
pIX又はpVIIへの融合を介したヒトデノボpIXファージディスプレイライブラリの設計及び作製、ベクター、抗体、及び方法
本発明は、抗体又は抗原断片を作製するためのpIXファージディスプレイライブラリを作製するための組成物及び方法に関する。

【発明の詳細な説明】
【技術分野】
【0001】
本発明は、抗体又は抗体断片を産生するための、pIX又はpVIIファージディスプレイライブラリを作製及び使用するための、組成物及び方法に関する。
【背景技術】
【0002】
ファージ又はファージミド系における融合パートナーとしてpIII及びpVIIIを用いる繊維状ファージディスプレイは、タンパク質工学、とりわけデノボ抗体単離及び親和性成熟のための技術として用いられている。ヒト様fab配列は、テンプレートとしての既知の抗体配列から作製することができ、ランダム又は突然変異相補性決定領域(CDR)又は重鎖CDR3(H3)のような抗原結合領域は、抗原又は免疫付与していない他の関心タンパク質標的に対するパニングを介して抗体断片配列の変異型を提示するファージライブラリから作製及び単離することができる。既に用いられているヒトデノボ抗体ライブラリは、合成的に、又は天然源に由来するIgG遺伝子の分子クローニングにより創製されてきた。合成ライブラリでは、可変重鎖及び軽鎖フレームワーク及びCDR領域を含む抗体DNA配列は、1)規定のIgG遺伝子、2)特定のIg生殖細胞系遺伝子、及び3)Ig生殖細胞系遺伝子のファミリーに由来する共通配列、並びに4)天然源に由来するPCRにより得られたIgG断片に基づき設計及び合成される。合成抗体ライブラリに加えて、ライブラリはまた、ヒト組織、例えば、骨髄及び末梢血細胞に由来するIgG DNAのコンビナトリアルクローニングにより創製することもできる。このようなライブラリは、fab抗体断片を提供するため、並びに選択された標的タンパク質の高親和性又は阻害性生物活性のような所望の特性を有するfab抗体断片を見つけることを試みるためにパニング及び成熟又は修正の連続するラウンドを実行するために用いられてきた。
【0003】
いくつかの標的タンパク質に対するヒト様fab抗体が、ファージディスプレイpIII又はpVIII抗体ライブラリから単離されている。特定の標的に対するfab抗体の単離には成功しているが、このようなファージディスプレイライブラリアプローチは、所望の特徴を有するfab抗体を単離するために数回、ライブラリ作製、パニング、及び成熟プロセスを繰り返さなければならないという問題に悩まされている。このようなファージライブラリはまた、VH/VL対合の寄与、抗原認識に関連する様々な生殖細胞系ファミリーのトポロジー特性、アミノ酸変異の位置及び程度、並びに様々なヒト生殖細胞系遺伝子に由来する抗体の相対的存在量を含む、ヒトに存在するヒト免疫多様性の範囲を完全に包含又は模倣していないという問題にも悩まされている。合成抗体の天然のレパートリーからのずれは、ヒトの治療用に用いられる場合、好ましくない生化学的特性、及び免疫原性のリスクを高める恐れがある。
【発明の概要】
【発明が解決しようとする課題】
【0004】
高親和性及び活性、高生産性、良好な溶解特性、並びに人間に投与されたときに低免疫反応傾向であるヒト治療抗体の重要な要素を同時にもたらす合成抗体ライブラリ及び方法に対する要求が存在する。更に、現在の方法に比べて、抗体発見の資源コストを低減し、生物学的評価のための抗体の供給を加速するために、合成ライブラリからの抗体単離効率を高める必要性が存在する。本発明のライブラリ及び方法は、総合設計、アセンブリ技術、及びファージpIX Fabライブラリを組み合わせることにより、これらの要求を満たす。
【課題を解決するための手段】
【0005】
先行技術の教示とは対照的に、現在、pVII及びpIXは、例えば、fab及び抗体断片作製並びに治療抗体の選択のための効率的かつ迅速なプラットフォームを提供するために、突然変異誘発又は他の多様性を生み出す技術(所望により、インライン成熟とともに)を用いて、高親和性fabライブラリの作製のためにうまく用い得ることが発見されている。本発明により、pVII及びpIXに融合する抗体可変又はfab領域は、ファージ表面上に動的相互作用で係合して、機能的抗体断片、代表的なヘテロ二量体モチーフを提示する。したがって、抗体重鎖及び軽鎖可変領域のファージ上への提示は、メンバーが二量体人工抗体種として機能することができ、新規又は所望の生物活性の選択を可能にする、コンビナトリアルヘテロ二量体アレイの多様なライブラリの提示及びアッセイに好適かつ好ましい方法である。
【0006】
本発明は、種々の改善されたかつ新規のpIX及びpVIIファージディスプレイデノボライブラリ作製方法、並びに(i)pIX又はpVIIファージタンパク質に融合する設計及び提示された抗体Fabデノボライブラリ、(ii)広く用いられているM13ファージのpIII及びpVIIIとは異なるファージ表面タンパク質の使用、(iii)ヒトレパートリーの配列及び構造を表す生殖細胞系VH及びVL遺伝子の小さなアレイの使用、(iv)Vh及びVl領域の相補性領域における、改善された、設計された、コンビナトリアル多様性を提供するための、ライブラリのスカフォールドとしてのこのようなファージ構成要素の使用、(v)抗原認識のための設計された配列及び構造トポロジーの効果の系統的試験を可能にする抗体選択プロセス、(vi)ライブラリ選択の一部としての、効率化された親和性成熟及びインライン成熟プロセス、のうちの1つ以上であるが、これらに限定されない、構成要素を提供する。ライブラリの個々の又は群のライブラリ設計、選択、最適化及び成熟のこのような新規系は、うまく抗体を新規発見するための再現可能かつ信頼できる系を提供し、また抗体の抗原に対する相互作用の構造機能関係の理解を促進する。
【0007】
上記ヒトFabデノボライブラリは、M13ファージのpIX遺伝子を介して、その提示により現在の抗体ライブラリの現況技術とは区別できる。ライブラリスカフォールドとしての代表的なヒト生殖細胞系及び構造配列の使用、及び天然のアミノ酸分布を模倣するCDR配列の設計は、ヒトレパートリーを包括的に網羅し、ライブラリ抗体は天然由来のヒト抗体に対して高い配列同一性を有する。ヒト免疫レパートリーの包括的網羅は、文献に報告されている単一生殖細胞系又はIgG遺伝子スカフォールド上に構築されたライブラリに比べて、抗体の新規発見の機会を増やす。別々に作製されたサブライブラリ(それぞれ独自のスカフォールド(VH/VL対)及び/又はH−CDR3長を保有する)は、独自の抗体を同定する機会を最大化し、抗体/抗原結合構造及び機能関係を系統的に試験するための機序を提供する。統合された親和性成熟プロセスは、多様かつ高親和性抗体を発見するために必要な時間を低減する。
【0008】
人工抗体は、本明細書では、(1)配列相同性及び標的抗原の毒性、(2)抗体を回収するために用いられる宿主又はハイブリドーマ培養において作製された抗体の生物学的影響、及び(3)所望の活性の選択に対するスクリーニングを含む、抗体分子の機能的戦略を用いるが、インビボ拘束を含まず作製することができる多様性の大きなタンパク質モチーフとして定義される。抗体分子は、3次元空間におけるペプチド要素のコンビナトリアルアレイを表現するための生物学的デバイスである。本質的な特徴は、CDR(相補性決定領域)が協力して結合部位を形成するが、その相互作用はCDR自体の間に構造的会合をほとんど有しない動的及び機能的なものであることである。この方法では、アミノ酸残基の完全な相補体は、結合のための最低エネルギーコストで抗原認識に利用可能である。配列空間だけでなく、3次元空間においてもコンビナトリアル設計を制御する能力は、免疫レパートリーの天然設計を再現し、最終的にはそれをしのぐ。
【0009】
したがって、本発明は、多様性に富むヘテロ二量体ポリペプチドアレイの構築のためのコンビナトリアルファージディスプレイフォーマットを記載する。具体的には、本発明は、融合ポリペプチドをコード化するゲノムを封入する繊維状ファージ粒子であって、融合ポリペプチドが繊維状ファージpVII又はpIXタンパク質のアミノ末端に融合する外来ポリペプチドを含むファージ粒子を記載する。好ましくは、ファージ粒子は、ファージ粒子の表面上に発現した融合タンパク質を含む。
【0010】
好ましい実施形態では、ファージゲノムは、第2の融合ポリペプチドを更にコード化し、第2の融合ポリペプチドはpIXのアミノ末端に融合する第2の外来ポリペプチドを含み、第1の融合ポリペプチド中の第1の外来ポリペプチドはpVIIタンパク質のアミノ末端に融合する。この実施形態では、第1及び第2の融合ポリペプチドは会合して、免疫グロブリンFv、触媒性Fv、受容体、核酸結合タンパク質又は酵素のような、ヘテロ二量体タンパク質複合体を形成することができる。
【0011】
関連する実施形態では、本発明は、融合タンパク質を発現するためのカセットを含む繊維状ファージの表面上で融合タンパク質を発現するためのベクターを記載する。カセットは、インサートDNAの指向性ライゲーションに適合したヌクレオチドの配列を介して機能的に連結した上流及び下流の翻訳可能なDNA配列、すなわち、ポリリンカーを含み、この場合、上流配列は原核生物の分泌シグナルをコード化し、下流配列はpVII又はpIX繊維状ファージタンパク質をコード化する。翻訳可能なDNA配列は、融合ポリペプチドの一部として翻訳可能なDNA配列を発現するためのDNA発現シグナル一式に機能的に連結する。好ましい変形では、ベクターは更に繊維状ファージの表面上で第2の融合タンパク質を発現するための第2のカセットを含み、ここで、第2のカセットは第1のカセットの構造を有するが、ただし第1の融合タンパク質発現カセットはpVIIタンパク質をコード化し、第2の融合タンパク質発現カセットはpIXタンパク質をコード化する。ベクターは、ヘテロ二量体の2つの外来ポリペプチドが、それぞれ第1及び第2のファージタンパク質、pVII及びpIXへの融合によりファージ粒子上に固定されるファージ粒子の表面上でヘテロ二量体タンパク質複合体を発現させるためのファージゲノムとして用いられる。
【0012】
別の実施形態では、本発明は、ライブラリ中の代表的な粒子がそれぞれ異なる融合タンパク質を提示する、本発明によるファージ粒子のライブラリ、すなわち、コンビナトリアルライブラリを意図する。粒子がヘテロ二量体タンパク質複合体を提示する場合、ライブラリは、Fv分子のライブラリの形態の抗体のような、ヘテロ二量体のコンビナトリアルライブラリを含む。好ましいライブラリは、少なくとも103、104、105、106、107、108、109、1010、1011、1012、1013、又はこの中の任意の範囲若しくは値のコンビナトリアル多様性、融合タンパク質の異なる種を有する。
【0013】
関連する実施形態は、第1のポリペプチドが外来タンパク質であり、第2のポリペプチドが繊維状ファージpVII又はpIXタンパク質であり、外来タンパク質が繊維状ファージタンパク質のアミノ末端に融合する、第1及び第2のポリペプチドを含む融合タンパク質を記載する。
【0014】
なお更に、本発明は、外来ポリペプチドをコード化する遺伝子のレパートリーを本発明のベクターにクローニングし、突然変異誘発により、第1及び第2の融合タンパク質ライブラリの集団のランダムな組み合わせにより、ライブラリの多様性を変化させるための標的及び親和性選択(「パニング」)等により、ライブラリ中の外来ポリペプチドの構造を修正することにより、ファージのコンビナトリアルライブラリを作製する種々の方法を意図する。
【0015】
改善された又は新規の機能を有するタンパク質の設計は、種々の医療、工業、環境、及び基礎研究用途の重要な目的である。コンビナトリアル抗体ライブラリの開発に続いて、強力な次の段階は、人工抗体構築、並びに二量体種がネイティブである、又は機能的である他のタンパク質モチーフへの進化である。
【0016】
本発明は、pVII及びpIXが二量体種を形成する融合タンパク質の提示に利用されるコンビナトリアルヘテロ二量体ポリペプチドアレイの構築のためのファージディスプレイフォーマットを提供することにより、これらの問題に取り組む。機能的相互作用のライブラリを作製するために非常に近接する2つのタンパク質モチーフを独立に提示することができるため、これは完全に新しい方法論であることに留意することが重要である。
【0017】
技術の範囲及び能力に固有なのは、二量体相互作用で係合できる種々のタンパク質を提示する能力である。これらは、抗体だけではなく、いくつかの酵素、ホルモン及びホルモン受容体、並びにDNA結合タンパク質を含む。本明細書に記載する提示技術は、抗体フレームワーク領域のコンビナトリアルな変化のために、並びに抗体構造を認識及び小型化するために、又はリプレッサのようなDNA結合タンパク質を提示するために、臨床的にかつ治療的に重要な特定のDNA配列に対する選択のためのヘテロ二量体のライブラリとして使用することができる。
【0018】
したがって、本技術は、突然変異体二量体タンパク質の提示及び選択、並びにメンバーがヘテロ二量体アレイからなるコンビナトリアルライブラリを提供する。この技術を用いて、ネイティブな免疫グロブリン構造(本明細書に示すヘテロ二量体VH−VL Fvフォーマットにおける)は、様々な方法で修正され、特異性及び活性についてスクリーニングされ得る。例えば、フレームワーク領域(FR)のコンビナトリアルな変化、又は「相補性決定領域(CDR)シャフリング」及び「ツインボディ」形成と呼ばれるプロセスにより抗体構造を再編成及び小型化するための他の操作により、新たなパラトープ又は完全に異なる構造的要素を含む抗体様2次構造が生じる。天然抗原及び/又は基質、並びにいくつかの関連化合物に対する、結合及び/又は触媒作用の選択は、ヘテロ二量体タンパク質のライブラリをスクリーニングするために用いられるであろう。
【0019】
更に、ライブラリを形成するための配列のランダム化、及びハイブリッド種を形成するための鎖シャフリングプロトコルは、新たなタンパク質の部分集合を導くことができる。例えば、ホモ二量体又はヘテロ二量体型におけるジンクフィンガードメインのアレイの提示及び修正は、特異的DNA相互作用を有する構造を生じさせる。加えて、完全に新たな構築は、抗体鎖のような予形成スカフォールド内の所望のコード化断片の挿入を介して可能である。可能なインサートとしては、酵素の特徴的な配列又はリプレッサ結合タンパク質が挙げられる。
【0020】
前述の一般的な説明及び以下の詳細な説明は、両方とも例示及び解説のためだけのものであり、請求されるような本発明を制限するものではないことを理解すべきである。
【図面の簡単な説明】
【0021】
【図1】スカフォールドVH及びVLアミノ酸配列。
【図2】ファージディスプレイ及びFab発現ベクターのベクター図:pCNTO−Fab−pIX−lacI。
【図3A】ライブラリスカフォールドFab発現及び提示のグラフ表示及び画像。3Aは、M13の表面上におけるFab発現の相対レベルを示す。3Bは、検出試薬として抗Fab’(2)を用いるウエスタンブロットにおけるFabの相対発現を示す。3Cは、既知の濃度のFabと比較することにより大腸菌培地上清におけるFabスカフォールドのおおよその発現を示す。これは、カッパ遺伝子スカフォールド(塗りつぶされたバー)及びラムダ遺伝子スカフォールド(陰影をつけられたバー)を含む。
【図3B】ライブラリスカフォールドFab発現及び提示のグラフ表示及び画像。3Aは、M13の表面上におけるFab発現の相対レベルを示す。3Bは、検出試薬として抗Fab’(2)を用いるウエスタンブロットにおけるFabの相対発現を示す。3Cは、既知の濃度のFabと比較することにより大腸菌培地上清におけるFabスカフォールドのおおよその発現を示す。これは、カッパ遺伝子スカフォールド(塗りつぶされたバー)及びラムダ遺伝子スカフォールド(陰影をつけられたバー)を含む。
【図3C】ライブラリスカフォールドFab発現及び提示のグラフ表示及び画像。3Aは、M13の表面上におけるFab発現の相対レベルを示す。3Bは、検出試薬として抗Fab’(2)を用いるウエスタンブロットにおけるFabの相対発現を示す。3Cは、既知の濃度のFabと比較することにより大腸菌培地上清におけるFabスカフォールドのおおよその発現を示す。これは、カッパ遺伝子スカフォールド(塗りつぶされたバー)及びラムダ遺伝子スカフォールド(陰影をつけられたバー)を含む。
【図4】H−CDR3設計パターンの概略。
【図5】H−CDR3オリゴヌクレオチド設計の概略。
【図6】H1〜69テンプレート用のH−CDR3位置におけるアミノ酸分布A.393D位置における分布(長さ9〜14)B.2163N位置における分布(長さ9〜14)C.H−CDR3の973Yにおける分布(長さ9〜14);D.NNSコドンを用いる長さ7及び8のH−CDR3の全ての位置における分布。
【図7】ライブラリ構築の要約図。
【図8】パリンドローム補助親ストランド脱離の概略。
【図9】V領域の全領域を標的とするメガプライマーの概略。
【図10】ヒットからのVH断片がPCRにより増幅され、VL−ループベクターにクローニングされる。得られたプラスミドはssDNAとして産生され、メガプライマーのハイブリダーゼーションと分解突然変異誘発のテンプレートとして機能する。Lcライブラリメガプライマーを用いてプラスミドを複製することができ、これによって、単離されたHc配列に関連するLcライブラリの多様性を組み込むことができる。この親和性成熟ライブラリは、M13ファージとしてパッケージ化され、初期抗原に対するファージパニングに利用され、親和性成熟したヒットを得られる。
【発明を実施するための形態】
【0022】
本発明は、新規ファージディスプレイデノボライブラリ作製方法、並びに(i)pIX又は他のファージタンパク質に融合する設計及び提示された抗体Fabデノボライブラリタンパク質、(ii)広く用いられているM13ファージのpIII及びpVIIIとは異なるファージ表面タンパク質の使用、(iii)ヒトレパートリーの配列及び構造を表す生殖細胞系VH及びVL遺伝子の小さなアレイの使用、(iv)Vh及びVl領域の相補性領域における、改善された、設計された、コンビナトリアル多様性を提供するための、ライブラリのスカフォールドとしてのこのようなファージ構成要素の使用、(v)抗原認識のための設計された配列及び構造トポロジーの効果の系統的試験を可能にする抗体選択プロセス、(vi)ライブラリ選択の一部としての効率化された親和性成熟プロセス等であるが、これらに限定されない、構成要素を提供する。ライブラリの個々の又は群のライブラリ設計、選択、最適化及び成熟のこのような新規系は、うまく抗体を新規発見するための再現可能かつ信頼できる系を提供し、また抗体の抗原に対する相互作用の構造機能関係の理解を促進する。
【0023】
上記ヒトFabデノボライブラリは、M13ファージのpIX遺伝子を介して、その提示により現在の抗体ライブラリの現況技術とは区別できる。ライブラリのスカフォールドとしての代表的なヒト生殖細胞系及び構造配列の使用、並びに天然の長さ及びアミノ酸分布を模倣するCDR配列の設計は、ヒト免疫レパートリーを包括的に網羅し、ライブラリ抗体は天然由来のヒト抗体に対して高い相同性を有する。免疫レパートリーの包括的網羅は、文献に報告されている単一生殖細胞系又はIgG遺伝子スカフォールド上に構築されたライブラリに比べて、抗体の新規発見の機会を増やす。別々に作製されたサブライブラリ(それぞれ独自のスカフォールド(例えばVH/VL対及び/又はH−CDR3長)を保有する)は、独自の抗体を同定する機会を最大化し、抗体/抗原結合構造及び機能関係を系統的に試験するための機序を提供する。統合された親和性成熟プロセスは、多様かつ高親和性抗体を発見するために必要な時間を低減する。
【0024】
定義:
抗体:種々の文法的な形の抗体という用語は、本明細書で使用されるとき、免疫グロブリン分子及び免疫グロブリン分子の免疫的に活性のある部分、すなわち、抗体結合部位又はパラトープを含む分子を指す。代表的な抗体分子は、インタクトな免疫グロブリン分子、実質的にインタクトな免疫グロブリン分子、並びにFab、Fab’、F(ab’)2及びFvのような当該技術分野において既知である部分を含む、免疫グロブリン分子の一部である。
【0025】
抗体結合部位:抗体結合部位は、抗原に特異的に結合する(免疫反応する)重鎖及び軽鎖可変並びに超可変領域から構成される抗体分子の構造部分である。種々の形の免疫反応という用語は、抗原決定基含有分子と、抗体分子全体又はその一部のような抗原結合部位を含む分子との間の特異的結合を意味する。
【0026】
融合ポリペプチド:少なくとも2つのポリペプチドと、その2つのポリペプチドを1つの連続するポリペプチドに機能的に連結する連結配列から構成されるポリペプチド。融合ポリペプチドに連結する2つのポリペプチドは、典型的には、2つの独立した源に由来し、したがって、融合ポリペプチドは自然界では通常連結した状態では見られない2つの連結したポリペプチドを含む。
【0027】
シストロン:アミノ酸残基配列をコードし、上流及び下流にDNA発現制御要素を含むDNA分子中のヌクレオチドの配列。
【0028】
繊維状ファージ
本発明は、融合タンパク質(タンパク質)をコード化するゲノムを封入するタンパク質のマトリックスを含む繊維状ファージを意図する。融合タンパク質は、繊維状ファージpVII又はpIXタンパク質のアミノ末端に融合する外来ポリペプチド部分を含む。
【0029】
「外来」とは、ファージタンパク質に融合するポリペプチドが、繊維状ファージの野性型変異におけるファージpVII又はpIXタンパク質と通常は関連せず、正常ファージタンパク質とは異なることを意味する。
【0030】
典型的な外来ポリペプチドは、免疫グロブリン重鎖可変ドメイン(VH)、免疫グロブリン軽鎖可変ドメイン(VL)、天然又は合成ポリペプチド、単鎖抗体(scFV)等を含む、任意の関心ポリペプチドである。
【0031】
好ましい実施形態では、繊維状ファージは第1及び第2の融合タンパク質をコード化するゲノムを封入し、ここで、第1の融合タンパク質はpVIIに融合する第1の外来ポリペプチドを含み、第2の融合タンパク質はpIXに融合する第2の外来ポリペプチドを含む。
【0032】
繊維状ファージは、実施例に記載のような、ファージ粒子の表面上に提示される融合タンパク質を更に含有する。したがって、第1及び第2の融合タンパク質が存在する場合、ファージは、第1及び第2の外来ポリペプチドがヘテロ二量体として相互作用して、ファージ表面上に機能的な2本の鎖のタンパク質複合体を形成するように、機能的方式でこれらのタンパク質を提示できる。
【0033】
発現したヘテロ二量体タンパク質がリガンドに結合する能力を有する場合、それはあるいは本明細書ではリガンド結合ヘテロ二量体受容体と呼ばれる。
【0034】
好ましい実施形態におけるヘテロ二量体受容体は、エピトープ結合複合体である。すなわち、第1及び第2のポリペプチドの複合体は、エピトープに結合することができる。好ましくは、第1及び第2のポリペプチドは、抗体重鎖及び軽鎖ポリペプチドである。具体的には、好ましい実施形態はVH及びVLを利用してFv複合体を形成する。他のヘテロ二量体タンパク質複合体としては、触媒性Fv、受容体、核酸結合タンパク質、及び酵素等のヘテロ二量体タンパク質が挙げられる。
【0035】
本発明のファージ上に存在する融合タンパク質では、外来ポリペプチドと繊維状ファージpVII又はpIXタンパク質との間の「融合」は、典型的なアミド連結を含んでよく、又は実施例に記載のようなリンカーポリペプチド(すなわち、「リンカー」)を含んでもよい。典型的には長さ約5〜50アミノ酸のストレッチである種々のリンカーのいずれを用いてよい。特に好ましいリンカーは、リンカーの点で融合タンパク質に高度な可動性をもたらす。
【0036】
ライブラリ設計:先行技術の合成ライブラリは、以下のいくつかを組み込んでいたが、包括的方式で全てを含むものは存在しない。
【0037】
生殖細胞系V領域ファミリー多様性重鎖及び軽鎖のヒト可変レパートリーは、配列相同性及び長さにより定義される関連配列のファミリーからなる。このような任意のファミリーでは、個々のメンバーは主に相補性決定領域の配列及び長さが異なる。これらの差は、既知の構造的変異パターン(カノニカル構造)、並びに抗原と相互作用しやすくする構造的に類似の位置における生殖細胞系アミノ酸変異を導く。ライブラリに単一Vh及びVI遺伝子テンプレートのみを選択することは、捕捉され得る天然生殖細胞系多様性の範囲を狭める(ジェネンティックライブラリを参照)。全てのVh及びVl遺伝子を選択することは、ライブラリ作製の効率を無効にする。本発明のライブラリは、(1)再編成されたヒト抗体における優性カノニカル構造群を表す少数の生殖細胞系Vh、Vk、及びV−ラムダを同定し(IMGT、Kabat、NCBI)、(2)コンビナトリアルオリゴヌクレオチド突然変異誘発により、Vk及びV−ラムダ領域のCDR1及び2、又はCDR1〜3をもコード化するVH遺伝子における関連ファミリーメンバーの天然ヒト生殖細胞系多様性を組み込むことにより生殖細胞系多様性を効率よく捕捉する。
【0038】
VH−CDR3多様性。VH−CDR3は、V、D及びJセグメントの接合により創製され、両端付加及びヌクレオチド鎖分解性事象により達成される。約25の生殖細胞系D領域が存在し、この数は複合体接合事象を伴い、体細胞変異は抗体の最も多様な領域を創製する。これらの事象は、生殖細胞系配列の規定のセットで生じ、ランダムではないが、予測することは困難である。しかしながら、再編成されたヒト抗体配列のデータベースは、現在、VH−CDR3のそれぞれの位置における長さ及びアミノ酸分布の両方の統計的評価を適用するのに十分な大きさに達している(約5000VH領域)。本発明のライブラリは、この領域を集めるために、設計された変性オリゴヌクレオチドを利用することによりこの天然のヒト多様性を再現する。
【0039】
体細胞多様性の位置及び性質。体細胞多様性は、いかにヒト抗体が高親和性、選択的結合実体に成熟するかの証明である。この体細胞突然変異の作製及び蓄積はランダムではない。ヌクレオチド突然変異の部位及び種類は、DNA配列によりバイアスがかかるが、しばしば中性置換とともに、結合及び機能的利点をもたらす突然変異のみが選択され、保存される。機序からの予測には従わないが、再編成されたヒト抗体配列のデータベース及び構造−機能解析は、タンパク質、ペプチド及び小分子抗原の間の分化を含む、CDR領域における抗原の認識に最も高い頻度で関連する位置及びアミノ酸置換を同定する。本発明のライブラリは、VH CDR1及び2、又はVk及びV−ラムダ領域のCDR1〜3にも置換を組み込むために、設計された変性オリゴヌクレオチドを利用することにより、天然のヒト多様性を再現する。
【0040】
