
cDNA合成の改良
【課題】cDNA分子およびcDNAライブラリーの作製方法を提供する。
【解決手段】少なくとも1つのmRNAもしくはポリA RNA鋳型、またはこのような鋳型の集団を、逆転写活性を有する少なくとも1つのポリペプチドと混合する工程;および該混合物を、合成された全長のcDNA分子の量または百分率を増大させるために十分な条件下でインキュベートする工程、を包含する、1つ以上のcDNA分子またはcDNA分子の集団を合成するための方法、前記方法に従って産生されたcDNA分子およびcDNAライブラリー、前記cDNA分子およびライブラリーを含有しているベクターおよび宿主細胞、および前記cDA分子およびライブラリーを作製するためのキット。
【解決手段】少なくとも1つのmRNAもしくはポリA RNA鋳型、またはこのような鋳型の集団を、逆転写活性を有する少なくとも1つのポリペプチドと混合する工程;および該混合物を、合成された全長のcDNA分子の量または百分率を増大させるために十分な条件下でインキュベートする工程、を包含する、1つ以上のcDNA分子またはcDNA分子の集団を合成するための方法、前記方法に従って産生されたcDNA分子およびcDNAライブラリー、前記cDNA分子およびライブラリーを含有しているベクターおよび宿主細胞、および前記cDA分子およびライブラリーを作製するためのキット。
【発明の詳細な説明】
【技術分野】
【0001】
(発明の背景)
本発明は、分子生物学および細胞生物学の分野に関する。本発明は一般に、cDNAの合成方法に関する。より詳細には、本発明は、平均のcDNAインサートの大きさを増大させる方法に関し、さらに具体的にはcDNAライブラリー中に存在する全長cDNAの百分率を増大させる方法に関する。それゆえ、本発明は遺伝子の発見に有用な改良されたcDNAライブラリーを提供する。
【背景技術】
【0002】
生物体、組織または細胞の構造および生理機能を検査する際に、その遺伝内容を決定することがしばしば望まれる。生物体の遺伝子のフレームワークは、生物体の体細胞および生殖細胞中に含まれるデオキシリボ核酸(DNA)中のヌクレオチド塩基の二本鎖配列でコードされる。DNAの特定のセグメント、すなわち遺伝子の遺伝内容は、その遺伝子がコードするタンパク質の産生でのみ明らかになる。タンパク質を産生するために、DNA二重らせんの一本の鎖(「コード」鎖)の相補的なコピーがポリメラーゼ酵素によって産生され、その結果、リボ核酸(RNA)の特異的配列がもたらされる。この特定のタイプのRNAは、タンパク質を産生するためのDNAからの遺伝メッセージを含んでいるので、メッセンジャーRNA(mRNA)と呼ばれる。
【0003】
所定の細胞、組織または生物体の中には、多くのmRNA種が存在し、各々が別個の特異的タンパク質をコードしている。この事実は、組織または細胞における遺伝子の発現の研究に興味を抱く研究者に対して強力なツールを提供する。mRNA分子を単離し、そして種々の分子生物学的技術によってさらに操作することができ、それにより細胞、組織または生物体の機能的遺伝内容の全体を解明することが可能となる。
【0004】
遺伝子発現の研究に対する共通のアプローチは相補的DNA(cDNA)クローンの産生である。この技術においては、生物体由来のmRNA分子が、その生物体の細胞または組織の抽出物から単離される。この単離には、チミジン(T)のオリゴマーを複合体化した、セルロースまたはアガロースのような固体クロマトグラフィーマトリックスを使用することが多い。大部分の真核mRNA分子の3’末端は一連のアデノシン(A)塩基を含み、そしてAはTに結合するので、mRNA分子は組織または細胞抽出物中の他の分子および物質から迅速に精製され得る。これらの精製されたmRNA分子から、cDNAコピーが、逆転写酵素(RT)またはRT活性を有するDNAポリメラーゼを用いて作製され得、これにより、一本鎖cDNA分子が産生される。次に、この一本鎖cDNAは、DNAポリメラーゼの作用により、元のmRNAの(従って、このmRNAをコードする、生物体のゲノムに含まれる元の二本鎖DNA配列の)完全な二本鎖DNAコピー(すなわち二本鎖cDNA)に転換され得る。このタンパク質特異的二本鎖cDNAは次に、ベクター中に挿入され、これが次に宿主の細菌、酵母、動物または植物細胞中に導入され、このプロセスは形質転換またはトランスフェクションと呼ばれる。宿主細胞を次に培地中で増殖させ、その結果、目的の遺伝子または目的の遺伝子の一部を含む(あるいは、多くの場合、それを発現する)宿主細胞の集団がもたらされる。
【0005】
この全体のプロセスは、mRNAの単離から、cDNAのベクター(例えば、プラスミド、ウイルスベクター、コスミド等)への挿入、単離された遺伝子または遺伝子の一部を含む宿主細胞集団の増殖までが「cDNAクローニング」と呼ばれる。cDNAが多数の異なるmRNAから調製されるならば、得られるcDNAのセットは「cDNAライブラリー」と呼ばれ、これは適切な用語である。なぜなら、cDNAのセットは、供給源細胞、組織または生物体中に存在する機能的遺伝情報を含む遺伝子または遺伝子の一部の「集団」を表すからである。これらのcDNAライブラリーの遺伝子型分析は、それが由来する生物体の構造および機能に対する多くの情報を与え得る。
【0006】
産生されるcDNAの総量を増大させる能力、より詳細には、cDNA分子の平均の大きさが増大したcDNAライブラリーを生成する能力、および/または全長cDNA分子の百分率が増大したcDNAライブラリーを生成する能力は、cDNAライブラリーの構築に顕著な進歩を与える。特に、このような進歩は、目的の全長遺伝子を見いだす確率を大幅に向上させる。
【0007】
理想的には、cDNA分子の合成を、mRNA分子の3’末端またはその近傍から開始する。cDNA合成をポリAテールの3’末端でオリゴ(dT)プライマーを用いてプライミングすることにより、mRNAの3’メッセージが、産生されるcDNA分子中に示されることを確実にする。mRNA分子内で生じるプライミング(内部プライミング)は、目的の遺伝子についての全長メッセージを含まないcDNA分子の合成をもたらす。すなわち、内部プライミングは目的の遺伝子(単数または複数)の一部のみを含む短縮型cDNA分子をもたらす。典型的には、内部プライミングは、メッセージ集団から3’配列の損失を引き起こす。それゆえ、内部プライミングは、産生されるcDNAの総量を減少させ、cDNAライブラリーについてのcDNA分子の平均のインサートの大きさを減少させ、および/または所定のcDNAライブラリーにおける全長cDNA分子の百分率を低下させる。配列決定分析は、多くの真核生物mRNAが内部ポリアデニル化区間(stretch)を有することを示し、これはオリゴ(dT)プライマーが逆転写酵素による第1鎖cDNA合成のために用いられる場合にプライミング部位として働き得る。さらに、いくつかのmRNAは16個もの内部プライミング部位を有し得ることが研究によって示されている(Lovett,M.ら、The construction of full−length cDNA libraries by conventional methods and a novel double capture technique、University of Texas Southwestern Medical Center, Dallas,TX、The American Society of Human Genetics主催、第48回年会、1998年10月27日〜31日、Denver、Coloradoにて発表)。それゆえ、このような内部ポリA配列に対するプライマーの内部プライミングはcDNA合成に悪影響を与え得る。
【発明の概要】
【0008】
本発明は、内部プライミングを軽減、防止、低減または実質的に低減し、それによりcDNAおよびcDNAライブラリー構築の改良を提供する。従って、本発明は、全長遺伝子を高百分率で含むcDNAライブラリーを提供することによって、遺伝子の発見を大幅に促進する。
【0009】
従って、本発明は、mRNA鋳型またはmRNA鋳型の集団からcDNA分子(単数または複数)を、生成されるcDNAの総量を増大させる、生成されるcDNA分子の長さを増大させる、および/または生成される全長cDNA分子の量または百分率を増大させるのに十分な条件下で合成することに関する。本発明によれば、内部プライミングを阻害、防止、低減または実質的に低減する任意の条件が使用され得る。このような条件は、好ましくは、プライマー濃度の至適化、反応温度の至適化および/またはプライマーの長さもしくは特異性の至適化を包含するが、これらに限定されない。このような結果はまた、本発明によれば、至適化することによって、好ましくは至適または所望の反応条件が達成されるまで逆転写反応を阻害または防止することによって、達成され得る。
【0010】
cDNAライブラリーを構築するための従来法は、第1鎖cDNA合成のために15:1のオリゴ(dT)プライマー/mRNA鋳型のモル比を用いる。このような過剰量のオリゴ(dT)プライマーの使用は、反応中に、1つ以上のプライマーの、1つ以上のmRNA鋳型に対する内部プライミングを可能にする。本発明の好適な局面によれば、第1鎖cDNAの合成のためのオリゴ(dT)プライマーの量を低減して、内部プライミングを阻害、防止、低減または実質的に低減する。プライマー対鋳型の好ましいモル比は約12:1;10:1;9:1;8:1;7:1;6:1;5:1;4:1;3:1;2:1;1:1;1:2;1:3;1:4;1:5;1:6;1:7;1:8;1:9;1:10および1:12の範囲である。好ましくは、プライマー(例えば、オリゴ(dT))対鋳型(例えば、mRNA)のモル比は約5:1から約1:20の範囲であるが、それより低いプライマー対鋳型のモル比も本発明に従って用いられ得る。特に、プライマー対鋳型のモル比は、約1:10;1:15;1:20;1:25;1:50;1:75および1:100未満であり得る。好ましくは、モル比の範囲は、約5:1;4:1;3:1;2:1;1:1;1:2;1:3;1:4;および1:5未満である。最も好ましくは、プライマー対鋳型の比は、約10:1から1:10;5:1から1:10;4:1から1:10;3:1から1:10;2.5:1から1:10;2:1から1:10;1.5:1から1:10;および1:1から1:10の範囲である。プライマー対鋳型の至適な比は、プライマー、mRNA、逆転写酵素および反応条件(アニーリング温度、緩衝剤塩など)に応じて変化し得る。所望のプライマー対鋳型の比は当業者によって容易に決定され得る。
【0011】
cDNAライブラリー構築の従来法において、プライマーの鋳型へのアニールまたはハイブリダイズは、内部プライミングを防止、阻害、低減または実質的に低減する温度では行われない。典型的には、混合物(例えば、mRNAおよびオリゴ(dT)プライマー)は変性または加熱の後、氷上で冷却される。このプロセスは典型的には、内部部位へのプライマーのアニーリングまたはハイブリダイゼーションを引き起こす。本発明の好適な局面によれば、プライマーと鋳型との間のアニーリングまたはハイブリダイゼーションの際の温度は、内部プライミングが阻害、防止、低減または実質的に低減するように維持される。本発明によれば、このような結果は、プライマーのアニーリングまたはハイブリダイゼーションをより高い温度で行うことによって達成される。このような条件はまたcDNA合成の際のmRNA二次構造の形成を低減し得る。好ましくは、プライマーを鋳型にアニールまたはハイブリダイズさせるための温度は、約10℃から約90℃;より好ましくは約10℃から約80℃;さらに好ましくは約20℃から約75℃;より好ましくは約25℃から約75℃;さらになお好ましくは約30℃から約65℃;さらになお好ましくは約37℃から約60℃;さらになお好ましくは約40℃から約60℃;さらになお好ましくは約45℃から約60℃;さらになお好ましくは約45℃から約55℃;そして最も好ましくは約45℃から約65℃の範囲である。用いられる温度はプライマーおよび鋳型のタイプおよび量に応じ、そして逆転写酵素の至適温度に応じて、変化し得る。至適温度または温度範囲は当業者によって容易に決定され得る。
【0012】
cDNA合成のための従来法は、典型的には、特定の長さ(12〜18塩基またはマー)のオリゴ(dT)プライマーの使用を必要とする。しかし、このようなプライマー長はプライマーの特異性を低下させ、これにより内部プライミングを可能にする。それゆえ、本発明はまた、プライマーの特異性を高めて、内部プライミングを防止、阻害、低減または実質的に低減することに関する。好適な局面において、プライマーの特異性はプライマーの長さを延ばすことによって高められる。それゆえ、本発明によれば、cDNA合成のために、長めのオリゴ(dT)プライマーが用いられ得る。好ましくは、プライマー長は約20から約100塩基、約20から約75塩基、約20から約60塩基、そして約20から約50塩基の範囲であり;より好ましくは約20から約45塩基;より好ましくは約20から約40塩基;そして最も好ましくは約25から約35塩基の範囲である。好適な局面において、プライマーの長さは19塩基より長く;より好ましくは約20塩基より長く;より好ましくは約25塩基より長く;そしてさらにより好ましくは約30塩基より長い。このようなプライマー長は、鋳型にアニールまたはハイブリダイズするプライマーの長さに関連している。プライマーの至適な長さおよび内容(ヌクレオチド配列)は鋳型のタイプ、所望の反応条件、および逆転写酵素に応じて変化し得る。本発明によれば、追加の配列および/または修飾ヌクレオチドを本発明のプライマーに含めることができる。例えば、追加配列(これは必ずしも鋳型にアニールまたはハイブリダイズする必要はない)は、cDNA合成を補助するために本発明のプライマー中に含めることができ、このような追加配列は、1つ以上の制限エンドヌクレアーゼ部位、1つ以上の誘導体ヌクレオチド(例えば、ビオチン化ヌクレオチドのようなハプテン含有ヌクレオチド)などを含む配列を包含する。本発明に従って用いられるプライマーのタイプおよび長さは当業者によって容易に決定され得る。
【0013】
従来のcDNA合成法は、逆転写酵素反応を至適化するために逆転写酵素を制御したり、あるいはその活性を変化させたりしない。本発明によれば、逆転写酵素の活性は、好ましくは、反応中の所望の時間に合成を開始するために、制御される。好適な局面において、逆転写酵素活性は、至適または所望の反応条件に到達するまで、阻害または防止される。このような結果は、本発明によれば、逆転写酵素活性を阻害するインヒビター(例えば、抗体または抗体フラグメント)の使用によって達成される。このような逆転写酵素インヒビターは、内部プライミングが防止、阻害、低減または実質的に低減されるように、逆転写酵素活性を低温で防止または阻害する。本発明によれば、このようなインヒビターは好ましくは逆転写酵素活性を35℃未満、40℃未満、45℃未満、50℃未満、55℃未満、60℃未満、65℃未満、70℃未満、75℃未満、80℃未満、85℃未満、および90℃未満で防止する。逆転写酵素活性を有する酵素の熱安定性に応じて、インヒビターは、目的の酵素について至適な温度またはその近傍の点で酵素の活性を阻害するように設計され得る。好ましくは、インヒビターは、使用する酵素の至適温度未満または近傍の温度で不活化され、それにより逆転写が起こることを可能にする。それゆえ、本発明は一般に、cDNA合成における逆転写酵素インヒビターの使用に関する。インヒビターのタイプおよび量は逆転写酵素のタイプおよび量に応じ、そして用いられる反応条件に応じて変化し得る。インヒビターのタイプならびにこのようなインヒビターについて使用する条件は当業者により容易に決定され得る。
【0014】
本発明によれば、cDNA合成に対する上記の改良のいずれか1つあるいは組合せが用いられ得る。これらの改良のいずれか1つあるいは組合せの使用は、改良された第1鎖cDNA合成を提供する(例えば、総cDNAの増加、cDNAの増加および/または全長cDNAの増加)。本発明によれば、第1鎖cDNA分子は、1つ以上の二本鎖核酸分子(例えば、二本鎖cDNA分子)を、本発明の方法によって産生される1つ以上の第1鎖cDNA分子をこの第1鎖cDNA分子の全てまたは一部と相補的な1つ以上の核酸分子の作製に十分な条件下でインキュベートすることによって作製するための鋳型として用いられ得る。二本鎖核酸分子の作製のための条件は、好ましくは、1つ以上のDNAポリメラーゼ、1つ以上のヌクレオチド、1つ以上の緩衝化塩、および1つ以上のプライマーからなる1つ以上の成分と共にインキュベートすることを含む。本発明の別の局面において、このような条件は、生成される二本鎖cDNAの総量の増大、生成される二本鎖cDNA分子の長さまたは大きさの増大、および/あるいは生成される全長二本鎖cDNA分子の百分率の増大をもたらすように改変される。好ましくは、このような条件は、第1鎖cDNA合成の後のリボヌクレアーゼ(RNase)消化の至適化に関連する。第1鎖cDNA合成の間に、mRNA鋳型に対して相補的な全長cDNA分子が作製されないならば、キャップ構造を含む一本鎖mRNAがmRNA/cDNAハイブリッドのmRNAの5’末端に存在する。全長cDNAが生成されるならば、二本鎖mRNA/cDNAハイブリッドが生成され、一本鎖mRNAは存在しない。好ましくは、このような消化条件は、第1鎖cDNA合成の間に形成されるmRNA/cDNA二本鎖分子の一本鎖mRNAがRNase消化に供されるように至適化される。このようにして、全長ではないmRNA/cDNAハイブリッド由来のキャップ捕捉構造は除去されるが、全長mRNA/cDNAハイブリッドはキャップ構造を維持する。それゆえ、キャップ捕捉を用いて、全長分子について選択し、そして全長ではない分子に対して選択することができる。好適な局面において、この条件は、mRNA/cDNAハイブリッドの一本鎖mRNAが消化または分解され、他方、二本鎖mRNA/cDNAハイブリッドのmRNAが分解されないかまたは実質的に分解されないような条件である。それゆえ、このようなRNase消化は、第2鎖合成に実質的に悪影響を与えない条件下で行われる。すなわち、本発明に従う第2鎖合成は従来技術と比べてより大きな二本鎖cDNA分子を生成する。従来のRNaseI条件は典型的には37℃で25u/μgから40u/μg mRNAの範囲であり、そしてRNaseA条件は典型的には37℃で1000ng/μg mRNAである。従来のRNase消化を用いると、生成される二本鎖cDNA分子の平均の大きさは約200塩基である。本発明によれば、生成される二本鎖cDNA分子の平均の大きさは好ましくは約300塩基を超え、約400塩基を超え、約500塩基を超え、約600塩基を超え、約700塩基を超え、約800塩基を超え、約900塩基を超え、約1キロ塩基を超え、約1.5キロ塩基を超え、そして約2キロ塩基を超える。本発明の1つの実施形態において、リボヌクレアーゼの濃度、リボヌクレアーゼのタイプおよび反応条件は、本発明に従う二本鎖cDNA合成を改良するように至適化される。リボヌクレアーゼ消化において使用するための好ましいリボヌクレアーゼは、リボヌクレアーゼA(RNaseA)および/またはリボヌクレアーゼI(RNaseI)を包含する。一般に、低めの温度(約4℃から約50℃)および高めの塩濃度(約5mMから約5M)は、本発明によるRNase消化の阻害または制御を補助する。使用される塩は塩化ナトリウム、塩化カリウム、塩化マグネシウム、酢酸ナトリウムなどを包含し得る。従って、RNase量または濃度の低下を用いて、所望の結果が達成され得る。RNaseAについてのこのような濃度は約0.001ng/μg mRNAから約500ng/μg mRNAまでの範囲であり得、そしてRNaseIについては約0.001u/μg mRNAから約500u/μg mRNAの範囲であり得る。インキュベーション温度、RNase濃度および塩濃度は当業者によって容易に決定され得る。好適な局面において、RNaseAの濃度は、TE緩衝液(10mM Tris、pH7.5、1mM EDTA)中、37℃で、0.1ng/μg mRNAから10ng/μg mRNAの範囲を包含する。あるいは、RNaseAの濃度は、250mMのNaClを含む10mM Tris(pH7.5)緩衝液中、25℃で30分間、0.1ng/μg mRNAから500ng/μg mRNAの範囲を包含し得る。好ましくは、用いられるRNaseIの濃度は、10mM Tris−HCl(pH7.5)、5mM EDTA(pH8.0)、200mM酢酸ナトリウム中、37℃で、0.1単位/μg mRNAから1.0単位/μg mRNAの範囲である。あるいは、RNaseIの濃度は、同じ緩衝液中、25℃で30分間、1.0単位/μg mRNAから100単位/μg mRNAの範囲で用いられ得る。
【0015】
別の局面において、本発明は、第1鎖cDNA合成の前、間あるいは後での、mRNAのキャップ構造(例えば、m7GpppN)の捕捉または結合に関連する。それゆえ、本発明は、本発明の方法の実施において、キャップ構造を有するmRNA(第1鎖合成の前)またはmRNA/cDNAハイブリッド(第1鎖合成の後または間)の選択に関連する。このような選択または捕捉は、eIF4E、eIF4Eペプチド、eIF4Eペプチドフラグメント(WO 98/08865)のような任意のキャップ結合分子、およびキャップ構造に対して特異的な抗体または抗体フラグメントを用いて達成され得る。好適な局面において、キャップ構造の選択は第1鎖合成の後に達成される。より好ましくは、このようなキャップ捕捉は本発明の方法によるリボヌクレアーゼ消化の後に行われる。例えば、リボヌクレアーゼ消化に供されるmRNA/cDNAハイブリッドは捕捉され、そして次に本発明による第2鎖cDNA合成のために使用される。
【0016】
それゆえ、本発明は一般に核酸分子の合成方法に関する。本発明はより詳細には、1つ以上の核酸分子、特にcDNA分子またはcDNAライブラリーの作製方法に関し、この方法は、1つ以上の核酸鋳型(好ましくはmRNA、ポリA RNAまたはmRNA分子の集団)を逆転写酵素活性を有する少なくとも1つのポリペプチドと混合する工程、およびこの混合物を、1つ以上の核酸鋳型の全てまたは一部と相補的な1つ以上の第1の核酸分子(例えば、第1鎖cDNA)を作製するのに十分な条件下でインキュベートする工程を包含する。本発明に従うと、このような条件は、改良された改変も本発明の条件をも使用しない従来の手順と比較して増大した総量で産生された核酸分子(cDNA)を提供する。本発明はまた、改良された改変も本発明の条件をも使用しない従来の手順と比較して、産生された核酸分子(cDNA)の長さまたは平均の大きさの増大、および/あるいは産生された全長の核酸分子(cDNA)の百分率または量における増大を提供する。産生されたcDNAの量、長さ、および全長の含有量の決定は、当業者に周知であり、そして本明細書中に記載されるような従来技術によって決定され得る。本発明に従って産生されたcDNAライブラリー中の全長のcDNAの百分率または平均の百分率は、好ましくは、約15%より高く、より好ましくは約20%より高く、より好ましくは約25%より高く、より好ましくは約30%より高く、より好ましくは約40%より高く、より好ましくは約50%より高く、より好ましくは約60%より高く、より好ましくは約70%より高く、より好ましくは約80%より高く、そして最も好ましくは約90%より高い。このような全長の百分率は、好ましくは、目的のcDNAライブラリーのクローンの一部(例えば、100から1000クローン)のランダムな選択、そのクローンの配列決定、および公知の配列データベースに対する配列の比較によって決定される。
【0017】
