
bcl−XLタンパク質レベルを調節することによる、T細胞生存の調節法
【課題】T細胞を細胞死から防御する。
【解決手段】本発明は、T細胞を細胞死から防御する方法を提供する。この方法は、T細胞を、T細胞中のbcl−XLタンパク質レベルを増大する作用物質と接触させて、T細胞を細胞死から防御することを含んでなる。本発明は、さらに、細胞死に対するT細胞の感受性を増大させる方法を提供し、T細胞を、T細胞中のbcl−XLタンパク質レベルを減少する少なくとも1つの作用物質と接触させることを含む、インビトロ法およびインビボ法の両方を提供する。
【解決手段】本発明は、T細胞を細胞死から防御する方法を提供する。この方法は、T細胞を、T細胞中のbcl−XLタンパク質レベルを増大する作用物質と接触させて、T細胞を細胞死から防御することを含んでなる。本発明は、さらに、細胞死に対するT細胞の感受性を増大させる方法を提供し、T細胞を、T細胞中のbcl−XLタンパク質レベルを減少する少なくとも1つの作用物質と接触させることを含む、インビトロ法およびインビボ法の両方を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
bcl−XLタンパク質レベルを調節することによる、T細胞生存の調節法
政府支持
本明細書に記載した研究は、NIH認可PO1AI35294およびNMRDS認可61153NAE.4120.001.1402により一部支持されている。従って、米国政府は、本発明の権利を有する。
【背景技術】
【0002】
発明の背景
末梢血T細胞生存の制御は、効果的な末梢性免疫レパトアーの維持に重要である。T細胞生存の幾つかの態様は、T細胞活性化の状態と関連しているようである。T細胞活性化は、抗原提示細胞(APC)表面で主要組織適合遺伝子複合体(MHC)分子に結合したペプチド−抗原によるT細胞受容体/CD3複合体(TCR/CD3)の結合により開始される(Schwartz,R.H.(1990)Science 248,1349)。これはT細胞活性化の一次シグナルであるが、APCとT細胞との間のその他の受容体リガンド相互作用が完全な活性化には要求される。例えば、他の分子相互作用なしでのTCR刺激は、アネルギー状態を誘発することがあり、そのため、これらの細胞は、再刺激時に完全な活性化シグナルに応答できない(Schwartz,R.H.(1990)Science 248,1349;Harding,F.A.,McArthur J.G.,Gross,J.A.,Raulet,D.H.,およびAllinson,J.P.(1992)Nature 356,607)。また、T細胞は、TCR結合(engagement)のみにより活性化される時、プログラムされた細胞死(PCD)により死滅することが示されている(Webb,S.,Morris,C.,およびSprent,J.(1990)Cell 63,1249;Kawabe,Y.,およびOchi,A.(1991)Nature 349,245;Kabelitz,D.,およびWesselborg,S.(1992)Int.Immunol 4,1381;Groux,H.,Monte,D.,Plouvier,B.,Capron,A.,およびAmeisen,J-C(1993)Eur.J.Immunol.23,1623)。
【0003】
T細胞とAPCの間には複合的な受容体−リガンド相互作用が起こる。多くの相互作用は、自然に密着したものであり、かつ2つの細胞間の接触を強化するものである(Spinger,T.A.,Dustin,M.L.,Kishimoto,T.K.,およびMarlin,S.D.(1987)Annual,Rev.Immunol.5,223)のに対し、その他の相互作用はT細胞に更なる活性化シグナルを形質導入するものである(Bierer,B.E.,およびBurakoff,S.J.(1991)Adv,Cancer Res.56,49)。CD28は、ヒトの末梢T細胞の80%に存在する表面糖タンパク質であり、重要な共刺激性(コスティミュラトリー)受容体であることが示されている(June,C.H.,Bluestone,J.A.,Nadler,L.M.およびThompson,C.B.(1994)Immunol.Today 15,321;Linsley,P.S.Ledbetter,J.A.(1993)Annu.Rev.Immunol.11,191)。コスティミュラトリー・シグナルは、T細胞がCD28リガンドB7−1またはB7−2のいずれかを発現する抗原提示細胞と遭遇するときにCD28を介して形質導入される。
【0004】
T細胞の共刺激(コスティミュレーション)は、T細胞活性化の多くの態様に影響を与えることが示されている(June,C.H.,Bluestone,J.A.,Nadler,L.M.およびThompson,C.B.(1994)Immunol.Today 15,321)。共刺激は、培養物中の増殖応答を誘導するのに必要な抗CD3の濃度を低減させる(Gimmi,C.D.Freeman,G.J.,Gribben,J.G.,Sugita,K.,Freedman,A.S.,Morimoto,C.,およびNadler,L.M.(1991) Proc.Natl.Acad.Sci.USA 88,6575)。CD28共刺激はまた、遺伝子発現の転写調節および転写後調節を介するヘルパーT細胞によるリンパ球産生を著しく高め(Lindsten,T.,June,C.H.,Ledbetter,J.A.,Stella,G.,およびThompson,C.B.(1989)Science 244,339;Fraser,J.D.,Irving,B.A.,Crabtree,G.R.,およびWeiss,A.(1991)Science 251,313)、細胞毒性T細胞の細胞溶解能を活性化し得る。CD28共刺激をインビトロで阻害することにより、異種移植片拒絶を遮断でき、また同種移植片拒絶が顕著に遅延する(Lenschow,D.J.,Zeng,Y.,Thistlethwaite,J.R.,Montag,A.,Brady,W.,Gibson,M.G.,Linsley,P.S.,およびBluestone,J.A.,(1992)Science257,789;Turka,L.A.,Linsley,P.S.,Lin,H.,Brady,W.,Leiden,J.M.Wei,R-Q.,Gibson,M.L.,Zheng,X-G.,Mydral,S.,Gordon,D.,Bailey,T.,Bolling,S.F.,およびThompson,C.B.(1992)Proc.Natl.Acad.Sci.USA 89,11102)。更に、腫瘍細胞系列へのB7のトランスフェクションにより、腫瘍増殖の認識および防止が容易になる(Chen,L.,Ashe,S.,Brady,W.A.,Hellstrom,I.,Hellstrom,K.e.,Ledbetter,J.A.,McGowan,P.,およびLinsley P.S.(1992)Cell 71,1093;Townsend,S.E.,およびAllinson,J.P.(1993)Science 259,368)。
【0005】
最近まで、T細胞生存がどのように制御されているかについてほとんど知られていなかった。各研究により、T細胞のマイトジェン活性化が、放射能などの作用物質での処理により開始されるプログラムされた細胞死(PCD)に対するその耐性を強化することが示された(Schrek,R.およびStefani,S.(1964)J.Nat.Cancer Inst.32,507;Lowenthal,J.W.およびHarris,A.W.(1985)J.Immunol.135,1119;Stewart,C.C.,Stevenson,A.P.,およびHabbersett,R.C.(1988)J.Radiat.Biol.53,77)。反対に、TCRのみによるT細胞活性化は、PCDを被るT細胞の感受性を増大させることが報告されている(Kabelitz,D.,およびWesselborg,S.(1992)Int.Immunol 4.1381;Groux,H.,Monte,D.,Plouvier,B.,Capron,A.,およびAmeisen,J-C(1993),Eur.J.Immunol.23,1623)。
【0006】
近年、T細胞生存を調節する役割を果すと思われる数種の遺伝子が同定された。マウスにおける休止リンパ球の生存は、bcl−2遺伝子の発現に依存する(Nakayama,K-I.,Nakayama,K.,Negishi,I.,Kuida,K.,Shinkai,Y.,Louie,M.C. Fields,L.E.,Lucas,P.J.Stewart,V.,Alt,F.W.,およびLoh,D.Y.(1993)Science 261,1584)。bcl−2遺伝子を欠く動物は、培養したときにPCDを被る感受性の高いT細胞を有する。Bcl−2欠損動物は寿命の最初の数週間以内に深刻なリンパ球減少状態になる(Veis,D.J.,Sorenson,C.M.,Shutter,J.R.,およびKorsmeyer,S.J.(1993a)Cell 75,229)。反対に、Fas細胞表面受容体に突然変異を持つ動物は、免疫応答過程に産生される過剰な免疫細胞を除去できない(Watanabe-Fugunaga,R.,Brannan,C.I.Copeland,N.G.,Jenkins,N.A.,およびNagata,S.(1992)Nature 356,314)。Fas欠損動物は、最終的に深刻な自己免疫疾患を発症する。これらのデータは、T細胞生存が、T細胞増殖と同じく厳密に調節され得ることを示している。
【0007】
T細胞死を不適切に調節すると免疫系異常(例えば、免疫不全または自己免疫)を引き起こすこともある。更に、T細胞がある種の感染性微生物に感染するとT細胞は殺傷される。特に、T細胞がヒト免疫不全ウイルス(HIV)に感染するとプログラムされた細胞死により誘発される細胞死が起こる。したがって、T細胞死を制御する方法および特に、そのような死を阻害する方法が必要とされている。
【発明の開示】
【0008】
概要
本発明は、T細胞生存を調整する方法、特に、T細胞を細胞死から防御する方法を提供する。本発明の方法は、少なくともその一部で、T細胞共刺激(例えば、CD28による)がT細胞中のタンパク質bcl−XLの産生増大およびT細胞生存強化をもたらすという発見に基づくものである。更に、その他の手段(例えば、T細胞へのbcl−XL遺伝子のトランスフェクション)によるT細胞中のbcl−XLタンパク質の産生増大もまた、T細胞生存強化をもたらす。したがって、T細胞を細胞死から保護するために、本発明の方法に従い、T細胞中のbcl−XLタンパク質の量を増大して、T細胞生存を強化する。
【0009】
一実施態様では、T細胞生存を、T細胞をbcl−XLタンパク質レベルを増大する作用物質と接触させることにより強化する。本方法の好ましい実施態様では、T細胞中のbcl−XLタンパク質レベルは、T細胞中にbcl−XLタンパク質をコードする核酸を導入することにより増大する。本方法の別の実施態様では、T細胞中のbcl−XLタンパク質レベルは、T細胞を、細胞内に作用して内生bcl−XLタンパク質レベルを増大する作用物質と接触させることにより増大する。bcl−XLタンパク質レベルを増大するその他の好ましい作用物質は、T細胞表面上の分子と相互作用する物質である。本発明の方法は、増大したまたは不適切なT細胞死に関連する異常または症状、例えば、HIVによるT細胞感染、を処置するのに有用である。本方法は、また、T細胞生存を強化し、それによって、免疫反応を刺激する、例えば、病原性微生物の排除を加速する、のにも有用である。
【0010】
本発明は、T細胞死を誘導する、またはT細胞を細胞死に対して感受性にする方法にも関連する。これらの方法は、T細胞を、T細胞中のbcl−XLタンパク質レベルを減少する作用物質と接触させることを含む。これらの方法は、免疫反応をダウンレギュレーションするのに有用である。
【0011】
図面の簡単な説明
図1は、γ−照射(パネルB)前、または未処理(パネルA)のままで、培地のみ(白抜き四角)、抗CD3の存在下(白抜き菱形)、または抗CD3と抗CD28の存在下(黒塗り四角)で12時間T細胞をインキュベーション後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【0012】
図2A−Dは、γ−照射(パネルBおよびD)前、または未処理(パネルAおよびC)のままで、抗CD3のみ(パネルAおよびB)または抗CD3と抗CD28(パネルCおよびD)と共に12時間T細胞をインキュベーションし、その後、それらをならし培地に残す(白抜き四角)か、洗浄して新たな培地に懸濁する(白抜き菱形)か、または洗浄して200U/mlのrIL−2を加えた新たな培地に懸濁した(黒塗り四角)後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【0013】
図3パネルB、CおよびDは、それぞれ、培地のみ(MED)でインキュベーションするか、抗CD3抗体(αCD3)の存在下で1、6または12時間インキュベーションするか、または抗CD3と抗CD28(αCD3+αCD28)の存在下でインキュベーションしたT細胞中でのbcl−XL発現、bcl−2発現およびHLA発現のレベルを示すノーザン・ブロットを表す。パネルAは、非変性アガロースゲルでの電気泳動後に各RNA試料のアリコートをエチジウムブロマイド染色したものを示す。
【0014】
図3Eは、単独で(0)または抗CD3(αCD3)と共に6または12時間、または抗CD3および抗CD28(αCD3+αCD28)と共に6または12時間インキュベーションしたT細胞中のbcl−XLおよびbcl−XSタンパク質の量を示すウエスタン・ブロットの写真である。
【0015】
図4は、単独で(0)または抗CD3(αCD3)と共に6、12または24時間、または抗CD3および抗CD28(αCD3+αCD28)と共に612、または24時間インキュベーションしたT細胞中のbcl−XLおよびBcl−2タンパク質の量を示すウエスタン・ブロットの写真である。
【0016】
図5は、培地単独で(Medium)または抗CD28(αCD28)、抗CD3(αCD3)、抗CD3+抗CD28(αCD3/αCD28)、または抗CD3+IL−2(100units/ml)(αCD3/IL−2)の存在下、24時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【0017】
図6は、培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。パネル下部は、培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションし、抗Fas抗体を添加してさらに24時間インキュベーションしたT細胞の生存能力パーセントを示す。
【0018】
図7A−Bは、抗Fas抗体(αFas、パネルA)または抗CD3抗体(αCD3、パネルB)で処理後0、12、24、48および72時間でbcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミドでトランスフェクションさせたJurkatクローンの生存能力パーセントのグラフ表示である。
【0019】
図7パネルCは、bcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミド(Neoクローン1、2および3)でトランスフェクションさせたJurkatクローン中のbcl−XLタンパク質の量を示すウェスタン・ブロットの写真である。
【0020】
図7パネルDは、IL−2回収後0、12、24、36および48時間でbcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローンの生存パーセントのグラフ表示である。
【0021】
図7パネルEは、bcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローン中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【0022】
図8は、培地単独で、抗CD3の存在下、抗CD3および抗CD28の存在下、および抗CD3およびCTLA4Igの存在下、24時間インキュベーションしたマウスリンパ節細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【0023】
図9は、bcl−XL発現ベクター(ウイルス希釈率v平均bcl−XL)、または対照ベクター(ウイルス希釈率v平均neo)でトランスフェクションし、さまざまな量のHIVで感染させたJurkat細胞の生存能力パーセントのグラフ表示である。
【0024】
図10パネルA−Dは、PWM(パネルAおよびB)でまたは抗CD3および抗CD28抗体(CD3/CD28、パネルCおよびD)で0、24、48または72時間処理した対照およびHIV感染個体由来のPBMCにおいて、アポトーシスを被った細胞の割合(%アポトーシス)およびbcl−Xタンパク質を発現する細胞の割合(%bcl−x)のグラフ表示である。
【0025】
詳細な説明
本発明はT細胞の生存を調節する方法を提供する。好ましい実施態様において、T細胞を細胞死から防御する。細胞死からT細胞を防御する本発明方法は、T細胞の生存を強化するように、bcl−XLタンパク質レベルを増大する少なくとも一つの作用物質でT細胞を処理することを含む。本発明の一つの実施態様において、bcl−XLタンパク質レベルを増大する物質はbcl−XLタンパク質をコードする核酸であって、これはT細胞中へ導入されると発現する。本発明の他の実施態様において、bcl−XLタンパク質レベルは、例えば外来bcl−XL遺伝子からのbcl−XLタンパク質産生を増大せしめる少なくとも一つの作用物質とT細胞を接触させることによりT細胞中において増加される。1つの実施態様において、bcl−XLタンパク質レベルを増加する少なくとも一つの作用物質は、CD28などのT細胞の表面上の分子と相互作用をする作用物質を含む。T細胞はさらに、T細胞に一次活性化シグナルを提供する作用物質と接触させ得る。他の実施態様において、少なくとも一つの作用物質は、bcl−XLタンパク質レベルを増大するために細胞内に作用する物質である。他の実施態様において、本発明は、さらに、T細胞におけるbcl−XLタンパク質レベルを低下することによりT細胞を細胞死に対して感受性にする方法に関する。bcl−XLタンパク質レベルが本発明方法により調節されているT細胞は、被験体に存在するT細胞またはエクスビボで培養されたT細胞であり得る。本発明の方法は、免疫応答の調節、すなわち免疫応答の強化または抑制のために有用である。このように本発明方法は、免疫反応を刺激する(例えば、感染に抵抗する)、あるいは生存寿命が短くなっている(例えば、ヒト免疫不全ウイルスによる感染などの感染結果として)T細胞の生存を刺激するなどの数多くの応用性を有する。
【0026】
bcl−XLがIL−3欠乏誘導化アポトーシスからネズミIL−3依存性前リンパ球細胞系を防御し得ることは、以前から知られている(Boise, H.B. et al.(1993)Cell 74,597 および PCT出願 WO95/00642)。アポトーシスが個体発生におけるネガティブおよびポジティブセレクションを調節する重要なメカニズムであるので、著者は、T細胞におけるbcl−XLmRNAレベルを解析した。胸腺細胞も、休止T細胞も、PMAおよびイオノマイシンで活性化したT細胞も、bcl−XLmRNAを含有していなかった。しかし、胸腺細胞および活性化T細胞は、アポトーシスに役立つことが示されている(Boise, H.B. et al,後出、およびPCT出願WO95/00642)タンパク質bcl−XLをコードするbcl−X遺伝子の別のスプライス形態、bcl−XSmRNAを含んでいた。これらの結果は、T細胞個体発生におけるアポトーシス誘導細胞死の制御がbcl−XLタンパク質レベルの制御によるよりもむしろ、bcl−XSタンパク質レベルの制御によって少なくとも部分的には調節されることを示唆している。
【0027】
本発明は、bcl−XLタンパク質レベルをT細胞中で増加する、ヒトbcl−XLをコードする核酸によるT細胞のトランスフェクションにより、T細胞が細胞死から防御されるという発見に少なくとも部分的に基づいている。このようにして修飾されたT細胞は、γ−照射、T細胞受容体架橋、Fas架橋および成長因子欠乏などの種々の刺激によって誘導される細胞死から防御される。T細胞が細胞死から防御され得る、または、細胞死に対し感受性にするように、T細胞におけるbcl−XLタンパク質レベルを調節する作用物質とT細胞とを接触させることによりT細胞におけるbcl−XLタンパク質レベルを調節することは、本発明の一つの目的である。本発明方法は、被験体のT細胞、例えば感染への抵抗に関与するT細胞の少なくともいくらかの生存を長引かせることを望む症状において医療上有用である。他方、本発明方法は、自己免疫疾患などの、T細胞の死が望まれる状態においても医療上有用である。
【0028】
1.細胞死からT細胞を防御する方法
本発明方法は、細胞死からT細胞を防御することを含む。“T細胞”は、一般的に認識されているものであって、胸腺細胞、未熟Tリンパ球、成熟Tリンパ球、休止Tリンパ球または活性化Tリンパ球を含むことを意図している。T細胞はTヘルパー(Th)細胞であることができ、例えばTヘルパー1(Th1)またはTヘルパー2(Th2)細胞である。T細胞は、CD4+T細胞、CD8+T細胞、CD4+CD8+T細胞、CD4−CD8−T細胞またはT細胞の他のサブセットである。“細胞死からT細胞を防御する”なる文言は、T細胞における細胞死が起きるのを阻害または少なくとも遅延することを含む。細胞死は、なんらかのメカニズムによって細胞死がおきることを含む。細胞死はプログラムされた細胞死(PCD)であって、“アポトーシス”と称することもできる。アポトーシスによるT細胞死は、核ヘテロクロマチンの凝縮、細胞の縮小、細胞質の濃縮、およびアポトーシスの後期段階においてはT細胞DNAの分離フラグメントへのエンドヌクレアーゼ仲介切断を含み、これらを特徴とするものである。アポトーシスが起きた細胞のDNAの電気泳動解析では、分離フラグメントの特徴的な“ラダー(ladder)”が見られる。
【0029】
本発明は、自然に起きる細胞死または細胞中に誘導されたシグナルに起因する細胞死からT細胞を防御する方法に関する。例えば、アポトーシスは、普通、T細胞中の特定のシグナルの誘導からもたらされる。このように本発明の方法は、コスティミュラトリー・シグナルの不在下でT細胞受容体の架橋に起因する細胞死からT細胞を防御することを提供する。T細胞受容体の架橋は、抗CD3抗体などのポリクローナル・アクチベーターにより、あるいは抗原提示細胞上の抗原によって、コスティミュラトリー・シグナルの不在下に(例えばCD28/CTLA4によるシグナルの不在下において)、T細胞アネルギーまたはT細胞死をもたらすことが当分野では知られている。
【0030】
本発明方法は、成長因子減少に起因する細胞死からT細胞を防御することも提供する。例えば、T細胞は増殖にIL−2を必要とし、IL−2の不在は細胞死をもたらす。このように、本発明方法によりT細胞中のbcl−XLタンパク質のレベルを増加することは、IL−2不在における細胞死から正常なIL−2依存性T細胞を防御する(すなわち、本発明はIL−2の不在下で生存できるT細胞を提供する)。本発明の範囲には、T細胞生存に普通に必要な他の成長因子類、サイトカイン類、リンホカイン類の不在に起因する細胞死からT細胞を防御する方法も含まれる。このような因子の例には、インターロイキン類、コロニー刺激因子類およびインターフェロン類がある。
【0031】
本発明方法はさらに、Fasまたは腫瘍壊死因子受容体(TNFR)架橋に起因する細胞死からT細胞を防御することを可能にする。T細胞上のFas受容体の架橋がT細胞のアポトーシス性細胞死をもたらすことが示されている。FasおよびTNFRタンパク質はかなりの相同性を共有しており、これらは架橋すると細胞死を誘導するタンパク質の拡大ファミリーに属せしめることができる。このように、本発明方法は、このファミリーに属するいかなる受容体の架橋に起因する細胞死からもT細胞を防御するのに有用である。
【0032】
本発明方法はまた、グルココルチコイドなどのある種のホルモン、例えばデキサメタゾンにより誘導される細胞死からT細胞を防御する手段を提供する。さらに細胞死は、被験体においてグルココルチコイドレベル上昇を誘導することが知られているストレスの結果としても起こり得る。このように本発明方法は、ストレスを被った被験体に起きるT細胞死に対する防御も提供する。
【0033】
さらに本発明方法は、DNA障害剤により誘導される細胞死からT細胞を防御することを可能にする。
【0034】
T細胞はまたT細胞個体発生中に細胞死を被る。T細胞の増殖および分化の過程は厳密に調節されており、T細胞のポジティブおよびネガティブセレクションを誘導する。この過程中のT細胞生存に影響を与える方法も本発明の範囲内である。
【0035】
T細胞死は、T細胞中のアポトーシス誘導タンパク質、例えば、bcl−XS,Bad,p53,c−myc,またはインターロイキン−1β変換酵素(ICE)(Savilel,J.(1994) Eur. J.Clin. Investigat. 24,715)の活性によっても生ずる。従って、本発明の範囲内の方法は、これらのタンパク質に関係する細胞死を防御する方法も含む。
【0036】
1.2.T細胞中にbcl−XLタンパク質をコードする核酸を導入することよりなる、T細胞を細胞死から防御する方法
【0037】
本発明の一実施態様では、T細胞を、その細胞中にbcl−XLタンパク質をコードする核酸を導入し、T細胞中のbcl−XLタンパク質レベルを増大させることにより、細胞死から防御する。このbcl−XLタンパク質をコードする核酸分子は、T細胞中で遺伝子を発現させる形態で存在するので、bcl−XLタンパク質がT細胞中で産生される。
【0038】
1.2.1.bcl−XLタンパク質をコードする核酸またはそれらの修飾形態
“bcl−XLをコードする核酸分子”なる用語は、T細胞中にその核酸分子を導入したときにbcl−XLタンパク質へ転写および翻訳される全ての核酸分子を含むことを意図している(例えば、この分子は、bcl−XLの発現を調節する適当な制御配列をも含み得る)。 このbcl−XLをコードする核酸分子は、対応するbcl−XL遺伝子のコード領域のみからなるものであってよく、あるいは、例えば5'または3'非翻訳領域、イントロン、それらのフラグメント、または他の配列のような、非コード領域を含むものであってもよい。
【0039】
bcl−XLタンパク質はbcl−X遺伝子によってコードされている。分化スプライシングの結果、このbcl−X遺伝子は2種の異なるmRNA分子を生成し、そのひとつはbcl−XL(bcl−Xの長い形態に対応)タンパク質をコードし、他のものは小さい方のbcl−XS(bcl−Xの短い形態に対応)をコードしている。このbcl−XSは、bcl−XLタンパク質とは63アミノ酸ストレッチ(stretch)を欠く点で異なる。この欠失は、bcl−XLに存在する第2コードエクソンを、第1コードエクソン内のより5'末端に近いスプライス供与体へとスプライシングする結果として起こる。このように、T細胞中のbcl−XLの発現には、bcl−XLとbcl−XSタンパク質の両方の産生をもたらし得る、bcl−X遺伝子を含有するゲノムフラグメントよりもむしろ、bcl−XL cDNAを使用する方が好ましい。
【0040】
ヒトの細胞処理では、bcl−XLをコードする核酸は、他の動物種由来の核酸も本発明の範囲内であるが、ヒト由来のものが好ましい。さらに、bcl−XL核酸は、その核酸をT細胞中に導入したとき、その核酸によってコードされるタンパク質がT細胞を細胞死から防御する限り、各種を通して使用できる。本発明の好ましい態様では、bcl−XLタンパク質をコードしている核酸は、ヒトcDNA(配列番号1)である。本発明の範囲内であるbcl−XLタンパク質をコードしている核酸は、PCT出願国際公開WO95/00642や下記の文献類に記載されている:Boise et al.(1993) Cell 74,597;およびFang et al.(1994) J. Immunol. 153,4388。さらに、ヒトbcl−XL cDNAのヌクレオチド配列、およびヒトbcl−XLタンパク質のアミノ酸配列は、配列番号1および2にそれぞれ示してある。この核酸分子は、bcl−XLタンパク質全長をコードするものであってもよく、あるいは、この核酸は、それをT細胞中に導入したとき、T細胞を細胞死から防御するに充分なだけのbcl−XLのペプチドフラグメントをコードしているものであってもよい。この核酸は、天然のbcl−XLまたはそのフラグメントをコードするものであってもよく、あるいは、bcl−XLタンパク質の修飾形態またはそのフラグメントであってもよい。本発明の範囲内である、天然bcl−XLタンパク質の修飾形態は、以下に記載する。
【0041】
本発明の方法は、T細胞を細胞死から防御する能力を保持している、bcl−XLタンパク質の各フラグメント、各変異体、または各変種(例えば、修飾形態)の使用を含むことを意図している。“bcl−XLタンパク質の形態”は、天然bcl−XLタンパク質との顕著な相同性を共有し、かつT細胞を細胞死から防御し得るタンパク質を意味することを意図している。“bcl−XLタンパク質の形態”は、タンパク質Bcl−2を含めることは意図していない。“生物学的に活性なbcl−XLタンパク質”または“bcl−XLタンパク質の生物学的に活性な形態”または“bcl−XLタンパク質の機能的に活性な形態”の用語は、本書中では、T細胞を細胞死から防御し得る、bcl−XLタンパク質類の各形態を含むことを意味している。当業者は、T細胞中にbcl−XLタンパク質をコードする核酸を導入した際に、T細胞を細胞死から防御する能力に基づき、bcl−XLタンパク質のそのような形態を選ぶことができる。T細胞を細胞死から防御するbcl−XLの特定形態の能力は、例えば、T細胞中にbcl−XLの特定形態をコードする核酸をトランスフェクションし、通常、T細胞中で細胞死が誘発される条件下でT細胞にてbcl−XLタンパク質を合成させることにより測定できる。細胞死の導入に際しても、もしbcl−XLの形態を発現するように修飾したT細胞集団中での細胞死が、bcl−XLの形態を発現するように修飾していないT細胞集団中よりも少なくなれば、bcl−XLタンパク質のその形態は、T細胞中の細胞死に対して防御効果を有する。細胞死は種々の手段でモニターできる。細胞集団中の細胞死の程度は、例えば、両方の集団中でのT細胞の数を、血球計またはコウルター計数器を用いて数えることにより測定できる。T細胞集団中の細胞死の程度を測定する好ましい方法は、プロピジウムヨージド(propidium iodide)排除アッセイによるものである。プロピジウムヨージド排除アッセイは、T細胞をプロピジウムヨージドとともに培養することにより実施でき、このプロピジウムヨージド染料は、死亡細胞に優先的に吸収され、生存細胞から排除される。細胞死の程度は、次いで、本願の実施例6に記載しているように、蛍光活性化セルソーター(FACS)分析により測定される。付加的に使用できる染料には、アクリジンオレンジやヘキスト33342が含まれる。T細胞集団中の細胞死の程度を測定する他の方法には、切断DNAの末端をラベルする(Gavrieli, Y. et al.(1992) J. Cell Biol. 119,493)の種々の方法が含まれる。T細胞集団中の細胞死の程度を測定する他の方法には、T細胞の核酸の電気泳動分析がある。この方法では、精製されているかまたは精製されていない形態のT細胞の核酸を、ゲル電気泳動に付し、次いで、エチジウムブロマイドによるゲル染色し、さらに紫外線下で核酸の可視化を行う。少なくとも相当数のT細胞がアポトーシスを受けているT細胞集団由来の核酸は、"ラダー(ladder)”即ち、DNAの分離フラグメントの集団、が現れるであろう。反面、アポトーシスを受けていないT細胞のDNAは、単一の高分子量バンドとして現れる。
【0042】
bcl−XLタンパク質の形態について、細胞死からT細胞を防御する能力を測定するために、数種のアッセイが使用できる。これらのアッセイには、T細胞集団を特定の作用物質と接触させて、T細胞集団がアポトーシスを受けるよう誘導するアッセイが含まれる。そのような作用物質には、T細胞レセプターを架橋する作用物質類、例えば抗CD3抗体、FasまたはTNFレセプターを架橋する作用物質類、グルココルチコイド類、が含まれる。場合によっては、成長因子欠乏、例えばIL−2欠乏により、T細胞に細胞死を誘導できる。これらのアッセイは本明細書中、殊に実施例6および7に記載する。
【0043】
T細胞中のbcl−XLフラグメントは、bcl−XLタンパク質フラグメントをコードする核酸フラグメントを導入することにより、産生できる。この核酸はcDNAであってもよく、またはゲノムDNAフラグメントであってもよい。bcl−XLの変異体は、例えば、bcl−XLタンパク質をコードする核酸分子(例えば、bcl−XL cDNA)中に、標準法、例えば部位指向性突然変異誘発またはポリメラーゼ連鎖反応経由突然変異誘発により、ヌクレオチド塩基対修飾(例えば、置換、欠失、付加等)を導入することにより調製できる。bcl−XLの好ましい修飾は、T細胞中のbcl−XLタンパク質の半減期を修飾するものを含む。従って、本方法の他の実施態様では、長い半減期を有するbcl−XLの形態をT細胞中に導入することが望ましい場合もあるが、具体的な実施態様では、非常に短い半減期を有するbcl−XLタンパク質の形態をT細胞中に導入することが望ましい。
【0044】
好ましい態様では、bcl−XLタンパク質を、“Bax”タンパク質との相互作用を促進し、そして“Bad”タンパク質との相互作用を阻害または減少させるように修飾する。bcl−XLの細胞中のアポトーシスに対する防御効果は、一つのbcl−XLタンパク質と他の“Bax”と名付けたタンパク質とにより構成されるヘテロ二量体により媒介され得ることが示されている。しかしながら、さらなるデータは、“Bad”と称したタンパク質がbcl−XLとも相互作用し得ること、およびbcl−XLとBadから構成されるヘテロ二量体がbcl−XLの細胞死に対する防御効果を不活性化し得ることを示している(Yang, et al.(1995) Cell 80,285)。さらに、Badは、Bax/bcl−XLヘテロ二量体のBaxタンパク質と置き換えて、bcl−XL/Badヘテロ二量体を形成する能力があることが示されている。このように、本方法の好ましい実施態様では、bcl−XLの修飾形態のBadへの結合を阻害するかまたは少なくとも減少するように修飾したが、野生型bcl−XLタンパク質と比較して、bcl−XLの修飾形態のBaxへの結合には有意に影響されない、bcl−XLタンパク質をコードしている核酸をT細胞中に導入することにより、T細胞を細胞死から防御する。本発明の好ましい実施態様においては、bcl−XLタンパク質の修飾は、Bcl−2相同性(BH)ドメイン1または2(それぞれ、“BH1”および“BH2”)中に位置している、bcl−XLの少なくとも1つのアミノ酸を置換することを含む。BH1は、ヒトbcl−XLタンパク質(配列番号2)のアミノ酸129と148の間に位置しており、BH2はヒトbcl−XLタンパク質(配列番号2)のアミノ酸180と191との間に位置している(Yang, E. et al.(1995) Cell 80,285)。
【0045】
さらに、bcl−XLの一次アミノ酸配列の変化は、細胞死からT細胞を防御するbcl−XL分子の能力を有意に損なうことなく許容されるようであることは、当業者により評価されるであろう。従って、天然に存在するbcl−XL分子のアミノ酸の配列と比較して、アミノ酸置換、欠失、および/または付加を有し、なお機能的活性を保持している、本書中に記載したbcl−XLの変異体形態は、本発明の範囲内に属するものである。bcl−XLの機能的性質を保持するために、好ましくは、保存性アミノ酸置換はひとつまたはそれ以上のアミノ酸残基で行われる。“保存性アミノ酸置換”とは、アミノ酸残基が同種の側鎖を有するアミノ酸残基で置換されることである。同種の側鎖を有するアミノ酸残基のファミリーは、当技術分野で、塩基性側鎖(例えば、リシン、アルギニン、ヒスチジン)、酸性側鎖(例えば、アスパラギン酸、グルタミン酸)、非荷電極性側鎖(例えば、グリシン、アスパラギン、グルタミン、セリン、トレオニン、チロシン、システイン)、非極性側鎖(例えば、アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン)、β−分枝側鎖(例えば、トレオニン、バリン、イソロイシン)、および芳香族側鎖(例えば、チロシン、フェニルアラニン、トリプトファン、ヒスチジン)を含むものとして定義されている。
【0046】
1.2.2.T細胞中でbcl−XLコード核酸の発現のための調節配列
T細胞中でbcl−XLをコードする核酸分子を発現させ、T細胞中でbcl−XLタンパク質のレベルを増大させる(細胞死からT細胞を防御する)ためには、該核酸を調節配列に作動可能に連結させなければならない。“作動可能に連結させた”とは、T細胞中でヌクレオチド配列が発現できるような態様でbcl−XLをコードするヌクレオチド配列を少なくとも一つの調節配列に連結させたという意味であることを意図している。調節配列は、適当なT細胞中で所望のタンパク質の発現を指向するように選ばれる。従って、“調節配列”の用語は、プロモーター類、エンハンサー類、および他の発現制御配列類を含む。そのような調節配列は当業者に知られており、またGoeddel, gene Expression Technology: Methods in Enzymology 185、 Academic Press, San Diego, CA(1990)に記載されている。
【0047】
これらの調節配列は、bcl−XLをコードする核酸の転写および翻訳に必要とされるものを含み、さらに、プロモーター類、エンハンサー類、ポリアデニル化シグナル類、および好ましくはミトコンドリア膜外である、適切な細胞コンパートメントへ分子を伝達するのに必要な配列を含み得る(Gonzales-Garcia, M. et al.(1994) Development 120,3033)。核酸が組換え発現ベクター中のcDNAである場合、cDNAの転写および/または翻訳を担う調節機能は、しばしばウイルスの配列により提供される。一般に使用されるウイルスのプロモーターの例には、ポリオーマ、アデノウイルス2、サイトメガロウイルスおよびシミアンウイルス40(SV40)、およびレトロウイルスLTRs由来のものが含まれる。
【0048】
cDNAに連結させる調節配列は、構成的または誘導的転写を与えるものを選択できる。誘導的転写は、例えば誘導性エンハンサーを使用することにより達成され得る。このように、本発明の具体的な実施態様において、bcl−XLの形態をコードする核酸分子は、誘導性制御配列に影響を与える作用物質を用いてbcl−XLの形態の発現をオンまたはオフにできる(またはその中間のレベルにする)ように、誘導性制御配列の制御下にある(例えば、発現は、T細胞の存在下で、誘導性作用物質の濃度を調節することにより調節できる)。このことにより、T細胞中での細胞死に対するbcl−XLの防御効果を有効または無効に切り替えることが可能である。実際には、ある症状においてのみ、またはある時間のみT細胞の生存を促進することが望ましい。例えば、被験体における感染部位で、限られた時間枠に感染作用物質を除去するために免疫反応を強化することが望ましい。しかしながら、感染作用物質を除去する際に、T細胞を除去することが望ましい。このように、感染部位に位置するT細胞中のbcl−XLの発現は、T細胞を誘導性作用物質と接触させることにより、誘導制御配列を介して刺激され得る。ついで、感染の除去時に、誘導性作用物質を除去してT細胞中のbcl−XLの産生を停止できる。
【0049】
哺乳動物細胞に使用する誘導性調節システムは、当分野では知られており、例えば、遺伝子発現を重金属イオン(Mayo et al.(1982)Cell29:99-108;Brinster et al.(1982)Nature296:39-42;Searle et al.(1985)Mol.Cell.Biol.5:1480-1489)、熱ショック(Nouer et al.(1991)in Heat Shock Response、Nouer,L.編、CRC、Boca Raton、FL、ppl167-220)、ホルモン(Lee et al.(1981)Nature294:228-232;Hynes et al.(1981)Proc.Natl.Acad.Sci.USA78:2038-2042;Klock et al.(1987)Nature329:734-736;Israel&Kaufman(1989)Nucl.Acids.Res17:2589-2604)またはテトラサイクリン(Gossen,M.およびBujard,H.(1992)Proc.Natl.Acad.Sci.USA89:5547-5551)により調節するシステムがある。T細胞を特定の誘導性作用物質に接触させることにより制御可能な誘導的遺伝子発現をもたらすその他の機構は、PCT公開公報WO94/18317およびPCT公開公報WO93/23431に記載されている。
【0050】
誘導性制御配列は、全てのT細胞において機能でき、または、例えばCD4+T細胞、CD8+T細胞、Tヘルパー1(Th1)、Tヘルパー2(Th2)細胞などの特定のT細胞サブセットにおいてのみ機能できる。誘導性制御配列は、また、1タイプのT細胞(例えばCD4+T細胞)においては1つの作用物質により調節されるが、他のタイプのT細胞(例えばCD8+T細胞)においては他の作用物質により調節されるように、選択し得る。
【0051】
