転写および翻訳)させることができる原核プロモーターのような適切なプロモーターを含むこともできる。
【0050】
プロモーターとは、RNAポリメラーゼを結合させ、転写を開始させるDNA配列によって形成された発現コントロール成分である。典型的な細菌性宿主と適合するプロモーター配列は、本発明のDNAセグメントの挿入に都合のよい制限部位を含むプラスミドベクターに設けられる。
【0051】
通常の原核生物のベクタープラスミドは、Biorad Laboratories(Richmond,CA,USA)から入手可能なpUC18、pUC19、pBR322およびpBR329と、Pharmacia, Piscataway,NJ,USAから入手可能なpTrc99AおよびpKK223−3である。
【0052】
通常の哺乳動物細胞ベクタープラスミドは、Pharmacia,Piscataway,NJ,USAから入手可能なpSVLである。このベクターは、クローン化遺伝子の発現を誘導するためにSV40後期プロモーターを用い、最高レベルの発現が、COS-1細胞のようなT抗原産生細胞に見られる。
【0053】
誘導できる哺乳動物発現ベクターの例はpMSGであり、これもPharmaciaから入手できる。このベクターは、クローン化遺伝子の発現を引き起こすために、マウス乳房腫瘍ウイルス長期リピートのグルココルチコイド誘導プロモーターを用いる。
【0054】
使用できる酵母プラスミドベクターは、pRS403−406およびpRS413−416であり、一般的に、Stratagene Cloning Systems, La Jolla, CA 92037, USAから入手できる。プラスミドpRS403、pRS404、pRS405、およびpRS406は、酵母統合プラスミド(YIp)であり、酵母選択マーカーHIS3、TRP1、LEU2およびURA3を取り込む。プラスミドpRS413−416は酵母セントロメアプラスミド(Ycp)である。
【0055】
他のベクターおよび発現システムは、種々の宿主細胞を用いた使用について当該技術分野において周知である。
【0056】
また、本発明は、本発明のポリヌクレオチドベクター構成物でトランスフォームされた宿主細胞に関する。宿主細胞は原核生物または真核生物とすることができる。細菌細胞は、一部の状況では原核生物宿主細胞が好ましく、通常は、E.coliの株、例えば、Bethesda Research Laboratories Inc.,Bethesda,MD,USAから入手可能なE.coli株DH5およびRockville,MD,USAのAmerican Type Culture Collection(ATCC)から入手できるRR1(No ATCC 31343)である。好ましい真核生物宿主細胞は、酵母、昆虫および哺乳動物細胞、好ましくは、マウス、ラット、サルまたはヒトの繊維芽細胞および腎臓細胞系統のような脊椎動物細胞を含む。酵母宿主細胞は、Stratagene Cloning Systems, La Jolla, CA 92037,USAから入手できるYPH499、YPH500およびYPH501を含む。好ましい哺乳動物宿主細胞は、CCL61としてATCCから入手可能なチャイニーズハムスター卵巣(CHO)細胞、CRL1658としてATCCから入手可能なNIH Swissマウス胚細胞NIH/3T3、CRL1650としてATCCから入手可能なサル腎臓誘導COS−1細胞、および、ヒト胚腎臓細胞である293細胞を含む。好ましい昆虫細胞は、バキュロウイルス発現ベクターでトランスフェクションされうるSf9細胞である。
【0057】
本発明のDNA構成物を用いた適切な細胞宿主のトランスフォーメーションは、用いられるベクターのタイプに依存する周知の方法によって行われる。原核生物宿主細胞のトランスフォーメーションに関しては、例えば、Cohenら(1972)Proc.Natl.Acad.Sci.USA69,2110およびSambrookら(1989)Molecular Cloning,A Laboratory Manual, Cold Spring Harbor Laboratory, Cold Spring Harbor,NYを参照。酵母細胞のトランスフォーメーションは、Shermanら(1986)Methods In Yeast Genetics, A Laboratory Manual, Cold Spring Harbor, NYに記載されている。Beggs(1978)Nature 275,104-109の方法も使用できる。脊椎動物細胞に関して、かかる細胞をトランスフェクションするのに使用できる試薬、例えば、リン酸カルシウムおよびDEAE-デキストランまたはリポソームフォーミュレーションは、Stratagene Cloning SystemsまたはLife Technologies Inc.,Gaithersburg,MD 20877, USAから入手できる。
【0058】
エレクトロポレーションも、細胞をトランスフォームおよび/またはトランスフェクトするのに使用でき、酵母細胞、細菌細胞、昆虫細胞および脊椎動物細胞をトランスフォームするのに、当該技術分野において周知である。
【0059】
例えば、多くの細菌種が、参照としてここに含めるLuchanskyら(1988)Mol.Microbiol.2,637-646に記載された方法によりトランスフォームされる。最大数のトランスフォーマントは、一貫して、25μFDにおいてcm当たり6250Vを用いて2.5X PEBに懸濁されたDNA-細胞混合物のエレクトロポレーション後に回収される。
【0060】
エレクトロポレーションによる酵母のトランスフォーメーション方法は、Becker&Guarente (1990) Metholds Enzymol.194,182に記載されている。
【0061】
首尾よくトランスフォームした細胞、すなわち本発明のDNA構成物を含む細胞は、周知の技術により同定することができる。例えば、本発明の発現構成物の導入から得られた細胞は、増殖して本発明のポリペプチドを生じる。細胞を回収して溶解し、そのDNA内容物を、Southern(1975)J.Mol.Biol.98,503またはBerentら(1985)Biotech.3,208に記載されている方法を用いてDNAの存在について試験することができる。あるいは、上清におけるタンパクの存在を、以下の抗体を用いて検出することができる。
【0062】
組み換えDNAの存在を直接アッセイすることに加えて、組み換えDNAがタンパクの発現に向けられる場合には、トランスフォーメーションの成功を、周知の免疫学的方法によって確認できる。例えば、発現ベクターで首尾よくトランスフォームした細胞は、適切な抗原性を示すタンパクを産生する。トランスフォームされたと思われる細胞のサンプルを、回収し、適切な抗体を用いてタンパクをアッセイする。
【0063】
かくして、トランスフォームされた宿主細胞自身の他に、本発明は、これらの細胞の培養物、好ましくはモノクローナル(クローンとして均質)培養物、または栄養培地におけるモノクローナル培養から誘導された培養物をも含む。
【0064】
本発明の一部の宿主細胞、例えば、細菌、酵母および昆虫の細胞が、本発明のペプチドの調製に使用できることも考えられる。しかしながら、他の宿主細胞は、一部の治療方法に使用できる。例えば、抗原提示細胞、例えば樹状細胞は、本発明のペプチドを発現し、適切なMHC分子に結合するために使用できる。
【0065】
本発明のさらなる態様は、本発明の第一、第二または第三の態様のペプチドを産生する方法を提供することであり、この方法は、ペプチドをコードするポリヌクレオチドまたは発現ベクターを含む宿主細胞を培養し、宿主細胞または培養培地からペプチドを得ることを含む。
【0066】
本発明のさらなる態様は、薬学的に許容できるキャリアーと、本発明の第一、第二または第三の態様にかかるペプチドまたはかかるペプチドをコードするポリヌクレオチドまたは発現ベクターを含む薬学的組成物を提供する。この薬学的製剤は、患者への投与に適した形態に調製され、無菌でありかつ発熱物質を含まない。本発明のさらなる態様は、本発明の第一、第二または第三の態様にかかるペプチド、またはかかるペプチドをコードするポリヌクレオチドまたは発現ベクターを、薬剤における使用のために提供する。このペプチドまたはポリヌクレオチドまたは発現ベクターは、収容されて、薬剤における使用のために提供される。
【0067】
薬学的組成物、または収容および提示は、適切な形態とすることができる。適切な形態が静脈(i.v.)注射、皮下(s.c.)注射、皮内(i.d.)注射、腹腔内(i.p.)注射、筋肉内(i.m.)注射用であることが考えられる。
【0068】
ペプチド注射の好ましい方法は、s.c.、i.d.、i.p.、i.m.そしてi.v.である。
【0069】
DNA注射の好ましい方法は、i.d.、i.m.、s.c.、i.p.そしてi.v.である。
【0070】
1ないし500mgのペプチドまたはDNAの投与量を与えることができる。
【0071】
本発明のさらなる態様は、本発明の第一、第二または第三態様のいずれかのアミノ酸配列を含むポリペプチドを異常に発現する、患者の標的細胞を殺す方法を提供するものであって、この方法は、患者に、有効量の、本発明の第一、第二または第三態様のいずれかにかかるペプチドを投与すること、あるいは、前記ペプチドをコードする発現ベクターまたはポリヌクレオチドの有効量を投与することを含み、前記ペプチドの量またはポリヌクレオチドまたは発現ベクターの量は、前記患者の抗ターゲット細胞免疫応答を誘発するのに有効な量である。
【0072】
ターゲット細胞は、通常、腫瘍またはガン細胞である。通常、腫瘍またはガン細胞は、WT1またはgata−1を異常に発現する細胞である。
【0073】
ペプチドまたはペプチドをコードする核酸は、腫瘍またはガンワクチンを構成する。患者に直接、影響を受けた器官または全身的に投与されてもよく、患者またはヒト細胞系統から誘導された細胞にex vivoで適用され、その後患者に投与されてもよく、また、患者から誘導された免疫細胞から部分集団を選別するためにin vitroで用いられ、次いで患者に再投与されてもよい。核酸がin vitroで細胞に投与される場合には、細胞が、インターロイキン-2のような免疫刺激サイトカインを共発現するようにトランスフェクションされることが有益である。ペプチドは実質的に純粋であっても、Detoxのような免疫刺激アジュバントと組み合わされてもよく、免疫刺激サイトカインと組み合わせて用いられてもよく、また、リポソームのような適切なデリバリーシステムと共に投与されてもよい。また、ペプチドは、キーホールリンペットヘモシアニン(KLH)またはマンナンのような適切なキャリアーと組み合わされてもよい(WO95/18145およびLongeneckerら(1993)Ann.NY Acad.Sci.690,276-291参照)。また、このペプチドは、ダグが付されるか、融合タンパクとされるか、ハイブリッド分子とされてもよい。本発明の第一、第二または第三態様にその配列が与えられたペプチドは、CD8 CTLを刺激することが期待される。しかしながら、刺激はCD4T細胞によって提供される助けがあるとさらに効率的である。かくして、ハイブリッド分子の融合パートナーまたはセクションは、CD4T細胞を刺激するエピトープを適切に提供する。CD4刺激エピトープは、当該技術分野において周知であり、破傷風トキソイドに同定されるものを含む。ポリヌクレオチドは、実質的に純粋であっても、適切なベクターまたはデリバリーシステムに含まれてもよい。適切なベクターおよびデリバリーシステムは、アデノウイルス、ワクシニアウイルス、レトロウイルス、ヘルペスウイルス、アデノ関連ウイルス、または一以上のウイルスの成分を含むハイブリッドに基づくシステムのようなウイルス性のものを含む。非ウイルス性デリバリーシステムは、DNAデリバリーの分野で周知であるようなカチオン脂質およびカチオンポリマーを含む。物理的デリバリー、例えば、“遺伝子銃”を介するものも使用できる。ペプチドま
たは核酸によってコードされるペプチドは、例えば、CD4T細胞を刺激する破傷風トキソイドのエピトープとの融合タンパクであってもよい。
【0074】