本発明の好ましい局面においては、本発明の改良された結果は、好ましくは、核酸またはcDNAの合成のための条件に対する改変の1つまたは組合せによって達成される。このような条件は、好ましくは、第1鎖のcDNA合成を改良するための改変、および/または第2鎖のcDNA合成を改良するための改変を含む。
【0018】
好ましい局面においては、本発明は、特に、1つ以上の二本鎖のcDNA分子を作製する方法に関する。この方法は、1つ以上のmRNA分子(好ましくは、mRNA分子の集団)を本発明の1つ以上のプライマーとともに、第1鎖のcDNAの合成の前またはその間の内部プライミングを防ぐ、阻害する、減少させる、または実質的に減少させるような温度およびプライマー濃度で、インキュベートする工程を包含する。このような反応は、好ましくは、本発明に従って、1つ以上の逆転写酵素活性のインヒビターの存在下で行われる。リボヌクレアーゼ消化は、好ましくは、第2鎖のcDNAの合成の前に、そして第2鎖の合成の間に産生される二本鎖のcDNA分子の長さ、量、および/または大きさを増大させるために十分なリボヌクレアーゼ濃度で、行われる。本発明に従うと、キャップ捕捉は好ましくは、リボヌクレアーゼ消化の間またはその後で達成される。
【0019】
本発明はまた、上記の方法に従って産生された核酸分子およびcDNA分子またはcDNA分子の集団(一本鎖または二本鎖)、ならびにこれらの核酸分子およびcDNA分子を含有しているベクター(詳細には、発現ベクター)に関する。本発明はまた、このようなcDNA分子および/またはベクターを含有している宿主細胞に関する。
【0020】
本発明はまた、本発明の方法における使用のためのキットに関する。このようなキットは、一本鎖または二本鎖の核酸分子を作製するために使用され得る。本発明のキットは、キャリア(例えば、箱またはカートン)を含み、これは、その中に1つ以上の容器(例えば、バイアル、チューブ、ボトルなど)を有する。このようなキットは、以下からなる群より選択される少なくとも1つの成分を含み得る:プライマー(好ましくは、より高い特異性を有するプライマーであり、そして最も好ましくは、20塩基以上の長さを有するオリゴ(dT)プライマー)、逆転写酵素活性を有する1つ以上のポリペプチド(逆転写酵素およびDNAポリメラーゼ)、1つ以上の逆転写のインヒビター(例えば、RT活性を有するポリペプチドに対して指向された抗体および抗体フラグメント)、1つ以上のキャップ結合分子(例えば、キャップ構造に対して指向された抗体または抗体フラグメント)、核酸合成反応の緩衝液、1つ以上のヌクレオチド、1つ以上のベクター、および本発明の方法を実行するための説明書。
【0021】
本発明はまた、本発明における使用のための組成物、または本発明の方法を実行しながら作製される組成物に関する。このような組成物は、少なくとも1つのプライマー(例えば、オリゴ(dT)またはその誘導体)および少なくとも1つの鋳型を、本発明に従う量または比でサンプルまたは反応混合物中に含み得る。このような組成物はさらに、逆転写酵素活性を有する1つ以上のポリペプチド、1つ以上の逆転写のインヒビター(例えば、抗RT抗体またはそのフラグメント)、1つ以上のヌクレオチド、1つ以上のキャップ結合分子(例えば、抗キャップ抗体またはそのフラグメント)、1つ以上の緩衝塩などを含み得る。このような組成物はまた、本発明に従って内部プライミングを回避するような温度で維持され得る。
【0022】
本発明の組成物はまた、本発明に従う量のリボヌクレアーゼを含み得る。このような組成物はさらに、1つ以上のmRNA/cDNAハイブリッド、1つ以上のヌクレオチド、逆転写酵素活性を有する1つ以上のポリペプチド、1つ以上の緩衝塩、1つ以上のキャップ結合分子(例えば、抗キャップ抗体またはそのフラグメント)などから選択される少なくとも1つの成分を含み得る。
【0023】
本発明はまた、本発明の方法、組成物、およびキットにおける使用のための、1つ以上の抗体(モノクローナルおよびポリクローナル)ならびにそれらのフラグメントに関する。このような抗体として、抗キャップおよび/または抗RT抗体、ならびに抗体フラグメントが挙げられる。
【0024】
本発明の他の好ましい実施形態は、以下の図面および本発明の記載を参照して当業者に明らかである。
【図面の簡単な説明】
【0025】
【図1】図1は、1:1、2.5:1、5:1、10:1、および50:1のオリゴ(dT)25-30/mRNAのモル比を用いて、5/6Kbの鋳型を用いて45℃でSuperScriptTMII(SSII)RTで合成した第1鎖のcDNAのオートラジオグラフである。
【図2】図2は、1:1、2.5:1、5:1、10:1、および50:1のオリゴ(dT)25-30/mRNAのモル比を用いて、5/6Kbの鋳型を用いて45℃、50℃、および55℃でThermoScriptTMII(TSII)RTで合成した第1鎖のcDNAのオートラジオグラフである。
【図3】図3は、0:1、1:1、および15:1のビオチン化されたNotI−オリゴ(dT)25/mRNAのモル比を用いて、標準的な反応温度を使用して、および反応温度を変化させて、SSII RTで合成した第1鎖のcDNAのオートラジオグラフである。
【図4】図4は、標準的な反応条件(ここでは、プライマー/鋳型のアニーリングは、cDNA合成の前に、氷上でインキュベートされる)を使用して、および本発明に従う条件(ここでは、アニーリングおよび合成反応の温度は、約30℃より高く(好ましくは、37℃より高く)維持される)を使用して、1:1および15:1のビオチン化されたNotI−オリゴ(dT)25/mRNAのモル比を用いてTSII RTで合成した第1鎖のcDNAのオートラジオグラフである。本発明に従って、30℃より高く(好ましくは、37℃より高く)アニーリングおよび反応の温度を維持することは、「ホットスタート」ともまた呼ばれ得る。
【図5】図5は、種々の量のRNaseAを使用して合成した第2鎖のcDNAのオートラジオグラフである。
【図6】図6は、種々の量のRNaseIを使用して合成した第2鎖のcDNAのオートラジオグラフである。
【発明を実施するための形態】
【0026】
(好ましい実施形態の詳細な説明)
(定義)
このような用語に対して与えられた範囲を含む、明細書および特許請求の範囲の明確でかつ一貫した理解を与えるために、以下の定義が提供される。
【0027】
「内部プライミング」は、本明細書中で使用される場合は、mRNA分子の3’末端に配置されたポリAテール以外の、1つ以上のmRNA分子中の1つ以上の部位での1つ以上のプライマーのハイブリダイゼーションまたはアニーリングをいう。
【0028】
「ライブラリー」は、本明細書中で使用される場合は、生物のDNA内容物の全てまたは一部またはかなりの部分の代表である核酸分子(環状および直鎖状)のセット(「ゲノムライブラリー」)、あるいは細胞、組織、器官、または生物体中で発現された遺伝子の全てまたは一部またはかなりの部分の代表である核酸分子のセット(「cDNAライブラリー」)をいう。このようなライブラリーは、1つ以上のベクター中に含まれてもよいし含まれなくともよい。
【0029】
「ベクター」は、本明細書中で使用される場合は、プラスミド、コスミド、ファージミド、またはファージDNA、あるいは、宿主細胞中で自律的に複製することが可能であり、そして、このようなDNA配列がベクターの必須の生物学的機能の損失を伴わずに決定可能な様式で切断され得、そしてその中にDNAがその複製およびクローニングを生じるように挿入され得る、1つまたは少数の制限エンドヌクレアーゼ認識部位によって特徴付けられる他のDNA分子をいう。ベクターは、さらに、ベクターで形質転換された細胞の同定における使用に適切な1つ以上のマーカーを含み得る。マーカーとして、例えば、テトラサイクリン耐性またはアンピシリン耐性が挙げられるが、これらに限定されない。このようなベクターはまた、1つ以上の組換え部位、1つ以上の終結部位、1つ以上の複製起点などを含み得る。
【0030】
「プライマー」は、本明細書中で使用される場合は、DNA分子の増幅または重合の間にヌクレオチドの単量体の共有結合によって伸張される、一本鎖のオリゴヌクレオチドをいう。本発明での使用に好ましいプライマーとして、オリゴ(dT)プライマー、またはその誘導体もしくは改変体が挙げられる。
【0031】
「オリゴヌクレオチド」は、本明細書中で使用される場合は、1つのヌクレオチドのデオキシリボースまたはリボースの3’位と、隣接するヌクレオチドのデオキシリボースまたはリボースの5’位との間でのホスホジエステル結合によって連結されるヌクレオチドの共有結合された配列を含む、合成の分子または天然の分子をいう。
【0032】
「鋳型」は、本明細書中で使用される場合は、増幅されるか、合成されるか、または配列決定されるべき、二本鎖または一本鎖の核酸分子をいう。二本鎖の分子の場合においては、第1鎖および第2鎖を形成するその鎖の変性は、好ましくは、これらの分子が増幅され得るか、合成され得るか、または配列決定され得る前に、あるいは、二本鎖の分子が鋳型として直接使用され得る前に、行われる。一本鎖の鋳型については、鋳型の一部に対して相補的なプライマーが、適切な条件下でハイブリダイズされるかまたはアニーリングされ、そして次いで1つ以上のポリメラーゼまたは逆転写酵素が、上記の鋳型の全体または一部に対して相補的な核酸分子を合成し得る。本発明に従って新しく合成された分子は、最初の鋳型と等しい長さであり得るか、または最初の鋳型の長さよりも短い長さであり得る。
【0033】
「取りこむ」は、本明細書中で使用される場合は、DNAおよび/またはRNAの分子またはプライマーの一部になることを意味する。
【0034】
「増幅」は、本明細書中で使用される場合は、ポリメラーゼを用いてヌクレオチド配列のコピー数を増大させるための任意のインビトロでの方法をいう。核酸の増幅は、DNAおよび/またはRNAの分子またはプライマーへのヌクレチドの取りこみを生じ、それによって鋳型に対して相補的である新しい分子を形成する。形成された核酸分子およびその鋳型は、さらなる核酸分子を合成するための鋳型として使用され得る。本明細書中で使用される場合は、1回の増幅反応は、複数回の複製から構成され得る。DNA増幅反応として、例えば、ポリメラーゼ連鎖反応(PCR)が挙げられる。1回のPCR反応は、DNA分子の変性および合成の5から100回の「サイクル」から構成され得る。
【0035】
「ヌクレオチド」は、本明細書中で使用される場合は、塩基−糖−リン酸の組合せをいう。ヌクレオチドは、核酸配列の単量体単位(DNAおよびRNA)である。用語ヌクレオチドは、リボヌクレシド三リン酸(ATP、UTP、CTP、GTP)、およびデオキシリボヌクレオシド三リン酸(例えば、dATP、dCTP、dITP、dUTP、dGTP、dTTP)、またはそれらの誘導体を含む。このような誘導体として、例えば、[αS]dATP、7−デアザ−dGTP、7−デアザ−dATP、およびビオチン化されたかまたはハプテン化されたヌクレオチドが挙げられる。用語ヌクレオチドは、本明細書中で使用される場合はまた、ジデオキシリボヌクレオシド三リン酸(ddNTP)、およびそれらの誘導体をいう。デオキシリボヌクレオシド三リン酸の説明的な例として、ddATP、ddCTP、ddGTP、ddITP、およびddTTPが挙げられるが、これらに限定されない。本発明に従うと、「ヌクレオチド」は、標識されなくともよいし、周知の技術によって検出可能に標識されてもよい。検出可能な標識として、例えば、放射性同位元素、蛍光標識、化学発光標識、生体発光標識、および酵素標識が挙げられる。
【0036】
「ハイブリダイゼーション」または「アニーリング」は、本明細書中で使用される場合は、二本鎖の分子を生じる2つの相補的な一本鎖の核酸分子(RNAおよび/またはDNA)の塩基対合をいう。本明細書中で使用される場合は、対合は完全には相補的ではないが、2つの核酸分子がハイブリダイズされ得るかまたはアニーリングされ得る。従って、不一致塩基は、当該分野で周知の適切な条件が使用される限りにおいては、2つの核酸分子のハイブリダイゼーションもアニーリングをも妨げない。本発明においては、用語ハイブリダイゼーションまたはアニーリングは、好ましくは、1つ以上の鋳型(例えば、mRNA)に対する1つ以上のプライマー(例えば、オリゴ(dT)またはその誘導体)のハイブリダイゼーションをいう。
【0037】
「宿主細胞」は、本明細書中で使用される場合には、複製可能な発現ベクターまたはクローニングベクターのレシピエントである、任意の原核生物細胞または真核生物細胞をいう。用語「宿主」または「宿主細胞」は、本明細書中では互換的に使用され得る。このような宿主の例については、Maniatisら、「Molecular Cloning:A Laboratory Manual」、Cold Spring Harbor Laboratory,Cold Spring Harbor、New York(1982)を参照のこと。好ましい原核生物宿主として、Escherichia(例えば、E.coli)、Bacillus、Staphylococcus、Agrobacter(例えば、A.tumefaciens)、Streptomyces、Pseudomonas、Salmonella、Serratia、Caryophanon属などの細菌が挙げられるが、これらに限定されない。最も好ましい原核生物宿主はE.coliである。本発明において特に目的の細菌宿主として、Ecoli K12株、DH10B株、DH5α株、Stbl2株、およびHB101株、ならびにLife Technologies,Inc.から入手可能な他の株が挙げられる。好ましい真核生物宿主として、真菌、魚の細胞、酵母細胞、植物細胞、および動物細胞が挙げられるが、これらに限定されない。特に好ましい動物細胞は、Drosophilla細胞、Spodoptera Sf9細胞、Sf21細胞、およびTrichoplusia High−Five細胞のような昆虫細胞;C.elegans細胞のような線虫細胞;ならびにCOS細胞、CHO細胞、VERO細胞、293細胞、PERC6細胞、BHK細胞、およびヒト細胞のような哺乳動物細胞である。
【0038】
「発現ベクター」は、本明細書中で使用される場合は、宿主細胞中への形質転換またはトランスフェクション後に、その中にクローン化されている遺伝子または遺伝子の一部の発現を増強することが可能なベクターをいう。クローン化された遺伝子は、通常は、プロモーター配列のような特定の制御配列の制御下に(すなわち、それに対して作動可能に連結される)配置される。このようなプロモーターとして、ファージλPLプロモーター、およびE.coli lac、trp、およびtacプロモーターが挙げられるが、これらに限定されない。他の適切なプロモーターが当業者に公知である。
【0039】
本発明に従う逆転写に適切な核酸の鋳型として、任意の核酸分子または核酸分子の集団(好ましくは、1つ以上のRNA分子(例えば、1つ以上のmRNA分子またはポリA+RNA分子、およびより好ましくはmRNA分子の集団)あるいは1つ以上のDNA分子)が挙げられ、特に、細胞または組織に由来するものが挙げられる。好ましい局面においては、mRNA分子の集団(多数の異なるmRNA分子)が、本発明に従ってcDNAライブラリーを作製するために使用される。
【0040】
1つ以上の鋳型に対して相補的である核酸分子(単数または複数)を作製するために、プライマー(例えば、オリゴ(dT)プライマー)および1つ以上のヌクレオチドが、代表的には3’から5’方向での核酸の合成のために使用される。本発明のこの局面に従う逆転写に適切な核酸分子として、任意の核酸分子、特に、原核生物細胞または真核生物細胞に由来する任意の核酸分子が挙げられる。このような細胞として、正常な細胞、疾患の細胞、形質転換された細胞、樹立された細胞、先祖細胞、前駆細胞、胎児の細胞、胚性の細胞、細菌の細胞、酵母細胞、動物細胞(ヒト細胞を含む)、鳥類の細胞、植物細胞など、あるいは植物(例えば、トウモロコシ、トマト、タバコ、ジャガイモ、ダイズなど)または動物(例えば、ヒト、ウシ、ブタ、マウス、ヒツジ、ウマ、サル、イヌ、ネコ、ラット、ウサギ、鳥類、魚、昆虫など)から単離された組織が挙げられ得る。このような核酸分子はまた、ウイルスからも単離され得る。
【0041】
本発明の方法に従ってcDNA分子を調製するために鋳型として使用される核酸分子は、好ましくは、種々の細胞、組織、器官、または生物体のような天然の供給源から得られる。核酸分子の供給源として使用され得る細胞は、原核生物細胞(細菌細胞(Escherichia、Bacillus、Serratia、Salmonella、Staphylococcus、Streptococcus、Clostridium、Clamydia、Neisseria、Treponema、Mycoplasma、Borrelia、Legionella、Pseudomonas、Mycobacterium、Helicobacter、Erwinia、Agrobacterium、Rhizobium、Xanthomonas、Streptomyces属の種を含むがこれらに限定されない))、または真核生物(真菌(特に、酵母)、植物、原生動物、および他の寄生生物、ならびに動物(昆虫(特に、Drosophila spp.細胞を含む)、線虫(特に、Caenorhabditis elegans細胞)、ならびに哺乳動物(特にヒト細胞)を含む)であり得る。
【0042】
核酸の供給源として使用され得る哺乳動物の体細胞として、血球(網状赤血球および白血球)、内皮細胞、上皮細胞、ニューロン細胞(中枢神経系または末梢神経系に由来する)、筋肉細胞(骨格筋、平滑筋、または心筋に由来する筋細胞および筋芽細胞を含む)、結合組織の細胞(線維芽細胞、脂肪細胞、軟骨細胞、軟骨芽細胞、骨細胞、および骨芽細胞を含む)、ならびに他の支質細胞(例えば、マクロファージ、樹状細胞、Schwann細胞)が挙げられる。哺乳動物の生殖細胞(精母細胞および卵母細胞)もまた、上記の体細胞および生殖細胞を生じる先祖細胞、前駆細胞、および幹細胞と同様に、本発明における使用のための核酸の供給源として使用され得る。脳、腎臓、肝臓、すい臓、血液、骨髄、筋肉、神経、皮膚、尿生殖器、循環、リンパ系、消化管、および結合組織の供給源に由来するもの、ならびに哺乳動物(ヒトを含む)の胚または胎児に由来するもののような、哺乳動物の組織または器官もまた、核酸の供給源としての使用に適切である。
【0043】
任意の上記の細胞、組織、および器官は、正常なもの、罹患したもの、形質転換されたもの、樹立されたもの、先祖、前駆体、胎児のもの、または胚性のものであり得る。罹患した細胞として、例えば、感染性の疾患(細菌、真菌、または酵母、ウイルス(AIDS、HIV、HTLV、ヘルペス、肝炎などを含む)、または寄生生物によって引き起こされる)、遺伝的もしくは生化学的な病因(例えば、嚢胞性線維症、血友病、アルツハイマー病、筋ジストロフィー、または多発性硬化症)、または癌性のプロセスに関連する罹患した細胞が挙げられ得る。形質転換されたかまたは樹立された動物細胞株として、例えば、COS細胞、CHO細胞、VERO細胞、BHK細胞、HeLa細胞、HepG2細胞、K562細胞、293細胞、L929細胞、F9細胞などが挙げられ得る。本発明における使用のための核酸の供給源として適切な他の細胞、細胞株、組織、器官、および生物体は、当業者に明らかである。
【0044】
一旦、細胞、組織、器官、または他の出発サンプルが得られると、核酸分子(例えば、mRNA)が、当該分野で周知の方法によってそれらから単離され得る(例えば、Maniatis,T.,ら、Cell 15:687−701(1978);Okayama,H.およびBerg,P.Mol.Cell.Biol.2:161−170(1982);Gubler,U.およびHoffman,B.J.,Gene 25:263−269(1983);ならびにLife Technologies,Inc.から入手可能なMessage MakerTMmRNA Isolation Systemを参照のこと)。このように単離された核酸分子は、次いで、本発明に従ってcDNA分子およびcDNAライブラリーを調製するために使用され得る。本発明に従って産生されたcDNA分子および/またはcDNAライブラリーは、好ましくは、1つ以上のベクター中に含まれる。このようなベクターは、当該分野で周知の標準的な形質転換またはトランスフェクションの技術によって1つ以上の宿主細胞中に導入され得る。好ましい宿主細胞として、Escherichia属、特に、E.coliの細胞のような原核生物宿主細胞が挙げられる。
【0045】
本発明の組成物、方法、およびキット中での使用のための酵素として、逆転写酵素活性を有する任意の酵素が挙げられる。このような酵素として、レトロウイルス逆転写酵素、レトロトランスポゾン逆転写酵素、B型肝炎逆転写酵素、カリフラワーモザイクウイルス逆転写酵素、細菌の逆転写酵素、Tth DNAポリメラーゼ、Taq DNAポリメラーゼ(Saiki,R.K.ら,Science 239:487−491(1988);米国特許第4,889,818号および同第4,965,188号)、Tne DNAポリメラーゼ(WO96/10640)、Tma DNAポリメラーゼ(米国特許第5,374,553号)、およびそれらの変異体、フラグメント、改変体、または誘導体が挙げられるが、これらに限定されない(例えば、共有に係る、同時係属中の米国特許出願番号第08/706,702号および08/706,706号(両方とも、1996年9月9日に出願され、これらはそれらの全体において本明細書中で参考として援用されている)を参照のこと)。当業者によって理解されているように、改変された逆転写酵素およびRT活性を有するDNAポリメラーゼは、当該分野で周知である組換えまたは遺伝子操作技術によって得ることができる。変異体の逆転写酵素またはポリメラーゼは、例えば、部位特異的またはランダムな変異誘発によって、目的の逆転写酵素またはポリメラーゼをコードする遺伝子(単数または複数)を変異させることによって得ることができる。このような変異は、点変異、欠失変異、および挿入変異を含み得る。好ましくは、1つ以上の点変異(例えば、1つ以上のアミノ酸の、1つ以上の異なるアミノ酸での置換)が、本発明での使用のための変異体の逆転写酵素またはポリメラーゼを構築するために使用される。逆転写酵素またはポリメラーゼのフラグメントもまた、当該分野で周知である組換え技術による欠失変異によるか、あるいは任意の多数の周知のタンパク質分解酵素を使用する目的の逆転写酵素(単数または複数)またはポリメラーゼ(単数または複数)の酵素消化によって得ることができる。
【0046】
本発明での使用に好ましい酵素として、RNaseH活性が減少しているかまたは実質的に減少している酵素が挙げられる。RNaseH活性を減少させるかまたは実質的に減少させるこのような酵素は、目的の逆転写酵素中のRNaseHドメインを変異させることによって、好ましくは、上記のような1つ以上の点変異、1つ以上の欠失変異、および/または1つ以上の挿入変異によって、得ることができる。「RNaseH活性が実質的に減少している」酵素によって、野生型のモロニーマウ白血病ウイルス(M−MLV)、鳥類骨髄芽球症ウイルス(AMV)またはラウス肉腫ウイルス(RSV)の逆転写酵素の場合のような対応する野生型またはRNaseH+酵素のRNaseH活性の、約30%未満、約25%未満、約20%未満、より好ましくは約15%未満、約10%未満、約7.5%未満、または約5%未満、そして最も好ましくは約5%未満または2%未満を有する酵素を意味する。任意の酵素のRNaseH活性は、例えば、米国特許第5,244,797号、Kotewicz,M.L.ら、Nucl.Acids Res.16:265(1988)、Gerard,G.F.ら、FOCUS 14(5):91(1992)、および米国特許第5,668,005号に記載されているような、種々のアッセイによって決定され得る。これらの開示の全ては、本明細書中で参考として完全に援用されている。