本発明のその他の態様においては、bcl−XLタンパク質をコードする核酸分子は、核酸分子の発現を構成的に起こす調節配列の制御下にある。本発明の具体的な実施態様において、HIV感染した被験体由来のT細胞を修飾してbcl−XLタンパク質を発現させ、被験体のT細胞を細胞死から構成的に防御するようにする。核酸分子の構成的発現を起こすために核酸分子に作動可能に連結させた制御配列は、好ましくはウイルスプロモーターである。一般的に使用されるウイルスプロモーターの例には、ポリオーマ、アデノウイルス2、サイトメガロウイルスおよびシミアンウイルス40、およびレトロウイルスLTR由来のものがある。その他に、例えばT細胞受容体エンハンサー(例えば、WinotoおよびBaltimore(1989)EMBOJ.8:729-733)などのT細胞特異的エンハンサーを使用できる。
調節配列に作動可能に連結させたbcl−XLタンパク質をコードする核酸分子は、典型的にベクターに移し得る。ベクターの例には、プラスミド、ウイルスまたは、例えば細菌中の核酸分子の選択および増幅に必要な配列を含むその他の核酸分子がある。このように、調節制御配列に作動可能に連結させたbcl−XLタンパク質をコードするヌクレオチド配列を含む核酸分子はまた、本明細書中では“bcl−XL発現ベクター”と呼ぶ。例えばウイルスベクターなどのベクターは、下記で更に議論する。
【0052】
1.2.3.bcl−XLタンパク質をコードする核酸分子をT細胞に導入する方法
bcl−XLタンパク質をコードする核酸分子は、典型的にトランスフェクションと呼ばれる様々な方法によりT細胞に導入され得る。“トランスフェクション”または“でトランスフェクションさせる”なる語は、外来核酸を哺乳動物細胞に導入することを意味し、エレクトロポレーション、リン酸カルシウム沈澱、DEAE−デキストラン処理、リポフェクション、マイクロインジェクション、およびウイルス感染を含む、核酸の哺乳動物細胞への導入に有用な数多くの技術を含むことを意図する。哺乳動物細胞をトランスフェクションする適当な方法は、Sambrook et al.(Molecular Cloning:A Laboratory Manual第2版、Cold Spring Harbor Laboratory press(1989))およびその他の実験書に見ることができる。
【0053】
本発明の好ましい実施態様において、bcl−XLタンパク質をコードする核酸分子はウイルスベクターを用いてT細胞に導入される。このようなウイルスベクターには、例えば、組換えレトロウイルス、アデノウイルス、アデノ随伴ウイルスおよび単純ヘルペスウイルス1がある。レトロウイルスベクターおよびアデノ随伴ウイルスベクターは、インビボでの特にヒトへの外来遺伝子の導入に優れた組換え遺伝子運搬系であると一般的に考えられている。その他に、そのようなベクターは、外来遺伝子をエクスビボでT細胞に導入するのにも使用できる。これらのベクターは、効果的に遺伝子をT細胞に運搬し、導入された核酸分子は、安定に宿主細胞の染色体DNAに組込まれる。レトルウイルスの使用における重要な必要条件は、特に細胞集団において野生型ウイルスが蔓延する可能性について、その使用の安全性を確認することである。複製欠損性レトロウイルスのみを産生する分化した細胞系(“パッケージング細胞”と呼ぶ)の開発により、遺伝子療法におけるレトロウイルスの用途が拡大し、欠損レトロウイルスは遺伝子療法の目的における遺伝子導入における使用について十分に特徴づけられている(Miller,A.D.(1990)Blood76:271参照)。このように、レトロウイルスコード配列の一部(gag、pol、env)を本発明のbcl−XLタンパク質をコードする核酸で置換えて、レトロウイルス複製欠損性にすることにより、組換えレトロウイルスを構築し得る。この場合、複製欠損性レトロウイルスはビリオン中にパッケージングさせるので、これを標準的な技術によりヘルパーウイルスを使用して標的細胞を感染させるのに使用できる。組換えレトロウイルスの産生およびインビトロまたはインビボでこのようなウイルスで細胞を感染させる方法は、Current Protocols in Molecular Biology、Ausubel,F.M.et al.編Greene Publishing Associates、(1989)、9.10−9.14章および標準的な実験マニュアルにおいて見いだされ得る。適当なレトロウイルスの例は、当業者に良く知られているpLJ、pZIP、pWEおよびpEMを含む。同種指向性および両種指向性レトロウイルス系を両方調製するための適当なパーケージングウイルス系の例は、ψCrip、φCre、ψ2およびψAmを含む。
【0054】
さらに、ウイルス粒子の表面上のウイルスパッケージングタンパク質を修飾することにより、レトロウイルスの、更にはレトロウイルスを基礎としたベクターの感染スペクトルを制限することが可能であることが示されている(例えばPCT発行物WO93/25234およびWO94/06920参照)。例えば、レトロウイルスベクターの感染スペクトルの修飾方法は:細胞表面抗原に特異的な抗体をウイルスのenvタンパク質に結合させる(Roux et al.(1989)PNAS86:9079-9083;Julan et al.(1992)J.Gen Virol73:3251-3255;およびGoud et al.(1983)Virology163:251-254);または細胞表面受容体リガンド類をウイルスのenvタンパク質に結合させる(Nada et al.(1991)J.Biol.Chem.266:14143-14146)ことを含む。結合は、タンパク質またはその他のさまざまなもの(例えば、envタンパク質をアジアロ糖タンパク質に変換するラクトース)との化学的架橋形態であることができ、並びに融合タンパク質(例えば、単鎖抗体/env融合タンパク質)を作成することによる。このように、本発明の具体的な実施態様では、bcl−XLの形態をコードする核酸分子を含むウイルス粒子を、例えば、上記した方法に従い、それらが特異的にT細胞のサブセットを標的とすることができるように修飾する。例えば、ウイルス粒子をあるタイプのT細胞に特異的な表面分子に対する抗体で被覆できる。特に、T細胞のCD4分子を認識するウイルス粒子抗体に連結させることにより選択的にCD4+T細胞を標的とすることが可能である。このように、CD4+細胞の感染は、CD8+T細胞の感染よりも優先的に起こる。本法は、T細胞の特定のサブセットのみを細胞死から防御することを望む場合に、特に有用である。さらに、本発明の方法をインビボで使用する具体的な実施態様において、bcl−XLタンパク質をコードする核酸分子のT細胞または特定のサブセットへの導入を制限することが望ましい。
【0055】
一次T細胞を含むT細胞のbcl−XLタンパク質をコードする核酸分子を導入し、かつ発現させるための更なるレトロウイルス系は、Kasid,A.et al.(1990)Proc.Natl.Acad.Sci.U.S.A.87,473;Morecki,S.et al(1991)Cancer Immunol.Immunother.32,342;Culver,K.et al.(1991)Proc.Natl.Acad.Sci.U.S.A.88,3155;およびFiner,M.H.et al.(1994)Blood、83,43に記載されている。
【0056】
本発明で有用な他のウイルス遺伝子運搬システムは、アデノウイルス由来のベクターを使用する。アデノウイルスのゲノムは、対象となる遺伝子産物をコードし、発現するが、正常細胞溶解ウイルスライフサイクルで複製する能力に関しては不活性化されているように、操作できる。例えば、Berkner et al. (1988) BioTechniques 6:616;Rosenfeld et al. (1991) Science 252:431-434;およびRosenfeld et al. (1992) Cell 68:143-155参照。アデノウイルス株Adタイプ5dl324またはアデノウイルスの他の株(例えば、Ad2、Ad3、Ad7等)由来の適当なアデノウイルスベクターは、当業者に既知である。組換えアデノウイルスは、それらが細胞分裂していない細胞に感染できないため、ある環境で有利である。更に、ウイルス粒子は、相対的に安定であり、精製および濃縮を受けることができ、上記のように、感染スペクトルに影響を与えるように修飾できる。更に、導入アデノウイルスDNA(およびそれを含む外来DNA)は、宿主細胞のゲノムに組み込まれないが、エピソームのままであり、それにより、導入DNAが宿主ゲノム(例えば、レトロウイルスDNA)に組み込まれる状況下、挿入突然変異誘発の結果起こり得る可能性のある問題を回避できる。更に、アデノウイルスゲノムが外来DNAを運搬する能力は、他の遺伝子運搬ベクターと比較して大きい(8キロ塩基まで)(Berkner et al. 前掲;Haj-AhmandおよびGraham(1986) J. Virol. 57;267)。現在使用され、そして本発明の好ましい殆どの複製欠損性アデノウイルスベクターは、ウイルスE1およびE3ゲノムの全てまたは一部を欠失しているが、アデノウイルス遺伝物質の80%程を残している(例えば、Jones et al. (1979) Cell 16:683;Berkner et al., 前掲;およびGraham et al., Methods in Molecular Biology, E. J. Murray編(Humana, Clifton, NL, 1991) vol. 7 pp. 109-127参照)。bcl−XLタンパク質をコードする対象となる核酸分子の発現は、例えば、E1Aプロモーター、主要遅延プロモーター(MLP)および関連リーダー配列、E3プロモーターまたは外来的に付加したプロモーター配列の制御下にあり得る。
【0057】
bcl−XLタンパク質をコードする核酸分子のインビトロ運搬に有用な更に別のウイルスベクター系はアデノ随伴ウイルス(AAV)である。アデノ随伴ウイルスは、アデノウイルスまたはヘルペスウイルスのような他のウイルスを有効な複製および増殖的ライフサイクルのためのヘルパーウイルスとして必要とする、天然に存在する欠損ウイルスである。(レビューのために、Muzyczka et al. Curr. Topics in Micro. and Immunol. (1992) 158:97-129参照)。そのDNAを細胞分裂していない細胞に組み込むことができ、安定な高頻度の組み込み(インテグレーション)を示す幾つかのウイルスの一つがまた存在する(例えば、Flotte et al. (1992) Am. J. Respir. Cell. Mol. Biol. 7:349-356;Samulski et al. (1989) J. Virol. 63:3822-3828;およびMcLaughlin et al. (1989) J. Virol. 62:1963-1973参照)。AAVの300塩基対程を含有するベクターは、パッケージでき、組み込みことができる。外来DNAのための領域は、約4.5kbに限定される。Tratschin et al. (1985) Mol. Cell. Biol. 5:3251-3260に記載のようなAAVベクターは、DNAを細胞に導入するのに使用できる。種々の核酸が、AAVベクターを使用して異なる細胞タイプに導入されている(例えば、Hermonat et al. (1984) Proc. Natl. Acad. Sci. USA 81:6466-6470;Tratschin et al. (1985) Mol. Cell. Biol. 4:2072-2081;Wondisford et al. (1988) Mol. Endocrinol. 2:32-39;Tratschin et al. (1984) J. Virol. 51:611-619;およびFlotte et al. (1993) J. Biol. Chem. 268:3781-3790参照)。遺伝子治療に適用し得る他のウイルスベクター系は、ヘルペスウイルス、ワクシニアウイルスおよび幾つかのRNAウイルスに由来する。
【0058】
bcl−XL−コード配列に加えて、発現ベクターはまた選択マーカーをコードする遺伝子も含み得る。好ましい選択マーカーは、G418、ヒグロマイシンおよびメトトレキサートのような医薬に対する耐性を付与するものを含む。選択マーカーは、bcl−XLタンパク質をコードする核酸分子と同じベクター(例えば、プラスミド)に導入し得るか、または別のベクター(例えば、プラスミド)に導入し得る。
【0059】
あるいは、bcl−XLタンパク質をコードする核酸分子は、細胞運搬媒体により運ばれ、細胞に輸送され得る。このような媒体は、例えば、カチオン性リポソーム(リポフェクチン)または誘導体化(例えば、抗体コンジュゲート)ポリリジンコンジュゲート、グラミシジンS、人工ウイルスエンベロープを含む。これらの媒体は、ベクター、例えばプラスミドまたはウイルスDNAにより運ばれるbcl−XLタンパク質をコードする核酸を、運搬できる。具体的態様において、一次Tリンパ球、特にCD3+、CT4+およびCT8+T細胞で発現する有効な遺伝子は、Philip R. et al. (1994) Mol. Cell. Biol. 14, 2411に記載のように、カチオン性リポソームと複合体形成させたアデノ随伴ウイルスプラスミドDNAを使用して得られる。
【0060】
本発明の他の態様において、bcl−XLタンパク質をコードする核酸分子を、可溶性分子複合体の形態で特定の細胞に運搬する。複合体は、特定の細胞の表面分子に結合し、細胞により続いてインターナリゼーションされ得るサイズである、核酸結合剤および細胞特異的結合剤を含む担体に解離可能に結合した核酸を含む。このような複合体は、米国特許出願第5,166,320号に記載されている。
本発明の他の態様において、bcl−XLタンパク質をコードする核酸は、Yang, N.−S.およびSun, W. H. (1995) Nature Medicine 1, 481に記載のように、T細胞に粒子衝撃により挿入する。
【0061】
1.1 blc−XLタンパク質レベルを増加させる作用物質を使用する方法
一つの態様において、本発明の方法は、T細胞内のbcl−XLタンパク質レベルを増加する少なくとも一つの作用物質にT細胞を接触させることにより、T細胞の生存を強化させる方法を含む。本発明の好ましい態様において、T細胞と相互作用して、bcl−XLタンパク質レベルを増加させる少なくとも一つの作用物質は、T細胞受容体およびCD28のような、T細胞の表面の分子と相互作用する、1またはそれ以上の作用物質を含む。本発明の他の態様において、T細胞内のbcl−XLタンパク質レベルを増加させる少なくとも一つの作用物質は、例えば、bcl−X遺伝子の発現の増加により、細胞内に作用する作用物質である。"T細胞内のbcl−XLタンパク質レベルを増加するために細胞内に作用する作用物質”なる用語は、T細胞の表面受容体には結合しないが、T細胞内のbcl−XLタンパク質レベルの増加をもたらす、T細胞の受容体の結合により誘導される細胞内シグナル(例えば、第2メッセンジャー)を模倣するか、または誘導する作用物質を含むことを意図する。作用物質は、T細胞内のbcl−XLタンパク質の製造を、bcl−X遺伝子転写の増加、bcl−XLmRNAの安定化またはbcl−XLmRNAの翻訳の増加による、種々の機構により刺激し得る。好ましい態様において、bcl−XLタンパク質レベルは、選択的に、すなわち、Bcl−XSタンパク質レベルの付随的増加なしに増加される。
【0062】
"内生遺伝子の発現を刺激する”なる用語は、遺伝子によりコードされるbcl−XLタンパク質のレベルが細胞内で増加するように、細胞内のbcl−X遺伝子に作用することを含むことを意図する。“転写を刺激する”なる用語は、mRNA転写の量が増加するように、転写に影響を与えることを含むことを意図する。"内生bcl−X遺伝子”なる用語は、T細胞に本来あるbcl−X遺伝子を意味することを意図し、T細胞に導入された“外来bcl−X遺伝子”と反対である。
【0063】
本発明の好ましい態様において、T細胞を少なくとも一つの作用物質と接触させ、T細胞内のbcl−XLタンパク質レベルの増加および細胞死からのT細胞の防御をもたらす。好ましい態様において、T細胞の生存は、T細胞を、T細胞を刺激する作用物質の組み合わせと接触させることにより増加する。作用物質の好ましい組み合わせは、T細胞を活性化し、T細胞に共刺激性分子を提供する作用物質を含む作用物質の組み合わせである。例えば、T細胞生存は、T細胞を、T細胞に一次活性化シグナルを提供する第1作用物質およびT細胞にコスティミュラトリー・シグナルを提供する第2作用物質と接触させることにより促進される。より好ましい組み合わせは、T細胞内でbcl−XLタンパク質レベルが増加するようにT細胞受容体を刺激する作用物質およびT細胞へのコスティミュラトリー・シグナルを提供する作用物質を含む。
【0064】
"一次活性化シグナル”なる用語は、典型的にT細胞受容体(TCR)/CD3複合体により誘発される、T細胞の活性化を誘導するシグナルを含むことを意図する。T細胞の活性化は、T細胞が、コスティミュラトリー・シグナルのような第2シグナルを受けて、増殖および分化が誘導されるような、T細胞の修飾を含むことを意図する。具体的態様において、一次活性化シグナルは、T細胞受容体またはT細胞受容体に関連するCD3複合体と接触させる作用物質により提供される。好ましい態様において、作用物質は、モノクローナル抗体OKT3(American Type Culture Collection, Rockvill, MD;No. CRL 8001から入手可能)のようなCD3に対して反応性の抗体である。本発明の他の態様において、刺激性作用物質は、抗体の組み合わせ、例えば、T11.3+T11.1またはT11.3+T11.2(例えば、Meuer, S.C. et al. (1984) Cell 36:897-906参照)のような、T細胞のCD2複合体を刺激する作用物質である。更に別の態様において、一次活性化シグナルは、抗原または抗原提示細胞により提供される。従って、T細胞を1またはそれ以上の抗原または1またはそれ以上の抗原提示細胞と接触させることにより、および所望により、コスティミュラトリー・シグナルを提供する第2作用物質と接触させることにより、T細胞の集団における特定のT細胞クローンの生存を刺激することが可能である。
【0065】
本発明の好ましい態様において、T細胞を、T細胞の一次活性化シグナルおよびコスティミュラトリー・シグナルの両方を刺激する作用物質の組み合わせを用いて刺激する。“共刺激性作用物質”なる用語は、一次活性化シグナルを受けたT細胞(例えば、活性化T細胞)を刺激して、増殖させるか、IL−2、IL−4またはインターフェロン−γのようなサイトカインを分泌させる、コスティミュラトリー・シグナルをT細胞に提供する作用物質を含むことを意図する。具体的な態様において、共刺激性作用物質は、T細胞の表面のCD28またはCTLA4分子と相互作用する。更により具体的な態様において、コスティミュラトリー・シグナルは、B−リンパ球抗原B7−1またはB7−2のようなCD28またはCTLA4のリガンドである。“CD28の天然リガンドの刺激性形態”なる用語は、T細胞にコスティミュラトリー・シグナルを提供できる、B7−1およびB7−2分子、それらのフラグメントまたはそれらの修飾物を含むことを意図する。CD28の天然リガンドの刺激性形態は、例えば、活性化T細胞をCD28の天然リガンドの形態と接触させ、標準T細胞増殖アッセイを行うことにより同定できる。従って、CD28の天然リガンドの刺激性形態はT細胞の増殖を刺激できる。CD28/CTLA4の天然リガンドの刺激は、例えば、PCT公開第WO95/03408に記載される。
【0066】
T細胞を細胞死から防御するのに使用できる他の作用物質は、bcl−XLタンパク質レベルをT細胞内で増加させるような、T細胞活性化および/または共刺激に関与する1またはそれ以上の細胞内シグナル伝達経路を刺激する作用物質を含む。本発明の好ましい態様において、刺激性作用物質は、イオノマイシンまたはA23187のようなカルシウムイオン性透過体である。あるいは、刺激性作用物質は、ホルボールエステルのようなプロテインキナーゼCを刺激する作用物質であり得る。好ましいホルボールエステルは、ホルボール−12,13−ジブチレートである。本発明のより更に好ましい組み合わせにおいて、T細胞をカルシウムイオノホアおよびホルボールエステルの組み合わせと接触させる。刺激性作用物質は、プロテインチロシンキナーゼを活性化する作用物質であり得る。プロテインチロシンキナーゼを活性化する好ましい作用物質は、ペルバナデート(O'Shea, J. I., et al. (1992) Proc. Natl. Acad. Sci. USA 89:10306)である。
【0067】
T細胞生存を刺激するのに使用できる他の作用物質は、bcl−XLタンパク質レベルを増加させる、ポリクローナルアクチベーターのような作用物質を含む。ポリクローナルアクチベーターは、T細胞の原形質膜上で発現する糖タンパク質に結合する作用物質を含み、フィトヘマグルチニン(PHA)、コンカナバリン(ConA)およびアメリカヤマゴボウマイトジェン(PWM)を含む。
【0068】
T細胞内のbcl−XLタンパク質レベルを増加できるスーパー抗原は、また本発明の範囲内である。本明細書で定義される“スーパー抗原”なる用語は、細菌エンテロトキシンまたはT細胞の増殖を刺激できる他の細菌タンパク質を含むことを意図する。スーパー抗原は、SEA、SEB、SEC、SEDおよびSEEのようなスタフィロコッカスエンテロトキシン(SE)を含む。スーパー抗原はまたはレトロウイルススーパー抗原のようなウイルス起源であり得る。
【0069】
T細胞生存の刺激に使用し得る更に別の抗原は、リンホカインを含み、それは単独または他の作用物質と組み合わせて、T細胞内のbcl−XLタンパク質レベルを増加させる。従って、本発明の好ましい態様において、T細胞を、T細胞の一次活性化シグナルを提供する作用物質(例えば、抗CD3抗体)およびbcl−XLタンパク質レベルをT細胞内で増加させる有効量のIL−2と接触させる。
【0070】
単独または他の作用物質と組み合わせて、T細胞死をbcl−XLタンパク質レベルの増加により予防できる更なる作用物質は、T細胞を作用物質単独または他の作用物質と共に接触させ、bcl−XLタンパク質レベルを、本明細書に記載のように、例えば、ウエスタンブロット分析により追跡することにより同定し得る。
【0071】
本発明の範囲内の作用物質は、溶液で、または固体表面に結合させて使用できる。固体表面は、例えば、組織培養皿またはビーズの表面であり得る。刺激性作用物質の性質に依存して、固体表面への結合は、当分野で既知の方法により行うことができる。例えば、タンパク質を商業的に入手可能な架橋剤(Pierce, Rokford IL)を使用して化学的に細胞表面に架橋するか、または一晩4℃でインキュベーションすることによりプラスチック上に固定化できる。数個の作用物質をT細胞内のbcl−XLレベルの増加に使用する場合、ある作用物質は溶液状であってよく、ある作用物質は固体支持体に結合させてもよい。好ましい態様において、T細胞は固相結合抗CD3抗体および可溶性抗CD28抗体の組み合わせと接触させる。
【0072】
細胞内に作用してbcl−XLタンパク質レベルを増加させる作用物質は、細胞内のbcl−XLタンパク質レベルの欠乏についての標準アッセイを使用して同定できる。例えば、T細胞を試験作用物質の存在下または不在下にインキュベーションでき、異なる時間に細胞により製造されたbcl−XLタンパク質の量を、本発明に参考として包含する、公開PCT出願番号WO95/00642の実施例部分に記載のように、ウエスタンブロット分析により測定できる。従って、本発明の方法を行うのに好ましい作用物質は、ウエスタンブロット分析で測定した際に、bcl−XLタンパク質レベルの著しい増加を誘導するものを含む。
【0073】
更に、T細胞のbcl−XLのタンパク質レベルの増加に使用できる作用物質は、同定用のbcl−X遺伝子の調節領域の分析により同定できる。DNAは、特定の作用物質により、遺伝子の転写を調節するように配列している。例えば、これらの配列に特異的に結合する転写アクチベーターは、bd−X遺伝子発現の刺激に使用できる。次いで、これらの作用物質が細胞内のbcl−XLタンパク質レベルを増加するか、ウエスタンブロット分析により確認できる。
【0074】
本発明の具体的態様において、作用物質は、選択的に、または少なくとも優勢にT細胞に作用し、bcl−XLタンパク質レベルを増加させる。従って、作用物質の投与は、T細胞内のみのまたは少なくとも好ましくはT細胞におけるbcl−XLタンパク質レベルの増加を得るようにする。T細胞特異的作用物質は、インビトロで、異なる細胞タイプを作用物質に接触させ、細胞内のbcl−XLタンパク質レベルの測定をウエスタンブロット分析により行うことにより同定できる。従って、好ましい作用物質は、T細胞内のみのまたは少なくとも好ましくはT細胞のbcl−XLタンパク質レベルの増加をもたらすものである。
【0075】
本発明の好ましい態様において、細胞内に作用してbcl−XLタンパク質レベルを増加させる作用物質は、相対的に短い半減期を有するものである。このような作用物質により、作用物質の効果が良好に制御される。特に、このような作用物質は、T細胞の細胞生存を短期間、中期間、または長期間刺激できる。実際、本発明の具体的態様において、T細胞生存は、一時的に、例えば、作用物質を感染部位の免疫応答のブースターに使用する条件下のみで、増加するのが好ましい。免疫応答は、T細胞の半減期の制御により少なくとも一部分制御される。ある条件下ではT細胞死が起こらないと、実際的には、有害な作用をもたらす場合がある。しかしながら、本発明の他の態様において、細胞内に作用して、bcl−XLタンパク質レベルを増加させる作用物質は、作用物質の被験体へのより少ない投与でもT細胞死に対する有効な長期防御を得ることができるように、長い半減期を有する。
【0076】
1.3. T細胞にタンパク質を導入することにより、T細胞を細胞死から防御する方法
本発明の他の態様において、T細胞を、T細胞による摂取に適した形態のbcl−XLタンパク質と接触させることを含む方法により、T細胞を細胞死から防御する。従って、本発明の具体的態様において、bcl−XLタンパク質は、細菌発現システムおよび適当な媒体でのT細胞への輸送のような、慣用法によりインビトロで合成される。適当な媒体は、特定の細胞、特にT細胞またはT細胞の選択的サブセットを標的とするように修飾できるリポソームを含む。
【0077】
bcl−XLタンパク質は、bcl−XLタンパク質をコードする核酸分子またはbcl−XLの生理学的に活性な形態を、その後種々の宿主、特に哺乳動物または昆虫細胞培養物のような真核細胞だけでなく、E. coliのような原核細胞での対応するタンパク質の合成を指向する、種々の発現ベクターに挿入することによりインビトロで製造できる。本発明の範囲内の発現ベクターは、本明細書に記載の核酸および核酸に作動可能に連結させたプロモーターを含む。このような発現ベクターは、宿主細胞をトランスフェクションするのに使用でき、それにより本明細書に記載の核酸によりコードされるタンパク質が製造される。本明細書に記載の本発明の発現ベクターは、典型的に、少なくとも一つの調節配列に作動可能に連結させたbcl−XLタンパク質をコードするヌクレオチド配列を含む。調節配列は、上記である。発現ベクターの設計は、トランスフェクションすべき宿主細胞の選択および/または発現することが望ましいタンパク質のタイプおよび/または量のような因子に依存し得る。
【0078】
本発明の発現ベクターは、真核または原核(例えば、哺乳類、昆虫または酵母細胞)の細胞のトランスフェクションに使用でき、それにより、ベクターのヌクレオチド配列によりコードされるタンパク質を合成する。原核細胞における発現は、多くの場合、E. coliにて構成性または誘導性プロモーターと共に行う。あるE. coli発現ベクター(いわゆる融合ベクター)は、多くのアミノ酸残基を発現組換えタンパク質に、通常は、発現タンパク質のアミノ末端に付加するために設計される。このような融合ベクターは、3つの目的で働く:1)組換えタンパク質の発現の増加のため;2)標的組換えタンパク質の溶解性の増加のため;および3)アフィニティー精製におけるリガンドとして作用することにより、標的組換えタンパク質の精製における補助のため。融合発現ベクターの例には、グルタチオンS−トランスフェラーゼおよびマルトースE結合タンパク質を、それぞれ、標的組換えタンパク質に融合するpGEX(Amrad Corp., Melbourne, Australia)およびpMAL(New England Biolabs, Berberly, MA)がある。従って、bcl−XLタンパク質をコードする核酸分子は、真核融合ベクター中の更なるコード配列に結合でき、融合タンパク質の発現、溶解性または精製を補助する。しばしば、融合発現ベクターにおいて、タンパク質分解切断部位を、融合分子と標的組換えタンパク質の結合部で挿入することにより、融合タンパク質の精製に続く標的組換えタンパク質を融合分子から分離することが可能である。このような酵素およびそれらの連結認識配列は、ファクターXa、トロンビンおよびエンテロキナーゼを含む。
【0079】
誘導性非融合発現ベクターには、pTrc(Amann et al. (1988) Gene 69:301-315)およびpET11d(Studier et al., Gene Expression Technology:Methods in Enzymology 185, Academic Press, San Diego, California (1990)60-89)がある。pTrcベクター4からの標的遺伝子発現は、ハイブリッドtrp-lac融合プロモーターからの宿主RNAポリメラーゼ転写に依存する。pET11dベクターからの標的遺伝子発現は、共発現(coexpressed)ウイルスRNAポリメラーゼ(T7gn1)により媒介されるT7gn10-lac0融合プロモーターからの転写に依存する。このウイルスポリメラーゼは、T7gn1を有する非活性λプロファージから宿主株BL21(DE3)またはHMS174(DE3)により供給される。
【0080】
E. coli内のbcl−XLタンパク質の発現を最大にする一つの方法は、組換えタンパク質をタンパク質分解的に切断する能力を低下させて、宿主細菌中のタンパク質を発現させることである(Gottesman, S., Gene Expression Technology:Methods in Enzymology 185, Academic Press, San Diego, California (1990) 119-128)。他の方法は、各アミノ酸の個々のコドンが、高度に発現したE. coliタンパク質において優先的に利用されるものであるように、発現ベクター内に挿入されるbcl−XLタンパク質をコードする核酸分子のヌクレオチド配列を変えることである(Wada et al., (1992) Nuc. Acids Res. 20:2111-2118)。このような核酸配列の置換は、本発明に含まれ、標準DNA技術により行うことができる。
【0081】
あるいは、bcl−XLタンパク質は、哺乳動物細胞(例えば、チャイニーズハムスター卵巣細胞(CHO)またはNSO細胞)、昆虫細胞(例えば、バキュロウイルスベクターを使用して)または酵母細胞のような真核宿主細胞で発現できる。他の適当な宿主細胞は、Goeddel, (1990)前掲に見られるか、当業者に既知であり得る。真核細胞における真核細胞性タンパク質の発現が、組換えタンパク質の部分的または完全グリコシル化および/または対応する鎖内または鎖間ジスルフィド結合の形成を導くため、真核よりむしろ原核でのbcl−XLタンパク質の発現が好ましいことがある。哺乳動物細胞で発現させる場合、ベクターの制御機能は、ウイルス性の物質により提供されることが多い。例えば、通常使用されるプロモーターは、ポリオーマ、アデノウイルス2、サイトメガロウイルスおよびシミアンウイルス40に由来する。哺乳動物細胞でbcl−XLタンパク質を発現させる場合、一般にCOS細胞(Gluzman, Y. (1981) Cell 23:175-182)は一過性増幅/発現のためにpCDM8(Seed, B. (1987) Nature 329:740)のようなベクターとの複合体で使用するのに対し、CHO(dhfr Chinese Hamster Ovary)細胞は哺乳動物細胞での安定な増幅/発現のためにPMT2PC(Kaufman et al., (1987) EMBO J. 6:187-195)のようなベクターと共に使用する。組換えタンパク質の製造のための好ましい細胞系は、ECACC(カタログ番号85110503)から入手可能であり、Galfre, G.およびMilstein, C.((1981) Methods in Enzymology 73(13):3-46;およびPreparation of Monoclonal Antibodies:Strategies and Procedures, Academic Press, N.Y., N.Y.)記載のNSOミエローマ細胞である。酵母での組換えタンパク質の発現に適当なベクターの例は、pYepSec1(Baldari et al., (1987) Embo J. 6:229-234)、pMA(KurjanおよびHerskowitz, (1982) Cell 30:933-943)、pJRY88(Schultz et al., (1987) Gene 54:113-123)、pYES2(Invitrogen Corporation, San Diego, CA)を含む。培養昆虫細胞(SF9細胞)中のタンパク質の発現に利用可能なバキュロウイルスベクターは、pAcシリーズ(Smith et al., (1983) Mol. Cell Biol. 3:2156-2165)およびpVLシリーズ(Lucklow, V.A.,およびSummers, M.D., (1989) Virology 170:31-39)を含む。
【0082】
ベクターDNAは、リン酸カルシウムまたは塩化カルシウム共沈、DEAE−デキストラン媒体トンラスフェクション、リポフェクションまたはエレクトロポレーションのような慣用の形質転換またはトランスフェクション法により、真核または原核細胞に挿入できる。宿主細胞を形質転換する適当な方法は、Sambrook et al. (Molecular Cloning:A Laboratory Manual, 2nd Edition, Cold Spring Harbor Laboratory press(1989))および他の研究用テキストに見ることができる。
【0083】
哺乳動物細胞を安定にトランスフェクションさせる場合、使用する発現ベクターおよびトランスフェクション技術にもよるが、小さいフラクションの細胞のみがDNAをそのゲノムに組み込み得ることが知られている。これらの組み込み体(インテグレート)の同定および選択のために、一般的に、選択マーカー(例えば、抗生物質への耐性)をコードする遺伝子を、対象となる遺伝子と共に宿主細胞に導入する。好ましい選択マーカーは、G418、ヒグロマイシンおよびメトトレキサートのような医薬に対する耐性を付与するものを含む。選択マーカーをコードする核酸は、対象となる遺伝子と同じプラスミドにより宿主細胞内に挿入できるかまたは別のプラスミドにより挿入できる。これらの対象となる遺伝子を含む細胞は、医薬選択(例えば、選択マーカー遺伝子を取り込んた細胞は生存するが、他の細胞は死滅する)により同定できる。次いで生存細胞を、bcl−XLタンパク質の製造について、例えば、抗bcl−XL抗体の細胞上清からの免疫沈殿によりスクリーニングできる。
【0084】
組換え技術により製造されるbcl−XLタンパク質は、細胞の混合物およびタンパク質を含む培地から分泌および単離できる。bcl−XLタンパク質の選択のために、適当なシグナルペプチドをコードするDNA配列をbcl−XLをコードするヌクレオチド配列の5'末端に連結させて、bcl−XLタンパク質を、細胞からのタンパク質の分泌をもたらすシグナルペプチドに連結させる。あるいは、タンパク質を細胞質に残すこともでき、その細胞を回収し、溶解してタンパク質を単離し得る。細胞培養に適当な培地は当分野で既知である。タンパク質は、細胞培養培地、宿主細胞または両方から、タンパク質精製の分野で既知の技術を使用して単離できる。
【0085】
組換え的に製造されたbcl−XLタンパク質は、被験体に投与するための適当な医薬媒体にパッケージでき、そうして、bcl−XLタンパク質を被験体のT細胞に導入し、細胞死に対するT細胞の防御をもたらす。組換えbcl−XLタンパク質をT細胞へインビボおよびエクスビボ導入する場合、組換えタンパク質は、好ましくはリポソームにパッケージされる。しかしながら、他の担体システムも使用できる。
【0086】
この方法のその他の実施態様において、T細胞生存は、Badのbcl−XSタンパク質のようなbcl−XLのアンタゴニストのタンパク質レベルの減少により、促進される。bcl−XLアンタゴニストのタンパク質レベル減少は、T細胞を、アンタゴニストをコードする遺伝子の発現を減少させる少なくとも一つの作用物質と接触させることにより、またはT細胞に、アンタゴニストの生理学的活性を減少させる核酸または他の化合物を挿入することにより、達成できる。bcl−XSおよびBadタンパク質レベルをダウンレギュレーションする方法は、bcl−XLタンパク質レベルをダウンレギュレーションするための本明細書に記載の方法から改作できる。
【0087】
T細胞死を減少させるために、上記方法を組み合わせることができる。
【0088】
2.T細胞を細胞死に対して感受性にする方法
本発明の具体的な実施態様において、T細胞内のbcl−XLタンパク質のレベルを阻害または減少させることにより、T細胞を細胞死に対してより感受性にする。例えば、T細胞の細胞死への感受性は、T細胞を、bcl−XL機能を不活性にするかまたはbcl−XLの生物学的活性を減少させる作用物質と接触させることにより、増加できる。“T細胞を細胞死に対して感受性にする”なる用語は、T細胞の細胞死への感受性が、非修飾T細胞と比較して増加するように、T細胞内の生物学的に活性なbcl−XLタンパク質の量が減少されるようにT細胞を修飾することを含むことを意図する。"機能的bcl−XLタンパク質”および“生物学的活性bcl−XLタンパク質”なる用語は、野生型bcl−XLタンパク質または、あるいは、野生型bcl−XLタンパク質の生物学的機能を遂行できるbcl−XLタンパク質の修飾形態を含むことを意図する。T細胞を細胞死に対して感受性にする本発明の方法は、T細胞のポリクローナル集団または特異的T細胞クローンの欠失を引き起こしたい場合に多くの治療的応用性を有する。従って、本発明の方法は、例えば、自己免疫疾患の処置に有用である。
【0089】
T細胞内のbcl−XLタンパク質レベルを減少する方法は、T細胞を、bcl−X遺伝子の転写のレベルを減少させる作用物質、bcl−XLmRNAを脱安定化する作用物質、bcl−XpremRNAのbcl−XLmRNAへのスプライシングを阻止する作用物質、mRNAの翻訳を阻止する作用物質またはこれらの作用物質の組み合わせと接触させることから成る方法を含む。あるいは、bcl−XLと相互作用し、bcl−XLの生物学的活性を阻害または減少させるタンパク質のタンパク質レベルを増加させることによりbcl−XLタンパク質の防御効果に打ち勝つことができる。一つの実施態様において、Badまたはbcl−XSタンパク質のタンパク質レベルを、T細胞が細胞死に対して感受性となるように、T細胞内で増加させる。
【0090】
本発明の具体的実施態様において、T細胞内のbcl−XLタンパク質レベルは、T細胞に、bcl−XL機能を妨害するRNAまたはタンパク質を発現する核酸を挿入することにより減少する。例えば、一つの実施態様では、bcl−XLタンパク質の製造を阻害するアンチセンス核酸をT細胞に導入する。“アンチセンス”核酸は、“センス”核酸に相補的な、例えば、タンパク質をコードするmRNAに相補的な核酸配列を含み、ワトソンとクリックの塩基対形成規則に従い構築される。従って、アンチセンス核酸は、センス核酸に水素結合できる。mRNAの配列に相補的なアンチセンス配列は、mRNAのコード領域に相補的であり得るか、またはmRNAの5'または3'非翻訳領域に相補的であり得る。ヒトbcl−XLをコードするヌクレオチド配列のコード領域は配列番号1に示す。好ましくは、アンチセンス核酸は、開始コドンの前にあるかまたはそのコドンにまたがる領域と相補的であるか、またはmRNAの3'非翻訳領域にある。アンチセンス核酸は、配列番号1に示すヌクレオチド配列、または当分野で既知の他のbcl−XLコード配列に基づいて設計できる。例えば、核酸は、配列番号1のヌクレオチド配列のコード領域または非翻訳領域に相補的な配列を有するように設計する。
【0091】
本発明のアンチセンス核酸は、当分野で既知の方法を使用した化学的合成および酵素的ライゲーション反応を使用して構築できる。アンチセンス核酸(例えば、アンチセンスオリゴヌクレオチド)は、天然に存在するヌクレオチドまたは、分子の生物学的安定性を増加するためにまたはアンチセンスとセンス核酸の間に形成された二本鎖の物理的安定性を増加するために設計した種々の修飾ヌクレオチドを使用して化学的に合成でき、例えば、ホスホロチオエート誘導体およびアクリジン置換ヌクレオチドを使用できる。アンチセンスオリゴヌクレオチドは、培養中のT細胞に導入でき、bcl−XLの発現を阻害する。オリゴヌクレオチドのような1またはそれ以上のアンチセンス核酸を培養培地中の細胞に、典型的には約200μg/mlで添加できる。
【0092】
