
ZP15遺伝子座内への標的組込み
例えば対象のポリペプチドの発現のための、植物Zp15遺伝子座内への外因性配列の標的組込みのための方法および組成物を本明細書において開示する。
【発明の詳細な説明】
【技術分野】
【0001】
連邦支援の研究の下でなされた発明に対する権利の声明
適用されない。
【0002】
本開示は、植物ゲノム工学、特に植物Zp15遺伝子内への導入遺伝子の標的組込みの分野にある。
【背景技術】
【0003】
バイオテクノロジーは、食糧生産に対して増大する世界的需要の課題に対処するために不可欠なツールとして台頭してきた。農業の生産性を向上させるための従来のアプローチ、例えば収穫量の増大または遺伝子操作による害虫抵抗性は、突然変異育種または形質転換による作物種のゲノム内への新規遺伝子の導入のいずれかに依存している。どちらの方法も本質的に非特異的であり、かつ比較的非効率的である。例えば、従来の植物形質転換法は、ゲノム内のランダムな位置に組み込む外因性DNAを送達する。したがって、所望の特性を備えた形質転換株を同定かつ単離するために、何千もの固有のランダム組込み事象を作出し、その後所望の個体をスクリーニングする必要がある。結果として、従来の植物特質の遺伝子操作は、労力を要し、多大な時間を要し、かつ予測不可能な仕事である。さらに、この組込みのランダム性によって、意図しないゲノム破壊による多面発現効果が生じているかどうかを予測するのが難しくなる。結果として、遺伝子操作された遺伝子または特質を備えた植物株の作出、単離、および特徴付けは、成功の確率が低く、極めて労力を要し、かつコストの大きい手法となっている。
【発明の概要】
【発明が解決しようとする課題】
【0004】
標的遺伝子改変は、植物システムにおける従来的実施の合理的な課題を克服するものであり、ゆえに基礎的な植物生物学の研究および農業バイオテクノロジーのどちらにおいても、長年にわたる、しかしとらえどころのない目標である。しかしながら、イネにおける陽性−陰性の薬剤選択または事前に遺伝子操作された制限酵素部位の使用による「遺伝子ターゲティング」を除いて、すべての植物種における標的ゲノム改変は、モデルでも農作物でも、最近まで非常に困難であることが分かっている。Teradaら,(2002)Nat Biotechnol 20(10):1030;Teradaら,(2007)Plant Physiol 144(2):846;D’Halluinら,(2008)Plant Biotechnology J.6(1):93。
【0005】
最近、ゲノムDNAの標的切断のための方法および組成物が記載されている。そのような標的切断の事象を用いて、例えば、標的突然変異生成を誘発すること、細胞DNA配列の標的欠失を誘導すること、ならびに所定の染色体座位での標的組換えを容易にすることが可能である。例えば、米国特許公報第20030232410号;第20050208489号;第20050026157号;第20050064474号;および第20060188987号,ならびに国際公報WO2007/014275を参照されたく、その開示は参照することによりあらゆる目的でそれらの全体として組み入れられているものとする。米国特許公報第20080182332号は植物ゲノムの標的改変のための非標準ジンクフィンガーヌクレアーゼ(ZFN)の使用について記載しており、また米国特許出願第12/284,888号は植物EPSPS遺伝子座内へのZFNを介した標的組込みについて記載している。
【0006】
しかしながら、植物およびその後代における安定的で遺伝的な遺伝子改変を確立するために、植物ゲノム内のさらなる遺伝子座内への安定的な標的組込みのための組成物および方法の必要性が依然として残っている。
【課題を解決するための手段】
【0007】
本開示は、植物細胞におけるZp15遺伝子内に組み込まれている外因性核酸配列の1つ以上の産物(すなわち、タンパク質またはRNA分子)を発現させるための方法および組成物を提供する。本明細書に示されるように、Zp15遺伝子座におけるまたは近隣における1つ以上の外因性配列の組込みによって、宿主植物は再生、開花、または種をつける能力を損なわないと考えられ、場合によっては、世代にわたった外因性配列の遺伝的伝達が可能になる。前記外因性核酸配列は、例えば、1つ以上の遺伝子もしくはcDNA分子、または任意の種類のコードもしくは非コード配列、および1つ以上の制御エレメント(例えば、プロモーター)を含み得る。例えば、除草剤耐性遺伝子をこの遺伝子座内に組み込んで、所望の除草剤抵抗性を備えた作物を生産することができる。Zp15遺伝子座においてまたは近隣において外因性核酸を含有する細胞は、配偶子(生殖細胞系)にも寄与し得、ゆえに次世代の後代に伝達され得る。
【0008】
Zp15遺伝子内への外因性核酸配列の取込みは、選択されたZp15遺伝子座におけるゲノムの標的二本鎖切断によって容易になる。切断は、選択されたZp15遺伝子座内の配列に結合するように遺伝子操作された、例えば、メガヌクレアーゼDNA結合ドメイン、ロイシンジッパーDNA結合ドメイン、ジンクフィンガータンパク質(ZFP)、または前述のキメラ組み合わせ等のDNA結合ドメイン、および切断ドメインまたは切断ハーフドメインを含む融合タンパク質を用いることによってZp15遺伝子を標的とする。このような切断によって、切断部位におけるまたは近隣における外因性ポリヌクレオチド配列の組込みが刺激される。外因性配列の組込みは、相同性依存的および相同性非依存的メカニズムの両方によって進行し得る。
【0009】
一実施形態において、Zp15遺伝子における標的部位に結合する遺伝子操作されたDNA結合ドメイン(例えば、ZFP、メガヌクレアーゼ、またはロイシンジッパー)を本明細書において開示する。前記DNA結合ドメインは、例えば、表1に示した認識ヘリックスを含む任意の遺伝子操作されたジンクフィンガーDNA結合ドメインを含み得る。本明細書に記載される任意のDNA結合ドメインは、機能ドメイン、例えば切断ドメインまたは切断ハーフドメインをさらに含んでもよい。いくつかの実施態様では、前記切断ハーフドメインは、FokIまたはStsI等のIIS型制限エンドヌクレアーゼに由来するものであり得る。他の実施態様では、前記切断ドメインは、ホーミングエンドヌクレアーゼ、例えば改変されたDNA結合ドメインを有するホーミングエンドヌクレアーゼを含み得る。
【0010】
別の実施形態において、Zp15遺伝子座内に組み込まれた外因性配列を含む植物または種子を本明細書において開示する。ある実施態様では、前記外因性配列を植物の配偶子内に組み込む。
【0011】
別の実施形態において、細胞内で外因性核酸配列の産物を発現させるための方法であって、前記方法は:(a)前記細胞内で第一の融合タンパク質を発現させる工程であって、前記第一の融合タンパク質は第一のDNA結合ドメイン(例えば、ZFP)および第一の切断ハーフドメインを含んでおり、前記DNA結合ドメインは前記細胞のゲノムのZp15遺伝子における第一の標的部位に結合するように遺伝子操作されている工程;(b)前記細胞内で第二の融合タンパク質を発現させる工程であって、前記第二の融合タンパク質は第二のDNAドメインおよび第二の切断ハーフドメインを含んでおり、前記第二のDNAドメインは前記細胞のゲノムのZp15遺伝子における第二の標的部位に結合し、前記第二の標的部位は前記第一の標的部位とは異なる工程;ならびに(c)前記細胞を外因性核酸配列およびZp15遺伝子における第一の配列に相同である第一のヌクレオチド配列を含むポリヌクレオチドと接触させる工程であって、前記第一の融合タンパク質の前記第一の標的部位への結合、および前記第二の融合タンパク質の前記第二の標的部位への結合により、前記切断ハーフドメインは前記細胞のゲノムがZp15遺伝子内で切断されるように位置し、それによってZp15遺伝子における前記細胞のゲノム内への外因性配列の組込みおよび前記外因性配列の産物の発現がもたらされる工程;を含む方法を本明細書において開示する。
【0012】
前記外因性核酸配列は、植物に所望の特質を付与する、1つ以上のプロモーターの有無にかかわらず、1つ以上の機能性ポリペプチド(例えば、cDNA)をコードする配列を含んでいてもよく、かつ/あるいは1つ以上のRNA配列を(例えば、1つ以上のshRNA発現カセットによって)産生してもよい。そのような特質には、除草剤抵抗性または耐性;虫害抵抗性または耐性;病害抵抗性または耐性(ウイルス性、細菌性、菌性、線虫による);乾燥、熱、低温、凍結、過度の湿気、塩分ストレスに対する抵抗性または耐性などのストレス耐性および/または抵抗性;酸化ストレス;収穫量の増加;食物の含有量および構成;見た目;雄性不稔;ドライダウン;耐倒伏性;多産;デンプンの量および質;油分の量および質;タンパク質の質および量;アミノ酸組成等が含まれるが、これに制限されない。もちろん、除草剤、害虫、病害(ウイルス性、細菌性、菌性、線虫による)または乾燥抵抗性、雄性不稔、ドライダウン、耐倒伏性、多産、デンプンの性質、油分の量および質を与えるもの、あるいは収穫量または栄養価を増加させるもの等の、任意の記載の任意の2つ以上の外因性核酸を所望どおりに利用してよい。ある実施態様では、前記核酸配列は、除草剤抵抗性タンパク質(例えば、AAD−1(アリールオキシアルカノエート・ジオキシゲナーゼ)遺伝子、AAD−12遺伝子、またはホスフィノトリシン・アセチル・トランスフェラーゼ(PAT)遺伝子)および/またはその機能性フラグメントをコードする配列を含む。組み込まれた配列の発現は、前記組み込まれた配列に操作可能に連結されたプロモーターによって促進され得る。あるいは、組み込まれた配列にはプロモーターがなく、転写は内在性Zp15プロモーターによって促進される。
【0013】
ある実施態様では、前記ポリヌクレオチドはZp15遺伝子における第二の配列に相同である第二のヌクレオチド配列をさらに含む。前記第二のヌクレオチド配列は、Zp15遺伝子における前記第二の配列と同一であってもよい。さらに、第一および第二のヌクレオチド配列を含む実施態様では、前記第一のヌクレオチド配列はZp15遺伝子における第一の配列と同一であってよく、かつ前記第二のヌクレオチド配列はZp15遺伝子における第二の配列に相同であるが同一でないものであってよい。本明細書に記載される方法のいずれかにおいて、前記第一および第二のヌクレオチド配列は、外因性配列に隣接する。ある実施態様では、前記ポリヌクレオチドはプラスミドである。他の実施態様では、前記ポリヌクレオチドは線状のDNA分子である。
【0014】
別の実施形態において、細胞のゲノムにおけるZp15遺伝子内に外因性配列を組み込むための方法であって、前記方法は:(a)前記細胞内で第一の融合タンパク質を発現させる工程であって、前記第一の融合タンパク質は第一のDNA結合ドメイン(例えば、ZFP)および第一の切断ハーフドメインを含んでおり、前記第一のDNA結合ドメインは前記細胞のゲノムにおけるZp15遺伝子座における第一の標的部位に結合するように遺伝子操作されている工程;(b)前記細胞内で第二の融合タンパク質を発現させる工程であって、前記第二の融合タンパク質は第二のDNA結合ドメイン(例えば、ZFP)および第二の切断ハーフドメインを含んでおり、前記第二のDNA結合ドメインは前記細胞のゲノムにおけるZp15遺伝子座における第二の標的部位に結合し、前記第二の標的部位は前記第一の標的部位とは異なる工程;ならびに(c)前記細胞を外因性核酸配列を含むポリヌクレオチドと接触させる工程であって、前記第一の融合タンパク質の前記第一の標的部位への結合、および前記第二の融合タンパク質の前記第二の標的部位への結合により、前記切断ハーフドメインは前記細胞のゲノムがZp15遺伝子座内で切断されるように位置し、それによってZp15遺伝子座内の前記細胞のゲノム内への外因性配列の相同性依存的組込みがもたらされる工程;を含む方法を本明細書において提示する。ある実施態様では、機能性ポリペプチドをコードする外因性配列をZp15遺伝子内に挿入する。
【0015】
本明細書に記載される方法のいずれかにおいて、前記第一および第二の切断ハーフドメインは、IIS型制限エンドヌクレアーゼ、例えばFokIまたはStsIに由来するものであり得る。さらに、本明細書に記載される方法のいずれかにおいて、少なくとも1つの融合タンパク質は、例えば前記切断ハーフドメインの必須のヘテロ二量体が形成されるような、前記切断ハーフドメインの二量化接触面のアミノ酸配列の変更を含み得る。
【0016】
本明細書に記載される方法のいずれかにおいて、植物細胞は単子葉性または双子葉性の植物細胞を含み得る。ある実施態様では、前記植物細胞は作物、例えばトウモロコシである。
【図面の簡単な説明】
【0017】
【図1】図1は、ZFN対#25の一過性の発現を受けた細胞に由来するトウモロコシHiII gDNAからのZp15増幅産物についての例示的配列分析結果を表し(結合部位に下線)、期待される切断部位における6bpのNHEJ挿入(太字)を示す。
【図2】図2は、ZFN対#24の一過性の発現を受けた細胞に由来するトウモロコシHiII gDNAからのZp15増幅産物についての例示的配列分析結果を表し(結合部位に下線)、期待される切断部位における3bpの欠失を示す。
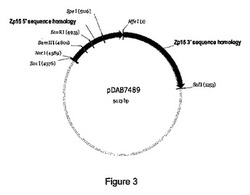
【図3】図3は、pDAB7489と称される構築物を図示した概略図である。
【図4】図4は、AAD遺伝子をコードする例示的な除草剤耐性遺伝子発現カセットを図示した概略図である。
【図5】図5は、pDAB7490と称される構築物を図示した概略図である。
【図6−1】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−2】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−3】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−4】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−5】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−6】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−7】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−8】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図7】図7は、pDAB104101と称される構築物を図示した概略図である。
【図8】図8は、PAT発現カセットを図示した概略図である。
【図9】図9は、pDAB104107と称される構築物を図示した概略図である。
【図10】図10は、pDAB104104と称される構築物を図示した概略図である。
【図11】図11は、pDAB104105と称される構築物を図示した概略図である。
【図12】図12は、pDAB104106と称される構築物を図示した概略図である。
【図13】図13は、pDAB104100と称される構築物を図示した概略図である。
【図14】図14は、pDAB104103と称される構築物を図示した概略図である。
【図15】図15は、pDAB104102と称される構築物を図示した概略図である。
【発明を実施するための形態】
【0018】
詳細な説明
本開示は、トウモロコシの第6染色体上に位置する植物Zp15遺伝子内への標的組込み(TI)のための方法および組成物に関する。DNA結合ドメイン(例えば、ZFP、メガヌクレアーゼ、またはロイシンジッパー)およびヌクレアーゼドメインを含む融合タンパク質を用いることによって、挿入された(ドナー)配列を外因性プロモーターに操作可能に連結させることができ、またはプロモーターがなくてもよい。プロモーターがない場合、組み込まれたオープン・リーディング・フレームの転写は、プロモーター特定の組織における内在性Zp15遺伝子プロモーターによって生じ得る。プロモーターがないドナーの使用によって、前記ドナーのランダムな組込みの可能性および/または前記ドナー上に保持されたプロモーターによる内在性遺伝子の疑似活性化は低下する。
【0019】
Zp15遺伝子内への標的切断および組換えに有用な組成物は、切断ドメイン(または切断ハーフドメイン)とDNA結合ドメイン(例えば、ZFP)とを含む融合タンパク質、これらのタンパク質をコードするポリヌクレオチド、およびポリペプチドとポリペプチドをコードするポリヌクレオチドとの組み合わせを含有する。ジンクフィンガー結合ドメインは、1つ以上のジンクフィンガー(例えば、2、3、4、5、6、7、8、9、またはそれ以上のジンクフィンガー)を含んでいてよく、かつZp15遺伝子内の任意の配列に結合するように遺伝子操作されていてもよい。細胞内におけるそのような融合タンパク質の存在は、前記融合タンパク質のその(それらの)結合部位への結合および内在性Zp15遺伝子内での切断をもたらす。
【0020】
概要
当該方法の実施、ならびに本明細書に開示される組成物の調製および使用は、特に指定のない限り、分子生物学、生化学、クロマチン構造および分析、計算化学、細胞培養、組換えDNA、ならびに当該技術分野の技術の範囲内にある関連分野における従来技術を利用する。これらの技術は文献で十分に説明されている。例えば、Sambrookら,MOLECULAR CLONING:A LABORATORY MANUAL,Second edition,Cold Spring Harbor Laboratory Press,1989およびThird edition,2001;Ausubelら,CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John Wiley&Sons,New York,1987および定期的に更新したもの;the series METHODS IN ENZYMOLOGY,Academic Press,San Diego;Wolffe,CHROMATIN STRUCTURE AND FUNCTION,Third edition,Academic Press,San Diego,1998;METHODS IN ENZYMOLOGY,Vol.304,“Chromatin”(P.M.WassarmanおよびA.P.Wolffe,eds.),Academic Press,San Diego,1999;ならびにMETHODS IN MOLECULAR BIOLOGY,Vol.119,“Chromatin Protocols”(P.B.Becker,ed.)Humana Press,Totowa,1999を参照されたい。
【0021】
定義
「核酸」、「ポリヌクレオチド」、および「オリゴヌクレオチド」という用語は、代替可能に用いられ、線状または環状の構造で、かつ一本鎖または二本鎖のいずれかの形態のデオキシリボヌクレオチドまたはリボヌクレオチドのポリマーを表す。本開示上、これらの用語は、ポリマーの長さについて制限するものとして解釈されるべきではない。前記用語は、天然ヌクレオチドの既知の類似体、ならびに塩基、糖および/またはリン酸塩部分(例えば、ホスホロチオエート骨格)において改変されているヌクレオチドを包含し得る。一般に、特定のヌクレオチドの類似体は同じ塩基対合特異性を有し、すなわちAの類似体はTと塩基対合する。
【0022】
「ポリペプチド」、「ペプチド」、および「タンパク質」という用語は、アミノ酸残基のポリマーを表すために代替可能に用いられる。前記用語はまた、1つ以上のアミノ酸が化学的類似体または対応する天然に存在しているアミノ酸の改変された誘導体であるアミノ酸ポリマーに適用される。
【0023】
「結合」とは、高分子間(例えば、タンパク質と核酸との間)の配列特異的、非共有相互作用を表す。全体としての相互作用が配列特異的であるならば、結合相互作用のすべての構成成分が配列特異的である(例えば、DNA骨格におけるリン酸残基と接触する)必要はない。そのような相互作用は、通常、10-6M-1以下の解離定数(Kd)によって特徴付けされる。「親和性」とは結合の強度を表し、結合親和性の増大はより低いKdと相関している。
【0024】
「結合タンパク質」とは、別の分子に非共有的に結合し得るタンパク質である。結合タンパク質は、例えば、DNA分子(DNA結合タンパク質)、RNA分子(RNA結合タンパク質)、および/またはタンパク質分子(タンパク質結合タンパク質)に結合し得る。タンパク質結合タンパク質の場合、それはそれ自体に結合し得(ホモ二量体、ホモ三量体等を形成する)、かつ/またはそれは1つ以上の異なるタンパク質の分子に結合し得る。結合タンパク質は、1種類より多い結合活性を有し得る。例えば、ジンクフィンガータンパク質は、DNA結合、RNA結合、およびタンパク質結合活性を有する。
【0025】
「ジンクフィンガーDNA結合タンパク質」(または結合ドメイン)とは、亜鉛イオンの配位によって構造が安定化する結合ドメイン内のアミノ酸配列の領域である、1つ以上のジンクフィンガーを介した配列特異的な様式でDNAに結合するタンパク質またはより大きなタンパク質内のドメインである。ジンクフィンガーDNA結合タンパク質という用語は、しばしば、ジンクフィンガータンパク質またはZFPと略記される。
【0026】
ジンクフィンガー結合ドメインを、所定のヌクレオチド配列に結合するように「遺伝子操作」することができる。ジンクフィンガータンパク質を遺伝子操作するための方法の制限されない例は、設計および選択である。設計されたジンクフィンガータンパク質とは、主に合理的基準に由来する設計/組成を有する天然には存在していないタンパク質である。設計のための合理的基準には、置換法則の適用、ならびに既存のZFP設計および結合データの情報を蓄積しているデータベース内の情報を処理するためのコンピュータアルゴリズムが含まれる。例えば、米国特許第6,140,081号;第6,453,242号;および第6,534,261号を参照されたく、またWO98/53058;WO98/53059;WO98/53060;WO02/016536およびWO03/016496も参照されたい。
【0027】
「選択された」ジンクフィンガータンパク質とは、ファージディスプレイ、相互作用トラップ、またはハイブリッド選択等の実験に基づいた手法に主に由来して産生される天然には見られないタンパク質である。例えば、米国第5,789,538号;米国第5,925,523号;米国第6,007,988号;米国第6,013,453号;米国第6,200,759号;WO95/19431;WO96/06166;WO98/53057;WO98/54311;WO00/27878;WO01/60970;WO01/88197、およびWO02/099084を参照されたい。
【0028】
「配列」という用語は、DNAまたはRNAであってよく;線状、環状、または分岐したものであってよく;かつ一本鎖または二本鎖のいずれかであってよい、任意の長さのヌクレオチド配列を表す。「ドナー配列」という用語は、ゲノム内に挿入されたヌクレオチド配列を表す。ドナー配列は任意の長さであってよく、例えば、2〜10,000ヌクレオチド長の間(またはその間のもしくはそれを上回る任意の整数値)、好ましくは約100〜1,000ヌクレオチド長の間(またはその間の任意の整数)、より好ましくは約200〜500ヌクレオチド長の間である。
【0029】
「相同であり、同一でない配列」とは、第二の配列とある程度の配列同一性を共有するが、その配列は前記第二の配列のものとは同一ではない第一の配列を表す。例えば、変異体遺伝子の野生型配列を含むポリヌクレオチドは、前記変異体遺伝子の配列に相同であり、かつ同一ではない。ある実施態様では、前記2つの配列間の相同性の程度は、通常の細胞メカニズムを利用することによってそれらの間で相同組換えが起こるのに十分である。2つの相同であり同一でない配列は任意の長さであってよく、その非相同性の程度は、1ヌクレオチド程度に小さくても(例えば、標的相同組換えによるゲノムの点変異の修正のための)または10キロベース以上程度に大きくてもよい(例えば、染色体の所定の異所性部位における遺伝子の挿入のための)。相同であり同一でない配列を含む2つのポリヌクレオチドは、同じ長さである必要はない。例えば、20〜10,000ヌクレオチドまたはヌクレオチド対の間の外因性ポリヌクレオチド(すなわち、ドナーポリヌクレオチド)を用いることができる。
【0030】
核酸およびアミノ酸の配列同一性を決定するための技術は、当該技術分野において既知である。典型的に、そのような技術には、遺伝子に対するmRNAのヌクレオチド配列を決定すること、および/またはそれによってコードされるアミノ酸配列を決定すること、およびこれらの配列を第二のヌクレオチドもしくはアミノ酸配列と比較することが含まれる。このやり方でゲノム配列を決定および比較することもできる。一般に、同一性とは、2つのポリヌクレオチドまたはポリペプチド配列の、それぞれヌクレオチド対ヌクレオチドまたはアミノ酸対アミノ酸の正確な対応を表す。2つ以上の配列(ポリヌクレオチドまたはアミノ酸)を、それらの同一性パーセントを決定することによって比較することができる。2つの配列の同一性パーセントは、核酸またはアミノ酸配列にかかわらず、2つの並んだ配列間の正確な一致をより短い配列の長さで割って、100をかけた数である。核酸配列のためのおおよそのアラインメントは、SmithおよびWatermanの局所相同性アルゴリズム、Advances in Applied Mathematics 2:482−489(1981)によって提供されている。このアルゴリズムを、Dayhoff,Atlas of Protein Sequences and Structure,M.O.Dayhoff ed.,5 suppl.3:353−358,National Biomedical Research Foundation,Washington,D.C.,USAによって開発され、Gribskov,Nucl.Acids Res.14(6):6745−6763(1986)によって正規化されたスコア行列を用いることによって、アミノ酸配列に適用することができる。配列の同一性パーセントを決定するためのこのアルゴリズムの例示的実施は、Genetics Computer Group(Madison,WI)による「BestFit」ユーティリティーアプリケーションにおいて提供されている。配列間の同一性または類似性パーセントを算出するのに適したプログラムは、当該技術分野において一般に既知であり、例えば、別のアラインメントプログラムはデフォルトパラメーターを用いて使用されるBLASTである。例えば、BLASTNおよびBLASTPを、以下のデフォルトパラメーター:遺伝子コード=標準;フィルター=なし;鎖=両方;カットオフ=60;期待数=10;行列=BLOSUM62;表示=50配列;分類=高スコア;データベース=非重複、GenBank+EMBL+DDBJ+PDB+GenBank CDS翻訳+Swissタンパク質+Spupdate+PIRを用いることによって使用することができる。これらのプログラムの詳細はインターネット上で見出すことができる。本明細書に記載される配列に関して、所望される配列同一性の程度の範囲は、およそ80%〜100%、およびその間の任意の整数値である。典型的には、配列間の同一性パーセントは、少なくとも70〜75%、好ましくは80〜82%、より好ましくは85〜90%、さらに好ましくは92%、なおいっそう好ましくは95%、および最も好ましくは98%の配列同一性である。
【0031】
あるいは、ポリヌクレオチド間の配列類似性の程度を、相同な領域間で安定した二重鎖が形成される条件下でのポリヌクレオチドのハイブリダイゼーション、続いて一本鎖特異的ヌクレアーゼを用いた消化、および消化されたフラグメントの大きさの決定によって決定することができる。2つの核酸または2つのポリペプチド配列は、配列が、前述の方法を用いることによって決定される、少なくとも約70〜75%、好ましくは80〜82%、より好ましくは85〜90%、さらに好ましくは92%、なおいっそう好ましくは95%、および最も好ましくは98%の配列同一性を当該分子の既定の長さにわたって示す場合、互いに実質的に相同である。本明細書において使用するとき、実質的に相同であるとは、特定のDNAまたはポリペプチド配列と完全な同一性を示す配列も表す。実質的に相同であるDNA配列を、特定のシステムに対して規定される、例えばストリンジェントな条件下でのサザンハイブリダイゼーション実験で同定することができる。適切なハイブリダイゼーション条件を規定することは、当該技術分野の技術の範囲内である。例えば、Sambrookら,前記参照;Nucleic Acid Hybridization:A Practical Approach,editors B.D.HamesおよびS.J.Higgins,(1985)Oxford;Washington,DC;IRL Pressを参照されたい。
【0032】
2つの核酸フラグメントの選択的ハイブリダイゼーションは、以下のように決定することができる。2つの核酸分子間の配列同一性の程度は、前記分子間のハイブリダイゼーション事象の効率および強度に影響を及ぼす。部分的に同一な核酸配列は、完全に同一な配列の標的分子へのハイブリダイゼーションを少なくとも部分的に阻害する。完全に同一な配列のハイブリダイゼーションの阻害を、当該技術分野において周知であるハイブリダイゼーションアッセイ(例えば、サザン(DNA)ブロット、ノーザン(RNA)ブロット、溶液ハイブリダイゼーション等、Sambrookら,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)を用いることによって評価することができる。そのようなアッセイは、様々な程度の選択性を用いることによって、例えば、低ストリンジェントから高ストリンジェントに変化する条件を用いることによって実施され得る。低ストリンジェントな条件を利用する場合、部分的な程度の配列同一性さえも欠いている第二のプローブ(例えば、標的分子と約30%未満の配列同一性を有するプローブ)を用いることによって、非特異的結合の事象がない場合、前記第二のプローブは標的にハイブリダイズしないというように、非特異的結合がないことを評価することができる。
【0033】
ハイブリダイゼーションに基づく検出システムを利用する場合、参照核酸配列に相補的である核酸プローブが選ばれ、次いで、適切な条件の選択によって前記プローブと前記参照配列が互いに選択的にハイブリダイズまたは結合して二重鎖分子を形成する。中程度にストリンジェントなハイブリダイゼーション条件下で、参照配列に選択的にハイブリダイズし得る核酸分子は、典型的に、選択された核酸プローブの配列と少なくともおよそ70%の配列同一性を有する少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にする条件下でハイブリダイズする。ストリンジェントなハイブリダイゼーション条件は、典型的に、選択された核酸プローブの配列と約90〜95%より高い配列同一性を有する少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にする。プローブと参照配列が特定程度の配列同一性を有する場合、プローブ/参照配列のハイブリダイゼーションに有用なハイブリダイゼーション条件を、当該技術分野において既知であるように決定することができる(例えば、Nucleic Acid Hybridization:A Practical Approach,editors B.D.HamesおよびS.J.Higgins,(1985)Oxford;Washington,DC;IRL Pressを参照されたい)。
【0034】
ハイブリダイゼーションのための条件は当業者に周知である。ハイブリダイゼーションのストリンジェンシーとは、ハイブリダイゼーション条件が不一致のヌクレオチドを含有する合成物の形成を嫌う程度を表し、より高いストリンジェンシーは不一致の合成物に対するより低い許容度と相関する。ハイブリダイゼーションのストリンジェンシーに影響を及ぼす要素は当業者に周知であり、温度、pH、イオン強度、および例えばホルムアミドおよびジメチルスルホキシド等の有機溶媒の濃度を含むが、これに制限されない。当業者に既知であるように、ハイブリダイゼーションのストリンジェンシーは、より高い温度、より低いイオン強度、およびより低い溶媒濃度によって上昇する。
【0035】
ハイブリダイゼーションのためのストリンジェントな条件に関して、多数の同等の条件を利用し、例えば以下の要素:配列の長さおよび性質、種々の配列の塩基組成、塩および他のハイブリダイゼーション溶液成分の濃度、ハイブリダイゼーション溶液中における遮断物質の有無(例えば、硫酸デキストランおよびポリエチレングリコール)、ハイブリダイゼーションの反応温度、および時間パラメーター、ならびに様々な洗浄条件を変えることによって、特定のストリンジェンシーを確立し得ることが当該技術分野において周知である。特定のハイブリダイゼーション条件のセットの選択は、当該技術分野における以下の標準的方法に従うことによって選択される(例えば、Sambrookら,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)。
【0036】
「組換え」とは、2つのポリヌクレオチド間での遺伝情報の交換のプロセスを表す。本開示上、「相同組換え(HR)」とは、例えば細胞内で二本鎖断裂の修復の間に起こるような交換の特殊な形態を表す。このプロセスは、ヌクレオチドの配列相同性を必要とし、「標的」分子(すなわち、二本鎖断裂を受けたもの)のテンプレート修復に対する「ドナー」分子を用い、かつドナーから標的への遺伝情報の転移をもたらすため、「非交差型の遺伝子変換」または「短路の遺伝子変換」として様々に知られている。任意の特定の理論に束縛されることを望むことなく、そのような転移は、切断された標的とドナーとの間で形成するヘテロ二重鎖のミスマッチ修正、および/またはドナーを用いて標的の一部になるであろう遺伝情報を再合成する「合成依存的な鎖アニーリング」、および/または関連プロセスを伴い得る。そのような特殊なHRは、しばしば、ドナーポリヌクレオチドの配列の一部またはすべてが標的ポリヌクレオチド内に組み入れられるような、標的分子の配列の変更をもたらす。
【0037】
「切断」とは、DNA分子の共有結合骨格の断裂を表す。切断は、ホスホジエステル結合の酵素的または化学的な加水分解を含むが、これに制限されない種々の方法によって開始され得る。一本鎖切断および二本鎖切断のいずれも可能であり、二本鎖切断は2つの別個の一本鎖切断事象の結果として生じ得る。DNA切断によって、平滑末端または互い違いの末端の産生がもたらされ得る。ある実施態様では、標的二本鎖DNA切断に融合ポリペプチドを用いる。
【0038】
「切断ドメイン」には、DNA切断に対する触媒活性を持つ1つ以上のポリペプチド配列が含まれる。切断ドメインは1つのポリペプチド鎖内に含有されていてもよく、あるいは2つ(またはそれ以上)のポリペプチドの会合によって切断活性が生じてもよい。
【0039】
「切断ハーフドメイン」とは、第二のポリペプチド(同一または異なる)と連動して切断活性(好ましくは二本鎖切断活性)を有する複合体を形成するポリペプチド配列である。
【0040】
「クロマチン」とは、細胞ゲノムを含む核タンパク質構造体である。細胞クロマチンは、核酸、主にDNA、ならびにヒストンおよび非ヒストン染色体タンパク質を含むタンパク質を含む。大部分の真核細胞のクロマチンはヌクレオソームの形態で存在し、ヌクレオソームコアは、ヒストンH2A、H2B、H3、およびH4のそれぞれ2つを含む八量体と会合したおよそ150塩基対のDNA、ならびにヌクレオソームコア間に伸びる(生物に依存した可変長の)リンカーDNAを含む。ヒストンH1の分子は、通常、リンカーDNAと会合する。本開示上、「クロマチン」という用語は、原核生物および真核生物の両方のあらゆる種類の細胞内核タンパク質を包含することを意図される。細胞クロマチンは、染色体クロマチンおよびエピソーム性クロマチンの両方を含む。
【0041】
「染色体」とは、細胞のゲノムのすべてまたは一部を含むクロマチン複合体である。細胞のゲノムは、しばしば、細胞のゲノムを含むすべての染色体の集合体であるその核型によって特徴付けされる。細胞のゲノムは、1つ以上の染色体を含み得る。
【0042】
「エピソーム」とは、複製核酸、核タンパク質複合体、または細胞の染色体核型の一部でない核酸を含む他の構造物である。エピソームの例には、プラスミドおよびある種のウイルスゲノムが含まれる。
【0043】
「接近可能領域」とは、核酸中に存在する標的部位に前記標的部位を認識する外因性分子が結合し得る、細胞クロマチン内の部位である。任意の特定の理論に束縛されることを望むことなく、接近可能領域はヌクレオソーム構造内にパッケージされていないものであると考えられている。接近可能領域の独特の構造を、しばしば、化学的および酵素的プローブ、例えばヌクレアーゼに対するその感度によって検出できる。
【0044】
「標的部位」または「標的配列」とは、結合に十分な条件が存在するという条件で結合分子が結合するであろう核酸の部分を規定する核酸配列である。例えば、5’−GAATTC−3’という配列は、EcoRI制限エンドヌクレアーゼに対する標的部位である。
【0045】
「外因性」分子とは、通常細胞内に存在しないが、1つ以上の遺伝学的、生化学的、または他の方法によって細胞内に導入され得る分子である。「細胞内での通常の存在」とは、前記細胞の特定の発生段階および環境条件に対して決定される。したがって、例えば筋肉の胚発生の間だけ存在する分子は、成体の筋細胞に対して外因性分子である。同様に、ヒートショックによって誘発された分子は、ヒートショックされていない細胞に対して外因性分子である。外因性分子には、例えば、任意のポリペプチドまたはそのフラグメントに対するコード配列、機能不全である内在性分子の機能型、または通常に機能する内在性分子の機能不全型が含まれ得る。さらに、外因性分子には、宿主細胞の内在性遺伝子のオルソログである別の種由来のコード配列が含まれ得る。
【0046】
外因性分子は、とりわけ、コンビナトリアルケミストリーの手法によって生成されるような小分子、あるいはタンパク質、核酸、炭水化物、脂質、糖タンパク質、リポタンパク質、多糖、前記分子の任意の改変された誘導体、または1つ以上の前記分子を含む任意の複合体等の高分子であってよい。核酸にはDNAおよびRNAが含まれ、一本鎖または二本鎖であってよく、線状、分岐したもの、または環状であってよく、かつ任意の長さのものであってよい。核酸には、二重鎖を形成し得るもの、ならびに三重鎖形成核酸が含まれる。例えば、米国特許第5,176,996号および第5,422,251号を参照されたい。タンパク質には、DNA結合タンパク質、転写因子、クロマチン再構築因子、メチル化DNA結合タンパク質、ポリメラーゼ、メチラーゼ、デメチラーゼ、アセチラーゼ、デアセチラーゼ、キナーゼ、ホスファターゼ、インテグラーゼ、リコンビナーゼ、リガーゼ、トポイソメラーゼ、ジャイレース、およびヘリカーゼが含まれるが、これに制限されない。
【0047】
外因性分子は、内在性分子と同じ種類の分子、例えば外因性タンパク質または核酸であってよい。例えば、外因性核酸には、感染ウイルスゲノム、細胞内に導入されたプラスミドまたはエピソーム、あるいは細胞内に通常存在しない染色体が含まれ得る。細胞内に外因性分子を導入するための方法は、当業者に既知であり、脂質を介した転移(すなわち、中性およびカチオン性脂質を含むリポソーム)、エレクトロポレーション、直接注入、細胞融合、粒子砲撃、リン酸カルシウム共沈殿、ナノ粒子による形質転換、DEAE−デキストランを介した転移、およびウイルスベクターを介した転移を含むが、これに制限されない。
【0048】
一方、「内在性」分子とは、特定の環境条件下での特定の発生段階において特定の細胞内に通常存在するものである。例えば、内在性核酸には、染色体、ミトコンドリアのゲノム、葉緑体または他の細胞内小器官、あるいは天然に存在しているエピソーム性核酸が含まれ得る。さらなる内在性分子には、タンパク質、例えば転写因子および酵素が含まれ得る。
【0049】
本明細書において使用するとき、「外因性核酸の産物」という用語は、ポリヌクレオチドおよびポリペプチド産物の両方、例えば転写産物(RNA等のポリヌクレオチド)および翻訳産物(ポリペプチド)を含む。
【0050】
「融合」分子とは、2つ以上のサブユニット分子が、例えば共有結合で連結している分子である。前記サブユニット分子は、同じ化学的分子種であってもよく、または異なる化学的分子種であってもよい。第一の種類の融合分子の例には、融合タンパク質(例えば、ZFP DNA結合ドメインと切断ドメインとの間の融合)および融合核酸(例えば、前記の融合タンパク質をコードする核酸)が含まれるが、これに制限されない。第二の種類の融合分子の例には、三重鎖形成核酸とポリペプチドとの間の融合、および小溝バインダーと核酸との間の融合が含まれるが、これに制限されない。
【0051】
細胞内での融合タンパク質の発現は、細胞への融合タンパク質の送達によって、または細胞への融合タンパク質をコードするポリヌクレオチドの送達であって、前記ポリヌクレオチドが転写され、かつ転写産物が翻訳されて、融合タンパク質を生成する送達によってもたらされ得る。トランススプライシング、ポリペプチドの切断、およびポリペプチドのライゲーションも、細胞内でのタンパク質の発現に関与し得る。細胞へのポリヌクレオチドおよびポリペプチドの送達のための方法は、本開示の別の部分で提供されている。
【0052】
本開示上、「遺伝子」とは、遺伝子産物をコードするDNA領域(下記を参照されたい)、ならびに遺伝子産物の産生を制御するすべてのDNA領域を含むものであって、そのような制御配列がコード配列および/または転写配列に隣接しているかどうかは問わない。したがって、遺伝子には、プロモーター配列、ターミネーター、リボソーム結合部位および内部リボソーム侵入部位等の翻訳制御配列、エンハンサー、サイレンサー、インシュレーター、境界エレメント、複製起点、マトリックス付着部位、ならびに遺伝子座調節領域が含まれるが、必ずしもこれに制限されない。
【0053】
「遺伝子発現」とは、遺伝子に含有される情報の遺伝子産物への転換を表す。遺伝子産物とは、遺伝子の直接的な転写産物(例えば、mRNA、tRNA、rRNA、アンチセンスRNA、干渉RNA、リボザイム、構造RNA、または他の任意の種類のRNA)、あるいはmRNAの翻訳によって産生されたタンパク質であり得る。遺伝子産物には、キャッピング、ポリアデニル化、メチル化、および編集等のプロセスによって修飾されているRNA、ならびに、例えばメチル化、アセチル化、リン酸化、ユビキチン化、ADPリボシル化、ミリストイル化(myristilation)、およびグリコシル化によって修飾されたタンパク質も含まれる。
【0054】
遺伝子発現の「調整」とは、遺伝子の活性の変化を表す。発現の調整には、遺伝子活性化および遺伝子抑制が含まれ得るが、これに制限されない。
【0055】
