
iPS細胞の生成および調節のための方法およびその組成物
本発明は、従来の方法と比べて誘導効率が高い誘導多能性幹(iPS)細胞生成方法を提供する。その方法は、体細胞を、その細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤と組み合わせて核初期化因子により処理することを含む。本発明はさらに、そのような方法により生成されたiPS細胞、ならびにそのようなiPS細胞の臨床使用および研究使用を提供する。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、一般に、誘導多能性幹(iPS)細胞の分野に関し、より具体的には、そのような細胞を体細胞から生成する方法、ならびにそのような方法により生成されたiPS細胞の臨床使用および研究使用に関する。
【背景技術】
【0002】
誘導多能性幹細胞(iPSC)は、胚性幹(ES)細胞の特性を示し、当初はマウス体細胞における4種の核初期化因子(4F):Oct4、Sox2、Klf4およびcMycの異所発現により生成された。ヒト細胞では、最初の4種の山中因子の他にも、4種の因子の別のセット、例えば、Oct4 Nanog Lin28およびSox2を用いてiPSCを生成することもできる。異なる組織由来の多くの細胞種が初期化可能であると確認されているが、典型的に0.01%〜0.2%という低初期化効率がiPSC誘導およびさらなる治療的使用のための主要な障害である。初期化効率を高める小分子についてのスクリーニングだけでなく、iPSC誘導の新しい方法の開発にも多大な努力が注ぎ込まれてきたが、初代繊維芽細胞がES様状態に初期化される機構はまだ大部分が不明である。
【0003】
細胞初期化の機構を理解するために、種々のアプローチが用いられてきた。小分子に基づく方法では、Dnmt1阻害剤で細胞を処理することによって、初期化プロセスを加速させることができることが確認された。またTGFβ阻害もより速く、かつ、より効率的なiPSC誘導を可能にすることが見出された。これはSox2およびcMycに取って代わることができる。さらにまたアレイ解析では、部分的に初期化されたiPSCは、メチルトランスフェラーゼ阻害剤などの因子での処理が行われるとさらに完全に初期化され得ることが示された。4種の初期化因子によるプロモーター結合および発現誘導の網羅的ゲノム解析では、これらの因子はiPSCおよびmES細胞において類似した標的を有し、おそらく類似した遺伝子セットを調節するであろうこと、さらに、部分的iPSCでは初期化因子のターゲッティングが変更されることも証明されている。
【0004】
ごく最近では、いくつかのグループによってp53媒介腫瘍抑制経路がiPSC誘導に拮抗し得ることが確認された。p53およびその下流のエフェクターp21の両方は初期化プロセス中に誘導され、両タンパク質の発現低下によってiPSCコロニー形成が促進され得る。これらのタンパク質は4種の初期化因子(4F)を発現する大部分の細胞においてアップレギュレートされ、cMycはp21発現を遮断すると報告されているが、これらの4種の因子(4F)の異所発現が癌遺伝子/導入遺伝子過剰発現に対する細胞応答をいかにして克服し、なぜ細胞のごく一部の集団だけが完全に初期化されるかについては依然として不明である。
【0005】
マイクロRNA(MicoRNA)は、RNA誘導サイレンシング複合体(RISC)と呼ばれるタンパク質複合体と関係がある18〜24ヌクレオチドの一本鎖低分子RNAである。これらの低分子RNAは、通常遺伝子転写物の非コード領域から生成され、翻訳抑制により遺伝子発現を抑制する働きをする。近年、マイクロRNAは多くの異なる重要なプロセス(多数ある中で、ヒトES細胞の自己複製遺伝子発現、胚性幹(ES)細胞の細胞周期制御、選択的スプライシング、心臓発生など)への関与が見出された。さらに、ES細胞特異的マイクロRNAは、マウスiPSC誘導を増強し、初期化中にcMycの機能に取って代わることができることが最近報告された。またhES特異的miR−302は、ヒト繊維芽細胞における4種の因子の発現により老化応答軽減することも示唆されている。しかしながら、これらのマイクロRNAは初期化プロセスの極めて終わりに近い段階まで発現されないため、マイクロRNAがiPSC誘導において重要な役割を果たすかどうかはこれまで分かっていなかった。
【発明の概要】
【0006】
本発明は、iPSC誘導中にマイクロRNAが必要とされるという重大な発見に基づくものである。マイクロRNA生合成機構へ干渉により初期化効率の著しい低下がもたらされる。初期化の初期段階中に高度に誘導されるマイクロRNAクラスターが確認され、機能試験では、そのようなマイクロRNAを体細胞に導入することにより誘導効率が高まることが示されている。加えて、細胞の初期化に使用される重要なレギュレーターも確認されたが、それらのレギュレーターを標的として、初期化効率ならびにiPS細胞の直接分化を著しく増加させ得ることが有利である。
【0007】
よって、一実施形態では、本発明は、iPS細胞を生成する方法を提供する。その方法は、体細胞を核初期化因子と接触させること、およびその細胞を、その細胞でRNAレベルまたは活性を変更するマイクロRNAと接触させ、それによってiPS細胞を生成することを含む。一態様では、マイクロRNAまたはRNAが修飾される。別の態様では、マイクロRNAはベクター中に存在する。別の態様では、マイクロRNAは、miR−17、miR−25、miR−106a、miR let−7ファミリーメンバー(例えば、let−7a、miR 98)またはmiR−302bクラスター中に存在する。別の態様では、マイクロRNAは、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである。
【0008】
一態様では、マイクロRNAは、配列番号1を含むポリヌクレオチド配列を有する。別の態様では、マイクロRNAは、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する。別の態様では、マイクロRNAは、p21、Tgfbr2、p53、またはそれらの組合せの発現または活性を調節する。別の態様では、マイクロRNAは、Spry 1/2、p85、CDC42またはERK1/2の経路を調節する。
【0009】
一態様では、核初期化因子は、ベクター中に含まれる遺伝子によりコードされる。別の態様では、核初期化因子は、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せである。別の態様では、核初期化因子は、OCT4、SOX2、KLF4、C−MYCの1種以上である。別の態様では、核初期化因子はc−Mycを含む。別の態様では、誘導効率は、マイクロRNAを使用しない場合と比べて少なくとも2倍となる。
【0010】
一態様では、マイクロRNAとの接触前に、接触と同時にまたは接触後に、体細胞に初期化因子を接触させる。別の態様では、体細胞は哺乳類細胞である。さらなる態様では、体細胞はヒト細胞またはマウス細胞である。
【0011】
別の実施形態では、本発明は、体細胞を核初期化因子と、p21発現または活性の阻害剤とに接触させることによりiPS細胞を生成する方法を提供する。
【0012】
別の実施形態では、本発明は、体細胞を、その細胞内でRNAレベルまたは活性を変更する作用物質と接触させ(ここで、該作用物質は該体細胞中で多能性を誘導し、ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することにより誘導多能性幹(iPS)細胞を生成する方法を提供する。様々な実施形態では、RNAは非コードRNA(ncRNA)であり、マイクロRNAが含まれる。
【0013】
上に記載した方法の一態様では、作用物質はポリヌクレオチド、ポリペプチドまたは小分子である。さらなる態様では、ポリヌクレオチドはアンチセンスオリゴヌクレオチド、化学修飾されたオリゴヌクレオチド、ロックド核酸(LNA)またはDNAである。別の態様では、ポリヌクレオチドはRNAである。さらなる態様では、RNAは、マイクロRNA、dsRNA、siRNA、stRNAまたはshRNAからなる群から選択される。別の態様では、体細胞はマウス胚性繊維芽細胞(MEF)である。
【0014】
様々な態様では、RNAを変更する作用物質は、p21、Tgfbr2、p53、またはそれらの組合せを、発現または活性について阻害することができる。一態様では、作用物質はポリヌクレオチド、ポリペプチドまたは小分子であってよい。別の態様では、作用物質またはp21、Tgfbr2および/またはp53の阻害剤はRNA分子であり、マイクロRNA、dsRNA、siRNA、stRNAまたはshRNA、またはアンチセンスオリゴヌクレオチドが含まれる。例示的態様では、作用物質またはp21、Tgfbr2および/またはp53の阻害剤は、マイクロRNA分子であり、前記細胞に導入された組換えベクター中に含まれるポリヌクレオチドによりコードされる。
【0015】
様々な態様では、マイクロRNAは、iPSCの誘導またはその分化中に活性または発現の増加または減少を示すクラスター中に含まれるマイクロRNAであってよい。一態様では、誘導効率は、作用物質を使用しない場合と比べて少なくとも2倍となる。別の態様では、誘導効率は、作用物質を使用しない場合と比べて少なくとも3倍である。別の態様では、誘導効率は、作用物質を使用しない場合と比べて少なくとも5倍である。一態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中の1種以上のマイクロRNAであってよく、miR−93、miR−106b、miR−21、miR−29a、miR−let−7ファミリーメンバー(例えば、let−7a;miR 98)またはそれらの組合せが含まれる。関連態様では、マイクロRNAは、miR−17、miR−25、miR−106aおよびmiR−302bクラスター中のマイクロRNA種に対応する様々なマイクロRNA(例えば、配列番号2〜11のもの)の間で保存されると判明している、配列番号1を含むポリヌクレオチド配列を有する。一態様では、マイクロRNAは、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する。
【0016】
様々な態様では、核初期化因子は、前記細胞に導入された組換えベクター中に含まれる遺伝子によりコードされる。別の態様では、作用物質は、p21、Tgfbr2、p53、またはそれらの組合せの発現または活性を阻害する。別の態様では、作用物質は、Spry 1/2、p85、CDC42またはERK1/2の経路を調節する。
【0017】
様々な態様では、核初期化因子は、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せの1種以上によりコードされる。例示的態様では、核初期化因子は、OCT4、SOX2、KLF4、C−MYCの1種以上である。別の態様では、少なくとも1種の核初期化因子がc−Mycを含む。さらなる態様では、c−Mycは、少なくとも1つのmiRNAを抑制することにより初期化を少なくともある程度まで促進する。
【0018】
別の実施形態では、本発明は、本明細書に記載した方法を用いて生産されたiPS細胞またはそのような細胞の集団を提供する。別の実施形態では、本発明は、本明細書に記載した方法により生産された誘導多能性幹(iPS)細胞の濃縮された集団を提供する。
【0019】
同様に、別の実施形態では、本発明は、本明細書に記載した方法を用いて生成されたiPSCの分化を誘導することにより得られた分化した細胞を提供する。一態様では、体細胞はiPSCをRNA分子またはアンチセンスオリゴヌクレオチドと接触させることによって分化を誘導することにより得られる。一態様では、RNA分子は、マイクロRNA、dsRNA、siRNA、stRNAまたはshRNAからなる群から選択される。
【0020】
別の実施形態では、本発明は、本明細書に記載した方法を用いて生成されたiPS細胞を用いて対象を治療する方法を提供する。その方法は、本明細書に記載した方法を用いて対象の体細胞を誘導多能性幹(iPS)細胞に誘導すること、そのiPS細胞の分化を誘導すること、およびその分化した細胞をその対象に導入し、それによってその症状を治療することを含む。
【0021】
別の実施形態では、本発明は、本明細書に記載した方法により生成されたiPS細胞またはそれから得られた体細胞を用いて作用物質の生理学的機能を評価するための方法を提供する。一態様では、その方法は、本明細書に記載した方法を用いて生産された誘導多能性幹(iPS)細胞を処理することおよびその作用物質による少なくとも1つの細胞機能の変化を評価することを含む。別の態様では、その方法は、本明細書に記載した多能性幹細胞の分化を誘導することにより得られた分化した細胞を作用物質により処理することおよびその作用物質による細胞機能の変化を評価することを含む。
【0022】
別の実施形態では、本発明は、本明細書に記載した方法により生成されたiPS細胞またはそれから得られた体細胞を用いて化合物の毒性を評価する方法を提供する。一態様では、その方法は、本明細書に記載した方法を用いて生産された誘導多能性幹(iPS)細胞を化合物により処理することおよびその化合物の毒性を評価することを含む。別の態様では、その方法は、本明細書に記載した多能性幹細胞の分化を誘導することにより得られた分化した細胞を化合物により処理することおよびその化合物の毒性を評価することを含む。
【0023】
別の実施形態では、本発明は、誘導多能性幹(iPS)細胞を生成する方法を提供する。その方法は、体細胞を少なくとも1種の核初期化因子と接触させること;およびその細胞をp21、Tgfbr2、p53、またはそれらの組合せの発現または活性についての阻害剤と接触させることを含む。一態様では、阻害剤はp21の発現および/または活性を阻害する。別の態様では、阻害剤はTgfbr2の発現および/または活性を阻害する。別の態様では、阻害剤はp53の発現および/または活性を阻害する。
【0024】
別の実施形態では、本発明は、誘導多能性幹(iPS)細胞を生成する方法を提供する。その方法は、体細胞を、その細胞内でRNAレベルまたは活性を変更する作用物質と接触させ(ここで、該作用物質は該体細胞中で多能性を誘導し、ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することを含む。
【0025】
別の実施形態では、本発明は、対象を治療する方法を提供する。その方法は、本明細書に記載した方法により対象の体細胞から誘導多能性幹(iPS)細胞を生成すること;そのiPS細胞の分化を誘導すること;およびその細胞をその対象に導入し、それによってその症状を治療することを含む。
【0026】
別の実施形態では、本発明は、iPS細胞の生成効率を高めるためのマイクロRNAの使用を提供する。一態様では、マイクロRNAは、miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、またはそれらの組合せからなる群から選択される。別の態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中に存在する。別の態様では、マイクロRNAは、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである。
【0027】
別の実施形態では、本発明は、miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、miR let−7ファミリーメンバーまたはそれらの組合せからなる群から選択されるmiR配列の組合せを提供する。別の態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中に存在する。別の態様では、マイクロRNAは、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである。
【図面の簡単な説明】
【0028】
【図1】マウスiPSC誘導へのRNAi機構の関与を示す図である。図1a、図1bおよび図1cは、それぞれ、shRNAによるマウスRNAi機構遺伝子Ago2、DroshaおよびDicerのノックダウンを示している。標的遺伝子のmRNAおよびタンパク質のレベルの両方をRT−qPCRにより解析し、ヒストグラムおよび対応するウエスタンブロットに示している。初代マウス胚性繊維芽細胞(MEF)に、4種の因子を、Drosha、DicerおよびAgo2をターゲッティングするshRNAとともに形質導入する。MEFにレンチウイルスshRNAを4μg/μlのポリブレンとともに形質導入し、形質導入後3日目にトータルRNAまたはタンパク質を採取する。標的遺伝子のmRNAおよびタンパク質のレベルを、それぞれ、RT−qPCRおよびウエスタンブロッティングにより解析する。pLKOは、shRNAレンチウイルスベクターに対する空ベクター対照である。pGIPZは、ターゲッティングしないshRNAを発現するレンチウイルスベクターである。図1dは、Ago2のノックダウンによりOSKによるiPSC誘導が減少するということを示している。形質導入後21日目にコロニーを染色し、APについて定量する。エラーバーは、2反復のウェルの標準偏差を表す。図1eは、shAgo2に関するiPSCのGFP+コロニーの定量を示している。形質導入後21日目にGFP+コロニーを定量する。エラーバーは、2反復のウェルの標準偏差を表す。図1fは、Ago2のノックダウンにより4FによるiPSC誘導が劇的に減少するということを示している。初代MEFに4種の初期化因子(OSKM(4F))をshRNA Ago2とともに形質導入する。形質導入後14日目にコロニーをアルカリ性ホスファターゼについて染色することができる。アルカリ性ホスファターゼはmES/iPS細胞のマーカーである。pLKOベクターおよびpGIPZベクターを陰性対照とした。
【図2】初期化の初期段階中のマイクロRNAクラスターmiR−17、25、106aおよび302bの誘導を示す図である。図2aは、4種の因子の形質導入後の、初期段階にある10種のマイクロRNAクラスターの発現誘導を示すグラフを示している。miR RT−qPCRを用いて、ES細胞において高度に発現される10種のクラスターの代表的なマイクロRNAの発現変化を定量する。感染後4日目に開始時のMEFおよび4Fを用いたMEFのトータルRNAを解析する。ヒストグラムの色の濃いバーは感染後4日目のMEFを示し、白色のバーは開始時のMEFを示す。アスタリスクは、誘導されたマイクロRNAを示す。図2bは、4F形質導入後4日目に誘導された異なるmiRクラスターのシード領域比較を示している。類似したシード領域に下線を施している。図2cは、マイクロRNAの誘導についてのグラフを示している。代表的なマイクロRNAは4種の因子の種々の組合せを用いて誘導され得る。形質導入後4日後にマイクロRNA発現を定量する。4F、OSK、OSおよび単一因子を用いて、miR発現の変化にどの因子が関与したかを解析する。
【図3】miR−93およびmiR−106bによるiPSCの誘導増強を示す図である。図3aは、初期化アッセイの時系列を示す絵画表現である。マイクロRNAミミックを0日目および5日目に終濃度50nMでトランスフェクトする。4F誘導については11日目に、OSK 3因子のiPSC誘導については15〜20日目にGFP+コロニーを定量する。図3bは、miR−93およびmiR−106bミミックが4F誘導に関するiPSC誘導を増強することを示すグラフである。Oct4−GFP MEFを50nMの表示マイクロRNAでトランスフェクトする。形質導入後11日目にGFP+コロニーを定量する。誘導倍率およびエラーバーは、3反復のウェルを用いた3つの独立した実験から計算した。図3cは、OSK系を用いたmiR−93およびmiR−106bの増強効果の識別を示すグラフである。4F実験の場合と同様に、マイクロRNAミミックをトランスフェクトする。15〜20日目にGFP+コロニーを定量する。エラーバーは、3反復のウェルでの3つの独立した実験の標準偏差を表す。図3dは、初期化効率に対するマイクロRNA阻害の影響を示すグラフである。miR−93およびmiR−106bの阻害剤は初期化効率を劇的に低下させる。マイクロRNA阻害剤もまた終濃度50nMでトランスフェクトし、miRミミックトランスフェクションと同じ実験時系列を維持する。エラーバーは、3反復のウェルでの3つの独立した実験の標準偏差を表す。
【図4】異なる内因性ESマーカーのRT−PCRによる発現を解析するmiRミミック実験から得られたiPSCクローンの特性評価を示す図である。継代後3日目にiPS細胞株からトータルRNAを単離する。ES細胞特異的マーカー(Eras、ECat I、Nanogおよび内因性Oct4など)の発現をRT−PCRにより解析する。
【図5】miR−93およびmiR−106bによるマウスp21およびTgfbr2のターゲッティングを示す図である。図5aは、miR−93および106bトランスフェクションによりp21タンパク質レベルが減少するということを示している。Oct4−GFP MEFを、50nMのmiRミミックでトランスフェクトし、ウエスタン解析のためにトランスフェクションの48時間後に採取する。ローディング対照としてアクチンを用いる。図5bは、p21がsiRNAにより効率的にノックダウンされるということを示している。P21 siRNAトランスフェクトMEFおよび対照トランスフェクトMEFを、48時間およびRT−qPCRの時点で採取し、ウエスタンブロッティングを行って、p21発現を確認する。p21 mRNAをGAPDHに対して正規化する。図5cは、siRNAによるp21のノックダウンによりiPSC誘導が増強されるということを示している。MEFを4Fウイルスに感染させ、マイクロRNAミミックトランスフェクションと同じ時系列に従ってsiRNAをトランスフェクトする。GFP+コロニーを11日目に定量する。エラーバーは、3反復のウェルを用いた少なくとも2つの独立した実験を表す。図5dは、miR−93および106bトランスフェクションによりTgfbr2発現が減少するということを示している。ウエスタンブロッティングのためにトランスフェクト細胞を48時間の時点で採取する。図5eは、siRNAによりTgfbr2がノックダウンされるということを示している。相対的Tgfbr2 mRNAレベルをGapdhのものに対して正規化する。図5fは、siRNAによるTgfbr2のノックダウンによりiPSC誘導が増強されるということを示している。エラーバーは、3反復のウェルを用いた少なくとも3つの独立した実験を表す。
【図6】マイクロRNAによる初期化の促進を示す図である。図6aは、miR−17およびmiR−106aにより初期化効率を高めることができるが、miR−16では初期化効率を高めることができないということを示している。miR−17およびmiR−106aミミックをMEFに終濃度50nMでトランスフェクトする。形質導入後11日目にGFP+コロニーを定量する。エラーバーは、3反復のウェルでの2つの独立した実験を表す。図6bは、miR−17および106aがp21をターゲッティングするということを示している。マイクロRNAミミックのMEFへのトランスフェクションの2日後にp21のウエスタンブロッティングを行う。miR−17およびmiR−106aはTgfbr2発現をターゲッティングする。マイクロRNAミミックをMEFに50nMの終濃度でトランスフェクトする。図6cは、miR−17および106aがTgfbr2をターゲッティングするということを示している。トランスフェクションの2日後にウエスタンブロッティングを行う。図6dは、iPSC誘導中のマイクロRNAの役割についてのモデルを示している。初期化の初期段階中に、miR−17、25および106aクラスターを含むいくつかのマイクロRNAが誘導される。これらのマイクロRNAは、初期化プロセスに拮抗する因子(p21および他の未同定タンパク質など)をターゲッティングすることにより完全初期化を促進する。上昇および下降は、初期化プロセス中の種々の潜在的な段階および障壁を表し、破線は、初期化された細胞におけるマイクロRNA誘導により低下した初期化の障壁を示す。
【図7】マウスiPSC誘導におけるmiR−93およびmiR−106bの用量応答を示すグラフである。Oct4−GFP MEFを異なる濃度(5nM、15nMおよび50nM)のマイクロRNAでトランスフェクトする。ミミック対照siRNAを対照として用いる。形質導入後11日目にGFP+コロニーを定量する。データは、12ウェルプレートでの3反復のウェルを表す。
【図8】iPSC誘導中に誘導されたp21発現を示す図である。図8aは、異なるp21発現系(左から右へ:OSKM、OSK、OS、Klf4、cMycおよびMEF対照)を用いたウエスタンブロット解析を示している。p21発現はKlf4およびcMycにより誘導される。ウエスタンブロッティング解析のために形質導入後5日目に4F、OSK、OS、Klf4およびcMycに感染させたMEFを採取する。図8bは、感染したMEFにおける異なる導入遺伝子の発現の確認を示すグラフを示している。
【図9】OSK 3因子を用いた初期化のp21過剰発現による阻害を示す図である。図9aは、OSK誘導およびp21過剰発現によるiPSCのAP+コロニーの定量のグラフである。21日目に誘導された細胞をアルカリ性ホスファターゼについて染色する。p21ウイルスは、OSKと同時に導入する。図9bは、OSK誘導およびp21過剰発現によるiPSCのGFP+コロニーの定量のグラフである。
【図10】miRNAによるp21発現の直接調節を示す図である。図10aは、p21 mRNA 3'UTRに見出される2つの潜在的部位を示す絵画表現である。突然変異を第1の部位(保存部位)に導入して、miR−93および106bの結合親和性を破壊する。図10bは、Hela細胞におけるpGL3−p21ルシフェラーゼレポーター発現の定量を示すグラフである。Hela細胞を、pGL3−p21およびpRL−TKと、マイクロRNAとで48時間トランスフェクトした後採取する。結果を、トランスフェクト細胞におけるpRL−TKレベルに対して正規化する。
【図11】miRNAによるTgfbr2発現の直接調節を示す図である。図11aは、Tgfbr2 mRNA 3'UTRに見出される2つの潜在的部位を示す絵画表現である。図11bは、p21実験と同様に実施した、Hela細胞におけるルシフェラーゼレポーター発現の定量を示すグラフである。結果を、トランスフェクト細胞におけるpRL−TKレベルに対して正規化する。
【図12】表示した様々なmiRNAの存在下での相対的Tgfbr2 mRNAレベルを示す図である。
【図13】shRNAがshAgo2感染MEFにおいて活発に発現されることを示す図である。図13aはshAgo2レベルを示し、図13bはshRNAレベルを示している。図13cは、Ago2感染MEFにおけるES特異的マーカーの発現を示している。
【図14】OSKM因子の形質導入後0日目、4日目、8日目および12日目の相対的miRNA発現を示す図である。
【図15】miR−93の相対的レベルに対するmiR−93ミミックの影響を示す図である。
【図16】図16aは、miR阻害剤が標的miR発現を減少させることができるということを示している。図16bは、初期化プロセスの種々の段階中のmiR阻害剤の影響をさらに示している。
【図17】miR−93またはmiR−106bが導入されるときの内因性Nanog遺伝子座のプロモーターメチル化レベルを示す図である。
【図18】図18aおよび図18bは、miR−93トランスフェクション後に著しく減少する遺伝子がiPSCにおいて低度に発現される遺伝子の3倍濃縮を示し得る一方で、miR−93トランスフェクション後に増加する遺伝子がそのような濃縮を示さないということを示している。
【図19】図19aは、mRNAアレイ解析またはRT−qPCR解析のいずれかを用いた、miR−93の導入後の相対的Tgfbr2 mRNAレベルを示している。図19bは、miR−25、miR−93またはmiR−106bの導入後の相対的mRNAレベルを示している。
【図20】MEFで濃縮されたマイクロRNA、miR−21およびmiR−29aの阻害によりiPS細胞初期化効率が高まることを示す図である。図20aは、MEFにおいてmiR−29a、miR−21およびlet7aが高度に発現されるということを示している。トータルRNAをOct4−EGFP MEFおよびマウスES細胞から単離し、ゲル電気泳動により分離する。表示miRNAに対して特異的な放射性標識プローブを用いて、シグナルを検出する。U6 snRNAをローディング対照とする。図20bは、miRNA阻害により初期化効率が高まるということを示している。Oct4−EGFP MEFにOSKMを形質導入する。形質導入後14日目に蛍光顕微鏡検査法によりGFP陽性コロニーを確認し、計数する。GFP+コロニー数を、anti miRターゲッティングしない対照処理の数に対して正規化し、変化倍率として報告する。エラーバーは、3つの独立した実験の標準偏差を表す。*p値<0.05。
【図21】c−Mycが初期化中の、MEFで濃縮されたmiRNAの主要なレプレッサーであることを示す図である。図21aは、初期化後5日目の選択miRNAのノーザン解析を示している。Oct4−EGFP MEFに、表示した、単一因子または様々な初期化因子組合せを形質導入する。1F、1種の因子;2F、2種の因子;3F、3種の因子;OSKM:Oct4、Sox2、Klf4およびc−Myc。U6をローディング対照RNAとして用いる。胚幹細胞(ES)のトータルRNAを、MEFおよび形質導入細胞に対する陰性対照とする。様々なプローブを用いて、右側に表示した特定miRNAを検出する。miR−291ブロッティングがES RNAの陽性対照である。 図21bは、様々な初期化因子の存在下でのmiRNA発現の定量的表現を示している。シグナル強度をU6 snRNAの強度に対して正規化する。発現比を、MEFにおける発現(これを100%に任意設定する)に対する各miRNAの発現割合として計算する。(パネルAの)様々なmiRNAを定量し、右側に表示する。 図21cは、OSKまたはOSKMによる初期化後の様々な時点におけるOct4−EGFP MEFにおける選択miRNAのリアルタイムRT−PCR解析を示している。リアルタイムRT−PCR解析のために形質導入後の表示した日(D)にRNAを単離する。シグナルをU6に対して正規化し、MEFにおいて発現されたmiRNA(これを100に任意設定する)に対する割合として示す。エラーバーは、2つの独立した実験の標準偏差を表す。
【図22】図22は、miR−21またはmiR−29aの阻害が、p53タンパク質レベルを低下させp85αおよびCDC42の経路をアップレギュレートすることによりiPS細胞初期化を促進することを示す図である。図22aは、様々なmiRNAの阻害後のp53、CDC42およびp85αの発現のウエスタン解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトする。5日後に細胞を採取し、解析する。図22bは、表示miR阻害剤の存在下でのタンパク質発現の定量的表現を示している。シグナル強度を、GAPDH強度に対して正規化し、対照(NT)細胞における発現(これを100に任意設定した)に対する割合として示す。エラーバーは、少なくとも3つの独立した実験の標準偏差を示す。*p値<0.05。 図22cは、様々なmiRNAの阻害およびOSKM形質導入の後のp53、CDC42およびp85αの発現のイムノブロット解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトする。5日後に細胞を採取し、イムノブロットにより解析する。(B)に記載のとおり、シグナル強度を正規化する。エラーバーは、少なくとも3つの独立した実験の標準偏差を示す。*p値<0.05。図22dは、miR−29aまたはp53の枯渇により初期化効率が高まるということを示している。4X104 Oct4−EGFP MEFを、表示siRNAおよびmiRNA阻害剤と、OSKM初期化因子とでトランスフェクトする。形質導入後12日目にGFP陽性細胞を計数する。エラーバーは、少なくとも3つの独立した実験の標準偏差を示す。*p値<0.05。
【図23】miR−21およびmiR−29aの枯渇が、ERK1/2経路をダウンレギュレートすることにより初期化効率を高めることを示す図である。図23aは、MEFにおける様々なmiRNAの阻害後のリン酸化ERK1/2およびトータルERK1/2のウエスタン解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトし、5日後に採取し、イムノブロットする。シグナル強度を、アクチンに対して正規化し、anti miR NT対照の発現に対する割合として示す。エラーバーは、3つの独立した実験の標準偏差を示す。*p値<0.05;**p値<0.005。図23bは、miR−21およびmiR−29aの枯渇によりSpry1タンパク質レベルが増加するということを示している。Spry1発現比のウエスタンブロット解析を示す。MEFを、表示した様々なmiRNA阻害剤でトランスフェクトする。ウエスタンブロット解析のためにトランスフェクション後5日目に細胞を採取する。シグナル強度を、アクチンに対して正規化し、図23aに記載したように示す。エラーバーは、3つの独立した実験の標準偏差を表す。*p値<0.05;**p値<0.005。 図23cは、ERK1/2またはGSK3βのノックダウン後の初期化効率における変化倍率を示している。4X104 Oct4−EGFP MEFを表示siRNAと、OSKMとでトランスフェクトする。2週間後にGFP陽性細胞を計数する。siNTでのトランスフェクションを初期化効率の対照とする。エラーバーは、3つの独立した実験の標準偏差を示す。**p値<0.005。図23dは、MEFにおける様々なmiRNAの阻害後のリン酸化GSK−3βおよびトータルGSK−3βのウエスタン解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトし、5日後に採取し、イムノブロットにより解析する。図23aに記載したように、シグナル強度を正規化する。エラーバーは、3つの独立した実験の標準偏差を示す。
【図24】c−Mycが、MEFで濃縮されたmiRNA、miR−21およびmiR−29aをダウンレギュレートすることにより初期化を促進することを示す模式図を示す図である。p53およびERK1/2の経路は初期化の障壁として働き、miR−21およびmiR−29aは、CDC42、p85αおよびSpry1をダウンレギュレートすることによりそれらの経路を間接的に活性化する。miR−21/p53経路とmiR−29a/ERK1/2経路との間のクロストークも示す。c−Mycは、これらのmiRNAの発現を抑制し、次にはERK1/2およびp53の誘導に支障をきたす。点線はiPS初期化に対するp53およびERK1/2の作用を示す。
【図25】miR−21の阻害が、OSKによるiPS細胞初期化を促進することを示す図である。miRNAの阻害剤は、OSKでの初期化中にOct4−MEFに導入される。形質導入後の様々な時点においてGFP陽性コロニーを計数する。エラーバーは、2つの独立した実験の標準偏差を表す。
【図26】miRNAの阻害が、初期化中にアポトーシスまたは増殖速度を変更しないことを示す図である。図26aは、miRNAの阻害剤が、OSKMでの初期化中にOct4−MEFに導入されるということを示している。形質導入後8〜9日目に細胞を収集する。PE Annexin V Apoptosis Detection Kit I(BD Pharmingen;カタログ番号559763)および7−アミノ−アクチノマイシン(7−AAD)を用いてアポトーシスを評価する。シグナルをFACSにより検出する。エラーバーは、3つの独立した実験の標準偏差を表す。図26bは、miRNA阻害剤が、OSKMでの初期化中にOct4−MEFに導入されるということを示している。形質導入後8〜9日目に細胞を収集する。収集の1日前に、Click−iT Edu Imaging Kits(Invitrogen;カタログ番号C10337)を用いて5−エチニル−2'−デオキシウリジン(Edu)で細胞を処理する。シグナルをFACSにより検出する。エラーバーは、3つの独立した実験の標準偏差を表す。
【発明を実施するための形態】
【0029】
本発明は、iPSC誘導に関与する重要な調節機構の発見に基づくものである。重要な態様は、iPSCの誘導に関する細胞マイクロRNA間の関連性の発見である。これは、重要なマイクロRNA経路タンパク質のノックダウンによるマイクロRNA生合成機構への干渉により初期化効率の著しい低下がもたらされ得るという観察結果から明らかである。特に、初期化の初期段階中に高度に誘導される、少なくとも3種のマイクロRNAクラスターが明らかとなっている、miR−17〜92、106b〜25および106a〜363。非常に類似したシード領域を有するいくつかのマイクロRNA(miR−93およびmiR−106bなど)は、p21発現をターゲッティングすることによりiPSC誘導を大幅に増強し、得られたクローンを完全初期化状態に到達させ得る。
【0030】
本発明によれば、マイクロRNAはiPSC誘導において直接働くことができ、マイクロRNA生合成機構への干渉により初期化効率が著しく低下することとなる。本発明は、初期化の初期段階中に高度に誘導される3種のマイクロRNAクラスター、miR−17〜92、miR−106b〜25およびmiR−106a〜363を提供する。機能解析では、これらのマイクロRNAをMEFに導入することによりOct4−GFP+iPSCコロニー形成が促進されることが示された。また、本発明によれば、Tgfbr2およびp21(これらは両方とも初期化を阻害する)はこれらのマイクロRNAにより直接ターゲッティングされ、それらの活性の遮断により初期化効率が著しく低下することとなる。本発明によれば、miR−93およびmiR−106bは初期化活性の重要なレギュレーターであることが提案される。
【0031】
本組成物および方法を記載する前に、記載する特定の組成物、方法および実験条件は変化し得ることから本発明はそのような組成物、方法および条件に限定されないことを理解すべきである。また、本発明の範囲は添付の特許請求の範囲によってのみ限定されるため、本明細書において用いられる用語は特定の実施形態を記載することを目的とするものにすぎず、限定するものではないことも理解すべきである。
【0032】
本明細書および添付の特許請求の範囲において用いられるように、単数形「a」、「an」および「the」は、特に明示されない限り、複数の参照語を包含する。従って、例えば、「方法」について述べる場合は、1以上の方法および/または、この開示内容などを読むことによって当業者には明らかとなるであろう、本明細書に記載した種類の工程も包含する。
【0033】
特に定義されない限り、本明細書において用いられる全ての技術用語および科学用語は、本発明が属する分野の通常の技術を有する人に一般に理解されている意味と同じ意味を有する。本発明の実施または試験において、本明細書に記載したものと類似または同等の方法および材料を使用することができるが、好ましい方法および材料を次に記載する。
【0034】
本明細書において論じるように、マイクロRNAが初期化プロセスおよびiPSC誘導効率に関与するという発見によって、それらの細胞中のこれらのマイクロRNAのレベルを操作することによりiPSC誘導効率を大幅に高める能力に至っている。よって、本発明は、既知方法と比べて誘導効率が改善されたiPS細胞生成方法を提供する。その方法は、体細胞を核初期化因子と、その細胞内でマイクロRNAレベルまたは活性を変更する作用物質とに接触させること(ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することを含む。
【0035】
本発明はまた、初期化プロセスおよびiPSC誘導効率に直接関与する調節タンパク質の発見にも基づく。そのような1つのタンパク質はp21(165アミノ酸しか含まない低分子タンパク質)であり、このタンパク質は、p53依存性G1増殖停止を引き起こし分化および細胞老化を促進することにより癌発生中の腫瘍抑制遺伝子として長年にわたり知られていた。iPSC誘導中のマイクロRNAによるp21発現阻害はこの点で誘導効率を高めることが証明された。よって、1つの実施形態では、本発明は、体細胞を核初期化因子と、p21発現または活性の阻害剤とに接触させることによるiPS細胞生成方法を提供する。
【0036】
iPSC生成へのRNAの調節関与を前提として、核初期化因子以外の、RNAレベルを調節する作用物質を用いて誘導が起こり得ると考えられる。よって、本発明は、体細胞を、その細胞内でRNAレベルまたは活性を変更する作用物質と接触させ(ここで、該作用物質は該体細胞中で多能性を誘導し、ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することにより誘導多能性幹(iPS)細胞を生成する方法を提供する。様々な実施形態では、RNAは非コードRNA(ncRNA)、例えば、マイクロRNAなどである。
【0037】
