
乳酸生産酵母および乳酸生産方法
【課題】酵母における乳酸生産に好ましい乳酸高発現系を構築する。
【解決手段】乳酸生産酵母であって、ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母が提供される。この有機酸生産酵母においては、ピルビン酸脱炭酸酵素遺伝子が破壊されていることが好ましい態様であり、さらに、アルコール脱水素酵素遺伝子が破壊されていることが好ましい態様である。
【解決手段】乳酸生産酵母であって、ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母が提供される。この有機酸生産酵母においては、ピルビン酸脱炭酸酵素遺伝子が破壊されていることが好ましい態様であり、さらに、アルコール脱水素酵素遺伝子が破壊されていることが好ましい態様である。
【発明の詳細な説明】
【技術分野】
【0001】
本発明は、乳酸生産酵母および乳酸生産方法に関する。
【背景技術】
【0002】
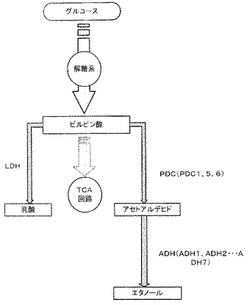
L−乳酸などの乳酸を酵母を利用して生産することが試みられている。例えば、図1に示すように、乳酸を生産するために、乳酸脱水素酵素(LDH)遺伝子を酵母サッカロマイセス・セレビシエ属に導入し、ピルビン酸から乳酸を生産させる試みがなされている。これらの試みの多くは、酵母本来のエタノール発酵を抑制して酵母における乳酸生産を高めるために、アルコール発酵経路の強力でかつ構成的酵素であるピルビン酸脱炭酸酵素(PDC)の発現を抑制するものである(特許文献1、2)。また、酵母染色体中のピルビン酸脱炭酸酵素1(PDC1)遺伝子プロモーターの下流に目的とする有用遺伝子(この場合は、L−乳酸脱水素酵素遺伝子)を結合させると同時に、酵母染色体中のPDC1遺伝子を破壊することによって、PDC1プロモーター機能を利用して乳酸脱水素酵素遺伝子を発現させつつ、当該プロモーターによって本来発現されるべきPDC1の発現を排除するシステムも開発されている(特許文献3)。このようなPDC遺伝子の破壊は乳酸生産量を明らかに増大させた。
【0003】
また、酵母のアルコール発酵経路においてピルビン酸脱炭酸酵素の後段の酵素であるアルコール脱水素酵素1(ADH1)遺伝子を破壊してこの酵素の発現を抑制する試みもなされている(非特許文献1)。この試みによれば、染色体上のADH1遺伝子を破壊しLDHをコードするDNAを発現するプラスミドが導入された形質転換酵母は、ADH1遺伝子を維持する野生型酵母に同様のプラスミドを導入した形質転換酵母よりもエタノール生産量は減ったものの乳酸生産量が顕著に低下していた。また、PDC活性が低下した酵母に同様のプラスミドを導入した形質転換酵母の乳酸生産量よりも低い生産量であった。すなわち、ADH1遺伝子の破壊によるADH1の発現抑制は必ずしも乳酸生産に有効ではなかった。
【0004】
【特許文献1】特表2001−516584号
【特許文献2】特表2003−500062号
【特許文献3】特開2003−164295号
【非特許文献1】J Ind Microbiol Biotechnol(2003)30:22−27(英国)
【発明の開示】
【発明が解決しようとする課題】
【0005】
このようにアルコール発酵経路のピルビン酸脱炭酸酵素の後段の酵素であるアルコール脱水素酵素活性の抑制はピルビン酸脱炭酸酵素活性の抑制による乳酸生産効果を上回るものではなく、逆に好ましくない結果をもたらしていた。以上のことから、その活性を抑制することが酵母において効率的な乳酸生産をさせるのに有効であろうと推測されるアルコール発酵系酵素やTCA回路系酵素は多数存在するものの、現在までのところ、ピルビン酸脱炭酸酵素以外に活性抑制が有効な酵素や組み合わせは見出されていない。
【0006】
そこで、本発明では、酵母における乳酸生産に好ましい乳酸高発現系を構築することをその目的とする。具体的には、乳酸の高生産が可能な乳酸生産酵母を提供することを一つの目的とし、さらに、効率的な乳酸の生産方法を提供することを他の一つの目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、PDC破壊酵母と同等あるいはそれを上回る乳酸生産を可能とする高発現系を確立すべく、酵母における乳酸生産のための活性抑制が有効な酵素を探索した。ADHは、PDCよりも後段の酵素であるため、既にPDCが破壊されてPDCの発現が抑制されている場合、ADHを破壊しても乳酸の生産性を向上させることは期待できないと考えられた。また、上記背景技術からもADHの破壊はアセトアルデヒドの蓄積により細胞の増殖に悪影響がある可能性が高かった。しかしながら、本発明者らがPDC遺伝子を破壊するとともにADH遺伝子をも破壊した上で、外来LDHをコードするDNAを発現させたところ、上記した推測に反し高率で乳酸生産することを見出した。この知見に基づき、本発明者らは発明を完成した。すなわち、本発明によれば、この知見に基づき以下の手段が提供される。
【0008】
本発明によれば、乳酸生産酵母であって、ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母が提供される。この有機酸生産酵母においては、ピルビン酸脱炭酸酵素遺伝子が破壊されていることが好ましい態様であり、さらに、アルコール脱水素酵素遺伝子が破壊されていることが好ましい態様である。さらにまた、前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、ピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられていることが好ましい態様であり、より好ましくは、酵母染色体上のピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられている。また、上記した有機酸生産酵母のいずれかにおいて、前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、アルコール脱水素酵素遺伝子のプロモーターの制御下で発現可能に備えられていることが好ましく、より好ましくは、酵母染色体上のアルコール脱水素酵素遺伝子の制御下で発現可能に備えられている。
【0009】
また、本発明によれば、乳酸生産酵母であって、染色体上のピルビン酸脱炭酸酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下で乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備え、染色体上のアルコール脱水素酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下に乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母が提供される。
【0010】
これらのいずれかの形態の乳酸生産酵母において、前記ピルビン酸脱炭酸酵素遺伝子は、ピルビン酸脱炭酸酵素1遺伝子であることが好ましく、前記アルコール脱水素酵素遺伝子はアルコール脱水素酵素1遺伝子であることが好ましい。
【0011】
また、上記いずれかの酵母において、前記乳酸脱水素酵素活性を有するタンパク質はウシ由来であり、前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAを複数有することが好ましく、前記酵母はサッカロマイセス属に属することが好ましい。さらに、サッカロマイセス・セレビジエであることが好ましい。
【0012】
また、本発明によれば、上記いずれかの乳酸生産酵母を培養する工程を備える、乳酸生産方法が提供される。
【発明を実施するための最良の形態】
【0013】
本発明の乳酸生産酵母は、ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備えている。この乳酸生産酵母によれば、アルコール発酵経路の酵素であるピルビン酸脱炭酸酵素とアルコール脱水素酵素とがそれぞれ低下されている一方、同じくピルビン酸を基質とする乳酸脱水素酵素がピルビン酸脱炭酸酵素プロモーターの制御下に発現可能に備えられているため、ピルビン酸は乳酸脱水素酵素によって代謝されやすくなっており、乳酸の生産が促進されている。アルコール発酵経路の第1の酵素であるピルビン酸脱炭酸酵素活性が低下されていることにより、酵母のアルコール発酵は効果的に抑制されることは従来からも知られている。こうしたピルビン酸脱炭酸酵素活性が低下した系においてさらにアルコール脱水素酵素活性が抑制されることは、基質であるピルビン酸の代謝経路をアルコール発酵経路から乳酸発酵経路へと大きく変化させるものとは考えにくいが、本発明によれば、意外にもアルコール生産量を低下させるとともに乳酸生産量を増大させることができる。すなわち、アルコール脱水素酵素活性を低下させてもアセトアルデヒドの生成による細胞の増殖阻害等を抑制して細胞増殖を確保し乳酸生産量を増大させることができる。
【0014】
以下、本発明の乳酸生産酵母について説明するとともに、この酵母を用いた乳酸生産方法について説明する。
【0015】
(乳酸生産酵母)
本発明の乳酸生産酵母は、外来の乳酸脱水素酵素遺伝子を有する形質転換酵母である。本乳酸生産酵母を得るための酵母としては、アルコール発酵を行う酵母であって、サッカロマイセス・セレビシエ、シゾサッカロマイセス・ポンベ(Scizosaccharomyces pombe)などのサッカロマイセス属酵母、ピキア・パストリス(Pichia pastoris)、などの酵母を挙げることができる。より好ましくは、サッカロマイセス・セレビシエなどのサッカロマイセス属を始めとする酵母である。好ましい酵母の一例としては、サッカロマイセス・セレビシエIFO2260株や同YPH株を例示できる。
【0016】
また、本乳酸生産酵母および乳酸生産方法において乳酸とは、L−乳酸、D−乳酸、及びDL−乳酸があるが、これらのいずれであってもよい。
【0017】
(ピルビン酸脱炭酸酵素及びアルコール脱水素酵素)
本乳酸生産酵母においては、ピルビン酸脱炭酸酵素(PDC)活性およびアルコール脱水素酵素(ADH)活性が低下されている。これらの酵素は、酵母のアルコール発酵経路の酵素であり、アルコール発酵を行う酵母がその染色体上に本来的に有している。ピルビン酸脱炭酸酵素は、いわゆるオートレギュレーション機構を有する酵素であり、ピルビン酸脱炭酸酵素1,5,6がある。これらのうちいずれか1種あるいは2種以上が破壊されていればよいが、好ましくは最も活性が高いあるいは大量に発現されているピルビン酸脱炭酸酵素1(PDC1)の活性が低下されていることが好ましい。また、アルコール脱水素酵素においても各種の亜種(ADH1,2,3,4,5,6,7)があるが、アルコール脱水素酵素1(ADH1)が大量に発現されている。したがってアルコール脱水素酵素1の活性が低下されていることが好ましい。
【0018】
ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性が低下されているとは、人工的な操作によりあるいはスクリーニングによって、野生型よりも低い活性の該酵素が生産されているかあるいは該酵素の生産量が野生型よりも少ないものであることが好ましい。このような酵素活性の低下のための人工的操作としては、この酵素遺伝子を破壊(ノックアウト)することが好ましい。染色体上の特定遺伝子を破壊する手法は、当業者において周知である。
【0019】
(乳酸脱水素酵素)
本乳酸生産酵母は、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを保持している。LDHをコードするDNAは、酵母においては外来性である。乳酸脱水素酵素(LDH)としては、生物の種類に応じてあるいは生体内においても各種同族体が存在する。本発明において使用する乳酸脱水素酵素としては、天然由来のLDHの他、化学合成的あるいは遺伝子工学的に人工的に合成されたLDHも包含している。また、LDHとしては、L−LDHであってもD−LDHであってもよい。LDHとしては、好ましくは、ラクトバシルス・ヘルベティカス、ラクトバシルス・カゼイ、クルイベロマイセス・サーモトレランス、トルラスポラ・デルブルッキ、シゾサッカロミセス・ポンビ、リゾプス・オリゼ、B.メガテリウムなどの原核生物もしくはカビなどの真核微生物由来であり、より好ましくは、植物、動物、昆虫などの高等真核生物由来であり、さらに好ましくは、ウシを始めとする哺乳類を含む高等真核生物由来である。例えば、ウシ由来のLDH(L−LDH)である。例えば、ウシ由来のLDHとして配列番号:2に示すアミノ酸配列からなるタンパク質を挙げることができる。また、かかるLDHをコードするDNAとしては、配列番号:1に記載される塩基配列からなるDNAを挙げることができる。さらに、本発明におけるLDHは、これらのLDHのホモログも包含している。LDHホモログは、天然由来のLDHのアミノ酸配列において、1もしくは数個のアミノ酸の置換、欠失、挿入および/または付加されたアミノ酸でありかつLDH活性を有しているタンパク質、および、天然由来のLDHとアミノ配列の相同性が少なくとも70%、好ましくは80%以上を有しかつLDH活性を有しているタンパク質を含んでいる。したがってLDHをコードするDNAとして、これらのいずれかのLDHをコードするDNAを用いることができる。本乳酸生産酵母は、LDHをコードするDNAを少なくとも一つ、好ましくは二以上保持している。より好ましくは三以上保持している。さらに好ましくは四つ以上保持している。
【0020】
LDHをコードするDNAは、酵母染色体外で維持されるプラスミドやYACなどにおいて保持されていてもよいが、好ましくは酵母染色体に組み込まれて保持される。
【0021】
(PDCプロモーター)
LDHをコードするDNAは、強力なプロモーター活性を有するプロモーターの制御下に発現可能に備えられていることが好ましい。例えば、ピルビン酸脱炭酸酵素遺伝子プロモーター(PDCプロモーター)の制御下に発現可能に備えられていることが好ましい。なかでも、十分に強力なプロモーターであるPDC1プロモーターを用いることが好ましい。より好ましくは、サッカロマイセス属(好ましくはセレビシエ)のPDC1プロモーターである。
【0022】
LDHをコードするDNAは、好ましくは、酵母染色体上のPDCプロモーターの制御下に発現可能である。既に述べたように、本乳酸生産酵母においては、PDC遺伝子が破壊されていることが好ましい。このため、LDHをコードするDNAは、染色体においてPDCプロモーターによって制御可能であってかつPDC遺伝子を破壊するように酵母染色体に組み込まれていることが好ましい。こうすることで、PDC活性を効果的に低下させると同時に、本来PDC遺伝子発現に作用していた強力なPDC遺伝子のプロモーター活性をLDHをコードするDNA発現にスイッチさせることができる。破壊されそしてそのプロモーターがLDHをコードするDNAの発現に利用されるPDC遺伝子は、好ましくはPDC1遺伝子である。最も強力なPDC1プロモーターによって制御されるPDC1遺伝子が破壊されてLDHをコードするDNAが代わりに発現されることで効果的にPDC活性低下とLDH活性発現とを実現できる。
【0023】
例えば、PDC1プロモーターとしては、配列番号:3に記載の塩基配列からなるDNAの他、同等の機能を有する限り、該DNAとストリンジェントな条件でハイブリダイズするDNA、該塩基配列において1あるいは2以上の塩基が置換、欠失、付加、及び/又は挿入された配列からなるDNA、あるいは、該DNAとの相同性が70%以上、80%以上、好ましくは90%以上、より好ましくは95%以上であるDNAを用いることができる。すなわち、酵母染色体上のPDC1プロモーターが、同一ではないが同等の機能を有するこれらのプロモーター活性を有するDNAによって相同組換え等を介して置換されていてもよい。
【0024】
ストリンジェントな条件としては、50%ホルムアミド存在下でハイブリダイゼーション温度が37℃であるハイブリダイゼーション条件あるいはこれと同様のストリンジェンシーのハイブリダイゼーション条件を意味している。よりストリンジェンシーの高い条件によれば、より相同性の高いDNAを単離できる。かかるハイブリダイゼーション条件としては、例えば、50%ホルムアミド存在下でハイブリダイゼーション温度が約42℃、さらにストリンジェンシーの高い条件としては、50%ホルムアミド存在下で約65℃のハイブリダイゼーション条件を挙げることができる。