「植物」細胞には、単子葉性(単子葉植物)または双子葉性(双子葉植物)植物の細胞が含まれるが、これに制限されない。単子葉植物の制限されない例には、トウモロコシ、イネ、オオムギ、オートムギ、コムギ、モロコシ、ライムギ、サトウキビ、パイナップル、タマネギ、バナナ、およびココナツ等の穀物用植物が含まれる。双子葉植物の制限されない例には、タバコ、トマト、ヒマワリ、コットン、テンサイ、ジャガイモ、レタス、メロン、大豆、キャノーラ(ナタネ)、およびアルファルファが含まれる。植物細胞は、植物の任意の部位に由来するもの、および/または植物の任意の発生段階に由来するものであってよい。
【0056】
「対象の領域」とは、外因性分子に結合することが所望される、例えば遺伝子あるいは遺伝子の内部または隣接する非コード配列等の細胞クロマチンの任意の領域である。結合は、標的DNA切断および/または標的組換えを目的とするものであってよい。対象の領域は、例えば染色体、エピソーム、細胞内小器官ゲノム(例えば、ミトコンドリア、葉緑体)、感染ウイルスゲノム内に存在し得る。対象の領域は、遺伝子のコード領域内、例えばリーダー配列、トレーラー配列、またはイントロン等の転写される非コード領域内、あるいはコード領域の上流または下流のいずれかの非転写領域内にあり得る。対象の領域は、1ヌクレオチド対程度に小さくても、または長さが最大2,000ヌクレオチド対であっても、または任意の整数値のヌクレオチド対であってもよい。
【0057】
「作動性の連結」および「作動可能に連結された」(または「操作可能に連結された」)という用語は、並列の2つ以上の構成成分(配列エレメント等)に関して代替可能に用いられるものであって、前記構成成分は、両方の構成成分が正常に機能し、かつ少なくとも1つの前記構成成分が、少なくとも1つの他の構成成分に対して影響を及ぼす機能を媒介し得る可能性を与えるように配置されている。実例として、転写制御配列が1つ以上の転写制御因子の有無に応答してコード配列の転写のレベルを調節している場合、プロモーター等の転写制御配列はコード配列に作動可能に連結されている。転写制御配列は、一般にコード配列とシスで作動可能に連結されているが、直接的にそれに隣接している必要はない。例えば、エンハンサーは、たとえそれらが連続していないとしても、コード配列に作動可能に連結されている転写制御配列である。
【0058】
融合ポリペプチドに関して、「作動可能に連結された」という用語は、構成成分のそれぞれが他の構成成分と連結して、それがそのように連結されていなかった場合になすのと同じ機能を果たすという事実を表す。例えば、ZFP DNA結合ドメインが切断ドメインと融合している融合ポリペプチドに関して、前記融合ポリペプチドにおいて、ZFP DNA結合ドメイン部分がその標的部位および/またはその結合部位に結合し得、一方で切断ドメインが標的部位の付近でDNAを切断し得る場合、前記ZFP DNA結合ドメインと前記切断ドメインは作動性の連結の状態にある。
【0059】
タンパク質、ポリペプチド、または核酸の「機能性フラグメント」とは、その配列が全長のタンパク質、ポリペプチド、または核酸と同一ではないが、全長のタンパク質、ポリペプチド、または核酸と同じ機能を維持しているタンパク質、ポリペプチド、または核酸である。機能性フラグメントは、対応する天然型分子より多い、少ない、または同じ数の残基を保有し得、かつ/あるいは1つ以上のアミノ酸またはヌクレオチド置換を含有し得る。核酸の機能(例えば、コード機能、別の核酸にハイブリダイズする能力)を判定するための方法は、当該技術分野において周知である。同様に、タンパク質機能を判定するための方法は周知である。例えば、ポリペプチドのDNA結合機能を、例えばフィルター結合アッセイ、電気泳動移動度シフトアッセイ、または免疫沈降アッセイによって判定することができる。DNA切断を、ゲル電気泳動によってアッセイすることができる。前記のAusubelらを参照されたい。タンパク質が別のタンパク質と相互作用する能力を、例えば共免疫沈降、ツーハイブリッドアッセイ、または遺伝学的かつ生化学的相補性の両方によって判定することができる。例えば、Fieldsら,(1989)Nature 340:245−246;米国特許第5,585,245号およびPCT WO98/44350を参照されたい。
【0060】
標的部位
開示される方法および組成物には、切断ドメイン(または切断ハーフドメイン)およびDNA結合ドメイン(例えば、ZFP、メガヌクレアーゼ、またはロイシンジッパー)を含み、前記DNA結合ドメイン(例えば、ジンクフィンガードメイン、メガヌクレアーゼ、またはロイシンジッパー)が、植物Zp15遺伝子座内の配列に結合することによって切断ドメイン(または切断ハーフドメイン)の活性を前記配列の付近に向けさせ、それによってZp15内での切断(例えば、二本鎖断裂)を誘導する融合タンパク質が含まれる。本開示の別の部分で示すように、ジンクフィンガードメインは、実質的に任意の所望の配列に結合するように遺伝子操作されていてよい。したがって、1つ以上のDNA結合ドメイン(例えば、ZFP)は、植物Zp15遺伝子内の1つ以上の配列に結合するように遺伝子操作されていてよい。DNA結合ドメイン(例えば、ZFP)および切断ドメインを含む融合タンパク質(あるいは、それぞれがDNA結合ドメインおよび切断ハーフドメインを含む2つの融合タンパク質)の細胞内での発現は、Zp15遺伝子における切断をもたらす。
【0061】
ジンクフィンガードメインによる結合のためのZp15内の配列(例えば、標的部位)の選択は、例えば、選択された配列に結合するようにZFPを設計するための方法も開示している共同所有の米国特許第6,453,242号(2002年9月17日)に開示されている方法に従って達成され得る。ヌクレオチド配列の簡単な目視検査も標的部位の選択に用いることができることは当業者に明白であろう。したがって、標的部位選択のための任意の手段を、本明細書に記載される方法において用いることができる。
【0062】
ZFP DNA結合ドメインに対して、標的部位は一般に複数の隣接する標的サブサイトから構成される。標的サブサイトとは、個々のジンクフィンガーによって結合される配列(通常、ヌクレオチドトリプレット、または隣接するクァドループレットと1ヌクレオチド重複し得るヌクレオチドクァドループレットのいずれか)を表す。例えば、WO02/077227を参照されたい。ジンクフィンガータンパク質が最も接触する鎖を標的鎖、「主要な認識鎖」、または「主要な接触鎖」と呼ぶ場合、いくつかのジンクフィンガータンパク質は標的鎖内の3塩基トリプレットおよび非標的鎖上の第4の塩基に結合する。標的部位は、一般に少なくとも9ヌクレオチドの長さを有し、したがって、少なくとも3つのジンクフィンガーを含むジンクフィンガー結合ドメインによって結合される。しかしながら、例えば4フィンガー結合ドメインの12ヌクレオチド標的部位への結合、5フィンガー結合ドメインの15ヌクレオチド標的部位への結合、または6フィンガー結合ドメインの18ヌクレオチド標的部位への結合も可能である。明白であろうが、より大きな結合ドメイン(例えば、7、8、9フィンガー以上)のより長い標的部位への結合も可能である。
【0063】
標的部位が3ヌクレオチドの倍数である必要はない。例えば、交差鎖相互作用が生じる場合(例えば、米国特許第6,453,242号およびWO02/077227を参照されたい)、マルチフィンガー結合ドメインの1つ以上の個々のジンクフィンガーは、重複しているクァドループレットサブサイトに結合し得る。結果として、3フィンガータンパク質は、10番目のヌクレオチドが末端フィンガーによって結合されるクァドループレットの一部である10ヌクレオチド配列に結合し得、4フィンガータンパク質は、13番目のヌクレオチドが末端フィンガーによって結合されるクァドループレットの一部である13ヌクレオチド配列に結合し得る、等々。
【0064】
マルチフィンガー結合ドメイン内の個々のジンクフィンガー間のアミノ酸リンカー配列の長さおよび性質も、標的配列への結合に影響を及ぼす。例えば、マルチフィンガー結合ドメイン内の隣接するジンクフィンガー間のいわゆる「非標準的リンカー」、「長いリンカー」、または「構造化リンカー」の存在によって、それらのフィンガーは直接に隣接していないサブサイトに結合することができる。そのようなリンカーの制限されない例は、例えば米国特許第6,479,626号およびWO01/53480に記載されている。したがって、ジンクフィンガー結合ドメインに対する標的部位内の1つ以上のサブサイトは、1、2、3、4、5、またはそれ以上のヌクレオチドずつ互いに分離し得る。ほんの一例として、4フィンガー結合ドメインは、配列中に2つの連続する3ヌクレオチドサブサイト、介在ヌクレオチド、および2つの連続するトリプレットサブサイトを含む13ヌクレオチド標的部位に結合し得る。
【0065】
配列(例えば、標的部位)間の距離とは、互いに最も近隣の配列の端から測定される、2つの配列間に介在するヌクレオチドまたはヌクレオチド対の数を表す。
【0066】
2つのジンクフィンガードメイン/切断ハーフドメイン融合分子の離れた標的部位への結合に切断が依存しているある実施態様では、前記2つの標的部位は向かい合ったDNA鎖上にあり得る。他の実施態様では、両方の標的部位は同じDNA鎖上にある。
【0067】
DNA結合ドメイン
任意のDNA結合ドメインを、本明細書に開示される方法において用いることができる。ある実施態様では、DNA結合ドメインはジンクフィンガータンパク質を含む。ジンクフィンガー結合ドメインは、1つ以上のジンクフィンガーを含む。Millerら,(1985)EMBO J.4:1609−1614;Rhodes(1993)Scientific American Feb.:56−65;米国特許第6,453,242号。本明細書に記載されるジンクフィンガー結合ドメインは、概して2、3、4、5、6、またはさらに多くのジンクフィンガーを含む。
【0068】
典型的には、1つのジンクフィンガードメインは、長さが約30アミノ酸である。構造研究によって、それぞれのジンクフィンガードメイン(モチーフ)は、2個のシステインおよび2個のヒスチジンによる亜鉛原子の配位を介した特定の構造に保持された、2つのβシート(2個の不変のシステイン残基を含有するβターンに保持されている)およびαヘリックス(2個の不変のヒスチジン残基を含有している)を含有することが証明されている。
【0069】
ジンクフィンガーには、標準的C2H2ジンクフィンガー(すなわち、亜鉛イオンが2個のシステインと2個のヒスチジン残基によって配位されているもの)、ならびに、例えばC3Hジンクフィンガー(亜鉛イオンが3個のシステイン残基と1個のヒスチジン残基によって配位されているもの)およびC4ジンクフィンガー(亜鉛イオンが4個のシステイン残基によって配位されているもの)等の非標準的ジンクフィンガーの両方が含まれる。植物における使用のための非標準的ZFPに関するWO02/057293および米国特許公報第20080182332号も参照されたい。
【0070】
遺伝子操作されたジンクフィンガー結合ドメインは、天然に存在しているジンクフィンガータンパク質と比較して新規な結合特異性を有し得る。遺伝子操作法には、合理的設計および様々な種類の選択が含まれるが、これに制限されない。合理的設計には、例えばトリプレット(またはクァドループレット)ヌクレオチド配列と個々のジンクフィンガーアミノ酸配列とを含むデータベースであって、それぞれのトリプレットまたはクァドループレットヌクレオチド配列が、特定のトリプレットまたはクァドループレット配列に結合するジンクフィンガーの1つ以上のアミノ酸配列と関連付けされているデータベースを用いることが含まれる。
【0071】
ファージディスプレイおよびツーハイブリッドシステムを含む例示的選択法は、米国特許第5,789,538号;第5,925,523号;第6,007,988号;第6,013,453号;第6,410,248号;第6,140,466号;第6,200,759号;および第6,242,568号;ならびにWO98/37186;WO98/53057;WO00/27878;WO01/88197、およびGB2,338,237に開示されている。
【0072】
ジンクフィンガー結合ドメインに対する結合特異性の増強は、例えば共同所有のWO02/077227に記載されている。
【0073】
個々のジンクフィンガーは3ヌクレオチド(すなわち、トリプレット)配列(または、隣接するジンクフィンガーの4ヌクレオチド結合部位と1ヌクレオチド重複し得る4ヌクレオチド配列)に結合するため、ジンクフィンガー結合ドメインが結合するように遺伝子操作されている配列(例えば、標的配列)の長さは、遺伝子操作されたジンクフィンガー結合ドメイン内のジンクフィンガーの数を決定することになる。例えば、フィンガーモチーフが重複サブサイトに結合しないZFPについて、6ヌクレオチド標的配列は2フィンガー結合ドメインによって結合され;9ヌクレオチド標的配列は3フィンガー結合ドメインによって結合される、等々。本明細書で述べているように、標的部位における個々のジンクフィンガーに対する結合部位(すなわち、サブサイト)は、連続している必要はないが、マルチフィンガー結合ドメイン内のジンクフィンガー間のアミノ酸配列(すなわち、フィンガー間リンカー)の長さおよび性質に依存して、1つまたは数個のヌクレオチドずつ分離され得る。
【0074】
マルチフィンガージンクフィンガー結合ドメインにおいて、隣接するジンクフィンガーは、およそ5アミノ酸のアミノ酸リンカー配列(いわゆる「標準的」フィンガー間リンカー)によって、あるいは1つ以上の非標準的リンカーによって分離され得る。例えば、共同所有の米国特許第6,453,242号および第6,534,261号を参照されたい。3つを上回るフィンガーを含む遺伝子操作されたジンクフィンガー結合ドメインについて、前記ジンクフィンガーの一部の間でのより長い(「非標準的」)フィンガー間リンカーの挿入は、それによって結合ドメインによる結合の親和性および/または特異性が増加し得るため、場合によっては所望され得る。例えば、米国特許第6,479,626号およびWO01/53480を参照されたい。したがって、マルチフィンガージンクフィンガー結合ドメインを、非標準的フィンガー間リンカーの存在および位置に関して特徴付けすることができる。例えば、3つのフィンガー(2個の標準的フィンガー間リンカーによって接合している)、長いリンカー、および3つのさらなるフィンガー(2個の標準的フィンガー間リンカーによって接合している)を含む6フィンガージンクフィンガー結合ドメインは、2×3配置と表示される。同様に、2つのフィンガー(その間に標準的リンカーを有する)、長いリンカー、および2つのさらなるフィンガー(標準的リンカーによって接合している)を含む結合ドメインは、2×2配置と表示される。3つの2フィンガー単位(そのそれぞれにおいて、2つのフィンガーが標準的リンカーによって接合している)を含み、かつそれぞれの2フィンガー単位が隣接する2つのフィンガー単位と長いリンカーによって接合しているタンパク質は、3×2配置として表される。
【0075】
マルチフィンガー結合ドメイン内の2つの隣接するジンクフィンガー間での長いまたは非標準的フィンガー間リンカーの存在によって、前記2つのフィンガーは、しばしば、標的配列における直接に連続していないサブサイトに結合する。したがって、標的部位におけるサブサイト間には1つ以上のヌクレオチドの差が存在し得、すなわち、標的部位はジンクフィンガーによって接触されていない1つ以上のヌクレオチドを含有し得る。例えば、2×2ジンクフィンガー結合ドメインは、1つのヌクレオチドによって分離された2つの6ヌクレオチド配列に結合し得、すなわち、それは13ヌクレオチド標的部位に結合する。Mooreら,(2001a)Proc.Natl.Acad.Sci.USA 98:1432−1436;Mooreら,(2001b)Proc.Natl.Acad.Sci.USA 98:1437−1441、およびWO01/53480も参照されたい。
【0076】
先に言及しているように、標的サブサイトは、1つのジンクフィンガーによって結合される3または4ヌクレオチド配列である。ある目的のために、2フィンガー単位を「結合モジュール」と表示する。結合モジュールを、例えば、マルチフィンガータンパク質(一般に3つのフィンガー)との関連で、特定の6ヌクレオチド標的配列に結合する2つの隣接するフィンガーを選択することによって獲得することができる。あるいは、個々のジンクフィンガーを集合させることによってモジュールを構築することができる。WO98/53057およびWO01/53480も参照されたい。
【0077】
あるいは、DNA結合ドメインはヌクレアーゼに由来してもよい。例えば、ホーミングエンドヌクレアーゼおよびメガヌクレアーゼの認識配列、例えばI−SceI、I−CeuI、PI−PspI、PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIII等が既知である。米国特許第5,420,032号;米国特許第6,833,252号;Belfortら,(1997)Nucleic Acids Res.25:3379−3388;Dujonら,(1989)Gene 82:115−118;Perlerら,(1994)Nucleic Acids Res.22,1125−1127;Jasin(1996)Trends Genet.12:224−228;Gimbleら,(1996)J.Mol.Biol.263:163−180;Argastら,(1998)J.Mol.Biol.280:345−353、およびNew England Biolabsカタログも参照されたい。さらに、ホーミングエンドヌクレアーゼおよびメガヌクレアーゼのDNA結合特異性を、天然でない標的部位に結合するように遺伝子操作することができる。例えば、Chevalierら,(2002)Molec.Cell 10:895−905;Epinatら,(2003)Nucleic Acids Res.31:2952−2962;Ashworthら,(2006)Nature 441:656−659;Paquesら,(2007)Current Gene Therapy 7:49−66;米国特許公報第20070117128号を参照されたい。
【0078】
別の代替法として、DNA結合ドメインはロイシンジッパータンパク質に由来してもよい。ロイシンジッパーとは、遺伝子発現に関連した重要な転写因子である多くの真核性制御タンパク質におけるタンパク質−タンパク質相互作用に関与するタンパク質のクラスである。ロイシンジッパーとは、動物、植物、酵母、等々を含むいくつかの界を越えて、これらの転写因子で共有されている共通の構造モチーフを表す。ロイシンジッパーは、ロイシン残基がαヘリックスのいたる所に均等に間隔をあけて配置され、それによって前記2つのポリペプチドのロイシン残基が最終的に前記ヘリックスの同じ面上にくることになる様式で特異的DNA配列に結合する、2つのポリペプチド(ホモ二量体またはヘテロ二量体)によって形成される。ロイシンジッパーのDNA結合特異性を、本明細書に開示されるDNA結合ドメインに利用することができる。
【0079】
いくつかの実施態様では、DNA結合ドメインは、植物病原菌キサントモナスに由来するTALエフェクターからの遺伝子操作されたドメインである(Bochら,(2009)Science 29,2009年10月(10.1126/science.117881)、ならびにMoscouおよびBogdanove,(2009)Science 29,2009年10月(10.1126/science.1178817)を参照されたい)。
【0080】
切断ドメイン
前述のように、DNA結合ドメインは、切断(ヌクレアーゼ)ドメインと関連していてもよい。例えば、ホーミングエンドヌクレアーゼは、ヌクレアーゼ機能を維持している一方で、そのDNA結合特異性を改変されていてもよい。さらに、ジンクフィンガータンパク質はまた、切断ドメインと融合して、ジンクフィンガーヌクレアーゼ(ZFN)を形成してもよい。本明細書に開示される融合タンパク質の切断ドメイン部分は、任意のエンドヌクレアーゼまたはエキソヌクレアーゼから獲得され得る。切断ドメインが由来し得る例示的エンドヌクレアーゼには、制限エンドヌクレアーゼおよびホーミングエンドヌクレアーゼが含まれるが、これに制限されない。例えば、2002−2003カタログ,New England Biolabs,Beverly,MA;およびBelfortら,(1997)Nucleic Acids Res.25:3379−3388を参照されたい。DNAを切断するさらなる酵素が既知である(例えば、S1ヌクレアーゼ;マングビーンヌクレアーゼ;膵臓DNaseI;ミクロコッカスヌクレアーゼ;酵母HOエンドヌクレアーゼ;Linnら,(eds.)Nucleases,Cold Spring Harbor Laboratory Press,1993も参照されたい)。ホーミングエンドヌクレアーゼおよびメガヌクレアーゼの制限されない例には、I−SceI、I−CeuI、PI−PspI、PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIIIが含まれる。米国特許第5,420,032号;米国特許第6,833,252号;Belfortら,(1997)Nucleic Acids Res.25:3379−3388;Dujonら,(1989)Gene 82:115−118;Perlerら,(1994)Nucleic Acids Res.22,1125−1127;Jasin(1996)Trends Genet.12:224−228;Gimbleら,(1996)J.Mol.Biol.263:163−180;Argastら,(1998)J.Mol.Biol.280:345−353;およびthe New England Biolabsカタログも参照されたい。1つ以上のこれらの酵素(またはその機能性フラグメント)を、切断ドメインおよび切断ハーフドメインの供給源として用いることができる。
【0081】
制限エンドヌクレアーゼ(制限酵素)は、多くの種に存在し、DNAに(認識部位において)配列特異的に結合し得、かつ結合の部位においてまたは近隣においてDNAを切断し得る。ある制限酵素(例えば、IIS型)は、認識部位から外れた部位でDNAを切断し、かつ分離可能な結合ドメインおよび切断ドメインを有する。例えば、IIS型酵素のFokIは、一方の鎖上にあるその認識部位から9ヌクレオチドの部位で、かつもう一方の鎖上のその認識部位から13ヌクレオチドの部位でDNAの二本鎖切断を触媒する。例えば、米国特許第5,356,802号;第5,436,150号および第5,487,994号;ならびにLiら,(1992)Proc.Natl.Acad.Sci.USA 89:4275−4279;Liら,(1993)Proc.Natl.Acad.Sci.USA 90:2764−2768;Kimら,(1994a)Proc.Natl.Acad.Sci.USA 91:883−887;Kimら,(1994b)J.Biol.Chem.269:31,978−31,982を参照されたい。したがって、一実施態様では、融合タンパク質は、少なくとも1種のIIS型制限酵素由来の切断ドメイン(または切断ハーフドメイン)、および遺伝子操作されていてもされていなくてもよい1つ以上のジンクフィンガー結合ドメインを含む。
【0082】
その切断ドメインが結合ドメインから分離可能である例示的IIS型制限酵素は、FokIである。この特定の酵素は、二量体として活性を有する。Bitinaiteら,(1998)Proc.Natl.Acad.Sci.USA 95:10,570−10,575。したがって、本開示上、開示される融合タンパク質に用いられたFokI酵素の一部は切断ハーフドメインと考えられる。したがって、ジンクフィンガー−FokI融合体を用いた細胞配列の標的二本鎖切断および/または標的置換のために、それぞれがFokI切断ハーフドメインを含む2つの融合タンパク質を用いて、触媒活性のある切断ドメインを再構築することができる。あるいは、ジンクフィンガー結合ドメインと2つのFokI切断ハーフドメインとを含有する1つのポリヌクレオチド分子も用いることができる。ジンクフィンガー−FokI融合体を用いた標的切断および標的配列変更に対するパラメーターは、本開示の別の部分で提供されている。
【0083】
切断ドメインまたは切断ハーフドメインは、切断活性を維持している、または多量化(例えば、二量化)して機能性切断ドメインを形成する能力を維持しているタンパク質の任意の部分であってよい。
【0084】
例示的IIS型制限酵素は、参照することによりその全体として本明細書に組み入れられている、共同所有の国際公報WO2007/014275に記載されている。
【0085】
切断特異性を増強させるために、切断ドメインを改変してもよい。ある実施態様では、切断ハーフドメインの変形型が利用され、これらの変形型は切断ハーフドメインのホモ二量化を最小限に抑えるまたは阻止する。そのような改変された切断ハーフドメインの制限されない例は、参照することによりその全体として本明細書に組み入れられている、WO2007/014275に詳細に記載されている。実施例も参照されたい。ある実施態様では、切断ドメインは、ホモ二量化を最小限に抑えるまたは阻止し、当業者に既知であり、かつ参照することによりそれらの全体として本明細書に組み入れられている、例えば米国特許公報第20050064474および第20060188987号に記載されている遺伝子操作された切断ハーフドメイン(二量化ドメイン変異体とも称される)を含む。FokIの446、447、479、483、484、486、487、490、491、496、498、499、500、531、534、537、および538位のアミノ酸残基はすべて、FokI切断ハーフドメインの二量化に影響を与えるための標的である。例えば、米国特許公報第20050064474号および第20060188987号;国際特許公報WO07/139898;Millerら,(2007)Nat.Biotechnol.25(7):778−785を参照されたい。
【0086】
必須のヘテロ二量体を形成するFokIのさらなる遺伝子操作された切断ハーフドメインも、本明細書に記載されるZFNに用いることができる。一実施態様では、第一の切断ハーフドメインはFokIの490および538位のアミノ酸残基における変異を含み、かつ第二の切断ハーフドメインはアミノ酸残基486および499における変異を含む。
【0087】
ある実施態様では、切断ドメインは、その両方が結合ドメイン、第一の切断ハーフドメイン、および第二の切断ハーフドメインを含む1つのポリペプチドの一部である、2つの切断ハーフドメインを含む。切断ハーフドメインは、それらがDNAを切断する機能を果たす限り、同じアミノ酸配列または異なるアミノ酸配列を有し得る。
【0088】
一般に、融合タンパク質が切断ハーフドメインを含む場合、2つの融合タンパク質が切断に必要とされる。あるいは、2つの切断ハーフドメインを含む1つのタンパク質を用いることができる。前記2つの切断ハーフドメインは同じエンドヌクレアーゼ(または、その機能性フラグメント)に由来してもよく、あるいはそれぞれの切断ハーフドメインは異なるエンドヌクレアーゼ(またはその機能性フラグメント)に由来してもよい。さらに、前記2つの融合タンパク質に対する標的部位は、好ましくは、2つの融合タンパク質のそれらそれぞれの標的部位への結合により、切断ハーフドメインが例えば二量化することによって機能性切断ドメインを形成し得る空間的方向に互いに切断ハーフドメインが位置付けられるように、互いに対して配置される。したがって、ある実施態様では、標的部位の近隣の端を、5〜8ヌクレオチドで、または15〜18ヌクレオチドで分離する。しかしながら、任意の整数値のヌクレオチドまたはヌクレオチド対を、2つの標的部位間に介在させることができる(例えば、2〜50ヌクレオチド以上)。一般に、切断の地点は標的部位の間にある。
【0089】
融合タンパク質
融合タンパク質(およびそれをコードするポリヌクレオチド)の設計および構築のための方法は、当業者に既知である。例えば、DNA結合ドメイン(例えば、ジンクフィンガードメイン)および制御または切断ドメイン(もしくは切断ハーフドメイン)、および融合タンパク質をコードするポリヌクレオチドを含む融合タンパク質の設計および構築のための方法は、参照することによりそれらの全体として本明細書に組み入れられている、共同所有の米国特許第6,453,242号および第6,534,261号、ならびに米国特許出願公報第2007/0134796号および第2005/0064474号に記載されている。ある実施態様では、融合タンパク質をコードするポリヌクレオチドを構築する。これらのポリヌクレオチドをベクター内に挿入することができ、かつ前記ベクターを細胞内に導入することができる(ベクターおよび細胞内にポリヌクレオチドを導入するための方法に関するさらなる開示のために、下記を参照されたい)。
【0090】
本明細書に記載される方法のある実施態様では、ジンクフィンガーヌクレアーゼは、ジンクフィンガー結合ドメインおよびFokI制限酵素由来の切断ハーフドメインを含む融合タンパク質を含み、かつ2つのそのような融合タンパク質を細胞内で発現させる。細胞内での2つの融合タンパク質の発現は、前記2つのタンパク質の前記細胞への送達;1つのタンパク質および前記タンパク質の一方をコードする1つの核酸の前記細胞への送達;それぞれが前記タンパク質の一方をコードする2つの核酸の前記細胞への送達;または両方のタンパク質をコードする1つの核酸の前記細胞への送達によって生じ得る。さらなる実施態様では、融合タンパク質は、2つの切断ハーフドメインとジンクフィンガー結合ドメインとを含む1本のポリペプチド鎖を含む。この場合、1つの融合タンパク質が細胞内で発現し、かつ理論に束縛されることを望むことなく、前記切断ハーフドメインの分子内二量体の形成の結果としてDNAを切断すると考えられている。
【0091】
ある実施態様では、融合タンパク質(例えば、ZFP−FokI融合体)の構成成分を、ジンクフィンガードメインが融合タンパク質のアミノ末端の最も近隣にあり、かつ切断ハーフドメインがカルボキシ末端の最も近隣にあるように配置する。これは、FokI酵素に由来するもの等の天然に存在している二量化切断ドメインにおける、DNA結合ドメインがアミノ末端の最も近隣にあり、かつ切断ハーフドメインがカルボキシ末端の最も近隣にある、切断ドメインの相対的方向を反映する。これらの実施態様では、機能性ヌクレアーゼを形成するための切断ハーフドメインの二量化は、融合タンパク質が向かい合ったDNA鎖上の部位に結合することによって引き起こされ、結合部位の5’末端は互いに隣接している。
【0092】
さらなる実施態様では、融合タンパク質(例えば、ZFP−FokI融合体)の構成成分を、切断ハーフドメインが融合タンパク質のアミノ末端の最も近隣にあり、かつジンクフィンガードメインがカルボキシ末端の最も近隣にあるように配置する。これらの実施態様では、機能性ヌクレアーゼを形成するための切断ハーフドメインの二量化は、融合タンパク質が向かい合ったDNA鎖上の部位に結合することによって引き起こされ、結合部位の3’末端は互いに隣接している。
【0093】
その上さらなる実施態様では、第一の融合タンパク質は、融合タンパク質のアミノ末端の最も近隣にある切断ハーフドメインおよびカルボキシ末端の最も近隣にあるジンクフィンガードメインを含有し、かつ第二の融合タンパク質を、ジンクフィンガードメインが融合タンパク質のアミノ末端の最も近隣にあり、かつ切断ハーフドメインがカルボキシ末端の最も近隣にあるように配置する。これらの実施態様では、両方の融合タンパク質は同じDNA鎖に結合し、カルボキシ末端の最も近隣にあるジンクフィンガードメインを含有する第一の融合タンパク質の結合部位が、アミノ末端の最も近隣にあるジンクフィンガードメインを含有する第二の融合タンパク質の結合部位の5’側に位置する。
【0094】
開示される融合タンパク質のある実施態様では、ジンクフィンガードメインおよび切断ドメイン(または切断ハーフドメイン)の間のアミノ酸配列を「ZCリンカー」と表示する。ZCリンカーは、前述のフィンガー間リンカーとは区別されるべきである。切断を最適化するZCリンカーの獲得に関する詳細については、例えば、米国特許公報第20050064474号A1および第20030232410号,ならびに国際特許公報WO05/084190を参照されたい。
【0095】
一実施態様では、本開示は、表1に示した1つ以上の認識ヘリックスアミノ酸配列を有するジンクフィンガータンパク質を含むZFNを提供する。別の実施態様では、表1に示した1つ以上の認識ヘリックスを有するZFPをコードするヌクレオチド配列を含むZFP発現ベクターを本明細書において提供する。
【0096】
標的組込み
開示される方法および組成物を用いて、植物の細胞クロマチンのZp15遺伝子におけるDNAを切断することができ、それによって前記遺伝子座への外因性配列の安定的な標的組込みが容易になる。本明細書に記載されるように、内在性Zp15遺伝子の機能の喪失は植物細胞に十分に許容され、かつこの遺伝子内に組み込まれた配列が広範に転写され、組み込まれた配列の遺伝的伝達のための生殖細胞系の改変を有する植物を作出する。したがって、Zp15は外因性配列の標的組込みのための所望の部位である。
【0097】
Zp15内への標的組込みのために、1つ以上のDNA結合ドメイン(例えば、ZFP)を、所定の切断部位におけるまたは近隣における標的部位に結合するように遺伝子操作し、かつ操作されたDNA結合ドメインと切断ドメインとを含む融合タンパク質を細胞内で発現させる。融合タンパク質のDNA結合(例えば、ジンクフィンガー)部分が標的部位へ結合すると、DNAは切断ドメインによって標的部位の近隣で好ましくは二本鎖断裂によって切断される。
【0098】
Zp15遺伝子座における二本鎖断裂の存在によって、相同組換えを介した外因性配列の組込みが容易になる。したがって、Zp15遺伝子内に挿入されるべき外因性配列を含むポリヌクレオチドは、相同組換えを容易にするためにZp15遺伝子に相同な1つ以上の領域を含むことになる。
【0099】
本明細書に記載されるように、対象の任意の配列(外因性配列)をZp15遺伝子座内に導入することができる。例示的外因性配列には、任意のポリペプチドコード配列(例えば、cDNA)、プロモーター、エンハンサー、および他の制御配列(例えば、干渉RNA配列、shRNA発現カセット、エピトープタグ、マーカー遺伝子、切断酵素認識部位、および様々な種類の発現構築物)が含まれるが、これに制限されない。そのような配列は、標準的な分子生物学的技術(クローニング、合成、等々)を用いることによって容易に獲得され得、かつ/あるいは市販されている。
【0100】
本明細書に記載される融合分子に加えて、選択されたゲノム配列の標的置換もまた、置換(またはドナー)配列の導入を伴う。ドナー配列を、融合タンパク質の発現の前に、同時に、またはそれに続いて、細胞内に導入することができる。ドナーポリヌクレオチドは、それとそれが相同性を有するZp15ゲノム配列との間の相同組換え(または相同指向修復)を支持するのに十分なZp15との相同性を含有している。ドナーおよびゲノム配列の間のおよそ25、50、100、200、500、750、1,000、1,500、2,000ヌクレオチド以上(または10〜2,000ヌクレオチド、もしくはそれ以上の間の任意の整数値)の配列相同性によって、それらの間の相同組換えが支持されることになる。ある実施態様では、ホモロジーアームは長さが1,000塩基対未満である。他の実施態様では、ホモロジーアームは長さが750塩基対未満である。参照することにより本明細書に組み入れられている、米国仮特許出願第61/124,047号も参照されたい。
【0101】
ドナー配列は、長さが10〜5,000ヌクレオチド(またはその間の任意の整数値のヌクレオチド)またはそれより長い範囲であってよい。典型的にドナー配列はそれが置換するゲノム配列と同一でないことは、容易に明白であろう。例えば、ドナーポリヌクレオチドの配列は、染色体配列との十分な相同性が存在する限り、ゲノム配列に対して1つ以上の一塩基の変化、挿入、欠失、逆位、または転位を含有してよい。あるいは、ドナー配列は、2つの相同な領域が隣接した非相同的配列を含有してよい。さらに、ドナー配列は、細胞クロマチン内の対象の領域に相同でない配列を含有するベクターを含んでよい。一般に、ドナー配列の相同領域は、組換えが所望されるゲノム配列と少なくとも50%の配列同一性を有する。ある実施態様では、60%、70%、80%、90%、95%、98%、99%、または99.9%の配列同一性が存在する。ドナーポリヌクレオチドの長さに依存して、1%〜100%の間の任意の値の配列同一性が存在し得る。
【0102】
ドナー分子は、細胞クロマチンに相同ないくつかの不連続領域を含有してよい。例えば、対象の領域に通常存在しない配列の標的挿入のために、前記配列は、ドナー核酸分子内に存在してよく、かつ対象の領域内の遺伝子配列に相同な領域によって隣接されていてもよい。
【0103】
ドナー分子は、Zp15遺伝子座内に挿入されて、後の使用のための貯蔵物としての役割を果たすこともできる。例えば、内在性遺伝子に相同であるが対象の変異を含有しているドナー分子を、Zp15遺伝子座内に挿入してもよい。次いで、内在性遺伝子座と対象の変異を含有するZp15遺伝子座内のドナー分子との両方を切断するであろう、前記内在性遺伝子に特異的なZFNを導入することができる。ゲノムにおいて結果として生じるDSBは、その後Zp15遺伝子座から解放されたドナー分子に対する組込み部位になり得る。このようにして、当該方法はZFNをコードする核酸およびそのドナー配列の両方の同時取込みに依存しないため、対象の任意の領域におけるドナー配列の標的組込みの効率を大幅に増加させることができる。
【0104】
ドナー分子は、Zp15遺伝子座内に挿入されて、それに続く挿入のための標的部位としての役割を果たすこともできる。例えば、さらなるZFN設計物のための認識部位を含有するDNA配列から構成されるドナー分子を、Zp15遺伝子座内に挿入してもよい。それに続き、さらなるZFN設計物を作出し、かつ細胞内で発現させてもよく、それによって、オリジナルのドナー分子は切断され、かつ修復または相同組換えによって改変される。このようにして、ドナー分子の反復組込みがZp15遺伝子座において生じ得る。
【0105】
ドナー配列の挿入の成功を判別するためのアッセイ(例えば、ハイブリダイゼーション、PCR、制限酵素消化)を簡略化するために、Zp15ゲノム配列と比較して、ある程度の配列の相違がドナー配列に存在してもよい。好ましくは、コード領域に位置する場合、そのようなヌクレオチド配列の相違は、アミノ酸配列を変化させない、またはサイレントなアミノ酸変化(すなわち、タンパク質の構造または機能に影響を及ぼさない変化)をもたらす。ドナーポリヌクレオチドは、場合によっては、対象の領域における部位に結合するDNA結合ドメインに対応する配列の変化を含有して、相同組換えによって細胞クロマチン内に導入されているドナー配列の切断を阻止することもできる。
【0106】
ドナーポリヌクレオチドは、DNAまたはRNA、一本鎖または二本鎖であってよく、かつ線状または環状の形態で細胞内に導入されてよい。線状の形態で導入された場合、ドナー配列の端を、当業者に既知の方法によって(例えば、エキソヌクレアーゼによる分解から)保護することができる。例えば、1つ以上のジオキシヌクレオチド残基を線状分子の3’末端に付加し、かつ/あるいは自己相補的オリゴヌクレオチドを一方または両方の端にライゲートさせる。例えば、Changら,(1987)Proc.Natl.Acad.Sci.USA 84:4959−4963;Nehlsら,(1996)Science 272:886−889を参照されたい。外因性ポリヌクレオチドを分解から保護するためのさらなる方法には、末端アミノ基の付加、ならびに、例えばホスホロチオエート、ホスホロアミダート、およびO−メチルリボースまたはデオキシリボース残基等の修飾されたヌクレオチド間連結の使用が含まれるが、これに制限されない。
【0107】
ポリヌクレオチドを、例えば複製起点、プロモーター、および抗生物質耐性をコードする遺伝子等のさらなる配列を有するベクター分子の一部として細胞内に導入することができる。さらに、ドナーポリヌクレオチドを、ネイキッド核酸として、ナノ粒子、リポソーム、またはポロキサマー等の物質と複合体を形成した核酸として導入することができ、あるいは細菌またはウイルス(例えば、アグロバクテリウム、リゾビウム種、NGR234、シノリゾビウム・メリロッティ(Sinorhizoboium meliloti)、メソリゾビウム・ロッティ、タバコモザイクウイルス、ジャガイモウイルスX、カリフラワーモザイクウイルス、およびキャッサバベインモザイクウイルス)によって送達することができる。例えば、Chungら,(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0108】
断裂に隣接したまたは周辺の領域との相同性を有する外因性DNA分子の存在を伴った細胞配列における二本鎖断裂の存在は、ドナー分子から細胞(例えば、ゲノムまたは染色体)配列への配列情報の転移によって、すなわち、「遺伝子変換」としても既知である相同指向修復のプロセスによって断裂を修復する細胞メカニズムを活性化するようである。本出願人の方法は、Zp15遺伝子等のパラロガス遺伝子を特異的に標的化する遺伝子操作されたZFPの強力な標的化能を切断ドメイン(または切断ハーフドメイン)と有利に組み合わせるものであって、それによって標的配列の切断により外因性配列の挿入が所望されるゲノムの領域において二本鎖断裂が引き起されるものである。
【0109】
染色体配列の変更のために、十分量のドナー配列が所望の配列変更をもたらすためにコピーされてさえいれば、ドナーの全配列が染色体内にコピーされる必要はない。
【0110】
相同組換えによるドナー配列の挿入の効率は、細胞DNAにおける二本鎖断裂と組換えが所望される部位との間の距離に反比例する。言い換えれば、組換えが所望される部位に二本鎖断裂がより近い場合ほど、より高い効率の相同組換えが観察される。組換えの正確な部位があらかじめ定められていない(例えば、所望の組換え事象がゲノム配列の間隔にわたって生じ得る)場合には、ドナー核酸の長さおよび配列は、切断の部位とともに、所望の組換え事象が得られるように選択される。ゲノム配列における1つのヌクレオチド対の配列を変化させるように所望の事象を設計する場合には、細胞クロマチンはそのヌクレオチド対のいずれかの側上の10,000ヌクレオチド以内で切断される。ある実施態様では、切断は、配列を変化させるべきヌクレオチド対のいずれかの側上で、1,000、500、200、100、90、80、70、60、50、40、30、20、10、5、または2ヌクレオチド、あるいは2〜1,000ヌクレオチドの間の任意の整数値の範囲内で起こる。
【0111】