ガンワクチンに使用されるペプチドは、適切なペプチドであってもよい。特に、適切な9マーのペプチド、または適切な7マーまたは8マーまたは10マーのペプチドとすることができる。また、より長いペプチドも適しているが、9マーまたは10マーのペプチドが好ましい。ガンワクチンが、WT1を発現するガンに関して用いられる場合には、本発明の第一および第二の態様の両方のペプチドを用いる、あるいは、本発明の第一および第二の態様に記載された両方の配列を含む一つのペプチドを用いることが有利である。
【0075】
患者に投与される核酸が無菌かつ発熱性物質を含まないことが適切である。裸のDNAは、筋肉内、皮内または皮下に投与できる。ペプチドは、筋肉内、皮内または皮下に投与できる。
【0076】
ワクチン接種は、プロフェッショナル抗原提示細胞により刺激されたCTL応答をもたらし、CTLが刺激されると、腫瘍細胞におけるMHC発現を増強する利点が存在する。
【0077】
注入部位、ターゲッティングベクターおよびデリバリーシステムの使用、または、患者からかかる細胞集団を選択的に精製してペプチドまたは核酸をex vivo投与することのいずれかによって、ワクチンを、例えば抗原提示細胞のような特異的細胞集団に向けることも有益である(例えば、樹状細胞をZhouら(1995) Blood86,3295-3301;Rothら(1996)Scand.J.Immunology 43,646-651に記載されているように選別してもよい)。例えば、ダーゲッティングベクターは、適切な場所において抗原を発現させる組織-または腫瘍-特異的プロモーターを含んでもよい。
【0078】
治療が施される患者は、WT1またはgata−1の過剰発現または異常発現(両方とも異常な発現)のタイプの腫瘍を有しうる。
【0079】
それゆえ、本発明のさらなる態様は、ガン、またはガンまたは腫瘍細胞に有効なワクチンを提供することであり、これは、本発明の第一、第二または第三の態様のいずれか一つにかかるペプチドの有効量を含むか、かかるペプチドをコードする核酸を含む。
【0080】
ワクチンが核酸ワクチンであることが特に好ましい。あるポリペプチドをコードするDNAワクチンのような核酸ワクチンを用いた接種が、T細胞反応を導くことが知られている。
【0081】
都合よく、核酸ワクチンは、適切な核酸デリバリー手段を含んでもよい。核酸、好ましくはDNAは、裸であってもよく(すなわち、投与される他の成分を実質的に含まない)、また、リポソーム中またはウイルスベクターデリバリーシステムの一部としてデリバリーされてもよい。
【0082】
樹状細胞による核酸の取り込みおよびコードされたポリペプチドの発現が、免疫応答の開示のメカニズムであってもよい;しかしながら、樹状細胞はトランスフェクションされなくてもよいが、組織のトランスフェクションされた細胞から発現されたペプチドをピックアップするかもしれないので、重要である。
【0083】
DNAワクチンのようなワクチンを筋肉内に投与することが好ましい。また、皮膚にワクチンが投与されることも好ましい。
【0084】
核酸ワクチンはアジュバントを含まずに投与されてもよい。また、核酸ワクチンは、BCGまたはミョウバンのようなアジュバントと共に投与されてもよい。他の適切なアジュバントは、サポニン、マイコバクテリウム抽出物、および合成細菌性細胞壁模造体から誘導されるAquila QS21スティミュロン(stimulon)(Aquila Biotech,Worcester,MA,USA)およびRibi's Detoxのようなプロプリエトリー(proprietory)アジュバントを含む。別のサポニン誘導アジュバントであるQuil Aも使用できる(Superfos,Denmark)。核酸ワクチンがアジュバントなしで投与されることが好ましい。
【0085】
Freund'sのような別のアジュバントも使用できる。また、好ましくはアジュバントと共に、キーホールリンペットヘモシアニンと複合体形成したペプチドを得ることも有益である。
【0086】
ガンのポリヌクレオチド媒介免疫治療は、Conryら(1996)Seminars in Oncology 23,135-147; Condonら (1996) Nature Medicine 2,1122-1127; Gongら(1997)Nature Medicine 3,558-561; Zhaiら(1996) J.Immunol.156,700-710; Grahamら(1996)Int J. Cancer 65,664-670;およびBurchellら(1996)pp 309-313 In:Breast Cancer, Advances in biology and therapeutics, Calvoら(eds),Jhon Libbey Eurotextに記載されており、これら全てが参照としてここに含められる。
【0087】
本発明のさらなる態様は、本発明の第一、第二または第三の態様にかかるペプチド、もしくはかかるペプチドをコードするポリヌクレオチドまたは発現ベクターの、本発明の第一、第二または第三の態様に定義されたアミノ酸配列を含むポリペプチドを異常に発現する患者のターゲット細胞を殺すための薬剤の製造における使用を提供するものである。
【0088】
かくして、薬剤は、WT1またはgata−1を異常に発現するガンを治療するために使用できる。
【0089】
本発明のさらなる態様は、in vitroにおいて活性化細胞毒性Tリンパ球(CTL)を産生する方法であり、この方法は、in vitroにおいて、前記CTLを抗原特異的に活性化するのに十分な時間、適切な抗原提示細胞の表面に発現された抗原-結合ヒトクラスI MHC分子とCTLとを接触させることを含み、ここで、前記抗原は、本発明の第一、第二または第三の態様にかかるペプチドである。
【0090】
CTLはCD8+細胞であるが、CD4+細胞であってもよい。MHCクラスI分子は、適切な細胞の表面に発現され、その細胞がMHCクラスI分子を自然に発現しないもの(この場合、かかる分子を発現するように細胞がトランスフェクションされる)であるか、抗原処理または抗原提示経路に欠陥を有するものが好ましい。このように、MHCクラスI分子を発現する細胞は、CTLを活性化する前に選択されたペプチド抗原で実質的に完全に開始されうる。
【0091】
抗原提示細胞(または刺激細胞)は、通常、その表面にMHCクラスI分子を有し、好ましくは、自然には、選択された抗原をMHCクラスI分子に結合することができない。以下に詳細に記載するように、MHCクラスI分子は、in vitroにおいて選択された抗原と容易に結合されてもよい。
【0092】
前記抗原提示細胞は、少なくとも一部の前記選択された分子がペプチドである場合、前記MHCクラスI分子に結合されないような、ペプチドトランスポーターの発現に欠陥のある哺乳動物細胞であると都合がよい。
【0093】
好ましくは、哺乳動物細胞が、TAPペプチドトランスポーターを欠失しているか、低下したレベル、または低下した機能で有する。TAPペプチドトランスポーターを欠失した適切な細胞は、T2、RMA-Sおよびショウジョウバエの細胞を含む。TAPとは、抗原処理に関連したトランスポーター(Transporter Associated with antigen Processing)である。
【0094】
かくして、細胞がショウジョウバエのような昆虫細胞であると都合がよい。
【0095】
ヒトペプチド結合欠陥細胞系統T2は、American Type Culture Collection, 12301 Parklawn Drive, Rockville, Maryland 20852, USAからCatalogue No CRL 1992のもとに入手でき、ショウジョウバエ細胞系統Schneider系統2は、ATCCからCatalogue No CRL 19863のもとに入手でき、マウスRMA−S細胞系統は、KarreとLjunggren (1985)J.Exp.Med.162,1745に記載されており、参照としてここに含める。
【0096】
好ましい実施態様では、刺激細胞は、MHCクラスI分子を発現することができる核酸分子でトランスフェクションされた宿主細胞(例えば、T2、RMA−Sまたはショウジョウバエ細胞)である。T2およびRMA−S細胞は、HLAクラスI分子をトランスフェクション前に発現するが、ペプチドと結合しない。
【0097】
哺乳動物細胞は、当該技術分野において周知の方法によってトランスフェクションされうる。ショウジョウバエ細胞は、Jacksonら(1992) proc.Natl.Acad.Sci.USA 89, 12117に記載されているようにトランスフェクションすることができ、参照としてここに含める。
【0098】
好ましくは、トランスフェクション前に前記宿主細胞は、実質的にMHCクラスI分子を全く発現しない。
【0099】
刺激細胞が、B7.1、B7.2、ICAM−1およびLFA3のいずれかのようなT細胞共刺激に重要な分子を発現することも好ましい。
【0100】
多数のMHCクラスI分子、および共刺激分子の核酸配列は、GenBankおよびEMBLデータベースから公的に入手可能である。
【0101】
前記刺激細胞の表面に発現された実質的に全てのMHCクラスI分子が、同じタイプであることが特に好ましい。
【0102】
ヒトのHLAクラスIおよび他の動物における等価物は、遺伝的に非常に複雑である。例えば、HLA−B座の少なくとも110のアレルと、HLA−A座の少なくとも90のアレルが存在する。HLAクラスI(または等価)分子は本発明の態様において使用できるが、ヒトの集団において適度に高頻度で生じるHLAクラスI分子において少なくとも一部の選択された分子を示すことが好ましい。HLAクラスIアレルの頻度が、カフカス人、アフリカ人、中国人等の異なる民族のグループ間で異なることが周知である。少なくともカフカス人に関する限り、HLAクラスI分子がHLA−A2アレル、HLA−A1アレル、HLA−A3アレル、またはHLA−B27アレルにコードされる。HLA−A2が特に好ましい。
【0103】
さらなる態様において、HLA分子の組み合わせも用いることができる。例えば、HLA−A2とHLA−A3との組み合わせは、カフカス人の74%をカバーする。
【0104】
多重CD8 CTLエピトープのデリバリーのための組み換えポリエピトープワクチンの使用は、Thomsonら(1996) J.Immunol.157, 822-826およびWO96/03144に記載されており、これらの両方を参照としてここに含める。本発明に関して、一つのワクチンにペプチド(またはペプチドをコードする核酸)を含むことが望ましく、ここでペプチドは、任意の順で、アミノ酸配列RMFPNAPYL、CMTWNQMNL、HLMPFPGPLLおよびCD4 T細胞刺激エピトープ(例えば破傷風トキソイド由来)を含む。かかるワクチンは、WT−1およびgata−1を発現するガンを治療するために特に有益である。かかる“ビーズ・オン・ストリング(bead on a string)”ワクチンは、通常、DNAワクチンである。
【0105】
CTL(CD8+細胞)を活性化する簡単な方法は、WO93/17095に記載されており、参照としてここに含める。WO93/17095の方法は、同系の(すなわち、自己の)HLAクラスI分子によって提示されるペプチドに対してCTLを生じる。
【0106】
別の多数の方法が、in vitroでCTLを生じるために用いられてもよい。例えば、Peoplesら(1995)Proc.Natl.Acad.Sci.USA 92,432-436およびKawakamiら(1992) J.Immunol.148,638-643に記載されている方法は、CTL産生において自己の腫瘍浸潤リンパ球を用いる。Plebanskiら(1995)Eur.J.Immunol.25,1783-1787は、CTLの調製において自己の末梢血液リンパ球(PLB)を利用する。Jochmusら(1997) J.Gen.Virol.78,1689-1695は、樹状細胞をペプチドまたはポリペプチドを用いてパルスすること、あるいは、組み換えウイルスで感染させることによる、自己のCTLの産生を記載する。
【0107】
Hillら(1995) J.Exp.Med.181, 2221-2228およびJeromeら(1993) J.Immunol.151,1654-1662は、自己CTLの産生にB細胞を利用する。さらに、ペプチドまたはポリペプチドでパルスした、あるいは、組み換えウイルスで感染したマクロファージが、自己CTLの調製に使用されてもよい。