【0047】
本発明での使用のための逆転写酵素活性を有するポリペプチドは、例えば、Life Technologies,Inc.(Rockville,Maryland)、Pharmacia(Piscataway,New Jersey)、Sigma(Saint Louis,Missouri)、またはBoehringer Mannheim Biochemicals(Indianapolis,Indiana)から商業的に入手可能であり得る。あるいは、逆転写酵素活性を有するポリペプチドは、当業者に周知の天然のタンパク質を単離および精製するための標準的な手順に従って、それらの天然のウイルスまたは細菌の供給源から単離され得る(例えば、Houts,G.E.ら、J.Virol.29:517(1979)を参照のこと)。さらに、逆転写酵素活性を有するポリペプチドは、当業者に良く知られている組換えDNA技術によって調製され得る(例えば、Kotewicz,M.L.ら、Nucl.Acids Res.16:265(1988);Soltis,D.A.およびSkalka,A.M.Proc.Natl.Acad.Sci.USA 85:3372−3376(1988)を参照のこと)。
【0048】
本発明での使用に好ましい逆転写酵素活性を有するポリペプチドとして、M−MLV逆転写酵素、RSV逆転写酵素、AMV逆転写酵素、ラウス関連(Rous Associated)ウイルス(RAV)の逆転写酵素、骨髄芽球症関連ウイルス(MAV)逆転写酵素、およびヒト免疫不全ウイルス(HIV)の逆転写酵素、およびWO98/47921に記載されている他のもの、ならびにそれらの誘導体、改変体、フラグメント、または変異体、ならびにそれらの組合せが挙げられる。さらに好ましい実施形態においては、逆転写酵素は、RNaseH活性が減少しているかまたは実質的に減少しており、そして最も好ましくは、M−MLV H-逆転写酵素、RSV H-逆転写酵素、AMV H-逆転写酵素、RAV H-逆転写酵素、MAV H-逆転写酵素、およびHIV H-逆転写酵素、ならびにそれらの誘導体、改変体、フラグメント、または変異体、ならびにそれらの組合せからなる群より選択される。特定の目的の逆転写酵素として、AMV RTおよびM−MLV RT、そしてより好ましくは、減少したかまたは実質的に減少したRNaseH活性を有するAMV RTおよびM−MLV RT(好ましくは、AMV RT αH-/BH+およびM−MLV RT H-)が挙げられる。本発明での使用に最も好ましい逆転写酵素として、Life Technologies,Inc.から入手可能な、SuperScriptTM、SuperScriptTMII、ThermoScriptTM、およびThermoScriptTMIIが挙げられる。一般的には、WO98/47921、米国特許第5,244,797号、および同第5,668,005号を参照のこと。それらのそれぞれの内容の全体は、本明細書中で参考として援用されている。
【0049】
種々のDNAポリメラーゼは、本発明に従って有用である。このようなポリメラーゼとして、以下が挙げられるが、これらに限定されない:Thermus thermophilus(Tth)DNAポリメラーゼ、Thermus aquaticus(Taq)DNAポリメラーゼ、Thermotoga neapolitana(Tne)DNAポリメラーゼ、Thermotoga maritima(Tma)DNAポリメラーゼ、Thermococcus litoralis(TliまたはVENTTM)DNAポリメラーゼ、Pyrococcus furiosis(Pfu)DNAポリメラーゼ、DEEPVENTTM DNAポリメラーゼ、Pyrococcus woosii(Pwo)DNAポリメラーゼ、Bacillus sterothermophilus(Bst)DNAポリメラーゼ、Bacillus caldophilus(Bca)DNAポリメラーゼ、Sulfolobus acidocaldarius(Sac)DNAポリメラーゼ、Thermoplasma acidophilum(Tac)DNAポリメラーゼ、Thermus flavus(Tfl/Tub)DNAポリメラーゼ、Thermus ruber(Tru)DNAポリメラーゼ、Thermus brockianus(DYNAZYMETM)DNAポリメラーゼ、Methanobacterium thermoautotrophicum(Mth)DNAポリメラーゼ、Mycobacterium spp.DNAポリメラーゼ(Mtb,Mlep)、ならびにそれらの変異体、改変体、および誘導体。
【0050】
本発明に従って使用されるDNAポリメラーゼは、代表的には5’から3’の方向で、核酸の鋳型からDNA分子を合成し得る任意の酵素であり得る。このようなポリメラーゼは、中温性または高温性であり得る。中温性のポリメラーゼとして、T4 DNAポリメラーゼ、T5 DNAポリメラーゼ、T7 DNAポリメラーゼ、KlenowフラグメントDNAポリメラーゼ、DNAポリメラーゼIII、DNAポリメラーゼIなどが挙げられる。熱安定性のDNAポリメラーゼとして、Taq、Tne、Tma、Pfu、VENTTM、DEEPVENTTM、Tth、ならびにそれらの変異体、改変体、および誘導体が挙げられる(米国特許第5,436,149号;米国特許第5,512,462号;第WO92/06188号;第WO92/06200号;第WO96/10640号;Barnes,W.M.,Gene 112:29−35(1992);Lawyer,F.C.,ら、PCR Meth.Appl.2:275−287(1993);Flaman,J.−M.ら、Nucl.Acids Res.22(15):3259−3260(1994))。
【0051】
本発明での使用のためのDNAポリメラーゼは、例えば、Life Technologies,Inc.(Rockville,Maryland)、Perkin−Elmer(Branchburg,New Jersey)、New England BioLabs(Beverly,Massachusetts)、またはBoehringer Mannheim Biochemicals(Indianapolis,Indiana)から商業的に入手することができる。
【0052】
本発明はまた、cDNA分子(特に、全長のcDNA分子)であり得る本発明の方法によって産生された核酸分子、これらの核酸分子およびcDNA分子を含有しているベクター(特に、発現ベクター)、ならびにこれらの核酸分子、cDNA分子、および/またはベクターを含有している宿主細胞に関する。
【0053】
組換えベクターは、当該分野で周知の方法を使用して、本発明の方法に従って調製された1つ以上のcDNA分子または核酸分子を1つ以上のベクター中に挿入することによって、本発明のこの局面に従って産生され得る。本発明のこの局面で使用されるベクターは、例えば、ファージまたはファージミドベクターであり得、そして好ましくは、プラスミドである。目的のポリペプチドをコードする核酸に対するシス作用性制御領域を含有しているベクターが、好ましい。適切なトランス作用性因子は、宿主によって供給され得るか、補完ベクターによって供給され得るか、または宿主への導入の際にベクター自体によって供給され得る。
【0054】
本発明において有用な発現ベクターとして、染色体に由来するベクター、エピソームに由来するベクター、およびウイルスに由来するベクター(例えば、細菌のプラスミドまたはバクテリオファージに由来するベクター)、ならびにそれらの組合せに由来するベクター(例えば、コスミド、およびファージミド)が挙げられ、そして好ましくは、細菌性宿主細胞中での培養のためにテトラサイクリンまたはアンピシリンの耐性遺伝子のような少なくとも1つの選択マーカーを含む。このような発現ベクターへの挿入の前に、本発明のcDNAまたは核酸分子は、適切なプロモーターに作動可能に連結されるべきである。
【0055】
本発明における使用に好ましいベクターの中には、Qiagenから入手可能なpQE70、pQE60、およびpQE−9;Stratageneから入手可能なpBSベクター、Phagescriptベクター、Bluescriptベクター、pNH8A、pNH16a、pNH18A、pNH46A;Invitrogenから入手可能なpcDNA3;Pharmaciaから入手可能なpGEX、pTrxfus、pTrc99a、pET−5、pET−9、pKK223−3、pKK233−3、pDR540、pRIT5;ならびにLife Technologies,Inc.から入手可能なpSPORT1、pSORT2、pSV・SPORT1、pCMVSPORT6、およびpCMVSPORTが挙げられる。他の適切なベクターは、当業者に容易に明らかである。
【0056】
本発明は、cDNA分子またはライブラリーを産生するために、当該分野で周知の任意のcDNAの合成方法(例えば、Gubler,U.およびHoffman,B.J.,Gene 25:263−269(1983);Krug,M.S.およびBerger,S.L.,Meth.Enzymol.152:316−325(1987);Sambrook,J.ら、Molecular Cloning:A Laboratory Manual,第2版、Cold Spring Harbor、NY:Cold Spring Harbor Laboratory Press、8.60−8.63頁(1989);PCT US98/19948;ならびにWO98/51699を参照のこと)と組合せて使用され得る。本発明を有利に使用し得る他のcDNA合成方法が、当業者に容易に明らかである。
【0057】
本発明の方法に従ってcDNA分子またはライブラリーを得たら、これらのcDNAは、さらなる分析または操作のために単離され得る。cDNAの精製のための詳細な方法論は、その全体において本明細書中で参考として援用されているGENETRAPPERTMマニュアル(Life Technologies)において教示されているが、当該分野で公知である別の標準的な技術(例えば、Sambrook,J.ら、Molecular Cloning:A Laboratory Manual,第2版、Cold Spring Harbor,NY:Cold Spring Harbor Laboratory Press、8.60−8.63頁(1989)を参照のこと)もまた使用され得る。本発明によって産生されたcDNA分子またはライブラリーもまた、ツーハイブリッド分析、cDNAの基準化、配列決定、および増幅のような標準的な分子生物学的技術によってさらに操作され得る。より詳細には、本発明の方法、およびこのような方法によって産生されたcDNA分子またはライブラリーは、RT−PCRおよび5’RACE技術(Life Technologies,Inc.)、ならびにディファレンシャルディスプレイとの組合せにおいて使用され得る。
【0058】
種々のインヒビターおよび結合分子が、本発明の方法における使用に適切である。逆転写酵素活性を有する上記のポリペプチドに対して結合する抗体(例えば、抗AMV RT抗体、抗M−MLV RT抗体、または抗RSV RT抗体を含む抗RT抗体)、またはキャップ構造に対して結合する抗体(例えば、抗キャップ抗体)、およびそれらのフラグメント(例えば、FabまたはF(ab’)2フラグメント)が、これらのインヒビターまたは結合分子に含まれる。このような抗体は、ポリクローナルまたはモノクローナルであり得、そして当該分野で周知の方法に従って種々の種において調製され得る。例えば、Sutcliffe,J.G.ら、Science 219:660−666(1983);Wilsonら、Cell 37:767(1984);およびBittle,F.J.ら、J.Gen.Virol.66:2347−2354(1985)を参照のこと。任意の上記の逆転写酵素またはキャップ構造について特異的な抗体は、インタクトなポリメラーゼポリペプチドまたはキャップ構造、あるいは1つ以上のそれらのフラグメントに対して惹起され得る。これらのポリペプチドまたはキャップ構造、あるいはそれらのフラグメントは、キャリアタンパク質(例えば、アルブミン)とともに動物の系(例えば、ウサギまたはマウス)に対して提示され得るか、またはこれらのポリペプチドが十分に長い(少なくとも約25アミノ酸)の場合には、キャリアを伴わずに動物の系に対して提示され得る。
【0059】
本明細書中で使用される場合は、用語「抗体」(Ab)は、以下のような特異的な状況が以下を除いて、用語「ポリクローナル抗体」または「モノクローナル抗体」(mAb)と互換的に使用され得る。これらの用語は、本明細書中で使用される場合は、インタクトな分子、および逆転写酵素活性を有するポリペプチド(DNAポリメラーゼまたは逆転写酵素)またはキャップ構造あるいはそれらの一部に対して特異的に結合し得る抗体フラグメント(例えば、FabおよびF(ab’)2フラグメントのような)を含むことを意味する。
【0060】
本発明の方法で使用される抗体は、ポリクローナルまたはモノクローナルであり得、そして任意の種々の方法によって調製され得る(例えば、米国特許第5,587,287号を参照のこと)。例えば、ポリクローナル抗体は、標準的な技術に従って、動物を、逆転写酵素活性またはキャップ構造あるいはそれらの一部を有する1つ以上のポリペプチドで免疫化することによって作製され得る(例えば、Harlow,E.およびLane,D.、Antibodies:A Laboratory Manual,Cold Spring Harbor,NY:Cold Spring Harbor Laboratory Press(1988);Kaufman,P.B.ら、Handbook of Molecular and Cellular Methods in Biology and Medicine、Boca Raton,Florida:CRC Press、468−469頁(1995)を参照のこと)。あるいは、本発明の方法において使用されるモノクローナル抗体(またはそのフラグメント)は、当該分野で周知のハイブリドーマ技術を使用して調製され得る(Koehlerら、Nature 256:495(1975);Koehlerら、Eur.J.Immunol.6:511(1976);Koehlerら、Eur.J.Immunol.6:292(1976);Hammerlingら、Monoclonal Antibodies and T−Cell Hybridomas、New York:Elsevier、563−681頁(1981);Kaufman,P.B.ら、Handbook of Molecular and Cellular Methods in Biology and Medicine,Boca Raton,Florida:CRC Press、444−467頁(1995))。
【0061】
上記の抗体のFab、F(ab’)2および他のフラグメントは、本明細書中に記載されている方法において使用され得ることが認識される。このようなフラグメントは、代表的には、パパイン(Fabフラグメントを産生するため)またはペプシン(F(ab’)2フラグメントを産生するため)のような酵素を使用して、タンパク質分解的切断によって産生される。抗体フラグメントはまた、組換えDNA技術の適用を通じて、または合成化学を通じて産生され得る。
【0062】
本発明はまた、本発明に従う使用のためのキットを提供する。このようなキットは、キャリア手段(例えば、箱またはカートン)を含み、その中には1つ以上の容器手段(例えば、バイアル、チューブ、ボトルなど)が密閉されている。ここでは、キットは、1つ以上の宿主細胞、1つ以上の逆転写酵素、1つ以上の逆転写のインヒビター、1つ以上のキャップ結合分子、1つ以上のDNAポリメラーゼ、適切な緩衝液、1つ以上のヌクレオチド、および/または1つ以上のプライマー(例えば、逆転写のためのオリゴ(dT))を、(同じ容器または別の容器中に)含み得る。本発明のこの局面によって含まれるキットは、標準的な核酸の逆転写プロトコールを実行するために必要なさらなる試薬および化合物をさらに含み得る。
【0063】
本明細書中に記載されている方法および出願に対する他の適切な改変および適応が明らかであり、そして本発明の範囲またはその任意の実施形態を逸脱することなく行われ得ることが、当業者に容易に明らかである。本発明は本明細書中で詳細に記載されているが、本発明が、以下の実施例を参照してより明確に理解される。以下の実施例は、本発明の説明の目的のために本発明中に含まれ、そして本発明を制限することは意図しない。
【実施例】
【0064】
(実施例1:オリゴ(dT)プライマー/mRNAの様々な比を用いた第1鎖cDNA合成の比較)
この実施例は、オリゴdTプライマー/出発mRNAの様々な比を用いた、MAP4遺伝子の第1鎖cDNA合成を比較する。全ての成分は、他に指定しなければ、Life Technologies,Inc.、Rockville、Marylandから入手可能である。
【0065】
Superscript II逆転写酵素(SSIIRT)のマスターミックスを、下記の表1で指定したように調製した。
【0066】
【表1】

ThermoScriptTMIIRT(TSRT)(AMV RT αH-βH+)(第WO98/47921号を参照のこと)のマスターミックスを、下記の表2で指定したように調製した。
【0067】
【表2】
マスターアニーリングミックスを、下記の表3で指定する量で5つのチューブに、5KbのMAP4 mRNA、オリゴ(dT)25-30および水を加えることによって調製した。
【0068】
【表3】
混合物を70℃で10分間加熱し、そして次いで氷上で5分間冷却した。
【0069】
下記の表4でまとめたように、全容量20μlで、9μlの適切な逆転写酵素マスターミックス、10μlのマスターアニーリングミックス、および1μlのSSIIRT(200単位/μl)またはTSIIRT(15単位/ul)のいずれかを加えることによって、第1鎖cDNAの合成を行った。
【0070】
【表4】
反応物を、SSIIRTでは45℃、TSIIRTでは45、50または55℃で1時間インキュベートした。チューブを氷上に置いて反応を終了させた。反応チューブの18μlの第1鎖cDNAを沈殿させ、そして10μlの水に再懸濁した。5μlの第1鎖cDNAを、5μlの標準ローディング緩衝液(60mMのNaOH、4mMのEDTA、0.1%のブロモフェノールブルー)と混合し、そして分析のために1.4%アルカリアガロースゲルにローディングした。これらの結果を図1および図2に示す。
【0071】
図1は、SSIIRTを用いて45℃で合成した第1鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1〜5はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比を用いた反応条件を示す。図2は、TSIIRTを用いて合成した第1鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1〜5はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比を用いた45℃での反応条件を示す。レーン6〜10はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比をを用いた50℃での反応条件を示す。レーン11〜15はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比を有する55℃での反応条件を示す。その結果は、オリゴ(dT)プライマー/mRNAのモル比を減少させることによって(好ましくは1:1)、逆転写酵素による内部プライミングがほとんど完全に除去されたことを示す。
【0072】
(実施例2:標準およびホットスタート条件下での第1鎖cDNA合成の比較)
標準反応およびホットスタート条件でのMAP4遺伝子の第1鎖cDNA合成を比較するために、この実験を設計した。
【0073】
1μgのMAP4 mRNAおよびビオチン化NotIオリゴ(dT)25プライマー((ビオチン)4 GACTAGTTCTAGATCGCGAGCGG CCGCCCTTTTT TTTTTTTTTTTT TTTTTTTT;第WO98/51699号を参照のこと)を、薄壁(thin−walled)PCRチューブに0:1、1:1、または15:1のオリゴ(dT)/mRNAの望ましいモル比で混合し、そして水で容量を10μlにすることによって、アニーリングミックスを調製した。いくつかのチューブが同じである場合、1つのバッチとして作成し、そしてしかるべく等分し得る。アニーリングミックスを氷上で保持した。
【0074】
Superscript II逆転写酵素(SSIIRT)についてのマスターミックスを、下記の表5で指定したように調製した。
【0075】
【表5】
次いでSSIIRTマスターミックスを2つの等量のアリコートに分け、その1つは標準反応温度で処理するためであり(バッチ1)、そして1つはホットスタート反応温度で処理するためである(バッチ2)。凝結を可能にするために、さらに10%容量の水をバッチ2に加えた。全てのミックスを氷上に保持した。
【0076】
小滴を回収するためにアニーリングミックスを含むチューブを短時間回転させ、このチューブをサーモサイクラー(thermocycler)中に置き、そして次いで70℃で10分間加熱することによって第1鎖cDNAの合成を開始した。この70℃で10分間のサイクルの後、バッチ1のアニーリングミックスのチューブを、すぐに氷上へ移動させた。バッチ2のマスターミックスをサーモサイクラー中に置き、そして45℃で5分間インキュベートしながら、バッチ2のアニーリングミックスのチューブを、サーモサイクラー中で45℃に冷却した。5分間のインキュベーションの後、11μlのバッチ2のマスターミックスを、バッチ2アニーリングチューブそれぞれに加え、そしてピペットで2回混合した。温度の低下を避けるために、チューブを回転させないように気をつけた。
【0077】
10μlのバッチ1のマスターミックスを、バッチ1アニーリングチューブそれぞれに加えた。バッチ1チューブを軽くボルテックスして、そして短時間遠心分離して凝結小滴を回収した。次いでバッチ1チューブを、サーモサイクラーに戻して、そしてバッチ1および2両方のチューブを45℃で1時間インキュベートした。
【0078】
各チューブからの5μlの第1鎖cDNAを、5μlの標準ローディング緩衝液(60mMのNaOH、4mMのEDTA、0.1%のブロモフェノールブルー)と混合し、そして分析のために1.4%のアルカリアガロースゲルにローディングした。結果を図3に示す。
【0079】
図3は、SSIIRTを用いて合成した第1鎖cDNAのオートラジオグラフである。レーン1、3および5はそれぞれ、0:1、1:1および15:1のビオチン化オリゴ(dT)/mRNAのモル比を用いたバッチ1反応条件を示す。レーン2、4および6はそれぞれ、0:1、1:1および15:1のビオチン化オリゴ(dT)/mRNAのモル比を用いたバッチ2反応条件を示す。
【0080】
第1鎖DNAをまた、1μgのmRNAあたり15単位のTSIIRTを用いて、1:1および15:1のビオチン化オリゴ(dT)/mRNA比を用いて、TSIIRTで合成した。温度を50℃に変えた以外は、上記で記載した同じプロトコールに従った。結果を図4に示す。図4は、TSIIRTを用いて合成した第1鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1および3はそれぞれ、1:1比および15:1比で標準反応温度を用いた反応条件を示す。レーン2および4は、上記で記載したように、それぞれ1:1比および15:1比でのホットスタート反応条件を示す。
【0081】