上で詳述されるように、それぞれがジンクフィンガー結合ドメインおよび切断ハーフドメインを含む2つの融合タンパク質に対する結合部位は、もう一方の結合部位の最も近隣にあるそれぞれの結合部位の端から測定して5〜8または15〜18ヌクレオチド離れて位置し得、かつ切断は前記結合部位間で起こる。切断されるゲノム配列はドナー配列によって置換されるため、切断が結合部位間の1つの部位または複数の部位で起こるかどうかは重要ではない。したがって、標的組換えによる1つのヌクレオチド対の配列の効率的な変更のために、結合部位間の領域の中間点は、そのヌクレオチド対の10,000ヌクレオチド以内、好ましくは1,000ヌクレオチド以内、または500ヌクレオチド以内、または200ヌクレオチド以内、または100ヌクレオチド以内、または50ヌクレオチド以内、または20ヌクレオチド以内、または10ヌクレオチド以内、または5ヌクレオチド以内、または2ヌクレオチド以内、または1ヌクレオチド以内、または対象のヌクレオチド対の地点にある。
【0112】
ある実施態様では、相同染色体はドナーポリヌクレオチドとしての役割を果たし得る。したがって、例えばヘテロ接合体における変異の修正は、1つの染色体上の変異配列に結合かつ切断するが、相同染色体上の野生型配列を切断しない融合タンパク質を遺伝子操作することによって達成され得る。変異を有する染色体上での二本鎖断裂は、切断された染色体に相同染色体由来の野生型配列がコピーされ、ゆえに野生型配列の2コピーが復元される、相同性に基づく「遺伝子変換」プロセスを刺激する。
【0113】
相同組換えに関与する遺伝子、例えば、RAD54エピスタシス群の植物遺伝子(例えば、AtRad54、AtRad51)、およびその産物が前述の遺伝子産物と相互作用する遺伝子等の発現を活性化するためのさらなるZFP−機能性ドメイン融合体の使用を含むが、これに制限されない標的組換えのレベルを増強し得る方法および組成物も提供される。例えば、Klutstein M.ら,Genetics.2008 Apr;178(4):2389−97を参照されたい。
【0114】
同様に、ZFP−機能性ドメイン融合体を本明細書に開示される方法および組成物と組み合わせて用いて、非相同末端の接合に関与する遺伝子(例えば、Ku70/80、XRCC4、ポリ(ADPリボース)ポリメラーゼ、DNAリガーゼ4)の発現を抑制することができる。例えば、Rihaら,(2002)EMBO 21:2819−2826;Freisnerら,(2003)Plant J.34:427−440;Chenら,(1994)European Journal of Biochemistry 224:135−142を参照されたい。ジンクフィンガー結合ドメインおよび機能性ドメインの間の融合体を用いた遺伝子発現の活性化および抑制のための方法は、例えば共同所有の米国特許第6,534,261号;第6,824,978号、および第6,933,113号に開示されている。さらなる抑制方法には、抑制されるべき遺伝子の配列を標的とした、アンチセンスオリゴヌクレオチドおよび/または低分子干渉RNA(siRNAもしくはRNAi)、またはshRNAが含まれる。
【0115】
相同組換えに関与する遺伝子産物の発現を活性化する代替法として、またはそれに加えて、Zp15を標的とするジンクフィンガー結合ドメインを有するこれらのタンパク質(またはその機能性フラグメント)の融合体を用いて、これらのタンパク質(組換えタンパク質)を対象の領域に誘導することができ、それによってそれらの局所的濃度を増加させ、さらに相同組換えプロセスを刺激することができる。あるいは、前述のような相同組換えに関与するポリペプチド(またはその機能性フラグメント)は、ジンクフィンガー結合ドメイン、切断ドメイン(または切断ハーフドメイン)、および組換えタンパク質(またはその機能性フラグメント)を含むトリプル融合タンパク質の一部であってよい。前述の方法および組成物に用いることができる、遺伝子変換および組換えに関連したクロマチン再構築に関与するさらなるタンパク質には、ヒストン・アセチルトランスフェラーゼ(例えば、Esa1p、Tip60)、ヒストン・メチルトランスフェラーゼ(例えば、Dot1p)、ヒストンキナーゼ、およびヒストンホスファターゼが含まれる。Bhatら,(1999)Plant J.33:455−469も参照されたい。
【0116】
ジンクフィンガー/ヌクレアーゼ融合分子およびドナーDNA分子を含む細胞における標的組換えの効率のさらなる上昇は、相同性駆動による修復プロセスが最大限に活発である細胞周期のG2期にある細胞を遮断することによって達成される。そのような停止は、多くの方法で達成され得る。例えば、細胞を例えば細胞周期の進行に影響を与える薬剤、化合物、および/または小分子で処理して、G2期で細胞を停止させることができる。この種類の例示的分子には、微小管重合に作用する化合物(例えば、ビンブラスチン、ノコダゾール、タキソール)、DNAと相互作用する化合物(例えば、シス−ジアンミン白金(II)ジクロリド(cis−platinum(II)diamine dichloride)、シスプラチン、ドキソルビシン)、および/またはDNA合成に作用する化合物(例えば、チミジン、ヒドロキシウレア、L−ミモシン、エトポシド、5−フルオロウラシル)が含まれるが、これに制限されない。組換え効率のさらなる上昇は、クロマチン構造を変更して、ゲノムDNAを細胞の組換え機構にさらに接近できるようにするヒストン・デアセチラーゼ(HDAC)阻害剤(例えば、酪酸ナトリウム、トリコスタチンA)の使用によって達成される。
【0117】
細胞周期の停止のためのさらなる方法には、例えば、当該タンパク質をコードするcDNAを細胞内に導入することによって、または当該タンパク質をコードする遺伝子の発現を活性化する遺伝子操作されたZFPを細胞内に導入することによって、CDK細胞周期キナーゼの活性を阻害するタンパク質の過剰発現が含まれる。細胞周期の停止は、例えばRNAi法を用いてサイクリンおよびCDKの活性を阻害することによって(例えば、米国特許第6,506,559号)、あるいは例えばサイクリンおよび/またはCDK遺伝子等の細胞周期の進行に関与する1つ以上の遺伝子の発現を抑制する遺伝子操作されたZFPを細胞内に導入することによっても達成される。遺伝子発現の制御のための遺伝子操作されたジンクフィンガータンパク質の合成のための方法について、例えば共同所有の米国特許第6,534,261号を参照されたい。
【0118】
あるいは、ある場合において、標的切断はドナーポリヌクレオチドの非存在下で行われ(好ましくはS期またはG2期に)、かつ組換えは相同染色体間で起こる。
【0119】
発現ベクター
本明細書に記載されるような1つ以上の融合タンパク質(例えば、ZFN)をコードする核酸を、複製および/または発現のために、原核細胞または真核細胞内への形質転換用ベクター内にクローニングすることができる。ベクターは、原核細胞用ベクター、例えばプラスミドまたはシャトルベクター、昆虫細胞用ベクター、あるいは真核細胞用ベクターであってよい。融合タンパク質をコードする核酸を、植物細胞への投与のための発現ベクター内にクローニングすることもできる。
【0120】
融合タンパク質(例えば、ZFN)を発現させるために、前記融合タンパク質をコードする配列を、典型的には、転写を指令するプロモーターを含有する発現ベクター内にサブクローニングする。適切な細菌および真核生物プロモーターは、当該技術分野において周知であり、例えばSambrookら,Molecular Cloning,A Laboratory Manual(2nd ed.1989;3rd ed.,2001);Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);ならびにCurrent Protocols in Molecular Biology(Ausubelら,前記参照)に記載されている。ZFPを発現させるための細菌発現システムは、例えば大腸菌、バチルス属菌、およびサルモネラ菌で利用可能である(Palvaら,Gene 22:229−235(1983)。そのような発現システム用のキットが市販されている。哺乳類細胞、酵母、および昆虫細胞のための真核細胞発現システムは当業者に周知であり、市販もされている。
【0121】
融合タンパク質をコードする核酸の発現を指令するのに用いられるプロモーターは、特定の用途に依存する。例えば、宿主細胞に適した強力な構成的プロモーターは、典型的に融合タンパク質の発現および精製のために用いられる。
【0122】
対照的に、融合タンパク質を植物遺伝子の制御のためにインビボで投与する場合(下記の「植物細胞への核酸の送達」の章を参照されたい)、前記融合タンパク質の特定の使用に応じて、構成的または誘導可能なプロモーターのいずれかが用いられる。植物プロモーターの制限されない例には、シロイヌナズナのユビキチン−3(ubi−3)(Callisら,1990,J.Biol.Chem.265−12486−12493);A.ツメファシエンス(A.tumifaciens)のマンノピン・シンターゼ(Δmas)(Petolinoら,米国特許第6,730,824号);および/またはキャッサバベインモザイクウイルス(CsVMV)(Verdaguerら,1996,Plant Molecular Biology 31:1129−1139)に由来するプロモーター配列が含まれる。実施例も参照されたい。
【0123】
プロモーターに加えて、発現ベクターは典型的に転写単位、あるいは原核または真核のいずれかの宿主細胞における核酸の発現に必要とされるすべてのさらなるエレメントを含有する発現カセットを含有する。したがって、典型的な発現カセットは、例えば融合タンパク質をコードする核酸配列に操作可能に連結されたプロモーター、および、例えば転写産物の効率的なポリアデニル化、転写終結、リボソーム結合部位、または翻訳終結に必要とされるシグナルを含有する。さらなるカセットのエレメントに、例えばエンハンサー、ヘテロスプライシングシグナル、および/または核局在化シグナル(NLS)が含まれてもよい。
【0124】
遺伝情報を細胞内へ輸送するのに用いられる特定の発現ベクターは、融合タンパク質の使用目的、例えば植物、動物、細菌、真菌、原生動物等々での発現(下記の発現ベクターを参照されたい)に関して選択される。標準的な細菌用および動物用発現ベクターは当該技術分野において既知であり、例えば米国特許公報第20050064474号A1、ならびに国際特許公報WO05/084190、WO05/014791、およびWO03/080809に詳細に記載されている。
【0125】
標準的トランスフェクション法を用いて、大量のタンパク質を発現する細菌、哺乳類、酵母、または昆虫の細胞株を作製することができ、次いでそれを標準的技術を用いることによって精製することができる(例えば、Colleyら,J.Biol.Chem.264:17619−17622(1989);Guide to Protein Purification, in Methods in Enzymology,vol.182(Deutscher,ed.,1990)を参照されたい)。真核および原核細胞の形質転換は、標準的技術に従って行われる(例えば、Morrison,J.Bact.132:349−351(1977);Clark−Curtiss&Curtiss,Methods in Enzymology 101:347−362(Wuら,eds.,1983)を参照されたい)。
【0126】
そのような宿主細胞内に外来ヌクレオチド配列を導入するための任意の周知の手法を用いてよい。これらには、リン酸カルシウムトランスフェクション、ポリブレン、プロトプラスト融合、エレクトロポレーション、超音波法(例えば、ソノポレーション)、リポソーム、マイクロインジェクション、ネイキッドDNA、プラスミドベクター、エピソーム型および組込み型の両方のウイルスベクター、ならびにクローン化されたゲノムDNA、cDNA、合成DNA、または他の外来遺伝物質を宿主細胞内に導入するための任意の他の周知の方法の使用が含まれる(例えば、前記のSambrookら,を参照されたい)。用いられる特定の遺伝子操作手法は、最適なタンパク質を発現し得る宿主細胞内に少なくとも1つの遺伝子を上手く導入できさえすればよい。
【0127】
植物細胞への核酸の送達
前述のように、DNA構築物を様々な従来技術によって所望の植物宿主内へ(例えば、そのゲノム内へ)導入することができる。そのような技術を見直すために、例えば、Weissbach&Weissbach,Methods for Plant Molecular Biology(1988,Academic Press,N.Y.)Section VIII,pp.421−463;およびGrierson&Corey,Plant Molecular Biology(1988,2d Ed.),Blackie,London,Ch.7−9を参照されたい。
【0128】
例えば、DNA構築物を、植物細胞プロトプラストのエレクトロポレーションおよびマイクロインジェクション等の技術を用いて植物細胞のゲノムDNA内に直接的に導入してもよく、あるいはDNA構築物を、DNA粒子砲撃等の微粒子銃法を用いて植物組織へ直接的に導入することができる(例えば、Kleinら,(1987)Nature 327:70−73を参照されたい)。あるいは、DNA構築物を、ナノ粒子による形質転換を介して植物細胞内に導入することもできる(例えば、参照することによりその全体として本明細書に組み入れられている、米国特許出願第12/245,685号を参照されたい)。あるいは、DNA構築物を適切なT−DNA境界/隣接領域とつなぎ合わせて、従来のアグロバクテリウム・ツメファシエンス宿主ベクター内に導入してもよい。バイナリーベクターを安全化および使用することを含む、アグロバクテリウム・ツメファシエンスを介した形質転換技術は、科学文献に十分に記載されている。例えば、Horschら,(1984)Science 233:496−498;およびFraleyら,(1983)Proc.Nat’l.Acad.Sci.USA 80:4803を参照されたい。
【0129】
さらに、遺伝子移入は、非アグロバクテリウム性の細菌またはウイルス、例えばリゾビウム種、NGR234、シノリゾビウム・メリロッティ、メソリゾビウム・ロッティ、ジャガイモウイルスX、カリフラワーモザイクウイルス、およびキャッサバベインモザイクウイルス、および/またはタバコモザイクウイルス等を用いて達成され得る。例えば、Chungら,(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0130】
バイナリーT DNAベクター(Bevan(1984)Nuc.Acid Res.12:8711−8721)または共培養手法(Horschら,(1985)Science 227:1229−1231)を用いることによって細胞を細菌に感染させる場合、アグロバクテリウム・ツメファシエンス宿主の毒性機能は、当該構築物および隣接マーカーを含有するT鎖の植物細胞DNA内への挿入を指令すると考えられる。一般に、アグロバクテリウム形質転換システムは、双子葉植物を遺伝子操作するために用いられる(Bevanら,(1982)Ann.Rev.Genet 16:357−384;Rogersら,(1986)Methods Enzymol.118:627−641)。アグロバクテリウム形質転換システムを用いて、DNAを単子葉性の植物および植物細胞に形質転換ならびに移入することもできる。米国特許第5,591,616号;Hernalsteenら,(1984)EMBO J 3:3039−3041;Hooykass−Van Slogterenら,(1984)Nature 311:763−764;Grimsleyら,(1987)Nature 325:1677−179;Boultonら,(1989)Plant Mol.Biol.12:31−40;およびGouldら,(1991)Plant Physiol.95:426−434を参照されたい。
【0131】
代替的な遺伝子移入および形質転換方法には、カルシウムを介した、ポリエチレングリコール(PEG)を介した、またはエレクトロポレーションを介したネイキッドDNAの取込みによるプロトプラスト形質転換(Paszkowskiら,(1984)EMBO J 3:2717−2722,Potrykusら,(1985)Molec.Gen.Genet.199:169−177;Frommら,(1985)Proc.Nat.Acad.Sci.USA 82:5824−5828;およびShimamoto(1989)Nature 338:274−276)、ならびに植物組織のエレクトロポレーション(D’Halluinら,(1992)Plant Cell 4:1495−1505)が含まれるが、これに制限されない。植物細胞の形質転換のためのさらなる方法には、マイクロインジェクション、炭化ケイ素を介したDNAの取込み(Kaepplerら,(1990)Plant Cell Reporter 9:415−418)、ならびにマイクロプロジェクタイル砲撃(Kleinら,(1988)Proc.Nat.Acad.Sci.USA 85:4305−4309;およびGordon−Kammら,(1990)Plant Cell 2:603−618)が含まれる。
【0132】
開示される方法および組成物を用いて、外因性配列を植物細胞ゲノムにおける所定の位置(例えば、Zp15遺伝子)内に挿入することができる。これは、植物ゲノム内に導入された導入遺伝子の発現はその組込み部位に決定的に依存するため有用である。したがって、例えば除草剤耐性、虫害抵抗性、栄養素、抗生物質、または治療用分子をコードする遺伝子を、標的組換えによってそれらの発現に有利な植物ゲノムの領域内に挿入することができる。
【0133】
前記の形質転換技術のいずれかによって作製される形質転換した植物細胞を培養して、形質転換した遺伝子型、ゆえに所望の表現型を有する植物全体を再生することができる。そのような再生技術は、典型的に、所望のヌクレオチド配列とともに導入されている殺生物剤および/または除草剤マーカーに依存している、組織培養成長培地におけるある植物ホルモンの操作に依存する。培養したプロトプラストからの植物再生は、Evansら,“Protoplasts Isolation and Culture”in Handbook of Plant Cell Culture,pp.124−176,Macmillian Publishing Company,New York,1983;およびBinding,Regeneration of Plants,Plant Protoplasts,pp.21−73,CRC Press,Boca Raton,1985に記載されている。植物のカルス、外植片、器官、花粉、胚、またはその一部から再生することもできる。そのような再生技術はKleeら,(1987)Ann.Rev.of Plant Phys.38:467−486に大まかに記載されている。
【0134】
植物細胞に導入された核酸を用いて、実質的に任意の植物に所望の特質を与えることができる。多種多様な植物および植物細胞システムを、本開示の核酸構築物および前述の種々の形質転換法を用いることによって、本明細書に記載される所望の生理学的かつ農学的特徴のために遺伝子操作することができる。好ましい実施態様では、遺伝子操作のための標的植物および植物細胞には、その単子葉および双子葉植物、例えば穀類作物(例えば、コムギ、トウモロコシ、イネ、キビ、オオムギ)、果実作物(例えば、トマト、リンゴ、ナシ、イチゴ、オレンジ)、飼料作物(例えば、アルファルファ)、根菜作物(例えば、ニンジン、ジャガイモ、サトウキビ、ヤムイモ)、葉菜作物(例えば、レタス、ホウレンソウ)を含む作物;顕花植物(例えば、ペチュニア、バラ、キク)、針葉樹、および松の木(例えば、パインファー、トウヒ);ファイトレメディエーションに用いられる植物(例えば、重金属を蓄積する植物);油料作物(例えば、ヒマワリ、ナタネ)、ならびに実験的目的のために用いられる植物(例えば、シロイヌナズナ)等が含まれるが、これに制限されない。したがって、開示される方法および組成物は、アスパラガス属、カラスムギ属、アブラナ属、ミカン属、スイカ属、トウガラシ属、カボチャ属、ニンジン属、ムカシヨモギ属、ダイズ属、ワタ属、オオムギ属、アキノノゲシ属、ドクムギ属、トマト属、リンゴ属、キャッサバ属、タバコ属、オオアラセイトウ(Orychophragmus)属、イネ属、ワニナシ属、インゲンマメ属、エンドウ属、ナシ属、サクラ属、ダイコン属、ライムギ属、ナス属、モロコシ属、コムギ属、ブドウ属、ササゲ属、およびトウモロコシ属由来の種を含むが、これに制限されない広範囲にわたる植物を使用する。
【0135】
当業者であれば、外因性配列をトランスジェニック植物に安定的に組み込み、操作可能であると確認した後に、それを生殖交配によって他の植物内に導入することができるということを理解するであろう。多くの標準的な育種技術のうちのいずれかを、交配される種に応じて用いることができる。
【0136】
形質転換するDNA上に存在するマーカー遺伝子によってコードされる特質について、遺伝子操作された植物物質を選択またはスクリーニングすることによって、形質転換した植物細胞、カルス、組織、または植物を同定かつ単離することができる。例えば、形質転換する遺伝子構築物によって耐性を与えられている、阻害量の抗生物質または除草剤を含有する培地上で遺伝子操作された植物物質を成長させることによって、選択を行うことができる。さらに、組換え核酸構築物上に存在し得る任意の可視的マーカー遺伝子(例えば、β−グルクロニダーゼ、ルシフェラーゼ、BまたはC1遺伝子)の活性についてスクリーニングすることによっても、形質転換した植物および植物細胞を同定することができる。そのような選択およびスクリーニング方法論は当業者に周知である。
【0137】
物理学的かつ生化学的方法を用いて、挿入した遺伝子構築物を含有する植物または植物細胞の形質転換体を同定することもできる。これらの方法には、:1)組換えDNAインサートの構造を検出かつ決定するためのサザン分析またはPCR増幅;2)遺伝子構築物のRNA転写産物を検出かつ調査するためのノーザンブロット、S1 RNaseプロテクション、プライマー伸長によるもしくは逆転写酵素によるPCR増幅;3)酵素またはリボザイムの活性を検出するための酵素アッセイであって、そのような遺伝子産物は遺伝子構築物によってコードされているアッセイ;4)遺伝子構築物の産物がタンパク質である場合、タンパク質ゲル電気泳動、ウェスタンブロット技術、免疫沈降、または酵素結合免疫測定法(ELISA)が含まれるが、これに制限されない。in situハイブリダイゼーション、酵素染色、および免疫染色等のさらなる技術を用いて、特定の植物器官および組織における組換え構築物の存在または発現を検出することもできる。これらすべてのアッセイを行うための方法は当業者に周知である。
【0138】
本明細書に開示される方法を用いた遺伝子操作の効果を、例えば、対象の組織から単離したRNA(例えば、mRNA)のノーザンブロットによって観察することができる。典型的には、mRNAが存在しているまたはmRNAの量が増加している場合、対応する導入遺伝子が発現していると推測することができる。遺伝子および/またはコードされたポリペプチドの活性を測定する他の方法を用いてもよい。用いられた基質、および反応産物または副産物の増加または減少を検出する方法に応じて、様々な種類の酵素アッセイを用いることができる。さらに、発現させたポリペプチドの量を、免疫化学的に、すなわちELISA、RIA、EIA、および当業者に周知の他の抗体に基づくアッセイ、例えば電気泳動検出アッセイ(染色またはウェスタンブロッティングのいずれかを伴う)等によって測定することができる。制限されない一例として、ELISAアッセイを用いたAAD−1およびPATタンパク質の検出は、参照することによりその全体として本明細書に組み入れられている、米国特許出願第11/587,893号に記載されている。植物のある組織においてまたはある発生段階において、導入遺伝子を選択的に発現させることができ、あるいは実質的にすべての植物組織において、実質的にその生活環全体にわたって、導入遺伝子を発現させることができる。しかしながら、任意の組み合わせ発現様式も適用可能である。
【0139】
本開示はまた、前記のトランスジェニック植物の種子を包含するものであって、前記種子は導入遺伝子または遺伝子構築物を有する。本開示はさらに、前記のトランスジェニック植物の後代、クローン、細胞株、または細胞を包含するものであって、前記後代、クローン、細胞株、または細胞は導入遺伝子または遺伝子構築物を有する。
【0140】
融合タンパク質(例えば、ZFN)および融合タンパク質をコードする発現ベクターを、遺伝子制御、標的切断および/または組換えのために、植物に直接投与することができる。ある実施態様では、植物は複数のパラロガス標的遺伝子を含有する。植物が複数のパラロガス遺伝子を含有し得ることは既知である。したがって、1つ以上の異なる融合タンパク質または融合タンパク質をコードする発現ベクターを植物に投与して、植物における1つ以上のZp15遺伝子を標的とすることができる。
【0141】
有効量の投与は、処理される植物細胞と最大限に接触して融合タンパク質を導入するために通常用いられる経路のいずれかによるものである。ZFPを、任意の適切な様式で、好ましくは許容可能なキャリアとともに投与する。そのようなモジュレーターを投与する適切な方法が使用可能で当業者に周知であり、また1つより多い経路を用いて特定の組成物を投与することができるが、特定の経路はしばしば別の経路より即時かつ効果的な反応を提供し得る。
【0142】
キャリアは、用いてもよく、投与される特定の組成物によって、ならびに前記組成物を投与するのに用いられる特定の方法によって、ある程度決定される。したがって、使用可能である多種多様なキャリアの適切な処方が存在する。
【実施例】
【0143】
以下は、本開示を実施するための特定の態様の実施例である。実施例は、例示を目的として提示されるものであって、決して本開示の範囲を制限することを意図されるものではない。
【0144】
用いた数(例えば、量、温度、等々)に関する正確性を確保するための努力はなされているが、いくらかの実験誤差および偏差は当然許容されるべきである。
【0145】
実施例1:Zp15標的遺伝子座の同定および特徴付け
公的に入手可能な遺伝子地図(Lawrence,C.ら,(2004)NAR 32:393−397;トウモロコシのインターネットのデータベース)およびトウモロコシのドラフトゲノム配列に基づき、第6染色体の短腕上にあるZp15遺伝子座を、位置およびZp15について入手可能な情報に基づくZFNを用いた改変の標的として選択した。トウモロコシ由来の15kDのβゼインをコードする遺伝子(Zp15)のゲノム構造および配列は、公に記載されかつ注釈が付されている(Wooら,(2001)Plant Cell 13(10):2297−2313)。Zp15遺伝子に対する配列は、参照することにより本明細書に組み入れられている、GenBankアクセス番号AF371264に記載されている。
【0146】
Zp15ゲノム配列を用いて、BLASTアルゴリズムを用いたTIGRおよびトウモロコシGDBデータベースへのクエリーを実行した。2つのコンティグ(AZM5_16782およびZmGSStuc11−12−04.8785.1)を含むがこれに制限されない、Zp15との重複相同性を有するいくつかの配列、ならびにいくつかのEST(M72708、M13507、M12147、AY103640、およびAF371264)が同定された。これらの登録の配列ならびにZp15配列に基づき、プライマー3プログラム(Rozen,S.およびSkaletsky,H.J.(2000)Primer3 on the WWW for general users and for biologist programmers.In:Krawetz S,Misener S(eds.)Bioinformatics Methods and Protocols:Methods in Molecular Biology.Humana Press,Totowa,NJ,pp365−386;インターネット上でも利用可能)を用いることによって、複数の短いオリゴヌクレオチドをPCRプライマーとしての使用のために設計した。
【0147】
これらのプライマーには、以下の正方向オリゴヌクレオチド:
P67F 5’−CGTATGAATTCATTGACAACC−3’(配列番号:1)
P68F 5’−ATGATCTATCTGTAAATCC−3’(配列番号:2)
P69F 5’−CGTCATGCAACGCAACATTCC−3’(配列番号:3)
P73F 5’−AAGAACATCACAAGTTATGC−3’(配列番号:4)
P74F 5’−TCATGTGGATCCAAGGCATC−3’(配列番号:5)
が含まれるが、これに制限されない。
【0148】
さらに、プライマーには、以下の逆方向オリゴヌクレオチド:
P70R 5’−ATGTGTGTCGTCTTACTGC−3’(配列番号:6)
P71R 5’−CAGTAGTAGGGCGGAATG−3’(配列番号:7)
P72R 5’−GGGCAGCTGGTACTG−3’(配列番号:8)
P75R 5’−CTATAATCGATGTAGAGC−3’(配列番号:9)
P76R 5’−CTATGCTTTGTCTATAGTCG−3’(配列番号:10)
が含まれるが、これに制限されない。
【0149】
すべてのオリゴヌクレオチドプライマーを、Integrated DNA Technologies(IDT,Coralville,IA)によって合成し、そこから購入した。トウモロコシ亜種Hi−II由来のgDNAの増幅を、30ngのgDNAを用いてPCRサーマルサイクラーで行った。Hi−II由来のZp15遺伝子に相当する2,215bpの増幅フラグメントを単離し、pCR2.1プラスミド(Invitrogen,Carlsbad,CA)内にクローニングした。このフラグメントの配列分析により、トウモロコシ亜種Hi−II由来のZp15のゲノム構造には、2つのエクソンおよび31bpの1つの小さなイントロン(配列番号:126)が含有されていることが明らかになった。Zp15標的ZFNの設計は、トウモロコシ亜種Hi−II由来のZp15遺伝子のコード領域に集中させた。
【0150】
実施例2:トウモロコシZp15遺伝子を標的とするジンクフィンガーヌクレアーゼの設計
ZFNに対する発現ベクターを組み立てるために、ライブラリーアーカイブから選択したまたは新たに合成したZFNコード遺伝子の任意の所定の対に対して適用可能である、段階的なモジュラークローニング方式を考案した。
【0151】
まず、Doyonら,(2008)Nature Biotechnology 26(6):702および米国特許出願第12/284,887号に記載されているような酵母アッセイ選別を用いて、Zp15標的ZFNをスクリーニングした。簡潔には、Zp15遺伝子座全体を出芽酵母ゲノムのHO遺伝子座内に導入して、標的遺伝子内の種々の結合部位におけるZFN活性を直接比較し、Doyonらに記載されているようなレポーターアッセイ(MEL1)を用いることによって、レポーター遺伝子においてDSBを誘導し得るそれらの能力についてZFNをスクリーニングした。
【0152】
これらのプロキシシステムアッセイの結果に基づき、試験された様々なZFN対がZp15内でDSBを誘導し得ることが確認された。
【0153】
酵母プレスクリーニングに続き、次いでZFN対をトウモロコシ特異的発現ベクター内にサブクローニングした。米国特許公報第20080182332号に記載されているように、トウモロコシop−2に由来する再設計かつ合成された核局在化シグナル(NLS)のセグメント、およびトウモロコシのコドンバイアスを使用しているFokIヌクレアーゼドメインを含むベクターを、固有のXhoI部位の下流に1ヌクレオチド(C)を挿入して改変し、追加のSacI部位を創出した。同様のベクターを、Thosea asignaウイルス由来の2Aリボソーム断続(stuttering)配列を含むように改変した。個々のジンクフィンガータンパク質のORFをコードする遺伝子カセットを、これらのベクターのいずれかの中にKpnIおよびBamHI制限酵素認識部位を用いてクローニングし、続いて前記2つのベクターをBglII/XhoI制限酵素認識部位を用いて組み合わせ、NcoIおよびSacI制限酵素認識部位が隣接した2つのZFNコードドメインを含むカセットを含有する中間構築物を産生した。
【0154】
NcoI/SacIカセットをこの中間構築物から制限酵素消化によって切り出し、トウモロコシのユビキチン−1遺伝子由来のプロモーター(Sharrockら,(1992)Plant Mol Biol.18(4):675)、およびトウモロコシ根の選択的カチオン性ペルオキシダーゼ遺伝子由来のターミネーター配列(米国特許第7,179,902号)を含有する、プラスミド骨格のpDAB3872内にライゲーションした。
【0155】
結果として生じたプラスミドには、ZFN遺伝子、さらにプラスミド維持のための関連する選択可能なマーカー、およびInvitrogen(Carlsbad,CA)製のGATEWAY(商標)システムを用いた便利な操作のための隣接attL部位が含まれる。このクローニング方式を用いて作製されたZFN構築物のそれぞれを、大腸菌DH5α細胞内に形質転換し、その後適切な選択の下で維持した。
【0156】
表1は、Zp15遺伝子座内への標的組込み実験に用いた例示的なZp15を標的とするZFNを示す。ZFNに対するDNA標的配列を2列目に示し(DNA標的部位を大文字で表示;接触しないヌクレオチドを小文字で表示)、3〜6列目は、前記タンパク質におけるジンクフィンガーのそれぞれ(F1〜F4)の認識領域のアミノ酸配列(ヘリックスの開始に対してアミノ酸−1〜+6)を示す。また、表1の1列目にはそれぞれのタンパク質に対する識別番号を提示する。
【表1】

【表2】
【0157】
ZFNをC2H2またはC3H骨格内に容易に挿入できること、および種々の配列を用いてジンクフィンガータンパク質と切断ドメインを接合することができることは明白であろう。米国特許公報第20080182332号、特にそのような配列に関する表6を参照されたく、この参考文献は参照することによりその全体として本明細書に組み入れられている。
【0158】
実施例3:トウモロコシ細胞におけるZFNを介したZp15の破壊
ZFN対によるDSBの誘導を試験した。内在性Zp15遺伝子の意図される標的部位においてDSBを効率的に誘導し得るZFN対を同定した。ZFN誘導性断裂の部位において小さなDNA欠失/挿入をもたらすことが既知である非相同末端結合(NHEJ)によるDSB修復のエラーを起こしやすい性質を利用して、内在性Zp15遺伝子標的部位に効率的に結合かつ切断するZFN対を選択した。
【0159】
ZFNを培養トウモロコシ細胞で一過性に発現させ、予測される切断部位における標的遺伝子座の配列分析を行った。例えば、ZFN対#25(前記で示した11768/11766認識ヘリックス)をコードするプラスミドpDAB7468を、参照することによりその全体として本明細書に組み入れられている、米国特許出願第12/001,939号に記載されているような、WHISKERS(商標)を介した形質転換によってトウモロコシHi−II細胞培養物内へ送達した。一過性の発現の24時間または72時間後のいずれかに、結果として生じた破壊されたZFN標的配列を、単離したゲノムDNAから増幅し、プラスミドベクターpCR2.1内にクローニングした。gDNAを酵素Bsu36Iを用いた制限酵素消化にかけ、その後にZp15標的配列の増幅およびPCR産物のプラスミドベクターpCR2.1内へのクローニングが続いた。クローン化された増幅産物の個々のコロニーを、プラスミドDNAの制限酵素消化によって分析し、その後にアガロースゲル電気泳動が続いた(制限酵素Bsu36Iによる切断に対して耐性を示したクローン化された増幅産物は、ZFN切断部位に関連した制限酵素認識部位を破壊する変異を含有していると考えられた)。
【0160】
192クローンの直接配列分析によって、6bpの挿入が明らかになった(図1)。別の実施例では、ZFN対#24(前記で示した11753/11750認識ヘリックス)をコードするプラスミドpDAB7467を、トウモロコシ培養細胞物内に直接送達し、一過性の発現の24時間または72時間後のいずれかに、ZFN標的配列を増幅し、プラスミドベクターpCR2.1内にクローニングした。192クローンの直接配列分析によって、正確な切断部位における3bpの欠失が明らかになった(図2)。ここに記載される挿入および欠失は、標的部位において誘導されたDSBのNHEJ修復の結果であり、かつZFN24および25がトウモロコシ細胞内の内在性Zp15遺伝子座において切断活性を有することを示す。
【0161】
ZFN25または28を用いて同じ手法を行ったが、コロニーを制限酵素消化によってスクリーニングする代わりに、192の独立したクローンを直接シークエンシングした。6bpの挿入が検出された(図2、上)
【0162】
まとめると、これらのデータは、ZFNへの一過性の暴露は培養トウモロコシ細胞内のZp15遺伝子座において標的DSBを誘導するのに十分であることを証明している。
【0163】
実施例4:Zp15遺伝子座内への標的組込み
Zp15における切断活性を有する設計されたZFNが、外因性配列の組込みを促進し得るかどうかを試験するために、本発明者らは、例示的な外因性の除草剤耐性遺伝子であるスフィンゴモナス・ヘルビシドボランス由来のAAD−1(ATCC700291)をコードする自律的遺伝子カセットを保持するドナーDNA分子を構築した。AAD−1は、アリールオキシアルカノエート・ジオキシゲナーゼ酵素をコードし、アリールオキシフェノキシプロピオン酸系除草剤に対する耐性を与える(国際特許WO2005/107437,WO2008141154A2)。当業者であれば、関連したADD−12遺伝子等の他の除草剤耐性遺伝子を含むが、これに制限されない他の外因性核酸を、ドナーDNA分子内に同様に組み入れることができるということを理解するであろう。この除草剤耐性遺伝子ドナーにおいて、プロモーター配列はO.サティバのアクチンに由来しており(GenBankアクセス番号S44221およびX63830)、かつターミネーター配列はZ.メイスのL.リパーゼに由来している(GenBankアクセス番号L35913)。
【0164】
A.ドナーDNA分子の構築
Zp15に相同な領域を含有するドナー構築物を以下のように作製した。Zp15遺伝子に対して相同な隣接部を含有するプラスミド骨格を、Zp15遺伝子の対応する標的部位内への任意のドナーDNA配列の組込みが可能になるように遺伝子操作した。ここに例示したプラスミド骨格は、基本プラスミドベクターpBC SK(−)ファージミド(3.4kbp)(Stratagene,La Jolla,CA)に由来したものであった。この手法には4つの工程があった。
【0165】
第一に、3μgのpBC SK(−)をSpeIおよびSalI(New England Biolabs,Beverly,MA)制限エンドヌクレアーゼを用いて線状化することによって、基本プラスミドを調製した。3.3kbpのSpeI/SalI消化したサブクローニングベクターのpBC SK(−)をゲルから切り出し、QIAQUICKゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。
【0166】
第二に、Integrated DNA Technologies社(Coralville,IA)によって合成された以下のオリゴヌクレオチドプライマー:
Zp15HRドナーNotI(205)F:5’−GCGGCCGCATGCAAGAGCTGTTGATC−3’(配列番号:97)
Zp15HRドナーMfeI(1025)R:5’−CAATTGCCGGCGTAGTAGGGCGCCGCCGCCAGC−3’(配列番号:98)
Zp15HRドナーMfeI(1025)F:5’−CAATTGGTGTGGGCAGCCGAGCGCCATGTTCCAG−3’(配列番号:99)
Zp15HRドナーSalI(2270)R:5’−GTCGACCGATACTGATGCGGACCGTCCACCTTGTC−3’(配列番号:100)
を用いて、Zp15から5’および3’相同隣接部を単離した。
【0167】
PCR増幅反応を、LA TAQ PCRキット(TaKaRa Biotechnology社,Otsu,Shiga,Japan)に備わった試薬を用いて行った。PCR反応カクテルは、5μLの10×LA PCR(商標)バッファーII(Mg2+)、20ngの二本鎖テンプレート(Hi−IIトウモロコシゲノムDNA)、10pmolの正方向オリゴヌクレオチドプライマー、10pmolの逆方向オリゴヌクレオチドプライマー、8μLのdNTPミックス(2.5mMの各dNTP)、33.5μLのH2O、0.5μL(2.5単位)のTaKaRa LA Taq(商標)DNAポリメラーゼ、1滴のミネラルオイルからなるものであった。プライマーZp15HRドナーNotI(205)FおよびZp15HRドナーMfeI(1025)Rを一反応に用い、プライマーZp15HRドナーMfeI(1025)FおよびZp15HRドナーSalI(2270)Rを第二の反応に用いた。Perkin−Elmer Cetus,48サンプルDNAサーマルサイクラー(Norwalk,CT)を用いて、以下のサイクル条件:94℃,4分間/1サイクル;98℃ 20秒間,65℃ 1分間,68℃ 1分間/30サイクル;72℃,5分間/1サイクル;4℃/ホールドの下でPCR反応を行った。15μlの各PCR反応物を電気泳動し、増幅されたフラグメントを紫外線で可視化し、フラグメントの大きさを1kbpのDNAラダーと比較することによって概算した。期待されるプラスミドクローンを、5’フラグメントについては825bp、または3’フラグメントについては1,250bpのDNAフラグメントの存在によって診断した。これらのフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、精製したフラグメントをTOPO TAクローニング(登録商標)キットを用いてpCR2.1プラスミド内にクローニングし、化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)内にメーカーのプロトコールに従って形質転換した。
【0168】
825bpの5’フラグメントまたは1,250bpの3’フラグメントを含有する個々のコロニーを、制限酵素消化およびシークエンシングデータによって同定かつ確認した。825bpの5’フラグメントを含有するコロニーは、MfeIおよびNotI(New England Biolabs,Beverly,MA)の制限酵素消化によって確認された。1,250bpの3’フラグメントを含有するコロニーは、SalI(New England Biolabs,Beverly,MA)を用いた制限酵素消化によって同定かつ確認された。期待されるプラスミドクローンを、3.9kbpのpCR(登録商標)2.1ベクターに加えて、825bp(5’フラグメント)または1,250bp(3’フラグメント)の挿入されたDNAフラグメントの存在によって診断した。プラスミドクローンの二本鎖シークエンシング反応を、CEQ(商標)DTCS−クイックスタートキット(Beckman−Coulter,Palo Alto,CA)を用いて、メーカーによって記載されるとおりに行った。反応物を、Performa DTRゲル濾過カートリッジ(Edge BioSystems,Gaithersburg,MD)を用いて、メーカーのプロトコールに記載されるとおりに精製した。シークエンシング反応物をBeckman−Coulter CEQ(商標)2000 XL DNA分析システム上で分析し、ヌクレオチドの特徴付けをSEQUENCHER(商標)バージョン4.1.4(Gene Codes社,Ann Arbor,MI)を用いて行った。Zp15由来の5’および3’相同フラグメントの配列を、配列番号127および配列番号:128に示す。