生殖細胞系遺伝子の利用。ヒト生殖細胞系レパートリーは、約30のV−カッパ、56のV−ラムダ、及び40のV−重鎖機能遺伝子からなる。しかしながら、再編成された抗体におけるそれらの表現は強くバイアスがかかっており、これは異なる軽重鎖V−領域の対合の頻度において反映される(de Wildt et al.,J Mol Bio 285:895〜901(1999))。本発明のライブラリは、上記(a)に記載された多様性部類のそれぞれの優性生殖細胞系V−領域を選択することにより、このバイアスを取り込む。
【0041】
発現、生化学的、及び生物物理的特性。好ましいヒト抗体は、所望の生物学的及び結合活性を有するが、また種々の宿主から効率よく産生され、安定であり、良好な溶解特性を有する。高頻度の生殖細胞系遺伝子使用(1d)はまた、哺乳類系における良好な発現を示す。加えて、選択又はスクリーニングの細菌ファージディスプレイ法によりライブラリから回収される抗体は、細菌宿主においてよく発現するはずである。本発明のライブラリは、よく発現するヒト生殖細胞系由来のテンプレートに基づき、標準的な組み換え哺乳類宿主(例えばHEK293及びCHO細胞)並びに細菌宿主から精製され、高い安定性及び良好な溶解特性を有する。
【0042】
成熟。それぞれの位置で最大20の異なるアミノ酸の潜在的変異と合わせて、抗原認識に影響を与える場合のあるV−領域配列における多数の位置は、単一ライブラリにおいて全ての変異を含むことを実用的に不可能にする。ヒト抗体は、体細胞突然変異の進行性過程により高親和性及び特異性を獲得する。本発明のライブラリは、ヒト抗体を反映するようにそれぞれの抗体鎖の配列一体性を維持しながら、平行選択及び標的化変異を可能にするために設計及び指示される。
【0043】
代替設計。上記設計は、天然のヒト抗体を再現する。系のモジュラー性は、位置の任意の集積におけるアミノ酸の任意の集積を組み込みやすい。
【0044】
ライブラリアセンブリ技術。好ましいデノボ抗体ライブラリは、高い多様性を有し(>1010)、変化しやすく、組み立てが容易であり、望ましくない配列のバックグラウンドが低い。これらのバックグラウンド配列は親テンプレート及び低標的化多様性を含む。以下の方法を組み合わせると、ライブラリの組み立てが加速し、低バックグラウンドを導く。(a)Kunkle−ベースの単鎖突然変異誘発、(b)制限酵素部位を有するパリンドロームループ、(c)メガプライマー。
【0045】
pIX Fabファージディスプレイ法。全ての先行技術の繊維状デノボヒト抗体ライブラリは、単一Fvライブラリ(Scripps参照)を除いて、提示のためにpIII又はpVIIIファージコートタンパク質を利用する。pIXと選択されたFabテンプレートとの組み合わせは、mAb及び他の関連分子に変換するとき、その選択された特性を保持する抗体を回収するためのより効率のよい選択系である。
【0046】
Fabディスプレイ。scFvとは異なり、Fabはヒト抗体の天然セグメントであり、それらは完全抗体に操作されるとき活性をより再現する。効率のよいFabの繊維状提示は、細菌宿主における良好な発現を超える特性を必要とする場合がある。本発明のV−領域テンプレートは、繊維状ファージ上のpIXによる効率のよい提示について選択した。
【0047】
ファージミドディスプレイ。Fab分子はファージpIXコートタンパク質に比べて大きく、よって細菌宿主において産生される全てのpIXタンパク質に連結する場合、組み換えファージ粒子の組み立てに干渉する恐れがある。この干渉をバイパスするためのアプローチの1つは、(Scripps参照)により記載されているような、pIXファージミド系を使用することであり、それにより野性型及びFab連結pIXタンパク質の両方を組み換えファージ粒子に組み込むことができる。好ましい用途では、本発明のライブラリは、ファージミド系においてpIXにより提示される。
【0048】
提示のためのファージコートタンパク質pIX。pIIIのように、pIXはファージ上に少コピー数存在し、提示されたFabを親和性選択しやすい。しかしながら、pIIIタンパク質は、感染過程において決定的に関与し、このタンパク質上に提示されるタンパク質は感染効率に干渉する恐れがある。更に、重鎖Fd又は軽鎖セグメントのいずれかが提示のためにpIXに融合することができる。pIXタンパク質上に提示される本発明のライブラリは、効率よく複製されると予測され、選択及び/又はスクリーニングのために提示される。
【0049】
Fab−pIX発現。ファージライブラリから回収されるFabをスクリーニングするアプローチの1つは、提示のためにFab分子に連結するファージのコートタンパク質を取り除くことである。小さなサイズのpIXタンパク質は、この工程を行うことなく直接Fabのスクリーニングを作製する選択肢を提供する。
【0050】
ライブラリスカフォールドの設計。ライブラリスカフォールドは、一式のヒト生殖細胞系VH及びVL遺伝子から作製される。分析文献並びに独占抗体情報は、ヒトIgG遺伝子ファミリーを表す生殖細胞系遺伝子の同定、IgGレパートリーにおける使用、及びヒト抗体カノニカル構造を導いた。VhとVLとの間の良好な対合及び生殖細胞系遺伝子に由来する抗体の成熟成功の確率もまた、分析において考慮された。ヒトの性質及びスカフォールドVH及びVLの良好な対合の設計は、ライブラリスカフォールドとしてヒト生殖細胞系遺伝子のコンセンサスを用いる設計(MorphoSys HuCAL GOLD)より優れており、ライブラリスカフォールドとして単一生殖細胞系遺伝子に基づいた設計(Dyax VH and Affytech)よりヒトレパートリーをより包括的に表す。
【0051】
ライブラリスカフォールドの発現及び提示能力。ライブラリスカフォールドFabの良好な発現及び提示能力は、直接、スカフォールド遺伝子上に発生するライブラリの品質に関連する。ライブラリスカフォールドFab発現及び提示能力は、ライブラリ構築前に調べられた。発現したが、全く又はそれほど提示しなかったいくつかのスカフォールドFabは、ライブラリ構築から除外した。良好に発現し提示したライブラリスカフォールドは、ライブラリ内の高い割合のFabが機能的であり、天然源から遺伝的に増幅されたVH及びVL遺伝子のコンビナトリアルクローニングに由来するライブラリ(CAT)より優れている。
【0052】
H−CDR3多様性の設計。ライブラリVH−CDR3を、長さ及び配列の両方において多様化した。CDRの長さに依存する約109〜1018の総配列可能性を、抗原結合におけるVH−CDR3の重要性を反映するVH−CDR3において設計した。アミノ酸多様化の全てのアミノ酸コドンの使用(例えば、Genentech 2004によるNNK)、及び天然源からのVH−CDR3配列を操作するための包括的クローニング法の使用(CAT、Dyax、Affytech)とは異なり、我々は、ヒトIgGレパートリーにおけるアミノ酸使用パターンを模倣するアミノ酸をコード化する設計されたオリゴヌクレオチドを使用することによりVH−CDR3を創製した。設計されたVH−CDR3は、より少ない、望ましくない(例えば、終止コドン)及びより少ない、好ましくないアミノ酸(例えば、システイン)を含むが、より高い割合のIgG様CDR3配列を含む。変性されたオリゴヌクレオチドは、突然変異誘発反応において大きなサイズの遺伝子ライブラリを作製するために容易に用いることができる。
【0053】
H−CDR1及びH−CDR2多様性の設計。VH−CDR1及びVH−CDR2におけるアミノ酸多様性は、生殖細胞系配列内の変異を模倣するよう設計される。VH−CDR1及びVH−CDR2における組み合わせられた総配列多様性は、102〜105の範囲である。大きなVH−CDR3配列の変異と併せて、これは、ライブラリ中の総配列変異を増加させ、したがって、抗原結合抗体を同定する機会を増やす。小さな、生殖細胞系様多様性の独自の設計は、生殖細胞系又は天然様抗体配列を発見することを好み、ライブラリ作製に対するコンビナトリアル効果のために非天然CDRの組み合わせを単離する可能性を最小化する。この設計は、全ての3つのVH CDR(Genentech、Dyax、及びMorphoSys)におけるより大きな配列多様性を含む他の合成又は半合成抗体ライブラリの多くとは異なり、更に大きな理論的配列多様性のほんの一部のみがライブラリに取り込まれ得る。
【0054】
LCにおける多様性の設計。ライブラリ軽鎖は、デノボライブラリにおいて変動するよう戦略的に設計され、Lc多様性はFabがファージパニングを通して選択された後CDRに組み込まれた。抗原結合FabにおけるLc多様性は結合親和性を改善するために設計されるため、既知の構造の抗原−抗体複合体において頻繁に抗原と接触することが見出されているCDR位置は、多様化のために選択された。段階的CDR多様化戦略は、大きな理論的CDR配列多様性を有効に管理し、生物学的特徴により好適な、高結合親和性抗体を作製する。
【0055】
ライブラリ作製方法。修正されたKunkel突然変異誘発法(それぞれ異なるFab配列を持つ数十億の大腸菌コロニーを効率よく作製する)は、大きなFabライブラリを作製するために用いられる。効率的でありながら、高配列複合体ライブラリの作製に適合するとき、突然変異誘発されていない親DNAの割合が増加する。加えて、合成の長いオリゴヌクレオチドの技術的限界は、離れた領域において配列多様性を含むライブラリを作製するための方法の効率を低下させる。この限界を打開するために、>350ベースのオリゴヌクレオチド(メガプライマー)の作製、及び突然変異誘発テンプレートに制限酵素認識部位を含むステムループ配列の創製の更なる技術が、標準的なKunkel突然変異誘発法と併用された。ライブラリ作製のために他者により使用される、制限クローニング(MorphoSys、CAT及びDyax)、ファージ組み換え(Giggapack、Invitrogene)、及び配列特異的組み換え(CAT)のような他のライブラリ技術と比べて、改善されたKunkelに基づく方法は、>109配列多様性ライブラリの作製において著しくより有効であり、標的化DNAの任意の位置における配列多様性を導入するためにより可変性である。
【0056】
インライン親和性成熟。統合された親和性成熟プロセス、又はインライン親和性成熟は、ライブラリから選択される全てのFabの結合親和性を改善するために設計される。ライブラリは種々のFabのVLを残すために戦略的に設計されるため、追加の抗原結合部位はCDR配列多様化を介してVLにおいて容易に創製されるべきである。パニング後、全てのFabの改善された結合親和性は、治療的抗体リード同定の成功を増加させる。ライブラリ作製のためのKunkel法の使用は、単純かつ連続プロセスにおけるVL配列多様化戦略の有効な実施を保証する。改善されたKunkel突然変異誘発法の設計戦略及び技術的利点は、このアプローチを、目的の効率及び利益を低減する冗長なライブラリ作製法が使用される場合、CAT及びMorphoSysにより報告される他のプールされた成熟戦略より優れたものにする。
【0057】
平行ライブラリパニング。現状技術、半自動化設備を用いる平行パニングプロセスは、個々にサブライブラリを作製するプロセスのために開発される。平行パニングは、独自に設計され作製されたライブラリを支持する、抗体の多様なセットを発見する可能性を最大化する。インライン親和性成熟における平行パニングの有効な使用はまた、同時に改善されるべき広範な親和性を有する抗体を可能にする。機械に基づくパニングの開発はまた、所望の特性の抗体を発見するために異なるパニング条件を系統液に評価することを可能にする。
【0058】
親和性ランキング。親和性に基づく結合アッセイは、更に特徴付けするために最良の結合抗体を選択するために大きく、多様で、高い親和性の抗原特異的結合抗体に適用される。多数のサンプルを処理するのに好適な、ELISAのような標準的な生化学的方法、並びに例えば、BIAcore、Octet、及びBINDS等の親和性測定器具は、この目的のために単独で又は組み合わせて用いられる。
【0059】
一般的な用語で本発明を記載してきたが、本発明の実施形態は更に、特許請求の範囲を限定するものとして解釈されるべきではない以下の実施例に開示される。
【実施例】
【0060】
実施例1:実施例1ライブラリスカフォールドの設計
ヒト生殖細胞系遺伝子をライブラリ骨格として設計した。生殖細胞系VH及びVL遺伝子のパネルを、最初に、1)天然に発現しているIgGの使用、2)タンパク質及びペプチド抗原認識を好む構造(主鎖立体構造)トポロジー、3)生殖細胞系遺伝子に由来する抗体の生化学的及び生物物理学的特性、及び4)抗体としてVH及びVLヘテロ二量体を形成する可能性の特性に基づいて同定した。パネルから上記列挙した特性を最もよく表す4つのVH及び4つのVL生殖細胞系遺伝子を更に選択した。配列改変を含む既知の抗体に由来する10アミノ酸の単一、人工H−CDR3配列、及びヒトJH4セグメントをそれぞれの生殖細胞系VHと併用して、完全なVH配列を完成させた。VLでは、Jκ1セグメントを用いて選択された生殖細胞系VLのそれぞれを組み合わせた。これらの4つのVH及び4つのVL生殖細胞系に基づく遺伝子を化学的に合成する。定義されたCH及びCL配列と組み合わせて、それらは16の組み換えヒトFabを構成した。VL−λでは、Jλ2セグメントを選択されたVL−λ生殖細胞系スカフォールドのそれぞれと組み合わせた。3つのVH(169、323、及び551)生殖細胞系遺伝子を、VL−λスカフォールドと組み合わせて、CH1及びCλ配列を含むベクターと組み合わせて、12の組み換えヒトFabを得た。これらの28のFabをライブラリスカフォールド又はテンプレートとして用いて、重鎖又はLc鎖相補性決定領域(CDR)における配列多様性を提示した。図1は、設計された4つのVH及び4つのVLの配列を示す。
【0061】
実施例2.ライブラリスカフォールドの発現及び提示
合成FabDNAをpCNTO−lacI−pIXベクター(図−−)にクローニングする。VH遺伝子をNcoI及びApaI部位を介してクローニングした。VL遺伝子(ompAシグナル配列及び上流配列を含む)を、NheI及びBsiWI部位を介してクローニングした(図2)。スカフォールドFabの発現及び提示を、それぞれウエスタンブロット分析、及びファージELISAで調べた。
【0062】
提示を調べるために、ファージを調製し、ファージELISAで試験した。簡潔に述べると、pCNTO−Fab−pIX−lacI構築をMC1061F’細胞に形質転換し、振盪しながら37℃で2xYT/Carb(100μg/ml)/TET(15μg/ml)/1%グルコース培地中で一晩増殖させた。次の朝、50μlのこの増殖物を用いて、5mlの2xYT/Carb(100μg/ml)を播種し、OD600が0.6〜0.8になるまで増殖させた。次いで培養物に、振盪せずに37℃で40分間ヘルパーファージ(ファージの#)を感染させた。感染した細胞を遠心沈殿させ、5mlの2xYT/Carb(100μg/ml)/TET(15μg/ml)/Kan(35μg/ml)/0.5mM IPTG培地に再懸濁し、振盪しながら30℃で一晩増殖させた。次の朝、細胞を遠心沈殿させ、ファージ上清を回収し、ファージELISAに用いた。50μlのファージ上清(未希釈)、並びに1:5の3種の連続希釈液を抗Fd(Hc特異的)又は抗カッパ(Lc特異的)抗体でコーティングされたELISAウェルに添加した。インキュベーション後、結合していないファージを洗い流し、結合しているファージを抗M13抗体により検出した。H3−53/L−B3及びH3−53/L−A27を除く全てのスカフォールドの組み合わせが抗Fd及び抗カッパ抗体に対して良好な結合を示し、これはファージ表面上のHc及びLcの十分な提示を示す(図3)。
【0063】
Fab発現を調べるために、pIX遺伝子をまずSpeI及びNheI制限酵素消化によりまずpCNTO−Fab−lacI−pIXベクターから切り取り、続いて消化されたベクターDNAをセルフライゲーションさせる。これはFab発現の構築、pCNTO−Fab−lacIを作製する(図2)。pCNTO−Fab−lacIファージミドを有するM1061F’細胞を、振盪しながら37℃で2 X YT/1%glucose/Amp(100μg/ml)培地中で一晩増殖させた。一晩培養物の5mLのアリコートを未誘導対照として用いた。0.1mLの一晩培養物を10ml 2XYT/0.1%グルコース/Amp(100μg/ml)培養培地に接種した。培養物を振盪しながら37℃でOD600nmが0.8〜1.0になるまで増殖させた。IPTGを最終濃度0.5mMまで添加して、Fab発現を誘導した。振盪しながら30℃で更に16〜20時間培養を続けて増殖させた。誘導した培養物を遠心沈殿させ、それぞれの細胞のペレットを0.4mLのBPER II試薬(Pierce)を添加することにより溶解させた。使用済み細胞可溶化物の上清を回収し、Fab発現をウエスタンブロットにより分析した。約10倍低いレベルで発現したB3LCを含むものを除いて、全てのFabが(>2μg/ml)で発現した。またFab発現をELISAを用いて調べた。50μlの使用済み細胞可溶化物、並びに1:5の6種の連続希釈液を、抗Fd(Hc特異的)でコーティングしたELISAウェルに添加した。Fabを抗カッパ又は抗ラムダHRP結合抗体のいずれかで検出した。Fab対照標準曲線と比べてFabの量を定量化した(図3C)。
【0064】
実施例3.H−CDR3多様性の設計
天然免疫レパートリーを模倣するH−CDR3における大きな配列多様性を設計した。合計約5250の重複しない、完全(v領域、CDR3及びフレームワーク4)ヒト抗体配列を、種々の公的源から集めた。データセットは全ての7Vhファミリーを含んでいた。独自に開発したプログラムを書いて、Kabatの定義に基づいてCDR3セグメントを抽出した。抗体配列を、CDR3の長さに基づいて更に分析した。H−CDR3の長さは1〜27アミノ酸の範囲であり、正規分布を示した。最大数の抗体は、H−CDR3に12aaを含んでいた。同じ長さを有する抗体配列を整列させ、グループ化した。JH6由来の配列を長さの群から引いて、JH6に関連する、複数の、連続するチロシンを含有する配列を最小化した。プログラムを書いて、それぞれの長さのアミノ酸分布を計算した。H−CDR3長さ群のそれぞれの位置における残基を、その頻度に基づいて並べ替えた。内部にインストールされていたWeblogo(9−Crooks GE,Hon G,Chandonia JM,Brenner SE WebLogo:A sequence logo generator,Genome Research,14:1188〜1190,(2004))を用いて、それぞれのHCDR3の長さ分布をプロットした。およそ全てのヒト抗体レパートリーの約65%を網羅する、H−CDR3の7〜14アミノ酸を含む配列で作業することを決定した。
【0065】
アミノ酸分布を分析することにより、7〜14アミノ酸を含むH−CDR3のアミノ酸分布を説明するパターンを同定する。図4は、H−CDR3のパターンを示した。高度に多様なアミノ酸がH−CDR3で観察されたにもかからわず、アミノ酸グリシン及びアラニンは全ての位置で頻繁に用いられる。加えて、アスパラギン酸(D)は位置95で頻繁に用いられ、チロシン(Y)はH−CDR3におけるJセグメントにより近い位置で頻繁にコード化される。位置99〜101の配列は変化するが、アミノ酸フェニルアラニン(F)、アスパラギン酸(D)及びチロシン(Y)は、これらの位置でIgGにおいて主に用いられる。これらの位置はH−CDR3に対する構造的支持体として機能することが多く、抗原及び/又はIgGの表面に接近しにくいため、それぞれ99はF/L、100はD、101はYのアミノ酸に固定した。
【0066】
H−CDR3における高度に多様なアミノ酸分布を模倣するために、H−CDR3をコード化する混合された塩基オリゴヌクレオチドを設計して、それらをライブラリ構築で用いた。Wang & Saven(10−Wang W,Saven JG.Designing gene libraries from protein profiles for combinatorial protein experiments.Nucleic Acids Res.Vol 30,No 21,e120.2002)に記載されている手順を用いて、コドントリプレットに由来するコードアミノ酸が標的とするアミノ酸分布を模倣するように、複数のランダム試験におけるコドントリプレットのそれぞれの位置でヌクレオチド混合比の初期値を決定する。終止コドンを最小化し(<3%)、混合塩基オリゴヌクレオチドによりシステインコドンを低減するために、Wang & Saven手順のように同じ標的機能に対して、MicroSoft Excel(商標)のSolverにおけるスクリプトを用いてコドントリプレットのそれぞれの位置におけるヌクレオチド混合比の値を更に絞り込んだ。D及びN位置に用いられるヌクレオチド組成を図5に示す。より短いH−CDR3長さ、例えば7〜8アミノ酸では、NNSコドンを代わりに用いる。
【0067】
次いで、コドン設計に基づいて混合塩基オリゴヌクレオチドを作製した。D及びN位置に加えて、チロシン(〜18%Y)に富む位置は、別の方法でN位置と類似のaa分布を共有する。N位置におけるN設計コドンと、それぞれのチロシン位置における固定されたコドンTATを有する別個のオリゴを作製し、それを用いて11〜14アミノ酸のCDR長さを有するライブラリのN設計コドンオリゴと混合した。Nコドンオリゴ及びNと、特定のY位置用TATコドンオリゴを混合するために用いた比は約7:1である。設計者はNコドンに約4.7%のチロシンを与えるため、この混合物によりY位置における固定されたTATコドンから13%のチロシンが更に得られる。
【0068】
実施例6に記載の方法を用いてデノボFabライブラリを構築するためにオリゴを用いた。それぞれのサブライブラリから約100個のコロニーのDNA配列を得、分析した。H−CDR3におけるTAA終止コドンから突然変異したクローンを陽性クローンとみなした。翻訳されたH−CDR3配列を用いて、個々の又は組み合わせられたD、N及びY位置におけるアミノ酸分布を決定した。結果は、これらの位置で観察されたアミノ酸分布は、データベース及び設計で見出された分布を非常に模倣していることを示した(図6)。また、設計された混合塩基オリゴは、NNS変性オリゴよりも、設計及びデータベースに対するこれらの位置のアミノ酸分布をより模倣していることを示した(図6)。
【0069】
実施例4.H−CDR1及びH−CDR2多様性の設計
生殖細胞系遺伝子レパートリーを模倣するためにH−CDR1及びH−CDR2における多様性を設計する。多様化のために標的とされるH−CDR1&CDR2位置を、1)生殖細胞系遺伝子における多様性(11−Vbase)、及び2)溶媒曝露分析及び文献により決定された既知の構造の抗体−抗原複合体における抗原との接触が頻繁に見られること(12−Almagro 2004)により決定する。その位置におけるアミノ酸多様性は、1)生殖細胞系における利用(11)、2)生殖細胞系遺伝子(包括的データベース)に由来するIgGにおいて最も頻繁に用いられるアミノ酸、3)体細胞突然変異の結果として誘導されると予測されるアミノ酸、及び4)抗原認識に寄与するアミノ酸の生化学的及び生物物理学的特性により決定する。H−CDR1&CDR2における組み合わせられた配列多様性は、102〜105の範囲である。使用頻度を、IgGにおいて頻繁には用いられないアミノ酸を制限するためのフィルタとして用いた。これは、コンビナトリアル突然変異により作製される非天然配列を最小化する。表1は、配列分析及びH−CDR1及びCDR2多様性の設計を示した。
【0070】
H−CDR1及びH−CDR2設計を最もよく網羅する変性オリゴヌクレオチドを設計し、合成する(表1)。オリゴヌクレオチドを、単一Kunkle ssDNA依存性突然変異誘発反応におけるプライマーとして用いる。したがって、定義されたH−CDR1&CDR2位置における設計された縮重は、予測通りH−CDR1及びH−CDR2に導入される。Kunkel突然変異誘発反応に従ってランダムに選ばれたクローンの配列分析は、31%〜55.7%のクローン配列がH−CDR1及び/又はH−CDR2領域における突然変異を有することを示した(表2)。H−CDR1及びH−CDR2の両方に突然変異を有するクローンの百分率は、1つのCDR領域にのみ突然変異を有するクローンと等しいか又はそれより高いことが見出された。
【0071】
実施例5.Lcにおける多様性の設計
以下のパラメータは、設計軽鎖CDR多様性において考慮される。まず、我々は、組み合わせられたLc多様性が108を超えるべきではないことを定義した、なぜなら、Lcライブラリは親和性成熟で用いられ、したがってライブラリのパニングの第1段階中、ファージ選択に由来するおよそ102の独特のHcクローンのプールと組み合わせられるからである。したがって、組み合わせられたHc及びLc複雑度は約<1010であり、これは単一Kunkel突然変異誘発反応により生じるライブラリにおいて有効に捕捉され得る。2番目に、我々は、既知の構造の抗原−抗体複合体における抗原と接触することが見出されている多様化位置、又はいわゆる特異性決定残基(SDR;12)、つまり、それぞれL−CDR1、L−CDR2及びL−CDR3について位置30〜32、50及び91〜94を標的とした。3番目に、位置96はまた、それが接合(J)遺伝子及びその位置が異なる全ての5つのヒトJ遺伝子にコード化されているため、変化に富んでいる。4番目に、設計H−CDR1及びH−CDR2で用いられている概念と同じように、多様化されるべき再編成されたIgGでも頻繁に見られる生殖細胞系にコード化されているアミノ酸を選択することにより、自然に発生する可能性の低い配列多様性を最小化した。最後に、所与の位置における抗原結合に好適なアミノ酸の生化学的及び生物物理学的特性を評価し((13−Tomlinson et al.,1995)、108基準に達するためにそれらのアミノ酸を有するV生殖細胞系遺伝子多様性を補完した。個々に設計したLc−CDR多様性を表5に示す。
【0072】
重複PCR法を用いて、特定の位置における設計された多様性を含むLcを作製した。変性コドンを、個々のCDRに及ぶ増幅オリゴ内の位置に置くことができる。これらの変性コドンは、CDR位置における設計されたアミノ酸をコード化する。あるいは、それぞれの位置における特定の所望のコドンをコード化しているいくつかのオリゴを合成することができる。次いで、これらのオリゴを混合して、重複PCRで用いられるライブラリのDNAを増幅するために用いられるライブラリを作製することができる。設計されたオリゴを用いて、Lcの断片を増幅し、所望のSDR位置における配列変異のプールを作製する。フレームワーク#1領域をコード化するLc断片、CDR1−2突然変異領域、及びフレームワーク#3CDR3突然変異領域を、変性オリゴを用いるPCRにより合成した。既に言及したVLライブラリ断片を、次いで、隣接する増幅プライマーを用いる重複PCRで組み合わせた。この反応は、所望の位置で3つ全てのCDRに配列変異を有する完全長VLライブラリを生じさせる。