その結果は、プライマーおよびmRNA混合物の変性後、反応温度を逆転写酵素反応温度まで下げることによって、反応が直接開始され、そして内部プライミングが完全に避けられたことを示した。
【0082】
(実施例3:反応温度ならびに塩およびRNaseの濃度を制御することによる二本鎖cDNAの合成)
この実施例は、第1鎖cDNA合成の後、cDNA/mRNAハイブリッドの処理中に、反応温度ならびに塩および異なるリボヌクレアーゼ(RNase)の濃度を制御することによる、二本鎖cDNAの合成を記載する。
【0083】
第1鎖cDNAを、上記の実施例2で記載したように合成し、そして下記でさらに記載するようにRNaseIまたはRNaseAのいずれかで消化した。
【0084】
第1鎖cDNAを、180μlの水および20μlの10×RNaseI緩衝液(100mMのTris−HCl(pH7.5)、50mMのEDTA、2Mの酢酸ナトリウム)に再懸濁することによって、第1鎖cDNAのRNaseI消化を行った。2.5単位のRNaseI(1単位/μg mRNA)を加え、そして混合物をよく混合した。RNaseI消化混合物を25℃で30分インキュベートし、そしてフェノール/クロロホルムを用いて1回抽出した。上清を1μlのグリコーゲン、100μlの酢酸アンモニウム、および800μlのエタノールを用いて沈殿させた。
【0085】
第1鎖cDNAを、200μlの消化緩衝液(10mMのTris−HCl(pH7.5)、250mMのNaCl)に再懸濁することによって、第1鎖cDNAのRNaseA消化を行った。12.5ngのRNaseA(5ng/μg mRNA)を加え、そして混合物をよく混合した。RNaseA消化混合物を25℃で30分インキュベートし、そしてフェノール/クロロホルムを用いて1回抽出した。上清を1μlのグリコーゲン、100μlの酢酸アンモニウム、および800μlのエタノールを用いて沈殿させた。
【0086】
(実施例4:キャップ結合タンパク質を用いた全長cDNAクローンの富化)
この実施例は、キャップ結合タンパク質eIF4Eを用いた全長cDNAクローンの富化を記載する。
【0087】
上記の実施例3で記載したRNaseIで処理した第1鎖cDNAを沈殿させ、そして70%エタノールを用いて洗浄することによってcDNAを調製した。得られたペレットを室温で5分間乾燥させ、そして210μlの10mM KPO4、100mMのKCl、2mMのEDTA、6mMのDTTおよび5%のグリセロールに再懸濁させた。cDNAを氷上に保存した。
【0088】
eIF4Eグルタチオンセファロース4Bビーズを、最初にグルタチオンセファロース4Bビーズ(Pharmacia、Sweden)をよく混合することによって調製した。eIF4Eビーズを調製するために、GSTタグeIF4Eタンパク質を発現する組換え宿主細胞(eIF4E遺伝子をGST融合ベクターにクローニングし、N末端GST−eIF4E融合遺伝子を作成した)を増殖させ、そして融合タンパク質を標準的な技術によって精製した。従って、本発明はまた、eIF4Eタンパク質(特に融合タンパク質として)を発現する組換え宿主細胞、そのようなタンパク質または融合タンパク質を発現する遺伝子を含むベクター、および産生される組換えタンパク質または融合タンパク質に関連する。本発明において、あらゆるタグを使用し得る(例えば、Hisタグ、GSTタグ、HAタグ、Trxタグ等)。そのようなタグは、eIF4E遺伝子のカルボキシ末端領域および/またはN末端領域に位置し得る。
【0089】
GST−eIF4E融合タンパク質を、製造会社のプロトコールに従って、グルタチオンセファロース4Bビーズ(Pharmacia Biotech)を用いて、グルタチオン結合によって、セファロース4Bビーズと複合体化した。200μlのビーズを、1.5mlの微量遠心(microcentrifuge)チューブに移し、1秒間遠心分離し、そして75μlの上清を除去した。ビーズを1mlの反応緩衝液(10mMのKPO4、100mMのKCl、2mMのEDTA、6mMのDTTおよび5%のグリセロール)を用いて2回洗浄し、そして258μlの反応緩衝液に再懸濁し、続いて42μl(18pmol/μl)のeIF4Eタンパク質(600pmol/100μlビーズ)を加えた。混合物を、接近(head to head)ローラーで、4℃で30分間混合した。混合物を次いで1秒間遠心分離し、そして上清を除去した。ビーズを1mlの反応緩衝液を用いて2回、そして反応緩衝液中1mlの25μg/ml酵母tRNAを用いて1回洗浄した。次いで20μlの反応緩衝液および5μgの酵母tRNAをビーズに加えた。200μlのRNaseI処理cDNAをビーズに加え、そして内容物をローラーで、室温で1時間混合した。1時間後、混合物を1秒間遠心分離し、そして上清を除去した。ビーズを1mlの反応緩衝液を用いて2回、そして反応緩衝液中1mlの500μM GDPを用いて1回洗浄した。cDNAを、反応緩衝液中250μlの500μM GDPで2回溶出した。溶出溶液をプールし、そして1分間遠心分離してビーズを除去した。溶出したcDNAを、等容量のフェノール/クロロホルムで2回抽出した。cDNAを2つのチューブに分け、そして1μlのグリコーゲン、0.5容量の7.5M酢酸アンモニウムおよび2.5容量のエタノールを用いて沈殿させた。
【0090】
(実施例5:cDNAライブラリーの評価)
上記で記載した全長法で構築したcDNAライブラリーの質を評価するために、MAP4遺伝子(5〜6kb)および他の遺伝子を標的遺伝子として選択した。MAP4および他のcDNAクローンを、当該分野で周知の標準的な方法(SuperScriptTM Plasmid Manual、Life Technologies,Inc.を参照のこと)ならびに3’および5’GeneTrapper cDNA Positive Selection System(Life Technologies,Inc.、Rockville、Maryland)を用いた上記で記載した全長法によって構築したライブラリーから単離した。陽性のクローンを、PCRによって大きさを分析した。下記の表6および7は、当該分野で周知の方法(コントロール)および上記で記載した全長法(全長法)で構築したヒト線維芽細胞cDNAライブラリーにおける全長cDNAクローンの富化の結果をまとめる。
【0091】
【表6】
コントロールライブラリーを、公知の方法を用いてSSIIRTで構築した。
【0092】
【表7】
これらの結果は、上記で記載した全長法は、標準的な方法を用いた<13%に比べて、5’GeneTrapperシステムによって>90%の全長cDNAクローンを産生したことを示す。さらに、上記で記載した全長法は、標準的な方法を用いた<7%に比べて、3’GeneTrapperシステムによって>37%の全長クローンを産生した。
【0093】
(実施例6:第1鎖cDNA合成、RNaseI消化、およびeIF−4E捕捉)
以下を除いて、上記の実施例2、3(RNaseI)および4で記載した全ての条件およびパラメーターに従った:1反応あたり10μgのヒト線維芽細胞細胞質mRNAという4つの反応を使用した(第WO98/45311号を参照のこと);ビオチン化プライマー−アダプター(ビオチン)4−GACTAGTTCTAGATCGCGAGCGGCCGCCC(T)25を、1:1のプライマー/mRNAモル比で使用した;TSIIRTを50℃で使用した;そしてSSIIRTを45℃で使用した。下記の表8は、第1鎖cDNAおよびeIF−4E捕捉の結果をまとめる。
【0094】
(実施例7:第2鎖cDNA合成)
最初に上記の実施例6で得た4つの反応ペレットそれぞれを、104μlのDEPC処理水に溶解し、そして次いで以下の試薬を各反応に加えることによって、第2鎖cDNAを合成した:
4μlの5×第1鎖緩衝液*
30μlの5×第2鎖緩衝液*
2μlの0.1M DTT
4μlの10mM dNTP
1μlのE.coli DNAリガーゼ(10単位/μl)
1μlのE.coli RNase H(2単位/μl)
4μlのE.coli DNAポリメラーゼ(10単位/μl)
SuperScript Plasmid Systemマニュアル(Life Technologies,Inc.、Rockville、Maryland)を参照のこと。
【0095】
これらの反応混合物を、次いで16℃で2時間インキュベートした。2μlのT4 DNAポリメラーゼ(5単位/μl)を加え、そして16℃でのインキュベーションをさらに5分間続けた。
【0096】
(実施例8:ストレプトアビジンビーズの調製)
上記の実施例7で記載した2時間の第2鎖反応の最後の30分間に、ストレプトアビジン常磁性体ビーズを以下のように調製した。
【0097】
ストレプトアビジン常磁性ビーズ(Seradyn)を、ビーズが完全に再懸濁するまでピペッティングによって静かに混合した。150μlの混合ビーズを、各反応のために微量遠心チューブの底に移した。チューブをMagna−Sep Magnetic Particle Separator(Life Technologies,Inc.、Rockville、Maryland)(磁石)に挿入して、そして2分間静置した。チューブがマグネット中にある間に、上清をピペッティングによって除去し、そして100μlのTE緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA)をすぐにビーズに加えた。
【0098】
次いでチューブを磁石から取り出し、そして指で軽くたたくか、または一番低い設定でボルテックスして、ビーズを静かに再懸濁した。チューブを磁石に再び挿入した。2分後、上清を除去し、ビーズを160μlの結合緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA、1MのNaCl)に再懸濁し、そしてチューブを微量遠心チューブラックに置いた。
【0099】
(実施例9:二本鎖cDNAライブラリーの捕捉)
上記の実施例7で記載したように、第2鎖反応物とT4 DNAポリメラーゼとをインキュベートした後、反応混合物を氷上に置き、そして10μlの0.5M EDTAを加えた。次いでcDNAライブラリーを以下の手順に従って捕捉した(一般的には第WO98/51699号を参照のこと)。
【0100】
実施例8に従って調製した常磁性ビーズを、第2鎖反応混合物チューブへ移し、そしてピペッティングによって静かに混合し、そして懸濁液を室温で60分間インキュベートした。次いでチューブを磁石に挿入した。2分後、上清を除去および廃棄した。
【0101】
100μlの洗浄緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA、500mMのNaCl)をビーズに加え、指で軽くたたくか、または一番低い設定でボルテックスしてビーズを再懸濁し、そしてチューブを2分間磁石へ再び挿入した。上清を除去および廃棄した。この洗浄工程をもう1回反復し、そして次いで100μlの洗浄緩衝液をビーズに加えた。次いでチューブを5分間磁石に再び挿入した。
【0102】
(実施例10:NotI消化)
実施例9の最終工程で記載した5分間のインキュベーション後、常磁性ビーズから上清を除去および廃棄し、そして41μlのオートクレーブ処理蒸留水、5μlのREact3緩衝液、4μlのNotIを加え、そしてビーズをピペッティングによってよく混合した。次いで反応物を37℃で2時間インキュベートした。次いでチューブを2分間磁石に挿入し、そしてcDNAライブラリーを含む上清を新しいチューブに移した。
【0103】
50μlのフェノール:クロロホルム:イソアミルアルコール(25:24:1)を上清に加え、溶液を完全にボルテックスし、そして次いで14,000×gで5分間、室温で遠心分離した。45μlの上部の水層を注意深く取り出し、そして新しい微量遠心チューブへ移した。23μlの7.5M酢酸アンモニウム、1μlのグリコーゲン(20μg)および172μlのエタノール(−20℃)を加えた。溶液をよく混合して、そしてドライアイス上(または−70℃冷凍庫)で15分間保存した。
【0104】
次いでエタノール溶液を14,000×gで30分間、4℃で遠心分離した。上清を小さなペレットから注意深く除去した。100μlの70%エタノールを加え、そしてチューブを14,000×gで2分間、室温で遠心分離した。エタノールを除去し、そしてペレットをスピードバック(speed−vac)で2分間、または乾燥するまで乾燥した。次いでペレットを20μlのTE緩衝液(10mMのTris−HCl(pH7.5)、0.1mMのEDTA)に溶解した。cDNAの最終的な収量を、チェレンコフカウント(Cerenkov counts)によって決定した(下記の表8を参照のこと)。
【0105】
【表8】
(実施例11:cDNAのベクターへの連結およびE.coliへの導入)
10から30ngの非分画またはサイズ分画(低融点ゲル電気泳動によって≧1.5kb)cDNAを、ベクターpCMVSPORT6(Life Technologies,Inc.)に連結した。この連結を、クローニングベクターをNotIおよびEcoRVで予め消化した以外は、SuperScript Plasmid Systemマニュアル(Life Technologies,Inc.、Rockville、Maryland)で記載されたように、エレクトロポレーションによってE.coliに導入した。
【0106】
構築したcDNAライブラリーから無作為に選んだクローン(304クローン)の配列分析を、5’および3’配列決定によって分析し、cDNAライブラリー中の全長ランダムクローンの全百分率を決定した。配列をGeneBank配列と相同性に関して比較した。結果を下記の表9にまとめる。その結果に基づいて、約68%のランダムクローンが全長(公知の全長クローンおよび未知の全長クローンを含む)であった。従って、ヒト線維芽細胞細胞質mRNAライブラリーから、約17%の未知全長クローンが得られた。
【0107】
【表9】
(実施例12:RNaseアッセイ)
第1鎖cDNAを、TE緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA)中で1000ng/μg mRNAのRNaseAで、およびTEN(10mMのTris−HCl(pH7.5)、5mMのEDTA(pH8.0)、200mMの酢酸ナトリウム)中で25から40u/μg mRNAのRNaseIで、本質的に実施例3で記載したように37℃で処理した。しかし、上昇した温度での大量のRNaseを用いたこの処理は、非常に小さい平均cDNAインサートの大きさ(約200bp)を含むライブラリーを生じた。従って、必要なRNaseの最適量を決定するために、第2鎖cDNAアッセイを開発した。
【0108】
第1鎖cDNA(放射性標識および非放射性標識)を、500ngのRNA/反応でHeLa mRNAを用いて合成した。第1鎖cDNAを、エタノールを用いて沈殿させ、そしてDEPC−処理水に溶解した。コールドの第1鎖cDNAを、種々の量のRNaseを含むRNase緩衝液に加えた。25℃で30分間インキュベートした後、処理したcDNAをフェノール:クロロホルムを用いて抽出し、そしてエタノールを用いて沈殿させた。処理したcDNAをDEPC処理水に溶解し、32P−dCTPプラスおよびマイナスRNase Hを用いて第2鎖cDNA反応を行った。反応物をフェノール:クロロホルムを用いて抽出し、そしてエタノールを用いて沈殿させた。等量のcpmを1.4%アルカリアガロースゲルに電気泳動した。結果を図5および図6に示す。
【0109】
図5は、種々の量のRNaseAを用いて合成した第2鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1は、未処理の第1鎖cDNAを示す。レーン2は、未処理の第2鎖cDNAを示す。レーン3、5、7、および9は、RNase H無しで、そしてそれぞれ0、1.25ng、2.5ng、および5ngの濃度のRNaseAを用いて合成した第2鎖cDNAを示す。レーン4、6、8、および10は、RNase Hを用いて、そしてそれぞれ0、1.25ng、2.5ng、および5ngの濃度のRNaseAを用いて合成した第2鎖cDNAを示す。
【0110】
図6は、種々の量のRNaseIを用いて合成した第2鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1は、未処理の第1鎖cDNAを示す。レーン2は、未処理の第2鎖cDNAを示す。レーン3、5、7、および9は、RNaseH無しで、そしてそれぞれ0、0.5u、1.25u、および2.5uの濃度のRNaseIを用いて合成した第2鎖cDNAを示す。レーン4、6、8、および10は、RNase Hを用いて、そしてそれぞれ0、0.5u、1.25u、および2.5uの濃度のRNaseIを用いて合成した第2鎖cDNAを示す。
【0111】
これらのゲル分析は、1.25ngの濃度のRNaseA(図5を参照のこと)または0.5単位のRNaseI(図6を参照のこと)が、500ngの出発mRNAについて用いるのに最適であり得ることを証明した。
【0112】
(実施例13:キャップ構造に対する抗体の調製)
キャップに対する抗体を、m7グアノシン−KLHを抗原として用いて産生した。1200個のハイブリドーマをプレーティングし、そして120個のコロニーのみが産生された。これらのうち6個のコロニーのみがキャップに関して陽性であった。さらなる分析の後、3つが必要な親和性を有すると決定された。最初のスクリーニングELISAは、m7グアノシン−BSAのELISAプレートへの結合、BSAを用いたブロック、ハイブリドーマ上清の結合、2次抗体との反応、およびBCIP/NPTを用いた比色定量反応による陽性の決定からなる。2次スクリーニングは、ハイブリドーマ上清の適切な希釈物を、0.1mMのm7GTP、0.1mMのキャップアナログM7G5'ppp5'G、0.5mMのm7グアノシンまたは0.5mMのGTPのいずれかとインキュベートすることを含んでいた。前処理した上清を次いで標準的なELISA手順で使用した。GTPはm7グアノシン−BSAと競合しなかったが、m7バージョンは全て効率よく競合した。
【0113】
理解の明解さのために、説明および例によってここで本発明をある程度詳細にわたって完全に記載したが、本発明またはそのあらゆる特定の実施態様の範囲に影響を与えることなく、条件、処方および他のパラメーターの広いおよび同じ範囲内で、本発明を改変または変化させることによって本発明を実施し得ること、およびそのような改変または変化は、添付した特許請求の範囲内に含まれると意図されることが、当業者に明らかである。
【0114】
本明細書中で言及された全ての出版物、特許および特許出願は、本発明が属する、当該分野の技術水準を示し、そして本明細書中で、個々の出版物、特許または特許出願が、具体的かつ個々に参考として援用されると示されたのと同じ程度、参考として援用される。
【技術分野】
【0001】
(発明の背景)
本発明は、分子生物学および細胞生物学の分野に関する。本発明は一般に、cDNAの合成方法に関する。より詳細には、本発明は、平均のcDNAインサートの大きさを増大させる方法に関し、さらに具体的にはcDNAライブラリー中に存在する全長cDNAの百分率を増大させる方法に関する。それゆえ、本発明は遺伝子の発見に有用な改良されたcDNAライブラリーを提供する。
【背景技術】
【0002】
生物体、組織または細胞の構造および生理機能を検査する際に、その遺伝内容を決定することがしばしば望まれる。生物体の遺伝子のフレームワークは、生物体の体細胞および生殖細胞中に含まれるデオキシリボ核酸(DNA)中のヌクレオチド塩基の二本鎖配列でコードされる。DNAの特定のセグメント、すなわち遺伝子の遺伝内容は、その遺伝子がコードするタンパク質の産生でのみ明らかになる。タンパク質を産生するために、DNA二重らせんの一本の鎖(「コード」鎖)の相補的なコピーがポリメラーゼ酵素によって産生され、その結果、リボ核酸(RNA)の特異的配列がもたらされる。この特定のタイプのRNAは、タンパク質を産生するためのDNAからの遺伝メッセージを含んでいるので、メッセンジャーRNA(mRNA)と呼ばれる。
【0003】
所定の細胞、組織または生物体の中には、多くのmRNA種が存在し、各々が別個の特異的タンパク質をコードしている。この事実は、組織または細胞における遺伝子の発現の研究に興味を抱く研究者に対して強力なツールを提供する。mRNA分子を単離し、そして種々の分子生物学的技術によってさらに操作することができ、それにより細胞、組織または生物体の機能的遺伝内容の全体を解明することが可能となる。
【0004】
遺伝子発現の研究に対する共通のアプローチは相補的DNA(cDNA)クローンの産生である。この技術においては、生物体由来のmRNA分子が、その生物体の細胞または組織の抽出物から単離される。この単離には、チミジン(T)のオリゴマーを複合体化した、セルロースまたはアガロースのような固体クロマトグラフィーマトリックスを使用することが多い。大部分の真核mRNA分子の3’末端は一連のアデノシン(A)塩基を含み、そしてAはTに結合するので、mRNA分子は組織または細胞抽出物中の他の分子および物質から迅速に精製され得る。これらの精製されたmRNA分子から、cDNAコピーが、逆転写酵素(RT)またはRT活性を有するDNAポリメラーゼを用いて作製され得、これにより、一本鎖cDNA分子が産生される。次に、この一本鎖cDNAは、DNAポリメラーゼの作用により、元のmRNAの(従って、このmRNAをコードする、生物体のゲノムに含まれる元の二本鎖DNA配列の)完全な二本鎖DNAコピー(すなわち二本鎖cDNA)に転換され得る。このタンパク質特異的二本鎖cDNAは次に、ベクター中に挿入され、これが次に宿主の細菌、酵母、動物または植物細胞中に導入され、このプロセスは形質転換またはトランスフェクションと呼ばれる。宿主細胞を次に培地中で増殖させ、その結果、目的の遺伝子または目的の遺伝子の一部を含む(あるいは、多くの場合、それを発現する)宿主細胞の集団がもたらされる。
【0005】
この全体のプロセスは、mRNAの単離から、cDNAのベクター(例えば、プラスミド、ウイルスベクター、コスミド等)への挿入、単離された遺伝子または遺伝子の一部を含む宿主細胞集団の増殖までが「cDNAクローニング」と呼ばれる。cDNAが多数の異なるmRNAから調製されるならば、得られるcDNAのセットは「cDNAライブラリー」と呼ばれ、これは適切な用語である。なぜなら、cDNAのセットは、供給源細胞、組織または生物体中に存在する機能的遺伝情報を含む遺伝子または遺伝子の一部の「集団」を表すからである。これらのcDNAライブラリーの遺伝子型分析は、それが由来する生物体の構造および機能に対する多くの情報を与え得る。
【0006】
産生されるcDNAの総量を増大させる能力、より詳細には、cDNA分子の平均の大きさが増大したcDNAライブラリーを生成する能力、および/または全長cDNA分子の百分率が増大したcDNAライブラリーを生成する能力は、cDNAライブラリーの構築に顕著な進歩を与える。特に、このような進歩は、目的の全長遺伝子を見いだす確率を大幅に向上させる。
【0007】