あるいは、bcl−XLタンパク質をコードするヌクレオチド配列の少なくともフラグメントに対応する核酸が、アンチセンス配向(すなわち、挿入した核酸から転写されるRNAが対象となる標的核酸に対してアンチセンス配向である)でサブクローンされている発現ベクターを使用して、アンチセンス核酸をT細胞内で生物学的に製造できる。アンチセンス発現ベクターは、アンチセンス核酸を非常に有効な調節領域の制御下に製造する、組換えプラスミド、ファージミドまたは弱毒化ウイルスの形態であり得る。調節領域は、核酸分子の構成的または誘導的発現を刺激できる。核酸分子の発現を制御する調節領域、このような配列を有するベクター、および核酸分子をT細胞に導入する方法は上記である。bcl−XLアンチセンスmRNAをコードする核酸分子は、T細胞にエクスビボまたはインビボで導入できる。核酸をT細胞にインビボで導入する方法は上記である。アンチセンス遺伝子を使用した遺伝子発現の調節の記載は、Weintraub, H. et al., Antisense RNA as a molecular tool for genetic analysis, Reviews-Trends in Genetics, Vol. 1(1) 1986参照。
【0093】
本発明の他の態様において、T細胞中のbcl−XLタンパク質レベルは、T細胞に、リボザイムであるアンチセンス核酸の形態をコードする核酸を導入することにより減少する。リボザイムは、リボヌクレアーゼ活性を有する触媒的RNA分子であり、mRNAのような一本鎖核酸に対して相補的な領域を有してそれらを切断する能力がある。bcl−XLコード配列に特異性を有するリボザイムは、bcl−XLコードmRNAのヌクレオチド配列に基づいて設計できる。例えば、テトラヒメナL-19IVS RNAの誘導体は、活性部位の塩基配列が、bcl−XLコードmRNAで切断すべき塩基配列と相補的であるように構築できる。例えば、Cech et al.米国特許第4,987,071号;およびChech et al.米国特許第5,116,742号参照。あるいは、bcl−XLコード配列は、RNA分子のプールから、特異的リボヌクレアーゼ活性を有する触媒的RNAを選択するのに使用できる、例えば、Bartel, D.およびSzostak, J. W. (1993) Science 261:1411-1418参照。
【0094】
この方法の更に別の態様において、T細胞にbcl−XLと相互作用してbcl−XLの生理学的機能を阻害する阻害性タンパク質をコードする核酸分子を導入することにより、bcl−XLタンパク質のレベルを減少させることにより、T細胞を細胞死に対して感受性にする。本発明の具体的実施態様において、阻害性タンパク質はBadタンパク質または、bcl−XLの生理学的活性の減少をできるそれらのフラグメントである(Yang E. et al.(1995) Cell 80, 285)。他の態様において、阻害性タンパク質は、bcl−XLと相互作用する細胞内抗体である。細胞内抗体分子は、T細胞内に一本鎖抗体分子を本明細書に記載の方法により導入および発現することを含む方法でT細胞に導入できる。このような方法は、例えば、Biocca, S. et al. (1993) Biochemical and Biophysical Research Communications 197, 422;Biocca, S. et al. (1994) Bio/Technology 12, 396;Marasco, W. A., et al. (1993) Proc. Natl. Acad. Sci. USA 90, pp. 7889;およびWerge et al. (1990) FEBS 274, 193に記載されている。
【0095】
本発明の他の実施態様では、T細胞内のbcl−XLタンパク質レベルは、T細胞を、内因性bcl−XLタンパク質レベルをダウンレギュレーションするする作用物質と接触させることにより減少させる。具体的実施態様において、作用物質は、bcl−XL遺伝子の転写を減少させるものである。T細胞内のbcl−XLタンパク質レベルをダウンレギュレーションする作用物質は、試験すべき作用物質をT細胞とインキュベーションし、bcl−XLタンパク質レベルを本明細書に記載のようにウエスタンブロット分析で測定するアッセイを使用して同定できる。
【0096】
bcl−XSまたはBadのようなbcl−XLのアンタゴニストのレベルを増加させることにより、T細胞を細胞死に対して感受性にする方法もまた本発明の範囲内である。これらのタンパク質のいずれかまたは両方のタンパク質レベルは、T細胞を例えば、bcl−XSおよび/またはBadをコードする遺伝子の発現を刺激する作用物質と接触させることにより、T細胞内で増加させることができる。一つの実施態様において、bcl−XSをコードするbcl−X遺伝子の発現を刺激する作用物質は、bcl−XL産生を刺激しない。あるいは、これらのタンパク質をコードする核酸は、bcl−XLタンパク質レベルの増加について本明細書に記載の方法と同様にしてT細胞に導入できる。bcl−XLのアンタゴニストをコードする核酸配列は、例えば、Yang, E.et al. (1995) Cell 80, 285に記載されている。bcl−XSまたはBadタンパク質レベルを増加させる方法は、bcl−XLタンパク質レベルの増加について本明細書に記載した方法を応用できる。
【0097】
本発明の一つの態様では、T細胞を、T細胞内のbcl−XLタンパク質レベルを減少する作用物質と一次T細胞活性化シグナルを提供してT細胞死を刺激する作用物質と共に接触させることにより、T細胞を細胞死に対して感受性にする。一次活性化シグナルを提供する作用物質は、抗CD3抗体のようなポリクローナルアクチベーターであり得る。本発明の好ましい態様において、一次活性化シグナルを提供する作用物質は、抗原提示細胞上の抗原である。本発明の更に別の態様では、少なくとも一つのT細胞クローンを細胞死に対して感受性にするように、T細胞をさらに抗原提示細胞の更なる抗原と接触させる。更なる態様では、T細胞を、コスティミュラトリー・シグナルを阻止する作用物質と接触させる。従って、本発明の一つの実施態様において、T細胞死は、本発明の方法に従ったT細胞における一次活性化シグナルの刺激、T細胞の共刺激の阻害およびT細胞におけるbcl−XLタンパク質レベルの減少により誘発される。本発明の方法は、従って、T細胞のポリクローナル集団のT細胞死を可能にし、または、あるいは、限定された数のT細胞クローン(例えば、T細胞集団中の抗原特異的T細胞クローン)のT細胞死の誘導を可能にする。
【0098】
T細胞の細胞死に対する感受性を増加する上記の方法は、組み合わせることができる。
【0099】
3.医薬組成物
本発明は、T細胞内のbcl−XLタンパク質レベルを増加することにより、T細胞を細胞死から防御する方法を提供する。T細胞内のbcl−XLのレベルの増加は、内生bcl−XLタンパク質のレベルの増加、またはT細胞へのbcl−XLタンパク質をコードする核酸の導入、または両方の組み合わせにより達成できる。具体的態様では、内生bcl−XLタンパク質のレベルを、T細胞を細胞内に作用する作用物質と接触させることにより増加させる。本発明の他の態様では、T細胞の細胞死に対する感受性は、T細胞を、T細胞内のbcl−XLタンパク質レベルを減少させる作用物質と接触させることにより増加させる。本発明の方法は、インビボ、エクスビボまたは両方の組み合わせにより実施できる。本発明の方法をエクスビボで実施するには、T細胞集団を被験体から得、インビトロでbcl−XLタンパク質レベルを増加または減少させる作用物質と接触させ、所望により、被験体に再投与する(この態様は更に下に記載する)。bcl−XLタンパク質のレベルは、本明細書に記載のようにウエスタンブロット分析により追跡できる。
【0100】
本発明の方法をインビボで実施する場合、作用物質をインビボの医薬投与に適した生物学的適合形態で被験体に投与する。“インビボで投与に適した生物学的適合形態”は、作用物質の治療的効果が毒性効果より重いような、投与すべき作用物質の形態を意味する。投与すべき作用物質は、bcl−XLタンパク質をコードする核酸分子または細胞内のbcl−XLタンパク質の産生を阻害するアンチセンス核酸分子をコードする核酸分子、細胞内に作用してbcl−XLタンパク質レベルを増加または減少する作用物質、およびbcl−XLタンパク質をコードする核酸分子にまたはbcl−XLアンチセンス核酸分子をコードする核酸分子に作動可能に連結させた誘導性制御配列を調節する作用物質、を含む。
【0101】
“被験体”なる用語は、免疫応答を誘導できる生存生物、例えば哺乳動物を含むことを意図する。被験体の例は、ヒト、イヌ、ネコ、マウス、ラットおよびそれらのトランスジェニック種である。例えば、本発明の範囲内の動物は、家畜および家禽のような農業的対象となる動物を含む。あるいは、本発明の方法はまた植物に応用できる。
【0102】
本発明の作用物質の治療的有効量の投与は、所望の結果を達成するのに必要な投与量および期間で有効な量と定義する。例えば、作用物質の治療的有効量は、病気の状態、年令、性別、および被験体の体重および被験体に所望の応答を誘発する作用物質の能力などの要因に依存して変化し得る。用量レジメは、最適な治療的応答を提供するように調節し得る。例えば、数回に分割した用量を、毎日投与してもよく、またその用量を治療的状態の緊急性に合わせて徐々に減少させてもよい。
【0103】
作用物質は、注射(皮下、静脈内等)、経口投与、吸入、経皮投与または直腸投与のような慣用法で投与し得る。投与経路によるが、作用物質を不活性化し得る、酵素、酸および他の自然条件の作用から防御するために、作用物質をある物質で被覆してもよい。
【0104】
非経口投与以外で作用物質を投与するために、作用物質を、その不活性化を防御する物質でコートするか、その作用物質と同時投与する。例えば、bcl−XLタンパク質をコードする核酸分子を含む発現プラスミドは、酵素阻害剤と共に適当な担体または希釈剤で、またはリポソームのような適当な担体で、被験体に投与し得る。薬学的に許容される希釈剤は、食塩水および水性緩衝化溶液を含む、酵素阻害剤は、膵臓トリプシン阻害剤、ジイソプロピルフルオロホスフェート(DEP)およびトラシロールを含む。リポソームは、水中油中水(water-in-oil-in-water)エマルジョンおよび慣用のリポソームを含む(Strejan et al., (1984) J. Neuroimmunol. 7:27)。分散剤もまた、グリセロール、液体ポリエチレングリコールおよびこれらの混合物中、および油中で製造できる。貯蔵および使用の慣用条件下で、これらの製剤は微生物の生長を防ぐ防腐剤を含み得る。
注射使用に適当な医薬組成物は、滅菌水性溶液(水溶性の場合)または分散剤および滅菌注射溶液または分散剤の即席製造用の滅菌粉末を含む。全ての場合、組成物は滅菌状態でなければならず、容易な注射可能性が存在する程度に液体でなければならない。製造および貯蔵の条件下で安定でなければならず、細菌および真菌のような微生物の汚染作用から防御されていなければならない。担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコールおよび液体プロピレングリコール等)およびそれらの適当な混合物を含む溶剤または分散媒体であり得る。適当な溶解性は、例えば、レシチンコーティングの使用により、分散剤の場合では、必要な粒子サイズの維持により、および界面活性剤の使用により維持できる。微生物活性の阻止は、種々の抗細菌および抗真菌剤、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサール等により達成できる。多くの場合、等張剤、例えば、糖、マンニトール、ソルビトールのようなポリアルコール、塩化ナトリウムを組成物中に含むのが好ましい。注射組成物の吸収延長は、吸収を遅延する作用物質、例えば、モノステアリン酸アルミニウムおよびゼラチンを組成物中に含むことにより達成できる。
【0105】
滅菌注射溶液は、必要な量の作用物質を、適当な溶媒に、必要に応じて上述の1またはそれ以上の成分と組み合わせて包含させ、続いて滅菌濾過することにより製造できる。一般に、分散剤は、作用物質を塩基性分散媒体および上述からの必要な他の成分を含む滅菌媒体に包含させることにより製造する。滅菌注射溶液の製造における滅菌粉末の場合、好ましい製法は、真空乾燥および凍結乾燥であり、予め滅菌濾過した溶液から、有効成分(例えば、ペプチド)+他の所望の成分の粉末を製造する。
【0106】
作用物質が上記のように適切に保護されている場合、例えば、不活性希釈剤または吸収可能食用担体と共に、経口投与し得る。本明細書で使用する限り、"薬学的に許容される担体”は、任意のおよび全ての溶媒、分散媒体、コーティング、抗細菌および抗真菌剤、等張剤および吸収遅延剤等を含む。このような媒体および作用物質の薬学的活性物質への使用は当分野で既知である。慣用の媒体または試薬が作用物質と不適合である場合を除いて、治療用組成物へのその使用が期待される。補足的有効化合物もまた組成物に包含できる。
【0107】
非経口投与組成物を、投与が容易であり、均一の量である投与単位形態に調剤するのが特に有利である。本明細書で使用する投与単位形態は、処置すべき哺乳動物被験体に均一に投与するのに適した物質的に別の単位を意味する;各単位は、必要な薬学的担体と共に、所望の治療効果を生むと計算された予定量の有効化合物を含む。本発明の投与単位形態の詳細は、(a)作用物質の独自の特性および達成すべき具体的な治療効果、およびb)被験体における感受性処置剤のような調剤の分野で固有の限界により規定されており、それに直接依存している。
【0108】
4.本発明の方法の応用
本発明は、T細胞内のbcl−XLタンパク質レベルを増加することにより、T細胞を細胞死から防御する方法に関する。本発明はまたT細胞内のbcl−XLタンパク質レベルを減少させることにより、T細胞を細胞死に対して感受性にすることにも関する。これらの方法は、インビボおよびエクソビボの両方で実施できる。エクソビボで実施する時、末梢血単核細胞を被験体から得、密度勾配遠心、例えば、フィコール/ハイパークにより単離できる。具体的態様において、純粋な末梢血細胞を次いでbcl−XLタンパク質のレベルを修飾する作用物質と接触させる。この方法の他の態様において、末梢血単核細胞をbcl−XLタンパク質のレベルを調節する作用物質と接触させる前に特異的細胞タイプで豊富化させる。単核細胞は、例えば、プラスチック上への接着により枯渇できる。所望により、CD4+T細胞集団を、残りの単球、B細胞、NK細胞およびCT8+T細胞から、モノクローナル抗体(mAb)および商業的に入手可能なmAb(例えば、抗CD14(Mo2)、抗CD11b(Mo1)、抗CD20(B1)、抗CD16(3G8)および抗CD8(7PT3F9)mAb)を使用した抗マウスIg被覆磁気ビーズを使用して分離できる。本発明の方法は、CD4+CD45RA+(自生CD4+T細胞)およびCD4+CD45RO+(記憶T細胞)T細胞サブセットのようなCD4+T細胞のサブセットに適応できる。これらは、CD4+CD45RA+細胞の製造については、抗CD45RO抗体(UCHLI)の更なる使用と共に、CD4+CD45RO+T細胞の製造については抗CD45RA抗体(2H4)の更なる使用と共に、上記のように製造できる。
【0109】
精製の効果は、抗CD3、抗CD4、抗CD8および抗CD14mAbまたはT細胞の特異的サブセットを認識する付加的抗体、続くフルオレセインイソチオシアネート接合ヤギ抗マウス免疫グロブリン(Fisher, Pittsburgh, PA)または他の二次抗体を使用したフローサイトメトリー(Coulter, EPICS Elite)により分析できる。
【0110】
エクスビボで使用する細胞集団のタイプは、bcl−XLタンパク質レベルの修飾に使用する作用物質のタイプ、作用物質をT細胞に運搬するのに使用する媒体のタイプ、bcl−XLタンパク質レベルの増加が望ましいT細胞のサブセットを含む種々の因子に依存する。従って、作用物質がT細胞のサブセットに特異的に影響を与える場合、(例えば、T細胞の特異的サブセットのみを標的にする運搬媒体の使用により)、T細胞の特異的サブセットの精製は必要ではない。作用物質が細胞の特異的サブセットを標的とするようにさせる媒体は、リポソームまたは所望の細胞タイプを認識する分子が結合した組換えウイルス粒子を含む。このような分子には、表面分子に対する抗体または受容体に対するリガンドがある。T細胞の選択的サブセットのみを、例えばCD4+T細胞のみを標的としたい場合、および使用する作用物質がこのT細胞のサブセットを選択的に標的とできない場合、細胞を作用物質と接触させる前にT細胞の特異的サブセットを単離する必要があり得る。
【0111】
被験体から得るT細胞のような末梢血細胞またはその精製サブセットを、次いで、インビトロで、T細胞内のbcl−XLタンパク質レベルを調節する作用物質の存在下にインキュベーションする。作用物質の量は、作用物質のタイプ、所望の効果および作用物質と接触させる細胞の集団などの種々の要因によって変わる。細胞集団に添加すべき作用物質の適当な量は、作用物質の種々の量を細胞培養物に添加し、種々の時間でのbcl−XLタンパク質の量を、本明細書に記載のようにウエスタンブロット分析により測定するアッセイを行うことにより決定できる。細胞の異種集団を作用物質と接触させる場合、細胞をウエスタンブロット分析にかける前に、bcl−XLの修飾が望まれるT細胞のサブセットを最初に単離することが必要であり得る。T細胞の特異的サブセットは、上記のようにネガティブセレクションにより細胞集団から単離できるか、またはあるいは、T細胞の特異的サブセットをFACSを使用して単離できる。
【0112】
本発明の具体的態様において、被験体から単離されたT細胞を、bcl−XLのタンパク質レベルを修飾する作用物質と接触させ、更に、インビトロでT細胞の集団を拡張する(即ち、集団内のT細胞の数を増加させる)ために培養する。T細胞の集団のインビトロ拡張は、公開PCT出願番号WO94/29436に記載され、その内容は本明細書に参考として包含させる。
【0113】
T細胞またはそのサブセット内のbcl−XLタンパク質レベルの修飾に続いて、細胞を被験体に再投与できる。具体的態様では、まず細胞を精製して、被験体への投与に望ましくない培養培地中の物質を除去する。精製は、例えば、フィコールハイパーク勾配遠心により行うことができる。
【0114】
あるいは、本発明の方法はインビボで実施できる。この態様において、T細胞内のbcl−XLタンパク質レベルを修飾する作用物質は、薬学的に許容可能な媒体中で、所望の治療効果を得るのに充分な量で被験体に投与する。作用物質は、本発明の方法により処置すべき症状タイプによって、局所的または全身的に投与する。作用物質の投与に適当な媒体および経路は、本明細書3項に記載している。
【0115】
4.1.本発明の方法のT細胞死からの防御が有利な状態への適用
本発明の方法は、T細胞の細胞死を防止するのに有用である。本発明の方法により、全てのT細胞で細胞死が防止予防できるか、またはあるいは、細胞死をT細胞の特異的サブセット中で選択的に防止できる。更に、T細胞を、長期間、または短期間、細胞死から防御できる。局所的に存在するT細胞または全身的に(例えば、末梢血に)存在するT細胞を、細胞死から防御できる。
【0116】
好ましい態様において、本発明の方法は、HIV感染個体のCD4+T細胞の細胞死を防止し、HIV感染からT細胞を防御するのに使用する。HIV感染中、ウイルスはCD4+T細胞に感染し、これを殺傷する。従って、個体中のCD4+T細胞の数は、被験体の微生物からの感染を防止するのに不十分な量まで段々に減少する。bcl−XLタンパク質レベルが増加しているT細胞は、T細胞の生存率増加を測定したところ、HIV感染に対して著しい防御を示すことが観察される(実施例9参照)。従って、T細胞内のbcl−XLタンパク質レベル増加は、T細胞をHIV誘導細胞死から防御する。従って、T細胞内のbcl−XL過発現は、HIV感染被験体におけるCD4+T細胞の数を維持する有効な方法を提供する。更に、本発明の方法は、HIV感染被験体のCD4+T細胞の数の増加および/またはHIV感染個体におけるT細胞枯渇の速度減少をもたらし得る。
【0117】
本発明の方法は、HIV感染、またはT細胞を細胞死に対して感受性にする他の感染物の感染ような、T細胞関連疾患を有する個体由来のT細胞の集団の拡張を可能にする方法を提供する。従って、この方法は、インビトロで細胞を拡張する方法を提供する。この場合、拡張T細胞集団を、被験体に投与し戻すことができる。好ましい態様において、T細胞を、T細胞に一次活性化シグナルを提供する作用物質およびT細胞にコスティミュラトリー・シグナルを提供する作用物質の組み合わせで刺激する。より好ましい態様において、T細胞をT細胞上のCD28と相互作用する作用物質およびT細胞受容体を介してbcl−XLタンパク質レベルが増加され、細胞が細胞死から防御されるようにT細胞を刺激する作用物質の組み合わせと共に培養する。
【0118】
HIV感染被験体のT細胞死を防止する本発明の具体的実施態様では、末梢血細胞を被験体から入手し、インビトロで修飾して、CD4+T細胞中のbcl−XLタンパク質の量を増加させ、個体に再投与する。本発明の好ましい態様では、bcl−XLタンパク質をコードする核酸分子をT細胞に挿入することにより、被験体由来のCD4+T細胞を修飾して、bcl−XLタンパク質レベルを増加するようにする。より更に好ましい態様では、bcl−XLをコードする核酸分子は、細胞をウイルス粒子で感染させるような方法により、T細胞に導入されるウイルスベクター内に含有される。方法の他の好ましい態様は、CD4+T細胞を、被験体に再投与する前に培養で更に拡張する。従って、本発明の方法は、HIVに感染した被験体の免疫系をCD4+T細胞で再集合させ、同時にこれらのCD4+T細胞をウイルスによる感染に対して耐性にすることを可能にする。
【0119】
被験体に、個体のCD4+T細胞内のbcl−XLタンパク質のレベルを増加させる作用物質を投与することにより、HIVに感染した被験体のCD4+T細胞を、細胞死から防御することもまた可能である。この作用物質は、生理学的に許容される媒体に含まれたbcl−XLタンパク質をコードする核酸分子であり得る。本発明のより好ましい態様において、媒体を更に核酸がCD4+T細胞を標的とするように操作する。あるいは、作用物質は、CD4+T細胞のbcl−XLタンパク質レベルを増加させるように細胞内に作用する作用物質である。
【0120】
他の態様において、HIVで感染させた被験体のT細胞を、上記のエクスビボおよびインビボ法を組み合わせた方法により細胞死から防御する。本方法は、個体のCD4+T細胞を、インビボおよび/またはエクスビボでbcl−XLタンパク質レベルを増加する幾つかの作用物質と接触させることを含む。例えば、bcl−XLタンパク質をコードする核酸をT細胞に導入し、T細胞を更に内生bcl−XLタンパク質レベルを増加するために細胞内に作用する作用物質と接触させる。
【0121】
あるいは、本発明の方法は、より迅速な感染の除去のために、免疫応答を強化するのに有用である。従って、本発明の具体的態様は、活性化T細胞をT細胞内のbcl−XLタンパク質レベルを増加させる作用物質と接触させて、細胞死から防御する。T細胞の活性化に続き、CT4+T細胞を細胞死から防御することにより、ヘルパーT細胞が、Tヘルパー細胞が通常提供し得る以上の有効な“ヘルプ”を提供できる。同様に、延長寿命を有するCD8+T細胞は、通常の寿命のCD8+T細胞よりも標的細胞を溶解できる。本発明の範囲内の方法は、全身感染および局所感染を処置する方法である。従って、作用物質を全身または局所的に投与できる。本発明の好ましい態様において、T細胞内のbcl−XLタンパク質レベルを増加する作用物質は、bcl−XLタンパク質レベルを増加するために細胞内に作用する作用物質である。本発明のより更に好ましい態様において、作用物質は、活性化T細胞の寿命が必要以上に(例えば、活性化T細胞が記憶T細胞になるかまたは感染の除去後に死滅する)長くならないような短い半減期を有する作用物質である。
【0122】
4.2.T細胞死に対する感受性増大が有利な状態への本発明方法の応用
本発明の方法はまた、T細胞の細胞死に対する感受性の増大にも有用である。従って、通常、内生bcl−XLタンパク質レベル増加により細胞死から防御される状態に遭遇するT細胞は、T細胞内のbcl−XLタンパク質のレベルが著しく減少すれば死滅する。本発明の方法は、T細胞関連疾患の被験体の処置に有用である。“T細胞関連疾患”は、延長寿命を有するT細胞に関連する疾患を意味する。
【0123】
具体的実施態様において、本発明の方法は、被験者の自己免疫疾患の処置にも有用である。細胞死に対する感受性を自己反応性T細胞中で増大させ、自己免疫疾患の作用を改善する。処置し得る自己免疫疾患の例は、多発性硬化症、インシュリン依存性糖尿病、関節炎(例えば、リウマチ性関節炎、若年性リウマチ性関節炎、骨関節炎)、ミエスシェニア・グラビス(myesthenia gravis)、心筋炎、ギラン・バレー症候群、全身性紅斑狼瘡、自己免疫甲状腺炎、皮膚炎、乾癬、ショーグレン症候群、円形脱毛症、クーロン病、アフタ性潰瘍、虹彩炎、結膜炎、角結膜炎、潰瘍性大腸炎、アレルギー、皮膚性紅斑狼瘡、硬皮症、膣炎、直腸肛門炎、薬疹、癩反転反応、癩性結節性紅斑、自己免疫葡萄膜炎、アレルギー性脳脊髄炎、急性壊死性出血性脳疾患、特発性両側性進行性感音難聴、形成不全貧血、赤芽球癆、特発性血小板減少症、多発性軟骨炎、ウェゲナー肉芽腫症、慢性活動性肝炎、スティーブン・ジョンソン症候群、特発性スプルー、偏平苔癬、グレーブス眼病、結節症、原発性胆汁性肝硬変、後部葡萄膜炎および肺内部線維症を含む。
【0124】
他の態様において、本発明の方法は、移植片拒絶反応の低減に使用する。例えば、移植細胞を、移植片に反応性のT細胞を細胞死に対して感受性にするように、T細胞内のbcl−XLタンパク質レベルを減少する作用物質と共に宿主に投与する。この方法は、宿主に、CTLA4Igのような共刺激を遮断する作用物質を投与することも含み得る。同様に、本発明の方法は、移植片対宿主病の予防にも使用できる。好ましい方法において、ドナー骨髄を移植前に宿主細胞およびT細胞内のbcl−XLタンパクを減少する作用物質と接触させる。こうして、ドナー骨髄のT細胞を細胞死に対して感受性にし、共刺激を阻害する作用物質と併用することで宿主特異的抗原に反応性のT細胞の細胞死を増大できる。
【0125】
以下は、本発明を更に下記実施例により説明するものであり、これは更に限定するものと見なすべきではない。本出願明細書に引用の全ての参考文献、継続特許出願および公開特許は、参考として本明細書に明示包含する。
【実施例】
【0126】
実施例1: 活性化がγ−照射後のT細胞の生存を高める
T細胞生存に対するT細胞活性化経路の効果を調べるために、休止T細胞をヒト末梢血から前から知られているネガティブセレクション法(June,C.H.et.al.(1987)Mol.Cell.Biol.7:4472−4481)により、分離した。簡単に言うと、細胞を抗体の反応混液の対象とし、休止CD28−ポジティブT細胞以外のすべての細胞を除去する。ウシ胎児血清(10%)、L−グルタミン(2mM)ペニシリン/ストレプトマイシン(100U/ml,100μg/ml)およびHEPES(20mM)を補充したRPMI 1640中でT細胞を培養した。細胞は、活性化または照射の前に一夜置いた。
【0127】
CD28+T細胞は、CD28特異モノクローナル抗体により提供される共刺激の存在または不在下に、培地のみで培養するか、あるいはTCR/CD3複合体を12時間架橋することにより刺激した。T細胞の架橋はプレート固定化抗−CD3(G19.4〔1μg/mlで〕)を用いて行い、T細胞の共刺激はCD28に対する可溶性抗体(モノクローナル抗体(mAb)9.3)により1μg/mlで実施された。これらの各グループからの細胞はグループに分けた。1つのグループはさらに操作を行わないで培養し、他のグループはセシウム源γ−照射(J.L.Shepherd Inc.)による15Gyのγ照射を行った。照射後、細胞は96ウエル培養皿(Costar)中に100,000細胞/ウエルで入れ、細胞の生存能力および全数をトリパンブルー排除法およびプロピジウムヨージド排除法により調べた。
【0128】
プロピジウムヨージド排除法について、2×105細胞の2つの分離サンプルを1%BSAおよび0.01%ナトリウムアジドを補充したPBS 0.5mlに入れて、再懸濁した。プロピリジウムヨージド2ml(0.5mg/ml)を細胞に加え、サンプルをFACSortおよびLysis IIソフトウエアを用いたFACSで分析した。生存能力パーセントは、プロピジウムヨージドを排除した細胞数を全細胞数で割ることにより求めた。解析による破片を除くために、生存細胞の前進光散布特性を利用した。
【0129】
結果を図1に図示する。全3集団の細胞が4日以上の培養期間において高い生存能力を保持した(図1、パネルA)。休止および活性化T細胞の生存もγ−照射後に調べた(図1、パネルB)。γ−照射誘発アポトーシスは、特殊なレセプターリガンド干渉を必要とせず、特殊レセプターレベルについてのコントロールの必要性なしに細胞の生存に細胞の活性化がどのように影響するのかを調べる手段とし得る。これらのアッセイに用いた照射量(15Gy)は、実際に全群のすべての細胞に致命的なDNA損傷を与えるのに充分である。従って、細胞が確保された日数を越えて死ぬ率は、DNA障害に対する応答において被ったPCDに抵抗する能力を測定するのに用いられる。
【0130】
図1、パネルBに示されるように、照射後、休止T細胞の生存能力は、刺激されたリンパ球のいかなる集団にみられるよりも、非常に速く低下した。照射4日後、いずれかの活性化集団におけるT細胞は、休止集団の細胞よりも統計的により生存し得た(p<0.02)。CD28レセプターによって共刺激された細胞は、抗−CD3のみで刺激された細胞よりも、生存能力の僅かな、しかし再現性ある上昇を示した。抗−CD3刺激および抗−CD3+抗−CD28刺激細胞における細胞生存性が保持されることは、生存能力アッセイに平行してなされた細胞数の測定が細胞の絶対数の変化を示さなかったから、後のT細胞増殖の結果ではなかった。さらに、活性化された集団における全細胞は、遅G1またはG2のいずれかにおける細胞周期内に止まった。3母集団における細胞死は、細胞が最初に円鋸歯状になり、次いで核の凝縮およびDNAフラグメンテーションをおこす典型的なアポトーシス性パターンに続いておきる。
【0131】
このように、本実施例は、T細胞の活性化がγ−照射後のT細胞生存を高めることを示す。
【0132】
実施例2: CD28共刺激は抗−CD3活性化T細胞の生存能力を増加する
CD28共刺激の存在または不在により刺激された細胞間における一つの相違は、これらの細胞により産生されたリンホカインのレベルである(Lindsten,T.et.al.(1989)Since244:339−343)。従来から、生長因子が多様な細胞タイプにおける細胞生存の外的調節で重要な役割を果すことが示唆されている(Groux,H.et.al.(1993)Eur.J.Immunol.23:1623−1629),Nunez,G.et.al.(1990)J.Immunol.144:3602−3610)。生長因子が共刺激の保護作用の因であるかどうかを決めるために、次の実施例が行われた。
【0133】
CD28+T細胞は、実施例1記載のように、12時間、抗−CD3の存在下にCD28モノクローナル抗体との共刺激の存在または不在で培養された。細胞は、そのならし培地にそのまま残し、洗い、次いで新しい培地に再懸濁するか、あるいは洗って、組換えIL−2(rIL−2)(Boehringer-Mannheim)200μ/mlを補充した新しい培地に再懸濁された。各グループからのアリコートを無操作のままにするか、または12Gyのγ−照射にさらした。細胞生存能力は、実施例1に記載したように、操作後4日間毎日、プロピジウムヨージド排除法により測定した。
【0134】
結果を図2に図示する。図2のパネルAによると、TCR/CD3複合体のみで活性化されたT細胞は、活性化後培養において高い生存能力を保持していた。しかし、刺激細胞を水洗することによるならし培地の除去は、培養中に生存する細胞の能力を著しく低下せしめることにつながった。このインビトロ培養中に生存する能力の低下は、IL−2の添加により逆転され得る。抗−CD3刺激細胞に15Gyのγ−照射がなされたとき(図2、パネルB)、一夜刺激後のならし培地の除去は、IL−2添加により逆転された細胞生存性の激しい低下をもたらした。このことは、生存が抗原レセプター活性化細胞における外的生長因子のレベルによって第一義的に決定されることを示している。従って、活性T細胞が培養中で生存し得る能力は、それがIL−2などの生長因子を生産するオートクリン能力に基づいている。これらのデータは、IL−2がT細胞生存の外的レギュレーターであることを確認する。
【0135】
TCR/CD3複合体のみで刺激されたT細胞についての結果と対照的に、抗−CD3+抗−CD28併用で刺激されたT細胞は内因リンホカインを洗出しても、さらにIL−2を培養上澄液に加えても、その生存に変わりはない(図2、パネルCおよびD)。
【0136】
本実施例の結果は、CD−28仲介共刺激が細胞生存の外的メディエーターの産生に影響を及ぼさないだけでなく、PCDを受けた細胞の内因的感受性を調節する役割を演じる(次の実施例でさらに調べる)ことを表している。
【0137】
実施例3: T細胞活性化中のbcl−2およびbcl−X mRNA発現
PCDを受けたT細胞の内因的感受性に関与する遺伝子の発現を調節するときのCD28の役割について調べるために、休止ヒトT細胞および抗−CD28の存在または不在下のTCR抗原レセプター複合物の架橋により活性化されたT細胞についてbcl−Xおよびbcl−2発現レベルを解析した。
【0138】
RNAは、培地のみで培養されたCD28+T細胞から単離され、次いで1,6および12時間、抗−CD3または抗−CD3と抗−CD28で刺激され、ノーザンブロット・ハイブリダイゼイションにより解析された。RNAは、すでに記述されているように(June,C.H.et.al.(1987)Mol.Cell.Biol.7:4472-4481)、勾配グアニジウム/CsCl2による遠心分離でT細胞から分離した。等量のRNA(非変性アガロースゲル上で電気泳動した28SリボソームRNAのエチジウムブロマイド着色で検定)をアガロース/ホルムアルデヒド変性ゲル上に充填し、大きさによって分離した。ゲルをニトロセルロース(SchleicherおよびSchuell)に移して、真空で2時間80℃に加熱する。ブロットは、42℃6時間Stark液(50%ホルムアミド、5×SSC(1×SSCは0.15M Nacl,0.015Mクエン酸ナトリウム)1×Denhardt液,25mM リン酸ナトリウム,pH6.5,Torula RNA 250μg/ml)中でプレハイブリダイズし、次いでStark液中で10%硫酸デキストランおよび32P−ラベルニック翻訳プローブ(1×106dpm/ml)で一夜42℃ハイブリダイズした。ブロットをプローブし、次いでヒトbcl−XL cDNAおよびネズミbcl−2 cDNAに特異なプローブ(S.Korsmeyer)で剥離した。
【0139】
ノーザンブロット・ハイブリダイゼーションの結果を図3、パネルAに示す。その結果によると、bcl−Xまたはbcl−2 mRNAのいずれも、休止T細胞において検出レベルでは発現されなかった。しかし、両bcl−2およびbcl−X mRNAの発現はT細胞活性化6時間後に誘導される。CD28共刺激はbcl−2 mRNAの発現に顕著な作用を有しなかった。一方CD28共刺激はのbcl−X mRNA発現を高めた。抗−CD3および抗−CD28で刺激された細胞は、レベルが低下し始めた後48時間、bcl−Xの高レベルの発現を維持した。bcl−2機能調節等に負の役割を有すと思われるbcl−2ファミリーの第3ナンバーは、遺伝子baxである。共刺激もbax mRNAレベルに影響を与えなかった。
【0140】
bcl−Xの第1エクソンにおける2つの5'スプライス供与体の代替使用は2つの明白なmRNA種を生成する。bcl−XLはエクソン1の完全コード領域を保持し、細胞の生存を高めるように機能する。一方、エクソン1内の上流スプライス供与体部位の利用は、エクソン1コード領域内で189bp欠失を含むbcl−XS mRNAをもたらす。Bcl−XSタンパク質は、両機能の優性ネガティブレギュレーターとして作用する(Boise,L.H.et.al.(1993)Cell 74:597−608)。bcl−XSおよびbcl−XL mRNAは189bpによってのみ相違するので、RNase保護は、T細胞活性および共刺激においてどのbcl−X mRNAが誘導されたかを決定するために、行われた。
【0141】
Rnase保護検定は、休止細胞あるいは抗−CD3または抗−CD3および抗−CD28で6または12時間刺激されたT細胞から分離されたRNAについて実施された。Rnase保護アッセイは、市販キット(Ambion)の説明書に従って実施された。T細胞RNA 3μgを放射性標識リボプローブでハイブリダイズした。このプローブは、bcl−XL cDNA+プラスミド中のpBluesript SKをAccIで切断し、Promegaからのキットを用いてプロモータよりインビトロで転写体を生成せしめて、つくられた。336ヌクレオチド・リボプローブはゲル電気泳動により精製され、抽出され、次いで、RNaseAおよびRNaseT1の反応混液の添加前に16時間42℃でT細胞RNAにハイブリダイズされた。bcl−XL mRNAによるプローブの保護は264ヌクレオチドのフラグメントを生成し、bcl−XSメッセージへのハイブリダイゼイションは放射性標識プローブの163ヌクレオチドフラグメントを保護する。保護産物を5%アクリルアミド/7M尿素シーフェンス・ゲル上で分離した。末端ラベルpBluescript SKII+HpaII消化はマーカーとして用いた。ゲルを乾燥し、XAR−5フィルム(Kodak)に感光した。
【0142】
Rnase保護アッセイの結果を図3パネルBに示す。その結果によると、CD28共刺激により調節が上昇された主なbcl−X mRNAは、bcl−XLである。小さい誘導もbcl−XSのレベル上でみられる。
【0143】
そのように、本実施例の結果は、CD28による活性T細胞の共刺激がbcl−XL mRNAに顕著な増加をもたらすことを示している。
【0144】
実施例4: T細胞がBcl−2タンパク質の構成的発現を発揮する
bcl−2 mRNAのレベルがbcl−2タンパク質のレベルとあまり相関しないことは前に報告されているので(Chleq-Dechamps,C.M.et.al.(1993)Blood,81:293−298)、抗−CD28で共刺激されるか、またはされない活性T細胞中のbcl−2タンパク質の量を測定した。
【0145】
この実施例において、CD28+T細胞は上記のように抗−CD3または抗−CD3および抗−CD28抗体と共に0、6、12または24時間培養され、bc−2タンパク質レベルが免疫沈降法およびウエスタンブロット法により測定された。各時点で、各培養条件の細胞4×107が分離され、1.0ml NET−N(100mM NaCl,1mM EDTA 20mM Tris,pH8.0,0.2%NP−40)中に溶解された。核および細片は2分間4℃の微遠心で除去された。上澄液を30分間パンソルビン(Calbiochem)50mlで処理した。遠心でパンソルビンを除いた後、上澄液を2つの等量(450μl)に分け、それに抗−bcl−Xウサギ血清(実施例5参照)1μgまたはハムスター抗−Bcl−2モノクローナル6C8(Hockenbery,D,et.al.(1990)Nature348:334−336)2.5μgのいずれかを加えた。溶解生成物を4℃で1時間振動し、次いで30分間でタンパク質Aアガロース(Gibco BRL)25μlを加えた。免疫沈降物を採取し、NET−Nで2回、NETで1回洗った。2×SDS充填緩衝液100μlを加え、素早く凍らせ、ゲル電気泳動を行うまで−20℃に保持した。
【0146】
免疫沈降物を煮沸し、15%ゲルでSDS−PAGEにかけた。ゲルは、200mAで3時間、BioRad移送器を用いて電気ブロットによりニトロセルロースに移した。ブロットを一夜4℃で5%非脂肪ミルク/0.2%ツイン20中でブロックし、上記のブロット液中で精製6C8 mAb(1:200)とハイブリダイズした。ウエスタン・ブロットをHyperfilm(Amersham)を有するECLシステム(Amersham)を用いて展開した。
【0147】
ウエスタン・ブロットの結果を図4に示す。休止末梢血T細胞がBcl−2タンパク質を高レベルで発現することが分かる。さらに、Bcl−2タンパク質のレベルがTCR/CD3レセプターの抗−CD3抗体での架橋後の最初の24時間以後あまり変動しない。CD28レセプターでの共刺激もBcl−2タンパク質レベルに影響を与えないようであった。
【0148】
本実施例は、休止T細胞がBcl−2タンパク質を発現し、共刺激あり、または、なしのT細胞レセプターでのT細胞刺激がBcl−2タンパク質レベルを上昇しないことを示す。
【0149】
実施例5:CD28共刺激はbcl−X1タンパク質発現を高める
本実施例において、休止T細胞およびCD282の共刺激の存在または不在でT細胞レセプターで活性化されたT細胞におけるbcl−X1のタンパク質レベルを解析した。
【0150】
bcl−X1のタンパク質レベルは、Bcl−2のタンパク質レベルの測定と平行して、実施例4に記載されたアッセイ法によって測定された。あらかじめ清澄化した後、上澄液を2等量(450μl)に分け、抗−bcl−Xウサギ血清1μlをこれらのアリコートの1つに加えた。タンパク質レベルは、モノクローナル抗−bcl−X抗体2A1(1:1希釈)を用いて実施例4のように測定した。
【0151】