【0108】
同種細胞もCTLの調製に使用でき、この方法はWO97/26328に詳細に記載されており、参照としてここに取り込む。例えば、ショウジョウバエ細胞およびT2細胞に加えて、CHO細胞、バキュロウイルス感染昆虫細胞、細菌、酵母、ワクシニア感染ターゲット細胞のような他の細胞が、抗原を提示するために用いられてもよい。さらに、植物ウイルスを用いてもよい(例えば、Portaら(1994) Virology 202,449-955参照、外因ペプチドの提示に関する高い収量のシステムとしてカウピーモザイクウイルスの発達について記載)。
【0109】
CTLが本発明のペプチドに関してアロMHC制限されるように、同種細胞がCTLの調製に用いられることが好ましい。CTLがHLA−A0201陰性患者に由来し、かつ、ペプチドが抗原提示細胞によりHLA−A0201クラスI分子によって提示されることが特に好ましい。
【0110】
外因的に適用されたペプチドは、CTLによる提示のためのMHCクラスI経路に向けるために、HIV tatペプチドに連結されてもよい(例えば、Kimら(1997) J.Immunol. 159, 1666-1668参照)。
【0111】
本発明のペプチドに向けられた活性化CTLを、治療に使用することができる。かくして、本発明のさらなる態様は、本発明の上記方法により得られた活性化CTLを提供する。
【0112】
本発明のさらなる態様は、本発明の第一、第二または第三の態様に記載されたアミノ酸配列を含むポリペプチドを異常に発現する細胞を選択的に認識する活性化CTLを提供する。好ましくは、CTLは、本発明の第一、第二または第三の態様のいずれかに定義されたペプチドに結合することにより前記細胞を認識する。
【0113】
CTLは、患者においてターゲット細胞を殺す方法に使用できる。この方法において、ターゲット細胞は、本発明の第一、第二または第三の態様のいずれか一つに記載されたアミノ酸配列を含むポリペプチドを異常に発現するものであり、患者には、有効数の活性化CTLが投与される。
【0114】
患者に投与されるCTLは、患者から誘導され、上記のように活性化される(すなわち、これらは自己のCTLである)。あるいは、CTLは患者由来ではないが、別の個人から由来するものである。もちろん、その個人が健康な個人であることが好ましい。“健康な個人”により、その個人が一般的に健康であり、好ましくは、反応性の免疫系を備え、さらに好ましくは、容易に試験または検出できるあらゆる疾病を患っていないことを意味する。この実施態様において、CTLは、HLAクラスI分子が患者のそれとミスマッチである個人から誘導されたものである。かくして、CTLがアロ制限(allo-restricted)されることが好ましい。アロ制限されたCTLを用いた治療は、本願出願人の先の出願WO97/26328に記載されており、参照としてここに取り込む。
【0115】
かくして、本発明の方法は、養子免疫療法を含む。
【0116】
活性化CTLは、T細胞レセプター(TCR)を含み、これは、異常なポリペプチドを発現する細胞を認識することに関係する。TCRをコードするcDNAが活性化CTLからクローン化され、発現のためのさらなるCTLに導入されることが有益である。
【0117】
本発明の第一、第二または第三の態様のペプチドに特異的な本発明のCTLクローンのTCR(アロ制限または自己制限)がクローン化される。CTLクローンにおけるTCR用途は、(i)TCR可変領域特異的モノクローナル抗体および(ii)VαおよびVβ遺伝子ファミリーに特異的なプライマーを用いたRT−PCRを用いて調べられる。cDNAライブラリーは、CTLクローンから抽出されたポリA mRNAから調製される。TCRαおよびβ鎖のC末端部位および同定されたVαおよびβセグメントのN末端部位に特異的なプライマーが用いられる。TCRαおよびβ鎖の完全なcDNAを、高い忠実度のDNAポリメラーゼを用いて増幅し、その増幅産物を適切なクローニングベクターにクローン化する。クローン化αおよびβ鎖遺伝子は、Chungら(1994)Proc.Natl.Acad.Sci.USA 91, 12654-12658に記載された方法によって一本鎖TCRにアセンブルされてもよい。この一本鎖構成物において、VαJセグメントの後にVβDJセグメントが存在し、さらにその後にCβセグメント、CD3ζ鎖のトランスメンブレンおよび細胞質セグメントが続く。次いで、この一本鎖TCRを、レトロウイルス発現ベクターに挿入する(ベクターのパネルが、成熟ヒトCD8+Tリンパ球を感染および遺伝子発現を媒介する能力に基づいて用いられてもよい:レトロウイルスベクターシステムKatが好ましい(Finerら(1994) Blood 83,43参照))。高力価両栄養性レトロウイルスが、参照としてここに含めるRobertsら(1994) Blood 84, 2878-2889に記載されているプロトコールに従って、腫瘍患者の末梢血液から単離された精製されたCD8+Tリンパ球を感染するために用いられる。抗CD3抗体は、精製されたCD8+T細胞の増殖を開始するために用いられ、レトロウイルスの統合と一本鎖TCRの安定な発現を容易にする。レトロウイルス形質導入の効率は、一本鎖TCRに特異的な抗体で感染CD8+T細胞を染色することにより調べられる。形質導入されたCD8+T細胞のin vitro分析は、それらが、TCR鎖が最初にクローン化されたアロ制限CTLクローンで見られるものと同じ腫瘍特異的殺傷を示すことを確証する。予期される特異性を備えた形質導入された
CD8+T細胞の集団は、腫瘍患者の養子免疫療法に使用できる。患者は、108〜1011(好ましくは109−1010)の間の自己の、形質導入されたCTLで治療されうる。
【0118】
CTLに遺伝子を導入するのに適した他のシステムは、Moritzら(1994) Proc.Natl.Acad.Sci.USA 91, 4318-4322に記載されており、参照としてここに含める。Eshharら(1993) Proc.Natl.Acad.Sci.USA 90, 720-724とHwuら(1993) J.Exp.Med.178,361-366もCTLのトランスフェクションについて記載している。
【0119】
かくして本発明のさらなる態様は、本発明の第一、第二または第三の態様のいずれか一つに与えられたアミノ酸配列を含むポリペプチドを異常に発現する細胞を認識するTCRを提供するものであり、TCRは活性化されたCTLから得られたものである。
【0120】
TCRに加えて、TCRと機能的に等価な分子が、本発明に含まれる。これらは、TCRと同じ機能を行うことができるTCRに機能的に等価な分子を含む。特に、かかる分子は、参照としてここに含めるChungら(1994)Proc.Natl.Acad.Sci.USA 91,12654-12658に記載された方法により製造された遺伝的に操作された3ドメイン一本鎖TCRを含む。
【0121】
また、本発明は、TCRまたは機能的に等価な分子をコードするポリヌクレオチド、TCRまたは機能的に等価な分子をコードする発現ベクターを含む。本発明のTCRを発現するのに適した発現ベクターは、本発明のペプチドの発現に関して上述したものを含む。しかしながら、発現ベクターが、トランスフェクション後にCTLにおいてTCRを発現できるものであることが好ましい。
【0122】
本発明のさらなる態様は、本発明の第一、第二または第三の態様のいずれかに記載されたアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺す方法を提供するものであり、この方法は、(1)患者からCTLを得る工程、(2)上記の、TCRをコードするポリヌクレオチド、または機能的に等価な分子を、前記細胞に導入する工程、および(3)患者において工程(2)で産生された細胞を導入する工程を含む。
【0123】
本発明のさらなる態様は、本発明の第一、第二または第三の態様に定義されたアミノ酸配列を含むポリペプチドを異常に発現する標的細胞を患者において殺す方法を提供するものであり、この方法は、(1)樹状細胞のような抗原提示細胞を患者から得る工程、(2)前記抗原提示細胞を本発明の第一、第二または第三の態様に記載されたペプチド、またはかかるペプチドをコードするポリヌクレオチドと、ex vivoで接触させる工程、および(3)このように処理された抗原提示細胞を患者に再導入する工程を含む。
【0124】
好ましくは、抗原提示細胞は、樹状細胞である。
【0125】
樹状細胞は、抗原ペプチドでパルスされた自己樹状細胞であることが適切である。抗原ペプチドは、適切なT細胞応答を生じる適切な抗原ペプチドとすることができる。腫瘍関連抗原からのペプチドでパルスされた自己樹状細胞を用いたT細胞治療は、Murphyら(1996)The Prostate 29, 371-380とTjuaら(1997) The Prostate 32, 272-278に記載されている。
【0126】
さらなる態様では、樹状細胞のような抗原提示細胞は、本発明のペプチドをコードするポリヌクレオチドと接触される。ポリヌクレオチドは、適切なポリヌクレオチドとすることができ、樹状細胞を形質導入し、かくしてペプチドの提示および免疫誘導をもたらすことができることが好ましい。
【0127】
都合よく、ポリヌクレオチドは、ウイルスポリヌクレオチドまたはウイルスに含まれてもよい。例えば、アデノウイルス形質導入樹状細胞は、MUC1に関して抗原特異的抗腫瘍免疫を誘導することが示された(Gongら(1997)Gene Ther.4,1023-1028参照)。同様に、アデノウイルスをベースとするシステムが用いられてもよい(例えば、Wanら(1997)Hum.Gene Ther.8, 1355-1363参照)。レトロウイルスシステムが用いられてもよい(Spechtら(1997) J.Exp.Med.186,1213-1221およびSzabolcsら(1997)Blood 90, 2160-2167)。樹状細胞への粒子媒介輸送も使用できる(Tutingら(1997) Eur.J.Immunol.27,2702-2707)。また、RNAも使用できる(Ashleyら(1997) J.Exp.Med.186, 1177-1182)。
【0128】
患者におけるターゲット細胞を殺す方法に関して、ターゲット細胞がガン細胞であることが特に好ましい。
【0129】
WT1ポリペプチドは、アミノ酸配列RMFPNAPYLおよびCMTWNQMNLを含み、白血病、乳ガン、メラノーマおよび卵巣ガンにおいて異常に発現される。
【0130】
gata−1ポリペプチドは、アミノ酸配列HLMPFPGPLLを含み、白血病において異常に発現される。
【0131】
特に、本発明の方法によって治療される患者が、HLA−A0201ハプロタイプを有することが好ましい。かくして、好ましい実施態様では、患者のHLAハプロタイプが、治療前に調べられる。HLAハプロタイプは、適切な方法を用いて行われ、かかる方法は当該技術分野において周知である。
【0132】
本発明は、特に、活性なin vivoでのワクチン接種;in vitroでの自己樹状細胞の操作、その後かかる操作がされた樹状細胞のin vivoでの導入して、CTL応答を活性化;in vitroで自己CTLを活性化して、養子治療(すなわち、このように操作されたCTLを患者に導入する);かつ、健康なドナー(MHC適合またはミスマッチ)のCTLをin vitroで活性化して、養子治療するために、本発明のペプチド(またはそれらをコードするポリヌクレオチド)の使用を含む。
【0133】
本発明を、図面および実施例を参照してより詳細に記載する。
【実施例】
【0134】
実施例1:WT1およびgata−1におけるHLA−A0201提示CTLエピトープの同定 HLA−A2結合モチーフを備えた8つのWT−1ペプチドを分析し、二つが天然のCTLエピトープであることが見出された。HLA−A2結合モチーフを備えた12のgata−1ペプチドを分析し、一つが天然のCTLエピトープであることが見出された。
【0135】
以下のアプローチを用いた:i)HLA−A0201結合アッセイにおける合成ペプチドの分析;iii)HLA−A0201陰性のヒト由来のCTL応答を刺激するためにHLA−A0201結合ぺぷちどを使用;iv)WT−1またはgata−1を内因的に発現する腫瘍細胞に対するペプチド特異的CTLの試験。