様々な実施形態では、1種以上の核初期化因子を用いて、卵子、胚またはES細胞を使用せずに分化した細胞の初期化を誘導することができる。誘導プロセスの効率は、誘導プロセス中にその細胞内でマイクロRNAレベルまたは活性を変更する作用物質を利用することにより高められる。その方法を用いて、ES細胞と類似した多能性と増殖能を有する誘導多能性幹細胞を便宜にかつ高度に再現性良く樹立し得る。例えば、核初期化因子は、RNA分子(マイクロRNAなど)をコードするポリヌクレオチドを含む組換えベクターとともに、核初期化因子をコードする遺伝子を含む組換えベクターを細胞に形質導入することにより、細胞に導入され得る。よって、その細胞は、組換えベクター中に含まれる遺伝子の産物として発現される核初期化因子を発現することができ、同様に組換えベクター中に含まれるポリヌクレオチドの産物として発現されるマイクロRNAを発現し、それによって核初期化因子だけを使用する場合と比べて高い効率(割合)で分化した細胞の初期化を誘導する。
【0038】
本明細書において用いられるように、多能性細胞とは、in vitroで長期間(1年より長い期間)分裂する可能性があり、内胚葉、中胚葉および外胚葉を含む3つの胚葉全てから得られる細胞に分化する独特の能力を有する細胞を包含する。
【0039】
本発明で用いる体細胞は初代細胞または不死化細胞であってよい。そのような細胞は、初代細胞(非不死化細胞)(動物から新たに単離したものなど)であってよく、または細胞株(不死化細胞)から得てもよい。例示的態様では、体細胞は、例えば、ヒト細胞またはマウス細胞などの哺乳類細胞である。体細胞は、周知の方法により、種々の器官(限定されるものではないが、皮膚、肺、膵臓、肝臓、胃、腸、心臓、生殖器、膀胱、腎臓、尿道および他の泌尿器など)から、または一般的には生存体細胞を含有する任意の器官または組織から得てもよい。本発明において有用な哺乳類体細胞には、例として、成体幹細胞、セルトリ細胞、内皮細胞、顆粒膜上皮細胞、ニューロン、膵島細胞、表皮細胞、上皮細胞、肝細胞、毛包細胞、ケラチノサイト、造血細胞、メラノサイト、軟骨細胞、リンパ球(Bリンパ球およびTリンパ球)、赤血球、マクロファージ、単球、単核細胞、繊維芽細胞、心筋細胞、他の公知の筋肉細胞、一般的には任意の生存体細胞が挙げられる。特定の実施形態では、繊維芽細胞が用いられる。本明細書において用いられる体細胞という用語はまた、成体幹細胞を含むようにも意図されている。成体幹細胞は、特定組織のあらゆる細胞種を生じさせることが可能な細胞である。成体幹細胞の例としては、造血幹細胞、神経幹細胞および間葉系幹細胞が挙げられる。
【0040】
本明細書において用いられるように、初期化とは、一部分化したまたは最終分化した体細胞の分化状態を変更するまたは後退させるプロセスを指すよう意図されている。体細胞の初期化は、体細胞の分化状態の一部後退または完全後退であってよい。例示的態様では、初期化は完全なものであり、この場合、体細胞は誘導多能性幹細胞に初期化される。しかしながら、初期化は、一部であってもよい(低分化状態への後退など)。例えば、最終分化した細胞の低分化状態の細胞(多分化性細胞など)への後退。
【0041】
本発明の様々な態様では、核初期化因子は、多能性を誘導する遺伝子であり、分化した細胞または半分化した細胞を、最初の細胞より原始的な表現型(多能性幹細胞の表現型など)に初期化するために利用される。そのような遺伝子は、細胞中でマイクロRNAレベルまたは活性を変更するおよび/またはp21の発現または活性を阻害して誘導効率を高める作用物質とともに利用される。そのような遺伝子および作用物質は、体細胞のゲノムに組み込まれた1種以上の該遺伝子の発現により、体細胞からの多能性幹細胞の生成が可能である。本明細書において用いられるように、多能性を誘導する遺伝子とは、多能性と関係がある遺伝子であって、該遺伝子の組込みおよび発現により体細胞からの低分化細胞(多能性幹細胞など)の生成が可能である遺伝子を指すよう意図されている。多能性遺伝子の発現は、典型的には多能性幹細胞に限定され、多能性幹細胞の機能的同一性に不可欠である。
【0042】
細胞中でマイクロRNAのレベルまたは活性を変更するまたはp21の発現または活性を阻害する作用物質には、様々な異なる種類の分子が含まれることは当業者ならば理解するであろう。本発明の方法のいずれかにおいて有用な作用物質は、あらゆる種類の分子、例えば、ポリヌクレオチド、ペプチド、ペプチドミメティック、ペプトイド(ビニローグペプトイド(vinylogous peptoids)など)、化学化合物(有機分子または小有機分子など)などであり得る。よって、一態様では、本発明の方法に用いる作用物質は、ポリヌクレオチド(アンチセンスオリゴヌクレオチドまたはRNA分子など)である。様々な態様では、作用物質は、ポリヌクレオチド(アンチセンスオリゴヌクレオチドまたはRNA分子(マイクロRNA、dsRNA、siRNA、stRNAおよびshRNAなど)など)であってよい。例示的な態様では、作用物質は、細胞に導入されるマイクロRNAであり、その導入の結果、その細胞中でマイクロRNAのレベルおよび活性が増加しかつ/またはp21が阻害される。
【0043】
マイクロRNA(miRNA)は、遺伝子発現を調節する一本鎖RNA分子である。miRNAは、遺伝子によりコードされておりそのDNAから転写されるがmiRNAはタンパク質へ翻訳されない;その代わりに一次転写物(pri−miRNA)は各々、pre−miRNAと呼ばれる短いステムループ構造にプロセシングされ、最終的には機能性miRNAにプロセシングされる。成熟したmiRNA分子は1以上のメッセンジャーRNA(mRNA)分子と完全にまたは部分的に相補的であり、それらの主な機能は遺伝子発現をダウンレギュレートすることである。マイクロRNAは、独立した遺伝子によりコードされ得るが、また、様々な異なるRNA種(イントロン、mRNAの3'UTR、長い非コードRNA、snoRNAおよびトランスポゾンを含む)から(酵素Dicerによって)プロセシングを受け得る。本明細書において用いられるように、マイクロRNAとは、「ミミック」マイクロRNAも包含し、「ミミック」マイクロRNAは、それらの内因性対応物と同じまたは実質的に同じ機能を有する、細胞に外から導入されるマイクロRNAを意味するよう意図されている。よって、当業者ならば作用物質が外から導入されるRNAであってよいことを理解する上で、作用物質はまた、細胞中でマイクロRNAの発現を増加または減少させる化合物なども包含する。
【0044】
様々な態様では、マイクロRNAは、iPSCの誘導またはその分化中に活性または発現の増加または減少を示すクラスター中に含まれるマイクロRNAであってよい。一態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中の1種以上のマイクロRNA(miR−93、miR−106b、またはそれらの任意の組合せなど)であってよい。miR−17〜92、miR−106b〜25およびmiR−106a〜363クラスターの誘導は、正確な初期化に重要であることが示されている。そのプロセス中にそのようなマイクロRNAは初期化障壁を低くするように思われるため、それらの細胞中のこれらのマイクロRNAのレベルを操作して、初期化効率を改善し得る。マイクロRNAは重要な調節分子であることが示されていることから、また、マイクロRNAを操作して、iPSCの分化を指示し得る。
【0045】
3種のマイクロRNAクラスターは、iPSC誘導中に誘導されることがここで確認され、これらのクラスター内のいくつかのマイクロRNAは、同じヌクレオチドシード領域配列を有すると判明しており、それらは類似したmRNAをターゲッティングすることが示された。また、同じシード領域のヌクレオチド配列を共有するそのようなマイクロRNAはiPSC誘導を増強する一方で、p21発現を減少させることも判明している。よって、一態様では、マイクロRNAは、様々なマイクロRNA(例えば、配列番号2〜11のもの)の間で保存されると判明している、配列番号1を含むポリヌクレオチド配列、5'−AAGUGC−3'を有する。よって、関連態様では、マイクロRNAは、配列番号2〜11のいずれかのヌクレオチド配列を有する。
【0046】
「低分子干渉RNA」および「siRNA」という用語はまた、様々な生物学的役割を果たす短鎖二本鎖RNA分子の種類である、短鎖干渉RNAまたはサイレンシングRNAを指すように、本明細書において用いられる。とりわけ、siRNAはRNA干渉(RNAi)経路に関与し、そこでsiRNAは特異的遺伝子の発現に干渉する。RNAi経路におけるそれらの役割に加え、siRNAはRNAi関連経路においても(例えば、抗ウイルス機構としてまたはゲノムのクロマチン構造形成において)作用する。
【0047】
本発明のポリヌクレオチド(アンチセンスオリゴヌクレオチドおよびRNA分子など)は任意の好適な長さのものであってよい。例えば、遺伝子発現を調節するためにアンチセンスオリゴヌクレオチドまたはRNA分子を使用するにはどのような長さが好適であるかは当業者ならば理解するであろう。そのような分子は、典型的には、約5〜100ヌクレオチド長、5〜50ヌクレオチド長、5〜45ヌクレオチド長、5〜40ヌクレオチド長、5〜35ヌクレオチド長、5〜30ヌクレオチド長、5〜25ヌクレオチド長、5〜20ヌクレオチド長または10〜20ヌクレオチド長である。例えば、その分子は、約5ヌクレオチド長、10ヌクレオチド長、15ヌクレオチド長、16ヌクレオチド長、17ヌクレオチド長、18ヌクレオチド長、19ヌクレオチド長、20ヌクレオチド長、21ヌクレオチド長、22ヌクレオチド長、23ヌクレオチド長、24ヌクレオチド長、25ヌクレオチド長、26ヌクレオチド長、27ヌクレオチド長、28ヌクレオチド長、29ヌクレオチド長、30ヌクレオチド長、31ヌクレオチド長、32ヌクレオチド長、33ヌクレオチド長、34ヌクレオチド長、35ヌクレオチド長、40ヌクレオチド長、45ヌクレオチド長または50ヌクレオチド長であってよい。そのようなポリヌクレオチドは、少なくとも約15ヌクレオチド〜約120を超えるヌクレオチドを含んでよい(少なくとも約16ヌクレオチド、少なくとも約17ヌクレオチド、少なくとも約18ヌクレオチド、少なくとも約19ヌクレオチド、少なくとも約20ヌクレオチド、少なくとも約21ヌクレオチド、少なくとも約22ヌクレオチド、少なくとも約23ヌクレオチド、少なくとも約24ヌクレオチド、少なくとも約25ヌクレオチド、少なくとも約26ヌクレオチド、少なくとも約27ヌクレオチド、少なくとも約28ヌクレオチド、少なくとも約29ヌクレオチド、少なくとも約30ヌクレオチド、少なくとも約35ヌクレオチド、少なくとも約40ヌクレオチド、少なくとも約45ヌクレオチド、少なくとも約50ヌクレオチド、少なくとも約55ヌクレオチド、少なくとも約60ヌクレオチド、少なくとも約65ヌクレオチド、少なくとも約70ヌクレオチド、少なくとも約75ヌクレオチド、少なくとも約80ヌクレオチド、少なくとも約85ヌクレオチド、少なくとも約90ヌクレオチド、少なくとも約95ヌクレオチド、少なくとも約100ヌクレオチド、少なくとも約110ヌクレオチド、少なくとも約120ヌクレオチドまたは120を超えるヌクレオチドを含む)。
【0048】
「ポリヌクレオチド」または「ヌクレオチド配列」または「核酸分子」という用語は、ホスホジエステル結合によって相互に連結される2種以上のデオキシリボヌクレオチドまたはリボヌクレオチドの配列を意味するように、本明細書において広く用いられる。そのようなものとして、それらの用語は、RNAおよびDNA(遺伝子またはその部分、cDNA、合成ポリデオキシリボ核酸配列などであってよく、かつ一本鎖または二本鎖であってよい)、ならびにDNA/RNAハイブリッドを包含する。さらに、本明細書において用いられるそれらの用語は、細胞から単離され得る天然に存在する核酸分子、ならびに例えば、化学合成法によりまたは酵素法により(ポリメラーゼ連鎖反応(PCR)によるなど)調製され得る合成ポリヌクレオチドを包含する。例えば、組成物の異なる成分を区別するために、議論の便宜上異なる用語を用いているにすぎないことを認識すべきである。
【0049】
一般的に、ポリヌクレオチドを含むヌクレオチドは、天然に存在するデオキシリボヌクレオチド(2'−デオキシリボースと連結されたアデニン、シトシン、グアニンまたはチミンなど)またはリボヌクレオチド(リボースと連結されたアデニン、シトシン、グアニンまたはウラシルなど)である。しかしながら、使用に応じて、ポリヌクレオチドは、天然に存在しない合成ヌクレオチドまたは修飾された天然に存在するヌクレオチドを含むヌクレオチド類似体も含み得る。ヌクレオチド類似体は、当技術分野で周知であり、市販されており、そのようなヌクレオチド類似体を含有するポリヌクレオチドと同様である。ポリヌクレオチドのヌクレオチドを連結している共有結合は、一般的にはホスホジエステル結合である。しかしながら、ポリヌクレオチドを使用する目的に応じて、その共有結合は多数の他の結合のいずれかであってもよく、チオジエステル結合、ホスホロチオエート結合、ペプチド様結合または合成ポリヌクレオチドを生産するためのヌクレオチドの連結に有用な、当業者に公知の任意の他の結合が含まれる。
【0050】
天然に存在するヌクレオチドおよびホスホジエステル結合を含むポリヌクレオチドまたはオリゴヌクレオチドは、化学合成することができ、または鋳型として適当なポリヌクレオチドを用い、組換えDNA法を用いて生産することができる。それに対し、ヌクレオチド類似体またはホスホジエステル結合以外の共有結合を含むポリヌクレオチドは、一般的に化学合成されるが、T7ポリメラーゼなどの酵素によりある特定の種類のヌクレオチド類似体をポリヌクレオチドに組み込むことができるため、そのような酵素を用いて、適当な鋳型から組換えによりそのようなポリヌクレオチドを生産することができる。
【0051】
様々な実施形態では、アンチセンスオリゴヌクレオチドまたはRNA分子には、修飾を含むオリゴヌクレオチドが含まれる。様々な修飾が当技術分野で公知であり、本発明における使用のために考えられる。例えば、修飾主鎖または非天然ヌクレオシド間結合を含むオリゴヌクレオチドが考えられる。本明細書において用いられるように、修飾主鎖を有するオリゴヌクレオチドとは、主鎖中にリン原子を有するものおよび主鎖中にリン原子がないものを包含する。本明細書の目的において、当技術分野において言及されることがあるように、ヌクレオシド間主鎖中にリン原子がない修飾オリゴヌクレオチドもまた、オリゴヌクレオシドであると考えることができる。
【0052】
様々な態様では、修飾オリゴヌクレオチド主鎖としては、例えば、ホスホロチオエート、キラルホスホロチオエート、ホスホロジチオエート、ホスホトリエステル、アミノアルキルホスホトリエステル、メチルホスホネートおよび他のアルキルホスホネート(3'−アルキレンホスホネート、5'−アルキレンホスホネートおよびキラルホスホネートを含む)、ホスフィネート、ホスホルアミダート(3'−アミノホスホルアミダートおよびアミノアルキルホスホルアミダートを含む)、チオノホスホルアミダート、チオノアルキルホスホネート、チオノアルキルホスホトリエステル、通常の3'−5'結合を有するセレノホスフェートおよびボラノホスフェート、これらの2'−5'結合した類似体、および1以上のヌクレオチド間結合が3'−3'結合、5'−5'結合または2'−2'結合である逆の極性を有するものが挙げられる。逆の極性を有するある特定のオリゴヌクレオチドは、最も3'側のヌクレオチド間結合に単一の3'−3'結合、すなわち、脱塩基であってよい(核酸塩基が欠けているかまたはその代わりにヒドロキシル基を有する)単一の逆のヌクレオシド残基を含む。様々な塩、混合塩および遊離酸形も含まれる。
【0053】
様々な態様では、内部にリン原子を含まない修飾オリゴヌクレオチド主鎖は、短鎖アルキルまたはシクロアルキルヌクレオシド間結合、混合ヘテロ原子およびアルキルまたはシクロアルキルヌクレオシド間結合、または1以上の短鎖ヘテロ原子ヌクレオシド間結合または複素環式ヌクレオシド間結合によって形成される主鎖を有する。これらには、モルホリノ結合(ヌクレオシドの糖部分から一部形成される);シロキサン主鎖;スルフィド、スルホキシドおよびスルホン主鎖;ホルムアセチルおよびチオホルムアセチル主鎖;メチレンホルムアセチルおよびメチレンチオホルムアセチル主鎖;リボアセチル主鎖;アルケン含有主鎖;スルファメート主鎖;メチレンイミノおよびメチレンヒドラジノ主鎖;スルホネートおよびスルホンアミド主鎖;アミド主鎖;および混合N、O、SおよびCH2成分を有する他のものを有するものが含まれる。
【0054】
様々な態様では、オリゴヌクレオチドミメティック、糖およびヌクレオシド間結合の両方、すなわち、ヌクレオチド単位の主鎖は、新規な基に置換されている。塩基単位は、適当な核酸標的化合物とのハイブリダイゼーションのために維持される。そのような1つのオリゴマー化合物、優れたハイブリダイゼーション特性を示すことが示されたオリゴヌクレオチドミメティックは、ペプチド核酸(PNA)と呼ばれる。PNA化合物では、オリゴヌクレオチドの糖−主鎖は、アミド含有主鎖、特に、アミノエチルグリシン主鎖に置換されている。核酸塩基は、保持され、主鎖のアミド部分のアザ窒素原子に直接または間接的に結合している。様々な態様では、オリゴヌクレオチドはホスホロチオエート主鎖およびヘテロ原子主鎖を含むオリゴヌクレオシドを含み得る。修飾オリゴヌクレオチドはまた、1以上の置換された糖部分も含み得る。いくつかの実施形態では、オリゴヌクレオチドは、2'位に次のものの1つを含む:OH;F;O−、S−、もしくはN−アルキル;O−、S−、もしくはN−アルケニル;O−、S−もしくはN−アルキニル;またはO−アルキル−O−アルキル(ここで、該アルキル、アルケニルおよびアルキニルは、置換されているかまたは非置換のC1〜C10アルキルまたはC2〜C10アルケニルおよびアルキニルであってよい)。特に好ましいのは、O[(CH2)nO]mCH3、O(CH2)nOCH3、O(CH2)nNH2、O(CH2)nCH3、O(CH2)nONH2およびO(CH2)nON[(CH2)nCH3]]2(ここで、nおよびmは1〜約10である)である。他の好ましいオリゴヌクレオチドは、2'位に次のものの1つを含む:C1〜C10低級アルキル、置換低級アルキル、アルケニル、アルキニル、アルカリル、アラルキル、O−アルカリルもしくはO−アラルキル、SH、SCH3、OCN、Cl、Br、CN、CF3、OCF3、SOCH3、SO2CH3、ONO2、NO2、N3、NH2、ヘテロシクロアルキル、ヘテロシクロアルカリル、アミノアルキルアミノ、ポリアルキルアミノ、置換シリル、RNA切断基、レポーター基、インターカレーター、オリゴヌクレオチドの薬物動態特性を向上させるための基、またはオリゴヌクレオチドの薬力学的特性を向上させるための基、および類似した特性を有する他の置換基。別の修飾としては、2'−メトキシエトキシ(2'OCH2CH2OCH3、2'−O−(2−メトキシエチル)または2'−MOEとしても知られる)が挙げられる。
【0055】
関連態様では、本発明は、標的ポリヌクレオチドに対する親和性および特異性が増強されたアンチセンス核酸を生成するためのロックド核酸(LNA)の使用を含む。LNAは、2'−ヒドロキシル基が糖環の3'または4'炭素原子と結合し、それによって二環式糖部分を形成する核酸である。その結合は、2'酸素原子と4'炭素原子を架橋するメチレン (methelyne)(−CH2−)n基(ここで、nは1または2である)であることが好ましい。
【0056】
他の修飾としては、2'−メトキシ(2'−O−CH3)、2'−アミノプロポキシ(2'−OCH2CH2CH2NH2)、2'−アリル(2'−CH−CH−CH2)、2'−O−アリル(2'−O−CH2−CH−CH2)、2'−フルオロ(2'−F)、2'−アミノ、2'−チオ、2'−Oメチル、2'−メトキシメチル、2'−プロピルなどが挙げられる。2'−修飾は、アラビノ(上)位またはリボ(下)位に存在してよい。好ましい2'−アラビノ修飾は2'−Fである。また、類似した修飾も、オリゴヌクレオチドの他の位置で、特に、3'末端ヌクレオチド上または2'−5'結合オリゴヌクレオチド中の糖の3'位および5'末端ヌクレオチドの5'位で行ってよい。オリゴヌクレオチドはまた、ペントフラノシル糖の代わりにシクロブチル部分などの糖ミメティックも有していてよい。
【0057】
オリゴヌクレオチドはまた、核酸塩基の修飾または置換も含み得る。本明細書において用いられるように、「非修飾」または「天然」核酸塩基としては、プリン塩基であるアデニン(A)およびグアニン(G)、ならびにピリミジン塩基であるチミン(T)、シトシン(C)およびウラシル(U)が挙げられる。修飾された核酸塩基としては、他の合成および天然核酸塩基、例えば、5−メチルシトシン、5−ヒドロキシメチルシトシン、キサンチン、ヒポキサンチン、2−アミノアデニン、アデニンおよびグアニンの6−メチル誘導体および他のアルキル誘導体、アデニンおよびグアニンの2−プロピル誘導体および他のアルキル誘導体、2−チオウラシル、2−チオチミンおよび2−チオシトシン、5−ハロウラシルおよび5−ハロシトシン、5−プロピニルウラシルおよび5−プロピニルシトシン、ならびにピリミジン塩基の他のアルキニル誘導体、6−アゾウラシル、6−アゾシトシンおよび6−アゾチミン、5−ウラシル(シュードウラシル)、4−チオウラシル、8−ハロ、8−アミノ、8−チオール、8−チオアルキル、8−ヒドロキシルおよび他の8−置換のアデニンおよびグアニン、5−ハロ、特に、5−ブロモ、5−トリフルオロメチルおよび他の5−置換のウラシルおよびシトシン、7−メチルグアニンおよび7−メチルアデニン、2−F−アデニン、2−アミノ−アデニン、8−アザグアニンおよび8−アザアデニン、7−デアザグアニンおよび7−デアザアデニン、ならびに3−デアザグアニンおよび3−デアザアデニンなどが挙げられる。さらなる修飾核酸塩基としては、三環式ピリミジン、例えば、フェノキサジンシチジン(1H−ピリミド[5,4−b][1,4]ベンゾオキサジン−2(3H)−オン)、フェノチアジンシチジン(1H−ピリミド[5,4−b][1,4]ベンゾチアジン−2(3H)−オン)、置換フェノキサジンシチジン(例えば、9−(2−アミノエトキシ)−H−ピリミド[5,4−b][1,4]ベンゾオキサジン−2(3H)−オン)などのGクランプ、カルバゾールシチジン(2H−ピリミド[4,5−b]インドール−2−オン)、ピリドインドールシチジン(H−ピリミド[3',2':4,5]ピロロ[2,3−d]ピリミジン−2−オン)などが挙げられる。修飾核酸塩基としては、プリン塩基またはピリミジン塩基が他の複素環、例えば、7−デアザ−アデニン、7−デアザグアノシン、2−アミノピリジンおよび2−ピリドンで置換されたものも挙げられ得る。さらなる核酸塩基は当技術分野で公知である。これらの核酸塩基のいくつかは、本明細書に記載したオリゴマー化合物の結合親和性を高めるために特に有用である。これらには、5−置換ピリミジン、6−アザピリミジンおよびN−2、N−6およびO−6置換プリン(2−アミノプロピルアデニンを含む)、5−プロピニルウラシルおよび5−プロピニルシトシンが含まれる。5−メチルシトシン置換は、0.6〜1.2Cにより核酸二本鎖安定性を高めることが示され、現在好ましい塩基置換であり、さらに特には、2'−O−メトキシエチル糖修飾と組み合わせられる場合である。
【0058】
本明細書に記載したアンチセンスオリゴヌクレオチドの別の修飾は、オリゴヌクレオチドの活性、細胞分布または細胞取込みを高めるオリゴヌクレオチドの1以上の部分またはコンジュゲートとの化学的結合を含む。アンチセンスオリゴヌクレオチドは、第一級または第二級ヒドロキシル基などの官能基と共有結合されたコンジュゲート群を含み得る。コンジュゲート群としては、インターカレーター、レポーター分子、ポリアミン、ポリアミド、ポリエチレングリコール、ポリエーテル、オリゴマーの薬力学的特性を増強する群、およびオリゴマーの薬物動態特性を増強する群が挙げられる。典型的なコンジュゲート群としては、コレステロール、脂質、リン脂質、ビオチン、フェナジン、葉酸、フェナントリジン、アントラキノン、アクリジン、フルオレセイン、ローダミン、クマリンおよび色素が挙げられる。薬力学的特性を増強する群とは、本発明の文脈において、オリゴマーの取込みを向上させ、オリゴマーの分解耐性を増強し、および/またはRNAとの配列特異的ハイブリダイゼーションを強化する群を包含する。薬物動態特性を増強する群とは、本発明の文脈において、オリゴマーの取込み、分布、代謝または排出を向上させる群を包含する。コンジュゲート部分としては、限定されるものではないが、コレステロール部分、コール酸、チオエーテル、例えば、ヘキシル−5−トリチルチオール、チオコレステロール、脂肪族鎖、例えば、ドデカンジオールまたはウンデシル残基、リン脂質、例えば、ジヘキサデシル−rac−グリセロールまたはトリエチルアンモニウム 1,2−ジ−O−ヘキサデシル−rac−グリセロ−3−H−ホスホネート、ポリアミンまたはポリエチレングリコール鎖、あるいはアダマンタン酢酸、パルミチル部分、あるいはオクタデシルアミンまたはヘキシルアミノカルボニルオキシコレステロール部分などの脂質部分が挙げられる。
【0059】
いくつかの遺伝子は、多能性と関係があり、初期化因子としての本発明での使用に好適であることが見出された。そのような遺伝子は当技術分野で公知であり、それらの例として、SOXファミリー遺伝子(SOX1、SOX2、SOX3、SOX15、SOX18)、KLFファミリー遺伝子(KLF1、KLF2、KLF4、KLF5)、MYCファミリー遺伝子(C−MYC、L−MYC、N−MYC)、SALL4、OCT4、NANOG、LIN28、STELLA、NOBOXまたはSTATファミリー遺伝子が挙げられる。STATファミリーメンバーには、例えば、STAT1、STAT2、STAT3、STAT4、STAT5(STAT5AおよびSTAT5B)、およびSTAT6が含まれ得る。場合によっては、多能性を誘導するために1種の遺伝子だけを使用することも可能であるが、一般的には、多能性を誘導するために2種以上の遺伝子の発現が必要である。例えば、2種、3種、4種またはそれ以上の遺伝子をポリシストロン性構築物として体細胞ゲノムに同時に組み込んで、そのような遺伝子の同時発現を可能にし得る。例示的態様では、多能性を誘導するために、OCT4、SOX2、KLF4およびC−MYCを含む4種の遺伝子が利用される。本発明での使用に好適な初期化因子として知られるさらなる遺伝子は、米国特許出願第10/997,146号および米国特許出願第12/289,873号において開示されており、これらの特許出願は引用することにより本明細書の一部とされる。
【0060】
これらの遺伝子の全ては、ヒトを含む哺乳類に一般に存在するため、任意の哺乳類由来の相同体も本発明において用いてよい(限定されるものではないが、マウス、ラット、ウシ、ヒツジ、ウマおよびサルを含む哺乳類から得られた遺伝子など)。さらに、野生型遺伝子産物に加えて、いくつかの(例えば、1〜10個、1〜6個、1〜4個、1〜3個および1個または2個)のアミノ酸の置換、挿入および/または欠失を含み野生型遺伝子産物と類似した機能を有する変異遺伝子産物も使用することができる。さらに、因子の組合せは野生型の遺伝子または遺伝子産物の使用に限定されない。例えば、野生型Mycの代わりにMycキメラまたは他のMyc変異体を使用することができる。
【0061】
本発明は、任意の特定の核初期化因子組合せに限定されない。本明細書において論じるように、核初期化因子は、1種以上の遺伝子産物を含んでよい。核初期化因子はまた、本明細書において論じるように、遺伝子産物の組合せも含んでよい。本明細書において開示するように、各核初期化因子を単独でまたは他の核初期化因子と組み合わせて用いてよい。さらに、本発明の核初期化因子は、スクリーニング方法により、例えば、米国特許出願第10/997,146号において論じられているように、同定することができ、この特許出願は引用することにより本明細書の一部とされる。加えて、本発明の核初期化因子は、分化、発生、増殖などに関連する1種以上の因子および他の生理学的活性を有する因子、ならびに核初期化因子として機能し得る他の遺伝子産物を含み得る。
【0062】
核初期化因子は、タンパク質またはペプチドを含んでよい。そのタンパク質は、本明細書において論じるように、遺伝子から生産されてよく、あるいは、そのタンパク質と別のタンパク質、ペプチドなどとの融合遺伝子産物の形であってよい。そのタンパク質またはペプチドは、蛍光タンパク質および/または融合タンパク質であってよい。例えば、緑色蛍光タンパク質(GFP)との融合タンパク質またはヒスチジンタグなどのペプチドとの融合遺伝子産物を用いることもできる。さらに、ウイルスHIVから得られたTATペプチドとの融合タンパク質を調製し使用することによって、細胞膜を通じての核初期化因子の細胞内取込みを促進することができ、それによって、融合タンパク質を培地に加え遺伝子形質導入などの複雑な操作を行わないことから、初期化の誘導だけが可能になる。そのような融合遺伝子産物の調製方法は当業者には周知であるため、当業者ならば、目的に応じて適当な融合遺伝子産物を容易に設計し調製することができる。
【0063】
本明細書において論じるように、iPSCは、体細胞に核初期化因子と、その細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤とを組み合わせて接触させることにより誘導され得る。当業者には理解されるように、体細胞への送達は当技術分野で公知の任意の好適な方法により行われ得る。一態様では、核初期化因子は、核初期化因子をコードする遺伝子を含む組換えベクターを用いて細胞に導入され得る。同様に、マイクロRNAを変更する作用物質は、RNA分子(例えば、マイクロRNA、shRNA、アンチセンスオリゴヌクレオチドなど)をコードするポリヌクレオチドを含む組換えベクターを用いて細胞に導入され得る。同様に、p21の阻害剤は、ペプチド阻害剤またはRNA分子(例えば、マイクロRNA、shRNA、アンチセンスオリゴヌクレオチドなど)をコードするポリヌクレオチドを含む組換えベクターを用いて細胞に導入され得る。よって、その細胞は、組換えベクター中に含まれる遺伝子の産物として発現される核初期化因子を発現することができ、同様に組換えベクター中に含まれるポリヌクレオチドの産物として作用物質またはp21阻害剤を発現し、それによって核初期化因子だけを使用する場合と比べて高い効率(割合)で分化した細胞の初期化を誘導する。
【0064】
本発明の核酸構築物(組換えベクターなど)は、様々な周知の技術(細胞の非ウイルス性トランスフェクションなど)を用いて細胞に導入され得る。例示的態様では、構築物は、ベクターに組み込まれ、細胞に導入されて、その構築物の発現が可能になる。細胞への導入は、当技術分野で公知の任意のウイルス性または非ウイルス性トランスフェクション(限定されるものではないが、エレクトロポレーション、リン酸カルシウム法、ヌクレオフェクション、ソノポレーション、熱ショック、マグネトフェクション、リポソーム法、マイクロインジェクション、微粒子銃法(ナノ粒子)、陽イオン性ポリマー法(DEAE−デキストラン、ポリエチレンイミン、ポリエチレングリコール(PEG)など)または細胞融合など)により行われ得る。他のトランスフェクション方法としては、専売のトランスフェクション試薬(Lipofectamine(商標)、Dojindo Hilymax(商標)、Fugene(商標)、jetPEI(商標)、Effectene(商標)およびDreamFect(商標)など)が挙げられる。
【0065】
他の態様では、体細胞への、核初期化因子と、その細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤とを組み合わせての誘導中の接触は、当技術分野で公知の任意の方法、例えば、細胞膜を介したタンパク質、RNA分子などの直接送達により行われ得る。
【0066】
細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤と組み合わせた核初期化因子の使用では、初期化因子だけを使用する場合と比べて誘導効率は高まる。様々な態様では、誘導効率は、従来の方法と比べて10%、20%、30%、40%、50%、60%、70%、80%、90%、100%、150%、200%、300%、400%または500%までも高まり得る。例えば、誘導効率は、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、15%、20%、25%または50%(例えば、開始時の体細胞総数に対する誘導された細胞の割合)ほどの高さであり得る。
【0067】
誘導プロセス中、体細胞に、その細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤を接触させると同時にまたはその前に、体細胞に核初期化因子を接触させ得る。様々な態様では、体細胞に任意の他の作用物質または阻害剤を接触させる約1日、2日、3日、4日、5日、7日、8日、9日、10日、11日、12日、13日、14日またはそれ以上前に、体細胞に初期化因子を接触させる。例示的態様では、体細胞に任意の他の作用物質または阻害剤を接触させる約1日、2日、3日、4日または5日前に、体細胞に初期化因子を接触させる。
【0068】
初期化細胞の多能性を評価するためにさらなる解析を行ってよい。細胞を異なる増殖特性および胚幹細胞様形態について解析し得る。例えば、特異的細胞種への分化を推進することが知られているある特定の増殖因子を加えることによって細胞をin vitroで分化させ得る。体の数種の細胞種だけを形成可能な初期化細胞は多分化性であり、一方、体のいかなる細胞種をも形成可能な初期化細胞は多能性である。
【0069】
多能性を評価するための初期化体細胞の発現プロファイリングも行ってよい。多能性と関係がある個別遺伝子の発現もまた調査し得る。加えて、胚幹細胞表面マーカーの発現も解析し得る。多能性幹細胞と関係があることが当技術分野で公知の様々な遺伝子の検出および解析としては、限定されるものではないが、OCT4、NANOG、SALL4、SSEA−1、SSEA−3、SSEA−4、TRA−1−60、TRA−1−81、またはそれらの組合せなどの遺伝子の解析が挙げられ得る。iPS細胞はいくつもの多能性細胞マーカーを発現し得、それらには以下が挙げられる:アルカリ性ホスファターゼ(alkaline phosphatase)(AP);ABCG2;ステージ特異的胚抗原−1(stage specific embryonic antigen-1)(SSEA−1);SSEA−3;SSEA−4;TRA−1−60;TRA−1−81;Tra−2−49/6E;ERas/ECAT5、E−カドヘリン(E-cadherin);β−チュブリンIII(β-tubulin III);α−平滑筋アクチン(α-smooth muscle actin)(α−SMA);繊維芽細胞増殖因子4(fibroblast growth factor 4)(FGF4)、Cripto、Dax1;亜鉛フィンガータンパク質296(zinc finger protein 296)(Zfp296);N−アセチルトランスフェラーゼ−1(N-acetyltransferase-1)(Nat1);ES細胞関連転写物1(ES cell associated transcript 1)(ECAT1);ESG1/DPPA5/ECAT2;ECAT3;ECAT6;ECAT7;ECAT8;ECAT9;ECAT10;ECAT15−1;ECAT15−2;Fthll7;Sall4;未分化胚細胞転写因子(undifferentiated embryonic cell transcription factor)(Utf1);Rex1;p53;G3PDH;テロメラーゼ(telomerase)(TERTを含む);サイレントX染色体遺伝子(silent X chromosome genes);Dnmt3a;Dnmt3b;TRIM28;F-box containing protein 15(Fbx15);Nanog/ECAT4;Oct3/4;Sox2;Klf4;c−Myc;Esrrb;TDGF1;GABRB3;Zfp42、FoxD3;GDF3;CYP25A1;developmental pluripotency-associated 2(DPPA2);T-cell lymphoma breakpoint 1(Tcl1);DPPA3/Stella;DPPA4;ならびに他の一般的な多能性マーカー、例えば、細胞を初期化するために誘導中に用いられる任意の遺伝子。iPS細胞はまた、iPS細胞が誘導される分化細胞に特有のマーカーのダウンレギュレーションによっても特徴付けることができる。
【0070】
本発明はさらに、本明細書に記載した方法を用いて生産されたiPS細胞、ならびにそのような細胞の集団を提供する。様々な細胞種へ分化可能な、本発明の初期化細胞には、様々な用途および治療的使用がある。幹細胞の基本特性である、無限に自己複製する能力および体のあらゆる細胞種に分化する能力は治療的使用に理想的となる。
【0071】
よって、一態様では、本発明は、本明細書に記載した方法を用いて生成された誘導多能性幹細胞を用いて対象における障害および/または症状の治療または予防方法をさらに提供する。その方法は、対象から体細胞を得ることおよび本明細書に記載した方法を用いてその体細胞を誘導多能性幹(iPS)細胞に初期化することを含む。その細胞は、次いで、症状の治療に好適な所望の細胞種にその細胞を分化させるのに好適な条件下で培養される。分化した細胞は、次いで、症状を治療または予防するために対象に導入され得る。
【0072】
一態様では、本明細書に記載した方法を用いて生産されたiPS細胞、ならびにそのような細胞の集団は、それらの細胞を、それらの細胞中でマイクロRNAレベルまたは活性を変更する作用物質により処理するかまたはそのような作用物質と接触させることによりin vitroで分化させ得る。マイクロRNAはiPSC誘導において重要なレギュレーターとして確認されているため、そのようなiPSCの分化を指示するために個別マイクロRNAまたはマイクロRNA集団の操作を用い得ると思われる。そのような処理を、増殖因子または当技術分野で一般的に公知の他の作用物質および刺激と組み合わせて用いて、特異的細胞種への分化を推進し得る。
【0073】
本発明の1つの利点は、移植に好適な同系(isogenic or synegenic)ヒト細胞について本質的に無制限の供給を提供することである。iPS細胞は、患者に特異的に合わせられ、免疫拒絶が回避される。従って、現行の移植方法に関する重大な問題(宿主対移植片または移植片対宿主の拒絶によって起こり得る移植組織の拒絶など)がうまく回避される。健常なヒトから得られた体細胞由来のiPS細胞またはiPS細胞から調製された完全に分化した細胞のいくつかの種類を、iPS細胞バンクに細胞のライブラリーとして保存することができ、幹細胞療法を受ける患者による拒絶が起こらない体細胞、組織または器官の調製に、そのライブラリー中の1種類以上のiPS細胞を用いることができる。
【0074】
本発明のiPS細胞は、当技術分野で公知の方法により様々な障害を治療するための複数の異なる細胞種に分化させ得る。例えば、iPS細胞を誘導して、造血幹細胞(hematopoetic stem cells)、筋肉細胞、心筋細胞、肝臓細胞、軟骨細胞、上皮細胞、尿路細胞、神経細胞などに分化させ得る。分化した細胞は、症状を予防または治療するために、次いで、患者の体に移植して戻され得る。よって、本発明の方法は、特定の細胞種/組織の増加もしくは置換または細胞の脱分化が望ましい、心筋梗塞、鬱血性心不全、卒中、虚血、末梢血管疾患、アルコール性肝疾患、硬変、パーキンソン病、アルツハイマー病、糖尿病、癌、関節炎、創傷治癒、免疫不全、再生不良性貧血、貧血、ハンチントン病、筋萎縮性側索硬化症(ALS)、リソソーム蓄積症、多発性硬化症、脊髄損傷、遺伝性障害および類似疾患を有する対象を治療するために使用し得る。
【0075】
様々な実施形態では、その方法によって、組織または器官の細胞数が、対応する未処理の対照組織または器官と比べて少なくとも約5%、10%、25%、50%、75%またはそれ以上増加する。さらに別の実施形態では、その方法によって、組織または器官の生物活性が、対応する未処理の対照組織または器官と比べて少なくとも約5%、10%、25%、50%、75%またはそれ以上増加する。さらに別の実施形態では、その方法によって、組織または器官における血管形成が、対応する未処理の対照組織または器官と比べて少なくとも約5%、10%、25%、50%、75%またはそれ以上増加する。さらに別の実施形態では、前記細胞は対象に対して、細胞数の増加が望まれる部位に直接投与される。
【0076】
本発明はさらに、本明細書に記載した方法により得られた様々な細胞を用いることにより作用物質、化合物、医薬、毒物などの生理学的機能または毒性を評価するための方法を提供する。
【0077】
4種の転写因子、Oct4、Sox2、Klf4およびcMycの異所発現により体細胞をES様状態に初期化して、誘導多能性幹細胞(iPSC)を作出することができる。本発明によれば、細胞のマイクロRNAはiPSC生成を調節することとなる。重要なマイクロRNA経路タンパク質のノックダウンにより初期化効率の著しい低下がもたらされ得る。3種のマイクロRNAクラスター、miR−17〜92、106b〜25および106a〜363は、初期化の初期段階中に高度に誘導されることが示されている。非常に類似したシード領域を有するいくつかのマイクロRNA(miR−93およびmiR−106bを含む)は、iPSC誘導を大幅に増強し、これらのマイクロRNAの阻害により初期化効率が著しく低下した。さらに、miR−iPSCクローンは完全初期化状態に到達し得る。本発明によれば、Tgfbr2およびp21は、これらのマイクロRNAにより直接ターゲッティングされることとなり、両遺伝子のsiRNAノックダウンによりiPSC誘導が実際に増強されることとなる。また、本発明によれば、miR−93およびそのファミリーメンバーはTGF−β受容体IIを直接ターゲッティングして、iPSC初期化を促進することとなる。本発明によれば、マイクロRNAは初期化プロセスにおいて作用することとなり、細胞中のマイクロRNAレベルをモジュレートすることによりiPSC誘導効率は大幅に高められ得ることとなる。