【0025】
また、本明細書において、DNAなどの核酸の相同性については、なお、DNAの塩基配列のホモロジーは、遺伝子解析プログラムBLASTなどによって決定することができる。なお、DNAの塩基配列のホモロジーは、遺伝子解析プログラムBLAST(http://blast.genome.ad.jp),FASTA(http://fasta.genome.ad.jp/SIT/FASTA.html)などによって決定することができる。
【0026】
PDC1遺伝子は、本発明者らが既に開示しているように、オートレギュレーション機構が存在する遺伝子である(特開2003−164295号)。オートレギュレーション機構とは、同じ機能を有する遺伝子が同一生物において複数存在し、通常、そのうちの少なくとも一つは発現しているが、残りは抑制されており、通常発現している遺伝子が破壊などにより機能しなくなった場合にのみ、残りの遺伝子が発現されてその機能を継続する機構を意味している。かかる機構が存在するため、例えば、酵母のPDC1遺伝子が破壊されたとしても、PDC5遺伝子が発現し、酵母の生理的機能が維持されることになる。このようなオートレギュレーション機構が存在する遺伝子を破壊することで、生物自体の生存、増殖機能を維持することができて、結果として、外来DNAを保持する形質転換体の増殖を維持しながら目的産物を効果的に生産させることができる。
【0027】
(ADHプロモーター)
また、LDHをコードするDNAは、ADH遺伝子のプロモーター(ADHプロモーター)の制御下に発現可能に備えることもできる。ADHは既に述べたようにアルコール発酵経路においてPDCのすぐ後続の酵素である。ADHプロモーターとしては、十分に強力なプロモーターであるADH1プロモーターを用いることが好ましい。
【0028】
LDHをコードするDNAは、好ましくは、酵母染色体上のADHプロモーターの制御下に発現可能である。また、既に述べたように、本乳酸生産酵母においては、ADH遺伝子が破壊されていることが好ましい。このため、LDHをコードするDNAは、ADHプロモーターによって制御可能であってかつADH遺伝子を破壊するように酵母染色体に組み込まれていることが好ましい。こうすることで、アルコール発酵経路における代謝活性を効果的に低下させると同時に、本来ADH遺伝子発現に作用していたADHプロモーター活性をLDHをコードするDNA発現にスイッチさせることができる。また、ADH遺伝子は、好ましくはADH1遺伝子である。強力なあるいは構成的なADH1プロモーターによって制御されるADH1遺伝子が破壊されてLDHをコードするDNAが代わりに発現されることで効果的にADH活性低下とLDH活性発現とを実現できる。
【0029】
なお、ADH1プロモーターは、酵母の染色体上のADH1プロモーターと同等の機能を有する限り、該プロモーターのDNAとストリンジェントな条件でハイブリダイズするDNA、該塩基配列において1あるいは2以上の塩基が置換、欠失、付加、及び/又は挿入された配列からなるDNA、あるいは、ADH1プロモーターとの相同性が80%、好ましくは90%、より好ましくは95%以上であるDNAを用いることができる。すなわち、酵母染色体上のPDC1プロモーターが、同一ではないが同等の機能を有するこれらのプロモーターによって相同組換え等により置換されていてもよい。
【0030】
(他のプロモーター)
LDHをコードするDNAは、酵母サッカロマイセス・セレビジエの高浸透圧応答7遺伝子(HOR7遺伝子)、グリセルアルデヒド3リン酸脱水素酵素2遺伝子(TDH2遺伝子)、ヘキソース輸送タンパク質7遺伝子(HXT7遺伝子)、熱ショックタンパク質30遺伝子(HSP30遺伝子)、チオレドキシンペルオキシダーゼ1遺伝子(AHP1遺伝子)、膜タンパク質1関連遺伝子(MRH1遺伝子)、グリセルアルデヒド三リン酸脱水素酵素3(TDH3)遺伝子、ガラストース一リン酸キナーゼ1(GAL1)遺伝子、ホスホグリセレートキナーゼ(PGK)遺伝子及び転写エンハンサー因子−1(TEF)遺伝子のプロモーターの制御下に発現可能に導入することもできる。これらの他のプロモーターとしては、酵母、あるいはサッカロマイセス属酵母のこれらの遺伝子のプロモーター活性を有するDNAのほかかかるプロモーター領域に一定以上(70%以上、好ましくは80%以上、より好ましくは90%、さらに好ましくは95%以上)の相同性を有し、各プロモーター活性を有するDNAであってもよい。
【0031】
本発明者らによれば、これらの他の酵母由来プロモーターのいずれもが乳酸存在時において高い遺伝子発現を示すプロモーターであるという知見を有している。したがって、かかるプロモーターの制御下にLDHをコードするDNAを導入することで、乳酸の生産量の増大によって乳酸の生産が抑制されない、あるいは乳酸の生産量の増大によってさらに乳酸の生産が促進される実用的な乳酸生産酵母などの形質転換体を得ることがおおいに期待される。
【0032】
LDHをコードするDNAは、PDCプロモーターやADHプロモーターにおけるのと同様、酵母染色体上にあるこれらの他のプロモーターの制御下にある内在性遺伝子を破壊するとともに該プロモーターの制御下に発現可能に組み込むこともできる。また、これらの他のプロモーターの制御下に発現可能に連結したLDHをコードするDNAを、PDC遺伝子および/またはADH遺伝子を破壊するように酵母染色体に組み込みこともできる。
【0033】
なお、明らかなように、PDCプロモーター、ADHプロモーター及び他のプロモーターのDNAは、ゲノムDNAであってもよく、また、化学的に合成されたDNAであってもよい。
【0034】
本乳酸生産酵母においては、上記したPDC活性及びADH活性を低下させる態様、各種のLDHをコードするDNA発現態様を各種組み合わせて採ることができる。好ましい乳酸生産酵母は、染色体上のPDC(好ましくはPDC1)遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下にLDHをコードするDNAを発現可能に備えるとともに、染色体上のADH(好ましくはADH1)遺伝子が破壊されている。また、この態様において、破壊された該ADH(好ましくはADH1)遺伝子のプロモーターの制御下にLDHをコードするDNAを発現可能に備えることができる。こうした乳酸生産酵母においては、好ましくは2以上、より好ましくは3以上、さらに好ましくは4以上のLDHをコードするDNAを染色体上に備えている。また、さらに、酵母染色体上の上記他のプロモーターの制御下にLDHをコードするDNAを発現可能に備えることができる。
【0035】
本乳酸生産酵母を取得するには、宿主酵母に対して、PDC遺伝子及びADH遺伝子の破壊を行うとともに、LDHをコードするDNAを発現可能に導入する。酵母に対してこのような遺伝子修飾を行うには、組換え用DNA構築物を利用する。組換え用DNA構築物は、特に限定しないで、線状等のDNA断片、プラスミド(DNA)、ウイルス(DNA)、レトロトランスポゾン(DNA)、人工染色体(YAC)を、外来遺伝子の導入形態(染色体外あるいは染色体内)等に応じて選択してベクターとしての形態をとることができる。
【0036】
PDC遺伝子及びADH遺伝子の破壊のためのDNA構築物は、これらの遺伝子部位に導入して遺伝子を破壊するために相同組換え用の配列を備えている。相同組換え用DNA配列は、破壊しようとするPDC遺伝子及びADH遺伝子であるターゲット部位あるいはその近傍のDNA配列と相同なDNA配列である。相同組換え用DNA配列は、一つのターゲット遺伝子あるいはその近傍の少なくとも1箇所に相同である1の配列を有しており、好ましくは、ターゲット遺伝子あるいはその近傍の少なくとも2箇所にそれぞれ相同な配列を備えている。例えば、2個の相同組換え用DNA配列を、染色体上のターゲット遺伝子の上流側と下流側のDNAとのそれぞれに相同なDNA配列とし、これらの相同組換え用DNA配列の間に遺伝子を破壊するためのDNAを備えるDNA構築物を酵母染色体に相同組換えにより導入することでターゲット部位の遺伝子を破壊することができる。
【0037】
このような染色体上への組み込みを実現するための相同組換え用DNAの選択は、当業者において周知であり、当業者であれば必要に応じて適切な相同組換え用DNAを選択して相同組換え用DNA構築物を構成することができる。例えば、PDCプロモーターの少なくとも一部と、PDC遺伝子あるいはターミネーターの少なくとも一部とにそれぞれ相同なDNA配列を相同組換え用DNA配列として用いることができる。同様に、ADHプロモーターの少なくとも一部とADH遺伝子あるいはターミネーターの少なくとも一部とにそれぞれ相同なDNA配列を相同組換え用DNA配列として用いることができる。
【0038】
PDC遺伝子を破壊するDNA構築物には、PDC遺伝子をターゲットとするための相同組換え用DNAとともにPDC遺伝子を置換可能な形態でLDHをコードするDNAを有することができる。このようなDNA構築物によれば、PDC遺伝子の破壊とともにPDCプロモーターの制御下にLDHをコードするDNAを発現可能に導入できる。
【0039】
また、PDC遺伝子を破壊するとともに、破壊した該PDC遺伝子のプロモーターの制御下にLDHをコードするDNAを導入することもできるが、相同組換えにより染色体上のPDCプロモーターに替えて同等のプロモーターを導入することもできる。例えば、既に述べたPDC1プロモーターのホモログ等である。
【0040】
なお、ADH遺伝子を破壊するとともに、破壊した該ADH遺伝子のプロモーターの制御下にLDHをコードするDNAを導入するためのDNA構築物も、上記したPDC遺伝子についてのDNA構築物と同様に構築することができる。
【0041】
なお、DNA構築物には、CYC1ターミネーターやTDH3ターミネーターなどのターミネーター他、必要に応じてエンハンサーなどのシスエレメント、スプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(SD配列)を連結することができる。選択マーカーとしては、特に限定しないで、薬剤抵抗性遺伝子、栄養要求性遺伝子などを始めとする公知の各種選択マーカー遺伝子を利用できる。例えば、アンピシリン耐性遺伝子、カナマイシン耐性G418遺伝子、ハイグロマイシン耐性遺伝子、ブレオマイシン耐性遺伝子、ネオマイシン耐性遺伝子、ジヒドロ葉酸還元酵素遺伝子、クロラムフェニコール耐性遺伝子、シクロヘキサミド耐性遺伝子等を使用することができる。
【0042】
DNA構築物を、酵母に、トランスフォーメーション法や、トランスフェクション法、接合法、プロトプラスト融合、エレクトロポレーション法、リポフェクション法、酢酸リチウム法、パーティクルガン法、リン酸カルシウム沈殿法、アグロバクテリウム法、PEG法、直接マイクロインジェクション法等の各種の適切な手段のいずれかにより、これを導入することができる。DNA構築物の導入後、酵母は、選択培地で培養される。
【0043】
なお、DNA構築物が酵母に導入されたか否か、あるいは染色体上の所望の部位に本DNA構築物が導入されたか否かの確認は、PCR法やサザンハイブリダイゼーション法により行うことができる。例えば、形質転換体からDNAを調製し、導入部位特異的プライマーによりPCRを行い、PCR産物について、電気泳動において予期されるバンドを検出することによって確認できる。あるいは蛍光色素などで標識したプライマーでPCRを行うことでも確認できる。これらの方法は、当業者において周知である。
【0044】
(乳酸の生産方法)
本乳酸生産酵母を適当な炭素源の存在下で培養することにより、培養物中にLDHをコードするDNAの発現産物である乳酸を生成させることができる。本乳酸生産方法によれば、培養系から乳酸を分離する工程を実施することにより、乳酸を得ることができる。なお、本発明において培養物とは、培養上清の他、培養細胞あるいは菌体、細胞若しくは菌体の破砕物を包含している。
【0045】
本発明の乳酸生産酵母の培養にあたっては、酵母の種類に応じて培養条件を選択することができる。このような培養条件は、当業者においては周知である。乳酸の生産にあたっては、必要に応じて産物である乳酸等の中和を行うかあるいは、連続的に乳酸を除去する等の処理を行うこともできる。酵母を培養する培地としては、微生物が資化可能な炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行うことができる培地であれば、天然培地、合成培地のいずれも使用することができる。炭素源としては、グルコース、フルクトース、スクロース、デンプン、セルロース等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノール等のアルコールを用いることができる。窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の無機酸もしくは有機酸のアンモニウム塩またはその他の含窒素化合物の他、ペプトン、肉エキス、コーンスティープリカー等を用いることができる。無機物としては、リン酸第一カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウムなどを用いることができる。
【0046】
培養は、通常、静置培養、振とう培養または通気攪拌培養等の、嫌気条件下または微好気条件下、30℃〜35℃で6〜72時間行う。培養期間中、pHは2.0〜7.0に保持することが好ましい。また、pHの調整は、無機あるいは有機酸、アルカリ溶液等を用いて行うことができる。培養中は、必要に応じてアンピシリン、テトラサイクリンなどの抗生物質を培地に添加することができる。
【0047】
培養終了後、培養物から乳酸を分離するには、通常の乳酸精製手段を使用することができる。例えば、酵母内に生産された場合は、常法により菌体を超音波破壊処理、摩砕処理、加圧破砕などで細胞を破壊して、乳酸を細胞と分離することができる。この場合、必要に応じてプロテアーゼを添加する。また、菌体外に乳酸が生産された場合には、この培養液等を、ろ過、遠心分離などにより固形分を除去する。
【0048】
これらの粗抽出画分に対しては、従来公知の各種精製分離法等を利用して、乳酸を精製することができる。また、必要に応じて、当該粗抽出画分及びその精製物に対してエステル化等を行うことにより、各種の乳酸誘導体を得ることができる。
【実施例1】
【0049】
以下に、本発明の具体例を記載するが、本発明を以下の具体例に限定する趣旨ではなく、本発明の要旨を逸脱しない範囲で種々の態様で実施できる。
【0050】
以下に述べる遺伝子組換え操作はMolecular Cloning: A Laboratory Manual (T. Maniatis, et al., Cold Spring Harbor Laboratory) に従い行った。PCRによる遺伝子増幅は、特に述べない限り、KOD plus DNA polymerase(TOYOBO)を用い、添付のプロトコルに従って行った。PCR増幅反応は94℃で2分間の熱処理を行った後、94℃で15秒と53℃で30秒と68℃でX分(X:増幅遺伝子の大きさが1kbにつき1分とした)との3つの温度変化を1サイクルとし、これを25サイクル繰り返し、最後に4℃とした。PCR増幅装置はGene Amp PCR system 9700(PE Applied Biosystems)を使用した。ライゲーション反応にはLigaFast Rapid DNA Ligation System(Promega)を用いた。大腸菌の形質転換は、JM109株のコンピテント細胞(TOYOBO)を用い、添付のプロトコルに従って行った。大腸菌の形質転換体の選抜はアンピシリン100μg/mlを含むLBプレートを用いて行った。大腸菌からのプラスミドの抽出にはQIA Prep Spin Miniprepkit (QIAGEN)を用いた。DNA断片末端の平滑化にはT4 DNA polymerase(TaKaRa)を用いた。酵母のゲノムDNAの調製はFast DNA Kit(Bio 101)を用い、添付のプロトコルに従って行った。酵母の形質転換はFrozen-EZ Yeast Transformation II(ZYMO RESEARCH)を用い、添付のプロトコルに従って行った。