【0073】
あるいは、DNA合成法「GeneWriter」を用いて、設計に従って個々のLcライブラリを合成することができる。ヌクレオチド配列は、大腸菌の好ましいコドンを用いて設計されたアミノ酸配列から逆翻訳することができる。それぞれ設計された配列の一部を網羅し、設計されたヌクレオチド変異を含むオリゴヌクレオチドを合成し、他の部分に記載した技術を用いて遺伝子として組み立てることができる(14−米国特許出願公開第0165946 A1号)。簡潔に述べると、オリゴヌクレオチド配列及び設計された変異を、オリゴヌクレオチド作製のためにソフトウェア「MultiWriter」を通して加工することができる。種々の数のライブラリオリゴヌクレオチドを、センス鎖に対して作製して、所望の配列変異に基づく全ての組み合わせを表すことができる。同じソフトウェアを用いて、アンチセンス鎖を設計し、合成することができる。VL遺伝子は、特許(14)に「遺伝子アセンブラ」として記載されているものより小さな合成オリゴマーの一連のアニーリング−ライゲーションを介して組み立てることができる。完全長VL DNAをPCRにより増幅し、PCR産物をKunkel法におけるプライマーとして、又はライブラリ作製のためのpCNTO−pIX−lacIベクターにおける設計された部位に直接クローニングするための源として用いることができる。また、代替DNA合成及び大きな遺伝子を組み立てる方法を用いて、設計されたVL遺伝子を作製することができる。
【0074】
実施例6.ライブラリ作製法
修正されたKunkelの単鎖突然変異誘発法を用いて、それぞれのスカフォールドFabに基づいて個々にライブラリを作製した(15−Genentechの論文、16− Henry Lowmanの書籍)。最終的なライブラリの機能に対する非突然変異誘発スカフォールドFabテンプレートの効果を最小化するために、TAA終止コドンをテンプレートのH−CDR3領域に挿入した。それぞれの終止コドンを含有するテンプレートの単鎖DNAを、CJ236株から調製した。オリゴは、それぞれ設計されたH−CDR1及びCDR2多様性をコード化し、T7 DNAポリメラーゼ及びT4リガーゼを用いてDNA重合反応におけるプライマーとして同時に用いられる。反応混合物を用いて、MC1061F’コンピーテントセルを形質転換し、典型的にはライブラリコンストラクションあたり>109の独立コロニーを得た。コロニーをプレートから擦り取り、新鮮培地に接種し、二本鎖プラスミドDNAを調製し、それを用いてCJ236細胞を形質転換した。形質転換された細胞にヘルパーファージを感染させ、単鎖DNAを一晩細胞培養物から調製し、反応テンプレートとして用いてH−CDR3多様性を導入した。個々のFabスカフォールド/テンプレートのライブラリを個々に構築した。8種の異なる長さのH−CDR3を4つの反応で構築し、ここでH−CDR3の長さ7及び8、9及び10、11及び12、並びに13及び14aaをそれぞれグループ化した。図7はライブラリ構造を示す。
【0075】
有効ではあるものの、我々は現在のKunkel法が、高度配列複合体ライブラリを構築するとき、20〜50%の範囲で、突然変異していない親ssDNAのみを除くことが多いことを見出した。ライブラリ作製中の段階的Kunkel反応において使用される短いオリゴヌクレオチドは、更にこの方法の効率を低下させる。更に親DNAを除くために、6つのカッター制限酵素部位(XbaI)を含むパリンドロームを、標的化突然変異配列領域、例えば、VH及び/又はVLに操作した。プライマーのssDNAへのアニーリング中、パリンドロームは、二本鎖ステムループ構造を形成し、突然変異プライマーは隣接する配列へのハイブリダーゼーションによりこのパリンドロームに及ぶ(図8)。DNA複製は、T7 DNAポリメラーゼを用いてアニーリングされたプライマーから進行する。クローニングされた環状DNAは、T4DNAリガーゼにより作製される。この産物をパリンドロームループ内の部位(XbaI)で制限エンドヌクレアーゼを用いて消化し、よって親DNA鎖に切れ目を入れ、そのインビボ複製能を破壊する。組み合わせられたパリンドローム配列削除及びKunkel法は95%を超える親DNAを除き、現在のKunkel法のみに比べて著しく改善されている。我々はまた、重複PCRにより配列多様化されるべき3つの全ての標的化CDRを網羅するメガプライマーを作製し、突然変異誘発反応で用いた。これは、1つの反応で達成されるべき全てのコンビナトリアル配列多様化を可能にし、短いオリゴヌクレオチドを用いる段階的反応より効率的である。例えば、我々は、重複PCRにより、及び/又はGeneWriter(商標)を用いる化学合成により、VLライブラリ断片を作製し、それをハイブリダイゼーション突然変異誘発のメガプライマーとして用いた。メガプライマーをPCRにより作製し、マイナス鎖をビオチン化プライマーを用いて増幅する(図9)。ビオチン化鎖はストレプトアビジン磁気ビーズ(Dynal)上に捕捉され、ビオチン化されていない鎖は0.15MのNaOH中における変性により精製された。溶出された鎖を、次いで、ssDNA(REStr420jw)上におけるメガプライマー媒介DNA複製で用いた。
【0076】
VH及びVLにおける多様性を有するライブラリはまた、代替法及び源により作製することができる。例えば、VH及びVL遺伝子を免疫された動物組織から増幅し、定常領域とともにインフレームでクローニングして、Fabライブラリを作製した(17−winter)。あるいは、免疫された動物からのCDR領域、例えば、H−CDR3をPCRで増幅し、デノボH−CDR1、H−CDR2及びVLライブラリバックグラウンドにおいて免疫化H−CDR3のハイブリッドを形成するデノボライブラリにおける対応領域にクローニングできる。このようなライブラリのファージパニングは、高親和性結合抗体を単離できる。
【0077】
実施例7インライン親和性成熟
ファージパニングプロセスの一部としてデノボライブラリから選択された結合材の親和性成熟を設計し、そのプロセスを「インライン成熟」と名付けた。プロセスは、詳細な特徴付けの有り無しで、結合抗体のプールを用いて開始し、続いて結合剤のLcにおけるCDR配列を多様化し、これはライブラリスカフォールドとして用いられた4つの生殖細胞系Lcの1つに由来していた。LcCDR多様性は、更なるファージパニングを介して選択できる、追加の結合活性を作製する。プロセスを以下に記載し、また以下に詳述する(図10)。まず、リード抗体のVH領域をパニングを通して単離し、FabデノボライブラリをVL−パリンドロームループベクターにサブクローニングした。これらのベクターのssDNAを、実施例6に記載のVLライブラリメガプライマー又は好適な方法を用いて、ハイブリダイゼーション突然変異誘発のために作製した。例えば、組み合わせられたパリンドローム配列削除、及びメガプライマーKunkel突然変異誘発法を有効に用いて、インライン親和性成熟のためのVL CDR多様化2次ライブラリを作製することができる。親DNAをXbaI消化、続いて非許容状態の宿主株に形質転換することにより除く。この方法は、設計された配列多様性を網羅するライブラリの高度に効率的な作製を可能にする。次いで、ファージライブラリを、突然変異ライブラリにヘルパーファージを感染させることにより作製した。次に、ライブラリを高度な選択厳密性でパニングの更なるラウンドに供して、1次ライブラリから最初に選択された結合剤よりも改善された結合親和性を有する結合剤を増やす。これらは、抗原濃度を低下させ、洗浄時間及び頻度を増加させることを含む。あるいは、結合競合物又は他の結合ストレスコンポーネント、例えば、結合温度の変更及び剤の添加は、所望の特性を有する結合剤を選択するための結合及び/又は洗浄緩衝液に含まれてよい。ファージ結合及びその後の大腸菌感染に好適な他の生化学的及び生物物理学的条件もまた、パニングに含まれてもよい。パニング後、DNAをファージから回収する。Fabは発現し、結合、並びに他の生化学的、生物物理学的、及び生物学的特徴付けに供される。ファージから回収したDNAはまた、IgGベクターに直接クローニングすることもできる。この場合、Fabの代わりにmAbが発現し、特徴付けされる。例えば、重複PCR法によりVLライブラリ断片を作製し、それをハイブリダイゼーション突然変異誘発のメガプライマーとして用いた。メガプライマーは、PCRにより作製され、マイナス鎖はビオチン化プライマーを用いて増幅される(実施例6を参照)。ビオチン化鎖はストレプトアビジン磁気ビーズ(Dynal)上に捕捉され、ビオチン化されていない鎖は0.15MのNaOH中における変性により精製された。溶出された鎖を、次いで、ssDNAにおけるメガプライマー媒介DNA複製で用いた。Fabデノボライブラリのパニングを通して単離されたリード抗体のVH領域を、VL−ループベクターにサブクローニングした。これらの選択されたVH/VL−ループベクターを、VLライブラリメガプライマーとともにハイブリダイゼーション突然変異誘発のためのssDNAにした。XbaI含有パリンドロームの喪失により述べたように、親DNAの削除は、ランダムに選ばれたライブラリクローンのサンプルの配列により100%決定された。これらのCDRのライブラリの変異型への突然変異誘発は75%であった。この方法によりライブラリの4×108変異型の高度に効率的な作製が可能になった。我々は、Fabデノボライブラリにおいて用いられる他の3つのVL−k鎖におけるライブラリの作製と同様の結果を見出した。ライブラリを2×108に形質転換し、ファージを1011cfu/mLにした。BSAをデノボpIXライブラリでパニングし、ヒットを単離した(表3)。2つのLCライブラリを、上記方法を用いてBSAに対する1次ヒット上に構築した。ライブラリは、A27LC多様性を有するF4HC、及びB3LC多様性を有するD6、F6、C2、C6HCのプールからなっていた。
【0078】
【表1】
【0079】
【表2】
【0080】
3ラウンドのパニングを、非ストリンジェントな又はストリンジェントな洗浄条件のいずれかを用いて、それぞれ10nM、1nM、及び1nMの抗原で実施した。抗原陽性クローンの配列を決定し、選択クローンを精製して親和性データを得た。より低ストリンジェントな条件でパニングしたB3ライブラリでは、増やされたクローンの大部分が野性型であった。それらの野性型ではないクローンのうち、豊富さを示したものはなく、Biacoreデータはそれらが親和性を改善しなかったことを確認した。よりストリンジェントなパニング条件をこの同じライブラリに適用したとき、LCCDR1における突然変異を有するクローンの豊富さが見られ、その後Biacore分析でおよそ3倍の親和性の改善が示された(表4)。このライブラリから単離された全てのクローンはD6重鎖を含んでおり、これは他の重鎖に比べてBSAに対する親和性が最も高かった。A27ライブラリでは、パニング法ではクローンの濃縮も野性型の回復も見られなかった。単一クローンの濃縮は観察されなかったが、アミノ酸又は特定のCDR位置におけるアミノ酸特徴の濃縮は見られた。選択クローンは、親に比べて最大10倍の親和性改善を示した(表5及び図示せず)。
【0081】
【表3】
【0082】
【表4】
【0083】
実施例8.平行ライブラリパニング
我々のライブラリは、それぞれのサブライブラリがスカフォールドHC又はLCに基づいて個々に用いられ得るような方法で設計及び構築されるため、我々は、平行パニング戦略を設計して、個々並びにサブプールライブラリセットをパニングして、多様な結合抗体を同定する機会を最大化する。
【0084】
BSA及びリゾチームを、初期ライブラリ検証のモデル抗原として選択した。我々は、Kingfisher( )を用いる平行パニングとは別にLCフレームワークを保つことにより4つのサブセットに1〜69及び3〜23HCフレームワークサブライブラリをプールした。比較として、我々はまた、全てのサブライブラリ1〜69及び3〜23HC、並びに関連LCを1つのプールに完全にプールし、それを用いてBSAに対してパニングした。ビオチン化抗原でコーティングされた常磁性ビーズ(Dyal)を、室温で1時間ファージとともにインキュベートした。結合及び洗浄を96ウェルブロック中で、ハイスループットフォーマットで実施した。いくつかの洗浄工程後、結合したファージを溶出し、次の選択ラウンドのためにMC1061F’細胞を感染させることにより増幅した(詳細は実施例2)。選択ラウンド後、DNAを単離し、pIXを制限酵素消化により除去し、ライゲーションさせ、MC1061F’に形質転換して、可溶性Fabを発現させる。単離した単一クローンをFab発現及び抗原結合についてELISAにより試験した。抗Fd抗体でコーティングされたELISAプレート上でFab含有細胞抽出物を、続いてHRP結合抗カッパ抗体をインキュベートすることにより、Fab発現を検出した。抗原特異性を、Fab含有細胞可溶化物をその後添加したストレプトアビジンでコーティングされたプレート上でビオチン化抗原を捕捉することにより試験した。競合ELISAでは、過剰量の非ビオチン化抗原が含まれ、Fab結合に競合した。結合したFabをHRP結合抗カッパ抗体を用いて検出した。
【0085】
平行パニングの結果は、90のスクリーニングされたクローンのうち30がBSAに結合したことを示し、これは、この実験のヒット率が33%であることを示す。ヒット率は、完全にプールされたライブラリを用いてパニングを完了したときの7%のヒット率より非常に高い(表6)。リゾチーム用の2つのパニング戦略の直接比較は存在しないが、完全にプールされたライブラリパニングのみが実施された場合、ヒット率は4%である(表6)。BSAについて6つの独自の抗体及びリゾチームについて3つを同定した配列分析を、図11に示す。
【0086】
独自のBSA及びリゾチーム結合剤を精製して、結合親和性(KD)を決定した。2段階Fab精製手順(REStm334bw.doc)を用いてFabを精製した。簡潔に述べると、大腸菌溶菌液を最初の1段階IMAC手順に供した。透析後、IMACカラム溶出液を2番目の精製工程、QFFセファロース樹脂を用いるアニオン交換に供し、これは、Fabの純度を〜80%〜90%改善した。図12は、精製されたBSA及びリゾチームFabを示す。
【0087】
BIAcore分析を用いて、6つのBSA及び3つのリゾチーム結合剤の親和性を決定した。結果は、それぞれ、BSA結合剤について0.1〜28nM、リゾチーム結合剤について6〜260nMに及ぶFabの結合親和性を示した。BSAへのFab群の非特異的結合は存在せず、リゾチーム、又は参照表面(空の、活性化され、ブロックされたCM5マトリックス)は検出されなかった。表7及び8は、BSA及びリゾチーム結合剤の結合反応速度を示す。
【0088】
実施例9.親和性ランキング
結合解離定数KoffによりFabを順位付けするために、粗大腸菌上清をOctetシステムを用いて測定した(例えば、forteo.comのような公的ウェブサイトに記載されているように)。SAセンサプローブを、試験ビオチン化抗原でコーティングした。一晩大腸菌誘導培養物を、2xBBS/リゾチーム緩衝液で溶解し、96アッセイウェルに入れた。較正したセンサチップを、大腸菌溶菌液を収容しているアッセイウェルに浸漬し、結合会合を測定した。センサチップを次いで移動させ、緩衝液のみの中に入れて、結合解離を測定した。結合反応速度を、製造者のソフトウェアを用いて分析した。参照のために、表6は、同じ抗原に対して、Octet及びBIAcoreに由来する順位付け結果と、3つの異なるFabサンプルを比較する。結合反応速度はまた、BIND機器(例えば、srubio systemsによる)を用いて評価し、Fab発現細胞可溶化物をまず濾過して溶菌していない細胞、大きな細胞残屑、脂質、及び剪断された核酸を除去した。ストレプトアビジンでコーティングされたセンサプレートを較正し、ビオチン化抗原とともにインキュベートした。ウェル30秒間隔で2分間読み取り、100μL/ウェルの1xPBSで3回洗浄した。次いで、フィルタ処理したFab溶解液をアッセイウェルに添加し、ストレプトアビジンセンサプレート表面上に提示された抗原に結合することを可能にした。ウェルを空にし、200μLの1xPBSを添加した。ビオチン化抗原に対する結合シグナルを、30秒間隔で2〜5分間測定した。センサ表面に結合した複数の既知の量の抗原についての解離データを適合させることによりKoff値を得た。会合速度を得ることはできなかったが、解離速度(Fab濃度には依存しない)は、Biacore結果に相当した。
【0089】
上記代替アッセイフォーマットは、Fabタンパク質上に含まれるタグに適切な抗−F(ab’)2、抗−Fd、又は抗−HIS又は抗FLAGを提示する。抗体はセンサプレートのアミン反応性表面に結合させることができる。濾過した大腸菌溶菌液又はFabタンパク質を含有するペリプラズム画分を、センサプレート上に結合された抗−F(ab’)2により捕捉した。抗原とともにインキュベートした後、結合反応速度を上記のように測定することができる。
【0090】
粗大腸菌上清を用いて結合解離定数(Koff)を測定するための2つの方法を開発した。1つは、粗大腸菌培地上清を用いるELISAに基づく方法であり、他方は同時に複数のサンプルの結合反応速度を測定することができるベンチトップ機器であるOctet system(Fortebio website)である。親和性ランキングELISAではマイクロエクスプレッション(500μL)により作製されたFabの量を定量化した。Fab発現ELISAは、細胞可溶化物画分及び使用済み培地画分におけるFabタンパク質の量を示す。両方の画分は同程度の量のFabタンパク質を有するため、細胞可溶化物は作製するのがより面倒であり、使用済み培地画分をランキングELISAにおける使用のために選択した。3つの抗原を用いてELISAランキング法を確立した。それらは、マウスST2L、ヒトレジスチン、及びマウスMCP−5である。配列が独特であるFabを用いて、その反応性抗原に結合する能力を試験した。それぞれのサンプルについて発現したFabを含有する使用済み培地の複数の希釈液をそれぞれのELISAに適用した。Fabのそれぞれの希釈液を2つに分割し、一方のアリコートは抗原に結合するため、他方のアリコートはそれぞれの希釈したサンプルに含有されるFabタンパク質の相対量を測定するためである。抗原結合及び発現ELISAの結果を計算して、それぞれの希釈点の特異的活性比を確立した。対応するクローンに対する特異的活性比をプロットすることは、全てのFabサンプルの全体的視野を提供する。この視野から、希釈が曲線の直線点上に位置し、ランキングのための大部分のFab候補を取り込むという測定をすることができる。53のmST2Lサンプルを、1:2及び1:4希釈で試験した。
【0091】
1:4希釈を用いて、これらのFabの順位を決定した、なぜならそれは希釈曲線の線状セクター中にあるように思われるためである。8つのmMCP−5 Fabの順位付けは、より広い希釈範囲、1:2〜1:10を用いて行われた。ここで用いられた希釈範囲は、結合曲線の線状範囲の外側であった。80のレジスチンFabを1:2、1:4、及び1:8で希釈して、3種の希釈が線状範囲をよりよく確立するかどうかを測定した。これらのFabの部分集合のみが、この希釈範囲における線状活性:濃度挙動を有していた。レジスチンFabを再び発現させ、使用済み上清を1:5、1:10、及び1:20で希釈した。1:5及び1:10希釈は、線状滴定曲線内にサンプルを有さなかったが、1:20は線状範囲内であった。特異的活性比を全てのサンプルについて計算し、順位を確立した(表9A、B&C)。Biacoreによる親和性測定値は、それらの結合反応速度に関してmST2L Fabの一部とほとんど違いを示さなかったが、ランキングELISAデータは少し違いを示した(表9A)。Fab28の順位付けにおけるELISAとBiacore結果との間の不一致は、抗原配向に用いられたフォーマットによるものである可能性がある。ELISAは溶液中に抗原を有し、一方Biacoreフォーマットは表面上に提示された抗原を有していた。Biacore結果におけるFab40は、全体で最低のKDを与えるfast on−rate(ka)を示す。これは、不正確なタンパク質濃度によるものである可能性がある。mMCP−5Fabは、ELISAとBiacore結果との間の順位付けにおいて良好な相関を示す。両方のアッセイにおいてFab S10、S14、及びS30は最も低い順位の結合剤であり、Fab P9、P12、P20、及びS1は最も高い順位の結合剤であった(表9B)。FabP1はELISAランキングアッセイにおいて良好な結合剤であったにもかかわらず、それはまた、1:10希釈で低下したシグナルを示し、Biacoreでは並の結合剤であった。
【0092】
レジスチンFabは、ELISAとBiacoreランキングの間に相関を示す(表9C)。ELISA及びBiacoreランキングにおいて、クローン551−22が最良であり、クローン169−7が最悪であった。ELISA及びBiacoreランキングの両方において、更に323−7及び323−32は、クローン551−22に次いで良い順位である。
【0093】
【表5】
【0094】
【表6】
【0095】
【表7】
【0096】
ELISAランキングデータは、いくつかの例外を除いてBiacore相互作用分析から推定される解離値に最もよく相関していた。最適化されたELISAに基づくランキング法は、多数のFab候補の適切なスクリーニング及び管理を支持する。Octet機器を用いて、動力学結合活性によりFab候補を順位付けするための2つのフォーマットを開発した。溶液フォーマットは、チップセンサ表面に適用されたFab及び溶液中に存在するAgを用いて実施される。ある期間中センサ表面に添加されたバイオマスの測定を通してAgがFabに結合するときの会合速度を測定する。AgがFabに結合した後、センサが緩衝液(すなわち1xPBS)中に置かれたとき解離速度を計算し、経時的なセンサ上のバイオマスの低下をもたらす。固定化された動力学結合アッセイでは、抗原をチップセンサ表面に適用し、Fabは溶液中に存在する。会合及び解離速度を、経時的なチップ上のFab増加及び、続くバイオマスの低下により決定する。
【0097】
アミン反応性センサチップ用の基本的なプロトコルは、まず初期ベースラインの確立から始まり、続いて第1の分子(すなわち2次rgt、Fab、又はAg)がチップセンサに適用される活性化工程を行う。クエンチ工程を、第1の分子がチップセンサに結合した後に実施され、別の分子との結合相互作用が生じることが可能になる前に別のベースラインが確立される。ロード(すなわち2次試薬でコーティングされたチップセンサにFab又はAgを添加)又は会合工程(すなわちFab又はAgでコーティングされたセンサ)のいずれかを次に実施する。これがロード工程である場合、別のベースラインが会合工程が行われる前に確立される。いったん会合工程が完了すると、解離工程が実施され、実験が完了する。溶液動力学結合アッセイは、高結合SAコーティングされたセンサチップを用いる。サンプル96ウェルプレート(カタログ番号675076、Greiner Bio−One)を以下のように調製する:カラム1に100μLの1xPBSをチップセンサのベースラインまで充填し、カラム2に、1xPBS中10μg/mLの濃度でBt−抗ヒトF(Ab)’2(カタログ番号109−066−097、Jackson Immunological)を充填して、SAでコーティングされたセンサチップ表面にロードし、カラム3に第2のベースラインとして100μlの1xPBSを充填し、カラム4に第2のロード工程として1xPBS中10μg/mlのFabサンプルを100μl充填し、カラム5に第3のベースラインとして100μg/mlの1xPBSを充填し、カラム6に会合工程として1xPBS中10μg/mlのAgを充填し、カラム6のウェルは解離工程として1xPBSを収容する。動力学プロトコルをそれぞれの工程につき設定し、30℃で実行し、1000rpmで振盪する。第1のベースラインを100秒間実施する。第1のロード工程(抗ヒトF(Ab)’2)を600秒間実行する。第2のベースラインを200秒間実行する。第2のロード工程(Fab候補)を600又は900秒間実行する。第3のベースラインを300秒以内実行する。会合工程(Ag)を600秒間実行し、解離工程を最大900秒間実行する。固定化動力学結合アッセイは、アミン反応性センサチップを用いる。チップセンサボックスを、まず10分間室温で適切なMES pH溶液(Amine Coupling Kit,Fortebio)中でインキュベートする。サンプル96ウェルプレート(カタログ番号675076,Greiner Bio−One)を以下のように調製する:カラム1に、チップセンサのベースラインまで適切なMESpH溶液を充填し、カラム2に、ベースラインのMES pH溶液に1:50比のEDC/NHS(アミンカップリングキット、Fortebio)を充填して、センサ表面を活性化し、カラム3にMES pH溶液中10μg/mLのAgを充填して、センサチップをロードし、カラム4にエタノールアミン溶液(アミンカップリングキット、Fortebio)を充填して、Agのロードされたセンサの非占有表面をクエンチし、カラム5に第2のベースラインとして100μLの1xPBSを充填し、カラム6に会合工程として1xPBS中1μg/mLのFabサンプルを充填し、カラム7ウェルに解離工程として1xPBSを充填する。動力学プロトコルをそれぞれの工程につき設定し、30℃で実行し、1000rpmで振盪する。第1のベースラインを100秒間実行する。活性化工程を最大900秒間実行する。Agロード工程を600秒間実行する。クエンチ工程を最大300秒間実行する。第2のベースラインを最大300秒間実行する。会合工程(Ag)を600秒間実行し、解離工程を最大900秒間実行する。溶液動力学アッセイを、3つの異なるサイズの抗原に対して実施した。この追加の変数(抗原のサイズ)を用いて、いかに異なるバイオマスの増加がFab候補のランキングの最終的な計算結果に影響を与えるかについての理解が得られた。我々は、より大きなサイズの抗原から、より良いシグナル(y軸)及びR2値が得られることを見出した(表13A〜C)。Fabのセンサへの直接的なアミンカップリングは、センサ表面から100kDaの分子、抗F(Ab)’2を除くことにより、溶液動力学アッセイにおける会合シグナルの増加を補助し、したがって、最後の分子の質量を付加する効果を見る能力を高める可能性がある。Octetは、その会合及び解離シグナル曲線に従ってFab間の差を示し、それらは計算された速度定数と相関する。mST2L Fab 16、26、及び28は、Fab 1及び38より良好な結合速度を有する(図13a)。レジスチンFab323−11及び551−7は最高の、Fab 169−1及び551−38は最低の結合剤である(図13b)。mMCP−5Fabについて、それは、Fab 14が最低であることを除いて、Ag結合に関する順位付けをこのフォーマットで区別することはできない(図13c)。既に論じたように、mMCP−5を測定するためのこのフォーマットは最良ではない可能性がある。mMCP−5のサイズはFabより著しく小さく(9kDa対50kDa)、加えて100kDaの2次試薬でコーティングされたチップは、シグナルの差を測定できることをより悪化させる。