【0078】
誘導多能性幹細胞(iPSC)はオーダーメイド再生医療に大いに有望であるが、初期化の分子基盤は大部分が不明である。細胞同一性を維持する障壁を克服することが分化した細胞の初期化における重要な段階である。マイクロRNA(miRNA)は標的遺伝子を組織特異的にモジュレートするため、本発明によれば、異なるマウス胚性繊維芽細胞(MEF)濃縮miRNAは、初期化障壁として働くタンパク質を転写後にモジュレートすることとなる。これらのmiRNAの阻害は、細胞シグナル伝達に影響を及ぼして、それらの障壁を低くするはずである。本発明によれば、miR−21およびmiR−29aの枯渇によりMEFにおける初期化効率が高まることとなる。本発明によれば、p53およびERK1/2の経路はmiR−21およびmiR−29aによって調節され、初期化において作用することとなる。さらに、本発明によれば、c−Mycは、MEFで濃縮されたmiRNA(miR−21およびmiR−29aなど)を抑制することにより初期化をある程度まで促進することとなる。本発明は、iPSC初期化に関与する複数のシグナル伝達ネットワークの調節におけるmiRNA機能を提供する。
【0079】
C−Mycは、4種の初期化因子(4F:Oct3/4、Sox2、Klf4およびc−Myc)のうちの1つであり、細胞増殖および腫瘍発生において重要な役割を果たす。C−Mycは、サイクリン依存性キナーゼ(CDK)阻害剤p21Cip1の抑制を通じて細胞性塞栓およびアポトーシスの重要なレギュレーターである。Miz−1機能を無効にし、p15INK4bを抑制することにより、c−Mycは、初代細胞の不死化において重要な役割を果たす。c−Mycの多くの転写機能はMaxまたはMiz−1との協同作用を必要とする。原癌遺伝子としてのc−Mycは初期化効率を大幅に高めるが、初期化では重要でない。従って、初期化中のc−Mycの下流の分子経路を定義することにより初期化効率を損なうことなくiPS細胞の治療用途を増すことができる。
【0080】
Oct4−GFPマウス胚性繊維芽細胞(MEF)は、D13.5に、pou5f1の停止コドンの下流にIRES−EGFP融合カセットを有するマウス(Jackson lab、ストック番号008214)から得られる。これらのMEFは、10%FBS(Invitrogen)とグルタミンおよびNEAAとを含有するDMEM(Invitrogen、11995−065)で培養される。iPSC誘導では、0〜4代継代のMEFだけを用いる。
【0081】
C−Mycは、マイクロRNA(miRNA)発現を調節することによってES細胞再生を維持するように働く側面があると報告されている。マイクロRNAは、22−ヌクレオチドの非コード低分子RNAであり、RNA誘導サイレンシング複合体(RISC)に装入され、全体的な遺伝子サイレンシング機能を発揮する。miR−141、miR−200およびmiR−429の発現はES細胞においてc−Mycにより誘導され、分化に拮抗する。C−Mycはまた、miR−17〜92マイクロRNAクラスターをアップレギュレートすることによりまたは既知腫瘍抑制遺伝子(let−7ファミリー、miR−15a/16−1、miR−29ファミリーおよびmiR−34aなど)を抑制することにより腫瘍形成も促進する。
【0082】
体細胞同一性を固定する、Ink4−Arf、p53およびp21などの因子により媒介される障壁を克服することは、初期化における律速段階である。miRNAは標的遺伝子を組織特異的にモジュレートするため、本発明によれば、異なるMEF miRNAは、初期化レギュレーターとして働くタンパク質を転写後にモジュレートすることとなる。これらのmiRNAの阻害により、細胞シグナル伝達に影響を及ぼして、それらの障壁を低くすることができる。
【0083】
本発明によれば、MEFにおいて豊富なmiRNA(miR−21およびmiR−29a)の枯渇により初期化効率が約2.1倍〜2.8倍高まることとなる。また、本発明によれば、c−MycはmiRNA(miR−21およびmiR−29a)を抑制して、MEFの初期化を促進することとなる。さらに、本発明によれば、miR−21およびmiR−29aは、初期化プロセス中にp53レベルおよびERK1/2リン酸化を間接的にダウンレギュレートすることによりp53およびERK1/2の経路を調節することとなる。
【0084】
次の実施例を示して、本発明の実施形態をさらに説明するが、それらの実施例は本発明の範囲を限定するものではない。それらの実施例は用いられる可能性のある典型的なものであるが、当業者に公知の他の手順、方法論または技術を代わりに用いてもよい。
【実施例】
【0085】
実施例1
細胞培養物、ベクターおよびウイルス形質導入
Oct4−GFPマウス胚性繊維芽細胞(MEF)は、D13.5に、pou5f1の停止コドンの下流にIRES−EGFP融合カセットを有するマウス(Jackson lab、ストック番号008214)から得られる。これらのMEFは、10%FBS(Invitrogen)とグルタミンおよびNEAAとを含有するDMEM(Invitrogen、11995−065)で培養される。iPSC誘導では、0〜4代継代のMEFだけを用いる。
【0086】
プラスミドpMXs−Oct4、Sox2、Klf4およびcMycはAddgeneから購入する。プラスミドpMX−HA−p21は、N末端タグ付きp21をpMXベクターのEcoRI部位に挿入することにより生成する。pLKO−shRNAのクローンはOpen-Biosystemsから購入する。
【0087】
レトロウイルスを生成するために、PLAT−E細胞を10cmプレートに播種し、翌日Lipofectamine(商標)(Invitrogen、18324−012)およびPLUS(商標)(Invitrogen、11514−015)を用いて各因子9μgをトランスフェクトする。ウイルスを採取し、2日後に合わせる。iPSC誘導では、MEFを12ウェルプレートに播種し、翌日4種の因子ウイルスを4μg/mlポリブレンとともに形質導入する。形質導入の1日後、培地を新鮮なMEF培地に換え、3日後にLIF(Millipore、ESG1107)を補給したmES培養培地に替える。形質導入後14日目からGFP+コロニーを採取し、拡大に成功したクローンを、15%FBS(Hyclone)とLIF、チオグリセロール、グルタミンおよびNEAAとを含有するDMEMで培養した。照射CF1 MEFをmESおよび得られたiPSCクローンの培養用のフィーダー細胞層として用いる。
【0088】
shRNAレンチウイルスを生成するために、shRNAレンチウイルスベクターを293FT細胞にpPACKH1パッケージングシステム(SBI、カタログ番号LV500A−1)とともに共トランスフェクトする。トランスフェクション後2日目にレンチウイルスを採取し、室温で4,000rpmで5分間遠心分離する。ウイルスを生産するために、10cm組織培養プレートで4μgのpLKOまたはpGIPZベクターおよび10μgのパッケージングミックスを293FT細胞(Invitrogen)にトランスフェクトした。トランスフェクションの2日後に、ウイルスを含有する上清を採取し、4μg/μlポリブレンとのさらなる形質導入に用いた。ShRNAウイルスを4種の因子ウイルスとともに体積比1:1:1:1:1で加える。
【0089】
マイクロRNA、siRNAおよびMEFのトランスフェクションは次のとおり行う:マイクロRNAミミックおよび阻害剤、siRNAはDharmaconから購入する。MEFをトランスフェクトするために、マイクロRNAミミックをOpti−MEM(Invitrogen、11058−021)で所望の終濃度に希釈する。次いで、その混合物にLipofectamine(商標)2000(Invitrogen、11668−019)を2μl/ウェルで加え、室温で20分インキュベートする。12ウェルトランスフェクションでは、80μlのmiR混合物を、320μlのOpti−MEMが入った各ウェルに加える。3時間後に、各ウェルに0.8mlのウイルス混合物(iPSCの場合)または新鮮培地を加え、翌日その培地を新鮮なMEF培地に換える。
【0090】
ウエスタンブロッティングは次のとおり行う:全細胞溶解物を、MPERバッファー(PIERCE、78503)を氷上で20分間用いて調製し、13,000rpmで10分間遠心分離することにより清澄にする。10%SDS−PAGEゲル中に同量の溶解物をローディングし、セミドライ式システム(Bio-Rad)を用いてタンパク質をPVDF膜(Bio-Rad、1620177)に転写する。次いで、PVDF膜を、TBST中5%ミルクにより室温で少なくとも1時間または4℃で一晩ブロッキングする。
【0091】
使用する抗体には、抗p21(BD、556430)、抗mNanog(R&D、AF2729)、抗h/mSSEA1(R&D、MAB2156)、抗HA(Roche、11867423001)、抗mAgo2(Wako、01422023)、抗Dicer(Abcam、ab13502)、抗Drosha(Abcam、ab12286)、抗アクチン(Thermo、MS1295P0)、抗AFP(Abcam、ab7751)、抗βチュブリンIII(R&D systems、MAB1368)、抗αアクチニン(Sigma、A7811)を含む。
【0092】
mRNAおよびマイクロRNAのRTおよび定量PCRは次のとおり行う:トータルRNAをTrizol法(Invitrogen)により抽出する。抽出後、1μgのトータルRNAを、スーパースクリプトII(商標)(Invitrogen)によるRTに用いる。定量PCRは、Roche LightCycler480 II(商標)およびAbgeneのSybrgreen混合物(Ab−4166)を用いることにより行う。マウスAgo2、Dicer、Drosha、Graphおよびp21のプライマーを下の表1に記載する。他のプライマーは、Takahashi, K. and S. Yamanaka (2006) Cell 126(4): 663-76に記載されている。
【0093】
マイクロRNAの定量解析では、トータルRNAを、上に記載した方法を用いて抽出する。抽出後、1.5〜3μgのトータルRNAを、QuantiMir(商標)キット(SBI、RA420A−1)を用い製造業者のプロトコールに従うマイクロRNA逆転写に用いる。次いで、RT産物を、正方向プライマーとしての成熟マイクロRNA配列およびキットで提供されるユニバーサルプライマーを用いる定量PCRに用いる。
【0094】
免疫染色は次のとおり行う:細胞をPBSで2回洗浄し、4%パラホルムアルデヒドにより室温で20分間固定する。固定した細胞を0.1%Triton X−100により5分間透過処理する。次いで、それらの細胞を、0.1%Triton X−100を含有するPBS中5%BSAにより室温で1時間ブロッキングする。一次抗体を、製造業者の提示に従って、0.1%Triton X−100を含有する2.5%BSA PBSにより1:100〜1:400に希釈する。次いで、一次抗体で細胞を1時間染色し、その後、PBSで3回洗浄する。二次抗体を1:400希釈し、室温で45分間細胞を染色する。
【0095】
胚様体(EB)形成および分化のアッセイは次のとおり行う:iPS細胞をトリプシン処理して単一の細胞懸濁液にし、懸滴法を用いて胚体を生成する。各液滴には、20μlのEB分化培地中4000個のiPS細胞を用いる。EBを3日間懸滴培養した後、ゼラチンコーティングプレートに再播種する。再播種後、収縮を繰り返す領域がはっきりと確認される14日目まで細胞をさらに培養する。
【0096】
(表1)qPCR解析用プライマー

【0097】
プロモーターメチル化解析は次のとおり行う:NanogおよびPou5f1プロモーターのCpGメチル化を、これまでに記載されている同じ手法に従って解析する(Takahashi, K. and S. Yamanaka (2006) Cell 126(4): 663-76)。簡潔には、得られたクローンのゲノムDNAを、Qiagen(商標)キットを用いて抽出する。次いで、1μgのDNAを、製造業者のプロトコールに従うゲノム修飾解析(EZ DNA Methylation−Direct kit、Zymo Research、D5020)に用いる。修飾後、選択した領域のPCRを行い、それらの産物をpCR2.1−TOPO(商標)(Invitrogen)にクローニングする。各遺伝子について10のクローンを配列決定する。
【0098】
実施例2
奇形腫形成、キメラ作製およびマイクロアレイ解析
奇形腫形成およびキメラ作製は次のとおり行う:奇形腫を生成するために、iPS細胞をトリプシン処理し、濃度1×107細胞/mlで再懸濁する。無胸腺ヌードマウスにまずアベルチンで麻酔をかけ、次いで、およそ150μlの細胞懸濁液を各マウスに注射する。3〜4週間、週1回マウスを腫瘍について調べる。腫瘍を採取し、パラフィン包理およびH&E染色の前に亜鉛ホルマリン溶液により室温で24時間固定する。得られたiPSCクローンがキメラに寄与する能力を試験するために、iPS細胞をC57BL/6J−Tyr(C−2J)/J(アルビノ)胚盤胞に注射する。一般的に、各胚盤胞は12〜18個のiPS細胞を受ける。胚移植にはICRレシピエント雌を用いる。ドナーiPS細胞はアグーチまたは黒色のいずれかである。
【0099】
mRNA マイクロアレイ解析を次のとおり行う:miR−93およびsiControlをMEFにトランスフェクトし、トランスフェクションの48時間後にトータルRNAを採取する。mRNAマイクロアレイをサンフォード・バーナム研究所(Sanford-Burnham institute)内のマイクロアレイ施設により実施する。潜在的機能標的(変化倍率>2、p<0.05)および全標的(変化倍率>25%、p<0.05)の両方についての遺伝子一覧表を、ボルカノマップ(volcano maps)でフィルタリングすることにより作成する。次いで、遺伝子一覧表を、GeneGoソフトウェアを用いその会社のガイドラインに従うオントロジー解析に用いる。
【0100】
デュアルルシフェラーゼアッセイは次のとおり行う:p21およびTgfbr2両方の3'UTRを、pGL3対照ベクターのXbaI部位にクローニングする。12ウェルプレートの各ウェルに、200ngの得られたベクターおよび50ngのpRL−TK(ウミシイタケルシフェラーゼ)を、トランスフェクションの1日前に播種した1×105個のHela細胞にトランスフェクトする。各処理に50nMのマイクロRNAを用い、トランスフェクション後2日目に細胞溶解物を採取する。次いで、20μlの溶解物を、製造業者のプロトコール(Dual−Luciferase(登録商標)Reporter Assay System Promega、E1910)に従うデュアルルシフェラーゼアッセイに用いる。
【0101】
細胞増殖アッセイは次のとおり行う:3000個のMEFを96ウェルプレートの各ウェルに播種し、4FウイルスおよびshRNAレンチウイルスを形質導入する(またはマイクロRNA阻害剤でトランスフェクトする)。形質導入/トランスフェクション後1日目から、2日おきに、細胞をCelltiter 96 Aqueous one Solution(Promega、G3580)を含有するmES培地とともに組織培養インキュベーター中で1時間インキュベートする。次いで、490nmでの吸光度を各ウェルについてプレートリーダーを使用して測定し、収集データを用いて相対的増殖曲線を作成し、形質導入/トランスフェクション後1日目のシグナルを標準として用いる。
【0102】
実施例3
転写後調節経路は体細胞の初期化に関与する
転写後調節経路は、MEFのiPS細胞への初期化に関与することが確認された。iPSC誘導中の転写後遺伝子調節の役割を調査するために、マウスDicer、DroshaおよびAgo2をターゲッティングするレンチウイルスshRNAベクターを、初代Oct4−GFP MEFにおける安定なノックダウンに用いる。これらのshRNA構築物のノックダウン効率をウエスタンおよびRT−qPCRの両方により検証する(図1a、図1bおよび図1c)。各shRNAではおよそ70%〜80%のmRNAレベルノックダウンが常に観察され、タンパク質レベルの著しい低下も観察される。
【0103】
次いで、shRNAを個別に用いて、MEFを4種の因子OSKM(Oct4、Sox2、Klf4およびcMyc)を発現するウイルスとともに体積比1:1:1:1:1で形質導入する(tranduce)。14日後に、コロニーを固定し、アルカリ性ホスファターゼ(AP)活性(これは広く用いられているES細胞マーカーである)について染色する。各処理についてAP+コロニーを定量し、重要なRNAi機構タンパク質Dicer、DroshaおよびAgo2のノックダウンによりAP+コロニーがpLKOおよびpGIPZ対照と比べて劇的に減少する。OSK(3種の因子 3F)形質導入を用いることによっても類似した結果が観察される。
【0104】
GFP+およびAP+コロニーの両方の定量では、Ago2のノックダウンは初期化効率を劇的に低下させるが、一方、形質導入繊維芽細胞の増殖には影響を及ぼさないことが確認された。(図1d、図1eおよび図1f)。Ago2ノックダウンにより初期化効率が低下するにもかかわらず、shAgo2でのいくつかのGFP+コロニーは感染MEFであり、さらなる特性評価では、これらのコロニーはshRNA組込みについて陽性でありそこではshRNAが活発に発現されることが判明した(図13aおよび図13b)。これらの細胞はまた、試験したES特異的マーカー全てを発現するが、内因性Oct4遺伝子座で示した(図13c)。これらのデータは、転写後調節、とりわけ、マイクロRNAが初期化プロセスにおいて重要な役割を果たすことを強く示唆している。
【0105】
実施例4
マイクロRNAクラスターは体細胞の初期化中に誘導される
マイクロRNA miR−17、25、106aおよび302bクラスターは初期化の初期段階中に誘導されることが判明している。4種の転写因子はiPSC誘導中に多くの遺伝子発現変化を誘導するため、これらの因子によっていくつかのES特異的マイクロRNAが誘導され、そして、これらの因子がMEFの初期化の成功を手助けし得ると推測される。iPSC誘導を増強するES特異的マイクロRNAに関する最新の刊行物でも、その仮説が裏付けられているが、報告されているマイクロRNAは初期化の極めて終わりに近い段階まで発現が見出されなかった。公表されている結果を解析することにより、解析には、マウスES細胞において高度に発現されると判明している9種のマイクロRNAクラスターを選択し、それらを表2に示す。
【0106】
各クラスターからの2種の代表的なマイクロRNAを、miR qPCRに基づく方法を用いて評価して、異なる初期化段階(OSKM因子の形質導入後0日目、4日目、8日目および12日目を含む)における発現変化を定量する。多くのES特異的マイクロRNA(miR−290クラスターおよびmiR−293クラスターなど)は、8日目まで誘導されず(図14)、この段階においてGFP+コロニーはすでに検出できる。いくつかの他のマイクロRNAクラスター(miR−17〜92、25〜106b、106a〜363および302b〜367を含む)は、4種の因子の形質導入後4日目までに様々な程度に発現される(図2a)。これらの4種のマイクロRNAクラスターの間では、MEFにおけるmiR−302b〜367のレベルが最も低い。初期化4日目に高度に誘導される3種のクラスターの間では、一部に非常に類似したシード領域の共有があり(図2b)、それらが初期化において作用し、類似した遺伝子セットをターゲッティングすることができることを示唆している。
【0107】
(表2)iPSC実験に用いたマイクロRNAの一覧表
【0108】
次いで、4種の因子のどの因子がこれらのマイクロRNAの誘導に関与しているかを判定するために解析を行う。同じ用量の4種の因子の種々の組合せをMEFに形質導入することにより、miR qPCR解析のために感染後4日目にトータルRNAを採取する(図2c)。この解析により、cMyc単独でmiR−17〜92、miR−25〜106bおよびmiR−106a〜363クラスターの発現を誘導することができることが確認される。しかしながら、全ての場合において、4種の初期化因子全ての組合せがマイクロRNAクラスターの最も豊富な発現を誘導し、その強い発現は最大の初期化効率と相関している(図2c)。
【0109】
これらの結果により、3種のマイクロRNAクラスター(miR−17〜92、25〜106b、106a〜363を含む)は初期化の初期段階中に誘導されること、さらに、これらのマイクロRNAの発現は4種の因子組によって最も高度に誘導されるが、単一因子でも程度は劣るもののそれらの発現を誘導することができることが確認された。
【0110】
実施例5
マイクロRNAはiPSC誘導を増強する
マイクロRNA miR−93およびmiR−106bは、マウスiPSC誘導を増強することが判明している。確認されている4種のマイクロRNAクラスターは、類似したシード領域を有するいくつかのマイクロRNAを含むため、miR−106b〜25クラスターが3種のマイクロRNA(すなわち、miR−25、miR−93およびmiR−106b)を含むことからこのクラスターをさらに解析する。miR−93およびmiR−106bは同一シード領域を有し、両方とも4種の初期化因子によって高度に誘導される(図2a)。これらのマイクロRNAが初期化細胞で機能しているならば、そのプロセス中にこれらのマイクロRNAを導入することによりiPSC誘導の効率向上が期待されることとなる。
【0111】
これらの誘導されたマイクロRNAの機能試験には、マイクロRNAミミックをMEFに直接トランスフェクトする戦略を用いる(図3a)。マイクロRNAを4種の因子(またはOSK)ウイルスとともに2回、0日目および5日目に導入し、内因性Oct4プロモーターの制御下でGFP発現するレポーターMEFを用いた。例えば、0日目および5日目に、マイクロRNAミミックを、4種の因子(4F、OSKM)全てまたは単独のOct4、Sox2およびKlf4(OSK)のいずれかを発現するベクターとともに、Oct−4−GFPを保有しているMEFに直接トランスフェクトし、GFP発現に基づいて初期化をアッセイする。これらの細胞がiPSCへの初期化に成功したら、それらはGFP陽性(+)となる。11日目頃にGFP+コロニーを定量して、初期化効率を評価する(図3b;表3)。実際には、miR−93およびmiR−106bミミックのトランスフェクションによって4F形質導入およびOSK形質導入の両方でGFP+コロニーの約4〜6倍増加となり(図3c)、iPSC誘導中に誘導されるこれらのマイクロRNAはMEF初期化を促進することが確認された。
【0112】
(表3)iPSC誘導に関するmiRによるGFP+コロニー数
* 12ウェルプレート(ゼラチンコーティング処理)に4x104個MEF/ウェル
【0113】
用量依存実験は、5〜15nMの範囲ほどの低いmiRで初期化効率の向上が見られることを示している(図7)。コロニーをアルカリ性ホスファターゼ基質で染色すると、miRミミックトランスフェクションでのAP+コロニーの著しい増加はないようであり、miR−93およびmiR−106bはiPSCコロニーの成熟プロセスを促進することができることが示唆される。このことは、OSK系を用いて観察される現象(miRミミックトランスフェクト細胞ではOSK形質導入後15日目に多くのGFP+コロニーが現われるが、一方、対照ウェルではこの段階において成熟したiPSCコロニーを全く示さなかった)によっても裏付けられる。
【0114】
これらのマイクロRNAがiPSC誘導において重要であることを確認するために、miR阻害剤を用いて、そのプロセス中に標的とするマイクロRNAのノックダウンも行う。試験したmiR阻害剤の全ては標的miR発現を効率的に減少させることができ、それらのトランスフェクションは増殖に影響を及ぼさない(図16aおよび図16b)。miRミミック実験と一致して、miR−93およびmiR−106bのノックダウンによってGFP+コロニーの劇的な減少が促され得る(図3d)。miR−25ミミックはMEFのiPSC誘導を増強しないが、このマイクロRNAのノックダウンにより初期化効率が約40%低下し(図3d)、miR−25は初期化プロセス中にも作用し得ることが示唆される。対照として、Let7a阻害剤は、初期化効率に対していかなる影響も及ぼさなかった。これらのデータは、miR−93およびmiR−106bがMEFのiPSCへの初期化を促進することを強く示している。初期化効率は、iPSC誘導中にこれらのマイクロRNAをモジュレートすることによってさらに高め得る。
【0115】
実施例6
マイクロRNAにより誘導されたクローンは完全な多能性を有する
誘導された細胞が完全な多能性状態に達するかどうかを調べるために、各マイクロRNAならびにmiR対照についていくつかのiPSCクローンを得、多能性マーカーの発現について解析する。全てのクローンはGFP+(再活性化されるOct4発現を示す)である。免疫染色により、NanogおよびSSEA1も全てのクローンにおいて活性化されることが確認された。他のmESマーカー(Eras、ECat Iおよび内因性(endogeneous)Oct4など)についてのRT−qPCRは類似した結果を示す。全ゲノムmRNA発現プロファイリングでも、得られたクローンがMEFよりもマウスES細胞に類似した遺伝子発現パターンを示すことを示す。内因性Nanog遺伝子座のプロモーターメチル化を解析し、試験したクローンは全て、マウスES細胞で観察されるように、脱メチル化プロモーターを示した(図17)。
【0116】
得られたクローンがmES細胞の完全分化能を示すかどうかを調査するために、胚様体(EB)形成を評価する。得られたクローンは全て、効率的なEB形成を示し、EBは系統マーカー(β−チュブリンIII(外胚葉)、AFP(内胚葉)およびα−アクチニン(中胚葉)など)について陽性染色を示す。これらの細胞から収縮を繰り返すEBも得られ、これは、これらのmiR−iPSCクローンから機能的な心筋細胞が誘導され得ることを示す。これらのmiR−iPSCを無胸腺ヌードマウスに注射すると、3〜4週間で奇形腫が容易に誘導される。最後に、より厳しい試験として、miRにより誘導されたiPSCクローンをアルビノ/黒色B6胚盤胞に注射し、キメラマウスを作製する。さらに、これらの細胞は、得られたE13.5胚の生殖隆起に寄与し得た。これらの結果は、初期化に対するmiR−93およびmiR−106bの促進作用は誘導された多能性細胞の分化能を変更しないこと、そして得られたクローンは3種の生殖細胞系全てに分化することができることを示す。
【0117】
実施例7
miR−93およびmiR−106bはマウスにおいてTgfbr2およびP21をターゲッティングする
miR−93およびmiR−106bによる初期化効率向上の基礎にある機構をさらに理解するために、これらのマイクロRNAの細胞標的を調査する。miR−93がmiR−106bと同じシード領域を共有することから、まず解析にそれを選択する。miR−93ミミックをMEFにトランスフェクトし、mRNA発現プロフィール解析のために2日目にトータルRNAを採取する。その解析は、公表されているMEFおよびiPSCの発現プロフィールと比べてmiR−93の潜在的機能標的を同定する。miR−93トランスフェクションにより著しく減少した遺伝子は、iPSCにおいて低度に発現される遺伝子の3倍の濃縮を示し(図18a)、一方、miR−93トランスフェクションにより増加した遺伝子はそのような濃縮を示さない。加えて、miR−93トランスフェクトMEFの発現プロフィールについて経路オントロジー解析を行う。興味深いことに、iPSC誘導の2つの重要な経路は、miR−93:TGF−βシグナル伝達経路およびG1/S移行経路により調節される。
【0118】
TGF−βシグナル伝達では、miR−93トランスフェクションにより最も著しく減少した遺伝子の1つにTgfbr2がある。Tgfbr2は、TGF−βシグナル伝達において重要な役割を果たす構成的に活性な受容体キナーゼであり、最近の小分子スクリーニングにより、そのヘテロ二量体パートナーであるTgfbr1の阻害剤がiPSC誘導を増強することが示されている。マイクロRNA標的部位予測により、miR−93およびそのファミリーであるマイクロRNAでは2つの保存されたターゲッティング部位がその3'UTRに存在することが示されている。そのため、さらなる調査のために、miR−93が第1の標的候補として選択される。
【0119】
G1/S移行に関しては、ヒト固形腫瘍サンプル(乳房、結腸、腎臓、胃および肺)および胃癌細胞株における最近の結果により、miR−106b〜25クラスターは細胞周期調節因子(CDK阻害剤p21およびp57など)をターゲッティングすることができること、そしてヒトおよびマウスp21が3'UTRの保存されたmiR−93/106b標的部位を共有することが示されているため、潜在的標的としてp21が選択される。
【0120】
さらに、マウスES細胞特異的マイクロRNAクラスター(miR−290およびmiR−293クラスターを含む)はまた、いくつかのG1−S期移行負調節因子(p21を含む)をターゲッティングすることも提示された。加えて、miR−290および293クラスターマイクロRNAはmiR−93およびmiR−106bと非常に類似したシード領域を共有する。そのため、p21も標的候補として解析する。さらに、p21は、iPSC誘導の初期段階中に4種の因子OSKMにより大幅に誘導される(図8a)。詳細な解析により、Oct4およびSox2の組合せはp21レベルの著しい変化を示さないため、p21の誘導が主にKlf4およびcMycの過剰発現によることが明らかになっている(図8a)。
【0121】
マウスTgfbr2およびp21がmiR−93およびmiR−106bによってターゲッティングされるかどうかを確認するために、miRミミックをMEFにトランスフェクトし、48時間後にウエスタンブロッティングにより全細胞溶解物を解析する。実際には、miR−93およびmiR−106bは、Tgfbr2およびp21両方のタンパク質レベルを効率的に低下させ(図5aおよび図5d)、さらにp21 mRNAレベルを約25〜30%低下させ、Tgfbr2 mRNAレベルを約60〜70%低下させる(図19)。p21がmiR−93およびmiR−106bの直接標的であるかどうかをさらに調査するために、ルシフェラーゼアッセイ(p21 3'UTR配列を有するルシフェラーゼレポーターがホタルルシフェラーゼコード配列の下流に挿入される)を行う。ルシフェラーゼアッセイによって、miRミミックをHela細胞にトランスフェクトすることによりルシフェラーゼ活性の一貫した約40%の抑制が達成され得ることが明らかになっている。また、保存されたp21 3'UTR標的部位のシード領域に突然変異が導入されると、マイクロRNAミミックの抑制が完全に破壊され得ることも判明している(図10)。Tgfbr2については、ルシフェラーゼアッセイでGL活性の約50%減少も示し、一方、miR−93変異株にはそのような影響はない(図11)。
【0122】
p21により促進される細胞周期停止によって初期化に必要な後成的修飾が阻害され得るが、増殖中の細胞においてそのような修飾がより起こりやすいためである。p21発現がiPSC誘導に支障をきたすかどうかを判定するために、HAタグ付きp21 cDNAをpMXレトロウイルス主鎖にクローニングし、MEF細胞で過剰発現させる。HA−p21ウイルスを4種のOSKM因子とともにMEFに導入すると、アルカリ性ホスファターゼ染色とOct4−GFP陽性コロニー形成に基づいて、iPSC誘導のほぼ完全な阻害が観察される(図9a)。初期化に3種のOSK因子を用いる場合も類似した結果が得られる(図9b)。
【0123】
解析により、miR−93およびmiR−106bがTgfbr2およびp21の発現両方を効率的に抑制することが示されているため、Tgfbr2およびp21の両方を、それらの活性が初期化に拮抗し得るかどうかをさらに調査する。マイクロRNAミミックで使用した同じ実験時系列を用いて、Tgfbr2またはp21 siRNAをMEFにトランスフェクトする。ウエスタンブロッティングおよびRT−qPCRでは、ウイルス形質導入を行わずにsiRNAによって、それぞれ、タンパク質およびmRNAレベルの両方が効率的にノックダウンされることが確認される(図5bおよび図5e)。次いで、OSKM形質導入によりMEF初期化を開始し、形質導入後11日目にOct4−GFP+コロニーを定量する。各遺伝子についてコロニー数の約2倍の誘導が観察される(図5cおよび図5f)。同時に、我々のデータによって、Tgfbr2およびp21がmiR−93およびmiR−106bの直接標的であり、これらの遺伝子のダウンレギュレーションにより初期化プロセスを促進し得ることが確認される。
【0124】
実施例8
miR−93およびmiR−106bトランスフェクションから得られたiPSCクローンの多能性
miR−93および106bは、マウスiPSC誘導を増強する能力について確認されたが、誘導された細胞が完全な多能性状態に達するかどうかという問題が残っている。この問題に答えるために、各マイクロRNAならびにmiR対照についていくつかのiPSCクローンを得て、多能性マーカーおよび分化能を解析する。これらの得られたクローンは全てGFP+(Oct4遺伝子座の再活性化を示す)である。免疫染色により、NanogおよびSSEA1もこれらの細胞において活性化されることも確認された。他のmESマーカーについてのRT−qPCRは類似した結果を示す。全ゲノムmRNA発現プロフィールでも、これらの得られたクローンがMEFではなくmESと非常に類似した遺伝子発現パターンを有することを示す。内因性Oct4およびNanog遺伝子座のプロモーターメチル化も解析し、試験したクローンは全て、脱メチル化プロモーター有することが観察された。
【0125】
得られたクローンがmES細胞の完全分化能を有するかどうかを調査するために、胚体形成を最初の試験として用いる。得られたクローンは全て、効率的なEB形成を生じ、それらのEBは系統マーカー染色について陽性であると判明している。これらの細胞から収縮を繰り返すEBも得られる。
【0126】
最後に、より厳しい試験として、これらの得られたクローンを注射して、それらがキメラマウスに寄与するかどうかを調べる。実際には、試験したクローン全てからキメラが得られる。これらの結果は、初期化に対するmiR−93およびmiR−106bの促進作用は誘導された細胞の能力を変化させないこと、さらにES様状態に達した誘導クローンは3種の系統全てに分化することができることを示す。
【0127】
実施例9
他のマイクロRNAのアップレギュレーションもiPSC誘導を増強する
本明細書において論じるように、3種のマイクロRNAクラスターは、iPSC誘導中に4種の因子により誘導されると確認され、これらのクラスター内のいくつかのマイクロRNAは、同じシード領域を有すると判明しており、それらは類似したmRNAをターゲッティングすることが示されている(図2)。miR−93およびmiR−106bと同じシード領域を共有する他のマイクロRNAがiPSC誘導を同様に増強することができるかどうかを調査するために、miR−17およびmiR−106aのマイクロRNAミミックを、miR−93ミミック処理およびiPSC誘導について上に記載したものと類似した試験手法を用いて試験する。実際には、これらのマイクロRNAは、miR−106b〜25クラスターで見られたものと同様に初期化を促進し(図6a)、これらのmiRのトランスフェクション全てで、Tgfbr2およびp21タンパク質レベルの低下が起こる(図6bおよび図6c)。
【0128】
同時に、これらの結果は、miR−17〜92、miR−106b〜25およびmiR−106a〜363クラスターの誘導は正確な初期化に重要であること、そしてこれらのマイクロRNAのアップレギュレーションはiPSC生成プロセスへの初期化障壁を低くすることを示唆している(図6d)。よって、これらのマイクロRNAの細胞内レベルを操作して、初期化効率を向上させ得る。
【0129】
実施例10
iPSC初期化機構
得られたクローンは、内因性Oct4−GFP発現を活性化することは分かっている。マイクロRNAミミックとのOSKM形質導入後12日目からコロニーを採取し、照射MEFフィーダープレート上で維持する。内因性pou5f1遺伝子座のGFPシグナルとして緑色蛍光を観察することができる。クローンは、ES特異的マーカーのアルカリ性ホスファターゼ染色および免疫染色(NanogおよびSSEA1染色に基づく)を用いて示すことができる。核染色にはHoechst 33342を用いることができる。3胚葉全てからの細胞は、誘導iPSCクローンを用いる胚様体(EB)アッセイで得ることができる。EB形成のためにiPS細胞を約4000個の細胞/20μl液滴で3日間培養し、次いで、収縮を繰り返す心筋細胞が観察される12〜14日目までさらに培養するために、EBをゼラチンコーティングプレート上に再播種する。異なる系統マーカー(外胚葉マーカーのβ−チュブリンIII;内胚葉マーカーのAFP;および中胚葉マーカーのα−アクチニンを含む)で細胞を免疫染色することができる。奇形腫は、iPS細胞の注射により生成することができ、150万個の細胞を各マウスに注射し、パラフィン包理およびH&E染色のために注射の3〜4週間後に腫瘍を採取する。誘導クローンを用いて、キメラマウスを作製することもできる。iPS細胞をアルビノまたは黒色C57B6マウス(NCI)の胚盤胞に注射し、iPSCの寄与はアグーチまたは黒色の毛色により見ることができる。
【0130】
12日目の初期化細胞はアルカリ性ホスファターゼ基質によって染色され得る。本発明によれば、miRミミックトランスフェクションによりAP+コロニーの著しい増加は起こらないが、miR−93および106bのノックダウンによってAP+コロニーだけでなくGFP+コロニーの著しい減少が生じることとなる。マイクロRNAミミックはAP+コロニー形成全体に影響を及ぼさないが、一方、阻害剤は影響を及ぼす。
【0131】
MEFはiPS細胞に初期化することができるという発見以降、このすばらしいプロセスの基本的機構の理解に多くの努力が傾けられてきた。本明細書に記載した結果によって、転写後遺伝子調節が初期化中に直接関与すること、そしてRNAi機構への干渉により初期化効率を著しく変更することができることが初めて確認された。加えて、前の実施例に示されるように、3種のマイクロRNAクラスターは、iPS細胞を誘導するために用いられる4種の因子によって著しくアップレギュレートされ、これらのクラスター内のマイクロRNAは少なくとも2つの重要な経路:TGF−βシグナル伝達および細胞周期制御をおそらくターゲッティングする。
【0132】
この研究は続行されているが、いくつかの最近の報告では数種の下流腫瘍抑制遺伝子(p21など)を含むp53経路がiPSC誘導中の大きな障壁の1つであることも確認された。4種の因子(OSKM)の異所発現によりp53が容易にアップレギュレートされ、細胞防御プログラムの連続反応(細胞周期停止、アポトーシスまたはDNA損傷応答など)が開始されることを示す多くの証拠もある。これらの防御応答はおそらく低初期化効率(約0.1%前後と考えられる)の根底にある。しかしながら、これらのデータは、初期化に成功した細胞がいかにして細胞障壁を克服してiPS細胞になるかを説明していない。本明細書に記載した実施例は、初期化の成功に拮抗する経路をターゲッティングするマイクロRNAの発現を誘導することによって、これらの細胞が、全てではないが少なくとも一部において障壁を克服し得ることを示している。初代繊維芽細胞におけるマイクロRNAレベルをモジュレートすることによって、初期化効率の著しい向上が達成され得る。
【0133】
TGF−βシグナル伝達は、原腸形成、器官特異的形態形成および組織ホメオスタシスのような多様なプロセスにおいて機能する重要な経路である。標準的なTGF−β形質導入の現行モデルによって、TGF−βリガンドはTGF−β受容体II(Tgfbr2)と結合し、次いで、それが、Tgfbr1とヘテロ二量体を形成して受容体関連Smadsによりシグナルを伝達することが示されている。TGF−βシグナル伝達は、ヒトおよびマウスES細胞自己複製、の両方で機能することが報告されており、FGF2は、ES細胞培養に広く用いられている増殖因子であり、TGF−βリガンド発現を誘導し、BMP様活性を抑制する。化学阻害剤によるTGF−β受容体Iファミリーキナーゼの遮断はES細胞自己複製に支障をきたす。そのような阻害剤は初期化中に全く異なる役割を果たすと思われるため、これらの発見はiPSC誘導に特に重要である。最近の化学スクリーニングでは、TGF−β受容体I(Tgfbr1)の小分子阻害剤はiPSC誘導を実際に増強し、Nanog発現を誘導することによってSox2の必要条件の代わりとし得ることが示された。さらに、TGF−βリガンドによる初期化中の細胞の処理はiPSC誘導に対して悪影響を及ぼす。よって、TGF−βシグナル伝達はES細胞自己複製に重要であるが、初期化にとっては障壁である。本発明によれば、Tgfbr1に加えて、構成的に活性なキナーゼTgfbr2の活性もまた初期化に拮抗することとなる。また、本発明によれば、miR−93およびそのファミリーメンバーはTgfbr2を直接ターゲッティングして、そのシグナル伝達と初期化をモジュレートすることとなる。