bcl−Xに対するポリクローナルおよびモノクローナル抗体を次のように製造した。bcl−XSのオープン・リーディング・フレーム(転写解読枠)をプライマー(5'−GGA GAT ATA CAT ATG TCT CAG AGC AAC CGG GAG CTG GTG−3’および5'−CGG GAT CCC GTC ATT TCC GAC TGA AGA GTG AGC CCA GCA G−3’)でのPCRで増幅し、pET−3bのNdeIおよびBamHI部位(Novagen)にクローン化した。組換えタンパク質を0.4mM IPTGでの誘導によりBL21細胞中で製造した。このタンパク質は、細菌溶解の不可溶フラクション中にあると測定され、次のように部分的に精製された。不可溶フラクションを2M尿素で洗い、6Mグアニジン−HClで安定化した。タンパク質は、3Mおよび1Mグアニジン−HClによる段階透析により再生し、次いでpH8.3で500mM KClでPBSに対する透析を行った。タンパク質濃度は、市販キット(BioRad)を用いたBradfordアッセイ法により測定し、タンパク質純度はSDS−PAGEおよびコーマシックブルー着色法によりアッセイした。
【0152】
ウサギポリクローナルの抗体の製造のために、組換えタンパク質1mgを完全フロイント・アジュバント(CFA)に懸濁し、ウサギの脊中の数箇所に皮下注射した。不完全フロイント・アジュバント中のタンパク質200μgで追加免疫した。インビトロ翻訳タンパク質およびトランスフェクトション細胞系列を用いて、組換えタンパク質に対して、および特異的免疫沈降についてのウエスタンブロット法で血清を選別した。
【0153】
モノクローナル抗体の製造のために、CFA中に乳化したポリアクリルアミドゲル中の組換えbcl−XSを後足の皮下および腹腔内投与して、BALB/Cマウスを免疫化した。マウスの免疫は、不完全フロイント・アジュバント中に乳化した可溶性のアクリルアミド基底ゲル結合タンパク質の混合物で、30日の間隔をおいて2度、追加免疫した。最後の追加免疫から3日後に、脾臓およびリンパ節細胞を採取し、標準的技術(Kearney,J.F.(1984)In Fundamental Immunology,W.E.Paul,ed.,Raven Press,NY,751-766)を用いて、P3X63−Ag8.653ミエローマ細胞と融合せしめた。融合15日後、ハイブリドーマ上澄液について、ホウ酸塩緩衝液中(pH8.4)5μg/mlで組換えbcl−XSによりコートされたウエルにおいてELISAによる活性を調べた。ELISAに陽性であったハイブリドーマをbcl−XLまたはbcl−2でトランスフェクションさせたFL5.12に対するウエスタンブロットにより選別した。陽性ハイブリドーマ系は、制限希釈によりクローンし、再選別し、腹水製造のためにプリスタン−初回免疫BALB/Cマウスに注射した。
【0154】
bcl−Xのタンパク質レベルを表すウエスタンブロット解析の結果を図4に示す。bcl−Xのタンパク質レベルは、T細胞活性化のレベルによって非常に変化する。検出可能なbcl−Xタンパク質産物は休止末梢T細胞において測定されなかった。ウエスタンブロットで抗体により認識されたタンパク質は、対応cDNAでトランスフェクトされた細胞中に合成されるbcl−XLと同様にインビトロ翻訳bcl−XLが約29kDaであるので(Boise,L.H.et al.(1993)Cell 74:597-608)、bcl−XLに対応し、bcl−XSに対応していない。
【0155】
図4はさらに、休止T細胞の抗−CD3刺激が、刺激6時間後に最初にみられ、次いで分析の24時間を通して細胞中に蓄積する検出可能なbcl−XLタンパク質の発現を誘発することを表している。さらに抗−CD3刺激細胞のCD28共刺激は、bcl−XLの発現を有意に高めた。bcl−XSではいかなる時点でもみられなかった。
【0156】
他の例では、休止T細胞は、培地のみ、抗−CD28,抗−CD3,抗−CD3,および抗−CD28または抗−CD3およびIL−2(100μ/ml)により24時間で活性化された。細胞質抽出物を記載のように調製し、タンパク質100μgをSDS−PAGEおよびポリクローナルbcl−X血清と共にウエスタンブロット分析に供した。
【0157】
その結果を図5に示すが、休止T細胞および抗−CD28でインキュベートしたT細胞は、bcl−XLタンパク質を発現しなかった。しかし、抗−CD3刺激細胞のCD28共刺激は、bcl−XLタンパク質の発現を有意に高めた。24時間点の抗−CD3+抗−CD28刺激細胞におけるbcl−XL発現レベルは、脾臓ホーカス形成ウイルス長期リピートのコントロール下で、bcl−XLの発現がなされている安定なトランスフェクタントのレベルと同じであった(図5の最終欄)。
【0158】
CD28共刺激は、早ければ刺激6時間後に観測され、24時間培養中継続するbcl−XLタンパク質の蓄積を高めた(図4および5)。一方、抗−CD3およびIL−2(100units/ml)での細胞処理は、抗−CD3のみで処理されたレベル以上にはbcl−XL発現をさらに高めなかった(図5)。さらに、CD2,CD5,CD11a,CD18に対するまたはMHCクラスIに対するモノクローナル抗体で抗−CD3−活性T細胞を処理しても、bcl−XLレベルは抗−CD3のみで処理した細胞以上には高まらない。
【0159】
これらの結果は、休止T細胞がbcl−Xタンパク質を発現せず、抗−CD3で活性化されたT細胞がbcl−Xタンパク質を発現し、抗−CD3で活性化され、および抗−CD28で共刺激されたT細胞がより顕著にbcl−Xタンパク質を発現することを示している。
【0160】
実施例6:bcl−XLはJurkat T細胞におけるFas−および抗−CD3誘導PCDを防止する
T細胞系に対するFas架橋はアポトーシスの速い誘導をもたらす。しかし、正常T細胞は、長い時間がかかって活性化されるまでは、Fas−架橋に対する応答において細胞死に対し感受性にならない(Klas,C.et al.(1993)Int. Immunol. 5:625-630)。細胞は迅速に誘導されて、抗−CD3+抗−CD28による活性化の24時間中に細胞表面に高レベルのFasを発現する。Fasレベルは、培養中、次の数日間一定である。これにもかかわらず、アポトーシスを誘導するFas−架橋の能力は、刺激72時間後まで明らかでなく、次いで培養中次の数日間以上でより効果的に増大する。本例は、抗−CD3および抗−CD28で刺激されたT細胞におけるbcl−XLタンパク質レベルがFas誘導細胞死に対する抵抗性と相関することを表している。
【0161】
T細胞は、120時間かけて培地のみまたは抗−CD3および抗−CD28で活性化された。細胞質溶解産物が調製され、上記のようにポリクローナル抗−bcl−XL抗体で着色したウエスタンブロット法で分析された。同時に、抗−CD3および抗−CD28刺激T細胞を0.5μg/ml CH−11抗−Fasモノクローナル抗体(Panvera)またはアイソタイプ・コントロール(IgM)で120時間の24時間の間隔で24時間、処理した。生存性を上記のようにプロピジウムヨージド排除法により調べた。Fas表面発現は、30分間のFITC共役抗マウスIgM(Sigma)に続けて、30分間CH−11で着色することにより、確認された。着色細胞は、LysisIIソフトウェアを用いた流細胞計算(FACSort, Becton-Dickinson)により分析した。
【0162】
抗Fas抗体で架橋したT細胞のbcl−XLタンパク質レベルよび生存性を図6に示す。抗−CD3+抗−CD28共刺激後、T細胞のbcl−XLタンパク質レベルのピークは24−48時間にあって、その後徐々に低下した。図の下半分に示すように、抗−CD3および抗−CD28で24または48時間処理され、次いでFasで架橋された細胞は、細胞死に対して完全に耐性である。しかし、抗−CD3および抗−CD28での刺激後のかなり遅い時点で、抗−Fas抗体でT細胞が架橋されたときは、細胞生存能力が低下した。このようにbcl−XLの存在はFas誘導細胞死に対するT細胞の防御と相関する。
【0163】
bcl−XLタンパク質がFas−架橋誘導細胞死に対する耐性を誘導することをさらに明らかにするために、Jurkat細胞をbcl−XLコードする発現ベクターでトランスフェクトし、細胞生存能力を抗Fas抗体を加えて測定した。
【0164】
Jurkat細胞は上記のように培地中に保持し、pSFFVNeo−bcl−XL(Boise,L.H.et.al.(1993)Cell 74:597−608)またはGene PulserでのエレクトロポレーションによるpSFFVNeo(BioRad)で250Vおよび960μFにおいてトランスフェクションした。トランスフェクション体をG418(Sigma)で1mg/mlにおいて選択し、いくつかの独立クローンを制限希釈によって分離した。ウエスタン・ブロット分析は、高いbcl−XLタンパク質レベルがbcl−XL発現ベクターでトランスフェクションさせたクローン中に存在すること、および対照ベクター(Neoクローン)でトランスフェクションさせたクローン(Neoクローン)がbcl−XLタンパク質を発現しないことを表している(図7、パネルC)。bcl−XLトランスフェクション体中のbcl−XLレベルは、共刺激24時間にT細胞中の発現レベル匹敵した(参照、例えば図5)。クローン上のFas発現は上記した着色によって確認された。
【0165】
表面で比較可能レベルのFasを発現した3つのbcl−XLおよび3つのNeoは、培地中2.5×105細胞/mlでインキュベートされ、10ng/mlのFas抗体またはアイソタイプ適合対照で処理され、次いで生存性がプロピジウムヨージド排除法により調べられた。Jurkat細胞クローンの生存能力を図7、パネルAに示す。グラフから、Fasが全3対照トランスフェクション体に迅速な細胞死(48時間で10%の生存能力)をもたらし、一方、3bcl−XLトランスフェクション体は実験中を通じて60%以上の生存能力であることが分かる。neo−およびbcl−XL−トランスフェクション体のいずれも、抗体もあるいはアイソタイプ適合対照mAb(IgM)が用いられなかったときは、生存能力が低下することを示している。このようにbcl−XLはJurkat細胞におけるFas−誘導細胞死を実質的に阻止する。
【0166】
抗−CD3−架橋がT細胞クローンおよび細胞ラインにおいてアポトーシスを誘導することは報告されている(Shi,Y.et.al.(1989) Nature339:625−626),Ucker,D.S.et.al.(1989)J.Immunol.143:3461−3469)。TCR誘導細胞死を防止するbcl−XLの能力を検討するために、Jurkatトランスフェクション体を抗−CD3で刺激し、その生存を調べた。抗−CD3誘導細胞死は1μg/mlでプレート結合抗−CD3(OKT3)でもって行われ、プロピジウムヨージド排除法により毎日、調べられた。
【0167】
結果を図7、パネルBに図示する。84時間で抗−CD3処理は、Neoクローン培養において30−60%の細胞死をもたらした。一方、抗−CD3誘導細胞死は、bcl−XLの存在によってほとんど完全に阻止された(図7、パネルB)。
【0168】
このようにbcl−XLタンパク質は、Fas−およびT細胞レセプター架橋誘導細胞死からT細胞を保護することができる。
【0169】
実施例7:bcl−XLはIL−2とは無関係に機能してT細胞生存を強化し得る
TCR/CD3複合体の架橋により活性化されたT細胞の生存に対するIL−2の効果のアッセイに基づくと、IL−2は抗原活性化T細胞の生存能力の維持における生存因子として機能し得るようである。CD28共刺激は、上清中の成長因子レベルとは無関係にT細胞の生存をもたらすようであるが、CD28刺激化細胞の生存強化は、単にCD28共刺激により産生された高いレベルのIL−2の結果だけではないということを除外することは難しい。CD28共刺激細胞中で産生された高いレベルのIL−2があると、充分に洗浄してもリンホカインの効果を完全に除くことは不可能である。CD28−共刺激細胞により産生された高いレベルのリンホカインは、培養上清中に検知可能なリンホカインの蓄積がなくても、オートクリン様式で細胞に影響を与えることができる場合もある。本実施例は、bcl−XLタンパク質は、IL−2依存性T細胞系がIL−2使用中止によリPCDを受けるのを防御し得ることを実証する。
【0170】
CTLL−2は、培養中での生存および増殖がIL−2に依存しているT細胞系である(Gillis,S.およびSmith,K.A.(1977)Nature 268:154-156)。これらの細胞は、それら自身のIL−2を分泌するように誘導され得ないので、IL−2のバイオアッセイにしばしば使用される。CTLL−2細胞はβ−メルカプトエタノール(50μM)および組換えIL−2(100単位/ml)を加えて、上記成長培地中で維持した(Nunez,G.et al.(1990)J.Immunol.144:3602-3610)。細胞を、250Vおよび960μFにて、ジーンパルサー(Gene Pulser、Bio Rad)を用いた電気処理により発現ベクターpSFFVNeo−bcl−XL(Boise,L.H.et al.(1993)Cell 74:597-608)を用いてトランスフェクションさせた。トランスフェクション体を、250μg/mlG418で選別し、bcl−XL陽性クローンをウエスタンブロット解析により同定した。CTLL−2細胞(2.5×106)を50mlNET−Nに溶解し、核および残骸を上記のように除去した。50μlの2×SDSローディング緩衝液を上清に加え、SDS−PAGEおよびウエスタンブロットを上記のように行った。
【0171】
高いレベルのbcl−XLタンパク質を発現する2つのbcl−XLクローン(図7パネルE)をIL−2の不在下、培養液中での生存能力について試験した。これらのクローンは、抗−CD3+抗−CD28活性化T細胞と似たレベルのbcl−XLを発現した。bcl−XL発現細胞を欠乏の1日前に3×105細胞/mlの新鮮培地中で培養した。ついで細胞をIL−2を含まない新鮮培地中で3回洗浄し、同培地に再懸濁した。指定された日にプロピジウムヨージドを排除することにより生存能力を測定した。結果は、図7のパネルDにグラフで示す。親細胞系と比較した場合に比べて、IL−2の不在下で両方のbcl−XLトランスフェクションクローンの生存が強化された(図7、パネルD)。このように、bcl−XLはIL−2の不在下でT細胞の生存を強化するように機能し得る。またこれらのクローンについて、図2で示されたアッセイと似たアッセイ系において、照射誘導死に関する試験を行った。IL−2の不在下または存在下のいずれかで試験した対照細胞と比較した場合、bcl−XLを発現するクローンは、照射誘導死から顕著に防御されることを示した。このように、本実施例は、bcl−XLタンパク質は、T細胞をIL−2欠乏により誘導された細胞死から防御することを実証する。
【0172】
実施例8:CD28共刺激によるbcl−XLの発現は進化的に保存されている
bcl−XL発現を調節するCD28共刺激の能力は、マウスおよびヒトの間で保存されているかどうかを決定するために、ネズミリンパ節から単離した細胞を、可溶性抗−CD3を用いて活性化し、bcl−XL発現を測定した。
【0173】
リンパ節をC57BL/6マウス(Jackson laboratories)から採取し、単一の細胞懸濁液をナイロン網を通過させて調製した。細胞を、10%ウシ胎児血清アルブミン、ペニシリン(100U/ml)、ストレプトマイシン(100U/ml)、10mMHEPES、50mMβ−MEおよび0.1mM非必須アミノ酸を追加したDMEM(GIBCO BRL)からなる完全培地中2×106/mlで培養した。可溶性抗−CD3(145−2C11、10μg/ml J.Bluestoneから)を単独、または抗−CD28(10μg/ml)またはCTLA4Ig(100μg/ml、Repligenから)との組み合わせで加えた。培養液を24時間で37℃、7%CO2でインキューベートし、ウエスタンブロット解析用に採取した。ネズミリンパ球(5×106)を、50μlNET−Nで溶解し、核および残骸を上記のように除去した。50μlの2×SDSローディング緩衝液を上清に加え、SDS−PAGEおよびウエスタンブロットを上記のように行った。生存能力を研究するために、細胞を上記のように処理した。活性化後のT細胞の生存能力を、Thy−1陽性細胞からプロピジウムヨージドを排除することにより測定した。
【0174】
結果は、図8に示されているように、ヒトT細胞の場合と同じように、休止ネズミリンパ球は全く検知可能なbcl−XLを発現しないことを示す。共刺激シグナルを伝達するためにアクセサリー細胞が存在する場合、24時間の可溶性抗−CD3処置により、T細胞にbcl−XL発現を誘導した。このbcl−XLのレベルは、この共刺激シグナルが抗−CD28の添加により増幅される場合に上昇した。対照的に、アクセサリー細胞の存在下における可溶性抗−CD3のbcl−XL発現をアップレギュレーションする能力は、CD28リガンドB7−1およびB7−2がCD28と相互作用するのを競争的に阻害することによりCD28の共刺激を妨げる試薬である、CTLA4Igを加えることによりほぼ完全に阻害される。これらのデータは、マウスT細胞におけるbcl−XL発現は細胞が活性化される間に誘導されることを実証する。さらに、抗CD3の副次的な細胞分裂誘導性用量において、bcl−XL発現の誘導はほぼ完全にCD28共刺激に依存する。この発現パターンは、bcl−XLがマウスの活性化T細胞生存において役割を果していることを実証する。この可能性と合致して、抗−CD3のみで刺激した未分離のリンパ節細胞集合由来のThy−1陽性細胞の生存能力は、72時間後に62%であった。対照的に、抗−CD28の添加により、Thy−1陽性細胞の生存能力は88%に上昇した。逆に、CTLA4Igを用いてリンパ節細胞内のCD28/B7仲介共刺激を阻害することにより、活性化T細胞の生存能力は72時間で20%に減少する。
【0175】
このように、本実施例は、bcl−XLはヒトT細胞と同様に、マウスT細胞においても同じ発現パターンを有すことが示され、すなわち、bcl−XLタンパク質は休止T細胞に存在せず、bcl−XLタンパク質はT細胞受容体の架橋を介して活性化されたT細胞中に発現され、bcl−XLタンパク質レベルはCD28を通した共刺激によりさらに増加することを示す。
【0176】
実施例9:bcl−XL発現はHIV−1−誘導細胞死を妨げ得る
bcl−XL発現はT細胞をHIV−1−誘導細胞死から防御するかどうかを決定するために、上記のpSFFVNeo−bcl−XL(bcl−XL)、またはベクターのみ(Neo)を用いてトランスフェクションさせたJurkat細胞クローンをHIVを用いて感染させ、生存能力を測定した。
【0177】
bcl−XL発現ベクターを用いてトランスフェクションさせた3つのJurkat細胞クローンおよび対照ベクターを用いてトランスフェクションさせた3つのJurkat細胞クローンをHIV−RF単離物の細胞を含まないウイルスストックの様々な希釈液を用いて感染させた。感染の12日後、細胞死をテトラゾリン/ホルマザンアッセイにより定量した(Shearman,M.S.et al.(1994)Proc.Natl.Acad.Sci.USA 91:1470-1474;Hansen,M.B.et al.(1989)J.Immun.Methods 119:203-210 参照)。
【0178】
3つの細胞系の平均生存能力を図9にグラフで示す。1:4から1:256で希釈したウイルスストックを用いて感染させた場合、対照Jurkat細胞において細胞死の割合が高かった。対照的に、bcl−XL遺伝子を発現するトランスフェクションJurkat細胞はこれらの同じウイルス希釈において、生存能力における欠陥は全くなかった。
【0179】
bcl−XL発現の防御効果は、HIV−1感染への耐性に起因するか、または細胞死の誘導における、より特定した効果に起因するかどうかを決定するために、感染細胞の上清中のウイルス量をSpearmanおよびKarberの方法により感染7日後に定量した。Spearman-Karber法は、『Richman D.B.,Johnson V.A.,Mayrs V.L.(1993)抗HIV活性に対する実験作用物質のin vitro評価』に記載されている。Current Protocols in Immunology ch.12.9.,Colligan J.E.et al.、GreeneおよびWiley編、Interscience NY。
【0180】
結果は表1に示す。対照細胞に比較し、bcl−XL発現細胞の上清中のウイルス量は約4倍減少した。
【0181】
【表1】

【0182】
これらの例の結果は、bcl−XLタンパク質は、HIV誘導細胞死からT細胞を防御することを実証する。この効果はHIV感染の間のCD4細胞の減少を防御するのに有益であり得る。本結果は、HIV感染をもつ患者における過剰細胞死の1つの機構は、これらの患者におけるT細胞活性化のある形は非感染個体と匹敵するレベルのbcl−XL発現を誘導できないであろうことを示唆している。このように、HIV感染の治療法の1つは、ex vivoでの細胞活性化および増幅、またはCD28シグナル形質導入のin vivo誘導により、bcl−XL発現の誘導を回復することである。その他に、bcl−XL発現の誘導は、CD28を刺激する作用物質以外の作用物質を用いて刺激することにより達成され得る。これらのデータにより、CD28のbcl−XL発現を亢進する能力は、過剰リンパ細胞死が起こる免疫不全の状態時、特にプログラム化細胞死が末梢血T細胞およびリンパ節T細胞で起こるHIV感染時における臨床用途になり得ることを示す。
【0183】
実施例10:HIV感染個体由来のPBMCの抗−CD3および抗−CD28刺激は、細胞を細胞死から防御するに充分なレベルのbcl−XLタンパク質を増加させる結果となる
本実施例は、PBMCまたはHIV感染個体由来のリンパ球を、抗−CD3および抗−CD28で刺激すると、細胞をアポトーシスから防御するに充分なレベルのbcl−XLタンパク質になるように増加することを実証する。
【0184】
無症候性のHIV感染個体における臨床的免疫不全の経緯は、CD4+T細胞の段階的なしかし進行性の欠失により特徴づけられる。HIV感染患者におけるCD4+T細胞の減少機構は現在のところ知られていないが、最近の証拠により、これらの細胞は活性化シグナルに応答してアポトーシスに対して異常に感受性をもつことが示される。HIV疾患におけるアポトーシス防御タンパク質であるbcl−2およびbcl−xの役割を、HIV感染無症候性個体由来の非刺激アメリカヤマゴボウマイトジェン(PWM)刺激および抗−CD3に抗−CD28を加えた刺激試料において測定した。
【0185】
HIV感染個体由来のPBMCを以下のように得た。ヘパリン化血液サンプルを、インフォームドコンセントの後に、HIV感染(n=25)およびHIV負の対照(n=15)から静脈穿刺により得た。患者を、ウォルターリードステージングシステム(Redfield,R.R.et al.(1986)New Engl.J.Med.314:131-132)に基づく、無症候性初期および中期段階の患者(>500および200−500 CD4細胞/mm3)として特徴づけた。サンプルをすぐに処理した。新しく単離した単核細胞を、無菌HBSS(Bio Whittaker、Walkersvile MD)で1:1に希釈しておいた血液軟膜から得、フィコール−ハイパック(Ficoll-Hypaque)で遠心分離した。サンプルの生存能力は、一般的に95%よりも大きかった。特記しない限り、PBMCは、24穴プレート中で10%FCS(T細胞培地)を追加したRPMI1640中で1×106細胞/mlで48時間培養した。PWM(Sigma Chemical Company、セントルイス MO)を用いて10μg/mlで細胞を刺激した。CD3のみ、またはCD28を介する細胞の刺激は、Weng,N.P.et al.(1995)PNAS USA92:11091-11094に記載されているように、磁気ビーズに結合しているヤギ抗−マウスIgG抗体により架橋された抗−CD3、または抗−CD3およびCD28mAbの組み合わせを用いて行なった。Jurkat細胞を、ここで記載されているpSFFNeo−bcl−XLまたはpSFFVNeo構築物のいずれかでトランスフェクションさせ、1mg/mlG418(Sigma Chemical Company)を含有するT細胞培地に維持し、bcl−x染色の陽性対照として保存した。
【0186】
細胞中のbcl−Xおよびbcl−2タンパク質の量を、以下のように行う分子内染色およびフローサイトメーターに基づいたbcl−Xの単一の細胞の検知を可能とする方法により決定した。PBMCを採取し、1×106細胞をPBS中で洗浄し、1%パラホルムアルデヒド溶液(pH7.2)で30分間室温で固定した。サンプルをPBS中で洗浄し、ついで、細胞を浸透させるために5分間PBS+0.05%Triton−X100中に再懸濁した。PBSで洗浄後、20%ヒトAB型血清(Sigma Chemical Company)および1μg/ml抗−bcl−Xモノクローナル抗体7B2(Craig Thompson、シカゴ大学)、または1μg/mlハムスター抗−bcl−2mAb 6C8(PharMingen、Inc.、サンディエゴ、カリフォルニア)を含む100μlPBS中に1時間4℃で再懸濁した。IgEアイソタイプ適合mAbsは、抗−Bcl−xおよび抗−Bcl−2で染色したサンプルの対照として使用した。一次抗体を用いてインキュベーションした後、サンプルをPBS+0.05Triton−X100で洗浄し、100μlPBS+0.05%ヤギF(ab)2抗−マウス(H+L)中に再懸濁した。FITC(Gibro Life Technologies)を30分間4℃で加えた。抗−Bcl−2で染色したサンプルを30分間4℃で、20μg/mlFITC−抗−ハムスターIgG混合(PharMingen)を用いてインキュベーションした。最後に、サンプルをPBS中で洗浄し、染色後、2〜48時間フローサイトメーターにより解析した。この時間枠の中では、bcl−xで染色した細胞のパーセンテージが減少した事実はなく、また、平均蛍光強度も変化はなかった。
【0187】
このアッセイは実質的にbcl−xLのレベルよりもむしろ、bcl−x(すなわちbcl−xLおよびbcl−xS)のレベルを測定するものであるが、免疫沈降実験により示されたところによると、これらの条件下で測定したbcl−xタンパク質の大部分はbcl−xLに対応する。
【0188】
HIV感染サンプル由来の非刺激PBMCにおけるbcl−xで染色した細胞の平均蛍光強度およびパーセンテージは、HIV非感染対照においてみられるレベルと差はないが、bcl−X発現は、PWMおよび抗−CD3に抗−CD28を加えたものを用いて活性化後の非感染対照におけるものよりも、HIV感染個体由来のPBMCにおける方が低かったことを示す。しかしながら、このbcl−Xタンパク質レベルは、細胞を細胞死から防御するのに十分である。これは、例えば、下記のように、抗−CD3および抗−CD28またはPWMを用いて様々な場合に刺激した後にHIV感染個体および非HIV感染対照個体由来のbcl−Xタンパク質の量およびPBMCのアポトーシスのレベルを比較することにより実証された。
【0189】
PBMCにおけるアポトーシスは、TdTにより触媒されたdUTPを用いてDNA一本鎖崩壊をラベルすることにより測定した(Li、X、et al(1995)Cytometry20:172-180)。フローサイトメーター解析を使用してPWMまたは抗CD3に抗CD28を加えたもののいずれかを用いて刺激したPBMCの培養中における消滅細胞を定量した。このアッセイは以下のように行った。PBMCを採取し、トリパンブルー排除により生存能力を測定し、1×106細胞を10分間、氷上で1%ホルムアルデヒド(pH7.4)溶液を用いて固定した。サンプルをHBSS中で洗浄し、細胞ペレットを70%エタノール中に再懸濁し、−20℃で2時間〜3日貯蔵した。HBSSで1回洗浄した後サンプルを30分間37℃で、0.5nMビオチン16−dUTP(Boehringer、Indianapolis IN)および10単位のTdT(Boehringer)を含有する50μlターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)反応混合物(0.1Mカルボン酸、1mMCoCl2、0.1mMジチオトレイトールおよび50μlBSA)中に再懸濁した。HBSSで洗浄後、2.5μg/mlFITCアビジン(Gibco Life Technologies、Gaithersburg、MD)を染色溶液(4×SSC、0.1%TritonX−100および5%非脂肪乾燥ミルク)に加え、サンプルを15分間室温でインキュベーションした。0.5μg/mlヒト抗−Fas抗体(Upstate Biotechnology Inc.Lake Placid NY)で24時間処理したJurkat細胞をアッセイの陽性対照として使用し、一方、TdTを欠損しているサンプルを負の対照として使用した。サンプルをELITE-ESP(Coulter Inc.、Kindal FL)フローサイトメーターを用いた細胞蛍光アッセイにより解析した。5千から1万の細胞を、log90度散乱とlogFIFC強度のパラメーターを用いて計算した。消滅細胞を、低い前角および低い90度散乱を有するFITC陽性細胞として定義した。トリパンブルー排除に基づく生存能力により、TdTを基礎とするアッセイの信頼性が確認された。
【0190】
PWMおよび抗−CD3を抗−CD28に加えた刺激サンプルに起こるアポトーシスに関連したbcl−x誘導の動力学を測定するために、3つのHIV感染および3つのHIV負の対照を培養の0時間、24時間、48時間および72時間目に実験した。平均パーセンテージとして示したように、結果は図10、パネルA−Dに示す。PWMで処理した対照サンプルでは、bcl−x発現は、24時間から48時間の間で増加したが、アポトーシスのレベルは低いままであった。抗−CD3に抗−CD28を加えたもので処理した対照サンプルにおいて、アポトーシスの低いレベルのみは明白であるが(図10、パネルA)、bcl−xのレベルは急激に増加した。これらのHIV負の対照においては、芽球化反応および細胞数の増加が両方の培養条件下で起こり、これは細胞が増殖していることを示す。HIV感染患者由来のPBMCのPWM刺激培養は、48時間で高いレベルのアポトーシスを有し、下降する前に40%でピークに達した(図10、パネルB)。HIV感染個体由来のPWM刺激PBMCは、0から48時間の間にわずかな量のbcl−xしか発現しなかった。興味深いことに、bcl−x発現細胞の分画におけるわずかな増加が、48時間から72時間の間で見られ、おそらくこれは、これは48時間以上生存した細胞が増えたことを反映する(図10、パネルB)。bcl−x発現は低く、HIV負の対照に比べて、抗−CD3に抗−CD28を加えたもので培養したHIV感染患者由来のPBMCにおいて誘導の遅延がみられた(図10、パネルD)。アポトーシスレベルは抗−CD3に抗−CD28を加えた刺激培養において低いが、それらはHIV負の対照におけるものより高かった(図10、パネルCおよびD)。PWMで培養したHIV感染患者由来のPBMCにおいては、ほとんど増殖が見られなかったが、細胞数および細胞の大きさの増大が、抗−CD3に抗−CD28を加えた培養中で見られた。
【0191】
アポトーシスおよびbcl−x誘導を、48時間培養後にHIV負の対照(n=5)およびHIV感染個体(n=20)においてさらに解析した。サンプルを、培地のみ、PWMを加えて、または上記のように抗−CD3に抗−CD28を加えて培養した。結果を表2に示す。HIV陰性の対照における、アポトーシスレベルは様々な刺激に応答して類似であった。アポトーシスレベルはPWM処理に応答して基線よりわずかに上昇し、抗−CD3に抗−CD28を加えたものに応答して幾分減少した。同サンプルにおいて、bcl−x発現はPWM処理培養中で穏やかに増加し、抗−CD3に抗−CD28を加えて処理した培養中で劇的に増加した。対照的に、アポトーシスの基線レベルを越えた顕著な増加は、PWMと共に培養した48時間後にHIV感染個体由来PBMC中で見られた。アポトーシスにおける増加は抗−CD3に抗−CD28を加えたもので培養したHIV感染患者由来のPBMC中においては観察されなかった。
【0192】
【表2】
【0193】
結論として、bcl−x発現は、刺激非感染サンプルにおいて経時的に劇的に増加するが、刺激HIV感染サンプルにおいて遅延し減少した応答が、観察された。加えて、bcl−xの単一細胞内染色は、PWM誘導bcl−x発現およびアポトーシスの間に強い逆相関がみられた(r=0.695;p=0.005)。同様な相関が抗−CD3および抗−CD28で刺激したPBMC中では観察されず、これは、共刺激はbcl−x誘導の防御レベルへと導くことを示唆している。結果はまた、PBMCの代わりにリンパ球を用いたときにも得られ、すなわち、抗−CD3に抗−CD28を加えたもので刺激したHIV感染個体由来のリンパ球は、非感染個体由来のリンパ球に比較して低いbcl−x発現を示した。遅延し減少したbcl−xの発現は試験サンプル中のCD3+CD28+細胞のパーセンテージの減少に起因するものではない。なぜなら、bcl−xの発現の減少はPWMで刺激したCD28+HIV感染部分集合において明白だからである。さらに、低いbcl−x発現はリンパ球活性化の減少に起因するものではない。なぜなら、PWM刺激はHIV感染患者において早い活性化マーカーであるCD69の発現を誘導するからである。
【0194】
このように、HIV感染個体由来のリンパ球は、抗−CD3および抗−CD28で刺激時に、非HIV感染個体よりも、bcl−XLタンパク質レベルが低いが、このレベルは細胞を細胞死から防御するに必要なbcl−XLタンパク質の限界値を上回っている。
等価体
【0195】
当業者は慣用的な実験法以外を用いて、ここで記載された本発明の特別の態様に関する数多くの等価体が存在すると認識または確認することができるだろう。このような等価体は以下の請求の範囲に含まれる。
【0196】
【表3】
【0197】
【表4】
【0198】
【表5】
【0199】
【表6】
【0200】
【表7】
【図面の簡単な説明】
【0201】
【図1】γ−照射(パネルB)前、または未処理(パネルA)のままで、培地のみ(白抜き四角)、抗CD3の存在下(白抜き菱形)、または抗CD3と抗CD28の存在下(黒塗り四角)で12時間T細胞をインキュベーション後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【図2】γ−照射(パネルBおよびD)前、または未処理(パネルAおよびC)のままで、抗CD3のみ(パネルAおよびB)または抗CD3と抗CD28(パネルCおよびD)と共に12時間T細胞をインキュベーションし、その後、それらをならし培地に残す(白抜き四角)か、洗浄して新たな培地に懸濁する(白抜き菱形)か、または洗浄して200U/mlのrIL−2を加えた新たな培地に懸濁した(黒塗り四角)後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【図3】図3パネルB、CおよびDは、それぞれ、培地のみ(MED)でインキュベーションするか、抗CD3抗体(αCD3)の存在下で1、6または12時間インキュベーションするか、または抗CD3と抗CD28(αCD3+αCD28)の存在下でインキュベーションしたT細胞中でのbcl−XL発現、bcl−2発現およびHLA発現のレベルを示すノーザン・ブロットを表す。パネルAは、非変性アガロースゲルでの電気泳動後に各RNA試料のアリコートをエチジウムブロマイド染色したものを示す。 図3Eは、単独で(0)または抗CD3(αCD3)と共に6または12時間、または抗CD3および抗CD28(αCD3+αCD28)と共に6または12時間インキュベーションしたT細胞中のbcl−XLおよびbcl−XSタンパク質の量を示すウエスタン・ブロットの写真である。
【図4】単独で(0)または抗CD3(αCD3)と共に6、12または24時間、または抗CD3および抗CD28(αCD3+αCD28)と共に612、または24時間インキュベーションしたT細胞中のbcl−XLおよびBcl−2タンパク質の量を示すウエスタン・ブロットの写真である。
【図5】培地単独で(Medium)または抗CD28(αCD28)、抗CD3(αCD3)、抗CD3+抗CD28(αCD3/αCD28)、または抗CD3+IL−2(100units/ml)(αCD3/IL−2)の存在下、24時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【図6】培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。パネル下部は、培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションし、抗Fas抗体を添加してさらに24時間インキュベーションしたT細胞の生存能力パーセントを示す。
【図7】図7A−Bは、抗Fas抗体(αFas、パネルA)または抗CD3抗体(αCD3、パネルB)で処理後0、12、24、48および72時間でbcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミドでトランスフェクションさせたJurkatクローンの生存能力パーセントのグラフ表示である。 図7パネルCは、bcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミド(Neoクローン1、2および3)でトランスフェクションさせたJurkatクローン中のbcl−XLタンパク質の量を示すウェスタン・ブロットの写真である。 図7パネルDは、IL−2回収後0、12、24、36および48時間でbcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローンの生存パーセントのグラフ表示である。 図7パネルEは、bcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローン中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【図8】培地単独で、抗CD3の存在下、抗CD3および抗CD28の存在下、および抗CD3およびCTLA4Igの存在下、24時間インキュベーションしたマウスリンパ節細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【図9】bcl−XL発現ベクター(ウイルス希釈率v平均bcl−XL)、または対照ベクター(ウイルス希釈率v平均neo)でトランスフェクションし、さまざまな量のHIVで感染させたJurkat細胞の生存能力パーセントのグラフ表示である。
【図10】図10パネルA−Dは、PWM(パネルAおよびB)でまたは抗CD3および抗CD28抗体(CD3/CD28、パネルCおよびD)で0、24、48または72時間処理した対照およびHIV感染個体由来のPBMCにおいて、アポトーシスを被った細胞の割合(%アポトーシス)およびbcl−Xタンパク質を発現する細胞の割合(%bcl−x)のグラフ表示である。
【図1A】
【図1B】
【図2A】
【図2B】
【図2C】
【図2D】
【図3A−D】
【図3E】
【技術分野】
【0001】
bcl−XLタンパク質レベルを調節することによる、T細胞生存の調節法
政府支持
本明細書に記載した研究は、NIH認可PO1AI35294およびNMRDS認可61153NAE.4120.001.1402により一部支持されている。従って、米国政府は、本発明の権利を有する。
【背景技術】
【0002】
発明の背景
末梢血T細胞生存の制御は、効果的な末梢性免疫レパトアーの維持に重要である。T細胞生存の幾つかの態様は、T細胞活性化の状態と関連しているようである。T細胞活性化は、抗原提示細胞(APC)表面で主要組織適合遺伝子複合体(MHC)分子に結合したペプチド−抗原によるT細胞受容体/CD3複合体(TCR/CD3)の結合により開始される(Schwartz,R.H.(1990)Science 248,1349)。これはT細胞活性化の一次シグナルであるが、APCとT細胞との間のその他の受容体リガンド相互作用が完全な活性化には要求される。例えば、他の分子相互作用なしでのTCR刺激は、アネルギー状態を誘発することがあり、そのため、これらの細胞は、再刺激時に完全な活性化シグナルに応答できない(Schwartz,R.H.(1990)Science 248,1349;Harding,F.A.,McArthur J.G.,Gross,J.A.,Raulet,D.H.,およびAllinson,J.P.(1992)Nature 356,607)。また、T細胞は、TCR結合(engagement)のみにより活性化される時、プログラムされた細胞死(PCD)により死滅することが示されている(Webb,S.,Morris,C.,およびSprent,J.(1990)Cell 63,1249;Kawabe,Y.,およびOchi,A.(1991)Nature 349,245;Kabelitz,D.,およびWesselborg,S.(1992)Int.Immunol 4,1381;Groux,H.,Monte,D.,Plouvier,B.,Capron,A.,およびAmeisen,J-C(1993)Eur.J.Immunol.23,1623)。
【0003】
T細胞とAPCの間には複合的な受容体−リガンド相互作用が起こる。多くの相互作用は、自然に密着したものであり、かつ2つの細胞間の接触を強化するものである(Spinger,T.A.,Dustin,M.L.,Kishimoto,T.K.,およびMarlin,S.D.(1987)Annual,Rev.Immunol.5,223)のに対し、その他の相互作用はT細胞に更なる活性化シグナルを形質導入するものである(Bierer,B.E.,およびBurakoff,S.J.(1991)Adv,Cancer Res.56,49)。CD28は、ヒトの末梢T細胞の80%に存在する表面糖タンパク質であり、重要な共刺激性(コスティミュラトリー)受容体であることが示されている(June,C.H.,Bluestone,J.A.,Nadler,L.M.およびThompson,C.B.(1994)Immunol.Today 15,321;Linsley,P.S.Ledbetter,J.A.(1993)Annu.Rev.Immunol.11,191)。コスティミュラトリー・シグナルは、T細胞がCD28リガンドB7−1またはB7−2のいずれかを発現する抗原提示細胞と遭遇するときにCD28を介して形質導入される。