【0051】
(PDC1破壊株の作製)
PDC1破壊株は、図2に示すpBTrp-PDC1-LDHKCBベクターを用いて作製した。すなわち、pBTrp-PDC1-LDHKCBベクターを制限酵素SacIおよびApaIで消化した断片をもちいて、宿主である酵母IFO2260株のトリプトファン要求性変異株の形質転換を行った。形質転換処理を行った菌体をトリプトファン選択培地(SD-Trp)に塗沫し、生育してきたコロニーを新たなトリプトファン選択培地に画線培養してコロニーを純化した。純化された選抜株について、1 mlのYPA培地(2% 酢酸カリウム、2% ペプトン、1% 酵母エキス)に接種し、一晩振とう培養を行った。増殖した菌体を滅菌水で2回洗浄した後、胞子誘導プレート(1% 酢酸カリウム、0.1% ペプトン、0.05% グルコース、1.5% アガー)に塗布し、室温で4日間培養して胞子を誘導した。プレート上の菌体をかきとり、0.25 u/mlのZymolyase(ZYMO RESERCH)溶液中で15分間処理して子嚢壁を消化した。次にマイクロマニピュレータを用いて胞子を解剖し、YPDプレート上で分離した。30℃で4日間培養して生育したクローンをトリプトファン選択培地にレプリカし、同時にゲノムDNAを調製した。このゲノムDNAを鋳型として、ゲノム上のPCRプライマーであるPDC1-1000Fとベクター上のPCRプライマーであるLDHKCB-Rを用いてPCRを行ったところ、トリプトファン選択培地で生育したコロニーのみで2kbのバンド増幅された。さらにゲノム上のPCRプライマーであるPDC1+1300Rとベクター上のPCRプライマーであるTRP1-Fを用いてPCRを行ったところ、トリプトファン選択培地で生育したコロニーのみで1.5kbのバンド増幅された。このことから、トリプトファン選択培地で生育したコロニーはPDC1構造遺伝子部分が破壊され、PDC1プロモータの下流にLDHが挿入されていることが確認された。この操作で得られた株はPDC1構造遺伝子がホモ破壊されており、破壊されたPDC1構造遺伝子上流のPDC1プロモータによって発現するLDH遺伝子が2コピー導入されている。そこでこの株をPDC1破壊株(PDC1p-LDH2コピー体)と称した。用いたPCRプライマーの配列は以下の通りである。
【0052】
PDC1-1000F(配列番号6):5'- aaa atg aag gcc aaa tca agg cgg gaa ggg -3'
LDHKCB-R(配列番号7):5'- tta tta aaa ttg caa ttc ttt ttg aat acc -3'
TRP1-F(配列番号8):5'- ctc tgc aag ccg caa act ttc acc aat gga -3'
PDC1+1300R(配列番号9):5'- gat caa ttt ctt cag cag cga aag cag cac -3'
【0053】
なお、pBTrp-PDC1-LDHKCBベクターは、以下の方法で構築したものである。
1.ウシ由来のL-乳酸脱水素酵素を、酵母サッカロマイセス・セレビシエにおいて効率的に生産するために、ウシ由来のL-乳酸脱水素酵素のアミノ酸配列をコードするDNAに対して使用コドンを酵母の発現に最適化した遺伝子配列(配列番号:4)を設計し、長鎖DNAの合成方法として知られている藤本らの方法(藤本英也、合成遺伝子の作成法、植物細胞工学シリーズ7、植物のPCR実験プロトコール、1997、集潤社、p95-100)を用いて該DNAを全合成した(合成したDNAをLDHKCBと称した)。LDHKCBを制限酵素EcoRIで消化したものをLDHKCB断片と称した。
2. PDC1プロモータ断片(PDC1P, 0.7kb)は、サッカロマイセス・セレビシエYPH499株のゲノムDNAを鋳型として使用したPCR反応により増幅した。使用したPCRプライマーの配列は以下の通りであった。
・PDC1P-LDH-U(配列番号10)ata tat gga tcc gcg ttt att tac cta tct c(BamHIサイトを付加)
・PDC1P-LDH-D(配列番号11)ata tat gaa ttc ttt gat tga ttt gac tgt g(EcoRIサイトを付加)
得られたPCR増幅断片を制限酵素BamHIおよびEcoRIにより消化したものをPDC1P断片と称した。
3. PDC1遺伝子下流領域断片(PDC1D, 0.5kb)は、サッカロマイセス・セレビシエYPH499株のゲノムDNAを鋳型として使用したPCR反応により増幅した。使用したPCRプライマーの配列は以下の通りであった。
・PDC1D-LDH-U(配列番号12)ata tat ctc gag gcc agc taa ctt ctt ggt cga c(XhoIサイトを付加)
・PDC1D-LDH-D(配列番号13)ata tat gaa ttc ttt gat tga ttt gac tgt g(ApaIサイトを付加)
得られたPCR増幅断片を制限酵素XhoIおよびApaIにより消化したものをPDC1D断片と称した。
4. トリプトファン要求性マーカー(TRP1, 1.3kb)は、pRS404ベクター(Stratagene)を制限酵素AatIIおよびSspIで消化して切り出し、末端を平滑化した。得られた断片をTRP1断片と称した。
5. TDH3ターミネーター(TDH3t, 0.15kb)はサッカロマイセス・セレビシエYPH499株のゲノムDNAを鋳型として使用したPCR反応により増幅した。使用したPCRプライマーの配列は以下の通りであった。
・TDH3t-F(配列番号14)tcg act tgg ttg aac acg ttg cca agg ctt a
・TDH3t-R(配列番号15)aaa gct ttc aat caa tga atc gaa aa
得られたPCR増幅断片をTDH3t断片と称した。
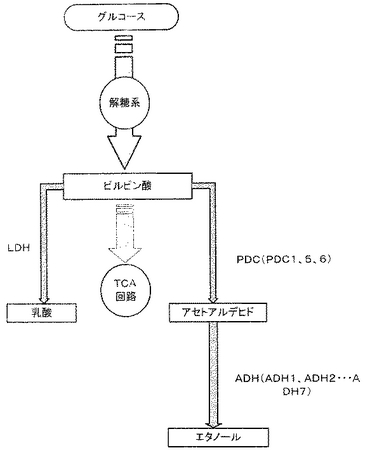
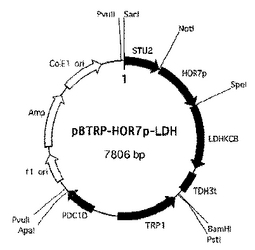
6. 上記操作で得られた各断片(PDC1P断片、LDHKCB断片、TDH3t断片、TRP1断片およびPDC1D断片)を、順次pBluescriptII SK+ベクター(TOYOBO)のマルチクローニングサイトに連結して、染色体導入型ベクターpBTrp-PDC1-LDHKCBを構築した(図2)。
【実施例2】
【0054】
(染色体導入型ベクターの構築)
他の遺伝子を破壊することなく、染色体上にLDH遺伝子を導入するための染色体導入型ベクターpBble-LDHKCB(図3)を以下の方法で構築した。
(フレオマイシン耐性マーカーカセットの構築)
抗生物質フレオマイシン耐性遺伝子を大腸菌XL1-Blue MRF’ Kan株(STRATAGENE)のゲノムDNAを鋳型にしてPCRにより増幅した。大腸菌のゲノムDNAは、LB培地にて一晩振盪培養を行った後、集菌、洗浄したものを98℃ 10分間の加熱処理したのち遠心分離した上清を、エタノール沈殿し、滅菌水50μlに溶解することにより調製した。使用したPCRプライマーの配列は以下の通りであった。
・Tn5ble-U(配列番号16)ata tat gaa ttc atg acc gac caa gcg acg c (EcoRIサイトを付加)
・Tn5ble-D(配列番号17)ata tat aag ctt tca tga gat gcc tgc aag c(HindIIIサイトを付加)
得られたPCR増幅断片(0.4kb)を制限酵素EcoRIおよびHindIIIで消化したものをTn5 ble断片と称した。
【0055】
Tn5 bleを酵母菌体内で発現できるようにするために、CYC1プロモータ配列およびCYC1ターミネーター配列を酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・CYCP-U(配列番号18)ata tat gga tcc gac agc atc gtc gaa tat g(BamHIサイトを付加)
・CYCP-D(配列番号19)ata tat gaa ttc tat taa ttt agt gtg tgt att tg(EcoRIサイトを付加)
・CYCT-U(配列番号20)ata tat aag ctt aca ggc ccc ttt tcc ttt g(HindIIIサイトを付加)
・CYCT-D(配列番号21)ata tat gtc gac gtt aca tgc gta cac gcg (SalIサイトを付加)
【0056】
PCRプライマーCYCP-UおよびCYCP-Dを用いて増幅されたCYC1プロモータ配列(0.5kb)を制限酵素BamHIおよびEcoRIで消化したものをCYC1P断片と称した。PCRプライマーCYCT-UおよびCYCT-Dをもちいて増幅されたCYC1ターミネーター配列(0.2kb)を制限酵素HindIIIおよびSalIで消化したものをCYC1T断片と称した。
【0057】
上記操作で得られたCYC1P断片、Tn5 ble断片およびCYC1T断片を、この順番で並ぶように順次pBluescriptII SK+ベクターのマルチクローニングサイトに連結してフレオマイシン耐性マーカーカセットを構築した。得られたベクターを制限酵素SpeIおよびApaIで消化することによりフレオマイシン耐性マーカーカセットを切り出し、末端を平滑化したものをフレオマイシン耐性マーカーカセット断片と称した。
【0058】
(染色体導入型ベクターpBble-LDHKCBの構築)
先に構築したpBTrp-PDC1-LDHKCBを制限酵素BamHIおよびPstIにより消化して切り出したものをLDHKCB発現カセット断片(PDC1P断片、LDHKCB断片およびTDH3t断片がこの順番で連結された断片)と称した。
【0059】
相同組み換え配列であるPDC5遺伝子下流領域配列およびSLX4遺伝子上流領域配列を、酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・PDC5D-U(配列番号22) cca tga tta gat ggg gtt tg
・PDC5D-D(配列番号23) ctg gaa gac agg aca gaa
・SLX4-F(配列番号24)aat tat gtc gac ggt taa aga tta gct tct aat (SalIサイトを付加)
・SLX4-R(配列番号25)ata taa ggt acc caa ctg aac tac tgg tta tta tta tg(KpnIサイトを付加)
PCRプライマーPDC5D-UおよびPDC5D-Dをもちいて増幅されたPDC5遺伝子下流領域配列(0.6kb)をPDC5D断片と称した。PCRプライマーSLX4-FおよびSLX4-Rをもちいて増幅されたSLX4遺伝子上流領域配列(0.7kb)を制限酵素SalIおよびKpnIで消化したものをSLX4断片と称した。
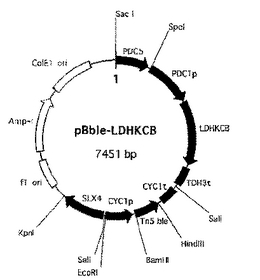
上記操作で得られた各断片(PDC5D断片、LDHKCB発現カセット断片、フレオマイシン耐性マーカーカセット断片およびSLX4断片)を、順次pBluescriptII SK+ベクターのマルチクローニングサイトに連結して、染色体導入型ベクターpBble-LDHKCB(図3)を構築した。
【0060】
(4コピー体の作製)
pBble-LDHKCBベクターを制限酵素SacIおよびKpnIで消化した断片をもちいて、実施例1で作製されたPDC1破壊株(PDC1p-LDH2コピー体)の形質転換を行った。形質転換処理を行った菌体をフレオマイシン選択培地(10μg/mlのフレオマイシンを含むYPD培地)に塗沫し、生育してきたコロニーを新たなフレオマイシン選択培地に画線培養してコロニーを純化した。純化された選抜株について、実施例1と同様な方法で胞子を誘導し、胞子解剖を行った。YPDプレート上で分離した胞子を30℃で4日間培養して、生育したクローンをフレオマイシン選択培地にレプリカし、同時にゲノムDNAを調製した。このゲノムDNAを鋳型として、ゲノム上のPCRプライマーであるPDC5-320Fとベクター上のPCRプライマーであるLDHKCB-R(配列番号7)を用いてPCRを行ったところ、トリプトファン選択培地で生育したコロニーのみで4kbのバンド増幅された。さらにゲノム上のPCRプライマーであるPDC5+2788Rとベクター上のPCRプライマーであるTCYC-F1を用いてPCRを行ったところ、フレオマイシン選択培地で生育したコロニーのみで1.9kbのバンド増幅された。このことから、フレオマイシン選択培地で生育したコロニーは酵母染色体上のPDC5遺伝子とSLX4遺伝子の間に、両遺伝子を破壊することなく挿入されていることが確認された。この操作で得られた株はPDC1構造遺伝子がホモ破壊されており、PDC1プロモータによって発現するLDH遺伝子が4コピー導入されている。そこでこの株をPDC1破壊株(PDC1p-LDH4コピー体)と称した。用いたPCRプライマーの配列は以下の通りであった。
・PDC5-320F(配列番号26)5'- atg gga aaa gcc tcc ata tcc aaa ggt c -3'
・PDC5+2788R(配列番号27)5'- ttc gta gtc tct tgc gtc ata g -3'
・TCYC-F1(配列番号28)5'- ttg tct aac tcc ttc ctt ttc g -3'
【実施例3】
【0061】
(ADH1遺伝子破壊用ベクターの構築)
本実施例では、酵母の染色体上に存在するアルコール脱水素酵素をコードするADH1遺伝子を破壊すると同時に、破壊された該ADH1プロモーターの制御下にウシ由来LDH遺伝子を挿入することのできるDNA構築物を構築した。操作過程を図4に示す。
【0062】
まず、既に取得したウシ由来のLDH遺伝子にTDH3ターミネーターを接続し、さらに酵母用マーカーセット(ハイグロマイシン耐性遺伝子カセットを含むベクター(pBHPH-LDHKCBpreと称する。)を作製した(図5)。作製方法を以下に示す。
【0063】
既知文献(K.R.Kaster, S.G.Burgett, R.N.Rao, T.D.Ingolia Analysis of a bacterial hygromycin B resistance gene by transcriptional and translational fusions and by DNA sequencing. Nucleic Acids Research. Vol.11 (19), p6895-6911 (1983)、および L.Gritz, J.Davies Plasmid-encoded hygromycin B resitance; the sequence of hygromycin B phospotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene. Vol.25, p179-188 (1983))に基づいてHPH遺伝子配列情報を取得して、以下のPCRプライマーを合成した。
・HPH-U(配列番号29)5’- atg aaa aag cct gaa ctc acc -3’
・HPH-D (配列番号30)5’- cta ttc ctt tgc cct cgg acg -3’
次に大腸菌K12株ゲノムDNAを鋳型としたPCR反応によりHPH遺伝子(1kb)を増幅したものをHPH断片と称した。
【0064】
HPH遺伝子を酵母菌体内で発現できるようにするためにTDH3プロモーターを酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・TDH3P-U(配列番号31)5’-ata tat gga tcc tag cgt tga atg tta gcg tca ac -3’(BamHIサイトを付加)
・TDH3P-D(配列番号32)5’-ata tat ccc ggg ttt gtt tgt tta tgt gtg ttt att cg -3’(SmaIサイトを付加)
増幅されたTDH3プロモータ配列(0.8kb)を制限酵素BamHIおよびSmaIで消化したものをTDH3P断片と称した。
