
サノフイにより出願された特許
41 - 50 / 433
[4[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−(1H−ピロロ−ピリジン−イル)−メタノン類及びその合成
本明細書において本発明は、炎症性疾患の処置及び寛解のための化合物及び組成物に関する。詳細には、本発明は、トリプターゼ阻害活性を有する化合物及びその中間体、このような化合物を含む医薬組成物、並びにトリプターゼの阻害剤の投与により寛解され得る状態疾患又は障害(限定されないが、例えば喘息、及び加齢性黄斑変性を含む他の炎症性疾患)に罹患している被験体を処置する方法に関する。 (もっと読む)
炎症性腸疾患の処置
化合物:[4−(5−アミノメチル−2−フルオロフェニル)ピペリジン−1−イル][7−フルオロ−1−(2−メトキシエチル)−4−トリフルオロメトキシ−1H−インドール−3−イル]メタノンを用いる、炎症性腸疾患の処置が開示されている。 (もっと読む)
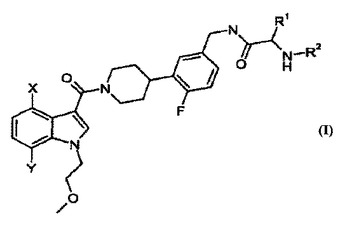
[4[4−(5−アミノメチル−2−フルオロ−フェニル)−ピペリジン−1−イル]−(1H−ピロロ−ピリジン−イル)−メタノンのプロドラッグ及びその合成
この発明は、本明細書中で炎症性疾患の処置及び改善のための化合物及び組成物に関する。具体的には、この発明はトリプターゼ阻害活性を有する化合物、及びその中間体、そうした化合物を含んでなる医薬組成物、及びトリプターゼインヒビターを投与することによって改善することができる状態疾患又は障害[例えば、喘息及び急性黄斑変性症を含めて他の炎症性疾患を含むが、それらには限定されない]に罹患している対象を処置する方法に関する。
【化1】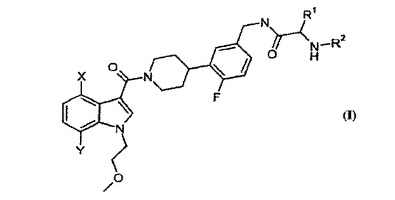
 (もっと読む)
(もっと読む)
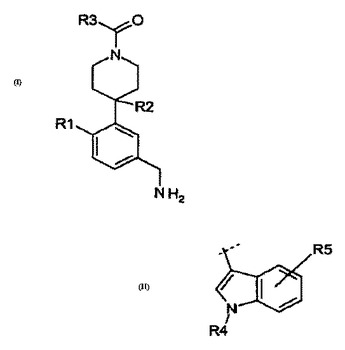
β−トリプターゼインヒビターとしてのインドリル−ピペリジニルベンジルアミン
この発明は、式(I)[ここで、R1、R2及びR3は本明細書中の記載の通りである]の一連の置換インドリル−ピペリジニルベンジルアミンを開示し、請求している。より詳しくは、この発明の化合物はβ−トリプターゼのインヒビターであり、それ故、医薬品として有用である。これに加えて、本発明はまた、置換インドリル−ピペリジニルベンジルアミンの製造方法を開示している。一実施形態では、R3が(II)である式(I)の化合物が提供されている。
【化1】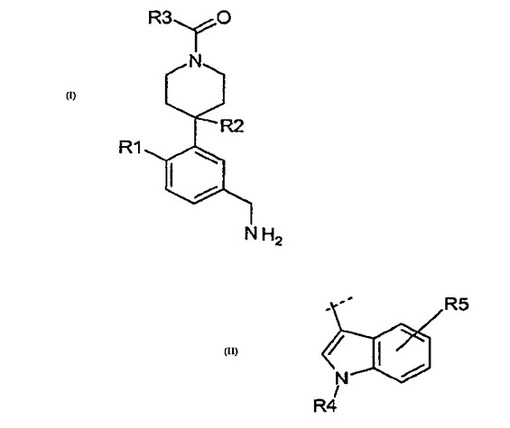
 (もっと読む)
(もっと読む)
GPVIに対する新規アンタゴニスト抗体および該抗体のFabフラグメントならびにこれらの使用
FR2009/113 PCT特許出願 名称 GPVIに対する新規アンタゴニスト抗体および該抗体のFabフラグメントならびにこれらの使用 SANOFI−AVENTIS 要約 本発明は、ヒト血小板膜タンパク質・糖タンパク質VI(GPVI)に特異的に結合する新規抗体および該抗体の一価フラグメントまたは誘導体を開示する。本発明の抗体は、GPVI枯渇表現型を誘導することができる、ハイブリドーマクローン390からの抗体および該抗体のフラグメント抗体である。これらの抗体およびFabフラグメントは、コラーゲン結合を遮断することができ、従って、コラーゲンによる血小板活性化を防止することができる。本発明は、開示する抗体およびFabフラグメントの生産のためのハイブリドーマクローンおよび発現プラスミドにも関する。本発明は、さらに、血栓症および他の血管疾患の治療用の研究、診断および免疫療法薬を製造するための一価抗体フラグメントの使用に関する。本発明は、C末端先端部に分子を有するFabにも関し、およびこのような修飾Fabを使用して抗体によるFabの認識を防止する方法にも関する。本発明は、抗GPVI Fabを使用するときの血小板活性化の防止方法に関する。 (もっと読む)
4−ブロモメチル−[1,1’−ビフェニル]−2’−カルボニトリルの調製方法
一般式(I)の化合物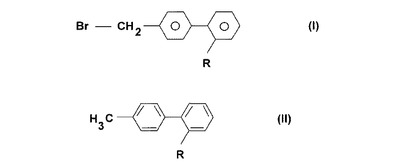