【0169】
第三に、3’相同隣接部を、以下のように基本プラスミド内にライゲーションした。正確な3’相同隣接部配列を含有するクローンを制限酵素で消化し、DNAフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いて精製した。これらのフラグメントを、あらかじめSpeI/SalI(前記参照)で消化された精製した基本プラスミド内に、16℃のウォータバス中で16時間のインキュベーションの条件下、20μLの反応容量中に500単位のT4 DNAリガーゼ(Invitrogen Life Technologies,Carlsbad,CA)を用いて、1:5のベクター:インサートの割合でライゲーションした。その後、5μLのライゲーション反応物をワンショット(登録商標)Top10化学的コンピテントセル(Invitrogen Life Technologies,Carlsbad,CA)内に形質転換し、抗生物質選択を含有する培地上にプレーティングした。推定コロニーを単離し、SpeIおよびSalI制限酵素(New England Biolabs,Beverly,MA)で消化して、ライゲーションされた3’フラグメントを含有するクローンを同定した。
【0170】
第四に、5’相同隣接部を、3’相同隣接部を含有するプラスミド内にライゲーションした。前記の第三の工程で記載した3’相同隣接部を含有するプラスミドを、MfeIおよびNotI(New England Biolabs,Beverly,MA)制限エンドヌクレアーゼで37℃にて1時間消化した。3’相同隣接部を含有するNotI/MfeI消化されたプラスミドをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。
【0171】
MfeIおよびNotI制限酵素を用いた制限酵素消化によって生成された5’相同隣接部ドナーの単離したフラグメントを作製し、1:5のベクター:インサートの割合および500単位のT4 DNAリガーゼ(Invitrogen Life Technologies,Carlsbad,CA)を用いた20μLのライゲーション反応液中で、3’相同隣接部を含有するプラスミドとライゲーションした。ライゲーション反応液を16℃のウォータバス中で16時間インキュベーションした。
【0172】
ライゲーションの後、5μLのライゲーション反応物を、MAX EFFICIENCY(登録商標)DH5α(商標)化学的コンピテントセル(Invitrogen Life Technologies,Carlsbad,CA)内にメーカーの推奨どおりに形質転換した。個々のコロニーを選択し、プラスミドDNAを単離し、かつNotI制限酵素(New England Biolabs,Beverly,MA)で消化して、5’相同隣接部ドナーの組み込まれたフラグメントを含有するプラスミドを同定した。結果として生じたプラスミドをpDAB7489と命名した(図3)。
【0173】
プロモーターを含有する植物転写単位(PTU)、除草剤耐性遺伝子、およびポリアデニル化(ポリA)終結配列を含む除草剤耐性遺伝子発現カセットを構築した。前記プロモーター配列は、O.サティバのアクチンに由来する(McElroyら,(1990)Plant Cell 2,163−171;GenBankアクセス番号S44221およびGenBankアクセス番号X63830)。前記除草剤耐性遺伝子は、アリールオキシフェノキシプロピオン酸系除草剤に対する耐性を与える(WO2005/107437)、AAD−1(アリールオキシアルカノエート・ジオキシゲナーゼ)遺伝子を含んでいた。利用した遺伝子のバージョンは、植物における発現に対してコドン最適化配列を含むバージョン#3であった。ターミネーター配列は、Z.メイスのL.リパーゼ(GenBankアクセス番号L35913のトウモロコシリパーゼcDNAクローン)に由来する。このトウモロコシ配列は、AAD−1遺伝子に対する3’非翻訳領域/転写ターミネーター領域を含む。前記除草剤耐性遺伝子発現カセットを図4に示す。
【0174】
このカセットを作製するために、以下のオリゴヌクレオチドプライマー:
OsActAad1v.3ZmLipMfeIF 5’−CAATTGGTCATTCATATGCTTGAGAAGAG−3’(配列番号:101)
OsActAad1v.3ZmLipMfeIR 5’−CAATTGAGCACTTAAAGATCTTTAGAAG−3’(配列番号:102)
をIntegrated DNA Technologies社(Coralville,IA)によって標準的脱塩の条件下で合成し、0.125μg/μLの濃度まで水で希釈した。
【0175】
PCR増幅反応を、LA TAQ PCRキット(TaKaRa Biotechnology社,Otsu,Shiga,Japan)を用いて行った。PCR反応カクテルは、5μLの10×LA PCR(商標)バッファーII(Mg2+)、20ngの二本鎖テンプレート(pDAB3878プラスミドDNA)、10pmolの正方向オリゴヌクレオチドプライマー、10pmolの逆方向オリゴヌクレオチドプライマー、8μLのdNTPミックス(それぞれ2.5mM)、33.5μLのH2O、0.5μL(2.5単位)のTaKaRa LA Taq(商標)DNAポリメラーゼ、1滴のミネラルオイルを含んでいた。Perkin−Elmer Cetus,48サンプルDNAサーマルサイクラー(Norwalk,CT)を用いて、以下のサイクル条件:94℃,4分間/1サイクル;98℃ 20秒間,55℃ 1分間,68℃ 3分間/30サイクル;72℃,5分間/1サイクル;4℃/ホールドの下でPCR反応を行った。15μlの各PCR反応物を、0.5%エチジウムブロマイドを補充した1.0%TAEアガロースゲル中で1時間100Vで電気泳動した。増幅されたフラグメントを紫外線で可視化し、フラグメントの大きさを1kbpのDNAラダーと比較することによって概算した。
【0176】
期待されるPCR産物を、2.7kbpのDNAフラグメント(AAD−1 PTU)の存在によって診断した。これらのフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、精製したフラグメントを、TOPO TAクローニング(登録商標)キット(pCR(登録商標)2.1ベクターを備えた)および化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR2.1プラスミド内にクローニングした。
【0177】
個々のコロニーを選択し、プラスミドDNAを単離し、かつ制限酵素MfeI(New England Biolabs,Beverly,MA)で消化した。期待されるプラスミドクローンを、3.9kbpのpCR(登録商標)2.1ベクターに加えて、2,674bp(AAD−1 PTU)の挿入されたDNAフラグメントの存在によって診断した。プラスミドクローンの二本鎖シークエンシング反応を、CEQ(商標)DTCS−クイックスタートキット(Beckman−Coulter,Palo Alto,CA)を用いて、メーカーによって記載されるとおりに行った。反応物を、Performa DTRゲル濾過カートリッジ(Edge BioSystems,Gaithersburg,MD)を用いて、メーカーのプロトコールに記載されるとおりに精製した。シークエンシング反応物をBeckman−Coulter CEQ(商標)2000 XL DNA分析システム上で分析し、ヌクレオチドの特徴付けをSEQUENCHER(商標)バージョン4.1.4(Gene Codes社,Ann Arbor,MI)を用いて行った。
【0178】
正確な2,674bpの配列を含有するクローン由来の制限酵素消化されたフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、このフラグメントを、制限酵素MfeIで消化され、かつその後脱リン酸化されている、精製したpDAB7489(プラスミド骨格)とライゲーション反応でつなぎ合わせた。以下の条件下:1:5のベクター:インサートの割合、および16℃のウォータバス中で16時間のインキュベーションの条件下、20μLの反応容量中に500単位のT4 DNAリガーゼ(Invitrogen Life Technologies,Carlsbad,CA)でライゲーションを行った。その後、5μLのライゲーション反応物を、50μlの大腸菌MAX EFFICIENCY(登録商標)DH5α(商標)化学的コンピテントセル(Invitrogen Life Technologies,Carlsbad,CA)内に形質転換し、メーカーによって記載される選択条件下でプレーティングした。
【0179】
個々のコロニーを選択し、プラスミドDNAを単離し、かつ制限酵素MfeI(New England Biolabs,Beverly,MA)で消化した。期待されるプラスミドクローンは、DNAフラグメント2,674bp(AAD−1 PTU)および5,413bp(pDAB7489ベクター)を含有していた。結果として生じたプラスミドをpDAB7490と命名した(図5)。
【0180】
トウモロコシ亜種Hi−IIの胚発生細胞培養物(Armstrongら,(1991)Maize Genet Coop Newsletter 65:92−93)を作製し、維持し、かつZFN24およびドナー分子pDAB7490をコードするプラスミドの同時形質転換にかけた。カルス組織の形質転換および選択、ならびにその後の形質転換体の再生は、米国特許出願第12/001,939号に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。形質転換および選択のプロトコールに関するさらなるガイダンスのために、Petolinoら,(2000)Plant Cell Rept.19:781−786を参照されたい。開花後、植物を自己授粉またはトウモロコシ亜種DAS5XH751と異種交配させた。結果として生じた後代種子を収穫し、乾燥させた。無傷の、稔性のトウモロコシ植物へのカルスの再生は、米国特許出願第12/001,939号、特に実施例22に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。
【0181】
B.Zp15遺伝子座内へのAAD−1遺伝子カセットの標的組込み
除草剤耐性遺伝子カセットをコードする組み込まれたドナーDNA分子を含有する除草剤耐性事象のうち、前記事象のある割合は、ZFN誘導性二本鎖断裂の部位へのドナーDNAの標的組込みの産物であると予想されている。これらの標的組込み事象を除草剤耐性遺伝子カセットのランダムな組込みに由来するものと区別するために、ゲノム特異的およびそれに続くゲノム特異的、さらにドナー特異的PCRプライマーを用いたPCRに基づく遺伝子型同定ストラテジーを利用した。
【0182】
すべての除草剤耐性形質転換事象におけるAAD−1導入遺伝子の標的組込み対ランダムな組込みについての示差的遺伝子型同定を、Zp15遺伝子座およびAAD−1遺伝子に特異的なPCRに基づくアッセイを用いて行った。ここに提示される実施例では、すべてのオリゴヌクレオチドプライマーを、標準的脱塩の条件下でIntegrated DNA Technologies社(Coralville,IA)によって合成し、100μMの濃度まで水で希釈した。以下の正方向および逆方向オリゴヌクレオチドプライマーのセット;
HB501f:5’−AAGGTCCCAAATCTGAGGCATACTGTTGCT−3’(配列番号:103)
HB502r:5’−GAGGTCCTATGCTTTGTCTATAGTCGGCAG−3’(配列番号:104)
を、ドナーDNA配列の境界の外側に位置するZp15遺伝子標的に特異的なgDNAにアニールするように設計した。
【0183】
正方向および逆方向オリゴヌクレオチドプライマーの第二のセット:
HB503f:5’−GGCATACTGTTGCTGCCCTGCTGGAA−3’(配列番号:105)
HB504r:5’−GACACCTATAATCGATGTAGAGCCGAAGAG−3’(配列番号:106)
も、ドナーDNA配列の境界の外側のZp15遺伝子標的に特異的なgDNAにアニールするように設計し、しかしながら第一の対の中に入れ子にした。
【0184】
正方向および逆方向オリゴヌクレオチドプライマーを、AAD−1除草剤耐性遺伝子のコード領域に対応するドナーDNAに特異的にアニールするようにさらに設計した。
HB505f:5’−AGTCCACCCCAGTGATCTCAGCACCA−3’(配列番号:107)
HB506f:5’−AGTGGCTGGACAGCTATTCTCTCAAAGCGT−3’(配列番号:108)
HB507r:5’−ACGCTTTGAGAGAATAGCTGTCCAGCCACT−3’(配列番号:109)
HB508r:5’−TGGTGCTGAGATCACTGGGGTGGACT−3’(配列番号:110)
【0185】
2つの別個なプライマリー増幅反応を、Zp15ゲノム領域で結合するプライマーおよびドナー分子を利用して行い、ゲノムとドナーとの間の組込みの境界にまたがるアンプリコンを生じた。第一の反応は、ゲノムとドナーとの間の5’境界に焦点を合わせ、プライマーセットHB501fおよびHB507rを用いた。第二の反応は、ドナーとゲノムとの間の3’境界に焦点を合わせ、プライマーセットHB505fおよびHB502rを用いた。形質転換したトウモロコシHi−II事象からゲノムDNAを単離した。プライマリーPCR増幅反応を、LA TAQ PCRキット(TaKaRa Biotechnology社,Otsu,Shiga,Japan)によって提供されている試薬を用いて行った。PCR反応カクテルは、2.5μlの10×La Taq PCR(商標)バッファー、40〜200ngの二本鎖ゲノムDNAテンプレート、10μMの正方向オリゴヌクレオチドプライマー、10μMの逆方向オリゴヌクレオチドプライマー、2μlのdNTPミックス(それぞれ2.5mM)、16.25μlのH2O、0.25μl(1.25単位)のLA Taq(商標)DNAポリメラーゼからなるものであった。Bio−Rad,96サンプルDNA Engine Tetrad2,Peltierサーマルサイクラー(Hercules,CA)を用いて、以下のサイクル条件:94℃,2分間/1サイクル;94℃ 30秒間,62℃ 30秒間,68℃ 5分間/30サイクル;4℃/ホールドの下でPCR反応を行った。
【0186】
続いて、プライマリーPCRの反応産物を水で1:100に希釈し、2つの別個なセカンドPCR反応のためのテンプレートDNAとして用いた。セカンド反応も、Zp15ゲノム領域で結合するプライマーおよびドナー分子を利用し、ゲノムとドナーとの間の組込みの境界にまたがるアンプリコンを生じる。特異的プライマーの同一性によって、増幅がゲノムとドナーとの間の5’または3’境界のいずれかに焦点を合わせるかが決定される。第一の反応は、ゲノムとドナーとの間の5’境界に焦点を合わせ、プライマーセットHB503fおよびHB508rを用いた。第二の反応は、ドナーとゲノムとの間の3’境界に焦点を合わせ、プライマーセットHB506fおよびHB504rを用いた。両方の反応は、以下の:2.5μlの10×La Taq PCR(商標)バッファー、1μlのテンプレート[1oPCR反応の1:50希釈]、10μMの正方向オリゴヌクレオチドプライマー、10μMの逆方向オリゴヌクレオチドプライマー、2μlのdNTPミックス(それぞれ2.5mM)、16.25μlのH2O、0.25μl(1.25単位)のLA Taq(商標)DNAポリメラーゼからなるものであった。Bio−Rad,96サンプルDNA Engine Tetrad2,Peltierサーマルサイクラー(Hercules,CA)を用いて、以下のサイクル条件:94℃,2分間/1サイクル;94℃ 30秒間,62℃ 30秒間,68℃ 5分間/30サイクル;4℃/ホールドの下でPCR反応を行った。5’境界に対する2,180bpまたは3’境界に対する2,980bpの期待されるPCRアンプリコンフラグメントが観察された。
【0187】
これらのフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。続いて、精製したフラグメントを、TOPO TAクローニング(登録商標)キット(pCR(登録商標)2.1ベクターを備えた)および化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR2.1プラスミド内にクローニングした。
【0188】
個々のコロニーを選択し、プラスミドDNAを単離し、かつ制限酵素EcoRI(New England Biolabs,Beverly,MA)で消化した。期待されるプラスミドクローンを、3.9kbpのpCR(登録商標)2.1ベクターに加えて、適切な大きさの挿入されたDNAフラグメントの存在によって診断した。
【0189】
プラスミドクローンの二本鎖シークエンシング反応を行い、ヌクレオチドの特徴付けおよびアラインメントをSEQUENCHER(商標)バージョン4.1.4(Gene Codes社,Ann Arbor,MI)を用いて行った。
【0190】
Zp15標的遺伝子内に挿入されたAAD−1ドナー遺伝子カセットの標的組込み事象(事象#147)から得られた、選択した配列データを図6のアラインメントに示す。
【0191】
ゲノムとドナーとの間の5’または3’境界のいずれかに焦点を合わせたプライマリーPCR増幅産物を、同様にゲノムとドナーとの間の5’または3’境界のいずれかに焦点を合わせたセカンド増幅にかけた。これらのセカンド増幅産物に対応するクローン化フラグメントと、野生型Zp15ゲノム配列、ならびに標的組込み事象の期待される配列とのアラインメントは、標的部位においてドナーDNAの正確な組込みが起こったことをはっきりと示している。Zp15ゲノム遺伝子座のヌクレオチド配列、ゲノム/ドナーの境界、Zp15相同隣接部に対応するドナー領域のヌクレオチド配列、および除草剤耐性カセットのヌクレオチド配列は、この事象に由来する複数のクローン化PCR産物においてすべて保存されていた。したがって、この事象は、ZFNを介した二本鎖断裂の相同性駆動による修復、および特定の遺伝子標的におけるドナーDNAの標的組込みが起きているゲノムを表す。図6において、本発明者らは1つの代表的な単離した形質転換トウモロコシカルスから得られた配列アラインメントデータを示す(事象#147)。
【0192】
固有の標的組込みの発生を表すさらなる形質転換事象が得られており、本明細書で教示された方法がトウモロコシカルスにおいて再現可能であることを証明している。
【0193】
実施例5:Zp15遺伝子座内へのPATの標的組込み
本明細書に開示される標的遺伝子座内への選択された外因性ポリヌクレオチドの標的組込みのための方法のさらなる例示として、PATをコードする自律的遺伝子カセットを保持するさらなるDNAドナー分子を設計かつ構築し、内在性Zp15標的遺伝子座内への外因性ドナー配列の組込みを試験した。この遺伝子座でDSBを引き起こすために、内在性Zp15遺伝子標的の特異的切断活性を有するZFNを配置した。続いて、ストレプトマイセス・ビリドクロモゲネス由来のPATをコードする自律的遺伝子カセットを保持するドナーDNA分子およびZp15に相同な隣接配列を、Zp15標的のZFN誘導性DSB内に組み込んだ。PATは、ホスフィノトリシン・アセチル・トランスフェラーゼ酵素をコードし、アセチル化によって除草活性化合物ホスフィノトリシン(PPT)に対する耐性を与える(米国特許第5,633,434号)。ホスフィノトリシンは、除草剤のリバティ、バスタ、およびイグナイトの活性成分である。PATコード配列を植物転写単位(PTU)として構築し、O.サティバのアクチン(GenBankアクセス番号S44221およびX63830)に由来するプロモーター配列、およびZ.メイスのL.リパーゼ(GenBankアクセス番号L35913)に由来するターミネーター配列を含有させた。
【0194】
A.ドナーDNA分子の構築
Zp15に相同な領域を含有するZp15ドナー構築物を、以下のように合成して作製した。pDAB7489由来のヌクレオチド4595−5346(5’相同配列;配列番号:111)およびヌクレオチド21−796(3’相同配列;配列番号112)を含むZp15相同領域を設計した。この相同領域は、5’および3’相同エレメントの間にMfeIクローニング部位、ならびに5’および3’末端にNotI制限酵素認識部位を含んでいた。このDNA配列(配列番号:113)を合成し、カナマイシン耐性ColE1型プラスミドであるpMK内のSacIおよびKpnIクローニング部位(Gene Art Ag,Regensburg,Germany)に挿入した。結果として生じたドナープラスミドはpDAB104101と称され(図7)、Zp15遺伝子の対応する標的部位内への対象の任意のDNA配列の組込みを可能にする、Zp15遺伝子に対する相同隣接部を含有している。
【0195】
プロモーター、除草剤耐性遺伝子、およびポリアデニル化(ポリA)終結配列を含有する完全なPTUを含む除草剤耐性遺伝子発現カセットを構築した。前記プロモーター配列は、O.サティバのアクチンに由来する(McElroyら,(1990)Plant Cell 2:163−171)。GenBankアクセス番号S44221およびGenBankアクセス番号X63830。前記除草剤耐性遺伝子はPATであった。前記終結配列は、Z.メイスのL.リパーゼ(GenBankアクセス番号L35913のトウモロコシリパーゼcDNAクローン)に由来する。このトウモロコシ配列は、PAT遺伝子に対する3’非翻訳領域/転写ターミネーター領域を含む。PAT除草剤耐性遺伝子発現カセットを図8に示す。
【0196】
PAT遺伝子カセットを、Integrated DNA Technologies社(Coralville,IA)によって合成されたプライマー:
DC001 5’−CCAGTGCAATTGGGTCATTCATATGCTTGAGAAG−3’(配列番号:114)
DC002 5’ −CCAGTGCAATTGAATTCAGCACTTAAAGATCTTTAG−3’(配列番号:115)
を用いたPCRによって、プラスミドpDAB102256から増幅した。
【0197】
PCR増幅反応を、PHUSION(商標)DNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,60℃ 20秒間,72℃ 45秒間/9サイクル;98℃,10秒間,72℃ 60秒間/24サイクル;72℃,10分間/1サイクル;4℃/ホールドの下で行った。PCR反応物を、1.0%TAEアガロースゲル中での電気泳動によって分析した。
【0198】
期待されるPCR産物を、2.3kbpのDNAフラグメント(PAT PTU)の存在によって診断した。このフラグメントを切り出し、QIAQUICK(商標)ゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従ってゲルから精製した。次いで、精製したフラグメントを、ゼロブラントTOPO PCRクローニングキットおよび化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、pCR−ブラントII−TOPOベクター内にクローニングした。
【0199】
個々のコロニーを選び、プラスミドDNAを単離し、かつプラスミドDNA制限酵素消化および配列分析にかけた。クローン化したPATインサートをシークエンシングして、クローン化したPCR産物(配列番号:116)の同一性および配列忠実性を証明した。1つのそのようなプラスミドクローンをpDAB104107と称し(図9)、続いて新しいZp15相同ドナーベクター内への挿入のためのPAT遺伝子カセットの供給源として用いた。
【0200】
2.3kbpのPAT遺伝子フラグメントを、MfeIを用いた消化によってpDAB104107から回収し、その後にゲル電気泳動、切り出し、および精製が続いた。Zp15相同ドナープラスミドpDAB104101もMfeIで消化し、ゲルから精製した。pDAB104101内へのPAT遺伝子フラグメントのライゲーションによって、PAT遺伝子が、示差的制限酵素消化によって決定される、Zp15遺伝子配列に対する2つの方向のいずれかにあるMfeI部位に挿入されているクローンが産生された。pDAB104104(図10)は、Zp15遺伝子と同じ転写方向に挿入されたPAT遺伝子を含んでいた。pDAB104105(図11)は、Zp15遺伝子に対して反対方向に挿入されたPAT遺伝子を含んでいた。
【0201】
B.さらなるドナーDNA分子の構築
pDAB7489内のZp15 3’相同配列を切り取ることによって改変し、一方でpDAB7489内のZp15 5’相同配列は同じ状態のままである、Zp15に相同な領域を含有する別のドナー構築物を作製した。切り取った3’相同領域を、Integrated DNA Technologies社(Coralville,IA)によって合成されたプライマー:
DC003 5’−CTAATCGTCGACTCGTCAAGCCCCGCCTTTAAAT−3’(配列番号:117)
DC004 5’ −CTAATCCAATTGGTGTGGGCAGCCGAGCG−3’(配列番号:118)
を用いてPCRによってpDAB7489から作製した。
【0202】
PCR増幅反応を、PHUSIONホットスタートDNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,72℃ 15秒間/33サイクル;72℃,5分間/1サイクル;4℃/ホールドの下で行った。PCR反応物を、1.0%TAEアガロースゲル中での電気泳動にかけた。
【0203】
期待されるPCR産物を、0.8kbpのDNAフラグメント(Zp15 3’相同)の存在によって診断した。このフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、精製したフラグメントを、ゼロブラントTOPO PCRクローニングキットおよび化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR−ブラントII−TOPOベクター内にクローニングした。
【0204】
個々のコロニーを選び、プラスミドDNAの単離および制限酵素消化による確認にかけた。Zp15 3’相同インサートをシークエンシングして、クローン化したPCR産物(配列番号:119)の同一性および配列忠実性を証明した。1つのそのようなプラスミドクローンをpDAB104106と称し(図12)、続いてpDAB7489内への置換のための新しいZp15 3’相同配列の供給源として用いて、新しいZp15相同ドナーベクターを作出した。
【0205】
pDAB7489をMfeIおよびSalIで連続して消化し、4.2kbpのベクターフラグメントをゲルから精製した。pDAB104106もMfeIおよびSalIで消化し、切り取られたZp15 3’相同配列を含む0.8kbpのフラグメントをゲルから精製した。これらのゲルから精製したフラグメントのライゲーションおよび形質転換によって、切り取ったZp15 3’相同配列でオリジナルのZp15 3’相同配列5が置換されたクローンが産生された。このプラスミドをpDAB104100と称し(図13)、PAT遺伝子のレシピエントとして用いた。
【0206】
PAT遺伝子を、MfeIを用いた消化によってpDAB104107から取り出した。ゲル電気泳動の後、2.3kbpのPAT遺伝子フラグメントをゲルから精製した。Zp15相同ドナープラスミドpDAB104100もMfeIで消化し、ゲルから精製した。pDAB104100内へのPAT遺伝子のライゲーションによって、PAT遺伝子が、示差的制限酵素消化によって決定される、Zp15遺伝子に対する2つの方向のいずれかにあるMfeI部位に挿入されているクローンが産生された。pDAB104103(図14)は、Zp15遺伝子と同じ転写方向に挿入されたPAT遺伝子を含んでいた。pDAB104102(図15)は、Zp15遺伝子に対して反対方向に挿入されたPAT遺伝子を含んでいた。
【0207】
C.トウモロコシの形質転換およびZp15標的PAT挿入の回収
トウモロコシ亜種Hi−IIの胚発生細胞培養物(Armstrongら,(1991)Maize Genet Coop Newsletter 65:92−93)を作製し、維持し、かつZFN24およびドナー分子をコードするプラスミドの同時形質転換にかけた。ドナー分子には、ここに記載されるもの;pDAB104102、pDAB104103、pDAB104104、およびpDAB104105が含まれる。カルス組織の形質転換および選択、ならびにその後の形質転換体の再生は、米国特許出願第12/001,939号、特に実施例19に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。形質転換および選択のプロトコールに関するさらなるガイダンスのために、Petolinoら,(2000)Plant Cell Rept.19:781−786を参照されたい。開花後、植物を自己授粉またはDAS5XH751等のトウモロコシ亜種と異種交配させることができる。結果として生じた後代種子を収穫かつ乾燥させることができ、これらの種子由来の植物を分析して、標的組込み事象の遺伝性を証明することができる。無傷の、稔性のトウモロコシ植物へのカルスの再生は、米国特許出願第12/001,939号、特に実施例22に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。
【0208】
D.Zp15を標的としたPAT挿入の同定
形質転換したカルス組織におけるZp15を標的としたPAT挿入を、PCRによって検出する。テンプレートゲノムDNAを、周知かつ植物DNEASYキット(QIAGEN社,Valencia,CA)等のよく用いられる方法、またはDellaporta(Dellaportaら,(1983)Plant Mol.Biol.Rep.1;19−21)の方法によって、カルス組織から抽出する。Zp15遺伝子座内へのAAD−1標的組込みを検出するためにすでに用いられているPAT特異的プライマーとZp15隣接配列プライマーとの併用によって、5’および3’Zp15相同領域におけるPAT標的挿入接合部の増幅がもたらされる。前記PAT特異的プライマーは、:
DC013 5’−CAATCGTAAGCGTTCCTAGCCTTCCAG−3’ (配列番号:120)
DC014 5’ −CTGGAAGGCTAGGAACGCTTACGATTG−3’(配列番号:121)
であってよい。具体的には、ドナーDNAがZp15遺伝子に対して直列方向(direct orientation)にPAT遺伝子を有する場合、ドナーDNA 5’Zp15相同配列に隣接するゲノム領域内のプライマーHB501fまたはHB503fを、PATタンパク質コード領域内のプライマーDC013(配列番号:120)とともに用いて、PAT−Zp15 5’インサート接合部を検出する。同様に、ドナーDNAがZp15遺伝子に対して非直列方向(indirect orientation)にPAT遺伝子を有する場合、プライマーHB501fまたはHB503fを、PATタンパク質コード領域内のプライマーDC014(配列番号:121)とともに用いて、PAT−Zp15 5’インサート接合部を検出する。ドナーDNAがZp15遺伝子に対して非直列方向にPAT遺伝子を有する場合、PAT−Zp15 3’インサート接合部の検出のために、プライマーHB502rまたはHB504rをプライマーDC013(配列番号:120)とともに用いて、インサート接合部を検出する。同様に、ドナーDNAがZp15遺伝子に対して直列方向にPAT遺伝子を有する場合、プライマーHB502rまたはHB504rをプライマーDC014(配列番号:121)とともに用いて、PAT−Zp15 3’インサート接合部を検出する。
【0209】
PCR増幅反応を、PHUSIONホットスタートDNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,72℃ 15秒間/33サイクル;72℃,5分間/1サイクル;4℃/ホールドの下で行う。TAEアガロースゲル電気泳動を用いて、PCR産物を分解(resolved)かつ同定する。種々のPAT−Zp15ドナーDNAを用いることによって作製されたトランスジェニックカルスにおける、PAT−Zp15標的組込み事象由来のPCR産物に対して期待されるゲルフラグメントの大きさは、以下のとおり:
HB501f/HB503f+DC013(5’)=2.6kbp(pDAB104103),2.6kbp(pDAB104104)
HB501f/HB503f+DC014(5’)=1.6kbp(pDAB104105),1.7kbp(pDAB104102)
HB502r/HB504r+DC013(3’)=3.2kbp(pDAB104105),3.2kbp(pDAB104102)
HB502r/HB504r+DC014(3’)=1.2kbp(pDAB104103),1.2kbp(pDAB104104)
である。
【0210】
5’および3’PAT−Zp15標的組込み接合部を含むPCR産物を、当業者に既知の標準的方法を用いることによってクローニングおよびシークエンシングする。例えば、PCR産物をアガロースゲルから精製し、TOPOブラントクローニング(登録商標)キットおよび化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR−ブラントII−TOPOプラスミド内にクローニングする。
【0211】
次いで、クローン化した組込み接合部をシークエンシングして、ドナー形質転換ベクター内に組み入れられている5’および3’Zp15相同配列を介した相同組換えによって、PAT遺伝子がZp15遺伝子座においてトウモロコシゲノム内に挿入されたことを証明する。TOPOベクター特異的プライマーM13正方向およびM13逆方向に加えて、PAT遺伝子カセット特異的プライマーを用いて、標的組込みクローンの完全配列を得る。PATタンパク質コード配列に特異的であるPCRプライマーの配列番号:120および配列番号:121は、シークエンシングプライマーとしても用いられる。さらに、他のプライマーもシークエンシングに用いることができる。これらには、PATカセットのイネアクチンプロモーターエレメントに特異的であるもの:
DC−S1 5’−CCAACTGGACAATAGTCTCCAC−3’(配列番号:122)
DC−S2 5’−CATCGCCACTATATACATACC-3’(配列番号:123)
およびPATタンパク質コード配列に特異的であるもの:
DC−S3 5’−CGTCTCAATGTAATGGTTAACG−3’(配列番号:124)
DC−S4 5’−GCCCAGCGTAAGCAATACCAG−3’(配列番号:125)
を含むが、これに制限されない。
【0212】
実施例6:Zp15遺伝子座におけるAAD−1標的組込みの遺伝性
Zp15遺伝子座を標的とするAAD−1遺伝子カセットを保持するトランスジェニックカルス事象を作出した(事象138)。事象138のT0植物を再生し、雌性植物としてDAS5XH751雄性植物と交配させた。結果として生じたT1種子を植え付け、T1植物を成長させた。
【0213】
T1植物をPCRによって分析して、AAD−1−Zp15標的組込みの発生を証明した。PCR増幅反応を、PHUSIONホットスタートDNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,72℃ 15秒間/33サイクル;72℃,5分間/1サイクル;4℃/ホールドの下で行った。PCR反応物を、1.0%TAEアガロースゲル中での電気泳動によって分析した。ゲノムDNAをT1植物から抽出し、AAD−1 Zp15 5’組込み接合部を検出するように設計されたネストプライマーを用いたPCR反応におけるテンプレートDNAとして用いた。カルス事象147の分析で以前用いた同じプライマーを用いて、プライマリーおよびセカンドPCR反応を行った。プライマリーPCRに対し、プライマーHB501fおよびHB507rを用いた。プライマリーPCRによって、2.2kbpの期待される大きさにバンドが生じた。プライマリーPCR反応物のアリコートを1:100希釈し、ネストプライマーHB503fおよびHB508rを用いたセカンドPCRにおけるテンプレートとして用いた。セカンドPCRによっても、2.2kbpの期待される大きさにバンドが生じた。
【0214】
2.2kbpのセカンドPCR産物を、ゼロブラントTOPO PCRクローニングキットおよび化学的にコンピテントな大腸菌細胞であるワンショットTOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR−ブラントII−TOPO内にクローニングした。クローン化したDNAを、隣接するベクター特異的プライマー(M13正方向およびM13逆方向)を用いてシークエンシングした。配列は、Zp15遺伝子座におけるAAD−1の標的組込みに期待されるものと同一であることが判明した。
【0215】
本明細書において言及されるすべての特許、特許出願、および刊行物は、参照することによりあらゆる目的でそれらの全体として本明細書によって組み入れられているものとする。
【0216】
明確な理解のために、図解および実施例によって多少詳しく開示が提供されているが、本開示の精神または範囲から逸脱することなく、多様な変化および修正を実施し得ることは当業者に明白であろう。したがって、前述の記載および実施例は制限するものとして解釈されるべきでない。
【技術分野】
【0001】
連邦支援の研究の下でなされた発明に対する権利の声明
適用されない。
【0002】
本開示は、植物ゲノム工学、特に植物Zp15遺伝子内への導入遺伝子の標的組込みの分野にある。
【背景技術】
【0003】
バイオテクノロジーは、食糧生産に対して増大する世界的需要の課題に対処するために不可欠なツールとして台頭してきた。農業の生産性を向上させるための従来のアプローチ、例えば収穫量の増大または遺伝子操作による害虫抵抗性は、突然変異育種または形質転換による作物種のゲノム内への新規遺伝子の導入のいずれかに依存している。どちらの方法も本質的に非特異的であり、かつ比較的非効率的である。例えば、従来の植物形質転換法は、ゲノム内のランダムな位置に組み込む外因性DNAを送達する。したがって、所望の特性を備えた形質転換株を同定かつ単離するために、何千もの固有のランダム組込み事象を作出し、その後所望の個体をスクリーニングする必要がある。結果として、従来の植物特質の遺伝子操作は、労力を要し、多大な時間を要し、かつ予測不可能な仕事である。さらに、この組込みのランダム性によって、意図しないゲノム破壊による多面発現効果が生じているかどうかを予測するのが難しくなる。結果として、遺伝子操作された遺伝子または特質を備えた植物株の作出、単離、および特徴付けは、成功の確率が低く、極めて労力を要し、かつコストの大きい手法となっている。
【発明の概要】
【発明が解決しようとする課題】
【0004】
標的遺伝子改変は、植物システムにおける従来的実施の合理的な課題を克服するものであり、ゆえに基礎的な植物生物学の研究および農業バイオテクノロジーのどちらにおいても、長年にわたる、しかしとらえどころのない目標である。しかしながら、イネにおける陽性−陰性の薬剤選択または事前に遺伝子操作された制限酵素部位の使用による「遺伝子ターゲティング」を除いて、すべての植物種における標的ゲノム改変は、モデルでも農作物でも、最近まで非常に困難であることが分かっている。Teradaら,(2002)Nat Biotechnol 20(10):1030;Teradaら,(2007)Plant Physiol 144(2):846;D’Halluinら,(2008)Plant Biotechnology J.6(1):93。
【0005】
最近、ゲノムDNAの標的切断のための方法および組成物が記載されている。そのような標的切断の事象を用いて、例えば、標的突然変異生成を誘発すること、細胞DNA配列の標的欠失を誘導すること、ならびに所定の染色体座位での標的組換えを容易にすることが可能である。例えば、米国特許公報第20030232410号;第20050208489号;第20050026157号;第20050064474号;および第20060188987号,ならびに国際公報WO2007/014275を参照されたく、その開示は参照することによりあらゆる目的でそれらの全体として組み入れられているものとする。米国特許公報第20080182332号は植物ゲノムの標的改変のための非標準ジンクフィンガーヌクレアーゼ(ZFN)の使用について記載しており、また米国特許出願第12/284,888号は植物EPSPS遺伝子座内へのZFNを介した標的組込みについて記載している。
【0006】
しかしながら、植物およびその後代における安定的で遺伝的な遺伝子改変を確立するために、植物ゲノム内のさらなる遺伝子座内への安定的な標的組込みのための組成物および方法の必要性が依然として残っている。
【課題を解決するための手段】
【0007】
本開示は、植物細胞におけるZp15遺伝子内に組み込まれている外因性核酸配列の1つ以上の産物(すなわち、タンパク質またはRNA分子)を発現させるための方法および組成物を提供する。本明細書に示されるように、Zp15遺伝子座におけるまたは近隣における1つ以上の外因性配列の組込みによって、宿主植物は再生、開花、または種をつける能力を損なわないと考えられ、場合によっては、世代にわたった外因性配列の遺伝的伝達が可能になる。前記外因性核酸配列は、例えば、1つ以上の遺伝子もしくはcDNA分子、または任意の種類のコードもしくは非コード配列、および1つ以上の制御エレメント(例えば、プロモーター)を含み得る。例えば、除草剤耐性遺伝子をこの遺伝子座内に組み込んで、所望の除草剤抵抗性を備えた作物を生産することができる。Zp15遺伝子座においてまたは近隣において外因性核酸を含有する細胞は、配偶子(生殖細胞系)にも寄与し得、ゆえに次世代の後代に伝達され得る。
【0008】
Zp15遺伝子内への外因性核酸配列の取込みは、選択されたZp15遺伝子座におけるゲノムの標的二本鎖切断によって容易になる。切断は、選択されたZp15遺伝子座内の配列に結合するように遺伝子操作された、例えば、メガヌクレアーゼDNA結合ドメイン、ロイシンジッパーDNA結合ドメイン、ジンクフィンガータンパク質(ZFP)、または前述のキメラ組み合わせ等のDNA結合ドメイン、および切断ドメインまたは切断ハーフドメインを含む融合タンパク質を用いることによってZp15遺伝子を標的とする。このような切断によって、切断部位におけるまたは近隣における外因性ポリヌクレオチド配列の組込みが刺激される。外因性配列の組込みは、相同性依存的および相同性非依存的メカニズムの両方によって進行し得る。