【0136】
ペプチド結合アッセイ:5x105 T2細胞を、96ウェルプレートにおいて、5%の煮沸したFCS(プロテアーゼを破壊するため)と種々の濃度の合成ペプチドを含む100mlのRPMI1640培地中で、一晩(o/n)インキュベートした。ペプチドまたは周知のA2−結合ペプチドを含まないT2細胞を含むウェルを、それぞれ、陰性および陽性コントロールとして用いた。一晩インキュベーションした後、細胞を洗浄し、A-2特異的モノクローナル抗体HB54およびHB117(American Type Culture Collection, ATCC)を用いて表面HLA−A2について間接免疫蛍光により染色した。FACS分析を、Coulter Corporationフローサイトメーターで行った(Haiteah, Florida)。
【0137】
アロ制限CTL系統およびクローンの産生:HLA−A2陰性バフィーコート血液パックからのPBMCを、レスポンダーとして用いた。24ウェルプレートの各ウェルに、2x106Ficoll分離PBMCと2x105刺激細胞を与えた。刺激細胞は、5%の煮沸FCSを含むRPMI中の100μMペプチドでT2細胞を一晩インキュベートすることによって調製された。5日目に、T細胞を回収し、フィーダーとして2x106の自己の照射したPBMC、2x105ペプチドコート照射T2またはC1R−A2細胞、500nMペプチドおよび10%QS4120培養上清を加えた24ウェルプレートにおいてウェル当たり5x105の密度で新鮮なT細胞培地中に移し、CD4T細胞の成長を抑制する。この培養物を、刺激因子として免疫ペプチドで被覆されたHLA−A2陽性細胞系統を用いて、2週間毎にフィードした。このバルク培養を、T細胞培地に、ウェル当たり1、10および30細胞の密度でクローン化した。104ペプチド被覆T2細胞、2x105HLA−A2陰性フィーダー、および2UのIL−2を各ウェルに添加した。ウェル当たり1細胞でまかれたミクロ培養から得られたCTLを、ウェル増殖細胞のパーセントが30%を越えない場合には、さらにクローン化される。
【0138】
CTLアッセイ:106T2細胞を37℃で100μMの合成ペプチドを含む200μlのアッセイ培地(5% 熱不活性化FCSを含むRPMI 1640)において1時間インキュベートした。ペプチド被覆および未被覆の細胞を、さらに1時間51Crラベルし、洗浄し、丸底96ウェルプレートにおいて連続する二倍希釈のエフェクター細胞に添加して、200μl/ウェルの全体量とした。CTLバルク系統を、51Crラベル化ターゲット細胞当たり30のコールドK562細胞の存在下で分析し、NK細胞により引き起こされたバックグラウンド殺傷を低減を低減した。T細胞クローンの感度を試験するために、アッセイ培地における一連の希釈のペプチドを、96ウェルプレートに調製した。5x103の51Crラベル化T2細胞を各ウェルに添加し、100μlの全体量として、1時間インキュベートした。エフェクター細胞を、最大CTL殺傷に十分なE:T比で添加した。アッセイプレートを37℃、5%CO2でインキュベートし、4時間後、100μlの上清を各ウェルから回収し、Wallac Gamma Counterを用いてカウントした。比溶解(specific lysis)を、式(実験放出自然放出)/(最大放出自然放出)x100%によって計算した。
【0139】
結果
WT−1タンパクにおいて、WT−1発現腫瘍細胞において発現された二つのCTL認識ペプチドエピトープが同定された:WT126−34:RMFPNAPYLWT235−43:CMTWNQMNL
【0140】
これらは9アミノ酸の長さのペプチドであり、CTLによる有効な認識に必要な最小エピトープを示すと考えられる。
【0141】
WT126−34に対するCTLの殺傷活性を、以下の表に記載する。
【0142】
【表1】
【0143】
表のデータは、WT126−34ペプチドに対するCTLが、WT−1およびHLA−A0201を発現する腫瘍細胞を殺傷することを示す。WT−1陰性Bリンパ芽球細胞は殺傷されない。図1および2は、抗WT126−34CTLの殺傷活性を示す代表例を示す。
【0144】
抗WT235−43CTLを用いて得られたデータは、WT126−34CTLを用いて得られたデータほど大規模ではない。しかしながら、WT235−43CTLがWT−1を内因的に発現するHLA−A0201陽性腫瘍細胞を殺傷することが明らかである。
【0145】
gata−1タンパクにおいて、gata−1発現腫瘍細胞において発現されたCTL認識ペプチドエピトープが同定された:Hug378−87 HLMPFPGPLL。
【0146】
これは10アミノ酸の長さのペプチドであり、CTLによる有効な認識に必要とされる最小エピトープを示すと考えられる。
【0147】
このペプチドに対するCTLは、内因的にgata−1を発現するHLA−A0201形質転換K562白血病細胞系統を認識できるが、形質転換されていないK562細胞は認識されない。
【0148】
結論
WT−1およびgata−1のCTL認識ペプチドエピトープが同定された。これらのエピトープは、これらのタンパクを異常に発現する腫瘍細胞に提示される。WT−1およびgata−1の生理学的発現は、比較的少数の正常細胞に制限されている。かくして、自己のCTLがこれらのタンパクに対して制限された寛容を示すことができ、かつ、同定されたCTLエピトープが、異常なWT−1およびgata−1発現を有する腫瘍に対してワクチン接種、および別の免疫治療法に使用することができる。
【0149】
実施例2:クラスI分子とWT1ペプチド抗原RMFPNAPYLを用いた活性化された細胞傷害性リンパ球(CTL)の産生およびその投与 活性化された細胞傷害性Tリンパ球(CTL)を、HLA−A2クラスI分子とWT1由来のノナマーペプチド:RMFPNAPYLを用いて産生した。
【0150】
PCT特許出願WO93/17095に記載された方法を、CTLを作製するために使用した。ショウジョウバエ細胞をCTLにペプチド抗原を提示するために使用した。HLA−A2分子は、ショウジョウバエ細胞において発現された。
【0151】
ペプチドは、Applied Biosystems synthesiser, ABI 431A(Foster City, CA, USA)で合成し、HPLCで精製した。
【0152】
WO93/17095に詳細に記載されているように、特異的な細胞傷害性T細胞の産生のためのin vitro条件を最適化するために、刺激細胞の培養が適切な培地で維持された。刺激細胞は、WO93/17095に記載されているようなショウジョウバエ細胞であり、好ましくは血清を含まない培地に維持される(例えば、Excell 400)。
【0153】
前駆体CD8細胞のような活性化される細胞と刺激細胞のインキュベーションの前に、ヒトクラスI分子に結合して、刺激細胞の表面に提示されるのに十分な量の所定量の抗原性ペプチドが、刺激細胞培養物に添加される。十分な量のペプチドとは、ペプチドを結合したヒトクラスI MHC分子が約200,好ましくは200以上、各刺激細胞の表面に提示される量である。刺激細胞は通常、>20μg/mlのペプチドとインキュベートされる。
【0154】
休止または前駆体CD8細胞を、CD8細胞を活性化するのに十分な時間、適切な刺激細胞と培養においてインキュベートした。CD8細胞は、抗原特異的方法で活性化される。休止または前駆体CD8(エフェクター)細胞の刺激細胞に対する比率は、個々に異なってよく、培養条件に対する個人のリンパ球の感度のような変異性に依存してもよい。リンパ球:刺激細胞(ショウジョウバエ細胞)比は、通常、約30:1から300:1の範囲である。例えば、3x107ヒトPBLと1x106ショウジョウバエ細胞が混合され、20mlのRPMI 1640培養培地に維持される。
【0155】
エフェクター/刺激培養物を、治療に使用できるまたは有効な数のCD8細胞を刺激するのに必要な時間にわたって維持する。最適な時間は、通常、“プラトー(plateau)”、すなわち“最大”特異的CD8活性化レベルであり、通常培養の5日後に見られる、で約1〜5日の間である。CD8細胞のin vitro活性化は、通常、細胞系統のトランスフェクション後、短い期間内に検出される。CD8細胞を活性化することができるトランスフェクションされた細胞系統における一過性の発現は、トランスフェクションから48時間以内に検出される。これは、ヒトクラスI MHC分子を発現する形質転換された細胞の安定または一過性の培養が、CD8細胞を活性化するのに有効であることを、はっきりと示す。
【0156】
活性化されたCD8細胞は、刺激細胞、刺激細胞上に結合されたペプチド、またはCD8細胞(またはそのセグメント)に特異的なモノクローナル抗体を用いて、その適切な相補的リガンドに結合させることにより、刺激(ショウジョウバエ)細胞から有効に分離することができる。次いで、抗体タグ化した分子は、免疫沈降またはイムノアッセイ方法を解して刺激−エフェクター細胞混合物から抽出される。
【0157】
有効な、細胞傷害的な量の活性化CD8細胞は、in vitroおよびin vivoでの使用の間で、これらの傷害細胞の究極のターゲットである細胞の量およびタイプによって変化することができ、約1x106および1x1012の活性化されたCTLが、成人に対して用いられる。
【0158】
活性化されたCD8細胞は、処置される個人にCD8細胞を投与する前に、ショウジョウバエ細胞培養から回収される。しかしながら、他の存在しかつ提案された治療モダリティーとは異なって、この実施例に記載した方法が、腫瘍形成性でない細胞培養システム(すなわちショウジョウバエ細胞)を用いないことに注意することが重要である。それゆえ、ショウジョウバエ細胞と活性化されたCD8細胞との完全な分離が達成されない場合でも、少量のショウジョウバエ細胞の投与に関連することが知られた固有の危険性は存在しない。一方、哺乳動物の腫瘍促進細胞の投与は危険である。
【0159】
例えば、Honsikらの米国特許第4844893号およびRosenbergの米国特許第4690915号に例示されているような、細胞性成分を再導入する方法が用いられる。例えば、静脈注入による活性化CD8細胞の投与が適切である。
【0160】
実施例3:乳ガンを処置するための、WT1ペプチドCMTWNQMNLを用いてパルスした樹状細胞 乳ガンは、真に局在かしている場合にのみ治療可能性がある。最も一般的な問題は、明確な転移に関する遅い発表か、多くは、局所治癒後の全身的転移の発達である。転移性乳ガンは、高度に化学的感受性であり、有効な化学療法は、きまって疾患の軽減を誘導し、二次疾患の開始を遅延し、広範囲の疾患の症状を緩和する。
【0161】
この種の免疫治療は、腫瘍増殖および転移が、新生細胞による抗原提示の低下または患者の免疫における一般的な衰えのいずれかによる、免疫学的監視の欠陥を反映するという考えに基づく。両方のメカニズムが乳ガンにおいて生じ、特に樹状細胞(DC)機能に重要な欠陥が存在するという証拠が存在する(Gabrilovichら(1997) Clin.Cancer Res.3, 483-490)。細胞傷害性T細胞応答は、WT1ペプチド:CMTWNQMNLのような免疫原性ペプチドに対してin vitroで証明されている。DCは、プロフェッショナル抗原-処理および-提示細胞である。これは、腫瘍MHC制限T細胞免疫の発達に重要である。これらは、骨髄のCD34+前駆体に由来するが、末梢血液中のポストコロニー形成ユニットCD14+中間体からも誘導される。DCは、皮膚、粘膜、脾臓および胸腺において末梢部位に移動する。これらは、同種移植片拒絶、アトピー性疾患、自己免疫および抗腫瘍免疫を含む、種々の臨床的に重要な処理に関係している。
【0162】
患者は、HLA−A2に分類される。
【0163】