【0098】
固定化アッセイフォーマットは、Fab結合動力学を評価するための別の手段を提供する。図13Cの溶液フォーマットアッセイに見られるように、AgがFabより小さいとき(例えばmMCP−5)、シグナルは低下する。センサ上で既に確立されている大きなマス上の小さなタンパク質を読み取ろうとすることは、小さな効果を有する。より小さな分子、mMCP−5がセンサ上にある場合、結合パートナーを回転させ、次いでより大きな分子、例えばFabの結合活性を読み取ることは、より大きなシグナルを生じさせるはずである。この効果は図14で観察され、ここで、mMCP−5はセンサ表面にアミンカップリングして、Fabが添加されたとき会合を測定する。このアッセイフォーマットにおけるシグナル増加は、溶液フォーマットで見られるものの少なくとも2倍大きい。加えて、会合R2値はここで0.85より大きく、一方それらは溶液フォーマットで平均0.57であった(表14)。
【0099】
Biacoreは現在、結合動力学を決定するための標準である。我々は、上述のFabについてOctetから得られたデータを、Biacoreのデータと比較した。BiacoreとOctetとの間に感受性の差が見られるように思われる(図15)。Biacoreの親和定数KDは、一般に、Octet親和定数より高い。Octet実験では、分析物を10μg/mLの濃度で、Biacoreでは1μg/mLで用いた。10回用いたところ、Octetの分析物は会合速度定数をより早くすることができる。また、解離速度定数は、アッセイにおける過剰分析物競合のために間接的に影響を受ける場合がある。この原因と併せて、親和定数はBiacoreに比べてOctetの方が悪いように見える。直接立証されてはいないが、この感受性問題は同様に順位付けに対して影響を与える可能性がある。順位付けに関しては、レジスチンFabはBiacoreとOctetとの間で若干相関を示す。レジスチンと他のAg実験との間の唯一の差が、FabとAgとの間の分子サイズの差であった。Fabは50kDaであり、レジスチンは66kDaであり、これはサイズをより近くし、100kDAのmST2Lと9kDaのmMCP−5は異なるサイズである。
【0100】
結合動力学はまた、同時に96サンプルを測定できるBIND(www.srubiosystems.com)(別のベンチトップ機器)を用いて測定した。Fab発現細胞可溶化物をまず濾過して可溶化していない細胞、大きな細胞残屑、及び剪断された核酸を取り除いた。ストレプトアビジンでコーティングされたセンサプレートを較正し、ビオチン化抗原とともにインキュベートした。ウェル30秒間隔で2分間読み取り、100μL/ウェルの1xPBSで3回洗浄した。次いで、フィルタ処理したFab溶解液をアッセイウェルに添加し、ストレプトアビジンセンサプレート表面上に提示された抗原に結合することを可能にした。ウェルを空にし、200μLの1xPBSを添加した。ビオチン化抗原に結合するためのシグナルを、30秒間隔で2〜5分間測定した。SRU−BINDソフトウェアは参照細胞(Fabを含まない細胞可溶化物を添加した)から得たバルクシグナルを引く。センサ表面に結合した複数の既知の量の抗原についての解離データを適合させることによりKoff値を得た。会合速度を得ることは非常に困難であったが、解離速度(Fab濃度には依存しない)は、Biacore結果に相当した。
【0101】
上記に対するいくつかの代替アッセイフォーマットが存在する。例えば、BIND機器では、Fabは、必要に応じてセンサプレート上に固定化された抗−F(ab’)2、抗−Fd、又は抗−HIS又は抗FLAGにより捕捉され得る。濾過した大腸菌溶菌液又はFabタンパク質を含有するペリプラズム画分を、センサプレート上に結合された抗−F(ab’)2により捕捉され得る。抗原とともにインキュベートした後、結合反応速度を上記のように測定することができる。また、精製されたFabをセンサプレートのアミン反応性表面に直接結合させて、抗原結合を測定するためのこのフォーマットで用いることができる。
【0102】
結合動力学はまた、同時に96サンプルを測定できるBIND(商標)(Srubiosystems website)(別のベンチトップ機器)を用いて測定した。Fab発現細胞可溶化物をまず濾過して可溶化していない細胞、大きな細胞残屑、及び剪断された核酸を取り除いた。ストレプトアビジンでコーティングされたセンサプレートを較正し、ビオチン化抗原とともにインキュベートした。ウェル30秒間隔で2分間読み取り、100μL/ウェルの1xPBSで3回洗浄した。次いで、フィルタ処理したFab溶解液をアッセイウェルに添加し、ストレプトアビジンセンサプレート表面上に提示された抗原に結合することを可能にした。ウェルを空にし、200μLの1xPBSを添加した。ビオチン化抗原に結合するためのシグナルを、30秒間隔で2〜5分間測定した。SRU−BIND(商標)ソフトウェアは参照細胞(Fabを含まない細胞可溶化物を添加した)から得たバルクシグナルを引く。センサ表面に結合した複数の既知の量の抗原についての解離データを適合させることによりKoff値を得た。会合速度を得ることは非常に困難であったが、解離速度(Fab濃度には依存しない)は、Biacore結果に相当した。
【0103】
上記に対するいくつかの代替アッセイフォーマットが存在する。例えば、BIND機器では、Fabは、必要に応じてセンサプレート上に固定化された抗−F(ab’)2、抗−Fd、又は抗−HIS又は抗FLAGにより捕捉され得る。濾過した大腸菌溶菌液又はFabタンパク質を含有するペリプラズム画分は、センサプレート上に結合された抗−F(ab’)2により捕捉され得る。抗原とともにインキュベートした後、結合反応速度を上記のように測定することができる。また、精製されたFabをセンサプレートのアミン反応性表面に直接結合させて、抗原結合を測定するためのこのフォーマットで用いることができる。
【0104】
利点
上記ヒトFabデノボライブラリは他のこのようなライブラリとは異なる、なぜなら提示がM13ファージのpIXタンパク質を介しているためである。代表的なヒト生殖細胞系及びライブラリスカフォールドとしてのカノニカル構造配列の使用、並びにH−CDR3におけるアミノ酸の天然分布を模倣するCDR配列の設計は、ヒト免疫レパートリーの網羅においてライブラリを包括的にする。このライブラリ設計は、単一又は共通生殖細胞系遺伝子又は成熟mAbスカフォールド上に構築される、文献に報告されている他のデノボライブラリと比べて、標的抗原に対する抗体発見の可能性が高い。ライブラリの平行パニングに関与する別個のサブライブラリ(それぞれ独自のスカフォールド(VH/VL対)又はH−CDR3長さから構成される)は、標的抗原に対する多様な配列の抗体の発見の可能性を最大化する。更に、これはそれぞれのVH/VL対の機能性、及びそのそれぞれの配列、及び抗体/抗原結合における構造の詳細な研究のための機序を提供する。統合インライン親和性成熟プロセスは、多様性を維持しながら高親和性抗体を単離するのに有効な方法である。
【0105】
参考文献:
1.Kretzschmar & von Ruden,Current Opin Biotechnol.13:598〜602,2002)
2.Crooks GE,Hon G,Chandonia JM,Brenner SE WebLogo:A sequence logo generator,Genome Research,14:1188〜1190,(2004)
3.Wang W,Saven JG.Designing gene libraries from protein profiles for combinatorial protein experiments.Nucleic Acids Res.Vol 30,No 21,e120.2002
4.Hennie R Hoogenboom.Nature Biotech.(2005)23:1105.Selecting and screening antibody libraries.
5.V.Lee,Wei−Ching Liang,Mark S.Dennis,Charles Eigenbrot,Sachdev S.Sidhu and Germaine Fuh JMB(2004)340:1073〜1093.High−affinity Human Antibodies from Phage−displayed Synthetic Fab Libraries with a Single Framework Scaffold.
6.Michela Silacci,Simon Brack,Giulia Schirru,Jessica Marlind,Anna Ettorre,Adrian Merlo,Francesca Viti and Dario Neri.Proteomics(2005)5:2340〜2350.Design,construction,and characterization of a large synthetic human antibody phage display library
7.Eskil Soderlind and Carl A.K.Borrebaek.Nature Biotech.(2000).18:852.Recombining germline−derived CDR sequences for creating diverse single−framework antibody library.
8.Achim Knappik,Liming Ge,Annemarie Honegger,Peter Pack,Melanie Fischer,Gunter Wellnhofer,Adolf Hoess,Joachim Wolle, Andreas Pluckthunand Bernhard Virnekas JMB(2000)296:57〜86.Fully Synthetic Human Combinatorial Antibody Libraries(HuCAL) Based on Modular Consensus Frameworks and CDRs Randomized with Trinucleotides.
9.Griffiths AD,Williams SC,Hartley O,Tomlinson IM,Waterhouse P,Crosby WL,Kontermann RE,Jones PT,Low NM,Allison TJ,Prospero TD,Hoogenboom HR,Missim A,Cox JPL,Harrison JL,Zaccolo,Gherardi E and Winter G.EMBO J 1994:13(14):3245〜3260.Isolation of high affinity human antibodies directly from large synthetic repertoires
10.Hans J.de Haard,Nicole van Neer,Anneke Reurs,Simon E.Hufton,Rob C.Roovers,
Paula Henderikx,Adriaan P.de Bruyne,Jan−Willem Arends,and Hennie R.Hoogenboom JBC(1999).274:18218〜18230.A Large Non−immunized Human Fab Fragment Phage Library That Permits Rapid Isolation and Kinetic Analysis of High Affinity Antibodies*
11.Rene Michael Hoet and Robert ladner,et al.,Nature Biotech.(2005)23:344.Generation of high affinity human antibodies by combining donor−derived and syntheric complementarity−determining region diversity.
12.Crooks GE,Hon G,Chandonia JM,Brenner SE WebLogo:A sequence logo generator,Genome Research,14:1188〜1190,(2004)
13.Wang W,Saven JG.Designing gene libraries from protein profiles for combinatorial protein experiments.Nucleic Acids Res.Vol 30,No 21,e120.2002
14.Vbase:The database of human antibody genes.Public website:vbase.mrc−cpe.cam.ac.uk
15.Juan Carlos Almagro.J.Mol.Recognit.(2004)17:132〜143.Identification of differences in the specificity−determining residues of antibodies that recognize antigens of different size:implications for the rational design of antibody repertoires.
16.Ian M.Tomlinson,Jonathan RL.Cox,Ermanno Gherardi,Arthur M.Lesk and Cyrus Chothia.The EMBO Journal(1995)14:4628〜4638.The structural repertoire of the human VK domain.
17.米国特許出願公開第0165946 A1号、参照することにより全文が本明細書に組み込まれる
18.欧州特許出願第90201671.6号、参照することにより全文が本明細書に組み込まれる
19.米国特許第5,942,609号、参照することにより全文が本明細書に組み込まれる
20.Dillon et al.,BioTechniques 9(3):298〜300(1990);
21.Prodomou et al.,Protein Engineering 5(8):827〜829(1992);
22.Chen et al.,JACS 116:8799〜8800(1994);
23.Hayashi et al.,BioTechniques 17(2):310〜315(1994).
24.Stemmer,Nature 370:370:389〜391(1994)
25.Stemmer,Proc.Natl.Acad.Sci.USA 91:10747〜10751(1994).
26.Sidhu et al,Methods in Enzymology.(2000)328:333−363.Phage display for selection of novel binding peptides。
27.Tim Clackson and Henry B Lowma.2004.Phage display−a practical approach.Oxford University Press
28.Figini et al,J.Mol.Bio.(1994)239:68〜78.In vitro assembly of repertoires of antibody chains on the surface of phage by renaturation
29.William F.Dall’Acqua,Melissa M.Damschroder,Jingli Zhang,Robert M.Woods,Lusiana Widjaja,Julie Yu,Herren Wu.Methods(2005)36:43〜60.Antibody humanization by framework shuffling.
【0106】
表
【表8】
【0107】
【表9】
【0108】
【表10】
【0109】
【表11】
太字:同じファミリーの生殖細胞系配列に現れる
イタリック:ヌクレオチド
【0110】
【表12】
【0111】
【表13】
【0112】
【表14】
【0113】
【表15】
【0114】
NB:結合せず
【0115】
【表16】
【0116】
【表17】
【0117】
【表18】
【0118】
【表19】
【0119】
表13A〜Cは、Octetからの溶液フォーマット動力学データを示す。A)100kDのST2Lに結合するFab、Fab 26、Fab 28、Fab 1、Fab 38を、会合及び解離速度定数によりOctetを用いて順位付けした。B)66kDaのレジスチンに結合するFab 169−3、Fab 169−7、Fab 323−11、Fab 323−32、Fab 551−7、Fab551−22及びFab 551−38を、会合及び解離速度定数によりOctetを用いて順位付けした。C)9kDaのMCP−5に結合するFab P1、Fab P9、Fab P12、Fab P20、Fab S1、Fab S14及びFab S30を会合及び解離速度定数によりOctetを用いて順位付けした。
【0120】
【表20】
【0121】
表14:Octetからの固定化フォーマット動力学データ9kDaのMCP−5に結合するFab P1、Fab P9、Fab P12、Fab P20、Fab S1、Fab S14及びFab S30を、会合及び解離速度定数によりOctetを用いて順位付けした。
【0122】
【表21】
【0123】
【表22】
【0124】
【表23】
【0125】
表15A〜C:BiacoreとOctetとの間の反応速度チャートの比較。
上から下のチャートへ、mST2L、レジスチン、及びmMCP−5 Fab群である。
【技術分野】
【0001】
本発明は、抗体又は抗体断片を産生するための、pIX又はpVIIファージディスプレイライブラリを作製及び使用するための、組成物及び方法に関する。
【背景技術】
【0002】
ファージ又はファージミド系における融合パートナーとしてpIII及びpVIIIを用いる繊維状ファージディスプレイは、タンパク質工学、とりわけデノボ抗体単離及び親和性成熟のための技術として用いられている。ヒト様fab配列は、テンプレートとしての既知の抗体配列から作製することができ、ランダム又は突然変異相補性決定領域(CDR)又は重鎖CDR3(H3)のような抗原結合領域は、抗原又は免疫付与していない他の関心タンパク質標的に対するパニングを介して抗体断片配列の変異型を提示するファージライブラリから作製及び単離することができる。既に用いられているヒトデノボ抗体ライブラリは、合成的に、又は天然源に由来するIgG遺伝子の分子クローニングにより創製されてきた。合成ライブラリでは、可変重鎖及び軽鎖フレームワーク及びCDR領域を含む抗体DNA配列は、1)規定のIgG遺伝子、2)特定のIg生殖細胞系遺伝子、及び3)Ig生殖細胞系遺伝子のファミリーに由来する共通配列、並びに4)天然源に由来するPCRにより得られたIgG断片に基づき設計及び合成される。合成抗体ライブラリに加えて、ライブラリはまた、ヒト組織、例えば、骨髄及び末梢血細胞に由来するIgG DNAのコンビナトリアルクローニングにより創製することもできる。このようなライブラリは、fab抗体断片を提供するため、並びに選択された標的タンパク質の高親和性又は阻害性生物活性のような所望の特性を有するfab抗体断片を見つけることを試みるためにパニング及び成熟又は修正の連続するラウンドを実行するために用いられてきた。
【0003】
いくつかの標的タンパク質に対するヒト様fab抗体が、ファージディスプレイpIII又はpVIII抗体ライブラリから単離されている。特定の標的に対するfab抗体の単離には成功しているが、このようなファージディスプレイライブラリアプローチは、所望の特徴を有するfab抗体を単離するために数回、ライブラリ作製、パニング、及び成熟プロセスを繰り返さなければならないという問題に悩まされている。このようなファージライブラリはまた、VH/VL対合の寄与、抗原認識に関連する様々な生殖細胞系ファミリーのトポロジー特性、アミノ酸変異の位置及び程度、並びに様々なヒト生殖細胞系遺伝子に由来する抗体の相対的存在量を含む、ヒトに存在するヒト免疫多様性の範囲を完全に包含又は模倣していないという問題にも悩まされている。合成抗体の天然のレパートリーからのずれは、ヒトの治療用に用いられる場合、好ましくない生化学的特性、及び免疫原性のリスクを高める恐れがある。
【発明の概要】
【発明が解決しようとする課題】
【0004】
高親和性及び活性、高生産性、良好な溶解特性、並びに人間に投与されたときに低免疫反応傾向であるヒト治療抗体の重要な要素を同時にもたらす合成抗体ライブラリ及び方法に対する要求が存在する。更に、現在の方法に比べて、抗体発見の資源コストを低減し、生物学的評価のための抗体の供給を加速するために、合成ライブラリからの抗体単離効率を高める必要性が存在する。本発明のライブラリ及び方法は、総合設計、アセンブリ技術、及びファージpIX Fabライブラリを組み合わせることにより、これらの要求を満たす。
【課題を解決するための手段】
【0005】
先行技術の教示とは対照的に、現在、pVII及びpIXは、例えば、fab及び抗体断片作製並びに治療抗体の選択のための効率的かつ迅速なプラットフォームを提供するために、突然変異誘発又は他の多様性を生み出す技術(所望により、インライン成熟とともに)を用いて、高親和性fabライブラリの作製のためにうまく用い得ることが発見されている。本発明により、pVII及びpIXに融合する抗体可変又はfab領域は、ファージ表面上に動的相互作用で係合して、機能的抗体断片、代表的なヘテロ二量体モチーフを提示する。したがって、抗体重鎖及び軽鎖可変領域のファージ上への提示は、メンバーが二量体人工抗体種として機能することができ、新規又は所望の生物活性の選択を可能にする、コンビナトリアルヘテロ二量体アレイの多様なライブラリの提示及びアッセイに好適かつ好ましい方法である。
【0006】
本発明は、種々の改善されたかつ新規のpIX及びpVIIファージディスプレイデノボライブラリ作製方法、並びに(i)pIX又はpVIIファージタンパク質に融合する設計及び提示された抗体Fabデノボライブラリ、(ii)広く用いられているM13ファージのpIII及びpVIIIとは異なるファージ表面タンパク質の使用、(iii)ヒトレパートリーの配列及び構造を表す生殖細胞系VH及びVL遺伝子の小さなアレイの使用、(iv)Vh及びVl領域の相補性領域における、改善された、設計された、コンビナトリアル多様性を提供するための、ライブラリのスカフォールドとしてのこのようなファージ構成要素の使用、(v)抗原認識のための設計された配列及び構造トポロジーの効果の系統的試験を可能にする抗体選択プロセス、(vi)ライブラリ選択の一部としての、効率化された親和性成熟及びインライン成熟プロセス、のうちの1つ以上であるが、これらに限定されない、構成要素を提供する。ライブラリの個々の又は群のライブラリ設計、選択、最適化及び成熟のこのような新規系は、うまく抗体を新規発見するための再現可能かつ信頼できる系を提供し、また抗体の抗原に対する相互作用の構造機能関係の理解を促進する。
【0007】
上記ヒトFabデノボライブラリは、M13ファージのpIX遺伝子を介して、その提示により現在の抗体ライブラリの現況技術とは区別できる。ライブラリスカフォールドとしての代表的なヒト生殖細胞系及び構造配列の使用、及び天然のアミノ酸分布を模倣するCDR配列の設計は、ヒトレパートリーを包括的に網羅し、ライブラリ抗体は天然由来のヒト抗体に対して高い配列同一性を有する。ヒト免疫レパートリーの包括的網羅は、文献に報告されている単一生殖細胞系又はIgG遺伝子スカフォールド上に構築されたライブラリに比べて、抗体の新規発見の機会を増やす。別々に作製されたサブライブラリ(それぞれ独自のスカフォールド(VH/VL対)及び/又はH−CDR3長を保有する)は、独自の抗体を同定する機会を最大化し、抗体/抗原結合構造及び機能関係を系統的に試験するための機序を提供する。統合された親和性成熟プロセスは、多様かつ高親和性抗体を発見するために必要な時間を低減する。
【0008】
人工抗体は、本明細書では、(1)配列相同性及び標的抗原の毒性、(2)抗体を回収するために用いられる宿主又はハイブリドーマ培養において作製された抗体の生物学的影響、及び(3)所望の活性の選択に対するスクリーニングを含む、抗体分子の機能的戦略を用いるが、インビボ拘束を含まず作製することができる多様性の大きなタンパク質モチーフとして定義される。抗体分子は、3次元空間におけるペプチド要素のコンビナトリアルアレイを表現するための生物学的デバイスである。本質的な特徴は、CDR(相補性決定領域)が協力して結合部位を形成するが、その相互作用はCDR自体の間に構造的会合をほとんど有しない動的及び機能的なものであることである。この方法では、アミノ酸残基の完全な相補体は、結合のための最低エネルギーコストで抗原認識に利用可能である。配列空間だけでなく、3次元空間においてもコンビナトリアル設計を制御する能力は、免疫レパートリーの天然設計を再現し、最終的にはそれをしのぐ。
【0009】
したがって、本発明は、多様性に富むヘテロ二量体ポリペプチドアレイの構築のためのコンビナトリアルファージディスプレイフォーマットを記載する。具体的には、本発明は、融合ポリペプチドをコード化するゲノムを封入する繊維状ファージ粒子であって、融合ポリペプチドが繊維状ファージpVII又はpIXタンパク質のアミノ末端に融合する外来ポリペプチドを含むファージ粒子を記載する。好ましくは、ファージ粒子は、ファージ粒子の表面上に発現した融合タンパク質を含む。
【0010】
好ましい実施形態では、ファージゲノムは、第2の融合ポリペプチドを更にコード化し、第2の融合ポリペプチドはpIXのアミノ末端に融合する第2の外来ポリペプチドを含み、第1の融合ポリペプチド中の第1の外来ポリペプチドはpVIIタンパク質のアミノ末端に融合する。この実施形態では、第1及び第2の融合ポリペプチドは会合して、免疫グロブリンFv、触媒性Fv、受容体、核酸結合タンパク質又は酵素のような、ヘテロ二量体タンパク質複合体を形成することができる。