【0009】
一実施形態において、Zp15遺伝子における標的部位に結合する遺伝子操作されたDNA結合ドメイン(例えば、ZFP、メガヌクレアーゼ、またはロイシンジッパー)を本明細書において開示する。前記DNA結合ドメインは、例えば、表1に示した認識ヘリックスを含む任意の遺伝子操作されたジンクフィンガーDNA結合ドメインを含み得る。本明細書に記載される任意のDNA結合ドメインは、機能ドメイン、例えば切断ドメインまたは切断ハーフドメインをさらに含んでもよい。いくつかの実施態様では、前記切断ハーフドメインは、FokIまたはStsI等のIIS型制限エンドヌクレアーゼに由来するものであり得る。他の実施態様では、前記切断ドメインは、ホーミングエンドヌクレアーゼ、例えば改変されたDNA結合ドメインを有するホーミングエンドヌクレアーゼを含み得る。
【0010】
別の実施形態において、Zp15遺伝子座内に組み込まれた外因性配列を含む植物または種子を本明細書において開示する。ある実施態様では、前記外因性配列を植物の配偶子内に組み込む。
【0011】
別の実施形態において、細胞内で外因性核酸配列の産物を発現させるための方法であって、前記方法は:(a)前記細胞内で第一の融合タンパク質を発現させる工程であって、前記第一の融合タンパク質は第一のDNA結合ドメイン(例えば、ZFP)および第一の切断ハーフドメインを含んでおり、前記DNA結合ドメインは前記細胞のゲノムのZp15遺伝子における第一の標的部位に結合するように遺伝子操作されている工程;(b)前記細胞内で第二の融合タンパク質を発現させる工程であって、前記第二の融合タンパク質は第二のDNAドメインおよび第二の切断ハーフドメインを含んでおり、前記第二のDNAドメインは前記細胞のゲノムのZp15遺伝子における第二の標的部位に結合し、前記第二の標的部位は前記第一の標的部位とは異なる工程;ならびに(c)前記細胞を外因性核酸配列およびZp15遺伝子における第一の配列に相同である第一のヌクレオチド配列を含むポリヌクレオチドと接触させる工程であって、前記第一の融合タンパク質の前記第一の標的部位への結合、および前記第二の融合タンパク質の前記第二の標的部位への結合により、前記切断ハーフドメインは前記細胞のゲノムがZp15遺伝子内で切断されるように位置し、それによってZp15遺伝子における前記細胞のゲノム内への外因性配列の組込みおよび前記外因性配列の産物の発現がもたらされる工程;を含む方法を本明細書において開示する。
【0012】
前記外因性核酸配列は、植物に所望の特質を付与する、1つ以上のプロモーターの有無にかかわらず、1つ以上の機能性ポリペプチド(例えば、cDNA)をコードする配列を含んでいてもよく、かつ/あるいは1つ以上のRNA配列を(例えば、1つ以上のshRNA発現カセットによって)産生してもよい。そのような特質には、除草剤抵抗性または耐性;虫害抵抗性または耐性;病害抵抗性または耐性(ウイルス性、細菌性、菌性、線虫による);乾燥、熱、低温、凍結、過度の湿気、塩分ストレスに対する抵抗性または耐性などのストレス耐性および/または抵抗性;酸化ストレス;収穫量の増加;食物の含有量および構成;見た目;雄性不稔;ドライダウン;耐倒伏性;多産;デンプンの量および質;油分の量および質;タンパク質の質および量;アミノ酸組成等が含まれるが、これに制限されない。もちろん、除草剤、害虫、病害(ウイルス性、細菌性、菌性、線虫による)または乾燥抵抗性、雄性不稔、ドライダウン、耐倒伏性、多産、デンプンの性質、油分の量および質を与えるもの、あるいは収穫量または栄養価を増加させるもの等の、任意の記載の任意の2つ以上の外因性核酸を所望どおりに利用してよい。ある実施態様では、前記核酸配列は、除草剤抵抗性タンパク質(例えば、AAD−1(アリールオキシアルカノエート・ジオキシゲナーゼ)遺伝子、AAD−12遺伝子、またはホスフィノトリシン・アセチル・トランスフェラーゼ(PAT)遺伝子)および/またはその機能性フラグメントをコードする配列を含む。組み込まれた配列の発現は、前記組み込まれた配列に操作可能に連結されたプロモーターによって促進され得る。あるいは、組み込まれた配列にはプロモーターがなく、転写は内在性Zp15プロモーターによって促進される。
【0013】
ある実施態様では、前記ポリヌクレオチドはZp15遺伝子における第二の配列に相同である第二のヌクレオチド配列をさらに含む。前記第二のヌクレオチド配列は、Zp15遺伝子における前記第二の配列と同一であってもよい。さらに、第一および第二のヌクレオチド配列を含む実施態様では、前記第一のヌクレオチド配列はZp15遺伝子における第一の配列と同一であってよく、かつ前記第二のヌクレオチド配列はZp15遺伝子における第二の配列に相同であるが同一でないものであってよい。本明細書に記載される方法のいずれかにおいて、前記第一および第二のヌクレオチド配列は、外因性配列に隣接する。ある実施態様では、前記ポリヌクレオチドはプラスミドである。他の実施態様では、前記ポリヌクレオチドは線状のDNA分子である。
【0014】
別の実施形態において、細胞のゲノムにおけるZp15遺伝子内に外因性配列を組み込むための方法であって、前記方法は:(a)前記細胞内で第一の融合タンパク質を発現させる工程であって、前記第一の融合タンパク質は第一のDNA結合ドメイン(例えば、ZFP)および第一の切断ハーフドメインを含んでおり、前記第一のDNA結合ドメインは前記細胞のゲノムにおけるZp15遺伝子座における第一の標的部位に結合するように遺伝子操作されている工程;(b)前記細胞内で第二の融合タンパク質を発現させる工程であって、前記第二の融合タンパク質は第二のDNA結合ドメイン(例えば、ZFP)および第二の切断ハーフドメインを含んでおり、前記第二のDNA結合ドメインは前記細胞のゲノムにおけるZp15遺伝子座における第二の標的部位に結合し、前記第二の標的部位は前記第一の標的部位とは異なる工程;ならびに(c)前記細胞を外因性核酸配列を含むポリヌクレオチドと接触させる工程であって、前記第一の融合タンパク質の前記第一の標的部位への結合、および前記第二の融合タンパク質の前記第二の標的部位への結合により、前記切断ハーフドメインは前記細胞のゲノムがZp15遺伝子座内で切断されるように位置し、それによってZp15遺伝子座内の前記細胞のゲノム内への外因性配列の相同性依存的組込みがもたらされる工程;を含む方法を本明細書において提示する。ある実施態様では、機能性ポリペプチドをコードする外因性配列をZp15遺伝子内に挿入する。
【0015】
本明細書に記載される方法のいずれかにおいて、前記第一および第二の切断ハーフドメインは、IIS型制限エンドヌクレアーゼ、例えばFokIまたはStsIに由来するものであり得る。さらに、本明細書に記載される方法のいずれかにおいて、少なくとも1つの融合タンパク質は、例えば前記切断ハーフドメインの必須のヘテロ二量体が形成されるような、前記切断ハーフドメインの二量化接触面のアミノ酸配列の変更を含み得る。
【0016】
本明細書に記載される方法のいずれかにおいて、植物細胞は単子葉性または双子葉性の植物細胞を含み得る。ある実施態様では、前記植物細胞は作物、例えばトウモロコシである。
【図面の簡単な説明】
【0017】
【図1】図1は、ZFN対#25の一過性の発現を受けた細胞に由来するトウモロコシHiII gDNAからのZp15増幅産物についての例示的配列分析結果を表し(結合部位に下線)、期待される切断部位における6bpのNHEJ挿入(太字)を示す。
【図2】図2は、ZFN対#24の一過性の発現を受けた細胞に由来するトウモロコシHiII gDNAからのZp15増幅産物についての例示的配列分析結果を表し(結合部位に下線)、期待される切断部位における3bpの欠失を示す。
【図3】図3は、pDAB7489と称される構築物を図示した概略図である。
【図4】図4は、AAD遺伝子をコードする例示的な除草剤耐性遺伝子発現カセットを図示した概略図である。
【図5】図5は、pDAB7490と称される構築物を図示した概略図である。
【図6−1】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−2】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−3】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−4】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−5】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−6】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−7】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図6−8】図6は、トウモロコシ野生型(WT)、Zp15ドナーフラグメント、ならびに組み込まれたドナー配列に隣接している5’および3’境界領域を並べた、標的組込み(TI)事象のアラインメントを表す。
【図7】図7は、pDAB104101と称される構築物を図示した概略図である。
【図8】図8は、PAT発現カセットを図示した概略図である。
【図9】図9は、pDAB104107と称される構築物を図示した概略図である。
【図10】図10は、pDAB104104と称される構築物を図示した概略図である。
【図11】図11は、pDAB104105と称される構築物を図示した概略図である。
【図12】図12は、pDAB104106と称される構築物を図示した概略図である。
【図13】図13は、pDAB104100と称される構築物を図示した概略図である。
【図14】図14は、pDAB104103と称される構築物を図示した概略図である。
【図15】図15は、pDAB104102と称される構築物を図示した概略図である。
【発明を実施するための形態】
【0018】
詳細な説明
本開示は、トウモロコシの第6染色体上に位置する植物Zp15遺伝子内への標的組込み(TI)のための方法および組成物に関する。DNA結合ドメイン(例えば、ZFP、メガヌクレアーゼ、またはロイシンジッパー)およびヌクレアーゼドメインを含む融合タンパク質を用いることによって、挿入された(ドナー)配列を外因性プロモーターに操作可能に連結させることができ、またはプロモーターがなくてもよい。プロモーターがない場合、組み込まれたオープン・リーディング・フレームの転写は、プロモーター特定の組織における内在性Zp15遺伝子プロモーターによって生じ得る。プロモーターがないドナーの使用によって、前記ドナーのランダムな組込みの可能性および/または前記ドナー上に保持されたプロモーターによる内在性遺伝子の疑似活性化は低下する。
【0019】
Zp15遺伝子内への標的切断および組換えに有用な組成物は、切断ドメイン(または切断ハーフドメイン)とDNA結合ドメイン(例えば、ZFP)とを含む融合タンパク質、これらのタンパク質をコードするポリヌクレオチド、およびポリペプチドとポリペプチドをコードするポリヌクレオチドとの組み合わせを含有する。ジンクフィンガー結合ドメインは、1つ以上のジンクフィンガー(例えば、2、3、4、5、6、7、8、9、またはそれ以上のジンクフィンガー)を含んでいてよく、かつZp15遺伝子内の任意の配列に結合するように遺伝子操作されていてもよい。細胞内におけるそのような融合タンパク質の存在は、前記融合タンパク質のその(それらの)結合部位への結合および内在性Zp15遺伝子内での切断をもたらす。
【0020】
概要
当該方法の実施、ならびに本明細書に開示される組成物の調製および使用は、特に指定のない限り、分子生物学、生化学、クロマチン構造および分析、計算化学、細胞培養、組換えDNA、ならびに当該技術分野の技術の範囲内にある関連分野における従来技術を利用する。これらの技術は文献で十分に説明されている。例えば、Sambrookら,MOLECULAR CLONING:A LABORATORY MANUAL,Second edition,Cold Spring Harbor Laboratory Press,1989およびThird edition,2001;Ausubelら,CURRENT PROTOCOLS IN MOLECULAR BIOLOGY,John Wiley&Sons,New York,1987および定期的に更新したもの;the series METHODS IN ENZYMOLOGY,Academic Press,San Diego;Wolffe,CHROMATIN STRUCTURE AND FUNCTION,Third edition,Academic Press,San Diego,1998;METHODS IN ENZYMOLOGY,Vol.304,“Chromatin”(P.M.WassarmanおよびA.P.Wolffe,eds.),Academic Press,San Diego,1999;ならびにMETHODS IN MOLECULAR BIOLOGY,Vol.119,“Chromatin Protocols”(P.B.Becker,ed.)Humana Press,Totowa,1999を参照されたい。
【0021】
定義
「核酸」、「ポリヌクレオチド」、および「オリゴヌクレオチド」という用語は、代替可能に用いられ、線状または環状の構造で、かつ一本鎖または二本鎖のいずれかの形態のデオキシリボヌクレオチドまたはリボヌクレオチドのポリマーを表す。本開示上、これらの用語は、ポリマーの長さについて制限するものとして解釈されるべきではない。前記用語は、天然ヌクレオチドの既知の類似体、ならびに塩基、糖および/またはリン酸塩部分(例えば、ホスホロチオエート骨格)において改変されているヌクレオチドを包含し得る。一般に、特定のヌクレオチドの類似体は同じ塩基対合特異性を有し、すなわちAの類似体はTと塩基対合する。
【0022】
「ポリペプチド」、「ペプチド」、および「タンパク質」という用語は、アミノ酸残基のポリマーを表すために代替可能に用いられる。前記用語はまた、1つ以上のアミノ酸が化学的類似体または対応する天然に存在しているアミノ酸の改変された誘導体であるアミノ酸ポリマーに適用される。
【0023】
「結合」とは、高分子間(例えば、タンパク質と核酸との間)の配列特異的、非共有相互作用を表す。全体としての相互作用が配列特異的であるならば、結合相互作用のすべての構成成分が配列特異的である(例えば、DNA骨格におけるリン酸残基と接触する)必要はない。そのような相互作用は、通常、10-6M-1以下の解離定数(Kd)によって特徴付けされる。「親和性」とは結合の強度を表し、結合親和性の増大はより低いKdと相関している。
【0024】
「結合タンパク質」とは、別の分子に非共有的に結合し得るタンパク質である。結合タンパク質は、例えば、DNA分子(DNA結合タンパク質)、RNA分子(RNA結合タンパク質)、および/またはタンパク質分子(タンパク質結合タンパク質)に結合し得る。タンパク質結合タンパク質の場合、それはそれ自体に結合し得(ホモ二量体、ホモ三量体等を形成する)、かつ/またはそれは1つ以上の異なるタンパク質の分子に結合し得る。結合タンパク質は、1種類より多い結合活性を有し得る。例えば、ジンクフィンガータンパク質は、DNA結合、RNA結合、およびタンパク質結合活性を有する。
【0025】
「ジンクフィンガーDNA結合タンパク質」(または結合ドメイン)とは、亜鉛イオンの配位によって構造が安定化する結合ドメイン内のアミノ酸配列の領域である、1つ以上のジンクフィンガーを介した配列特異的な様式でDNAに結合するタンパク質またはより大きなタンパク質内のドメインである。ジンクフィンガーDNA結合タンパク質という用語は、しばしば、ジンクフィンガータンパク質またはZFPと略記される。
【0026】
ジンクフィンガー結合ドメインを、所定のヌクレオチド配列に結合するように「遺伝子操作」することができる。ジンクフィンガータンパク質を遺伝子操作するための方法の制限されない例は、設計および選択である。設計されたジンクフィンガータンパク質とは、主に合理的基準に由来する設計/組成を有する天然には存在していないタンパク質である。設計のための合理的基準には、置換法則の適用、ならびに既存のZFP設計および結合データの情報を蓄積しているデータベース内の情報を処理するためのコンピュータアルゴリズムが含まれる。例えば、米国特許第6,140,081号;第6,453,242号;および第6,534,261号を参照されたく、またWO98/53058;WO98/53059;WO98/53060;WO02/016536およびWO03/016496も参照されたい。
【0027】
「選択された」ジンクフィンガータンパク質とは、ファージディスプレイ、相互作用トラップ、またはハイブリッド選択等の実験に基づいた手法に主に由来して産生される天然には見られないタンパク質である。例えば、米国第5,789,538号;米国第5,925,523号;米国第6,007,988号;米国第6,013,453号;米国第6,200,759号;WO95/19431;WO96/06166;WO98/53057;WO98/54311;WO00/27878;WO01/60970;WO01/88197、およびWO02/099084を参照されたい。
【0028】
「配列」という用語は、DNAまたはRNAであってよく;線状、環状、または分岐したものであってよく;かつ一本鎖または二本鎖のいずれかであってよい、任意の長さのヌクレオチド配列を表す。「ドナー配列」という用語は、ゲノム内に挿入されたヌクレオチド配列を表す。ドナー配列は任意の長さであってよく、例えば、2〜10,000ヌクレオチド長の間(またはその間のもしくはそれを上回る任意の整数値)、好ましくは約100〜1,000ヌクレオチド長の間(またはその間の任意の整数)、より好ましくは約200〜500ヌクレオチド長の間である。
【0029】
「相同であり、同一でない配列」とは、第二の配列とある程度の配列同一性を共有するが、その配列は前記第二の配列のものとは同一ではない第一の配列を表す。例えば、変異体遺伝子の野生型配列を含むポリヌクレオチドは、前記変異体遺伝子の配列に相同であり、かつ同一ではない。ある実施態様では、前記2つの配列間の相同性の程度は、通常の細胞メカニズムを利用することによってそれらの間で相同組換えが起こるのに十分である。2つの相同であり同一でない配列は任意の長さであってよく、その非相同性の程度は、1ヌクレオチド程度に小さくても(例えば、標的相同組換えによるゲノムの点変異の修正のための)または10キロベース以上程度に大きくてもよい(例えば、染色体の所定の異所性部位における遺伝子の挿入のための)。相同であり同一でない配列を含む2つのポリヌクレオチドは、同じ長さである必要はない。例えば、20〜10,000ヌクレオチドまたはヌクレオチド対の間の外因性ポリヌクレオチド(すなわち、ドナーポリヌクレオチド)を用いることができる。
【0030】
核酸およびアミノ酸の配列同一性を決定するための技術は、当該技術分野において既知である。典型的に、そのような技術には、遺伝子に対するmRNAのヌクレオチド配列を決定すること、および/またはそれによってコードされるアミノ酸配列を決定すること、およびこれらの配列を第二のヌクレオチドもしくはアミノ酸配列と比較することが含まれる。このやり方でゲノム配列を決定および比較することもできる。一般に、同一性とは、2つのポリヌクレオチドまたはポリペプチド配列の、それぞれヌクレオチド対ヌクレオチドまたはアミノ酸対アミノ酸の正確な対応を表す。2つ以上の配列(ポリヌクレオチドまたはアミノ酸)を、それらの同一性パーセントを決定することによって比較することができる。2つの配列の同一性パーセントは、核酸またはアミノ酸配列にかかわらず、2つの並んだ配列間の正確な一致をより短い配列の長さで割って、100をかけた数である。核酸配列のためのおおよそのアラインメントは、SmithおよびWatermanの局所相同性アルゴリズム、Advances in Applied Mathematics 2:482−489(1981)によって提供されている。このアルゴリズムを、Dayhoff,Atlas of Protein Sequences and Structure,M.O.Dayhoff ed.,5 suppl.3:353−358,National Biomedical Research Foundation,Washington,D.C.,USAによって開発され、Gribskov,Nucl.Acids Res.14(6):6745−6763(1986)によって正規化されたスコア行列を用いることによって、アミノ酸配列に適用することができる。配列の同一性パーセントを決定するためのこのアルゴリズムの例示的実施は、Genetics Computer Group(Madison,WI)による「BestFit」ユーティリティーアプリケーションにおいて提供されている。配列間の同一性または類似性パーセントを算出するのに適したプログラムは、当該技術分野において一般に既知であり、例えば、別のアラインメントプログラムはデフォルトパラメーターを用いて使用されるBLASTである。例えば、BLASTNおよびBLASTPを、以下のデフォルトパラメーター:遺伝子コード=標準;フィルター=なし;鎖=両方;カットオフ=60;期待数=10;行列=BLOSUM62;表示=50配列;分類=高スコア;データベース=非重複、GenBank+EMBL+DDBJ+PDB+GenBank CDS翻訳+Swissタンパク質+Spupdate+PIRを用いることによって使用することができる。これらのプログラムの詳細はインターネット上で見出すことができる。本明細書に記載される配列に関して、所望される配列同一性の程度の範囲は、およそ80%〜100%、およびその間の任意の整数値である。典型的には、配列間の同一性パーセントは、少なくとも70〜75%、好ましくは80〜82%、より好ましくは85〜90%、さらに好ましくは92%、なおいっそう好ましくは95%、および最も好ましくは98%の配列同一性である。
【0031】
あるいは、ポリヌクレオチド間の配列類似性の程度を、相同な領域間で安定した二重鎖が形成される条件下でのポリヌクレオチドのハイブリダイゼーション、続いて一本鎖特異的ヌクレアーゼを用いた消化、および消化されたフラグメントの大きさの決定によって決定することができる。2つの核酸または2つのポリペプチド配列は、配列が、前述の方法を用いることによって決定される、少なくとも約70〜75%、好ましくは80〜82%、より好ましくは85〜90%、さらに好ましくは92%、なおいっそう好ましくは95%、および最も好ましくは98%の配列同一性を当該分子の既定の長さにわたって示す場合、互いに実質的に相同である。本明細書において使用するとき、実質的に相同であるとは、特定のDNAまたはポリペプチド配列と完全な同一性を示す配列も表す。実質的に相同であるDNA配列を、特定のシステムに対して規定される、例えばストリンジェントな条件下でのサザンハイブリダイゼーション実験で同定することができる。適切なハイブリダイゼーション条件を規定することは、当該技術分野の技術の範囲内である。例えば、Sambrookら,前記参照;Nucleic Acid Hybridization:A Practical Approach,editors B.D.HamesおよびS.J.Higgins,(1985)Oxford;Washington,DC;IRL Pressを参照されたい。
【0032】
2つの核酸フラグメントの選択的ハイブリダイゼーションは、以下のように決定することができる。2つの核酸分子間の配列同一性の程度は、前記分子間のハイブリダイゼーション事象の効率および強度に影響を及ぼす。部分的に同一な核酸配列は、完全に同一な配列の標的分子へのハイブリダイゼーションを少なくとも部分的に阻害する。完全に同一な配列のハイブリダイゼーションの阻害を、当該技術分野において周知であるハイブリダイゼーションアッセイ(例えば、サザン(DNA)ブロット、ノーザン(RNA)ブロット、溶液ハイブリダイゼーション等、Sambrookら,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)を用いることによって評価することができる。そのようなアッセイは、様々な程度の選択性を用いることによって、例えば、低ストリンジェントから高ストリンジェントに変化する条件を用いることによって実施され得る。低ストリンジェントな条件を利用する場合、部分的な程度の配列同一性さえも欠いている第二のプローブ(例えば、標的分子と約30%未満の配列同一性を有するプローブ)を用いることによって、非特異的結合の事象がない場合、前記第二のプローブは標的にハイブリダイズしないというように、非特異的結合がないことを評価することができる。
【0033】
ハイブリダイゼーションに基づく検出システムを利用する場合、参照核酸配列に相補的である核酸プローブが選ばれ、次いで、適切な条件の選択によって前記プローブと前記参照配列が互いに選択的にハイブリダイズまたは結合して二重鎖分子を形成する。中程度にストリンジェントなハイブリダイゼーション条件下で、参照配列に選択的にハイブリダイズし得る核酸分子は、典型的に、選択された核酸プローブの配列と少なくともおよそ70%の配列同一性を有する少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にする条件下でハイブリダイズする。ストリンジェントなハイブリダイゼーション条件は、典型的に、選択された核酸プローブの配列と約90〜95%より高い配列同一性を有する少なくとも約10〜14ヌクレオチド長の標的核酸配列の検出を可能にする。プローブと参照配列が特定程度の配列同一性を有する場合、プローブ/参照配列のハイブリダイゼーションに有用なハイブリダイゼーション条件を、当該技術分野において既知であるように決定することができる(例えば、Nucleic Acid Hybridization:A Practical Approach,editors B.D.HamesおよびS.J.Higgins,(1985)Oxford;Washington,DC;IRL Pressを参照されたい)。
【0034】
ハイブリダイゼーションのための条件は当業者に周知である。ハイブリダイゼーションのストリンジェンシーとは、ハイブリダイゼーション条件が不一致のヌクレオチドを含有する合成物の形成を嫌う程度を表し、より高いストリンジェンシーは不一致の合成物に対するより低い許容度と相関する。ハイブリダイゼーションのストリンジェンシーに影響を及ぼす要素は当業者に周知であり、温度、pH、イオン強度、および例えばホルムアミドおよびジメチルスルホキシド等の有機溶媒の濃度を含むが、これに制限されない。当業者に既知であるように、ハイブリダイゼーションのストリンジェンシーは、より高い温度、より低いイオン強度、およびより低い溶媒濃度によって上昇する。
【0035】
ハイブリダイゼーションのためのストリンジェントな条件に関して、多数の同等の条件を利用し、例えば以下の要素:配列の長さおよび性質、種々の配列の塩基組成、塩および他のハイブリダイゼーション溶液成分の濃度、ハイブリダイゼーション溶液中における遮断物質の有無(例えば、硫酸デキストランおよびポリエチレングリコール)、ハイブリダイゼーションの反応温度、および時間パラメーター、ならびに様々な洗浄条件を変えることによって、特定のストリンジェンシーを確立し得ることが当該技術分野において周知である。特定のハイブリダイゼーション条件のセットの選択は、当該技術分野における以下の標準的方法に従うことによって選択される(例えば、Sambrookら,Molecular Cloning:A Laboratory Manual,Second Edition,(1989)Cold Spring Harbor,N.Y.を参照されたい)。
【0036】
「組換え」とは、2つのポリヌクレオチド間での遺伝情報の交換のプロセスを表す。本開示上、「相同組換え(HR)」とは、例えば細胞内で二本鎖断裂の修復の間に起こるような交換の特殊な形態を表す。このプロセスは、ヌクレオチドの配列相同性を必要とし、「標的」分子(すなわち、二本鎖断裂を受けたもの)のテンプレート修復に対する「ドナー」分子を用い、かつドナーから標的への遺伝情報の転移をもたらすため、「非交差型の遺伝子変換」または「短路の遺伝子変換」として様々に知られている。任意の特定の理論に束縛されることを望むことなく、そのような転移は、切断された標的とドナーとの間で形成するヘテロ二重鎖のミスマッチ修正、および/またはドナーを用いて標的の一部になるであろう遺伝情報を再合成する「合成依存的な鎖アニーリング」、および/または関連プロセスを伴い得る。そのような特殊なHRは、しばしば、ドナーポリヌクレオチドの配列の一部またはすべてが標的ポリヌクレオチド内に組み入れられるような、標的分子の配列の変更をもたらす。
【0037】
「切断」とは、DNA分子の共有結合骨格の断裂を表す。切断は、ホスホジエステル結合の酵素的または化学的な加水分解を含むが、これに制限されない種々の方法によって開始され得る。一本鎖切断および二本鎖切断のいずれも可能であり、二本鎖切断は2つの別個の一本鎖切断事象の結果として生じ得る。DNA切断によって、平滑末端または互い違いの末端の産生がもたらされ得る。ある実施態様では、標的二本鎖DNA切断に融合ポリペプチドを用いる。
【0038】
「切断ドメイン」には、DNA切断に対する触媒活性を持つ1つ以上のポリペプチド配列が含まれる。切断ドメインは1つのポリペプチド鎖内に含有されていてもよく、あるいは2つ(またはそれ以上)のポリペプチドの会合によって切断活性が生じてもよい。
【0039】
「切断ハーフドメイン」とは、第二のポリペプチド(同一または異なる)と連動して切断活性(好ましくは二本鎖切断活性)を有する複合体を形成するポリペプチド配列である。
【0040】
「クロマチン」とは、細胞ゲノムを含む核タンパク質構造体である。細胞クロマチンは、核酸、主にDNA、ならびにヒストンおよび非ヒストン染色体タンパク質を含むタンパク質を含む。大部分の真核細胞のクロマチンはヌクレオソームの形態で存在し、ヌクレオソームコアは、ヒストンH2A、H2B、H3、およびH4のそれぞれ2つを含む八量体と会合したおよそ150塩基対のDNA、ならびにヌクレオソームコア間に伸びる(生物に依存した可変長の)リンカーDNAを含む。ヒストンH1の分子は、通常、リンカーDNAと会合する。本開示上、「クロマチン」という用語は、原核生物および真核生物の両方のあらゆる種類の細胞内核タンパク質を包含することを意図される。細胞クロマチンは、染色体クロマチンおよびエピソーム性クロマチンの両方を含む。
【0041】
「染色体」とは、細胞のゲノムのすべてまたは一部を含むクロマチン複合体である。細胞のゲノムは、しばしば、細胞のゲノムを含むすべての染色体の集合体であるその核型によって特徴付けされる。細胞のゲノムは、1つ以上の染色体を含み得る。
【0042】
「エピソーム」とは、複製核酸、核タンパク質複合体、または細胞の染色体核型の一部でない核酸を含む他の構造物である。エピソームの例には、プラスミドおよびある種のウイルスゲノムが含まれる。
【0043】
「接近可能領域」とは、核酸中に存在する標的部位に前記標的部位を認識する外因性分子が結合し得る、細胞クロマチン内の部位である。任意の特定の理論に束縛されることを望むことなく、接近可能領域はヌクレオソーム構造内にパッケージされていないものであると考えられている。接近可能領域の独特の構造を、しばしば、化学的および酵素的プローブ、例えばヌクレアーゼに対するその感度によって検出できる。
【0044】
「標的部位」または「標的配列」とは、結合に十分な条件が存在するという条件で結合分子が結合するであろう核酸の部分を規定する核酸配列である。例えば、5’−GAATTC−3’という配列は、EcoRI制限エンドヌクレアーゼに対する標的部位である。
【0045】
「外因性」分子とは、通常細胞内に存在しないが、1つ以上の遺伝学的、生化学的、または他の方法によって細胞内に導入され得る分子である。「細胞内での通常の存在」とは、前記細胞の特定の発生段階および環境条件に対して決定される。したがって、例えば筋肉の胚発生の間だけ存在する分子は、成体の筋細胞に対して外因性分子である。同様に、ヒートショックによって誘発された分子は、ヒートショックされていない細胞に対して外因性分子である。外因性分子には、例えば、任意のポリペプチドまたはそのフラグメントに対するコード配列、機能不全である内在性分子の機能型、または通常に機能する内在性分子の機能不全型が含まれ得る。さらに、外因性分子には、宿主細胞の内在性遺伝子のオルソログである別の種由来のコード配列が含まれ得る。
【0046】
外因性分子は、とりわけ、コンビナトリアルケミストリーの手法によって生成されるような小分子、あるいはタンパク質、核酸、炭水化物、脂質、糖タンパク質、リポタンパク質、多糖、前記分子の任意の改変された誘導体、または1つ以上の前記分子を含む任意の複合体等の高分子であってよい。核酸にはDNAおよびRNAが含まれ、一本鎖または二本鎖であってよく、線状、分岐したもの、または環状であってよく、かつ任意の長さのものであってよい。核酸には、二重鎖を形成し得るもの、ならびに三重鎖形成核酸が含まれる。例えば、米国特許第5,176,996号および第5,422,251号を参照されたい。タンパク質には、DNA結合タンパク質、転写因子、クロマチン再構築因子、メチル化DNA結合タンパク質、ポリメラーゼ、メチラーゼ、デメチラーゼ、アセチラーゼ、デアセチラーゼ、キナーゼ、ホスファターゼ、インテグラーゼ、リコンビナーゼ、リガーゼ、トポイソメラーゼ、ジャイレース、およびヘリカーゼが含まれるが、これに制限されない。
【0047】
外因性分子は、内在性分子と同じ種類の分子、例えば外因性タンパク質または核酸であってよい。例えば、外因性核酸には、感染ウイルスゲノム、細胞内に導入されたプラスミドまたはエピソーム、あるいは細胞内に通常存在しない染色体が含まれ得る。細胞内に外因性分子を導入するための方法は、当業者に既知であり、脂質を介した転移(すなわち、中性およびカチオン性脂質を含むリポソーム)、エレクトロポレーション、直接注入、細胞融合、粒子砲撃、リン酸カルシウム共沈殿、ナノ粒子による形質転換、DEAE−デキストランを介した転移、およびウイルスベクターを介した転移を含むが、これに制限されない。
【0048】
一方、「内在性」分子とは、特定の環境条件下での特定の発生段階において特定の細胞内に通常存在するものである。例えば、内在性核酸には、染色体、ミトコンドリアのゲノム、葉緑体または他の細胞内小器官、あるいは天然に存在しているエピソーム性核酸が含まれ得る。さらなる内在性分子には、タンパク質、例えば転写因子および酵素が含まれ得る。
【0049】
本明細書において使用するとき、「外因性核酸の産物」という用語は、ポリヌクレオチドおよびポリペプチド産物の両方、例えば転写産物(RNA等のポリヌクレオチド)および翻訳産物(ポリペプチド)を含む。
【0050】
「融合」分子とは、2つ以上のサブユニット分子が、例えば共有結合で連結している分子である。前記サブユニット分子は、同じ化学的分子種であってもよく、または異なる化学的分子種であってもよい。第一の種類の融合分子の例には、融合タンパク質(例えば、ZFP DNA結合ドメインと切断ドメインとの間の融合)および融合核酸(例えば、前記の融合タンパク質をコードする核酸)が含まれるが、これに制限されない。第二の種類の融合分子の例には、三重鎖形成核酸とポリペプチドとの間の融合、および小溝バインダーと核酸との間の融合が含まれるが、これに制限されない。
【0051】
細胞内での融合タンパク質の発現は、細胞への融合タンパク質の送達によって、または細胞への融合タンパク質をコードするポリヌクレオチドの送達であって、前記ポリヌクレオチドが転写され、かつ転写産物が翻訳されて、融合タンパク質を生成する送達によってもたらされ得る。トランススプライシング、ポリペプチドの切断、およびポリペプチドのライゲーションも、細胞内でのタンパク質の発現に関与し得る。細胞へのポリヌクレオチドおよびポリペプチドの送達のための方法は、本開示の別の部分で提供されている。
【0052】
本開示上、「遺伝子」とは、遺伝子産物をコードするDNA領域(下記を参照されたい)、ならびに遺伝子産物の産生を制御するすべてのDNA領域を含むものであって、そのような制御配列がコード配列および/または転写配列に隣接しているかどうかは問わない。したがって、遺伝子には、プロモーター配列、ターミネーター、リボソーム結合部位および内部リボソーム侵入部位等の翻訳制御配列、エンハンサー、サイレンサー、インシュレーター、境界エレメント、複製起点、マトリックス付着部位、ならびに遺伝子座調節領域が含まれるが、必ずしもこれに制限されない。
【0053】
「遺伝子発現」とは、遺伝子に含有される情報の遺伝子産物への転換を表す。遺伝子産物とは、遺伝子の直接的な転写産物(例えば、mRNA、tRNA、rRNA、アンチセンスRNA、干渉RNA、リボザイム、構造RNA、または他の任意の種類のRNA)、あるいはmRNAの翻訳によって産生されたタンパク質であり得る。遺伝子産物には、キャッピング、ポリアデニル化、メチル化、および編集等のプロセスによって修飾されているRNA、ならびに、例えばメチル化、アセチル化、リン酸化、ユビキチン化、ADPリボシル化、ミリストイル化(myristilation)、およびグリコシル化によって修飾されたタンパク質も含まれる。
【0054】
遺伝子発現の「調整」とは、遺伝子の活性の変化を表す。発現の調整には、遺伝子活性化および遺伝子抑制が含まれ得るが、これに制限されない。
【0055】
「植物」細胞には、単子葉性(単子葉植物)または双子葉性(双子葉植物)植物の細胞が含まれるが、これに制限されない。単子葉植物の制限されない例には、トウモロコシ、イネ、オオムギ、オートムギ、コムギ、モロコシ、ライムギ、サトウキビ、パイナップル、タマネギ、バナナ、およびココナツ等の穀物用植物が含まれる。双子葉植物の制限されない例には、タバコ、トマト、ヒマワリ、コットン、テンサイ、ジャガイモ、レタス、メロン、大豆、キャノーラ(ナタネ)、およびアルファルファが含まれる。植物細胞は、植物の任意の部位に由来するもの、および/または植物の任意の発生段階に由来するものであってよい。
【0056】
「対象の領域」とは、外因性分子に結合することが所望される、例えば遺伝子あるいは遺伝子の内部または隣接する非コード配列等の細胞クロマチンの任意の領域である。結合は、標的DNA切断および/または標的組換えを目的とするものであってよい。対象の領域は、例えば染色体、エピソーム、細胞内小器官ゲノム(例えば、ミトコンドリア、葉緑体)、感染ウイルスゲノム内に存在し得る。対象の領域は、遺伝子のコード領域内、例えばリーダー配列、トレーラー配列、またはイントロン等の転写される非コード領域内、あるいはコード領域の上流または下流のいずれかの非転写領域内にあり得る。対象の領域は、1ヌクレオチド対程度に小さくても、または長さが最大2,000ヌクレオチド対であっても、または任意の整数値のヌクレオチド対であってもよい。
【0057】
「作動性の連結」および「作動可能に連結された」(または「操作可能に連結された」)という用語は、並列の2つ以上の構成成分(配列エレメント等)に関して代替可能に用いられるものであって、前記構成成分は、両方の構成成分が正常に機能し、かつ少なくとも1つの前記構成成分が、少なくとも1つの他の構成成分に対して影響を及ぼす機能を媒介し得る可能性を与えるように配置されている。実例として、転写制御配列が1つ以上の転写制御因子の有無に応答してコード配列の転写のレベルを調節している場合、プロモーター等の転写制御配列はコード配列に作動可能に連結されている。転写制御配列は、一般にコード配列とシスで作動可能に連結されているが、直接的にそれに隣接している必要はない。例えば、エンハンサーは、たとえそれらが連続していないとしても、コード配列に作動可能に連結されている転写制御配列である。
【0058】
融合ポリペプチドに関して、「作動可能に連結された」という用語は、構成成分のそれぞれが他の構成成分と連結して、それがそのように連結されていなかった場合になすのと同じ機能を果たすという事実を表す。例えば、ZFP DNA結合ドメインが切断ドメインと融合している融合ポリペプチドに関して、前記融合ポリペプチドにおいて、ZFP DNA結合ドメイン部分がその標的部位および/またはその結合部位に結合し得、一方で切断ドメインが標的部位の付近でDNAを切断し得る場合、前記ZFP DNA結合ドメインと前記切断ドメインは作動性の連結の状態にある。
【0059】
タンパク質、ポリペプチド、または核酸の「機能性フラグメント」とは、その配列が全長のタンパク質、ポリペプチド、または核酸と同一ではないが、全長のタンパク質、ポリペプチド、または核酸と同じ機能を維持しているタンパク質、ポリペプチド、または核酸である。機能性フラグメントは、対応する天然型分子より多い、少ない、または同じ数の残基を保有し得、かつ/あるいは1つ以上のアミノ酸またはヌクレオチド置換を含有し得る。核酸の機能(例えば、コード機能、別の核酸にハイブリダイズする能力)を判定するための方法は、当該技術分野において周知である。同様に、タンパク質機能を判定するための方法は周知である。例えば、ポリペプチドのDNA結合機能を、例えばフィルター結合アッセイ、電気泳動移動度シフトアッセイ、または免疫沈降アッセイによって判定することができる。DNA切断を、ゲル電気泳動によってアッセイすることができる。前記のAusubelらを参照されたい。タンパク質が別のタンパク質と相互作用する能力を、例えば共免疫沈降、ツーハイブリッドアッセイ、または遺伝学的かつ生化学的相補性の両方によって判定することができる。例えば、Fieldsら,(1989)Nature 340:245−246;米国特許第5,585,245号およびPCT WO98/44350を参照されたい。
【0060】
標的部位
開示される方法および組成物には、切断ドメイン(または切断ハーフドメイン)およびDNA結合ドメイン(例えば、ZFP、メガヌクレアーゼ、またはロイシンジッパー)を含み、前記DNA結合ドメイン(例えば、ジンクフィンガードメイン、メガヌクレアーゼ、またはロイシンジッパー)が、植物Zp15遺伝子座内の配列に結合することによって切断ドメイン(または切断ハーフドメイン)の活性を前記配列の付近に向けさせ、それによってZp15内での切断(例えば、二本鎖断裂)を誘導する融合タンパク質が含まれる。本開示の別の部分で示すように、ジンクフィンガードメインは、実質的に任意の所望の配列に結合するように遺伝子操作されていてよい。したがって、1つ以上のDNA結合ドメイン(例えば、ZFP)は、植物Zp15遺伝子内の1つ以上の配列に結合するように遺伝子操作されていてよい。DNA結合ドメイン(例えば、ZFP)および切断ドメインを含む融合タンパク質(あるいは、それぞれがDNA結合ドメインおよび切断ハーフドメインを含む2つの融合タンパク質)の細胞内での発現は、Zp15遺伝子における切断をもたらす。
【0061】