【0134】
P21(これは165アミノ酸しか含まない低分子タンパク質である)は、p53依存性G1増殖停止を引き起こし分化および細胞老化を促進することにより癌発生中の腫瘍抑制遺伝子として長年にわたり認められてきた。本発明によれば、4種の因子(OSKM)がMEF細胞に導入されるとp21発現はアップレギュレートされるが、p21の過剰発現によりiPSC誘導がほぼ完全に遮断される(図9)ため、このアップレギュレーションは初期化プロセスに拮抗することとなる(図8)。Klf4初期化因子がp21プロモーターと結合しp21転写を増加させるため、初期化中の細胞におけるp21の誘導は p53に依存することがあるがp53に依存しないこともある。
【0135】
同じ転写因子がiPSC誘導を促進することがあり、iPSC誘導に拮抗することもあることから、これは4種の初期化因子の機能についての興味深い問題を提起している。実際には、ある特定レベルのp21誘導が初期化プロセスにとって有益であるという可能性を除外することができる証拠は現在ない。p21は、p53依存性細胞周期停止における周知の役割の他に、若干の発癌活性を有することも報告されている。例えば、p21は、アポトーシス(細胞周期制御におけるその通常の機能とは無関係の機能)から細胞を保護することにより発癌活性を有する。
【0136】
初期化におけるp21の潜在的利益は、タンパク質−タンパク質相互作用を通じて遺伝子発現を調節する能力に依存し得る。例えば、p21は、アポトーシスに関与するいくつかのタンパク質(カスパーゼ8、カスパーゼ10およびプロカスパーゼ3など)と直接結合することができる。別の例では、p21は、Myc N末端と結合してMyc−Maxヘテロ二量体形成を遮断することにより、Mycのアポトーシス促進作用のサプレッサーでもある。実際には、Myc自体がMEFにおいて過剰発現されると、細胞培養物では細胞死の著しい増加が認められるが、一方、4種の因子を形質導入した細胞では、細胞死はmycだけのサンプルと比べて最小限にとどまる。従って、p21の誘導は、初期化プロセスの障壁となり得るが、細胞アポトーシスを減少させるのに必要なある特定レベルのp21を維持し得るため初期化効率を向上させ得る。
【0137】
miR−93およびmiR−106bのトランスフェクションはp21 siRNAトランスフェクションよりも初期化に対する促進効果が高く、その際、miR−93および106bはp21 siRNAほどp21の発現を抑制しなかったため、本明細書において示すデータはこの仮説を裏付ける部分的証拠ともいえる。しかしながら、この効果は、これらのマイクロRNAによる、Tgfbr2およびp21を含む複数のタンパク質のターゲッティングによるものである可能性もある。
【0138】
マイクロRNAは通常複数の細胞タンパク質をターゲッティングするため、miR−93およびmiR−106bの促進効果は、初期化に関与するさらなる遺伝子を見つける機会を与え、そのプロセスがより理解される。実際には、p21の他に、G1−S期移行の負の調節因子であると報告されているいくつかの他の遺伝子もmRNA転写物の3'UTR領域にmiR−93およびmiR−106b標的部位を有する(Rb1、Rbl1、Rbl2およびLats2など)。miR−93およびmiR−106bの、報告されている別の興味深い標的は転写因子E2F1であり、この転写因子は多くのヒト腫瘍サンプルにおいて脱制御され過活性化されることがよく見られる。E2F1の1つの重大な機能は、ARFおよびINK4aをコードするCDKN2A遺伝子座の発現を活性化することである。Ink4a/Arf遺伝子座もまた初期化効率を阻害し得る。よって、本発明によれば、miR−93およびmiR−106bのトランスフェクションはE2F1をターゲッティングし、CDKN2A遺伝子座を活性化する可能性を低くして初期化の障壁を下げることができることとなる。
【0139】
最後に、miR−17〜92、miR−106b〜25およびmiR−106a〜363クラスターはマウスとヒトとの間で完全に保存される。よって、本発明によれば、miR−93およびmiR−106bの促進効果はヒト初期化にも適用し得ることとなる。
【0140】
実施例11
マイクロRNAはiPS細胞初期化をモジュレートする
マウス胚性繊維芽細胞(MEF)誘導:Oct4−EGFP MEFは、マウス株B6;129S4−Pou5f1tm2(EGFP)Jae/J(Jackson laboratory;ストック番号008214)から、WiCell研究所のウェブサイト(www.wiCell.org/)で提供されているプロトコールを用いて得る。Oct4−EGFP MEFを、MEF完全培地(10%FBS、非必須アミノ酸、L−グルタミン含有、ピルビン酸ナトリウム不含のDMEM)が入った0.1%ゼラチンコーティングプレートで維持する。
【0141】
レトロウイルスを用いた初期化:4X104個のOct4−EGFP MEFにpMXレトロウイルスを形質導入して、Oct4、Sox2、Klf4およびc−Myc(Addgene)を誤発現させる。2日後に、形質導入Oct4−EGFP MEFにES培地(15%ES−screened FBS、非必須アミノ酸、L−グルタミン、モノチオグリセロールおよび1000U/ml LIF含有のDMEM)を供給し、1日おきに培地を換える。特に断りのない限り、形質導入の約2週間後に、蛍光顕微鏡検査法によって初期化幹細胞(EGFP+iPSCコロニーと定義される)にスコアをつける。iPSCを得るために、実体顕微鏡(Leica)下でEGFP+コロニーを手作業で採取する。
【0142】
マイクロRNA阻害剤またはsiRNAトランスフェクション:let−7a、miR−21、およびmiR−29aマイクロRNAの阻害剤は、Dharmaconから購入する。4X104個のOct4−EGFP MEFをLipofectamineおよび阻害剤で、製造業者の教示(Invitrogen)に従いトランスフェクトする。3〜5時間後に、培地を捨て、MEF完全培地に替える;初期化では、初期化因子(Oct4、Sox2、Klf4およびc−Myc)をコードするレトロウイルスを加え、翌日培地を完全培地に替える。特に断りのない限り、トランスフェクション/形質導入後5日目に、阻害剤またはsiRNAを再度導入する。
【0143】
ノーザン解析では、1×105個のOct4−EGFP MEFをトランスフェクトし、5日後に採取する。トータルRNAをTRIZOL(Invitrogen)により単離し、約9μgのトータルRNAを14%変性ポリアクリルアミドゲル(National Diagnostics)上で分離する。RNAをHybond−XL膜(GE healthcare)上に転写し、マイクロRNAを同位体標識した特異的DNAプローブにより検出する。シグナル強度をホスホイメージャーにより視覚化し、Multi Gauge V3.0(FUJIFILM)を用いて解析する。マイクロRNAシグナル強度を、U6 snRNAのものに対して正規化する。実験は3反復で実施する。
【0144】
ウエスタン解析では、1×105個のOct4−EGFP MEFをトランスフェクトし、5日後に採取する。トータルタンパク質をM−PERバッファー(Pierce)で調製し、同量のトータルタンパク質を10%SDS−PAGEゲル上で分離する。タンパク質をPVDF膜に転写し、次の抗体を用いてバンドを検出する:GAPDH(Santa Cruz;カタログ番号sc−20357)、p53(Santa Cruz;カタログ番号sc−55476)、PI3キナーゼp85(Cell Signaling;カタログ番号4257);Cdc42(Santa Cruz;カタログ番号sc−8401);p−ERK1/2(Cell Signaling;カタログ番号9101);ERK1/2(Cell Signaling;カタログ番号9102);p−GSK3β(Cell Signaling;カタログ番号9323);GSK3β(Cell Signaling;カタログ番号9315);βアクチン(Thermo Scientific;カタログ番号MS−1295)。シグナル強度を、Multi Gauge V3.0(FUJIFILM)により定量し、GAPDHまたはβアクチンに対して正規化する。実験は3〜5回繰り返す。
【0145】
in vitro分化および奇形腫形成アッセイ:in vitro分化では、iPSCをトリプシン/EDTAにより解離し、胚様体(EB)培地(15%FBS、非必須アミノ酸、L−グルタミン含有のDMEM)中に終濃度5X104細胞/mlに再懸濁する。EB形成を誘導するために、逆さにしたペトリ皿の蓋で20μl中1000個のiPS細胞を懸滴培養する。3〜5日後に、EBを採取し、ウェル当たり約10個のEBで0.1%ゼラチンコーティングした6ウェルプレートに転写する。EB形成の2週間後に、収縮を繰り返す心筋細胞(中胚葉)が顕微鏡により確認される。内胚葉および外胚葉から得られた細胞は、それぞれ、α−フェトプロテイン(R&D;カタログ番号MAB1368)抗体およびニューロン特異的βIII−チュブリン(abcam;カタログ番号ab7751)抗体によって確認した。
【0146】
奇形腫アッセイでは、1.5X106個のiSPCをトリプシン処理し、150μlに再懸濁し、次いで、アベルチンで麻酔をかけた無胸腺ヌードマウスの後肢背面に皮下注射する。3週間後に、マウスを犠牲にして、奇形腫を採取する。腫瘍塊を固定し、切開し、サンフォード・バーナム研究所にあるCell Imaging−Histology core施設内で解析する。
【0147】
キメラ解析:採取の2時間前にiPSC培地を換える。トリプシン処理したiPSCを0.1%ゼラチンコーティングプレートで30分間培養して、フィーダー細胞を除去する。iPSCをE3.5 C57BL/6−cBrd/cBrd胚盤胞に注射し、次いで、偽妊娠したレシピエント雌へ移す。生後、子の毛色によりiPSCの寄与を評価する:黒色はiPSCに由来する。
【0148】
免疫蛍光およびアルカリ性ホスファターゼ(AP)染色:iPSCを0.1%ゼラチンコーティングした6ウェルプレートに播種し培養する。4日後に、細胞を4%パラホルムアルデヒド(Electron Microscopy Sciences;カタログ番号15710−S)により固定する。免疫蛍光染色では、固定した細胞をPBS中0.1%Trixton X−100により透過処理し、5%BSA/PBSによりブロッキングする。SSEA−1(R&D;カタログ番号MAB2155)およびNanog(R&D;カタログ番号AF2729)に対する抗体をESマーカーとする。核をHoechst 33342染色(Invitrogen)により視覚化する。AP染色では、アルカリ性ホスファターゼ基質を用い製造業者の教示に従って(Vector Laboratories;カタログ番号SK−5100)固定した細胞を処理する。
【0149】
実施例12
miR−21またはmiR−29aの阻害は初期化効率を高める
マウス胚性繊維芽細胞(MEF)誘導:Oct4−EGFP MEFは、マウス株B6;129S4−Pou5f1tm2(EGFP)Jae/J(Jackson laboratory;ストック番号008214)から、WiCell研究所のウェブサイト(www.wiCell.org/)で提供されているプロトコールを用いて得る。Oct4−EGFP MEFを、MEF完全培地(10%FBS、非必須アミノ酸、L−グルタミン含有、ピルビン酸ナトリウム不含のDMEM)が入った0.1%ゼラチンコーティングプレートで維持する。
【0150】
MEF特異的miRNAの阻害が初期化障壁を低くするかどうかを判定するために、MEFで濃縮されたmiRNAを解析し、それらのレベルを、マウスES(mES)細胞で見られるものと比較する。図20aに示されるように、let−7a、miR−21およびmiR−29aは、mES細胞と比べ、MEFにおいて高度に発現される。一方、miR 291は、mESでは非常に豊富であるがMEFでは存在しない(図20a)。次に、let−7a、miR−21、およびmiR−29aに対し、miRNA阻害剤を、Oct3/4、Sox2、Klf4およびc−Myc(OSKM)を発現するレトロウイルスとともにOct4−EGFP MEF(Oct4−EGFPレポーターを保有するMEF)に導入する。形質導入後14日目に、miR−21阻害剤で処理した細胞はターゲッティングしない(NT)対照と比べて初期化効率の約2.1倍向上を示す(図20b)。同様に、miR−29aの阻害後、初期化効率は有意に約2.8倍向上する(図20b)。miR 29aまたは21阻害で用いた同様のantagomir処理に従って、let−7a阻害後にOSKM初期化に対して軽度の効果が観察される(図20b)。c−Mycの不在下でmiRNA阻害により3種の因子による初期化が促進されるかどうかをさらに試験するために、miRNA阻害剤を、OSKMよりずっと低い効率で細胞を初期化するOSKとともに細胞に形質導入する。OSKにより初期化したiPS細胞コロニーの数は、OSK単独での処理と比べ、miR−21阻害剤の存在下で増加する(図25)。これらの結果は、MEFで濃縮されたmiRNA miR−21およびmiR−29の枯渇により4Fによる初期化が著しく促進されること、そしてmiR−21の遮断により3種の因子(OSK)による初期化の効率が中等度に向上することを示す。
【0151】
C−Mycは、初期化中にmiRNA let−7a、miR−16、miR−21、miR−29aおよびmiR−143の発現を抑制する:最近の研究により、OSKM因子は、後成的機構と転写機構の両方を通じて細胞同一性を変更することが示されている。本発明によれば、OSKM初期化因子は、MEFで濃縮されたmiRNAをダウンレギュレートし得ることとなる。miRNA発現に対する各初期化因子の潜在的影響を評価するために、MEFにOSKM因子の様々な組合せを形質導入し、それらに対してノーザンブロット解析を行う(図21a)。興味深いことに、Sox2だけが、miR−21、miR−29aおよびlet−7aの発現レベルが、MEF対照と比べて2倍より高く誘導する(図21b、左のパネル)。Klf4は、選択したmiRNAに対して軽度であるがSox2と類似した影響を及ぼす(図21b、左のパネル)。Oct4過剰発現に関してのみ、miRNAは発現レベルが変化しない(図21b、左のパネル)。Oct4、Sox2およびKlf4とは対照的に、単一因子のc−MycはmiR−21およびmiR−29a(MEFにおいて最も豊富なmiRNA)の発現をMEF対照の約70%ダウンレギュレートする(図21aおよび図21b、左のパネル)。さらに、図21b(中央のパネル)に示す2種の因子(2F)の様々な組合せの間では、c−Mycを含む組合せは、3種のmiRNA全て(miR−21、miR−29aおよびlet−7aを含む)における減少を、約25〜80%増大し得る(図21b、中央のパネル)。Sox2とOct4とでは、miRNAに対する1Fの影響と同様に、miR−21およびmiR−29aを、MEF対照の1.5倍および2.3倍増加させ、OKおよびSKではmiRNA発現に対して及ぼす明らかな影響はない。さらに、3種の因子(3F)の様々な組合せの間では、miRNA−21の発現は、SKM細胞およびOKM細胞において、MEFにおいて見られる発現と比べて、それぞれ約70%および78%減少し、同様に、miR−29a発現は、c−Mycを含む3F組合せで約48〜70%減少する(図21b、右のパネル)。3F組合せにc−Mycを含むことによりlet−7aレベルも少し減少する(図21b、右のパネル)。c−Mycを含まないOSKでは、miRNA発現に対して及ぼす影響はほとんどなかった(図21b、右のパネル)。よって、これらの結果は、c−Mycが初期化中のmiRNA発現の調節において重要な役割を果たすことを強く示唆している。
【0152】
c−MycがmiRNA発現に拮抗する第1因子であることをさらに確認するために、OSKをc−Mycとともにまたはc−Mycを含めずに細胞に形質導入し、miRNA発現を形質導入後の様々な時点においてリアルタイム定量的逆転写ポリメラーゼ連鎖反応(RT−qPCR)により調査する。OSKに対し、OSKM形質導入により初期化中のlet−7a、miR−16、miR−21、miR−29a、miR−143の発現は大幅に減少し(図21c)、c−Mycは、MEFで濃縮されたmiRNA(最も豊富なものである、let−7a、miR−21およびmiR−29aを含む)の発現の調節において中心的な役割を果たすことが分かる。これらのデータは、c−MycはmiRNAダウンレギュレーションを通じてある程度初期化を促進することも示唆している。
【0153】
実施例13
miRNA枯渇によって誘導されたiPS細胞は多能性を獲得する
本発明によれば、miR−21およびmiR−29a阻害剤を用いて得られたマウスiPS細胞は多能性であることとなる。OSKM/anti miR−29a iPS細胞についてES細胞マーカー染色を行うことができる。さらなる解析のために、OSKMおよびmiR−29a阻害剤での処理後に得られたGFP+コロニーを採取する。胚幹細胞マーカーNanogおよびSSEA1を発現する代表的なコロニーを同定する。内因性Oct4も活性化し、それはEGFP染色により示すことができる。強いアルカリ性ホスファターゼ(AP)活性はESマーカーの1つとして観察することができる。
【0154】
OSKM/anti miR−29a iPS細胞のin vitro分化を行うことができる。胚様体をin vitroで形成し、2週間培養することができる。細胞を固定し、抗αフェトプロテイン(中胚葉について)および抗βチュブリンIII(外胚葉について)で染色することができる。核は、Hoescht染色による対比染色として観察することができる。OSKM/anti miR−29a iPS細胞の奇形腫形成解析も行うことができる。1.5X106個のiPSCを無胸腺ヌード雌マウスに皮下注射する。注射の3週間後に腫瘍塊を採取し、組織病理学的解析のために固定する。3胚葉から得られた様々な組織を確認することができ、それらには、腸管様上皮(内胚葉)、脂肪組織、軟骨および筋肉(中胚葉)、ならびに神経組織および表皮(外胚葉)が含まれる。OSKM/anti miR−29a iPS細胞およびOSK/anti miR−21 iPS細胞のキメラ解析も行うことができる。8〜14個のiPS細胞をE3.5マウス胚盤胞に注射することができる。各キメラへのiPS細胞の寄与は、黒色の毛色を評価することにより推定することができ、割合として観察することができる。
【0155】
miR−21またはmiR−29aの遮断がiPS細胞の多能性に支障をきたすかどうかを調査するために、OSKM/anti miR−29aまたはOSK/anti miR−21を用いたiPS細胞を多能性について評価する。まず、初期化のおよそ2週間後に細胞を手作業で採取し、拡大して、形態およびES特異的マーカーの発現を調べる。細胞はES様形態および高度に発現されたOct4−EGFP示す(内因性ES細胞シグナル伝達の確立を示す。加えて、OSKM/anti miR−29a iPS細胞またはOSK/anti miR−21 iPS細胞は、NanogおよびSSEA1を含むES細胞特異的マーカーを発現し、アルカリ性ホスファターゼ活性を示した。それらのiPS細胞が、通常誘導されるiPS細胞と同等の多能性潜在能力を示すかどうかを試験するために、OSKM/anti miR−29a iPS細胞およびOSK/anti miR−21 iPS細胞を、誘導して胚様体(EB)を形成するかまたはヌードマウスに注射し、様々な組織に分化させる。in vitro分化の2週間後に、3胚葉全てから得られた典型的な細胞種が観察される。注射の3週間後に形成された奇形腫腫瘍に対して、組織病理学的解析を行う。3胚葉全てに由来する様々な組織が生成され、iPS細胞が多能性を得たことが確認される。多能性についての最も厳しい試験を用いるために、iPS細胞をE3.5胚盤胞に注射して、キメラマウスを作出する。miR枯渇によるiPS細胞から得られたマウスは、iPS細胞に起因する、有意の約15%〜25%黒色毛色を示す。これらのデータは、miR−21およびmiR−29aの枯渇が、得られたiPS細胞の多能性に対して悪影響を及ぼさないことを示している。
【0156】
実施例14
miR−29aの阻害はP85αおよびCDC42の経路を通じてP53をダウンレギュレートする
初期化に及ぼすmiR−29aの作用の基礎にある機構を理解するために、p85αおよびCDC42の発現を調査する。p85αおよびCDC42はHeLa細胞においてmiR−29の直接標的であると報告されている。その目的で、miRNA阻害剤をMEFにトランスフェクトし、トランスフェクション後5日目にp85αおよびCDC42タンパク質発現をウエスタンブロットにより評価する。P85αおよびCDC42タンパク質レベルはmiR−29a遮断によって少し増加するが、それに対し、let−7a阻害剤ではほとんど影響がない(図22aおよび図22b)。また、形質転換関連タンパク質53(Trp53またはp53)はp85αおよびCDC4の直接標的であると報告されている。よって、p53についても、p53がMEFにおいてmiR−29aによって間接的に調節されるかどうかを調査する。それを試験するために、MEFをmiRNA阻害剤でトランスフェクトし、免疫ブロッティングのために5日採取して、p53の発現を評価する。P53タンパク質レベルはmiR−29a阻害によって約30%低下する(図22aおよび図22b)がNT対照やlet−7a阻害では変化しない。miR−21の枯渇によってもp85αおよびCDC42タンパク質抑制から解放され、それゆえにp85αおよびCDC42のレベルが増加し、その結果、p53発現の約25%ダウンレギュレーションが起こることは重要である(図22aおよび図22b)。
【0157】
初期化中にmiR−21またはmiR−29aの阻害によってp53レベルが低下することをさらに確認するために、初期化5日目にp53発現をウエスタンブロット解析により調査する。初期化を開始するために、miRNA阻害剤をOSKMとともに導入する。MEF単独での観察と一致して、初期化中に、miR−21またはmiR−29aの枯渇によって、p53タンパク質レベルがOSKM対照と比べてそれぞれ約25%または約40%低下する(図22c)。要するに、我々のデータによって、初期化中、miR−29aの遮断によってp85αおよびCDC42の経路を通じてp53タンパク質レベルが約30〜40%低下することがわかった。加えて、miR−21の枯渇はp85αおよびCDC42の両方に対して類似した影響を及ぼし、p53タンパク質レベルを約25%〜約30%低下させた。
【0158】
miR−29aの阻害は、p53ダウンレギュレーションを通じて初期化効率を向上させる:p53の欠乏によって初期化効率が大幅に向上し得ることは報告されている。miR−29aの枯渇はp53レベルを著しく低下させ、初期化効率を約2.8倍向上させることから、本発明によれば、miR−29aノックダウンの効果は主にp53ダウンレギュレーションによって媒介されることとなる。これを受けて、p53 siRNAおよび/またはmiR−29a阻害剤をOSKMとともにOct4−EGFP MEFにトランスフェクトして、初期化を開始する。siRNAによるp53のダウンレギュレーション(約80%)は、初期化効率に対して、miR−29a阻害の場合と類似した良い影響を及ぼす(図22d)。p53 siRNAの存在下でmiR阻害剤を加えた場合には初期化効率の向上は見られない(図22d)。これらの結果は、miR−29aの阻害はp53のダウンレギュレーションを通じて初期化効率を向上させるようにある程度作用することを示唆している。
【0159】
実施例15
miR−21およびmiR−29aの阻害はGSK3βではなくERK1/2のリン酸化を減少させて初期化を促進する
miR21は、心臓繊維芽細胞においてsprouty homologue 1(Spry1)の阻害を通じてMAPK/ERKを活性化することが報告されている。MAPK/ERK活性の遮断は、神経幹細胞の初期化を促進し、ESC自己複製の基底状態を保証する。よって、本発明によれば、miRNA阻害剤導入後にMEFにおけるERK1/2リン酸化を評価することによってmiR−21は初期化中にMAPK/ERK経路を調節することとなる。それを試験するために、MEFをmiRNA阻害剤でトランスフェクトし、次いで、ウエスタンブロット解析のために採取して、リン酸化ERK1/2レベルを決定する。ウエスタンブロット解析は、miR−21の遮断によってERK1/2リン酸化がNT対照と比べて約45%有意に低下したが、それに対し、let−7a阻害剤ではそのような影響がないことを示している(図23a)。興味深いことに、miR−29a枯渇MEFでもERK1/2リン酸化がNT対照と比べて60%有意に低下する(図23a)。また、本発明によれば、miR−21およびmiR−29aは、Spry1レベルを変更することによりERK1/2リン酸化に影響を及ぼし得ることとなる。様々なmiRNA阻害剤をトランスフェクトすることによりMEFにおいてmiR−21またはmiR−29aを枯渇させ、Spry1発現レベルを免疫ブロッティングにより定量し、それらの結果は、miR−21およびmiR−29aの阻害によりSpry1発現レベルが高まった(図23b)ことを示している。ゆえに、miR−21およびmiR−29aの枯渇は、Spry1タンパク質レベルをモジュレートすることによってERK1/2のリン酸化をダウンレギュレートする。
【0160】
ERK1/2ダウンレギュレーションによって初期化効率が高まるかどうかについて取り組むために、4F初期化の過程でERK1または2をターゲッティングするsiRNAをOct4−EGFP MEFに導入する。いずれの枯渇も成熟したiPS細胞の生成を促進する(図23c)。本発明によれば、miR−21の遮断によってERK1/2リン酸化が減少することからmiR−21はMEFにおけるERK1/2活性化のインデューサーとして働くこととなる。miR−29aの枯渇によってもERK1/2リン酸化は著しく減少する。これらの結果は、miR−21およびmiR−29aがERK1/2活性を調節して初期化効率を高めることを強く示唆している(図23a、図23bおよび図23c)。
【0161】
GSK3β経路もES自己複製および神経幹細胞の初期化を抑制する。GSK3βの枯渇により成熟iPS細胞生成は大幅に増加する(図23c)。本発明によれば、miRNA枯渇がGSK3β活性化を調節したこととなる。免疫ブロッティングは、Oct4−EGFP MEFにおけるmiRNAの遮断がGSK3β活性化に対して大きな影響を及ぼさないことを示している(図23d)。本発明によれば、細胞生存率および複製速度を評価するためにフローサイトメトリーを用いることによってmiRNA枯渇が初期化中にアポトーシスまたは細胞増殖を変更することとなる。OSKMでの初期化中のmiRNA 21、29aまたはlet−7の遮断ではアポトーシスまたは増殖速度は変更されない(図26)。全体として、miR−29aおよびmiR−21はp53およびERK1/2の経路をモジュレートして、iPS細胞初期化効率を調節する。
【0162】
実施例16
C−MycはmiR−21およびmiR−29aのダウンレギュレーションを通じてP53レベルを低下させERK1/2活性化に拮抗することにより初期化の閾値を下げる
初期化誘導に現在用いられている導入遺伝子の代替物を開発するために、これらの因子によってシグナル伝達経路がどのように調節されるかを理解することが重要である。本発明によれば、c−Mycは初期化中に、MEFで濃縮されたmiRNA(miR−21、let−7a、およびmiR−29aなど)を抑制することとなる(図20)。阻害剤によるmiR−29aの枯渇は、p85αおよびCDC42抑制を解放することによってp53タンパク質レベルを低下させる可能性が高い(図22)。加えて、miR−21の枯渇はERK1/2リン酸化も減少させる(図23)。本発明によれば、miR−21阻害はp53タンパク質レベルを低下させることとなり、そしてmiR−29aの阻害もERK1/2リン酸化レベルを低下させることとなる。p53およびERK1/2のシグナル伝達はいずれも初期化に拮抗する。miR−21およびmiR−29aの遮断またはp53およびERK1/2のノックダウンにより初期化効率を高めることができる(図22および図23)。本発明によれば、c−Mycは、p53タンパク質レベルおよびERK1/2活性化の誘導を通じて初期化障壁として働く、MEFで濃縮されたmiRNA、miR−21およびmiR−29aを抑制することによって、ある程度初期化を促進することとなる(図24)。
【0163】
初期化を促進するために、miR−290ファミリーのES特異的miRNAの強制発現をc−Mycの代わりにすることができる。また、C−MycはmiR−290クラスターのプロモーター領域と結合する。しかしながら、c−Myc導入遺伝子の初期発現は、初期化を開始するのに有効であるが、miR−290クラスターが発現を開始する成熟iPS細胞の移行段階またはそれ以降では重要なものでない。よって、c−MycがmiR−290ファミリーの活性化を通じて初期化の初期段階を促進するという可能性は低い。
【0164】
本発明によれば、初期化のためにc−Mycが導入されると、MEFで濃縮されたmiRNA(miR−29a、miR−21、miR−143およびlet−7aを含む)の発現レベルは低下することとなる。C−Mycは、腫瘍形成の促進または多能性基底状態の維持においてmiRNAに深刻な転写の影響を及ぼす。よって、miRNA発現のc−Myc抑制は初期化の基礎にあると考えられる機構である。
【0165】
miR−21は、正のメディエーターとして、TGFβ1およびERK1/2の経路(これらの両経路は初期化およびES細胞基底状態に影響を及ぼすことがわかっている)を通じて繊維形成活性を増強するように働く。特に、確認したmiR−29a標的の間では、p53がmiR−29aによって正に調節される。加えて、最近の研究では、Ink4−Arf/p53/p21経路が初期化に支障をきたし、p53の欠乏によって初期化効率が大幅に高まることが示されている。これらのことから、これらのシグナル伝達経路は初期化プロセスに対する第1の障壁であるといえよう。
【0166】
c−MycをターゲッティングするmiRNA、miR−21およびmiR−29aの枯渇により、初期化効率が約2.1倍〜約2.8倍高まり(図20)、MEFで濃縮されたmiRNAも初期化障壁として働くことが示唆される。let−7阻害は初期化を促進することが最近報告されたが、しかしながら、何度かの試みによると、antagomirによりlet−7が阻害された場合、観察される初期化への影響は軽度なものでしかない(図20)。さらに、本発明によれば、初期化中のp53の誘導はmiR−29a阻害によって支障をきたし初期化効率が高まることとなる。同様に、初期化は、miR−21の枯渇またはERK1/2のいずれかによって大幅に促進され得る。C−Mycは、初期化の初期段階の大きな要因であり、そのプロセスを移行段階および終わりに近い段階で維持するには必要でなく、これによれば、高効率の初期化を開始するためにc−Mycを調節するmiRNAを使用し得ることとなる。
【0167】
C−Mycは、miR−29aプロモーターと直接結合し抑制することが報告されている。本発明によれば、c−Mycは、miR−21の枯渇と部分的にしか置き換えることができないこととなり、これはc−Mycが初期化において他の機能を有することを示唆している。よって、初期化中のc−Myc機能に代わる、複数の経路の調節またはMEFで濃縮されたmiRNAの広い抑制が必要とされ得る。
【0168】
本発明によれば、c−MycはmiR−21およびmiR−29aのダウンレギュレーションを通じてp53レベルを低下させERK1/2活性化に拮抗することにより初期化の閾値を下げることとなる。加えて、c−Mycの下流の因子は、初期化を改善するために、siRNA、miRNAまたは小分子による操作の対象となり得る。これらのアプローチは、4種の初期化因子全ての代わりとするために広げることができる。
【0169】
上記の実施例により本発明を記載してきたが、修飾および変形が本発明の精神および範囲内に含まれることは理解されるであろう。よって、本発明は次の特許請求の範囲によってのみ限定される。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図1F】
【図2A】
【図2B】
【図2C】
【技術分野】
【0001】
本発明は、一般に、誘導多能性幹(iPS)細胞の分野に関し、より具体的には、そのような細胞を体細胞から生成する方法、ならびにそのような方法により生成されたiPS細胞の臨床使用および研究使用に関する。
【背景技術】
【0002】
誘導多能性幹細胞(iPSC)は、胚性幹(ES)細胞の特性を示し、当初はマウス体細胞における4種の核初期化因子(4F):Oct4、Sox2、Klf4およびcMycの異所発現により生成された。ヒト細胞では、最初の4種の山中因子の他にも、4種の因子の別のセット、例えば、Oct4 Nanog Lin28およびSox2を用いてiPSCを生成することもできる。異なる組織由来の多くの細胞種が初期化可能であると確認されているが、典型的に0.01%〜0.2%という低初期化効率がiPSC誘導およびさらなる治療的使用のための主要な障害である。初期化効率を高める小分子についてのスクリーニングだけでなく、iPSC誘導の新しい方法の開発にも多大な努力が注ぎ込まれてきたが、初代繊維芽細胞がES様状態に初期化される機構はまだ大部分が不明である。
【0003】
細胞初期化の機構を理解するために、種々のアプローチが用いられてきた。小分子に基づく方法では、Dnmt1阻害剤で細胞を処理することによって、初期化プロセスを加速させることができることが確認された。またTGFβ阻害もより速く、かつ、より効率的なiPSC誘導を可能にすることが見出された。これはSox2およびcMycに取って代わることができる。さらにまたアレイ解析では、部分的に初期化されたiPSCは、メチルトランスフェラーゼ阻害剤などの因子での処理が行われるとさらに完全に初期化され得ることが示された。4種の初期化因子によるプロモーター結合および発現誘導の網羅的ゲノム解析では、これらの因子はiPSCおよびmES細胞において類似した標的を有し、おそらく類似した遺伝子セットを調節するであろうこと、さらに、部分的iPSCでは初期化因子のターゲッティングが変更されることも証明されている。
【0004】
ごく最近では、いくつかのグループによってp53媒介腫瘍抑制経路がiPSC誘導に拮抗し得ることが確認された。p53およびその下流のエフェクターp21の両方は初期化プロセス中に誘導され、両タンパク質の発現低下によってiPSCコロニー形成が促進され得る。これらのタンパク質は4種の初期化因子(4F)を発現する大部分の細胞においてアップレギュレートされ、cMycはp21発現を遮断すると報告されているが、これらの4種の因子(4F)の異所発現が癌遺伝子/導入遺伝子過剰発現に対する細胞応答をいかにして克服し、なぜ細胞のごく一部の集団だけが完全に初期化されるかについては依然として不明である。
【0005】
マイクロRNA(MicoRNA)は、RNA誘導サイレンシング複合体(RISC)と呼ばれるタンパク質複合体と関係がある18〜24ヌクレオチドの一本鎖低分子RNAである。これらの低分子RNAは、通常遺伝子転写物の非コード領域から生成され、翻訳抑制により遺伝子発現を抑制する働きをする。近年、マイクロRNAは多くの異なる重要なプロセス(多数ある中で、ヒトES細胞の自己複製遺伝子発現、胚性幹(ES)細胞の細胞周期制御、選択的スプライシング、心臓発生など)への関与が見出された。さらに、ES細胞特異的マイクロRNAは、マウスiPSC誘導を増強し、初期化中にcMycの機能に取って代わることができることが最近報告された。またhES特異的miR−302は、ヒト繊維芽細胞における4種の因子の発現により老化応答軽減することも示唆されている。しかしながら、これらのマイクロRNAは初期化プロセスの極めて終わりに近い段階まで発現されないため、マイクロRNAがiPSC誘導において重要な役割を果たすかどうかはこれまで分かっていなかった。
【発明の概要】
【0006】
本発明は、iPSC誘導中にマイクロRNAが必要とされるという重大な発見に基づくものである。マイクロRNA生合成機構へ干渉により初期化効率の著しい低下がもたらされる。初期化の初期段階中に高度に誘導されるマイクロRNAクラスターが確認され、機能試験では、そのようなマイクロRNAを体細胞に導入することにより誘導効率が高まることが示されている。加えて、細胞の初期化に使用される重要なレギュレーターも確認されたが、それらのレギュレーターを標的として、初期化効率ならびにiPS細胞の直接分化を著しく増加させ得ることが有利である。
【0007】
よって、一実施形態では、本発明は、iPS細胞を生成する方法を提供する。その方法は、体細胞を核初期化因子と接触させること、およびその細胞を、その細胞でRNAレベルまたは活性を変更するマイクロRNAと接触させ、それによってiPS細胞を生成することを含む。一態様では、マイクロRNAまたはRNAが修飾される。別の態様では、マイクロRNAはベクター中に存在する。別の態様では、マイクロRNAは、miR−17、miR−25、miR−106a、miR let−7ファミリーメンバー(例えば、let−7a、miR 98)またはmiR−302bクラスター中に存在する。別の態様では、マイクロRNAは、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである。
【0008】
一態様では、マイクロRNAは、配列番号1を含むポリヌクレオチド配列を有する。別の態様では、マイクロRNAは、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する。別の態様では、マイクロRNAは、p21、Tgfbr2、p53、またはそれらの組合せの発現または活性を調節する。別の態様では、マイクロRNAは、Spry 1/2、p85、CDC42またはERK1/2の経路を調節する。
【0009】
一態様では、核初期化因子は、ベクター中に含まれる遺伝子によりコードされる。別の態様では、核初期化因子は、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せである。別の態様では、核初期化因子は、OCT4、SOX2、KLF4、C−MYCの1種以上である。別の態様では、核初期化因子はc−Mycを含む。別の態様では、誘導効率は、マイクロRNAを使用しない場合と比べて少なくとも2倍となる。
【0010】
一態様では、マイクロRNAとの接触前に、接触と同時にまたは接触後に、体細胞に初期化因子を接触させる。別の態様では、体細胞は哺乳類細胞である。さらなる態様では、体細胞はヒト細胞またはマウス細胞である。
【0011】
別の実施形態では、本発明は、体細胞を核初期化因子と、p21発現または活性の阻害剤とに接触させることによりiPS細胞を生成する方法を提供する。
【0012】
別の実施形態では、本発明は、体細胞を、その細胞内でRNAレベルまたは活性を変更する作用物質と接触させ(ここで、該作用物質は該体細胞中で多能性を誘導し、ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することにより誘導多能性幹(iPS)細胞を生成する方法を提供する。様々な実施形態では、RNAは非コードRNA(ncRNA)であり、マイクロRNAが含まれる。
【0013】
上に記載した方法の一態様では、作用物質はポリヌクレオチド、ポリペプチドまたは小分子である。さらなる態様では、ポリヌクレオチドはアンチセンスオリゴヌクレオチド、化学修飾されたオリゴヌクレオチド、ロックド核酸(LNA)またはDNAである。別の態様では、ポリヌクレオチドはRNAである。さらなる態様では、RNAは、マイクロRNA、dsRNA、siRNA、stRNAまたはshRNAからなる群から選択される。別の態様では、体細胞はマウス胚性繊維芽細胞(MEF)である。
【0014】
様々な態様では、RNAを変更する作用物質は、p21、Tgfbr2、p53、またはそれらの組合せを、発現または活性について阻害することができる。一態様では、作用物質はポリヌクレオチド、ポリペプチドまたは小分子であってよい。別の態様では、作用物質またはp21、Tgfbr2および/またはp53の阻害剤はRNA分子であり、マイクロRNA、dsRNA、siRNA、stRNAまたはshRNA、またはアンチセンスオリゴヌクレオチドが含まれる。例示的態様では、作用物質またはp21、Tgfbr2および/またはp53の阻害剤は、マイクロRNA分子であり、前記細胞に導入された組換えベクター中に含まれるポリヌクレオチドによりコードされる。
【0015】