DCは、主にGM-CSF、IL−4およびTNFαのようなサイトカインを用いて、CD34+ステム細胞またはCD14+末梢血液単球からex vivoで培養される。これら両方の源に由来するDCは免疫応答性であり、外因的に提示された抗原を取り込み、処理して、細胞傷害性T細胞にそれを提示することができる(Grabbeら (1995) Immunology Today 16, 117-121; Girolomoni & Ricciardi-Castagnoli (1997) Immunology Today 18, 102-104)。最近の研究は、DCがin vivoで生成された抗原特異的腫瘍免疫を運搬しうること(Kwakら(1995) Lancet 345, 1016-1020)、および、ex vivoで腫瘍抗原を用いてパルスされた自己のDCが、無視できない抗腫瘍効果を誘導しうること(Hsuら (1996) Nature Medicine 2, 52-58)を証明した。DCは、粗製腫瘍膜溶解物、精製されたペプチドまたはペプチドフラグメントを用いて効率的にパルスされうる。
【0164】
WT1は、乳ガンによって発現されるポリペプチドである。
【0165】
キーホールリンペットヘモシアニン(KLH)は、これと類似した研究において患者の免疫応答性に対する無害の陽性コントロールとして用いられる免疫原性タンパクである(Hsuら (1996) Nature Medicine 2, 52-58)。
【0166】
WT1ペプチドCMTWNQMNLの精製された調製物と結合し、かつ、養子免疫療法として再注入された、再発性乳ガン患者由来のex vivo拡大自己樹状細胞を用いる実現性は、以下のように確立される。
【0167】
記載された研究は、再発性乳ガン患者の末梢血液からのex vivo拡大(expansion)による自己DCの最適な生成方法論を確立し;外因性WT1ペプチドをDCに載せる(結合させる:load)可能性を評価し;自己再注入の急性寛容および毒性を評価し;WT1ペプチドまたはKLHディベロップに対して免疫が反応するか否かを調べ;かつ、無視できない腫瘍バルクへの効果を調べる。
【0168】
養子免疫療法は、バルク疾患の低減よりも、最小の残余疾患のコントロールまたは消失において最も効果的であろう。免疫療法は、細胞傷害性Tリンパ球の流入によりバルク疾患の大きさを一時的に増大させうると思われる。疾患の程度およびバルクは、治療後に監視されるが、正式な終点として用いられない。患者は、決まった方法で長期にわたり追跡調査され、長期にわたって、不利な症状が現れないことを確認する。
【0169】
正常な志願者由来の樹状細胞培養 CD14+末梢血液単球を組織培養フラスコに付着させ、1%AB血清、GM−CSF(400ng/ml)およびIL−4(400IU/ml)の存在下で7日間培養した。これは、DCの多型性を備えた細胞を与え、抗原を取り込んで提示するDCの未熟形態を示すCD1a+マーカーで49%の平均を与えた。次いで、これらの細胞を、DCを細胞傷害性T細胞に対して抗原提示可能にする、TNFα(15ng/ml)の添加によりCD83+細胞に成熟化される。7%の細胞が、1日以内にCD83+になるが、最大効果のためには少なくとも3日必要である。単球調整培地が、1%AB血清と置換できる。
【0170】
再発した乳ガン患者由来の樹状細胞培養 救助療法前後の、再発した転移性疾患の6人の患者からDCを生成した(末梢血液の12サンプルの合計、それぞれ50ml)。
【0171】
臨床研究 標準的なプロトコールに従って、単一ユニットの自己の血液を患者が献血した。血液輸血サービス医師によって献血前に患者を評価した。自己の献血を、決まったウイルスマーカー(HIV、HBV、HCVおよび梅毒)について同種献血と同様に選別した。患者は、参加を望めば、適切なカウンセリングの後に、これに同意した。この警戒は、臨床および実験室スタッフを可能性のある感染から保護し、交差汚染の可能性から決まった血液供給を保護する。この血液を、決まったクワドパックに入れる。これは、赤血球、バフィコートおよびプラスマの自動化された分離を可能にする。このバフィコートは、約670x106の単核白血球を与え、これは現在の技術を用いて約47x106のDCを与える。患者当たり8−128x106のDCの投与範囲を用いる。末梢血液単球を二つの分注量に分け、1から10日間、WT1ペプチドおよびKLHを用いてパルスした。血清を含まない培養条件または自己プラスマを、同種AB血清に優先して用いた。培養されたDCを集め、洗浄し、100mlの生理食塩水に再懸濁した後に、1時間かけて注入した。自己赤血球濃縮物は、標準臨床示度以外、患者に戻さない。ex vivo DC培養方法を、以下の良好な製造処理に従って実施した。
【0172】
最初の血液サンプルを献血する患者は、このときまでに、救助療法を受けており、臨床的寛解にあってもそうでなくてもよい。再発転移性疾患患者は、化学療法を受ける前に処置を受ける。二つの処置プログラムがある。
(1)転移性再発、標準治療後、養子免疫療法;
(2)転移性再発、養子免疫療法後、標準治療。
【0173】
治療のための患者を含める基準:限局性の再発または転移性乳ガン患者。
細胞毒性化学療法またはホルモン治療を用いて以前に処置。
評価可能な疾患(UICC基準)。
>12週であると予想される生存。
自己血液献血の基準を満たす(HgB>120g/Iを含む)。
告知に基づく同意。
年齢が18歳〜70歳。
【0174】
処置から患者を除く基準:妊婦。
CNS転移。
以前または共同性転移。
告知に基づく同意が得られない。
同意拒否。
年齢が<18歳または>70歳。
【0175】
産物の注入は、必要であれば蘇生および支持的な便宜を利用できる寝椅子の上で、監視のもとで、経験のある医師の直接的監督のもとに行われた。
【0176】
実施例4:WT1に特異的な細胞傷害性Tリンパ球による白血病CD34+始原細胞の選択的除去 例えば、急性および慢性骨髄性白血病のような血液学悪性腫瘍は、未熟なCD34+始原細胞の悪性トランスフォーメーションによって特徴付けされる。トランスフォーメーションは、WT1転写因子の増加した発現と関係する。ここで、WT1が、白血病性始原細胞に対する鋭敏な特異性を備えたCTLのターゲットとして作用しうることを証明した。WT1に特異的なHLA−A0201制限CTLは白血病性細胞系統を殺し、CML患者から単離された形質転換されたCD34+始原細胞によるコロニー形成を阻害したが、正常なCD34+始原細胞によるコロニー形成は影響されなかった。
【0177】
かくして、組織特異的転写因子WT1は、in vitroでの白血病性始原細胞のCTL媒介パージング、および、in vivoにおける白血病およびWT1発現悪性腫瘍の抗原特異的治療の、理想のターゲットである。
【0178】
造血系の細胞を、自己再生および分化可能な幹細胞(HSC)から誘導した。ヒトとマウスにおける移植実験は、CD34+細胞集団が、骨髄切除受容者において赤血球、脊髄およびリンパ球の直系を再構成可能であるHSCを含むことを示した1。さらに、マウスの宿主を再構成することができるHSCが、CD34-/lin-骨髄細胞の稀な集団において最近証明された2。
【0179】
CMLとAMLにおける重要なトランスフォーメーションが、未熟なCD34+始原細胞に影響するという強い証拠がある。大多数の白血病性芽細胞が、制限された増殖能を有するので、悪性疾患は、強い増殖力と自己新生力を備えた白血病性始原細胞の部分集団によって維持されなければならない3、4。AML患者由来の精製された細胞を用いた移植研究は、未熟なCD34+細胞のみが、免疫無防備状態のマウス受容者において白血病を開始することができることを示した5。同様に、CML患者由来の精製されたCD34+細胞は、マウス受容者において効率的に白血病を開始した6、7。
【0180】
未調整始原細胞増殖を導く分子事象は完全には理解されていない。t(9;22)染色体転座に関連するBCR/ABL融合タンパクはCMLの証明であるが、BCR/ABL転写物は、健康な個人にも観察され、さらなるファクターが白血病の発達に必要であることを示唆する8。WT1転写因子は、白血病誘発に寄与する候補タンパクである。この転写因子は、通常、未熟なCD34+始原細胞に発現され、分化はWT1ダウンレギュレーションに関与する9、10。WT1発現の高められたレベルは、AMLおよびCML患者由来の精製されたCD34+細胞および未分離の単核細胞において観察された11、12。増大したWT1発現が正常な分化をブロックし、造血始原細胞の増殖を増強することを示すin vitro研究は、白血病誘発に寄与するWT1の能力の説明を与える13、14。
【0181】
最近の研究結果は、CD34+始原細胞に特異的なTリンパ球が、CML患者における抗白血病性効果を媒介することに極めて重要であるということを示唆した15。この研究において、白血病性始原細胞に対して細胞傷害性Tリンパ球を向けるために、ターゲット分子としてWT1を開発する可能性を調査した。正常なCD34+始原細胞ではなくCMLが、CTL攻撃を開始するのに十分なWT1タンパクを発現するという仮説を調べた。
【0182】
方法:細胞系:急性転化状態の女性のCML患者の胸水からK562細胞を確立した16。急性転化状態の男性のCML患者の末梢血からBV173細胞を確立した17。急性リンパ性白血病を有する12歳の少年の骨髄から細胞系697を確立した18。C1R細胞系は、HLA−A0201陰性EBVトランスフォームリンパ芽球細胞系である19。T2細胞系は、TAP(transporter associated with antigen processing)をコードする遺伝子の欠失について選択され、内因性ペプチドを有するHLAクラスI分子を不十分にしか運搬しないことを引き起こす20。従って、T2細胞のHLA−A0201分子は、外因性ペプチドを効率的に運搬する。HLA−A0201、ヒトβ−2ミクログロブリン、B7.1及びICAM−1でトランスフェクトされたDrosophila細胞は、Dr. M. Jacksonからの好意で得た。
合成ペプチド:ヒトウィルムス主要抗原1P126から由来するペプチド(RMFPNAPYL)及びヒトパピローマウイルスタイプ16のE7タンパク質から由来するコントロールHLA−A0201結合ペプチドを、フルオレニルメトキシカルボニル化学を使用して、Imperial College Medical Schoolの中央ペプチド合成研究室によって合成した。ペプチドの質をHPLC分析によって評価し、予想される分子量をマトリックスアシストレーザー放出マススペクトロメトリーを使用して観察した。ペプチドをPBS(pH7.4)に溶解し、2mMの濃度を得て−20℃で貯蔵した。
【0183】
アロ−HLA−制限CTL系の世代:PBMCをFicoll勾配遠心分離を使用してバッフィーコートパックから分離し、モノクローナル抗体HB54(抗HLA−A2,B17)及びHB117(抗HLA−A2,A28)で染色した。A2陰性PBMCを応答因子として使用した。HLA−A0201、ヒトβ2−ミクログロブリン、B7.1及びICAM−1でトランスフェクトされたペプチドコートDrosophila細胞を、初期刺激因子として使用した。Drosophila細胞を48時間100mMのCuSO4中で誘導し、培地で3回洗浄し、4時間100μMの濃度でペプチドを乗せた。24穴プレートの各ウェルは、2mlのT細胞培地中に2×106の応答因子PBMCと2×105の刺激因子細胞を受け取った。5日目でT細胞を回収し、餌として2×106の自己発光PBMC、2×105の発光ペプチドコートT2細胞、10%QS4120培養上清(抗CD4抗体を含む)、及び10U/mlhu−rIL−2(Boehringer)を添加して、ウェル当たり5×105の密度で新鮮なT細胞培地中に蒔いた。刺激因子として免疫化ペプチドでコートされたT2細部を使用して、培養物を弱く再刺激化した。2−3サイクルの刺激の後、大きな培養物がウェル当たり1,10及び30細胞の密度で96穴プレート中にクローン化した。104ペプチドコートT2細胞、2×105HLA−A2陰性PBMCフィーダー、及び2U/mlIL−2を、各ウェルに加えた。各ウェルの細胞毒性を、免疫化ペプチドまたはコントロールHLA−A0201結合ペプチドでコートされたT2標的細胞に対して試験した。ペプチド特異的ミクロ培養物を増殖させ、2×106のフィーダー、2×105の刺激因子細胞、及び10U/mlIL−2を加えることによって、24穴プレート中で弱く再刺激化した。T細胞系77(図4B参照)を、培養物中で1年間維持し、この研究でのほとんどの実験についてCTLのソースとして機能した。この系はCD4+及びCD8+細胞より成るため、特異的な殺傷活性がCD8+CTLによって介在されることを示すため、CD8+サブクローンを使用した。親の77系とは異なり、CD8+サブクローンのin vitroでのライフスパンは数カ月に制限された。