理想的には、cDNA分子の合成を、mRNA分子の3’末端またはその近傍から開始する。cDNA合成をポリAテールの3’末端でオリゴ(dT)プライマーを用いてプライミングすることにより、mRNAの3’メッセージが、産生されるcDNA分子中に示されることを確実にする。mRNA分子内で生じるプライミング(内部プライミング)は、目的の遺伝子についての全長メッセージを含まないcDNA分子の合成をもたらす。すなわち、内部プライミングは目的の遺伝子(単数または複数)の一部のみを含む短縮型cDNA分子をもたらす。典型的には、内部プライミングは、メッセージ集団から3’配列の損失を引き起こす。それゆえ、内部プライミングは、産生されるcDNAの総量を減少させ、cDNAライブラリーについてのcDNA分子の平均のインサートの大きさを減少させ、および/または所定のcDNAライブラリーにおける全長cDNA分子の百分率を低下させる。配列決定分析は、多くの真核生物mRNAが内部ポリアデニル化区間(stretch)を有することを示し、これはオリゴ(dT)プライマーが逆転写酵素による第1鎖cDNA合成のために用いられる場合にプライミング部位として働き得る。さらに、いくつかのmRNAは16個もの内部プライミング部位を有し得ることが研究によって示されている(Lovett,M.ら、The construction of full−length cDNA libraries by conventional methods and a novel double capture technique、University of Texas Southwestern Medical Center, Dallas,TX、The American Society of Human Genetics主催、第48回年会、1998年10月27日〜31日、Denver、Coloradoにて発表)。それゆえ、このような内部ポリA配列に対するプライマーの内部プライミングはcDNA合成に悪影響を与え得る。
【発明の概要】
【0008】
本発明は、内部プライミングを軽減、防止、低減または実質的に低減し、それによりcDNAおよびcDNAライブラリー構築の改良を提供する。従って、本発明は、全長遺伝子を高百分率で含むcDNAライブラリーを提供することによって、遺伝子の発見を大幅に促進する。
【0009】
従って、本発明は、mRNA鋳型またはmRNA鋳型の集団からcDNA分子(単数または複数)を、生成されるcDNAの総量を増大させる、生成されるcDNA分子の長さを増大させる、および/または生成される全長cDNA分子の量または百分率を増大させるのに十分な条件下で合成することに関する。本発明によれば、内部プライミングを阻害、防止、低減または実質的に低減する任意の条件が使用され得る。このような条件は、好ましくは、プライマー濃度の至適化、反応温度の至適化および/またはプライマーの長さもしくは特異性の至適化を包含するが、これらに限定されない。このような結果はまた、本発明によれば、至適化することによって、好ましくは至適または所望の反応条件が達成されるまで逆転写反応を阻害または防止することによって、達成され得る。
【0010】
cDNAライブラリーを構築するための従来法は、第1鎖cDNA合成のために15:1のオリゴ(dT)プライマー/mRNA鋳型のモル比を用いる。このような過剰量のオリゴ(dT)プライマーの使用は、反応中に、1つ以上のプライマーの、1つ以上のmRNA鋳型に対する内部プライミングを可能にする。本発明の好適な局面によれば、第1鎖cDNAの合成のためのオリゴ(dT)プライマーの量を低減して、内部プライミングを阻害、防止、低減または実質的に低減する。プライマー対鋳型の好ましいモル比は約12:1;10:1;9:1;8:1;7:1;6:1;5:1;4:1;3:1;2:1;1:1;1:2;1:3;1:4;1:5;1:6;1:7;1:8;1:9;1:10および1:12の範囲である。好ましくは、プライマー(例えば、オリゴ(dT))対鋳型(例えば、mRNA)のモル比は約5:1から約1:20の範囲であるが、それより低いプライマー対鋳型のモル比も本発明に従って用いられ得る。特に、プライマー対鋳型のモル比は、約1:10;1:15;1:20;1:25;1:50;1:75および1:100未満であり得る。好ましくは、モル比の範囲は、約5:1;4:1;3:1;2:1;1:1;1:2;1:3;1:4;および1:5未満である。最も好ましくは、プライマー対鋳型の比は、約10:1から1:10;5:1から1:10;4:1から1:10;3:1から1:10;2.5:1から1:10;2:1から1:10;1.5:1から1:10;および1:1から1:10の範囲である。プライマー対鋳型の至適な比は、プライマー、mRNA、逆転写酵素および反応条件(アニーリング温度、緩衝剤塩など)に応じて変化し得る。所望のプライマー対鋳型の比は当業者によって容易に決定され得る。
【0011】
cDNAライブラリー構築の従来法において、プライマーの鋳型へのアニールまたはハイブリダイズは、内部プライミングを防止、阻害、低減または実質的に低減する温度では行われない。典型的には、混合物(例えば、mRNAおよびオリゴ(dT)プライマー)は変性または加熱の後、氷上で冷却される。このプロセスは典型的には、内部部位へのプライマーのアニーリングまたはハイブリダイゼーションを引き起こす。本発明の好適な局面によれば、プライマーと鋳型との間のアニーリングまたはハイブリダイゼーションの際の温度は、内部プライミングが阻害、防止、低減または実質的に低減するように維持される。本発明によれば、このような結果は、プライマーのアニーリングまたはハイブリダイゼーションをより高い温度で行うことによって達成される。このような条件はまたcDNA合成の際のmRNA二次構造の形成を低減し得る。好ましくは、プライマーを鋳型にアニールまたはハイブリダイズさせるための温度は、約10℃から約90℃;より好ましくは約10℃から約80℃;さらに好ましくは約20℃から約75℃;より好ましくは約25℃から約75℃;さらになお好ましくは約30℃から約65℃;さらになお好ましくは約37℃から約60℃;さらになお好ましくは約40℃から約60℃;さらになお好ましくは約45℃から約60℃;さらになお好ましくは約45℃から約55℃;そして最も好ましくは約45℃から約65℃の範囲である。用いられる温度はプライマーおよび鋳型のタイプおよび量に応じ、そして逆転写酵素の至適温度に応じて、変化し得る。至適温度または温度範囲は当業者によって容易に決定され得る。
【0012】
cDNA合成のための従来法は、典型的には、特定の長さ(12〜18塩基またはマー)のオリゴ(dT)プライマーの使用を必要とする。しかし、このようなプライマー長はプライマーの特異性を低下させ、これにより内部プライミングを可能にする。それゆえ、本発明はまた、プライマーの特異性を高めて、内部プライミングを防止、阻害、低減または実質的に低減することに関する。好適な局面において、プライマーの特異性はプライマーの長さを延ばすことによって高められる。それゆえ、本発明によれば、cDNA合成のために、長めのオリゴ(dT)プライマーが用いられ得る。好ましくは、プライマー長は約20から約100塩基、約20から約75塩基、約20から約60塩基、そして約20から約50塩基の範囲であり;より好ましくは約20から約45塩基;より好ましくは約20から約40塩基;そして最も好ましくは約25から約35塩基の範囲である。好適な局面において、プライマーの長さは19塩基より長く;より好ましくは約20塩基より長く;より好ましくは約25塩基より長く;そしてさらにより好ましくは約30塩基より長い。このようなプライマー長は、鋳型にアニールまたはハイブリダイズするプライマーの長さに関連している。プライマーの至適な長さおよび内容(ヌクレオチド配列)は鋳型のタイプ、所望の反応条件、および逆転写酵素に応じて変化し得る。本発明によれば、追加の配列および/または修飾ヌクレオチドを本発明のプライマーに含めることができる。例えば、追加配列(これは必ずしも鋳型にアニールまたはハイブリダイズする必要はない)は、cDNA合成を補助するために本発明のプライマー中に含めることができ、このような追加配列は、1つ以上の制限エンドヌクレアーゼ部位、1つ以上の誘導体ヌクレオチド(例えば、ビオチン化ヌクレオチドのようなハプテン含有ヌクレオチド)などを含む配列を包含する。本発明に従って用いられるプライマーのタイプおよび長さは当業者によって容易に決定され得る。
【0013】
従来のcDNA合成法は、逆転写酵素反応を至適化するために逆転写酵素を制御したり、あるいはその活性を変化させたりしない。本発明によれば、逆転写酵素の活性は、好ましくは、反応中の所望の時間に合成を開始するために、制御される。好適な局面において、逆転写酵素活性は、至適または所望の反応条件に到達するまで、阻害または防止される。このような結果は、本発明によれば、逆転写酵素活性を阻害するインヒビター(例えば、抗体または抗体フラグメント)の使用によって達成される。このような逆転写酵素インヒビターは、内部プライミングが防止、阻害、低減または実質的に低減されるように、逆転写酵素活性を低温で防止または阻害する。本発明によれば、このようなインヒビターは好ましくは逆転写酵素活性を35℃未満、40℃未満、45℃未満、50℃未満、55℃未満、60℃未満、65℃未満、70℃未満、75℃未満、80℃未満、85℃未満、および90℃未満で防止する。逆転写酵素活性を有する酵素の熱安定性に応じて、インヒビターは、目的の酵素について至適な温度またはその近傍の点で酵素の活性を阻害するように設計され得る。好ましくは、インヒビターは、使用する酵素の至適温度未満または近傍の温度で不活化され、それにより逆転写が起こることを可能にする。それゆえ、本発明は一般に、cDNA合成における逆転写酵素インヒビターの使用に関する。インヒビターのタイプおよび量は逆転写酵素のタイプおよび量に応じ、そして用いられる反応条件に応じて変化し得る。インヒビターのタイプならびにこのようなインヒビターについて使用する条件は当業者により容易に決定され得る。
【0014】
本発明によれば、cDNA合成に対する上記の改良のいずれか1つあるいは組合せが用いられ得る。これらの改良のいずれか1つあるいは組合せの使用は、改良された第1鎖cDNA合成を提供する(例えば、総cDNAの増加、cDNAの増加および/または全長cDNAの増加)。本発明によれば、第1鎖cDNA分子は、1つ以上の二本鎖核酸分子(例えば、二本鎖cDNA分子)を、本発明の方法によって産生される1つ以上の第1鎖cDNA分子をこの第1鎖cDNA分子の全てまたは一部と相補的な1つ以上の核酸分子の作製に十分な条件下でインキュベートすることによって作製するための鋳型として用いられ得る。二本鎖核酸分子の作製のための条件は、好ましくは、1つ以上のDNAポリメラーゼ、1つ以上のヌクレオチド、1つ以上の緩衝化塩、および1つ以上のプライマーからなる1つ以上の成分と共にインキュベートすることを含む。本発明の別の局面において、このような条件は、生成される二本鎖cDNAの総量の増大、生成される二本鎖cDNA分子の長さまたは大きさの増大、および/あるいは生成される全長二本鎖cDNA分子の百分率の増大をもたらすように改変される。好ましくは、このような条件は、第1鎖cDNA合成の後のリボヌクレアーゼ(RNase)消化の至適化に関連する。第1鎖cDNA合成の間に、mRNA鋳型に対して相補的な全長cDNA分子が作製されないならば、キャップ構造を含む一本鎖mRNAがmRNA/cDNAハイブリッドのmRNAの5’末端に存在する。全長cDNAが生成されるならば、二本鎖mRNA/cDNAハイブリッドが生成され、一本鎖mRNAは存在しない。好ましくは、このような消化条件は、第1鎖cDNA合成の間に形成されるmRNA/cDNA二本鎖分子の一本鎖mRNAがRNase消化に供されるように至適化される。このようにして、全長ではないmRNA/cDNAハイブリッド由来のキャップ捕捉構造は除去されるが、全長mRNA/cDNAハイブリッドはキャップ構造を維持する。それゆえ、キャップ捕捉を用いて、全長分子について選択し、そして全長ではない分子に対して選択することができる。好適な局面において、この条件は、mRNA/cDNAハイブリッドの一本鎖mRNAが消化または分解され、他方、二本鎖mRNA/cDNAハイブリッドのmRNAが分解されないかまたは実質的に分解されないような条件である。それゆえ、このようなRNase消化は、第2鎖合成に実質的に悪影響を与えない条件下で行われる。すなわち、本発明に従う第2鎖合成は従来技術と比べてより大きな二本鎖cDNA分子を生成する。従来のRNaseI条件は典型的には37℃で25u/μgから40u/μg mRNAの範囲であり、そしてRNaseA条件は典型的には37℃で1000ng/μg mRNAである。従来のRNase消化を用いると、生成される二本鎖cDNA分子の平均の大きさは約200塩基である。本発明によれば、生成される二本鎖cDNA分子の平均の大きさは好ましくは約300塩基を超え、約400塩基を超え、約500塩基を超え、約600塩基を超え、約700塩基を超え、約800塩基を超え、約900塩基を超え、約1キロ塩基を超え、約1.5キロ塩基を超え、そして約2キロ塩基を超える。本発明の1つの実施形態において、リボヌクレアーゼの濃度、リボヌクレアーゼのタイプおよび反応条件は、本発明に従う二本鎖cDNA合成を改良するように至適化される。リボヌクレアーゼ消化において使用するための好ましいリボヌクレアーゼは、リボヌクレアーゼA(RNaseA)および/またはリボヌクレアーゼI(RNaseI)を包含する。一般に、低めの温度(約4℃から約50℃)および高めの塩濃度(約5mMから約5M)は、本発明によるRNase消化の阻害または制御を補助する。使用される塩は塩化ナトリウム、塩化カリウム、塩化マグネシウム、酢酸ナトリウムなどを包含し得る。従って、RNase量または濃度の低下を用いて、所望の結果が達成され得る。RNaseAについてのこのような濃度は約0.001ng/μg mRNAから約500ng/μg mRNAまでの範囲であり得、そしてRNaseIについては約0.001u/μg mRNAから約500u/μg mRNAの範囲であり得る。インキュベーション温度、RNase濃度および塩濃度は当業者によって容易に決定され得る。好適な局面において、RNaseAの濃度は、TE緩衝液(10mM Tris、pH7.5、1mM EDTA)中、37℃で、0.1ng/μg mRNAから10ng/μg mRNAの範囲を包含する。あるいは、RNaseAの濃度は、250mMのNaClを含む10mM Tris(pH7.5)緩衝液中、25℃で30分間、0.1ng/μg mRNAから500ng/μg mRNAの範囲を包含し得る。好ましくは、用いられるRNaseIの濃度は、10mM Tris−HCl(pH7.5)、5mM EDTA(pH8.0)、200mM酢酸ナトリウム中、37℃で、0.1単位/μg mRNAから1.0単位/μg mRNAの範囲である。あるいは、RNaseIの濃度は、同じ緩衝液中、25℃で30分間、1.0単位/μg mRNAから100単位/μg mRNAの範囲で用いられ得る。
【0015】
別の局面において、本発明は、第1鎖cDNA合成の前、間あるいは後での、mRNAのキャップ構造(例えば、m7GpppN)の捕捉または結合に関連する。それゆえ、本発明は、本発明の方法の実施において、キャップ構造を有するmRNA(第1鎖合成の前)またはmRNA/cDNAハイブリッド(第1鎖合成の後または間)の選択に関連する。このような選択または捕捉は、eIF4E、eIF4Eペプチド、eIF4Eペプチドフラグメント(WO 98/08865)のような任意のキャップ結合分子、およびキャップ構造に対して特異的な抗体または抗体フラグメントを用いて達成され得る。好適な局面において、キャップ構造の選択は第1鎖合成の後に達成される。より好ましくは、このようなキャップ捕捉は本発明の方法によるリボヌクレアーゼ消化の後に行われる。例えば、リボヌクレアーゼ消化に供されるmRNA/cDNAハイブリッドは捕捉され、そして次に本発明による第2鎖cDNA合成のために使用される。
【0016】
それゆえ、本発明は一般に核酸分子の合成方法に関する。本発明はより詳細には、1つ以上の核酸分子、特にcDNA分子またはcDNAライブラリーの作製方法に関し、この方法は、1つ以上の核酸鋳型(好ましくはmRNA、ポリA RNAまたはmRNA分子の集団)を逆転写酵素活性を有する少なくとも1つのポリペプチドと混合する工程、およびこの混合物を、1つ以上の核酸鋳型の全てまたは一部と相補的な1つ以上の第1の核酸分子(例えば、第1鎖cDNA)を作製するのに十分な条件下でインキュベートする工程を包含する。本発明に従うと、このような条件は、改良された改変も本発明の条件をも使用しない従来の手順と比較して増大した総量で産生された核酸分子(cDNA)を提供する。本発明はまた、改良された改変も本発明の条件をも使用しない従来の手順と比較して、産生された核酸分子(cDNA)の長さまたは平均の大きさの増大、および/あるいは産生された全長の核酸分子(cDNA)の百分率または量における増大を提供する。産生されたcDNAの量、長さ、および全長の含有量の決定は、当業者に周知であり、そして本明細書中に記載されるような従来技術によって決定され得る。本発明に従って産生されたcDNAライブラリー中の全長のcDNAの百分率または平均の百分率は、好ましくは、約15%より高く、より好ましくは約20%より高く、より好ましくは約25%より高く、より好ましくは約30%より高く、より好ましくは約40%より高く、より好ましくは約50%より高く、より好ましくは約60%より高く、より好ましくは約70%より高く、より好ましくは約80%より高く、そして最も好ましくは約90%より高い。このような全長の百分率は、好ましくは、目的のcDNAライブラリーのクローンの一部(例えば、100から1000クローン)のランダムな選択、そのクローンの配列決定、および公知の配列データベースに対する配列の比較によって決定される。
【0017】
本発明の好ましい局面においては、本発明の改良された結果は、好ましくは、核酸またはcDNAの合成のための条件に対する改変の1つまたは組合せによって達成される。このような条件は、好ましくは、第1鎖のcDNA合成を改良するための改変、および/または第2鎖のcDNA合成を改良するための改変を含む。
【0018】
好ましい局面においては、本発明は、特に、1つ以上の二本鎖のcDNA分子を作製する方法に関する。この方法は、1つ以上のmRNA分子(好ましくは、mRNA分子の集団)を本発明の1つ以上のプライマーとともに、第1鎖のcDNAの合成の前またはその間の内部プライミングを防ぐ、阻害する、減少させる、または実質的に減少させるような温度およびプライマー濃度で、インキュベートする工程を包含する。このような反応は、好ましくは、本発明に従って、1つ以上の逆転写酵素活性のインヒビターの存在下で行われる。リボヌクレアーゼ消化は、好ましくは、第2鎖のcDNAの合成の前に、そして第2鎖の合成の間に産生される二本鎖のcDNA分子の長さ、量、および/または大きさを増大させるために十分なリボヌクレアーゼ濃度で、行われる。本発明に従うと、キャップ捕捉は好ましくは、リボヌクレアーゼ消化の間またはその後で達成される。
【0019】
本発明はまた、上記の方法に従って産生された核酸分子およびcDNA分子またはcDNA分子の集団(一本鎖または二本鎖)、ならびにこれらの核酸分子およびcDNA分子を含有しているベクター(詳細には、発現ベクター)に関する。本発明はまた、このようなcDNA分子および/またはベクターを含有している宿主細胞に関する。
【0020】
本発明はまた、本発明の方法における使用のためのキットに関する。このようなキットは、一本鎖または二本鎖の核酸分子を作製するために使用され得る。本発明のキットは、キャリア(例えば、箱またはカートン)を含み、これは、その中に1つ以上の容器(例えば、バイアル、チューブ、ボトルなど)を有する。このようなキットは、以下からなる群より選択される少なくとも1つの成分を含み得る:プライマー(好ましくは、より高い特異性を有するプライマーであり、そして最も好ましくは、20塩基以上の長さを有するオリゴ(dT)プライマー)、逆転写酵素活性を有する1つ以上のポリペプチド(逆転写酵素およびDNAポリメラーゼ)、1つ以上の逆転写のインヒビター(例えば、RT活性を有するポリペプチドに対して指向された抗体および抗体フラグメント)、1つ以上のキャップ結合分子(例えば、キャップ構造に対して指向された抗体または抗体フラグメント)、核酸合成反応の緩衝液、1つ以上のヌクレオチド、1つ以上のベクター、および本発明の方法を実行するための説明書。
【0021】
本発明はまた、本発明における使用のための組成物、または本発明の方法を実行しながら作製される組成物に関する。このような組成物は、少なくとも1つのプライマー(例えば、オリゴ(dT)またはその誘導体)および少なくとも1つの鋳型を、本発明に従う量または比でサンプルまたは反応混合物中に含み得る。このような組成物はさらに、逆転写酵素活性を有する1つ以上のポリペプチド、1つ以上の逆転写のインヒビター(例えば、抗RT抗体またはそのフラグメント)、1つ以上のヌクレオチド、1つ以上のキャップ結合分子(例えば、抗キャップ抗体またはそのフラグメント)、1つ以上の緩衝塩などを含み得る。このような組成物はまた、本発明に従って内部プライミングを回避するような温度で維持され得る。
【0022】
本発明の組成物はまた、本発明に従う量のリボヌクレアーゼを含み得る。このような組成物はさらに、1つ以上のmRNA/cDNAハイブリッド、1つ以上のヌクレオチド、逆転写酵素活性を有する1つ以上のポリペプチド、1つ以上の緩衝塩、1つ以上のキャップ結合分子(例えば、抗キャップ抗体またはそのフラグメント)などから選択される少なくとも1つの成分を含み得る。
【0023】
本発明はまた、本発明の方法、組成物、およびキットにおける使用のための、1つ以上の抗体(モノクローナルおよびポリクローナル)ならびにそれらのフラグメントに関する。このような抗体として、抗キャップおよび/または抗RT抗体、ならびに抗体フラグメントが挙げられる。
【0024】
本発明の他の好ましい実施形態は、以下の図面および本発明の記載を参照して当業者に明らかである。
【図面の簡単な説明】
【0025】
【図1】図1は、1:1、2.5:1、5:1、10:1、および50:1のオリゴ(dT)25-30/mRNAのモル比を用いて、5/6Kbの鋳型を用いて45℃でSuperScriptTMII(SSII)RTで合成した第1鎖のcDNAのオートラジオグラフである。
【図2】図2は、1:1、2.5:1、5:1、10:1、および50:1のオリゴ(dT)25-30/mRNAのモル比を用いて、5/6Kbの鋳型を用いて45℃、50℃、および55℃でThermoScriptTMII(TSII)RTで合成した第1鎖のcDNAのオートラジオグラフである。
【図3】図3は、0:1、1:1、および15:1のビオチン化されたNotI−オリゴ(dT)25/mRNAのモル比を用いて、標準的な反応温度を使用して、および反応温度を変化させて、SSII RTで合成した第1鎖のcDNAのオートラジオグラフである。
【図4】図4は、標準的な反応条件(ここでは、プライマー/鋳型のアニーリングは、cDNA合成の前に、氷上でインキュベートされる)を使用して、および本発明に従う条件(ここでは、アニーリングおよび合成反応の温度は、約30℃より高く(好ましくは、37℃より高く)維持される)を使用して、1:1および15:1のビオチン化されたNotI−オリゴ(dT)25/mRNAのモル比を用いてTSII RTで合成した第1鎖のcDNAのオートラジオグラフである。本発明に従って、30℃より高く(好ましくは、37℃より高く)アニーリングおよび反応の温度を維持することは、「ホットスタート」ともまた呼ばれ得る。
【図5】図5は、種々の量のRNaseAを使用して合成した第2鎖のcDNAのオートラジオグラフである。
【図6】図6は、種々の量のRNaseIを使用して合成した第2鎖のcDNAのオートラジオグラフである。
【発明を実施するための形態】
【0026】
(好ましい実施形態の詳細な説明)
(定義)
このような用語に対して与えられた範囲を含む、明細書および特許請求の範囲の明確でかつ一貫した理解を与えるために、以下の定義が提供される。
【0027】