ジンクフィンガードメインによる結合のためのZp15内の配列(例えば、標的部位)の選択は、例えば、選択された配列に結合するようにZFPを設計するための方法も開示している共同所有の米国特許第6,453,242号(2002年9月17日)に開示されている方法に従って達成され得る。ヌクレオチド配列の簡単な目視検査も標的部位の選択に用いることができることは当業者に明白であろう。したがって、標的部位選択のための任意の手段を、本明細書に記載される方法において用いることができる。
【0062】
ZFP DNA結合ドメインに対して、標的部位は一般に複数の隣接する標的サブサイトから構成される。標的サブサイトとは、個々のジンクフィンガーによって結合される配列(通常、ヌクレオチドトリプレット、または隣接するクァドループレットと1ヌクレオチド重複し得るヌクレオチドクァドループレットのいずれか)を表す。例えば、WO02/077227を参照されたい。ジンクフィンガータンパク質が最も接触する鎖を標的鎖、「主要な認識鎖」、または「主要な接触鎖」と呼ぶ場合、いくつかのジンクフィンガータンパク質は標的鎖内の3塩基トリプレットおよび非標的鎖上の第4の塩基に結合する。標的部位は、一般に少なくとも9ヌクレオチドの長さを有し、したがって、少なくとも3つのジンクフィンガーを含むジンクフィンガー結合ドメインによって結合される。しかしながら、例えば4フィンガー結合ドメインの12ヌクレオチド標的部位への結合、5フィンガー結合ドメインの15ヌクレオチド標的部位への結合、または6フィンガー結合ドメインの18ヌクレオチド標的部位への結合も可能である。明白であろうが、より大きな結合ドメイン(例えば、7、8、9フィンガー以上)のより長い標的部位への結合も可能である。
【0063】
標的部位が3ヌクレオチドの倍数である必要はない。例えば、交差鎖相互作用が生じる場合(例えば、米国特許第6,453,242号およびWO02/077227を参照されたい)、マルチフィンガー結合ドメインの1つ以上の個々のジンクフィンガーは、重複しているクァドループレットサブサイトに結合し得る。結果として、3フィンガータンパク質は、10番目のヌクレオチドが末端フィンガーによって結合されるクァドループレットの一部である10ヌクレオチド配列に結合し得、4フィンガータンパク質は、13番目のヌクレオチドが末端フィンガーによって結合されるクァドループレットの一部である13ヌクレオチド配列に結合し得る、等々。
【0064】
マルチフィンガー結合ドメイン内の個々のジンクフィンガー間のアミノ酸リンカー配列の長さおよび性質も、標的配列への結合に影響を及ぼす。例えば、マルチフィンガー結合ドメイン内の隣接するジンクフィンガー間のいわゆる「非標準的リンカー」、「長いリンカー」、または「構造化リンカー」の存在によって、それらのフィンガーは直接に隣接していないサブサイトに結合することができる。そのようなリンカーの制限されない例は、例えば米国特許第6,479,626号およびWO01/53480に記載されている。したがって、ジンクフィンガー結合ドメインに対する標的部位内の1つ以上のサブサイトは、1、2、3、4、5、またはそれ以上のヌクレオチドずつ互いに分離し得る。ほんの一例として、4フィンガー結合ドメインは、配列中に2つの連続する3ヌクレオチドサブサイト、介在ヌクレオチド、および2つの連続するトリプレットサブサイトを含む13ヌクレオチド標的部位に結合し得る。
【0065】
配列(例えば、標的部位)間の距離とは、互いに最も近隣の配列の端から測定される、2つの配列間に介在するヌクレオチドまたはヌクレオチド対の数を表す。
【0066】
2つのジンクフィンガードメイン/切断ハーフドメイン融合分子の離れた標的部位への結合に切断が依存しているある実施態様では、前記2つの標的部位は向かい合ったDNA鎖上にあり得る。他の実施態様では、両方の標的部位は同じDNA鎖上にある。
【0067】
DNA結合ドメイン
任意のDNA結合ドメインを、本明細書に開示される方法において用いることができる。ある実施態様では、DNA結合ドメインはジンクフィンガータンパク質を含む。ジンクフィンガー結合ドメインは、1つ以上のジンクフィンガーを含む。Millerら,(1985)EMBO J.4:1609−1614;Rhodes(1993)Scientific American Feb.:56−65;米国特許第6,453,242号。本明細書に記載されるジンクフィンガー結合ドメインは、概して2、3、4、5、6、またはさらに多くのジンクフィンガーを含む。
【0068】
典型的には、1つのジンクフィンガードメインは、長さが約30アミノ酸である。構造研究によって、それぞれのジンクフィンガードメイン(モチーフ)は、2個のシステインおよび2個のヒスチジンによる亜鉛原子の配位を介した特定の構造に保持された、2つのβシート(2個の不変のシステイン残基を含有するβターンに保持されている)およびαヘリックス(2個の不変のヒスチジン残基を含有している)を含有することが証明されている。
【0069】
ジンクフィンガーには、標準的C2H2ジンクフィンガー(すなわち、亜鉛イオンが2個のシステインと2個のヒスチジン残基によって配位されているもの)、ならびに、例えばC3Hジンクフィンガー(亜鉛イオンが3個のシステイン残基と1個のヒスチジン残基によって配位されているもの)およびC4ジンクフィンガー(亜鉛イオンが4個のシステイン残基によって配位されているもの)等の非標準的ジンクフィンガーの両方が含まれる。植物における使用のための非標準的ZFPに関するWO02/057293および米国特許公報第20080182332号も参照されたい。
【0070】
遺伝子操作されたジンクフィンガー結合ドメインは、天然に存在しているジンクフィンガータンパク質と比較して新規な結合特異性を有し得る。遺伝子操作法には、合理的設計および様々な種類の選択が含まれるが、これに制限されない。合理的設計には、例えばトリプレット(またはクァドループレット)ヌクレオチド配列と個々のジンクフィンガーアミノ酸配列とを含むデータベースであって、それぞれのトリプレットまたはクァドループレットヌクレオチド配列が、特定のトリプレットまたはクァドループレット配列に結合するジンクフィンガーの1つ以上のアミノ酸配列と関連付けされているデータベースを用いることが含まれる。
【0071】
ファージディスプレイおよびツーハイブリッドシステムを含む例示的選択法は、米国特許第5,789,538号;第5,925,523号;第6,007,988号;第6,013,453号;第6,410,248号;第6,140,466号;第6,200,759号;および第6,242,568号;ならびにWO98/37186;WO98/53057;WO00/27878;WO01/88197、およびGB2,338,237に開示されている。
【0072】
ジンクフィンガー結合ドメインに対する結合特異性の増強は、例えば共同所有のWO02/077227に記載されている。
【0073】
個々のジンクフィンガーは3ヌクレオチド(すなわち、トリプレット)配列(または、隣接するジンクフィンガーの4ヌクレオチド結合部位と1ヌクレオチド重複し得る4ヌクレオチド配列)に結合するため、ジンクフィンガー結合ドメインが結合するように遺伝子操作されている配列(例えば、標的配列)の長さは、遺伝子操作されたジンクフィンガー結合ドメイン内のジンクフィンガーの数を決定することになる。例えば、フィンガーモチーフが重複サブサイトに結合しないZFPについて、6ヌクレオチド標的配列は2フィンガー結合ドメインによって結合され;9ヌクレオチド標的配列は3フィンガー結合ドメインによって結合される、等々。本明細書で述べているように、標的部位における個々のジンクフィンガーに対する結合部位(すなわち、サブサイト)は、連続している必要はないが、マルチフィンガー結合ドメイン内のジンクフィンガー間のアミノ酸配列(すなわち、フィンガー間リンカー)の長さおよび性質に依存して、1つまたは数個のヌクレオチドずつ分離され得る。
【0074】
マルチフィンガージンクフィンガー結合ドメインにおいて、隣接するジンクフィンガーは、およそ5アミノ酸のアミノ酸リンカー配列(いわゆる「標準的」フィンガー間リンカー)によって、あるいは1つ以上の非標準的リンカーによって分離され得る。例えば、共同所有の米国特許第6,453,242号および第6,534,261号を参照されたい。3つを上回るフィンガーを含む遺伝子操作されたジンクフィンガー結合ドメインについて、前記ジンクフィンガーの一部の間でのより長い(「非標準的」)フィンガー間リンカーの挿入は、それによって結合ドメインによる結合の親和性および/または特異性が増加し得るため、場合によっては所望され得る。例えば、米国特許第6,479,626号およびWO01/53480を参照されたい。したがって、マルチフィンガージンクフィンガー結合ドメインを、非標準的フィンガー間リンカーの存在および位置に関して特徴付けすることができる。例えば、3つのフィンガー(2個の標準的フィンガー間リンカーによって接合している)、長いリンカー、および3つのさらなるフィンガー(2個の標準的フィンガー間リンカーによって接合している)を含む6フィンガージンクフィンガー結合ドメインは、2×3配置と表示される。同様に、2つのフィンガー(その間に標準的リンカーを有する)、長いリンカー、および2つのさらなるフィンガー(標準的リンカーによって接合している)を含む結合ドメインは、2×2配置と表示される。3つの2フィンガー単位(そのそれぞれにおいて、2つのフィンガーが標準的リンカーによって接合している)を含み、かつそれぞれの2フィンガー単位が隣接する2つのフィンガー単位と長いリンカーによって接合しているタンパク質は、3×2配置として表される。
【0075】
マルチフィンガー結合ドメイン内の2つの隣接するジンクフィンガー間での長いまたは非標準的フィンガー間リンカーの存在によって、前記2つのフィンガーは、しばしば、標的配列における直接に連続していないサブサイトに結合する。したがって、標的部位におけるサブサイト間には1つ以上のヌクレオチドの差が存在し得、すなわち、標的部位はジンクフィンガーによって接触されていない1つ以上のヌクレオチドを含有し得る。例えば、2×2ジンクフィンガー結合ドメインは、1つのヌクレオチドによって分離された2つの6ヌクレオチド配列に結合し得、すなわち、それは13ヌクレオチド標的部位に結合する。Mooreら,(2001a)Proc.Natl.Acad.Sci.USA 98:1432−1436;Mooreら,(2001b)Proc.Natl.Acad.Sci.USA 98:1437−1441、およびWO01/53480も参照されたい。
【0076】
先に言及しているように、標的サブサイトは、1つのジンクフィンガーによって結合される3または4ヌクレオチド配列である。ある目的のために、2フィンガー単位を「結合モジュール」と表示する。結合モジュールを、例えば、マルチフィンガータンパク質(一般に3つのフィンガー)との関連で、特定の6ヌクレオチド標的配列に結合する2つの隣接するフィンガーを選択することによって獲得することができる。あるいは、個々のジンクフィンガーを集合させることによってモジュールを構築することができる。WO98/53057およびWO01/53480も参照されたい。
【0077】
あるいは、DNA結合ドメインはヌクレアーゼに由来してもよい。例えば、ホーミングエンドヌクレアーゼおよびメガヌクレアーゼの認識配列、例えばI−SceI、I−CeuI、PI−PspI、PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIII等が既知である。米国特許第5,420,032号;米国特許第6,833,252号;Belfortら,(1997)Nucleic Acids Res.25:3379−3388;Dujonら,(1989)Gene 82:115−118;Perlerら,(1994)Nucleic Acids Res.22,1125−1127;Jasin(1996)Trends Genet.12:224−228;Gimbleら,(1996)J.Mol.Biol.263:163−180;Argastら,(1998)J.Mol.Biol.280:345−353、およびNew England Biolabsカタログも参照されたい。さらに、ホーミングエンドヌクレアーゼおよびメガヌクレアーゼのDNA結合特異性を、天然でない標的部位に結合するように遺伝子操作することができる。例えば、Chevalierら,(2002)Molec.Cell 10:895−905;Epinatら,(2003)Nucleic Acids Res.31:2952−2962;Ashworthら,(2006)Nature 441:656−659;Paquesら,(2007)Current Gene Therapy 7:49−66;米国特許公報第20070117128号を参照されたい。
【0078】
別の代替法として、DNA結合ドメインはロイシンジッパータンパク質に由来してもよい。ロイシンジッパーとは、遺伝子発現に関連した重要な転写因子である多くの真核性制御タンパク質におけるタンパク質−タンパク質相互作用に関与するタンパク質のクラスである。ロイシンジッパーとは、動物、植物、酵母、等々を含むいくつかの界を越えて、これらの転写因子で共有されている共通の構造モチーフを表す。ロイシンジッパーは、ロイシン残基がαヘリックスのいたる所に均等に間隔をあけて配置され、それによって前記2つのポリペプチドのロイシン残基が最終的に前記ヘリックスの同じ面上にくることになる様式で特異的DNA配列に結合する、2つのポリペプチド(ホモ二量体またはヘテロ二量体)によって形成される。ロイシンジッパーのDNA結合特異性を、本明細書に開示されるDNA結合ドメインに利用することができる。
【0079】
いくつかの実施態様では、DNA結合ドメインは、植物病原菌キサントモナスに由来するTALエフェクターからの遺伝子操作されたドメインである(Bochら,(2009)Science 29,2009年10月(10.1126/science.117881)、ならびにMoscouおよびBogdanove,(2009)Science 29,2009年10月(10.1126/science.1178817)を参照されたい)。
【0080】
切断ドメイン
前述のように、DNA結合ドメインは、切断(ヌクレアーゼ)ドメインと関連していてもよい。例えば、ホーミングエンドヌクレアーゼは、ヌクレアーゼ機能を維持している一方で、そのDNA結合特異性を改変されていてもよい。さらに、ジンクフィンガータンパク質はまた、切断ドメインと融合して、ジンクフィンガーヌクレアーゼ(ZFN)を形成してもよい。本明細書に開示される融合タンパク質の切断ドメイン部分は、任意のエンドヌクレアーゼまたはエキソヌクレアーゼから獲得され得る。切断ドメインが由来し得る例示的エンドヌクレアーゼには、制限エンドヌクレアーゼおよびホーミングエンドヌクレアーゼが含まれるが、これに制限されない。例えば、2002−2003カタログ,New England Biolabs,Beverly,MA;およびBelfortら,(1997)Nucleic Acids Res.25:3379−3388を参照されたい。DNAを切断するさらなる酵素が既知である(例えば、S1ヌクレアーゼ;マングビーンヌクレアーゼ;膵臓DNaseI;ミクロコッカスヌクレアーゼ;酵母HOエンドヌクレアーゼ;Linnら,(eds.)Nucleases,Cold Spring Harbor Laboratory Press,1993も参照されたい)。ホーミングエンドヌクレアーゼおよびメガヌクレアーゼの制限されない例には、I−SceI、I−CeuI、PI−PspI、PI−Sce、I−SceIV、I−CsmI、I−PanI、I−SceII、I−PpoI、I−SceIII、I−CreI、I−TevI、I−TevII、およびI−TevIIIが含まれる。米国特許第5,420,032号;米国特許第6,833,252号;Belfortら,(1997)Nucleic Acids Res.25:3379−3388;Dujonら,(1989)Gene 82:115−118;Perlerら,(1994)Nucleic Acids Res.22,1125−1127;Jasin(1996)Trends Genet.12:224−228;Gimbleら,(1996)J.Mol.Biol.263:163−180;Argastら,(1998)J.Mol.Biol.280:345−353;およびthe New England Biolabsカタログも参照されたい。1つ以上のこれらの酵素(またはその機能性フラグメント)を、切断ドメインおよび切断ハーフドメインの供給源として用いることができる。
【0081】
制限エンドヌクレアーゼ(制限酵素)は、多くの種に存在し、DNAに(認識部位において)配列特異的に結合し得、かつ結合の部位においてまたは近隣においてDNAを切断し得る。ある制限酵素(例えば、IIS型)は、認識部位から外れた部位でDNAを切断し、かつ分離可能な結合ドメインおよび切断ドメインを有する。例えば、IIS型酵素のFokIは、一方の鎖上にあるその認識部位から9ヌクレオチドの部位で、かつもう一方の鎖上のその認識部位から13ヌクレオチドの部位でDNAの二本鎖切断を触媒する。例えば、米国特許第5,356,802号;第5,436,150号および第5,487,994号;ならびにLiら,(1992)Proc.Natl.Acad.Sci.USA 89:4275−4279;Liら,(1993)Proc.Natl.Acad.Sci.USA 90:2764−2768;Kimら,(1994a)Proc.Natl.Acad.Sci.USA 91:883−887;Kimら,(1994b)J.Biol.Chem.269:31,978−31,982を参照されたい。したがって、一実施態様では、融合タンパク質は、少なくとも1種のIIS型制限酵素由来の切断ドメイン(または切断ハーフドメイン)、および遺伝子操作されていてもされていなくてもよい1つ以上のジンクフィンガー結合ドメインを含む。
【0082】
その切断ドメインが結合ドメインから分離可能である例示的IIS型制限酵素は、FokIである。この特定の酵素は、二量体として活性を有する。Bitinaiteら,(1998)Proc.Natl.Acad.Sci.USA 95:10,570−10,575。したがって、本開示上、開示される融合タンパク質に用いられたFokI酵素の一部は切断ハーフドメインと考えられる。したがって、ジンクフィンガー−FokI融合体を用いた細胞配列の標的二本鎖切断および/または標的置換のために、それぞれがFokI切断ハーフドメインを含む2つの融合タンパク質を用いて、触媒活性のある切断ドメインを再構築することができる。あるいは、ジンクフィンガー結合ドメインと2つのFokI切断ハーフドメインとを含有する1つのポリヌクレオチド分子も用いることができる。ジンクフィンガー−FokI融合体を用いた標的切断および標的配列変更に対するパラメーターは、本開示の別の部分で提供されている。
【0083】
切断ドメインまたは切断ハーフドメインは、切断活性を維持している、または多量化(例えば、二量化)して機能性切断ドメインを形成する能力を維持しているタンパク質の任意の部分であってよい。
【0084】
例示的IIS型制限酵素は、参照することによりその全体として本明細書に組み入れられている、共同所有の国際公報WO2007/014275に記載されている。
【0085】
切断特異性を増強させるために、切断ドメインを改変してもよい。ある実施態様では、切断ハーフドメインの変形型が利用され、これらの変形型は切断ハーフドメインのホモ二量化を最小限に抑えるまたは阻止する。そのような改変された切断ハーフドメインの制限されない例は、参照することによりその全体として本明細書に組み入れられている、WO2007/014275に詳細に記載されている。実施例も参照されたい。ある実施態様では、切断ドメインは、ホモ二量化を最小限に抑えるまたは阻止し、当業者に既知であり、かつ参照することによりそれらの全体として本明細書に組み入れられている、例えば米国特許公報第20050064474および第20060188987号に記載されている遺伝子操作された切断ハーフドメイン(二量化ドメイン変異体とも称される)を含む。FokIの446、447、479、483、484、486、487、490、491、496、498、499、500、531、534、537、および538位のアミノ酸残基はすべて、FokI切断ハーフドメインの二量化に影響を与えるための標的である。例えば、米国特許公報第20050064474号および第20060188987号;国際特許公報WO07/139898;Millerら,(2007)Nat.Biotechnol.25(7):778−785を参照されたい。
【0086】
必須のヘテロ二量体を形成するFokIのさらなる遺伝子操作された切断ハーフドメインも、本明細書に記載されるZFNに用いることができる。一実施態様では、第一の切断ハーフドメインはFokIの490および538位のアミノ酸残基における変異を含み、かつ第二の切断ハーフドメインはアミノ酸残基486および499における変異を含む。
【0087】
ある実施態様では、切断ドメインは、その両方が結合ドメイン、第一の切断ハーフドメイン、および第二の切断ハーフドメインを含む1つのポリペプチドの一部である、2つの切断ハーフドメインを含む。切断ハーフドメインは、それらがDNAを切断する機能を果たす限り、同じアミノ酸配列または異なるアミノ酸配列を有し得る。
【0088】
一般に、融合タンパク質が切断ハーフドメインを含む場合、2つの融合タンパク質が切断に必要とされる。あるいは、2つの切断ハーフドメインを含む1つのタンパク質を用いることができる。前記2つの切断ハーフドメインは同じエンドヌクレアーゼ(または、その機能性フラグメント)に由来してもよく、あるいはそれぞれの切断ハーフドメインは異なるエンドヌクレアーゼ(またはその機能性フラグメント)に由来してもよい。さらに、前記2つの融合タンパク質に対する標的部位は、好ましくは、2つの融合タンパク質のそれらそれぞれの標的部位への結合により、切断ハーフドメインが例えば二量化することによって機能性切断ドメインを形成し得る空間的方向に互いに切断ハーフドメインが位置付けられるように、互いに対して配置される。したがって、ある実施態様では、標的部位の近隣の端を、5〜8ヌクレオチドで、または15〜18ヌクレオチドで分離する。しかしながら、任意の整数値のヌクレオチドまたはヌクレオチド対を、2つの標的部位間に介在させることができる(例えば、2〜50ヌクレオチド以上)。一般に、切断の地点は標的部位の間にある。
【0089】
融合タンパク質
融合タンパク質(およびそれをコードするポリヌクレオチド)の設計および構築のための方法は、当業者に既知である。例えば、DNA結合ドメイン(例えば、ジンクフィンガードメイン)および制御または切断ドメイン(もしくは切断ハーフドメイン)、および融合タンパク質をコードするポリヌクレオチドを含む融合タンパク質の設計および構築のための方法は、参照することによりそれらの全体として本明細書に組み入れられている、共同所有の米国特許第6,453,242号および第6,534,261号、ならびに米国特許出願公報第2007/0134796号および第2005/0064474号に記載されている。ある実施態様では、融合タンパク質をコードするポリヌクレオチドを構築する。これらのポリヌクレオチドをベクター内に挿入することができ、かつ前記ベクターを細胞内に導入することができる(ベクターおよび細胞内にポリヌクレオチドを導入するための方法に関するさらなる開示のために、下記を参照されたい)。
【0090】
本明細書に記載される方法のある実施態様では、ジンクフィンガーヌクレアーゼは、ジンクフィンガー結合ドメインおよびFokI制限酵素由来の切断ハーフドメインを含む融合タンパク質を含み、かつ2つのそのような融合タンパク質を細胞内で発現させる。細胞内での2つの融合タンパク質の発現は、前記2つのタンパク質の前記細胞への送達;1つのタンパク質および前記タンパク質の一方をコードする1つの核酸の前記細胞への送達;それぞれが前記タンパク質の一方をコードする2つの核酸の前記細胞への送達;または両方のタンパク質をコードする1つの核酸の前記細胞への送達によって生じ得る。さらなる実施態様では、融合タンパク質は、2つの切断ハーフドメインとジンクフィンガー結合ドメインとを含む1本のポリペプチド鎖を含む。この場合、1つの融合タンパク質が細胞内で発現し、かつ理論に束縛されることを望むことなく、前記切断ハーフドメインの分子内二量体の形成の結果としてDNAを切断すると考えられている。
【0091】
ある実施態様では、融合タンパク質(例えば、ZFP−FokI融合体)の構成成分を、ジンクフィンガードメインが融合タンパク質のアミノ末端の最も近隣にあり、かつ切断ハーフドメインがカルボキシ末端の最も近隣にあるように配置する。これは、FokI酵素に由来するもの等の天然に存在している二量化切断ドメインにおける、DNA結合ドメインがアミノ末端の最も近隣にあり、かつ切断ハーフドメインがカルボキシ末端の最も近隣にある、切断ドメインの相対的方向を反映する。これらの実施態様では、機能性ヌクレアーゼを形成するための切断ハーフドメインの二量化は、融合タンパク質が向かい合ったDNA鎖上の部位に結合することによって引き起こされ、結合部位の5’末端は互いに隣接している。
【0092】
さらなる実施態様では、融合タンパク質(例えば、ZFP−FokI融合体)の構成成分を、切断ハーフドメインが融合タンパク質のアミノ末端の最も近隣にあり、かつジンクフィンガードメインがカルボキシ末端の最も近隣にあるように配置する。これらの実施態様では、機能性ヌクレアーゼを形成するための切断ハーフドメインの二量化は、融合タンパク質が向かい合ったDNA鎖上の部位に結合することによって引き起こされ、結合部位の3’末端は互いに隣接している。
【0093】
その上さらなる実施態様では、第一の融合タンパク質は、融合タンパク質のアミノ末端の最も近隣にある切断ハーフドメインおよびカルボキシ末端の最も近隣にあるジンクフィンガードメインを含有し、かつ第二の融合タンパク質を、ジンクフィンガードメインが融合タンパク質のアミノ末端の最も近隣にあり、かつ切断ハーフドメインがカルボキシ末端の最も近隣にあるように配置する。これらの実施態様では、両方の融合タンパク質は同じDNA鎖に結合し、カルボキシ末端の最も近隣にあるジンクフィンガードメインを含有する第一の融合タンパク質の結合部位が、アミノ末端の最も近隣にあるジンクフィンガードメインを含有する第二の融合タンパク質の結合部位の5’側に位置する。
【0094】
開示される融合タンパク質のある実施態様では、ジンクフィンガードメインおよび切断ドメイン(または切断ハーフドメイン)の間のアミノ酸配列を「ZCリンカー」と表示する。ZCリンカーは、前述のフィンガー間リンカーとは区別されるべきである。切断を最適化するZCリンカーの獲得に関する詳細については、例えば、米国特許公報第20050064474号A1および第20030232410号,ならびに国際特許公報WO05/084190を参照されたい。
【0095】
一実施態様では、本開示は、表1に示した1つ以上の認識ヘリックスアミノ酸配列を有するジンクフィンガータンパク質を含むZFNを提供する。別の実施態様では、表1に示した1つ以上の認識ヘリックスを有するZFPをコードするヌクレオチド配列を含むZFP発現ベクターを本明細書において提供する。
【0096】
標的組込み
開示される方法および組成物を用いて、植物の細胞クロマチンのZp15遺伝子におけるDNAを切断することができ、それによって前記遺伝子座への外因性配列の安定的な標的組込みが容易になる。本明細書に記載されるように、内在性Zp15遺伝子の機能の喪失は植物細胞に十分に許容され、かつこの遺伝子内に組み込まれた配列が広範に転写され、組み込まれた配列の遺伝的伝達のための生殖細胞系の改変を有する植物を作出する。したがって、Zp15は外因性配列の標的組込みのための所望の部位である。
【0097】
Zp15内への標的組込みのために、1つ以上のDNA結合ドメイン(例えば、ZFP)を、所定の切断部位におけるまたは近隣における標的部位に結合するように遺伝子操作し、かつ操作されたDNA結合ドメインと切断ドメインとを含む融合タンパク質を細胞内で発現させる。融合タンパク質のDNA結合(例えば、ジンクフィンガー)部分が標的部位へ結合すると、DNAは切断ドメインによって標的部位の近隣で好ましくは二本鎖断裂によって切断される。
【0098】
Zp15遺伝子座における二本鎖断裂の存在によって、相同組換えを介した外因性配列の組込みが容易になる。したがって、Zp15遺伝子内に挿入されるべき外因性配列を含むポリヌクレオチドは、相同組換えを容易にするためにZp15遺伝子に相同な1つ以上の領域を含むことになる。
【0099】
本明細書に記載されるように、対象の任意の配列(外因性配列)をZp15遺伝子座内に導入することができる。例示的外因性配列には、任意のポリペプチドコード配列(例えば、cDNA)、プロモーター、エンハンサー、および他の制御配列(例えば、干渉RNA配列、shRNA発現カセット、エピトープタグ、マーカー遺伝子、切断酵素認識部位、および様々な種類の発現構築物)が含まれるが、これに制限されない。そのような配列は、標準的な分子生物学的技術(クローニング、合成、等々)を用いることによって容易に獲得され得、かつ/あるいは市販されている。
【0100】
本明細書に記載される融合分子に加えて、選択されたゲノム配列の標的置換もまた、置換(またはドナー)配列の導入を伴う。ドナー配列を、融合タンパク質の発現の前に、同時に、またはそれに続いて、細胞内に導入することができる。ドナーポリヌクレオチドは、それとそれが相同性を有するZp15ゲノム配列との間の相同組換え(または相同指向修復)を支持するのに十分なZp15との相同性を含有している。ドナーおよびゲノム配列の間のおよそ25、50、100、200、500、750、1,000、1,500、2,000ヌクレオチド以上(または10〜2,000ヌクレオチド、もしくはそれ以上の間の任意の整数値)の配列相同性によって、それらの間の相同組換えが支持されることになる。ある実施態様では、ホモロジーアームは長さが1,000塩基対未満である。他の実施態様では、ホモロジーアームは長さが750塩基対未満である。参照することにより本明細書に組み入れられている、米国仮特許出願第61/124,047号も参照されたい。
【0101】
ドナー配列は、長さが10〜5,000ヌクレオチド(またはその間の任意の整数値のヌクレオチド)またはそれより長い範囲であってよい。典型的にドナー配列はそれが置換するゲノム配列と同一でないことは、容易に明白であろう。例えば、ドナーポリヌクレオチドの配列は、染色体配列との十分な相同性が存在する限り、ゲノム配列に対して1つ以上の一塩基の変化、挿入、欠失、逆位、または転位を含有してよい。あるいは、ドナー配列は、2つの相同な領域が隣接した非相同的配列を含有してよい。さらに、ドナー配列は、細胞クロマチン内の対象の領域に相同でない配列を含有するベクターを含んでよい。一般に、ドナー配列の相同領域は、組換えが所望されるゲノム配列と少なくとも50%の配列同一性を有する。ある実施態様では、60%、70%、80%、90%、95%、98%、99%、または99.9%の配列同一性が存在する。ドナーポリヌクレオチドの長さに依存して、1%〜100%の間の任意の値の配列同一性が存在し得る。
【0102】
ドナー分子は、細胞クロマチンに相同ないくつかの不連続領域を含有してよい。例えば、対象の領域に通常存在しない配列の標的挿入のために、前記配列は、ドナー核酸分子内に存在してよく、かつ対象の領域内の遺伝子配列に相同な領域によって隣接されていてもよい。
【0103】
ドナー分子は、Zp15遺伝子座内に挿入されて、後の使用のための貯蔵物としての役割を果たすこともできる。例えば、内在性遺伝子に相同であるが対象の変異を含有しているドナー分子を、Zp15遺伝子座内に挿入してもよい。次いで、内在性遺伝子座と対象の変異を含有するZp15遺伝子座内のドナー分子との両方を切断するであろう、前記内在性遺伝子に特異的なZFNを導入することができる。ゲノムにおいて結果として生じるDSBは、その後Zp15遺伝子座から解放されたドナー分子に対する組込み部位になり得る。このようにして、当該方法はZFNをコードする核酸およびそのドナー配列の両方の同時取込みに依存しないため、対象の任意の領域におけるドナー配列の標的組込みの効率を大幅に増加させることができる。
【0104】
ドナー分子は、Zp15遺伝子座内に挿入されて、それに続く挿入のための標的部位としての役割を果たすこともできる。例えば、さらなるZFN設計物のための認識部位を含有するDNA配列から構成されるドナー分子を、Zp15遺伝子座内に挿入してもよい。それに続き、さらなるZFN設計物を作出し、かつ細胞内で発現させてもよく、それによって、オリジナルのドナー分子は切断され、かつ修復または相同組換えによって改変される。このようにして、ドナー分子の反復組込みがZp15遺伝子座において生じ得る。
【0105】
ドナー配列の挿入の成功を判別するためのアッセイ(例えば、ハイブリダイゼーション、PCR、制限酵素消化)を簡略化するために、Zp15ゲノム配列と比較して、ある程度の配列の相違がドナー配列に存在してもよい。好ましくは、コード領域に位置する場合、そのようなヌクレオチド配列の相違は、アミノ酸配列を変化させない、またはサイレントなアミノ酸変化(すなわち、タンパク質の構造または機能に影響を及ぼさない変化)をもたらす。ドナーポリヌクレオチドは、場合によっては、対象の領域における部位に結合するDNA結合ドメインに対応する配列の変化を含有して、相同組換えによって細胞クロマチン内に導入されているドナー配列の切断を阻止することもできる。
【0106】
ドナーポリヌクレオチドは、DNAまたはRNA、一本鎖または二本鎖であってよく、かつ線状または環状の形態で細胞内に導入されてよい。線状の形態で導入された場合、ドナー配列の端を、当業者に既知の方法によって(例えば、エキソヌクレアーゼによる分解から)保護することができる。例えば、1つ以上のジオキシヌクレオチド残基を線状分子の3’末端に付加し、かつ/あるいは自己相補的オリゴヌクレオチドを一方または両方の端にライゲートさせる。例えば、Changら,(1987)Proc.Natl.Acad.Sci.USA 84:4959−4963;Nehlsら,(1996)Science 272:886−889を参照されたい。外因性ポリヌクレオチドを分解から保護するためのさらなる方法には、末端アミノ基の付加、ならびに、例えばホスホロチオエート、ホスホロアミダート、およびO−メチルリボースまたはデオキシリボース残基等の修飾されたヌクレオチド間連結の使用が含まれるが、これに制限されない。
【0107】
ポリヌクレオチドを、例えば複製起点、プロモーター、および抗生物質耐性をコードする遺伝子等のさらなる配列を有するベクター分子の一部として細胞内に導入することができる。さらに、ドナーポリヌクレオチドを、ネイキッド核酸として、ナノ粒子、リポソーム、またはポロキサマー等の物質と複合体を形成した核酸として導入することができ、あるいは細菌またはウイルス(例えば、アグロバクテリウム、リゾビウム種、NGR234、シノリゾビウム・メリロッティ(Sinorhizoboium meliloti)、メソリゾビウム・ロッティ、タバコモザイクウイルス、ジャガイモウイルスX、カリフラワーモザイクウイルス、およびキャッサバベインモザイクウイルス)によって送達することができる。例えば、Chungら,(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0108】
断裂に隣接したまたは周辺の領域との相同性を有する外因性DNA分子の存在を伴った細胞配列における二本鎖断裂の存在は、ドナー分子から細胞(例えば、ゲノムまたは染色体)配列への配列情報の転移によって、すなわち、「遺伝子変換」としても既知である相同指向修復のプロセスによって断裂を修復する細胞メカニズムを活性化するようである。本出願人の方法は、Zp15遺伝子等のパラロガス遺伝子を特異的に標的化する遺伝子操作されたZFPの強力な標的化能を切断ドメイン(または切断ハーフドメイン)と有利に組み合わせるものであって、それによって標的配列の切断により外因性配列の挿入が所望されるゲノムの領域において二本鎖断裂が引き起されるものである。
【0109】
染色体配列の変更のために、十分量のドナー配列が所望の配列変更をもたらすためにコピーされてさえいれば、ドナーの全配列が染色体内にコピーされる必要はない。
【0110】
相同組換えによるドナー配列の挿入の効率は、細胞DNAにおける二本鎖断裂と組換えが所望される部位との間の距離に反比例する。言い換えれば、組換えが所望される部位に二本鎖断裂がより近い場合ほど、より高い効率の相同組換えが観察される。組換えの正確な部位があらかじめ定められていない(例えば、所望の組換え事象がゲノム配列の間隔にわたって生じ得る)場合には、ドナー核酸の長さおよび配列は、切断の部位とともに、所望の組換え事象が得られるように選択される。ゲノム配列における1つのヌクレオチド対の配列を変化させるように所望の事象を設計する場合には、細胞クロマチンはそのヌクレオチド対のいずれかの側上の10,000ヌクレオチド以内で切断される。ある実施態様では、切断は、配列を変化させるべきヌクレオチド対のいずれかの側上で、1,000、500、200、100、90、80、70、60、50、40、30、20、10、5、または2ヌクレオチド、あるいは2〜1,000ヌクレオチドの間の任意の整数値の範囲内で起こる。
【0111】
上で詳述されるように、それぞれがジンクフィンガー結合ドメインおよび切断ハーフドメインを含む2つの融合タンパク質に対する結合部位は、もう一方の結合部位の最も近隣にあるそれぞれの結合部位の端から測定して5〜8または15〜18ヌクレオチド離れて位置し得、かつ切断は前記結合部位間で起こる。切断されるゲノム配列はドナー配列によって置換されるため、切断が結合部位間の1つの部位または複数の部位で起こるかどうかは重要ではない。したがって、標的組換えによる1つのヌクレオチド対の配列の効率的な変更のために、結合部位間の領域の中間点は、そのヌクレオチド対の10,000ヌクレオチド以内、好ましくは1,000ヌクレオチド以内、または500ヌクレオチド以内、または200ヌクレオチド以内、または100ヌクレオチド以内、または50ヌクレオチド以内、または20ヌクレオチド以内、または10ヌクレオチド以内、または5ヌクレオチド以内、または2ヌクレオチド以内、または1ヌクレオチド以内、または対象のヌクレオチド対の地点にある。
【0112】
ある実施態様では、相同染色体はドナーポリヌクレオチドとしての役割を果たし得る。したがって、例えばヘテロ接合体における変異の修正は、1つの染色体上の変異配列に結合かつ切断するが、相同染色体上の野生型配列を切断しない融合タンパク質を遺伝子操作することによって達成され得る。変異を有する染色体上での二本鎖断裂は、切断された染色体に相同染色体由来の野生型配列がコピーされ、ゆえに野生型配列の2コピーが復元される、相同性に基づく「遺伝子変換」プロセスを刺激する。
【0113】
相同組換えに関与する遺伝子、例えば、RAD54エピスタシス群の植物遺伝子(例えば、AtRad54、AtRad51)、およびその産物が前述の遺伝子産物と相互作用する遺伝子等の発現を活性化するためのさらなるZFP−機能性ドメイン融合体の使用を含むが、これに制限されない標的組換えのレベルを増強し得る方法および組成物も提供される。例えば、Klutstein M.ら,Genetics.2008 Apr;178(4):2389−97を参照されたい。
【0114】
同様に、ZFP−機能性ドメイン融合体を本明細書に開示される方法および組成物と組み合わせて用いて、非相同末端の接合に関与する遺伝子(例えば、Ku70/80、XRCC4、ポリ(ADPリボース)ポリメラーゼ、DNAリガーゼ4)の発現を抑制することができる。例えば、Rihaら,(2002)EMBO 21:2819−2826;Freisnerら,(2003)Plant J.34:427−440;Chenら,(1994)European Journal of Biochemistry 224:135−142を参照されたい。ジンクフィンガー結合ドメインおよび機能性ドメインの間の融合体を用いた遺伝子発現の活性化および抑制のための方法は、例えば共同所有の米国特許第6,534,261号;第6,824,978号、および第6,933,113号に開示されている。さらなる抑制方法には、抑制されるべき遺伝子の配列を標的とした、アンチセンスオリゴヌクレオチドおよび/または低分子干渉RNA(siRNAもしくはRNAi)、またはshRNAが含まれる。
【0115】
相同組換えに関与する遺伝子産物の発現を活性化する代替法として、またはそれに加えて、Zp15を標的とするジンクフィンガー結合ドメインを有するこれらのタンパク質(またはその機能性フラグメント)の融合体を用いて、これらのタンパク質(組換えタンパク質)を対象の領域に誘導することができ、それによってそれらの局所的濃度を増加させ、さらに相同組換えプロセスを刺激することができる。あるいは、前述のような相同組換えに関与するポリペプチド(またはその機能性フラグメント)は、ジンクフィンガー結合ドメイン、切断ドメイン(または切断ハーフドメイン)、および組換えタンパク質(またはその機能性フラグメント)を含むトリプル融合タンパク質の一部であってよい。前述の方法および組成物に用いることができる、遺伝子変換および組換えに関連したクロマチン再構築に関与するさらなるタンパク質には、ヒストン・アセチルトランスフェラーゼ(例えば、Esa1p、Tip60)、ヒストン・メチルトランスフェラーゼ(例えば、Dot1p)、ヒストンキナーゼ、およびヒストンホスファターゼが含まれる。Bhatら,(1999)Plant J.33:455−469も参照されたい。
【0116】
ジンクフィンガー/ヌクレアーゼ融合分子およびドナーDNA分子を含む細胞における標的組換えの効率のさらなる上昇は、相同性駆動による修復プロセスが最大限に活発である細胞周期のG2期にある細胞を遮断することによって達成される。そのような停止は、多くの方法で達成され得る。例えば、細胞を例えば細胞周期の進行に影響を与える薬剤、化合物、および/または小分子で処理して、G2期で細胞を停止させることができる。この種類の例示的分子には、微小管重合に作用する化合物(例えば、ビンブラスチン、ノコダゾール、タキソール)、DNAと相互作用する化合物(例えば、シス−ジアンミン白金(II)ジクロリド(cis−platinum(II)diamine dichloride)、シスプラチン、ドキソルビシン)、および/またはDNA合成に作用する化合物(例えば、チミジン、ヒドロキシウレア、L−ミモシン、エトポシド、5−フルオロウラシル)が含まれるが、これに制限されない。組換え効率のさらなる上昇は、クロマチン構造を変更して、ゲノムDNAを細胞の組換え機構にさらに接近できるようにするヒストン・デアセチラーゼ(HDAC)阻害剤(例えば、酪酸ナトリウム、トリコスタチンA)の使用によって達成される。
【0117】