【0184】
CTLアッセイ:CTLアッセイを記載されているように実施した。略記すると、106T2細胞を、37℃で100μMの合成ペプチドを有する200μlのアッセイ培地(5%熱不活性化FCSを有するRPMI1640)中で1時間培養した。ペプチドコートT2細胞または腫瘍細胞を1時間51Crラベルし、洗浄し、丸底96穴プレート中に連続的な2倍希釈のエフェクター細胞に加え、200μl/ウェルの全容量を得た。アッセイプレートを37℃で5%CO2で4時間インキュベートした。100μlの上清を回収し、Wallac Gamma Counterを使用してカウントした。特異的な溶解を、以下の式:(実験的放出−自発的放出)/(最大放出−自発的放出)×100%によって計算した。
【0185】
造血CD34+細胞の精製:正常CD34+細胞のソースとして、成人で健康なドナー由来のヒト骨髄(n=5)、疾患回復期の幹細胞不朽固化腫瘍患者からの白血球搬出産物(n=2)及び臍帯血(n=1)を使用した。臍帯血のサンプルは、滅菌回収チューブ内への血液の排出によって廃棄された胎盤及びへそのお組織から得られた。これらの細胞の使用についてのインフォームドコンセントを、ドナー及び患者から適切なように取得した。白血病CD34+細胞のソースとして、少なくとも3ヶ月間インターフェロンで処理されていない慢性状態のCML患者から末梢血を得た。
【0186】
サンプルをHanks緩衝塩溶液(HBSS)中で1:2に希釈し、密度勾配遠心分離(Lymphoprep 1.077g/ml, Nycomed, UK)によって単核細胞で富化し、回収した単核分画を、Minimacsシステムを使用して製造者の説明書に従って(Miltenyi Biotec, UK)、CD34+分画の単離のための磁性ミクロビーズ選択にかけた。抗ヒトCD34フィコエリスリン(PE)マウスモノクローナル抗体(Becton Dickinson)を使用したFACS分析によって見積もると、細胞集団の純度は80−95%の範囲であった。
【0187】
RNA抽出及びRT−PCR:106細胞の全RNAを、RNAzolTMBプロトコール(AMS Bio, UK)に従って単離した。全RNAペレットのcDNA合成を、40μl反応物で実施した。溶解RNAペレットを初めに65℃で10分2μgのオリゴdT12−18プライマー(Life Technologies, Scotland)でインキュベートし、次いで50Uの齧歯類白血病ウイルス(MuLV)逆転写酵素、10mMのジチオスレイトール、1mMのdNTP(Boehringer Mannheim, UK)、40UのRAアーゼインヒビター(Promega, UK)の混合物で、42℃で1時間インキュベーションした。5μlのcDNA調製物を、2.5UのTaqDNAポリメラーゼ(Qiagen, UK)、1mMdNTP、20OD/mlのプライマーを含む最終反応混合物の50μl容量中でPCR増幅のために使用した。ヒトWT1コード領域の増幅は、エクソン7に位置するセンスプライマー(21マー、5'-ggc atc tga gac cag tga gaa-3')及びエクソン10のアンチセンスプライマー(22マー、5'-gag agt cag act tga aag cag t-3')を使用して達成した。WT1PCR産物の予想サイズは482bpである。RNAの完全性を、22マーセンス5'-ccc aac ctt ttc gtt gca ctg t-3';22マーアンチセンス5'-cgg ctc tcg gag gag acg atg a-3'といったイントロンにまたがるプライマーを使用して全てのサンプル中のヒトc−abl遺伝子を増幅することによって確認した。c−ABLPCR産物の予想サイズは385bpである。ホットスタートPCRを、以下の条件(ABL及びWT1増幅について同じ):95℃で1分の変性、56℃で1分のプライマーアニリング、及び72℃で2分の鎖伸張の下で、熱サイクラー(Techne Genius, Cambridge)で35サイクル実施した。サイクリングは、95℃で5分の変性工程で開始し、逆転写酵素を熱不活性化し、72℃で10分の最終伸張によって終結した。全てのRT−PCRを少なくとも2回実施し、陰性コントロール(cDNA含まず)及び陽性コントロール(WT1発現白血病細胞系BV173から得たcDNA)を全ての実験で含ませた。PCR産物を1.5%アガロースゲルで電気泳動した。
【0188】
ウエスタンブロット分析:分離したCD34+細胞(2×105細胞)をPBSで洗浄し、Laemmli Buffer中で溶解した。細胞溶解物を12%SDSポリアクリルアミドゲル電気泳動(PAGE)によって分画し、湿潤トランスファーによりニトロセルロース膜(Amersham, UK)にトランスファーした。次いで膜を、0.01%Tween20及び5%ノンファットドライミルクを含むPBS中で室温で1時間ブロックし、ウサギ抗ヒトWT−1C19ポリクローナル抗体(Santa Cruz CA, ブロッカー中で1:200)で最初に4℃で一晩インキュベーションし、次いでウサギ抗アクチンポリクローナル血清(Sigma UK, ブロッカー中で1:500)で室温で30分インキュベーションした。セイヨウワサビペルオキシダーゼ接合ブタ抗ウサギ抗体(DAKO UK, 1:1000)での膜のインキュベーションと製造者の説明書に従ったECL反応(Amersham UK)により、シグナルを検出した。
【0189】
祖先アッセイ(CFUアッセイ):以下の組換えヒト成長因子(Stem Cell Technologies):幹細胞因子(SCF、50ng/ml)、インターロイキン−3(IL−3,20ng/ml)、インターロイキン−6(IL−6,20ng/ml)、顆粒球マクロファージコロニー刺激因子(GM−CSF、20ng/ml)、顆粒球コロニー刺激因子(G−CSF)を補ったメチルセルロース培地中に、1000−3000CD34+細胞を蒔くことによってCFUアッセイを実施した。培養物を5%CO2の湿潤環境で37℃で14日間インキュベートし、コロニー形成単位顆粒球マクロファージ(CFU−GM)の発達を可能にした。
【0190】
結果:WT−1特異的CTLの世代:成人におけるWT1転写因子の発現は、腎有足突起、精巣セルトーリ細胞、卵巣顆粒層細胞、及びCD34+骨髄細胞で検出可能である21。WT1に対する可能性のある免疫寛容を避けるために、MHCミスマッチドナーから得られたペプチド特異的CTLを生産する以前に記載されたアプローチを使用した。このアプローチは、免疫寛容と独立した腫瘍細胞で過剰発現されるいずれかのタンパク質に対してCTLを生産するのに適している22,23。9のアミノ酸長のWT−1由来ペプチドエピトープP126(RMFPNAPYL)を、CVTL標的として選択した。このペプチドは、コーカサス人で見出される最も顕著なクラスI対立遺伝子であるHLA−A0201クラスI分子(実施例1参照)に結合する。HLA−A0201-ドナーから得た応答因子白血球を、P126ペプチドを提示するHLA−A0201+刺激因子細胞と共にin vitroで培養し、希釈培養物の制限を、ペプチド特異的CTL細胞を単離するために使用した。ペプチドコートT2標的細胞での実験により、CTLがP126ペプチドに対して非常に特異的であることが示された(図4A)。ペプチド滴定により、CTLが低いピコモーラーのペプチド濃度を認識可能な非常に渇望した系と、低いピコモーラーのペプチド濃度を認識するあまり渇望していない系に分割できることが示された(図4B)。非常に渇望したCTL系を、さらなる実験のために選択した。
【0191】
WT−1特異的CTL殺傷白血病細胞系:白血病細胞系のパネルの分析により、P126特異的CTLが、HLA−A0201+細胞BV173及び697を殺傷することが明らかとなった(図4C)。HLA−A0201-白血病細胞系K562は、HLA−A0201でのトランスフェクションの後のみで殺傷される。対照的に、細胞がP126ペプチドでコートされていないと、HLA−A0201+EBVトランスフォームB細胞系C1R−A2は殺傷されなかった(図4C;同じ結果は他のEBVトランスフォーム細胞でも見られた)。CTL標的細胞中でのWT1の発現を、RNA及びタンパク質レベルで分析した。RT−PCRにより、白血病細胞系はWT1RNAを発現するが、EBVトランスフォームC1R−A2細胞は発現しないことが示された(図5A)。同じ結果は、WT1タンパク質が白血病細胞でのみ発現され、C1R−A2では発現されないことを示すウエスタンブロットによっても得られた(図5C)。このデータを総合して、CTLはA0201+白血病細胞系を認識し、CTL殺傷はWT1発現と相関することが示された。
【0192】
WT1特異的CTL殺傷新鮮白血病CD34+細胞:慢性状態のCML患者の末梢血から得た単核細胞を、未成熟CD34+と成熟CD34-集団に分類した。予想されたように、HLA−A0201-患者から単離された細胞は、P126特異的CTLによって認識されなかった(図6A)。HLA−A0201+CML患者から得られた細胞を分析した場合、CTLはCD34+細胞集団を選択的に認識する一方で、より成熟したCD34-集団の殺傷は観察されなかった(図6A)。CD34-細胞の殺傷の欠如は、外因性P126ペプチドを有するこれらの細胞のコーティングがCTL殺傷を引き起こすため、WT1由来標的ペプチドの不十分な発現のためである可能性が高い(図6A)。RT−PCR分析により、CD34+細胞における強力なWT1RNA発現と、CD34-細胞における低い発現が明らかとなった(図5B)。ウエスタンブロッティングにより検出可能なWT1タンパク質の発現はCD34+に制限され、CD34-細胞ではタンパク質は検出不可能であった(図5C)。RT−PCRよウエスタン分析の両者により、白血病CD34+細胞集団におけるWT1発現のレベルの変異が示された。かくして、WT1発現のレベルにおける観察された変異が、CTL殺傷のレベルの変異を生ずるかどうかを調査した。しかしながら11の異なるCML患者の分析により、CTLはCD34+集団の20%(SD5%)を一貫して溶解することが示された(図5B)。この結果は、WT1発現がCD34+細胞の約20%のサブ集団に制限され、全ての11のCML患者における発現レベルが、これらの細胞のほとんどをCTL殺傷に感受性にするのには不十分である可能性を示唆した。それ故、CTLによって認識されるサブ集団が、顆粒球/マクロファージ直系のコロニーに高めることができるクローン原性の始原細胞を含むかどうかを調査した。実際、9のCML患者から単離されたCD34+集団をP126特異的CTLで処理した場合、これはコロニー形成の80−100%の阻害を生ずる(図5D)。このことは、コロニー形成始原細胞の大多数がP126特異的CTLによって除去されることを示した。CTL処理サンプルで見られる「エスケープ」コロニーは、非処理サンプル中のコロニーと比較した場合小さかった。これは、「エスケープ」コロニーが、WT1発現の下流調節と関連する顆粒球/マクロファージ直系に向けた分化をすでに開始している始原細胞から由来する可能性と一致する。上記部分的に分化した始原細胞は、WT1特異的CTLによる認識を避け、CFUアッセイで観察される小さいサイズのコロニーは、これらの始原細胞の減少したクローン増大サイズを反映しているであろう。
【0193】
重要なことに、HLA−A0201-CML患者から得たCD34+細胞によるコロニー形成は影響されず、コロニー形成活性を有する始原細胞の消失は、CTLによるHLA制限抗原認識に依存することを示した。
【0194】
最後に、CTLが白血病細胞と正常なCD34+始原細胞の間を識別するかどうかを調査した。正常CD34+細胞を、HLA−A0201+ドナーの骨髄、末梢血または臍帯血から単離し、CTL標的として使用した。CD34+細胞のソースとは独立して、抗P126CTLはこれらの細胞によってコロニー形成を阻害しなかった(図5D)。さらに正常CD34+細胞は、細胞毒性アッセイにおいて標的として使用された場合殺傷されなかった(図6B及びC)。白血病対正常CD34+細胞の選択的なCTL殺傷は、WT1発現における差異によって説明できる。WT1RNA発現は、正常CD34+細胞と比較して白血病細胞で高く(図5B)、ウエスタンブロット分析は白血病細胞でのみWT1タンパク質を検出し、正常CD34+細胞集団においては検出しなかった(図5C)。