「内部プライミング」は、本明細書中で使用される場合は、mRNA分子の3’末端に配置されたポリAテール以外の、1つ以上のmRNA分子中の1つ以上の部位での1つ以上のプライマーのハイブリダイゼーションまたはアニーリングをいう。
【0028】
「ライブラリー」は、本明細書中で使用される場合は、生物のDNA内容物の全てまたは一部またはかなりの部分の代表である核酸分子(環状および直鎖状)のセット(「ゲノムライブラリー」)、あるいは細胞、組織、器官、または生物体中で発現された遺伝子の全てまたは一部またはかなりの部分の代表である核酸分子のセット(「cDNAライブラリー」)をいう。このようなライブラリーは、1つ以上のベクター中に含まれてもよいし含まれなくともよい。
【0029】
「ベクター」は、本明細書中で使用される場合は、プラスミド、コスミド、ファージミド、またはファージDNA、あるいは、宿主細胞中で自律的に複製することが可能であり、そして、このようなDNA配列がベクターの必須の生物学的機能の損失を伴わずに決定可能な様式で切断され得、そしてその中にDNAがその複製およびクローニングを生じるように挿入され得る、1つまたは少数の制限エンドヌクレアーゼ認識部位によって特徴付けられる他のDNA分子をいう。ベクターは、さらに、ベクターで形質転換された細胞の同定における使用に適切な1つ以上のマーカーを含み得る。マーカーとして、例えば、テトラサイクリン耐性またはアンピシリン耐性が挙げられるが、これらに限定されない。このようなベクターはまた、1つ以上の組換え部位、1つ以上の終結部位、1つ以上の複製起点などを含み得る。
【0030】
「プライマー」は、本明細書中で使用される場合は、DNA分子の増幅または重合の間にヌクレオチドの単量体の共有結合によって伸張される、一本鎖のオリゴヌクレオチドをいう。本発明での使用に好ましいプライマーとして、オリゴ(dT)プライマー、またはその誘導体もしくは改変体が挙げられる。
【0031】
「オリゴヌクレオチド」は、本明細書中で使用される場合は、1つのヌクレオチドのデオキシリボースまたはリボースの3’位と、隣接するヌクレオチドのデオキシリボースまたはリボースの5’位との間でのホスホジエステル結合によって連結されるヌクレオチドの共有結合された配列を含む、合成の分子または天然の分子をいう。
【0032】
「鋳型」は、本明細書中で使用される場合は、増幅されるか、合成されるか、または配列決定されるべき、二本鎖または一本鎖の核酸分子をいう。二本鎖の分子の場合においては、第1鎖および第2鎖を形成するその鎖の変性は、好ましくは、これらの分子が増幅され得るか、合成され得るか、または配列決定され得る前に、あるいは、二本鎖の分子が鋳型として直接使用され得る前に、行われる。一本鎖の鋳型については、鋳型の一部に対して相補的なプライマーが、適切な条件下でハイブリダイズされるかまたはアニーリングされ、そして次いで1つ以上のポリメラーゼまたは逆転写酵素が、上記の鋳型の全体または一部に対して相補的な核酸分子を合成し得る。本発明に従って新しく合成された分子は、最初の鋳型と等しい長さであり得るか、または最初の鋳型の長さよりも短い長さであり得る。
【0033】
「取りこむ」は、本明細書中で使用される場合は、DNAおよび/またはRNAの分子またはプライマーの一部になることを意味する。
【0034】
「増幅」は、本明細書中で使用される場合は、ポリメラーゼを用いてヌクレオチド配列のコピー数を増大させるための任意のインビトロでの方法をいう。核酸の増幅は、DNAおよび/またはRNAの分子またはプライマーへのヌクレチドの取りこみを生じ、それによって鋳型に対して相補的である新しい分子を形成する。形成された核酸分子およびその鋳型は、さらなる核酸分子を合成するための鋳型として使用され得る。本明細書中で使用される場合は、1回の増幅反応は、複数回の複製から構成され得る。DNA増幅反応として、例えば、ポリメラーゼ連鎖反応(PCR)が挙げられる。1回のPCR反応は、DNA分子の変性および合成の5から100回の「サイクル」から構成され得る。
【0035】
「ヌクレオチド」は、本明細書中で使用される場合は、塩基−糖−リン酸の組合せをいう。ヌクレオチドは、核酸配列の単量体単位(DNAおよびRNA)である。用語ヌクレオチドは、リボヌクレシド三リン酸(ATP、UTP、CTP、GTP)、およびデオキシリボヌクレオシド三リン酸(例えば、dATP、dCTP、dITP、dUTP、dGTP、dTTP)、またはそれらの誘導体を含む。このような誘導体として、例えば、[αS]dATP、7−デアザ−dGTP、7−デアザ−dATP、およびビオチン化されたかまたはハプテン化されたヌクレオチドが挙げられる。用語ヌクレオチドは、本明細書中で使用される場合はまた、ジデオキシリボヌクレオシド三リン酸(ddNTP)、およびそれらの誘導体をいう。デオキシリボヌクレオシド三リン酸の説明的な例として、ddATP、ddCTP、ddGTP、ddITP、およびddTTPが挙げられるが、これらに限定されない。本発明に従うと、「ヌクレオチド」は、標識されなくともよいし、周知の技術によって検出可能に標識されてもよい。検出可能な標識として、例えば、放射性同位元素、蛍光標識、化学発光標識、生体発光標識、および酵素標識が挙げられる。
【0036】
「ハイブリダイゼーション」または「アニーリング」は、本明細書中で使用される場合は、二本鎖の分子を生じる2つの相補的な一本鎖の核酸分子(RNAおよび/またはDNA)の塩基対合をいう。本明細書中で使用される場合は、対合は完全には相補的ではないが、2つの核酸分子がハイブリダイズされ得るかまたはアニーリングされ得る。従って、不一致塩基は、当該分野で周知の適切な条件が使用される限りにおいては、2つの核酸分子のハイブリダイゼーションもアニーリングをも妨げない。本発明においては、用語ハイブリダイゼーションまたはアニーリングは、好ましくは、1つ以上の鋳型(例えば、mRNA)に対する1つ以上のプライマー(例えば、オリゴ(dT)またはその誘導体)のハイブリダイゼーションをいう。
【0037】
「宿主細胞」は、本明細書中で使用される場合には、複製可能な発現ベクターまたはクローニングベクターのレシピエントである、任意の原核生物細胞または真核生物細胞をいう。用語「宿主」または「宿主細胞」は、本明細書中では互換的に使用され得る。このような宿主の例については、Maniatisら、「Molecular Cloning:A Laboratory Manual」、Cold Spring Harbor Laboratory,Cold Spring Harbor、New York(1982)を参照のこと。好ましい原核生物宿主として、Escherichia(例えば、E.coli)、Bacillus、Staphylococcus、Agrobacter(例えば、A.tumefaciens)、Streptomyces、Pseudomonas、Salmonella、Serratia、Caryophanon属などの細菌が挙げられるが、これらに限定されない。最も好ましい原核生物宿主はE.coliである。本発明において特に目的の細菌宿主として、Ecoli K12株、DH10B株、DH5α株、Stbl2株、およびHB101株、ならびにLife Technologies,Inc.から入手可能な他の株が挙げられる。好ましい真核生物宿主として、真菌、魚の細胞、酵母細胞、植物細胞、および動物細胞が挙げられるが、これらに限定されない。特に好ましい動物細胞は、Drosophilla細胞、Spodoptera Sf9細胞、Sf21細胞、およびTrichoplusia High−Five細胞のような昆虫細胞;C.elegans細胞のような線虫細胞;ならびにCOS細胞、CHO細胞、VERO細胞、293細胞、PERC6細胞、BHK細胞、およびヒト細胞のような哺乳動物細胞である。
【0038】
「発現ベクター」は、本明細書中で使用される場合は、宿主細胞中への形質転換またはトランスフェクション後に、その中にクローン化されている遺伝子または遺伝子の一部の発現を増強することが可能なベクターをいう。クローン化された遺伝子は、通常は、プロモーター配列のような特定の制御配列の制御下に(すなわち、それに対して作動可能に連結される)配置される。このようなプロモーターとして、ファージλPLプロモーター、およびE.coli lac、trp、およびtacプロモーターが挙げられるが、これらに限定されない。他の適切なプロモーターが当業者に公知である。
【0039】
本発明に従う逆転写に適切な核酸の鋳型として、任意の核酸分子または核酸分子の集団(好ましくは、1つ以上のRNA分子(例えば、1つ以上のmRNA分子またはポリA+RNA分子、およびより好ましくはmRNA分子の集団)あるいは1つ以上のDNA分子)が挙げられ、特に、細胞または組織に由来するものが挙げられる。好ましい局面においては、mRNA分子の集団(多数の異なるmRNA分子)が、本発明に従ってcDNAライブラリーを作製するために使用される。
【0040】
1つ以上の鋳型に対して相補的である核酸分子(単数または複数)を作製するために、プライマー(例えば、オリゴ(dT)プライマー)および1つ以上のヌクレオチドが、代表的には3’から5’方向での核酸の合成のために使用される。本発明のこの局面に従う逆転写に適切な核酸分子として、任意の核酸分子、特に、原核生物細胞または真核生物細胞に由来する任意の核酸分子が挙げられる。このような細胞として、正常な細胞、疾患の細胞、形質転換された細胞、樹立された細胞、先祖細胞、前駆細胞、胎児の細胞、胚性の細胞、細菌の細胞、酵母細胞、動物細胞(ヒト細胞を含む)、鳥類の細胞、植物細胞など、あるいは植物(例えば、トウモロコシ、トマト、タバコ、ジャガイモ、ダイズなど)または動物(例えば、ヒト、ウシ、ブタ、マウス、ヒツジ、ウマ、サル、イヌ、ネコ、ラット、ウサギ、鳥類、魚、昆虫など)から単離された組織が挙げられ得る。このような核酸分子はまた、ウイルスからも単離され得る。
【0041】
本発明の方法に従ってcDNA分子を調製するために鋳型として使用される核酸分子は、好ましくは、種々の細胞、組織、器官、または生物体のような天然の供給源から得られる。核酸分子の供給源として使用され得る細胞は、原核生物細胞(細菌細胞(Escherichia、Bacillus、Serratia、Salmonella、Staphylococcus、Streptococcus、Clostridium、Clamydia、Neisseria、Treponema、Mycoplasma、Borrelia、Legionella、Pseudomonas、Mycobacterium、Helicobacter、Erwinia、Agrobacterium、Rhizobium、Xanthomonas、Streptomyces属の種を含むがこれらに限定されない))、または真核生物(真菌(特に、酵母)、植物、原生動物、および他の寄生生物、ならびに動物(昆虫(特に、Drosophila spp.細胞を含む)、線虫(特に、Caenorhabditis elegans細胞)、ならびに哺乳動物(特にヒト細胞)を含む)であり得る。
【0042】
核酸の供給源として使用され得る哺乳動物の体細胞として、血球(網状赤血球および白血球)、内皮細胞、上皮細胞、ニューロン細胞(中枢神経系または末梢神経系に由来する)、筋肉細胞(骨格筋、平滑筋、または心筋に由来する筋細胞および筋芽細胞を含む)、結合組織の細胞(線維芽細胞、脂肪細胞、軟骨細胞、軟骨芽細胞、骨細胞、および骨芽細胞を含む)、ならびに他の支質細胞(例えば、マクロファージ、樹状細胞、Schwann細胞)が挙げられる。哺乳動物の生殖細胞(精母細胞および卵母細胞)もまた、上記の体細胞および生殖細胞を生じる先祖細胞、前駆細胞、および幹細胞と同様に、本発明における使用のための核酸の供給源として使用され得る。脳、腎臓、肝臓、すい臓、血液、骨髄、筋肉、神経、皮膚、尿生殖器、循環、リンパ系、消化管、および結合組織の供給源に由来するもの、ならびに哺乳動物(ヒトを含む)の胚または胎児に由来するもののような、哺乳動物の組織または器官もまた、核酸の供給源としての使用に適切である。
【0043】
任意の上記の細胞、組織、および器官は、正常なもの、罹患したもの、形質転換されたもの、樹立されたもの、先祖、前駆体、胎児のもの、または胚性のものであり得る。罹患した細胞として、例えば、感染性の疾患(細菌、真菌、または酵母、ウイルス(AIDS、HIV、HTLV、ヘルペス、肝炎などを含む)、または寄生生物によって引き起こされる)、遺伝的もしくは生化学的な病因(例えば、嚢胞性線維症、血友病、アルツハイマー病、筋ジストロフィー、または多発性硬化症)、または癌性のプロセスに関連する罹患した細胞が挙げられ得る。形質転換されたかまたは樹立された動物細胞株として、例えば、COS細胞、CHO細胞、VERO細胞、BHK細胞、HeLa細胞、HepG2細胞、K562細胞、293細胞、L929細胞、F9細胞などが挙げられ得る。本発明における使用のための核酸の供給源として適切な他の細胞、細胞株、組織、器官、および生物体は、当業者に明らかである。
【0044】
一旦、細胞、組織、器官、または他の出発サンプルが得られると、核酸分子(例えば、mRNA)が、当該分野で周知の方法によってそれらから単離され得る(例えば、Maniatis,T.,ら、Cell 15:687−701(1978);Okayama,H.およびBerg,P.Mol.Cell.Biol.2:161−170(1982);Gubler,U.およびHoffman,B.J.,Gene 25:263−269(1983);ならびにLife Technologies,Inc.から入手可能なMessage MakerTMmRNA Isolation Systemを参照のこと)。このように単離された核酸分子は、次いで、本発明に従ってcDNA分子およびcDNAライブラリーを調製するために使用され得る。本発明に従って産生されたcDNA分子および/またはcDNAライブラリーは、好ましくは、1つ以上のベクター中に含まれる。このようなベクターは、当該分野で周知の標準的な形質転換またはトランスフェクションの技術によって1つ以上の宿主細胞中に導入され得る。好ましい宿主細胞として、Escherichia属、特に、E.coliの細胞のような原核生物宿主細胞が挙げられる。
【0045】
本発明の組成物、方法、およびキット中での使用のための酵素として、逆転写酵素活性を有する任意の酵素が挙げられる。このような酵素として、レトロウイルス逆転写酵素、レトロトランスポゾン逆転写酵素、B型肝炎逆転写酵素、カリフラワーモザイクウイルス逆転写酵素、細菌の逆転写酵素、Tth DNAポリメラーゼ、Taq DNAポリメラーゼ(Saiki,R.K.ら,Science 239:487−491(1988);米国特許第4,889,818号および同第4,965,188号)、Tne DNAポリメラーゼ(WO96/10640)、Tma DNAポリメラーゼ(米国特許第5,374,553号)、およびそれらの変異体、フラグメント、改変体、または誘導体が挙げられるが、これらに限定されない(例えば、共有に係る、同時係属中の米国特許出願番号第08/706,702号および08/706,706号(両方とも、1996年9月9日に出願され、これらはそれらの全体において本明細書中で参考として援用されている)を参照のこと)。当業者によって理解されているように、改変された逆転写酵素およびRT活性を有するDNAポリメラーゼは、当該分野で周知である組換えまたは遺伝子操作技術によって得ることができる。変異体の逆転写酵素またはポリメラーゼは、例えば、部位特異的またはランダムな変異誘発によって、目的の逆転写酵素またはポリメラーゼをコードする遺伝子(単数または複数)を変異させることによって得ることができる。このような変異は、点変異、欠失変異、および挿入変異を含み得る。好ましくは、1つ以上の点変異(例えば、1つ以上のアミノ酸の、1つ以上の異なるアミノ酸での置換)が、本発明での使用のための変異体の逆転写酵素またはポリメラーゼを構築するために使用される。逆転写酵素またはポリメラーゼのフラグメントもまた、当該分野で周知である組換え技術による欠失変異によるか、あるいは任意の多数の周知のタンパク質分解酵素を使用する目的の逆転写酵素(単数または複数)またはポリメラーゼ(単数または複数)の酵素消化によって得ることができる。
【0046】
本発明での使用に好ましい酵素として、RNaseH活性が減少しているかまたは実質的に減少している酵素が挙げられる。RNaseH活性を減少させるかまたは実質的に減少させるこのような酵素は、目的の逆転写酵素中のRNaseHドメインを変異させることによって、好ましくは、上記のような1つ以上の点変異、1つ以上の欠失変異、および/または1つ以上の挿入変異によって、得ることができる。「RNaseH活性が実質的に減少している」酵素によって、野生型のモロニーマウ白血病ウイルス(M−MLV)、鳥類骨髄芽球症ウイルス(AMV)またはラウス肉腫ウイルス(RSV)の逆転写酵素の場合のような対応する野生型またはRNaseH+酵素のRNaseH活性の、約30%未満、約25%未満、約20%未満、より好ましくは約15%未満、約10%未満、約7.5%未満、または約5%未満、そして最も好ましくは約5%未満または2%未満を有する酵素を意味する。任意の酵素のRNaseH活性は、例えば、米国特許第5,244,797号、Kotewicz,M.L.ら、Nucl.Acids Res.16:265(1988)、Gerard,G.F.ら、FOCUS 14(5):91(1992)、および米国特許第5,668,005号に記載されているような、種々のアッセイによって決定され得る。これらの開示の全ては、本明細書中で参考として完全に援用されている。
【0047】
本発明での使用のための逆転写酵素活性を有するポリペプチドは、例えば、Life Technologies,Inc.(Rockville,Maryland)、Pharmacia(Piscataway,New Jersey)、Sigma(Saint Louis,Missouri)、またはBoehringer Mannheim Biochemicals(Indianapolis,Indiana)から商業的に入手可能であり得る。あるいは、逆転写酵素活性を有するポリペプチドは、当業者に周知の天然のタンパク質を単離および精製するための標準的な手順に従って、それらの天然のウイルスまたは細菌の供給源から単離され得る(例えば、Houts,G.E.ら、J.Virol.29:517(1979)を参照のこと)。さらに、逆転写酵素活性を有するポリペプチドは、当業者に良く知られている組換えDNA技術によって調製され得る(例えば、Kotewicz,M.L.ら、Nucl.Acids Res.16:265(1988);Soltis,D.A.およびSkalka,A.M.Proc.Natl.Acad.Sci.USA 85:3372−3376(1988)を参照のこと)。
【0048】
本発明での使用に好ましい逆転写酵素活性を有するポリペプチドとして、M−MLV逆転写酵素、RSV逆転写酵素、AMV逆転写酵素、ラウス関連(Rous Associated)ウイルス(RAV)の逆転写酵素、骨髄芽球症関連ウイルス(MAV)逆転写酵素、およびヒト免疫不全ウイルス(HIV)の逆転写酵素、およびWO98/47921に記載されている他のもの、ならびにそれらの誘導体、改変体、フラグメント、または変異体、ならびにそれらの組合せが挙げられる。さらに好ましい実施形態においては、逆転写酵素は、RNaseH活性が減少しているかまたは実質的に減少しており、そして最も好ましくは、M−MLV H-逆転写酵素、RSV H-逆転写酵素、AMV H-逆転写酵素、RAV H-逆転写酵素、MAV H-逆転写酵素、およびHIV H-逆転写酵素、ならびにそれらの誘導体、改変体、フラグメント、または変異体、ならびにそれらの組合せからなる群より選択される。特定の目的の逆転写酵素として、AMV RTおよびM−MLV RT、そしてより好ましくは、減少したかまたは実質的に減少したRNaseH活性を有するAMV RTおよびM−MLV RT(好ましくは、AMV RT αH-/BH+およびM−MLV RT H-)が挙げられる。本発明での使用に最も好ましい逆転写酵素として、Life Technologies,Inc.から入手可能な、SuperScriptTM、SuperScriptTMII、ThermoScriptTM、およびThermoScriptTMIIが挙げられる。一般的には、WO98/47921、米国特許第5,244,797号、および同第5,668,005号を参照のこと。それらのそれぞれの内容の全体は、本明細書中で参考として援用されている。
【0049】
種々のDNAポリメラーゼは、本発明に従って有用である。このようなポリメラーゼとして、以下が挙げられるが、これらに限定されない:Thermus thermophilus(Tth)DNAポリメラーゼ、Thermus aquaticus(Taq)DNAポリメラーゼ、Thermotoga neapolitana(Tne)DNAポリメラーゼ、Thermotoga maritima(Tma)DNAポリメラーゼ、Thermococcus litoralis(TliまたはVENTTM)DNAポリメラーゼ、Pyrococcus furiosis(Pfu)DNAポリメラーゼ、DEEPVENTTM DNAポリメラーゼ、Pyrococcus woosii(Pwo)DNAポリメラーゼ、Bacillus sterothermophilus(Bst)DNAポリメラーゼ、Bacillus caldophilus(Bca)DNAポリメラーゼ、Sulfolobus acidocaldarius(Sac)DNAポリメラーゼ、Thermoplasma acidophilum(Tac)DNAポリメラーゼ、Thermus flavus(Tfl/Tub)DNAポリメラーゼ、Thermus ruber(Tru)DNAポリメラーゼ、Thermus brockianus(DYNAZYMETM)DNAポリメラーゼ、Methanobacterium thermoautotrophicum(Mth)DNAポリメラーゼ、Mycobacterium spp.DNAポリメラーゼ(Mtb,Mlep)、ならびにそれらの変異体、改変体、および誘導体。
【0050】
本発明に従って使用されるDNAポリメラーゼは、代表的には5’から3’の方向で、核酸の鋳型からDNA分子を合成し得る任意の酵素であり得る。このようなポリメラーゼは、中温性または高温性であり得る。中温性のポリメラーゼとして、T4 DNAポリメラーゼ、T5 DNAポリメラーゼ、T7 DNAポリメラーゼ、KlenowフラグメントDNAポリメラーゼ、DNAポリメラーゼIII、DNAポリメラーゼIなどが挙げられる。熱安定性のDNAポリメラーゼとして、Taq、Tne、Tma、Pfu、VENTTM、DEEPVENTTM、Tth、ならびにそれらの変異体、改変体、および誘導体が挙げられる(米国特許第5,436,149号;米国特許第5,512,462号;第WO92/06188号;第WO92/06200号;第WO96/10640号;Barnes,W.M.,Gene 112:29−35(1992);Lawyer,F.C.,ら、PCR Meth.Appl.2:275−287(1993);Flaman,J.−M.ら、Nucl.Acids Res.22(15):3259−3260(1994))。
【0051】
本発明での使用のためのDNAポリメラーゼは、例えば、Life Technologies,Inc.(Rockville,Maryland)、Perkin−Elmer(Branchburg,New Jersey)、New England BioLabs(Beverly,Massachusetts)、またはBoehringer Mannheim Biochemicals(Indianapolis,Indiana)から商業的に入手することができる。
【0052】