【0011】
関連する実施形態では、本発明は、融合タンパク質を発現するためのカセットを含む繊維状ファージの表面上で融合タンパク質を発現するためのベクターを記載する。カセットは、インサートDNAの指向性ライゲーションに適合したヌクレオチドの配列を介して機能的に連結した上流及び下流の翻訳可能なDNA配列、すなわち、ポリリンカーを含み、この場合、上流配列は原核生物の分泌シグナルをコード化し、下流配列はpVII又はpIX繊維状ファージタンパク質をコード化する。翻訳可能なDNA配列は、融合ポリペプチドの一部として翻訳可能なDNA配列を発現するためのDNA発現シグナル一式に機能的に連結する。好ましい変形では、ベクターは更に繊維状ファージの表面上で第2の融合タンパク質を発現するための第2のカセットを含み、ここで、第2のカセットは第1のカセットの構造を有するが、ただし第1の融合タンパク質発現カセットはpVIIタンパク質をコード化し、第2の融合タンパク質発現カセットはpIXタンパク質をコード化する。ベクターは、ヘテロ二量体の2つの外来ポリペプチドが、それぞれ第1及び第2のファージタンパク質、pVII及びpIXへの融合によりファージ粒子上に固定されるファージ粒子の表面上でヘテロ二量体タンパク質複合体を発現させるためのファージゲノムとして用いられる。
【0012】
別の実施形態では、本発明は、ライブラリ中の代表的な粒子がそれぞれ異なる融合タンパク質を提示する、本発明によるファージ粒子のライブラリ、すなわち、コンビナトリアルライブラリを意図する。粒子がヘテロ二量体タンパク質複合体を提示する場合、ライブラリは、Fv分子のライブラリの形態の抗体のような、ヘテロ二量体のコンビナトリアルライブラリを含む。好ましいライブラリは、少なくとも103、104、105、106、107、108、109、1010、1011、1012、1013、又はこの中の任意の範囲若しくは値のコンビナトリアル多様性、融合タンパク質の異なる種を有する。
【0013】
関連する実施形態は、第1のポリペプチドが外来タンパク質であり、第2のポリペプチドが繊維状ファージpVII又はpIXタンパク質であり、外来タンパク質が繊維状ファージタンパク質のアミノ末端に融合する、第1及び第2のポリペプチドを含む融合タンパク質を記載する。
【0014】
なお更に、本発明は、外来ポリペプチドをコード化する遺伝子のレパートリーを本発明のベクターにクローニングし、突然変異誘発により、第1及び第2の融合タンパク質ライブラリの集団のランダムな組み合わせにより、ライブラリの多様性を変化させるための標的及び親和性選択(「パニング」)等により、ライブラリ中の外来ポリペプチドの構造を修正することにより、ファージのコンビナトリアルライブラリを作製する種々の方法を意図する。
【0015】
改善された又は新規の機能を有するタンパク質の設計は、種々の医療、工業、環境、及び基礎研究用途の重要な目的である。コンビナトリアル抗体ライブラリの開発に続いて、強力な次の段階は、人工抗体構築、並びに二量体種がネイティブである、又は機能的である他のタンパク質モチーフへの進化である。
【0016】
本発明は、pVII及びpIXが二量体種を形成する融合タンパク質の提示に利用されるコンビナトリアルヘテロ二量体ポリペプチドアレイの構築のためのファージディスプレイフォーマットを提供することにより、これらの問題に取り組む。機能的相互作用のライブラリを作製するために非常に近接する2つのタンパク質モチーフを独立に提示することができるため、これは完全に新しい方法論であることに留意することが重要である。
【0017】
技術の範囲及び能力に固有なのは、二量体相互作用で係合できる種々のタンパク質を提示する能力である。これらは、抗体だけではなく、いくつかの酵素、ホルモン及びホルモン受容体、並びにDNA結合タンパク質を含む。本明細書に記載する提示技術は、抗体フレームワーク領域のコンビナトリアルな変化のために、並びに抗体構造を認識及び小型化するために、又はリプレッサのようなDNA結合タンパク質を提示するために、臨床的にかつ治療的に重要な特定のDNA配列に対する選択のためのヘテロ二量体のライブラリとして使用することができる。
【0018】
したがって、本技術は、突然変異体二量体タンパク質の提示及び選択、並びにメンバーがヘテロ二量体アレイからなるコンビナトリアルライブラリを提供する。この技術を用いて、ネイティブな免疫グロブリン構造(本明細書に示すヘテロ二量体VH−VL Fvフォーマットにおける)は、様々な方法で修正され、特異性及び活性についてスクリーニングされ得る。例えば、フレームワーク領域(FR)のコンビナトリアルな変化、又は「相補性決定領域(CDR)シャフリング」及び「ツインボディ」形成と呼ばれるプロセスにより抗体構造を再編成及び小型化するための他の操作により、新たなパラトープ又は完全に異なる構造的要素を含む抗体様2次構造が生じる。天然抗原及び/又は基質、並びにいくつかの関連化合物に対する、結合及び/又は触媒作用の選択は、ヘテロ二量体タンパク質のライブラリをスクリーニングするために用いられるであろう。
【0019】
更に、ライブラリを形成するための配列のランダム化、及びハイブリッド種を形成するための鎖シャフリングプロトコルは、新たなタンパク質の部分集合を導くことができる。例えば、ホモ二量体又はヘテロ二量体型におけるジンクフィンガードメインのアレイの提示及び修正は、特異的DNA相互作用を有する構造を生じさせる。加えて、完全に新たな構築は、抗体鎖のような予形成スカフォールド内の所望のコード化断片の挿入を介して可能である。可能なインサートとしては、酵素の特徴的な配列又はリプレッサ結合タンパク質が挙げられる。
【0020】
前述の一般的な説明及び以下の詳細な説明は、両方とも例示及び解説のためだけのものであり、請求されるような本発明を制限するものではないことを理解すべきである。
【図面の簡単な説明】
【0021】
【図1】スカフォールドVH及びVLアミノ酸配列。
【図2】ファージディスプレイ及びFab発現ベクターのベクター図:pCNTO−Fab−pIX−lacI。
【図3A】ライブラリスカフォールドFab発現及び提示のグラフ表示及び画像。3Aは、M13の表面上におけるFab発現の相対レベルを示す。3Bは、検出試薬として抗Fab’(2)を用いるウエスタンブロットにおけるFabの相対発現を示す。3Cは、既知の濃度のFabと比較することにより大腸菌培地上清におけるFabスカフォールドのおおよその発現を示す。これは、カッパ遺伝子スカフォールド(塗りつぶされたバー)及びラムダ遺伝子スカフォールド(陰影をつけられたバー)を含む。
【図3B】ライブラリスカフォールドFab発現及び提示のグラフ表示及び画像。3Aは、M13の表面上におけるFab発現の相対レベルを示す。3Bは、検出試薬として抗Fab’(2)を用いるウエスタンブロットにおけるFabの相対発現を示す。3Cは、既知の濃度のFabと比較することにより大腸菌培地上清におけるFabスカフォールドのおおよその発現を示す。これは、カッパ遺伝子スカフォールド(塗りつぶされたバー)及びラムダ遺伝子スカフォールド(陰影をつけられたバー)を含む。
【図3C】ライブラリスカフォールドFab発現及び提示のグラフ表示及び画像。3Aは、M13の表面上におけるFab発現の相対レベルを示す。3Bは、検出試薬として抗Fab’(2)を用いるウエスタンブロットにおけるFabの相対発現を示す。3Cは、既知の濃度のFabと比較することにより大腸菌培地上清におけるFabスカフォールドのおおよその発現を示す。これは、カッパ遺伝子スカフォールド(塗りつぶされたバー)及びラムダ遺伝子スカフォールド(陰影をつけられたバー)を含む。
【図4】H−CDR3設計パターンの概略。
【図5】H−CDR3オリゴヌクレオチド設計の概略。
【図6】H1〜69テンプレート用のH−CDR3位置におけるアミノ酸分布A.393D位置における分布(長さ9〜14)B.2163N位置における分布(長さ9〜14)C.H−CDR3の973Yにおける分布(長さ9〜14);D.NNSコドンを用いる長さ7及び8のH−CDR3の全ての位置における分布。
【図7】ライブラリ構築の要約図。
【図8】パリンドローム補助親ストランド脱離の概略。
【図9】V領域の全領域を標的とするメガプライマーの概略。
【図10】ヒットからのVH断片がPCRにより増幅され、VL−ループベクターにクローニングされる。得られたプラスミドはssDNAとして産生され、メガプライマーのハイブリダーゼーションと分解突然変異誘発のテンプレートとして機能する。Lcライブラリメガプライマーを用いてプラスミドを複製することができ、これによって、単離されたHc配列に関連するLcライブラリの多様性を組み込むことができる。この親和性成熟ライブラリは、M13ファージとしてパッケージ化され、初期抗原に対するファージパニングに利用され、親和性成熟したヒットを得られる。
【発明を実施するための形態】
【0022】
本発明は、新規ファージディスプレイデノボライブラリ作製方法、並びに(i)pIX又は他のファージタンパク質に融合する設計及び提示された抗体Fabデノボライブラリタンパク質、(ii)広く用いられているM13ファージのpIII及びpVIIIとは異なるファージ表面タンパク質の使用、(iii)ヒトレパートリーの配列及び構造を表す生殖細胞系VH及びVL遺伝子の小さなアレイの使用、(iv)Vh及びVl領域の相補性領域における、改善された、設計された、コンビナトリアル多様性を提供するための、ライブラリのスカフォールドとしてのこのようなファージ構成要素の使用、(v)抗原認識のための設計された配列及び構造トポロジーの効果の系統的試験を可能にする抗体選択プロセス、(vi)ライブラリ選択の一部としての効率化された親和性成熟プロセス等であるが、これらに限定されない、構成要素を提供する。ライブラリの個々の又は群のライブラリ設計、選択、最適化及び成熟のこのような新規系は、うまく抗体を新規発見するための再現可能かつ信頼できる系を提供し、また抗体の抗原に対する相互作用の構造機能関係の理解を促進する。
【0023】
上記ヒトFabデノボライブラリは、M13ファージのpIX遺伝子を介して、その提示により現在の抗体ライブラリの現況技術とは区別できる。ライブラリのスカフォールドとしての代表的なヒト生殖細胞系及び構造配列の使用、並びに天然の長さ及びアミノ酸分布を模倣するCDR配列の設計は、ヒト免疫レパートリーを包括的に網羅し、ライブラリ抗体は天然由来のヒト抗体に対して高い相同性を有する。免疫レパートリーの包括的網羅は、文献に報告されている単一生殖細胞系又はIgG遺伝子スカフォールド上に構築されたライブラリに比べて、抗体の新規発見の機会を増やす。別々に作製されたサブライブラリ(それぞれ独自のスカフォールド(例えばVH/VL対及び/又はH−CDR3長)を保有する)は、独自の抗体を同定する機会を最大化し、抗体/抗原結合構造及び機能関係を系統的に試験するための機序を提供する。統合された親和性成熟プロセスは、多様かつ高親和性抗体を発見するために必要な時間を低減する。
【0024】
定義:
抗体:種々の文法的な形の抗体という用語は、本明細書で使用されるとき、免疫グロブリン分子及び免疫グロブリン分子の免疫的に活性のある部分、すなわち、抗体結合部位又はパラトープを含む分子を指す。代表的な抗体分子は、インタクトな免疫グロブリン分子、実質的にインタクトな免疫グロブリン分子、並びにFab、Fab’、F(ab’)2及びFvのような当該技術分野において既知である部分を含む、免疫グロブリン分子の一部である。
【0025】
抗体結合部位:抗体結合部位は、抗原に特異的に結合する(免疫反応する)重鎖及び軽鎖可変並びに超可変領域から構成される抗体分子の構造部分である。種々の形の免疫反応という用語は、抗原決定基含有分子と、抗体分子全体又はその一部のような抗原結合部位を含む分子との間の特異的結合を意味する。
【0026】
融合ポリペプチド:少なくとも2つのポリペプチドと、その2つのポリペプチドを1つの連続するポリペプチドに機能的に連結する連結配列から構成されるポリペプチド。融合ポリペプチドに連結する2つのポリペプチドは、典型的には、2つの独立した源に由来し、したがって、融合ポリペプチドは自然界では通常連結した状態では見られない2つの連結したポリペプチドを含む。
【0027】
シストロン:アミノ酸残基配列をコードし、上流及び下流にDNA発現制御要素を含むDNA分子中のヌクレオチドの配列。
【0028】
繊維状ファージ
本発明は、融合タンパク質(タンパク質)をコード化するゲノムを封入するタンパク質のマトリックスを含む繊維状ファージを意図する。融合タンパク質は、繊維状ファージpVII又はpIXタンパク質のアミノ末端に融合する外来ポリペプチド部分を含む。
【0029】
「外来」とは、ファージタンパク質に融合するポリペプチドが、繊維状ファージの野性型変異におけるファージpVII又はpIXタンパク質と通常は関連せず、正常ファージタンパク質とは異なることを意味する。
【0030】
典型的な外来ポリペプチドは、免疫グロブリン重鎖可変ドメイン(VH)、免疫グロブリン軽鎖可変ドメイン(VL)、天然又は合成ポリペプチド、単鎖抗体(scFV)等を含む、任意の関心ポリペプチドである。
【0031】
好ましい実施形態では、繊維状ファージは第1及び第2の融合タンパク質をコード化するゲノムを封入し、ここで、第1の融合タンパク質はpVIIに融合する第1の外来ポリペプチドを含み、第2の融合タンパク質はpIXに融合する第2の外来ポリペプチドを含む。
【0032】
繊維状ファージは、実施例に記載のような、ファージ粒子の表面上に提示される融合タンパク質を更に含有する。したがって、第1及び第2の融合タンパク質が存在する場合、ファージは、第1及び第2の外来ポリペプチドがヘテロ二量体として相互作用して、ファージ表面上に機能的な2本の鎖のタンパク質複合体を形成するように、機能的方式でこれらのタンパク質を提示できる。
【0033】
発現したヘテロ二量体タンパク質がリガンドに結合する能力を有する場合、それはあるいは本明細書ではリガンド結合ヘテロ二量体受容体と呼ばれる。
【0034】
好ましい実施形態におけるヘテロ二量体受容体は、エピトープ結合複合体である。すなわち、第1及び第2のポリペプチドの複合体は、エピトープに結合することができる。好ましくは、第1及び第2のポリペプチドは、抗体重鎖及び軽鎖ポリペプチドである。具体的には、好ましい実施形態はVH及びVLを利用してFv複合体を形成する。他のヘテロ二量体タンパク質複合体としては、触媒性Fv、受容体、核酸結合タンパク質、及び酵素等のヘテロ二量体タンパク質が挙げられる。
【0035】
本発明のファージ上に存在する融合タンパク質では、外来ポリペプチドと繊維状ファージpVII又はpIXタンパク質との間の「融合」は、典型的なアミド連結を含んでよく、又は実施例に記載のようなリンカーポリペプチド(すなわち、「リンカー」)を含んでもよい。典型的には長さ約5〜50アミノ酸のストレッチである種々のリンカーのいずれを用いてよい。特に好ましいリンカーは、リンカーの点で融合タンパク質に高度な可動性をもたらす。
【0036】
ライブラリ設計:先行技術の合成ライブラリは、以下のいくつかを組み込んでいたが、包括的方式で全てを含むものは存在しない。
【0037】
生殖細胞系V領域ファミリー多様性重鎖及び軽鎖のヒト可変レパートリーは、配列相同性及び長さにより定義される関連配列のファミリーからなる。このような任意のファミリーでは、個々のメンバーは主に相補性決定領域の配列及び長さが異なる。これらの差は、既知の構造的変異パターン(カノニカル構造)、並びに抗原と相互作用しやすくする構造的に類似の位置における生殖細胞系アミノ酸変異を導く。ライブラリに単一Vh及びVI遺伝子テンプレートのみを選択することは、捕捉され得る天然生殖細胞系多様性の範囲を狭める(ジェネンティックライブラリを参照)。全てのVh及びVl遺伝子を選択することは、ライブラリ作製の効率を無効にする。本発明のライブラリは、(1)再編成されたヒト抗体における優性カノニカル構造群を表す少数の生殖細胞系Vh、Vk、及びV−ラムダを同定し(IMGT、Kabat、NCBI)、(2)コンビナトリアルオリゴヌクレオチド突然変異誘発により、Vk及びV−ラムダ領域のCDR1及び2、又はCDR1〜3をもコード化するVH遺伝子における関連ファミリーメンバーの天然ヒト生殖細胞系多様性を組み込むことにより生殖細胞系多様性を効率よく捕捉する。
【0038】
VH−CDR3多様性。VH−CDR3は、V、D及びJセグメントの接合により創製され、両端付加及びヌクレオチド鎖分解性事象により達成される。約25の生殖細胞系D領域が存在し、この数は複合体接合事象を伴い、体細胞変異は抗体の最も多様な領域を創製する。これらの事象は、生殖細胞系配列の規定のセットで生じ、ランダムではないが、予測することは困難である。しかしながら、再編成されたヒト抗体配列のデータベースは、現在、VH−CDR3のそれぞれの位置における長さ及びアミノ酸分布の両方の統計的評価を適用するのに十分な大きさに達している(約5000VH領域)。本発明のライブラリは、この領域を集めるために、設計された変性オリゴヌクレオチドを利用することによりこの天然のヒト多様性を再現する。
【0039】
体細胞多様性の位置及び性質。体細胞多様性は、いかにヒト抗体が高親和性、選択的結合実体に成熟するかの証明である。この体細胞突然変異の作製及び蓄積はランダムではない。ヌクレオチド突然変異の部位及び種類は、DNA配列によりバイアスがかかるが、しばしば中性置換とともに、結合及び機能的利点をもたらす突然変異のみが選択され、保存される。機序からの予測には従わないが、再編成されたヒト抗体配列のデータベース及び構造−機能解析は、タンパク質、ペプチド及び小分子抗原の間の分化を含む、CDR領域における抗原の認識に最も高い頻度で関連する位置及びアミノ酸置換を同定する。本発明のライブラリは、VH CDR1及び2、又はVk及びV−ラムダ領域のCDR1〜3にも置換を組み込むために、設計された変性オリゴヌクレオチドを利用することにより、天然のヒト多様性を再現する。
【0040】
生殖細胞系遺伝子の利用。ヒト生殖細胞系レパートリーは、約30のV−カッパ、56のV−ラムダ、及び40のV−重鎖機能遺伝子からなる。しかしながら、再編成された抗体におけるそれらの表現は強くバイアスがかかっており、これは異なる軽重鎖V−領域の対合の頻度において反映される(de Wildt et al.,J Mol Bio 285:895〜901(1999))。本発明のライブラリは、上記(a)に記載された多様性部類のそれぞれの優性生殖細胞系V−領域を選択することにより、このバイアスを取り込む。
【0041】
発現、生化学的、及び生物物理的特性。好ましいヒト抗体は、所望の生物学的及び結合活性を有するが、また種々の宿主から効率よく産生され、安定であり、良好な溶解特性を有する。高頻度の生殖細胞系遺伝子使用(1d)はまた、哺乳類系における良好な発現を示す。加えて、選択又はスクリーニングの細菌ファージディスプレイ法によりライブラリから回収される抗体は、細菌宿主においてよく発現するはずである。本発明のライブラリは、よく発現するヒト生殖細胞系由来のテンプレートに基づき、標準的な組み換え哺乳類宿主(例えばHEK293及びCHO細胞)並びに細菌宿主から精製され、高い安定性及び良好な溶解特性を有する。
【0042】
成熟。それぞれの位置で最大20の異なるアミノ酸の潜在的変異と合わせて、抗原認識に影響を与える場合のあるV−領域配列における多数の位置は、単一ライブラリにおいて全ての変異を含むことを実用的に不可能にする。ヒト抗体は、体細胞突然変異の進行性過程により高親和性及び特異性を獲得する。本発明のライブラリは、ヒト抗体を反映するようにそれぞれの抗体鎖の配列一体性を維持しながら、平行選択及び標的化変異を可能にするために設計及び指示される。
【0043】
代替設計。上記設計は、天然のヒト抗体を再現する。系のモジュラー性は、位置の任意の集積におけるアミノ酸の任意の集積を組み込みやすい。
【0044】
ライブラリアセンブリ技術。好ましいデノボ抗体ライブラリは、高い多様性を有し(>1010)、変化しやすく、組み立てが容易であり、望ましくない配列のバックグラウンドが低い。これらのバックグラウンド配列は親テンプレート及び低標的化多様性を含む。以下の方法を組み合わせると、ライブラリの組み立てが加速し、低バックグラウンドを導く。(a)Kunkle−ベースの単鎖突然変異誘発、(b)制限酵素部位を有するパリンドロームループ、(c)メガプライマー。
【0045】
pIX Fabファージディスプレイ法。全ての先行技術の繊維状デノボヒト抗体ライブラリは、単一Fvライブラリ(Scripps参照)を除いて、提示のためにpIII又はpVIIIファージコートタンパク質を利用する。pIXと選択されたFabテンプレートとの組み合わせは、mAb及び他の関連分子に変換するとき、その選択された特性を保持する抗体を回収するためのより効率のよい選択系である。
【0046】
Fabディスプレイ。scFvとは異なり、Fabはヒト抗体の天然セグメントであり、それらは完全抗体に操作されるとき活性をより再現する。効率のよいFabの繊維状提示は、細菌宿主における良好な発現を超える特性を必要とする場合がある。本発明のV−領域テンプレートは、繊維状ファージ上のpIXによる効率のよい提示について選択した。
【0047】
ファージミドディスプレイ。Fab分子はファージpIXコートタンパク質に比べて大きく、よって細菌宿主において産生される全てのpIXタンパク質に連結する場合、組み換えファージ粒子の組み立てに干渉する恐れがある。この干渉をバイパスするためのアプローチの1つは、(Scripps参照)により記載されているような、pIXファージミド系を使用することであり、それにより野性型及びFab連結pIXタンパク質の両方を組み換えファージ粒子に組み込むことができる。好ましい用途では、本発明のライブラリは、ファージミド系においてpIXにより提示される。
【0048】
提示のためのファージコートタンパク質pIX。pIIIのように、pIXはファージ上に少コピー数存在し、提示されたFabを親和性選択しやすい。しかしながら、pIIIタンパク質は、感染過程において決定的に関与し、このタンパク質上に提示されるタンパク質は感染効率に干渉する恐れがある。更に、重鎖Fd又は軽鎖セグメントのいずれかが提示のためにpIXに融合することができる。pIXタンパク質上に提示される本発明のライブラリは、効率よく複製されると予測され、選択及び/又はスクリーニングのために提示される。
【0049】
Fab−pIX発現。ファージライブラリから回収されるFabをスクリーニングするアプローチの1つは、提示のためにFab分子に連結するファージのコートタンパク質を取り除くことである。小さなサイズのpIXタンパク質は、この工程を行うことなく直接Fabのスクリーニングを作製する選択肢を提供する。
【0050】
ライブラリスカフォールドの設計。ライブラリスカフォールドは、一式のヒト生殖細胞系VH及びVL遺伝子から作製される。分析文献並びに独占抗体情報は、ヒトIgG遺伝子ファミリーを表す生殖細胞系遺伝子の同定、IgGレパートリーにおける使用、及びヒト抗体カノニカル構造を導いた。VhとVLとの間の良好な対合及び生殖細胞系遺伝子に由来する抗体の成熟成功の確率もまた、分析において考慮された。ヒトの性質及びスカフォールドVH及びVLの良好な対合の設計は、ライブラリスカフォールドとしてヒト生殖細胞系遺伝子のコンセンサスを用いる設計(MorphoSys HuCAL GOLD)より優れており、ライブラリスカフォールドとして単一生殖細胞系遺伝子に基づいた設計(Dyax VH and Affytech)よりヒトレパートリーをより包括的に表す。
【0051】
ライブラリスカフォールドの発現及び提示能力。ライブラリスカフォールドFabの良好な発現及び提示能力は、直接、スカフォールド遺伝子上に発生するライブラリの品質に関連する。ライブラリスカフォールドFab発現及び提示能力は、ライブラリ構築前に調べられた。発現したが、全く又はそれほど提示しなかったいくつかのスカフォールドFabは、ライブラリ構築から除外した。良好に発現し提示したライブラリスカフォールドは、ライブラリ内の高い割合のFabが機能的であり、天然源から遺伝的に増幅されたVH及びVL遺伝子のコンビナトリアルクローニングに由来するライブラリ(CAT)より優れている。
【0052】
H−CDR3多様性の設計。ライブラリVH−CDR3を、長さ及び配列の両方において多様化した。CDRの長さに依存する約109〜1018の総配列可能性を、抗原結合におけるVH−CDR3の重要性を反映するVH−CDR3において設計した。アミノ酸多様化の全てのアミノ酸コドンの使用(例えば、Genentech 2004によるNNK)、及び天然源からのVH−CDR3配列を操作するための包括的クローニング法の使用(CAT、Dyax、Affytech)とは異なり、我々は、ヒトIgGレパートリーにおけるアミノ酸使用パターンを模倣するアミノ酸をコード化する設計されたオリゴヌクレオチドを使用することによりVH−CDR3を創製した。設計されたVH−CDR3は、より少ない、望ましくない(例えば、終止コドン)及びより少ない、好ましくないアミノ酸(例えば、システイン)を含むが、より高い割合のIgG様CDR3配列を含む。変性されたオリゴヌクレオチドは、突然変異誘発反応において大きなサイズの遺伝子ライブラリを作製するために容易に用いることができる。
【0053】
H−CDR1及びH−CDR2多様性の設計。VH−CDR1及びVH−CDR2におけるアミノ酸多様性は、生殖細胞系配列内の変異を模倣するよう設計される。VH−CDR1及びVH−CDR2における組み合わせられた総配列多様性は、102〜105の範囲である。大きなVH−CDR3配列の変異と併せて、これは、ライブラリ中の総配列変異を増加させ、したがって、抗原結合抗体を同定する機会を増やす。小さな、生殖細胞系様多様性の独自の設計は、生殖細胞系又は天然様抗体配列を発見することを好み、ライブラリ作製に対するコンビナトリアル効果のために非天然CDRの組み合わせを単離する可能性を最小化する。この設計は、全ての3つのVH CDR(Genentech、Dyax、及びMorphoSys)におけるより大きな配列多様性を含む他の合成又は半合成抗体ライブラリの多くとは異なり、更に大きな理論的配列多様性のほんの一部のみがライブラリに取り込まれ得る。
【0054】
LCにおける多様性の設計。ライブラリ軽鎖は、デノボライブラリにおいて変動するよう戦略的に設計され、Lc多様性はFabがファージパニングを通して選択された後CDRに組み込まれた。抗原結合FabにおけるLc多様性は結合親和性を改善するために設計されるため、既知の構造の抗原−抗体複合体において頻繁に抗原と接触することが見出されているCDR位置は、多様化のために選択された。段階的CDR多様化戦略は、大きな理論的CDR配列多様性を有効に管理し、生物学的特徴により好適な、高結合親和性抗体を作製する。