【0195】
以前の自己幹細胞移植の後に再発した患者に対するドナーリンパ球(DLI)の注射は、強力なグラフト対白血病効果(GVL)を引き起こすことができる24,25。最近の研究により、DLIを経験したCML患者における完全な回復は、白血病CD34+始原細胞を認識するT細胞の増大した割合と関連することが示された15。対照的に、CML患者におけるより成熟したCD34-細胞のT細胞認識は、好ましい臨床上の応答と関連しなかった。このことはCD34+CML始原細胞に対して特異性を有するTリンパ球が、in vivoでの抗白血病効果を介在する点で必須に重要であることを示唆した。しかしながら、白血病CD34+始原細胞に対して選択的にT細胞応答を向けることができる標的抗原の性質に関する情報は現在存在しない。
【0196】
上記抗原を同定するために、免疫寛容と独立して、トランスフォーム細胞において上昇したレベルで発現されるいずれかのタンパク質に対してCTLを与えるのに適した、自己制限CTLアプローチを使用した22,23,26。細胞毒性エフェクター機能の引き金を引くことは初めの現象であるために、トランスフォーム細胞における上昇した標的タンパク質レベルによってのみ引き金を引かれ、正常細胞におけるタンパク質の生理学的レベルによっては引かれないCTLを選択することが可能である23。
【0197】
白血病CD34+細胞におけるWT1の過剰発現の証拠が存在する12。上昇したWT1発現は、トランスフォーメーションに寄与できる13,14。正常なWT1発現は、出生後の人生において少数の細胞に制限される21。白血病に加えて、上昇したWT1発現はまた、腎細胞カルシノーマ、卵巣ガン、進行性乳ガン、及びメラノーマにおいて観察されている27-30。それ故、WT1過剰発現悪性細胞を選択的に認識するCTLは、白血病、並びに他のより一般的な悪性疾患の抗原特異的な治療に対して貴重な試薬である。さらに、WT1発現の下流調節による腫瘍エスケープは、もし過剰発現がトランスフォームされた表現型を維持するために必要である場合生じないようである31-33。
【0198】
ここに記載される自己制限CTLは、HLA−A0201-ドナーから単離され、それらはHLA−A0201クラスI分子に関してWT1由来P126ペプチドを提示する白血病始原細胞に対して特異的であった。異なるHLA−A0201-ドナーから由来するリンパ球のin vitroの刺激は、ペプチド特異的HLA−A0201制限CTLを一貫して誘導するため、P126ペプチドは非常に免疫原性である。それ故P126特異的CTLは、HLA−A0201対立遺伝子に関して一つのローカスHLAミスマッチを提示するドナーから幹細胞移植を受けているHLA−A0201+白血病患者の抗原特異的治療のための新規な試薬である。一つのローカスHLAミスマッチは、一つのローカスミスマッチを有しHLAマッチを有する非関連ドナーから移植を受けた白血病患者における比較可能な予後を示す最近の研究に示されているように、臨床上許容可能である34。かくして一つのローカスミスマッチ移植は、ドナーから由来する自己制限CTLでの抗原特異的治療のための理想的なセッティングを提供する。P126ペプチドを含むHLA−A0201テトラマーでの染色による、関連CTLの選択と組み合わせたここに記載されるin vitro刺激プロトコールは、採用可能な治療のためのP126特異的CTLの迅速な単離を可能にするであろう。
【0199】
さらに、WT1は自己セッティングにおける抗原特異的治療について調査可能である。これは、P126特異的CTLがHLA−A0201+ドナーから単離できるという観察によって支持される。成人におけるWT1発現は、比較的小さい数の細胞(例えばCD34+骨髄細胞、腎有足突起、精巣セリトール細胞及び卵巣顆粒層細胞)に制限されるため、WT1に対する自己Tリンパ球の寛容はおそらく不完全である。それ故、白血病、並びに腎細胞カルシノーマ、卵巣ガン、メラノーマ、及び乳ガンのような、上昇したWT1発現を有する他の悪性疾患に対してCTL応答を刺激するために、抗WT1ワクチン調製物のデザインのために同定されたP126エピトープを利用することが可能であろう。
【0200】
in vivoでの治療に加えて、WT1特異的CTLは、白血病患者から回収された自己骨髄細胞のin vitroでの浄化のためのツールを提供する。CTLは、顆粒球/マクロファージ直系の白血病始原細胞(図6D)及び赤血球直系の始原細胞(示さず)を除去する。対照的にCTLは、これらの直系の正常の始原細胞は認識しない。トランスフォームCD34+始原細胞の選択的な除去は、白血病始原細胞を再注射する危険を減少するはずであり、かくして自己幹細胞移植の主要な制限を解消する35。
【0201】
今日まで、組織特異的マイナー組織適合性抗原及びプロテイナーゼ3のような直系特異的抗原は、白血病反応性CTLに対する潜在的案標的として研究されている36-39。WT−1転写因子は、白血病始原細胞に対してCTL応答を選択的に向けることが可能な最初の標的抗原である。
【0202】
実施例4の参考文献
[参考文献]
【特許請求の範囲】
【請求項1】
ペプチドが完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列RMFPNAPYLを含むペプチドまたはその部分または異型。
【請求項2】
ペプチドが完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列CMTWNQMNLを含むペプチドまたはその部分または異型。
【請求項3】
ペプチドが完全なヒトgata−1ポリペプチドではないという条件で、アミノ酸配列HLMPFPGPLLを含むペプチドまたはその部分または異型。
【請求項4】
ペプチドがHLA−A0201に結合できる、請求項1ないし3のいずれか一項に記載のペプチド。
【請求項5】
HLA−A0201に結合した際に、ペプチド結合HLA−A0201が、前記アミノ酸配列を含むポリペプチドを異常に発現する細胞を認識する細胞傷害性Tリンパ球(CTL)の産生を導くことができる、請求項4記載のペプチド。
【請求項6】
ペプチドが非ペプチド結合を含む、請求項1ないし5のいずれか一項に記載のペプチド。
【請求項7】
アミノ酸配列RMFPNAPYLからなるペプチド。
【請求項8】
アミノ酸配列CMTWNQMNLからなるペプチド。
【請求項9】
アミノ酸配列HLMPFPGPLLからなるペプチド。
【請求項10】
請求項1ないし5および7ないし9のいずれか一項に記載のペプチドをコードするポリヌクレオチド。
【請求項11】
DNAである請求項10記載のポリヌクレオチド。
【請求項12】
請求項1ないし5および7ないし9のいずれか一項に記載のポリペプチドを発現することができる発現ベクター。
【請求項13】
請求項10または11記載のポリヌクレオチドまたは請求項12記載の発現ベクターを含む宿主細胞。
【請求項14】
請求項1ないし5および7ないし9のいずれか一項に記載のペプチドを産生する方法であって、請求項13記載の宿主細胞を培養し、かつ、宿主細胞またはその培養培地からペプチドを得ることを含む方法。
【請求項15】
請求項1ないし9のいずれか一項に記載のペプチドおよび薬学的に許容できるキャリアーを含む薬学的組成物。
【請求項16】
請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクター、および薬学的に許容できるキャリアーを含む薬学的組成物。
【請求項17】
薬剤に用いる請求項1ないし9のいずれか一項に記載のペプチド。
【請求項18】
薬剤に用いる請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクター。
【請求項19】
請求項1ないし9のいずれか一項に記載のペプチドまたは請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクターを含むガンワクチン。
【請求項20】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、請求項1ないし9のいずれか一項に記載のペプチドまたは請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクターの有効量を患者に投与することを含み、前記ペプチドまたはポリヌクレオチドまたは発現ベクターの量は、患者において抗ターゲット細胞免疫応答を誘発するのに十分な量とされる方法。
【請求項21】
請求項1ないし9のいずれか一項に記載のペプチドまたは請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクターの、請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における使用。
【請求項22】
in vitroにおいて活性化細胞傷害性Tリンパ球(CTL)を産生する方法であって、抗原特異的に前記CTLを活性するのに十分な時間、適切な抗原提示細胞の表面に発現された抗原結合ヒトクラスI MHC分子とCTLとをin vitroで接触させることを含み、前記抗原が請求項1ないし9のいずれか一項に記載のペプチドである方法。
【請求項23】
CTLと抗原提示細胞が、クラスI MHC分子に関して同種異系(アロ制限)である、請求項22記載の方法。
【請求項24】
CTLと抗原提示細胞が、クラスI MHC分子に関して同系(自己制限)である、請求項22記載の方法。
【請求項25】
抗原提示細胞と十分量の抗原とを接触させることにより適切な抗原提示細胞の表面に発現されたクラスI MHC分子に抗原が結合される、請求項22ないし24のいずれか一項に記載の方法であって、接触前には、抗原提示細胞のクラスI MHC分子が実質的に占有されてなく、かつ、接触後には、クラスI MHC分子が実質的に十分に占有される方法。
【請求項26】
抗原提示細胞が、請求項12記載の発現ベクターを含む、請求項22ないし24のいずれか一項に記載の方法。
【請求項27】
クラスI MHC分子がHLA−A0201である、請求項22ないし26のいずれか一項に記載の方法。
【請求項28】
請求項22ないし27のいずれか一項に記載の方法により得られた活性化細胞傷害性Tリンパ球(CTL)。
【請求項29】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現する細胞を選択的に認識する活性化細胞傷害性Tリンパ球(CTL)。
【請求項30】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現する細胞を認識するT細胞レセプター(TCR)であって、請求項28または29記載の細胞傷害性Tリンパ球(CTL)から得られるか、あるいは、TCRに機能的に等価な分子であるTCR。
【請求項31】
請求項30に記載されたT細胞レセプター(TCR)をコードするポリヌクレオチド。
【請求項32】
請求項30に記載されたT細胞レセプター(TCR)を発現することができる発現ベクター。
【請求項33】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、患者に、請求項28または29に記載された細胞傷害性Tリンパ球(CTL)の有効数を投与することを含む方法。
【請求項34】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、(1)患者から細胞傷害性Tリンパ球(CTL)を得る工程;(2)請求項30に記載した、T細胞レセプター(TCR)、または機能的に等価な分子をコードするポリヌクレオチドを、前記細胞に導入する工程;および(3)患者において工程(2)で産生された細胞を導入する工程を含む方法。
【請求項35】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、(1)患者から樹状細胞を得る工程;(2)前記樹状細胞を、請求項1ないし9のいずれか一項に記載されたペプチドまたは請求項10ないし12記載のポリヌクレオチドまたは発現ベクターとex vivoで接触させる工程;および(3)このように処理された樹状細胞を患者に再導入する工程を含む方法。