【0004】
T細胞の共刺激(コスティミュレーション)は、T細胞活性化の多くの態様に影響を与えることが示されている(June,C.H.,Bluestone,J.A.,Nadler,L.M.およびThompson,C.B.(1994)Immunol.Today 15,321)。共刺激は、培養物中の増殖応答を誘導するのに必要な抗CD3の濃度を低減させる(Gimmi,C.D.Freeman,G.J.,Gribben,J.G.,Sugita,K.,Freedman,A.S.,Morimoto,C.,およびNadler,L.M.(1991) Proc.Natl.Acad.Sci.USA 88,6575)。CD28共刺激はまた、遺伝子発現の転写調節および転写後調節を介するヘルパーT細胞によるリンパ球産生を著しく高め(Lindsten,T.,June,C.H.,Ledbetter,J.A.,Stella,G.,およびThompson,C.B.(1989)Science 244,339;Fraser,J.D.,Irving,B.A.,Crabtree,G.R.,およびWeiss,A.(1991)Science 251,313)、細胞毒性T細胞の細胞溶解能を活性化し得る。CD28共刺激をインビトロで阻害することにより、異種移植片拒絶を遮断でき、また同種移植片拒絶が顕著に遅延する(Lenschow,D.J.,Zeng,Y.,Thistlethwaite,J.R.,Montag,A.,Brady,W.,Gibson,M.G.,Linsley,P.S.,およびBluestone,J.A.,(1992)Science257,789;Turka,L.A.,Linsley,P.S.,Lin,H.,Brady,W.,Leiden,J.M.Wei,R-Q.,Gibson,M.L.,Zheng,X-G.,Mydral,S.,Gordon,D.,Bailey,T.,Bolling,S.F.,およびThompson,C.B.(1992)Proc.Natl.Acad.Sci.USA 89,11102)。更に、腫瘍細胞系列へのB7のトランスフェクションにより、腫瘍増殖の認識および防止が容易になる(Chen,L.,Ashe,S.,Brady,W.A.,Hellstrom,I.,Hellstrom,K.e.,Ledbetter,J.A.,McGowan,P.,およびLinsley P.S.(1992)Cell 71,1093;Townsend,S.E.,およびAllinson,J.P.(1993)Science 259,368)。
【0005】
最近まで、T細胞生存がどのように制御されているかについてほとんど知られていなかった。各研究により、T細胞のマイトジェン活性化が、放射能などの作用物質での処理により開始されるプログラムされた細胞死(PCD)に対するその耐性を強化することが示された(Schrek,R.およびStefani,S.(1964)J.Nat.Cancer Inst.32,507;Lowenthal,J.W.およびHarris,A.W.(1985)J.Immunol.135,1119;Stewart,C.C.,Stevenson,A.P.,およびHabbersett,R.C.(1988)J.Radiat.Biol.53,77)。反対に、TCRのみによるT細胞活性化は、PCDを被るT細胞の感受性を増大させることが報告されている(Kabelitz,D.,およびWesselborg,S.(1992)Int.Immunol 4.1381;Groux,H.,Monte,D.,Plouvier,B.,Capron,A.,およびAmeisen,J-C(1993),Eur.J.Immunol.23,1623)。
【0006】
近年、T細胞生存を調節する役割を果すと思われる数種の遺伝子が同定された。マウスにおける休止リンパ球の生存は、bcl−2遺伝子の発現に依存する(Nakayama,K-I.,Nakayama,K.,Negishi,I.,Kuida,K.,Shinkai,Y.,Louie,M.C. Fields,L.E.,Lucas,P.J.Stewart,V.,Alt,F.W.,およびLoh,D.Y.(1993)Science 261,1584)。bcl−2遺伝子を欠く動物は、培養したときにPCDを被る感受性の高いT細胞を有する。Bcl−2欠損動物は寿命の最初の数週間以内に深刻なリンパ球減少状態になる(Veis,D.J.,Sorenson,C.M.,Shutter,J.R.,およびKorsmeyer,S.J.(1993a)Cell 75,229)。反対に、Fas細胞表面受容体に突然変異を持つ動物は、免疫応答過程に産生される過剰な免疫細胞を除去できない(Watanabe-Fugunaga,R.,Brannan,C.I.Copeland,N.G.,Jenkins,N.A.,およびNagata,S.(1992)Nature 356,314)。Fas欠損動物は、最終的に深刻な自己免疫疾患を発症する。これらのデータは、T細胞生存が、T細胞増殖と同じく厳密に調節され得ることを示している。
【0007】
T細胞死を不適切に調節すると免疫系異常(例えば、免疫不全または自己免疫)を引き起こすこともある。更に、T細胞がある種の感染性微生物に感染するとT細胞は殺傷される。特に、T細胞がヒト免疫不全ウイルス(HIV)に感染するとプログラムされた細胞死により誘発される細胞死が起こる。したがって、T細胞死を制御する方法および特に、そのような死を阻害する方法が必要とされている。
【発明の開示】
【0008】
概要
本発明は、T細胞生存を調整する方法、特に、T細胞を細胞死から防御する方法を提供する。本発明の方法は、少なくともその一部で、T細胞共刺激(例えば、CD28による)がT細胞中のタンパク質bcl−XLの産生増大およびT細胞生存強化をもたらすという発見に基づくものである。更に、その他の手段(例えば、T細胞へのbcl−XL遺伝子のトランスフェクション)によるT細胞中のbcl−XLタンパク質の産生増大もまた、T細胞生存強化をもたらす。したがって、T細胞を細胞死から保護するために、本発明の方法に従い、T細胞中のbcl−XLタンパク質の量を増大して、T細胞生存を強化する。
【0009】
一実施態様では、T細胞生存を、T細胞をbcl−XLタンパク質レベルを増大する作用物質と接触させることにより強化する。本方法の好ましい実施態様では、T細胞中のbcl−XLタンパク質レベルは、T細胞中にbcl−XLタンパク質をコードする核酸を導入することにより増大する。本方法の別の実施態様では、T細胞中のbcl−XLタンパク質レベルは、T細胞を、細胞内に作用して内生bcl−XLタンパク質レベルを増大する作用物質と接触させることにより増大する。bcl−XLタンパク質レベルを増大するその他の好ましい作用物質は、T細胞表面上の分子と相互作用する物質である。本発明の方法は、増大したまたは不適切なT細胞死に関連する異常または症状、例えば、HIVによるT細胞感染、を処置するのに有用である。本方法は、また、T細胞生存を強化し、それによって、免疫反応を刺激する、例えば、病原性微生物の排除を加速する、のにも有用である。
【0010】
本発明は、T細胞死を誘導する、またはT細胞を細胞死に対して感受性にする方法にも関連する。これらの方法は、T細胞を、T細胞中のbcl−XLタンパク質レベルを減少する作用物質と接触させることを含む。これらの方法は、免疫反応をダウンレギュレーションするのに有用である。
【0011】
図面の簡単な説明
図1は、γ−照射(パネルB)前、または未処理(パネルA)のままで、培地のみ(白抜き四角)、抗CD3の存在下(白抜き菱形)、または抗CD3と抗CD28の存在下(黒塗り四角)で12時間T細胞をインキュベーション後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【0012】
図2A−Dは、γ−照射(パネルBおよびD)前、または未処理(パネルAおよびC)のままで、抗CD3のみ(パネルAおよびB)または抗CD3と抗CD28(パネルCおよびD)と共に12時間T細胞をインキュベーションし、その後、それらをならし培地に残す(白抜き四角)か、洗浄して新たな培地に懸濁する(白抜き菱形)か、または洗浄して200U/mlのrIL−2を加えた新たな培地に懸濁した(黒塗り四角)後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【0013】
図3パネルB、CおよびDは、それぞれ、培地のみ(MED)でインキュベーションするか、抗CD3抗体(αCD3)の存在下で1、6または12時間インキュベーションするか、または抗CD3と抗CD28(αCD3+αCD28)の存在下でインキュベーションしたT細胞中でのbcl−XL発現、bcl−2発現およびHLA発現のレベルを示すノーザン・ブロットを表す。パネルAは、非変性アガロースゲルでの電気泳動後に各RNA試料のアリコートをエチジウムブロマイド染色したものを示す。
【0014】
図3Eは、単独で(0)または抗CD3(αCD3)と共に6または12時間、または抗CD3および抗CD28(αCD3+αCD28)と共に6または12時間インキュベーションしたT細胞中のbcl−XLおよびbcl−XSタンパク質の量を示すウエスタン・ブロットの写真である。
【0015】
図4は、単独で(0)または抗CD3(αCD3)と共に6、12または24時間、または抗CD3および抗CD28(αCD3+αCD28)と共に612、または24時間インキュベーションしたT細胞中のbcl−XLおよびBcl−2タンパク質の量を示すウエスタン・ブロットの写真である。
【0016】
図5は、培地単独で(Medium)または抗CD28(αCD28)、抗CD3(αCD3)、抗CD3+抗CD28(αCD3/αCD28)、または抗CD3+IL−2(100units/ml)(αCD3/IL−2)の存在下、24時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【0017】
図6は、培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。パネル下部は、培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションし、抗Fas抗体を添加してさらに24時間インキュベーションしたT細胞の生存能力パーセントを示す。
【0018】
図7A−Bは、抗Fas抗体(αFas、パネルA)または抗CD3抗体(αCD3、パネルB)で処理後0、12、24、48および72時間でbcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミドでトランスフェクションさせたJurkatクローンの生存能力パーセントのグラフ表示である。
【0019】
図7パネルCは、bcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミド(Neoクローン1、2および3)でトランスフェクションさせたJurkatクローン中のbcl−XLタンパク質の量を示すウェスタン・ブロットの写真である。
【0020】
図7パネルDは、IL−2回収後0、12、24、36および48時間でbcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローンの生存パーセントのグラフ表示である。
【0021】
図7パネルEは、bcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローン中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【0022】
図8は、培地単独で、抗CD3の存在下、抗CD3および抗CD28の存在下、および抗CD3およびCTLA4Igの存在下、24時間インキュベーションしたマウスリンパ節細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【0023】
図9は、bcl−XL発現ベクター(ウイルス希釈率v平均bcl−XL)、または対照ベクター(ウイルス希釈率v平均neo)でトランスフェクションし、さまざまな量のHIVで感染させたJurkat細胞の生存能力パーセントのグラフ表示である。
【0024】
図10パネルA−Dは、PWM(パネルAおよびB)でまたは抗CD3および抗CD28抗体(CD3/CD28、パネルCおよびD)で0、24、48または72時間処理した対照およびHIV感染個体由来のPBMCにおいて、アポトーシスを被った細胞の割合(%アポトーシス)およびbcl−Xタンパク質を発現する細胞の割合(%bcl−x)のグラフ表示である。
【0025】
詳細な説明
本発明はT細胞の生存を調節する方法を提供する。好ましい実施態様において、T細胞を細胞死から防御する。細胞死からT細胞を防御する本発明方法は、T細胞の生存を強化するように、bcl−XLタンパク質レベルを増大する少なくとも一つの作用物質でT細胞を処理することを含む。本発明の一つの実施態様において、bcl−XLタンパク質レベルを増大する物質はbcl−XLタンパク質をコードする核酸であって、これはT細胞中へ導入されると発現する。本発明の他の実施態様において、bcl−XLタンパク質レベルは、例えば外来bcl−XL遺伝子からのbcl−XLタンパク質産生を増大せしめる少なくとも一つの作用物質とT細胞を接触させることによりT細胞中において増加される。1つの実施態様において、bcl−XLタンパク質レベルを増加する少なくとも一つの作用物質は、CD28などのT細胞の表面上の分子と相互作用をする作用物質を含む。T細胞はさらに、T細胞に一次活性化シグナルを提供する作用物質と接触させ得る。他の実施態様において、少なくとも一つの作用物質は、bcl−XLタンパク質レベルを増大するために細胞内に作用する物質である。他の実施態様において、本発明は、さらに、T細胞におけるbcl−XLタンパク質レベルを低下することによりT細胞を細胞死に対して感受性にする方法に関する。bcl−XLタンパク質レベルが本発明方法により調節されているT細胞は、被験体に存在するT細胞またはエクスビボで培養されたT細胞であり得る。本発明の方法は、免疫応答の調節、すなわち免疫応答の強化または抑制のために有用である。このように本発明方法は、免疫反応を刺激する(例えば、感染に抵抗する)、あるいは生存寿命が短くなっている(例えば、ヒト免疫不全ウイルスによる感染などの感染結果として)T細胞の生存を刺激するなどの数多くの応用性を有する。
【0026】
bcl−XLがIL−3欠乏誘導化アポトーシスからネズミIL−3依存性前リンパ球細胞系を防御し得ることは、以前から知られている(Boise, H.B. et al.(1993)Cell 74,597 および PCT出願 WO95/00642)。アポトーシスが個体発生におけるネガティブおよびポジティブセレクションを調節する重要なメカニズムであるので、著者は、T細胞におけるbcl−XLmRNAレベルを解析した。胸腺細胞も、休止T細胞も、PMAおよびイオノマイシンで活性化したT細胞も、bcl−XLmRNAを含有していなかった。しかし、胸腺細胞および活性化T細胞は、アポトーシスに役立つことが示されている(Boise, H.B. et al,後出、およびPCT出願WO95/00642)タンパク質bcl−XLをコードするbcl−X遺伝子の別のスプライス形態、bcl−XSmRNAを含んでいた。これらの結果は、T細胞個体発生におけるアポトーシス誘導細胞死の制御がbcl−XLタンパク質レベルの制御によるよりもむしろ、bcl−XSタンパク質レベルの制御によって少なくとも部分的には調節されることを示唆している。
【0027】
本発明は、bcl−XLタンパク質レベルをT細胞中で増加する、ヒトbcl−XLをコードする核酸によるT細胞のトランスフェクションにより、T細胞が細胞死から防御されるという発見に少なくとも部分的に基づいている。このようにして修飾されたT細胞は、γ−照射、T細胞受容体架橋、Fas架橋および成長因子欠乏などの種々の刺激によって誘導される細胞死から防御される。T細胞が細胞死から防御され得る、または、細胞死に対し感受性にするように、T細胞におけるbcl−XLタンパク質レベルを調節する作用物質とT細胞とを接触させることによりT細胞におけるbcl−XLタンパク質レベルを調節することは、本発明の一つの目的である。本発明方法は、被験体のT細胞、例えば感染への抵抗に関与するT細胞の少なくともいくらかの生存を長引かせることを望む症状において医療上有用である。他方、本発明方法は、自己免疫疾患などの、T細胞の死が望まれる状態においても医療上有用である。
【0028】
1.細胞死からT細胞を防御する方法
本発明方法は、細胞死からT細胞を防御することを含む。“T細胞”は、一般的に認識されているものであって、胸腺細胞、未熟Tリンパ球、成熟Tリンパ球、休止Tリンパ球または活性化Tリンパ球を含むことを意図している。T細胞はTヘルパー(Th)細胞であることができ、例えばTヘルパー1(Th1)またはTヘルパー2(Th2)細胞である。T細胞は、CD4+T細胞、CD8+T細胞、CD4+CD8+T細胞、CD4−CD8−T細胞またはT細胞の他のサブセットである。“細胞死からT細胞を防御する”なる文言は、T細胞における細胞死が起きるのを阻害または少なくとも遅延することを含む。細胞死は、なんらかのメカニズムによって細胞死がおきることを含む。細胞死はプログラムされた細胞死(PCD)であって、“アポトーシス”と称することもできる。アポトーシスによるT細胞死は、核ヘテロクロマチンの凝縮、細胞の縮小、細胞質の濃縮、およびアポトーシスの後期段階においてはT細胞DNAの分離フラグメントへのエンドヌクレアーゼ仲介切断を含み、これらを特徴とするものである。アポトーシスが起きた細胞のDNAの電気泳動解析では、分離フラグメントの特徴的な“ラダー(ladder)”が見られる。
【0029】
本発明は、自然に起きる細胞死または細胞中に誘導されたシグナルに起因する細胞死からT細胞を防御する方法に関する。例えば、アポトーシスは、普通、T細胞中の特定のシグナルの誘導からもたらされる。このように本発明の方法は、コスティミュラトリー・シグナルの不在下でT細胞受容体の架橋に起因する細胞死からT細胞を防御することを提供する。T細胞受容体の架橋は、抗CD3抗体などのポリクローナル・アクチベーターにより、あるいは抗原提示細胞上の抗原によって、コスティミュラトリー・シグナルの不在下に(例えばCD28/CTLA4によるシグナルの不在下において)、T細胞アネルギーまたはT細胞死をもたらすことが当分野では知られている。
【0030】
本発明方法は、成長因子減少に起因する細胞死からT細胞を防御することも提供する。例えば、T細胞は増殖にIL−2を必要とし、IL−2の不在は細胞死をもたらす。このように、本発明方法によりT細胞中のbcl−XLタンパク質のレベルを増加することは、IL−2不在における細胞死から正常なIL−2依存性T細胞を防御する(すなわち、本発明はIL−2の不在下で生存できるT細胞を提供する)。本発明の範囲には、T細胞生存に普通に必要な他の成長因子類、サイトカイン類、リンホカイン類の不在に起因する細胞死からT細胞を防御する方法も含まれる。このような因子の例には、インターロイキン類、コロニー刺激因子類およびインターフェロン類がある。
【0031】
本発明方法はさらに、Fasまたは腫瘍壊死因子受容体(TNFR)架橋に起因する細胞死からT細胞を防御することを可能にする。T細胞上のFas受容体の架橋がT細胞のアポトーシス性細胞死をもたらすことが示されている。FasおよびTNFRタンパク質はかなりの相同性を共有しており、これらは架橋すると細胞死を誘導するタンパク質の拡大ファミリーに属せしめることができる。このように、本発明方法は、このファミリーに属するいかなる受容体の架橋に起因する細胞死からもT細胞を防御するのに有用である。
【0032】
本発明方法はまた、グルココルチコイドなどのある種のホルモン、例えばデキサメタゾンにより誘導される細胞死からT細胞を防御する手段を提供する。さらに細胞死は、被験体においてグルココルチコイドレベル上昇を誘導することが知られているストレスの結果としても起こり得る。このように本発明方法は、ストレスを被った被験体に起きるT細胞死に対する防御も提供する。
【0033】
さらに本発明方法は、DNA障害剤により誘導される細胞死からT細胞を防御することを可能にする。
【0034】
T細胞はまたT細胞個体発生中に細胞死を被る。T細胞の増殖および分化の過程は厳密に調節されており、T細胞のポジティブおよびネガティブセレクションを誘導する。この過程中のT細胞生存に影響を与える方法も本発明の範囲内である。
【0035】
T細胞死は、T細胞中のアポトーシス誘導タンパク質、例えば、bcl−XS,Bad,p53,c−myc,またはインターロイキン−1β変換酵素(ICE)(Savilel,J.(1994) Eur. J.Clin. Investigat. 24,715)の活性によっても生ずる。従って、本発明の範囲内の方法は、これらのタンパク質に関係する細胞死を防御する方法も含む。
【0036】
1.2.T細胞中にbcl−XLタンパク質をコードする核酸を導入することよりなる、T細胞を細胞死から防御する方法
【0037】
本発明の一実施態様では、T細胞を、その細胞中にbcl−XLタンパク質をコードする核酸を導入し、T細胞中のbcl−XLタンパク質レベルを増大させることにより、細胞死から防御する。このbcl−XLタンパク質をコードする核酸分子は、T細胞中で遺伝子を発現させる形態で存在するので、bcl−XLタンパク質がT細胞中で産生される。
【0038】
1.2.1.bcl−XLタンパク質をコードする核酸またはそれらの修飾形態
“bcl−XLをコードする核酸分子”なる用語は、T細胞中にその核酸分子を導入したときにbcl−XLタンパク質へ転写および翻訳される全ての核酸分子を含むことを意図している(例えば、この分子は、bcl−XLの発現を調節する適当な制御配列をも含み得る)。 このbcl−XLをコードする核酸分子は、対応するbcl−XL遺伝子のコード領域のみからなるものであってよく、あるいは、例えば5'または3'非翻訳領域、イントロン、それらのフラグメント、または他の配列のような、非コード領域を含むものであってもよい。
【0039】
bcl−XLタンパク質はbcl−X遺伝子によってコードされている。分化スプライシングの結果、このbcl−X遺伝子は2種の異なるmRNA分子を生成し、そのひとつはbcl−XL(bcl−Xの長い形態に対応)タンパク質をコードし、他のものは小さい方のbcl−XS(bcl−Xの短い形態に対応)をコードしている。このbcl−XSは、bcl−XLタンパク質とは63アミノ酸ストレッチ(stretch)を欠く点で異なる。この欠失は、bcl−XLに存在する第2コードエクソンを、第1コードエクソン内のより5'末端に近いスプライス供与体へとスプライシングする結果として起こる。このように、T細胞中のbcl−XLの発現には、bcl−XLとbcl−XSタンパク質の両方の産生をもたらし得る、bcl−X遺伝子を含有するゲノムフラグメントよりもむしろ、bcl−XL cDNAを使用する方が好ましい。
【0040】
ヒトの細胞処理では、bcl−XLをコードする核酸は、他の動物種由来の核酸も本発明の範囲内であるが、ヒト由来のものが好ましい。さらに、bcl−XL核酸は、その核酸をT細胞中に導入したとき、その核酸によってコードされるタンパク質がT細胞を細胞死から防御する限り、各種を通して使用できる。本発明の好ましい態様では、bcl−XLタンパク質をコードしている核酸は、ヒトcDNA(配列番号1)である。本発明の範囲内であるbcl−XLタンパク質をコードしている核酸は、PCT出願国際公開WO95/00642や下記の文献類に記載されている:Boise et al.(1993) Cell 74,597;およびFang et al.(1994) J. Immunol. 153,4388。さらに、ヒトbcl−XL cDNAのヌクレオチド配列、およびヒトbcl−XLタンパク質のアミノ酸配列は、配列番号1および2にそれぞれ示してある。この核酸分子は、bcl−XLタンパク質全長をコードするものであってもよく、あるいは、この核酸は、それをT細胞中に導入したとき、T細胞を細胞死から防御するに充分なだけのbcl−XLのペプチドフラグメントをコードしているものであってもよい。この核酸は、天然のbcl−XLまたはそのフラグメントをコードするものであってもよく、あるいは、bcl−XLタンパク質の修飾形態またはそのフラグメントであってもよい。本発明の範囲内である、天然bcl−XLタンパク質の修飾形態は、以下に記載する。
【0041】
本発明の方法は、T細胞を細胞死から防御する能力を保持している、bcl−XLタンパク質の各フラグメント、各変異体、または各変種(例えば、修飾形態)の使用を含むことを意図している。“bcl−XLタンパク質の形態”は、天然bcl−XLタンパク質との顕著な相同性を共有し、かつT細胞を細胞死から防御し得るタンパク質を意味することを意図している。“bcl−XLタンパク質の形態”は、タンパク質Bcl−2を含めることは意図していない。“生物学的に活性なbcl−XLタンパク質”または“bcl−XLタンパク質の生物学的に活性な形態”または“bcl−XLタンパク質の機能的に活性な形態”の用語は、本書中では、T細胞を細胞死から防御し得る、bcl−XLタンパク質類の各形態を含むことを意味している。当業者は、T細胞中にbcl−XLタンパク質をコードする核酸を導入した際に、T細胞を細胞死から防御する能力に基づき、bcl−XLタンパク質のそのような形態を選ぶことができる。T細胞を細胞死から防御するbcl−XLの特定形態の能力は、例えば、T細胞中にbcl−XLの特定形態をコードする核酸をトランスフェクションし、通常、T細胞中で細胞死が誘発される条件下でT細胞にてbcl−XLタンパク質を合成させることにより測定できる。細胞死の導入に際しても、もしbcl−XLの形態を発現するように修飾したT細胞集団中での細胞死が、bcl−XLの形態を発現するように修飾していないT細胞集団中よりも少なくなれば、bcl−XLタンパク質のその形態は、T細胞中の細胞死に対して防御効果を有する。細胞死は種々の手段でモニターできる。細胞集団中の細胞死の程度は、例えば、両方の集団中でのT細胞の数を、血球計またはコウルター計数器を用いて数えることにより測定できる。T細胞集団中の細胞死の程度を測定する好ましい方法は、プロピジウムヨージド(propidium iodide)排除アッセイによるものである。プロピジウムヨージド排除アッセイは、T細胞をプロピジウムヨージドとともに培養することにより実施でき、このプロピジウムヨージド染料は、死亡細胞に優先的に吸収され、生存細胞から排除される。細胞死の程度は、次いで、本願の実施例6に記載しているように、蛍光活性化セルソーター(FACS)分析により測定される。付加的に使用できる染料には、アクリジンオレンジやヘキスト33342が含まれる。T細胞集団中の細胞死の程度を測定する他の方法には、切断DNAの末端をラベルする(Gavrieli, Y. et al.(1992) J. Cell Biol. 119,493)の種々の方法が含まれる。T細胞集団中の細胞死の程度を測定する他の方法には、T細胞の核酸の電気泳動分析がある。この方法では、精製されているかまたは精製されていない形態のT細胞の核酸を、ゲル電気泳動に付し、次いで、エチジウムブロマイドによるゲル染色し、さらに紫外線下で核酸の可視化を行う。少なくとも相当数のT細胞がアポトーシスを受けているT細胞集団由来の核酸は、"ラダー(ladder)”即ち、DNAの分離フラグメントの集団、が現れるであろう。反面、アポトーシスを受けていないT細胞のDNAは、単一の高分子量バンドとして現れる。
【0042】
bcl−XLタンパク質の形態について、細胞死からT細胞を防御する能力を測定するために、数種のアッセイが使用できる。これらのアッセイには、T細胞集団を特定の作用物質と接触させて、T細胞集団がアポトーシスを受けるよう誘導するアッセイが含まれる。そのような作用物質には、T細胞レセプターを架橋する作用物質類、例えば抗CD3抗体、FasまたはTNFレセプターを架橋する作用物質類、グルココルチコイド類、が含まれる。場合によっては、成長因子欠乏、例えばIL−2欠乏により、T細胞に細胞死を誘導できる。これらのアッセイは本明細書中、殊に実施例6および7に記載する。
【0043】
T細胞中のbcl−XLフラグメントは、bcl−XLタンパク質フラグメントをコードする核酸フラグメントを導入することにより、産生できる。この核酸はcDNAであってもよく、またはゲノムDNAフラグメントであってもよい。bcl−XLの変異体は、例えば、bcl−XLタンパク質をコードする核酸分子(例えば、bcl−XL cDNA)中に、標準法、例えば部位指向性突然変異誘発またはポリメラーゼ連鎖反応経由突然変異誘発により、ヌクレオチド塩基対修飾(例えば、置換、欠失、付加等)を導入することにより調製できる。bcl−XLの好ましい修飾は、T細胞中のbcl−XLタンパク質の半減期を修飾するものを含む。従って、本方法の他の実施態様では、長い半減期を有するbcl−XLの形態をT細胞中に導入することが望ましい場合もあるが、具体的な実施態様では、非常に短い半減期を有するbcl−XLタンパク質の形態をT細胞中に導入することが望ましい。
【0044】
好ましい態様では、bcl−XLタンパク質を、“Bax”タンパク質との相互作用を促進し、そして“Bad”タンパク質との相互作用を阻害または減少させるように修飾する。bcl−XLの細胞中のアポトーシスに対する防御効果は、一つのbcl−XLタンパク質と他の“Bax”と名付けたタンパク質とにより構成されるヘテロ二量体により媒介され得ることが示されている。しかしながら、さらなるデータは、“Bad”と称したタンパク質がbcl−XLとも相互作用し得ること、およびbcl−XLとBadから構成されるヘテロ二量体がbcl−XLの細胞死に対する防御効果を不活性化し得ることを示している(Yang, et al.(1995) Cell 80,285)。さらに、Badは、Bax/bcl−XLヘテロ二量体のBaxタンパク質と置き換えて、bcl−XL/Badヘテロ二量体を形成する能力があることが示されている。このように、本方法の好ましい実施態様では、bcl−XLの修飾形態のBadへの結合を阻害するかまたは少なくとも減少するように修飾したが、野生型bcl−XLタンパク質と比較して、bcl−XLの修飾形態のBaxへの結合には有意に影響されない、bcl−XLタンパク質をコードしている核酸をT細胞中に導入することにより、T細胞を細胞死から防御する。本発明の好ましい実施態様においては、bcl−XLタンパク質の修飾は、Bcl−2相同性(BH)ドメイン1または2(それぞれ、“BH1”および“BH2”)中に位置している、bcl−XLの少なくとも1つのアミノ酸を置換することを含む。BH1は、ヒトbcl−XLタンパク質(配列番号2)のアミノ酸129と148の間に位置しており、BH2はヒトbcl−XLタンパク質(配列番号2)のアミノ酸180と191との間に位置している(Yang, E. et al.(1995) Cell 80,285)。
【0045】
さらに、bcl−XLの一次アミノ酸配列の変化は、細胞死からT細胞を防御するbcl−XL分子の能力を有意に損なうことなく許容されるようであることは、当業者により評価されるであろう。従って、天然に存在するbcl−XL分子のアミノ酸の配列と比較して、アミノ酸置換、欠失、および/または付加を有し、なお機能的活性を保持している、本書中に記載したbcl−XLの変異体形態は、本発明の範囲内に属するものである。bcl−XLの機能的性質を保持するために、好ましくは、保存性アミノ酸置換はひとつまたはそれ以上のアミノ酸残基で行われる。“保存性アミノ酸置換”とは、アミノ酸残基が同種の側鎖を有するアミノ酸残基で置換されることである。同種の側鎖を有するアミノ酸残基のファミリーは、当技術分野で、塩基性側鎖(例えば、リシン、アルギニン、ヒスチジン)、酸性側鎖(例えば、アスパラギン酸、グルタミン酸)、非荷電極性側鎖(例えば、グリシン、アスパラギン、グルタミン、セリン、トレオニン、チロシン、システイン)、非極性側鎖(例えば、アラニン、バリン、ロイシン、イソロイシン、プロリン、フェニルアラニン、メチオニン、トリプトファン)、β−分枝側鎖(例えば、トレオニン、バリン、イソロイシン)、および芳香族側鎖(例えば、チロシン、フェニルアラニン、トリプトファン、ヒスチジン)を含むものとして定義されている。
【0046】
1.2.2.T細胞中でbcl−XLコード核酸の発現のための調節配列
T細胞中でbcl−XLをコードする核酸分子を発現させ、T細胞中でbcl−XLタンパク質のレベルを増大させる(細胞死からT細胞を防御する)ためには、該核酸を調節配列に作動可能に連結させなければならない。“作動可能に連結させた”とは、T細胞中でヌクレオチド配列が発現できるような態様でbcl−XLをコードするヌクレオチド配列を少なくとも一つの調節配列に連結させたという意味であることを意図している。調節配列は、適当なT細胞中で所望のタンパク質の発現を指向するように選ばれる。従って、“調節配列”の用語は、プロモーター類、エンハンサー類、および他の発現制御配列類を含む。そのような調節配列は当業者に知られており、またGoeddel, gene Expression Technology: Methods in Enzymology 185、 Academic Press, San Diego, CA(1990)に記載されている。
【0047】
これらの調節配列は、bcl−XLをコードする核酸の転写および翻訳に必要とされるものを含み、さらに、プロモーター類、エンハンサー類、ポリアデニル化シグナル類、および好ましくはミトコンドリア膜外である、適切な細胞コンパートメントへ分子を伝達するのに必要な配列を含み得る(Gonzales-Garcia, M. et al.(1994) Development 120,3033)。核酸が組換え発現ベクター中のcDNAである場合、cDNAの転写および/または翻訳を担う調節機能は、しばしばウイルスの配列により提供される。一般に使用されるウイルスのプロモーターの例には、ポリオーマ、アデノウイルス2、サイトメガロウイルスおよびシミアンウイルス40(SV40)、およびレトロウイルスLTRs由来のものが含まれる。
【0048】
cDNAに連結させる調節配列は、構成的または誘導的転写を与えるものを選択できる。誘導的転写は、例えば誘導性エンハンサーを使用することにより達成され得る。このように、本発明の具体的な実施態様において、bcl−XLの形態をコードする核酸分子は、誘導性制御配列に影響を与える作用物質を用いてbcl−XLの形態の発現をオンまたはオフにできる(またはその中間のレベルにする)ように、誘導性制御配列の制御下にある(例えば、発現は、T細胞の存在下で、誘導性作用物質の濃度を調節することにより調節できる)。このことにより、T細胞中での細胞死に対するbcl−XLの防御効果を有効または無効に切り替えることが可能である。実際には、ある症状においてのみ、またはある時間のみT細胞の生存を促進することが望ましい。例えば、被験体における感染部位で、限られた時間枠に感染作用物質を除去するために免疫反応を強化することが望ましい。しかしながら、感染作用物質を除去する際に、T細胞を除去することが望ましい。このように、感染部位に位置するT細胞中のbcl−XLの発現は、T細胞を誘導性作用物質と接触させることにより、誘導制御配列を介して刺激され得る。ついで、感染の除去時に、誘導性作用物質を除去してT細胞中のbcl−XLの産生を停止できる。
【0049】
哺乳動物細胞に使用する誘導性調節システムは、当分野では知られており、例えば、遺伝子発現を重金属イオン(Mayo et al.(1982)Cell29:99-108;Brinster et al.(1982)Nature296:39-42;Searle et al.(1985)Mol.Cell.Biol.5:1480-1489)、熱ショック(Nouer et al.(1991)in Heat Shock Response、Nouer,L.編、CRC、Boca Raton、FL、ppl167-220)、ホルモン(Lee et al.(1981)Nature294:228-232;Hynes et al.(1981)Proc.Natl.Acad.Sci.USA78:2038-2042;Klock et al.(1987)Nature329:734-736;Israel&Kaufman(1989)Nucl.Acids.Res17:2589-2604)またはテトラサイクリン(Gossen,M.およびBujard,H.(1992)Proc.Natl.Acad.Sci.USA89:5547-5551)により調節するシステムがある。T細胞を特定の誘導性作用物質に接触させることにより制御可能な誘導的遺伝子発現をもたらすその他の機構は、PCT公開公報WO94/18317およびPCT公開公報WO93/23431に記載されている。
【0050】
誘導性制御配列は、全てのT細胞において機能でき、または、例えばCD4+T細胞、CD8+T細胞、Tヘルパー1(Th1)、Tヘルパー2(Th2)細胞などの特定のT細胞サブセットにおいてのみ機能できる。誘導性制御配列は、また、1タイプのT細胞(例えばCD4+T細胞)においては1つの作用物質により調節されるが、他のタイプのT細胞(例えばCD8+T細胞)においては他の作用物質により調節されるように、選択し得る。
【0051】
本発明のその他の態様においては、bcl−XLタンパク質をコードする核酸分子は、核酸分子の発現を構成的に起こす調節配列の制御下にある。本発明の具体的な実施態様において、HIV感染した被験体由来のT細胞を修飾してbcl−XLタンパク質を発現させ、被験体のT細胞を細胞死から構成的に防御するようにする。核酸分子の構成的発現を起こすために核酸分子に作動可能に連結させた制御配列は、好ましくはウイルスプロモーターである。一般的に使用されるウイルスプロモーターの例には、ポリオーマ、アデノウイルス2、サイトメガロウイルスおよびシミアンウイルス40、およびレトロウイルスLTR由来のものがある。その他に、例えばT細胞受容体エンハンサー(例えば、WinotoおよびBaltimore(1989)EMBOJ.8:729-733)などのT細胞特異的エンハンサーを使用できる。
調節配列に作動可能に連結させたbcl−XLタンパク質をコードする核酸分子は、典型的にベクターに移し得る。ベクターの例には、プラスミド、ウイルスまたは、例えば細菌中の核酸分子の選択および増幅に必要な配列を含むその他の核酸分子がある。このように、調節制御配列に作動可能に連結させたbcl−XLタンパク質をコードするヌクレオチド配列を含む核酸分子はまた、本明細書中では“bcl−XL発現ベクター”と呼ぶ。例えばウイルスベクターなどのベクターは、下記で更に議論する。
【0052】
1.2.3.bcl−XLタンパク質をコードする核酸分子をT細胞に導入する方法
bcl−XLタンパク質をコードする核酸分子は、典型的にトランスフェクションと呼ばれる様々な方法によりT細胞に導入され得る。“トランスフェクション”または“でトランスフェクションさせる”なる語は、外来核酸を哺乳動物細胞に導入することを意味し、エレクトロポレーション、リン酸カルシウム沈澱、DEAE−デキストラン処理、リポフェクション、マイクロインジェクション、およびウイルス感染を含む、核酸の哺乳動物細胞への導入に有用な数多くの技術を含むことを意図する。哺乳動物細胞をトランスフェクションする適当な方法は、Sambrook et al.(Molecular Cloning:A Laboratory Manual第2版、Cold Spring Harbor Laboratory press(1989))およびその他の実験書に見ることができる。
【0053】
本発明の好ましい実施態様において、bcl−XLタンパク質をコードする核酸分子はウイルスベクターを用いてT細胞に導入される。このようなウイルスベクターには、例えば、組換えレトロウイルス、アデノウイルス、アデノ随伴ウイルスおよび単純ヘルペスウイルス1がある。レトロウイルスベクターおよびアデノ随伴ウイルスベクターは、インビボでの特にヒトへの外来遺伝子の導入に優れた組換え遺伝子運搬系であると一般的に考えられている。その他に、そのようなベクターは、外来遺伝子をエクスビボでT細胞に導入するのにも使用できる。これらのベクターは、効果的に遺伝子をT細胞に運搬し、導入された核酸分子は、安定に宿主細胞の染色体DNAに組込まれる。レトルウイルスの使用における重要な必要条件は、特に細胞集団において野生型ウイルスが蔓延する可能性について、その使用の安全性を確認することである。複製欠損性レトロウイルスのみを産生する分化した細胞系(“パッケージング細胞”と呼ぶ)の開発により、遺伝子療法におけるレトロウイルスの用途が拡大し、欠損レトロウイルスは遺伝子療法の目的における遺伝子導入における使用について十分に特徴づけられている(Miller,A.D.(1990)Blood76:271参照)。このように、レトロウイルスコード配列の一部(gag、pol、env)を本発明のbcl−XLタンパク質をコードする核酸で置換えて、レトロウイルス複製欠損性にすることにより、組換えレトロウイルスを構築し得る。この場合、複製欠損性レトロウイルスはビリオン中にパッケージングさせるので、これを標準的な技術によりヘルパーウイルスを使用して標的細胞を感染させるのに使用できる。組換えレトロウイルスの産生およびインビトロまたはインビボでこのようなウイルスで細胞を感染させる方法は、Current Protocols in Molecular Biology、Ausubel,F.M.et al.編Greene Publishing Associates、(1989)、9.10−9.14章および標準的な実験マニュアルにおいて見いだされ得る。適当なレトロウイルスの例は、当業者に良く知られているpLJ、pZIP、pWEおよびpEMを含む。同種指向性および両種指向性レトロウイルス系を両方調製するための適当なパーケージングウイルス系の例は、ψCrip、φCre、ψ2およびψAmを含む。