【0055】
ライブラリ作製方法。修正されたKunkel突然変異誘発法(それぞれ異なるFab配列を持つ数十億の大腸菌コロニーを効率よく作製する)は、大きなFabライブラリを作製するために用いられる。効率的でありながら、高配列複合体ライブラリの作製に適合するとき、突然変異誘発されていない親DNAの割合が増加する。加えて、合成の長いオリゴヌクレオチドの技術的限界は、離れた領域において配列多様性を含むライブラリを作製するための方法の効率を低下させる。この限界を打開するために、>350ベースのオリゴヌクレオチド(メガプライマー)の作製、及び突然変異誘発テンプレートに制限酵素認識部位を含むステムループ配列の創製の更なる技術が、標準的なKunkel突然変異誘発法と併用された。ライブラリ作製のために他者により使用される、制限クローニング(MorphoSys、CAT及びDyax)、ファージ組み換え(Giggapack、Invitrogene)、及び配列特異的組み換え(CAT)のような他のライブラリ技術と比べて、改善されたKunkelに基づく方法は、>109配列多様性ライブラリの作製において著しくより有効であり、標的化DNAの任意の位置における配列多様性を導入するためにより可変性である。
【0056】
インライン親和性成熟。統合された親和性成熟プロセス、又はインライン親和性成熟は、ライブラリから選択される全てのFabの結合親和性を改善するために設計される。ライブラリは種々のFabのVLを残すために戦略的に設計されるため、追加の抗原結合部位はCDR配列多様化を介してVLにおいて容易に創製されるべきである。パニング後、全てのFabの改善された結合親和性は、治療的抗体リード同定の成功を増加させる。ライブラリ作製のためのKunkel法の使用は、単純かつ連続プロセスにおけるVL配列多様化戦略の有効な実施を保証する。改善されたKunkel突然変異誘発法の設計戦略及び技術的利点は、このアプローチを、目的の効率及び利益を低減する冗長なライブラリ作製法が使用される場合、CAT及びMorphoSysにより報告される他のプールされた成熟戦略より優れたものにする。
【0057】
平行ライブラリパニング。現状技術、半自動化設備を用いる平行パニングプロセスは、個々にサブライブラリを作製するプロセスのために開発される。平行パニングは、独自に設計され作製されたライブラリを支持する、抗体の多様なセットを発見する可能性を最大化する。インライン親和性成熟における平行パニングの有効な使用はまた、同時に改善されるべき広範な親和性を有する抗体を可能にする。機械に基づくパニングの開発はまた、所望の特性の抗体を発見するために異なるパニング条件を系統液に評価することを可能にする。
【0058】
親和性ランキング。親和性に基づく結合アッセイは、更に特徴付けするために最良の結合抗体を選択するために大きく、多様で、高い親和性の抗原特異的結合抗体に適用される。多数のサンプルを処理するのに好適な、ELISAのような標準的な生化学的方法、並びに例えば、BIAcore、Octet、及びBINDS等の親和性測定器具は、この目的のために単独で又は組み合わせて用いられる。
【0059】
一般的な用語で本発明を記載してきたが、本発明の実施形態は更に、特許請求の範囲を限定するものとして解釈されるべきではない以下の実施例に開示される。
【実施例】
【0060】
実施例1:実施例1ライブラリスカフォールドの設計
ヒト生殖細胞系遺伝子をライブラリ骨格として設計した。生殖細胞系VH及びVL遺伝子のパネルを、最初に、1)天然に発現しているIgGの使用、2)タンパク質及びペプチド抗原認識を好む構造(主鎖立体構造)トポロジー、3)生殖細胞系遺伝子に由来する抗体の生化学的及び生物物理学的特性、及び4)抗体としてVH及びVLヘテロ二量体を形成する可能性の特性に基づいて同定した。パネルから上記列挙した特性を最もよく表す4つのVH及び4つのVL生殖細胞系遺伝子を更に選択した。配列改変を含む既知の抗体に由来する10アミノ酸の単一、人工H−CDR3配列、及びヒトJH4セグメントをそれぞれの生殖細胞系VHと併用して、完全なVH配列を完成させた。VLでは、Jκ1セグメントを用いて選択された生殖細胞系VLのそれぞれを組み合わせた。これらの4つのVH及び4つのVL生殖細胞系に基づく遺伝子を化学的に合成する。定義されたCH及びCL配列と組み合わせて、それらは16の組み換えヒトFabを構成した。VL−λでは、Jλ2セグメントを選択されたVL−λ生殖細胞系スカフォールドのそれぞれと組み合わせた。3つのVH(169、323、及び551)生殖細胞系遺伝子を、VL−λスカフォールドと組み合わせて、CH1及びCλ配列を含むベクターと組み合わせて、12の組み換えヒトFabを得た。これらの28のFabをライブラリスカフォールド又はテンプレートとして用いて、重鎖又はLc鎖相補性決定領域(CDR)における配列多様性を提示した。図1は、設計された4つのVH及び4つのVLの配列を示す。
【0061】
実施例2.ライブラリスカフォールドの発現及び提示
合成FabDNAをpCNTO−lacI−pIXベクター(図−−)にクローニングする。VH遺伝子をNcoI及びApaI部位を介してクローニングした。VL遺伝子(ompAシグナル配列及び上流配列を含む)を、NheI及びBsiWI部位を介してクローニングした(図2)。スカフォールドFabの発現及び提示を、それぞれウエスタンブロット分析、及びファージELISAで調べた。
【0062】
提示を調べるために、ファージを調製し、ファージELISAで試験した。簡潔に述べると、pCNTO−Fab−pIX−lacI構築をMC1061F’細胞に形質転換し、振盪しながら37℃で2xYT/Carb(100μg/ml)/TET(15μg/ml)/1%グルコース培地中で一晩増殖させた。次の朝、50μlのこの増殖物を用いて、5mlの2xYT/Carb(100μg/ml)を播種し、OD600が0.6〜0.8になるまで増殖させた。次いで培養物に、振盪せずに37℃で40分間ヘルパーファージ(ファージの#)を感染させた。感染した細胞を遠心沈殿させ、5mlの2xYT/Carb(100μg/ml)/TET(15μg/ml)/Kan(35μg/ml)/0.5mM IPTG培地に再懸濁し、振盪しながら30℃で一晩増殖させた。次の朝、細胞を遠心沈殿させ、ファージ上清を回収し、ファージELISAに用いた。50μlのファージ上清(未希釈)、並びに1:5の3種の連続希釈液を抗Fd(Hc特異的)又は抗カッパ(Lc特異的)抗体でコーティングされたELISAウェルに添加した。インキュベーション後、結合していないファージを洗い流し、結合しているファージを抗M13抗体により検出した。H3−53/L−B3及びH3−53/L−A27を除く全てのスカフォールドの組み合わせが抗Fd及び抗カッパ抗体に対して良好な結合を示し、これはファージ表面上のHc及びLcの十分な提示を示す(図3)。
【0063】
Fab発現を調べるために、pIX遺伝子をまずSpeI及びNheI制限酵素消化によりまずpCNTO−Fab−lacI−pIXベクターから切り取り、続いて消化されたベクターDNAをセルフライゲーションさせる。これはFab発現の構築、pCNTO−Fab−lacIを作製する(図2)。pCNTO−Fab−lacIファージミドを有するM1061F’細胞を、振盪しながら37℃で2 X YT/1%glucose/Amp(100μg/ml)培地中で一晩増殖させた。一晩培養物の5mLのアリコートを未誘導対照として用いた。0.1mLの一晩培養物を10ml 2XYT/0.1%グルコース/Amp(100μg/ml)培養培地に接種した。培養物を振盪しながら37℃でOD600nmが0.8〜1.0になるまで増殖させた。IPTGを最終濃度0.5mMまで添加して、Fab発現を誘導した。振盪しながら30℃で更に16〜20時間培養を続けて増殖させた。誘導した培養物を遠心沈殿させ、それぞれの細胞のペレットを0.4mLのBPER II試薬(Pierce)を添加することにより溶解させた。使用済み細胞可溶化物の上清を回収し、Fab発現をウエスタンブロットにより分析した。約10倍低いレベルで発現したB3LCを含むものを除いて、全てのFabが(>2μg/ml)で発現した。またFab発現をELISAを用いて調べた。50μlの使用済み細胞可溶化物、並びに1:5の6種の連続希釈液を、抗Fd(Hc特異的)でコーティングしたELISAウェルに添加した。Fabを抗カッパ又は抗ラムダHRP結合抗体のいずれかで検出した。Fab対照標準曲線と比べてFabの量を定量化した(図3C)。
【0064】
実施例3.H−CDR3多様性の設計
天然免疫レパートリーを模倣するH−CDR3における大きな配列多様性を設計した。合計約5250の重複しない、完全(v領域、CDR3及びフレームワーク4)ヒト抗体配列を、種々の公的源から集めた。データセットは全ての7Vhファミリーを含んでいた。独自に開発したプログラムを書いて、Kabatの定義に基づいてCDR3セグメントを抽出した。抗体配列を、CDR3の長さに基づいて更に分析した。H−CDR3の長さは1〜27アミノ酸の範囲であり、正規分布を示した。最大数の抗体は、H−CDR3に12aaを含んでいた。同じ長さを有する抗体配列を整列させ、グループ化した。JH6由来の配列を長さの群から引いて、JH6に関連する、複数の、連続するチロシンを含有する配列を最小化した。プログラムを書いて、それぞれの長さのアミノ酸分布を計算した。H−CDR3長さ群のそれぞれの位置における残基を、その頻度に基づいて並べ替えた。内部にインストールされていたWeblogo(9−Crooks GE,Hon G,Chandonia JM,Brenner SE WebLogo:A sequence logo generator,Genome Research,14:1188〜1190,(2004))を用いて、それぞれのHCDR3の長さ分布をプロットした。およそ全てのヒト抗体レパートリーの約65%を網羅する、H−CDR3の7〜14アミノ酸を含む配列で作業することを決定した。
【0065】
アミノ酸分布を分析することにより、7〜14アミノ酸を含むH−CDR3のアミノ酸分布を説明するパターンを同定する。図4は、H−CDR3のパターンを示した。高度に多様なアミノ酸がH−CDR3で観察されたにもかからわず、アミノ酸グリシン及びアラニンは全ての位置で頻繁に用いられる。加えて、アスパラギン酸(D)は位置95で頻繁に用いられ、チロシン(Y)はH−CDR3におけるJセグメントにより近い位置で頻繁にコード化される。位置99〜101の配列は変化するが、アミノ酸フェニルアラニン(F)、アスパラギン酸(D)及びチロシン(Y)は、これらの位置でIgGにおいて主に用いられる。これらの位置はH−CDR3に対する構造的支持体として機能することが多く、抗原及び/又はIgGの表面に接近しにくいため、それぞれ99はF/L、100はD、101はYのアミノ酸に固定した。
【0066】
H−CDR3における高度に多様なアミノ酸分布を模倣するために、H−CDR3をコード化する混合された塩基オリゴヌクレオチドを設計して、それらをライブラリ構築で用いた。Wang & Saven(10−Wang W,Saven JG.Designing gene libraries from protein profiles for combinatorial protein experiments.Nucleic Acids Res.Vol 30,No 21,e120.2002)に記載されている手順を用いて、コドントリプレットに由来するコードアミノ酸が標的とするアミノ酸分布を模倣するように、複数のランダム試験におけるコドントリプレットのそれぞれの位置でヌクレオチド混合比の初期値を決定する。終止コドンを最小化し(<3%)、混合塩基オリゴヌクレオチドによりシステインコドンを低減するために、Wang & Saven手順のように同じ標的機能に対して、MicroSoft Excel(商標)のSolverにおけるスクリプトを用いてコドントリプレットのそれぞれの位置におけるヌクレオチド混合比の値を更に絞り込んだ。D及びN位置に用いられるヌクレオチド組成を図5に示す。より短いH−CDR3長さ、例えば7〜8アミノ酸では、NNSコドンを代わりに用いる。
【0067】
次いで、コドン設計に基づいて混合塩基オリゴヌクレオチドを作製した。D及びN位置に加えて、チロシン(〜18%Y)に富む位置は、別の方法でN位置と類似のaa分布を共有する。N位置におけるN設計コドンと、それぞれのチロシン位置における固定されたコドンTATを有する別個のオリゴを作製し、それを用いて11〜14アミノ酸のCDR長さを有するライブラリのN設計コドンオリゴと混合した。Nコドンオリゴ及びNと、特定のY位置用TATコドンオリゴを混合するために用いた比は約7:1である。設計者はNコドンに約4.7%のチロシンを与えるため、この混合物によりY位置における固定されたTATコドンから13%のチロシンが更に得られる。
【0068】
実施例6に記載の方法を用いてデノボFabライブラリを構築するためにオリゴを用いた。それぞれのサブライブラリから約100個のコロニーのDNA配列を得、分析した。H−CDR3におけるTAA終止コドンから突然変異したクローンを陽性クローンとみなした。翻訳されたH−CDR3配列を用いて、個々の又は組み合わせられたD、N及びY位置におけるアミノ酸分布を決定した。結果は、これらの位置で観察されたアミノ酸分布は、データベース及び設計で見出された分布を非常に模倣していることを示した(図6)。また、設計された混合塩基オリゴは、NNS変性オリゴよりも、設計及びデータベースに対するこれらの位置のアミノ酸分布をより模倣していることを示した(図6)。
【0069】
実施例4.H−CDR1及びH−CDR2多様性の設計
生殖細胞系遺伝子レパートリーを模倣するためにH−CDR1及びH−CDR2における多様性を設計する。多様化のために標的とされるH−CDR1&CDR2位置を、1)生殖細胞系遺伝子における多様性(11−Vbase)、及び2)溶媒曝露分析及び文献により決定された既知の構造の抗体−抗原複合体における抗原との接触が頻繁に見られること(12−Almagro 2004)により決定する。その位置におけるアミノ酸多様性は、1)生殖細胞系における利用(11)、2)生殖細胞系遺伝子(包括的データベース)に由来するIgGにおいて最も頻繁に用いられるアミノ酸、3)体細胞突然変異の結果として誘導されると予測されるアミノ酸、及び4)抗原認識に寄与するアミノ酸の生化学的及び生物物理学的特性により決定する。H−CDR1&CDR2における組み合わせられた配列多様性は、102〜105の範囲である。使用頻度を、IgGにおいて頻繁には用いられないアミノ酸を制限するためのフィルタとして用いた。これは、コンビナトリアル突然変異により作製される非天然配列を最小化する。表1は、配列分析及びH−CDR1及びCDR2多様性の設計を示した。
【0070】
H−CDR1及びH−CDR2設計を最もよく網羅する変性オリゴヌクレオチドを設計し、合成する(表1)。オリゴヌクレオチドを、単一Kunkle ssDNA依存性突然変異誘発反応におけるプライマーとして用いる。したがって、定義されたH−CDR1&CDR2位置における設計された縮重は、予測通りH−CDR1及びH−CDR2に導入される。Kunkel突然変異誘発反応に従ってランダムに選ばれたクローンの配列分析は、31%〜55.7%のクローン配列がH−CDR1及び/又はH−CDR2領域における突然変異を有することを示した(表2)。H−CDR1及びH−CDR2の両方に突然変異を有するクローンの百分率は、1つのCDR領域にのみ突然変異を有するクローンと等しいか又はそれより高いことが見出された。
【0071】
実施例5.Lcにおける多様性の設計
以下のパラメータは、設計軽鎖CDR多様性において考慮される。まず、我々は、組み合わせられたLc多様性が108を超えるべきではないことを定義した、なぜなら、Lcライブラリは親和性成熟で用いられ、したがってライブラリのパニングの第1段階中、ファージ選択に由来するおよそ102の独特のHcクローンのプールと組み合わせられるからである。したがって、組み合わせられたHc及びLc複雑度は約<1010であり、これは単一Kunkel突然変異誘発反応により生じるライブラリにおいて有効に捕捉され得る。2番目に、我々は、既知の構造の抗原−抗体複合体における抗原と接触することが見出されている多様化位置、又はいわゆる特異性決定残基(SDR;12)、つまり、それぞれL−CDR1、L−CDR2及びL−CDR3について位置30〜32、50及び91〜94を標的とした。3番目に、位置96はまた、それが接合(J)遺伝子及びその位置が異なる全ての5つのヒトJ遺伝子にコード化されているため、変化に富んでいる。4番目に、設計H−CDR1及びH−CDR2で用いられている概念と同じように、多様化されるべき再編成されたIgGでも頻繁に見られる生殖細胞系にコード化されているアミノ酸を選択することにより、自然に発生する可能性の低い配列多様性を最小化した。最後に、所与の位置における抗原結合に好適なアミノ酸の生化学的及び生物物理学的特性を評価し((13−Tomlinson et al.,1995)、108基準に達するためにそれらのアミノ酸を有するV生殖細胞系遺伝子多様性を補完した。個々に設計したLc−CDR多様性を表5に示す。
【0072】
重複PCR法を用いて、特定の位置における設計された多様性を含むLcを作製した。変性コドンを、個々のCDRに及ぶ増幅オリゴ内の位置に置くことができる。これらの変性コドンは、CDR位置における設計されたアミノ酸をコード化する。あるいは、それぞれの位置における特定の所望のコドンをコード化しているいくつかのオリゴを合成することができる。次いで、これらのオリゴを混合して、重複PCRで用いられるライブラリのDNAを増幅するために用いられるライブラリを作製することができる。設計されたオリゴを用いて、Lcの断片を増幅し、所望のSDR位置における配列変異のプールを作製する。フレームワーク#1領域をコード化するLc断片、CDR1−2突然変異領域、及びフレームワーク#3CDR3突然変異領域を、変性オリゴを用いるPCRにより合成した。既に言及したVLライブラリ断片を、次いで、隣接する増幅プライマーを用いる重複PCRで組み合わせた。この反応は、所望の位置で3つ全てのCDRに配列変異を有する完全長VLライブラリを生じさせる。
【0073】
あるいは、DNA合成法「GeneWriter」を用いて、設計に従って個々のLcライブラリを合成することができる。ヌクレオチド配列は、大腸菌の好ましいコドンを用いて設計されたアミノ酸配列から逆翻訳することができる。それぞれ設計された配列の一部を網羅し、設計されたヌクレオチド変異を含むオリゴヌクレオチドを合成し、他の部分に記載した技術を用いて遺伝子として組み立てることができる(14−米国特許出願公開第0165946 A1号)。簡潔に述べると、オリゴヌクレオチド配列及び設計された変異を、オリゴヌクレオチド作製のためにソフトウェア「MultiWriter」を通して加工することができる。種々の数のライブラリオリゴヌクレオチドを、センス鎖に対して作製して、所望の配列変異に基づく全ての組み合わせを表すことができる。同じソフトウェアを用いて、アンチセンス鎖を設計し、合成することができる。VL遺伝子は、特許(14)に「遺伝子アセンブラ」として記載されているものより小さな合成オリゴマーの一連のアニーリング−ライゲーションを介して組み立てることができる。完全長VL DNAをPCRにより増幅し、PCR産物をKunkel法におけるプライマーとして、又はライブラリ作製のためのpCNTO−pIX−lacIベクターにおける設計された部位に直接クローニングするための源として用いることができる。また、代替DNA合成及び大きな遺伝子を組み立てる方法を用いて、設計されたVL遺伝子を作製することができる。
【0074】
実施例6.ライブラリ作製法
修正されたKunkelの単鎖突然変異誘発法を用いて、それぞれのスカフォールドFabに基づいて個々にライブラリを作製した(15−Genentechの論文、16− Henry Lowmanの書籍)。最終的なライブラリの機能に対する非突然変異誘発スカフォールドFabテンプレートの効果を最小化するために、TAA終止コドンをテンプレートのH−CDR3領域に挿入した。それぞれの終止コドンを含有するテンプレートの単鎖DNAを、CJ236株から調製した。オリゴは、それぞれ設計されたH−CDR1及びCDR2多様性をコード化し、T7 DNAポリメラーゼ及びT4リガーゼを用いてDNA重合反応におけるプライマーとして同時に用いられる。反応混合物を用いて、MC1061F’コンピーテントセルを形質転換し、典型的にはライブラリコンストラクションあたり>109の独立コロニーを得た。コロニーをプレートから擦り取り、新鮮培地に接種し、二本鎖プラスミドDNAを調製し、それを用いてCJ236細胞を形質転換した。形質転換された細胞にヘルパーファージを感染させ、単鎖DNAを一晩細胞培養物から調製し、反応テンプレートとして用いてH−CDR3多様性を導入した。個々のFabスカフォールド/テンプレートのライブラリを個々に構築した。8種の異なる長さのH−CDR3を4つの反応で構築し、ここでH−CDR3の長さ7及び8、9及び10、11及び12、並びに13及び14aaをそれぞれグループ化した。図7はライブラリ構造を示す。
【0075】
有効ではあるものの、我々は現在のKunkel法が、高度配列複合体ライブラリを構築するとき、20〜50%の範囲で、突然変異していない親ssDNAのみを除くことが多いことを見出した。ライブラリ作製中の段階的Kunkel反応において使用される短いオリゴヌクレオチドは、更にこの方法の効率を低下させる。更に親DNAを除くために、6つのカッター制限酵素部位(XbaI)を含むパリンドロームを、標的化突然変異配列領域、例えば、VH及び/又はVLに操作した。プライマーのssDNAへのアニーリング中、パリンドロームは、二本鎖ステムループ構造を形成し、突然変異プライマーは隣接する配列へのハイブリダーゼーションによりこのパリンドロームに及ぶ(図8)。DNA複製は、T7 DNAポリメラーゼを用いてアニーリングされたプライマーから進行する。クローニングされた環状DNAは、T4DNAリガーゼにより作製される。この産物をパリンドロームループ内の部位(XbaI)で制限エンドヌクレアーゼを用いて消化し、よって親DNA鎖に切れ目を入れ、そのインビボ複製能を破壊する。組み合わせられたパリンドローム配列削除及びKunkel法は95%を超える親DNAを除き、現在のKunkel法のみに比べて著しく改善されている。我々はまた、重複PCRにより配列多様化されるべき3つの全ての標的化CDRを網羅するメガプライマーを作製し、突然変異誘発反応で用いた。これは、1つの反応で達成されるべき全てのコンビナトリアル配列多様化を可能にし、短いオリゴヌクレオチドを用いる段階的反応より効率的である。例えば、我々は、重複PCRにより、及び/又はGeneWriter(商標)を用いる化学合成により、VLライブラリ断片を作製し、それをハイブリダイゼーション突然変異誘発のメガプライマーとして用いた。メガプライマーをPCRにより作製し、マイナス鎖をビオチン化プライマーを用いて増幅する(図9)。ビオチン化鎖はストレプトアビジン磁気ビーズ(Dynal)上に捕捉され、ビオチン化されていない鎖は0.15MのNaOH中における変性により精製された。溶出された鎖を、次いで、ssDNA(REStr420jw)上におけるメガプライマー媒介DNA複製で用いた。
【0076】
VH及びVLにおける多様性を有するライブラリはまた、代替法及び源により作製することができる。例えば、VH及びVL遺伝子を免疫された動物組織から増幅し、定常領域とともにインフレームでクローニングして、Fabライブラリを作製した(17−winter)。あるいは、免疫された動物からのCDR領域、例えば、H−CDR3をPCRで増幅し、デノボH−CDR1、H−CDR2及びVLライブラリバックグラウンドにおいて免疫化H−CDR3のハイブリッドを形成するデノボライブラリにおける対応領域にクローニングできる。このようなライブラリのファージパニングは、高親和性結合抗体を単離できる。
【0077】
実施例7インライン親和性成熟
ファージパニングプロセスの一部としてデノボライブラリから選択された結合材の親和性成熟を設計し、そのプロセスを「インライン成熟」と名付けた。プロセスは、詳細な特徴付けの有り無しで、結合抗体のプールを用いて開始し、続いて結合剤のLcにおけるCDR配列を多様化し、これはライブラリスカフォールドとして用いられた4つの生殖細胞系Lcの1つに由来していた。LcCDR多様性は、更なるファージパニングを介して選択できる、追加の結合活性を作製する。プロセスを以下に記載し、また以下に詳述する(図10)。まず、リード抗体のVH領域をパニングを通して単離し、FabデノボライブラリをVL−パリンドロームループベクターにサブクローニングした。これらのベクターのssDNAを、実施例6に記載のVLライブラリメガプライマー又は好適な方法を用いて、ハイブリダイゼーション突然変異誘発のために作製した。例えば、組み合わせられたパリンドローム配列削除、及びメガプライマーKunkel突然変異誘発法を有効に用いて、インライン親和性成熟のためのVL CDR多様化2次ライブラリを作製することができる。親DNAをXbaI消化、続いて非許容状態の宿主株に形質転換することにより除く。この方法は、設計された配列多様性を網羅するライブラリの高度に効率的な作製を可能にする。次いで、ファージライブラリを、突然変異ライブラリにヘルパーファージを感染させることにより作製した。次に、ライブラリを高度な選択厳密性でパニングの更なるラウンドに供して、1次ライブラリから最初に選択された結合剤よりも改善された結合親和性を有する結合剤を増やす。これらは、抗原濃度を低下させ、洗浄時間及び頻度を増加させることを含む。あるいは、結合競合物又は他の結合ストレスコンポーネント、例えば、結合温度の変更及び剤の添加は、所望の特性を有する結合剤を選択するための結合及び/又は洗浄緩衝液に含まれてよい。ファージ結合及びその後の大腸菌感染に好適な他の生化学的及び生物物理学的条件もまた、パニングに含まれてもよい。パニング後、DNAをファージから回収する。Fabは発現し、結合、並びに他の生化学的、生物物理学的、及び生物学的特徴付けに供される。ファージから回収したDNAはまた、IgGベクターに直接クローニングすることもできる。この場合、Fabの代わりにmAbが発現し、特徴付けされる。例えば、重複PCR法によりVLライブラリ断片を作製し、それをハイブリダイゼーション突然変異誘発のメガプライマーとして用いた。メガプライマーは、PCRにより作製され、マイナス鎖はビオチン化プライマーを用いて増幅される(実施例6を参照)。ビオチン化鎖はストレプトアビジン磁気ビーズ(Dynal)上に捕捉され、ビオチン化されていない鎖は0.15MのNaOH中における変性により精製された。溶出された鎖を、次いで、ssDNAにおけるメガプライマー媒介DNA複製で用いた。Fabデノボライブラリのパニングを通して単離されたリード抗体のVH領域を、VL−ループベクターにサブクローニングした。これらの選択されたVH/VL−ループベクターを、VLライブラリメガプライマーとともにハイブリダイゼーション突然変異誘発のためのssDNAにした。XbaI含有パリンドロームの喪失により述べたように、親DNAの削除は、ランダムに選ばれたライブラリクローンのサンプルの配列により100%決定された。これらのCDRのライブラリの変異型への突然変異誘発は75%であった。この方法によりライブラリの4×108変異型の高度に効率的な作製が可能になった。我々は、Fabデノボライブラリにおいて用いられる他の3つのVL−k鎖におけるライブラリの作製と同様の結果を見出した。ライブラリを2×108に形質転換し、ファージを1011cfu/mLにした。BSAをデノボpIXライブラリでパニングし、ヒットを単離した(表3)。2つのLCライブラリを、上記方法を用いてBSAに対する1次ヒット上に構築した。ライブラリは、A27LC多様性を有するF4HC、及びB3LC多様性を有するD6、F6、C2、C6HCのプールからなっていた。