様々な態様では、マイクロRNAは、iPSCの誘導またはその分化中に活性または発現の増加または減少を示すクラスター中に含まれるマイクロRNAであってよい。一態様では、誘導効率は、作用物質を使用しない場合と比べて少なくとも2倍となる。別の態様では、誘導効率は、作用物質を使用しない場合と比べて少なくとも3倍である。別の態様では、誘導効率は、作用物質を使用しない場合と比べて少なくとも5倍である。一態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中の1種以上のマイクロRNAであってよく、miR−93、miR−106b、miR−21、miR−29a、miR−let−7ファミリーメンバー(例えば、let−7a;miR 98)またはそれらの組合せが含まれる。関連態様では、マイクロRNAは、miR−17、miR−25、miR−106aおよびmiR−302bクラスター中のマイクロRNA種に対応する様々なマイクロRNA(例えば、配列番号2〜11のもの)の間で保存されると判明している、配列番号1を含むポリヌクレオチド配列を有する。一態様では、マイクロRNAは、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する。
【0016】
様々な態様では、核初期化因子は、前記細胞に導入された組換えベクター中に含まれる遺伝子によりコードされる。別の態様では、作用物質は、p21、Tgfbr2、p53、またはそれらの組合せの発現または活性を阻害する。別の態様では、作用物質は、Spry 1/2、p85、CDC42またはERK1/2の経路を調節する。
【0017】
様々な態様では、核初期化因子は、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せの1種以上によりコードされる。例示的態様では、核初期化因子は、OCT4、SOX2、KLF4、C−MYCの1種以上である。別の態様では、少なくとも1種の核初期化因子がc−Mycを含む。さらなる態様では、c−Mycは、少なくとも1つのmiRNAを抑制することにより初期化を少なくともある程度まで促進する。
【0018】
別の実施形態では、本発明は、本明細書に記載した方法を用いて生産されたiPS細胞またはそのような細胞の集団を提供する。別の実施形態では、本発明は、本明細書に記載した方法により生産された誘導多能性幹(iPS)細胞の濃縮された集団を提供する。
【0019】
同様に、別の実施形態では、本発明は、本明細書に記載した方法を用いて生成されたiPSCの分化を誘導することにより得られた分化した細胞を提供する。一態様では、体細胞はiPSCをRNA分子またはアンチセンスオリゴヌクレオチドと接触させることによって分化を誘導することにより得られる。一態様では、RNA分子は、マイクロRNA、dsRNA、siRNA、stRNAまたはshRNAからなる群から選択される。
【0020】
別の実施形態では、本発明は、本明細書に記載した方法を用いて生成されたiPS細胞を用いて対象を治療する方法を提供する。その方法は、本明細書に記載した方法を用いて対象の体細胞を誘導多能性幹(iPS)細胞に誘導すること、そのiPS細胞の分化を誘導すること、およびその分化した細胞をその対象に導入し、それによってその症状を治療することを含む。
【0021】
別の実施形態では、本発明は、本明細書に記載した方法により生成されたiPS細胞またはそれから得られた体細胞を用いて作用物質の生理学的機能を評価するための方法を提供する。一態様では、その方法は、本明細書に記載した方法を用いて生産された誘導多能性幹(iPS)細胞を処理することおよびその作用物質による少なくとも1つの細胞機能の変化を評価することを含む。別の態様では、その方法は、本明細書に記載した多能性幹細胞の分化を誘導することにより得られた分化した細胞を作用物質により処理することおよびその作用物質による細胞機能の変化を評価することを含む。
【0022】
別の実施形態では、本発明は、本明細書に記載した方法により生成されたiPS細胞またはそれから得られた体細胞を用いて化合物の毒性を評価する方法を提供する。一態様では、その方法は、本明細書に記載した方法を用いて生産された誘導多能性幹(iPS)細胞を化合物により処理することおよびその化合物の毒性を評価することを含む。別の態様では、その方法は、本明細書に記載した多能性幹細胞の分化を誘導することにより得られた分化した細胞を化合物により処理することおよびその化合物の毒性を評価することを含む。
【0023】
別の実施形態では、本発明は、誘導多能性幹(iPS)細胞を生成する方法を提供する。その方法は、体細胞を少なくとも1種の核初期化因子と接触させること;およびその細胞をp21、Tgfbr2、p53、またはそれらの組合せの発現または活性についての阻害剤と接触させることを含む。一態様では、阻害剤はp21の発現および/または活性を阻害する。別の態様では、阻害剤はTgfbr2の発現および/または活性を阻害する。別の態様では、阻害剤はp53の発現および/または活性を阻害する。
【0024】
別の実施形態では、本発明は、誘導多能性幹(iPS)細胞を生成する方法を提供する。その方法は、体細胞を、その細胞内でRNAレベルまたは活性を変更する作用物質と接触させ(ここで、該作用物質は該体細胞中で多能性を誘導し、ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することを含む。
【0025】
別の実施形態では、本発明は、対象を治療する方法を提供する。その方法は、本明細書に記載した方法により対象の体細胞から誘導多能性幹(iPS)細胞を生成すること;そのiPS細胞の分化を誘導すること;およびその細胞をその対象に導入し、それによってその症状を治療することを含む。
【0026】
別の実施形態では、本発明は、iPS細胞の生成効率を高めるためのマイクロRNAの使用を提供する。一態様では、マイクロRNAは、miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、またはそれらの組合せからなる群から選択される。別の態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中に存在する。別の態様では、マイクロRNAは、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである。
【0027】
別の実施形態では、本発明は、miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、miR let−7ファミリーメンバーまたはそれらの組合せからなる群から選択されるmiR配列の組合せを提供する。別の態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中に存在する。別の態様では、マイクロRNAは、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである。
【図面の簡単な説明】
【0028】
【図1】マウスiPSC誘導へのRNAi機構の関与を示す図である。図1a、図1bおよび図1cは、それぞれ、shRNAによるマウスRNAi機構遺伝子Ago2、DroshaおよびDicerのノックダウンを示している。標的遺伝子のmRNAおよびタンパク質のレベルの両方をRT−qPCRにより解析し、ヒストグラムおよび対応するウエスタンブロットに示している。初代マウス胚性繊維芽細胞(MEF)に、4種の因子を、Drosha、DicerおよびAgo2をターゲッティングするshRNAとともに形質導入する。MEFにレンチウイルスshRNAを4μg/μlのポリブレンとともに形質導入し、形質導入後3日目にトータルRNAまたはタンパク質を採取する。標的遺伝子のmRNAおよびタンパク質のレベルを、それぞれ、RT−qPCRおよびウエスタンブロッティングにより解析する。pLKOは、shRNAレンチウイルスベクターに対する空ベクター対照である。pGIPZは、ターゲッティングしないshRNAを発現するレンチウイルスベクターである。図1dは、Ago2のノックダウンによりOSKによるiPSC誘導が減少するということを示している。形質導入後21日目にコロニーを染色し、APについて定量する。エラーバーは、2反復のウェルの標準偏差を表す。図1eは、shAgo2に関するiPSCのGFP+コロニーの定量を示している。形質導入後21日目にGFP+コロニーを定量する。エラーバーは、2反復のウェルの標準偏差を表す。図1fは、Ago2のノックダウンにより4FによるiPSC誘導が劇的に減少するということを示している。初代MEFに4種の初期化因子(OSKM(4F))をshRNA Ago2とともに形質導入する。形質導入後14日目にコロニーをアルカリ性ホスファターゼについて染色することができる。アルカリ性ホスファターゼはmES/iPS細胞のマーカーである。pLKOベクターおよびpGIPZベクターを陰性対照とした。
【図2】初期化の初期段階中のマイクロRNAクラスターmiR−17、25、106aおよび302bの誘導を示す図である。図2aは、4種の因子の形質導入後の、初期段階にある10種のマイクロRNAクラスターの発現誘導を示すグラフを示している。miR RT−qPCRを用いて、ES細胞において高度に発現される10種のクラスターの代表的なマイクロRNAの発現変化を定量する。感染後4日目に開始時のMEFおよび4Fを用いたMEFのトータルRNAを解析する。ヒストグラムの色の濃いバーは感染後4日目のMEFを示し、白色のバーは開始時のMEFを示す。アスタリスクは、誘導されたマイクロRNAを示す。図2bは、4F形質導入後4日目に誘導された異なるmiRクラスターのシード領域比較を示している。類似したシード領域に下線を施している。図2cは、マイクロRNAの誘導についてのグラフを示している。代表的なマイクロRNAは4種の因子の種々の組合せを用いて誘導され得る。形質導入後4日後にマイクロRNA発現を定量する。4F、OSK、OSおよび単一因子を用いて、miR発現の変化にどの因子が関与したかを解析する。
【図3】miR−93およびmiR−106bによるiPSCの誘導増強を示す図である。図3aは、初期化アッセイの時系列を示す絵画表現である。マイクロRNAミミックを0日目および5日目に終濃度50nMでトランスフェクトする。4F誘導については11日目に、OSK 3因子のiPSC誘導については15〜20日目にGFP+コロニーを定量する。図3bは、miR−93およびmiR−106bミミックが4F誘導に関するiPSC誘導を増強することを示すグラフである。Oct4−GFP MEFを50nMの表示マイクロRNAでトランスフェクトする。形質導入後11日目にGFP+コロニーを定量する。誘導倍率およびエラーバーは、3反復のウェルを用いた3つの独立した実験から計算した。図3cは、OSK系を用いたmiR−93およびmiR−106bの増強効果の識別を示すグラフである。4F実験の場合と同様に、マイクロRNAミミックをトランスフェクトする。15〜20日目にGFP+コロニーを定量する。エラーバーは、3反復のウェルでの3つの独立した実験の標準偏差を表す。図3dは、初期化効率に対するマイクロRNA阻害の影響を示すグラフである。miR−93およびmiR−106bの阻害剤は初期化効率を劇的に低下させる。マイクロRNA阻害剤もまた終濃度50nMでトランスフェクトし、miRミミックトランスフェクションと同じ実験時系列を維持する。エラーバーは、3反復のウェルでの3つの独立した実験の標準偏差を表す。
【図4】異なる内因性ESマーカーのRT−PCRによる発現を解析するmiRミミック実験から得られたiPSCクローンの特性評価を示す図である。継代後3日目にiPS細胞株からトータルRNAを単離する。ES細胞特異的マーカー(Eras、ECat I、Nanogおよび内因性Oct4など)の発現をRT−PCRにより解析する。
【図5】miR−93およびmiR−106bによるマウスp21およびTgfbr2のターゲッティングを示す図である。図5aは、miR−93および106bトランスフェクションによりp21タンパク質レベルが減少するということを示している。Oct4−GFP MEFを、50nMのmiRミミックでトランスフェクトし、ウエスタン解析のためにトランスフェクションの48時間後に採取する。ローディング対照としてアクチンを用いる。図5bは、p21がsiRNAにより効率的にノックダウンされるということを示している。P21 siRNAトランスフェクトMEFおよび対照トランスフェクトMEFを、48時間およびRT−qPCRの時点で採取し、ウエスタンブロッティングを行って、p21発現を確認する。p21 mRNAをGAPDHに対して正規化する。図5cは、siRNAによるp21のノックダウンによりiPSC誘導が増強されるということを示している。MEFを4Fウイルスに感染させ、マイクロRNAミミックトランスフェクションと同じ時系列に従ってsiRNAをトランスフェクトする。GFP+コロニーを11日目に定量する。エラーバーは、3反復のウェルを用いた少なくとも2つの独立した実験を表す。図5dは、miR−93および106bトランスフェクションによりTgfbr2発現が減少するということを示している。ウエスタンブロッティングのためにトランスフェクト細胞を48時間の時点で採取する。図5eは、siRNAによりTgfbr2がノックダウンされるということを示している。相対的Tgfbr2 mRNAレベルをGapdhのものに対して正規化する。図5fは、siRNAによるTgfbr2のノックダウンによりiPSC誘導が増強されるということを示している。エラーバーは、3反復のウェルを用いた少なくとも3つの独立した実験を表す。
【図6】マイクロRNAによる初期化の促進を示す図である。図6aは、miR−17およびmiR−106aにより初期化効率を高めることができるが、miR−16では初期化効率を高めることができないということを示している。miR−17およびmiR−106aミミックをMEFに終濃度50nMでトランスフェクトする。形質導入後11日目にGFP+コロニーを定量する。エラーバーは、3反復のウェルでの2つの独立した実験を表す。図6bは、miR−17および106aがp21をターゲッティングするということを示している。マイクロRNAミミックのMEFへのトランスフェクションの2日後にp21のウエスタンブロッティングを行う。miR−17およびmiR−106aはTgfbr2発現をターゲッティングする。マイクロRNAミミックをMEFに50nMの終濃度でトランスフェクトする。図6cは、miR−17および106aがTgfbr2をターゲッティングするということを示している。トランスフェクションの2日後にウエスタンブロッティングを行う。図6dは、iPSC誘導中のマイクロRNAの役割についてのモデルを示している。初期化の初期段階中に、miR−17、25および106aクラスターを含むいくつかのマイクロRNAが誘導される。これらのマイクロRNAは、初期化プロセスに拮抗する因子(p21および他の未同定タンパク質など)をターゲッティングすることにより完全初期化を促進する。上昇および下降は、初期化プロセス中の種々の潜在的な段階および障壁を表し、破線は、初期化された細胞におけるマイクロRNA誘導により低下した初期化の障壁を示す。
【図7】マウスiPSC誘導におけるmiR−93およびmiR−106bの用量応答を示すグラフである。Oct4−GFP MEFを異なる濃度(5nM、15nMおよび50nM)のマイクロRNAでトランスフェクトする。ミミック対照siRNAを対照として用いる。形質導入後11日目にGFP+コロニーを定量する。データは、12ウェルプレートでの3反復のウェルを表す。
【図8】iPSC誘導中に誘導されたp21発現を示す図である。図8aは、異なるp21発現系(左から右へ:OSKM、OSK、OS、Klf4、cMycおよびMEF対照)を用いたウエスタンブロット解析を示している。p21発現はKlf4およびcMycにより誘導される。ウエスタンブロッティング解析のために形質導入後5日目に4F、OSK、OS、Klf4およびcMycに感染させたMEFを採取する。図8bは、感染したMEFにおける異なる導入遺伝子の発現の確認を示すグラフを示している。
【図9】OSK 3因子を用いた初期化のp21過剰発現による阻害を示す図である。図9aは、OSK誘導およびp21過剰発現によるiPSCのAP+コロニーの定量のグラフである。21日目に誘導された細胞をアルカリ性ホスファターゼについて染色する。p21ウイルスは、OSKと同時に導入する。図9bは、OSK誘導およびp21過剰発現によるiPSCのGFP+コロニーの定量のグラフである。
【図10】miRNAによるp21発現の直接調節を示す図である。図10aは、p21 mRNA 3'UTRに見出される2つの潜在的部位を示す絵画表現である。突然変異を第1の部位(保存部位)に導入して、miR−93および106bの結合親和性を破壊する。図10bは、Hela細胞におけるpGL3−p21ルシフェラーゼレポーター発現の定量を示すグラフである。Hela細胞を、pGL3−p21およびpRL−TKと、マイクロRNAとで48時間トランスフェクトした後採取する。結果を、トランスフェクト細胞におけるpRL−TKレベルに対して正規化する。
【図11】miRNAによるTgfbr2発現の直接調節を示す図である。図11aは、Tgfbr2 mRNA 3'UTRに見出される2つの潜在的部位を示す絵画表現である。図11bは、p21実験と同様に実施した、Hela細胞におけるルシフェラーゼレポーター発現の定量を示すグラフである。結果を、トランスフェクト細胞におけるpRL−TKレベルに対して正規化する。
【図12】表示した様々なmiRNAの存在下での相対的Tgfbr2 mRNAレベルを示す図である。
【図13】shRNAがshAgo2感染MEFにおいて活発に発現されることを示す図である。図13aはshAgo2レベルを示し、図13bはshRNAレベルを示している。図13cは、Ago2感染MEFにおけるES特異的マーカーの発現を示している。
【図14】OSKM因子の形質導入後0日目、4日目、8日目および12日目の相対的miRNA発現を示す図である。
【図15】miR−93の相対的レベルに対するmiR−93ミミックの影響を示す図である。
【図16】図16aは、miR阻害剤が標的miR発現を減少させることができるということを示している。図16bは、初期化プロセスの種々の段階中のmiR阻害剤の影響をさらに示している。
【図17】miR−93またはmiR−106bが導入されるときの内因性Nanog遺伝子座のプロモーターメチル化レベルを示す図である。
【図18】図18aおよび図18bは、miR−93トランスフェクション後に著しく減少する遺伝子がiPSCにおいて低度に発現される遺伝子の3倍濃縮を示し得る一方で、miR−93トランスフェクション後に増加する遺伝子がそのような濃縮を示さないということを示している。
【図19】図19aは、mRNAアレイ解析またはRT−qPCR解析のいずれかを用いた、miR−93の導入後の相対的Tgfbr2 mRNAレベルを示している。図19bは、miR−25、miR−93またはmiR−106bの導入後の相対的mRNAレベルを示している。
【図20】MEFで濃縮されたマイクロRNA、miR−21およびmiR−29aの阻害によりiPS細胞初期化効率が高まることを示す図である。図20aは、MEFにおいてmiR−29a、miR−21およびlet7aが高度に発現されるということを示している。トータルRNAをOct4−EGFP MEFおよびマウスES細胞から単離し、ゲル電気泳動により分離する。表示miRNAに対して特異的な放射性標識プローブを用いて、シグナルを検出する。U6 snRNAをローディング対照とする。図20bは、miRNA阻害により初期化効率が高まるということを示している。Oct4−EGFP MEFにOSKMを形質導入する。形質導入後14日目に蛍光顕微鏡検査法によりGFP陽性コロニーを確認し、計数する。GFP+コロニー数を、anti miRターゲッティングしない対照処理の数に対して正規化し、変化倍率として報告する。エラーバーは、3つの独立した実験の標準偏差を表す。*p値<0.05。
【図21】c−Mycが初期化中の、MEFで濃縮されたmiRNAの主要なレプレッサーであることを示す図である。図21aは、初期化後5日目の選択miRNAのノーザン解析を示している。Oct4−EGFP MEFに、表示した、単一因子または様々な初期化因子組合せを形質導入する。1F、1種の因子;2F、2種の因子;3F、3種の因子;OSKM:Oct4、Sox2、Klf4およびc−Myc。U6をローディング対照RNAとして用いる。胚幹細胞(ES)のトータルRNAを、MEFおよび形質導入細胞に対する陰性対照とする。様々なプローブを用いて、右側に表示した特定miRNAを検出する。miR−291ブロッティングがES RNAの陽性対照である。 図21bは、様々な初期化因子の存在下でのmiRNA発現の定量的表現を示している。シグナル強度をU6 snRNAの強度に対して正規化する。発現比を、MEFにおける発現(これを100%に任意設定する)に対する各miRNAの発現割合として計算する。(パネルAの)様々なmiRNAを定量し、右側に表示する。 図21cは、OSKまたはOSKMによる初期化後の様々な時点におけるOct4−EGFP MEFにおける選択miRNAのリアルタイムRT−PCR解析を示している。リアルタイムRT−PCR解析のために形質導入後の表示した日(D)にRNAを単離する。シグナルをU6に対して正規化し、MEFにおいて発現されたmiRNA(これを100に任意設定する)に対する割合として示す。エラーバーは、2つの独立した実験の標準偏差を表す。
【図22】図22は、miR−21またはmiR−29aの阻害が、p53タンパク質レベルを低下させp85αおよびCDC42の経路をアップレギュレートすることによりiPS細胞初期化を促進することを示す図である。図22aは、様々なmiRNAの阻害後のp53、CDC42およびp85αの発現のウエスタン解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトする。5日後に細胞を採取し、解析する。図22bは、表示miR阻害剤の存在下でのタンパク質発現の定量的表現を示している。シグナル強度を、GAPDH強度に対して正規化し、対照(NT)細胞における発現(これを100に任意設定した)に対する割合として示す。エラーバーは、少なくとも3つの独立した実験の標準偏差を示す。*p値<0.05。 図22cは、様々なmiRNAの阻害およびOSKM形質導入の後のp53、CDC42およびp85αの発現のイムノブロット解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトする。5日後に細胞を採取し、イムノブロットにより解析する。(B)に記載のとおり、シグナル強度を正規化する。エラーバーは、少なくとも3つの独立した実験の標準偏差を示す。*p値<0.05。図22dは、miR−29aまたはp53の枯渇により初期化効率が高まるということを示している。4X104 Oct4−EGFP MEFを、表示siRNAおよびmiRNA阻害剤と、OSKM初期化因子とでトランスフェクトする。形質導入後12日目にGFP陽性細胞を計数する。エラーバーは、少なくとも3つの独立した実験の標準偏差を示す。*p値<0.05。
【図23】miR−21およびmiR−29aの枯渇が、ERK1/2経路をダウンレギュレートすることにより初期化効率を高めることを示す図である。図23aは、MEFにおける様々なmiRNAの阻害後のリン酸化ERK1/2およびトータルERK1/2のウエスタン解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトし、5日後に採取し、イムノブロットする。シグナル強度を、アクチンに対して正規化し、anti miR NT対照の発現に対する割合として示す。エラーバーは、3つの独立した実験の標準偏差を示す。*p値<0.05;**p値<0.005。図23bは、miR−21およびmiR−29aの枯渇によりSpry1タンパク質レベルが増加するということを示している。Spry1発現比のウエスタンブロット解析を示す。MEFを、表示した様々なmiRNA阻害剤でトランスフェクトする。ウエスタンブロット解析のためにトランスフェクション後5日目に細胞を採取する。シグナル強度を、アクチンに対して正規化し、図23aに記載したように示す。エラーバーは、3つの独立した実験の標準偏差を表す。*p値<0.05;**p値<0.005。 図23cは、ERK1/2またはGSK3βのノックダウン後の初期化効率における変化倍率を示している。4X104 Oct4−EGFP MEFを表示siRNAと、OSKMとでトランスフェクトする。2週間後にGFP陽性細胞を計数する。siNTでのトランスフェクションを初期化効率の対照とする。エラーバーは、3つの独立した実験の標準偏差を示す。**p値<0.005。図23dは、MEFにおける様々なmiRNAの阻害後のリン酸化GSK−3βおよびトータルGSK−3βのウエスタン解析を示している。1×105 Oct4−EGFP MEFを表示miRNA阻害剤でトランスフェクトし、5日後に採取し、イムノブロットにより解析する。図23aに記載したように、シグナル強度を正規化する。エラーバーは、3つの独立した実験の標準偏差を示す。
【図24】c−Mycが、MEFで濃縮されたmiRNA、miR−21およびmiR−29aをダウンレギュレートすることにより初期化を促進することを示す模式図を示す図である。p53およびERK1/2の経路は初期化の障壁として働き、miR−21およびmiR−29aは、CDC42、p85αおよびSpry1をダウンレギュレートすることによりそれらの経路を間接的に活性化する。miR−21/p53経路とmiR−29a/ERK1/2経路との間のクロストークも示す。c−Mycは、これらのmiRNAの発現を抑制し、次にはERK1/2およびp53の誘導に支障をきたす。点線はiPS初期化に対するp53およびERK1/2の作用を示す。
【図25】miR−21の阻害が、OSKによるiPS細胞初期化を促進することを示す図である。miRNAの阻害剤は、OSKでの初期化中にOct4−MEFに導入される。形質導入後の様々な時点においてGFP陽性コロニーを計数する。エラーバーは、2つの独立した実験の標準偏差を表す。
【図26】miRNAの阻害が、初期化中にアポトーシスまたは増殖速度を変更しないことを示す図である。図26aは、miRNAの阻害剤が、OSKMでの初期化中にOct4−MEFに導入されるということを示している。形質導入後8〜9日目に細胞を収集する。PE Annexin V Apoptosis Detection Kit I(BD Pharmingen;カタログ番号559763)および7−アミノ−アクチノマイシン(7−AAD)を用いてアポトーシスを評価する。シグナルをFACSにより検出する。エラーバーは、3つの独立した実験の標準偏差を表す。図26bは、miRNA阻害剤が、OSKMでの初期化中にOct4−MEFに導入されるということを示している。形質導入後8〜9日目に細胞を収集する。収集の1日前に、Click−iT Edu Imaging Kits(Invitrogen;カタログ番号C10337)を用いて5−エチニル−2'−デオキシウリジン(Edu)で細胞を処理する。シグナルをFACSにより検出する。エラーバーは、3つの独立した実験の標準偏差を表す。
【発明を実施するための形態】
【0029】
本発明は、iPSC誘導に関与する重要な調節機構の発見に基づくものである。重要な態様は、iPSCの誘導に関する細胞マイクロRNA間の関連性の発見である。これは、重要なマイクロRNA経路タンパク質のノックダウンによるマイクロRNA生合成機構への干渉により初期化効率の著しい低下がもたらされ得るという観察結果から明らかである。特に、初期化の初期段階中に高度に誘導される、少なくとも3種のマイクロRNAクラスターが明らかとなっている、miR−17〜92、106b〜25および106a〜363。非常に類似したシード領域を有するいくつかのマイクロRNA(miR−93およびmiR−106bなど)は、p21発現をターゲッティングすることによりiPSC誘導を大幅に増強し、得られたクローンを完全初期化状態に到達させ得る。
【0030】
本発明によれば、マイクロRNAはiPSC誘導において直接働くことができ、マイクロRNA生合成機構への干渉により初期化効率が著しく低下することとなる。本発明は、初期化の初期段階中に高度に誘導される3種のマイクロRNAクラスター、miR−17〜92、miR−106b〜25およびmiR−106a〜363を提供する。機能解析では、これらのマイクロRNAをMEFに導入することによりOct4−GFP+iPSCコロニー形成が促進されることが示された。また、本発明によれば、Tgfbr2およびp21(これらは両方とも初期化を阻害する)はこれらのマイクロRNAにより直接ターゲッティングされ、それらの活性の遮断により初期化効率が著しく低下することとなる。本発明によれば、miR−93およびmiR−106bは初期化活性の重要なレギュレーターであることが提案される。
【0031】
本組成物および方法を記載する前に、記載する特定の組成物、方法および実験条件は変化し得ることから本発明はそのような組成物、方法および条件に限定されないことを理解すべきである。また、本発明の範囲は添付の特許請求の範囲によってのみ限定されるため、本明細書において用いられる用語は特定の実施形態を記載することを目的とするものにすぎず、限定するものではないことも理解すべきである。
【0032】
本明細書および添付の特許請求の範囲において用いられるように、単数形「a」、「an」および「the」は、特に明示されない限り、複数の参照語を包含する。従って、例えば、「方法」について述べる場合は、1以上の方法および/または、この開示内容などを読むことによって当業者には明らかとなるであろう、本明細書に記載した種類の工程も包含する。
【0033】
特に定義されない限り、本明細書において用いられる全ての技術用語および科学用語は、本発明が属する分野の通常の技術を有する人に一般に理解されている意味と同じ意味を有する。本発明の実施または試験において、本明細書に記載したものと類似または同等の方法および材料を使用することができるが、好ましい方法および材料を次に記載する。
【0034】
本明細書において論じるように、マイクロRNAが初期化プロセスおよびiPSC誘導効率に関与するという発見によって、それらの細胞中のこれらのマイクロRNAのレベルを操作することによりiPSC誘導効率を大幅に高める能力に至っている。よって、本発明は、既知方法と比べて誘導効率が改善されたiPS細胞生成方法を提供する。その方法は、体細胞を核初期化因子と、その細胞内でマイクロRNAレベルまたは活性を変更する作用物質とに接触させること(ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することを含む。
【0035】
本発明はまた、初期化プロセスおよびiPSC誘導効率に直接関与する調節タンパク質の発見にも基づく。そのような1つのタンパク質はp21(165アミノ酸しか含まない低分子タンパク質)であり、このタンパク質は、p53依存性G1増殖停止を引き起こし分化および細胞老化を促進することにより癌発生中の腫瘍抑制遺伝子として長年にわたり知られていた。iPSC誘導中のマイクロRNAによるp21発現阻害はこの点で誘導効率を高めることが証明された。よって、1つの実施形態では、本発明は、体細胞を核初期化因子と、p21発現または活性の阻害剤とに接触させることによるiPS細胞生成方法を提供する。
【0036】
iPSC生成へのRNAの調節関与を前提として、核初期化因子以外の、RNAレベルを調節する作用物質を用いて誘導が起こり得ると考えられる。よって、本発明は、体細胞を、その細胞内でRNAレベルまたは活性を変更する作用物質と接触させ(ここで、該作用物質は該体細胞中で多能性を誘導し、ただし、該作用物質は核初期化因子ではない)、それによってiPS細胞を生成することにより誘導多能性幹(iPS)細胞を生成する方法を提供する。様々な実施形態では、RNAは非コードRNA(ncRNA)、例えば、マイクロRNAなどである。
【0037】
様々な実施形態では、1種以上の核初期化因子を用いて、卵子、胚またはES細胞を使用せずに分化した細胞の初期化を誘導することができる。誘導プロセスの効率は、誘導プロセス中にその細胞内でマイクロRNAレベルまたは活性を変更する作用物質を利用することにより高められる。その方法を用いて、ES細胞と類似した多能性と増殖能を有する誘導多能性幹細胞を便宜にかつ高度に再現性良く樹立し得る。例えば、核初期化因子は、RNA分子(マイクロRNAなど)をコードするポリヌクレオチドを含む組換えベクターとともに、核初期化因子をコードする遺伝子を含む組換えベクターを細胞に形質導入することにより、細胞に導入され得る。よって、その細胞は、組換えベクター中に含まれる遺伝子の産物として発現される核初期化因子を発現することができ、同様に組換えベクター中に含まれるポリヌクレオチドの産物として発現されるマイクロRNAを発現し、それによって核初期化因子だけを使用する場合と比べて高い効率(割合)で分化した細胞の初期化を誘導する。
【0038】
本明細書において用いられるように、多能性細胞とは、in vitroで長期間(1年より長い期間)分裂する可能性があり、内胚葉、中胚葉および外胚葉を含む3つの胚葉全てから得られる細胞に分化する独特の能力を有する細胞を包含する。
【0039】
本発明で用いる体細胞は初代細胞または不死化細胞であってよい。そのような細胞は、初代細胞(非不死化細胞)(動物から新たに単離したものなど)であってよく、または細胞株(不死化細胞)から得てもよい。例示的態様では、体細胞は、例えば、ヒト細胞またはマウス細胞などの哺乳類細胞である。体細胞は、周知の方法により、種々の器官(限定されるものではないが、皮膚、肺、膵臓、肝臓、胃、腸、心臓、生殖器、膀胱、腎臓、尿道および他の泌尿器など)から、または一般的には生存体細胞を含有する任意の器官または組織から得てもよい。本発明において有用な哺乳類体細胞には、例として、成体幹細胞、セルトリ細胞、内皮細胞、顆粒膜上皮細胞、ニューロン、膵島細胞、表皮細胞、上皮細胞、肝細胞、毛包細胞、ケラチノサイト、造血細胞、メラノサイト、軟骨細胞、リンパ球(Bリンパ球およびTリンパ球)、赤血球、マクロファージ、単球、単核細胞、繊維芽細胞、心筋細胞、他の公知の筋肉細胞、一般的には任意の生存体細胞が挙げられる。特定の実施形態では、繊維芽細胞が用いられる。本明細書において用いられる体細胞という用語はまた、成体幹細胞を含むようにも意図されている。成体幹細胞は、特定組織のあらゆる細胞種を生じさせることが可能な細胞である。成体幹細胞の例としては、造血幹細胞、神経幹細胞および間葉系幹細胞が挙げられる。
【0040】
本明細書において用いられるように、初期化とは、一部分化したまたは最終分化した体細胞の分化状態を変更するまたは後退させるプロセスを指すよう意図されている。体細胞の初期化は、体細胞の分化状態の一部後退または完全後退であってよい。例示的態様では、初期化は完全なものであり、この場合、体細胞は誘導多能性幹細胞に初期化される。しかしながら、初期化は、一部であってもよい(低分化状態への後退など)。例えば、最終分化した細胞の低分化状態の細胞(多分化性細胞など)への後退。
【0041】
本発明の様々な態様では、核初期化因子は、多能性を誘導する遺伝子であり、分化した細胞または半分化した細胞を、最初の細胞より原始的な表現型(多能性幹細胞の表現型など)に初期化するために利用される。そのような遺伝子は、細胞中でマイクロRNAレベルまたは活性を変更するおよび/またはp21の発現または活性を阻害して誘導効率を高める作用物質とともに利用される。そのような遺伝子および作用物質は、体細胞のゲノムに組み込まれた1種以上の該遺伝子の発現により、体細胞からの多能性幹細胞の生成が可能である。本明細書において用いられるように、多能性を誘導する遺伝子とは、多能性と関係がある遺伝子であって、該遺伝子の組込みおよび発現により体細胞からの低分化細胞(多能性幹細胞など)の生成が可能である遺伝子を指すよう意図されている。多能性遺伝子の発現は、典型的には多能性幹細胞に限定され、多能性幹細胞の機能的同一性に不可欠である。
【0042】
細胞中でマイクロRNAのレベルまたは活性を変更するまたはp21の発現または活性を阻害する作用物質には、様々な異なる種類の分子が含まれることは当業者ならば理解するであろう。本発明の方法のいずれかにおいて有用な作用物質は、あらゆる種類の分子、例えば、ポリヌクレオチド、ペプチド、ペプチドミメティック、ペプトイド(ビニローグペプトイド(vinylogous peptoids)など)、化学化合物(有機分子または小有機分子など)などであり得る。よって、一態様では、本発明の方法に用いる作用物質は、ポリヌクレオチド(アンチセンスオリゴヌクレオチドまたはRNA分子など)である。様々な態様では、作用物質は、ポリヌクレオチド(アンチセンスオリゴヌクレオチドまたはRNA分子(マイクロRNA、dsRNA、siRNA、stRNAおよびshRNAなど)など)であってよい。例示的な態様では、作用物質は、細胞に導入されるマイクロRNAであり、その導入の結果、その細胞中でマイクロRNAのレベルおよび活性が増加しかつ/またはp21が阻害される。
【0043】
マイクロRNA(miRNA)は、遺伝子発現を調節する一本鎖RNA分子である。miRNAは、遺伝子によりコードされておりそのDNAから転写されるがmiRNAはタンパク質へ翻訳されない;その代わりに一次転写物(pri−miRNA)は各々、pre−miRNAと呼ばれる短いステムループ構造にプロセシングされ、最終的には機能性miRNAにプロセシングされる。成熟したmiRNA分子は1以上のメッセンジャーRNA(mRNA)分子と完全にまたは部分的に相補的であり、それらの主な機能は遺伝子発現をダウンレギュレートすることである。マイクロRNAは、独立した遺伝子によりコードされ得るが、また、様々な異なるRNA種(イントロン、mRNAの3'UTR、長い非コードRNA、snoRNAおよびトランスポゾンを含む)から(酵素Dicerによって)プロセシングを受け得る。本明細書において用いられるように、マイクロRNAとは、「ミミック」マイクロRNAも包含し、「ミミック」マイクロRNAは、それらの内因性対応物と同じまたは実質的に同じ機能を有する、細胞に外から導入されるマイクロRNAを意味するよう意図されている。よって、当業者ならば作用物質が外から導入されるRNAであってよいことを理解する上で、作用物質はまた、細胞中でマイクロRNAの発現を増加または減少させる化合物なども包含する。
【0044】
様々な態様では、マイクロRNAは、iPSCの誘導またはその分化中に活性または発現の増加または減少を示すクラスター中に含まれるマイクロRNAであってよい。一態様では、マイクロRNAは、miR−17、miR−25、miR−106aまたはmiR−302bクラスター中の1種以上のマイクロRNA(miR−93、miR−106b、またはそれらの任意の組合せなど)であってよい。miR−17〜92、miR−106b〜25およびmiR−106a〜363クラスターの誘導は、正確な初期化に重要であることが示されている。そのプロセス中にそのようなマイクロRNAは初期化障壁を低くするように思われるため、それらの細胞中のこれらのマイクロRNAのレベルを操作して、初期化効率を改善し得る。マイクロRNAは重要な調節分子であることが示されていることから、また、マイクロRNAを操作して、iPSCの分化を指示し得る。
【0045】
3種のマイクロRNAクラスターは、iPSC誘導中に誘導されることがここで確認され、これらのクラスター内のいくつかのマイクロRNAは、同じヌクレオチドシード領域配列を有すると判明しており、それらは類似したmRNAをターゲッティングすることが示された。また、同じシード領域のヌクレオチド配列を共有するそのようなマイクロRNAはiPSC誘導を増強する一方で、p21発現を減少させることも判明している。よって、一態様では、マイクロRNAは、様々なマイクロRNA(例えば、配列番号2〜11のもの)の間で保存されると判明している、配列番号1を含むポリヌクレオチド配列、5'−AAGUGC−3'を有する。よって、関連態様では、マイクロRNAは、配列番号2〜11のいずれかのヌクレオチド配列を有する。
【0046】
「低分子干渉RNA」および「siRNA」という用語はまた、様々な生物学的役割を果たす短鎖二本鎖RNA分子の種類である、短鎖干渉RNAまたはサイレンシングRNAを指すように、本明細書において用いられる。とりわけ、siRNAはRNA干渉(RNAi)経路に関与し、そこでsiRNAは特異的遺伝子の発現に干渉する。RNAi経路におけるそれらの役割に加え、siRNAはRNAi関連経路においても(例えば、抗ウイルス機構としてまたはゲノムのクロマチン構造形成において)作用する。
【0047】