【0054】
さらに、ウイルス粒子の表面上のウイルスパッケージングタンパク質を修飾することにより、レトロウイルスの、更にはレトロウイルスを基礎としたベクターの感染スペクトルを制限することが可能であることが示されている(例えばPCT発行物WO93/25234およびWO94/06920参照)。例えば、レトロウイルスベクターの感染スペクトルの修飾方法は:細胞表面抗原に特異的な抗体をウイルスのenvタンパク質に結合させる(Roux et al.(1989)PNAS86:9079-9083;Julan et al.(1992)J.Gen Virol73:3251-3255;およびGoud et al.(1983)Virology163:251-254);または細胞表面受容体リガンド類をウイルスのenvタンパク質に結合させる(Nada et al.(1991)J.Biol.Chem.266:14143-14146)ことを含む。結合は、タンパク質またはその他のさまざまなもの(例えば、envタンパク質をアジアロ糖タンパク質に変換するラクトース)との化学的架橋形態であることができ、並びに融合タンパク質(例えば、単鎖抗体/env融合タンパク質)を作成することによる。このように、本発明の具体的な実施態様では、bcl−XLの形態をコードする核酸分子を含むウイルス粒子を、例えば、上記した方法に従い、それらが特異的にT細胞のサブセットを標的とすることができるように修飾する。例えば、ウイルス粒子をあるタイプのT細胞に特異的な表面分子に対する抗体で被覆できる。特に、T細胞のCD4分子を認識するウイルス粒子抗体に連結させることにより選択的にCD4+T細胞を標的とすることが可能である。このように、CD4+細胞の感染は、CD8+T細胞の感染よりも優先的に起こる。本法は、T細胞の特定のサブセットのみを細胞死から防御することを望む場合に、特に有用である。さらに、本発明の方法をインビボで使用する具体的な実施態様において、bcl−XLタンパク質をコードする核酸分子のT細胞または特定のサブセットへの導入を制限することが望ましい。
【0055】
一次T細胞を含むT細胞のbcl−XLタンパク質をコードする核酸分子を導入し、かつ発現させるための更なるレトロウイルス系は、Kasid,A.et al.(1990)Proc.Natl.Acad.Sci.U.S.A.87,473;Morecki,S.et al(1991)Cancer Immunol.Immunother.32,342;Culver,K.et al.(1991)Proc.Natl.Acad.Sci.U.S.A.88,3155;およびFiner,M.H.et al.(1994)Blood、83,43に記載されている。
【0056】
本発明で有用な他のウイルス遺伝子運搬システムは、アデノウイルス由来のベクターを使用する。アデノウイルスのゲノムは、対象となる遺伝子産物をコードし、発現するが、正常細胞溶解ウイルスライフサイクルで複製する能力に関しては不活性化されているように、操作できる。例えば、Berkner et al. (1988) BioTechniques 6:616;Rosenfeld et al. (1991) Science 252:431-434;およびRosenfeld et al. (1992) Cell 68:143-155参照。アデノウイルス株Adタイプ5dl324またはアデノウイルスの他の株(例えば、Ad2、Ad3、Ad7等)由来の適当なアデノウイルスベクターは、当業者に既知である。組換えアデノウイルスは、それらが細胞分裂していない細胞に感染できないため、ある環境で有利である。更に、ウイルス粒子は、相対的に安定であり、精製および濃縮を受けることができ、上記のように、感染スペクトルに影響を与えるように修飾できる。更に、導入アデノウイルスDNA(およびそれを含む外来DNA)は、宿主細胞のゲノムに組み込まれないが、エピソームのままであり、それにより、導入DNAが宿主ゲノム(例えば、レトロウイルスDNA)に組み込まれる状況下、挿入突然変異誘発の結果起こり得る可能性のある問題を回避できる。更に、アデノウイルスゲノムが外来DNAを運搬する能力は、他の遺伝子運搬ベクターと比較して大きい(8キロ塩基まで)(Berkner et al. 前掲;Haj-AhmandおよびGraham(1986) J. Virol. 57;267)。現在使用され、そして本発明の好ましい殆どの複製欠損性アデノウイルスベクターは、ウイルスE1およびE3ゲノムの全てまたは一部を欠失しているが、アデノウイルス遺伝物質の80%程を残している(例えば、Jones et al. (1979) Cell 16:683;Berkner et al., 前掲;およびGraham et al., Methods in Molecular Biology, E. J. Murray編(Humana, Clifton, NL, 1991) vol. 7 pp. 109-127参照)。bcl−XLタンパク質をコードする対象となる核酸分子の発現は、例えば、E1Aプロモーター、主要遅延プロモーター(MLP)および関連リーダー配列、E3プロモーターまたは外来的に付加したプロモーター配列の制御下にあり得る。
【0057】
bcl−XLタンパク質をコードする核酸分子のインビトロ運搬に有用な更に別のウイルスベクター系はアデノ随伴ウイルス(AAV)である。アデノ随伴ウイルスは、アデノウイルスまたはヘルペスウイルスのような他のウイルスを有効な複製および増殖的ライフサイクルのためのヘルパーウイルスとして必要とする、天然に存在する欠損ウイルスである。(レビューのために、Muzyczka et al. Curr. Topics in Micro. and Immunol. (1992) 158:97-129参照)。そのDNAを細胞分裂していない細胞に組み込むことができ、安定な高頻度の組み込み(インテグレーション)を示す幾つかのウイルスの一つがまた存在する(例えば、Flotte et al. (1992) Am. J. Respir. Cell. Mol. Biol. 7:349-356;Samulski et al. (1989) J. Virol. 63:3822-3828;およびMcLaughlin et al. (1989) J. Virol. 62:1963-1973参照)。AAVの300塩基対程を含有するベクターは、パッケージでき、組み込みことができる。外来DNAのための領域は、約4.5kbに限定される。Tratschin et al. (1985) Mol. Cell. Biol. 5:3251-3260に記載のようなAAVベクターは、DNAを細胞に導入するのに使用できる。種々の核酸が、AAVベクターを使用して異なる細胞タイプに導入されている(例えば、Hermonat et al. (1984) Proc. Natl. Acad. Sci. USA 81:6466-6470;Tratschin et al. (1985) Mol. Cell. Biol. 4:2072-2081;Wondisford et al. (1988) Mol. Endocrinol. 2:32-39;Tratschin et al. (1984) J. Virol. 51:611-619;およびFlotte et al. (1993) J. Biol. Chem. 268:3781-3790参照)。遺伝子治療に適用し得る他のウイルスベクター系は、ヘルペスウイルス、ワクシニアウイルスおよび幾つかのRNAウイルスに由来する。
【0058】
bcl−XL−コード配列に加えて、発現ベクターはまた選択マーカーをコードする遺伝子も含み得る。好ましい選択マーカーは、G418、ヒグロマイシンおよびメトトレキサートのような医薬に対する耐性を付与するものを含む。選択マーカーは、bcl−XLタンパク質をコードする核酸分子と同じベクター(例えば、プラスミド)に導入し得るか、または別のベクター(例えば、プラスミド)に導入し得る。
【0059】
あるいは、bcl−XLタンパク質をコードする核酸分子は、細胞運搬媒体により運ばれ、細胞に輸送され得る。このような媒体は、例えば、カチオン性リポソーム(リポフェクチン)または誘導体化(例えば、抗体コンジュゲート)ポリリジンコンジュゲート、グラミシジンS、人工ウイルスエンベロープを含む。これらの媒体は、ベクター、例えばプラスミドまたはウイルスDNAにより運ばれるbcl−XLタンパク質をコードする核酸を、運搬できる。具体的態様において、一次Tリンパ球、特にCD3+、CT4+およびCT8+T細胞で発現する有効な遺伝子は、Philip R. et al. (1994) Mol. Cell. Biol. 14, 2411に記載のように、カチオン性リポソームと複合体形成させたアデノ随伴ウイルスプラスミドDNAを使用して得られる。
【0060】
本発明の他の態様において、bcl−XLタンパク質をコードする核酸分子を、可溶性分子複合体の形態で特定の細胞に運搬する。複合体は、特定の細胞の表面分子に結合し、細胞により続いてインターナリゼーションされ得るサイズである、核酸結合剤および細胞特異的結合剤を含む担体に解離可能に結合した核酸を含む。このような複合体は、米国特許出願第5,166,320号に記載されている。
本発明の他の態様において、bcl−XLタンパク質をコードする核酸は、Yang, N.−S.およびSun, W. H. (1995) Nature Medicine 1, 481に記載のように、T細胞に粒子衝撃により挿入する。
【0061】
1.1 blc−XLタンパク質レベルを増加させる作用物質を使用する方法
一つの態様において、本発明の方法は、T細胞内のbcl−XLタンパク質レベルを増加する少なくとも一つの作用物質にT細胞を接触させることにより、T細胞の生存を強化させる方法を含む。本発明の好ましい態様において、T細胞と相互作用して、bcl−XLタンパク質レベルを増加させる少なくとも一つの作用物質は、T細胞受容体およびCD28のような、T細胞の表面の分子と相互作用する、1またはそれ以上の作用物質を含む。本発明の他の態様において、T細胞内のbcl−XLタンパク質レベルを増加させる少なくとも一つの作用物質は、例えば、bcl−X遺伝子の発現の増加により、細胞内に作用する作用物質である。"T細胞内のbcl−XLタンパク質レベルを増加するために細胞内に作用する作用物質”なる用語は、T細胞の表面受容体には結合しないが、T細胞内のbcl−XLタンパク質レベルの増加をもたらす、T細胞の受容体の結合により誘導される細胞内シグナル(例えば、第2メッセンジャー)を模倣するか、または誘導する作用物質を含むことを意図する。作用物質は、T細胞内のbcl−XLタンパク質の製造を、bcl−X遺伝子転写の増加、bcl−XLmRNAの安定化またはbcl−XLmRNAの翻訳の増加による、種々の機構により刺激し得る。好ましい態様において、bcl−XLタンパク質レベルは、選択的に、すなわち、Bcl−XSタンパク質レベルの付随的増加なしに増加される。
【0062】
"内生遺伝子の発現を刺激する”なる用語は、遺伝子によりコードされるbcl−XLタンパク質のレベルが細胞内で増加するように、細胞内のbcl−X遺伝子に作用することを含むことを意図する。“転写を刺激する”なる用語は、mRNA転写の量が増加するように、転写に影響を与えることを含むことを意図する。"内生bcl−X遺伝子”なる用語は、T細胞に本来あるbcl−X遺伝子を意味することを意図し、T細胞に導入された“外来bcl−X遺伝子”と反対である。
【0063】
本発明の好ましい態様において、T細胞を少なくとも一つの作用物質と接触させ、T細胞内のbcl−XLタンパク質レベルの増加および細胞死からのT細胞の防御をもたらす。好ましい態様において、T細胞の生存は、T細胞を、T細胞を刺激する作用物質の組み合わせと接触させることにより増加する。作用物質の好ましい組み合わせは、T細胞を活性化し、T細胞に共刺激性分子を提供する作用物質を含む作用物質の組み合わせである。例えば、T細胞生存は、T細胞を、T細胞に一次活性化シグナルを提供する第1作用物質およびT細胞にコスティミュラトリー・シグナルを提供する第2作用物質と接触させることにより促進される。より好ましい組み合わせは、T細胞内でbcl−XLタンパク質レベルが増加するようにT細胞受容体を刺激する作用物質およびT細胞へのコスティミュラトリー・シグナルを提供する作用物質を含む。
【0064】
"一次活性化シグナル”なる用語は、典型的にT細胞受容体(TCR)/CD3複合体により誘発される、T細胞の活性化を誘導するシグナルを含むことを意図する。T細胞の活性化は、T細胞が、コスティミュラトリー・シグナルのような第2シグナルを受けて、増殖および分化が誘導されるような、T細胞の修飾を含むことを意図する。具体的態様において、一次活性化シグナルは、T細胞受容体またはT細胞受容体に関連するCD3複合体と接触させる作用物質により提供される。好ましい態様において、作用物質は、モノクローナル抗体OKT3(American Type Culture Collection, Rockvill, MD;No. CRL 8001から入手可能)のようなCD3に対して反応性の抗体である。本発明の他の態様において、刺激性作用物質は、抗体の組み合わせ、例えば、T11.3+T11.1またはT11.3+T11.2(例えば、Meuer, S.C. et al. (1984) Cell 36:897-906参照)のような、T細胞のCD2複合体を刺激する作用物質である。更に別の態様において、一次活性化シグナルは、抗原または抗原提示細胞により提供される。従って、T細胞を1またはそれ以上の抗原または1またはそれ以上の抗原提示細胞と接触させることにより、および所望により、コスティミュラトリー・シグナルを提供する第2作用物質と接触させることにより、T細胞の集団における特定のT細胞クローンの生存を刺激することが可能である。
【0065】
本発明の好ましい態様において、T細胞を、T細胞の一次活性化シグナルおよびコスティミュラトリー・シグナルの両方を刺激する作用物質の組み合わせを用いて刺激する。“共刺激性作用物質”なる用語は、一次活性化シグナルを受けたT細胞(例えば、活性化T細胞)を刺激して、増殖させるか、IL−2、IL−4またはインターフェロン−γのようなサイトカインを分泌させる、コスティミュラトリー・シグナルをT細胞に提供する作用物質を含むことを意図する。具体的な態様において、共刺激性作用物質は、T細胞の表面のCD28またはCTLA4分子と相互作用する。更により具体的な態様において、コスティミュラトリー・シグナルは、B−リンパ球抗原B7−1またはB7−2のようなCD28またはCTLA4のリガンドである。“CD28の天然リガンドの刺激性形態”なる用語は、T細胞にコスティミュラトリー・シグナルを提供できる、B7−1およびB7−2分子、それらのフラグメントまたはそれらの修飾物を含むことを意図する。CD28の天然リガンドの刺激性形態は、例えば、活性化T細胞をCD28の天然リガンドの形態と接触させ、標準T細胞増殖アッセイを行うことにより同定できる。従って、CD28の天然リガンドの刺激性形態はT細胞の増殖を刺激できる。CD28/CTLA4の天然リガンドの刺激は、例えば、PCT公開第WO95/03408に記載される。
【0066】
T細胞を細胞死から防御するのに使用できる他の作用物質は、bcl−XLタンパク質レベルをT細胞内で増加させるような、T細胞活性化および/または共刺激に関与する1またはそれ以上の細胞内シグナル伝達経路を刺激する作用物質を含む。本発明の好ましい態様において、刺激性作用物質は、イオノマイシンまたはA23187のようなカルシウムイオン性透過体である。あるいは、刺激性作用物質は、ホルボールエステルのようなプロテインキナーゼCを刺激する作用物質であり得る。好ましいホルボールエステルは、ホルボール−12,13−ジブチレートである。本発明のより更に好ましい組み合わせにおいて、T細胞をカルシウムイオノホアおよびホルボールエステルの組み合わせと接触させる。刺激性作用物質は、プロテインチロシンキナーゼを活性化する作用物質であり得る。プロテインチロシンキナーゼを活性化する好ましい作用物質は、ペルバナデート(O'Shea, J. I., et al. (1992) Proc. Natl. Acad. Sci. USA 89:10306)である。
【0067】
T細胞生存を刺激するのに使用できる他の作用物質は、bcl−XLタンパク質レベルを増加させる、ポリクローナルアクチベーターのような作用物質を含む。ポリクローナルアクチベーターは、T細胞の原形質膜上で発現する糖タンパク質に結合する作用物質を含み、フィトヘマグルチニン(PHA)、コンカナバリン(ConA)およびアメリカヤマゴボウマイトジェン(PWM)を含む。
【0068】
T細胞内のbcl−XLタンパク質レベルを増加できるスーパー抗原は、また本発明の範囲内である。本明細書で定義される“スーパー抗原”なる用語は、細菌エンテロトキシンまたはT細胞の増殖を刺激できる他の細菌タンパク質を含むことを意図する。スーパー抗原は、SEA、SEB、SEC、SEDおよびSEEのようなスタフィロコッカスエンテロトキシン(SE)を含む。スーパー抗原はまたはレトロウイルススーパー抗原のようなウイルス起源であり得る。
【0069】
T細胞生存の刺激に使用し得る更に別の抗原は、リンホカインを含み、それは単独または他の作用物質と組み合わせて、T細胞内のbcl−XLタンパク質レベルを増加させる。従って、本発明の好ましい態様において、T細胞を、T細胞の一次活性化シグナルを提供する作用物質(例えば、抗CD3抗体)およびbcl−XLタンパク質レベルをT細胞内で増加させる有効量のIL−2と接触させる。
【0070】
単独または他の作用物質と組み合わせて、T細胞死をbcl−XLタンパク質レベルの増加により予防できる更なる作用物質は、T細胞を作用物質単独または他の作用物質と共に接触させ、bcl−XLタンパク質レベルを、本明細書に記載のように、例えば、ウエスタンブロット分析により追跡することにより同定し得る。
【0071】
本発明の範囲内の作用物質は、溶液で、または固体表面に結合させて使用できる。固体表面は、例えば、組織培養皿またはビーズの表面であり得る。刺激性作用物質の性質に依存して、固体表面への結合は、当分野で既知の方法により行うことができる。例えば、タンパク質を商業的に入手可能な架橋剤(Pierce, Rokford IL)を使用して化学的に細胞表面に架橋するか、または一晩4℃でインキュベーションすることによりプラスチック上に固定化できる。数個の作用物質をT細胞内のbcl−XLレベルの増加に使用する場合、ある作用物質は溶液状であってよく、ある作用物質は固体支持体に結合させてもよい。好ましい態様において、T細胞は固相結合抗CD3抗体および可溶性抗CD28抗体の組み合わせと接触させる。
【0072】
細胞内に作用してbcl−XLタンパク質レベルを増加させる作用物質は、細胞内のbcl−XLタンパク質レベルの欠乏についての標準アッセイを使用して同定できる。例えば、T細胞を試験作用物質の存在下または不在下にインキュベーションでき、異なる時間に細胞により製造されたbcl−XLタンパク質の量を、本発明に参考として包含する、公開PCT出願番号WO95/00642の実施例部分に記載のように、ウエスタンブロット分析により測定できる。従って、本発明の方法を行うのに好ましい作用物質は、ウエスタンブロット分析で測定した際に、bcl−XLタンパク質レベルの著しい増加を誘導するものを含む。
【0073】
更に、T細胞のbcl−XLのタンパク質レベルの増加に使用できる作用物質は、同定用のbcl−X遺伝子の調節領域の分析により同定できる。DNAは、特定の作用物質により、遺伝子の転写を調節するように配列している。例えば、これらの配列に特異的に結合する転写アクチベーターは、bd−X遺伝子発現の刺激に使用できる。次いで、これらの作用物質が細胞内のbcl−XLタンパク質レベルを増加するか、ウエスタンブロット分析により確認できる。
【0074】
本発明の具体的態様において、作用物質は、選択的に、または少なくとも優勢にT細胞に作用し、bcl−XLタンパク質レベルを増加させる。従って、作用物質の投与は、T細胞内のみのまたは少なくとも好ましくはT細胞におけるbcl−XLタンパク質レベルの増加を得るようにする。T細胞特異的作用物質は、インビトロで、異なる細胞タイプを作用物質に接触させ、細胞内のbcl−XLタンパク質レベルの測定をウエスタンブロット分析により行うことにより同定できる。従って、好ましい作用物質は、T細胞内のみのまたは少なくとも好ましくはT細胞のbcl−XLタンパク質レベルの増加をもたらすものである。
【0075】
本発明の好ましい態様において、細胞内に作用してbcl−XLタンパク質レベルを増加させる作用物質は、相対的に短い半減期を有するものである。このような作用物質により、作用物質の効果が良好に制御される。特に、このような作用物質は、T細胞の細胞生存を短期間、中期間、または長期間刺激できる。実際、本発明の具体的態様において、T細胞生存は、一時的に、例えば、作用物質を感染部位の免疫応答のブースターに使用する条件下のみで、増加するのが好ましい。免疫応答は、T細胞の半減期の制御により少なくとも一部分制御される。ある条件下ではT細胞死が起こらないと、実際的には、有害な作用をもたらす場合がある。しかしながら、本発明の他の態様において、細胞内に作用して、bcl−XLタンパク質レベルを増加させる作用物質は、作用物質の被験体へのより少ない投与でもT細胞死に対する有効な長期防御を得ることができるように、長い半減期を有する。
【0076】
1.3. T細胞にタンパク質を導入することにより、T細胞を細胞死から防御する方法
本発明の他の態様において、T細胞を、T細胞による摂取に適した形態のbcl−XLタンパク質と接触させることを含む方法により、T細胞を細胞死から防御する。従って、本発明の具体的態様において、bcl−XLタンパク質は、細菌発現システムおよび適当な媒体でのT細胞への輸送のような、慣用法によりインビトロで合成される。適当な媒体は、特定の細胞、特にT細胞またはT細胞の選択的サブセットを標的とするように修飾できるリポソームを含む。
【0077】
bcl−XLタンパク質は、bcl−XLタンパク質をコードする核酸分子またはbcl−XLの生理学的に活性な形態を、その後種々の宿主、特に哺乳動物または昆虫細胞培養物のような真核細胞だけでなく、E. coliのような原核細胞での対応するタンパク質の合成を指向する、種々の発現ベクターに挿入することによりインビトロで製造できる。本発明の範囲内の発現ベクターは、本明細書に記載の核酸および核酸に作動可能に連結させたプロモーターを含む。このような発現ベクターは、宿主細胞をトランスフェクションするのに使用でき、それにより本明細書に記載の核酸によりコードされるタンパク質が製造される。本明細書に記載の本発明の発現ベクターは、典型的に、少なくとも一つの調節配列に作動可能に連結させたbcl−XLタンパク質をコードするヌクレオチド配列を含む。調節配列は、上記である。発現ベクターの設計は、トランスフェクションすべき宿主細胞の選択および/または発現することが望ましいタンパク質のタイプおよび/または量のような因子に依存し得る。
【0078】
本発明の発現ベクターは、真核または原核(例えば、哺乳類、昆虫または酵母細胞)の細胞のトランスフェクションに使用でき、それにより、ベクターのヌクレオチド配列によりコードされるタンパク質を合成する。原核細胞における発現は、多くの場合、E. coliにて構成性または誘導性プロモーターと共に行う。あるE. coli発現ベクター(いわゆる融合ベクター)は、多くのアミノ酸残基を発現組換えタンパク質に、通常は、発現タンパク質のアミノ末端に付加するために設計される。このような融合ベクターは、3つの目的で働く:1)組換えタンパク質の発現の増加のため;2)標的組換えタンパク質の溶解性の増加のため;および3)アフィニティー精製におけるリガンドとして作用することにより、標的組換えタンパク質の精製における補助のため。融合発現ベクターの例には、グルタチオンS−トランスフェラーゼおよびマルトースE結合タンパク質を、それぞれ、標的組換えタンパク質に融合するpGEX(Amrad Corp., Melbourne, Australia)およびpMAL(New England Biolabs, Berberly, MA)がある。従って、bcl−XLタンパク質をコードする核酸分子は、真核融合ベクター中の更なるコード配列に結合でき、融合タンパク質の発現、溶解性または精製を補助する。しばしば、融合発現ベクターにおいて、タンパク質分解切断部位を、融合分子と標的組換えタンパク質の結合部で挿入することにより、融合タンパク質の精製に続く標的組換えタンパク質を融合分子から分離することが可能である。このような酵素およびそれらの連結認識配列は、ファクターXa、トロンビンおよびエンテロキナーゼを含む。
【0079】
誘導性非融合発現ベクターには、pTrc(Amann et al. (1988) Gene 69:301-315)およびpET11d(Studier et al., Gene Expression Technology:Methods in Enzymology 185, Academic Press, San Diego, California (1990)60-89)がある。pTrcベクター4からの標的遺伝子発現は、ハイブリッドtrp-lac融合プロモーターからの宿主RNAポリメラーゼ転写に依存する。pET11dベクターからの標的遺伝子発現は、共発現(coexpressed)ウイルスRNAポリメラーゼ(T7gn1)により媒介されるT7gn10-lac0融合プロモーターからの転写に依存する。このウイルスポリメラーゼは、T7gn1を有する非活性λプロファージから宿主株BL21(DE3)またはHMS174(DE3)により供給される。
【0080】
E. coli内のbcl−XLタンパク質の発現を最大にする一つの方法は、組換えタンパク質をタンパク質分解的に切断する能力を低下させて、宿主細菌中のタンパク質を発現させることである(Gottesman, S., Gene Expression Technology:Methods in Enzymology 185, Academic Press, San Diego, California (1990) 119-128)。他の方法は、各アミノ酸の個々のコドンが、高度に発現したE. coliタンパク質において優先的に利用されるものであるように、発現ベクター内に挿入されるbcl−XLタンパク質をコードする核酸分子のヌクレオチド配列を変えることである(Wada et al., (1992) Nuc. Acids Res. 20:2111-2118)。このような核酸配列の置換は、本発明に含まれ、標準DNA技術により行うことができる。
【0081】
あるいは、bcl−XLタンパク質は、哺乳動物細胞(例えば、チャイニーズハムスター卵巣細胞(CHO)またはNSO細胞)、昆虫細胞(例えば、バキュロウイルスベクターを使用して)または酵母細胞のような真核宿主細胞で発現できる。他の適当な宿主細胞は、Goeddel, (1990)前掲に見られるか、当業者に既知であり得る。真核細胞における真核細胞性タンパク質の発現が、組換えタンパク質の部分的または完全グリコシル化および/または対応する鎖内または鎖間ジスルフィド結合の形成を導くため、真核よりむしろ原核でのbcl−XLタンパク質の発現が好ましいことがある。哺乳動物細胞で発現させる場合、ベクターの制御機能は、ウイルス性の物質により提供されることが多い。例えば、通常使用されるプロモーターは、ポリオーマ、アデノウイルス2、サイトメガロウイルスおよびシミアンウイルス40に由来する。哺乳動物細胞でbcl−XLタンパク質を発現させる場合、一般にCOS細胞(Gluzman, Y. (1981) Cell 23:175-182)は一過性増幅/発現のためにpCDM8(Seed, B. (1987) Nature 329:740)のようなベクターとの複合体で使用するのに対し、CHO(dhfr Chinese Hamster Ovary)細胞は哺乳動物細胞での安定な増幅/発現のためにPMT2PC(Kaufman et al., (1987) EMBO J. 6:187-195)のようなベクターと共に使用する。組換えタンパク質の製造のための好ましい細胞系は、ECACC(カタログ番号85110503)から入手可能であり、Galfre, G.およびMilstein, C.((1981) Methods in Enzymology 73(13):3-46;およびPreparation of Monoclonal Antibodies:Strategies and Procedures, Academic Press, N.Y., N.Y.)記載のNSOミエローマ細胞である。酵母での組換えタンパク質の発現に適当なベクターの例は、pYepSec1(Baldari et al., (1987) Embo J. 6:229-234)、pMA(KurjanおよびHerskowitz, (1982) Cell 30:933-943)、pJRY88(Schultz et al., (1987) Gene 54:113-123)、pYES2(Invitrogen Corporation, San Diego, CA)を含む。培養昆虫細胞(SF9細胞)中のタンパク質の発現に利用可能なバキュロウイルスベクターは、pAcシリーズ(Smith et al., (1983) Mol. Cell Biol. 3:2156-2165)およびpVLシリーズ(Lucklow, V.A.,およびSummers, M.D., (1989) Virology 170:31-39)を含む。
【0082】
ベクターDNAは、リン酸カルシウムまたは塩化カルシウム共沈、DEAE−デキストラン媒体トンラスフェクション、リポフェクションまたはエレクトロポレーションのような慣用の形質転換またはトランスフェクション法により、真核または原核細胞に挿入できる。宿主細胞を形質転換する適当な方法は、Sambrook et al. (Molecular Cloning:A Laboratory Manual, 2nd Edition, Cold Spring Harbor Laboratory press(1989))および他の研究用テキストに見ることができる。
【0083】
哺乳動物細胞を安定にトランスフェクションさせる場合、使用する発現ベクターおよびトランスフェクション技術にもよるが、小さいフラクションの細胞のみがDNAをそのゲノムに組み込み得ることが知られている。これらの組み込み体(インテグレート)の同定および選択のために、一般的に、選択マーカー(例えば、抗生物質への耐性)をコードする遺伝子を、対象となる遺伝子と共に宿主細胞に導入する。好ましい選択マーカーは、G418、ヒグロマイシンおよびメトトレキサートのような医薬に対する耐性を付与するものを含む。選択マーカーをコードする核酸は、対象となる遺伝子と同じプラスミドにより宿主細胞内に挿入できるかまたは別のプラスミドにより挿入できる。これらの対象となる遺伝子を含む細胞は、医薬選択(例えば、選択マーカー遺伝子を取り込んた細胞は生存するが、他の細胞は死滅する)により同定できる。次いで生存細胞を、bcl−XLタンパク質の製造について、例えば、抗bcl−XL抗体の細胞上清からの免疫沈殿によりスクリーニングできる。
【0084】
組換え技術により製造されるbcl−XLタンパク質は、細胞の混合物およびタンパク質を含む培地から分泌および単離できる。bcl−XLタンパク質の選択のために、適当なシグナルペプチドをコードするDNA配列をbcl−XLをコードするヌクレオチド配列の5'末端に連結させて、bcl−XLタンパク質を、細胞からのタンパク質の分泌をもたらすシグナルペプチドに連結させる。あるいは、タンパク質を細胞質に残すこともでき、その細胞を回収し、溶解してタンパク質を単離し得る。細胞培養に適当な培地は当分野で既知である。タンパク質は、細胞培養培地、宿主細胞または両方から、タンパク質精製の分野で既知の技術を使用して単離できる。
【0085】
組換え的に製造されたbcl−XLタンパク質は、被験体に投与するための適当な医薬媒体にパッケージでき、そうして、bcl−XLタンパク質を被験体のT細胞に導入し、細胞死に対するT細胞の防御をもたらす。組換えbcl−XLタンパク質をT細胞へインビボおよびエクスビボ導入する場合、組換えタンパク質は、好ましくはリポソームにパッケージされる。しかしながら、他の担体システムも使用できる。
【0086】
この方法のその他の実施態様において、T細胞生存は、Badのbcl−XSタンパク質のようなbcl−XLのアンタゴニストのタンパク質レベルの減少により、促進される。bcl−XLアンタゴニストのタンパク質レベル減少は、T細胞を、アンタゴニストをコードする遺伝子の発現を減少させる少なくとも一つの作用物質と接触させることにより、またはT細胞に、アンタゴニストの生理学的活性を減少させる核酸または他の化合物を挿入することにより、達成できる。bcl−XSおよびBadタンパク質レベルをダウンレギュレーションする方法は、bcl−XLタンパク質レベルをダウンレギュレーションするための本明細書に記載の方法から改作できる。
【0087】
T細胞死を減少させるために、上記方法を組み合わせることができる。
【0088】
2.T細胞を細胞死に対して感受性にする方法
本発明の具体的な実施態様において、T細胞内のbcl−XLタンパク質のレベルを阻害または減少させることにより、T細胞を細胞死に対してより感受性にする。例えば、T細胞の細胞死への感受性は、T細胞を、bcl−XL機能を不活性にするかまたはbcl−XLの生物学的活性を減少させる作用物質と接触させることにより、増加できる。“T細胞を細胞死に対して感受性にする”なる用語は、T細胞の細胞死への感受性が、非修飾T細胞と比較して増加するように、T細胞内の生物学的に活性なbcl−XLタンパク質の量が減少されるようにT細胞を修飾することを含むことを意図する。"機能的bcl−XLタンパク質”および“生物学的活性bcl−XLタンパク質”なる用語は、野生型bcl−XLタンパク質または、あるいは、野生型bcl−XLタンパク質の生物学的機能を遂行できるbcl−XLタンパク質の修飾形態を含むことを意図する。T細胞を細胞死に対して感受性にする本発明の方法は、T細胞のポリクローナル集団または特異的T細胞クローンの欠失を引き起こしたい場合に多くの治療的応用性を有する。従って、本発明の方法は、例えば、自己免疫疾患の処置に有用である。
【0089】
T細胞内のbcl−XLタンパク質レベルを減少する方法は、T細胞を、bcl−X遺伝子の転写のレベルを減少させる作用物質、bcl−XLmRNAを脱安定化する作用物質、bcl−XpremRNAのbcl−XLmRNAへのスプライシングを阻止する作用物質、mRNAの翻訳を阻止する作用物質またはこれらの作用物質の組み合わせと接触させることから成る方法を含む。あるいは、bcl−XLと相互作用し、bcl−XLの生物学的活性を阻害または減少させるタンパク質のタンパク質レベルを増加させることによりbcl−XLタンパク質の防御効果に打ち勝つことができる。一つの実施態様において、Badまたはbcl−XSタンパク質のタンパク質レベルを、T細胞が細胞死に対して感受性となるように、T細胞内で増加させる。
【0090】
本発明の具体的実施態様において、T細胞内のbcl−XLタンパク質レベルは、T細胞に、bcl−XL機能を妨害するRNAまたはタンパク質を発現する核酸を挿入することにより減少する。例えば、一つの実施態様では、bcl−XLタンパク質の製造を阻害するアンチセンス核酸をT細胞に導入する。“アンチセンス”核酸は、“センス”核酸に相補的な、例えば、タンパク質をコードするmRNAに相補的な核酸配列を含み、ワトソンとクリックの塩基対形成規則に従い構築される。従って、アンチセンス核酸は、センス核酸に水素結合できる。mRNAの配列に相補的なアンチセンス配列は、mRNAのコード領域に相補的であり得るか、またはmRNAの5'または3'非翻訳領域に相補的であり得る。ヒトbcl−XLをコードするヌクレオチド配列のコード領域は配列番号1に示す。好ましくは、アンチセンス核酸は、開始コドンの前にあるかまたはそのコドンにまたがる領域と相補的であるか、またはmRNAの3'非翻訳領域にある。アンチセンス核酸は、配列番号1に示すヌクレオチド配列、または当分野で既知の他のbcl−XLコード配列に基づいて設計できる。例えば、核酸は、配列番号1のヌクレオチド配列のコード領域または非翻訳領域に相補的な配列を有するように設計する。
【0091】
本発明のアンチセンス核酸は、当分野で既知の方法を使用した化学的合成および酵素的ライゲーション反応を使用して構築できる。アンチセンス核酸(例えば、アンチセンスオリゴヌクレオチド)は、天然に存在するヌクレオチドまたは、分子の生物学的安定性を増加するためにまたはアンチセンスとセンス核酸の間に形成された二本鎖の物理的安定性を増加するために設計した種々の修飾ヌクレオチドを使用して化学的に合成でき、例えば、ホスホロチオエート誘導体およびアクリジン置換ヌクレオチドを使用できる。アンチセンスオリゴヌクレオチドは、培養中のT細胞に導入でき、bcl−XLの発現を阻害する。オリゴヌクレオチドのような1またはそれ以上のアンチセンス核酸を培養培地中の細胞に、典型的には約200μg/mlで添加できる。
【0092】
あるいは、bcl−XLタンパク質をコードするヌクレオチド配列の少なくともフラグメントに対応する核酸が、アンチセンス配向(すなわち、挿入した核酸から転写されるRNAが対象となる標的核酸に対してアンチセンス配向である)でサブクローンされている発現ベクターを使用して、アンチセンス核酸をT細胞内で生物学的に製造できる。アンチセンス発現ベクターは、アンチセンス核酸を非常に有効な調節領域の制御下に製造する、組換えプラスミド、ファージミドまたは弱毒化ウイルスの形態であり得る。調節領域は、核酸分子の構成的または誘導的発現を刺激できる。核酸分子の発現を制御する調節領域、このような配列を有するベクター、および核酸分子をT細胞に導入する方法は上記である。bcl−XLアンチセンスmRNAをコードする核酸分子は、T細胞にエクスビボまたはインビボで導入できる。核酸をT細胞にインビボで導入する方法は上記である。アンチセンス遺伝子を使用した遺伝子発現の調節の記載は、Weintraub, H. et al., Antisense RNA as a molecular tool for genetic analysis, Reviews-Trends in Genetics, Vol. 1(1) 1986参照。
【0093】
本発明の他の態様において、T細胞中のbcl−XLタンパク質レベルは、T細胞に、リボザイムであるアンチセンス核酸の形態をコードする核酸を導入することにより減少する。リボザイムは、リボヌクレアーゼ活性を有する触媒的RNA分子であり、mRNAのような一本鎖核酸に対して相補的な領域を有してそれらを切断する能力がある。bcl−XLコード配列に特異性を有するリボザイムは、bcl−XLコードmRNAのヌクレオチド配列に基づいて設計できる。例えば、テトラヒメナL-19IVS RNAの誘導体は、活性部位の塩基配列が、bcl−XLコードmRNAで切断すべき塩基配列と相補的であるように構築できる。例えば、Cech et al.米国特許第4,987,071号;およびChech et al.米国特許第5,116,742号参照。あるいは、bcl−XLコード配列は、RNA分子のプールから、特異的リボヌクレアーゼ活性を有する触媒的RNAを選択するのに使用できる、例えば、Bartel, D.およびSzostak, J. W. (1993) Science 261:1411-1418参照。
【0094】
この方法の更に別の態様において、T細胞にbcl−XLと相互作用してbcl−XLの生理学的機能を阻害する阻害性タンパク質をコードする核酸分子を導入することにより、bcl−XLタンパク質のレベルを減少させることにより、T細胞を細胞死に対して感受性にする。本発明の具体的実施態様において、阻害性タンパク質はBadタンパク質または、bcl−XLの生理学的活性の減少をできるそれらのフラグメントである(Yang E. et al.(1995) Cell 80, 285)。他の態様において、阻害性タンパク質は、bcl−XLと相互作用する細胞内抗体である。細胞内抗体分子は、T細胞内に一本鎖抗体分子を本明細書に記載の方法により導入および発現することを含む方法でT細胞に導入できる。このような方法は、例えば、Biocca, S. et al. (1993) Biochemical and Biophysical Research Communications 197, 422;Biocca, S. et al. (1994) Bio/Technology 12, 396;Marasco, W. A., et al. (1993) Proc. Natl. Acad. Sci. USA 90, pp. 7889;およびWerge et al. (1990) FEBS 274, 193に記載されている。
【0095】
本発明の他の実施態様では、T細胞内のbcl−XLタンパク質レベルは、T細胞を、内因性bcl−XLタンパク質レベルをダウンレギュレーションするする作用物質と接触させることにより減少させる。具体的実施態様において、作用物質は、bcl−XL遺伝子の転写を減少させるものである。T細胞内のbcl−XLタンパク質レベルをダウンレギュレーションする作用物質は、試験すべき作用物質をT細胞とインキュベーションし、bcl−XLタンパク質レベルを本明細書に記載のようにウエスタンブロット分析で測定するアッセイを使用して同定できる。