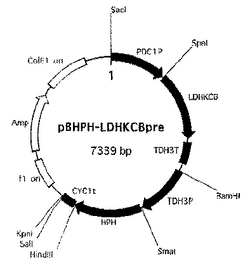
上記操作で得られたTDH3P断片と、HPH遺伝子断片および実施例2のCYC1T断片を、この順番で並ぶように順次pBluescriptII SK+ベクターのマルチクローニングサイトに連結して得られたベクターをpBHPH-PTベクターと称した。次にpBHPH-PTベクター中に実施例2のLDHKCB発現カセット断片を連結して得られたベクターをpBHPH-LDHKCBpre(図5)と称した。
【0065】
次に、pBHPH-LDHKCBpreベクターを鋳型とするPCR法により、ADH1プロモーターの直後にウシ由来のLDH遺伝子とTDH3ターミネーターと上記マーカーとが導入されて、ADH1遺伝子の全長が破壊されるとともに、ADH1プロモーターの制御下にLDH遺伝子が導入されるような染色体導入を可能とするDNA断片が得られるような相同組換え用DNA配列を含むPCRプライマー1(配列番号33)及びプライマー2(配列番号34)を合成した。これらのプライマー1、2を用いpBHPH-LDHKCBpreベクターを鋳型としてPCR法を行うことで目的のDNA断片を増幅した。
【0066】
なお、上記プライマー配列1、2においては、それぞれ5'側の75mer(上流側)と77mer(下流側)とが付加された相同組換え用配列である。Ex-Taq(TaKaRa)の付属のプロトコールに従って300μLのPCR反応液を調製した。PCR反応サイクルは、94℃で1分の後、94℃で15分と50℃で30分および72℃で3.7分を1サイクルとし、合計30サイクル実施した。PCR反応後、反応液1μLをアガロースゲル電気泳動にかけて3.7Kbの目的のDNA断片がシングルバンドで確認することができた。
【実施例4】
【0067】
(PDC1/ADH1破壊株の作製)
実施例2で得たPCR反応液の残液の全量をエタノール沈殿して得られたDNA約10μgを用いて、実施例1で作製したPDC1破壊株(PDC1p-LDH/2コピー体)を形質転換した。
【0068】
形質転換を行った菌体をYPD培地で2日間振とう培養し、その培養液50μlを、ハイグロマイシン選択プレート(150μg/mlハイグロマイシンを含有するYPDプレート)に塗布した。30℃で3日間培養して生育したコロニーを、新しいハイグロマイシン選択プレートに画線培養してコロニーを純化した。純化された選抜株について、実施例1と同様な方法で胞子を誘導し、胞子解剖を行った。YPDプレート上で分離した胞子を30℃で4日間培養して、生育したクローンをハイグロマイシン選択培地にレプリカし、同時にゲノムDNAを調製した。
【0069】
このゲノムDNAを鋳型とし、PCRをプライマーとして図4に示す3種(ADH1-206F、ADH1+857R、LDH-R)を(A)ADH1-206FとADH1+857R、(B)ADH1-206FとLDH-Rの組み合わせで用いてPCRを行ったところ、(A)の組み合わせではハイグロマイシン選択プレートで生育できなかったクローンのみで1.1Kbのバンドが増幅され、(B)の組み合わせではハイグロマイシン選択プレートで生育したクローンのみで1.2Kbのバンドが増幅された。このことから、ハイグロマイシン選択プレートで生育したクローンは、ADH1が破壊され、ADH1プロモーターの下流にLDHが挿入されていることが確認された。以上により、PDC1/ADH1破壊株を取得した。PDC1/ADH1破壊株では、染色体上のPDC1遺伝子の全長が破壊されるとともにPDC1プロモーターの制御下にLDH遺伝子が導入されていると同時に染色体上のADH1遺伝子の全長が破壊されているとともにADH1プロモーターの制御下にLDH遺伝子が導入されていた。この破壊株においてはLDH遺伝子は計4コピー存在することになる。なお、用いたプライマー配列は以下のとおりであった。
・ADH1-206F(配列番号:35)5’- taa tga gca acg gta tac gg -3’
・ADH1+857R(配列番号:36)5’- acg act tgg ttg aag aca tca g -3’
・LDH-R(配列番号:37)5’- tta tta aaa ttg caa ttc ttt ttg aat -3’
【実施例5】
【0070】
(発酵試験)
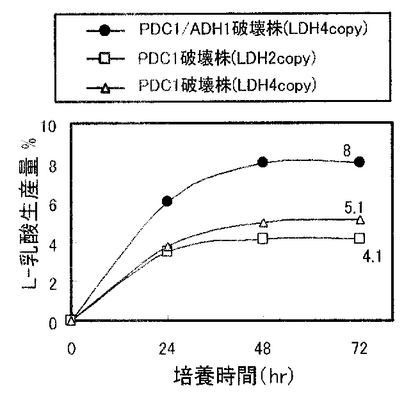
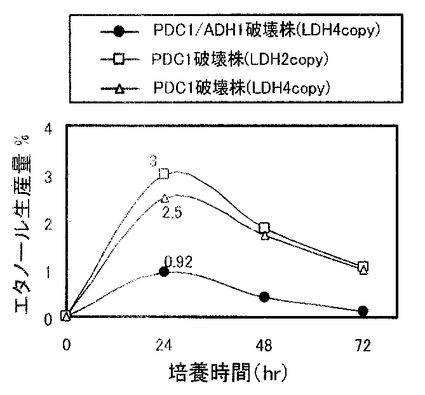
実施例4で作製したPDC1/ADH1破壊株を、5mlのYPD2(グルコース濃度2%のYPDである。)を入れた試験管に接種し、30℃で1日間回転振とう培養(130rpm)して前培養を行った。生育した菌体を遠心分離により集菌して前培養菌体とした。本培養は次のように行った。100ml容の三角フラスコに20mlのYPD10(グルコース濃度10%のYPDである。)を加え、中和剤として5%(w/v)の炭酸カルシウムを添加した。そこに前培養菌体を初発菌体濃度がOD600=3.8となるように接種し、シリコン栓をして30℃、80rpmで回転振とう培養を行った。経時的に培養液を採取して遠心分離した上清中のL−乳酸、エタノール濃度を酵素センサー(王子計測機器、バイオセンサーBF−4)により測定した。なお、同様に、実施例1および実施例2で作製したPDC1破壊株(PDC1p-LDH2コピー体および4コピー体)についても発酵試験を行った。各株の乳酸生産量の経時変化を図6に示し、各株のエタノール生産量の経時変化を図7に示す。
【0071】
図6及び図7に示すように、PDC1破壊株(PDC1p-LDH2コピー体および4コピー体)(最大4.1%及び5.1%)よりもPDC1/ADH1破壊株の方が高い乳酸生産量(8%)を示した。すなわち、PDC1破壊株においてコピー数を倍にしてもほとんど乳酸生産量は増加しなかったが、ADH1遺伝子を破壊し、該プロモーターの制御下にLDH遺伝子を導入することにより、約2倍の乳酸生産量となった。また、PDC1/ADH1破壊株においては、エタノール生産量もPDC1破壊株の約1/3となっており、酵母のエタノール発酵が効果的にかつ十分に低く抑制されていることがわかった。
【0072】
図6からも明らかなように、PDC1破壊株においては、LDH遺伝子のコピー数は倍になっているものの、ほとんど乳酸生産量は増えておらず、PDC1破壊株においては必ずしもLDH遺伝子のコピー数に応じて乳酸生産量が増加するとはいえなかった。このような状況下、ADH1破壊によって乳酸生産量がおおよそ倍加しているということは予想を越えるものであり、本実施例のPDC1/ADH1破壊株は、乳酸生産に高度に最適化された高発現系を備えているといえる。
【実施例6】
【0073】
(HOR7プロモーターによるLDH発現株の作製)
HOR7プロモーターによるLDH発現株は、図8に示すpBTrp-HOR7p-LDHベクターを用いて作製した。すなわち、pBTrp-HOR7p-LDHベクターを制限酵素SacIおよびApaIで消化した断片をもちいて、宿主である酵母IFO2260株のトリプトファン要求性変異株の形質転換を行った。形質転換処理を行った菌体について、実施例1と同様な操作によってPDC1破壊株(HOR7p-LDH2コピー体)を作製した。この株は、染色体上のPDC1構造遺伝子部分が破壊され、HOR7プロモータの制御下でLDHが発現されている。
【0074】
なお、pBTrp-HOR7p-LDHベクターは、以下の方法で構築したものである。
1.pBTRP-PDC1-LDHKCBベクターを制限酵素ApaIとPstIで消化して得られたTRP1-PDC1D断片を、同じ制限酵素で消化したpBluescriptII SK+ベクターに導入し、pBTRP-PDC1Dベクターを構築した。
2. 相同組み換え配列であるSTU2配列を、酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・STU2-U(配列番号:38)ata tat gag ctc gga cgt caa cta tga aa(SacIサイトを付加)
・STU2-D(配列番号:39)ata tat gcg gcc gcg gta tgg gtg cag tg(NotIサイトを付加)
増幅されたSTU2配列(0.7kb)を制限酵素SacIとNotIで消化したものをSTU2断片と称した。
3. HOR7遺伝子プロモータ配列を、酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・HOR7P-U(配列番号:40)ata tat gcg gcc gct cgc agc cac ggg tca acc cg(NotIサイトを付加)
・HOR7P-D(配列番号:41)ata tat act agt ttt tat tat tag tct ttt ttt ttt ttg ac(SpeIサイトを付加)
増幅されたHOR7遺伝子プロモータ配列(0.8kb)を制限酵素NotIとSpeIで消化したものをHOR7P断片と称した。
4. LDHKCB-TDH3T配列を、pBTrp-PDC1-LDHKCB鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・LDHKCB-F(配列番号:42)ata att act agt aca atg gct act ttg aaa gat caa (SpeIサイトを付加)
・TDH3t-BR(配列番号:43)tta att gga tcc aaa gct ttc aat caa tga atc gaa aa(BamHIサイトを付加)
増幅されたLDHKCB-TDH3T配列を制限酵素SpeIとBamHIで消化したものをLDHKCB-TDH3T断片と称した。
5. 上記操作で得られた各断片(STU2断片、HOR7P断片、LDHKCB-TDH3T断片)を、順次前記のpBTRP-PDC1Dベクターに連結して、染色体導入型ベクターpBTrp-HOR7-LDHを構築した(図8)。
【実施例7】
【0075】
(PDC1/ADH1破壊株の作製)
実施例4と同様の操作により、実施例6で作製したPDC1破壊株(HOR7p-LDH2コピー体)のADH1遺伝子を破壊すると同時に、破壊された該ADH1プロモーターの制御下にウシ由来LDH遺伝子が発現する組換え体を作製した。本実施例で作製されたPDC1/ADH1破壊株では、染色体上のPDC1遺伝子の全長が破壊されるとともにHOR7プロモーターの制御下にLDH遺伝子が導入されていると同時に染色体上のADH1遺伝子の全長が破壊されているとともにADH1プロモーターの制御下にLDH遺伝子が導入されていた。この破壊株においてはLDH遺伝子は計4コピー存在することになる。
【実施例8】
【0076】
(発酵試験)
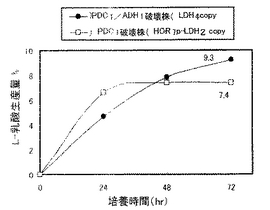
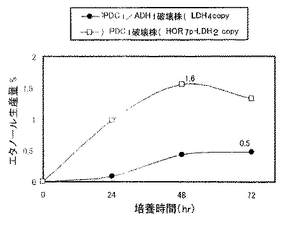
実施例7で作製したPDC1/ADH1破壊株を、5mlのYPD2(グルコース濃度2%のYPDである。)を入れた試験管に接種し、30℃で1日間回転振とう培養(130rpm)して前培養を行った。生育した菌体を遠心分離により集菌して前培養菌体とした。本培養は次のように行った。100ml容の三角フラスコに20mlのYPD10(グルコース濃度10%のYPDである。)を加え、中和剤として5%(w/v)の炭酸カルシウムを添加した。そこに前培養菌体を初発菌体濃度がOD600=3.8となるように接種し、シリコン栓をして30℃、80rpmで回転振とう培養を行った。経時的に培養液を採取して遠心分離した上清中のL−乳酸、エタノール濃度を酵素センサー(王子計測機器、バイオセンサーBF−4)により測定した。なお、同様に、実施例6で作製したPDC1破壊株(HOR71p-LDH2コピー体)についても発酵試験を行った。両株の乳酸生産量の経時変化を図9に示し、両株のエタノール生産量の経時変化を図10に示す。
【0077】
図9及び図10に示すように、PDC1破壊株(HOR7p-LDH2コピー体)(最大7.4%)よりもPDC1/ADH1破壊株の方が高い乳酸生産量(9.3%)を示した。すなわち、PDC1破壊株(HOR7p-LDH2コピー体)のADH1遺伝子を破壊し、該プロモーターの制御下にLDH遺伝子を導入することにより、約1.25倍の乳酸生産量となり、糖から乳酸への変換効率(対糖収率)は93%と非常に高い値を示した。また、PDC1/ADH1破壊株においては、エタノール生産量もPDC1破壊株の約1/3となっており、酵母のエタノール発酵が効果的にかつ十分に低く抑制されていることがわかった。
【配列表フリーテキスト】
【0078】
配列番号:4 合成DNA
配列番号:6〜43
プライマー
【図面の簡単な説明】
【0079】
【図1】酵母におけるピルビン酸からのアルコール発酵経路と乳酸生産の経路とを示す図。
【図2】染色体導入型ベクターpBTrp-PDC1-LDHKCBを示す図。
【図3】染色体導入型ベクターpBble-LDHKCBを示す図。
【図4】PDC1/ADH1破壊株の作製スキームを示す図。
【図5】ベクターpBHPH-LDHKCBpreを示す図。
【図6】PDC1破壊株およびPDC1/ADH1破壊株のL−乳酸生産量を示すグラフ。
【図7】PDC1破壊株およびPDC1/ADH1破壊株のエタノール生産量を示すグラフ。
【図8】染色体導入型ベクターpBTrp-HOR7p-LDHを示す図。
【図9】PDC1破壊株およびPDC1/ADH1破壊株のL−乳酸生産量を示すグラフ。
【図10】PDC1破壊株およびPDC1/ADH1破壊株のエタノール生産量を示すグラフ。
【技術分野】
【0001】
本発明は、乳酸生産酵母および乳酸生産方法に関する。
【背景技術】
【0002】
L−乳酸などの乳酸を酵母を利用して生産することが試みられている。例えば、図1に示すように、乳酸を生産するために、乳酸脱水素酵素(LDH)遺伝子を酵母サッカロマイセス・セレビシエ属に導入し、ピルビン酸から乳酸を生産させる試みがなされている。これらの試みの多くは、酵母本来のエタノール発酵を抑制して酵母における乳酸生産を高めるために、アルコール発酵経路の強力でかつ構成的酵素であるピルビン酸脱炭酸酵素(PDC)の発現を抑制するものである(特許文献1、2)。また、酵母染色体中のピルビン酸脱炭酸酵素1(PDC1)遺伝子プロモーターの下流に目的とする有用遺伝子(この場合は、L−乳酸脱水素酵素遺伝子)を結合させると同時に、酵母染色体中のPDC1遺伝子を破壊することによって、PDC1プロモーター機能を利用して乳酸脱水素酵素遺伝子を発現させつつ、当該プロモーターによって本来発現されるべきPDC1の発現を排除するシステムも開発されている(特許文献3)。このようなPDC遺伝子の破壊は乳酸生産量を明らかに増大させた。
【0003】
また、酵母のアルコール発酵経路においてピルビン酸脱炭酸酵素の後段の酵素であるアルコール脱水素酵素1(ADH1)遺伝子を破壊してこの酵素の発現を抑制する試みもなされている(非特許文献1)。この試みによれば、染色体上のADH1遺伝子を破壊しLDHをコードするDNAを発現するプラスミドが導入された形質転換酵母は、ADH1遺伝子を維持する野生型酵母に同様のプラスミドを導入した形質転換酵母よりもエタノール生産量は減ったものの乳酸生産量が顕著に低下していた。また、PDC活性が低下した酵母に同様のプラスミドを導入した形質転換酵母の乳酸生産量よりも低い生産量であった。すなわち、ADH1遺伝子の破壊によるADH1の発現抑制は必ずしも乳酸生産に有効ではなかった。
【0004】
【特許文献1】特表2001−516584号
【特許文献2】特表2003−500062号
【特許文献3】特開2003−164295号
【非特許文献1】J Ind Microbiol Biotechnol(2003)30:22−27(英国)
【発明の開示】
【発明が解決しようとする課題】
【0005】
このようにアルコール発酵経路のピルビン酸脱炭酸酵素の後段の酵素であるアルコール脱水素酵素活性の抑制はピルビン酸脱炭酸酵素活性の抑制による乳酸生産効果を上回るものではなく、逆に好ましくない結果をもたらしていた。以上のことから、その活性を抑制することが酵母において効率的な乳酸生産をさせるのに有効であろうと推測されるアルコール発酵系酵素やTCA回路系酵素は多数存在するものの、現在までのところ、ピルビン酸脱炭酸酵素以外に活性抑制が有効な酵素や組み合わせは見出されていない。