【0078】
【表1】
【0079】
【表2】
【0080】
3ラウンドのパニングを、非ストリンジェントな又はストリンジェントな洗浄条件のいずれかを用いて、それぞれ10nM、1nM、及び1nMの抗原で実施した。抗原陽性クローンの配列を決定し、選択クローンを精製して親和性データを得た。より低ストリンジェントな条件でパニングしたB3ライブラリでは、増やされたクローンの大部分が野性型であった。それらの野性型ではないクローンのうち、豊富さを示したものはなく、Biacoreデータはそれらが親和性を改善しなかったことを確認した。よりストリンジェントなパニング条件をこの同じライブラリに適用したとき、LCCDR1における突然変異を有するクローンの豊富さが見られ、その後Biacore分析でおよそ3倍の親和性の改善が示された(表4)。このライブラリから単離された全てのクローンはD6重鎖を含んでおり、これは他の重鎖に比べてBSAに対する親和性が最も高かった。A27ライブラリでは、パニング法ではクローンの濃縮も野性型の回復も見られなかった。単一クローンの濃縮は観察されなかったが、アミノ酸又は特定のCDR位置におけるアミノ酸特徴の濃縮は見られた。選択クローンは、親に比べて最大10倍の親和性改善を示した(表5及び図示せず)。
【0081】
【表3】
【0082】
【表4】
【0083】
実施例8.平行ライブラリパニング
我々のライブラリは、それぞれのサブライブラリがスカフォールドHC又はLCに基づいて個々に用いられ得るような方法で設計及び構築されるため、我々は、平行パニング戦略を設計して、個々並びにサブプールライブラリセットをパニングして、多様な結合抗体を同定する機会を最大化する。
【0084】
BSA及びリゾチームを、初期ライブラリ検証のモデル抗原として選択した。我々は、Kingfisher( )を用いる平行パニングとは別にLCフレームワークを保つことにより4つのサブセットに1〜69及び3〜23HCフレームワークサブライブラリをプールした。比較として、我々はまた、全てのサブライブラリ1〜69及び3〜23HC、並びに関連LCを1つのプールに完全にプールし、それを用いてBSAに対してパニングした。ビオチン化抗原でコーティングされた常磁性ビーズ(Dyal)を、室温で1時間ファージとともにインキュベートした。結合及び洗浄を96ウェルブロック中で、ハイスループットフォーマットで実施した。いくつかの洗浄工程後、結合したファージを溶出し、次の選択ラウンドのためにMC1061F’細胞を感染させることにより増幅した(詳細は実施例2)。選択ラウンド後、DNAを単離し、pIXを制限酵素消化により除去し、ライゲーションさせ、MC1061F’に形質転換して、可溶性Fabを発現させる。単離した単一クローンをFab発現及び抗原結合についてELISAにより試験した。抗Fd抗体でコーティングされたELISAプレート上でFab含有細胞抽出物を、続いてHRP結合抗カッパ抗体をインキュベートすることにより、Fab発現を検出した。抗原特異性を、Fab含有細胞可溶化物をその後添加したストレプトアビジンでコーティングされたプレート上でビオチン化抗原を捕捉することにより試験した。競合ELISAでは、過剰量の非ビオチン化抗原が含まれ、Fab結合に競合した。結合したFabをHRP結合抗カッパ抗体を用いて検出した。
【0085】
平行パニングの結果は、90のスクリーニングされたクローンのうち30がBSAに結合したことを示し、これは、この実験のヒット率が33%であることを示す。ヒット率は、完全にプールされたライブラリを用いてパニングを完了したときの7%のヒット率より非常に高い(表6)。リゾチーム用の2つのパニング戦略の直接比較は存在しないが、完全にプールされたライブラリパニングのみが実施された場合、ヒット率は4%である(表6)。BSAについて6つの独自の抗体及びリゾチームについて3つを同定した配列分析を、図11に示す。
【0086】
独自のBSA及びリゾチーム結合剤を精製して、結合親和性(KD)を決定した。2段階Fab精製手順(REStm334bw.doc)を用いてFabを精製した。簡潔に述べると、大腸菌溶菌液を最初の1段階IMAC手順に供した。透析後、IMACカラム溶出液を2番目の精製工程、QFFセファロース樹脂を用いるアニオン交換に供し、これは、Fabの純度を〜80%〜90%改善した。図12は、精製されたBSA及びリゾチームFabを示す。
【0087】
BIAcore分析を用いて、6つのBSA及び3つのリゾチーム結合剤の親和性を決定した。結果は、それぞれ、BSA結合剤について0.1〜28nM、リゾチーム結合剤について6〜260nMに及ぶFabの結合親和性を示した。BSAへのFab群の非特異的結合は存在せず、リゾチーム、又は参照表面(空の、活性化され、ブロックされたCM5マトリックス)は検出されなかった。表7及び8は、BSA及びリゾチーム結合剤の結合反応速度を示す。
【0088】
実施例9.親和性ランキング
結合解離定数KoffによりFabを順位付けするために、粗大腸菌上清をOctetシステムを用いて測定した(例えば、forteo.comのような公的ウェブサイトに記載されているように)。SAセンサプローブを、試験ビオチン化抗原でコーティングした。一晩大腸菌誘導培養物を、2xBBS/リゾチーム緩衝液で溶解し、96アッセイウェルに入れた。較正したセンサチップを、大腸菌溶菌液を収容しているアッセイウェルに浸漬し、結合会合を測定した。センサチップを次いで移動させ、緩衝液のみの中に入れて、結合解離を測定した。結合反応速度を、製造者のソフトウェアを用いて分析した。参照のために、表6は、同じ抗原に対して、Octet及びBIAcoreに由来する順位付け結果と、3つの異なるFabサンプルを比較する。結合反応速度はまた、BIND機器(例えば、srubio systemsによる)を用いて評価し、Fab発現細胞可溶化物をまず濾過して溶菌していない細胞、大きな細胞残屑、脂質、及び剪断された核酸を除去した。ストレプトアビジンでコーティングされたセンサプレートを較正し、ビオチン化抗原とともにインキュベートした。ウェル30秒間隔で2分間読み取り、100μL/ウェルの1xPBSで3回洗浄した。次いで、フィルタ処理したFab溶解液をアッセイウェルに添加し、ストレプトアビジンセンサプレート表面上に提示された抗原に結合することを可能にした。ウェルを空にし、200μLの1xPBSを添加した。ビオチン化抗原に対する結合シグナルを、30秒間隔で2〜5分間測定した。センサ表面に結合した複数の既知の量の抗原についての解離データを適合させることによりKoff値を得た。会合速度を得ることはできなかったが、解離速度(Fab濃度には依存しない)は、Biacore結果に相当した。
【0089】
上記代替アッセイフォーマットは、Fabタンパク質上に含まれるタグに適切な抗−F(ab’)2、抗−Fd、又は抗−HIS又は抗FLAGを提示する。抗体はセンサプレートのアミン反応性表面に結合させることができる。濾過した大腸菌溶菌液又はFabタンパク質を含有するペリプラズム画分を、センサプレート上に結合された抗−F(ab’)2により捕捉した。抗原とともにインキュベートした後、結合反応速度を上記のように測定することができる。
【0090】
粗大腸菌上清を用いて結合解離定数(Koff)を測定するための2つの方法を開発した。1つは、粗大腸菌培地上清を用いるELISAに基づく方法であり、他方は同時に複数のサンプルの結合反応速度を測定することができるベンチトップ機器であるOctet system(Fortebio website)である。親和性ランキングELISAではマイクロエクスプレッション(500μL)により作製されたFabの量を定量化した。Fab発現ELISAは、細胞可溶化物画分及び使用済み培地画分におけるFabタンパク質の量を示す。両方の画分は同程度の量のFabタンパク質を有するため、細胞可溶化物は作製するのがより面倒であり、使用済み培地画分をランキングELISAにおける使用のために選択した。3つの抗原を用いてELISAランキング法を確立した。それらは、マウスST2L、ヒトレジスチン、及びマウスMCP−5である。配列が独特であるFabを用いて、その反応性抗原に結合する能力を試験した。それぞれのサンプルについて発現したFabを含有する使用済み培地の複数の希釈液をそれぞれのELISAに適用した。Fabのそれぞれの希釈液を2つに分割し、一方のアリコートは抗原に結合するため、他方のアリコートはそれぞれの希釈したサンプルに含有されるFabタンパク質の相対量を測定するためである。抗原結合及び発現ELISAの結果を計算して、それぞれの希釈点の特異的活性比を確立した。対応するクローンに対する特異的活性比をプロットすることは、全てのFabサンプルの全体的視野を提供する。この視野から、希釈が曲線の直線点上に位置し、ランキングのための大部分のFab候補を取り込むという測定をすることができる。53のmST2Lサンプルを、1:2及び1:4希釈で試験した。
【0091】
1:4希釈を用いて、これらのFabの順位を決定した、なぜならそれは希釈曲線の線状セクター中にあるように思われるためである。8つのmMCP−5 Fabの順位付けは、より広い希釈範囲、1:2〜1:10を用いて行われた。ここで用いられた希釈範囲は、結合曲線の線状範囲の外側であった。80のレジスチンFabを1:2、1:4、及び1:8で希釈して、3種の希釈が線状範囲をよりよく確立するかどうかを測定した。これらのFabの部分集合のみが、この希釈範囲における線状活性:濃度挙動を有していた。レジスチンFabを再び発現させ、使用済み上清を1:5、1:10、及び1:20で希釈した。1:5及び1:10希釈は、線状滴定曲線内にサンプルを有さなかったが、1:20は線状範囲内であった。特異的活性比を全てのサンプルについて計算し、順位を確立した(表9A、B&C)。Biacoreによる親和性測定値は、それらの結合反応速度に関してmST2L Fabの一部とほとんど違いを示さなかったが、ランキングELISAデータは少し違いを示した(表9A)。Fab28の順位付けにおけるELISAとBiacore結果との間の不一致は、抗原配向に用いられたフォーマットによるものである可能性がある。ELISAは溶液中に抗原を有し、一方Biacoreフォーマットは表面上に提示された抗原を有していた。Biacore結果におけるFab40は、全体で最低のKDを与えるfast on−rate(ka)を示す。これは、不正確なタンパク質濃度によるものである可能性がある。mMCP−5Fabは、ELISAとBiacore結果との間の順位付けにおいて良好な相関を示す。両方のアッセイにおいてFab S10、S14、及びS30は最も低い順位の結合剤であり、Fab P9、P12、P20、及びS1は最も高い順位の結合剤であった(表9B)。FabP1はELISAランキングアッセイにおいて良好な結合剤であったにもかかわらず、それはまた、1:10希釈で低下したシグナルを示し、Biacoreでは並の結合剤であった。
【0092】
レジスチンFabは、ELISAとBiacoreランキングの間に相関を示す(表9C)。ELISA及びBiacoreランキングにおいて、クローン551−22が最良であり、クローン169−7が最悪であった。ELISA及びBiacoreランキングの両方において、更に323−7及び323−32は、クローン551−22に次いで良い順位である。
【0093】
【表5】
【0094】
【表6】
【0095】
【表7】
【0096】
ELISAランキングデータは、いくつかの例外を除いてBiacore相互作用分析から推定される解離値に最もよく相関していた。最適化されたELISAに基づくランキング法は、多数のFab候補の適切なスクリーニング及び管理を支持する。Octet機器を用いて、動力学結合活性によりFab候補を順位付けするための2つのフォーマットを開発した。溶液フォーマットは、チップセンサ表面に適用されたFab及び溶液中に存在するAgを用いて実施される。ある期間中センサ表面に添加されたバイオマスの測定を通してAgがFabに結合するときの会合速度を測定する。AgがFabに結合した後、センサが緩衝液(すなわち1xPBS)中に置かれたとき解離速度を計算し、経時的なセンサ上のバイオマスの低下をもたらす。固定化された動力学結合アッセイでは、抗原をチップセンサ表面に適用し、Fabは溶液中に存在する。会合及び解離速度を、経時的なチップ上のFab増加及び、続くバイオマスの低下により決定する。
【0097】
アミン反応性センサチップ用の基本的なプロトコルは、まず初期ベースラインの確立から始まり、続いて第1の分子(すなわち2次rgt、Fab、又はAg)がチップセンサに適用される活性化工程を行う。クエンチ工程を、第1の分子がチップセンサに結合した後に実施され、別の分子との結合相互作用が生じることが可能になる前に別のベースラインが確立される。ロード(すなわち2次試薬でコーティングされたチップセンサにFab又はAgを添加)又は会合工程(すなわちFab又はAgでコーティングされたセンサ)のいずれかを次に実施する。これがロード工程である場合、別のベースラインが会合工程が行われる前に確立される。いったん会合工程が完了すると、解離工程が実施され、実験が完了する。溶液動力学結合アッセイは、高結合SAコーティングされたセンサチップを用いる。サンプル96ウェルプレート(カタログ番号675076、Greiner Bio−One)を以下のように調製する:カラム1に100μLの1xPBSをチップセンサのベースラインまで充填し、カラム2に、1xPBS中10μg/mLの濃度でBt−抗ヒトF(Ab)’2(カタログ番号109−066−097、Jackson Immunological)を充填して、SAでコーティングされたセンサチップ表面にロードし、カラム3に第2のベースラインとして100μlの1xPBSを充填し、カラム4に第2のロード工程として1xPBS中10μg/mlのFabサンプルを100μl充填し、カラム5に第3のベースラインとして100μg/mlの1xPBSを充填し、カラム6に会合工程として1xPBS中10μg/mlのAgを充填し、カラム6のウェルは解離工程として1xPBSを収容する。動力学プロトコルをそれぞれの工程につき設定し、30℃で実行し、1000rpmで振盪する。第1のベースラインを100秒間実施する。第1のロード工程(抗ヒトF(Ab)’2)を600秒間実行する。第2のベースラインを200秒間実行する。第2のロード工程(Fab候補)を600又は900秒間実行する。第3のベースラインを300秒以内実行する。会合工程(Ag)を600秒間実行し、解離工程を最大900秒間実行する。固定化動力学結合アッセイは、アミン反応性センサチップを用いる。チップセンサボックスを、まず10分間室温で適切なMES pH溶液(Amine Coupling Kit,Fortebio)中でインキュベートする。サンプル96ウェルプレート(カタログ番号675076,Greiner Bio−One)を以下のように調製する:カラム1に、チップセンサのベースラインまで適切なMESpH溶液を充填し、カラム2に、ベースラインのMES pH溶液に1:50比のEDC/NHS(アミンカップリングキット、Fortebio)を充填して、センサ表面を活性化し、カラム3にMES pH溶液中10μg/mLのAgを充填して、センサチップをロードし、カラム4にエタノールアミン溶液(アミンカップリングキット、Fortebio)を充填して、Agのロードされたセンサの非占有表面をクエンチし、カラム5に第2のベースラインとして100μLの1xPBSを充填し、カラム6に会合工程として1xPBS中1μg/mLのFabサンプルを充填し、カラム7ウェルに解離工程として1xPBSを充填する。動力学プロトコルをそれぞれの工程につき設定し、30℃で実行し、1000rpmで振盪する。第1のベースラインを100秒間実行する。活性化工程を最大900秒間実行する。Agロード工程を600秒間実行する。クエンチ工程を最大300秒間実行する。第2のベースラインを最大300秒間実行する。会合工程(Ag)を600秒間実行し、解離工程を最大900秒間実行する。溶液動力学アッセイを、3つの異なるサイズの抗原に対して実施した。この追加の変数(抗原のサイズ)を用いて、いかに異なるバイオマスの増加がFab候補のランキングの最終的な計算結果に影響を与えるかについての理解が得られた。我々は、より大きなサイズの抗原から、より良いシグナル(y軸)及びR2値が得られることを見出した(表13A〜C)。Fabのセンサへの直接的なアミンカップリングは、センサ表面から100kDaの分子、抗F(Ab)’2を除くことにより、溶液動力学アッセイにおける会合シグナルの増加を補助し、したがって、最後の分子の質量を付加する効果を見る能力を高める可能性がある。Octetは、その会合及び解離シグナル曲線に従ってFab間の差を示し、それらは計算された速度定数と相関する。mST2L Fab 16、26、及び28は、Fab 1及び38より良好な結合速度を有する(図13a)。レジスチンFab323−11及び551−7は最高の、Fab 169−1及び551−38は最低の結合剤である(図13b)。mMCP−5Fabについて、それは、Fab 14が最低であることを除いて、Ag結合に関する順位付けをこのフォーマットで区別することはできない(図13c)。既に論じたように、mMCP−5を測定するためのこのフォーマットは最良ではない可能性がある。mMCP−5のサイズはFabより著しく小さく(9kDa対50kDa)、加えて100kDaの2次試薬でコーティングされたチップは、シグナルの差を測定できることをより悪化させる。
【0098】
固定化アッセイフォーマットは、Fab結合動力学を評価するための別の手段を提供する。図13Cの溶液フォーマットアッセイに見られるように、AgがFabより小さいとき(例えばmMCP−5)、シグナルは低下する。センサ上で既に確立されている大きなマス上の小さなタンパク質を読み取ろうとすることは、小さな効果を有する。より小さな分子、mMCP−5がセンサ上にある場合、結合パートナーを回転させ、次いでより大きな分子、例えばFabの結合活性を読み取ることは、より大きなシグナルを生じさせるはずである。この効果は図14で観察され、ここで、mMCP−5はセンサ表面にアミンカップリングして、Fabが添加されたとき会合を測定する。このアッセイフォーマットにおけるシグナル増加は、溶液フォーマットで見られるものの少なくとも2倍大きい。加えて、会合R2値はここで0.85より大きく、一方それらは溶液フォーマットで平均0.57であった(表14)。
【0099】
Biacoreは現在、結合動力学を決定するための標準である。我々は、上述のFabについてOctetから得られたデータを、Biacoreのデータと比較した。BiacoreとOctetとの間に感受性の差が見られるように思われる(図15)。Biacoreの親和定数KDは、一般に、Octet親和定数より高い。Octet実験では、分析物を10μg/mLの濃度で、Biacoreでは1μg/mLで用いた。10回用いたところ、Octetの分析物は会合速度定数をより早くすることができる。また、解離速度定数は、アッセイにおける過剰分析物競合のために間接的に影響を受ける場合がある。この原因と併せて、親和定数はBiacoreに比べてOctetの方が悪いように見える。直接立証されてはいないが、この感受性問題は同様に順位付けに対して影響を与える可能性がある。順位付けに関しては、レジスチンFabはBiacoreとOctetとの間で若干相関を示す。レジスチンと他のAg実験との間の唯一の差が、FabとAgとの間の分子サイズの差であった。Fabは50kDaであり、レジスチンは66kDaであり、これはサイズをより近くし、100kDAのmST2Lと9kDaのmMCP−5は異なるサイズである。
【0100】
結合動力学はまた、同時に96サンプルを測定できるBIND(www.srubiosystems.com)(別のベンチトップ機器)を用いて測定した。Fab発現細胞可溶化物をまず濾過して可溶化していない細胞、大きな細胞残屑、及び剪断された核酸を取り除いた。ストレプトアビジンでコーティングされたセンサプレートを較正し、ビオチン化抗原とともにインキュベートした。ウェル30秒間隔で2分間読み取り、100μL/ウェルの1xPBSで3回洗浄した。次いで、フィルタ処理したFab溶解液をアッセイウェルに添加し、ストレプトアビジンセンサプレート表面上に提示された抗原に結合することを可能にした。ウェルを空にし、200μLの1xPBSを添加した。ビオチン化抗原に結合するためのシグナルを、30秒間隔で2〜5分間測定した。SRU−BINDソフトウェアは参照細胞(Fabを含まない細胞可溶化物を添加した)から得たバルクシグナルを引く。センサ表面に結合した複数の既知の量の抗原についての解離データを適合させることによりKoff値を得た。会合速度を得ることは非常に困難であったが、解離速度(Fab濃度には依存しない)は、Biacore結果に相当した。
【0101】
上記に対するいくつかの代替アッセイフォーマットが存在する。例えば、BIND機器では、Fabは、必要に応じてセンサプレート上に固定化された抗−F(ab’)2、抗−Fd、又は抗−HIS又は抗FLAGにより捕捉され得る。濾過した大腸菌溶菌液又はFabタンパク質を含有するペリプラズム画分を、センサプレート上に結合された抗−F(ab’)2により捕捉され得る。抗原とともにインキュベートした後、結合反応速度を上記のように測定することができる。また、精製されたFabをセンサプレートのアミン反応性表面に直接結合させて、抗原結合を測定するためのこのフォーマットで用いることができる。
【0102】
結合動力学はまた、同時に96サンプルを測定できるBIND(商標)(Srubiosystems website)(別のベンチトップ機器)を用いて測定した。Fab発現細胞可溶化物をまず濾過して可溶化していない細胞、大きな細胞残屑、及び剪断された核酸を取り除いた。ストレプトアビジンでコーティングされたセンサプレートを較正し、ビオチン化抗原とともにインキュベートした。ウェル30秒間隔で2分間読み取り、100μL/ウェルの1xPBSで3回洗浄した。次いで、フィルタ処理したFab溶解液をアッセイウェルに添加し、ストレプトアビジンセンサプレート表面上に提示された抗原に結合することを可能にした。ウェルを空にし、200μLの1xPBSを添加した。ビオチン化抗原に結合するためのシグナルを、30秒間隔で2〜5分間測定した。SRU−BIND(商標)ソフトウェアは参照細胞(Fabを含まない細胞可溶化物を添加した)から得たバルクシグナルを引く。センサ表面に結合した複数の既知の量の抗原についての解離データを適合させることによりKoff値を得た。会合速度を得ることは非常に困難であったが、解離速度(Fab濃度には依存しない)は、Biacore結果に相当した。
【0103】
上記に対するいくつかの代替アッセイフォーマットが存在する。例えば、BIND機器では、Fabは、必要に応じてセンサプレート上に固定化された抗−F(ab’)2、抗−Fd、又は抗−HIS又は抗FLAGにより捕捉され得る。濾過した大腸菌溶菌液又はFabタンパク質を含有するペリプラズム画分は、センサプレート上に結合された抗−F(ab’)2により捕捉され得る。抗原とともにインキュベートした後、結合反応速度を上記のように測定することができる。また、精製されたFabをセンサプレートのアミン反応性表面に直接結合させて、抗原結合を測定するためのこのフォーマットで用いることができる。
【0104】
利点
上記ヒトFabデノボライブラリは他のこのようなライブラリとは異なる、なぜなら提示がM13ファージのpIXタンパク質を介しているためである。代表的なヒト生殖細胞系及びライブラリスカフォールドとしてのカノニカル構造配列の使用、並びにH−CDR3におけるアミノ酸の天然分布を模倣するCDR配列の設計は、ヒト免疫レパートリーの網羅においてライブラリを包括的にする。このライブラリ設計は、単一又は共通生殖細胞系遺伝子又は成熟mAbスカフォールド上に構築される、文献に報告されている他のデノボライブラリと比べて、標的抗原に対する抗体発見の可能性が高い。ライブラリの平行パニングに関与する別個のサブライブラリ(それぞれ独自のスカフォールド(VH/VL対)又はH−CDR3長さから構成される)は、標的抗原に対する多様な配列の抗体の発見の可能性を最大化する。更に、これはそれぞれのVH/VL対の機能性、及びそのそれぞれの配列、及び抗体/抗原結合における構造の詳細な研究のための機序を提供する。統合インライン親和性成熟プロセスは、多様性を維持しながら高親和性抗体を単離するのに有効な方法である。
【0105】
参考文献:
1.Kretzschmar & von Ruden,Current Opin Biotechnol.13:598〜602,2002)
2.Crooks GE,Hon G,Chandonia JM,Brenner SE WebLogo:A sequence logo generator,Genome Research,14:1188〜1190,(2004)
3.Wang W,Saven JG.Designing gene libraries from protein profiles for combinatorial protein experiments.Nucleic Acids Res.Vol 30,No 21,e120.2002
4.Hennie R Hoogenboom.Nature Biotech.(2005)23:1105.Selecting and screening antibody libraries.