本発明のポリヌクレオチド(アンチセンスオリゴヌクレオチドおよびRNA分子など)は任意の好適な長さのものであってよい。例えば、遺伝子発現を調節するためにアンチセンスオリゴヌクレオチドまたはRNA分子を使用するにはどのような長さが好適であるかは当業者ならば理解するであろう。そのような分子は、典型的には、約5〜100ヌクレオチド長、5〜50ヌクレオチド長、5〜45ヌクレオチド長、5〜40ヌクレオチド長、5〜35ヌクレオチド長、5〜30ヌクレオチド長、5〜25ヌクレオチド長、5〜20ヌクレオチド長または10〜20ヌクレオチド長である。例えば、その分子は、約5ヌクレオチド長、10ヌクレオチド長、15ヌクレオチド長、16ヌクレオチド長、17ヌクレオチド長、18ヌクレオチド長、19ヌクレオチド長、20ヌクレオチド長、21ヌクレオチド長、22ヌクレオチド長、23ヌクレオチド長、24ヌクレオチド長、25ヌクレオチド長、26ヌクレオチド長、27ヌクレオチド長、28ヌクレオチド長、29ヌクレオチド長、30ヌクレオチド長、31ヌクレオチド長、32ヌクレオチド長、33ヌクレオチド長、34ヌクレオチド長、35ヌクレオチド長、40ヌクレオチド長、45ヌクレオチド長または50ヌクレオチド長であってよい。そのようなポリヌクレオチドは、少なくとも約15ヌクレオチド〜約120を超えるヌクレオチドを含んでよい(少なくとも約16ヌクレオチド、少なくとも約17ヌクレオチド、少なくとも約18ヌクレオチド、少なくとも約19ヌクレオチド、少なくとも約20ヌクレオチド、少なくとも約21ヌクレオチド、少なくとも約22ヌクレオチド、少なくとも約23ヌクレオチド、少なくとも約24ヌクレオチド、少なくとも約25ヌクレオチド、少なくとも約26ヌクレオチド、少なくとも約27ヌクレオチド、少なくとも約28ヌクレオチド、少なくとも約29ヌクレオチド、少なくとも約30ヌクレオチド、少なくとも約35ヌクレオチド、少なくとも約40ヌクレオチド、少なくとも約45ヌクレオチド、少なくとも約50ヌクレオチド、少なくとも約55ヌクレオチド、少なくとも約60ヌクレオチド、少なくとも約65ヌクレオチド、少なくとも約70ヌクレオチド、少なくとも約75ヌクレオチド、少なくとも約80ヌクレオチド、少なくとも約85ヌクレオチド、少なくとも約90ヌクレオチド、少なくとも約95ヌクレオチド、少なくとも約100ヌクレオチド、少なくとも約110ヌクレオチド、少なくとも約120ヌクレオチドまたは120を超えるヌクレオチドを含む)。
【0048】
「ポリヌクレオチド」または「ヌクレオチド配列」または「核酸分子」という用語は、ホスホジエステル結合によって相互に連結される2種以上のデオキシリボヌクレオチドまたはリボヌクレオチドの配列を意味するように、本明細書において広く用いられる。そのようなものとして、それらの用語は、RNAおよびDNA(遺伝子またはその部分、cDNA、合成ポリデオキシリボ核酸配列などであってよく、かつ一本鎖または二本鎖であってよい)、ならびにDNA/RNAハイブリッドを包含する。さらに、本明細書において用いられるそれらの用語は、細胞から単離され得る天然に存在する核酸分子、ならびに例えば、化学合成法によりまたは酵素法により(ポリメラーゼ連鎖反応(PCR)によるなど)調製され得る合成ポリヌクレオチドを包含する。例えば、組成物の異なる成分を区別するために、議論の便宜上異なる用語を用いているにすぎないことを認識すべきである。
【0049】
一般的に、ポリヌクレオチドを含むヌクレオチドは、天然に存在するデオキシリボヌクレオチド(2'−デオキシリボースと連結されたアデニン、シトシン、グアニンまたはチミンなど)またはリボヌクレオチド(リボースと連結されたアデニン、シトシン、グアニンまたはウラシルなど)である。しかしながら、使用に応じて、ポリヌクレオチドは、天然に存在しない合成ヌクレオチドまたは修飾された天然に存在するヌクレオチドを含むヌクレオチド類似体も含み得る。ヌクレオチド類似体は、当技術分野で周知であり、市販されており、そのようなヌクレオチド類似体を含有するポリヌクレオチドと同様である。ポリヌクレオチドのヌクレオチドを連結している共有結合は、一般的にはホスホジエステル結合である。しかしながら、ポリヌクレオチドを使用する目的に応じて、その共有結合は多数の他の結合のいずれかであってもよく、チオジエステル結合、ホスホロチオエート結合、ペプチド様結合または合成ポリヌクレオチドを生産するためのヌクレオチドの連結に有用な、当業者に公知の任意の他の結合が含まれる。
【0050】
天然に存在するヌクレオチドおよびホスホジエステル結合を含むポリヌクレオチドまたはオリゴヌクレオチドは、化学合成することができ、または鋳型として適当なポリヌクレオチドを用い、組換えDNA法を用いて生産することができる。それに対し、ヌクレオチド類似体またはホスホジエステル結合以外の共有結合を含むポリヌクレオチドは、一般的に化学合成されるが、T7ポリメラーゼなどの酵素によりある特定の種類のヌクレオチド類似体をポリヌクレオチドに組み込むことができるため、そのような酵素を用いて、適当な鋳型から組換えによりそのようなポリヌクレオチドを生産することができる。
【0051】
様々な実施形態では、アンチセンスオリゴヌクレオチドまたはRNA分子には、修飾を含むオリゴヌクレオチドが含まれる。様々な修飾が当技術分野で公知であり、本発明における使用のために考えられる。例えば、修飾主鎖または非天然ヌクレオシド間結合を含むオリゴヌクレオチドが考えられる。本明細書において用いられるように、修飾主鎖を有するオリゴヌクレオチドとは、主鎖中にリン原子を有するものおよび主鎖中にリン原子がないものを包含する。本明細書の目的において、当技術分野において言及されることがあるように、ヌクレオシド間主鎖中にリン原子がない修飾オリゴヌクレオチドもまた、オリゴヌクレオシドであると考えることができる。
【0052】
様々な態様では、修飾オリゴヌクレオチド主鎖としては、例えば、ホスホロチオエート、キラルホスホロチオエート、ホスホロジチオエート、ホスホトリエステル、アミノアルキルホスホトリエステル、メチルホスホネートおよび他のアルキルホスホネート(3'−アルキレンホスホネート、5'−アルキレンホスホネートおよびキラルホスホネートを含む)、ホスフィネート、ホスホルアミダート(3'−アミノホスホルアミダートおよびアミノアルキルホスホルアミダートを含む)、チオノホスホルアミダート、チオノアルキルホスホネート、チオノアルキルホスホトリエステル、通常の3'−5'結合を有するセレノホスフェートおよびボラノホスフェート、これらの2'−5'結合した類似体、および1以上のヌクレオチド間結合が3'−3'結合、5'−5'結合または2'−2'結合である逆の極性を有するものが挙げられる。逆の極性を有するある特定のオリゴヌクレオチドは、最も3'側のヌクレオチド間結合に単一の3'−3'結合、すなわち、脱塩基であってよい(核酸塩基が欠けているかまたはその代わりにヒドロキシル基を有する)単一の逆のヌクレオシド残基を含む。様々な塩、混合塩および遊離酸形も含まれる。
【0053】
様々な態様では、内部にリン原子を含まない修飾オリゴヌクレオチド主鎖は、短鎖アルキルまたはシクロアルキルヌクレオシド間結合、混合ヘテロ原子およびアルキルまたはシクロアルキルヌクレオシド間結合、または1以上の短鎖ヘテロ原子ヌクレオシド間結合または複素環式ヌクレオシド間結合によって形成される主鎖を有する。これらには、モルホリノ結合(ヌクレオシドの糖部分から一部形成される);シロキサン主鎖;スルフィド、スルホキシドおよびスルホン主鎖;ホルムアセチルおよびチオホルムアセチル主鎖;メチレンホルムアセチルおよびメチレンチオホルムアセチル主鎖;リボアセチル主鎖;アルケン含有主鎖;スルファメート主鎖;メチレンイミノおよびメチレンヒドラジノ主鎖;スルホネートおよびスルホンアミド主鎖;アミド主鎖;および混合N、O、SおよびCH2成分を有する他のものを有するものが含まれる。
【0054】
様々な態様では、オリゴヌクレオチドミメティック、糖およびヌクレオシド間結合の両方、すなわち、ヌクレオチド単位の主鎖は、新規な基に置換されている。塩基単位は、適当な核酸標的化合物とのハイブリダイゼーションのために維持される。そのような1つのオリゴマー化合物、優れたハイブリダイゼーション特性を示すことが示されたオリゴヌクレオチドミメティックは、ペプチド核酸(PNA)と呼ばれる。PNA化合物では、オリゴヌクレオチドの糖−主鎖は、アミド含有主鎖、特に、アミノエチルグリシン主鎖に置換されている。核酸塩基は、保持され、主鎖のアミド部分のアザ窒素原子に直接または間接的に結合している。様々な態様では、オリゴヌクレオチドはホスホロチオエート主鎖およびヘテロ原子主鎖を含むオリゴヌクレオシドを含み得る。修飾オリゴヌクレオチドはまた、1以上の置換された糖部分も含み得る。いくつかの実施形態では、オリゴヌクレオチドは、2'位に次のものの1つを含む:OH;F;O−、S−、もしくはN−アルキル;O−、S−、もしくはN−アルケニル;O−、S−もしくはN−アルキニル;またはO−アルキル−O−アルキル(ここで、該アルキル、アルケニルおよびアルキニルは、置換されているかまたは非置換のC1〜C10アルキルまたはC2〜C10アルケニルおよびアルキニルであってよい)。特に好ましいのは、O[(CH2)nO]mCH3、O(CH2)nOCH3、O(CH2)nNH2、O(CH2)nCH3、O(CH2)nONH2およびO(CH2)nON[(CH2)nCH3]]2(ここで、nおよびmは1〜約10である)である。他の好ましいオリゴヌクレオチドは、2'位に次のものの1つを含む:C1〜C10低級アルキル、置換低級アルキル、アルケニル、アルキニル、アルカリル、アラルキル、O−アルカリルもしくはO−アラルキル、SH、SCH3、OCN、Cl、Br、CN、CF3、OCF3、SOCH3、SO2CH3、ONO2、NO2、N3、NH2、ヘテロシクロアルキル、ヘテロシクロアルカリル、アミノアルキルアミノ、ポリアルキルアミノ、置換シリル、RNA切断基、レポーター基、インターカレーター、オリゴヌクレオチドの薬物動態特性を向上させるための基、またはオリゴヌクレオチドの薬力学的特性を向上させるための基、および類似した特性を有する他の置換基。別の修飾としては、2'−メトキシエトキシ(2'OCH2CH2OCH3、2'−O−(2−メトキシエチル)または2'−MOEとしても知られる)が挙げられる。
【0055】
関連態様では、本発明は、標的ポリヌクレオチドに対する親和性および特異性が増強されたアンチセンス核酸を生成するためのロックド核酸(LNA)の使用を含む。LNAは、2'−ヒドロキシル基が糖環の3'または4'炭素原子と結合し、それによって二環式糖部分を形成する核酸である。その結合は、2'酸素原子と4'炭素原子を架橋するメチレン (methelyne)(−CH2−)n基(ここで、nは1または2である)であることが好ましい。
【0056】
他の修飾としては、2'−メトキシ(2'−O−CH3)、2'−アミノプロポキシ(2'−OCH2CH2CH2NH2)、2'−アリル(2'−CH−CH−CH2)、2'−O−アリル(2'−O−CH2−CH−CH2)、2'−フルオロ(2'−F)、2'−アミノ、2'−チオ、2'−Oメチル、2'−メトキシメチル、2'−プロピルなどが挙げられる。2'−修飾は、アラビノ(上)位またはリボ(下)位に存在してよい。好ましい2'−アラビノ修飾は2'−Fである。また、類似した修飾も、オリゴヌクレオチドの他の位置で、特に、3'末端ヌクレオチド上または2'−5'結合オリゴヌクレオチド中の糖の3'位および5'末端ヌクレオチドの5'位で行ってよい。オリゴヌクレオチドはまた、ペントフラノシル糖の代わりにシクロブチル部分などの糖ミメティックも有していてよい。
【0057】
オリゴヌクレオチドはまた、核酸塩基の修飾または置換も含み得る。本明細書において用いられるように、「非修飾」または「天然」核酸塩基としては、プリン塩基であるアデニン(A)およびグアニン(G)、ならびにピリミジン塩基であるチミン(T)、シトシン(C)およびウラシル(U)が挙げられる。修飾された核酸塩基としては、他の合成および天然核酸塩基、例えば、5−メチルシトシン、5−ヒドロキシメチルシトシン、キサンチン、ヒポキサンチン、2−アミノアデニン、アデニンおよびグアニンの6−メチル誘導体および他のアルキル誘導体、アデニンおよびグアニンの2−プロピル誘導体および他のアルキル誘導体、2−チオウラシル、2−チオチミンおよび2−チオシトシン、5−ハロウラシルおよび5−ハロシトシン、5−プロピニルウラシルおよび5−プロピニルシトシン、ならびにピリミジン塩基の他のアルキニル誘導体、6−アゾウラシル、6−アゾシトシンおよび6−アゾチミン、5−ウラシル(シュードウラシル)、4−チオウラシル、8−ハロ、8−アミノ、8−チオール、8−チオアルキル、8−ヒドロキシルおよび他の8−置換のアデニンおよびグアニン、5−ハロ、特に、5−ブロモ、5−トリフルオロメチルおよび他の5−置換のウラシルおよびシトシン、7−メチルグアニンおよび7−メチルアデニン、2−F−アデニン、2−アミノ−アデニン、8−アザグアニンおよび8−アザアデニン、7−デアザグアニンおよび7−デアザアデニン、ならびに3−デアザグアニンおよび3−デアザアデニンなどが挙げられる。さらなる修飾核酸塩基としては、三環式ピリミジン、例えば、フェノキサジンシチジン(1H−ピリミド[5,4−b][1,4]ベンゾオキサジン−2(3H)−オン)、フェノチアジンシチジン(1H−ピリミド[5,4−b][1,4]ベンゾチアジン−2(3H)−オン)、置換フェノキサジンシチジン(例えば、9−(2−アミノエトキシ)−H−ピリミド[5,4−b][1,4]ベンゾオキサジン−2(3H)−オン)などのGクランプ、カルバゾールシチジン(2H−ピリミド[4,5−b]インドール−2−オン)、ピリドインドールシチジン(H−ピリミド[3',2':4,5]ピロロ[2,3−d]ピリミジン−2−オン)などが挙げられる。修飾核酸塩基としては、プリン塩基またはピリミジン塩基が他の複素環、例えば、7−デアザ−アデニン、7−デアザグアノシン、2−アミノピリジンおよび2−ピリドンで置換されたものも挙げられ得る。さらなる核酸塩基は当技術分野で公知である。これらの核酸塩基のいくつかは、本明細書に記載したオリゴマー化合物の結合親和性を高めるために特に有用である。これらには、5−置換ピリミジン、6−アザピリミジンおよびN−2、N−6およびO−6置換プリン(2−アミノプロピルアデニンを含む)、5−プロピニルウラシルおよび5−プロピニルシトシンが含まれる。5−メチルシトシン置換は、0.6〜1.2Cにより核酸二本鎖安定性を高めることが示され、現在好ましい塩基置換であり、さらに特には、2'−O−メトキシエチル糖修飾と組み合わせられる場合である。
【0058】
本明細書に記載したアンチセンスオリゴヌクレオチドの別の修飾は、オリゴヌクレオチドの活性、細胞分布または細胞取込みを高めるオリゴヌクレオチドの1以上の部分またはコンジュゲートとの化学的結合を含む。アンチセンスオリゴヌクレオチドは、第一級または第二級ヒドロキシル基などの官能基と共有結合されたコンジュゲート群を含み得る。コンジュゲート群としては、インターカレーター、レポーター分子、ポリアミン、ポリアミド、ポリエチレングリコール、ポリエーテル、オリゴマーの薬力学的特性を増強する群、およびオリゴマーの薬物動態特性を増強する群が挙げられる。典型的なコンジュゲート群としては、コレステロール、脂質、リン脂質、ビオチン、フェナジン、葉酸、フェナントリジン、アントラキノン、アクリジン、フルオレセイン、ローダミン、クマリンおよび色素が挙げられる。薬力学的特性を増強する群とは、本発明の文脈において、オリゴマーの取込みを向上させ、オリゴマーの分解耐性を増強し、および/またはRNAとの配列特異的ハイブリダイゼーションを強化する群を包含する。薬物動態特性を増強する群とは、本発明の文脈において、オリゴマーの取込み、分布、代謝または排出を向上させる群を包含する。コンジュゲート部分としては、限定されるものではないが、コレステロール部分、コール酸、チオエーテル、例えば、ヘキシル−5−トリチルチオール、チオコレステロール、脂肪族鎖、例えば、ドデカンジオールまたはウンデシル残基、リン脂質、例えば、ジヘキサデシル−rac−グリセロールまたはトリエチルアンモニウム 1,2−ジ−O−ヘキサデシル−rac−グリセロ−3−H−ホスホネート、ポリアミンまたはポリエチレングリコール鎖、あるいはアダマンタン酢酸、パルミチル部分、あるいはオクタデシルアミンまたはヘキシルアミノカルボニルオキシコレステロール部分などの脂質部分が挙げられる。
【0059】
いくつかの遺伝子は、多能性と関係があり、初期化因子としての本発明での使用に好適であることが見出された。そのような遺伝子は当技術分野で公知であり、それらの例として、SOXファミリー遺伝子(SOX1、SOX2、SOX3、SOX15、SOX18)、KLFファミリー遺伝子(KLF1、KLF2、KLF4、KLF5)、MYCファミリー遺伝子(C−MYC、L−MYC、N−MYC)、SALL4、OCT4、NANOG、LIN28、STELLA、NOBOXまたはSTATファミリー遺伝子が挙げられる。STATファミリーメンバーには、例えば、STAT1、STAT2、STAT3、STAT4、STAT5(STAT5AおよびSTAT5B)、およびSTAT6が含まれ得る。場合によっては、多能性を誘導するために1種の遺伝子だけを使用することも可能であるが、一般的には、多能性を誘導するために2種以上の遺伝子の発現が必要である。例えば、2種、3種、4種またはそれ以上の遺伝子をポリシストロン性構築物として体細胞ゲノムに同時に組み込んで、そのような遺伝子の同時発現を可能にし得る。例示的態様では、多能性を誘導するために、OCT4、SOX2、KLF4およびC−MYCを含む4種の遺伝子が利用される。本発明での使用に好適な初期化因子として知られるさらなる遺伝子は、米国特許出願第10/997,146号および米国特許出願第12/289,873号において開示されており、これらの特許出願は引用することにより本明細書の一部とされる。
【0060】
これらの遺伝子の全ては、ヒトを含む哺乳類に一般に存在するため、任意の哺乳類由来の相同体も本発明において用いてよい(限定されるものではないが、マウス、ラット、ウシ、ヒツジ、ウマおよびサルを含む哺乳類から得られた遺伝子など)。さらに、野生型遺伝子産物に加えて、いくつかの(例えば、1〜10個、1〜6個、1〜4個、1〜3個および1個または2個)のアミノ酸の置換、挿入および/または欠失を含み野生型遺伝子産物と類似した機能を有する変異遺伝子産物も使用することができる。さらに、因子の組合せは野生型の遺伝子または遺伝子産物の使用に限定されない。例えば、野生型Mycの代わりにMycキメラまたは他のMyc変異体を使用することができる。
【0061】
本発明は、任意の特定の核初期化因子組合せに限定されない。本明細書において論じるように、核初期化因子は、1種以上の遺伝子産物を含んでよい。核初期化因子はまた、本明細書において論じるように、遺伝子産物の組合せも含んでよい。本明細書において開示するように、各核初期化因子を単独でまたは他の核初期化因子と組み合わせて用いてよい。さらに、本発明の核初期化因子は、スクリーニング方法により、例えば、米国特許出願第10/997,146号において論じられているように、同定することができ、この特許出願は引用することにより本明細書の一部とされる。加えて、本発明の核初期化因子は、分化、発生、増殖などに関連する1種以上の因子および他の生理学的活性を有する因子、ならびに核初期化因子として機能し得る他の遺伝子産物を含み得る。
【0062】
核初期化因子は、タンパク質またはペプチドを含んでよい。そのタンパク質は、本明細書において論じるように、遺伝子から生産されてよく、あるいは、そのタンパク質と別のタンパク質、ペプチドなどとの融合遺伝子産物の形であってよい。そのタンパク質またはペプチドは、蛍光タンパク質および/または融合タンパク質であってよい。例えば、緑色蛍光タンパク質(GFP)との融合タンパク質またはヒスチジンタグなどのペプチドとの融合遺伝子産物を用いることもできる。さらに、ウイルスHIVから得られたTATペプチドとの融合タンパク質を調製し使用することによって、細胞膜を通じての核初期化因子の細胞内取込みを促進することができ、それによって、融合タンパク質を培地に加え遺伝子形質導入などの複雑な操作を行わないことから、初期化の誘導だけが可能になる。そのような融合遺伝子産物の調製方法は当業者には周知であるため、当業者ならば、目的に応じて適当な融合遺伝子産物を容易に設計し調製することができる。
【0063】
本明細書において論じるように、iPSCは、体細胞に核初期化因子と、その細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤とを組み合わせて接触させることにより誘導され得る。当業者には理解されるように、体細胞への送達は当技術分野で公知の任意の好適な方法により行われ得る。一態様では、核初期化因子は、核初期化因子をコードする遺伝子を含む組換えベクターを用いて細胞に導入され得る。同様に、マイクロRNAを変更する作用物質は、RNA分子(例えば、マイクロRNA、shRNA、アンチセンスオリゴヌクレオチドなど)をコードするポリヌクレオチドを含む組換えベクターを用いて細胞に導入され得る。同様に、p21の阻害剤は、ペプチド阻害剤またはRNA分子(例えば、マイクロRNA、shRNA、アンチセンスオリゴヌクレオチドなど)をコードするポリヌクレオチドを含む組換えベクターを用いて細胞に導入され得る。よって、その細胞は、組換えベクター中に含まれる遺伝子の産物として発現される核初期化因子を発現することができ、同様に組換えベクター中に含まれるポリヌクレオチドの産物として作用物質またはp21阻害剤を発現し、それによって核初期化因子だけを使用する場合と比べて高い効率(割合)で分化した細胞の初期化を誘導する。
【0064】
本発明の核酸構築物(組換えベクターなど)は、様々な周知の技術(細胞の非ウイルス性トランスフェクションなど)を用いて細胞に導入され得る。例示的態様では、構築物は、ベクターに組み込まれ、細胞に導入されて、その構築物の発現が可能になる。細胞への導入は、当技術分野で公知の任意のウイルス性または非ウイルス性トランスフェクション(限定されるものではないが、エレクトロポレーション、リン酸カルシウム法、ヌクレオフェクション、ソノポレーション、熱ショック、マグネトフェクション、リポソーム法、マイクロインジェクション、微粒子銃法(ナノ粒子)、陽イオン性ポリマー法(DEAE−デキストラン、ポリエチレンイミン、ポリエチレングリコール(PEG)など)または細胞融合など)により行われ得る。他のトランスフェクション方法としては、専売のトランスフェクション試薬(Lipofectamine(商標)、Dojindo Hilymax(商標)、Fugene(商標)、jetPEI(商標)、Effectene(商標)およびDreamFect(商標)など)が挙げられる。
【0065】
他の態様では、体細胞への、核初期化因子と、その細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤とを組み合わせての誘導中の接触は、当技術分野で公知の任意の方法、例えば、細胞膜を介したタンパク質、RNA分子などの直接送達により行われ得る。
【0066】
細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤と組み合わせた核初期化因子の使用では、初期化因子だけを使用する場合と比べて誘導効率は高まる。様々な態様では、誘導効率は、従来の方法と比べて10%、20%、30%、40%、50%、60%、70%、80%、90%、100%、150%、200%、300%、400%または500%までも高まり得る。例えば、誘導効率は、1%、2%、3%、4%、5%、6%、7%、8%、9%、10%、15%、20%、25%または50%(例えば、開始時の体細胞総数に対する誘導された細胞の割合)ほどの高さであり得る。
【0067】
誘導プロセス中、体細胞に、その細胞中でマイクロRNAレベルまたは活性を変更する作用物質および/またはp21阻害剤を接触させると同時にまたはその前に、体細胞に核初期化因子を接触させ得る。様々な態様では、体細胞に任意の他の作用物質または阻害剤を接触させる約1日、2日、3日、4日、5日、7日、8日、9日、10日、11日、12日、13日、14日またはそれ以上前に、体細胞に初期化因子を接触させる。例示的態様では、体細胞に任意の他の作用物質または阻害剤を接触させる約1日、2日、3日、4日または5日前に、体細胞に初期化因子を接触させる。
【0068】
初期化細胞の多能性を評価するためにさらなる解析を行ってよい。細胞を異なる増殖特性および胚幹細胞様形態について解析し得る。例えば、特異的細胞種への分化を推進することが知られているある特定の増殖因子を加えることによって細胞をin vitroで分化させ得る。体の数種の細胞種だけを形成可能な初期化細胞は多分化性であり、一方、体のいかなる細胞種をも形成可能な初期化細胞は多能性である。
【0069】
多能性を評価するための初期化体細胞の発現プロファイリングも行ってよい。多能性と関係がある個別遺伝子の発現もまた調査し得る。加えて、胚幹細胞表面マーカーの発現も解析し得る。多能性幹細胞と関係があることが当技術分野で公知の様々な遺伝子の検出および解析としては、限定されるものではないが、OCT4、NANOG、SALL4、SSEA−1、SSEA−3、SSEA−4、TRA−1−60、TRA−1−81、またはそれらの組合せなどの遺伝子の解析が挙げられ得る。iPS細胞はいくつもの多能性細胞マーカーを発現し得、それらには以下が挙げられる:アルカリ性ホスファターゼ(alkaline phosphatase)(AP);ABCG2;ステージ特異的胚抗原−1(stage specific embryonic antigen-1)(SSEA−1);SSEA−3;SSEA−4;TRA−1−60;TRA−1−81;Tra−2−49/6E;ERas/ECAT5、E−カドヘリン(E-cadherin);β−チュブリンIII(β-tubulin III);α−平滑筋アクチン(α-smooth muscle actin)(α−SMA);繊維芽細胞増殖因子4(fibroblast growth factor 4)(FGF4)、Cripto、Dax1;亜鉛フィンガータンパク質296(zinc finger protein 296)(Zfp296);N−アセチルトランスフェラーゼ−1(N-acetyltransferase-1)(Nat1);ES細胞関連転写物1(ES cell associated transcript 1)(ECAT1);ESG1/DPPA5/ECAT2;ECAT3;ECAT6;ECAT7;ECAT8;ECAT9;ECAT10;ECAT15−1;ECAT15−2;Fthll7;Sall4;未分化胚細胞転写因子(undifferentiated embryonic cell transcription factor)(Utf1);Rex1;p53;G3PDH;テロメラーゼ(telomerase)(TERTを含む);サイレントX染色体遺伝子(silent X chromosome genes);Dnmt3a;Dnmt3b;TRIM28;F-box containing protein 15(Fbx15);Nanog/ECAT4;Oct3/4;Sox2;Klf4;c−Myc;Esrrb;TDGF1;GABRB3;Zfp42、FoxD3;GDF3;CYP25A1;developmental pluripotency-associated 2(DPPA2);T-cell lymphoma breakpoint 1(Tcl1);DPPA3/Stella;DPPA4;ならびに他の一般的な多能性マーカー、例えば、細胞を初期化するために誘導中に用いられる任意の遺伝子。iPS細胞はまた、iPS細胞が誘導される分化細胞に特有のマーカーのダウンレギュレーションによっても特徴付けることができる。
【0070】
本発明はさらに、本明細書に記載した方法を用いて生産されたiPS細胞、ならびにそのような細胞の集団を提供する。様々な細胞種へ分化可能な、本発明の初期化細胞には、様々な用途および治療的使用がある。幹細胞の基本特性である、無限に自己複製する能力および体のあらゆる細胞種に分化する能力は治療的使用に理想的となる。
【0071】
よって、一態様では、本発明は、本明細書に記載した方法を用いて生成された誘導多能性幹細胞を用いて対象における障害および/または症状の治療または予防方法をさらに提供する。その方法は、対象から体細胞を得ることおよび本明細書に記載した方法を用いてその体細胞を誘導多能性幹(iPS)細胞に初期化することを含む。その細胞は、次いで、症状の治療に好適な所望の細胞種にその細胞を分化させるのに好適な条件下で培養される。分化した細胞は、次いで、症状を治療または予防するために対象に導入され得る。
【0072】
一態様では、本明細書に記載した方法を用いて生産されたiPS細胞、ならびにそのような細胞の集団は、それらの細胞を、それらの細胞中でマイクロRNAレベルまたは活性を変更する作用物質により処理するかまたはそのような作用物質と接触させることによりin vitroで分化させ得る。マイクロRNAはiPSC誘導において重要なレギュレーターとして確認されているため、そのようなiPSCの分化を指示するために個別マイクロRNAまたはマイクロRNA集団の操作を用い得ると思われる。そのような処理を、増殖因子または当技術分野で一般的に公知の他の作用物質および刺激と組み合わせて用いて、特異的細胞種への分化を推進し得る。
【0073】
本発明の1つの利点は、移植に好適な同系(isogenic or synegenic)ヒト細胞について本質的に無制限の供給を提供することである。iPS細胞は、患者に特異的に合わせられ、免疫拒絶が回避される。従って、現行の移植方法に関する重大な問題(宿主対移植片または移植片対宿主の拒絶によって起こり得る移植組織の拒絶など)がうまく回避される。健常なヒトから得られた体細胞由来のiPS細胞またはiPS細胞から調製された完全に分化した細胞のいくつかの種類を、iPS細胞バンクに細胞のライブラリーとして保存することができ、幹細胞療法を受ける患者による拒絶が起こらない体細胞、組織または器官の調製に、そのライブラリー中の1種類以上のiPS細胞を用いることができる。
【0074】
本発明のiPS細胞は、当技術分野で公知の方法により様々な障害を治療するための複数の異なる細胞種に分化させ得る。例えば、iPS細胞を誘導して、造血幹細胞(hematopoetic stem cells)、筋肉細胞、心筋細胞、肝臓細胞、軟骨細胞、上皮細胞、尿路細胞、神経細胞などに分化させ得る。分化した細胞は、症状を予防または治療するために、次いで、患者の体に移植して戻され得る。よって、本発明の方法は、特定の細胞種/組織の増加もしくは置換または細胞の脱分化が望ましい、心筋梗塞、鬱血性心不全、卒中、虚血、末梢血管疾患、アルコール性肝疾患、硬変、パーキンソン病、アルツハイマー病、糖尿病、癌、関節炎、創傷治癒、免疫不全、再生不良性貧血、貧血、ハンチントン病、筋萎縮性側索硬化症(ALS)、リソソーム蓄積症、多発性硬化症、脊髄損傷、遺伝性障害および類似疾患を有する対象を治療するために使用し得る。
【0075】
様々な実施形態では、その方法によって、組織または器官の細胞数が、対応する未処理の対照組織または器官と比べて少なくとも約5%、10%、25%、50%、75%またはそれ以上増加する。さらに別の実施形態では、その方法によって、組織または器官の生物活性が、対応する未処理の対照組織または器官と比べて少なくとも約5%、10%、25%、50%、75%またはそれ以上増加する。さらに別の実施形態では、その方法によって、組織または器官における血管形成が、対応する未処理の対照組織または器官と比べて少なくとも約5%、10%、25%、50%、75%またはそれ以上増加する。さらに別の実施形態では、前記細胞は対象に対して、細胞数の増加が望まれる部位に直接投与される。
【0076】
本発明はさらに、本明細書に記載した方法により得られた様々な細胞を用いることにより作用物質、化合物、医薬、毒物などの生理学的機能または毒性を評価するための方法を提供する。
【0077】
4種の転写因子、Oct4、Sox2、Klf4およびcMycの異所発現により体細胞をES様状態に初期化して、誘導多能性幹細胞(iPSC)を作出することができる。本発明によれば、細胞のマイクロRNAはiPSC生成を調節することとなる。重要なマイクロRNA経路タンパク質のノックダウンにより初期化効率の著しい低下がもたらされ得る。3種のマイクロRNAクラスター、miR−17〜92、106b〜25および106a〜363は、初期化の初期段階中に高度に誘導されることが示されている。非常に類似したシード領域を有するいくつかのマイクロRNA(miR−93およびmiR−106bを含む)は、iPSC誘導を大幅に増強し、これらのマイクロRNAの阻害により初期化効率が著しく低下した。さらに、miR−iPSCクローンは完全初期化状態に到達し得る。本発明によれば、Tgfbr2およびp21は、これらのマイクロRNAにより直接ターゲッティングされることとなり、両遺伝子のsiRNAノックダウンによりiPSC誘導が実際に増強されることとなる。また、本発明によれば、miR−93およびそのファミリーメンバーはTGF−β受容体IIを直接ターゲッティングして、iPSC初期化を促進することとなる。本発明によれば、マイクロRNAは初期化プロセスにおいて作用することとなり、細胞中のマイクロRNAレベルをモジュレートすることによりiPSC誘導効率は大幅に高められ得ることとなる。
【0078】
誘導多能性幹細胞(iPSC)はオーダーメイド再生医療に大いに有望であるが、初期化の分子基盤は大部分が不明である。細胞同一性を維持する障壁を克服することが分化した細胞の初期化における重要な段階である。マイクロRNA(miRNA)は標的遺伝子を組織特異的にモジュレートするため、本発明によれば、異なるマウス胚性繊維芽細胞(MEF)濃縮miRNAは、初期化障壁として働くタンパク質を転写後にモジュレートすることとなる。これらのmiRNAの阻害は、細胞シグナル伝達に影響を及ぼして、それらの障壁を低くするはずである。本発明によれば、miR−21およびmiR−29aの枯渇によりMEFにおける初期化効率が高まることとなる。本発明によれば、p53およびERK1/2の経路はmiR−21およびmiR−29aによって調節され、初期化において作用することとなる。さらに、本発明によれば、c−Mycは、MEFで濃縮されたmiRNA(miR−21およびmiR−29aなど)を抑制することにより初期化をある程度まで促進することとなる。本発明は、iPSC初期化に関与する複数のシグナル伝達ネットワークの調節におけるmiRNA機能を提供する。
【0079】
C−Mycは、4種の初期化因子(4F:Oct3/4、Sox2、Klf4およびc−Myc)のうちの1つであり、細胞増殖および腫瘍発生において重要な役割を果たす。C−Mycは、サイクリン依存性キナーゼ(CDK)阻害剤p21Cip1の抑制を通じて細胞性塞栓およびアポトーシスの重要なレギュレーターである。Miz−1機能を無効にし、p15INK4bを抑制することにより、c−Mycは、初代細胞の不死化において重要な役割を果たす。c−Mycの多くの転写機能はMaxまたはMiz−1との協同作用を必要とする。原癌遺伝子としてのc−Mycは初期化効率を大幅に高めるが、初期化では重要でない。従って、初期化中のc−Mycの下流の分子経路を定義することにより初期化効率を損なうことなくiPS細胞の治療用途を増すことができる。
【0080】
Oct4−GFPマウス胚性繊維芽細胞(MEF)は、D13.5に、pou5f1の停止コドンの下流にIRES−EGFP融合カセットを有するマウス(Jackson lab、ストック番号008214)から得られる。これらのMEFは、10%FBS(Invitrogen)とグルタミンおよびNEAAとを含有するDMEM(Invitrogen、11995−065)で培養される。iPSC誘導では、0〜4代継代のMEFだけを用いる。
【0081】
C−Mycは、マイクロRNA(miRNA)発現を調節することによってES細胞再生を維持するように働く側面があると報告されている。マイクロRNAは、22−ヌクレオチドの非コード低分子RNAであり、RNA誘導サイレンシング複合体(RISC)に装入され、全体的な遺伝子サイレンシング機能を発揮する。miR−141、miR−200およびmiR−429の発現はES細胞においてc−Mycにより誘導され、分化に拮抗する。C−Mycはまた、miR−17〜92マイクロRNAクラスターをアップレギュレートすることによりまたは既知腫瘍抑制遺伝子(let−7ファミリー、miR−15a/16−1、miR−29ファミリーおよびmiR−34aなど)を抑制することにより腫瘍形成も促進する。
【0082】
体細胞同一性を固定する、Ink4−Arf、p53およびp21などの因子により媒介される障壁を克服することは、初期化における律速段階である。miRNAは標的遺伝子を組織特異的にモジュレートするため、本発明によれば、異なるMEF miRNAは、初期化レギュレーターとして働くタンパク質を転写後にモジュレートすることとなる。これらのmiRNAの阻害により、細胞シグナル伝達に影響を及ぼして、それらの障壁を低くすることができる。
【0083】
本発明によれば、MEFにおいて豊富なmiRNA(miR−21およびmiR−29a)の枯渇により初期化効率が約2.1倍〜2.8倍高まることとなる。また、本発明によれば、c−MycはmiRNA(miR−21およびmiR−29a)を抑制して、MEFの初期化を促進することとなる。さらに、本発明によれば、miR−21およびmiR−29aは、初期化プロセス中にp53レベルおよびERK1/2リン酸化を間接的にダウンレギュレートすることによりp53およびERK1/2の経路を調節することとなる。
【0084】
次の実施例を示して、本発明の実施形態をさらに説明するが、それらの実施例は本発明の範囲を限定するものではない。それらの実施例は用いられる可能性のある典型的なものであるが、当業者に公知の他の手順、方法論または技術を代わりに用いてもよい。
【実施例】
【0085】
実施例1
細胞培養物、ベクターおよびウイルス形質導入
Oct4−GFPマウス胚性繊維芽細胞(MEF)は、D13.5に、pou5f1の停止コドンの下流にIRES−EGFP融合カセットを有するマウス(Jackson lab、ストック番号008214)から得られる。これらのMEFは、10%FBS(Invitrogen)とグルタミンおよびNEAAとを含有するDMEM(Invitrogen、11995−065)で培養される。iPSC誘導では、0〜4代継代のMEFだけを用いる。
【0086】
プラスミドpMXs−Oct4、Sox2、Klf4およびcMycはAddgeneから購入する。プラスミドpMX−HA−p21は、N末端タグ付きp21をpMXベクターのEcoRI部位に挿入することにより生成する。pLKO−shRNAのクローンはOpen-Biosystemsから購入する。
【0087】
レトロウイルスを生成するために、PLAT−E細胞を10cmプレートに播種し、翌日Lipofectamine(商標)(Invitrogen、18324−012)およびPLUS(商標)(Invitrogen、11514−015)を用いて各因子9μgをトランスフェクトする。ウイルスを採取し、2日後に合わせる。iPSC誘導では、MEFを12ウェルプレートに播種し、翌日4種の因子ウイルスを4μg/mlポリブレンとともに形質導入する。形質導入の1日後、培地を新鮮なMEF培地に換え、3日後にLIF(Millipore、ESG1107)を補給したmES培養培地に替える。形質導入後14日目からGFP+コロニーを採取し、拡大に成功したクローンを、15%FBS(Hyclone)とLIF、チオグリセロール、グルタミンおよびNEAAとを含有するDMEMで培養した。照射CF1 MEFをmESおよび得られたiPSCクローンの培養用のフィーダー細胞層として用いる。
【0088】
shRNAレンチウイルスを生成するために、shRNAレンチウイルスベクターを293FT細胞にpPACKH1パッケージングシステム(SBI、カタログ番号LV500A−1)とともに共トランスフェクトする。トランスフェクション後2日目にレンチウイルスを採取し、室温で4,000rpmで5分間遠心分離する。ウイルスを生産するために、10cm組織培養プレートで4μgのpLKOまたはpGIPZベクターおよび10μgのパッケージングミックスを293FT細胞(Invitrogen)にトランスフェクトした。トランスフェクションの2日後に、ウイルスを含有する上清を採取し、4μg/μlポリブレンとのさらなる形質導入に用いた。ShRNAウイルスを4種の因子ウイルスとともに体積比1:1:1:1:1で加える。
【0089】
マイクロRNA、siRNAおよびMEFのトランスフェクションは次のとおり行う:マイクロRNAミミックおよび阻害剤、siRNAはDharmaconから購入する。MEFをトランスフェクトするために、マイクロRNAミミックをOpti−MEM(Invitrogen、11058−021)で所望の終濃度に希釈する。次いで、その混合物にLipofectamine(商標)2000(Invitrogen、11668−019)を2μl/ウェルで加え、室温で20分インキュベートする。12ウェルトランスフェクションでは、80μlのmiR混合物を、320μlのOpti−MEMが入った各ウェルに加える。3時間後に、各ウェルに0.8mlのウイルス混合物(iPSCの場合)または新鮮培地を加え、翌日その培地を新鮮なMEF培地に換える。
【0090】
ウエスタンブロッティングは次のとおり行う:全細胞溶解物を、MPERバッファー(PIERCE、78503)を氷上で20分間用いて調製し、13,000rpmで10分間遠心分離することにより清澄にする。10%SDS−PAGEゲル中に同量の溶解物をローディングし、セミドライ式システム(Bio-Rad)を用いてタンパク質をPVDF膜(Bio-Rad、1620177)に転写する。次いで、PVDF膜を、TBST中5%ミルクにより室温で少なくとも1時間または4℃で一晩ブロッキングする。
【0091】
使用する抗体には、抗p21(BD、556430)、抗mNanog(R&D、AF2729)、抗h/mSSEA1(R&D、MAB2156)、抗HA(Roche、11867423001)、抗mAgo2(Wako、01422023)、抗Dicer(Abcam、ab13502)、抗Drosha(Abcam、ab12286)、抗アクチン(Thermo、MS1295P0)、抗AFP(Abcam、ab7751)、抗βチュブリンIII(R&D systems、MAB1368)、抗αアクチニン(Sigma、A7811)を含む。