【0006】
そこで、本発明では、酵母における乳酸生産に好ましい乳酸高発現系を構築することをその目的とする。具体的には、乳酸の高生産が可能な乳酸生産酵母を提供することを一つの目的とし、さらに、効率的な乳酸の生産方法を提供することを他の一つの目的とする。
【課題を解決するための手段】
【0007】
本発明者らは、PDC破壊酵母と同等あるいはそれを上回る乳酸生産を可能とする高発現系を確立すべく、酵母における乳酸生産のための活性抑制が有効な酵素を探索した。ADHは、PDCよりも後段の酵素であるため、既にPDCが破壊されてPDCの発現が抑制されている場合、ADHを破壊しても乳酸の生産性を向上させることは期待できないと考えられた。また、上記背景技術からもADHの破壊はアセトアルデヒドの蓄積により細胞の増殖に悪影響がある可能性が高かった。しかしながら、本発明者らがPDC遺伝子を破壊するとともにADH遺伝子をも破壊した上で、外来LDHをコードするDNAを発現させたところ、上記した推測に反し高率で乳酸生産することを見出した。この知見に基づき、本発明者らは発明を完成した。すなわち、本発明によれば、この知見に基づき以下の手段が提供される。
【0008】
本発明によれば、乳酸生産酵母であって、ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母が提供される。この有機酸生産酵母においては、ピルビン酸脱炭酸酵素遺伝子が破壊されていることが好ましい態様であり、さらに、アルコール脱水素酵素遺伝子が破壊されていることが好ましい態様である。さらにまた、前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、ピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられていることが好ましい態様であり、より好ましくは、酵母染色体上のピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられている。また、上記した有機酸生産酵母のいずれかにおいて、前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、アルコール脱水素酵素遺伝子のプロモーターの制御下で発現可能に備えられていることが好ましく、より好ましくは、酵母染色体上のアルコール脱水素酵素遺伝子の制御下で発現可能に備えられている。
【0009】
また、本発明によれば、乳酸生産酵母であって、染色体上のピルビン酸脱炭酸酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下で乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備え、染色体上のアルコール脱水素酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下に乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母が提供される。
【0010】
これらのいずれかの形態の乳酸生産酵母において、前記ピルビン酸脱炭酸酵素遺伝子は、ピルビン酸脱炭酸酵素1遺伝子であることが好ましく、前記アルコール脱水素酵素遺伝子はアルコール脱水素酵素1遺伝子であることが好ましい。
【0011】
また、上記いずれかの酵母において、前記乳酸脱水素酵素活性を有するタンパク質はウシ由来であり、前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAを複数有することが好ましく、前記酵母はサッカロマイセス属に属することが好ましい。さらに、サッカロマイセス・セレビジエであることが好ましい。
【0012】
また、本発明によれば、上記いずれかの乳酸生産酵母を培養する工程を備える、乳酸生産方法が提供される。
【発明を実施するための最良の形態】
【0013】
本発明の乳酸生産酵母は、ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備えている。この乳酸生産酵母によれば、アルコール発酵経路の酵素であるピルビン酸脱炭酸酵素とアルコール脱水素酵素とがそれぞれ低下されている一方、同じくピルビン酸を基質とする乳酸脱水素酵素がピルビン酸脱炭酸酵素プロモーターの制御下に発現可能に備えられているため、ピルビン酸は乳酸脱水素酵素によって代謝されやすくなっており、乳酸の生産が促進されている。アルコール発酵経路の第1の酵素であるピルビン酸脱炭酸酵素活性が低下されていることにより、酵母のアルコール発酵は効果的に抑制されることは従来からも知られている。こうしたピルビン酸脱炭酸酵素活性が低下した系においてさらにアルコール脱水素酵素活性が抑制されることは、基質であるピルビン酸の代謝経路をアルコール発酵経路から乳酸発酵経路へと大きく変化させるものとは考えにくいが、本発明によれば、意外にもアルコール生産量を低下させるとともに乳酸生産量を増大させることができる。すなわち、アルコール脱水素酵素活性を低下させてもアセトアルデヒドの生成による細胞の増殖阻害等を抑制して細胞増殖を確保し乳酸生産量を増大させることができる。
【0014】
以下、本発明の乳酸生産酵母について説明するとともに、この酵母を用いた乳酸生産方法について説明する。
【0015】
(乳酸生産酵母)
本発明の乳酸生産酵母は、外来の乳酸脱水素酵素遺伝子を有する形質転換酵母である。本乳酸生産酵母を得るための酵母としては、アルコール発酵を行う酵母であって、サッカロマイセス・セレビシエ、シゾサッカロマイセス・ポンベ(Scizosaccharomyces pombe)などのサッカロマイセス属酵母、ピキア・パストリス(Pichia pastoris)、などの酵母を挙げることができる。より好ましくは、サッカロマイセス・セレビシエなどのサッカロマイセス属を始めとする酵母である。好ましい酵母の一例としては、サッカロマイセス・セレビシエIFO2260株や同YPH株を例示できる。
【0016】
また、本乳酸生産酵母および乳酸生産方法において乳酸とは、L−乳酸、D−乳酸、及びDL−乳酸があるが、これらのいずれであってもよい。
【0017】
(ピルビン酸脱炭酸酵素及びアルコール脱水素酵素)
本乳酸生産酵母においては、ピルビン酸脱炭酸酵素(PDC)活性およびアルコール脱水素酵素(ADH)活性が低下されている。これらの酵素は、酵母のアルコール発酵経路の酵素であり、アルコール発酵を行う酵母がその染色体上に本来的に有している。ピルビン酸脱炭酸酵素は、いわゆるオートレギュレーション機構を有する酵素であり、ピルビン酸脱炭酸酵素1,5,6がある。これらのうちいずれか1種あるいは2種以上が破壊されていればよいが、好ましくは最も活性が高いあるいは大量に発現されているピルビン酸脱炭酸酵素1(PDC1)の活性が低下されていることが好ましい。また、アルコール脱水素酵素においても各種の亜種(ADH1,2,3,4,5,6,7)があるが、アルコール脱水素酵素1(ADH1)が大量に発現されている。したがってアルコール脱水素酵素1の活性が低下されていることが好ましい。
【0018】
ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性が低下されているとは、人工的な操作によりあるいはスクリーニングによって、野生型よりも低い活性の該酵素が生産されているかあるいは該酵素の生産量が野生型よりも少ないものであることが好ましい。このような酵素活性の低下のための人工的操作としては、この酵素遺伝子を破壊(ノックアウト)することが好ましい。染色体上の特定遺伝子を破壊する手法は、当業者において周知である。
【0019】
(乳酸脱水素酵素)
本乳酸生産酵母は、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを保持している。LDHをコードするDNAは、酵母においては外来性である。乳酸脱水素酵素(LDH)としては、生物の種類に応じてあるいは生体内においても各種同族体が存在する。本発明において使用する乳酸脱水素酵素としては、天然由来のLDHの他、化学合成的あるいは遺伝子工学的に人工的に合成されたLDHも包含している。また、LDHとしては、L−LDHであってもD−LDHであってもよい。LDHとしては、好ましくは、ラクトバシルス・ヘルベティカス、ラクトバシルス・カゼイ、クルイベロマイセス・サーモトレランス、トルラスポラ・デルブルッキ、シゾサッカロミセス・ポンビ、リゾプス・オリゼ、B.メガテリウムなどの原核生物もしくはカビなどの真核微生物由来であり、より好ましくは、植物、動物、昆虫などの高等真核生物由来であり、さらに好ましくは、ウシを始めとする哺乳類を含む高等真核生物由来である。例えば、ウシ由来のLDH(L−LDH)である。例えば、ウシ由来のLDHとして配列番号:2に示すアミノ酸配列からなるタンパク質を挙げることができる。また、かかるLDHをコードするDNAとしては、配列番号:1に記載される塩基配列からなるDNAを挙げることができる。さらに、本発明におけるLDHは、これらのLDHのホモログも包含している。LDHホモログは、天然由来のLDHのアミノ酸配列において、1もしくは数個のアミノ酸の置換、欠失、挿入および/または付加されたアミノ酸でありかつLDH活性を有しているタンパク質、および、天然由来のLDHとアミノ配列の相同性が少なくとも70%、好ましくは80%以上を有しかつLDH活性を有しているタンパク質を含んでいる。したがってLDHをコードするDNAとして、これらのいずれかのLDHをコードするDNAを用いることができる。本乳酸生産酵母は、LDHをコードするDNAを少なくとも一つ、好ましくは二以上保持している。より好ましくは三以上保持している。さらに好ましくは四つ以上保持している。
【0020】
LDHをコードするDNAは、酵母染色体外で維持されるプラスミドやYACなどにおいて保持されていてもよいが、好ましくは酵母染色体に組み込まれて保持される。
【0021】
(PDCプロモーター)
LDHをコードするDNAは、強力なプロモーター活性を有するプロモーターの制御下に発現可能に備えられていることが好ましい。例えば、ピルビン酸脱炭酸酵素遺伝子プロモーター(PDCプロモーター)の制御下に発現可能に備えられていることが好ましい。なかでも、十分に強力なプロモーターであるPDC1プロモーターを用いることが好ましい。より好ましくは、サッカロマイセス属(好ましくはセレビシエ)のPDC1プロモーターである。
【0022】
LDHをコードするDNAは、好ましくは、酵母染色体上のPDCプロモーターの制御下に発現可能である。既に述べたように、本乳酸生産酵母においては、PDC遺伝子が破壊されていることが好ましい。このため、LDHをコードするDNAは、染色体においてPDCプロモーターによって制御可能であってかつPDC遺伝子を破壊するように酵母染色体に組み込まれていることが好ましい。こうすることで、PDC活性を効果的に低下させると同時に、本来PDC遺伝子発現に作用していた強力なPDC遺伝子のプロモーター活性をLDHをコードするDNA発現にスイッチさせることができる。破壊されそしてそのプロモーターがLDHをコードするDNAの発現に利用されるPDC遺伝子は、好ましくはPDC1遺伝子である。最も強力なPDC1プロモーターによって制御されるPDC1遺伝子が破壊されてLDHをコードするDNAが代わりに発現されることで効果的にPDC活性低下とLDH活性発現とを実現できる。
【0023】
例えば、PDC1プロモーターとしては、配列番号:3に記載の塩基配列からなるDNAの他、同等の機能を有する限り、該DNAとストリンジェントな条件でハイブリダイズするDNA、該塩基配列において1あるいは2以上の塩基が置換、欠失、付加、及び/又は挿入された配列からなるDNA、あるいは、該DNAとの相同性が70%以上、80%以上、好ましくは90%以上、より好ましくは95%以上であるDNAを用いることができる。すなわち、酵母染色体上のPDC1プロモーターが、同一ではないが同等の機能を有するこれらのプロモーター活性を有するDNAによって相同組換え等を介して置換されていてもよい。
【0024】
ストリンジェントな条件としては、50%ホルムアミド存在下でハイブリダイゼーション温度が37℃であるハイブリダイゼーション条件あるいはこれと同様のストリンジェンシーのハイブリダイゼーション条件を意味している。よりストリンジェンシーの高い条件によれば、より相同性の高いDNAを単離できる。かかるハイブリダイゼーション条件としては、例えば、50%ホルムアミド存在下でハイブリダイゼーション温度が約42℃、さらにストリンジェンシーの高い条件としては、50%ホルムアミド存在下で約65℃のハイブリダイゼーション条件を挙げることができる。
【0025】
また、本明細書において、DNAなどの核酸の相同性については、なお、DNAの塩基配列のホモロジーは、遺伝子解析プログラムBLASTなどによって決定することができる。なお、DNAの塩基配列のホモロジーは、遺伝子解析プログラムBLAST(http://blast.genome.ad.jp),FASTA(http://fasta.genome.ad.jp/SIT/FASTA.html)などによって決定することができる。
【0026】
PDC1遺伝子は、本発明者らが既に開示しているように、オートレギュレーション機構が存在する遺伝子である(特開2003−164295号)。オートレギュレーション機構とは、同じ機能を有する遺伝子が同一生物において複数存在し、通常、そのうちの少なくとも一つは発現しているが、残りは抑制されており、通常発現している遺伝子が破壊などにより機能しなくなった場合にのみ、残りの遺伝子が発現されてその機能を継続する機構を意味している。かかる機構が存在するため、例えば、酵母のPDC1遺伝子が破壊されたとしても、PDC5遺伝子が発現し、酵母の生理的機能が維持されることになる。このようなオートレギュレーション機構が存在する遺伝子を破壊することで、生物自体の生存、増殖機能を維持することができて、結果として、外来DNAを保持する形質転換体の増殖を維持しながら目的産物を効果的に生産させることができる。
【0027】
(ADHプロモーター)
また、LDHをコードするDNAは、ADH遺伝子のプロモーター(ADHプロモーター)の制御下に発現可能に備えることもできる。ADHは既に述べたようにアルコール発酵経路においてPDCのすぐ後続の酵素である。ADHプロモーターとしては、十分に強力なプロモーターであるADH1プロモーターを用いることが好ましい。
【0028】
LDHをコードするDNAは、好ましくは、酵母染色体上のADHプロモーターの制御下に発現可能である。また、既に述べたように、本乳酸生産酵母においては、ADH遺伝子が破壊されていることが好ましい。このため、LDHをコードするDNAは、ADHプロモーターによって制御可能であってかつADH遺伝子を破壊するように酵母染色体に組み込まれていることが好ましい。こうすることで、アルコール発酵経路における代謝活性を効果的に低下させると同時に、本来ADH遺伝子発現に作用していたADHプロモーター活性をLDHをコードするDNA発現にスイッチさせることができる。また、ADH遺伝子は、好ましくはADH1遺伝子である。強力なあるいは構成的なADH1プロモーターによって制御されるADH1遺伝子が破壊されてLDHをコードするDNAが代わりに発現されることで効果的にADH活性低下とLDH活性発現とを実現できる。
【0029】
なお、ADH1プロモーターは、酵母の染色体上のADH1プロモーターと同等の機能を有する限り、該プロモーターのDNAとストリンジェントな条件でハイブリダイズするDNA、該塩基配列において1あるいは2以上の塩基が置換、欠失、付加、及び/又は挿入された配列からなるDNA、あるいは、ADH1プロモーターとの相同性が80%、好ましくは90%、より好ましくは95%以上であるDNAを用いることができる。すなわち、酵母染色体上のPDC1プロモーターが、同一ではないが同等の機能を有するこれらのプロモーターによって相同組換え等により置換されていてもよい。
【0030】
(他のプロモーター)