本発明はまた、cDNA分子(特に、全長のcDNA分子)であり得る本発明の方法によって産生された核酸分子、これらの核酸分子およびcDNA分子を含有しているベクター(特に、発現ベクター)、ならびにこれらの核酸分子、cDNA分子、および/またはベクターを含有している宿主細胞に関する。
【0053】
組換えベクターは、当該分野で周知の方法を使用して、本発明の方法に従って調製された1つ以上のcDNA分子または核酸分子を1つ以上のベクター中に挿入することによって、本発明のこの局面に従って産生され得る。本発明のこの局面で使用されるベクターは、例えば、ファージまたはファージミドベクターであり得、そして好ましくは、プラスミドである。目的のポリペプチドをコードする核酸に対するシス作用性制御領域を含有しているベクターが、好ましい。適切なトランス作用性因子は、宿主によって供給され得るか、補完ベクターによって供給され得るか、または宿主への導入の際にベクター自体によって供給され得る。
【0054】
本発明において有用な発現ベクターとして、染色体に由来するベクター、エピソームに由来するベクター、およびウイルスに由来するベクター(例えば、細菌のプラスミドまたはバクテリオファージに由来するベクター)、ならびにそれらの組合せに由来するベクター(例えば、コスミド、およびファージミド)が挙げられ、そして好ましくは、細菌性宿主細胞中での培養のためにテトラサイクリンまたはアンピシリンの耐性遺伝子のような少なくとも1つの選択マーカーを含む。このような発現ベクターへの挿入の前に、本発明のcDNAまたは核酸分子は、適切なプロモーターに作動可能に連結されるべきである。
【0055】
本発明における使用に好ましいベクターの中には、Qiagenから入手可能なpQE70、pQE60、およびpQE−9;Stratageneから入手可能なpBSベクター、Phagescriptベクター、Bluescriptベクター、pNH8A、pNH16a、pNH18A、pNH46A;Invitrogenから入手可能なpcDNA3;Pharmaciaから入手可能なpGEX、pTrxfus、pTrc99a、pET−5、pET−9、pKK223−3、pKK233−3、pDR540、pRIT5;ならびにLife Technologies,Inc.から入手可能なpSPORT1、pSORT2、pSV・SPORT1、pCMVSPORT6、およびpCMVSPORTが挙げられる。他の適切なベクターは、当業者に容易に明らかである。
【0056】
本発明は、cDNA分子またはライブラリーを産生するために、当該分野で周知の任意のcDNAの合成方法(例えば、Gubler,U.およびHoffman,B.J.,Gene 25:263−269(1983);Krug,M.S.およびBerger,S.L.,Meth.Enzymol.152:316−325(1987);Sambrook,J.ら、Molecular Cloning:A Laboratory Manual,第2版、Cold Spring Harbor、NY:Cold Spring Harbor Laboratory Press、8.60−8.63頁(1989);PCT US98/19948;ならびにWO98/51699を参照のこと)と組合せて使用され得る。本発明を有利に使用し得る他のcDNA合成方法が、当業者に容易に明らかである。
【0057】
本発明の方法に従ってcDNA分子またはライブラリーを得たら、これらのcDNAは、さらなる分析または操作のために単離され得る。cDNAの精製のための詳細な方法論は、その全体において本明細書中で参考として援用されているGENETRAPPERTMマニュアル(Life Technologies)において教示されているが、当該分野で公知である別の標準的な技術(例えば、Sambrook,J.ら、Molecular Cloning:A Laboratory Manual,第2版、Cold Spring Harbor,NY:Cold Spring Harbor Laboratory Press、8.60−8.63頁(1989)を参照のこと)もまた使用され得る。本発明によって産生されたcDNA分子またはライブラリーもまた、ツーハイブリッド分析、cDNAの基準化、配列決定、および増幅のような標準的な分子生物学的技術によってさらに操作され得る。より詳細には、本発明の方法、およびこのような方法によって産生されたcDNA分子またはライブラリーは、RT−PCRおよび5’RACE技術(Life Technologies,Inc.)、ならびにディファレンシャルディスプレイとの組合せにおいて使用され得る。
【0058】
種々のインヒビターおよび結合分子が、本発明の方法における使用に適切である。逆転写酵素活性を有する上記のポリペプチドに対して結合する抗体(例えば、抗AMV RT抗体、抗M−MLV RT抗体、または抗RSV RT抗体を含む抗RT抗体)、またはキャップ構造に対して結合する抗体(例えば、抗キャップ抗体)、およびそれらのフラグメント(例えば、FabまたはF(ab’)2フラグメント)が、これらのインヒビターまたは結合分子に含まれる。このような抗体は、ポリクローナルまたはモノクローナルであり得、そして当該分野で周知の方法に従って種々の種において調製され得る。例えば、Sutcliffe,J.G.ら、Science 219:660−666(1983);Wilsonら、Cell 37:767(1984);およびBittle,F.J.ら、J.Gen.Virol.66:2347−2354(1985)を参照のこと。任意の上記の逆転写酵素またはキャップ構造について特異的な抗体は、インタクトなポリメラーゼポリペプチドまたはキャップ構造、あるいは1つ以上のそれらのフラグメントに対して惹起され得る。これらのポリペプチドまたはキャップ構造、あるいはそれらのフラグメントは、キャリアタンパク質(例えば、アルブミン)とともに動物の系(例えば、ウサギまたはマウス)に対して提示され得るか、またはこれらのポリペプチドが十分に長い(少なくとも約25アミノ酸)の場合には、キャリアを伴わずに動物の系に対して提示され得る。
【0059】
本明細書中で使用される場合は、用語「抗体」(Ab)は、以下のような特異的な状況が以下を除いて、用語「ポリクローナル抗体」または「モノクローナル抗体」(mAb)と互換的に使用され得る。これらの用語は、本明細書中で使用される場合は、インタクトな分子、および逆転写酵素活性を有するポリペプチド(DNAポリメラーゼまたは逆転写酵素)またはキャップ構造あるいはそれらの一部に対して特異的に結合し得る抗体フラグメント(例えば、FabおよびF(ab’)2フラグメントのような)を含むことを意味する。
【0060】
本発明の方法で使用される抗体は、ポリクローナルまたはモノクローナルであり得、そして任意の種々の方法によって調製され得る(例えば、米国特許第5,587,287号を参照のこと)。例えば、ポリクローナル抗体は、標準的な技術に従って、動物を、逆転写酵素活性またはキャップ構造あるいはそれらの一部を有する1つ以上のポリペプチドで免疫化することによって作製され得る(例えば、Harlow,E.およびLane,D.、Antibodies:A Laboratory Manual,Cold Spring Harbor,NY:Cold Spring Harbor Laboratory Press(1988);Kaufman,P.B.ら、Handbook of Molecular and Cellular Methods in Biology and Medicine、Boca Raton,Florida:CRC Press、468−469頁(1995)を参照のこと)。あるいは、本発明の方法において使用されるモノクローナル抗体(またはそのフラグメント)は、当該分野で周知のハイブリドーマ技術を使用して調製され得る(Koehlerら、Nature 256:495(1975);Koehlerら、Eur.J.Immunol.6:511(1976);Koehlerら、Eur.J.Immunol.6:292(1976);Hammerlingら、Monoclonal Antibodies and T−Cell Hybridomas、New York:Elsevier、563−681頁(1981);Kaufman,P.B.ら、Handbook of Molecular and Cellular Methods in Biology and Medicine,Boca Raton,Florida:CRC Press、444−467頁(1995))。
【0061】
上記の抗体のFab、F(ab’)2および他のフラグメントは、本明細書中に記載されている方法において使用され得ることが認識される。このようなフラグメントは、代表的には、パパイン(Fabフラグメントを産生するため)またはペプシン(F(ab’)2フラグメントを産生するため)のような酵素を使用して、タンパク質分解的切断によって産生される。抗体フラグメントはまた、組換えDNA技術の適用を通じて、または合成化学を通じて産生され得る。
【0062】
本発明はまた、本発明に従う使用のためのキットを提供する。このようなキットは、キャリア手段(例えば、箱またはカートン)を含み、その中には1つ以上の容器手段(例えば、バイアル、チューブ、ボトルなど)が密閉されている。ここでは、キットは、1つ以上の宿主細胞、1つ以上の逆転写酵素、1つ以上の逆転写のインヒビター、1つ以上のキャップ結合分子、1つ以上のDNAポリメラーゼ、適切な緩衝液、1つ以上のヌクレオチド、および/または1つ以上のプライマー(例えば、逆転写のためのオリゴ(dT))を、(同じ容器または別の容器中に)含み得る。本発明のこの局面によって含まれるキットは、標準的な核酸の逆転写プロトコールを実行するために必要なさらなる試薬および化合物をさらに含み得る。
【0063】
本明細書中に記載されている方法および出願に対する他の適切な改変および適応が明らかであり、そして本発明の範囲またはその任意の実施形態を逸脱することなく行われ得ることが、当業者に容易に明らかである。本発明は本明細書中で詳細に記載されているが、本発明が、以下の実施例を参照してより明確に理解される。以下の実施例は、本発明の説明の目的のために本発明中に含まれ、そして本発明を制限することは意図しない。
【実施例】
【0064】
(実施例1:オリゴ(dT)プライマー/mRNAの様々な比を用いた第1鎖cDNA合成の比較)
この実施例は、オリゴdTプライマー/出発mRNAの様々な比を用いた、MAP4遺伝子の第1鎖cDNA合成を比較する。全ての成分は、他に指定しなければ、Life Technologies,Inc.、Rockville、Marylandから入手可能である。
【0065】
Superscript II逆転写酵素(SSIIRT)のマスターミックスを、下記の表1で指定したように調製した。
【0066】
【表1】
ThermoScriptTMIIRT(TSRT)(AMV RT αH-βH+)(第WO98/47921号を参照のこと)のマスターミックスを、下記の表2で指定したように調製した。
【0067】
【表2】
マスターアニーリングミックスを、下記の表3で指定する量で5つのチューブに、5KbのMAP4 mRNA、オリゴ(dT)25-30および水を加えることによって調製した。
【0068】
【表3】
混合物を70℃で10分間加熱し、そして次いで氷上で5分間冷却した。
【0069】
下記の表4でまとめたように、全容量20μlで、9μlの適切な逆転写酵素マスターミックス、10μlのマスターアニーリングミックス、および1μlのSSIIRT(200単位/μl)またはTSIIRT(15単位/ul)のいずれかを加えることによって、第1鎖cDNAの合成を行った。
【0070】
【表4】
反応物を、SSIIRTでは45℃、TSIIRTでは45、50または55℃で1時間インキュベートした。チューブを氷上に置いて反応を終了させた。反応チューブの18μlの第1鎖cDNAを沈殿させ、そして10μlの水に再懸濁した。5μlの第1鎖cDNAを、5μlの標準ローディング緩衝液(60mMのNaOH、4mMのEDTA、0.1%のブロモフェノールブルー)と混合し、そして分析のために1.4%アルカリアガロースゲルにローディングした。これらの結果を図1および図2に示す。
【0071】
図1は、SSIIRTを用いて45℃で合成した第1鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1〜5はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比を用いた反応条件を示す。図2は、TSIIRTを用いて合成した第1鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1〜5はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比を用いた45℃での反応条件を示す。レーン6〜10はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比をを用いた50℃での反応条件を示す。レーン11〜15はそれぞれ、1:1、2.5:1、5:1、10:1および50:1のオリゴ(dT)25-30/mRNAのモル比を有する55℃での反応条件を示す。その結果は、オリゴ(dT)プライマー/mRNAのモル比を減少させることによって(好ましくは1:1)、逆転写酵素による内部プライミングがほとんど完全に除去されたことを示す。
【0072】
(実施例2:標準およびホットスタート条件下での第1鎖cDNA合成の比較)
標準反応およびホットスタート条件でのMAP4遺伝子の第1鎖cDNA合成を比較するために、この実験を設計した。
【0073】
1μgのMAP4 mRNAおよびビオチン化NotIオリゴ(dT)25プライマー((ビオチン)4 GACTAGTTCTAGATCGCGAGCGG CCGCCCTTTTT TTTTTTTTTTTT TTTTTTTT;第WO98/51699号を参照のこと)を、薄壁(thin−walled)PCRチューブに0:1、1:1、または15:1のオリゴ(dT)/mRNAの望ましいモル比で混合し、そして水で容量を10μlにすることによって、アニーリングミックスを調製した。いくつかのチューブが同じである場合、1つのバッチとして作成し、そしてしかるべく等分し得る。アニーリングミックスを氷上で保持した。
【0074】
Superscript II逆転写酵素(SSIIRT)についてのマスターミックスを、下記の表5で指定したように調製した。
【0075】
【表5】
次いでSSIIRTマスターミックスを2つの等量のアリコートに分け、その1つは標準反応温度で処理するためであり(バッチ1)、そして1つはホットスタート反応温度で処理するためである(バッチ2)。凝結を可能にするために、さらに10%容量の水をバッチ2に加えた。全てのミックスを氷上に保持した。
【0076】
小滴を回収するためにアニーリングミックスを含むチューブを短時間回転させ、このチューブをサーモサイクラー(thermocycler)中に置き、そして次いで70℃で10分間加熱することによって第1鎖cDNAの合成を開始した。この70℃で10分間のサイクルの後、バッチ1のアニーリングミックスのチューブを、すぐに氷上へ移動させた。バッチ2のマスターミックスをサーモサイクラー中に置き、そして45℃で5分間インキュベートしながら、バッチ2のアニーリングミックスのチューブを、サーモサイクラー中で45℃に冷却した。5分間のインキュベーションの後、11μlのバッチ2のマスターミックスを、バッチ2アニーリングチューブそれぞれに加え、そしてピペットで2回混合した。温度の低下を避けるために、チューブを回転させないように気をつけた。
【0077】
10μlのバッチ1のマスターミックスを、バッチ1アニーリングチューブそれぞれに加えた。バッチ1チューブを軽くボルテックスして、そして短時間遠心分離して凝結小滴を回収した。次いでバッチ1チューブを、サーモサイクラーに戻して、そしてバッチ1および2両方のチューブを45℃で1時間インキュベートした。
【0078】
各チューブからの5μlの第1鎖cDNAを、5μlの標準ローディング緩衝液(60mMのNaOH、4mMのEDTA、0.1%のブロモフェノールブルー)と混合し、そして分析のために1.4%のアルカリアガロースゲルにローディングした。結果を図3に示す。
【0079】
図3は、SSIIRTを用いて合成した第1鎖cDNAのオートラジオグラフである。レーン1、3および5はそれぞれ、0:1、1:1および15:1のビオチン化オリゴ(dT)/mRNAのモル比を用いたバッチ1反応条件を示す。レーン2、4および6はそれぞれ、0:1、1:1および15:1のビオチン化オリゴ(dT)/mRNAのモル比を用いたバッチ2反応条件を示す。
【0080】
第1鎖DNAをまた、1μgのmRNAあたり15単位のTSIIRTを用いて、1:1および15:1のビオチン化オリゴ(dT)/mRNA比を用いて、TSIIRTで合成した。温度を50℃に変えた以外は、上記で記載した同じプロトコールに従った。結果を図4に示す。図4は、TSIIRTを用いて合成した第1鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1および3はそれぞれ、1:1比および15:1比で標準反応温度を用いた反応条件を示す。レーン2および4は、上記で記載したように、それぞれ1:1比および15:1比でのホットスタート反応条件を示す。
【0081】
その結果は、プライマーおよびmRNA混合物の変性後、反応温度を逆転写酵素反応温度まで下げることによって、反応が直接開始され、そして内部プライミングが完全に避けられたことを示した。
【0082】
(実施例3:反応温度ならびに塩およびRNaseの濃度を制御することによる二本鎖cDNAの合成)
この実施例は、第1鎖cDNA合成の後、cDNA/mRNAハイブリッドの処理中に、反応温度ならびに塩および異なるリボヌクレアーゼ(RNase)の濃度を制御することによる、二本鎖cDNAの合成を記載する。
【0083】
第1鎖cDNAを、上記の実施例2で記載したように合成し、そして下記でさらに記載するようにRNaseIまたはRNaseAのいずれかで消化した。
【0084】
第1鎖cDNAを、180μlの水および20μlの10×RNaseI緩衝液(100mMのTris−HCl(pH7.5)、50mMのEDTA、2Mの酢酸ナトリウム)に再懸濁することによって、第1鎖cDNAのRNaseI消化を行った。2.5単位のRNaseI(1単位/μg mRNA)を加え、そして混合物をよく混合した。RNaseI消化混合物を25℃で30分インキュベートし、そしてフェノール/クロロホルムを用いて1回抽出した。上清を1μlのグリコーゲン、100μlの酢酸アンモニウム、および800μlのエタノールを用いて沈殿させた。
【0085】
第1鎖cDNAを、200μlの消化緩衝液(10mMのTris−HCl(pH7.5)、250mMのNaCl)に再懸濁することによって、第1鎖cDNAのRNaseA消化を行った。12.5ngのRNaseA(5ng/μg mRNA)を加え、そして混合物をよく混合した。RNaseA消化混合物を25℃で30分インキュベートし、そしてフェノール/クロロホルムを用いて1回抽出した。上清を1μlのグリコーゲン、100μlの酢酸アンモニウム、および800μlのエタノールを用いて沈殿させた。
【0086】
(実施例4:キャップ結合タンパク質を用いた全長cDNAクローンの富化)
この実施例は、キャップ結合タンパク質eIF4Eを用いた全長cDNAクローンの富化を記載する。
【0087】
上記の実施例3で記載したRNaseIで処理した第1鎖cDNAを沈殿させ、そして70%エタノールを用いて洗浄することによってcDNAを調製した。得られたペレットを室温で5分間乾燥させ、そして210μlの10mM KPO4、100mMのKCl、2mMのEDTA、6mMのDTTおよび5%のグリセロールに再懸濁させた。cDNAを氷上に保存した。
【0088】
eIF4Eグルタチオンセファロース4Bビーズを、最初にグルタチオンセファロース4Bビーズ(Pharmacia、Sweden)をよく混合することによって調製した。eIF4Eビーズを調製するために、GSTタグeIF4Eタンパク質を発現する組換え宿主細胞(eIF4E遺伝子をGST融合ベクターにクローニングし、N末端GST−eIF4E融合遺伝子を作成した)を増殖させ、そして融合タンパク質を標準的な技術によって精製した。従って、本発明はまた、eIF4Eタンパク質(特に融合タンパク質として)を発現する組換え宿主細胞、そのようなタンパク質または融合タンパク質を発現する遺伝子を含むベクター、および産生される組換えタンパク質または融合タンパク質に関連する。本発明において、あらゆるタグを使用し得る(例えば、Hisタグ、GSTタグ、HAタグ、Trxタグ等)。そのようなタグは、eIF4E遺伝子のカルボキシ末端領域および/またはN末端領域に位置し得る。
【0089】
GST−eIF4E融合タンパク質を、製造会社のプロトコールに従って、グルタチオンセファロース4Bビーズ(Pharmacia Biotech)を用いて、グルタチオン結合によって、セファロース4Bビーズと複合体化した。200μlのビーズを、1.5mlの微量遠心(microcentrifuge)チューブに移し、1秒間遠心分離し、そして75μlの上清を除去した。ビーズを1mlの反応緩衝液(10mMのKPO4、100mMのKCl、2mMのEDTA、6mMのDTTおよび5%のグリセロール)を用いて2回洗浄し、そして258μlの反応緩衝液に再懸濁し、続いて42μl(18pmol/μl)のeIF4Eタンパク質(600pmol/100μlビーズ)を加えた。混合物を、接近(head to head)ローラーで、4℃で30分間混合した。混合物を次いで1秒間遠心分離し、そして上清を除去した。ビーズを1mlの反応緩衝液を用いて2回、そして反応緩衝液中1mlの25μg/ml酵母tRNAを用いて1回洗浄した。次いで20μlの反応緩衝液および5μgの酵母tRNAをビーズに加えた。200μlのRNaseI処理cDNAをビーズに加え、そして内容物をローラーで、室温で1時間混合した。1時間後、混合物を1秒間遠心分離し、そして上清を除去した。ビーズを1mlの反応緩衝液を用いて2回、そして反応緩衝液中1mlの500μM GDPを用いて1回洗浄した。cDNAを、反応緩衝液中250μlの500μM GDPで2回溶出した。溶出溶液をプールし、そして1分間遠心分離してビーズを除去した。溶出したcDNAを、等容量のフェノール/クロロホルムで2回抽出した。cDNAを2つのチューブに分け、そして1μlのグリコーゲン、0.5容量の7.5M酢酸アンモニウムおよび2.5容量のエタノールを用いて沈殿させた。
【0090】
(実施例5:cDNAライブラリーの評価)
上記で記載した全長法で構築したcDNAライブラリーの質を評価するために、MAP4遺伝子(5〜6kb)および他の遺伝子を標的遺伝子として選択した。MAP4および他のcDNAクローンを、当該分野で周知の標準的な方法(SuperScriptTM Plasmid Manual、Life Technologies,Inc.を参照のこと)ならびに3’および5’GeneTrapper cDNA Positive Selection System(Life Technologies,Inc.、Rockville、Maryland)を用いた上記で記載した全長法によって構築したライブラリーから単離した。陽性のクローンを、PCRによって大きさを分析した。下記の表6および7は、当該分野で周知の方法(コントロール)および上記で記載した全長法(全長法)で構築したヒト線維芽細胞cDNAライブラリーにおける全長cDNAクローンの富化の結果をまとめる。
【0091】