【0096】
bcl−XSまたはBadのようなbcl−XLのアンタゴニストのレベルを増加させることにより、T細胞を細胞死に対して感受性にする方法もまた本発明の範囲内である。これらのタンパク質のいずれかまたは両方のタンパク質レベルは、T細胞を例えば、bcl−XSおよび/またはBadをコードする遺伝子の発現を刺激する作用物質と接触させることにより、T細胞内で増加させることができる。一つの実施態様において、bcl−XSをコードするbcl−X遺伝子の発現を刺激する作用物質は、bcl−XL産生を刺激しない。あるいは、これらのタンパク質をコードする核酸は、bcl−XLタンパク質レベルの増加について本明細書に記載の方法と同様にしてT細胞に導入できる。bcl−XLのアンタゴニストをコードする核酸配列は、例えば、Yang, E.et al. (1995) Cell 80, 285に記載されている。bcl−XSまたはBadタンパク質レベルを増加させる方法は、bcl−XLタンパク質レベルの増加について本明細書に記載した方法を応用できる。
【0097】
本発明の一つの態様では、T細胞を、T細胞内のbcl−XLタンパク質レベルを減少する作用物質と一次T細胞活性化シグナルを提供してT細胞死を刺激する作用物質と共に接触させることにより、T細胞を細胞死に対して感受性にする。一次活性化シグナルを提供する作用物質は、抗CD3抗体のようなポリクローナルアクチベーターであり得る。本発明の好ましい態様において、一次活性化シグナルを提供する作用物質は、抗原提示細胞上の抗原である。本発明の更に別の態様では、少なくとも一つのT細胞クローンを細胞死に対して感受性にするように、T細胞をさらに抗原提示細胞の更なる抗原と接触させる。更なる態様では、T細胞を、コスティミュラトリー・シグナルを阻止する作用物質と接触させる。従って、本発明の一つの実施態様において、T細胞死は、本発明の方法に従ったT細胞における一次活性化シグナルの刺激、T細胞の共刺激の阻害およびT細胞におけるbcl−XLタンパク質レベルの減少により誘発される。本発明の方法は、従って、T細胞のポリクローナル集団のT細胞死を可能にし、または、あるいは、限定された数のT細胞クローン(例えば、T細胞集団中の抗原特異的T細胞クローン)のT細胞死の誘導を可能にする。
【0098】
T細胞の細胞死に対する感受性を増加する上記の方法は、組み合わせることができる。
【0099】
3.医薬組成物
本発明は、T細胞内のbcl−XLタンパク質レベルを増加することにより、T細胞を細胞死から防御する方法を提供する。T細胞内のbcl−XLのレベルの増加は、内生bcl−XLタンパク質のレベルの増加、またはT細胞へのbcl−XLタンパク質をコードする核酸の導入、または両方の組み合わせにより達成できる。具体的態様では、内生bcl−XLタンパク質のレベルを、T細胞を細胞内に作用する作用物質と接触させることにより増加させる。本発明の他の態様では、T細胞の細胞死に対する感受性は、T細胞を、T細胞内のbcl−XLタンパク質レベルを減少させる作用物質と接触させることにより増加させる。本発明の方法は、インビボ、エクスビボまたは両方の組み合わせにより実施できる。本発明の方法をエクスビボで実施するには、T細胞集団を被験体から得、インビトロでbcl−XLタンパク質レベルを増加または減少させる作用物質と接触させ、所望により、被験体に再投与する(この態様は更に下に記載する)。bcl−XLタンパク質のレベルは、本明細書に記載のようにウエスタンブロット分析により追跡できる。
【0100】
本発明の方法をインビボで実施する場合、作用物質をインビボの医薬投与に適した生物学的適合形態で被験体に投与する。“インビボで投与に適した生物学的適合形態”は、作用物質の治療的効果が毒性効果より重いような、投与すべき作用物質の形態を意味する。投与すべき作用物質は、bcl−XLタンパク質をコードする核酸分子または細胞内のbcl−XLタンパク質の産生を阻害するアンチセンス核酸分子をコードする核酸分子、細胞内に作用してbcl−XLタンパク質レベルを増加または減少する作用物質、およびbcl−XLタンパク質をコードする核酸分子にまたはbcl−XLアンチセンス核酸分子をコードする核酸分子に作動可能に連結させた誘導性制御配列を調節する作用物質、を含む。
【0101】
“被験体”なる用語は、免疫応答を誘導できる生存生物、例えば哺乳動物を含むことを意図する。被験体の例は、ヒト、イヌ、ネコ、マウス、ラットおよびそれらのトランスジェニック種である。例えば、本発明の範囲内の動物は、家畜および家禽のような農業的対象となる動物を含む。あるいは、本発明の方法はまた植物に応用できる。
【0102】
本発明の作用物質の治療的有効量の投与は、所望の結果を達成するのに必要な投与量および期間で有効な量と定義する。例えば、作用物質の治療的有効量は、病気の状態、年令、性別、および被験体の体重および被験体に所望の応答を誘発する作用物質の能力などの要因に依存して変化し得る。用量レジメは、最適な治療的応答を提供するように調節し得る。例えば、数回に分割した用量を、毎日投与してもよく、またその用量を治療的状態の緊急性に合わせて徐々に減少させてもよい。
【0103】
作用物質は、注射(皮下、静脈内等)、経口投与、吸入、経皮投与または直腸投与のような慣用法で投与し得る。投与経路によるが、作用物質を不活性化し得る、酵素、酸および他の自然条件の作用から防御するために、作用物質をある物質で被覆してもよい。
【0104】
非経口投与以外で作用物質を投与するために、作用物質を、その不活性化を防御する物質でコートするか、その作用物質と同時投与する。例えば、bcl−XLタンパク質をコードする核酸分子を含む発現プラスミドは、酵素阻害剤と共に適当な担体または希釈剤で、またはリポソームのような適当な担体で、被験体に投与し得る。薬学的に許容される希釈剤は、食塩水および水性緩衝化溶液を含む、酵素阻害剤は、膵臓トリプシン阻害剤、ジイソプロピルフルオロホスフェート(DEP)およびトラシロールを含む。リポソームは、水中油中水(water-in-oil-in-water)エマルジョンおよび慣用のリポソームを含む(Strejan et al., (1984) J. Neuroimmunol. 7:27)。分散剤もまた、グリセロール、液体ポリエチレングリコールおよびこれらの混合物中、および油中で製造できる。貯蔵および使用の慣用条件下で、これらの製剤は微生物の生長を防ぐ防腐剤を含み得る。
注射使用に適当な医薬組成物は、滅菌水性溶液(水溶性の場合)または分散剤および滅菌注射溶液または分散剤の即席製造用の滅菌粉末を含む。全ての場合、組成物は滅菌状態でなければならず、容易な注射可能性が存在する程度に液体でなければならない。製造および貯蔵の条件下で安定でなければならず、細菌および真菌のような微生物の汚染作用から防御されていなければならない。担体は、例えば、水、エタノール、ポリオール(例えば、グリセロール、プロピレングリコールおよび液体プロピレングリコール等)およびそれらの適当な混合物を含む溶剤または分散媒体であり得る。適当な溶解性は、例えば、レシチンコーティングの使用により、分散剤の場合では、必要な粒子サイズの維持により、および界面活性剤の使用により維持できる。微生物活性の阻止は、種々の抗細菌および抗真菌剤、例えば、パラベン、クロロブタノール、フェノール、アスコルビン酸、チメロサール等により達成できる。多くの場合、等張剤、例えば、糖、マンニトール、ソルビトールのようなポリアルコール、塩化ナトリウムを組成物中に含むのが好ましい。注射組成物の吸収延長は、吸収を遅延する作用物質、例えば、モノステアリン酸アルミニウムおよびゼラチンを組成物中に含むことにより達成できる。
【0105】
滅菌注射溶液は、必要な量の作用物質を、適当な溶媒に、必要に応じて上述の1またはそれ以上の成分と組み合わせて包含させ、続いて滅菌濾過することにより製造できる。一般に、分散剤は、作用物質を塩基性分散媒体および上述からの必要な他の成分を含む滅菌媒体に包含させることにより製造する。滅菌注射溶液の製造における滅菌粉末の場合、好ましい製法は、真空乾燥および凍結乾燥であり、予め滅菌濾過した溶液から、有効成分(例えば、ペプチド)+他の所望の成分の粉末を製造する。
【0106】
作用物質が上記のように適切に保護されている場合、例えば、不活性希釈剤または吸収可能食用担体と共に、経口投与し得る。本明細書で使用する限り、"薬学的に許容される担体”は、任意のおよび全ての溶媒、分散媒体、コーティング、抗細菌および抗真菌剤、等張剤および吸収遅延剤等を含む。このような媒体および作用物質の薬学的活性物質への使用は当分野で既知である。慣用の媒体または試薬が作用物質と不適合である場合を除いて、治療用組成物へのその使用が期待される。補足的有効化合物もまた組成物に包含できる。
【0107】
非経口投与組成物を、投与が容易であり、均一の量である投与単位形態に調剤するのが特に有利である。本明細書で使用する投与単位形態は、処置すべき哺乳動物被験体に均一に投与するのに適した物質的に別の単位を意味する;各単位は、必要な薬学的担体と共に、所望の治療効果を生むと計算された予定量の有効化合物を含む。本発明の投与単位形態の詳細は、(a)作用物質の独自の特性および達成すべき具体的な治療効果、およびb)被験体における感受性処置剤のような調剤の分野で固有の限界により規定されており、それに直接依存している。
【0108】
4.本発明の方法の応用
本発明は、T細胞内のbcl−XLタンパク質レベルを増加することにより、T細胞を細胞死から防御する方法に関する。本発明はまたT細胞内のbcl−XLタンパク質レベルを減少させることにより、T細胞を細胞死に対して感受性にすることにも関する。これらの方法は、インビボおよびエクソビボの両方で実施できる。エクソビボで実施する時、末梢血単核細胞を被験体から得、密度勾配遠心、例えば、フィコール/ハイパークにより単離できる。具体的態様において、純粋な末梢血細胞を次いでbcl−XLタンパク質のレベルを修飾する作用物質と接触させる。この方法の他の態様において、末梢血単核細胞をbcl−XLタンパク質のレベルを調節する作用物質と接触させる前に特異的細胞タイプで豊富化させる。単核細胞は、例えば、プラスチック上への接着により枯渇できる。所望により、CD4+T細胞集団を、残りの単球、B細胞、NK細胞およびCT8+T細胞から、モノクローナル抗体(mAb)および商業的に入手可能なmAb(例えば、抗CD14(Mo2)、抗CD11b(Mo1)、抗CD20(B1)、抗CD16(3G8)および抗CD8(7PT3F9)mAb)を使用した抗マウスIg被覆磁気ビーズを使用して分離できる。本発明の方法は、CD4+CD45RA+(自生CD4+T細胞)およびCD4+CD45RO+(記憶T細胞)T細胞サブセットのようなCD4+T細胞のサブセットに適応できる。これらは、CD4+CD45RA+細胞の製造については、抗CD45RO抗体(UCHLI)の更なる使用と共に、CD4+CD45RO+T細胞の製造については抗CD45RA抗体(2H4)の更なる使用と共に、上記のように製造できる。
【0109】
精製の効果は、抗CD3、抗CD4、抗CD8および抗CD14mAbまたはT細胞の特異的サブセットを認識する付加的抗体、続くフルオレセインイソチオシアネート接合ヤギ抗マウス免疫グロブリン(Fisher, Pittsburgh, PA)または他の二次抗体を使用したフローサイトメトリー(Coulter, EPICS Elite)により分析できる。
【0110】
エクスビボで使用する細胞集団のタイプは、bcl−XLタンパク質レベルの修飾に使用する作用物質のタイプ、作用物質をT細胞に運搬するのに使用する媒体のタイプ、bcl−XLタンパク質レベルの増加が望ましいT細胞のサブセットを含む種々の因子に依存する。従って、作用物質がT細胞のサブセットに特異的に影響を与える場合、(例えば、T細胞の特異的サブセットのみを標的にする運搬媒体の使用により)、T細胞の特異的サブセットの精製は必要ではない。作用物質が細胞の特異的サブセットを標的とするようにさせる媒体は、リポソームまたは所望の細胞タイプを認識する分子が結合した組換えウイルス粒子を含む。このような分子には、表面分子に対する抗体または受容体に対するリガンドがある。T細胞の選択的サブセットのみを、例えばCD4+T細胞のみを標的としたい場合、および使用する作用物質がこのT細胞のサブセットを選択的に標的とできない場合、細胞を作用物質と接触させる前にT細胞の特異的サブセットを単離する必要があり得る。
【0111】
被験体から得るT細胞のような末梢血細胞またはその精製サブセットを、次いで、インビトロで、T細胞内のbcl−XLタンパク質レベルを調節する作用物質の存在下にインキュベーションする。作用物質の量は、作用物質のタイプ、所望の効果および作用物質と接触させる細胞の集団などの種々の要因によって変わる。細胞集団に添加すべき作用物質の適当な量は、作用物質の種々の量を細胞培養物に添加し、種々の時間でのbcl−XLタンパク質の量を、本明細書に記載のようにウエスタンブロット分析により測定するアッセイを行うことにより決定できる。細胞の異種集団を作用物質と接触させる場合、細胞をウエスタンブロット分析にかける前に、bcl−XLの修飾が望まれるT細胞のサブセットを最初に単離することが必要であり得る。T細胞の特異的サブセットは、上記のようにネガティブセレクションにより細胞集団から単離できるか、またはあるいは、T細胞の特異的サブセットをFACSを使用して単離できる。
【0112】
本発明の具体的態様において、被験体から単離されたT細胞を、bcl−XLのタンパク質レベルを修飾する作用物質と接触させ、更に、インビトロでT細胞の集団を拡張する(即ち、集団内のT細胞の数を増加させる)ために培養する。T細胞の集団のインビトロ拡張は、公開PCT出願番号WO94/29436に記載され、その内容は本明細書に参考として包含させる。
【0113】
T細胞またはそのサブセット内のbcl−XLタンパク質レベルの修飾に続いて、細胞を被験体に再投与できる。具体的態様では、まず細胞を精製して、被験体への投与に望ましくない培養培地中の物質を除去する。精製は、例えば、フィコールハイパーク勾配遠心により行うことができる。
【0114】
あるいは、本発明の方法はインビボで実施できる。この態様において、T細胞内のbcl−XLタンパク質レベルを修飾する作用物質は、薬学的に許容可能な媒体中で、所望の治療効果を得るのに充分な量で被験体に投与する。作用物質は、本発明の方法により処置すべき症状タイプによって、局所的または全身的に投与する。作用物質の投与に適当な媒体および経路は、本明細書3項に記載している。
【0115】
4.1.本発明の方法のT細胞死からの防御が有利な状態への適用
本発明の方法は、T細胞の細胞死を防止するのに有用である。本発明の方法により、全てのT細胞で細胞死が防止予防できるか、またはあるいは、細胞死をT細胞の特異的サブセット中で選択的に防止できる。更に、T細胞を、長期間、または短期間、細胞死から防御できる。局所的に存在するT細胞または全身的に(例えば、末梢血に)存在するT細胞を、細胞死から防御できる。
【0116】
好ましい態様において、本発明の方法は、HIV感染個体のCD4+T細胞の細胞死を防止し、HIV感染からT細胞を防御するのに使用する。HIV感染中、ウイルスはCD4+T細胞に感染し、これを殺傷する。従って、個体中のCD4+T細胞の数は、被験体の微生物からの感染を防止するのに不十分な量まで段々に減少する。bcl−XLタンパク質レベルが増加しているT細胞は、T細胞の生存率増加を測定したところ、HIV感染に対して著しい防御を示すことが観察される(実施例9参照)。従って、T細胞内のbcl−XLタンパク質レベル増加は、T細胞をHIV誘導細胞死から防御する。従って、T細胞内のbcl−XL過発現は、HIV感染被験体におけるCD4+T細胞の数を維持する有効な方法を提供する。更に、本発明の方法は、HIV感染被験体のCD4+T細胞の数の増加および/またはHIV感染個体におけるT細胞枯渇の速度減少をもたらし得る。
【0117】
本発明の方法は、HIV感染、またはT細胞を細胞死に対して感受性にする他の感染物の感染ような、T細胞関連疾患を有する個体由来のT細胞の集団の拡張を可能にする方法を提供する。従って、この方法は、インビトロで細胞を拡張する方法を提供する。この場合、拡張T細胞集団を、被験体に投与し戻すことができる。好ましい態様において、T細胞を、T細胞に一次活性化シグナルを提供する作用物質およびT細胞にコスティミュラトリー・シグナルを提供する作用物質の組み合わせで刺激する。より好ましい態様において、T細胞をT細胞上のCD28と相互作用する作用物質およびT細胞受容体を介してbcl−XLタンパク質レベルが増加され、細胞が細胞死から防御されるようにT細胞を刺激する作用物質の組み合わせと共に培養する。
【0118】
HIV感染被験体のT細胞死を防止する本発明の具体的実施態様では、末梢血細胞を被験体から入手し、インビトロで修飾して、CD4+T細胞中のbcl−XLタンパク質の量を増加させ、個体に再投与する。本発明の好ましい態様では、bcl−XLタンパク質をコードする核酸分子をT細胞に挿入することにより、被験体由来のCD4+T細胞を修飾して、bcl−XLタンパク質レベルを増加するようにする。より更に好ましい態様では、bcl−XLをコードする核酸分子は、細胞をウイルス粒子で感染させるような方法により、T細胞に導入されるウイルスベクター内に含有される。方法の他の好ましい態様は、CD4+T細胞を、被験体に再投与する前に培養で更に拡張する。従って、本発明の方法は、HIVに感染した被験体の免疫系をCD4+T細胞で再集合させ、同時にこれらのCD4+T細胞をウイルスによる感染に対して耐性にすることを可能にする。
【0119】
被験体に、個体のCD4+T細胞内のbcl−XLタンパク質のレベルを増加させる作用物質を投与することにより、HIVに感染した被験体のCD4+T細胞を、細胞死から防御することもまた可能である。この作用物質は、生理学的に許容される媒体に含まれたbcl−XLタンパク質をコードする核酸分子であり得る。本発明のより好ましい態様において、媒体を更に核酸がCD4+T細胞を標的とするように操作する。あるいは、作用物質は、CD4+T細胞のbcl−XLタンパク質レベルを増加させるように細胞内に作用する作用物質である。
【0120】
他の態様において、HIVで感染させた被験体のT細胞を、上記のエクスビボおよびインビボ法を組み合わせた方法により細胞死から防御する。本方法は、個体のCD4+T細胞を、インビボおよび/またはエクスビボでbcl−XLタンパク質レベルを増加する幾つかの作用物質と接触させることを含む。例えば、bcl−XLタンパク質をコードする核酸をT細胞に導入し、T細胞を更に内生bcl−XLタンパク質レベルを増加するために細胞内に作用する作用物質と接触させる。
【0121】
あるいは、本発明の方法は、より迅速な感染の除去のために、免疫応答を強化するのに有用である。従って、本発明の具体的態様は、活性化T細胞をT細胞内のbcl−XLタンパク質レベルを増加させる作用物質と接触させて、細胞死から防御する。T細胞の活性化に続き、CT4+T細胞を細胞死から防御することにより、ヘルパーT細胞が、Tヘルパー細胞が通常提供し得る以上の有効な“ヘルプ”を提供できる。同様に、延長寿命を有するCD8+T細胞は、通常の寿命のCD8+T細胞よりも標的細胞を溶解できる。本発明の範囲内の方法は、全身感染および局所感染を処置する方法である。従って、作用物質を全身または局所的に投与できる。本発明の好ましい態様において、T細胞内のbcl−XLタンパク質レベルを増加する作用物質は、bcl−XLタンパク質レベルを増加するために細胞内に作用する作用物質である。本発明のより更に好ましい態様において、作用物質は、活性化T細胞の寿命が必要以上に(例えば、活性化T細胞が記憶T細胞になるかまたは感染の除去後に死滅する)長くならないような短い半減期を有する作用物質である。
【0122】
4.2.T細胞死に対する感受性増大が有利な状態への本発明方法の応用
本発明の方法はまた、T細胞の細胞死に対する感受性の増大にも有用である。従って、通常、内生bcl−XLタンパク質レベル増加により細胞死から防御される状態に遭遇するT細胞は、T細胞内のbcl−XLタンパク質のレベルが著しく減少すれば死滅する。本発明の方法は、T細胞関連疾患の被験体の処置に有用である。“T細胞関連疾患”は、延長寿命を有するT細胞に関連する疾患を意味する。
【0123】
具体的実施態様において、本発明の方法は、被験者の自己免疫疾患の処置にも有用である。細胞死に対する感受性を自己反応性T細胞中で増大させ、自己免疫疾患の作用を改善する。処置し得る自己免疫疾患の例は、多発性硬化症、インシュリン依存性糖尿病、関節炎(例えば、リウマチ性関節炎、若年性リウマチ性関節炎、骨関節炎)、ミエスシェニア・グラビス(myesthenia gravis)、心筋炎、ギラン・バレー症候群、全身性紅斑狼瘡、自己免疫甲状腺炎、皮膚炎、乾癬、ショーグレン症候群、円形脱毛症、クーロン病、アフタ性潰瘍、虹彩炎、結膜炎、角結膜炎、潰瘍性大腸炎、アレルギー、皮膚性紅斑狼瘡、硬皮症、膣炎、直腸肛門炎、薬疹、癩反転反応、癩性結節性紅斑、自己免疫葡萄膜炎、アレルギー性脳脊髄炎、急性壊死性出血性脳疾患、特発性両側性進行性感音難聴、形成不全貧血、赤芽球癆、特発性血小板減少症、多発性軟骨炎、ウェゲナー肉芽腫症、慢性活動性肝炎、スティーブン・ジョンソン症候群、特発性スプルー、偏平苔癬、グレーブス眼病、結節症、原発性胆汁性肝硬変、後部葡萄膜炎および肺内部線維症を含む。
【0124】
他の態様において、本発明の方法は、移植片拒絶反応の低減に使用する。例えば、移植細胞を、移植片に反応性のT細胞を細胞死に対して感受性にするように、T細胞内のbcl−XLタンパク質レベルを減少する作用物質と共に宿主に投与する。この方法は、宿主に、CTLA4Igのような共刺激を遮断する作用物質を投与することも含み得る。同様に、本発明の方法は、移植片対宿主病の予防にも使用できる。好ましい方法において、ドナー骨髄を移植前に宿主細胞およびT細胞内のbcl−XLタンパクを減少する作用物質と接触させる。こうして、ドナー骨髄のT細胞を細胞死に対して感受性にし、共刺激を阻害する作用物質と併用することで宿主特異的抗原に反応性のT細胞の細胞死を増大できる。
【0125】
以下は、本発明を更に下記実施例により説明するものであり、これは更に限定するものと見なすべきではない。本出願明細書に引用の全ての参考文献、継続特許出願および公開特許は、参考として本明細書に明示包含する。
【実施例】
【0126】
実施例1: 活性化がγ−照射後のT細胞の生存を高める
T細胞生存に対するT細胞活性化経路の効果を調べるために、休止T細胞をヒト末梢血から前から知られているネガティブセレクション法(June,C.H.et.al.(1987)Mol.Cell.Biol.7:4472−4481)により、分離した。簡単に言うと、細胞を抗体の反応混液の対象とし、休止CD28−ポジティブT細胞以外のすべての細胞を除去する。ウシ胎児血清(10%)、L−グルタミン(2mM)ペニシリン/ストレプトマイシン(100U/ml,100μg/ml)およびHEPES(20mM)を補充したRPMI 1640中でT細胞を培養した。細胞は、活性化または照射の前に一夜置いた。
【0127】
CD28+T細胞は、CD28特異モノクローナル抗体により提供される共刺激の存在または不在下に、培地のみで培養するか、あるいはTCR/CD3複合体を12時間架橋することにより刺激した。T細胞の架橋はプレート固定化抗−CD3(G19.4〔1μg/mlで〕)を用いて行い、T細胞の共刺激はCD28に対する可溶性抗体(モノクローナル抗体(mAb)9.3)により1μg/mlで実施された。これらの各グループからの細胞はグループに分けた。1つのグループはさらに操作を行わないで培養し、他のグループはセシウム源γ−照射(J.L.Shepherd Inc.)による15Gyのγ照射を行った。照射後、細胞は96ウエル培養皿(Costar)中に100,000細胞/ウエルで入れ、細胞の生存能力および全数をトリパンブルー排除法およびプロピジウムヨージド排除法により調べた。
【0128】
プロピジウムヨージド排除法について、2×105細胞の2つの分離サンプルを1%BSAおよび0.01%ナトリウムアジドを補充したPBS 0.5mlに入れて、再懸濁した。プロピリジウムヨージド2ml(0.5mg/ml)を細胞に加え、サンプルをFACSortおよびLysis IIソフトウエアを用いたFACSで分析した。生存能力パーセントは、プロピジウムヨージドを排除した細胞数を全細胞数で割ることにより求めた。解析による破片を除くために、生存細胞の前進光散布特性を利用した。
【0129】
結果を図1に図示する。全3集団の細胞が4日以上の培養期間において高い生存能力を保持した(図1、パネルA)。休止および活性化T細胞の生存もγ−照射後に調べた(図1、パネルB)。γ−照射誘発アポトーシスは、特殊なレセプターリガンド干渉を必要とせず、特殊レセプターレベルについてのコントロールの必要性なしに細胞の生存に細胞の活性化がどのように影響するのかを調べる手段とし得る。これらのアッセイに用いた照射量(15Gy)は、実際に全群のすべての細胞に致命的なDNA損傷を与えるのに充分である。従って、細胞が確保された日数を越えて死ぬ率は、DNA障害に対する応答において被ったPCDに抵抗する能力を測定するのに用いられる。
【0130】
図1、パネルBに示されるように、照射後、休止T細胞の生存能力は、刺激されたリンパ球のいかなる集団にみられるよりも、非常に速く低下した。照射4日後、いずれかの活性化集団におけるT細胞は、休止集団の細胞よりも統計的により生存し得た(p<0.02)。CD28レセプターによって共刺激された細胞は、抗−CD3のみで刺激された細胞よりも、生存能力の僅かな、しかし再現性ある上昇を示した。抗−CD3刺激および抗−CD3+抗−CD28刺激細胞における細胞生存性が保持されることは、生存能力アッセイに平行してなされた細胞数の測定が細胞の絶対数の変化を示さなかったから、後のT細胞増殖の結果ではなかった。さらに、活性化された集団における全細胞は、遅G1またはG2のいずれかにおける細胞周期内に止まった。3母集団における細胞死は、細胞が最初に円鋸歯状になり、次いで核の凝縮およびDNAフラグメンテーションをおこす典型的なアポトーシス性パターンに続いておきる。
【0131】
このように、本実施例は、T細胞の活性化がγ−照射後のT細胞生存を高めることを示す。
【0132】
実施例2: CD28共刺激は抗−CD3活性化T細胞の生存能力を増加する
CD28共刺激の存在または不在により刺激された細胞間における一つの相違は、これらの細胞により産生されたリンホカインのレベルである(Lindsten,T.et.al.(1989)Since244:339−343)。従来から、生長因子が多様な細胞タイプにおける細胞生存の外的調節で重要な役割を果すことが示唆されている(Groux,H.et.al.(1993)Eur.J.Immunol.23:1623−1629),Nunez,G.et.al.(1990)J.Immunol.144:3602−3610)。生長因子が共刺激の保護作用の因であるかどうかを決めるために、次の実施例が行われた。
【0133】
CD28+T細胞は、実施例1記載のように、12時間、抗−CD3の存在下にCD28モノクローナル抗体との共刺激の存在または不在で培養された。細胞は、そのならし培地にそのまま残し、洗い、次いで新しい培地に再懸濁するか、あるいは洗って、組換えIL−2(rIL−2)(Boehringer-Mannheim)200μ/mlを補充した新しい培地に再懸濁された。各グループからのアリコートを無操作のままにするか、または12Gyのγ−照射にさらした。細胞生存能力は、実施例1に記載したように、操作後4日間毎日、プロピジウムヨージド排除法により測定した。
【0134】
結果を図2に図示する。図2のパネルAによると、TCR/CD3複合体のみで活性化されたT細胞は、活性化後培養において高い生存能力を保持していた。しかし、刺激細胞を水洗することによるならし培地の除去は、培養中に生存する細胞の能力を著しく低下せしめることにつながった。このインビトロ培養中に生存する能力の低下は、IL−2の添加により逆転され得る。抗−CD3刺激細胞に15Gyのγ−照射がなされたとき(図2、パネルB)、一夜刺激後のならし培地の除去は、IL−2添加により逆転された細胞生存性の激しい低下をもたらした。このことは、生存が抗原レセプター活性化細胞における外的生長因子のレベルによって第一義的に決定されることを示している。従って、活性T細胞が培養中で生存し得る能力は、それがIL−2などの生長因子を生産するオートクリン能力に基づいている。これらのデータは、IL−2がT細胞生存の外的レギュレーターであることを確認する。
【0135】
TCR/CD3複合体のみで刺激されたT細胞についての結果と対照的に、抗−CD3+抗−CD28併用で刺激されたT細胞は内因リンホカインを洗出しても、さらにIL−2を培養上澄液に加えても、その生存に変わりはない(図2、パネルCおよびD)。
【0136】
本実施例の結果は、CD−28仲介共刺激が細胞生存の外的メディエーターの産生に影響を及ぼさないだけでなく、PCDを受けた細胞の内因的感受性を調節する役割を演じる(次の実施例でさらに調べる)ことを表している。
【0137】
実施例3: T細胞活性化中のbcl−2およびbcl−X mRNA発現
PCDを受けたT細胞の内因的感受性に関与する遺伝子の発現を調節するときのCD28の役割について調べるために、休止ヒトT細胞および抗−CD28の存在または不在下のTCR抗原レセプター複合物の架橋により活性化されたT細胞についてbcl−Xおよびbcl−2発現レベルを解析した。
【0138】
RNAは、培地のみで培養されたCD28+T細胞から単離され、次いで1,6および12時間、抗−CD3または抗−CD3と抗−CD28で刺激され、ノーザンブロット・ハイブリダイゼイションにより解析された。RNAは、すでに記述されているように(June,C.H.et.al.(1987)Mol.Cell.Biol.7:4472-4481)、勾配グアニジウム/CsCl2による遠心分離でT細胞から分離した。等量のRNA(非変性アガロースゲル上で電気泳動した28SリボソームRNAのエチジウムブロマイド着色で検定)をアガロース/ホルムアルデヒド変性ゲル上に充填し、大きさによって分離した。ゲルをニトロセルロース(SchleicherおよびSchuell)に移して、真空で2時間80℃に加熱する。ブロットは、42℃6時間Stark液(50%ホルムアミド、5×SSC(1×SSCは0.15M Nacl,0.015Mクエン酸ナトリウム)1×Denhardt液,25mM リン酸ナトリウム,pH6.5,Torula RNA 250μg/ml)中でプレハイブリダイズし、次いでStark液中で10%硫酸デキストランおよび32P−ラベルニック翻訳プローブ(1×106dpm/ml)で一夜42℃ハイブリダイズした。ブロットをプローブし、次いでヒトbcl−XL cDNAおよびネズミbcl−2 cDNAに特異なプローブ(S.Korsmeyer)で剥離した。
【0139】
ノーザンブロット・ハイブリダイゼーションの結果を図3、パネルAに示す。その結果によると、bcl−Xまたはbcl−2 mRNAのいずれも、休止T細胞において検出レベルでは発現されなかった。しかし、両bcl−2およびbcl−X mRNAの発現はT細胞活性化6時間後に誘導される。CD28共刺激はbcl−2 mRNAの発現に顕著な作用を有しなかった。一方CD28共刺激はのbcl−X mRNA発現を高めた。抗−CD3および抗−CD28で刺激された細胞は、レベルが低下し始めた後48時間、bcl−Xの高レベルの発現を維持した。bcl−2機能調節等に負の役割を有すと思われるbcl−2ファミリーの第3ナンバーは、遺伝子baxである。共刺激もbax mRNAレベルに影響を与えなかった。
【0140】
bcl−Xの第1エクソンにおける2つの5'スプライス供与体の代替使用は2つの明白なmRNA種を生成する。bcl−XLはエクソン1の完全コード領域を保持し、細胞の生存を高めるように機能する。一方、エクソン1内の上流スプライス供与体部位の利用は、エクソン1コード領域内で189bp欠失を含むbcl−XS mRNAをもたらす。Bcl−XSタンパク質は、両機能の優性ネガティブレギュレーターとして作用する(Boise,L.H.et.al.(1993)Cell 74:597−608)。bcl−XSおよびbcl−XL mRNAは189bpによってのみ相違するので、RNase保護は、T細胞活性および共刺激においてどのbcl−X mRNAが誘導されたかを決定するために、行われた。
【0141】
Rnase保護検定は、休止細胞あるいは抗−CD3または抗−CD3および抗−CD28で6または12時間刺激されたT細胞から分離されたRNAについて実施された。Rnase保護アッセイは、市販キット(Ambion)の説明書に従って実施された。T細胞RNA 3μgを放射性標識リボプローブでハイブリダイズした。このプローブは、bcl−XL cDNA+プラスミド中のpBluesript SKをAccIで切断し、Promegaからのキットを用いてプロモータよりインビトロで転写体を生成せしめて、つくられた。336ヌクレオチド・リボプローブはゲル電気泳動により精製され、抽出され、次いで、RNaseAおよびRNaseT1の反応混液の添加前に16時間42℃でT細胞RNAにハイブリダイズされた。bcl−XL mRNAによるプローブの保護は264ヌクレオチドのフラグメントを生成し、bcl−XSメッセージへのハイブリダイゼイションは放射性標識プローブの163ヌクレオチドフラグメントを保護する。保護産物を5%アクリルアミド/7M尿素シーフェンス・ゲル上で分離した。末端ラベルpBluescript SKII+HpaII消化はマーカーとして用いた。ゲルを乾燥し、XAR−5フィルム(Kodak)に感光した。
【0142】
Rnase保護アッセイの結果を図3パネルBに示す。その結果によると、CD28共刺激により調節が上昇された主なbcl−X mRNAは、bcl−XLである。小さい誘導もbcl−XSのレベル上でみられる。
【0143】
そのように、本実施例の結果は、CD28による活性T細胞の共刺激がbcl−XL mRNAに顕著な増加をもたらすことを示している。
【0144】
実施例4: T細胞がBcl−2タンパク質の構成的発現を発揮する
bcl−2 mRNAのレベルがbcl−2タンパク質のレベルとあまり相関しないことは前に報告されているので(Chleq-Dechamps,C.M.et.al.(1993)Blood,81:293−298)、抗−CD28で共刺激されるか、またはされない活性T細胞中のbcl−2タンパク質の量を測定した。
【0145】
この実施例において、CD28+T細胞は上記のように抗−CD3または抗−CD3および抗−CD28抗体と共に0、6、12または24時間培養され、bc−2タンパク質レベルが免疫沈降法およびウエスタンブロット法により測定された。各時点で、各培養条件の細胞4×107が分離され、1.0ml NET−N(100mM NaCl,1mM EDTA 20mM Tris,pH8.0,0.2%NP−40)中に溶解された。核および細片は2分間4℃の微遠心で除去された。上澄液を30分間パンソルビン(Calbiochem)50mlで処理した。遠心でパンソルビンを除いた後、上澄液を2つの等量(450μl)に分け、それに抗−bcl−Xウサギ血清(実施例5参照)1μgまたはハムスター抗−Bcl−2モノクローナル6C8(Hockenbery,D,et.al.(1990)Nature348:334−336)2.5μgのいずれかを加えた。溶解生成物を4℃で1時間振動し、次いで30分間でタンパク質Aアガロース(Gibco BRL)25μlを加えた。免疫沈降物を採取し、NET−Nで2回、NETで1回洗った。2×SDS充填緩衝液100μlを加え、素早く凍らせ、ゲル電気泳動を行うまで−20℃に保持した。
【0146】
免疫沈降物を煮沸し、15%ゲルでSDS−PAGEにかけた。ゲルは、200mAで3時間、BioRad移送器を用いて電気ブロットによりニトロセルロースに移した。ブロットを一夜4℃で5%非脂肪ミルク/0.2%ツイン20中でブロックし、上記のブロット液中で精製6C8 mAb(1:200)とハイブリダイズした。ウエスタン・ブロットをHyperfilm(Amersham)を有するECLシステム(Amersham)を用いて展開した。
【0147】
ウエスタン・ブロットの結果を図4に示す。休止末梢血T細胞がBcl−2タンパク質を高レベルで発現することが分かる。さらに、Bcl−2タンパク質のレベルがTCR/CD3レセプターの抗−CD3抗体での架橋後の最初の24時間以後あまり変動しない。CD28レセプターでの共刺激もBcl−2タンパク質レベルに影響を与えないようであった。
【0148】
本実施例は、休止T細胞がBcl−2タンパク質を発現し、共刺激あり、または、なしのT細胞レセプターでのT細胞刺激がBcl−2タンパク質レベルを上昇しないことを示す。
【0149】
実施例5:CD28共刺激はbcl−X1タンパク質発現を高める
本実施例において、休止T細胞およびCD282の共刺激の存在または不在でT細胞レセプターで活性化されたT細胞におけるbcl−X1のタンパク質レベルを解析した。
【0150】
bcl−X1のタンパク質レベルは、Bcl−2のタンパク質レベルの測定と平行して、実施例4に記載されたアッセイ法によって測定された。あらかじめ清澄化した後、上澄液を2等量(450μl)に分け、抗−bcl−Xウサギ血清1μlをこれらのアリコートの1つに加えた。タンパク質レベルは、モノクローナル抗−bcl−X抗体2A1(1:1希釈)を用いて実施例4のように測定した。
【0151】
bcl−Xに対するポリクローナルおよびモノクローナル抗体を次のように製造した。bcl−XSのオープン・リーディング・フレーム(転写解読枠)をプライマー(5'−GGA GAT ATA CAT ATG TCT CAG AGC AAC CGG GAG CTG GTG−3’および5'−CGG GAT CCC GTC ATT TCC GAC TGA AGA GTG AGC CCA GCA G−3’)でのPCRで増幅し、pET−3bのNdeIおよびBamHI部位(Novagen)にクローン化した。組換えタンパク質を0.4mM IPTGでの誘導によりBL21細胞中で製造した。このタンパク質は、細菌溶解の不可溶フラクション中にあると測定され、次のように部分的に精製された。不可溶フラクションを2M尿素で洗い、6Mグアニジン−HClで安定化した。タンパク質は、3Mおよび1Mグアニジン−HClによる段階透析により再生し、次いでpH8.3で500mM KClでPBSに対する透析を行った。タンパク質濃度は、市販キット(BioRad)を用いたBradfordアッセイ法により測定し、タンパク質純度はSDS−PAGEおよびコーマシックブルー着色法によりアッセイした。
【0152】
ウサギポリクローナルの抗体の製造のために、組換えタンパク質1mgを完全フロイント・アジュバント(CFA)に懸濁し、ウサギの脊中の数箇所に皮下注射した。不完全フロイント・アジュバント中のタンパク質200μgで追加免疫した。インビトロ翻訳タンパク質およびトランスフェクトション細胞系列を用いて、組換えタンパク質に対して、および特異的免疫沈降についてのウエスタンブロット法で血清を選別した。
【0153】
モノクローナル抗体の製造のために、CFA中に乳化したポリアクリルアミドゲル中の組換えbcl−XSを後足の皮下および腹腔内投与して、BALB/Cマウスを免疫化した。マウスの免疫は、不完全フロイント・アジュバント中に乳化した可溶性のアクリルアミド基底ゲル結合タンパク質の混合物で、30日の間隔をおいて2度、追加免疫した。最後の追加免疫から3日後に、脾臓およびリンパ節細胞を採取し、標準的技術(Kearney,J.F.(1984)In Fundamental Immunology,W.E.Paul,ed.,Raven Press,NY,751-766)を用いて、P3X63−Ag8.653ミエローマ細胞と融合せしめた。融合15日後、ハイブリドーマ上澄液について、ホウ酸塩緩衝液中(pH8.4)5μg/mlで組換えbcl−XSによりコートされたウエルにおいてELISAによる活性を調べた。ELISAに陽性であったハイブリドーマをbcl−XLまたはbcl−2でトランスフェクションさせたFL5.12に対するウエスタンブロットにより選別した。陽性ハイブリドーマ系は、制限希釈によりクローンし、再選別し、腹水製造のためにプリスタン−初回免疫BALB/Cマウスに注射した。
【0154】
bcl−Xのタンパク質レベルを表すウエスタンブロット解析の結果を図4に示す。bcl−Xのタンパク質レベルは、T細胞活性化のレベルによって非常に変化する。検出可能なbcl−Xタンパク質産物は休止末梢T細胞において測定されなかった。ウエスタンブロットで抗体により認識されたタンパク質は、対応cDNAでトランスフェクトされた細胞中に合成されるbcl−XLと同様にインビトロ翻訳bcl−XLが約29kDaであるので(Boise,L.H.et al.(1993)Cell 74:597-608)、bcl−XLに対応し、bcl−XSに対応していない。
【0155】
図4はさらに、休止T細胞の抗−CD3刺激が、刺激6時間後に最初にみられ、次いで分析の24時間を通して細胞中に蓄積する検出可能なbcl−XLタンパク質の発現を誘発することを表している。さらに抗−CD3刺激細胞のCD28共刺激は、bcl−XLの発現を有意に高めた。bcl−XSではいかなる時点でもみられなかった。
【0156】
他の例では、休止T細胞は、培地のみ、抗−CD28,抗−CD3,抗−CD3,および抗−CD28または抗−CD3およびIL−2(100μ/ml)により24時間で活性化された。細胞質抽出物を記載のように調製し、タンパク質100μgをSDS−PAGEおよびポリクローナルbcl−X血清と共にウエスタンブロット分析に供した。
【0157】