LDHをコードするDNAは、酵母サッカロマイセス・セレビジエの高浸透圧応答7遺伝子(HOR7遺伝子)、グリセルアルデヒド3リン酸脱水素酵素2遺伝子(TDH2遺伝子)、ヘキソース輸送タンパク質7遺伝子(HXT7遺伝子)、熱ショックタンパク質30遺伝子(HSP30遺伝子)、チオレドキシンペルオキシダーゼ1遺伝子(AHP1遺伝子)、膜タンパク質1関連遺伝子(MRH1遺伝子)、グリセルアルデヒド三リン酸脱水素酵素3(TDH3)遺伝子、ガラストース一リン酸キナーゼ1(GAL1)遺伝子、ホスホグリセレートキナーゼ(PGK)遺伝子及び転写エンハンサー因子−1(TEF)遺伝子のプロモーターの制御下に発現可能に導入することもできる。これらの他のプロモーターとしては、酵母、あるいはサッカロマイセス属酵母のこれらの遺伝子のプロモーター活性を有するDNAのほかかかるプロモーター領域に一定以上(70%以上、好ましくは80%以上、より好ましくは90%、さらに好ましくは95%以上)の相同性を有し、各プロモーター活性を有するDNAであってもよい。
【0031】
本発明者らによれば、これらの他の酵母由来プロモーターのいずれもが乳酸存在時において高い遺伝子発現を示すプロモーターであるという知見を有している。したがって、かかるプロモーターの制御下にLDHをコードするDNAを導入することで、乳酸の生産量の増大によって乳酸の生産が抑制されない、あるいは乳酸の生産量の増大によってさらに乳酸の生産が促進される実用的な乳酸生産酵母などの形質転換体を得ることがおおいに期待される。
【0032】
LDHをコードするDNAは、PDCプロモーターやADHプロモーターにおけるのと同様、酵母染色体上にあるこれらの他のプロモーターの制御下にある内在性遺伝子を破壊するとともに該プロモーターの制御下に発現可能に組み込むこともできる。また、これらの他のプロモーターの制御下に発現可能に連結したLDHをコードするDNAを、PDC遺伝子および/またはADH遺伝子を破壊するように酵母染色体に組み込みこともできる。
【0033】
なお、明らかなように、PDCプロモーター、ADHプロモーター及び他のプロモーターのDNAは、ゲノムDNAであってもよく、また、化学的に合成されたDNAであってもよい。
【0034】
本乳酸生産酵母においては、上記したPDC活性及びADH活性を低下させる態様、各種のLDHをコードするDNA発現態様を各種組み合わせて採ることができる。好ましい乳酸生産酵母は、染色体上のPDC(好ましくはPDC1)遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下にLDHをコードするDNAを発現可能に備えるとともに、染色体上のADH(好ましくはADH1)遺伝子が破壊されている。また、この態様において、破壊された該ADH(好ましくはADH1)遺伝子のプロモーターの制御下にLDHをコードするDNAを発現可能に備えることができる。こうした乳酸生産酵母においては、好ましくは2以上、より好ましくは3以上、さらに好ましくは4以上のLDHをコードするDNAを染色体上に備えている。また、さらに、酵母染色体上の上記他のプロモーターの制御下にLDHをコードするDNAを発現可能に備えることができる。
【0035】
本乳酸生産酵母を取得するには、宿主酵母に対して、PDC遺伝子及びADH遺伝子の破壊を行うとともに、LDHをコードするDNAを発現可能に導入する。酵母に対してこのような遺伝子修飾を行うには、組換え用DNA構築物を利用する。組換え用DNA構築物は、特に限定しないで、線状等のDNA断片、プラスミド(DNA)、ウイルス(DNA)、レトロトランスポゾン(DNA)、人工染色体(YAC)を、外来遺伝子の導入形態(染色体外あるいは染色体内)等に応じて選択してベクターとしての形態をとることができる。
【0036】
PDC遺伝子及びADH遺伝子の破壊のためのDNA構築物は、これらの遺伝子部位に導入して遺伝子を破壊するために相同組換え用の配列を備えている。相同組換え用DNA配列は、破壊しようとするPDC遺伝子及びADH遺伝子であるターゲット部位あるいはその近傍のDNA配列と相同なDNA配列である。相同組換え用DNA配列は、一つのターゲット遺伝子あるいはその近傍の少なくとも1箇所に相同である1の配列を有しており、好ましくは、ターゲット遺伝子あるいはその近傍の少なくとも2箇所にそれぞれ相同な配列を備えている。例えば、2個の相同組換え用DNA配列を、染色体上のターゲット遺伝子の上流側と下流側のDNAとのそれぞれに相同なDNA配列とし、これらの相同組換え用DNA配列の間に遺伝子を破壊するためのDNAを備えるDNA構築物を酵母染色体に相同組換えにより導入することでターゲット部位の遺伝子を破壊することができる。
【0037】
このような染色体上への組み込みを実現するための相同組換え用DNAの選択は、当業者において周知であり、当業者であれば必要に応じて適切な相同組換え用DNAを選択して相同組換え用DNA構築物を構成することができる。例えば、PDCプロモーターの少なくとも一部と、PDC遺伝子あるいはターミネーターの少なくとも一部とにそれぞれ相同なDNA配列を相同組換え用DNA配列として用いることができる。同様に、ADHプロモーターの少なくとも一部とADH遺伝子あるいはターミネーターの少なくとも一部とにそれぞれ相同なDNA配列を相同組換え用DNA配列として用いることができる。
【0038】
PDC遺伝子を破壊するDNA構築物には、PDC遺伝子をターゲットとするための相同組換え用DNAとともにPDC遺伝子を置換可能な形態でLDHをコードするDNAを有することができる。このようなDNA構築物によれば、PDC遺伝子の破壊とともにPDCプロモーターの制御下にLDHをコードするDNAを発現可能に導入できる。
【0039】
また、PDC遺伝子を破壊するとともに、破壊した該PDC遺伝子のプロモーターの制御下にLDHをコードするDNAを導入することもできるが、相同組換えにより染色体上のPDCプロモーターに替えて同等のプロモーターを導入することもできる。例えば、既に述べたPDC1プロモーターのホモログ等である。
【0040】
なお、ADH遺伝子を破壊するとともに、破壊した該ADH遺伝子のプロモーターの制御下にLDHをコードするDNAを導入するためのDNA構築物も、上記したPDC遺伝子についてのDNA構築物と同様に構築することができる。
【0041】
なお、DNA構築物には、CYC1ターミネーターやTDH3ターミネーターなどのターミネーター他、必要に応じてエンハンサーなどのシスエレメント、スプライシングシグナル、ポリA付加シグナル、選択マーカー、リボソーム結合配列(SD配列)を連結することができる。選択マーカーとしては、特に限定しないで、薬剤抵抗性遺伝子、栄養要求性遺伝子などを始めとする公知の各種選択マーカー遺伝子を利用できる。例えば、アンピシリン耐性遺伝子、カナマイシン耐性G418遺伝子、ハイグロマイシン耐性遺伝子、ブレオマイシン耐性遺伝子、ネオマイシン耐性遺伝子、ジヒドロ葉酸還元酵素遺伝子、クロラムフェニコール耐性遺伝子、シクロヘキサミド耐性遺伝子等を使用することができる。
【0042】
DNA構築物を、酵母に、トランスフォーメーション法や、トランスフェクション法、接合法、プロトプラスト融合、エレクトロポレーション法、リポフェクション法、酢酸リチウム法、パーティクルガン法、リン酸カルシウム沈殿法、アグロバクテリウム法、PEG法、直接マイクロインジェクション法等の各種の適切な手段のいずれかにより、これを導入することができる。DNA構築物の導入後、酵母は、選択培地で培養される。
【0043】
なお、DNA構築物が酵母に導入されたか否か、あるいは染色体上の所望の部位に本DNA構築物が導入されたか否かの確認は、PCR法やサザンハイブリダイゼーション法により行うことができる。例えば、形質転換体からDNAを調製し、導入部位特異的プライマーによりPCRを行い、PCR産物について、電気泳動において予期されるバンドを検出することによって確認できる。あるいは蛍光色素などで標識したプライマーでPCRを行うことでも確認できる。これらの方法は、当業者において周知である。
【0044】
(乳酸の生産方法)
本乳酸生産酵母を適当な炭素源の存在下で培養することにより、培養物中にLDHをコードするDNAの発現産物である乳酸を生成させることができる。本乳酸生産方法によれば、培養系から乳酸を分離する工程を実施することにより、乳酸を得ることができる。なお、本発明において培養物とは、培養上清の他、培養細胞あるいは菌体、細胞若しくは菌体の破砕物を包含している。
【0045】
本発明の乳酸生産酵母の培養にあたっては、酵母の種類に応じて培養条件を選択することができる。このような培養条件は、当業者においては周知である。乳酸の生産にあたっては、必要に応じて産物である乳酸等の中和を行うかあるいは、連続的に乳酸を除去する等の処理を行うこともできる。酵母を培養する培地としては、微生物が資化可能な炭素源、窒素源、無機塩類等を含有し、形質転換体の培養を効率的に行うことができる培地であれば、天然培地、合成培地のいずれも使用することができる。炭素源としては、グルコース、フルクトース、スクロース、デンプン、セルロース等の炭水化物、酢酸、プロピオン酸等の有機酸、エタノール、プロパノール等のアルコールを用いることができる。窒素源としては、アンモニア、塩化アンモニウム、硫酸アンモニウム、酢酸アンモニウム、リン酸アンモニウム等の無機酸もしくは有機酸のアンモニウム塩またはその他の含窒素化合物の他、ペプトン、肉エキス、コーンスティープリカー等を用いることができる。無機物としては、リン酸第一カリウム、リン酸マグネシウム、硫酸マグネシウム、塩化ナトリウム、硫酸第一鉄、硫酸マンガン、硫酸銅、炭酸カルシウムなどを用いることができる。
【0046】
培養は、通常、静置培養、振とう培養または通気攪拌培養等の、嫌気条件下または微好気条件下、30℃〜35℃で6〜72時間行う。培養期間中、pHは2.0〜7.0に保持することが好ましい。また、pHの調整は、無機あるいは有機酸、アルカリ溶液等を用いて行うことができる。培養中は、必要に応じてアンピシリン、テトラサイクリンなどの抗生物質を培地に添加することができる。
【0047】
培養終了後、培養物から乳酸を分離するには、通常の乳酸精製手段を使用することができる。例えば、酵母内に生産された場合は、常法により菌体を超音波破壊処理、摩砕処理、加圧破砕などで細胞を破壊して、乳酸を細胞と分離することができる。この場合、必要に応じてプロテアーゼを添加する。また、菌体外に乳酸が生産された場合には、この培養液等を、ろ過、遠心分離などにより固形分を除去する。
【0048】
これらの粗抽出画分に対しては、従来公知の各種精製分離法等を利用して、乳酸を精製することができる。また、必要に応じて、当該粗抽出画分及びその精製物に対してエステル化等を行うことにより、各種の乳酸誘導体を得ることができる。
【実施例1】
【0049】
以下に、本発明の具体例を記載するが、本発明を以下の具体例に限定する趣旨ではなく、本発明の要旨を逸脱しない範囲で種々の態様で実施できる。
【0050】
以下に述べる遺伝子組換え操作はMolecular Cloning: A Laboratory Manual (T. Maniatis, et al., Cold Spring Harbor Laboratory) に従い行った。PCRによる遺伝子増幅は、特に述べない限り、KOD plus DNA polymerase(TOYOBO)を用い、添付のプロトコルに従って行った。PCR増幅反応は94℃で2分間の熱処理を行った後、94℃で15秒と53℃で30秒と68℃でX分(X:増幅遺伝子の大きさが1kbにつき1分とした)との3つの温度変化を1サイクルとし、これを25サイクル繰り返し、最後に4℃とした。PCR増幅装置はGene Amp PCR system 9700(PE Applied Biosystems)を使用した。ライゲーション反応にはLigaFast Rapid DNA Ligation System(Promega)を用いた。大腸菌の形質転換は、JM109株のコンピテント細胞(TOYOBO)を用い、添付のプロトコルに従って行った。大腸菌の形質転換体の選抜はアンピシリン100μg/mlを含むLBプレートを用いて行った。大腸菌からのプラスミドの抽出にはQIA Prep Spin Miniprepkit (QIAGEN)を用いた。DNA断片末端の平滑化にはT4 DNA polymerase(TaKaRa)を用いた。酵母のゲノムDNAの調製はFast DNA Kit(Bio 101)を用い、添付のプロトコルに従って行った。酵母の形質転換はFrozen-EZ Yeast Transformation II(ZYMO RESEARCH)を用い、添付のプロトコルに従って行った。
【0051】
(PDC1破壊株の作製)
PDC1破壊株は、図2に示すpBTrp-PDC1-LDHKCBベクターを用いて作製した。すなわち、pBTrp-PDC1-LDHKCBベクターを制限酵素SacIおよびApaIで消化した断片をもちいて、宿主である酵母IFO2260株のトリプトファン要求性変異株の形質転換を行った。形質転換処理を行った菌体をトリプトファン選択培地(SD-Trp)に塗沫し、生育してきたコロニーを新たなトリプトファン選択培地に画線培養してコロニーを純化した。純化された選抜株について、1 mlのYPA培地(2% 酢酸カリウム、2% ペプトン、1% 酵母エキス)に接種し、一晩振とう培養を行った。増殖した菌体を滅菌水で2回洗浄した後、胞子誘導プレート(1% 酢酸カリウム、0.1% ペプトン、0.05% グルコース、1.5% アガー)に塗布し、室温で4日間培養して胞子を誘導した。プレート上の菌体をかきとり、0.25 u/mlのZymolyase(ZYMO RESERCH)溶液中で15分間処理して子嚢壁を消化した。次にマイクロマニピュレータを用いて胞子を解剖し、YPDプレート上で分離した。30℃で4日間培養して生育したクローンをトリプトファン選択培地にレプリカし、同時にゲノムDNAを調製した。このゲノムDNAを鋳型として、ゲノム上のPCRプライマーであるPDC1-1000Fとベクター上のPCRプライマーであるLDHKCB-Rを用いてPCRを行ったところ、トリプトファン選択培地で生育したコロニーのみで2kbのバンド増幅された。さらにゲノム上のPCRプライマーであるPDC1+1300Rとベクター上のPCRプライマーであるTRP1-Fを用いてPCRを行ったところ、トリプトファン選択培地で生育したコロニーのみで1.5kbのバンド増幅された。このことから、トリプトファン選択培地で生育したコロニーはPDC1構造遺伝子部分が破壊され、PDC1プロモータの下流にLDHが挿入されていることが確認された。この操作で得られた株はPDC1構造遺伝子がホモ破壊されており、破壊されたPDC1構造遺伝子上流のPDC1プロモータによって発現するLDH遺伝子が2コピー導入されている。そこでこの株をPDC1破壊株(PDC1p-LDH2コピー体)と称した。用いたPCRプライマーの配列は以下の通りである。
【0052】
PDC1-1000F(配列番号6):5'- aaa atg aag gcc aaa tca agg cgg gaa ggg -3'
LDHKCB-R(配列番号7):5'- tta tta aaa ttg caa ttc ttt ttg aat acc -3'
TRP1-F(配列番号8):5'- ctc tgc aag ccg caa act ttc acc aat gga -3'
PDC1+1300R(配列番号9):5'- gat caa ttt ctt cag cag cga aag cag cac -3'
【0053】
なお、pBTrp-PDC1-LDHKCBベクターは、以下の方法で構築したものである。
1.ウシ由来のL-乳酸脱水素酵素を、酵母サッカロマイセス・セレビシエにおいて効率的に生産するために、ウシ由来のL-乳酸脱水素酵素のアミノ酸配列をコードするDNAに対して使用コドンを酵母の発現に最適化した遺伝子配列(配列番号:4)を設計し、長鎖DNAの合成方法として知られている藤本らの方法(藤本英也、合成遺伝子の作成法、植物細胞工学シリーズ7、植物のPCR実験プロトコール、1997、集潤社、p95-100)を用いて該DNAを全合成した(合成したDNAをLDHKCBと称した)。LDHKCBを制限酵素EcoRIで消化したものをLDHKCB断片と称した。
2. PDC1プロモータ断片(PDC1P, 0.7kb)は、サッカロマイセス・セレビシエYPH499株のゲノムDNAを鋳型として使用したPCR反応により増幅した。使用したPCRプライマーの配列は以下の通りであった。