(式中、Rは、シアノ基、−COOR1または−CONR2R3基を示し、ここでR1、R2およびR3は、以下の原子または基:水素原子、直鎖または分枝C1−7アルキル基、C3−6シクロアルキル基から選ばれる同じまたは同じでない置換基;または所定の場合において、C1−4アルキル基もしくはトリフェニルメチル基によって置換されたテトラゾリル基を表す。)を調製する方法であって、一般式(II)の化合物(式中、Rの意味は上で定義した通りである。)を以下の臭素源としての試薬対の1つ:臭素酸塩と亜硫酸水素塩、または臭素酸塩とピロ亜硫酸塩、または臭化物と過炭酸塩、または臭化物と過ホウ酸塩と、水性−有機2元系において反応させること、および場合により一般式(I)で得られた化合物を反応混合物からそれ自体公知の方法によって単離する方法。 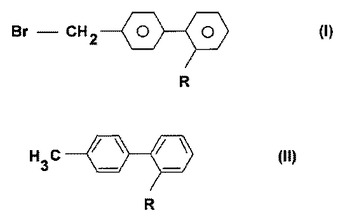 (もっと読む)
(もっと読む)
ドロネダロンの新規な調製方法
本発明の主題は、式IのN−[2−n−ブチル−3−{4−[(3−ジブチルアミノ)プロポキシ]ベンゾイル}−1−ベンゾフラン−5−イル]メタンスルホンアミド(ドロネダロン)の調製のための新規な方法および該調製方法の新しい中間体である。
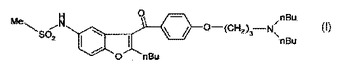 (もっと読む)
(もっと読む)
コンブレタスタチン誘導体調製方法
本発明はコンブレタスタチン誘導体(I)または(II)の調製方法に関し、該方法は以下の工程を含む:トリアリール(3,4,5−トリメトキシベンジル)ホスホニウムハライドP3(III)(式中、Arはフェニルまたはチエニルのうちから選択されるアリール基を示す。)を式(IV)を有するP2または式(V)を有するP’2と、それぞれ式(VI)および(VII)を有する、化合物P4またはP’4がそれぞれ得られるように反応させ:次に、脱保護工程の間、酸および/または塩基の存在下で、P4またはP’4を有する化合物が、任意の精製工程の後、式(I)または(II)を有する化合物を生じる。
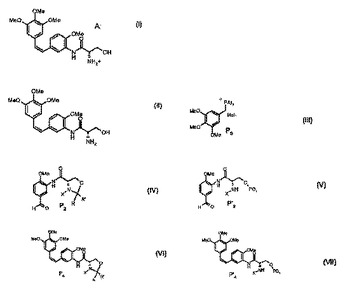 (もっと読む)
(もっと読む)
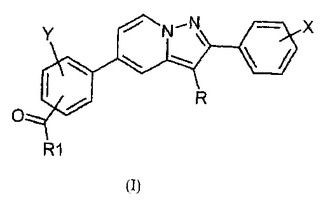
ジフェニル−ピラゾロピリジン誘導体、この誘導体の調製および調節因子ではなく核受容体としてのこの誘導体の使用
本発明は、式(I)に関し、式中、Rは、水素原子もしくはハロゲン原子であるか、または(C1−C6)アルキル基である;Xは、水素原子もしくはハロゲン原子、(C1−C6)アルキル基、ハロ(C1−C6)アルキル基、(C1−C6)アルコキシ基、ハロ(C1−C6)アルコキシ基、シアノ基、ヒドロキシ基、またはヒドロキシ(C1−C6)アルキル基より選択される1つ以上の置換基である;Yは、水素原子もしくはハロゲン原子または(C1−C6)アルキル基である;R1は、NR2R3基またはOR4基である;R2およびR3は、独立して、水素原子、(C1−C6)アルキル基、ヒドロキシ(C1−C6)アルキル基もしくはオキソ(C1−C6)アルキル基であるか、またはR2およびR3は、R2およびR3を支持する窒素原子と共に、(C1−C6)アルキル基、ヒドロキシ基もしくはオキソ基で場合により置換される複素環を形成し;ならびにR4は、(C1−C6)アルキル基、ヒドロキシ(C1−C6)アルキル基、またはオキソ(C1−C6)アルキル基であり、塩基または酸付加塩の状態である。上記式は、NR4A2、NOT、TINUR、RNR−1、およびHZF3としても公知である核受容体Nurr−1に関わる疾患を処理または予防するために治療的に使用され得る。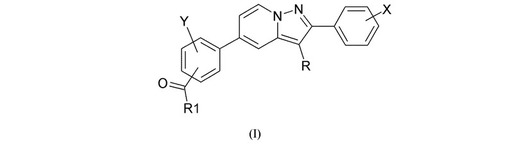
 (もっと読む)
(もっと読む)
N−スクシンイミジルN−ビオチニル−6−アミノカプロエートの調製方法
本発明は、N−スクシンイミジルN−ビオチニル−6−アミノカプロエートの調製方法であって、N−ビオチニル−6−カプロン酸を混合無水物の形態で活性化した後、該混合無水物をN−ヒドロキシスクシンイミドとカップリングさせる工程を含むことを特徴とする方法に関する。 (もっと読む)
41 - 50 / 433
[ Back to top ]