その結果を図5に示すが、休止T細胞および抗−CD28でインキュベートしたT細胞は、bcl−XLタンパク質を発現しなかった。しかし、抗−CD3刺激細胞のCD28共刺激は、bcl−XLタンパク質の発現を有意に高めた。24時間点の抗−CD3+抗−CD28刺激細胞におけるbcl−XL発現レベルは、脾臓ホーカス形成ウイルス長期リピートのコントロール下で、bcl−XLの発現がなされている安定なトランスフェクタントのレベルと同じであった(図5の最終欄)。
【0158】
CD28共刺激は、早ければ刺激6時間後に観測され、24時間培養中継続するbcl−XLタンパク質の蓄積を高めた(図4および5)。一方、抗−CD3およびIL−2(100units/ml)での細胞処理は、抗−CD3のみで処理されたレベル以上にはbcl−XL発現をさらに高めなかった(図5)。さらに、CD2,CD5,CD11a,CD18に対するまたはMHCクラスIに対するモノクローナル抗体で抗−CD3−活性T細胞を処理しても、bcl−XLレベルは抗−CD3のみで処理した細胞以上には高まらない。
【0159】
これらの結果は、休止T細胞がbcl−Xタンパク質を発現せず、抗−CD3で活性化されたT細胞がbcl−Xタンパク質を発現し、抗−CD3で活性化され、および抗−CD28で共刺激されたT細胞がより顕著にbcl−Xタンパク質を発現することを示している。
【0160】
実施例6:bcl−XLはJurkat T細胞におけるFas−および抗−CD3誘導PCDを防止する
T細胞系に対するFas架橋はアポトーシスの速い誘導をもたらす。しかし、正常T細胞は、長い時間がかかって活性化されるまでは、Fas−架橋に対する応答において細胞死に対し感受性にならない(Klas,C.et al.(1993)Int. Immunol. 5:625-630)。細胞は迅速に誘導されて、抗−CD3+抗−CD28による活性化の24時間中に細胞表面に高レベルのFasを発現する。Fasレベルは、培養中、次の数日間一定である。これにもかかわらず、アポトーシスを誘導するFas−架橋の能力は、刺激72時間後まで明らかでなく、次いで培養中次の数日間以上でより効果的に増大する。本例は、抗−CD3および抗−CD28で刺激されたT細胞におけるbcl−XLタンパク質レベルがFas誘導細胞死に対する抵抗性と相関することを表している。
【0161】
T細胞は、120時間かけて培地のみまたは抗−CD3および抗−CD28で活性化された。細胞質溶解産物が調製され、上記のようにポリクローナル抗−bcl−XL抗体で着色したウエスタンブロット法で分析された。同時に、抗−CD3および抗−CD28刺激T細胞を0.5μg/ml CH−11抗−Fasモノクローナル抗体(Panvera)またはアイソタイプ・コントロール(IgM)で120時間の24時間の間隔で24時間、処理した。生存性を上記のようにプロピジウムヨージド排除法により調べた。Fas表面発現は、30分間のFITC共役抗マウスIgM(Sigma)に続けて、30分間CH−11で着色することにより、確認された。着色細胞は、LysisIIソフトウェアを用いた流細胞計算(FACSort, Becton-Dickinson)により分析した。
【0162】
抗Fas抗体で架橋したT細胞のbcl−XLタンパク質レベルよび生存性を図6に示す。抗−CD3+抗−CD28共刺激後、T細胞のbcl−XLタンパク質レベルのピークは24−48時間にあって、その後徐々に低下した。図の下半分に示すように、抗−CD3および抗−CD28で24または48時間処理され、次いでFasで架橋された細胞は、細胞死に対して完全に耐性である。しかし、抗−CD3および抗−CD28での刺激後のかなり遅い時点で、抗−Fas抗体でT細胞が架橋されたときは、細胞生存能力が低下した。このようにbcl−XLの存在はFas誘導細胞死に対するT細胞の防御と相関する。
【0163】
bcl−XLタンパク質がFas−架橋誘導細胞死に対する耐性を誘導することをさらに明らかにするために、Jurkat細胞をbcl−XLコードする発現ベクターでトランスフェクトし、細胞生存能力を抗Fas抗体を加えて測定した。
【0164】
Jurkat細胞は上記のように培地中に保持し、pSFFVNeo−bcl−XL(Boise,L.H.et.al.(1993)Cell 74:597−608)またはGene PulserでのエレクトロポレーションによるpSFFVNeo(BioRad)で250Vおよび960μFにおいてトランスフェクションした。トランスフェクション体をG418(Sigma)で1mg/mlにおいて選択し、いくつかの独立クローンを制限希釈によって分離した。ウエスタン・ブロット分析は、高いbcl−XLタンパク質レベルがbcl−XL発現ベクターでトランスフェクションさせたクローン中に存在すること、および対照ベクター(Neoクローン)でトランスフェクションさせたクローン(Neoクローン)がbcl−XLタンパク質を発現しないことを表している(図7、パネルC)。bcl−XLトランスフェクション体中のbcl−XLレベルは、共刺激24時間にT細胞中の発現レベル匹敵した(参照、例えば図5)。クローン上のFas発現は上記した着色によって確認された。
【0165】
表面で比較可能レベルのFasを発現した3つのbcl−XLおよび3つのNeoは、培地中2.5×105細胞/mlでインキュベートされ、10ng/mlのFas抗体またはアイソタイプ適合対照で処理され、次いで生存性がプロピジウムヨージド排除法により調べられた。Jurkat細胞クローンの生存能力を図7、パネルAに示す。グラフから、Fasが全3対照トランスフェクション体に迅速な細胞死(48時間で10%の生存能力)をもたらし、一方、3bcl−XLトランスフェクション体は実験中を通じて60%以上の生存能力であることが分かる。neo−およびbcl−XL−トランスフェクション体のいずれも、抗体もあるいはアイソタイプ適合対照mAb(IgM)が用いられなかったときは、生存能力が低下することを示している。このようにbcl−XLはJurkat細胞におけるFas−誘導細胞死を実質的に阻止する。
【0166】
抗−CD3−架橋がT細胞クローンおよび細胞ラインにおいてアポトーシスを誘導することは報告されている(Shi,Y.et.al.(1989) Nature339:625−626),Ucker,D.S.et.al.(1989)J.Immunol.143:3461−3469)。TCR誘導細胞死を防止するbcl−XLの能力を検討するために、Jurkatトランスフェクション体を抗−CD3で刺激し、その生存を調べた。抗−CD3誘導細胞死は1μg/mlでプレート結合抗−CD3(OKT3)でもって行われ、プロピジウムヨージド排除法により毎日、調べられた。
【0167】
結果を図7、パネルBに図示する。84時間で抗−CD3処理は、Neoクローン培養において30−60%の細胞死をもたらした。一方、抗−CD3誘導細胞死は、bcl−XLの存在によってほとんど完全に阻止された(図7、パネルB)。
【0168】
このようにbcl−XLタンパク質は、Fas−およびT細胞レセプター架橋誘導細胞死からT細胞を保護することができる。
【0169】
実施例7:bcl−XLはIL−2とは無関係に機能してT細胞生存を強化し得る
TCR/CD3複合体の架橋により活性化されたT細胞の生存に対するIL−2の効果のアッセイに基づくと、IL−2は抗原活性化T細胞の生存能力の維持における生存因子として機能し得るようである。CD28共刺激は、上清中の成長因子レベルとは無関係にT細胞の生存をもたらすようであるが、CD28刺激化細胞の生存強化は、単にCD28共刺激により産生された高いレベルのIL−2の結果だけではないということを除外することは難しい。CD28共刺激細胞中で産生された高いレベルのIL−2があると、充分に洗浄してもリンホカインの効果を完全に除くことは不可能である。CD28−共刺激細胞により産生された高いレベルのリンホカインは、培養上清中に検知可能なリンホカインの蓄積がなくても、オートクリン様式で細胞に影響を与えることができる場合もある。本実施例は、bcl−XLタンパク質は、IL−2依存性T細胞系がIL−2使用中止によリPCDを受けるのを防御し得ることを実証する。
【0170】
CTLL−2は、培養中での生存および増殖がIL−2に依存しているT細胞系である(Gillis,S.およびSmith,K.A.(1977)Nature 268:154-156)。これらの細胞は、それら自身のIL−2を分泌するように誘導され得ないので、IL−2のバイオアッセイにしばしば使用される。CTLL−2細胞はβ−メルカプトエタノール(50μM)および組換えIL−2(100単位/ml)を加えて、上記成長培地中で維持した(Nunez,G.et al.(1990)J.Immunol.144:3602-3610)。細胞を、250Vおよび960μFにて、ジーンパルサー(Gene Pulser、Bio Rad)を用いた電気処理により発現ベクターpSFFVNeo−bcl−XL(Boise,L.H.et al.(1993)Cell 74:597-608)を用いてトランスフェクションさせた。トランスフェクション体を、250μg/mlG418で選別し、bcl−XL陽性クローンをウエスタンブロット解析により同定した。CTLL−2細胞(2.5×106)を50mlNET−Nに溶解し、核および残骸を上記のように除去した。50μlの2×SDSローディング緩衝液を上清に加え、SDS−PAGEおよびウエスタンブロットを上記のように行った。
【0171】
高いレベルのbcl−XLタンパク質を発現する2つのbcl−XLクローン(図7パネルE)をIL−2の不在下、培養液中での生存能力について試験した。これらのクローンは、抗−CD3+抗−CD28活性化T細胞と似たレベルのbcl−XLを発現した。bcl−XL発現細胞を欠乏の1日前に3×105細胞/mlの新鮮培地中で培養した。ついで細胞をIL−2を含まない新鮮培地中で3回洗浄し、同培地に再懸濁した。指定された日にプロピジウムヨージドを排除することにより生存能力を測定した。結果は、図7のパネルDにグラフで示す。親細胞系と比較した場合に比べて、IL−2の不在下で両方のbcl−XLトランスフェクションクローンの生存が強化された(図7、パネルD)。このように、bcl−XLはIL−2の不在下でT細胞の生存を強化するように機能し得る。またこれらのクローンについて、図2で示されたアッセイと似たアッセイ系において、照射誘導死に関する試験を行った。IL−2の不在下または存在下のいずれかで試験した対照細胞と比較した場合、bcl−XLを発現するクローンは、照射誘導死から顕著に防御されることを示した。このように、本実施例は、bcl−XLタンパク質は、T細胞をIL−2欠乏により誘導された細胞死から防御することを実証する。
【0172】
実施例8:CD28共刺激によるbcl−XLの発現は進化的に保存されている
bcl−XL発現を調節するCD28共刺激の能力は、マウスおよびヒトの間で保存されているかどうかを決定するために、ネズミリンパ節から単離した細胞を、可溶性抗−CD3を用いて活性化し、bcl−XL発現を測定した。
【0173】
リンパ節をC57BL/6マウス(Jackson laboratories)から採取し、単一の細胞懸濁液をナイロン網を通過させて調製した。細胞を、10%ウシ胎児血清アルブミン、ペニシリン(100U/ml)、ストレプトマイシン(100U/ml)、10mMHEPES、50mMβ−MEおよび0.1mM非必須アミノ酸を追加したDMEM(GIBCO BRL)からなる完全培地中2×106/mlで培養した。可溶性抗−CD3(145−2C11、10μg/ml J.Bluestoneから)を単独、または抗−CD28(10μg/ml)またはCTLA4Ig(100μg/ml、Repligenから)との組み合わせで加えた。培養液を24時間で37℃、7%CO2でインキューベートし、ウエスタンブロット解析用に採取した。ネズミリンパ球(5×106)を、50μlNET−Nで溶解し、核および残骸を上記のように除去した。50μlの2×SDSローディング緩衝液を上清に加え、SDS−PAGEおよびウエスタンブロットを上記のように行った。生存能力を研究するために、細胞を上記のように処理した。活性化後のT細胞の生存能力を、Thy−1陽性細胞からプロピジウムヨージドを排除することにより測定した。
【0174】
結果は、図8に示されているように、ヒトT細胞の場合と同じように、休止ネズミリンパ球は全く検知可能なbcl−XLを発現しないことを示す。共刺激シグナルを伝達するためにアクセサリー細胞が存在する場合、24時間の可溶性抗−CD3処置により、T細胞にbcl−XL発現を誘導した。このbcl−XLのレベルは、この共刺激シグナルが抗−CD28の添加により増幅される場合に上昇した。対照的に、アクセサリー細胞の存在下における可溶性抗−CD3のbcl−XL発現をアップレギュレーションする能力は、CD28リガンドB7−1およびB7−2がCD28と相互作用するのを競争的に阻害することによりCD28の共刺激を妨げる試薬である、CTLA4Igを加えることによりほぼ完全に阻害される。これらのデータは、マウスT細胞におけるbcl−XL発現は細胞が活性化される間に誘導されることを実証する。さらに、抗CD3の副次的な細胞分裂誘導性用量において、bcl−XL発現の誘導はほぼ完全にCD28共刺激に依存する。この発現パターンは、bcl−XLがマウスの活性化T細胞生存において役割を果していることを実証する。この可能性と合致して、抗−CD3のみで刺激した未分離のリンパ節細胞集合由来のThy−1陽性細胞の生存能力は、72時間後に62%であった。対照的に、抗−CD28の添加により、Thy−1陽性細胞の生存能力は88%に上昇した。逆に、CTLA4Igを用いてリンパ節細胞内のCD28/B7仲介共刺激を阻害することにより、活性化T細胞の生存能力は72時間で20%に減少する。
【0175】
このように、本実施例は、bcl−XLはヒトT細胞と同様に、マウスT細胞においても同じ発現パターンを有すことが示され、すなわち、bcl−XLタンパク質は休止T細胞に存在せず、bcl−XLタンパク質はT細胞受容体の架橋を介して活性化されたT細胞中に発現され、bcl−XLタンパク質レベルはCD28を通した共刺激によりさらに増加することを示す。
【0176】
実施例9:bcl−XL発現はHIV−1−誘導細胞死を妨げ得る
bcl−XL発現はT細胞をHIV−1−誘導細胞死から防御するかどうかを決定するために、上記のpSFFVNeo−bcl−XL(bcl−XL)、またはベクターのみ(Neo)を用いてトランスフェクションさせたJurkat細胞クローンをHIVを用いて感染させ、生存能力を測定した。
【0177】
bcl−XL発現ベクターを用いてトランスフェクションさせた3つのJurkat細胞クローンおよび対照ベクターを用いてトランスフェクションさせた3つのJurkat細胞クローンをHIV−RF単離物の細胞を含まないウイルスストックの様々な希釈液を用いて感染させた。感染の12日後、細胞死をテトラゾリン/ホルマザンアッセイにより定量した(Shearman,M.S.et al.(1994)Proc.Natl.Acad.Sci.USA 91:1470-1474;Hansen,M.B.et al.(1989)J.Immun.Methods 119:203-210 参照)。
【0178】
3つの細胞系の平均生存能力を図9にグラフで示す。1:4から1:256で希釈したウイルスストックを用いて感染させた場合、対照Jurkat細胞において細胞死の割合が高かった。対照的に、bcl−XL遺伝子を発現するトランスフェクションJurkat細胞はこれらの同じウイルス希釈において、生存能力における欠陥は全くなかった。
【0179】
bcl−XL発現の防御効果は、HIV−1感染への耐性に起因するか、または細胞死の誘導における、より特定した効果に起因するかどうかを決定するために、感染細胞の上清中のウイルス量をSpearmanおよびKarberの方法により感染7日後に定量した。Spearman-Karber法は、『Richman D.B.,Johnson V.A.,Mayrs V.L.(1993)抗HIV活性に対する実験作用物質のin vitro評価』に記載されている。Current Protocols in Immunology ch.12.9.,Colligan J.E.et al.、GreeneおよびWiley編、Interscience NY。
【0180】
結果は表1に示す。対照細胞に比較し、bcl−XL発現細胞の上清中のウイルス量は約4倍減少した。
【0181】
【表1】
【0182】
これらの例の結果は、bcl−XLタンパク質は、HIV誘導細胞死からT細胞を防御することを実証する。この効果はHIV感染の間のCD4細胞の減少を防御するのに有益であり得る。本結果は、HIV感染をもつ患者における過剰細胞死の1つの機構は、これらの患者におけるT細胞活性化のある形は非感染個体と匹敵するレベルのbcl−XL発現を誘導できないであろうことを示唆している。このように、HIV感染の治療法の1つは、ex vivoでの細胞活性化および増幅、またはCD28シグナル形質導入のin vivo誘導により、bcl−XL発現の誘導を回復することである。その他に、bcl−XL発現の誘導は、CD28を刺激する作用物質以外の作用物質を用いて刺激することにより達成され得る。これらのデータにより、CD28のbcl−XL発現を亢進する能力は、過剰リンパ細胞死が起こる免疫不全の状態時、特にプログラム化細胞死が末梢血T細胞およびリンパ節T細胞で起こるHIV感染時における臨床用途になり得ることを示す。
【0183】
実施例10:HIV感染個体由来のPBMCの抗−CD3および抗−CD28刺激は、細胞を細胞死から防御するに充分なレベルのbcl−XLタンパク質を増加させる結果となる
本実施例は、PBMCまたはHIV感染個体由来のリンパ球を、抗−CD3および抗−CD28で刺激すると、細胞をアポトーシスから防御するに充分なレベルのbcl−XLタンパク質になるように増加することを実証する。
【0184】
無症候性のHIV感染個体における臨床的免疫不全の経緯は、CD4+T細胞の段階的なしかし進行性の欠失により特徴づけられる。HIV感染患者におけるCD4+T細胞の減少機構は現在のところ知られていないが、最近の証拠により、これらの細胞は活性化シグナルに応答してアポトーシスに対して異常に感受性をもつことが示される。HIV疾患におけるアポトーシス防御タンパク質であるbcl−2およびbcl−xの役割を、HIV感染無症候性個体由来の非刺激アメリカヤマゴボウマイトジェン(PWM)刺激および抗−CD3に抗−CD28を加えた刺激試料において測定した。
【0185】
HIV感染個体由来のPBMCを以下のように得た。ヘパリン化血液サンプルを、インフォームドコンセントの後に、HIV感染(n=25)およびHIV負の対照(n=15)から静脈穿刺により得た。患者を、ウォルターリードステージングシステム(Redfield,R.R.et al.(1986)New Engl.J.Med.314:131-132)に基づく、無症候性初期および中期段階の患者(>500および200−500 CD4細胞/mm3)として特徴づけた。サンプルをすぐに処理した。新しく単離した単核細胞を、無菌HBSS(Bio Whittaker、Walkersvile MD)で1:1に希釈しておいた血液軟膜から得、フィコール−ハイパック(Ficoll-Hypaque)で遠心分離した。サンプルの生存能力は、一般的に95%よりも大きかった。特記しない限り、PBMCは、24穴プレート中で10%FCS(T細胞培地)を追加したRPMI1640中で1×106細胞/mlで48時間培養した。PWM(Sigma Chemical Company、セントルイス MO)を用いて10μg/mlで細胞を刺激した。CD3のみ、またはCD28を介する細胞の刺激は、Weng,N.P.et al.(1995)PNAS USA92:11091-11094に記載されているように、磁気ビーズに結合しているヤギ抗−マウスIgG抗体により架橋された抗−CD3、または抗−CD3およびCD28mAbの組み合わせを用いて行なった。Jurkat細胞を、ここで記載されているpSFFNeo−bcl−XLまたはpSFFVNeo構築物のいずれかでトランスフェクションさせ、1mg/mlG418(Sigma Chemical Company)を含有するT細胞培地に維持し、bcl−x染色の陽性対照として保存した。
【0186】
細胞中のbcl−Xおよびbcl−2タンパク質の量を、以下のように行う分子内染色およびフローサイトメーターに基づいたbcl−Xの単一の細胞の検知を可能とする方法により決定した。PBMCを採取し、1×106細胞をPBS中で洗浄し、1%パラホルムアルデヒド溶液(pH7.2)で30分間室温で固定した。サンプルをPBS中で洗浄し、ついで、細胞を浸透させるために5分間PBS+0.05%Triton−X100中に再懸濁した。PBSで洗浄後、20%ヒトAB型血清(Sigma Chemical Company)および1μg/ml抗−bcl−Xモノクローナル抗体7B2(Craig Thompson、シカゴ大学)、または1μg/mlハムスター抗−bcl−2mAb 6C8(PharMingen、Inc.、サンディエゴ、カリフォルニア)を含む100μlPBS中に1時間4℃で再懸濁した。IgEアイソタイプ適合mAbsは、抗−Bcl−xおよび抗−Bcl−2で染色したサンプルの対照として使用した。一次抗体を用いてインキュベーションした後、サンプルをPBS+0.05Triton−X100で洗浄し、100μlPBS+0.05%ヤギF(ab)2抗−マウス(H+L)中に再懸濁した。FITC(Gibro Life Technologies)を30分間4℃で加えた。抗−Bcl−2で染色したサンプルを30分間4℃で、20μg/mlFITC−抗−ハムスターIgG混合(PharMingen)を用いてインキュベーションした。最後に、サンプルをPBS中で洗浄し、染色後、2〜48時間フローサイトメーターにより解析した。この時間枠の中では、bcl−xで染色した細胞のパーセンテージが減少した事実はなく、また、平均蛍光強度も変化はなかった。
【0187】
このアッセイは実質的にbcl−xLのレベルよりもむしろ、bcl−x(すなわちbcl−xLおよびbcl−xS)のレベルを測定するものであるが、免疫沈降実験により示されたところによると、これらの条件下で測定したbcl−xタンパク質の大部分はbcl−xLに対応する。
【0188】
HIV感染サンプル由来の非刺激PBMCにおけるbcl−xで染色した細胞の平均蛍光強度およびパーセンテージは、HIV非感染対照においてみられるレベルと差はないが、bcl−X発現は、PWMおよび抗−CD3に抗−CD28を加えたものを用いて活性化後の非感染対照におけるものよりも、HIV感染個体由来のPBMCにおける方が低かったことを示す。しかしながら、このbcl−Xタンパク質レベルは、細胞を細胞死から防御するのに十分である。これは、例えば、下記のように、抗−CD3および抗−CD28またはPWMを用いて様々な場合に刺激した後にHIV感染個体および非HIV感染対照個体由来のbcl−Xタンパク質の量およびPBMCのアポトーシスのレベルを比較することにより実証された。
【0189】
PBMCにおけるアポトーシスは、TdTにより触媒されたdUTPを用いてDNA一本鎖崩壊をラベルすることにより測定した(Li、X、et al(1995)Cytometry20:172-180)。フローサイトメーター解析を使用してPWMまたは抗CD3に抗CD28を加えたもののいずれかを用いて刺激したPBMCの培養中における消滅細胞を定量した。このアッセイは以下のように行った。PBMCを採取し、トリパンブルー排除により生存能力を測定し、1×106細胞を10分間、氷上で1%ホルムアルデヒド(pH7.4)溶液を用いて固定した。サンプルをHBSS中で洗浄し、細胞ペレットを70%エタノール中に再懸濁し、−20℃で2時間〜3日貯蔵した。HBSSで1回洗浄した後サンプルを30分間37℃で、0.5nMビオチン16−dUTP(Boehringer、Indianapolis IN)および10単位のTdT(Boehringer)を含有する50μlターミナルデオキシヌクレオチジルトランスフェラーゼ(TdT)反応混合物(0.1Mカルボン酸、1mMCoCl2、0.1mMジチオトレイトールおよび50μlBSA)中に再懸濁した。HBSSで洗浄後、2.5μg/mlFITCアビジン(Gibco Life Technologies、Gaithersburg、MD)を染色溶液(4×SSC、0.1%TritonX−100および5%非脂肪乾燥ミルク)に加え、サンプルを15分間室温でインキュベーションした。0.5μg/mlヒト抗−Fas抗体(Upstate Biotechnology Inc.Lake Placid NY)で24時間処理したJurkat細胞をアッセイの陽性対照として使用し、一方、TdTを欠損しているサンプルを負の対照として使用した。サンプルをELITE-ESP(Coulter Inc.、Kindal FL)フローサイトメーターを用いた細胞蛍光アッセイにより解析した。5千から1万の細胞を、log90度散乱とlogFIFC強度のパラメーターを用いて計算した。消滅細胞を、低い前角および低い90度散乱を有するFITC陽性細胞として定義した。トリパンブルー排除に基づく生存能力により、TdTを基礎とするアッセイの信頼性が確認された。
【0190】
PWMおよび抗−CD3を抗−CD28に加えた刺激サンプルに起こるアポトーシスに関連したbcl−x誘導の動力学を測定するために、3つのHIV感染および3つのHIV負の対照を培養の0時間、24時間、48時間および72時間目に実験した。平均パーセンテージとして示したように、結果は図10、パネルA−Dに示す。PWMで処理した対照サンプルでは、bcl−x発現は、24時間から48時間の間で増加したが、アポトーシスのレベルは低いままであった。抗−CD3に抗−CD28を加えたもので処理した対照サンプルにおいて、アポトーシスの低いレベルのみは明白であるが(図10、パネルA)、bcl−xのレベルは急激に増加した。これらのHIV負の対照においては、芽球化反応および細胞数の増加が両方の培養条件下で起こり、これは細胞が増殖していることを示す。HIV感染患者由来のPBMCのPWM刺激培養は、48時間で高いレベルのアポトーシスを有し、下降する前に40%でピークに達した(図10、パネルB)。HIV感染個体由来のPWM刺激PBMCは、0から48時間の間にわずかな量のbcl−xしか発現しなかった。興味深いことに、bcl−x発現細胞の分画におけるわずかな増加が、48時間から72時間の間で見られ、おそらくこれは、これは48時間以上生存した細胞が増えたことを反映する(図10、パネルB)。bcl−x発現は低く、HIV負の対照に比べて、抗−CD3に抗−CD28を加えたもので培養したHIV感染患者由来のPBMCにおいて誘導の遅延がみられた(図10、パネルD)。アポトーシスレベルは抗−CD3に抗−CD28を加えた刺激培養において低いが、それらはHIV負の対照におけるものより高かった(図10、パネルCおよびD)。PWMで培養したHIV感染患者由来のPBMCにおいては、ほとんど増殖が見られなかったが、細胞数および細胞の大きさの増大が、抗−CD3に抗−CD28を加えた培養中で見られた。
【0191】
アポトーシスおよびbcl−x誘導を、48時間培養後にHIV負の対照(n=5)およびHIV感染個体(n=20)においてさらに解析した。サンプルを、培地のみ、PWMを加えて、または上記のように抗−CD3に抗−CD28を加えて培養した。結果を表2に示す。HIV陰性の対照における、アポトーシスレベルは様々な刺激に応答して類似であった。アポトーシスレベルはPWM処理に応答して基線よりわずかに上昇し、抗−CD3に抗−CD28を加えたものに応答して幾分減少した。同サンプルにおいて、bcl−x発現はPWM処理培養中で穏やかに増加し、抗−CD3に抗−CD28を加えて処理した培養中で劇的に増加した。対照的に、アポトーシスの基線レベルを越えた顕著な増加は、PWMと共に培養した48時間後にHIV感染個体由来PBMC中で見られた。アポトーシスにおける増加は抗−CD3に抗−CD28を加えたもので培養したHIV感染患者由来のPBMC中においては観察されなかった。
【0192】
【表2】
【0193】
結論として、bcl−x発現は、刺激非感染サンプルにおいて経時的に劇的に増加するが、刺激HIV感染サンプルにおいて遅延し減少した応答が、観察された。加えて、bcl−xの単一細胞内染色は、PWM誘導bcl−x発現およびアポトーシスの間に強い逆相関がみられた(r=0.695;p=0.005)。同様な相関が抗−CD3および抗−CD28で刺激したPBMC中では観察されず、これは、共刺激はbcl−x誘導の防御レベルへと導くことを示唆している。結果はまた、PBMCの代わりにリンパ球を用いたときにも得られ、すなわち、抗−CD3に抗−CD28を加えたもので刺激したHIV感染個体由来のリンパ球は、非感染個体由来のリンパ球に比較して低いbcl−x発現を示した。遅延し減少したbcl−xの発現は試験サンプル中のCD3+CD28+細胞のパーセンテージの減少に起因するものではない。なぜなら、bcl−xの発現の減少はPWMで刺激したCD28+HIV感染部分集合において明白だからである。さらに、低いbcl−x発現はリンパ球活性化の減少に起因するものではない。なぜなら、PWM刺激はHIV感染患者において早い活性化マーカーであるCD69の発現を誘導するからである。
【0194】
このように、HIV感染個体由来のリンパ球は、抗−CD3および抗−CD28で刺激時に、非HIV感染個体よりも、bcl−XLタンパク質レベルが低いが、このレベルは細胞を細胞死から防御するに必要なbcl−XLタンパク質の限界値を上回っている。
等価体
【0195】
当業者は慣用的な実験法以外を用いて、ここで記載された本発明の特別の態様に関する数多くの等価体が存在すると認識または確認することができるだろう。このような等価体は以下の請求の範囲に含まれる。
【0196】
【表3】
【0197】
【表4】
【0198】
【表5】
【0199】
【表6】
【0200】
【表7】
【図面の簡単な説明】
【0201】
【図1】γ−照射(パネルB)前、または未処理(パネルA)のままで、培地のみ(白抜き四角)、抗CD3の存在下(白抜き菱形)、または抗CD3と抗CD28の存在下(黒塗り四角)で12時間T細胞をインキュベーション後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【図2】γ−照射(パネルBおよびD)前、または未処理(パネルAおよびC)のままで、抗CD3のみ(パネルAおよびB)または抗CD3と抗CD28(パネルCおよびD)と共に12時間T細胞をインキュベーションし、その後、それらをならし培地に残す(白抜き四角)か、洗浄して新たな培地に懸濁する(白抜き菱形)か、または洗浄して200U/mlのrIL−2を加えた新たな培地に懸濁した(黒塗り四角)後1、2、3、4および5日目のCD28+T細胞の生存能力パーセントのグラフ表示である。
【図3】図3パネルB、CおよびDは、それぞれ、培地のみ(MED)でインキュベーションするか、抗CD3抗体(αCD3)の存在下で1、6または12時間インキュベーションするか、または抗CD3と抗CD28(αCD3+αCD28)の存在下でインキュベーションしたT細胞中でのbcl−XL発現、bcl−2発現およびHLA発現のレベルを示すノーザン・ブロットを表す。パネルAは、非変性アガロースゲルでの電気泳動後に各RNA試料のアリコートをエチジウムブロマイド染色したものを示す。 図3Eは、単独で(0)または抗CD3(αCD3)と共に6または12時間、または抗CD3および抗CD28(αCD3+αCD28)と共に6または12時間インキュベーションしたT細胞中のbcl−XLおよびbcl−XSタンパク質の量を示すウエスタン・ブロットの写真である。
【図4】単独で(0)または抗CD3(αCD3)と共に6、12または24時間、または抗CD3および抗CD28(αCD3+αCD28)と共に612、または24時間インキュベーションしたT細胞中のbcl−XLおよびBcl−2タンパク質の量を示すウエスタン・ブロットの写真である。
【図5】培地単独で(Medium)または抗CD28(αCD28)、抗CD3(αCD3)、抗CD3+抗CD28(αCD3/αCD28)、または抗CD3+IL−2(100units/ml)(αCD3/IL−2)の存在下、24時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【図6】培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションしたT細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。パネル下部は、培地単独(Resting)でインキュベーションした、または抗CD3および抗CD28抗体の存在下で24、48、72、96または120時間インキュベーションし、抗Fas抗体を添加してさらに24時間インキュベーションしたT細胞の生存能力パーセントを示す。
【図7】図7A−Bは、抗Fas抗体(αFas、パネルA)または抗CD3抗体(αCD3、パネルB)で処理後0、12、24、48および72時間でbcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミドでトランスフェクションさせたJurkatクローンの生存能力パーセントのグラフ表示である。 図7パネルCは、bcl−XL発現プラスミド(bcl−XLクローン1、2および3)または対照プラスミド(Neoクローン1、2および3)でトランスフェクションさせたJurkatクローン中のbcl−XLタンパク質の量を示すウェスタン・ブロットの写真である。 図7パネルDは、IL−2回収後0、12、24、36および48時間でbcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローンの生存パーセントのグラフ表示である。 図7パネルEは、bcl−XL発現プラスミドでトランスフェクション(CTLL−2bcl−XLクローン1および2)させた、またはトランスフェクションさせなかった(CTLL−2)CTLL−2細胞クローン中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【図8】培地単独で、抗CD3の存在下、抗CD3および抗CD28の存在下、および抗CD3およびCTLA4Igの存在下、24時間インキュベーションしたマウスリンパ節細胞中のbcl−XLタンパク質の量を示すウエスタン・ブロットの写真である。
【図9】bcl−XL発現ベクター(ウイルス希釈率v平均bcl−XL)、または対照ベクター(ウイルス希釈率v平均neo)でトランスフェクションし、さまざまな量のHIVで感染させたJurkat細胞の生存能力パーセントのグラフ表示である。
【図10】図10パネルA−Dは、PWM(パネルAおよびB)でまたは抗CD3および抗CD28抗体(CD3/CD28、パネルCおよびD)で0、24、48または72時間処理した対照およびHIV感染個体由来のPBMCにおいて、アポトーシスを被った細胞の割合(%アポトーシス)およびbcl−Xタンパク質を発現する細胞の割合(%bcl−x)のグラフ表示である。
【図1A】
【図1B】
【図2A】
【図2B】
【図2C】
【図2D】
【図3A−D】
【図3E】
【特許請求の範囲】
【請求項1】
T細胞受容体およびCD28を介して共刺激されることにより活性化されたT細胞の集団において、エクスビボで細胞死を誘発する方法であって、活性化されたT細胞の集団においてT細胞死を誘発するために、活性化されたT細胞の集団に、T細胞中のbcl−XLタンパク質の産生を阻害する少なくとも一つの作用物質を接触させることを含む、方法。
【請求項2】
少なくとも一つの作用物質がアンチセンス核酸分子である、請求項1に記載の方法。
【請求項3】
少なくとも一つの作用物質が、細胞内抗体である、請求項2に記載の方法。
【請求項4】
さらに、T細胞の集団に一次活性化シグナルを提供する作用物質を接触させることを含む、請求項1に記載の方法。
【請求項5】
T細胞に一次活性化シグナルを提供する作用物質が、抗原提示細胞上に提示された抗原である、請求項4に記載の方法。
【請求項6】
T細胞に一次活性化シグナルを提供する作用物質が、抗CD3抗体である、請求項2に記載の方法。
【請求項7】
さらに、T細胞の集団に、T細胞においてコスティミュラトリー・シグナルを遮断する第二作用物質を接触させることを含む、請求項4に記載の方法。
【請求項8】
少なくとも一つの作用物質が、bcl−XLタンパク質のアンタゴニストである、請求項1に記載の方法。
【請求項9】
作用物質が、bcl−XSまたはBadである、請求項8に記載の方法。
【請求項10】
T細胞において細胞死を誘発する医薬組成物の調製のための、T細胞の使用、ただし、該T細胞は、T細胞受容体およびCD28を介した共刺激により活性化されるものであり、そして、被験体から入手された該T細胞は、インビトロでT細胞においてbcl−XLタンパク質の産生を阻害する少なくとも一つの作用物質で処理されるものである。
【請求項11】
さらに、該被験体から入手された該T細胞を、インビトロでT細胞に一次活性化シグナルを提供する作用物質で処理することを含む、請求項10に記載の使用。
【請求項12】
作用物質が抗原である、請求項11に記載の使用。
【請求項13】
さらに、該被験体から入手された該T細胞を、T細胞においてコスティミュラトリー・シグナルを遮断する第二作用物質で処理することを含む、請求項11に記載の使用。
【請求項1】
T細胞受容体およびCD28を介して共刺激されることにより活性化されたT細胞の集団において、エクスビボで細胞死を誘発する方法であって、活性化されたT細胞の集団においてT細胞死を誘発するために、活性化されたT細胞の集団に、T細胞中のbcl−XLタンパク質の産生を阻害する少なくとも一つの作用物質を接触させることを含む、方法。
【請求項2】
少なくとも一つの作用物質がアンチセンス核酸分子である、請求項1に記載の方法。
【請求項3】
少なくとも一つの作用物質が、細胞内抗体である、請求項2に記載の方法。
【請求項4】
さらに、T細胞の集団に一次活性化シグナルを提供する作用物質を接触させることを含む、請求項1に記載の方法。
【請求項5】
T細胞に一次活性化シグナルを提供する作用物質が、抗原提示細胞上に提示された抗原である、請求項4に記載の方法。
【請求項6】
T細胞に一次活性化シグナルを提供する作用物質が、抗CD3抗体である、請求項2に記載の方法。
【請求項7】
さらに、T細胞の集団に、T細胞においてコスティミュラトリー・シグナルを遮断する第二作用物質を接触させることを含む、請求項4に記載の方法。
【請求項8】
少なくとも一つの作用物質が、bcl−XLタンパク質のアンタゴニストである、請求項1に記載の方法。
【請求項9】
作用物質が、bcl−XSまたはBadである、請求項8に記載の方法。
【請求項10】
T細胞において細胞死を誘発する医薬組成物の調製のための、T細胞の使用、ただし、該T細胞は、T細胞受容体およびCD28を介した共刺激により活性化されるものであり、そして、被験体から入手された該T細胞は、インビトロでT細胞においてbcl−XLタンパク質の産生を阻害する少なくとも一つの作用物質で処理されるものである。
【請求項11】
さらに、該被験体から入手された該T細胞を、インビトロでT細胞に一次活性化シグナルを提供する作用物質で処理することを含む、請求項10に記載の使用。
【請求項12】
作用物質が抗原である、請求項11に記載の使用。
【請求項13】
さらに、該被験体から入手された該T細胞を、T細胞においてコスティミュラトリー・シグナルを遮断する第二作用物質で処理することを含む、請求項11に記載の使用。
【図4】

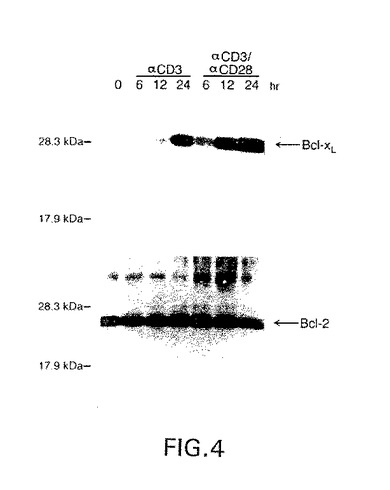
【図5】
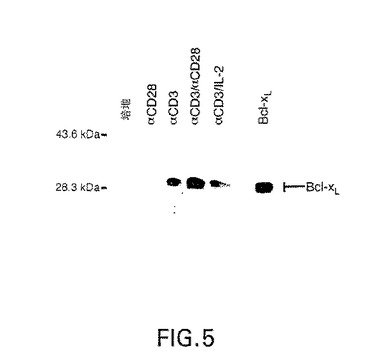
【図6】
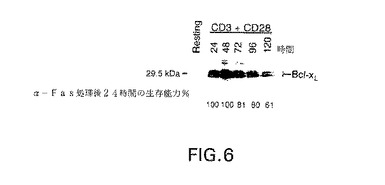
【図7A−C】
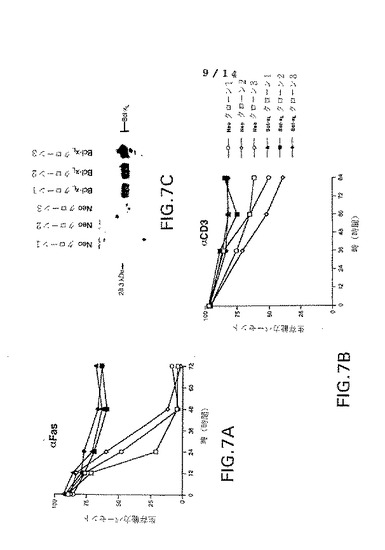
【図7D−E】
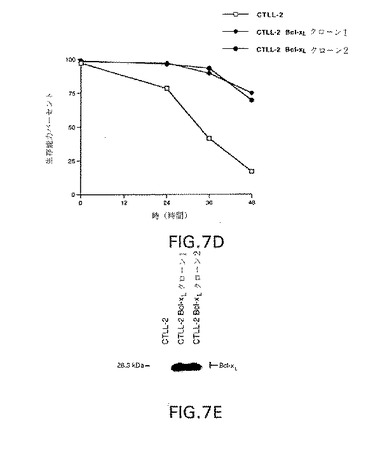
【図8】
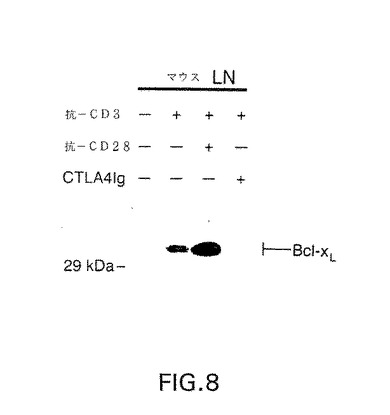
【図9】
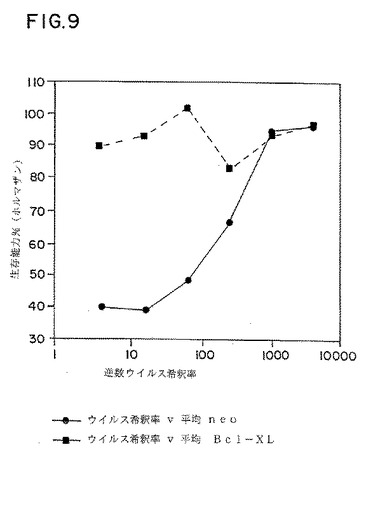
【図10A−B】
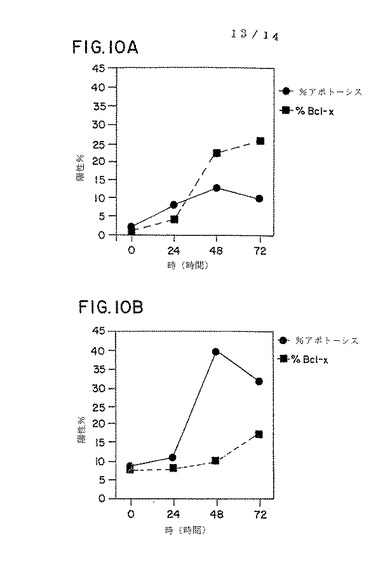
【図10C−D】
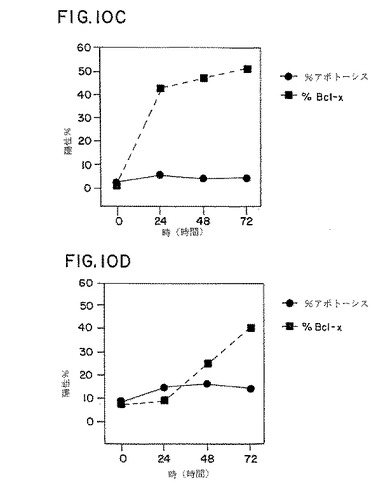
【図5】
【図6】
【図7A−C】
【図7D−E】
【図8】
【図9】
【図10A−B】
【図10C−D】
【公開番号】特開2008−101003(P2008−101003A)
【公開日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願番号】特願2007−277882(P2007−277882)
【出願日】平成19年10月25日(2007.10.25)
【分割の表示】特願平8−533512の分割
【原出願日】平成8年5月2日(1996.5.2)
【出願人】(505477408)アメリカ合衆国 (6)
【氏名又は名称原語表記】THE UNITED STATES OF AMERICA
【出願人】(503316824)アーチ・ディベロプメント・コーポレイション (1)
【Fターム(参考)】
【公開日】平成20年5月1日(2008.5.1)
【国際特許分類】
【出願日】平成19年10月25日(2007.10.25)
【分割の表示】特願平8−533512の分割
【原出願日】平成8年5月2日(1996.5.2)
【出願人】(505477408)アメリカ合衆国 (6)
【氏名又は名称原語表記】THE UNITED STATES OF AMERICA
【出願人】(503316824)アーチ・ディベロプメント・コーポレイション (1)
【Fターム(参考)】
[ Back to top ]