・PDC1P-LDH-U(配列番号10)ata tat gga tcc gcg ttt att tac cta tct c(BamHIサイトを付加)
・PDC1P-LDH-D(配列番号11)ata tat gaa ttc ttt gat tga ttt gac tgt g(EcoRIサイトを付加)
得られたPCR増幅断片を制限酵素BamHIおよびEcoRIにより消化したものをPDC1P断片と称した。
3. PDC1遺伝子下流領域断片(PDC1D, 0.5kb)は、サッカロマイセス・セレビシエYPH499株のゲノムDNAを鋳型として使用したPCR反応により増幅した。使用したPCRプライマーの配列は以下の通りであった。
・PDC1D-LDH-U(配列番号12)ata tat ctc gag gcc agc taa ctt ctt ggt cga c(XhoIサイトを付加)
・PDC1D-LDH-D(配列番号13)ata tat gaa ttc ttt gat tga ttt gac tgt g(ApaIサイトを付加)
得られたPCR増幅断片を制限酵素XhoIおよびApaIにより消化したものをPDC1D断片と称した。
4. トリプトファン要求性マーカー(TRP1, 1.3kb)は、pRS404ベクター(Stratagene)を制限酵素AatIIおよびSspIで消化して切り出し、末端を平滑化した。得られた断片をTRP1断片と称した。
5. TDH3ターミネーター(TDH3t, 0.15kb)はサッカロマイセス・セレビシエYPH499株のゲノムDNAを鋳型として使用したPCR反応により増幅した。使用したPCRプライマーの配列は以下の通りであった。
・TDH3t-F(配列番号14)tcg act tgg ttg aac acg ttg cca agg ctt a
・TDH3t-R(配列番号15)aaa gct ttc aat caa tga atc gaa aa
得られたPCR増幅断片をTDH3t断片と称した。
6. 上記操作で得られた各断片(PDC1P断片、LDHKCB断片、TDH3t断片、TRP1断片およびPDC1D断片)を、順次pBluescriptII SK+ベクター(TOYOBO)のマルチクローニングサイトに連結して、染色体導入型ベクターpBTrp-PDC1-LDHKCBを構築した(図2)。
【実施例2】
【0054】
(染色体導入型ベクターの構築)
他の遺伝子を破壊することなく、染色体上にLDH遺伝子を導入するための染色体導入型ベクターpBble-LDHKCB(図3)を以下の方法で構築した。
(フレオマイシン耐性マーカーカセットの構築)
抗生物質フレオマイシン耐性遺伝子を大腸菌XL1-Blue MRF’ Kan株(STRATAGENE)のゲノムDNAを鋳型にしてPCRにより増幅した。大腸菌のゲノムDNAは、LB培地にて一晩振盪培養を行った後、集菌、洗浄したものを98℃ 10分間の加熱処理したのち遠心分離した上清を、エタノール沈殿し、滅菌水50μlに溶解することにより調製した。使用したPCRプライマーの配列は以下の通りであった。
・Tn5ble-U(配列番号16)ata tat gaa ttc atg acc gac caa gcg acg c (EcoRIサイトを付加)
・Tn5ble-D(配列番号17)ata tat aag ctt tca tga gat gcc tgc aag c(HindIIIサイトを付加)
得られたPCR増幅断片(0.4kb)を制限酵素EcoRIおよびHindIIIで消化したものをTn5 ble断片と称した。
【0055】
Tn5 bleを酵母菌体内で発現できるようにするために、CYC1プロモータ配列およびCYC1ターミネーター配列を酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・CYCP-U(配列番号18)ata tat gga tcc gac agc atc gtc gaa tat g(BamHIサイトを付加)
・CYCP-D(配列番号19)ata tat gaa ttc tat taa ttt agt gtg tgt att tg(EcoRIサイトを付加)
・CYCT-U(配列番号20)ata tat aag ctt aca ggc ccc ttt tcc ttt g(HindIIIサイトを付加)
・CYCT-D(配列番号21)ata tat gtc gac gtt aca tgc gta cac gcg (SalIサイトを付加)
【0056】
PCRプライマーCYCP-UおよびCYCP-Dを用いて増幅されたCYC1プロモータ配列(0.5kb)を制限酵素BamHIおよびEcoRIで消化したものをCYC1P断片と称した。PCRプライマーCYCT-UおよびCYCT-Dをもちいて増幅されたCYC1ターミネーター配列(0.2kb)を制限酵素HindIIIおよびSalIで消化したものをCYC1T断片と称した。
【0057】
上記操作で得られたCYC1P断片、Tn5 ble断片およびCYC1T断片を、この順番で並ぶように順次pBluescriptII SK+ベクターのマルチクローニングサイトに連結してフレオマイシン耐性マーカーカセットを構築した。得られたベクターを制限酵素SpeIおよびApaIで消化することによりフレオマイシン耐性マーカーカセットを切り出し、末端を平滑化したものをフレオマイシン耐性マーカーカセット断片と称した。
【0058】
(染色体導入型ベクターpBble-LDHKCBの構築)
先に構築したpBTrp-PDC1-LDHKCBを制限酵素BamHIおよびPstIにより消化して切り出したものをLDHKCB発現カセット断片(PDC1P断片、LDHKCB断片およびTDH3t断片がこの順番で連結された断片)と称した。
【0059】
相同組み換え配列であるPDC5遺伝子下流領域配列およびSLX4遺伝子上流領域配列を、酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・PDC5D-U(配列番号22) cca tga tta gat ggg gtt tg
・PDC5D-D(配列番号23) ctg gaa gac agg aca gaa
・SLX4-F(配列番号24)aat tat gtc gac ggt taa aga tta gct tct aat (SalIサイトを付加)
・SLX4-R(配列番号25)ata taa ggt acc caa ctg aac tac tgg tta tta tta tg(KpnIサイトを付加)
PCRプライマーPDC5D-UおよびPDC5D-Dをもちいて増幅されたPDC5遺伝子下流領域配列(0.6kb)をPDC5D断片と称した。PCRプライマーSLX4-FおよびSLX4-Rをもちいて増幅されたSLX4遺伝子上流領域配列(0.7kb)を制限酵素SalIおよびKpnIで消化したものをSLX4断片と称した。
上記操作で得られた各断片(PDC5D断片、LDHKCB発現カセット断片、フレオマイシン耐性マーカーカセット断片およびSLX4断片)を、順次pBluescriptII SK+ベクターのマルチクローニングサイトに連結して、染色体導入型ベクターpBble-LDHKCB(図3)を構築した。
【0060】
(4コピー体の作製)
pBble-LDHKCBベクターを制限酵素SacIおよびKpnIで消化した断片をもちいて、実施例1で作製されたPDC1破壊株(PDC1p-LDH2コピー体)の形質転換を行った。形質転換処理を行った菌体をフレオマイシン選択培地(10μg/mlのフレオマイシンを含むYPD培地)に塗沫し、生育してきたコロニーを新たなフレオマイシン選択培地に画線培養してコロニーを純化した。純化された選抜株について、実施例1と同様な方法で胞子を誘導し、胞子解剖を行った。YPDプレート上で分離した胞子を30℃で4日間培養して、生育したクローンをフレオマイシン選択培地にレプリカし、同時にゲノムDNAを調製した。このゲノムDNAを鋳型として、ゲノム上のPCRプライマーであるPDC5-320Fとベクター上のPCRプライマーであるLDHKCB-R(配列番号7)を用いてPCRを行ったところ、トリプトファン選択培地で生育したコロニーのみで4kbのバンド増幅された。さらにゲノム上のPCRプライマーであるPDC5+2788Rとベクター上のPCRプライマーであるTCYC-F1を用いてPCRを行ったところ、フレオマイシン選択培地で生育したコロニーのみで1.9kbのバンド増幅された。このことから、フレオマイシン選択培地で生育したコロニーは酵母染色体上のPDC5遺伝子とSLX4遺伝子の間に、両遺伝子を破壊することなく挿入されていることが確認された。この操作で得られた株はPDC1構造遺伝子がホモ破壊されており、PDC1プロモータによって発現するLDH遺伝子が4コピー導入されている。そこでこの株をPDC1破壊株(PDC1p-LDH4コピー体)と称した。用いたPCRプライマーの配列は以下の通りであった。
・PDC5-320F(配列番号26)5'- atg gga aaa gcc tcc ata tcc aaa ggt c -3'
・PDC5+2788R(配列番号27)5'- ttc gta gtc tct tgc gtc ata g -3'
・TCYC-F1(配列番号28)5'- ttg tct aac tcc ttc ctt ttc g -3'
【実施例3】
【0061】
(ADH1遺伝子破壊用ベクターの構築)
本実施例では、酵母の染色体上に存在するアルコール脱水素酵素をコードするADH1遺伝子を破壊すると同時に、破壊された該ADH1プロモーターの制御下にウシ由来LDH遺伝子を挿入することのできるDNA構築物を構築した。操作過程を図4に示す。
【0062】
まず、既に取得したウシ由来のLDH遺伝子にTDH3ターミネーターを接続し、さらに酵母用マーカーセット(ハイグロマイシン耐性遺伝子カセットを含むベクター(pBHPH-LDHKCBpreと称する。)を作製した(図5)。作製方法を以下に示す。
【0063】
既知文献(K.R.Kaster, S.G.Burgett, R.N.Rao, T.D.Ingolia Analysis of a bacterial hygromycin B resistance gene by transcriptional and translational fusions and by DNA sequencing. Nucleic Acids Research. Vol.11 (19), p6895-6911 (1983)、および L.Gritz, J.Davies Plasmid-encoded hygromycin B resitance; the sequence of hygromycin B phospotransferase gene and its expression in Escherichia coli and Saccharomyces cerevisiae. Gene. Vol.25, p179-188 (1983))に基づいてHPH遺伝子配列情報を取得して、以下のPCRプライマーを合成した。
・HPH-U(配列番号29)5’- atg aaa aag cct gaa ctc acc -3’
・HPH-D (配列番号30)5’- cta ttc ctt tgc cct cgg acg -3’
次に大腸菌K12株ゲノムDNAを鋳型としたPCR反応によりHPH遺伝子(1kb)を増幅したものをHPH断片と称した。
【0064】
HPH遺伝子を酵母菌体内で発現できるようにするためにTDH3プロモーターを酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・TDH3P-U(配列番号31)5’-ata tat gga tcc tag cgt tga atg tta gcg tca ac -3’(BamHIサイトを付加)
・TDH3P-D(配列番号32)5’-ata tat ccc ggg ttt gtt tgt tta tgt gtg ttt att cg -3’(SmaIサイトを付加)
増幅されたTDH3プロモータ配列(0.8kb)を制限酵素BamHIおよびSmaIで消化したものをTDH3P断片と称した。
上記操作で得られたTDH3P断片と、HPH遺伝子断片および実施例2のCYC1T断片を、この順番で並ぶように順次pBluescriptII SK+ベクターのマルチクローニングサイトに連結して得られたベクターをpBHPH-PTベクターと称した。次にpBHPH-PTベクター中に実施例2のLDHKCB発現カセット断片を連結して得られたベクターをpBHPH-LDHKCBpre(図5)と称した。
【0065】
次に、pBHPH-LDHKCBpreベクターを鋳型とするPCR法により、ADH1プロモーターの直後にウシ由来のLDH遺伝子とTDH3ターミネーターと上記マーカーとが導入されて、ADH1遺伝子の全長が破壊されるとともに、ADH1プロモーターの制御下にLDH遺伝子が導入されるような染色体導入を可能とするDNA断片が得られるような相同組換え用DNA配列を含むPCRプライマー1(配列番号33)及びプライマー2(配列番号34)を合成した。これらのプライマー1、2を用いpBHPH-LDHKCBpreベクターを鋳型としてPCR法を行うことで目的のDNA断片を増幅した。
【0066】
なお、上記プライマー配列1、2においては、それぞれ5'側の75mer(上流側)と77mer(下流側)とが付加された相同組換え用配列である。Ex-Taq(TaKaRa)の付属のプロトコールに従って300μLのPCR反応液を調製した。PCR反応サイクルは、94℃で1分の後、94℃で15分と50℃で30分および72℃で3.7分を1サイクルとし、合計30サイクル実施した。PCR反応後、反応液1μLをアガロースゲル電気泳動にかけて3.7Kbの目的のDNA断片がシングルバンドで確認することができた。
【実施例4】
【0067】
(PDC1/ADH1破壊株の作製)
実施例2で得たPCR反応液の残液の全量をエタノール沈殿して得られたDNA約10μgを用いて、実施例1で作製したPDC1破壊株(PDC1p-LDH/2コピー体)を形質転換した。
【0068】
形質転換を行った菌体をYPD培地で2日間振とう培養し、その培養液50μlを、ハイグロマイシン選択プレート(150μg/mlハイグロマイシンを含有するYPDプレート)に塗布した。30℃で3日間培養して生育したコロニーを、新しいハイグロマイシン選択プレートに画線培養してコロニーを純化した。純化された選抜株について、実施例1と同様な方法で胞子を誘導し、胞子解剖を行った。YPDプレート上で分離した胞子を30℃で4日間培養して、生育したクローンをハイグロマイシン選択培地にレプリカし、同時にゲノムDNAを調製した。
【0069】
このゲノムDNAを鋳型とし、PCRをプライマーとして図4に示す3種(ADH1-206F、ADH1+857R、LDH-R)を(A)ADH1-206FとADH1+857R、(B)ADH1-206FとLDH-Rの組み合わせで用いてPCRを行ったところ、(A)の組み合わせではハイグロマイシン選択プレートで生育できなかったクローンのみで1.1Kbのバンドが増幅され、(B)の組み合わせではハイグロマイシン選択プレートで生育したクローンのみで1.2Kbのバンドが増幅された。このことから、ハイグロマイシン選択プレートで生育したクローンは、ADH1が破壊され、ADH1プロモーターの下流にLDHが挿入されていることが確認された。以上により、PDC1/ADH1破壊株を取得した。PDC1/ADH1破壊株では、染色体上のPDC1遺伝子の全長が破壊されるとともにPDC1プロモーターの制御下にLDH遺伝子が導入されていると同時に染色体上のADH1遺伝子の全長が破壊されているとともにADH1プロモーターの制御下にLDH遺伝子が導入されていた。この破壊株においてはLDH遺伝子は計4コピー存在することになる。なお、用いたプライマー配列は以下のとおりであった。
・ADH1-206F(配列番号:35)5’- taa tga gca acg gta tac gg -3’
・ADH1+857R(配列番号:36)5’- acg act tgg ttg aag aca tca g -3’
・LDH-R(配列番号:37)5’- tta tta aaa ttg caa ttc ttt ttg aat -3’
【実施例5】
【0070】