【請求項36】
ターゲット細胞がガン細胞である請求項20または33ないし35のいずれか一項に記載の、患者におけるターゲット細胞を殺す方法。
【請求項37】
ガンが、アミノ酸配列RMFPNAPYLおよびCMTWNQMNLを含むWT1ポリペプチドを異常に発現する、白血病、乳ガン、メラノーマおよび卵巣ガンのいずれか一つである請求項36記載の方法。
【請求項38】
ガンが、アミノ酸配列HLMPFPGPLLを含むgata−1ポリペプチドを異常に発現する白血病である、請求項36記載の方法。
【請求項39】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における、請求項28または29に記載の細胞傷害性Tリンパ球の使用。
【請求項40】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における、請求項34の工程(2)で産生される細胞傷害性Tリンパ球の使用。
【請求項41】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における、請求項35の工程(2)において産生される樹状細胞の使用。
【請求項42】
ここに記載されるガンを治療するための新規方法。
【請求項1】
ペプチドが完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列RMFPNAPYLを含むペプチドまたはその部分または異型。
【請求項2】
ペプチドが完全なヒトWT−1ポリペプチドではないという条件で、アミノ酸配列CMTWNQMNLを含むペプチドまたはその部分または異型。
【請求項3】
ペプチドが完全なヒトgata−1ポリペプチドではないという条件で、アミノ酸配列HLMPFPGPLLを含むペプチドまたはその部分または異型。
【請求項4】
ペプチドがHLA−A0201に結合できる、請求項1ないし3のいずれか一項に記載のペプチド。
【請求項5】
HLA−A0201に結合した際に、ペプチド結合HLA−A0201が、前記アミノ酸配列を含むポリペプチドを異常に発現する細胞を認識する細胞傷害性Tリンパ球(CTL)の産生を導くことができる、請求項4記載のペプチド。
【請求項6】
ペプチドが非ペプチド結合を含む、請求項1ないし5のいずれか一項に記載のペプチド。
【請求項7】
アミノ酸配列RMFPNAPYLからなるペプチド。
【請求項8】
アミノ酸配列CMTWNQMNLからなるペプチド。
【請求項9】
アミノ酸配列HLMPFPGPLLからなるペプチド。
【請求項10】
請求項1ないし5および7ないし9のいずれか一項に記載のペプチドをコードするポリヌクレオチド。
【請求項11】
DNAである請求項10記載のポリヌクレオチド。
【請求項12】
請求項1ないし5および7ないし9のいずれか一項に記載のポリペプチドを発現することができる発現ベクター。
【請求項13】
請求項10または11記載のポリヌクレオチドまたは請求項12記載の発現ベクターを含む宿主細胞。
【請求項14】
請求項1ないし5および7ないし9のいずれか一項に記載のペプチドを産生する方法であって、請求項13記載の宿主細胞を培養し、かつ、宿主細胞またはその培養培地からペプチドを得ることを含む方法。
【請求項15】
請求項1ないし9のいずれか一項に記載のペプチドおよび薬学的に許容できるキャリアーを含む薬学的組成物。
【請求項16】
請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクター、および薬学的に許容できるキャリアーを含む薬学的組成物。
【請求項17】
薬剤に用いる請求項1ないし9のいずれか一項に記載のペプチド。
【請求項18】
薬剤に用いる請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクター。
【請求項19】
請求項1ないし9のいずれか一項に記載のペプチドまたは請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクターを含むガンワクチン。
【請求項20】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、請求項1ないし9のいずれか一項に記載のペプチドまたは請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクターの有効量を患者に投与することを含み、前記ペプチドまたはポリヌクレオチドまたは発現ベクターの量は、患者において抗ターゲット細胞免疫応答を誘発するのに十分な量とされる方法。
【請求項21】
請求項1ないし9のいずれか一項に記載のペプチドまたは請求項10または11に記載のポリヌクレオチドまたは請求項12に記載の発現ベクターの、請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における使用。
【請求項22】
in vitroにおいて活性化細胞傷害性Tリンパ球(CTL)を産生する方法であって、抗原特異的に前記CTLを活性するのに十分な時間、適切な抗原提示細胞の表面に発現された抗原結合ヒトクラスI MHC分子とCTLとをin vitroで接触させることを含み、前記抗原が請求項1ないし9のいずれか一項に記載のペプチドである方法。
【請求項23】
CTLと抗原提示細胞が、クラスI MHC分子に関して同種異系(アロ制限)である、請求項22記載の方法。
【請求項24】
CTLと抗原提示細胞が、クラスI MHC分子に関して同系(自己制限)である、請求項22記載の方法。
【請求項25】
抗原提示細胞と十分量の抗原とを接触させることにより適切な抗原提示細胞の表面に発現されたクラスI MHC分子に抗原が結合される、請求項22ないし24のいずれか一項に記載の方法であって、接触前には、抗原提示細胞のクラスI MHC分子が実質的に占有されてなく、かつ、接触後には、クラスI MHC分子が実質的に十分に占有される方法。
【請求項26】
抗原提示細胞が、請求項12記載の発現ベクターを含む、請求項22ないし24のいずれか一項に記載の方法。
【請求項27】
クラスI MHC分子がHLA−A0201である、請求項22ないし26のいずれか一項に記載の方法。
【請求項28】
請求項22ないし27のいずれか一項に記載の方法により得られた活性化細胞傷害性Tリンパ球(CTL)。
【請求項29】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現する細胞を選択的に認識する活性化細胞傷害性Tリンパ球(CTL)。
【請求項30】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現する細胞を認識するT細胞レセプター(TCR)であって、請求項28または29記載の細胞傷害性Tリンパ球(CTL)から得られるか、あるいは、TCRに機能的に等価な分子であるTCR。
【請求項31】
請求項30に記載されたT細胞レセプター(TCR)をコードするポリヌクレオチド。
【請求項32】
請求項30に記載されたT細胞レセプター(TCR)を発現することができる発現ベクター。
【請求項33】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、患者に、請求項28または29に記載された細胞傷害性Tリンパ球(CTL)の有効数を投与することを含む方法。
【請求項34】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、(1)患者から細胞傷害性Tリンパ球(CTL)を得る工程;(2)請求項30に記載した、T細胞レセプター(TCR)、または機能的に等価な分子をコードするポリヌクレオチドを、前記細胞に導入する工程;および(3)患者において工程(2)で産生された細胞を導入する工程を含む方法。
【請求項35】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷する方法であって、(1)患者から樹状細胞を得る工程;(2)前記樹状細胞を、請求項1ないし9のいずれか一項に記載されたペプチドまたは請求項10ないし12記載のポリヌクレオチドまたは発現ベクターとex vivoで接触させる工程;および(3)このように処理された樹状細胞を患者に再導入する工程を含む方法。
【請求項36】
ターゲット細胞がガン細胞である請求項20または33ないし35のいずれか一項に記載の、患者におけるターゲット細胞を殺す方法。
【請求項37】
ガンが、アミノ酸配列RMFPNAPYLおよびCMTWNQMNLを含むWT1ポリペプチドを異常に発現する、白血病、乳ガン、メラノーマおよび卵巣ガンのいずれか一つである請求項36記載の方法。
【請求項38】
ガンが、アミノ酸配列HLMPFPGPLLを含むgata−1ポリペプチドを異常に発現する白血病である、請求項36記載の方法。
【請求項39】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における、請求項28または29に記載の細胞傷害性Tリンパ球の使用。
【請求項40】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における、請求項34の工程(2)で産生される細胞傷害性Tリンパ球の使用。
【請求項41】
請求項1ないし3のいずれか一項に記載のアミノ酸配列を含むポリペプチドを異常に発現するターゲット細胞を患者において殺傷するための薬剤の製造における、請求項35の工程(2)において産生される樹状細胞の使用。
【請求項42】
ここに記載されるガンを治療するための新規方法。
【図1】

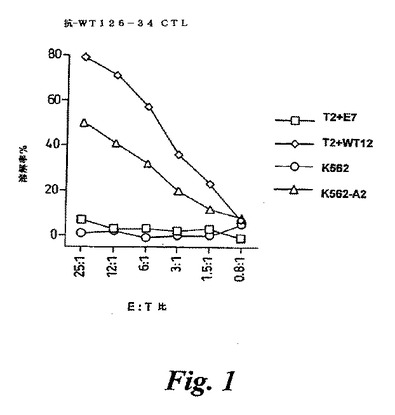
【図2】
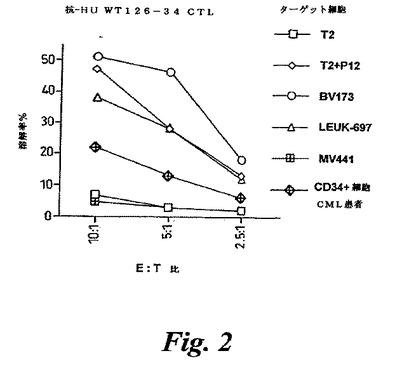
【図3】
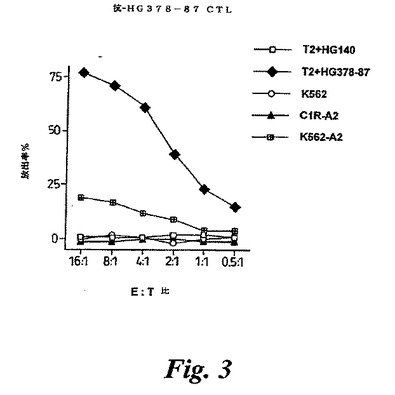
【図4A】
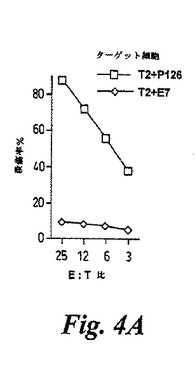
【図4B】
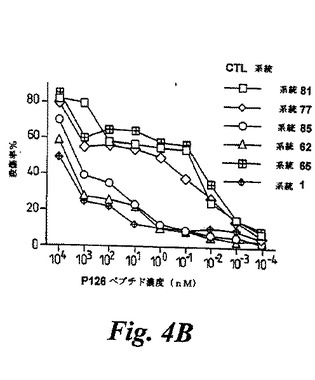
【図4C】
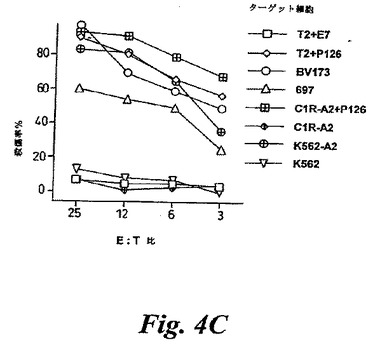
【図5】
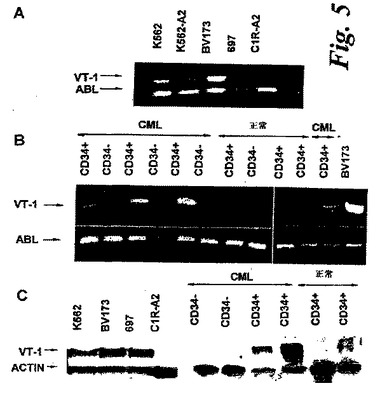
【図6A】
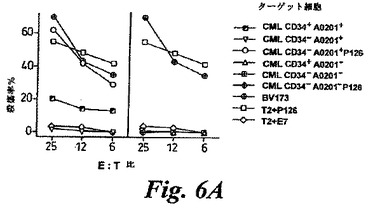
【図6B】
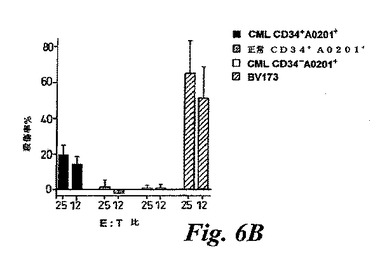
【図6C】
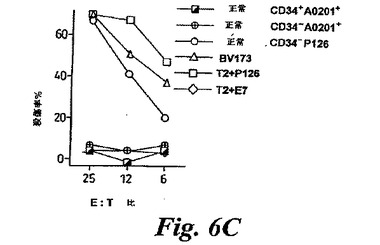
【図6D】
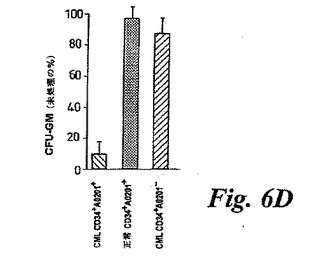
【図6E】
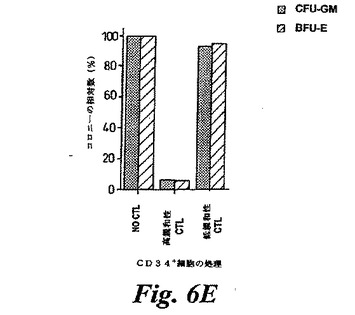
【図6F】
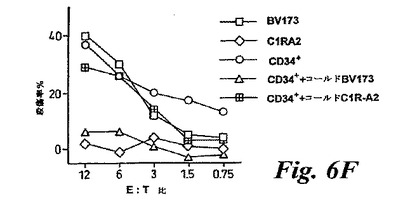
【図2】
【図3】
【図4A】
【図4B】
【図4C】
【図5】
【図6A】
【図6B】
【図6C】
【図6D】
【図6E】
【図6F】
【公開番号】特開2012−183067(P2012−183067A)
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願番号】特願2012−105984(P2012−105984)
【出願日】平成24年5月7日(2012.5.7)
【分割の表示】特願2000−579636(P2000−579636)の分割
【原出願日】平成11年11月2日(1999.11.2)
【出願人】(506233140)ガニメド・ファーマスーティカルズ・アーゲー (1)
【Fターム(参考)】
【公開日】平成24年9月27日(2012.9.27)
【国際特許分類】
【出願日】平成24年5月7日(2012.5.7)
【分割の表示】特願2000−579636(P2000−579636)の分割
【原出願日】平成11年11月2日(1999.11.2)
【出願人】(506233140)ガニメド・ファーマスーティカルズ・アーゲー (1)
【Fターム(参考)】
[ Back to top ]