【0092】
mRNAおよびマイクロRNAのRTおよび定量PCRは次のとおり行う:トータルRNAをTrizol法(Invitrogen)により抽出する。抽出後、1μgのトータルRNAを、スーパースクリプトII(商標)(Invitrogen)によるRTに用いる。定量PCRは、Roche LightCycler480 II(商標)およびAbgeneのSybrgreen混合物(Ab−4166)を用いることにより行う。マウスAgo2、Dicer、Drosha、Graphおよびp21のプライマーを下の表1に記載する。他のプライマーは、Takahashi, K. and S. Yamanaka (2006) Cell 126(4): 663-76に記載されている。
【0093】
マイクロRNAの定量解析では、トータルRNAを、上に記載した方法を用いて抽出する。抽出後、1.5〜3μgのトータルRNAを、QuantiMir(商標)キット(SBI、RA420A−1)を用い製造業者のプロトコールに従うマイクロRNA逆転写に用いる。次いで、RT産物を、正方向プライマーとしての成熟マイクロRNA配列およびキットで提供されるユニバーサルプライマーを用いる定量PCRに用いる。
【0094】
免疫染色は次のとおり行う:細胞をPBSで2回洗浄し、4%パラホルムアルデヒドにより室温で20分間固定する。固定した細胞を0.1%Triton X−100により5分間透過処理する。次いで、それらの細胞を、0.1%Triton X−100を含有するPBS中5%BSAにより室温で1時間ブロッキングする。一次抗体を、製造業者の提示に従って、0.1%Triton X−100を含有する2.5%BSA PBSにより1:100〜1:400に希釈する。次いで、一次抗体で細胞を1時間染色し、その後、PBSで3回洗浄する。二次抗体を1:400希釈し、室温で45分間細胞を染色する。
【0095】
胚様体(EB)形成および分化のアッセイは次のとおり行う:iPS細胞をトリプシン処理して単一の細胞懸濁液にし、懸滴法を用いて胚体を生成する。各液滴には、20μlのEB分化培地中4000個のiPS細胞を用いる。EBを3日間懸滴培養した後、ゼラチンコーティングプレートに再播種する。再播種後、収縮を繰り返す領域がはっきりと確認される14日目まで細胞をさらに培養する。
【0096】
(表1)qPCR解析用プライマー
【0097】
プロモーターメチル化解析は次のとおり行う:NanogおよびPou5f1プロモーターのCpGメチル化を、これまでに記載されている同じ手法に従って解析する(Takahashi, K. and S. Yamanaka (2006) Cell 126(4): 663-76)。簡潔には、得られたクローンのゲノムDNAを、Qiagen(商標)キットを用いて抽出する。次いで、1μgのDNAを、製造業者のプロトコールに従うゲノム修飾解析(EZ DNA Methylation−Direct kit、Zymo Research、D5020)に用いる。修飾後、選択した領域のPCRを行い、それらの産物をpCR2.1−TOPO(商標)(Invitrogen)にクローニングする。各遺伝子について10のクローンを配列決定する。
【0098】
実施例2
奇形腫形成、キメラ作製およびマイクロアレイ解析
奇形腫形成およびキメラ作製は次のとおり行う:奇形腫を生成するために、iPS細胞をトリプシン処理し、濃度1×107細胞/mlで再懸濁する。無胸腺ヌードマウスにまずアベルチンで麻酔をかけ、次いで、およそ150μlの細胞懸濁液を各マウスに注射する。3〜4週間、週1回マウスを腫瘍について調べる。腫瘍を採取し、パラフィン包理およびH&E染色の前に亜鉛ホルマリン溶液により室温で24時間固定する。得られたiPSCクローンがキメラに寄与する能力を試験するために、iPS細胞をC57BL/6J−Tyr(C−2J)/J(アルビノ)胚盤胞に注射する。一般的に、各胚盤胞は12〜18個のiPS細胞を受ける。胚移植にはICRレシピエント雌を用いる。ドナーiPS細胞はアグーチまたは黒色のいずれかである。
【0099】
mRNA マイクロアレイ解析を次のとおり行う:miR−93およびsiControlをMEFにトランスフェクトし、トランスフェクションの48時間後にトータルRNAを採取する。mRNAマイクロアレイをサンフォード・バーナム研究所(Sanford-Burnham institute)内のマイクロアレイ施設により実施する。潜在的機能標的(変化倍率>2、p<0.05)および全標的(変化倍率>25%、p<0.05)の両方についての遺伝子一覧表を、ボルカノマップ(volcano maps)でフィルタリングすることにより作成する。次いで、遺伝子一覧表を、GeneGoソフトウェアを用いその会社のガイドラインに従うオントロジー解析に用いる。
【0100】
デュアルルシフェラーゼアッセイは次のとおり行う:p21およびTgfbr2両方の3'UTRを、pGL3対照ベクターのXbaI部位にクローニングする。12ウェルプレートの各ウェルに、200ngの得られたベクターおよび50ngのpRL−TK(ウミシイタケルシフェラーゼ)を、トランスフェクションの1日前に播種した1×105個のHela細胞にトランスフェクトする。各処理に50nMのマイクロRNAを用い、トランスフェクション後2日目に細胞溶解物を採取する。次いで、20μlの溶解物を、製造業者のプロトコール(Dual−Luciferase(登録商標)Reporter Assay System Promega、E1910)に従うデュアルルシフェラーゼアッセイに用いる。
【0101】
細胞増殖アッセイは次のとおり行う:3000個のMEFを96ウェルプレートの各ウェルに播種し、4FウイルスおよびshRNAレンチウイルスを形質導入する(またはマイクロRNA阻害剤でトランスフェクトする)。形質導入/トランスフェクション後1日目から、2日おきに、細胞をCelltiter 96 Aqueous one Solution(Promega、G3580)を含有するmES培地とともに組織培養インキュベーター中で1時間インキュベートする。次いで、490nmでの吸光度を各ウェルについてプレートリーダーを使用して測定し、収集データを用いて相対的増殖曲線を作成し、形質導入/トランスフェクション後1日目のシグナルを標準として用いる。
【0102】
実施例3
転写後調節経路は体細胞の初期化に関与する
転写後調節経路は、MEFのiPS細胞への初期化に関与することが確認された。iPSC誘導中の転写後遺伝子調節の役割を調査するために、マウスDicer、DroshaおよびAgo2をターゲッティングするレンチウイルスshRNAベクターを、初代Oct4−GFP MEFにおける安定なノックダウンに用いる。これらのshRNA構築物のノックダウン効率をウエスタンおよびRT−qPCRの両方により検証する(図1a、図1bおよび図1c)。各shRNAではおよそ70%〜80%のmRNAレベルノックダウンが常に観察され、タンパク質レベルの著しい低下も観察される。
【0103】
次いで、shRNAを個別に用いて、MEFを4種の因子OSKM(Oct4、Sox2、Klf4およびcMyc)を発現するウイルスとともに体積比1:1:1:1:1で形質導入する(tranduce)。14日後に、コロニーを固定し、アルカリ性ホスファターゼ(AP)活性(これは広く用いられているES細胞マーカーである)について染色する。各処理についてAP+コロニーを定量し、重要なRNAi機構タンパク質Dicer、DroshaおよびAgo2のノックダウンによりAP+コロニーがpLKOおよびpGIPZ対照と比べて劇的に減少する。OSK(3種の因子 3F)形質導入を用いることによっても類似した結果が観察される。
【0104】
GFP+およびAP+コロニーの両方の定量では、Ago2のノックダウンは初期化効率を劇的に低下させるが、一方、形質導入繊維芽細胞の増殖には影響を及ぼさないことが確認された。(図1d、図1eおよび図1f)。Ago2ノックダウンにより初期化効率が低下するにもかかわらず、shAgo2でのいくつかのGFP+コロニーは感染MEFであり、さらなる特性評価では、これらのコロニーはshRNA組込みについて陽性でありそこではshRNAが活発に発現されることが判明した(図13aおよび図13b)。これらの細胞はまた、試験したES特異的マーカー全てを発現するが、内因性Oct4遺伝子座で示した(図13c)。これらのデータは、転写後調節、とりわけ、マイクロRNAが初期化プロセスにおいて重要な役割を果たすことを強く示唆している。
【0105】
実施例4
マイクロRNAクラスターは体細胞の初期化中に誘導される
マイクロRNA miR−17、25、106aおよび302bクラスターは初期化の初期段階中に誘導されることが判明している。4種の転写因子はiPSC誘導中に多くの遺伝子発現変化を誘導するため、これらの因子によっていくつかのES特異的マイクロRNAが誘導され、そして、これらの因子がMEFの初期化の成功を手助けし得ると推測される。iPSC誘導を増強するES特異的マイクロRNAに関する最新の刊行物でも、その仮説が裏付けられているが、報告されているマイクロRNAは初期化の極めて終わりに近い段階まで発現が見出されなかった。公表されている結果を解析することにより、解析には、マウスES細胞において高度に発現されると判明している9種のマイクロRNAクラスターを選択し、それらを表2に示す。
【0106】
各クラスターからの2種の代表的なマイクロRNAを、miR qPCRに基づく方法を用いて評価して、異なる初期化段階(OSKM因子の形質導入後0日目、4日目、8日目および12日目を含む)における発現変化を定量する。多くのES特異的マイクロRNA(miR−290クラスターおよびmiR−293クラスターなど)は、8日目まで誘導されず(図14)、この段階においてGFP+コロニーはすでに検出できる。いくつかの他のマイクロRNAクラスター(miR−17〜92、25〜106b、106a〜363および302b〜367を含む)は、4種の因子の形質導入後4日目までに様々な程度に発現される(図2a)。これらの4種のマイクロRNAクラスターの間では、MEFにおけるmiR−302b〜367のレベルが最も低い。初期化4日目に高度に誘導される3種のクラスターの間では、一部に非常に類似したシード領域の共有があり(図2b)、それらが初期化において作用し、類似した遺伝子セットをターゲッティングすることができることを示唆している。
【0107】
(表2)iPSC実験に用いたマイクロRNAの一覧表
【0108】
次いで、4種の因子のどの因子がこれらのマイクロRNAの誘導に関与しているかを判定するために解析を行う。同じ用量の4種の因子の種々の組合せをMEFに形質導入することにより、miR qPCR解析のために感染後4日目にトータルRNAを採取する(図2c)。この解析により、cMyc単独でmiR−17〜92、miR−25〜106bおよびmiR−106a〜363クラスターの発現を誘導することができることが確認される。しかしながら、全ての場合において、4種の初期化因子全ての組合せがマイクロRNAクラスターの最も豊富な発現を誘導し、その強い発現は最大の初期化効率と相関している(図2c)。
【0109】
これらの結果により、3種のマイクロRNAクラスター(miR−17〜92、25〜106b、106a〜363を含む)は初期化の初期段階中に誘導されること、さらに、これらのマイクロRNAの発現は4種の因子組によって最も高度に誘導されるが、単一因子でも程度は劣るもののそれらの発現を誘導することができることが確認された。
【0110】
実施例5
マイクロRNAはiPSC誘導を増強する
マイクロRNA miR−93およびmiR−106bは、マウスiPSC誘導を増強することが判明している。確認されている4種のマイクロRNAクラスターは、類似したシード領域を有するいくつかのマイクロRNAを含むため、miR−106b〜25クラスターが3種のマイクロRNA(すなわち、miR−25、miR−93およびmiR−106b)を含むことからこのクラスターをさらに解析する。miR−93およびmiR−106bは同一シード領域を有し、両方とも4種の初期化因子によって高度に誘導される(図2a)。これらのマイクロRNAが初期化細胞で機能しているならば、そのプロセス中にこれらのマイクロRNAを導入することによりiPSC誘導の効率向上が期待されることとなる。
【0111】
これらの誘導されたマイクロRNAの機能試験には、マイクロRNAミミックをMEFに直接トランスフェクトする戦略を用いる(図3a)。マイクロRNAを4種の因子(またはOSK)ウイルスとともに2回、0日目および5日目に導入し、内因性Oct4プロモーターの制御下でGFP発現するレポーターMEFを用いた。例えば、0日目および5日目に、マイクロRNAミミックを、4種の因子(4F、OSKM)全てまたは単独のOct4、Sox2およびKlf4(OSK)のいずれかを発現するベクターとともに、Oct−4−GFPを保有しているMEFに直接トランスフェクトし、GFP発現に基づいて初期化をアッセイする。これらの細胞がiPSCへの初期化に成功したら、それらはGFP陽性(+)となる。11日目頃にGFP+コロニーを定量して、初期化効率を評価する(図3b;表3)。実際には、miR−93およびmiR−106bミミックのトランスフェクションによって4F形質導入およびOSK形質導入の両方でGFP+コロニーの約4〜6倍増加となり(図3c)、iPSC誘導中に誘導されるこれらのマイクロRNAはMEF初期化を促進することが確認された。
【0112】
(表3)iPSC誘導に関するmiRによるGFP+コロニー数
* 12ウェルプレート(ゼラチンコーティング処理)に4x104個MEF/ウェル
【0113】
用量依存実験は、5〜15nMの範囲ほどの低いmiRで初期化効率の向上が見られることを示している(図7)。コロニーをアルカリ性ホスファターゼ基質で染色すると、miRミミックトランスフェクションでのAP+コロニーの著しい増加はないようであり、miR−93およびmiR−106bはiPSCコロニーの成熟プロセスを促進することができることが示唆される。このことは、OSK系を用いて観察される現象(miRミミックトランスフェクト細胞ではOSK形質導入後15日目に多くのGFP+コロニーが現われるが、一方、対照ウェルではこの段階において成熟したiPSCコロニーを全く示さなかった)によっても裏付けられる。
【0114】
これらのマイクロRNAがiPSC誘導において重要であることを確認するために、miR阻害剤を用いて、そのプロセス中に標的とするマイクロRNAのノックダウンも行う。試験したmiR阻害剤の全ては標的miR発現を効率的に減少させることができ、それらのトランスフェクションは増殖に影響を及ぼさない(図16aおよび図16b)。miRミミック実験と一致して、miR−93およびmiR−106bのノックダウンによってGFP+コロニーの劇的な減少が促され得る(図3d)。miR−25ミミックはMEFのiPSC誘導を増強しないが、このマイクロRNAのノックダウンにより初期化効率が約40%低下し(図3d)、miR−25は初期化プロセス中にも作用し得ることが示唆される。対照として、Let7a阻害剤は、初期化効率に対していかなる影響も及ぼさなかった。これらのデータは、miR−93およびmiR−106bがMEFのiPSCへの初期化を促進することを強く示している。初期化効率は、iPSC誘導中にこれらのマイクロRNAをモジュレートすることによってさらに高め得る。
【0115】
実施例6
マイクロRNAにより誘導されたクローンは完全な多能性を有する
誘導された細胞が完全な多能性状態に達するかどうかを調べるために、各マイクロRNAならびにmiR対照についていくつかのiPSCクローンを得、多能性マーカーの発現について解析する。全てのクローンはGFP+(再活性化されるOct4発現を示す)である。免疫染色により、NanogおよびSSEA1も全てのクローンにおいて活性化されることが確認された。他のmESマーカー(Eras、ECat Iおよび内因性(endogeneous)Oct4など)についてのRT−qPCRは類似した結果を示す。全ゲノムmRNA発現プロファイリングでも、得られたクローンがMEFよりもマウスES細胞に類似した遺伝子発現パターンを示すことを示す。内因性Nanog遺伝子座のプロモーターメチル化を解析し、試験したクローンは全て、マウスES細胞で観察されるように、脱メチル化プロモーターを示した(図17)。
【0116】
得られたクローンがmES細胞の完全分化能を示すかどうかを調査するために、胚様体(EB)形成を評価する。得られたクローンは全て、効率的なEB形成を示し、EBは系統マーカー(β−チュブリンIII(外胚葉)、AFP(内胚葉)およびα−アクチニン(中胚葉)など)について陽性染色を示す。これらの細胞から収縮を繰り返すEBも得られ、これは、これらのmiR−iPSCクローンから機能的な心筋細胞が誘導され得ることを示す。これらのmiR−iPSCを無胸腺ヌードマウスに注射すると、3〜4週間で奇形腫が容易に誘導される。最後に、より厳しい試験として、miRにより誘導されたiPSCクローンをアルビノ/黒色B6胚盤胞に注射し、キメラマウスを作製する。さらに、これらの細胞は、得られたE13.5胚の生殖隆起に寄与し得た。これらの結果は、初期化に対するmiR−93およびmiR−106bの促進作用は誘導された多能性細胞の分化能を変更しないこと、そして得られたクローンは3種の生殖細胞系全てに分化することができることを示す。
【0117】
実施例7
miR−93およびmiR−106bはマウスにおいてTgfbr2およびP21をターゲッティングする
miR−93およびmiR−106bによる初期化効率向上の基礎にある機構をさらに理解するために、これらのマイクロRNAの細胞標的を調査する。miR−93がmiR−106bと同じシード領域を共有することから、まず解析にそれを選択する。miR−93ミミックをMEFにトランスフェクトし、mRNA発現プロフィール解析のために2日目にトータルRNAを採取する。その解析は、公表されているMEFおよびiPSCの発現プロフィールと比べてmiR−93の潜在的機能標的を同定する。miR−93トランスフェクションにより著しく減少した遺伝子は、iPSCにおいて低度に発現される遺伝子の3倍の濃縮を示し(図18a)、一方、miR−93トランスフェクションにより増加した遺伝子はそのような濃縮を示さない。加えて、miR−93トランスフェクトMEFの発現プロフィールについて経路オントロジー解析を行う。興味深いことに、iPSC誘導の2つの重要な経路は、miR−93:TGF−βシグナル伝達経路およびG1/S移行経路により調節される。
【0118】
TGF−βシグナル伝達では、miR−93トランスフェクションにより最も著しく減少した遺伝子の1つにTgfbr2がある。Tgfbr2は、TGF−βシグナル伝達において重要な役割を果たす構成的に活性な受容体キナーゼであり、最近の小分子スクリーニングにより、そのヘテロ二量体パートナーであるTgfbr1の阻害剤がiPSC誘導を増強することが示されている。マイクロRNA標的部位予測により、miR−93およびそのファミリーであるマイクロRNAでは2つの保存されたターゲッティング部位がその3'UTRに存在することが示されている。そのため、さらなる調査のために、miR−93が第1の標的候補として選択される。
【0119】
G1/S移行に関しては、ヒト固形腫瘍サンプル(乳房、結腸、腎臓、胃および肺)および胃癌細胞株における最近の結果により、miR−106b〜25クラスターは細胞周期調節因子(CDK阻害剤p21およびp57など)をターゲッティングすることができること、そしてヒトおよびマウスp21が3'UTRの保存されたmiR−93/106b標的部位を共有することが示されているため、潜在的標的としてp21が選択される。
【0120】
さらに、マウスES細胞特異的マイクロRNAクラスター(miR−290およびmiR−293クラスターを含む)はまた、いくつかのG1−S期移行負調節因子(p21を含む)をターゲッティングすることも提示された。加えて、miR−290および293クラスターマイクロRNAはmiR−93およびmiR−106bと非常に類似したシード領域を共有する。そのため、p21も標的候補として解析する。さらに、p21は、iPSC誘導の初期段階中に4種の因子OSKMにより大幅に誘導される(図8a)。詳細な解析により、Oct4およびSox2の組合せはp21レベルの著しい変化を示さないため、p21の誘導が主にKlf4およびcMycの過剰発現によることが明らかになっている(図8a)。
【0121】
マウスTgfbr2およびp21がmiR−93およびmiR−106bによってターゲッティングされるかどうかを確認するために、miRミミックをMEFにトランスフェクトし、48時間後にウエスタンブロッティングにより全細胞溶解物を解析する。実際には、miR−93およびmiR−106bは、Tgfbr2およびp21両方のタンパク質レベルを効率的に低下させ(図5aおよび図5d)、さらにp21 mRNAレベルを約25〜30%低下させ、Tgfbr2 mRNAレベルを約60〜70%低下させる(図19)。p21がmiR−93およびmiR−106bの直接標的であるかどうかをさらに調査するために、ルシフェラーゼアッセイ(p21 3'UTR配列を有するルシフェラーゼレポーターがホタルルシフェラーゼコード配列の下流に挿入される)を行う。ルシフェラーゼアッセイによって、miRミミックをHela細胞にトランスフェクトすることによりルシフェラーゼ活性の一貫した約40%の抑制が達成され得ることが明らかになっている。また、保存されたp21 3'UTR標的部位のシード領域に突然変異が導入されると、マイクロRNAミミックの抑制が完全に破壊され得ることも判明している(図10)。Tgfbr2については、ルシフェラーゼアッセイでGL活性の約50%減少も示し、一方、miR−93変異株にはそのような影響はない(図11)。
【0122】
p21により促進される細胞周期停止によって初期化に必要な後成的修飾が阻害され得るが、増殖中の細胞においてそのような修飾がより起こりやすいためである。p21発現がiPSC誘導に支障をきたすかどうかを判定するために、HAタグ付きp21 cDNAをpMXレトロウイルス主鎖にクローニングし、MEF細胞で過剰発現させる。HA−p21ウイルスを4種のOSKM因子とともにMEFに導入すると、アルカリ性ホスファターゼ染色とOct4−GFP陽性コロニー形成に基づいて、iPSC誘導のほぼ完全な阻害が観察される(図9a)。初期化に3種のOSK因子を用いる場合も類似した結果が得られる(図9b)。
【0123】
解析により、miR−93およびmiR−106bがTgfbr2およびp21の発現両方を効率的に抑制することが示されているため、Tgfbr2およびp21の両方を、それらの活性が初期化に拮抗し得るかどうかをさらに調査する。マイクロRNAミミックで使用した同じ実験時系列を用いて、Tgfbr2またはp21 siRNAをMEFにトランスフェクトする。ウエスタンブロッティングおよびRT−qPCRでは、ウイルス形質導入を行わずにsiRNAによって、それぞれ、タンパク質およびmRNAレベルの両方が効率的にノックダウンされることが確認される(図5bおよび図5e)。次いで、OSKM形質導入によりMEF初期化を開始し、形質導入後11日目にOct4−GFP+コロニーを定量する。各遺伝子についてコロニー数の約2倍の誘導が観察される(図5cおよび図5f)。同時に、我々のデータによって、Tgfbr2およびp21がmiR−93およびmiR−106bの直接標的であり、これらの遺伝子のダウンレギュレーションにより初期化プロセスを促進し得ることが確認される。
【0124】
実施例8
miR−93およびmiR−106bトランスフェクションから得られたiPSCクローンの多能性
miR−93および106bは、マウスiPSC誘導を増強する能力について確認されたが、誘導された細胞が完全な多能性状態に達するかどうかという問題が残っている。この問題に答えるために、各マイクロRNAならびにmiR対照についていくつかのiPSCクローンを得て、多能性マーカーおよび分化能を解析する。これらの得られたクローンは全てGFP+(Oct4遺伝子座の再活性化を示す)である。免疫染色により、NanogおよびSSEA1もこれらの細胞において活性化されることも確認された。他のmESマーカーについてのRT−qPCRは類似した結果を示す。全ゲノムmRNA発現プロフィールでも、これらの得られたクローンがMEFではなくmESと非常に類似した遺伝子発現パターンを有することを示す。内因性Oct4およびNanog遺伝子座のプロモーターメチル化も解析し、試験したクローンは全て、脱メチル化プロモーター有することが観察された。
【0125】
得られたクローンがmES細胞の完全分化能を有するかどうかを調査するために、胚体形成を最初の試験として用いる。得られたクローンは全て、効率的なEB形成を生じ、それらのEBは系統マーカー染色について陽性であると判明している。これらの細胞から収縮を繰り返すEBも得られる。
【0126】
最後に、より厳しい試験として、これらの得られたクローンを注射して、それらがキメラマウスに寄与するかどうかを調べる。実際には、試験したクローン全てからキメラが得られる。これらの結果は、初期化に対するmiR−93およびmiR−106bの促進作用は誘導された細胞の能力を変化させないこと、さらにES様状態に達した誘導クローンは3種の系統全てに分化することができることを示す。
【0127】
実施例9
他のマイクロRNAのアップレギュレーションもiPSC誘導を増強する
本明細書において論じるように、3種のマイクロRNAクラスターは、iPSC誘導中に4種の因子により誘導されると確認され、これらのクラスター内のいくつかのマイクロRNAは、同じシード領域を有すると判明しており、それらは類似したmRNAをターゲッティングすることが示されている(図2)。miR−93およびmiR−106bと同じシード領域を共有する他のマイクロRNAがiPSC誘導を同様に増強することができるかどうかを調査するために、miR−17およびmiR−106aのマイクロRNAミミックを、miR−93ミミック処理およびiPSC誘導について上に記載したものと類似した試験手法を用いて試験する。実際には、これらのマイクロRNAは、miR−106b〜25クラスターで見られたものと同様に初期化を促進し(図6a)、これらのmiRのトランスフェクション全てで、Tgfbr2およびp21タンパク質レベルの低下が起こる(図6bおよび図6c)。
【0128】
同時に、これらの結果は、miR−17〜92、miR−106b〜25およびmiR−106a〜363クラスターの誘導は正確な初期化に重要であること、そしてこれらのマイクロRNAのアップレギュレーションはiPSC生成プロセスへの初期化障壁を低くすることを示唆している(図6d)。よって、これらのマイクロRNAの細胞内レベルを操作して、初期化効率を向上させ得る。
【0129】
実施例10
iPSC初期化機構
得られたクローンは、内因性Oct4−GFP発現を活性化することは分かっている。マイクロRNAミミックとのOSKM形質導入後12日目からコロニーを採取し、照射MEFフィーダープレート上で維持する。内因性pou5f1遺伝子座のGFPシグナルとして緑色蛍光を観察することができる。クローンは、ES特異的マーカーのアルカリ性ホスファターゼ染色および免疫染色(NanogおよびSSEA1染色に基づく)を用いて示すことができる。核染色にはHoechst 33342を用いることができる。3胚葉全てからの細胞は、誘導iPSCクローンを用いる胚様体(EB)アッセイで得ることができる。EB形成のためにiPS細胞を約4000個の細胞/20μl液滴で3日間培養し、次いで、収縮を繰り返す心筋細胞が観察される12〜14日目までさらに培養するために、EBをゼラチンコーティングプレート上に再播種する。異なる系統マーカー(外胚葉マーカーのβ−チュブリンIII;内胚葉マーカーのAFP;および中胚葉マーカーのα−アクチニンを含む)で細胞を免疫染色することができる。奇形腫は、iPS細胞の注射により生成することができ、150万個の細胞を各マウスに注射し、パラフィン包理およびH&E染色のために注射の3〜4週間後に腫瘍を採取する。誘導クローンを用いて、キメラマウスを作製することもできる。iPS細胞をアルビノまたは黒色C57B6マウス(NCI)の胚盤胞に注射し、iPSCの寄与はアグーチまたは黒色の毛色により見ることができる。
【0130】
12日目の初期化細胞はアルカリ性ホスファターゼ基質によって染色され得る。本発明によれば、miRミミックトランスフェクションによりAP+コロニーの著しい増加は起こらないが、miR−93および106bのノックダウンによってAP+コロニーだけでなくGFP+コロニーの著しい減少が生じることとなる。マイクロRNAミミックはAP+コロニー形成全体に影響を及ぼさないが、一方、阻害剤は影響を及ぼす。
【0131】
MEFはiPS細胞に初期化することができるという発見以降、このすばらしいプロセスの基本的機構の理解に多くの努力が傾けられてきた。本明細書に記載した結果によって、転写後遺伝子調節が初期化中に直接関与すること、そしてRNAi機構への干渉により初期化効率を著しく変更することができることが初めて確認された。加えて、前の実施例に示されるように、3種のマイクロRNAクラスターは、iPS細胞を誘導するために用いられる4種の因子によって著しくアップレギュレートされ、これらのクラスター内のマイクロRNAは少なくとも2つの重要な経路:TGF−βシグナル伝達および細胞周期制御をおそらくターゲッティングする。
【0132】
この研究は続行されているが、いくつかの最近の報告では数種の下流腫瘍抑制遺伝子(p21など)を含むp53経路がiPSC誘導中の大きな障壁の1つであることも確認された。4種の因子(OSKM)の異所発現によりp53が容易にアップレギュレートされ、細胞防御プログラムの連続反応(細胞周期停止、アポトーシスまたはDNA損傷応答など)が開始されることを示す多くの証拠もある。これらの防御応答はおそらく低初期化効率(約0.1%前後と考えられる)の根底にある。しかしながら、これらのデータは、初期化に成功した細胞がいかにして細胞障壁を克服してiPS細胞になるかを説明していない。本明細書に記載した実施例は、初期化の成功に拮抗する経路をターゲッティングするマイクロRNAの発現を誘導することによって、これらの細胞が、全てではないが少なくとも一部において障壁を克服し得ることを示している。初代繊維芽細胞におけるマイクロRNAレベルをモジュレートすることによって、初期化効率の著しい向上が達成され得る。
【0133】
TGF−βシグナル伝達は、原腸形成、器官特異的形態形成および組織ホメオスタシスのような多様なプロセスにおいて機能する重要な経路である。標準的なTGF−β形質導入の現行モデルによって、TGF−βリガンドはTGF−β受容体II(Tgfbr2)と結合し、次いで、それが、Tgfbr1とヘテロ二量体を形成して受容体関連Smadsによりシグナルを伝達することが示されている。TGF−βシグナル伝達は、ヒトおよびマウスES細胞自己複製、の両方で機能することが報告されており、FGF2は、ES細胞培養に広く用いられている増殖因子であり、TGF−βリガンド発現を誘導し、BMP様活性を抑制する。化学阻害剤によるTGF−β受容体Iファミリーキナーゼの遮断はES細胞自己複製に支障をきたす。そのような阻害剤は初期化中に全く異なる役割を果たすと思われるため、これらの発見はiPSC誘導に特に重要である。最近の化学スクリーニングでは、TGF−β受容体I(Tgfbr1)の小分子阻害剤はiPSC誘導を実際に増強し、Nanog発現を誘導することによってSox2の必要条件の代わりとし得ることが示された。さらに、TGF−βリガンドによる初期化中の細胞の処理はiPSC誘導に対して悪影響を及ぼす。よって、TGF−βシグナル伝達はES細胞自己複製に重要であるが、初期化にとっては障壁である。本発明によれば、Tgfbr1に加えて、構成的に活性なキナーゼTgfbr2の活性もまた初期化に拮抗することとなる。また、本発明によれば、miR−93およびそのファミリーメンバーはTgfbr2を直接ターゲッティングして、そのシグナル伝達と初期化をモジュレートすることとなる。
【0134】
P21(これは165アミノ酸しか含まない低分子タンパク質である)は、p53依存性G1増殖停止を引き起こし分化および細胞老化を促進することにより癌発生中の腫瘍抑制遺伝子として長年にわたり認められてきた。本発明によれば、4種の因子(OSKM)がMEF細胞に導入されるとp21発現はアップレギュレートされるが、p21の過剰発現によりiPSC誘導がほぼ完全に遮断される(図9)ため、このアップレギュレーションは初期化プロセスに拮抗することとなる(図8)。Klf4初期化因子がp21プロモーターと結合しp21転写を増加させるため、初期化中の細胞におけるp21の誘導は p53に依存することがあるがp53に依存しないこともある。
【0135】
同じ転写因子がiPSC誘導を促進することがあり、iPSC誘導に拮抗することもあることから、これは4種の初期化因子の機能についての興味深い問題を提起している。実際には、ある特定レベルのp21誘導が初期化プロセスにとって有益であるという可能性を除外することができる証拠は現在ない。p21は、p53依存性細胞周期停止における周知の役割の他に、若干の発癌活性を有することも報告されている。例えば、p21は、アポトーシス(細胞周期制御におけるその通常の機能とは無関係の機能)から細胞を保護することにより発癌活性を有する。
【0136】
初期化におけるp21の潜在的利益は、タンパク質−タンパク質相互作用を通じて遺伝子発現を調節する能力に依存し得る。例えば、p21は、アポトーシスに関与するいくつかのタンパク質(カスパーゼ8、カスパーゼ10およびプロカスパーゼ3など)と直接結合することができる。別の例では、p21は、Myc N末端と結合してMyc−Maxヘテロ二量体形成を遮断することにより、Mycのアポトーシス促進作用のサプレッサーでもある。実際には、Myc自体がMEFにおいて過剰発現されると、細胞培養物では細胞死の著しい増加が認められるが、一方、4種の因子を形質導入した細胞では、細胞死はmycだけのサンプルと比べて最小限にとどまる。従って、p21の誘導は、初期化プロセスの障壁となり得るが、細胞アポトーシスを減少させるのに必要なある特定レベルのp21を維持し得るため初期化効率を向上させ得る。
【0137】
miR−93およびmiR−106bのトランスフェクションはp21 siRNAトランスフェクションよりも初期化に対する促進効果が高く、その際、miR−93および106bはp21 siRNAほどp21の発現を抑制しなかったため、本明細書において示すデータはこの仮説を裏付ける部分的証拠ともいえる。しかしながら、この効果は、これらのマイクロRNAによる、Tgfbr2およびp21を含む複数のタンパク質のターゲッティングによるものである可能性もある。
【0138】
マイクロRNAは通常複数の細胞タンパク質をターゲッティングするため、miR−93およびmiR−106bの促進効果は、初期化に関与するさらなる遺伝子を見つける機会を与え、そのプロセスがより理解される。実際には、p21の他に、G1−S期移行の負の調節因子であると報告されているいくつかの他の遺伝子もmRNA転写物の3'UTR領域にmiR−93およびmiR−106b標的部位を有する(Rb1、Rbl1、Rbl2およびLats2など)。miR−93およびmiR−106bの、報告されている別の興味深い標的は転写因子E2F1であり、この転写因子は多くのヒト腫瘍サンプルにおいて脱制御され過活性化されることがよく見られる。E2F1の1つの重大な機能は、ARFおよびINK4aをコードするCDKN2A遺伝子座の発現を活性化することである。Ink4a/Arf遺伝子座もまた初期化効率を阻害し得る。よって、本発明によれば、miR−93およびmiR−106bのトランスフェクションはE2F1をターゲッティングし、CDKN2A遺伝子座を活性化する可能性を低くして初期化の障壁を下げることができることとなる。
【0139】
最後に、miR−17〜92、miR−106b〜25およびmiR−106a〜363クラスターはマウスとヒトとの間で完全に保存される。よって、本発明によれば、miR−93およびmiR−106bの促進効果はヒト初期化にも適用し得ることとなる。
【0140】
実施例11
マイクロRNAはiPS細胞初期化をモジュレートする
マウス胚性繊維芽細胞(MEF)誘導:Oct4−EGFP MEFは、マウス株B6;129S4−Pou5f1tm2(EGFP)Jae/J(Jackson laboratory;ストック番号008214)から、WiCell研究所のウェブサイト(www.wiCell.org/)で提供されているプロトコールを用いて得る。Oct4−EGFP MEFを、MEF完全培地(10%FBS、非必須アミノ酸、L−グルタミン含有、ピルビン酸ナトリウム不含のDMEM)が入った0.1%ゼラチンコーティングプレートで維持する。
【0141】
レトロウイルスを用いた初期化:4X104個のOct4−EGFP MEFにpMXレトロウイルスを形質導入して、Oct4、Sox2、Klf4およびc−Myc(Addgene)を誤発現させる。2日後に、形質導入Oct4−EGFP MEFにES培地(15%ES−screened FBS、非必須アミノ酸、L−グルタミン、モノチオグリセロールおよび1000U/ml LIF含有のDMEM)を供給し、1日おきに培地を換える。特に断りのない限り、形質導入の約2週間後に、蛍光顕微鏡検査法によって初期化幹細胞(EGFP+iPSCコロニーと定義される)にスコアをつける。iPSCを得るために、実体顕微鏡(Leica)下でEGFP+コロニーを手作業で採取する。
【0142】
マイクロRNA阻害剤またはsiRNAトランスフェクション:let−7a、miR−21、およびmiR−29aマイクロRNAの阻害剤は、Dharmaconから購入する。4X104個のOct4−EGFP MEFをLipofectamineおよび阻害剤で、製造業者の教示(Invitrogen)に従いトランスフェクトする。3〜5時間後に、培地を捨て、MEF完全培地に替える;初期化では、初期化因子(Oct4、Sox2、Klf4およびc−Myc)をコードするレトロウイルスを加え、翌日培地を完全培地に替える。特に断りのない限り、トランスフェクション/形質導入後5日目に、阻害剤またはsiRNAを再度導入する。
【0143】
ノーザン解析では、1×105個のOct4−EGFP MEFをトランスフェクトし、5日後に採取する。トータルRNAをTRIZOL(Invitrogen)により単離し、約9μgのトータルRNAを14%変性ポリアクリルアミドゲル(National Diagnostics)上で分離する。RNAをHybond−XL膜(GE healthcare)上に転写し、マイクロRNAを同位体標識した特異的DNAプローブにより検出する。シグナル強度をホスホイメージャーにより視覚化し、Multi Gauge V3.0(FUJIFILM)を用いて解析する。マイクロRNAシグナル強度を、U6 snRNAのものに対して正規化する。実験は3反復で実施する。
【0144】
ウエスタン解析では、1×105個のOct4−EGFP MEFをトランスフェクトし、5日後に採取する。トータルタンパク質をM−PERバッファー(Pierce)で調製し、同量のトータルタンパク質を10%SDS−PAGEゲル上で分離する。タンパク質をPVDF膜に転写し、次の抗体を用いてバンドを検出する:GAPDH(Santa Cruz;カタログ番号sc−20357)、p53(Santa Cruz;カタログ番号sc−55476)、PI3キナーゼp85(Cell Signaling;カタログ番号4257);Cdc42(Santa Cruz;カタログ番号sc−8401);p−ERK1/2(Cell Signaling;カタログ番号9101);ERK1/2(Cell Signaling;カタログ番号9102);p−GSK3β(Cell Signaling;カタログ番号9323);GSK3β(Cell Signaling;カタログ番号9315);βアクチン(Thermo Scientific;カタログ番号MS−1295)。シグナル強度を、Multi Gauge V3.0(FUJIFILM)により定量し、GAPDHまたはβアクチンに対して正規化する。実験は3〜5回繰り返す。
【0145】
in vitro分化および奇形腫形成アッセイ:in vitro分化では、iPSCをトリプシン/EDTAにより解離し、胚様体(EB)培地(15%FBS、非必須アミノ酸、L−グルタミン含有のDMEM)中に終濃度5X104細胞/mlに再懸濁する。EB形成を誘導するために、逆さにしたペトリ皿の蓋で20μl中1000個のiPS細胞を懸滴培養する。3〜5日後に、EBを採取し、ウェル当たり約10個のEBで0.1%ゼラチンコーティングした6ウェルプレートに転写する。EB形成の2週間後に、収縮を繰り返す心筋細胞(中胚葉)が顕微鏡により確認される。内胚葉および外胚葉から得られた細胞は、それぞれ、α−フェトプロテイン(R&D;カタログ番号MAB1368)抗体およびニューロン特異的βIII−チュブリン(abcam;カタログ番号ab7751)抗体によって確認した。
【0146】
奇形腫アッセイでは、1.5X106個のiSPCをトリプシン処理し、150μlに再懸濁し、次いで、アベルチンで麻酔をかけた無胸腺ヌードマウスの後肢背面に皮下注射する。3週間後に、マウスを犠牲にして、奇形腫を採取する。腫瘍塊を固定し、切開し、サンフォード・バーナム研究所にあるCell Imaging−Histology core施設内で解析する。