(発酵試験)
実施例4で作製したPDC1/ADH1破壊株を、5mlのYPD2(グルコース濃度2%のYPDである。)を入れた試験管に接種し、30℃で1日間回転振とう培養(130rpm)して前培養を行った。生育した菌体を遠心分離により集菌して前培養菌体とした。本培養は次のように行った。100ml容の三角フラスコに20mlのYPD10(グルコース濃度10%のYPDである。)を加え、中和剤として5%(w/v)の炭酸カルシウムを添加した。そこに前培養菌体を初発菌体濃度がOD600=3.8となるように接種し、シリコン栓をして30℃、80rpmで回転振とう培養を行った。経時的に培養液を採取して遠心分離した上清中のL−乳酸、エタノール濃度を酵素センサー(王子計測機器、バイオセンサーBF−4)により測定した。なお、同様に、実施例1および実施例2で作製したPDC1破壊株(PDC1p-LDH2コピー体および4コピー体)についても発酵試験を行った。各株の乳酸生産量の経時変化を図6に示し、各株のエタノール生産量の経時変化を図7に示す。
【0071】
図6及び図7に示すように、PDC1破壊株(PDC1p-LDH2コピー体および4コピー体)(最大4.1%及び5.1%)よりもPDC1/ADH1破壊株の方が高い乳酸生産量(8%)を示した。すなわち、PDC1破壊株においてコピー数を倍にしてもほとんど乳酸生産量は増加しなかったが、ADH1遺伝子を破壊し、該プロモーターの制御下にLDH遺伝子を導入することにより、約2倍の乳酸生産量となった。また、PDC1/ADH1破壊株においては、エタノール生産量もPDC1破壊株の約1/3となっており、酵母のエタノール発酵が効果的にかつ十分に低く抑制されていることがわかった。
【0072】
図6からも明らかなように、PDC1破壊株においては、LDH遺伝子のコピー数は倍になっているものの、ほとんど乳酸生産量は増えておらず、PDC1破壊株においては必ずしもLDH遺伝子のコピー数に応じて乳酸生産量が増加するとはいえなかった。このような状況下、ADH1破壊によって乳酸生産量がおおよそ倍加しているということは予想を越えるものであり、本実施例のPDC1/ADH1破壊株は、乳酸生産に高度に最適化された高発現系を備えているといえる。
【実施例6】
【0073】
(HOR7プロモーターによるLDH発現株の作製)
HOR7プロモーターによるLDH発現株は、図8に示すpBTrp-HOR7p-LDHベクターを用いて作製した。すなわち、pBTrp-HOR7p-LDHベクターを制限酵素SacIおよびApaIで消化した断片をもちいて、宿主である酵母IFO2260株のトリプトファン要求性変異株の形質転換を行った。形質転換処理を行った菌体について、実施例1と同様な操作によってPDC1破壊株(HOR7p-LDH2コピー体)を作製した。この株は、染色体上のPDC1構造遺伝子部分が破壊され、HOR7プロモータの制御下でLDHが発現されている。
【0074】
なお、pBTrp-HOR7p-LDHベクターは、以下の方法で構築したものである。
1.pBTRP-PDC1-LDHKCBベクターを制限酵素ApaIとPstIで消化して得られたTRP1-PDC1D断片を、同じ制限酵素で消化したpBluescriptII SK+ベクターに導入し、pBTRP-PDC1Dベクターを構築した。
2. 相同組み換え配列であるSTU2配列を、酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・STU2-U(配列番号:38)ata tat gag ctc gga cgt caa cta tga aa(SacIサイトを付加)
・STU2-D(配列番号:39)ata tat gcg gcc gcg gta tgg gtg cag tg(NotIサイトを付加)
増幅されたSTU2配列(0.7kb)を制限酵素SacIとNotIで消化したものをSTU2断片と称した。
3. HOR7遺伝子プロモータ配列を、酵母IFO2260株のゲノムDNAを鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・HOR7P-U(配列番号:40)ata tat gcg gcc gct cgc agc cac ggg tca acc cg(NotIサイトを付加)
・HOR7P-D(配列番号:41)ata tat act agt ttt tat tat tag tct ttt ttt ttt ttg ac(SpeIサイトを付加)
増幅されたHOR7遺伝子プロモータ配列(0.8kb)を制限酵素NotIとSpeIで消化したものをHOR7P断片と称した。
4. LDHKCB-TDH3T配列を、pBTrp-PDC1-LDHKCB鋳型にしてPCRにより増幅した。使用したPCRプライマーの配列は以下の通りであった。
・LDHKCB-F(配列番号:42)ata att act agt aca atg gct act ttg aaa gat caa (SpeIサイトを付加)
・TDH3t-BR(配列番号:43)tta att gga tcc aaa gct ttc aat caa tga atc gaa aa(BamHIサイトを付加)
増幅されたLDHKCB-TDH3T配列を制限酵素SpeIとBamHIで消化したものをLDHKCB-TDH3T断片と称した。
5. 上記操作で得られた各断片(STU2断片、HOR7P断片、LDHKCB-TDH3T断片)を、順次前記のpBTRP-PDC1Dベクターに連結して、染色体導入型ベクターpBTrp-HOR7-LDHを構築した(図8)。
【実施例7】
【0075】
(PDC1/ADH1破壊株の作製)
実施例4と同様の操作により、実施例6で作製したPDC1破壊株(HOR7p-LDH2コピー体)のADH1遺伝子を破壊すると同時に、破壊された該ADH1プロモーターの制御下にウシ由来LDH遺伝子が発現する組換え体を作製した。本実施例で作製されたPDC1/ADH1破壊株では、染色体上のPDC1遺伝子の全長が破壊されるとともにHOR7プロモーターの制御下にLDH遺伝子が導入されていると同時に染色体上のADH1遺伝子の全長が破壊されているとともにADH1プロモーターの制御下にLDH遺伝子が導入されていた。この破壊株においてはLDH遺伝子は計4コピー存在することになる。
【実施例8】
【0076】
(発酵試験)
実施例7で作製したPDC1/ADH1破壊株を、5mlのYPD2(グルコース濃度2%のYPDである。)を入れた試験管に接種し、30℃で1日間回転振とう培養(130rpm)して前培養を行った。生育した菌体を遠心分離により集菌して前培養菌体とした。本培養は次のように行った。100ml容の三角フラスコに20mlのYPD10(グルコース濃度10%のYPDである。)を加え、中和剤として5%(w/v)の炭酸カルシウムを添加した。そこに前培養菌体を初発菌体濃度がOD600=3.8となるように接種し、シリコン栓をして30℃、80rpmで回転振とう培養を行った。経時的に培養液を採取して遠心分離した上清中のL−乳酸、エタノール濃度を酵素センサー(王子計測機器、バイオセンサーBF−4)により測定した。なお、同様に、実施例6で作製したPDC1破壊株(HOR71p-LDH2コピー体)についても発酵試験を行った。両株の乳酸生産量の経時変化を図9に示し、両株のエタノール生産量の経時変化を図10に示す。
【0077】
図9及び図10に示すように、PDC1破壊株(HOR7p-LDH2コピー体)(最大7.4%)よりもPDC1/ADH1破壊株の方が高い乳酸生産量(9.3%)を示した。すなわち、PDC1破壊株(HOR7p-LDH2コピー体)のADH1遺伝子を破壊し、該プロモーターの制御下にLDH遺伝子を導入することにより、約1.25倍の乳酸生産量となり、糖から乳酸への変換効率(対糖収率)は93%と非常に高い値を示した。また、PDC1/ADH1破壊株においては、エタノール生産量もPDC1破壊株の約1/3となっており、酵母のエタノール発酵が効果的にかつ十分に低く抑制されていることがわかった。
【配列表フリーテキスト】
【0078】
配列番号:4 合成DNA
配列番号:6〜43
プライマー
【図面の簡単な説明】
【0079】
【図1】酵母におけるピルビン酸からのアルコール発酵経路と乳酸生産の経路とを示す図。
【図2】染色体導入型ベクターpBTrp-PDC1-LDHKCBを示す図。
【図3】染色体導入型ベクターpBble-LDHKCBを示す図。
【図4】PDC1/ADH1破壊株の作製スキームを示す図。
【図5】ベクターpBHPH-LDHKCBpreを示す図。
【図6】PDC1破壊株およびPDC1/ADH1破壊株のL−乳酸生産量を示すグラフ。
【図7】PDC1破壊株およびPDC1/ADH1破壊株のエタノール生産量を示すグラフ。
【図8】染色体導入型ベクターpBTrp-HOR7p-LDHを示す図。
【図9】PDC1破壊株およびPDC1/ADH1破壊株のL−乳酸生産量を示すグラフ。
【図10】PDC1破壊株およびPDC1/ADH1破壊株のエタノール生産量を示すグラフ。
【特許請求の範囲】
【請求項1】
乳酸生産酵母であって、
ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母。
【請求項2】
ピルビン酸脱炭酸酵素遺伝子が破壊されている、請求項1に記載の酵母。
【請求項3】
アルコール脱水素酵素遺伝子が破壊されている、請求項1または2に記載の酵母。
【請求項4】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、ピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項1〜3のいずれかに記載の酵母。
【請求項5】
前記乳酸脱水素酵素活性を有するタンパク質のコードするDNAは、酵母染色体上のピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項4に記載の酵母。
【請求項6】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、アルコール脱水素酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項1〜4のいずれかに記載の酵母。
【請求項7】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、酵母染色体上のアルコール脱水素酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項6に記載の酵母。
【請求項8】
乳酸生産酵母であって、
染色体上のピルビン酸脱炭酸酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下で乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備え、
染色体上のアルコール脱水素酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下で乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母。
【請求項9】
前記ピルビン酸脱炭酸酵素遺伝子は、ピルビン酸脱炭酸酵素1遺伝子である、請求項1〜8のいずれかに記載の酵母。
【請求項10】
前記アルコール脱水素酵素遺伝子は、アルコール脱水素酵素1遺伝子である、請求項1〜9のいずれかに記載の酵母。
【請求項11】
前記乳酸脱水素酵素活性を有するタンパク質はウシ由来である、請求項1〜10のいずれかに記載の酵母。
【請求項12】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAを複数有する、請求項1〜11のいずれかに記載の酵母。
【請求項13】
前記酵母はサッカロマイセス属に属する、請求項1〜12のいずれかに記載の酵母。
【請求項14】
前記酵母は、サッカロマイセス・セレビジエである、請求項13に記載の酵母。
【請求項15】
有機酸の生産方法であって、
請求項1〜14のいずれかに記載の乳酸生産酵母を培養する工程を備える、生産方法。
【請求項1】
乳酸生産酵母であって、
ピルビン酸脱炭酸酵素活性およびアルコール脱水素酵素活性がそれぞれ低下され、乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母。
【請求項2】
ピルビン酸脱炭酸酵素遺伝子が破壊されている、請求項1に記載の酵母。
【請求項3】
アルコール脱水素酵素遺伝子が破壊されている、請求項1または2に記載の酵母。
【請求項4】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、ピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項1〜3のいずれかに記載の酵母。
【請求項5】
前記乳酸脱水素酵素活性を有するタンパク質のコードするDNAは、酵母染色体上のピルビン酸脱炭酸酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項4に記載の酵母。
【請求項6】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、アルコール脱水素酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項1〜4のいずれかに記載の酵母。
【請求項7】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAは、酵母染色体上のアルコール脱水素酵素遺伝子のプロモーターの制御下で発現可能に備えられている、請求項6に記載の酵母。
【請求項8】
乳酸生産酵母であって、
染色体上のピルビン酸脱炭酸酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下で乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備え、
染色体上のアルコール脱水素酵素遺伝子が破壊されるとともに破壊された該遺伝子のプロモーターの制御下で乳酸脱水素酵素活性を有するタンパク質をコードするDNAを発現可能に備える、酵母。
【請求項9】
前記ピルビン酸脱炭酸酵素遺伝子は、ピルビン酸脱炭酸酵素1遺伝子である、請求項1〜8のいずれかに記載の酵母。
【請求項10】
前記アルコール脱水素酵素遺伝子は、アルコール脱水素酵素1遺伝子である、請求項1〜9のいずれかに記載の酵母。
【請求項11】
前記乳酸脱水素酵素活性を有するタンパク質はウシ由来である、請求項1〜10のいずれかに記載の酵母。
【請求項12】
前記乳酸脱水素酵素活性を有するタンパク質をコードするDNAを複数有する、請求項1〜11のいずれかに記載の酵母。
【請求項13】
前記酵母はサッカロマイセス属に属する、請求項1〜12のいずれかに記載の酵母。
【請求項14】
前記酵母は、サッカロマイセス・セレビジエである、請求項13に記載の酵母。
【請求項15】
有機酸の生産方法であって、
請求項1〜14のいずれかに記載の乳酸生産酵母を培養する工程を備える、生産方法。
【図1】


【図2】

【図3】
【図4】

【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【図2】
【図3】
【図4】
【図5】
【図6】
【図7】
【図8】
【図9】
【図10】
【公開番号】特開2006−6271(P2006−6271A)
【公開日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願番号】特願2004−191562(P2004−191562)
【出願日】平成16年6月29日(2004.6.29)
【出願人】(000003609)株式会社豊田中央研究所 (4,200)
【出願人】(000003207)トヨタ自動車株式会社 (59,920)
【Fターム(参考)】
【公開日】平成18年1月12日(2006.1.12)
【国際特許分類】
【出願日】平成16年6月29日(2004.6.29)
【出願人】(000003609)株式会社豊田中央研究所 (4,200)
【出願人】(000003207)トヨタ自動車株式会社 (59,920)
【Fターム(参考)】
[ Back to top ]