細胞周期の停止のためのさらなる方法には、例えば、当該タンパク質をコードするcDNAを細胞内に導入することによって、または当該タンパク質をコードする遺伝子の発現を活性化する遺伝子操作されたZFPを細胞内に導入することによって、CDK細胞周期キナーゼの活性を阻害するタンパク質の過剰発現が含まれる。細胞周期の停止は、例えばRNAi法を用いてサイクリンおよびCDKの活性を阻害することによって(例えば、米国特許第6,506,559号)、あるいは例えばサイクリンおよび/またはCDK遺伝子等の細胞周期の進行に関与する1つ以上の遺伝子の発現を抑制する遺伝子操作されたZFPを細胞内に導入することによっても達成される。遺伝子発現の制御のための遺伝子操作されたジンクフィンガータンパク質の合成のための方法について、例えば共同所有の米国特許第6,534,261号を参照されたい。
【0118】
あるいは、ある場合において、標的切断はドナーポリヌクレオチドの非存在下で行われ(好ましくはS期またはG2期に)、かつ組換えは相同染色体間で起こる。
【0119】
発現ベクター
本明細書に記載されるような1つ以上の融合タンパク質(例えば、ZFN)をコードする核酸を、複製および/または発現のために、原核細胞または真核細胞内への形質転換用ベクター内にクローニングすることができる。ベクターは、原核細胞用ベクター、例えばプラスミドまたはシャトルベクター、昆虫細胞用ベクター、あるいは真核細胞用ベクターであってよい。融合タンパク質をコードする核酸を、植物細胞への投与のための発現ベクター内にクローニングすることもできる。
【0120】
融合タンパク質(例えば、ZFN)を発現させるために、前記融合タンパク質をコードする配列を、典型的には、転写を指令するプロモーターを含有する発現ベクター内にサブクローニングする。適切な細菌および真核生物プロモーターは、当該技術分野において周知であり、例えばSambrookら,Molecular Cloning,A Laboratory Manual(2nd ed.1989;3rd ed.,2001);Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);ならびにCurrent Protocols in Molecular Biology(Ausubelら,前記参照)に記載されている。ZFPを発現させるための細菌発現システムは、例えば大腸菌、バチルス属菌、およびサルモネラ菌で利用可能である(Palvaら,Gene 22:229−235(1983)。そのような発現システム用のキットが市販されている。哺乳類細胞、酵母、および昆虫細胞のための真核細胞発現システムは当業者に周知であり、市販もされている。
【0121】
融合タンパク質をコードする核酸の発現を指令するのに用いられるプロモーターは、特定の用途に依存する。例えば、宿主細胞に適した強力な構成的プロモーターは、典型的に融合タンパク質の発現および精製のために用いられる。
【0122】
対照的に、融合タンパク質を植物遺伝子の制御のためにインビボで投与する場合(下記の「植物細胞への核酸の送達」の章を参照されたい)、前記融合タンパク質の特定の使用に応じて、構成的または誘導可能なプロモーターのいずれかが用いられる。植物プロモーターの制限されない例には、シロイヌナズナのユビキチン−3(ubi−3)(Callisら,1990,J.Biol.Chem.265−12486−12493);A.ツメファシエンス(A.tumifaciens)のマンノピン・シンターゼ(Δmas)(Petolinoら,米国特許第6,730,824号);および/またはキャッサバベインモザイクウイルス(CsVMV)(Verdaguerら,1996,Plant Molecular Biology 31:1129−1139)に由来するプロモーター配列が含まれる。実施例も参照されたい。
【0123】
プロモーターに加えて、発現ベクターは典型的に転写単位、あるいは原核または真核のいずれかの宿主細胞における核酸の発現に必要とされるすべてのさらなるエレメントを含有する発現カセットを含有する。したがって、典型的な発現カセットは、例えば融合タンパク質をコードする核酸配列に操作可能に連結されたプロモーター、および、例えば転写産物の効率的なポリアデニル化、転写終結、リボソーム結合部位、または翻訳終結に必要とされるシグナルを含有する。さらなるカセットのエレメントに、例えばエンハンサー、ヘテロスプライシングシグナル、および/または核局在化シグナル(NLS)が含まれてもよい。
【0124】
遺伝情報を細胞内へ輸送するのに用いられる特定の発現ベクターは、融合タンパク質の使用目的、例えば植物、動物、細菌、真菌、原生動物等々での発現(下記の発現ベクターを参照されたい)に関して選択される。標準的な細菌用および動物用発現ベクターは当該技術分野において既知であり、例えば米国特許公報第20050064474号A1、ならびに国際特許公報WO05/084190、WO05/014791、およびWO03/080809に詳細に記載されている。
【0125】
標準的トランスフェクション法を用いて、大量のタンパク質を発現する細菌、哺乳類、酵母、または昆虫の細胞株を作製することができ、次いでそれを標準的技術を用いることによって精製することができる(例えば、Colleyら,J.Biol.Chem.264:17619−17622(1989);Guide to Protein Purification, in Methods in Enzymology,vol.182(Deutscher,ed.,1990)を参照されたい)。真核および原核細胞の形質転換は、標準的技術に従って行われる(例えば、Morrison,J.Bact.132:349−351(1977);Clark−Curtiss&Curtiss,Methods in Enzymology 101:347−362(Wuら,eds.,1983)を参照されたい)。
【0126】
そのような宿主細胞内に外来ヌクレオチド配列を導入するための任意の周知の手法を用いてよい。これらには、リン酸カルシウムトランスフェクション、ポリブレン、プロトプラスト融合、エレクトロポレーション、超音波法(例えば、ソノポレーション)、リポソーム、マイクロインジェクション、ネイキッドDNA、プラスミドベクター、エピソーム型および組込み型の両方のウイルスベクター、ならびにクローン化されたゲノムDNA、cDNA、合成DNA、または他の外来遺伝物質を宿主細胞内に導入するための任意の他の周知の方法の使用が含まれる(例えば、前記のSambrookら,を参照されたい)。用いられる特定の遺伝子操作手法は、最適なタンパク質を発現し得る宿主細胞内に少なくとも1つの遺伝子を上手く導入できさえすればよい。
【0127】
植物細胞への核酸の送達
前述のように、DNA構築物を様々な従来技術によって所望の植物宿主内へ(例えば、そのゲノム内へ)導入することができる。そのような技術を見直すために、例えば、Weissbach&Weissbach,Methods for Plant Molecular Biology(1988,Academic Press,N.Y.)Section VIII,pp.421−463;およびGrierson&Corey,Plant Molecular Biology(1988,2d Ed.),Blackie,London,Ch.7−9を参照されたい。
【0128】
例えば、DNA構築物を、植物細胞プロトプラストのエレクトロポレーションおよびマイクロインジェクション等の技術を用いて植物細胞のゲノムDNA内に直接的に導入してもよく、あるいはDNA構築物を、DNA粒子砲撃等の微粒子銃法を用いて植物組織へ直接的に導入することができる(例えば、Kleinら,(1987)Nature 327:70−73を参照されたい)。あるいは、DNA構築物を、ナノ粒子による形質転換を介して植物細胞内に導入することもできる(例えば、参照することによりその全体として本明細書に組み入れられている、米国特許出願第12/245,685号を参照されたい)。あるいは、DNA構築物を適切なT−DNA境界/隣接領域とつなぎ合わせて、従来のアグロバクテリウム・ツメファシエンス宿主ベクター内に導入してもよい。バイナリーベクターを安全化および使用することを含む、アグロバクテリウム・ツメファシエンスを介した形質転換技術は、科学文献に十分に記載されている。例えば、Horschら,(1984)Science 233:496−498;およびFraleyら,(1983)Proc.Nat’l.Acad.Sci.USA 80:4803を参照されたい。
【0129】
さらに、遺伝子移入は、非アグロバクテリウム性の細菌またはウイルス、例えばリゾビウム種、NGR234、シノリゾビウム・メリロッティ、メソリゾビウム・ロッティ、ジャガイモウイルスX、カリフラワーモザイクウイルス、およびキャッサバベインモザイクウイルス、および/またはタバコモザイクウイルス等を用いて達成され得る。例えば、Chungら,(2006)Trends Plant Sci.11(1):1−4を参照されたい。
【0130】
バイナリーT DNAベクター(Bevan(1984)Nuc.Acid Res.12:8711−8721)または共培養手法(Horschら,(1985)Science 227:1229−1231)を用いることによって細胞を細菌に感染させる場合、アグロバクテリウム・ツメファシエンス宿主の毒性機能は、当該構築物および隣接マーカーを含有するT鎖の植物細胞DNA内への挿入を指令すると考えられる。一般に、アグロバクテリウム形質転換システムは、双子葉植物を遺伝子操作するために用いられる(Bevanら,(1982)Ann.Rev.Genet 16:357−384;Rogersら,(1986)Methods Enzymol.118:627−641)。アグロバクテリウム形質転換システムを用いて、DNAを単子葉性の植物および植物細胞に形質転換ならびに移入することもできる。米国特許第5,591,616号;Hernalsteenら,(1984)EMBO J 3:3039−3041;Hooykass−Van Slogterenら,(1984)Nature 311:763−764;Grimsleyら,(1987)Nature 325:1677−179;Boultonら,(1989)Plant Mol.Biol.12:31−40;およびGouldら,(1991)Plant Physiol.95:426−434を参照されたい。
【0131】
代替的な遺伝子移入および形質転換方法には、カルシウムを介した、ポリエチレングリコール(PEG)を介した、またはエレクトロポレーションを介したネイキッドDNAの取込みによるプロトプラスト形質転換(Paszkowskiら,(1984)EMBO J 3:2717−2722,Potrykusら,(1985)Molec.Gen.Genet.199:169−177;Frommら,(1985)Proc.Nat.Acad.Sci.USA 82:5824−5828;およびShimamoto(1989)Nature 338:274−276)、ならびに植物組織のエレクトロポレーション(D’Halluinら,(1992)Plant Cell 4:1495−1505)が含まれるが、これに制限されない。植物細胞の形質転換のためのさらなる方法には、マイクロインジェクション、炭化ケイ素を介したDNAの取込み(Kaepplerら,(1990)Plant Cell Reporter 9:415−418)、ならびにマイクロプロジェクタイル砲撃(Kleinら,(1988)Proc.Nat.Acad.Sci.USA 85:4305−4309;およびGordon−Kammら,(1990)Plant Cell 2:603−618)が含まれる。
【0132】
開示される方法および組成物を用いて、外因性配列を植物細胞ゲノムにおける所定の位置(例えば、Zp15遺伝子)内に挿入することができる。これは、植物ゲノム内に導入された導入遺伝子の発現はその組込み部位に決定的に依存するため有用である。したがって、例えば除草剤耐性、虫害抵抗性、栄養素、抗生物質、または治療用分子をコードする遺伝子を、標的組換えによってそれらの発現に有利な植物ゲノムの領域内に挿入することができる。
【0133】
前記の形質転換技術のいずれかによって作製される形質転換した植物細胞を培養して、形質転換した遺伝子型、ゆえに所望の表現型を有する植物全体を再生することができる。そのような再生技術は、典型的に、所望のヌクレオチド配列とともに導入されている殺生物剤および/または除草剤マーカーに依存している、組織培養成長培地におけるある植物ホルモンの操作に依存する。培養したプロトプラストからの植物再生は、Evansら,“Protoplasts Isolation and Culture”in Handbook of Plant Cell Culture,pp.124−176,Macmillian Publishing Company,New York,1983;およびBinding,Regeneration of Plants,Plant Protoplasts,pp.21−73,CRC Press,Boca Raton,1985に記載されている。植物のカルス、外植片、器官、花粉、胚、またはその一部から再生することもできる。そのような再生技術はKleeら,(1987)Ann.Rev.of Plant Phys.38:467−486に大まかに記載されている。
【0134】
植物細胞に導入された核酸を用いて、実質的に任意の植物に所望の特質を与えることができる。多種多様な植物および植物細胞システムを、本開示の核酸構築物および前述の種々の形質転換法を用いることによって、本明細書に記載される所望の生理学的かつ農学的特徴のために遺伝子操作することができる。好ましい実施態様では、遺伝子操作のための標的植物および植物細胞には、その単子葉および双子葉植物、例えば穀類作物(例えば、コムギ、トウモロコシ、イネ、キビ、オオムギ)、果実作物(例えば、トマト、リンゴ、ナシ、イチゴ、オレンジ)、飼料作物(例えば、アルファルファ)、根菜作物(例えば、ニンジン、ジャガイモ、サトウキビ、ヤムイモ)、葉菜作物(例えば、レタス、ホウレンソウ)を含む作物;顕花植物(例えば、ペチュニア、バラ、キク)、針葉樹、および松の木(例えば、パインファー、トウヒ);ファイトレメディエーションに用いられる植物(例えば、重金属を蓄積する植物);油料作物(例えば、ヒマワリ、ナタネ)、ならびに実験的目的のために用いられる植物(例えば、シロイヌナズナ)等が含まれるが、これに制限されない。したがって、開示される方法および組成物は、アスパラガス属、カラスムギ属、アブラナ属、ミカン属、スイカ属、トウガラシ属、カボチャ属、ニンジン属、ムカシヨモギ属、ダイズ属、ワタ属、オオムギ属、アキノノゲシ属、ドクムギ属、トマト属、リンゴ属、キャッサバ属、タバコ属、オオアラセイトウ(Orychophragmus)属、イネ属、ワニナシ属、インゲンマメ属、エンドウ属、ナシ属、サクラ属、ダイコン属、ライムギ属、ナス属、モロコシ属、コムギ属、ブドウ属、ササゲ属、およびトウモロコシ属由来の種を含むが、これに制限されない広範囲にわたる植物を使用する。
【0135】
当業者であれば、外因性配列をトランスジェニック植物に安定的に組み込み、操作可能であると確認した後に、それを生殖交配によって他の植物内に導入することができるということを理解するであろう。多くの標準的な育種技術のうちのいずれかを、交配される種に応じて用いることができる。
【0136】
形質転換するDNA上に存在するマーカー遺伝子によってコードされる特質について、遺伝子操作された植物物質を選択またはスクリーニングすることによって、形質転換した植物細胞、カルス、組織、または植物を同定かつ単離することができる。例えば、形質転換する遺伝子構築物によって耐性を与えられている、阻害量の抗生物質または除草剤を含有する培地上で遺伝子操作された植物物質を成長させることによって、選択を行うことができる。さらに、組換え核酸構築物上に存在し得る任意の可視的マーカー遺伝子(例えば、β−グルクロニダーゼ、ルシフェラーゼ、BまたはC1遺伝子)の活性についてスクリーニングすることによっても、形質転換した植物および植物細胞を同定することができる。そのような選択およびスクリーニング方法論は当業者に周知である。
【0137】
物理学的かつ生化学的方法を用いて、挿入した遺伝子構築物を含有する植物または植物細胞の形質転換体を同定することもできる。これらの方法には、:1)組換えDNAインサートの構造を検出かつ決定するためのサザン分析またはPCR増幅;2)遺伝子構築物のRNA転写産物を検出かつ調査するためのノーザンブロット、S1 RNaseプロテクション、プライマー伸長によるもしくは逆転写酵素によるPCR増幅;3)酵素またはリボザイムの活性を検出するための酵素アッセイであって、そのような遺伝子産物は遺伝子構築物によってコードされているアッセイ;4)遺伝子構築物の産物がタンパク質である場合、タンパク質ゲル電気泳動、ウェスタンブロット技術、免疫沈降、または酵素結合免疫測定法(ELISA)が含まれるが、これに制限されない。in situハイブリダイゼーション、酵素染色、および免疫染色等のさらなる技術を用いて、特定の植物器官および組織における組換え構築物の存在または発現を検出することもできる。これらすべてのアッセイを行うための方法は当業者に周知である。
【0138】
本明細書に開示される方法を用いた遺伝子操作の効果を、例えば、対象の組織から単離したRNA(例えば、mRNA)のノーザンブロットによって観察することができる。典型的には、mRNAが存在しているまたはmRNAの量が増加している場合、対応する導入遺伝子が発現していると推測することができる。遺伝子および/またはコードされたポリペプチドの活性を測定する他の方法を用いてもよい。用いられた基質、および反応産物または副産物の増加または減少を検出する方法に応じて、様々な種類の酵素アッセイを用いることができる。さらに、発現させたポリペプチドの量を、免疫化学的に、すなわちELISA、RIA、EIA、および当業者に周知の他の抗体に基づくアッセイ、例えば電気泳動検出アッセイ(染色またはウェスタンブロッティングのいずれかを伴う)等によって測定することができる。制限されない一例として、ELISAアッセイを用いたAAD−1およびPATタンパク質の検出は、参照することによりその全体として本明細書に組み入れられている、米国特許出願第11/587,893号に記載されている。植物のある組織においてまたはある発生段階において、導入遺伝子を選択的に発現させることができ、あるいは実質的にすべての植物組織において、実質的にその生活環全体にわたって、導入遺伝子を発現させることができる。しかしながら、任意の組み合わせ発現様式も適用可能である。
【0139】
本開示はまた、前記のトランスジェニック植物の種子を包含するものであって、前記種子は導入遺伝子または遺伝子構築物を有する。本開示はさらに、前記のトランスジェニック植物の後代、クローン、細胞株、または細胞を包含するものであって、前記後代、クローン、細胞株、または細胞は導入遺伝子または遺伝子構築物を有する。
【0140】
融合タンパク質(例えば、ZFN)および融合タンパク質をコードする発現ベクターを、遺伝子制御、標的切断および/または組換えのために、植物に直接投与することができる。ある実施態様では、植物は複数のパラロガス標的遺伝子を含有する。植物が複数のパラロガス遺伝子を含有し得ることは既知である。したがって、1つ以上の異なる融合タンパク質または融合タンパク質をコードする発現ベクターを植物に投与して、植物における1つ以上のZp15遺伝子を標的とすることができる。
【0141】
有効量の投与は、処理される植物細胞と最大限に接触して融合タンパク質を導入するために通常用いられる経路のいずれかによるものである。ZFPを、任意の適切な様式で、好ましくは許容可能なキャリアとともに投与する。そのようなモジュレーターを投与する適切な方法が使用可能で当業者に周知であり、また1つより多い経路を用いて特定の組成物を投与することができるが、特定の経路はしばしば別の経路より即時かつ効果的な反応を提供し得る。
【0142】
キャリアは、用いてもよく、投与される特定の組成物によって、ならびに前記組成物を投与するのに用いられる特定の方法によって、ある程度決定される。したがって、使用可能である多種多様なキャリアの適切な処方が存在する。
【実施例】
【0143】
以下は、本開示を実施するための特定の態様の実施例である。実施例は、例示を目的として提示されるものであって、決して本開示の範囲を制限することを意図されるものではない。
【0144】
用いた数(例えば、量、温度、等々)に関する正確性を確保するための努力はなされているが、いくらかの実験誤差および偏差は当然許容されるべきである。
【0145】
実施例1:Zp15標的遺伝子座の同定および特徴付け
公的に入手可能な遺伝子地図(Lawrence,C.ら,(2004)NAR 32:393−397;トウモロコシのインターネットのデータベース)およびトウモロコシのドラフトゲノム配列に基づき、第6染色体の短腕上にあるZp15遺伝子座を、位置およびZp15について入手可能な情報に基づくZFNを用いた改変の標的として選択した。トウモロコシ由来の15kDのβゼインをコードする遺伝子(Zp15)のゲノム構造および配列は、公に記載されかつ注釈が付されている(Wooら,(2001)Plant Cell 13(10):2297−2313)。Zp15遺伝子に対する配列は、参照することにより本明細書に組み入れられている、GenBankアクセス番号AF371264に記載されている。
【0146】
Zp15ゲノム配列を用いて、BLASTアルゴリズムを用いたTIGRおよびトウモロコシGDBデータベースへのクエリーを実行した。2つのコンティグ(AZM5_16782およびZmGSStuc11−12−04.8785.1)を含むがこれに制限されない、Zp15との重複相同性を有するいくつかの配列、ならびにいくつかのEST(M72708、M13507、M12147、AY103640、およびAF371264)が同定された。これらの登録の配列ならびにZp15配列に基づき、プライマー3プログラム(Rozen,S.およびSkaletsky,H.J.(2000)Primer3 on the WWW for general users and for biologist programmers.In:Krawetz S,Misener S(eds.)Bioinformatics Methods and Protocols:Methods in Molecular Biology.Humana Press,Totowa,NJ,pp365−386;インターネット上でも利用可能)を用いることによって、複数の短いオリゴヌクレオチドをPCRプライマーとしての使用のために設計した。
【0147】
これらのプライマーには、以下の正方向オリゴヌクレオチド:
P67F 5’−CGTATGAATTCATTGACAACC−3’(配列番号:1)
P68F 5’−ATGATCTATCTGTAAATCC−3’(配列番号:2)
P69F 5’−CGTCATGCAACGCAACATTCC−3’(配列番号:3)
P73F 5’−AAGAACATCACAAGTTATGC−3’(配列番号:4)
P74F 5’−TCATGTGGATCCAAGGCATC−3’(配列番号:5)
が含まれるが、これに制限されない。
【0148】
さらに、プライマーには、以下の逆方向オリゴヌクレオチド:
P70R 5’−ATGTGTGTCGTCTTACTGC−3’(配列番号:6)
P71R 5’−CAGTAGTAGGGCGGAATG−3’(配列番号:7)
P72R 5’−GGGCAGCTGGTACTG−3’(配列番号:8)
P75R 5’−CTATAATCGATGTAGAGC−3’(配列番号:9)
P76R 5’−CTATGCTTTGTCTATAGTCG−3’(配列番号:10)
が含まれるが、これに制限されない。
【0149】
すべてのオリゴヌクレオチドプライマーを、Integrated DNA Technologies(IDT,Coralville,IA)によって合成し、そこから購入した。トウモロコシ亜種Hi−II由来のgDNAの増幅を、30ngのgDNAを用いてPCRサーマルサイクラーで行った。Hi−II由来のZp15遺伝子に相当する2,215bpの増幅フラグメントを単離し、pCR2.1プラスミド(Invitrogen,Carlsbad,CA)内にクローニングした。このフラグメントの配列分析により、トウモロコシ亜種Hi−II由来のZp15のゲノム構造には、2つのエクソンおよび31bpの1つの小さなイントロン(配列番号:126)が含有されていることが明らかになった。Zp15標的ZFNの設計は、トウモロコシ亜種Hi−II由来のZp15遺伝子のコード領域に集中させた。
【0150】
実施例2:トウモロコシZp15遺伝子を標的とするジンクフィンガーヌクレアーゼの設計
ZFNに対する発現ベクターを組み立てるために、ライブラリーアーカイブから選択したまたは新たに合成したZFNコード遺伝子の任意の所定の対に対して適用可能である、段階的なモジュラークローニング方式を考案した。
【0151】
まず、Doyonら,(2008)Nature Biotechnology 26(6):702および米国特許出願第12/284,887号に記載されているような酵母アッセイ選別を用いて、Zp15標的ZFNをスクリーニングした。簡潔には、Zp15遺伝子座全体を出芽酵母ゲノムのHO遺伝子座内に導入して、標的遺伝子内の種々の結合部位におけるZFN活性を直接比較し、Doyonらに記載されているようなレポーターアッセイ(MEL1)を用いることによって、レポーター遺伝子においてDSBを誘導し得るそれらの能力についてZFNをスクリーニングした。
【0152】
これらのプロキシシステムアッセイの結果に基づき、試験された様々なZFN対がZp15内でDSBを誘導し得ることが確認された。
【0153】
酵母プレスクリーニングに続き、次いでZFN対をトウモロコシ特異的発現ベクター内にサブクローニングした。米国特許公報第20080182332号に記載されているように、トウモロコシop−2に由来する再設計かつ合成された核局在化シグナル(NLS)のセグメント、およびトウモロコシのコドンバイアスを使用しているFokIヌクレアーゼドメインを含むベクターを、固有のXhoI部位の下流に1ヌクレオチド(C)を挿入して改変し、追加のSacI部位を創出した。同様のベクターを、Thosea asignaウイルス由来の2Aリボソーム断続(stuttering)配列を含むように改変した。個々のジンクフィンガータンパク質のORFをコードする遺伝子カセットを、これらのベクターのいずれかの中にKpnIおよびBamHI制限酵素認識部位を用いてクローニングし、続いて前記2つのベクターをBglII/XhoI制限酵素認識部位を用いて組み合わせ、NcoIおよびSacI制限酵素認識部位が隣接した2つのZFNコードドメインを含むカセットを含有する中間構築物を産生した。
【0154】
NcoI/SacIカセットをこの中間構築物から制限酵素消化によって切り出し、トウモロコシのユビキチン−1遺伝子由来のプロモーター(Sharrockら,(1992)Plant Mol Biol.18(4):675)、およびトウモロコシ根の選択的カチオン性ペルオキシダーゼ遺伝子由来のターミネーター配列(米国特許第7,179,902号)を含有する、プラスミド骨格のpDAB3872内にライゲーションした。
【0155】
結果として生じたプラスミドには、ZFN遺伝子、さらにプラスミド維持のための関連する選択可能なマーカー、およびInvitrogen(Carlsbad,CA)製のGATEWAY(商標)システムを用いた便利な操作のための隣接attL部位が含まれる。このクローニング方式を用いて作製されたZFN構築物のそれぞれを、大腸菌DH5α細胞内に形質転換し、その後適切な選択の下で維持した。
【0156】
表1は、Zp15遺伝子座内への標的組込み実験に用いた例示的なZp15を標的とするZFNを示す。ZFNに対するDNA標的配列を2列目に示し(DNA標的部位を大文字で表示;接触しないヌクレオチドを小文字で表示)、3〜6列目は、前記タンパク質におけるジンクフィンガーのそれぞれ(F1〜F4)の認識領域のアミノ酸配列(ヘリックスの開始に対してアミノ酸−1〜+6)を示す。また、表1の1列目にはそれぞれのタンパク質に対する識別番号を提示する。
【表1】
【表2】
【0157】
ZFNをC2H2またはC3H骨格内に容易に挿入できること、および種々の配列を用いてジンクフィンガータンパク質と切断ドメインを接合することができることは明白であろう。米国特許公報第20080182332号、特にそのような配列に関する表6を参照されたく、この参考文献は参照することによりその全体として本明細書に組み入れられている。
【0158】
実施例3:トウモロコシ細胞におけるZFNを介したZp15の破壊
ZFN対によるDSBの誘導を試験した。内在性Zp15遺伝子の意図される標的部位においてDSBを効率的に誘導し得るZFN対を同定した。ZFN誘導性断裂の部位において小さなDNA欠失/挿入をもたらすことが既知である非相同末端結合(NHEJ)によるDSB修復のエラーを起こしやすい性質を利用して、内在性Zp15遺伝子標的部位に効率的に結合かつ切断するZFN対を選択した。
【0159】
ZFNを培養トウモロコシ細胞で一過性に発現させ、予測される切断部位における標的遺伝子座の配列分析を行った。例えば、ZFN対#25(前記で示した11768/11766認識ヘリックス)をコードするプラスミドpDAB7468を、参照することによりその全体として本明細書に組み入れられている、米国特許出願第12/001,939号に記載されているような、WHISKERS(商標)を介した形質転換によってトウモロコシHi−II細胞培養物内へ送達した。一過性の発現の24時間または72時間後のいずれかに、結果として生じた破壊されたZFN標的配列を、単離したゲノムDNAから増幅し、プラスミドベクターpCR2.1内にクローニングした。gDNAを酵素Bsu36Iを用いた制限酵素消化にかけ、その後にZp15標的配列の増幅およびPCR産物のプラスミドベクターpCR2.1内へのクローニングが続いた。クローン化された増幅産物の個々のコロニーを、プラスミドDNAの制限酵素消化によって分析し、その後にアガロースゲル電気泳動が続いた(制限酵素Bsu36Iによる切断に対して耐性を示したクローン化された増幅産物は、ZFN切断部位に関連した制限酵素認識部位を破壊する変異を含有していると考えられた)。
【0160】
192クローンの直接配列分析によって、6bpの挿入が明らかになった(図1)。別の実施例では、ZFN対#24(前記で示した11753/11750認識ヘリックス)をコードするプラスミドpDAB7467を、トウモロコシ培養細胞物内に直接送達し、一過性の発現の24時間または72時間後のいずれかに、ZFN標的配列を増幅し、プラスミドベクターpCR2.1内にクローニングした。192クローンの直接配列分析によって、正確な切断部位における3bpの欠失が明らかになった(図2)。ここに記載される挿入および欠失は、標的部位において誘導されたDSBのNHEJ修復の結果であり、かつZFN24および25がトウモロコシ細胞内の内在性Zp15遺伝子座において切断活性を有することを示す。
【0161】
ZFN25または28を用いて同じ手法を行ったが、コロニーを制限酵素消化によってスクリーニングする代わりに、192の独立したクローンを直接シークエンシングした。6bpの挿入が検出された(図2、上)
【0162】
まとめると、これらのデータは、ZFNへの一過性の暴露は培養トウモロコシ細胞内のZp15遺伝子座において標的DSBを誘導するのに十分であることを証明している。
【0163】
実施例4:Zp15遺伝子座内への標的組込み
Zp15における切断活性を有する設計されたZFNが、外因性配列の組込みを促進し得るかどうかを試験するために、本発明者らは、例示的な外因性の除草剤耐性遺伝子であるスフィンゴモナス・ヘルビシドボランス由来のAAD−1(ATCC700291)をコードする自律的遺伝子カセットを保持するドナーDNA分子を構築した。AAD−1は、アリールオキシアルカノエート・ジオキシゲナーゼ酵素をコードし、アリールオキシフェノキシプロピオン酸系除草剤に対する耐性を与える(国際特許WO2005/107437,WO2008141154A2)。当業者であれば、関連したADD−12遺伝子等の他の除草剤耐性遺伝子を含むが、これに制限されない他の外因性核酸を、ドナーDNA分子内に同様に組み入れることができるということを理解するであろう。この除草剤耐性遺伝子ドナーにおいて、プロモーター配列はO.サティバのアクチンに由来しており(GenBankアクセス番号S44221およびX63830)、かつターミネーター配列はZ.メイスのL.リパーゼに由来している(GenBankアクセス番号L35913)。
【0164】
A.ドナーDNA分子の構築
Zp15に相同な領域を含有するドナー構築物を以下のように作製した。Zp15遺伝子に対して相同な隣接部を含有するプラスミド骨格を、Zp15遺伝子の対応する標的部位内への任意のドナーDNA配列の組込みが可能になるように遺伝子操作した。ここに例示したプラスミド骨格は、基本プラスミドベクターpBC SK(−)ファージミド(3.4kbp)(Stratagene,La Jolla,CA)に由来したものであった。この手法には4つの工程があった。
【0165】
第一に、3μgのpBC SK(−)をSpeIおよびSalI(New England Biolabs,Beverly,MA)制限エンドヌクレアーゼを用いて線状化することによって、基本プラスミドを調製した。3.3kbpのSpeI/SalI消化したサブクローニングベクターのpBC SK(−)をゲルから切り出し、QIAQUICKゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。
【0166】
第二に、Integrated DNA Technologies社(Coralville,IA)によって合成された以下のオリゴヌクレオチドプライマー:
Zp15HRドナーNotI(205)F:5’−GCGGCCGCATGCAAGAGCTGTTGATC−3’(配列番号:97)
Zp15HRドナーMfeI(1025)R:5’−CAATTGCCGGCGTAGTAGGGCGCCGCCGCCAGC−3’(配列番号:98)
Zp15HRドナーMfeI(1025)F:5’−CAATTGGTGTGGGCAGCCGAGCGCCATGTTCCAG−3’(配列番号:99)
Zp15HRドナーSalI(2270)R:5’−GTCGACCGATACTGATGCGGACCGTCCACCTTGTC−3’(配列番号:100)
を用いて、Zp15から5’および3’相同隣接部を単離した。
【0167】
PCR増幅反応を、LA TAQ PCRキット(TaKaRa Biotechnology社,Otsu,Shiga,Japan)に備わった試薬を用いて行った。PCR反応カクテルは、5μLの10×LA PCR(商標)バッファーII(Mg2+)、20ngの二本鎖テンプレート(Hi−IIトウモロコシゲノムDNA)、10pmolの正方向オリゴヌクレオチドプライマー、10pmolの逆方向オリゴヌクレオチドプライマー、8μLのdNTPミックス(2.5mMの各dNTP)、33.5μLのH2O、0.5μL(2.5単位)のTaKaRa LA Taq(商標)DNAポリメラーゼ、1滴のミネラルオイルからなるものであった。プライマーZp15HRドナーNotI(205)FおよびZp15HRドナーMfeI(1025)Rを一反応に用い、プライマーZp15HRドナーMfeI(1025)FおよびZp15HRドナーSalI(2270)Rを第二の反応に用いた。Perkin−Elmer Cetus,48サンプルDNAサーマルサイクラー(Norwalk,CT)を用いて、以下のサイクル条件:94℃,4分間/1サイクル;98℃ 20秒間,65℃ 1分間,68℃ 1分間/30サイクル;72℃,5分間/1サイクル;4℃/ホールドの下でPCR反応を行った。15μlの各PCR反応物を電気泳動し、増幅されたフラグメントを紫外線で可視化し、フラグメントの大きさを1kbpのDNAラダーと比較することによって概算した。期待されるプラスミドクローンを、5’フラグメントについては825bp、または3’フラグメントについては1,250bpのDNAフラグメントの存在によって診断した。これらのフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、精製したフラグメントをTOPO TAクローニング(登録商標)キットを用いてpCR2.1プラスミド内にクローニングし、化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)内にメーカーのプロトコールに従って形質転換した。
【0168】
825bpの5’フラグメントまたは1,250bpの3’フラグメントを含有する個々のコロニーを、制限酵素消化およびシークエンシングデータによって同定かつ確認した。825bpの5’フラグメントを含有するコロニーは、MfeIおよびNotI(New England Biolabs,Beverly,MA)の制限酵素消化によって確認された。1,250bpの3’フラグメントを含有するコロニーは、SalI(New England Biolabs,Beverly,MA)を用いた制限酵素消化によって同定かつ確認された。期待されるプラスミドクローンを、3.9kbpのpCR(登録商標)2.1ベクターに加えて、825bp(5’フラグメント)または1,250bp(3’フラグメント)の挿入されたDNAフラグメントの存在によって診断した。プラスミドクローンの二本鎖シークエンシング反応を、CEQ(商標)DTCS−クイックスタートキット(Beckman−Coulter,Palo Alto,CA)を用いて、メーカーによって記載されるとおりに行った。反応物を、Performa DTRゲル濾過カートリッジ(Edge BioSystems,Gaithersburg,MD)を用いて、メーカーのプロトコールに記載されるとおりに精製した。シークエンシング反応物をBeckman−Coulter CEQ(商標)2000 XL DNA分析システム上で分析し、ヌクレオチドの特徴付けをSEQUENCHER(商標)バージョン4.1.4(Gene Codes社,Ann Arbor,MI)を用いて行った。Zp15由来の5’および3’相同フラグメントの配列を、配列番号127および配列番号:128に示す。
【0169】
第三に、3’相同隣接部を、以下のように基本プラスミド内にライゲーションした。正確な3’相同隣接部配列を含有するクローンを制限酵素で消化し、DNAフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いて精製した。これらのフラグメントを、あらかじめSpeI/SalI(前記参照)で消化された精製した基本プラスミド内に、16℃のウォータバス中で16時間のインキュベーションの条件下、20μLの反応容量中に500単位のT4 DNAリガーゼ(Invitrogen Life Technologies,Carlsbad,CA)を用いて、1:5のベクター:インサートの割合でライゲーションした。その後、5μLのライゲーション反応物をワンショット(登録商標)Top10化学的コンピテントセル(Invitrogen Life Technologies,Carlsbad,CA)内に形質転換し、抗生物質選択を含有する培地上にプレーティングした。推定コロニーを単離し、SpeIおよびSalI制限酵素(New England Biolabs,Beverly,MA)で消化して、ライゲーションされた3’フラグメントを含有するクローンを同定した。
【0170】
第四に、5’相同隣接部を、3’相同隣接部を含有するプラスミド内にライゲーションした。前記の第三の工程で記載した3’相同隣接部を含有するプラスミドを、MfeIおよびNotI(New England Biolabs,Beverly,MA)制限エンドヌクレアーゼで37℃にて1時間消化した。3’相同隣接部を含有するNotI/MfeI消化されたプラスミドをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。
【0171】
MfeIおよびNotI制限酵素を用いた制限酵素消化によって生成された5’相同隣接部ドナーの単離したフラグメントを作製し、1:5のベクター:インサートの割合および500単位のT4 DNAリガーゼ(Invitrogen Life Technologies,Carlsbad,CA)を用いた20μLのライゲーション反応液中で、3’相同隣接部を含有するプラスミドとライゲーションした。ライゲーション反応液を16℃のウォータバス中で16時間インキュベーションした。
【0172】
ライゲーションの後、5μLのライゲーション反応物を、MAX EFFICIENCY(登録商標)DH5α(商標)化学的コンピテントセル(Invitrogen Life Technologies,Carlsbad,CA)内にメーカーの推奨どおりに形質転換した。個々のコロニーを選択し、プラスミドDNAを単離し、かつNotI制限酵素(New England Biolabs,Beverly,MA)で消化して、5’相同隣接部ドナーの組み込まれたフラグメントを含有するプラスミドを同定した。結果として生じたプラスミドをpDAB7489と命名した(図3)。
【0173】
プロモーターを含有する植物転写単位(PTU)、除草剤耐性遺伝子、およびポリアデニル化(ポリA)終結配列を含む除草剤耐性遺伝子発現カセットを構築した。前記プロモーター配列は、O.サティバのアクチンに由来する(McElroyら,(1990)Plant Cell 2,163−171;GenBankアクセス番号S44221およびGenBankアクセス番号X63830)。前記除草剤耐性遺伝子は、アリールオキシフェノキシプロピオン酸系除草剤に対する耐性を与える(WO2005/107437)、AAD−1(アリールオキシアルカノエート・ジオキシゲナーゼ)遺伝子を含んでいた。利用した遺伝子のバージョンは、植物における発現に対してコドン最適化配列を含むバージョン#3であった。ターミネーター配列は、Z.メイスのL.リパーゼ(GenBankアクセス番号L35913のトウモロコシリパーゼcDNAクローン)に由来する。このトウモロコシ配列は、AAD−1遺伝子に対する3’非翻訳領域/転写ターミネーター領域を含む。前記除草剤耐性遺伝子発現カセットを図4に示す。
【0174】
このカセットを作製するために、以下のオリゴヌクレオチドプライマー:
OsActAad1v.3ZmLipMfeIF 5’−CAATTGGTCATTCATATGCTTGAGAAGAG−3’(配列番号:101)
OsActAad1v.3ZmLipMfeIR 5’−CAATTGAGCACTTAAAGATCTTTAGAAG−3’(配列番号:102)
をIntegrated DNA Technologies社(Coralville,IA)によって標準的脱塩の条件下で合成し、0.125μg/μLの濃度まで水で希釈した。
【0175】