【0147】
キメラ解析:採取の2時間前にiPSC培地を換える。トリプシン処理したiPSCを0.1%ゼラチンコーティングプレートで30分間培養して、フィーダー細胞を除去する。iPSCをE3.5 C57BL/6−cBrd/cBrd胚盤胞に注射し、次いで、偽妊娠したレシピエント雌へ移す。生後、子の毛色によりiPSCの寄与を評価する:黒色はiPSCに由来する。
【0148】
免疫蛍光およびアルカリ性ホスファターゼ(AP)染色:iPSCを0.1%ゼラチンコーティングした6ウェルプレートに播種し培養する。4日後に、細胞を4%パラホルムアルデヒド(Electron Microscopy Sciences;カタログ番号15710−S)により固定する。免疫蛍光染色では、固定した細胞をPBS中0.1%Trixton X−100により透過処理し、5%BSA/PBSによりブロッキングする。SSEA−1(R&D;カタログ番号MAB2155)およびNanog(R&D;カタログ番号AF2729)に対する抗体をESマーカーとする。核をHoechst 33342染色(Invitrogen)により視覚化する。AP染色では、アルカリ性ホスファターゼ基質を用い製造業者の教示に従って(Vector Laboratories;カタログ番号SK−5100)固定した細胞を処理する。
【0149】
実施例12
miR−21またはmiR−29aの阻害は初期化効率を高める
マウス胚性繊維芽細胞(MEF)誘導:Oct4−EGFP MEFは、マウス株B6;129S4−Pou5f1tm2(EGFP)Jae/J(Jackson laboratory;ストック番号008214)から、WiCell研究所のウェブサイト(www.wiCell.org/)で提供されているプロトコールを用いて得る。Oct4−EGFP MEFを、MEF完全培地(10%FBS、非必須アミノ酸、L−グルタミン含有、ピルビン酸ナトリウム不含のDMEM)が入った0.1%ゼラチンコーティングプレートで維持する。
【0150】
MEF特異的miRNAの阻害が初期化障壁を低くするかどうかを判定するために、MEFで濃縮されたmiRNAを解析し、それらのレベルを、マウスES(mES)細胞で見られるものと比較する。図20aに示されるように、let−7a、miR−21およびmiR−29aは、mES細胞と比べ、MEFにおいて高度に発現される。一方、miR 291は、mESでは非常に豊富であるがMEFでは存在しない(図20a)。次に、let−7a、miR−21、およびmiR−29aに対し、miRNA阻害剤を、Oct3/4、Sox2、Klf4およびc−Myc(OSKM)を発現するレトロウイルスとともにOct4−EGFP MEF(Oct4−EGFPレポーターを保有するMEF)に導入する。形質導入後14日目に、miR−21阻害剤で処理した細胞はターゲッティングしない(NT)対照と比べて初期化効率の約2.1倍向上を示す(図20b)。同様に、miR−29aの阻害後、初期化効率は有意に約2.8倍向上する(図20b)。miR 29aまたは21阻害で用いた同様のantagomir処理に従って、let−7a阻害後にOSKM初期化に対して軽度の効果が観察される(図20b)。c−Mycの不在下でmiRNA阻害により3種の因子による初期化が促進されるかどうかをさらに試験するために、miRNA阻害剤を、OSKMよりずっと低い効率で細胞を初期化するOSKとともに細胞に形質導入する。OSKにより初期化したiPS細胞コロニーの数は、OSK単独での処理と比べ、miR−21阻害剤の存在下で増加する(図25)。これらの結果は、MEFで濃縮されたmiRNA miR−21およびmiR−29の枯渇により4Fによる初期化が著しく促進されること、そしてmiR−21の遮断により3種の因子(OSK)による初期化の効率が中等度に向上することを示す。
【0151】
C−Mycは、初期化中にmiRNA let−7a、miR−16、miR−21、miR−29aおよびmiR−143の発現を抑制する:最近の研究により、OSKM因子は、後成的機構と転写機構の両方を通じて細胞同一性を変更することが示されている。本発明によれば、OSKM初期化因子は、MEFで濃縮されたmiRNAをダウンレギュレートし得ることとなる。miRNA発現に対する各初期化因子の潜在的影響を評価するために、MEFにOSKM因子の様々な組合せを形質導入し、それらに対してノーザンブロット解析を行う(図21a)。興味深いことに、Sox2だけが、miR−21、miR−29aおよびlet−7aの発現レベルが、MEF対照と比べて2倍より高く誘導する(図21b、左のパネル)。Klf4は、選択したmiRNAに対して軽度であるがSox2と類似した影響を及ぼす(図21b、左のパネル)。Oct4過剰発現に関してのみ、miRNAは発現レベルが変化しない(図21b、左のパネル)。Oct4、Sox2およびKlf4とは対照的に、単一因子のc−MycはmiR−21およびmiR−29a(MEFにおいて最も豊富なmiRNA)の発現をMEF対照の約70%ダウンレギュレートする(図21aおよび図21b、左のパネル)。さらに、図21b(中央のパネル)に示す2種の因子(2F)の様々な組合せの間では、c−Mycを含む組合せは、3種のmiRNA全て(miR−21、miR−29aおよびlet−7aを含む)における減少を、約25〜80%増大し得る(図21b、中央のパネル)。Sox2とOct4とでは、miRNAに対する1Fの影響と同様に、miR−21およびmiR−29aを、MEF対照の1.5倍および2.3倍増加させ、OKおよびSKではmiRNA発現に対して及ぼす明らかな影響はない。さらに、3種の因子(3F)の様々な組合せの間では、miRNA−21の発現は、SKM細胞およびOKM細胞において、MEFにおいて見られる発現と比べて、それぞれ約70%および78%減少し、同様に、miR−29a発現は、c−Mycを含む3F組合せで約48〜70%減少する(図21b、右のパネル)。3F組合せにc−Mycを含むことによりlet−7aレベルも少し減少する(図21b、右のパネル)。c−Mycを含まないOSKでは、miRNA発現に対して及ぼす影響はほとんどなかった(図21b、右のパネル)。よって、これらの結果は、c−Mycが初期化中のmiRNA発現の調節において重要な役割を果たすことを強く示唆している。
【0152】
c−MycがmiRNA発現に拮抗する第1因子であることをさらに確認するために、OSKをc−Mycとともにまたはc−Mycを含めずに細胞に形質導入し、miRNA発現を形質導入後の様々な時点においてリアルタイム定量的逆転写ポリメラーゼ連鎖反応(RT−qPCR)により調査する。OSKに対し、OSKM形質導入により初期化中のlet−7a、miR−16、miR−21、miR−29a、miR−143の発現は大幅に減少し(図21c)、c−Mycは、MEFで濃縮されたmiRNA(最も豊富なものである、let−7a、miR−21およびmiR−29aを含む)の発現の調節において中心的な役割を果たすことが分かる。これらのデータは、c−MycはmiRNAダウンレギュレーションを通じてある程度初期化を促進することも示唆している。
【0153】
実施例13
miRNA枯渇によって誘導されたiPS細胞は多能性を獲得する
本発明によれば、miR−21およびmiR−29a阻害剤を用いて得られたマウスiPS細胞は多能性であることとなる。OSKM/anti miR−29a iPS細胞についてES細胞マーカー染色を行うことができる。さらなる解析のために、OSKMおよびmiR−29a阻害剤での処理後に得られたGFP+コロニーを採取する。胚幹細胞マーカーNanogおよびSSEA1を発現する代表的なコロニーを同定する。内因性Oct4も活性化し、それはEGFP染色により示すことができる。強いアルカリ性ホスファターゼ(AP)活性はESマーカーの1つとして観察することができる。
【0154】
OSKM/anti miR−29a iPS細胞のin vitro分化を行うことができる。胚様体をin vitroで形成し、2週間培養することができる。細胞を固定し、抗αフェトプロテイン(中胚葉について)および抗βチュブリンIII(外胚葉について)で染色することができる。核は、Hoescht染色による対比染色として観察することができる。OSKM/anti miR−29a iPS細胞の奇形腫形成解析も行うことができる。1.5X106個のiPSCを無胸腺ヌード雌マウスに皮下注射する。注射の3週間後に腫瘍塊を採取し、組織病理学的解析のために固定する。3胚葉から得られた様々な組織を確認することができ、それらには、腸管様上皮(内胚葉)、脂肪組織、軟骨および筋肉(中胚葉)、ならびに神経組織および表皮(外胚葉)が含まれる。OSKM/anti miR−29a iPS細胞およびOSK/anti miR−21 iPS細胞のキメラ解析も行うことができる。8〜14個のiPS細胞をE3.5マウス胚盤胞に注射することができる。各キメラへのiPS細胞の寄与は、黒色の毛色を評価することにより推定することができ、割合として観察することができる。
【0155】
miR−21またはmiR−29aの遮断がiPS細胞の多能性に支障をきたすかどうかを調査するために、OSKM/anti miR−29aまたはOSK/anti miR−21を用いたiPS細胞を多能性について評価する。まず、初期化のおよそ2週間後に細胞を手作業で採取し、拡大して、形態およびES特異的マーカーの発現を調べる。細胞はES様形態および高度に発現されたOct4−EGFP示す(内因性ES細胞シグナル伝達の確立を示す。加えて、OSKM/anti miR−29a iPS細胞またはOSK/anti miR−21 iPS細胞は、NanogおよびSSEA1を含むES細胞特異的マーカーを発現し、アルカリ性ホスファターゼ活性を示した。それらのiPS細胞が、通常誘導されるiPS細胞と同等の多能性潜在能力を示すかどうかを試験するために、OSKM/anti miR−29a iPS細胞およびOSK/anti miR−21 iPS細胞を、誘導して胚様体(EB)を形成するかまたはヌードマウスに注射し、様々な組織に分化させる。in vitro分化の2週間後に、3胚葉全てから得られた典型的な細胞種が観察される。注射の3週間後に形成された奇形腫腫瘍に対して、組織病理学的解析を行う。3胚葉全てに由来する様々な組織が生成され、iPS細胞が多能性を得たことが確認される。多能性についての最も厳しい試験を用いるために、iPS細胞をE3.5胚盤胞に注射して、キメラマウスを作出する。miR枯渇によるiPS細胞から得られたマウスは、iPS細胞に起因する、有意の約15%〜25%黒色毛色を示す。これらのデータは、miR−21およびmiR−29aの枯渇が、得られたiPS細胞の多能性に対して悪影響を及ぼさないことを示している。
【0156】
実施例14
miR−29aの阻害はP85αおよびCDC42の経路を通じてP53をダウンレギュレートする
初期化に及ぼすmiR−29aの作用の基礎にある機構を理解するために、p85αおよびCDC42の発現を調査する。p85αおよびCDC42はHeLa細胞においてmiR−29の直接標的であると報告されている。その目的で、miRNA阻害剤をMEFにトランスフェクトし、トランスフェクション後5日目にp85αおよびCDC42タンパク質発現をウエスタンブロットにより評価する。P85αおよびCDC42タンパク質レベルはmiR−29a遮断によって少し増加するが、それに対し、let−7a阻害剤ではほとんど影響がない(図22aおよび図22b)。また、形質転換関連タンパク質53(Trp53またはp53)はp85αおよびCDC4の直接標的であると報告されている。よって、p53についても、p53がMEFにおいてmiR−29aによって間接的に調節されるかどうかを調査する。それを試験するために、MEFをmiRNA阻害剤でトランスフェクトし、免疫ブロッティングのために5日採取して、p53の発現を評価する。P53タンパク質レベルはmiR−29a阻害によって約30%低下する(図22aおよび図22b)がNT対照やlet−7a阻害では変化しない。miR−21の枯渇によってもp85αおよびCDC42タンパク質抑制から解放され、それゆえにp85αおよびCDC42のレベルが増加し、その結果、p53発現の約25%ダウンレギュレーションが起こることは重要である(図22aおよび図22b)。
【0157】
初期化中にmiR−21またはmiR−29aの阻害によってp53レベルが低下することをさらに確認するために、初期化5日目にp53発現をウエスタンブロット解析により調査する。初期化を開始するために、miRNA阻害剤をOSKMとともに導入する。MEF単独での観察と一致して、初期化中に、miR−21またはmiR−29aの枯渇によって、p53タンパク質レベルがOSKM対照と比べてそれぞれ約25%または約40%低下する(図22c)。要するに、我々のデータによって、初期化中、miR−29aの遮断によってp85αおよびCDC42の経路を通じてp53タンパク質レベルが約30〜40%低下することがわかった。加えて、miR−21の枯渇はp85αおよびCDC42の両方に対して類似した影響を及ぼし、p53タンパク質レベルを約25%〜約30%低下させた。
【0158】
miR−29aの阻害は、p53ダウンレギュレーションを通じて初期化効率を向上させる:p53の欠乏によって初期化効率が大幅に向上し得ることは報告されている。miR−29aの枯渇はp53レベルを著しく低下させ、初期化効率を約2.8倍向上させることから、本発明によれば、miR−29aノックダウンの効果は主にp53ダウンレギュレーションによって媒介されることとなる。これを受けて、p53 siRNAおよび/またはmiR−29a阻害剤をOSKMとともにOct4−EGFP MEFにトランスフェクトして、初期化を開始する。siRNAによるp53のダウンレギュレーション(約80%)は、初期化効率に対して、miR−29a阻害の場合と類似した良い影響を及ぼす(図22d)。p53 siRNAの存在下でmiR阻害剤を加えた場合には初期化効率の向上は見られない(図22d)。これらの結果は、miR−29aの阻害はp53のダウンレギュレーションを通じて初期化効率を向上させるようにある程度作用することを示唆している。
【0159】
実施例15
miR−21およびmiR−29aの阻害はGSK3βではなくERK1/2のリン酸化を減少させて初期化を促進する
miR21は、心臓繊維芽細胞においてsprouty homologue 1(Spry1)の阻害を通じてMAPK/ERKを活性化することが報告されている。MAPK/ERK活性の遮断は、神経幹細胞の初期化を促進し、ESC自己複製の基底状態を保証する。よって、本発明によれば、miRNA阻害剤導入後にMEFにおけるERK1/2リン酸化を評価することによってmiR−21は初期化中にMAPK/ERK経路を調節することとなる。それを試験するために、MEFをmiRNA阻害剤でトランスフェクトし、次いで、ウエスタンブロット解析のために採取して、リン酸化ERK1/2レベルを決定する。ウエスタンブロット解析は、miR−21の遮断によってERK1/2リン酸化がNT対照と比べて約45%有意に低下したが、それに対し、let−7a阻害剤ではそのような影響がないことを示している(図23a)。興味深いことに、miR−29a枯渇MEFでもERK1/2リン酸化がNT対照と比べて60%有意に低下する(図23a)。また、本発明によれば、miR−21およびmiR−29aは、Spry1レベルを変更することによりERK1/2リン酸化に影響を及ぼし得ることとなる。様々なmiRNA阻害剤をトランスフェクトすることによりMEFにおいてmiR−21またはmiR−29aを枯渇させ、Spry1発現レベルを免疫ブロッティングにより定量し、それらの結果は、miR−21およびmiR−29aの阻害によりSpry1発現レベルが高まった(図23b)ことを示している。ゆえに、miR−21およびmiR−29aの枯渇は、Spry1タンパク質レベルをモジュレートすることによってERK1/2のリン酸化をダウンレギュレートする。
【0160】
ERK1/2ダウンレギュレーションによって初期化効率が高まるかどうかについて取り組むために、4F初期化の過程でERK1または2をターゲッティングするsiRNAをOct4−EGFP MEFに導入する。いずれの枯渇も成熟したiPS細胞の生成を促進する(図23c)。本発明によれば、miR−21の遮断によってERK1/2リン酸化が減少することからmiR−21はMEFにおけるERK1/2活性化のインデューサーとして働くこととなる。miR−29aの枯渇によってもERK1/2リン酸化は著しく減少する。これらの結果は、miR−21およびmiR−29aがERK1/2活性を調節して初期化効率を高めることを強く示唆している(図23a、図23bおよび図23c)。
【0161】
GSK3β経路もES自己複製および神経幹細胞の初期化を抑制する。GSK3βの枯渇により成熟iPS細胞生成は大幅に増加する(図23c)。本発明によれば、miRNA枯渇がGSK3β活性化を調節したこととなる。免疫ブロッティングは、Oct4−EGFP MEFにおけるmiRNAの遮断がGSK3β活性化に対して大きな影響を及ぼさないことを示している(図23d)。本発明によれば、細胞生存率および複製速度を評価するためにフローサイトメトリーを用いることによってmiRNA枯渇が初期化中にアポトーシスまたは細胞増殖を変更することとなる。OSKMでの初期化中のmiRNA 21、29aまたはlet−7の遮断ではアポトーシスまたは増殖速度は変更されない(図26)。全体として、miR−29aおよびmiR−21はp53およびERK1/2の経路をモジュレートして、iPS細胞初期化効率を調節する。
【0162】
実施例16
C−MycはmiR−21およびmiR−29aのダウンレギュレーションを通じてP53レベルを低下させERK1/2活性化に拮抗することにより初期化の閾値を下げる
初期化誘導に現在用いられている導入遺伝子の代替物を開発するために、これらの因子によってシグナル伝達経路がどのように調節されるかを理解することが重要である。本発明によれば、c−Mycは初期化中に、MEFで濃縮されたmiRNA(miR−21、let−7a、およびmiR−29aなど)を抑制することとなる(図20)。阻害剤によるmiR−29aの枯渇は、p85αおよびCDC42抑制を解放することによってp53タンパク質レベルを低下させる可能性が高い(図22)。加えて、miR−21の枯渇はERK1/2リン酸化も減少させる(図23)。本発明によれば、miR−21阻害はp53タンパク質レベルを低下させることとなり、そしてmiR−29aの阻害もERK1/2リン酸化レベルを低下させることとなる。p53およびERK1/2のシグナル伝達はいずれも初期化に拮抗する。miR−21およびmiR−29aの遮断またはp53およびERK1/2のノックダウンにより初期化効率を高めることができる(図22および図23)。本発明によれば、c−Mycは、p53タンパク質レベルおよびERK1/2活性化の誘導を通じて初期化障壁として働く、MEFで濃縮されたmiRNA、miR−21およびmiR−29aを抑制することによって、ある程度初期化を促進することとなる(図24)。
【0163】
初期化を促進するために、miR−290ファミリーのES特異的miRNAの強制発現をc−Mycの代わりにすることができる。また、C−MycはmiR−290クラスターのプロモーター領域と結合する。しかしながら、c−Myc導入遺伝子の初期発現は、初期化を開始するのに有効であるが、miR−290クラスターが発現を開始する成熟iPS細胞の移行段階またはそれ以降では重要なものでない。よって、c−MycがmiR−290ファミリーの活性化を通じて初期化の初期段階を促進するという可能性は低い。
【0164】
本発明によれば、初期化のためにc−Mycが導入されると、MEFで濃縮されたmiRNA(miR−29a、miR−21、miR−143およびlet−7aを含む)の発現レベルは低下することとなる。C−Mycは、腫瘍形成の促進または多能性基底状態の維持においてmiRNAに深刻な転写の影響を及ぼす。よって、miRNA発現のc−Myc抑制は初期化の基礎にあると考えられる機構である。
【0165】
miR−21は、正のメディエーターとして、TGFβ1およびERK1/2の経路(これらの両経路は初期化およびES細胞基底状態に影響を及ぼすことがわかっている)を通じて繊維形成活性を増強するように働く。特に、確認したmiR−29a標的の間では、p53がmiR−29aによって正に調節される。加えて、最近の研究では、Ink4−Arf/p53/p21経路が初期化に支障をきたし、p53の欠乏によって初期化効率が大幅に高まることが示されている。これらのことから、これらのシグナル伝達経路は初期化プロセスに対する第1の障壁であるといえよう。
【0166】
c−MycをターゲッティングするmiRNA、miR−21およびmiR−29aの枯渇により、初期化効率が約2.1倍〜約2.8倍高まり(図20)、MEFで濃縮されたmiRNAも初期化障壁として働くことが示唆される。let−7阻害は初期化を促進することが最近報告されたが、しかしながら、何度かの試みによると、antagomirによりlet−7が阻害された場合、観察される初期化への影響は軽度なものでしかない(図20)。さらに、本発明によれば、初期化中のp53の誘導はmiR−29a阻害によって支障をきたし初期化効率が高まることとなる。同様に、初期化は、miR−21の枯渇またはERK1/2のいずれかによって大幅に促進され得る。C−Mycは、初期化の初期段階の大きな要因であり、そのプロセスを移行段階および終わりに近い段階で維持するには必要でなく、これによれば、高効率の初期化を開始するためにc−Mycを調節するmiRNAを使用し得ることとなる。
【0167】
C−Mycは、miR−29aプロモーターと直接結合し抑制することが報告されている。本発明によれば、c−Mycは、miR−21の枯渇と部分的にしか置き換えることができないこととなり、これはc−Mycが初期化において他の機能を有することを示唆している。よって、初期化中のc−Myc機能に代わる、複数の経路の調節またはMEFで濃縮されたmiRNAの広い抑制が必要とされ得る。
【0168】
本発明によれば、c−MycはmiR−21およびmiR−29aのダウンレギュレーションを通じてp53レベルを低下させERK1/2活性化に拮抗することにより初期化の閾値を下げることとなる。加えて、c−Mycの下流の因子は、初期化を改善するために、siRNA、miRNAまたは小分子による操作の対象となり得る。これらのアプローチは、4種の初期化因子全ての代わりとするために広げることができる。
【0169】
上記の実施例により本発明を記載してきたが、修飾および変形が本発明の精神および範囲内に含まれることは理解されるであろう。よって、本発明は次の特許請求の範囲によってのみ限定される。
【図1A】
【図1B】
【図1C】
【図1D】
【図1E】
【図1F】
【図2A】
【図2B】
【図2C】
【特許請求の範囲】
【請求項1】
誘導多能性幹(iPS)細胞を生成する方法であって、
a)体細胞を核初期化因子と接触させること;および
b)(a)の細胞を、該細胞内でRNAレベルまたは活性を変更するマイクロRNAと接触させ、それによってiPS細胞を生成すること
を含む、前記生成する方法。
【請求項2】
前記マイクロRNAまたはRNAが修飾される、請求項1に記載の方法。
【請求項3】
前記マイクロRNAがベクター中に存在する、請求項1に記載の方法。
【請求項4】
前記マイクロRNAが、miR−17、miR−25、miR−106a、miR let−7ファミリーメンバーまたはmiR−302bクラスター中に存在する、請求項1に記載の方法。
【請求項5】
前記マイクロRNAが、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである、請求項1に記載の方法。
【請求項6】
前記マイクロRNAが、配列番号1を含むポリヌクレオチド配列を有する、請求項1に記載の方法
【請求項7】
前記マイクロRNAが、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する、請求項1に記載の方法。
【請求項8】
前記マイクロRNAが、p21、Tgfbr2、p53、Ago2、またはそれらの組合せの発現または活性を調節する、請求項1に記載の方法。
【請求項9】
前記マイクロRNAが、Spry 1/2、p85、CDC42またはERK1/2の経路を調節する、請求項1に記載の方法。
【請求項10】
前記核初期化因子が、ベクター中に含まれる遺伝子によりコードされる、請求項1に記載の方法。
【請求項11】
前記核初期化因子が、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せである、請求項1に記載の方法。
【請求項12】
前記核初期化因子が、OCT4、SOX2、KLF4、C−MYCの1種以上である、請求項1に記載の方法。
【請求項13】
前記核初期化因子がc−Mycを含む、請求項1に記載の方法。
【請求項14】
前記マイクロRNAとの接触前に、接触と同時に、または接触後に、前記体細胞に前記初期化因子を接触させる、請求項1に記載の方法。
【請求項15】
前記体細胞が哺乳類細胞である、請求項1に記載の方法。
【請求項16】
請求項1に記載の方法を用いて生産された誘導多能性幹(iPS)細胞。
【請求項17】
請求項1に記載の方法により生産された誘導多能性幹(iPS)細胞の濃縮された集団。
【請求項18】
請求項1に記載の方法により生産された多能性幹細胞の分化を誘導することにより得られた分化した細胞。
【請求項19】
対象を治療する方法であって、
a)請求項1に記載の方法により対象の体細胞から誘導多能性幹(iPS)細胞を生成すること;
b)工程(a)のiPS細胞の分化を誘導すること;および
c)(b)の細胞を該対象に導入し、それによって症状を治療すること
を含む、前記治療する方法。
【請求項20】
iPS細胞の生成効率を高めるためのマイクロRNAの使用。
【請求項21】
前記マイクロRNAが、miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、miR let−7ファミリーメンバー、またはそれらの組合せからなる群から選択される、請求項20に記載の使用。
【請求項22】
miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、miR let−7ファミリーメンバー、またはそれらの組合せの少なくとも2種以上からなる群から選択されるmiR配列の組合せ。
【請求項23】
誘導多能性幹(iPS)細胞を生成する方法であって、
a)体細胞を核初期化因子と接触させること;および
b)(a)の細胞を、マイクロRNAの阻害剤と接触させ、それによってiPS細胞を生成すること
を含む方法。
【請求項24】
前記マイクロRNAが、miR−17、miR−25、miR−106a、miR let−7ファミリーメンバー、またはmiR−302bクラスター中に存在する、請求項23に記載の方法。
【請求項25】
前記マイクロRNAが、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである、請求項23に記載の方法。
【請求項26】
前記マイクロRNAが、配列番号1に示されるポリヌクレオチド配列を有する、請求項23に記載の方法。
【請求項27】
前記マイクロRNAが、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する、請求項23に記載の方法。
【請求項28】
前記マイクロRNAが、p21、Tgfbr2、p53、Ago2、またはそれらの組合せの発現または活性を調節する、請求項23に記載の方法。
【請求項29】
前記マイクロRNAが、Spry 1/2、p85、CDC42、またはERK1/2の経路を調節する、請求項23に記載の方法。
【請求項30】
前記核初期化因子が、ベクター中に含まれる遺伝子によりコードされる、請求項23に記載の方法。
【請求項31】
前記核初期化因子が、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せである、請求項23に記載の方法。
【請求項32】
前記核初期化因子が、OCT4、SOX2、KLF4、C−MYCの1種以上である、請求項23に記載の方法。
【請求項33】
初期化効率が、前記マイクロRNA阻害剤を使用しない対照と比べて少なくとも2倍である、請求項23に記載の方法。
【請求項34】
前記体細胞が繊維芽細胞を含む、請求項23に記載の方法。
【請求項35】
前記マイクロRNA阻害剤との接触前に、接触と同時に、または接触後に、前記体細胞に前記初期化因子を接触させる、請求項23に記載の方法。
【請求項36】
前記体細胞が哺乳類細胞である、請求項23に記載の方法。
【請求項37】
請求項23に記載の方法を用いて生産された誘導多能性幹(iPS)細胞。
【請求項38】
請求項23に記載の方法により生産された誘導多能性幹(iPS)細胞の濃縮された集団。
【請求項39】
請求項38に記載の多能性幹細胞の分化を誘導することにより得られた分化した細胞。
【請求項1】
誘導多能性幹(iPS)細胞を生成する方法であって、
a)体細胞を核初期化因子と接触させること;および
b)(a)の細胞を、該細胞内でRNAレベルまたは活性を変更するマイクロRNAと接触させ、それによってiPS細胞を生成すること
を含む、前記生成する方法。
【請求項2】
前記マイクロRNAまたはRNAが修飾される、請求項1に記載の方法。
【請求項3】
前記マイクロRNAがベクター中に存在する、請求項1に記載の方法。
【請求項4】
前記マイクロRNAが、miR−17、miR−25、miR−106a、miR let−7ファミリーメンバーまたはmiR−302bクラスター中に存在する、請求項1に記載の方法。
【請求項5】
前記マイクロRNAが、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである、請求項1に記載の方法。
【請求項6】
前記マイクロRNAが、配列番号1を含むポリヌクレオチド配列を有する、請求項1に記載の方法
【請求項7】
前記マイクロRNAが、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する、請求項1に記載の方法。
【請求項8】
前記マイクロRNAが、p21、Tgfbr2、p53、Ago2、またはそれらの組合せの発現または活性を調節する、請求項1に記載の方法。
【請求項9】
前記マイクロRNAが、Spry 1/2、p85、CDC42またはERK1/2の経路を調節する、請求項1に記載の方法。
【請求項10】
前記核初期化因子が、ベクター中に含まれる遺伝子によりコードされる、請求項1に記載の方法。
【請求項11】
前記核初期化因子が、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せである、請求項1に記載の方法。
【請求項12】
前記核初期化因子が、OCT4、SOX2、KLF4、C−MYCの1種以上である、請求項1に記載の方法。
【請求項13】
前記核初期化因子がc−Mycを含む、請求項1に記載の方法。
【請求項14】
前記マイクロRNAとの接触前に、接触と同時に、または接触後に、前記体細胞に前記初期化因子を接触させる、請求項1に記載の方法。
【請求項15】
前記体細胞が哺乳類細胞である、請求項1に記載の方法。
【請求項16】
請求項1に記載の方法を用いて生産された誘導多能性幹(iPS)細胞。
【請求項17】
請求項1に記載の方法により生産された誘導多能性幹(iPS)細胞の濃縮された集団。
【請求項18】
請求項1に記載の方法により生産された多能性幹細胞の分化を誘導することにより得られた分化した細胞。
【請求項19】
対象を治療する方法であって、
a)請求項1に記載の方法により対象の体細胞から誘導多能性幹(iPS)細胞を生成すること;
b)工程(a)のiPS細胞の分化を誘導すること;および
c)(b)の細胞を該対象に導入し、それによって症状を治療すること
を含む、前記治療する方法。
【請求項20】
iPS細胞の生成効率を高めるためのマイクロRNAの使用。
【請求項21】
前記マイクロRNAが、miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、miR let−7ファミリーメンバー、またはそれらの組合せからなる群から選択される、請求項20に記載の使用。
【請求項22】
miR−17、miR−25、miR−93、miR−106a、miR−106b、miR−21、miR−29a、miR−302bクラスター、miR let−7ファミリーメンバー、またはそれらの組合せの少なくとも2種以上からなる群から選択されるmiR配列の組合せ。
【請求項23】
誘導多能性幹(iPS)細胞を生成する方法であって、
a)体細胞を核初期化因子と接触させること;および
b)(a)の細胞を、マイクロRNAの阻害剤と接触させ、それによってiPS細胞を生成すること
を含む方法。
【請求項24】
前記マイクロRNAが、miR−17、miR−25、miR−106a、miR let−7ファミリーメンバー、またはmiR−302bクラスター中に存在する、請求項23に記載の方法。
【請求項25】
前記マイクロRNAが、miR−93、miR−106b、miR−21、miR−29a、またはそれらの組合せである、請求項23に記載の方法。
【請求項26】
前記マイクロRNAが、配列番号1に示されるポリヌクレオチド配列を有する、請求項23に記載の方法。
【請求項27】
前記マイクロRNAが、配列番号2〜11からなる群から選択されるポリヌクレオチド配列を有する、請求項23に記載の方法。
【請求項28】
前記マイクロRNAが、p21、Tgfbr2、p53、Ago2、またはそれらの組合せの発現または活性を調節する、請求項23に記載の方法。
【請求項29】
前記マイクロRNAが、Spry 1/2、p85、CDC42、またはERK1/2の経路を調節する、請求項23に記載の方法。
【請求項30】
前記核初期化因子が、ベクター中に含まれる遺伝子によりコードされる、請求項23に記載の方法。
【請求項31】
前記核初期化因子が、SOXファミリー遺伝子、KLFファミリー遺伝子、MYCファミリー遺伝子、SALL4、OCT4、NANOG、LIN28、またはそれらの組合せである、請求項23に記載の方法。
【請求項32】
前記核初期化因子が、OCT4、SOX2、KLF4、C−MYCの1種以上である、請求項23に記載の方法。
【請求項33】
初期化効率が、前記マイクロRNA阻害剤を使用しない対照と比べて少なくとも2倍である、請求項23に記載の方法。
【請求項34】
前記体細胞が繊維芽細胞を含む、請求項23に記載の方法。
【請求項35】
前記マイクロRNA阻害剤との接触前に、接触と同時に、または接触後に、前記体細胞に前記初期化因子を接触させる、請求項23に記載の方法。
【請求項36】
前記体細胞が哺乳類細胞である、請求項23に記載の方法。
【請求項37】
請求項23に記載の方法を用いて生産された誘導多能性幹(iPS)細胞。
【請求項38】
請求項23に記載の方法により生産された誘導多能性幹(iPS)細胞の濃縮された集団。
【請求項39】
請求項38に記載の多能性幹細胞の分化を誘導することにより得られた分化した細胞。
【図3】

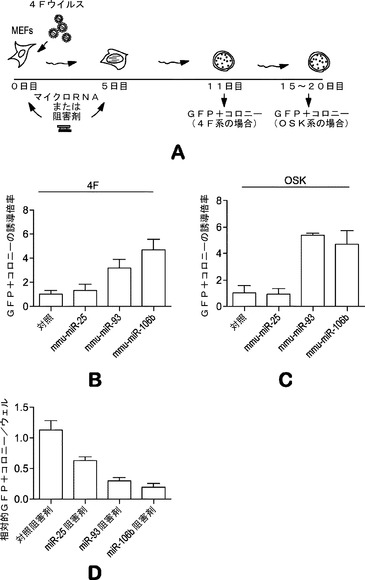
【図4】
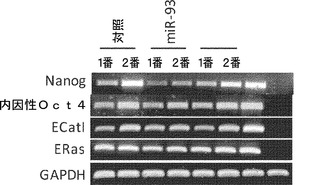
【図5A】

【図5B】
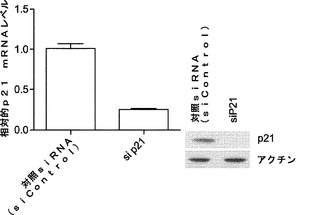
【図5C】
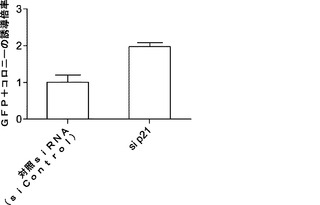
【図5D】
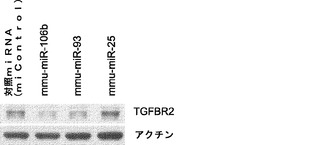
【図5E】
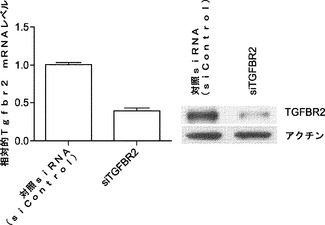
【図5F】
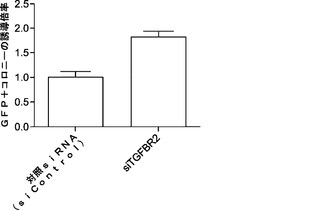
【図6A】
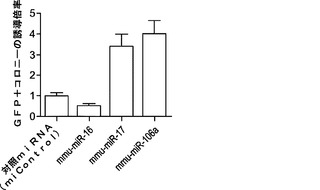
【図6B】
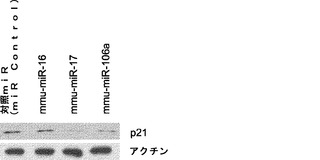
【図6C】
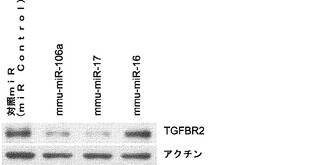
【図6D】
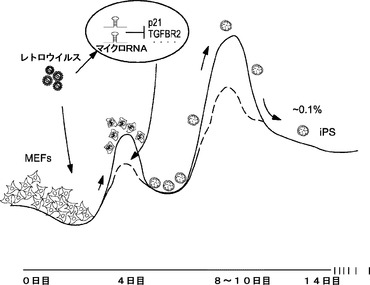
【図7】
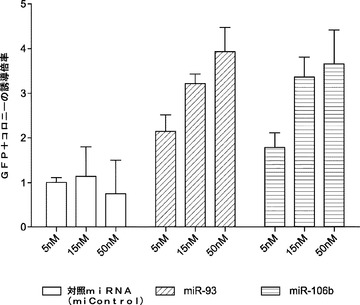
【図8】
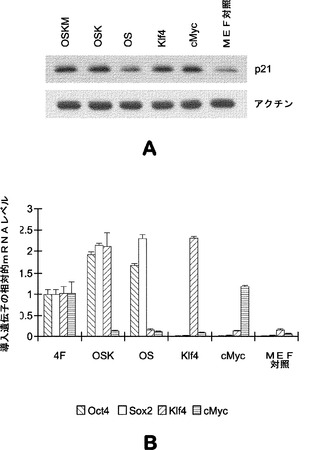
【図9】
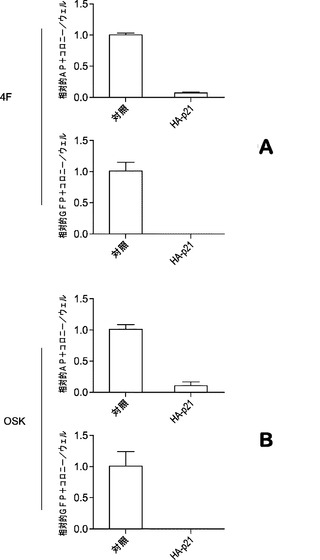
【図10A】
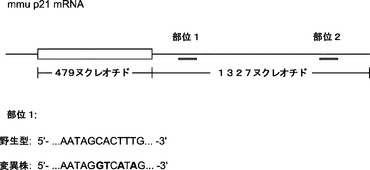
【図10B】
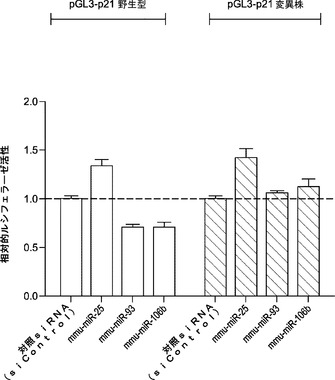
【図11】
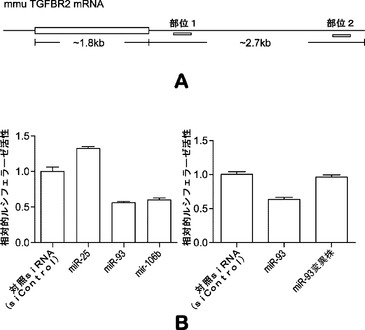
【図12】
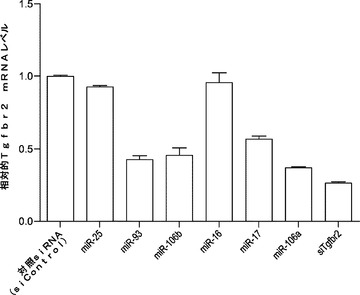
【図13】
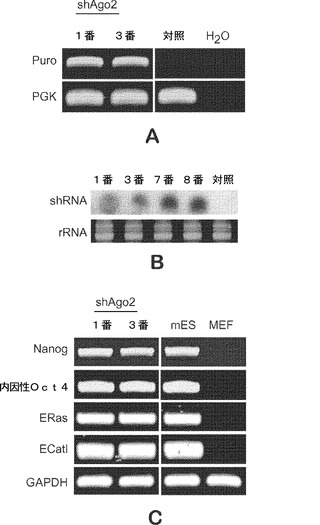
【図14】
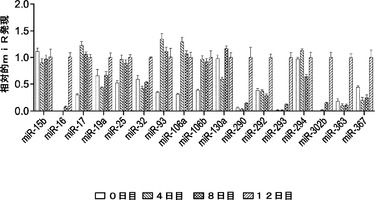
【図15】
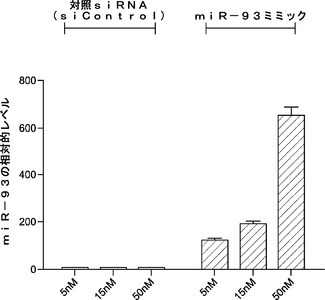
【図16A】
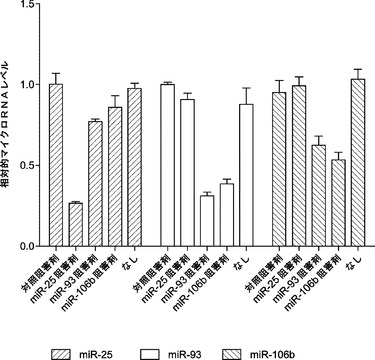
【図16B】
【図17】

【図18A】
【図18B】
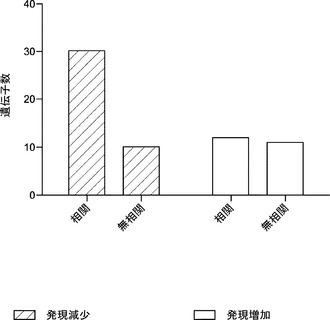
【図19】
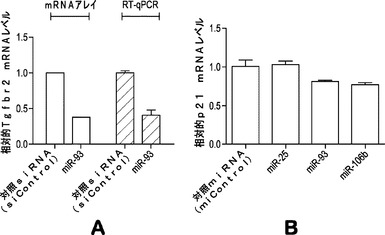
【図20】
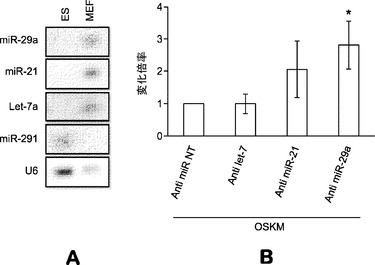
【図21A】
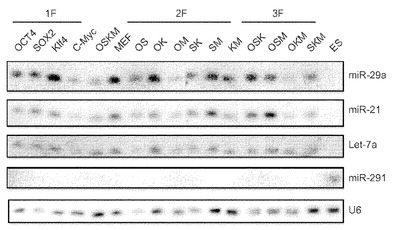
【図21B】
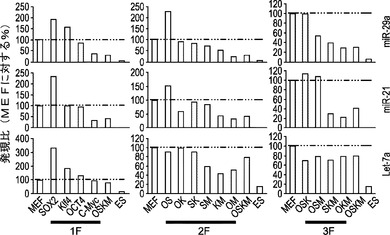
【図21C】
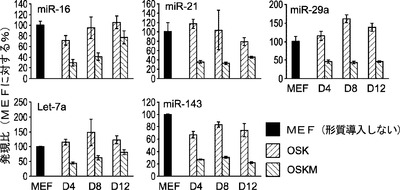
【図22A】
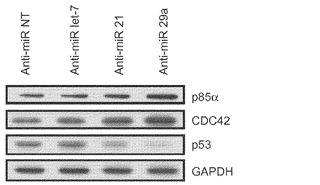
【図22B】
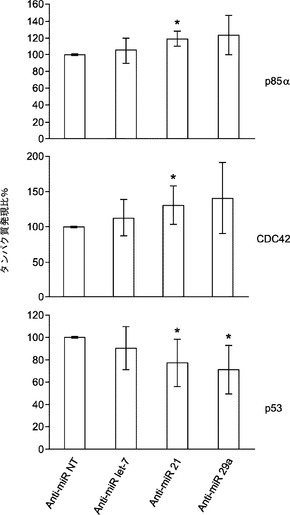
【図22C】
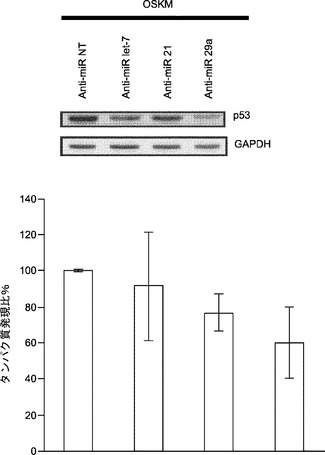
【図22D】
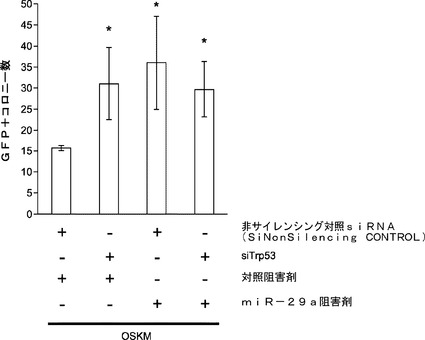
【図23A】
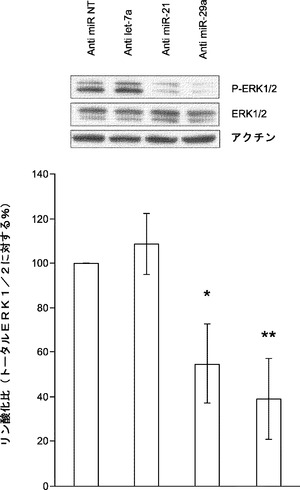
【図23B】
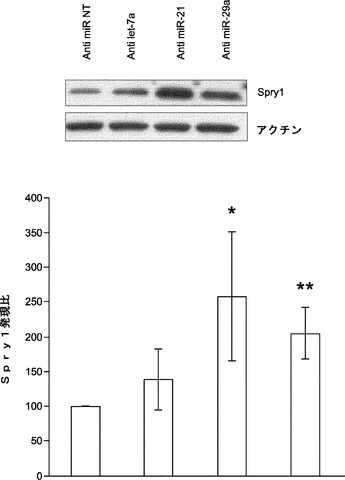
【図23C】
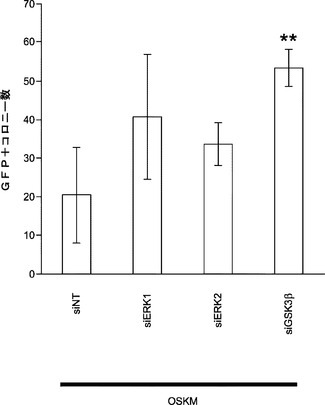
【図23D】
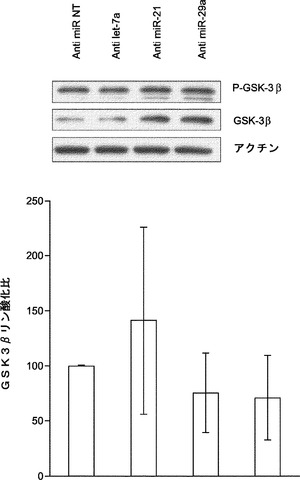
【図24】
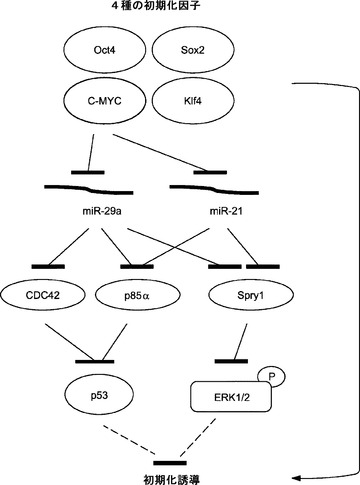
【図25】
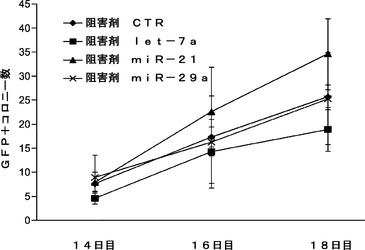
【図26A】
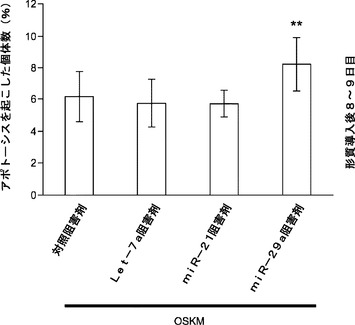
【図26B】
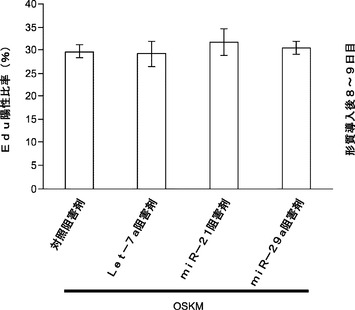
【図4】
【図5A】
【図5B】
【図5C】
【図5D】
【図5E】
【図5F】
【図6A】
【図6B】
【図6C】
【図6D】
【図7】
【図8】
【図9】
【図10A】
【図10B】
【図11】
【図12】
【図13】
【図14】
【図15】
【図16A】
【図16B】
【図17】
【図18A】
【図18B】
【図19】
【図20】
【図21A】
【図21B】
【図21C】
【図22A】
【図22B】
【図22C】
【図22D】
【図23A】
【図23B】
【図23C】
【図23D】
【図24】
【図25】
【図26A】
【図26B】
【公表番号】特表2013−510576(P2013−510576A)
【公表日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願番号】特願2012−538957(P2012−538957)
【出願日】平成22年11月10日(2010.11.10)
【国際出願番号】PCT/US2010/056273
【国際公開番号】WO2011/060100
【国際公開日】平成23年5月19日(2011.5.19)
【出願人】(511150953)サンフォード−バーンハム メディカル リサーチ インスティテュート (4)
【Fターム(参考)】
【公表日】平成25年3月28日(2013.3.28)
【国際特許分類】
【出願日】平成22年11月10日(2010.11.10)
【国際出願番号】PCT/US2010/056273
【国際公開番号】WO2011/060100
【国際公開日】平成23年5月19日(2011.5.19)
【出願人】(511150953)サンフォード−バーンハム メディカル リサーチ インスティテュート (4)
【Fターム(参考)】
[ Back to top ]