5.V.Lee,Wei−Ching Liang,Mark S.Dennis,Charles Eigenbrot,Sachdev S.Sidhu and Germaine Fuh JMB(2004)340:1073〜1093.High−affinity Human Antibodies from Phage−displayed Synthetic Fab Libraries with a Single Framework Scaffold.
6.Michela Silacci,Simon Brack,Giulia Schirru,Jessica Marlind,Anna Ettorre,Adrian Merlo,Francesca Viti and Dario Neri.Proteomics(2005)5:2340〜2350.Design,construction,and characterization of a large synthetic human antibody phage display library
7.Eskil Soderlind and Carl A.K.Borrebaek.Nature Biotech.(2000).18:852.Recombining germline−derived CDR sequences for creating diverse single−framework antibody library.
8.Achim Knappik,Liming Ge,Annemarie Honegger,Peter Pack,Melanie Fischer,Gunter Wellnhofer,Adolf Hoess,Joachim Wolle, Andreas Pluckthunand Bernhard Virnekas JMB(2000)296:57〜86.Fully Synthetic Human Combinatorial Antibody Libraries(HuCAL) Based on Modular Consensus Frameworks and CDRs Randomized with Trinucleotides.
9.Griffiths AD,Williams SC,Hartley O,Tomlinson IM,Waterhouse P,Crosby WL,Kontermann RE,Jones PT,Low NM,Allison TJ,Prospero TD,Hoogenboom HR,Missim A,Cox JPL,Harrison JL,Zaccolo,Gherardi E and Winter G.EMBO J 1994:13(14):3245〜3260.Isolation of high affinity human antibodies directly from large synthetic repertoires
10.Hans J.de Haard,Nicole van Neer,Anneke Reurs,Simon E.Hufton,Rob C.Roovers,
Paula Henderikx,Adriaan P.de Bruyne,Jan−Willem Arends,and Hennie R.Hoogenboom JBC(1999).274:18218〜18230.A Large Non−immunized Human Fab Fragment Phage Library That Permits Rapid Isolation and Kinetic Analysis of High Affinity Antibodies*
11.Rene Michael Hoet and Robert ladner,et al.,Nature Biotech.(2005)23:344.Generation of high affinity human antibodies by combining donor−derived and syntheric complementarity−determining region diversity.
12.Crooks GE,Hon G,Chandonia JM,Brenner SE WebLogo:A sequence logo generator,Genome Research,14:1188〜1190,(2004)
13.Wang W,Saven JG.Designing gene libraries from protein profiles for combinatorial protein experiments.Nucleic Acids Res.Vol 30,No 21,e120.2002
14.Vbase:The database of human antibody genes.Public website:vbase.mrc−cpe.cam.ac.uk
15.Juan Carlos Almagro.J.Mol.Recognit.(2004)17:132〜143.Identification of differences in the specificity−determining residues of antibodies that recognize antigens of different size:implications for the rational design of antibody repertoires.
16.Ian M.Tomlinson,Jonathan RL.Cox,Ermanno Gherardi,Arthur M.Lesk and Cyrus Chothia.The EMBO Journal(1995)14:4628〜4638.The structural repertoire of the human VK domain.
17.米国特許出願公開第0165946 A1号、参照することにより全文が本明細書に組み込まれる
18.欧州特許出願第90201671.6号、参照することにより全文が本明細書に組み込まれる
19.米国特許第5,942,609号、参照することにより全文が本明細書に組み込まれる
20.Dillon et al.,BioTechniques 9(3):298〜300(1990);
21.Prodomou et al.,Protein Engineering 5(8):827〜829(1992);
22.Chen et al.,JACS 116:8799〜8800(1994);
23.Hayashi et al.,BioTechniques 17(2):310〜315(1994).
24.Stemmer,Nature 370:370:389〜391(1994)
25.Stemmer,Proc.Natl.Acad.Sci.USA 91:10747〜10751(1994).
26.Sidhu et al,Methods in Enzymology.(2000)328:333−363.Phage display for selection of novel binding peptides。
27.Tim Clackson and Henry B Lowma.2004.Phage display−a practical approach.Oxford University Press
28.Figini et al,J.Mol.Bio.(1994)239:68〜78.In vitro assembly of repertoires of antibody chains on the surface of phage by renaturation
29.William F.Dall’Acqua,Melissa M.Damschroder,Jingli Zhang,Robert M.Woods,Lusiana Widjaja,Julie Yu,Herren Wu.Methods(2005)36:43〜60.Antibody humanization by framework shuffling.
【0106】
表
【表8】
【0107】
【表9】
【0108】
【表10】
【0109】
【表11】
太字:同じファミリーの生殖細胞系配列に現れる
イタリック:ヌクレオチド
【0110】
【表12】
【0111】
【表13】
【0112】
【表14】
【0113】
【表15】
【0114】
NB:結合せず
【0115】
【表16】
【0116】
【表17】
【0117】
【表18】
【0118】
【表19】
【0119】
表13A〜Cは、Octetからの溶液フォーマット動力学データを示す。A)100kDのST2Lに結合するFab、Fab 26、Fab 28、Fab 1、Fab 38を、会合及び解離速度定数によりOctetを用いて順位付けした。B)66kDaのレジスチンに結合するFab 169−3、Fab 169−7、Fab 323−11、Fab 323−32、Fab 551−7、Fab551−22及びFab 551−38を、会合及び解離速度定数によりOctetを用いて順位付けした。C)9kDaのMCP−5に結合するFab P1、Fab P9、Fab P12、Fab P20、Fab S1、Fab S14及びFab S30を会合及び解離速度定数によりOctetを用いて順位付けした。
【0120】
【表20】
【0121】
表14:Octetからの固定化フォーマット動力学データ9kDaのMCP−5に結合するFab P1、Fab P9、Fab P12、Fab P20、Fab S1、Fab S14及びFab S30を、会合及び解離速度定数によりOctetを用いて順位付けした。
【0122】
【表21】
【0123】
【表22】
【0124】
【表23】
【0125】
表15A〜C:BiacoreとOctetとの間の反応速度チャートの比較。
上から下のチャートへ、mST2L、レジスチン、及びmMCP−5 Fab群である。
【特許請求の範囲】
【請求項1】
選択された生物学的に活性のあるリガンドに結合する、ファージディスプレイpIX又はpVII融合抗体fab断片アミノ酸配列を発現させるための操作された組み換え核酸ファージベクターであって、
a. b.に機能的に連結した、組み換えファージリーダーコード核酸配列と、
b. c.に機能的に連結した、組み換えタグ、プロモータ、又は選択コード核酸配列と、
c. d.に機能的に連結した、前記生物学的に活性のあるリガンドに選択的に結合する可変重鎖生殖細胞系コード化核酸配列と、
d. e.に機能的に連結した、前記生物学的に活性のあるリガンドに選択的に結合する定常重鎖生殖細胞系コード化核酸配列の一部と、
e. f.に機能的に連結した、ペプチドリンカーコード化核酸配列と、
f. 組み換えpIX又はpVIIコード化核酸配列と、
g. h.に機能的に連結した、前記生物学的に活性のあるリガンドに選択的に結合する可変軽鎖生殖細胞系コード化核酸配列と、
h. 前記生物学的に活性のあるリガンドに選択的に結合する定常軽鎖生殖系コード化核酸配列と、を含むベクター。
【請求項2】
前記ファージリーダーコード配列が、pelB配列である、請求項1に記載の操作された核酸ファージベクター。
【請求項3】
組み換えタグ又は選択配列が、FLAGタグ配列である、請求項1に記載の操作された核酸ファージベクター。
【請求項4】
前記プロモータが、誘導可能なプロモータである、請求項1に記載の操作された核酸ファージベクター。
【請求項5】
前記誘導可能なプロモータが、lacプロモータである、請求項1に記載の操作された核酸ファージベクター。
【請求項6】
前記生物学的に活性のあるリガンドが、少なくとも1つの生物学的インビボ活性を媒介する、請求項9に記載の操作された核酸ファージベクター。
【請求項7】
請求項1に記載の操作された核酸ファージベクターを含む細菌宿主細胞。
【請求項8】
請求項11に記載の細菌宿主細胞により発現される生物学的に活性のある融合タンパク質。
【請求項9】
請求項13に記載の前記融合タンパク質に由来する、生物学的に活性のあるfab抗体断片変異型。
【請求項10】
請求項1に記載の複数の操作された核酸ファージベクターを含む細菌宿主細胞のファージライブラリ。
【請求項11】
前記生物学的に活性のあるfab抗体断片変異型が、複数のfab抗体断片として発現する、請求項14に記載のファージライブラリ。
【請求項12】
(a)請求項11に記載のファージライブラリからfab抗体断片を発現させる工程と、(b)前記所望の生物活性を有する前記fab抗体を発現する細菌細胞を選択する工程と、を含む、所望の生物活性を有するfab抗体断片のためにファージfab抗体断片ライブラリをスクリーニングする方法。
【請求項13】
請求項12に記載の方法から得られる、fab抗体断片コード化核酸配列。
【請求項14】
請求項13に記載のfab抗体断片を含むIgG又はIgAクラスの免疫グロブリン。
【請求項15】
請求項13に記載のfab抗体断片を含む製薬学的組成物。
【請求項16】
前記複数が、少なくとも103、104、105、106、107、108、109、1010、1011、1012、又は1013の前記変異型から選択される少なくとも1つである、請求項11に記載のファージライブラリ。
【請求項17】
前記変異型が、Kunkel突然変異誘発法を用いて作製される、請求項11に記載のファージライブラリ。
【請求項18】
前記変異型が、前記fab抗体断片に含まれる複数の多様な重鎖相補性決定領域(CDR)3変異型を含む、請求項11に記載のファージライブラリ。
【請求項19】
前記多様なCDR3変異型が、約109〜1018の配列変異型を含む、請求項18に記載のファージライブラリ。
【請求項20】
前記変異型が、前記fab抗体断片に含まれる複数の多様な重鎖相補性決定領域(CDR)1及び2変異型を含む、請求項11に記載のファージライブラリ。
【請求項21】
前記多様なCDR3変異型が、約102〜105の配列変異型を含む、請求項20に記載のファージライブラリ。
【請求項22】
前記ファージライブラリが、前記ファージライブラリの先行バージョンに比べて、結合親和性が改善された、親和性成熟fab抗体断片を更に含む、請求項11に記載のファージライブラリ。
【請求項23】
前記fab抗体断片が、親和性又は生物活性を高めるためにパニングされる、請求項22に記載のファージライブラリ。
【請求項24】
前記パニングが、ライブラリ作製プロセスの一部として、又は前記ファージライブラリの部分集合を用いて並行して実施される、請求項23に記載のファージライブラリ。
【請求項25】
請求項11に記載の前記ファージライブラリの一部を含む、部分集合ファージライブラリであって、親和性又は生物活性に関する選択された特徴を有するfab抗体断片を含む部分集合ファージライブラリ。
【請求項1】
選択された生物学的に活性のあるリガンドに結合する、ファージディスプレイpIX又はpVII融合抗体fab断片アミノ酸配列を発現させるための操作された組み換え核酸ファージベクターであって、
a. b.に機能的に連結した、組み換えファージリーダーコード核酸配列と、
b. c.に機能的に連結した、組み換えタグ、プロモータ、又は選択コード核酸配列と、
c. d.に機能的に連結した、前記生物学的に活性のあるリガンドに選択的に結合する可変重鎖生殖細胞系コード化核酸配列と、
d. e.に機能的に連結した、前記生物学的に活性のあるリガンドに選択的に結合する定常重鎖生殖細胞系コード化核酸配列の一部と、
e. f.に機能的に連結した、ペプチドリンカーコード化核酸配列と、
f. 組み換えpIX又はpVIIコード化核酸配列と、
g. h.に機能的に連結した、前記生物学的に活性のあるリガンドに選択的に結合する可変軽鎖生殖細胞系コード化核酸配列と、
h. 前記生物学的に活性のあるリガンドに選択的に結合する定常軽鎖生殖系コード化核酸配列と、を含むベクター。
【請求項2】
前記ファージリーダーコード配列が、pelB配列である、請求項1に記載の操作された核酸ファージベクター。
【請求項3】
組み換えタグ又は選択配列が、FLAGタグ配列である、請求項1に記載の操作された核酸ファージベクター。
【請求項4】
前記プロモータが、誘導可能なプロモータである、請求項1に記載の操作された核酸ファージベクター。
【請求項5】
前記誘導可能なプロモータが、lacプロモータである、請求項1に記載の操作された核酸ファージベクター。
【請求項6】
前記生物学的に活性のあるリガンドが、少なくとも1つの生物学的インビボ活性を媒介する、請求項9に記載の操作された核酸ファージベクター。
【請求項7】
請求項1に記載の操作された核酸ファージベクターを含む細菌宿主細胞。
【請求項8】
請求項11に記載の細菌宿主細胞により発現される生物学的に活性のある融合タンパク質。
【請求項9】
請求項13に記載の前記融合タンパク質に由来する、生物学的に活性のあるfab抗体断片変異型。
【請求項10】
請求項1に記載の複数の操作された核酸ファージベクターを含む細菌宿主細胞のファージライブラリ。
【請求項11】
前記生物学的に活性のあるfab抗体断片変異型が、複数のfab抗体断片として発現する、請求項14に記載のファージライブラリ。
【請求項12】
(a)請求項11に記載のファージライブラリからfab抗体断片を発現させる工程と、(b)前記所望の生物活性を有する前記fab抗体を発現する細菌細胞を選択する工程と、を含む、所望の生物活性を有するfab抗体断片のためにファージfab抗体断片ライブラリをスクリーニングする方法。
【請求項13】
請求項12に記載の方法から得られる、fab抗体断片コード化核酸配列。
【請求項14】
請求項13に記載のfab抗体断片を含むIgG又はIgAクラスの免疫グロブリン。
【請求項15】
請求項13に記載のfab抗体断片を含む製薬学的組成物。
【請求項16】
前記複数が、少なくとも103、104、105、106、107、108、109、1010、1011、1012、又は1013の前記変異型から選択される少なくとも1つである、請求項11に記載のファージライブラリ。
【請求項17】
前記変異型が、Kunkel突然変異誘発法を用いて作製される、請求項11に記載のファージライブラリ。
【請求項18】
前記変異型が、前記fab抗体断片に含まれる複数の多様な重鎖相補性決定領域(CDR)3変異型を含む、請求項11に記載のファージライブラリ。
【請求項19】
前記多様なCDR3変異型が、約109〜1018の配列変異型を含む、請求項18に記載のファージライブラリ。
【請求項20】
前記変異型が、前記fab抗体断片に含まれる複数の多様な重鎖相補性決定領域(CDR)1及び2変異型を含む、請求項11に記載のファージライブラリ。
【請求項21】
前記多様なCDR3変異型が、約102〜105の配列変異型を含む、請求項20に記載のファージライブラリ。
【請求項22】
前記ファージライブラリが、前記ファージライブラリの先行バージョンに比べて、結合親和性が改善された、親和性成熟fab抗体断片を更に含む、請求項11に記載のファージライブラリ。
【請求項23】
前記fab抗体断片が、親和性又は生物活性を高めるためにパニングされる、請求項22に記載のファージライブラリ。
【請求項24】
前記パニングが、ライブラリ作製プロセスの一部として、又は前記ファージライブラリの部分集合を用いて並行して実施される、請求項23に記載のファージライブラリ。
【請求項25】
請求項11に記載の前記ファージライブラリの一部を含む、部分集合ファージライブラリであって、親和性又は生物活性に関する選択された特徴を有するfab抗体断片を含む部分集合ファージライブラリ。
【図1】


【図2】
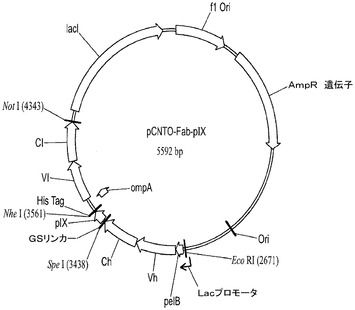
【図3A】

【図3B】
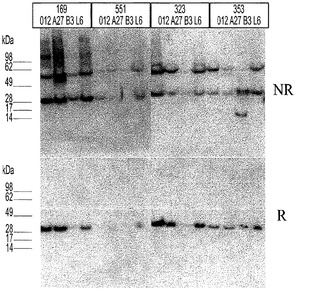
【図3C】
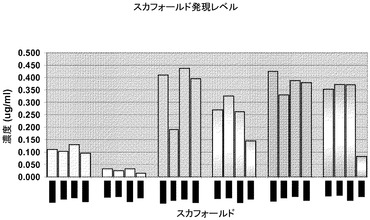
【図4】

【図5】
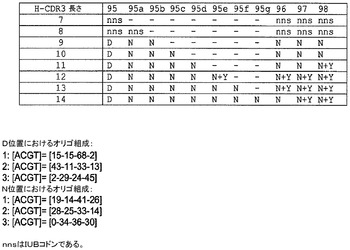
【図6A】
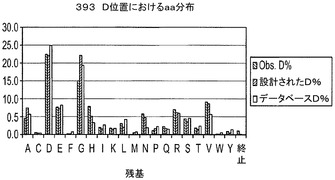
【図6B】
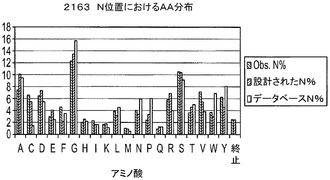
【図6C】
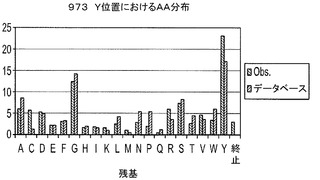
【図6D】
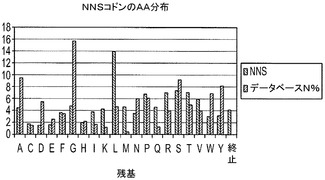
【図7】
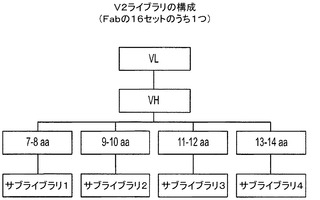
【図8】
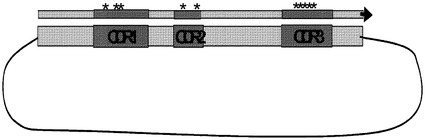
【図9】
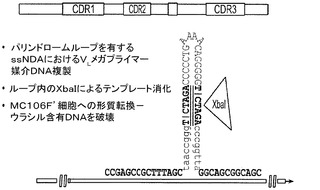
【図10】
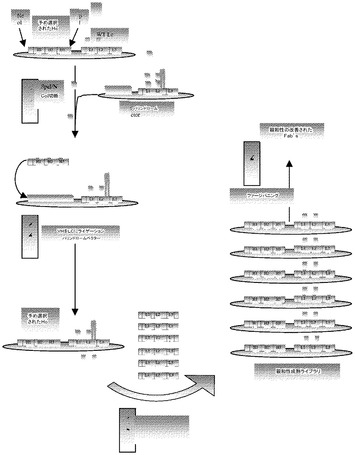
【図2】
【図3A】
【図3B】
【図3C】
【図4】
【図5】
【図6A】
【図6B】
【図6C】
【図6D】
【図7】
【図8】
【図9】
【図10】
【公表番号】特表2011−507519(P2011−507519A)
【公表日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願番号】特願2010−539575(P2010−539575)
【出願日】平成20年11月21日(2008.11.21)
【国際出願番号】PCT/US2008/084255
【国際公開番号】WO2009/085462
【国際公開日】平成21年7月9日(2009.7.9)
【出願人】(509087759)セントコア・オーソ・バイオテツク・インコーポレーテツド (77)
【Fターム(参考)】
【公表日】平成23年3月10日(2011.3.10)
【国際特許分類】
【出願日】平成20年11月21日(2008.11.21)
【国際出願番号】PCT/US2008/084255
【国際公開番号】WO2009/085462
【国際公開日】平成21年7月9日(2009.7.9)
【出願人】(509087759)セントコア・オーソ・バイオテツク・インコーポレーテツド (77)
【Fターム(参考)】
[ Back to top ]