PCR増幅反応を、LA TAQ PCRキット(TaKaRa Biotechnology社,Otsu,Shiga,Japan)を用いて行った。PCR反応カクテルは、5μLの10×LA PCR(商標)バッファーII(Mg2+)、20ngの二本鎖テンプレート(pDAB3878プラスミドDNA)、10pmolの正方向オリゴヌクレオチドプライマー、10pmolの逆方向オリゴヌクレオチドプライマー、8μLのdNTPミックス(それぞれ2.5mM)、33.5μLのH2O、0.5μL(2.5単位)のTaKaRa LA Taq(商標)DNAポリメラーゼ、1滴のミネラルオイルを含んでいた。Perkin−Elmer Cetus,48サンプルDNAサーマルサイクラー(Norwalk,CT)を用いて、以下のサイクル条件:94℃,4分間/1サイクル;98℃ 20秒間,55℃ 1分間,68℃ 3分間/30サイクル;72℃,5分間/1サイクル;4℃/ホールドの下でPCR反応を行った。15μlの各PCR反応物を、0.5%エチジウムブロマイドを補充した1.0%TAEアガロースゲル中で1時間100Vで電気泳動した。増幅されたフラグメントを紫外線で可視化し、フラグメントの大きさを1kbpのDNAラダーと比較することによって概算した。
【0176】
期待されるPCR産物を、2.7kbpのDNAフラグメント(AAD−1 PTU)の存在によって診断した。これらのフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、精製したフラグメントを、TOPO TAクローニング(登録商標)キット(pCR(登録商標)2.1ベクターを備えた)および化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR2.1プラスミド内にクローニングした。
【0177】
個々のコロニーを選択し、プラスミドDNAを単離し、かつ制限酵素MfeI(New England Biolabs,Beverly,MA)で消化した。期待されるプラスミドクローンを、3.9kbpのpCR(登録商標)2.1ベクターに加えて、2,674bp(AAD−1 PTU)の挿入されたDNAフラグメントの存在によって診断した。プラスミドクローンの二本鎖シークエンシング反応を、CEQ(商標)DTCS−クイックスタートキット(Beckman−Coulter,Palo Alto,CA)を用いて、メーカーによって記載されるとおりに行った。反応物を、Performa DTRゲル濾過カートリッジ(Edge BioSystems,Gaithersburg,MD)を用いて、メーカーのプロトコールに記載されるとおりに精製した。シークエンシング反応物をBeckman−Coulter CEQ(商標)2000 XL DNA分析システム上で分析し、ヌクレオチドの特徴付けをSEQUENCHER(商標)バージョン4.1.4(Gene Codes社,Ann Arbor,MI)を用いて行った。
【0178】
正確な2,674bpの配列を含有するクローン由来の制限酵素消化されたフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、このフラグメントを、制限酵素MfeIで消化され、かつその後脱リン酸化されている、精製したpDAB7489(プラスミド骨格)とライゲーション反応でつなぎ合わせた。以下の条件下:1:5のベクター:インサートの割合、および16℃のウォータバス中で16時間のインキュベーションの条件下、20μLの反応容量中に500単位のT4 DNAリガーゼ(Invitrogen Life Technologies,Carlsbad,CA)でライゲーションを行った。その後、5μLのライゲーション反応物を、50μlの大腸菌MAX EFFICIENCY(登録商標)DH5α(商標)化学的コンピテントセル(Invitrogen Life Technologies,Carlsbad,CA)内に形質転換し、メーカーによって記載される選択条件下でプレーティングした。
【0179】
個々のコロニーを選択し、プラスミドDNAを単離し、かつ制限酵素MfeI(New England Biolabs,Beverly,MA)で消化した。期待されるプラスミドクローンは、DNAフラグメント2,674bp(AAD−1 PTU)および5,413bp(pDAB7489ベクター)を含有していた。結果として生じたプラスミドをpDAB7490と命名した(図5)。
【0180】
トウモロコシ亜種Hi−IIの胚発生細胞培養物(Armstrongら,(1991)Maize Genet Coop Newsletter 65:92−93)を作製し、維持し、かつZFN24およびドナー分子pDAB7490をコードするプラスミドの同時形質転換にかけた。カルス組織の形質転換および選択、ならびにその後の形質転換体の再生は、米国特許出願第12/001,939号に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。形質転換および選択のプロトコールに関するさらなるガイダンスのために、Petolinoら,(2000)Plant Cell Rept.19:781−786を参照されたい。開花後、植物を自己授粉またはトウモロコシ亜種DAS5XH751と異種交配させた。結果として生じた後代種子を収穫し、乾燥させた。無傷の、稔性のトウモロコシ植物へのカルスの再生は、米国特許出願第12/001,939号、特に実施例22に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。
【0181】
B.Zp15遺伝子座内へのAAD−1遺伝子カセットの標的組込み
除草剤耐性遺伝子カセットをコードする組み込まれたドナーDNA分子を含有する除草剤耐性事象のうち、前記事象のある割合は、ZFN誘導性二本鎖断裂の部位へのドナーDNAの標的組込みの産物であると予想されている。これらの標的組込み事象を除草剤耐性遺伝子カセットのランダムな組込みに由来するものと区別するために、ゲノム特異的およびそれに続くゲノム特異的、さらにドナー特異的PCRプライマーを用いたPCRに基づく遺伝子型同定ストラテジーを利用した。
【0182】
すべての除草剤耐性形質転換事象におけるAAD−1導入遺伝子の標的組込み対ランダムな組込みについての示差的遺伝子型同定を、Zp15遺伝子座およびAAD−1遺伝子に特異的なPCRに基づくアッセイを用いて行った。ここに提示される実施例では、すべてのオリゴヌクレオチドプライマーを、標準的脱塩の条件下でIntegrated DNA Technologies社(Coralville,IA)によって合成し、100μMの濃度まで水で希釈した。以下の正方向および逆方向オリゴヌクレオチドプライマーのセット;
HB501f:5’−AAGGTCCCAAATCTGAGGCATACTGTTGCT−3’(配列番号:103)
HB502r:5’−GAGGTCCTATGCTTTGTCTATAGTCGGCAG−3’(配列番号:104)
を、ドナーDNA配列の境界の外側に位置するZp15遺伝子標的に特異的なgDNAにアニールするように設計した。
【0183】
正方向および逆方向オリゴヌクレオチドプライマーの第二のセット:
HB503f:5’−GGCATACTGTTGCTGCCCTGCTGGAA−3’(配列番号:105)
HB504r:5’−GACACCTATAATCGATGTAGAGCCGAAGAG−3’(配列番号:106)
も、ドナーDNA配列の境界の外側のZp15遺伝子標的に特異的なgDNAにアニールするように設計し、しかしながら第一の対の中に入れ子にした。
【0184】
正方向および逆方向オリゴヌクレオチドプライマーを、AAD−1除草剤耐性遺伝子のコード領域に対応するドナーDNAに特異的にアニールするようにさらに設計した。
HB505f:5’−AGTCCACCCCAGTGATCTCAGCACCA−3’(配列番号:107)
HB506f:5’−AGTGGCTGGACAGCTATTCTCTCAAAGCGT−3’(配列番号:108)
HB507r:5’−ACGCTTTGAGAGAATAGCTGTCCAGCCACT−3’(配列番号:109)
HB508r:5’−TGGTGCTGAGATCACTGGGGTGGACT−3’(配列番号:110)
【0185】
2つの別個なプライマリー増幅反応を、Zp15ゲノム領域で結合するプライマーおよびドナー分子を利用して行い、ゲノムとドナーとの間の組込みの境界にまたがるアンプリコンを生じた。第一の反応は、ゲノムとドナーとの間の5’境界に焦点を合わせ、プライマーセットHB501fおよびHB507rを用いた。第二の反応は、ドナーとゲノムとの間の3’境界に焦点を合わせ、プライマーセットHB505fおよびHB502rを用いた。形質転換したトウモロコシHi−II事象からゲノムDNAを単離した。プライマリーPCR増幅反応を、LA TAQ PCRキット(TaKaRa Biotechnology社,Otsu,Shiga,Japan)によって提供されている試薬を用いて行った。PCR反応カクテルは、2.5μlの10×La Taq PCR(商標)バッファー、40〜200ngの二本鎖ゲノムDNAテンプレート、10μMの正方向オリゴヌクレオチドプライマー、10μMの逆方向オリゴヌクレオチドプライマー、2μlのdNTPミックス(それぞれ2.5mM)、16.25μlのH2O、0.25μl(1.25単位)のLA Taq(商標)DNAポリメラーゼからなるものであった。Bio−Rad,96サンプルDNA Engine Tetrad2,Peltierサーマルサイクラー(Hercules,CA)を用いて、以下のサイクル条件:94℃,2分間/1サイクル;94℃ 30秒間,62℃ 30秒間,68℃ 5分間/30サイクル;4℃/ホールドの下でPCR反応を行った。
【0186】
続いて、プライマリーPCRの反応産物を水で1:100に希釈し、2つの別個なセカンドPCR反応のためのテンプレートDNAとして用いた。セカンド反応も、Zp15ゲノム領域で結合するプライマーおよびドナー分子を利用し、ゲノムとドナーとの間の組込みの境界にまたがるアンプリコンを生じる。特異的プライマーの同一性によって、増幅がゲノムとドナーとの間の5’または3’境界のいずれかに焦点を合わせるかが決定される。第一の反応は、ゲノムとドナーとの間の5’境界に焦点を合わせ、プライマーセットHB503fおよびHB508rを用いた。第二の反応は、ドナーとゲノムとの間の3’境界に焦点を合わせ、プライマーセットHB506fおよびHB504rを用いた。両方の反応は、以下の:2.5μlの10×La Taq PCR(商標)バッファー、1μlのテンプレート[1oPCR反応の1:50希釈]、10μMの正方向オリゴヌクレオチドプライマー、10μMの逆方向オリゴヌクレオチドプライマー、2μlのdNTPミックス(それぞれ2.5mM)、16.25μlのH2O、0.25μl(1.25単位)のLA Taq(商標)DNAポリメラーゼからなるものであった。Bio−Rad,96サンプルDNA Engine Tetrad2,Peltierサーマルサイクラー(Hercules,CA)を用いて、以下のサイクル条件:94℃,2分間/1サイクル;94℃ 30秒間,62℃ 30秒間,68℃ 5分間/30サイクル;4℃/ホールドの下でPCR反応を行った。5’境界に対する2,180bpまたは3’境界に対する2,980bpの期待されるPCRアンプリコンフラグメントが観察された。
【0187】
これらのフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。続いて、精製したフラグメントを、TOPO TAクローニング(登録商標)キット(pCR(登録商標)2.1ベクターを備えた)および化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR2.1プラスミド内にクローニングした。
【0188】
個々のコロニーを選択し、プラスミドDNAを単離し、かつ制限酵素EcoRI(New England Biolabs,Beverly,MA)で消化した。期待されるプラスミドクローンを、3.9kbpのpCR(登録商標)2.1ベクターに加えて、適切な大きさの挿入されたDNAフラグメントの存在によって診断した。
【0189】
プラスミドクローンの二本鎖シークエンシング反応を行い、ヌクレオチドの特徴付けおよびアラインメントをSEQUENCHER(商標)バージョン4.1.4(Gene Codes社,Ann Arbor,MI)を用いて行った。
【0190】
Zp15標的遺伝子内に挿入されたAAD−1ドナー遺伝子カセットの標的組込み事象(事象#147)から得られた、選択した配列データを図6のアラインメントに示す。
【0191】
ゲノムとドナーとの間の5’または3’境界のいずれかに焦点を合わせたプライマリーPCR増幅産物を、同様にゲノムとドナーとの間の5’または3’境界のいずれかに焦点を合わせたセカンド増幅にかけた。これらのセカンド増幅産物に対応するクローン化フラグメントと、野生型Zp15ゲノム配列、ならびに標的組込み事象の期待される配列とのアラインメントは、標的部位においてドナーDNAの正確な組込みが起こったことをはっきりと示している。Zp15ゲノム遺伝子座のヌクレオチド配列、ゲノム/ドナーの境界、Zp15相同隣接部に対応するドナー領域のヌクレオチド配列、および除草剤耐性カセットのヌクレオチド配列は、この事象に由来する複数のクローン化PCR産物においてすべて保存されていた。したがって、この事象は、ZFNを介した二本鎖断裂の相同性駆動による修復、および特定の遺伝子標的におけるドナーDNAの標的組込みが起きているゲノムを表す。図6において、本発明者らは1つの代表的な単離した形質転換トウモロコシカルスから得られた配列アラインメントデータを示す(事象#147)。
【0192】
固有の標的組込みの発生を表すさらなる形質転換事象が得られており、本明細書で教示された方法がトウモロコシカルスにおいて再現可能であることを証明している。
【0193】
実施例5:Zp15遺伝子座内へのPATの標的組込み
本明細書に開示される標的遺伝子座内への選択された外因性ポリヌクレオチドの標的組込みのための方法のさらなる例示として、PATをコードする自律的遺伝子カセットを保持するさらなるDNAドナー分子を設計かつ構築し、内在性Zp15標的遺伝子座内への外因性ドナー配列の組込みを試験した。この遺伝子座でDSBを引き起こすために、内在性Zp15遺伝子標的の特異的切断活性を有するZFNを配置した。続いて、ストレプトマイセス・ビリドクロモゲネス由来のPATをコードする自律的遺伝子カセットを保持するドナーDNA分子およびZp15に相同な隣接配列を、Zp15標的のZFN誘導性DSB内に組み込んだ。PATは、ホスフィノトリシン・アセチル・トランスフェラーゼ酵素をコードし、アセチル化によって除草活性化合物ホスフィノトリシン(PPT)に対する耐性を与える(米国特許第5,633,434号)。ホスフィノトリシンは、除草剤のリバティ、バスタ、およびイグナイトの活性成分である。PATコード配列を植物転写単位(PTU)として構築し、O.サティバのアクチン(GenBankアクセス番号S44221およびX63830)に由来するプロモーター配列、およびZ.メイスのL.リパーゼ(GenBankアクセス番号L35913)に由来するターミネーター配列を含有させた。
【0194】
A.ドナーDNA分子の構築
Zp15に相同な領域を含有するZp15ドナー構築物を、以下のように合成して作製した。pDAB7489由来のヌクレオチド4595−5346(5’相同配列;配列番号:111)およびヌクレオチド21−796(3’相同配列;配列番号112)を含むZp15相同領域を設計した。この相同領域は、5’および3’相同エレメントの間にMfeIクローニング部位、ならびに5’および3’末端にNotI制限酵素認識部位を含んでいた。このDNA配列(配列番号:113)を合成し、カナマイシン耐性ColE1型プラスミドであるpMK内のSacIおよびKpnIクローニング部位(Gene Art Ag,Regensburg,Germany)に挿入した。結果として生じたドナープラスミドはpDAB104101と称され(図7)、Zp15遺伝子の対応する標的部位内への対象の任意のDNA配列の組込みを可能にする、Zp15遺伝子に対する相同隣接部を含有している。
【0195】
プロモーター、除草剤耐性遺伝子、およびポリアデニル化(ポリA)終結配列を含有する完全なPTUを含む除草剤耐性遺伝子発現カセットを構築した。前記プロモーター配列は、O.サティバのアクチンに由来する(McElroyら,(1990)Plant Cell 2:163−171)。GenBankアクセス番号S44221およびGenBankアクセス番号X63830。前記除草剤耐性遺伝子はPATであった。前記終結配列は、Z.メイスのL.リパーゼ(GenBankアクセス番号L35913のトウモロコシリパーゼcDNAクローン)に由来する。このトウモロコシ配列は、PAT遺伝子に対する3’非翻訳領域/転写ターミネーター領域を含む。PAT除草剤耐性遺伝子発現カセットを図8に示す。
【0196】
PAT遺伝子カセットを、Integrated DNA Technologies社(Coralville,IA)によって合成されたプライマー:
DC001 5’−CCAGTGCAATTGGGTCATTCATATGCTTGAGAAG−3’(配列番号:114)
DC002 5’ −CCAGTGCAATTGAATTCAGCACTTAAAGATCTTTAG−3’(配列番号:115)
を用いたPCRによって、プラスミドpDAB102256から増幅した。
【0197】
PCR増幅反応を、PHUSION(商標)DNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,60℃ 20秒間,72℃ 45秒間/9サイクル;98℃,10秒間,72℃ 60秒間/24サイクル;72℃,10分間/1サイクル;4℃/ホールドの下で行った。PCR反応物を、1.0%TAEアガロースゲル中での電気泳動によって分析した。
【0198】
期待されるPCR産物を、2.3kbpのDNAフラグメント(PAT PTU)の存在によって診断した。このフラグメントを切り出し、QIAQUICK(商標)ゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従ってゲルから精製した。次いで、精製したフラグメントを、ゼロブラントTOPO PCRクローニングキットおよび化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、pCR−ブラントII−TOPOベクター内にクローニングした。
【0199】
個々のコロニーを選び、プラスミドDNAを単離し、かつプラスミドDNA制限酵素消化および配列分析にかけた。クローン化したPATインサートをシークエンシングして、クローン化したPCR産物(配列番号:116)の同一性および配列忠実性を証明した。1つのそのようなプラスミドクローンをpDAB104107と称し(図9)、続いて新しいZp15相同ドナーベクター内への挿入のためのPAT遺伝子カセットの供給源として用いた。
【0200】
2.3kbpのPAT遺伝子フラグメントを、MfeIを用いた消化によってpDAB104107から回収し、その後にゲル電気泳動、切り出し、および精製が続いた。Zp15相同ドナープラスミドpDAB104101もMfeIで消化し、ゲルから精製した。pDAB104101内へのPAT遺伝子フラグメントのライゲーションによって、PAT遺伝子が、示差的制限酵素消化によって決定される、Zp15遺伝子配列に対する2つの方向のいずれかにあるMfeI部位に挿入されているクローンが産生された。pDAB104104(図10)は、Zp15遺伝子と同じ転写方向に挿入されたPAT遺伝子を含んでいた。pDAB104105(図11)は、Zp15遺伝子に対して反対方向に挿入されたPAT遺伝子を含んでいた。
【0201】
B.さらなるドナーDNA分子の構築
pDAB7489内のZp15 3’相同配列を切り取ることによって改変し、一方でpDAB7489内のZp15 5’相同配列は同じ状態のままである、Zp15に相同な領域を含有する別のドナー構築物を作製した。切り取った3’相同領域を、Integrated DNA Technologies社(Coralville,IA)によって合成されたプライマー:
DC003 5’−CTAATCGTCGACTCGTCAAGCCCCGCCTTTAAAT−3’(配列番号:117)
DC004 5’ −CTAATCCAATTGGTGTGGGCAGCCGAGCG−3’(配列番号:118)
を用いてPCRによってpDAB7489から作製した。
【0202】
PCR増幅反応を、PHUSIONホットスタートDNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,72℃ 15秒間/33サイクル;72℃,5分間/1サイクル;4℃/ホールドの下で行った。PCR反応物を、1.0%TAEアガロースゲル中での電気泳動にかけた。
【0203】
期待されるPCR産物を、0.8kbpのDNAフラグメント(Zp15 3’相同)の存在によって診断した。このフラグメントをゲルから切り出し、QIAquickゲル抽出キット(QIAGEN社,Valencia,CA)を用いてメーカーの指示書に従って精製した。次いで、精製したフラグメントを、ゼロブラントTOPO PCRクローニングキットおよび化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR−ブラントII−TOPOベクター内にクローニングした。
【0204】
個々のコロニーを選び、プラスミドDNAの単離および制限酵素消化による確認にかけた。Zp15 3’相同インサートをシークエンシングして、クローン化したPCR産物(配列番号:119)の同一性および配列忠実性を証明した。1つのそのようなプラスミドクローンをpDAB104106と称し(図12)、続いてpDAB7489内への置換のための新しいZp15 3’相同配列の供給源として用いて、新しいZp15相同ドナーベクターを作出した。
【0205】
pDAB7489をMfeIおよびSalIで連続して消化し、4.2kbpのベクターフラグメントをゲルから精製した。pDAB104106もMfeIおよびSalIで消化し、切り取られたZp15 3’相同配列を含む0.8kbpのフラグメントをゲルから精製した。これらのゲルから精製したフラグメントのライゲーションおよび形質転換によって、切り取ったZp15 3’相同配列でオリジナルのZp15 3’相同配列5が置換されたクローンが産生された。このプラスミドをpDAB104100と称し(図13)、PAT遺伝子のレシピエントとして用いた。
【0206】
PAT遺伝子を、MfeIを用いた消化によってpDAB104107から取り出した。ゲル電気泳動の後、2.3kbpのPAT遺伝子フラグメントをゲルから精製した。Zp15相同ドナープラスミドpDAB104100もMfeIで消化し、ゲルから精製した。pDAB104100内へのPAT遺伝子のライゲーションによって、PAT遺伝子が、示差的制限酵素消化によって決定される、Zp15遺伝子に対する2つの方向のいずれかにあるMfeI部位に挿入されているクローンが産生された。pDAB104103(図14)は、Zp15遺伝子と同じ転写方向に挿入されたPAT遺伝子を含んでいた。pDAB104102(図15)は、Zp15遺伝子に対して反対方向に挿入されたPAT遺伝子を含んでいた。
【0207】
C.トウモロコシの形質転換およびZp15標的PAT挿入の回収
トウモロコシ亜種Hi−IIの胚発生細胞培養物(Armstrongら,(1991)Maize Genet Coop Newsletter 65:92−93)を作製し、維持し、かつZFN24およびドナー分子をコードするプラスミドの同時形質転換にかけた。ドナー分子には、ここに記載されるもの;pDAB104102、pDAB104103、pDAB104104、およびpDAB104105が含まれる。カルス組織の形質転換および選択、ならびにその後の形質転換体の再生は、米国特許出願第12/001,939号、特に実施例19に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。形質転換および選択のプロトコールに関するさらなるガイダンスのために、Petolinoら,(2000)Plant Cell Rept.19:781−786を参照されたい。開花後、植物を自己授粉またはDAS5XH751等のトウモロコシ亜種と異種交配させることができる。結果として生じた後代種子を収穫かつ乾燥させることができ、これらの種子由来の植物を分析して、標的組込み事象の遺伝性を証明することができる。無傷の、稔性のトウモロコシ植物へのカルスの再生は、米国特許出願第12/001,939号、特に実施例22に記載されており、この参考文献は参照することによりその全体として本明細書に組み入れられている。
【0208】
D.Zp15を標的としたPAT挿入の同定
形質転換したカルス組織におけるZp15を標的としたPAT挿入を、PCRによって検出する。テンプレートゲノムDNAを、周知かつ植物DNEASYキット(QIAGEN社,Valencia,CA)等のよく用いられる方法、またはDellaporta(Dellaportaら,(1983)Plant Mol.Biol.Rep.1;19−21)の方法によって、カルス組織から抽出する。Zp15遺伝子座内へのAAD−1標的組込みを検出するためにすでに用いられているPAT特異的プライマーとZp15隣接配列プライマーとの併用によって、5’および3’Zp15相同領域におけるPAT標的挿入接合部の増幅がもたらされる。前記PAT特異的プライマーは、:
DC013 5’−CAATCGTAAGCGTTCCTAGCCTTCCAG−3’ (配列番号:120)
DC014 5’ −CTGGAAGGCTAGGAACGCTTACGATTG−3’(配列番号:121)
であってよい。具体的には、ドナーDNAがZp15遺伝子に対して直列方向(direct orientation)にPAT遺伝子を有する場合、ドナーDNA 5’Zp15相同配列に隣接するゲノム領域内のプライマーHB501fまたはHB503fを、PATタンパク質コード領域内のプライマーDC013(配列番号:120)とともに用いて、PAT−Zp15 5’インサート接合部を検出する。同様に、ドナーDNAがZp15遺伝子に対して非直列方向(indirect orientation)にPAT遺伝子を有する場合、プライマーHB501fまたはHB503fを、PATタンパク質コード領域内のプライマーDC014(配列番号:121)とともに用いて、PAT−Zp15 5’インサート接合部を検出する。ドナーDNAがZp15遺伝子に対して非直列方向にPAT遺伝子を有する場合、PAT−Zp15 3’インサート接合部の検出のために、プライマーHB502rまたはHB504rをプライマーDC013(配列番号:120)とともに用いて、インサート接合部を検出する。同様に、ドナーDNAがZp15遺伝子に対して直列方向にPAT遺伝子を有する場合、プライマーHB502rまたはHB504rをプライマーDC014(配列番号:121)とともに用いて、PAT−Zp15 3’インサート接合部を検出する。
【0209】
PCR増幅反応を、PHUSIONホットスタートDNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,72℃ 15秒間/33サイクル;72℃,5分間/1サイクル;4℃/ホールドの下で行う。TAEアガロースゲル電気泳動を用いて、PCR産物を分解(resolved)かつ同定する。種々のPAT−Zp15ドナーDNAを用いることによって作製されたトランスジェニックカルスにおける、PAT−Zp15標的組込み事象由来のPCR産物に対して期待されるゲルフラグメントの大きさは、以下のとおり:
HB501f/HB503f+DC013(5’)=2.6kbp(pDAB104103),2.6kbp(pDAB104104)
HB501f/HB503f+DC014(5’)=1.6kbp(pDAB104105),1.7kbp(pDAB104102)
HB502r/HB504r+DC013(3’)=3.2kbp(pDAB104105),3.2kbp(pDAB104102)
HB502r/HB504r+DC014(3’)=1.2kbp(pDAB104103),1.2kbp(pDAB104104)
である。
【0210】
5’および3’PAT−Zp15標的組込み接合部を含むPCR産物を、当業者に既知の標準的方法を用いることによってクローニングおよびシークエンシングする。例えば、PCR産物をアガロースゲルから精製し、TOPOブラントクローニング(登録商標)キットおよび化学的にコンピテントな大腸菌細胞であるワンショット(登録商標)TOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR−ブラントII−TOPOプラスミド内にクローニングする。
【0211】
次いで、クローン化した組込み接合部をシークエンシングして、ドナー形質転換ベクター内に組み入れられている5’および3’Zp15相同配列を介した相同組換えによって、PAT遺伝子がZp15遺伝子座においてトウモロコシゲノム内に挿入されたことを証明する。TOPOベクター特異的プライマーM13正方向およびM13逆方向に加えて、PAT遺伝子カセット特異的プライマーを用いて、標的組込みクローンの完全配列を得る。PATタンパク質コード配列に特異的であるPCRプライマーの配列番号:120および配列番号:121は、シークエンシングプライマーとしても用いられる。さらに、他のプライマーもシークエンシングに用いることができる。これらには、PATカセットのイネアクチンプロモーターエレメントに特異的であるもの:
DC−S1 5’−CCAACTGGACAATAGTCTCCAC−3’(配列番号:122)
DC−S2 5’−CATCGCCACTATATACATACC-3’(配列番号:123)
およびPATタンパク質コード配列に特異的であるもの:
DC−S3 5’−CGTCTCAATGTAATGGTTAACG−3’(配列番号:124)
DC−S4 5’−GCCCAGCGTAAGCAATACCAG−3’(配列番号:125)
を含むが、これに制限されない。
【0212】
実施例6:Zp15遺伝子座におけるAAD−1標的組込みの遺伝性
Zp15遺伝子座を標的とするAAD−1遺伝子カセットを保持するトランスジェニックカルス事象を作出した(事象138)。事象138のT0植物を再生し、雌性植物としてDAS5XH751雄性植物と交配させた。結果として生じたT1種子を植え付け、T1植物を成長させた。
【0213】
T1植物をPCRによって分析して、AAD−1−Zp15標的組込みの発生を証明した。PCR増幅反応を、PHUSIONホットスタートDNAポリメラーゼ(New England Biolabs,Beverly,MA)を用いて、以下のサイクル条件:98℃,30秒間/1サイクル;98℃ 10秒間,72℃ 15秒間/33サイクル;72℃,5分間/1サイクル;4℃/ホールドの下で行った。PCR反応物を、1.0%TAEアガロースゲル中での電気泳動によって分析した。ゲノムDNAをT1植物から抽出し、AAD−1 Zp15 5’組込み接合部を検出するように設計されたネストプライマーを用いたPCR反応におけるテンプレートDNAとして用いた。カルス事象147の分析で以前用いた同じプライマーを用いて、プライマリーおよびセカンドPCR反応を行った。プライマリーPCRに対し、プライマーHB501fおよびHB507rを用いた。プライマリーPCRによって、2.2kbpの期待される大きさにバンドが生じた。プライマリーPCR反応物のアリコートを1:100希釈し、ネストプライマーHB503fおよびHB508rを用いたセカンドPCRにおけるテンプレートとして用いた。セカンドPCRによっても、2.2kbpの期待される大きさにバンドが生じた。
【0214】
2.2kbpのセカンドPCR産物を、ゼロブラントTOPO PCRクローニングキットおよび化学的にコンピテントな大腸菌細胞であるワンショットTOP10(Invitrogen Life Technologies,Carlsbad,CA)を用いて、メーカーのプロトコールに従ってpCR−ブラントII−TOPO内にクローニングした。クローン化したDNAを、隣接するベクター特異的プライマー(M13正方向およびM13逆方向)を用いてシークエンシングした。配列は、Zp15遺伝子座におけるAAD−1の標的組込みに期待されるものと同一であることが判明した。
【0215】
本明細書において言及されるすべての特許、特許出願、および刊行物は、参照することによりあらゆる目的でそれらの全体として本明細書によって組み入れられているものとする。
【0216】
明確な理解のために、図解および実施例によって多少詳しく開示が提供されているが、本開示の精神または範囲から逸脱することなく、多様な変化および修正を実施し得ることは当業者に明白であろう。したがって、前述の記載および実施例は制限するものとして解釈されるべきでない。
【特許請求の範囲】
【請求項1】
植物細胞のゲノム内に1つ以上の外因性核酸配列を組み込む方法であって、前記植物細胞のゲノムにおけるZp15遺伝子座においてまたは近隣において二本鎖切断を引き起こし、それによって前記Zp15遺伝子座におけるまたは近隣における前記細胞のゲノム内への前記1つ以上の外因性配列を含むポリヌクレオチドの組込みをもたらす工程を含む方法。
【請求項2】
前記1つ以上の外因性配列の産物を発現させる工程をさらに含む、請求項1に記載の方法。
【請求項3】
前記二本鎖切断が、
(a)前記細胞内で第一の融合タンパク質を発現させる工程であって、前記第一の融合タンパク質は第一のジンクフィンガー結合ドメインおよび第一の切断ハーフドメインを含んでおり、前記第一のジンクフィンガー結合ドメインは前記植物細胞のゲノム内のZp15遺伝子座におけるまたは近隣における第一の標的部位に結合するように遺伝子操作されている工程;ならびに
(b)前記細胞内で第二の融合タンパク質を発現させる工程であって、前記第二の融合タンパク質は第二のジンクフィンガー結合ドメインおよび第二の切断ハーフドメインを含んでおり、前記第二のジンクフィンガー結合ドメインは前記植物細胞のゲノム内のZp15遺伝子座におけるまたは近隣における第二の標的部位に結合し、前記第二の標的部位は前記第一の標的部位とは異なり、かつ前記第一の融合タンパク質の前記第一の標的部位への結合および前記第二の融合タンパク質の前記第二の標的部位への結合により、前記切断ハーフドメインは前記植物細胞のゲノムが前記Zp15遺伝子座においてまたは近隣において切断されるように位置する工程
によって引き起こされる、請求項1または2に記載の方法。
【請求項4】
前記ジンクフィンガー結合ドメインが表1に示した群から選択される、請求項3に記載の方法。
【請求項5】
前記切断ハーフドメインが天然に存在しているまたは天然には存在していないものである、請求項3または4に記載の方法。
【請求項6】
前記二本鎖切断が、天然に存在しているまたは天然には存在していないメガヌクレアーゼ、IIS型制限酵素、あるいは前記メガヌクレアーゼおよび前記IIS型制限酵素を含む融合タンパク質によって引き起こされる、請求項1または2に記載の方法。
【請求項7】
前記1つ以上の外因性核酸配列が、コード配列、制御配列、またはDNA結合ドメインに対する標的部位を含む、請求項1から6のいずれか一項に記載の方法。
【請求項8】
前記コード配列が、:除草剤抵抗性;除草剤耐性;害虫抵抗性;害虫耐性;病害抵抗性;病害耐性;ストレス耐性;ストレス抵抗性;酸化ストレスの変化;油分の収量増大;食物の含有量および構成の変化;見た目の変化;雄性不稔;ドライダウン;耐倒伏性;多産;デンプンの量または質の変化;油分の質の変化;タンパク質の質または量の変化;アミノ酸組成の変化、あるいはその組み合わせを与える産物をコードする、請求項7に記載の方法。
【請求項9】
前記ポリヌクレオチドが前記Zp15遺伝子座内の配列に相同であるヌクレオチド配列をさらに含む、請求項1から8のいずれか一項に記載の方法。
【請求項10】
前記相同なヌクレオチド配列が前記外因性配列に隣接する、請求項9に記載の方法。
【請求項11】
前記ポリヌクレオチドがプロモーターをさらに含む、請求項1から10のいずれか一項に記載の方法。
【請求項12】
1つ以上の組み込まれた外因性配列が次世代の後代に伝達される、請求項1から11のいずれか一項に記載の方法。
【請求項13】
前記植物細胞が単子葉植物細胞である、請求項1から12のいずれか一項に記載の方法。
【請求項14】
前記植物細胞がトウモロコシ細胞である、請求項13に記載の方法。
【請求項15】
請求項1から14のいずれか一項に記載の方法によってZp15遺伝子座内に組み込まれた1つ以上の外因性配列を含む、植物または植物部位。
【請求項16】
請求項1から14のいずれか一項に記載の方法によってZp15遺伝子座内に組み込まれた1つ以上の外因性配列を含む、種子。
【請求項1】
植物細胞のゲノム内に1つ以上の外因性核酸配列を組み込む方法であって、前記植物細胞のゲノムにおけるZp15遺伝子座においてまたは近隣において二本鎖切断を引き起こし、それによって前記Zp15遺伝子座におけるまたは近隣における前記細胞のゲノム内への前記1つ以上の外因性配列を含むポリヌクレオチドの組込みをもたらす工程を含む方法。
【請求項2】
前記1つ以上の外因性配列の産物を発現させる工程をさらに含む、請求項1に記載の方法。
【請求項3】
前記二本鎖切断が、
(a)前記細胞内で第一の融合タンパク質を発現させる工程であって、前記第一の融合タンパク質は第一のジンクフィンガー結合ドメインおよび第一の切断ハーフドメインを含んでおり、前記第一のジンクフィンガー結合ドメインは前記植物細胞のゲノム内のZp15遺伝子座におけるまたは近隣における第一の標的部位に結合するように遺伝子操作されている工程;ならびに
(b)前記細胞内で第二の融合タンパク質を発現させる工程であって、前記第二の融合タンパク質は第二のジンクフィンガー結合ドメインおよび第二の切断ハーフドメインを含んでおり、前記第二のジンクフィンガー結合ドメインは前記植物細胞のゲノム内のZp15遺伝子座におけるまたは近隣における第二の標的部位に結合し、前記第二の標的部位は前記第一の標的部位とは異なり、かつ前記第一の融合タンパク質の前記第一の標的部位への結合および前記第二の融合タンパク質の前記第二の標的部位への結合により、前記切断ハーフドメインは前記植物細胞のゲノムが前記Zp15遺伝子座においてまたは近隣において切断されるように位置する工程
によって引き起こされる、請求項1または2に記載の方法。
【請求項4】
前記ジンクフィンガー結合ドメインが表1に示した群から選択される、請求項3に記載の方法。
【請求項5】
前記切断ハーフドメインが天然に存在しているまたは天然には存在していないものである、請求項3または4に記載の方法。
【請求項6】
前記二本鎖切断が、天然に存在しているまたは天然には存在していないメガヌクレアーゼ、IIS型制限酵素、あるいは前記メガヌクレアーゼおよび前記IIS型制限酵素を含む融合タンパク質によって引き起こされる、請求項1または2に記載の方法。
【請求項7】
前記1つ以上の外因性核酸配列が、コード配列、制御配列、またはDNA結合ドメインに対する標的部位を含む、請求項1から6のいずれか一項に記載の方法。
【請求項8】
前記コード配列が、:除草剤抵抗性;除草剤耐性;害虫抵抗性;害虫耐性;病害抵抗性;病害耐性;ストレス耐性;ストレス抵抗性;酸化ストレスの変化;油分の収量増大;食物の含有量および構成の変化;見た目の変化;雄性不稔;ドライダウン;耐倒伏性;多産;デンプンの量または質の変化;油分の質の変化;タンパク質の質または量の変化;アミノ酸組成の変化、あるいはその組み合わせを与える産物をコードする、請求項7に記載の方法。
【請求項9】
前記ポリヌクレオチドが前記Zp15遺伝子座内の配列に相同であるヌクレオチド配列をさらに含む、請求項1から8のいずれか一項に記載の方法。
【請求項10】
前記相同なヌクレオチド配列が前記外因性配列に隣接する、請求項9に記載の方法。
【請求項11】
前記ポリヌクレオチドがプロモーターをさらに含む、請求項1から10のいずれか一項に記載の方法。
【請求項12】
1つ以上の組み込まれた外因性配列が次世代の後代に伝達される、請求項1から11のいずれか一項に記載の方法。
【請求項13】
前記植物細胞が単子葉植物細胞である、請求項1から12のいずれか一項に記載の方法。
【請求項14】
前記植物細胞がトウモロコシ細胞である、請求項13に記載の方法。
【請求項15】
請求項1から14のいずれか一項に記載の方法によってZp15遺伝子座内に組み込まれた1つ以上の外因性配列を含む、植物または植物部位。
【請求項16】
請求項1から14のいずれか一項に記載の方法によってZp15遺伝子座内に組み込まれた1つ以上の外因性配列を含む、種子。
【図1】


【図2】
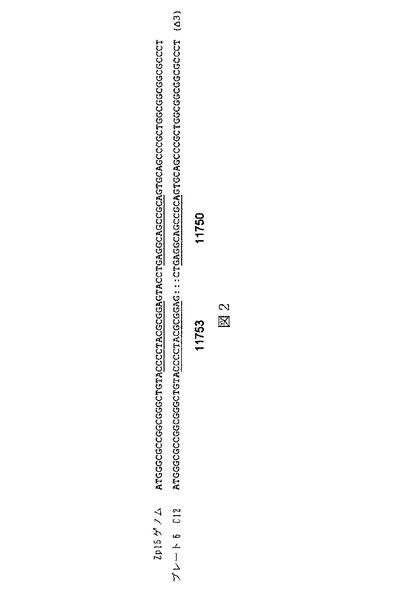
【図3】
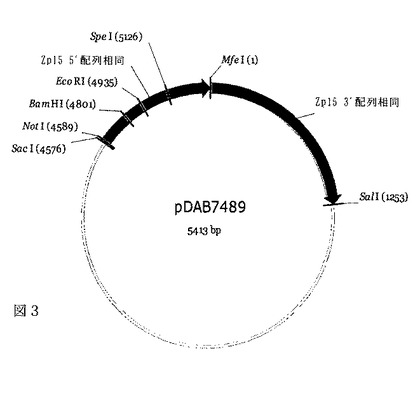
【図4】

【図5】

【図6−1】

【図6−2】

【図6−3】

【図6−4】

【図6−5】

【図6−6】

【図6−7】

【図6−8】

【図7】
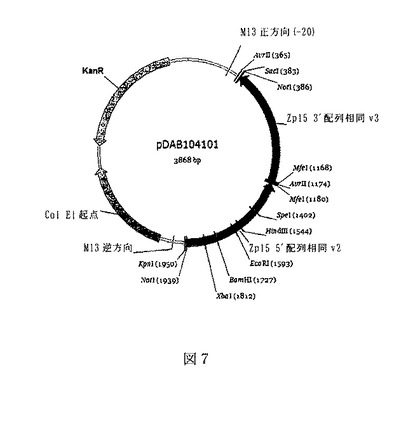
【図8】
【図9】
【図10】

【図11】
【図12】
【図13】

【図14】
【図15】
【図2】
【図3】
【図4】
【図5】
【図6−1】
【図6−2】
【図6−3】
【図6−4】
【図6−5】
【図6−6】
【図6−7】
【図6−8】
【図7】
【図8】
【図9】
【図10】
【図11】
【図12】
【図13】
【図14】
【図15】
【公表番号】特表2012−511926(P2012−511926A)
【公表日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願番号】特願2011−542133(P2011−542133)
【出願日】平成21年12月17日(2009.12.17)
【国際出願番号】PCT/US2009/006606
【国際公開番号】WO2010/077319
【国際公開日】平成22年7月8日(2010.7.8)
【出願人】(390039192)ダウ・アグロサイエンス・エル・エル・シー (20)
【氏名又は名称原語表記】Dow AgroSciences LLC
【出願人】(508241200)サンガモ バイオサイエンシーズ, インコーポレイテッド (28)
【Fターム(参考)】
【公表日】平成24年5月31日(2012.5.31)
【国際特許分類】
【出願日】平成21年12月17日(2009.12.17)
【国際出願番号】PCT/US2009/006606
【国際公開番号】WO2010/077319
【国際公開日】平成22年7月8日(2010.7.8)
【出願人】(390039192)ダウ・アグロサイエンス・エル・エル・シー (20)
【氏名又は名称原語表記】Dow AgroSciences LLC
【出願人】(508241200)サンガモ バイオサイエンシーズ, インコーポレイテッド (28)
【Fターム(参考)】
[ Back to top ]