【表6】
コントロールライブラリーを、公知の方法を用いてSSIIRTで構築した。
【0092】
【表7】
これらの結果は、上記で記載した全長法は、標準的な方法を用いた<13%に比べて、5’GeneTrapperシステムによって>90%の全長cDNAクローンを産生したことを示す。さらに、上記で記載した全長法は、標準的な方法を用いた<7%に比べて、3’GeneTrapperシステムによって>37%の全長クローンを産生した。
【0093】
(実施例6:第1鎖cDNA合成、RNaseI消化、およびeIF−4E捕捉)
以下を除いて、上記の実施例2、3(RNaseI)および4で記載した全ての条件およびパラメーターに従った:1反応あたり10μgのヒト線維芽細胞細胞質mRNAという4つの反応を使用した(第WO98/45311号を参照のこと);ビオチン化プライマー−アダプター(ビオチン)4−GACTAGTTCTAGATCGCGAGCGGCCGCCC(T)25を、1:1のプライマー/mRNAモル比で使用した;TSIIRTを50℃で使用した;そしてSSIIRTを45℃で使用した。下記の表8は、第1鎖cDNAおよびeIF−4E捕捉の結果をまとめる。
【0094】
(実施例7:第2鎖cDNA合成)
最初に上記の実施例6で得た4つの反応ペレットそれぞれを、104μlのDEPC処理水に溶解し、そして次いで以下の試薬を各反応に加えることによって、第2鎖cDNAを合成した:
4μlの5×第1鎖緩衝液*
30μlの5×第2鎖緩衝液*
2μlの0.1M DTT
4μlの10mM dNTP
1μlのE.coli DNAリガーゼ(10単位/μl)
1μlのE.coli RNase H(2単位/μl)
4μlのE.coli DNAポリメラーゼ(10単位/μl)
SuperScript Plasmid Systemマニュアル(Life Technologies,Inc.、Rockville、Maryland)を参照のこと。
【0095】
これらの反応混合物を、次いで16℃で2時間インキュベートした。2μlのT4 DNAポリメラーゼ(5単位/μl)を加え、そして16℃でのインキュベーションをさらに5分間続けた。
【0096】
(実施例8:ストレプトアビジンビーズの調製)
上記の実施例7で記載した2時間の第2鎖反応の最後の30分間に、ストレプトアビジン常磁性体ビーズを以下のように調製した。
【0097】
ストレプトアビジン常磁性ビーズ(Seradyn)を、ビーズが完全に再懸濁するまでピペッティングによって静かに混合した。150μlの混合ビーズを、各反応のために微量遠心チューブの底に移した。チューブをMagna−Sep Magnetic Particle Separator(Life Technologies,Inc.、Rockville、Maryland)(磁石)に挿入して、そして2分間静置した。チューブがマグネット中にある間に、上清をピペッティングによって除去し、そして100μlのTE緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA)をすぐにビーズに加えた。
【0098】
次いでチューブを磁石から取り出し、そして指で軽くたたくか、または一番低い設定でボルテックスして、ビーズを静かに再懸濁した。チューブを磁石に再び挿入した。2分後、上清を除去し、ビーズを160μlの結合緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA、1MのNaCl)に再懸濁し、そしてチューブを微量遠心チューブラックに置いた。
【0099】
(実施例9:二本鎖cDNAライブラリーの捕捉)
上記の実施例7で記載したように、第2鎖反応物とT4 DNAポリメラーゼとをインキュベートした後、反応混合物を氷上に置き、そして10μlの0.5M EDTAを加えた。次いでcDNAライブラリーを以下の手順に従って捕捉した(一般的には第WO98/51699号を参照のこと)。
【0100】
実施例8に従って調製した常磁性ビーズを、第2鎖反応混合物チューブへ移し、そしてピペッティングによって静かに混合し、そして懸濁液を室温で60分間インキュベートした。次いでチューブを磁石に挿入した。2分後、上清を除去および廃棄した。
【0101】
100μlの洗浄緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA、500mMのNaCl)をビーズに加え、指で軽くたたくか、または一番低い設定でボルテックスしてビーズを再懸濁し、そしてチューブを2分間磁石へ再び挿入した。上清を除去および廃棄した。この洗浄工程をもう1回反復し、そして次いで100μlの洗浄緩衝液をビーズに加えた。次いでチューブを5分間磁石に再び挿入した。
【0102】
(実施例10:NotI消化)
実施例9の最終工程で記載した5分間のインキュベーション後、常磁性ビーズから上清を除去および廃棄し、そして41μlのオートクレーブ処理蒸留水、5μlのREact3緩衝液、4μlのNotIを加え、そしてビーズをピペッティングによってよく混合した。次いで反応物を37℃で2時間インキュベートした。次いでチューブを2分間磁石に挿入し、そしてcDNAライブラリーを含む上清を新しいチューブに移した。
【0103】
50μlのフェノール:クロロホルム:イソアミルアルコール(25:24:1)を上清に加え、溶液を完全にボルテックスし、そして次いで14,000×gで5分間、室温で遠心分離した。45μlの上部の水層を注意深く取り出し、そして新しい微量遠心チューブへ移した。23μlの7.5M酢酸アンモニウム、1μlのグリコーゲン(20μg)および172μlのエタノール(−20℃)を加えた。溶液をよく混合して、そしてドライアイス上(または−70℃冷凍庫)で15分間保存した。
【0104】
次いでエタノール溶液を14,000×gで30分間、4℃で遠心分離した。上清を小さなペレットから注意深く除去した。100μlの70%エタノールを加え、そしてチューブを14,000×gで2分間、室温で遠心分離した。エタノールを除去し、そしてペレットをスピードバック(speed−vac)で2分間、または乾燥するまで乾燥した。次いでペレットを20μlのTE緩衝液(10mMのTris−HCl(pH7.5)、0.1mMのEDTA)に溶解した。cDNAの最終的な収量を、チェレンコフカウント(Cerenkov counts)によって決定した(下記の表8を参照のこと)。
【0105】
【表8】
(実施例11:cDNAのベクターへの連結およびE.coliへの導入)
10から30ngの非分画またはサイズ分画(低融点ゲル電気泳動によって≧1.5kb)cDNAを、ベクターpCMVSPORT6(Life Technologies,Inc.)に連結した。この連結を、クローニングベクターをNotIおよびEcoRVで予め消化した以外は、SuperScript Plasmid Systemマニュアル(Life Technologies,Inc.、Rockville、Maryland)で記載されたように、エレクトロポレーションによってE.coliに導入した。
【0106】
構築したcDNAライブラリーから無作為に選んだクローン(304クローン)の配列分析を、5’および3’配列決定によって分析し、cDNAライブラリー中の全長ランダムクローンの全百分率を決定した。配列をGeneBank配列と相同性に関して比較した。結果を下記の表9にまとめる。その結果に基づいて、約68%のランダムクローンが全長(公知の全長クローンおよび未知の全長クローンを含む)であった。従って、ヒト線維芽細胞細胞質mRNAライブラリーから、約17%の未知全長クローンが得られた。
【0107】
【表9】
(実施例12:RNaseアッセイ)
第1鎖cDNAを、TE緩衝液(10mMのTris−HCl(pH7.5)、1mMのEDTA)中で1000ng/μg mRNAのRNaseAで、およびTEN(10mMのTris−HCl(pH7.5)、5mMのEDTA(pH8.0)、200mMの酢酸ナトリウム)中で25から40u/μg mRNAのRNaseIで、本質的に実施例3で記載したように37℃で処理した。しかし、上昇した温度での大量のRNaseを用いたこの処理は、非常に小さい平均cDNAインサートの大きさ(約200bp)を含むライブラリーを生じた。従って、必要なRNaseの最適量を決定するために、第2鎖cDNAアッセイを開発した。
【0108】
第1鎖cDNA(放射性標識および非放射性標識)を、500ngのRNA/反応でHeLa mRNAを用いて合成した。第1鎖cDNAを、エタノールを用いて沈殿させ、そしてDEPC−処理水に溶解した。コールドの第1鎖cDNAを、種々の量のRNaseを含むRNase緩衝液に加えた。25℃で30分間インキュベートした後、処理したcDNAをフェノール:クロロホルムを用いて抽出し、そしてエタノールを用いて沈殿させた。処理したcDNAをDEPC処理水に溶解し、32P−dCTPプラスおよびマイナスRNase Hを用いて第2鎖cDNA反応を行った。反応物をフェノール:クロロホルムを用いて抽出し、そしてエタノールを用いて沈殿させた。等量のcpmを1.4%アルカリアガロースゲルに電気泳動した。結果を図5および図6に示す。
【0109】
図5は、種々の量のRNaseAを用いて合成した第2鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1は、未処理の第1鎖cDNAを示す。レーン2は、未処理の第2鎖cDNAを示す。レーン3、5、7、および9は、RNase H無しで、そしてそれぞれ0、1.25ng、2.5ng、および5ngの濃度のRNaseAを用いて合成した第2鎖cDNAを示す。レーン4、6、8、および10は、RNase Hを用いて、そしてそれぞれ0、1.25ng、2.5ng、および5ngの濃度のRNaseAを用いて合成した第2鎖cDNAを示す。
【0110】
図6は、種々の量のRNaseIを用いて合成した第2鎖cDNAのオートラジオグラフである。レーンMは1kbのDNAラダーである。レーン1は、未処理の第1鎖cDNAを示す。レーン2は、未処理の第2鎖cDNAを示す。レーン3、5、7、および9は、RNaseH無しで、そしてそれぞれ0、0.5u、1.25u、および2.5uの濃度のRNaseIを用いて合成した第2鎖cDNAを示す。レーン4、6、8、および10は、RNase Hを用いて、そしてそれぞれ0、0.5u、1.25u、および2.5uの濃度のRNaseIを用いて合成した第2鎖cDNAを示す。
【0111】
これらのゲル分析は、1.25ngの濃度のRNaseA(図5を参照のこと)または0.5単位のRNaseI(図6を参照のこと)が、500ngの出発mRNAについて用いるのに最適であり得ることを証明した。
【0112】
(実施例13:キャップ構造に対する抗体の調製)
キャップに対する抗体を、m7グアノシン−KLHを抗原として用いて産生した。1200個のハイブリドーマをプレーティングし、そして120個のコロニーのみが産生された。これらのうち6個のコロニーのみがキャップに関して陽性であった。さらなる分析の後、3つが必要な親和性を有すると決定された。最初のスクリーニングELISAは、m7グアノシン−BSAのELISAプレートへの結合、BSAを用いたブロック、ハイブリドーマ上清の結合、2次抗体との反応、およびBCIP/NPTを用いた比色定量反応による陽性の決定からなる。2次スクリーニングは、ハイブリドーマ上清の適切な希釈物を、0.1mMのm7GTP、0.1mMのキャップアナログM7G5'ppp5'G、0.5mMのm7グアノシンまたは0.5mMのGTPのいずれかとインキュベートすることを含んでいた。前処理した上清を次いで標準的なELISA手順で使用した。GTPはm7グアノシン−BSAと競合しなかったが、m7バージョンは全て効率よく競合した。
【0113】
理解の明解さのために、説明および例によってここで本発明をある程度詳細にわたって完全に記載したが、本発明またはそのあらゆる特定の実施態様の範囲に影響を与えることなく、条件、処方および他のパラメーターの広いおよび同じ範囲内で、本発明を改変または変化させることによって本発明を実施し得ること、およびそのような改変または変化は、添付した特許請求の範囲内に含まれると意図されることが、当業者に明らかである。
【0114】
本明細書中で言及された全ての出版物、特許および特許出願は、本発明が属する、当該分野の技術水準を示し、そして本明細書中で、個々の出版物、特許または特許出願が、具体的かつ個々に参考として援用されると示されたのと同じ程度、参考として援用される。
【特許請求の範囲】
【請求項1】
1つ以上のcDNA分子またはcDNA分子の集団を合成するための方法であって、以下の工程:
少なくとも1つのmRNAもしくはポリA RNA鋳型、またはこのような鋳型の集団を、逆転写活性を有する少なくとも1つのポリペプチドと混合する工程;および
該混合物を、合成された全長のcDNA分子の量または百分率を増大させるために十分な条件下でインキュベートする工程、
を包含する、方法。
【請求項2】
前記条件が、内部プライミングを減少させるかまたは実質的に減少させる、請求項1に記載の方法。
【請求項3】
前記ポリペプチドが、M−MLV RT、RSV RT、AMV RT、RAV RT、MAV RT、およびHIV RT、ならびにそれらの誘導体、フラグメント、変異体および改変体からなる群より選択される逆転写酵素である、請求項1に記載の方法。
【請求項4】
前記逆転写酵素が、RNase H活性が減少しているかまたは実質的に減少している、請求項3に記載の方法。
【請求項5】
前記条件が、上昇させた温度で、前記鋳型に対して1つ以上のプライマーをアニーリングまたはハイブリダイズすることを含む、請求項2に記載の方法。
【請求項6】
前記上昇させた温度が、約20℃から約90℃までの範囲である、請求項5に記載の方法。
【請求項7】
前記条件が、前記鋳型の量と比較して少ない量のプライマーを含む、請求項2に記載の方法。
【請求項8】
前記プライマーの前記鋳型に対する比が、約5:1から約1:20までの範囲である、請求項7に記載の方法。
【請求項9】
前記条件が、逆転写酵素活性を有する前記ポリペプチドのインヒビターの使用を含む、請求項1に記載の方法。
【請求項10】
前記インヒビターが、抗体または抗体フラグメントである、請求項9に記載の方法。
【請求項11】
前記抗体または抗体フラグメントが、ポリクローナルまたはモノクローナルである、請求項10に記載の方法。
【請求項12】
前記条件が、高い特異性を有するプライマーの使用を含む、請求項2に記載の方法。
【請求項13】
前記条件が、前記プライマーの長さを増大させることを含む、請求項2に記載の方法。
【請求項14】
前記鋳型にハイブリダイズする前記プライマーの長さが、約20塩基から約60塩基までの範囲である、請求項13に記載の方法。
【請求項15】
前記方法がさらに、前記少なくとも1つのcDNA分子の全てまたは一部に対して相補的である少なくとも1つの第2の核酸分子を作製するために十分な条件下で、少なくとも1つの該cDNA分子をインキュベートし、それによって1つ以上の二本鎖のcDNA分子を産生する工程を包含する、請求項1に記載の方法。
【請求項16】
前記第2の核酸分子を作製するための前記条件が、全長の二本鎖のcDNA分子の量または百分率を増大させる、請求項15に記載の方法。
【請求項17】
前記条件が、リボヌクレアーゼ消化を最適化する工程を包含する、請求項16に記載の方法。
【請求項18】
前記条件が、第1鎖のcDNA合成の後で形成されるmRNA/cDNAハイブリッドに含まれる一本鎖のmRNAの消化を可能にする、請求項17に記載の方法。
【請求項19】
前記条件が、二本鎖のmRNA/cDNAハイブリッドにおけるmRNAの消化を妨げる、阻害する、減少させる、または実質的に減少させる、請求項18に記載の方法。
【請求項20】
前記リボヌクレアーゼが、RNaseAおよびRNaseI、またはそれらの組合せからなる群より選択される、請求項17に記載の方法。
【請求項21】
請求項1に記載される方法に従って作製された、cDNA分子またはcDNA分子の集団。
【請求項22】
請求項15に記載される方法に従って作製された、cDNA分子またはcDNA分子の集団。
【請求項23】
請求項21に記載の核酸分子を含有している、ベクター。
【請求項24】
請求項22に記載の核酸分子を含有している、ベクター。
【請求項25】
請求項23に記載のベクターを含有している、宿主細胞。
【請求項26】
請求項24に記載のベクターを含有している、宿主細胞。
【請求項27】
請求項21に記載の分子を含有している、宿主細胞。
【請求項28】
請求項22に記載の分子を含有している、宿主細胞。
【請求項29】
増大した量または百分率の全長のcDNAを作製するためのキットであって、1つ以上のプライマー、1つ以上の逆転写のインヒビター、1つ以上の逆転写酵素、1つ以上のヌクレオチド、1つ以上のキャップ結合分子、1つ以上の逆転写緩衝液、および全長のcDNAを作製するための説明書からなる群より選択される少なくとも1つの成分を含む、キット。
【請求項30】
増大した量または百分率の全長cDNAを作製するための、組成物。
【請求項31】
逆転写活性酵素を有するポリペプチドに対して特異的に結合する、抗体またはそのフラグメント。
【請求項32】
mRNAキャップ構造に対して特異的に結合する、抗体またはそのフラグメント。
【請求項1】
1つ以上のcDNA分子またはcDNA分子の集団を合成するための方法であって、以下の工程:
少なくとも1つのmRNAもしくはポリA RNA鋳型、またはこのような鋳型の集団を、逆転写活性を有する少なくとも1つのポリペプチドと混合する工程;および
該混合物を、合成された全長のcDNA分子の量または百分率を増大させるために十分な条件下でインキュベートする工程、
を包含する、方法。
【請求項2】
前記条件が、内部プライミングを減少させるかまたは実質的に減少させる、請求項1に記載の方法。
【請求項3】
前記ポリペプチドが、M−MLV RT、RSV RT、AMV RT、RAV RT、MAV RT、およびHIV RT、ならびにそれらの誘導体、フラグメント、変異体および改変体からなる群より選択される逆転写酵素である、請求項1に記載の方法。
【請求項4】
前記逆転写酵素が、RNase H活性が減少しているかまたは実質的に減少している、請求項3に記載の方法。
【請求項5】
前記条件が、上昇させた温度で、前記鋳型に対して1つ以上のプライマーをアニーリングまたはハイブリダイズすることを含む、請求項2に記載の方法。
【請求項6】
前記上昇させた温度が、約20℃から約90℃までの範囲である、請求項5に記載の方法。
【請求項7】
前記条件が、前記鋳型の量と比較して少ない量のプライマーを含む、請求項2に記載の方法。
【請求項8】
前記プライマーの前記鋳型に対する比が、約5:1から約1:20までの範囲である、請求項7に記載の方法。
【請求項9】
前記条件が、逆転写酵素活性を有する前記ポリペプチドのインヒビターの使用を含む、請求項1に記載の方法。
【請求項10】
前記インヒビターが、抗体または抗体フラグメントである、請求項9に記載の方法。
【請求項11】
前記抗体または抗体フラグメントが、ポリクローナルまたはモノクローナルである、請求項10に記載の方法。
【請求項12】
前記条件が、高い特異性を有するプライマーの使用を含む、請求項2に記載の方法。
【請求項13】
前記条件が、前記プライマーの長さを増大させることを含む、請求項2に記載の方法。
【請求項14】
前記鋳型にハイブリダイズする前記プライマーの長さが、約20塩基から約60塩基までの範囲である、請求項13に記載の方法。
【請求項15】
前記方法がさらに、前記少なくとも1つのcDNA分子の全てまたは一部に対して相補的である少なくとも1つの第2の核酸分子を作製するために十分な条件下で、少なくとも1つの該cDNA分子をインキュベートし、それによって1つ以上の二本鎖のcDNA分子を産生する工程を包含する、請求項1に記載の方法。
【請求項16】
前記第2の核酸分子を作製するための前記条件が、全長の二本鎖のcDNA分子の量または百分率を増大させる、請求項15に記載の方法。
【請求項17】
前記条件が、リボヌクレアーゼ消化を最適化する工程を包含する、請求項16に記載の方法。
【請求項18】
前記条件が、第1鎖のcDNA合成の後で形成されるmRNA/cDNAハイブリッドに含まれる一本鎖のmRNAの消化を可能にする、請求項17に記載の方法。
【請求項19】
前記条件が、二本鎖のmRNA/cDNAハイブリッドにおけるmRNAの消化を妨げる、阻害する、減少させる、または実質的に減少させる、請求項18に記載の方法。
【請求項20】
前記リボヌクレアーゼが、RNaseAおよびRNaseI、またはそれらの組合せからなる群より選択される、請求項17に記載の方法。
【請求項21】
請求項1に記載される方法に従って作製された、cDNA分子またはcDNA分子の集団。
【請求項22】
請求項15に記載される方法に従って作製された、cDNA分子またはcDNA分子の集団。
【請求項23】
請求項21に記載の核酸分子を含有している、ベクター。
【請求項24】
請求項22に記載の核酸分子を含有している、ベクター。
【請求項25】
請求項23に記載のベクターを含有している、宿主細胞。
【請求項26】
請求項24に記載のベクターを含有している、宿主細胞。
【請求項27】
請求項21に記載の分子を含有している、宿主細胞。
【請求項28】
請求項22に記載の分子を含有している、宿主細胞。
【請求項29】
増大した量または百分率の全長のcDNAを作製するためのキットであって、1つ以上のプライマー、1つ以上の逆転写のインヒビター、1つ以上の逆転写酵素、1つ以上のヌクレオチド、1つ以上のキャップ結合分子、1つ以上の逆転写緩衝液、および全長のcDNAを作製するための説明書からなる群より選択される少なくとも1つの成分を含む、キット。
【請求項30】
増大した量または百分率の全長cDNAを作製するための、組成物。
【請求項31】
逆転写活性酵素を有するポリペプチドに対して特異的に結合する、抗体またはそのフラグメント。
【請求項32】
mRNAキャップ構造に対して特異的に結合する、抗体またはそのフラグメント。
【図1】

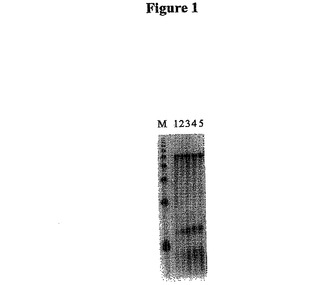
【図2】
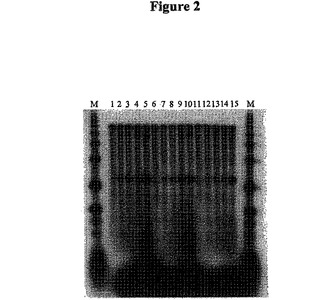
【図3】

【図4】
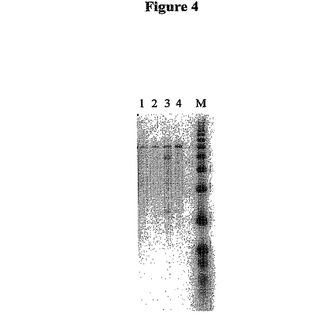
【図5】
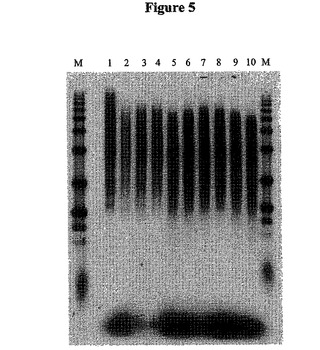
【図6】
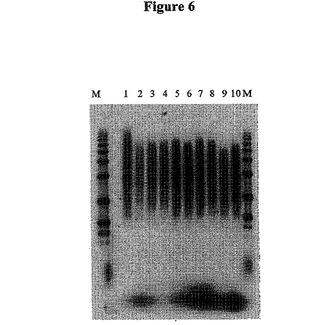
【図2】
【図3】
【図4】
【図5】
【図6】
【公開番号】特開2010−263905(P2010−263905A)
【公開日】平成22年11月25日(2010.11.25)
【国際特許分類】
【出願番号】特願2010−152752(P2010−152752)
【出願日】平成22年7月5日(2010.7.5)
【分割の表示】特願2000−602801(P2000−602801)の分割
【原出願日】平成12年3月1日(2000.3.1)
【出願人】(502221282)ライフ テクノロジーズ コーポレーション (113)
【Fターム(参考)】
【公開日】平成22年11月25日(2010.11.25)
【国際特許分類】
【出願日】平成22年7月5日(2010.7.5)
【分割の表示】特願2000−602801(P2000−602801)の分割
【原出願日】平成12年3月1日(2000.3.1)
【出願人】(502221282)ライフ テクノロジーズ コーポレーション (113)
【Fターム(参考)】
[ Back to top ]